WO2025042742A1 - Compositions comprising antibodies that bind bcma and cd3 and methods of treatment - Google Patents
Compositions comprising antibodies that bind bcma and cd3 and methods of treatment Download PDFInfo
- Publication number
- WO2025042742A1 WO2025042742A1 PCT/US2024/042698 US2024042698W WO2025042742A1 WO 2025042742 A1 WO2025042742 A1 WO 2025042742A1 US 2024042698 W US2024042698 W US 2024042698W WO 2025042742 A1 WO2025042742 A1 WO 2025042742A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- pharmaceutical composition
- antibody
- seq
- dose
- region
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39591—Stabilisation, fragmentation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2809—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against the T-cell receptor (TcR)-CD3 complex
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2878—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the NGF-receptor/TNF-receptor superfamily, e.g. CD27, CD30, CD40, CD95
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/31—Immunoglobulins specific features characterized by aspects of specificity or valency multispecific
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/35—Valency
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/52—Constant or Fc region; Isotype
- C07K2317/524—CH2 domain
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/52—Constant or Fc region; Isotype
- C07K2317/526—CH3 domain
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/60—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments
- C07K2317/64—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments comprising a combination of variable region and constant region components
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/71—Decreased effector function due to an Fc-modification
Definitions
- compositions comprising antibodies that bind BCMA and CD3, as well as their use in treating disorder associated with BCMA expression (e.g. BCMA-expressing B-cell cancers, such as multiple myeloma).
- BCMA-expressing B-cell cancers such as multiple myeloma
- Alnuctamab is a bispecific antibody that binds BCMA and CD3 and is currently in clinical trials for treatment of cancer, including multiple myeloma. There is a need for formulations of alnuctamab that provide convenient dosing and enhanced stability.
- compositions comprising alnuctamab and methods of using such compositions to treat a patient having a disorder associated with BCMA expression (e.g. BCMA-expressing B-cell cancers, such as multiple myeloma).
- a disorder associated with BCMA expression e.g. BCMA-expressing B-cell cancers, such as multiple myeloma.
- Embodiment 1 A pharmaceutical composition comprising:
- Embodiment 2 The pharmaceutical composition of embodiment 1, comprising about 4.5 mg/mL to 7.5 mg/mL of the multispecific antibody.
- Embodiment 3 The pharmaceutical composition of embodiment 1, comprising about 6 mg/mL of the multispecific antibody.
- Embodiment 4 The pharmaceutical composition of embodiment 1, comprising about 22.5 mg/mL to 37.5 mg/mL of the multispecific antibody.
- Embodiment 5 The pharmaceutical composition of embodiment 1, comprising about 30 mg/mL of the multispecific antibody.
- Embodiment 6 The pharmaceutical composition of embodiment 1, comprising:
- Embodiment 7 The pharmaceutical composition of embodiment 1, comprising:
- Embodiment 8 The pharmaceutical composition of any one of embodiments 1-7, wherein the pH of the composition is from about 5.7 to about 6.3.
- Embodiment 9 The pharmaceutical composition of any one of embodiments 1-8, wherein the pH of the composition is about 6.0.
- Embodiment 10 The pharmaceutical composition of any one of embodiments 1-
- the multispecific antibody comprises: an anti-BCMA antibody, or antigen binding fragment thereof, comprising a VH region comprising a CDR1H region of SEQ ID NO:21, a CDR2H region of SEQ ID NO:22 and a CDR3H region of SEQ ID NO: 17 and a VL region comprising a CDR1L region of SEQ ID NO:27, a CDR2L region of SEQ ID NO:28 and a CDR3L region of SEQ ID NO:20; and an anti-CD3 antibody, or antigen binding fragment thereof, comprising a CDR1H region of SEQ ID NO: 1, a CDR2H region of SEQ ID NO: 2, a CDR3H region of SEQ ID NO: 3, a CDR1L region of SEQ ID NO:4, a CDR2L region of SEQ ID NO:5 and a CDR3L region of SEQ ID NO:6).
- an anti-BCMA antibody, or antigen binding fragment thereof comprising a VH region comprising
- Embodiment 11 The pharmaceutical composition of any one of embodiments 1-
- the multispecific antibody comprises: an anti-BCMA antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO: 10 and a VL region of SEQ ID NO: 14; and an anti-CD3 antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO: 7 and a VL region of SEQ ID NO: 8.
- Embodiment 12 The pharmaceutical composition of any one of embodiments 1-11, wherein the multispecific antibody is a trivalent bispecific antibody comprising two Fab fragments of an anti-BCMA antibody, one Fab fragment of an anti-CD3 antibody, and one Fc portion, wherein the bispecific antibody is in the format BCMA Fab - Fc - CD3 Fab - BCMA Fab.
- Embodiment 13 The pharmaceutical composition of embodiment 12, wherein each Fab fragment of an anti-BCMA antibody, or antigen binding fragment thereof, comprises a VH region comprising a CDR1H region of SEQ ID NO:21, a CDR2H region of SEQ ID NO:22 and a CDR3H region of SEQ ID NO: 17 and a VL region comprising a CDR1L region of SEQ ID NO:27, a CDR2L region of SEQ ID NO:28 and a CDR3L region of SEQ ID NO:20.
- Embodiment 14 The pharmaceutical composition of embodiment 12 or embodiment 13, wherein each Fab fragment of an anti-BCMA antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO: 10 and a VL region of SEQ ID NO: 14.
- Embodiment 15 The pharmaceutical composition of any one of embodiments 12-14, wherein the Fab fragment of the anti-CD3 antibody, or antigen binding fragment thereof, comprises a CDR1H region of SEQ ID NO: 1, a CDR2H region of SEQ ID NO:2, a CDR3H region of SEQ ID NO:3, a CDR1L region of SEQ ID NO:4, a CDR2L region of SEQ ID NO:5 and a CDR3L region of SEQ ID NO:6.
- Embodiment 16 The pharmaceutical composition of any one of embodiments 12-15, wherein the Fab fragment of the anti-CD3 antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO: 7 and a VL region of SEQ ID NO: 8.
- Embodiment 17 The pharmaceutical composition of any one of embodiments 1-16, wherein the multispecific antibody comprises a first polypeptide comprising the amino acid sequence of SEQ ID NO: 48, a second polypeptide comprising the amino acid sequence of SEQ ID NO: 55 or 58, a third polypeptide comprising the amino acid sequence of SEQ ID NO: 56 or 59, and fourth and fifth polypeptides each comprising the amino acid sequence of SEQ ID NO: 57.
- Embodiment 18 A unit dose comprising the pharmaceutical composition of any one of embodiments 1-17.
- Embodiment 19 The unit dose of embodiment 18, wherein the volume of the unit dose is
- Embodiment 20 A unit dose comprising a pharmaceutical composition comprising:
- Embodiment 21 A unit dose comprising a pharmaceutical composition comprising:
- Embodiment 22 The unit dose of embodiment 20 or 21, wherein the multispecific antibody comprises: an anti-BCMA antibody, or antigen binding fragment thereof, comprising a VH region comprising a CDR1H region of SEQ ID NO:21, a CDR2H region of SEQ ID NO:22 and a CDR3H region of SEQ ID NO: 17 and a VL region comprising a CDR1L region of SEQ ID NO:27, a CDR2L region of SEQ ID NO:28 and a CDR3L region of SEQ ID NO:20; and an anti-CD3 antibody, or antigen binding fragment thereof, comprising a CDR1H region of SEQ ID NO: 1, a CDR2H region of SEQ ID NO: 2, a CDR3H region of SEQ ID NO: 3, a CDR1L region of SEQ ID NO:4, a CDR2L region of SEQ ID NO:5 and a CDR3L region of SEQ ID NO:6).
- Embodiment 23 The unit dose of any one of embodiments 20-22, wherein the multispecific antibody comprises: an anti-BCMA antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO: 10 and a VL region of SEQ ID NO: 14; and an anti-CD3 antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO: 7 and a VL region of SEQ ID NO: 8.
- the multispecific antibody comprises: an anti-BCMA antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO: 10 and a VL region of SEQ ID NO: 14; and an anti-CD3 antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO: 7 and a VL region of SEQ ID NO: 8.
- Embodiment 24 The unit dose of any one of embodiments 20-23, wherein the multispecific antibody is a trivalent bispecific antibody comprising two Fab fragments of an anti- BCMA antibody, one Fab fragment of an anti-CD3 antibody, and one Fc portion, wherein the bispecific antibody is in the format BCMA Fab - Fc - CD3 Fab - BCMA Fab.
- Embodiment 25 The unit dose of embodiment 24, wherein each Fab fragment of an anti-BCMA antibody, or antigen binding fragment thereof, comprises a VH region comprising a CDR1H region of SEQ ID NO:21, a CDR2H region of SEQ ID NO:22 and a CDR3H region of SEQ ID NO: 17 and a VL region comprising a CDR1L region of SEQ ID NO:27, a CDR2L region of SEQ ID NO:28 and a CDR3L region of SEQ ID NO:20.
- Embodiment 26 The unit dose of embodiment 24 or embodiment 25, wherein each Fab fragment of an anti-BCMA antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO: 10 and a VL region of SEQ ID NO: 14
- Embodiment 27 The unit dose of any one of embodiments 24-26, wherein the Fab fragment of the anti-CD3 antibody, or antigen binding fragment thereof, comprises a CDR1H region of SEQ ID NO: 1, a CDR2H region of SEQ ID NO: 2, a CDR3H region of SEQ ID NO: 3, a CDR1L region of SEQ ID NO:4, a CDR2L region of SEQ ID NO:5 and a CDR3L region of SEQ ID NO:6.
- Embodiment 28 The unit dose of any one of embodiments 24-27, wherein the Fab fragment of the anti-CD3 antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO: 7 and a VL region of SEQ ID NO: 8.
- Embodiment 29 The unit dose of any one of embodiments 20-28, wherein the multispecific antibody comprises a first polypeptide comprising the amino acid sequence of SEQ ID NO: 48, a second polypeptide comprising the amino acid sequence of SEQ ID NO: 55 or 58, a third polypeptide comprising the amino acid sequence of SEQ ID NO: 56 or 59, and fourth and fifth polypeptides each comprising the amino acid sequence of SEQ ID NO: 57.
- Embodiment 30 The unit dose of any one of embodiments 20-29, wherein the volume of the unit dose is 0.5-2.5 mb, or 0.5-0.9 mb, or 1.0-1.5 mb, or 2.0-2.5 mb, or 0.5, 0.6, 0.7, 0.8, 0.9. 1.0, 1.1, 1.2, 1.3, 1.4, 1.5, 1.6, 1.7, 1.8, 1.9, 2.0, 2.1, 2.2, 2.3, 2.4, or 2.5 mL.
- Embodiment 31 A vial comprising the pharmaceutical composition of any one of embodiments 1-17 or the unit dose of any one of embodiments 18-30.
- Embodiment 32 A method of treating a subject in need thereof, comprising subcutaneously administering to the subject at least one dose of the pharmaceutical composition of any one of embodiments 1-17 or at least one unit dose of any one of embodiments 18-30.
- Embodiment 33 A method of treating multiple myeloma or an autoimmune disease in a subject, comprising subcutaneously administering to the subject at least one dose of the pharmaceutical composition of any one of embodiments 1-17 or at least one unit dose of any one of embodiments 18-30.
- Embodiment 34 The method of embodiment 32 or embodiment 33, wherein the treatment comprises the administration of the pharmaceutical composition or unit dose in a dosing regimen comprising:
- a maintenance phase wherein a first maintenance dose of the pharmaceutical composition or unit dose is administered to the subject, optionally followed by at least one additional maintenance dose of the pharmaceutical composition or unit dose, wherein each maintenance dose is greater than the one or more starting doses.
- the starting phase comprises a single fixed dose of the pharmaceutical composition or unit dose comprising about 1.5 mg to 4.5 mg of the multispecific antibody.
- Embodiment 36 The method of embodiment 34, wherein the starting phase comprises a single fixed dose of the pharmaceutical composition or unit dose comprising about 3 mg of the multispecific antibody.
- Embodiment 37 The method of any one of embodiments 34-36, wherein the first maintenance dose is a fixed dose of the pharmaceutical composition or unit dose comprising about 4.5 mg to about 7.5 mg of the multi-specific antibody.
- Embodiment 38 The method of any one of embodiments 34-36, wherein the first maintenance dose is a fixed dose of the pharmaceutical composition or unit dose comprising about 6 mg of the multispecific antibody.
- Embodiment 39 The method of any one of embodiments 34-38, wherein the at least one additional maintenance dose is a fixed dose of the pharmaceutical composition or unit dose comprising about 28 to about 32 mg of the multispecific antibody.
- Embodiment 40 The method of any one of embodiments 34-38, wherein the at least one additional maintenance dose is a fixed dose of the pharmaceutical composition or unit dose comprising about 30 mg of the multispecific antibody.
- Embodiment 41 The method of any one of embodiments 34-40, wherein the treatment comprises:
- Embodiment 42 The method of embodiment 41, wherein the maintenance doses are administered on days 1, 8, 15 and 22 for the second and third treatment cycle, on days 1 and 15 for the fourth to sixth treatment cycle, and on day 1 for the seventh and subsequent cycle.
- Embodiment 43 The method of any one of embodiments 34-42, wherein the treatment comprises:
- a first treatment cycle wherein the starting dose comprises a single fixed dose of the pharmaceutical composition or unit dose comprising about 3 mg of the multi-specific antibody is administered on day 1, the maintenance dose of the pharmaceutical composition or unit dose comprising about 6 mg of the multispecific antibody is administered on day 4, and the maintenance doses of the pharmaceutical composition or unit dose comprising about 30 mg of the multispecific antibody are administered on days 8, 15 and 22,
- Embodiment 44 The pharmaceutical composition of any one of embodiments 1-17 or the unit dose of any one of embodiments 18-30 for use in treating a disorder associated with BCMA expression in a subject.
- Embodiment 45 The pharmaceutical composition or unit dose for use of embodiment 44, wherein the disorder is multiple myeloma or an autoimmune disease.
- Embodiment 46 The pharmaceutical composition or unit dose for use of embodiment 44 or embodiment 45, wherein the treatment comprises the administration of the pharmaceutical composition or unit dose in a dosing regimen comprising:
- a maintenance phase wherein a first maintenance dose of the pharmaceutical composition or unit dose is administered to the subject, optionally followed by at least one additional maintenance dose of the pharmaceutical composition or unit dose, wherein each maintenance dose is greater than the one or more starting doses.
- Embodiment 47 The pharmaceutical composition or unit dose for use of embodiment 46, wherein the starting phase comprises a single fixed dose of the pharmaceutical composition or unit dose comprising about 1.5 mg to 4.5 mg of the multispecific antibody.
- Embodiment 48 The pharmaceutical composition or unit dose for use of embodiment 46, wherein the starting phase comprises a single fixed dose of the pharmaceutical composition or unit dose comprising about 3 mg of the multi-specific antibody.
- Embodiment 49 The pharmaceutical composition or unit dose for use of any one of embodiments 46-48, wherein the first maintenance dose is a fixed dose of the pharmaceutical composition or unit dose comprising about 4.5 mg to about 7.5 mg of the multi-specific antibody.
- Embodiment 50 The pharmaceutical composition or unit dose for use of any one of embodiments 46-48, wherein the first maintenance dose is a fixed dose of the pharmaceutical composition or unit dose comprising about 6 mg of the multi-specific antibody.
- Embodiment 51 The pharmaceutical composition or unit dose for use of any one of embodiments 46-50, wherein the at least one additional maintenance dose is a fixed dose of the pharmaceutical composition or unit dose comprising about 28 to about 32 mg of the multispecific antibody.
- Embodiment 52 The pharmaceutical composition or unit dose for use of any one of embodiments 46-50, wherein the at least one additional maintenance dose is a fixed dose of the pharmaceutical composition or unit dose comprising about 30 mg of the multi-specific antibody.
- Embodiment 53 The pharmaceutical composition or unit dose for use of any one of embodiments 46-52, wherein the treatment comprises:
- Embodiment 54 The pharmaceutical composition or unit dose for use of embodiment 53, wherein the maintenance doses are administered on days 1, 8, 15 and 22 for the second and third treatment cycle, on days 1 and 15 for the fourth to sixth treatment cycle, and on day 1 for the seventh and subsequent cycle.
- Embodiment 55 The pharmaceutical composition or unit dose for use of any one of embodiments 46-54, wherein the treatment comprises:
- a first treatment cycle wherein the starting dose of a single fixed dose of the pharmaceutical composition or unit dose comprising about 3 mg of the multi-specific antibody is administered on day 1, the maintenance dose of the pharmaceutical composition or unit dose comprising about 6 mg of the multispecific antibody is administered on day 4, and the maintenance doses of the pharmaceutical composition or unit dose comprising about 30 mg of the multispecific antibody are administered on days 8, 15 and 22,
- a second and third treatment cycle wherein the maintenance doses of the pharmaceutical composition or unit dose comprising about 30 mg of the multispecific antibody are administered on days 1, 8, 15, and 22 in a weekly dosing interval
- a fourth to sixth treatment cycle wherein the maintenance doses of the pharmaceutical composition or unit dose comprising about 30 mg of the multispecific antibody are administered on days 1 and 15 in a biweekly dosing interval
- Embodiment 56 Use of the pharmaceutical composition of any one of embodiments 1-17 or the unit dose of any one of embodiments 18-30 for the preparation of a medicament for treating a disorder associated with BCMA expression.
- Embodiment 57 The use of embodiment 56, wherein the disorder is multiple myeloma.
- Embodiment 58 The use of embodiment 56 or embodiment 57, wherein the medicament is administered to a subject in a dosing regimen comprising:
- a maintenance phase wherein a first maintenance dose of the medicament is administered to the subject, optionally followed by at least one additional maintenance dose of the medicament, wherein each maintenance dose is greater than the one or more starting doses.
- Embodiment 59 The use of embodiment 58, wherein the starting phase comprises a single fixed dose of the medicament comprising about 1.5 mg to 4.5 mg of the multispecific antibody.
- Embodiment 60 The use of embodiment 58, wherein the starting phase comprises a single fixed dose of the medicament comprising about 3 mg of the multi-specific antibody.
- Embodiment 61 The use of any one of embodiments 58-60, wherein the first maintenance dose is a fixed dose of the medicament comprising about 4.5 mg to about 7.5 mg of the multi-specific antibody.
- Embodiment 63 The use of any one of embodiments 58-62, wherein the at least one additional maintenance dose is a fixed dose of the medicament comprising about 28 to about 32 mg of the multi-specific antibody.
- Embodiment 64 The use of any one of embodiments 58-62, wherein the at least one additional maintenance dose is a fixed dose of the medicament comprising about 30 mg of the multispecific antibody.
- Embodiment 65 The use of any one of embodiments 58-64, wherein the medicament is administered in a dosing regimen comprising: (i) a first treatment cycle, wherein the starting dose is administered on day 1, and the maintenance doses are administered on days 4, 8, 15 and 22,
- Embodiment 66 The use of embodiment 65, wherein the maintenance doses are administered on days 1, 8, 15 and 22 for the second and third treatment cycle, on days 1 and 15 for the fourth to sixth treatment cycle, and on day 1 for the seventh and subsequent cycle.
- Embodiment 67 The use of any one of embodiments 58-66, wherein the medicament is administered in a dosing regimen comprising:
- a first treatment cycle wherein the starting dose comprises a single fixed dose of the medicament comprising about 3 mg of the multi-specific antibody is administered on day 1, the maintenance dose of the medicament comprising about 6 mg of the multispecific antibody is administered on day 4, and the maintenance doses of the medicament comprising about 30 mg of the multispecific antibody are administered on days 8, 15 and 22,
- Embodiment 68 A pharmaceutical composition comprising (a) about 6 mg/mL of alnuctamab; (b) about 20 mM histidine; (c) about 250 mM sucrose; (d) about 0.04% w/v polysorbate 80; and (e) about 50 pM pentetic acid.
- Embodiment 69 A pharmaceutical composition comprising (a) about 30 mg/mL of alnuctamab; (b) about 20 mM histidine; (c) about 250 mM sucrose; (d) about 0.04% w/v polysorbate 80; and (e) about 50 pM pentetic acid Embodiment 70.
- a pharmaceutical composition comprising (a) 6 mg/mL of alnuctamab; (b) 20 mM histidine; (c) 250 mM sucrose; (d) 0.04% w/v polysorbate 80; and (e) 50 pM pentetic acid.
- a pharmaceutical composition comprises (a) 30 mg/mL of alnuctamab; (b) 20 mM histidine; (c) 250 mM sucrose; (d) 0.04% w/v polysorbate 80; and (e) 50 pM pentetic acid.
- Embodiment 72 The vial of embodiment 31, wherein the vial is a pre-filled syringe or an autoinjector.
- Embodiment 74 The method of embodiment 73, wherein pentetic acid binds to the anti- CD3 domain of alnuctamab.
- Embodiment 75 The method of embodiment 73 or 74, wherein binding alnuctamab to pentetic acid prevents or reduces tryptophan residue oxidation and/or asparagine residue deamidation in alnuctamab.
- Embodiment 76 The method of embodiment 75, wherein the tryptophan residue(s) are located in the CDR of the anti-CD3 domain of alnuctamab.
- Embodiment 77 The method of embodiment 75 or 76, wherein the asparagine residue(s) are located in the CDR of the anti-CD3 domain of alnuctamab.
- Embodiment 78 The method of any of embodiments 73-77, wherein the pharmaceutical composition is the pharmaceutical composition of any of embodiments 1-17.
- FIG. 1 illustrates a format of bispecific trivalent antibodies for use in the present invention, which comprise Fab fragments binding to CD3 and BCMA in the following formats: Fab BCMA - Fc - Fab CD3 - Fab BCMA.
- the CD3 Fab may include a VH-VL crossover to reduce light chain mispairing and side -products. Amino acid substitutions “RK/EE” may be introduced in CL-CH1 to reduce light chain mispairing/side products in production.
- the CD3 Fab and BCMA Fab may be linked to each other with flexible linkers.
- FIG. 2A-2B show % high molecular weight species formed during agitation (FIG. 2A, each set of three bars, left to right: time 0, 400 rpm for 24 hours, 400 rpm for 70 hours) and freeze-thaw (FIG. 2B, each set of three bars, from left to right: time 0, three freeze-thaw cycles, five freeze-thaw cycles) of a compositions comprising 100 mg/mL of alnuctamab with differing pHs (5.7-6.3) and surfactant levels (0.02%-0.06%), as measured by size-exclusion chromatography.
- FIG. 3A-3C show HMW% (FIG. 3A), main peak% (FIG. 3B), and LMW% (FIG.
- FIG. 5A-5B acidic species % (FIG. 5A) and main peak % (FIG. 5B) as assessed by cation exchange chromatography (CEX) for vials of formulations comprising 10 mg/mL, 50 mg/mL, and 100 mg/mL of alnuctamab at various temperatures (5 °C, 25 °C, and 40°C) over a period of 24 months.
- CEX cation exchange chromatography
- FIG. 6 shows potency data for formulations comprising 10 mg/mL, 50 mg/mL, and 100 mg/mL of alnuctamab at 5 °C over a period of 18 months.
- FIG. 8 is a table showing changes in PS 80 concentration for formulations comprising 10 mg/mL, 50 mg/mL, and 100 mg/mL of alnuctamab at 5 °C, 25 °C and 60% relative humidity, and 40°C and 75% relative humidity, over a period of up to 24 months.
- FIG. 9A-9C show HMW % (FIG. 9A), monomer % (FIG. 9B) and LMW % (FIG. 9C) as assessed by SEC for formulations comprising 44 mg/mL alnuctamab with and without 50 pm DTPA at 5 °C, 25 °C, or 40°C over 12 months.
- FIG. 10 shows main peak % as assessed by CE-SDS NR for formulations comprising 44 mg/mL alnuctamab with and without 50 pm DTPA at 5°C, 25°C, and 40°C over 12 months.
- FIG. 11A-11C show acidic species % (FIG. 11A), main peak % (FIG. 1 IB), and basic species % (FIG. 11C) as assessed by CEX for formulations comprising 44 mg/mL alnuctamab with and without 50 pm DTPA at 5°C, 25°C, and 40°C over 12 months.
- FIG. 12A-12B show HMW % (FIG. 12A) and monomer % (FIG. 12B) as assessed by SEC for formulations comprising 1 mg/mL alnuctamab with and without 50 pm DTPA at 5 °C, 25 °C, and 40°C over 6 months.
- FIG. 13 shows main peak % as assessed by CE-SDS-NR for formulations comprising 1 mg/mL alnuctamab with and without 50 pm DTPA at 5°C, 25°C, and 40°C over 6 months.
- FIG. 14A-14B show acidic species % (FIG. 14A) and main peak % (FIG. 14B) as assessed by CEX for formulations comprising 1 mg/mL alnuctamab with and without 50 pm DTPA at 5 °C, 25 °C, and 40°C over 6 months.
- FIG. 15 shows PS-80 concentration for formulations comprising 1 mg/mL alnuctamab with and without 50 pm DTPA at 5°C, 25°C, and 40°C.
- FIG. 16A-16F show HMW (FIG. 16A-16C) and main peak (FIG. 16D-16F) as assessed by SEC for formulations comprising 5 mg/mL alnuctamab and PS-80 (with or without DTPA and with or without metal spiking) at various temperatures: 25 °C (FIG. 16A and 16D), RT/RL (room temperature/room light) (FIG. 16B and 16E), and 40°C (FIG. 16C and 16F).
- Each set of four bars, from left to right all are 5 mg/mL alnuctamab: DTPA+PS80; PS80; DTPA+PS80+metal; PS80+metal.
- FIG. 17A-17F show HMW (FIG. 17A-C) and main peak (FIG. 17D-F) as assessed by SEC for formulations comprising 60 mg/mL alnuctamab and PS-80 (with or without DTPA and with or without metal spiking) at various temperatures: 25°C (FIG. 17A and 17D), RT/RL (room temperature/room light) (FIG. 17B and 17E), and 40°C (FIG. 17C and 17F).
- Each set of four bars, from left to right all are 60 mg/mL alnuctamab: DTPA+PS80; PS80; DTPA+PS80+metal; PS80+metal.
- FIG. 18A-18F show acidic species (FIG. 18A-18C) and main peak (FIG. 18D-18F) as assessed by CEX for formulations comprising 5 mg/mL alnuctamab and PS-80 (with or without DTPA and with or without metal spiking) at various temperatures: 25°C (FIG. 18A and 18D), RT/RL (room temperature/room light) (FIG. 18B and 18E), and 40°C (FIG. 18C and 18F).
- Each set of four bars, from left to right all are 5 mg/mL alnuctamab: DTPA+PS80; PS80; DTPA+PS80+metal; PS80+metal.
- FIG. 19A-19F show acidic species (FIG. 19A-19C) and main peak (FIG. 189-19F) as assessed by CEX for formulations comprising 60 mg/mL alnuctamab and PS-80 (with or without DTPA and with or without metal spiking) at various temperatures: 25°C (FIG. 19A and 19D), RT/RL (room temperature/room light) (FIG. 19B and 19E), and 40°C (FIG. 19C and 19F).
- Each set of four bars, from left to right all are 60 mg/mL alnuctamab: DTPA+PS80; PS80; DTPA+PS80+metal; PS80+metal.
- FIG. 20A-20C show main peak % as assessed by CE-SDS NR for formulations comprising 5 mg/mL alnuctamab and PS-80 (with or without DTPA and with or without metal spiking) at various temperatures: 25°C (FIG. 20A), RT/RL (room temperature/room light) (FIG. 20B), and 40°C (FIG. 20C).
- FIG. 21A-21C show main peak% as assessed by CE-SDS NR for formulations comprising 60 mg/mL alnuctamab and PS-80 (with or without DTPA and with or without metal spiking) at various temperatures: 25°C (FIG. 21 A), RT/RL (room temperature/room light) (FIG. 2 IB), and 40°C (FIG. 21C).
- Each set of four bars, from left to right all are 60 mg/mL alnuctamab: DTPA+PS80; PS80; DTPA+PS80+metal; PS80+metal.
- FIG. 22A-22B shows PS-80 oxidation in formulations comprising 5 mg/mL alnuctamab (FIG. 22A) and 60 mg/mL alnuctamab (FIG. 22B) and PS80 (with or without DTPA and with or without metal spking) at 25 °C and 40°C.
- the articles “a” and “an” may refer to one or to more than one (e.g. to at least one) of the grammatical object of the article.
- “About” may generally mean an acceptable degree of error for the quantity measured given the nature or precision of the measurements. Exemplary degrees of error are within 20 percent (%), within 10%, or within 5% of a given value or range of values.
- Embodiments described herein as “comprising” one or more features may also be considered as disclosure of the corresponding embodiments “consisting of’ and/or “consisting essentially of’ such features.
- an “antibody” of the present disclosure is capable of binding to more than one antigen, e.g., a “multispecific” antibody.
- a “bispecific” antibody is an antibody that is capable of specifically binding two antigens, wherein the first and second antigen are the same or different.
- the multispecific (e.g. bispecific) antibodies of the invention specifically bind to BCMA and to CD3.
- the terms “antibody against BCMA and CD3”, “anti-BCMA anti-CD3 antibody” or “an antibody that binds to BCMA and CD3,” refer to a multispecific antibody (e.g., a bispecific antibody) that is capable of binding to BCMA and CD3 with sufficient affinity such that the antibody is useful as a therapeutic agent. This is achieved by making a molecule which comprises a first antibody, or antigenbinding fragment, that binds to BCMA and a second antibody, or antigen-binding fragment, that binds to CD3.
- Such multispecific antibodies may be trispecific antibodies or bispecific antibodies.
- the multispecific antibodies are bispecific antibodies.
- BCMA human B cell maturation antigen
- TR17_HUMAN human B cell maturation antigen
- TNFRSF17 UniProt Q02223
- the extracellular domain of BCMA consists according to UniProt of amino acids 1 - 54 (or 5-51).
- antibody against BCMA “anti BCMA antibody” or “an antibody that binds to BCMA” as used herein relate to an antibody specifically binding to the extracellular domain of BCMA.
- the term “specifically binds to BCMA” refers to an antibody that is capable of binding to the defined target with sufficient affinity such that the antibody is useful as a therapeutic agent in targeting BCMA.
- an antibody that specifically binds to BCMA does not bind to other antigens, or does not bind to other antigens with sufficient affinity to produce a physiological effect.
- the extent of binding of an anti-BCMA antibody to an unrelated, non-BCMA protein is about 10-fold preferably > 100-fold less than the binding of the antibody to BCMA as measured, e.g., by surface plasmon resonance (SPR) e.g. Biacore®, enzyme-linked immunosorbent (ELISA) or flow cytometry (FACS).
- SPR surface plasmon resonance
- ELISA enzyme-linked immunosorbent
- FACS flow cytometry
- the antibody that binds to BCMA has a dissociation constant (Kd) of 10' 8 M or less, preferably from 10' 8 M to 10' 13 M, preferably from 10' 9 M to IO' 13 M.
- the anti -BCMA antibody binds to an epitope of BCMA that is conserved among BCMA from different species, preferably among human and cynomolgus, and in addition preferably also to mouse and rat BCMA.
- the anti-BCMA antibody specifically binds to a group of BCMA, consisting of human BCMA and BCMA of non-human mammalian origin, preferably BCMA from cynomolgus, mouse and/or rat.
- Anti-BCMA antibodies are analyzed by ELISA for binding to human BCMA using plate-bound BCMA.
- an amount of plate-bound BCMA preferably 1.5 pg/mL and concentration(s) ranging from 0.1 pM to 200 nM of anti-BCMA antibody are used.
- the term “specifically binds to CD3” refers to an antibody that is capable of binding to the defined target with sufficient affinity such that the antibody is useful as a therapeutic agent in targeting CD3.
- an antibody that specifically binds to CD3 does not bind to other antigens, or does not bind to other antigens with sufficient affinity to produce a physiological effect.
- the multispecific (e.g. bispecific) antibodies of the invention can be analysed by surface plasmon resonance (SPR), e.g. Biacore®, for binding to CD3.
- SPR surface plasmon resonance
- the bispecific antibodies bind to human CD3 with a dissociation constant (KD) of about 10' 7 M or less, a KD of about IO -8 M or less, a KD of about 10' 9 M or less, a KD of about IO -10 M or less, a KD of about 10 11 M or less, or a KD of about IO -12 M or less, as determined by a surface plasmon resonance assay, preferably measured using Biacore 8K at 25°C.
- the bispecific antibodies bind to human CD3 with a dissociation constant (KD) of about KF 8 M or less.
- a “heavy chain” comprises a heavy chain variable region (abbreviated herein as “VH”) and a heavy chain constant region (abbreviated herein as “CH”).
- the heavy chain constant region comprises the heavy chain constant domains CHI, CH2 and CH3 (antibody classes IgA, IgD, and IgG) and optionally the heavy chain constant domain CH4 (antibody classes IgE and IgM).
- a “light chain” comprises a light chain variable domain (abbreviated herein as “VL”) and a light chain constant domain (abbreviated herein as “CL”).
- VL variable chain variable domain
- CL light chain constant domain
- the variable regions VH and VL can be further subdivided into regions of hypervariability, termed complementarity determining regions (CDR), interspersed with regions that are more conserved, termed framework regions (ER).
- CDR complementarity determining regions
- ER framework regions
- Each VH and VL is composed of three CDRs and four L Rs, arranged from amino-terminus to carboxy-terminus in the following order: FR1, CDR1, LR2, CDR2, LR3, CDR3, LR4.
- the “constant domains” of the heavy chain and of the light chain are not involved directly in binding of an antibody to a target, but exhibit various effector functions.
- CDRs Complementarity Determining Regions
- the CDRs are regions of high sequence variability, located within the variable region of the antibody heavy chain and light chain, where they form the antigen-binding site.
- the CDRs are the main determinants of antigen specificity.
- the antibody heavy chain and light chain each comprise three CDRs which are arranged non-consecutively.
- the antibody heavy and light chain CDR3 regions play a particularly important role in the binding specificity/affinity of the antibodies according to the invention and therefore provide a further aspect of the invention.
- antigen binding fragment incudes any naturally-occurring or artificially-constructed configuration of an antigen-binding polypeptide comprising three light chain CDRs, and three heavy chain CDRs, wherein the polypeptide is capable of binding to the antigen.
- the term refers to a molecule other than an intact antibody that comprises a portion of an intact antibody that binds the antigen to which the intact antibody binds.
- antibody fragments include but are not limited to Fv, Fab, Fab’, Fab’-SH, F(ab’)2; diabodies; linear antibodies; single-chain antibody molecules (e.g. scFv); and multispecific antibodies formed from antibody fragments.
- Fab fragment and “Fab” are used interchangeably herein and contain a single light chain (i.e. a constant domain CL and a VL) and a single heavy chain (i.e. the constant domain CHI and a VH).
- the heavy chain of a Fab fragment is not capable of forming a disulfide bond with another heavy chain.
- a “Fab 1 fragment” contains a single light chain and a single heavy chain but in addition to the CHI and the VH, a “Fab 1 fragment” contains the region of the heavy chain between the CHI and CH2 domains that is required for the formation of an inter-chain disulfide bond. Thus, two “Fab 1 fragments” can associate via the formation of a disulphide bond to form a F(ab')2 molecule.
- a “F(ab')2 fragment” contains two light chains and two heavy chains. Each chain includes a portion of the constant region necessary for the formation of an inter-chain disulfide bond between two heavy chains.
- an “Fv fragment” contains only the variable regions of the heavy and light chain. It contains no constant regions.
- a “single-chain Fv” (“scFv”) is antibody fragment containing the VH and VL domain of an antibody, linked together to form a single chain. A polypeptide linker is commonly used to connect the VH and VL domains of the scFv.
- a “tandem scFv”, also known as a TandAb®, is a single-chain Fv molecule formed by covalent bonding of two scFvs in a tandem orientation with a flexible peptide linker.
- a “bi-specific T cell engager” (BiTE®) is a fusion protein consisting of two single-chain variable fragments (scFvs) on a single peptide chain. One of the scFvs binds to T cells via the CD3 receptor, and the other to a tumour cell antigen.
- the sequence of a CDR may be identified by reference to any number system known in the art, for example, the Kabat system (Kabat, E. A., et al., Sequences of Proteins of Immunological Interest, 5th ed., Public Health Service, National Institutes of Health, Bethesda, MD (1991); the Chothia system (Chothia &, Lesk, “Canonical Structures for the Hypervariable Regions of Immunoglobulins,” J. Mol. Biol. 196, 901-917 (1987)); or the IMGT system (Lefranc et al., “IMGT Unique Numbering for Immunoglobulin and Cell Receptor Variable Domains and Ig superfamily V-like domains,” Dev. Comp. Immunol. 27, 55-77 (2003)).
- Kabat system Kabat system
- Chothia system Chothia &, Lesk, “Canonical Structures for the Hypervariable Regions of Immunoglobulins”
- humanized antibody refers to antibodies in which the framework or “complementarity determining regions” (CDRs) have been modified to comprise the CDR of an immunoglobulin of different specificity as compared to that of the parent immunoglobulin.
- CDRs complementarity determining regions
- a murine CDR may be grafted into the framework region of a human antibody to prepare the “humanized antibody.” See, e.g., Riechmann, L., et al., Nature 332 (1988) 323-327; and Neuberger, M.S., et al., Nature 314 (1985) 268-270.
- “humanized antibodies” are those in which the constant region has been additionally modified or changed from that of the original antibody to generate the properties of the antibodies according to the invention, especially in regard to Clq binding and/or Fc receptor (FcR) binding.
- human antibody is one which possesses an amino acid sequence which corresponds to that of an antibody produced by a human or a human cell or derived from a non-human source that utilizes human antibody repertoires or other human antibody-encoding sequences. This definition of a human antibody specifically excludes a humanized antibody comprising non-human antigen-binding residues. Human antibodies can be produced using various techniques known in the art, including phage-display libraries.
- chimeric antibody refers to an antibody comprising a variable region, i.e., binding region, from one source or species and at least a portion of a constant region derived from a different source or species, usually prepared by recombinant DNA techniques. Chimeric antibodies comprising a murine variable region and a human constant region are preferred. Other preferred forms of “chimeric antibodies” encompassed by the present invention are those in which the constant region has been modified or changed from that of the original antibody to generate the properties of the antibodies according to the invention, especially in regard to Clq binding and/or Fc receptor (FcR) binding. Such chimeric antibodies are also referred to as “class-switched antibodies”.
- Chimeric antibodies are the product of expressed immunoglobulin genes comprising DNA segments encoding immunoglobulin variable regions and DNA segments encoding immunoglobulin constant regions.
- Methods for producing chimeric antibodies involving conventional recombinant DNA and gene transfection techniques are well known in the art. See, e.g., Morrison, S.L., et al., Proc. Natl. Acad. Sci. USA 81 (1984) 6851-6855; US Patent Nos. 5,202,238 and 5,204,244.
- Fc region and “Fc” are used interchangeably herein and refer to the portion of a native immunoglobulin that is formed by two Fc chains.
- Each “Fc chain” comprises a constant domain CH2 and a constant domain CH3.
- Each Fc chain may also comprise a hinge region.
- a native Fc region is homodimeric.
- the Fc region may contain modifications to enforce Fc heterodimerization.
- Fc part refers to the portion of an antibody of the invention, or antigen binding fragment thereof, which corresponds to the Fc region.
- IgA heavy chain constant region
- IgG is separated into four subclasses known as IgGl, IgG2, IgG3, and IgG4.
- Ig molecules interact with multiple classes of cellular receptors.
- IgG molecules interact with three classes of Fey receptors (FcyR) specific for the IgG class of antibody, namely FcyRI, FcyRII, and FcyRIII.
- FcyR Fey receptors
- the antibodies of the invention or antigen-binding fragments thereof may be any isotype, i.e. IgA, IgD, IgE, IgG and IgM, and synthetic multimers of the four-chain immunoglobulin (Ig) structure.
- the antibodies or antigen-binding fragments thereof are IgG isotype.
- the antibodies or antigen-binding fragments can be any IgG subclass, for example IgGl, IgG2, IgG3, or IgG4 isotype.
- the antibodies or antigen-binding fragments thereof are of an IgGl isotype.
- the antibodies comprise a heavy chain constant region that is of IgG isotype. In some embodiments, the antibodies comprise a portion of a heavy chain constant region that is of IgG isotype. In some embodiments, the IgG constant region or portion thereof is an IgGl, IgG2, IgG3, or IgG4 constant region. Preferably, the IgG constant region or portion thereof is an IgGl constant region.
- the antibodies of the invention or antigen-binding fragments thereof may comprise a lambda light chain or a kappa light chain.
- the antibodies or antigen-binding fragments thereof comprise a light chain that is a kappa light chain.
- the antibody or antigen-binding fragment comprises a light chain comprising a light chain constant region (CL) that is a kappa constant region.
- the antibody comprises a light chain comprising a light chain variable region (VL) that is a kappa variable region.
- VL light chain variable region
- the kappa light chain comprises a VL that is a kappa VL and a CL that is a kappa CL.
- the antibodies or antigen-binding fragments thereof may comprise a light chain that is a lambda light chain.
- the antibody or antigen-binding fragment comprises a light chain comprising a light chain constant region (CL) that is a lambda constant region.
- the antibody comprises a light chain comprising a light chain variable region (VL) that is a lambda variable region.
- bispecific antibody formats are described in Kontermann RE, mAbs 4:2 1-16 (2012); Holliger P., Hudson PJ, Nature Biotech.23 (2005) 1126- 1136, Chan AC, Carter PJ Nature Reviews Immunology 10, 301-316 (2010) and Cuesta AM etal., Trends Biotech 28 (2011) 355-362.
- the multispecific, e.g. bispecific, antibodies of the invention may have any format.
- Multispecific and bispecific antibody formats include, for example, multivalent single chain antibodies, diabodies and triabodies, and antibodies having the constant domain structure of full length antibodies to which further antigen-binding domains (e.g., single chain Fv, a tandem scFv, a VH domain and/or a VL domain, Fab, or (Fab)2,) are linked via one or more peptide-linkers.
- the multispecific, e.g. bispecific, antibodies of the invention have the format of an scFv such as a bispecific T cell engager (BITE®).
- the antibodies of the invention are single chain antibodies which comprise a first domain which binds to BCMA, a second domain which binds to a T cell antigen (e.g. CD3), and a third domain which comprises two polypeptide monomers, each comprising a hinge, a CH2 domain and a CH3 domain, wherein the two polypeptide monomers are fused to each other via a peptide linker (e.g. (hinge-CH2-CH3-linker-hinge-CH2-CH3).
- a peptide linker e.g. (hinge-CH2-CH3-linker-hinge-CH2-CH3
- the “valency” of an antibody denotes the number of binding domains.
- the terms “bivalent”, “trivalenf ’, and “multivalent” denote the presence of two binding domains, three binding domains, and multiple binding domains, respectively.
- the multispecific, e.g. bispecific, antibodies of the invention may have more than one binding domain capable of binding to each target antigen (i.e., the antibody is trivalent or multivalent).
- the multispecific, e.g. bispecific, antibodies of the invention have more than one binding domain capable of binding to the same epitope of each target antigen.
- the multispecific, e.g. bispecific, antibodies of the invention have more than one binding domain capable of binding to different epitopes on each target antigen.
- the multispecific, e.g. bispecific, antibodies of the invention may be bivalent, trivalent or tetravalent.
- the multispecific, e.g. bispecific, antibody is trivalent, preferably wherein the trivalent antibody is bivalent for BCMA.
- the bispecific antibody may be trivalent, wherein the trivalent antibody is bivalent for BCMA.
- the multispecific, e.g. bispecific, antibodies can be full length from a single species, or can be chimerized or humanized. For an antibody with more than two antigen-binding domains, some binding domains may be identical, as long as the protein has binding domains for two different antigens.
- the multispecific, e.g. bispecific, antibodies of the invention can have a bispecific heterodimeric format.
- the bispecific antibody comprises two different heavy chains and two different light chains.
- the multispecific, e.g. bispecific, antibody comprises two identical light chains and two different heavy chains.
- one of the two pairs of heavy chain and light chain (HC/LC) specifically binds to CD3 and the other one specifically binds to BCMA.
- bispecific antibodies of the invention may comprise one anti-BCMA antibody and one anti-CD3 antibody (referred to herein as the “1+1” format).
- the bivalent bispecific antibodies in the 1+1 format may have the format: CD3 Fab - BCMA Fab (i.e. when no Fc is present).
- the bispecific antibodies may have the format: Fc - CD3 Fab - BCMA Fab; Fc- BCMA Fab - CD3 Fab; or BCMA Fab - Fc - CD3 Fab (i.e. when an Fc is present).
- the bivalent bispecific antibodies have the format BCMA Fab - Fc - CD3 Fab.
- CD3 Fab - BCMA Fab means that the CD3 Fab is bound via its N-terminus to the C- terminus of the BCMA Fab.
- Fc - BCMA Fab - CD3 Fab means that the BCMA Fab is bound via its C-terminus to the N-terminus of the Fc, and the CD3 Fab is bound via its C-terminus to the N-terminus of the BCMA Fab.
- Fc - CD3 Fab - BCMA Fab means that the CD3 Fab is bound via its C-terminus to the N-terminus of the Fc, and the BCMA Fab is bound via its C-terminus to the N-terminus of the CD3 Fab.
- BCMA Fab - Fc - CD3 Fab means that the BCMA and CD3 Fab fragments are bound via their C-terminus to the N-terminus of the Fc.
- bispecific antibodies of the invention may comprise two anti -BCMA antibodies and one anti-CD3 antibody (referred to herein as the “2+1” format).
- the trivalent bispecific antibodies in the 2+1 format may have the format: CD3 Fab - BCMA Fab - BCMA Fab; or BCMA Fab - CD3 Fab - BCMA Fab (i.e. when no Fc is present).
- the bispecific antibodies may have the format: BCMA Fab - Fc - CD3 Fab - BCMA Fab; BCMA Fab - Fc - BCMA Fab - CD3 Fab; or CD3 Fab - Fc - BCMA Fab - BCMA Fab (i.e. when an Fc is present).
- the trivalent bispecific antibodies have the format BCMA Fab - Fc - CD3 Fab - BCMA Fab.
- BCMA Fab - CD3 Fab - BCMA Fab means that the first BCMA Fab is bound via its C-terminus to the N-terminus of the CD3 Fab, and the CD3 Fab is bound via its C-terminus to the N- terminus of the second BCMA Fab.
- BCMA Fab - Fc - CD3 Fab - BCMA Fab means that the first BCMA Fab and the CD3 Fab are bound via their C-terminus to the N-terminus of the Fc, and the second BCMA Fab is bound via its C-terminus to the N-terminus of the CD3 Fab.
- BCMA Fab - Fc - BCMA Fab - CD3 Fab means that the first BCMA Fab and the second BCMA Fab are bound via their C-terminus to the N-terminus of the Fc, and the CD3 Fab is bound via its C-terminus to the N-terminus of the second BCMA Fab.
- CD3 Fab - Fc - BCMA Fab - BCMA Fab means that the CD3 Fab and the first BCMA Fab are bound via their C-terminus to the N-terminus of the Fc, and the second BCMA Fab is bound via its C-terminus to the N-terminus of the first BCMA Fab.
- the bispecific antibodies of the invention may comprise not more than one BCMA Fab specifically binding to BCMA, and not more than one CD3 Fab specifically binding to CD3 and not more than one Fc part.
- the bispecific antibody comprises not more than one CD3 Fab specifically binding to CD3, not more than two BCMA Fabs specifically binding to BCMA and not more than one Fc part.
- not more than one CD3 Fab and not more than one BCMA Fab are linked to the Fc part and linking is performed via C-terminal binding of the Fab(s) to the hinge region of the Fc part.
- the second BCMA Fab is linked via its C-terminus either to the N- terminus of the CD3 Fab or to the hinge region of the Fc part and is therefore between the Fc part of the bispecific antibody and the CD3 Fab.
- the BCMA Fabs are preferably derived from the same antibody and are preferably identical in the CDR sequences, variable domain sequences VH and VL and/or the constant domain sequences CHI and CL.
- the amino acid sequences of the two BCMA Fab are identical.
- bispecific antibodies of the invention can also comprise scFvs instead of the Fabs.
- the bispecific antibodies have any one of the above formats, wherein each Fab is replaced with a corresponding scFv.
- the components, e.g. the Fab fragments, of the bispecific antibodies of the invention may be chemically linked together by the use of an appropriate linker according to the state of the art.
- a (Gly4-Serl)2 linker (SEQ ID NO: 60) is used (Desplancq DK et al., Protein Eng. 1994 Aug;7(8): 1027-33 and Mack M. et al., PNAS July 18, 1995 vol. 92 no. 15 7021-7025).
- “Chemically linked” (or “linked”) as used herein means that the components are linked by covalent binding.
- the linker is a peptidic linker, such covalent binding is usually performed by biochemical recombinant means.
- the binding may be performed using a nucleic acid encoding the VL and/or VH domains of the respective Fab fragments, the linker and the Fc part chain if the antibody comprises an Fc.
- this linker may be of a length and sequence sufficient to ensure that each of the first and second domains can, independently from each other, retain their differential binding specificities.
- the multispecific (e.g. bispecific) antibody comprises an anti- BCMA antibody, or antigen binding fragment thereof, comprising a VH region comprising a CDR1H region of SEQ ID NO:21, a CDR2H region of SEQ ID NO:22 and a CDR3H region of SEQ ID NO: 17 and a VL region comprising a CDR1L region of SEQ ID NO:27, a CDR2L region of SEQ ID NO:28 and a CDR3L region of SEQ ID NO:20.
- an anti- BCMA antibody or antigen binding fragment thereof, comprising a VH region comprising a CDR1H region of SEQ ID NO:21, a CDR2H region of SEQ ID NO:22 and a CDR3H region of SEQ ID NO: 17 and a VL region comprising a CDR1L region of SEQ ID NO:27, a CDR2L region of SEQ ID NO:28 and a CDR3L region of SEQ ID NO:20.
- the anti-BCMA antibody, or antigen binding fragment thereof comprises a VH region of SEQ ID NOTO and a VL region of SEQ ID NO: 14. 1
- the multispecific (e.g. bispecific) antibody comprises an anti-CD3 antibody, or antigen binding fragment thereof.
- the multispecific (e.g. bispecific) antibody of the invention comprises a humanized SP34 antibody or antigen-binding fragment thereof.
- the anti-CD3 antibody, or antigen binding fragment thereof may be derived from SP34 and may have similar sequences and the same properties with regard to epitope binding as antibody SP34.
- the multispecific (e.g. bispecific) antibody comprises an anti-CD3 antibody, or antigen binding fragment thereof, comprising a variable domain VH comprising the heavy chain CDRs of SEQ ID NO: 1, 2 and 3 as respectively heavy chain CDR1H, CDR2H and CDR3H and a variable domain VL comprising the light chain CDRs of SEQ ID NO: 4, 5 and 6 as respectively light chain CDR1L, CDR2L and CDR3L.
- the multispecific (e.g. bispecific) antibody comprises an anti-CD3 antibody, or antigen binding fragment thereof, comprising the variable domains of SEQ ID NO:7 (VH) and SEQ ID NO:8 (VL).
- the multispecific (e.g. bispecific) antibody comprises: an anti-BCMA antibody, or antigen binding fragment thereof, comprising a VH region comprising a CDR1H region of SEQ ID NO:21, a CDR2H region of SEQ ID NO:22 and a CDR3H region of SEQ ID NO: 17 and a VL region comprising a CDR1L region of SEQ ID NO:27, a CDR2L region of SEQ ID NO:28 and a CDR3L region of SEQ ID NO:20; and an anti-CD3 antibody, or antigen binding fragment thereof, comprising a CDR1H region of SEQ ID NO: 1, a CDR2H region of SEQ ID NO:2, a CDR3H region of SEQ ID NO:3, a CDR1L region of SEQ ID NO:4, a CDR2L region of SEQ ID NO:5 and a CDR3L region of SEQ ID NO:6.
- an anti-BCMA antibody, or antigen binding fragment thereof comprising
- the multispecific (e.g. bispecific) antibody comprises an anti-BCMA antibody, or antigen binding fragment thereof, comprising a VH region of SEQ ID NO: 10 and a VL region of SEQ ID NO: 14, and an anti-CD3 antibody, or antigen binding fragment thereof, comprising a VH region of SEQ ID NO:7 and a VL region of SEQ ID NO:8.
- the multispecific, e.g. bispecific, antibodies of the invention may have an Fc or may not have an Fc.
- the multispecific antibodies of the invention comprise an Fc, preferably a human Fc.
- the Fc is a variant Fc, e.g., an Fc sequence that has been modified (for example by amino acid substitution, deletion and/or insertion) relative to a parent Fc sequence (for example an unmodified Fc polypeptide that is subsequently modified to generate a variant), to provide desirable structural features and/or biological activity,
- a variant Fc e.g., an Fc sequence that has been modified (for example by amino acid substitution, deletion and/or insertion) relative to a parent Fc sequence (for example an unmodified Fc polypeptide that is subsequently modified to generate a variant), to provide desirable structural features and/or biological activity
- the multispecific antibodies e.g. bispecific antibodies, of the invention may comprise an Fc comprising one or more modifications, typically to alter one or more functional properties of the antibody, such as serum half-life, complement fixation, Fc receptor binding, and/or antigen-dependent cellular cytotoxicity.
- the Fc may be linked to the anti-BCMA and/or anti-CD3 Fab fragments in the antibodies of the invention.
- the presence of an Fc has the advantage of extending the elimination half-life of the antibody.
- the antibodies, e.g. bispecific antibodies, of the invention may have an elimination half-life in mice or cynomolgus monkeys, preferably cynomolgus monkeys, of longer than 12 hours, preferably 3 days or longer.
- the antibodies, e.g. bispecific antibodies, of the invention have an elimination half-life of about 1 to 12 days, which allows at least once or twice/week administration.
- the bispecific antibodies of the invention comprise an Fc region (e.g. of IgGl subclass) that comprises modifications to avoid FcR and Clq binding and minimize ADCC/CDC.
- Fc region e.g. of IgGl subclass
- modifications to avoid FcR and Clq binding and minimize ADCC/CDC e.g. of IgGl subclass
- the bispecific antibody mediates its tumour cell killing efficacy by the effector cell, e.g. T cell, redirection/activation. Therefore, additional mechanisms of action, such as effects on the complement system and on effector cells expressing FcR, are avoided and the risk of sideeffects, such as infusion-related reactions, is decreased.
- the antibodies, e.g. bispecific antibodies, of the invention comprise an IgG, particularly IgGl, Fc region comprising the modifications L234A, L235A and P329G (numbered according to EU numbering).
- the multispecific, e.g. bispecific, antibodies of the invention may be heteromultimeric antibodies.
- Such heteromultimeric antibodies may comprise modifications in regions involved in interactions between antibody chains to promote correct assembly of the antibodies.
- the bispecific antibodies of the invention may comprise an Fc having one or more modification(s) in the CH2 and CH3 domain to enforce Fc heterodimerization.
- the bispecific antibodies of the invention may comprise modifications in the CHI and CL region to promote preferential pairing between the heavy chain and light chain of a Fab fragment.
- a number of strategies exist for promoting heterodimerization may include the introduction of asymmetric complementary modifications into each of two antibody chains, such that both chains are compatible with each other and thus able to form a heterodimer, but each chain is not able to dimerize with itself. Such modifications may encompass insertions, deletions, conservative and non-conservative substitutions and rearrangements.
- Heterodimerization may be promoted by the introduction of charged residues to create favourable electrostatic interactions between a first antibody chain and a second antibody chain.
- one or more positively charged amino acids amino acid may be introduced into a first antibody chain
- one or more negatively charged amino acids may be introduced into a corresponding positions in a second antibody chain.
- heterodimerization may be promoted by the introduction of steric hindrance between contacting residues.
- one or more residues with a bulky side chain may be introduced into a first antibody chain, and a one or more residues able to accommodate the bulky side chain may be introduced into the second antibody chain.
- heterodimerization may be promoted by the introduction of one or more modification(s) to the hydrophilic and hydrophobic residues at the interface between chains, in order make heterodimer formation more entropically and enthalpically favourable than homodimer formation.
- a further strategy for promoting heterodimerization is to rearrange portions of the antibody chains such that each chain remains compatible only with a chain comprising corresponding rearrangements.
- CrossMAb technology is based on the crossover of antibody domains in order to enable correct chain association.
- the bispecific antibodies of the invention may comprise an exchange of the VH and VL.
- the antibodies, e.g. bispecific antibodies, of the invention may comprise an exchange of the CHI and CL.
- the antibodies, e.g. bispecific antibodies, of the invention may comprise an exchange of the VH and VL and an exchange of the CHI and CL.
- the antibodies, e.g. bispecific antibodies, of the invention comprise an exchange of the VH and VL.
- a combination of the above strategies may be used to maximise the efficiency of assembly while minimising the impact on antibody stability.
- multispecific antibodies e.g. bispecific antibodies
- multispecific antibodies may have a heterodimeric Fc, for example they may comprise one heavy chain originating from an anti-BCMA antibody, and one heavy chain originating from an anti-CD3 antibody.
- the antibodies, e.g. bispecific antibodies, of the invention may comprise a heterodimeric Fc which comprises one or more modification s) which promotes the association of the first CH2 and/or CH3 domain with the second CH2 and/or CH3 domain.
- the one or more modification(s) promote the association of the first CH3 domain with the second CH3 domain, for example by resulting in asymmetric modifications to the CH3 domain.
- the one or more modification(s) may comprise modifications selected from amino acid insertions, deletions, conservative and nonconservative substitutions and rearrangements, and combinations thereof.
- first CH3 domain and the second CH3 domain are both engineered in a complementary manner so that each CH3 domain (or the heavy chain comprising it) can no longer homodimerize with itself but is forced to heterodimerize with the complementary engineered other CH3 domain (so that the first and second CH3 domain heterodimerize and no homodimers between the two first or the two second CH3 domains are formed).
- the multispecific, e.g. bispecific, antibodies of the invention may comprise an Fc having one or more of “knob-into-holes” modification s), which are described in detail with several examples in e.g. WO 96/027011, Ridgway, J.B., et al., Protein Eng. 9 (1996) 617-621, Merchant, A.M. et al., Nat. Biotechnol. 16 (1998) 677-68, and WO 98/050431.
- the interaction surfaces of the two CH3 domains are altered to increase the heterodimerization of both Fc chains containing these two CH3 domains.
- One of the two CH3 domains (of the two Fc chains) can be the “knob”, while the other is the “hole”.
- the bispecific antibodies of the invention may comprise two CH3 domains, wherein the first CH3 domain of the first Fc chain and the second CH3 domain of the second Fc chain each meet at an interface which comprises an original interface between the antibody CH3 domains, wherein said interface is altered to promote the formation of the antibody.
- the CH3 domain of one Fc chain is altered, so that within the original interface of the CH3 domain of the one Fc chain that meets the original interface of the CH3 domain of the other Fc chain, an amino acid residue is replaced with an amino acid residue having a larger side chain volume, thereby generating a protuberance within the interface of the CH3 domain of one Fc chain which is positionable in a cavity within the interface of the CH3 domain of the other Fc chain; and ii) the CH3 domain of the other Fc chain is altered, so that within the original interface of the CH3 domain of the other Fc chain that meets the original interface of the CH3 domain of the one Fc chain, an amino acid residue is replaced with an amino acid residue having a smaller side chain volume, thereby generating a cavity within the interface of the CH3 domain of the other Fc chain within which a protuberance within the interface of the CH3 domain of the one Fc chain is positionable.
- said amino acid residue having a larger side chain volume is selected from the group consisting of arginine (R), phenylalanine (F), tyrosine (Y), tryptophan (W).
- the multispecific, e.g. bispecific, antibodies of the invention comprise a first CH3 domain comprising modification(s) at positions T366, L368 and Y407, e.g. T366S, L368A, and Y407V (numbered according to EU numbering).
- the multispecific, e.g. bispecific, antibodies of the invention comprise a second CH3 domain comprising a modification at position T366 (“knob modification”), e.g. T366W (numbered according to EU numbering).
- knock modification e.g. T366W (numbered according to EU numbering).
- the multispecific, e.g. bispecific, antibodies of the invention comprise a first CH3 domain comprising the modifications T366S, L368A, and Y407V, or conservative substitutions thereof, and a second CH3 domain comprising the modification T366W, or a conservative substitution thereof (numbered according to EU numbering).
- the multispecific, e.g. bispecific, antibodies of the invention comprise a first CH3 domain comprising the modification set forth in Table 2 and a second CH3 domain comprising the modifications set forth in Table 2.
- the bispecific antibody according to the invention is of IgG2 isotype and the heterodimerization approach described in W02010/129304 can be used.
- the bispecific antibodies of the invention may comprise an Fc, wherein both CH3 domains are altered by the introduction of cysteine (C) as the amino acid in the corresponding positions of each CH3 domain such that a disulphide bridge between both CH3 domains can be formed.
- C cysteine
- the cysteines may be introduced at position 349 in one of the CH3 domains and at position 354 in the other CH3 domain (numbered according to EU numbering).
- the cysteine introduced at position 354 is in the first CH3 domain and the cysteine introduced at position 349 is in the second CH3 domain (numbered according to EU numbering).
- the Fc may comprise modifications, such as D356E, L358M, N384S, K392N, V397M, and V422I (numbered according to EU numbering).
- both CH3 domains comprise D356E and L358M (numbered according to EU numbering).
- one or more of the immunoglobulin heavy chains and light chains may comprise one or more modification(s), e.g. amino acid modifications that are capable of promoting preferential pairing of a specific heavy chain with a specific light chain when heavy chains and light chains are co-expressed or co-produced.
- modification(s) e.g. amino acid modifications that are capable of promoting preferential pairing of a specific heavy chain with a specific light chain when heavy chains and light chains are co-expressed or co-produced.
- modification(s) e.g. amino acid modifications that are capable of promoting preferential pairing of a specific heavy chain with a specific light chain when heavy chains and light chains are co-expressed or co-produced.
- modification(s) e.g. amino acid modifications that are capable of promoting preferential pairing of a specific heavy chain with a specific light chain when heavy chains and light chains are co-expressed or co-produced.
- modification(s) such as amino acid exchanges
- the amino acid exchanges may be substitutions of charged amino acids with opposite charges (for example in the CH1/CL interface) which reduce light chain mispairing, e.g. Bence-Jones type side products.
- the one or more modification(s) assist light and heavy chain heterodimerization are amino acid modifications in the light and heavy chains outside of the CDRs.
- the one or more modification(s) may be present in the anti-BCMA antibody or antigenbinding fragment thereof. Alternatively, the one or more modification(s) may be present in the anti-CD3 antibody or antigen-binding fragment thereof. In preferred embodiments, the one or more modification(s) are present in the anti-BCMA antibody or antigen-binding fragment thereof.
- the multispecific, e.g. bispecific, antibodies of the invention comprise an immunoglobulin heavy chain comprising a CHI domain having amino acid modifications K147E/D and K213E/D (numbered according to EU numbering) and a corresponding immunoglobulin light chain comprising a CL domain having amino acid modifications E123K/R/H and Q124K/R/H (numbered according to Kabat).
- the CHI domain comprises the amino acid modifications K147E and K213E (numbered according to EU numbering) or conservative substitutions thereof
- the corresponding CL domain comprises the amino acid modifications E123R and Q124K or conservative substitutions thereof (numbered according to Kabat).
- Such multispecific, e.g. bispecific, antibodies can be produced in high yield and can be easily purified.
- amino acid modifications described in Table 3 can be in the BCMA antibody or in the CD3 antibody.
- the bispecific antibodies of the invention are bivalent, and comprise one anti-BCMA antibody or antigen-binding fragment thereof and one anti-CD3 antibody or antigenbinding fragment thereof (the “1+1” format), wherein:
- the BCMA antibody or antigen-binding fragment thereof comprises a CHI domain having amino acid modifications set forth in Table 3 and a corresponding CL domain having the amino acid modifications Table 3;
- the CD3 antibody or antigen-binding fragment thereof comprises a CHI domain having amino acid modifications set forth in Table 3 and a corresponding CL domain having the amino acid modifications Table 3.
- the bispecific antibodies of the invention are trivalent and comprise two anti-BCMA antibodies or antigen-binding fragments thereof and one anti-CD3 antibody or antigenbinding fragment thereof (the “2+1” format), wherein:
- BCMA antibodies or antigen-binding fragments thereof comprises a CHI domain having amino acid modifications set forth in Table 3 and a corresponding CL domain having the amino acid modifications Table 3;
- the CD3 antibody (e.g. CD3 Fab) comprises a CHI domain having amino acid modifications set forth in Table 3 and a corresponding CL domain having the amino acid modifications Table 3.
- each BCMA antibody (e.g. BCMA Fab) may comprise a CHI domain having amino acid modifications set forth in Table 3 and a corresponding CL domain having the amino acid modifications Table 3.
- the multispecific, e.g. bispecific, antibodies of the invention comprise the modifications set forth in Table 3 in combination with the modifications set forth in Table 2.
- the bispecific antibodies of the invention are bivalent, and comprise:
- the BCMA antibody or antigenbinding fragment thereof comprises a CHI domain that comprises the amino acid modifications K147E and K213E, and a corresponding CL domain that comprises the amino acid modifications E123R and Q124K (i.e. the modifications set forth in Table 3)
- the CD3 antibody or antigen-binding fragment thereof comprises a CHI domain that comprises the amino acid modifications K147E and K213E, and a corresponding CL domain that comprises the amino acid modifications E123R and Q124K (i.e. the modifications set forth in Table 3); and
- the bispecific antibodies of the invention are trivalent and comprise: (a) two anti-BCMA antibodies or antigen-binding fragments thereof and one anti-CD3 antibody or antigen-binding fragment thereof (the “2+1” format), wherein (i) one or both BCMA antibodies or antigen-binding fragments thereof (e.g. BCMA Fabs) comprises a CHI domain that comprises the amino acid modifications K147E and K213E, and a corresponding CL domain that comprises the amino acid modifications E123Rand Q124K (i.e. the modifications set forth in Table 3), or (ii) the CD3 antibody or antigen-binding fragment thereof (e.g. CD3 Fab) comprises a CHI domain that comprises the amino acid modifications K147E and K213E, and a corresponding CL domain that comprises the amino acid modifications E123R and Q124K (i.e. the modifications set forth in Table 3); and
- each BCMA antibody (e.g. BCMA Fab) may comprise a CHI domain having amino acid modifications set forth in Table 3 and a corresponding CL domain having the amino acid modifications Table 3.
- the first Fc chain is bound at the N-terminus of the Fc to the C-terminus of the first anti-BCMA antibody
- the second Fc chain is bound at the N- terminus of the Fc to the C-terminus of the anti-CD3 antibody.
- the multispecific, e.g. bispecific, antibodies of the invention may additionally comprise an amino acid substitution at position 49 of the VL region selected from the group of amino acids tyrosine (Y), glutamic acid (E), serine (S), and histidine (H) and/or an amino acid substitution at position 74 of the VL region that is threonine (T) or alanine (A).
- the multispecific, e.g. bispecific, antibodies of the invention may comprise CrossMAb technology.
- CrossMAb technology is based on the crossover of antibody domains in order to enable correct chain association. It is used to facilitate multispecific antibody formation.
- the variable domains VL and VH or the constant domains CL and CHI may be replaced by each other.
- the antibodies, e.g. bispecific antibodies, of the invention may comprise an exchange of the VH and VL and an exchange of the CHI and CL.
- the multispecific, e.g. bispecific, antibodies of the invention may comprise a crossover light chain and a crossover heavy chain.
- a multispecific, e.g. bispecific, antibody comprising an anti-BCMA antibody of the invention, or an antigen-binding fragment thereof, and an anti-CD3 antibody, or antigen-binding fragment thereof, wherein the multispecific, e.g. bispecific, antibody comprises:
- variable domains VL and VH and/or the constant domains CL and CHI are replaced by each other in (i) the anti-BCMA antibody; and/or (ii) the anti-CD3 antibody.
- variable domains VL and VH or the constant domains CL and CHI of the anti-CD3 antibody or antigen binding fragment thereof are replaced by each other. More preferably, the variable domains VL and VH of the anti-CD3 antibody or antigen binding fragment thereof are replaced by each other.
- the bispecific antibodies in the 1+1 format have the format: CD3 Fab - BCMA Fab (i.e. when no Fc is present); Fc - CD3 Fab - BCMA Fab; Fc- BCMA Fab - CD3 Fab; or BCMA Fab - Fc - CD3 Fab
- the bispecific antibodies may comprise the CrossMAb format, e.g. CrossMAb Fab , CrossMAb VH ’ VL or CrossMAb CH1 CL .
- the BCMA Fab may have the CrossMAb format, e.g. CrossMAb Fab , CrossMAb VH ’ VL or CrossMAb CH1 CL .
- the CD3 Fab may have the CrossMAb format, e.g. CrossMAb Fab , CrossMAb VH VL or CrossMAb CH1 CL .
- the CD3 Fab of the bispecific antibody comprises the CrossMAb VH VL format.
- the bispecific antibodies of the invention having the 2+1 format may comprise CrossMAb technology.
- the trivalent bispecific antibodies in the 2+1 format have the format: CD3 Fab - BCMA Fab - BCMA Fab; BCMA Fab - CD3 Fab - BCMA Fab (i.e. when no Fc is present); BCMA Fab - Fc - CD3 Fab - BCMA Fab; BCMA Fab - Fc - BCMA Fab - CD3 Fab; or CD3 Fab - Fc - BCMA Fab - BCMA Fab
- the bispecific antibodies may comprise the CrossMAb format, e.g.
- the BCMA Fab may have the CrossMAb format, e.g. CrossMAb Fab , CrossMAb VH VL or CrossMAb CH1 CL .
- the CD3 Fab may have the CrossMAb format, e.g. CrossMAb Fab , CrossMAb VH ’ VL or CrossMAb CH1 CL .
- the CD3 Fab of the bispecific antibody comprises the CrossMAb VH ’ VL format.
- the bispecific antibodies of the invention having the 1+1 format do not comprise CrossMAb technology, i.e. neither the anti-BCMA antibody nor the anti-CD3 antibody have the variable domains VL and VH or the constant domains CL and CHI replaced by each other.
- FIG. 1 Exemplary embodiments are set out in FIG. 1.
- the bispecific antibodies according to the invention are trivalent bispecific antibodies comprising one Fab fragment of an anti-CD3 antibody, two Fab fragments of an anti-BCMA antibody and one Fc part according to the format BCMA Fab - Fc - CD3 Fab - BCMA Fab.
- Each anti-BCMA Fab fragment comprises the amino acid modifications set forth in Table 3.
- the anti- CD3 Fab fragment comprises a light chain and heavy chain, wherein the light chain is a crossover light chain that comprises a variable domain VH and a constant domain CL, and wherein the heavy chain is a crossover heavy chain that comprises a variable domain VL and a constant domain CHI . This embodiment is illustrated in FIG. 1.
- the antibodies illustrated in FIG. 1 additionally comprise the modifications set forth in Table 2.
- the bispecific antibodies according to the invention are trivalent bispecific antibodies comprising one Fab fragment of an anti-CD3 antibody, two Fab fragments of an anti-BCMA antibody and one Fc part according to the format BCMA Fab - Fc - CD3 Fab - BCMA Fab.
- the anti- CD3 Fab fragment comprises a light chain and heavy chain, wherein the light chain is a crossover light chain that comprises a variable domain VH and a constant domain CL, and wherein the heavy chain is a crossover heavy chain that comprises a variable domain VL and a constant domain CHI .
- Each anti- BCMA Fab fragment comprises a light chain and heavy chain, wherein the heavy chain comprises a CHI domain which comprises the amino acid modifications K147E and K213E (numbered according to EU numbering) and wherein the light chain comprises a corresponding CL domain which comprises the amino acid modifications E123R and Q124K (numbered according to Kabat) (i.e. the modifications set forth in Table 3).
- the Fc part comprises a first Fc chain and a second Fc chain, wherein the first Fc chain comprises a first constant domain CH2 and a first constant domain CH3, and the second Fc chain comprises a second constant domain CH2 and a second constant domain CH3.
- the first Fc chain is bound at the N-terminus of the Fc to the C-terminus of the first anti-BCMA Fab
- the second Fc chain is bound at the N-terminus of the Fc to the C-terminus of the anti-CD3 Fab.
- the first CH3 domain comprises the modifications T366S, L368A, and Y407V (“hole modifications”) and the second CH3 domain comprises the modification T366W (“knob modification”) (numbered according to EU numbering) (i.e. the modifications set forth in Table 2).
- both Fc chains further comprise the modifications L234A, L235A and P329G, and optionally D356E and L358M (numbered according to EU numbering).
- the first CH3 domain further comprises the amino acid modification S354C
- the second CH3 domain further comprises the amino acid modification Y349C (numbered according to EU numbering) such that a disulphide bridge between both CH3 domains is formed.
- the anti-BCMA Fab fragment comprises a a CDR1H region of SEQ ID NO: 21, a CDR2H region of SEQ ID NO: 22 and a CDR3H region of SEQ ID NO: 17 and a VL region comprising a CDR1L region of SEQ ID NO:27, a CDR2L region of SEQ ID NO:28 and a CDR3L region of SEQ ID NO:20; and the anti-CD3 Fab fragment comprises a CDR1H region of SEQ ID NO: 1, a CDR2H region of SEQ ID NO:2, a CDR3H region of SEQ ID NO:3, a CDR1L region of SEQ ID NO:4, a CDR2L region of SEQ ID NO:5 and a CDR3L region of SEQ ID NO:6.
- the anti-BCMA Fab fragment comprises a VH region of SEQ ID NO: 1
- the anti-CD3 Fab fragment comprises a VH region of SEQ ID NO : 7 and a VL region of SEQ ID NO : 8.
- the bispecific antibody according to the invention comprises the SEQ ID NOs (as mentioned in Tables 7A and 8B below) of Alnuctamab (42-TCBcv): 48, 55 or 58, 56 or 59, and 57 (x2) (FIG. 1).
- alnuctamab (also referred to as 42-TCBcv” and “Mab42”) as used herein refers to the bispecific antibody comprising a first polypeptide comprising the amino acid sequence of SEQ ID NO: 48, a second polypeptide comprising the amino acid sequence of SEQ ID NO: 55 or 58, a third polypeptide comprising the amino acid sequence of SEQ ID NO: 56 or 59, and fourth and fifth polypeptides each comprising the amino acid sequence of SEQ ID NO: 57, and as shown in FIG. 1 and described in WO 2017/021450.
- the bispecific antibody is 42-TCBcv (alnuctamab).
- the multispecific, e.g. bispecific, antibodies of the invention can be administered to the patient as a pharmaceutical composition. Accordingly, the present invention also provides a pharmaceutical composition comprising the multispecific, e.g. bispecific, antibodies of the invention discussed herein.
- pharmaceutical compositions described herein comprise 42- TCBcv (alnuctamab).
- the pharmaceutical composition comprises at least about 0.05 mg/mL to at least about 37.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 1 mg/mL to at least about 37.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.5 mg/mL to at least about 37.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 1 mg/mL to at least about 37.5 mg/mL of the multispecific antibody, e.g. alnuctamab.
- the pharmaceutical composition comprises at least about 4.5 mg/mL to at least about 37.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 4.5 mg/mL to at least about 37.5 mg/mL, at least about 5 mg/mL to at least about 35 mg/mL, at least about 5.5 mg/mL to at least about 32 mg/mL, at least about 6 mg/mL to at least about 30 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.05 mg/mL of the multispecific antibody, e.g. alnuctamab..
- the pharmaceutical composition comprises at least about 0.06 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.07 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.08 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.09 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0. 1 mg/mL of the multispecific antibody, e.g. alnuctamab.
- the pharmaceutical composition comprises at least about 0.2 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.3 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.4 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.6 mg/mL of the multispecific antibody, e.g. alnuctamab.
- the pharmaceutical composition comprises at least about 0.7 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.8 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.9 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 1 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 1.5 mg/mL of the multispecific antibody, e.g. alnuctamab.
- the pharmaceutical composition comprises at least about 2 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 3 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 3.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 4 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 4.5 mg/mL of the multispecific antibody, e.g. alnuctamab.
- the pharmaceutical composition comprises at least about 5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 5.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 6 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 6.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 7 mg/mL of the multispecific antibody, e.g. alnuctamab.
- the pharmaceutical composition comprises at least about 7.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 22.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 25 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 27 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 28 mg/mL of the multispecific antibody, e.g. alnuctamab.
- the pharmaceutical composition comprises at least about 29 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 29.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 30 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 30.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 31 mg/mL of the multispecific antibody, e.g. alnuctamab.
- the pharmaceutical composition comprises at least about 32 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 35 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 37 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 37.5 mg/mL of the multispecific antibody, e.g. alnuctamab.
- the pharmaceutical composition further comprises a metal chelator.
- the metal chelator prevents oxidation of the formulation components and/or improves stability of the antibody, e.g. alnuctamab.
- the metal chelator is pentetic acid (DTP A) or ethylenediaminetetraacetic acid (EDTA).
- the metal chelator is pentetic acid (DTP A).
- the pharmaceutical composition comprises from at least about 1 mM to at least about 250 pM pentetic acid (DTP A).
- the pharmaceutical composition comprises at least about 1 pM, at least about 5 pM, at least about 10 pM, at least about 15 pM, at least about 20 pM, at least about 25 pM, at least about 30 pM, at least about 35 pM, at least about 40 pM, at least about 45 pM, at least about 50 pM, at least about 55 pM, at least about 60 pM, at least about 65 pM, at least about 70 pM, at least about 75 pM, at least about 80 pM, at least about 85 pM, at least about 90 pM, at least about 95 pM, or at least about 100 pM, at least about 110 pM, at least about 120 pM, at least about 130 pM, at least about 140 pM, at least about 150 pM, at least about 160 pM, at least about 170 pM, at least about 180 pM, at least about 190 pM, or at least about
- the pharmaceutical composition comprises at least about 75 pM pentetic acid (DTPA). In certain aspects, the pharmaceutical composition comprises at least about 70 pM pentetic acid (DTP A). In certain aspects, the pharmaceutical composition comprises at least about 65 pM pentetic acid (DTP A). In certain aspects, the pharmaceutical composition comprises at least about 60 pM pentetic acid (DTPA). In certain aspects, the pharmaceutical composition comprises at least about 55 pM pentetic acid (DTPA). In certain aspects, the pharmaceutical composition comprises at least about 50 pM pentetic acid (DTPA). In certain aspects, the pharmaceutical composition comprises at least about 45 pM pentetic acid (DTPA). In certain aspects, the pharmaceutical composition comprises at least about 40 pM pentetic acid (DTPA).
- DTPA pM pentetic acid
- the pharmaceutical composition comprises at least about 35 pM pentetic acid (DTPA). In certain aspects, the pharmaceutical composition comprises at least about 30 pM pentetic acid (DTPA). In certain aspects, the pharmaceutical composition comprises at least about 25 pM pentetic acid (DTPA).
- the pharmaceutical composition further comprises a tonicity modifier and/or stabilizer.
- Any tonicity modifier and/or any stabilizer can be used in the pharmaceutical compositions disclosed herein.
- the tonicity modifier and/or stabilizer comprises a sugar, an amino acid, a polyol, a salt, or any combination thereof.
- the tonicity modifier and/or stabilizer is selected from the group consisting of sucrose, sorbitol, trehalose, mannitol, glycerol, glycine, leucine, isoleucine, sodium chloride, proline, arginine, polyols, amino acids, and salts.
- the pharmaceutical composition comprises sucrose.
- the pharmaceutical composition comprises from at least about 1 mM to at least about 500 mM sucrose. In some aspects, the pharmaceutical compositions comprises from at least about 10 mM to at least about 400 mM, at least about 50 mM to at least about 400 mM, at least about 100 mM to at least about 400 mM, at least about 150 mM to at least about 400 mM, at least about 200 mM to at least about 400 mM, at least about 250 mM to at least about 400 mM, at least about 300 mM to at least about 400 mM, at least about 350 mM to at least about 400 mM, at least about 50 mM to at least about 350 mM, at least about 100 mM to at least about 300 mM, at least about 100 mM to at least about 250 mM, at least about 100 mM to at least about 200 mM, at least about 100 mM to at least about 150 mM, at least about 200 mM to at least
- the pharmaceutical compositions comprises at least about 10 mM, at least about 20 mM, at least about 30 mM, at least about 40 mM, at least about 50 mM, at least about 60 mM, at least about 70 mM, at least about 80 mM, at least about 90 mM, at least about 100 mM, at least about 110 mM, at least about 120 mM, at least about 130 mM, at least about 140 mM, at least about 150 mM, at least about 160 mM, at least about 170 mM, at least about 180 mM, at least about 190 mM, at least about 200 mM, at least about 210 mM, at least about 220 mM, at least about 230 mM, at least about 240 mM, at least about 250 mM, at least about 260 mM, at least about 270 mM, at least about 280 mM, at least about 290 mM, at least about 300
- the pharmaceutical composition comprises at least about 200 mM sucrose. In certain aspects, the pharmaceutical composition comprises at least about 210 mM sucrose. In certain aspects, the pharmaceutical composition comprises at least about 220 mM sucrose. In certain aspects, the pharmaceutical composition comprises at least about 230 mM sucrose. In certain aspects, the pharmaceutical composition comprises at least about 240 mM sucrose. In certain aspects, the pharmaceutical composition comprises at least about 250 mM sucrose. In certain aspects, the pharmaceutical composition comprises at least about 260 mM sucrose. In certain aspects, the pharmaceutical composition comprises at least about 270 mM sucrose. In certain aspects, the pharmaceutical composition comprises at least about 280 mM sucrose. In certain aspects, the pharmaceutical composition comprises at least about 290 mM sucrose. In certain aspects, the pharmaceutical composition comprises at least about 300 mM sucrose.
- the pharmaceutical composition further comprises a buffering agent.
- the buffering agent is selected from histidine, succinate, tromethamine, sodium phosphate, sodium acetate, and sodium citrate.
- the pharmaceutical composition comprises histidine.
- the pharmaceutical composition comprises from at least about 1 mM to at least about 100 mM histidine.
- the pharmaceutical composition comprises from at least about 5 mM to at least about 100 mM, at least about 10 mM to at least about 100 mM, at least about 15 mM to at least about 100 mM, at least about 20 mM to at least about 100 mM, at least about 25 mM to at least about 100 mM, at least about 30 mM to at least about 100 mM, at least about 35 mM to at least about 100 mM, at least about 40 mM to at least about 100 mM, at least about 45 mM to at least about 100 mM, at least about 50 mM to at least about 100 mM, at least about 10 mM to at least about 75 mM, at least about 10 mM to at least about 50 mM, at least about 10 mM to at least about 40 mM, at least about 10 mM to at least about 30 mM, at least about 15 mM to at least about 30 mM, at least about 10 mM, at
- the pharmaceutical composition comprises at least about 5 mM, at least about 10 mM, at least about 15 mM, at least about 20 mM, at least about 25 mM, at least about 30 mM, at least about 35 mM, at least about 40 mM, at least about 45 mM, at least about 50 mM, at least about 60 mM, at least about 70 mM, at least about 80 mM, at least about 90 mM, or at least about 100 mM histidine.
- the pharmaceutical composition comprises at least about 10 mM histidine.
- the pharmaceutical composition comprises at least about 15 mM histidine.
- the pharmaceutical composition comprises at least about 20 mM histidine.
- the pharmaceutical composition comprises at least about 25 mM histidine. In certain aspects, the pharmaceutical composition comprises at least about 30 mM histidine. In certain aspects, the pharmaceutical composition comprises at least about 35 mM histidine. In certain aspects, the pharmaceutical composition comprises at least about 40 mM histidine. In certain aspects, the pharmaceutical composition comprises at least about 45 mM histidine. In certain aspects, the pharmaceutical composition comprises at least about 50 mM histidine.
- the pharmaceutical composition comprises a pH of about 5.2 to about 6.8.
- the pH of the pharmaceutical composition is about 5.2.
- the pH of the pharmaceutical composition is about 5.3.
- the pH of the pharmaceutical composition is about 5.4.
- the pH of the pharmaceutical composition is about 5.5.
- the pH of the pharmaceutical composition is about 5.6.
- the pH of the pharmaceutical composition is about 5.7.
- the pH of the pharmaceutical composition is about 5.8.
- the pH of the pharmaceutical composition is about 5.9.
- the pH of the pharmaceutical composition is about 6.0.
- the pH of the pharmaceutical composition is about 6. 1.
- the pH of the pharmaceutical composition is about 6.2.
- the pH of the pharmaceutical composition is about 6.3. In some aspects, the pH of the pharmaceutical composition is about 6.4. In some aspects, the pH of the pharmaceutical composition is about 6.5. In some aspects, the pH of the pharmaceutical composition is about 6.6. In some aspects, the pH of the pharmaceutical composition is about 6.7. In some aspects, the pH of the pharmaceutical composition is about 6.8.
- the pharmaceutical composition further comprises a surfactant.
- a surfactant can be used in the pharmaceutical compositions disclosed herein.
- the surfactant is selected from the group consisting of polysorbate 20, polysorbate 80, and poloxamer 188.
- the pharmaceutical composition comprises polysorbate 80.
- the pharmaceutical composition comprises from at least about 0.001% to at least about 1% w/v polysorbate 80.
- the pharmaceutical compositions comprises at least about 0.01% to at least about 0.1%, at least about 0.02% to at least about 0.1%, at least about 0.03% to at least about 0.1%, at least about 0.04% to at least about 0.1%, at least about 0.05% to at least about 0.1%, at least about 0.01% to at least about 0.09%, at least about 0.01% to at least about 0.8%, at least about 0.01% to at least about 0.7%, at least about 0.01% to at least about 0.6%, at least about 0.01% to at least about 0.5%, at least about 0.02% to at least about 0.09%, at least about 0.03% to at least about 0.08%, at least about 0.04% to at least about 0.07%, or at least about 0.04% to at least about 0.06% w/v polysorbate 80.
- the pharmaceutical compositions comprises at least about 0.01% to at least about 0.1% w/v polysorbate 80. In some aspects, the pharmaceutical composition comprises at least about 0.01% w/v, at least about 0.02% w/v, at least about 0.03% w/v, at least about 0.04% w/v, at least about 0.05% w/v, at least about 0.06% w/v, at least about 0.07% w/v, at least about 0.08% w/v, at least about 0.09% w/v, or at least about 0.1% w/v polysorbate 80. In certain aspects, the pharmaceutical composition comprises at least about 0.03% w/v polysorbate 80. In certain aspects, the pharmaceutical composition comprises at least about
- the pharmaceutical composition comprises at least about
- the pharmaceutical composition comprises at least about
- the pharmaceutical composition comprises at least about
- the pharmaceutical composition comprises: (a) at least about 4.5 mg/mL to at least about 37.5 mg/mL of a multispecific antibody that binds to BCMA and CD3, e.g. alnuctamab, (b) at least about 5 mM to at least about 100 mM histidine, (c) at least about 10 mM to at least about 500 mM sucrose, (d) at least about 0.01% w/v to at least about 0. 1% w/v polysorbate 80, and (e) at least about 10 pM to about 200 pM pentetic acid (DTP A).
- the pharmaceutical composition comprises (a) about 6 mg/mL of the multispecific antibody, e.g.
- the pharmaceutical composition comprises (a) about 30 mg/mL of the multispecific antibody, e.g. alnuctamab; (b) about 20 mM histidine; (c) about 250 mM sucrose; (d) about 0.04% w/v polysorbate 80; and (e) about 50 pM pentetic acid.
- the pharmaceutical composition comprises (a) about 30 mg/mL of the multispecific antibody, e.g. alnuctamab; (b) about 20 mM histidine; (c) about 250 mM sucrose; (d) about 0.04% w/v polysorbate 80; and (e) about 50 pM pentetic acid.
- the pharmaceutical composition comprises (a) about 6 mg/mL of alnuctamab; (b) about 20 mM histidine; (c) about 250 mM sucrose; (d) about 0.04% w/v polysorbate 80; and (e) about 50 pM pentetic acid.
- the pharmaceutical composition comprises (a) about 30 mg/mL of alnuctamab; (b) about 20 mM histidine; (c) about 250 mM sucrose; (d) about 0.04% w/v polysorbate 80; and (e) about 50 pM pentetic acid.
- the pharmaceutical composition comprises (a) 6 mg/mL of alnuctamab; (b) 20 mM histidine; (c) 250 mM sucrose; (d) 0.04% w/v polysorbate 80; and (e) 50 pM pentetic acid.
- the pharmaceutical composition comprises (a) 30 mg/mL of alnuctamab; (b) 20 mM histidine; (c) 250 mM sucrose; (d) 0.04% w/v polysorbate 80; and (e) 50 pM pentetic acid.
- the vial comprises a unit dose of the pharmaceutical composition.
- the vial or unit dose comprises (a) about 6 mg/mL of the multispecific antibody, e.g. alnuctamab; (b) about 20 mM histidine; (c) about 250 mM sucrose; (d) about 0.04% w/v polysorbate 80; and (e) about 50 pM pentetic acid.
- the vial or unit dose comprises (a) about 30 mg/mL of the multispecific antibody, e.g. alnuctamab; (b) about 20 mM histidine; (c) about 250 mM sucrose; (d) about 0.04% w/v polysorbate 80; and (e) about 50 pM pentetic acid.
- Certain embodiments are directed to a method of preventing or reducing oxidation and/or deamidation of alnuctamab in a pharmaceutical composition comprising binding alnuctamab to pentetic acid in the pharmaceutical composition, thereby preventing or reducing oxidation and/or deamidation of alnuctamab in the pharmaceutical composition.
- the method of preventing or reducing oxidation and/or deamidation of alnuctamab in a pharmaceutical composition comprises binding the anti- CD3 domain of alnuctamab to pentetic acid.
- binding alnuctamab to pentetic acid prevents or reduces tryptophan residue oxidation and/or asparagine residue deamidation in alnuctamab.
- the tryptophan residue(s) are located in the CDR of the anti-CD3 domain of alnuctamab.
- the asparagine residue(s) are located in the CDR of the anti-CD3 domain of alnuctamab.
- binding alnuctamab to pentetic acid in the pharmaceutical composition comprises formulating about 6 mg/mL of alnuctamab with about 50 pM pentetic acid in the pharmaceutical composition.
- binding alnuctamab to pentetic acid in the pharmaceutical composition comprises formulating about 30 mg/mL of alnuctamab with about 50 pM pentetic acid in the pharmaceutical composition.
- the pharmaceutical composition comprising the multispecific, e.g. bispecific, antibody of the invention, e.g. alnuctamab, to the patient as a monotherapy.
- the pharmaceutical composition further comprises one or more additional therapeutic agents as a combination therapy, wherein the combination therapy comprises the administration of the multispecific, e.g. bispecific, antibody of the invention, e.g. alnuctamab, and one or more additional therapeutic agents.
- combination therapy is meant to encompass administration of the selected therapeutic agents to a single patient, and is intended to include treatments in which the agents are administered by the same or different route of administration or at the same or different time.
- the one or more additional therapeutic agents are selected from the group consisting of thalidomide and an immunotherapeutic derivative thereof, an anti-CD38 antibody, an anti-PD-1 antibody, an anti-PD-Ll antibody, a gamma secretase inhibitor (GSI), an anti- BCMA antibody drug conjugate and anti-BCMA CAR T-cell therapy.
- thalidomide an immunotherapeutic derivative thereof, an anti-CD38 antibody, an anti-PD-1 antibody, an anti-PD-Ll antibody, a gamma secretase inhibitor (GSI), an anti- BCMA antibody drug conjugate and anti-BCMA CAR T-cell therapy.
- anti-CD38 antibody as used herein relates to an antibody specifically binding to human CD38.
- the anti-CD38 antibody is daratumumab (US20150246123).
- the anti-CD38 antibody is isatuximab (SAR650984, US8877899).
- the anti-CD38 antibody is MOR202 (WO 2012041800).
- the anti-CD38 antibody is Ab79 (US8362211).
- the anti-CD38 antibody is Abl9 (US8362211).
- the dosage of such anti- CD38 antibody is performed according to the state of the art and described in the respective prescribing information. For example, Daratumumab dosage is usually 16mg/kg (www.ema.europa.eu).
- thalidomide compound or “thalidomide and an immunotherapeutic derivative” as used herein relates to 2-(2,6-dioxopiperidin-3-yl)-2,3-dihydro-lH-isoindole-l, 3-dione and immunotherapeutic derivatives thereof.
- the thalidomide compound is selected from the group consisting of, but not limited to, thalidomide (CAS Registry Number 50-35-1), lenalidomide (CAS Registry Number 191732-72-6), pomalidomide (CAS Registry Number 19171-19-8), CC122 (CAS Registry Number 1398053-45-6) and CC-220 (CAS Registry Number 1323403-33-3) and the respective salts (preferably HC1 salts 1: 1).
- CC-122 The chemical formula of CC-122 is 2,6-piperidinedione,3- (5 -amino-2 -methyl -4-oxo-3(4H-quinazolinyl), hydrochloride (1: 1) and of CC-220 it is 2,6- piperidinedione, 3-[l,3-dihydro-4-[[4-(4-morpholinyhnethyl)phenyl]methoxy]-l-oxo-2H-isoindol-2-yl]-, (3S)-, hydrochloride (1: 1).
- Methods of preparing CC-220 are described, e.g., in US 20110196150, the entirety of which is incorporated herein by reference.
- thalidomide compounds are performed according to the state of the art and described in the respective prescribing information.
- Revlimid® (lenalidomide) dosage is usually 25 mg once daily orally on days 1-21 of repeated 28- day cycles (www.revlimid.com)
- POMALYST® (pomalidomide) dosage for the treatment of Multiple Myeloma is usually 4 mg per day taken orally on days 1-21 of repeated 28-day cycles (www.celgene.com).
- 3-(5- amino-2 -methyl -4-oxo-4H-quinazolin-3-yl)-piperidine-2, 6-dione is administered in an amount of about 5 to about 50 mg per day.
- CC-122 and CC-220 are administered in an amount of about 5 to about 25 mg per day. In another embodiment, CC-122 and CC-220 are administered in an amount of about 5, 10, 15, 25, 30 or 50 mg per day. In another embodiment, 10 or 25 mg of CC-122 and CC-220 are administered per day. In one embodiment, CC-122 and CC-220 are administered twice per day.
- anti-PD-1 antibody as used herein relates to an antibody specifically binding to human PD-1.
- Such antibodies are e.g. described in WO2015026634 (MK-3475, pembrolizumab), US7521051, US8008449, and US8354509.
- Pembrolizumab Keytruda®, MK-3475
- WO 2009/114335 Poole, R.M. Drugs (2014) 74: 1973; Seiwert, Tpens et al., J. Clin. Oncol. 32,5s (suppl; abstr 6011).
- the PD-1 antibody is MK-3475 (WHO Drug Information, Vol. 27, No.
- the PD-1 antibody is nivolumab (BMS-936558, MDX 1106; WHO Drug Information, Vol. 27, No. 1, pages 68-69 (2013), W02006/121168 amino acid sequences shown in WO 2015026634).
- the PD-1 antibody is; pidilizumab (CT-011, also known as hBAT or hBAT-1; amino acid sequence see W02003/099196; WO 2009/101611, Fried l, et al.; Neuro Oncol (2014) 16 (suppl 5): vl 11 -v 112.).
- the PD-1 antibody is MEDI-0680 (AMP-514, W02010/027423, WO2010/027827, WO2010/027828, Hamid O. et al.; J Clin Oncol 33, 2015 (suppl; abstr TPS3087).
- the PD-1 antibody is PDR001 (Naing A.
- the PD-1 antibody is REGN2810 (Papadopoulos KPet al.; J Clin Oncol 34, 2016 (suppl; abstr 3024).
- the PD-1 antibody is pembrolizumab (WO2008/156712).
- the PD-1 antibody is h409Al 1, h409A16 or h409A17, which are described in WO2008/156712.
- the dosage of such anti-PD-1 antibody is performed according to the state of the art and described in the respective prescribing information. For example, Keytruda® is administered usually in a concentration of 2mg/kg body weight every three weeks (ec .europa. eu/health/documents) .
- anti-PD-Ll antibody as used herein relates to an antibody specifically binding to human PD-L1.
- Such antibodies are e.g. described in WO2015026634, W02013/019906, W02010/077634 and US8383796.
- the PD-L1 antibody is MPDL3280A (atezolizumab, YW243.55.S70, WO2010/077634, McDermott DF. Et al., JCO March 10, 2016 vol. 34 no. 8 833-842).
- the PD-L1 antibody is MDX-1105 (BMS-936559, W02007/005874, Patrick A.
- the PD-L1 antibody is MEDI4736 (durvalumab, WO 2016/040238 Gilbert J. et al., Journal for ImmunoTherapy of Cancer 20153(Suppl 2):P152).
- the PD-L1 antibody is MSB001071 8C (avelumab, Disis ML. et al., Journal of Clinical Oncology, Vol 33, No 15_suppl (May 20 Supplement), 2015: 5509).
- the PD-L1 antibody is the anti-PD-Ll antibody comprising a VH sequence of SEQ ID NO: 16 and a VL sequence of SEQ ID NO: 17 as described in W02016007235.
- the dosage of such anti-PD-Ll antibody is performed according to the state of the art and described in the respective prescribing information. For example, atezolizumab is administered usually in a concentration of 1200 mg as an intravenous infusion over 60 minutes every 3 weeks (www.accessdata.fda.gov).
- gamma secretase refers to any protein or protein complex that exhibits gamma secretase activities including binding to a substrate having a gamma secretase cleavage sequence, and catalyzing the cleavage of the gamma secretase cleavage sequence, at a gamma secretase cleavage site, to produce substrate cleavage products.
- gamma secretase is a protein complex comprising one or more of the following subunits: presenilin, nicastrin, gamma-secretase subunit APH-1, and gamma-secretase subunit PEN-2.
- gamma secretase inhibitor refers to any molecule capable of inhibiting or reducing expression and/or function of gamma secretase.
- the GSI reduces expression and/or function of a subunit of gamma secretase (e.g., presenilin, nicastrin, APH-1, or PEN-2). Any form of a “gamma secretase inhibitor” such as a salt, a co-crystal, a crystalline form, a pro-drug, etc., is included within this term.
- the GSI is selected from an antibody or antigen-binding fragment, a small molecule, a protein or peptide and a nucleic acid.
- the invention is based, in part, on methods of treating a patient having a disorder associated with BCMA expression (e.g. BCMA-expressing B-cell cancers, such as multiple myeloma) using dose-escalation dosing regimens with pharmaceutical compositions comprising multispecific (e.g. bispecific) antibodies that bind to CD3 and BCMA, e.g. alnuctamab.
- the methods are expected to reduce or inhibit unwanted treatment effects, such as cytokine release syndrome (CRS), thereby treating the patient while achieving a more favorable benefit-risk profile.
- a “subject” includes any human or nonhuman animal.
- nonhuman animal includes, but is not limited to, vertebrates such as nonhuman primates, sheep, dogs, and rodents such as mice, rats and guinea pigs.
- the subject is a human.
- subject and patient are used interchangeably herein.
- a “disorder associated with BCMA expression” is a plasma cell disorder or a B cell disorder which correlates with enhanced BCMA expression.
- Plasma cell disorders include BCMA-expressing B-cell cancer, plasmacytoma, plasma cell leukemia, multiple myeloma, macroglobulinemia, amyloidosis, Waldenstrom's macroglobulinemia, solitary bone plasmacytoma, extramedullar plasmacytoma, osteosclerotic myeloma (POEMS Syndrome) and heavy chain diseases as well as the clinically unclear monoclonal gammopathy of undetermined significance/smoldering multiple myeloma.
- POEMS Syndrome osteosclerotic myeloma
- the B cell disorder is a BCMA-expressing B-cell cancer, such as multiple myeloma.
- Multiple myeloma is a plasma cell malignancy characterized by a monoclonal expansion and accumulation of abnormal plasma cells in the bone marrow compartment.
- Multiple myeloma also involves circulating clonal plasma cells with same IgG gene rearrangement and somatic hypermutation.
- Multiple myeloma arises from an asymptomatic, premalignant condition called monoclonal gammopathy of unknown significance (MGUS), characterized by low levels of bone marrow plasma cells and a monoclonal protein. Multiple myeloma cells proliferate at low rate.
- MGUS monoclonal gammopathy of unknown significance
- Multiple myeloma results from a progressive occurrence of multiple structural chromosomal changes (e.g. unbalanced translocations).
- Multiple myeloma involves the mutual interaction of malignant plasma cells and bone marrow microenvironment (e.g. normal bone marrow stromal cells).
- Clinical signs of active multiple myeloma include monoclonal antibody spike, plasma cells overcrowding the bone marrow, lytic bone lesions and bone destruction resulting from overstimulation of osteoclasts (Dimopulos & Terpos, Ann Oncol 2010; 21 suppl 7: vii 143-150).
- a method comprises treating a patient having an autoimmune disease by administering a pharmaceutical composition provided herein.
- the autoimmune disease is associated with plasma cells or B cells expressing BCMA.
- the autoimmune disease is systemic lupus erythematosus (SLE), IgA nephropathy, IgG4 related disease, membranous nephropathy, Myasthenia gravis, Neuromyelitis optica, Pemphigus vulgaris, anti-PAD4- activating rheumatoid arthritis, Sensitized / preformed antibodies in solid organ transplant, Guillain-Barre Syndrome (Acute inflammatory demyelinating polyneuropathy - AIDP), Chronic inflammatory demyelinating polyneuropathy (CIDP), Immune thrombocytopenic purpura, rheumatoid arthritis (RA), or ANCA-associated vasculitis (AAV).
- SLE systemic lupus erythematosus
- the autoimmune disease is idiopathic inflammatory myopathy (IIM) or systemic sclerosis (SSc).
- the pharmaceutical composition comprises at least about 0.05 mg/mL of the multispecific antibody, e.g. alnuctamab.. In some aspects, the pharmaceutical composition comprises at least about 0.06 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.07 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.08 mg/mL of the multispecific antibody, e.g. alnuctamab.
- the pharmaceutical composition comprises at least about 0.09 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.1 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.2 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.3 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.4 mg/mL of the multispecific antibody, e.g. alnuctamab.
- the pharmaceutical composition comprises at least about 0.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.6 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.7 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.8 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.9 mg/mL of the multispecific antibody, e.g. alnuctamab.
- the pharmaceutical composition comprises at least about 1 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 1.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 2 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 3 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 3.5 mg/mL of the multispecific antibody, e.g. alnuctamab.
- the pharmaceutical composition comprises at least about 4 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 4.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 5.5 mg/mL of the multispecific antibody, e.g. alnuctamab.
- the terms “treatment,” “treating,” and the like refer to obtaining a desired pharmacologic and/or physiologic effect.
- the effect is therapeutic, i.e., the effect partially or completely cures a disease and/or adverse symptom attributable to the disease.
- the pharmacologic and/or physiologic effect may be prophylactic, i.e., the effect completely or partially prevents a disease or symptom thereof.
- administering refers to the physical introduction of a composition comprising a therapeutic agent to a subject, using any of the various methods and delivery systems known to those skilled in the art.
- Administration can refer to any form of administration for the pharmaceutical composition comprising multispecific (e.g. bispecific) antibody, include intravenous, intramuscular, subcutaneous, intraperitoneal, spinal or other parenteral routes of administration, for example by injection or infusion.
- the phrases “subcutaneous administration” and “subcutaneous injection” are used interchangeably and refer to modes of administration wherein a substance, e.g., a pharmaceutical composition comprising multispecific (e.g. bispecific) antibody is delivered to a subject under the skin, between the dermis and, e.g., the muscle.
- Administering can be performed, for example, once, a plurality of times, and/or over one or more extended periods.
- administering can refer to a single unit dose or more than one unit dose.
- dose is defined as an amount of a therapeutic agent that can be administered at a given point.
- the dose or dosage can be an amount sufficient to achieve or at least partially achieve a desired effect, but such a desired effect may not be visible or detectable.
- a “therapeutically effective amount” or “therapeutically effective dosage” of a drug or therapeutic agent is any amount of the drug that, when used alone or in combination with another therapeutic agent, promotes disease regression evidenced by a decrease in severity of disease symptoms, an increase in frequency and duration of disease symptom-free periods, an increase in overall survival (the length of time from either the date of diagnosis or the start of treatment for a disease, such as cancer, that patients diagnosed with the disease are still alive), or a prevention of impairment or disability due to the disease affliction.
- An amount or dosage of a drug includes a “prophylactically effective amount” or a “prophylactically effective dosage”, which is any amount of the drug that, when administered alone or in combination with another therapeutic agent to a subject at risk of developing a disease or of suffering a recurrence of disease, inhibits the development or recurrence of the disease.
- a therapeutically effective amount or a “prophylactically effective dosage”
- the ability of a therapeutic agent to promote disease regression or inhibit the development or recurrence of the disease can be evaluated using a variety of methods available to the skilled practitioner, such as in human subjects during clinical trials, in animal model systems predictive of efficacy in humans, or by assaying the activity of the agent in in vitro assays.
- a “dose” can comprise a single unit dose or multiple unit doses.
- the dose comprises a single unit dose. In some aspects, the dose comprises multiple unit doses. In some aspects, multiple subcutaneous doses are administered to achieve a therapeutically effective dose. When multiple unit doses are administered, individual unit doses can be administered at the same time or sequentially.
- a subcutaneous “unit dose” refers to a single amount of a substance delivered by a subcutaneous injection, e.g., from a single vial, a single auto-injector, and/or a single syringe.
- at least one of the subcutaneous unit doses has a total volume of less than about 5 mb (e.g., about 4.5 mb, about 4.0 mb, about 3.5 mb, about 3.0 mb, about 3 mb, about 2.5 mb, about 2.0 mb, about 1.5 mb, about 1.0 mb, or about 0.5 mb).
- the volume of a unit dose is 0.5-2.5 mL, or 0.5-0.9 mL, or 1.0-1.5 mL, or 2.0-2.5 mb, or 0.5, 0.6, 0.7, 0.8, 0.9. 1.0, 1.1, 1.2, 1.3, 1.4, 1.5, 1.6, 1.7, 1.8, 1.9, 2.0, 2.1, 2.2, 2.3, 2.4, or 2.5 mL.
- the use of the term “fixed dose” with regard to the methods and dosages of the disclosure means a dose that is administered to a patient without regard for the weight or body surface area (BSA) of the patient.
- the fixed dose is therefore not provided as a mg/kg dose, but rather as an absolute amount of the agent (e.g., the multispecific (e.g. bispecific) antibody).
- the agent e.g., the multispecific (e.g. bispecific) antibody
- an antibody e.g., 3 mg of a multispecific (e.g. bispecific) antibody.
- the present invention provides a method for treating a disorder associated with BCMA expression (e.g. BCMA-expressing B-cell cancers, such as multiple myeloma) in a patient (e.g. a human), wherein the treatment comprises the administration of a pharmaceutical composition comprising multispecific (e.g. bispecific) antibody that binds to BCMA and CD3, e.g. alnuctamab, in a dosing regimen comprising:
- a maintenance phase wherein a first maintenance dose of the pharmaceutical composition is administered to the patient, optionally followed by at least one additional maintenance dose of the pharmaceutical composition; wherein each maintenance dose is greater than the one or more starting doses.
- the present invention provides a pharmaceutical composition comprising multispecific (e.g. bispecific) antibody that binds to BCMA and CD3, e.g. alnuctamab, for use in treating a disorder associated with BCMA expression (e.g. BCMA-expressing B-cell cancers, such as multiple myeloma) in a patient (e.g. a human), wherein the treatment comprises the administration of the pharmaceutical composition in a dosing regimen which comprises:
- a maintenance phase wherein a first maintenance dose of the pharmaceutical composition is administered to the patient, optionally followed by at least one additional maintenance dose of the pharmaceutical composition; wherein each maintenance dose is greater than the one or more starting doses.
- the starting phase comprises a single fixed dose.
- the starting dose of the pharmaceutical composition is a single fixed dose comprising about 1.5 mg to about 4.5 mg of the multispecific (e.g. bispecific) antibody, e.g. alnuctamab; from about 2 mg to about 4 mg; from about 2.5 mg to about 3.5 mg, e.g. about 3 mg.
- the starting phase comprises two or more starting starting doses of the same concentration.
- the starting dose of the pharmaceutical composition may be administered at a fixed dose of about 1.5 mg to 4.5 mg dose of the multispecific (e.g. bispecific) antibody, e.g. alnuctamab, from about 2 mg to 4 mg, from about 2.5 mg to 3.5 mg, e.g. about 3 mg.
- the multispecific antibody e.g. bispecific
- the subsequent starting dose may be administered to the patient up to 12 weeks after the starting dose that triggered the adverse event.
- the subsequent starting dose may be administered up to 10 weeks after, up to 8 weeks after, up to 6 weeks after, up to 4 weeks after, up to two weeks after, e.g. up to one week after the starting dose that triggered the adverse event.
- the subsequent starting dose may be of the same concentration or lower concentration than the starting dose that triggered the adverse event.
- the starting phase may comprise an additional starting dose administered to the patient up to 12 weeks after the starting dose that triggered the adverse event.
- the additional starting dose may be administered up to 10 weeks after, up to 8 weeks after, up to 6 weeks after, up to 4 weeks after, up to two weeks after, e.g. up to one week after the starting dose that triggered the adverse event.
- the additional starting dose may be of the same concentration or lower concentration than the starting dose that triggered the adverse event.
- the first maintenance dose of the pharmaceutical composition may be administered at a fixed dose of about 4.5 mg to about 7.5 mg of the multispecific (e.g. bispecific) antibody, e.g. alnuctamab; from about 5 mg to about 6 mg; from about 5.5 mg to about 6.5 mg, e.g. about 6 mg.
- the starting dose of the pharmaceutical composition is a (e.g. single) fixed dose of about 3 mg of the multispecific (e.g. bispecific) antibody, e.g. alnuctamab
- the first maintenance dose of the pharmaceutical composition is a fixed dose of about 6 mg of multispecific (e.g. bispecific) antibody, e.g. alnuctamab.
- the maintenance phase comprises two or more maintenance doses of the same concentration or of escalating concentration.
- the maintenance dose of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody is administered as two or more doses of escalating concentration (i.e. increasing doses).
- a subsequent dose can be increased by a particular increment, or by variable increments, until a maximum dose is reached, at which point administration may cease or may continue at the maximum dose.
- the first maintenance dose of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab, is greater than the starting dose and the second (and optionally subsequent) maintenance dose(s) of the pharmaceutical composition is greater than the first maintenance dose.
- subsequent (e.g. third, fourth or fifth) optional maintenance dose(s) of pharmaceutical composition may be the same or greater than the second maintenance dose.
- subsequent (e.g. third, fourth or fifth) optional maintenance dose(s) of the pharmaceutical composition may be the same as the second maintenance dose.
- the first maintenance dose of the pharmaceutical composition may be administered at a fixed dose of about 4.5 mg to 7.5 mg of the multspecific (e.g. bispecific) antibody, e.g. alnuctamab; from about 5 mg to 6 mg; from about 5.5 mg to 6.5 mg, e.g. about 6 mg, and a subsequent (e.g. second, third, fourth or fifth) maintenance dose of the pharmaceutical composition may be administered at a fixed dose greater than the first maintenance dose.
- the multspecific (e.g. bispecific) antibody e.g. alnuctamab
- a subsequent (e.g. second, third, fourth or fifth) maintenance dose of the pharmaceutical composition may be administered at a fixed dose greater than the first maintenance dose.
- the first maintenance dose of the pharmaceutical composition may be administered at a fixed dose of about 4.5 mg to 7.5 mg of the multispecific (e.g. bispecific) antibody, e.g. alnuctamab; from about 5 mg to 6 mg; from about 5.5 mg to 6.5 mg, e.g. about 6 mg, and a subsequent (e.g. second, third, fourth or fifth) maintenance dose of the composition antibody may be administered at a fixed dose of about 25 mg to about 35 mg, about 28 mg to about 32 mg, about 29 mg to about 31 mg, e.g. about 30 mg of the multispecific (e.g. bispecific) antibody, e.g. alnuctamab.
- the multispecific (e.g. bispecific) antibody e.g. alnuctamab.
- the starting dose is a (e.g. single) fixed dose of about 3 mg of the multispecific (e.g. bispecific) antibody, e.g. alnuctamab
- the first maintenance dose of the is a fixed dose of about 6 mg of the multispecific (e.g. bispecific) antibody
- a second (and optionally subsequent) maintenance dose(s) is a fixed dose of about 30 mg of the multispecific (e.g. bispecific) antibody, e.g. alnuctamab.
- the first maintenance dose of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab is administered to the patient 1- 21 days, e.g. 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, or 21 days, after the starting dose.
- the first maintenance dose of the pharmaceutical composition may be administered to the patient 2 days, after the starting dose.
- the first maintenance dose of the pharmaceutical composition may be administered to the patient 3 days after the starting dose.
- the first maintenance dose of the pharmacentical composition may be administered to the patient 7 days after the starting dose.
- the first maintenance dose of the pharmaceutical composition may be administered to the patient 14 days after the starting dose.
- the second maintenance dose of the pharmaceutical composition is administered to the patient 1-21 days, e.g. 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, or 21 days, after the first maintenance dose.
- the second maintenance dose may be administered 4 days after the first maintenance dose.
- the next maintenance dose may be administered to the patient up to 12 weeks after the maintenance dose that triggered the adverse event.
- the next maintenance starting dose may be administered up to 10 weeks after, up to 8 weeks after, up to 6 weeks after, up to 4 weeks after, up to two weeks after, e.g. up to one week after the starting dose that triggered the adverse event.
- the next maintenance dose may be of the same concentration or lower concentration than the maintenance dose that triggered the adverse event.
- the third and subsequent maintenance doses are administered at about a once weekly or longer dosing interval.
- a “dosing interval” means the amount of time that elapses between multiple doses being administered to a patient. If an adverse event (e.g. CRS or infection) occurs following administration of a maintenance dose (e.g. third or subsequent maintenance dose) of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab, the dosing interval may reset on the day the next maintenance dose is administered to the patient.
- an adverse event e.g. CRS or infection
- a maintenance dose e.g. third or subsequent maintenance dose
- the dosing interval may reset on the day the next maintenance dose is administered to the patient.
- the dosing interval for the third and subsequent maintenance doses may be about once weekly.
- a “weekly dosing interval” includes every 5-9, 6-9, 7-9, 5-8, 5-7, 6-8, 6-7, 7-8, preferably 7 days.
- the dosing interval for the third and subsequent maintenance doses may be about once biweekly.
- a “biweekly dosing interval” includes every 12-16, 13-16, 14-16, 12-15, 12-14, 13-15, 13-14, 14-15, preferably 14 days.
- the dosing interval for the third and subsequent maintenance dose may be about once every three weeks.
- a “three week dosing interval” includes every 19-23, 20-23, 21-23, 19-22, 19-21, 20-22, 20-21, 21-22, preferably 21 days.
- the dosing interval for the third and subsequent maintenance dose may be about once every four weeks.
- a “four week dosing interval” includes every 26-30, 27-30, 28-30, 26- 29, 26-28, 27-29, 27-28, 28-29, preferably 28 days.
- the dosing interval for the third and subsequent maintenance doses may be about once monthly.
- the dosing interval for the third and subsequent maintenance dose may be a combination of one or more of a weekly dosing interval, a biweekly dosing interval, a three week dosing interval and a four week dosing interval.
- the dosing interval for the third and subsequent maintenance dose may be a combination of a weekly dosing interval, a biweekly dosing interval, and a four week dosing interval.
- the third and subsequent maintenance doses are administered in a weekly dosing interval (e.g. every 7 days), then a biweekly dosing interval (e.g. every 14 days), then a three week dosing interval (e.g. every 21 days) and then a four week dosing interval (e.g. every 28 days).
- the third and subsequent maintenance doses are administered in a weekly dosing interval (e.g. every 7 days), then a biweekly dosing interval (e.g. every 14 days) and then a four week dosing interval (e.g. every 28 days).
- the treatment comprises at least one treatment cycle of 28 days.
- a “treatment cycle” is 28 days. If a starting dose is administered beyond day 28 of the first treatment cycle as a result of an adverse event (e.g. CRS or infection), the first treatment cycle may restart on the day the starting dose is administered to the patient. If a maintenance dose is administered beyond day 28 of the current treatment cycle as a result of an adverse event (e.g. CRS or infection), the next treatment cycle may begin on the day the maintenance dose is administered to the patient.
- an adverse event e.g. CRS or infection
- the treatment comprises a first treatment cycle, wherein the starting dose is administered to the patient as a fixed dose on day 1, the first maintenance dose is administered to the patient as a fixed dose on day 4, the second maintenance dose is administered to the patient as a fixed dose on day 8, and the subsequent maintenance doses are subsequently administered in a weekly dosing interval (e.g. every 7 days) for three consecutive weeks (e.g. on days 15 and 22).
- the maintenance doses may continue to be administered in a weekly or longer dosing interval in subsequent treatment cycles.
- the treatment comprises a second treatment cycle, wherein the maintenance doses are administered in a weekly dosing interval (e.g. on days 1, 8, 15 and 22).
- the patient remains on a weekly dosing interval for between 1-5, 1-3, 1-2, 2-3 further treatment cycles, preferably 2 further treatment cycles (in addition to the first treatment cycle).
- the treatment comprises a second and third treatment cycle, wherein the maintenance doses are administered in a weekly dosing interval (e.g. on days 1, 8, 15 and 22).
- the maintenance doses may be administered in a biweekly dosing interval in treatment cycles (e.g. on days 1 and 15) after completion of the weekly treatment cycle(s).
- the patient remains on a biweekly dosing interval for between 1-5, 1-3, 1-2, 2-3 biweekly treatment cycles, preferably 3 biweekly treatment cycles.
- the treatment comprises a fourth, fifth and sixth treatment cycle, wherein the maintenance doses are administered in a biweekly dosing interval (e.g. on days 1 and 15).
- the maintenance doses may be administered in a four week dosing interval in subsequent treatment cycles (e.g. on day 1) after completion of the biweekly treatment cycle(s).
- the patient remains on a four week dosing interval for at least one cycle. Some patients continue to receive treatment for the rest of their lives.
- the treatment comprises a first treatment cycle, wherein the starting dose is administered to the patient as a fixed dose on day 1, the first maintenance dose is administered 3 days after the starting dose (e.g. on day 4), the second maintenance dose is administered 4 days after the first maintenance dose (e.g. on day 8), and the third and fourth maintenance doses are administered in a weekly interval (e.g. on days 15 and 22).
- the maintenance doses may continue to be administered in a weekly or longer dosing interval in subsequent treatment cycles.
- the treatment comprises a second treatment cycle, wherein the maintenance doses are administered in a weekly dosing interval (e.g. on days 1, 8, 15 and 22).
- the patient remains on a weekly dosing interval for between 1-5, 1-3, 1-2, 2-3 further treatment cycles, preferably 2 further treatment cycles (in addition to the first treatment cycle).
- the treatment comprises a second and third treatment cycle, wherein the maintenance doses are administered in a weekly dosing interval (e.g. on days 1, 8, 15 and 22).
- the maintenance doses may be administered in a biweekly dosing interval in treatment cycles (e.g. on days 1 and 15) after completion of the weekly treatment cycle(s).
- the patient remains on a biweekly dosing interval for between 1-5, 1-3, 1-2, 2-3 weekly treatment cycles, preferably 3 biweekly treatment cycles.
- the treatment comprises a fourth, fifth and sixth treatment cycle, wherein the maintenance doses are administered in a biweekly dosing interval (e.g. on days 1 and 15).
- the maintenance doses may be administered in a four week dosing interval in subsequent treatment cycles (e.g. on day 1) after completion of the biweekly treatment cycle(s).
- the patient remains on a four week dosing interval for at least one cycle. Some patients continue to receive treatment for the rest of their lives.
- the treatment comprises:
- a fourth to sixth treatment cycle wherein the maintenance doses are administered in a biweekly dosing interval (e.g. on days 1 and 15); and (iv) a seventh and subsequent cycle, wherein the maintenance doses are administered in a four week dosing interval (e.g. on day 1)
- the pharmaceutical composition comprising the multispecific
- bispecific antibody e.g. “42-TCBcv”
- 42-TCBcv may be administered to the patient in accordance with the regimen set out in Table 4.
- the maintenance doses of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody are administered as two or more doses of escalating concentration.
- the starting dose of the pharmaceutical composition is a fixed dose of about 1.5 mg to 4.5 mg; about 2 mg to 4 mg; about 2.5 mg to 3.5 mg, e.g. about 3 mg, of the multispecific (e.g. bispecific) antibody, e.g. alnuctamab.
- the first maintenance dose may be administered at a fixed dose of about 4.5 mg to 7.5 mg; about 5 mg to 7 mg; about 5.5 mg to 6.5 mg, e.g. about 6 mg, of the multispecific (e.g. bispecific) antibody, e.g. alnuctamab
- the second (and optionally subsequent) maintenance dose(s) of the pharmaceutical composition may be administered at a fixed dose greater than the first maintenance dose.
- the second (and optionally subsequent) maintenance dose(s) of the pharmaceutical composition may be administered at a fixed dose of about 25 mg to about 35 mg, about 28 mg to about 32 mg, about 29 mg to about 31 mg, e.g. about 30 mg, of the multispecific (e.g. bispecific) antibody, e.g. alnuctamab.
- the starting dose of the pharmaceutical composition is a (e.g. single) fixed dose of about 3 mg of the multispecific (e.g. bispecific) antibody
- the first maintenance dose of the pharmaceutical composition is a fixed dose of about 6 mg of the multispecific (e.g. bispecific) antibody
- the second (and optionally subsequent) maintenance dose(s) of the pharmaceutical composition is a fixed dose of about 30 mg of the multispecific (e.g. bispecific) antibody, e.g. alnuctamab.
- the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody may be administered to the patient in accordance with the regiment set out in Table 5.
- the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody discussed herein is administered intravenously or subcutaneously. In some embodiments, the pharmaceutical composition is administered subcutaneously. In some embodiments, the multispecific (e.g. bispecific) antibody is “42-TCBcv”, and the pharmaceutical composition is administered subcutaneously.
- the starting dose and the first maintenance dose of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab may be administered subcutaneously, and a subsequent (e.g. second, third, fourth or fifth) maintenance dose of the pharmaceutical composition may be administered subcutaneously.
- the multispecific antibody e.g. bispecific
- a subsequent (e.g. second, third, fourth or fifth) maintenance dose of the pharmaceutical composition may be administered subcutaneously.
- the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody may be administered to the patient in accordance with any of the regimens set out in Tables 4-5, wherein the starting dose and the first maintenance dose of the multispecific (e.g. bispecific) antibody may be administered subcutaneously, and a subsequent (e.g. second, third, fourth or fifth) maintenance dose of the multispecific (e.g. bispecific) may be administered subcutaneously.
- the starting dose and the first maintenance dose of the multispecific (e.g. bispecific) antibody may be administered subcutaneously
- a subsequent (e.g. second, third, fourth or fifth) maintenance dose of the multispecific (e.g. bispecific) may be administered subcutaneously.
- the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody may be administered to the patient in accordance with any of the regimens set out in Tables 4-5, wherein cycles 1-2 may be administered subcutaneously, and cycles 3+ may be administered subcutaneously.
- the patient develops, or is at risk of developing, an adverse event associated with the administration of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab.
- the adverse event may be cytokine-driven toxicities (e.g. cytokine release syndrome (CRS)), infusion-related reactions (IRRs), macrophage activation syndrome (MAS), neurologic toxicities, severe tumor lysis syndrome (TLS), neutropenia, thrombocytopenia, elevated liver enzymes, bacterial infections, viral infections, and/or central nervous system (CNS) toxicities.
- CRS central nervous system
- the adverse event is CRS.
- the treatment according to any aspect of the invention further comprises the administration of an agent capable of treating, preventing, delaying, reducing or attenuating the development or risk of development of the adverse event.
- the agent may be administered to the patient prior to the initiation of the treatment with the pharmaceutical composition comprsing the multispecific (e.g. bispecific) antibody (e.g. as a prophylaxis in order to prevent or reduce the risk of an adverse event developing) or during treatment with the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody (e.g.
- the agent comprises a steroid, such as a corticosteroid.
- corticosteroid means any naturally occurring or synthetic steroid hormone that can be derived from cholesterol and is characterized by a hydrogenated cyclopentanoperhydrophenanthrene ring system.
- Naturally occurring corticosteroids are generally produced by the adrenal cortex. Synthetic corticosteroids may be halogenated. Functional groups required for activity include a double bond at A4, a C3 ketone, and a C20 ketone.
- Corticosteroids may have glucocorticoid and/or mineralocorticoid activity.
- Examples of exemplary corticosteroids include prednisolone, methylprednisolone, prednisone, triamcinolone, betamethasone, budesonide, and dexamethasone.
- the agent is dexamethasone.
- the agent comprises an antagonist of a cytokine receptor or cytokine selected from among GM-CSF, IL- 10, IL-10R, IL-6, IL-6 receptor (IL-6R), IFNy, IFNGR, IL-2, IL-2R/CD25, MCP-1, CCR2, CCR4, MIPip, CCR5, TNFalpha, TNFR1, IL-1 (e g. IL-la, IL-lp, IL- 1RA), and IL-1 receptor (IL-1R), wherein the antagonist is selected from an antibody or antigen-binding fragment, a small molecule, a protein or peptide and a nucleic acid.
- a cytokine receptor or cytokine selected from among GM-CSF, IL- 10, IL-10R, IL-6, IL-6 receptor (IL-6R), IFNy, IFNGR, IL-2, IL-2R/CD25, MCP-1, CCR2, CCR4, MIPip, CCR5, TNF
- the antagonist may be an anti-IL-6 antibody and/or an anti-IL6R antibody.
- the antagonist may be selected from tocilizumab, siltuximab, clazakizumab, sarilumab, olokizumab, elsilimomab, ALD518/BMS-945429, sirukumab (CNTO 136), CPSI-2634, ARGX-109, lenzilumab, FE301 and FM101.
- the antagonist is tocilizumab and/or siltuximab.
- the antagonist may be an anti -IL- 1 antagonist and/or an anti-IL-lR antagonist e.g. anakinra.
- the agent comprises a molecule that decreases the regulatory T cell (Treg) population.
- Agents that decrease the number of (e.g., deplete) Treg cells are known in the art and include, e.g., CD25 depletion, cyclophosphamide administration, anti-CTLA4 antibody and modulating Glucocorticoid-induced TNLR family related gene (GITR) function.
- GITR is a member of the TNLR superfamily that is upregulated on activated T cells, which enhances the immune system.
- the treatment comprises the administration of cyclophosphamide.
- the agent capable of treating, preventing, delaying, reducing or attenuating the development or risk of development of the adverse event is administered as one or more doses to the patient prior to the initiation of the treatment with the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody as a prophylactic treatment for the adverse event.
- the agent capable of treating, preventing, delaying, reducing or attenuating the development or risk of development of the adverse event is administered to the patient in combination with one or more dose of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody as a prophylactic treatment for the adverse event.
- the agent may be administered as one or more doses consecutively (before and/or after), and/or concurrently with the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody.
- the agent capable of treating, preventing, delaying, reducing or attenuating the development or risk of development of the adverse event is administered to the patient in combination with the first dose of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab, as a prophylactic treatment for the adverse event.
- the agent may be administered as one or more doses consecutively (before and/or after), and/or concurrently with the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab.
- the agent capable of treating, preventing, delaying, reducing or attenuating the development or risk of development of the adverse event is administered to the patient in combination with each increase in dose of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab, as a prophylactic treatment for the adverse event.
- the agent may be administered as one or more doses consecutively (before and/or after), and/or concurrently with the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab.
- the prophylactic treatment comprises administration of the agent (e.g. CRS agent) at an amount sufficient to prevent, delay, reduce or attenuate the development or risk of development of the adverse event (e.g. CRS).
- the agent e.g. CRS agent
- the prophylactic treatment comprises the administration of a corticosteroid, such as dexamethasone.
- a corticosteroid such as dexamethasone.
- the dexamethasone is administered at a dose of about 10-20 mg, preferably intravenously.
- dexamethasone is administered as a prophylactic treatment for a cytokine-driven toxicity (e.g. CRS)
- cytokines e.g. GM-CSF, IL-2 and/or TNF-a
- the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody of the invention, e.g. alnuctamab.
- the prophylactic treatment comprises the administration of an antagonist of a cytokine receptor or cytokine, such as an antagonist of IL-6, an IL-6 receptor (IL-6R), IL-1 (e.g. IL- la, IL- 1 , IL- IRA) and/or an IL-1 receptor (IL-1R) wherein the antagonist is selected from an antibody or antigen-binding fragment, a small molecule, a protein or peptide and a nucleic acid.
- an antagonist of a cytokine receptor or cytokine such as an antagonist of IL-6, an IL-6 receptor (IL-6R), IL-1 (e.g. IL- la, IL- 1 , IL- IRA) and/or an IL-1 receptor (IL-1R) wherein the antagonist is selected from an antibody or antigen-binding fragment, a small molecule, a protein or peptide and a nucleic acid.
- an antagonist of a cytokine receptor or cytokine such as an antagonist of
- the prophylactic treatment comprises an anti -IL-6 antagonist antibody and/or an anti-IL-6R antagonist antibody, e.g. tocilizumab.
- tocilizumab is administered to the patient as a one or more doses of about 8 mg/kg, preferably intravenously.
- tocilizumab is administered at least 30 minutes prior to the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody.
- tocilizumab is administered as a prophylactic treatment for a cytokine-driven toxicity (e.g. CRS)
- tocilizumab is administered at an amount sufficient to attenuate IL-6 receptor signalling induced by the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody of the invention, e.g. alnuctamab.
- the prophylactic treatment comprises an anti-IL-1 antagonist and/or an anti-IL-lR antagonist, e.g. anakinra.
- anakinra is administered as a prophylactic treatment for a cytokine -driven toxicity (e.g. CRS), preferably at an amount sufficient to attenuate IL-1 receptor signalling induced by the multi specific (e.g. bispecific) antibody of the invention.
- Anakinra may be administered at a dose of about 100 mg (e.g. 100 mg ⁇ 20%), preferably subcutaneously.
- anakinra is administered to the patient as a dose of about 100 mg, preferably subcutaneously.
- the prophylactic treatment comprises at least one dose of anakinra administered before the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, and at least one dose of anakinra administered after the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody.
- Anakinra may be administered to the patient as one or more fixed dose(s) between about 16 hours to about 2 hours prior to the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, and optionally a fixed dose between about 20 hours to about 22 hours after the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody.
- anakinra is administered as:
- a fixed dose between about 16 hours to about 8 hours prior to the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab; and/or
- a fixed dose between about 4 hours to about 2 hours prior to the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab, optionally wherein an additional fixed dose of anakinra is administered between about 20 hours to about 22 hours after the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody.
- the multispecific antibody e.g. bispecific
- an additional fixed dose of anakinra is administered between about 20 hours to about 22 hours after the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody.
- the prophylactic treatment comprises the administration of dexamethasone (e.g. about 10-20 mg, preferably intravenously) with tocilizumab (e.g. about 8 mg/kg, preferably intravenously).
- the prophylactic treatment comprises the administration of dexamethasone (e.g. about 10-20 mg, preferably intravenously) with anakinra (e.g. about 100 mg, preferably subcutaneously).
- the prophylactic treatment comprises the administration of symptomatic support, including administration of antipyretics, analgesics, antivirals and/or antibiotics.
- the symptomatic support comprises the administration of antivirals (e.g. acyclovir, oseltamivir, zanamivir and/or equivalents) and/or antibiotics (e.g. trimethoprim-sulfamethoxazole, levofloxacin and/or equivalents).
- the prophylactic treatment comprises the administration of seizure prophylaxis (e.g. levetiracetam).
- the symptomatic support and/or seizure prophylaxis may be administered in addition to the agent capable of treating, preventing, delaying, reducing or attenuating the development or risk of development of the adverse event.
- the agent capable of treating, preventing, delaying, reducing or attenuating the development or risk of development of the adverse event is administered to the patient in the event that the patient develops an adverse event associated with the administration of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody.
- the treatment comprises administration of the agent at a therapeutic amount, or an amount sufficient to partially or completely alleviate or ameliorate the adverse event (e.g. CRS) or symptoms thereof.
- the treatment may further comprise the administration of an anti-IL-6R antagonist antibody, e.g., tocilizumab.
- an anti-IL-6R antagonist antibody e.g., tocilizumab.
- tocilizumab is administered to the patient as a single dose of about 8 mg/kg, preferably intravenously.
- the treatment may further include administering to the patient one or more additional doses of an IL-6R antagonist antibody, e.g., tocilizumab.
- tocilizumab is administered to the patient in one or more additional doses of about 8 mg/kg, preferably intravenously.
- the treatment may further comprise the administration of an anti -IL- 1 antagonist and/or an anti-IL-lR antagonist, e.g. anakinra.
- anakinra is administered to the patient as one more fixed doses of about 100 mg, preferably subcutaneously.
- anakinra is administered to the patient twice daily, preferably as fixed doses of about 100 mg, preferably subcutaneously.
- the treatment may further comprise the administration of an IL-6 antagonist antibody, e.g., siltuximab.
- an IL-6 antagonist antibody e.g., siltuximab.
- siltuximab is administered to the patient as a single dose of about 11 mg/kg, preferably intravenously.
- the treatment may further comprise administering to the patient a corticosteroid, such as methylprednisolone or dexamethasone.
- a corticosteroid such as methylprednisolone or dexamethasone.
- the dexamethasone is administered at a dose of about 10-20 mg, preferably intravenously.
- the methylprednisolone is administered at a dose of about 1 mg/kg per day to about 5 mg/kg per day, e.g., about 2 mg/kg per day.
- the additional treatments may be based on the stage of the CRS.
- a modification of the common CTCAE CRS grading scale has been established for the grading and treatment of CRS, and is detailed in Table 6: Table 6: Grading and Treatment of Cytokine Release Syndrome
- the treatment may further comprise the administration of a first line treatment comprising the administration of a first dose of an anti-IL-6 antagonist antibody and/or an anti-IL-6R antagonist antibody, e.g., tocilizumab.
- tocilizumab is administered intravenously to the patient as a single dose of about 8 mg/kg.
- the treatment may further comprise the administration of a first line treatment comprising the administration of one or more fixed dose(s) of anti-IL-1 antagonist and/or an anti-IL-lR antagonist, e.g. anakinra.
- Anakinra may be administered at a dose of about 100 mg (e.g. 100 mg ⁇ 20%), preferably subcutaneously.
- anakinra is administered to the patient as one more fixed dose(s) of about 100 mg, preferably subcutaneously.
- anakinra is administered to the patient twice daily, preferably as fixed doses of about 100 mg, preferably subcutaneously.
- the treatment may further comprise the administration of a first line treatment comprising:
- an anti-IL-6 antagonist antibody and/or an anti-IL-6R antagonist antibody e.g., tocilizumab
- tocilizumab is administered intravenously to the patient at a dose of about 8 mg/kg.
- the treatment may further comprise the administration of a first line treatment comprising:
- an anti-IL-1 antagonist and/or an anti-IL-lR antagonist e.g. anakinra
- a corticosteroid e.g. dexamethasone or methylprednisolone.
- anakinra is administered to the patient as one more fixed dose(s) of about 100 mg, preferably subcutaneously. In some embodiments, anakinra is administered to the patient twice daily, preferably as fixed doses of about 100 mg, preferably subcutaneously.
- the corticosteroid may be administered consecutively (before or after) or concurrently with the (i) anti-IL-6 antagonist antibody and/or an anti-IL-6R antagonist antibody, e.g., tocilizumab, or (ii) anti-IL-1 antagonist and/or an anti-IL-lR antagonist, e.g. anakinra.
- the corticosteroid is dexamethasone.
- the dexamethasone is administered at a dose of about 10-20 mg, preferably intravenously.
- the corticosteroid is methylprednisolone.
- methylprednisolone is administered at a dose of about 1 mg/kg per day to about 5 mg/kg per day, e.g., about 2 mg/kg per day.
- the first line treatment comprises the administration of symptomatic support for CRS, including administration of antipyretics, analgesics and/or antibiotics.
- the first line treatment comprises the administration of seizure prophylaxis (e.g. levetiracetam).
- seizure prophylaxis e.g. levetiracetam
- the symptomatic support and/or seizure prophylaxis may be administered in addition to the agent capable of treating, preventing, delaying, reducing or attenuating the development or risk of development of the adverse event.
- the next dose (e.g. next starting dose or next maintenance dose) may be administered to the patient when toxicity reaches Grade ⁇ 1 as described herein.
- the next dose may be administered to the patient when toxicity reaches baseline levels.
- the treatment may further comprise the administration of a second line treatment comprising:
- one or more e.g., one, two, three, four, or five or more
- additional doses of the corticosteroid e.g. dexamethasone or methylprednisolone.
- the one or more additional doses of tocilizumab are administered intravenously to the patient at a dose of about 8 mg/kg.
- the corticosteroid may be administered consecutively (before or after) or concurrently with the anti-IL-6 antagonist antibody and/or an anti-IL- 6R antagonist antibody, e.g., tocilizumab.
- the corticosteroid is dexamethasone.
- the dexamethasone is administered at a dose of about 10-20 mg, preferably intravenously.
- the corticosteroid is methylprednisolone.
- methylprednisolone is administered at a dose of about 1 mg/kg per day to about 5 mg/kg per day, e.g., about 2 mg/kg per day.
- the treatment may further comprise the administration of a third line treatment comprising the administration of an antagonist of a cytokine receptor or cytokine selected from among GM-CSF, IL- 10, IL-10R, IL-6, IL-6 receptor (IL-6R), IFNy, IFNGR, IL-2, IL-2R/CD25, MCP-1, CCR2, CCR4, MIPip, CCR5, TNFalpha, TNFR1, IL-1 (e.g.
- IL-la IL-la
- IL- ip IL-IRA
- IL-1 receptor IL-1 receptor
- the antagonist is selected from an antibody or antigen-binding fragment, a small molecule, a protein or peptide and a nucleic acid.
- the antagonist may be an anti-IL-6 antibody and/or an anti-IL6R antibody.
- the antagonist may be selected from tocilizumab, siltuximab, clazakizumab, sarilumab, olokizumab, elsilimomab, ALD518/BMS-945429, sirukumab (CNTO 136), CPSI-2634, ARGX-109, lenzilumab, FE301 and FM101.
- the third line treatment comprises the administration of siltuximab.
- siltuximab is administered to the patient as a single dose of about 11 mg/kg, preferably intravenously.
- the antagonist may be an anti -IL- 1 antagonist and/or an anti-IL-lR antagonist e.g. anakinra.
- the treatment may further comprise the administration of a fourth line treatment comprising the administration of a molecule that decreases the regulatory T cell (Treg) population.
- a fourth line treatment comprising the administration of a molecule that decreases the regulatory T cell (Treg) population.
- Molecules that decrease the number of (e.g., deplete) Treg cells are known in the art and include, e.g., CD25 depletion, cyclophosphamide administration, anti-CTLA4 antibody and modulating Glucocorticoid-induced TNLR family related gene (GITR) function.
- GITR is a member of the TNLR superfamily that is upregulated on activated T cells, which enhances the immune system.
- the fourth line treatment comprises the administration of cyclophosphamide.
- the treatment may further comprise symptomatic support, including administration of antipyretics, analgesics, antivirals and/or antibiotics.
- seizure prophylaxis e.g. levetiracetam
- the treatment may further comprise administration of antibiotics (e.g. levofloxacin or equivalent).
- the treatment may further comprise administration of oseltamivir, zanamivir and/or equivalents.
- the next dose (e.g. next starting dose or next maintenance dose) may be administered to the patient when symptoms of the infection resolve.
- tire next dose may be administered after a negative test for tire viral infection, e.g. a negative PCJR viral panel, and/or at least 14 days after a positive test for the viral infection, e.g. a positive PCJR viral panel.
- a viral panel (e.g. PCR viral panel) may test for influenza A/B, respiratory syncytial virus, parainfluenza virus, metapneumovirus, adenovirus and/or SARS-CoV-2.
- Clinical formulations for the pharmaceutical composition injection 12 mg/2.0 mL, 60 mg/2.0 mL, 3 mg/0.5 mL, and 30 mg/1.0 mL are sterile, non-pyrogenic, single-use, preservative-free, isotonic aqueous solutions for subcutaneous administration. They are also referred to as alnuctamab injection.
- the clinical drug products shown in Table 9 are packaged in a 6-cc Type I glass vial, stoppered with a 20-mm chlorobutyl rubber serum stopper, and sealed with a 20-mm flip-off aluminum seal.
- DTPA dipentetic acid
- HMW High molecular weight forms
- LMW low molecular weight forms
- purity purity, and charge variant were monitored for up to 24 months.
- CE-SDS Capillary electrophoresis sodium dodecyl sulphate
- NR non-reducing
- R reducing
- Cation exchange chromatography CEX was used to assess acidic species and % main peak under each condition and concentration. No concentration-dependent change in charge profile was observed, and acceptable stability was observed at 5°C across all concentrations. See FIG. 5A-5B.
- alnuctamab for formulations comprising alnuctamab at 10 mg/mL, 50 mg/mL, and 100 mg/mL, at the storage condition of 5 °C, alnuctamab showed good stability in current clinical formulation up to 24 months at 10 mg/mL and 18 months at 50 and 100 mg/mL.
- compositions containing 44 mg/mL or 1 mg/mL alnuctamab, 20mM histidine, 250mM sucrose, and 0.04% (w/v) polysorbate 80, at pH 6.0, with or without 50 pM DTPA were prepared.
- DTPA evaluation was performed in 2R vials and formulations were placed at 5°C, 25°C, or 40°C.
- High molecular weight forms (HMW), monomer (single molecule alnuctamab), low molecular weight forms (LMW), main peak, acidic species, and polysorbate 80 concentrations were monitored for up to 12 months for the 44 mg/mL alnuctamab composition and up to 6 months for the 1 mg/mL alnuctamab composition.
- the formulations comprising 1 mg/mL of alnuctamab with or without 50 pM DTPA were inspected for visual precipitation / aggregation at 0, 1, and 3 months. Visual precipitation / aggregation was observed for the Hl formulation (no DTPA) at 40°C after 3 months.
- the formulations were also tested for subvisible particulate matter by HIAC particle counter for particles > 10pm (See Table 13 A) or particles > 25 pm at 0 months, 1 month, 3 months, and 6 months represented as number of particulate/mL (See Table 13B).
- formulations that include DTPA were shown to have better physical and chemical stability at 25 °C and 40°C, and the benefit of adding DTPA is more evident at 1 mg/mL anuctamab formulations.
- formulation with DTPA showed better physical and chemical stability under stress conditions tested.
- Formulations without DTPA in various protein concentrations showed acceptable stability at 5 °C (up to 24 months), but stability studies at accelerated conditions (25 °C and 40°C) showed that formulations with 50 pM of DTPA have stabilizing effects on both physical and chemical stability.
- Polysorbate 80 degradation/oxidation was mitigated with 50 pM DTPA in metal spiked formulations comprising 60 mg/mL and 5 mg/mL alnuctamab.
- Metal spiking studies showed beneficial effect on both physical and chemical stability in metal spiked samples. No detrimental effects of DTPA addition were observed on quality attributes monitored in the stability studies.
- 50 pM DTPA was included in the formulation, to arrive at final formulation comprising 20 mM histidine, 250 mM sucrose, 0.04% polysorbate 80, 50 pM DTPA, at pH 6.0.
- the two Arg residues are sterically close to the CDR, containing two Trp residues which could be oxidized, and two Asn residues which could be deamidated. Prediction of binding site was supported by native fluorescence spectroscopy in which Trp fluorescence blue shift was observed, as an indicator of decreased hydrophobicity. Also, Raman spectroscopy revealed a minor rotation of the Trp side chain. The binding site was confirmed by carbene footprints. After adding DTPA, label uptake in anti-CD3 CDR was found to decrease, indicating anti-CD3 CDR overlaps with binding sites. Label uptake in most other regions of the protein did not decrease, indicating anti-CD3 CDR is most likely the only binding site of DTPA. In summary, it was discovered DTPA binds to alnuctamab, binding is likely to be driven by charge-charge interaction, and the binding site is most likely in the anti-CD3 CDR.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Immunology (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Mycology (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
Provided herein are compositions comprising multispecific (e.g. bispecific) antibodies that bind to CD3 and BCMA, such as alnuctamab, and methods of using such compositions to treat a patient having a disorder associated with BCMA expression (e.g. BCMA-expressing B-cell cancers, such as multiple myeloma).
Description
COMPOSITIONS COMPRISING ANTIBODIES THAT BIND BCMA AND CD3 AND METHODS OF TREATMENT
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] This application claims the benefit of priority of US Provisional Application No. 63/520,561, filed August 18, 2023, which is incorporated by reference herein in its entirety for any purpose.
REFERENCE TO SEQUENCE LISTING
[0002] The present application is being filed with a Sequence Listing in electronic format. The sequence listing filed, entitled 01277-0047-00PCT.xml was created on August 7, 2024, and is 34,418 bytes in size. The information in electronic format of the Sequence Listing is incorporated herein by reference in its entirety.
FIELD
[0003] Provided herein are compositions comprising antibodies that bind BCMA and CD3, as well as their use in treating disorder associated with BCMA expression (e.g. BCMA-expressing B-cell cancers, such as multiple myeloma).
BACKGROUND
[0004] Alnuctamab is a bispecific antibody that binds BCMA and CD3 and is currently in clinical trials for treatment of cancer, including multiple myeloma. There is a need for formulations of alnuctamab that provide convenient dosing and enhanced stability.
SUMMARY
[0005] The present disclosure provides pharmaceutical compositions comprising alnuctamab and methods of using such compositions to treat a patient having a disorder associated with BCMA expression (e.g. BCMA-expressing B-cell cancers, such as multiple myeloma).
Embodiment 1. A pharmaceutical composition comprising:
(a) at least about 0.05 mg/mL to at least about 37.5 mg/mL of a multispecific antibody that binds to BCMA and CD3, at least about 0.5 mg/mL to at least about 37.5 mg/mL of a multispecific antibody that binds to BCMA and CD3, or at least about 4.5 mg/mL to at least about 37.5 mg/mL of a multispecific antibody that binds to BCMA and CD3,
(b) at lest about 5 mM to at least about 100 mM histidine,
(c) at least about 10 mM to at least about 500 mM sucrose,
(d) at least about 0.01% w/v to at least about 0.1% w/v polysorbate 80, and
(e) at least about 10 pM to about 200 pM pentetic acid (DTPA).
Embodiment 2. The pharmaceutical composition of embodiment 1, comprising about 4.5 mg/mL to 7.5 mg/mL of the multispecific antibody.
1
SUBSTITUTE SHEET (RULE 26)
Embodiment 3. The pharmaceutical composition of embodiment 1, comprising about 6 mg/mL of the multispecific antibody.
Embodiment 4. The pharmaceutical composition of embodiment 1, comprising about 22.5 mg/mL to 37.5 mg/mL of the multispecific antibody.
Embodiment 5. The pharmaceutical composition of embodiment 1, comprising about 30 mg/mL of the multispecific antibody.
Embodiment 6. The pharmaceutical composition of embodiment 1, comprising:
(a) about 6 mg/mL of the multispecific antibody;
(b) about 20 mM histidine;
(c) about 250 mM sucrose;
(d) about 0.04% w/v polysorbate 80; and
(e) about 50 pM pentetic acid.
Embodiment 7. The pharmaceutical composition of embodiment 1, comprising:
(a) about 30 mg/mL of the multispecific antibody;
(b) about 20 mM histidine;
(c) about 250 mM sucrose;
(d) about 0.04% w/v polysorbate 80; and
(e) about 50 pM pentetic acid.
Embodiment 8. The pharmaceutical composition of any one of embodiments 1-7, wherein the pH of the composition is from about 5.7 to about 6.3.
Embodiment 9. The pharmaceutical composition of any one of embodiments 1-8, wherein the pH of the composition is about 6.0.
Embodiment 10. The pharmaceutical composition of any one of embodiments 1-
9, wherein the multispecific antibody comprises: an anti-BCMA antibody, or antigen binding fragment thereof, comprising a VH region comprising a CDR1H region of SEQ ID NO:21, a CDR2H region of SEQ ID NO:22 and a CDR3H region of SEQ ID NO: 17 and a VL region comprising a CDR1L region of SEQ ID NO:27, a CDR2L region of SEQ ID NO:28 and a CDR3L region of SEQ ID NO:20; and an anti-CD3 antibody, or antigen binding fragment thereof, comprising a CDR1H region of SEQ ID NO: 1, a CDR2H region of SEQ ID NO: 2, a CDR3H region of SEQ ID NO: 3, a CDR1L region of SEQ ID NO:4, a CDR2L region of SEQ ID NO:5 and a CDR3L region of SEQ ID NO:6).
Embodiment 11. The pharmaceutical composition of any one of embodiments 1-
10, wherein the multispecific antibody comprises: an anti-BCMA antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO: 10 and a VL region of SEQ ID NO: 14; and
an anti-CD3 antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO: 7 and a VL region of SEQ ID NO: 8.
Embodiment 12. The pharmaceutical composition of any one of embodiments 1-11, wherein the multispecific antibody is a trivalent bispecific antibody comprising two Fab fragments of an anti-BCMA antibody, one Fab fragment of an anti-CD3 antibody, and one Fc portion, wherein the bispecific antibody is in the format BCMA Fab - Fc - CD3 Fab - BCMA Fab.
Embodiment 13. The pharmaceutical composition of embodiment 12, wherein each Fab fragment of an anti-BCMA antibody, or antigen binding fragment thereof, comprises a VH region comprising a CDR1H region of SEQ ID NO:21, a CDR2H region of SEQ ID NO:22 and a CDR3H region of SEQ ID NO: 17 and a VL region comprising a CDR1L region of SEQ ID NO:27, a CDR2L region of SEQ ID NO:28 and a CDR3L region of SEQ ID NO:20.
Embodiment 14. The pharmaceutical composition of embodiment 12 or embodiment 13, wherein each Fab fragment of an anti-BCMA antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO: 10 and a VL region of SEQ ID NO: 14.
Embodiment 15. The pharmaceutical composition of any one of embodiments 12-14, wherein the Fab fragment of the anti-CD3 antibody, or antigen binding fragment thereof, comprises a CDR1H region of SEQ ID NO: 1, a CDR2H region of SEQ ID NO:2, a CDR3H region of SEQ ID NO:3, a CDR1L region of SEQ ID NO:4, a CDR2L region of SEQ ID NO:5 and a CDR3L region of SEQ ID NO:6.
Embodiment 16. The pharmaceutical composition of any one of embodiments 12-15, wherein the Fab fragment of the anti-CD3 antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO: 7 and a VL region of SEQ ID NO: 8.
Embodiment 17. The pharmaceutical composition of any one of embodiments 1-16, wherein the multispecific antibody comprises a first polypeptide comprising the amino acid sequence of SEQ ID NO: 48, a second polypeptide comprising the amino acid sequence of SEQ ID NO: 55 or 58, a third polypeptide comprising the amino acid sequence of SEQ ID NO: 56 or 59, and fourth and fifth polypeptides each comprising the amino acid sequence of SEQ ID NO: 57.
Embodiment 18. A unit dose comprising the pharmaceutical composition of any one of embodiments 1-17.
Embodiment 19. The unit dose of embodiment 18, wherein the volume of the unit dose is
0.5-2.5 mL, or 0.5-0.9 mL, or 1.0-1.5 mL, or 2.0-2.5 mL, or 0.5, 0.6, 0.7, 0.8, 0.9. 1.0, 1.1, 1.2, 1.3, 1.4, 1.5, 1.6, 1.7, 1.8, 1.9, 2.0, 2.1, 2.2, 2.3, 2.4, or 2.5 mL.
Embodiment 20. A unit dose comprising a pharmaceutical composition comprising:
(a) about 6 mg/mL of a multispecific antibody that binds to BCMA and CD3;
(b) about 20 mM histidine;
(c) about 250 mM sucrose;
(d) about 0.04% w/v polysorbate 80; and
(e) about 50 pM pentetic acid; wherein the pH of the pharmaceutical composition is from about 5.7 to about 6.3.
Embodiment 21. A unit dose comprising a pharmaceutical composition comprising:
(a) about 30 mg/mL of a multispecific antibody that binds to BCMA and CD3 ;
(b) about 20 mM histidine;
(c) about 250 mM sucrose;
(d) about 0.04% w/v polysorbate 80; and
(e) about 50 pM pentetic acid; and wherein the pH of the pharmaceutical composition is from about 5.7 to about 6.3.
Embodiment 22. The unit dose of embodiment 20 or 21, wherein the multispecific antibody comprises: an anti-BCMA antibody, or antigen binding fragment thereof, comprising a VH region comprising a CDR1H region of SEQ ID NO:21, a CDR2H region of SEQ ID NO:22 and a CDR3H region of SEQ ID NO: 17 and a VL region comprising a CDR1L region of SEQ ID NO:27, a CDR2L region of SEQ ID NO:28 and a CDR3L region of SEQ ID NO:20; and an anti-CD3 antibody, or antigen binding fragment thereof, comprising a CDR1H region of SEQ ID NO: 1, a CDR2H region of SEQ ID NO: 2, a CDR3H region of SEQ ID NO: 3, a CDR1L region of SEQ ID NO:4, a CDR2L region of SEQ ID NO:5 and a CDR3L region of SEQ ID NO:6).
Embodiment 23. The unit dose of any one of embodiments 20-22, wherein the multispecific antibody comprises: an anti-BCMA antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO: 10 and a VL region of SEQ ID NO: 14; and an anti-CD3 antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO: 7 and a VL region of SEQ ID NO: 8.
Embodiment 24. The unit dose of any one of embodiments 20-23, wherein the multispecific antibody is a trivalent bispecific antibody comprising two Fab fragments of an anti- BCMA antibody, one Fab fragment of an anti-CD3 antibody, and one Fc portion, wherein the bispecific antibody is in the format BCMA Fab - Fc - CD3 Fab - BCMA Fab.
Embodiment 25. The unit dose of embodiment 24, wherein each Fab fragment of an anti-BCMA antibody, or antigen binding fragment thereof, comprises a VH region comprising a CDR1H region of SEQ ID NO:21, a CDR2H region of SEQ ID NO:22 and a CDR3H region of SEQ ID NO: 17 and a VL region comprising a CDR1L region of SEQ ID NO:27, a CDR2L region of SEQ ID NO:28 and a CDR3L region of SEQ ID NO:20.
Embodiment 26. The unit dose of embodiment 24 or embodiment 25, wherein each Fab fragment of an anti-BCMA antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO: 10 and a VL region of SEQ ID NO: 14
Embodiment 27. The unit dose of any one of embodiments 24-26, wherein the Fab fragment of the anti-CD3 antibody, or antigen binding fragment thereof, comprises a CDR1H region of SEQ ID NO: 1, a CDR2H region of SEQ ID NO: 2, a CDR3H region of SEQ ID NO: 3, a CDR1L region of SEQ ID NO:4, a CDR2L region of SEQ ID NO:5 and a CDR3L region of SEQ ID NO:6.
Embodiment 28. The unit dose of any one of embodiments 24-27, wherein the Fab fragment of the anti-CD3 antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO: 7 and a VL region of SEQ ID NO: 8.
Embodiment 29. The unit dose of any one of embodiments 20-28, wherein the multispecific antibody comprises a first polypeptide comprising the amino acid sequence of SEQ ID NO: 48, a second polypeptide comprising the amino acid sequence of SEQ ID NO: 55 or 58, a third polypeptide comprising the amino acid sequence of SEQ ID NO: 56 or 59, and fourth and fifth polypeptides each comprising the amino acid sequence of SEQ ID NO: 57.
Embodiment 30. The unit dose of any one of embodiments 20-29, wherein the volume of the unit dose is 0.5-2.5 mb, or 0.5-0.9 mb, or 1.0-1.5 mb, or 2.0-2.5 mb, or 0.5, 0.6, 0.7, 0.8, 0.9. 1.0, 1.1, 1.2, 1.3, 1.4, 1.5, 1.6, 1.7, 1.8, 1.9, 2.0, 2.1, 2.2, 2.3, 2.4, or 2.5 mL.
Embodiment 31. A vial comprising the pharmaceutical composition of any one of embodiments 1-17 or the unit dose of any one of embodiments 18-30.
Embodiment 32. A method of treating a subject in need thereof, comprising subcutaneously administering to the subject at least one dose of the pharmaceutical composition of any one of embodiments 1-17 or at least one unit dose of any one of embodiments 18-30.
Embodiment 33. A method of treating multiple myeloma or an autoimmune disease in a subject, comprising subcutaneously administering to the subject at least one dose of the pharmaceutical composition of any one of embodiments 1-17 or at least one unit dose of any one of embodiments 18-30.
Embodiment 34. The method of embodiment 32 or embodiment 33, wherein the treatment comprises the administration of the pharmaceutical composition or unit dose in a dosing regimen comprising:
(a) a starting phase, wherein one or more starting doses of the pharmaceutical composition or unit dose are administered to the subject; and
(b) a maintenance phase, wherein a first maintenance dose of the pharmaceutical composition or unit dose is administered to the subject, optionally followed by at least one additional maintenance dose of the pharmaceutical composition or unit dose, wherein each maintenance dose is greater than the one or more starting doses.
Embodiment 35. The method of embodiment 34, wherein the starting phase comprises a single fixed dose of the pharmaceutical composition or unit dose comprising about 1.5 mg to 4.5 mg of the multispecific antibody.
Embodiment 36. The method of embodiment 34, wherein the starting phase comprises a single fixed dose of the pharmaceutical composition or unit dose comprising about 3 mg of the multispecific antibody.
Embodiment 37. The method of any one of embodiments 34-36, wherein the first maintenance dose is a fixed dose of the pharmaceutical composition or unit dose comprising about 4.5 mg to about 7.5 mg of the multi-specific antibody.
Embodiment 38. The method of any one of embodiments 34-36, wherein the first maintenance dose is a fixed dose of the pharmaceutical composition or unit dose comprising about 6 mg of the multispecific antibody.
Embodiment 39. The method of any one of embodiments 34-38, wherein the at least one additional maintenance dose is a fixed dose of the pharmaceutical composition or unit dose comprising about 28 to about 32 mg of the multispecific antibody.
Embodiment 40. The method of any one of embodiments 34-38, wherein the at least one additional maintenance dose is a fixed dose of the pharmaceutical composition or unit dose comprising about 30 mg of the multispecific antibody.
Embodiment 41. The method of any one of embodiments 34-40, wherein the treatment comprises:
(i) a first treatment cycle, wherein the starting dose is administered on day 1, and the maintenance doses are administered on days 4, 8, 15 and 22,
(ii) a second and third treatment cycle, wherein the maintenance doses are administered in a weekly dosing interval,
(iii) a fourth to sixth treatment cycle, wherein the maintenance doses are administered in a biweekly dosing interval, and
(iv) a seventh and subsequent cycle, wherein the maintenance doses are administered in a four week dosing interval; wherein each treatment cycle is 28 days.
Embodiment 42. The method of embodiment 41, wherein the maintenance doses are administered on days 1, 8, 15 and 22 for the second and third treatment cycle, on days 1 and 15 for the fourth to sixth treatment cycle, and on day 1 for the seventh and subsequent cycle.
Embodiment 43. The method of any one of embodiments 34-42, wherein the treatment comprises:
(i) a first treatment cycle, wherein the starting dose comprises a single fixed dose of the pharmaceutical composition or unit dose comprising about 3 mg of the multi-specific antibody is administered on day 1, the maintenance dose of the pharmaceutical composition or unit dose
comprising about 6 mg of the multispecific antibody is administered on day 4, and the maintenance doses of the pharmaceutical composition or unit dose comprising about 30 mg of the multispecific antibody are administered on days 8, 15 and 22,
(ii) a second and third treatment cycle, wherein the maintenance doses of the pharmaceutical composition or unit dose comprising about 30 mg of the multispecific antibody are administered on days 1, 8, 15, and 22 in a weekly dosing interval,
(iii) a fourth to sixth treatment cycle, wherein the maintenance doses of the pharmaceutical composition or unit dose comprising about 30 mg of the multispecific antibody are administered on days 1 and 15 in a biweekly dosing interval, and
(iv) a seventh and subsequent cycle, wherein the maintenance doses of the pharmaceutical composition or unit dose comprising about 30 mg of the multispecific antibody are administered on day 1 in a four week dosing interval; wherein each treatment cycle is 28 days.
Embodiment 44. The pharmaceutical composition of any one of embodiments 1-17 or the unit dose of any one of embodiments 18-30 for use in treating a disorder associated with BCMA expression in a subject.
Embodiment 45. The pharmaceutical composition or unit dose for use of embodiment 44, wherein the disorder is multiple myeloma or an autoimmune disease.
Embodiment 46. The pharmaceutical composition or unit dose for use of embodiment 44 or embodiment 45, wherein the treatment comprises the administration of the pharmaceutical composition or unit dose in a dosing regimen comprising:
(i) a starting phase, wherein one or more starting doses of the pharmaceutical composition or unit dose are administered to the subject; and
(ii) a maintenance phase, wherein a first maintenance dose of the pharmaceutical composition or unit dose is administered to the subject, optionally followed by at least one additional maintenance dose of the pharmaceutical composition or unit dose, wherein each maintenance dose is greater than the one or more starting doses.
Embodiment 47. The pharmaceutical composition or unit dose for use of embodiment 46, wherein the starting phase comprises a single fixed dose of the pharmaceutical composition or unit dose comprising about 1.5 mg to 4.5 mg of the multispecific antibody.
Embodiment 48. The pharmaceutical composition or unit dose for use of embodiment 46, wherein the starting phase comprises a single fixed dose of the pharmaceutical composition or unit dose comprising about 3 mg of the multi-specific antibody.
Embodiment 49. The pharmaceutical composition or unit dose for use of any one of embodiments 46-48, wherein the first maintenance dose is a fixed dose of the pharmaceutical composition or unit dose comprising about 4.5 mg to about 7.5 mg of the multi-specific antibody.
Embodiment 50. The pharmaceutical composition or unit dose for use of any one of embodiments 46-48, wherein the first maintenance dose is a fixed dose of the pharmaceutical composition or unit dose comprising about 6 mg of the multi-specific antibody.
Embodiment 51. The pharmaceutical composition or unit dose for use of any one of embodiments 46-50, wherein the at least one additional maintenance dose is a fixed dose of the pharmaceutical composition or unit dose comprising about 28 to about 32 mg of the multispecific antibody.
Embodiment 52. The pharmaceutical composition or unit dose for use of any one of embodiments 46-50, wherein the at least one additional maintenance dose is a fixed dose of the pharmaceutical composition or unit dose comprising about 30 mg of the multi-specific antibody.
Embodiment 53. The pharmaceutical composition or unit dose for use of any one of embodiments 46-52, wherein the treatment comprises:
(i) a first treatment cycle, wherein the starting dose is administered on day 1, and the maintenance doses are administered on days 4, 8, 15 and 22,
(ii) a second and third treatment cycle, wherein the maintenance doses are administered in a weekly dosing interval,
(iii) a fourth to sixth treatment cycle, wherein the maintenance doses are administered in a biweekly dosing interval, and
(iv) a seventh and subsequent cycle, wherein the maintenance doses are administered in a four week dosing interval; wherein each treatment cycle is 28 days.
Embodiment 54. The pharmaceutical composition or unit dose for use of embodiment 53, wherein the maintenance doses are administered on days 1, 8, 15 and 22 for the second and third treatment cycle, on days 1 and 15 for the fourth to sixth treatment cycle, and on day 1 for the seventh and subsequent cycle.
Embodiment 55. The pharmaceutical composition or unit dose for use of any one of embodiments 46-54, wherein the treatment comprises:
(i) a first treatment cycle, wherein the starting dose of a single fixed dose of the pharmaceutical composition or unit dose comprising about 3 mg of the multi-specific antibody is administered on day 1, the maintenance dose of the pharmaceutical composition or unit dose comprising about 6 mg of the multispecific antibody is administered on day 4, and the maintenance doses of the pharmaceutical composition or unit dose comprising about 30 mg of the multispecific antibody are administered on days 8, 15 and 22,
(ii) a second and third treatment cycle, wherein the maintenance doses of the pharmaceutical composition or unit dose comprising about 30 mg of the multispecific antibody are administered on days 1, 8, 15, and 22 in a weekly dosing interval,
(iii) a fourth to sixth treatment cycle, wherein the maintenance doses of the pharmaceutical composition or unit dose comprising about 30 mg of the multispecific antibody are administered on days 1 and 15 in a biweekly dosing interval, and
(iv) a seventh and subsequent cycle, wherein the maintenance doses of the pharmaceutical composition or unit dose comprising about 30 mg of the multispecific antibody are administered on day 1 in a four week dosing interval; wherein each treatment cycle is 28 days.
Embodiment 56. Use of the pharmaceutical composition of any one of embodiments 1-17 or the unit dose of any one of embodiments 18-30 for the preparation of a medicament for treating a disorder associated with BCMA expression.
Embodiment 57. The use of embodiment 56, wherein the disorder is multiple myeloma.
Embodiment 58. The use of embodiment 56 or embodiment 57, wherein the medicament is administered to a subject in a dosing regimen comprising:
(i) a starting phase, wherein one or more starting doses of the medicament are administered to the subject; and
(ii) a maintenance phase, wherein a first maintenance dose of the medicament is administered to the subject, optionally followed by at least one additional maintenance dose of the medicament, wherein each maintenance dose is greater than the one or more starting doses.
Embodiment 59. The use of embodiment 58, wherein the starting phase comprises a single fixed dose of the medicament comprising about 1.5 mg to 4.5 mg of the multispecific antibody.
Embodiment 60. The use of embodiment 58, wherein the starting phase comprises a single fixed dose of the medicament comprising about 3 mg of the multi-specific antibody.
Embodiment 61. The use of any one of embodiments 58-60, wherein the first maintenance dose is a fixed dose of the medicament comprising about 4.5 mg to about 7.5 mg of the multi-specific antibody.
Embodiment 62. The use of any one of embodiments 58-60, wherein the first maintenance dose is a fixed dose of the medicament comprising about 6 mg of the multi-specific antibody.
Embodiment 63. The use of any one of embodiments 58-62, wherein the at least one additional maintenance dose is a fixed dose of the medicament comprising about 28 to about 32 mg of the multi-specific antibody.
Embodiment 64. The use of any one of embodiments 58-62, wherein the at least one additional maintenance dose is a fixed dose of the medicament comprising about 30 mg of the multispecific antibody.
Embodiment 65. The use of any one of embodiments 58-64, wherein the medicament is administered in a dosing regimen comprising:
(i) a first treatment cycle, wherein the starting dose is administered on day 1, and the maintenance doses are administered on days 4, 8, 15 and 22,
(ii) a second and third treatment cycle, wherein the maintenance doses are administered in a weekly dosing interval,
(iii) a fourth to sixth treatment cycle, wherein the maintenance doses are administered in a biweekly dosing interval, and
(iv) a seventh and subsequent cycle, wherein the maintenance doses are administered in a four week dosing interval; wherein each treatment cycle is 28 days.
Embodiment 66. The use of embodiment 65, wherein the maintenance doses are administered on days 1, 8, 15 and 22 for the second and third treatment cycle, on days 1 and 15 for the fourth to sixth treatment cycle, and on day 1 for the seventh and subsequent cycle.
Embodiment 67. The use of any one of embodiments 58-66, wherein the medicament is administered in a dosing regimen comprising:
(i) a first treatment cycle, wherein the starting dose comprises a single fixed dose of the medicament comprising about 3 mg of the multi-specific antibody is administered on day 1, the maintenance dose of the medicament comprising about 6 mg of the multispecific antibody is administered on day 4, and the maintenance doses of the medicament comprising about 30 mg of the multispecific antibody are administered on days 8, 15 and 22,
(ii) a second and third treatment cycle, wherein the maintenance doses of the medicament comprising about 30 mg of the multispecific antibody are administered on days 1, 8, 15, and 22 in a weekly dosing interval,
(iii) a fourth to sixth treatment cycle, wherein the maintenance doses of the medicament comprising about 30 mg of the multispecific antibody are administered on days 1 and 15 in a biweekly dosing interval, and
(iv) a seventh and subsequent cycle, wherein the maintenance doses of the medicament comprising about 30 mg of the multispecific antibody are administered on day 1 in a four week dosing interval; wherein each treatment cycle is 28 days.
Embodiment 68. A pharmaceutical composition comprising (a) about 6 mg/mL of alnuctamab; (b) about 20 mM histidine; (c) about 250 mM sucrose; (d) about 0.04% w/v polysorbate 80; and (e) about 50 pM pentetic acid.
Embodiment 69. A pharmaceutical composition comprising (a) about 30 mg/mL of alnuctamab; (b) about 20 mM histidine; (c) about 250 mM sucrose; (d) about 0.04% w/v polysorbate 80; and (e) about 50 pM pentetic acid
Embodiment 70. A pharmaceutical composition comprising (a) 6 mg/mL of alnuctamab; (b) 20 mM histidine; (c) 250 mM sucrose; (d) 0.04% w/v polysorbate 80; and (e) 50 pM pentetic acid.
Embodiment 71. A pharmaceutical composition comprises (a) 30 mg/mL of alnuctamab; (b) 20 mM histidine; (c) 250 mM sucrose; (d) 0.04% w/v polysorbate 80; and (e) 50 pM pentetic acid.
Embodiment 72. The vial of embodiment 31, wherein the vial is a pre-filled syringe or an autoinjector.
Embodiment 73. A method of preventing or reducing oxidation and/or deamidation of alnuctamab in a pharmaceutical composition comprising binding alnuctamab to pentetic acid in the pharmaceutical composition, thereby preventing or reducing oxidation and/or deamidation of alnuctamab in the pharmaceutical composition.
Embodiment 74. The method of embodiment 73, wherein pentetic acid binds to the anti- CD3 domain of alnuctamab.
Embodiment 75. The method of embodiment 73 or 74, wherein binding alnuctamab to pentetic acid prevents or reduces tryptophan residue oxidation and/or asparagine residue deamidation in alnuctamab.
Embodiment 76. The method of embodiment 75, wherein the tryptophan residue(s) are located in the CDR of the anti-CD3 domain of alnuctamab.
Embodiment 77. The method of embodiment 75 or 76, wherein the asparagine residue(s) are located in the CDR of the anti-CD3 domain of alnuctamab.
Embodiment 78. The method of any of embodiments 73-77, wherein the pharmaceutical composition is the pharmaceutical composition of any of embodiments 1-17.
BRIEF DESCRIPTION OF FIGURES
[0006] The present invention will now be described in more detail with reference to the attached Figures, in which:
[0007] FIG. 1 illustrates a format of bispecific trivalent antibodies for use in the present invention, which comprise Fab fragments binding to CD3 and BCMA in the following formats: Fab BCMA - Fc - Fab CD3 - Fab BCMA. The CD3 Fab may include a VH-VL crossover to reduce light chain mispairing and side -products. Amino acid substitutions “RK/EE” may be introduced in CL-CH1 to reduce light chain mispairing/side products in production. The CD3 Fab and BCMA Fab may be linked to each other with flexible linkers.
[0008] FIG. 2A-2B show % high molecular weight species formed during agitation (FIG. 2A, each set of three bars, left to right: time 0, 400 rpm for 24 hours, 400 rpm for 70 hours) and freeze-thaw (FIG. 2B, each set of three bars, from left to right: time 0, three freeze-thaw cycles, five freeze-thaw cycles) of a compositions comprising 100 mg/mL of alnuctamab with differing pHs (5.7-6.3) and surfactant levels (0.02%-0.06%), as measured by size-exclusion chromatography.
[0009] FIG. 3A-3C show HMW% (FIG. 3A), main peak% (FIG. 3B), and LMW% (FIG. 3C) as assessed by size exclusion chromatograph (SEC) for vials of formulations comprising 10 mg/mL, 50 mg/mL, and 100 mg/mL of alnuctamab at various temperatures (5°C, 25°C, and 40°C) over a period of 24 months.
[00010] FIG. 4A-4B show main peak % as assessed by capillary electrophoresis sodium dodecyl sulphate (CE-SDS) under non-reducing (NR) (FIG. 4A) and purity % under reducing (R) conditions (FIG. 4B) for vials of formulations comprising 10 mg/mL, 50 mg/mL, and 100 mg/mL of alnuctamab at various temperatures (5 °C, 25 °C, and 40°C) over a period of 24 months.
[00011] FIG. 5A-5B acidic species % (FIG. 5A) and main peak % (FIG. 5B) as assessed by cation exchange chromatography (CEX) for vials of formulations comprising 10 mg/mL, 50 mg/mL, and 100 mg/mL of alnuctamab at various temperatures (5 °C, 25 °C, and 40°C) over a period of 24 months.
[00012] FIG. 6 shows potency data for formulations comprising 10 mg/mL, 50 mg/mL, and 100 mg/mL of alnuctamab at 5 °C over a period of 18 months.
[00013] FIG. 7 shows change in PS80 concentration for formulations comprising 10 mg/mL, 50 mg/mL, and 100 mg/mL of alnuctamab at 5 °C over a period of 24 months.
[00014] FIG. 8 is a table showing changes in PS 80 concentration for formulations comprising 10 mg/mL, 50 mg/mL, and 100 mg/mL of alnuctamab at 5 °C, 25 °C and 60% relative humidity, and 40°C and 75% relative humidity, over a period of up to 24 months.
[00015] FIG. 9A-9C show HMW % (FIG. 9A), monomer % (FIG. 9B) and LMW % (FIG. 9C) as assessed by SEC for formulations comprising 44 mg/mL alnuctamab with and without 50 pm DTPA at 5 °C, 25 °C, or 40°C over 12 months.
[00016] FIG. 10 shows main peak % as assessed by CE-SDS NR for formulations comprising 44 mg/mL alnuctamab with and without 50 pm DTPA at 5°C, 25°C, and 40°C over 12 months.
[00017] FIG. 11A-11C show acidic species % (FIG. 11A), main peak % (FIG. 1 IB), and basic species % (FIG. 11C) as assessed by CEX for formulations comprising 44 mg/mL alnuctamab with and without 50 pm DTPA at 5°C, 25°C, and 40°C over 12 months.
[00018] FIG. 12A-12B show HMW % (FIG. 12A) and monomer % (FIG. 12B) as assessed by SEC for formulations comprising 1 mg/mL alnuctamab with and without 50 pm DTPA at 5 °C, 25 °C, and 40°C over 6 months.
[00019] FIG. 13 shows main peak % as assessed by CE-SDS-NR for formulations comprising 1 mg/mL alnuctamab with and without 50 pm DTPA at 5°C, 25°C, and 40°C over 6 months.
[00020] FIG. 14A-14B show acidic species % (FIG. 14A) and main peak % (FIG. 14B) as assessed by CEX for formulations comprising 1 mg/mL alnuctamab with and without 50 pm DTPA at 5 °C, 25 °C, and 40°C over 6 months.
[00021] FIG. 15 shows PS-80 concentration for formulations comprising 1 mg/mL alnuctamab with and without 50 pm DTPA at 5°C, 25°C, and 40°C.
[00022] FIG. 16A-16F show HMW (FIG. 16A-16C) and main peak (FIG. 16D-16F) as assessed by SEC for formulations comprising 5 mg/mL alnuctamab and PS-80 (with or without DTPA and with or without metal spiking) at various temperatures: 25 °C (FIG. 16A and 16D), RT/RL (room temperature/room light) (FIG. 16B and 16E), and 40°C (FIG. 16C and 16F). Each set of four bars, from left to right (all are 5 mg/mL alnuctamab): DTPA+PS80; PS80; DTPA+PS80+metal; PS80+metal.
[00023] FIG. 17A-17F show HMW (FIG. 17A-C) and main peak (FIG. 17D-F) as assessed by SEC for formulations comprising 60 mg/mL alnuctamab and PS-80 (with or without DTPA and with or without metal spiking) at various temperatures: 25°C (FIG. 17A and 17D), RT/RL (room temperature/room light) (FIG. 17B and 17E), and 40°C (FIG. 17C and 17F). Each set of four bars, from left to right (all are 60 mg/mL alnuctamab): DTPA+PS80; PS80; DTPA+PS80+metal; PS80+metal.
[00024] FIG. 18A-18F show acidic species (FIG. 18A-18C) and main peak (FIG. 18D-18F) as assessed by CEX for formulations comprising 5 mg/mL alnuctamab and PS-80 (with or without DTPA and with or without metal spiking) at various temperatures: 25°C (FIG. 18A and 18D), RT/RL (room temperature/room light) (FIG. 18B and 18E), and 40°C (FIG. 18C and 18F). Each set of four bars, from left to right (all are 5 mg/mL alnuctamab): DTPA+PS80; PS80; DTPA+PS80+metal; PS80+metal.
[00025] FIG. 19A-19F show acidic species (FIG. 19A-19C) and main peak (FIG. 189-19F) as assessed by CEX for formulations comprising 60 mg/mL alnuctamab and PS-80 (with or without DTPA and with or without metal spiking) at various temperatures: 25°C (FIG. 19A and 19D), RT/RL (room temperature/room light) (FIG. 19B and 19E), and 40°C (FIG. 19C and 19F). Each set of four bars, from left to right (all are 60 mg/mL alnuctamab): DTPA+PS80; PS80; DTPA+PS80+metal; PS80+metal.
[00026] FIG. 20A-20C show main peak % as assessed by CE-SDS NR for formulations comprising 5 mg/mL alnuctamab and PS-80 (with or without DTPA and with or without metal spiking) at various temperatures: 25°C (FIG. 20A), RT/RL (room temperature/room light) (FIG. 20B), and 40°C (FIG. 20C). Each set of four bars, from left to right (all are 5 mg/mL alnuctamab): DTPA+PS80; PS80; DTPA+PS80+metal; PS80+metal.
[00027] FIG. 21A-21C show main peak% as assessed by CE-SDS NR for formulations comprising 60 mg/mL alnuctamab and PS-80 (with or without DTPA and with or without metal spiking) at various temperatures: 25°C (FIG. 21 A), RT/RL (room temperature/room light) (FIG. 2 IB), and 40°C (FIG. 21C). Each set of four bars, from left to right (all are 60 mg/mL alnuctamab): DTPA+PS80; PS80; DTPA+PS80+metal; PS80+metal.
[00028] FIG. 22A-22B shows PS-80 oxidation in formulations comprising 5 mg/mL alnuctamab (FIG. 22A) and 60 mg/mL alnuctamab (FIG. 22B) and PS80 (with or without DTPA and with or without metal spking) at 25 °C and 40°C.
DETAILED DESCRIPTION
[00029] As used herein, the articles “a” and “an” may refer to one or to more than one (e.g. to at least one) of the grammatical object of the article.
[00030] “About” may generally mean an acceptable degree of error for the quantity measured given the nature or precision of the measurements. Exemplary degrees of error are within 20 percent (%), within 10%, or within 5% of a given value or range of values.
[00031] Embodiments described herein as “comprising” one or more features may also be considered as disclosure of the corresponding embodiments “consisting of’ and/or “consisting essentially of’ such features.
[00032] Concentrations, amounts, volumes, percentages and other numerical values may be presented herein in a range format. It is also to be understood that such range format is used merely for convenience and brevity and should be interpreted flexibly to include not only the numerical values explicitly recited as the limits of the range but also to include all the individual numerical values or subranges encompassed within that range as if each numerical value and sub-range is explicitly recited.
The Multispecific Antibody
[00033] In some aspects, an “antibody” of the present disclosure is capable of binding to more than one antigen, e.g., a “multispecific” antibody. As used herein, a “bispecific” antibody is an antibody that is capable of specifically binding two antigens, wherein the first and second antigen are the same or different.
[00034] The multispecific (e.g. bispecific) antibodies of the invention specifically bind to BCMA and to CD3. The terms “antibody against BCMA and CD3”, “anti-BCMA anti-CD3 antibody” or “an antibody that binds to BCMA and CD3,” refer to a multispecific antibody (e.g., a bispecific antibody) that is capable of binding to BCMA and CD3 with sufficient affinity such that the antibody is useful as a therapeutic agent. This is achieved by making a molecule which comprises a first antibody, or antigenbinding fragment, that binds to BCMA and a second antibody, or antigen-binding fragment, that binds to CD3. Such multispecific antibodies may be trispecific antibodies or bispecific antibodies. In preferred embodiments, the multispecific antibodies are bispecific antibodies.
[00035] The term “BCMA” as used herein relate to human B cell maturation antigen, also known as BCMA; TR17_HUMAN, TNFRSF17 (UniProt Q02223), which is a member of the tumor necrosis receptor superfamily that is preferentially expressed in differentiated plasma cells. The extracellular domain of BCMA consists according to UniProt of amino acids 1 - 54 (or 5-51). The terms “antibody against BCMA”, “anti BCMA antibody” or “an antibody that binds to BCMA” as used herein relate to an antibody specifically binding to the extracellular domain of BCMA.
[00036] The term “specifically binds to BCMA” refers to an antibody that is capable of binding to the defined target with sufficient affinity such that the antibody is useful as a therapeutic agent in targeting BCMA. In some embodiments, an antibody that specifically binds to BCMA does not bind to other antigens, or does not bind to other antigens with sufficient affinity to produce a physiological effect.
[00037] In some embodiments, the extent of binding of an anti-BCMA antibody to an unrelated,
non-BCMA protein is about 10-fold preferably > 100-fold less than the binding of the antibody to BCMA as measured, e.g., by surface plasmon resonance (SPR) e.g. Biacore®, enzyme-linked immunosorbent (ELISA) or flow cytometry (FACS). In one embodiment the antibody that binds to BCMA has a dissociation constant (Kd) of 10'8 M or less, preferably from 10'8 M to 10'13 M, preferably from 10'9 M to IO'13 M.
[00038] In one embodiment the anti -BCMA antibody binds to an epitope of BCMA that is conserved among BCMA from different species, preferably among human and cynomolgus, and in addition preferably also to mouse and rat BCMA.
[00039] Preferably the anti-BCMA antibody specifically binds to a group of BCMA, consisting of human BCMA and BCMA of non-human mammalian origin, preferably BCMA from cynomolgus, mouse and/or rat. Anti-BCMA antibodies are analyzed by ELISA for binding to human BCMA using plate-bound BCMA. For this assay, an amount of plate-bound BCMA preferably 1.5 pg/mL and concentration(s) ranging from 0.1 pM to 200 nM of anti-BCMA antibody are used.
[00040] The term “CD3” refers to the human CD3 protein multi-subunit complex. The CD3 protein multi-subunit complex is composed to 6 distinctive polypeptide chains. Thus the term includes a CD3y chain (SwissProt P09693), a CD35 chain (SwissProt P04234), two CD3a chains (SwissProt P07766), and one CD3^ chain homodimer (SwissProt 20963), and which is associated with the T cell receptor a and P chain. The term encompasses “full-length,” unprocessed CD3, as well as any CD3 variant, isoform and species homolog which is naturally expressed by cells (including T cells) or can be expressed on cells transfected with genes or cDNA encoding those polypeptides.
[00041] The term “specifically binds to CD3” refers to an antibody that is capable of binding to the defined target with sufficient affinity such that the antibody is useful as a therapeutic agent in targeting CD3. In some embodiments, an antibody that specifically binds to CD3 does not bind to other antigens, or does not bind to other antigens with sufficient affinity to produce a physiological effect.
[00042] The multispecific (e.g. bispecific) antibodies of the invention can be analysed by surface plasmon resonance (SPR), e.g. Biacore®, for binding to CD3. In some embodiments, the bispecific antibodies bind to human CD3 with a dissociation constant (KD) of about 10'7 M or less, a KD of about IO-8 M or less, a KD of about 10'9 M or less, a KD of about IO-10 M or less, a KD of about 10 11 M or less, or a KD of about IO-12 M or less, as determined by a surface plasmon resonance assay, preferably measured using Biacore 8K at 25°C. In preferred embodiments, the bispecific antibodies bind to human CD3 with a dissociation constant (KD) of about KF8 M or less.
[00043] The term “antibody” herein encompasses various antibody structures, including but not limited to monoclonal antibodies, polyclonal antibodies, multispecific antibodies (e.g., bispecific antibodies), and antibody fragments so long as they exhibit the desired antigen-binding activity.
[00044] A “heavy chain” comprises a heavy chain variable region (abbreviated herein as “VH”) and a heavy chain constant region (abbreviated herein as “CH”). The heavy chain constant region comprises the heavy chain constant domains CHI, CH2 and CH3 (antibody classes IgA, IgD, and IgG)
and optionally the heavy chain constant domain CH4 (antibody classes IgE and IgM).
[00045] A “light chain” comprises a light chain variable domain (abbreviated herein as “VL”) and a light chain constant domain (abbreviated herein as “CL”). The variable regions VH and VL can be further subdivided into regions of hypervariability, termed complementarity determining regions (CDR), interspersed with regions that are more conserved, termed framework regions (ER). Each VH and VL is composed of three CDRs and four L Rs, arranged from amino-terminus to carboxy-terminus in the following order: FR1, CDR1, LR2, CDR2, LR3, CDR3, LR4. The “constant domains” of the heavy chain and of the light chain are not involved directly in binding of an antibody to a target, but exhibit various effector functions.
[00046] Binding between an antibody and its target antigen or epitope is mediated by the Complementarity Determining Regions (CDRs). The CDRs are regions of high sequence variability, located within the variable region of the antibody heavy chain and light chain, where they form the antigen-binding site. The CDRs are the main determinants of antigen specificity. Typically, the antibody heavy chain and light chain each comprise three CDRs which are arranged non-consecutively. The antibody heavy and light chain CDR3 regions play a particularly important role in the binding specificity/affinity of the antibodies according to the invention and therefore provide a further aspect of the invention.
[00047] The term “antigen binding fragment” as used herein incudes any naturally-occurring or artificially-constructed configuration of an antigen-binding polypeptide comprising three light chain CDRs, and three heavy chain CDRs, wherein the polypeptide is capable of binding to the antigen. Thus, the term refers to a molecule other than an intact antibody that comprises a portion of an intact antibody that binds the antigen to which the intact antibody binds. Examples of antibody fragments include but are not limited to Fv, Fab, Fab’, Fab’-SH, F(ab’)2; diabodies; linear antibodies; single-chain antibody molecules (e.g. scFv); and multispecific antibodies formed from antibody fragments.
[00048] The terms “Fab fragment” and “Fab” are used interchangeably herein and contain a single light chain (i.e. a constant domain CL and a VL) and a single heavy chain (i.e. the constant domain CHI and a VH). The heavy chain of a Fab fragment is not capable of forming a disulfide bond with another heavy chain.
[00049] A “Fab1 fragment” contains a single light chain and a single heavy chain but in addition to the CHI and the VH, a “Fab1 fragment” contains the region of the heavy chain between the CHI and CH2 domains that is required for the formation of an inter-chain disulfide bond. Thus, two “Fab1 fragments” can associate via the formation of a disulphide bond to form a F(ab')2 molecule.
[00050] A “F(ab')2 fragment” contains two light chains and two heavy chains. Each chain includes a portion of the constant region necessary for the formation of an inter-chain disulfide bond between two heavy chains.
[00051] An “Fv fragment” contains only the variable regions of the heavy and light chain. It contains no constant regions.
[00052] A “single-chain Fv” (“scFv”) is antibody fragment containing the VH and VL domain of an antibody, linked together to form a single chain. A polypeptide linker is commonly used to connect the VH and VL domains of the scFv.
[00053] A “tandem scFv”, also known as a TandAb®, is a single-chain Fv molecule formed by covalent bonding of two scFvs in a tandem orientation with a flexible peptide linker.
[00054] A “bi-specific T cell engager” (BiTE®) is a fusion protein consisting of two single-chain variable fragments (scFvs) on a single peptide chain. One of the scFvs binds to T cells via the CD3 receptor, and the other to a tumour cell antigen.
[00055] The sequence of a CDR may be identified by reference to any number system known in the art, for example, the Kabat system (Kabat, E. A., et al., Sequences of Proteins of Immunological Interest, 5th ed., Public Health Service, National Institutes of Health, Bethesda, MD (1991); the Chothia system (Chothia &, Lesk, “Canonical Structures for the Hypervariable Regions of Immunoglobulins,” J. Mol. Biol. 196, 901-917 (1987)); or the IMGT system (Lefranc et al., “IMGT Unique Numbering for Immunoglobulin and Cell Receptor Variable Domains and Ig superfamily V-like domains,” Dev. Comp. Immunol. 27, 55-77 (2003)).
Table 1: CDR definitions

