WO2025045826A1 - Compounds for activation of fgfr1 signaling - Google Patents
Compounds for activation of fgfr1 signaling Download PDFInfo
- Publication number
- WO2025045826A1 WO2025045826A1 PCT/EP2024/073830 EP2024073830W WO2025045826A1 WO 2025045826 A1 WO2025045826 A1 WO 2025045826A1 EP 2024073830 W EP2024073830 W EP 2024073830W WO 2025045826 A1 WO2025045826 A1 WO 2025045826A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cells
- nmr
- mhz
- mmol
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/81—Amides; Imides
- C07D213/82—Amides; Imides in position 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C281/00—Derivatives of carbonic acid containing functional groups covered by groups C07C269/00 - C07C279/00 in which at least one nitrogen atom of these functional groups is further bound to another nitrogen atom not being part of a nitro or nitroso group
- C07C281/16—Compounds containing any of the groups, e.g. aminoguanidine
- C07C281/18—Compounds containing any of the groups, e.g. aminoguanidine the other nitrogen atom being further doubly-bound to a carbon atom, e.g. guanylhydrazones
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/0068—General culture methods using substrates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/02—Systems containing only non-condensed rings with a three-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/04—Systems containing only non-condensed rings with a four-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2602/00—Systems containing two condensed rings
- C07C2602/36—Systems containing two condensed rings the rings having more than two atoms in common
- C07C2602/38—Systems containing two condensed rings the rings having more than two atoms in common the bicyclo ring system containing five carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/113—Acidic fibroblast growth factor (aFGF, FGF-1)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/999—Small molecules not provided for elsewhere
Definitions
- the present invention relates to compounds of formula (I) for activating the Fibroblast Growth Factor Receptor 1 (FGFR1) signaling pathway. Moreover, it refers to the use of such compounds (I) as a culture media component for the cultivation of living cells or as a medicament for treatment and/or prevention of different diseases.
- FGFR1 Fibroblast Growth Factor Receptor 1
- animal and human cell cultures i.e., the cultivation of cells of animal or human origin in vitro
- industrial productions e.g., of biopharmaceuticals such as, e.g., antibodies, hormones and other cellular products
- stem cell techniques in in vitro fertilization, in preparation of implantable cells, and in the production of cultivated substitutes for meat and other animal-like comestible goods
- animal and human cell cultures are of high interest.
- animal cells including human cells
- stimulation typically proliferate. This is typically achieved by the addition of serum, which is obtained from processed animal blood.
- the amount of serum is typically set to a high level to reach the sufficient content of one or more stimulating growth factors.
- the present invention is, inter alia, based on the surprising finding that compounds of formula (I) and salts thereof as defined in the present invention when added to cell culture significantly enhance proliferation of the cells. It is understood that the compounds of formula (I) are efficient activators of the FGFR1 signalling pathway leading to this proliferation. Accordingly, the compounds of formula (I) are useful for supporting the cultivation of cells. Thereby, the compounds may fully or partly substitute FGF2. Some of these compounds have already been described in the art for different applications (see below).
- the core moiety A is selected from o an optionally substituted C5-18 aromatic group, with 1 to 3 optionally fused rings; wherein 0 to 4 C atoms are replaced by heteroatoms, wherein the heteroatoms are individually selected from N, O, and S; o an optionally substituted Ce-is arylalkyl group wherein 0 to 4 C atoms are replaced by heteroatoms, wherein the heteroatoms are individually selected from N, O, and S, an optionally substituted C3-12 cycloalkyl group, with 1 to 6 rings optionally fused, bridged or strained, wherein 0 to 4 C atoms are replaced by heteroatoms, wherein the heteroatoms are individually selected from N, O, S and Si; o an optionally substituted, straight or branched, saturated or unsaturated C2-20 aliphatic group, wherein 0 to 4 C atoms are replaced by heteroatoms, wherein the heteroatoms are individually selected from N, O, S, and Si; o an aromatic or ali
- linkers Bi and B2 are individually selected from an ester, an ether, an amine, a thioether, an amide; and a sulfonamide;
- tripod moieties Ci and C2 are individually selected from: o an optionally substituted C5-6 aromatic group, wherein 0 to 3 C atoms are replaced by heteroatoms, wherein the heteroatoms are individually selected from N, O, and S; o an optionally substituted C3-6 cycloalkyl group, wherein 0 to 3 C atoms are replaced by heteroatoms, wherein the heteroatoms are individually selected from N, O, and S;
- Ci and C2 may contain one to three substituents, which are independently selected from the group consisting of hydrogen, alkyl and alkoxy,
- a compound of formula (I) or a pharmaceutically acceptable salt thereof may be used in various medical applications, in particular for the treatment of diseases or conditions of patients, which may be treated or ameliorated by activation of the FGFR1 signaling pathway in cells of the patient.
- the invention provides a compound of formula (I) as defined in the first aspect, or a pharmaceutically acceptable salt thereof for use in medical treatment, wherein the medical treatment is a method of wound healing, a method of organ regeneration in transplantation medicine, a method of treating a burned lesion, a disease associated with disturbed cell renewal, or a disease associated with muscle wasting, wherein the treatment or prevention may optionally comprise tissue engineering.
- the invention provides a compound (I) as defined in the first aspect, provided that the compound is not 1-1 , I-8, 1-16, 1-17, I-75, I-76, I- 116 or 1-117.
- the compounds of formula (I) may be used as cell culture medium supplement, such as, e.g., for cultivated meat production.
- nutrients sufficient for cell growth including at least one carbon source and one nitrogen source,
- (C) at least one compound of formula (I) as defined in the first aspect or a pharmaceutically acceptable salt thereof, preferably from in a concentration of 0.05 to 10 pM, preferably in a concentration of 0.1 to 5 pM, more preferably, in a concentration of 0.2 to 3 pM, and
- the present invention also allows efficient cultivation of cells.
- the invention provides a method for the cultivation of cells, comprising the steps of
- step (iii) subjecting the cell culture medium of step (ii) to conditions sufficient for establishing cell growth and/or for cell differentiation;
- the invention provides a food product comprising cultivated cells obtainable by the process according the fifth aspect or material derived therefrom, preferably wherein the food product is cultivated meat or a drinkable composition.
- Fig. 3 Optimization of proliferative activity of 1-1 in NIH/3T3 cells using I-53 as an example. This compound showed an approx. 4.5-fold EC50 improvement over 1-1 from 0.9 to 0.2 pM.
- Fig. 4 Dose-response with 1-1 using the Luciferase reporter assay in HEK293T cells. Upon treatment with 1-1 alone, an increase in luminescence was observed at compound concentrations higher than 3 pM. When treated with 1-1 and FGFR1 inhibitor PD166866 a signal decrease was observed. This indicates that 1-1 works partly through the FGFR1 pathway.
- the x-axis depicts the concentration of 1-1 in pM, the y-axis shows the normalized luminescence signal (Signal/ Negative Control).
- Fig. 5 Determination of FGFR1 dependency in NIH/ 3t3 cells.
- PD166866 did not change the proliferative effect observed for 100 ng/ mL EGF and 2 ng/ mL TGF-pi , underlining the specificity of the observed effect.
- the x-axis depicts the concentration of PD166866 in pM, the y-axis shows the normalized luciferase signal (signal/ negative control).
- Fig. 7 Competition assay between recombinant FGFR1-Fc and cellular FGFR1 for FGF2 or 1-1.
- Recombinant FGFR1-Fc inhibited the proliferative effect of (A) 1.4 pM 1-1 and (B) 100 ng/ mL FGF2, while Fc alone did not have any effect.
- Fig. 8 Determination of 1-1 stability assessed by loss of proliferative activity and compared to FGF2.
- 1-1 was stable over the course of 14 days in the cell culture incubator.
- FGF2 wildtype showed decreased stability over the course of 14 days in the cell culture incubator. This reveals that 1-1 is more stable than FGF2 wildtype at 37°C/ 5% CO2.
- Fig. 9 Measurement of cytotoxicity by LDH release and cell viability by ATP quantification upon treatment with 1-1. A change in the dead and live cell fraction was observed at 30
- the x-axis depicts the concentration of compound in pM in logarithmic scale, the left y-axis shows the cytotoxicity calculated as: (Experimental LDH Release - Medium Background)/ (Maximum LDH Release Control - Medium Background); and the right y-axis show the viability calculated as: Treated cells/ DMSO control.
- the moieties in the compound of the invention may optionally be substituted with one or more substituents, such as are illustrated generally herein, or as exemplified by particular classes, subclasses, and species of the invention. It will be appreciated that the phrase “optionally substituted” is used interchangeably with the phrase “substituted or unsubstituted.” In general, the term “substituted”, whether preceded by the term “optionally” or not, refers to the replacement of hydrogen radicals in a given structure with the radical of a specified substituent.
- an optionally substituted group may have a substituent at each substitutable position of the group, and when more than one position in any given structure may be substituted with more than one substituent selected from a specified group, the substituent may be either the same or different at every position.
- Combinations of substituents envisioned by this invention are preferably those that result in the formation of stable or chemically feasible compounds.
- a substituent connected by a bond drawn from the center of a ring means that the substituent can be bonded to any position in the ring.
- stable refers to compounds that are not substantially altered when subjected to conditions to allow for their production, detection, recovery, purification, and use for one or more of the purposes disclosed herein.
- a stable compound or chemically feasible compound is one that is not substantially altered when kept at a temperature of 40° C. or less, in the absence of moisture or other chemically reactive conditions, for at least a week.
- aliphatic or “aliphatic group”, as used herein, means a straight-chain (i.e., unbranched), branched, or cyclic, substituted or unsubstituted hydrocarbon chain that is completely saturated or that contains one or more units of unsaturation that has a single point of attachment to the rest of the molecule.
- aliphatic groups contain 1-20 aliphatic carbon atoms. In some embodiments, aliphatic groups contain 1-10 aliphatic carbon atoms. In other embodiments, aliphatic groups contain 1-8 aliphatic carbon atoms. In still other embodiments, aliphatic groups contain 1-6 aliphatic carbon atoms, and in yet other embodiments aliphatic groups contain 1-4 aliphatic carbon atoms. Aliphatic groups may be linear or branched, substituted or unsubstituted alkyl, alkenyl, or alkynyl groups.
- Aliphatic groups may also be cyclic or have a combination of linear or branched and cyclic groups. Examples of such types of aliphatic groups include, but are not limited to cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyclohexenyl, CH2-cyclopropyl, CH2CH2CH(CH3)- cyclohexyl.
- cycloaliphatic refers to a monocyclic Ca-Cs hydrocarbon or bicyclic C8-C12 hydrocarbon that is completely saturated or that contains one or more units of unsaturation, but which is not aromatic, that has a single point of attachment to the rest of the molecule wherein any individual ring in said bicyclic ring system has 3-7 members.
- cycloaliphatic groups include, but are not limited to, cycloalkyl and cycloalkenyl groups. Specific examples include, but are not limited to, cyclohexyl, cyclopropenyl, and cyclobutyl.
- heterocycle means nonaromatic, monocyclic, bicyclic, or tricyclic ring systems in which one or more ring members are an independently selected heteroatom.
- the “heterocycle”, “heterocyclyl”, or “heterocyclic” group has three to fourteen ring members in which one or more ring members is a heteroatom independently selected from oxygen, sulfur, nitrogen, or phosphorus, and each ring in the system contains 3 to 7 ring members.
- heterocycles include, but are not limited to, 3-1 H-benzimidazol-2-one,
- 2-imidazolidinyl 4-imidazolidinyl, 5-imidazolidinyl, indolinyl, tetrahydroquinolinyl, tetrahydroisoquinolinyl, benzothiolane, benzodithiane, and
- Cyclic groups (e.g. cycloaliphatic and heterocycles), can be linearly fused, bridged, or spirocyclic.
- heteroatom means one or more of oxygen, sulfur, nitrogen, phosphorus, or silicon (including, any oxidized form of nitrogen, sulfur, phosphorus, or silicon; the quaternized form of any basic nitrogen or a substitutable nitrogen of a heterocyclic ring, for example N (as in 3,4-dihydro-2H-pyrrolyl), NH (as in pyrrolidinyl) or NR+ (as in N-substituted pyrrolidinyl)).
- unsaturated means that a moiety has one or more units of unsaturation.
- unsaturated groups can be partially unsaturated or fully unsaturated. Examples of partially unsaturated groups include, but are not limited to, butene, cyclohexene, and tetrahydropyridine.
- Fully unsaturated groups can be aromatic, anti-aromatic, or non-aromatic. Examples of fully unsaturated groups include, but are not limited to, phenyl, cyclooctatetraene, pyridyl, thienyl, and 1-methylpyridin-2(1 H)-one.
- alkoxy refers to an alkyl group, as previously defined, attached through an oxygen (“alkoxy”) or sulfur (“thioalkyl”) atom.
- haloalkyl refers to alkyl, alkenyl or alkoxy, as the case may be, substituted with one or more halogen atoms. This term includes perfluorinated alkyl groups, such as — CF3 and — CF2CF3.
- halogen means F, Cl, Br, or I.
- aryl used alone or as part of a larger moiety as in “aralkyl”, “aralkoxy”, or “aryloxyalkyl”, refers to monocyclic, bicyclic, and tricyclic ring systems having a total of five to fourteen ring members, wherein at least one ring in the system is aromatic and wherein each ring in the system contains 3 to 7 ring members.
- aryl may be used interchangeably with the term “aryl ring”.
- heteroaryl used alone or as part of a larger moiety as in “heteroaralkyl” or “heteroarylalkoxy”, refers to monocyclic, bicyclic, and tricyclic ring systems having a total of five to fourteen ring members, wherein at least one ring in the system is aromatic, at least one ring in the system contains one or more heteroatoms, and wherein each ring in the system contains 3 to 7 ring members.
- heteroaryl may be used interchangeably with the term “heteroaryl ring” or the term “heteroaromatic”.
- heteroaryl rings include, but are not limited to, 2-furanyl,
- in silico describes experiments or studies conducted through computational methods, using computer software and algorithms to analyze and predict biological, chemical, or physical processes. In silico methods are often employed to complement or guide in vitro and in vivo experiments, providing insights into complex systems or facilitating the design of new inventions. For example, computational simulations of protein interactions or drug docking studies are considered in silico experiments.
- orthosteric refers to the primary or main active site on a receptor or enzyme where a ligand or substrate typically binds.
- Each receptor can be activated by several FGFs. In many cases, the FGFs themselves can also activate more than one receptor (i.e., FGF1 , which binds all seven principal FGFRs (Ornitz et al. 1996). FGF7, however, can only activate FGFR2b (Duchesne et al. 2006), and FGF18 was recently shown to activate FGFR3 (Davidson et al. 2005).
- FGF2 (or b-FGF) is an 18 kD protein and is one of the most studied natural modulators of FGFR1 activity. FGF2 mediates a broad spectrum of mitogenic and pro-survival effects (Nawrocka et al. 2020) and, thus, promotes different cellular phenotypes (e.g. survival, proliferation, migration, invasion, angiogenesis, sternness, maturation) in a multitude of cell types (e.g. stem cells, fibroblasts, skeletal muscle cells, endothelial cells) in a context-dependent manner. FGF2 binding to FGFR1 may lead to conformational changes of FGFR1 that allow receptor dimerization.
- b-FGF is an 18 kD protein and is one of the most studied natural modulators of FGFR1 activity. FGF2 mediates a broad spectrum of mitogenic and pro-survival effects (Nawrocka et al. 2020) and, thus, promotes different cellular phenotypes (e.g
- the compounds of the general formula (I) are composed of core moiety A, which is connected by two linkers Bi and B2 to the two tripod moieties Ci and C2. Each tripod moiety is connected to two active moieties Di and D2 or D3 and D4 respectively.
- the compound of formula 1-1 was discovered via structure-based virtual screening using a proprietary algorithm as a molecule that potentially binds to and activates FGFR1. Based on the in silico study, it is inferred that the guanidines are among the key chemical groups responsible for activity. This is experimentally confirmed in Example 2 showing that a further compound of the formula (I), namely compound 1-17, which has a very different core structure as compared to compound 1-1 , has a comparable effect.
- the active moieties Di, D2, D3 and D4 are H or wherein Di and D2 may not both be hydrogen and D3 and D4 may not both be hydrogen. According to a preferred embodiment, all four the active moieties Di , D2, D3 and D4 are
- the core moiety A is optionally substituted Cs-is aryl, i.e. C 5 , Cg, C7, Cg , C9 , C 10 , C11, C12 , C13 , C14, C15, Ci6, C17, or Cig . with 1 , 2 or 3 optionally fused rings.
- the rings may be fused or bound.
- 0 to 4 C atoms i.e. 0, 1 , 2, 3, or 4 C atoms, may replac-ed by heteroatoms, wherein the heteroatoms are individually selected from N, O, and S.
- the core moiety A is an optionally substituted Cs-saryl wherein 0 to 3 C atoms are replaced by heteroatoms, wherein the heteroatoms are individually selected from N, O, and S.
- the core moiety A is an optionally substituted six-membered aryl or heteroaryl.
- the Cs heteroaryl may have one or two heteroatoms.
- the heteroatom is N.
- the B groups are in meta position of the six-membered aryl or heteroaryl. Accordingly, the B groups are separated by one carbon atom on the six-membered aryl or heteroaryl. This corresponds to the 1 ,3-positions.
- the B groups are in para position of the six-membered aryl or heteroaryl.
- A is a benzene ring.
- the core moiety A is an optionally substituted Ce-is, arylalkyl group, i.e. a Cg, C7, C 8 , C 9 , C10 , On, Ci 2 , C13 , C14, C15, Ci6, C17, or Cig, aralkyl.
- arylalkyl group 0 to 4 C atoms, i.e. 0, 1 , 2, 3, or 4 C atoms, may be replaced by heteroatoms.
- the heteroatoms are individually selected from N, O, and S.
- the aralkyl is a six-membered aryl or heteroaryl ring with two C1-3 alkyl groups attached.
- the alkyl groups may be methyl, ethyl or propyl, preferably methyl.
- A contains two benzene rings.
- A is selected from a dibenzyne, biphenyl, diphenylmethane or diphenyl ether.
- the core moiety A is an optionally substituted C3-12 cycloalkyl group i.e. a C 3 , C 4 , C 5 , C 6 , C 7 , C 8 , C 9 , C 10 , CH, or C 12 , with 1 to 6 rings, i.e. 1 , 2, 3, 4, 5, or 6 rings.
- the rings may optionally be fused, bridged or strained.
- 0 to 4 C atoms i.e. 0, 1 , 2, 3, or 4 C atoms, may be replaced by heteroatoms,
- the heteroatoms may be individually selected from N, O, S and Si.
- the core moiety A is an optionally substituted C3-6 cycloalkyl, i.e. an optionally substituted cyclopropane, cyclobutane, cyclohexane or cyclopentane.
- the C3-6 cycloalkyl may be bridged.
- 0 to 1 C atoms may be replaced by a heteroatom.
- the heteroatoms are individually selected from N, O, and S.
- the C3-6 cycloalkyl does not contain heteroatoms.
- the core moiety A is an optionally substituted, straight or branched, saturated or unsaturated C2-20 aliphatic group, i.e. C 2 , C3, C 4 , C5, C 6 , C 7 , C 8 , C 9 , C10 , C11, C12 , C13 , C14, Cis, Ci6, C17, Cig, Ci9, or C20.
- C2-20 aliphatic group i.e. C 2 , C3, C 4 , C5, C 6 , C 7 , C 8 , C 9 , C10 , C11, C12 , C13 , C14, Cis, Ci6, C17, Cig, Ci9, or C20.
- 0 to 4 C atoms i.e. 0, 1 , 2, 3, or 4 C atoms, are replaced by heteroatoms.
- the heteroatoms are individually selected from N, O, S, and Si.
- the C2-20 aliphatic group is saturated.
- the core moiety A is an optionally substituted, straight or branched, saturated or unsaturated C2-12 aliphatic group, wherein 0 to 4 C atoms are replaced by heteroatoms, wherein the heteroatoms are individually selected from N, O, and S.
- the C2-12 aliphatic group is saturated.
- the C2-12 aliphatic group is straight.
- a substituent is in ortho position to Bi or B 2 .
- a substituent is in ortho position to Bi and B 2 .
- first substituent is in meta position to Bi and B 2 .
- the core moiety A is an aromatic or aliphatic crown ether.
- the core moiety A is an aromatic crown ether.
- the crown ether may be a 18-crown-6 ether.
- the core moiety A is optionally substituted, which means that the core moiety A may contain one to three substituents, which are independently selected from the group consisting of halogen, amide, amine, nitro, cyano, hydroxyl or hydrocarbyloxy, or aldehyde, ketone, cycloalkyl, carboxyl, ether, ester, optionally halogenated alkyl, alkenyl, alkenylaryl, alkenyl aryl, alkynyl, alkoxy, alkylthio, sulfonyl, sulfonylamide.
- A may contain one to three substituents, which are independently selected from the group consisting of halogen, hydrogen, hydroxyl, methyl, methoxy, optionally halogenated Ci- 6 alkyl, optionally halogenated Ci- 6 alkoxy, C5-6 cycloalkyl, in which 0 to 2 C atoms are replaced by heteroatoms and amine.
- the substituent is a polyether.
- the substituent is selected from: wherein X is N, O, S, preferably O.
- A contains three halogen substituents.
- the halogens are F.
- the Ci- 6 alkyl is selected from methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, pentyl, isopentyl, neopentyl, hexyl, isohexyl, neohexyl.
- the substituent is a tert-butyl, preferably in meta position to both Bi and B2.
- the Ci- 6 alkyl is halogenated by one to three of fluorine, chlorine, bromine or iodine.
- the halogenated alkyl is trifluoromethyl, preferably in meta position to both Bi and B2.
- the halogenated alkyl is difluoromethyl, preferably in meta position to both Bi and B2.
- the halogenated alkoxy is trifluoromethoxy, preferably in meta position to both Bi and B2.
- the substituent is morpholine, preferably in ortho or meta postion to Bi.
- the alkylthio group is methylthio, preferably in meta position to both Bi and B2.
- the sulfonyl group is methylsulfonyl, preferably in meta position to both Bi and B2.
- the substituent is styryl, preferably in ortho or meta position to Bi.
- the cycloal kylethynyl is selected from cyclohexylethynyl or a cyclopropylethynyl and piperidineethynyl, preferably in meta position to both Bi and B2.
- the alkynyl is methylacetylene, preferably in meta position to both Bi and B2.
- the core moiety A is selected from the following residues (the dashed lines indicate the bonds to Bi and B2):
- X may be N, O, or S.
- X is N.
- Rs may be O, S, or NRTM, with R w being H or C1-6 alkyl.
- Ra is NRTM.
- R3 is H.
- R2 may be H, or C1-6 alkyl.
- the amine NR5R6 may be selected from:-
- R? may be halogen, methyl, methoxy, tert-butyl, NO2, CN, CF3, OCF3, CF2, COOH, COOCH3, SCH 3 , CO2NH2, N(CH 3 ) 2 , NH 2 , SO2CH3, NHCOOCH3, NHSO 2 CH3, and NHSO2CF3, and CONR11R12 with R11 and R12 being independently selected from H and C1-6 alkyl.
- Rn and R12 are both H.
- Rn and/or R12 are methyl.
- Rs may be H or phenyl. Preferably, Rs is H.
- R9 may be methyl, Cs-Cs cycloalkyl, piperidinyl or hydroxyisopropyl.
- Rg is methyl.
- Xi may be oxygen or methylene.
- Xi is oxygen.
- A is not a polyether. According to one embodiment, A is not selected from: wherein X is N, O, S, preferably O.
- At least one of Ci and C2 and preferably both of Ci and C2 are both pyridinyl groups.
- the compound of formula (I) is selected from 1-1 , I-2, I-3, I-4, I-5, I-6, I-7, I-8, I-9, 1-10, 1-11 , 1-15, 1-16, 1-17, 1-18, 1-19, I-20, I-22, I-23, I-24, I-25, I-26, I-27, I-28, I-29, I-30, 1-31 , I-32, I-33, I-34, I-35, I-36, I-37, I-38, I-39, I-40, 1-41 , I-42, I-43, I-44, I-45, 1-46, 1-47, 1-48, 1-49, 1-50, 1-51 , I-52, I-53, I-54, I-55, I-56, I-57, I-58, I-59, I-60, 1-61 , I-62, I-63, I-64, I-65, I-66, I-67, I-68, I-69, I-70, 1-71
- a pharmaceutically acceptable salt may be understood in the broadest sense as any salt of the respective compound of formula (I) that is reasonably usable in a pharmaceutical context.
- pharmaceutically acceptable does not necessarily mean that the respective component is indeed used in a pharmaceutical context. This rather refers to the suitability to do so.
- a pharmaceutically acceptable salt or other component or composition will typically also be inherently usable as being cosmetically acceptable.
- a pharmaceutically acceptable salt or other component or composition typically bears a low toxicity and can be administered to a human or non-human animal (typically mammal or avian) body without seriously harming this human or non-human animal as well as to a cell culture without seriously harming this cell culture, when administered in reasonable concentrations as used for the compound of formula (I).
- a concentration higher than 10 pM could lead to wastage of expensive reagents without necessarily improving cell culture outcomes.
- a concentration below 0.05 pM might not stimulate enough cell growth and proliferation.
- the compound of formula (I) is present in a concentration of 0.1 to 5 pM. Maintaining concentrations within a defined range can be cost-effective as it ensures that the appropriate amount of growth factor is used - not too much (wasteful) or too little (ineffective)
- the concentration of the compound of formula (I) is 0.2 to 3 pM. A concentration in this range ensures that cells have sufficient quantities of the growth factor to promote cell proliferation and growth.
- the actual concentration of the compound of formula (I) can depend on various factors, including the intrinsic factors such as cell type, genetic factors, age and passage number, and health status and extrinsic factors such as nutrient availability, oxygen levels, temperature, CO2 concentration, culture vessel and surface cell density. Therefore, the most suitable concentration can be chosen based on these considerations to optimize the therapeutic effects of the compound.
- the compounds of the invention may be prepared as described in Example 1. Furthermore, compounds 1-1 , I-8, 1-16 1-17, 1-117 and their methods of synthesis have been described in US 5,599,984 A, which is incorporated herein by reference. Compounds I-75, I-76 and 1-116 as well as their methods of synthesis have been described in Schroder et al. 2016, which is incorporated herein by reference. Compound 1-117 and its method of synthesis has been described in US 5,750,573, which is incorporated herein by reference.
- the compounds of formula (I) may be used for treatment of diseases or conditions of patients, which may be treated or ameliorated by activation of the FGFR1 signaling pathway in cells of the patient. This activation leads to an increase in cell proliferation in the patient.
- the compounds may in particular be useful for the treatment of the following disorders of conditions:
- Osteoporosis is characterized by a reduction in bone density due to an imbalance in the bone remodeling process. Compounds that promote the proliferation of osteoblasts (bone-forming cells) can potentially restore this balance, leading to increased bone formation and improved bone density. In osteoporotic patients, fractures are a significant concern. Enhancing cell proliferation might also accelerate the fracture healing process. FGFR1 signaling has a role in bone formation and maintenance. Increasing its signaling could potentially promote osteoblast activity, which may be beneficial in conditions of decreased bone density or osteoporosis.
- FGFR1 signaling has been implicated in the survival and regeneration of certain neuron populations. Hence, enhancing its activity could be explored in the context of promoting neural regeneration or protecting neurons in diseases like Parkinson's. In conditions like Parkinson's disease, Alzheimer's disease, or Huntington's disease, where specific neuronal populations are lost, stimulating the proliferation of neural progenitor cells or specific subsets of neurons might have therapeutic benefits. In the case of nerve injuries or certain degenerative conditions like peripheral neuropathies, compounds that promote the proliferation of neurons or supporting cells (like Schwann cells) can aid in neural tissue regeneration.
- the compounds of formula (I) that increase FGFR1 signaling will have a role in enhancing organ regeneration or repair in transplantation medicine.
- FGFR1 signaling can promote angiogenesis, which is the formation of new blood vessels. Compounds that enhance this signaling pathway could potentially speed up the process of revascularization in the transplanted organ. Stimulating cell proliferation and differentiation: Proper organ function often requires the growth and differentiation of specific cell types. FGFR1 signaling can support the proliferation of progenitor cells and guide the differentiation of certain cell lineages, aiding in tissue repair and regeneration. Reduction of fibrosis: FGFR signaling might help reduce tissue fibrosis, a process where normal tissue is replaced with scar tissue.
- fibrosis In the context of organ transplantation, controlling fibrosis can be crucial for the long-term success of the transplant, as excessive fibrosis can impair organ function. Protection against ischemia-reperfusion injury: This type of injury can occur when blood supply returns to the tissue after a period of lack of oxygen (ischemia). It's a significant concern in organ transplantation. FGFR signaling has the potential to protect tissues against such injuries.
- Pre-transplant Organ Cultivation For organs grown ex vivo (outside the body), such as in bioengineered organs or tissues, promoting cell proliferation can accelerate the growth and maturation of the organ, making it suitable for transplantation sooner.
- Posttransplant Organ Integration After an organ is transplanted, it is crucial that it integrates well with the recipient's body. Compounds that promote cell proliferation could aid in the faster establishment and growth of the transplanted organ, improving its function and longevity.
- Enhancing FGFR1 signaling will have therapeutic effects in the contexts of burned lesions: Wound Healing: FGFR signaling plays roles in promoting tissue repair and wound healing. Upregulating FGFR1 signaling could potentially accelerate the healing process in burn wounds. Promoting cell proliferation can accelerate the healing process of the burn wound by enhancing tissue regeneration. Angiogenesis: Restoration of blood flow to burned tissue is essential for healing. FGFR1 can promote angiogenesis (formation of new blood vessels), which may speed up revascularization of the affected tissue. Reduction of Scarring: FGFR signaling might help modulate the wound healing response in a manner that reduces excessive fibrosis and scarring, which are common complications of severe burns.
- Promoting cell proliferation can by facilitating a more efficient regenerative process, the formation of scar tissue might be minimized.
- Enhancing FGFR1 signaling will have a therapeutic effect in the contexts of diseases with disturbed cell renewal: Skin disorders: For conditions like psoriasis, where there is disturbed skin cell renewal, modulating FGFR1 signaling could help normalize the skin cell growth cycle. Moreover, conditions like psoriasis or eczema can benefit from compounds that promote skin cell proliferation, leading to the renewal and repair of skin layers.
- Hair Growth FGFR signaling is involved in hair follicle regulation. If disturbances in hair growth are related to compromised FGFR signaling, enhancing this pathway might be beneficial. Certain hair loss disorders result from a disturbance in hair follicle cell proliferation. Promoting cell growth can potentially stimulate hair regrowth.
- Enhancing FGFR1 signaling will have a therapeutic effect in the contexts of Diseases associated with muscle wasting.
- Promotion of Muscle Cell Growth FGFR1 signaling has roles in muscle cell differentiation and proliferation. In diseases such as muscular dystrophy or in conditions like cachexia (seen in some cancer patients), increasing FGFR1 signaling could potentially promote muscle regeneration. Promoting muscle cell proliferation can potentially counteract muscle atrophy seen in diseases like muscular dystrophy or conditions like cachexia. Stimulation of satellite cells: Satellite cells are muscle stem cells that play a crucial role in muscle repair. Compounds that promote their proliferation can be beneficial for muscle recovery. FGFR1 signaling might help in the activation and differentiation of these cells, aiding muscle repair and regeneration.
- the invention provides a compound of formula (I) as defined in the first aspect, or a pharmaceutically acceptable salt thereof for use in medical treatment, wherein the medical treatment is a method of wound healing, a method of organ regeneration in transplantation medicine, a method of treating a burned lesion, a disease associated with disturbed cell renewal, or a disease associated with muscle wasting, wherein the treatment or prevention may optionally comprise tissue engineering.
- the second aspect relates to a method of treatment wherein the medical treatment is a method of wound healing, a method of treating osteoporosis, a method of organ regeneration in transplantation medicine, a method of treating a burned lesion, a disease associated with disturbed cell renewal, or a disease associated with muscle wasting, wherein the treatment or prevention may optionally comprise tissue engineering.
- a medicament of the present invention may have any galenic form.
- a medicament may be in any dosage for e.g., an ingestible composition (e.g., a pill, a dragee, a syrup, a drinkable liquid), an inhalable composition (e.g., a spray), a dosage form penetrating the skin (e.g., a cream, a lotion, a plaster, a suppository, an eye drop, a spray), or an injectable composition. It is preferably (essentially) sterile and is preferably a-pyrogenic.
- the patient suffers from a disease associated with (dysregulated) cellular growth and/or FGF-mediated (in particular FGF2-mediated) activity, and/or disturbed FGFR signaling (in particular FGFR1 signaling).
- a disease associated with (dysregulated) cellular growth and/or FGF-mediated (in particular FGF2-mediated) activity and/or disturbed FGFR signaling (in particular FGFR1 signaling).
- the invention provides a compound (I) as defined in the first aspect, provided that the compound is not 1-1 , I-8.I-9 1-16,1-17, I-75, I-76, 1-116 or 1-117.
- the core moiety A is not a Ge aryl or C2-8 unsubstituted alkane. According to one embodiment, Ci and C2 are not both benzyl rings. According to one embodiment, the core moiety A is not a six-membered aryl or heteroaryl or a C2-8 unsubstituted alkane.
- the cell culture medium The cell culture medium
- the compounds of formula (I) may be used in specialized cell culture media for survival, proliferation, migration, invasion, angiogenesis, sternness, and differentiation of animal cells.
- a cell culture may benefit from the use of one or more compounds of formula (I) or pharmaceutically acceptable salt thereof.
- Cells of interest may be contacted with the one or more compounds of formula (I) or pharmaceutically acceptable salts thereof by any means.
- the cells of interest may be contacted with the one or more compounds of formula (I) or pharmaceutically acceptable salts thereof by the presence of the latter in a cell culture medium.
- Such cell culture medium bears technically special characteristics.
- the present invention relates further to a cell culture medium comprising at least one compound of formula (I) or a pharmaceutically acceptable salt thereof as defined in the first aspect.
- the invention provides a cell culture medium comprising at least one compound of formula (I) or a pharmaceutically acceptable salt thereof as defined in the first aspect, preferably wherein the cell culture medium comprises or consists of:
- nutrients sufficient for cell growth including at least one carbon source and one nitrogen source,
- (C) at least one compound of formula (I) as defined in the first aspect or a pharmaceutically acceptable salt thereof, preferably from in a concentration of 0.05 to 10 pM, preferably in a concentration of 0.1 to 5 pM, more preferably, in a concentration of 0.2 to 3 pM, and
- a cell culture medium may be any medium that is suitable to allow maintenance and viability and preferably proliferation of cells of interest.
- the person skilled in the art knows a variety of commercially available cell culture media.
- a cell culture medium at least comprises water and nutrients.
- the cell culture medium of the present invention may comprise any contents of at least one compound of formula (I) or a pharmaceutically acceptable salt thereof.
- it may comprise 0.01 nM to 10 mM, or 0.05 nM to 1 mM, or 0.1 nM to 100 pM, or 0.5 nM to 50 pM, or 1 nM to 10 pM, or 5 nM to 1 pM, or 10 nM to 500 pM, of at least one compound of formula (I) or a pharmaceutically acceptable salt thereof.
- a cell culture medium typically has an osmolarity that is near by the isotonic osmolarity to avoid disturbance of cells.
- a cell culture medium typically has a pH range that is near by the neutral range and slightly basic such as, e.g., in the range of pH 6.0 to 8.0, or pH 6.5 to 7.8, or pH 7.0 to 7.7, or pH 7.2 to 7.5, or (approximately) pH 7.4.
- the person skilled in the art knows adequate buffers to achieve such pH range such a hydrogen phosphate/phosphate buffers, MES (2-(N-morpholino)ethanesulfonic acid) buffers, etc.
- the buffer system may also comprise an open buffer based on, e.g., addition of gaseous CO2 (e.g., approximately 5% during cultivation of the cells).
- the person skilled in the art will be able to choose and use commercially available cell culture media accordingly.
- the compounds of formula (I) may be used as cell culture medium supplement such as, e.g., for cultivated meat production.
- the compounds may particularly support serum-free cell culture media and may partly replace growth factor activities, in particular FGF activities and thereby of serum.
- the cell culture medium may be liquid, viscous, or solid.
- a cell culture medium is a liquid cell culture medium.
- it may be a (hydro)gel on or in which cells are cultivated, either in contact with air or covered by another cell culture medium, which is preferably liquid.
- one or more compounds of formula (I) or a pharmaceutically acceptable salts thereof may partly or completely substitute one or more growth factors such as FGF, in particular FGF2. Therefore, culturing cells when omitting such one or more growth factors may be enabled.
- the cell culture medium is further characterized in that it does (essentially) not contain serum of animal origin, any subtype of FGF of animal origin, any peptide growth factor of animal origin, in particular any peptide growth factor or steroid growth factor of animal origin, in particular any steroid growth factor.
- the composition of serum is complex and not fully defined. It contains various growth factors, hormones, and other components that can influence cell behavior, but their exact concentrations and roles may not be well-understood. This lack of definition can lead to difficulties in interpreting experimental results. Serum composition can vary significantly between different batches and sources, leading to inconsistencies in cell culture conditions. This variability can affect cell growth, differentiation, and experimental outcomes, making it challenging to reproduce results. Proteins derived from animal sources, such as bovine serum albumin (BSA) or bovine transferrin, can also exhibit batch-to- batch variability. Similar to serum, proteins from animal origin can be expensive, especially if large quantities are required for extensive cell culture studies.
- BSA bovine serum albumin
- the cell culture medium does (essentially) not contain FGF2, in particular no FGF2 of animal origin.
- FGF2 is the compound to be replaced by the compounds of the invention.
- the combination of FGF2 and the compounds of the invention does not have an additive effect.
- the present invention also allows efficient cultivation of cells.
- the invention provides a method for the cultivation of cells, comprising the steps of
- step (iii) subjecting the cell culture medium of step (ii) to conditions sufficient for establishing cell growth and/or for cell differentiation;
- the cells preferably comprise FGFR1 , which signaling pathway is activated by a compound of formula (I) as defined in the first aspect.
- the cells may be from human or animal source.
- the cells may be for example obtained from the following animals: cattle (bovine), the cells of which are one of the primary sources for cultivated beef production; Chicken (poultry), the cells of which are used to produce cultivated chicken meat; pigs (swine), the cells of which are used for cultivated pork production; fish (e.g., salmon, tuna), the cells of can be used to produce cultivated fish fillets; ducks, the cells of which can be a potential source for cultivated duck meat; sheep, the cells of which can be used to produce cultivated lamb or mutton; rabbits, the cells of which can be used to produce cultivated rabbit meat; quails: quails are a potential source for cultivated quail meat; turkey, the cells of which can be used to produce cultivated turkey meat; deer, the cells of which cells can be used to produce cultivated venison.
- Myoblasts are muscle precursor cells that can differentiate and fuse to form multinucleated myotubes, which eventually mature into muscle fibers. Myoblasts are a primary choice for cultivated meat production due to their natural ability to develop into muscle tissue. Satellite cells are a type of stem cell found in skeletal muscle. These cells play a crucial role in muscle regeneration and repair. When activated, satellite cells can proliferate and differentiate into myoblasts, making them suitable candidates for cultivated meat.
- Addition of the compound of formula (I) or a pharmaceutically acceptable salt thereof may be performed by any means.
- the compound of formula (I) or a pharmaceutically acceptable salt thereof may be added during cultivation. It may be added as such, i.e., as pure substance, may be added in a dilution in a stock solution of higher concentration, or may be added by replacing the cell culture medium. Cultivating with the compound of formula (I) or a pharmaceutically acceptable salt thereof may be conducted once, twice or more often or may be constantly.
- the concentrations of compound of formula (I) or a pharmaceutically acceptable salt thereof may be maintained constant during cultivation of cells or may be varied by any profile, e.g., increased or decreased over time.
- Separating the cultivated cells from the cell culture medium may be conducted by any means. If the cells are adherently grown, these may, for instance, be detached by mechanical scraping and/or enzymatically (e.g., by using a trypsin or other digestive enzyme composition). Suspended cells may be separated from the cell culture medium by centrifugation and/or filtration (e.g., dead end filtration or cross-flow filtration).
- the method can be included in a method of producing a food product, in particular cultivated meat.
- the method of cell expansion further contains a step of separating the cultivated cells from the cell culture medium.
- the method of producing cultivated meat may contain the following steps:
- the first step in cell cultivation is to obtain cells from a living donor animal. This can be done through a biopsy or tissue sample, which typically contains muscle cells, also known as myocytes.
- the donor animal's welfare is a crucial consideration, and efforts are made to minimize any harm during the cell collection process.
- the next step in the generation of cultivated meat is to develop the cultured cells into functional muscle tissue.
- the cultured cells need to differentiate into specific cell types. This process involves exposing the cells to specific biochemical and mechanical cues that mimic the natural environment of muscle tissue development.
- a scaffold made of biocompatible materials can be used to provide structural support for the growing cells. Additionally, 3D bioprinting techniques can be employed to create complex tissue structures, enhancing the organization and functionality of the cultured meat.
- the cultured cells are further cultivated and matured to promote the formation of muscle tissue.
- This maturation process aims to develop a product that closely resembles traditional meat in terms of texture, taste, and nutritional content.
- the cultivated meat reaches the desired level of maturity, it is harvested and processed into various meat products, such as burgers, sausages, or nuggets, using conventional food processing techniques.
- the cultivated meat products are packaged and prepared for distribution to consumers or food outlets.
- the invention provides a food product comprising cultivated cells obtainable by the process according the fifth aspect or material derived therefrom, preferably wherein the food product is cultivated meat or a drinkable composition.
- the cultivated meat according to the invention may be for example cultivated beef, cultivated chicken, cultivated pork, cultivated fish (e.g., salmon, tuna), cultivated duck, cultivated lamb or mutton, cultivated rabbit, cultivated quail, cultivated turkey, cultivated venison.
- cultivated beef cultivated chicken
- cultivated pork cultivated fish
- cultivated duck cultivated lamb or mutton
- cultivated rabbit cultivated quail
- cultivated turkey cultivated venison.
- the food product is cultivated meat or a drinkable composition.
- the cell culture medium and the food product of the present invention may be provided in any package. Depending on the intended use, it may be provided in different packaging. It may be stored at any condition suitable for this purpose such as, e.g., at ambient temperature (e.g., 18 to 30°C, preferably 18 to 25°C), in a fridge (e.g., at 0 to 15°C, preferably 3 to 10°C), in a freezer (e.g., -30 to 0°C, preferably -25 to -10°C), in a deep freezer (e.g., -100 to -300°C, preferably -90 to -55°C), on liquid nitrogen, on dry ice, or even one or more liquid noble gases. For instance, it may be provided in a flask, a bottle, another container, or a package for solid material. It may be stored at dry state, as a (hydro)gel, as a suspension, emul
- reaction apparatuses were dried under dynamic vacuum using a heat gun, and anhydrous solvents (Sure-SealTM products from Merck KGaAr AcrosealTM products from Thermo Fischer Scientific inc.) were employed. Commercial solvents and reagents were used without further purification.
- reaction conditions reaction time and temperature may vary.
- TLC thin-layer chromatography
- UHPLC-MS ultra-high performance liquid chromatography-mass spectrometry
- the column eluate was analysed using a 1260 Infinity II Diode Array Detector WR scanning from 200 to 400 nm and LC/MSD iQ mass spectrometer scanning in both positive and negative ion modes from 140 to 1000 Da. Purifications were performed by automated flash chromatography (FC) using Pure C-810 Buchi instrument and pre-packed BGB Scorpius C18-HP 100 A or Silica 60 A cartridges or HPLC Agilent Infinity II Preparative HPLC using InfinityLab ZORBAX Eclipse Plus C18 column. UV detection was used to trigger fraction collection.
- FC automated flash chromatography
- Purifications may vary in general, solvents and the solvent ratios used for eluents/gradients were chosen to provide appropriate Rfs or retention times.
- Mass spectrometry data are reported from UHPLC-MS analyses. Mass spectrometry (MS) was performed via electrospray ionization (ESI).
- concentration refers to the removal of solvent at reduced pressure on a rotary evaporator with a bath temperature less than 60° C.
- the abbreviations “min” and “h” stand for “minutes” and “hours,” respectively.
- the term “TLC” refers to thin-layer chromatography, “room temperature or ambient temperature” means a temperature between 18 to 25° C., “, “UHPLC” refers to ultra-high performance liquid chromatography, “HPLC” refers to high-performance liquid chromatography, FC refers to automated flash chromatography.
- ACN acetonitrile
- DIPEA N,N-Diisopropylethylamine.
- Dppf 1 , 1 -ferrocenediyl-bis(diphenylphosphine).
- PCya tricyclohexylphosphine
- PyBOP benzotriazol- 1-yloxytripyrrolidinophosphonium hexafluorophosphate.
- Rpm revolution per minute.
- TCFH Chloro-N,N,N',N'-tetramethylformamidinium hexafluorophosphate
- Methyl ester was dissolved in mixture of THF/MeOH/Water (1 :1 :1) and LiOH was added. The resulting mixture was stirred overnight. After the completion of the reaction, 1 M HCI was added (5-10 ml) and the mixture was extracted with EtOAc. The organic layers were combined, dried over sodium sulfate and evaporated under the reduced pressure obtaining intermediate.
- Step 1
- Step 1
- Step 1
- Step 1
- Step 1
- f-BuBrettPhos Pd G3 43 mg, 0.05 mmol
- f-BuBrettPhos 24 mg, 0.05 mmol
- K4[Fe(CN)e]*3H2O 184 mg, 0.5 mmol
- 5-bromoisophthalic acid dimethyl ester 273 mg, 1 mmol
- the vessel was evacuated and refilled with nitrogen (three cycles).
- Degassed dioxane (2.5 mL), KOAc (12 mg, 0.125 mmol) and water (2.5 mL) were then added to the reaction tube via syringe.
- test tube was placed in an oil bath preheated to 100 °C. After 4 h of stirring at 100 °C, the reaction mixture was then cooled to room temperature. The contents of the test tube were transferred to a separation funnel using EtOAc (15 mL) and brine (15 mL), and the organic layer was separated from the aqueous layer. The solvents were evaporated and the residue was purified by chromatography on silica gel eluting with cyclohexane to cyclohexane:ethylacetate 50:50 to obtain the product in 72% yield (158 mg).
- Step 1
- Step 1
- Step 1
- Step 1
- Trimethyl 1 ,3,5-benzenetricarboxylate (2.00 g, 7.93 mmol) was suspended in methanol (180 mL) and I M sodium hydroxide solution (7.14 mL, 280 mg of sodium hydroxide) was added. The mixture was stirred at room temperature for 18 h. The resulting solution was concentrated under reduced pressure to afford a white solid which was partitioned between dichloromethane (150 mL) and saturated aqueous sodium, bicarbonate solution (150 mL). The organic phase was separated and was extracted with saturated aqueous sodium bicarbonate solution (150 mL) before being discarded.
- Step 1
- Step 1
- Step 1 Method B.
- Step 1
- Step l Method B. Starting materials: 5-bromoisophthalic acid dimethyl ester (400 mg, 1.46 mmol), 6,6-difluoro-3-azabicyclo[3.1.0]hexane hydrochloride (227 mg, 1.46 mmol), CS2CO3 (0.716 mg, 2.20 mmol), 15 ml of toluene, Pd(OAc)2 (16.6 mg, 0.074 mmol), BINAP (45 mg, 0.074 mmol). MS (M+H) + : 312.1.
- Step 1
- Step 1
- Step 1
- Step 2 Synthesized according to general procedure 1. Starting materials: THF/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (400 mg).
- Step 1
- Step 1
- Step 1
- Step 1
- Step 1
- Step 1 MS (M+H) + : 300.9.
- Step 1
- Step 1
- Step 1 anhydride (12.5 mL) was cooled to -40 °C prior to the dropwise addition of 69% nitric acid (0.82 mL). The reaction mixture was warmed to room temperature and stirred for 1.5 h. The solution was then poured onto ice water and neutralized with sat. sodium bicarbonate solution. The aqueous layer was then separated and extracted with ethyl acetate. The combined organic extracts were then dried using MgSCU, filtered, then concentrated in vacuo. The residue was purified by silica gel column chromatography eluting with cyclohexane to cyclohexane: ethylacetate 50:50 to obtain the desired product as a yellow solid in 23% yield (320 mg). MS (M+H) + : 154.8.
- tin(ll)chloride (1.05 g, 5 mmol) was dissolved in 3 of mL cone. HCI and warmed up to 60 °C followed by the slow addition of product from the previous step (250 mg). Afterwards, the solution was stirred for further 15 min and then poured, under gas formation, in a mixture of 5 g potassium carbonate and 20 mL ice/water. The product was extracted with EtOAc and the combined organic layers were dried over sodium sulfate. Finally, the solvent was removed in vacuo obtaining the product as black solid, which was immediately used in the next step.
- Step 1
- Step 1
- Step 1 Additional step between Step 1 and 2 was added if Boc-deprotection was necessary.
- step 1 The product from step 1 was dissolved in mixture of DCM:TFA (1 ml/1ml) and stirred for 3 h at rt. After the completion of the reaction the solvents were evaporated, and the residue was used in the next step.
- Step 1-2
- the compounds was synthesized according to general procedure 2 using the product from the previous step.
- Step 1
- the compound was synthesized according to general procedure 2 using the product from the previous step.
- Step 1
- the compounds were synthesized according to general procedure 2 using the product from the previous step. 1-67.
- Step 1
- the compound was synthesized according to general procedure 2 using the product from the previous step.
- Step 1
- the compound was synthesized according to general procedure 2 using the product from the previous step.
- Step 1
- the compound was synthesized according to general procedure 2 using the product from the previous step.
- Step 1
- the compound was synthesized according to general procedure 2 using the product from the previous step.
- Step 1
- the intermediate 53 (58 mg, 0.14 mmol) and diamine (0.14 mmol) were dissolved in 1 ml of MeCN. N-methyl imidazole (0.06 ml) and TCFH (73 mg, 0.28 mmol) were added sequentially. After the completion of the reaction, the solids were filtrated, washed with ethanol and used in the next step without additional purification.
- the compounds were synthesized according to general procedure 2 using the product from the previous step. 1-121.
- Step 1
- the compound was synthesized according to general procedure 2 using the product from the previous step.
- Step 1
- the compound was synthesized according to general procedure 2 using the product from the previous step.
- Step 1
- the compound was synthesized according to general procedure 2 using the product from the previous step.
- Step 1
- the compound was synthesized according to general procedure 2 using the product from the previous step.
- Step 1
- the compound was synthesized according to general procedure 2 using the product from the previous step.
- Step 1
- the compounds were synthesized according to general procedure 2 using the product from the previous step.
- Step 1
- Step 1
- Step 1
- the compound was synthesized according to general procedure 2 using the product from the previous step.
- Step 1
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Health & Medical Sciences (AREA)
- Biomedical Technology (AREA)
- Biotechnology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Genetics & Genomics (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Cell Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention relates to compound of formula (I) and their use for cultivating cells in vitro, wherein the compound has the general formula (I). Furthermore, the invention to the compound of formula (I) for use in medical treatment, wherein the medical treatment is a method of wound healing, a method of organ regeneration in transplantation medicine, a method of treating a burned lesion, a disease associated with disturbed cell renewal, or a disease associated with muscle wasting, wherein the treatment or prevention may optionally comprise tissue engineering. The compounds of formula (I) may be used as cell culture medium supplement. Thus, the invention also relates to a cell culture medium comprising at least one compound of formula (I) a method for the cultivation of cells and a food product comprising cultivated cells.
Description
COMPOUNDS FOR ACTIVATION OF FGFR1 SIGNALING
FIELD OF THE INVENTION
The present invention relates to compounds of formula (I) for activating the Fibroblast Growth Factor Receptor 1 (FGFR1) signaling pathway. Moreover, it refers to the use of such compounds (I) as a culture media component for the cultivation of living cells or as a medicament for treatment and/or prevention of different diseases.
BACKGROUND OF THE INVENTION
Today, animal and human cell cultures (i.e., the cultivation of cells of animal or human origin in vitro) are of high interest for numerous applications. For instance, in research, in industrial productions (e.g., of biopharmaceuticals such as, e.g., antibodies, hormones and other cellular products), in stem cell techniques, in in vitro fertilization, in preparation of implantable cells, and in the production of cultivated substitutes for meat and other animal-like comestible goods, animal and human cell cultures are of high interest.
It is known that animal cells (including human cells) that are cultivated in cell culture typically require stimulation to proliferate. This is typically achieved by the addition of serum, which is obtained from processed animal blood. The amount of serum is typically set to a high level to reach the sufficient content of one or more stimulating growth factors.
Such high contents of serum are technically undesirable. Such sera bear residual risks that these can comprise pathogens, undesired hormones, remaining contents of xenobiotics and medicinal agents (e.g., antibiotics), etc. Though such risks can essentially be excluded by comprehensive controls, this is still undesired, because such controls require laborious additional efforts. Standardization and reproducibility may be hampered, because sera from different animals or in different states of female and other hormone cycles may bear varying properties. In some applications, standardization and reproducibility is, however, of high interest to achieve a well-defined product. In addition, there may be ethical discussions when using high amounts of animal-derived sera obtained from animal bodies, typically from slaughtered animals. Further stimulating proliferation of cells can also be of interest in vivo such as, e.g., for wound healing.
Thus, there is a desire to reduce the amount of required serum and/or to support growth factors contained therein.
There were attempts to provide growth factors obtained by gene technology. This is, however, complicated and laborious and requires high efforts and is not particularly effective. The low stability of several growth factors such as, e.g., Fibroblast Growth Factor 2 (FGF2) may result in a comparably low half-life (e.g., in human cells of <10 hours) at 37°C (Koledova et al. 2019). The use of growth factors such as FGF2 has several disadvantages. The production of recombinant growth factors can be expensive, especially for large-scale cell culture applications. Achieving high purity and stability in recombinant growth factor production can be challenging. Impurities or variations in the protein structure can affect their activity and reliability in cell culture. Despite efforts to standardize production, recombinant growth factors can still exhibit batch-to-batch variability, which may lead to inconsistent results and difficulties in reproducibility. Animal-derived growth factors, especially those from nonqualified sources, may contain contaminants, such as viruses, bacteria, or endotoxins. These contaminants can negatively impact cell cultures and experimental results.
So far, development of biologically active molecules towards extracellular targets besides native ligands and their mutants often focused on antibodies (Jin et al. 2022). In this context, the activity of a natural ligand is often blocked by binding of an antibody to its receptor (Zahavi and Weiner, Antibodies, 2020, 9:34; doi:10.3390/antib9030034). A major downside of antibody-based approaches is high costs of development and production. Moreover, the use of antibodies in the therapeutic context is typically blocking activities mediated by the targeted cell surface receptor. The development of small molecules for modulating activity of cellsurface receptors concentrated mostly on intracellular domains so far. Only a few small molecules were found to modulate growth factor receptors such as Fibroblast Growth Factor Receptor 1 (FGFR1) activity, such as the inhibitors SSR128129E (Bono et al., 2013; Herbert et al., 2013) and rosmarinic acid (Pagano et al., 2021). Limited success was achieved in designing modulators, in particular agonists, for FGFR1. For example, US, 9120819B2 describes synthetic heterocyclic molecules that are supposed to be capable of activating the formation of new blood vessels (or angiogenesis) by inducing FGF receptor dimerization.
Thus, there is still an unmet need for further compounds that promote proliferation of the cells, in particular by activation of cell surface receptors thereby presenting a valuable replacement for growth factors.
SUMMARY OF THE INVENTION
The present invention is, inter alia, based on the surprising finding that compounds of formula (I) and salts thereof as defined in the present invention when added to cell culture significantly
enhance proliferation of the cells. It is understood that the compounds of formula (I) are efficient activators of the FGFR1 signalling pathway leading to this proliferation. Accordingly, the compounds of formula (I) are useful for supporting the cultivation of cells. Thereby, the compounds may fully or partly substitute FGF2. Some of these compounds have already been described in the art for different applications (see below).
Thus, according to a first aspect, the invention relates to the use of at least one compound or a pharmaceutically acceptable salt thereof for cultivating cells in vitro, wherein the compound has the general formula (I):
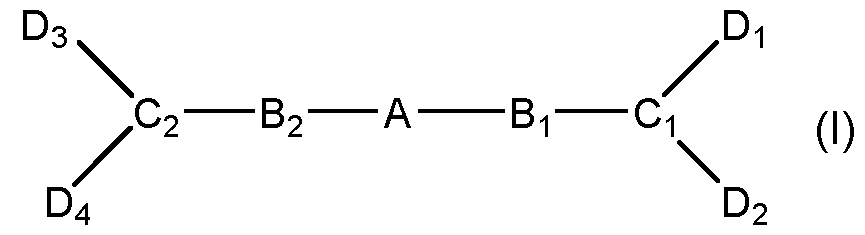 wherein:
wherein:
• the core moiety A is selected from o an optionally substituted C5-18 aromatic group, with 1 to 3 optionally fused rings; wherein 0 to 4 C atoms are replaced by heteroatoms, wherein the heteroatoms are individually selected from N, O, and S; o an optionally substituted Ce-is arylalkyl group wherein 0 to 4 C atoms are replaced by heteroatoms, wherein the heteroatoms are individually selected from N, O, and S, an optionally substituted C3-12 cycloalkyl group, with 1 to 6 rings optionally fused, bridged or strained, wherein 0 to 4 C atoms are replaced by heteroatoms, wherein the heteroatoms are individually selected from N, O, S and Si; o an optionally substituted, straight or branched, saturated or unsaturated C2-20 aliphatic group, wherein 0 to 4 C atoms are replaced by heteroatoms, wherein the heteroatoms are individually selected from N, O, S, and Si; o an aromatic or aliphatic crown ether; o or a bond; wherein A may contain one to three substituents, which are independently selected from the group consisting of halogen, amide, amine, nitro, cyano, hydroxyl or hydrocarbyloxy, or aldehyde, ketone, carboxyl, ether, ester, alkyl, alkenyl, alkynyl, sulfonyl, sulfonylamide;
• the linkers Bi and B2 are individually selected from an ester, an ether, an amine, a thioether, an amide; and a sulfonamide;
• wherein the tripod moieties Ci and C2 are individually selected from:
o an optionally substituted C5-6 aromatic group, wherein 0 to 3 C atoms are replaced by heteroatoms, wherein the heteroatoms are individually selected from N, O, and S; o an optionally substituted C3-6 cycloalkyl group, wherein 0 to 3 C atoms are replaced by heteroatoms, wherein the heteroatoms are individually selected from N, O, and S;
• wherein Ci and C2 may contain one to three substituents, which are independently selected from the group consisting of hydrogen, alkyl and alkoxy,
• the active moieties Di, D2, D3 and D4 are independently selected from hydrogen or
 with R1 being X or CH3; wherein Di and D2 may not both be hydrogen and D3 and D4 may not both be hydrogen.
with R1 being X or CH3; wherein Di and D2 may not both be hydrogen and D3 and D4 may not both be hydrogen.
Furthermore, a compound of formula (I) or a pharmaceutically acceptable salt thereof may be used in various medical applications, in particular for the treatment of diseases or conditions of patients, which may be treated or ameliorated by activation of the FGFR1 signaling pathway in cells of the patient.
Thus, according to a second aspect, the invention provides a compound of formula (I) as defined in the first aspect, or a pharmaceutically acceptable salt thereof for use in medical treatment, wherein the medical treatment is a method of wound healing, a method of organ regeneration in transplantation medicine, a method of treating a burned lesion, a disease associated with disturbed cell renewal, or a disease associated with muscle wasting, wherein the treatment or prevention may optionally comprise tissue engineering.
While some to the compounds of formula (I), such as 1-1 , I-8, 1-16, 1-17, I-75, I-76, 1-116 or I- 117, have been previously described as compounds for treating cachexia or the inhibition of arginine uptake in the treatment of tumors or infections, other compounds are described for the first time. Thus, according to a third aspect, the invention provides a compound (I) as defined in the first aspect, provided that the compound is not 1-1 , I-8, 1-16, 1-17, I-75, I-76, I- 116 or 1-117. The compounds of formula (I) may be used as cell culture medium supplement, such as, e.g., for cultivated meat production. The compounds may particularly support serum-
free cell culture media and may partly replace growth factor activities, in particular FGF activities. Thus, according to a fourth aspect, the invention provides a cell culture medium comprising at least one compound of formula (I) or a pharmaceutically acceptable salt thereof as defined in the first aspect, preferably wherein the cell culture medium comprises or consists of:
(A) water,
(B) nutrients sufficient for cell growth, including at least one carbon source and one nitrogen source,
(C) at least one compound of formula (I) as defined in the first aspect or a pharmaceutically acceptable salt thereof, preferably from in a concentration of 0.05 to 10 pM, preferably in a concentration of 0.1 to 5 pM, more preferably, in a concentration of 0.2 to 3 pM, and
(D) one or more salts acceptable in cell culture,
(E) optionally one or more buffer agents,
(F) optionally one or more gel-forming materials, and
(G) optionally one or more colorings acceptable in cell culture, preferably wherein the cell culture medium is a liquid cell culture medium.
As laid out above, the present invention also allows efficient cultivation of cells. Thus, a fifth aspect, the invention provides a method for the cultivation of cells, comprising the steps of
(i) providing:
(A) a cell culture medium; and
(B) cells
(ii) adding the compound of formula (I) or a pharmaceutically active salt as defined in the first aspect to the cell culture medium;
(iii) subjecting the cell culture medium of step (ii) to conditions sufficient for establishing cell growth and/or for cell differentiation; and
(iv) obtaining cultivated cells,
And optionally further comprising one or more of the following steps:
(v) separating the cultivated cells from the cell culture medium.
The cultivated cells or - in case of a further tissue engineering step - material derived therefrom may further be treated to prepare a food product.
Thus, according to a sixth aspect, the invention provides a food product comprising cultivated cells obtainable by the process according the fifth aspect or material derived therefrom, preferably wherein the food product is cultivated meat or a drinkable composition.
FIGURES
Fig. 1 Proliferation of NIH/ 3t3 cells upon treatment with different doses of 1-1 in graphical depiction. (A) ATP quantification by CellTiter-Glo® 2.0 reagent showed increased proliferation for concentrations between 0.1-1.4 pM after treatment with 1-1. (B) Confluency measurements based on images showed the same increase in proliferation as observed for ATP quantification. This correlation of two different readouts manifests that 1-1 has a proliferative effect on NIH/ 3t3 cells. The x-axis depicts the concentration of compound in pM in logarithmic scale, the y-axis shows either the normalized luciferase signal (0= DMSO control/ 1= 100 ng/ mL FGF2) or the normalized confluency (timepoint / 1= 0 h).
The individual data points in this and subsequent CellTiter Gio® experiments are plotted using Prism (Graphpad). The standard deviation is based on four replicates.
Fig. 2 Proliferation of NIH/ 3T3 cells upon treatment with different doses of 11-1 or Aminoguanidine hydrochloride (AGH). Both molecules did not show cell proliferation at tested concentration. This indicates that neither aminoguanidine moieties alone nor the “monomeric version of 1-1 are sufficient to induce response observed for 1-1. The x-axis depicts the concentration of compound in pM in logarithmic scale, the y-axis shows the normalized luciferase signal (0= DMSO control/ 1= 100 ng/ mL FGF2).
Fig. 3 Optimization of proliferative activity of 1-1 in NIH/3T3 cells using I-53 as an example. This compound showed an approx. 4.5-fold EC50 improvement over 1-1 from 0.9 to 0.2 pM. The x-axis depicts the concentration of compound in pM in logarithmic scale, the y-axis shows the normalized luciferase signal (0= DMSO control/ 1= 100 ng/ mL FGF2).
Fig. 4 Dose-response with 1-1 using the Luciferase reporter assay in HEK293T cells. Upon treatment with 1-1 alone, an increase in luminescence was observed at compound concentrations higher than 3 pM. When treated with 1-1 and FGFR1 inhibitor PD166866 a signal decrease was observed. This indicates that 1-1 works partly through the FGFR1 pathway. The x-axis depicts the concentration of 1-1 in pM, the y-axis shows the normalized luminescence signal (Signal/ Negative Control).
Fig. 5 Determination of FGFR1 dependency in NIH/ 3t3 cells. Treatment of 40 ng/ mL FGF2 and 1 pM 1-1 with increasing concentrations of FGFR1 inhibitor PD166866 led to a loss of their proliferative effect (IC50 of approx. 0.16 and 0.04 pM). In contrast, PD166866 did not change the proliferative effect observed for 100 ng/ mL EGF and 2 ng/ mL TGF-pi , underlining the specificity of the observed effect. The x-axis depicts the concentration of PD166866 in pM, the y-axis shows the normalized luciferase signal (signal/ negative control). The x-axis depicts the concentration of compound in pM in logarithmic scale, the y-axis shows the normalized luciferase signal (0= DMSO control/ 1= 100 ng/ mL FGF2).
Fig. 6 Delineation of the signalling pathway activated by 1-1 in NIH/ 3t3 cells by treatment with various inhibitors. Proliferative activity of (A) 1.4 pM 1-1 and (B) 100 ng/ mL FGF2 was inhibited by FGFR1-specific and ERK1/2-specific inhibitors. In contrast, using inhibitors against AKT and PKC, two other branches of the FGFR1-signaling pathway, did not have an effect. This shows that 1-1 acts through the FGFR1- ERK1/2 axis similar to FGF2. The x-axis depicts the concentration of inhibitor in pM in logarithmic scale, the y-axis shows the normalized luciferase signal (0= DMSO control/ 1= 100 ng/ mL FGF2).
Fig. 7 Competition assay between recombinant FGFR1-Fc and cellular FGFR1 for FGF2 or 1-1. Recombinant FGFR1-Fc inhibited the proliferative effect of (A) 1.4 pM 1-1 and (B) 100 ng/ mL FGF2, while Fc alone did not have any effect. This shows that the presence of recombinant FGFR1-Fc inhibits function of 1-1 on cellular FGFR1. The x-axis depicts the concentration of the protein in pM in logarithmic scale, the y-axis shows the normalized luciferase signal (0= DMSO control/ 1= 100 ng/ mL FGF2).
Fig. 8 Determination of 1-1 stability assessed by loss of proliferative activity and compared to FGF2. (A) 1-1 was stable over the course of 14 days in the cell culture incubator. (B) In contrast, FGF2 wildtype showed decreased stability over the course of 14 days in the cell culture incubator. This reveals that 1-1 is more stable than FGF2 wildtype at 37°C/ 5% CO2. The x-axis depicts the concentration of compound in pM in logarithmic scale, the y-axis shows the normalized luciferase signal (0= DMSO control/ 1= 100 ng/ mL FGF2).
Fig. 9 Measurement of cytotoxicity by LDH release and cell viability by ATP quantification upon treatment with 1-1. A change in the dead and live cell fraction was observed
at 30 |iM , showing that the compound is not toxic at concentrations < 10 pM in HT- 1080 cells as measured by LDH release. The x-axis depicts the concentration of compound in pM in logarithmic scale, the left y-axis shows the cytotoxicity calculated as: (Experimental LDH Release - Medium Background)/ (Maximum LDH Release Control - Medium Background); and the right y-axis show the viability calculated as: Treated cells/ DMSO control.
DETAILED DESCRIPTION OF THE INVENTION
Definitions
Compounds of this invention include those described generally herein, and are further illustrated by the classes, subclasses, and species disclosed herein. As used herein, the following definitions shall apply unless otherwise indicated. For purposes of this invention, the chemical elements are identified in accordance with the Periodic Table of the Elements, CAS version, Handbook of Chemistry and Physics, 75th Ed. Additionally, general principles of organic chemistry are described in “Organic Chemistry”, Thomas Sorrell, University Science Books, Sausalito: 1999, and “March's Advanced Organic Chemistry”, 5th Ed., Ed.: Smith, M. B. and March, J., John Wiley & Sons, New York: 2001 , the entire contents of which are hereby incorporated by reference.
As described herein, a specified number range of atoms includes any integer therein. For example, a group having from 1-4 atoms could have 1 , 2, 3, or 4 atoms.
As used herein, the moieties in the compound of the invention may optionally be substituted with one or more substituents, such as are illustrated generally herein, or as exemplified by particular classes, subclasses, and species of the invention. It will be appreciated that the phrase “optionally substituted” is used interchangeably with the phrase “substituted or unsubstituted.” In general, the term “substituted”, whether preceded by the term “optionally” or not, refers to the replacement of hydrogen radicals in a given structure with the radical of a specified substituent. Unless otherwise indicated, an optionally substituted group may have a substituent at each substitutable position of the group, and when more than one position in any given structure may be substituted with more than one substituent selected from a specified group, the substituent may be either the same or different at every position. Combinations of substituents envisioned by this invention are preferably those that result in the formation of stable or chemically feasible compounds.
Unless otherwise indicated, a substituent connected by a bond drawn from the center of a ring means that the substituent can be bonded to any position in the ring.
The term “stable”, as used herein, refers to compounds that are not substantially altered when subjected to conditions to allow for their production, detection, recovery, purification, and use for one or more of the purposes disclosed herein. In some embodiments, a stable compound or chemically feasible compound is one that is not substantially altered when kept at a temperature of 40° C. or less, in the absence of moisture or other chemically reactive conditions, for at least a week.
The term “aliphatic” or “aliphatic group”, as used herein, means a straight-chain (i.e., unbranched), branched, or cyclic, substituted or unsubstituted hydrocarbon chain that is completely saturated or that contains one or more units of unsaturation that has a single point of attachment to the rest of the molecule.
Unless otherwise specified, aliphatic groups contain 1-20 aliphatic carbon atoms. In some embodiments, aliphatic groups contain 1-10 aliphatic carbon atoms. In other embodiments, aliphatic groups contain 1-8 aliphatic carbon atoms. In still other embodiments, aliphatic groups contain 1-6 aliphatic carbon atoms, and in yet other embodiments aliphatic groups contain 1-4 aliphatic carbon atoms. Aliphatic groups may be linear or branched, substituted or unsubstituted alkyl, alkenyl, or alkynyl groups. Specific examples include, but are not limited to, methyl, ethyl, isopropyl, n-propyl, sec-butyl, vinyl, n-butenyl, ethynyl, and tert-butyl. Aliphatic groups may also be cyclic or have a combination of linear or branched and cyclic groups. Examples of such types of aliphatic groups include, but are not limited to cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyclohexenyl, CH2-cyclopropyl, CH2CH2CH(CH3)- cyclohexyl.
The term “cycloaliphatic” (or “carbocycle” or “carbocyclyl”) refers to a monocyclic Ca-Cs hydrocarbon or bicyclic C8-C12 hydrocarbon that is completely saturated or that contains one or more units of unsaturation, but which is not aromatic, that has a single point of attachment to the rest of the molecule wherein any individual ring in said bicyclic ring system has 3-7 members. Examples of cycloaliphatic groups include, but are not limited to, cycloalkyl and cycloalkenyl groups. Specific examples include, but are not limited to, cyclohexyl, cyclopropenyl, and cyclobutyl.
The terms “heterocycle”, “heterocyclyl”, or “heterocyclic” as used herein means nonaromatic, monocyclic, bicyclic, or tricyclic ring systems in which one or more ring members are an independently selected heteroatom. In some embodiments, the “heterocycle”, “heterocyclyl”, or “heterocyclic” group has three to fourteen ring members in which one or more ring members is a heteroatom independently selected from oxygen, sulfur, nitrogen, or phosphorus, and each ring in the system contains 3 to 7 ring members.
Examples of heterocycles include, but are not limited to, 3-1 H-benzimidazol-2-one,
3-(1-alkyl)-benzimidazol-2-one, 2-tetrahydrofuranyl, 3-tetrahydrofuranyl,
2-tetrahydrothiophenyl, 3-tetrahydrothiophenyl, 2-morpholino, 3-morpholino,
4-morpholino, 2-thiomorpholino, 3-thiomorpholino, 4-thiomorpholino, 1 -pyrrolidinyl,
2-pyrrolidinyl, 3-pyrrolidinyl, 1-tetrahydropiperazinyl, 2-tetrahydropiperazinyl,
3-tetrahydropiperazinyl, 1-piperidinyl, 2-piperidinyl, 3-piperidinyl, 1-pyrazolinyl,
3-pyrazolinyl, 4-pyrazolinyl, 5-pyrazolinyl, 1-piperidinyl, 2-piperidinyl, 3-piperidinyl,
4-piperidinyl, 2-thiazolidinyl, 3-thiazolidinyl, 4-thiazolidinyl, 1-imidazolidinyl,
2-imidazolidinyl, 4-imidazolidinyl, 5-imidazolidinyl, indolinyl, tetrahydroquinolinyl, tetrahydroisoquinolinyl, benzothiolane, benzodithiane, and
1 ,3-dihydro-imidazol-2-one.
Cyclic groups, (e.g. cycloaliphatic and heterocycles), can be linearly fused, bridged, or spirocyclic.
The term “heteroatom” means one or more of oxygen, sulfur, nitrogen, phosphorus, or silicon (including, any oxidized form of nitrogen, sulfur, phosphorus, or silicon; the quaternized form of any basic nitrogen or a substitutable nitrogen of a heterocyclic ring, for example N (as in 3,4-dihydro-2H-pyrrolyl), NH (as in pyrrolidinyl) or NR+ (as in N-substituted pyrrolidinyl)).
The term “unsaturated”, as used herein, means that a moiety has one or more units of unsaturation. As would be known by one of skill in the art, unsaturated groups can be partially unsaturated or fully unsaturated. Examples of partially unsaturated groups include, but are not limited to, butene, cyclohexene, and tetrahydropyridine. Fully unsaturated groups can be aromatic, anti-aromatic, or non-aromatic. Examples of fully unsaturated groups include, but are not limited to, phenyl, cyclooctatetraene, pyridyl, thienyl, and 1-methylpyridin-2(1 H)-one.
The term “alkoxy”, or “thioalkyl”, as used herein, refers to an alkyl group, as previously defined, attached through an oxygen (“alkoxy”) or sulfur (“thioalkyl”) atom.
The terms “haloalkyl”, “haloalkenyl”, “haloaliphatic”, and “haloalkoxy” mean alkyl, alkenyl or alkoxy, as the case may be, substituted with one or more halogen atoms. This term includes perfluorinated alkyl groups, such as — CF3 and — CF2CF3.
The terms “halogen”, “halo”, and “hal” mean F, Cl, Br, or I.
The term “aryl” used alone or as part of a larger moiety as in “aralkyl”, “aralkoxy”, or “aryloxyalkyl”, refers to monocyclic, bicyclic, and tricyclic ring systems having a total of five to fourteen ring members, wherein at least one ring in the system is aromatic and wherein each ring in the system contains 3 to 7 ring members. The term “aryl” may be used interchangeably with the term “aryl ring”.
The term “heteroaryl”, used alone or as part of a larger moiety as in “heteroaralkyl” or “heteroarylalkoxy”, refers to monocyclic, bicyclic, and tricyclic ring systems having a total of five to fourteen ring members, wherein at least one ring in the system is aromatic, at least one ring in the system contains one or more heteroatoms, and wherein each ring in the system contains 3 to 7 ring members.
The term “heteroaryl” may be used interchangeably with the term “heteroaryl ring” or the term “heteroaromatic”. Examples of heteroaryl rings include, but are not limited to, 2-furanyl,
3-furanyl, N-imidazolyl, 2-imidazolyl, 4-imidazolyl, 5-imidazolyl, benzimidazolyl, 3-isoxazolyl,
4-isoxazolyl, 5-isoxazolyl, 2-oxazolyl, 4-oxazolyl, 5-oxazolyl, N-pyrrolyl, 2-pyrrolyl, 3-pyrrolyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 2-pyrimidinyl, 4-pyrimidinyl, 5-pyrimidinyl, pyridazinyl (e.g., 3-pyridazinyl), 2-thiazolyl, 4-thiazolyl, 5-thiazolyl, tetrazolyl (e.g., 5-tetrazolyl), triazolyl (e.g., 2-triazolyl and 5-triazolyl), 2-thienyl, 3-thienyl, benzofuryl, benzothiophenyl, indolyl (e.g., 2-indolyl), pyrazolyl (e.g., 2-pyrazolyl), isothiazolyl, 1 ,2,3-oxadiazolyl, 1 ,2,5-oxadiazolyl, 1 ,2,4-oxadiazolyl, 1 ,2, 3- triazolyl, 1 ,2,3-thiadiazolyl, 1 ,3,4-thiadiazolyl, 1 ,2,5-thiadiazolyl, purinyl, pyrazinyl, 1 ,3,5-triazinyl, quinolinyl (e.g., 2-quinolinyl, 3-quinolinyl, 4-quinolinyl), and isoquinolinyl (e.g., 1 -isoquinolinyl, 3-isoquinolinyl, or 4-isoquinolinyl).
It shall be understood that the term “heteroaryl” includes certain types of heteroaryl rings that exist in equilibrium between two different forms. More specifically, for example, species such hydropyridine and pyridinone (and likewise hydroxypyrimidine and pyrimidinone) are meant to be encompassed within the definition of “heteroaryl”.
A “pharmaceutically acceptable salt” means any non-toxic salt of a compound of this invention that, upon administration to a recipient, is capable of providing, a compound of this invention.
As used herein, the terms “approximately” and “about” may be understood as a scope including a deviation of up +/- 10% of the respective number value. It will be understood that the specific values are also explicitly disclosed.
As used herein, "in vitro", literally translated "in glass”, refers to experiments or studies conducted outside of a living organism in a controlled laboratory environment, typically using isolated cells, tissues, or biomolecules. In vitro experiments are designed to investigate biological or chemical processes under controlled conditions and can be used to test the effects of a particular substance or treatment on cells or tissues. For example, cell culture studies or biochemical assays performed in test tubes or petri dishes are considered in vitro experiments.
As used herein, "in vivo", literally translated "in the living", refers to experiments or studies conducted within a living organism, such as animals or humans. In vivo experiments are used to assess the effects of a drug, treatment, or innovation on a whole organism's physiology, behavior, or health. These studies are often necessary to evaluate the safety and efficacy of a new medical treatment or intervention. For example, animal testing or clinical trials involving human participants are considered in vivo studies.
As used herein, "in silico" describes experiments or studies conducted through computational methods, using computer software and algorithms to analyze and predict biological, chemical, or physical processes. In silico methods are often employed to complement or guide in vitro and in vivo experiments, providing insights into complex systems or facilitating the design of new inventions. For example, computational simulations of protein interactions or drug docking studies are considered in silico experiments.
As used herein, the terms “approximately” and “about” may be understood as a scope including a deviation of up +/- 10% of the respective number value. It will be understood that the specific values are also explicitly disclosed.
As used herein, "activating a signaling pathway" refers to the process of initiating or triggering a series of biochemical events within a cell or organism in response to the binding of a ligand (molecule) to a specific receptor on the cell's surface or a specific protein inside
the cell. This activation (binding) leads to a cascade of intracellular signaling events that ultimately result in various cellular responses or physiological effects.
As used herein, and in agreement with the use of the term in art, “orthosteric” refers to the primary or main active site on a receptor or enzyme where a ligand or substrate typically binds.
The use of compounds of formula (I) for use in cell culture
A compound of formula (I) or a pharmaceutically acceptable salt thereof may be used as cell culture medium supplement such as, e.g., for cultivated meat production. It may particularly support serum-free cell culture media and may partly replace growth factor activities, in particular FGF activities and thereby serum. This may be of interest in research as well as in industrial processes. However, it may also be used in combination with serum, growth promoting molecules or serum substitutes. Furthermore, a compound (I) or a pharmaceutically acceptable salt thereof may be used in biopharmaceutical productions such as, e.g. antibody production. Such production may be cheaper, more efficient and safer.
The compounds of formula (I) enhance proliferation of cells in culture, according to the present understanding by binding to the extracellular domain of FGFR1. They are more stable than a native ligand such as, e.g., FGF2 (including thermostability), and may be produced at high yields and comparably low costs.
According to the first aspect, a compound of formula (I) or a pharmaceutically acceptable salt thereof is used for cultivating cells in vitro.

