WO2025049450A1 - Assays for detecting bk virus (bkv) - Google Patents
Assays for detecting bk virus (bkv) Download PDFInfo
- Publication number
- WO2025049450A1 WO2025049450A1 PCT/US2024/044000 US2024044000W WO2025049450A1 WO 2025049450 A1 WO2025049450 A1 WO 2025049450A1 US 2024044000 W US2024044000 W US 2024044000W WO 2025049450 A1 WO2025049450 A1 WO 2025049450A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- bkv
- amplification
- microparticles
- seq
- well
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/70—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving virus or bacteriophage
- C12Q1/701—Specific hybridization probes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6806—Preparing nucleic acids for analysis, e.g. for polymerase chain reaction [PCR] assay
Definitions
- BKV BK virus
- Viral infections are a major cause of morbidity and mortality in a diversity of clinical settings including, for example, care of immunocompromised patients and postoperative care of solid organ transplant recipients.
- Chronic immunosuppression required to maintain allograft function post-organ transplant predisposes transplant patients to viral infections. These may arise at each stage post-transplantation.
- Certain infections, such as BK virus (BKV) may occur within months after transplantation.
- BKV infections may be managed by decreasing immunosuppression. Accordingly, methods, compositions, kits and systems for detecting nucleic acid sequences from BKV in samples including, for example, plasma, whole blood and urine samples are needed.
- a set of oligonucleotides comprises at least one first amplification oligonucleotide, at least one second amplification oligonucleotide, and at least one probe oligonucleotide.
- the probe oligonucleotide may comprise a detectable label (e.g., a fluorophore).
- the set of oligonucleotides is for polymerase amplification and detection of BKV in a sample.
- the reagents comprise a group of oligonucleotides comprising one or more sets of oligonucleotides.
- the present invention provides dual-target PCR primer and probe sets for each of BKV nucleic acid sequences.
- the present in invention provides methods for detecting BKV in a plasma or urine sample comprising one or more of: instrument preparation comprising filling an integrated reaction unit with one or more of a lysis buffer and a wash buffer, an Internal Control, 55 pL +/- 20% CuTi microparticles, and 50 pL +/- 20% elution buffer; pretreatment comprising addition of 500pL +/- 20% of a sample to a lysis well at 72°C +/- 20%; lysis comprising transfer of the pretreated sample to a lysis well comprising 1500pL +/- 20% lysis buffer and 3x CuTi microparticles with mixing by a magnetic plunger sheathed with a disposable plastic sheath during incubation; washing comprising transfer of the CuTi microparticles to a wash well comprising 900pL +/- 20% lysis buffer; transfer of the CuTi microparticles to 2 successive washes with 1400pL +/- 20% H2O and lOOpL +/
- the present in invention provides methods for detecting BKV in a plasma or urine sample comprising one or more of: instrument preparation comprising filling an integrated reaction unit with one or more of a lysis buffer and a wash buffer, an Internal Control, CuTi microparticles, and an elution buffer; pretreatment comprising addition of the sample to a lysis well at 72°C; lysis comprising transfer of the pretreated sample to a lysis well comprising lysis buffer and CuTi microparticles with mixing by a magnetic plunger sheathed with a disposable plastic sheath during incubation; washing comprising transfer of the CuTi microparticles to a wash well comprising lysis buffer; transfer of the CuTi microparticles to 2 successive washes with H2O; capture of the CuTi microparticles by the plunger magnet; transfer of the CuTi microparticles to an elution well comprising elution buffer with elution of purified DNA at 80 °C; transfer of the e
- the present invention provides a method for detecting BKV in a plasma or urine sample, comprising: a) transfer of the sample to a lysis well comprising lysis buffer and CuTi microparticles followed by mixing and incubation at 72°C in the well; b) transfer of the CuTi microparticles of step a) to a first wash well comprising lysis buffer; c) transfer of the CuTi microparticles of step b) to a second wash well and washing the microparticles with water; d) capture of the magnetic microparticles of step c) with a plunger magnet; e) transfer of the magnetic microparticles of step d) to an elution well comprising an elution buffer; f) elution of purified DNA from the CuTi microparticles of step e) at 80°C; g) transfer of the purified DNA to a reaction vessel comprising an activator, one or more amplification primers, one or more probes comprising
- the one or more amplification primers comprise one or more of SEQ ID NO: 1, SEQ ID NO: 2, SEQ ID NO: 3, SEQ ID NO: 6, and SEQ ID NO: 7.
- the one or more probes comprise one or more of SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 8, and SEQ ID NO: 9.
- the present invention provides a system, comprising: a) a pipettor; b) an integrated reaction unit (IRU) comprising one or more lanes comprising in linear order of sample transfer: 1) a lysis well comprising an internal control, lysis buffer, CuTi microparticles at 72°C and a magnetic microparticle transfer plunger sheathed with a disposable plastic sheath; 3) a wash well comprising lysis buffer: 4) a first wash well comprising water: 5) a second wash well comprising water;
- IRU integrated reaction unit
- an elution well comprising elution buffer 2 at 80°C; and 7) a PCR reaction vessel comprising activator formulation, Mastermix, and one or more BKV primer pairs and probes; c) a thermocycler: and d) a fluorescence detector.
- the disclosure also provides methods for detecting BKV in a sample.
- the sample may comprise a nasal swab or brush, saliva, mucus, blood, serum, plasma, urine or feces.
- kits for detecting BKV in a sample comprising at least one set of oligonucleotides, any of the oligonucleotides as disclosed herein, reagents for amplifying and detecting nucleic acid sequences, and/or instructions for use.
- kits for detecting BKV in a sample comprising at least one set of oligonucleotides, any of the oligonucleotides as disclosed herein, reagents for amplifying and detecting nucleic acid sequences, and/or instructions for use.
- Figure 1 shows the Alinity m BKV dual target VP2/VP3 and small-t antigen assay design.
- Figures 2A and 2B show BKV in plasma and urine sensitivity results. Each panel member was tested in 10 replicates per day on 3 days with each of 3 AMP kit lots.
- Figures 3 A and 3B show a BKV quantification range from 50 to IxlO 19 lU/mL (1.7 to 9 Log lU/mL). 24 replicates were tested at each target level.
- Figures 4A and 4B show BKV in plasma and urine precision results. Each panel member was tested in multiple replicates twice each day for 5 days in Alinity m Systems by 3 operators using 3 AMP kit lots.
- Figures 5A and 5B show BKV urine specimen quantification and correlation with a comparator. 165 quantified fresh (never frozen) urine samples from transplant recipients were collected at 4 sites.
- Figures 6A and 6B show BKV plasma specimen quantification and correlation with a comparator. 171 quantified fresh and frozen specimens were collected at 6 sites.
- Figure 7 shows BKV urine specimen agreement vs. a comparator. 453 fresh (never frozen) specimens were collected from transplant patients at 4 sites.
- Figure 8 shows BKV plasma specimen evaluation agreement vs. a comparator. 521 fresh and frozen plasma samples were collected at 6 sites.
- Figure 9 shows BKV plasma sample specimen analytical sensitivity evaluation agreement vs. a comparator. Analytical sensitivity was assessed using a panel prepared by diluting a verification panel member to 40 lU/mL (1.60 Log lU/mL) and 50 lU/mL (1.70 Log lU/mL) in negative plasma.
- Figure 10 shows BKV plasma sample specimen linearity. Linearity was evaluated with a commercially available BKV panel in plasma with concentration ranging from 2.00 Log lU/mL to 7.30 Log lU/mL (ExactDx, Ft. Worth, Tx) that was tested at 2 sites.
- Figure 11 shows BKV plasma sample specimen precision. Precision was evaluated by testing panels ranging from 2.00 to 7.30 Log lU/mL across 5 separate runs over 5 days at two sites.
- Figure 12 shows BKV plasma sample specimen regression and mean bias evaluated by comparing the Alinity m BKV IUO with the ELITech MGV Alert BKV run on the Abbott 7712000 using DNA extraction.
- PPA 98.6%
- NPA 91.7%. 4 samples not detected by ELITech were quantitated ⁇ LLOQ by Alinity m BKV
- Figure 13 shows regression analysis of Figure 12 samples.
- Figure 14 shows mean bias analysis of Figure 12samples.
- Figure 16 shows regression analysis of Figure 15 samples.
- Figure 17 shows mean bias analysis of Figure 15 samples.
- Figure 18 shows BKV urine sample specimen regression and mean bias evaluated by comparing the Alinity m BKV IUO with the ELITech MGV Alert BKV run on the Abbott ///2000 using total nucleic acid (TNA) extraction.
- PPA 96.6%
- NPA 81.3%.
- Figure 19 shows regression analysis of Figure 18 samples.
- Figure 20 shows mean bias analysis of Figure 18 samples.
- BKV BK virus
- BKV The BK virus
- BKV The BK virus
- BKV is a polyomavirus with seroprevalence over 90% by 4 years of age.
- Immunocompromised individuals such as transplant recipients and those with other acquired immune system deficiencies are at risk of complications due to BKV reactivation or new infections from BKV positive donors
- BKV causes BKV-associated nephropathy (BKV AN) occurring in up to 10% of kidney transplant recipients and ureteral stenosis.
- BKV-associated hemorrhagic cystitis BK-HC
- BK-HC BKV-associated hemorrhagic cystitis
- BK viruria and viremia precede the histologic diagnosis of polyomavirus- associated nephropathy (PVAN) by 12 weeks and 8 weeks, respectively, screening for BKV replication in urine and plasma I recommended at least every 3 months during the first 2 years after transplantation, or if allograft dysfunction occurs prompting an allograft biopsy for workup of BKV-associated kidney injury.
- PVAN polyomavirus- associated nephropathy
- BKV loads persisting at levels greater than 4 log geq/ml for 4 weeks or longer defines presumptive PVAN for which intervention is considered even in the absence of positive histologic findings.
- BKV genomic variants may emerge that are associated with higher replicative capacity and more extensive cytopathic damage. Because of earlier manifestations, biweekly or monthly screening may be used during the first 6 months as a presumptive intervention strategy.
- the terms “comprise(s)”, “include(s)”, “having”, “has”, “can”, “contain(s)”, and variants thereof are intended to be open-ended transitional phrases, terms, or words that do not preclude the possibility of additional acts or structures.
- the singular forms “a”, “and” and “the” include plural references unless the context clearly dictates otherwise.
- the present disclosure also contemplates other embodiments “comprising,” “consisting of’ and “consisting essentially of,” the embodiments or elements presented herein, whether explicitly set forth or not.
- each intervening number there between with the same degree of precision is explicitly contemplated.
- the numbers 7 and 8 are contemplated in addition to 6 and 9, and for the range 6.0-7.0, the number 6.0, 6.1, 6.2, 6.3, 6.4, 6.5, 6.6, 6.7, 6.8, 6.9, and 7.0 are explicitly contemplated.
- oligonucleotide refers to a short nucleic acid sequence comprising from about 2 to about 100 nucleotides (e.g., about 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 99, or 100 nucleotides, or a range defined by any of the foregoing values).
- nucleic acid and polynucleotide refer to a polymeric form of nucleotides of any length, either ribonucleotides (RNA) or deoxyribonucleotides (DNA).
- RNA and DNA refer to the primary structure of the molecule, and thus include double- and single-stranded DNA, and double- and single-stranded RNA.
- the terms include, as equivalents, analogs of either RNA or DNA made from nucleotide analogs and modified polynucleotides such as, for example, methylated and/or capped polynucleotides.
- Nucleic acids are typically linked via phosphate bonds to form nucleic acid sequences or polynucleotides, though many other linkages are known in the art (e.g., phosphorothioates, boranophosphates, and the like).
- Oligonucleotides can be single-stranded or double-stranded or can contain portions of both double-stranded and single-stranded sequences.
- the oligonucleotide can be DNA, both genomic and complimentary DNA (cDNA), RNA, or a hybrid, where the nucleic acid can contain combinations of deoxyribo- and ribonucleotides, and combinations of bases including uracil, adenine, thymine, cytosine, guanine, inosine, xanthine hypoxanthine, isocytosine and isoguanine.
- Oligonucleotides can be obtained by chemical synthesis methods or by recombinant methods.
- percent sequence identity refers to the percentage of nucleotides or nucleotide analogs in a nucleic acid sequence, or amino acids in an amino acid sequence, that is identical with the corresponding nucleotides or amino acids in a reference sequence after aligning the two sequences and introducing gaps, if necessary, to achieve the maximum percent identity.
- nucleic acid according to the technology is longer than a reference sequence, additional nucleotides in the nucleic acid, that do not align with the reference sequence, are not taken into account for determining sequence identity.
- Methods and computer programs for alignment are well known in the art, including BLAST, Align 2, and FASTA.
- the amplification oligonucleotides of the present disclosure can be of any suitable size, and desirably comprise, consist essentially of, or consist of about 15 to 50 nucleotides, preferably about 20 to 40 nucleotides.
