WO2025038857A1 - TETRALINS TARGETING MUTANT HIF-2α - Google Patents
TETRALINS TARGETING MUTANT HIF-2α Download PDFInfo
- Publication number
- WO2025038857A1 WO2025038857A1 PCT/US2024/042509 US2024042509W WO2025038857A1 WO 2025038857 A1 WO2025038857 A1 WO 2025038857A1 US 2024042509 W US2024042509 W US 2024042509W WO 2025038857 A1 WO2025038857 A1 WO 2025038857A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cancer
- compound
- membered heteroaryl
- alkyl
- pharmaceutically acceptable
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4192—1,2,3-Triazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/02—Halogenated hydrocarbons
- A61K31/025—Halogenated hydrocarbons carbocyclic
- A61K31/03—Halogenated hydrocarbons carbocyclic aromatic
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/35—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom
- A61K31/351—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom not condensed with another ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/426—1,3-Thiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/428—Thiazoles condensed with carbocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- HIFs are heterodimeric transcription factors consisting of a common constitutive subunit called the aryl hydrocarbon receptor nuclear translocator (ARNT, or HIF- ⁇ ) and one of three HIF- ⁇ subunits.
- ARNT aryl hydrocarbon receptor nuclear translocator
- the ⁇ -subunits are hydroxylated at conserved proline residues by prolyl-4-hydroxylases, and subsequently targeted for degradation by the von Hippel-Lindau ubiquitin E3 ligase complex.
- HIF- ⁇ accumulates and enters the nucleus to activate the expression of genes that regulate metabolism, angiogenesis, cell proliferation and survival, immune evasion, and inflammatory response.
- HIF-1 ⁇ , HIF-2 ⁇ and the less characterized HIF-3 ⁇ , HIF-1 ⁇ and HIF-2 ⁇ overexpression have been associated with poor clinical outcomes in patients with various cancers.
- HIF-2 ⁇ has been found to be a marker of poor prognosis in glioblastoma, neuroblastoma, head and neck squamous carcinoma, and non-small cell lung cancer.
- Hypoxia is also prevalent in many acute and chronic inflammatory disorders, such as inflammatory bowel disease and rheumatoid arthritis.
- HIF-2 ⁇ inhibitors have been described in the literature, belzutifan being the first approved for the treatment of renal cell carcinoma associated with von-Hippel-Lindau disease, central nervous system hemangioblastomas, and pancreatic neuroendocrine tumors.
- a mutation within the internal cavity of the PASB domain of HIF-2 ⁇ was reported in a patient undergoing treatment with a HIF-2 ⁇ inhibitor. This mutation resulted in an acquired resistance to treatment with the HIF-2 ⁇ inhibitor. This resistance has been associated with a gatekeeper mutation (G323E) in HIF-2 ⁇ , which interferes with drug binding.
- G323E gatekeeper mutation
- HIF-2 ⁇ plays a significant role in cancer, inflammation, and other disorders.
- the present disclosure relates to compounds that inhibit the activity of mutant HIF-2 ⁇ . In some embodiments, the present disclosure relates to compounds that inhibit the activity of G323E mutant HIF-2 ⁇ .
- the compounds are represented by Formula I: (Formula I) or a pharmaceutically acceptable salt thereof, wherein: n is 0, 1, or 2; each R 1 when present is independently halo; R 2 and R 3 are independently halo, C1-C6 alkyl or -CN; or R 2 and R 3 combine with the atoms to which they are attached to form a C4-C6 cycloalkyl, wherein said C 4 -C 6 cycloalkyl is unsubstituted or substituted with 1-3 R a ; each R a when present is independently halo or -OH; R 4 is halo, C 1 -C 6 haloalkyl, -CN, -S(O) 2 -R 4a , 4- to 8-membered heterocycloalkyl, or 5- to 10-membered heteroaryl; wherein said 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatom or heteroatom groups independently selected from N, O, S, and S(O
- this disclosure is directed to methods of inhibiting the activity of mutant HIF-2 ⁇ (e.g., G323E mutant HIF-2 ⁇ ) in a subject comprising administering to the subject an effective amount of a compound or pharmaceutically acceptable salt thereof described herein.
- this disclosure provides methods for treating a disease, disorder, and/or condition mediated at least in part by the activity of mutant HIF-2 ⁇ (e.g., G323E mutant HIF-2 ⁇ ) in a subject, comprising administering to the subject a therapeutically effective amount of a compound or pharmaceutically acceptable salt thereof described herein.
- mutant HIF-2 ⁇ e.g., G323E mutant HIF-2 ⁇
- this disclosure provides a method of treating a subject with resistance (e.g., partial or complete) to treatment with a HIF-2 ⁇ inhibitor, said method comprising administering a compound or a pharmaceutically acceptable salt thereof to a subject in need thereof.
- Certain aspects of the present disclosure further comprise the administration of one or more additional therapeutic agents as set forth herein below.
- the term “about” refers to the usual error range for the respective value readily known to the skilled person in this technical field. If the degree of approximation is not otherwise clear from the context, “about” means either within plus or minus 10% of the provided value, or rounded to the nearest significant figure, in all cases inclusive of the provided value. Where ranges are provided, they are inclusive of the boundary values.
- Alkyl can include any number of carbons, such as C1-2, C1-3, C1-4, C1-5, C1-6, C1-7, C1-8, C1-9, C1-10, C2-3, C2-4, C2-5, C2-6, C3-4, C3-5, C3-6, C4-5, C4-6, and C5-6.
- alkyl groups include methyl (Me), ethyl (Et), n-propyl, isopropyl, n- butyl, t-butyl, isobutyl, sec-butyl, n-pentyl, n-hexyl, n-heptyl, n-octyl, and the like.
- alkylene refers to a straight or branched, saturated, aliphatic radical having the number of carbon atoms indicated, and linking at least two other groups, i.e., a divalent hydrocarbon radical.
- the two moieties linked to the alkylene can be linked to the same atom or different atoms of the alkylene group.
- a straight chain alkylene can be the bivalent radical of -(CH2)n-, where n is 1, 2, 3, 4, 5, or 6.
- Representative alkylene groups include, but are not limited to, methylene, ethylene, propylene, isopropylene, butylene, isobutylene, sec-butylene, pentylene and hexylene.
- Alkylene groups in some embodiments, can be substituted or unsubstituted. When a group comprising an alkylene is optionally substituted, it is understood that the optional substitutions may be on the alkylene portion of the moiety.
- cycloalkyl “carbocycle,” or “carbocyclic ring” refers to a hydrocarbon ring having the indicated number of ring atoms (e.g., C 3-6 cycloalkyl) and being fully saturated or having no more than one double bond between ring vertices.
- Cycloalkyl is also meant to refer to bicyclic and polycyclic hydrocarbon rings such as, for example, bicyclo[2.2.1]heptane, bicyclo[2.2.2]octane, etc.
- the cycloalkyl compounds of the present disclosure are monocyclic C 3-6 cycloalkyl moieties (e.g., cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl).
- heterocycloalkyl refers to a cycloalkyl ring having the indicated number of ring vertices ( or members) and having from one to five heteroatoms selected from N, O, and S, which replace one to five of the carbon vertices, and wherein the nitrogen and sulfur atoms are optionally oxidized, and the nitrogen atom(s) are optionally quaternized.
- the heterocycloalkyl may be a monocyclic, a bicyclic or a polycyclic ring system, and may have one or two double bonds connecting ring vertices.
- heterocycloalkyl groups include pyrrolidine, imidazolidine, pyrazolidine, butyrolactam, valerolactam, imidazolidinone, hydantoin, dioxolane, phthalimide, piperidine, 1,4- dioxane, morpholine, thiomorpholine, thiomorpholine-S-oxide, thiomorpholine-S,S-oxide, piperazine, pyran, pyridone, 3-pyrroline, thiopyran, pyrone, tetrahydrofuran, tetrahydrothiophene, quinuclidine, and the like.
- a heterocycloalkyl group can be attached to the remainder of the molecule through a ring carbon or a heteroatom.
- the heterocycle is a 5- to 6-membered heterocycle (e.g., pyrrolidine, tetrahydrofuran, tetrahydropyran, piperidine, piperazine, morpholine, and the like).
- a wavy line, “ ”, that intersects a single, double or triple bond in any chemical structure depicted herein, represents that the point of attachment of the single, double, or triple bond to the remainder of the molecule.
- a bond extending to the center of a ring is meant to indicate attachment at any of the available ring vertices.
- a representation is meant to include either orientation (forward or reverse).
- the group “-C(O)NH-” is meant to include a linkage in either orientation: -C(O)NH- or -NHC(O)-, and similarly, "-O-CH2CH2-” is meant to include both -O-CH2CH2- and -CH2CH2-O-.
- halo or “halogen,” by themselves or as part of another substituent, mean, unless otherwise stated, a fluorine, chlorine, bromine, or iodine atom. Additionally, terms such as “haloalkyl,” are meant to include monohaloalkyl and polyhaloalkyl. For example, the term “C 1 -C 4 haloalkyl” is meant to include trifluoromethyl, 2,2,2-trifluoroethyl, 4-chlorobutyl, 3- bromopropyl, and the like.
- aryl means, unless otherwise stated, an aromatic, hydrocarbon group which can be a single ring or multiple rings (up to three rings) which are fused together or linked covalently.
- Non-limiting examples of aryl groups include phenyl, naphthyl, and biphenyl.
- the term is also meant to include fused cycloalkylphenyl, and heterocycloalkylphenyl ring systems such as, for example, indane, tetrahydronaphthalene, chromane, and isochromane rings.
- the point of attachment to the remainder of the molecule, for a fused ring system can be through a carbon atom on the aromatic portion, a carbon atom on the cycloalkyl portion, or an atom on the heterocycloalkyl portion.
- the aryl groups are phenyl.
- heteroaryl refers to monocyclic or fused bicyclic aryl groups (or rings) that contain from one to five heteroatoms selected from N, O, and S, wherein the nitrogen and sulfur atoms are optionally oxidized, and the nitrogen atom(s) are optionally quaternized.
- heteroaryl group can be attached to the remainder of the molecule through a heteroatom, when chemically permissible.
- heteroaryl also embraces heteroaryl groups fused to phenyl rings.
- the heteroaryl group may be attached to the remainder of the molecule via the heteroaryl ring portion or the phenyl ring portion of the fused bicyclic heteroaryl group.
- heteroaryl groups include pyridyl, pyridazinyl, pyrazinyl, pyrimindinyl, triazinyl, quinolinyl, quinoxalinyl, quinazolinyl, cinnolinyl, phthalazinyl, benzotriazinyl, purinyl, benzimidazolyl, benzopyrazolyl, benzotriazolyl, benzisoxazolyl, benzoxazolyl, isobenzofuryl, isoindolyl, indolizinyl, benzotriazinyl, thienopyridinyl, thienopyrimidinyl, pyrrolopyridinyl, pyrazolopyrimidinyl, imidazolopyridinyl, imidazolopyridazinyl, benzothiazolyl, benzofuranyl, benzothienyl, indolyl, quinoly
- heteroaryl groups are 5- to 9-membered heteroaryl groups having 1-3 ring heteroatoms independently selected from N, O and S (e.g., imidazolyl, pyrazolyl, triazolyl, thiazolyl, pyridinyl, pyrimidinyl, pyrrolopyridinyl, imidazolopyridinyl, imidazolopyridazinyl, benzothiazolyl, etc.).
- alkyl e.g., “alkyl,” “aryl” and “heteroaryl”
- Selected substituents for each type of radical are provided below.
- R', R", and R"' each independently refer to hydrogen, unsubstituted C 1-8 alkyl, unsubstituted aryl, aryl substituted with 1-3 halogens, C 1-8 alkoxy, C 1-8 thioalkoxy groups, or unsubstituted aryl-C 1-4 alkyl groups.
- R' and R" When R' and R" are attached to the same nitrogen atom, they can be combined with the nitrogen atom to form a 3-, 4-, 5-, 6-, or 7-membered ring.
- -NR'R is meant to include 1-pyrrolidinyl and 4-morpholinyl.
- R', R" and R"' each independently refer to hydrogen, unsubstituted C1-8 alkyl, unsubstituted aryl, aryl substituted with 1-3 halogens, C1-8 alkoxy, C1-8 thioalkoxy groups, or unsubstituted aryl-C1-4 alkyl groups.
- substituents include each of the above aryl substituents attached to a ring atom by an alkylene tether of from 1-6 carbon atoms.
- Two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -T-C(O)-(CH2)q-U-, wherein T and U are independently -NH-, -O-, -CH 2 - or a single bond, and q is an integer of from 0 to 2.
- two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -A-(CR f R g )r-B-, wherein A and B are independently -CH 2 -, -O-, -NH-, -S-, -S(O)-, -S(O) 2 -, -S(O) 2 NR'-, or a single bond, r is an integer of from 1 to 3, and R f and R g are each independently H or halogen.
- One of the single bonds of the new ring so formed may optionally be replaced with a double bond.
- two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -(CH 2 ) s -X-(CH 2 ) t -, where s and t are independently integers of from 0 to 3, and X is -O-, -NR'-, -S-, -S(O)-, -S(O)2-, or -S(O)2NR' -.
- the substituent R' in -NR'- and -S(O)2NR'- is selected from hydrogen or unsubstituted C1-6 alkyl.
- heteroatom is meant to include oxygen (O), nitrogen (N), sulfur (S), and silicon (Si).
- the compounds of the present disclosure can be present in their neutral form, or as a pharmaceutically acceptable salt, isomer, polymorph or solvate thereof, and may be present in a crystalline form, amorphous form, or mixtures thereof.
- pharmaceutically acceptable salt is meant to include salts of the compounds according to this disclosure that are prepared with relatively nontoxic acids or bases, depending on the particular substituents found on the compounds described herein.
- base addition salts can be obtained by contacting the neutral form of such compounds with a sufficient amount of the desired base, either neat or in a suitable inert solvent.
- salts derived from pharmaceutically acceptable inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic, manganous, potassium, sodium, zinc, and the like.
- Salts derived from pharmaceutically-acceptable organic bases include salts of primary, secondary and tertiary amines, including substituted amines, cyclic amines, naturally-occurring amines and the like, such as arginine, betaine, caffeine, choline, N,N’-dibenzylethylenediamine, diethylamine, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine and the like.
- acid addition salts can be obtained by contacting the neutral form of such compounds with a sufficient amount of the desired acid, either neat or in a suitable inert solvent.
- pharmaceutically acceptable acid addition salts include those derived from inorganic acids like hydrochloric, hydrobromic, nitric, carbonic, monohydrogencarbonic, phosphoric, monohydrogenphosphoric, dihydrogenphosphoric, sulfuric, monohydrogensulfuric, hydriodic, or phosphorous acids and the like, as well as the salts derived from relatively nontoxic organic acids like acetic, propionic, isobutyric, malonic, benzoic, succinic, suberic, fumaric, mandelic, phthalic, benzenesulfonic, p-tolylsulfonic, citric, tartaric, methanesulfonic, maleic, oxalic, trans- cinnamic, and the like.
- salts of amino acids such as arginate and the like, and salts of organic acids like glucuronic or galactunoric acids, and the like (see, for example, Berge, S.M., et al, “Pharmaceutical Salts”, Journal of Pharmaceutical Science, 1977, 66, 1-19).
- Certain specific compounds of the present disclosure contain both basic and acidic functionalities that allow the compounds to be converted into either base or acid addition salts.
- the neutral forms of the compounds may be regenerated by contacting the salt with a base or acid and isolating the parent compound in the conventional manner.
- the parent form of the compound differs from the various salt forms in certain physical properties, such as solubility in polar solvents, but otherwise the salts are equivalent to the parent form of the compound for the purposes of the present disclosure.
- This disclosure also contemplates isomers of the compounds described herein (e.g., stereoisomers, and atropisomers).
- certain compounds of the present disclosure possess asymmetric carbon atoms (chiral centers), or hindered rotation about a single bond; the racemates, diastereomers, enantiomers, and atropisomers (e.g., R a , S a , P, and M isomers) of which are all intended to be encompassed within the scope of the present disclosure.
- Stereoisomeric forms may be defined, in terms of absolute stereochemistry, as (R) or (S), and/or depicted uses dashes and/or wedges.
- a stereochemical depiction e.g., using dashes, , and/or wedges,
- a stereochemical assignment e.g., using (R) and (S) notation
- is made in a chemical name it is meant to indicate that the depicted isomer is present and substantially free of one or more other isomer(s) (e.g., enantiomers and diastereomers, when present).
- “Substantially free of” other isomer(s) indicates at least an 70/30 ratio of the indicated isomer to the other isomer(s), more preferably 80/20, 90/10, or 95/5 or more. In some embodiments, the indicated isomer will be present in an amount of at least 99%.
- a chemical bond to an asymmetric carbon that is depicted as a solid line ( ) indicates that all possible stereoisomers (e.g., enantiomers, diastereomers, racemic mixtures, etc.) at that carbon atom are included. In such instances, the compound may be present as a racemic mixture, scalemic mixture, or a mixture of diastereomers.
- the compounds of the present disclosure may also contain unnatural proportions of atomic isotopes at one or more of the atoms that constitute such compounds.
- Unnatural proportions of an isotope may be defined as ranging from the amount found in nature to an amount consisting of 100% of the atom in question.
- the compounds may incorporate radioactive isotopes, such as for example tritium ( 3 H), iodine-125 ( 125 I), or carbon-14 ( 14 C), or non-radioactive isotopes, such as deuterium ( 2 H) or carbon-13 ( 13 C).
- radioactive isotopes such as for example tritium ( 3 H), iodine-125 ( 125 I), or carbon-14 ( 14 C), or non-radioactive isotopes, such as deuterium ( 2 H) or carbon-13 ( 13 C).
- isotopic variations can provide additional utilities to those described elsewhere herein.
- isotopic variants of the compounds of the disclosure may find additional utility, including but not limited to, as diagnostic and/or imaging reagents, or as cytotoxic/radiotoxic therapeutic agents. Additionally, isotopic variants of the compounds of the disclosure can have altered pharmacokinetic and pharmacodynamic characteristics which can contribute to enhanced safety, tolerability, or efficacy during treatment. All isotopic variations of the compounds of the present disclosure, whether radioactive or not, are intended to be encompassed within the scope of the present disclosure. In some embodiments, the compounds according to this disclosure are characterized by one or more deuterium atoms.
- administering includes any route of introducing or delivering to a subject a compound to perform its intended function. Administration can be carried out by any suitable route, including but not limited to, orally, intranasally, parenterally (intravenously, intramuscularly, intraperitoneally, or subcutaneously), rectally, intrathecally, intratumorally or topically. Administration includes self- administration and the administration by another. In one embodiment, administration is oral.
- treat refers to a course of action that eliminates, reduces, suppresses, mitigates, ameliorates, or prevents the worsening of, either temporarily or permanently, a disease, disorder, or condition to which the term applies, or at least one of the symptoms associated therewith.
- Treatment includes alleviation of symptoms, diminishment of extent of disease, inhibiting (e.g., arresting the development or further development of the disease, disorder or condition or clinical symptoms association therewith) an active disease, delaying or slowing of disease progression, improving the quality of life, and/or prolonging survival of a subject as compared to expected survival if not receiving treatment or as compared to a published standard of care therapy for a particular disease.
- the term “in need of treatment” as used herein refers to a judgment made by a physician or similar professional that a subject requires or will benefit from treatment. This judgment is made based on a variety of factors that are in the realm of the physician’s expertise, which may include a positive diagnosis of a disease, disorder, or condition.
- the terms “prevent”, “preventing”, “prevention”, “prophylaxis”, and the like refer to a course of action initiated in a manner (e.g., prior to the onset of a disease, disorder, condition, or symptom thereof) so as to prevent, suppress, inhibit or reduce, either temporarily or permanently, a subject’s risk of developing a disease, disorder, condition, or the like (as determined by, for example, the absence of clinical symptoms) or delaying the onset thereof, generally in the context of a subject predisposed to having a particular disease, disorder, or condition. In certain instances, the terms also refer to slowing the progression of the disease, disorder, or condition or inhibiting progression thereof to a harmful or otherwise undesired state.
- Prevention also refers to a course of action initiated in a subject after the subject has been treated for a disease, disorder, condition, or a symptom associated therewith in order to prevent relapse of that disease, disorder, condition, or symptom.
- the term “in need of prevention” as used herein refers to a judgment made by a physician or other caregiver that a subject requires or will benefit from preventative care. This judgment is made based on a variety of factors that are in the realm of a physician’s or caregiver’s expertise.
- “Substantially pure” indicates that a component (e.g., a compound according to this disclosure) makes up greater than about 50% of the total content of the composition, and typically greater than about 60% of the total content.
- “substantially pure” refers to compositions in which at least 75%, at least 85%, at least 90%, or more of the total composition is the component of interest. In some cases, the component of interest will make up greater than about 90%, or greater than about 95%, of the total content of the composition.
- the term “mutant HIF-2 ⁇ ” as used herein refers to human HIF-2 ⁇ protein that is characterized by one or more amino acid mutations in its sequence. Exemplary amino acid mutations include amino acid substitutions, insertions, or deletions. In some embodiments, the one or more mutations occur in the binding pocket within the PAS-B domain of the HIF-2 ⁇ protein.
- the mutation occurs as a substitution, insertion, or deletion of one or more amino acids listed in Table 1.
- Table 1 Wild Type HIF-2 ⁇ Binding Pocket Within the PAS-B Domain
- G323E mutant HIF-2 ⁇ refers to human HIF-2 ⁇ protein with a G323E amino acid substitution, i.e., glycine (G) at position 323 of the HIF-2 ⁇ protein is substituted with glutamic acid (E).
- Mutant HIF-2 ⁇ may be identified by nucleic acid sequencing methods known in the art. For example, mutations can be detected using polymerase chain reaction (PCR).
- PCR steps amplify DNA regions of interest and multiplex single-base primer extension with dideoxynucleotides. Extension products can then be analyzed by, for example, mass spectrometry, which can distinguish different bases according to their mass-to-charge (m/z) ratio (Sequenom Mass Spectrometry system) or by capillary electrophoresis, which distinguishes bases by size and by the color of fluorescently labeled nucleotides (Applied Biosystems SNaPshot system). See Fumagalli et al., BMC Cancer, 10:101 (2010). Protein-based methods may also be used, including but not limited to sequencing HIF-2 ⁇ protein purified (partially or completely) from a suitable sample.
- G323E mutant HIF-2 ⁇ may be identified, for example by whole genome sequencing or targeted sequencing of the HIF-2 ⁇ gene (EPAS1) and identification of a c.968G >A heterozygous mutation resulting in a G323E substitution, or by transcriptome analysis.
- EAS1 whole genome sequencing or targeted sequencing of the HIF-2 ⁇ gene
- c.968G >A heterozygous mutation resulting in a G323E substitution or by transcriptome analysis.
- Compounds of the Disclosure [0046] The present disclosure relates to compounds that inhibit the activity of mutant HIF-2 ⁇ (e.g., G323E mutant HIF-2 ⁇ ), as well as compositions, methods, and uses including such compounds.
- the present disclosure is directed to a compound having a structure according to Formula I: (Formula I) or a pharmaceutically acceptable salt thereof, wherein: n is 0, 1, or 2; each R 1 when present is independently halo; R 2 and R 3 are independently halo, C1-C6 alkyl or -CN; or R 2 and R 3 combine with the atoms to which they are attached to form a C4-C6 cycloalkyl, wherein said C 4 -C 6 cycloalkyl is unsubstituted or substituted with 1-3 R a ; each R a when present is independently halo or -OH; R 4 is halo, C 1 -C 6 haloalkyl, -CN, -S(O) 2 -R 4a , 4- to 8-membered heterocycloalkyl, or 5- to 10-membered heteroaryl; wherein said 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatom
- n is 1 or 2 and at least one R 1 is F. In some embodiments, n is be 1 and R 1 is F. In some embodiments, n is 2 and both R 1 groups are independently F. In some embodiments, n is 1 or 2 and at least one R 1 is Cl. In some embodiments, n is 1 or 2 and at least one R 1 is Br. In some embodiments, n is 1 or 2 and at least one R 1 is I. In some embodiments, n is 2 and both R 1 groups are the same type of halo group. In some embodiments, n is 2 and each R 1 is a different type of halo. In some embodiments, n is 0 and R 1 is not present.
- R 2 is selected from the group consisting of halo, C1-C6 alkyl or -CN. In some embodiments, R 2 is halo. In some embodiments, R 2 is F. In some embodiments, R 2 is Cl. In some embodiments, R 2 is Br. In some embodiments, R 2 is I. In some embodiments, R 2 is a C1-C6 alkyl. In some embodiments, R 2 is methyl. In some embodiments, R 2 is -CN. [0050] In some embodiments, R 3 is selected from the group consisting of halo, C 1 -C 6 alkyl, and -CN. In some embodiments, R 3 is halo. In some embodiments, R 3 is F.
- R 3 is Cl. In some embodiments, R 3 is Br. In some embodiments, R 3 is I. In some embodiments, R 3 is C 1 -C 6 alkyl. In some embodiments, R 3 is -CN. [0051] In some embodiments, R 2 is C1-C6 alkyl and R 3 is -CN. In some embodiments, R 2 is methyl and R 3 is -CN. In some embodiments, R 2 is -CN and R 3 is C1-C6 alkyl. In some embodiments, R 2 is C 1 -C 6 alkyl and R 3 is halo. In some embodiments, R 2 is halo and R 3 is a C 1 - C 6 alkyl.
- R 2 is -CN and R 3 is halo. In some embodiments, R 2 is halo and R 3 is -CN. [0052] In some embodiments, R 2 and R 3 combine with the atoms to which they are attached to form a C 4 -C 6 cycloalkyl. In some embodiments, the C 4 -C 6 cycloalkyl is unsubstituted. In some embodiments, R 2 and R 3 combine to form an unsubstituted cyclobutyl. In some embodiments, R 2 and R 3 combine to form an unsubstituted cyclopentyl. In some embodiments, R 2 and R 3 combine to form an unsubstituted cyclohexyl.
- R 2 and R 3 combine with the atoms to which they are attached to form a C4-C6 cycloalkyl substituted with 1-3 R a .
- the C4-C6 cycloalkyl is substituted with 2-3 R a .
- the C 4 -C 6 cycloalkyl is substituted with 1 R a .
- the C 4 -C 6 cycloalkyl is substituted with 2 R a .
- the C 4 - C6 cycloalkyl is substituted with 3 R a .
- R 2 and R 3 combine to form a C4 cycloalkyl substituted with 1-3 R a or 2-3 R a . In some embodiments, R 2 and R 3 combine to form a C 5 cycloalkyl substituted with 1-3 R a or 2-3 R a . In some embodiments, R 2 and R 3 combine to form a C6 cycloalkyl substituted with 1-3 R a or 2-3 R a .
- each R a is independently halo or -OH. In some embodiments, at least one R a is halo. In some embodiments, at least one R a is fluoro. In some embodiments, at least one R a is chloro.
- At least one R a is bromo. In some embodiments, at least one R a is iodo. In some embodiments, at least one R a is -OH. In some embodiments, at least one R a is fluoro and at least one R a is-OH. In some embodiments, each R a is independently halo. In some embodiments, each R a is independently -OH.
- R 4 is selected from the group consisting of halo, C 1 -C 6 haloalkyl, -CN, -S(O)2-R 4a , 4- to 8-membered heterocycloalkyl, and 5- to 10-membered heteroaryl; wherein said 4- to 8-membered heterocycloalkyl has 1-4 ring heteroatom or heteroatom groups or 1-3 heteroatom or heteroatom groups independently selected from N, O, S, and S(O) 2 , and said 4- to 8-membered heterocycloalkyl is unsubstituted or substituted with 1-3 R b ; and wherein said 5- to 10-membered heteroaryl has 1-4 ring heteroatoms or 1-3 ring heteroatoms independently selected from N, O, and S, and said 5- to 10-membered heteroaryl is unsubstituted or substituted with 1-3 R b ; wherein R 4a is C 1 -C 3 alkyl, C 1 -C 3 hal
- At least one ring heteroatom of the 4- to 8-membered heterocycloalkyl or 5- to 10- membered heteroaryl is N. In some embodiments, at least one ring heteroatom of the 4- to 8- membered heterocycloalkyl or 5- to 10-membered heteroaryl is O. In some embodiments, at least one ring heteroatom in the 4- to 8-membered heterocycloalkyl or 5- to 10-membered heteroaryl is S. In some embodiments, the 4- to 8-membered heterocycloalkyl or 5- to 10- membered heteroaryl has at least two ring heteroatoms and at least two ring heteroatoms are different types of heteroatoms.
- the 4- to 8-membered heterocycloalkyl or 5- to 10-membered heteroaryl has at least two ring heteroatoms and at least two ring heteroatoms are the same type of heteroatom.
- R 4 is halo, -S(O) 2 -R 4a , 4- to 8-membered heterocycloalkyl, or 5- to 10-membered heteroaryl; wherein said 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatom or heteroatom groups independently selected from N, O, S, and S(O)2; said 5- to 10- membered heteroaryl has 1-3 ring heteroatoms independently selected from N, O, and S; and said 4- to 8-membered heterocycloalkyl and 5- to 10-membered heteroaryl are unsubstituted, or substituted with 1-3 R b .
- R 4 is halo or a 5- to 6-membered heteroaryl having 1-3 ring heteroatoms independently selected from N, O, and S; wherein said 5- to 6-membered heteroaryl is unsubstituted, or substituted with 1-2 R b .
- R b is independently -CN, C1- C6 alkyl, -N(R c )2, or -C(O)-N(R c )2; and each R c is independently -H or C1-C6 alkyl.
- R 4 is a 5- to 10-membered heteroaryl, 5- to 8-membered heteroaryl, or 5- to 6-membered heteroaryl, wherein said 5- to 10-membered heteroaryl, 5- to 8- membered heteroaryl, or 5- to 6-membered heteroaryl has 1-3 ring heteroatoms independently selected from N, O, and S, and the heteroaryl is unsubstituted.
- R 4 is a 5- to 10-membered heteroaryl, 5- to 8-membered heteroaryl, or 5- to 6-membered heteroaryl, wherein said 5- to 10-membered heteroaryl, 5- to 8-membered heteroaryl, or 5- to 6-membered heteroaryl has 1-3 ring heteroatoms independently selected from N, O, and S, and the heteroaryl is substituted with 1-2 R b , wherein R b is independently -CN, C 1 -C 6 alkyl, -N(R c ) 2 , or -C(O)- N(R c )2; and each R c is independently -H or C1-C6 alkyl.
- R 4 is halo, -S(O)2-R 4a , 4- to 8-membered heterocycloalkyl, or 5- to 10-membered heteroaryl, wherein said 4- to 8-membered heterocycloalkyl and 5- to 10- membered heteroaryl have 1-4 ring heteroatoms independently selected from N, O, and S; and wherein said 4- to 8-membered heterocycloalkyl and 5- to 10-membered heteroaryl are unsubstituted or substituted with 1-2 R b .
- R 4a is C 1 -C 3 alkyl, or -NR 4b R 4c , wherein R 4b and R 4c are independently C1-C3 alkyl.
- R 4 is halo, 5- to 6-membered heteroaryl, or 5- to 8-membered heterocycloalkyl; wherein said 5- to 6-membered heteroaryl has 1-3 ring nitrogen atoms; said 5- to 8-membered heterocycloalkyl has 1-2 ring heteroatoms independently selected from N, and O; and said 5- to 6-membered heteroaryl and 5- to 8-membered heterocycloalkyl are unsubstituted or substituted with 1-2 R b .
- R 4 is fluoro.
- R 4 is chloro.
- R 4 is bromo.
- R 4 is iodo.
- R 4 is selected from the group consisting of halo, C1-C6 haloalkyl, -CN, or -S(O)2-R 4a ; wherein R 4a is selected from the group consisting of C1-C3 alkyl, C 1 -C 3 haloalkyl, C 3 -C 6 cycloalkyl, -NR 4b R 4c , and 4- to 6-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S; wherein R 4b and R 4c are each independently -H or C1-C3 alkyl.
- R 4a is C1-C3 alkyl or -NR 4b R 4c , wherein R 4b and R 4c are each independently C 1 -C 3 alkyl.
- R 4a is a 5- to 6- membered heterocycloalkyl having 1-4 ring heteroatoms, 1-3 ring heteroatoms, or 2-3 ring heteroatoms, wherein each heteroatom is independently selected from N, O, and S.
- R 4 is selected from the group consisting of unsubstituted triazolyl, triazolyl substituted with 1-2 R b , unsubstituted imidazolyl, imidazolyl substituted with 1-2 R b , unsubstituted pyrazolyl, pyrazolyl substituted with 1-2 R b , unsubstituted pyridyl, and pyridyl substituted with 1-2 R b .
- each R b when present, is independently -CN, C 1 -C 6 alkyl, -N(R c ) 2 , or -C(O)-N(R c ) 2 ; and each R c is independently -H or C 1 -C 6 alkyl.
- R 4 is an unsubstituted tetrahydropyranyl. In some embodiments, R 4 is tetrahydropyranyl substituted with 1-2 R b .
- R 4 is Cl; -S(O) 2 Me; -S(O) 2 NMe 2 ; a heteroaryl selected from the group consisting of or a heterocycloalkyl selected from the group consisting of wherein each heteroaryl and heterocycloalkyl is unsubstituted, or substituted with 1-2 R b .
- R b is methyl, -NH2, or -C(O)-NH2.
- R 4 is Cl, -S(O)2Me, - [0063] In some embodiments, R 4 is Cl; -S(O)2Me; -S(O)2NMe2; a heteroaryl selected from the g , the group consisting of wherein each heteroaryl and heterocycloalkyl is unsubstituted, or substituted with 1-2 R b . In some embodiments, R b is methyl, -CN, -NH 2 , or -C(O)-NH 2 .
- R 4 is Cl, -S(O) 2 Me, -S(O) 2 NMe 2 ,
- X is CR 5 or N; and R 5 is -H or halo.
- X is CH.
- X is C-halo.
- X is C-F.
- X is N.
- the compound or pharmaceutically acceptable salt thereof is a compound of Formula IA: (Formula IA) where n is 0 or 1.
- the compound or pharmaceutically acceptable salt thereof is a compound of Formula IB: (Formula Ib) where n is 0 or 1; and m is 2 or 3.
- the compound or pharmaceutically acceptable salt thereof according to this disclosure is selected from the compounds provided in Table 2. Table 2.
- the compound or pharmaceutically acceptable salt thereof according to this disclosure is selected from a compound provided in Table 3. Table 3.
- the compound or pharmaceutically acceptable salt thereof according to this disclosure is selected from the compounds provided in Table 4. Table 4.
- the compound according to this disclosure is selected from the compounds provided in any one of Tables 2-4.
- the present disclosure provides methods for using the compounds described herein in the preparation of a medicament for inhibiting mutant HIF-2 ⁇ .
- the mutant HIF-2 ⁇ is G323E mutant HIF-2 ⁇ .
- the terms “inhibit”, ‘inhibition” and the like refer to the ability of an antagonist to decrease the function or activity of a particular target, e.g., G323E mutant HIF-2 ⁇ .
- the decrease is preferably at least a 50% and may be, for example, at least about 55%, at least about 60%, at least about 65%, at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, or at least about 95%.
- the present disclosure provides methods of treating a disease, disorder, and/or condition mediated by mutant HIF-2 ⁇ , said method comprising administering a compound according to this disclosure, or pharmaceutically acceptable salt thereof, to a subject in need thereof.
- the mutant HIF-2 ⁇ is G323E mutant HIF-2 ⁇ .
- Diseases, disorders and/or conditions contemplated are described in further detail below.
- the present disclosure also encompasses the use of the compounds described herein in the preparation of a medicament for the treatment or prevention of diseases, disorders, and/or conditions that would benefit from inhibition of mutant HIF-2 ⁇ (e.g., G323E mutant HIF-2 ⁇ ).
- the present disclosure encompasses the use of the compounds described herein in the preparation of a medicament for the treatment of cancer.
- the compounds described herein are used in combination with at least one additional therapy, examples of which are set forth elsewhere herein.
- diseases, disorders, and/or conditions that would benefit from mutant HIF-2 ⁇ (e.g., G323E mutant HIF-2 ⁇ ) inhibition may be characterized by detectable mutant HIF-2 ⁇ (e.g., G323E mutant HIF-2 ⁇ ) expression in one or more suitable samples (e.g., a tumor biopsy, surgical resection sample, etc.).
- diseases, disorders, and/or conditions that would benefit from mutant HIF-2 ⁇ (e.g., G323E mutant HIF-2 ⁇ ) inhibition may be characterized by increased mutant HIF-2 ⁇ (e.g., G323E HIF-2 ⁇ ) expression in one or more suitable samples (e.g., a tumor biopsy, surgical resection sample, etc.) as compared to a similar sample from (a) a healthy control, (b) a sample taken from the same subject at an earlier timepoint, or (c) a sample from a subject with another disease, disorder and/or condition not responsive to mutant HIF-2 ⁇ (e.g., G323E HIF-2 ⁇ ) inhibition.
- the disease, disorder and/or condition is cancer.
- the disease, disorder and/or condition is Von Hippel-Lindau (VHL) disease (including VHL disease associated with renal cell carcinoma (RCC), central nervous system (CNS) hemangioblastomas, pancreatic neuroendocrine tumors (pNET), or solid tumors).
- VHL Von Hippel-Lindau
- RRC renal cell carcinoma
- CNS central nervous system
- pNET pancreatic neuroendocrine tumors
- compounds described herein are administered to a subject in need thereof in an amount effective to inhibit mutant HIF-2 ⁇ (e.g., G323E mutant HIF-2 ⁇ ).
- Mutant HIF-2 ⁇ (e.g., G323E mutant HIF-2 ⁇ ) activity may be assessed using a peripheral blood sample or a tissue sample (e.g., a tumor sample) obtained from the subject.
- the present disclosure further provides a method of inhibiting mutant HIF-2 ⁇ (e.g., G323E mutant HIF-2 ⁇ ), said method comprising contacting a cell of a subject with a compound according to this disclosure, or a pharmaceutically acceptable salt thereof.
- the cell comprises mutant HIF-2 ⁇ .
- the cell comprises G323E HIF-2 ⁇ .
- the cell is a cancer cell.
- the cancer cell is from a solid tumor, examples of which are provided in further detail below.
- the compounds described herein are administered to a subject in need thereof to treat and/or prevent cancer or a cancer-related disease, disorder or condition.
- the compounds described herein are administered to a subject in need thereof to treat cancer, optionally in combination with at least one additional therapy, examples of which are set forth elsewhere herein.
- the compounds described herein are administered to a subject with von Hippel-Lindau (VHL) disease to treat and/or prevent a VHL- associated disease, disorder or condition.
- VHL von Hippel-Lindau
- the compounds described herein are administered to a subject with von Hippel-Lindau (VHL) disease to treat and/or prevent associated renal cell carcinoma, central nervous system hemangioblastomas, or pancreatic neuroendocrine tumors, optionally wherein immediate surgery is not required.
- VHL von Hippel-Lindau
- the compounds described herein are useful in the treatment and/or prophylaxis of cancer (e.g., carcinomas, sarcomas, leukemias, lymphomas, myelomas, etc.).
- the cancer may be locally advanced and/or unresectable, metastatic, or at risk of becoming metastatic.
- the cancer may be recurrent or no longer responding to a treatment, such as a standard of care treatment known to one of skill in the art.
- a treatment such as a standard of care treatment known to one of skill in the art.
- the cancer is resistant treatment with a previous therapy (e.g., treatment with an inhibitor of HIF-2 ⁇ ).
- Exemplary types of cancer contemplated by this disclosure include cancer of the genitourinary tract (e.g., bladder, kidney, renal cell, penile, prostate, testicular, Von Hippel- Lindau disease, etc.), uterus, cervix, ovary, breast, gastrointestinal tract (e.g., esophagus, oropharynx, stomach, small or large intestines, colon, or rectum), bone, bone marrow, skin (e.g., melanoma), head and neck, liver, gall bladder, bile ducts, heart, lung, pancreas, salivary gland, adrenal gland, thyroid, brain (e.g., gliomas), ganglia, central nervous system (CNS), peripheral nervous system (PNS), the hematopoietic system (i.e., hematological malignancies), and the immune system (e.g., spleen or thymus).
- genitourinary tract e.g., bladder,
- the compounds according to this disclosure are useful in the treatment and/or prophylaxis of hematological malignancies.
- Exemplary types of cancer affecting the hematopoietic system include leukemias, lymphomas and myelomas, including acute myeloid leukemia, adult T-cell leukemia, T-cell large granular lymphocyte leukemia, acute lymphoblastic leukemia, chronic lymphocytic leukemia, chronic myelogenous leukemia, acute monocytic leukemia, Hodgkin’s and Non-Hodgkin’s lymphoma, Diffuse large B Cell lymphoma, and multiple myeloma.
- the compounds according to this disclosure are useful in the treatment and/or prophylaxis of solid tumors.
- the solid tumor may be, for example, ovarian cancer, endometrial cancer, breast cancer, lung cancer (small cell or non-small cell), colon cancer, prostate cancer, cervical cancer, biliary cancer, pancreatic cancer, gastric cancer, esophageal cancer, liver cancer (hepatocellular carcinoma), kidney cancer (renal cell carcinoma), head-and-neck tumors, mesothelioma, melanoma, sarcomas, central nervous system (CNS) hemangioblastomas, and brain tumors (e.g., gliomas, such as astrocytoma, oligodendroglioma and glioblastomas).
- gliomas such as astrocytoma, oligodendroglioma and glioblastomas.
- the compounds according to this disclosure are useful in the treatment and/or prophylaxis of lung cancer, genitourinary cancer, gastrointestinal cancer, or a combination thereof.
- the compounds according to this disclosure are useful in the treatment and/or prophylaxis of gastrointestinal cancer, genitourinary cancer, gynecological cancer, lung cancer, or a combination thereof.
- the compounds according to this disclosure are useful in the treatment of gastrointestinal (GI) cancer.
- the GI cancer is colorectal cancer, pancreatic cancer, or liver cancer.
- the GI cancer is an upper GI cancer, such as esophageal or gastric cancer.
- the upper GI cancer is an adenocarcinoma, a squamous cell carcinoma, or any combination thereof.
- the upper GI cancer is esophageal adenocarcinoma (EAC), esophageal squamous cell carcinoma (ESCC), gastroesophageal junction adenocarcinoma (GEJ), gastric adenocarcinoma (also referred to herein as “gastric cancer”) or any combination thereof.
- EAC esophageal adenocarcinoma
- ESCC esophageal squamous cell carcinoma
- GEJ gastroesophageal junction adenocarcinoma
- gastric cancer gastric adenocarcinoma
- the compounds according to this disclosure are useful in the treatment of pancreatic cancer.
- the pancreatic cancer is pancreatic neuroendocrine tumor or pancreatic adenocarcinoma.
- the compounds according to this disclosure are useful in the treatment and/or prophylaxis of liver cancer.
- the liver cancer is hepatocellular carcinoma.
- the liver cancer is liver metastases.
- the compounds according to this disclosure are useful in the treatment of genitourinary cancer.
- the genitourinary cancer is bladder cancer, kidney cancer or prostate cancer.
- the compounds according to this disclosure are useful in the treatment of kidney cancer.
- the kidney cancer is renal cell carcinoma.
- the renal cell carcinoma is clear cell renal carcinoma.
- the compounds according to this disclosure are useful in the treatment of gynecological cancer.
- the gynecological cancer is breast cancer, endometrial cancer, or ovarian cancer.
- the gynecological cancer is hormone receptor positive (e.g., ER ⁇ -positive cancer, PR-positive cancer, ER ⁇ -positive and PR-positive cancer), HER2 positive cancer, HER2 over-expressing cancer, or any combination thereof.
- the cancer is triple negative cancer (e.g., ER, PR and HER2 negative).
- the compounds according to this disclosure are useful in the treatment of lung cancer.
- the lung cancer is mesothelioma, small cell lung cancer (SCLC) or non-small cell lung cancer (NSCLC).
- SCLC small cell lung cancer
- NSCLC non-small cell lung cancer
- the lung cancer is NSCLC, optionally lung squamous cell carcinoma or lung adenocarcinoma.
- the compounds according to this disclosure are useful in the treatment of a neuroendocrine tumor.
- the neuroendocrine tumor is pancreatic neuroendocrine tumor, pheochromocytoma, paraganglioma, or a tumor of the adrenal gland (e.g., neuroblastoma).
- the compounds according to this disclosure are useful in the treatment of brain cancer.
- the brain cancer is a glioma.
- the glioma is an astrocytoma, an oligodendroglioma, or a glioblastoma.
- the compounds according to this disclosure are useful in the treatment and/or prophylaxis of renal cell carcinoma or hepatocellular carcinoma.
- the methods of the present disclosure may be practiced in an adjuvant setting or neoadjuvant setting. Alternatively or in addition, the methods described herein may be indicated as a first line treatment, optionally in the treatment of locally advanced, unresectable, or metastatic cancer.
- the methods described herein may be indicated as a second line, third line, or greater line of treatment, optionally in the treatment of locally advanced, unresectable, or metastatic cancer.
- an earlier line of therapy may have included an inhibitor of HIF-2 ⁇ .
- cancer-related diseases, disorders and conditions refer broadly to conditions that are associated, directly or indirectly, with cancer and non-cancerous proliferative disease, and includes, e.g., angiogenesis, precancerous conditions such as dysplasia, and non-cancerous proliferative diseases disorders or conditions, such as benign proliferative breast disease and papillomas.
- angiogenesis precancerous conditions
- non-cancerous proliferative diseases disorders or conditions such as benign proliferative breast disease and papillomas.
- the term(s) cancer-related disease, disorder and condition do not include cancer per se.
- the disclosed methods for treating or preventing cancer, or a cancer-related disease, disorder or condition, in a subject in need thereof comprise administering to the subject a compound disclosed here.
- the present disclosure provides methods for treating or preventing cancer, or a cancer-related disease, disorder or condition with a compound disclosed herein and at least one additional therapy, examples of which are set forth elsewhere herein.
- Von-Hippel-Lindau Disease In one or more embodiments, the compounds described herein are useful in the treatment and/or prophylaxis of von Hippel-Lindau (VHL) disease or VHL disease associated tumors.
- the disease, disorder, and/or condition is VHL disease associated with renal cell carcinoma (RCC), central nervous system (CNS) hemangioblastomas, pancreatic neuroendocrine tumors (pNET), or solid tumors).
- RCC renal cell carcinoma
- CNS central nervous system
- pNET pancreatic neuroendocrine tumors
- solid tumors solid tumors
- the methods according to this disclosure may be provided in selected patients, for example subjects identified as having, e.g., detectable mutant HIF-2 ⁇ expression.
- the subject is identified as having an increased mutant HIF-2 ⁇ expression as compared to a suitable control.
- the detectable and/or increased mutant HIF-2 ⁇ expression is measured from one or more suitable samples, including, e.g., a tumor biopsy, or sample obtained by surgical resection.
- a suitable control is selected from the group consisting of a sample from (a) a healthy subject, (b) a sample taken from the same subject at an earlier timepoint, or (c) a sample from a subject with another disease, disorder and/or condition not responsive to mutant HIF-2 ⁇ inhibition.
- the methods according to this disclosure may be provided in selected patients, for example subjects identified as having, e.g., detectable G323E mutant HIF- 2 ⁇ expression.
- the subject is identified as having an increased G323E HIF-2 ⁇ expression as compared to a suitable control.
- the detectable and/or increased G323E HIF-2 ⁇ expression is measured from one or more suitable samples, including, e.g., a tumor biopsy, or sample obtained by surgical resection.
- a suitable control is selected from the group consisting of a sample from (a) a healthy subject, (b) a sample taken from the same subject at an earlier timepoint, or (c) a sample from a subject with another disease, disorder and/or condition not responsive to G323E HIF-2 ⁇ inhibition.
- the methods according to this disclosure may be used in patients identified or previously identified as having a biomarker of hypoxia or pseudohypoxia, microsatellite instability, high tumor mutational burden as measured in a relevant tissue or sample, or malignancies with HIF stabilizing mutations.
- the methods according to this disclosure may be used in patients having VHL disease (e.g., renal cell carcinoma, central nervous system hemangioblastomas, pancreatic neuroendocrine tumors, or solid tumors associated with VHL disease) who have undergone treatment with a HIF-2 ⁇ inhibitor, but have developed resistance to said treatment.
- VHL disease e.g., renal cell carcinoma, central nervous system hemangioblastomas, pancreatic neuroendocrine tumors, or solid tumors associated with VHL disease
- the methods according to this disclosure may be used in patients identified as having partial or complete resistance to treatment with a HIF-2 ⁇ inhibitor.
- the subject having partial or complete resistance to treatment with a HIF-2 ⁇ inhibitor was previously administered a HIF-2 ⁇ inhibitor.
- the patient is no longer responding to treatment with the HIF-2 ⁇ inhibitor, or the extent of the response has diminished.
- the patient has a detectable amount of mutant HIF-2 ⁇ expression.
- the patient has a detectable amount of G323E mutant HIF-2 ⁇ expression.
- the patient has an increased amount of mutant HIF-2 ⁇ expression as compared to a suitable control.
- the patient has an increased amount of G323E mutant HIF-2 ⁇ expression as compared to a suitable control.
- the detectable and/or increased mutant HIF-2 ⁇ expression is measured from one or more suitable samples, including, e.g., a tumor biopsy, or sample obtained by surgical resection.
- a suitable control is selected from the group consisting of a sample from (a) a healthy subject, (b) a sample taken from the same subject at an earlier timepoint, or (c) a sample from a subject with another disease, disorder and/or condition not responsive to mutant HIF-2 ⁇ inhibition (e.g., G323E HIF-2 ⁇ inhibition).
- Combination Therapy [0104] The present disclosure contemplates the use of compounds described herein alone or in combination with one or more additional therapy.
- Each additional therapy can be a therapeutic agent or another treatment modality.
- each agent may target a different, but complementary, mechanism of action.
- the additional therapeutic agents can be small chemical molecules; macromolecules such as proteins, antibodies, peptibodies, peptides, DNA, RNA or fragments of such macromolecules; or cellular or gene therapies.
- Non-limiting examples of additional treatment modalities include surgical resection of a tumor, bone marrow transplant, radiation therapy, and photodynamic therapy.
- the use of a compound described herein in combination with one or more additional therapies may have a synergistic therapeutic or prophylactic effect on the underlying disease, disorder, or condition.
- the combination therapy may allow for a dose reduction of one or more of the therapies, thereby ameliorating, reducing or eliminating adverse effects associated with one or more of the agents.
- the compound can be administered before, after or during treatment with the additional treatment modality.
- the therapeutic agents used in such combination therapy can be formulated as a single composition or as separate compositions. If administered separately, each therapeutic agent in the combination can be given at or around the same time, or at different times.
- the therapeutic agents are administered “in combination” even if they have different forms of administration (e.g., oral capsule and intravenous), they are given at different dosing intervals, one therapeutic agent is given at a constant dosing regimen while another is titrated up, titrated down or discontinued, or each therapeutic agent in the combination is independently titrated up, titrated down, increased or decreased in dosage, or discontinued and/or resumed during a patient’s course of therapy. If the combination is formulated as separate compositions, in some embodiments, the separate compositions are provided together in a kit. Cancer Therapies [0106] The present disclosure contemplates the use of the compounds described herein in combination with one or more additional therapies useful in the treatment of cancer.
- one or more of the additional therapies is an additional treatment modality.
- Exemplary treatment modalities include but are not limited to surgical resection of a tumor, bone marrow transplant, radiation therapy, and photodynamic therapy.
- one or more of the additional therapeutic agents is a chemotherapeutic agent.
- chemotherapeutic agents include, but are not limited to, alkylating agents such as thiotepa and cyclophosphamide; alkyl sulfonates such as busulfan, improsulfan and piposulfan; aziridines such as benzodopa, carboquone, meturedopa, and uredopa; ethylenimines and methylamelamines including altretamine, triethylenemelamine, triethylenephosphoramide, triethylenethiophosphoramide and trimethylolomelamime; nitrogen mustards such as chlorambucil, chlornaphazine, cholophosphamide, estramustine, ifosfamide, mechlorethamine, mechlorethamine oxide hydrochloride, melphalan, novembichin, phenesterine, prednimustine, trofosfamide, uracil mustard; nitrosureas such as carmustine, chlorozotocin
- combination therapy comprises a chemotherapy regimen that includes one or more chemotherapeutic agents.
- combination therapy comprises a chemotherapeutic regimen comprising one or more of FOLFOX (folinic acid, fluorouracil, and oxaliplatin), FOLFIRI (e.g., folinic acid, fluorouracil, and irinotecan), FOLFIRINOX (e.g., fluorouracil, leucovorin, irinotecan, and oxaliplatin), CAPOX (capecitabine and oxaliplatin), a taxoid (e.g., docetaxel, paclitaxel, nab- paclitaxel,etc.), a fluoropyrimidine-containing chemotherapeutic agent (e.g., fluorouracil, capecitabine, floxuridine), a platinum-containing chemotherapeutic agent, and/or gemcitabine.
- FOLFOX folinic acid, fluorouraci
- one or more of the additional therapeutic agents is an inhibitor of a hypoxia-inducible factor (HIF) transcription factor, particularly HIF-2 ⁇ .
- HIF hypoxia-inducible factor
- an inhibitor if HIF-2 ⁇ is administered in a prior line of therapy.
- HIF- 2 ⁇ inhibitors include belzutifan, AND021, BPI-452080, ARO-HIF2, PT-2385, AB521 (casdatifan), NKT-2152 (HS-10516), SMP-215, DFF332, and those described in WO 2021113436, WO 2021188769, and WO 2023077046, each of which is incorporated by reference herein.
- one or more of the additional therapeutic agents is an immune checkpoint inhibitor.
- immune checkpoint inhibitor refers to an antagonist of an inhibitory or co-inhibitory immune checkpoint.
- checkpoint inhibitor checkpoint inhibitor
- CPI CPI
- Immune checkpoint inhibitors may antagonize an inhibitory or co-inhibitory immune checkpoint by interfering with receptor -ligand binding and/or altering receptor signaling.
- immune checkpoints ligands and receptors
- PD-1 programmed cell death protein 1
- PD-L1 PD1 ligand
- BTLA B and T lymphocyte attenuator
- CTLA-4 cytotoxic T-lymphocyte associated antigen 4
- TIM-3 T cell immunoglobulin and mucin domain containing protein 3
- LAG-3 lymphocyte activation gene 3
- TIGIT T cell immunoreceptor with Ig and ITIM domains
- CD276 B7-H3
- PD-L2 Galectin 9, CEACAM-1, CD69, Galectin-1, CD113, GPR56, VISTA, 2B4, CD48, GARP, PD1H, LAIR1, TIM-1, and TIM-4, and Killer Inhibitory Receptors, which can be divided into two classes based on their structural features: i) killer cell immunoglobulin-like receptors (KIRs), and
- an immune checkpoint inhibitor is a CTLA-4 antagonist.
- the CTLA-4 antagonist can be an antagonistic CTLA-4 antibody.
- Suitable antagonistic CTLA-4 antibodies include, for example, monospecific antibodies such as ipilimumab or tremelimumab, as well as bispecific antibodies such as MEDI5752 and KN046.
- an immune checkpoint inhibitor is a PD-1 antagonist.
- the PD-1 antagonist can be an antagonistic PD-1 antibody, small molecule or peptide.
- Suitable antagonistic PD-1 antibodies include, for example, monospecific antibodies such as balstilimab, budigalimab, camrelizumab, cosibelimab, dostarlimab, cemiplimab, ezabenlimab (BI-754091), MEDI-0680 (AMP-514; as described in WO2012/145493, which is incorporated by reference herein), nivolumab, pembrolizumab, pidilizumab (CT-011), pimivalimab, retifanlimab, sasanlimab, spartalizumab, sintilmab, tislelizumab, toripalimab, and zimberelimab; as well as bi-specific antibodies such as LY3434172.
- monospecific antibodies such as balstilimab, budigalimab, camrelizumab, cosibelimab, dostarlimab, cemiplima
- the PD-1 antagonist can be a recombinant protein composed of the extracellular domain of PD- L2 (B7-DC) fused to the Fc portion of IgGl (AMP-224).
- an immune checkpoint inhibitor is zimberelimab.
- an immune checkpoint inhibitor is a PD-L1 antagonist.
- the PD-L1 antagonist can be an antagonistic PD-L1 antibody.
- Suitable antagonistic PD-Ll antibodies include, for example, monospecific antibodies such as avelumab, atezolizumab, durvalumab, BMS-936559, and envafolimab as well as bi-specific antibodies such as LY3434172 and KN046.
- an immune checkpoint inhibitor is a TIGIT antagonist.
- the TIGIT antagonist can be an antagonistic TIGIT antibody.
- Suitable antagonistic anti-TIGIT antibodies include monospecific antibodies such as AGEN1327, AB308 (as described in WO2021247591, which is incorporated by reference herein), BMS 986207, COM902, domvanalimab, belrestotug, etigilimab, IBI-929, JS006, dargistotug, ociperlimab, SEA-TGT, tiragolumab, vibostolimab; as well as bi-specific antibodies such as AGEN1777 and AZD2936.
- an immune checkpoint inhibitor is an antagonistic anti- TIGIT antibody disclosed in WO2017152088 or WO2021247591, which are incorporated by reference herein.
- an immune checkpoint inhibitor is domvanalimab or AB308.
- an immune checkpoint inhibitor is a LAG-3 antagonist.
- the LAG-3 antagonist can be an antagonistic LAG-3 antibody.
- Suitable antagonistic LAG-3 antibodies include, for example, BMS-986016 (as described in WO10/19570 and WO14/08218, each of which is incorporated by reference herein), or IMP-731 or IMP-321 (as described in WO08/132601 and WO09/44273, each of which is incorporated by reference herein).
- an immune checkpoint inhibitor is a B7-H3 antagonist.
- the B7-H3 antagonist is an antagonistic B7-H3 antibody.
- Suitable antagonist B7-H3 antibodies include, for example, enoblituzumab (MGA271; as described in WO11/109400, which is incorporated by reference herein), omburtumab, DS-7300a, ABBV-155, and SHR-A1811.
- an immune checkpoint inhibitor is a TIM-3 antagonist.
- the TIM-3 antagonist can be an antagonistic TIM-3 antibody.
- Suitable antagonistic TIM-3 antibodies include, for example, dostarlimab, sabatolimab, BMS-986258. And RG7769/RO7121661.
- a compound according to this disclosure is administered with one or more than one additional therapy.
- each additional therapy is independently selected from an immune checkpoint inhibitor, a chemotherapeutic agent, and radiation therapy.
- the immune checkpoint inhibitor antagonizes PD-1, PD-L1, BTLA, LAG-3, a B7 family member, TIM-3, TIGIT, or CTLA-4, or a combination of any two or more thereof;
- the immune checkpoint inhibitor antagonizes PD-1 or PD-L1;
- the immune checkpoint inhibitor that antagonizes PD-1 or PD-L1 is selected from the group consisting of avelumab, atezolizumab, balstilimab, budigalimab, camrelizumab, cosibelimab, dostarlimab, durvalumab, emiplimab, envafolimab ezaben
- NCCN National Comprehensive Cancer Network
- compositions containing a compound according to this disclosure may be in a form suitable for oral administration.
- Oral administration may involve swallowing the formulation thereby allowing the compound to be absorbed into the bloodstream in the gastrointestinal tract.
- oral administration may involve buccal, lingual or sublingual administration, thereby allowing the compound to be absorbed into the blood stream through oral mucosa.
- the pharmaceutical compositions containing a compound according to this disclosure may be in a form suitable for parenteral administration.
- parenteral administration include, but are not limited to, intravenous, intraarterial, intramuscular, intradermal, intraperitoneal, intrathecal, intracisternal, intracerebral, intracerebroventricular, intraventricular, and subcutaneous.
- Pharmaceutical compositions suitable for parenteral administration may be formulated using suitable aqueous or non-aqueous carriers. Depot injections, which are generally administered subcutaneously or intramuscularly, may also be utilized to release the compounds disclosed herein over a defined period of time.
- Other routes of administration are also contemplated by this disclosure, including, but not limited to, nasal, vaginal, intraocular, rectal, topical (e.g., transdermal), and inhalation.
- compositions suitable for administration to a subject are pharmaceutical compositions comprising a compound according to this disclosure or a pharmaceutically acceptable salt thereof and one or more pharmaceutically acceptable excipients.
- the compound, or pharmaceutically acceptable salt thereof may be present in an effective amount.
- the pharmaceutical compositions may be used in the methods of the present disclosure; thus, for example, the pharmaceutical compositions comprising a compound according to this disclosure can be administered to a subject in order to practice the therapeutic and prophylactic methods and uses described herein.
- compositions of the present disclosure can be formulated to be compatible with the intended method or route of administration. Routes of administration may include those known in the art. Exemplary routes of administration are oral and parenteral. Furthermore, the pharmaceutical compositions may be used in combination with one or more other therapies described herein in order to treat or prevent the diseases, disorders and conditions as contemplated by the present disclosure. In one embodiment, one or more other therapeutic agents contemplated by this disclosure are included in the same pharmaceutical composition that comprises a compound according to this disclosure. In another embodiment, the one or more other therapeutical agents are in a composition that is separate from the pharmaceutical composition comprising the compound according to this disclosure. [0126] In one aspect, the compounds described herein may be administered orally. Oral administration may be via, for example, capsule or tablets.
- the tablet or capsule typically includes at least one pharmaceutically acceptable excipient.
- pharmaceutically acceptable excipients include lactose, dextrose, sucrose, sorbitol, mannitol, starches, gum acacia, calcium phosphate, alginates, tragacanth, gelatin, calcium silicate, microcrystalline cellulose, polyvinylpyrrolidone, polyethylene glycol, cellulose, sterile water, syrup, and methyl cellulose.
- Additional pharmaceutically acceptable excipients include lubricating agents such as talc, magnesium stearate, and mineral oil; wetting agents; emulsifying and suspending agents; preserving agents such as methyl and propylhydroxy-benzoates.
- lubricating agents such as talc, magnesium stearate, and mineral oil
- wetting agents such as talc, magnesium stearate, and mineral oil
- emulsifying and suspending agents such as methyl and propylhydroxy-benzoates.
- preserving agents such as methyl and propylhydroxy-benzoates.
- the compounds described herein may be administered parenterally, for example by intravenous injection.
- a pharmaceutical composition appropriate for parenteral administration may be formulated in solution for injection or may be reconstituted for injection in an appropriate system such as a physiological solution.
- Such solutions may include sterile water for injection, salts, buffers, and tonicity excipients in amounts appropriate to achieve isotonicity with the appropriate physiology.
- the container is designed to maintain stability for the pharmaceutical composition over a given period of time.
- Administering [0129]
- the disclosed methods comprise administering a compound described herein, or a composition thereof, in an effective amount to a subject in need thereof.
- An “effective amount” with reference to a mutant HIF-2 ⁇ inhibitor (e.g., a G323E mutant HIF-2 ⁇ inhibitor), of the present disclosure means an amount of the compound that is sufficient to engage the target (by inhibiting, agonizing or antagonizing the target) at a level that is indicative of the potency of the compound.
- target engagement can be determined by one or more biochemical or cellular assays resulting in an EC50, ED50, EC90, IC50, or similar value which can be used as one assessment of the potency of the compound.
- Assays for determining target engagement include, but are not limited to, those described in the Examples.
- the effective amount may be administered as a single quantity or as multiple, smaller quantities (e.g., as one tablet with “x” amount, as two tablets each with “x/2” amount, etc.).
- the disclosed methods comprise administering a therapeutically effective amount of a compound described herein to a subject in need thereof.
- a therapeutically effective amount with reference to compound means a dose regimen (i.e., amount and interval) of the compound that provides the specific pharmacological effect for which the compound is administered to a subject in need of such treatment.
- a therapeutically effective amount may be effective to eliminate or reduce the risk, lessen the severity, or delay the onset of the disease, including biochemical, histological and/or behavioral signs or symptoms of the disease.
- a therapeutically effective amount may be effective to reduce, ameliorate, or eliminate one or more signs or symptoms associated with a disease, delay disease progression, prolong survival, decrease the dose of other medication(s) required to treat the disease, or a combination thereof.
- a therapeutically effective amount may, for example, result in the killing of cancer cells, reduce cancer cell counts, reduce tumor burden, eliminate tumors or metastasis, or reduce metastatic spread.
- a therapeutically effective amount may vary based on, for example, one or more of the following: the age and weight of the subject, the subject’s overall health, the stage of the subject’s disease, the route of administration, and prior or concomitant treatments. [0131] Administration may comprise one or more (e.g., one, two, or three or more) dosing cycles.
- the compounds contemplated by the present disclosure may be administered (e.g., orally, parenterally, etc.) at about 0.01 mg/kg to about 50 mg/kg, or about 1 mg/kg to about 25 mg/kg, of subject’s body weight per day, one or more times a day, a week, or a month, to obtain the desired effect.
- a suitable weight-based dose of a compound contemplated by the present disclosure is used to determine a dose that is administered independent of a subject’s body weight
- the compounds of the present disclosure are administered (e.g., orally, parenterally, etc.) at fixed dosage levels of about 1 mg to about 1000 mg, particularly 1, 3, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, 500, 600, 750, 800, 900, or 1000 mg, one or more times a day, a week, or a month, to obtain the desired effect.
- the compound is contained in a “unit dosage form”.
- unit dosage form refers to physically discrete units, each unit containing a predetermined amount of the compound, either alone or in combination with one or more additional agents, sufficient to produce the desired effect. It will be appreciated that the parameters of a unit dosage form will depend on the particular agent and the effect to be achieved.
- Methods of Synthesis General methods for compound preparation [0134] Compounds of Formula I, including the exemplary molecules described herein and represented in the claims, may be synthesized using various standard synthetic methods and techniques that are known to those skilled in the art. Compounds of Formula I and compounds having the structures described in the prior sections were synthesized from starting materials that are available from common commercial sources, or from starting materials that can be synthesized using standard synthetic methods.
- All assayed compounds were purified to ⁇ 95% purity as determined by 1 H NMR or LCMS (AGILENT® 1100 or 1200 series LCMS with UV detection at 254 or 280 nm using a binary solvent system [0.1% formic acid in MeCN/0.1% formic acid in H2O] using one of the following columns: AGILENT® Eclipse Plus C18 [3.5 ⁇ m, 4.6 mm i.d. ⁇ 100 mm], WATERSTM XSelect HSS C18 [3.5 ⁇ m, 2.1 mm i.d. ⁇ 75 mm]).
- Example 5 (5R,6S,8R)-8-(4-Chloro-3-cyano-5-fluoro-2-methylphenyl)-3,5,6-trifluoro- 5,6,7,8-tetrahydronaphthalene-1-carbonitrile [0140]
- Step a Into a 3L round bottom flask, 4-bromo-2-fluoro-5-methylaniline (50.0 g, 245 mmol, 1.0 equiv.), glacial acetic acid (1L, 0.25 M), N-iodosuccinimide (57.8 g, 257.7 mmol, 1.05 equiv.) were sequentially added. After completion of the addition, the reaction was stirred at room temperature for 3 h.
- step a Into a 500 mL round bottom flask, product of step a (16.0 g, 69.85 mmol, 1.0 equiv.), copper(I) chloride (20.75 g, 209.56 mmol, 3.0 equiv.) and copper(II) chloride (32.87 g, 244.48 mmol, 3.5 equiv.) and ACN (150 mL, 0.5M) were added. The mixture was cooled to 0 °C and tert-butyl nitrite (33.23 mL, 279.40 mmol, 4.0 equiv.) was added. The resulting mixture was stirred at 23°C for 6 h.
- Step c A 250 mL flask was charged with the product of step b (5.47 g, 22.02 mmol, 1.0 equiv.), B2Pin2 (7.27 g, 28.62 mmol, 1.3 equiv.), Pd(dppf)Cl2 (1.29 g, 1.76 mmol, 8 mol%), KOAc (4.75 g, 48.44 mmol, 2.2 equiv.) and 1,4-dioxane (110 mL). The reaction mixture was degassed with N2 bubbling for 10 min before being heated to 90 °C.