[00056] For heavy chain constant region amino acid positions discussed in the invention, numbering is according to the EU index first described in Edelman, G.M., et al., Proc. Natl. Acad. Sci. USA 63 (1969) 78-85). The EU numbering of Edelman is also set forth in Kabat et al. (1991) (supra . Thus, the terms “EU index as set forth in Kabat”, “EU Index”. “EU index of Kabat” or “EU numbering” in the context of the heavy chain refers to the residue numbering system based on the human IgGl EU antibody of Edelman et al. as set forth in Kabat et al. (1991). The numbering system used forthe light chain constant region amino acid sequence is similarly set forth in Kabat et al. (supra.). Thus, as used herein, “numbered according to Kabat” refers to the Kabat set forth in Kabat et al. (supra .
[00057] Especially preferred are human or humanized antibodies, especially as recombinant human or humanized antibodies.
[00058] The term “humanized antibody” refers to antibodies in which the framework or “complementarity determining regions” (CDRs) have been modified to comprise the CDR of an immunoglobulin of different specificity as compared to that of the parent immunoglobulin. For example, a murine CDR may be grafted into the framework region of a human antibody to prepare the “humanized antibody.” See, e.g., Riechmann, L., et al., Nature 332 (1988) 323-327; and Neuberger, M.S., et al., Nature 314 (1985) 268-270. In some embodiments, “humanized antibodies” are those in which the constant region has been additionally modified or changed from that of the original antibody to generate the properties of the antibodies according to the invention, especially in regard to Clq binding and/or Fc receptor (FcR) binding.
[00059] The term “human antibody” is one which possesses an amino acid sequence which corresponds to that of an antibody produced by a human or a human cell or derived from a non-human source that utilizes human antibody repertoires or other human antibody-encoding sequences. This definition of a human antibody specifically excludes a humanized antibody comprising non-human antigen-binding residues. Human antibodies can be produced using various techniques known in the art, including phage-display libraries.
[00060] The term “chimeric antibody” refers to an antibody comprising a variable region, i.e., binding region, from one source or species and at least a portion of a constant region derived from a different source or species, usually prepared by recombinant DNA techniques. Chimeric antibodies comprising a murine variable region and a human constant region are preferred. Other preferred forms of “chimeric antibodies” encompassed by the present invention are those in which the constant region has been modified or changed from that of the original antibody to generate the properties of the antibodies according to the invention, especially in regard to Clq binding and/or Fc receptor (FcR) binding. Such chimeric antibodies are also referred to as “class-switched antibodies”. Chimeric antibodies are the product of expressed immunoglobulin genes comprising DNA segments encoding immunoglobulin variable regions and DNA segments encoding immunoglobulin constant regions. Methods for producing chimeric antibodies involving conventional recombinant DNA and gene transfection techniques are well known in the art. See, e.g., Morrison, S.L., et al., Proc. Natl. Acad. Sci. USA 81 (1984) 6851-6855; US Patent Nos. 5,202,238 and 5,204,244.
[00061] The terms “Fc region” and “Fc” are used interchangeably herein and refer to the portion of a native immunoglobulin that is formed by two Fc chains. Each “Fc chain” comprises a constant domain CH2 and a constant domain CH3. Each Fc chain may also comprise a hinge region. A native Fc region is homodimeric. In some embodiments, the Fc region may contain modifications to enforce Fc heterodimerization.
[00062] The term “Fc part” refers to the portion of an antibody of the invention, or antigen binding fragment thereof, which corresponds to the Fc region.
[00063] There are five major classes of heavy chain constant region, classified as IgA, IgG, IgD, IgE and IgM, each with characteristic effector functions designated by isotype. For example, IgG is
separated into four subclasses known as IgGl, IgG2, IgG3, and IgG4. Ig molecules interact with multiple classes of cellular receptors. For example, IgG molecules interact with three classes of Fey receptors (FcyR) specific for the IgG class of antibody, namely FcyRI, FcyRII, and FcyRIII. The important sequences for the binding of IgG to the FcyR receptors have been reported to be located in the CH2 and CH3 domains.
[00064] The antibodies of the invention or antigen-binding fragments thereof may be any isotype, i.e. IgA, IgD, IgE, IgG and IgM, and synthetic multimers of the four-chain immunoglobulin (Ig) structure. In preferred embodiments, the antibodies or antigen-binding fragments thereof are IgG isotype. The antibodies or antigen-binding fragments can be any IgG subclass, for example IgGl, IgG2, IgG3, or IgG4 isotype. In preferred embodiments, the antibodies or antigen-binding fragments thereof are of an IgGl isotype.
[00065] In some embodiments, the antibodies comprise a heavy chain constant region that is of IgG isotype. In some embodiments, the antibodies comprise a portion of a heavy chain constant region that is of IgG isotype. In some embodiments, the IgG constant region or portion thereof is an IgGl, IgG2, IgG3, or IgG4 constant region. Preferably, the IgG constant region or portion thereof is an IgGl constant region.
[00066] The antibodies of the invention or antigen-binding fragments thereof may comprise a lambda light chain or a kappa light chain.
[00067] In preferred embodiments, the antibodies or antigen-binding fragments thereof comprise a light chain that is a kappa light chain. In some embodiments, the antibody or antigen-binding fragment comprises a light chain comprising a light chain constant region (CL) that is a kappa constant region.
[00068] In some embodiments, the antibody comprises a light chain comprising a light chain variable region (VL) that is a kappa variable region. Preferably, the kappa light chain comprises a VL that is a kappa VL and a CL that is a kappa CL.
[00069] Alternatively, the antibodies or antigen-binding fragments thereof may comprise a light chain that is a lambda light chain. In some embodiments, the antibody or antigen-binding fragment comprises a light chain comprising a light chain constant region (CL) that is a lambda constant region. In some embodiments, the antibody comprises a light chain comprising a light chain variable region (VL) that is a lambda variable region.
Antibody format
[00070] Formats for multispecific, e.g. bispecific, antibodies are known in the state of the art. For example, bispecific antibody formats are described in Kontermann RE, mAbs 4:2 1-16 (2012); Holliger P., Hudson PJ, Nature Biotech.23 (2005) 1126- 1136, Chan AC, Carter PJ Nature Reviews Immunology 10, 301-316 (2010) and Cuesta AM etal., Trends Biotech 28 (2011) 355-362.
[00071] The multispecific, e.g. bispecific, antibodies of the invention may have any format.
Multispecific and bispecific antibody formats include, for example, multivalent single chain antibodies,
diabodies and triabodies, and antibodies having the constant domain structure of full length antibodies to which further antigen-binding domains (e.g., single chain Fv, a tandem scFv, a VH domain and/or a VL domain, Fab, or (Fab)2,) are linked via one or more peptide-linkers. In some embodiments, the multispecific, e.g. bispecific, antibodies of the invention have the format of an scFv such as a bispecific T cell engager (BITE®). In some embodiments, the antibodies of the invention are single chain antibodies which comprise a first domain which binds to BCMA, a second domain which binds to a T cell antigen (e.g. CD3), and a third domain which comprises two polypeptide monomers, each comprising a hinge, a CH2 domain and a CH3 domain, wherein the two polypeptide monomers are fused to each other via a peptide linker (e.g. (hinge-CH2-CH3-linker-hinge-CH2-CH3).
[00072] The “valency” of an antibody denotes the number of binding domains. As such, the terms “bivalent”, “trivalenf ’, and “multivalent” denote the presence of two binding domains, three binding domains, and multiple binding domains, respectively. The multispecific, e.g. bispecific, antibodies of the invention may have more than one binding domain capable of binding to each target antigen (i.e., the antibody is trivalent or multivalent). In preferred embodiments, the multispecific, e.g. bispecific, antibodies of the invention have more than one binding domain capable of binding to the same epitope of each target antigen. In some embodiments, the multispecific, e.g. bispecific, antibodies of the invention have more than one binding domain capable of binding to different epitopes on each target antigen.
[00073] The multispecific, e.g. bispecific, antibodies of the invention may be bivalent, trivalent or tetravalent. In preferred embodiments, the multispecific, e.g. bispecific, antibody is trivalent, preferably wherein the trivalent antibody is bivalent for BCMA. Thus, the bispecific antibody may be trivalent, wherein the trivalent antibody is bivalent for BCMA.
[00074] The multispecific, e.g. bispecific, antibodies can be full length from a single species, or can be chimerized or humanized. For an antibody with more than two antigen-binding domains, some binding domains may be identical, as long as the protein has binding domains for two different antigens. [00075] The multispecific, e.g. bispecific, antibodies of the invention can have a bispecific heterodimeric format. In some embodiments, the bispecific antibody comprises two different heavy chains and two different light chains. In other embodiments, the multispecific, e.g. bispecific, antibody comprises two identical light chains and two different heavy chains. In some embodiments, in the multispecific, e.g. bispecific, antibodies of the invention one of the two pairs of heavy chain and light chain (HC/LC) specifically binds to CD3 and the other one specifically binds to BCMA.
[00076] In embodiments in which the bispecific antibodies of the invention are bivalent, they may comprise one anti-BCMA antibody and one anti-CD3 antibody (referred to herein as the “1+1” format).
[00077] In embodiments in which the BCMA and CD3 antibodies are Fabs, the bivalent bispecific antibodies in the 1+1 format may have the format: CD3 Fab - BCMA Fab (i.e. when no Fc is present). Alternatively, the bispecific antibodies may have the format: Fc - CD3 Fab - BCMA Fab; Fc-
BCMA Fab - CD3 Fab; or BCMA Fab - Fc - CD3 Fab (i.e. when an Fc is present). In preferred embodiments, the bivalent bispecific antibodies have the format BCMA Fab - Fc - CD3 Fab.
[00078] “CD3 Fab - BCMA Fab” means that the CD3 Fab is bound via its N-terminus to the C- terminus of the BCMA Fab.
[00079] “Fc - BCMA Fab - CD3 Fab” means that the BCMA Fab is bound via its C-terminus to the N-terminus of the Fc, and the CD3 Fab is bound via its C-terminus to the N-terminus of the BCMA Fab.
[00080] “Fc - CD3 Fab - BCMA Fab” means that the CD3 Fab is bound via its C-terminus to the N-terminus of the Fc, and the BCMA Fab is bound via its C-terminus to the N-terminus of the CD3 Fab. [00081] “BCMA Fab - Fc - CD3 Fab” means that the BCMA and CD3 Fab fragments are bound via their C-terminus to the N-terminus of the Fc.
[00082] In embodiments in which the bispecific antibodies of the invention are trivalent, they may comprise two anti -BCMA antibodies and one anti-CD3 antibody (referred to herein as the “2+1” format).
[00083] In embodiments in which the BCMA and CD3 antibodies are Fabs, the trivalent bispecific antibodies in the 2+1 format may have the format: CD3 Fab - BCMA Fab - BCMA Fab; or BCMA Fab - CD3 Fab - BCMA Fab (i.e. when no Fc is present). Alternatively, the bispecific antibodies may have the format: BCMA Fab - Fc - CD3 Fab - BCMA Fab; BCMA Fab - Fc - BCMA Fab - CD3 Fab; or CD3 Fab - Fc - BCMA Fab - BCMA Fab (i.e. when an Fc is present). In preferred embodiments, the trivalent bispecific antibodies have the format BCMA Fab - Fc - CD3 Fab - BCMA Fab.
[00084] “CD3 Fab - BCMA Fab - BCMA Fab” means that the CD3 Fab is bound via its C- terminus to the N-terminus of the first BCMA Fab, and the first BCMA Fab is bound via its C-terminus to the N-terminus of the second BCMA Fab.
[00085] “BCMA Fab - CD3 Fab - BCMA Fab” means that the first BCMA Fab is bound via its C-terminus to the N-terminus of the CD3 Fab, and the CD3 Fab is bound via its C-terminus to the N- terminus of the second BCMA Fab.
[00086] “BCMA Fab - Fc - CD3 Fab - BCMA Fab” means that the first BCMA Fab and the CD3 Fab are bound via their C-terminus to the N-terminus of the Fc, and the second BCMA Fab is bound via its C-terminus to the N-terminus of the CD3 Fab.
[00087] “BCMA Fab - Fc - BCMA Fab - CD3 Fab” means that the first BCMA Fab and the second BCMA Fab are bound via their C-terminus to the N-terminus of the Fc, and the CD3 Fab is bound via its C-terminus to the N-terminus of the second BCMA Fab.
[00088] “CD3 Fab - Fc - BCMA Fab - BCMA Fab” means that the CD3 Fab and the first BCMA Fab are bound via their C-terminus to the N-terminus of the Fc, and the second BCMA Fab is bound via its C-terminus to the N-terminus of the first BCMA Fab.
[00089] In some embodiments, the bispecific antibodies of the invention may comprise not more
than one BCMA Fab specifically binding to BCMA, and not more than one CD3 Fab specifically binding to CD3 and not more than one Fc part.
[00090] In some embodiments, the bispecific antibody comprises not more than one CD3 Fab specifically binding to CD3, not more than two BCMA Fabs specifically binding to BCMA and not more than one Fc part. In some embodiments, not more than one CD3 Fab and not more than one BCMA Fab are linked to the Fc part and linking is performed via C-terminal binding of the Fab(s) to the hinge region of the Fc part. In some embodiments, the second BCMA Fab is linked via its C-terminus either to the N- terminus of the CD3 Fab or to the hinge region of the Fc part and is therefore between the Fc part of the bispecific antibody and the CD3 Fab.
[00091] In embodiments comprising two BCMA Fabs, the BCMA Fabs are preferably derived from the same antibody and are preferably identical in the CDR sequences, variable domain sequences VH and VL and/or the constant domain sequences CHI and CL. Preferably, the amino acid sequences of the two BCMA Fab are identical.
[00092] The bispecific antibodies of the invention can also comprise scFvs instead of the Fabs.
Thus, in some embodiments, the bispecific antibodies have any one of the above formats, wherein each Fab is replaced with a corresponding scFv.
[00093] The components, e.g. the Fab fragments, of the bispecific antibodies of the invention may be chemically linked together by the use of an appropriate linker according to the state of the art. In preferred embodiments, a (Gly4-Serl)2 linker (SEQ ID NO: 60) is used (Desplancq DK et al., Protein Eng. 1994 Aug;7(8): 1027-33 and Mack M. et al., PNAS July 18, 1995 vol. 92 no. 15 7021-7025). “Chemically linked” (or “linked”) as used herein means that the components are linked by covalent binding. As the linker is a peptidic linker, such covalent binding is usually performed by biochemical recombinant means. For example, the binding may be performed using a nucleic acid encoding the VL and/or VH domains of the respective Fab fragments, the linker and the Fc part chain if the antibody comprises an Fc.
[00094] In the event that a linker is used, this linker may be of a length and sequence sufficient to ensure that each of the first and second domains can, independently from each other, retain their differential binding specificities.
Antibody sequences
[00095] In some embodiments, the multispecific (e.g. bispecific) antibody comprises an anti- BCMA antibody, or antigen binding fragment thereof, comprising a VH region comprising a CDR1H region of SEQ ID NO:21, a CDR2H region of SEQ ID NO:22 and a CDR3H region of SEQ ID NO: 17 and a VL region comprising a CDR1L region of SEQ ID NO:27, a CDR2L region of SEQ ID NO:28 and a CDR3L region of SEQ ID NO:20.
[00096] In particularly preferred embodiments, the anti-BCMA antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NOTO and a VL region of SEQ ID NO: 14. 1
[00097] In some embodiments, the multispecific (e.g. bispecific) antibody comprises an anti-CD3 antibody, or antigen binding fragment thereof.
[00098] In some embodiments, the multispecific (e.g. bispecific) antibody of the invention comprises a humanized SP34 antibody or antigen-binding fragment thereof.
[00099] In some preferred embodiments, the anti-CD3 antibody, or antigen binding fragment thereof, may be derived from SP34 and may have similar sequences and the same properties with regard to epitope binding as antibody SP34.
[000100] In some embodiments, the multispecific (e.g. bispecific) antibody comprises an anti-CD3 antibody, or antigen binding fragment thereof, comprising a variable domain VH comprising the heavy chain CDRs of SEQ ID NO: 1, 2 and 3 as respectively heavy chain CDR1H, CDR2H and CDR3H and a variable domain VL comprising the light chain CDRs of SEQ ID NO: 4, 5 and 6 as respectively light chain CDR1L, CDR2L and CDR3L. In some embodiments, the multispecific (e.g. bispecific) antibody comprises an anti-CD3 antibody, or antigen binding fragment thereof, comprising the variable domains of SEQ ID NO:7 (VH) and SEQ ID NO:8 (VL).
[000101] In particularly preferred embodiments, the multispecific (e.g. bispecific) antibody comprises: an anti-BCMA antibody, or antigen binding fragment thereof, comprising a VH region comprising a CDR1H region of SEQ ID NO:21, a CDR2H region of SEQ ID NO:22 and a CDR3H region of SEQ ID NO: 17 and a VL region comprising a CDR1L region of SEQ ID NO:27, a CDR2L region of SEQ ID NO:28 and a CDR3L region of SEQ ID NO:20; and an anti-CD3 antibody, or antigen binding fragment thereof, comprising a CDR1H region of SEQ ID NO: 1, a CDR2H region of SEQ ID NO:2, a CDR3H region of SEQ ID NO:3, a CDR1L region of SEQ ID NO:4, a CDR2L region of SEQ ID NO:5 and a CDR3L region of SEQ ID NO:6.
[000102] In particularly preferred embodiments, the multispecific (e.g. bispecific) antibody comprises an anti-BCMA antibody, or antigen binding fragment thereof, comprising a VH region of SEQ ID NO: 10 and a VL region of SEQ ID NO: 14, and an anti-CD3 antibody, or antigen binding fragment thereof, comprising a VH region of SEQ ID NO:7 and a VL region of SEQ ID NO:8.
Fc
[000103] The multispecific, e.g. bispecific, antibodies of the invention may have an Fc or may not have an Fc. In preferred embodiments, the multispecific antibodies of the invention comprise an Fc, preferably a human Fc.
[000104] In certain embodiments, the Fc is a variant Fc, e.g., an Fc sequence that has been modified (for example by amino acid substitution, deletion and/or insertion) relative to a parent Fc sequence (for example an unmodified Fc polypeptide that is subsequently modified to generate a variant), to provide desirable structural features and/or biological activity,
[000105] Accordingly, the multispecific antibodies, e.g. bispecific antibodies, of the invention
may comprise an Fc comprising one or more modifications, typically to alter one or more functional properties of the antibody, such as serum half-life, complement fixation, Fc receptor binding, and/or antigen-dependent cellular cytotoxicity. The Fc may be linked to the anti-BCMA and/or anti-CD3 Fab fragments in the antibodies of the invention.
[000106] The presence of an Fc has the advantage of extending the elimination half-life of the antibody. The antibodies, e.g. bispecific antibodies, of the invention may have an elimination half-life in mice or cynomolgus monkeys, preferably cynomolgus monkeys, of longer than 12 hours, preferably 3 days or longer. In some embodiments, the antibodies, e.g. bispecific antibodies, of the invention have an elimination half-life of about 1 to 12 days, which allows at least once or twice/week administration.
Reduced effector function
[000107] Preferably, the bispecific antibodies of the invention comprise an Fc region (e.g. of IgGl subclass) that comprises modifications to avoid FcR and Clq binding and minimize ADCC/CDC. This provides the advantage that the bispecific antibody mediates its tumour cell killing efficacy by the effector cell, e.g. T cell, redirection/activation. Therefore, additional mechanisms of action, such as effects on the complement system and on effector cells expressing FcR, are avoided and the risk of sideeffects, such as infusion-related reactions, is decreased.
[000108] In preferred embodiments, the antibodies, e.g. bispecific antibodies, of the invention comprise an IgG, particularly IgGl, Fc region comprising the modifications L234A, L235A and P329G (numbered according to EU numbering).
Heterodimerization
[000109] The multispecific, e.g. bispecific, antibodies of the invention may be heteromultimeric antibodies. Such heteromultimeric antibodies may comprise modifications in regions involved in interactions between antibody chains to promote correct assembly of the antibodies.
[000110] For example, the bispecific antibodies of the invention may comprise an Fc having one or more modification(s) in the CH2 and CH3 domain to enforce Fc heterodimerization. Alternatively or in addition, the bispecific antibodies of the invention may comprise modifications in the CHI and CL region to promote preferential pairing between the heavy chain and light chain of a Fab fragment.
[000111] A number of strategies exist for promoting heterodimerization. These strategies may include the introduction of asymmetric complementary modifications into each of two antibody chains, such that both chains are compatible with each other and thus able to form a heterodimer, but each chain is not able to dimerize with itself. Such modifications may encompass insertions, deletions, conservative and non-conservative substitutions and rearrangements.
[000112] Heterodimerization may be promoted by the introduction of charged residues to create favourable electrostatic interactions between a first antibody chain and a second antibody chain. For example, one or more positively charged amino acids amino acid may be introduced into a first antibody chain, and one or more negatively charged amino acids may be introduced into a corresponding positions
in a second antibody chain.
[000113] Alternatively or in addition, heterodimerization may be promoted by the introduction of steric hindrance between contacting residues. For example, one or more residues with a bulky side chain may be introduced into a first antibody chain, and a one or more residues able to accommodate the bulky side chain may be introduced into the second antibody chain.
[000114] Alternatively or in addition, heterodimerization may be promoted by the introduction of one or more modification(s) to the hydrophilic and hydrophobic residues at the interface between chains, in order make heterodimer formation more entropically and enthalpically favourable than homodimer formation.
[000115] A further strategy for promoting heterodimerization is to rearrange portions of the antibody chains such that each chain remains compatible only with a chain comprising corresponding rearrangements. For example, CrossMAb technology is based on the crossover of antibody domains in order to enable correct chain association. There are three main CrossMAb formats, these are: (i) CrossMAbFab in which the VH and VL are exchanged and the CHI and CL are exchanged; (ii) CrossMAbVH’VL in which the VH and VL are exchanged; and (iii) CrossMAbCH1 CL in which the CHI and CL are exchanged (Klein et al., 2016. MABS, 8(6): 1010-1020).
[000116] jn some embodiments, the bispecific antibodies of the invention may comprise an exchange of the VH and VL. In some embodiments, the antibodies, e.g. bispecific antibodies, of the invention may comprise an exchange of the CHI and CL. In some embodiments, the antibodies, e.g. bispecific antibodies, of the invention may comprise an exchange of the VH and VL and an exchange of the CHI and CL.
[000117] In preferred embodiments, the antibodies, e.g. bispecific antibodies, of the invention comprise an exchange of the VH and VL.
[000118] Other approaches to promoting heterodimerization include the use of a strand exchange engineered domain (SEED) (Davis et al., 2010. Protein Eng Des Sei, 23 (4); 195- 202).
[000119] A combination of the above strategies may be used to maximise the efficiency of assembly while minimising the impact on antibody stability.
Fc Heterodimerization
[000120] In some embodiments, multispecific antibodies, e.g. bispecific antibodies, of the invention may have a heterodimeric Fc, for example they may comprise one heavy chain originating from an anti-BCMA antibody, and one heavy chain originating from an anti-CD3 antibody.
[000121] The antibodies, e.g. bispecific antibodies, of the invention may comprise a heterodimeric Fc which comprises one or more modification s) which promotes the association of the first CH2 and/or CH3 domain with the second CH2 and/or CH3 domain. In preferred embodiments, the one or more modification(s) promote the association of the first CH3 domain with the second CH3 domain, for example by resulting in asymmetric modifications to the CH3 domain. The one or more modification(s)
may comprise modifications selected from amino acid insertions, deletions, conservative and nonconservative substitutions and rearrangements, and combinations thereof.
[000122] Typically the first CH3 domain and the second CH3 domain are both engineered in a complementary manner so that each CH3 domain (or the heavy chain comprising it) can no longer homodimerize with itself but is forced to heterodimerize with the complementary engineered other CH3 domain (so that the first and second CH3 domain heterodimerize and no homodimers between the two first or the two second CH3 domains are formed).
[000123] The multispecific, e.g. bispecific, antibodies of the invention may comprise an Fc having one or more of “knob-into-holes” modification s), which are described in detail with several examples in e.g. WO 96/027011, Ridgway, J.B., et al., Protein Eng. 9 (1996) 617-621, Merchant, A.M. et al., Nat. Biotechnol. 16 (1998) 677-68, and WO 98/050431.
[000124] In this method, the interaction surfaces of the two CH3 domains are altered to increase the heterodimerization of both Fc chains containing these two CH3 domains. One of the two CH3 domains (of the two Fc chains) can be the “knob”, while the other is the “hole”.
[000125] Accordingly, the bispecific antibodies of the invention may comprise two CH3 domains, wherein the first CH3 domain of the first Fc chain and the second CH3 domain of the second Fc chain each meet at an interface which comprises an original interface between the antibody CH3 domains, wherein said interface is altered to promote the formation of the antibody.
[000126] In some embodiments:
(i) the CH3 domain of one Fc chain is altered, so that within the original interface of the CH3 domain of the one Fc chain that meets the original interface of the CH3 domain of the other Fc chain, an amino acid residue is replaced with an amino acid residue having a larger side chain volume, thereby generating a protuberance within the interface of the CH3 domain of one Fc chain which is positionable in a cavity within the interface of the CH3 domain of the other Fc chain; and ii) the CH3 domain of the other Fc chain is altered, so that within the original interface of the CH3 domain of the other Fc chain that meets the original interface of the CH3 domain of the one Fc chain, an amino acid residue is replaced with an amino acid residue having a smaller side chain volume, thereby generating a cavity within the interface of the CH3 domain of the other Fc chain within which a protuberance within the interface of the CH3 domain of the one Fc chain is positionable.
[000127] Preferably, said amino acid residue having a larger side chain volume is selected from the group consisting of arginine (R), phenylalanine (F), tyrosine (Y), tryptophan (W).
[000128] In some embodiments, the multispecific, e.g. bispecific, antibodies of the invention comprise a first CH3 domain comprising modification(s) at positions T366, L368 and Y407, e.g. T366S, L368A, and Y407V (numbered according to EU numbering).
[000129] In some embodiments, the multispecific, e.g. bispecific, antibodies of the invention comprise a second CH3 domain comprising a modification at position T366 (“knob modification”), e.g.
T366W (numbered according to EU numbering).
[000130] In particularly preferred embodiments, the multispecific, e.g. bispecific, antibodies of the invention comprise a first CH3 domain comprising the modifications T366S, L368A, and Y407V, or conservative substitutions thereof, and a second CH3 domain comprising the modification T366W, or a conservative substitution thereof (numbered according to EU numbering).
[000131] In one embodiment, the multispecific, e.g. bispecific, antibodies of the invention comprise a first CH3 domain comprising the modification set forth in Table 2 and a second CH3 domain comprising the modifications set forth in Table 2.
Table 2: “Knob-into-holes” modification