- the oligonucleotides of the present disclosure can contain additional nucleotides in addition to those described herein.
- the amplification oligonucleotides can include, for example, a nicking enzyme site and a stabilizing region upstream (see, e.g., U.S. Patent Nos 9,689,031; 9,617,586; 9,562,264; and 9,562,263, each of which is incorporated herein by reference in its entirety).
- the set of oligonucleotides described herein may be used to amplify and detect one or more target BKV sequences in a sample.
- target sequence and “target nucleic acid” are used interchangeably herein and refer to a specific nucleic acid sequence, the presence or absence of which is to be detected by the disclosed method.
- a target sequence preferably includes a nucleic acid sequence to which one or more oligonucleotides will hybridize and from which amplification will initiate.
- the target sequence can also include a probe-hybridizing region with which a probe may form a stable hybrid under appropriate amplification conditions.
- a target sequence may be single-stranded or double-stranded.
- the target BKV sequence may be within any portion of the BKV genome.
- the set comprises a first amplification oligonucleotide, a second amplification oligonucleotide, and a probe oligonucleotide.
- any of the oligonucleotides described herein may be modified in any suitable manner so as to stabilize or enhance the binding affinity of the oligonucleotide for its target.
- an oligonucleotide sequence as described herein may comprise one or more modified oligonucleotide.
- any of the sequences listed which include internal spacers or modifications may be used without the modifications or spacers.
- any of the oligonucleotides described herein may include, for example, spacers, blocking groups, and modified nucleotides.
- Modified nucleotides are nucleotides or nucleotide triphosphates that differ in composition and/or structure from natural nucleotides and nucleotide triphosphates. Modifications include those naturally occurring that result from modification by enzymes that modify nucleotides, such as methyltransferases. Modified nucleotides also include synthetic or non-naturally occurring nucleotides. For example, modified nucleotides include those with 2' modifications, such as 2'-O-methyl and 2'-fluoro.
- Modified nucleotides or nucleotide triphosphates used herein may, for example, be modified in such a way that, when the modifications are present on one strand of a double-stranded nucleic acid where there is a restriction endonuclease recognition site, the modified nucleotide or nucleotide triphosphates protect the modified strand against cleavage by restriction enzymes.
- Blocking groups or polymerase-arresting molecules are chemical moieties that inhibit target sequence-independent nucleic acid polymerization by the polymerase.
- the blocking group may render the oligonucleotide capable of binding a target nucleic acid molecule, but incapable of supporting template extension utilizing the detectable oligonucleotide probe as a target.
- the presence of one or more moieties that do not allow polymerase progression likely causes polymerase arrest in non-nucleic acid backbone additions to the oligonucleotide or through stalling of a replicative polymerase.
- Oligonucleotides with these moieties may prevent or reduce illegitimate amplification of the probe during the course of the amplification reaction.
- blocking groups include, for example, alkyl groups, non-nucleotide linkers, phosphorothioate, alkane-diol residues, peptide nucleic acid, and nucleotide derivatives lacking a 3'-OH, including, for example, cordycepin, spacer moieties, damaged DNA bases and the like.
- spacers include, for example, C3 spacers. Spacers may be used, for example, within the oligonucleotide, and also, for example, at the ends to attach other groups, such as, for example, labels.
- any of the oligonucleotide sequences described herein may comprise, consist essentially of, or consist of a complement of any of the sequences disclosed herein.
- the terms “complement” or “complementary sequence” as used herein refer to a nucleic acid sequence that forms a stable duplex with an oligonucleotide described herein via Watson-Crick base pairing rules, and typically shares about 80%, about 81%, about 82%, about 83%, about 84%, about 85%, about 86%, about 87%, about 88%, about 89%, about 90%, about 91%, about 92%, about 93%, about 94%, about 95%, about 96%, about 97%, about 98%, or about 99% greater identity with the disclosed oligonucleotide.
- Nucleic acid sequence identity can be determined using any suitable mathematical algorithm or computer software known in the art, such as, for example, CLUSTAL-W, T-Coffee, and ALIGN (for alignment of nucleic acid and amino acid sequences), BLAST programs (e.g., BLAST 2.1, BL2SEQ, and later versions thereof) and FASTA programs (e.g., FASTA3*, FASTM, and SSEARCH) (for sequence alignment and sequence similarity searches). Sequence alignment algorithms also are disclosed in, for example, Altschul et al., J. Molecular Biol., 215(3) 403-410 (1990); Beigert et al., Proc. Natl. Acad. Sci.
- oligonucleotides described herein may be prepared using any suitable method, a variety of which are known in the art (see, for example, Sambrook et al., Molecular Cloning. A Laboratory Manual, 1989, 2. Supp. Ed., Cold Spring Harbour Laboratory Press: New York, N.Y.; M. A. Innis (Ed.), PCR Protocols. A Guide to Methods and Applications, Academic Press: New York, N.Y. (1990); P. Tijssen, Hybridization with Nucleic Acid Probes - Laboratory Techniques in Biochemistry and Molecular Biology (Parts I and II), Elsevier Science (1993); M. A. Innis (Ed.), PCR Strategies, Academic Press: New York, N.Y. (1995); and F.
- Oligonucleotide pairs also can be designed using a variety of tools, such as the Primer-BLAST tool provided by the National Center of Biotechnology Information (NCBI).
- Oligonucleotide synthesis may be performed on oligo synthesizers such as those commercially available from Perkin Elmer/ Applied Biosystems, Inc. (Foster City, CA), DuPont (Wilmington, DE), or Milligen (Bedford, MA).
- oligonucleotides can be custom made and obtained from a variety of commercial sources well-known in the art, including, for example, the Midland Certified Reagent Company (Midland, TX), Eurofins Scientific (Louisville, KY), BioSearch Technologies, Inc. (Novato, CA), and the like.
- Oligonucleotides may be purified using any suitable method known in the art, such as, for example, native acrylamide gel electrophoresis, anion-exchange HPLC (see, e.g., Pearson et al., J. Chrom., 255: 137-149 (1983), incorporated herein by reference), and reverse phase HPLC (see, e.g., McFarland et al., Nucleic Acids Res., 7: 1067-1080 (1979), incorporated herein by reference).
- suitable method known in the art such as, for example, native acrylamide gel electrophoresis, anion-exchange HPLC (see, e.g., Pearson et al., J. Chrom., 255: 137-149 (1983), incorporated herein by reference), and reverse phase HPLC (see, e.g., McFarland et al., Nucleic Acids Res., 7: 1067-1080 (1979), incorporated herein by reference).
- any suitable detectable label that can be conjugated or linked to an oligonucleotide in order to detect binding of the oligonucleotide to a target sequence can be used, many of which are known in the art.
- the detectable label may be detected indirectly.
- Indirectly detectable labels are typically specific binding members used in conjunction with a “conjugate” that is attached or coupled to a directly detectable label. Coupling chemistries for synthesizing such conjugates are well-known in the art and are designed such that the specific binding property of the specific binding member and the detectable property of the label remain intact.
- binding member and “conjugate” refer to the two members of a binding pair, e.g., two different molecules, where the specific binding member binds specifically to the polynucleotide of the present disclosure, and the “conjugate” specifically binds to the specific binding member. Binding between the two members of the pair is typically chemical or physical in nature. Examples of such binding pairs include, but are not limited to, antigens and antibodies, avidin/streptavidin and biotin, haptens and antibodies specific for haptens, complementary nucleotide sequences, enzyme cofactors/substrates and enzymes, and the like.
- Each of the probe oligonucleotide sequences desirably comprises a detectable label.
- Each of the probes may be labeled with the same detectable label or different detectable labels.
- the detectable label may be directly detected.
- directly detectable labels include, for example, radioisotopes, fluorophores, chemiluminophores, enzymes, colloidal particles, fluorescent microparticles, intercalating dyes (e.g., SYBR Green or ethidium bromide), and the like.
- the detectable label may be a fluorophore, such as a fluorescein-family dye, polyhalofluorescein-family dye, hexachlorofluorescein-family dye, coumarin-family dye, rhodamine-family dye, cyanine- family dye, oxazine-family dye, thiazin-family dye, squaraine-family dye, chelated lanthanide-family dye, azo-family dye, triphenylmethane-family dye, or a BODIPY®-family dye.
- a fluorescein-family dye such as a fluorescein-family dye, polyhalofluorescein-family dye, hexachlorofluorescein-family dye, coumarin-family dye, rhodamine-family dye, cyanine- family dye, oxazine-family dye, thiazin-family dye, squaraine-family dye, chelated lanthanide-family dye, azo
- fluorophores examples include, but are not limited to, FAMTM, CAL-FLUOR®, QUASAR®, HEXTM, JOETM, NEDTM, PET®, ROXTM, TAMRATM, TETTM, TEXAS RED®, and VIC®.
- FAMTM FAMTM
- CAL-FLUOR® QUASAR®
- HEXTM JOETM
- NEDTM NEDTM
- PET® ROXTM
- TAMRATM TAMRATM
- TETTM TEXAS RED®
- VIC® VIC®
- Methods for labeling oligonucleotides, such as probes are well-known in the art and described in, e.g., L. J. Kricka, Ann. Clin.
- any one or more of the oligonucleotides described herein may also comprise a quencher moiety.
- a detectable label e.g., a fluorophore
- quencher moiety prevents detection of a signal (e.g., fluorescence) from the detectable label.
- a signal e.g., fluorescence
- the quencher may be selected from any suitable quencher known in the art, such as, for example, BLACK HOLE QUENCHER® 1 (BHQ-1®), BLACK HOLE QUENCHER® 2 (BHQ-2®), BLACK HOLE QUENCHER® 3 (BHQ-3®), IOWA BLACK® FQ, and IOWA BLACK® RQ.
- the oligonucleotide probe may comprise a FAM fluorophore, CAL-FLUOR®, or QUASAR fluorophore and a BHQ-1 or BHQ-2 quencher.
- the present disclosure provides a method for detecting BKV in a sample.
- the method comprises: contacting a sample with the set of oligonucleotides disclosed herein and reagents for amplification; amplifying one or more target BKV nucleic acid sequences present in the sample; hybridizing one or more of the oligonucleotide probes to one or more amplified target BKV nucleic acid sequences; and detecting hybridization of the one or more probe oligonucleotide sequences to the one or more amplified BKV target nucleic acid sequences by measuring a signal from the detectable labels.
- Descriptions of the oligonucleotides set forth herein with respect to the aforementioned set of oligonucleotides also are applicable to the disclosed method.
- the sample can be any suitable sample obtained from any suitable subject, typically a mammal (e.g., dogs, cats, rabbits, mice, rats, goats, sheep, cows, pigs, horses, non -human primates, or humans).
- the subject is a human.
- the sample may be obtained from any suitable biological source, such as, a nasal swab or brush, or a physiological fluid including, but not limited to, whole blood, serum, plasma, interstitial fluid, saliva, ocular lens fluid, cerebral spinal fluid, sweat, urine, milk, ascites fluid, mucous, synovial fluid, peritoneal fluid, vaginal fluid, menses, amniotic fluid, semen, feces, and the like.
- the sample can be obtained from the subject using routine techniques known to those skilled in the art, and the sample may be used directly as obtained from the biological source or following a pretreatment to modify the character of the sample.
- a pretreatment may include, for example, preparing plasma from blood, diluting viscous fluids, filtration, precipitation, dilution, distillation, mixing, concentration, inactivation of interfering components, the addition of reagents, lysing, and the like.
- the sample may be contacted with the set of oligonucleotides comprising amplification oligonucleotides and probes as described herein to form a reaction mixture.
- the reaction mixture is then placed under amplification conditions.
- amplification conditions refers to conditions that promote annealing and/or extension of the amplification oligonucleotides. Amplification conditions encompass all reaction conditions including, but not limited to, temperature and/or temperature cycling, buffer, salt, ionic strength, pH, and the like.
- Amplifying a BKV nucleic acid sequence in the sample can be performed using any suitable nucleic acid sequence amplification method known in the art.
- the amplification includes, but is not limited to, polymerase chain reaction (PCR), reversetranscriptase PCR (RT-PCR), real-time PCR, transcription-mediated amplification (TMA), rolling circle amplification, nucleic acid sequence-based amplification (NASBA), strand displacement amplification (SDA), Transcription-Mediated Amplification (TMA), Single Primer Isothermal Amplification (SPIA), Helicase-dependent amplification (HD A), Loop mediated amplification (LAMP), Recombinase-Polymerase Amplification (RPA), and ligase chain reaction (LCR).
- PCR polymerase chain reaction
- RT-PCR reversetranscriptase PCR
- TMA transcription-mediated amplification
- NASBA nucleic acid sequence-based amplification
- SDA strand displacement amplification
- amplification of BKV nucleic acid sequences is performed using isothermal amplification(e.g., RPA or NEAR). In some embodiments, amplification and detection of BKV nucleic acid sequences is performed using a point of care device (e.g., the ID NOW system (Abbott)).
