WO2025030002A2 - Dgk targeting compounds and uses thereof - Google Patents
Dgk targeting compounds and uses thereof Download PDFInfo
- Publication number
- WO2025030002A2 WO2025030002A2 PCT/US2024/040518 US2024040518W WO2025030002A2 WO 2025030002 A2 WO2025030002 A2 WO 2025030002A2 US 2024040518 W US2024040518 W US 2024040518W WO 2025030002 A2 WO2025030002 A2 WO 2025030002A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- chloro
- compound
- pyridyl
- methyl
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/38—Nitrogen atoms
- C07D215/42—Nitrogen atoms attached in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
- C07D491/107—Spiro-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/12—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains three hetero rings
- C07D491/20—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Definitions
- DGKs Diacylglycerol kinases
- DGKs are lipid kinases that mediate the conversion of diacylglycerol to phosphatidic acid thereby terminating T-cell functions propagated through the TCR signaling pathway.
- DGKs serve as intracellular checkpoints, and inhibition of DGKs is expected to enhance T-cell signaling pathways and T-cell activation.
- Knock-out mouse models of DGK ⁇ have shown a hyper-responsive T-cell phenotype and improved anti- tumor immune activity (Zha Y et al. Nature Immunology, (2006) 12:1343; Olenchock B. A. et al., Nature (2006) 11: 1174–81).
- the permissible substituents include acyclic and cyclic, branched, and unbranched, carbocyclic and heterocyclic, aromatic and non-aromatic substituents of organic compounds.
- the permissible substituents can be one or more and the same or different for appropriate organic compounds.
- the heteroatoms such as nitrogen may have hydrogen substituents and/or any permissible substituents of organic compounds described herein which satisfy the valences of the heteroatoms.
- the dosage regimen can vary widely, but may be routinely determined using standard methods.
- the daily dose can be administered in one to four doses divided per day.
- Other dosing schedules include one dose per week and one dose per two-day cycle.
- Methods of Treatment The compounds of the invention as defined hereinbefore, or a pharmaceutically acceptable salt thereof, are useful for the treatment of cancer.
- the compounds of the invention, or a pharmaceutically acceptable salt thereof can be used in the treatment of diseases or disorders associated with DGK target inhibition in T-cells.
- the compound of the invention is prepared in combination with one or more additional therapeutic agents for conjoint administration for treating diseases or disorders associated with DGK target inhibition in T- cells.
- the compounds described herein may be used to treat or prevent viral infections and proliferative diseases such as cancer.
- disease or conditions that are associated with DGK target inhibition in T cells include viral and other infections (e.g., skin infections, GI infection, urinary tract infections, genito-urinary infections, systemic infections), and proliferative diseases (e.g., cancer).
- the subject is being treated for cancer.
- Types of cancers that may be treated with a compound of the invention include, but are not limited to, brain cancers, skin cancers, bladder cancers, ovarian cancers, breast cancers, gastric cancers, pancreatic cancers, prostate cancers, colon cancers, blood cancers, lung cancers and bone cancers.
- cancer types include neuroblastoma, intestine carcinoma such as rectum carcinoma, colon carcinoma, familiar adenomatous polyposis carcinoma and hereditary non-polyposis colorectal cancer, esophageal carcinoma, labial carcinoma, larynx carcinoma, hypopharynx carcinoma, tongue carcinoma, salivary gland carcinoma, gastric carcinoma, adenocarcinoma, medullary thyroid carcinoma, papillary thyroid carcinoma, renal carcinoma, kidney parenchymal carcinoma, ovarian carcinoma, cervix carcinoma, uterine corpus carcinoma, endometrium carcinoma, chorion carcinoma, pancreatic carcinoma, prostate carcinoma, testis carcinoma, breast carcinoma, urinary carcinoma, melanoma, brain tumors such as glioblastoma, astrocytoma, meningioma, medulloblastoma and peripheral neuroectodermal tumors, Hodgkin lymphoma, non-Hodgkin lymphoma, Burkitt lymphoma, acute lymphatic leuk
- B7 family which includes B7-1, B7-2, B7-H1 (PD-L1), B7- DC (PD-L2), B7-H2 (ICOS-L), B7-H3, B7-H4, B7-H5 (VISTA), and B7-H6.
- agents that can be combined with the compounds of the invention for the treatment of cancer include antagonists of inhibitory receptors on NK cells or agonists of activating receptors on NK cells.
- the compounds of the invention can be combined with antagonists of KIR, such as lirilumab.
- agents for combination therapies include agents that inhibit or deplete macrophages or monocytes, including but not limited to CSF-1R antagonists such as CSF-1R antagonist antibodies including RG7155 (e.g., International Patent Publication Nos.
- the additional anticancer agent is a CTLA-4 antagonist, such as an antagonistic CTLA-4 antibody.