As shown in Example 5, the compounds of formula (I) activate the FGFR1 signaling pathway. Without wanting to be bound to theory, it is understood that the compound of formula (I) binds to and specifically activates FGFR1. According to one embodiment, the compound of formula (I) activates the FGFR1 signaling pathway. According to one embodiment, the compound of formula (I) binds to and specifically activates FGFR1. According to one embodiment, the compound of formula (I) may bind to an extracellular orthosteric site of FGFR1 .
Due to the structural similarity, it is expected that the compounds of formula (I) not only bind to FGFR1 , but also other fibroblast growth factor receptors (FGFR) are, as their name implies, receptors that bind to members of the fibroblast growth factor (FGF) family of proteins. So far, five distinct membrane FGFRs have been identified in vertebrates and all of them belong to the tyrosine kinase superfamily (FGFR1 to FGFR5).
The fibroblast growth factor receptors consist of an extracellular ligand domain composed of three immunoglobulin-like domains, a single transmembrane helix domain, and an intracellular domain with tyrosine kinase activity. These receptors bind fibroblast growth factors, members of the largest family of growth factor ligands, comprising 23 members (Ornitz and Itoh 2001 and Belov and Mohammadi 2013).
The natural alternate splicing of four fibroblast growth factor receptor (FGFR) genes results in the production of over 48 different isoforms of FGFR (Duchesne et al. 2006). These isoforms vary in their ligand-binding properties and kinase domains; however, all share the common extracellular region composed of three immunoglobulin(lg)-like domains (D1-D3), and thus belong to the immunoglobulin superfamily (Coutts and Gallagher 1995).
The three immunoglobulin(lg)-like domains — D1 , D2, and D3 — present a stretch of acidic amino acids ("the acid box") between D1 and D2 (Kalinina et al. 2012). This "acid box" can participate in the regulation of FGF binding to the FGFR. Immunoglobulin-like domains D2 and D3 are sufficient for FGF binding. Each receptor can be activated by several FGFs. In many cases, the FGFs themselves can also activate more than one receptor (i.e., FGF1 , which binds all seven principal FGFRs (Ornitz et al. 1996). FGF7, however, can only activate FGFR2b (Duchesne et al. 2006), and FGF18 was recently shown to activate FGFR3 (Davidson et al. 2005).
A gene for a fifth FGFR protein, FGFR5, has also been identified. In contrast to FGFRs 1-4, it lacks a cytoplasmic tyrosine kinase domain and one isoform, FGFR5y, only contains the extracellular domains D1 and D2 (Sleeman et al. 2001). The FGFRs are known to dimerize as heterodimers and homodimers.
According to one embodiment, the compound of formula (I) competes with the natural ligand Fibroblast Growth Factor (FGF), in particular Fibroblast Growth Factor 2 (FGF2).
Several natural growth factors bind to FGFR1 and modulate its activity. FGF2 (or b-FGF) is an 18 kD protein and is one of the most studied natural modulators of FGFR1 activity. FGF2
mediates a broad spectrum of mitogenic and pro-survival effects (Nawrocka et al. 2020) and, thus, promotes different cellular phenotypes (e.g. survival, proliferation, migration, invasion, angiogenesis, sternness, maturation) in a multitude of cell types (e.g. stem cells, fibroblasts, skeletal muscle cells, endothelial cells) in a context-dependent manner. FGF2 binding to FGFR1 may lead to conformational changes of FGFR1 that allow receptor dimerization. The close proximity of the intracellular kinase domains may result in their cross-phosphorylation and activation (Sarabipour and Hristova 2015). Intracellularly, the FGF2 signal may be predominantly transmitted via Ras/Raf/MAPKs signaling, in particular the ERK pathway (Xie et al. 2020). Perturbation of the FGF2/FGFR1-ERK signaling axis may affect the mitogenic effect on the cells (e.g. proliferation and survival of melanoma cells) (Lefevre et al. 2009).
As shown in Examples 2 and 4, addition of the compounds of formula (I) to cultured cells leads to a significant increase in proliferation. Specifically, 14 compounds included in the definition of formula (I) had this effect. Moreover, as shown in Example 5, the compounds of formula (I) activate the FGFR1 signaling pathway.
The FGFR, in particular FGFR1 , in context of the present invention may be of any origin. Preferably, the FGFR, in particular FGFR1 , in context of the present invention may be of human or other mammal or avian origin, in particular of human origin for biomedical uses and of mammal e.g. pig, cow, sheep, etc. or avian such as, e.g., chicken, duck, quail, etc. origin for food or feed uses. Preferably, it is of the same species origin as the cell it is expressed in. It can, however, also be heterologously expressed in another species.
In a preferred embodiment, FGFR1 is defined as in Bono et al. (Cancer Cell, 2013, 23:477- 488) and Herbert et al. (Cancer Cell, 2013, 23:489-501). In a preferred embodiment, FGFR1 may be one of the following: FGFR1_pig: K7GQJ1 from Uniprot; or FGFR1_human: P11362 from Uniprot.
The bovine receptor FGFR1 may be equivalent to that of pig, i.e., may be essentially or completely the same as FGFR1_pig: K7GQJ1 from Uniprot.
With the activation of the FGFR1 signaling pathway and the consequent activation of proliferation, the compounds of the invention substitute natural FGFR1 ligands, such as FGF2. According to one embodiment, the compound of formula (I) competes with the natural ligand of FGFR1 , in particular Fibroblast Growth Factor 2 (FGF2). The amino acid sequence of the human FGF2 is identified by 1 ii4 from Protein Data Bank. The amino acid sequence of
the FGF2 from pig FGF2 is identified by A0A287BGK8 from UniProtKB. FGF2 is also commercially available such as FGF2-G3 (Defined Bioscience Cat No LSR-101).
The compounds of the general formula (I) are composed of core moiety A, which is connected by two linkers Bi and B2 to the two tripod moieties Ci and C2. Each tripod moiety is connected to two active moieties Di and D2 or D3 and D4 respectively.
The compound of formula 1-1 was discovered via structure-based virtual screening using a proprietary algorithm as a molecule that potentially binds to and activates FGFR1. Based on the in silico study, it is inferred that the guanidines are among the key chemical groups responsible for activity. This is experimentally confirmed in Example 2 showing that a further compound of the formula (I), namely compound 1-17, which has a very different core structure as compared to compound 1-1 , has a comparable effect.
It is conceivable that active groups are required on both sides of the molecule, i.e., bound to Ci and C2. Possibly, the active groups D1/D2/D3/D4 are required on the two sides to interact with two monomers of FGFR1 , thereby facilitating the dimerization. This hypothesis is supported by the finding that compound 11-1 , which contains the same C1/D1/D2 setup as compound 1-1 but does not contain the part B1-A-B2-C2-D3D4, does not activate FGFR1 signaling as it did not lead to any measurable increase in proliferation (see Example 3). Further, it is understood that one active group, either one of Di , and D2, and either one of D3 and D4 is sufficient for binding to and activating FGFR1 and that the second copy only increases the binding rate. Therefore, one of Di, and D2, and one of D3 and D4 may be a hydrogen.
Thus, the active moieties Di, D2, D3 and D4 are independently selected from or hydrogen,
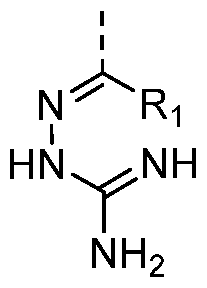 with R1 being H or CH3. Importantly Di and D2 may not both be hydrogen and likewise D3 and D4 may not both be hydrogen. Based on in silico binding studies with the proprietary algorithm, compounds with the thiourea or urea groups are expected to bind to FGFR1 with a comparable affinity as the compounds 1-1 to 1-17 with the guanidine group. More important appears to be the presence of the further three nitrogen groups. The methyl group, which is
present in compounds 1-1 to 1-17 is most likely not involved in the binding. Therefore, Ri may either be CH3 or H.
with R1 being H or CH3. Importantly Di and D2 may not both be hydrogen and likewise D3 and D4 may not both be hydrogen. Based on in silico binding studies with the proprietary algorithm, compounds with the thiourea or urea groups are expected to bind to FGFR1 with a comparable affinity as the compounds 1-1 to 1-17 with the guanidine group. More important appears to be the presence of the further three nitrogen groups. The methyl group, which is
present in compounds 1-1 to 1-17 is most likely not involved in the binding. Therefore, Ri may either be CH3 or H.
According to one embodiment, the active moieties Di, D2, D3 and D4 are H or
 wherein Di and D2 may not both be hydrogen and D3 and D4 may not both be hydrogen. According to a preferred embodiment, all four the active moieties Di , D2, D3 and D4 are
wherein Di and D2 may not both be hydrogen and D3 and D4 may not both be hydrogen. According to a preferred embodiment, all four the active moieties Di , D2, D3 and D4 are

As shown in Example 4, compounds with highly different core moieties A have comparable activities in increasing proliferation. Based on the tested compounds, the following embodiments of core moiety A are possible.
According to one embodiment, the core moiety A is optionally substituted Cs-is aryl, i.e. C5, Cg, C7, Cg , C9 , C10, C11, C12 , C13 , C14, C15, Ci6, C17, or Cig . with 1 , 2 or 3 optionally fused rings. The rings may be fused or bound. In the Cs-is aryl, 0 to 4 C atoms, i.e. 0, 1 , 2, 3, or 4 C atoms, may replac-ed by heteroatoms, wherein the heteroatoms are individually selected from N, O, and S.
According to one embodiment, the core moiety A is an optionally substituted Cs-saryl wherein 0 to 3 C atoms are replaced by heteroatoms, wherein the heteroatoms are individually selected from N, O, and S. According to one embodiment, the core moiety A is an optionally substituted six-membered aryl or heteroaryl. The Cs heteroaryl may have one or two heteroatoms. According to a preferred embodiment, the heteroatom is N. According to one embodiment, the B groups are in meta position of the six-membered aryl or heteroaryl. Accordingly, the B groups are separated by one carbon atom on the six-membered aryl or heteroaryl. This corresponds to the 1 ,3-positions. According to one embodiment, the B groups are in para position of the six-membered aryl or heteroaryl. According to a preferred embodiment, A is a benzene ring.
According to one embodiment, the core moiety A is an optionally substituted Ce-is, arylalkyl group, i.e. a Cg, C7, C8, C9, C10 , On, Ci2, C13 , C14, C15, Ci6, C17, or Cig, aralkyl. In the C -is arylalkyl group 0 to 4 C atoms, i.e. 0, 1 , 2, 3, or 4 C atoms, may be replaced by heteroatoms. The heteroatoms are individually selected from N, O, and S. According to one embodiment, the aralkyl is a six-membered aryl or heteroaryl ring with two C1-3 alkyl groups attached. The alkyl groups may be methyl, ethyl or propyl, preferably methyl. According to one embodiment, A contains two benzene rings. According to one embodiment, A is selected from a dibenzyne, biphenyl, diphenylmethane or diphenyl ether.
According to one embodiment, the core moiety A is an optionally substituted C3-12 cycloalkyl group i.e. a C3, C4, C5, C6, C7, C8, C9, C10, CH, or C12, with 1 to 6 rings, i.e. 1 , 2, 3, 4, 5, or 6 rings. The rings may optionally be fused, bridged or strained. In the C3-12 cycloalkyl group, 0 to 4 C atoms, i.e. 0, 1 , 2, 3, or 4 C atoms, may be replaced by heteroatoms, The heteroatoms may be individually selected from N, O, S and Si. According to one embodiment, the core moiety A is an optionally substituted C3-6 cycloalkyl, i.e. an optionally substituted cyclopropane, cyclobutane, cyclohexane or cyclopentane. The C3-6 cycloalkyl may be bridged. In the C3-6 cycloalkyl 0 to 1 C atoms may be replaced by a heteroatom. The heteroatoms are individually selected from N, O, and S. According to one embodiment, the C3-6 cycloalkyl does not contain heteroatoms.
According to one embodiment, the core moiety A is an optionally substituted, straight or branched, saturated or unsaturated C2-20 aliphatic group, i.e. C2, C3, C4, C5, C6, C7, C8, C9, C10 , C11, C12 , C13 , C14, Cis, Ci6, C17, Cig, Ci9, or C20. , In the C2.2o aliphatic group, 0 to 4 C atoms, i.e. 0, 1 , 2, 3, or 4 C atoms, are replaced by heteroatoms. The heteroatoms are individually selected from N, O, S, and Si. According to one embodiment, the C2-20 aliphatic group is saturated.
According to one embodiment, the core moiety A is an optionally substituted, straight or branched, saturated or unsaturated C2-12 aliphatic group, wherein 0 to 4 C atoms are replaced by heteroatoms, wherein the heteroatoms are individually selected from N, O, and S. According to one embodiment, the C2-12 aliphatic group is saturated. According to one embodiment, the C2-12 aliphatic group is straight. According to one embodiment, a substituent is in ortho position to Bi or B2. According to one embodiment, a substituent is in ortho position to Bi and B2. According to one embodiment, first substituent is in meta position to Bi and B2. According to one embodiment, the core moiety A is an aromatic or aliphatic crown ether. According to one embodiment, the core moiety A is an aromatic crown ether. The crown ether may be a 18-crown-6 ether.
The core moiety A is optionally substituted, which means that the core moiety A may contain one to three substituents, which are independently selected from the group consisting of halogen, amide, amine, nitro, cyano, hydroxyl or hydrocarbyloxy, or aldehyde, ketone, cycloalkyl, carboxyl, ether, ester, optionally halogenated alkyl, alkenyl, alkenylaryl, alkenyl aryl, alkynyl, alkoxy, alkylthio, sulfonyl, sulfonylamide. According to one embodiment, A may contain one to three substituents, which are independently selected from the group consisting of halogen, hydrogen, hydroxyl, methyl, methoxy, optionally halogenated Ci-6alkyl, optionally halogenated Ci-6 alkoxy, C5-6 cycloalkyl, in which 0 to 2 C atoms are replaced by heteroatoms and amine. According to one embodiment, the substituent is a polyether. According to one embodiment, the substituent is selected from:
 wherein X is N, O, S, preferably O.
wherein X is N, O, S, preferably O.
According to one embodiment A contains three halogen substituents. According to one embodiment, the halogens are F.
According to one embodiment, the Ci-6 alkyl is selected from methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, pentyl, isopentyl, neopentyl, hexyl, isohexyl, neohexyl. According to one embodiment, the substituent is a tert-butyl, preferably in meta position to both Bi and B2. According to one embodiment, the Ci-6 alkyl is halogenated by one to three of fluorine, chlorine, bromine or iodine. According to one embodiment, the halogenated alkyl is trifluoromethyl, preferably in meta position to both Bi and B2. According to one embodiment, the halogenated alkyl is difluoromethyl, preferably in meta position to both Bi and B2. According to one embodiment, the halogenated alkoxy is trifluoromethoxy, preferably in meta position to both Bi and B2. According to one embodiment, the substituent is morpholine, preferably in ortho or meta postion to Bi. According to one embodiment, the alkylthio group is methylthio, preferably in meta position to both Bi and B2. According to one embodiment, the sulfonyl group is methylsulfonyl, preferably in meta position to both Bi and B2.
According to one embodiment, the substituent is styryl, preferably in ortho or meta position to Bi. According to one embodiment, the cycloal kylethynyl is selected from cyclohexylethynyl or a cyclopropylethynyl and piperidineethynyl, preferably in meta position to both Bi and B2. According to one embodiment, the alkynyl is methylacetylene, preferably in meta position to both Bi and B2.
According to one embodiment, the core moiety A is selected from the following residues (the dashed lines indicate the bonds to Bi and B2):
 In these residues, X may be N, O, or S. Preferably, X is N. Rs may be O, S, or NR™, with Rw being H or C1-6 alkyl. Preferably, Ra is NR™. More preferably, R3 is H. R2 may be H, or C1-6 alkyl. The amine NR5R6 may be selected from:-
In these residues, X may be N, O, or S. Preferably, X is N. Rs may be O, S, or NR™, with Rw being H or C1-6 alkyl. Preferably, Ra is NR™. More preferably, R3 is H. R2 may be H, or C1-6 alkyl. The amine NR5R6 may be selected from:-
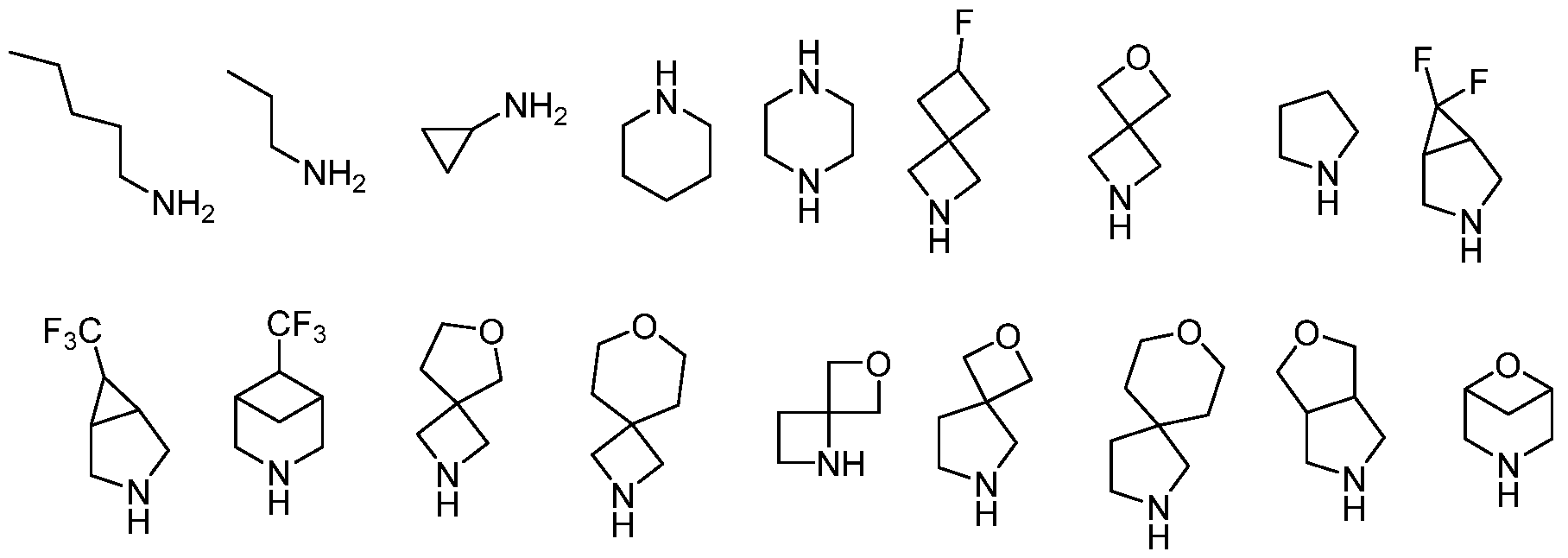
R? may be halogen, methyl, methoxy, tert-butyl, NO2, CN, CF3, OCF3, CF2, COOH, COOCH3, SCH3, CO2NH2, N(CH3)2, NH2, SO2CH3, NHCOOCH3, NHSO2CH3, and NHSO2CF3, and CONR11R12 with R11 and R12 being independently selected from H and C1-6 alkyl. According to one embodiment, Rn and R12 are both H. According to one embodiment, Rn and/or R12 are methyl.
Rs may be H or phenyl. Preferably, Rs is H.
R9 may be methyl, Cs-Cs cycloalkyl, piperidinyl or hydroxyisopropyl. Preferably, Rg is methyl. Xi may be oxygen or methylene. Preferably, Xi is oxygen.
According to one embodiment, A is not a polyether. According to one embodiment, A is not selected from:
 wherein X is N, O, S, preferably O.
wherein X is N, O, S, preferably O.
According to one embodiment, the linkers Bi and B2 are individually selected from an ester, an ether, an amine, a thioether, an amide; and a sulfonamide.
One of Bi and B2 may be absent. If A is not a bond, both Bi and B2 may be absent. According to one embodiment, the linkers Bi and B2 are selected from:

According to one embodiment of the use, the linkers Bi and B2 are different. According to one embodiment, the linkers Bi and B2 are identical. According to one embodiment of the use, at least one Bi and B2 is an amide. According to a preferred embodiment, the linkers Bi and B2 are both amides. Preferably, the nitrogen is connected to the Ci or C2 and the oxygen is connected to A.
The tripod moieties Ci and C2 may be individually an optionally substituted Cs-e aryl, wherein 0 to 3 C atoms, i.e. 0, 1 , 2, or 3 C atoms are replaced by heteroatoms. The heteroatoms are individually selected from N, O, and S.
Alternatively, the tripod moieties Ci and C2 may be individually an optionally substituted C3-6 cycloalkyl group, wherein 0 to 3 C atoms are replaced by a heteroatom. Ci and C2 may be individually an optionally substituted cyclopropane, cyclobutane, cyclohexane or cyclopentane, wherein 0 to 3 C atoms are replaced by a heteroatom. This means 0, 1 , 2 or 3 C atoms may be replaced by a heteroatom. The heteroatoms are individually selected from N, O, and S.
The tripod moieties Ci and C2 are optionally substituted, which means that the wherein the Ci and C2 may individually contain 1 , 2 or 3 substituents, which are independently selected from the group consisting of hydrogen, alkyl and alkoxy. According to one embodiment, the substituents are selected from hydrogen, methyl and methoxy. Alternatively, Ci and/or C2 are not substituted.
According to one embodiment, the tripod moieties Ci and C2 are independently selected from benzene, pyridine, pyrimidine, substituted pyrrole, and piperidine. According to a preferred embodiment, Ci and C2 are individually selected from:
 According to one embodiment, the tripod moieties Ci and C2 are different. According to one embodiment, Ci and C2 are identical. According to one embodiment of the use, at least one Ci and C2 is a six-membered aryl or heteroaryl ring. According to a preferred embodiment, at least one of Ci and C2 and preferably both of Ci and C2 are selected from benzyl, pyridinyl and pyrimidinyl, more preferably with the following binding positions.
According to one embodiment, the tripod moieties Ci and C2 are different. According to one embodiment, Ci and C2 are identical. According to one embodiment of the use, at least one Ci and C2 is a six-membered aryl or heteroaryl ring. According to a preferred embodiment, at least one of Ci and C2 and preferably both of Ci and C2 are selected from benzyl, pyridinyl and pyrimidinyl, more preferably with the following binding positions.
According to a preferred embodiment, at least one of Ci and C2 and preferably both of Ci and C2 are both pyridinyl groups.
According to one embodiment, the compound of formula (I) is selected from 1-1 , I-2, I-3, I-4, I-5, I-6, I-7, I-8, I-9, 1-10, 1-11 , 1-15, 1-16, 1-17, 1-18, 1-19, I-20, I-22, I-23, I-24, I-25, I-26, I-27, I-28, I-29, I-30, 1-31 , I-32, I-33, I-34, I-35, I-36, I-37, I-38, I-39, I-40, 1-41 , I-42, I-43, I-44, I-45, 1-46, 1-47, 1-48, 1-49, 1-50, 1-51 , I-52, I-53, I-54, I-55, I-56, I-57, I-58, I-59, I-60, 1-61 , I-62, I-63, I-64, I-65, I-66, I-67, I-68, I-69, I-70, 1-71 , I-72, I-73, I-74, I-75, I-76, I-77, I-78, I-79, I-80, 1-81 , I-82, I-83, I-84, I-85, I-86, I-87, I-88, I-89, I-90, 1-91 , I-92, I-93, I-94, I-95, I-96, I-97, I-98, I-99, 1-100, 1-101 , 1-102, 1-103, 1-104, 1-105, 1-106, 1-107, 1-108, 1-109, 1-110, 1-111 , 1-112, 1-113, I- 115, 1-116, 1-117, 1-118, 1-119, 1-120, 1-121 , 1-122, 1-123, 1-124, 1-125, 1-126, 1-127, 1-128, I- 129, 1-130, 1-131 , 1-132, 1-133, 1-134, 1-135, 1-136, 1-137, 1-138, 1-139, 1-140, 1-141 , 1-142, I- 143.
A pharmaceutically acceptable salt may be understood in the broadest sense as any salt of the respective compound of formula (I) that is reasonably usable in a pharmaceutical context. The term “pharmaceutically acceptable” does not necessarily mean that the respective component is indeed used in a pharmaceutical context. This rather refers to the suitability to do so. It will be understood that a pharmaceutically acceptable salt or other component or composition will typically also be inherently usable as being cosmetically acceptable. A pharmaceutically acceptable salt or other component or composition typically bears a low toxicity and can be administered to a human or non-human animal (typically mammal or avian) body without seriously harming this human or non-human animal as well as to a cell culture without seriously harming this cell culture, when administered in reasonable concentrations as used for the compound of formula (I). Typically, a respective counterion of compound of formula (I) bears no higher toxicity as compound of formula (I) to the cell culture or organism of interest.
The counter ion may be monovalent, bivalent or have a higher valency. For instance, a pharmaceutically acceptable salt of the compound of formula (I) may comprise one or more counter ions selected from the group consisting of a proton (H+), sodium ion, potassium ion, magnesium ion, calcium ion, zinc ion, chloride ion, sulfate ion, a (hydrogen) phosphate ion, a nitrate ion, an ion of an organic acid (e.g., an acetate, a fatty acid ion), an ammonium ion, an organic sulfate, and an organic amine.
According to one embodiment, the use is included in a method of in vitro cultivating cells, preferably human or animal cells. According to one embodiment the use is included in one or more of: (a) a method of producing cultivated meat, (b) a method of maintaining, proliferating and differentiating animal cells, (c) a method of producing biopharmaceuticals, and (d) a method of differentiating cells, such as stem cells or muscle cells or fiber cells or fat cells.
According to one embodiment of the use, the compound of formula (I) is present in a concentration of 0.05 to 10 pM. The concentration of the compound may be, for example, 0.05 pM, 0.1 pM, 0.2 pM, 0.3 pM, 0.4 pM, 0.5 pM, 0.6 pM, 0.7 pM, 0.8 pM, 0.9 pM, 1 pM, 2 pM, 3 pM, 4 pM, 5 pM, 6 pM, 7 pM, 8 pM, 9 pM, or 10 pM.
A concentration higher than 10 pM could lead to wastage of expensive reagents without necessarily improving cell culture outcomes. A concentration below 0.05 pM might not stimulate enough cell growth and proliferation.
In one embodiment, the compound of formula (I) is present in a concentration of 0.1 to 5 pM. Maintaining concentrations within a defined range can be cost-effective as it ensures that the appropriate amount of growth factor is used - not too much (wasteful) or too little (ineffective)
According to a further embodiment, the concentration of the compound of formula (I) is 0.2 to 3 pM. A concentration in this range ensures that cells have sufficient quantities of the growth factor to promote cell proliferation and growth.
It is important to consider that the actual concentration of the compound of formula (I) can depend on various factors, including the intrinsic factors such as cell type, genetic factors, age and passage number, and health status and extrinsic factors such as nutrient availability, oxygen levels, temperature, CO2 concentration, culture vessel and surface cell density. Therefore, the most suitable concentration can be chosen based on these considerations to optimize the therapeutic effects of the compound.
The compounds of the invention may be prepared as described in Example 1. Furthermore, compounds 1-1 , I-8, 1-16 1-17, 1-117 and their methods of synthesis have been described in US 5,599,984 A, which is incorporated herein by reference. Compounds I-75, I-76 and 1-116 as well as their methods of synthesis have been described in Schroder et al. 2016, which is incorporated herein by reference. Compound 1-117 and its method of synthesis has been described in US 5,750,573, which is incorporated herein by reference.
The medical use
The compounds of formula (I) may be used for treatment of diseases or conditions of patients, which may be treated or ameliorated by activation of the FGFR1 signaling pathway in cells of the patient. This activation leads to an increase in cell proliferation in the patient.
The compounds may in particular be useful for the treatment of the following disorders of conditions:
Wound healing and tissue regeneration: Compounds that promote cell proliferation can accelerate the wound healing process by stimulating the growth of skin cells (keratinocytes), fibroblasts, and endothelial cells. This can lead to faster wound closure, granulation tissue formation, and angiogenesis (formation of new blood vessels). In injuries involving tendons, ligaments, or cartilage, promoting cell proliferation can potentially aid in quicker tissue regeneration and recovery. FGFR signaling, in general, has been shown to play roles in promoting tissue repair and wound healing. Upregulating FGFR1 signaling may have potential benefits in this context.
Osteoporosis: Osteoporosis is characterized by a reduction in bone density due to an imbalance in the bone remodeling process. Compounds that promote the proliferation of osteoblasts (bone-forming cells) can potentially restore this balance, leading to increased bone formation and improved bone density. In osteoporotic patients, fractures are a significant concern. Enhancing cell proliferation might also accelerate the fracture healing process. FGFR1 signaling has a role in bone formation and maintenance. Increasing its signaling could potentially promote osteoblast activity, which may be beneficial in conditions of decreased bone density or osteoporosis.
Certain types of neural damage or degenerative diseases: FGFR1 signaling has been implicated in the survival and regeneration of certain neuron populations. Hence, enhancing its activity could be explored in the context of promoting neural regeneration or protecting neurons in diseases like Parkinson's. In conditions like Parkinson's disease, Alzheimer's disease, or Huntington's disease, where specific neuronal populations are lost, stimulating the proliferation of neural progenitor cells or specific subsets of neurons might have
therapeutic benefits. In the case of nerve injuries or certain degenerative conditions like peripheral neuropathies, compounds that promote the proliferation of neurons or supporting cells (like Schwann cells) can aid in neural tissue regeneration.
The compounds of formula (I) that increase FGFR1 signaling will have a role in enhancing organ regeneration or repair in transplantation medicine.
Promotion of angiogenesis: After an organ transplant, re-establishing blood supply is critical. FGFR1 signaling can promote angiogenesis, which is the formation of new blood vessels. Compounds that enhance this signaling pathway could potentially speed up the process of revascularization in the transplanted organ. Stimulating cell proliferation and differentiation: Proper organ function often requires the growth and differentiation of specific cell types. FGFR1 signaling can support the proliferation of progenitor cells and guide the differentiation of certain cell lineages, aiding in tissue repair and regeneration. Reduction of fibrosis: FGFR signaling might help reduce tissue fibrosis, a process where normal tissue is replaced with scar tissue. In the context of organ transplantation, controlling fibrosis can be crucial for the long-term success of the transplant, as excessive fibrosis can impair organ function. Protection against ischemia-reperfusion injury: This type of injury can occur when blood supply returns to the tissue after a period of lack of oxygen (ischemia). It's a significant concern in organ transplantation. FGFR signaling has the potential to protect tissues against such injuries. Pre-transplant Organ Cultivation: For organs grown ex vivo (outside the body), such as in bioengineered organs or tissues, promoting cell proliferation can accelerate the growth and maturation of the organ, making it suitable for transplantation sooner. Posttransplant Organ Integration: After an organ is transplanted, it is crucial that it integrates well with the recipient's body. Compounds that promote cell proliferation could aid in the faster establishment and growth of the transplanted organ, improving its function and longevity.
Enhancing FGFR1 signaling will have therapeutic effects in the contexts of burned lesions: Wound Healing: FGFR signaling plays roles in promoting tissue repair and wound healing. Upregulating FGFR1 signaling could potentially accelerate the healing process in burn wounds. Promoting cell proliferation can accelerate the healing process of the burn wound by enhancing tissue regeneration. Angiogenesis: Restoration of blood flow to burned tissue is essential for healing. FGFR1 can promote angiogenesis (formation of new blood vessels), which may speed up revascularization of the affected tissue. Reduction of Scarring: FGFR signaling might help modulate the wound healing response in a manner that reduces excessive fibrosis and scarring, which are common complications of severe burns. Promoting cell proliferation can by facilitating a more efficient regenerative process, the formation of scar tissue might be minimized.
Enhancing FGFR1 signaling will have a therapeutic effect in the contexts of diseases with disturbed cell renewal: Skin disorders: For conditions like psoriasis, where there is disturbed skin cell renewal, modulating FGFR1 signaling could help normalize the skin cell growth cycle. Moreover, conditions like psoriasis or eczema can benefit from compounds that promote skin cell proliferation, leading to the renewal and repair of skin layers. Hair Growth: FGFR signaling is involved in hair follicle regulation. If disturbances in hair growth are related to compromised FGFR signaling, enhancing this pathway might be beneficial. Certain hair loss disorders result from a disturbance in hair follicle cell proliferation. Promoting cell growth can potentially stimulate hair regrowth.
Enhancing FGFR1 signaling will have a therapeutic effect in the contexts of Diseases associated with muscle wasting. Promotion of Muscle Cell Growth: FGFR1 signaling has roles in muscle cell differentiation and proliferation. In diseases such as muscular dystrophy or in conditions like cachexia (seen in some cancer patients), increasing FGFR1 signaling could potentially promote muscle regeneration. Promoting muscle cell proliferation can potentially counteract muscle atrophy seen in diseases like muscular dystrophy or conditions like cachexia. Stimulation of satellite cells: Satellite cells are muscle stem cells that play a crucial role in muscle repair. Compounds that promote their proliferation can be beneficial for muscle recovery. FGFR1 signaling might help in the activation and differentiation of these cells, aiding muscle repair and regeneration.
According to a second aspect, the invention provides a compound of formula (I) as defined in the first aspect, or a pharmaceutically acceptable salt thereof for use in medical treatment, wherein the medical treatment is a method of wound healing, a method of organ regeneration in transplantation medicine, a method of treating a burned lesion, a disease associated with disturbed cell renewal, or a disease associated with muscle wasting, wherein the treatment or prevention may optionally comprise tissue engineering.
In other words, the second aspect relates to a method of treatment wherein the medical treatment is a method of wound healing, a method of treating osteoporosis, a method of organ regeneration in transplantation medicine, a method of treating a burned lesion, a disease associated with disturbed cell renewal, or a disease associated with muscle wasting, wherein the treatment or prevention may optionally comprise tissue engineering.
In the use of the method, the patient is administered with a sufficient amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof. As used herein, in the context of
treatment or prevention, the terms “sufficient amount” and “appropriate amount” should be understood interchangeably.
A medicament of the present invention may have any galenic form. A medicament may be in any dosage for e.g., an ingestible composition (e.g., a pill, a dragee, a syrup, a drinkable liquid), an inhalable composition (e.g., a spray), a dosage form penetrating the skin (e.g., a cream, a lotion, a plaster, a suppository, an eye drop, a spray), or an injectable composition. It is preferably (essentially) sterile and is preferably a-pyrogenic.
In this context, preferably, the patient suffers from a disease associated with (dysregulated) cellular growth and/or FGF-mediated (in particular FGF2-mediated) activity, and/or disturbed FGFR signaling (in particular FGFR1 signaling).
The terms “patient”, “individual” and “subject” may be understood interchangeably in the broadest sense as any animal or human that expresses FGFR1 that can be modulated, in particular stimulated or a pharmaceutically acceptable salt thereof. Preferably, such patient is a human or other mammal, in particular a human.
The compounds of formula (I)
While some to the compounds of formula (I), such as 1-1 , I-8, 1-16, 1-17, I-75, I-76, 1-116 or I- 117, have been previously described as compounds for treating cachexia or the inhibition of arginine uptake in the treatment of tumors or infections other compounds are described for the first time. Thus, according to a third aspect, the invention provides a compound (I) as defined in the first aspect, provided that the compound is not 1-1 , I-8.I-9 1-16,1-17, I-75, I-76, 1-116 or 1-117.
According to one embodiment, if Ci and C2 are both benzyl, and/or all four of Di , D2, D3 and D4 are
 or only Di and D3 and both D2 and D4 are hydrogen, the core moiety A is not a Ge aryl or C2-8 unsubstituted alkane.
According to one embodiment, Ci and C2 are not both benzyl rings. According to one embodiment, the core moiety A is not a six-membered aryl or heteroaryl or a C2-8 unsubstituted alkane.
or only Di and D3 and both D2 and D4 are hydrogen, the core moiety A is not a Ge aryl or C2-8 unsubstituted alkane.
According to one embodiment, Ci and C2 are not both benzyl rings. According to one embodiment, the core moiety A is not a six-membered aryl or heteroaryl or a C2-8 unsubstituted alkane.
The cell culture medium
The compounds of formula (I) may be used in specialized cell culture media for survival, proliferation, migration, invasion, angiogenesis, sternness, and differentiation of animal cells.
As indicated above, a cell culture may benefit from the use of one or more compounds of formula (I) or pharmaceutically acceptable salt thereof. Cells of interest may be contacted with the one or more compounds of formula (I) or pharmaceutically acceptable salts thereof by any means. Preferably, the cells of interest may be contacted with the one or more compounds of formula (I) or pharmaceutically acceptable salts thereof by the presence of the latter in a cell culture medium. Such cell culture medium bears technically special characteristics.
Accordingly, the present invention relates further to a cell culture medium comprising at least one compound of formula (I) or a pharmaceutically acceptable salt thereof as defined in the first aspect.
According to a fourth aspect, the invention provides a cell culture medium comprising at least one compound of formula (I) or a pharmaceutically acceptable salt thereof as defined in the first aspect, preferably wherein the cell culture medium comprises or consists of:
(A) water,
(B) nutrients sufficient for cell growth, including at least one carbon source and one nitrogen source,
(C) at least one compound of formula (I) as defined in the first aspect or a pharmaceutically acceptable salt thereof, preferably from in a concentration of 0.05 to 10 pM, preferably in a concentration of 0.1 to 5 pM, more preferably, in a concentration of 0.2 to 3 pM, and
(D) one or more salts acceptable in cell culture,
(E) optionally one or more buffer agents,
(F) optionally one or more gel-forming materials, and
(G) optionally one or more colorings acceptable in cell culture, preferably wherein the cell culture medium is a liquid cell culture medium.
A cell culture medium may be any medium that is suitable to allow maintenance and viability and preferably proliferation of cells of interest. The person skilled in the art knows a variety of commercially available cell culture media. Typically, a cell culture medium at least comprises water and nutrients.
The cell culture medium of the present invention may comprise any contents of at least one compound of formula (I) or a pharmaceutically acceptable salt thereof. For instance, it may comprise 0.01 nM to 10 mM, or 0.05 nM to 1 mM, or 0.1 nM to 100 pM, or 0.5 nM to 50 pM, or 1 nM to 10 pM, or 5 nM to 1 pM, or 10 nM to 500 pM, of at least one compound of formula (I) or a pharmaceutically acceptable salt thereof.
It will be understood that a cell culture medium typically has an osmolarity that is near by the isotonic osmolarity to avoid disturbance of cells.
It will be understood that a cell culture medium typically has a pH range that is near by the neutral range and slightly basic such as, e.g., in the range of pH 6.0 to 8.0, or pH 6.5 to 7.8, or pH 7.0 to 7.7, or pH 7.2 to 7.5, or (approximately) pH 7.4. The person skilled in the art knows adequate buffers to achieve such pH range such a hydrogen phosphate/phosphate buffers, MES (2-(N-morpholino)ethanesulfonic acid) buffers, etc. The buffer system may also comprise an open buffer based on, e.g., addition of gaseous CO2 (e.g., approximately 5% during cultivation of the cells).
The person skilled in the art will be able to choose and use commercially available cell culture media accordingly. The compounds of formula (I) may be used as cell culture medium supplement such as, e.g., for cultivated meat production. The compounds may particularly support serum-free cell culture media and may partly replace growth factor activities, in particular FGF activities and thereby of serum.
The cell culture medium may be liquid, viscous, or solid. Preferably, a cell culture medium is a liquid cell culture medium. Alternatively, it may be a (hydro)gel on or in which cells are cultivated, either in contact with air or covered by another cell culture medium, which is preferably liquid.
As indicated above, one or more compounds of formula (I) or a pharmaceutically acceptable salts thereof may partly or completely substitute one or more growth factors such as FGF, in particular FGF2. Therefore, culturing cells when omitting such one or more growth factors may be enabled.
In one embodiment, the cell culture medium is further characterized in that it does (essentially) not contain serum of animal origin, any subtype of FGF of animal origin, any peptide growth factor of animal origin, in particular any peptide growth factor or steroid growth factor of animal origin, in particular any steroid growth factor.
The composition of serum is complex and not fully defined. It contains various growth factors, hormones, and other components that can influence cell behavior, but their exact concentrations and roles may not be well-understood. This lack of definition can lead to difficulties in interpreting experimental results. Serum composition can vary significantly between different batches and sources, leading to inconsistencies in cell culture conditions. This variability can affect cell growth, differentiation, and experimental outcomes, making it challenging to reproduce results. Proteins derived from animal sources, such as bovine serum albumin (BSA) or bovine transferrin, can also exhibit batch-to- batch variability. Similar to serum, proteins from animal origin can be expensive, especially if large quantities are required for extensive cell culture studies.
In one embodiment, the cell culture medium does (essentially) not contain FGF2, in particular no FGF2 of animal origin. FGF2 is the compound to be replaced by the compounds of the invention. Moreover, the combination of FGF2 and the compounds of the invention does not have an additive effect.
The method of cultivation
As laid out above, the present invention also allows efficient cultivation of cells. Thus, a fifth aspect, the invention provides a method for the cultivation of cells, comprising the steps of
(i) providing:
(A) a cell culture medium; and
(B) cells;
(ii) adding the compound of formula (I) or a pharmaceutically active salt as defined in the first aspect to the cell culture medium;
(iii) subjecting the cell culture medium of step (ii) to conditions sufficient for establishing cell growth and/or for cell differentiation; and
(iv) obtaining cultivated cells, and
(v) optionally separating the cultivated cells from the cell culture medium.
The cells preferably comprise FGFR1 , which signaling pathway is activated by a compound of formula (I) as defined in the first aspect. Depending on the application of the cell culture, the cells may be from human or animal source. For the production of food products, the cells may be for example obtained from the following animals: cattle (bovine), the cells of which are one of the primary sources for cultivated beef production; Chicken (poultry), the cells of which are used to produce cultivated chicken meat; pigs (swine), the cells of which are used for cultivated pork production; fish (e.g., salmon, tuna), the cells of can be used to produce cultivated fish fillets; ducks, the cells of which can be a potential source for cultivated duck meat; sheep, the cells of which can be used to produce cultivated lamb or mutton; rabbits, the cells of which can be used to produce cultivated rabbit meat; quails: quails are a potential source for cultivated quail meat; turkey, the cells of which can be used to produce cultivated turkey meat; deer, the cells of which cells can be used to produce cultivated venison.
In the field of cultivated meat production, several cell types can be used to produce meat-like products. These cells are isolated from living animals and then cultured in vitro to grow and multiply, ultimately forming muscle tissue. Some common cell types used for cultivated meat production are introduced in the following. Myoblasts are muscle precursor cells that can differentiate and fuse to form multinucleated myotubes, which eventually mature into muscle fibers. Myoblasts are a primary choice for cultivated meat production due to their natural ability to develop into muscle tissue. Satellite cells are a type of stem cell found in skeletal muscle. These cells play a crucial role in muscle regeneration and repair. When activated, satellite cells can proliferate and differentiate into myoblasts, making them suitable candidates for cultivated meat. Fibroblasts are connective tissue cells responsible for synthesizing the extracellular matrix. They can be reprogrammed into myogenic (muscle-forming) cells using genetic or chemical methods, making them a potential cell source for cultivated meat production. Adipocytes are fat cells, and their inclusion in cultivated meat production is essential for producing meat products with appropriate marbling and fat content. Endothelial cells form the inner lining of blood vessels. Their presence is crucial for establishing a vascular network within the cultured meat to supply nutrients and oxygen and remove waste products. Pericytes are cells found in association with blood vessels. They play a role in vascular development and regulation and can contribute to the formation of vascular structures within the cultured meat. Embryonic stem cells (ESCs) or induced pluripotent stem cells (iPSCs) have the potential to differentiate into various cell types, including muscle cells, making them versatile candidates for cultivated meat production.
In a preferred embodiment, the cell culture medium is serum-free, more preferably wherein the cell culture medium is free of FGF2, in particular free of all subtypes of FGF.
The cells may be cultivated at any conditions suitable for this purpose. For instance, these may be cultivated at a temperature between 20°C and 45°C, preferably between 25°C and 42°C, more preferably between 28°C and 40°C, even more preferably between 30°C and 39°C, even more preferably between 32°C and 39°C, even more preferably between 34°C and 39°C, even more preferably between 35°C and 39°C, even more preferably between 36°C and 38°C, in particular at (approximately) 37°C. The temperature may be adapted to the used cell type. For avian cells, for instance, the temperature range may be adapted accordingly.
Cell culture may be conducted on a short time scale or on a long time scale. For instance, cells may be contacted with a cell culture medium comprising compound of formula (I) or a pharmaceutically acceptable salt thereof for a tame range of less than 30 min, between 30 min and 1 hour, between 1 and 2 hours, between 2 and 12 hours, between 6 and 24 hours, between 1 and 2 days, between 2 and 7 days, between 1 and 2 weeks, between 1 week and 1 month, between 2 weeks and 2 months, between 1 and 6 months, or even more than 6 months.
Addition of the compound of formula (I) or a pharmaceutically acceptable salt thereof may be performed by any means. The compound of formula (I) or a pharmaceutically acceptable salt thereof may be added during cultivation. It may be added as such, i.e., as pure substance, may be added in a dilution in a stock solution of higher concentration, or may be added by replacing the cell culture medium. Cultivating with the compound of formula (I) or a pharmaceutically acceptable salt thereof may be conducted once, twice or more often or may be constantly. The concentrations of compound of formula (I) or a pharmaceutically acceptable salt thereof may be maintained constant during cultivation of cells or may be varied by any profile, e.g., increased or decreased over time.
Separating the cultivated cells from the cell culture medium may be conducted by any means. If the cells are adherently grown, these may, for instance, be detached by mechanical scraping and/or enzymatically (e.g., by using a trypsin or other digestive enzyme composition). Suspended cells may be separated from the cell culture medium by centrifugation and/or filtration (e.g., dead end filtration or cross-flow filtration).
The method can be included in a method of producing a food product, in particular cultivated meat. In this context, the method of cell expansion further contains a step of separating the cultivated cells from the cell culture medium.
The method of producing cultivated meat may contain the following steps:
(1) Cell Isolation;
(2) Cell Expansion;
(3) Cell Differentiation;
(4) Scaffold and 3D Bioprinting (Optional);
(5) Harvesting and Processing;
(6) Packaging and Distribution;
The first step in cell cultivation is to obtain cells from a living donor animal. This can be done through a biopsy or tissue sample, which typically contains muscle cells, also known as myocytes. The donor animal's welfare is a crucial consideration, and efforts are made to minimize any harm during the cell collection process.
Starting from the cultured cells, the next step in the generation of cultivated meat, is to develop the cultured cells into functional muscle tissue. For this, the cultured cells need to differentiate into specific cell types. This process involves exposing the cells to specific biochemical and mechanical cues that mimic the natural environment of muscle tissue development.
In some cases, a scaffold made of biocompatible materials can be used to provide structural support for the growing cells. Additionally, 3D bioprinting techniques can be employed to create complex tissue structures, enhancing the organization and functionality of the cultured meat.
The cultured cells are further cultivated and matured to promote the formation of muscle tissue. This maturation process aims to develop a product that closely resembles traditional meat in terms of texture, taste, and nutritional content. Once the cultivated meat reaches the desired level of maturity, it is harvested and processed into various meat products, such as burgers, sausages, or nuggets, using conventional food processing techniques. The cultivated meat products are packaged and prepared for distribution to consumers or food outlets.
The food product
The cultivated cells or - in case of a further tissue engineering step - material derived therefrom may further be treated to prepare a food product. Thus, according to a sixth aspect, the invention provides a food product comprising cultivated cells obtainable by the process
according the fifth aspect or material derived therefrom, preferably wherein the food product is cultivated meat or a drinkable composition.
The cultivated meat according to the invention may be for example cultivated beef, cultivated chicken, cultivated pork, cultivated fish (e.g., salmon, tuna), cultivated duck, cultivated lamb or mutton, cultivated rabbit, cultivated quail, cultivated turkey, cultivated venison.
According to one embodiment, the food product is cultivated meat or a drinkable composition. The cell culture medium and the food product of the present invention may be provided in any package. Depending on the intended use, it may be provided in different packaging. It may be stored at any condition suitable for this purpose such as, e.g., at ambient temperature (e.g., 18 to 30°C, preferably 18 to 25°C), in a fridge (e.g., at 0 to 15°C, preferably 3 to 10°C), in a freezer (e.g., -30 to 0°C, preferably -25 to -10°C), in a deep freezer (e.g., -100 to -300°C, preferably -90 to -55°C), on liquid nitrogen, on dry ice, or even one or more liquid noble gases. For instance, it may be provided in a flask, a bottle, another container, or a package for solid material. It may be stored at dry state, as a (hydro)gel, as a suspension, emulsion, colloid or solution.
EXAMPLES
Example 1 - Synthesis of the aminoguanidine compounds
The following illustrates the synthesis of various compounds of the present invention. Additional compounds within the scope of this invention may be prepared using the methods illustrated in these Examples, either alone or in combination with techniques generally known in the art. All starting materials in these Preparations and Examples are either commercially available or can be prepared by methods known in the art or as described herein.
All reactions were carried out using continuous stirring unless otherwise noted. When appropriate, reaction apparatuses were dried under dynamic vacuum using a heat gun, and anhydrous solvents (Sure-Seal™ products from Merck KGaAr Acroseal™ products from Thermo Fischer Scientific inc.) were employed. Commercial solvents and reagents were used without further purification. For syntheses referencing procedures in other Examples or Methods, reaction conditions (reaction time and temperature) may vary.
Reaction progress was monitored using thin-layer chromatography (TLC), or ultra-high performance liquid chromatography-mass spectrometry (UHPLC-MS). TLC was performed
on pre-coated silica gel plates with a fluorescence indicator (254 nm excitation wavelength) and visualized under UV light. UHPLC-MS data were acquired on an Agilent Infinity II 1260 instrument with a 1260 Infinity II liquid autosampler, Poroshell 120 EC-C18 columns, acetonitrile/water gradients, and formic acid modifier. The column eluate was analysed using a 1260 Infinity II Diode Array Detector WR scanning from 200 to 400 nm and LC/MSD iQ mass spectrometer scanning in both positive and negative ion modes from 140 to 1000 Da. Purifications were performed by automated flash chromatography (FC) using Pure C-810 Buchi instrument and pre-packed BGB Scorpius C18-HP 100 A or Silica 60 A cartridges or HPLC Agilent Infinity II Preparative HPLC using InfinityLab ZORBAX Eclipse Plus C18 column. UV detection was used to trigger fraction collection.
Purifications may vary in general, solvents and the solvent ratios used for eluents/gradients were chosen to provide appropriate Rfs or retention times.
Mass spectrometry data are reported from UHPLC-MS analyses. Mass spectrometry (MS) was performed via electrospray ionization (ESI).
Proton nuclear magnetic resonance spectroscopy (1 H NMR) chemical shifts are given in parts per million downfield from tetramethylsilane and were recorded on 300 or 400 MHz Bruker spectrometers. Chemical shifts are expressed in parts per million (ppm, 5) referenced to the deuterated solvent residual peaks (chloroform, 7.26 ppm; dimethyl sulfoxide-, 2.50 ppm). The peak shapes are described as follows: s, singlet; d, doublet; t, triplet; q, quartet; quin, quintet; m, multiplet; br s, broad singlet; app, apparent.
Unless otherwise noted, chemical reactions were performed at room temperature (about 23 °C).
Unless noted otherwise, all reactants were obtained commercially and used without further purification or were prepared using methods known in the literature.
The terms “concentrated”, “evaporated”, and “concentrated in vacuo” refer to the removal of solvent at reduced pressure on a rotary evaporator with a bath temperature less than 60° C. The abbreviations “min” and “h” stand for “minutes” and “hours,” respectively. The term “TLC” refers to thin-layer chromatography, “room temperature or ambient temperature” means a temperature between 18 to 25° C., “, “UHPLC” refers to ultra-high performance liquid chromatography, “HPLC” refers to high-performance liquid chromatography, FC refers to automated flash chromatography.
Chemical abbreviations:
Ac: acetyl.
ACN: acetonitrile.
AC2O: acetic anhydride.
AcOH: acetic acid.
Bl NAP: (2,2'-bis(diphenylphosphino)-1 ,1 '-binaphthyl)
Boc: terf-butyloxycarbonyl
(BPin)2: Bis(pinacolato)diboron
BTMG: 2-tert-Butyl-1 , 1 ,3,3-tetramethylguanidine.
Cone.: concentrated.
DCM: Dichloromethane.
DIPEA: N,N-Diisopropylethylamine.
Dippf: 1 , 1 '-bis(di-isopropylphosphino)ferrocene.
DMF: dimethylformamide.
DMSO: dimethylsulfoxide.
Dppf: 1 , 1 -ferrocenediyl-bis(diphenylphosphine).
EtsN: triethylamine.
EtOAc: ethylacetate.
EtOH: ethanol.
[lr{dF(CF3)ppy}2(dtbpy)]PF6: [4,4'-S/s(1 , 1 -dimethylethyl)-2,2'-bipyridine-A/1 , A/1 ']£>/s[3,5- difluoro-2-[5-(trifluoromethyl)-2-pyridinyl-A/]phenyl-C]lridium(l 11) hexafluorophosphate.
MCPBA: mefa-chloroperoxybenzoic acid.
MeOH: methanol.
MeCN: acetonitrile.
NMI: M-methylimidazole.
PCya: tricyclohexylphosphine
PPha: triphenylphosphine.
PyBOP: benzotriazol- 1-yloxytripyrrolidinophosphonium hexafluorophosphate.
Rpm: revolution per minute.
Rt: room temperature
TCFH: Chloro-N,N,N',N'-tetramethylformamidinium hexafluorophosphate
THF: tetrahydrofuran.
1.2 Synthesis of Intermediates
General procedure 1. Deprotection of methyl esters.
Methyl ester was dissolved in mixture of THF/MeOH/Water (1 :1 :1) and LiOH was added. The resulting mixture was stirred overnight. After the completion of the reaction, 1 M HCI was added (5-10 ml) and the mixture was extracted with EtOAc. The organic layers were
combined, dried over sodium sulfate and evaporated under the reduced pressure obtaining intermediate.
Intermediate 1.
Step 1 :

To a suspension of 5-nitroisophthalic acid (10 g, 60 mmol) in 20 mL of thionyl chloride, 5 drops of dry DMF were added and the reaction mixture was refluxed for 2 h giving a paleyellow solution. The solvent was removed in vacuo yielding a yellow solid, which was used without further purification for the next step.
Diethyl malonate (17.5 ml, 0.11 mmol) was dissolved in 225 mL of THF, 60% sodium hydride in mineral oil (5 g, 0.12 mol) of was added portion wise and the mixture was heated for 3 h under reflux. To this solution 5-Nitroisophthalic acid dichloride acquired in the previous step was dissolved in 40 ml of THF and was added dropwise. After complete addition, the solution was refluxed overnight, and then the solvent was evaporated under reduced pressure. The brown slurry was dissolved in 100 mL of DCM and 100 ml of water, separated and aqueous layers were extracted with DCM (2*100 ml). DCM was removed in vacuo yielding a yellow oil. The oil was dissolved in a mixture of glacial acetic acid/conc. sulfuric acid/water (100 mL: 5 mL: 20 mL) and refluxed overnight. After that, 250 mL water was added, and the mixture was extracted with DCM (3*200 mL). The organic layers combined, the solvent was removed in vacuo and the resulting residue was purified by flash chromatography on silica gel eluting with 0-60% Cyclohexane- EtOAc obtaining the desired product as a slightly yellow solid in 25% yield (2.5 g).
MS (M+H) +: 208.4
1H NMR (300 MHz, CDCI3) 6 8.87 (d, J = 1.6 Hz, 2H), 8.74 (t, J = 1.6 Hz, 1 H), 2.68 (s, 6H). 13C NMR (75 MHz, CDCI3) 6 194.96, 148.82, 138.78, 132.73, 126.63, 26.80.
Step 2.

In a round-bottom flask tin (ll)chloride (10.2 g, 45 mmol) was dissolved in 33 of mL cone. HCI and warmed up to 50 °C followed by the slow addition of 2.5 g of product of the step 1. Afterwards, the solution was stirred for further 15 minutes and then poured, under gas formation, in a mixture of 55 g potassium carbonate and 200 mL ice/water. The product was extracted with EtOAc and the combined organic layers were dried over sodium sulfate. Finally, the solvent was removed in vacuo obtaining a bright yellow solid in 54% yield (1.2 g). MS (M+H) +: 177.9
1H NMR (300 MHz, CDCI3) 7.63 (s, 1 H), 7.36 (s, 2H), 5.63 (s, 2H), 2.55 (s, 6H)
13C NMR (75 MHz, CDCI3) 197.85, 149.44, 137.90, 116.96, 115.48, 26.81
Intermediate 2.

Step 1 :
Catalytic R11CI3 hydrate (60 mg) and NalC>4 (6.7 g, 31 mmol) were added to a stirred solution of a-pinene (1.25 mL, 8 mmol) in 2:2:3 mixture of chloroform/acetonitrile/water (65 mL). After the mixture was stirred at room temperature for 24 h, diethylether (100 mL) was added and the mixture was stirred for 5 min and extracted with diethylether. Both organic and water layers were filtered through celite, and water layer was extracted with 100 ml diethylether more. The combined organic extracts were dried (Na2SO4) and concentrated under reduced pressure affording quantitatively crude pinonic acid, which was used without additional purification.
MS (M+H) +: 185.0.
1H NMR (300 MHz, CDCI3) 5 2.80 (dd, J = 10.4, 7.8 Hz, 1 H), 2.51 - 2.28 (m, 3H), 2.24 - 2.06 (m, 1 H), 2.03 - 1.85 (m, 1 H), 1.27 (s, 3H), 1.03 (s, 3H).
13C NMR (75 MHz, CDCI3) 6 178.99, 178.75, 46.10, 42.94, 37.98, 35.09, 29.91 , 24.25, 17.56. Step 2:
To a stirred solution of pinonic acid (1 .5 g, 8 mmol) in dioxane (25 mL), cooled at -15 °C was added an aqueous solution of NaOBr prepared from bromine (1.2 mL, 24 mmol), NaOH (4.1 g, 102 mmol), and water (98 mL), and previously cooled to 0 °C. After being stirred at 0 °C for 2 h and at room temperature overnight, the solution was extracted with dichloromethane. Aqueous Na2SOs and concentrated HCI were subsequently added to the aqueous phase. The resulting acid solution was extracted with diethylether, the extracts were dried (Na2SC>4), and the solvent was removed to afford the product as brown solid pure enough to be used in the next step without further purification in 67% yield (1.05 g).
MS (M+H) +: 187.0.
1H NMR (300 MHz, CDCI3) 6 2.80 (dd, J = 10.4, 7.8 Hz, 1 H), 2.51 - 2.28 (m, 3H), 2.24 - 2.06 (m, 1 H), 2.03 - 1.85 (m, 1 H), 1.27 (s, 3H), 1.03 (s, 3H).
13C NMR (75 MHz, CDCI3) 6 178.99, 178.75, 46.10, 42.94, 37.98, 35.09, 29.91 , 24.25, 17.56.
Intermediate 3.

To 4-bromoisophthalic acid (1.98 g, 8.01 mmol) in methanol (30 mL) was added concentrated H2SO4 (1.5 mL). The mixture was refluxed for 24 h, and then cooled to room temperature. After the solvent was evaporated, CH2CI2 (150 mL) and H2O (100 mL) were added. The organic phase was separated, and the aqueous phase was extracted with CH2CI2. The organic phase was combined, washed with saturated NaHCOs solution, dried over anhydrous MgSCL and filtered. The solvents were removed under reduced pressure obtaining intermediate 3 as white solid in 87% yield (1.9 g).
MS (M+H) +: 272.9.
1H NMR (300 MHz, CDCI3) 6 8.44 (d, J = 2.1 Hz, 1 H), 7.97 (dd, J = 8.4, 2.2 Hz, 1 H), 7.76 (d, J = 8.4 Hz, 1 H), 3.97 (s, 3H), 3.94 (s, 3H).
13C NMR (75 MHz, CDCI3) 6 165.75, 165.54, 134.73, 133.02, 132.36, 129.37, 127.08, 52.70, 52.51.
Intermediate 4.

To a solution of dimethyl 2-hydroxybenzene-1 , 3-dioate (0.5 g, 2.21 mmol) and K2CC>3 (500 mg, 3.6 mmol) in DMF (10 mL) was added CH3I (0.826 ml, 13.26 mmol). The resulting mixture was refluxed at 60 °C overnight. The reaction mixture was diluted with EtOAc and washed with 1 M HCI. After drying over Na2SC>4, the organic layer was filtered and concentrated under reduced pressure. The deprotection was carried out according to general procedure 1. Starting materials: THF/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (500 mg).
Yield: 64% (320 mg).
MS (M+H)+: 222.9.
1H NMR (300 MHz, DMSO) 5 12.44 (s, 2H), 8.22 (s, 1 H), 6.72 (s, 1 H), 3.94 (s, 6H). 13C NMR (75 MHz, DMSO) 5 165.69, 163.42, 136.09, 111.61 , 96.92, 56.17.
General scheme for intermediate 5-7.
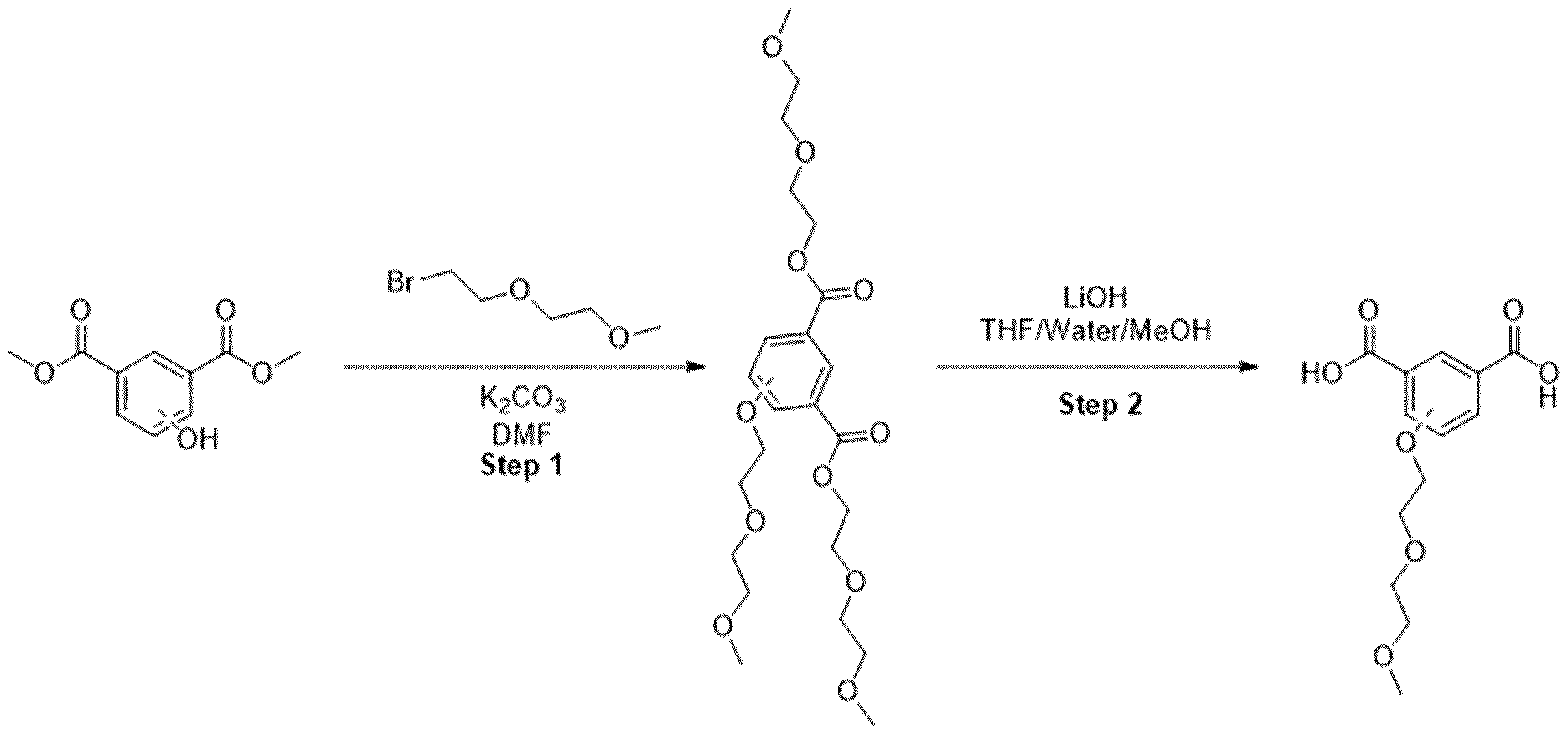
To a solution of dimethyl hydroxybenzene-1 , 3-dioate and K2CO3 (500 mg, 3.6 mmol) in DMF (10 mL) 1-bromo-2-(2-methoxyethoxy)ethane was added. The resulting mixture was refluxed at 60 °C overnight. The reaction mixture was diluted with EtOAc and washed with 1 M HCI. After drying over Na2SO4, the organic layer was filtered and concentrated under reduced pressure. The deprotection was done according to general procedure 1. Starting materials: THF/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (500 mg).
Intermediate 5.
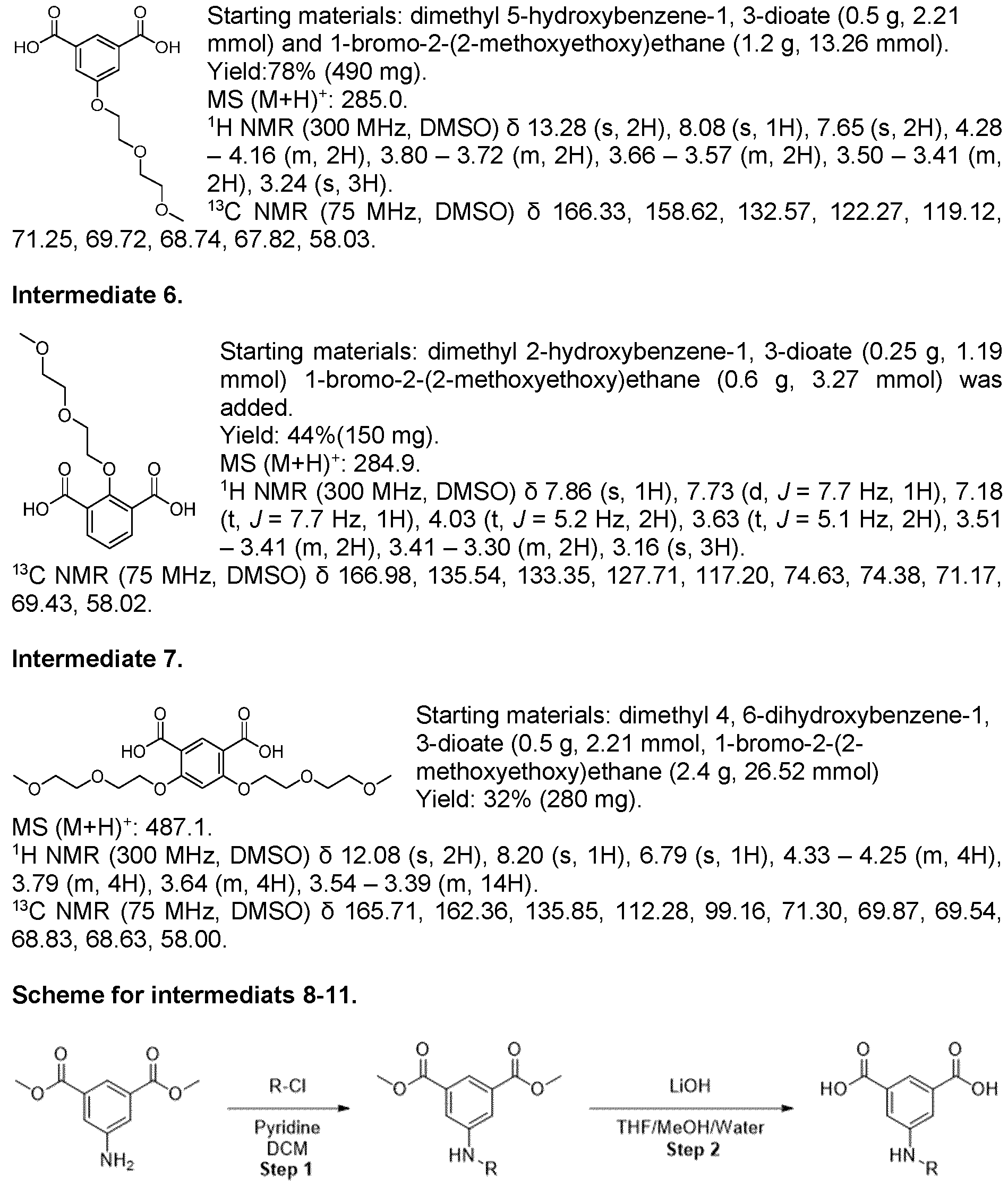
Step 1 :
To a stirred solution of dimethyl 5-aminoisophthalate (500 mg, 2.4 mmol) in dichloromethane (8 mL), pyridine (600 mL, 8 mmol) was added at room temperature. At 0 °C, corresponding anhydride or chloranhydride (2.75 mmol) was added and the resulting mixture was stirred overnight at room temperature. The solvent was evaporated under the reduced pressure, and the residue was used in the next step or purified by silica gel column chromatography eluting with cyclohexane to cyclohexane:ethylacetate 50:50 to obtain the desired product.
Step 2:
Synthesized according to general procedure 1. Starting materials: THF/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (400 mg).
Intermediate 8.

52.60, 39.78.
Step 2:
MS (M-H)-: 257.8.
1H NMR (300 MHz, DMSO) 5 13.35 (s, 2H), 10.20 (s, 1 H), 8.19 (t, J = 1.5 Hz, 1 H), 8.02 (d, J = 1.5 Hz, 2H), 3.04 (s, 3H).
13C NMR (75 MHz, DMSO) 5 166.17, 139.20, 132.31 , 124.94, 123.63. One peak was not observed due to similar shift with DMSO-d6.
Intermediate 11. Starting material: triflic anhydride (760 mg). Yield, over two steps: 45 mg (6%). Step 1 : MS (M+H)+: 341.9. 1H NMR (300 MHz, CDCI3) 6 8.60 (t, J = 1 .4 Hz, 1 H), 8.31 (s, 1 H),
 8.28 (s, 1 H), 4.01 (s, 6H).
8.28 (s, 1 H), 4.01 (s, 6H).
13C NMR (75 MHz, CDCI3) 6 165.64, 135.66, 132.11 , 128.64, 127.03, 53.07 (CF3 carbon was not observed due to low intensity).
Step 2:
1H NMR (300 MHz, DMSO) 5 13.47 (s, 2H), 8.94 (s, 1 H), 8.28 (t, J = 1.5 Hz, 1 H), 8.01 (d, J = 1.5 Hz, 2H).
13C NMR (75 MHz, DMSO) 5 165.81 , 132.53, 126.19 (three carbon signals were not observed due to low intensity).
Intermediate 15.

Step 1 :
A suspension of 5-bromoisophthalic acid dimethyl ester (2.0 g, 7,32 mmol), cyclopropylboronic acid (1 ,0 g, 11 .7 mmol) and potassium phosphate (4.66 g, 21 .97 mmol) in toluene (45 mL) was purged with nitrogen. Pd(OAc)2 (0,49 g, 2.2 mmol) was added, followed by tricyclohexylphosphine (616 mg, 2.25 mmol) and water (1.0 mL). The resulting reaction mixture was heated at 100 °C overnight. After the completion of the reaction, the solvent was evaporated and the residue was purified by flash column chromatography on silica gel eluting with cyclohexane to cyclohexane:ethylacetate 85:15 to obtain the desired product as a white solid.
MS (M+H)+: 234.9.
1H NMR (300 MHz, CDCI3) 6 8.45 (t, J = 1 .6 Hz, 1 H), 7.93 (d, J = 1 .6 Hz, 2H), 3.94 (s, 6H), 2.00 (tt, J = 8.4, 5.1 Hz, 1 H), 1.13 - 0.96 (m, 2H), 0.88 - 0.70 (m, 2H).
13C NMR (75 MHz, CDCI3) 6 166.42, 145.15, 131.09, 130.49, 127.75, 52.27, 15.22, 9.61.
Step 2:
Synthesized according to general procedure 1. Starting materials: THF/MeOH/Water (2 ml/2 ml/2 ml) and LiOH (800 mg).
Yield, over two steps: 630 mg (42%).
MS (M+H)+: 206.8.
1H NMR (300 MHz, DMSO) 5 12.99 (s, 2H), 8.04 (t, J = 1.6 Hz, 1 H), 7.64 (d, J = 1.6 Hz, H), 1.92 (ddd, J = 13.4, 8.4, 5.1 Hz, 1 H), 0.92 - 0.68 (m, 2H), 0.63 - 0.40 (m, 2H).
13C NMR (75 MHz, DMSO) 5 166.62, 145.04, 131.24, 130.08, 127.06, 14.65, 9.79.
Intermediate 16.
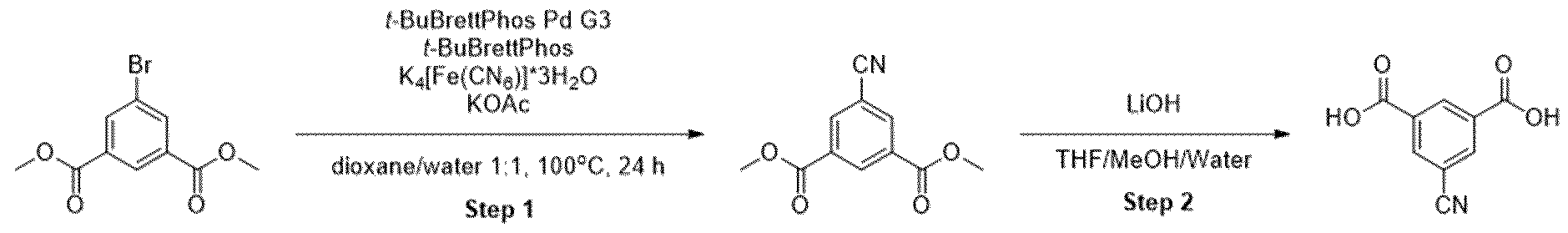
Step 1 :
To a screw-top test tube equipped with a magnetic stir bar was added f-BuBrettPhos Pd G3 (43 mg, 0.05 mmol), f-BuBrettPhos (24 mg, 0.05 mmol), K4[Fe(CN)e]*3H2O (184 mg, 0.5 mmol) and 5-bromoisophthalic acid dimethyl ester (273 mg, 1 mmol). After sealing with a Teflon-lined screw-cap septum, the vessel was evacuated and refilled with nitrogen (three cycles). Degassed dioxane (2.5 mL), KOAc (12 mg, 0.125 mmol) and water (2.5 mL) were then added to the reaction tube via syringe. The test tube was placed in an oil bath preheated to 100 °C. After 4 h of stirring at 100 °C, the reaction mixture was then cooled to room temperature. The contents of the test tube were transferred to a separation funnel using EtOAc (15 mL) and brine (15 mL), and the organic layer was separated from the aqueous layer. The solvents were evaporated and the residue was purified by chromatography on silica gel eluting with cyclohexane to cyclohexane:ethylacetate 50:50 to obtain the product in 72% yield (158 mg).
MS (M+H)+: 210.5.
1H NMR (300 MHz, CDCI3) 6 8.87 (s, 1 H), 8.48 (d, J = 1.6 Hz, 2H), 3.99 (s, 6H).
13C NMR (75 MHz, CDCI3) 6 164.45, 136.93, 134.50, 132.20, 117.14, 113.72, 53.12.
Step 2:
Synthesized according to general procedure 1. Starting materials: THF/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (200 mg).
Yield: 91 % (125 mg).
MS (M-H)-: 189.9.
1H NMR (300 MHz, DMSO) 5 13.65 (s, 1 H), 8.65 (s, 1 H), 8.51 (d, J = 1.6 Hz, 2H). 13C NMR (75 MHz, DMSO) 5 165.00, 136.49, 133.72, 132.66, 117.31 , 112.72.
Intermediate 17.
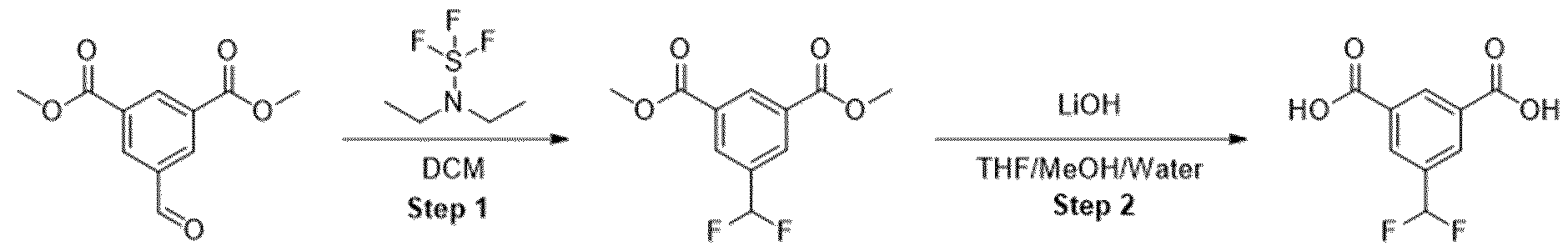
Step 1 :
To a solution of 1 ,3-dimethyl 5-formylbenzene-1 ,3-dicarboxylate (444 mg, 2 mmol) in dichloromethane (13 ml) was added (diethylamino)sulphur trifluoride (0.5 ml, 38.6 mmol) and the reaction mixture was stirred at room temperature for 18 h. To the mixture was added additional dichloromethane and saturated aqueous sodium hydrogen carbonate solution and the two layers were separated. The solvent was evaporated, and the residue was purified by silica gel column chromatography eluting with cyclohexane to cyclohexane:ethylacetate 60:40 to obtain the desired product.
MS (M+H)+: 244.9.
1H NMR (300 MHz, CDCI3) 6 8.71 (s, 1 H), 8.29 (s, 2H), 6.66 (t, J = 56.0 Hz, 1 H), 3.90 (s, 6H). 19F NMR (283 MHz, CDCI3) 6 -111.77 (s, 2F).
13C NMR (75 MHz, CDCI3) 6 165.33, 135.35 (t, J = 23.3 Hz), 132.69 (t, J = 1.6 Hz), 131.43, 130.87 (t, J = 5.9 Hz), 113.46 (t, J = 240.4 Hz), 52.64.
Step 2:
Synthesized according to general procedure 1. Starting materials: THF/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (400 mg).
Yield, over two steps: 370 mg (89%).
MS (M+H)+: 216.9.
1H NMR (300 MHz, DMSO) 5 13.52 (s, 1 H), 8.59 (s, 2H), 8.33 (s, 2H), 7.23 (t, J = 55.4 Hz, 1 H).
19F NMR (283 MHz, DMSO) 5 -106.19 (s, 2F).
13C NMR (75 MHz, DMSO) 5 165.74, 135.12 (t, J = 30.3 Hz), 132.12, 131.98, 130.25 (t, J = 6.0 Hz), 113.85 (t, J = 240.4 Hz).
Intermediate 18.

Step 1 :
A 25 mL round bottom flask equipped with a magnetic stir bar and a condenser was charged with the 1 ,3-dibromo-5-(trifluoromethoxy)benzene (0.6 g, 1.9 mmol) and DMF (4 ml). The reaction mixture was degassed using nitrogen gas. Pd(PPh3)4 (0.65 g, 0.56 mmol) and Zn(CN)2 (0.2422 g, 2.06 mmol) were added afterwards. The mixture was stirred at 85 °C overnight. The solvent was evaporated under the reduced pressure, and the residue was purified by silica gel column chromatography eluting with cyclohexane to cyclohexane: ethylacetate 50:50 to obtain the desired product.
Mass was not observed in ESI.
1H NMR (300 MHz, CDCI3) 6 7.91 (t, J = 1.4 Hz, 1 H), 7.76 (s, 1 H).
19F NMR (283 MHz, CDCI3) 6 -58.06 (s, 3F).
13C NMR (75 MHz, CDCI3) 5 149.62, 147.49, 133.49, 128.30, 119.98 (q, J = 268.0 Hz), 115.98, 115.23.
Step 2:
The product from the previous step in EtOH (6 ml) was treated with 1 N KOH (6 ml, 336 mg of KOH) and refluxed at 80 °C overnight. After cooling to room temperature, the volatiles were removed via rotary evaporation. The mixture was adjusted to pH=l-2 with concentrated HC1 and the mixture was extracted with EtOAc. The combined organics were dried over Na2S04. The organic layers were combined, dried over the sodium sulfate and evaporated under the reduced pressure yielding the product as white solid.
Yield, over two steps: 205 mg (41%).
MS (M-H)-: 248.8.
1H NMR (300 MHz, DMSO) 5 13.41 (s, 2H), 8.45 (t, J = 1.5 Hz, 1 H), 8.03 (s, 2H).
19F NMR (283 MHz, DMSO) 5 -57.05 (s, 1 F).
13C NMR (75 MHz, DMSO) 5 165.19, 148.36, 133.69, 128.61 , 125.35, 119.92 (d, J = 257.8 Hz).
Intermediate 19.
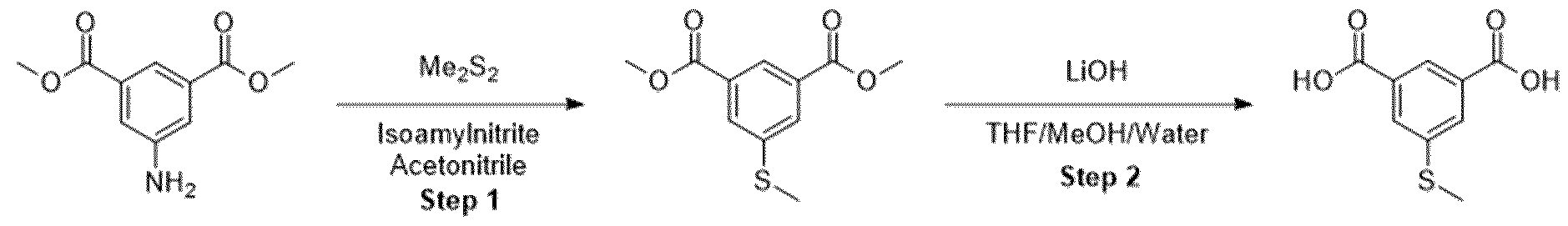 Step 1 :
Step 1 :
To a solution of starting aniline (5 g, 23.8 mmol) in anhydrous acetonitrile (50 mL) at RT was added dimethylsulfide (3.2 ml, 36 mmol) followed by slow addition of isoamyl nitrite (1.6 ml, 12 mmol). The reaction was stirred and heated to reflux for 4 hours then cooled to 60°C and stirred overnight. The solvents were evaporated under the reduced pressure, and the residue was purified by silica gel column chromatography eluting with cyclohexane to cyclohexane:ethylacetate 0:100 obtaining the intermediate as a light orange solid in 8% yield (500 mg).
MS (M+H)+: 240.9.
1H NMR (300 MHz, CDCI3) 6 8.33 (t, J = 1.6 Hz, 1 H), 7.99 (d, J = 1.5 Hz, 2H), 3.87 (s, 6H), 2.48 (s, 3H).
13C NMR (75 MHz, CDCI3) 6 165.91 , 140.42, 131.07, 130.94, 126.96, 52.47, 15.51.
Step 2:
Synthesized according to general procedure 1. Starting materials: product from the previous step (223 mg, 1 mmol), THF/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (400 mg).
Yield: 90% (200 mg).
MS (M+H)+: 212.9.
1H NMR (300 MHz, DMSO) 5 13.42 (s, 2H), 8.26 (t, J = 1.6 Hz, 1 H), 8.01 (d, J = 1.5 Hz, 2H), 2.63 (s, 3H).
13C NMR (75 MHz, DMSO) 5 166.16, 140.05, 131.89, 129.74, 126.13, 14.40.
Intermediate 20.