[000132] Other techniques for CH3 modifications to enforce heterodimerization are contemplated as alternatives of the invention and are described e.g. in WO96/27011, W098/050431, EP1870459, W02007/110205, W02007/ 147901, W02009/089004, W02010/129304, WO2011/90754, WO2011/143545, WO2012/058768, WO2013/157954, WO2013/157953, and WO2013/096291.
[000133] In some embodiments, the bispecific antibody according to the invention is of IgG2 isotype and the heterodimerization approach described in W02010/129304 can be used.
Other Fc modifications
[000134] In some embodiments, the bispecific antibodies of the invention may comprise an Fc, wherein both CH3 domains are altered by the introduction of cysteine (C) as the amino acid in the corresponding positions of each CH3 domain such that a disulphide bridge between both CH3 domains can be formed. The cysteines may be introduced at position 349 in one of the CH3 domains and at position 354 in the other CH3 domain (numbered according to EU numbering).
[000135] Preferably, the cysteine introduced at position 354 is in the first CH3 domain and the cysteine introduced at position 349 is in the second CH3 domain (numbered according to EU numbering).
[000136] The Fc may comprise modifications, such as D356E, L358M, N384S, K392N, V397M, and V422I (numbered according to EU numbering). Preferably, both CH3 domains comprise D356E and L358M (numbered according to EU numbering).
Light and heavy chain heterodimerization
[000137] In the multispecific, e.g. bispecific, antibodies of the invention, one or more of the immunoglobulin heavy chains and light chains may comprise one or more modification(s), e.g. amino acid modifications that are capable of promoting preferential pairing of a specific heavy chain with a specific light chain when heavy chains and light chains are co-expressed or co-produced. Such modifications can provide considerably improved production/purification without changing biological properties such as binding to BCMA. In particular, by introduction of one or more modification(s) such as amino acid exchanges, light chain mispairing and the formation of side products in production can be significantly reduced and therefore yield is increased and purification is facilitated.
[000138] The amino acid exchanges may be substitutions of charged amino acids with opposite charges (for example in the CH1/CL interface) which reduce light chain mispairing, e.g. Bence-Jones type side products.
[000139] In preferred embodiments, the one or more modification(s) assist light and heavy chain heterodimerization are amino acid modifications in the light and heavy chains outside of the CDRs.
[000140] The one or more modification(s) may be present in the anti-BCMA antibody or antigenbinding fragment thereof. Alternatively, the one or more modification(s) may be present in the anti-CD3 antibody or antigen-binding fragment thereof. In preferred embodiments, the one or more modification(s) are present in the anti-BCMA antibody or antigen-binding fragment thereof.
[000141] In some embodiments, the multispecific, e.g. bispecific, antibodies of the invention comprise an immunoglobulin heavy chain comprising a CHI domain having amino acid modifications K147E/D and K213E/D (numbered according to EU numbering) and a corresponding immunoglobulin light chain comprising a CL domain having amino acid modifications E123K/R/H and Q124K/R/H (numbered according to Kabat). Preferably, the CHI domain comprises the amino acid modifications K147E and K213E (numbered according to EU numbering) or conservative substitutions thereof, and the corresponding CL domain comprises the amino acid modifications E123R and Q124K or conservative substitutions thereof (numbered according to Kabat). Such multispecific, e.g. bispecific, antibodies can be produced in high yield and can be easily purified.
[000142] In one embodiment, the amino acid modifications described in Table 3 can be in the BCMA antibody or in the CD3 antibody.
[000143] In one embodiment, the bispecific antibodies of the invention are bivalent, and comprise one anti-BCMA antibody or antigen-binding fragment thereof and one anti-CD3 antibody or antigenbinding fragment thereof (the “1+1” format), wherein:
(a) the BCMA antibody or antigen-binding fragment thereof (e.g. BCMA Fab) comprises a CHI domain having amino acid modifications set forth in Table 3 and a corresponding CL domain having the amino acid modifications Table 3; or
(b) the CD3 antibody or antigen-binding fragment thereof (e.g. CD3 Fab) comprises a CHI domain
having amino acid modifications set forth in Table 3 and a corresponding CL domain having the amino acid modifications Table 3.
[000144] In one embodiment, the bispecific antibodies of the invention are trivalent and comprise two anti-BCMA antibodies or antigen-binding fragments thereof and one anti-CD3 antibody or antigenbinding fragment thereof (the “2+1” format), wherein:
(a) one or both BCMA antibodies or antigen-binding fragments thereof (e.g. BCMA Fabs) comprises a CHI domain having amino acid modifications set forth in Table 3 and a corresponding CL domain having the amino acid modifications Table 3; or
(b) the CD3 antibody (e.g. CD3 Fab) comprises a CHI domain having amino acid modifications set forth in Table 3 and a corresponding CL domain having the amino acid modifications Table 3.
[000145] In particular, each BCMA antibody (e.g. BCMA Fab) may comprise a CHI domain having amino acid modifications set forth in Table 3 and a corresponding CL domain having the amino acid modifications Table 3.
Table 3: Light and heavy chain heterodimerization modifications