- Suitable CTLA-4 antibodies include, for example, YERVOY (ipilimumab), or tremelimumab.
- the additional anticancer agent is a PD-1 antagonist, such as an antagonistic PD-1 antibody.
- Suitable PD-1 antibodies include, for example, OPDIVO (nivolumab), KEYTRUDA (pembrolizumab), or MEDI-0680 (AMP-514; e.g., International Patent Publication No. WO2012/145493).
- the additional anticancer agent is a LAG-3 antagonist, such as an antagonistic LAG-3 antibody.
- LAG3 antibodies include, for example, BMS-986016 (e.g., International Patent Publication Nos. WO10/19570, WO14/08218), or IMP-731 or IMP-321 (e.g., International Patent Publication Nos. WO08/132601, WO09/44273).
- the additional anticancer agent is a CD137 (4-1BB) agonist, such as an agonistic CD137 antibody.
- Suitable CD137 antibodies include, for example, urelumab, and PF-05082566 (e.g., International Patent Publication No. WO12/32433).
- the additional anticancer agent is a GITR agonist, such as an agonistic GITR antibody.
- Suitable GITR antibodies include, for example, BMS-986153, BMS-986156, TRX- 518 (e.g., International Patent Publication Nos. WO06/105021, WO09/009116), and MK- 4166 (e.g., International Patent Publication No. WO11/028683).
- the additional anticancer agent is an IDO antagonist.
- Suitable IDO antagonists include, for example, INCB-024360 e.g., International Patent Publication Nos. (WO2006/122150, WO07/75598, WO08/36653, WO08/36642), indoximod, BMS-986205, or NLG-919 (e.g., International Patent Publication No. WO09/73620, WO09/1156652, WO11/56652, WO12/142237).
- the additional anticancer agent is an OX40 agonist, such as an agonistic OX40 antibody.
- Suitable OX40 antibodies include, for example, MEDI-6383 or MEDI-6469.
- the additional anticancer agent is an OX4OL antagonist, such as an antagonistic OX40 antibody.
- OX4OL antagonists include, for example, RG-7888 (e.g., International Patent Publication No. WO06/029879).
- the additional anticancer agent is a CD40 agonist, such as an agonistic CD40 antibody.
- the anticancer agent is a CD40 antagonist, such as an antagonistic CD40 antibody.
- Suitable CD40 antibodies include, for example, lucatumumab or dacetuzumab.
- the additional anticancer agent agent is a CD27 agonist, such as an agonistic CD27 antibody.
- Suitable CD27 antibodies include, for example, varlilumab.
- the additional anticancer agent is MGA271 (to B7H3) (e.g., International Patent Publication No. WO11/109400).
- Combination therapies, as disclosed herein, are intended to embrace conjoint administration of these therapeutic agents; for example, administration of said therapeutic agents in a sequential manner, wherein each therapeutic agent is administered at a different time, as well as administration of these therapeutic agents, or at least two of the therapeutic agents in a substantially simultaneous manner.
- Substantially simultaneous administration can be accomplished, for example, by administering to the subject a single dosage form having a fixed ratio of each therapeutic agent or in multiple, single dosage forms for each of the therapeutic agents.
- Sequential or substantially simultaneous administration of each therapeutic agent can be affected by any appropriate route including, but not limited to, oral routes, parental routes, intravenous routes, intramuscular routes, and direct absorption through mucous membrane tissues.
- the therapeutic agents can be administered by the same route or by different routes.
- a first therapeutic agent of the combination selected may be administered by intravenous injection while the other therapeutic agents of the combination may be administered orally.
- all therapeutic agents may be administered orally, or both therapeutic agents may be administered by parentally, e.g., by intravenous injection.
- Combination therapy also can embrace the administration of the therapeutic agents as described above in further combination with other biologically active ingredients and non-drug therapies (e.g., surgery or radiation treatment).
- the non-drug treatment may be conducted at any suitable time so long as a beneficial effect from the co-action of the combination of the therapeutic agents and non-drug treatment is achieved.
- the beneficial effect is still achieved when the non-drug treatment is temporally removed from the administration of the therapeutic agents, perhaps by days or even weeks.
- One or more additional pharmaceutical agents or treatment methods such as, for example, anti-viral agents, chemotherapeutics or other anti-cancer agents, immune enhancers, immunosuppressants, radiation, anti-tumor and anti-viral vaccines, cytokine therapy (e.g., IL2 and GM-CSF), and/or tyrosine kinase inhibitors can be optionally used in combination with the compounds of the invention for treatment of DGK ⁇ associated diseases, disorders, or conditions.
- the agents can be combined with the present compounds in a single dosage form, or the agents can be administered simultaneously or sequentially as separate dosage forms.