Step 1 :
To a solution of product from step 1 in the synthesis of intermediate 19 (223 mg, 1 mmol) in DCM (10 mL) was added 70% mCPBA (614 mg, 2.5 mmol) and the reaction was stirred at RT overnight. The final mixture was diluted with saturated aqueous sodium bicarbonate and extracted with DCM. The combined organic layers were dried over Na SO and the solvents were evaporated under the reduced pressure. The residue was purified by silica gel column chromatography eluting with cyclohexane to cyclohexane:ethylacetate 0:100 obtaining the intermediate as a white solid.
MS (M+H)+: 272.9.
1H NMR (300 MHz, CDCI3) 6 8.96 (t, J = 1.6 Hz, 1 H), 8.80 (d, J = 1.6 Hz, 2H), 4.03 (s, 6H), 3.14 (s, 3H).
13C NMR (75 MHz, CDCI3) 6 161.81 , 144.16, 132.31 , 125.17, 52.97, 44.33.
Step 2:
Synthesized according to general procedure 1. Starting materials: THF/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (400 mg).
Yield, over two steps: 95 mg (39%).
MS (M+H)+: 244.8.
1H NMR (300 MHz, DMSO) 5 13.81 (s, 2H), 8.70 (t, J = 1.6 Hz, 1 H), 8.60 (d, J = 1.6 Hz, 2H), 3.36 (s, 3H).
13C NMR (75 MHz, DMSO) 5 165.24, 142.03, 134.10, 132.71 , 131.43, 43.22.
Intermediate 22.
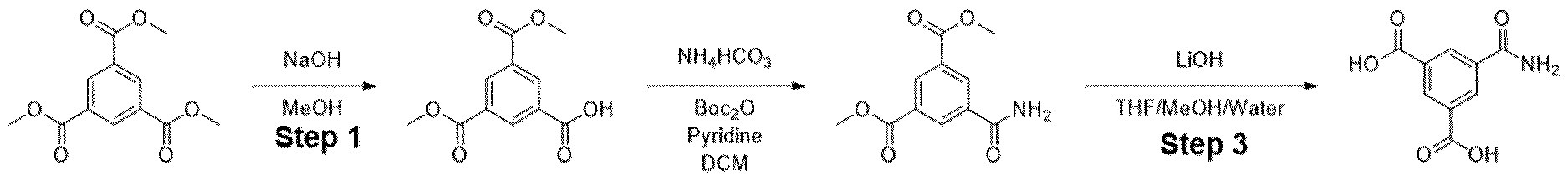
Step 2
Step 1 :
Trimethyl 1 ,3,5-benzenetricarboxylate (2.00 g, 7.93 mmol) was suspended in methanol (180 mL) and I M sodium hydroxide solution (7.14 mL, 280 mg of sodium hydroxide) was added. The mixture was stirred at room temperature for 18 h. The resulting solution was concentrated under reduced pressure to afford a white solid which was partitioned between dichloromethane (150 mL) and saturated aqueous sodium, bicarbonate solution (150 mL). The organic phase was separated and was extracted with saturated aqueous sodium bicarbonate solution (150 mL) before being discarded. The combined aqueous layers were acidified to pH 1-2 using concentrated hydrochloric acid and were extracted with ethyl acetate (2*150 mL). The combined organic layers were dried (MgSCL) and concentrated to give the desired product as a white solid in 58% yield (1 .09 g), MS (M+H)+: 238.9.
1H NMR (300 MHz, DMSO) 5 13.59 (s, 1 H), 8.76 - 8.48 (m, 3H), 3.92 (s, 6H).
13C NMR (75 MHz, DMSO) 5 165.51 , 164.66, 133.62, 133.08, 132.18, 130.82, 52.72.
Step 2:
NH4HCO3 (0.15 g, 1.89 mmol, 1.26 eq) was added to a stirred solution of the product from step 1 (0.42 g, 1.5 mmol), pyridine (0.24 mL, 0.237 g), and (Boc)2O (0.43 g, 1.95 mmol) in 2 mL of dioxane. The reaction was stirred over the weekend to form a white solid. This solid was filtered, washed with concentrated HCI and ethylacetate yielding the desired product as a white solid.
MS (M+H)+: 237.9.
1H NMR (300 MHz, DMSO) 5 8.69 (s, 2H), 8.66 (d, J = 1.8 Hz, 1 H), 8.43 (s, 1 H), 7.66 (s, 1 H), 3.92 (s, 6H).
Step 3:
Synthesized according to general procedure 1. Starting materials: THF/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (400 mg).
Yield, over two steps: 210 mg (59%).
MS (M+H)+: 209.8.
1 H NMR (300 MHz, DMSO) 5 13.47 (s, 1 H), 8.67 - 8.62 (m, 2H), 8.57 (t, J = 1.6 Hz, 1 H), 8.35 (s, 1 H), 7.59 (s, 1 H).
13C NMR (75 MHz, DMSO) 5 166.26, 166.14, 135.27, 133.54, 132.18, 131.50.
Intermediate 23.
 (methoxycarbonyl)benzene-1 ,3-dicarboxylic acid (250 mg, 1.1 mmol) and intermediate 1 (400 mg, 2.3 mmol) were dissolved in 8 ml of MeCN. 0.47 mL N-methylimidazole and TCFH (640 mg, 2.2 mmol) were added sequentially. The reaction was stirred overnight. After the completion of the reaction the product was filtered and washed with ethanol to obtain the product as white solid in 27% yield (150 mg).
MS (M+H)+: 543.2.
(methoxycarbonyl)benzene-1 ,3-dicarboxylic acid (250 mg, 1.1 mmol) and intermediate 1 (400 mg, 2.3 mmol) were dissolved in 8 ml of MeCN. 0.47 mL N-methylimidazole and TCFH (640 mg, 2.2 mmol) were added sequentially. The reaction was stirred overnight. After the completion of the reaction the product was filtered and washed with ethanol to obtain the product as white solid in 27% yield (150 mg).
MS (M+H)+: 543.2.
1H NMR (300 MHz, DMSO) 5 11.01 (s, 2H), 8.94 (s, 1 H), 8.81 (d, J = 1.8 Hz, 2H), 8.68 (d, J = 1.6 Hz, 4H), 8.27 (s, 2H), 3.99 (s, 3H), 2.69 (s, 12H).
Intermediate 24.
 stirred overnight. After completion of the reaction, 1 M HCI was added (5 ml) and solid was filtered and dried yielding the product in 81 % yield (81%).
stirred overnight. After completion of the reaction, 1 M HCI was added (5 ml) and solid was filtered and dried yielding the product in 81 % yield (81%).
MS (M+H)+: 528.8.
1H NMR (300 MHz, DMSO) 5 11.02 (s, 1 H), 8.91 (s, 1 H), 8.81 (d, J = 1.7 Hz, 2H), 8.69 (d, J = 1.5 Hz, 4H), 8.26 (s, 2H), 2.68 (d, J = 2.1 Hz, 12H).13C NMR (75 MHz, DMSO) 5 165.86, 164.96, 133.81 , 132.32, 131.80, 130.43, 52.57.
Scheme for intermediats 25-26.
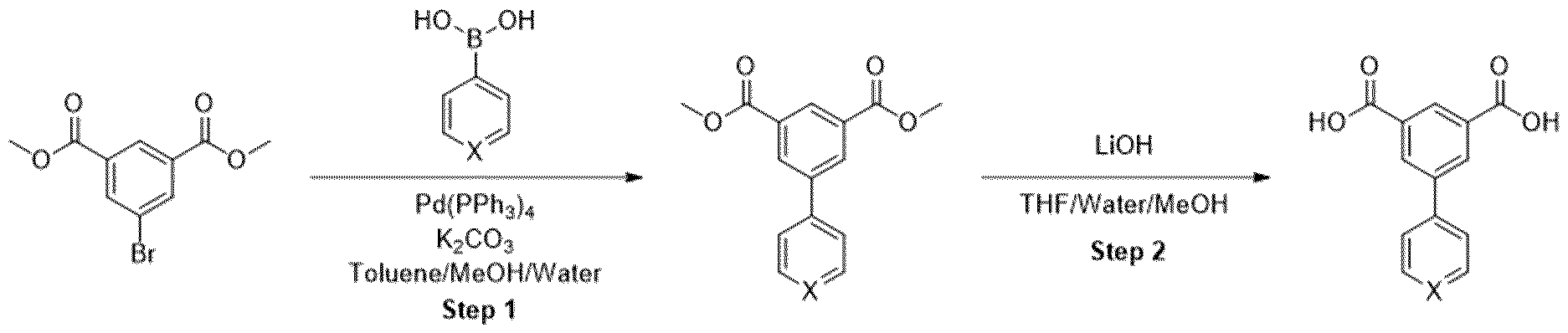
Step 1 :
A 100 mL round bottom flask equipped with a magnetic stir bar and a condenser was charged with 5-bromoisophthalic acid dimethyl ester (272 mg, 1 mmol), aryllboronic acid (1 mmol), K2CO3 (555 mg, 4 mmol). The solids were suspended in 12 ml/5 ml/1.5 ml toluene/MeOH/H2O, degassed, and the atmosphere was exchanged by nitrogen. Afterwards, Pd(PPh3)4 (42 mg, 72 pmol) was added. The mixture was stirred at 80 °C overnight. After the completion of the reaction, the solvent was evaporated and the residue was purified using flash chromatography on silica gel eluting with cyclohexane to cyclohexane: ethylacetate 60:40 to obtain the desired product as a white solid.
Step 2:
Synthesized according to general procedure 1. Starting materials: THF/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (200 mg).
Intermediate 25. Step 1 :
Starting material: phenylboronic acid (121 mg). MS (M+H)+: 271.1. 1H NMR (300 MHz, CDCI3) 6 8.59 (s, 1 H), 8.39 (s, 2H), 7.59 (d, J = 6.9 Hz, 2H), 7.41 (t, J = 8.0 Hz, 2H), 7.35 (d, J = 8.6 Hz, 1 H), 3.91 (s, 5H).
 13C NMR (75 MHz, CDCI3) 6 166.26, 141.96, 139.05, 132.31 , 131.17,
13C NMR (75 MHz, CDCI3) 6 166.26, 141.96, 139.05, 132.31 , 131.17,
129.33, 129.04, 128.24, 127.19, 52.46.
Step 2:
Yield, over two steps: 60 mg (25%).
MS (M-H)-: 240.9.
1H NMR (300 MHz, DMSO) 6 8.35 (s, 1 H), 8.26 (d, J = 1.5 Hz, 2H), 7.63 (d, J = 8.6 Hz, 2H), 7.47 - 7.36 (m, 2H), 7.35 (s, 1 H).
13C NMR (75 MHz, DMSO) 5 166.51 , 141.02, 138.44, 132.26, 131.17, 129.21 , 128.76, 128.28, 126.89.
Intermediate 26. Step 1 :
Starting material: 4-pyridinylboronic acid (121 mg) MS (M+H)+: 271.8. 1H NMR (300 MHz, CDCI3) 6 8.78 (m, 3H), 8.53 (s, 2H), 8.02 (d, J = 6.7 Hz, 2H), 3.96 (s, 6H).
 Step 2: The product was not extracted but filtered and addition of HCI. Yield, over two steps: 75 mg (31%).
Step 2: The product was not extracted but filtered and addition of HCI. Yield, over two steps: 75 mg (31%).
MS (M+H)+: 243.9
1H NMR (300 MHz, DMSO) 5 8.94 (d, J = 6.7 Hz, 2H), 8.62 (s, 3H), 8.36 (d, J = 6.7 Hz, 2H). 13C NMR (75 MHz, DMSO) 5 165.99, 144.43, 132.60, 132.27, 131.58, 123.98, 122.32.
General procedure for intermediates 27-29.
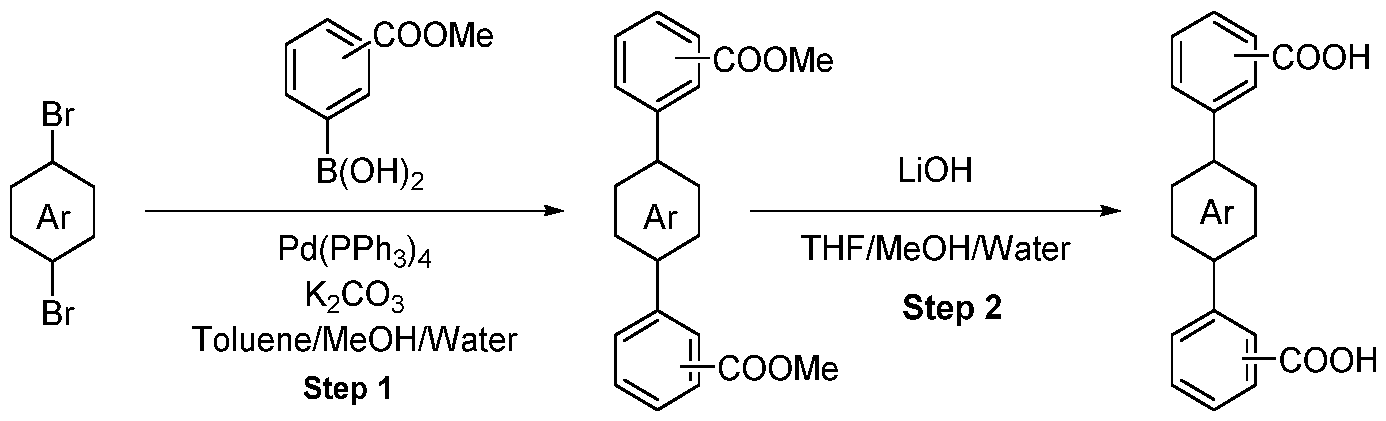
Step 1 :
A 100 mL round bottom flask equipped with a magnetic stir bar and a condenser was charged with dibromoarene (1 mmol), methoxycarbonylphenylboronic acid (380 mg, 2 mmol) and K2COs (1.1 g, 8 mmol). The solids were suspended in 12 ml/5 ml/1.5 ml toluene/MeOH/H2O (8.8 mL), degassed, and the atmosphere was exchanged with nitrogen. Afterwards, Pd(PPh3)4 (84 mg, 72 pmol) was added. The mixture was stirred at 80 °C overnight. The solvent was evaporated and the residue was purified by silica gel column chromatography eluting with cyclohexane to cyclohexane: ethylacetate 20:80 to obtain the desired product as a white solid.
Step 2:
Synthesized according to general procedure 1. Starting materials: THF/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (200 mg). The product was filtered after addition of HCI.

1H NMR (300 MHz, CDCI3) 6 8.26 (s, 2H), 7.97 (d, J = 7.8 Hz,
2H), 7.97 (d, J = 7.8 Hz, 2H), 7.66 (s, 2H), 7.47 (t, J = 7.8 Hz, 4H), 3.89 (s, 6H).
13C NMR (75 MHz, CDCI3) 6 167.03, 140.80, 139.49, 131.42, 130.81 , 128.96, 128.55, 128.17, 127.65, 52.25.
Step 2:
MS (M+H)+: 319.4.
1H NMR (300 MHz, DMSO) 5 13.12 (s, 2H), 8.26 (s, 2H), 7.99 (t, J = 8.5 Hz, 4H), 7.85 (s, 4H), 7.64 (t, J = 7.8 Hz, 2H).
13C NMR (75 MHz, DMSO) 5 167.17, 139.77, 138.61 , 131.54, 130.99, 129.39, 128.40, 127.43, 127.17.
Intermediate 28.
Starting materials:_1 ,3-dibromobenzene (235 mg), 4- methoxycarbonylphenylboronic acid. Yield, over two steps: 250 mg (78%).
 Step 1 :
Step 1 :
1H NMR (300 MHz, CDCI3) 5 8.06 (d, J = 8.4 Hz, 4H), 7.77 (s, 1 H), 7.64 (d, J = 8.5 Hz, 4H), 7.57 (d, J = 8.8 Hz, 2H), 7.49 (dd, J = 8.8, 6.3 Hz, 1 H), 3.88 (s, 6H).
13C NMR (75 MHz, CDCI3) 5 166.93, 145.30, 140.83, 130.19, 129.54, 129.22, 127.18, 127.06, 126.31 , 52.18.
Step 2:
MS (M-H)’: 317.3.
1H NMR (300 MHz, DMSO) 5 8.10 - 8.01 (m, 5H), 7.93 (d, J = 8.5 Hz, 4H), 7.80 (d, J = 5.8 Hz, 2H), 7.69 - 7.57 (m, 1 H).
13C NMR (75 MHz, DMSO) 5 167.06, 144.00, 139.85, 129.90, 129.82, 127.08, 126.87, 125.60.
 7.91 - 7.80 (m, 2H), 7.54 (d, J= 8.4 Hz,4H), 7.46 - 7.33 (m, 4H), 3.91
7.91 - 7.80 (m, 2H), 7.54 (d, J= 8.4 Hz,4H), 7.46 - 7.33 (m, 4H), 3.91
(s, 6H).
13C NMR (75 MHz, CDCI3) 6 167.01 , 145.42, 139.36, 131.61 , 130.16, 129.67, 129.22, 126.39,
126.14, 52.23.
Step 2:
MS (M-H)’: 367.3.
1H NMR (300 MHz, DMSO) 5 13.11 (s, 2H), 8.12 (d, J = 8.2 Hz, 4H), 7.87 (dd, J = 6.5, 3.4 Hz, 2H), 7.66 (d, J = 8.4 Hz, 4H), 7.60 - 7.53 (m, 4H).
13C NMR (75 MHz, DMSO) 5 167.13, 144.25, 138.71 , 130.89, 130.04, 129.96, 129.50, 126.68, 126.53, 125.68.
General scheme for intermediates 30-41.
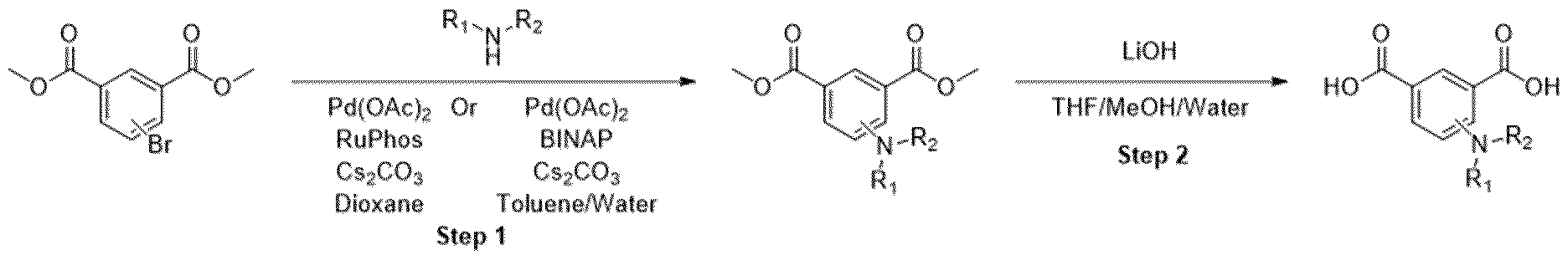
Procedure for examples:
Step 1 :
Method A:
A 100 mL round bottom flask equipped with a magnetic stir bar and a condenser was charged with 5-bromoisophthalic acid dimethyl ester in dioxane, CS2CO3 and amine. The reaction mixture was degassed using nitrogen gas. Pd(OAc)2 were added. The reaction mixture was stirred at 90° C overnight. The solvent was evaporated, and the residue was purified by silica gel column chromatography eluting with cyclohexane to cyclohexane: ethylacetate 60:40 to obtain the desired product..
Or
Method B:
A 100 mL round bottom flask equipped with a magnetic stir bar and a condenser was charged with the bromide, amine, CS2CO3 and toluene. The reaction mixture was degassed using nitrogen gas. Pd(OAc)2 and BINAP were added. The mixture was stirred at 100 °C overnight. The solvent was evaporated and the residue was purified by silica gel column chromatography eluting with cyclohexane to cyclohexane: ethylacetate 60:40 to obtain the desired product.
Step 2:
Synthesized according to general procedure 1.
Intermediate 30.
Step 1 :
Method A.
Starting materials: 5-bromoisophthalic acid dimethyl ester (500 mg, 1.83 mmol), dioxane (4 mL), CS2CO3 (1.49 g, 4.58 mmol), hexylamine (277.25 mg, 2.75 mmol), Pd(OAc)2 (8.2 mg, 0.040 mmol), RuPhos (42.7 mg, 0.090 mmol).
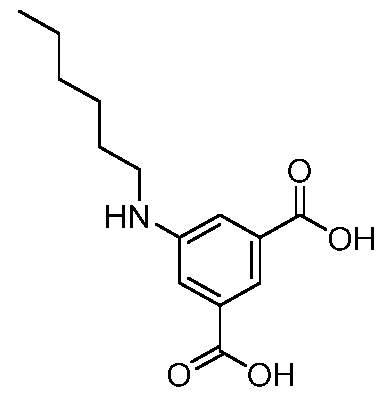 MS (M+H)+: 294.1.
MS (M+H)+: 294.1.
1H NMR (300 MHz, CDCI3) 6 8.08 (s, 1 H), 7.60 (s, 2H), 3.86 (s, 6H), 3.12
(d, J = 7.4 Hz, 2H), 1 .59 (m, 2H), 1 .36 (s, 6H), 0.89 - 0.71 (m, 3H).
Step 2:
Starting materials: THF/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (200 mg).
Yield, over two steps: 39 mg (15%).
1H NMR (300 MHz, DMSO) 5 12.90 (s, 2H), 7.67 (s, 1 H), 7.34 (s, 2H), 6.17 (s, 1 H), 3.04 (t, J = 7.0 Hz, 2H), 1.67 - 1.48 (m, 2H), 1.43 - 1.11 (m, 6H), 1.01 - 0.77 (m, 3H).
13C NMR (75 MHz, DMSO) 5 167.21 , 149.24, 131.62, 116.96, 115.99, 42.71 , 31.04, 28.31 , 26.25, 22.08, 13.89.
Intermediate 31. Step 1 : Method B.
Starting materials: 5-bromoisophthalic acid dimethyl ester (1.99 g, 7.32 mmol), cyclopropylamine (0.413 ml, 7.32 mmol), Cs2CC>3(3.58 g, 10.98 mmol), 15 ml of toluene, Pd(OAc)2 (83 mg, 0.37 mmol), BINAP (228 mg, 0.37 mmol).
MS (M+H)+: 249.9.
 z, CDCI3) 6 8.10 (s, 1 H), 7.78 - 7.59 (m, 2H), 3.87 (s, 4H), 2.60 - 2.43 (m, 1 H), 0.86 - 0.74 (m, 2H), 0.68 - 0.59 (m, 2H).
z, CDCI3) 6 8.10 (s, 1 H), 7.78 - 7.59 (m, 2H), 3.87 (s, 4H), 2.60 - 2.43 (m, 1 H), 0.86 - 0.74 (m, 2H), 0.68 - 0.59 (m, 2H).
Step 2:
Starting materials: THF/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (200 mg).
Yield, over two steps: 40 mg (3%).
1H NMR (300 MHz, DMSO) 5 7.78 (s, 1 H), 7.54 (s, 2H), 6.67 (s, 1 H), 2.48 - 2.37 (m, 1 H), 0.89 - 0.71 (m, 2H), 0.51 - 0.35 (m, 2H).
13C NMR (75 MHz, DMSO) 5 167.17, 149.63, 131.56, 116.61 , 24.31 , 6.77.
Intermediate 32. Step 1 : Method A.
Starting materials: 5-bromoisophthalic acid dimethyl ester (500 mg, 1.83 mmol), dioxane (4 mL), CS2CO3 (1.49 g, 4.58 mmol), cyclohexylamine (272 mg, 2.75 mmol), Pd(OAc)2 (8.2 mg, 0.040 mmol), RuPhos (42.7 mg, 0.090 mmol). MS (M+H)+: 292.0.
 1H NMR (300 MHz, CDCI3) 6 8.03 (s, 1 H), 7.54 (s, 2H), 3.84 (s,6H), 3.26 (m, 1 H), 2.07 - 1.91 (m, 2H), 1.77 - 1.01 (m, 8H).
1H NMR (300 MHz, CDCI3) 6 8.03 (s, 1 H), 7.54 (s, 2H), 3.84 (s,6H), 3.26 (m, 1 H), 2.07 - 1.91 (m, 2H), 1.77 - 1.01 (m, 8H).
Step 2:
Starting materials: THF/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (200 mg).
Yield, over two steps: 120 mg (45%).
MS (M+H)+: 264.0
1H NMR (300 MHz, DMSO) 5 12.88 (s, 1 H), 7.64 (s, 1 H), 7.34 (s, 2H), 3.24 (s, 1 H), 1.96 - 1.85 (m, 2H), 1.78 - 1.68 (m, 2H), 1.65 - 1.53 (m, 1 H),1.46 - 1.25 (m, 2H), 1.26 - 1.07 (m, 3H).
13C NMR (75 MHz, DMSO) 5 167.24, 148.20, 131.68, 116.76, 116.37, 50.46, 32.24, 25.48, 24.35.
Intermediate 33. Step 1 : Method A. Starting materials: 5-bromoisophthalic acid dimethyl ester (500 mg, 1.83 mmol), dioxane (4 mL), CS2CO3 (1.49 g, 4.58 mmol), pyrrolidine (233 mg, 2.75 mmol), Pd(OAc)2 (8.2 mg, 0.040 mmol), RuPhos (42.7 mg, 0.090 mmol).
MS (M+H)+: 264.0. DCI3) 6 7.97 (s, 1 H), 7.42 (s, 2H), 3.94 (s, 6H), 3.39 (m, 4H), 2.07 (m, DCI3) 6 167.09, 147.68, 131.08, 117.54, 116.59, 52.17, 25.45. F/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (200 mg). 102 mg (43%). MSO) 5 12.97 (s, 2H), 7.73 (s, 1 H), 7.25 (s, 2H), 3.28 (d, J = 6.6 Hz, z, 4H). MSO) 5 167.23, 147.54, 131.69, 116.57, 115.60, 47.38, 24.96. Step 1 : Method B:
Starting materials: 5-bromoisophthalic acid dimethyl ester (1.99 g, 7.32 mmol), piperidine (0.73 ml, 7.32 mmol), CS2CO3 (3.58 g, 10.98 mmol), 5 ml of toluene, Pd(OAc)2 (83 mg, 0.37 mmol), BINAP (228 mg, 0.37 mmol)