- a point of care device e.g., the ID NOW system (Abbott)
- amplification of BKV nucleic acid sequences is performed using real-time PCR.
- “Real-time PCR,” as used herein, refers to a PCR method in which the accumulation of amplification product is measured as the reaction progresses, in real time, with product quantification after each cycle, in contrast to conventional PCR in which the amplified DNA product is detected in an end-point analysis.
- Real-time PCR also is known in the art as “quantitative PCR (qPCR).”
- Real-time detection of PCR products typically involves the use of non-specific fluorescent dyes that intercalate with any double-stranded DNA and sequence-specific fluorescently-labeled DNA probes.
- Real-time PCR techniques and systems are known in the art (see, e.g., Dorak, M. Tevfik, ed. Real-time PCR. Taylor & Francis (2007); and Fraga et al., “Real-time PCR,” Current protocols essential laboratory techniques'.
- m2000rt REALTLWETM PCR system (Abbott Molecular, Inc., Des Plaines, IL); CFX Real-Time PCR Detection Systems (Bio-Rad Laboratories, Inc., Hercules, CA); and TAQMANTM Real-Time PCR System (ThermoFisher Scientific, Waltham, MA)), any of which can be employed in the methods described herein.
- sources e.g., m2000rt REALTLWETM PCR system (Abbott Molecular, Inc., Des Plaines, IL); CFX Real-Time PCR Detection Systems (Bio-Rad Laboratories, Inc., Hercules, CA); and TAQMANTM Real-Time PCR System (ThermoFisher Scientific, Waltham, MA)), any of which can be employed in the methods described herein.
- the isothermal amplification methods may rely on nicking and extension reactions, “nicking and extension amplification,” to amplify shorter sequences in a quicker timeframe than traditional amplification reactions.
- nicking and extension amplification may include, for example, reactions that use only two amplification oligonucleotides, one or two nicking enzymes, and a polymerase, under isothermal conditions.
- a target nucleic acid sequence having a sense and antisense strand
- a pair of amplification oligonucleotides is contacted with a pair of amplification oligonucleotides.
- the first amplification oligonucleotide comprises a nucleic acid sequence comprising a recognition region at the 3' end that is complementary to the 3' end of the target sequence antisense strand, a nicking enzyme site upstream of said recognition region, and a stabilizing region upstream of said nicking enzyme site.
- the second amplification oligonucleotide comprises a nucleotide sequence comprising a recognition region at the 3' end that is complementary to the 3' end of the target sequence sense strand, a nicking enzyme site upstream of said recognition region, and a stabilizing region upstream of said nicking enzyme site.
- Two nicking enzymes are provided.
- One nicking enzyme is capable of nicking at the nicking enzyme site of the first amplification oligonucleotide but incapable of nicking within said target sequence.
- the other nicking enzyme is capable of nicking at the nicking enzyme site of the second amplification oligonucleotide but incapable of nicking within said target sequence.
- a DNA polymerase is employed under conditions for amplification which involves multiple cycles of extension of the amplification oligonucleotides thereby producing a double-stranded nicking enzyme site which are nicked by the nicking enzymes to produce the amplification product.
- a DNA polymerase is employed under conditions for amplification which involves multiple cycles of extension of the amplification oligonucleotides thereby producing a double-stranded nicking enzyme site which are nicked by the nicking enzymes to produce the amplification product.
- amplification of BKV nucleic acid sequences is performed using Recombinase-Polymerase Amplification (RPA), which relies on the properties of recombinases and related proteins, to invade double-stranded DNA with single stranded homologous DNA permitting sequence specific priming of DNA polymerase reactions.
- RPA Recombinase-Polymerase Amplification
- a recombinase agent is contacted with first and second amplification oligonucleotides to form nucleoproteins.
- nucleoproteins contact the target sequence to form a first double stranded structure at a first portion of said first strand and form a double stranded structure at a second portion of said second strand so the 3' ends of said first amplification oligonucleotide and the second amplification oligonucleotide are oriented towards each other on the DNA comprising the target sequence.
- the 3' end of the amplification oligonucleotides in the nucleoprotein are extended by DNA polymerases to generate first and second double stranded nucleic acids, and first and second displaced strands of nucleic acid. The steps are repeated until the desired level of amplification is achieved.
- suitable recombinase agents include the E. coli RecA protein, the T4 uvsX protein, or any homologous protein or protein complex from any phyla.
- Other non-homologous recombinase agents may be utilized in place of RecA, for example as RecT, or RecO.
- Suitable recombinase loading proteins may include, for example, T4uvsY, E. coli recO, E. coli recR and derivatives and combinations of these proteins.
- Suitable single stranded DNA binding proteins may be the E. coli SSB or the T4 gp32 or a derivative or a combination of these proteins.
- the DNA polymerase may be a eukaryotic or prokaryotic polymerase.
- Examples of eukaryotic polymerases include pol-a, pol-P, pol-5, pol-s and derivatives and combinations thereof.
- Examples of prokaryotic polymerase include E. coli DNA polymerase I Klenow fragment, bacteriophage T4 gp43 DNA polymerase, Bacillus stearothermophilus polymerase I large fragment, Phi-29 DNA polymerase, T7 DNA polymerase, Bacillus subtilis Pol I, E. coli DNA polymerase I, E. coli DNA polymerase II, E. coli DNA polymerase III, E. coli DNA polymerase IV, E. coli DNA polymerase V and derivatives and combinations thereof.
- RPA Reactive protein kinase
- ATP ATP
- ATP analog ATP
- ATP-P-S ATP-P-S
- ddATP phosphate
- the RPA reaction may also include a system to regenerate ADP from AMP and a to convert pyrophosphate to phosphate (pyrophosphate).
- Suitable crowding agents used in RPA include polyethylene glycol (PEG), dextran and ficoll.
- the disclosed method may further comprise hybridizing one or more of the probe oligonucleotide sequences disclosed herein to the one or more amplified target BKV nucleic acid sequences.
- the method comprises detecting hybridization of the one or more probe oligonucleotide sequences to the one or more amplified target nucleic acid sequences by assessing a signal from each of the detectable labels, whereby (i) the presence of one or more signals indicates hybridization of the one or more probe oligonucleotide sequences to the one or more target BKV nucleic acid sequences and the presence of BKV in the sample, and (ii) the absence of signals indicates the absence of BKV in the sample.
- Detection of signals from the one or more probe oligonucleotide sequences may be performed using a variety of well-known methodologies, depending on the type of detectable label. For example, detection may be done using solution real-time fluorescence or using a solid surface method.
- a subject identified according to the methods described herein as having BKV may be treated, monitored (e.g., for the presence of a BKV nucleic acid determined in a sample from the subject), treated and monitored, and/or monitored and treated using routine techniques known in the art.
- the methods described herein further include treating the subject when the presence of BKV nucleic acid is determined in one or more samples obtained from the subject using the present methods.
- the disclosure also provides a kit for amplifying and detecting BKV in a sample.
- the kit comprises at least one oligonucleotide as described herein.
- the kit comprises a set or group of oligonucleotides described herein.
- the kit may further comprise reagents for amplifying and detecting nucleic acid sequences, and instructions for amplifying and detecting BKV. Descriptions of the oligonucleotides and sets of oligonucleotides set forth herein with respect to the aforementioned methods also are applicable to those same aspects of the kit described herein. Many such reagents are described herein or otherwise known in the art and commercially available.
- suitable reagents for inclusion in the kit include conventional reagents employed in nucleic acid amplification reactions, such as, for example, one or more enzymes having polymerase activity, enzyme cofactors (such as magnesium or nicotinamide adenine dinucleotide (NAD)), salts, buffers, deoxyribonucleotide, or ribonucleotide triphosphates (dNTPs/rNTPs; for example, deoxyadenosine triphosphate, deoxyguanosine triphosphate, deoxycytidine triphosphate, and deoxythymidine triphosphate) blocking agents, labeling agents, and the like.
- enzyme cofactors such as magnesium or nicotinamide adenine dinucleotide (NAD)
- NAD nicotinamide adenine dinucleotide
- salts such as magnesium or nicotinamide adenine dinucleotide (NAD)
- NAD nic
- kits of the present invention comprise one or more of a calibrator, a positive control, a negative control and/or an internal control.
- Activation Reagent was prepared by mixing molecular biology grade water, ProCiin 950, magnesium chloride, and tetramethyl ammonium chloride (TMAC). The activator solution provides reactions with the necessary salts to activate PCR enzymes and establish an ionic strength environment that is conducive to efficient amplification.
- TMAC tetramethyl ammonium chloride
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Immunology (AREA)
- Analytical Chemistry (AREA)
- Molecular Biology (AREA)
- Microbiology (AREA)
- Biotechnology (AREA)
- Physics & Mathematics (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Virology (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
Provided herein are methods, compositions, kits and systems for detecting target nucleic acid sequences from BK virus (BKV).
Description
ASSAYS FOR DETECTING BK VIRUS (BKV)
CROSS REFERENCE TO RELATED APPLICATIONS
This application claims priority to U.S. Provisional Application Serial Numbers 63/534,934, filed August 28, 2023, and 63/555,259, filed February 19, 2024, the entirety each of which is incorporated by reference herein.
SEQUENCE LISTING
The text of the computer readable sequence listing filed herewith, titled “42589- 601_SEQUENCE_LISTING”, created August 27, 2024, having a file size of 11,609 bytes, is hereby incorporated by reference in its entirety.
FIELD
Provided herein are methods, compositions, kits and systems for detecting target nucleic acid sequences from BK virus (BKV).
BACKGROUND OF THE INVENTION
Viral infections are a major cause of morbidity and mortality in a diversity of clinical settings including, for example, care of immunocompromised patients and postoperative care of solid organ transplant recipients. Chronic immunosuppression required to maintain allograft function post-organ transplant predisposes transplant patients to viral infections. These may arise at each stage post-transplantation. Certain infections, such as BK virus (BKV) may occur within months after transplantation. BKV infections may be managed by decreasing immunosuppression. Accordingly, methods, compositions, kits and systems for detecting nucleic acid sequences from BKV in samples including, for example, plasma, whole blood and urine samples are needed.
SUMMARY OF THE INVENTION
Provided herein are methods, compositions, kits and systems for detecting target nucleic acid sequences from BK virus (BKV). The present invention provides reagents, including oligonucleotides, for amplifying and detecting BKV in a sample. In some embodiments, a set of oligonucleotides comprises at least one first amplification oligonucleotide, at least one second amplification oligonucleotide, and at least one probe
oligonucleotide. The probe oligonucleotide may comprise a detectable label (e.g., a fluorophore). In some embodiments, the set of oligonucleotides is for polymerase amplification and detection of BKV in a sample. In some embodiments, the reagents comprise a group of oligonucleotides comprising one or more sets of oligonucleotides. In some embodiments, the present invention provides dual-target PCR primer and probe sets for each of BKV nucleic acid sequences.
In some embodiments, the present in invention provides methods for detecting BKV in a plasma or urine sample comprising one or more of: instrument preparation comprising filling an integrated reaction unit with one or more of a lysis buffer and a wash buffer, an Internal Control, 55 pL +/- 20% CuTi microparticles, and 50 pL +/- 20% elution buffer; pretreatment comprising addition of 500pL +/- 20% of a sample to a lysis well at 72°C +/- 20%; lysis comprising transfer of the pretreated sample to a lysis well comprising 1500pL +/- 20% lysis buffer and 3x CuTi microparticles with mixing by a magnetic plunger sheathed with a disposable plastic sheath during incubation; washing comprising transfer of the CuTi microparticles to a wash well comprising 900pL +/- 20% lysis buffer; transfer of the CuTi microparticles to 2 successive washes with 1400pL +/- 20% H2O and lOOpL +/- 20% H2O, respectively; capture of the CuTi microparticles by the plunger magnet; transfer of the CuTi microparticles to an elution well comprising 50pL +/- 20% elution buffer with elution of purified DNA at 80 °C +/- 20%;transfer of 15.6pL +/- 20% eluate with 5pL +/- 20% activator and 9.4pL +/- 20% Mastermix into a PCR reaction vessel (RV); layering 14pL +/- 20% of vapor barrier on top of the reaction mixture, and capping and transfer for thermocycling and signal detection.
In some embodiments, the present in invention provides methods for detecting BKV in a plasma or urine sample comprising one or more of: instrument preparation comprising filling an integrated reaction unit with one or more of a lysis buffer and a wash buffer, an Internal Control, CuTi microparticles, and an elution buffer; pretreatment comprising addition of the sample to a lysis well at 72°C; lysis comprising transfer of the pretreated sample to a lysis well comprising lysis buffer and CuTi microparticles with mixing by a magnetic plunger sheathed with a disposable plastic sheath during incubation; washing comprising transfer of the CuTi microparticles to a wash well comprising lysis buffer; transfer of the CuTi microparticles to 2 successive washes with H2O; capture of the CuTi microparticles by the plunger magnet; transfer of the CuTi microparticles to an elution well comprising elution buffer with elution of purified DNA at 80 °C; transfer of the eluate with activator and 9.4pL +/- Mastermix into a PCR reaction vessel (RV); layering a vapor barrier
on top of the reaction mixture, and capping and transfer for thermocycling and signal detection.