- Suitable additional anti-cancer agents include, for example, alkylating agents (including, without limitation, nitrogen mustards, ethylenimine derivatives, alkyl sulfonates, nitrosoureas and triazenes) such as uracil mustard, chlormethine, cyclophosphamide (CYTOXAN®), ifosfamide, melphalan, chlorambucil, pipobroman, triethylene-melamine, triethylenethiophosphoramine, busulfan, carmustine, lomustine, streptozocin, dacarbazine, and temozolomide.
- alkylating agents including, without limitation, nitrogen mustards, ethylenimine derivatives, alkyl sulfonates, nitrosoureas and triazenes
- alkylating agents including, without limitation, nitrogen mustards, ethylenimine derivatives, alkyl sulfonates, nitrosoureas and triazene
- suitable additional agents for use in combination with the compounds of the invention include: dacarbazine (DTIC), optionally, along with other chemotherapy drugs such as carmustine (BCNU) and cisplatin; the “Dartmouth regimen,” which consists of DTIC, BCNU, cisplatin and tamoxifen; a combination of cisplatin, vinblastine, and DTIC, temozolomide or YERVOYTM.
- DTIC dacarbazine
- BCNU carmustine
- cisplatin the “Dartmouth regimen,” which consists of DTIC, BCNU, cisplatin and tamoxifen
- a combination of cisplatin, vinblastine, and DTIC, temozolomide or YERVOYTM a combination of cisplatin, vinblastine, and DTIC, temozolomide or YERVOYTM.
- immunotherapy drugs including cytokines such as interferon alpha, interleukin 2, and tumor necrosis
- the compounds of the invention also can be used in combination with vaccine therapy in the treatment of cancer (e.g., melanoma).
- Antimelanoma vaccines are, in some ways, similar to the anti-virus vaccines that are used to prevent diseases caused by viruses such as polio, measles, and mumps. Weakened melanoma cells or parts of melanoma cells called antigens may be injected into a patient to stimulate the body’s immune system to destroy melanoma cells.
- Suitable additional anti-cancer agents also include, for example, anti-metabolites (including, without limitation, folic acid antagonists, pyrimidine analogs, purine analogs and adenosine deaminase inhibitors) such as methotrexate, 5-fluorouracil, floxuridine, cytarabine, 6- mercaptopurine, 6-thioguanine, fludarabine phosphate, pentostatine, and gemcitabine.
- anti-metabolites including, without limitation, folic acid antagonists, pyrimidine analogs, purine analogs and adenosine deaminase inhibitors
- methotrexate including, without limitation, folic acid antagonists, pyrimidine analogs, purine analogs and adenosine deaminase inhibitors
- methotrexate including, without limitation, folic acid antagonists, pyrimidine analogs, purine analogs and adenosine deaminase inhibitors
- Suitable additional anti-cancer agents further include, for example, certain natural products and their derivatives (for example, vinca alkaloids, antitumor antibiotics, enzymes, lymphokines and epipodophyllotoxins) such as vinblastine, vincristine, vindesine, bleomycin, dactinomycin, daunorubicin, doxorubicin, epirubicin, idarubicin, ara-C, paclitaxel (Taxol), mithramycin, deoxyco-formycin, mitomycin-C, L-asparaginase, interferons (especially IFN- a), etoposide, and teniposide.
- certain natural products and their derivatives for example, vinca alkaloids, antitumor antibiotics, enzymes, lymphokines and epipodophyllotoxins
- vinblastine vincristine, vindesine
- bleomycin dactinomycin
- daunorubicin daunorubicin
- cytotoxic agents include navelbene, CPT-11, anastrazole, letrazole, capecitabine, reloxafine, and droloxafine.
- cytotoxic agents such as epidophyllotoxin; an antineoplastic enzyme; a topoisomerase inhibitor; procarbazine; mitoxantrone; platinum coordination complexes such as cisplatin and carboplatin; biological response modifiers; growth inhibitors; antihormonal therapeutic agents; leucovorin; tegafur; and haematopoietic growth factors.
- additional anti-cancer agent(s) include antibody therapeutics such as trastuzumab (HERCEPTIN®), antibodies to costimulatory molecules such as CTLA-4, 4-1BB and PD-1, or antibodies to cytokines (IL-10 or TGF- ⁇ ).
- additional anti-cancer agents also include those that block immune cell migration such as antagonists to chemokine receptors, including CCR 2 and CCR 4.
- additional anti-cancer agents also include those that augment the immune system such as adjuvants or adoptive T-cell transfer.
- Additional anti-cancer agents also include anti-cancer vaccines, such as, for example, dendritic cells, synthetic peptides, DNA vaccines, and recombinant viruses.
- the treatment methods of the invention may optionally include conjointly administering at least one signal transduction inhibitor (STI).
- STI signal transduction inhibitor
- a “signal transduction inhibitor” is an agent that selectively inhibits one or more vital steps in signaling pathways, in the normal function of cancer cells, thereby leading to apoptosis.