MS (M+H)+: 278.0.
1H NMR (300 MHz, CDCI3) 6 8.02 (s, 1 H), 7.70 (s, 2H), 3.85 (s, 6H), 3.19 (t, J = 5.5 Hz, 4H), 1.79 - 1.43 (m, 6H).
13C NMR (75 MHz, CDCI3) 6 166.77, 151.85, 136.61 , 131.18, 120.95, 52.25, 50.11 , 25.48, 24.08.
Step 2:
Starting materials: THF/MeOH/Water (2 ml/2 ml/2 ml) and LiOH (800 mg.
Yield, over two steps: 320 mg (17%).
MS (M+H)+: 250.0.
1H NMR (300 MHz, DMSO) 5 13.07 (s, 2H), 7.87 (t, J = 1.4 Hz, 1 H), 7.65 (d, J = 1.4 Hz, 2H), 3.24 (t, J = 5.4 Hz, 4H), 1 .63 - 1.50 (m, 6H).
13C NMR (75 MHz, DMSO) 5 166.97, 151.43, 131.87, 119.69, 49.15, 24.90, 23.63.
Inte Vrmediate 35. o o Step 1 : Method A.
0H Starting materials: 5-bromoisophthalic acid dimethyl ester (500 mg, 1.83 mmol), dioxane (4 mL), CS2CO3 (1.49 g, 4.58 mmol),1-Boc-piperazine N (511 mg, 2.75 mmol), Pd(OAc)2 (8.2 mg, 0.040 mmol), RuPhos (42.7 mg,
T ''I 0.090 mmol). r MS (M+H)+: 379.0.
Boo 1H NMR (300 MHz, CDCI3) 6 8.14 (s, 1 H), 7.77 (s, 2H), 3.86 (s, 6H), 3.64 - 3.48 (m, 4H), 3.27 - 3.00 (m, 4H), 1.42 (s, 9H).
13C NMR (75 MHz, CDCI3) 6 166.36, 154.57, 131.56, 122.69, 121.52, 80.23, 77.44, 77.22, 76.59, 52.42, 49.37, 28.41.
Step 2:
Starting materials: THF/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (200 mg)
Yield, over two steps: 82 mg (23%).
MS (M+H)+: 350.9.
1H NMR (300 MHz, DMSO) 5 13.13 (s, 2H), 7.96 (s, 1 H), 7.71 (s, 2H), 3.51 (m, 4H), 3.31 - 3.05 (m, 4H), 1.46 (s, 9H).
13C NMR (75 MHz, DMSO) 5 166.86, 153.85, 151.02, 131.95, 120.46, 119.80, 79.02, 47.77, 28.03.
Intermediate 36.
Step 1 :
Method A. Starting materials: 5-bromoisophthalic acid dimethyl ester (500 mg, 1.83 mmol) in dioxane (4 mL), CS2CO3 (1.49 g, 4.58 mmol) and morpholine (0.237 ml, 2.75 mmol), Pd(OAc)2 (8.2 mg, 0.040 mmol), RuPhos (42.7 mg, 0.090 mmol) MS (M+H)+: 279.8.
 1H NMR (300 MHz, CDCI3) 5 8.15 (s, 1 H), 7.77 (s, 2H), 3.88-3.85 (m, 10H), 3.24 - 3.21 (m, 4H).
1H NMR (300 MHz, CDCI3) 5 8.15 (s, 1 H), 7.77 (s, 2H), 3.88-3.85 (m, 10H), 3.24 - 3.21 (m, 4H).
13C NMR (75 MHz, CDCI3) 5 166.38, 150.47, 131.55, 122.67, 120.86, 66.38, 52.43, 49.40.
Step 2:
Starting materials: THF/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (200 mg)
Yield, over two steps: 130 mg (28%).
MS (M+H)+: 251.8.
1H NMR (300 MHz, DMSO) 5 7.93 (s, 1 H), 7.68 (s, 2H), 3.86 - 3.59 (m, 4H), 3.29 - 3.05 (m, 4H).
13C NMR (75 MHz, DMSO) 5 166.86, 151.18, 131.93, 120.43, 119.12, 65.90, 47.95.
Intermediate 37.
Step 1 : Method B.
Starting materials: 4-bromoisophthalic acid dimethyl ester (400 mg, 1 46 mmol), morpholine (0.127 ml, 1.46 mmol), CS2CO3 (0.716 mg, 2.20 mmol), 15 ml of toluene. The reaction mixture was degassed using nitrogen gas. Pd(OAc)2 (16.6 mg, 0.074 mmol), BINAP (45 mg,
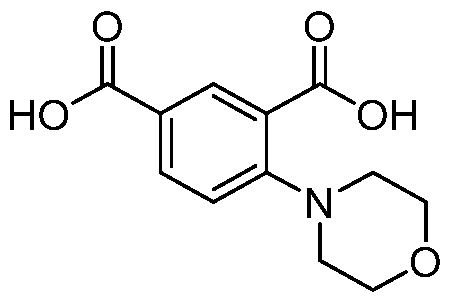 0.074 mmol)
0.074 mmol)
MS (M+H)+: 280.0.
1H NMR (300 MHz, CDCI3) 5 8.44 (d, J = 2.2 Hz, 1 H), 8.06 (dd, J = 8.6, 2.2 Hz, 1 H), 7.02 (d,
J = 8.6 Hz, 1 H), 3.95 - 3.83 (m, 10H), 3.18 (t, J = 4.7 Hz, 4H).
13C NMR (75 MHz, CDCI3) 6 167.44, 166.15, 154.94, 134.07, 122.41 , 121.90, 117.77,
66.65, 52.29, 52.19, 52.04, 26.91.
Step 2:
Starting materials:THF/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (400 mg)
Yield, over two steps: 100 mg (28%).
MS (M+H)+: 251.9.
1H NMR (300 MHz, DMSO) 5 13.27 (s, 2H), 8.25 (d, J = 2.2 Hz, 1 H), 8.00 - 7.97 (m, 1H), 7.25 (d, J = 8.6 Hz, 1H), 3.75 - 3.72 (m, 4H), 3.14 - 3.11 (m, 4H).
13C NMR (75 MHz, DMSO) 5 167.87, 166.42, 154.13, 146.94, 133.41 , 132.60, 123.13, 122.89, 118.90, 65.96, 51.51.
Intermediate 38.
Step 1 :
0 Method B: ""l Starting materials: 2-bromoisophthalic acid dimethyl ester (400 mg, 1.46 0 0 mmol), morpholine (0.127 ml, 1.46 mmol), CS2CO3 (0.716 mg, 2.20
LI 1 U mmol) 15 ml of toluene, Pd(OAc)2 (16.6 mg, 0.074 mmol), BINAP (45 mg,
HO^ ^OM 0.074 mmol).
MS (M+H)+: 280.1.
1H NMR (300 MHz, CDCI3) 6 7.55 (d, J = 7.7 Hz, 2H), 7.10 (t, J = 7.7 Hz, 1 H), 3.86 (s, 6H),
3.74 - 3.61 (m, 4H), 3.08 (t, J = 4.7 Hz, 4H).
13C NMR (75 MHz, CDCI3) 5 168.64, 148.42, 132.39, 131.54, 124.03, 67.63, 52.51 , 51.53.
Step 2:
Starting materials: THF/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (400 mg).
Yield, over two steps: 105 mg (29%).
MS (M+H)+: 251.9.
1H NMR (300 MHz, DMSO) 5 7.79 (d, J = 7.7 Hz, 2H), 7.36 (t, J = 7.7 Hz, 1H), 3.72 (s, 4H), 3.30 - 3.12 (m, 4H).
13C NMR (75 MHz, DMSO) 5 168.59, 146.46, 132.48, 131.14, 125.56, 66.61, 50.73.
Intermediate 39. Step l : Method B. Starting materials: 5-bromoisophthalic acid dimethyl ester (400 mg, 1.46 mmol), 6,6-difluoro-3-azabicyclo[3.1.0]hexane hydrochloride (227 mg, 1.46 mmol), CS2CO3 (0.716 mg, 2.20 mmol), 15 ml of toluene, Pd(OAc)2 (16.6 mg, 0.074 mmol), BINAP (45 mg, 0.074 mmol). MS (M+H)+: 312.1. 1H NMR (300 MHz, CDCI3) 5 7.95 (t, J = 1.5 Hz, 1H), 7.27 (d, J = 1.5 Hz,
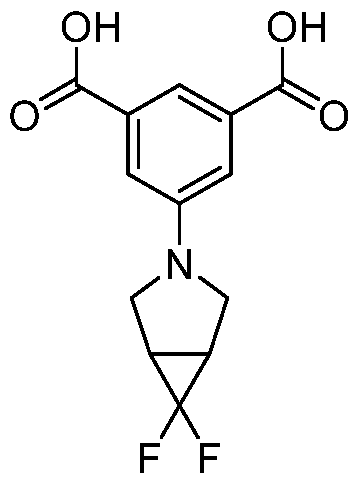 2H), 3.86 (s, 6H), 3.72 - 3.58 (m, 4H), 2.48 - 2.29 (m, 1 H).
2H), 3.86 (s, 6H), 3.72 - 3.58 (m, 4H), 2.48 - 2.29 (m, 1 H).
19F NMR (283 MHz, CDCI3) 6 -129.67 (d, J = 160.3 Hz, 1 F), -156.60 (d, J
= 160.6 Hz, 1 F).
13C NMR (75 MHz, CDCI3) 6 166.82, 147.53, 131.27, 118.56, 116.73, 52.29, 47.37, 26.24 (t, J = 12.4 Hz). CF2 carbon was not observed due to low intensity.
Step 2:
Starting materials: THF/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (400 mg).
Yield, over two steps: 180 mg (43%).
MS (M+H)+: 284.1.
1H NMR (300 MHz, DMSO) 5 13.07 (s, 2H), 7.80 (t, J = 1.4 Hz, 1 H), 7.25 (d, J = 1.5 Hz, 2H), 3.73 - 3.58 (m, 4H), 2.85 - 2.61 (m, 2H).
19F NMR (283 MHz, DMSO) 5 -129.67 (d, J = 160.3 Hz, 1 F), -156.60 (d, J = 160.6 Hz, 1 F).
13C NMR (75 MHz, DMSO) 5 167.02, 147.01 , 131.87, 117.60, 115.99, 47.12, 25.69 (t, J = 11 .2 Hz). One peak was not observed due to similar shift with DMSO-d6 and CF2 carbon was not observed due to low intensity.
Intermediate 40. Step 1 :
Method B.
Starting materials: 5-bromoisophthalic acid dimethyl ester (400 mg, 1.46 mmol), 7-oxa-2-azaspiro[3.5]nonane hydrochloride (237 mg, 1.46 mmol), CS2CO3 (0.716 mg, 2.20 mmol), 15 ml of toluene, Pd(OAc)2 (16.6 mg, 0.074 mmol), BINAP (45 mg, 0.074 mmol).
MS (M+H)+: 320.1.
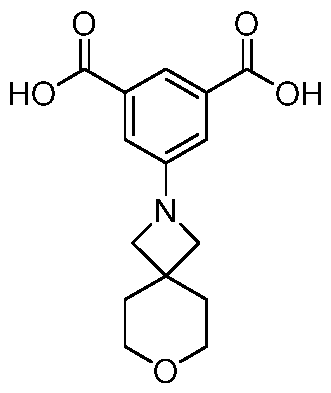 1H NMR (300 MHz, CDCI3) 5 7.95 (t, J = 1.4 Hz, 1 H), 7.19 (s, 2H), 3.85 (s,
1H NMR (300 MHz, CDCI3) 5 7.95 (t, J = 1.4 Hz, 1 H), 7.19 (s, 2H), 3.85 (s,
6H), 3.66 (s, 4H), 3.62 - 3.59 (m, 4H), 1.80 - 1.76 (m, 4H).
13C NMR (75 MHz, CDCI3) 6 166.71 , 151.38, 131.17, 119.20, 116.02, 64.94, 62.19, 52.27, 36.47, 34.09.
Step 2:
Starting materials: THF/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (400 mg).
Yield, over two steps: 345 mg (81%).
MS (M+H)+: 291.9.
1H NMR (300 MHz, DMSO) 5 12.97 (s, 2H), 7.80 (s, 1 H), 7.14 (s, 2H), 3.70 (s, 4H), 3.58 - 3.55 (m, 4H), 1.77 - 1.74 (m, 4H).
13C NMR (75 MHz, DMSO) 5 166.91 , 151.52, 131.75, 118.11 , 115.13, 64.03, 61.54, 35.90, 33.60.
Intermediate 41.
Step 1 :
Method B.
Starting materials: 5-bromoisophthalic acid dimethyl ester (400 mg, 1.46 mmol), 8-oxa-2-azaspiro[4.5]decane (205 mg, 1.46 mmol), CS2CO3 (0.716 mg, 2.20 mmol), 15 ml of toluene, Pd(OAc)2 (16.6 mg, 0.074 mmol), BINAP (45 mg, 0.074 mmol).
MS (M+H) +: 333.9.
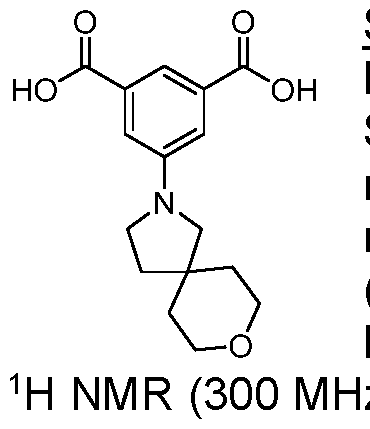 z, DMSO) 5 7.90 (t, J = 1.5 Hz, 1 H), 7.30 (d, J = 1.4 Hz, 2H), 3.86 (s, 4H), 3.78 - 3.53 (m, 1 H), 3.38 (t, J = 7.0 Hz, 2H), 3.21 (s, 2H), 1 .91 (d, J = 7.0 Hz, 1 H), 1 .59 (t, J = 5.5 Hz, 3H).
z, DMSO) 5 7.90 (t, J = 1.5 Hz, 1 H), 7.30 (d, J = 1.4 Hz, 2H), 3.86 (s, 4H), 3.78 - 3.53 (m, 1 H), 3.38 (t, J = 7.0 Hz, 2H), 3.21 (s, 2H), 1 .91 (d, J = 7.0 Hz, 1 H), 1 .59 (t, J = 5.5 Hz, 3H).
13C NMR (75 MHz, DMSO) 5 167.02, 147.66, 131.15, 117.71 , 116.35, 65.32, 58.24, 52.24, 46.34, 39.95, 36.43, 35.79.
Step 2:
Starting materials: THF/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (400 mg) Yield, over two steps: 305 mg (56%).
MS (M+H)+: 305.9.
1H NMR (300 MHz, DMSO) 6 13.00 (s, 2H), 7.74 (t, J = 1.4 Hz, 1 H), 7.27 (d, J = 1.4 Hz, 2H), 3.70 - 3.56 (m, 4H), 3.39 (t, J = 6.9 Hz, 2H), 3.23 (s, 2H), 1.93 (t, J = 6.9 Hz, 2H), 1.67 - 1.46 (m, 4H).
13C NMR (75 MHz, DMSO) 5 167.23, 146.98, 131.72, 116.63, 115.47, 64.32, 57.71 , 45.82, 35.56, 35.20 (one peak was not observed due to similar shift with DMSO-d6).
Intermediate 42.
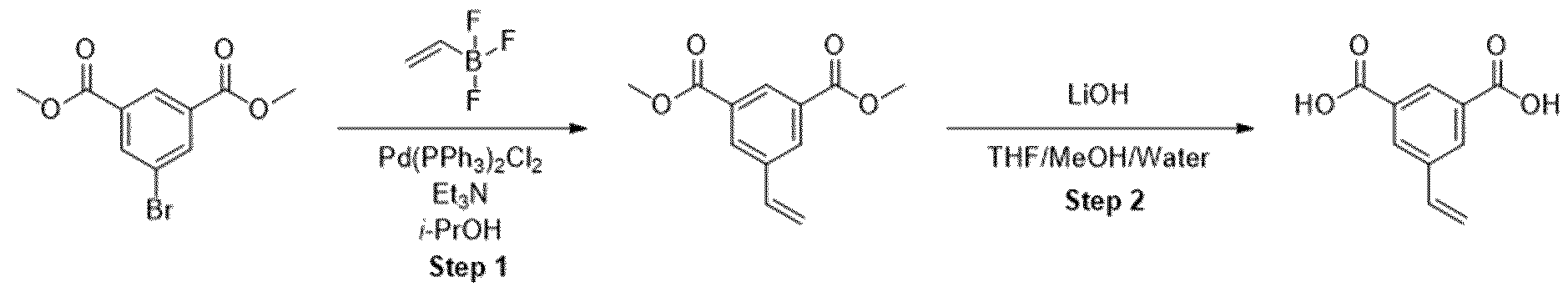
Step 1 :
To a stirred in nitrogen atmosphere solution of 5-bromoisophthalic acid dimethyl ester (1.00 g, 3.7 mmol), potassium trifluoro(vinyl)borate (0.98 g, 7.3 mmol) and triethylamine (1.12 g, 11.1 mmol) in degassed isopropanol (15 mL) was added bis(triphenylphosphine)palladium(ll) chloride (0.26 g, 0.37 mmol) and refluxed for 18 h. Afterwards, the solvent was removed under the reduced pressure and the residue was purified by silica gel column chromatography eluting with cyclohexane to cyclohexane:ethylacetate 70:30 to obtain the desired product as a yellow oil in 82% yield (670 mg).
MS (M+H)+: 220.8.
1H NMR (301 MHz, CDCI3) 6 8.48 (t, J = 1.7 Hz, 1 H), 8.17 (d, J = 1.7 Hz, 2H), 6.70 (dd, J = 17.6, 10.9 Hz, 1 H), 5.84 (d, J = 17.3 Hz, 1 H), 5.33 (d, J = 11.0 Hz, 1 H), 3.88 (s, 6H).
13C NMR (76 MHz, CDCI3) 6 166.20, 138.39, 135.05, 131.32, 130.92, 129.70, 116.48, 52.42. Step 2:
Synthesized according to general procedure 1. Starting materials: product from the previous step (220 mg, 1 mmol), THF/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (80 mg, 2 mmol).
Yield: 88% (170 mg).
MS (M+H)+: 192.9.
1H NMR (300 MHz, DMSO) 5 13.25 (s, 2H), 8.36 (t, J = 1.6 Hz, 1 H), 8.22 (d, J = 1.6 Hz, 2H), 6.91 (dd, J = 17.7, 11.1 Hz, 1 H), 5.99 (d, J = 17.7 Hz, 1 H), 5.41 (d, J = 11.1 Hz, 1 H).
13C NMR (75 MHz, DMSO) 5 166.44, 138.04, 135.10, 131.71 , 130.56, 129.11 , 116.64.
Genral scheme for intermediats 43-44.
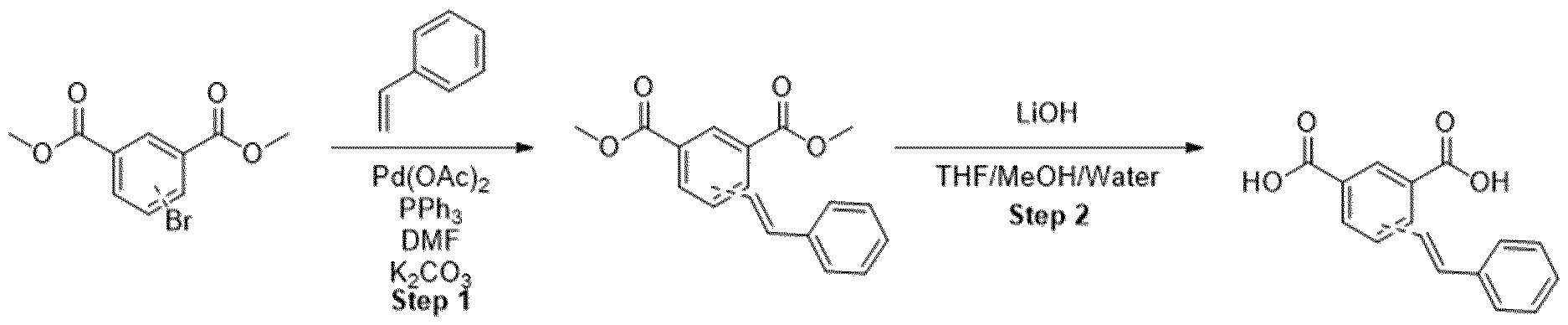
Step 1 :
A 25 mL round bottom flask equipped with a magnetic stir bar and a condenser was charged with the bromoisophthalic acid dimethyl ester (546 mg, 2 mmol), triphenylphosphine (4.2 mg, 0.016 mmol), styrene (229 mg, 2.2 mmol), potassium carbonate (300 mg, 2.20 mmol) and 5 ml of DMF. The reaction mixture was degassed using nitrogen gas. Palladium (II) acetate (16.6 mg, 0.08 mmol) was added. The mixture was stirred at 100 °C overnight. The solvent was evaporated, and the residue was purified by silica gel column chromatography eluting with cyclohexane to cyclohexane:ethylacetate 60:40 to obtain the desired product as a light yellow solid.
Step 2:
Synthesized according to general procedure 1. Starting materials: THF/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (400 mg).
Intermediate 43. Starting material: 5-bromoisophthalic acid dimethyl ester. Yield, over two steps: 100 mg (18%).
Step 1 : MS (M+H)+: 296.9. 1H NMR (300 MHz, CDCI3) 5 8.48 (t, J = 1.6 Hz, 1 H), 8.29 (s, 2H), 7.48 (d, J = 10.0 Hz, 2H), 7.37 - 7.28 (m, 1 H), 7.25 (d, J = 6.3 Hz, 2H), 7.15 (d, J = 16.5
 Hz, 1 H), 7.08 (d, J = 16.5 Hz, 1 H), 3.91 (s, 6H).
Hz, 1 H), 7.08 (d, J = 16.5 Hz, 1 H), 3.91 (s, 6H).
13C NMR (75 MHz, CDCI3) 5 166.25, 138.27, 136.56, 131.47, 131.17, 131.06, 129.32, 128.81 , 128.30, 126.77, 126.50, 52.46.
Step 2:
MS (M+H)+: 268.9.
1H NMR (300 MHz, DMSO) 5 13.25 (s, 2H), 8.44 (d, J = 1.5 Hz, 2H), 8.42 (t, J = 1.6 Hz, 1 H), 7.81 - 7.67 (m, 2H), 7.51 (s, 2H), 7.48 (d, J = 8.4 Hz, 2H), 7.37 (t, J = 8.0 Hz, 1 H).
13C NMR (75 MHz, DMSO) 5 166.53, 138.15, 136.57, 131.78, 130.82, 130.54, 128.68, 128.10, 126.82, 126.63.
“ Intermediate 44.
Starting material: 2-bromoisophthalic acid dimethyl ester.
Yield, over two steps: 410 mg (76%).
Step 1 :
MS (M+H)+: 296.9.
1H NMR (300 MHz, CDCI3) 6 7.82 (d, J = 7.8 Hz, 2H), 7.70 (d, J = 16.5 Hz, 1 H), 7.45 - 7.37 (m, 2H), 7.36 - 7.13 (m, 4H), 6.39 (d, J = 16.5 Hz, 1 H), 3.75 (s, 6H).
13C NMR (75 MHz, CDCI3) 6 168.44, 147.37, 139.31 , 137.21 , 133.14, 132.43, 132.09, 128.62, 127.86, 127.22, 126.82, 126.64, 52.38.
Step 2:
MS (M+H)+: 268.9.
1H NMR (300 MHz, DMSO) 5 13.11 (s, 1 H), 7.84 (d, J = 7.8 Hz, 2H), 7.69 (d, J = 16.5 Hz, 1 H), 7.54 - 7.42 (m, 2H), 7.38 (t, J = 7.3 Hz, 2H), 7.33 - 7.23 (m, 1 H), 6.51 (d, J = 16.4 Hz, 1 H).
13C NMR (75 MHz, DMSO) 5 169.03, 146.88, 136.97, 136.95, 133.25, 131.97, 131.30, 128.67, 127.82, 127.32, 127.12.
Intermediate 45.
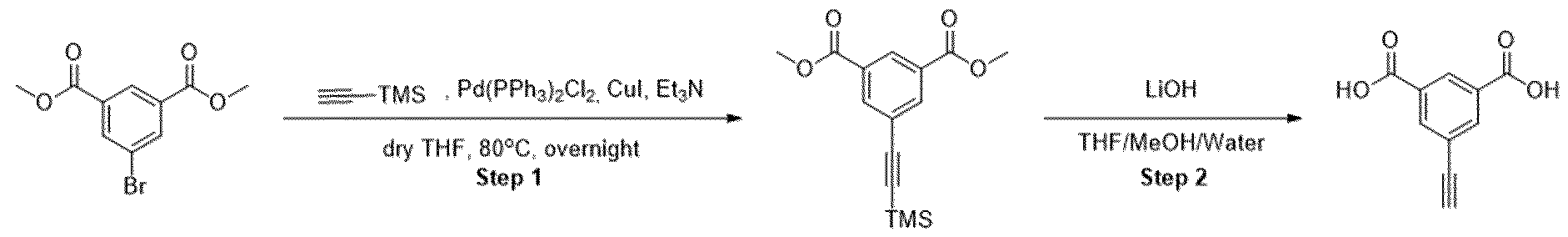
Step 1 :
A 25 mL round bottom flask equipped with a magnetic stir bar and a condenser was charged with the 5-bromoisophthalic acid dimethyl ester (819 mg, 3 mmol), trimethylsilylacetylene (589 mg, 6 mmol), THF (10 ml) and Et3N (2 ml). The reaction mixture was degassed using nitrogen gas. Pd(PPh3)2CI2 (84 mg, 0.12 mmol) and Cui (46 mg, 0.24 mmol) were added afterwards. The mixture was stirred at 70 °C overnight. The solvent was evaporated, and the residue was purified by silica gel column chromatography eluting with cyclohexane to cyclohexane: ethylacetate 60:40 to obtain the desired product as light orange solid in 72% yield (617 mg).
MS (M+H)+: 291.6.
1H NMR (300 MHz, CDCI3) 6 8.60 (s, 1 H), 8.29 (s, 2H), 3.95 (s, 6H), 0.26 (s, 9H)
13C NMR (75 MHz, CDCI3) 6 165.71 , 137.00, 130.98, 130.42, 124.38, 102.86, 96.88, 52.66, - 0.07
Step 2:
The product from the previous step was dissolved in mixture of THF/MeOH/Water (5 ml/5 ml/5 ml) and LiOH (1 g) was added. The resulting mixture was stirred overnight. After the completion of the reaction, 1M HCI was added (25 ml) and the mixture was extracted with EtOAc. The organic layers were combined, dried over the sodium sulfate and evaporated under the reduced pressure yielding intermediate 40 as white solid in 91 % yield (530 mg). MS (M+H)+: 190.7.
1H NMR (300 MHz, DMSO) 5 13.55 (s, 2H), 8.48 (t, J = 1.7 Hz, 1 H), 8.19 (d, J = 1.6 Hz, 2H), 4.45 (s, 1 H).
13C NMR (75 MHz, DMSO) 5 165.69, 135.84, 131.97, 129.98, 122.66, 82.61 , 81.50.
Scheme for intermediates 46-51.
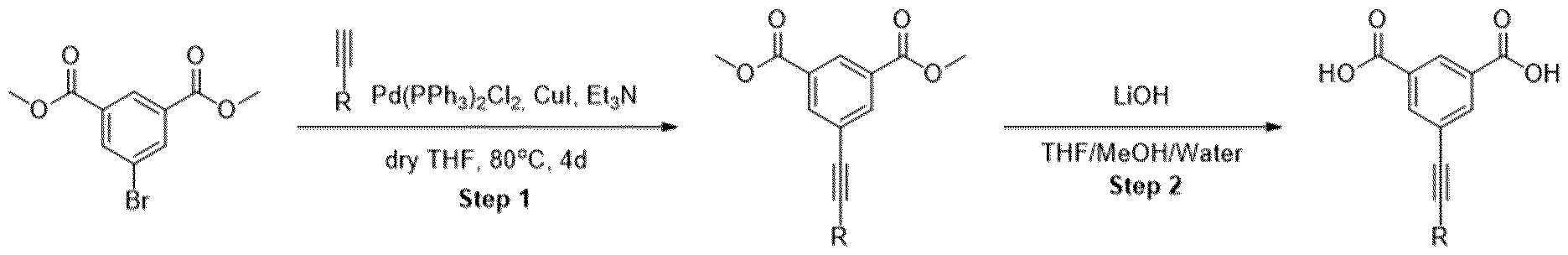
Step 1 :
A 25 mL round bottom flask equipped with a magnetic stir bar and a condenser was charged with the 5-bromoisophthalic acid dimethyl ester (400 mg, 1.46 mmol), acetylene (1.5 mmol), THF (7.5 ml) and Et3N (1.5 ml). The reaction mixture was degassed using nitrogen gas. Pd(PPh3)2CI2 (45 mg, 0.064 mmol) and Cui (16.6 mg, 0.09 mmol) were added afterwards. The mixture was stirred at 70 °C overnight. The solvent was evaporated, and the residue was purified by silica gel column chromatography eluting with cyclohexane to cyclohexane: ethylacetate 80:20 to obtain the desired product.
Step 2:
Synthesized according to general procedure 1. Starting materials: THF/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (400 mg).
Intermediate 46. Starting material: propyne (1 M solution in THF, 1.5 ml).
Yield, over two steps: 133 mg (44%). Step 1 :
MS (M+H)+: 233.0.
1H NMR (300 MHz, CDCI3) 6 8.48 (t, J = 1.7 Hz, 1 H), 8.15 (d, J = 1.6 Hz, 2H), 3.87 (s, 6H), 2.00 (s, 3H).
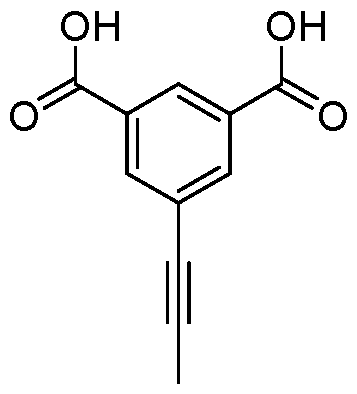 13C NMR (75 MHz, CDCI3) 6 165.73, 136.51 , 130.75, 129.42, 125.10,
13C NMR (75 MHz, CDCI3) 6 165.73, 136.51 , 130.75, 129.42, 125.10,
88.16, 78.00, 52.44, 4.30.
Step 2:
MS (M+H)+: 204.9.
1H NMR (300 MHz, DMSO) 5 13.43 (s, 2H), 8.37 (t, J = 1.6 Hz, 1 H), 8.06 (d, J = 1.6 Hz, 2H),
2.08 (s, 2H).
13C NMR (75 MHz, DMSO) 5 165.85, 135.36, 131.80, 128.94, 124.25, 88.88, 77.87, 3.85.
Intermediate 47.
Starting material: tert-butylacetylene (126 mg).
Yield, over two steps: 280 mg (79%).
Step 1 :
MS (M+H)+: 274.9.
1H NMR (300 MHz, CDCI3) 5 8.47 (t, J = 1.6 Hz, 1 H), 8.14 (d, J = 1.6 Hz, 2H), 3.87 (s, 6H), 1.25 (s, 9H).
13C NMR (75 MHz, CDCI3) 5 165.77, 136.55, 130.66, 129.30, 125.17,
 100.69, 77.38, 52.43, 30.83, 27.98.
100.69, 77.38, 52.43, 30.83, 27.98.
Step 2:
MS (M+H)+: 246.9.
1H NMR (300 MHz, DMSO) 6 13.50 (s, 2H), 8.42 (t, J = 1.6 Hz, 1 H), 8.09 (d, J = 1.6 Hz, 2H), 1.37 (s, 9H).
13C NMR (75 MHz, DMSO) 5 165.85, 135.35, 131.78, 128.94, 124.04, 100.48, 77.21 , 30.45, 27.58.
Intermediate 48. Starting material: cyclopropylacetylene (100 mg).
Yield, over two steps: 90 mg (39%).
Step 1 :
MS (M+H)+: 258.9.
1H NMR (300 MHz, CDCI3) 6 8.47 (t, J = 1.6 Hz, 1 H), 8.13 (d, J = 1.6 Hz,
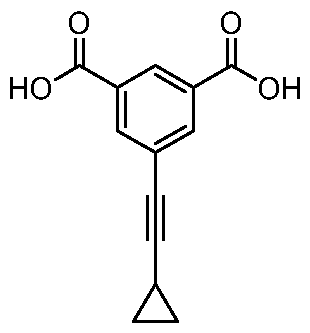 2H), 3.87 (s, 6H), 1 .48 - 1.28 (m, 1 H), 0.90 - 0.65 (m, 4H).
2H), 3.87 (s, 6H), 1 .48 - 1.28 (m, 1 H), 0.90 - 0.65 (m, 4H).
13C NMR (75 MHz, CDCI3) 6 165.63, 136.45, 130.61 , 129.18, 124.95, 95.70,
73.89, 52.34, 8.63, 0.00.
Step 2:
MS (M+H)+: 230.9.
1H NMR (300 MHz, DMSO) 5 13.48 (s, 2H), 8.41 (t, J = 1.7 Hz, 1 H), 8.09 (d, J = 1.6 Hz, 2H), 1.68 - 1.45 (m, 1 H), 1.05 - 0.71 (m, 4H).
13C NMR (75 MHz, DMSO) 5 166.18, 135.73, 132.08, 129.14, 124.49, 96.34, 74.04, 8.86, 0.00.
Intermediate 49. Starting material: cyclohexylacetylene (162 mg).
Yield, over two steps: 140 mg (52%). Step 1 : MS (M+H)+: 300.9. 1H NMR (300 MHz, CDCI3) 5 8.55 (t, J = 1.6 Hz, 1 H), 8.23 (d, J = 1.6 Hz, 2H), 2.72 - 2.53 (m, 1 H), 1.95 - 1.84 (m, 2H), 1.82 - 1.70 (m, 2H), 1.65 - 1.49 (m, 3H), 1.46 - 1.30 (m, 3H).
 13C NMR (75 MHz, CDCI3) 5 165.79, 136.58, 130.69, 129.30, 125.24, 96.70, 78.82, 52.44, 32.47, 29.59, 25.85, 24.82.
13C NMR (75 MHz, CDCI3) 5 165.79, 136.58, 130.69, 129.30, 125.24, 96.70, 78.82, 52.44, 32.47, 29.59, 25.85, 24.82.
Step 2:
MS (M+H)+: 273.0.
1H NMR (300 MHz, DMSO) 5 13.42 (s, 2H), 8.37 (t, J = 1.6 Hz, 1 H), 8.04 (d, J = 1.6 Hz, 2H), 2.75 - 2.59 (m, 1 H), 1.90 - 1.77 (m, 2H), 1.74 - 1.63 (m, 2H), 1.59 - 1.44 (m, 3H), 1.42 - 1.28 (m, 3H).
13C NMR (75 MHz, DMSO) 5 165.86, 135.39, 131.79, 128.94, 124.15, 96.42, 78.80, 31.87, 28.64, 25.27, 24.16.
Intermediate 50. Starting material: tert-butyl 4-ethynylpiperidine-1 -carboxylate (313 mg). Yield, over two steps: 280 mg (51%). Step 1 : MS (M-Boc)+: 302.0. 1H NMR (300 MHz, CDCI3) 6 8.50 (t, J = 1.6 Hz, 1 H), 8.15 (d, J = 1.6 Hz, 2H), 3.88 (s, 6H), 3.74 - 3.60 (m, 2H), 3.26 - 3.09 (m, 2H), 2.82 - 2.67 (m, 1 H), 1.92 - 1.70 (m, 2H), 1.67 - 1.55 (m, 2H), 1.35 (s, 9H).
 13C NMR (75 MHz, CDCI3) 6 165.67, 154.77, 136.57, 130.80, 129.65, 124.63, 94.01 , 80.20, 79.57, 52.48, 31.23, 28.44, 27.58.
13C NMR (75 MHz, CDCI3) 6 165.67, 154.77, 136.57, 130.80, 129.65, 124.63, 94.01 , 80.20, 79.57, 52.48, 31.23, 28.44, 27.58.
Step 2:
MS (M-Boc)+: 273.9.
1H NMR (300 MHz, DMSO) 5 13.42 (s, 2H), 8.38 (t, J = 1.6 Hz, 1 H), 8.08 (d, J = 1.6 Hz, 2H), 3.71 - 3.57 (m, 2H), 3.24 - 3.09 (m, 2H), 2.96 - 2.82 (m, 1 H), 1.88 - 1.76 (m, 2H), 1.65 - 1.46 (m, 2H), 1.40 (s, 9H).
13C NMR (75 MHz, DMSO) 5 165.82, 153.80, 135.51 , 131.80, 129.14, 123.82, 94.51 , 79.66, 78.64, 30.82, 28.03, 26.66.
Intermediate 51.
 150 mL of water and 150 mL of 3 M HCI were added, and the reaction mixture was extracted with ethyl acetate (3 x 200 mL). The organic extracts were combined and the solvents were evaporated under reduced pressure. The residue was purified by silica gel column chromatography eluting with cyclohexane to cyclohexane: ethylacetate 40:60 obtaining the intermediate 50 as a light orange solid in 50% yield (1.2 g).
150 mL of water and 150 mL of 3 M HCI were added, and the reaction mixture was extracted with ethyl acetate (3 x 200 mL). The organic extracts were combined and the solvents were evaporated under reduced pressure. The residue was purified by silica gel column chromatography eluting with cyclohexane to cyclohexane: ethylacetate 40:60 obtaining the intermediate 50 as a light orange solid in 50% yield (1.2 g).
MS (M+H)+: 242.5.
1H NMR (300 MHz, CDCI3) 6 8.34 (s, 1 H), 8.19 (s, 2H), 2.58 (s, 6H).
13C NMR (75 MHz, CDCI3) 6 195.87, 138.97, 135.36, 126.45, 123.51 , 26.72.
Intermediate 53.
A 50 mL Schlenk tube equipped with a stir bar was
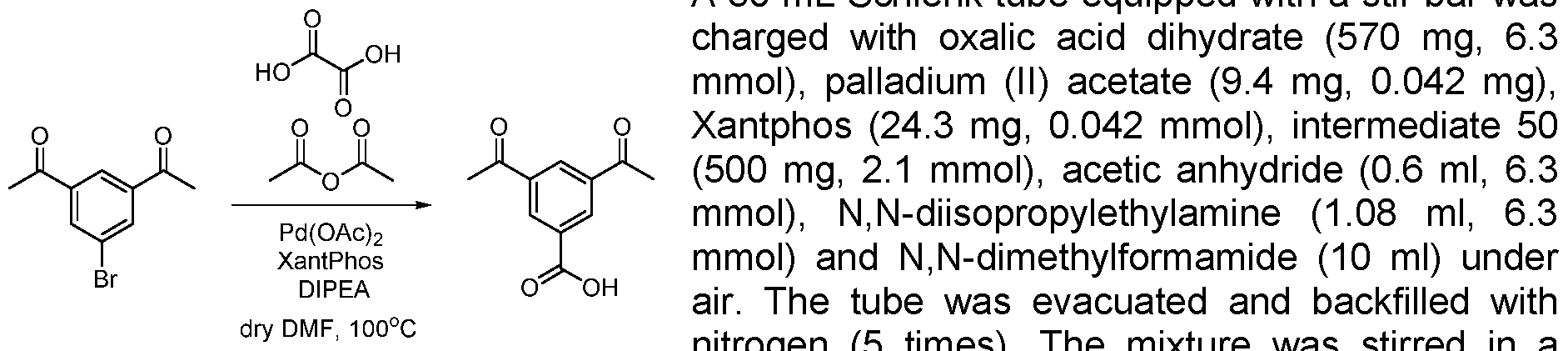 nitrogen (5 times). The mixture was stirred in a preheated oil bath (100 °C) for 21 h. The reaction mixture was diluted with ethyl acetate (10 mL), acidified with 2 M hydrochloric acid, and washed with brine. The organic phase was dried over anhydrous MgSCL and concentrated in vacuo. The residue was purified by silica gel column chromatography eluting with cyclohexane to cyclohexane: ethylacetate 50:50 to obtain the desired product as a yellow solid in 60% yield (260 mg).
nitrogen (5 times). The mixture was stirred in a preheated oil bath (100 °C) for 21 h. The reaction mixture was diluted with ethyl acetate (10 mL), acidified with 2 M hydrochloric acid, and washed with brine. The organic phase was dried over anhydrous MgSCL and concentrated in vacuo. The residue was purified by silica gel column chromatography eluting with cyclohexane to cyclohexane: ethylacetate 50:50 to obtain the desired product as a yellow solid in 60% yield (260 mg).
MS (M+H)+: 206.8.
1H NMR (300 MHz, DMSO) 5 8.63 (s, 3H), 2.70 (s, 6H)
13C NMR (75 MHz, DMSO) 5 196.99, 166.09, 137.56, 132.45, 132.01 , 131.29, 27.00
Intermediate 54.
 warmed to room temperature and then was heated to 80 °C over 30 min. The mixture was cooled and extracted with EtOAc. The organic solvents were evaporated under reduced pressure obtaining the product as red solid in 80% yield (210 mg).
warmed to room temperature and then was heated to 80 °C over 30 min. The mixture was cooled and extracted with EtOAc. The organic solvents were evaporated under reduced pressure obtaining the product as red solid in 80% yield (210 mg).
MS (M+H)+: 178.7.
1H NMR (300 MHz, CDCI3) 6 8.01 (s, 1 H), 7.63 (s, 1 H), 2.58 (s, 5H).
13C NMR (75 MHz, CDCI3) 6 197.71 , 156.75, 138.80, 120.69, 119.56, 26.84.
Intermediate 55.
 , stirring at room temperature overnight.
, stirring at room temperature overnight.
At this point, 30 mL of 1 M HCI (aq) was added then heated at for 3 h. The reaction was diluted with water and extracted with EtOAc. The combined organic layers were dried, filtered, and evaporated. The residue was purified by silica gel column chromatography eluting with cyclohexane to cyclohexane: ethylacetate 50:50 to obtain the desired product as a yellow solid in 53% yield (550 mg).
MS (M+H)+: 207.9.
1H NMR (300 MHz, CDCI3) 6 8.57 (d, J = 1.5 Hz, 1 H), 8.21 (dd, J = 8.0, 1.6 Hz, 1 H), 7.47 (d, J = 7.8 Hz, 1 H), 2.63 (s, 2H), 2.51 (s, 3H).
13C NMR (75 MHz, CDCI3) 6 198.98, 194.80, 141.41 , 138.75, 133.51 , 127.87, 124.18, 30.15, 26.75.
Step 2:
In a round-bottom flask tin(ll)chloride (2.1 g, 10 mmol) was dissolved in 6 of mL cone. HCI and warmed up to 60 °C followed by the slow addition of product from step 1 (550 mg, 2.65 mmol). Afterwards, the solution was stirred for further 15 min and then poured, under gas formation, in a mixture of 11 g potassium carbonate and 40 mL ice/water. The product was extracted with EtOAc and the combined organic layers were dried over sodium sulfate. The solvent was removed under the reduced pressure obtaining intermediate 53 as a yellow solid in 93% yield (400 mg).
MS (M+H)+: 178.0.
1H NMR (300 MHz, CDCI3) 6 7.73 (d, J = 8.4 Hz, 1 H), 7.20 - 7.17 (m, 1 H), 7.12 (dd, J = 8.4, 1.8 Hz, 1 H), 2.54 (s, 3H), 2.51 (s, 3H).
13C NMR (75 MHz, CDCI3) 6200.62, 197.85, 149.53, 141.17, 132.36, 120.88, 117.36, 115.28, 28.15, 26.89.
Intermediate 56. To a mixture of 4-Amino-2 6-
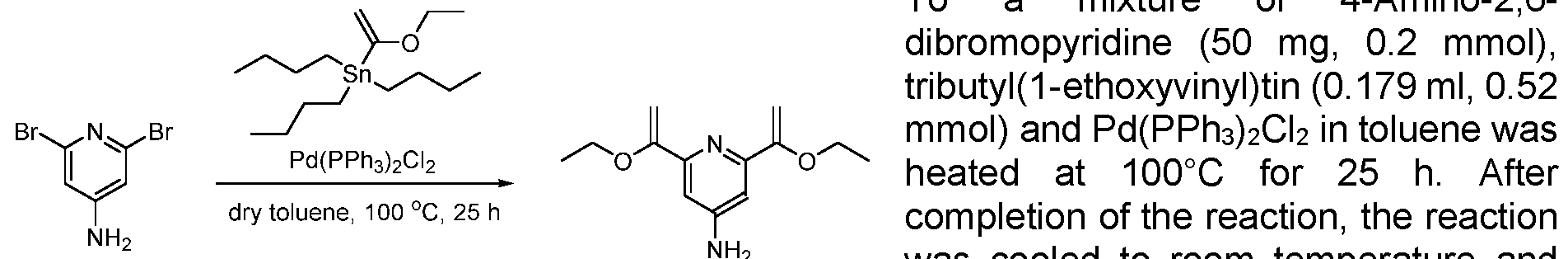 2 was cooled to room temperature and concentrated in vacuo. The residue was purified by silica gel column chromatography eluting with cyclohexane to cyclohexane:ethylacetate 50:50. The solvent was removed to obtain the enol ether product with the impurity of tin byproduct (72.9 mg). Yield was not calculated.
2 was cooled to room temperature and concentrated in vacuo. The residue was purified by silica gel column chromatography eluting with cyclohexane to cyclohexane:ethylacetate 50:50. The solvent was removed to obtain the enol ether product with the impurity of tin byproduct (72.9 mg). Yield was not calculated.
MS (M+H)+: 323.5
1H NMR (300 MHz, DMSO) 5 6.78 (s, 2H), 6.12 (s, 2H), 5.32 (d, J = 1.2 Hz, 2H), 4.27 (m, 2H), 3.88 (q, J = 7.0 Hz, 4H), 0.88 (t, J = 7.3 Hz, 6H).
13C NMR (75 MHz, DMSO) 5 158.47, 155.62, 151.94, 103.15, 83.98, 62.91 , 14.47.
Intermediate 57.
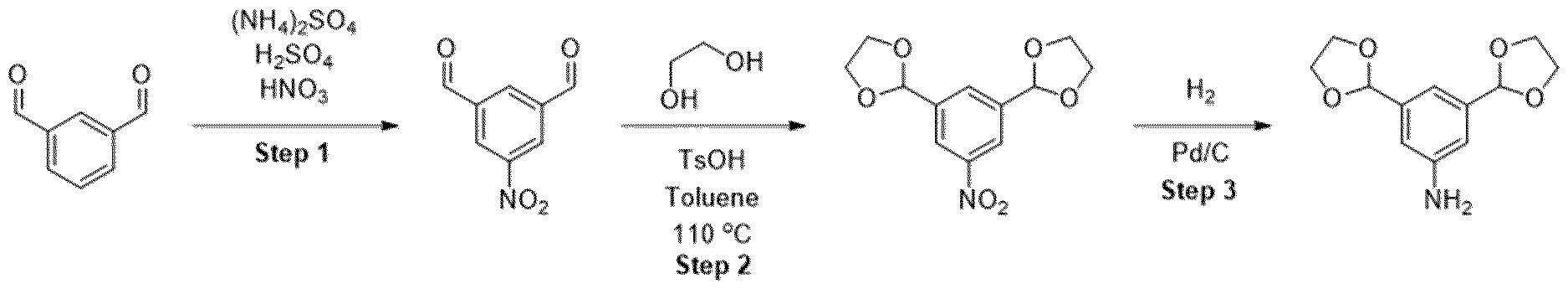
Step 1 :
To a mixture of 10 g of ammonium sulfate in 16.5 ml of sulfuric and and 3.5 ml of nitric acid the solution of isophthalaldehyde (2.5 g, 18.6 mmol) in 15 ml of sulfuric acid was added. The mixture was stirred for two days and then poured in ice. The precipitate was filtered and washed with water to afford the desired product as a white solid in 40% yield (1 .32 g).
MS (M+H)+: 179.9.
1H NMR (300 MHz, CDCI3) 6 10.23 (s, 2H), 8.95 (s, 2H), 8.74 (m, 1 H).
13C NMR (75 MHz, CDCI3) 6 188.59, 138.27, 134.60, 128.56.
Step 2:
The mixture of the product from step 1 (820 mg, 4.8 mmol), ethylene glycol (500 mg), and p- toluenesulfonic acid (4 mg) in toluene (25 ml) was refluxed overnight in flask connected to Dean-Stark apparatus. Afterwards, the solvent was removed under the reduced pressure and the residue was purified by silica gel column chromatography eluting with cyclohexane to cyclohexane: ethylacetate 0:100 to obtain the desired product as a yellow oil in 59% yield (760 mg).
MS (M+H)+: 267.7.
1H NMR (300 MHz, CDCI3) 6 8.27 (s, 2H), 7.84 (s, 1 H), 5.82 (s, 2H), 4.16 - 3.89 (m, 8H). 13C NMR (75 MHz, CDCI3) 6 148.32, 140.68, 130.67, 122.11 , 102.07, 65.46.
Step 3:
Product from the step 2 was dissolved in EtOH (20mL). 10% Pd/C (40 mg) was added afterwards, and the mixture was stirred in hydrogen atmosphere (1 atm) in stainless steel miniclave. After the end of the reaction, the catalyst was filtered, solvent removed under the reduced pressure to obtain the desired product as a yellow oil in 81 % yield (550 mg).
MS (M+H)+: 237.9.
1H NMR (300 MHz, CDCI3) 6 6.99 (s, 1 H), 6.81 (d, J = 1.5 Hz, 2H), 5.76 (s, 2H), 4.22 - 3.90 (m, 8H).
13C NMR (75 MHz, CDCI3) 6 146.62, 139.46, 114.82, 113.57, 103.46, 65.19.
Intermediate 58.
Step 1 :
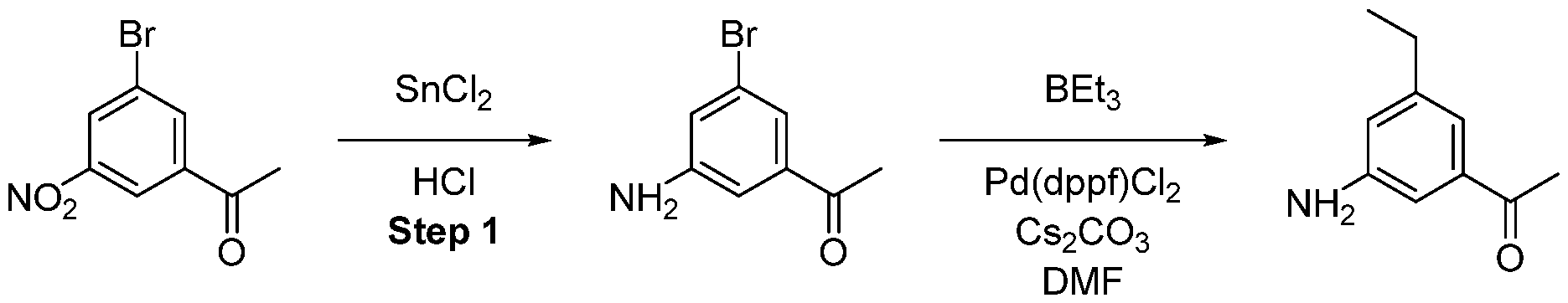
Step 2
In a round-bottom flask tin(ll)chloride (2.1 g, 10 mmol) was dissolved in 6 of mL cone. HCI and warmed up to 50 °C followed by the slow addition of 1-(3-bromo-5-nitrophenyl)ethenone (500 mg, 2.04 mmol). Afterwards, the solution was stirred for further 15 min and then poured, under gas formation, in a mixture of 11 g potassium carbonate and 40 mL ice/water. The product was extracted with EtOAc and the combined organic layers were dried over sodium sulfate. Finally, the solvent was removed in vacuo obtaining a bright yellow solid in 85% yield (340 mg).
1H NMR (300 MHz, CDCI3) 6 7.34 (s, 1 H), 7.08 (s, 1 H), 6.93 (s, 1 H), 3.62 (s, 2H), 2.46 (s, 4H).
13C NMR (75 MHz, CDCI3) 6 197.08, 153.18, 147.96, 139.49, 123.25, 121.83, 121.45, 112.86, 26.70.
Step 2:
In a three-necked flask, product from step 1 (330 mg, 1.6 mmol) and triethylborane (1 M in THF, 2.3 ml), CS2CO3 (2.7 g. 6.3 mmol) and 13 ml of DMF were placed. The mixture was evacuated and flushed with nitrogen three times and PddppfCh (30 mg, 0.04 mmol) was added. The reaction was stirred overnight at 70 °C. After the end of the reaction, it was cooled to room temperature and diluted with 15 ml of EtOAc. The organic layer was washed with water, separated and concentrated under reduced pressure. The residue was purified by silica gel column chromatography eluting with cyclohexane to cyclohexane: ethylacetate 40:60 obtaining the intermediate 57 as a light orange solid in 46% yield (120 mg).
MS (M+H)+: 163.9.
1H NMR (300 MHz, CDCI3) 6 7.18 (s, 1 H), 7.11 (s, 1 H), 6.76 (s, 1 H), 3.68 (s, 2H), 2.60 (q, J = 7.6 Hz, 2H), 2.54 (s, 3H), 1.22 (t, J = 7.5 Hz, 3H).
13C NMR (75 MHz, CDCI3) 6 198.63, 146.14, 145.86, 138.32, 124.70, 119.67, 118.83, 112.26, 31.46, 26.76, 15.41.
Intermediate 59.
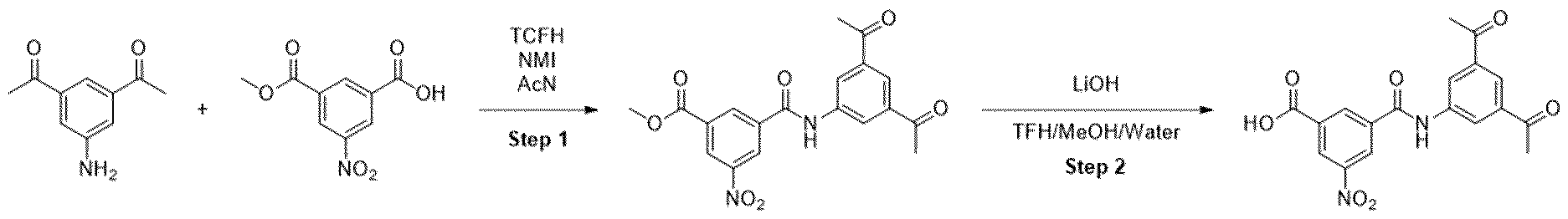
Step 1 :
The 5-Nitroisophthalic acid monomethyl ester (1.22 g, 5.6 mmol), intermediate 1 (1 g, 5.6 mmol) were dissolved in 20 ml of acetonitrile. /V-methylimidazole (1.2 ml) and TCFH (1.46 g) were added sequentially in a single portion. The reaction was stirred overnight. After the completion of the reaction. The product was filtered and used in the next step without additional purification.
MS (M+H)+: 384.9.
1H NMR (300 MHz, CDCI3) 6 9.92 (s, 1 H), 9.16 (t, J = 2.0 Hz, 1 H), 8.99 (t, J = 1.6 Hz, 1 H), 8.97 - 8.90 (m, 1 H), 8.71 (d, J = 1.6 Hz, 2H), 8.30 (t, J = 1.5 Hz, 1 H), 4.02 (s, 3H), 2.69 (s, 3H).
13C NMR (75 MHz, CDCI3) 6 197.48, 164.28, 163.23, 139.16, 138.01 , 136.43, 134.26, 132.38, 127.18, 126.81 , 124.49, 124.07, 121.79, 53.10, 26.89.
Step 2:
The product from the previous step was dissolved in a mixture of THF/MeOH/Water (3 ml/3 ml/3 ml) and LiOH (800 mg) was added. The resulting mixture was stirred overnight. After completion of the reaction, 1 M HCI was added (5 ml) and the mixture was extracted with EtOAc. The organic layers were combined, dried over sodium sulfate and evaporated under reduced pressure yielding the product as green solid.
Yield, over two steps: 1.13 g (55%).
MS (M+H)+: 371.0.
1H NMR (300 MHz, DMSO) 5 11.06 (s, 1 H), 9.06 (t, J = 1.9 Hz, 1 H), 8.96 (d, J = 1.6 Hz, 1 H), 8.78 (d, J = 1.5 Hz, 1 H), 8.63 (d, J = 1.6 Hz, 2H), 8.25 (s, 1 H), 2.68 (s, 6H).
13C NMR (75 MHz, DMSO) 5 197.13, 164.99, 162.75, 148.06, 139.42, 137.59, 136.04, 134.02, 132.91 , 126.51 , 126.28, 123.79, 123.65, 26.88.
Scheme for intermediates 60-63.
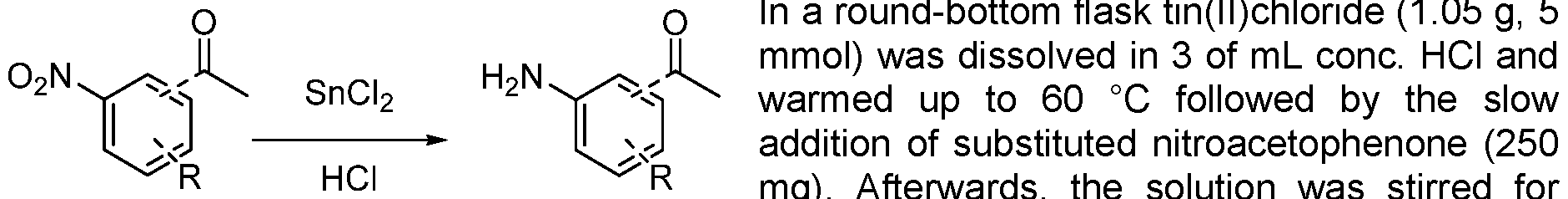 g . , r further 15 min and then poured, under gas formation, in a mixture of 5 g potassium carbonate and 20 mL ice/water. The product was extracted with EtOAc and the combined organic layers were dried over sodium sulfate. Finally, the solvent was removed in vacuo obtaining a product.
Intermediate 60.
g . , r further 15 min and then poured, under gas formation, in a mixture of 5 g potassium carbonate and 20 mL ice/water. The product was extracted with EtOAc and the combined organic layers were dried over sodium sulfate. Finally, the solvent was removed in vacuo obtaining a product.
Intermediate 60.
Starting material: 1-(4-methoxy-3-nitrophenyl)-1 -ethanone.
 mmol) in acetic
mmol) in acetic
Step 1 anhydride (12.5 mL) was cooled to -40 °C prior to the dropwise addition of 69% nitric acid (0.82 mL). The reaction mixture was warmed to room temperature and stirred for 1.5 h. The solution was then poured onto ice water and neutralized with sat. sodium bicarbonate solution. The aqueous layer was then separated and extracted with ethyl acetate. The combined organic extracts were then dried using MgSCU, filtered, then concentrated in vacuo. The residue was purified by silica gel column chromatography eluting with cyclohexane to cyclohexane: ethylacetate 50:50 to obtain the desired product as a yellow solid in 23% yield (320 mg).
MS (M+H)+: 154.8.
1H NMR (300 MHz, DMSO) 5 13.01 (s, 1 H), 8.14 (dd, J = 3.7, 1.6 Hz, 1 H), 7.68 (dd, J = 2.6, 1.6 Hz, 2H), 2.50 (s, 5H).
13C NMR (75 MHz, DMSO) 5 188.31 , 136.63, 131.42, 125.08, 111.05, 25.72.
Step 2:
In a round-bottom flask tin(ll)chloride (1.05 g, 5 mmol) was dissolved in 3 of mL cone. HCI and warmed up to 60 °C followed by the slow addition of product from the previous step (250 mg). Afterwards, the solution was stirred for further 15 min and then poured, under gas formation, in a mixture of 5 g potassium carbonate and 20 mL ice/water. The product was extracted with EtOAc and the combined organic layers were dried over sodium sulfate. Finally, the solvent was removed in vacuo obtaining the product as black solid, which was immediately used in the next step.
NMR was not measured due to fast degradation of the starting material.
1.2 Synthesis of compounds 1-1 to 1-144.
General procedure 2 for synthesis of guanilhydrazones.
Corresponding ketone was dissolved in 5 ml 96% ethanol, 100 mg of aminoguanidine hydrochloride and two drops of concentrated HCI were added. The reaction was heated to 80 °C and stirred at this temperature overnight. After the completion of the reaction the solvent was evaporated, and the residue was purified using reverse-phase column chromatography eluting with water-acetonitrile with formic acid modifier. The resulting solution was lyophilized yielding the product as a solid di, tri, tetra or pentaformate salt.
Method A:
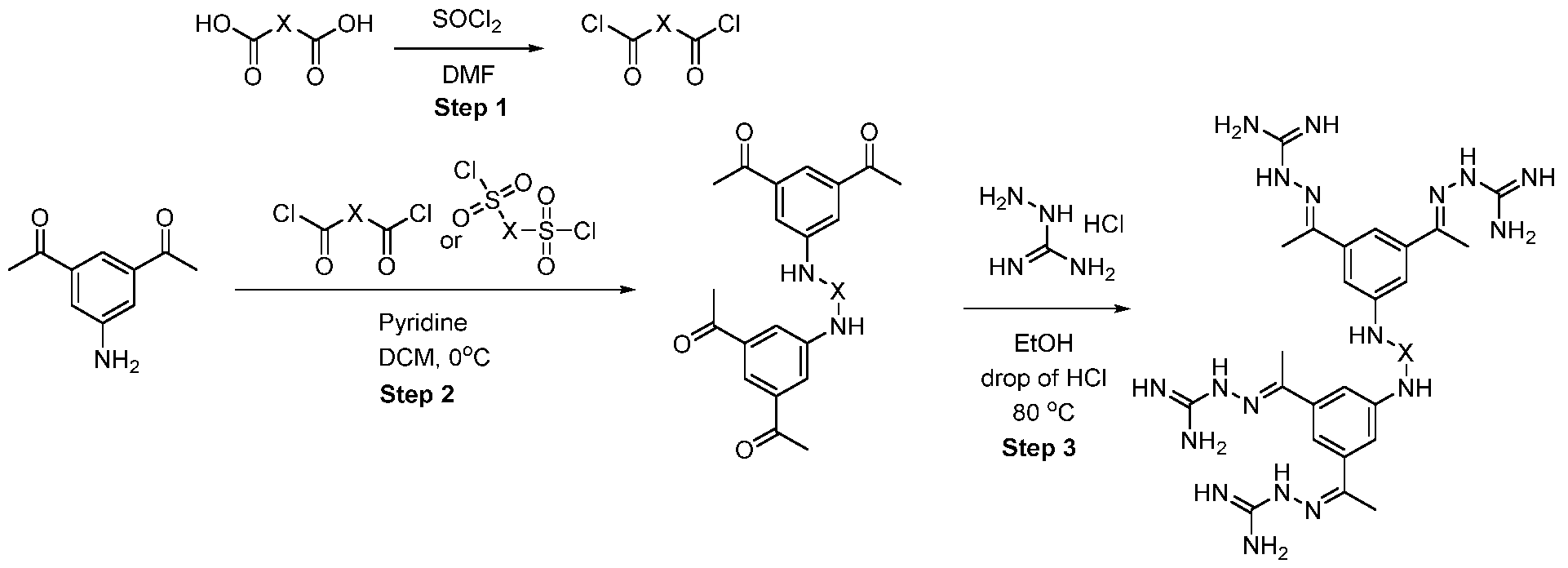
General procedure for examples:
The corresponding sulfonyl chlorides were purchased from vendors and dichloranhydrides were synthesized according to the procedure below:
Step 1 :
The corresponding diacid (0.14 mmol) was suspended in thionyl chloride (0.5 mL) and five drops of dry DMF were added. The mixture was heated to 80 °C and stirred for 2 h. The solvent was removed in vacuo yielding the diacid dichloride. Respective products were used without further purification.
Step 2:
Intermediate 1 (50 mg, 0.28 mmol) was dissolved in 2 ml of dry CH2CI2, 0.06 ml pyridine was added and the reaction mixture cooled down to 0 °C using an ice bath. The corresponding diacid chloride (dissolved in 1 ml dry DCM) was added slowly. Then, the reaction mixture was warmed up to rt and stirred overnight. The solvent was evaporated, and the residue was used in the next reaction without additional purification.
Step 3:
The synthesis was carried out according to general procedure 2 using the residue from the previous step.
Method B:
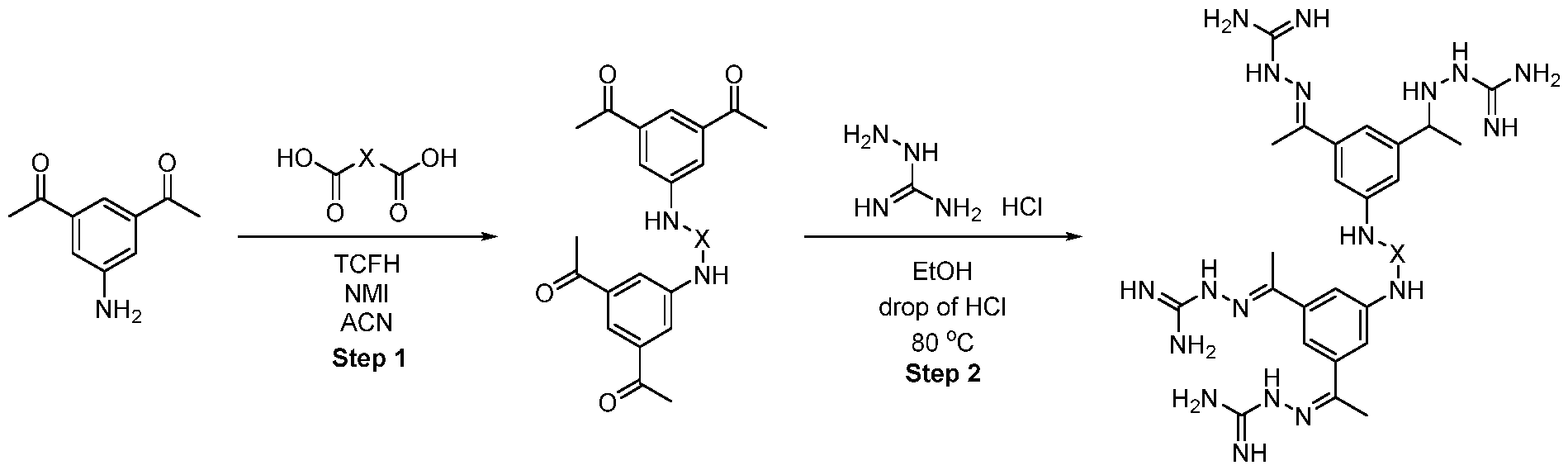
Step 1 :
The acid (0.14 mmol) and intermediate 1 (50 mg, 0.28 mmol) were dissolved in 1 ml of MeCN.
0.06 mL N-methylimidazole and TCFH (73 mg, 0.28 mmol) were added sequentially. The reaction was stirred overnight. After the completion of the reaction,
I: the product was filtered (if precipitated)
II: solvents were evaporated, and the residue was purified by silica gel column chromatography eluting with Cyclohexane: Ethylacetate 100:0% to Cyclohexane: Ethylacetate 0: 100% to obtain the desired product.
Step 2:
The synthesis was carried out according to general procedure 2 using the product from the previous step.
Additional step between Step 1 and 2 was added if Boc-deprotection was necessary.
The product from step 1 was dissolved in mixture of DCM:TFA (1 ml/1ml) and stirred for 3 h at rt. After the completion of the reaction the solvents were evaporated, and the residue was used in the next step.
1-1.
Starting material: isophthalic acid (23 mg).
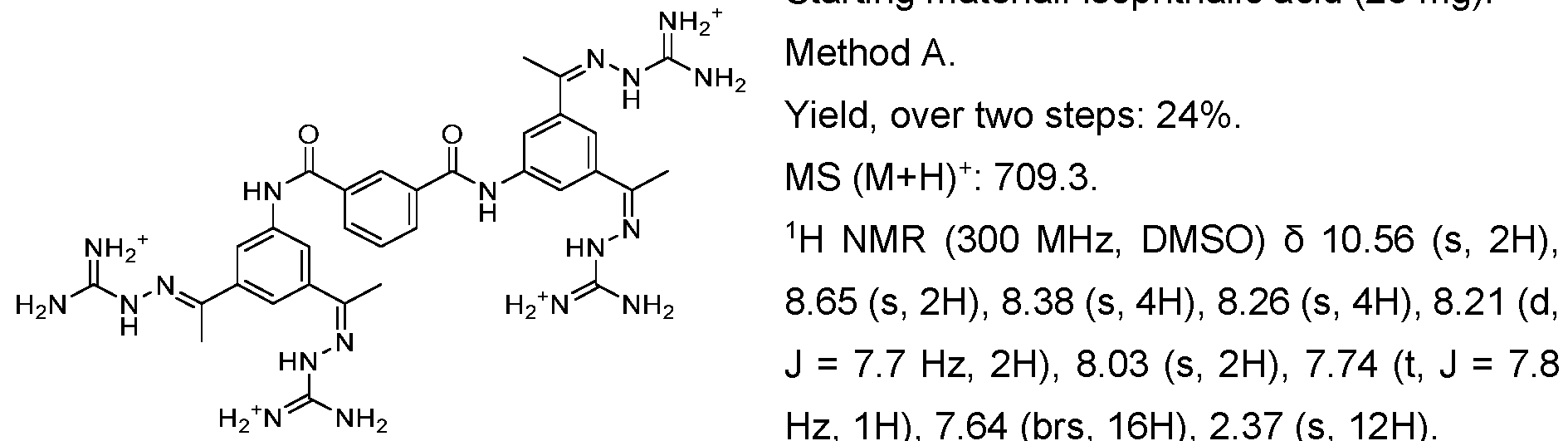 , , . , , . , .
1-2.
, , . , , . , .
1-2.
S i i l l h 13
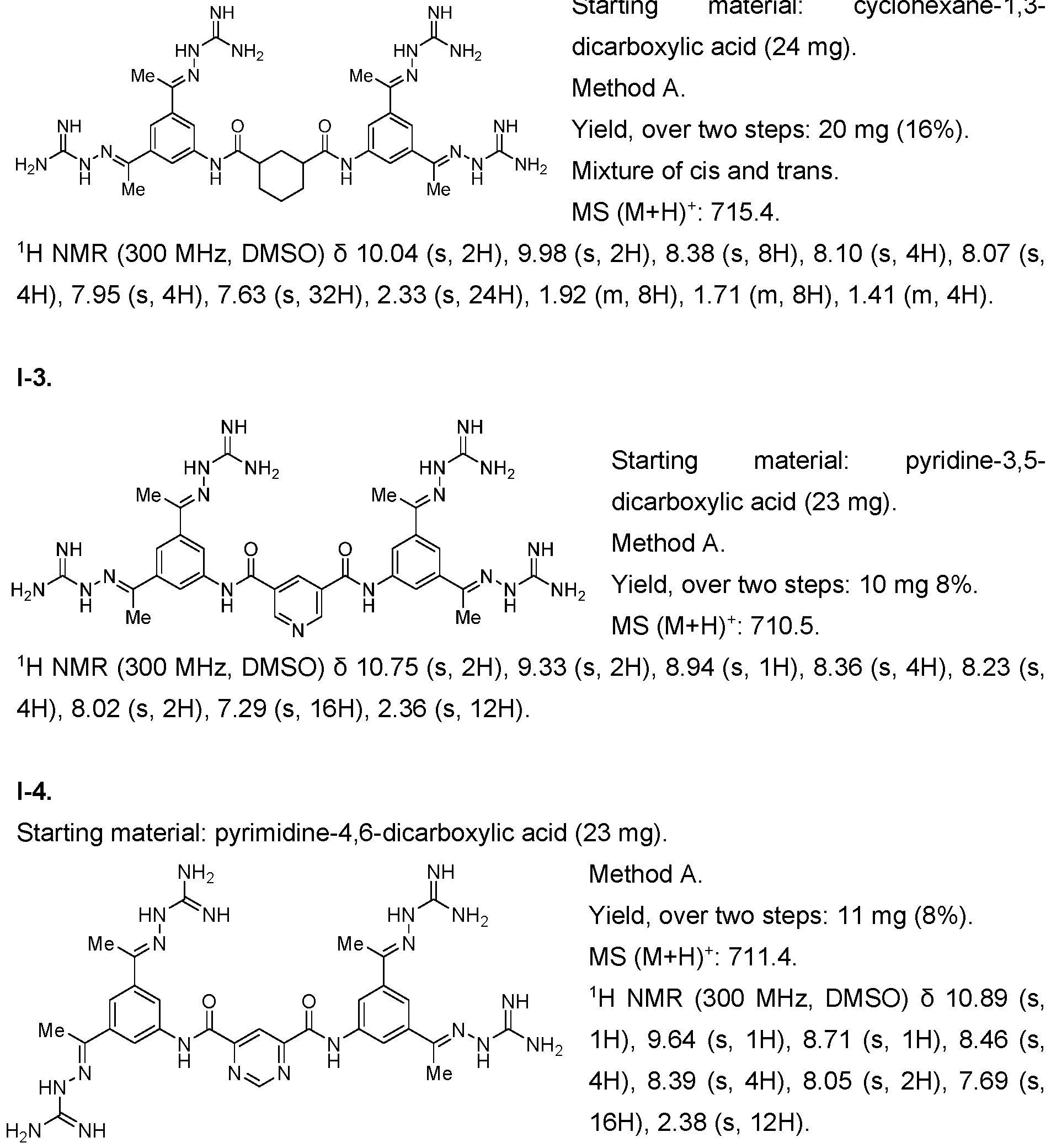 1-5.
1-5.
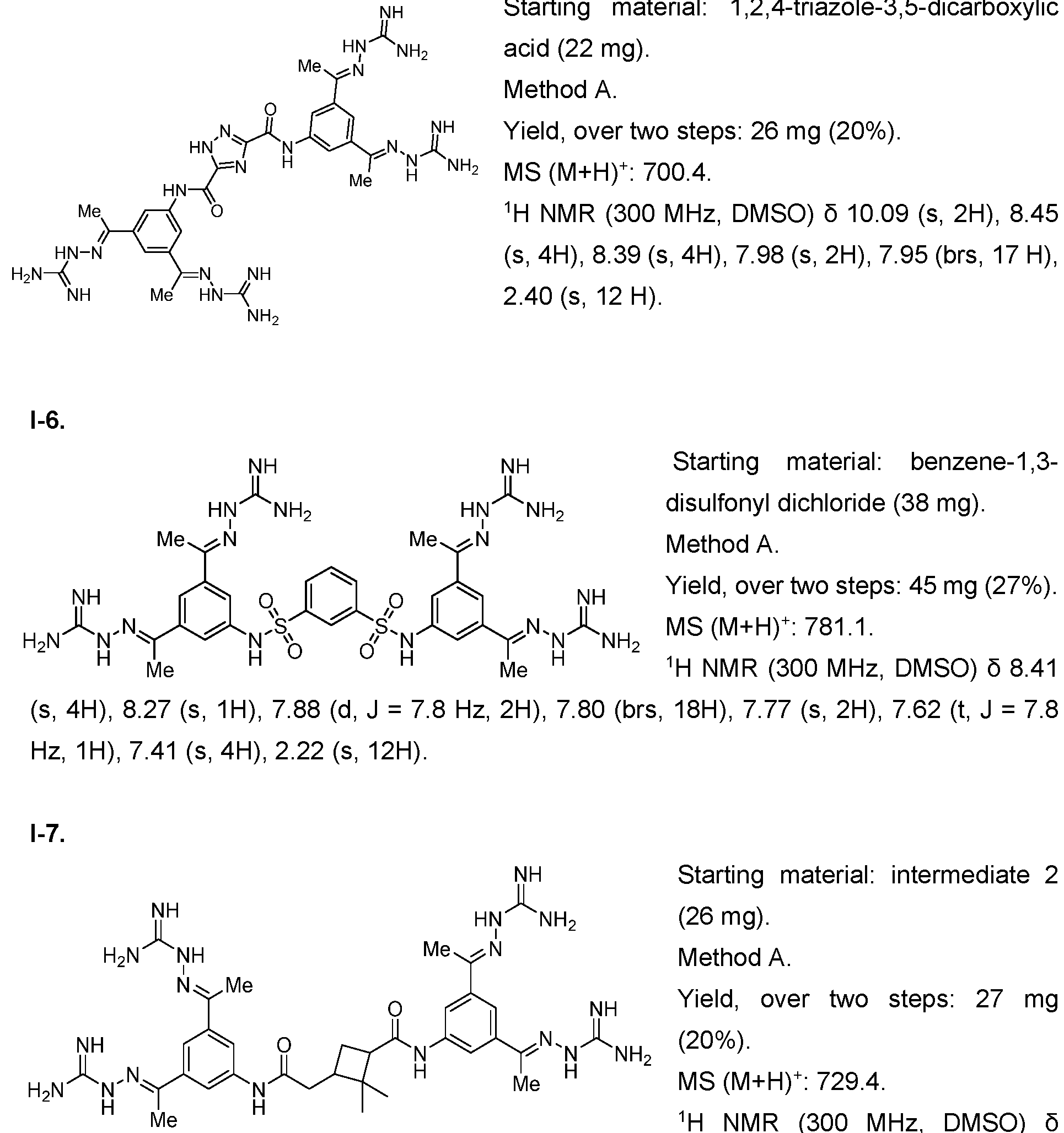 ,
,
10.07 (s, 1 H), 9.71 (s, 1 H), 8.41 (s, 4H), 8.08 (m, 2H), 8.04 (s, 2H), 7.97 (s, 2H) 7.99 - 7.95 (m, 3H), 7.88 (brs, 16H), 2.87 (t, J = 8.7 Hz, 1 H), 2.51-2.31 (m, 3H), 2.34 (s, 12H), 2.13-1.91 (m, 2 H) 1.28 (s, 3H), 0.96 (s, 3H).
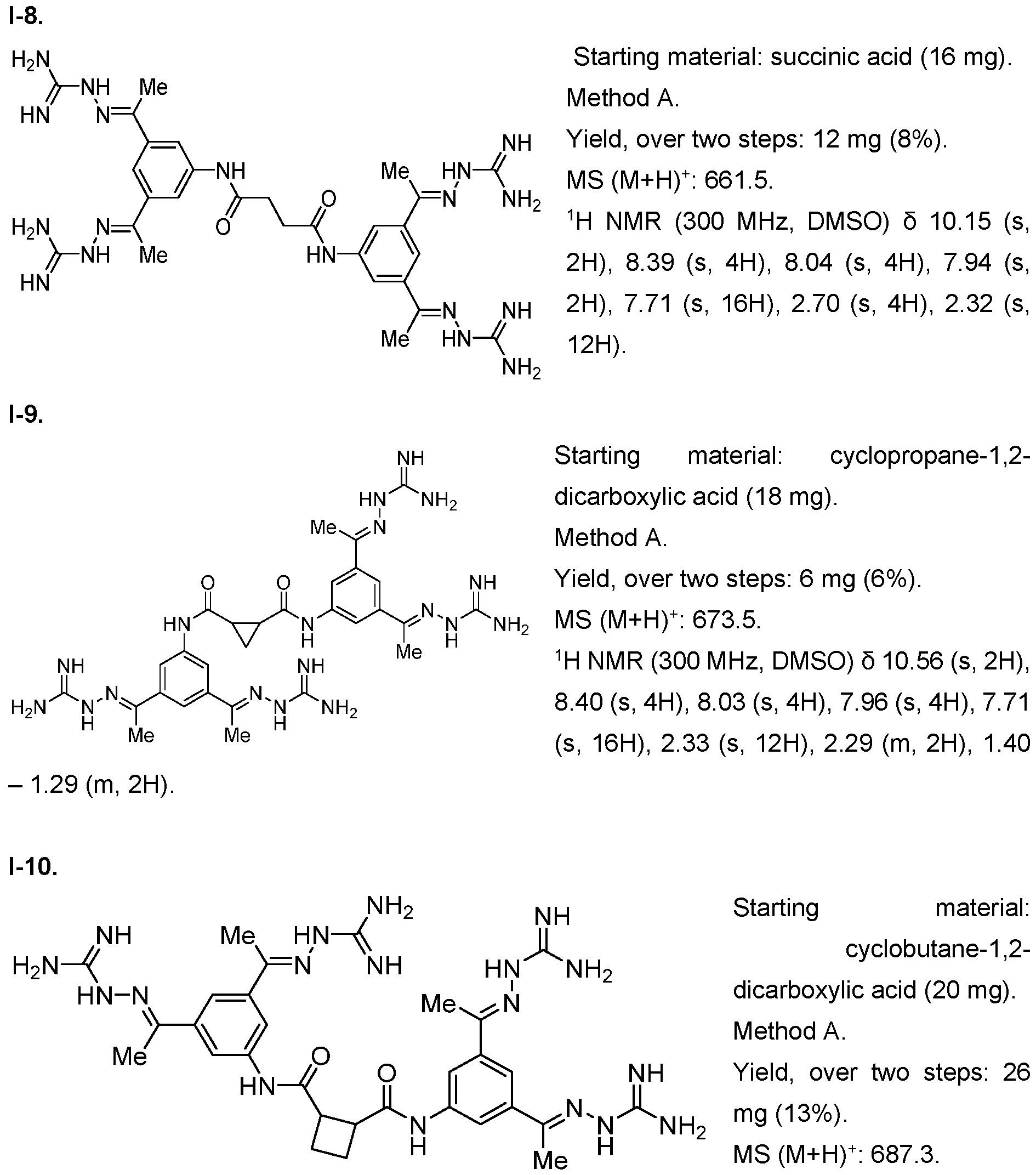
1H NMR (300 MHz, DMSO)
5 10.04 (s, 2H), 8.37 (s, 4H), 8.07 (s, 4H), 7.94 (s, 2H), 7.54 (brs, 16H), 2.32 (s, 14H), 2.15 (s, 4H).
1-11.
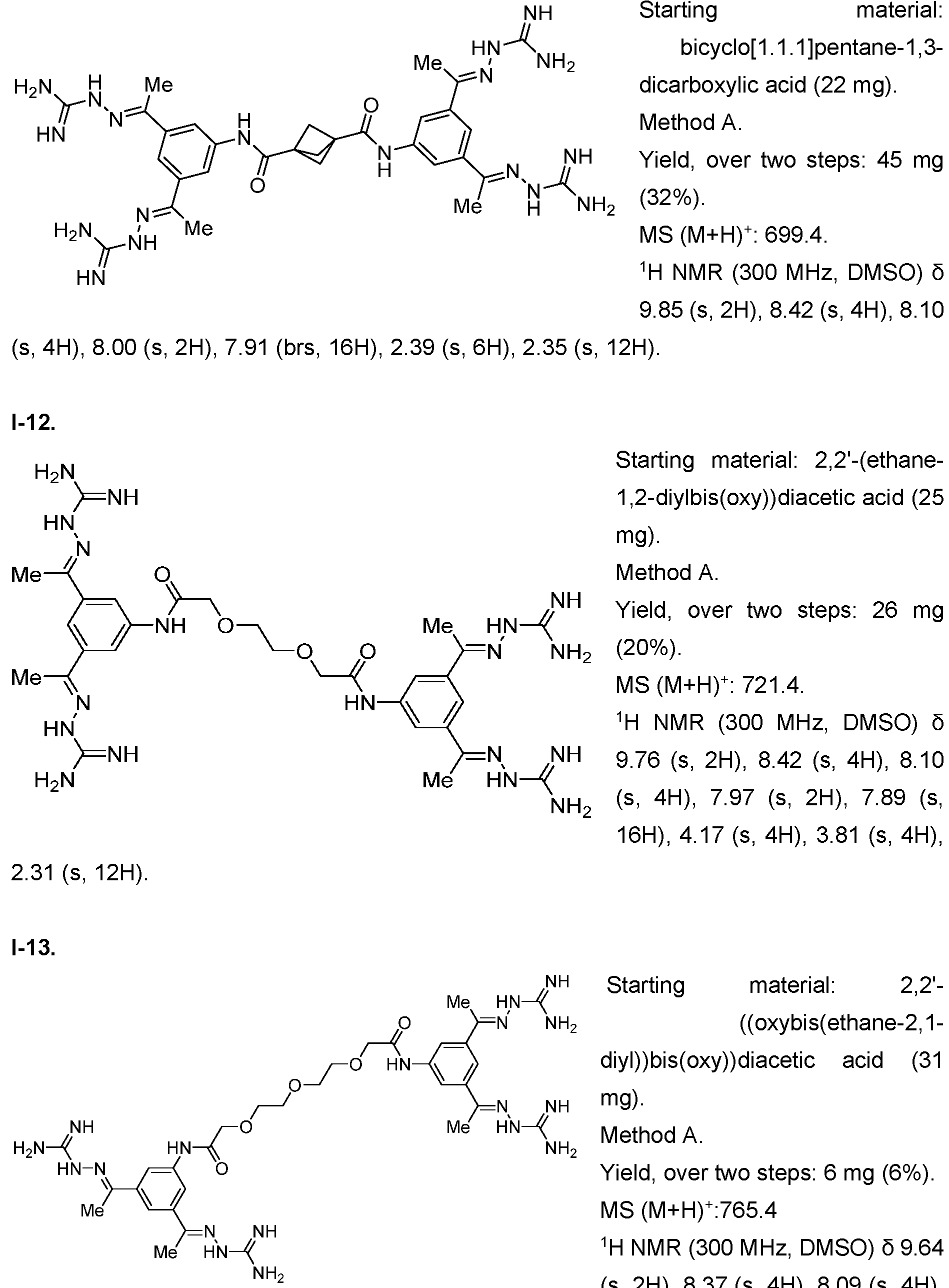 (s, 2H), 8.37 (s, 4H), 8.09 (s, 4H),
(s, 2H), 8.37 (s, 4H), 8.09 (s, 4H),
7.93 (s, 2H), 7.44 (brs, 16H), 4.10 (s, 4H), 3.70 (m, 8H), 2.31 (s, 12H).
1-14.
Startin material: 3 6 9 12-

10.03 (s, 2H), 8.37 (s, 4H),
8.03 (s, 4H), 7.92 (s, 2H), 7.42 (s, 16H), 2.43 (t, J = 7.3 Hz, 4H), 2.31 (s, 12H), 2.03 - 1.90 (m, 2H).
1-17.

To be synthesized. 1-19.