[000146] In a preferred embodiment, the multispecific, e.g. bispecific, antibodies of the invention comprise the modifications set forth in Table 3 in combination with the modifications set forth in Table 2. Thus, in one embodiment, the bispecific antibodies of the invention are bivalent, and comprise:
(a) one anti-BCMA antibody or antigen-binding fragment thereof and one anti-CD3 antibody or antigen-binding fragment thereof (the “1+1” format), wherein (i) the BCMA antibody or antigenbinding fragment thereof (e.g. BCMA Fab) comprises a CHI domain that comprises the amino acid modifications K147E and K213E, and a corresponding CL domain that comprises the amino acid modifications E123R and Q124K (i.e. the modifications set forth in Table 3), or (ii) the CD3 antibody or antigen-binding fragment thereof (e.g. CD3 Fab) comprises a CHI domain that comprises the amino acid modifications K147E and K213E, and a corresponding CL domain that comprises the amino acid modifications E123R and Q124K (i.e. the modifications set forth in Table 3); and
(b) a first CH3 domain comprising the modifications T366S, L368A, and Y407V, and a second CH3 domain comprising the modification T366W (i.e. the modifications set forth in Table 2).
[000147] In one embodiment, the bispecific antibodies of the invention are trivalent and comprise:
(a) two anti-BCMA antibodies or antigen-binding fragments thereof and one anti-CD3 antibody or antigen-binding fragment thereof (the “2+1” format), wherein (i) one or both BCMA antibodies or antigen-binding fragments thereof (e.g. BCMA Fabs) comprises a CHI domain that comprises the amino acid modifications K147E and K213E, and a corresponding CL domain that comprises the amino acid modifications E123Rand Q124K (i.e. the modifications set forth in Table 3), or (ii) the CD3 antibody or antigen-binding fragment thereof (e.g. CD3 Fab) comprises a CHI domain that comprises the amino acid modifications K147E and K213E, and a corresponding CL domain that comprises the amino acid modifications E123R and Q124K (i.e. the modifications set forth in Table 3); and
(b) a first CH3 domain comprising the modifications T366S, L368A, and Y407V, and a second CH3 domain comprising the modification T366W (i.e. the modifications set forth in Table 2).
[000148] In particular, each BCMA antibody (e.g. BCMA Fab) may comprise a CHI domain having amino acid modifications set forth in Table 3 and a corresponding CL domain having the amino acid modifications Table 3. In preferred embodiments, the first Fc chain is bound at the N-terminus of the Fc to the C-terminus of the first anti-BCMA antibody, and the second Fc chain is bound at the N- terminus of the Fc to the C-terminus of the anti-CD3 antibody.
[000149] The multispecific, e.g. bispecific, antibodies of the invention may additionally comprise an amino acid substitution at position 49 of the VL region selected from the group of amino acids tyrosine (Y), glutamic acid (E), serine (S), and histidine (H) and/or an amino acid substitution at position 74 of the VL region that is threonine (T) or alanine (A).
CrossMAb
[000150] The multispecific, e.g. bispecific, antibodies of the invention may comprise CrossMAb technology. CrossMAb technology is based on the crossover of antibody domains in order to enable correct chain association. It is used to facilitate multispecific antibody formation. There are three main CrossMAb formats, these are: (i) CrossMAbFab in which the VH and VL are exchanged and the CHI and CL are exchanged; (ii) CrossMAbVH’VL in which the VH and VL are exchanged; and (iii) CrossMAbCH1 CL in which the CHI and CL are exchanged (Klein et al., 2016. MABS, 8(6): 1010-1020).
[000151] CrossMAb technology is known in the state of the art. Bispecific antibodies wherein the variable domains VL and VH or the constant domains CL and CHI are replaced by each other are described in W02009080251 and W02009080252.
[000152] In one or more of the antibodies or antigen-binding fragments within the multispecific, e.g. bispecific, antibodies of the invention, the variable domains VL and VH or the constant domains CL and CHI may be replaced by each other. In some embodiments, the antibodies, e.g. bispecific antibodies, of the invention may comprise an exchange of the VH and VL and an exchange of the CHI and CL. Thus, the multispecific, e.g. bispecific, antibodies of the invention may comprise a crossover light chain and a crossover heavy chain.
[000153] In some aspects, there is provided a multispecific, e.g. bispecific, antibody comprising an anti-BCMA antibody of the invention, or an antigen-binding fragment thereof, and an anti-CD3 antibody, or antigen-binding fragment thereof, wherein the multispecific, e.g. bispecific, antibody comprises:
(a) a light chain and a heavy chain of an antibody specifically binding to CD3; and
(b) a light chain and heavy chain of an antibody specifically binding to BCMA, wherein the variable domains VL and VH and/or the constant domains CL and CHI are replaced by each other in (i) the anti-BCMA antibody; and/or (ii) the anti-CD3 antibody.
[000154] In some embodiments, the variable domains VL and VH or the constant domains CL and CHI of the anti-CD3 antibody or antigen binding fragment thereof are replaced by each other. More preferably, the variable domains VL and VH of the anti-CD3 antibody or antigen binding fragment thereof are replaced by each other.
[000155] In embodiments in which the bispecific antibodies in the 1+1 format have the format: CD3 Fab - BCMA Fab (i.e. when no Fc is present); Fc - CD3 Fab - BCMA Fab; Fc- BCMA Fab - CD3 Fab; or BCMA Fab - Fc - CD3 Fab, the bispecific antibodies may comprise the CrossMAb format, e.g. CrossMAbFab, CrossMAbVH’VL or CrossMAbCH1 CL. The BCMA Fab may have the CrossMAb format, e.g. CrossMAbFab, CrossMAbVH’VL or CrossMAbCH1 CL. Alternatively, the CD3 Fab may have the CrossMAb format, e.g. CrossMAbFab, CrossMAb VH VL or CrossMAbCH1 CL. In preferred embodiments, the CD3 Fab of the bispecific antibody comprises the CrossMAb VH VL format.
[000156] It is especially preferred for the bispecific antibodies of the invention having the 2+1 format to comprise CrossMAb technology. Thus, in embodiments in which the trivalent bispecific antibodies in the 2+1 format have the format: CD3 Fab - BCMA Fab - BCMA Fab; BCMA Fab - CD3 Fab - BCMA Fab (i.e. when no Fc is present); BCMA Fab - Fc - CD3 Fab - BCMA Fab; BCMA Fab - Fc - BCMA Fab - CD3 Fab; or CD3 Fab - Fc - BCMA Fab - BCMA Fab, the bispecific antibodies may comprise the CrossMAb format, e.g. CrossMAbFab, CrossMAb " ' l or CrossMAbCH1 CL. The BCMA Fab may have the CrossMAb format, e.g. CrossMAbFab, CrossMAb VH VL or CrossMAbCH1 CL. Alternatively, the CD3 Fab may have the CrossMAb format, e.g. CrossMAbFab, CrossMAbVH’VL or CrossMAb CH1 CL. In preferred embodiments, the CD3 Fab of the bispecific antibody comprises the CrossMAbVH’VL format. [000157] In some embodiments, the bispecific antibodies of the invention having the 1+1 format do not comprise CrossMAb technology, i.e. neither the anti-BCMA antibody nor the anti-CD3 antibody have the variable domains VL and VH or the constant domains CL and CHI replaced by each other.
Exemplary Embodiments
[000158] Exemplary embodiments are set out in FIG. 1.
[000159] In one embodiment, the bispecific antibodies according to the invention are trivalent bispecific antibodies comprising one Fab fragment of an anti-CD3 antibody, two Fab fragments of an anti-BCMA antibody and one Fc part according to the format BCMA Fab - Fc - CD3 Fab - BCMA Fab. Each anti-BCMA Fab fragment comprises the amino acid modifications set forth in Table 3. The anti-
CD3 Fab fragment comprises a light chain and heavy chain, wherein the light chain is a crossover light chain that comprises a variable domain VH and a constant domain CL, and wherein the heavy chain is a crossover heavy chain that comprises a variable domain VL and a constant domain CHI . This embodiment is illustrated in FIG. 1.
[000160] In one embodiment, the antibodies illustrated in FIG. 1 additionally comprise the modifications set forth in Table 2.
[000161] In one aspect, the bispecific antibodies according to the invention are trivalent bispecific antibodies comprising one Fab fragment of an anti-CD3 antibody, two Fab fragments of an anti-BCMA antibody and one Fc part according to the format BCMA Fab - Fc - CD3 Fab - BCMA Fab. The anti- CD3 Fab fragment comprises a light chain and heavy chain, wherein the light chain is a crossover light chain that comprises a variable domain VH and a constant domain CL, and wherein the heavy chain is a crossover heavy chain that comprises a variable domain VL and a constant domain CHI . Each anti- BCMA Fab fragment comprises a light chain and heavy chain, wherein the heavy chain comprises a CHI domain which comprises the amino acid modifications K147E and K213E (numbered according to EU numbering) and wherein the light chain comprises a corresponding CL domain which comprises the amino acid modifications E123R and Q124K (numbered according to Kabat) (i.e. the modifications set forth in Table 3). The Fc part comprises a first Fc chain and a second Fc chain, wherein the first Fc chain comprises a first constant domain CH2 and a first constant domain CH3, and the second Fc chain comprises a second constant domain CH2 and a second constant domain CH3. The first Fc chain is bound at the N-terminus of the Fc to the C-terminus of the first anti-BCMA Fab, and the second Fc chain is bound at the N-terminus of the Fc to the C-terminus of the anti-CD3 Fab. The first CH3 domain comprises the modifications T366S, L368A, and Y407V (“hole modifications”) and the second CH3 domain comprises the modification T366W (“knob modification”) (numbered according to EU numbering) (i.e. the modifications set forth in Table 2). Additionally, both Fc chains further comprise the modifications L234A, L235A and P329G, and optionally D356E and L358M (numbered according to EU numbering). Optionally, the first CH3 domain further comprises the amino acid modification S354C, and the second CH3 domain further comprises the amino acid modification Y349C (numbered according to EU numbering) such that a disulphide bridge between both CH3 domains is formed.
[000162] In some embodiments, the anti-BCMA Fab fragment comprises a a CDR1H region of SEQ ID NO: 21, a CDR2H region of SEQ ID NO: 22 and a CDR3H region of SEQ ID NO: 17 and a VL region comprising a CDR1L region of SEQ ID NO:27, a CDR2L region of SEQ ID NO:28 and a CDR3L region of SEQ ID NO:20; and the anti-CD3 Fab fragment comprises a CDR1H region of SEQ ID NO: 1, a CDR2H region of SEQ ID NO:2, a CDR3H region of SEQ ID NO:3, a CDR1L region of SEQ ID NO:4, a CDR2L region of SEQ ID NO:5 and a CDR3L region of SEQ ID NO:6.
[000163] In some embodiments, the anti-BCMA Fab fragment comprises a VH region of SEQ ID
NO: 10 and a VL region of SEQ ID NO: 14; and the anti-CD3 Fab fragment comprises a VH region of
SEQ ID NO : 7 and a VL region of SEQ ID NO : 8.
[000164] In various embodiments, the bispecific antibody according to the invention comprises the SEQ ID NOs (as mentioned in Tables 7A and 8B below) of Alnuctamab (42-TCBcv): 48, 55 or 58, 56 or 59, and 57 (x2) (FIG. 1).
[000165] The term “alnuctamab” (also referred to as 42-TCBcv” and “Mab42”) as used herein refers to the bispecific antibody comprising a first polypeptide comprising the amino acid sequence of SEQ ID NO: 48, a second polypeptide comprising the amino acid sequence of SEQ ID NO: 55 or 58, a third polypeptide comprising the amino acid sequence of SEQ ID NO: 56 or 59, and fourth and fifth polypeptides each comprising the amino acid sequence of SEQ ID NO: 57, and as shown in FIG. 1 and described in WO 2017/021450.
[000166] In preferred embodiments, the bispecific antibody is 42-TCBcv (alnuctamab). Pharmaceutical Compositions
[000167] The multispecific, e.g. bispecific, antibodies of the invention can be administered to the patient as a pharmaceutical composition. Accordingly, the present invention also provides a pharmaceutical composition comprising the multispecific, e.g. bispecific, antibodies of the invention discussed herein. In preferred embodiments, pharmaceutical compositions described herein comprise 42- TCBcv (alnuctamab).
[000168] In some aspects, the pharmaceutical composition comprises at least about 0.05 mg/mL to at least about 37.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 1 mg/mL to at least about 37.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.5 mg/mL to at least about 37.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 1 mg/mL to at least about 37.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 4.5 mg/mL to at least about 37.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 4.5 mg/mL to at least about 37.5 mg/mL, at least about 5 mg/mL to at least about 35 mg/mL, at least about 5.5 mg/mL to at least about 32 mg/mL, at least about 6 mg/mL to at least about 30 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.05 mg/mL of the multispecific antibody, e.g. alnuctamab.. In some aspects, the pharmaceutical composition comprises at least about 0.06 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.07 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.08 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.09 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0. 1 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.2 mg/mL of the
multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.3 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.4 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.6 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.7 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.8 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.9 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 1 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 1.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 2 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 3 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 3.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 4 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 4.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 5.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 6 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 6.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 7 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 7.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 22.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 25 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 27 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 28 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 29 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 29.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 30 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 30.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the
pharmaceutical composition comprises at least about 31 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 32 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 35 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 37 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 37.5 mg/mL of the multispecific antibody, e.g. alnuctamab.
[000169] In some aspects, the pharmaceutical composition further comprises a metal chelator. In some aspects, the metal chelator prevents oxidation of the formulation components and/or improves stability of the antibody, e.g. alnuctamab. In some aspects, the metal chelator is pentetic acid (DTP A) or ethylenediaminetetraacetic acid (EDTA). In some aspects, the metal chelator is pentetic acid (DTP A). In some aspects, the pharmaceutical composition comprises from at least about 1 mM to at least about 250 pM pentetic acid (DTP A). In some aspects, the pharmaceutical composition comprises at least about 1 pM, at least about 5 pM, at least about 10 pM, at least about 15 pM, at least about 20 pM, at least about 25 pM, at least about 30 pM, at least about 35 pM, at least about 40 pM, at least about 45 pM, at least about 50 pM, at least about 55 pM, at least about 60 pM, at least about 65 pM, at least about 70 pM, at least about 75 pM, at least about 80 pM, at least about 85 pM, at least about 90 pM, at least about 95 pM, or at least about 100 pM, at least about 110 pM, at least about 120 pM, at least about 130 pM, at least about 140 pM, at least about 150 pM, at least about 160 pM, at least about 170 pM, at least about 180 pM, at least about 190 pM, or at least about 200 pM DTPA. In certain aspects, the pharmaceutical composition comprises at least about 75 pM pentetic acid (DTPA). In certain aspects, the pharmaceutical composition comprises at least about 70 pM pentetic acid (DTP A). In certain aspects, the pharmaceutical composition comprises at least about 65 pM pentetic acid (DTP A). In certain aspects, the pharmaceutical composition comprises at least about 60 pM pentetic acid (DTPA). In certain aspects, the pharmaceutical composition comprises at least about 55 pM pentetic acid (DTPA). In certain aspects, the pharmaceutical composition comprises at least about 50 pM pentetic acid (DTPA). In certain aspects, the pharmaceutical composition comprises at least about 45 pM pentetic acid (DTPA). In certain aspects, the pharmaceutical composition comprises at least about 40 pM pentetic acid (DTPA). In certain aspects, the pharmaceutical composition comprises at least about 35 pM pentetic acid (DTPA). In certain aspects, the pharmaceutical composition comprises at least about 30 pM pentetic acid (DTPA). In certain aspects, the pharmaceutical composition comprises at least about 25 pM pentetic acid (DTPA).
[000170] In some aspects, the pharmaceutical composition further comprises a tonicity modifier and/or stabilizer. Any tonicity modifier and/or any stabilizer can be used in the pharmaceutical compositions disclosed herein. In some aspects, the tonicity modifier and/or stabilizer comprises a sugar, an amino acid, a polyol, a salt, or any combination thereof. In some aspects, the tonicity modifier and/or stabilizer is selected from the group consisting of sucrose, sorbitol, trehalose, mannitol, glycerol, glycine, leucine, isoleucine, sodium chloride, proline, arginine, polyols, amino acids, and salts. In certain aspects,
the pharmaceutical composition comprises sucrose. In some aspects, the pharmaceutical composition comprises from at least about 1 mM to at least about 500 mM sucrose. In some aspects, the pharmaceutical compositions comprises from at least about 10 mM to at least about 400 mM, at least about 50 mM to at least about 400 mM, at least about 100 mM to at least about 400 mM, at least about 150 mM to at least about 400 mM, at least about 200 mM to at least about 400 mM, at least about 250 mM to at least about 400 mM, at least about 300 mM to at least about 400 mM, at least about 350 mM to at least about 400 mM, at least about 50 mM to at least about 350 mM, at least about 100 mM to at least about 300 mM, at least about 100 mM to at least about 250 mM, at least about 100 mM to at least about 200 mM, at least about 100 mM to at least about 150 mM, at least about 200 mM to at least about 400 mM, at least about 200 mM to at least about 300 mM sucrose, or at least about 200 mM to at least about 250 mM. In some aspects, the pharmaceutical compositions comprises at least about 10 mM, at least about 20 mM, at least about 30 mM, at least about 40 mM, at least about 50 mM, at least about 60 mM, at least about 70 mM, at least about 80 mM, at least about 90 mM, at least about 100 mM, at least about 110 mM, at least about 120 mM, at least about 130 mM, at least about 140 mM, at least about 150 mM, at least about 160 mM, at least about 170 mM, at least about 180 mM, at least about 190 mM, at least about 200 mM, at least about 210 mM, at least about 220 mM, at least about 230 mM, at least about 240 mM, at least about 250 mM, at least about 260 mM, at least about 270 mM, at least about 280 mM, at least about 290 mM, at least about 300 mM, at least about 310 mM, at least about 320 mM, at least about 330 mM, at least about 340 mM, at least about 350 mM, at least about 360 mM, at least about 370 mM, at least about 380 mM, at least about 390 mM, at least about 400 mM, at least about 410 mM, at least about 420 mM, at least about 430 mM, at least about 440 mM, at least about 450 mM, at least about 460 mM, at least about 470 mM, at least about 480 mM, at least about 490 mM, or at least about 500 mM sucrose. In certain aspects, the pharmaceutical composition comprises at least about 200 mM sucrose. In certain aspects, the pharmaceutical composition comprises at least about 210 mM sucrose. In certain aspects, the pharmaceutical composition comprises at least about 220 mM sucrose. In certain aspects, the pharmaceutical composition comprises at least about 230 mM sucrose. In certain aspects, the pharmaceutical composition comprises at least about 240 mM sucrose. In certain aspects, the pharmaceutical composition comprises at least about 250 mM sucrose. In certain aspects, the pharmaceutical composition comprises at least about 260 mM sucrose. In certain aspects, the pharmaceutical composition comprises at least about 270 mM sucrose. In certain aspects, the pharmaceutical composition comprises at least about 280 mM sucrose. In certain aspects, the pharmaceutical composition comprises at least about 290 mM sucrose. In certain aspects, the pharmaceutical composition comprises at least about 300 mM sucrose.
[000171] In some aspects, the pharmaceutical composition further comprises a buffering agent. In some aspects, the buffering agent is selected from histidine, succinate, tromethamine, sodium phosphate, sodium acetate, and sodium citrate. In certain aspects, the pharmaceutical composition comprises histidine. In some aspects, the pharmaceutical composition comprises from at least about 1 mM to at least
about 100 mM histidine. In some aspects, the pharmaceutical composition comprises from at least about 5 mM to at least about 100 mM, at least about 10 mM to at least about 100 mM, at least about 15 mM to at least about 100 mM, at least about 20 mM to at least about 100 mM, at least about 25 mM to at least about 100 mM, at least about 30 mM to at least about 100 mM, at least about 35 mM to at least about 100 mM, at least about 40 mM to at least about 100 mM, at least about 45 mM to at least about 100 mM, at least about 50 mM to at least about 100 mM, at least about 10 mM to at least about 75 mM, at least about 10 mM to at least about 50 mM, at least about 10 mM to at least about 40 mM, at least about 10 mM to at least about 30 mM, at least about 15 mM to at least about 30 mM, at least about 10 mM to at least about 25 mM, or at least about 15 mM to at least about 25 mM, histidine. In some aspects, the pharmaceutical composition comprises at least about 5 mM, at least about 10 mM, at least about 15 mM, at least about 20 mM, at least about 25 mM, at least about 30 mM, at least about 35 mM, at least about 40 mM, at least about 45 mM, at least about 50 mM, at least about 60 mM, at least about 70 mM, at least about 80 mM, at least about 90 mM, or at least about 100 mM histidine. In certain aspects, the pharmaceutical composition comprises at least about 10 mM histidine. In certain aspects, the pharmaceutical composition comprises at least about 15 mM histidine. In certain aspects, the pharmaceutical composition comprises at least about 20 mM histidine. In certain aspects, the pharmaceutical composition comprises at least about 25 mM histidine. In certain aspects, the pharmaceutical composition comprises at least about 30 mM histidine. In certain aspects, the pharmaceutical composition comprises at least about 35 mM histidine. In certain aspects, the pharmaceutical composition comprises at least about 40 mM histidine. In certain aspects, the pharmaceutical composition comprises at least about 45 mM histidine. In certain aspects, the pharmaceutical composition comprises at least about 50 mM histidine.
[000172] In some aspects, the pharmaceutical composition comprises a pH of about 5.2 to about 6.8. In some aspects, the pH of the pharmaceutical composition is about 5.2. In some aspects, the pH of the pharmaceutical composition is about 5.3. In some aspects, the pH of the pharmaceutical composition is about 5.4. In some aspects, the pH of the pharmaceutical composition is about 5.5. In some aspects, the pH of the pharmaceutical composition is about 5.6. In some aspects, the pH of the pharmaceutical composition is about 5.7. In some aspects, the pH of the pharmaceutical composition is about 5.8. In some aspects, the pH of the pharmaceutical composition is about 5.9. In some aspects, the pH of the pharmaceutical composition is about 6.0. In some aspects, the pH of the pharmaceutical composition is about 6. 1. In some aspects, the pH of the pharmaceutical composition is about 6.2. In some aspects, the pH of the pharmaceutical composition is about 6.3. In some aspects, the pH of the pharmaceutical composition is about 6.4. In some aspects, the pH of the pharmaceutical composition is about 6.5. In some aspects, the pH of the pharmaceutical composition is about 6.6. In some aspects, the pH of the pharmaceutical composition is about 6.7. In some aspects, the pH of the pharmaceutical composition is about 6.8.
[000173] In some aspects, the pharmaceutical composition further comprises a surfactant. Any
surfactant can be used in the pharmaceutical compositions disclosed herein. In some aspects, the surfactant is selected from the group consisting of polysorbate 20, polysorbate 80, and poloxamer 188. In certain aspects, the pharmaceutical composition comprises polysorbate 80. In some aspects, the pharmaceutical composition comprises from at least about 0.001% to at least about 1% w/v polysorbate 80. In some aspects, the pharmaceutical compositions comprises at least about 0.01% to at least about 0.1%, at least about 0.02% to at least about 0.1%, at least about 0.03% to at least about 0.1%, at least about 0.04% to at least about 0.1%, at least about 0.05% to at least about 0.1%, at least about 0.01% to at least about 0.09%, at least about 0.01% to at least about 0.8%, at least about 0.01% to at least about 0.7%, at least about 0.01% to at least about 0.6%, at least about 0.01% to at least about 0.5%, at least about 0.02% to at least about 0.09%, at least about 0.03% to at least about 0.08%, at least about 0.04% to at least about 0.07%, or at least about 0.04% to at least about 0.06% w/v polysorbate 80. In some aspects, the pharmaceutical compositions comprises at least about 0.01% to at least about 0.1% w/v polysorbate 80. In some aspects, the pharmaceutical composition comprises at least about 0.01% w/v, at least about 0.02% w/v, at least about 0.03% w/v, at least about 0.04% w/v, at least about 0.05% w/v, at least about 0.06% w/v, at least about 0.07% w/v, at least about 0.08% w/v, at least about 0.09% w/v, or at least about 0.1% w/v polysorbate 80. In certain aspects, the pharmaceutical composition comprises at least about 0.03% w/v polysorbate 80. In certain aspects, the pharmaceutical composition comprises at least about
0.04% w/v polysorbate 80. In certain aspects, the pharmaceutical composition comprises at least about
0.05% w/v polysorbate 80. In certain aspects, the pharmaceutical composition comprises at least about
0.06% w/v polysorbate 80. In certain aspects, the pharmaceutical composition comprises at least about
0.07% w/v polysorbate 80.
[000174] In some aspects, the pharmaceutical composition comprises: (a) at least about 4.5 mg/mL to at least about 37.5 mg/mL of a multispecific antibody that binds to BCMA and CD3, e.g. alnuctamab, (b) at least about 5 mM to at least about 100 mM histidine, (c) at least about 10 mM to at least about 500 mM sucrose, (d) at least about 0.01% w/v to at least about 0. 1% w/v polysorbate 80, and (e) at least about 10 pM to about 200 pM pentetic acid (DTP A). In some aspects, the pharmaceutical composition comprises (a) about 6 mg/mL of the multispecific antibody, e.g. alnuctamab; (b) about 20 mM histidine; (c) about 250 mM sucrose; (d) about 0.04% w/v polysorbate 80; and (e) about 50 pM pentetic acid. In some aspects, the pharmaceutical composition comprises (a) about 30 mg/mL of the multispecific antibody, e.g. alnuctamab; (b) about 20 mM histidine; (c) about 250 mM sucrose; (d) about 0.04% w/v polysorbate 80; and (e) about 50 pM pentetic acid. In some aspects, the pharmaceutical composition comprises (a) about 6 mg/mL of alnuctamab; (b) about 20 mM histidine; (c) about 250 mM sucrose; (d) about 0.04% w/v polysorbate 80; and (e) about 50 pM pentetic acid. In some aspects, the pharmaceutical composition comprises (a) about 30 mg/mL of alnuctamab; (b) about 20 mM histidine; (c) about 250 mM sucrose; (d) about 0.04% w/v polysorbate 80; and (e) about 50 pM pentetic acid. In some aspects, the pharmaceutical composition comprises (a) 6 mg/mL of alnuctamab; (b) 20 mM histidine; (c) 250 mM sucrose; (d) 0.04% w/v polysorbate 80; and (e) 50 pM pentetic acid. In some
aspects, the pharmaceutical composition comprises (a) 30 mg/mL of alnuctamab; (b) 20 mM histidine; (c) 250 mM sucrose; (d) 0.04% w/v polysorbate 80; and (e) 50 pM pentetic acid.
[000175] Some aspects of the present disclosure are directed to a vial comprising the pharmaceutical composition disclosed herein. In some aspects, the vial comprises a unit dose of the pharmaceutical composition. In some aspects, the vial or unit dose comprises (a) about 6 mg/mL of the multispecific antibody, e.g. alnuctamab; (b) about 20 mM histidine; (c) about 250 mM sucrose; (d) about 0.04% w/v polysorbate 80; and (e) about 50 pM pentetic acid. In some aspects, the vial or unit dose comprises (a) about 30 mg/mL of the multispecific antibody, e.g. alnuctamab; (b) about 20 mM histidine; (c) about 250 mM sucrose; (d) about 0.04% w/v polysorbate 80; and (e) about 50 pM pentetic acid.
[000176] Certain embodiments are directed to a method of preventing or reducing oxidation and/or deamidation of alnuctamab in a pharmaceutical composition comprising binding alnuctamab to pentetic acid in the pharmaceutical composition, thereby preventing or reducing oxidation and/or deamidation of alnuctamab in the pharmaceutical composition. In some aspects, the method of preventing or reducing oxidation and/or deamidation of alnuctamab in a pharmaceutical composition comprises binding the anti- CD3 domain of alnuctamab to pentetic acid. In some aspects, binding alnuctamab to pentetic acid prevents or reduces tryptophan residue oxidation and/or asparagine residue deamidation in alnuctamab. In some aspects, the tryptophan residue(s) are located in the CDR of the anti-CD3 domain of alnuctamab. In some aspects, the asparagine residue(s) are located in the CDR of the anti-CD3 domain of alnuctamab. In certain embodiments, binding alnuctamab to pentetic acid in the pharmaceutical composition comprises formulating about 6 mg/mL of alnuctamab with about 50 pM pentetic acid in the pharmaceutical composition. In certain embodiments, binding alnuctamab to pentetic acid in the pharmaceutical composition comprises formulating about 30 mg/mL of alnuctamab with about 50 pM pentetic acid in the pharmaceutical composition.
Additional Therapies
[000177] In some embodiments, the pharmaceutical composition comprising the multispecific, e.g. bispecific, antibody of the invention, e.g. alnuctamab, to the patient as a monotherapy. In some embodiments, the pharmaceutical composition further comprises one or more additional therapeutic agents as a combination therapy, wherein the combination therapy comprises the administration of the multispecific, e.g. bispecific, antibody of the invention, e.g. alnuctamab, and one or more additional therapeutic agents. The term “combination therapy” is meant to encompass administration of the selected therapeutic agents to a single patient, and is intended to include treatments in which the agents are administered by the same or different route of administration or at the same or different time.
[000178] In some embodiments, the one or more additional therapeutic agents are selected from the group consisting of thalidomide and an immunotherapeutic derivative thereof, an anti-CD38 antibody, an anti-PD-1 antibody, an anti-PD-Ll antibody, a gamma secretase inhibitor (GSI), an anti-
BCMA antibody drug conjugate and anti-BCMA CAR T-cell therapy.
[000179] The term “anti-CD38 antibody” as used herein relates to an antibody specifically binding to human CD38. In an embodiment of the invention the anti-CD38 antibody is daratumumab (US20150246123). In an embodiment of the invention the anti-CD38 antibody is isatuximab (SAR650984, US8877899). In an embodiment of the invention the anti-CD38 antibody is MOR202 (WO 2012041800). In an embodiment of the invention the anti-CD38 antibody is Ab79 (US8362211). In an embodiment of the invention the anti-CD38 antibody is Abl9 (US8362211). The dosage of such anti- CD38 antibody is performed according to the state of the art and described in the respective prescribing information. For example, Daratumumab dosage is usually 16mg/kg (www.ema.europa.eu).
[000180] The term “thalidomide compound” or “thalidomide and an immunotherapeutic derivative” as used herein relates to 2-(2,6-dioxopiperidin-3-yl)-2,3-dihydro-lH-isoindole-l, 3-dione and immunotherapeutic derivatives thereof. In an embodiment of the invention the thalidomide compound is selected from the group consisting of, but not limited to, thalidomide (CAS Registry Number 50-35-1), lenalidomide (CAS Registry Number 191732-72-6), pomalidomide (CAS Registry Number 19171-19-8), CC122 (CAS Registry Number 1398053-45-6) and CC-220 (CAS Registry Number 1323403-33-3) and the respective salts (preferably HC1 salts 1: 1). The chemical formula of CC-122 is 2,6-piperidinedione,3- (5 -amino-2 -methyl -4-oxo-3(4H-quinazolinyl), hydrochloride (1: 1) and of CC-220 it is 2,6- piperidinedione, 3-[l,3-dihydro-4-[[4-(4-morpholinyhnethyl)phenyl]methoxy]-l-oxo-2H-isoindol-2-yl]-, (3S)-, hydrochloride (1: 1). Methods of preparing CC-220 are described, e.g., in US 20110196150, the entirety of which is incorporated herein by reference.
[000181] The dosage of thalidomide compounds is performed according to the state of the art and described in the respective prescribing information. For example, Revlimid® (lenalidomide) dosage is usually 25 mg once daily orally on days 1-21 of repeated 28- day cycles (www.revlimid.com) and POMALYST® (pomalidomide) dosage for the treatment of Multiple Myeloma is usually 4 mg per day taken orally on days 1-21 of repeated 28-day cycles (www.celgene.com). In one embodiment, 3-(5- amino-2 -methyl -4-oxo-4H-quinazolin-3-yl)-piperidine-2, 6-dione is administered in an amount of about 5 to about 50 mg per day.
[000182] In one embodiment, CC-122 and CC-220 are administered in an amount of about 5 to about 25 mg per day. In another embodiment, CC-122 and CC-220 are administered in an amount of about 5, 10, 15, 25, 30 or 50 mg per day. In another embodiment, 10 or 25 mg of CC-122 and CC-220 are administered per day. In one embodiment, CC-122 and CC-220 are administered twice per day.
[000183] The term “anti-PD-1 antibody” as used herein relates to an antibody specifically binding to human PD-1. Such antibodies are e.g. described in WO2015026634 (MK-3475, pembrolizumab), US7521051, US8008449, and US8354509. Pembrolizumab (Keytruda®, MK-3475) is also described in WO 2009/114335, Poole, R.M. Drugs (2014) 74: 1973; Seiwert, T„ et al., J. Clin. Oncol. 32,5s (suppl; abstr 6011). In an embodiment of the invention the PD-1 antibody is MK-3475 (WHO Drug Information, Vol. 27, No. 2, pages 161-162 (2013)) and which comprises the heavy and light chain amino acid
sequences shown in Figure 6 of WO 2015026634 The amino acid sequence of pembrolizumab is described in WO2008156712 ( light chain CDRs SEQ ID NOS: 15, 16 and 17 and heavy chain CDRs SEQ ID NOS: 18, 19 and 20)., In an embodiment of the invention the PD-1 antibody is nivolumab (BMS-936558, MDX 1106; WHO Drug Information, Vol. 27, No. 1, pages 68-69 (2013), W02006/121168 amino acid sequences shown in WO 2015026634). In an embodiment of the invention the PD-1 antibody is; pidilizumab (CT-011, also known as hBAT or hBAT-1; amino acid sequence see W02003/099196; WO 2009/101611, Fried l, et al.; Neuro Oncol (2014) 16 (suppl 5): vl 11 -v 112.). In an embodiment of the invention the PD-1 antibody is MEDI-0680 (AMP-514, W02010/027423, WO2010/027827, WO2010/027828, Hamid O. et al.; J Clin Oncol 33, 2015 (suppl; abstr TPS3087). In an embodiment of the invention the PD-1 antibody is PDR001 (Naing A. et al.; J Clin Oncol 34, 2016 (suppl; abstr 3060). In an embodiment of the invention the PD-1 antibody is REGN2810 (Papadopoulos KPet al.; J Clin Oncol 34, 2016 (suppl; abstr 3024). In an embodiment of the invention the PD-1 antibody is pembrolizumab (WO2008/156712). In an embodiment of the invention the PD-1 antibody is h409Al 1, h409A16 or h409A17, which are described in WO2008/156712. The dosage of such anti-PD-1 antibody is performed according to the state of the art and described in the respective prescribing information. For example, Keytruda® is administered usually in a concentration of 2mg/kg body weight every three weeks (ec .europa. eu/health/documents) .
[000184] The term “anti-PD-Ll antibody” as used herein relates to an antibody specifically binding to human PD-L1. Such antibodies are e.g. described in WO2015026634, W02013/019906, W02010/077634 and US8383796. In an embodiment of the invention the PD-L1 antibody is MPDL3280A (atezolizumab, YW243.55.S70, WO2010/077634, McDermott DF. Et al., JCO March 10, 2016 vol. 34 no. 8 833-842). In an embodiment of the invention the PD-L1 antibody is MDX-1105 (BMS-936559, W02007/005874, Patrick A. Ott PA et al., DOI: 10.1158/1078-0432, Clinical Cancer Research-13-0143). In an embodiment of the invention the PD-L1 antibody is MEDI4736 (durvalumab, WO 2016/040238 Gilbert J. et al., Journal for ImmunoTherapy of Cancer 20153(Suppl 2):P152). In an embodiment of the invention the PD-L1 antibody is MSB001071 8C (avelumab, Disis ML. et al., Journal of Clinical Oncology, Vol 33, No 15_suppl (May 20 Supplement), 2015: 5509). In an embodiment of the invention the PD-L1 antibody is the anti-PD-Ll antibody comprising a VH sequence of SEQ ID NO: 16 and a VL sequence of SEQ ID NO: 17 as described in W02016007235. The dosage of such anti-PD-Ll antibody is performed according to the state of the art and described in the respective prescribing information. For example, atezolizumab is administered usually in a concentration of 1200 mg as an intravenous infusion over 60 minutes every 3 weeks (www.accessdata.fda.gov).
[000185] The term “gamma secretase” as used herein refers to any protein or protein complex that exhibits gamma secretase activities including binding to a substrate having a gamma secretase cleavage sequence, and catalyzing the cleavage of the gamma secretase cleavage sequence, at a gamma secretase cleavage site, to produce substrate cleavage products. In one embodiment, gamma secretase is a protein complex comprising one or more of the following subunits: presenilin, nicastrin, gamma-secretase
subunit APH-1, and gamma-secretase subunit PEN-2.
[000186] The term “gamma secretase inhibitor” or “GSI” as used herein refers to any molecule capable of inhibiting or reducing expression and/or function of gamma secretase. In certain embodiment, the GSI reduces expression and/or function of a subunit of gamma secretase (e.g., presenilin, nicastrin, APH-1, or PEN-2). Any form of a “gamma secretase inhibitor” such as a salt, a co-crystal, a crystalline form, a pro-drug, etc., is included within this term. In some embodiments, the GSI is selected from an antibody or antigen-binding fragment, a small molecule, a protein or peptide and a nucleic acid.
Therapeutic Methods
[000187] The invention is based, in part, on methods of treating a patient having a disorder associated with BCMA expression (e.g. BCMA-expressing B-cell cancers, such as multiple myeloma) using dose-escalation dosing regimens with pharmaceutical compositions comprising multispecific (e.g. bispecific) antibodies that bind to CD3 and BCMA, e.g. alnuctamab. The methods are expected to reduce or inhibit unwanted treatment effects, such as cytokine release syndrome (CRS), thereby treating the patient while achieving a more favorable benefit-risk profile. A “subject” includes any human or nonhuman animal. The term “nonhuman animal” includes, but is not limited to, vertebrates such as nonhuman primates, sheep, dogs, and rodents such as mice, rats and guinea pigs. In some embodiments, the subject is a human. The terms, “subject” and “patient” are used interchangeably herein.
[000188] As used herein, a “disorder associated with BCMA expression” is a plasma cell disorder or a B cell disorder which correlates with enhanced BCMA expression. Plasma cell disorders include BCMA-expressing B-cell cancer, plasmacytoma, plasma cell leukemia, multiple myeloma, macroglobulinemia, amyloidosis, Waldenstrom's macroglobulinemia, solitary bone plasmacytoma, extramedullar plasmacytoma, osteosclerotic myeloma (POEMS Syndrome) and heavy chain diseases as well as the clinically unclear monoclonal gammopathy of undetermined significance/smoldering multiple myeloma.
[000189] In some embodiments, the B cell disorder is a BCMA-expressing B-cell cancer, such as multiple myeloma. Multiple myeloma is a plasma cell malignancy characterized by a monoclonal expansion and accumulation of abnormal plasma cells in the bone marrow compartment. Multiple myeloma also involves circulating clonal plasma cells with same IgG gene rearrangement and somatic hypermutation. Multiple myeloma arises from an asymptomatic, premalignant condition called monoclonal gammopathy of unknown significance (MGUS), characterized by low levels of bone marrow plasma cells and a monoclonal protein. Multiple myeloma cells proliferate at low rate. Multiple myeloma results from a progressive occurrence of multiple structural chromosomal changes (e.g. unbalanced translocations). Multiple myeloma involves the mutual interaction of malignant plasma cells and bone marrow microenvironment (e.g. normal bone marrow stromal cells). Clinical signs of active multiple myeloma include monoclonal antibody spike, plasma cells overcrowding the bone marrow, lytic bone lesions and bone destruction resulting from overstimulation of osteoclasts (Dimopulos & Terpos,
Ann Oncol 2010; 21 suppl 7: vii 143-150).
[000190] In some embodiments, a method comprises treating a patient having an autoimmune disease by administering a pharmaceutical composition provided herein. In some embodiments, the autoimmune disease is associated with plasma cells or B cells expressing BCMA. In some embodiments, the autoimmune disease is systemic lupus erythematosus (SLE), IgA nephropathy, IgG4 related disease, membranous nephropathy, Myasthenia gravis, Neuromyelitis optica, Pemphigus vulgaris, anti-PAD4- activating rheumatoid arthritis, Sensitized / preformed antibodies in solid organ transplant, Guillain-Barre Syndrome (Acute inflammatory demyelinating polyneuropathy - AIDP), Chronic inflammatory demyelinating polyneuropathy (CIDP), Immune thrombocytopenic purpura, rheumatoid arthritis (RA), or ANCA-associated vasculitis (AAV). In some embodiments, the autoimmune disease is idiopathic inflammatory myopathy (IIM) or systemic sclerosis (SSc). In some aspects, the pharmaceutical composition comprises at least about 0.05 mg/mL of the multispecific antibody, e.g. alnuctamab.. In some aspects, the pharmaceutical composition comprises at least about 0.06 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.07 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.08 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.09 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.1 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.2 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.3 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.4 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.6 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.7 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.8 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 0.9 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 1 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 1.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 2 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 3 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 3.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 4 mg/mL of the multispecific antibody, e.g.
alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 4.5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 5 mg/mL of the multispecific antibody, e.g. alnuctamab. In some aspects, the pharmaceutical composition comprises at least about 5.5 mg/mL of the multispecific antibody, e.g. alnuctamab.
[000191] As used herein, the terms “treatment,” “treating,” and the like refer to obtaining a desired pharmacologic and/or physiologic effect. Preferably, the effect is therapeutic, i.e., the effect partially or completely cures a disease and/or adverse symptom attributable to the disease. Alternatively, the pharmacologic and/or physiologic effect may be prophylactic, i.e., the effect completely or partially prevents a disease or symptom thereof.
[000192] As used herein, “administering” refers to the physical introduction of a composition comprising a therapeutic agent to a subject, using any of the various methods and delivery systems known to those skilled in the art. Administration can refer to any form of administration for the pharmaceutical composition comprising multispecific (e.g. bispecific) antibody, include intravenous, intramuscular, subcutaneous, intraperitoneal, spinal or other parenteral routes of administration, for example by injection or infusion. The phrases “subcutaneous administration” and “subcutaneous injection” are used interchangeably and refer to modes of administration wherein a substance, e.g., a pharmaceutical composition comprising multispecific (e.g. bispecific) antibody is delivered to a subject under the skin, between the dermis and, e.g., the muscle.
[000193] Administering can be performed, for example, once, a plurality of times, and/or over one or more extended periods. Thus, as used herein, administering can refer to a single unit dose or more than one unit dose.
[000194] As used herein, the term “dose” or “dosage” is defined as an amount of a therapeutic agent that can be administered at a given point. The dose or dosage can be an amount sufficient to achieve or at least partially achieve a desired effect, but such a desired effect may not be visible or detectable. A “therapeutically effective amount” or “therapeutically effective dosage” of a drug or therapeutic agent is any amount of the drug that, when used alone or in combination with another therapeutic agent, promotes disease regression evidenced by a decrease in severity of disease symptoms, an increase in frequency and duration of disease symptom-free periods, an increase in overall survival (the length of time from either the date of diagnosis or the start of treatment for a disease, such as cancer, that patients diagnosed with the disease are still alive), or a prevention of impairment or disability due to the disease affliction. An amount or dosage of a drug includes a “prophylactically effective amount” or a “prophylactically effective dosage”, which is any amount of the drug that, when administered alone or in combination with another therapeutic agent to a subject at risk of developing a disease or of suffering a recurrence of disease, inhibits the development or recurrence of the disease. The ability of a therapeutic agent to promote disease regression or inhibit the development or recurrence of the disease can be evaluated using a variety of methods available to the skilled practitioner, such as in human subjects during clinical trials, in animal model systems predictive of efficacy in humans, or by assaying the
activity of the agent in in vitro assays. A “dose” can comprise a single unit dose or multiple unit doses. In some aspects, the dose comprises a single unit dose. In some aspects, the dose comprises multiple unit doses. In some aspects, multiple subcutaneous doses are administered to achieve a therapeutically effective dose. When multiple unit doses are administered, individual unit doses can be administered at the same time or sequentially.
[000195] As used herein, a subcutaneous “unit dose” refers to a single amount of a substance delivered by a subcutaneous injection, e.g., from a single vial, a single auto-injector, and/or a single syringe. In some aspects, at least one of the subcutaneous unit doses has a total volume of less than about 5 mb (e.g., about 4.5 mb, about 4.0 mb, about 3.5 mb, about 3.0 mb, about 3 mb, about 2.5 mb, about 2.0 mb, about 1.5 mb, about 1.0 mb, or about 0.5 mb). In some embodiments, the volume of a unit dose is 0.5-2.5 mL, or 0.5-0.9 mL, or 1.0-1.5 mL, or 2.0-2.5 mb, or 0.5, 0.6, 0.7, 0.8, 0.9. 1.0, 1.1, 1.2, 1.3, 1.4, 1.5, 1.6, 1.7, 1.8, 1.9, 2.0, 2.1, 2.2, 2.3, 2.4, or 2.5 mL.
[000196] The use of the term “fixed dose” with regard to the methods and dosages of the disclosure means a dose that is administered to a patient without regard for the weight or body surface area (BSA) of the patient. The fixed dose is therefore not provided as a mg/kg dose, but rather as an absolute amount of the agent (e.g., the multispecific (e.g. bispecific) antibody). For example, a 60 kg person and a 100 kg person would receive the same dose of an antibody (e.g., 3 mg of a multispecific (e.g. bispecific) antibody).
[000197] Thus, in one aspect, the present invention provides a method for treating a disorder associated with BCMA expression (e.g. BCMA-expressing B-cell cancers, such as multiple myeloma) in a patient (e.g. a human), wherein the treatment comprises the administration of a pharmaceutical composition comprising multispecific (e.g. bispecific) antibody that binds to BCMA and CD3, e.g. alnuctamab, in a dosing regimen comprising:
(i) a starting phase, wherein one or more starting doses of the pharmaceutical composition are administered to the patient; and
(ii) a maintenance phase, wherein a first maintenance dose of the pharmaceutical composition is administered to the patient, optionally followed by at least one additional maintenance dose of the pharmaceutical composition; wherein each maintenance dose is greater than the one or more starting doses.
[000198] In another aspect, the present invention provides a pharmaceutical composition comprising multispecific (e.g. bispecific) antibody that binds to BCMA and CD3, e.g. alnuctamab, for use in treating a disorder associated with BCMA expression (e.g. BCMA-expressing B-cell cancers, such as multiple myeloma) in a patient (e.g. a human), wherein the treatment comprises the administration of the pharmaceutical composition in a dosing regimen which comprises:
(i) a starting phase, wherein one or more starting doses of the pharmaceutical composition are administered to the patient; and
(ii) a maintenance phase, wherein a first maintenance dose of the pharmaceutical composition
is administered to the patient, optionally followed by at least one additional maintenance dose of the pharmaceutical composition; wherein each maintenance dose is greater than the one or more starting doses.
[000199] Administration of the starting dose of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab, significantly reduces toxicity due to attenuation of cytokine release.
[000200] In some embodiments, the starting phase comprises a single fixed dose. In some embodiments, the starting dose of the pharmaceutical composition is a single fixed dose comprising about 1.5 mg to about 4.5 mg of the multispecific (e.g. bispecific) antibody, e.g. alnuctamab; from about 2 mg to about 4 mg; from about 2.5 mg to about 3.5 mg, e.g. about 3 mg.
[000201] In other embodiments, the starting phase comprises two or more starting starting doses of the same concentration. In embodiments in which the starting doses are administered as two or more doses of the same concentration, the starting dose of the pharmaceutical composition may be administered at a fixed dose of about 1.5 mg to 4.5 mg dose of the multispecific (e.g. bispecific) antibody, e.g. alnuctamab, from about 2 mg to 4 mg, from about 2.5 mg to 3.5 mg, e.g. about 3 mg.
[000202] If the patient develops an adverse event (e.g. CRS or infection) following administration of a starting dose (e.g. first starting dose) of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab, the subsequent starting dose (e.g. second starting dose) may be administered to the patient up to 12 weeks after the starting dose that triggered the adverse event. In some embodiments, the subsequent starting dose may be administered up to 10 weeks after, up to 8 weeks after, up to 6 weeks after, up to 4 weeks after, up to two weeks after, e.g. up to one week after the starting dose that triggered the adverse event. In some embodiments, the subsequent starting dose may be of the same concentration or lower concentration than the starting dose that triggered the adverse event. [000203] If the patient develops an adverse event (e.g. CRS or infection) following administration of the last starting dose of the starting phase, the starting phase may comprise an additional starting dose administered to the patient up to 12 weeks after the starting dose that triggered the adverse event. In some embodiments, the additional starting dose may be administered up to 10 weeks after, up to 8 weeks after, up to 6 weeks after, up to 4 weeks after, up to two weeks after, e.g. up to one week after the starting dose that triggered the adverse event. In some embodiments, the additional starting dose may be of the same concentration or lower concentration than the starting dose that triggered the adverse event.
[000204] In some embodiments, the first maintenance dose of the pharmaceutical composition may be administered at a fixed dose of about 4.5 mg to about 7.5 mg of the multispecific (e.g. bispecific) antibody, e.g. alnuctamab; from about 5 mg to about 6 mg; from about 5.5 mg to about 6.5 mg, e.g. about 6 mg. Thus, in some embodiments, the starting dose of the pharmaceutical composition is a (e.g. single) fixed dose of about 3 mg of the multispecific (e.g. bispecific) antibody, e.g. alnuctamab, and the first maintenance dose of the pharmaceutical composition is a fixed dose of about 6 mg of multispecific (e.g. bispecific) antibody, e.g. alnuctamab.
[000205] In some embodiments, the maintenance phase comprises two or more maintenance doses of the same concentration or of escalating concentration.
[000206] In some embodiments, the maintenance dose of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody is administered as two or more doses of escalating concentration (i.e. increasing doses). In this case, a subsequent dose can be increased by a particular increment, or by variable increments, until a maximum dose is reached, at which point administration may cease or may continue at the maximum dose.
[000207] Thus, in embodiments in which the maintenance doses are administered at escalating concentrations, the first maintenance dose of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab, is greater than the starting dose and the second (and optionally subsequent) maintenance dose(s) of the pharmaceutical composition is greater than the first maintenance dose. In some embodiments, subsequent (e.g. third, fourth or fifth) optional maintenance dose(s) of pharmaceutical composition may be the same or greater than the second maintenance dose. In some embodiments, subsequent (e.g. third, fourth or fifth) optional maintenance dose(s) of the pharmaceutical composition may be the same as the second maintenance dose.
[000208] In some embodiments, the first maintenance dose of the pharmaceutical composition may be administered at a fixed dose of about 4.5 mg to 7.5 mg of the multspecific (e.g. bispecific) antibody, e.g. alnuctamab; from about 5 mg to 6 mg; from about 5.5 mg to 6.5 mg, e.g. about 6 mg, and a subsequent (e.g. second, third, fourth or fifth) maintenance dose of the pharmaceutical composition may be administered at a fixed dose greater than the first maintenance dose.
[000209] In some embodiments, the first maintenance dose of the pharmaceutical composition may be administered at a fixed dose of about 4.5 mg to 7.5 mg of the multispecific (e.g. bispecific) antibody, e.g. alnuctamab; from about 5 mg to 6 mg; from about 5.5 mg to 6.5 mg, e.g. about 6 mg, and a subsequent (e.g. second, third, fourth or fifth) maintenance dose of the composition antibody may be administered at a fixed dose of about 25 mg to about 35 mg, about 28 mg to about 32 mg, about 29 mg to about 31 mg, e.g. about 30 mg of the multispecific (e.g. bispecific) antibody, e.g. alnuctamab.
[000210] Thus, in some embodiments, the starting dose is a (e.g. single) fixed dose of about 3 mg of the multispecific (e.g. bispecific) antibody, e.g. alnuctamab, the first maintenance dose of the is a fixed dose of about 6 mg of the multispecific (e.g. bispecific) antibody, and a second (and optionally subsequent) maintenance dose(s) is a fixed dose of about 30 mg of the multispecific (e.g. bispecific) antibody, e.g. alnuctamab.
[000211] In some embodiments, the first maintenance dose of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab, is administered to the patient 1- 21 days, e.g. 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, or 21 days, after the starting dose. In some embodiments, the first maintenance dose of the pharmaceutical composition may be administered to the patient 2 days, after the starting dose. In some embodiments, the first maintenance dose of the pharmaceutical composition may be administered to the patient 3 days after the starting dose.
In some embodiments, the first maintenance dose of the pharmacentical composition may be administered to the patient 7 days after the starting dose. In some embodiments, the first maintenance dose of the pharmaceutical composition may be administered to the patient 14 days after the starting dose.
[000212] In some embodiments, the second maintenance dose of the pharmaceutical composition is administered to the patient 1-21 days, e.g. 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, or 21 days, after the first maintenance dose.
[000213] In embodiments in which the first maintenance dose is administered 3 days after the starting dose, the second maintenance dose may be administered 4 days after the first maintenance dose. [000214] In some embodiments of any aspect of the invention, if the patient develops an adverse event (e.g. CRS or infection) following administration of a maintenance dose (e.g. first, second, third or subsequent maintenance dose) of the phamarceutical composition comprising the multispecific (e.g. bispecific) antibody, the next maintenance dose may be administered to the patient up to 12 weeks after the maintenance dose that triggered the adverse event. In some embodiments, the next maintenance starting dose may be administered up to 10 weeks after, up to 8 weeks after, up to 6 weeks after, up to 4 weeks after, up to two weeks after, e.g. up to one week after the starting dose that triggered the adverse event. In some embodiments, the next maintenance dose may be of the same concentration or lower concentration than the maintenance dose that triggered the adverse event.
[000215] In some embodiments of any aspect of the invention, the third and subsequent maintenance doses are administered at about a once weekly or longer dosing interval. As used herein, a “dosing interval” means the amount of time that elapses between multiple doses being administered to a patient. If an adverse event (e.g. CRS or infection) occurs following administration of a maintenance dose (e.g. third or subsequent maintenance dose) of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab, the dosing interval may reset on the day the next maintenance dose is administered to the patient.
[000216] In some embodiments of any aspect of the invention, the dosing interval for the third and subsequent maintenance doses may be about once weekly. As used herein, a “weekly dosing interval” includes every 5-9, 6-9, 7-9, 5-8, 5-7, 6-8, 6-7, 7-8, preferably 7 days. In some embodiments, the dosing interval for the third and subsequent maintenance doses may be about once biweekly. As used herein, a “biweekly dosing interval” includes every 12-16, 13-16, 14-16, 12-15, 12-14, 13-15, 13-14, 14-15, preferably 14 days. In some embodiments, the dosing interval for the third and subsequent maintenance dose may be about once every three weeks. As used herein, a “three week dosing interval” includes every 19-23, 20-23, 21-23, 19-22, 19-21, 20-22, 20-21, 21-22, preferably 21 days. In some embodiments, the dosing interval for the third and subsequent maintenance dose may be about once every four weeks. As used herein, a “four week dosing interval” includes every 26-30, 27-30, 28-30, 26- 29, 26-28, 27-29, 27-28, 28-29, preferably 28 days. In some embodiments, the dosing interval for the third and subsequent maintenance doses may be about once monthly.
[000217] In some embodiments of any aspect of the invention, the dosing interval for the third and subsequent maintenance dose may be a combination of one or more of a weekly dosing interval, a biweekly dosing interval, a three week dosing interval and a four week dosing interval. In some embodiments, the dosing interval for the third and subsequent maintenance dose may be a combination of a weekly dosing interval, a biweekly dosing interval, and a four week dosing interval.
[000218] In some embodiments of any aspect of the invention, the third and subsequent maintenance doses are administered in a weekly dosing interval (e.g. every 7 days), then a biweekly dosing interval (e.g. every 14 days), then a three week dosing interval (e.g. every 21 days) and then a four week dosing interval (e.g. every 28 days). In some embodiments, the third and subsequent maintenance doses are administered in a weekly dosing interval (e.g. every 7 days), then a biweekly dosing interval (e.g. every 14 days) and then a four week dosing interval (e.g. every 28 days).
[000219] In some embodiments of any aspect of the invention, the treatment comprises at least one treatment cycle of 28 days. As used herein, a “treatment cycle” is 28 days. If a starting dose is administered beyond day 28 of the first treatment cycle as a result of an adverse event (e.g. CRS or infection), the first treatment cycle may restart on the day the starting dose is administered to the patient. If a maintenance dose is administered beyond day 28 of the current treatment cycle as a result of an adverse event (e.g. CRS or infection), the next treatment cycle may begin on the day the maintenance dose is administered to the patient.
[000220] In some embodiments, the treatment comprises a first treatment cycle, wherein the starting dose is administered to the patient as a fixed dose on day 1, the first maintenance dose is administered to the patient as a fixed dose on day 4, the second maintenance dose is administered to the patient as a fixed dose on day 8, and the subsequent maintenance doses are subsequently administered in a weekly dosing interval (e.g. every 7 days) for three consecutive weeks (e.g. on days 15 and 22). The maintenance doses may continue to be administered in a weekly or longer dosing interval in subsequent treatment cycles.
[000221] In some embodiments of any aspect of the invention, the treatment comprises a second treatment cycle, wherein the maintenance doses are administered in a weekly dosing interval (e.g. on days 1, 8, 15 and 22). In further embodiments, the patient remains on a weekly dosing interval for between 1-5, 1-3, 1-2, 2-3 further treatment cycles, preferably 2 further treatment cycles (in addition to the first treatment cycle). In some embodiments, the treatment comprises a second and third treatment cycle, wherein the maintenance doses are administered in a weekly dosing interval (e.g. on days 1, 8, 15 and 22).
[000222] In some embodiments of any aspect of the invention, the maintenance doses may be administered in a biweekly dosing interval in treatment cycles (e.g. on days 1 and 15) after completion of the weekly treatment cycle(s). In further embodiments, the patient remains on a biweekly dosing interval for between 1-5, 1-3, 1-2, 2-3 biweekly treatment cycles, preferably 3 biweekly treatment cycles. In some embodiments, the treatment comprises a fourth, fifth and sixth treatment cycle, wherein the maintenance
doses are administered in a biweekly dosing interval (e.g. on days 1 and 15).
[000223] In some embodiments of any aspect of the invention, the maintenance doses may be administered in a four week dosing interval in subsequent treatment cycles (e.g. on day 1) after completion of the biweekly treatment cycle(s). In further embodiments, the patient remains on a four week dosing interval for at least one cycle. Some patients continue to receive treatment for the rest of their lives.
[000224] In some embodiments, the treatment comprises a first treatment cycle, wherein the starting dose is administered to the patient as a fixed dose on day 1, the first maintenance dose is administered 3 days after the starting dose (e.g. on day 4), the second maintenance dose is administered 4 days after the first maintenance dose (e.g. on day 8), and the third and fourth maintenance doses are administered in a weekly interval (e.g. on days 15 and 22). The maintenance doses may continue to be administered in a weekly or longer dosing interval in subsequent treatment cycles.
[000225] In some embodiments of any aspect of the invention, the treatment comprises a second treatment cycle, wherein the maintenance doses are administered in a weekly dosing interval (e.g. on days 1, 8, 15 and 22). In further embodiments, the patient remains on a weekly dosing interval for between 1-5, 1-3, 1-2, 2-3 further treatment cycles, preferably 2 further treatment cycles (in addition to the first treatment cycle). In some embodiments, the treatment comprises a second and third treatment cycle, wherein the maintenance doses are administered in a weekly dosing interval (e.g. on days 1, 8, 15 and 22).
[000226] In some embodiments of any aspect of the invention, the maintenance doses may be administered in a biweekly dosing interval in treatment cycles (e.g. on days 1 and 15) after completion of the weekly treatment cycle(s). In further embodiments, the patient remains on a biweekly dosing interval for between 1-5, 1-3, 1-2, 2-3 weekly treatment cycles, preferably 3 biweekly treatment cycles. In some embodiments, the treatment comprises a fourth, fifth and sixth treatment cycle, wherein the maintenance doses are administered in a biweekly dosing interval (e.g. on days 1 and 15).
[000227] In some embodiments of any aspect of the invention, the maintenance doses may be administered in a four week dosing interval in subsequent treatment cycles (e.g. on day 1) after completion of the biweekly treatment cycle(s). In further embodiments, the patient remains on a four week dosing interval for at least one cycle. Some patients continue to receive treatment for the rest of their lives.
[000228] In some embodiments, the treatment comprises:
(i) a first treatment cycle, wherein the starting dose is administered on day 1, and the maintenance doses are administered on days 4, 8, 15 and 22;
(ii) a second and third treatment cycle, wherein the maintenance doses are administered in a weekly dosing interval (e.g. on days 1, 8, 15 and 22);
(iii) a fourth to sixth treatment cycle, wherein the maintenance doses are administered in a biweekly dosing interval (e.g. on days 1 and 15); and
(iv) a seventh and subsequent cycle, wherein the maintenance doses are administered in a four week dosing interval (e.g. on day 1)
[000229] In some embodiments, the pharmaceutical composition comprising the multispecific
(e.g. bispecific) antibody (e.g. “42-TCBcv”) may be administered to the patient in accordance with the regimen set out in Table 4.
Table 4:
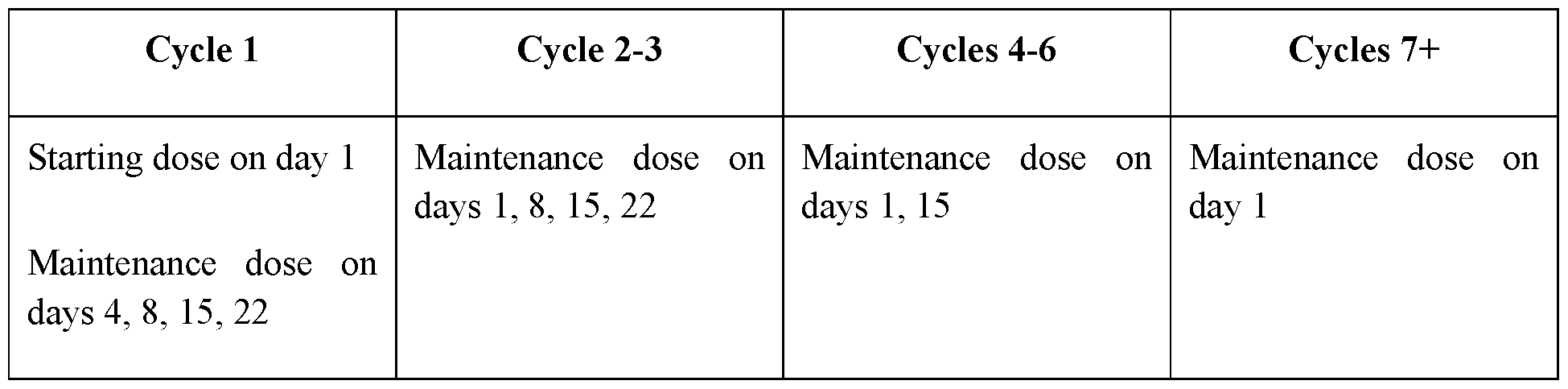
[000230] In some embodiments, in the regimen set out in Table 4, the maintenance doses of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody are administered as two or more doses of escalating concentration.
[000231] In some embodiments, the starting dose of the pharmaceutical composition is a fixed dose of about 1.5 mg to 4.5 mg; about 2 mg to 4 mg; about 2.5 mg to 3.5 mg, e.g. about 3 mg, of the multispecific (e.g. bispecific) antibody, e.g. alnuctamab. In some embodiments, the first maintenance dose may be administered at a fixed dose of about 4.5 mg to 7.5 mg; about 5 mg to 7 mg; about 5.5 mg to 6.5 mg, e.g. about 6 mg, of the multispecific (e.g. bispecific) antibody, e.g. alnuctamab, and the second (and optionally subsequent) maintenance dose(s) of the pharmaceutical composition may be administered at a fixed dose greater than the first maintenance dose. The second (and optionally subsequent) maintenance dose(s) of the pharmaceutical composition may be administered at a fixed dose of about 25 mg to about 35 mg, about 28 mg to about 32 mg, about 29 mg to about 31 mg, e.g. about 30 mg, of the multispecific (e.g. bispecific) antibody, e.g. alnuctamab. Thus, in some embodiments, the starting dose of the pharmaceutical composition is a (e.g. single) fixed dose of about 3 mg of the multispecific (e.g. bispecific) antibody, the first maintenance dose of the pharmaceutical composition is a fixed dose of about 6 mg of the multispecific (e.g. bispecific) antibody, and the second (and optionally subsequent) maintenance dose(s) of the pharmaceutical composition is a fixed dose of about 30 mg of the multispecific (e.g. bispecific) antibody, e.g. alnuctamab.
[000232] In some embodiments, the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody (e.g. “42-TCBcv”) may be administered to the patient in accordance with the regiment set out in Table 5.
Table 5:
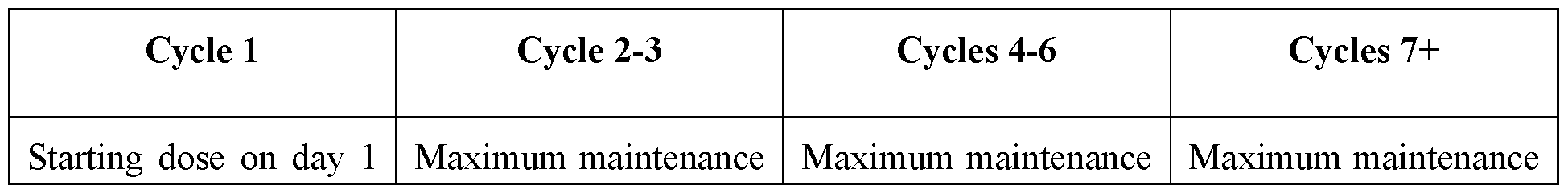