- Suitable STIs include, but are not limited to: (i) bcr/abl kinase inhibitors such as, for example, STI 571 (GLEEVEC®); (ii) epidermal growth factor (EGF) receptor inhibitors such as, for example, kinase inhibitors ORES SA®, SSI-774) and antibodies (Imclone: C225 [Goldstein et al. Clin.
- her-2/neu receptor inhibitors such as farnesyl transferase inhibitors (FTI) such as, for example, L-744,832 (Kohl et al. Nat. Med., 1(8):792–97 (1995));
- FTI farnesyl transferase inhibitors
- inhibitors of Akt family kinases or the Akt pathway such as, for example, rapamycin
- cell cycle kinase inhibitors such as, for example, flavopiridol and UCN-01 (see, for example, Sausville Curr. Med. Chem.
- At least one STI and at least one compound of Formula (I) may be in separate pharmaceutical compositions.
- at least one compound of the invention and at least one STI may be administered to the patient conjointly.
- At least one compound of the invention may be administered first or at least one STI may be administered first and the other is administered next; or at least one compound of the invention and at least one STI may be administered at the same time. Additionally, when more than one compound of invention and/or STI is used, the compounds may be administered in any order.
- pharmaceutical compositions for the treatment of a chronic viral infections in a subject comprising administering a therapeutically effective amount of at least one compound of the invention, optionally, at least one chemotherapeutic drug, and, optionally, at least one antiviral agent, in a pharmaceutically acceptable carrier.
- one or more compounds of the invention, one or more chemotherapeutic drugs, and/or one or more antiviral agents are administered conjointly.
- At least one compound of the invention may be administered first or at least one chemotherapeutic agent may be administered first.
- at least one compound of the invention and the at least one STI may be administered at the same time.
- the compounds may be administered in any order.
- any antiviral agent or STI may also be administered at any point in relation to the administration of the compound of the invention.
- Chronic viral infections that may be treated using the present combinatorial treatment include, but are not limited to, diseases caused by hepatitis C virus (HCV), human papilloma virus (HPV), cytomegalovirus (CMV), herpes simplex virus (HSV), Epstein-Barr virus (EBV), varicella zoster virus, coxsackie virus, human immunodeficiency virus (HIV).
- HCV hepatitis C virus
- HPV human papilloma virus
- CMV cytomegalovirus
- HSV herpes simplex virus
- EBV Epstein-Barr virus
- varicella zoster virus coxsackie virus
- coxsackie virus human immunodeficiency virus
- HCV hepatitis C virus
- HCV hepatitis C virus
- HPV human papilloma virus
- CMV cytomegalovirus
- HSV herpes simplex virus
- EBV Epstein-Barr virus
- Suitable antiviral agents contemplated for use in combination with the compound of Formula (I) can comprise nucleoside and nucleotide reverse transcriptase inhibitors (NRTIs), non- nucleoside reverse transcriptase inhibitors (NNRTIs), protease inhibitors and other antiviral drugs.
- NRTIs nucleoside and nucleotide reverse transcriptase inhibitors
- NRTIs non- nucleoside reverse transcriptase inhibitors
- protease inhibitors and other antiviral drugs.
- NRTIs examples include zidovudine (AZT); didanosine (ddl); zalcitabine (ddC); stavudine (d4T); lamivudine (3TC); abacavir (1592U89); adefovir dipivoxil [bis(P0M)- PMEA]; lobucavir; BCH-I0652; emitricitabine [(-)-FTC]; beta-L-FD4 (also called beta-L- D4C and named beta-L-2′,3′-dicleoxy-5-fluoro-cytidene); DAPD, (( ⁇ )-beta-D-2,6-diamino- purine dioxolane); and lodenosine (FddA).
- ZT zidovudine
- ddl didanosine
- ddC zalcitabine
- d4T stavudine
- lamivudine lami
- NNRTIs include nevirapine (BI- RG-587); delaviradine (BHAP, U-90152); efavirenz (DMP-266); PNU-142721; AG-1549; MKC-442 (1-(ethoxy-methyl)-5-(1-methylethyl)-6-(phenylmethyl)-(2,4(1H,3H)- pyrimidinedione); and (+)-calanolide A (NSC-675451) and B.
- Typical suitable protease inhibitors include saquinavir (Ro 31-8959); ritonavir (ABT-538); indinavir (MK-639); nelfnavir (AG-1343); amprenavir (141W94); lasinavir; DMP-450; BMS-2322623; ABT-378; and AG-1549.
- Other antiviral agents include hydroxyurea, ribavirin, IL-2, IL-12, pentafuside and Yissum Project No.11607.
- kits useful, for example, in the treatment or prevention of DGK ⁇ -associated diseases or disorders referred to herein which include one or more containers containing a pharmaceutical composition comprising a therapeutically effective amount of a compound of the invention.