In some embodiments, the present invention provides a method for detecting BKV in a plasma or urine sample, comprising: a) transfer of the sample to a lysis well comprising lysis buffer and CuTi microparticles followed by mixing and incubation at 72°C in the well; b) transfer of the CuTi microparticles of step a) to a first wash well comprising lysis buffer; c) transfer of the CuTi microparticles of step b) to a second wash well and washing the microparticles with water; d) capture of the magnetic microparticles of step c) with a plunger magnet; e) transfer of the magnetic microparticles of step d) to an elution well comprising an elution buffer; f) elution of purified DNA from the CuTi microparticles of step e) at 80°C; g) transfer of the purified DNA to a reaction vessel comprising an activator, one or more amplification primers, one or more probes comprising a detectable label, and one or more reagents for amplification; i) thermocycling the reaction vessel to generate a signal from the detectable label; and j) detecting the BKV by measuring the signal from the detectable label. In some embodiments, the one or more amplification primers comprise one or more of SEQ ID NO: 1, SEQ ID NO: 2, SEQ ID NO: 3, SEQ ID NO: 6, and SEQ ID NO: 7. In some embodiments, the one or more probes comprise one or more of SEQ ID NO: 4, SEQ ID NO: 5, SEQ ID NO: 8, and SEQ ID NO: 9.
In some embodiments, the present invention provides a system, comprising: a) a pipettor; b) an integrated reaction unit (IRU) comprising one or more lanes comprising in linear order of sample transfer: 1) a lysis well comprising an internal control, lysis buffer, CuTi microparticles at 72°C and a magnetic microparticle transfer plunger sheathed with a disposable plastic sheath; 3) a wash well comprising lysis buffer: 4) a first wash well comprising water: 5) a second wash well comprising water;
6) an elution well comprising elution buffer 2 at 80°C; and 7) a PCR reaction vessel comprising activator formulation, Mastermix, and one or more BKV primer pairs and probes; c) a thermocycler: and d) a fluorescence detector.
The disclosure also provides methods for detecting BKV in a sample. The sample may comprise a nasal swab or brush, saliva, mucus, blood, serum, plasma, urine or feces.
The disclosure further provides kits for detecting BKV in a sample comprising at least one set of oligonucleotides, any of the oligonucleotides as disclosed herein, reagents for amplifying and detecting nucleic acid sequences, and/or instructions for use.
Other aspects and embodiments of the disclosure will be apparent in light of the following detailed description and accompanying figures.
BRIEF DESCRIPTIONS OF THE DRAWINGS
Figure 1 shows the Alinity m BKV dual target VP2/VP3 and small-t antigen assay design.
Figures 2A and 2B show BKV in plasma and urine sensitivity results. Each panel member was tested in 10 replicates per day on 3 days with each of 3 AMP kit lots.
Figures 3 A and 3B show a BKV quantification range from 50 to IxlO19 lU/mL (1.7 to 9 Log lU/mL). 24 replicates were tested at each target level.
Figures 4A and 4B show BKV in plasma and urine precision results. Each panel member was tested in multiple replicates twice each day for 5 days in Alinity m Systems by 3 operators using 3 AMP kit lots.
Figures 5A and 5B show BKV urine specimen quantification and correlation with a comparator. 165 quantified fresh (never frozen) urine samples from transplant recipients were collected at 4 sites.
Figures 6A and 6B show BKV plasma specimen quantification and correlation with a comparator. 171 quantified fresh and frozen specimens were collected at 6 sites.
Figure 7 shows BKV urine specimen agreement vs. a comparator. 453 fresh (never frozen) specimens were collected from transplant patients at 4 sites.
Figure 8 shows BKV plasma specimen evaluation agreement vs. a comparator. 521 fresh and frozen plasma samples were collected at 6 sites.
Figure 9 shows BKV plasma sample specimen analytical sensitivity evaluation agreement vs. a comparator. Analytical sensitivity was assessed using a panel prepared by diluting a verification panel member to 40 lU/mL (1.60 Log lU/mL) and 50 lU/mL (1.70 Log lU/mL) in negative plasma.
Figure 10 shows BKV plasma sample specimen linearity. Linearity was evaluated with a commercially available BKV panel in plasma with concentration ranging from 2.00 Log lU/mL to 7.30 Log lU/mL (ExactDx, Ft. Worth, Tx) that was tested at 2 sites.
Figure 11 shows BKV plasma sample specimen precision. Precision was evaluated by testing panels ranging from 2.00 to 7.30 Log lU/mL across 5 separate runs over 5 days at two sites.
Figure 12 shows BKV plasma sample specimen regression and mean bias evaluated by comparing the Alinity m BKV IUO with the ELITech MGV Alert BKV run on the Abbott
7712000 using DNA extraction. Plasma samples (n=190) were tested with TOR and Alinity m BKV IUO assay. PPA = 98.6%, NPA = 91.7%. 4 samples not detected by ELITech were quantitated <LLOQ by Alinity m BKV
A 2 samples not detected by Alinity m BKV were quantitated <LLOQ by ELITech BKV.
Figure 13 shows regression analysis of Figure 12 samples.
Figure 14 shows mean bias analysis of Figure 12samples.
Figure 15 shows BKV plasma sample specimen regression and mean bias evaluated by comparing the Alinity m BKV IUO with the ELITech MGV Alert BKV run on the Abbott /7/2000 using total nucleic acid (TNA) extraction. Plasma samples (n=170) were tested with TOR and Alinity m BKV IUO assay. PPA = 100.0%, NPA = 84.0%. *Samples detected <LLOQ by Alinity m BKV.
Figure 16 shows regression analysis of Figure 15 samples.
Figure 17 shows mean bias analysis of Figure 15 samples.
Figure 18 shows BKV urine sample specimen regression and mean bias evaluated by comparing the Alinity m BKV IUO with the ELITech MGV Alert BKV run on the Abbott ///2000 using total nucleic acid (TNA) extraction. Urine samples (n=245) were tested with TOR and Alinity m BKV IUO assay. PPA = 96.6%, NPA = 81.3%. *Samples detected by Alinity m BKV with a range <LLOQ to 4.83 Log lU/mL A Samples detected <LLOQ and 5.41 Log lU/mL by ELITech BKV
Figure 19 shows regression analysis of Figure 18 samples.
Figure 20 shows mean bias analysis of Figure 18 samples.
DETAILED DESCRIPTION OF THE INVENTION
Provided herein are methods, compositions, kits and systems for detecting target nucleic acid sequences from BK virus (BKV).
BK virus
The BK virus (BKV) is a polyomavirus with seroprevalence over 90% by 4 years of age. Immunocompromised individuals such as transplant recipients and those with other acquired immune system deficiencies are at risk of complications due to BKV reactivation or new infections from BKV positive donors BKV causes BKV-associated nephropathy (BKV AN) occurring in up to 10% of kidney transplant recipients and ureteral stenosis. In allogeneic hematopoietic stem cell transplant recipients, BKV-associated hemorrhagic cystitis (BK-HC) occurs in up to 25% of pediatric and 54% of adult recipients.
Accordingly, monitoring BKV levels in transplant recipients and other immunocompromised patients plays an important role in their post-transplant and ongoing care. Because BK viruria and viremia precede the histologic diagnosis of polyomavirus- associated nephropathy (PVAN) by 12 weeks and 8 weeks, respectively, screening for BKV replication in urine and plasma I recommended at least every 3 months during the first 2 years after transplantation, or if allograft dysfunction occurs prompting an allograft biopsy for workup of BKV-associated kidney injury. The absence of BK viruria has a high negative predictive value to exclude PVAN. Confirmed BK viruria triggers testing for BK viremia. Increasing plasma BKV loads persisting at levels greater than 4 log geq/ml for 4 weeks or longer defines presumptive PVAN for which intervention is considered even in the absence of positive histologic findings. In patients with persistent high plasma BKV loads, BKV genomic variants may emerge that are associated with higher replicative capacity and more extensive cytopathic damage. Because of earlier manifestations, biweekly or monthly screening may be used during the first 6 months as a presumptive intervention strategy.
Definitions
As used herein, the terms “comprise(s)”, “include(s)”, “having”, “has”, “can”, “contain(s)”, and variants thereof are intended to be open-ended transitional phrases, terms, or words that do not preclude the possibility of additional acts or structures. The singular forms “a”, “and” and “the” include plural references unless the context clearly dictates otherwise. The present disclosure also contemplates other embodiments “comprising,” “consisting of’ and “consisting essentially of,” the embodiments or elements presented herein, whether explicitly set forth or not.
For the recitation of numeric ranges herein, each intervening number there between with the same degree of precision is explicitly contemplated. For example, for the range of 6- 9, the numbers 7 and 8 are contemplated in addition to 6 and 9, and for the range 6.0-7.0, the number 6.0, 6.1, 6.2, 6.3, 6.4, 6.5, 6.6, 6.7, 6.8, 6.9, and 7.0 are explicitly contemplated.
The terms “first” and “second” are used in this disclosure in their relative sense only. It will be understood that, unless otherwise noted, those terms are used merely as a matter of convenience in the description of one or more of the embodiments. The terms “first” and “second” are only used to distinguish one element from another element, and the scope of the rights of the disclosed technology should not be limited by these terms. For example, a first
element may be designated as a second element, and similarly the second element may be designated as the first element.
The term “oligonucleotide,” as used herein, refers to a short nucleic acid sequence comprising from about 2 to about 100 nucleotides (e.g., about 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 99, or 100 nucleotides, or a range defined by any of the foregoing values). The terms “nucleic acid” and “polynucleotide” as used herein refer to a polymeric form of nucleotides of any length, either ribonucleotides (RNA) or deoxyribonucleotides (DNA). These terms refer to the primary structure of the molecule, and thus include double- and single-stranded DNA, and double- and single-stranded RNA. The terms include, as equivalents, analogs of either RNA or DNA made from nucleotide analogs and modified polynucleotides such as, for example, methylated and/or capped polynucleotides. Nucleic acids are typically linked via phosphate bonds to form nucleic acid sequences or polynucleotides, though many other linkages are known in the art (e.g., phosphorothioates, boranophosphates, and the like).
Oligonucleotides can be single-stranded or double-stranded or can contain portions of both double-stranded and single-stranded sequences. The oligonucleotide can be DNA, both genomic and complimentary DNA (cDNA), RNA, or a hybrid, where the nucleic acid can contain combinations of deoxyribo- and ribonucleotides, and combinations of bases including uracil, adenine, thymine, cytosine, guanine, inosine, xanthine hypoxanthine, isocytosine and isoguanine. Oligonucleotides can be obtained by chemical synthesis methods or by recombinant methods.
As used herein, the term “percent sequence identity” refers to the percentage of nucleotides or nucleotide analogs in a nucleic acid sequence, or amino acids in an amino acid sequence, that is identical with the corresponding nucleotides or amino acids in a reference sequence after aligning the two sequences and introducing gaps, if necessary, to achieve the maximum percent identity. Hence, in case a nucleic acid according to the technology is longer than a reference sequence, additional nucleotides in the nucleic acid, that do not align with the reference sequence, are not taken into account for determining sequence identity. Methods and computer programs for alignment are well known in the art, including BLAST, Align 2, and FASTA.
Unless otherwise defined herein, scientific, and technical terms used in connection with the present disclosure shall have the meanings that are commonly understood by those of ordinary skill in the art. The meaning and scope of the terms should be clear; in the event, however of any latent ambiguity, definitions provided herein take precedent over any
dictionary or extrinsic definition. Further, unless otherwise required by context, singular terms shall include pluralities and plural terms shall include the singular.
Exemplary methods and materials are described below, although methods and materials similar or equivalent to those described herein can be used in practice or testing of the present disclosure. All publications, patent applications, patents and other references mentioned herein are incorporated by reference in their entirety. The materials, methods, and examples disclosed herein are illustrative only and not intended to be limiting. The section headings used herein are for organizational purposes only and are not to be construed as limiting the described subject matter in any way.
Amplification and Probe Oligonucleotides
In some embodiments, the oligonucleotides described herein may be used for nucleic acid amplification (e.g., primers) or as probes for nucleic acid hybridization and detection. The terms “primer”, “primer sequence”, “primer oligonucleotide”, and “amplification oligonucleotide” as used herein, refer to an oligonucleotide which is capable of acting as a point of initiation of synthesis of an extension product that is a complementary strand of nucleic acid (all types of DNA or RNA) when placed under suitable amplification conditions (e.g, buffer, salt, temperature and pH) in the presence of nucleotides and an agent for nucleic acid polymerization (e.g., a DNA-dependent or RNA-dependent polymerase). The amplification oligonucleotides of the present disclosure can be of any suitable size, and desirably comprise, consist essentially of, or consist of about 15 to 50 nucleotides, preferably about 20 to 40 nucleotides. The oligonucleotides of the present disclosure can contain additional nucleotides in addition to those described herein. Depending on the type of amplification process employed, the amplification oligonucleotides can include, for example, a nicking enzyme site and a stabilizing region upstream (see, e.g., U.S. Patent Nos 9,689,031; 9,617,586; 9,562,264; and 9,562,263, each of which is incorporated herein by reference in its entirety).