- reaction mixture was degassed with N2 bubbling for 10 min before it was stirred at 80 °C for 1 h.
- the reaction mixture was quenched with sat. sol. NaCl and extracted with EtOAc.
- the combined organic extract was washed with sat. sol. NaCl, dried over Na2SO4, filtered, and concentrated.
- the residue was purified using column chromatography on silica gel (EtOAc in hexane – 0% ⁇ 20%).
- the obtained orange foam was stirred with SilicaMetS dimercaptotriazine (16 g) and activated charcoal (4 g) in EtOAc (75 mL, 0.3M) at 23 °C for 16h.
- Step e Product of step d (7.8 g, 16.56 mmol, 1.0 equiv.) was dissolved in EtOAc (210 mL) and MeOH (70 mL) under nitrogen, and Et3N (6.9 mL, 49.68 mmol, 3.0 equiv.) and Pd/C (1.56 g, 20 wt%, 10% Pd) were added to the reaction vessel.
- Step g To a suspension of product of step f (1.52 g, 4.23 mmol, 1.0 equiv.) and NaHCO3 (0.39 g, 4.65 mmol, 1.1 equiv.) in CH2Cl2 (22 mL, 0.2M) at 0 °C was added DMP (1.97 g, 4.65 mmol, 1.1 equiv.) slowly in portions.
- the reaction was stirred for 30 min and was analyzed by LC/MS. Upon completion, the reaction was quenched by addition of a saturated sol. NaHCO 3 (20 mL) and saturated sol. Na 2 S 2 O 3 (20 mL). Celite® was added and the solution was stirred for 20 min before it was filtered over Celite® and washed with CH2Cl2. The layers were separated, and the aqueous layer was back extracted with CH 2 Cl 2 twice. The combined organic extract was washed with saturated sol. Na 2 S 2 O 3 , then dried over Na 2 SO 4 , filtered, and concentrated to dryness.
- Step h Product of step g (1.51 g, 4.23 mmol, 1.0 equiv.) was dissolved in CH2Cl2 (22 mL, 0.2M) and the reaction mixture was cooled to 0 °C.
- Step i Product of step f (1.99 g, 4.23 mmol, 1.0 equiv.) was dissolved in MeCN (22 mL, 0.2M) at 23 °C and SelectFluorTM (3.00 g, 8.46 mmol, 2.0 equiv.) was added slowly over 15 min.
- Step j Product of step i (1.67 g, 4.46 mmol, 1.0 equiv.) was dissolved in CH 2 Cl 2 (22 mL, 0.2M) and cooled to 0 °C.
- step j Product of step j (1.06 g, 2.81 mmol, 1.0 equiv.) in DCM (2 mL) was then added dropwise over 5 min and the orange suspension was warmed to 23 °C for 16h. Upon completion, the reaction was quenched with sat. sol. NaHCO 3 (50 mL) and diluted with EtOAc (25 mL). The organic layer was separated, and the aqueous layer was extracted with EtOAc (50 mL). The combined organic layers were washed with HCl (1M, 50 mL), dried with Na 2 SO 4 , filtered, and concentrated.
- the resulting mixture was heated at reflux for 3 h. After cooling to room temperature, 0.3 M H2SO4 (aq., 50 mL) was added to the reaction mixture. The resulting mixture was heated at reflux for another 1 h. After cooling to room temperature, large amount of the product was precipitated out and collected via filtration. The filtrate was concentrated and diluted with DCM (250 mL) and washed with H 2 O and brine. The organic phase was dried over Na2SO4 and concentrated. The product was directly used in the next step.
- Step b HCO 2 H (2.5 mL, 66 mmol, 3.0 equiv.) was added to a solution of Et 3 N (6.2 mL, 44 mmol, 2.0 equiv.) in DMF (20 mL) dropwise. The resulting solution was stirred at room temperature for 30 min, and then added to a solution of the product from step a ( ⁇ 22 mmol, 1.0 equiv.) and RuCl(p-cymene)[(R,R)-TsDPEN] (557 mg, 0.88 mmol, 4.0 mol%) in DMF (20 mL) at 0 °C.
- Step c To a solution of the product from step b (2.94 g, 10.4 mmol, 1.0 equiv.) and diisopropylethylamine (3.7 mL, 2.0 equiv.) in DCM (35 mL) was added and MOMBr (1.3 mL, 1.5 equiv.) at 0 °C. The resulting mixture was heated at 35 °C for overnight, and then quenched with saturated NaHCO 3 aqueous solution. The aqueous phase was extracted with DCM ⁇ 2.
- Step d To a solution of the product from step c (2.30 g, 7.0 mmol, 1.0 equiv.) in 1,4- dioxane (30 mL) was added B2Pin2 (2.14 g, 8.4 mmol, 1.2 equiv.), Pd(dppf)Cl2 (513 mg, 0.70 mmol, 10 mol%) and KOAc (1.37 g, 14 mmol, 2.0 equiv.).
- Step e To a mixture of (4R)-4-[tert-butyl(dimethyl)silyl]oxy-8-cyano-6-fluoro-3,4- dihydronaphthalen-1-yl] trifluoromethanesulfonate (2.85 g, 6.3 mmol, 0.9 equiv.) and the product from step d ( ⁇ 7.0 mmol, 1.0 equiv.) in 1,4-dioxane (30 mL) was added Pd(dppf)Cl 2 (513 mg, 0.70 mmol, 10 mol%) and Na2CO3 (1M in H2O, 14 mL, 2.0 equiv.).
- Step f A mixture of the product from step e (2.36 g, 4.3 mmol, 1.0 equiv.), Pd/C (10 wt% Pd, 760 mg) in MeOH (30 mL) was shaken in parr hydrogenator under H 2 (40 psi) for 2.5 h. After this time LCMS showed no remaining starting material.
- Step g To a solution of the product from step f (1.88 g, 3.4 mmol, 1.0 equiv.) in THF (15 mL) was added TBAF (1M in THF, 5.1 mL, 1.5 equiv.) at 0 °C. The resulting solution was stirred at room temperature for 1 h, and then quenched by saturated NH4Cl (aq.). The aqueous phase was extracted with EtOAc twice.
- Step h To a solution of the product from step g (1.24 g, 2.8 mmol, 1.0 equiv.) in DCM (14 mL) was added NaHCO 3 (470 mg, 5.6 mmol, 2.0 equiv.) and DMP (1.32 g, 3.1 mmol, 1.1 equiv.) at 0 °C. The resulting mixture was stirred at room temperature for overnight.
- Step i To a solution of the crude product from step h ( ⁇ 2.8 mmol) and triethylamine (3.2 mL, 22 mmol, 8.0 equiv.) in DCM (9 mL) was added TBSOTf (2.2 mL, 11 mmol, 4.0 equiv.) at 0 °C. The resulting mixture was then stirred at room temperature for 1.5 h and quenched with saturated NaHCO3 aqueous solution.
- Step j To a solution of the product from step i ( ⁇ 2.8 mmol, 1.0 equiv.) in MeCN (10 mL), SelectfluorTM (2.20 g, 6.2 mmol, 2.2 equiv.) was added portion-wise at room temperature. The resulting mixture was stirred at 60 °C for 30 min and then quenched with saturated NaHCO3 aqueous solution at 0 °C.
- Step k HCO 2 H (0.20 mL, 5.3 mmol, 3.0 equiv.) was added to a solution of Et 3 N (0.50 mL, 3.6 mmol, 2.0 equiv.) in DCM (5 mL) dropwise.
- Step l To a solution of the product from step k (620 mg, 1.4 mmol, 1.0 equiv.) and 3- nitrobenzoic acid (681 mg, 4.1 mmol, 3.0 equiv.) in THF (12 mL) was added PPh 3 (856 mg, 3.3 mmol, 2.4 equiv.) and DIAD (0.65 mL, 3.3 mmol, 2.4 equiv.) at 0 °C. The resulting mixture was stirred at this temperature until the TLC showed a full consumption of the substrate. The reaction mixture was then quenched with H 2 O and extracted with EtOAc twice.
- the combined organic layer was then washed with brine, dried over Na2SO4, concentrated, and purified by flash chromatography (SiO 2 , 0 to 30% EtOAc/hexane) to afford the ester intermediate.
- the ester compound was then dissolved in THF/MeOH (2:1 v/v, 6 mL) and treated with a solution of LiOH ⁇ H2O (115 mg, 2.7 mmol, 2.0 equiv.) in H2O (2 mL). The resulting mixture was stirred at room temperature for 3 h and then diluted with H2O. The aqueous phase was extracted with EtOAc twice. The combined organic layer was then washed with brine, dried over Na 2 SO 4 , and concentrated.
- Step m To a solution of the product from step l ( ⁇ 1.4 mmol) in DCM (10 mL) was added DAST (0.90 mL, 6.8 mmol, 5.0 equiv.) at -40 °C. The resulting mixture was then raised to -10 °C and stirred at this temperature until TLC showed a full conversion of the substrate. The reaction mixture was then quenched with saturated NaHCO3 aqueous solution at -10 °C. The aqueous phase was extracted with DCM twice.
- Step n To a solution of the product from step m (90.0 mg, 0.20 mmol, 1.0 equiv.) and 1,2-dimethyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)imidazole (66.6 mg, 0.30 mmol, 1.5 equiv.) in 1,4-dioxane (2.0 mL) was added SPhos Pd G2 (14.4 mg, 0.020 mmol, 10 mol%) and Na 2 CO 3 (1M in H 2 O, 0.40 mL, 2.0 equiv.).
- Step o To a solution of the product from step n ( ⁇ 0.20 mmol) in DCM (3 mL) was added TFA (0.30 mL). The resulting mixture was heated at 40 °C for 1 h and then concentrated. The crude mixture was then purified by HPLC to afford the title compound.
- Step a To a solution of 4-bromo-6,7-dichloro-2,3-dihydroinden-1-one (20.0 g, 81 mmol, 1.0 equiv.) in MeOH (400 mL) was added SelectfluorTM (31.5 g, 89 mmol, 1.1 equiv.) and concentrated H 2 SO 4 (1.0 mL).
- the resulting mixture was heated at reflux for 3 h. After cooling to room temperature, 0.3 M H2SO4 (aq., 100 mL) was added to the reaction mixture. The resulting mixture was heated at reflux for another 1 h. After cooling to room temperature, large amount of the product was precipitated out and collected via filtration. The filtrate was concentrated and diluted with DCM (250 mL) and washed with H 2 O and brine. The organic phase was dried over Na2SO4 and concentrated. The product was directly used in the next step.
- Step b To a solution of the product from step a (7.19 g, 27 mmol, 1.0 equiv.) in MeOH (100 mL) was added NaBH 4 (1.33 g, 35 mmol, 1.3 equiv.) at 0 °C. The resulting mixture was stirred at this temperature until TLC showed a full conversion. The reaction mixture was then quenched with water, diluted with EtOAc, and separated. The aqueous phase was extracted with EtOAc twice. The combined organic layer was then washed with brine, dried over Na 2 SO 4 , and concentrated. The crude product was directly used in the next step.
- Step c To a solution of the crude product from step b ( ⁇ 27 mmol) and diisopropylethylamine (14.2 mL, 3.0 equiv.) in DCM (70 mL) was added and MOMBr (4.4 mL, 2.0 equiv.) at 0 °C. The resulting mixture was heated at 30 °C for overnight, and then quenched with saturated NaHCO3 aqueous solution. The aqueous phase was extracted with DCM ⁇ 2.
- Step d To a solution of the product from step c (4.70 g, 15 mmol, 1.0 equiv.) in 1,4- dioxane (75 mL) was added B 2 Pin 2 (4.60 g, 18 mmol, 1.2 equiv.), Pd(dppf)Cl 2 (1.10 g, 1.5 mmol, 10 mol%) and KOAc (2.13 g, 22 mmol, 1.5 equiv.).
- reaction mixture was degassed with N2 bubbling for 10 min before being heated to 100 °C. After stirring at 100 °C overnight, the reaction mixture was cooled, concentrated on Celite® and purified by flash chromatography (SiO2, 0 to 15% EtOAc/hexane) to afford the product.
- Step e To a mixture of (4R)-4-[tert-butyl(dimethyl)silyl]oxy-8-cyano-6-fluoro-3,4- dihydronaphthalen-1-yl] trifluoromethanesulfonate (6.13 g, 13.6 mmol, 1.05 equiv.) and the product from step d (4.64 g, 12.9 mmol, 1.0 equiv.) in 1,4-dioxane (25 mL) was added Pd(dppf)Cl2 (471 mg, 0.64 mmol, 5 mol%) and Na2CO3 (1M in H2O, 25 mL, 2.0 equiv.).
- Step f A mixture of the product from step e (5.24 g, 9.8 mmol, 1.0 equiv.), Pd/C (10 wt% Pd, 1.00 g) in MeOH (50 mL) was shaken in parr hydrogenator under H 2 (50 psi) for 4 h, when LCMS showed no remaining starting material.
- Step g To a solution of the product from step f (3.58 g, 6.7 mmol, 1.0 equiv.) in THF (30 mL) was added TBAF (1M in THF, 10 mL, 1.5 equiv.) at 0 °C. The resulting solution was stirred at 0 °C for 15 min, and then quenched by saturated NH 4 Cl (aq.). The aqueous phase was extracted with EtOAc ⁇ 2.
- the combined organic layer was then washed with brine, dried over Na2SO4, and concentrated.
- the crude material was then dissolved with DCM (30 mL) and treated with NaHCO 3 (620 mg, 7.4 mmol, 1.1 equiv.) and DMP (3.12 g, 7.4 mmol, 1.1 equiv.) at 0 °C. The resulting mixture was stirred at room temperature for overnight. The reaction mixture was then quenched with saturated NaHCO3 and Na2S2O3 aqueous solution and extracted with DCM twice. The combined organic layer was then washed with brine, dried over Na2SO4, concentrated. The crude product was directly used in the next step.
- Step h To a solution of the crude product from step g ( ⁇ 6.7 mmol) and triethylamine (4.2 mL, 30 mmol, 4.5 equiv.) in DCM (30 mL) was added TBSOTf (5.4 mL, 24 mmol, 3.5 equiv.) at 0 °C. The resulting mixture was then stirred at room temperature for overnight and quenched with saturated NaHCO3 (aq.) and kept stirring for 1 h. The resulting mixture was then separated, and the aqueous phase was extracted with DCM twice.
- Step i To a solution of the product from step h (2.21 g, 4.1 mmol, 1.0 equiv.) in MeCN (20 mL), SelectfluorTM (2.50 g, 7.1 mmol, 1.7 equiv.) was added portion-wise at room temperature. The resulting mixture was stirred at room temperature for 30 min and then quenched with saturated NaHCO 3 aqueous solution.
- Step j HCO 2 H (0.47 mL, 12 mmol, 3.0 equiv.) was added to a solution of Et 3 N (1.2 mL, 8.3 mmol, 2.0 equiv.) in DCM (10 mL) dropwise.
- Step k To a solution of 4-(trimethylsilyl)morpholine (1.44 g, 9.0 mmol, 3.0 equiv.) in toluene (10 mL) was added deoxofluor (2.7 M in toluene, 3.3 mL, 9.0 mmol, 3.0 equiv.) dropwise at ⁇ 78 °C. The resulting solution was then stirred at this temperature for 5 min and warmed to room temperature for 1.5 h. The reaction mixture was diluted with another 40 mL of toluene and then cooled back to ⁇ 78 °C.
- deoxofluor 2.7 M in toluene, 3.3 mL, 9.0 mmol, 3.0 equiv.
- Step l To a solution of the crude product from step k ( ⁇ 3.0 mmol) in THF (20 mL) was added 6M HCl aqueous solution (20 mL).
- Step m To a solution of the crude product from step l ( ⁇ 3.0 mmol, 1.0 equiv.) in DCM (20 mL) was added NaHCO3 (519 mg, 6.0 mmol, 2.0 equiv.) and DMP (1.44 g, 3.4 mmol, 1.1 equiv.) at 0 °C.
- Step n To a solution of the product from step m (55 mg, 0.14 mmol, 1.0 equiv.) and imidazole (34 mg, 0.42 mmol, 3.0 equiv.) in MeCN (1.2 mL) was added diisopropylethylamine (0.13 mL, 0.70 mmol, 5.0 equiv.). The resulting mixture was heated at 80 °C for 4.5 h and then concentrated. The crude mixture was then purified by flash chromatography (SiO 2 , 0 to 10% MeOH/DCM) to afford the product.
- Step o HCO2H (7.3 ⁇ L, 0.19 mmol, 4.0 equiv.) was added to a solution of Et3N (20 ⁇ L, 0.14 mmol, 3.0 equiv.) in DMF (0.4 mL). The resulting solution was stirred at room temperature for 30 min, and then added to a solution of the product from step n (21 mg, 47.4 ⁇ mol, 1.0 equiv.) and RuCl(p-cymene)[(R,R)-TsDPEN] (0.9 mg, 1.4 ⁇ mol, 3.0 mol%) in DMF (0.1 mL) at 0 °C.
- Example 12 (5R,6S,8R)-8-[(1S,2R)-2,6-difluoro-1-hydroxy-7-(1-methyl-5-pyrazolyl)-4- indanyl]-3,5,6-trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile [0187]
- the title compound was prepared using a similar procedure as that described for Example 10 from the appropriate starting materials.
- Example 20 (5R,6S,8R)-8- ⁇ (1S,2R)-7-[(R)-3-methyl-4-morpholinyl]-2,6-difluoro-1- hydroxy-4-indanyl ⁇ -3,5,6-trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile [0195]
- the title compound was prepared using a similar procedure as that described for Example 10 from the appropriate starting materials.
- pcDNA3.1 expression vectors were generated by GenScript through gene synthesis technology.
- Stable HEK-293 cell lines expressing stabilized HIF-2 ⁇ with G323E mutation were generated by plasmid transfection followed by G418 selection. Single cell cloning was achieved by limited dilution method, and clones expressing the HiBit tagged HIF-2 ⁇ protein were identified utilizing the Nano-Glo® HiBiT Lytic Detection System (N3040, Promega).
- Stabilized HIF-2 ⁇ with G323E mutation positive clones were further transfected with Cignal Lenti HIF Luc Reporter lentivirus (CLS-007L, Qiagen) according to the manufacturer’s guidelines.
- 0.3x10 6 of the respective HEK 293 cells were transduced with lentivirus at a Multiplicity of Infection (MOI) of 25 for 24 hours.
- MOI Multiplicity of Infection
- cells were replenished with fresh RPMI 1640 Medium (Cat. No. 11875085, Thermo Fisher,) supplemented with 10% FBS (Cat. No. A3160502, Gibco), 2mM GlutaMax (Cat. No. 35050-061, Invitrogen), 100 units of penicillin and 100 ⁇ g/mL of streptomycin (Cat. No 15070063, Thermo Fisher) and G418 (Cat. No J63871, Thermo Fisher) for another 24 hours.
- No.31985088 Thermo Fisher was seeded into its respective 384 well white opaque plate (Corning 3570) and incubated at 37°C and 5% CO 2 . After a 4-hour incubation, 20 ⁇ L of 2x compound is added to each cell plate. Final assay conditions comprised 10,000 cells per well for the G323E cell line in 1% DMSO with test compound concentrations ranging from 50 ⁇ M to 0 ⁇ M. After a 20-hour incubation at 37°C and 5% CO 2 , luciferase activity was determined using ONE-Glo Luciferase Assay Reagent (E6110, Promega) following the manufacture’s recommended procedure.
- E6110 ONE-Glo Luciferase Assay Reagent
- luciferase reagents 40 ⁇ L of ONE-Glo luciferase reagents were added to each well and luciferase signals were measured using an Envision 2102 Multilabel Reader. Percentage maximum activity in each test well was calculated based on DMSO (maximum activity) and no cell control wells (baseline activity). The IC50 values of the test compounds were determined from compound dose response curves fitted using a standard four parameter fit equation (Table 5).
Landscapes
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Disclosed herein are compounds or pharmaceutically acceptable salts thereof that inhibit the activity of mutant HIF-2a (e.g., G323E mutant HIF-2a), where the compounds are represented by Formula I, as well as compositions containing the compounds or pharmaceutically acceptable salts thereof. Also disclosed are methods of preparing the compounds or pharmaceutically acceptable salts thereof and methods of using the compounds or pharmaceutically acceptable salts thereof for the treatment of diseases, disorders, or conditions.
Description
TETRALINS TARGETING MUTANT HIF-2α CROSS-REFERENCE TO RELATED APPLICATIONS [0001] This application claims the benefit of priority to US Provisional Patent Application No. 63/533,085, filed on August 16, 2023, the entire content of which is incorporated by reference herein. BACKGROUND [0002] The following discussion is provided to aid the reader in understanding the disclosure and is not admitted to describe or constitute prior art thereto. [0003] Hypoxia-inducible factor (HIF) transcription factors play an integral role in cellular response to low oxygen availability. HIFs are heterodimeric transcription factors consisting of a common constitutive subunit called the aryl hydrocarbon receptor nuclear translocator (ARNT, or HIF-β) and one of three HIF-α subunits. Under normal conditions, the α-subunits are hydroxylated at conserved proline residues by prolyl-4-hydroxylases, and subsequently targeted for degradation by the von Hippel-Lindau ubiquitin E3 ligase complex. However, under hypoxic conditions, HIF-α accumulates and enters the nucleus to activate the expression of genes that regulate metabolism, angiogenesis, cell proliferation and survival, immune evasion, and inflammatory response. [0004] Of the three different α-subunit isoforms, HIF-1α, HIF-2α and the less characterized HIF-3α, HIF-1α and HIF-2α overexpression have been associated with poor clinical outcomes in patients with various cancers. Specifically, HIF-2α has been found to be a marker of poor prognosis in glioblastoma, neuroblastoma, head and neck squamous carcinoma, and non-small cell lung cancer. Hypoxia is also prevalent in many acute and chronic inflammatory disorders, such as inflammatory bowel disease and rheumatoid arthritis. [0005] HIF-2α inhibitors have been described in the literature, belzutifan being the first approved for the treatment of renal cell carcinoma associated with von-Hippel-Lindau disease, central nervous system hemangioblastomas, and pancreatic neuroendocrine tumors. However, a mutation within the internal cavity of the PASB domain of HIF-2α was reported in a patient undergoing treatment with a HIF-2α inhibitor. This mutation resulted in an acquired resistance to
treatment with the HIF-2α inhibitor. This resistance has been associated with a gatekeeper mutation (G323E) in HIF-2α, which interferes with drug binding. [0006] HIF-2α plays a significant role in cancer, inflammation, and other disorders. In view of the resistance of G323E mutant HIF-2α to treatment with existing HIF-2α inhibitors, there is a need for HIF-2α inhibitors that are active against mutant forms of HIF-2α. The present invention addresses this need and provides related advantages as well. SUMMARY [0007] In one aspect, the present disclosure relates to compounds that inhibit the activity of mutant HIF-2α. In some embodiments, the present disclosure relates to compounds that inhibit the activity of G323E mutant HIF-2α. The compounds are represented by Formula I:
 (Formula I) or a pharmaceutically acceptable salt thereof, wherein: n is 0, 1, or 2; each R1 when present is independently halo; R2 and R3 are independently halo, C1-C6 alkyl or -CN; or R2 and R3 combine with the atoms to which they are attached to form a C4-C6 cycloalkyl, wherein said C4-C6 cycloalkyl is unsubstituted or substituted with 1-3 Ra; each Ra when present is independently halo or -OH; R4 is halo, C1-C6 haloalkyl, -CN, -S(O)2-R4a, 4- to 8-membered heterocycloalkyl, or 5- to 10-membered heteroaryl; wherein said 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatom or heteroatom groups independently selected from N, O, S, and S(O)2; said 5- to 10-membered heteroaryl has 1-3 ring heteroatoms independently selected from N, O, and S; and said 4- to 8-membered heterocycloalkyl and 5- to 10-membered heteroaryl are unsubstituted or substituted with 1-3 Rb;
R4a is C1-C3 alkyl, C1-C3 haloalkyl, C3-C6 cycloalkyl, -NR4bR4c, or 4- to 6- membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S; R4b and R4c are each independently -H or C1-C3 alkyl; each Rb when present is independently -CN, C1-C6 alkyl, -N(Rc)2, or -C(O)- N(Rc)2; each Rc is independently -H or C1-C6 alkyl; X is CR5 or N; and R5 is -H or halo. [0008] In another aspect, this disclosure is directed to methods of inhibiting the activity of mutant HIF-2α (e.g., G323E mutant HIF-2α) in a subject comprising administering to the subject an effective amount of a compound or pharmaceutically acceptable salt thereof described herein. [0009] In yet another aspect, this disclosure provides methods for treating a disease, disorder, and/or condition mediated at least in part by the activity of mutant HIF-2α (e.g., G323E mutant HIF-2α) in a subject, comprising administering to the subject a therapeutically effective amount of a compound or pharmaceutically acceptable salt thereof described herein. Diseases, disorders, and conditions mediated by the activity of mutant HIF-2α (e.g., G323E mutant HIF-2α) include cancer and cancer-related disorders. [0010] In yet another aspect, this disclosure provides a method of treating a subject with resistance (e.g., partial or complete) to treatment with a HIF-2α inhibitor, said method comprising administering a compound or a pharmaceutically acceptable salt thereof to a subject in need thereof. [0011] Certain aspects of the present disclosure further comprise the administration of one or more additional therapeutic agents as set forth herein below. DETAILED DESCRIPTION OF THE DISCLUSRE [0012] Before the present disclosure is further described, it is to be understood that the disclosure is not limited to the particular embodiments set forth herein, and it is also to be
understood that the terminology used herein is for the purpose of describing particular embodiments only, and is not intended to be limiting. Definitions [0013] Unless otherwise defined, all terms of art, notations and other scientific terms or terminology used herein are intended to have the meanings commonly understood by those of skill in the art to which this disclosure pertains. [0014] The term “about” as used herein has its original meaning of approximately and is to provide literal support for the exact number that it precedes, as well as a number that is near to or approximately the number that the term precedes. In general, the term “about” refers to the usual error range for the respective value readily known to the skilled person in this technical field. If the degree of approximation is not otherwise clear from the context, “about” means either within plus or minus 10% of the provided value, or rounded to the nearest significant figure, in all cases inclusive of the provided value. Where ranges are provided, they are inclusive of the boundary values. [0015] The phrase “and/or” as used in the present disclosure will be understood to mean any one of the recited members individually or a combination of any two or more thereof¾for example, “A, B, and/or C” would mean “A, or B, or C”, “A and B”, “A and C”, “B and C”, or the combination of “A, B, and C.” [0016] The term “alkyl”, by itself or as part of another substituent, means, unless otherwise stated, a saturated or branched chain hydrocarbon radical, having the number of carbon atoms designated (i.e., C1-8 means one to eight carbons). Alkyl can include any number of carbons, such as C1-2, C1-3, C1-4, C1-5, C1-6, C1-7, C1-8, C1-9, C1-10, C2-3, C2-4, C2-5, C2-6, C3-4, C3-5, C3-6, C4-5, C4-6, and C5-6. Examples of alkyl groups include methyl (Me), ethyl (Et), n-propyl, isopropyl, n- butyl, t-butyl, isobutyl, sec-butyl, n-pentyl, n-hexyl, n-heptyl, n-octyl, and the like. [0017] The term “alkylene” refers to a straight or branched, saturated, aliphatic radical having the number of carbon atoms indicated, and linking at least two other groups, i.e., a divalent hydrocarbon radical. The two moieties linked to the alkylene can be linked to the same atom or different atoms of the alkylene group. For instance, a straight chain alkylene can be the bivalent radical of -(CH2)n-, where n is 1, 2, 3, 4, 5, or 6. Representative alkylene groups include, but are
not limited to, methylene, ethylene, propylene, isopropylene, butylene, isobutylene, sec-butylene, pentylene and hexylene. Alkylene groups, in some embodiments, can be substituted or unsubstituted. When a group comprising an alkylene is optionally substituted, it is understood that the optional substitutions may be on the alkylene portion of the moiety. [0018] The term “cycloalkyl,” “carbocycle,” or “carbocyclic ring” refers to a hydrocarbon ring having the indicated number of ring atoms (e.g., C3-6 cycloalkyl) and being fully saturated or having no more than one double bond between ring vertices. “Cycloalkyl” is also meant to refer to bicyclic and polycyclic hydrocarbon rings such as, for example, bicyclo[2.2.1]heptane, bicyclo[2.2.2]octane, etc. In some embodiments, the cycloalkyl compounds of the present disclosure are monocyclic C3-6 cycloalkyl moieties (e.g., cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl). [0019] The term “heterocycloalkyl,” “heterocycle,” or “heterocyclic ring” refers to a cycloalkyl ring having the indicated number of ring vertices ( or members) and having from one to five heteroatoms selected from N, O, and S, which replace one to five of the carbon vertices, and wherein the nitrogen and sulfur atoms are optionally oxidized, and the nitrogen atom(s) are optionally quaternized. The heterocycloalkyl may be a monocyclic, a bicyclic or a polycyclic ring system, and may have one or two double bonds connecting ring vertices. Non limiting examples of heterocycloalkyl groups include pyrrolidine, imidazolidine, pyrazolidine, butyrolactam, valerolactam, imidazolidinone, hydantoin, dioxolane, phthalimide, piperidine, 1,4- dioxane, morpholine, thiomorpholine, thiomorpholine-S-oxide, thiomorpholine-S,S-oxide, piperazine, pyran, pyridone, 3-pyrroline, thiopyran, pyrone, tetrahydrofuran, tetrahydrothiophene, quinuclidine, and the like. A heterocycloalkyl group can be attached to the remainder of the molecule through a ring carbon or a heteroatom. In some embodiments, the heterocycle is a 5- to 6-membered heterocycle (e.g., pyrrolidine, tetrahydrofuran, tetrahydropyran, piperidine, piperazine, morpholine, and the like). [0020] As used herein, a wavy line, “ ”, that intersects a single, double or triple bond in any chemical structure depicted herein, represents that the point of attachment of the single, double, or triple bond to the remainder of the molecule. Additionally, a bond extending to the center of a ring (e.g., a phenyl ring) is meant to indicate attachment at any of the available ring vertices. One of skill in the art will understand that multiple substituents shown as being attached to a ring
will occupy ring vertices that provide stable compounds and are otherwise sterically compatible. For a divalent component, a representation is meant to include either orientation (forward or reverse). For example, the group “-C(O)NH-” is meant to include a linkage in either orientation: -C(O)NH- or -NHC(O)-, and similarly, "-O-CH2CH2-" is meant to include both -O-CH2CH2- and -CH2CH2-O-. [0021] The terms “halo” or “halogen,” by themselves or as part of another substituent, mean, unless otherwise stated, a fluorine, chlorine, bromine, or iodine atom. Additionally, terms such as “haloalkyl,” are meant to include monohaloalkyl and polyhaloalkyl. For example, the term “C1-C4 haloalkyl” is meant to include trifluoromethyl, 2,2,2-trifluoroethyl, 4-chlorobutyl, 3- bromopropyl, and the like. [0022] The term “aryl” means, unless otherwise stated, an aromatic, hydrocarbon group which can be a single ring or multiple rings (up to three rings) which are fused together or linked covalently. Non-limiting examples of aryl groups include phenyl, naphthyl, and biphenyl. The term is also meant to include fused cycloalkylphenyl, and heterocycloalkylphenyl ring systems such as, for example, indane, tetrahydronaphthalene, chromane, and isochromane rings. As a substituent group, the point of attachment to the remainder of the molecule, for a fused ring system can be through a carbon atom on the aromatic portion, a carbon atom on the cycloalkyl portion, or an atom on the heterocycloalkyl portion. In some embodiments, the aryl groups are phenyl. [0023] The term “heteroaryl” refers to monocyclic or fused bicyclic aryl groups (or rings) that contain from one to five heteroatoms selected from N, O, and S, wherein the nitrogen and sulfur atoms are optionally oxidized, and the nitrogen atom(s) are optionally quaternized. A heteroaryl group can be attached to the remainder of the molecule through a heteroatom, when chemically permissible. The term “heteroaryl” also embraces heteroaryl groups fused to phenyl rings. For fused bicyclic 9- to 10-membered heteroaryl groups containing a fused benzene ring, the heteroaryl group may be attached to the remainder of the molecule via the heteroaryl ring portion or the phenyl ring portion of the fused bicyclic heteroaryl group. Non-limiting examples of heteroaryl groups include pyridyl, pyridazinyl, pyrazinyl, pyrimindinyl, triazinyl, quinolinyl, quinoxalinyl, quinazolinyl, cinnolinyl, phthalazinyl, benzotriazinyl, purinyl, benzimidazolyl, benzopyrazolyl, benzotriazolyl, benzisoxazolyl, benzoxazolyl, isobenzofuryl, isoindolyl,
indolizinyl, benzotriazinyl, thienopyridinyl, thienopyrimidinyl, pyrrolopyridinyl, pyrazolopyrimidinyl, imidazolopyridinyl, imidazolopyridazinyl, benzothiazolyl, benzofuranyl, benzothienyl, indolyl, quinolyl, isoquinolyl, isothiazolyl, pyrazolyl, indazolyl, pteridinyl, imidazolyl, triazolyl, tetrazolyl, oxazolyl, isoxazolyl, thiadiazolyl, pyrrolyl, thiazolyl, furyl, thienyl and the like. Substituents for a heteroaryl ring can be selected from the group of acceptable substituents described below. In some embodiments, the heteroaryl groups are 5- to 9-membered heteroaryl groups having 1-3 ring heteroatoms independently selected from N, O and S (e.g., imidazolyl, pyrazolyl, triazolyl, thiazolyl, pyridinyl, pyrimidinyl, pyrrolopyridinyl, imidazolopyridinyl, imidazolopyridazinyl, benzothiazolyl, etc.). [0024] The above terms (e.g., “alkyl,” “aryl” and “heteroaryl”), in some embodiments, will be optionally substituted. Selected substituents for each type of radical are provided below. [0025] Optional substituents for the alkyl radicals (including those groups often referred to as alkylene, alkenyl, and alkynyl) can be a variety of groups, for example, groups selected from: halogen, -OR', -NR'R", -SR', -SiR'R"R"', -OC(O)R', -C(O)R', -CO2R', -CONR'R", -OC(O)NR'R", -NR"C(O)R', -NR'-C(O)NR"R"', -NR"C(O)2R', -NH-C(NH2)=NH, -NR'C(NH2)=NH, -NH-C(NH2)=NR', -S(O)R', -S(O)2R', -S(O)2NR'R", -NR'S(O)2R", -CN (cyano), -NO2, aryl, aryloxy, oxo (=O), cycloalkyl and heterocycloalkyl in a number ranging from zero to (2m'+ 1), where m' is the total number of carbon atoms in such radical. R', R", and R"' each independently refer to hydrogen, unsubstituted C1-8 alkyl, unsubstituted aryl, aryl substituted with 1-3 halogens, C1-8 alkoxy, C1-8 thioalkoxy groups, or unsubstituted aryl-C1-4 alkyl groups. When R' and R" are attached to the same nitrogen atom, they can be combined with the nitrogen atom to form a 3-, 4-, 5-, 6-, or 7-membered ring. For example, -NR'R" is meant to include 1-pyrrolidinyl and 4-morpholinyl. [0026] Optional substituents for the cycloalkyl and heterocycloalkyl radicals can be a variety of groups, for example, groups selected from: alkyl optionally substituted with -C(O)OR', halogen, -OR', -NR'R", -SR', -SiR'R"R"', -OC(O)R', -C(O)R', -CO2R', -CONR'R", -OC(O)NR'R", -NR"C(O)R', -NR'-C(O)NR"R"', -NR"C(O)2R', -NH-C(NH2)=NH, -NR'C(NH2)=NH, -NH-C(NH2)=NR', -S(O)R', -S(O)2R', -S(O)2NR'R", -NR'S(O)2R", -CN (cyano), -NO2, aryl, aryloxy, and oxo (=O). R', R" and R"' each independently refer to hydrogen,
unsubstituted C1-8 alkyl, unsubstituted aryl, aryl substituted with 1-3 halogens, C1-8 alkoxy, C1-8 thioalkoxy groups, or unsubstituted aryl-C1-4 alkyl groups. [0027] The optional substituents for the cycloalkyl and heterocycloalkyl radicals may also include olefins (=CR'R''), wherein R' and R" each independently refer to hydrogen, unsubstituted C1-8 alkyl, unsubstituted aryl, aryl substituted with 1-3 halogens, C1-8 alkoxy, C1-8 thioalkoxy groups, or unsubstituted aryl-C1-4 alkyl groups. For example, the olefin can be an unsubstituted olefin (=CH2). [0028] Similarly, optional substituents for the aryl and heteroaryl groups are varied and, for example, can be selected from: -halogen, -OR', -OC(O)R', -NR'R", -SR', -R', -CN, -NO2, -CO2R', -CONR'R", -C(O)R', -OC(O)NR'R", -NR"C(O)R', -NR"C(O)2R', -NR'-C(O)NR"R"', -NH- C(NH2)=NH, -NR'C(NH2)=NH, -NH-C(NH2)=NR', -S(O)R', -S(O)2R', -S(O)2NR'R", -NR'S(O)2R", -N3, perfluoro(C1-4)alkoxy, and perfluoro(C1-4)alkyl, in a number ranging from zero to the total number of open valences on the aromatic ring system; and where R', R" and R"' are independently selected from hydrogen, C1-8 alkyl, C1-8 haloalkyl, C3-6 cycloalkyl, C2-8 alkenyl, and C2-8 alkynyl. Other suitable substituents include each of the above aryl substituents attached to a ring atom by an alkylene tether of from 1-6 carbon atoms. [0029] Two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -T-C(O)-(CH2)q-U-, wherein T and U are independently -NH-, -O-, -CH2- or a single bond, and q is an integer of from 0 to 2. Alternatively, two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -A-(CRfRg)r-B-, wherein A and B are independently -CH2-, -O-, -NH-, -S-, -S(O)-, -S(O)2-, -S(O)2NR'-, or a single bond, r is an integer of from 1 to 3, and Rf and Rg are each independently H or halogen. One of the single bonds of the new ring so formed may optionally be replaced with a double bond. Alternatively, two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -(CH2)s-X-(CH2)t-, where s and t are independently integers of from 0 to 3, and X is -O-, -NR'-, -S-, -S(O)-, -S(O)2-, or -S(O)2NR' -. The substituent R' in -NR'- and -S(O)2NR'- is selected from hydrogen or unsubstituted C1-6 alkyl.
[0030] As used herein, the term “heteroatom” is meant to include oxygen (O), nitrogen (N), sulfur (S), and silicon (Si). [0031] The compounds of the present disclosure can be present in their neutral form, or as a pharmaceutically acceptable salt, isomer, polymorph or solvate thereof, and may be present in a crystalline form, amorphous form, or mixtures thereof. [0032] As referred to herein, “pharmaceutically acceptable salt” is meant to include salts of the compounds according to this disclosure that are prepared with relatively nontoxic acids or bases, depending on the particular substituents found on the compounds described herein. When compounds of the present disclosure contain relatively acidic functionalities, base addition salts can be obtained by contacting the neutral form of such compounds with a sufficient amount of the desired base, either neat or in a suitable inert solvent. Examples of salts derived from pharmaceutically acceptable inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic, manganous, potassium, sodium, zinc, and the like. Salts derived from pharmaceutically-acceptable organic bases include salts of primary, secondary and tertiary amines, including substituted amines, cyclic amines, naturally-occurring amines and the like, such as arginine, betaine, caffeine, choline, N,N’-dibenzylethylenediamine, diethylamine, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine and the like. When compounds of the present disclosure contain relatively basic functionalities, acid addition salts can be obtained by contacting the neutral form of such compounds with a sufficient amount of the desired acid, either neat or in a suitable inert solvent. Examples of pharmaceutically acceptable acid addition salts include those derived from inorganic acids like hydrochloric, hydrobromic, nitric, carbonic, monohydrogencarbonic, phosphoric, monohydrogenphosphoric, dihydrogenphosphoric, sulfuric, monohydrogensulfuric, hydriodic, or phosphorous acids and the like, as well as the salts derived from relatively nontoxic organic acids like acetic, propionic, isobutyric, malonic, benzoic, succinic, suberic, fumaric, mandelic, phthalic, benzenesulfonic, p-tolylsulfonic, citric, tartaric, methanesulfonic, maleic, oxalic, trans- cinnamic, and the like. Also included are salts of amino acids such as arginate and the like, and
salts of organic acids like glucuronic or galactunoric acids, and the like (see, for example, Berge, S.M., et al, “Pharmaceutical Salts”, Journal of Pharmaceutical Science, 1977, 66, 1-19). Certain specific compounds of the present disclosure contain both basic and acidic functionalities that allow the compounds to be converted into either base or acid addition salts. [0033] The neutral forms of the compounds may be regenerated by contacting the salt with a base or acid and isolating the parent compound in the conventional manner. The parent form of the compound differs from the various salt forms in certain physical properties, such as solubility in polar solvents, but otherwise the salts are equivalent to the parent form of the compound for the purposes of the present disclosure. [0034] This disclosure also contemplates isomers of the compounds described herein (e.g., stereoisomers, and atropisomers). For example, certain compounds of the present disclosure possess asymmetric carbon atoms (chiral centers), or hindered rotation about a single bond; the racemates, diastereomers, enantiomers, and atropisomers (e.g., Ra, Sa, P, and M isomers) of which are all intended to be encompassed within the scope of the present disclosure. Stereoisomeric forms may be defined, in terms of absolute stereochemistry, as (R) or (S), and/or depicted uses dashes and/or wedges. When a stereochemical depiction (e.g., using dashes, , and/or wedges, ) is shown in a chemical structure, or a stereochemical assignment (e.g., using (R) and (S) notation) is made in a chemical name, it is meant to indicate that the depicted isomer is present and substantially free of one or more other isomer(s) (e.g., enantiomers and diastereomers, when present). “Substantially free of” other isomer(s) indicates at least an 70/30 ratio of the indicated isomer to the other isomer(s), more preferably 80/20, 90/10, or 95/5 or more. In some embodiments, the indicated isomer will be present in an amount of at least 99%. A chemical bond to an asymmetric carbon that is depicted as a solid line ( ) indicates that all possible stereoisomers (e.g., enantiomers, diastereomers, racemic mixtures, etc.) at that carbon atom are included. In such instances, the compound may be present as a racemic mixture, scalemic mixture, or a mixture of diastereomers. [0035] The compounds of the present disclosure may also contain unnatural proportions of atomic isotopes at one or more of the atoms that constitute such compounds. Unnatural proportions of an isotope may be defined as ranging from the amount found in nature to an amount consisting of 100% of the atom in question. For example, the compounds may
incorporate radioactive isotopes, such as for example tritium (3H), iodine-125 (125I), or carbon-14 (14C), or non-radioactive isotopes, such as deuterium (2H) or carbon-13 (13C). Such isotopic variations can provide additional utilities to those described elsewhere herein. For instance, isotopic variants of the compounds of the disclosure may find additional utility, including but not limited to, as diagnostic and/or imaging reagents, or as cytotoxic/radiotoxic therapeutic agents. Additionally, isotopic variants of the compounds of the disclosure can have altered pharmacokinetic and pharmacodynamic characteristics which can contribute to enhanced safety, tolerability, or efficacy during treatment. All isotopic variations of the compounds of the present disclosure, whether radioactive or not, are intended to be encompassed within the scope of the present disclosure. In some embodiments, the compounds according to this disclosure are characterized by one or more deuterium atoms. [0036] The terms “patient” or “subject” are used interchangeably to refer to a human or a non- human animal (e.g., a mammal). [0037] As used herein, the term “administration” or "administering" of a compound or drug to a subject includes any route of introducing or delivering to a subject a compound to perform its intended function. Administration can be carried out by any suitable route, including but not limited to, orally, intranasally, parenterally (intravenously, intramuscularly, intraperitoneally, or subcutaneously), rectally, intrathecally, intratumorally or topically. Administration includes self- administration and the administration by another. In one embodiment, administration is oral. [0038] The terms “treat”, “treating”, treatment”, and the like refer to a course of action that eliminates, reduces, suppresses, mitigates, ameliorates, or prevents the worsening of, either temporarily or permanently, a disease, disorder, or condition to which the term applies, or at least one of the symptoms associated therewith. Treatment includes alleviation of symptoms, diminishment of extent of disease, inhibiting (e.g., arresting the development or further development of the disease, disorder or condition or clinical symptoms association therewith) an active disease, delaying or slowing of disease progression, improving the quality of life, and/or prolonging survival of a subject as compared to expected survival if not receiving treatment or as compared to a published standard of care therapy for a particular disease. [0039] The term “in need of treatment” as used herein refers to a judgment made by a physician or similar professional that a subject requires or will benefit from treatment. This
judgment is made based on a variety of factors that are in the realm of the physician’s expertise, which may include a positive diagnosis of a disease, disorder, or condition. [0040] The terms “prevent”, “preventing”, “prevention”, “prophylaxis”, and the like refer to a course of action initiated in a manner (e.g., prior to the onset of a disease, disorder, condition, or symptom thereof) so as to prevent, suppress, inhibit or reduce, either temporarily or permanently, a subject’s risk of developing a disease, disorder, condition, or the like (as determined by, for example, the absence of clinical symptoms) or delaying the onset thereof, generally in the context of a subject predisposed to having a particular disease, disorder, or condition. In certain instances, the terms also refer to slowing the progression of the disease, disorder, or condition or inhibiting progression thereof to a harmful or otherwise undesired state. Prevention also refers to a course of action initiated in a subject after the subject has been treated for a disease, disorder, condition, or a symptom associated therewith in order to prevent relapse of that disease, disorder, condition, or symptom. [0041] The term “in need of prevention” as used herein refers to a judgment made by a physician or other caregiver that a subject requires or will benefit from preventative care. This judgment is made based on a variety of factors that are in the realm of a physician’s or caregiver’s expertise. [0042] “Substantially pure” indicates that a component (e.g., a compound according to this disclosure) makes up greater than about 50% of the total content of the composition, and typically greater than about 60% of the total content. More typically, “substantially pure” refers to compositions in which at least 75%, at least 85%, at least 90%, or more of the total composition is the component of interest. In some cases, the component of interest will make up greater than about 90%, or greater than about 95%, of the total content of the composition. [0043] The term “mutant HIF-2α” as used herein refers to human HIF-2α protein that is characterized by one or more amino acid mutations in its sequence. Exemplary amino acid mutations include amino acid substitutions, insertions, or deletions. In some embodiments, the one or more mutations occur in the binding pocket within the PAS-B domain of the HIF-2α protein. In some embodiments, the mutation occurs as a substitution, insertion, or deletion of one or more amino acids listed in Table 1.
Table 1: Wild Type HIF-2α Binding Pocket Within the PAS-B Domain
(Formula I) or a pharmaceutically acceptable salt thereof, wherein: n is 0, 1, or 2; each R1 when present is independently halo; R2 and R3 are independently halo, C1-C6 alkyl or -CN; or R2 and R3 combine with the atoms to which they are attached to form a C4-C6 cycloalkyl, wherein said C4-C6 cycloalkyl is unsubstituted or substituted with 1-3 Ra; each Ra when present is independently halo or -OH; R4 is halo, C1-C6 haloalkyl, -CN, -S(O)2-R4a, 4- to 8-membered heterocycloalkyl, or 5- to 10-membered heteroaryl; wherein said 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatom or heteroatom groups independently selected from N, O, S, and S(O)2; said 5- to 10-membered heteroaryl has 1-3 ring heteroatoms independently selected from N, O, and S; and said 4- to 8-membered heterocycloalkyl and 5- to 10-membered heteroaryl are unsubstituted or substituted with 1-3 Rb;
R4a is C1-C3 alkyl, C1-C3 haloalkyl, C3-C6 cycloalkyl, -NR4bR4c, or 4- to 6- membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S; R4b and R4c are each independently -H or C1-C3 alkyl; each Rb when present is independently -CN, C1-C6 alkyl, -N(Rc)2, or -C(O)- N(Rc)2; each Rc is independently -H or C1-C6 alkyl; X is CR5 or N; and R5 is -H or halo. [0008] In another aspect, this disclosure is directed to methods of inhibiting the activity of mutant HIF-2α (e.g., G323E mutant HIF-2α) in a subject comprising administering to the subject an effective amount of a compound or pharmaceutically acceptable salt thereof described herein. [0009] In yet another aspect, this disclosure provides methods for treating a disease, disorder, and/or condition mediated at least in part by the activity of mutant HIF-2α (e.g., G323E mutant HIF-2α) in a subject, comprising administering to the subject a therapeutically effective amount of a compound or pharmaceutically acceptable salt thereof described herein. Diseases, disorders, and conditions mediated by the activity of mutant HIF-2α (e.g., G323E mutant HIF-2α) include cancer and cancer-related disorders. [0010] In yet another aspect, this disclosure provides a method of treating a subject with resistance (e.g., partial or complete) to treatment with a HIF-2α inhibitor, said method comprising administering a compound or a pharmaceutically acceptable salt thereof to a subject in need thereof. [0011] Certain aspects of the present disclosure further comprise the administration of one or more additional therapeutic agents as set forth herein below. DETAILED DESCRIPTION OF THE DISCLUSRE [0012] Before the present disclosure is further described, it is to be understood that the disclosure is not limited to the particular embodiments set forth herein, and it is also to be
understood that the terminology used herein is for the purpose of describing particular embodiments only, and is not intended to be limiting. Definitions [0013] Unless otherwise defined, all terms of art, notations and other scientific terms or terminology used herein are intended to have the meanings commonly understood by those of skill in the art to which this disclosure pertains. [0014] The term “about” as used herein has its original meaning of approximately and is to provide literal support for the exact number that it precedes, as well as a number that is near to or approximately the number that the term precedes. In general, the term “about” refers to the usual error range for the respective value readily known to the skilled person in this technical field. If the degree of approximation is not otherwise clear from the context, “about” means either within plus or minus 10% of the provided value, or rounded to the nearest significant figure, in all cases inclusive of the provided value. Where ranges are provided, they are inclusive of the boundary values. [0015] The phrase “and/or” as used in the present disclosure will be understood to mean any one of the recited members individually or a combination of any two or more thereof¾for example, “A, B, and/or C” would mean “A, or B, or C”, “A and B”, “A and C”, “B and C”, or the combination of “A, B, and C.” [0016] The term “alkyl”, by itself or as part of another substituent, means, unless otherwise stated, a saturated or branched chain hydrocarbon radical, having the number of carbon atoms designated (i.e., C1-8 means one to eight carbons). Alkyl can include any number of carbons, such as C1-2, C1-3, C1-4, C1-5, C1-6, C1-7, C1-8, C1-9, C1-10, C2-3, C2-4, C2-5, C2-6, C3-4, C3-5, C3-6, C4-5, C4-6, and C5-6. Examples of alkyl groups include methyl (Me), ethyl (Et), n-propyl, isopropyl, n- butyl, t-butyl, isobutyl, sec-butyl, n-pentyl, n-hexyl, n-heptyl, n-octyl, and the like. [0017] The term “alkylene” refers to a straight or branched, saturated, aliphatic radical having the number of carbon atoms indicated, and linking at least two other groups, i.e., a divalent hydrocarbon radical. The two moieties linked to the alkylene can be linked to the same atom or different atoms of the alkylene group. For instance, a straight chain alkylene can be the bivalent radical of -(CH2)n-, where n is 1, 2, 3, 4, 5, or 6. Representative alkylene groups include, but are
not limited to, methylene, ethylene, propylene, isopropylene, butylene, isobutylene, sec-butylene, pentylene and hexylene. Alkylene groups, in some embodiments, can be substituted or unsubstituted. When a group comprising an alkylene is optionally substituted, it is understood that the optional substitutions may be on the alkylene portion of the moiety. [0018] The term “cycloalkyl,” “carbocycle,” or “carbocyclic ring” refers to a hydrocarbon ring having the indicated number of ring atoms (e.g., C3-6 cycloalkyl) and being fully saturated or having no more than one double bond between ring vertices. “Cycloalkyl” is also meant to refer to bicyclic and polycyclic hydrocarbon rings such as, for example, bicyclo[2.2.1]heptane, bicyclo[2.2.2]octane, etc. In some embodiments, the cycloalkyl compounds of the present disclosure are monocyclic C3-6 cycloalkyl moieties (e.g., cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl). [0019] The term “heterocycloalkyl,” “heterocycle,” or “heterocyclic ring” refers to a cycloalkyl ring having the indicated number of ring vertices ( or members) and having from one to five heteroatoms selected from N, O, and S, which replace one to five of the carbon vertices, and wherein the nitrogen and sulfur atoms are optionally oxidized, and the nitrogen atom(s) are optionally quaternized. The heterocycloalkyl may be a monocyclic, a bicyclic or a polycyclic ring system, and may have one or two double bonds connecting ring vertices. Non limiting examples of heterocycloalkyl groups include pyrrolidine, imidazolidine, pyrazolidine, butyrolactam, valerolactam, imidazolidinone, hydantoin, dioxolane, phthalimide, piperidine, 1,4- dioxane, morpholine, thiomorpholine, thiomorpholine-S-oxide, thiomorpholine-S,S-oxide, piperazine, pyran, pyridone, 3-pyrroline, thiopyran, pyrone, tetrahydrofuran, tetrahydrothiophene, quinuclidine, and the like. A heterocycloalkyl group can be attached to the remainder of the molecule through a ring carbon or a heteroatom. In some embodiments, the heterocycle is a 5- to 6-membered heterocycle (e.g., pyrrolidine, tetrahydrofuran, tetrahydropyran, piperidine, piperazine, morpholine, and the like). [0020] As used herein, a wavy line, “ ”, that intersects a single, double or triple bond in any chemical structure depicted herein, represents that the point of attachment of the single, double, or triple bond to the remainder of the molecule. Additionally, a bond extending to the center of a ring (e.g., a phenyl ring) is meant to indicate attachment at any of the available ring vertices. One of skill in the art will understand that multiple substituents shown as being attached to a ring
will occupy ring vertices that provide stable compounds and are otherwise sterically compatible. For a divalent component, a representation is meant to include either orientation (forward or reverse). For example, the group “-C(O)NH-” is meant to include a linkage in either orientation: -C(O)NH- or -NHC(O)-, and similarly, "-O-CH2CH2-" is meant to include both -O-CH2CH2- and -CH2CH2-O-. [0021] The terms “halo” or “halogen,” by themselves or as part of another substituent, mean, unless otherwise stated, a fluorine, chlorine, bromine, or iodine atom. Additionally, terms such as “haloalkyl,” are meant to include monohaloalkyl and polyhaloalkyl. For example, the term “C1-C4 haloalkyl” is meant to include trifluoromethyl, 2,2,2-trifluoroethyl, 4-chlorobutyl, 3- bromopropyl, and the like. [0022] The term “aryl” means, unless otherwise stated, an aromatic, hydrocarbon group which can be a single ring or multiple rings (up to three rings) which are fused together or linked covalently. Non-limiting examples of aryl groups include phenyl, naphthyl, and biphenyl. The term is also meant to include fused cycloalkylphenyl, and heterocycloalkylphenyl ring systems such as, for example, indane, tetrahydronaphthalene, chromane, and isochromane rings. As a substituent group, the point of attachment to the remainder of the molecule, for a fused ring system can be through a carbon atom on the aromatic portion, a carbon atom on the cycloalkyl portion, or an atom on the heterocycloalkyl portion. In some embodiments, the aryl groups are phenyl. [0023] The term “heteroaryl” refers to monocyclic or fused bicyclic aryl groups (or rings) that contain from one to five heteroatoms selected from N, O, and S, wherein the nitrogen and sulfur atoms are optionally oxidized, and the nitrogen atom(s) are optionally quaternized. A heteroaryl group can be attached to the remainder of the molecule through a heteroatom, when chemically permissible. The term “heteroaryl” also embraces heteroaryl groups fused to phenyl rings. For fused bicyclic 9- to 10-membered heteroaryl groups containing a fused benzene ring, the heteroaryl group may be attached to the remainder of the molecule via the heteroaryl ring portion or the phenyl ring portion of the fused bicyclic heteroaryl group. Non-limiting examples of heteroaryl groups include pyridyl, pyridazinyl, pyrazinyl, pyrimindinyl, triazinyl, quinolinyl, quinoxalinyl, quinazolinyl, cinnolinyl, phthalazinyl, benzotriazinyl, purinyl, benzimidazolyl, benzopyrazolyl, benzotriazolyl, benzisoxazolyl, benzoxazolyl, isobenzofuryl, isoindolyl,
indolizinyl, benzotriazinyl, thienopyridinyl, thienopyrimidinyl, pyrrolopyridinyl, pyrazolopyrimidinyl, imidazolopyridinyl, imidazolopyridazinyl, benzothiazolyl, benzofuranyl, benzothienyl, indolyl, quinolyl, isoquinolyl, isothiazolyl, pyrazolyl, indazolyl, pteridinyl, imidazolyl, triazolyl, tetrazolyl, oxazolyl, isoxazolyl, thiadiazolyl, pyrrolyl, thiazolyl, furyl, thienyl and the like. Substituents for a heteroaryl ring can be selected from the group of acceptable substituents described below. In some embodiments, the heteroaryl groups are 5- to 9-membered heteroaryl groups having 1-3 ring heteroatoms independently selected from N, O and S (e.g., imidazolyl, pyrazolyl, triazolyl, thiazolyl, pyridinyl, pyrimidinyl, pyrrolopyridinyl, imidazolopyridinyl, imidazolopyridazinyl, benzothiazolyl, etc.). [0024] The above terms (e.g., “alkyl,” “aryl” and “heteroaryl”), in some embodiments, will be optionally substituted. Selected substituents for each type of radical are provided below. [0025] Optional substituents for the alkyl radicals (including those groups often referred to as alkylene, alkenyl, and alkynyl) can be a variety of groups, for example, groups selected from: halogen, -OR', -NR'R", -SR', -SiR'R"R"', -OC(O)R', -C(O)R', -CO2R', -CONR'R", -OC(O)NR'R", -NR"C(O)R', -NR'-C(O)NR"R"', -NR"C(O)2R', -NH-C(NH2)=NH, -NR'C(NH2)=NH, -NH-C(NH2)=NR', -S(O)R', -S(O)2R', -S(O)2NR'R", -NR'S(O)2R", -CN (cyano), -NO2, aryl, aryloxy, oxo (=O), cycloalkyl and heterocycloalkyl in a number ranging from zero to (2m'+ 1), where m' is the total number of carbon atoms in such radical. R', R", and R"' each independently refer to hydrogen, unsubstituted C1-8 alkyl, unsubstituted aryl, aryl substituted with 1-3 halogens, C1-8 alkoxy, C1-8 thioalkoxy groups, or unsubstituted aryl-C1-4 alkyl groups. When R' and R" are attached to the same nitrogen atom, they can be combined with the nitrogen atom to form a 3-, 4-, 5-, 6-, or 7-membered ring. For example, -NR'R" is meant to include 1-pyrrolidinyl and 4-morpholinyl. [0026] Optional substituents for the cycloalkyl and heterocycloalkyl radicals can be a variety of groups, for example, groups selected from: alkyl optionally substituted with -C(O)OR', halogen, -OR', -NR'R", -SR', -SiR'R"R"', -OC(O)R', -C(O)R', -CO2R', -CONR'R", -OC(O)NR'R", -NR"C(O)R', -NR'-C(O)NR"R"', -NR"C(O)2R', -NH-C(NH2)=NH, -NR'C(NH2)=NH, -NH-C(NH2)=NR', -S(O)R', -S(O)2R', -S(O)2NR'R", -NR'S(O)2R", -CN (cyano), -NO2, aryl, aryloxy, and oxo (=O). R', R" and R"' each independently refer to hydrogen,
unsubstituted C1-8 alkyl, unsubstituted aryl, aryl substituted with 1-3 halogens, C1-8 alkoxy, C1-8 thioalkoxy groups, or unsubstituted aryl-C1-4 alkyl groups. [0027] The optional substituents for the cycloalkyl and heterocycloalkyl radicals may also include olefins (=CR'R''), wherein R' and R" each independently refer to hydrogen, unsubstituted C1-8 alkyl, unsubstituted aryl, aryl substituted with 1-3 halogens, C1-8 alkoxy, C1-8 thioalkoxy groups, or unsubstituted aryl-C1-4 alkyl groups. For example, the olefin can be an unsubstituted olefin (=CH2). [0028] Similarly, optional substituents for the aryl and heteroaryl groups are varied and, for example, can be selected from: -halogen, -OR', -OC(O)R', -NR'R", -SR', -R', -CN, -NO2, -CO2R', -CONR'R", -C(O)R', -OC(O)NR'R", -NR"C(O)R', -NR"C(O)2R', -NR'-C(O)NR"R"', -NH- C(NH2)=NH, -NR'C(NH2)=NH, -NH-C(NH2)=NR', -S(O)R', -S(O)2R', -S(O)2NR'R", -NR'S(O)2R", -N3, perfluoro(C1-4)alkoxy, and perfluoro(C1-4)alkyl, in a number ranging from zero to the total number of open valences on the aromatic ring system; and where R', R" and R"' are independently selected from hydrogen, C1-8 alkyl, C1-8 haloalkyl, C3-6 cycloalkyl, C2-8 alkenyl, and C2-8 alkynyl. Other suitable substituents include each of the above aryl substituents attached to a ring atom by an alkylene tether of from 1-6 carbon atoms. [0029] Two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -T-C(O)-(CH2)q-U-, wherein T and U are independently -NH-, -O-, -CH2- or a single bond, and q is an integer of from 0 to 2. Alternatively, two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -A-(CRfRg)r-B-, wherein A and B are independently -CH2-, -O-, -NH-, -S-, -S(O)-, -S(O)2-, -S(O)2NR'-, or a single bond, r is an integer of from 1 to 3, and Rf and Rg are each independently H or halogen. One of the single bonds of the new ring so formed may optionally be replaced with a double bond. Alternatively, two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -(CH2)s-X-(CH2)t-, where s and t are independently integers of from 0 to 3, and X is -O-, -NR'-, -S-, -S(O)-, -S(O)2-, or -S(O)2NR' -. The substituent R' in -NR'- and -S(O)2NR'- is selected from hydrogen or unsubstituted C1-6 alkyl.
[0030] As used herein, the term “heteroatom” is meant to include oxygen (O), nitrogen (N), sulfur (S), and silicon (Si). [0031] The compounds of the present disclosure can be present in their neutral form, or as a pharmaceutically acceptable salt, isomer, polymorph or solvate thereof, and may be present in a crystalline form, amorphous form, or mixtures thereof. [0032] As referred to herein, “pharmaceutically acceptable salt” is meant to include salts of the compounds according to this disclosure that are prepared with relatively nontoxic acids or bases, depending on the particular substituents found on the compounds described herein. When compounds of the present disclosure contain relatively acidic functionalities, base addition salts can be obtained by contacting the neutral form of such compounds with a sufficient amount of the desired base, either neat or in a suitable inert solvent. Examples of salts derived from pharmaceutically acceptable inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic, manganous, potassium, sodium, zinc, and the like. Salts derived from pharmaceutically-acceptable organic bases include salts of primary, secondary and tertiary amines, including substituted amines, cyclic amines, naturally-occurring amines and the like, such as arginine, betaine, caffeine, choline, N,N’-dibenzylethylenediamine, diethylamine, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine and the like. When compounds of the present disclosure contain relatively basic functionalities, acid addition salts can be obtained by contacting the neutral form of such compounds with a sufficient amount of the desired acid, either neat or in a suitable inert solvent. Examples of pharmaceutically acceptable acid addition salts include those derived from inorganic acids like hydrochloric, hydrobromic, nitric, carbonic, monohydrogencarbonic, phosphoric, monohydrogenphosphoric, dihydrogenphosphoric, sulfuric, monohydrogensulfuric, hydriodic, or phosphorous acids and the like, as well as the salts derived from relatively nontoxic organic acids like acetic, propionic, isobutyric, malonic, benzoic, succinic, suberic, fumaric, mandelic, phthalic, benzenesulfonic, p-tolylsulfonic, citric, tartaric, methanesulfonic, maleic, oxalic, trans- cinnamic, and the like. Also included are salts of amino acids such as arginate and the like, and
salts of organic acids like glucuronic or galactunoric acids, and the like (see, for example, Berge, S.M., et al, “Pharmaceutical Salts”, Journal of Pharmaceutical Science, 1977, 66, 1-19). Certain specific compounds of the present disclosure contain both basic and acidic functionalities that allow the compounds to be converted into either base or acid addition salts. [0033] The neutral forms of the compounds may be regenerated by contacting the salt with a base or acid and isolating the parent compound in the conventional manner. The parent form of the compound differs from the various salt forms in certain physical properties, such as solubility in polar solvents, but otherwise the salts are equivalent to the parent form of the compound for the purposes of the present disclosure. [0034] This disclosure also contemplates isomers of the compounds described herein (e.g., stereoisomers, and atropisomers). For example, certain compounds of the present disclosure possess asymmetric carbon atoms (chiral centers), or hindered rotation about a single bond; the racemates, diastereomers, enantiomers, and atropisomers (e.g., Ra, Sa, P, and M isomers) of which are all intended to be encompassed within the scope of the present disclosure. Stereoisomeric forms may be defined, in terms of absolute stereochemistry, as (R) or (S), and/or depicted uses dashes and/or wedges. When a stereochemical depiction (e.g., using dashes, , and/or wedges, ) is shown in a chemical structure, or a stereochemical assignment (e.g., using (R) and (S) notation) is made in a chemical name, it is meant to indicate that the depicted isomer is present and substantially free of one or more other isomer(s) (e.g., enantiomers and diastereomers, when present). “Substantially free of” other isomer(s) indicates at least an 70/30 ratio of the indicated isomer to the other isomer(s), more preferably 80/20, 90/10, or 95/5 or more. In some embodiments, the indicated isomer will be present in an amount of at least 99%. A chemical bond to an asymmetric carbon that is depicted as a solid line ( ) indicates that all possible stereoisomers (e.g., enantiomers, diastereomers, racemic mixtures, etc.) at that carbon atom are included. In such instances, the compound may be present as a racemic mixture, scalemic mixture, or a mixture of diastereomers. [0035] The compounds of the present disclosure may also contain unnatural proportions of atomic isotopes at one or more of the atoms that constitute such compounds. Unnatural proportions of an isotope may be defined as ranging from the amount found in nature to an amount consisting of 100% of the atom in question. For example, the compounds may
incorporate radioactive isotopes, such as for example tritium (3H), iodine-125 (125I), or carbon-14 (14C), or non-radioactive isotopes, such as deuterium (2H) or carbon-13 (13C). Such isotopic variations can provide additional utilities to those described elsewhere herein. For instance, isotopic variants of the compounds of the disclosure may find additional utility, including but not limited to, as diagnostic and/or imaging reagents, or as cytotoxic/radiotoxic therapeutic agents. Additionally, isotopic variants of the compounds of the disclosure can have altered pharmacokinetic and pharmacodynamic characteristics which can contribute to enhanced safety, tolerability, or efficacy during treatment. All isotopic variations of the compounds of the present disclosure, whether radioactive or not, are intended to be encompassed within the scope of the present disclosure. In some embodiments, the compounds according to this disclosure are characterized by one or more deuterium atoms. [0036] The terms “patient” or “subject” are used interchangeably to refer to a human or a non- human animal (e.g., a mammal). [0037] As used herein, the term “administration” or "administering" of a compound or drug to a subject includes any route of introducing or delivering to a subject a compound to perform its intended function. Administration can be carried out by any suitable route, including but not limited to, orally, intranasally, parenterally (intravenously, intramuscularly, intraperitoneally, or subcutaneously), rectally, intrathecally, intratumorally or topically. Administration includes self- administration and the administration by another. In one embodiment, administration is oral. [0038] The terms “treat”, “treating”, treatment”, and the like refer to a course of action that eliminates, reduces, suppresses, mitigates, ameliorates, or prevents the worsening of, either temporarily or permanently, a disease, disorder, or condition to which the term applies, or at least one of the symptoms associated therewith. Treatment includes alleviation of symptoms, diminishment of extent of disease, inhibiting (e.g., arresting the development or further development of the disease, disorder or condition or clinical symptoms association therewith) an active disease, delaying or slowing of disease progression, improving the quality of life, and/or prolonging survival of a subject as compared to expected survival if not receiving treatment or as compared to a published standard of care therapy for a particular disease. [0039] The term “in need of treatment” as used herein refers to a judgment made by a physician or similar professional that a subject requires or will benefit from treatment. This
judgment is made based on a variety of factors that are in the realm of the physician’s expertise, which may include a positive diagnosis of a disease, disorder, or condition. [0040] The terms “prevent”, “preventing”, “prevention”, “prophylaxis”, and the like refer to a course of action initiated in a manner (e.g., prior to the onset of a disease, disorder, condition, or symptom thereof) so as to prevent, suppress, inhibit or reduce, either temporarily or permanently, a subject’s risk of developing a disease, disorder, condition, or the like (as determined by, for example, the absence of clinical symptoms) or delaying the onset thereof, generally in the context of a subject predisposed to having a particular disease, disorder, or condition. In certain instances, the terms also refer to slowing the progression of the disease, disorder, or condition or inhibiting progression thereof to a harmful or otherwise undesired state. Prevention also refers to a course of action initiated in a subject after the subject has been treated for a disease, disorder, condition, or a symptom associated therewith in order to prevent relapse of that disease, disorder, condition, or symptom. [0041] The term “in need of prevention” as used herein refers to a judgment made by a physician or other caregiver that a subject requires or will benefit from preventative care. This judgment is made based on a variety of factors that are in the realm of a physician’s or caregiver’s expertise. [0042] “Substantially pure” indicates that a component (e.g., a compound according to this disclosure) makes up greater than about 50% of the total content of the composition, and typically greater than about 60% of the total content. More typically, “substantially pure” refers to compositions in which at least 75%, at least 85%, at least 90%, or more of the total composition is the component of interest. In some cases, the component of interest will make up greater than about 90%, or greater than about 95%, of the total content of the composition. [0043] The term “mutant HIF-2α” as used herein refers to human HIF-2α protein that is characterized by one or more amino acid mutations in its sequence. Exemplary amino acid mutations include amino acid substitutions, insertions, or deletions. In some embodiments, the one or more mutations occur in the binding pocket within the PAS-B domain of the HIF-2α protein. In some embodiments, the mutation occurs as a substitution, insertion, or deletion of one or more amino acids listed in Table 1.
Table 1: Wild Type HIF-2α Binding Pocket Within the PAS-B Domain