Step 1-2:
Isophthalic acid (23 mg, 0.14 mmol) and intermediate 56 (73 mg) were dissolved in 1 ml of MeCN. 0.06 mL of /V-methylimidazole and TCFH (73 mg, 0.28 mmol) were added sequentially. The reaction was stirred overnight. After the completion of the reaction 1 ml of 4M HCI in dioxane was added and stirred for 3 h at rt. Afterwards, the solvents were evaporated, and the residue was purified by silica gel column chromatography eluting with
Cyclohexane: Ethylacetate 100:0% to Cyclohexane: Ethylacetate 0:100% and used in the next step.
MS (M-H)-: 485.3.
1H NMR (300 MHz, DMSO) 5 11.11 (s, 1 H), 8.61 (s, 1 H), 8.60 (s, 4H), 8.21 (d, J = 9.6 Hz, 2H), 7.73 (t, J = 7.8 Hz, 1 H), 2.68 (s, 12H).
Step 3.
The compounds was synthesized according to general procedure 2 using the product from the previous step.
Yield, over two steps: 2 mg (1%).
1H NMR (300 MHz, DMSO) 5 10.86 (s, 2H), 8.72 (s, 1 H), 8.54 (s, 4H), 8.35 (s, 4H), 8.27 (d, J = 7.8 Hz, 2H), 7.76 (t, J = 7.8 Hz, 1 H), 7.28 (s, 16H), 2.43 (s, 12H).
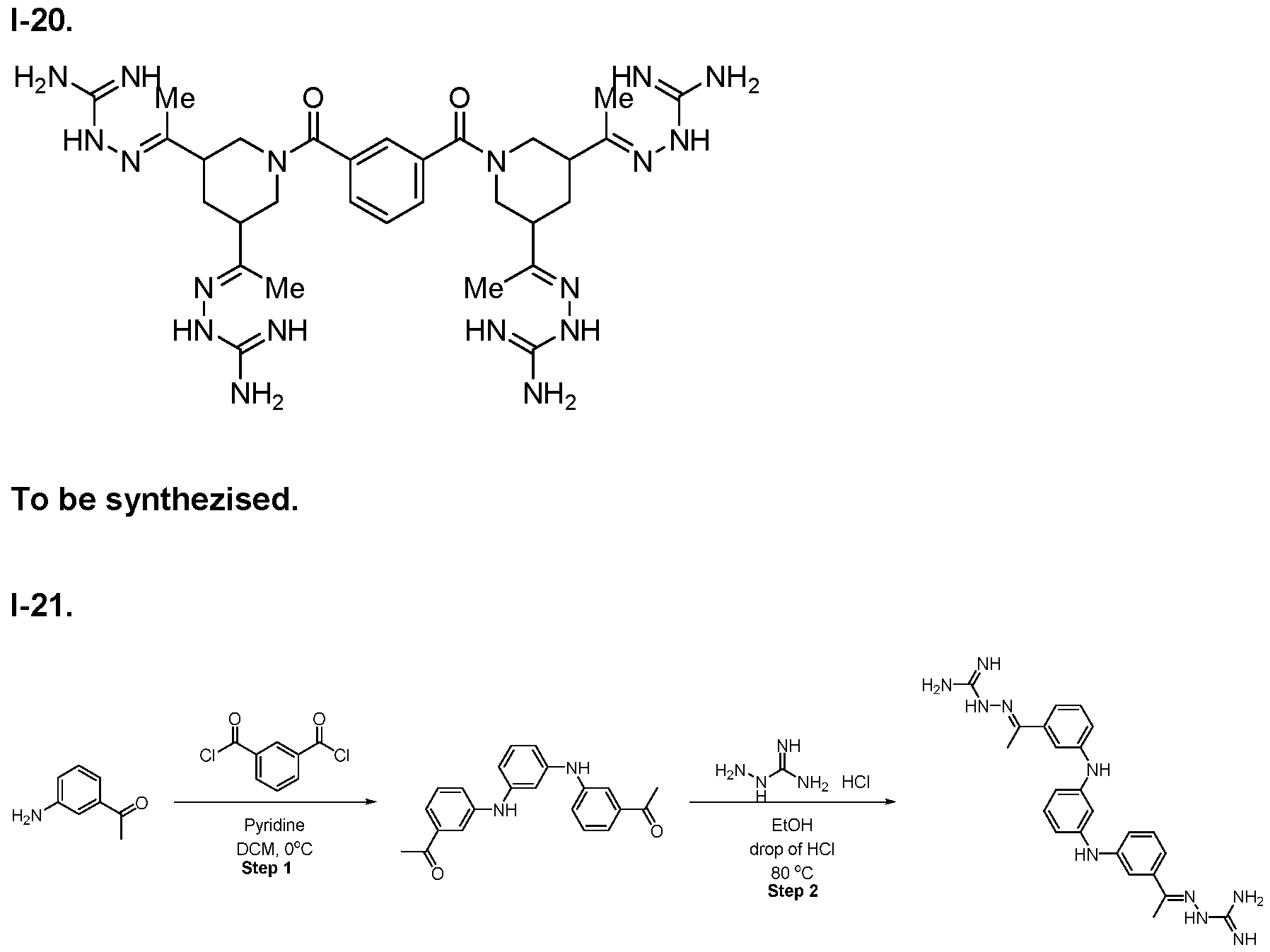
Step 1 :
3-aminoacethophenone (38 mg, 0.28 mmol) was dissolved in 2 ml of dry CH2CI2, 0.06 ml of pyridine were added, and the reaction mixture cooled down to 0 °C using an ice cooling bath. After that isophthaloyl chloride (28 mg, 0.14 mmol, dissolved in 1 ml of dry DCM) was added slowly. Then, the reaction mixture was warmed up to rt and stirred overnight. The solvent was evaporated, and the residue was used in the next reaction without additional purification.
Step 2:
The compound was synthesized according to general procedure 2 using the product from the previous step.
Yield, over two steps: 40 mg (48%).
MS (M+H)+: 512.9.
1H NMR (300 MHz, DMSO) 5 10.52 (s, 2H), 8.61 (s, 1 H), 8.41 (s, 2H), 8.23 - 8.14 (m, 4H), 7.92 (d, J = 7.5 Hz, 2H), 7.74 (m, 10H), 7.40 (t, J = 8.0 Hz, 2H), 2.33 (s, 6H).
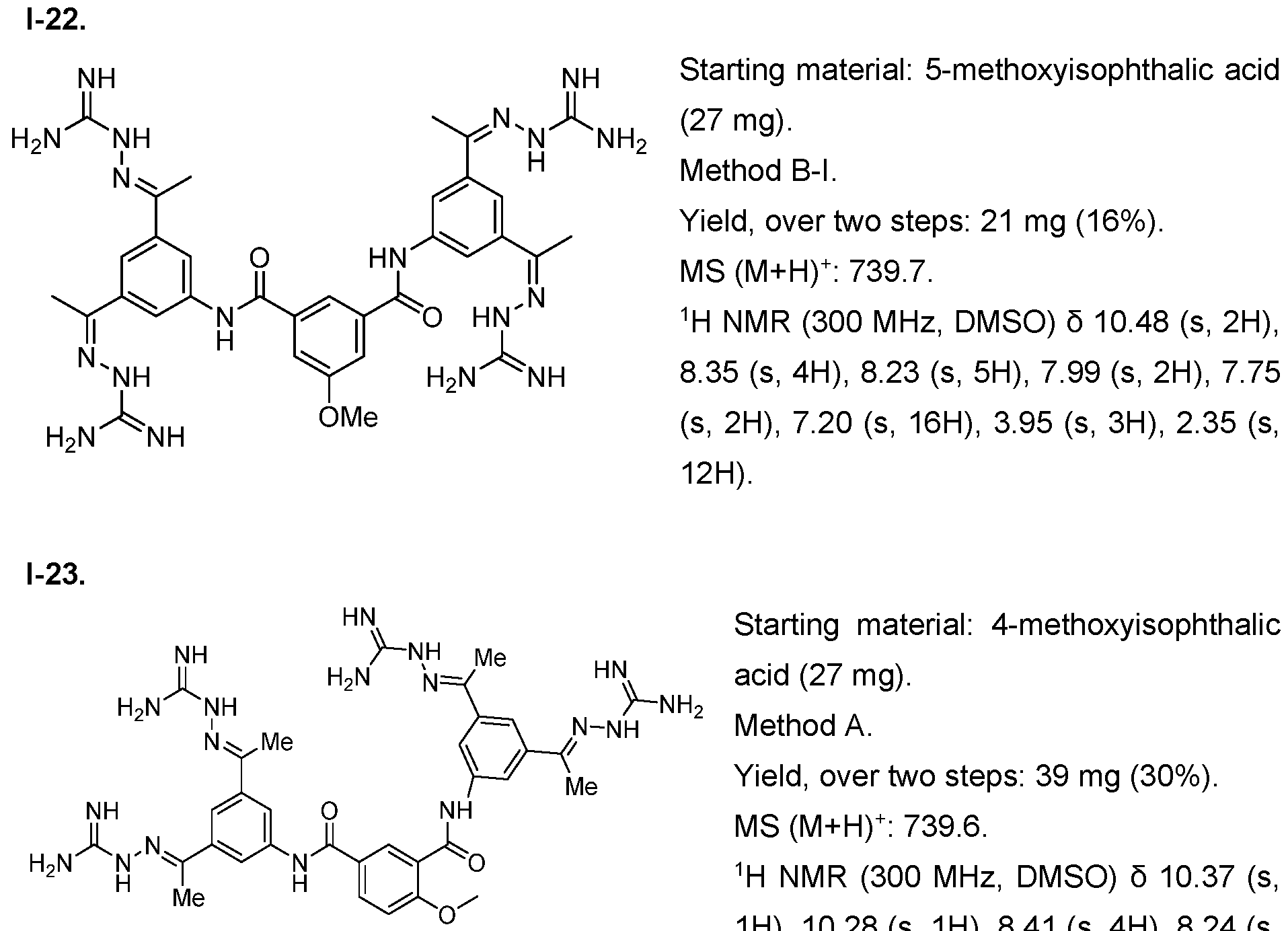 1 H), 10.28 (s, 1 H), 8.41 (s, 4H), 8.24 (s,
1 H), 10.28 (s, 1 H), 8.41 (s, 4H), 8.24 (s,
5H), 8.03 (s, 2H), 7.82 (brs, 17H), 7.37 (d, J = 9.1 Hz, 1 H), 4.02 (s, 3H), 2.38 (s, 12H).
1-24.
Starting material: 2-
 , . , , . , ,
, . , , . , ,
4.31 (m, 2H), 3.83 (m, 2H), 3.68 - 3.60 (m, 2H), 3.53 - 3.43 (m, 2H), 3.27 (s, 3H), 2.35 (s, 12H).
1-27.
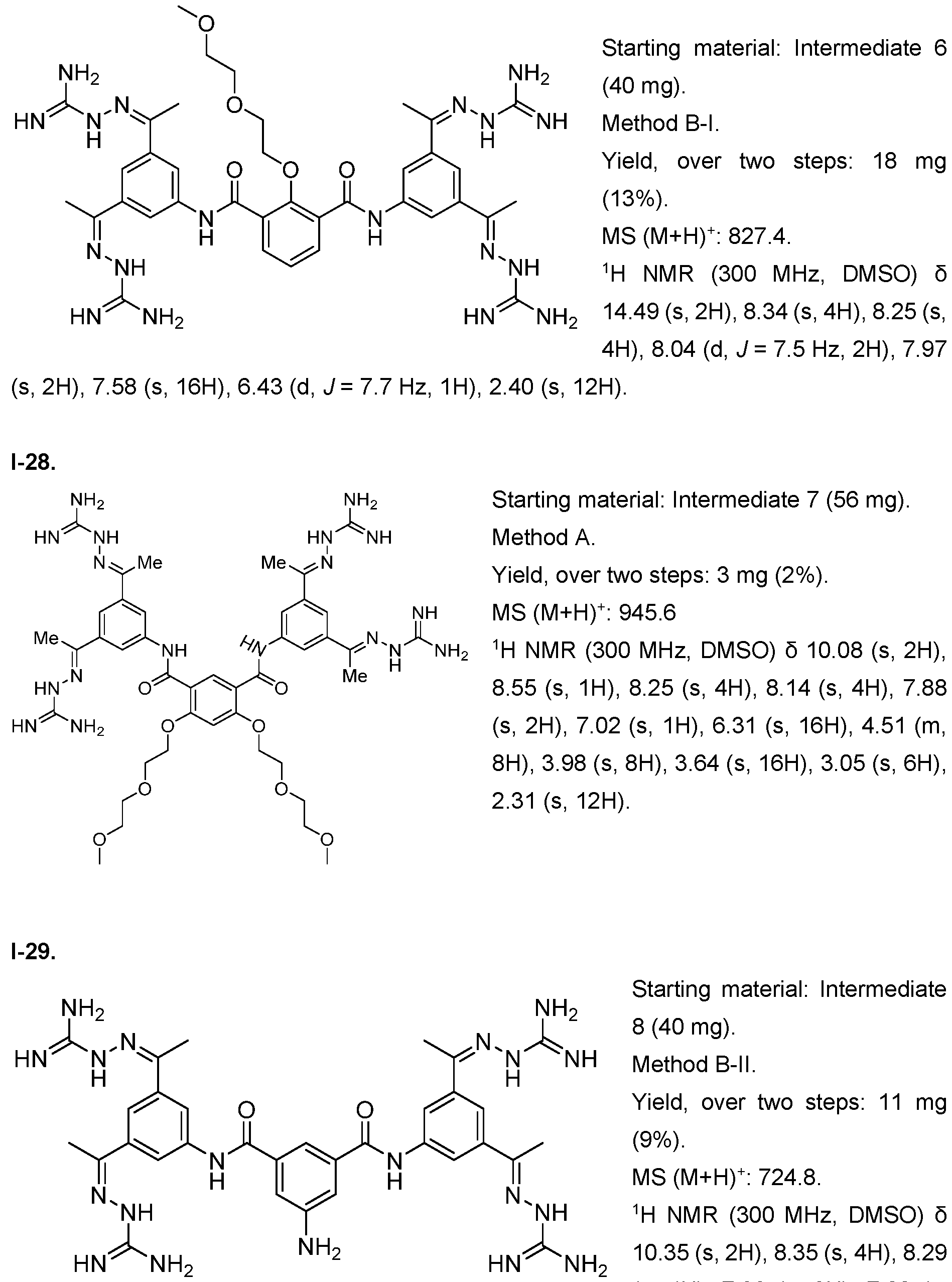 (s, 4H), 7.99 (s, 2H), 7.80 (s,
(s, 4H), 7.99 (s, 2H), 7.80 (s,
1 H), 7.39 (s, 16H), 7.32 (s, 2H), 5.65 (s, 2H), 2.36 (s, 12H).
1-30.
 7.78 (s, 2H), 7.30 (s, 16H), 2.36 (s, 12H).
7.78 (s, 2H), 7.30 (s, 16H), 2.36 (s, 12H).
 5 10.59 (s, 1H), 10.46 (s,
5 10.59 (s, 1H), 10.46 (s,
1H), 8.40 (d, J= 4.5 Hz, 2H), 8.32 (s, 4H), 8.19 (m, 5H), 7.98 (s, 2H), .7.57 (t, J= 9.4 Hz, 1H), 7.06 (m, 16H), 2.34 (s, 12H).
1-36.
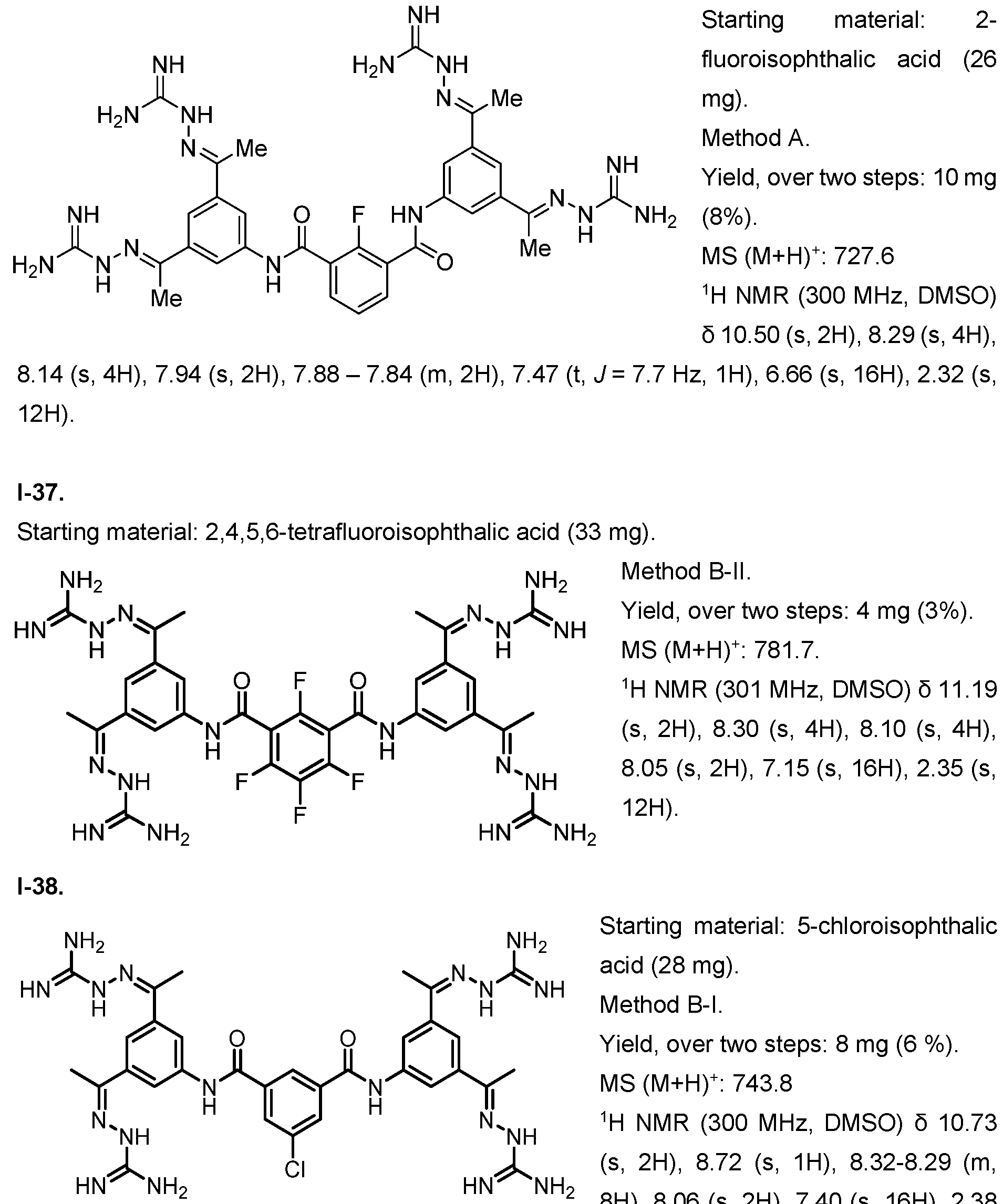 8H), 8.06 (s, 2H), 7.40 (s, 16H), 2.38
8H), 8.06 (s, 2H), 7.40 (s, 16H), 2.38
(s, 12H).
1-39.
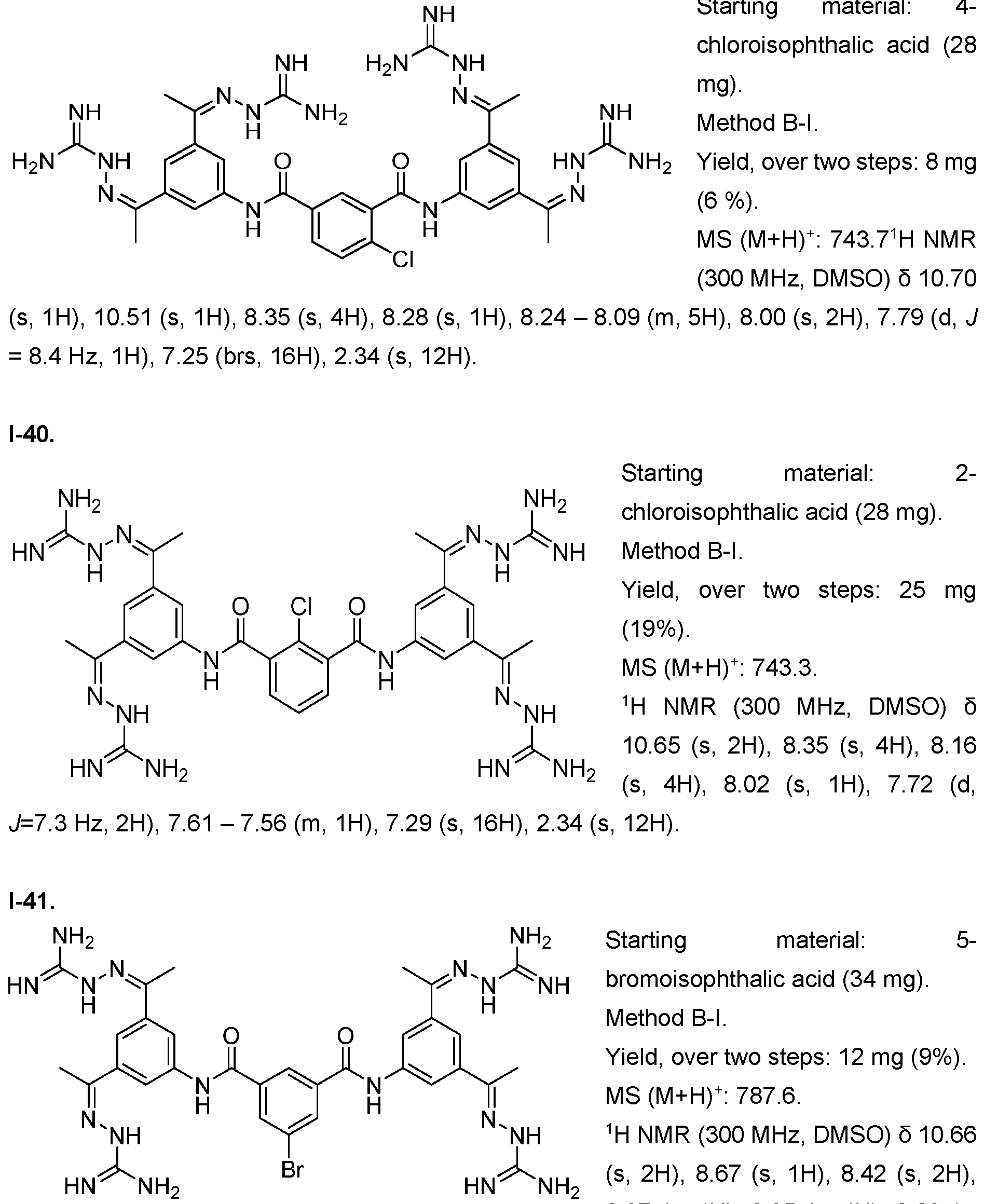 8.37 (s, 4H), 8.25 (s, 4H), 8.03 (s,
8.37 (s, 4H), 8.25 (s, 4H), 8.03 (s,
2H), 7.49 (s, 16H), 2.37 (s, 12H).
1-42.
Starting material: 4-bromoisophthalic
 (s, 4H), 7.98 (s, 2H), 6.86 (s, 16H), 2.34 (s,
(s, 4H), 7.98 (s, 2H), 6.86 (s, 16H), 2.34 (s,
12H).
1-45
 1H NMR (300 MHz, DMSO) 6 10.45 (s, 2H), 8.37 (s, 4H), 8.20 (s, 4H), 8.01 (s, 2H), 7.65 -
1H NMR (300 MHz, DMSO) 6 10.45 (s, 2H), 8.37 (s, 4H), 8.20 (s, 4H), 8.01 (s, 2H), 7.65 -
7.42 (m, 19H), 2.46 (s, 3H), 2.36 (s, 12H).
I-49.
 7.54 (s, 16H), 2.79 (t, J = 7.5
7.54 (s, 16H), 2.79 (t, J = 7.5
Hz, 2H), 2.37 (s, 12H), 1.71 (s, 2H), 1.35 (s, 4H), 1.01 - 0.80 (m, 3H).
1-52.
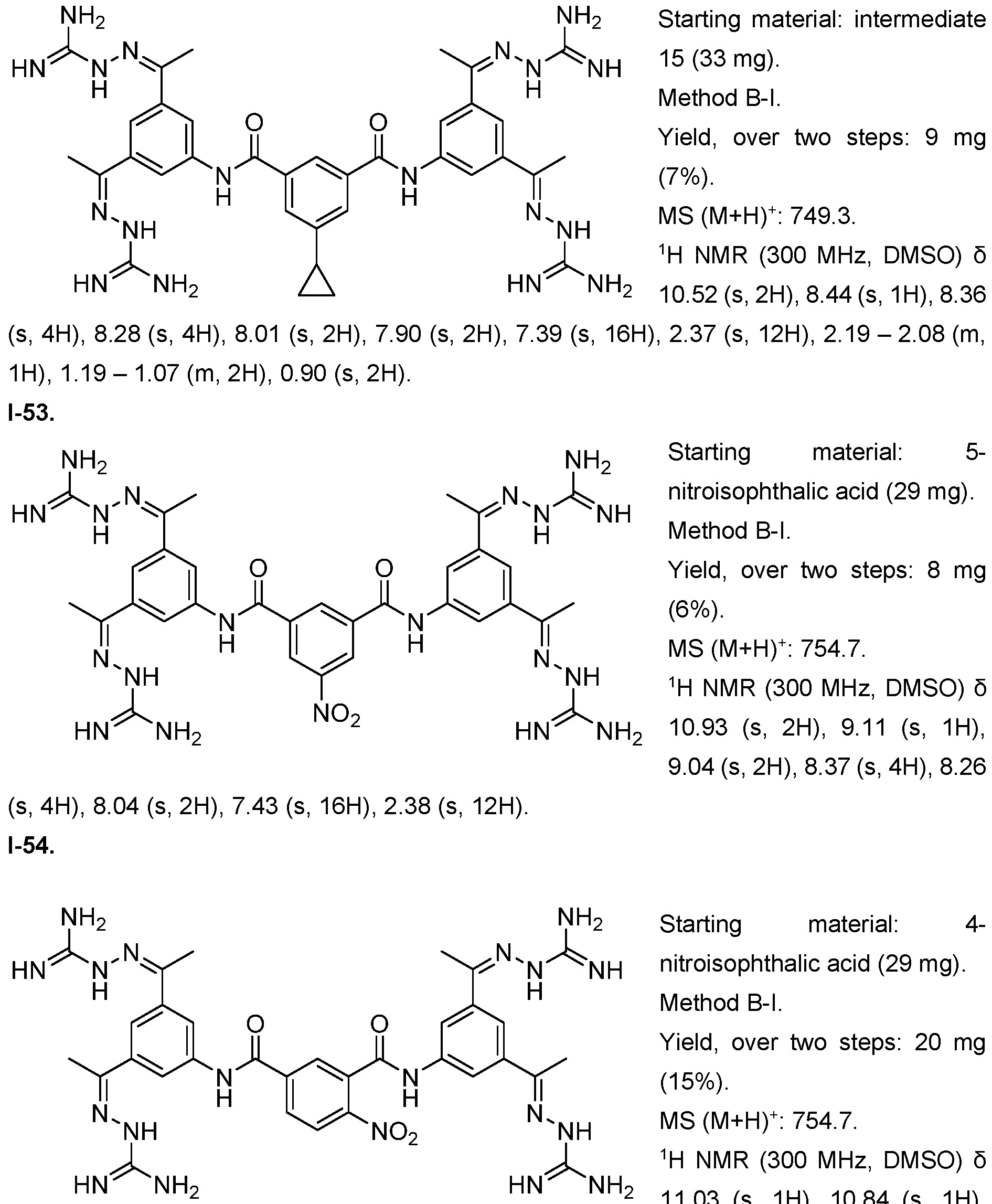 11.03 (s, 1 H), 10.84 (s, 1 H),
11.03 (s, 1 H), 10.84 (s, 1 H),
8.46 (s, 1 H), 8.36 (s, 4H), 8.37 - 8.29 (m, 2H), 8.26 (s, 2H), 8.16 (s, 2H), 8.11 - 8.02 (m, 2H),
7.50 (s, 16H), 2.37 (s, 6H), 2.36 (s, 6H).
1-55.
Starting material: 2-nitroisophthalic acid (29 mg).
 , , . , , . , ,
, , . , , . , ,
7.99 (s, 2H), 7.01 (s, 16H), 2.35 (s, 12H).
1-58.
 2H), 8.03 (s, 2H), 7.49 (s, 16H), 2.37 (s,
2H), 8.03 (s, 2H), 7.49 (s, 16H), 2.37 (s,
9H).
1-61.
 , . , , . , . , ,
, . , , . , . , ,
8.37 (s, 4H), 8.30 (s, 4H), 8.04 (s, 2H), 7.67 (s, 2H), 7.49 (s, 16H), 2.38 (s, 12H).
1-64.
Starting material: intermediate 22 (30
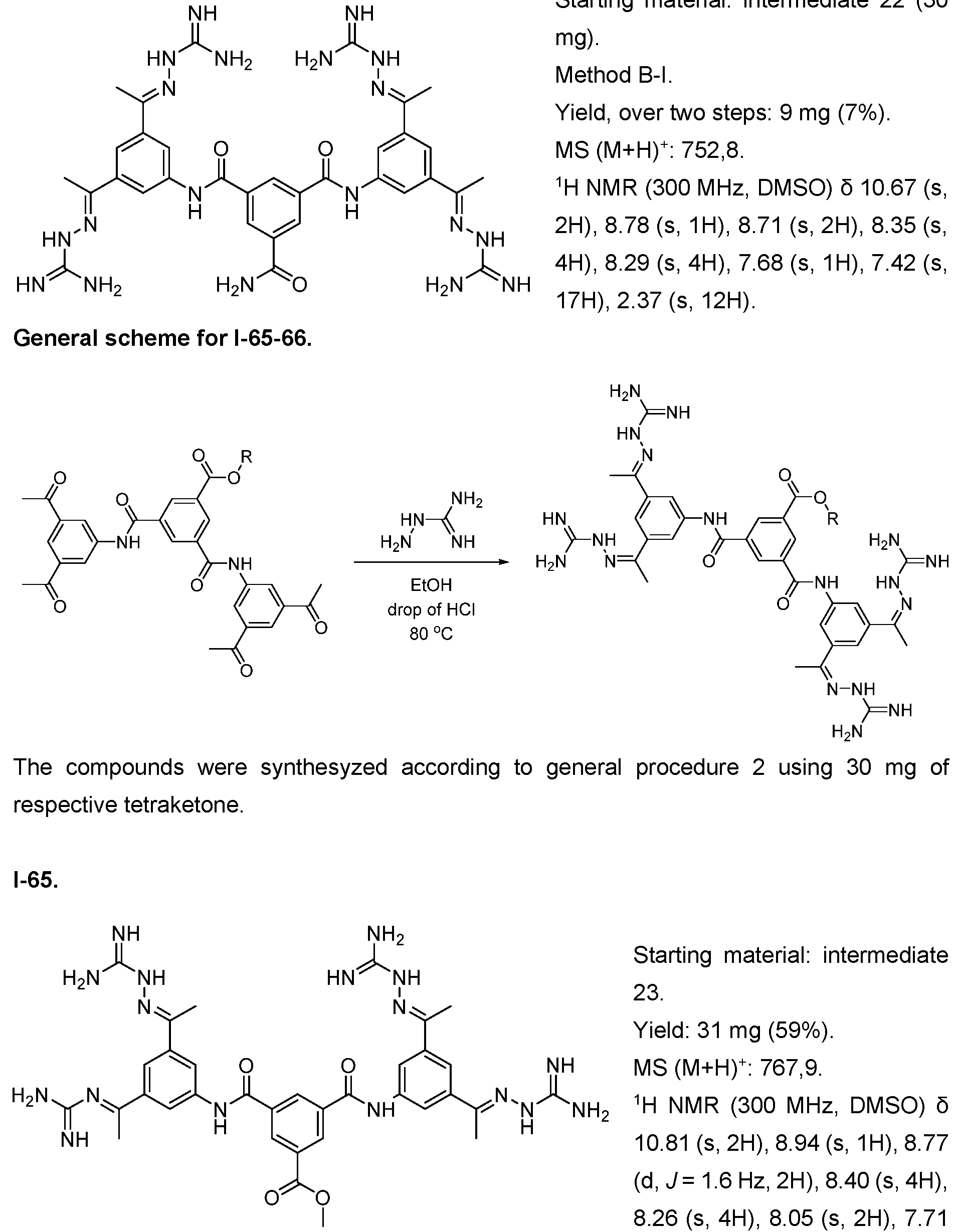
(s, 16H), 3.98 (s, 3H), 2.38 (s, 12H).
1-66.
 4H), 8.09 (s, 2H), 7.71 (s, 16H),
4H), 8.09 (s, 2H), 7.71 (s, 16H),
2.44 (s, 12H).
General scheme for synthesis of I-67-68.

Step 1 :
Intermediate 24 (30 mg, 0.056 mmol) and amine hydrochloride 1 (0.056 mmol) were dissolved in 0.25 ml of MeCN. 0.03 mL N-methylimidazole and TCFH (30 mg, 0.10 mmol) were added sequentially. The reaction was stirred overnight. After the completion of the reaction, the solvents were evaporated, and the residue was purified by silica gel column chromatography eluting with cyclohexane: ethylacetate 100:0% to cyclohexane: ethylacetate 0:100% to obtain the desired product as white solid.
Step 2:
The compounds were synthesized according to general procedure 2 using the product from the previous step.
1-67.
Starting material: dimethylamine

(s, 2H), 8.02 (s, 2H), 7.47 (s, 16H),
2.37 (s, 12H).
1-70.
 12H).
1-73.
12H).
1-73.
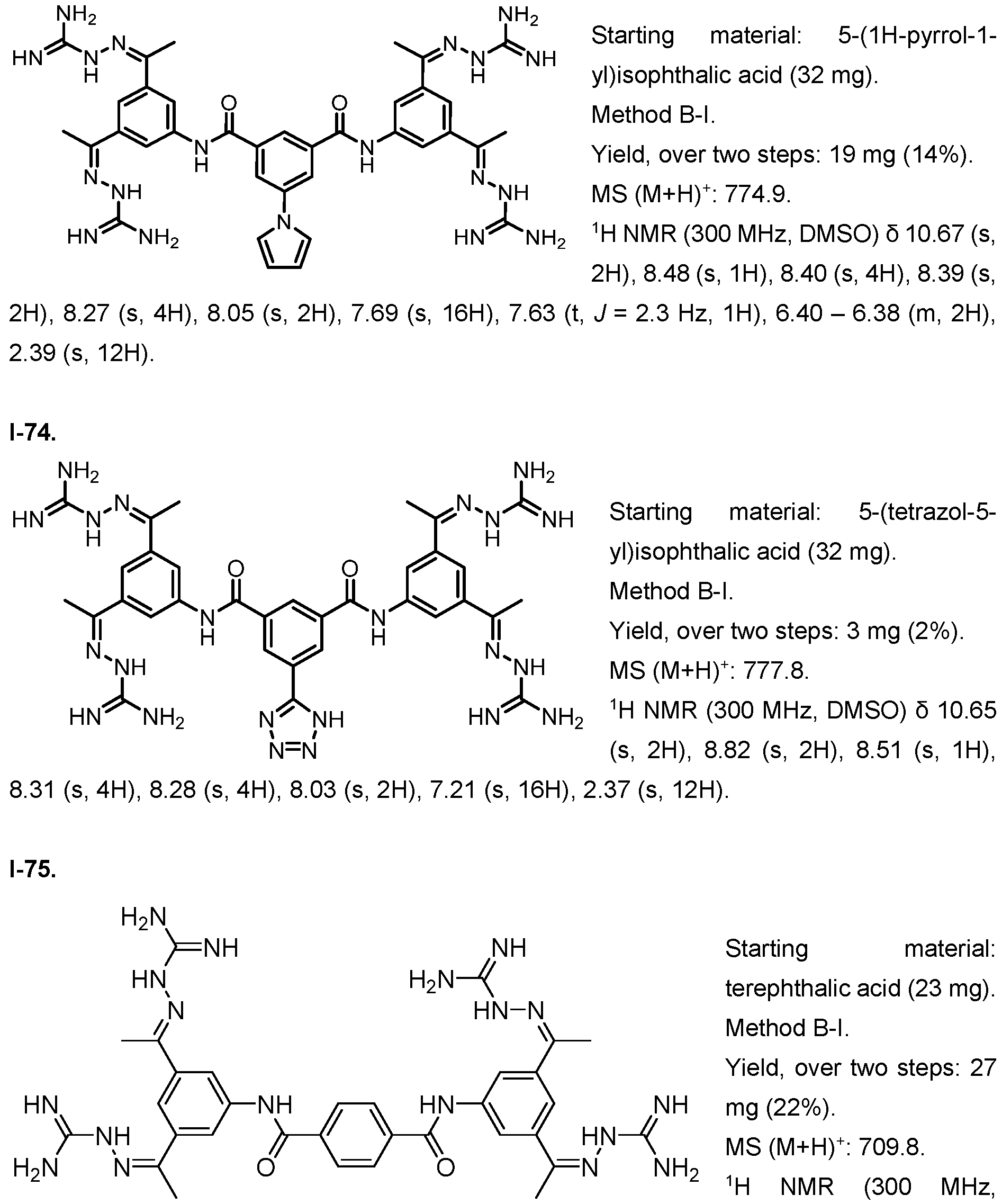
DMSO) 5 10.49 (s, 2H), 8.38 (s, 4H), 8.24 (s, 4H), 8.16 (s, 4H), 8.03 (s, 2H), 7.52 (s, 16H),
2.37 (s, 12H).
1-76.
Starting material: 2,2'-(1 ,3-phenylene)diacetic acid (27 mg).

1H NMR (300 MHz, DMSO) 5
10.49 (s, 2H), 8.41 (s, 2H), 8.33 (s, 4H), 8.25 (s, 4H), 8.07-8.01 (m, 6H), 7.71 (t, J = 7.8 Hz, 2H), 7.32 (s, 16H), 2.36 (s, 12H).
1-79.

MS (M+H)+: 799.8.
1H NMR (300 MHz, DMSO) 5 10.18 (s, 2H), 8.30 (s, 4H), 8.12 (d, J = 1.6 Hz, 4H), 7.92 (d, J
= 3.3 Hz, 2H), 7.86 (d, J = 8.2 Hz, 4H), 7.67 (s, 16H), 7.35 (d, J = 8.4 Hz, 4H), 2.27 (s, 12H).
1-82.
Starting material: 4,4'-oxydibenzoic acid (36 mg).
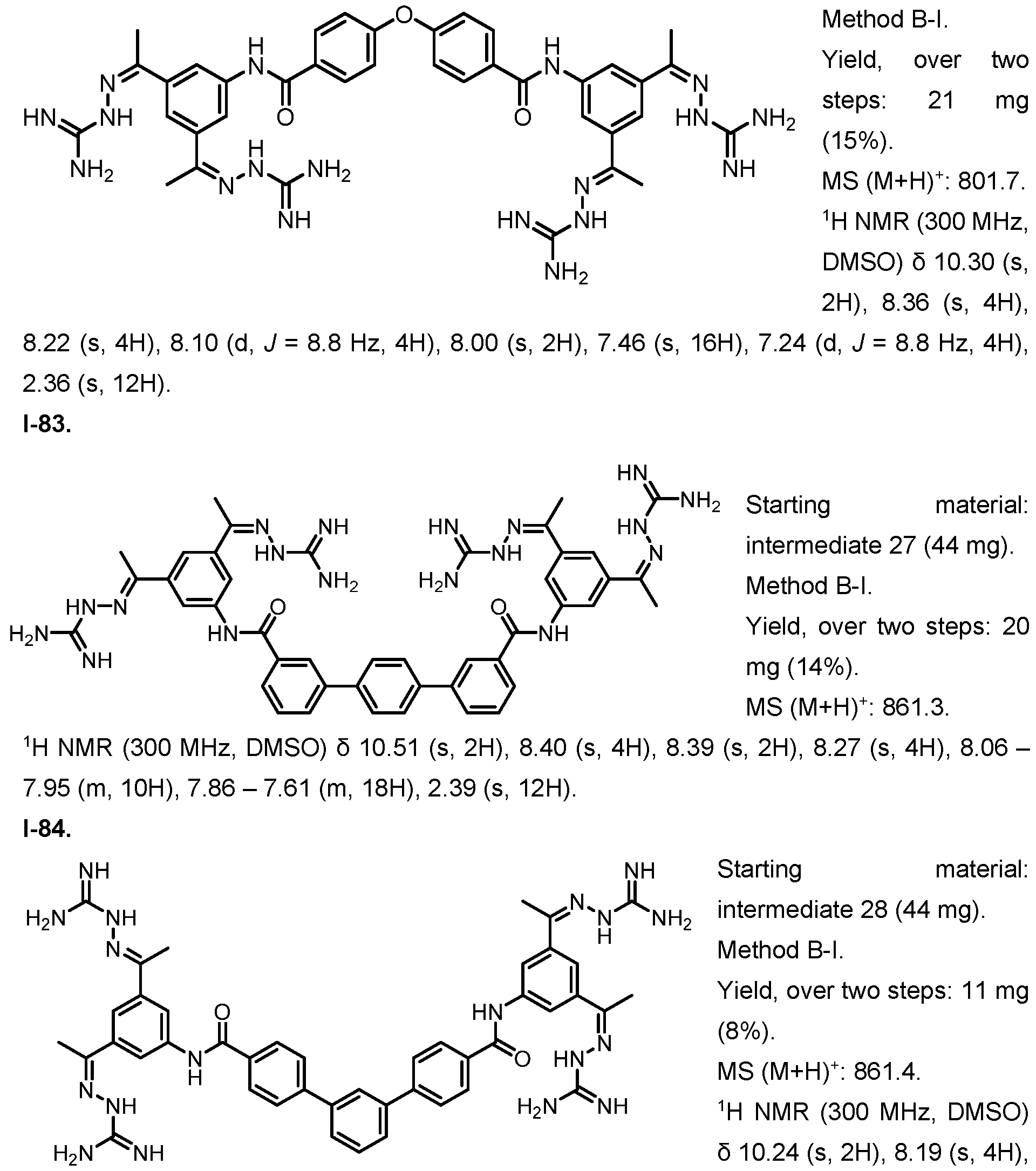 8.10 (s, 4H), 7.98 (m, 8H),
8.10 (s, 4H), 7.98 (m, 8H),
7.84 (m, 8H), 7.28 (s, 16H), 2.21 (s, 12H).
1-85.
Starting material:
 , , . , , . ,
, , . , , . ,
6H), 0.95-0.80 (m, 3H).
1-87.
Starting material: intermediate

(m, 18H), 3.40 (s, 4H), 2.35 (s, 12H),
2.04 (s, 4H).
1-90.
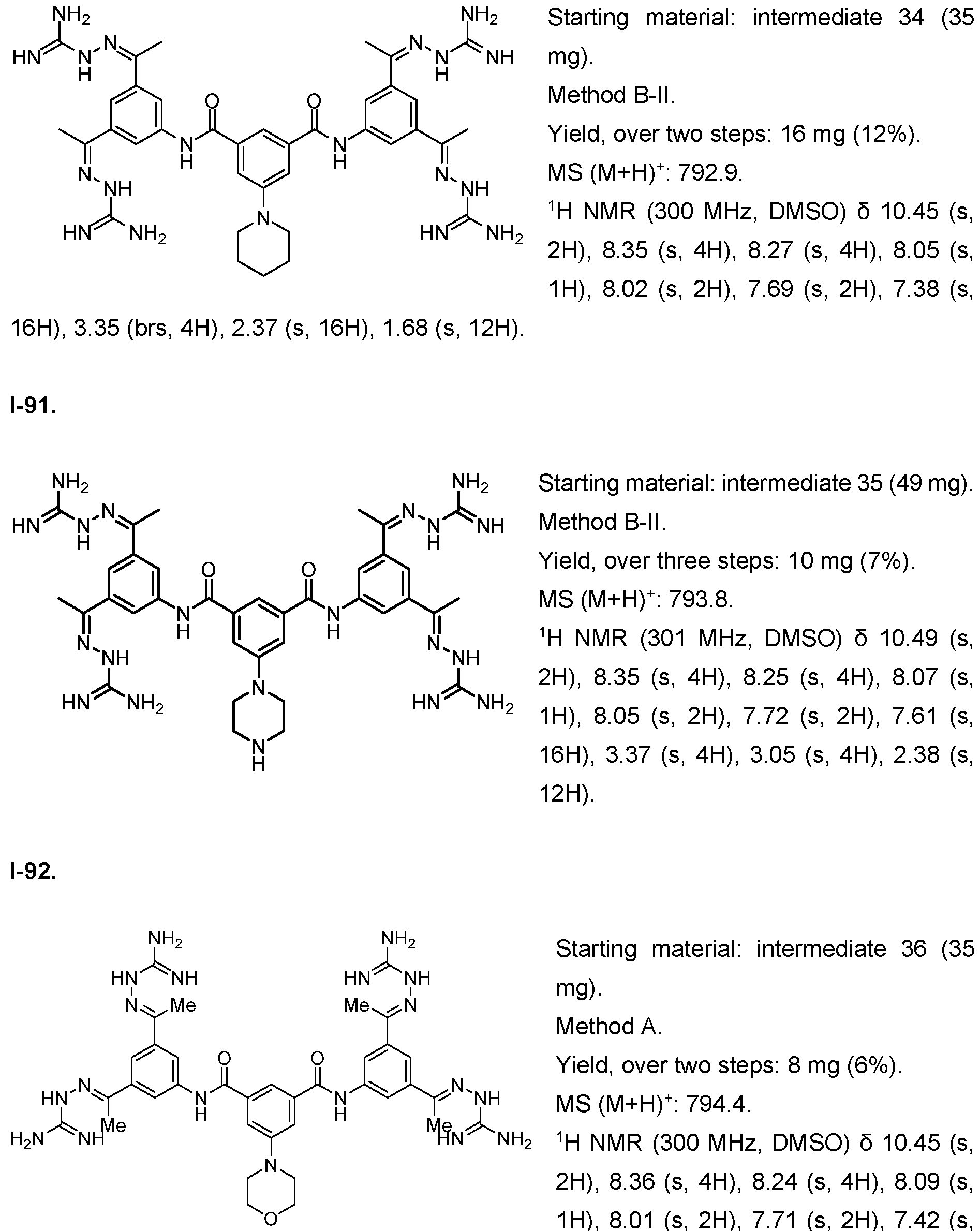 , . , , . , , . ,
, . , , . , , . ,
16H), 3.82 (t, J = 4.9 Hz, 4H), 3.33 (d, J = 4.7 Hz, 4H), 2.36 (s, 12H).
1-93.
 , , . , , . , ,
, , . , , . , ,
7.48 (s, 16H), 7.29 (s, 2H), 3.87 - 3.64 (m, 4H), 2.84 - 2.73 (m, 2H), 2.37 (s, 12H).
1-96.

2H), 8.27 (d, J = 1.6 Hz, 4H), 8.04 (s,
2H), 7.74 (s, 16H), 6.96 (dd, J= 17.6, 11.2 Hz, 1H), 6.13 (d, J= 17.6 Hz, 1H), 5.51 (d, J =
11.4 Hz, 1H), 2.38 (s, 12H).
1-99.
Starting material: intermediate 43
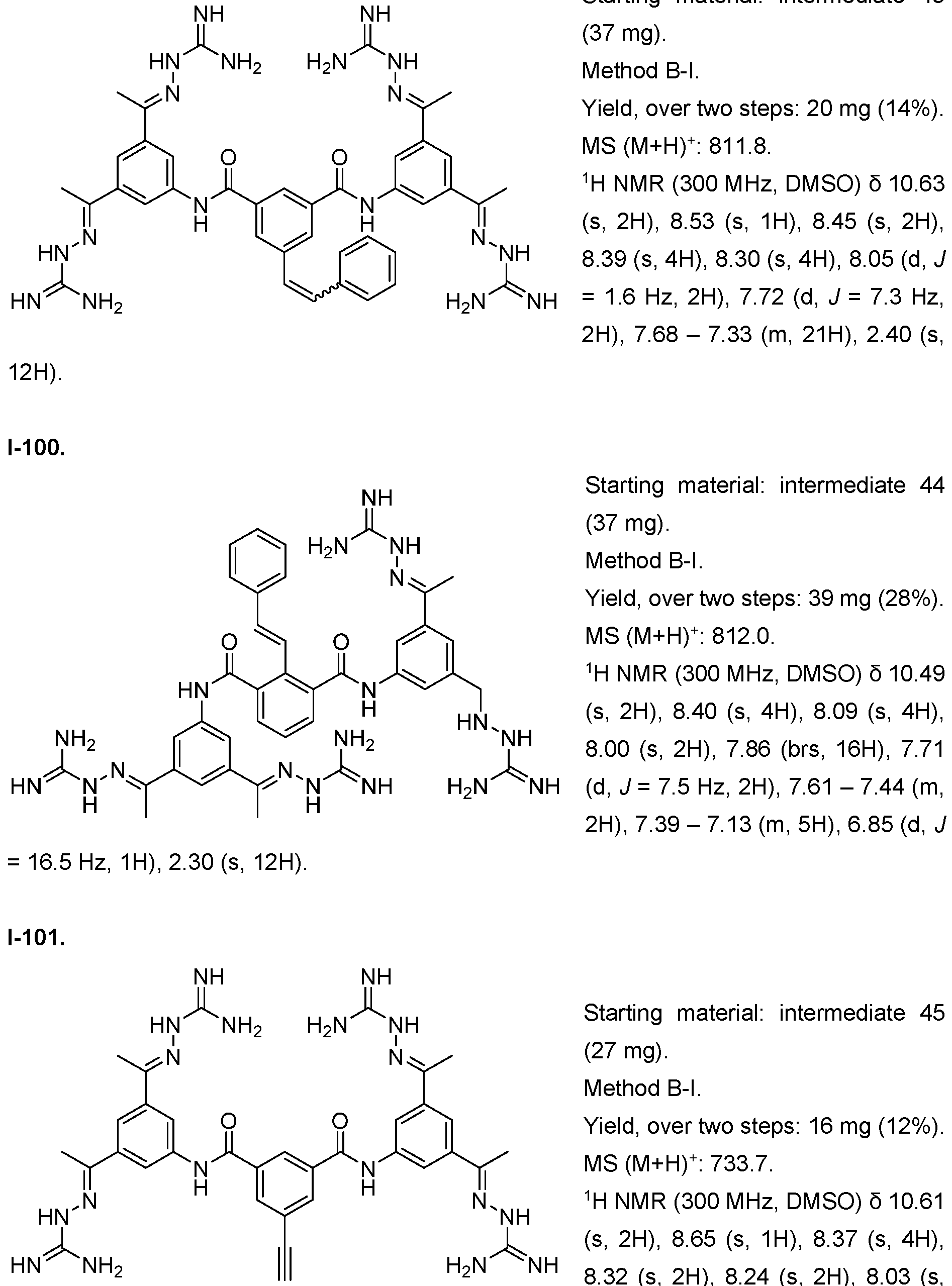 . , , . , , . ,
. , , . , , . ,
2H), 7.51 (s, 16H), 4.49 (s, 1H), 2.37 (s, 12H).
-102.
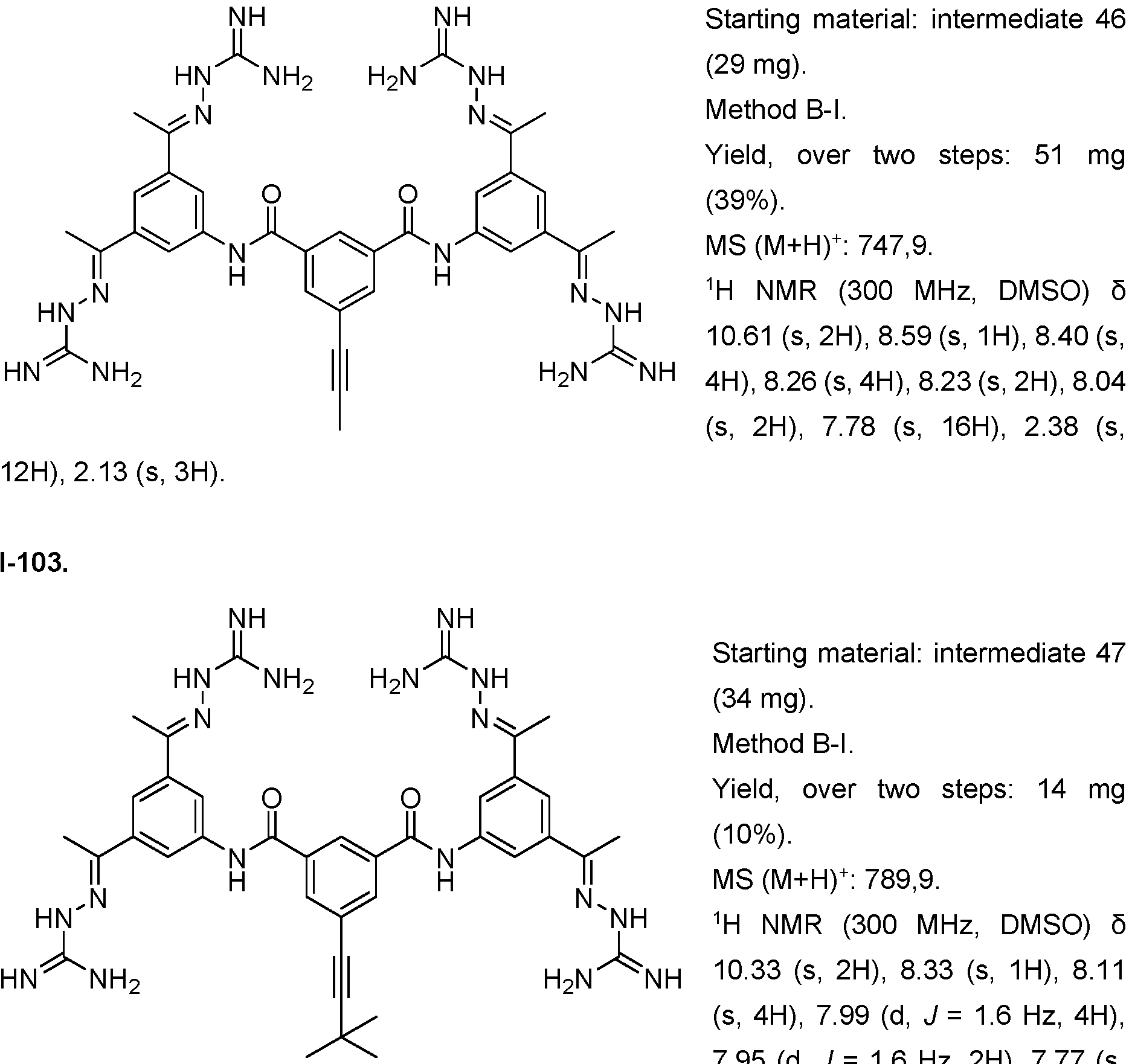 7.95 (d, J = 1.6 Hz, 2H), 7.77 (s, H), 7.10 (s, 16H), 2.12 (s, 12H), 1.12 (s, 9H).
7.95 (d, J = 1.6 Hz, 2H), 7.77 (s, H), 7.10 (s, 16H), 2.12 (s, 12H), 1.12 (s, 9H).
-104.
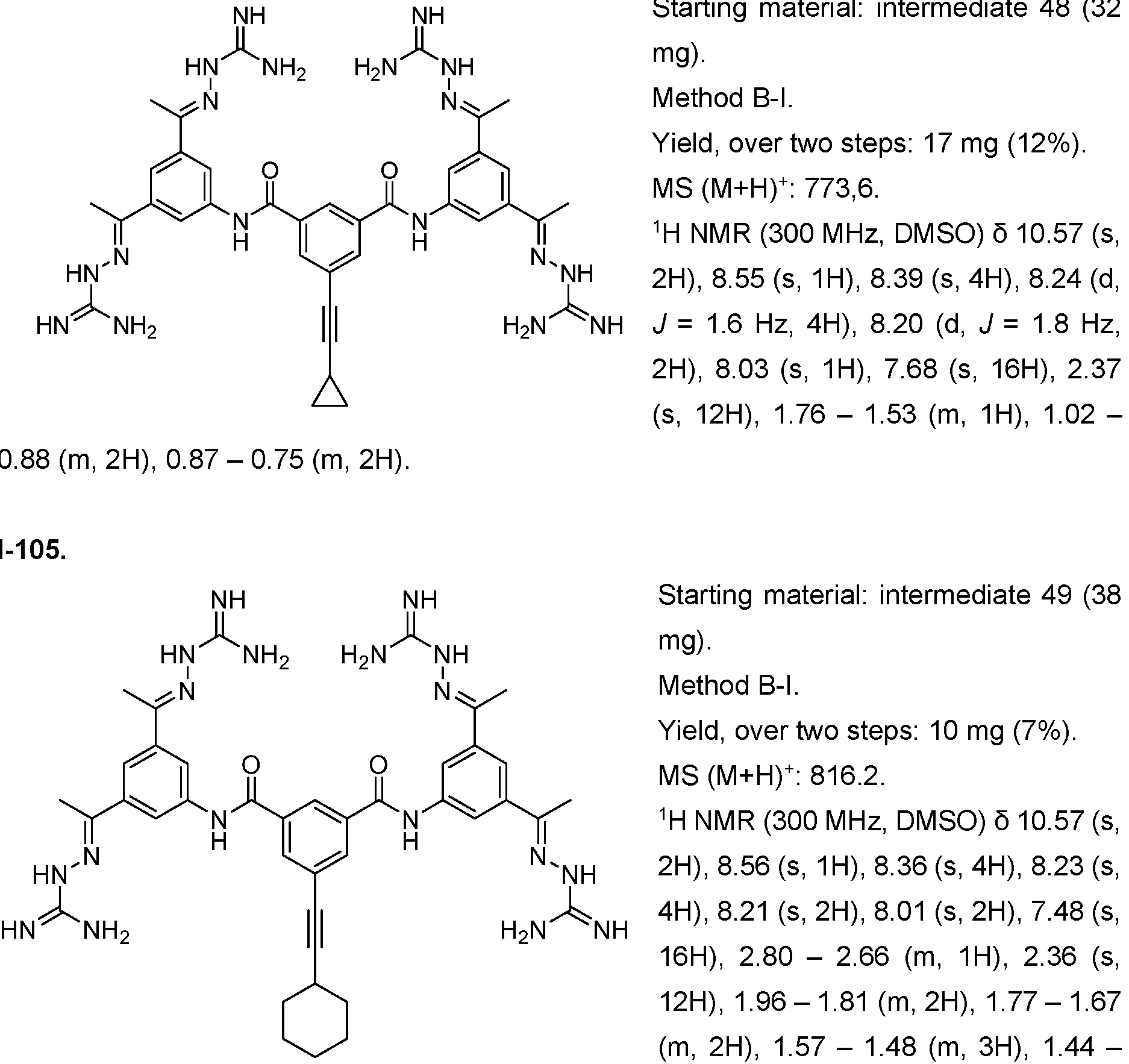 .35 (m, 3H).
.35 (m, 3H).
-106.
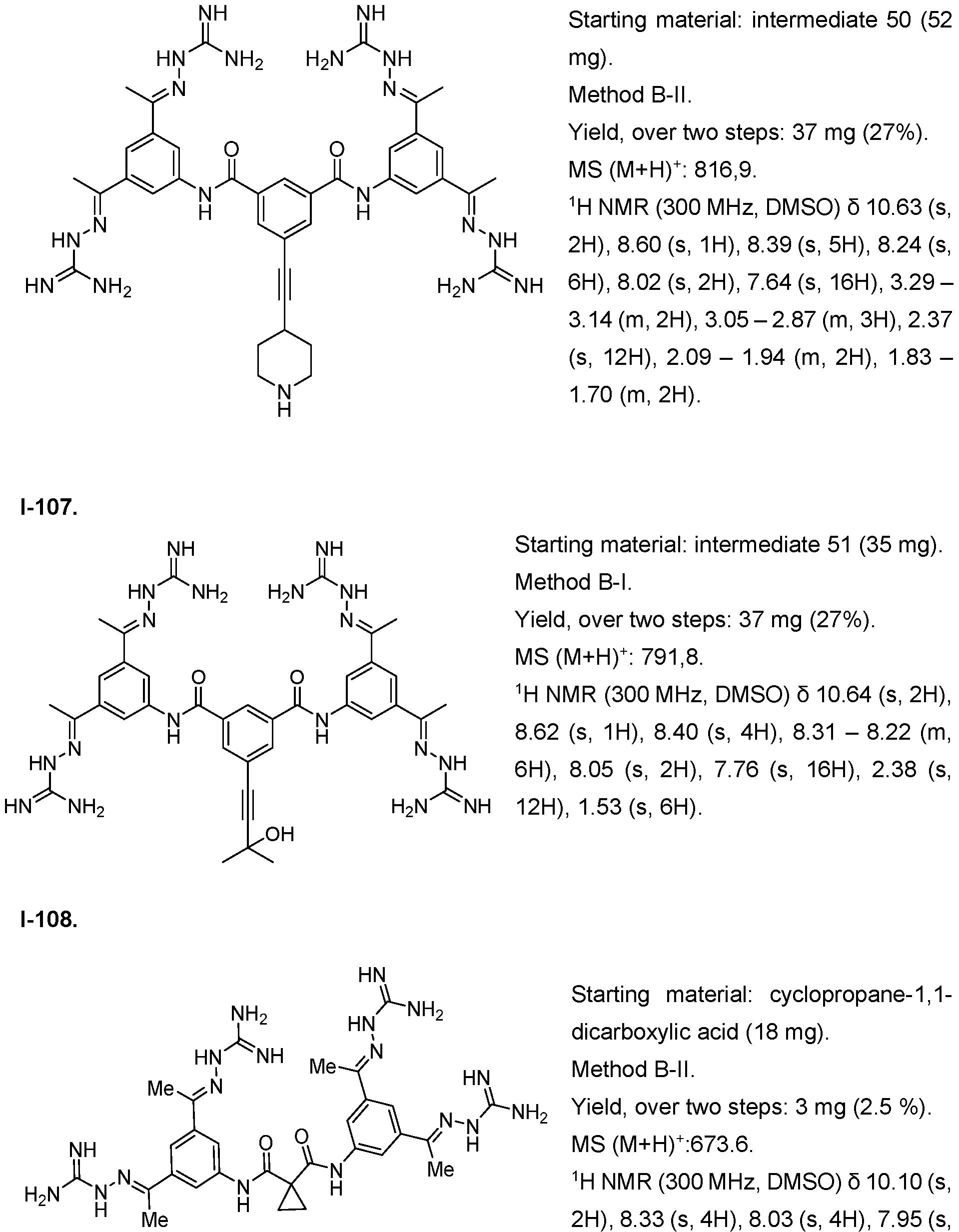 , , , , , , H), 7.35 (s, 16H), 2.30 (s, 12H), 1.53 (s, 4H).
-109.
, , , , , , H), 7.35 (s, 16H), 2.30 (s, 12H), 1.53 (s, 4H).
-109.
 1-112.
1-112.
Starting material: 2,2'-((tert-butoxycarbonyl)azanediyl)diacetic acid (32 mg).
Method B-ll.
Yield over three ste s: 3 m (2 5
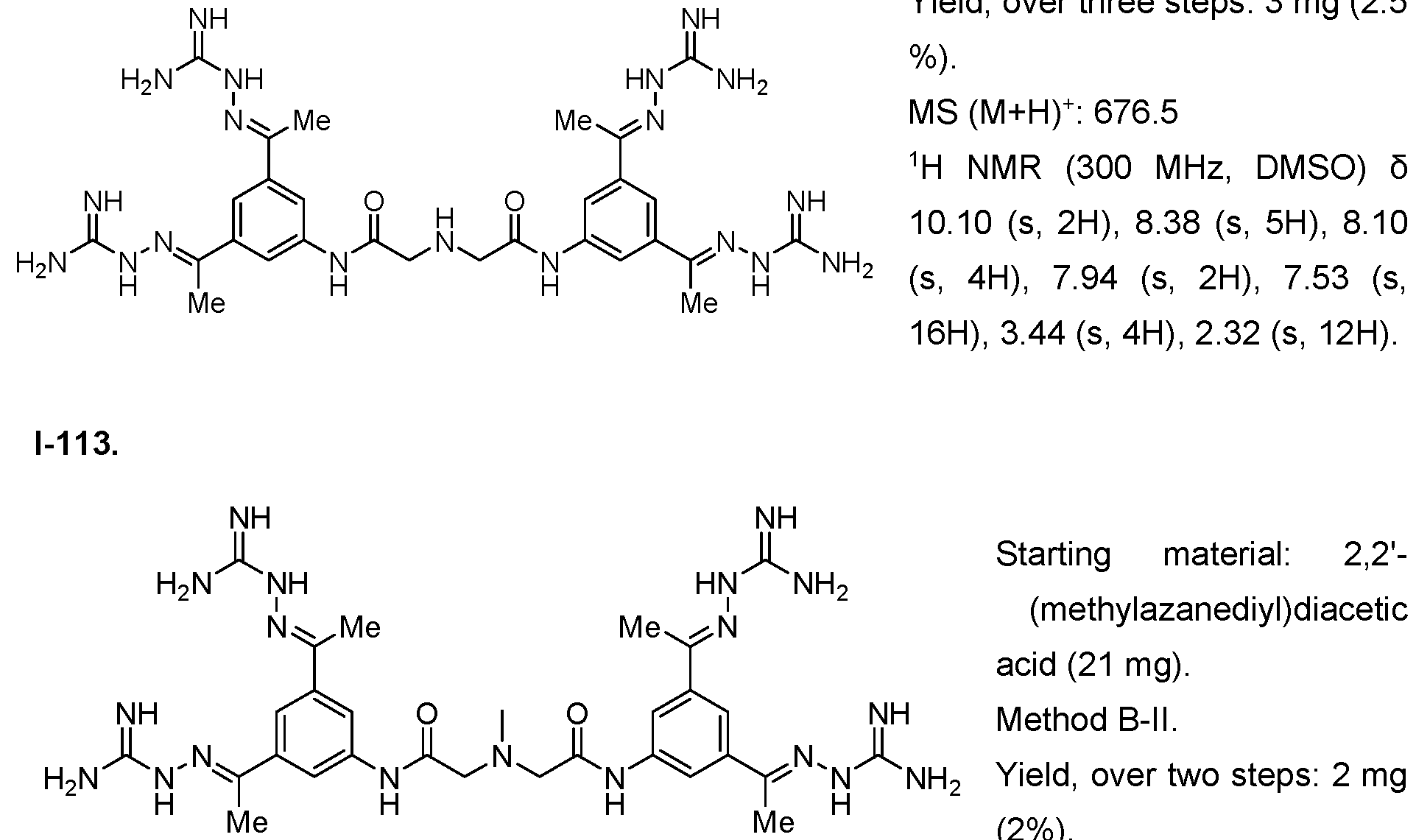 .
.
MS (M+H)+: 690.8
1H NMR (300 MHz, DMSO) 5 10.13 (s, 2H), 8.30 (s, 4H), 8.09 (s, 4H), 7.97 (s, 2H), 7.12 (s,
16H), 3.43 (s, 4H), 2.55 (s, 3H), 2.31 (s, 12H).
1-114.
Starting material: 1 ,3-Bis(3-carboxypropyl)tetramethyldisiloxane (42 mg).
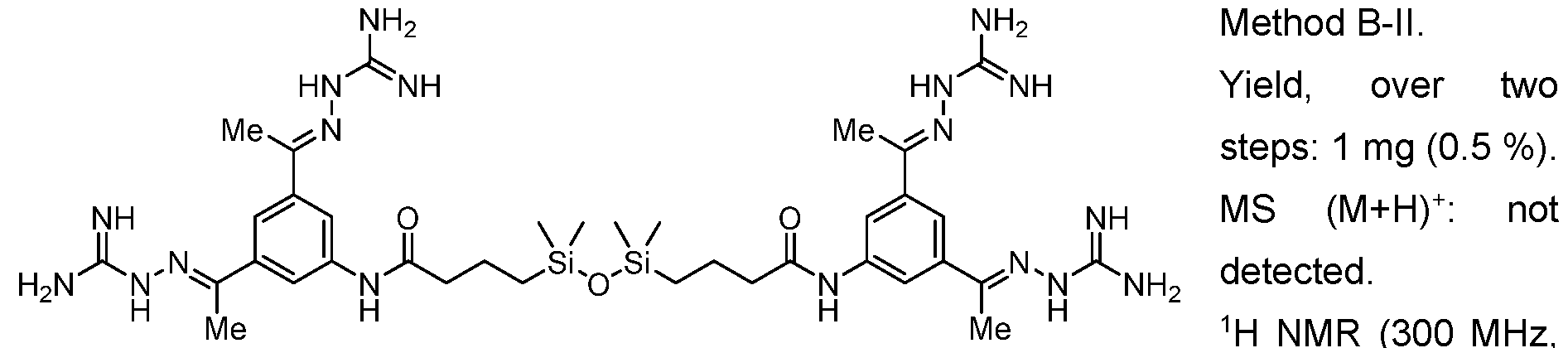 ,
,
DMSO) 5 9.92 (s, 2H), 8.35 (s, 4H), 8.01 (s, 4H), 7.90 (s, 2H), 7.42 (s, 16H), 2.31 (s, 12H),
1.64 (t, J = 7.7 Hz, 4H), 0.62 - 0.42 (m, 4H), 0.06-0.00 (m, 16H).
1-115.