[000233] In some embodiments of any aspect of the invention, the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody discussed herein is administered intravenously or subcutaneously. In some embodiments, the pharmaceutical composition is administered subcutaneously. In some embodiments, the multispecific (e.g. bispecific) antibody is “42-TCBcv”, and the pharmaceutical composition is administered subcutaneously.
[000234] In some embodiments, the starting dose and the first maintenance dose of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab, may be administered subcutaneously, and a subsequent (e.g. second, third, fourth or fifth) maintenance dose of the pharmaceutical composition may be administered subcutaneously.
[000235] In some embodiments, the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody (e.g. “42-TCBcv”) may be administered to the patient in accordance with any of the regimens set out in Tables 4-5, wherein the starting dose and the first maintenance dose of the multispecific (e.g. bispecific) antibody may be administered subcutaneously, and a subsequent (e.g. second, third, fourth or fifth) maintenance dose of the multispecific (e.g. bispecific) may be administered subcutaneously.
[000236] In some embodiments, the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody (e.g. “42-TCBcv”) may be administered to the patient in accordance with any of the regimens set out in Tables 4-5, wherein cycles 1-2 may be administered subcutaneously, and cycles 3+ may be administered subcutaneously.
Adverse Events
[000237] In some embodiments of any aspect of the invention, the patient develops, or is at risk of developing, an adverse event associated with the administration of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab. The adverse event may be cytokine-driven toxicities (e.g. cytokine release syndrome (CRS)), infusion-related reactions (IRRs), macrophage activation syndrome (MAS), neurologic toxicities, severe tumor lysis syndrome (TLS), neutropenia, thrombocytopenia, elevated liver enzymes, bacterial infections, viral infections, and/or central nervous system (CNS) toxicities. In particular embodiments, the adverse event is CRS.
[000238] In the event that the patient develops, or is at risk of developing, an adverse event associated with the administration of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab, the treatment according to any aspect of the invention further comprises the administration of an agent capable of treating, preventing, delaying, reducing or attenuating the development or risk of development of the adverse event. The agent may be administered to the patient prior to the initiation of the treatment with the pharmaceutical composition comprsing the multispecific (e.g. bispecific) antibody (e.g. as a prophylaxis in order to prevent or reduce the risk of an adverse event developing) or during treatment with the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody (e.g. in response to the development of an adverse event). In some embodiments, the agent comprises a steroid, such as a corticosteroid. As used herein, “corticosteroid” means any naturally occurring or synthetic steroid hormone that can be derived from cholesterol and is characterized by a hydrogenated cyclopentanoperhydrophenanthrene ring system. Naturally occurring corticosteroids are generally produced by the adrenal cortex. Synthetic corticosteroids may be halogenated. Functional groups required for activity include a double bond at A4, a C3 ketone, and a C20 ketone. Corticosteroids may have glucocorticoid and/or mineralocorticoid activity. Examples of exemplary corticosteroids include prednisolone, methylprednisolone, prednisone, triamcinolone, betamethasone, budesonide, and dexamethasone. In some embodiments, the agent is dexamethasone.
[000239] In some embodiments, the agent comprises an antagonist of a cytokine receptor or cytokine selected from among GM-CSF, IL- 10, IL-10R, IL-6, IL-6 receptor (IL-6R), IFNy, IFNGR, IL-2, IL-2R/CD25, MCP-1, CCR2, CCR4, MIPip, CCR5, TNFalpha, TNFR1, IL-1 (e g. IL-la, IL-lp, IL- 1RA), and IL-1 receptor (IL-1R), wherein the antagonist is selected from an antibody or antigen-binding fragment, a small molecule, a protein or peptide and a nucleic acid. The antagonist may be an anti-IL-6 antibody and/or an anti-IL6R antibody. For example, the antagonist may be selected from tocilizumab, siltuximab, clazakizumab, sarilumab, olokizumab, elsilimomab, ALD518/BMS-945429, sirukumab (CNTO 136), CPSI-2634, ARGX-109, lenzilumab, FE301 and FM101. In some embodiments, the antagonist is tocilizumab and/or siltuximab. Alternatively, the antagonist may be an anti -IL- 1 antagonist and/or an anti-IL-lR antagonist e.g. anakinra.
[000240] In some embodiments, the agent comprises a molecule that decreases the regulatory T cell (Treg) population. Agents that decrease the number of (e.g., deplete) Treg cells are known in the art and include, e.g., CD25 depletion, cyclophosphamide administration, anti-CTLA4 antibody and modulating Glucocorticoid-induced TNLR family related gene (GITR) function. GITR is a member of the TNLR superfamily that is upregulated on activated T cells, which enhances the immune system. In some embodiments, the treatment comprises the administration of cyclophosphamide.
[000241] In some embodiments, the agent capable of treating, preventing, delaying, reducing or attenuating the development or risk of development of the adverse event is administered as one or more doses to the patient prior to the initiation of the treatment with the pharmaceutical composition
comprising the multispecific (e.g. bispecific) antibody as a prophylactic treatment for the adverse event. [000242] In some embodiments, the agent capable of treating, preventing, delaying, reducing or attenuating the development or risk of development of the adverse event is administered to the patient in combination with one or more dose of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody as a prophylactic treatment for the adverse event. The agent may be administered as one or more doses consecutively (before and/or after), and/or concurrently with the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody.
[000243] In some embodiments, the agent capable of treating, preventing, delaying, reducing or attenuating the development or risk of development of the adverse event is administered to the patient in combination with the first dose of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab, as a prophylactic treatment for the adverse event. The agent may be administered as one or more doses consecutively (before and/or after), and/or concurrently with the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab. [000244] In some embodiments, the agent capable of treating, preventing, delaying, reducing or attenuating the development or risk of development of the adverse event is administered to the patient in combination with each increase in dose of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab, as a prophylactic treatment for the adverse event. The agent may be administered as one or more doses consecutively (before and/or after), and/or concurrently with the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab.
[000245] In some embodiments of any aspect of the invention, the prophylactic treatment comprises administration of the agent (e.g. CRS agent) at an amount sufficient to prevent, delay, reduce or attenuate the development or risk of development of the adverse event (e.g. CRS).
[000246] In some embodiments of any aspect of the invention, the prophylactic treatment comprises the administration of a corticosteroid, such as dexamethasone. In some embodiments, the dexamethasone is administered at a dose of about 10-20 mg, preferably intravenously. In embodiments in which dexamethasone is administered as a prophylactic treatment for a cytokine-driven toxicity (e.g. CRS), preferably dexamethasone is administered at an amount sufficient to attenuate secretion of cytokines (e.g. GM-CSF, IL-2 and/or TNF-a) induced by the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody of the invention, e.g. alnuctamab.
[000247] In some embodiments of any aspect of the invention, the prophylactic treatment comprises the administration of an antagonist of a cytokine receptor or cytokine, such as an antagonist of IL-6, an IL-6 receptor (IL-6R), IL-1 (e.g. IL- la, IL- 1 , IL- IRA) and/or an IL-1 receptor (IL-1R) wherein the antagonist is selected from an antibody or antigen-binding fragment, a small molecule, a protein or peptide and a nucleic acid.
[000248] In some embodiments of any aspect of the invention, the prophylactic treatment comprises an anti -IL-6 antagonist antibody and/or an anti-IL-6R antagonist antibody, e.g. tocilizumab. In some embodiments, tocilizumab is administered to the patient as a one or more doses of about 8 mg/kg,
preferably intravenously. In preferred embodiments, tocilizumab is administered at least 30 minutes prior to the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody. In embodiments in which tocilizumab is administered as a prophylactic treatment for a cytokine-driven toxicity (e.g. CRS), preferably tocilizumab is administered at an amount sufficient to attenuate IL-6 receptor signalling induced by the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody of the invention, e.g. alnuctamab.
[000249] In some embodiments of any aspect of the invention, the prophylactic treatment comprises an anti-IL-1 antagonist and/or an anti-IL-lR antagonist, e.g. anakinra. In some embodiments, anakinra is administered as a prophylactic treatment for a cytokine -driven toxicity (e.g. CRS), preferably at an amount sufficient to attenuate IL-1 receptor signalling induced by the multi specific (e.g. bispecific) antibody of the invention. Anakinra may be administered at a dose of about 100 mg (e.g. 100 mg ± 20%), preferably subcutaneously. In some embodiments, anakinra is administered to the patient as a dose of about 100 mg, preferably subcutaneously. In some embodiments, the prophylactic treatment comprises at least one dose of anakinra administered before the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, and at least one dose of anakinra administered after the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody.
[000250] Anakinra may be administered to the patient as one or more fixed dose(s) between about 16 hours to about 2 hours prior to the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, and optionally a fixed dose between about 20 hours to about 22 hours after the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody. In some embodiments of any aspect of the invention, anakinra is administered as:
(i) a fixed dose between about 16 hours to about 8 hours prior to the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab; and/or
(ii) a fixed dose between about 4 hours to about 2 hours prior to the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab, optionally wherein an additional fixed dose of anakinra is administered between about 20 hours to about 22 hours after the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody.
[000251] In some embodiments of any aspect of the invention, the prophylactic treatment comprises the administration of dexamethasone (e.g. about 10-20 mg, preferably intravenously) with tocilizumab (e.g. about 8 mg/kg, preferably intravenously). In some embodiments, the prophylactic treatment comprises the administration of dexamethasone (e.g. about 10-20 mg, preferably intravenously) with anakinra (e.g. about 100 mg, preferably subcutaneously).
[000252] In some embodiments of any aspect of the invention, the prophylactic treatment comprises the administration of symptomatic support, including administration of antipyretics, analgesics, antivirals and/or antibiotics. In some embodiments, the symptomatic support comprises the administration of antivirals (e.g. acyclovir, oseltamivir, zanamivir and/or equivalents) and/or antibiotics (e.g. trimethoprim-sulfamethoxazole, levofloxacin and/or equivalents). In some embodiments, the
prophylactic treatment comprises the administration of seizure prophylaxis (e.g. levetiracetam). The symptomatic support and/or seizure prophylaxis may be administered in addition to the agent capable of treating, preventing, delaying, reducing or attenuating the development or risk of development of the adverse event.
[000253] In some embodiments of any aspect of the invention, the agent capable of treating, preventing, delaying, reducing or attenuating the development or risk of development of the adverse event is administered to the patient in the event that the patient develops an adverse event associated with the administration of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody. In some embodiments, the treatment comprises administration of the agent at a therapeutic amount, or an amount sufficient to partially or completely alleviate or ameliorate the adverse event (e.g. CRS) or symptoms thereof.
[000254] If the patient develops an adverse event (e.g. CRS) following administration of a pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody of the invention, e.g. alnuctamab, the treatment may further comprise the administration of an anti-IL-6R antagonist antibody, e.g., tocilizumab. In some embodiments, tocilizumab is administered to the patient as a single dose of about 8 mg/kg, preferably intravenously. In some embodiments, the treatment may further include administering to the patient one or more additional doses of an IL-6R antagonist antibody, e.g., tocilizumab. In some embodiments, tocilizumab is administered to the patient in one or more additional doses of about 8 mg/kg, preferably intravenously.
[000255] In some embodiments, if the patient develops an adverse event (e.g. CRS) following administration of a pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody of the invention, e.g. alnuctamab, the treatment may further comprise the administration of an anti -IL- 1 antagonist and/or an anti-IL-lR antagonist, e.g. anakinra. In some embodiments, anakinra is administered to the patient as one more fixed doses of about 100 mg, preferably subcutaneously. In some embodiments, anakinra is administered to the patient twice daily, preferably as fixed doses of about 100 mg, preferably subcutaneously.
[000256] In some embodiments, if an adverse event (e.g. CRS) occurs the treatment may further comprise the administration of an IL-6 antagonist antibody, e.g., siltuximab. In some embodiments, siltuximab is administered to the patient as a single dose of about 11 mg/kg, preferably intravenously.
[000257] In some embodiments, if an adverse event (e.g. CRS) occurs the treatment may further comprise administering to the patient a corticosteroid, such as methylprednisolone or dexamethasone. In some embodiments, the dexamethasone is administered at a dose of about 10-20 mg, preferably intravenously. In some embodiments, the methylprednisolone is administered at a dose of about 1 mg/kg per day to about 5 mg/kg per day, e.g., about 2 mg/kg per day.
[000258] In some embodiments, the additional treatments may be based on the stage of the CRS. A modification of the common CTCAE CRS grading scale has been established for the grading and treatment of CRS, and is detailed in Table 6:
Table 6: Grading and Treatment of Cytokine Release Syndrome