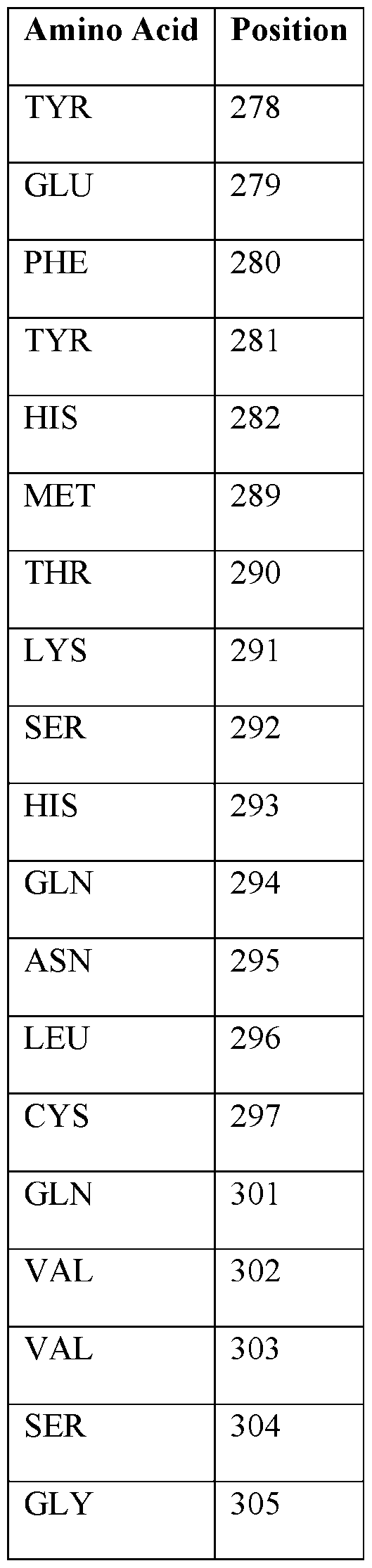
 [0044] The term “G323E mutant HIF-2α” as used herein refers to human HIF-2α protein with a G323E amino acid substitution, i.e., glycine (G) at position 323 of the HIF-2α protein is substituted with glutamic acid (E). [0045] Mutant HIF-2α may be identified by nucleic acid sequencing methods known in the art. For example, mutations can be detected using polymerase chain reaction (PCR). PCR steps amplify DNA regions of interest and multiplex single-base primer extension with dideoxynucleotides. Extension products can then be analyzed by, for example, mass spectrometry, which can distinguish different bases according to their mass-to-charge (m/z) ratio (Sequenom Mass Spectrometry system) or by capillary electrophoresis, which distinguishes bases by size and by the color of fluorescently labeled nucleotides (Applied Biosystems SNaPshot system). See Fumagalli et al., BMC Cancer, 10:101 (2010). Protein-based methods may also be used, including but not limited to sequencing HIF-2α protein purified (partially or completely) from a suitable sample. As another specific example, G323E mutant HIF-2α may be identified, for example by whole genome sequencing or targeted sequencing of the HIF-2α gene (EPAS1) and identification of a c.968G >A heterozygous mutation resulting in a G323E substitution, or by transcriptome analysis. Compounds of the Disclosure [0046] The present disclosure relates to compounds that inhibit the activity of mutant HIF-2α (e.g., G323E mutant HIF-2α), as well as compositions, methods, and uses including such compounds. [0047] In one aspect, the present disclosure is directed to a compound having a structure according to Formula I:
[0044] The term “G323E mutant HIF-2α” as used herein refers to human HIF-2α protein with a G323E amino acid substitution, i.e., glycine (G) at position 323 of the HIF-2α protein is substituted with glutamic acid (E). [0045] Mutant HIF-2α may be identified by nucleic acid sequencing methods known in the art. For example, mutations can be detected using polymerase chain reaction (PCR). PCR steps amplify DNA regions of interest and multiplex single-base primer extension with dideoxynucleotides. Extension products can then be analyzed by, for example, mass spectrometry, which can distinguish different bases according to their mass-to-charge (m/z) ratio (Sequenom Mass Spectrometry system) or by capillary electrophoresis, which distinguishes bases by size and by the color of fluorescently labeled nucleotides (Applied Biosystems SNaPshot system). See Fumagalli et al., BMC Cancer, 10:101 (2010). Protein-based methods may also be used, including but not limited to sequencing HIF-2α protein purified (partially or completely) from a suitable sample. As another specific example, G323E mutant HIF-2α may be identified, for example by whole genome sequencing or targeted sequencing of the HIF-2α gene (EPAS1) and identification of a c.968G >A heterozygous mutation resulting in a G323E substitution, or by transcriptome analysis. Compounds of the Disclosure [0046] The present disclosure relates to compounds that inhibit the activity of mutant HIF-2α (e.g., G323E mutant HIF-2α), as well as compositions, methods, and uses including such compounds. [0047] In one aspect, the present disclosure is directed to a compound having a structure according to Formula I:
 (Formula I) or a pharmaceutically acceptable salt thereof, wherein: n is 0, 1, or 2; each R1 when present is independently halo;
R2 and R3 are independently halo, C1-C6 alkyl or -CN; or R2 and R3 combine with the atoms to which they are attached to form a C4-C6 cycloalkyl, wherein said C4-C6 cycloalkyl is unsubstituted or substituted with 1-3 Ra; each Ra when present is independently halo or -OH; R4 is halo, C1-C6 haloalkyl, -CN, -S(O)2-R4a, 4- to 8-membered heterocycloalkyl, or 5- to 10-membered heteroaryl; wherein said 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatom or heteroatom groups independently selected from N, O, S, and S(O)2; said 5- to 10-membered heteroaryl has 1-3 ring heteroatoms independently selected from N, O, and S; and said 4- to 8-membered heterocycloalkyl and 5- to 10-membered heteroaryl are unsubstituted or substituted with 1-3 Rb; R4a is C1-C3 alkyl, C1-C3 haloalkyl, C3-C6 cycloalkyl, -NR4bR4c, or 4- to 6- membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S; R4b and R4c are each independently -H or C1-C3 alkyl; each Rb when present is independently -CN, C1-C6 alkyl, -N(Rc)2, or -C(O)- N(Rc)2; each Rc is independently -H or C1-C6 alkyl; X is CR5 or N; and R5 is -H or halo. [0048] In some embodiments, n is 1 or 2 and at least one R1 is F. In some embodiments, n is be 1 and R1 is F. In some embodiments, n is 2 and both R1 groups are independently F. In some embodiments, n is 1 or 2 and at least one R1 is Cl. In some embodiments, n is 1 or 2 and at least one R1 is Br. In some embodiments, n is 1 or 2 and at least one R1 is I. In some embodiments, n is 2 and both R1 groups are the same type of halo group. In some embodiments, n is 2 and each R1 is a different type of halo. In some embodiments, n is 0 and R1 is not present. [0049] In some embodiments, R2 is selected from the group consisting of halo, C1-C6 alkyl or -CN. In some embodiments, R2 is halo. In some embodiments, R2 is F. In some embodiments,
R2 is Cl. In some embodiments, R2 is Br. In some embodiments, R2 is I. In some embodiments, R2 is a C1-C6 alkyl. In some embodiments, R2 is methyl. In some embodiments, R2 is -CN. [0050] In some embodiments, R3 is selected from the group consisting of halo, C1-C6 alkyl, and -CN. In some embodiments, R3 is halo. In some embodiments, R3 is F. In some embodiments, R3 is Cl. In some embodiments, R3 is Br. In some embodiments, R3 is I. In some embodiments, R3 is C1-C6 alkyl. In some embodiments, R3 is -CN. [0051] In some embodiments, R2 is C1-C6 alkyl and R3 is -CN. In some embodiments, R2 is methyl and R3 is -CN. In some embodiments, R2 is -CN and R3 is C1-C6 alkyl. In some embodiments, R2 is C1-C6 alkyl and R3 is halo. In some embodiments, R2 is halo and R3 is a C1- C6 alkyl. In some embodiments, R2 is -CN and R3 is halo. In some embodiments, R2 is halo and R3 is -CN. [0052] In some embodiments, R2 and R3 combine with the atoms to which they are attached to form a C4-C6 cycloalkyl. In some embodiments, the C4-C6 cycloalkyl is unsubstituted. In some embodiments, R2 and R3 combine to form an unsubstituted cyclobutyl. In some embodiments, R2 and R3 combine to form an unsubstituted cyclopentyl. In some embodiments, R2 and R3 combine to form an unsubstituted cyclohexyl. [0053] In some embodiments, R2 and R3 combine with the atoms to which they are attached to form a C4-C6 cycloalkyl substituted with 1-3 Ra. In some embodiments, the C4-C6 cycloalkyl is substituted with 2-3 Ra. In some embodiments, the C4-C6 cycloalkyl is substituted with 1 Ra. In some embodiments, the C4-C6 cycloalkyl is substituted with 2 Ra. In some embodiments, the C4- C6 cycloalkyl is substituted with 3 Ra. In some embodiments, R2 and R3 combine to form a C4 cycloalkyl substituted with 1-3 Ra or 2-3 Ra. In some embodiments, R2 and R3 combine to form a C5 cycloalkyl substituted with 1-3 Ra or 2-3 Ra. In some embodiments, R2 and R3 combine to form a C6 cycloalkyl substituted with 1-3 Ra or 2-3 Ra. In some embodiments, each Ra is independently halo or -OH. In some embodiments, at least one Ra is halo. In some embodiments, at least one Ra is fluoro. In some embodiments, at least one Ra is chloro. In some embodiments, at least one Ra is bromo. In some embodiments, at least one Ra is iodo. In some embodiments, at least one Ra is -OH. In some embodiments, at least one Ra is fluoro and at least
one Ra is-OH. In some embodiments, each Ra is independently halo. In some embodiments, each Ra is independently -OH. [0054] In some embodiments, R4 is selected from the group consisting of halo, C1-C6 haloalkyl, -CN, -S(O)2-R4a, 4- to 8-membered heterocycloalkyl, and 5- to 10-membered heteroaryl; wherein said 4- to 8-membered heterocycloalkyl has 1-4 ring heteroatom or heteroatom groups or 1-3 heteroatom or heteroatom groups independently selected from N, O, S, and S(O)2, and said 4- to 8-membered heterocycloalkyl is unsubstituted or substituted with 1-3 Rb; and wherein said 5- to 10-membered heteroaryl has 1-4 ring heteroatoms or 1-3 ring heteroatoms independently selected from N, O, and S, and said 5- to 10-membered heteroaryl is unsubstituted or substituted with 1-3 Rb; wherein R4a is C1-C3 alkyl, C1-C3 haloalkyl, C3-C6 cycloalkyl, -NR4bR4c, or 4- to 6-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S; and each Rb when present is independently -CN, C1-C6 alkyl, -N(Rc)2, or -C(O)-N(Rc)2; and each Rc is independently -H or C1-C6 alkyl. In some embodiments, at least one ring heteroatom of the 4- to 8-membered heterocycloalkyl or 5- to 10- membered heteroaryl is N. In some embodiments, at least one ring heteroatom of the 4- to 8- membered heterocycloalkyl or 5- to 10-membered heteroaryl is O. In some embodiments, at least one ring heteroatom in the 4- to 8-membered heterocycloalkyl or 5- to 10-membered heteroaryl is S. In some embodiments, the 4- to 8-membered heterocycloalkyl or 5- to 10- membered heteroaryl has at least two ring heteroatoms and at least two ring heteroatoms are different types of heteroatoms. In some embodiments, the 4- to 8-membered heterocycloalkyl or 5- to 10-membered heteroaryl has at least two ring heteroatoms and at least two ring heteroatoms are the same type of heteroatom. [0055] In some embodiments, R4 is halo, -S(O)2-R4a, 4- to 8-membered heterocycloalkyl, or 5- to 10-membered heteroaryl; wherein said 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatom or heteroatom groups independently selected from N, O, S, and S(O)2; said 5- to 10- membered heteroaryl has 1-3 ring heteroatoms independently selected from N, O, and S; and said 4- to 8-membered heterocycloalkyl and 5- to 10-membered heteroaryl are unsubstituted, or substituted with 1-3 Rb. [0056] In some embodiments, R4 is halo or a 5- to 6-membered heteroaryl having 1-3 ring heteroatoms independently selected from N, O, and S; wherein said 5- to 6-membered heteroaryl
is unsubstituted, or substituted with 1-2 Rb. In some embodiments, Rb is independently -CN, C1- C6 alkyl, -N(Rc)2, or -C(O)-N(Rc)2; and each Rc is independently -H or C1-C6 alkyl. [0057] In some embodiments, R4 is a 5- to 10-membered heteroaryl, 5- to 8-membered heteroaryl, or 5- to 6-membered heteroaryl, wherein said 5- to 10-membered heteroaryl, 5- to 8- membered heteroaryl, or 5- to 6-membered heteroaryl has 1-3 ring heteroatoms independently selected from N, O, and S, and the heteroaryl is unsubstituted. In some embodiments, R4 is a 5- to 10-membered heteroaryl, 5- to 8-membered heteroaryl, or 5- to 6-membered heteroaryl, wherein said 5- to 10-membered heteroaryl, 5- to 8-membered heteroaryl, or 5- to 6-membered heteroaryl has 1-3 ring heteroatoms independently selected from N, O, and S, and the heteroaryl is substituted with 1-2 Rb, wherein Rb is independently -CN, C1-C6 alkyl, -N(Rc)2, or -C(O)- N(Rc)2; and each Rc is independently -H or C1-C6 alkyl. [0058] In some embodiments, R4 is halo, -S(O)2-R4a, 4- to 8-membered heterocycloalkyl, or 5- to 10-membered heteroaryl, wherein said 4- to 8-membered heterocycloalkyl and 5- to 10- membered heteroaryl have 1-4 ring heteroatoms independently selected from N, O, and S; and wherein said 4- to 8-membered heterocycloalkyl and 5- to 10-membered heteroaryl are unsubstituted or substituted with 1-2 Rb. In some embodiments, R4a is C1-C3 alkyl, or -NR4bR4c, wherein R4b and R4c are independently C1-C3 alkyl. [0059] In some embodiments, R4 is halo, 5- to 6-membered heteroaryl, or 5- to 8-membered heterocycloalkyl; wherein said 5- to 6-membered heteroaryl has 1-3 ring nitrogen atoms; said 5- to 8-membered heterocycloalkyl has 1-2 ring heteroatoms independently selected from N, and O; and said 5- to 6-membered heteroaryl and 5- to 8-membered heterocycloalkyl are unsubstituted or substituted with 1-2 Rb. In some embodiments, R4 is fluoro. In some embodiments, R4 is chloro. In some embodiments, R4 is bromo. In some embodiments, R4 is iodo. [0060] In some embodiments, R4 is selected from the group consisting of halo, C1-C6 haloalkyl, -CN, or -S(O)2-R4a; wherein R4a is selected from the group consisting of C1-C3 alkyl, C1-C3 haloalkyl, C3-C6 cycloalkyl, -NR4bR4c, and 4- to 6-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S; wherein R4b and R4c are each independently -H or C1-C3 alkyl. In some embodiments, R4a is C1-C3 alkyl or -NR4bR4c, wherein R4b and R4c are each independently C1-C3 alkyl. In some embodiments, R4a is a 5- to 6-
membered heterocycloalkyl having 1-4 ring heteroatoms, 1-3 ring heteroatoms, or 2-3 ring heteroatoms, wherein each heteroatom is independently selected from N, O, and S. [0061] In some embodiments, R4 is selected from the group consisting of unsubstituted triazolyl, triazolyl substituted with 1-2 Rb, unsubstituted imidazolyl, imidazolyl substituted with 1-2 Rb, unsubstituted pyrazolyl, pyrazolyl substituted with 1-2 Rb, unsubstituted pyridyl, and pyridyl substituted with 1-2 Rb. In some embodiments, each Rb, when present, is independently -CN, C1-C6 alkyl, -N(Rc)2, or -C(O)-N(Rc)2; and each Rc is independently -H or C1-C6 alkyl. In some embodiments, R4 is an unsubstituted tetrahydropyranyl. In some embodiments, R4 is tetrahydropyranyl substituted with 1-2 Rb. [0062] In some embodiments, R4 is Cl; -S(O)2Me; -S(O)2NMe2; a heteroaryl selected from the group consisting of
(Formula I) or a pharmaceutically acceptable salt thereof, wherein: n is 0, 1, or 2; each R1 when present is independently halo;
R2 and R3 are independently halo, C1-C6 alkyl or -CN; or R2 and R3 combine with the atoms to which they are attached to form a C4-C6 cycloalkyl, wherein said C4-C6 cycloalkyl is unsubstituted or substituted with 1-3 Ra; each Ra when present is independently halo or -OH; R4 is halo, C1-C6 haloalkyl, -CN, -S(O)2-R4a, 4- to 8-membered heterocycloalkyl, or 5- to 10-membered heteroaryl; wherein said 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatom or heteroatom groups independently selected from N, O, S, and S(O)2; said 5- to 10-membered heteroaryl has 1-3 ring heteroatoms independently selected from N, O, and S; and said 4- to 8-membered heterocycloalkyl and 5- to 10-membered heteroaryl are unsubstituted or substituted with 1-3 Rb; R4a is C1-C3 alkyl, C1-C3 haloalkyl, C3-C6 cycloalkyl, -NR4bR4c, or 4- to 6- membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S; R4b and R4c are each independently -H or C1-C3 alkyl; each Rb when present is independently -CN, C1-C6 alkyl, -N(Rc)2, or -C(O)- N(Rc)2; each Rc is independently -H or C1-C6 alkyl; X is CR5 or N; and R5 is -H or halo. [0048] In some embodiments, n is 1 or 2 and at least one R1 is F. In some embodiments, n is be 1 and R1 is F. In some embodiments, n is 2 and both R1 groups are independently F. In some embodiments, n is 1 or 2 and at least one R1 is Cl. In some embodiments, n is 1 or 2 and at least one R1 is Br. In some embodiments, n is 1 or 2 and at least one R1 is I. In some embodiments, n is 2 and both R1 groups are the same type of halo group. In some embodiments, n is 2 and each R1 is a different type of halo. In some embodiments, n is 0 and R1 is not present. [0049] In some embodiments, R2 is selected from the group consisting of halo, C1-C6 alkyl or -CN. In some embodiments, R2 is halo. In some embodiments, R2 is F. In some embodiments,
R2 is Cl. In some embodiments, R2 is Br. In some embodiments, R2 is I. In some embodiments, R2 is a C1-C6 alkyl. In some embodiments, R2 is methyl. In some embodiments, R2 is -CN. [0050] In some embodiments, R3 is selected from the group consisting of halo, C1-C6 alkyl, and -CN. In some embodiments, R3 is halo. In some embodiments, R3 is F. In some embodiments, R3 is Cl. In some embodiments, R3 is Br. In some embodiments, R3 is I. In some embodiments, R3 is C1-C6 alkyl. In some embodiments, R3 is -CN. [0051] In some embodiments, R2 is C1-C6 alkyl and R3 is -CN. In some embodiments, R2 is methyl and R3 is -CN. In some embodiments, R2 is -CN and R3 is C1-C6 alkyl. In some embodiments, R2 is C1-C6 alkyl and R3 is halo. In some embodiments, R2 is halo and R3 is a C1- C6 alkyl. In some embodiments, R2 is -CN and R3 is halo. In some embodiments, R2 is halo and R3 is -CN. [0052] In some embodiments, R2 and R3 combine with the atoms to which they are attached to form a C4-C6 cycloalkyl. In some embodiments, the C4-C6 cycloalkyl is unsubstituted. In some embodiments, R2 and R3 combine to form an unsubstituted cyclobutyl. In some embodiments, R2 and R3 combine to form an unsubstituted cyclopentyl. In some embodiments, R2 and R3 combine to form an unsubstituted cyclohexyl. [0053] In some embodiments, R2 and R3 combine with the atoms to which they are attached to form a C4-C6 cycloalkyl substituted with 1-3 Ra. In some embodiments, the C4-C6 cycloalkyl is substituted with 2-3 Ra. In some embodiments, the C4-C6 cycloalkyl is substituted with 1 Ra. In some embodiments, the C4-C6 cycloalkyl is substituted with 2 Ra. In some embodiments, the C4- C6 cycloalkyl is substituted with 3 Ra. In some embodiments, R2 and R3 combine to form a C4 cycloalkyl substituted with 1-3 Ra or 2-3 Ra. In some embodiments, R2 and R3 combine to form a C5 cycloalkyl substituted with 1-3 Ra or 2-3 Ra. In some embodiments, R2 and R3 combine to form a C6 cycloalkyl substituted with 1-3 Ra or 2-3 Ra. In some embodiments, each Ra is independently halo or -OH. In some embodiments, at least one Ra is halo. In some embodiments, at least one Ra is fluoro. In some embodiments, at least one Ra is chloro. In some embodiments, at least one Ra is bromo. In some embodiments, at least one Ra is iodo. In some embodiments, at least one Ra is -OH. In some embodiments, at least one Ra is fluoro and at least
one Ra is-OH. In some embodiments, each Ra is independently halo. In some embodiments, each Ra is independently -OH. [0054] In some embodiments, R4 is selected from the group consisting of halo, C1-C6 haloalkyl, -CN, -S(O)2-R4a, 4- to 8-membered heterocycloalkyl, and 5- to 10-membered heteroaryl; wherein said 4- to 8-membered heterocycloalkyl has 1-4 ring heteroatom or heteroatom groups or 1-3 heteroatom or heteroatom groups independently selected from N, O, S, and S(O)2, and said 4- to 8-membered heterocycloalkyl is unsubstituted or substituted with 1-3 Rb; and wherein said 5- to 10-membered heteroaryl has 1-4 ring heteroatoms or 1-3 ring heteroatoms independently selected from N, O, and S, and said 5- to 10-membered heteroaryl is unsubstituted or substituted with 1-3 Rb; wherein R4a is C1-C3 alkyl, C1-C3 haloalkyl, C3-C6 cycloalkyl, -NR4bR4c, or 4- to 6-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S; and each Rb when present is independently -CN, C1-C6 alkyl, -N(Rc)2, or -C(O)-N(Rc)2; and each Rc is independently -H or C1-C6 alkyl. In some embodiments, at least one ring heteroatom of the 4- to 8-membered heterocycloalkyl or 5- to 10- membered heteroaryl is N. In some embodiments, at least one ring heteroatom of the 4- to 8- membered heterocycloalkyl or 5- to 10-membered heteroaryl is O. In some embodiments, at least one ring heteroatom in the 4- to 8-membered heterocycloalkyl or 5- to 10-membered heteroaryl is S. In some embodiments, the 4- to 8-membered heterocycloalkyl or 5- to 10- membered heteroaryl has at least two ring heteroatoms and at least two ring heteroatoms are different types of heteroatoms. In some embodiments, the 4- to 8-membered heterocycloalkyl or 5- to 10-membered heteroaryl has at least two ring heteroatoms and at least two ring heteroatoms are the same type of heteroatom. [0055] In some embodiments, R4 is halo, -S(O)2-R4a, 4- to 8-membered heterocycloalkyl, or 5- to 10-membered heteroaryl; wherein said 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatom or heteroatom groups independently selected from N, O, S, and S(O)2; said 5- to 10- membered heteroaryl has 1-3 ring heteroatoms independently selected from N, O, and S; and said 4- to 8-membered heterocycloalkyl and 5- to 10-membered heteroaryl are unsubstituted, or substituted with 1-3 Rb. [0056] In some embodiments, R4 is halo or a 5- to 6-membered heteroaryl having 1-3 ring heteroatoms independently selected from N, O, and S; wherein said 5- to 6-membered heteroaryl
is unsubstituted, or substituted with 1-2 Rb. In some embodiments, Rb is independently -CN, C1- C6 alkyl, -N(Rc)2, or -C(O)-N(Rc)2; and each Rc is independently -H or C1-C6 alkyl. [0057] In some embodiments, R4 is a 5- to 10-membered heteroaryl, 5- to 8-membered heteroaryl, or 5- to 6-membered heteroaryl, wherein said 5- to 10-membered heteroaryl, 5- to 8- membered heteroaryl, or 5- to 6-membered heteroaryl has 1-3 ring heteroatoms independently selected from N, O, and S, and the heteroaryl is unsubstituted. In some embodiments, R4 is a 5- to 10-membered heteroaryl, 5- to 8-membered heteroaryl, or 5- to 6-membered heteroaryl, wherein said 5- to 10-membered heteroaryl, 5- to 8-membered heteroaryl, or 5- to 6-membered heteroaryl has 1-3 ring heteroatoms independently selected from N, O, and S, and the heteroaryl is substituted with 1-2 Rb, wherein Rb is independently -CN, C1-C6 alkyl, -N(Rc)2, or -C(O)- N(Rc)2; and each Rc is independently -H or C1-C6 alkyl. [0058] In some embodiments, R4 is halo, -S(O)2-R4a, 4- to 8-membered heterocycloalkyl, or 5- to 10-membered heteroaryl, wherein said 4- to 8-membered heterocycloalkyl and 5- to 10- membered heteroaryl have 1-4 ring heteroatoms independently selected from N, O, and S; and wherein said 4- to 8-membered heterocycloalkyl and 5- to 10-membered heteroaryl are unsubstituted or substituted with 1-2 Rb. In some embodiments, R4a is C1-C3 alkyl, or -NR4bR4c, wherein R4b and R4c are independently C1-C3 alkyl. [0059] In some embodiments, R4 is halo, 5- to 6-membered heteroaryl, or 5- to 8-membered heterocycloalkyl; wherein said 5- to 6-membered heteroaryl has 1-3 ring nitrogen atoms; said 5- to 8-membered heterocycloalkyl has 1-2 ring heteroatoms independently selected from N, and O; and said 5- to 6-membered heteroaryl and 5- to 8-membered heterocycloalkyl are unsubstituted or substituted with 1-2 Rb. In some embodiments, R4 is fluoro. In some embodiments, R4 is chloro. In some embodiments, R4 is bromo. In some embodiments, R4 is iodo. [0060] In some embodiments, R4 is selected from the group consisting of halo, C1-C6 haloalkyl, -CN, or -S(O)2-R4a; wherein R4a is selected from the group consisting of C1-C3 alkyl, C1-C3 haloalkyl, C3-C6 cycloalkyl, -NR4bR4c, and 4- to 6-membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S; wherein R4b and R4c are each independently -H or C1-C3 alkyl. In some embodiments, R4a is C1-C3 alkyl or -NR4bR4c, wherein R4b and R4c are each independently C1-C3 alkyl. In some embodiments, R4a is a 5- to 6-
membered heterocycloalkyl having 1-4 ring heteroatoms, 1-3 ring heteroatoms, or 2-3 ring heteroatoms, wherein each heteroatom is independently selected from N, O, and S. [0061] In some embodiments, R4 is selected from the group consisting of unsubstituted triazolyl, triazolyl substituted with 1-2 Rb, unsubstituted imidazolyl, imidazolyl substituted with 1-2 Rb, unsubstituted pyrazolyl, pyrazolyl substituted with 1-2 Rb, unsubstituted pyridyl, and pyridyl substituted with 1-2 Rb. In some embodiments, each Rb, when present, is independently -CN, C1-C6 alkyl, -N(Rc)2, or -C(O)-N(Rc)2; and each Rc is independently -H or C1-C6 alkyl. In some embodiments, R4 is an unsubstituted tetrahydropyranyl. In some embodiments, R4 is tetrahydropyranyl substituted with 1-2 Rb. [0062] In some embodiments, R4 is Cl; -S(O)2Me; -S(O)2NMe2; a heteroaryl selected from the group consisting of
 or a heterocycloalkyl selected from the group consisting of wherein
or a heterocycloalkyl selected from the group consisting of wherein
 each heteroaryl and heterocycloalkyl is unsubstituted, or substituted with 1-2 Rb. In some embodiments, Rb is methyl, -NH2, or -C(O)-NH2. In some embodiments, R4 is Cl, -S(O)2Me, -
each heteroaryl and heterocycloalkyl is unsubstituted, or substituted with 1-2 Rb. In some embodiments, Rb is methyl, -NH2, or -C(O)-NH2. In some embodiments, R4 is Cl, -S(O)2Me, -
 [0063] In some embodiments, R4 is Cl; -S(O)2Me; -S(O)2NMe2; a heteroaryl selected from the g ,
[0063] In some embodiments, R4 is Cl; -S(O)2Me; -S(O)2NMe2; a heteroaryl selected from the g ,
 the group consisting of
the group consisting of
 wherein each heteroaryl and heterocycloalkyl is unsubstituted, or substituted with 1-2 Rb. In some embodiments, Rb is methyl, -CN, -NH2, or -C(O)-NH2. In some embodiments, R4 is Cl, -S(O)2Me, -S(O)2NMe2,
wherein each heteroaryl and heterocycloalkyl is unsubstituted, or substituted with 1-2 Rb. In some embodiments, Rb is methyl, -CN, -NH2, or -C(O)-NH2. In some embodiments, R4 is Cl, -S(O)2Me, -S(O)2NMe2,
 [0064] In some embodiments, X is CR5 or N; and R5 is -H or halo. In some embodiments, X is CH. In some embodiments, X is C-halo. In some embodiments, X is C-F. In some embodiments, X is N. [0065] In some embodiments, the compound or pharmaceutically acceptable salt thereof, is a compound of Formula IA:
[0064] In some embodiments, X is CR5 or N; and R5 is -H or halo. In some embodiments, X is CH. In some embodiments, X is C-halo. In some embodiments, X is C-F. In some embodiments, X is N. [0065] In some embodiments, the compound or pharmaceutically acceptable salt thereof, is a compound of Formula IA:
 (Formula IA) where n is 0 or 1. [0066] In some embodiments, the compound or pharmaceutically acceptable salt thereof, is a compound of Formula IB:
(Formula IA) where n is 0 or 1. [0066] In some embodiments, the compound or pharmaceutically acceptable salt thereof, is a compound of Formula IB:
 (Formula Ib) where n is 0 or 1; and m is 2 or 3. [0067] In one or more embodiments, the compound or pharmaceutically acceptable salt thereof according to this disclosure is selected from the compounds provided in Table 2. Table 2.
(Formula Ib) where n is 0 or 1; and m is 2 or 3. [0067] In one or more embodiments, the compound or pharmaceutically acceptable salt thereof according to this disclosure is selected from the compounds provided in Table 2. Table 2.