- kits can further include, if desired, one or more of various conventional pharmaceutical kit components, such as, for example, containers with one or more pharmaceutically acceptable carriers, additional containers, as will be readily apparent to those skilled in the art.
- Instructions, either as inserts or as labels, indicating quantities of the components to be administered, guidelines for administration, and/or guidelines for mixing the components, can also be included in the kit.
- Methods of Preparation The compounds of the present invention may be synthesized by many methods available to those skilled in the art in view of the present disclosure.
- PCT/US2023/012145 filed February 1, 2023, and titled DGK TARGETING COMPOUNDS AND USES THEREOF.
- the compouinds can be made similar to “4-[(3aR,7aS)-5-[5-(trifluoromethoxy)- 2-pyridyl]-3,3a,4,6,7,7a-hexahydro-2H-pyrrolo[3,2-c]pyridin-1-yl]-6-chloro-1-methyl-2-oxo- 1,5-naphthyridine-3-carbonitrile” of PCT/US2023/012145, described below: Exemplary Synthesis of 4-[(3aR,7aS)-5-[5-(trifluoromethoxy)-2-pyridyl]-3,3a,4,6,7,7a- hexahydro-2H-pyrrolo[3,2-c]pyridin-1-yl]-6-chloro-1-methyl-2-oxo-1,
- Step 2 Preparation of tert-butyl (3aR,7aS)-5-[5-(trifluoromethoxy)-2-pyridyl]- 3,3a,4,6,7,7a-hexahydro-2H-pyrrolo[3,2-c]pyridine-1-carboxylate and tert-butyl (3aS,7aR)-5-[5-(trifluoromethoxy)-2-pyridyl]-3,3a,4,6,7,7a-hexahydro-2H-pyrrolo[3,2- c]pyridine-1-carboxylate Racemic tert-butyl (3aR,7aS)-5-[5-(trifluoromethoxy)-2-pyridyl]-3,3a,4,6,7,7a-hexahydro- 2H-pyrrolo[3,2-c]pyridine-1-carboxylate (1.9 g, 4.90 mmol, 1 eq) was purified by SFC (column:
- Step 3 Preparation of WC-ARV-JM-047-A-2a, (3aR,7aS)-5-[5-(trifluoromethoxy)-2- pyridyl]-1,2,3,3a,4,6,7,7a-octahydropyrrolo[3,2-c]pyridine
- tert-butyl (3aR,7aS)-5-[5-(trifluoromethoxy)-2-pyridyl]-3,3a,4,6,7,7a- hexahydro-2H-pyrrolo[3,2-c]pyridine-1-carboxylate 500 mg, 1.29 mmol, 1 eq
- dichloromethane 5 mL
- trifluoroacetic acid 7.70 g, 67.53 mmol, 5.00 mL, 52.32 eq.
- Step 4 Preparation of 4-[(3aR,7aS)-5-[5-(trifluoromethoxy)-2-pyridyl]-3,3a,4,6,7,7a- hexahydro-2H-pyrrolo[3,2-c]pyridin-1-yl]-6-chloro-1-methyl-2-oxo-1,5-naphthyridine- 3-carbonitrile
- 3aR,7aS)-5-[5-(trifluoromethoxy)-2-pyridyl]-1,2,3,3a,4,6,7,7a- octahydropyrrolo[3,2-c]pyridine (517 mg, 1.29 mmol, 1 eq, trifluoroacetic acid) and 4,6- dichloro-1-methyl-2-oxo-1,5-naphthyridine-3-carbonitrile (327 mg, 1.29 mmol, 1 eq) in acetonitrile (5 mL) was added
- Step 2 Preparation of (3aR,7aS)-1-(5-isopropoxy-2-pyridyl)-2,3,3a,4,5,6,7,7a- octahydropyrrolo[3,2-c]pyridine
- reaction mixture was concentrated under reduced pressure, and the resulting residue was purified by prep-HPLC (column: YMC Triart C18250*50mm*7um; mobile phase: [water(formic acid) in acetonitrile]: 50%-95%, 25min) followed by SFC (column: DAICEL CHIRALCEL OJ(250mm*30mm,10um); mobile phase:40% [0.1% ammonium hydroxide in ethanol] in supercritical carbon dioxide; 40%- 40%,6.0;84min).
- the material was separated by SFC (condition: column: REGIS(S,S)WHELK- O1(250mm*25mm,10um); mobile phase: 10% [0.1% ammonium hydroxide in ethanol] in supercritical CO 2 : 10%-10%,C6; 160min), then further separated by SFC (column: DAICEL CHIRALPAK AD (250mm*30mm,10um); mobile phase:10% [0.1% ammonium hydroxide in ethanol] in supercritical CO 2 , 10%-10%,c10; 60min).