Starting material: 6,7,9,10,17,18,20,21-octahydrodibenzo[b,k ][1 ,4,7, 10, 13, 16] hexaoxacycloocta decine-2,14-disulfonyl dichloride (25 mg, 0.044 mmol).
Method A, but was performed on 20 mg scale of intermediate 1.
Yield, over two steps: 15 mg (25%).
MS (M+2H)2+: 532.6 1H NMR (300 MHz, DMSO) 5 8.18 (s, 4H), 7.60 (s, 2H), 7.35 (s, 4H), 7.20 - 6.98 (m, 18H), 6.87 - 6.73 (m, 4H), 3.84 (m, 8H), 3.58 (m, 8H), 2.04 (s, 12H).
1-116.

Step 1 :
Intermediate 1 (120 mg, 0.68 mmol) was suspended in toluene (2 mL) and stirred in an ice bath. A solution of triphosgene (66 mg, 0.2 mmol) in toluene (0.5 mL) was added. The
suspension was allowed to warm to rt, and was stirred overnight at rt. The product precipitated, was filtered and used in the next step without additional purification.
Step 2:
The compound was synthesized according to general procedure 2 using the product from the previous step.
Yield, over two steps: 15 mg (3%).
MS (M+H)+: 605.6.
1H NMR (300 MHz, DMSO) 5 9.74 (s, 2H), 8.39 (s, 4H), 7.96 (s, 4H), 7.85 (s, 2H), 7.45 (s, 16H), 2.34 (s, 12H).
1-118.

Step 1 :
Intermediate 1 (90 mg, 0,5 mmol), intermediate 52 (120 mg, 0,5 mmol), BrettPhos Pd G3 (43 mg, 0,05 mmol), Brettphos (26 mg, 0,03 mmol) and NaOf-Bu (115 mg, 1 ,2 mmol) were added to dry Dioxane (1 mL) and were purged with N2 for 15 min. After the completion of the reaction, the volatiles were evaporated under reduced pressure and the residue was purified using flash chromatography eluting with Cyclohexane: Ethylacetate 100:0% to Cyclohexane: Ethylacetate 0:100% yielding the desired product as brown solid.
MS (M+H)+: 339.9
Step 2:
The compound was synthesized according to general procedure 2 using the product from the previous step.
Yield, over two steps: 2 mg (0.5% yield).
MS (M+H)+: 562.2.
1H NMR (300 MHz, DMSO) 5 8.32 (s, 4H), 7.68 (s, 2H), 7.57 (s, 4H), 6.95 (brs, 16 H), 2.30 (s, 12H).
1-119.

Step 1 :
A dry flask was charged with intermediate 52 (120 mg, 0.5 mmol), bis(pinacolato)diboron (64 mg, 0.25 mmol), and K2CO3 (212 mg, 1.5 mmol) and 3 mL of DMSO. The atmosphere was exchanged with nitrogen and PddppfCh (15 mg, 0.02 mmol) was added. The reaction was stirred at 80 °C overnight on the oil bath. The reaction mixture was cooled and diluted with EtOAc. Silica was added, and the solvents were evaporated under reduced pressure. The residue was loaded on silica column and eluted with Cyclohexane: Ethylacetate 100:0% to Cyclohexane: Ethylacetate 0:100% yielding the desired product as brown solid.
MS (M+H)+: 323.5.
1H NMR (300 MHz, DMSO) 5 8.54 (s, 2H), 8.38 (s, 4H), 2.72 (s, 12H).
Step 2:
The compound was synthesized according to general procedure 2 using the product from the previous step.
Yield, over two steps: 6 mg (2%).
MS (M+H)+: 547.8.
1H NMR (300 MHz, DMSO) 5 8.40 (s, 4H), 8.23 (s, 4H), 8.20 (s, 2H), 7.88 (s, 16H), 2.45 (s, 12H).
1-120.

Step 1 :
A 100 mL round bottom flask equipped with a magnetic stir bar and a condenser was charged with intermediate 52 (120 mg, 0.5 mmol), 1 ,3-benzenediboronic acid (41 mg, 0.25 mmol) and
K2CO3 (275 mg, 4 mmol). The solids were suspended in 12 ml/5 ml/1 .5 ml toluene/MeOH/H2O (8.8 mL), degassed, and the atmosphere was exchanged with nitrogen. Afterwards, Pd(PPh3)4 (84 mg, 72 pmol) was added. The mixture was stirred at 80 °C overnight. The solvent was evaporated and the residue was purified by silica gel column chromatography eluting with cyclohexane to cyclohexane:ethylacetate 20:80 to obtain the desired product as a white solid.
MS (M+H)+: 399.3.
1H NMR (300 MHz, DMSO) 5 8.45 (d, J = 1.6 Hz, 4H), 8.38 (t, J = 1.6 Hz, 2H), 8.12 (s, 1 H), 7.84 - 7.81 (m, 2H), 7.66 - 7.61 (m, 1 H), 2.68 (s, 12H).
13C NMR (75 MHz, DMSO) 5 197.65, 147.03, 141.09, 139.64, 137.85, 131.02, 129.88, 127.23, 126.27, 126.11 , 27.13.
Step 2.
The compound was synthesized according to general procedure 2 using the product from the previous step.
Yield, over two steps: 6 mg (1%).
MS (M+H)+: 623.8.
1H NMR (300 MHz, DMSO) 5 8.39 (s, 4H), 8.21 (s, 6H), 8.07 (s, 1 H), 7.84 (d, J = 9.5 Hz, 2H), 7.76 - 7.55 (m, 17H), 2.44 (s, 12H).
General scheme for synthesis of 1-121-122.
Step 1 :

The intermediate 53 (58 mg, 0.14 mmol) and diamine (0.14 mmol) were dissolved in 1 ml of MeCN. N-methyl imidazole (0.06 ml) and TCFH (73 mg, 0.28 mmol) were added sequentially. After the completion of the reaction, the solids were filtrated, washed with ethanol and used in the next step without additional purification.
Step 2:
The compounds were synthesized according to general procedure 2 using the product from the previous step.
1-121.
Starting material: p-phenylenediamine (15 mg).
Yield, over two steps: 14 mg (11 %).
MS (M+H)+: 826.8.

Step 1 :
The isophtalic acid (23 mg, 0.14 mmol ) and intermediate 54 (50 mg, 0.29 mmol) were dissolved in 1 ml of DCM. Pyridine (90 mg) and TCFH (78 mg, 0.28 mmol) were added sequentially. The reaction was stirred overnight. After the completion of the reaction, volatiles
were evaporated under reduced pressure and the residue was purified using flash chromatography eluting with Cyclohexane: Ethylacetate 100:0% to Cyclohexane: Ethylacetate 0:100% yielding the desired product as white solid.
MS (M+H)+: 487.6.
1H NMR (300 MHz, DMSO) 5 8.81 (s, 1 H), 8.47 (dd, J = 7.8, 1.9 Hz, 2H), 8.33 (s, 2H), 8.15 (d, J = 1.5 Hz, 4H), 7.85 (t, J = 7.8 Hz, 1 H), 2.62 (s, 12H).
13C NMR (75 MHz, DMSO) 5 196.75, 163.67, 150.99, 138.57, 135.16, 130.95, 129.98, 129.53, 126.05, 125.21 , 27.00.
Step 2:
The compound was synthesized according to general procedure 2 using the product from the previous step.
Yield, over two steps: 21 mg (17%).
MS (M+H)+: 711.8
1H NMR (300 MHz, DMSO) 5 8.89 (s, 1 H), 8.55 (s, 2H), 8.36 (s, 4H), 8.07 (s, 2H), 7.96 (s, 4H), 7.92 (t, J = 7.9 Hz, 1 H), 7.57 (s, 16H), 2.38 (s, 12H).
1-124.
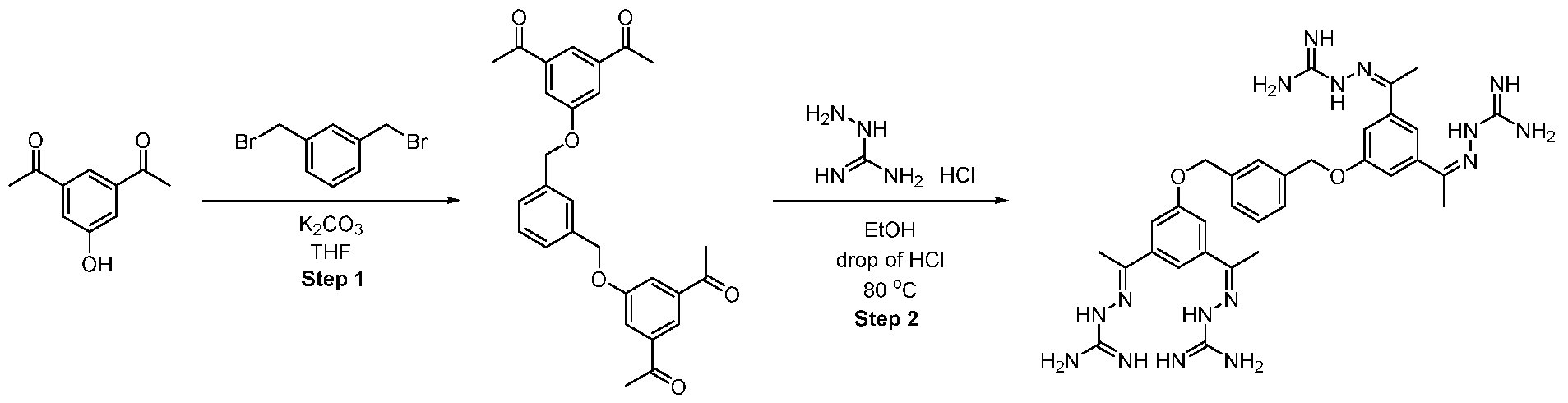
Step 1 :
To a solution of intermediate 54 (80 mg, 0.5 mmol) and K2CO3 (200 mg, 1.45 mmol) in DMF (1 mL), a,a'-dibromo-m-xylene (65 mg, 0.25 mmol) was added. The resulting mixture was stirred at 60 °C overnight. After the completion of the reaction, volatiles were evaporated under reduced pressure and the residue was purified using flash chromatography eluting with Cyclohexane: Ethylacetate 100:0% to Cyclohexane: Ethylacetate 0:100% yielding the desired product as white solid.
MS (M+H)+: 459.1
1H NMR (300 MHz, DMSO) 5 7.98 (s, 2H), 7.72 (s, 4H), 7.57 (s, 1 H), 7.41 (s, 3H), 5.23 (s, 4H), 2.57 (s, 12H).
13C NMR (75 MHz, DMSO) 5 197.27, 138.47, 136.80, 128.71 , 127.43, 126.96, 120.22, 118.47, 69.63, 26.96.
Step 2:
The compound was synthesized according to general procedure 2 using the product from the previous step.
Yield, over two steps: 28 mg (23%).
MS (M+H)+: 683.7.
1H NMR (300 MHz, DMSO) 5 8.39 (s, 4H), 7.77 (s, 18H), 7.63 (s, 1 H), 7.58 (s, 4H), 7.48 (m,
3H), 5.26 (s, 4H), 2.35 (s, 12H).
1-125.
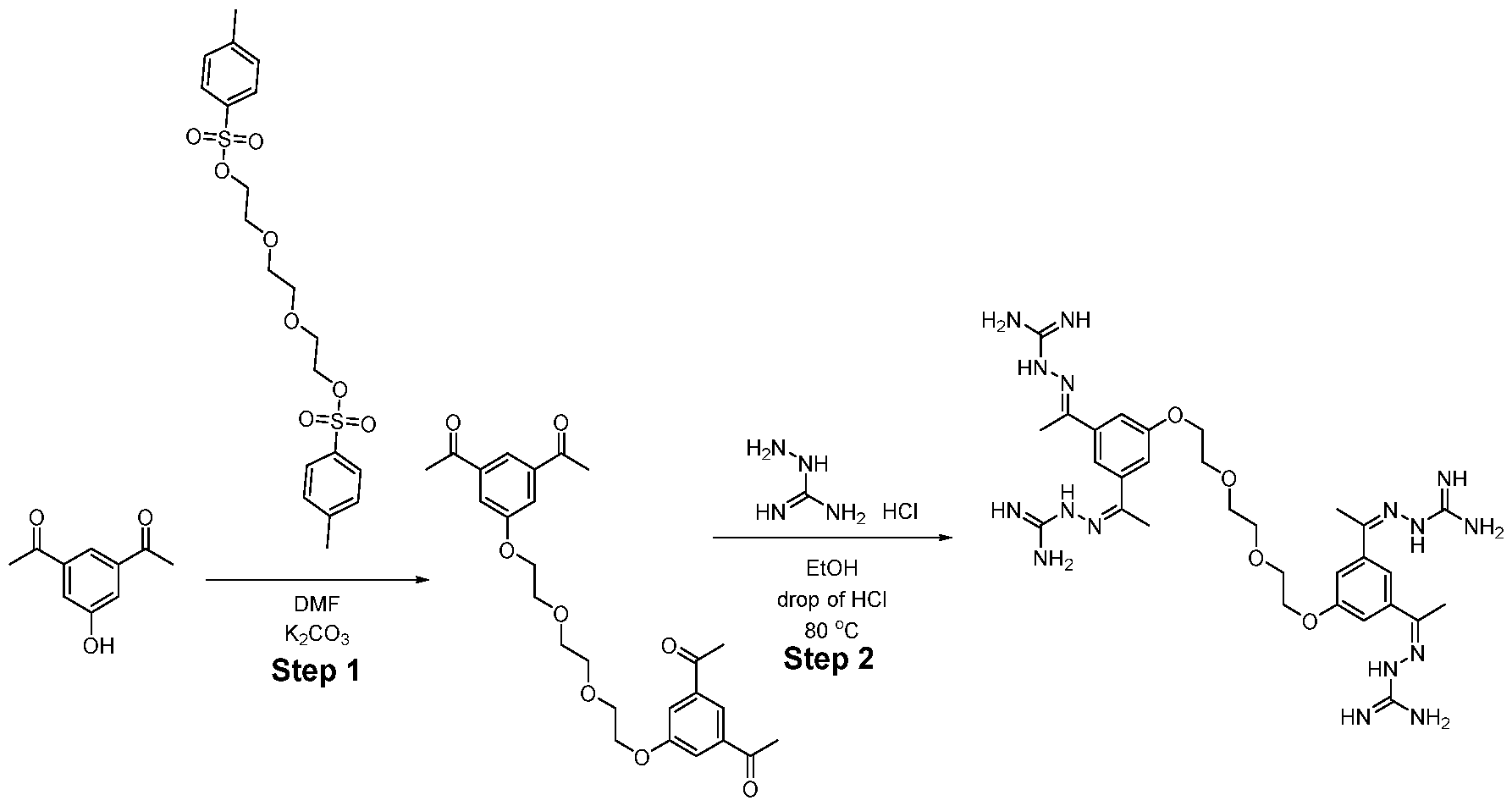
Step 1 :
To a solution of intermediate 54 (80 mg, 0.5 mmol) and K2CO3 (200 mg, 1.45 mmol) in DMF (1 mL), triethylene glycol di(p-toluenesulfonate (114 mg, 0.25 mmol) was added. The resulting mixture was stirred at 60 °C overnight. After the completion of the reaction, volatiles were evaporated under reduced pressure and the residue was purified using flash chromatography eluting with Cyclohexane: Ethylacetate 100:0% to Cyclohexane: Ethylacetate 0:100% yielding product as white solid.
MS (M+H)+: 471.6.
1H NMR (300 MHz, CDCI3) 6 8.02 (s, 2H), 7.61 (s, 4H), 4.17 - 4.14 (m, 4H), 3.85 - 3.81 (m, 4H), 3.70 (s, 4H), 2.55 (s, 12H).
13C NMR (75 MHz, CDCI3) 6 197.11 , 159.27, 138.62, 120.82, 118.47, 77.43, 77.00, 76.58, 70.91 , 69.63, 68.01 , 26.76.
Step 2:
The compound was synthesized according to general procedure 2 using the product from the previous step.
Yield, over two steps: 10 mg (8%).
MS (M+H)+: 695.9.
1H NMR (300 MHz, DMSO) 6 8.37 (s, 4H), 7.74 (s, 2H), 7.44 (s, 16H), 4.23 (s, 4H), 3.80 (s, 4H), 3.65 (s, 4H), 2.32 (s, 12H).
1-126.

Step 1 :
A 8 ml microwave vial was charged with intermediate 52 (120 mg, 0.5 mmol), 1 ,3- phenylendimethanthiol, (42.5 mg, 0.25 mmol), Pd(OAc)2 (2.24 mg, 0.01 mmol), dippf (5 mg, 0.011 mmol) , and f-BuONa (57 mg, 0.6 mmol). The reaction tube was sealed. The septum was pierced with a needle attached to a Schlenk line, and the tube was evacuated and backfilled with nitrogen (this process was repeated a total of three times). 1 ,4-dioxane (1.0 mL) were added via a syringe. The reaction was stirred at 85 °C overnight. After the completion of the reaction, the solvents were evaporated, and the residue was purified using flash chromatography eluting with Cyclohexane: Ethylacetate 100:0% to Cyclohexane: Ethylacetate 0:100% yielding product as white solid.
MS (M+H)+: 491.2.
1H NMR (300 MHz, CDCI3) 5 8.19 (s, 2H), 7.90 (s, 4H), 7.27 - 7.02 (m, 4H), 4.09 (s, 4H), 2.51 (s, 12H).
Step 2:
The compound was synthesized according to general procedure 2 using the product from the previous step.
Yield, over two steps: 2 mg (0.5%).
MS (M+H)+: 715.7.
1H NMR (300 MHz, DMSO) 5 8.32 (s, 4H), 7.90 (s, 2H), 7.74 (s, 4H), 7.49 (s, 1 H), 7.25 (m, 3H), 7.02 (s, 16H), 4.32 (s, 4H), 2.28 (s, 12H).
1-127.

Step 1 :
Isophthalic acid (22 mg, 0.14 mmol) and intermediate 55 (50 mg, 0.28 mmol) were dissolved in 1 ml of MeCN. N-methylimidazole (0.06 ml) and TCFH (73 mg, 0.28 mmol) were added sequentially. After the completion of the reaction, the solids were filtrated, washed with ethanol and used in the next step without additional purification.
Step 2:
The compound was synthesized according to general procedure 2 using the product from the previous step.
Yield, over two steps: 3 mg (2%).
MS (M+H)+: 709,4.
1H NMR (300 MHz, DMSO) 5 11.72 (s, 1 H), 8.63 - 8.47 (m, 3H), 8.32 (s, 4H), 8.10 (d, J = 9.6 Hz, 2H), 7.82 (d, J = 10.3 Hz, 2H), 7.63 - 7.34 (m, 11 H), 6.70 (s, 8H), 2.33 (s, 6H), 2.31 (s, 6H).
General scheme for synthesis of 1-128-130.
Step 1 :
The acid (0.5 mmol) and intermediate 57 (237 mg, 1 mmol) were dissolved in 4 ml of MeCN.

0.24 mL /V-methylimidazole and TCFH (300 mg, 1.07 mmol) were added sequentially. The reaction was stirred overnight. After the completion of the reaction, the solvent was evaporated and the residue was purified by silica gel column chromatography eluting with cyclohexane to cyclohexane: ethylacetate 0:100 to obtain the desired product as a light yellow oil.
Step 2:
The product from the previous step was dissolved in 4 M HCI in dioxane (3 ml) and stirred at room temperature for 3 h. After the completion of the reaction the solvents were evaporated, and the residue was used in the next steps without additional purification.
Step 3:
The compounds were synthesized according to general procedure 2 using the product from the previous step.
1-128.
Step 1 :
Starting material: isophthalic acid (88 mg).
MS M+H + 6050
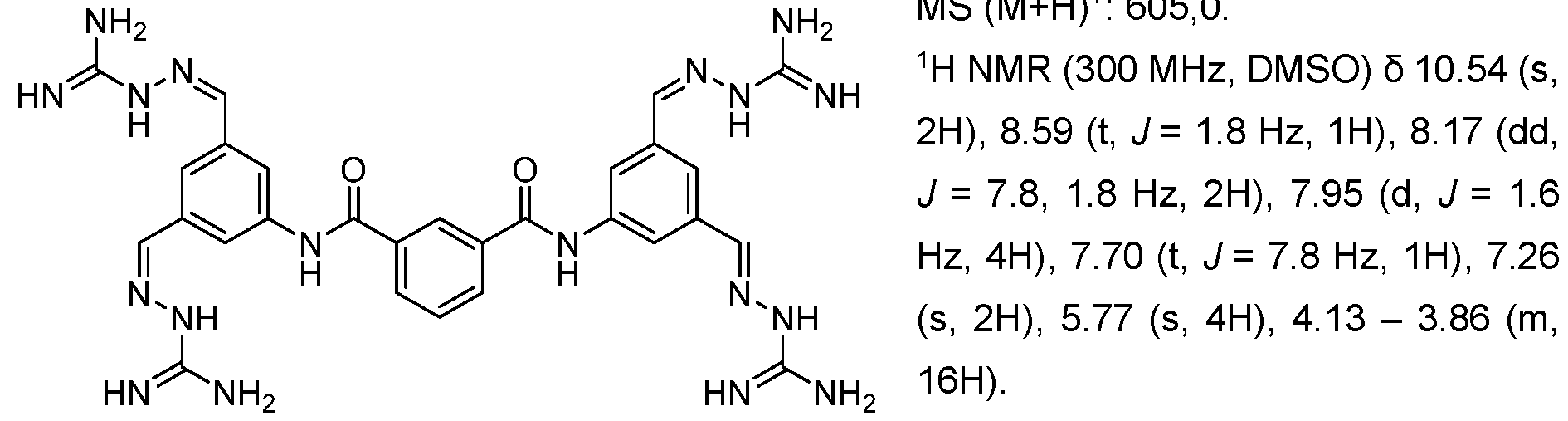
Step 2:
MS (M+H)+: 429,4.
1H NMR (300 MHz, DMSO) 511.06 (s, 2H), 10.14 (s, 4H), 8.75 (s, 1H), 8.71 (s, 4H), 8.26 (d,
J= 7.5 Hz, 1 H), 8.24 (s, 2H), 7.78 (t, J= 7.8 Hz, 1H).
Step 3:
Yield, over three steps: 20 mg (5%).
MS (M+H)+: 653,8.
1H NMR (300 MHz, DMSO) 510.59 (s, 2H), 8.64 (s, 1H), 8.37 (s, 4H), 8.24 - 8.17 (m, 2H),
8.11 (s, 4H), 8.07 (s, 6H), 7.74 (t, J= 7.8 Hz, 1H), 7.41 (s, 16H).
1-129.
The molecule was done on scale of 80 mg of intermediate 57 (0.25 mmol).
Step 1 :
Starting material: intermediate 40 (23
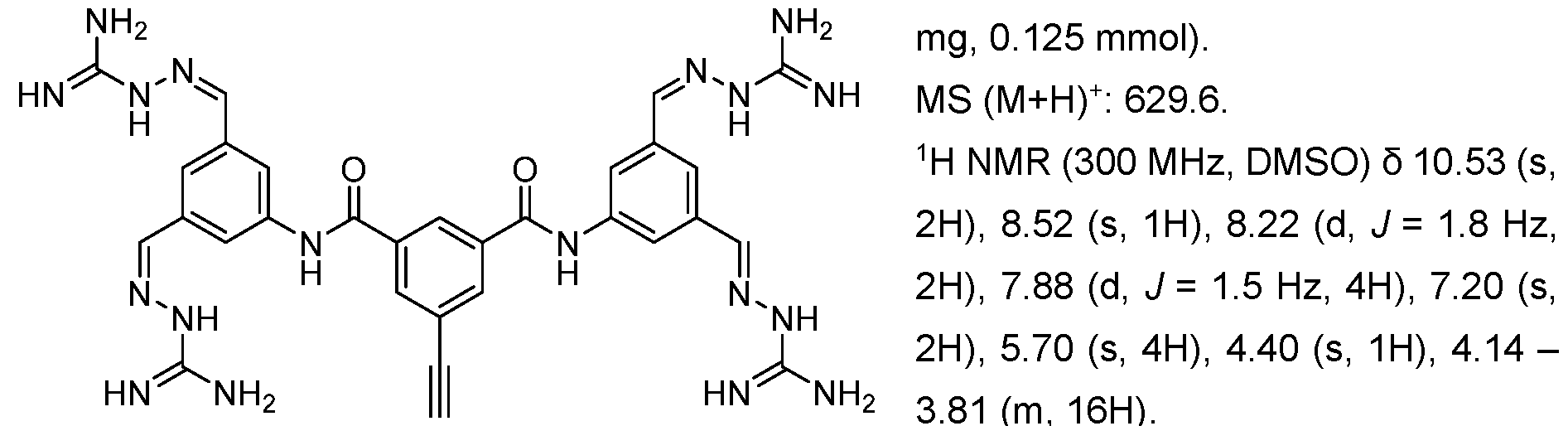 . , .
. , .
Step 2:
MS (M+H)+: 452,9.
Step 3:
Yield, over 3 steps: 6 mg (6%).
MS (M+H)+: 677.7.
1H NMR (300 MHz, DMSO) 510.67 (s, 2H), 8.64 (t, J= 1.7 Hz, 1H), 8.38 (s, 4H), 8.32 (d, J = 1.8 Hz, 2H), 8.11 (s, 2H), 8.10 (s, 4H), 8.07 (s, 2H), 7.57 (s, 4H), 4.49 (s, 1H).
1-130.
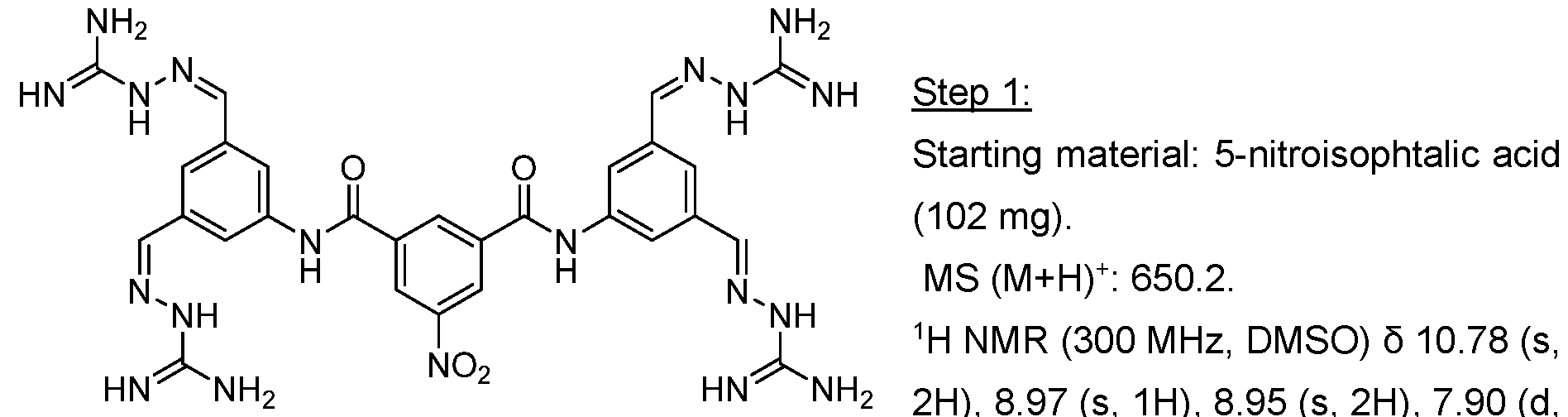 , . , , . , , . ,
, . , , . , , . ,
J = 1 .6 Hz, 4H), 7.23 (s, 2H), 5.72 (s, 4H), 4.06 - 3.82 (m, 16H).
Step 2:
MS (M+H)+: 474.4.
Step 3:
Yield, over 3 steps: 15 mg (2%).
MS (M+H)+: 698,2.
1H NMR (300 MHz, DMSO) 5 10.94 (s, 2H), 9.09 (s, 1 H), 9.05 (s, 2H), 8.36 (s, 4H), 8.12 (s, 3H), 8.09 (s, 2H), 8.08 (s, 4H), 7.37 (s, 16H).
111-1.

Step 1 :
Isophthalic acid (23 mg, 0.14 mmol) and intermediate 58 (50 mg, 0.28 mmol) were dissolved in 1 ml of ACN. 0.06 mL N-methylimidazole and TCFH (73 mg, 0.28 mmol) were added sequentially. The reaction was stirred overnight. After the completion of the reaction, the solvent was removed, and the residue was used in the next reaction without additional purification.
Step 2:
The compound was synthesized according to general procedure 2 using the product from the previous step.
Yield, over two steps: 2 mg (2%).
MS (M+H)+: 569.7.
1H NMR (300 MHz, DMSO) 5 10.40 (s, 2H), 8.60 (s, 1 H), 8.31 (s, 2H), 8.17 (d, J = 6.2 Hz, 2H), 8.00 (s, 2H), 7.80 - 7.69 (m, 3H), 7.56 (s, 2H), 7.10 (s, 8H), 2.77 - 2.58 (m, 4H), 2.30 (s, 6H), 1.24 (t, J = 7.5 Hz, 6H).
General scheme for examples 131-133.
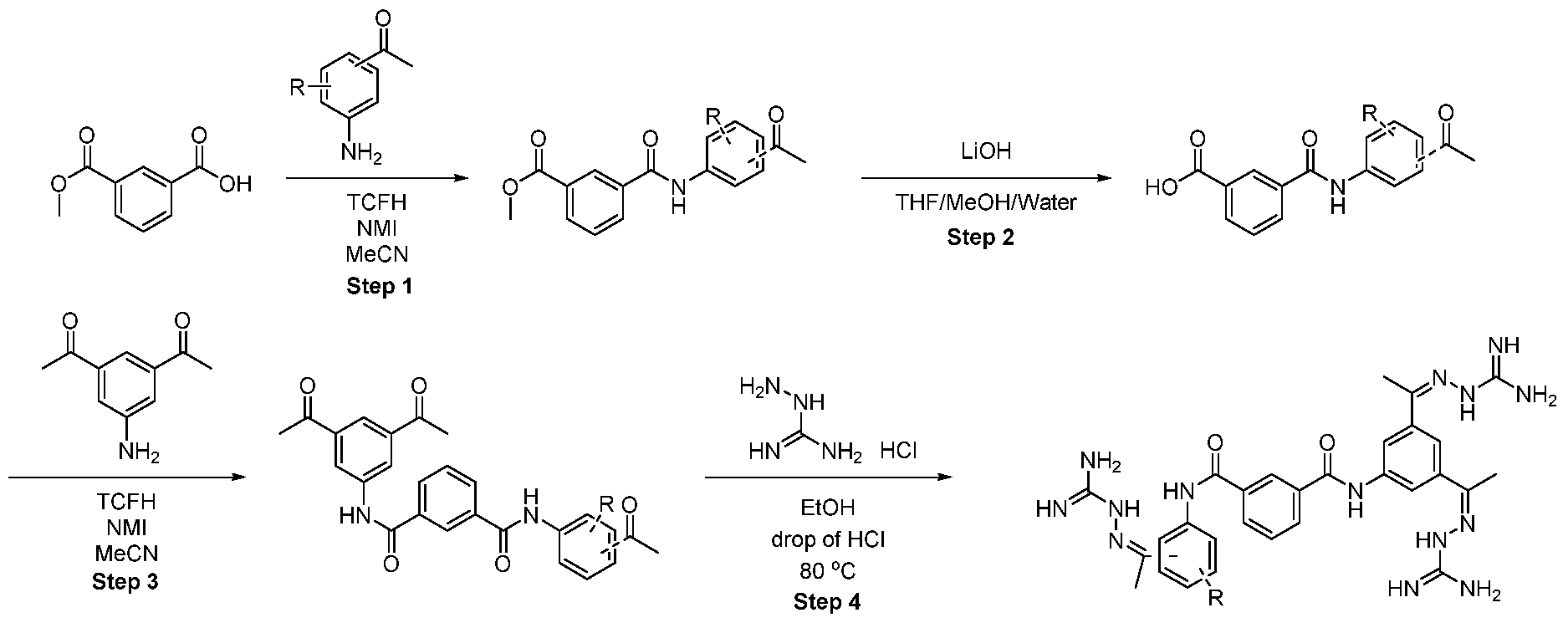
Step 1 :
The 3-(methoxycarbonyl)benzoic acid (180 mg, 1 mmol) and aminoacetophenone (1 mmol) were dissolved in 3 ml of ACN. 0.2 mL N-methylimidazole and TCFH (210 mg, 0.75 mmol) were added sequentially. The reaction was stirred overnight. After the completion of the reaction solvents were evaporated, and the residue was purified using flash chromatography eluting with Cyclohexane: Ethylacetate 100:0% to Cyclohexane: Ethylacetate 0:100% yielding product as white solid.
Step 2:
Synthesized according to general procedure 1. Starting materials: THF/MeOH/Water (1 ml/1 ml/1 ml) and LiOH (200 mg).
Step 3:
The product from previous step (0.14 mmol) and intermediate 1 (50 mg, 0.28 mmol) were dissolved in 1 ml of MeCN. 0.06 mL N-methylimidazole and TCFH (73 mg, 0.28 mmol) were added sequentially. The reaction was stirred overnight. After the completion of the reaction the product was filtered off yielding the desired product as white solid.
Step 4:
The compounds were synthesized according to general procedure 2 using the product from the previous step.
1-131.
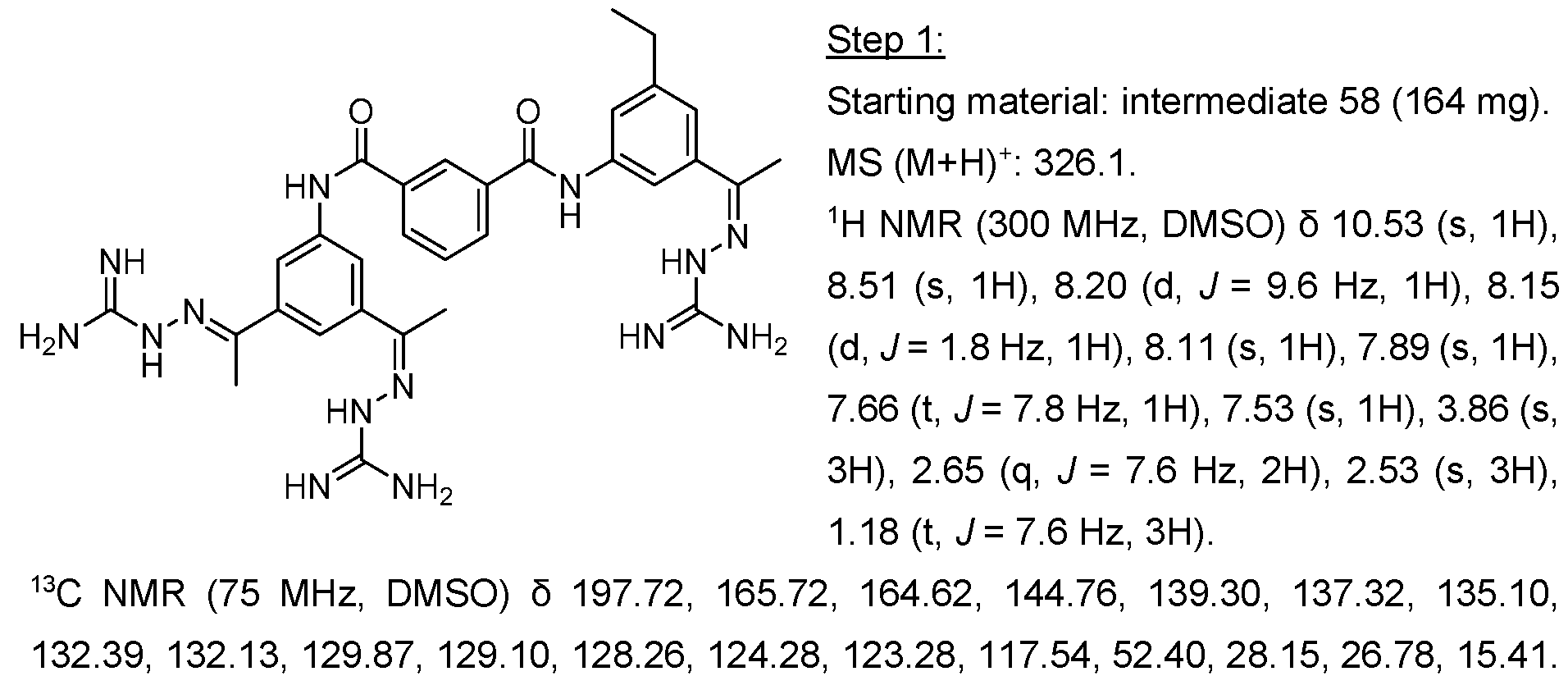 , , , , , , , , , , ,
, , , , , , , , , , ,
Step 2:
Yield, over step 1-2: 105 mg (33%).
MS (M+H)+: 312.1.
1H NMR (300 MHz, DMSO) 513.27 (s, 1H), 10.58 (s, 1H), 8.58 (s, 1H), 8.28-8.22 (m, 2H), 8.16 (d, J= 7.8 Hz, 1H), 7.97 (s, 1H), 7.68 (t, J= 7.8 Hz, 1H), 7.58 (s, 1H), 2.70 (q, J= 7.5 Hz, 2H), 2.59 (s, 3H), 1.24 (t, J= 7.6 Hz, 3H).
13C NMR (75 MHz, DMSO) 5 197.71, 166.78, 164.79, 144.73, 139.37, 137.31, 134.95, 132.29, 132.00, 131.04, 128.88, 128.41, 124.26, 123.19, 117.54, 28.15, 26.77, 15.39.
Step 3:
Starting material: The product from step 2 (40 mg).
Step 4:
Yield, over steps 3-4: 15 mg (14%).
MS (M+H)+: 639.6.
1H NMR (300 MHz, DMSO) 510.34 (s, 1H), 10.26 (s, 1H), 8.44 (s, 1H), 8.18 (s, 3H), 8.08 (s, 2H), 8.00 (m, 2H), 7.82 (s, 2H), 7.60 (s, 1H), 7.53 (t, J = 7.8 Hz, 1H), 7.60-7.25 (m, 13 H), 2.48 (q, J= 7.8 Hz, 1H), 2.18 (s, 6H), 2.12 (s, 3H), 1.05 (t, J= 7.6 Hz, 3H).
1-132.
 9.3 Hz, 1H), 8.04 (d, J= 8.1 Hz, 1H),
9.3 Hz, 1H), 8.04 (d, J= 8.1 Hz, 1H),
7.72 - 7.61 (m, 2H), 7.47 (t, J = 7.9 Hz, 1 H), 3.86 (s, 3H), 2.53 (s, 3H).
13C NMR (75 MHz, DMSO) 5 197.61, 165.72, 164.70, 146.65, 139.32, 137.24, 135.06, 132.40, 132.17, 129.88, 129.10, 128.30, 124.89, 123.87, 119.75, 52.40, 26.74.
Step 2:
Yield, oversteps 1-2: 118 mg (40%).
MS (M+H)+: 284.0.
1H NMR (300 MHz, DMSO) 510.56 (s, 1H), 8.49 (s, 1H), 8.29 (s, 1H), 8.15 (d, J= 9.3 Hz, 1H), 8.08 (d, J= 7.8 Hz, 1H), 8.01 (d, J= 10.4 Hz, 1H), 7.68-7.56 (m, 2H), 7.45 (t, J= 8.0 Hz, 1H), 2.42 (s, 3H).
13C NMR (75 MHz, DMSO) 5 197.62, 166.77, 164.87, 139.37, 137.23, 134.92, 132.33, 132.03, 131.04, 129.04, 128.89, 128.44, 124.88, 123.80, 119.75, 26.74.
Step 3:
Starting material: The product from step 2 (40 mg).
Step 4:
Yield, over steps 3-4: 25 mg (24%).
MS (M+H)+: 611.1.
1H NMR (300 MHz, DMSO) 510.57 (s, 1H), 10.54 (s, 1H), 8.64 (s, 1H), 8.41 (s, 3H), 8.28 (s, 2H), 8.23-8.18 (m, 3H), 8.04 (s, 1H), 7.93 (d, J= 7.4 Hz, 1H), 7.86-7.62 (m, 12H), 7.41 (t, J= 8.0 Hz, 1H), 2.38 (s, 6H), 2.33 (s, 3H).
1-133
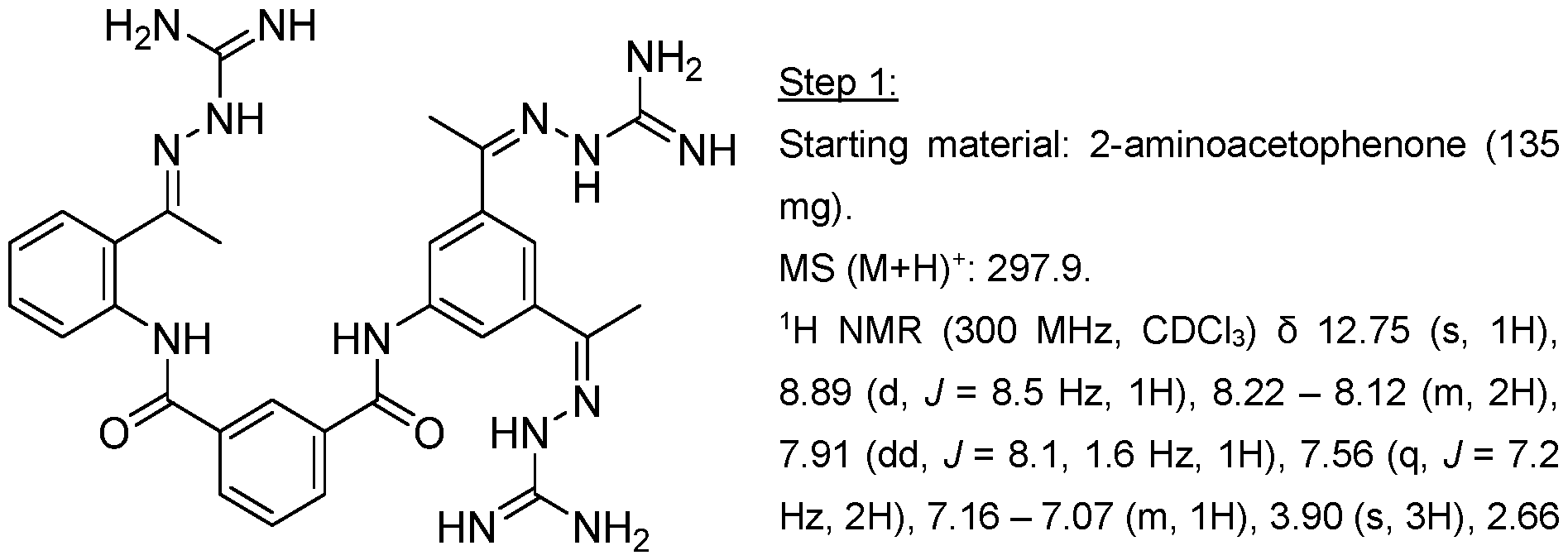 (s, 3H).
(s, 3H).
13C NMR (75 MHz, CDCI3) 6166.34, 164.71, 141.22, 135.45, 135.33, 132.85, 131.86, 131.56, 130.97, 129.02, 128.84, 122.78, 120.89, 52.43, 28.57.
Step 2:
Yield, oversteps 1-2: 120 mg (42%).
MS (M+H)+: 283.9.
1H NMR (300 MHz, DMSO) 513.28 (s, 1H), 12.41 (s, 1H), 8.61 (d, J = 9.7 Hz, 1H), 8.56 (s, 1H), 8.27-8.16 (m, 2H), 8.17-8.08 (m, 1H), 7.79-7.63 (m, 2H), 7.36-7.27 (m, 1H), 2.71 (s, 3H).
13C NMR (75 MHz, DMSO) 5 166.54, 164.20, 139.28, 132.74, 132.04, 131.51, 131.30, 129.50, 127.82, 124.02, 123.42, 120.59, 28.70.
Step 3:
Starting material: The product from step 2 (40 mg).
Step 4:
Yield, over steps 3-4: 20 mg (19%).
MS (M+H)+: 611.8.
1H NMR (300 MHz, DMSO) 511.65 (s, 1H), 10.51 (s, 1H), 8.57 (s, 1H), 8.35 (s, 3H), 8.25 (s, 2H), 8.20 (d, J= 8.0 Hz, 2H), 8.11 (d, J= 8.0 Hz, 2H), 8.04 (s, 1H), 7.81 -7.51 (m, 10H), 7.44 -7.35 (m, 1H), 7.31 -7.16 (m, 1H), 6.78 (s, 4H), 2.38 (s, 6H), 2.32 (s, 3H).
General scheme for synthesis of 1-134-143.
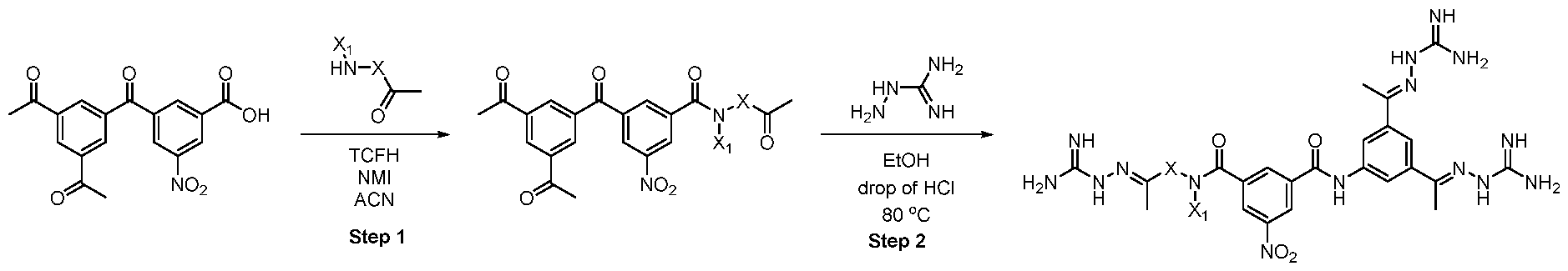 Step 1 :
Step 1 :
Intermediate 59 (50 mg, 0.14 mmol) and aminoketone (0.14 mmol) were dissolved in 1 ml of ACN. 0.06 mL N-methylimidazole and TCFH (73 mg, 0.28 mmol) were added sequentially. The reaction was stirred overnight. After the completion of the reaction:
I: the product was filtered (if precipitated)
II: solvents were evaporated, and the residue was purified by silica gel column chromatography eluting with Cyclohexane: Ethylacetate 100:0% to Cyclohexane: Ethylacetate 0:100% to obtain the desired product.
Step 2:
The compounds were synthesized according to general procedure 2 using the product from the previous step.
1-134
Starting material: 3-aminoacetophenone (19 mg).
Method I.
Yield, over two steps: 22 mg (20%).
MS (M+H)+: 656.7.
1H NMR (300 MHz, DMSO) 5 10.93 (s, 1 H), 10.88 (s, 1 H), 9.08 (s, 1 H), 9.01 (s, 2H), 8.38 (s, 3H), 8.25 (s, 2H), 8.17 (s, 1 H), 8.05 (s, 1 H), 7.91 (d, J = 8.0 Hz, 1 H), 7.76 (d, J = 8.0 Hz, 1 H), 7.61 (s, 12H), 7.42 (t, J = 8.0 Hz, 1 H), 2.38 (s, 6H), 2.33 (s, 3H).
1-135.
Starting material: 4-
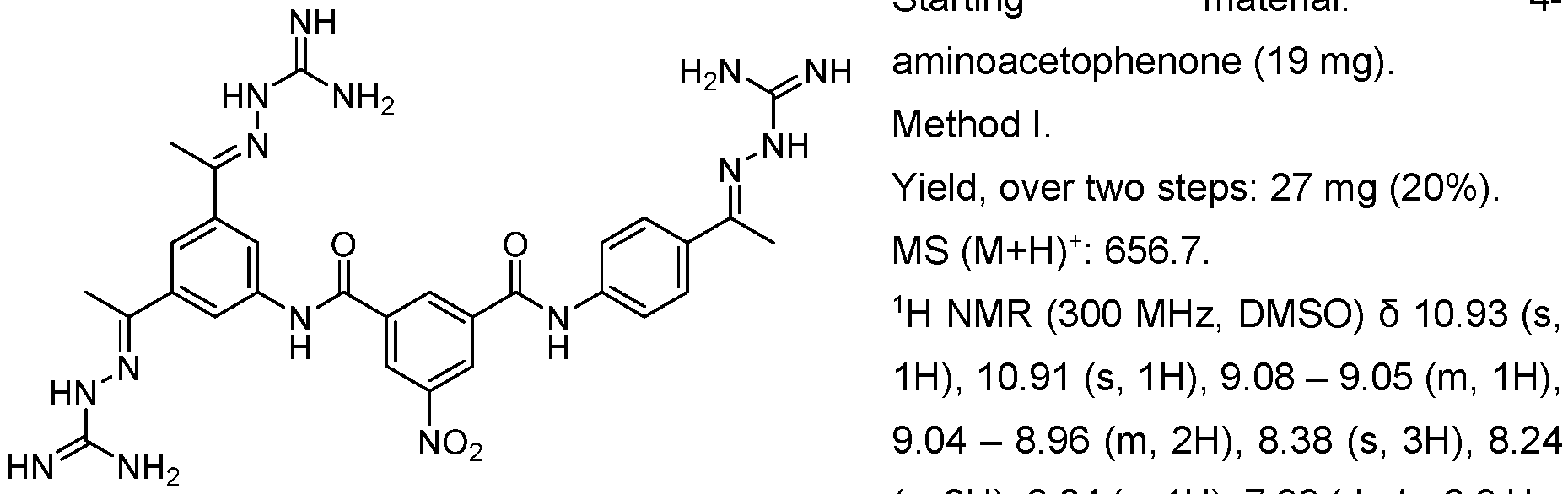 (s, 2H), 8.04 (s, 1 H), 7.99 (d, J= 8.9 Hz,
(s, 2H), 8.04 (s, 1 H), 7.99 (d, J= 8.9 Hz,
2H), 7.87 (d, J = 8.9 Hz, 2H), 7.80 (s, 4H), 7.52 (s, 8H), 2.37 (s, 6H), 2.31 (s, 3H).
1-136.
Starting material: intermediate 60 (23 mg).
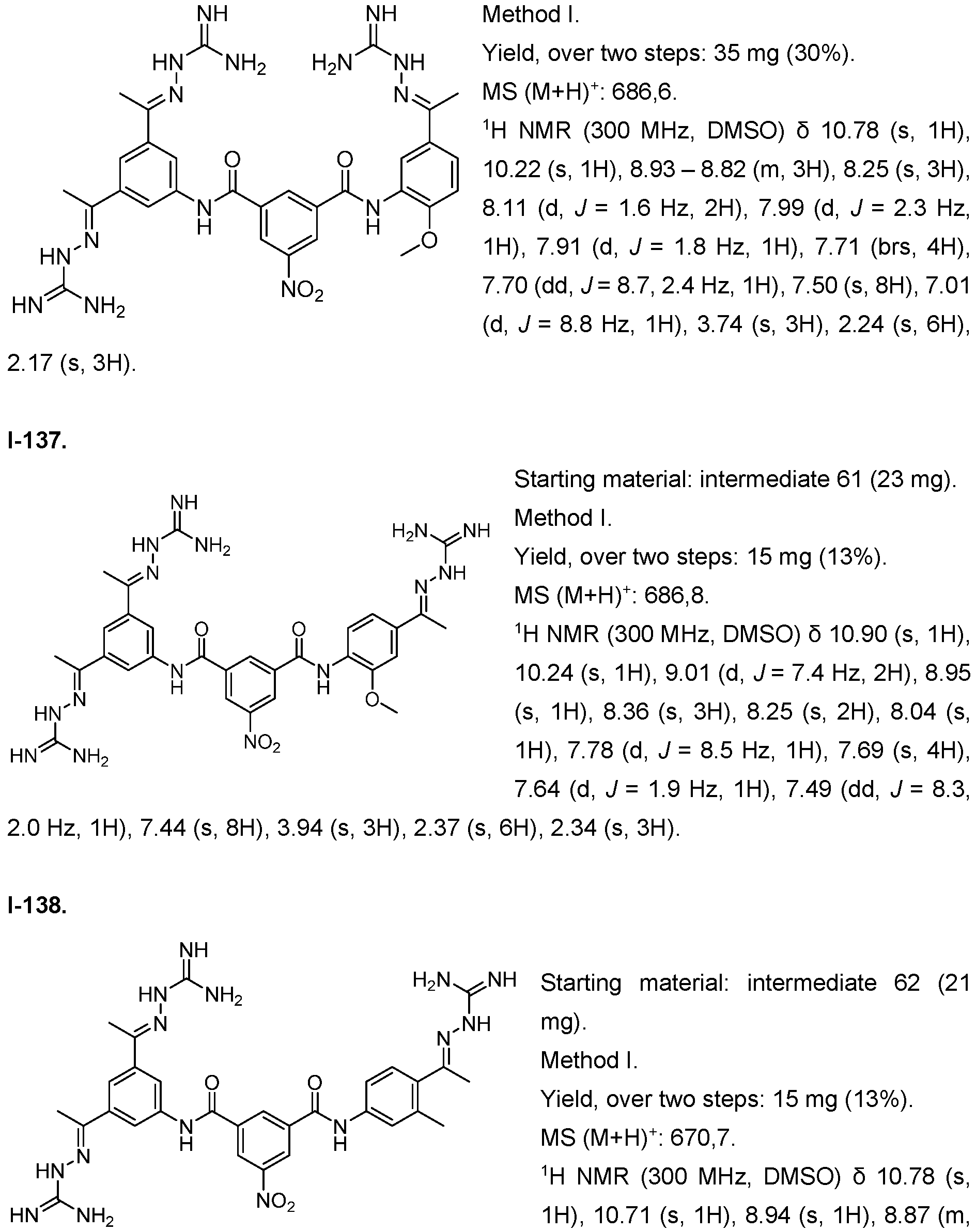
2H), 8.25 (s, 3H), 8.12 (s, 2H), 7.90 (s, 1 H), 7.68 - 7.49 (m, 6H), 7.40 - 7.15 (m, 9H), 2.25 (s, 3H), 2.24 (s, 6H), 2.15 (s, 3H).
1-139.
 H NMR (300 MHz, DMSO) 5
H NMR (300 MHz, DMSO) 5
10.81 (s, 1H), 8.90 (s, 1H), 8.56 (d, J = 5.4 Hz, 1H), 8.53 - 8.47 (m, 1H), 8.38 (s, 3H), 8.21 (s, 2H), 8.04 (s, 1 H), 7.90 (brs, 4H), 7.59 (brs, 6H), 3.81 - 3.45 (m, 5H), 3.20 - 3.03 (m, 1 H), 2.37 (s, 6H), 2.22-2.05 (m, 2H), 2.00 (s, 1.5 H), 1.92 (s, 1.5H).
1-141.
 8.36 (s, 3H), 8.20 (s, 2H), 8.02 (s,
8.36 (s, 3H), 8.20 (s, 2H), 8.02 (s,
1 H), 7.56 (s, 4H), 7.38 (s, 8H), 2.37 (s, 6H), 1 .98 (d, J = 7.2 Hz, 3H), 1.88 - 1.42 (m, 4H). 5H signals were not assigned due to similar shift with water.
1-143
 concentrated HCI were added. The reaction was heated to 80 °C and stirred at this temperature for 2 h. After the completion of the reaction the solvent was evaporated, and the residue was purified using reverse-phase chromatography and lyophilized yielding the product as a solid. Yield: 12% (8 mg).
concentrated HCI were added. The reaction was heated to 80 °C and stirred at this temperature for 2 h. After the completion of the reaction the solvent was evaporated, and the residue was purified using reverse-phase chromatography and lyophilized yielding the product as a solid. Yield: 12% (8 mg).
1H NMR (300 MHz, DMSO) 5 8.41 (s, 2H), 7.90 (s, 8H), 7.44 (s, 1 H), 7.12 (s, 2H), 2.29 (s, 6H) [M+2H]+145,8
Example 2 - Proliferation assay with compound 1-1
The compound 1-1 , for which an activation of the FGFR1 signaling pathway in cultured cells was found (see Example 5 and Example 6), was used in an assay for determining if the compound had a positive effect on proliferation of cultured cells.
NIH/ 3t3 cells were grown in standard growth medium supplemented with 10% Calf Serum (CS, VWR) in a T75-flask. At 80-90% confluency, medium was aspirated, cells were washed with 10 mL PBS and detached with 2 mL TrypLE Express (Gibco/ #12604-021). Cells were resuspended in 8 mL of Essential 6 medium (Gibco) without FGF2 and TGF-pi and counted
with Acridine Orange/Propidium iodide stain using the LUNA-FL Dual Fluorescence Cell Counter (Logos). Cells were diluted to 9.000 cells/ mL with Essential 6 medium. 50 pL of the cell dilution was dispensed to wells B2 to 023 of a collagen-coated white 384-well plate (Greiner/ #781983) using a 24-channel pipette. The outer wells were filled with 50 pL PBS. The plate was centrifuged for 30 s at 300 x g and incubated at 37°C and 5% CO2.
After 2-3 hours, the compound of the invention, compound 1-1 , was added to the well in varying concentrations and a maximum total volume of 0.2 pL DMSO (0.4% of total volume) using the l-DOT non-contact liquid handler (Dispendix). The plate was centrifuged for 30 s at 300 x g and incubated at 37°C and 5% CO2.
For live-cell imaging, the plate was imaged every 4 hours in the Incucyte S3 (Sartorius). The confluency was analyzed using “Al confluence” as segmentation method. Data was normalized to the first scan at time 0 and displayed as ratio.
The endpoint was measured after 64-70 hours by adding 30 pL of Cell-Titer Gio 2.0 reagent (Promega) to each cell-containing well. The plate was shaken for 2 min at 500 rpm and then incubated for 10 min at 22°C. Luminescence was recorded using the SpectraMax i3x Multimode microplate reader (Molecular Devices). Data was normalized by using the negative control (= DMSO) as 0, and the positive control (= 100 ng/ mL FGF2) as 1. For plate-to-plate comparison, the z’ factor was calculated as z’ = 1 - ((3* (SDpos - SDNeg))/ (Pos-Neg)), and the signal to noise ratio was determined as the ratio of the positive and negative control. Graphs were generated using Prism (GraphPad).
The results in Fig. 1 and Table 2 show correlation between ATP quantification and imaging data. A proliferative effect up to 70% of FGF2 activity was observed for 1-1.
Table 2 Cell-Titer Gio dose-response in NIH/ 3T3 cells upon treatment of compound 1-1 in a table format. Detailed description can be found in graph.