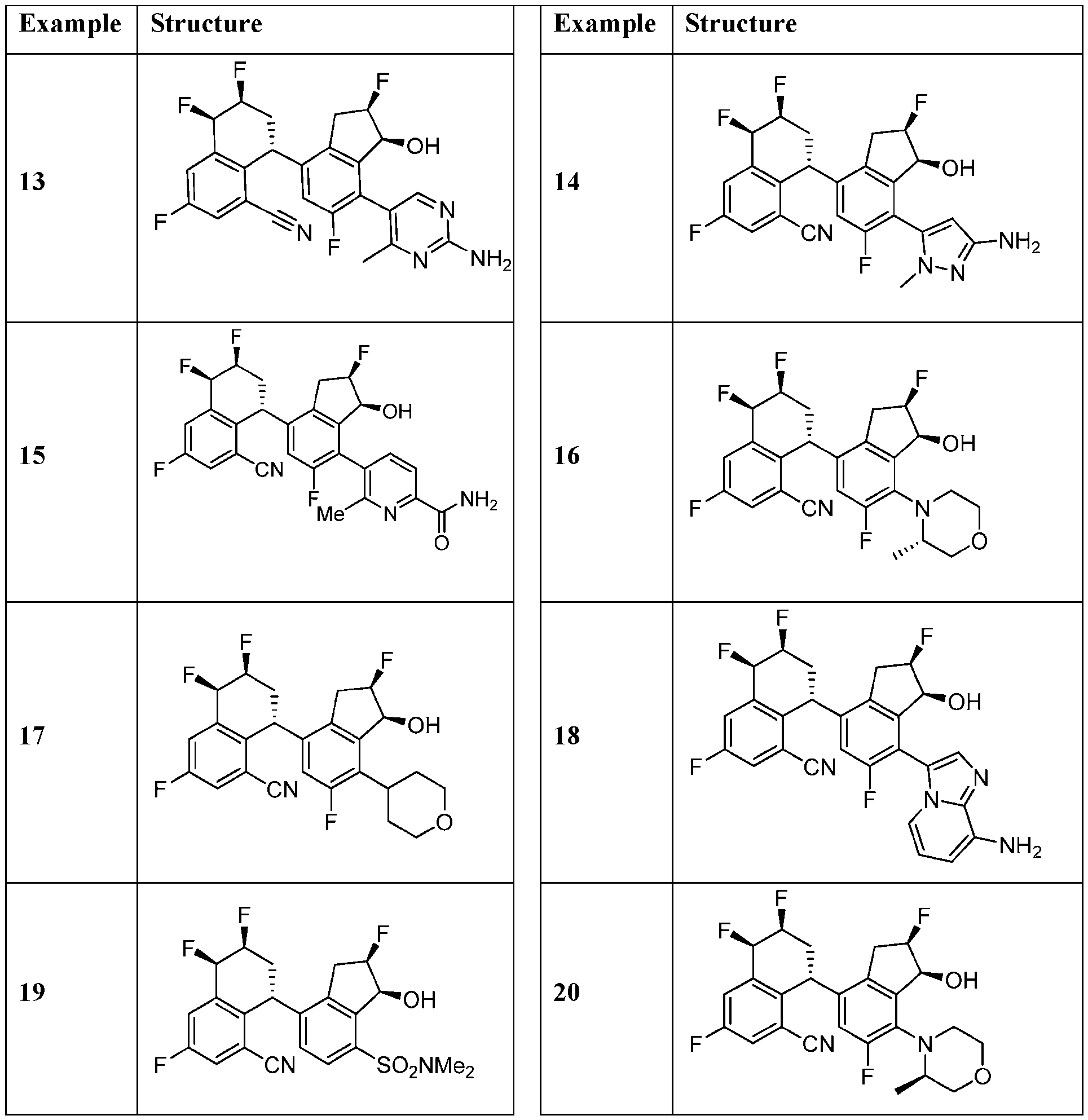 [0068] In one or more embodiments, the compound or pharmaceutically acceptable salt thereof according to this disclosure is selected from a compound provided in Table 3.
Table 3.
[0068] In one or more embodiments, the compound or pharmaceutically acceptable salt thereof according to this disclosure is selected from a compound provided in Table 3.
Table 3.
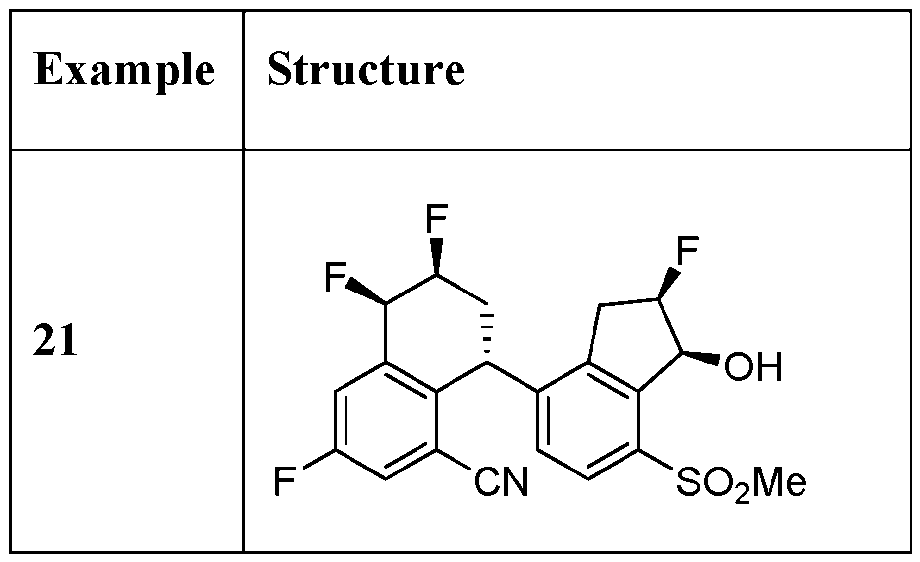 [0069] In one or more embodiments, the compound or pharmaceutically acceptable salt thereof according to this disclosure is selected from the compounds provided in Table 4. Table 4.
[0069] In one or more embodiments, the compound or pharmaceutically acceptable salt thereof according to this disclosure is selected from the compounds provided in Table 4. Table 4.