[000259] For example, in embodiments in which the patient has a grade 2 CRS following administration of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab, the treatment may further comprise the administration of a first line treatment comprising the administration of a first dose of an anti-IL-6 antagonist antibody and/or an anti-IL-6R antagonist antibody, e.g., tocilizumab. In some instances, tocilizumab is administered intravenously to the patient as a single dose of about 8 mg/kg.
[000260] In alternative embodiments in which the patient has a grade 2 CRS following administration of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, the treatment may further comprise the administration of a first line treatment comprising the administration of one or more fixed dose(s) of anti-IL-1 antagonist and/or an anti-IL-lR antagonist, e.g. anakinra. Anakinra may be administered at a dose of about 100 mg (e.g. 100 mg ± 20%), preferably subcutaneously. In some embodiments, anakinra is administered to the patient as one more fixed dose(s) of about 100 mg, preferably subcutaneously. In some embodiments, anakinra is administered to the patient twice daily, preferably as fixed doses of about 100 mg, preferably subcutaneously.
[000261] If the patient develops rapid onset of grade 2 CRS or develops grade >3 CSR onset following administration of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab, the treatment may further comprise the administration of a first line treatment comprising:
(i) an anti-IL-6 antagonist antibody and/or an anti-IL-6R antagonist antibody, e.g., tocilizumab; and
(ii) a corticosteroid, e.g. dexamethasone or methylprednisolone.
[000262] In some embodiments, tocilizumab is administered intravenously to the patient at a dose of about 8 mg/kg.
[000263] Alternatively, if the patient develops rapid onset of grade 2 CRS or develops grade >3 CRS onset following administration of the pharmaceutical composition comprising the multispecific (e.g. bispecific) antibody, e.g. alnuctamab, the treatment may further comprise the administration of a first line treatment comprising:
(i) an anti-IL-1 antagonist and/or an anti-IL-lR antagonist, e.g. anakinra; and
(ii) a corticosteroid, e.g. dexamethasone or methylprednisolone.
[000264] In some embodiments, anakinra is administered to the patient as one more fixed dose(s) of about 100 mg, preferably subcutaneously. In some embodiments, anakinra is administered to the patient twice daily, preferably as fixed doses of about 100 mg, preferably subcutaneously.
[000265] The corticosteroid may be administered consecutively (before or after) or concurrently with the (i) anti-IL-6 antagonist antibody and/or an anti-IL-6R antagonist antibody, e.g., tocilizumab, or (ii) anti-IL-1 antagonist and/or an anti-IL-lR antagonist, e.g. anakinra. In some embodiments, the corticosteroid is dexamethasone. In some embodiments, the dexamethasone is administered at a dose of about 10-20 mg, preferably intravenously. In some embodiments, the corticosteroid is methylprednisolone. In some embodiments, methylprednisolone is administered at a dose of about 1 mg/kg per day to about 5 mg/kg per day, e.g., about 2 mg/kg per day.
[000266] In some embodiments, the first line treatment comprises the administration of symptomatic support for CRS, including administration of antipyretics, analgesics and/or antibiotics. In some embodiments, the first line treatment comprises the administration of seizure prophylaxis (e.g. levetiracetam). The symptomatic support and/or seizure prophylaxis may be administered in addition to the agent capable of treating, preventing, delaying, reducing or attenuating the development or risk of development of the adverse event.
[000267] In some embodiments of any aspect of the invention, if the patient develops CRS following administration of a dose of the pharmaceutical composition comprising the multispecific (e g. bispecific) antibody (e.g. starting dose or maintenance dose), the next dose (e.g. next starting dose or next maintenance dose) may be administered to the patient when toxicity reaches Grade <1 as described herein. In alternative embodiments where the patient develops CRS, the next dose may be administered to the patient when toxicity reaches baseline levels.
[000268] In the event that the CRS does not resolve or worsens in response to first line treatment, the treatment may further comprise the administration of a second line treatment comprising:
(i) one or more (e.g., one, two, three, four, or five or more) additional doses of the anti-IL-6 antagonist antibody and/or an anti-IL-6R antagonist antibody, e.g., tocilizumab; and
(ii) one or more (e.g., one, two, three, four, or five or more) additional doses of the corticosteroid, e.g. dexamethasone or methylprednisolone.
[000269] In some embodiments, the one or more additional doses of tocilizumab are administered
intravenously to the patient at a dose of about 8 mg/kg. The corticosteroid may be administered consecutively (before or after) or concurrently with the anti-IL-6 antagonist antibody and/or an anti-IL- 6R antagonist antibody, e.g., tocilizumab. In some embodiments, the corticosteroid is dexamethasone. In some embodiments, the dexamethasone is administered at a dose of about 10-20 mg, preferably intravenously. In some embodiments, the corticosteroid is methylprednisolone. In some embodiments, methylprednisolone is administered at a dose of about 1 mg/kg per day to about 5 mg/kg per day, e.g., about 2 mg/kg per day.
[000270] In the event that the CRS does not resolve or worsens in response to second line treatment, the treatment may further comprise the administration of a third line treatment comprising the administration of an antagonist of a cytokine receptor or cytokine selected from among GM-CSF, IL- 10, IL-10R, IL-6, IL-6 receptor (IL-6R), IFNy, IFNGR, IL-2, IL-2R/CD25, MCP-1, CCR2, CCR4, MIPip, CCR5, TNFalpha, TNFR1, IL-1 (e.g. IL-la, IL- ip, IL-IRA), and IL-1 receptor (IL-1R), wherein the antagonist is selected from an antibody or antigen-binding fragment, a small molecule, a protein or peptide and a nucleic acid. The antagonist may be an anti-IL-6 antibody and/or an anti-IL6R antibody. For example, the antagonist may be selected from tocilizumab, siltuximab, clazakizumab, sarilumab, olokizumab, elsilimomab, ALD518/BMS-945429, sirukumab (CNTO 136), CPSI-2634, ARGX-109, lenzilumab, FE301 and FM101. In some embodiments, the third line treatment comprises the administration of siltuximab. In some embodiments, siltuximab is administered to the patient as a single dose of about 11 mg/kg, preferably intravenously. Alternatively, the antagonist may be an anti -IL- 1 antagonist and/or an anti-IL-lR antagonist e.g. anakinra.
[000271] In the event that the CRS does not resolve or worsens in response to third line treatment, the treatment may further comprise the administration of a fourth line treatment comprising the administration of a molecule that decreases the regulatory T cell (Treg) population. Molecules that decrease the number of (e.g., deplete) Treg cells are known in the art and include, e.g., CD25 depletion, cyclophosphamide administration, anti-CTLA4 antibody and modulating Glucocorticoid-induced TNLR family related gene (GITR) function. GITR is a member of the TNLR superfamily that is upregulated on activated T cells, which enhances the immune system. In some embodiments, the fourth line treatment comprises the administration of cyclophosphamide.
[000272] In some embodiments, if an adverse event occurs (e.g. neutropenia, infection) the treatment may further comprise symptomatic support, including administration of antipyretics, analgesics, antivirals and/or antibiotics. In some embodiments, seizure prophylaxis (e.g. levetiracetam) can be administered to the patient. If the patient develops neutropenia (e.g. at least grade 3 neutropenia), the treatment may further comprise administration of antibiotics (e.g. levofloxacin or equivalent). If the patient develops a viral infection (e.g. influenza), the treatment may further comprise administration of oseltamivir, zanamivir and/or equivalents.
[000273] In some embodiments of any aspect of the invention, if the patient develops a viral infection (e.g. influenza A/B, SARS-CoV-2) following administration of a dose of the pharmaceutical
composition comprising the multispecific (e.g. bispedfic) antibody (e.g. starting dose or maintenance dose), the next dose (e.g. next starting dose or next maintenance dose) may be administered to the patient when symptoms of the infection resolve. In alternative embodiments where the patient develops a viral infection, tire next dose may be administered after a negative test for tire viral infection, e.g. a negative PCJR viral panel, and/or at least 14 days after a positive test for the viral infection, e.g. a positive PCJR viral panel. A viral panel (e.g. PCR viral panel) may test for influenza A/B, respiratory syncytial virus, parainfluenza virus, metapneumovirus, adenovirus and/or SARS-CoV-2.
[000274] The above embodiments are to be understood as illustrative examples. Further embodiments are envisaged. It is to be understood that any feature described in relation to any one embodiment may be used alone, or in combination with other features described, and may also be used in combination with one or more features of any other of the embodiments, or any combination of any other of the embodiments. Furthermore, equivalents and modifications not described above may also be employed without departing from the scope of the invention, which is defined in the accompanying claims.
[000275] In the context of the present invention other examples and variations of the compositions and methods described herein will be apparent to a person of skill in the art. Other examples and variations are within the scope of the invention, as set out in the appended claims.
[000276] All documents cited herein are each entirely incorporated by reference herein, including all data, tables, figures, and text presented in the cited documents.
Table 7A: Antibody sequences