Example 3 - Proliferation assay with comparative compounds
As the compound 1-1 carried four copies of aminoguanidine groups, it was analyzed if an aminoguanidine is sufficient to cause the effect of increasing proliferation of cells in cell culture.
The experiment was carried out as described in Example 2, with aminoguanidine instead of the compounds according to the invention.
As shown in Fig. 2 with aminoguanidine (AGH), no increase in proliferation of the NIH/ 3t3 cells could be detected.
In a next step, it was analyzed if only one side of the formula (I), i.e. the tripod moiety Ci with two active moieties Di and D2 would be sufficient to cause the effect of increasing proliferation of cells in cell culture.
The experiment was carried out as described in Example 2, with the compound 11-1 instead of the compounds according to the invention. The compound 11-1 has the following structure:

As shown in Fig. 2, with compound 11-1 , no increase in proliferation of the NIH/ 3t3 cells could be detected.
Example 4 - Proliferation assay with further compounds of formula (I)
In this example, several compounds were tested using the proliferation assay as described in Example 2.
Table 4 shows all compounds that enhance proliferation with at least 5% of total FGF2 activity. The following classification was used for the EC50: A < 1 pM; B > 1 and < 1.4 pM; C > 1.4 and < 4.5 pM; D > 4.5 and < 10 pM; and for the FGF2 activity: 1 > 50%; 2 < 50 and > 40%; 3 < 40 and > 30%; 4 < 30 and > 5%.
A curve-fitting analysis is shown in Table 5 and was conducted for the compounds with the lowest EC50 and highest % of FGF2 activity that reached saturation. Prism was used to fit the curve using the equation: Y= Bottom + (XAHillslope)*(Top-Bottom)/ (XAHillslope + EC50AHillslope).
Fig. 3 depicts a graphical example of the curve-fitting analysis. It shows that the optimization of 1-1 led to an approx. 4.5-fold increase in EC50 as observed for compound I-53.
Table 4: Proliferation data in NIH/ 3t3 cells induced by compounds of the invention.



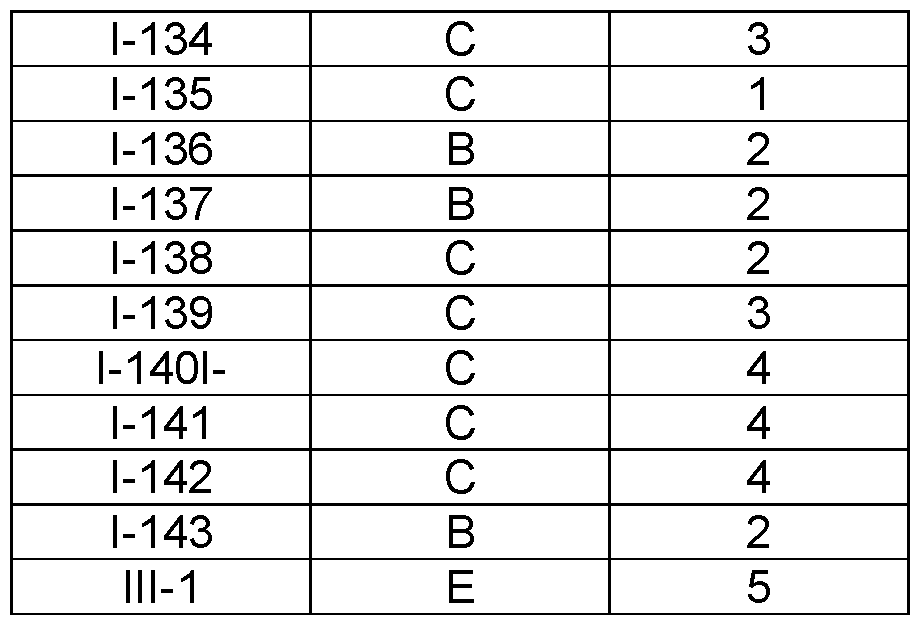
Table 5: Curve-fitting analysis of proliferation data of some of the 10 best performing 1-1 analogues.
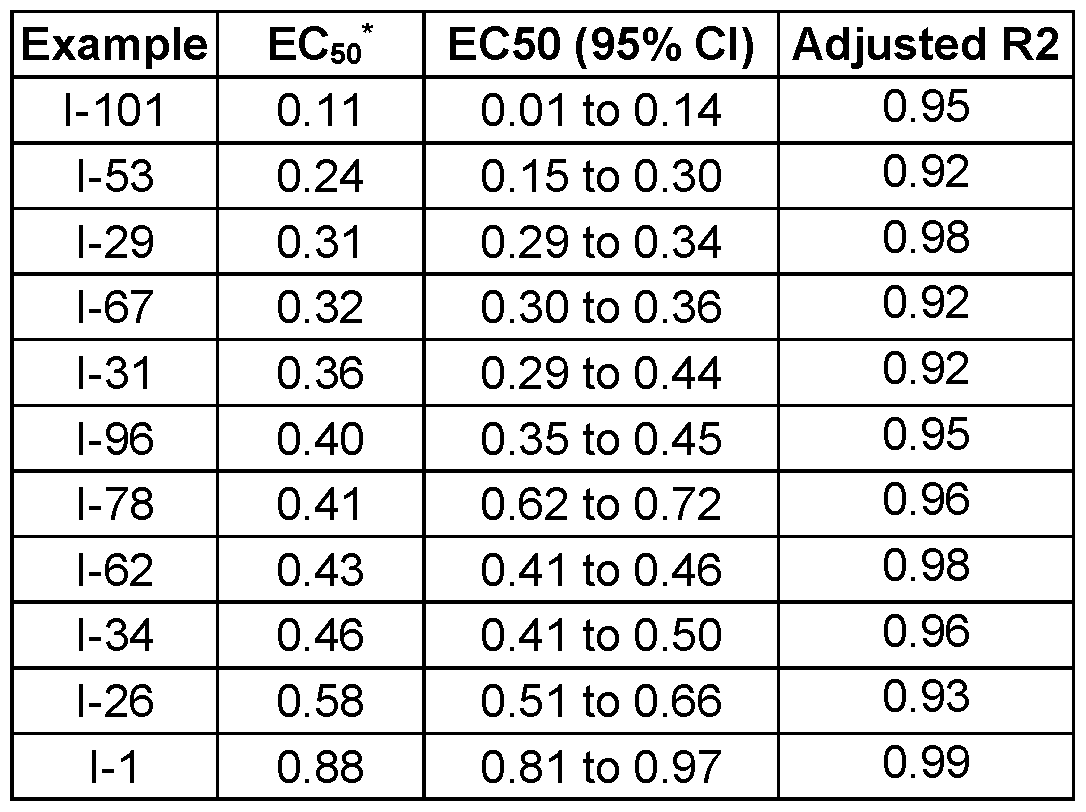
Example 5 - Luciferase Reporter Assay
The pGL4.33 [luc2P/ Hygro] vector contains a Serum Response Element (SRE) that drives transcription of the luciferase gene luc2P in response to activation of the MAPK/ ERK signaling pathway. This constitutes one of the pathways activated by FGFR1 and is thus a readout of FGFR1-activating substances.
HEK293T cells (ATCC, #CRL-316) were grown in standard growth medium supplemented with 10% Fetal Bovine Serum (FBS, Gibco) in a T75-flask. At 80-90% confluency, medium was aspirated, cells were washed with 10 mL PBS and incubated with 2 mL TrypLE Express. Cells were resuspended in 8 mL standard growth medium with 10% FBS, counted with Acridine Orange/ Propidium iodide stain using the LUNA-FL Dual Fluorescence Cell Counter, and diluted to 100.000 cells/ mL.
The pGL4.33 [luc2P/ SRE/ Hygro] vector (Promega) was used as reporter gene and added to cells by transient transfection. It contains a Serum Response Element (SRE) that drives transcription of the luciferase gene luc2P in response to activation of the MAPK/ ERK signaling pathway. For the reverse transfection mix, two vials containing per well (a) 100 ng of the vector and 0.2 pl P3000 (Invitrogen) in 5 pl OptiMEM, and (b) 0.2 pl Lipofectamine 3000 (Invitrogen) in 5 pl OptiMEM, were carefully mixed and incubated for 10 min. This transfection mix was then added to HEK293T cells diluted to 100.000 cells/ mL and carefully mixed by inversion.
A collagen-coated white 96-well plate (Greiner/ #655095) was prepared by dispensing 100 pL DMEM + 10% FBS to wells B2 to G11 , and 100 pL DMEM to the remaining outer wells. 100 pL of the cell and transfection mix suspension was dispensed to wells B2 to G11 of the previously prepared 96-well plate. After an attachment period of 6 hours, the media was removed and exchanged to 99.5 pl of a freshly prepared starvation media containing DMEM+ 0.5% FBS.
After incubation overnight, 1-1 or FGFR1 inhibitor PD166866 (MCE, #HY-101296) were added to the wells in varying concentrations and a maximum total volume of 0.5 pl DMSO using the l-DOT non-contact liquid handler.
After 6 h incubation at 37°C with 5% CO2, 100 pl ONE-Glo™ EX Luciferase Assay Substrate (Promega) was added, mixed using rocking motion, and incubated for 20 min at 22°C. Luminescence was recorded using the SpectraMax i3x Multimode microplate reader. Graphs were generated using Prism.
The results are shown in Fig. 4. Upon treatment with compound 1-1 alone, an increase in luminescence was observed at compound concentrations higher than 3 pM. When treated with 1-1 and PD166866 - a highly selective inhibitor of the FGFR1 tyrosine kinase domain - a signal decrease was observed. This indicates that 1-1 works partly through the FGFR1 pathway.
Example 6 - FGFR1 dependency - Proliferation assay with PD16686
PD166866 is a highly specific FGFR1 inhibitor that inhibits autophosphorylation of the intracellular tyrosine kinase. It is a tool to show FGFR1 -dependency of a growth factor or a small molecule acting upstream of FGFR1 kinase activity, e.g., ligand FGF2 binding to the extracellular FGFR1 domain.
TGF-P1 (PeproTech, #100-21), EGF (PeproTech, #100-15) and FGF2-G3 (Defined Bioscience, #LSR-101-10-L) were resuspended in PBS and 5% Trehalose to a concentration of 100 pg/ mL. 1-1 and PD166866 (MCE, #HY-101296) were prepared at 10 mM in DMSO. First, PD166866 was added to 450 N I HZ 3t3 cells in a 384-well plate at various concentrations using the l-DOT non-contact liquid handler and incubated for 1 hour. Then the various growth factors or 1-1 were added to the cells in their respective concentrations. The proliferation assay was conducted as described in Example 2.
The results are shown in Fig. 5. Treatment of 40 ng/ mL FGF2 and 1 pM 1-1 with increasing concentrations of FGFR1 inhibitor PD166866 led to a loss of proliferative effect (IC50 of approx. 0.16 and 0.04 pM). In contrast, PD166866 did not change the proliferative effect observed for 100 ng/ mL EGF and 2 ng/ mL TGF-pi , underlining the specificity of the observed effect.
Example 7 - FGFR1 signalling pathway
Several pathways are dependent on FGFR1 activation. To delineate the signalling pathways activated by 1-1 through FGFR1 , several key components downstream of the FGFR1 signalling pathway were inhibited: FGFR1 by PD166866, ERK1/2 by SCH772984, AKT by AZD5363 and PKC by Bisindolylmaleimide 1.
FGF2-G3 (Defined Bioscience, #LSR-101-10-L) was resuspended in PBS and 5% Trehalose to a concentration of 100 pg/ mL. 1-1 , PD166866 (MCE, #HY-101296), SCH772984 (Cayman, #199116), AZD5363 (Cayman, #Cay15406) and Bisindolylmaleimide 1 (Sigma, #202390) were prepared at 10 mM in DMSO. First, the various inhibitors were added to 450 NIH/ 3t3 cells/ well in a 384-well plate using the l-DOT non-contact liquid handler. After incubation for 1 hour, 1.4 pM 1-1 or 100 ng/ mL FGF2-G3 were added to the cells and the proliferation assay was conducted as described above in Example 2.
The results are shown in Fig. 6. Proliferative activity of (A) 1.4 pM 1-1 and (B) 100 ng/ mL FGF2 was inhibited by FGFR1 -specific inhibitor PD166866 and ERK1/2-specific inhibitor SCH772984. Inhibitors against AKT and PKC showed a significant effect only at 3 pM and are thus not the primary pathway activated by 1-1. This shows that 1-1 activates proliferation through FGFR1 and its downstream target ERK 1/2.
Example 8 - FGFR1 competition
To test if recombinant FGFR1 competes with cellular FGFR1 for 1-1 binding, first the recombinant protein was produced and then a cellular competition assay performed in NIH/ 3t3 cells.
The extracellular domain of porcine FGFR1 (aa 1-374) - tagged C-terminally with the Fc domain of human lgG1 and followed by a non-biotinylated Avi-Tag - was expressed using Gibco’s Expi293™ expression system according to the Expi293™ user guide. The supernatant was further purified by Protein A chromatography using a HiTrap® MabSelect Sure column (Cytiva) on an Akta Pure™ 25. The protein was eluted with elution buffer (20 mM MES, 3.6 M MgCI2, pH= 6.6) and buffer exchanged into TBS buffer (pH= 8.0) with either a Vivaspin20 10 kDa concentrator (Sartorius) or an Amicon® Ultra-15 10 kDa concentrator (Merck). A second buffer exchange into the final buffer (PBS, 10% Glycerol, pH= 7.4) was performed with an Amicon® Ultra-15 10 kDa concentrator (Merck). The concentration of the protein was determined by UVA/is spectrometry and its quality checked by reducing and nonreducing SDS-PAGE. The protein was aliquoted, flash frozen in liquid nitrogen and stored at -80°C.
The protein was then added to 400 NIH/ 3t3 cells/ well in a 384-well plate using the l-DOT non-contact liquid handler. As negative control, the Fc domain of human lgG1 (ACROBiosystems, #FCC-H5214) was used. After incubation for 30 minutes, 30 ng/ mL FGF2 and 1.4 pM 1-1 were added, and the proliferation assay was conducted as described above in Example 2.
The results of the ccompetition assay between recombinant FGFR1-Fc and cellular FGFR1 for FGF2 or 1-1 are shown in Fig. 7. Recombinant FGFR1-Fc inhibited the proliferative effect of (A) 1 .4 pM 1-1 and (B) 30 ng/ mL FGF2, while Fc alone did not have any effect. This shows that the presence of recombinant FGFR1 inhibits the function of 1-1 on cellular FGFR1 similar to FGF2.
Example 9 - Stability assay
The stability of 1-1 and FGF2 wildtype was assessed by incubating the samples in the incubator at different timepoints and then measuring the effect on proliferative activity.
1-1 was resuspended in DMSO to 10 mM, and porcine/ bovine FGF2 wildtype (Qkine, #Qk040) was resuspended in PBS and 5% Trehalose to 100 pg/ mL according to the manufacturer’s advice. Both reagents were then aliquoted and stored at -80°C. At designated time points, samples were taken out from the freezer, thawed, sealed with Parafilm, and transferred to an incubator at 37°C and 5% CO2. At the end of the incubation time, the proliferation assay was conducted as described in Example 2.
The results in Fig. 8 show that (A) 1-1 was stable over the course of 14 days in the cell culture incubator at all concentrations tested. In contrast (B) FGF2 wildtype showed decreased stability over the course of 14 days in the cell culture incubator. This underlines that 1-1 is more stable than FGF2 at 37°C and 5% CO2.
Example 10 LDH-Glo ™ Cytotoxicity assay
Lactate dehydrogenase (LDH) is an abundant cytosolic protein that is released upon disruption of the cell membrane. This established LDH as a widely used marker for cytotoxicity and is used here to assess the toxicity of 1-1.
HT-1080 cells (ATCC, #CCL-121) were grown in standard growth medium supplemented with 10% FBS in a T75-flask. At 80-90% confluency, medium was aspirated, cells were washed with 10 mL PBS and detached with 2 mL TrypLE Express. Cells were resuspended in 8 mL of standard growth medium and counted with Acridine Orange/ Propidium iodide stain using the LUNA-FL Dual Fluorescence Cell Counter. Cells were diluted to 24.000 cells/ mL in standard growth medium with 5% FBS. 50 pL of the cell dilution was dispensed to wells B2 to 023 of a white 384-well plate (Greiner/ #781983_32) using a 24-channel pipette. The outer wells were filled with 50 pL PBS. The plate was centrifuged for 30 s at 300 x g and incubated at 37°C and 5% CO2.
After 2-3 hours, 1-1 was added to the well in varying concentrations and a maximum total volume of 0.2 pL (0.4% of total volume) DMSO using the l-DOT non-contact liquid handler. The plate was centrifuged for 30 s at 300 x g and incubated at 37°C and 5% CO2.
Reagents of the LDH-Glo™ Cytotoxicity assay (Promega) were prepared according to the manufacturer’s advice. The LDH reaction reagent was prepared by mixing LDH Detection Enzyme Mix and Reductase Substrate at a ratio of 200 to 1. The maximum LDH release sample was prepared by adding 1 pL of Triton X-100 to 50 pL of Vehicle-Only Cells for 15 minutes before collecting the samples.
After 24 hours, 2 pL of the cell culture medium was diluted in 8 pL of LDH Storage Buffer (200 mM Tris-HCI (pH 7.3), 10% Glycerol, 15% BSA) in a white 384-well plate. 10 pL of the LDH reaction reagent was then added to the samples and incubated for 60 minutes at room temperature. Luminescence was recorded using the SpectraMax i3x Multimode microplate reader. Viability of the cells was determined using CellTiter® Gio reagent as described before. Graphs were generated using Prism. Cytotoxicity was calculated as: Cytotoxicity = (Experimental LDH Release - Medium Background)/ (Maximum LDH Release Control - Medium Background). Viability was calculated as: Viability = Treated sample/ DMSO Control. The standard deviation is based on 3 replicates.
The results in Figure 9 show that the compound is not toxic at concentrations < 10 pM in HT- 1080 cells as measured by LDH release.
Many modifications and other embodiments of the invention set forth herein will come to mind to the one skilled in the art to which the invention pertains having the benefit of the teachings presented in the foregoing description and the associated drawings. Therefore, it is to be understood that the invention is not to be limited to the specific embodiments disclosed and that modifications and other embodiments are intended to be included within the scope of the appended claims. Although specific terms are employed herein, they are used in a generic and descriptive sense only and not for purposes of limitation.
REFERENCES
Belov AA, Mohammadi M (June 2013). "Molecular mechanisms of fibroblast growth factor signaling in physiology and pathology". Cold Spring Harbor Perspectives in Biology. 5 (6): a015958. doi :10.1101 /cshperspect.aOl 5958. PMC 3660835. PM ID 23732477.
Duchesne L, Tissot B, Rudd TR, Dell A, Fernig DG (September 2006). "N-glycosylation of fibroblast growth factor receptor 1 regulates ligand and heparan sulfate co-receptor binding". The Journal of Biological Chemistry. 281 (37): 27178-89. doi:10.1074/jbc.M601248200. PMID 16829530.
Bono F, De Smet F, Herbert C, De Bock K, Georgiadou M, Fons P, Tjwa M, Alcouffe C, Ny A, Bianciotto M, Jonckx B, Murakami M, Lanahan AA, Michielsen C, Sibrac D, Dol-Gleizes F, Mazzone M, Zacchigna S, Herault JP, Fischer C, Rigon P, Ruiz de Almodovar C, Claes F, Blanc I, Poesen K, Zhang J, Segura I, Gueguen G, Bordes MF, Lambrechts D, Broussy R, van de Wouwer M, Michaux C, Shimada T, Jean I, Blacher S, Noel A, Motte P, Rom E, Rakic JM, Katsuma S, Schaeffer P, Yayon A, Van Schepdael A, Schwalbe H, Gervasio FL, Carmeliet G, Rozensky J, Dewerchin M, Simons M, Christopoulos A, Herbert JM, Carmeliet P. Inhibition of tumor angiogenesis and growth by a small-molecule multi-FGF receptor
blocker with allosteric properties. Cancer Cell. 2013 Apr 15;23(4):477-88. doi: 10.1016/j.ccr.2O13.02.019. PMID: 23597562.
Coutts JC, Gallagher JT (December 1995). "Receptors for fibroblast growth factors". Immunology and Cell Biology. 73 (6): 584-9. doi: 10.1038/icb.1995.92. PMID 8713482.
Davidson D, Blanc A, Filion D, Wang H, Plut P, Pfeffer G, et al. (May 2005). "Fibroblast growth factor (FGF) 18 signals through FGF receptor 3 to promote chondrogenesis". The Journal of Biological Chemistry. 280 (21): 20509-15. doi: 10.1074/jbc.M410148200. PMID 15781473.
Herbert C, Schieborr II, Saxena K, Juraszek J, De Smet F, Alcouffe C, Bianciotto M, Saladino G, Sibrac D, Kudlinzki D, Sreeramulu S, Brown A, Rigon P, Herault JP, Lassalle G, Blundell TL, Rousseau F, Gils A, Schymkowitz J, Tompa P, Herbert JM, Carmeliet P, Gervasio FL, Schwalbe H, Bono F. Molecular mechanism of SSR128129E, an extracellularly acting, smallmolecule, allosteric inhibitor of FGF receptor signaling. Cancer Cell. 2013 Apr 15;23(4):489- 501. doi: 10.1016/j.ccr.2013.02.018. Erratum in: Cancer Cell. 2016 Jul 11 ;30(1): 176-178. PMID: 23597563.
Jin S, Sun Y, Liang X, Gu X, Ning J, Xu Y, Chen S, Pan L. Emerging new therapeutic antibody derivatives for cancer treatment. Signal Transduct Target Ther. 2022 Feb 7;7(1):39. doi: 10.1038/S41392-021-00868-x. PMID: 35132063; PMCID: PMC8821599.
Kalinina J, Dutta K, llghari D, Beenken A, Goetz R, Eliseenkova AV, et al. (January 2012). "The alternatively spliced acid box region plays a key role in FGF receptor autoinhibition". Structure. 20 (1): 77-88. doi: 10.1016/j.str.2011.10.022. PMC 3378326. PMID 22244757.
Koledova Z, Sumbal J, Rabata A, de La Bourdonnaye G, Chaloupkova R, Hrdlickova B, Damborsky J, Stepankova V. Fibroblast Growth Factor 2 Protein Stability Provides Decreased Dependence on Heparin for Induction of FGFR Signaling and Alters ERK Signaling Dynamics. Front Cell Dev Biol. 2019 Dec 12;7:331. doi: 10.3389/fcell.2019.00331. PMID: 31921844; PMCID: PMC6924264.
Lefevre G, Babchia N, Calipel A, et al. Activation of the FGF2/FGFR1 autocrine loop for cell proliferation and survival in uveal melanoma cells. Investigative Ophthalmology & Visual Science. 2009 Mar;50(3): 1047-1057. DOI: 10.1167/iovs.08-2378. PMID: 19029025.
Nawrocka D, Krzyscik MA, Opalihski L, Zakrzewska M, Otlewski J. Stable Fibroblast Growth Factor 2 Dimers with High Pro-Survival and Mitogenic Potential. Int J Mol Sci. 2020 Jun 9;21 (11):4108. doi: 10.3390/ijms21114108. PMID: 32526859; PMCID: PMC7312490.
Ornitz DM, Itoh N (2001). "Fibroblast growth factors". Genome Biology. 2 (3): REVIEWS3005. doi:10.1186/gb-2001-2-3-reviews3005. PMC 138918. PMID 11276432.
Ornitz DM, Xu J, Colvin JS, McEwen DG, MacArthur CA, Coulier F, et al. (June 1996). "Receptor specificity of the fibroblast growth factor family". The Journal of Biological Chemistry. 271 (25): 15292-7. doi:10.1074/jbc.271.25.15292. PMID 8663044.
Pagano K, Carminati L, Tomaselli S, Molinari H, Taraboletti G, Ragona L. Molecular Basis of the Antiangiogenic Action of Rosmarinic Acid, a Natural Compound Targeting Fibroblast Growth Factor-2/FGFR Interactions. Chembiochem. 2021 Jan 5;22(1):160-169. doi: 10.1002/cbic.202000610. Epub 2020 Nov 6. PM ID: 32975328.
Sleeman M, Fraser J, McDonald M, Yuan S, White D, Grandison P, et al. (June 2001). "Identification of a new fibroblast growth factor receptor, FGFR5". Gene. 271 (2): 171-82. doi:10.1016/S0378-1119(01)00518-2. PMID 11418238.
Sarabipour S, Hristova K. Mechanism of FGF receptor dimerization and activation. Nat Commun. 2016 Jan 4;7:10262. doi: 10.1038/ncomms10262. PMID: 26725515; PMCID: PMC4725768.
Schroder, M., Kolodzik, A., Windshugel, B., Krepstakies, M., Priyadarshini, P., Hartjen, P., van Lunzen, J., Rarey, M., Hauber, J. and Meier, C. (2016), Linker-Region Modified Derivatives of the Deoxyhypusine Synthase Inhibitor CNI-1493 Suppress HIV-1 Replication. Arch. Pharm. Chem. Life Sci., 349: 91-103.
Xie Y, Su N, Yang J, Tan Q, Huang S, Jin M, Ni Z, Zhang B, Zhang D, Luo F, Chen H, Sun X, Feng JQ, Qi H, Chen L. FGF/FGFR signaling in health and disease. Signal Transduct Target Ther. 2020 Sep 2;5(1):181. doi: 10.1038/s41392-020-00222-7. PMID: 32879300; PMCID: PMC7468161.
Claims
1. Use of at least one compound or a pharmaceutically acceptable salt thereof for culturing cells in vitro, wherein the compound has the general formula (I):
 wherein:
wherein:
• the core moiety A is selected from o an optionally substituted Cs-is aryl, with 1 to 3 optionally fused rings; wherein 0 to 4 C atoms are replaced by heteroatoms, wherein the heteroatoms are individually selected from N, O, and S; o an optionally substituted Ce-is arylalkyl group wherein 0 to 4 C atoms are replaced by heteroatoms, wherein the heteroatoms are individually selected from N, O, and S, o an optionally substituted C3-12 cycloalkyl group, with 1 to 6 rings optionally fused, bridged or strained, wherein 0 to 4 C atoms are replaced by a heteroatoms, wherein the heteroatoms are individually selected from N, O, S and Si; o an optionally substituted, straight or branched, saturated or unsaturated C2-20 aliphatic group, wherein 0 to 4 C atoms are replaced by heteroatoms, wherein the heteroatoms are individually selected from N, O, S, and Si; o an aromatic or aliphatic crown ether; o or a bond; wherein A may contain one to three substituents, which are independently selected from the group consisting of halogen, amide, amine, nitro, cyano, hydroxyl or hydrocarbyloxy, or aldehyde, ketone, carboxyl, ether, ester, alkyl, alkenyl, alkynyl, sulfonyl, sulfonylamide;
• the linkers Bi and B2 are individually selected from an ester, an ether, an amine, a thioether, an amide; and a sulfonamide; wherein one of Bi and B2 may be absent and provided that A is not a bond both Bi and B2 may be absent;
• wherein the tripod moieties Ci and C2 are individually selected from: o an optionally substituted C5-6 aryl, wherein 0 to 3 C atoms are replaced by heteroatoms, wherein the heteroatoms are individually selected from N, O, and S;
o an optionally substituted C3-6 cycloalkyl group, wherein 0 to 3 C atoms are replaced by heteroatoms, wherein the heteroatoms are individually selected from N, O, and S;
• wherein Ci and C2 may contain one to three substituents, which are independently selected from the group consisting of hydrogen, alkyl and alkoxy,
• the active moieties Di, D2, D3 and D4 are independently selected from hydrogen or
 with R1 being H or CH3; wherein Di and D2 may not both be hydrogen and D3 and D4 may not both be hydrogen.
with R1 being H or CH3; wherein Di and D2 may not both be hydrogen and D3 and D4 may not both be hydrogen.
2. The use according to claim 1 , wherein
• the core moiety A is selected from o an optionally substituted C5-6 aryl wherein 0 to 3 C atoms are replaced by heteroatoms, wherein the heteroatoms are individually selected from N, O, and S; o an optionally substituted C3-6 cycloalkyl, optionally bridged or strained, wherein 0 to 1 C atoms are replaced by a heteroatoms, wherein the heteroatoms are individually selected from N, O, and S; o an optionally substituted, straight or branched, saturated or unsaturated C2-12 aliphatic group, wherein 0 to 4 C atoms are replaced by heteroatoms, wherein the heteroatoms are individually selected from N, O, and S; o or an aromatic crown ether; o wherein A may contain one to three substituents, which are independently selected from the group consisting of hydrogen, hydroxyl, methyl and methoxy, and an amine;
• preferably the core moiety A is selected from:

X is selected from N, O, and S
R2 is H or C1-6 alkyl;
R3 is selected from O, S, NR™, wherein Rw is H or C1-6 alkyl;
NR5R6 is selected from:

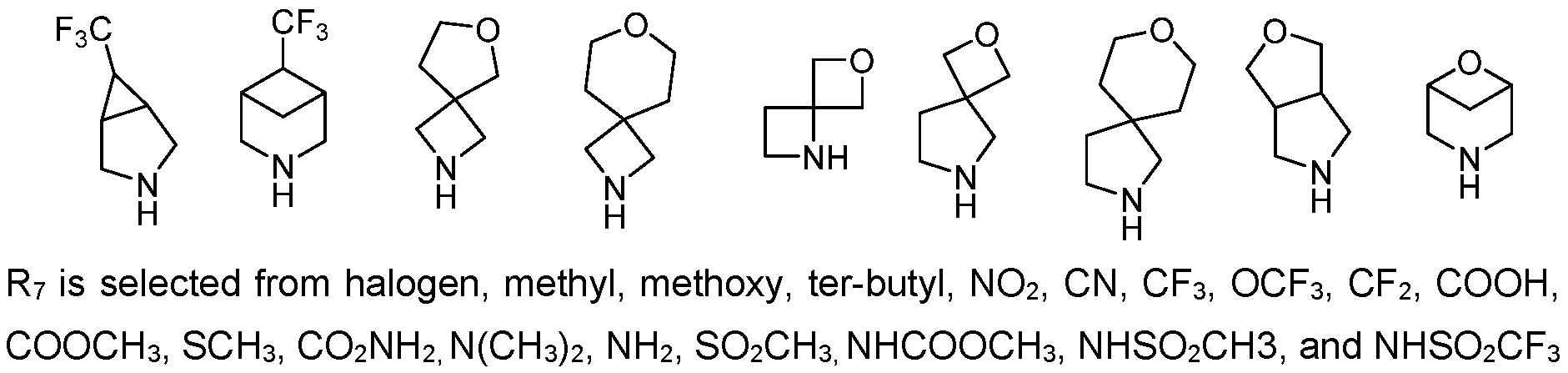 and CONR11R12 with Rn and R12 being independently selected from H and C1-6 alkyl;
and CONR11R12 with Rn and R12 being independently selected from H and C1-6 alkyl;
Rs is selected from H and C1-6 alkyl;
R9 is selected from methyl, Cs-Ce cycloalkyl, piperidinyl or hydroxyisopropyl; and
Xi is selected from oxygen or methylene.
3. The use according to claim 1 or 2, wherein the tripod moieties Ci and C2 are independently selected from benzene, pyridine, pyrimidine, substituted pyrrole, and piperidine; preferably the moieties Ci and C2 are individually selected from:

4. The use according to any one of claims 1 to 3, wherein the active moieties Di , D2, D3 and D4 are
 wherein Di and D2 may not both be hydrogen and D3 and D4 may not both be hydrogen; preferably, the active moieties Di, D2, D3 and D4 are
wherein Di and D2 may not both be hydrogen and D3 and D4 may not both be hydrogen; preferably, the active moieties Di, D2, D3 and D4 are
5. The use according to any one of claims 1 to 4, wherein the linkers Bi and B2 are selected from:

6. The use according to any one of claims 1 to 5, wherein
• the moieties Ci and C2 are identical; and/or
• the linkers Bi and B2 are identical.
7. The use according to any one of claims 1 to 6, wherein the compound of formula (I) is selected from 1-1 , I-2, I-3, I-4, I-5, I-6, I-7, I-8, I-9, 1-10, 1-11 , 1-15, 1-16, 1-17, 1-18, 1-19, I-20 I- 22, I-23, I-24, I-25, I-26, I-27, I-28, I-29, I-30, 1-31 , I-32, I-33, I-34, I-35, I-36, I-37, I-38, I-39, I-
40, 1-41 , I-42, I-43, I-44, I-45, I-46, I-47, I-48, I-49, I-50, 1-51 , I-52, I-53, I-54, I-55, I-56, I-57, I-
58, I-59, I-60, 1-61 , I-62, I-63, I-64, I-65, I-66, I-67, I-68, I-69, I-70, 1-71 , I-72, I-73, I-74, I-75, I-
76, I-77, I-78, I-79, I-80, 1-81 , I-82, I-83, I-84, I-85, I-86, I-87, I-88, I-89, I-90, 1-91 , I-92, I-93, I-
94, I-95, I-96, I-97, I-98, I-99, 1-100, 1-101 , 1-102, 1-103, 1-104, 1-105, 1-106, 1-107, 1-108, 1-109, 1-110, 1-111 , 1-112, 1-113, 1-115, 1-116, 1-117, 1-118, 1-119, 1-120, 1-121 , 1-122, 1-123, 1-124, I- 125, 1-126, 1-127, 1-128, 1-129, 1-130, 1-131 , 1-132, 1-133, 1-134, 1-135, 1-136, 1-137, 1-138, I- 139, 1-140, 1-141 , 1-142 and 1-143.
8. The use according to any of claims 1 to 7, wherein the compound of formula (I) activates the FGFR1 signaling pathway.
9. The use according to any of claims 1 to 8, wherein the compound of formula (I) increases the proliferation of said cells; preferably, wherein the use is included in one or more of:
(a) a method of producing cultivated meat,
(b) maintenance, proliferation and differentiation of animal cells,
(c) a method of producing biopharmaceuticals, and
(d) a method of differentiating cells, such as stem cells or muscle cells or fiber cells or fat cells.
10. The use according to claim 9, wherein the compound of formula (I) is present in a concentration of 0.05 to 10 pM, preferably in a concentration of 0.1 to 5 pM, more preferably, in a concentration of 0.2 to 3 pM.
11. A compound of formula (I) as defined in any of claims 1 to 10, provided that the compound is not 1-1 , I-8, 1-16, 1-17, I-75, I-76, 1-116 or 1-117.
12. A compound of formula (I) as defined in any of claims 1 to 10, or a pharmaceutically acceptable salt thereof for use in medical treatment, wherein the medical treatment is a method of wound healing, a method of organ regeneration in transplantation medicine, a method of treating a burned lesion, a disease associated with disturbed cell renewal, or a disease associated with muscle wasting, wherein the treatment or prevention may optionally comprise tissue engineering.
13. A cell culture medium comprising at least one compound of formula (I) or a pharmaceutically acceptable salt thereof as defined in any one of claims 1 to 10, preferably wherein the cell culture medium comprises or consists of:
(A) water,
(B) nutrients sufficient for cell growth, including at least one carbon source and one nitrogen source,
(C) at least one compound of formula (I) or a pharmaceutically acceptable salt thereof, preferably in a concentration of 0.05 to 10 pM, preferably in a concentration of 0.1 to 5 pM, more preferably, in a concentration of 0.2 to 3 pM, and
(D) one or more salts acceptable in cell culture,
(E) optionally one or more buffer agents,
(F) optionally one or more gel-forming materials, and
(G) optionally one or more colorings acceptable in cell culture, preferably wherein the cell culture medium is a liquid cell culture medium.
14. The cell culture medium according to claim 13, wherein the cell culture medium does not contain one or more of the following: serum of animal origin, FGF2, any subtype of FGF of animal origin, in particular any subtypes of FGF, any peptide growth factor of animal origin, in particular any peptide growth factor or steroid growth factor of animal origin, in particular any steroid growth factors.
15. The cell culture medium according to claim 13 or 14, for use in one or more of:
(a) a method of producing cultivated meat;
(b) maintaining, proliferation and differentiation of animal cells;
(c) a method of producing biopharmaceuticals; and
(d) a method of differentiating cells, such as stem cells or muscle cells or fiber cells or fat cells.
16. A method for the cultivation of living cells, comprising the steps of
(i) providing:
(A) a cell culture medium; and
(B) living cells comprising FGFR1 , which activity is activated by a compound (I) as defined in any one of claims 1 to 10;
(ii) adding a compound of formula (I) or a pharmaceutically acceptable salt thereof as defined in any one of claims 1 to 10 to the cell culture medium;
(iii) subjecting the cell culture medium of step (ii) to conditions sufficient for establishing cell growth and/or for cell differentiation; and
(iv) obtaining cultivated cells, and
(v) optionally separating the cultivated cells from the cell culture medium,
(vi) preparing a food product comprising the obtained cultivated cells or material derived therefrom.
17. A food product comprising cultivated cells obtainable by the process according to claim 16 or material derived therefrom, preferably wherein the food product is cultivated meat or a drinkable composition.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP23193402 | 2023-08-25 | ||
| EP23193402.7 | 2023-08-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025045826A1 true WO2025045826A1 (en) | 2025-03-06 |
Family
ID=87845506
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2024/073830 Pending WO2025045826A1 (en) | 2023-08-25 | 2024-08-26 | Compounds for activation of fgfr1 signaling |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025045826A1 (en) |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5599984A (en) | 1994-01-21 | 1997-02-04 | The Picower Institute For Medical Research | Guanylhydrazones and their use to treat inflammatory conditions |
| WO2006023586A2 (en) * | 2004-08-17 | 2006-03-02 | Cytokine Pharmasciences, Inc. | Guanylhydrazone compounds, compositions, methods of making and using |
| EP2799537A1 (en) * | 2011-12-28 | 2014-11-05 | Kyoto Prefectural Public University Corporation | Normalization of culture of corneal endothelial cells |
| US9120819B2 (en) | 2006-01-13 | 2015-09-01 | Sanofi | FGF-receptor agonist dimeric compounds |
| US20220364060A1 (en) * | 2021-05-13 | 2022-11-17 | Washington University | Enhanced methods for inducing and maintaining naive human pluripotent stem cells |
-
2024
- 2024-08-26 WO PCT/EP2024/073830 patent/WO2025045826A1/en active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5599984A (en) | 1994-01-21 | 1997-02-04 | The Picower Institute For Medical Research | Guanylhydrazones and their use to treat inflammatory conditions |
| US5750573A (en) | 1994-01-21 | 1998-05-12 | The Picower Institute For Medical Research | Guanylhydrazones and their use to treat inflammatory conditions |
| WO2006023586A2 (en) * | 2004-08-17 | 2006-03-02 | Cytokine Pharmasciences, Inc. | Guanylhydrazone compounds, compositions, methods of making and using |
| US9120819B2 (en) | 2006-01-13 | 2015-09-01 | Sanofi | FGF-receptor agonist dimeric compounds |
| EP2799537A1 (en) * | 2011-12-28 | 2014-11-05 | Kyoto Prefectural Public University Corporation | Normalization of culture of corneal endothelial cells |
| US20220364060A1 (en) * | 2021-05-13 | 2022-11-17 | Washington University | Enhanced methods for inducing and maintaining naive human pluripotent stem cells |
Non-Patent Citations (22)
| Title |
|---|
| BELOV AAMOHAMMADI M: "Molecular mechanisms of fibroblast growth factor signaling in physiology and pathology", COLD SPRING HARBOR PERSPECTIVES IN BIOLOGY, vol. 5, no. 6, June 2013 (2013-06-01), pages a015958 |
| BONO FDE SMET FHERBERT CDE BOCK KGEORGIADOU MFONS PTJWA MALCOUFFE CNY ABIANCIOTTO M: "Inhibition of tumor angiogenesis and growth by a small-molecule multi-FGF receptor blocker with allosteric properties", CANCER CELL, vol. 23, no. 4, 15 April 2013 (2013-04-15), pages 477 - 88, XP028578782, DOI: 10.1016/j.ccr.2013.02.019 |
| COUTTS JCGALLAGHER JT: "Receptors for fibroblast growth factors", IMMUNOLOGY AND CELL BIOLOGY., vol. 73, no. 6, December 1995 (1995-12-01), pages 584 - 9 |
| D'ARPA PETER ET AL: "Pharmaceutical Prophylaxis of Scarring with Emphasis on Burns: A Review of Preclinical and Clinical Studies", vol. 11, no. 8, 1 August 2022 (2022-08-01), pages 428 - 442, XP093088819, ISSN: 2162-1918, Retrieved from the Internet <URL:http://dx.doi.org/10.1089/wound.2020.1236> DOI: 10.1089/wound.2020.1236 * |
| DAVIDSON DBLANC AFILION DWANG HPLUT PPFEFFER G ET AL.: "Fibroblast growth factor (FGF) 18 signals through FGF receptor 3 to promote chondrogenesis", THE JOURNAL OF BIOLOGICAL CHEMISTRY, vol. 280, no. 21, May 2005 (2005-05-01), pages 20509 - 15, XP055682282, DOI: 10.1074/jbc.M410148200 |
| DUCHESNE LTISSOT BRUDD TRDELL AFERNIG DG: "N-glycosylation of fibroblast growth factor receptor 1 regulates ligand and heparan sulfate co-receptor binding", THE JOURNAL OF BIOLOGICAL CHEMISTRY, vol. 281, no. 37, September 2006 (2006-09-01), pages 27178 - 89, XP002429674, DOI: 10.1074/jbc.M601248200 |
| ERRATUM, CANCER CELL., vol. 30, no. 1, 11 July 2016 (2016-07-11), pages 176 - 178 |
| HERBERT CSCHIEBORR USAXENA KJURASZEK JDE SMET FALCOUFFE CBIANCIOTTO MSALADINO GSIBRAC DKUDLINZKI D: "Molecular mechanism of SSR128129E, an extracellularly acting, small-molecule, allosteric inhibitor of FGF receptor signaling", CANCER CELL, vol. 23, no. 4, 15 April 2013 (2013-04-15), pages 489 - 501, XP028578781, DOI: 10.1016/j.ccr.2013.02.018 |
| HERBERT ET AL., CANCER CELL, vol. 23, 2013, pages 489 - 501 |
| JIN SSUN YLIANG XGU XNING JXU YCHEN SPAN L: "Emerging new therapeutic antibody derivatives for cancer treatment", SIGNAL TRANSDUCT TARGET THER., vol. 7, no. 1, 7 February 2022 (2022-02-07), pages 39 |
| KALININA JDUTTA KILGHARI DBEENKEN AGOETZ RELISEENKOVA AV ET AL.: "The alternatively spliced acid box region plays a key role in FGF receptor autoinhibition", STRUCTURE, vol. 20, no. 1, January 2012 (2012-01-01), pages 77 - 88, XP028441924, DOI: 10.1016/j.str.2011.10.022 |
| KOLEDOVA ZSUMBAL JRABATA ADE LA BOURDONNAYE GCHALOUPKOVA RHRDLICKOVA BDAMBORSKY JSTEPANKOVA V: "Fibroblast Growth Factor 2 Protein Stability Provides Decreased Dependence on Heparin for Induction of FGFR Signaling and Alters ERK Signaling Dynamics", FRONT CELL DEV BIOL., vol. 7, 12 December 2019 (2019-12-12), pages 331 |
| LEFÈVRE GBABCHIA NCALIPEL A ET AL.: "Activation of the FGF2/FGFR1 autocrine loop for cell proliferation and survival in uveal melanoma cells", INVESTIGATIVE OPHTHALMOLOGY & VISUAL SCIENCE, vol. 50, no. 3, March 2009 (2009-03-01), pages 1047 - 1057 |
| NAWROCKA DKRZYSCIK MAOPALIRÍSKI LZAKRZEWSKA MOTLEWSKI J.: "Stable Fibroblast Growth Factor 2 Dimers with High Pro-Survival and Mitogenic Potential", INT J MOL SCI., vol. 21, no. 11, 9 June 2020 (2020-06-09), pages 4108 |
| ORNITZ DMITOH N: "Fibroblast growth factors", GENOME BIOLOGY., vol. 2, no. 3, 2001, XP055098812 |
| ORNITZ DMXU JCOLVIN JSMCEWEN DGMACARTHUR CACOULIER F ET AL.: "Receptor specificity of the fibroblast growth factor family", THE JOURNAL OF BIOLOGICAL CHEMISTRY, vol. 271, no. 25, June 1996 (1996-06-01), pages 15292 - 7, XP002919936, DOI: 10.1074/jbc.271.25.15292 |
| PAGANO KCARMINATI LTOMASELLI SMOLINARI HTARABOLETTI GRAGONA L.: "Molecular Basis of the Antiangiogenic Action of Rosmarinic Acid, a Natural Compound Targeting Fibroblast Growth Factor-2/FGFR Interactions", CHEMBIOCHEM, vol. 22, no. 1, 5 January 2021 (2021-01-05), pages 160 - 169 |
| SARABIPOUR SHRISTOVA K: "Mechanism of FGF receptor dimerization and activation", NAT COMMUN., vol. 7, 4 January 2016 (2016-01-04), pages 10262 |
| SCHRÖDER, M.KOLODZIK, A.WINDSHIGEL, B.KREPSTAKIES, M.PRIYADARSHINI, P.HARTJEN, P.VAN LUNZEN, J.RAREY, M.HAUBER, J.MEIER, C.: "Linker-Region Modified Derivatives of the Deoxyhypusine Synthase Inhibitor CNI-1493 Suppress HIV-1 Replication", ARCH. PHARM. CHEM. LIFE SCI., vol. 349, 2016, pages 91 - 103 |
| SLEEMAN MFRASER JMCDONALD MYUAN SWHITE DGRANDISON P ET AL.: "Identification of a new fibroblast growth factor receptor, FGFR5", GENE, vol. 271, no. 2, June 2001 (2001-06-01), pages 171 - 82, XP004246888, DOI: 10.1016/S0378-1119(01)00518-2 |
| THOMAS SORRELL: "Periodic Table of the Elements, CAS version, Handbook of Chemistry and Physics", 1999, UNIVERSITY SCIENCE BOOKS, article "Organic Chemistry" |
| XIE YSU NYANG JTAN QHUANG SJIN MNI ZZHANG BZHANG DLUO F: "FGF/FGFR signaling in health and disease", SIGNAL TRANSDUCT TARGET THER, vol. 5, no. 1, 2 September 2020 (2020-09-02), pages 181 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11897866B2 (en) | Heteroaryl inhibitors of PAD4 | |
| KR102099159B1 (en) | Acrylic acid derivatives, methods for their preparation and methods of use in medicine | |
| JP7382973B2 (en) | Pyridinyl and pyrazinyl-(aza)indole sulfonamides | |
| JP7126084B2 (en) | IRE1 small molecule inhibitor | |
| US10988465B2 (en) | CXCR4 inhibitors and uses thereof | |
| EA030001B1 (en) | Glucagon analogues | |
| Huang et al. | Induction of inactive TGF-β1 monomer formation by hydrogen sulfide contributes to its suppressive effects on Ang II-and TGF-β1-induced EMT in renal tubular epithelial cells | |
| BRPI0719650A2 (en) | AZABICYCLO-OCTANE DERIVATIVES, METHOD OF MANUFACTURING THEM AND USES OF THE SAME AS DIPEPTIDIL PEPTITY IV INHIBITORS | |
| CN116715625A (en) | Heteroaryl compounds as protein kinase inhibitors | |
| KR20190003685A (en) | Inhibitors of indoleamine 2,3-dioxygenase and methods of use thereof | |
| WO2018053373A1 (en) | Uses of satl-inducible kinase (sik) inhibitors for treating osteoporosis | |
| JP2023528637A (en) | Novel thyroid hormone beta receptor agonist | |
| RU2528333C2 (en) | Compounds and methods of treating pain and other diseases | |
| CN104072481B (en) | An anti-angiogenic compound | |
| TW200526625A (en) | Pharmaceutically active compounds | |
| JP6523267B2 (en) | Imidazole compound as regulator of FSHR and use thereof | |
| EP2382205A2 (en) | Derivatives of 2-pyridin-2-yl-pyrazol-3(2h)-one, preparation and therapeutic use thereof as hif activators | |
| WO2025045826A1 (en) | Compounds for activation of fgfr1 signaling | |
| WO2015184661A1 (en) | Compound and preparation method and use thereof | |
| CN117777042B (en) | A triazine hydroxamic acid derivative and its preparation method and application | |
| EP2640388A1 (en) | Quinolinone derivatives | |
| KR20090007352A (en) | Indole derivatives as inhibitors of soluble adenylate cyclase | |
| TW202320769A (en) | A kind of degradation agent and its application | |
| KR20090008261A (en) | Soluble adenylate cyclase inhibitor | |
| US20260028330A1 (en) | Heteroaryl carboxamide and related gpr84 antagonists and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24768495 Country of ref document: EP Kind code of ref document: A1 |