 [0070] In one or more embodiments, the compound according to this disclosure is selected from the compounds provided in any one of Tables 2-4. Method of Use [0071] The present disclosure provides methods for using the compounds described herein in the preparation of a medicament for inhibiting mutant HIF-2α. In some embodiments, the mutant HIF-2α is G323E mutant HIF-2α. As used herein, the terms “inhibit”, ‘inhibition” and the like refer to the ability of an antagonist to decrease the function or activity of a particular target, e.g., G323E mutant HIF-2α. The decrease is preferably at least a 50% and may be, for example, at least about 55%, at least about 60%, at least about 65%, at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, or at least about 95%. [0072] The present disclosure provides methods of treating a disease, disorder, and/or condition mediated by mutant HIF-2α, said method comprising administering a compound according to this disclosure, or pharmaceutically acceptable salt thereof, to a subject in need thereof. In some embodiments, the mutant HIF-2α is G323E mutant HIF-2α. Diseases, disorders and/or conditions contemplated are described in further detail below. [0073] The present disclosure also encompasses the use of the compounds described herein in the preparation of a medicament for the treatment or prevention of diseases, disorders, and/or conditions that would benefit from inhibition of mutant HIF-2α (e.g., G323E mutant HIF-2α). As one example, the present disclosure encompasses the use of the compounds described herein in the preparation of a medicament for the treatment of cancer. In some embodiments of the aforementioned methods, the compounds described herein are used in combination with at least one additional therapy, examples of which are set forth elsewhere herein. [0074] In some embodiments, diseases, disorders, and/or conditions that would benefit from mutant HIF-2α (e.g., G323E mutant HIF-2α) inhibition may be characterized by detectable mutant HIF-2α (e.g., G323E mutant HIF-2α) expression in one or more suitable samples (e.g., a tumor biopsy, surgical resection sample, etc.). In some embodiments, diseases, disorders, and/or conditions that would benefit from mutant HIF-2α (e.g., G323E mutant HIF-2α) inhibition may be characterized by increased mutant HIF-2α (e.g., G323E HIF-2α) expression in one or more
suitable samples (e.g., a tumor biopsy, surgical resection sample, etc.) as compared to a similar sample from (a) a healthy control, (b) a sample taken from the same subject at an earlier timepoint, or (c) a sample from a subject with another disease, disorder and/or condition not responsive to mutant HIF-2α (e.g., G323E HIF-2α) inhibition. In some embodiments, the disease, disorder and/or condition is cancer. In some embodiments, the disease, disorder and/or condition is Von Hippel-Lindau (VHL) disease (including VHL disease associated with renal cell carcinoma (RCC), central nervous system (CNS) hemangioblastomas, pancreatic neuroendocrine tumors (pNET), or solid tumors). [0075] Accordingly, in some embodiments, compounds described herein are administered to a subject in need thereof in an amount effective to inhibit mutant HIF-2α (e.g., G323E mutant HIF-2α). Mutant HIF-2α (e.g., G323E mutant HIF-2α) activity may be assessed using a peripheral blood sample or a tissue sample (e.g., a tumor sample) obtained from the subject. Activity may be determined, for example, by comparison to a previous sample obtained from the subject (i.e., prior to administration of the compound) or by comparison to a reference value for a control group (e.g., standard of care, a placebo, etc.). [0076] The present disclosure further provides a method of inhibiting mutant HIF-2α (e.g., G323E mutant HIF-2α), said method comprising contacting a cell of a subject with a compound according to this disclosure, or a pharmaceutically acceptable salt thereof. In some embodiments, the cell comprises mutant HIF-2α. In some embodiments, the cell comprises G323E HIF-2α. In some embodiments, the cell is a cancer cell. In further embodiments, the cancer cell is from a solid tumor, examples of which are provided in further detail below. [0077] Alternatively or in addition, in some embodiments, the compounds described herein are administered to a subject in need thereof to treat and/or prevent cancer or a cancer-related disease, disorder or condition. In some embodiments, the compounds described herein are administered to a subject in need thereof to treat cancer, optionally in combination with at least one additional therapy, examples of which are set forth elsewhere herein. [0078] Alternatively or in addition, in some embodiments, the compounds described herein are administered to a subject with von Hippel-Lindau (VHL) disease to treat and/or prevent a VHL- associated disease, disorder or condition. In some embodiments, the compounds described
herein are administered to a subject with von Hippel-Lindau (VHL) disease to treat and/or prevent associated renal cell carcinoma, central nervous system hemangioblastomas, or pancreatic neuroendocrine tumors, optionally wherein immediate surgery is not required. [0079] Oncology and Oncology-related Disorders. In one or more embodiments, the compounds described herein are useful in the treatment and/or prophylaxis of cancer (e.g., carcinomas, sarcomas, leukemias, lymphomas, myelomas, etc.). In certain embodiments, the cancer may be locally advanced and/or unresectable, metastatic, or at risk of becoming metastatic. Alternatively, or in addition, the cancer may be recurrent or no longer responding to a treatment, such as a standard of care treatment known to one of skill in the art. In some embodiments, the cancer is resistant treatment with a previous therapy (e.g., treatment with an inhibitor of HIF-2α). [0080] Exemplary types of cancer contemplated by this disclosure include cancer of the genitourinary tract (e.g., bladder, kidney, renal cell, penile, prostate, testicular, Von Hippel- Lindau disease, etc.), uterus, cervix, ovary, breast, gastrointestinal tract (e.g., esophagus, oropharynx, stomach, small or large intestines, colon, or rectum), bone, bone marrow, skin (e.g., melanoma), head and neck, liver, gall bladder, bile ducts, heart, lung, pancreas, salivary gland, adrenal gland, thyroid, brain (e.g., gliomas), ganglia, central nervous system (CNS), peripheral nervous system (PNS), the hematopoietic system (i.e., hematological malignancies), and the immune system (e.g., spleen or thymus). [0081] In some embodiments, the compounds according to this disclosure are useful in the treatment and/or prophylaxis of hematological malignancies. Exemplary types of cancer affecting the hematopoietic system include leukemias, lymphomas and myelomas, including acute myeloid leukemia, adult T-cell leukemia, T-cell large granular lymphocyte leukemia, acute lymphoblastic leukemia, chronic lymphocytic leukemia, chronic myelogenous leukemia, acute monocytic leukemia, Hodgkin’s and Non-Hodgkin’s lymphoma, Diffuse large B Cell lymphoma, and multiple myeloma. [0082] In another embodiment, the compounds according to this disclosure are useful in the treatment and/or prophylaxis of solid tumors. The solid tumor may be, for example, ovarian cancer, endometrial cancer, breast cancer, lung cancer (small cell or non-small cell), colon
cancer, prostate cancer, cervical cancer, biliary cancer, pancreatic cancer, gastric cancer, esophageal cancer, liver cancer (hepatocellular carcinoma), kidney cancer (renal cell carcinoma), head-and-neck tumors, mesothelioma, melanoma, sarcomas, central nervous system (CNS) hemangioblastomas, and brain tumors (e.g., gliomas, such as astrocytoma, oligodendroglioma and glioblastomas). [0083] In another embodiment, the compounds according to this disclosure are useful in the treatment and/or prophylaxis of lung cancer, genitourinary cancer, gastrointestinal cancer, or a combination thereof. [0084] In some embodiments, the compounds according to this disclosure are useful in the treatment and/or prophylaxis of gastrointestinal cancer, genitourinary cancer, gynecological cancer, lung cancer, or a combination thereof. [0085] In some embodiments, the compounds according to this disclosure are useful in the treatment of gastrointestinal (GI) cancer. In some embodiments, the GI cancer is colorectal cancer, pancreatic cancer, or liver cancer. In some embodiments, the GI cancer is an upper GI cancer, such as esophageal or gastric cancer. In further embodiments, the upper GI cancer is an adenocarcinoma, a squamous cell carcinoma, or any combination thereof. In still further embodiments, the upper GI cancer is esophageal adenocarcinoma (EAC), esophageal squamous cell carcinoma (ESCC), gastroesophageal junction adenocarcinoma (GEJ), gastric adenocarcinoma (also referred to herein as “gastric cancer”) or any combination thereof. [0086] In some embodiments, the compounds according to this disclosure are useful in the treatment of pancreatic cancer. In further embodiments, the pancreatic cancer is pancreatic neuroendocrine tumor or pancreatic adenocarcinoma. [0087] In some embodiments, the compounds according to this disclosure are useful in the treatment and/or prophylaxis of liver cancer. In further embodiments, the liver cancer is hepatocellular carcinoma. In other embodiments, the liver cancer is liver metastases. [0088] In some embodiments, the compounds according to this disclosure are useful in the treatment of genitourinary cancer. In some embodiments, the genitourinary cancer is bladder cancer, kidney cancer or prostate cancer.
[0089] In some embodiments, the compounds according to this disclosure are useful in the treatment of kidney cancer. In further embodiments, the kidney cancer is renal cell carcinoma. In still further embodiments, the renal cell carcinoma is clear cell renal carcinoma. [0090] In some embodiments, the compounds according to this disclosure are useful in the treatment of gynecological cancer. In some embodiments, the gynecological cancer is breast cancer, endometrial cancer, or ovarian cancer. In some embodiments, the gynecological cancer is hormone receptor positive (e.g., ERα-positive cancer, PR-positive cancer, ERα-positive and PR-positive cancer), HER2 positive cancer, HER2 over-expressing cancer, or any combination thereof. In still further embodiments, the cancer is triple negative cancer (e.g., ER, PR and HER2 negative). [0091] In some embodiments, the compounds according to this disclosure are useful in the treatment of lung cancer. In further embodiments, the lung cancer is mesothelioma, small cell lung cancer (SCLC) or non-small cell lung cancer (NSCLC). In still further embodiments, the lung cancer is NSCLC, optionally lung squamous cell carcinoma or lung adenocarcinoma. [0092] In some embodiments, the compounds according to this disclosure are useful in the treatment of a neuroendocrine tumor. In further embodiments, the neuroendocrine tumor is pancreatic neuroendocrine tumor, pheochromocytoma, paraganglioma, or a tumor of the adrenal gland (e.g., neuroblastoma). [0093] In some embodiments, the compounds according to this disclosure are useful in the treatment of brain cancer. In further embodiments, the brain cancer is a glioma. In still further embodiments, the glioma is an astrocytoma, an oligodendroglioma, or a glioblastoma. [0094] In some embodiments, the compounds according to this disclosure are useful in the treatment and/or prophylaxis of renal cell carcinoma or hepatocellular carcinoma. [0095] In the aforementioned embodiments, the methods of the present disclosure may be practiced in an adjuvant setting or neoadjuvant setting. Alternatively or in addition, the methods described herein may be indicated as a first line treatment, optionally in the treatment of locally advanced, unresectable, or metastatic cancer. In some embodiments, the methods described herein may be indicated as a second line, third line, or greater line of treatment, optionally in the
treatment of locally advanced, unresectable, or metastatic cancer. When indicated as a second line or greater treatment, in some embodiments an earlier line of therapy may have included an inhibitor of HIF-2α. [0096] The present disclosure also provides methods of treating or preventing other cancer- related diseases, disorders or conditions. The use of the term(s) cancer-related diseases, disorders and conditions is meant to refer broadly to conditions that are associated, directly or indirectly, with cancer and non-cancerous proliferative disease, and includes, e.g., angiogenesis, precancerous conditions such as dysplasia, and non-cancerous proliferative diseases disorders or conditions, such as benign proliferative breast disease and papillomas. For clarity, the term(s) cancer-related disease, disorder and condition do not include cancer per se. [0097] In general, the disclosed methods for treating or preventing cancer, or a cancer-related disease, disorder or condition, in a subject in need thereof comprise administering to the subject a compound disclosed here. In some embodiments, the present disclosure provides methods for treating or preventing cancer, or a cancer-related disease, disorder or condition with a compound disclosed herein and at least one additional therapy, examples of which are set forth elsewhere herein. [0098] Von-Hippel-Lindau Disease. In one or more embodiments, the compounds described herein are useful in the treatment and/or prophylaxis of von Hippel-Lindau (VHL) disease or VHL disease associated tumors. In some embodiments, the disease, disorder, and/or condition is VHL disease associated with renal cell carcinoma (RCC), central nervous system (CNS) hemangioblastomas, pancreatic neuroendocrine tumors (pNET), or solid tumors). [0099] Selection of patients. In some instances, the methods according to this disclosure may be provided in selected patients, for example subjects identified as having, e.g., detectable mutant HIF-2α expression. In some embodiments, the subject is identified as having an increased mutant HIF-2α expression as compared to a suitable control. In further embodiments, the detectable and/or increased mutant HIF-2α expression is measured from one or more suitable samples, including, e.g., a tumor biopsy, or sample obtained by surgical resection. In some embodiments a suitable control is selected from the group consisting of a sample from (a) a healthy subject, (b) a sample taken from the same subject at an earlier timepoint, or (c) a sample
from a subject with another disease, disorder and/or condition not responsive to mutant HIF-2α inhibition. [0100] In some embodiments, the methods according to this disclosure may be provided in selected patients, for example subjects identified as having, e.g., detectable G323E mutant HIF- 2α expression. In some embodiments, the subject is identified as having an increased G323E HIF-2α expression as compared to a suitable control. In further embodiments, the detectable and/or increased G323E HIF-2α expression is measured from one or more suitable samples, including, e.g., a tumor biopsy, or sample obtained by surgical resection. In some embodiments a suitable control is selected from the group consisting of a sample from (a) a healthy subject, (b) a sample taken from the same subject at an earlier timepoint, or (c) a sample from a subject with another disease, disorder and/or condition not responsive to G323E HIF-2α inhibition. [0101] Alternatively or in addition, in some embodiments, the methods according to this disclosure may be used in patients identified or previously identified as having a biomarker of hypoxia or pseudohypoxia, microsatellite instability, high tumor mutational burden as measured in a relevant tissue or sample, or malignancies with HIF stabilizing mutations. [0102] Alternatively or in addition, in some embodiments, the methods according to this disclosure may be used in patients having VHL disease (e.g., renal cell carcinoma, central nervous system hemangioblastomas, pancreatic neuroendocrine tumors, or solid tumors associated with VHL disease) who have undergone treatment with a HIF-2α inhibitor, but have developed resistance to said treatment. [0103] Alternatively or in addition, in some embodiments, the methods according to this disclosure may be used in patients identified as having partial or complete resistance to treatment with a HIF-2α inhibitor. In certain embodiments, the subject having partial or complete resistance to treatment with a HIF-2α inhibitor was previously administered a HIF-2α inhibitor. In certain such embodiments, the patient is no longer responding to treatment with the HIF-2α inhibitor, or the extent of the response has diminished. In some embodiments, the patient has a detectable amount of mutant HIF-2α expression. In some embodiments, the patient has a detectable amount of G323E mutant HIF-2α expression. In some embodiments, the patient has an increased amount of mutant HIF-2α expression as compared to a suitable control. In some
embodiments, the patient has an increased amount of G323E mutant HIF-2α expression as compared to a suitable control. In further embodiments, the detectable and/or increased mutant HIF-2α expression (e.g., G323E HIF-2α expression) is measured from one or more suitable samples, including, e.g., a tumor biopsy, or sample obtained by surgical resection. In some embodiments a suitable control is selected from the group consisting of a sample from (a) a healthy subject, (b) a sample taken from the same subject at an earlier timepoint, or (c) a sample from a subject with another disease, disorder and/or condition not responsive to mutant HIF-2α inhibition (e.g., G323E HIF-2α inhibition). Combination Therapy [0104] The present disclosure contemplates the use of compounds described herein alone or in combination with one or more additional therapy. Each additional therapy can be a therapeutic agent or another treatment modality. In embodiments comprising one or more additional therapeutic agents, each agent may target a different, but complementary, mechanism of action. The additional therapeutic agents can be small chemical molecules; macromolecules such as proteins, antibodies, peptibodies, peptides, DNA, RNA or fragments of such macromolecules; or cellular or gene therapies. Non-limiting examples of additional treatment modalities include surgical resection of a tumor, bone marrow transplant, radiation therapy, and photodynamic therapy. The use of a compound described herein in combination with one or more additional therapies may have a synergistic therapeutic or prophylactic effect on the underlying disease, disorder, or condition. In addition or alternatively, the combination therapy may allow for a dose reduction of one or more of the therapies, thereby ameliorating, reducing or eliminating adverse effects associated with one or more of the agents. [0105] In embodiments comprising one or more additional treatment modality, the compound can be administered before, after or during treatment with the additional treatment modality. In embodiments comprising one or more additional therapeutic agent, the therapeutic agents used in such combination therapy can be formulated as a single composition or as separate compositions. If administered separately, each therapeutic agent in the combination can be given at or around the same time, or at different times. Furthermore, the therapeutic agents are administered “in combination” even if they have different forms of administration (e.g., oral capsule and
intravenous), they are given at different dosing intervals, one therapeutic agent is given at a constant dosing regimen while another is titrated up, titrated down or discontinued, or each therapeutic agent in the combination is independently titrated up, titrated down, increased or decreased in dosage, or discontinued and/or resumed during a patient’s course of therapy. If the combination is formulated as separate compositions, in some embodiments, the separate compositions are provided together in a kit. Cancer Therapies [0106] The present disclosure contemplates the use of the compounds described herein in combination with one or more additional therapies useful in the treatment of cancer. [0107] In some embodiments, one or more of the additional therapies is an additional treatment modality. Exemplary treatment modalities include but are not limited to surgical resection of a tumor, bone marrow transplant, radiation therapy, and photodynamic therapy. [0108] In some embodiments, one or more of the additional therapeutic agents is a chemotherapeutic agent. Examples of chemotherapeutic agents include, but are not limited to, alkylating agents such as thiotepa and cyclophosphamide; alkyl sulfonates such as busulfan, improsulfan and piposulfan; aziridines such as benzodopa, carboquone, meturedopa, and uredopa; ethylenimines and methylamelamines including altretamine, triethylenemelamine, triethylenephosphoramide, triethylenethiophosphoramide and trimethylolomelamime; nitrogen mustards such as chlorambucil, chlornaphazine, cholophosphamide, estramustine, ifosfamide, mechlorethamine, mechlorethamine oxide hydrochloride, melphalan, novembichin, phenesterine, prednimustine, trofosfamide, uracil mustard; nitrosureas such as carmustine, chlorozotocin, fotemustine, lomustine, nimustine, ranimustine; antibiotics such as aclacinomysins, actinomycin, authramycin, azaserine, bleomycins, cactinomycin, calicheamicin, carabicin, caminomycin, carzinophilin, chromomycins, dactinomycin, daunorubicin, detorubicin, 6-diazo-5-oxo-L- norleucine, doxorubicin, epirubicin, esorubicin, idarubicin, marcellomycin, mitomycins, mycophenolic acid, nogalamycin, olivomycins, pomalidomide, peplomycin, potfiromycin, puromycin, quelamycin, rodorubicin, streptonigrin, streptozocin, tubercidin, ubenimex, zinostatin, zorubicin; anti-metabolites such as methotrexate and 5-fluorouracil (5-FU); folic acid analogs such as denopterin, methotrexate, pemetrexed, pteropterin, trimetrexate; purine analogs
such as fludarabine, 6-mercaptopurine, thiamiprine, thioguanine; pyrimidine analogs such as ancitabine, azacitidine, 6-azauridine, carmofur, cytarabine, dideoxyuridine, doxifluridine, enocitabine, floxuridine, 5-FU; androgens such as calusterone, dromostanolone propionate, epitiostanol, mepitiostane, testolactone; anti-adrenals such as aminoglutethimide, mitotane, trilostane; folic acid replenisher such as folinic acid; aceglatone; aldophosphamide glycoside; aminolevulinic acid; amsacrine; bestrabucil; bisantrene; edatraxate; defofamine; demecolcine; diaziquone; elformithine; elliptinium acetate; etoglucid; gallium nitrate; hydroxyurea; lentinan; lonidamine; mitoguazone; mitoxantrone; mopidamol; nitracrine; pentostatin; phenamet; pirarubicin; podophyllinic acid; 2-ethylhydrazide; procarbazine; razoxane; sizofiran; spirogermanium; tenuazonic acid; triaziquone; 2,2',2''-trichlorotriethylamine; urethan; vindesine; dacarbazine; mannomustine; mitobronitol; mitolactol; pipobroman; gacytosine; arabinoside (Ara-C); cyclophosphamide; thiotepa; taxoids, e.g., paclitaxel, nab paclitaxel, and docetaxel; chlorambucil; gemcitabine; 6-thioguanine; mercaptopurine; methotrexate; platinum and platinum coordination complexes such as cisplatin, carboplatin and oxaliplatin; vinblastine; etoposide (VP-16); ifosfamide; mitomycin C; mitoxantrone; vincristine; vinorelbine; navelbine; novantrone; teniposide; daunomycin; aminopterin; xeloda; ibandronate; CPT11; proteasome inhibitors such as bortezomib, carfilzomib and ixazomib; topoisomerase inhibitors such as irinotecan, topotecan, etoposide, mitoxantrone, teniposide; difluoromethylornithine (DMFO); retinoic acid; esperamicins; capecitabine; anthracyclines and pharmaceutically acceptable salts, acids or derivatives of any of the above. In certain embodiments, combination therapy comprises a chemotherapy regimen that includes one or more chemotherapeutic agents. In one embodiment, combination therapy comprises a chemotherapeutic regimen comprising one or more of FOLFOX (folinic acid, fluorouracil, and oxaliplatin), FOLFIRI (e.g., folinic acid, fluorouracil, and irinotecan), FOLFIRINOX (e.g., fluorouracil, leucovorin, irinotecan, and oxaliplatin), CAPOX (capecitabine and oxaliplatin), a taxoid (e.g., docetaxel, paclitaxel, nab- paclitaxel,etc.), a fluoropyrimidine-containing chemotherapeutic agent (e.g., fluorouracil, capecitabine, floxuridine), a platinum-containing chemotherapeutic agent, and/or gemcitabine. [0109] In some embodiments, one or more of the additional therapeutic agents is an inhibitor of a hypoxia-inducible factor (HIF) transcription factor, particularly HIF-2α. In some embodiments, an inhibitor if HIF-2α is administered in a prior line of therapy. Exemplary HIF-
2α inhibitors include belzutifan, AND021, BPI-452080, ARO-HIF2, PT-2385, AB521 (casdatifan), NKT-2152 (HS-10516), SMP-215, DFF332, and those described in WO 2021113436, WO 2021188769, and WO 2023077046, each of which is incorporated by reference herein. [0110] In some embodiments, one or more of the additional therapeutic agents is an immune checkpoint inhibitor. As used herein, the term “immune checkpoint inhibitor” refers to an antagonist of an inhibitory or co-inhibitory immune checkpoint. The terms “immune checkpoint inhibitor”, “checkpoint inhibitor” and “CPI” may be used herein interchangeably. Immune checkpoint inhibitors may antagonize an inhibitory or co-inhibitory immune checkpoint by interfering with receptor -ligand binding and/or altering receptor signaling. Examples of immune checkpoints (ligands and receptors), some of which are selectively upregulated in various types of cancer cells, that can be antagonized include PD-1 (programmed cell death protein 1); PD-L1 (PD1 ligand); BTLA (B and T lymphocyte attenuator); CTLA-4 (cytotoxic T-lymphocyte associated antigen 4); TIM-3 (T cell immunoglobulin and mucin domain containing protein 3); LAG-3 (lymphocyte activation gene 3); TIGIT (T cell immunoreceptor with Ig and ITIM domains); CD276 (B7-H3), PD-L2, Galectin 9, CEACAM-1, CD69, Galectin-1, CD113, GPR56, VISTA, 2B4, CD48, GARP, PD1H, LAIR1, TIM-1, and TIM-4, and Killer Inhibitory Receptors, which can be divided into two classes based on their structural features: i) killer cell immunoglobulin-like receptors (KIRs), and ii) C-type lectin receptors (members of the type II transmembrane receptor family). Also contemplated are other less well-defined immune checkpoints that have been described in the literature, including both receptors (e.g., the 2B4 (also known as CD244) receptor) and ligands (e.g., certain B7 family inhibitory ligands such B7- H3 (also known as CD276) and B7-H4 (also known as B7-S1, B7x and VCTN1)). [See Pardoll, (April 2012) Nature Rev. Cancer 12:252-64]. [0111] In some embodiments, an immune checkpoint inhibitor is a CTLA-4 antagonist. In further embodiments, the CTLA-4 antagonist can be an antagonistic CTLA-4 antibody. Suitable antagonistic CTLA-4 antibodies include, for example, monospecific antibodies such as ipilimumab or tremelimumab, as well as bispecific antibodies such as MEDI5752 and KN046.
[0112] In some embodiments, an immune checkpoint inhibitor is a PD-1 antagonist. In further embodiments, the PD-1 antagonist can be an antagonistic PD-1 antibody, small molecule or peptide. Suitable antagonistic PD-1 antibodies include, for example, monospecific antibodies such as balstilimab, budigalimab, camrelizumab, cosibelimab, dostarlimab, cemiplimab, ezabenlimab (BI-754091), MEDI-0680 (AMP-514; as described in WO2012/145493, which is incorporated by reference herein), nivolumab, pembrolizumab, pidilizumab (CT-011), pimivalimab, retifanlimab, sasanlimab, spartalizumab, sintilmab, tislelizumab, toripalimab, and zimberelimab; as well as bi-specific antibodies such as LY3434172. In still further embodiments, the PD-1 antagonist can be a recombinant protein composed of the extracellular domain of PD- L2 (B7-DC) fused to the Fc portion of IgGl (AMP-224). In certain embodiments, an immune checkpoint inhibitor is zimberelimab. [0113] In some embodiments, an immune checkpoint inhibitor is a PD-L1 antagonist. In further embodiments, the PD-L1 antagonist can be an antagonistic PD-L1 antibody. Suitable antagonistic PD-Ll antibodies include, for example, monospecific antibodies such as avelumab, atezolizumab, durvalumab, BMS-936559, and envafolimab as well as bi-specific antibodies such as LY3434172 and KN046. [0114] In some embodiments, an immune checkpoint inhibitor is a TIGIT antagonist. In further embodiments, the TIGIT antagonist can be an antagonistic TIGIT antibody. Suitable antagonistic anti-TIGIT antibodies include monospecific antibodies such as AGEN1327, AB308 (as described in WO2021247591, which is incorporated by reference herein), BMS 986207, COM902, domvanalimab, belrestotug, etigilimab, IBI-929, JS006, dargistotug, ociperlimab, SEA-TGT, tiragolumab, vibostolimab; as well as bi-specific antibodies such as AGEN1777 and AZD2936. In certain embodiments, an immune checkpoint inhibitor is an antagonistic anti- TIGIT antibody disclosed in WO2017152088 or WO2021247591, which are incorporated by reference herein. In certain embodiments, an immune checkpoint inhibitor is domvanalimab or AB308. [0115] In some embodiments, an immune checkpoint inhibitor is a LAG-3 antagonist. In further embodiments, the LAG-3 antagonist can be an antagonistic LAG-3 antibody. Suitable antagonistic LAG-3 antibodies include, for example, BMS-986016 (as described in WO10/19570
and WO14/08218, each of which is incorporated by reference herein), or IMP-731 or IMP-321 (as described in WO08/132601 and WO09/44273, each of which is incorporated by reference herein). [0116] In certain embodiments, an immune checkpoint inhibitor is a B7-H3 antagonist. In further embodiments, the B7-H3 antagonist is an antagonistic B7-H3 antibody. Suitable antagonist B7-H3 antibodies include, for example, enoblituzumab (MGA271; as described in WO11/109400, which is incorporated by reference herein), omburtumab, DS-7300a, ABBV-155, and SHR-A1811. [0117] In some embodiments, an immune checkpoint inhibitor is a TIM-3 antagonist. In further embodiments, the TIM-3 antagonist can be an antagonistic TIM-3 antibody. Suitable antagonistic TIM-3 antibodies include, for example, dostarlimab, sabatolimab, BMS-986258. And RG7769/RO7121661. [0118] In some embodiments, a compound according to this disclosure is administered with one or more than one additional therapy. In some embodiments, each additional therapy is independently selected from an immune checkpoint inhibitor, a chemotherapeutic agent, and radiation therapy. In further embodiments, of the above (a) the immune checkpoint inhibitor antagonizes PD-1, PD-L1, BTLA, LAG-3, a B7 family member, TIM-3, TIGIT, or CTLA-4, or a combination of any two or more thereof; (b) the immune checkpoint inhibitor antagonizes PD-1 or PD-L1; (c) the immune checkpoint inhibitor that antagonizes PD-1 or PD-L1 is selected from the group consisting of avelumab, atezolizumab, balstilimab, budigalimab, camrelizumab, cosibelimab, dostarlimab, durvalumab, emiplimab, envafolimab ezabenlimab, nivolumab, pembrolizumab, pidilizumab, pimivalimab, retifanlimab, sasanlimab, spartalizumab, sintilimab, tislelizumab, toripalimab, and zimberelimab; (d) the immune checkpoint inhibitor is zimberelimab; (e) the immune checkpoint inhibitor comprise an immune checkpoint inhibitor that antagonizes TIGIT; (f) the immune checkpoint inhibitor is domvanalimab or AB308; (g) the chemotherapeutic agent comprises a platinum-based, taxoid-based, or anthracycline-based chemotherapeutic agent, or a combination thereof; and (h) the chemotherapeutic agent is selected from the group consisting of gemcitabine, cisplatin, carboplatin, oxaliplatin, doxorubicin, docetaxel, paclitaxel, and nab-paclitaxel.
[0119] Selection of the additional therapeutic agent(s) may be informed by current standard of care for a particular cancer and/or mutational status of a subject’s cancer and/or stage of disease. Detailed standard of care guidelines are published, for example, by National Comprehensive Cancer Network (NCCN). See, for instance, NCCN Colon Cancer v1.2023, NCCN Rectal Cancer v1.2023, NCCN Hepatobiliary Cancers v1.2023, NCCN Hepatocellular Carcinoma v1.2023, NCCN Kidney Cancer, v4.2023, NCCN NSCLC v2.2023, NCCN Adenocarcinoma v1.2023. Routes of Administration [0120] In some embodiments, pharmaceutical compositions containing a compound according to this disclosure may be in a form suitable for oral administration. Oral administration may involve swallowing the formulation thereby allowing the compound to be absorbed into the bloodstream in the gastrointestinal tract. Alternatively, oral administration may involve buccal, lingual or sublingual administration, thereby allowing the compound to be absorbed into the blood stream through oral mucosa. [0121] In another embodiment, the pharmaceutical compositions containing a compound according to this disclosure may be in a form suitable for parenteral administration. Forms of parenteral administration include, but are not limited to, intravenous, intraarterial, intramuscular, intradermal, intraperitoneal, intrathecal, intracisternal, intracerebral, intracerebroventricular, intraventricular, and subcutaneous. Pharmaceutical compositions suitable for parenteral administration may be formulated using suitable aqueous or non-aqueous carriers. Depot injections, which are generally administered subcutaneously or intramuscularly, may also be utilized to release the compounds disclosed herein over a defined period of time. [0122] Other routes of administration are also contemplated by this disclosure, including, but not limited to, nasal, vaginal, intraocular, rectal, topical (e.g., transdermal), and inhalation. [0123] Particular embodiments of the present disclosure contemplate oral administration or parenteral administration.
Pharmaceutical Compositions [0124] The compounds of the present disclosure may be in the form of compositions suitable for administration to a subject. In general, such compositions are pharmaceutical compositions comprising a compound according to this disclosure or a pharmaceutically acceptable salt thereof and one or more pharmaceutically acceptable excipients. In certain embodiments, the compound, or pharmaceutically acceptable salt thereof, may be present in an effective amount. The pharmaceutical compositions may be used in the methods of the present disclosure; thus, for example, the pharmaceutical compositions comprising a compound according to this disclosure can be administered to a subject in order to practice the therapeutic and prophylactic methods and uses described herein. [0125] The pharmaceutical compositions of the present disclosure can be formulated to be compatible with the intended method or route of administration. Routes of administration may include those known in the art. Exemplary routes of administration are oral and parenteral. Furthermore, the pharmaceutical compositions may be used in combination with one or more other therapies described herein in order to treat or prevent the diseases, disorders and conditions as contemplated by the present disclosure. In one embodiment, one or more other therapeutic agents contemplated by this disclosure are included in the same pharmaceutical composition that comprises a compound according to this disclosure. In another embodiment, the one or more other therapeutical agents are in a composition that is separate from the pharmaceutical composition comprising the compound according to this disclosure. [0126] In one aspect, the compounds described herein may be administered orally. Oral administration may be via, for example, capsule or tablets. In making the pharmaceutical compositions that include the compound of Formula (I), or a pharmaceutically acceptable salt thereof, the tablet or capsule typically includes at least one pharmaceutically acceptable excipient. Non-limiting examples of pharmaceutically acceptable excipients include lactose, dextrose, sucrose, sorbitol, mannitol, starches, gum acacia, calcium phosphate, alginates, tragacanth, gelatin, calcium silicate, microcrystalline cellulose, polyvinylpyrrolidone, polyethylene glycol, cellulose, sterile water, syrup, and methyl cellulose. Additional pharmaceutically acceptable excipients include lubricating agents such as talc, magnesium
stearate, and mineral oil; wetting agents; emulsifying and suspending agents; preserving agents such as methyl and propylhydroxy-benzoates. [0127] In another aspect, the compounds described herein may be administered parenterally, for example by intravenous injection. A pharmaceutical composition appropriate for parenteral administration may be formulated in solution for injection or may be reconstituted for injection in an appropriate system such as a physiological solution. Such solutions may include sterile water for injection, salts, buffers, and tonicity excipients in amounts appropriate to achieve isotonicity with the appropriate physiology. [0128] The pharmaceutical compositions described herein may be stored in an appropriate sterile container or containers. In some embodiments, the container is designed to maintain stability for the pharmaceutical composition over a given period of time. Administering [0129] In general, the disclosed methods comprise administering a compound described herein, or a composition thereof, in an effective amount to a subject in need thereof. An “effective amount” with reference to a mutant HIF-2α inhibitor (e.g., a G323E mutant HIF-2α inhibitor), of the present disclosure means an amount of the compound that is sufficient to engage the target (by inhibiting, agonizing or antagonizing the target) at a level that is indicative of the potency of the compound. For mutant HIF-2α (e.g., G323E mutant HIF-2α), target engagement can be determined by one or more biochemical or cellular assays resulting in an EC50, ED50, EC90, IC50, or similar value which can be used as one assessment of the potency of the compound. Assays for determining target engagement include, but are not limited to, those described in the Examples. The effective amount may be administered as a single quantity or as multiple, smaller quantities (e.g., as one tablet with “x” amount, as two tablets each with “x/2” amount, etc.). [0130] In some embodiments, the disclosed methods comprise administering a therapeutically effective amount of a compound described herein to a subject in need thereof. As used herein, the phrase “therapeutically effective amount” with reference to compound means a dose regimen (i.e., amount and interval) of the compound that provides the specific pharmacological effect for
which the compound is administered to a subject in need of such treatment. For prophylactic use, a therapeutically effective amount may be effective to eliminate or reduce the risk, lessen the severity, or delay the onset of the disease, including biochemical, histological and/or behavioral signs or symptoms of the disease. For treatment, a therapeutically effective amount may be effective to reduce, ameliorate, or eliminate one or more signs or symptoms associated with a disease, delay disease progression, prolong survival, decrease the dose of other medication(s) required to treat the disease, or a combination thereof. With respect to cancer specifically, a therapeutically effective amount may, for example, result in the killing of cancer cells, reduce cancer cell counts, reduce tumor burden, eliminate tumors or metastasis, or reduce metastatic spread. A therapeutically effective amount may vary based on, for example, one or more of the following: the age and weight of the subject, the subject’s overall health, the stage of the subject’s disease, the route of administration, and prior or concomitant treatments. [0131] Administration may comprise one or more (e.g., one, two, or three or more) dosing cycles. [0132] In certain embodiments, the compounds contemplated by the present disclosure may be administered (e.g., orally, parenterally, etc.) at about 0.01 mg/kg to about 50 mg/kg, or about 1 mg/kg to about 25 mg/kg, of subject’s body weight per day, one or more times a day, a week, or a month, to obtain the desired effect. In some embodiments, a suitable weight-based dose of a compound contemplated by the present disclosure is used to determine a dose that is administered independent of a subject’s body weight In certain embodiments, the compounds of the present disclosure are administered (e.g., orally, parenterally, etc.) at fixed dosage levels of about 1 mg to about 1000 mg, particularly 1, 3, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, 500, 600, 750, 800, 900, or 1000 mg, one or more times a day, a week, or a month, to obtain the desired effect. [0133] In certain embodiments, the compound is contained in a “unit dosage form”. The phrase “unit dosage form” refers to physically discrete units, each unit containing a predetermined amount of the compound, either alone or in combination with one or more additional agents, sufficient to produce the desired effect. It will be appreciated that the
parameters of a unit dosage form will depend on the particular agent and the effect to be achieved. Methods of Synthesis General methods for compound preparation [0134] Compounds of Formula I, including the exemplary molecules described herein and represented in the claims, may be synthesized using various standard synthetic methods and techniques that are known to those skilled in the art. Compounds of Formula I and compounds having the structures described in the prior sections were synthesized from starting materials that are available from common commercial sources, or from starting materials that can be synthesized using standard synthetic methods. The compounds described herein and related compounds bearing different substituents can be prepared using methods that are derived from known chemical reactions and principals, which may be modified, as appropriate, to enable the introduction or modification of specific functional groups represented in the structures described herein. [0135] The following examples demonstrate the preparation of compounds of Formula I. EXPERIMENTAL [0136] The following examples are put forth so as to provide those of ordinary skill in the art with a complete disclosure and description of how to make and use the present disclosure, and are not intended to limit the scope of what the inventors regard as their invention. Efforts have been made to ensure accuracy with respect to numbers used (e.g., amounts, temperature, etc.), but some experimental errors and deviations should be accounted for. [0137] All reactions were performed using a Teflon-coated magnetic stir bar at the indicated temperature and were conducted under an inert atmosphere when stated. Purchased starting materials and reagents were generally used as received. Reactions were monitored by TLC (silica gel 60 with fluorescence F254, visualized with a short wave/long wave UV lamp) and/or LCMS (AGILENT® 1100 or 1200 series LCMS with UV detection at 254 or 280 nm using a binary solvent system [0.1% formic acid in MeCN/0.1% formic acid in H2O] using one of the following columns: AGILENT® Eclipse Plus C18 [3.5 μm, 4.6 mm i.d. × 100 mm], WATERS™ XSelect
HSS C18 [3.5 μm, 2.1 mm i.d. × 75 mm]). Flash chromatography was conducted on silica gel using an automated system (COMBIFLASH® RF+ manufactured by Teledyne ISCO), with detection wavelengths of 254 and 280 nm, and optionally equipped with an evaporative light scattering detector. Reverse phase preparative HPLC was conducted on an AGILENT® 1260 or 1290 Infinity series HPLC. Samples were eluted using a binary solvent system (MeCN/H2O with an acid modifier as needed – for example 0.1% TFA or 0.1% formic acid) with gradient elution on a Gemini C18110 Å column (21.2 mm i.d. ×x 250 mm) with variable wavelength detection. Final compounds obtained through preparative HPLC were concentrated through lyophilization. All assayed compounds were purified to ≥95% purity as determined by 1H NMR or LCMS (AGILENT® 1100 or 1200 series LCMS with UV detection at 254 or 280 nm using a binary solvent system [0.1% formic acid in MeCN/0.1% formic acid in H2O] using one of the following columns: AGILENT® Eclipse Plus C18 [3.5 μm, 4.6 mm i.d. × 100 mm], WATERS™ XSelect HSS C18 [3.5 μm, 2.1 mm i.d. × 75 mm]).1H NMR spectra were recorded on a Varian 400 MHz NMR spectrometer equipped with an Oxford AS400 magnet or a BRUKER® AVANCE NEO 400 MHz NMR. Chemical shifts (δ) are reported as parts per million (ppm) relative to residual undeuterated solvent as an internal reference. The abbreviations s, br s, d, t, q, dd, dt, ddd, and m stand for singlet, broad singlet, doublet, triplet, quartet, doublet of doublets, doublet of triplets, doublet of doublet of doublets, and multiplet, respectively. [0138] Unless indicated otherwise, temperature is in degrees Celsius (° C), and pressure is at or near atmospheric. Standard abbreviations are used, including the following: rt or RT=room temperature; min(s)=minute(s); h=hour(s); mg=milligram; g=gram; kg=kilogram; μL=microliter; ml or mL=milliliter; M=molar; mol=mole; mmol=millimole; sat.=saturated; sol.=solution; aq.=aqueous; calcd=calculated; equiv.=equivalents; H2=hydrogen gas ; H2SO4=sulfuric acid; DCM or CH2Cl2=dichloromethane; THF=tetrahydrofuran; PhMe=toluene; EtOAc=ethyl acetate; AcOEt=ethyl acetate; TFA=trifluoroacetic acid; MeCN or ACN=acetonitrile; DMF=N,N- dimethylformamide; DIAD=diisopropyl azodicarboxylate; DMP=Dess-Martin periodinane; DAST=diethylaminosulfur trifluoride; DMSO=dimethyl sulfoxide; Deoxo-Fluor=bis(2- methoxyethyl)aminosulfur trifluoride; Selectfluor™=(1-chloromethyl-4-fluoro-1,4- diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate); MeOH=methanol; Et3N=triethylamine; TMS-morpholine=4-(trimethylsilyl)morpholine; MOMBr=bromomethyl methyl ether; TBSOTf=tert-butyldimethylsilyl trifluoromethanesulfonate; TBAF=tetrabutylammonium
fluoride; HCO2H=formic acid; NaCl=sodium chloride; NH4Cl=ammonium chloride; Na2S2O3=sodiumthiosulfate; Na2SO4=sodium sulfate; NaHCO3=sodium bicarbonate; HCl=hydrochloric acid; KOAc=potassium acetate; B2Pin2=bis(pinacolato)diboron; Zn(CN)2=zinc cyanide; SPhos Pd 2nd Gen. or SPhos Pd G2= chloro(2-dicyclohexylphosphino- 2′,6′-dimethoxy-1,1′-biphenyl)[2-(2′-amino-1,1′-biphenyl)]palladium(II); Pd(dppf)Cl2=[1,1′- bis(diphenylphosphino)ferrocene] dichloropalladium(II); Pd/C=palladium on carbon; RuCl(p- cymene)[(R,R)-Ts-DPEN= [N-[(1R,2R)-2-(amino-κN)-1,2-diphenylethyl]-4- methylbenzenesulfonamidato-κN]chloro[(1,2,3,4,5,6-η)-1-methyl-4-(1-methylethyl)benzene]- ruthenium; RuCl(p-cymene)[(S,S)-TsDPEN]=[N-[(1S,2S)-2-(amino-κN)-1,2-diphenylethyl]-4- methylbenzenesulfonamidato-κN]chloro[(1,2,3,4,5,6-η)-1-methyl-4-(1-methylethyl)benzene]- ruthenium; PPh3=triphenylphospohine; MHz=megahertz; Hz=hertz; ppm=parts per million; ESI MS=electrospray ionization mass spectrometry; LCMS or LC/MS=liquid chromatography-mass spectrometry; TLC=thin layer chromatography; NMR=nuclear magnetic resonance; HPLC=high pressure liquid chromatography. Examples 1-4 [0139] The compounds of example numbers 1-4 and 21-32 were prepared as previously described in PCT publication number WO2021188769.
[0070] In one or more embodiments, the compound according to this disclosure is selected from the compounds provided in any one of Tables 2-4. Method of Use [0071] The present disclosure provides methods for using the compounds described herein in the preparation of a medicament for inhibiting mutant HIF-2α. In some embodiments, the mutant HIF-2α is G323E mutant HIF-2α. As used herein, the terms “inhibit”, ‘inhibition” and the like refer to the ability of an antagonist to decrease the function or activity of a particular target, e.g., G323E mutant HIF-2α. The decrease is preferably at least a 50% and may be, for example, at least about 55%, at least about 60%, at least about 65%, at least about 70%, at least about 75%, at least about 80%, at least about 85%, at least about 90%, or at least about 95%. [0072] The present disclosure provides methods of treating a disease, disorder, and/or condition mediated by mutant HIF-2α, said method comprising administering a compound according to this disclosure, or pharmaceutically acceptable salt thereof, to a subject in need thereof. In some embodiments, the mutant HIF-2α is G323E mutant HIF-2α. Diseases, disorders and/or conditions contemplated are described in further detail below. [0073] The present disclosure also encompasses the use of the compounds described herein in the preparation of a medicament for the treatment or prevention of diseases, disorders, and/or conditions that would benefit from inhibition of mutant HIF-2α (e.g., G323E mutant HIF-2α). As one example, the present disclosure encompasses the use of the compounds described herein in the preparation of a medicament for the treatment of cancer. In some embodiments of the aforementioned methods, the compounds described herein are used in combination with at least one additional therapy, examples of which are set forth elsewhere herein. [0074] In some embodiments, diseases, disorders, and/or conditions that would benefit from mutant HIF-2α (e.g., G323E mutant HIF-2α) inhibition may be characterized by detectable mutant HIF-2α (e.g., G323E mutant HIF-2α) expression in one or more suitable samples (e.g., a tumor biopsy, surgical resection sample, etc.). In some embodiments, diseases, disorders, and/or conditions that would benefit from mutant HIF-2α (e.g., G323E mutant HIF-2α) inhibition may be characterized by increased mutant HIF-2α (e.g., G323E HIF-2α) expression in one or more
suitable samples (e.g., a tumor biopsy, surgical resection sample, etc.) as compared to a similar sample from (a) a healthy control, (b) a sample taken from the same subject at an earlier timepoint, or (c) a sample from a subject with another disease, disorder and/or condition not responsive to mutant HIF-2α (e.g., G323E HIF-2α) inhibition. In some embodiments, the disease, disorder and/or condition is cancer. In some embodiments, the disease, disorder and/or condition is Von Hippel-Lindau (VHL) disease (including VHL disease associated with renal cell carcinoma (RCC), central nervous system (CNS) hemangioblastomas, pancreatic neuroendocrine tumors (pNET), or solid tumors). [0075] Accordingly, in some embodiments, compounds described herein are administered to a subject in need thereof in an amount effective to inhibit mutant HIF-2α (e.g., G323E mutant HIF-2α). Mutant HIF-2α (e.g., G323E mutant HIF-2α) activity may be assessed using a peripheral blood sample or a tissue sample (e.g., a tumor sample) obtained from the subject. Activity may be determined, for example, by comparison to a previous sample obtained from the subject (i.e., prior to administration of the compound) or by comparison to a reference value for a control group (e.g., standard of care, a placebo, etc.). [0076] The present disclosure further provides a method of inhibiting mutant HIF-2α (e.g., G323E mutant HIF-2α), said method comprising contacting a cell of a subject with a compound according to this disclosure, or a pharmaceutically acceptable salt thereof. In some embodiments, the cell comprises mutant HIF-2α. In some embodiments, the cell comprises G323E HIF-2α. In some embodiments, the cell is a cancer cell. In further embodiments, the cancer cell is from a solid tumor, examples of which are provided in further detail below. [0077] Alternatively or in addition, in some embodiments, the compounds described herein are administered to a subject in need thereof to treat and/or prevent cancer or a cancer-related disease, disorder or condition. In some embodiments, the compounds described herein are administered to a subject in need thereof to treat cancer, optionally in combination with at least one additional therapy, examples of which are set forth elsewhere herein. [0078] Alternatively or in addition, in some embodiments, the compounds described herein are administered to a subject with von Hippel-Lindau (VHL) disease to treat and/or prevent a VHL- associated disease, disorder or condition. In some embodiments, the compounds described
herein are administered to a subject with von Hippel-Lindau (VHL) disease to treat and/or prevent associated renal cell carcinoma, central nervous system hemangioblastomas, or pancreatic neuroendocrine tumors, optionally wherein immediate surgery is not required. [0079] Oncology and Oncology-related Disorders. In one or more embodiments, the compounds described herein are useful in the treatment and/or prophylaxis of cancer (e.g., carcinomas, sarcomas, leukemias, lymphomas, myelomas, etc.). In certain embodiments, the cancer may be locally advanced and/or unresectable, metastatic, or at risk of becoming metastatic. Alternatively, or in addition, the cancer may be recurrent or no longer responding to a treatment, such as a standard of care treatment known to one of skill in the art. In some embodiments, the cancer is resistant treatment with a previous therapy (e.g., treatment with an inhibitor of HIF-2α). [0080] Exemplary types of cancer contemplated by this disclosure include cancer of the genitourinary tract (e.g., bladder, kidney, renal cell, penile, prostate, testicular, Von Hippel- Lindau disease, etc.), uterus, cervix, ovary, breast, gastrointestinal tract (e.g., esophagus, oropharynx, stomach, small or large intestines, colon, or rectum), bone, bone marrow, skin (e.g., melanoma), head and neck, liver, gall bladder, bile ducts, heart, lung, pancreas, salivary gland, adrenal gland, thyroid, brain (e.g., gliomas), ganglia, central nervous system (CNS), peripheral nervous system (PNS), the hematopoietic system (i.e., hematological malignancies), and the immune system (e.g., spleen or thymus). [0081] In some embodiments, the compounds according to this disclosure are useful in the treatment and/or prophylaxis of hematological malignancies. Exemplary types of cancer affecting the hematopoietic system include leukemias, lymphomas and myelomas, including acute myeloid leukemia, adult T-cell leukemia, T-cell large granular lymphocyte leukemia, acute lymphoblastic leukemia, chronic lymphocytic leukemia, chronic myelogenous leukemia, acute monocytic leukemia, Hodgkin’s and Non-Hodgkin’s lymphoma, Diffuse large B Cell lymphoma, and multiple myeloma. [0082] In another embodiment, the compounds according to this disclosure are useful in the treatment and/or prophylaxis of solid tumors. The solid tumor may be, for example, ovarian cancer, endometrial cancer, breast cancer, lung cancer (small cell or non-small cell), colon
cancer, prostate cancer, cervical cancer, biliary cancer, pancreatic cancer, gastric cancer, esophageal cancer, liver cancer (hepatocellular carcinoma), kidney cancer (renal cell carcinoma), head-and-neck tumors, mesothelioma, melanoma, sarcomas, central nervous system (CNS) hemangioblastomas, and brain tumors (e.g., gliomas, such as astrocytoma, oligodendroglioma and glioblastomas). [0083] In another embodiment, the compounds according to this disclosure are useful in the treatment and/or prophylaxis of lung cancer, genitourinary cancer, gastrointestinal cancer, or a combination thereof. [0084] In some embodiments, the compounds according to this disclosure are useful in the treatment and/or prophylaxis of gastrointestinal cancer, genitourinary cancer, gynecological cancer, lung cancer, or a combination thereof. [0085] In some embodiments, the compounds according to this disclosure are useful in the treatment of gastrointestinal (GI) cancer. In some embodiments, the GI cancer is colorectal cancer, pancreatic cancer, or liver cancer. In some embodiments, the GI cancer is an upper GI cancer, such as esophageal or gastric cancer. In further embodiments, the upper GI cancer is an adenocarcinoma, a squamous cell carcinoma, or any combination thereof. In still further embodiments, the upper GI cancer is esophageal adenocarcinoma (EAC), esophageal squamous cell carcinoma (ESCC), gastroesophageal junction adenocarcinoma (GEJ), gastric adenocarcinoma (also referred to herein as “gastric cancer”) or any combination thereof. [0086] In some embodiments, the compounds according to this disclosure are useful in the treatment of pancreatic cancer. In further embodiments, the pancreatic cancer is pancreatic neuroendocrine tumor or pancreatic adenocarcinoma. [0087] In some embodiments, the compounds according to this disclosure are useful in the treatment and/or prophylaxis of liver cancer. In further embodiments, the liver cancer is hepatocellular carcinoma. In other embodiments, the liver cancer is liver metastases. [0088] In some embodiments, the compounds according to this disclosure are useful in the treatment of genitourinary cancer. In some embodiments, the genitourinary cancer is bladder cancer, kidney cancer or prostate cancer.
[0089] In some embodiments, the compounds according to this disclosure are useful in the treatment of kidney cancer. In further embodiments, the kidney cancer is renal cell carcinoma. In still further embodiments, the renal cell carcinoma is clear cell renal carcinoma. [0090] In some embodiments, the compounds according to this disclosure are useful in the treatment of gynecological cancer. In some embodiments, the gynecological cancer is breast cancer, endometrial cancer, or ovarian cancer. In some embodiments, the gynecological cancer is hormone receptor positive (e.g., ERα-positive cancer, PR-positive cancer, ERα-positive and PR-positive cancer), HER2 positive cancer, HER2 over-expressing cancer, or any combination thereof. In still further embodiments, the cancer is triple negative cancer (e.g., ER, PR and HER2 negative). [0091] In some embodiments, the compounds according to this disclosure are useful in the treatment of lung cancer. In further embodiments, the lung cancer is mesothelioma, small cell lung cancer (SCLC) or non-small cell lung cancer (NSCLC). In still further embodiments, the lung cancer is NSCLC, optionally lung squamous cell carcinoma or lung adenocarcinoma. [0092] In some embodiments, the compounds according to this disclosure are useful in the treatment of a neuroendocrine tumor. In further embodiments, the neuroendocrine tumor is pancreatic neuroendocrine tumor, pheochromocytoma, paraganglioma, or a tumor of the adrenal gland (e.g., neuroblastoma). [0093] In some embodiments, the compounds according to this disclosure are useful in the treatment of brain cancer. In further embodiments, the brain cancer is a glioma. In still further embodiments, the glioma is an astrocytoma, an oligodendroglioma, or a glioblastoma. [0094] In some embodiments, the compounds according to this disclosure are useful in the treatment and/or prophylaxis of renal cell carcinoma or hepatocellular carcinoma. [0095] In the aforementioned embodiments, the methods of the present disclosure may be practiced in an adjuvant setting or neoadjuvant setting. Alternatively or in addition, the methods described herein may be indicated as a first line treatment, optionally in the treatment of locally advanced, unresectable, or metastatic cancer. In some embodiments, the methods described herein may be indicated as a second line, third line, or greater line of treatment, optionally in the
treatment of locally advanced, unresectable, or metastatic cancer. When indicated as a second line or greater treatment, in some embodiments an earlier line of therapy may have included an inhibitor of HIF-2α. [0096] The present disclosure also provides methods of treating or preventing other cancer- related diseases, disorders or conditions. The use of the term(s) cancer-related diseases, disorders and conditions is meant to refer broadly to conditions that are associated, directly or indirectly, with cancer and non-cancerous proliferative disease, and includes, e.g., angiogenesis, precancerous conditions such as dysplasia, and non-cancerous proliferative diseases disorders or conditions, such as benign proliferative breast disease and papillomas. For clarity, the term(s) cancer-related disease, disorder and condition do not include cancer per se. [0097] In general, the disclosed methods for treating or preventing cancer, or a cancer-related disease, disorder or condition, in a subject in need thereof comprise administering to the subject a compound disclosed here. In some embodiments, the present disclosure provides methods for treating or preventing cancer, or a cancer-related disease, disorder or condition with a compound disclosed herein and at least one additional therapy, examples of which are set forth elsewhere herein. [0098] Von-Hippel-Lindau Disease. In one or more embodiments, the compounds described herein are useful in the treatment and/or prophylaxis of von Hippel-Lindau (VHL) disease or VHL disease associated tumors. In some embodiments, the disease, disorder, and/or condition is VHL disease associated with renal cell carcinoma (RCC), central nervous system (CNS) hemangioblastomas, pancreatic neuroendocrine tumors (pNET), or solid tumors). [0099] Selection of patients. In some instances, the methods according to this disclosure may be provided in selected patients, for example subjects identified as having, e.g., detectable mutant HIF-2α expression. In some embodiments, the subject is identified as having an increased mutant HIF-2α expression as compared to a suitable control. In further embodiments, the detectable and/or increased mutant HIF-2α expression is measured from one or more suitable samples, including, e.g., a tumor biopsy, or sample obtained by surgical resection. In some embodiments a suitable control is selected from the group consisting of a sample from (a) a healthy subject, (b) a sample taken from the same subject at an earlier timepoint, or (c) a sample
from a subject with another disease, disorder and/or condition not responsive to mutant HIF-2α inhibition. [0100] In some embodiments, the methods according to this disclosure may be provided in selected patients, for example subjects identified as having, e.g., detectable G323E mutant HIF- 2α expression. In some embodiments, the subject is identified as having an increased G323E HIF-2α expression as compared to a suitable control. In further embodiments, the detectable and/or increased G323E HIF-2α expression is measured from one or more suitable samples, including, e.g., a tumor biopsy, or sample obtained by surgical resection. In some embodiments a suitable control is selected from the group consisting of a sample from (a) a healthy subject, (b) a sample taken from the same subject at an earlier timepoint, or (c) a sample from a subject with another disease, disorder and/or condition not responsive to G323E HIF-2α inhibition. [0101] Alternatively or in addition, in some embodiments, the methods according to this disclosure may be used in patients identified or previously identified as having a biomarker of hypoxia or pseudohypoxia, microsatellite instability, high tumor mutational burden as measured in a relevant tissue or sample, or malignancies with HIF stabilizing mutations. [0102] Alternatively or in addition, in some embodiments, the methods according to this disclosure may be used in patients having VHL disease (e.g., renal cell carcinoma, central nervous system hemangioblastomas, pancreatic neuroendocrine tumors, or solid tumors associated with VHL disease) who have undergone treatment with a HIF-2α inhibitor, but have developed resistance to said treatment. [0103] Alternatively or in addition, in some embodiments, the methods according to this disclosure may be used in patients identified as having partial or complete resistance to treatment with a HIF-2α inhibitor. In certain embodiments, the subject having partial or complete resistance to treatment with a HIF-2α inhibitor was previously administered a HIF-2α inhibitor. In certain such embodiments, the patient is no longer responding to treatment with the HIF-2α inhibitor, or the extent of the response has diminished. In some embodiments, the patient has a detectable amount of mutant HIF-2α expression. In some embodiments, the patient has a detectable amount of G323E mutant HIF-2α expression. In some embodiments, the patient has an increased amount of mutant HIF-2α expression as compared to a suitable control. In some
embodiments, the patient has an increased amount of G323E mutant HIF-2α expression as compared to a suitable control. In further embodiments, the detectable and/or increased mutant HIF-2α expression (e.g., G323E HIF-2α expression) is measured from one or more suitable samples, including, e.g., a tumor biopsy, or sample obtained by surgical resection. In some embodiments a suitable control is selected from the group consisting of a sample from (a) a healthy subject, (b) a sample taken from the same subject at an earlier timepoint, or (c) a sample from a subject with another disease, disorder and/or condition not responsive to mutant HIF-2α inhibition (e.g., G323E HIF-2α inhibition). Combination Therapy [0104] The present disclosure contemplates the use of compounds described herein alone or in combination with one or more additional therapy. Each additional therapy can be a therapeutic agent or another treatment modality. In embodiments comprising one or more additional therapeutic agents, each agent may target a different, but complementary, mechanism of action. The additional therapeutic agents can be small chemical molecules; macromolecules such as proteins, antibodies, peptibodies, peptides, DNA, RNA or fragments of such macromolecules; or cellular or gene therapies. Non-limiting examples of additional treatment modalities include surgical resection of a tumor, bone marrow transplant, radiation therapy, and photodynamic therapy. The use of a compound described herein in combination with one or more additional therapies may have a synergistic therapeutic or prophylactic effect on the underlying disease, disorder, or condition. In addition or alternatively, the combination therapy may allow for a dose reduction of one or more of the therapies, thereby ameliorating, reducing or eliminating adverse effects associated with one or more of the agents. [0105] In embodiments comprising one or more additional treatment modality, the compound can be administered before, after or during treatment with the additional treatment modality. In embodiments comprising one or more additional therapeutic agent, the therapeutic agents used in such combination therapy can be formulated as a single composition or as separate compositions. If administered separately, each therapeutic agent in the combination can be given at or around the same time, or at different times. Furthermore, the therapeutic agents are administered “in combination” even if they have different forms of administration (e.g., oral capsule and
intravenous), they are given at different dosing intervals, one therapeutic agent is given at a constant dosing regimen while another is titrated up, titrated down or discontinued, or each therapeutic agent in the combination is independently titrated up, titrated down, increased or decreased in dosage, or discontinued and/or resumed during a patient’s course of therapy. If the combination is formulated as separate compositions, in some embodiments, the separate compositions are provided together in a kit. Cancer Therapies [0106] The present disclosure contemplates the use of the compounds described herein in combination with one or more additional therapies useful in the treatment of cancer. [0107] In some embodiments, one or more of the additional therapies is an additional treatment modality. Exemplary treatment modalities include but are not limited to surgical resection of a tumor, bone marrow transplant, radiation therapy, and photodynamic therapy. [0108] In some embodiments, one or more of the additional therapeutic agents is a chemotherapeutic agent. Examples of chemotherapeutic agents include, but are not limited to, alkylating agents such as thiotepa and cyclophosphamide; alkyl sulfonates such as busulfan, improsulfan and piposulfan; aziridines such as benzodopa, carboquone, meturedopa, and uredopa; ethylenimines and methylamelamines including altretamine, triethylenemelamine, triethylenephosphoramide, triethylenethiophosphoramide and trimethylolomelamime; nitrogen mustards such as chlorambucil, chlornaphazine, cholophosphamide, estramustine, ifosfamide, mechlorethamine, mechlorethamine oxide hydrochloride, melphalan, novembichin, phenesterine, prednimustine, trofosfamide, uracil mustard; nitrosureas such as carmustine, chlorozotocin, fotemustine, lomustine, nimustine, ranimustine; antibiotics such as aclacinomysins, actinomycin, authramycin, azaserine, bleomycins, cactinomycin, calicheamicin, carabicin, caminomycin, carzinophilin, chromomycins, dactinomycin, daunorubicin, detorubicin, 6-diazo-5-oxo-L- norleucine, doxorubicin, epirubicin, esorubicin, idarubicin, marcellomycin, mitomycins, mycophenolic acid, nogalamycin, olivomycins, pomalidomide, peplomycin, potfiromycin, puromycin, quelamycin, rodorubicin, streptonigrin, streptozocin, tubercidin, ubenimex, zinostatin, zorubicin; anti-metabolites such as methotrexate and 5-fluorouracil (5-FU); folic acid analogs such as denopterin, methotrexate, pemetrexed, pteropterin, trimetrexate; purine analogs
such as fludarabine, 6-mercaptopurine, thiamiprine, thioguanine; pyrimidine analogs such as ancitabine, azacitidine, 6-azauridine, carmofur, cytarabine, dideoxyuridine, doxifluridine, enocitabine, floxuridine, 5-FU; androgens such as calusterone, dromostanolone propionate, epitiostanol, mepitiostane, testolactone; anti-adrenals such as aminoglutethimide, mitotane, trilostane; folic acid replenisher such as folinic acid; aceglatone; aldophosphamide glycoside; aminolevulinic acid; amsacrine; bestrabucil; bisantrene; edatraxate; defofamine; demecolcine; diaziquone; elformithine; elliptinium acetate; etoglucid; gallium nitrate; hydroxyurea; lentinan; lonidamine; mitoguazone; mitoxantrone; mopidamol; nitracrine; pentostatin; phenamet; pirarubicin; podophyllinic acid; 2-ethylhydrazide; procarbazine; razoxane; sizofiran; spirogermanium; tenuazonic acid; triaziquone; 2,2',2''-trichlorotriethylamine; urethan; vindesine; dacarbazine; mannomustine; mitobronitol; mitolactol; pipobroman; gacytosine; arabinoside (Ara-C); cyclophosphamide; thiotepa; taxoids, e.g., paclitaxel, nab paclitaxel, and docetaxel; chlorambucil; gemcitabine; 6-thioguanine; mercaptopurine; methotrexate; platinum and platinum coordination complexes such as cisplatin, carboplatin and oxaliplatin; vinblastine; etoposide (VP-16); ifosfamide; mitomycin C; mitoxantrone; vincristine; vinorelbine; navelbine; novantrone; teniposide; daunomycin; aminopterin; xeloda; ibandronate; CPT11; proteasome inhibitors such as bortezomib, carfilzomib and ixazomib; topoisomerase inhibitors such as irinotecan, topotecan, etoposide, mitoxantrone, teniposide; difluoromethylornithine (DMFO); retinoic acid; esperamicins; capecitabine; anthracyclines and pharmaceutically acceptable salts, acids or derivatives of any of the above. In certain embodiments, combination therapy comprises a chemotherapy regimen that includes one or more chemotherapeutic agents. In one embodiment, combination therapy comprises a chemotherapeutic regimen comprising one or more of FOLFOX (folinic acid, fluorouracil, and oxaliplatin), FOLFIRI (e.g., folinic acid, fluorouracil, and irinotecan), FOLFIRINOX (e.g., fluorouracil, leucovorin, irinotecan, and oxaliplatin), CAPOX (capecitabine and oxaliplatin), a taxoid (e.g., docetaxel, paclitaxel, nab- paclitaxel,etc.), a fluoropyrimidine-containing chemotherapeutic agent (e.g., fluorouracil, capecitabine, floxuridine), a platinum-containing chemotherapeutic agent, and/or gemcitabine. [0109] In some embodiments, one or more of the additional therapeutic agents is an inhibitor of a hypoxia-inducible factor (HIF) transcription factor, particularly HIF-2α. In some embodiments, an inhibitor if HIF-2α is administered in a prior line of therapy. Exemplary HIF-
2α inhibitors include belzutifan, AND021, BPI-452080, ARO-HIF2, PT-2385, AB521 (casdatifan), NKT-2152 (HS-10516), SMP-215, DFF332, and those described in WO 2021113436, WO 2021188769, and WO 2023077046, each of which is incorporated by reference herein. [0110] In some embodiments, one or more of the additional therapeutic agents is an immune checkpoint inhibitor. As used herein, the term “immune checkpoint inhibitor” refers to an antagonist of an inhibitory or co-inhibitory immune checkpoint. The terms “immune checkpoint inhibitor”, “checkpoint inhibitor” and “CPI” may be used herein interchangeably. Immune checkpoint inhibitors may antagonize an inhibitory or co-inhibitory immune checkpoint by interfering with receptor -ligand binding and/or altering receptor signaling. Examples of immune checkpoints (ligands and receptors), some of which are selectively upregulated in various types of cancer cells, that can be antagonized include PD-1 (programmed cell death protein 1); PD-L1 (PD1 ligand); BTLA (B and T lymphocyte attenuator); CTLA-4 (cytotoxic T-lymphocyte associated antigen 4); TIM-3 (T cell immunoglobulin and mucin domain containing protein 3); LAG-3 (lymphocyte activation gene 3); TIGIT (T cell immunoreceptor with Ig and ITIM domains); CD276 (B7-H3), PD-L2, Galectin 9, CEACAM-1, CD69, Galectin-1, CD113, GPR56, VISTA, 2B4, CD48, GARP, PD1H, LAIR1, TIM-1, and TIM-4, and Killer Inhibitory Receptors, which can be divided into two classes based on their structural features: i) killer cell immunoglobulin-like receptors (KIRs), and ii) C-type lectin receptors (members of the type II transmembrane receptor family). Also contemplated are other less well-defined immune checkpoints that have been described in the literature, including both receptors (e.g., the 2B4 (also known as CD244) receptor) and ligands (e.g., certain B7 family inhibitory ligands such B7- H3 (also known as CD276) and B7-H4 (also known as B7-S1, B7x and VCTN1)). [See Pardoll, (April 2012) Nature Rev. Cancer 12:252-64]. [0111] In some embodiments, an immune checkpoint inhibitor is a CTLA-4 antagonist. In further embodiments, the CTLA-4 antagonist can be an antagonistic CTLA-4 antibody. Suitable antagonistic CTLA-4 antibodies include, for example, monospecific antibodies such as ipilimumab or tremelimumab, as well as bispecific antibodies such as MEDI5752 and KN046.
[0112] In some embodiments, an immune checkpoint inhibitor is a PD-1 antagonist. In further embodiments, the PD-1 antagonist can be an antagonistic PD-1 antibody, small molecule or peptide. Suitable antagonistic PD-1 antibodies include, for example, monospecific antibodies such as balstilimab, budigalimab, camrelizumab, cosibelimab, dostarlimab, cemiplimab, ezabenlimab (BI-754091), MEDI-0680 (AMP-514; as described in WO2012/145493, which is incorporated by reference herein), nivolumab, pembrolizumab, pidilizumab (CT-011), pimivalimab, retifanlimab, sasanlimab, spartalizumab, sintilmab, tislelizumab, toripalimab, and zimberelimab; as well as bi-specific antibodies such as LY3434172. In still further embodiments, the PD-1 antagonist can be a recombinant protein composed of the extracellular domain of PD- L2 (B7-DC) fused to the Fc portion of IgGl (AMP-224). In certain embodiments, an immune checkpoint inhibitor is zimberelimab. [0113] In some embodiments, an immune checkpoint inhibitor is a PD-L1 antagonist. In further embodiments, the PD-L1 antagonist can be an antagonistic PD-L1 antibody. Suitable antagonistic PD-Ll antibodies include, for example, monospecific antibodies such as avelumab, atezolizumab, durvalumab, BMS-936559, and envafolimab as well as bi-specific antibodies such as LY3434172 and KN046. [0114] In some embodiments, an immune checkpoint inhibitor is a TIGIT antagonist. In further embodiments, the TIGIT antagonist can be an antagonistic TIGIT antibody. Suitable antagonistic anti-TIGIT antibodies include monospecific antibodies such as AGEN1327, AB308 (as described in WO2021247591, which is incorporated by reference herein), BMS 986207, COM902, domvanalimab, belrestotug, etigilimab, IBI-929, JS006, dargistotug, ociperlimab, SEA-TGT, tiragolumab, vibostolimab; as well as bi-specific antibodies such as AGEN1777 and AZD2936. In certain embodiments, an immune checkpoint inhibitor is an antagonistic anti- TIGIT antibody disclosed in WO2017152088 or WO2021247591, which are incorporated by reference herein. In certain embodiments, an immune checkpoint inhibitor is domvanalimab or AB308. [0115] In some embodiments, an immune checkpoint inhibitor is a LAG-3 antagonist. In further embodiments, the LAG-3 antagonist can be an antagonistic LAG-3 antibody. Suitable antagonistic LAG-3 antibodies include, for example, BMS-986016 (as described in WO10/19570
and WO14/08218, each of which is incorporated by reference herein), or IMP-731 or IMP-321 (as described in WO08/132601 and WO09/44273, each of which is incorporated by reference herein). [0116] In certain embodiments, an immune checkpoint inhibitor is a B7-H3 antagonist. In further embodiments, the B7-H3 antagonist is an antagonistic B7-H3 antibody. Suitable antagonist B7-H3 antibodies include, for example, enoblituzumab (MGA271; as described in WO11/109400, which is incorporated by reference herein), omburtumab, DS-7300a, ABBV-155, and SHR-A1811. [0117] In some embodiments, an immune checkpoint inhibitor is a TIM-3 antagonist. In further embodiments, the TIM-3 antagonist can be an antagonistic TIM-3 antibody. Suitable antagonistic TIM-3 antibodies include, for example, dostarlimab, sabatolimab, BMS-986258. And RG7769/RO7121661. [0118] In some embodiments, a compound according to this disclosure is administered with one or more than one additional therapy. In some embodiments, each additional therapy is independently selected from an immune checkpoint inhibitor, a chemotherapeutic agent, and radiation therapy. In further embodiments, of the above (a) the immune checkpoint inhibitor antagonizes PD-1, PD-L1, BTLA, LAG-3, a B7 family member, TIM-3, TIGIT, or CTLA-4, or a combination of any two or more thereof; (b) the immune checkpoint inhibitor antagonizes PD-1 or PD-L1; (c) the immune checkpoint inhibitor that antagonizes PD-1 or PD-L1 is selected from the group consisting of avelumab, atezolizumab, balstilimab, budigalimab, camrelizumab, cosibelimab, dostarlimab, durvalumab, emiplimab, envafolimab ezabenlimab, nivolumab, pembrolizumab, pidilizumab, pimivalimab, retifanlimab, sasanlimab, spartalizumab, sintilimab, tislelizumab, toripalimab, and zimberelimab; (d) the immune checkpoint inhibitor is zimberelimab; (e) the immune checkpoint inhibitor comprise an immune checkpoint inhibitor that antagonizes TIGIT; (f) the immune checkpoint inhibitor is domvanalimab or AB308; (g) the chemotherapeutic agent comprises a platinum-based, taxoid-based, or anthracycline-based chemotherapeutic agent, or a combination thereof; and (h) the chemotherapeutic agent is selected from the group consisting of gemcitabine, cisplatin, carboplatin, oxaliplatin, doxorubicin, docetaxel, paclitaxel, and nab-paclitaxel.
[0119] Selection of the additional therapeutic agent(s) may be informed by current standard of care for a particular cancer and/or mutational status of a subject’s cancer and/or stage of disease. Detailed standard of care guidelines are published, for example, by National Comprehensive Cancer Network (NCCN). See, for instance, NCCN Colon Cancer v1.2023, NCCN Rectal Cancer v1.2023, NCCN Hepatobiliary Cancers v1.2023, NCCN Hepatocellular Carcinoma v1.2023, NCCN Kidney Cancer, v4.2023, NCCN NSCLC v2.2023, NCCN Adenocarcinoma v1.2023. Routes of Administration [0120] In some embodiments, pharmaceutical compositions containing a compound according to this disclosure may be in a form suitable for oral administration. Oral administration may involve swallowing the formulation thereby allowing the compound to be absorbed into the bloodstream in the gastrointestinal tract. Alternatively, oral administration may involve buccal, lingual or sublingual administration, thereby allowing the compound to be absorbed into the blood stream through oral mucosa. [0121] In another embodiment, the pharmaceutical compositions containing a compound according to this disclosure may be in a form suitable for parenteral administration. Forms of parenteral administration include, but are not limited to, intravenous, intraarterial, intramuscular, intradermal, intraperitoneal, intrathecal, intracisternal, intracerebral, intracerebroventricular, intraventricular, and subcutaneous. Pharmaceutical compositions suitable for parenteral administration may be formulated using suitable aqueous or non-aqueous carriers. Depot injections, which are generally administered subcutaneously or intramuscularly, may also be utilized to release the compounds disclosed herein over a defined period of time. [0122] Other routes of administration are also contemplated by this disclosure, including, but not limited to, nasal, vaginal, intraocular, rectal, topical (e.g., transdermal), and inhalation. [0123] Particular embodiments of the present disclosure contemplate oral administration or parenteral administration.
Pharmaceutical Compositions [0124] The compounds of the present disclosure may be in the form of compositions suitable for administration to a subject. In general, such compositions are pharmaceutical compositions comprising a compound according to this disclosure or a pharmaceutically acceptable salt thereof and one or more pharmaceutically acceptable excipients. In certain embodiments, the compound, or pharmaceutically acceptable salt thereof, may be present in an effective amount. The pharmaceutical compositions may be used in the methods of the present disclosure; thus, for example, the pharmaceutical compositions comprising a compound according to this disclosure can be administered to a subject in order to practice the therapeutic and prophylactic methods and uses described herein. [0125] The pharmaceutical compositions of the present disclosure can be formulated to be compatible with the intended method or route of administration. Routes of administration may include those known in the art. Exemplary routes of administration are oral and parenteral. Furthermore, the pharmaceutical compositions may be used in combination with one or more other therapies described herein in order to treat or prevent the diseases, disorders and conditions as contemplated by the present disclosure. In one embodiment, one or more other therapeutic agents contemplated by this disclosure are included in the same pharmaceutical composition that comprises a compound according to this disclosure. In another embodiment, the one or more other therapeutical agents are in a composition that is separate from the pharmaceutical composition comprising the compound according to this disclosure. [0126] In one aspect, the compounds described herein may be administered orally. Oral administration may be via, for example, capsule or tablets. In making the pharmaceutical compositions that include the compound of Formula (I), or a pharmaceutically acceptable salt thereof, the tablet or capsule typically includes at least one pharmaceutically acceptable excipient. Non-limiting examples of pharmaceutically acceptable excipients include lactose, dextrose, sucrose, sorbitol, mannitol, starches, gum acacia, calcium phosphate, alginates, tragacanth, gelatin, calcium silicate, microcrystalline cellulose, polyvinylpyrrolidone, polyethylene glycol, cellulose, sterile water, syrup, and methyl cellulose. Additional pharmaceutically acceptable excipients include lubricating agents such as talc, magnesium
stearate, and mineral oil; wetting agents; emulsifying and suspending agents; preserving agents such as methyl and propylhydroxy-benzoates. [0127] In another aspect, the compounds described herein may be administered parenterally, for example by intravenous injection. A pharmaceutical composition appropriate for parenteral administration may be formulated in solution for injection or may be reconstituted for injection in an appropriate system such as a physiological solution. Such solutions may include sterile water for injection, salts, buffers, and tonicity excipients in amounts appropriate to achieve isotonicity with the appropriate physiology. [0128] The pharmaceutical compositions described herein may be stored in an appropriate sterile container or containers. In some embodiments, the container is designed to maintain stability for the pharmaceutical composition over a given period of time. Administering [0129] In general, the disclosed methods comprise administering a compound described herein, or a composition thereof, in an effective amount to a subject in need thereof. An “effective amount” with reference to a mutant HIF-2α inhibitor (e.g., a G323E mutant HIF-2α inhibitor), of the present disclosure means an amount of the compound that is sufficient to engage the target (by inhibiting, agonizing or antagonizing the target) at a level that is indicative of the potency of the compound. For mutant HIF-2α (e.g., G323E mutant HIF-2α), target engagement can be determined by one or more biochemical or cellular assays resulting in an EC50, ED50, EC90, IC50, or similar value which can be used as one assessment of the potency of the compound. Assays for determining target engagement include, but are not limited to, those described in the Examples. The effective amount may be administered as a single quantity or as multiple, smaller quantities (e.g., as one tablet with “x” amount, as two tablets each with “x/2” amount, etc.). [0130] In some embodiments, the disclosed methods comprise administering a therapeutically effective amount of a compound described herein to a subject in need thereof. As used herein, the phrase “therapeutically effective amount” with reference to compound means a dose regimen (i.e., amount and interval) of the compound that provides the specific pharmacological effect for
which the compound is administered to a subject in need of such treatment. For prophylactic use, a therapeutically effective amount may be effective to eliminate or reduce the risk, lessen the severity, or delay the onset of the disease, including biochemical, histological and/or behavioral signs or symptoms of the disease. For treatment, a therapeutically effective amount may be effective to reduce, ameliorate, or eliminate one or more signs or symptoms associated with a disease, delay disease progression, prolong survival, decrease the dose of other medication(s) required to treat the disease, or a combination thereof. With respect to cancer specifically, a therapeutically effective amount may, for example, result in the killing of cancer cells, reduce cancer cell counts, reduce tumor burden, eliminate tumors or metastasis, or reduce metastatic spread. A therapeutically effective amount may vary based on, for example, one or more of the following: the age and weight of the subject, the subject’s overall health, the stage of the subject’s disease, the route of administration, and prior or concomitant treatments. [0131] Administration may comprise one or more (e.g., one, two, or three or more) dosing cycles. [0132] In certain embodiments, the compounds contemplated by the present disclosure may be administered (e.g., orally, parenterally, etc.) at about 0.01 mg/kg to about 50 mg/kg, or about 1 mg/kg to about 25 mg/kg, of subject’s body weight per day, one or more times a day, a week, or a month, to obtain the desired effect. In some embodiments, a suitable weight-based dose of a compound contemplated by the present disclosure is used to determine a dose that is administered independent of a subject’s body weight In certain embodiments, the compounds of the present disclosure are administered (e.g., orally, parenterally, etc.) at fixed dosage levels of about 1 mg to about 1000 mg, particularly 1, 3, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, 500, 600, 750, 800, 900, or 1000 mg, one or more times a day, a week, or a month, to obtain the desired effect. [0133] In certain embodiments, the compound is contained in a “unit dosage form”. The phrase “unit dosage form” refers to physically discrete units, each unit containing a predetermined amount of the compound, either alone or in combination with one or more additional agents, sufficient to produce the desired effect. It will be appreciated that the
parameters of a unit dosage form will depend on the particular agent and the effect to be achieved. Methods of Synthesis General methods for compound preparation [0134] Compounds of Formula I, including the exemplary molecules described herein and represented in the claims, may be synthesized using various standard synthetic methods and techniques that are known to those skilled in the art. Compounds of Formula I and compounds having the structures described in the prior sections were synthesized from starting materials that are available from common commercial sources, or from starting materials that can be synthesized using standard synthetic methods. The compounds described herein and related compounds bearing different substituents can be prepared using methods that are derived from known chemical reactions and principals, which may be modified, as appropriate, to enable the introduction or modification of specific functional groups represented in the structures described herein. [0135] The following examples demonstrate the preparation of compounds of Formula I. EXPERIMENTAL [0136] The following examples are put forth so as to provide those of ordinary skill in the art with a complete disclosure and description of how to make and use the present disclosure, and are not intended to limit the scope of what the inventors regard as their invention. Efforts have been made to ensure accuracy with respect to numbers used (e.g., amounts, temperature, etc.), but some experimental errors and deviations should be accounted for. [0137] All reactions were performed using a Teflon-coated magnetic stir bar at the indicated temperature and were conducted under an inert atmosphere when stated. Purchased starting materials and reagents were generally used as received. Reactions were monitored by TLC (silica gel 60 with fluorescence F254, visualized with a short wave/long wave UV lamp) and/or LCMS (AGILENT® 1100 or 1200 series LCMS with UV detection at 254 or 280 nm using a binary solvent system [0.1% formic acid in MeCN/0.1% formic acid in H2O] using one of the following columns: AGILENT® Eclipse Plus C18 [3.5 μm, 4.6 mm i.d. × 100 mm], WATERS™ XSelect
HSS C18 [3.5 μm, 2.1 mm i.d. × 75 mm]). Flash chromatography was conducted on silica gel using an automated system (COMBIFLASH® RF+ manufactured by Teledyne ISCO), with detection wavelengths of 254 and 280 nm, and optionally equipped with an evaporative light scattering detector. Reverse phase preparative HPLC was conducted on an AGILENT® 1260 or 1290 Infinity series HPLC. Samples were eluted using a binary solvent system (MeCN/H2O with an acid modifier as needed – for example 0.1% TFA or 0.1% formic acid) with gradient elution on a Gemini C18110 Å column (21.2 mm i.d. ×x 250 mm) with variable wavelength detection. Final compounds obtained through preparative HPLC were concentrated through lyophilization. All assayed compounds were purified to ≥95% purity as determined by 1H NMR or LCMS (AGILENT® 1100 or 1200 series LCMS with UV detection at 254 or 280 nm using a binary solvent system [0.1% formic acid in MeCN/0.1% formic acid in H2O] using one of the following columns: AGILENT® Eclipse Plus C18 [3.5 μm, 4.6 mm i.d. × 100 mm], WATERS™ XSelect HSS C18 [3.5 μm, 2.1 mm i.d. × 75 mm]).1H NMR spectra were recorded on a Varian 400 MHz NMR spectrometer equipped with an Oxford AS400 magnet or a BRUKER® AVANCE NEO 400 MHz NMR. Chemical shifts (δ) are reported as parts per million (ppm) relative to residual undeuterated solvent as an internal reference. The abbreviations s, br s, d, t, q, dd, dt, ddd, and m stand for singlet, broad singlet, doublet, triplet, quartet, doublet of doublets, doublet of triplets, doublet of doublet of doublets, and multiplet, respectively. [0138] Unless indicated otherwise, temperature is in degrees Celsius (° C), and pressure is at or near atmospheric. Standard abbreviations are used, including the following: rt or RT=room temperature; min(s)=minute(s); h=hour(s); mg=milligram; g=gram; kg=kilogram; μL=microliter; ml or mL=milliliter; M=molar; mol=mole; mmol=millimole; sat.=saturated; sol.=solution; aq.=aqueous; calcd=calculated; equiv.=equivalents; H2=hydrogen gas ; H2SO4=sulfuric acid; DCM or CH2Cl2=dichloromethane; THF=tetrahydrofuran; PhMe=toluene; EtOAc=ethyl acetate; AcOEt=ethyl acetate; TFA=trifluoroacetic acid; MeCN or ACN=acetonitrile; DMF=N,N- dimethylformamide; DIAD=diisopropyl azodicarboxylate; DMP=Dess-Martin periodinane; DAST=diethylaminosulfur trifluoride; DMSO=dimethyl sulfoxide; Deoxo-Fluor=bis(2- methoxyethyl)aminosulfur trifluoride; Selectfluor™=(1-chloromethyl-4-fluoro-1,4- diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate); MeOH=methanol; Et3N=triethylamine; TMS-morpholine=4-(trimethylsilyl)morpholine; MOMBr=bromomethyl methyl ether; TBSOTf=tert-butyldimethylsilyl trifluoromethanesulfonate; TBAF=tetrabutylammonium
fluoride; HCO2H=formic acid; NaCl=sodium chloride; NH4Cl=ammonium chloride; Na2S2O3=sodiumthiosulfate; Na2SO4=sodium sulfate; NaHCO3=sodium bicarbonate; HCl=hydrochloric acid; KOAc=potassium acetate; B2Pin2=bis(pinacolato)diboron; Zn(CN)2=zinc cyanide; SPhos Pd 2nd Gen. or SPhos Pd G2= chloro(2-dicyclohexylphosphino- 2′,6′-dimethoxy-1,1′-biphenyl)[2-(2′-amino-1,1′-biphenyl)]palladium(II); Pd(dppf)Cl2=[1,1′- bis(diphenylphosphino)ferrocene] dichloropalladium(II); Pd/C=palladium on carbon; RuCl(p- cymene)[(R,R)-Ts-DPEN= [N-[(1R,2R)-2-(amino-κN)-1,2-diphenylethyl]-4- methylbenzenesulfonamidato-κN]chloro[(1,2,3,4,5,6-η)-1-methyl-4-(1-methylethyl)benzene]- ruthenium; RuCl(p-cymene)[(S,S)-TsDPEN]=[N-[(1S,2S)-2-(amino-κN)-1,2-diphenylethyl]-4- methylbenzenesulfonamidato-κN]chloro[(1,2,3,4,5,6-η)-1-methyl-4-(1-methylethyl)benzene]- ruthenium; PPh3=triphenylphospohine; MHz=megahertz; Hz=hertz; ppm=parts per million; ESI MS=electrospray ionization mass spectrometry; LCMS or LC/MS=liquid chromatography-mass spectrometry; TLC=thin layer chromatography; NMR=nuclear magnetic resonance; HPLC=high pressure liquid chromatography. Examples 1-4 [0139] The compounds of example numbers 1-4 and 21-32 were prepared as previously described in PCT publication number WO2021188769.