The terms “probe”, “probe sequence”, and “probe oligonucleotide”, refer to an oligonucleotide that can selectively hybridize to at least a portion of a target sequence (e.g, a portion of a target sequence that has been amplified) under appropriate hybridization conditions. In general, a probe sequence is identified as being either “complementary” (e.g., complementary to the coding or sense strand (+)), or “reverse complementary” (e.g., complementary to the anti-sense strand (-)). The probes of the present disclosure can be of
any suitable size, and desirably comprise, consist essentially of, or consist of about 10-50 nucleotides, preferably about 12-35 nucleotides.
As used herein, the terms “set”, “primer set”, “probe set”, and “primer and probe set” refer to two or more oligonucleotides which together are capable of priming the amplification of a target sequence or target nucleic acid of interest (e.g., a target sequence within BKV) and/or at least one probe which can detect the target sequence or target nucleic acid. In certain embodiments, the term “set” refers to a pair of oligonucleotides including a first oligonucleotide that hybridizes with the 5 ’-end of the target sequence or target nucleic acid to be amplified and a second oligonucleotide that hybridizes with the complement of the target sequence or target nucleic acid to be amplified.
The set of oligonucleotides described herein may be used to amplify and detect one or more target BKV sequences in a sample. The terms “target sequence” and “target nucleic acid” are used interchangeably herein and refer to a specific nucleic acid sequence, the presence or absence of which is to be detected by the disclosed method. In the context of the present disclosure, a target sequence preferably includes a nucleic acid sequence to which one or more oligonucleotides will hybridize and from which amplification will initiate. The target sequence can also include a probe-hybridizing region with which a probe may form a stable hybrid under appropriate amplification conditions. A target sequence may be single-stranded or double-stranded. The target BKV sequence may be within any portion of the BKV genome.
In some embodiments, the set comprises a first amplification oligonucleotide, a second amplification oligonucleotide, and a probe oligonucleotide.
Any of the oligonucleotides described herein may be modified in any suitable manner so as to stabilize or enhance the binding affinity of the oligonucleotide for its target. For example, an oligonucleotide sequence as described herein may comprise one or more modified oligonucleotide. Furthermore, any of the sequences listed which include internal spacers or modifications may be used without the modifications or spacers.
Any of the oligonucleotides described herein may include, for example, spacers, blocking groups, and modified nucleotides. Modified nucleotides are nucleotides or nucleotide triphosphates that differ in composition and/or structure from natural nucleotides and nucleotide triphosphates. Modifications include those naturally occurring that result from modification by enzymes that modify nucleotides, such as methyltransferases. Modified nucleotides also include synthetic or non-naturally occurring nucleotides. For example, modified nucleotides include those with 2' modifications, such as 2'-O-methyl and 2'-fluoro.
Other 2'-modified nucleotides are known in the art and are described in, for example U.S. Pat. No. 9,096,897, which is incorporated herein by reference in its entirety. Modified nucleotides or nucleotide triphosphates used herein may, for example, be modified in such a way that, when the modifications are present on one strand of a double-stranded nucleic acid where there is a restriction endonuclease recognition site, the modified nucleotide or nucleotide triphosphates protect the modified strand against cleavage by restriction enzymes.
Blocking groups or polymerase-arresting molecules are chemical moieties that inhibit target sequence-independent nucleic acid polymerization by the polymerase. The blocking group may render the oligonucleotide capable of binding a target nucleic acid molecule, but incapable of supporting template extension utilizing the detectable oligonucleotide probe as a target. For example, the presence of one or more moieties that do not allow polymerase progression likely causes polymerase arrest in non-nucleic acid backbone additions to the oligonucleotide or through stalling of a replicative polymerase. Oligonucleotides with these moieties may prevent or reduce illegitimate amplification of the probe during the course of the amplification reaction. Examples of blocking groups include, for example, alkyl groups, non-nucleotide linkers, phosphorothioate, alkane-diol residues, peptide nucleic acid, and nucleotide derivatives lacking a 3'-OH, including, for example, cordycepin, spacer moieties, damaged DNA bases and the like. Examples of spacers include, for example, C3 spacers. Spacers may be used, for example, within the oligonucleotide, and also, for example, at the ends to attach other groups, such as, for example, labels.
Any of the oligonucleotide sequences described herein may comprise, consist essentially of, or consist of a complement of any of the sequences disclosed herein. The terms “complement” or “complementary sequence” as used herein, refer to a nucleic acid sequence that forms a stable duplex with an oligonucleotide described herein via Watson-Crick base pairing rules, and typically shares about 80%, about 81%, about 82%, about 83%, about 84%, about 85%, about 86%, about 87%, about 88%, about 89%, about 90%, about 91%, about 92%, about 93%, about 94%, about 95%, about 96%, about 97%, about 98%, or about 99% greater identity with the disclosed oligonucleotide. Nucleic acid sequence identity can be determined using any suitable mathematical algorithm or computer software known in the art, such as, for example, CLUSTAL-W, T-Coffee, and ALIGN (for alignment of nucleic acid and amino acid sequences), BLAST programs (e.g., BLAST 2.1, BL2SEQ, and later versions thereof) and FASTA programs (e.g., FASTA3*, FASTM, and SSEARCH) (for sequence alignment and sequence similarity searches). Sequence alignment algorithms also are disclosed in, for example, Altschul et al., J. Molecular Biol., 215(3) 403-410 (1990); Beigert
et al., Proc. Natl. Acad. Sci. USA, 106(10): 3770-3775 (2009), Durbin et al., eds., Biological Sequence Analysis: Probalistic Models of Proteins and Nucleic Acids, Cambridge University Press, Cambridge, UK (2009); Soding, Bioinformatics, 21(1)'. 951-960 (2005); Altschul et al., Nucleic Acids Res., 25(YT): 3389-3402 (1997); and Gusfield, Algorithms on Strings, Trees and Sequences, Cambridge University Press, Cambridge UK (1997), each of which is incorporated herein by reference in its entirety).
The oligonucleotides described herein may be prepared using any suitable method, a variety of which are known in the art (see, for example, Sambrook et al., Molecular Cloning. A Laboratory Manual, 1989, 2. Supp. Ed., Cold Spring Harbour Laboratory Press: New York, N.Y.; M. A. Innis (Ed.), PCR Protocols. A Guide to Methods and Applications, Academic Press: New York, N.Y. (1990); P. Tijssen, Hybridization with Nucleic Acid Probes - Laboratory Techniques in Biochemistry and Molecular Biology (Parts I and II), Elsevier Science (1993); M. A. Innis (Ed.), PCR Strategies, Academic Press: New York, N.Y. (1995); and F. M. Ausubel (Ed.), Short Protocols in Molecular Biology, John Wiley & Sons: Secaucus, N.J. (2002); Narang et al., Meth. Enzymol., 68: 90-98 (1979); Brown et al., Meth. Enzymol., 68: 109-151 (1979); and Belousov et al., Nucleic Acids Res., 25: 3440-3444 (1997), each of which is incorporated herein by reference in its entirety). Oligonucleotide pairs also can be designed using a variety of tools, such as the Primer-BLAST tool provided by the National Center of Biotechnology Information (NCBI). Oligonucleotide synthesis may be performed on oligo synthesizers such as those commercially available from Perkin Elmer/ Applied Biosystems, Inc. (Foster City, CA), DuPont (Wilmington, DE), or Milligen (Bedford, MA). Alternatively, oligonucleotides can be custom made and obtained from a variety of commercial sources well-known in the art, including, for example, the Midland Certified Reagent Company (Midland, TX), Eurofins Scientific (Louisville, KY), BioSearch Technologies, Inc. (Novato, CA), and the like. Oligonucleotides may be purified using any suitable method known in the art, such as, for example, native acrylamide gel electrophoresis, anion-exchange HPLC (see, e.g., Pearson et al., J. Chrom., 255: 137-149 (1983), incorporated herein by reference), and reverse phase HPLC (see, e.g., McFarland et al., Nucleic Acids Res., 7: 1067-1080 (1979), incorporated herein by reference).
The sequence of the oligonucleotides can be verified using any suitable sequencing method known in the art, including, but not limited to, chemical degradation (see, e.g., Maxam et al., Methods of Enzymology, 65: 499-560 (1980), incorporated herein by reference), matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry (see, e.g., Pieles et al., Nucleic Acids Res., 21: 3191-3196 (1993), incorporated
herein by reference), mass spectrometry following a combination of alkaline phosphatase and exonuclease digestions (Wu et al., Anal. Biochem., 290: 347-352 (2001), incorporated herein by reference), and the like.
Detectable Label
Any one or more of the oligonucleotide sequences described herein may comprise a detectable label, such that the amplification oligonucleotide(s) and/or the probe oligonucleotide can be measured. In one embodiment, each of the probe oligonucleotide sequences described herein comprise a detectable label. The term “detectable label” as used herein, refers to a moiety or compound that generates a signal which can be measured and whose intensity is related to (e.g., proportional to) the amount of entity bound thereto. Any suitable detectable label that can be conjugated or linked to an oligonucleotide in order to detect binding of the oligonucleotide to a target sequence can be used, many of which are known in the art. In one embodiment, the detectable label may be detected indirectly. Indirectly detectable labels are typically specific binding members used in conjunction with a “conjugate” that is attached or coupled to a directly detectable label. Coupling chemistries for synthesizing such conjugates are well-known in the art and are designed such that the specific binding property of the specific binding member and the detectable property of the label remain intact. As used herein, “specific binding member” and “conjugate” refer to the two members of a binding pair, e.g., two different molecules, where the specific binding member binds specifically to the polynucleotide of the present disclosure, and the “conjugate” specifically binds to the specific binding member. Binding between the two members of the pair is typically chemical or physical in nature. Examples of such binding pairs include, but are not limited to, antigens and antibodies, avidin/streptavidin and biotin, haptens and antibodies specific for haptens, complementary nucleotide sequences, enzyme cofactors/substrates and enzymes, and the like.
Each of the probe oligonucleotide sequences desirably comprises a detectable label. Each of the probes may be labeled with the same detectable label or different detectable labels.
In some embodiments, the detectable label may be directly detected. Such directly detectable labels include, for example, radioisotopes, fluorophores, chemiluminophores, enzymes, colloidal particles, fluorescent microparticles, intercalating dyes (e.g., SYBR Green or ethidium bromide), and the like. In select embodiments, the detectable label may be a fluorophore, such as a fluorescein-family dye, polyhalofluorescein-family dye,
hexachlorofluorescein-family dye, coumarin-family dye, rhodamine-family dye, cyanine- family dye, oxazine-family dye, thiazin-family dye, squaraine-family dye, chelated lanthanide-family dye, azo-family dye, triphenylmethane-family dye, or a BODIPY®-family dye. Examples of fluorophores include, but are not limited to, FAM™, CAL-FLUOR®, QUASAR®, HEX™, JOE™, NED™, PET®, ROX™, TAMRA™, TET™, TEXAS RED®, and VIC®. One skilled in the art will appreciate that directly detectable labels may require additional components, such as substrates, triggering reagents, light, and the like, to enable detection of the label. Methods for labeling oligonucleotides, such as probes, are well-known in the art and described in, e.g., L. J. Kricka, Ann. Clin. Biochem., 39: 114-129 (2002); van Gijlswijk et al., Expert Rev. Mol. Diagn., 1: 81-91 (2001); Joos et al., J. Biotechnol., 35: 135- 153 (1994); Smith et al., Nucl. Acids Res., 13: 2399-2412 (1985); Connoly et al., Nucl. Acids. Res., 13: 4485-4502 (1985); Broker et al., Nucl. Acids Res., 5: 363-384 (1978); Bayer et al., Methods of Biochem. Analysis, 26: 1-45 (1980); Langer et al., Proc. Natl. Acad. Sci. USA, 78: 6633-6637 (1981); Richardson et al., Nucl. Acids Res., 11: 6167-6184 (1983); Brigati et al., Virol., 126: 32-50 (1983); Tchen et al., Proc. Natl. Acad. Sci. USA, 81: 3466-3470 (1984); Landegent et al., Exp. Cell Res., 15: 61-72 (1984); A. H. Hopman et al., Exp. Cell Res., 169: 357-368 (1987); and Temsamani et al., Mol. Biotechnol., 5: 223-232 (1996), each of which is incorporated herein by reference in its entirety.