- tert-Butyl (3aR,7aS)-5-[5-(trifluoromethoxy)-2-pyridyl]-3,3a,4,6,7,7a-hexahydro-2H- pyrrolo[3,2-c]pyridine-1-carboxylate (66 mg, 19%) was obtained as a green oil.
- tert-Butyl (3aS,7aR)-5-[5-(trifluoromethoxy)-2-pyridyl]-3,3a,4,6,7,7a-hexahydro-2H- pyrrolo[3,2-c]pyridine-1-carboxylate 50 mg, 15% was obtained as a green oil.
- Step 2 Preparation of (3aS,7aR)-5-[5-(trifluoromethoxy)-2-pyridyl]-1,2,3,3a,4,6,7,7a- octahydropyrrolo[3,2-c]pyridine
- tert-butyl (3aS,7aR)-5-[5-(trifluoromethoxy)-2-pyridyl]-3,3a,4,6,7,7a- hexahydro-2H-pyrrolo[3,2-c]pyridine-1-carboxylate 80 mg, 0.21 mmol, 1 eq
- dichloromethane 1.5 mL
- trifluoroacetic acid 1.23 g, 10.80 mmol, 0.8mL, 52.32 eq.
- the reaction mixture was diluted with water 5 mL and extracted with ethyl acetate (2 x 10 mL). The combined organic layers were dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by flash silica gel chromatography (ISCO®; 12 g SepaFlash® Silica Flash Column, eluent of 0 ⁇ 40% ethyl acetate/petroleum ether gradient @ 60 mL/min) to afford ethyl 6-chloro-4-[4-hydroxy-4- [(1R)-1-[5-(trifluoromethoxy)-2-pyridyl]ethyl]-1-piperidyl]-1-methyl-2-oxo-quinoline-3- carboxylate (272 mg, 96% yield) was obtained as a yellow oil.
- ISCO® 12 g SepaFlash® Silica Flash Column, eluent of 0 ⁇ 40% ethyl acetate/petroleum ether
- Step 2 Preparation of tert-butyl 6-chloro-4-hydroxy-1-methyl-2-oxo-1,2- dihydroquinoline-3-carboxylate To a solution of di-tert-butyl malonate (6.35 mL, 1.5 eq) in N,N-dimethylacetamide (40 mL) was added sodium hydride (2.65 g, 60% purity, 3.5 eq) at 0 °C for 0.5 h.
- Step 3 Preparation of tert-butyl 6-chloro-1-methyl-2-oxo-4- (((trifluoromethyl)sulfonyl)oxy)-1,2-dihydroquinoline-3-carboxylate
- tert-butyl 6-chloro-4-hydroxy-1-methyl-2-oxo-quinoline-3-carboxylate (2 g, eq) in dimethylformamide (20 mL) was added sodium hydride (775 mg, 60% purity, 3 eq) at 0 °C.
- Step 4 Preparation of (5-chloro-2-pyridyl)-phenyl-methanol To a solution of copper iodide (10 g, 1.5 eq) in diethyl ether (50 mL) was added bromo(phenyl)magnesium (3 M, 17.66 mL, 1.5 eq) at -78 °C under nitrogen atmosphere and stirred for 0.5 h.
- 5-chloropyridine-2-carbaldehyde (5 g, 1 eq) in diethyl ether (50 mL) was added dropwise at -78°C under a nitrogen atmosphere. Then the mixture was stirred at 0°C for another 1h. The mixture was diluted with saturated ammonium chloride (100 mL), extracted with ethyl acetate (3 ⁇ 100 mL), washed with brine (3 ⁇ 100 mL), and dried over anhydrous sodium sulfate, filtered, and concentrated.
- Step 8 Preparation of 4-[(S)-(5-chloro-2-pyridyl)-phenyl-methyl]piperidin-4-ol
- trifluoroacetic acid (0.48 mL) in dichloromethane (0.5 mL) was degassed and purged with nitrogen three times, and then the mixture was stirred at 25 °C for 0.5 h under nitrogen atmosphere.
- reaction mixture was quenched by 5 mL saturated sodium sulfate, poured into water (15 mL) extracted with ethyl acetate (3 x 10 mL), the organic layer was washed with brine (2 x 15 mL), dried over anhydrous sodium sulfate, then concentrated under reduced pressure to afford the crude product.
- the crude product was added slowly to water, and it then saturated triethylamine was added to adjust the pH to 7.
- Step 12 Preparation of 6-chloro-4-[4-[(R)-(5-chloro-2-pyridyl)-phenyl-methyl]-4- hydroxy-1-piperidyl]-1-methyl-2-oxo-quinoline-3-carboxamide
- 6-chloro-4-[4-[(R)-(5-chloro-2-pyridyl)-phenyl-methyl]-4-hydroxy-1- piperidyl]-1-methyl-2-oxo-quinoline-3-carboxylic acid 140 mg, 1 eq
- N,N-dimethyl acetamide (2 mL) was added diisopropylethylamine (0.26 mL, 5 eq), O-(7-azabenzotriazol-1- yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate (247mg, 2.5 eq).