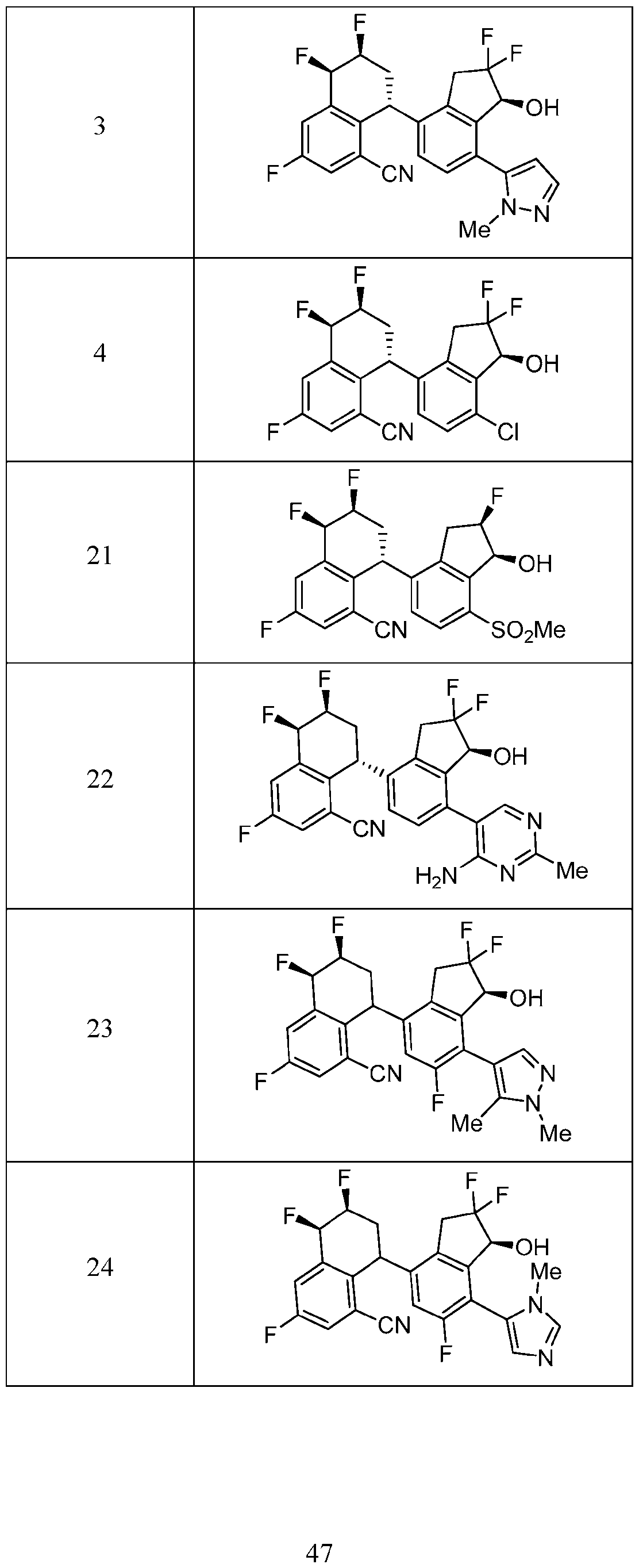


Example 5: (5R,6S,8R)-8-(4-Chloro-3-cyano-5-fluoro-2-methylphenyl)-3,5,6-trifluoro- 5,6,7,8-tetrahydronaphthalene-1-carbonitrile