Table 7B: Antibody sequences (short list)

Table 8A: Additional constructs


Table 8B: Additional constructs

EXAMPLES
[000277] The examples in this section are offered by way of illustration, and not by way of limitation.
Example 1- Alnuctamab Formulation
[000278] Clinical formulations for the pharmaceutical composition injection, 12 mg/2.0 mL, 60 mg/2.0 mL, 3 mg/0.5 mL, and 30 mg/1.0 mL are sterile, non-pyrogenic, single-use, preservative-free, isotonic aqueous solutions for subcutaneous administration. They are also referred to as alnuctamab injection. The clinical drug products shown in Table 9 are packaged in a 6-cc Type I glass vial, stoppered with a 20-mm chlorobutyl rubber serum stopper, and sealed with a 20-mm flip-off aluminum seal.
[000279] Commercial drug products shown in Table 10 are filled in 2-cc glass vials, stoppered with a 13 -mm chlorobutyl rubber serum stopper, and sealed with a 13 -mm flip-off aluminum seal.
Table 9. Alnuctamab Clinical Drug Product


Table 10. Alnuctamab Commercial Drug Product

Excess fill will be approx. 0.3 mL for both the commercial images, q.s. = quantity sufficient.
Example 2 - Formulation Development
[000280] As a part of the formulation studies, the effects of various pharmaceutically acceptable excipients on the stability of alnuctamab were evaluated in order to develop a stable, robust subcutaneous (SC) formulation suitable for commercial scale production.
A: Addition of a Metal Ion Chelator
[000281] DTPA (pentetic acid) was evaluated as a potential metal chelator to be incorporated into the formulation for alnuctamab SC injection.
[000282] Formulation robustness: Agitation and freeze-thaw studies:
[000283] In the initial part of the study, three different concentrations of polysorbate 80 (0.02%, 0.04%, and 0.06%) and three different pH levels (5.7, 6, and 6.3) were evaluated for their effect on the stability of alnuctamab compositions. Various compositions (containing 100 mg/mL of alnuctamab, 20 mM histidine, and 250 mM sucrose, along with various concentrations of polysorbate 80 and pHs) were prepared (see Table 11) and subjected to agitation and freeze-thaw studies.
[000284] Table 11: Formulations tested in agitation and freeze-thaw studies

[000285] All formulations in Table 11 showed good physical stability upon agitation at room temperature and freeze-thaw as measured by size exclusion chromatography (SEC), OD350 and sub- visible particle characterization. There was no discernable difference in physical stability between formulations with different pH (5.7-6.3) and surfactant levels (0.02%-0.06%). (Fig. 2A-B).
[000286] Stability data:
[000287] Stability was initiated in vials:
• 50 and 100 mg/mL of alnuctamab (2R Type I glass vials)
Both were formulated in 20mM Histidine, 250mM Sucrose and 0.04% (w/v) polysorbate 80, at pH 6.0.

[000288] The formulations were placed at 5 °C, 25 °C, and 40°C. High molecular weight forms (HMW), main peak, low molecular weight forms (LMW), purity, and charge variant were monitored for up to 24 months.
[000289] As assessed by size exclusion chromatography (SEC), the rate of HMW formation was concentration-dependent, while the rate of formation of LMW did not appear to be concentration dependent. See Figs. 3A and 3C. Acceptable stability was observed at 5°C across all concentration. See Fig. 3A-3C.
[000290] Capillary electrophoresis sodium dodecyl sulphate (CE-SDS) under non-reducing (NR) and reducing (R) conditions was used to assess the main peak (NR) and percent purity (R), and no concentration-dependent changes in main peak % / purity was observed. Acceptable stability was observed at 5 °C across all concentrations. See Figs. 4A-4B.
[000291] Cation exchange chromatography (CEX) was used to assess acidic species and % main peak under each condition and concentration. No concentration-dependent change in charge profile was observed, and acceptable stability was observed at 5°C across all concentrations. See FIG. 5A-5B.
[000292] The relative potency of the formulations was assessed over a period of 18 months using cell -based potency assay, and acceptable retention of potency was observed at 5 °C across all concentrations tested. See FIG. 6.
[000293] The formulations were also tested for polysorbate 80 concentration over a period of 24 months, and a drop in polysorbate 80 concentration was observed at 12 months at 5°C for formulations with 50 and 100 mg/mL of alnuctamab. See FIG. 7-8.
[000294] Thus, for formulations comprising alnuctamab at 10 mg/mL, 50 mg/mL, and 100 mg/mL, at the storage condition of 5 °C, alnuctamab showed good stability in current clinical formulation up to 24 months at 10 mg/mL and 18 months at 50 and 100 mg/mL.
[000295] DTPA Assessment:
[000296] To address the apparent polysorbate 80 degradation observed in samples stored at 5°C over time, metal chelator DTPA was assessed for improving stability of PS80. In this study, compositions containing 44 mg/mL or 1 mg/mL alnuctamab, 20mM histidine, 250mM sucrose, and 0.04% (w/v) polysorbate 80, at pH 6.0, with or without 50 pM DTPA were prepared. DTPA evaluation was performed in 2R vials and formulations were placed at 5°C, 25°C, or 40°C. High molecular weight forms (HMW), monomer (single molecule alnuctamab), low molecular weight forms (LMW), main peak, acidic species, and polysorbate 80 concentrations were monitored for up to 12 months for the 44 mg/mL alnuctamab composition and up to 6 months for the 1 mg/mL alnuctamab composition.
[000297] For formulations comprising 44 mg/mL anuctamab (without and without DTPA):
[000298] As assessed by SEC, no impact of DTPA inclusion at 5°C up to 12 months was observed, while reduction of the rate of HMW formation was observed at 25°C and 40°C with DTPA. FIG. 9A-9C.
[000299] As assessed by CE-SDS NR, no impact of DTPA addition at 5°C, 25°C, or 40°C was observed up to 12 months. FIG. 10.
[000300] As assessed by CEX, no significant impact of DTPA addition was observed at 5°C for acidic species, main peak, or basic species up to 12 months, although a slight reduction of the rate of acidic species formation was observed at 40°C at 3 months with DTPA. FIG. 11A-11C.
[000301] With regard to polysorbate 80 concentration, no polysorbate 80 degradation was observed at 5°C in the starting formulation, and no difference in polysorbate 80 concentration was observed between formulations with and without DTPA at all temperatures. See Table 12 (RH = Relative Humidity).
Table 12: PS-80 Concentration


[000302] Thus, comparable stability for formulations comprising 44 mg/mL alnuctamab (with and without DTP A) at 12 months at 5°C was observed. However, a faster increase in HMW was observed at 6 months at 25 °C and at 3 months at 40°C for formulation without DTPA, and a faster increase in acidic species was observed at 3 months at 40°C for formulation without DTPA. No difference in polysorbate 80 concentration was observed between formulations with and without DTPA.
[000303] For formulations comprising 1 mg/mL of alnuctamab (with or without 50 M DTPA)'.
[000304] As assessed by SEC, no impact of DTPA addition at 5 °C up to 6 months was observed, however, significantly greater rate of HMW formation was observed at 40°C at 3 months without DTPA, and faster decrease of monomer % was observed at 25°C and 40°C without DTPA. FIG. 12A-12B.
[000305] The formulations comprising 1 mg/mL of alnuctamab with or without 50 pM DTPA were inspected for visual precipitation / aggregation at 0, 1, and 3 months. Visual precipitation / aggregation was observed for the Hl formulation (no DTPA) at 40°C after 3 months. The formulations were also tested for subvisible particulate matter by HIAC particle counter for particles > 10pm (See Table 13 A) or particles > 25 pm at 0 months, 1 month, 3 months, and 6 months represented as number of particulate/mL (See Table 13B).
Table 13A-B: Sub Visible particles and protein concentration:

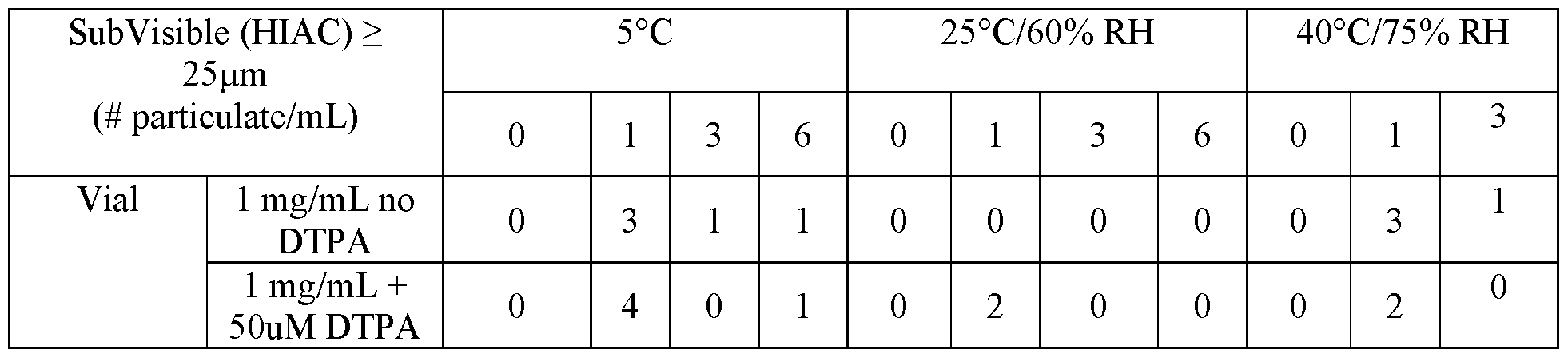
[000306] As assessed by CE-SDS NR, no impact of DTPA addition at 5°C up to 6 months was
observed, however, faster decrease of monomer % was observed at 25°C and 40°C without DTPA. FIG. 13.
[000307] As assessed by CEX, no impact of DTPA addition at 5°C up to 6 months was observed, however, faster rate of acidic species formation and main peak% decrease was observed at 25 °C and 40°C without DTPA. FIG. 14A-14B.
[000308] The formulations comprising 1 mg/mL of alnuctamab with or without DTPA (H2 and Hl, respectively) are aged (Hl- 7 months and H2- 2 months) and observed for 6 months at 5°C, 25°C, and 40°C. A faster rate of polysorbate 80 degradation/oxidation was observed at 25°C and 40°C for formulations without DTPA. DTPA was also shown to mitigate polysorbate 80 degradation in the formulations at all temperatures. See Table 14 and FIG. 15.
Table 14: PS-80 concentration