- Boc A mixture of 2-bromo-5-chloro-pyridine (1 g, 1 eq), tert-butyl 4-[(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)methylene]piperidine-1-carboxylate (2.02 g, 1.2 eq), sodium carbonate (2 M, 5.20 mL, 2 eq), tetrakis[triphenylphosphine]palladium(0) (600 mg, 0.1 eq) in dioxane (10 mL) was degassed and purged with nitrogen three times, and then the mixture was stirred at 80 °C for 3 h under a nitrogen atmosphere.
- reaction mixture was cooled to 0 °C and sodium hydroxide (4 M, 3.24 mL, 2 eq) was added dropwise. The ice-bath was removed and stirred at 25 °C for 2 h. To the reaction mixture was added water (10 mL), and then extracted with ethyl acetate (15 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure.
- sodium hydroxide 4 M, 3.24 mL, 2 eq
- the resultant residue was purified by flash silica gel chromatography (ISCO®; 40 g SepaFlash® Silica Flash Column, Eluent of 0 ⁇ 10% Ethyl acetate/Petroleum ether gradient @ 60mL/min) then further triturated with acetonitrile (10 mL) at 25 °C to produce tert-butyl 4-[(5-chloro-2-pyridyl)-cyclopropyl-methyl]-4-hydroxy-piperidine-1-carboxylate (800 mg, 2.18 mmol, 47% yield) as a white solid.
- ISCO® 40 g SepaFlash® Silica Flash Column, Eluent of 0 ⁇ 10% Ethyl acetate/Petroleum ether gradient @ 60mL/min
- the mixture was stirred at -78 °C for 1 h under a nitrogen atmosphere.
- the mixture was diluted with saturated ammonium chloride (150 mL), extracted with ethyl acetate (3 ⁇ 150 mL), washed with brine (500 mL), the combined organic portions were dried over anhydrous sodium sulfate, filtered and concentrated.
- the resultant residue was purified by prep-HPLC (column: Phenomenex luna C18 (250*70 mm, 10 um); mobile phase: [water (formic acid)- acetonitrile], 25%-60%, 26 min) to afford (5-chloro-2-pyridyl)-(4,4-difluorocyclohexyl)methanol (3.4 g, 21% yield) as a yellow oil.
- the mixture was stirred at -78 °C for 1 h under a nitrogen atmosphere.
- the mixture was diluted with saturated ammonium chloride (50 mL), extracted with ethyl acetate (3 ⁇ 50 mL), washed with brine (3 ⁇ 50 mL), and the combined organic portions were dried over anhydrous sodium sulfate, filtered, and concentrated.
- SFC: Rt peak 1 6.124 min.
- Step 6 Preparation of 6-chloro-4-[4-[(R)-(5-chloro-2-pyridyl)-(4,4- difluorocyclohexyl)methyl]-4-hydroxy-1-piperidyl]-1-methyl-2-oxo-1,5-naphthyridine-3- carbonitrile and 6-chloro-4-[4-[(S)-(5-chloro-2-pyridyl)-(4,4-difluorocyclohexyl)methyl]- 4-hydroxy-1-piperidyl]-1-methyl-2-oxo-1,5-naphthyridine-3-carbonitrile
- Step 1 Preparation of 1-(5-chloro-2-pyridyl)propan-1-ol To a solution of 2-bromo-5-chloro-pyridine (5 g, 1 eq) and propanal (3.78 mL, 2 eq) in tetrahydrofuran (50 mL) was added n-butyllithium (2.5 M, 15.59 mL, 1.5 eq) under a nitrogen atmosphere at -78°C.
- the resultant residue was purified by reverse- phase HPLC (column: Phenomenex luna C18250*50mm*15um; mobile phase: [water (formic acid) - acetonitrile]; gradient: 37%-67%, over 25 min) to produce tert-butyl 4-[1-(5-chloro-2- pyridyl)propyl]-4-hydroxy-piperidine-1-carboxylate (800 mg, 56% yield) as a yellow solid.
- Step 2 Preparation of tert-butyl 2-(5-chloro-2-pyridyl)-1-oxa-6-azaspiro[2.5]octane-6- carboxylate.
- tert-butyl 2-(5-chloro-2-pyridyl)-1-oxa-6-azaspiro[2.5]octane-6- carboxylate To a mixture of 5-methoxy-2-vinyl-pyridine tert-butyl 4-[(5-chloro-2- pyridyl)methylene]piperidine-1-carboxylate (4 g, 1 eq) in dioxane (40 mL) and water (40 mL) was added a solution of N-bromosuccinimide (2.77 g, 1.2 eq) in dioxane (40 mL) and water (40 mL) dropwise and stirred at 20 °C for 2 h under a nitrogen atmosphere.