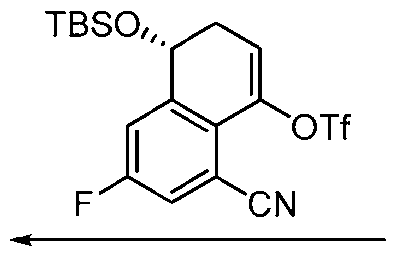



 [0140] Step a. Into a 3L round bottom flask, 4-bromo-2-fluoro-5-methylaniline (50.0 g, 245 mmol, 1.0 equiv.), glacial acetic acid (1L, 0.25 M), N-iodosuccinimide (57.8 g, 257.7 mmol, 1.05 equiv.) were sequentially added. After completion of the addition, the reaction was stirred at
room temperature for 3 h. After the reaction was completed, the reaction was concentrated under reduced pressure. The resulting residue was added with saturated aqueous NaHCO3 solution to adjust the pH to pH = 7 and extracted with EtOAc. The combined organic phase was concentrated under reduced pressure. The resulting residue was purified using silica gel column chromatography (EtOAc in hexane – 0% → 10%) to give 4-bromo-6-fluoro-2-iodo-3- methylaniline (70.0 g, 87%). A mixture of the obtained product (25 g, 75.7 mmol, 1.0 equiv.), 1,10-bis(triphenylphosphines)-ferrocene (1.68 g, 3.03 mmol, 0.04 equiv.), and Pd(dba)2 (1.30 g, 2.26 mmol, 0.03 equiv.) in degassed DMF (250 mL) and water (25 mL) was heated to 60 °C for 5 min before Zn(CN)2 (4.45 g, 37.89 mmol, 0.5 equiv.) was added. The reaction was stirred at 125 °C for 5 h under a nitrogen atmosphere. A saturated aqueous NaHCO3 solution was then added, and the resulting mixture was extracted with AcOEt. The combined organic phase was washed with brine, dried over Na2SO4, filtered, and concentrated. The resulting residue was purified using silica gel column chromatography (EtOAc in CH2Cl2 – 10% isocratic) to give 2- amino-5-bromo-3-fluoro-6-methylbenzonitrile. [0141] Step b. Into a 500 mL round bottom flask, product of step a (16.0 g, 69.85 mmol, 1.0 equiv.), copper(I) chloride (20.75 g, 209.56 mmol, 3.0 equiv.) and copper(II) chloride (32.87 g, 244.48 mmol, 3.5 equiv.) and ACN (150 mL, 0.5M) were added. The mixture was cooled to 0 °C and tert-butyl nitrite (33.23 mL, 279.40 mmol, 4.0 equiv.) was added. The resulting mixture was stirred at 23°C for 6 h. After complete conversion, the mixture was diluted with water and extracted with EtOAc. The combined organic layer was washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The resulting residue was purified using silica gel column chromatography (EtOAc in hexane – 0% → 12%) to give 5-bromo-2-chloro-3- fluoro-6-methyl-benzonitrile. [0142] Step c: A 250 mL flask was charged with the product of step b (5.47 g, 22.02 mmol, 1.0 equiv.), B2Pin2 (7.27 g, 28.62 mmol, 1.3 equiv.), Pd(dppf)Cl2 (1.29 g, 1.76 mmol, 8 mol%), KOAc (4.75 g, 48.44 mmol, 2.2 equiv.) and 1,4-dioxane (110 mL). The reaction mixture was degassed with N2 bubbling for 10 min before being heated to 90 °C. After stirring for 4h at 90 °C, the reaction mixture was cooled, filtered over Celite®, washed with EtOAc and evaporated. The resulting 2-chloro-3-fluoro-6-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)benzonitrile was used directly in the next step without further purification.
[0143] Step d. A round-bottomed flask was charged with [(4R)-4-[tert-butyl(dimethyl)silyl]- oxy-8-cyano-6-fluoro-3,4-dihydronaphthalen-1-yl] trifluoromethanesulfonate (9.94 g, 22.02 mmol, 1.0 equiv.), product of step c (6.51 g, 22.02 mmol, 1.0 equiv.), Pd(dppf)Cl2 (0.81 g, 1.10 mmol, 5 mol%), Na2CO3 (4.67 g, 44.07 mmol, 2.0 equiv.), 1,4-dioxane (80 mL) and H2O (20 mL). The reaction mixture was degassed with N2 bubbling for 10 min before it was stirred at 80 °C for 1 h. Upon completion, as shown by LC/MS, the reaction mixture was quenched with sat. sol. NaCl and extracted with EtOAc. The combined organic extract was washed with sat. sol. NaCl, dried over Na2SO4, filtered, and concentrated. The residue was purified using column chromatography on silica gel (EtOAc in hexane – 0% → 20%). The obtained orange foam was stirred with SilicaMetS dimercaptotriazine (16 g) and activated charcoal (4 g) in EtOAc (75 mL, 0.3M) at 23 °C for 16h. The solution was then filtered and concentrated to afford (5R)-5-[tert- butyl(dimethyl)silyl]oxy-8-(4-chloro-3-cyano-5-fluoro-2-methylphenyl)-3-fluoro-5,6- dihydronaphthalene-1-carbonitrile. [0144] Step e: Product of step d (7.8 g, 16.56 mmol, 1.0 equiv.) was dissolved in EtOAc (210 mL) and MeOH (70 mL) under nitrogen, and Et3N (6.9 mL, 49.68 mmol, 3.0 equiv.) and Pd/C (1.56 g, 20 wt%, 10% Pd) were added to the reaction vessel. The mixture was shaken in a Parr shaker apparatus under an atmosphere of H2 (50 psi) for 3h. At this point, Pd/C (0.39 g, 5 wt%, 10% Pd) was added the mixture was shaken in under an atmosphere of H2 (50 psi) for another 3h. Once the desired level of conversion was reached, Celite® and EtOAc were added to the reaction vessel, and the solution was stirred vigorously for 10 min. The mixture was then filtered through Celite®, washed with EtOAc (3 times), and concentrated to afford a dark grey solid. The residue was purified using column chromatography on silica gel (EtOAc in hexane – 0% → 10%) to afford (5R,8R)-5-[tert-butyl(dimethyl)silyl]oxy-8-(4-chloro-3-cyano-5-fluoro-2- methylphenyl)-3-fluoro-5,6,7,8-tetrahydronaphthalene-1-carbonitrile together with the starting material. [0145] Step f. TBAF (6.3 mL, 1M in THF, 6.34 mmol, 1.5 equiv.) was added over 10 min to a solution of product of step e (2.0 g, 4.23 mmol, 1.0 equiv.) in THF (28 mL) at 0 °C. After the addition, the reaction was analyzed by LC/MS, and upon completion it was partitioned between EtOAc and H2O. The solution was extracted with EtOAc, and the combined organic extract was washed with sat. sol. NaCl, dried over Na2SO4, filtered, and concentrated. The resulting (5R,8R)-
8-(4-chloro-3-cyano-5-fluoro-2-methylphenyl)-3-fluoro-5-hydroxy-5,6,7,8-tetrahydro- naphthalene-1-carbonitrile was used directly in the next step without further purification. [0146] Step g. To a suspension of product of step f (1.52 g, 4.23 mmol, 1.0 equiv.) and NaHCO3 (0.39 g, 4.65 mmol, 1.1 equiv.) in CH2Cl2 (22 mL, 0.2M) at 0 °C was added DMP (1.97 g, 4.65 mmol, 1.1 equiv.) slowly in portions. The reaction was stirred for 30 min and was analyzed by LC/MS. Upon completion, the reaction was quenched by addition of a saturated sol. NaHCO3 (20 mL) and saturated sol. Na2S2O3 (20 mL). Celite® was added and the solution was stirred for 20 min before it was filtered over Celite® and washed with CH2Cl2. The layers were separated, and the aqueous layer was back extracted with CH2Cl2 twice. The combined organic extract was washed with saturated sol. Na2S2O3, then dried over Na2SO4, filtered, and concentrated to dryness. The resulting (8R)-8-(4-chloro-3-cyano-5-fluoro-2-methylphenyl)-3- fluoro-5-oxo-7,8-dihydro-6H-naphthalene-1-carbonitrile was used directly in the next step without further purification. [0147] Step h. Product of step g (1.51 g, 4.23 mmol, 1.0 equiv.) was dissolved in CH2Cl2 (22 mL, 0.2M) and the reaction mixture was cooled to 0 °C. Et3N (4.1 mL, 29.60 mmol, 7.0 equiv.) was then added followed by TBSOTf (3.4 mL, 14.80 mmol, 3.5 equiv.) which was added dropwise over 15 min. The mixture was stirred for 2h at 23 °C and then analyzed by LC/MS. Upon completion, the reaction mixture was cooled in an ice-bath and quenched with sat. sol. NaHCO3 and extracted with CH2Cl2 twice. The combined organic extract was washed with sat. sol. NaCl, dried over Na2SO4, filtered, and concentrated. The residue was purified using column chromatography on silica gel (EtOAc in hexane – 0% → 18%) to afford (8R)-5-[tert- butyl(dimethyl)silyl]oxy-8-(4-chloro-3-cyano-5-fluoro-2-methylphenyl)-3-fluoro-7,8- dihydronaphthalene-1-carbonitrile. [0148] Step i. Product of step f (1.99 g, 4.23 mmol, 1.0 equiv.) was dissolved in MeCN (22 mL, 0.2M) at 23 °C and SelectFluor™ (3.00 g, 8.46 mmol, 2.0 equiv.) was added slowly over 15 min. During the course of the addition, the internal temperature increased to 40 °C. The mixture was then stirred for an additional 15 min at 40 °C, when complete conversion was observed by LC/MS. The reaction mixture was then quenched at 0 °C with sat. sol. NaHCO3 and extracted with EtOAc twice. The combined organic extract was washed water and then with sat. sol. NaCl, dried over Na2SO4, filtered, and concentrated. The resulting (6S,8R)-8-(4-chloro-3-cyano-5-
fluoro-2-methylphenyl)-3,6-difluoro-5-oxo-7,8-dihydro-6H-naphthalene-1-carbonitrile was used directly in the next step without further purification. [0149] Step j. Product of step i (1.67 g, 4.46 mmol, 1.0 equiv.) was dissolved in CH2Cl2 (22 mL, 0.2M) and cooled to 0 °C. Et3N (1.24 mL, 8.91 mmol, 2.0 equiv.) and HCO2H (0.50 mL, 13.37 mmol, 3.0 equiv.) were then added and the solution was degassed for 10 min. When the internal temperature of the reaction reached 0 °C, RuCl(p-cymene)[(R,R)-Ts-DPEN] (43 mg, 0.067 mmol, 0.015 equiv.) was added. A septum, a balloon, and a bubbler were attached to the flask, and the reaction was stirred at 4 °C for 16h. Upon completion, as shown by LC/MS, the reaction mixture was poured into a sat. sol. NaHCO3 and extracted with CH2Cl2. The combined organic extract was washed with sat. sol. NaCl, dried over Na2SO4, filtered, and concentrated. The residue was purified using column chromatography on silica gel (EtOAc in hexane – 0% → 30%) to afford the desired (5S,6S,8R)-8-(4-chloro-3-cyano-5-fluoro-2-methylphenyl)-3,6- difluoro-5-hydroxy-5,6,7,8-tetrahydronaphthalene-1-carbonitrile as a single isomer. [0150] Step k. A solution of Deoxo-Fluor (2.7 M in PhMe, 3.12 mL, 8.44 mmol, 3.0 equiv.) was added to a solution of PhMe (10 mL) and the mixture was cooled to 0 °C. TMS-morpholine (1.50 mL, 8.44 mmol, 3.0 equiv.) was added dropwise over 5 min. The mixture was warmed to room temperature over 2h, during which a white precipitate formed. The suspension was then diluted with PhMe (28 mL), and it was cooled to 0 °C. Product of step j (1.06 g, 2.81 mmol, 1.0 equiv.) in DCM (2 mL) was then added dropwise over 5 min and the orange suspension was warmed to 23 °C for 16h. Upon completion, the reaction was quenched with sat. sol. NaHCO3 (50 mL) and diluted with EtOAc (25 mL). The organic layer was separated, and the aqueous layer was extracted with EtOAc (50 mL). The combined organic layers were washed with HCl (1M, 50 mL), dried with Na2SO4, filtered, and concentrated. The residue was purified using column chromatography on silica gel (EtOAc in hexane – 0% → 22%) to afford the title compound.1H NMR (400 MHz, CDCl3) δ 7.58 (dd, J = 8.2, 2.8 Hz, 1H), 7.42 (ddt, J = 7.4, 2.8, 0.6 Hz, 1H), 6.34 (d, J = 9.3 Hz, 1H), 5.67 (ddd, J = 50.0, 16.7, 2.6 Hz, 1H), 5.13 (dddt, J = 48.6, 14.2, 8.8, 2.6 Hz, 1H), 4.83 (t, J = 6.8 Hz, 1H), 2.89 – 2.77 (m, 1H), 2.72 (d, J = 1.2 Hz, 3H), 1.94 – 1.79 (m, 1H). ESI MS [M+H]+ for C19H12Cl1F4N2, calcd 379.1, found 379.0.
Example 6: (5R,6S,8R)-8-[3-Cyano-5-fluoro-2-methyl-4-(2-methylpyrazol-3-yl)phenyl]- 3,5,6-trifluoro-5,6,7,8-tetrahydronaphthalene-1-carbonitrile
[0140] Step a. Into a 3L round bottom flask, 4-bromo-2-fluoro-5-methylaniline (50.0 g, 245 mmol, 1.0 equiv.), glacial acetic acid (1L, 0.25 M), N-iodosuccinimide (57.8 g, 257.7 mmol, 1.05 equiv.) were sequentially added. After completion of the addition, the reaction was stirred at
room temperature for 3 h. After the reaction was completed, the reaction was concentrated under reduced pressure. The resulting residue was added with saturated aqueous NaHCO3 solution to adjust the pH to pH = 7 and extracted with EtOAc. The combined organic phase was concentrated under reduced pressure. The resulting residue was purified using silica gel column chromatography (EtOAc in hexane – 0% → 10%) to give 4-bromo-6-fluoro-2-iodo-3- methylaniline (70.0 g, 87%). A mixture of the obtained product (25 g, 75.7 mmol, 1.0 equiv.), 1,10-bis(triphenylphosphines)-ferrocene (1.68 g, 3.03 mmol, 0.04 equiv.), and Pd(dba)2 (1.30 g, 2.26 mmol, 0.03 equiv.) in degassed DMF (250 mL) and water (25 mL) was heated to 60 °C for 5 min before Zn(CN)2 (4.45 g, 37.89 mmol, 0.5 equiv.) was added. The reaction was stirred at 125 °C for 5 h under a nitrogen atmosphere. A saturated aqueous NaHCO3 solution was then added, and the resulting mixture was extracted with AcOEt. The combined organic phase was washed with brine, dried over Na2SO4, filtered, and concentrated. The resulting residue was purified using silica gel column chromatography (EtOAc in CH2Cl2 – 10% isocratic) to give 2- amino-5-bromo-3-fluoro-6-methylbenzonitrile. [0141] Step b. Into a 500 mL round bottom flask, product of step a (16.0 g, 69.85 mmol, 1.0 equiv.), copper(I) chloride (20.75 g, 209.56 mmol, 3.0 equiv.) and copper(II) chloride (32.87 g, 244.48 mmol, 3.5 equiv.) and ACN (150 mL, 0.5M) were added. The mixture was cooled to 0 °C and tert-butyl nitrite (33.23 mL, 279.40 mmol, 4.0 equiv.) was added. The resulting mixture was stirred at 23°C for 6 h. After complete conversion, the mixture was diluted with water and extracted with EtOAc. The combined organic layer was washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The resulting residue was purified using silica gel column chromatography (EtOAc in hexane – 0% → 12%) to give 5-bromo-2-chloro-3- fluoro-6-methyl-benzonitrile. [0142] Step c: A 250 mL flask was charged with the product of step b (5.47 g, 22.02 mmol, 1.0 equiv.), B2Pin2 (7.27 g, 28.62 mmol, 1.3 equiv.), Pd(dppf)Cl2 (1.29 g, 1.76 mmol, 8 mol%), KOAc (4.75 g, 48.44 mmol, 2.2 equiv.) and 1,4-dioxane (110 mL). The reaction mixture was degassed with N2 bubbling for 10 min before being heated to 90 °C. After stirring for 4h at 90 °C, the reaction mixture was cooled, filtered over Celite®, washed with EtOAc and evaporated. The resulting 2-chloro-3-fluoro-6-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)benzonitrile was used directly in the next step without further purification.
[0143] Step d. A round-bottomed flask was charged with [(4R)-4-[tert-butyl(dimethyl)silyl]- oxy-8-cyano-6-fluoro-3,4-dihydronaphthalen-1-yl] trifluoromethanesulfonate (9.94 g, 22.02 mmol, 1.0 equiv.), product of step c (6.51 g, 22.02 mmol, 1.0 equiv.), Pd(dppf)Cl2 (0.81 g, 1.10 mmol, 5 mol%), Na2CO3 (4.67 g, 44.07 mmol, 2.0 equiv.), 1,4-dioxane (80 mL) and H2O (20 mL). The reaction mixture was degassed with N2 bubbling for 10 min before it was stirred at 80 °C for 1 h. Upon completion, as shown by LC/MS, the reaction mixture was quenched with sat. sol. NaCl and extracted with EtOAc. The combined organic extract was washed with sat. sol. NaCl, dried over Na2SO4, filtered, and concentrated. The residue was purified using column chromatography on silica gel (EtOAc in hexane – 0% → 20%). The obtained orange foam was stirred with SilicaMetS dimercaptotriazine (16 g) and activated charcoal (4 g) in EtOAc (75 mL, 0.3M) at 23 °C for 16h. The solution was then filtered and concentrated to afford (5R)-5-[tert- butyl(dimethyl)silyl]oxy-8-(4-chloro-3-cyano-5-fluoro-2-methylphenyl)-3-fluoro-5,6- dihydronaphthalene-1-carbonitrile. [0144] Step e: Product of step d (7.8 g, 16.56 mmol, 1.0 equiv.) was dissolved in EtOAc (210 mL) and MeOH (70 mL) under nitrogen, and Et3N (6.9 mL, 49.68 mmol, 3.0 equiv.) and Pd/C (1.56 g, 20 wt%, 10% Pd) were added to the reaction vessel. The mixture was shaken in a Parr shaker apparatus under an atmosphere of H2 (50 psi) for 3h. At this point, Pd/C (0.39 g, 5 wt%, 10% Pd) was added the mixture was shaken in under an atmosphere of H2 (50 psi) for another 3h. Once the desired level of conversion was reached, Celite® and EtOAc were added to the reaction vessel, and the solution was stirred vigorously for 10 min. The mixture was then filtered through Celite®, washed with EtOAc (3 times), and concentrated to afford a dark grey solid. The residue was purified using column chromatography on silica gel (EtOAc in hexane – 0% → 10%) to afford (5R,8R)-5-[tert-butyl(dimethyl)silyl]oxy-8-(4-chloro-3-cyano-5-fluoro-2- methylphenyl)-3-fluoro-5,6,7,8-tetrahydronaphthalene-1-carbonitrile together with the starting material. [0145] Step f. TBAF (6.3 mL, 1M in THF, 6.34 mmol, 1.5 equiv.) was added over 10 min to a solution of product of step e (2.0 g, 4.23 mmol, 1.0 equiv.) in THF (28 mL) at 0 °C. After the addition, the reaction was analyzed by LC/MS, and upon completion it was partitioned between EtOAc and H2O. The solution was extracted with EtOAc, and the combined organic extract was washed with sat. sol. NaCl, dried over Na2SO4, filtered, and concentrated. The resulting (5R,8R)-
8-(4-chloro-3-cyano-5-fluoro-2-methylphenyl)-3-fluoro-5-hydroxy-5,6,7,8-tetrahydro- naphthalene-1-carbonitrile was used directly in the next step without further purification. [0146] Step g. To a suspension of product of step f (1.52 g, 4.23 mmol, 1.0 equiv.) and NaHCO3 (0.39 g, 4.65 mmol, 1.1 equiv.) in CH2Cl2 (22 mL, 0.2M) at 0 °C was added DMP (1.97 g, 4.65 mmol, 1.1 equiv.) slowly in portions. The reaction was stirred for 30 min and was analyzed by LC/MS. Upon completion, the reaction was quenched by addition of a saturated sol. NaHCO3 (20 mL) and saturated sol. Na2S2O3 (20 mL). Celite® was added and the solution was stirred for 20 min before it was filtered over Celite® and washed with CH2Cl2. The layers were separated, and the aqueous layer was back extracted with CH2Cl2 twice. The combined organic extract was washed with saturated sol. Na2S2O3, then dried over Na2SO4, filtered, and concentrated to dryness. The resulting (8R)-8-(4-chloro-3-cyano-5-fluoro-2-methylphenyl)-3- fluoro-5-oxo-7,8-dihydro-6H-naphthalene-1-carbonitrile was used directly in the next step without further purification. [0147] Step h. Product of step g (1.51 g, 4.23 mmol, 1.0 equiv.) was dissolved in CH2Cl2 (22 mL, 0.2M) and the reaction mixture was cooled to 0 °C. Et3N (4.1 mL, 29.60 mmol, 7.0 equiv.) was then added followed by TBSOTf (3.4 mL, 14.80 mmol, 3.5 equiv.) which was added dropwise over 15 min. The mixture was stirred for 2h at 23 °C and then analyzed by LC/MS. Upon completion, the reaction mixture was cooled in an ice-bath and quenched with sat. sol. NaHCO3 and extracted with CH2Cl2 twice. The combined organic extract was washed with sat. sol. NaCl, dried over Na2SO4, filtered, and concentrated. The residue was purified using column chromatography on silica gel (EtOAc in hexane – 0% → 18%) to afford (8R)-5-[tert- butyl(dimethyl)silyl]oxy-8-(4-chloro-3-cyano-5-fluoro-2-methylphenyl)-3-fluoro-7,8- dihydronaphthalene-1-carbonitrile. [0148] Step i. Product of step f (1.99 g, 4.23 mmol, 1.0 equiv.) was dissolved in MeCN (22 mL, 0.2M) at 23 °C and SelectFluor™ (3.00 g, 8.46 mmol, 2.0 equiv.) was added slowly over 15 min. During the course of the addition, the internal temperature increased to 40 °C. The mixture was then stirred for an additional 15 min at 40 °C, when complete conversion was observed by LC/MS. The reaction mixture was then quenched at 0 °C with sat. sol. NaHCO3 and extracted with EtOAc twice. The combined organic extract was washed water and then with sat. sol. NaCl, dried over Na2SO4, filtered, and concentrated. The resulting (6S,8R)-8-(4-chloro-3-cyano-5-
fluoro-2-methylphenyl)-3,6-difluoro-5-oxo-7,8-dihydro-6H-naphthalene-1-carbonitrile was used directly in the next step without further purification. [0149] Step j. Product of step i (1.67 g, 4.46 mmol, 1.0 equiv.) was dissolved in CH2Cl2 (22 mL, 0.2M) and cooled to 0 °C. Et3N (1.24 mL, 8.91 mmol, 2.0 equiv.) and HCO2H (0.50 mL, 13.37 mmol, 3.0 equiv.) were then added and the solution was degassed for 10 min. When the internal temperature of the reaction reached 0 °C, RuCl(p-cymene)[(R,R)-Ts-DPEN] (43 mg, 0.067 mmol, 0.015 equiv.) was added. A septum, a balloon, and a bubbler were attached to the flask, and the reaction was stirred at 4 °C for 16h. Upon completion, as shown by LC/MS, the reaction mixture was poured into a sat. sol. NaHCO3 and extracted with CH2Cl2. The combined organic extract was washed with sat. sol. NaCl, dried over Na2SO4, filtered, and concentrated. The residue was purified using column chromatography on silica gel (EtOAc in hexane – 0% → 30%) to afford the desired (5S,6S,8R)-8-(4-chloro-3-cyano-5-fluoro-2-methylphenyl)-3,6- difluoro-5-hydroxy-5,6,7,8-tetrahydronaphthalene-1-carbonitrile as a single isomer. [0150] Step k. A solution of Deoxo-Fluor (2.7 M in PhMe, 3.12 mL, 8.44 mmol, 3.0 equiv.) was added to a solution of PhMe (10 mL) and the mixture was cooled to 0 °C. TMS-morpholine (1.50 mL, 8.44 mmol, 3.0 equiv.) was added dropwise over 5 min. The mixture was warmed to room temperature over 2h, during which a white precipitate formed. The suspension was then diluted with PhMe (28 mL), and it was cooled to 0 °C. Product of step j (1.06 g, 2.81 mmol, 1.0 equiv.) in DCM (2 mL) was then added dropwise over 5 min and the orange suspension was warmed to 23 °C for 16h. Upon completion, the reaction was quenched with sat. sol. NaHCO3 (50 mL) and diluted with EtOAc (25 mL). The organic layer was separated, and the aqueous layer was extracted with EtOAc (50 mL). The combined organic layers were washed with HCl (1M, 50 mL), dried with Na2SO4, filtered, and concentrated. The residue was purified using column chromatography on silica gel (EtOAc in hexane – 0% → 22%) to afford the title compound.1H NMR (400 MHz, CDCl3) δ 7.58 (dd, J = 8.2, 2.8 Hz, 1H), 7.42 (ddt, J = 7.4, 2.8, 0.6 Hz, 1H), 6.34 (d, J = 9.3 Hz, 1H), 5.67 (ddd, J = 50.0, 16.7, 2.6 Hz, 1H), 5.13 (dddt, J = 48.6, 14.2, 8.8, 2.6 Hz, 1H), 4.83 (t, J = 6.8 Hz, 1H), 2.89 – 2.77 (m, 1H), 2.72 (d, J = 1.2 Hz, 3H), 1.94 – 1.79 (m, 1H). ESI MS [M+H]+ for C19H12Cl1F4N2, calcd 379.1, found 379.0.
Example 6: (5R,6S,8R)-8-[3-Cyano-5-fluoro-2-methyl-4-(2-methylpyrazol-3-yl)phenyl]- 3,5,6-trifluoro-5,6,7,8-tetrahydronaphthalene-1-carbonitrile
 [0151] A vial was charged with (5R,6S,8R)-8-(4-chloro-3-cyano-5-fluoro-2-methylphenyl)- 3,5,6-trifluoro-5,6,7,8-tetrahydronaphthalene-1-carbonitrile (75 mg, 0.20 mmol, 1.0 equiv., the compound of example 5), 1-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole (62 mg, 0.30 mmol, 1.5 equiv.), SPhos Pd 2nd Gen. (29 mg, 0.040 mmol, 0.2 equiv.), aq. Na2CO3 (84 mg, 0.79 mmol, 4.0 equiv., 1M), and dioxane (2.0 mL). The reaction mixture was then sparged with N2 for 10 minutes before being heated to 100 °C for 1h. Upon completion, the solution was filtered over Celite®, and the solvent was evaporated. The residue was purified using column chromatography on silica gel (EtOAc in CH2Cl2 – 0% → 8%) to afford the tittle compound. 1H NMR (400 MHz, CDCl3) δ 7.64 – 7.57 (m, 2H), 7.46 (dd, J = 7.6, 2.7 Hz, 1H), 6.54 – 6.46 (m, 1H), 6.43 (d, J = 9.7 Hz, 1H), 5.82 – 5.59 (m, 1H), 5.20 (dddt, J = 48.6, 14.3, 9.1, 2.5 Hz, 1H), 4.93 (t, J = 6.9 Hz, 1H), 3.76 (s, 3H), 2.98 – 2.82 (m, 1H), 2.78 (s, 3H), 2.07 – 1.88 (m, 1H). ESI MS [M+H]+ for C23H17F4N4, calcd 425.1, found 425.0. Example 7: (5R,6S,8R)-8-[3-cyano-5-fluoro-4-(1-methyl-4-methyl-5-pyrazolyl)-2-tolyl]- 3,5,6-trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile
[0151] A vial was charged with (5R,6S,8R)-8-(4-chloro-3-cyano-5-fluoro-2-methylphenyl)- 3,5,6-trifluoro-5,6,7,8-tetrahydronaphthalene-1-carbonitrile (75 mg, 0.20 mmol, 1.0 equiv., the compound of example 5), 1-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole (62 mg, 0.30 mmol, 1.5 equiv.), SPhos Pd 2nd Gen. (29 mg, 0.040 mmol, 0.2 equiv.), aq. Na2CO3 (84 mg, 0.79 mmol, 4.0 equiv., 1M), and dioxane (2.0 mL). The reaction mixture was then sparged with N2 for 10 minutes before being heated to 100 °C for 1h. Upon completion, the solution was filtered over Celite®, and the solvent was evaporated. The residue was purified using column chromatography on silica gel (EtOAc in CH2Cl2 – 0% → 8%) to afford the tittle compound. 1H NMR (400 MHz, CDCl3) δ 7.64 – 7.57 (m, 2H), 7.46 (dd, J = 7.6, 2.7 Hz, 1H), 6.54 – 6.46 (m, 1H), 6.43 (d, J = 9.7 Hz, 1H), 5.82 – 5.59 (m, 1H), 5.20 (dddt, J = 48.6, 14.3, 9.1, 2.5 Hz, 1H), 4.93 (t, J = 6.9 Hz, 1H), 3.76 (s, 3H), 2.98 – 2.82 (m, 1H), 2.78 (s, 3H), 2.07 – 1.88 (m, 1H). ESI MS [M+H]+ for C23H17F4N4, calcd 425.1, found 425.0. Example 7: (5R,6S,8R)-8-[3-cyano-5-fluoro-4-(1-methyl-4-methyl-5-pyrazolyl)-2-tolyl]- 3,5,6-trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile
 [0152] The title compound was prepared using a similar procedure as that described for Example 6 from the appropriate starting materials to yield two atropisomers. The isomers were
separated using column chromatography on silica gel (EtOAc in hexane – 0% → 30% (atropoisomer A) → 35% (atropoisomer B) and EtOAc in CH2Cl2 – 0% → 8%). [0153] First eluting atropisomer A (ATROP-A) : 1H NMR (400 MHz, CDCl3) δ 7.60 (dd, J = 8.3, 2.7 Hz, 1H), 7.47 – 7.40 (m, 2H), 6.44 (d, J = 9.5 Hz, 1H), 5.70 (dd, J = 50.6, 17.4 Hz, 1H), 5.33 – 5.09 (m, 1H), 4.91 (t, J = 6.9 Hz, 1H), 3.66 (s, 3H), 2.94 – 2.83 (m, 1H), 2.77 (s, 3H), 2.05 – 1.91 (m, 4H). ESI MS [M+H]+ for C24H19F4N4, calcd 439.1, found 439.0. [0154] Second eluting atropisomer B (ATROP-B): 1H NMR (400 MHz, CDCl3) δ 7.59 (dd, J = 8.2, 2.7 Hz, 1H), 7.45 – 7.40 (m, 2H), 6.44 (d, J = 9.5 Hz, 1H), 5.79 – 5.60 (m, 1H), 5.32 – 5.06 (m, 1H), 4.91 (t, J = 6.8 Hz, 1H), 3.70 (s, 3H), 2.96 – 2.82 (m, 1H), 2.75 (s, 3H), 2.06 – 1.92 (m, 4H). ESI MS [M+H]+ for C24H19F4N4, calcd 439.1, found 439.0. Example 8: (5R,6S,8R)-8-[3-cyano-5-fluoro-4-(1-methyl-2-methyl-5-imidazolyl)-2-tolyl]- 3,5,6-trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile
[0152] The title compound was prepared using a similar procedure as that described for Example 6 from the appropriate starting materials to yield two atropisomers. The isomers were
separated using column chromatography on silica gel (EtOAc in hexane – 0% → 30% (atropoisomer A) → 35% (atropoisomer B) and EtOAc in CH2Cl2 – 0% → 8%). [0153] First eluting atropisomer A (ATROP-A) : 1H NMR (400 MHz, CDCl3) δ 7.60 (dd, J = 8.3, 2.7 Hz, 1H), 7.47 – 7.40 (m, 2H), 6.44 (d, J = 9.5 Hz, 1H), 5.70 (dd, J = 50.6, 17.4 Hz, 1H), 5.33 – 5.09 (m, 1H), 4.91 (t, J = 6.9 Hz, 1H), 3.66 (s, 3H), 2.94 – 2.83 (m, 1H), 2.77 (s, 3H), 2.05 – 1.91 (m, 4H). ESI MS [M+H]+ for C24H19F4N4, calcd 439.1, found 439.0. [0154] Second eluting atropisomer B (ATROP-B): 1H NMR (400 MHz, CDCl3) δ 7.59 (dd, J = 8.2, 2.7 Hz, 1H), 7.45 – 7.40 (m, 2H), 6.44 (d, J = 9.5 Hz, 1H), 5.79 – 5.60 (m, 1H), 5.32 – 5.06 (m, 1H), 4.91 (t, J = 6.8 Hz, 1H), 3.70 (s, 3H), 2.96 – 2.82 (m, 1H), 2.75 (s, 3H), 2.06 – 1.92 (m, 4H). ESI MS [M+H]+ for C24H19F4N4, calcd 439.1, found 439.0. Example 8: (5R,6S,8R)-8-[3-cyano-5-fluoro-4-(1-methyl-2-methyl-5-imidazolyl)-2-tolyl]- 3,5,6-trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile
 [0155] The title compound was prepared using a similar procedure as that described for Example 6 from the appropriate starting materials.1H NMR (400 MHz, CDCl3) δ 7.59 (dd, J = 8.2, 2.7 Hz, 1H), 7.43 (ddd, J = 7.3, 3.0, 0.9 Hz, 1H), 7.13 (s, 1H), 6.36 (d, J = 9.8 Hz, 1H), 5.79 – 5.59 (m, 1H), 5.28 – 5.06 (m, 1H), 4.90 (t, J = 6.7 Hz, 1H), 3.38 (d, J = 1.4 Hz, 3H), 2.93 – 2.81 (m, 1H), 2.76 – 2.71 (m, 3H), 2.44 (s, 3H), 2.03 – 1.89 (m, 1H). ESI MS [M+H]+ for C24H19F4N4, calcd 439.1, found 439.0.
Example 9: (5R,6S,8R)-8-[(1S,2R)-2,6-Difluoro-1-hydroxy-7-(1-methyl-2-methyl-5- imidazolyl)-4-indanyl]-3,5,6-trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile
[0155] The title compound was prepared using a similar procedure as that described for Example 6 from the appropriate starting materials.1H NMR (400 MHz, CDCl3) δ 7.59 (dd, J = 8.2, 2.7 Hz, 1H), 7.43 (ddd, J = 7.3, 3.0, 0.9 Hz, 1H), 7.13 (s, 1H), 6.36 (d, J = 9.8 Hz, 1H), 5.79 – 5.59 (m, 1H), 5.28 – 5.06 (m, 1H), 4.90 (t, J = 6.7 Hz, 1H), 3.38 (d, J = 1.4 Hz, 3H), 2.93 – 2.81 (m, 1H), 2.76 – 2.71 (m, 3H), 2.44 (s, 3H), 2.03 – 1.89 (m, 1H). ESI MS [M+H]+ for C24H19F4N4, calcd 439.1, found 439.0.
Example 9: (5R,6S,8R)-8-[(1S,2R)-2,6-Difluoro-1-hydroxy-7-(1-methyl-2-methyl-5- imidazolyl)-4-indanyl]-3,5,6-trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile
 [0156] Step a: To a solution of 4-bromo-6,7-dichloro-2,3-dihydroinden-1-one (5.20 g, 22 mmol, 1.0 equiv.) in MeOH (100 mL) was added Selectfluor™ (9.31 g, 26 mmol, 1.2 equiv.) and concentrated H2SO4 (5 drops). The resulting mixture was heated at reflux for 3 h. After cooling to room temperature, 0.3 M H2SO4 (aq., 50 mL) was added to the reaction mixture. The resulting mixture was heated at reflux for another 1 h. After cooling to room temperature, large amount of the product was precipitated out and collected via filtration. The filtrate was concentrated and diluted with DCM (250 mL) and washed with H2O and brine. The organic phase was dried over Na2SO4 and concentrated. The product was directly used in the next step. [0157] Step b: HCO2H (2.5 mL, 66 mmol, 3.0 equiv.) was added to a solution of Et3N (6.2 mL, 44 mmol, 2.0 equiv.) in DMF (20 mL) dropwise. The resulting solution was stirred at room temperature for 30 min, and then added to a solution of the product from step a (~22 mmol, 1.0 equiv.) and RuCl(p-cymene)[(R,R)-TsDPEN] (557 mg, 0.88 mmol, 4.0 mol%) in DMF (20 mL) at 0 °C. The resulting mixture was kept stirring at this temperature for overnight and then diluted with EtOAc and washed with H2O twice. The organic phase was then washed with brine, dried over Na2SO4, concentrated, and purified by flash chromatography (SiO2, 0 to 40% EtOAc/hexane) to afford the desired product (d.r.4.5:1, 75% e.e.). [0158] Step c: To a solution of the product from step b (2.94 g, 10.4 mmol, 1.0 equiv.) and diisopropylethylamine (3.7 mL, 2.0 equiv.) in DCM (35 mL) was added and MOMBr (1.3 mL, 1.5 equiv.) at 0 °C. The resulting mixture was heated at 35 °C for overnight, and then quenched with saturated NaHCO3 aqueous solution. The aqueous phase was extracted with DCM × 2. The combined organic layer was then washed with brine, dried over Na2SO4, concentrated, and purified by flash chromatography (SiO2, 10 to 70% DCM/hexane) to afford the desired product as single diastereomer. [0159] Step d: To a solution of the product from step c (2.30 g, 7.0 mmol, 1.0 equiv.) in 1,4- dioxane (30 mL) was added B2Pin2 (2.14 g, 8.4 mmol, 1.2 equiv.), Pd(dppf)Cl2 (513 mg, 0.70 mmol, 10 mol%) and KOAc (1.37 g, 14 mmol, 2.0 equiv.). The reaction mixture was degassed with N2 bubbling for 10 min before being heated. After stirring at 100 °C overnight, the reaction mixture was cooled, filtered through Celite®, and concentrated. The crude product was used directly in the next step.
[0160] Step e: To a mixture of (4R)-4-[tert-butyl(dimethyl)silyl]oxy-8-cyano-6-fluoro-3,4- dihydronaphthalen-1-yl] trifluoromethanesulfonate (2.85 g, 6.3 mmol, 0.9 equiv.) and the product from step d (~7.0 mmol, 1.0 equiv.) in 1,4-dioxane (30 mL) was added Pd(dppf)Cl2 (513 mg, 0.70 mmol, 10 mol%) and Na2CO3 (1M in H2O, 14 mL, 2.0 equiv.). The resulting mixture was degassed with N2 bubbling for 10 min before being heated to 100 °C and stirred overnight. The reaction mixture was cooled, concentrated onto Celite® and purified by flash chromatography (SiO2, 0 to 30% EtOAc/hexane) to afford the product. [0161] Step f: A mixture of the product from step e (2.36 g, 4.3 mmol, 1.0 equiv.), Pd/C (10 wt% Pd, 760 mg) in MeOH (30 mL) was shaken in parr hydrogenator under H2 (40 psi) for 2.5 h. After this time LCMS showed no remaining starting material. The reaction mixture was then filtered through Celite®, concentrated, and purified by flash chromatography (SiO2, 0 to 30% EtOAc/hexane) to afford the product. [0162] Step g: To a solution of the product from step f (1.88 g, 3.4 mmol, 1.0 equiv.) in THF (15 mL) was added TBAF (1M in THF, 5.1 mL, 1.5 equiv.) at 0 °C. The resulting solution was stirred at room temperature for 1 h, and then quenched by saturated NH4Cl (aq.). The aqueous phase was extracted with EtOAc twice. The combined organic layer was then washed with brine, dried over Na2SO4, concentrated, and purified by flash chromatography (SiO2, 0 to 50% EtOAc/hexane) to afford the product. [0163] Step h: To a solution of the product from step g (1.24 g, 2.8 mmol, 1.0 equiv.) in DCM (14 mL) was added NaHCO3 (470 mg, 5.6 mmol, 2.0 equiv.) and DMP (1.32 g, 3.1 mmol, 1.1 equiv.) at 0 °C. The resulting mixture was stirred at room temperature for overnight. The reaction mixture was then quenched with saturated NaHCO3 and Na2S2O3 aqueous solution and extracted with DCM twice. The combined organic layer was then washed with brine, dried over Na2SO4, and concentrated. The crude product was directly used in the next step. [0164] Step i: To a solution of the crude product from step h (~2.8 mmol) and triethylamine (3.2 mL, 22 mmol, 8.0 equiv.) in DCM (9 mL) was added TBSOTf (2.2 mL, 11 mmol, 4.0 equiv.) at 0 °C. The resulting mixture was then stirred at room temperature for 1.5 h and quenched with saturated NaHCO3 aqueous solution. The resulting mixture was then separated, and the aqueous phase was extracted with DCM twice. The combined organic phase was then
washed with brine, dried over Na2SO4, concentrated, and purified by flash chromatography (SiO2, 0 to 20% EtOAc/hexane) to afford the product. [0165] Step j: To a solution of the product from step i (~2.8 mmol, 1.0 equiv.) in MeCN (10 mL), Selectfluor™ (2.20 g, 6.2 mmol, 2.2 equiv.) was added portion-wise at room temperature. The resulting mixture was stirred at 60 °C for 30 min and then quenched with saturated NaHCO3 aqueous solution at 0 °C. The aqueous phase was extracted with EtOAc twice. The combined organic phase was then washed with brine, dried over Na2SO4, and concentrated. The crude product was directly used in the next step. [0166] Step k: HCO2H (0.20 mL, 5.3 mmol, 3.0 equiv.) was added to a solution of Et3N (0.50 mL, 3.6 mmol, 2.0 equiv.) in DCM (5 mL) dropwise. The resulting solution was stirred at room temperature for 30 min, and then added to a solution of the product from step j (800 mg, 1.8 mmol, 1.0 equiv.) and RuCl(p-cymene)[(S,S)-TsDPEN] (25 mg, 0.039 mmol, 2 mol%) in DCM (5 mL) at 0 °C. The resulting mixture was kept stirring at this temperature for overnight and then concentrated. The crude product was purified by flash chromatography (SiO2, 10 to 60% EtOAc/hexane) to afford the product. [0167] Step l: To a solution of the product from step k (620 mg, 1.4 mmol, 1.0 equiv.) and 3- nitrobenzoic acid (681 mg, 4.1 mmol, 3.0 equiv.) in THF (12 mL) was added PPh3 (856 mg, 3.3 mmol, 2.4 equiv.) and DIAD (0.65 mL, 3.3 mmol, 2.4 equiv.) at 0 °C. The resulting mixture was stirred at this temperature until the TLC showed a full consumption of the substrate. The reaction mixture was then quenched with H2O and extracted with EtOAc twice. The combined organic layer was then washed with brine, dried over Na2SO4, concentrated, and purified by flash chromatography (SiO2, 0 to 30% EtOAc/hexane) to afford the ester intermediate. The ester compound was then dissolved in THF/MeOH (2:1 v/v, 6 mL) and treated with a solution of LiOH·H2O (115 mg, 2.7 mmol, 2.0 equiv.) in H2O (2 mL). The resulting mixture was stirred at room temperature for 3 h and then diluted with H2O. The aqueous phase was extracted with EtOAc twice. The combined organic layer was then washed with brine, dried over Na2SO4, and concentrated. The crude product was directly used in the next step. [0168] Step m: To a solution of the product from step l (~1.4 mmol) in DCM (10 mL) was added DAST (0.90 mL, 6.8 mmol, 5.0 equiv.) at -40 °C. The resulting mixture was then raised to
-10 °C and stirred at this temperature until TLC showed a full conversion of the substrate. The reaction mixture was then quenched with saturated NaHCO3 aqueous solution at -10 °C. The aqueous phase was extracted with DCM twice. The combined organic layer was then washed with brine, dried over Na2SO4, concentrated, and purified by flash chromatography (SiO2, 0 to 30% EtOAc/hexane) to afford the product. [0169] Step n: To a solution of the product from step m (90.0 mg, 0.20 mmol, 1.0 equiv.) and 1,2-dimethyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)imidazole (66.6 mg, 0.30 mmol, 1.5 equiv.) in 1,4-dioxane (2.0 mL) was added SPhos Pd G2 (14.4 mg, 0.020 mmol, 10 mol%) and Na2CO3 (1M in H2O, 0.40 mL, 2.0 equiv.). The resulting mixture was degassed with N2 bubbling for 10 min before being heated to 100 °C and stirred overnight. The reaction mixture was cooled, concentrated onto Celite® and purified by flash chromatography (SiO2, 0 to 10% MeOH/DCM) to afford the product. [0170] Step o: To a solution of the product from step n (~0.20 mmol) in DCM (3 mL) was added TFA (0.30 mL). The resulting mixture was heated at 40 °C for 1 h and then concentrated. The crude mixture was then purified by HPLC to afford the title compound.1H NMR (400 MHz, DMSO-d6) δ 7.98 (ddd, J = 8.2, 2.8, 1.2 Hz, 1H), 7.89 (dd, J = 9.0, 2.8 Hz, 1H), 6.97 (s, 1H), 6.49 (d, J = 11.0 Hz, 1H), 5.99 (dd, J = 54.0, 14.3 Hz, 1H), 5.49 – 5.05 (m, 3H), 4.97 – 4.67 (m, 2H), 3.29 (d, J = 1.6 Hz, 3H), 3.14 (dtd, J = 44.5, 15.8, 15.1, 5.9 Hz, 2H), 2.78 – 2.62 (m, 1H), 2.35 (s, 3H), 2.12 – 1.94 (m, 1H). ESI MS [M+H]+ for C25H20F5N3O, calcd 474.2, found 474.1.
[0156] Step a: To a solution of 4-bromo-6,7-dichloro-2,3-dihydroinden-1-one (5.20 g, 22 mmol, 1.0 equiv.) in MeOH (100 mL) was added Selectfluor™ (9.31 g, 26 mmol, 1.2 equiv.) and concentrated H2SO4 (5 drops). The resulting mixture was heated at reflux for 3 h. After cooling to room temperature, 0.3 M H2SO4 (aq., 50 mL) was added to the reaction mixture. The resulting mixture was heated at reflux for another 1 h. After cooling to room temperature, large amount of the product was precipitated out and collected via filtration. The filtrate was concentrated and diluted with DCM (250 mL) and washed with H2O and brine. The organic phase was dried over Na2SO4 and concentrated. The product was directly used in the next step. [0157] Step b: HCO2H (2.5 mL, 66 mmol, 3.0 equiv.) was added to a solution of Et3N (6.2 mL, 44 mmol, 2.0 equiv.) in DMF (20 mL) dropwise. The resulting solution was stirred at room temperature for 30 min, and then added to a solution of the product from step a (~22 mmol, 1.0 equiv.) and RuCl(p-cymene)[(R,R)-TsDPEN] (557 mg, 0.88 mmol, 4.0 mol%) in DMF (20 mL) at 0 °C. The resulting mixture was kept stirring at this temperature for overnight and then diluted with EtOAc and washed with H2O twice. The organic phase was then washed with brine, dried over Na2SO4, concentrated, and purified by flash chromatography (SiO2, 0 to 40% EtOAc/hexane) to afford the desired product (d.r.4.5:1, 75% e.e.). [0158] Step c: To a solution of the product from step b (2.94 g, 10.4 mmol, 1.0 equiv.) and diisopropylethylamine (3.7 mL, 2.0 equiv.) in DCM (35 mL) was added and MOMBr (1.3 mL, 1.5 equiv.) at 0 °C. The resulting mixture was heated at 35 °C for overnight, and then quenched with saturated NaHCO3 aqueous solution. The aqueous phase was extracted with DCM × 2. The combined organic layer was then washed with brine, dried over Na2SO4, concentrated, and purified by flash chromatography (SiO2, 10 to 70% DCM/hexane) to afford the desired product as single diastereomer. [0159] Step d: To a solution of the product from step c (2.30 g, 7.0 mmol, 1.0 equiv.) in 1,4- dioxane (30 mL) was added B2Pin2 (2.14 g, 8.4 mmol, 1.2 equiv.), Pd(dppf)Cl2 (513 mg, 0.70 mmol, 10 mol%) and KOAc (1.37 g, 14 mmol, 2.0 equiv.). The reaction mixture was degassed with N2 bubbling for 10 min before being heated. After stirring at 100 °C overnight, the reaction mixture was cooled, filtered through Celite®, and concentrated. The crude product was used directly in the next step.
[0160] Step e: To a mixture of (4R)-4-[tert-butyl(dimethyl)silyl]oxy-8-cyano-6-fluoro-3,4- dihydronaphthalen-1-yl] trifluoromethanesulfonate (2.85 g, 6.3 mmol, 0.9 equiv.) and the product from step d (~7.0 mmol, 1.0 equiv.) in 1,4-dioxane (30 mL) was added Pd(dppf)Cl2 (513 mg, 0.70 mmol, 10 mol%) and Na2CO3 (1M in H2O, 14 mL, 2.0 equiv.). The resulting mixture was degassed with N2 bubbling for 10 min before being heated to 100 °C and stirred overnight. The reaction mixture was cooled, concentrated onto Celite® and purified by flash chromatography (SiO2, 0 to 30% EtOAc/hexane) to afford the product. [0161] Step f: A mixture of the product from step e (2.36 g, 4.3 mmol, 1.0 equiv.), Pd/C (10 wt% Pd, 760 mg) in MeOH (30 mL) was shaken in parr hydrogenator under H2 (40 psi) for 2.5 h. After this time LCMS showed no remaining starting material. The reaction mixture was then filtered through Celite®, concentrated, and purified by flash chromatography (SiO2, 0 to 30% EtOAc/hexane) to afford the product. [0162] Step g: To a solution of the product from step f (1.88 g, 3.4 mmol, 1.0 equiv.) in THF (15 mL) was added TBAF (1M in THF, 5.1 mL, 1.5 equiv.) at 0 °C. The resulting solution was stirred at room temperature for 1 h, and then quenched by saturated NH4Cl (aq.). The aqueous phase was extracted with EtOAc twice. The combined organic layer was then washed with brine, dried over Na2SO4, concentrated, and purified by flash chromatography (SiO2, 0 to 50% EtOAc/hexane) to afford the product. [0163] Step h: To a solution of the product from step g (1.24 g, 2.8 mmol, 1.0 equiv.) in DCM (14 mL) was added NaHCO3 (470 mg, 5.6 mmol, 2.0 equiv.) and DMP (1.32 g, 3.1 mmol, 1.1 equiv.) at 0 °C. The resulting mixture was stirred at room temperature for overnight. The reaction mixture was then quenched with saturated NaHCO3 and Na2S2O3 aqueous solution and extracted with DCM twice. The combined organic layer was then washed with brine, dried over Na2SO4, and concentrated. The crude product was directly used in the next step. [0164] Step i: To a solution of the crude product from step h (~2.8 mmol) and triethylamine (3.2 mL, 22 mmol, 8.0 equiv.) in DCM (9 mL) was added TBSOTf (2.2 mL, 11 mmol, 4.0 equiv.) at 0 °C. The resulting mixture was then stirred at room temperature for 1.5 h and quenched with saturated NaHCO3 aqueous solution. The resulting mixture was then separated, and the aqueous phase was extracted with DCM twice. The combined organic phase was then
washed with brine, dried over Na2SO4, concentrated, and purified by flash chromatography (SiO2, 0 to 20% EtOAc/hexane) to afford the product. [0165] Step j: To a solution of the product from step i (~2.8 mmol, 1.0 equiv.) in MeCN (10 mL), Selectfluor™ (2.20 g, 6.2 mmol, 2.2 equiv.) was added portion-wise at room temperature. The resulting mixture was stirred at 60 °C for 30 min and then quenched with saturated NaHCO3 aqueous solution at 0 °C. The aqueous phase was extracted with EtOAc twice. The combined organic phase was then washed with brine, dried over Na2SO4, and concentrated. The crude product was directly used in the next step. [0166] Step k: HCO2H (0.20 mL, 5.3 mmol, 3.0 equiv.) was added to a solution of Et3N (0.50 mL, 3.6 mmol, 2.0 equiv.) in DCM (5 mL) dropwise. The resulting solution was stirred at room temperature for 30 min, and then added to a solution of the product from step j (800 mg, 1.8 mmol, 1.0 equiv.) and RuCl(p-cymene)[(S,S)-TsDPEN] (25 mg, 0.039 mmol, 2 mol%) in DCM (5 mL) at 0 °C. The resulting mixture was kept stirring at this temperature for overnight and then concentrated. The crude product was purified by flash chromatography (SiO2, 10 to 60% EtOAc/hexane) to afford the product. [0167] Step l: To a solution of the product from step k (620 mg, 1.4 mmol, 1.0 equiv.) and 3- nitrobenzoic acid (681 mg, 4.1 mmol, 3.0 equiv.) in THF (12 mL) was added PPh3 (856 mg, 3.3 mmol, 2.4 equiv.) and DIAD (0.65 mL, 3.3 mmol, 2.4 equiv.) at 0 °C. The resulting mixture was stirred at this temperature until the TLC showed a full consumption of the substrate. The reaction mixture was then quenched with H2O and extracted with EtOAc twice. The combined organic layer was then washed with brine, dried over Na2SO4, concentrated, and purified by flash chromatography (SiO2, 0 to 30% EtOAc/hexane) to afford the ester intermediate. The ester compound was then dissolved in THF/MeOH (2:1 v/v, 6 mL) and treated with a solution of LiOH·H2O (115 mg, 2.7 mmol, 2.0 equiv.) in H2O (2 mL). The resulting mixture was stirred at room temperature for 3 h and then diluted with H2O. The aqueous phase was extracted with EtOAc twice. The combined organic layer was then washed with brine, dried over Na2SO4, and concentrated. The crude product was directly used in the next step. [0168] Step m: To a solution of the product from step l (~1.4 mmol) in DCM (10 mL) was added DAST (0.90 mL, 6.8 mmol, 5.0 equiv.) at -40 °C. The resulting mixture was then raised to
-10 °C and stirred at this temperature until TLC showed a full conversion of the substrate. The reaction mixture was then quenched with saturated NaHCO3 aqueous solution at -10 °C. The aqueous phase was extracted with DCM twice. The combined organic layer was then washed with brine, dried over Na2SO4, concentrated, and purified by flash chromatography (SiO2, 0 to 30% EtOAc/hexane) to afford the product. [0169] Step n: To a solution of the product from step m (90.0 mg, 0.20 mmol, 1.0 equiv.) and 1,2-dimethyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)imidazole (66.6 mg, 0.30 mmol, 1.5 equiv.) in 1,4-dioxane (2.0 mL) was added SPhos Pd G2 (14.4 mg, 0.020 mmol, 10 mol%) and Na2CO3 (1M in H2O, 0.40 mL, 2.0 equiv.). The resulting mixture was degassed with N2 bubbling for 10 min before being heated to 100 °C and stirred overnight. The reaction mixture was cooled, concentrated onto Celite® and purified by flash chromatography (SiO2, 0 to 10% MeOH/DCM) to afford the product. [0170] Step o: To a solution of the product from step n (~0.20 mmol) in DCM (3 mL) was added TFA (0.30 mL). The resulting mixture was heated at 40 °C for 1 h and then concentrated. The crude mixture was then purified by HPLC to afford the title compound.1H NMR (400 MHz, DMSO-d6) δ 7.98 (ddd, J = 8.2, 2.8, 1.2 Hz, 1H), 7.89 (dd, J = 9.0, 2.8 Hz, 1H), 6.97 (s, 1H), 6.49 (d, J = 11.0 Hz, 1H), 5.99 (dd, J = 54.0, 14.3 Hz, 1H), 5.49 – 5.05 (m, 3H), 4.97 – 4.67 (m, 2H), 3.29 (d, J = 1.6 Hz, 3H), 3.14 (dtd, J = 44.5, 15.8, 15.1, 5.9 Hz, 2H), 2.78 – 2.62 (m, 1H), 2.35 (s, 3H), 2.12 – 1.94 (m, 1H). ESI MS [M+H]+ for C25H20F5N3O, calcd 474.2, found 474.1.
Example 10: (5R,6S,8R)-8-[(1S,2R)-2,6-Difluoro-1-hydroxy-7-(1-imidazolyl)-4-indanyl]- 3,5,6-trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile
 [0171] Step a: To a solution of 4-bromo-6,7-dichloro-2,3-dihydroinden-1-one (20.0 g, 81 mmol, 1.0 equiv.) in MeOH (400 mL) was added Selectfluor™ (31.5 g, 89 mmol, 1.1 equiv.) and concentrated H2SO4 (1.0 mL). The resulting mixture was heated at reflux for 3 h. After cooling to room temperature, 0.3 M H2SO4 (aq., 100 mL) was added to the reaction mixture. The resulting mixture was heated at reflux for another 1 h. After cooling to room temperature, large amount of the product was precipitated out and collected via filtration. The filtrate was concentrated and diluted with DCM (250 mL) and washed with H2O and brine. The organic phase was dried over Na2SO4 and concentrated. The product was directly used in the next step. [0172] Step b: To a solution of the product from step a (7.19 g, 27 mmol, 1.0 equiv.) in MeOH (100 mL) was added NaBH4 (1.33 g, 35 mmol, 1.3 equiv.) at 0 °C. The resulting mixture was stirred at this temperature until TLC showed a full conversion. The reaction mixture was then quenched with water, diluted with EtOAc, and separated. The aqueous phase was extracted with EtOAc twice. The combined organic layer was then washed with brine, dried over Na2SO4, and concentrated. The crude product was directly used in the next step. [0173] Step c: To a solution of the crude product from step b (~27 mmol) and diisopropylethylamine (14.2 mL, 3.0 equiv.) in DCM (70 mL) was added and MOMBr (4.4 mL, 2.0 equiv.) at 0 °C. The resulting mixture was heated at 30 °C for overnight, and then quenched with saturated NaHCO3 aqueous solution. The aqueous phase was extracted with DCM × 2. The combined organic layer was then washed with brine, dried over Na2SO4, concentrated, and purified by flash chromatography (SiO2, 0 to 100% DCM/hexane) to afford the desired product as a racemic mixture. [0174] Step d: To a solution of the product from step c (4.70 g, 15 mmol, 1.0 equiv.) in 1,4- dioxane (75 mL) was added B2Pin2 (4.60 g, 18 mmol, 1.2 equiv.), Pd(dppf)Cl2 (1.10 g, 1.5 mmol, 10 mol%) and KOAc (2.13 g, 22 mmol, 1.5 equiv.). The reaction mixture was degassed with N2 bubbling for 10 min before being heated to 100 °C. After stirring at 100 °C overnight, the reaction mixture was cooled, concentrated on Celite® and purified by flash chromatography (SiO2, 0 to 15% EtOAc/hexane) to afford the product.
[0175] Step e: To a mixture of (4R)-4-[tert-butyl(dimethyl)silyl]oxy-8-cyano-6-fluoro-3,4- dihydronaphthalen-1-yl] trifluoromethanesulfonate (6.13 g, 13.6 mmol, 1.05 equiv.) and the product from step d (4.64 g, 12.9 mmol, 1.0 equiv.) in 1,4-dioxane (25 mL) was added Pd(dppf)Cl2 (471 mg, 0.64 mmol, 5 mol%) and Na2CO3 (1M in H2O, 25 mL, 2.0 equiv.). The resulting mixture was degassed with N2 bubbling for 10 min before being heated to 80 °C and stirred overnight. The reaction mixture was cooled, concentrated onto Celite® and purified by flash chromatography (SiO2, 0 to 15% EtOAc/hexane) to afford the desired product as a mixture of diastereomers (d.r.1:1). [0176] Step f: A mixture of the product from step e (5.24 g, 9.8 mmol, 1.0 equiv.), Pd/C (10 wt% Pd, 1.00 g) in MeOH (50 mL) was shaken in parr hydrogenator under H2 (50 psi) for 4 h, when LCMS showed no remaining starting material. The reaction mixture was then filtered through Celite®, concentrated, and purified by flash chromatography (SiO2, 0 to 15% EtOAc/Hex) to afford the product as a mixture of diastereomers (d.r.1:1). [0177] Step g: To a solution of the product from step f (3.58 g, 6.7 mmol, 1.0 equiv.) in THF (30 mL) was added TBAF (1M in THF, 10 mL, 1.5 equiv.) at 0 °C. The resulting solution was stirred at 0 °C for 15 min, and then quenched by saturated NH4Cl (aq.). The aqueous phase was extracted with EtOAc × 2. The combined organic layer was then washed with brine, dried over Na2SO4, and concentrated. The crude material was then dissolved with DCM (30 mL) and treated with NaHCO3 (620 mg, 7.4 mmol, 1.1 equiv.) and DMP (3.12 g, 7.4 mmol, 1.1 equiv.) at 0 °C. The resulting mixture was stirred at room temperature for overnight. The reaction mixture was then quenched with saturated NaHCO3 and Na2S2O3 aqueous solution and extracted with DCM twice. The combined organic layer was then washed with brine, dried over Na2SO4, concentrated. The crude product was directly used in the next step. [0178] Step h: To a solution of the crude product from step g (~6.7 mmol) and triethylamine (4.2 mL, 30 mmol, 4.5 equiv.) in DCM (30 mL) was added TBSOTf (5.4 mL, 24 mmol, 3.5 equiv.) at 0 °C. The resulting mixture was then stirred at room temperature for overnight and quenched with saturated NaHCO3 (aq.) and kept stirring for 1 h. The resulting mixture was then separated, and the aqueous phase was extracted with DCM twice. The combined organic phase was then washed with brine, dried over Na2SO4, concentrated, and purified by flash
chromatography (SiO2, 0 to 15% EtOAc/hexane) to afford the product as a mixture of diastereomers (d.r.1:1). [0179] Step i: To a solution of the product from step h (2.21 g, 4.1 mmol, 1.0 equiv.) in MeCN (20 mL), Selectfluor™ (2.50 g, 7.1 mmol, 1.7 equiv.) was added portion-wise at room temperature. The resulting mixture was stirred at room temperature for 30 min and then quenched with saturated NaHCO3 aqueous solution. The aqueous phase was extracted with EtOAc twice. The combined organic phase was then washed with brine, dried over Na2SO4, and concentrated. The crude product was directly used in the next step. [0180] Step j: HCO2H (0.47 mL, 12 mmol, 3.0 equiv.) was added to a solution of Et3N (1.2 mL, 8.3 mmol, 2.0 equiv.) in DCM (10 mL) dropwise. The resulting solution was stirred at room temperature for 30 min, and then added to a solution of the product from step i (~4.1 mmol, 1.0 equiv.) and RuCl(p-cymene)[(R,R)-TsDPEN] (79 mg, 0.12 mmol, 3.0 mol%) in DCM (10 mL) at 0 °C. The resulting mixture was kept stirring at this temperature for overnight and then concentrated. The crude product was purified by flash chromatography (SiO2, 10 to 60% EtOAc/hexane) to afford the product as a mixture of diastereomers. [0181] Step k: To a solution of 4-(trimethylsilyl)morpholine (1.44 g, 9.0 mmol, 3.0 equiv.) in toluene (10 mL) was added deoxofluor (2.7 M in toluene, 3.3 mL, 9.0 mmol, 3.0 equiv.) dropwise at −78 °C. The resulting solution was then stirred at this temperature for 5 min and warmed to room temperature for 1.5 h. The reaction mixture was diluted with another 40 mL of toluene and then cooled back to −78 °C. A solution of the product from step j (1.32 g, 3.0 mmol, 1.0 equiv.) in DCM (9 mL) was added dropwise. The resulting solution was then stirred at this temperature for 5 min, and then warmed to room temperature and stirred for overnight. The reaction was quenched with saturated NaHCO3 (aq.). The aqueous layer was extracted with DCM × 2. The combined organic layer was then washed with brine, dried over Na2SO4, and concentrated. The crude product was directly used in the next step. [0182] Step l: To a solution of the crude product from step k (~3.0 mmol) in THF (20 mL) was added 6M HCl aqueous solution (20 mL). The resulting mixture was heated at 40 °C until LCMS showed a full conversion. The reaction mixture was then cooled to room temperature and diluted with H2O. The aqueous phase was extracted with EtOAc× 3. The combined organic
phase was then washed with brine, dried over Na2SO4, and concentrated. The crude product was directly used in the next step. [0183] Step m: To a solution of the crude product from step l (~3.0 mmol, 1.0 equiv.) in DCM (20 mL) was added NaHCO3 (519 mg, 6.0 mmol, 2.0 equiv.) and DMP (1.44 g, 3.4 mmol, 1.1 equiv.) at 0 °C. The resulting mixture was stirred at this temperature for 1 h, before quenched with saturated NaHCO3 and Na2S2O3 aqueous solution and extracted with DCM twice. The combined organic layer was then washed with brine, dried over Na2SO4, concentrated, and purified by flash chromatography (SiO2, 10 to 60% EtOAc/hexane) to afford the product as a mixture of diastereomers (d.r.1:1). [0184] Step n: To a solution of the product from step m (55 mg, 0.14 mmol, 1.0 equiv.) and imidazole (34 mg, 0.42 mmol, 3.0 equiv.) in MeCN (1.2 mL) was added diisopropylethylamine (0.13 mL, 0.70 mmol, 5.0 equiv.). The resulting mixture was heated at 80 °C for 4.5 h and then concentrated. The crude mixture was then purified by flash chromatography (SiO2, 0 to 10% MeOH/DCM) to afford the product. [0185] Step o: HCO2H (7.3 µL, 0.19 mmol, 4.0 equiv.) was added to a solution of Et3N (20 µL, 0.14 mmol, 3.0 equiv.) in DMF (0.4 mL). The resulting solution was stirred at room temperature for 30 min, and then added to a solution of the product from step n (21 mg, 47.4 µmol, 1.0 equiv.) and RuCl(p-cymene)[(R,R)-TsDPEN] (0.9 mg, 1.4 µmol, 3.0 mol%) in DMF (0.1 mL) at 0 °C. The resulting mixture was kept stirring at this temperature for overnight and then diluted with EtOAc and washed with H2O twice. The organic phase was then washed with brine, dried over Na2SO4, and concentrated. The crude product was purified by HPLC to afford the title compound.1H NMR (400 MHz, DMSO-d6) δ 7.96 (dt, J = 8.2, 2.0 Hz, 1H), 7.91 – 7.85 (m, 2H), 7.43 – 7.37 (m, 1H), 7.06 – 7.00 (m, 1H), 6.55 (d, J = 11.4 Hz, 1H), 5.95 (dd, J = 53.0, 14.4 Hz, 1H), 5.53 (s, 1H), 5.34 – 5.03 (m, 3H), 4.80 (t, J = 5.7 Hz, 1H), 3.28 – 3.03 (m, 2H), 2.75 – 2.60 (m, 1H), 2.03 – 1.85 (m, 1H). ESI MS [M+H]+ for C23H16F5N3O, calcd 446.1, found 446.0.
Example 11: (5R,6S,8R)-8-[(1S,2R)-2,6-difluoro-1-hydroxy-7-mesyl-4-indanyl]-3,5,6- trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile
[0171] Step a: To a solution of 4-bromo-6,7-dichloro-2,3-dihydroinden-1-one (20.0 g, 81 mmol, 1.0 equiv.) in MeOH (400 mL) was added Selectfluor™ (31.5 g, 89 mmol, 1.1 equiv.) and concentrated H2SO4 (1.0 mL). The resulting mixture was heated at reflux for 3 h. After cooling to room temperature, 0.3 M H2SO4 (aq., 100 mL) was added to the reaction mixture. The resulting mixture was heated at reflux for another 1 h. After cooling to room temperature, large amount of the product was precipitated out and collected via filtration. The filtrate was concentrated and diluted with DCM (250 mL) and washed with H2O and brine. The organic phase was dried over Na2SO4 and concentrated. The product was directly used in the next step. [0172] Step b: To a solution of the product from step a (7.19 g, 27 mmol, 1.0 equiv.) in MeOH (100 mL) was added NaBH4 (1.33 g, 35 mmol, 1.3 equiv.) at 0 °C. The resulting mixture was stirred at this temperature until TLC showed a full conversion. The reaction mixture was then quenched with water, diluted with EtOAc, and separated. The aqueous phase was extracted with EtOAc twice. The combined organic layer was then washed with brine, dried over Na2SO4, and concentrated. The crude product was directly used in the next step. [0173] Step c: To a solution of the crude product from step b (~27 mmol) and diisopropylethylamine (14.2 mL, 3.0 equiv.) in DCM (70 mL) was added and MOMBr (4.4 mL, 2.0 equiv.) at 0 °C. The resulting mixture was heated at 30 °C for overnight, and then quenched with saturated NaHCO3 aqueous solution. The aqueous phase was extracted with DCM × 2. The combined organic layer was then washed with brine, dried over Na2SO4, concentrated, and purified by flash chromatography (SiO2, 0 to 100% DCM/hexane) to afford the desired product as a racemic mixture. [0174] Step d: To a solution of the product from step c (4.70 g, 15 mmol, 1.0 equiv.) in 1,4- dioxane (75 mL) was added B2Pin2 (4.60 g, 18 mmol, 1.2 equiv.), Pd(dppf)Cl2 (1.10 g, 1.5 mmol, 10 mol%) and KOAc (2.13 g, 22 mmol, 1.5 equiv.). The reaction mixture was degassed with N2 bubbling for 10 min before being heated to 100 °C. After stirring at 100 °C overnight, the reaction mixture was cooled, concentrated on Celite® and purified by flash chromatography (SiO2, 0 to 15% EtOAc/hexane) to afford the product.
[0175] Step e: To a mixture of (4R)-4-[tert-butyl(dimethyl)silyl]oxy-8-cyano-6-fluoro-3,4- dihydronaphthalen-1-yl] trifluoromethanesulfonate (6.13 g, 13.6 mmol, 1.05 equiv.) and the product from step d (4.64 g, 12.9 mmol, 1.0 equiv.) in 1,4-dioxane (25 mL) was added Pd(dppf)Cl2 (471 mg, 0.64 mmol, 5 mol%) and Na2CO3 (1M in H2O, 25 mL, 2.0 equiv.). The resulting mixture was degassed with N2 bubbling for 10 min before being heated to 80 °C and stirred overnight. The reaction mixture was cooled, concentrated onto Celite® and purified by flash chromatography (SiO2, 0 to 15% EtOAc/hexane) to afford the desired product as a mixture of diastereomers (d.r.1:1). [0176] Step f: A mixture of the product from step e (5.24 g, 9.8 mmol, 1.0 equiv.), Pd/C (10 wt% Pd, 1.00 g) in MeOH (50 mL) was shaken in parr hydrogenator under H2 (50 psi) for 4 h, when LCMS showed no remaining starting material. The reaction mixture was then filtered through Celite®, concentrated, and purified by flash chromatography (SiO2, 0 to 15% EtOAc/Hex) to afford the product as a mixture of diastereomers (d.r.1:1). [0177] Step g: To a solution of the product from step f (3.58 g, 6.7 mmol, 1.0 equiv.) in THF (30 mL) was added TBAF (1M in THF, 10 mL, 1.5 equiv.) at 0 °C. The resulting solution was stirred at 0 °C for 15 min, and then quenched by saturated NH4Cl (aq.). The aqueous phase was extracted with EtOAc × 2. The combined organic layer was then washed with brine, dried over Na2SO4, and concentrated. The crude material was then dissolved with DCM (30 mL) and treated with NaHCO3 (620 mg, 7.4 mmol, 1.1 equiv.) and DMP (3.12 g, 7.4 mmol, 1.1 equiv.) at 0 °C. The resulting mixture was stirred at room temperature for overnight. The reaction mixture was then quenched with saturated NaHCO3 and Na2S2O3 aqueous solution and extracted with DCM twice. The combined organic layer was then washed with brine, dried over Na2SO4, concentrated. The crude product was directly used in the next step. [0178] Step h: To a solution of the crude product from step g (~6.7 mmol) and triethylamine (4.2 mL, 30 mmol, 4.5 equiv.) in DCM (30 mL) was added TBSOTf (5.4 mL, 24 mmol, 3.5 equiv.) at 0 °C. The resulting mixture was then stirred at room temperature for overnight and quenched with saturated NaHCO3 (aq.) and kept stirring for 1 h. The resulting mixture was then separated, and the aqueous phase was extracted with DCM twice. The combined organic phase was then washed with brine, dried over Na2SO4, concentrated, and purified by flash
chromatography (SiO2, 0 to 15% EtOAc/hexane) to afford the product as a mixture of diastereomers (d.r.1:1). [0179] Step i: To a solution of the product from step h (2.21 g, 4.1 mmol, 1.0 equiv.) in MeCN (20 mL), Selectfluor™ (2.50 g, 7.1 mmol, 1.7 equiv.) was added portion-wise at room temperature. The resulting mixture was stirred at room temperature for 30 min and then quenched with saturated NaHCO3 aqueous solution. The aqueous phase was extracted with EtOAc twice. The combined organic phase was then washed with brine, dried over Na2SO4, and concentrated. The crude product was directly used in the next step. [0180] Step j: HCO2H (0.47 mL, 12 mmol, 3.0 equiv.) was added to a solution of Et3N (1.2 mL, 8.3 mmol, 2.0 equiv.) in DCM (10 mL) dropwise. The resulting solution was stirred at room temperature for 30 min, and then added to a solution of the product from step i (~4.1 mmol, 1.0 equiv.) and RuCl(p-cymene)[(R,R)-TsDPEN] (79 mg, 0.12 mmol, 3.0 mol%) in DCM (10 mL) at 0 °C. The resulting mixture was kept stirring at this temperature for overnight and then concentrated. The crude product was purified by flash chromatography (SiO2, 10 to 60% EtOAc/hexane) to afford the product as a mixture of diastereomers. [0181] Step k: To a solution of 4-(trimethylsilyl)morpholine (1.44 g, 9.0 mmol, 3.0 equiv.) in toluene (10 mL) was added deoxofluor (2.7 M in toluene, 3.3 mL, 9.0 mmol, 3.0 equiv.) dropwise at −78 °C. The resulting solution was then stirred at this temperature for 5 min and warmed to room temperature for 1.5 h. The reaction mixture was diluted with another 40 mL of toluene and then cooled back to −78 °C. A solution of the product from step j (1.32 g, 3.0 mmol, 1.0 equiv.) in DCM (9 mL) was added dropwise. The resulting solution was then stirred at this temperature for 5 min, and then warmed to room temperature and stirred for overnight. The reaction was quenched with saturated NaHCO3 (aq.). The aqueous layer was extracted with DCM × 2. The combined organic layer was then washed with brine, dried over Na2SO4, and concentrated. The crude product was directly used in the next step. [0182] Step l: To a solution of the crude product from step k (~3.0 mmol) in THF (20 mL) was added 6M HCl aqueous solution (20 mL). The resulting mixture was heated at 40 °C until LCMS showed a full conversion. The reaction mixture was then cooled to room temperature and diluted with H2O. The aqueous phase was extracted with EtOAc× 3. The combined organic
phase was then washed with brine, dried over Na2SO4, and concentrated. The crude product was directly used in the next step. [0183] Step m: To a solution of the crude product from step l (~3.0 mmol, 1.0 equiv.) in DCM (20 mL) was added NaHCO3 (519 mg, 6.0 mmol, 2.0 equiv.) and DMP (1.44 g, 3.4 mmol, 1.1 equiv.) at 0 °C. The resulting mixture was stirred at this temperature for 1 h, before quenched with saturated NaHCO3 and Na2S2O3 aqueous solution and extracted with DCM twice. The combined organic layer was then washed with brine, dried over Na2SO4, concentrated, and purified by flash chromatography (SiO2, 10 to 60% EtOAc/hexane) to afford the product as a mixture of diastereomers (d.r.1:1). [0184] Step n: To a solution of the product from step m (55 mg, 0.14 mmol, 1.0 equiv.) and imidazole (34 mg, 0.42 mmol, 3.0 equiv.) in MeCN (1.2 mL) was added diisopropylethylamine (0.13 mL, 0.70 mmol, 5.0 equiv.). The resulting mixture was heated at 80 °C for 4.5 h and then concentrated. The crude mixture was then purified by flash chromatography (SiO2, 0 to 10% MeOH/DCM) to afford the product. [0185] Step o: HCO2H (7.3 µL, 0.19 mmol, 4.0 equiv.) was added to a solution of Et3N (20 µL, 0.14 mmol, 3.0 equiv.) in DMF (0.4 mL). The resulting solution was stirred at room temperature for 30 min, and then added to a solution of the product from step n (21 mg, 47.4 µmol, 1.0 equiv.) and RuCl(p-cymene)[(R,R)-TsDPEN] (0.9 mg, 1.4 µmol, 3.0 mol%) in DMF (0.1 mL) at 0 °C. The resulting mixture was kept stirring at this temperature for overnight and then diluted with EtOAc and washed with H2O twice. The organic phase was then washed with brine, dried over Na2SO4, and concentrated. The crude product was purified by HPLC to afford the title compound.1H NMR (400 MHz, DMSO-d6) δ 7.96 (dt, J = 8.2, 2.0 Hz, 1H), 7.91 – 7.85 (m, 2H), 7.43 – 7.37 (m, 1H), 7.06 – 7.00 (m, 1H), 6.55 (d, J = 11.4 Hz, 1H), 5.95 (dd, J = 53.0, 14.4 Hz, 1H), 5.53 (s, 1H), 5.34 – 5.03 (m, 3H), 4.80 (t, J = 5.7 Hz, 1H), 3.28 – 3.03 (m, 2H), 2.75 – 2.60 (m, 1H), 2.03 – 1.85 (m, 1H). ESI MS [M+H]+ for C23H16F5N3O, calcd 446.1, found 446.0.
Example 11: (5R,6S,8R)-8-[(1S,2R)-2,6-difluoro-1-hydroxy-7-mesyl-4-indanyl]-3,5,6- trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile
 [0186] The title compound was prepared using a similar procedure as that described for Example 10 from the appropriate starting materials.1H NMR (400 MHz, DMSO-d6) 8.00 – 7.92 (m, 1H), 7.88 (dd, J = 9.2, 2.8 Hz, 1H), 6.61 (d, J = 11.8 Hz, 1H), 5.94 (dd, J = 51.0, 14.0 Hz, 1H), 5.62 (d, J = 5.6 Hz, 1H), 5.58 (q, J = 5.1 Hz, 1H), 5.30 – 5.21 (m, 1H), 5.19 – 5.08 (m, 1H), 4.79 (s, 1H), 3.31 (s, 3H), 3.14 (dd, J = 15.0, 6.5 Hz, 2H), 2.71 – 2.60 (m, 1H), 2.00 – 1.87 (m, 1H). ESI MS [M+NH4]+ for C21H20F5N2O3S, calcd 475.1, found 475.0. Example 12: (5R,6S,8R)-8-[(1S,2R)-2,6-difluoro-1-hydroxy-7-(1-methyl-5-pyrazolyl)-4- indanyl]-3,5,6-trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile
[0186] The title compound was prepared using a similar procedure as that described for Example 10 from the appropriate starting materials.1H NMR (400 MHz, DMSO-d6) 8.00 – 7.92 (m, 1H), 7.88 (dd, J = 9.2, 2.8 Hz, 1H), 6.61 (d, J = 11.8 Hz, 1H), 5.94 (dd, J = 51.0, 14.0 Hz, 1H), 5.62 (d, J = 5.6 Hz, 1H), 5.58 (q, J = 5.1 Hz, 1H), 5.30 – 5.21 (m, 1H), 5.19 – 5.08 (m, 1H), 4.79 (s, 1H), 3.31 (s, 3H), 3.14 (dd, J = 15.0, 6.5 Hz, 2H), 2.71 – 2.60 (m, 1H), 2.00 – 1.87 (m, 1H). ESI MS [M+NH4]+ for C21H20F5N2O3S, calcd 475.1, found 475.0. Example 12: (5R,6S,8R)-8-[(1S,2R)-2,6-difluoro-1-hydroxy-7-(1-methyl-5-pyrazolyl)-4- indanyl]-3,5,6-trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile
 [0187] The title compound was prepared using a similar procedure as that described for Example 10 from the appropriate starting materials.1H NMR (400 MHz, DMSO-d6) δ 7.98 (ddd, J = 8.3, 2.8, 1.2 Hz, 1H), 7.89 (dd, J = 9.0, 2.8 Hz, 1H), 7.48 (d, J = 1.9 Hz, 1H), 6.51 (d, J = 10.9 Hz, 1H), 6.40 (s, 1H), 6.00 (dd, J = 51.4, 14.4 Hz, 1H), 5.44 – 5.06 (m, 3H), 4.93 (s, 1H), 4.80 (t, J = 6.0 Hz, 1H), 3.60 (s, 3H), 3.29 – 2.98 (m, 2H), 2.76 – 2.62 (m, 1H), 2.11 – 1.92 (m, 1H). ESI MS [M+H]+ for C24H18F5N3O, calcd 460.1, found 460.0.
Example 13: (5R,6S,8R)-8-[(1S,2R)-7-(2-amino-4-methyl-5-pyrimidinyl)-2,6-difluoro-1- hydroxy-4-indanyl]-3,5,6-trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile
[0187] The title compound was prepared using a similar procedure as that described for Example 10 from the appropriate starting materials.1H NMR (400 MHz, DMSO-d6) δ 7.98 (ddd, J = 8.3, 2.8, 1.2 Hz, 1H), 7.89 (dd, J = 9.0, 2.8 Hz, 1H), 7.48 (d, J = 1.9 Hz, 1H), 6.51 (d, J = 10.9 Hz, 1H), 6.40 (s, 1H), 6.00 (dd, J = 51.4, 14.4 Hz, 1H), 5.44 – 5.06 (m, 3H), 4.93 (s, 1H), 4.80 (t, J = 6.0 Hz, 1H), 3.60 (s, 3H), 3.29 – 2.98 (m, 2H), 2.76 – 2.62 (m, 1H), 2.11 – 1.92 (m, 1H). ESI MS [M+H]+ for C24H18F5N3O, calcd 460.1, found 460.0.
Example 13: (5R,6S,8R)-8-[(1S,2R)-7-(2-amino-4-methyl-5-pyrimidinyl)-2,6-difluoro-1- hydroxy-4-indanyl]-3,5,6-trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile
 [0188] The title compound was prepared using a similar procedure as that described for Example 10 from the appropriate starting materials.1H NMR (400 MHz, Chloroform-d) δ 8.12 (d, J = 99.8 Hz, 1H), 7.58 (dd, J = 8.4, 2.3 Hz, 1H), 7.45 (t, J = 7.5 Hz, 1H), 6.25 (dd, J = 33.5, 10.0 Hz, 1H), 5.70 (dd, J = 50.6, 15.7 Hz, 1H), 5.47 – 4.84 (m, 5H), 4.72 (t, J = 6.5 Hz, 1H), 3.53 – 3.33 (m, 1H), 3.20 – 2.95 (m, 1H), 2.92 – 2.75 (m, 1H), 2.16 (d, J = 5.2 Hz, 3H), 2.13 – 1.93 (m, 1H). ESI MS [M+H]+ for C25H20F5N4O, calcd 487.2, found 487.3. Example 14: (5R,6S,8R)-8-[(1S,2R)-7-(3-amino-1-methyl-5-pyrazolyl)-2,6-difluoro-1- hydroxy-4-indanyl]-3,5,6-trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile
[0188] The title compound was prepared using a similar procedure as that described for Example 10 from the appropriate starting materials.1H NMR (400 MHz, Chloroform-d) δ 8.12 (d, J = 99.8 Hz, 1H), 7.58 (dd, J = 8.4, 2.3 Hz, 1H), 7.45 (t, J = 7.5 Hz, 1H), 6.25 (dd, J = 33.5, 10.0 Hz, 1H), 5.70 (dd, J = 50.6, 15.7 Hz, 1H), 5.47 – 4.84 (m, 5H), 4.72 (t, J = 6.5 Hz, 1H), 3.53 – 3.33 (m, 1H), 3.20 – 2.95 (m, 1H), 2.92 – 2.75 (m, 1H), 2.16 (d, J = 5.2 Hz, 3H), 2.13 – 1.93 (m, 1H). ESI MS [M+H]+ for C25H20F5N4O, calcd 487.2, found 487.3. Example 14: (5R,6S,8R)-8-[(1S,2R)-7-(3-amino-1-methyl-5-pyrazolyl)-2,6-difluoro-1- hydroxy-4-indanyl]-3,5,6-trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile
 [0189] The title compound was prepared using a similar procedure as that described for Example 10 from the appropriate starting materials.1H NMR (400 MHz, Methanol-d4) δ 7.71 (dd, J = 8.8, 2.8 Hz, 1H), 7.67 (ddd, J = 8.0, 2.8, 1.1 Hz, 1H), 6.47 (d, J = 10.6 Hz, 1H), 6.01 – 5.57 (m, 2H), 5.35 – 5.06 (m, 3H), 4.85 – 4.77 (m, 1H), 3.47 (s, 3H), 3.39 – 3.22 (m, 1H), 3.17 – 3.03 (m, 1H), 2.81 (dtd, J = 16.2, 9.2, 8.7, 4.5 Hz, 1H), 2.06 (ddd, J = 26.1, 14.1, 5.6 Hz, 1H). ESI MS [M+H]+ for C24H19F5N4O, calcd 475.2, found 475.1.
Example 15: 5-{(2R,3S)-7-[(1R,3S,4R)-8-cyano-3,4,6-trifluoro-1,2,3,4-tetrahydro-1- naphthyl]-2,5-difluoro-3-hydroxy-4-indanyl}-6-methyl-2-pyridinecarboxamide
[0189] The title compound was prepared using a similar procedure as that described for Example 10 from the appropriate starting materials.1H NMR (400 MHz, Methanol-d4) δ 7.71 (dd, J = 8.8, 2.8 Hz, 1H), 7.67 (ddd, J = 8.0, 2.8, 1.1 Hz, 1H), 6.47 (d, J = 10.6 Hz, 1H), 6.01 – 5.57 (m, 2H), 5.35 – 5.06 (m, 3H), 4.85 – 4.77 (m, 1H), 3.47 (s, 3H), 3.39 – 3.22 (m, 1H), 3.17 – 3.03 (m, 1H), 2.81 (dtd, J = 16.2, 9.2, 8.7, 4.5 Hz, 1H), 2.06 (ddd, J = 26.1, 14.1, 5.6 Hz, 1H). ESI MS [M+H]+ for C24H19F5N4O, calcd 475.2, found 475.1.
Example 15: 5-{(2R,3S)-7-[(1R,3S,4R)-8-cyano-3,4,6-trifluoro-1,2,3,4-tetrahydro-1- naphthyl]-2,5-difluoro-3-hydroxy-4-indanyl}-6-methyl-2-pyridinecarboxamide
 [0190] The title compound was prepared using a similar procedure as that described for Example 10 from the appropriate starting materials.1H NMR (400 MHz, DMSO-d6) δ 8.09 – 8.03 (m, 1H), 8.03 – 7.94 (m, 1H), 7.92 – 7.74 (m, 3H), 7.69 – 7.62 (m, 1H), 6.52 (dd, J = 59.8, 10.9 Hz, 1H), 6.02 (dt, J = 51.4, 16.3 Hz, 1H), 5.42 – 4.94 (m, 4H), 4.87 – 4.75 (m, 1H), 3.28 – 2.84 (m, 2H), 2.79 – 2.64 (m, 1H), 2.40 – 2.23 (m, 3H), 2.14 – 1.94 (m, 1H). ESI MS [M+H]+ for C27H20F5N3O2, calcd 514.2, found 514.0. Example 16: (5R,6S,8R)-8-{(1S,2R)-7-[(S)-3-methyl-4-morpholinyl]-2,6-difluoro-1- hydroxy-4-indanyl}-3,5,6-trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile
[0190] The title compound was prepared using a similar procedure as that described for Example 10 from the appropriate starting materials.1H NMR (400 MHz, DMSO-d6) δ 8.09 – 8.03 (m, 1H), 8.03 – 7.94 (m, 1H), 7.92 – 7.74 (m, 3H), 7.69 – 7.62 (m, 1H), 6.52 (dd, J = 59.8, 10.9 Hz, 1H), 6.02 (dt, J = 51.4, 16.3 Hz, 1H), 5.42 – 4.94 (m, 4H), 4.87 – 4.75 (m, 1H), 3.28 – 2.84 (m, 2H), 2.79 – 2.64 (m, 1H), 2.40 – 2.23 (m, 3H), 2.14 – 1.94 (m, 1H). ESI MS [M+H]+ for C27H20F5N3O2, calcd 514.2, found 514.0. Example 16: (5R,6S,8R)-8-{(1S,2R)-7-[(S)-3-methyl-4-morpholinyl]-2,6-difluoro-1- hydroxy-4-indanyl}-3,5,6-trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile
 [0191] The title compound was prepared using a similar procedure as that described for Example 10 from the appropriate starting materials.1H NMR (400 MHz, Methanol-d4) δ 7.69 (dd, J = 8.8, 2.8 Hz, 1H), 7.64 (ddd, J = 8.1, 2.9, 1.1 Hz, 1H), 6.41 (d, J = 13.3 Hz, 1H), 5.84 (ddd, J = 50.4, 16.8, 2.7 Hz, 1H), 5.28 – 5.18 (m, 2H), 5.16 – 5.04 (m, 1H), 4.70 (t, J = 6.7 Hz, 1H), 3.88 – 3.68 (m, 3H), 3.43 (ddt, J = 9.4, 6.1, 3.0 Hz, 1H), 3.29 – 3.04 (m, 4H), 2.99 – 2.88 (m, 1H), 2.83 – 2.70 (m, 1H), 2.11 – 1.94 (m, 1H), 0.76 (d, J = 6.3 Hz, 3H). ESI MS [M+H]+ for C25H23F5N2O2, calcd 479.2, found 479.0.
Example 17: (5R,6S,8R)-8-[(1S,2R)-2,6-difluoro-1-hydroxy-7-(tetrahydro-2H-pyran-4-yl)- 4-indanyl]-3,5,6-trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile
[0191] The title compound was prepared using a similar procedure as that described for Example 10 from the appropriate starting materials.1H NMR (400 MHz, Methanol-d4) δ 7.69 (dd, J = 8.8, 2.8 Hz, 1H), 7.64 (ddd, J = 8.1, 2.9, 1.1 Hz, 1H), 6.41 (d, J = 13.3 Hz, 1H), 5.84 (ddd, J = 50.4, 16.8, 2.7 Hz, 1H), 5.28 – 5.18 (m, 2H), 5.16 – 5.04 (m, 1H), 4.70 (t, J = 6.7 Hz, 1H), 3.88 – 3.68 (m, 3H), 3.43 (ddt, J = 9.4, 6.1, 3.0 Hz, 1H), 3.29 – 3.04 (m, 4H), 2.99 – 2.88 (m, 1H), 2.83 – 2.70 (m, 1H), 2.11 – 1.94 (m, 1H), 0.76 (d, J = 6.3 Hz, 3H). ESI MS [M+H]+ for C25H23F5N2O2, calcd 479.2, found 479.0.
Example 17: (5R,6S,8R)-8-[(1S,2R)-2,6-difluoro-1-hydroxy-7-(tetrahydro-2H-pyran-4-yl)- 4-indanyl]-3,5,6-trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile
 [0192] The title compound was prepared using a similar procedure as that described for Example 10 from the appropriate starting materials.1H NMR (400 MHz, CDCl3) δ 7.55 (dd, J = 8.2, 2.7 Hz, 1H), 7.42 (ddd, J = 7.5, 2.8, 1.3 Hz, 1H), 6.00 (d, J = 12.4 Hz, 1H), 5.66 (ddd, J = 51.0, 14.2, 2.7 Hz, 1H), 5.50 – 5.26 (m, 2H), 5.13 (dddt, J = 48.1, 16.6, 9.8, 2.9 Hz, 1H), 4.66 (t, J = 6.1 Hz, 1H), 4.05 (dd, J = 11.5, 4.4 Hz, 2H), 3.52 (qt, J = 11.8, 1.9 Hz, 2H), 3.47 – 3.32 (m, 2H), 3.11 – 2.96 (m, 1H), 2.76 (dddd, J = 19.9, 9.5, 5.6, 3.5 Hz, 1H), 2.65 – 2.51 (m, 1H), 2.32 – 2.10 (m, 2H), 1.95 (dddd, J = 22.6, 14.1, 5.4, 3.0 Hz, 1H), 1.71 – 1.48 (m, 2H). ESI MS [M- H2O+H]+ for C25H22F5NO2, calcd 446.2, found 446.0. Example 18: (5R,6S,8R)-8-[(1S,2R)-7-(7-amino-1,3a-diaza-3-indenyl)-2-fluoro-1-hydroxy-4- indanyl]-3,5,6-trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile
[0192] The title compound was prepared using a similar procedure as that described for Example 10 from the appropriate starting materials.1H NMR (400 MHz, CDCl3) δ 7.55 (dd, J = 8.2, 2.7 Hz, 1H), 7.42 (ddd, J = 7.5, 2.8, 1.3 Hz, 1H), 6.00 (d, J = 12.4 Hz, 1H), 5.66 (ddd, J = 51.0, 14.2, 2.7 Hz, 1H), 5.50 – 5.26 (m, 2H), 5.13 (dddt, J = 48.1, 16.6, 9.8, 2.9 Hz, 1H), 4.66 (t, J = 6.1 Hz, 1H), 4.05 (dd, J = 11.5, 4.4 Hz, 2H), 3.52 (qt, J = 11.8, 1.9 Hz, 2H), 3.47 – 3.32 (m, 2H), 3.11 – 2.96 (m, 1H), 2.76 (dddd, J = 19.9, 9.5, 5.6, 3.5 Hz, 1H), 2.65 – 2.51 (m, 1H), 2.32 – 2.10 (m, 2H), 1.95 (dddd, J = 22.6, 14.1, 5.4, 3.0 Hz, 1H), 1.71 – 1.48 (m, 2H). ESI MS [M- H2O+H]+ for C25H22F5NO2, calcd 446.2, found 446.0. Example 18: (5R,6S,8R)-8-[(1S,2R)-7-(7-amino-1,3a-diaza-3-indenyl)-2-fluoro-1-hydroxy-4- indanyl]-3,5,6-trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile
 [0193] The title compound was prepared using a similar procedure as that described for Example 10 from the appropriate starting materials.1H NMR (400 MHz, DMSO-d6) δ 7.97 (ddd, J = 8.3, 2.8, 1.4 Hz, 1H), 7.88 (m, 1H), 7.74 (s, 1H), 7.49 (dd, J = 6.8, 1.0 Hz, 1H), 7.24 (d, J = 8.0 Hz, 1H), 6.70 (t, J = 7.1 Hz, 1H), 6.51 (d, J = 7.9 Hz, 1H), 6.37 (d, J = 7.4 Hz, 1H), 5.98 (dd, J = 52.6, 12.8 Hz, 1H), 5.73 (bs, 2H), 5.29 – 5.21 (m, 2H), 5.13 (m, 1H), 4.97 (dt, J = 10.5, 5.3 Hz, 1H), 4.81 (m, 1H), 3.27 - 3.13 (m, 2H), 2.69 (m, 1H), 2.02 (m, 1H). ESI MS [M+H]+ for C27H20F4N4O, calcd 493.5, found 493.5
Example 19: (5R,6S,8R)-8-[(1S,2R)-7-(dimethylaminosulfonyl)-2-fluoro-1-hydroxy-4- indanyl]-3,5,6-trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile
[0193] The title compound was prepared using a similar procedure as that described for Example 10 from the appropriate starting materials.1H NMR (400 MHz, DMSO-d6) δ 7.97 (ddd, J = 8.3, 2.8, 1.4 Hz, 1H), 7.88 (m, 1H), 7.74 (s, 1H), 7.49 (dd, J = 6.8, 1.0 Hz, 1H), 7.24 (d, J = 8.0 Hz, 1H), 6.70 (t, J = 7.1 Hz, 1H), 6.51 (d, J = 7.9 Hz, 1H), 6.37 (d, J = 7.4 Hz, 1H), 5.98 (dd, J = 52.6, 12.8 Hz, 1H), 5.73 (bs, 2H), 5.29 – 5.21 (m, 2H), 5.13 (m, 1H), 4.97 (dt, J = 10.5, 5.3 Hz, 1H), 4.81 (m, 1H), 3.27 - 3.13 (m, 2H), 2.69 (m, 1H), 2.02 (m, 1H). ESI MS [M+H]+ for C27H20F4N4O, calcd 493.5, found 493.5
Example 19: (5R,6S,8R)-8-[(1S,2R)-7-(dimethylaminosulfonyl)-2-fluoro-1-hydroxy-4- indanyl]-3,5,6-trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile
 [0194] The title compound was prepared using a similar procedure as that described for Example 10 from the appropriate starting materials.1H NMR (400 MHz, Chloroform-d) δ 7.63 – 7.50 (m, 2H), 7.38 (ddd, J = 7.5, 2.7, 0.9 Hz, 1H), 6.72 (d, J = 8.1 Hz, 1H), 5.83 – 5.58 (m, 1H), 5.50 (dd, J = 6.1, 5.1 Hz, 1H), 5.38 – 5.06 (m, 2H), 4.71 (t, J = 7.0 Hz, 1H), 3.51 (s, 1H), 3.41 (dddd, J = 18.1, 15.9, 6.2, 0.9 Hz, 1H), 3.04 (ddd, J = 15.9, 11.2, 6.6 Hz, 1H), 2.79 (s, 7H), 2.01 – 1.85 (m, 1H). ESI MS [M+H]+ for C22H20F4N2O3S, calcd 469.1, found 469.1. Example 20: (5R,6S,8R)-8-{(1S,2R)-7-[(R)-3-methyl-4-morpholinyl]-2,6-difluoro-1- hydroxy-4-indanyl}-3,5,6-trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile
[0194] The title compound was prepared using a similar procedure as that described for Example 10 from the appropriate starting materials.1H NMR (400 MHz, Chloroform-d) δ 7.63 – 7.50 (m, 2H), 7.38 (ddd, J = 7.5, 2.7, 0.9 Hz, 1H), 6.72 (d, J = 8.1 Hz, 1H), 5.83 – 5.58 (m, 1H), 5.50 (dd, J = 6.1, 5.1 Hz, 1H), 5.38 – 5.06 (m, 2H), 4.71 (t, J = 7.0 Hz, 1H), 3.51 (s, 1H), 3.41 (dddd, J = 18.1, 15.9, 6.2, 0.9 Hz, 1H), 3.04 (ddd, J = 15.9, 11.2, 6.6 Hz, 1H), 2.79 (s, 7H), 2.01 – 1.85 (m, 1H). ESI MS [M+H]+ for C22H20F4N2O3S, calcd 469.1, found 469.1. Example 20: (5R,6S,8R)-8-{(1S,2R)-7-[(R)-3-methyl-4-morpholinyl]-2,6-difluoro-1- hydroxy-4-indanyl}-3,5,6-trifluoro-5,6,7,8-tetrahydro-1-naphthonitrile
 [0195] The title compound was prepared using a similar procedure as that described for Example 10 from the appropriate starting materials.1H NMR (400 MHz, Methanol-d4) δ 7.69 (dd, J = 8.8, 2.8 Hz, 1H), 7.64 (ddd, J = 8.0, 2.8, 1.1 Hz, 1H), 6.32 (d, J = 13.2 Hz, 1H), 5.83 (ddd, J = 50.6, 16.1, 2.7 Hz, 1H), 5.34 – 5.03 (m, 3H), 4.71 (t, J = 6.5 Hz, 1H), 3.89 – 3.79 (m, 2H), 3.74 (td, J = 10.8, 2.6 Hz, 1H), 3.56 – 3.43 (m, 1H), 3.33 – 3.12 (m, 3H), 3.06 – 2.90 (m, 2H), 2.76 (dddd, J = 18.7, 9.3, 6.1, 4.5 Hz, 1H), 2.09 – 1.92 (m, 1H), 0.84 (d, J = 6.4 Hz, 3H). ESI MS [M+H]+ for C25H23F5N2O2, calcd 479.2, found 479.1.
[0196] The compounds of Examples 33-47 were prepared using methods analogous to those described in Examples 5-20 above, and to methods previously described in PCT publication number WO2021188769 from the appropriate starting materials and precursors.
[0195] The title compound was prepared using a similar procedure as that described for Example 10 from the appropriate starting materials.1H NMR (400 MHz, Methanol-d4) δ 7.69 (dd, J = 8.8, 2.8 Hz, 1H), 7.64 (ddd, J = 8.0, 2.8, 1.1 Hz, 1H), 6.32 (d, J = 13.2 Hz, 1H), 5.83 (ddd, J = 50.6, 16.1, 2.7 Hz, 1H), 5.34 – 5.03 (m, 3H), 4.71 (t, J = 6.5 Hz, 1H), 3.89 – 3.79 (m, 2H), 3.74 (td, J = 10.8, 2.6 Hz, 1H), 3.56 – 3.43 (m, 1H), 3.33 – 3.12 (m, 3H), 3.06 – 2.90 (m, 2H), 2.76 (dddd, J = 18.7, 9.3, 6.1, 4.5 Hz, 1H), 2.09 – 1.92 (m, 1H), 0.84 (d, J = 6.4 Hz, 3H). ESI MS [M+H]+ for C25H23F5N2O2, calcd 479.2, found 479.1.
[0196] The compounds of Examples 33-47 were prepared using methods analogous to those described in Examples 5-20 above, and to methods previously described in PCT publication number WO2021188769 from the appropriate starting materials and precursors.