In some embodiments, any one or more of the oligonucleotides described herein may also comprise a quencher moiety. When a detectable label (e.g., a fluorophore) and quencher moiety are held in close proximity, such as at the ends of a probe, the quencher moiety prevents detection of a signal (e.g., fluorescence) from the detectable label. When the two moieties are physically separated, the signal becomes detectable. The quencher may be selected from any suitable quencher known in the art, such as, for example, BLACK HOLE QUENCHER® 1 (BHQ-1®), BLACK HOLE QUENCHER® 2 (BHQ-2®), BLACK HOLE QUENCHER® 3 (BHQ-3®), IOWA BLACK® FQ, and IOWA BLACK® RQ. For example, the oligonucleotide probe may comprise a FAM fluorophore, CAL-FLUOR®, or QUASAR fluorophore and a BHQ-1 or BHQ-2 quencher.
The selection of a particular label and labeling technique will depend on several factors, such as the ease and cost of the labeling method, spectral spacing between different detectable labels used, the quality of sample labeling desired, the effects of the detectable moiety on the hybridization reaction (e.g., on the rate and/or efficiency of the hybridization process), the nature of the amplification method used, the nature of the detection system, the nature and intensity of the signal generated by the detectable label, and the like.
Methods for Amplifying and Detecting BKV
The present disclosure provides a method for detecting BKV in a sample. The method comprises: contacting a sample with the set of oligonucleotides disclosed herein and reagents for amplification; amplifying one or more target BKV nucleic acid sequences present in the sample; hybridizing one or more of the oligonucleotide probes to one or more amplified target BKV nucleic acid sequences; and detecting hybridization of the one or more probe oligonucleotide sequences to the one or more amplified BKV target nucleic acid sequences by measuring a signal from the detectable labels. Descriptions of the oligonucleotides set forth herein with respect to the aforementioned set of oligonucleotides also are applicable to the disclosed method.
The sample can be any suitable sample obtained from any suitable subject, typically a mammal (e.g., dogs, cats, rabbits, mice, rats, goats, sheep, cows, pigs, horses, non -human primates, or humans). In some embodiments, the subject is a human. The sample may be obtained from any suitable biological source, such as, a nasal swab or brush, or a physiological fluid including, but not limited to, whole blood, serum, plasma, interstitial fluid, saliva, ocular lens fluid, cerebral spinal fluid, sweat, urine, milk, ascites fluid, mucous, synovial fluid, peritoneal fluid, vaginal fluid, menses, amniotic fluid, semen, feces, and the like.
The sample can be obtained from the subject using routine techniques known to those skilled in the art, and the sample may be used directly as obtained from the biological source or following a pretreatment to modify the character of the sample. Such pretreatment may include, for example, preparing plasma from blood, diluting viscous fluids, filtration, precipitation, dilution, distillation, mixing, concentration, inactivation of interfering components, the addition of reagents, lysing, and the like.
After the sample is obtained from a subject, the sample may be contacted with the set of oligonucleotides comprising amplification oligonucleotides and probes as described herein to form a reaction mixture. The reaction mixture is then placed under amplification conditions. The term “amplification conditions,” as used herein, refers to conditions that promote annealing and/or extension of the amplification oligonucleotides. Amplification conditions encompass all reaction conditions including, but not limited to, temperature and/or temperature cycling, buffer, salt, ionic strength, pH, and the like.
Amplifying a BKV nucleic acid sequence in the sample can be performed using any suitable nucleic acid sequence amplification method known in the art. In some embodiments,
the amplification includes, but is not limited to, polymerase chain reaction (PCR), reversetranscriptase PCR (RT-PCR), real-time PCR, transcription-mediated amplification (TMA), rolling circle amplification, nucleic acid sequence-based amplification (NASBA), strand displacement amplification (SDA), Transcription-Mediated Amplification (TMA), Single Primer Isothermal Amplification (SPIA), Helicase-dependent amplification (HD A), Loop mediated amplification (LAMP), Recombinase-Polymerase Amplification (RPA), and ligase chain reaction (LCR). In some embodiments, amplification of BKV nucleic acid sequences is performed using isothermal amplification(e.g., RPA or NEAR). In some embodiments, amplification and detection of BKV nucleic acid sequences is performed using a point of care device (e.g., the ID NOW system (Abbott)).
In some embodiments, amplification of BKV nucleic acid sequences is performed using real-time PCR. “Real-time PCR,” as used herein, refers to a PCR method in which the accumulation of amplification product is measured as the reaction progresses, in real time, with product quantification after each cycle, in contrast to conventional PCR in which the amplified DNA product is detected in an end-point analysis. Real-time PCR also is known in the art as “quantitative PCR (qPCR).” Real-time detection of PCR products typically involves the use of non-specific fluorescent dyes that intercalate with any double-stranded DNA and sequence-specific fluorescently-labeled DNA probes. Real-time PCR techniques and systems are known in the art (see, e.g., Dorak, M. Tevfik, ed. Real-time PCR. Taylor & Francis (2007); and Fraga et al., “Real-time PCR,” Current protocols essential laboratory techniques'. 10-3 (2008), each of which is incorporated herein by reference in its entirety) and are commercially available from a variety of sources (e.g., m2000rt REALTLWE™ PCR system (Abbott Molecular, Inc., Des Plaines, IL); CFX Real-Time PCR Detection Systems (Bio-Rad Laboratories, Inc., Hercules, CA); and TAQMAN™ Real-Time PCR System (ThermoFisher Scientific, Waltham, MA)), any of which can be employed in the methods described herein.
In select embodiments, the isothermal amplification methods may rely on nicking and extension reactions, “nicking and extension amplification,” to amplify shorter sequences in a quicker timeframe than traditional amplification reactions. These methods may include, for example, reactions that use only two amplification oligonucleotides, one or two nicking enzymes, and a polymerase, under isothermal conditions.
In nicking and extension amplification, a target nucleic acid sequence, having a sense and antisense strand, is contacted with a pair of amplification oligonucleotides. The first amplification oligonucleotide comprises a nucleic acid sequence comprising a recognition
region at the 3' end that is complementary to the 3' end of the target sequence antisense strand, a nicking enzyme site upstream of said recognition region, and a stabilizing region upstream of said nicking enzyme site. The second amplification oligonucleotide comprises a nucleotide sequence comprising a recognition region at the 3' end that is complementary to the 3' end of the target sequence sense strand, a nicking enzyme site upstream of said recognition region, and a stabilizing region upstream of said nicking enzyme site. Two nicking enzymes are provided. One nicking enzyme is capable of nicking at the nicking enzyme site of the first amplification oligonucleotide but incapable of nicking within said target sequence. The other nicking enzyme is capable of nicking at the nicking enzyme site of the second amplification oligonucleotide but incapable of nicking within said target sequence. A DNA polymerase is employed under conditions for amplification which involves multiple cycles of extension of the amplification oligonucleotides thereby producing a double-stranded nicking enzyme site which are nicked by the nicking enzymes to produce the amplification product. For example, see U.S. Patent Nos: 10,851,406; 9,689,031; 9,617,586; 9,562,264; and 9,562,263, and U.S. Patent Application Nos: 15/467,893; 15/600,951; and 16/243,829, each of which is incorporated herein by reference in its entirety.
In select embodiments, amplification of BKV nucleic acid sequences is performed using Recombinase-Polymerase Amplification (RPA), which relies on the properties of recombinases and related proteins, to invade double-stranded DNA with single stranded homologous DNA permitting sequence specific priming of DNA polymerase reactions. In RPA, a recombinase agent is contacted with first and second amplification oligonucleotides to form nucleoproteins. These nucleoproteins contact the target sequence to form a first double stranded structure at a first portion of said first strand and form a double stranded structure at a second portion of said second strand so the 3' ends of said first amplification oligonucleotide and the second amplification oligonucleotide are oriented towards each other on the DNA comprising the target sequence. The 3' end of the amplification oligonucleotides in the nucleoprotein are extended by DNA polymerases to generate first and second double stranded nucleic acids, and first and second displaced strands of nucleic acid. The steps are repeated until the desired level of amplification is achieved.
Methods and materials useful for RPA of a target nucleic acid sequence are known in the art. See U.S. Patent Nos: 7,270,981; 8,460,875; 7,399,590; 7,666,598; 8,030,000; 8,426,134; 8,945,845; 9,663,820; 10,329,603; 10,329,602; 8,017,339; 8,574,846; 8,962,255; 10,036,057; 8,071,308; 10,093,908; 11,339,382; 10,947,584: and 8,637,253, and U.S. Patent Application Nos: 15/099,754; and 14/705,150 each of which is incorporated herein by
reference in its entirety. For example, suitable recombinase agents include the E. coli RecA protein, the T4 uvsX protein, or any homologous protein or protein complex from any phyla. Other non-homologous recombinase agents may be utilized in place of RecA, for example as RecT, or RecO. Suitable recombinase loading proteins may include, for example, T4uvsY, E. coli recO, E. coli recR and derivatives and combinations of these proteins. Suitable single stranded DNA binding proteins may be the E. coli SSB or the T4 gp32 or a derivative or a combination of these proteins. The DNA polymerase may be a eukaryotic or prokaryotic polymerase. Examples of eukaryotic polymerases include pol-a, pol-P, pol-5, pol-s and derivatives and combinations thereof. Examples of prokaryotic polymerase include E. coli DNA polymerase I Klenow fragment, bacteriophage T4 gp43 DNA polymerase, Bacillus stearothermophilus polymerase I large fragment, Phi-29 DNA polymerase, T7 DNA polymerase, Bacillus subtilis Pol I, E. coli DNA polymerase I, E. coli DNA polymerase II, E. coli DNA polymerase III, E. coli DNA polymerase IV, E. coli DNA polymerase V and derivatives and combinations thereof. Other components of RPA include ATP, an ATP analog, or a system for ATP regeneration (convert ADP to ATP). Such system may utilize, for example, phosphocreatine and creatine kinase. The ATP or ATP analog may be any of ATP, ATP-y-S, ATP-P-S, ddATP or a combination thereof. The RPA reaction may also include a system to regenerate ADP from AMP and a to convert pyrophosphate to phosphate (pyrophosphate). Suitable crowding agents used in RPA include polyethylene glycol (PEG), dextran and ficoll.
Following amplification of one or more BKV virus nucleic acid sequences present in the sample, the disclosed method may further comprise hybridizing one or more of the probe oligonucleotide sequences disclosed herein to the one or more amplified target BKV nucleic acid sequences.
Following hybridization of the one or more of the probe oligonucleotide sequences to the one or more amplified target nucleic acid sequences, the method comprises detecting hybridization of the one or more probe oligonucleotide sequences to the one or more amplified target nucleic acid sequences by assessing a signal from each of the detectable labels, whereby (i) the presence of one or more signals indicates hybridization of the one or more probe oligonucleotide sequences to the one or more target BKV nucleic acid sequences and the presence of BKV in the sample, and (ii) the absence of signals indicates the absence of BKV in the sample. Detection of signals from the one or more probe oligonucleotide sequences may be performed using a variety of well-known methodologies, depending on the
type of detectable label. For example, detection may be done using solution real-time fluorescence or using a solid surface method.
Treatment and Monitoring of Subjects Identified as having BKV
A subject identified according to the methods described herein as having BKV may be treated, monitored (e.g., for the presence of a BKV nucleic acid determined in a sample from the subject), treated and monitored, and/or monitored and treated using routine techniques known in the art. In some embodiments, the methods described herein further include treating the subject when the presence of BKV nucleic acid is determined in one or more samples obtained from the subject using the present methods.
Kits
The disclosure also provides a kit for amplifying and detecting BKV in a sample. The kit comprises at least one oligonucleotide as described herein. In some embodiments, the kit comprises a set or group of oligonucleotides described herein. The kit may further comprise reagents for amplifying and detecting nucleic acid sequences, and instructions for amplifying and detecting BKV. Descriptions of the oligonucleotides and sets of oligonucleotides set forth herein with respect to the aforementioned methods also are applicable to those same aspects of the kit described herein. Many such reagents are described herein or otherwise known in the art and commercially available. Examples of suitable reagents for inclusion in the kit (in addition to the oligonucleotides described herein) include conventional reagents employed in nucleic acid amplification reactions, such as, for example, one or more enzymes having polymerase activity, enzyme cofactors (such as magnesium or nicotinamide adenine dinucleotide (NAD)), salts, buffers, deoxyribonucleotide, or ribonucleotide triphosphates (dNTPs/rNTPs; for example, deoxyadenosine triphosphate, deoxyguanosine triphosphate, deoxycytidine triphosphate, and deoxythymidine triphosphate) blocking agents, labeling agents, and the like. Other reagents used in amplification reactions include nicking enzymes, single-strand binding proteins, helicases, resolvases, and the like. In some embodiments, kits of the present invention comprise one or more of a calibrator, a positive control, a negative control and/or an internal control.