[000309] Thus, comparable stability at 1 mg/mL between formulations with and without DTPA at 6 months at 5 °C was observed, however, faster rate of HMW and acidic species formation at 6 months at 25°C and 3 months at 40°C was observed in formulations without DTPA. Further, visible aggregate was observed for formulation without DTPA at 3 months at 40°C, and faster decrease in main peak % by CE- SDS NR was observed at 6 months at 25°C and 3 months at 40°C in formulations without DTPA. Faster polysorbate 80 degradation/oxidation was observed in formulations without DTPA at both 25°C and 40°C, but not in formulations with DTPA.
[000310] In summary, formulations that include DTPA were shown to have better physical and chemical stability at 25 °C and 40°C, and the benefit of adding DTPA is more evident at 1 mg/mL anuctamab formulations.
[000311] Metal spiking study.
[000312] In this study, formulations were prepared containing 5 mg/mL and 60 mg/mL alnuctamab in 20mM histidine, 250 mM sucrose, and 0.04% Polysorbate 80, at pH 6.0. The samples were spiked with metal to a concentration of 250 ppb iron, 15 ppb chromium, 10 pb manganese, 15 ppb nickel, or 30 ppb copper. Unspiked samples were also prepared as controls. The following 4 formulations containing alnuctamab at 5 mg/mL were prepared: 1) No metal spike, DTPA; 2) no metal spike, no DTPA; 3) metal spike, DTPA; 4) metal spike, no DTPA. For formulations containing alnuctamab at 60 mg/mL, the following 4 formulations were prepared. 5) No metal spike, DTPA; 6) no metal spike, no DTPA; 7) metal spike, DTPA; 8) metal spike. The formulations were placed at 3 different conditions: 1) 25°C, 2) room temperature/ room light (RT/RL), and 3) 40°C for one month.
[000313] For 5 mg/mL of alnuctamab:
[000314] As assessed by SEC, metal spiking promoted HMW aggregate formation at 40°C, and 50 pM DTPA showed stabilizing effects in the presence of spiking metals. See FIG. 16A-16F.
[000315] As assessed by CEX, metal spiking promoted acidic species formation at 5 mg/mL at all stress conditions (40°C > 25°C ~ RT/RL), and 50pM DTPA showed stabilizing effects in the presence of spiking metals. For non-metal spiking samples, DTPA also showed promising stabilizing effect on acidic species increase. FIG. 18A-18F.
[000316] As assessed by CE-SDS NR, metal spiking resulted in faster drop in main peak at 5 mg/mL at 40°C, and 50pM DTPA showed stabilizing effects in the presence of spiking metals at 40°C. FIG. 20A-20C.
[000317] For 60 mg/mL of alnuctamcib :
[000318] As assessed by SEC, metal spiking promoted HMW aggregate formation at all stress conditions (40°C > RT/RL ~ 25°C), and 50pM DTPA showed stabilizing effects in the presence of spiking metals. The DTPA stabilizing effect on HMW aggregation was more evident at 60 mg/mL than 5 mg/mL. FIG. 17A-17F.
[000319] As assessed by CEX, no significant effect of metal spiking or DTPA was observed on levels of acidic species, and acidic species levels were primarily driven by storage condition. FIG. 19A- 19F.
[000320] As assessed by CE-SDS NR, metal spiking resulted in faster drop in main peak at 60 mg/mL at 40°C, and 50pM DTPA showed promising stabilizing effects in the presence of spiking metals at 40°C. FIG. 21A-21C.
[000321] As for polysorbate 80 levels at both 5 and 60 mg/mL, no difference in polysorbate 80 content was observed for any of the conditions. Table 15.
Table 15: PS80 concentration
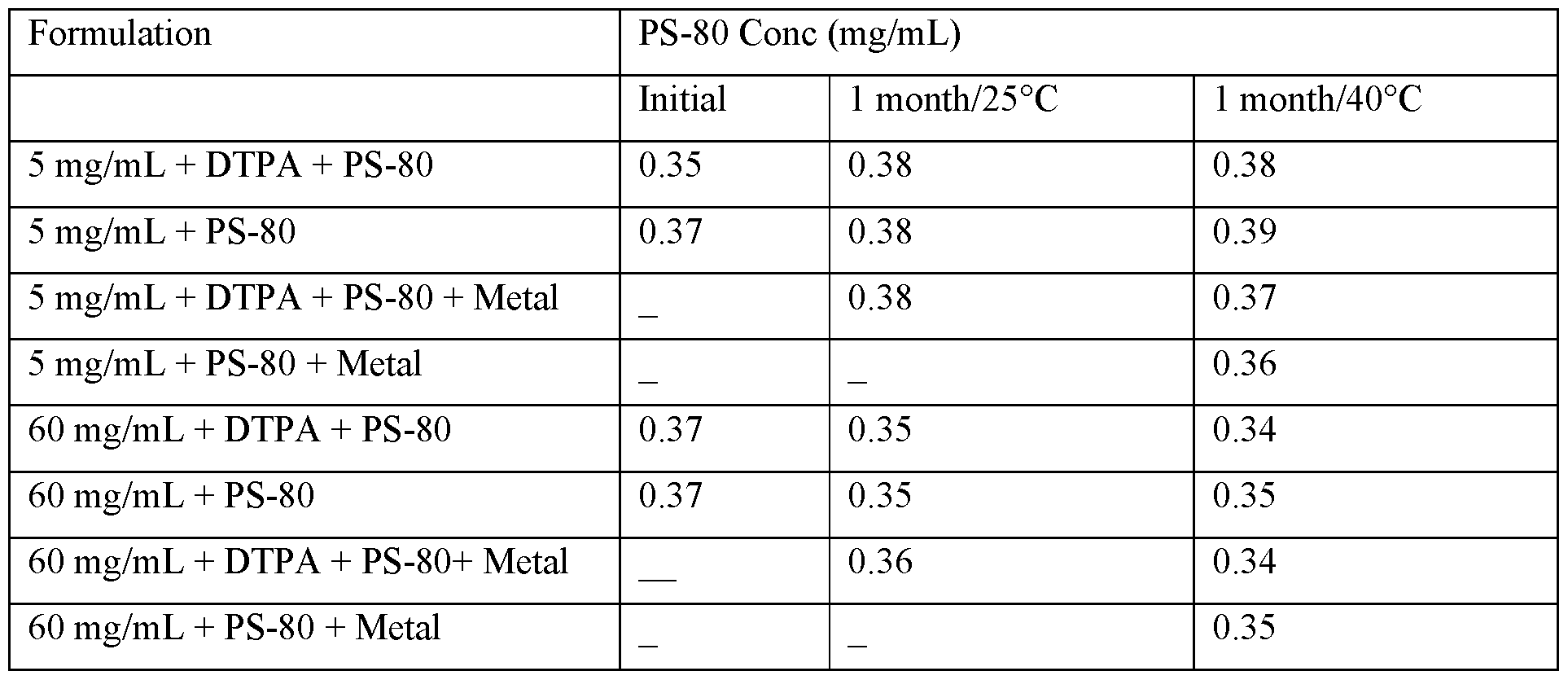
[000322] As for PS-80 oxidation, no change was observed at 60 mg/mL. At 5 mg/mL, no change
was observed in formulations with DTPA, but a slight increase in oxidation was observed at 40°C at one month in formulations without DTPA and without metal spiking, and a slight increase of oxidation was observed at 25 °C at one month in formulations without DTPA, but with metal spiking. At 5 mg/mL, significant increase of oxidation was observed at 40°C at 1 month in formulations without DTPA, with metal spiking. FIG. 22A-22B.
[000323] Thus, for the metal spiking study, 50 pM DTPA showed stabilizing effects in the presence of spiked metals. The DTPA stabilizing effect on HMW aggregation is more evident at 60 mg/mL than 5 mg/mL. On the contrary, the DTPA stabilizing effect on acidic species increase is more evident at 5mg/mL than 60 mg/mL. For non-metal spiking samples, DTPA also showed stabilizing effect on HMW aggregation increase and acidic species increase, which is more evident at 5 mg/mL than 60 mg/mL. High level of polysorbate 80 oxidation was also observed in metal spiked samples at 5mg/mL, which was mitigated by addition of DTPA.
[000324] In summary, formulation with DTPA showed better physical and chemical stability under stress conditions tested. Formulations without DTPA in various protein concentrations showed acceptable stability at 5 °C (up to 24 months), but stability studies at accelerated conditions (25 °C and 40°C) showed that formulations with 50 pM of DTPA have stabilizing effects on both physical and chemical stability. Polysorbate 80 degradation/oxidation was mitigated with 50 pM DTPA in metal spiked formulations comprising 60 mg/mL and 5 mg/mL alnuctamab. Metal spiking studies showed beneficial effect on both physical and chemical stability in metal spiked samples. No detrimental effects of DTPA addition were observed on quality attributes monitored in the stability studies. Thus, 50 pM DTPA was included in the formulation, to arrive at final formulation comprising 20 mM histidine, 250 mM sucrose, 0.04% polysorbate 80, 50 pM DTPA, at pH 6.0.
[000325] To understand the mechanism of DTPA protection and potential impacts of DTPA to alnuctamab protein structure, further characterization was performed. Near-UV CD spectroscopy data revealed that, following DTPA addition, alnuctamab protein tertiary structure around Trp residue changed, resulting to a more chiral environment. Differential scanning calorimetry (DSC) data indicated that DTPA binding increased the overall stability of the alnuctamab protein. By isothermal titration calorimetry (ITC), it was discovered that DTPA in formulation bound to alnuctamab with dissociation constant in micromolar range. Nuclear magnetic resonance (NMR) spectroscopy confirmed the binding event from three aspects. First, data from saturation transfer difference (STD) NMR demonstrated certain hydrogens on DTPA experienced differences in nearby chemical environment after adding alnuctamab. Second, ID fingerprinting of alnuctamab revealed differences in protein amide region before and after DTPA addition, indicating changes in chemical environment around protein backbone. Third, 2D fingerprinting by methyl heteronuclear multiple quantum coherence (Methyl-HMQC) NMR also revealed changes in chemical environment around certain methyl groups on alnuctamab. Homology modeling predicted the most likely binding site on alnuctamab is the CDR on anti-CD3 domain. The binding is likely to be driven by charge-charge interactions. More specifically, the interaction could be between two
positively charged arginine (Arg) residues and multiple negatively charged DTPA. The two Arg residues are sterically close to the CDR, containing two Trp residues which could be oxidized, and two Asn residues which could be deamidated. Prediction of binding site was supported by native fluorescence spectroscopy in which Trp fluorescence blue shift was observed, as an indicator of decreased hydrophobicity. Also, Raman spectroscopy revealed a minor rotation of the Trp side chain. The binding site was confirmed by carbene footprints. After adding DTPA, label uptake in anti-CD3 CDR was found to decrease, indicating anti-CD3 CDR overlaps with binding sites. Label uptake in most other regions of the protein did not decrease, indicating anti-CD3 CDR is most likely the only binding site of DTPA. In summary, it was discovered DTPA binds to alnuctamab, binding is likely to be driven by charge-charge interaction, and the binding site is most likely in the anti-CD3 CDR.
[000326] The invention is described in detail with reference to specific embodiments thereof, it will be understood that variations which are functionally equivalent are within the scope of this invention. Indeed, various modifications of the invention in addition to those shown and described herein will become apparent to those skilled in the art from the foregoing description and accompanying drawings. Such modifications are intended to fall within the scope of the appended claims. Those skilled in the art will recognize, or be able to ascertain using no more than routine experimentation, many equivalents to the specific embodiments of the invention described herein. Such equivalents are intended to be encompassed by the following claims.
EQUIVALENTS
[000327] Although the invention is described in detail with reference to specific embodiments thereof, it will be understood that variations which are functionally equivalent are within the scope of this invention. Indeed, various modifications of the invention in addition to those shown and described herein will become apparent to those skilled in the art from the foregoing description and accompanying drawings. Such modifications are intended to fall within the scope of the appended claims. Those skilled in the art will recognize, or be able to ascertain using no more than routine experimentation, many equivalents to the specific embodiments of the invention described herein. Such equivalents are intended to be encompassed by the following claims.
Claims
1. A pharmaceutical composition comprising:
(a) at least about 0.05 mg/mL to at least about 37.5 mg/mL of a multispecific antibody that binds to BCMA and CD3, at least about 0.5 mg/mL to at least about 37.5 mg/mL of a multispecific antibody that binds to BCMA and CD3, or at least about 4.5 mg/mL to at least about 37.5 mg/mL of a multispecific antibody that binds to BCMA and CD3,
(b) at lest about 5 mM to at least about 100 mM histidine,
(c) at least about 10 mM to at least about 500 mM sucrose,
(d) at least about 0.01% w/v to at least about 0.1% w/v polysorbate 80, and
(e) at least about 10 pM to about 200 pM pentetic acid (DTPA).
2. The pharmaceutical composition of claim 1, comprising about 4.5 mg/mL to 7.5 mg/mL of the multispecific antibody.
3. The pharmaceutical composition of claim 1, comprising about 6 mg/mL of the multispecific antibody.
4. The pharmaceutical composition of claim 1, comprising about 22.5 mg/mL to 37.5 mg/mL of the multispecific antibody.
5. The pharmaceutical composition of claim 1, comprising about 30 mg/mL of the multispecific antibody.
6. The pharmaceutical composition of claim 1, comprising:
(a) about 6 mg/mL of the multispecific antibody;
(b) about 20 mM histidine;
(c) about 250 mM sucrose;
(d) about 0.04% w/v polysorbate 80; and
(e) about 50 pM pentetic acid.
7. The pharmaceutical composition of claim 1, comprising:
(a) about 30 mg/mL of the multispecific antibody;
(b) about 20 mM histidine;
(c) about 250 mM sucrose;
(d) about 0.04% w/v polysorbate 80; and
(e) about 50 pM pentetic acid.
8. The pharmaceutical composition of any one of claims 1-7, wherein the pH of the composition is from about 5.7 to about 6.3.
9. The pharmaceutical composition of any one of claims 1-8, wherein the pH of the composition is about 6.0.
10. The pharmaceutical composition of any one of claims 1-9, wherein the multispecific antibody comprises:
an anti-BCMA antibody, or antigen binding fragment thereof, comprising a VH region comprising a CDR1H region of SEQ ID NO:21, a CDR2H region of SEQ ID NO:22 and a CDR3H region of SEQ ID NO: 17 and a VL region comprising a CDR1L region of SEQ ID NO:27, a CDR2L region of SEQ ID NO:28 and a CDR3L region of SEQ ID NO:20; and an anti-CD3 antibody, or antigen binding fragment thereof, comprising a CDR1H region of SEQ ID NO : 1 , a CDR2H region of SEQ ID NO : 2, a CDR3H region of SEQ ID NO : 3 , a CDR1 L region of SEQ ID NO:4, a CDR2L region of SEQ ID NO:5 and a CDR3L region of SEQ ID NO:6).
11. The pharmaceutical composition of any one of claims 1-10, wherein the multispecific antibody comprises: an anti-BCMA antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO: 10 and a VL region of SEQ ID NO: 14; and an anti-CD3 antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO: 7 and a VL region of SEQ ID NO: 8.
12. The pharmaceutical composition of any one of claims 1-11, wherein the multispecific antibody is a trivalent bispecific antibody comprising two Fab fragments of an anti-BCMA antibody, one Fab fragment of an anti-CD3 antibody, and one Fc portion, wherein the bispecific antibody is in the format BCMA Fab - Fc - CD3 Fab - BCMA Fab.
13. The pharmaceutical composition of claim 12, wherein each Fab fragment of an anti-BCMA antibody, or antigen binding fragment thereof, comprises a VH region comprising a CDR1H region of SEQ ID NO:21, a CDR2H region of SEQ ID NO:22 and a CDR3H region of SEQ ID NO: 17 and a VL region comprising a CDR1L region of SEQ ID NO:27, a CDR2L region of SEQ ID NO:28 and a CDR3L region of SEQ ID NO:20.
14. The pharmaceutical composition of claim 12 or claim 13, wherein each Fab fragment of an anti- BCMA antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO: 10 and a VL region of SEQ ID NO: 14.
15. The pharmaceutical composition of any one of claims 12-14, wherein the Fab fragment of the anti-CD3 antibody, or antigen binding fragment thereof, comprises a CDR1H region of SEQ ID NO : 1 , a CDR2H region of SEQ ID NO : 2, a CDR3H region of SEQ ID NO : 3 , a CDR1 L region of SEQ ID NO:4, a CDR2L region of SEQ ID NO:5 and a CDR3L region of SEQ ID NO:6.
16. The pharmaceutical composition of any one of claims 12-15, wherein the Fab fragment of the anti-CD3 antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO: 7 and a VL region of SEQ ID NO: 8.
17. The pharmaceutical composition of any one of claims 1-16, wherein the multispecific antibody comprises a first polypeptide comprising the amino acid sequence of SEQ ID NO: 48, a second polypeptide comprising the amino acid sequence of SEQ ID NO: 55 or 58, a third polypeptide comprising the amino acid sequence of SEQ ID NO: 56 or 59, and fourth and fifth polypeptides each comprising the amino acid sequence of SEQ ID NO: 57.
18. A unit dose comprising the pharmaceutical composition of any one of claims 1-17.
19. The unit dose of claim 18, wherein the volume of the unit dose is 0.5-2.5 mL, or 0.5-0.9 mL, or
1.0-1.5 mL, or 2.0-2.5 mL, or 0.5, 0.6, 0.7, 0.8, 0.9. 1.0, 1.1, 1.2, 1.3, 1.4, 1.5, 1.6, 1.7, 1.8, 1.9, 2.0, 2.1, 2.2, 2.3, 2.4, or 2.5 mL.
20. A unit dose comprising a pharmaceutical composition comprising:
(a) about 6 mg/mL of a multispecific antibody that binds to BCMA and CD3;
(b) about 20 mM histidine;
(c) about 250 mM sucrose;
(d) about 0.04% w/v polysorbate 80; and
(e) about 50 pM pentetic acid; wherein the pH of the pharmaceutical composition is from about 5.7 to about 6.3.
21. A unit dose comprising a pharmaceutical composition comprising:
(a) about 30 mg/mL of a multispecific antibody that binds to BCMA and CD3;
(b) about 20 mM histidine;
(c) about 250 mM sucrose;
(d) about 0.04% w/v polysorbate 80; and
(e) about 50 pM pentetic acid; and wherein the pH of the pharmaceutical composition is from about 5.7 to about 6.3.
22. The unit dose of claim 20 or 21, wherein the multispecific antibody comprises: an anti-BCMA antibody, or antigen binding fragment thereof, comprising a VH region comprising a CDR1H region of SEQ ID NO:21, a CDR2H region of SEQ ID NO:22 and a CDR3H region of SEQ ID NO: 17 and a VL region comprising a CDR1L region of SEQ ID NO:27, a CDR2L region of SEQ ID NO:28 and a CDR3L region of SEQ ID NO:20; and an anti-CD3 antibody, or antigen binding fragment thereof, comprising a CDR1H region of SEQ ID NO : 1 , a CDR2H region of SEQ ID NO : 2, a CDR3H region of SEQ ID NO : 3 , a CDR1 L region of SEQ ID NO: 4, a CDR2L region of SEQ ID NO: 5 and a CDR3L region of SEQ ID NO: 6).
23. The unit dose of any one of claims 20-22, wherein the multispecific antibody comprises: an anti-BCMA antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO:
10 and a VL region of SEQ ID NO: 14; and an anti-CD3 antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO: 7 and a VL region of SEQ ID NO: 8.
24. The unit dose of any one of claims 20-23, wherein the multispecific antibody is atrivalent bispecific antibody comprising two Fab fragments of an anti-BCMA antibody, one Fab fragment of an anti-CD3 antibody, and one Fc portion, wherein the bispecific antibody is in the format BCMA Fab - Fc - CD3 Fab - BCMA Fab.
25. The unit dose of claim 24, wherein each Fab fragment of an anti-BCMA antibody, or antigen binding fragment thereof, comprises a VH region comprising a CDR1H region of SEQ ID
NO : 21 , a CDR2H region of SEQ ID NO : 22 and a CDR3H region of SEQ ID NO : 17 and a VL region comprising a CDR1L region of SEQ ID NO:27, a CDR2L region of SEQ ID NO:28 and a CDR3L region of SEQ ID NO:20.
26. The unit dose of claim 24 or claim 25, wherein each Fab fragment of an anti-BCMA antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO: 10 and a VL region of SEQ ID NO: 14
27. The unit dose of any one of claims 24-26, wherein the Fab fragment of the anti-CD3 antibody, or antigen binding fragment thereof, comprises a CDR1H region of SEQ ID NO: 1, a CDR2H region of SEQ ID NO: 2, a CDR3H region of SEQ ID NO: 3, a CDR1L region of SEQ ID NO: 4, a CDR2L region of SEQ ID NO:5 and a CDR3L region of SEQ ID NO:6.
28. The unit dose of any one of claims 24-27, wherein the Fab fragment of the anti-CD3 antibody, or antigen binding fragment thereof, comprises a VH region of SEQ ID NO: 7 and a VL region of SEQ ID NO: 8.
29. The unit dose of any one of claims 20-28, wherein the multispecific antibody comprises a first polypeptide comprising the amino acid sequence of SEQ ID NO: 48, a second polypeptide comprising the amino acid sequence of SEQ ID NO: 55 or 58, a third polypeptide comprising the amino acid sequence of SEQ ID NO: 56 or 59, and fourth and fifth polypeptides each comprising the amino acid sequence of SEQ ID NO: 57.
30. The unit dose of any one of claims 20-29, wherein the volume of the unit dose is 0.5-2.5 mb, or 0.5-0.9 mL, or 1.0-1.5 mL, or 2.0-2.5 mL, or 0.5, 0.6, 0.7, 0.8, 0.9. 1.0, 1.1, 1.2, 1.3, 1.4, 1.5, 1.6, 1.7, 1.8, 1.9, 2.0, 2.1, 2.2, 2.3, 2.4, or 2.5 mL.
31. A vial comprising the pharmaceutical composition of any one of claims 1-17 or the unit dose of any one of claims 18-30.
32. A method of treating a subject in need thereof, comprising subcutaneously administering to the subject at least one dose of the pharmaceutical composition of any one of claims 1-17 or at least one unit dose of any one of claims 18-30.
33. A method of treating multiple myeloma or an autoimmune disease in a subject, comprising subcutaneously administering to the subject at least one dose of the pharmaceutical composition of any one of claims 1-17 or at least one unit dose of any one of claims 18-30.
34. The method of claim 32 or claim 33, wherein the treatment comprises the administration of the pharmaceutical composition or unit dose in a dosing regimen comprising:
(i) a starting phase, wherein one or more starting doses of the pharmaceutical composition or unit dose are administered to the subject; and
(ii) a maintenance phase, wherein a first maintenance dose of the pharmaceutical composition or unit dose is administered to the subject, optionally followed by at least one additional maintenance dose of the pharmaceutical composition or unit dose, wherein each maintenance dose is greater than the one or more starting doses.
35. The method of claim 34, wherein the starting phase comprises a single fixed dose of the pharmaceutical composition or unit dose comprising about 1.5 mg to 4.5 mg of the multispecific antibody.
36. The method of claim 34, wherein the starting phase comprises a single fixed dose of the pharmaceutical composition or unit dose comprising about 3 mg of the multispecific antibody.
37. The method of any one of claims 34-36, wherein the first maintenance dose is a fixed dose of the pharmaceutical composition or unit dose comprising about 4.5 mg to about 7.5 mg of the multispecific antibody.
38. The method of any one of claims 34-36, wherein the first maintenance dose is a fixed dose of the pharmaceutical composition or unit dose comprising about 6 mg of the multispecific antibody.
39. The method of any one of claims 34-38, wherein the at least one additional maintenance dose is a fixed dose of the pharmaceutical composition or unit dose comprising about 28 to about 32 mg of the multispecific antibody.
40. The method of any one of claims 34-38, wherein the at least one additional maintenance dose is a fixed dose of the pharmaceutical composition or unit dose comprising about 30 mg of the multispecific antibody.
41. The method of any one of claims 34-40, wherein the treatment comprises:
(i) a first treatment cycle, wherein the starting dose is administered on day 1, and the maintenance doses are administered on days 4, 8, 15 and 22,
(ii) a second and third treatment cycle, wherein the maintenance doses are administered in a weekly dosing interval,
(iii)a fourth to sixth treatment cycle, wherein the maintenance doses are administered in a biweekly dosing interval, and
(iv) a seventh and subsequent cycle, wherein the maintenance doses are administered in a four week dosing interval; wherein each treatment cycle is 28 days.
42. The method of claim 41, wherein the maintenance doses are administered on days 1, 8, 15 and 22 for the second and third treatment cycle, on days 1 and 15 for the fourth to sixth treatment cycle, and on day 1 for the seventh and subsequent cycle.
43. The method of any one of claims 34-42, wherein the treatment comprises:
(i) a first treatment cycle, wherein the starting dose comprises a single fixed dose of the pharmaceutical composition or unit dose comprising about 3 mg of the multi-specific antibody is administered on day 1, the maintenance dose of the pharmaceutical composition or unit dose comprising about 6 mg of the multispecific antibody is administered on day 4, and the maintenance doses of the pharmaceutical composition or unit dose comprising about 30 mg of the multispecific antibody are administered on days 8, 15 and 22,
(ii) a second and third treatment cycle, wherein the maintenance doses of the pharmaceutical composition or unit dose comprising about 30 mg of the multispecific antibody are administered on days 1, 8, 15, and 22 in a weekly dosing interval,
(iii) a fourth to sixth treatment cycle, wherein the maintenance doses of the pharmaceutical composition or unit dose comprising about 30 mg of the multispecific antibody are administered on days 1 and 15 in a biweekly dosing interval, and
(iv) a seventh and subsequent cycle, wherein the maintenance doses of the pharmaceutical composition or unit dose comprising about 30 mg of the multispecific antibody are administered on day 1 in a four week dosing interval; wherein each treatment cycle is 28 days.
44. The pharmaceutical composition of any one of claims 1-17 or the unit dose of any one of claims 18-30 for use in treating a disorder associated with BCMA expression in a subject.
45. The pharmaceutical composition or unit dose for use of claim 44, wherein the disorder is multiple myeloma or an autoimmune disease.
46. The pharmaceutical composition or unit dose for use of claim 44 or claim 45, wherein the treatment comprises the administration of the pharmaceutical composition or unit dose in a dosing regimen comprising:
(i) a starting phase, wherein one or more starting doses of the pharmaceutical composition or unit dose are administered to the subject; and
(ii) a maintenance phase, wherein a first maintenance dose of the pharmaceutical composition or unit dose is administered to the subject, optionally followed by at least one additional maintenance dose of the pharmaceutical composition or unit dose, wherein each maintenance dose is greater than the one or more starting doses.
47. The pharmaceutical composition or unit dose for use of claim 46, wherein the starting phase comprises a single fixed dose of the pharmaceutical composition or unit dose comprising about 1.5 mg to 4.5 mg of the multispecific antibody.
48. The pharmaceutical composition or unit dose for use of claim 46, wherein the starting phase comprises a single fixed dose of the pharmaceutical composition or unit dose comprising about 3 mg of the multi-specific antibody.
49. The pharmaceutical composition or unit dose for use of any one of claims 46-48, wherein the first maintenance dose is a fixed dose of the pharmaceutical composition or unit dose comprising about 4.5 mg to about 7.5 mg of the multi-specific antibody.
50. The pharmaceutical composition or unit dose for use of any one of claims 46-48, wherein the first maintenance dose is a fixed dose of the pharmaceutical composition or unit dose comprising about 6 mg of the multi-specific antibody.
51. The pharmaceutical composition or unit dose for use of any one of claims 46-50, wherein the at least one additional maintenance dose is a fixed dose of the pharmaceutical composition or unit dose comprising about 28 to about 32 mg of the multi-specific antibody.
52. The pharmaceutical composition or unit dose for use of any one of claims 46-50, wherein the at least one additional maintenance dose is a fixed dose of the pharmaceutical composition or unit dose comprising about 30 mg of the multi-specific antibody.
53. The pharmaceutical composition or unit dose for use of any one of claims 46-52, wherein the treatment comprises:
(i) a first treatment cycle, wherein the starting dose is administered on day 1, and the maintenance doses are administered on days 4, 8, 15 and 22,
(ii) a second and third treatment cycle, wherein the maintenance doses are administered in a weekly dosing interval,
(iii) a fourth to sixth treatment cycle, wherein the maintenance doses are administered in a biweekly dosing interval, and
(iv) a seventh and subsequent cycle, wherein the maintenance doses are administered in a four week dosing interval; wherein each treatment cycle is 28 days.
54. The pharmaceutical composition or unit dose for use of claim 53, wherein the maintenance doses are administered on days 1, 8, 15 and 22 for the second and third treatment cycle, on days 1 and 15 for the fourth to sixth treatment cycle, and on day 1 for the seventh and subsequent cycle.
55. The pharmaceutical composition or unit dose for use of any one of claims 46-54, wherein the treatment comprises:
(i) a first treatment cycle, wherein the starting dose of a single fixed dose of the pharmaceutical composition or unit dose comprising about 3 mg of the multi-specific antibody is administered on day 1, the maintenance dose of the pharmaceutical composition or unit dose comprising about 6 mg of the multispecific antibody is administered on day 4, and the maintenance doses of the pharmaceutical composition or unit dose comprising about 30 mg of the multispecific antibody are administered on days 8, 15 and 22,
(ii) a second and third treatment cycle, wherein the maintenance doses of the pharmaceutical composition or unit dose comprising about 30 mg of the multispecific antibody are administered on days 1, 8, 15, and 22 in a weekly dosing interval,
(iii)a fourth to sixth treatment cycle, wherein the maintenance doses of the pharmaceutical composition or unit dose comprising about 30 mg of the multispecific antibody are administered on days 1 and 15 in a biweekly dosing interval, and
(iv) a seventh and subsequent cycle, wherein the maintenance doses of the pharmaceutical composition or unit dose comprising about 30 mg of the multispecific antibody are administered on day 1 in a four week dosing interval;
wherein each treatment cycle is 28 days.
56. Use of the pharmaceutical composition of any one of claims 1-17 or the unit dose of any one of claims 18-30 for the preparation of a medicament for treating a disorder associated with BCMA expression.
57. The use of claim 56, wherein the disorder is multiple myeloma.
58. The use of claim 56 or claim 57, wherein the medicament is administered to a subject in a dosing regimen comprising:
(i) a starting phase, wherein one or more starting doses of the medicament are administered to the subject; and
(ii) a maintenance phase, wherein a first maintenance dose of the medicament is administered to the subject, optionally followed by at least one additional maintenance dose of the medicament, wherein each maintenance dose is greater than the one or more starting doses.
59. The use of claim 58, wherein the starting phase comprises a single fixed dose of the medicament comprising about 1.5 mg to 4.5 mg of the multispecific antibody.
60. The use of claim 58, wherein the starting phase comprises a single fixed dose of the medicament comprising about 3 mg of the multi-specific antibody.
61. The use of any one of claims 58-60, wherein the first maintenance dose is a fixed dose of the medicament comprising about 4.5 mg to about 7.5 mg of the multi-specific antibody.
62. The use of any one of claims 58-60, wherein the first maintenance dose is a fixed dose of the medicament comprising about 6 mg of the multi-specific antibody.
63. The use of any one of claims 58-62, wherein the at least one additional maintenance dose is a fixed dose of the medicament comprising about 28 to about 32 mg of the multi-specific antibody.
64. The use of any one of claims 58-62, wherein the at least one additional maintenance dose is a fixed dose of the medicament comprising about 30 mg of the multi-specific antibody.
65. The use of any one of claims 58-64, wherein the medicament is administered in a dosing regimen comprising:
(i) a first treatment cycle, wherein the starting dose is administered on day 1, and the maintenance doses are administered on days 4, 8, 15 and 22,
(ii) a second and third treatment cycle, wherein the maintenance doses are administered in a weekly dosing interval,
(iii)a fourth to sixth treatment cycle, wherein the maintenance doses are administered in a biweekly dosing interval, and
(iv) a seventh and subsequent cycle, wherein the maintenance doses are administered in a four week dosing interval; wherein each treatment cycle is 28 days.
66. The use of claim 65, wherein the maintenance doses are administered on days 1, 8, 15 and 22 for the second and third treatment cycle, on days 1 and 15 for the fourth to sixth treatment cycle, and on day 1 for the seventh and subsequent cycle.
67. The use of any one of claims 58-66, wherein the medicament is administered in a dosing regimen comprising:
(i) a first treatment cycle, wherein the starting dose comprises a single fixed dose of the medicament comprising about 3 mg of the multi-specific antibody is administered on day 1, the maintenance dose of the medicament comprising about 6 mg of the multispecific antibody is administered on day 4, and the maintenance doses of the medicament comprising about 30 mg of the multispecific antibody are administered on days 8, 15 and 22,
(ii) a second and third treatment cycle, wherein the maintenance doses of the medicament comprising about 30 mg of the multispecific antibody are administered on days 1, 8, 15, and 22 in a weekly dosing interval,
(iii) a fourth to sixth treatment cycle, wherein the maintenance doses of the medicament comprising about 30 mg of the multispecific antibody are administered on days 1 and 15 in a biweekly dosing interval, and
(iv) a seventh and subsequent cycle, wherein the maintenance doses of the medicament comprising about 30 mg of the multispecific antibody are administered on day 1 in a four week dosing interval; wherein each treatment cycle is 28 days.
68. A pharmaceutical composition comprising (a) about 6 mg/mL of alnuctamab; (b) about 20 mM histidine; (c) about 250 mM sucrose; (d) about 0.04% w/v polysorbate 80; and (e) about 50 pM pentetic acid.
69. A pharmaceutical composition comprising (a) about 30 mg/mL of alnuctamab; (b) about 20 mM histidine; (c) about 250 mM sucrose; (d) about 0.04% w/v polysorbate 80; and (e) about 50 pM pentetic acid.
70. A pharmaceutical composition comprising (a) 6 mg/mL of alnuctamab; (b) 20 mM histidine; (c) 250 mM sucrose; (d) 0.04% w/v polysorbate 80; and (e) 50 pM pentetic acid.
71. A pharmaceutical composition comprises (a) 30 mg/mL of alnuctamab; (b) 20 mM histidine; (c) 250 mM sucrose; (d) 0.04% w/v polysorbate 80; and (e) 50 pM pentetic acid.
72. The vial of claim 31, wherein the vial is a pre-filled syringe or an autoinjector.
73. A method of preventing or reducing oxidation and/or deamidation of alnuctamab in a pharmaceutical composition comprising binding alnuctamab to pentetic acid in the pharmaceutical composition, thereby preventing or reducing oxidation and/or deamidation of alnuctamab in the pharmaceutical composition.
74. The method of claim 73, wherein pentetic acid binds to the anti-CD3 domain of alnuctamab.
75. The method of claim 73 or 74, wherein binding alnuctamab to pentetic acid prevents or reduces tryptophan residue oxidation and/or asparagine residue deamidation in alnuctamab.
76. The method of claim 75, wherein the tryptophan residue(s) are located in the CDR of the anti- CD3 domain of alnuctamab.
77. The method of claim 75 or 76, wherein the asparagine residue(s) are located in the CDR of the anti-CD3 domain of alnuctamab.
78. The method of any of claims 73-77, wherein the pharmaceutical composition is the pharmaceutical composition of any of claims 1-17.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363520561P | 2023-08-18 | 2023-08-18 | |
| US63/520,561 | 2023-08-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025042742A1 true WO2025042742A1 (en) | 2025-02-27 |
Family
ID=92711172
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/042698 Pending WO2025042742A1 (en) | 2023-08-18 | 2024-08-16 | Compositions comprising antibodies that bind bcma and cd3 and methods of treatment |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025042742A1 (en) |
Citations (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5202238A (en) | 1987-10-27 | 1993-04-13 | Oncogen | Production of chimeric antibodies by homologous recombination |
| US5204244A (en) | 1987-10-27 | 1993-04-20 | Oncogen | Production of chimeric antibodies by homologous recombination |
| WO1996027011A1 (en) | 1995-03-01 | 1996-09-06 | Genentech, Inc. | A method for making heteromultimeric polypeptides |
| WO1998050431A2 (en) | 1997-05-02 | 1998-11-12 | Genentech, Inc. | A method for making multispecific antibodies having heteromultimeric and common components |
| WO2003099196A2 (en) | 2002-05-23 | 2003-12-04 | Cure Tech Ltd. | Humanized immunomodulatory monoclonal antibodies for the treatment of neoplastic disease or immunodeficiency |
| WO2006121168A1 (en) | 2005-05-09 | 2006-11-16 | Ono Pharmaceutical Co., Ltd. | Human monoclonal antibodies to programmed death 1(pd-1) and methods for treating cancer using anti-pd-1 antibodies alone or in combination with other immunotherapeutics |
| WO2007005874A2 (en) | 2005-07-01 | 2007-01-11 | Medarex, Inc. | Human monoclonal antibodies to programmed death ligand 1 (pd-l1) |
| WO2007110205A2 (en) | 2006-03-24 | 2007-10-04 | Merck Patent Gmbh | Engineered heterodimeric protein domains |
| EP1870459A1 (en) | 2005-03-31 | 2007-12-26 | Chugai Seiyaku Kabushiki Kaisha | Methods for producing polypeptides by regulating polypeptide association |
| WO2007147901A1 (en) | 2006-06-22 | 2007-12-27 | Novo Nordisk A/S | Production of bispecific antibodies |
| WO2008156712A1 (en) | 2007-06-18 | 2008-12-24 | N. V. Organon | Antibodies to human programmed death receptor pd-1 |
| US7521051B2 (en) | 2002-12-23 | 2009-04-21 | Medimmune Limited | Methods of upmodulating adaptive immune response using anti-PD-1 antibodies |
| WO2009080251A1 (en) | 2007-12-21 | 2009-07-02 | F. Hoffmann-La Roche Ag | Bivalent, bispecific antibodies |
| WO2009080252A1 (en) | 2007-12-21 | 2009-07-02 | F. Hoffmann-La Roche Ag | Bivalent, bispecific antibodies |
| WO2009089004A1 (en) | 2008-01-07 | 2009-07-16 | Amgen Inc. | Method for making antibody fc-heterodimeric molecules using electrostatic steering effects |
| WO2009101611A1 (en) | 2008-02-11 | 2009-08-20 | Curetech Ltd. | Monoclonal antibodies for tumor treatment |
| WO2009114335A2 (en) | 2008-03-12 | 2009-09-17 | Merck & Co., Inc. | Pd-1 binding proteins |
| WO2010027828A2 (en) | 2008-08-25 | 2010-03-11 | Amplimmune, Inc. | Pd-1 antagonists and methods of use thereof |
| WO2010027423A2 (en) | 2008-08-25 | 2010-03-11 | Amplimmune, Inc. | Compositions of pd-1 antagonists and methods of use |
| WO2010077634A1 (en) | 2008-12-09 | 2010-07-08 | Genentech, Inc. | Anti-pd-l1 antibodies and their use to enhance t-cell function |
| WO2010129304A2 (en) | 2009-04-27 | 2010-11-11 | Oncomed Pharmaceuticals, Inc. | Method for making heteromultimeric molecules |
| WO2011090754A1 (en) | 2009-12-29 | 2011-07-28 | Emergent Product Development Seattle, Llc | Polypeptide heterodimers and uses thereof |
| US20110196150A1 (en) | 2010-02-11 | 2011-08-11 | Hon-Wah Man | Arylmethoxy Isoindoline Derivatives and Compositions Comprising and Methods of Using the Same |
| WO2011143545A1 (en) | 2010-05-14 | 2011-11-17 | Rinat Neuroscience Corporation | Heterodimeric proteins and methods for producing and purifying them |
| WO2012058768A1 (en) | 2010-11-05 | 2012-05-10 | Zymeworks Inc. | Stable heterodimeric antibody design with mutations in the fc domain |
| US8362211B2 (en) | 2010-12-30 | 2013-01-29 | Takeda Pharmaceutical Company Limited | Anti-CD38 antibodies |
| WO2013019906A1 (en) | 2011-08-01 | 2013-02-07 | Genentech, Inc. | Methods of treating cancer using pd-1 axis binding antagonists and mek inhibitors |
| WO2013096291A2 (en) | 2011-12-20 | 2013-06-27 | Medimmune, Llc | Modified polypeptides for bispecific antibody scaffolds |
| WO2013157953A1 (en) | 2012-04-20 | 2013-10-24 | Merus B.V. | Methods and means for the production of ig-like molecules |
| US8877899B2 (en) | 2010-09-27 | 2014-11-04 | Morphosys Ag | Anti-CD38 antibody and lenalidomide or bortezomib for the treatment of multipe myeloma and NHL |
| WO2015026634A1 (en) | 2013-08-20 | 2015-02-26 | Merck Sharp & Dohme Corp. | Treating cancer with a combination of a pd-1 antagonist and dinaciclib |
| US20150246123A1 (en) | 2014-02-28 | 2015-09-03 | Janssen Biotech, Inc. | Combination Therapies with Anti-CD38 Antibodies |
| WO2016007235A1 (en) | 2014-07-11 | 2016-01-14 | Genentech, Inc. | Anti-pd-l1 antibodies and diagnostic uses thereof |
| EP2982692A1 (en) * | 2014-08-04 | 2016-02-10 | EngMab AG | Bispecific antibodies against CD3epsilon and BCMA |
| WO2016040238A1 (en) | 2014-09-08 | 2016-03-17 | Celgene Corporation | Methods for treating a disease or disorder using oral formulations of cytidine analogs in combination with an anti-pd1 or anti-pdl1 monoclonal antibody |
| WO2017021450A1 (en) | 2015-08-03 | 2017-02-09 | Engmab Ag | Monoclonal antibodies against bcma |
| WO2020261093A1 (en) * | 2019-06-24 | 2020-12-30 | Novartis Ag | Dosing regimen and combination therapies for multispecific antibodies targeting b-cell maturation antigen |
| WO2021092056A1 (en) * | 2019-11-05 | 2021-05-14 | Engmab Sàrl | Methods of treatment with antibodies against bcma and cd3 |
| WO2022146947A1 (en) * | 2020-12-28 | 2022-07-07 | Bristol-Myers Squibb Company | Antibody compositions and methods of use thereof |
| WO2023086817A1 (en) * | 2021-11-10 | 2023-05-19 | Janssen Biotech, Inc. | Stable formulations comprising a bispecific bcma/cd3 antibody |
-
2024
- 2024-08-16 WO PCT/US2024/042698 patent/WO2025042742A1/en active Pending
Patent Citations (45)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5204244A (en) | 1987-10-27 | 1993-04-20 | Oncogen | Production of chimeric antibodies by homologous recombination |
| US5202238A (en) | 1987-10-27 | 1993-04-13 | Oncogen | Production of chimeric antibodies by homologous recombination |
| WO1996027011A1 (en) | 1995-03-01 | 1996-09-06 | Genentech, Inc. | A method for making heteromultimeric polypeptides |
| WO1998050431A2 (en) | 1997-05-02 | 1998-11-12 | Genentech, Inc. | A method for making multispecific antibodies having heteromultimeric and common components |
| WO2003099196A2 (en) | 2002-05-23 | 2003-12-04 | Cure Tech Ltd. | Humanized immunomodulatory monoclonal antibodies for the treatment of neoplastic disease or immunodeficiency |
| US7521051B2 (en) | 2002-12-23 | 2009-04-21 | Medimmune Limited | Methods of upmodulating adaptive immune response using anti-PD-1 antibodies |
| EP1870459A1 (en) | 2005-03-31 | 2007-12-26 | Chugai Seiyaku Kabushiki Kaisha | Methods for producing polypeptides by regulating polypeptide association |
| WO2006121168A1 (en) | 2005-05-09 | 2006-11-16 | Ono Pharmaceutical Co., Ltd. | Human monoclonal antibodies to programmed death 1(pd-1) and methods for treating cancer using anti-pd-1 antibodies alone or in combination with other immunotherapeutics |
| US8008449B2 (en) | 2005-05-09 | 2011-08-30 | Medarex, Inc. | Human monoclonal antibodies to programmed death 1 (PD-1) and methods for treating cancer using anti-PD-1 antibodies alone or in combination with other immunotherapeutics |
| WO2007005874A2 (en) | 2005-07-01 | 2007-01-11 | Medarex, Inc. | Human monoclonal antibodies to programmed death ligand 1 (pd-l1) |
| US8383796B2 (en) | 2005-07-01 | 2013-02-26 | Medarex, Inc. | Nucleic acids encoding monoclonal antibodies to programmed death ligand 1 (PD-L1) |
| WO2007110205A2 (en) | 2006-03-24 | 2007-10-04 | Merck Patent Gmbh | Engineered heterodimeric protein domains |
| WO2007147901A1 (en) | 2006-06-22 | 2007-12-27 | Novo Nordisk A/S | Production of bispecific antibodies |
| WO2008156712A1 (en) | 2007-06-18 | 2008-12-24 | N. V. Organon | Antibodies to human programmed death receptor pd-1 |
| US8354509B2 (en) | 2007-06-18 | 2013-01-15 | Msd Oss B.V. | Antibodies to human programmed death receptor PD-1 |
| WO2009080251A1 (en) | 2007-12-21 | 2009-07-02 | F. Hoffmann-La Roche Ag | Bivalent, bispecific antibodies |
| WO2009080252A1 (en) | 2007-12-21 | 2009-07-02 | F. Hoffmann-La Roche Ag | Bivalent, bispecific antibodies |
| WO2009089004A1 (en) | 2008-01-07 | 2009-07-16 | Amgen Inc. | Method for making antibody fc-heterodimeric molecules using electrostatic steering effects |
| WO2009101611A1 (en) | 2008-02-11 | 2009-08-20 | Curetech Ltd. | Monoclonal antibodies for tumor treatment |
| WO2009114335A2 (en) | 2008-03-12 | 2009-09-17 | Merck & Co., Inc. | Pd-1 binding proteins |
| WO2010027828A2 (en) | 2008-08-25 | 2010-03-11 | Amplimmune, Inc. | Pd-1 antagonists and methods of use thereof |
| WO2010027827A2 (en) | 2008-08-25 | 2010-03-11 | Amplimmune, Inc. | Targeted costimulatory polypeptides and methods of use to treat cancer |
| WO2010027423A2 (en) | 2008-08-25 | 2010-03-11 | Amplimmune, Inc. | Compositions of pd-1 antagonists and methods of use |
| WO2010077634A1 (en) | 2008-12-09 | 2010-07-08 | Genentech, Inc. | Anti-pd-l1 antibodies and their use to enhance t-cell function |
| WO2010129304A2 (en) | 2009-04-27 | 2010-11-11 | Oncomed Pharmaceuticals, Inc. | Method for making heteromultimeric molecules |
| WO2011090754A1 (en) | 2009-12-29 | 2011-07-28 | Emergent Product Development Seattle, Llc | Polypeptide heterodimers and uses thereof |
| US20110196150A1 (en) | 2010-02-11 | 2011-08-11 | Hon-Wah Man | Arylmethoxy Isoindoline Derivatives and Compositions Comprising and Methods of Using the Same |
| WO2011143545A1 (en) | 2010-05-14 | 2011-11-17 | Rinat Neuroscience Corporation | Heterodimeric proteins and methods for producing and purifying them |
| US8877899B2 (en) | 2010-09-27 | 2014-11-04 | Morphosys Ag | Anti-CD38 antibody and lenalidomide or bortezomib for the treatment of multipe myeloma and NHL |
| WO2012058768A1 (en) | 2010-11-05 | 2012-05-10 | Zymeworks Inc. | Stable heterodimeric antibody design with mutations in the fc domain |
| US8362211B2 (en) | 2010-12-30 | 2013-01-29 | Takeda Pharmaceutical Company Limited | Anti-CD38 antibodies |
| WO2013019906A1 (en) | 2011-08-01 | 2013-02-07 | Genentech, Inc. | Methods of treating cancer using pd-1 axis binding antagonists and mek inhibitors |
| WO2013096291A2 (en) | 2011-12-20 | 2013-06-27 | Medimmune, Llc | Modified polypeptides for bispecific antibody scaffolds |
| WO2013157953A1 (en) | 2012-04-20 | 2013-10-24 | Merus B.V. | Methods and means for the production of ig-like molecules |
| WO2013157954A1 (en) | 2012-04-20 | 2013-10-24 | Merus B.V. | Methods and means for the production of ig-like molecules |
| WO2015026634A1 (en) | 2013-08-20 | 2015-02-26 | Merck Sharp & Dohme Corp. | Treating cancer with a combination of a pd-1 antagonist and dinaciclib |
| US20150246123A1 (en) | 2014-02-28 | 2015-09-03 | Janssen Biotech, Inc. | Combination Therapies with Anti-CD38 Antibodies |
| WO2016007235A1 (en) | 2014-07-11 | 2016-01-14 | Genentech, Inc. | Anti-pd-l1 antibodies and diagnostic uses thereof |
| EP2982692A1 (en) * | 2014-08-04 | 2016-02-10 | EngMab AG | Bispecific antibodies against CD3epsilon and BCMA |
| WO2016040238A1 (en) | 2014-09-08 | 2016-03-17 | Celgene Corporation | Methods for treating a disease or disorder using oral formulations of cytidine analogs in combination with an anti-pd1 or anti-pdl1 monoclonal antibody |
| WO2017021450A1 (en) | 2015-08-03 | 2017-02-09 | Engmab Ag | Monoclonal antibodies against bcma |
| WO2020261093A1 (en) * | 2019-06-24 | 2020-12-30 | Novartis Ag | Dosing regimen and combination therapies for multispecific antibodies targeting b-cell maturation antigen |
| WO2021092056A1 (en) * | 2019-11-05 | 2021-05-14 | Engmab Sàrl | Methods of treatment with antibodies against bcma and cd3 |
| WO2022146947A1 (en) * | 2020-12-28 | 2022-07-07 | Bristol-Myers Squibb Company | Antibody compositions and methods of use thereof |
| WO2023086817A1 (en) * | 2021-11-10 | 2023-05-19 | Janssen Biotech, Inc. | Stable formulations comprising a bispecific bcma/cd3 antibody |
Non-Patent Citations (28)
| Title |
|---|
| CHAN ACCARTER PJ, NATURE REVIEWS IMMUNOLOGY, vol. 10, 2010, pages 301 - 316 |
| CHOTHIALESK: "J. Mol. Biol.", vol. 196, 1987, article "Canonical Structures for the Hypervariable Regions of Immunoglobulins", pages: 901 - 917 |
| CUESTA AM ET AL., TRENDS BIOTECH, vol. 28, 2011, pages 355 - 362 |
| DAVIS ET AL., PROTEIN ENG DES SEL, vol. 23, no. 4, 2010, pages 195 - 202 |
| DESPLANCQ DK ET AL., ROTEIN ENG, vol. 7, no. 8, August 1994 (1994-08-01), pages 1027 - 33 |
| DISIS ML ET AL., JOURNAL OF CLINICAL ONCOLOGY, vol. 33, no. 15, pages 2015 |
| EDELMAN, G.M. ET AL., PROC. NATL. ACAD. SCI. USA, vol. 63, 1969, pages 78 - 85 |
| FRIED I ET AL., NEURO ONCOL, vol. 16, 2014 |
| GILBERT J. ET AL., JOURNAL FOR IMMUNOTHERAPY OF CANCER, vol. 20153, pages 152 |
| HAMID O ET AL., J CLIN ONCOL, vol. 33, 2015 |
| HOLLIGER P.HUDSON PJ, NATURE BIOTECH, vol. 23, no. 2005, pages 1126 - 1136 |
| KABAT, E. A. ET AL.: "Sequences of Proteins of Immunological Interest", 1991, PUBLIC HEALTH SERVICE, NATIONAL INSTITUTES OF HEALTH |
| KLEIN ET AL., MABS, vol. 8, no. 6, 2016, pages 1010 - 1020 |
| KONTERMANN RE, MABS, vol. 4, 2012, pages 2 1 - 16 |
| LEFRANC ET AL.: "MGT Unique Numbering for Immunoglobulin and Cell Receptor Variable Domains and Ig superfamily V-like domains", DEV. COMP. IMMUNOL., vol. 27, 2003, pages 55 - 77 |
| MACK M ET AL., PNAS, vol. 92, no. 15, 18 July 1995 (1995-07-18), pages 7021 - 7025 |
| MCDERMOTT DF ET AL., JCO, vol. 34, no. 8, 10 March 2016 (2016-03-10), pages 833 - 842 |
| MERCHANT, A.M ET AL., NAT. BIOTECHNOL., vol. 16, 1998, pages 677 - 68 |
| MORRISON, S.L. ET AL., PROC. NATL. ACAD. SCI. USA, vol. 81, 1984, pages 6851 - 6855 |
| NEUBERGER, M.S. ET AL., NATURE, vol. 314, 1985, pages 268 - 270 |
| PAPADOPOULOS KPET, J CLIN ONCOL, vol. 34, pages 2016 |
| PATRICK AOTT PA ET AL., CLINICAL CANCER RESEARCH, vol. 13, pages 0143 |
| POOLE, R.M. DRUGS, vol. 74, 2014, pages 1973 |
| RIDGWAY, J.B. ET AL., PROTEIN ENG, vol. 9, 1996, pages 617 - 621 |
| RIECHMANN, L. ET AL., NATURE, vol. 332, 1988, pages 323 - 327 |
| SEIWERT, T. ET AL., J. CLIN. ONCOL, vol. 32 |
| WHO DRUG INFORMATION, vol. 27, no. 1, 2013, pages 161 - 162 |
| WONG SANDY W ET AL: "P883 ALNUCTAMAB (ALNUC; BMS-986349; CC-93269), A BCMA x CD3 T-CELL ENGAGER, IN PATIENTS (PTS) WITH RELAPSED/REFRACTORY MULTIPLE MYELOMA (RRMM): LATEST RESULTS FROM A PHASE 1 FIRST-IN-HUMAN CLINICAL STUDY Topic: 14. Myeloma and other monoclonal gammopathies -Clinical", 8 August 2023 (2023-08-08), XP093218374, Retrieved from the Internet <URL:https://pmc.ncbi.nlm.nih.gov/articles/PMC10431068/pdf/hs9-7-e1220745.pdf> [retrieved on 20241025] * |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7792333B2 (en) | Methods of treatment using antibodies to BCMA and CD3 | |
| US20220332821A1 (en) | Dosing regimen and combination therapies for multispecific antibodies targeting b-cell maturation antigen | |
| KR20210077725A (en) | Methods of Providing Subcutaneous Administration of Anti-CD38 Antibodies | |
| US11987636B2 (en) | Dosing of a bispecific antibody that binds CD20 and CD3 | |
| WO2021092060A1 (en) | Methods of treatment | |
| JP7783183B2 (en) | Anti-BCMA Therapy in Autoimmune Disorders | |
| WO2025042742A1 (en) | Compositions comprising antibodies that bind bcma and cd3 and methods of treatment | |
| IL254335B (en) | Isolated peptides derived from the b7 ligand dimer interface and uses thereof | |
| US20230172923A1 (en) | Methods of treating cytokine-related adverse events | |
| EA049790B1 (en) | TREATMENT METHODS WITH ANTIBODIES TO BCMA AND CD3 | |
| HK40083315A (en) | Methods of treatment with antibodies against bcma and cd3 | |
| EA050324B1 (en) | ANTI-BCMA ANTIBODY THERAPY IN AUTOIMMUNE DISORDERS | |
| WO2025222102A1 (en) | Therapeutic combinations of acalabrutinib and a multispecific antibody to treat b-cell malignancies |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24768439 Country of ref document: EP Kind code of ref document: A1 |