 Biological Assay Examples Generation of Stabilized G323E Mutant HIF-2α HEK-293 Luciferase Reporter Cell Lines [0197] (P405A/P531A) double mutation was introduced to G323E mutant HIF-2α to achieve protein stability under normoxia condition. HIF-2α constructs were C-terminal HiBit tagged to monitor protein expression. pcDNA3.1 expression vectors were generated by GenScript through gene synthesis technology. Stable HEK-293 cell lines expressing stabilized HIF-2α with G323E mutation were generated by plasmid transfection followed by G418 selection. Single cell cloning was achieved by limited dilution method, and clones expressing the HiBit tagged HIF-2α protein were identified utilizing the Nano-Glo® HiBiT Lytic Detection System (N3040, Promega). [0198] Stabilized HIF-2α with G323E mutation positive clones were further transfected with Cignal Lenti HIF Luc Reporter lentivirus (CLS-007L, Qiagen) according to the manufacturer’s guidelines. In brief, 0.3x106 of the respective HEK 293 cells were transduced with lentivirus at a Multiplicity of Infection (MOI) of 25 for 24 hours. After transduction, cells were replenished with fresh RPMI 1640 Medium (Cat. No. 11875085, Thermo Fisher,) supplemented with 10% FBS (Cat. No. A3160502, Gibco), 2mM GlutaMax (Cat. No. 35050-061, Invitrogen), 100 units of penicillin and 100 μg/mL of streptomycin (Cat. No 15070063, Thermo Fisher) and G418 (Cat. No J63871, Thermo Fisher) for another 24 hours. Antibiotic selection was performed in cell media containing 4 μg/mL of Puromycin. After 7 days of antibiotic selection, stable pools of surviving cells were expanded and used in a luciferase reporter assay. Plasmid Sequence for pcDNA3.1-hHIF-2α (G323E)_st_Mut_HiBiT
Biological Assay Examples Generation of Stabilized G323E Mutant HIF-2α HEK-293 Luciferase Reporter Cell Lines [0197] (P405A/P531A) double mutation was introduced to G323E mutant HIF-2α to achieve protein stability under normoxia condition. HIF-2α constructs were C-terminal HiBit tagged to monitor protein expression. pcDNA3.1 expression vectors were generated by GenScript through gene synthesis technology. Stable HEK-293 cell lines expressing stabilized HIF-2α with G323E mutation were generated by plasmid transfection followed by G418 selection. Single cell cloning was achieved by limited dilution method, and clones expressing the HiBit tagged HIF-2α protein were identified utilizing the Nano-Glo® HiBiT Lytic Detection System (N3040, Promega). [0198] Stabilized HIF-2α with G323E mutation positive clones were further transfected with Cignal Lenti HIF Luc Reporter lentivirus (CLS-007L, Qiagen) according to the manufacturer’s guidelines. In brief, 0.3x106 of the respective HEK 293 cells were transduced with lentivirus at a Multiplicity of Infection (MOI) of 25 for 24 hours. After transduction, cells were replenished with fresh RPMI 1640 Medium (Cat. No. 11875085, Thermo Fisher,) supplemented with 10% FBS (Cat. No. A3160502, Gibco), 2mM GlutaMax (Cat. No. 35050-061, Invitrogen), 100 units of penicillin and 100 μg/mL of streptomycin (Cat. No 15070063, Thermo Fisher) and G418 (Cat. No J63871, Thermo Fisher) for another 24 hours. Antibiotic selection was performed in cell media containing 4 μg/mL of Puromycin. After 7 days of antibiotic selection, stable pools of surviving cells were expanded and used in a luciferase reporter assay. Plasmid Sequence for pcDNA3.1-hHIF-2α (G323E)_st_Mut_HiBiT
 LESKKTEPEHRPMSSIFFDAGSKASLPPCCGQASTPLSSMGGRSNTQWPPDPPLHFGPTK WAVGDQRTEFLGAAPLGPPVSPPHVSTFKTRSAKGFGARGPDVLSPAMVALSNKLKLK RQLEYEEQAFQDLSGGDPPGGSTSHLMWKRMKNLRGGSCPLMPDKPLSANVPNDKFT QNPMRGLGHPLRHLPLPQPPSAISPGENSKSRFPPQCYATQYQDYSLSSAHKVSGMASRL LGPSFESYLLPELTRYDCEVNVPVLGSSTLLQGGDLLRALDQATGGGGSGGGGSVSGW RLFKKIS (SEQ ID NO: 1) HEK 293 Stable G323E HIF-2α Luciferase Reporter Assay [0200] On day one, 20 µL of each of the cell-lines prepared in OptiMem (Cat. No.31985088, Thermo Fisher) was seeded into its respective 384 well white opaque plate (Corning 3570) and incubated at 37°C and 5% CO2. After a 4-hour incubation, 20 µL of 2x compound is added to each cell plate. Final assay conditions comprised 10,000 cells per well for the G323E cell line in 1% DMSO with test compound concentrations ranging from 50 µM to 0 µM. After a 20-hour incubation at 37°C and 5% CO2, luciferase activity was determined using ONE-Glo Luciferase Assay Reagent (E6110, Promega) following the manufacture’s recommended procedure. Briefly, 40 µL of ONE-Glo luciferase reagents were added to each well and luciferase signals were measured using an Envision 2102 Multilabel Reader. Percentage maximum activity in each test well was calculated based on DMSO (maximum activity) and no cell control wells (baseline activity). The IC50 values of the test compounds were determined from compound dose response curves fitted using a standard four parameter fit equation (Table 5). Table 5 Potency of Select Compounds Less than 100 nM (++++), 100 nM to less than 1 µM (+++), 1 µM to less than 5 µM (++), ≥ 5 µM (+)
LESKKTEPEHRPMSSIFFDAGSKASLPPCCGQASTPLSSMGGRSNTQWPPDPPLHFGPTK WAVGDQRTEFLGAAPLGPPVSPPHVSTFKTRSAKGFGARGPDVLSPAMVALSNKLKLK RQLEYEEQAFQDLSGGDPPGGSTSHLMWKRMKNLRGGSCPLMPDKPLSANVPNDKFT QNPMRGLGHPLRHLPLPQPPSAISPGENSKSRFPPQCYATQYQDYSLSSAHKVSGMASRL LGPSFESYLLPELTRYDCEVNVPVLGSSTLLQGGDLLRALDQATGGGGSGGGGSVSGW RLFKKIS (SEQ ID NO: 1) HEK 293 Stable G323E HIF-2α Luciferase Reporter Assay [0200] On day one, 20 µL of each of the cell-lines prepared in OptiMem (Cat. No.31985088, Thermo Fisher) was seeded into its respective 384 well white opaque plate (Corning 3570) and incubated at 37°C and 5% CO2. After a 4-hour incubation, 20 µL of 2x compound is added to each cell plate. Final assay conditions comprised 10,000 cells per well for the G323E cell line in 1% DMSO with test compound concentrations ranging from 50 µM to 0 µM. After a 20-hour incubation at 37°C and 5% CO2, luciferase activity was determined using ONE-Glo Luciferase Assay Reagent (E6110, Promega) following the manufacture’s recommended procedure. Briefly, 40 µL of ONE-Glo luciferase reagents were added to each well and luciferase signals were measured using an Envision 2102 Multilabel Reader. Percentage maximum activity in each test well was calculated based on DMSO (maximum activity) and no cell control wells (baseline activity). The IC50 values of the test compounds were determined from compound dose response curves fitted using a standard four parameter fit equation (Table 5). Table 5 Potency of Select Compounds Less than 100 nM (++++), 100 nM to less than 1 µM (+++), 1 µM to less than 5 µM (++), ≥ 5 µM (+)


 [0201] Particular embodiments of this disclosure are described herein, including the best mode known to the inventors for carrying out the disclosure. Upon reading the foregoing, description, variations of the disclosed embodiments may become apparent to individuals working in the art, and it is expected that those skilled artisans may employ such variations as appropriate. Accordingly, it is intended that the disclosure be practiced otherwise than as specifically described herein, and that the disclosure includes all modifications and equivalents of the subject matter recited in the claims appended hereto as permitted by applicable law. Moreover, any combination of the above-described elements in all possible variations thereof is encompassed by the disclosure unless otherwise indicated herein or otherwise clearly contradicted by context. [0202] All publications, patent applications, accession numbers, and other references cited in this specification are herein incorporated by reference for the purpose described herein.
[0201] Particular embodiments of this disclosure are described herein, including the best mode known to the inventors for carrying out the disclosure. Upon reading the foregoing, description, variations of the disclosed embodiments may become apparent to individuals working in the art, and it is expected that those skilled artisans may employ such variations as appropriate. Accordingly, it is intended that the disclosure be practiced otherwise than as specifically described herein, and that the disclosure includes all modifications and equivalents of the subject matter recited in the claims appended hereto as permitted by applicable law. Moreover, any combination of the above-described elements in all possible variations thereof is encompassed by the disclosure unless otherwise indicated herein or otherwise clearly contradicted by context. [0202] All publications, patent applications, accession numbers, and other references cited in this specification are herein incorporated by reference for the purpose described herein.
Claims
CLAIMS 1. A method of treating a disease, disorder, and/or condition mediated by G323E mutant HIF- 2α, said method comprising administering a compound or a pharmaceutically acceptable salt thereof to a subject in need thereof, wherein the compound has a structure according to Formula I
 (Formula I), or a pharmaceutically acceptable salt thereof, wherein n is 0, 1, or 2; each R1 when present is independently halo; R2 and R3 are independently halo, C1-C6 alkyl or -CN; or R2 and R3 combine with the atoms to which they are attached to form a C4-C6 cycloalkyl, wherein said C4-C6 cycloalkyl is unsubstituted or substituted with 1-3 Ra; each Ra when present is independently halo or -OH; R4 is halo, C1-C6 haloalkyl, -CN, -S(O)2-R4a, 4- to 8-membered heterocycloalkyl, or 5- to 10-membered heteroaryl; wherein said 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatom or heteroatom groups independently selected from N, O, S, and S(O)2; said 5- to 10-membered heteroaryl has 1-3 ring heteroatoms independently selected from N, O, and S; and said 4- to 8-membered heterocycloalkyl and 5- to 10-membered heteroaryl are unsubstituted or substituted with 1-3 Rb; R4a is C1-C3 alkyl, C1-C3 haloalkyl, C3-C6 cycloalkyl, -NR4bR4c, or 4- to 6- membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S; R4b and R4c are each independently -H or C1-C3 alkyl; each Rb when present is independently -CN, C1-C6 alkyl, -N(Rc)2, or -C(O)- N(Rc)2;
each Rc is independently -H or C1-C6 alkyl; X is CR5 or N; and R5 is -H or halo.
(Formula I), or a pharmaceutically acceptable salt thereof, wherein n is 0, 1, or 2; each R1 when present is independently halo; R2 and R3 are independently halo, C1-C6 alkyl or -CN; or R2 and R3 combine with the atoms to which they are attached to form a C4-C6 cycloalkyl, wherein said C4-C6 cycloalkyl is unsubstituted or substituted with 1-3 Ra; each Ra when present is independently halo or -OH; R4 is halo, C1-C6 haloalkyl, -CN, -S(O)2-R4a, 4- to 8-membered heterocycloalkyl, or 5- to 10-membered heteroaryl; wherein said 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatom or heteroatom groups independently selected from N, O, S, and S(O)2; said 5- to 10-membered heteroaryl has 1-3 ring heteroatoms independently selected from N, O, and S; and said 4- to 8-membered heterocycloalkyl and 5- to 10-membered heteroaryl are unsubstituted or substituted with 1-3 Rb; R4a is C1-C3 alkyl, C1-C3 haloalkyl, C3-C6 cycloalkyl, -NR4bR4c, or 4- to 6- membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S; R4b and R4c are each independently -H or C1-C3 alkyl; each Rb when present is independently -CN, C1-C6 alkyl, -N(Rc)2, or -C(O)- N(Rc)2;
each Rc is independently -H or C1-C6 alkyl; X is CR5 or N; and R5 is -H or halo.
2. The method of Claim 1, wherein the disease, disorder, and/or condition is cancer.
3. The method of Claim 2, wherein the cancer is a solid tumor, optionally wherein the solid tumor is associated with von Hippel-Lindau (VHL) disease.
4. The method of Claim 2, wherein the cancer is kidney cancer, liver cancer, prostate cancer, bladder cancer, breast cancer, gynecological cancer, gastrointestinal (GI) cancer, or is a neuroendocrine tumor.
5. The method of Claim 2, where the cancer is renal cell carcinoma (RCC), central nervous system (CNS) hemangioblastoma, pancreatic neuroendocrine tumors (pNETs), esophageal squamous cell carcinoma (ESCC), or hepatocellular carcinoma (HCC), optionally wherein the cancer is associated with von Hippel-Lindau (VHL) disease.
6. The method of any one of Claims 1-5, wherein the cancer is unresectable, locally advanced, or metastatic.
7. The method of any one of Claims 1-6, wherein the method comprises administering the compound or pharmaceutically acceptable salt thereof in combination with one or more than one additional therapy.
8. The method of Claim 7, wherein each additional therapy is independently selected from an immune checkpoint inhibitor, a chemotherapeutic agent; and radiation therapy.
9. The method of any one of Claims 1-8, wherein R2 is C1-C6 alkyl and R3 is -CN; or R2 and R3 combine with the atoms to which they are attached to form a C4-C6 cycloalkyl, wherein said C4-C6 cycloalkyl is unsubstituted or substituted with 2-3 Ra.
10. The method of any one of Claims 1-9, wherein R2 is C1-C6 alkyl.
11. The method of any one of Claims 1-10, wherein R3 is -CN.
12. The method of any one of Claims 1-11, wherein R2 and R3 combine with the atoms to which they are attached to form a C4-C6 cycloalkyl, wherein said C4-C6 cycloalkyl is unsubstituted or substituted with 2-3 Ra.
13. The method of Claim 12, wherein at least one Ra is fluoro.
14. The method of Claim 12 or Claim 13, wherein at least one Ra is -OH.
15. The method of any one of Claims 1-14, wherein R4 is halo, -S(O)2-R4a, 4- to 8-membered heterocycloalkyl, or 5- to 10-membered heteroaryl; wherein said 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatom or heteroatom groups independently selected from N, O, S, and S(O)2; said 5- to 10-membered heteroaryl has 1-3 ring heteroatoms independently selected from N, O, and S; and said 4- to 8-membered heterocycloalkyl and 5- to 10-membered heteroaryl are unsubstituted, or substituted with 1-3 Rb.
16. The method of any one of Claims 1-15, wherein R4 is halo or a 5- to 6-membered heteroaryl having 1-3 ring heteroatoms independently selected from N, O, and S; wherein said 5- to 6-membered heteroaryl is unsubstituted, or substituted with 1-2 Rb.
17. The method of any one of Claims 1-15, wherein R4 is halo, -S(O)2-R4a, 4- to 8-membered heterocycloalkyl, or 5- to 10-membered heteroaryl, wherein said 4- to 8-membered heterocycloalkyl and 5- to 10-membered heteroaryl have 1-4 ring heteroatoms independently selected from N, O, and S; and wherein said 4- to 8-membered heterocycloalkyl and 5- to 10-membered heteroaryl are unsubstituted or substituted with 1-2 Rb.
18. The method of Claim 17, wherein R4a is C1-C3 alkyl, or -NR4bR4c, wherein R4b and R4c are independently C1-C3 alkyl.
19. The method of any one of Claims 1-17, wherein R4 is halo, 5- to 6-membered heteroaryl, or 5- to 8-membered heterocycloalkyl; wherein said 5- to 6-membered heteroaryl has 1-3 ring nitrogen atoms; said 5- to 8-membered heterocycloalkyl has 1-2 ring heteroatoms
independently selected from N, and O; and said 5- to 6-membered heteroaryl and 5- to 8- membered heterocycloalkyl are unsubstituted or substituted with 1-2 Rb.
20. The method of any one of Claims 1-15 and 19, wherein R4 is selected from the group consisting of: (a) chloro; (b) triazolyl, imidazolyl, pyrazolyl, or pyridyl, each of which is unsubstituted or substituted with 1-2 Rb; and (c) unsubstituted tetrahydropyranyl.
21. The method of any one of Claims 1-20, wherein X is CR5.
22. The method of any one of Claims 1-21, wherein the compound is a compound of Formula Ia or a pharmaceutically acceptable salt thereof, wherein Formula Ia has the structure:
 (Formula Ia) where n is 0 or 1.
(Formula Ia) where n is 0 or 1.
23. The method of any one of Claims 1-22, wherein the compound is a compound of Formula Ib or a pharmaceutically acceptable salt thereof, wherein Formula Ib has the structure:
 (Formula Ib) where n is 0 or 1; and m is 2 or 3.
(Formula Ib) where n is 0 or 1; and m is 2 or 3.
24. The method of any one of Claims 1-8, wherein the compound or pharmaceutically acceptable salt thereof is a member or a pharmaceutically acceptable salt thereof selected f

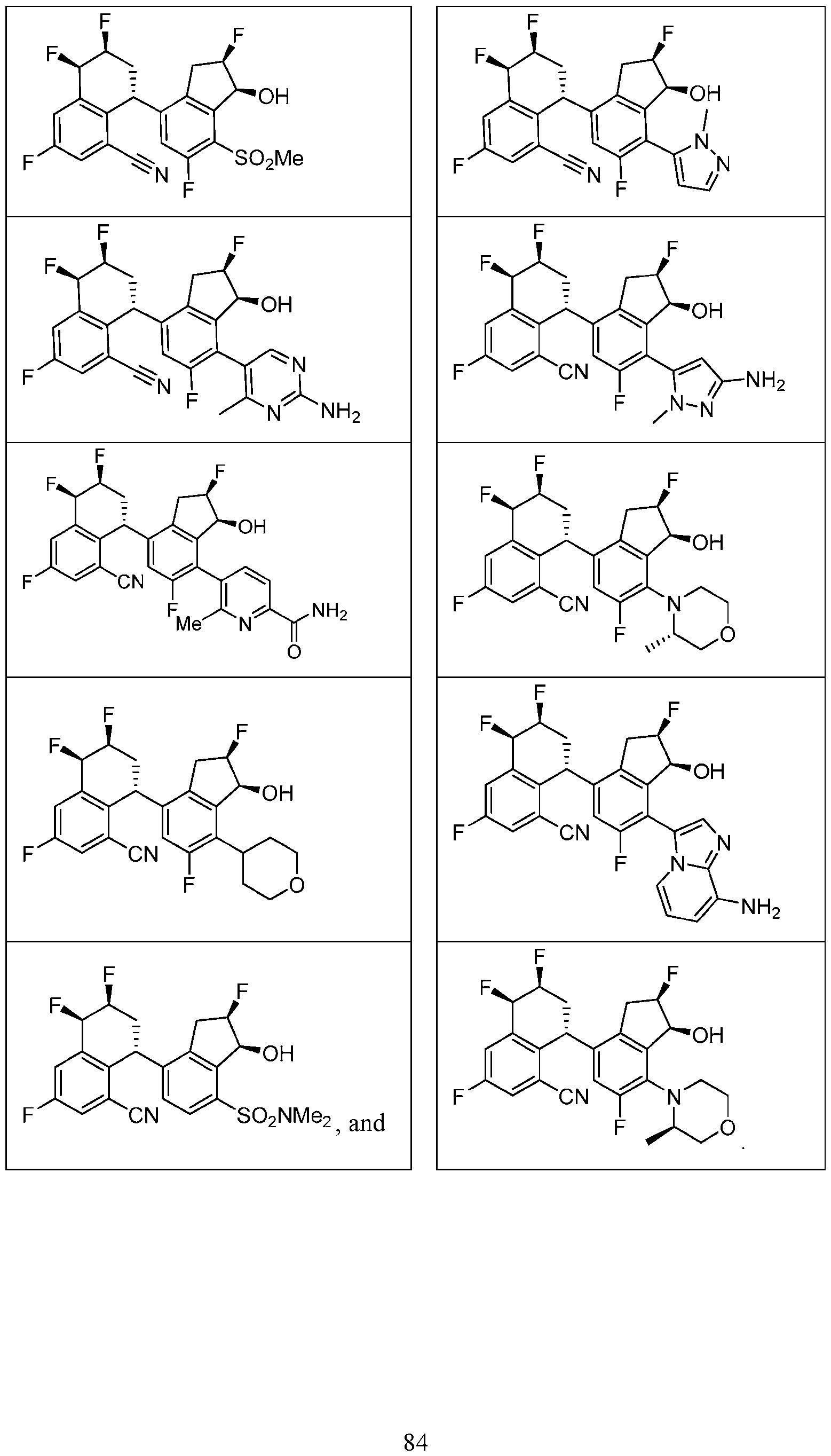
25. The method of any one of Claims 1-8, wherein the compound or pharmaceutically acceptable salt thereof is a member or a pharmaceutically acceptable salt thereof selected from the group consisting of: F F F F OH Me N F CN F N




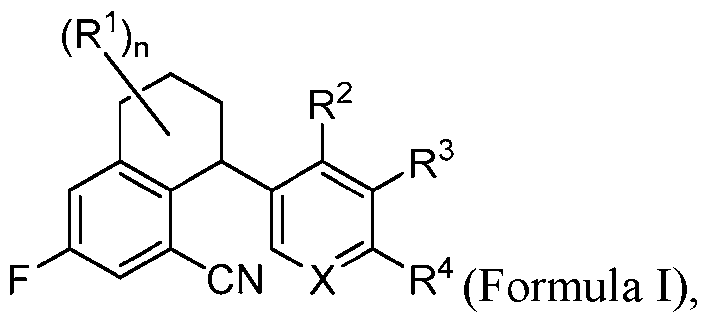
27. The method of Claim 26, wherein contacting a cell of a subject comprises administering the compound or pharmaceutically acceptable salt thereof to the subject.
28. The method of Claim 26, wherein the cell is a cancer cell.
29. The method of Claim 28, the cancer cell is a cell from a solid tumor, optionally wherein the solid tumor is associated with von Hippel-Lindau (VHL) disease.
30. The method of Claim 28, wherein the cancer cell is a kidney cancer cell, liver cancer cell, prostate cancer cell, bladder cancer cell, breast cancer cell, gynecological cancer cell, gastrointestinal (GI) cancer cell, or a neuroendocrine tumor cell.
31. The method of Claim 28, where the cancer cell is from renal cell carcinoma (RCC) cell,
central nervous system (CNS) hemangioblastoma cell, pancreatic neuroendocrine tumors (pNETs) cell, esophageal squamous cell carcinoma (ESCC) cell, or hepatocellular carcinoma (HCC) cell, optionally wherein the cancer cell is associated with von Hippel- Lindau (VHL) disease.
32. The method of any one of Claims 26-31, wherein the cancer cell is from an unresectable cancer, a locally advanced cancer, or a metastatic cancer.
33. The method of any one of Claims 26-32, wherein R2 is C1-C6 alkyl and R3 is -CN; or R2 and R3 combine with the atoms to which they are attached to form a C4-C6 cycloalkyl, wherein said C4-C6 cycloalkyl is unsubstituted or substituted with 2-3 Ra.
34. The method of any one of Claims 26-33, wherein R2 is C1-C6 alkyl.
35. The method of any one of Claims 26-34, wherein R3 is -CN.
36. The method of any one of Claims 26-35, wherein R2 and R3 combine with the atoms to which they are attached to form a C4-C6 cycloalkyl, wherein said C4-C6 cycloalkyl is unsubstituted or substituted with 2-3 Ra.
37. The method of Claim 36, wherein at least one Ra is fluoro.
38. The method of Claim 36 or Claim 37, wherein at least one Ra is -OH.
39. The method of any one of Claims 26-38, wherein R4 is halo, -S(O)2-R4a, 4- to 8-membered heterocycloalkyl, or 5- to 10-membered heteroaryl; wherein said 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatom or heteroatom groups independently selected from N, O, S, and S(O)2; said 5- to 10-membered heteroaryl has 1-3 ring heteroatoms independently selected from N, O, and S; and said 4- to 8-membered heterocycloalkyl and 5- to 10-membered heteroaryl are unsubstituted, or substituted with 1-3 Rb.
40. The method of any one of Claims 26-39, wherein R4 is halo or a 5- to 6-membered heteroaryl having 1-3 ring heteroatoms independently selected from N, O, and S; wherein said 5- to 6-membered heteroaryl is unsubstituted, or substituted with 1-2 Rb.
41. The method of any one of Claims 26-40, wherein R4 is halo, -S(O)2-R4a, 4- to 8-membered heterocycloalkyl, or 5- to 10-membered heteroaryl, wherein said 4- to 8-membered heterocycloalkyl and 5- to 10-membered heteroaryl have 1-4 ring heteroatoms independently selected from N, O, and S; and wherein said 4- to 8-membered heterocycloalkyl and 5- to 10-membered heteroaryl are unsubstituted or substituted with 1-2 Rb.
42. The method of Claim 41, wherein R4a is C1-C3 alkyl, or -NR4bR4c, wherein R4b and R4c are independently C1-C3 alkyl.
43. The method of any one of Claims 26-41, wherein R4 is halo, 5- to 6-membered heteroaryl, or 5- to 8-membered heterocycloalkyl; wherein said 5- to 6-membered heteroaryl has 1-3 ring nitrogen atoms; said 5- to 8-membered heterocycloalkyl has 1-2 ring heteroatoms independently selected from N, and O; and said 5- to 6-membered heteroaryl and 5- to 8- membered heterocycloalkyl are unsubstituted or substituted with 1-2 Rb.
44. The method of any one of Claims 26-38 and 43, wherein R4 is selected from the group consisting of: (a) chloro; (b) triazolyl, imidazolyl, pyrazolyl, or pyridyl, each of which is unsubstituted or substituted with 1-2 Rb; and (c) unsubstituted tetrahydropyranyl.
45. The method of any one of Claims 26-44, wherein X is CR5.
46. The method of any one of Claims 26-45, wherein the compound is a compound of Formula Ia or a pharmaceutically acceptable salt thereof, wherein Formula Ia has the structure:
 (Formula Ia) where n is 0 or 1.
(Formula Ia) where n is 0 or 1.
47. The method of any one of Claims 26-46, wherein the compound is a compound of Formula Ib or a pharmaceutically acceptable salt thereof, wherein Formula Ib has the structure:
 (Formula Ib) where n is 0 or 1; and m is 2 or 3.
(Formula Ib) where n is 0 or 1; and m is 2 or 3.
48. The method of any one of Claims 26-32, wherein the compound or pharmaceutically acceptable salt thereof is a member or a pharmaceutically acceptable salt thereof selected from the group consisting of



49. The method of any one of Claims 25-32, wherein the compound or pharmaceutically acceptable salt thereof is a member or a pharmaceutically acceptable salt thereof selected from the group consisting of



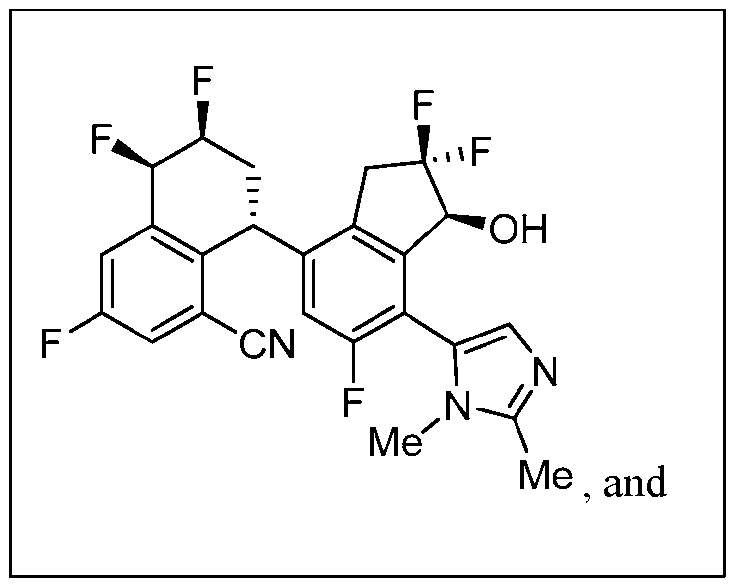

50. A compound or a pharmaceutically acceptable salt thereof of a member selected from the g


51. A compound or a pharmaceutically acceptable salt thereof of a member selected from the group consisting of




52. A pharmaceutical composition comprising the compound of Claim 50 or 51, or a pharmaceutically acceptable salt thereof, and one or more pharmaceutically acceptable excipients.
53. A method of treating a subject with resistance to treatment with a HIF-2α inhibitor, said method comprising administering a compound or a pharmaceutically acceptable salt
thereof to a subject in need thereof, wherein the compound has a structure according to Formula I:
 (Formula I), or a pharmaceutically acceptable salt thereof, wherein n is 0, 1, or 2; each R1 when present is independently halo; R2 and R3 are independently halo, C1-C6 alkyl or -CN; or R2 and R3 combine with the atoms to which they are attached to form a C4-C6 cycloalkyl, wherein said C4-C6 cycloalkyl is unsubstituted or substituted with 1-3 Ra; each Ra when present is independently halo or -OH; R4 is halo, C1-C6 haloalkyl, -CN, -S(O)2-R4a, 4- to 8-membered heterocycloalkyl, or 5- to 10-membered heteroaryl; wherein said 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatom or heteroatom groups independently selected from N, O, S, and S(O)2; said 5- to 10-membered heteroaryl has 1-3 ring heteroatoms independently selected from N, O, and S; and said 4- to 8-membered heterocycloalkyl and 5- to 10-membered heteroaryl are unsubstituted or substituted with 1-3 Rb; R4a is C1-C3 alkyl, C1-C3 haloalkyl, C3-C6 cycloalkyl, -NR4bR4c, or 4- to 6- membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S; R4b and R4c are each independently -H or C1-C3 alkyl; each Rb when present is independently -CN, C1-C6 alkyl, -N(Rc)2, or -C(O)- N(Rc)2; each Rc is independently -H or C1-C6 alkyl; X is CR5 or N; and R5 is -H or halo.
(Formula I), or a pharmaceutically acceptable salt thereof, wherein n is 0, 1, or 2; each R1 when present is independently halo; R2 and R3 are independently halo, C1-C6 alkyl or -CN; or R2 and R3 combine with the atoms to which they are attached to form a C4-C6 cycloalkyl, wherein said C4-C6 cycloalkyl is unsubstituted or substituted with 1-3 Ra; each Ra when present is independently halo or -OH; R4 is halo, C1-C6 haloalkyl, -CN, -S(O)2-R4a, 4- to 8-membered heterocycloalkyl, or 5- to 10-membered heteroaryl; wherein said 4- to 8-membered heterocycloalkyl has 1-3 ring heteroatom or heteroatom groups independently selected from N, O, S, and S(O)2; said 5- to 10-membered heteroaryl has 1-3 ring heteroatoms independently selected from N, O, and S; and said 4- to 8-membered heterocycloalkyl and 5- to 10-membered heteroaryl are unsubstituted or substituted with 1-3 Rb; R4a is C1-C3 alkyl, C1-C3 haloalkyl, C3-C6 cycloalkyl, -NR4bR4c, or 4- to 6- membered heterocycloalkyl having 1-3 ring heteroatoms independently selected from N, O, and S; R4b and R4c are each independently -H or C1-C3 alkyl; each Rb when present is independently -CN, C1-C6 alkyl, -N(Rc)2, or -C(O)- N(Rc)2; each Rc is independently -H or C1-C6 alkyl; X is CR5 or N; and R5 is -H or halo.
54. The method of Claim 53, wherein the subject was previously administered a HIF-2α inhibitor.
55. The method of Claims 53 or Claim 54, wherein G323E mutant HIF-2α is detectable in a sample obtained from the subject.
56. The method of any one of Claims 53-55, further comprising: testing for the presence of G323E mutant HIF-2α; and initiating administering the compound or the pharmaceutically acceptable salt thereof when the presence of G323E mutant HIF-2α is detected.
57. The method of Claim 56, wherein testing for the presence of G323E mutant HIF-2α comprises testing a sample obtained from the subject for the presence of G323E mutant HIF-2α, and wherein the sample comprises a tumor biopsy or surgical resection sample.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363533085P | 2023-08-16 | 2023-08-16 | |
| US63/533,085 | 2023-08-16 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025038857A1 true WO2025038857A1 (en) | 2025-02-20 |
Family
ID=92672231
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/042509 Pending WO2025038857A1 (en) | 2023-08-16 | 2024-08-15 | TETRALINS TARGETING MUTANT HIF-2α |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025038857A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2026006759A1 (en) * | 2024-06-28 | 2026-01-02 | Arcus Biosciences, Inc. | Pharmaceutical compositions, dosage forms, and methods of making and using same |
Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008132601A1 (en) | 2007-04-30 | 2008-11-06 | Immutep | Cytotoxic anti-lag-3 monoclonal antibody and its use in the treatment or prevention of organ transplant rejection and autoimmune disease |
| WO2009044273A2 (en) | 2007-10-05 | 2009-04-09 | Immutep | Use of recombinant lag-3 or the derivatives thereof for eliciting monocyte immune response |
| WO2010019570A2 (en) | 2008-08-11 | 2010-02-18 | Medarex, Inc. | Human antibodies that bind lymphocyte activation gene-3 (lag-3), and uses thereof |
| WO2011109400A2 (en) | 2010-03-04 | 2011-09-09 | Macrogenics,Inc. | Antibodies reactive with b7-h3, immunologically active fragments thereof and uses thereof |
| WO2012145493A1 (en) | 2011-04-20 | 2012-10-26 | Amplimmune, Inc. | Antibodies and other molecules that bind b7-h1 and pd-1 |
| WO2014008218A1 (en) | 2012-07-02 | 2014-01-09 | Bristol-Myers Squibb Company | Optimization of antibodies that bind lymphocyte activation gene-3 (lag-3), and uses thereof |
| WO2017152088A1 (en) | 2016-03-04 | 2017-09-08 | JN Biosciences, LLC | Antibodies to tigit |
| WO2021113436A1 (en) | 2019-12-04 | 2021-06-10 | Arcus Biosciences, Inc. | Inhibitors of hif-2alpha |
| WO2021188769A1 (en) | 2020-03-19 | 2021-09-23 | Arcus Biosciences, Inc. | Tetralin and tetrahydroquinoline compounds as inhibitors of hif-2alpha |
| WO2021220170A1 (en) * | 2020-04-29 | 2021-11-04 | Novartis Ag | Compounds and compositions for inhibiting the activity of hif2-alpha and their methods of use |
| WO2021247591A1 (en) | 2020-06-02 | 2021-12-09 | Arcus Biosciences, Inc. | Antibodies to tigit |
| WO2022086822A1 (en) * | 2020-10-19 | 2022-04-28 | Nikang Therapeutics, Inc. | Processes of preparing 3-fluoro-5-(((1s,2ar)-1,3,3,4,4-pentafluoro-2a-hydroxy-2,2a,3,4-tetrahydro-1h-cyclopenta[cd]inden-7-yl)oxy)-benzonitrile |
| WO2023077046A1 (en) | 2021-10-29 | 2023-05-04 | Arcus Biosciences, Inc. | Inhibitors of hif-2alpha and methods of use thereof |
-
2024
- 2024-08-15 WO PCT/US2024/042509 patent/WO2025038857A1/en active Pending
Patent Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008132601A1 (en) | 2007-04-30 | 2008-11-06 | Immutep | Cytotoxic anti-lag-3 monoclonal antibody and its use in the treatment or prevention of organ transplant rejection and autoimmune disease |
| WO2009044273A2 (en) | 2007-10-05 | 2009-04-09 | Immutep | Use of recombinant lag-3 or the derivatives thereof for eliciting monocyte immune response |
| WO2010019570A2 (en) | 2008-08-11 | 2010-02-18 | Medarex, Inc. | Human antibodies that bind lymphocyte activation gene-3 (lag-3), and uses thereof |
| WO2011109400A2 (en) | 2010-03-04 | 2011-09-09 | Macrogenics,Inc. | Antibodies reactive with b7-h3, immunologically active fragments thereof and uses thereof |
| WO2012145493A1 (en) | 2011-04-20 | 2012-10-26 | Amplimmune, Inc. | Antibodies and other molecules that bind b7-h1 and pd-1 |
| WO2014008218A1 (en) | 2012-07-02 | 2014-01-09 | Bristol-Myers Squibb Company | Optimization of antibodies that bind lymphocyte activation gene-3 (lag-3), and uses thereof |
| WO2017152088A1 (en) | 2016-03-04 | 2017-09-08 | JN Biosciences, LLC | Antibodies to tigit |
| WO2021113436A1 (en) | 2019-12-04 | 2021-06-10 | Arcus Biosciences, Inc. | Inhibitors of hif-2alpha |
| WO2021188769A1 (en) | 2020-03-19 | 2021-09-23 | Arcus Biosciences, Inc. | Tetralin and tetrahydroquinoline compounds as inhibitors of hif-2alpha |
| WO2021220170A1 (en) * | 2020-04-29 | 2021-11-04 | Novartis Ag | Compounds and compositions for inhibiting the activity of hif2-alpha and their methods of use |
| WO2021247591A1 (en) | 2020-06-02 | 2021-12-09 | Arcus Biosciences, Inc. | Antibodies to tigit |
| WO2022086822A1 (en) * | 2020-10-19 | 2022-04-28 | Nikang Therapeutics, Inc. | Processes of preparing 3-fluoro-5-(((1s,2ar)-1,3,3,4,4-pentafluoro-2a-hydroxy-2,2a,3,4-tetrahydro-1h-cyclopenta[cd]inden-7-yl)oxy)-benzonitrile |
| WO2023077046A1 (en) | 2021-10-29 | 2023-05-04 | Arcus Biosciences, Inc. | Inhibitors of hif-2alpha and methods of use thereof |
Non-Patent Citations (5)
| Title |
|---|
| BERGE, S.M. ET AL.: "Pharmaceutical Salts", JOURNAL OF PHARMACEUTICAL SCIENCE, vol. 66, 1977, pages 1 - 19, XP002675560, DOI: 10.1002/jps.2600660104 |
| COURTNEY KEVIN D. ET AL: "HIF-2 Complex Dissociation, Target Inhibition, and Acquired Resistance with PT2385, a First-in-Class HIF-2 Inhibitor, in Patients with Clear Cell Renal Cell Carcinoma", CLINICAL CANCER RESEARCH, vol. 26, no. 4, 14 February 2020 (2020-02-14), US, pages 793 - 803, XP093217817, ISSN: 1078-0432, Retrieved from the Internet <URL:https://aacrjournals.org/clincancerres/article-pdf/26/4/793/2064670/793.pdf> DOI: 10.1158/1078-0432.CCR-19-1459 * |
| FUMAGALLI ET AL., BMC CANCER, vol. 10, 2010, pages 101 |
| PARDOLL, NATURE REV. CANCER, vol. 12, April 2012 (2012-04-01), pages 252 - 64 |
| WEHN PAUL M. ET AL: "Design and Activity of Specific Hypoxia-Inducible Factor-2[alpha] (HIF-2[alpha]) Inhibitors for the Treatment of Clear Cell Renal Cell Carcinoma: Discovery of Clinical Candidate ( S )-3-((2,2-Difluoro-1-hydroxy-7-(methylsulfonyl)-2,3-dihydro-1 H -inden-4-yl)oxy)-5-fluorobenzonitrile (PT2385)", JOURNAL OF MEDICINAL CHEMISTRY, vol. 61, no. 21, 5 October 2018 (2018-10-05), US, pages 9691 - 9721, XP093091137, ISSN: 0022-2623, DOI: 10.1021/acs.jmedchem.8b01196 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2026006759A1 (en) * | 2024-06-28 | 2026-01-02 | Arcus Biosciences, Inc. | Pharmaceutical compositions, dosage forms, and methods of making and using same |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN116745288B (en) | Compounds and uses thereof | |
| AU2021213257B2 (en) | Compounds and uses thereof | |
| JP7693688B2 (en) | Compounds and their uses | |
| JP7600396B2 (en) | Compounds and their uses | |
| US12071411B2 (en) | Inhibitors of HIF-2α and methods of use thereof | |
| CA3173831A1 (en) | Tetralin and tetrahydroquinoline compounds as inhibitors of hif-2alpha | |
| JP2025525946A (en) | Heterocyclic compounds and uses thereof | |
| EP2739618B1 (en) | Quinazoline compounds as serine/threonine kinase inhibitors | |
| AU2020372881A1 (en) | Small molecule inhibitors of KRAS G12C mutant | |
| CN114760994A (en) | Inhibitors of HIF-2 alpha | |
| WO2020160192A1 (en) | Compounds and uses thereof | |
| WO2023244713A1 (en) | Quinazoline derivatives, compositions and methods thereof | |
| CN110407856B (en) | Macrocyclic compound and composition containing same | |
| KR20250122471A (en) | Heterocyclic rings and their uses | |
| WO2022109426A1 (en) | Compounds and uses thereof | |
| US20230150974A1 (en) | Compounds and uses thereof | |
| CN106488910A (en) | Inhibitors of KRAS G12C | |
| EP3283079A1 (en) | Targeted treatment of leiomyosarcoma | |
| TW202411228A (en) | Compounds and uses thereof | |
| CN117295727A (en) | Pyridazinone compounds as PARP7 inhibitors | |
| WO2025038857A1 (en) | TETRALINS TARGETING MUTANT HIF-2α | |
| CN114621256A (en) | Pyrazolo[1,5-a]pyridine compounds and preparation method and application thereof | |
| EP4293029B1 (en) | Azaheteroaryl compound, preparation method therefor, and application thereof | |
| CN115583938A (en) | Small molecule compounds targeting BCL9/β-catenin interaction | |
| CN113166148A (en) | Heterocyclic compounds as CDK-HDAC dual pathway inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24765872 Country of ref document: EP Kind code of ref document: A1 |