The kit may comprise instructions for using the amplification reagents and oligonucleotides described herein, e.g., for processing the test sample, extracting nucleic acid molecules, and/or performing the test; and for interpreting the results obtained. The
instructions may be printed or provided electronically (e.g., DVD, CD, or available for viewing or acquiring via internet resources).
The kit may be supplied in a solid (e.g., lyophilized) or liquid form. The various components of the kit of the present disclosure may optionally be contained within different containers (e.g., vial, ampoule, test tube, flask, or bottle) for each individual component (e.g., amplification oligonucleotides, probe oligonucleotides, or buffer). Each component will generally be suitable as aliquoted in its respective container or provided in a concentrated form. Other containers suitable for conducting certain steps of the amplification/detection assay may also be provided. The individual containers are preferably maintained in close confinement for commercial sale.
The kit may further comprise a swab for obtaining a biological sample. In some embodiments, the kit comprises reagents for gaining access to and/or extracting/isolating nucleic acid from a biological sample.
Systems
In some embodiments, the present invention provides systems for amplifying and detecting BKV in a sample. In some embodiments, the system performs sample preparation, amplification and detection of target nucleic acid sequences using, for example, real-time PCR. In some embodiments, a system operator interacts with a touch screen to select icons, buttons, menu commands, and other screen elements. In some embodiments, a monitor displays a user interface of the system and accepts on-screen /touchscreen selections from the operator. In some embodiments, optional components include a keyboard, a pointing mouse, system information, software function and updates and online help. In some embodiments, the two or more sample preparation steps of the system are executed automatically. In some embodiments, the system comprises sample preparation protocols that are shared between plasma and urine specimens.
In some embodiments, systems of the present invention perform continuous and random-access sample processing using multiple sample processors and thermocyclers in parallel. In some embodiments, an individual sample occupies a single sample lane followed by an associated PCR amplification-detection lane. In some embodiments, parallel lanes provide access to 300 or more samples per day in 2-8 hours from first aspiration to test result. In some embodiments, the system’s capacity incorporates all required reagents in integrated reaction units (IRUs). Samples and controls for a walk away time of 2-4 hours with continuous operator access to remove or replenish amplification reagents, ancillary reagents
and commodities without interfering with system processing. In some embodiments, the system comprises a plurality of Assay Processing Units configured for sample preparation and amplification-detection.
In some embodiments, the system provides individual fluorescent dye intensity changes over PCR cycles. In some embodiments, data reduction comprises a maxRatio algorithm. The maxRatio algorithm evaluates 2 features of assay results: 1) When the Reduced Fluorescence Intensity curves crosses a threshold, the algorithm determines whether a signal corresponds to PCR amplification of the true target’s existence (i.e., whether the PCR is reactive) based on maxRatio’s “peak PCR efficiency” analysis; 2) For rising signals, the algorithm determines whether there is an interference to the PCR (e.g., inhibition of noise) that potentially affects the accuracy of the assay result based on the maxRatio’s “curve shape” analysis. The cycle number (CN) corresponding to the peak maxRatio value (MR) is identified as the Fractional Cycle Number (FCN). Cycle threshold (CT) and FCN are inversely proportional to the Log of the target concentration in the sample prior to amplification. The algorithm provides an additional confirmation of the data’s quality and provides confidence for the reported results.
In some embodiments, systems and kits of the present invention comprise positive controls, negative controls, internal controls (for example, rehydrated from a lypholized unit dose), and calibrators. In some embodiments, systems, kits and methods comprise reagents and components configured to remove PCR inhibitors from sample DNA extract.
EXPERIMENTAL EXAMPLES
Example 1- BKV plasma and urine assay
BKV primers and probes of the present invention amplified and detected dual targets in the BKV genome. Amplification and detection of 2 targets ensures sensitive detection of the viral genome even at low levels. Due to BKV genetic variability, natural occurrences within primer/probe binding sites may result in inefficient hybridization and cause underquantitation and lack of detection. The dual primer/probe design enhances reliability of the BKV assay against new and emerging BKV variants. In addition to the BKV primers and probes, assays of the present invention prove an internal control (IC) primer/probe set for amplification and detection of IC target sequence that is unrelated to BKV and is labeled with a different fluorophore than the BKV probe. Amplification and detection of the internal control assures correct sample processing and assay validity.
Table 15. BKV Target Primers and Probes
 a FAM = Carboxyfluorescein b BHQ1 = Black Hole Quencher 1. The 3’ terminal (T) is part of the BHQ1 structure and is not complementary to the Probe target sequence.
Table 16. Internal Control Primers and Probes
a FAM = Carboxyfluorescein b BHQ1 = Black Hole Quencher 1. The 3’ terminal (T) is part of the BHQ1 structure and is not complementary to the Probe target sequence.
Table 16. Internal Control Primers and Probes
 a Q670 = fluorophore Quasar 670. b BHQ2 = Black Hole Quencher 2. The 3 ' terminal (T) is part of the BHQ2 structure and is not complementary to the IC target sequence.
a Q670 = fluorophore Quasar 670. b BHQ2 = Black Hole Quencher 2. The 3 ' terminal (T) is part of the BHQ2 structure and is not complementary to the IC target sequence.
RT-PCR BKV Mastermix Reagent Formuation
Mastermix Reagent was prepared by combining Platinum II Enzyme and BKV oligonucleotide mix (consisting of molecular biology grade water, PCR buffer components, dNTPs, oligonucleotide primers and probes, Cal Fluor Red 610 passive reference dye and ProCiin 950). The PCR BKV Mastermix Reagent Formulation is shown in Table 17.
Table 17. PCR BKV Mastermix Reagent Formulation

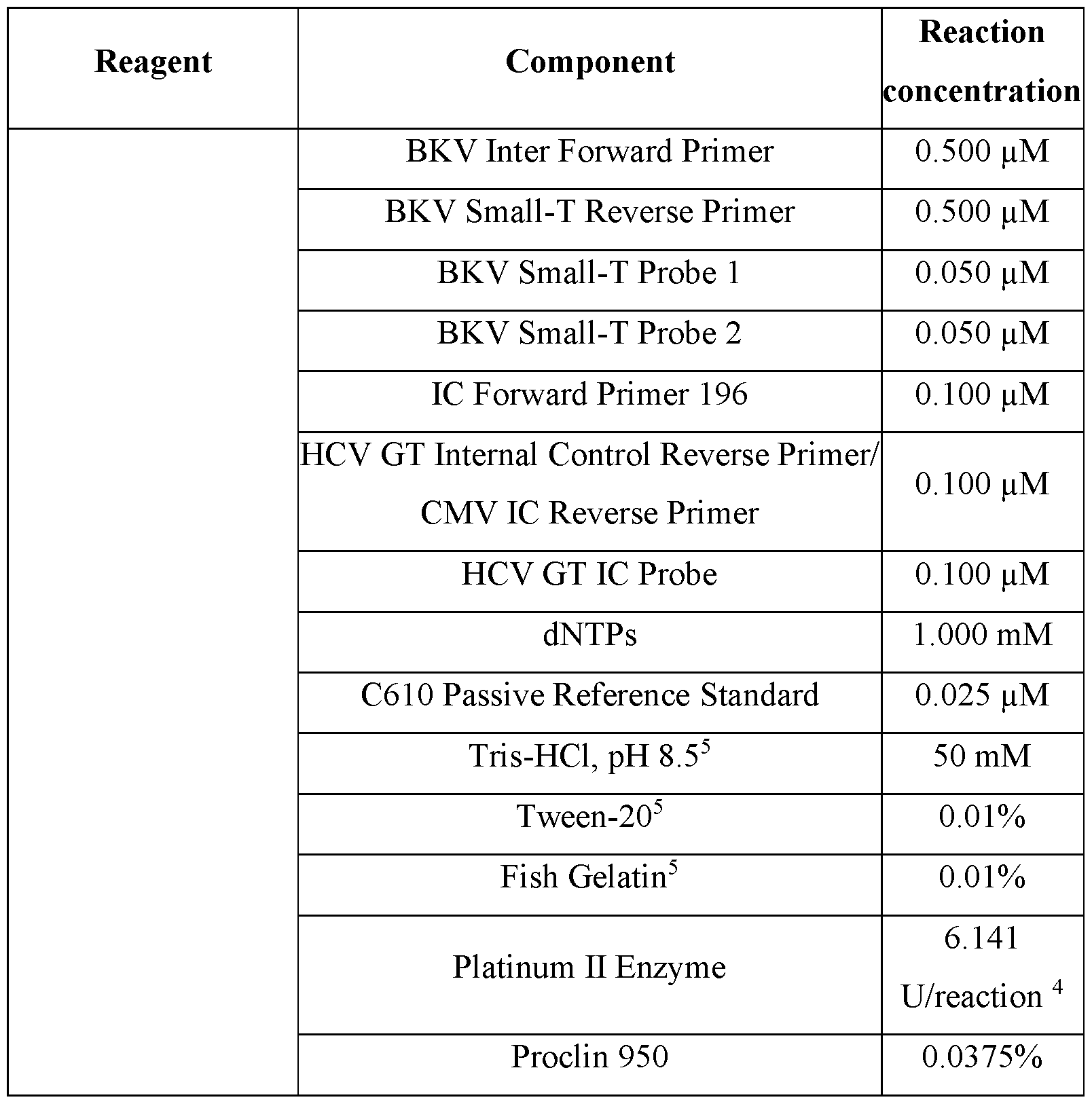
Activation Reagent was prepared by mixing molecular biology grade water, ProCiin 950, magnesium chloride, and tetramethyl ammonium chloride (TMAC). The activator solution provides reactions with the necessary salts to activate PCR enzymes and establish an ionic strength environment that is conducive to efficient amplification. The BKV Activation Reagent Formulation is shown in Table 18.
Table 18. BKV Activation Reagent Formulation
 Internal Control Reagent was prepared by mixing Internal Control plasmid DNA and an IC Diluent (consisting of Tris-based buffer containing salmon testes DNA and Proclin). The internal control target sequence was derived from the hydroxypyruvate reductase gene of the pumpkin plant, Cucurbita pepo, which is unrelated to BKV target sequences. The Internal Control Formulation is shown in Table 19.
Internal Control Reagent was prepared by mixing Internal Control plasmid DNA and an IC Diluent (consisting of Tris-based buffer containing salmon testes DNA and Proclin). The internal control target sequence was derived from the hydroxypyruvate reductase gene of the pumpkin plant, Cucurbita pepo, which is unrelated to BKV target sequences. The Internal Control Formulation is shown in Table 19.
Table 19. Internal Control Formulation:
 The PCR cycling conditions used in the BKV plasma/urine assay are provided in Table 20.
The PCR cycling conditions used in the BKV plasma/urine assay are provided in Table 20.
Table 20. PCR BKV Cycling Conditions
 Table 21. Workflow of BKV Plasma and urine Sample Prep Process
Table 21. Workflow of BKV Plasma and urine Sample Prep Process


To assess BKV plasma and urine assay performance, analytical sensitivity, linearity and precision was evaluated by comparison with a commercially available BKV assay. Regression and mean bias were evaluated comparing Alinity m BKV IUO with the ELITech MGV Alert BKV run on the Abbott /7/2000 using either total nucleic acid (TNA) or DNA extraction. As shown in Figures 9-20, the BKV assay of the present invention had high precision across the dynamic range with good overall correlation when compared with the
ELITech MGB Alert BKV assay thereby supporting the utility of the BKV assay in transplant patient management
It is understood that the foregoing detailed description and accompanying examples are merely illustrative and are not to be taken as limitations upon the scope of the disclosure, which is defined solely by the appended claims and their equivalents.
Various changes and modifications to the disclosed embodiments will be apparent to those skilled in the art and may be made without departing from the spirit and scope thereof.
Claims
1. A method for detecting BKV in a plasma or urine sample, comprising: a) transfer of said sample to a lysis well comprising lysis buffer and CuTi microparticles followed by mixing and incubation at 72°C in said well; b) transfer of said CuTi microparticles of step a) to a first wash well comprising lysis buffer; c) transfer of said CuTi microparticles of step b) to a second wash well and washing said microparticles with water; d) capture of said magnetic microparticles of step c) with a plunger magnet; e) transfer of said magnetic microparticles of step d) to an elution well comprising an elution buffer; f) elution of purified DNA from said CuTi microparticles of step e) at 80°C; g) transfer of said purified DNA to a reaction vessel comprising an activator, one or more amplification primers, one or more probes comprising a detectable label, and one or more reagents for amplification; i) thermocycling said reaction vessel to generate a signal from said detectable label; and j) detecting said BKV by measuring said signal from said detectable label.
2 The method of claim 1, wherein said one or more amplification primers comprise one or more of
SEQ ID NO: 1, SEQ ID NO: 2, SEQ ID NO: 3, SEQ ID NO: 6, and SEQ ID NO: 7.
3 The method of claim 1, wherein said one or more probes comprise one or more of SEQ ID NO:
4 SEQ ID NO: 5, SEQ ID NO: 8, and SEQ ID NO: 9.
4 A system, comprising: a) a pipettor; b) an integrated reaction unit (IRU) comprising one or more lanes comprising in linear order of sample transfer:
1) a lysis well comprising an internal control, lysis buffer, CuTi microparticles at 72°C and a magnetic microparticle transfer plunger sheathed with a disposable plastic sheath;
2) a wash well comprising lysis buffer:
3) a first wash well comprising water;
4) a second wash well comprising water;
5) an elution well comprising elution buffer 2 at 80°C; and
6) a PCR reaction vessel comprising activator formulation, Mastermix, and one or more BKV primer pairs and probes; c) a thermocycler: and d) a fluorescence detector.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363534934P | 2023-08-28 | 2023-08-28 | |
| US63/534,934 | 2023-08-28 | ||
| US202463555259P | 2024-02-19 | 2024-02-19 | |
| US63/555,259 | 2024-02-19 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025049450A1 true WO2025049450A1 (en) | 2025-03-06 |
Family
ID=92762424
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/044000 Pending WO2025049450A1 (en) | 2023-08-28 | 2024-08-27 | Assays for detecting bk virus (bkv) |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025049450A1 (en) |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7270981B2 (en) | 2002-02-21 | 2007-09-18 | Asm Scientific, Inc. | Recombinase polymerase amplification |
| US7399590B2 (en) | 2002-02-21 | 2008-07-15 | Asm Scientific, Inc. | Recombinase polymerase amplification |
| US8030000B2 (en) | 2002-02-21 | 2011-10-04 | Alere San Diego, Inc. | Recombinase polymerase amplification |
| US8071308B2 (en) | 2006-05-04 | 2011-12-06 | Alere San Diego, Inc. | Recombinase polymerase amplification |
| US9096897B2 (en) | 2012-04-09 | 2015-08-04 | Envirologix Inc. | Compositions and methods for quantifying a nucleic acid sequence in a sample comprising a primer oligonucleotide with a 3′-terminal region comprising a 2′-modified nucleotide |
| CN106065420A (en) * | 2016-07-29 | 2016-11-02 | 北京思尔成生物技术有限公司 | The detection method of BK virus, test kit and application thereof |
| US9562263B2 (en) | 2007-07-14 | 2017-02-07 | Ionian Technologies, Inc. | Nicking and extension amplification reaction for the exponential amplification of nucleic acids |
| WO2022232601A1 (en) * | 2021-04-29 | 2022-11-03 | Abbott Laboratories | High throughput nucleic acid testing of biological samples |
| WO2023278529A1 (en) * | 2021-07-01 | 2023-01-05 | Gen-Probe Incorporated | Enzyme formulations and reaction mixtures for nucleic acid amplification |
-
2024
- 2024-08-27 WO PCT/US2024/044000 patent/WO2025049450A1/en active Pending
Patent Citations (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10329603B2 (en) | 2002-02-21 | 2019-06-25 | Alere San Diego Inc. | Recombinase polymerase amplification |
| US10036057B2 (en) | 2002-02-21 | 2018-07-31 | Alere San Diego, Inc. | Recombinase polymerase amplification |
| US7666598B2 (en) | 2002-02-21 | 2010-02-23 | Twistdx, Inc. | Recombinase polymerase amplification |
| US8017339B2 (en) | 2002-02-21 | 2011-09-13 | Alere San Diego, Inc. | Compositions and kits for recombinase polymerase amplification |
| US10947584B2 (en) | 2002-02-21 | 2021-03-16 | Abbott Diagnostics Scarborough, Inc. | Recombinase polymerase amplification |
| US10329602B2 (en) | 2002-02-21 | 2019-06-25 | Alere San Diego, Inc. | Recombinase polymerase amplification |
| US8426134B2 (en) | 2002-02-21 | 2013-04-23 | Alere San Diego, Inc. | Recombinase polymerase amplification |
| US8460875B2 (en) | 2002-02-21 | 2013-06-11 | Alere San Diego, Inc. | Recombinase polymerase amplification |
| US8574846B2 (en) | 2002-02-21 | 2013-11-05 | Alere San Diego, Inc. | Recombinase polymerase amplification |
| US7270981B2 (en) | 2002-02-21 | 2007-09-18 | Asm Scientific, Inc. | Recombinase polymerase amplification |
| US8945845B2 (en) | 2002-02-21 | 2015-02-03 | Alere San Diego Inc. | Recombinase polymerase amplification |
| US8962255B2 (en) | 2002-02-21 | 2015-02-24 | Alere San Diego, Inc. | Recombinase polymerase amplification |
| US7399590B2 (en) | 2002-02-21 | 2008-07-15 | Asm Scientific, Inc. | Recombinase polymerase amplification |
| US9663820B2 (en) | 2002-02-21 | 2017-05-30 | Alere San Diego Inc. | Recombinase polymerase amplification |
| US8030000B2 (en) | 2002-02-21 | 2011-10-04 | Alere San Diego, Inc. | Recombinase polymerase amplification |
| US8071308B2 (en) | 2006-05-04 | 2011-12-06 | Alere San Diego, Inc. | Recombinase polymerase amplification |
| US8637253B2 (en) | 2006-05-04 | 2014-01-28 | Alere San Diego, Inc. | Recombinase polymerase amplification |
| US11339382B2 (en) | 2006-05-04 | 2022-05-24 | Abbott Diagnostics Scarborough, Inc. | Recombinase polymerase amplification |
| US10093908B2 (en) | 2006-05-04 | 2018-10-09 | Alere San Diego, Inc. | Recombinase polymerase amplification |
| US9562263B2 (en) | 2007-07-14 | 2017-02-07 | Ionian Technologies, Inc. | Nicking and extension amplification reaction for the exponential amplification of nucleic acids |
| US9562264B2 (en) | 2007-07-14 | 2017-02-07 | Ionian Technologies, Inc. | Nicking and extension amplification reaction for the exponential amplification of nucleic acids |
| US9689031B2 (en) | 2007-07-14 | 2017-06-27 | Ionian Technologies, Inc. | Nicking and extension amplification reaction for the exponential amplification of nucleic acids |
| US10851406B2 (en) | 2007-07-14 | 2020-12-01 | Ionian Technologies, Llc | Nicking and extension amplification reaction for the exponential amplification of nucleic acids |
| US9617586B2 (en) | 2007-07-14 | 2017-04-11 | Ionian Technologies, Inc. | Nicking and extension amplification reaction for the exponential amplification of nucleic acids |
| US9096897B2 (en) | 2012-04-09 | 2015-08-04 | Envirologix Inc. | Compositions and methods for quantifying a nucleic acid sequence in a sample comprising a primer oligonucleotide with a 3′-terminal region comprising a 2′-modified nucleotide |
| CN106065420A (en) * | 2016-07-29 | 2016-11-02 | 北京思尔成生物技术有限公司 | The detection method of BK virus, test kit and application thereof |
| WO2022232601A1 (en) * | 2021-04-29 | 2022-11-03 | Abbott Laboratories | High throughput nucleic acid testing of biological samples |
| WO2023278529A1 (en) * | 2021-07-01 | 2023-01-05 | Gen-Probe Incorporated | Enzyme formulations and reaction mixtures for nucleic acid amplification |
Non-Patent Citations (33)
| Title |
|---|
| "CFX Real-Time PCR Detection Systems", BIO-RAD LABORATORIES, INC. |
| "PCR Protocols. A Guide to Methods and Applications", 1990, ACADEMIC PRESS |
| "PCR Strategies", 1995, ACADEMIC PRESS |
| "Real-time PCR", 2007, TAYLOR & FRANCIS |
| A. H. HOPMAN ET AL., EXP. CELL RES., vol. 169, 1987, pages 357 - 368 |
| ALTSCHUL ET AL., J. MOLECULAR BIOL., vol. 215, no. 3, 1990, pages 403 - 410 |
| BAYER ET AL., METHODS OF BIOCHEM. ANALYSIS, vol. 26, 1980, pages 1 - 45 |
| BEIGERT ET AL., PROC. NATL. ACAD. SCI. USA, vol. 106, no. 10, 2009, pages 3770 - 3775 |
| BELOUSOV ET AL., NUCLEIC ACIDS RES., vol. 25, no. 17, 1997, pages 3440 - 3444 |
| BRIGATI ET AL., VIROL, vol. 126, 1983, pages 32 - 50 |
| BROKER ET AL., NUCL. ACIDS RES., vol. 5, 1978, pages 363 - 384 |
| CONNOLY ET AL., NUCL. ACIDS. RES., vol. 13, 1985, pages 4485 - 4502 |
| DURBIN ET AL.: "Biological Sequence Analysis: Probalistic Models of Proteins and Nucleic Acids", 2009, CAMBRIDGE UNIVERSITY PRESS |
| FRAGA ET AL.: "Real-time PCR", CURRENT PROTOCOLS ESSENTIAL LABORATORY TECHNIQUES, 2008, pages 10 - 3 |
| GIJLSWIJK ET AL., EXPERT REV. MOL. DIAGN., vol. 1, 2001, pages 81 - 91 |
| GUSFIELD: "Algorithms on Strings, Trees and Sequences", 1997, CAMBRIDGE UNIVERSITY PRESS |
| JOOS ET AL., J. BIOTECHNOL., vol. 35, 1994, pages 135 - 153 |
| L. J. KRICKA, ANN. CLIN. BIOCHEM., vol. 39, 2002, pages 114 - 129 |
| LANDEGENT ET AL., EXP. CELL RES., vol. 15, 1984, pages 61 - 72 |
| LANGER ET AL., PROC. NATL. ACAD. SCI. USA, vol. 78, 1981, pages 6633 - 6637 |
| MASSIMILIANO BERGALLO ET AL: "Evaluation of six methods for extraction and purification of viral DNA from urine and serum samples", NEW MICROBIOLOGICA, vol. 29, no. 2, 1 January 2006 (2006-01-01), IT, pages 111 - 119, XP055426235, ISSN: 1121-7138 * |
| MAXAM ET AL., METHODS OF ENZYMOLOGY, vol. 65, 1980, pages 499 - 560 |
| MCFARLAND ET AL., NUCLEIC ACIDS RES., vol. 7, 1979, pages 1067 - 1080 |
| NARANG ET AL., METH. ENZYMOL., vol. 68, 1979, pages 109 - 151 |
| PEARSON ET AL., J. CHROM., vol. 255, 1983, pages 137 - 149 |
| PIELES ET AL., NUCLEIC ACIDS RES., vol. 21, 1993, pages 3191 - 3196 |
| RICHARDSON ET AL., NUCL. ACIDS RES., vol. 11, 1983, pages 6167 - 6184 |
| SAMBROOK ET AL.: "Molecular Cloning. A Laboratory Manual", vol. 2, 1989, COLD SPRING HARBOUR LABORATORY PRESS |
| SMITH ET AL., NUCL. ACIDS RES., vol. 13, 1985, pages 2399 - 2412 |
| SODING, BIOINFORMATICS, vol. 21, no. 7, 2005, pages 951 - 960 |
| TCHEN ET AL., PROC. NATL. ACAD. SCI. USA, vol. 81, 1984, pages 3466 - 3470 |
| TEMSAMANI ET AL., MOL. BIOTECHNOL., vol. 5, 1996, pages 223 - 232 |
| WU ET AL., ANAL. BIOCHEM., vol. 290, 2001, pages 347 - 352 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8012685B2 (en) | Detection of analytes and nucleic acids | |
| EP2310527B1 (en) | Improved lysis and reverse transcription for mrna quantification | |
| EP2663654A1 (en) | Methods, compositions, and kits for detecting rare cells | |
| CN113508182B (en) | Assays for the detection of human papillomavirus (HPV) | |
| WO2021183941A1 (en) | Looped primer and loop-de-loop method for detecting target nucleic acid | |
| US12018324B2 (en) | Method of detecting minor BCR-ABL1 gene | |
| JP2022501073A (en) | Compositions and Methods for Amplifying or Detecting Varicella-Zoster Virus | |
| EP3628060B1 (en) | Assay for detecting hepatitis c virus (hcv) | |
| US11028439B2 (en) | Assay for detecting hepatitis B virus (HBV) | |
| WO2025049450A1 (en) | Assays for detecting bk virus (bkv) | |
| EP2625285B1 (en) | Method for cell lysis and pcr within the same reaction vessel | |
| WO2025049451A1 (en) | Assays for detecting cytomegalovirus (cmv) or epstein-bar virus (ebv) | |
| US20230313323A1 (en) | Assays for detecting coronavirus disease 2019 (covid-19) | |
| EP4565720A1 (en) | Assays for detecting monkeypox virus | |
| CN104711251B (en) | Helicase-dependent isothermal amplification additive and helicase-dependent isothermal amplification method for sputum nucleic acid | |
| US20170058364A1 (en) | Methods and kits for sequencing and characterizing protozoa | |
| WO2013102061A1 (en) | Actb primers and probes |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24769538 Country of ref document: EP Kind code of ref document: A1 |