- reaction mixture was cooled to 0 °C and sodium hydroxide (4 M, 6.48 mL, 2 eq) was added dropwise and purged with nitrogen three times. The mixture was then stirred at 20 °C for 12 h. The reaction mixture was added slowly to water (100ml), then extracted with ethyl acetate (3 x 100mL), the combined organic layers were dried over sodium sulfate anhydrous, filtered, and concentrated to afford tert-butyl 2-(5-chloro-2-pyridyl)-1-oxa-6-azaspiro[2.5]octane-6- carboxylate (3.29 g, 78% yield) as a colorless oil.
- Step 5 Preparation of 8-bromo-1-methyl-3, 1-benzoxazine-2, 4-dione
- a solution of 8-bromo-1H-3, 1-benzoxazine-2, 4-dione (4 g, 1 eq) in N, N- dimethylformamide (40 mL) was added diisopropylethylamine (5.76 mL, 2 eq), and then add iodomethane (3.09 mL, 3 eq) at 0 °C. The mixture was stirred at 25 °C for 12 h.
- Step 6 Preparation of 8-bromo-4-hydroxy-1-methyl-2-oxo-quinoline-3-carbonitrile
- ethyl 2-cyanoacetate 2.50 mL, 3 eq
- triethylamine 8.70 mL, 8 eq
- 8-bromo-1-methyl-3,1- benzoxazine-2,4-dione 2 g, 1 eq
- the mixture was stirred at 70 °C for 12 h.
- Step 7 Preparation of 8-bromo-4-chloro-1-methyl-2-oxo-quinoline-3-carbonitrile To a mixture of 8-bromo-4-hydroxy-1-methyl-2-oxo-quinoline-3-carbonitrile (1.7 g, 1 eq) in acetonitrile (20 mL) was added diisopropylethylamine (6.37 mL, 6 eq) then was added benzyl(triethyl)ammonium chloride (2.77 g, 2 eq) followed by addition of phosphoryl chloride (2.84 mL, 5 eq) in one portion at
- Step 8 Preparation of 8-bromo-4-[4-[1-(5-chloro-2-pyridyl)ethyl]-4-hydroxy-1- piperidyl]-1-methyl-2-oxo-quinoline-3-carbonitrile
- N,N-diisopropylethylamine (521 mg, 4.03 mmol, 0.7 mL, 3 eq) in acetonitrile (1 mL) was degassed and purged with nitrogen three times, and then the mixture was stirred at 40 °C for 12 h under a nitrogen atmosphere.
- Step 11 Preparation of 4-[4-[(1S)-1-(5-chloro-2-pyridyl)ethyl]-4-hydroxy-1-piperidyl]-1- methyl-8-(oxetan-3-yloxy)-2-oxo-quinoline-3-carbonitrile and 4-[4-[(1R)-1-(5-chloro-2- pyridyl)ethyl]-4-hydroxy-1-piperidyl]-1-methyl-8-(oxetan-3-yloxy)-2-oxo-quinoline-3- carbonitrile
- the reactant 4-[4-[1-[5-chloro-2-pyridyl)ethyl]-4-hydroxy-1-piperidyl]-1-methyl-8-(oxetan-3- yloxy)-2-oxo-quinoline-3-carbonitrile (50 mg, eq) was purified by SFC (column: DAICEL CHIRALPAK AS (250mm*
- Step 1 Preparation of 7-bromo-1-methyl-3,1-benzoxazine-2,4-dione
- N-ethyl-N-isopropyl- propan-2-amine (12.95 mL, 2 eq) in N,N-dimethylformamide (90 mL) was added methyl iodide (6.94 mL, 3 eq) at 0 °C.
- Step 3 Preparation of 7-bromo-4-chloro-1-methyl-2-oxo-quinoline-3-carbonitrile To a mixture of 7-bromo-4-hydroxy-1-methyl-2-oxo-quinoline-3-carbonitrile (2.77 g, 1 eq) and N-ethyl-N-isopropyl-propan-2-amine (10.37 mL, 6 eq) in acetonitrile (30 mL) was added phosphoryl trichloride (4.63 mL, 5 eq) and benzyl(triethyl)ammonium chloride (4.52 g, 2 eq) in one portion at 0 °C under nitrogen.
- Step 1 Preparation of (5-chloro-2-pyridyl)-tetrahydropyran-4-yl-methanol
- 2-bromo-5-chloro-pyridine (27.5 g, 1 eq) tetrahydropyran-4-carbaldehyde (17.94 g, 1.1 eq) in tetrahydrofuran (250 mL) was degassed and purged with N 2 three times, and to the mixture was added n-butyllithium (2.5 M, 114.32 mL, 2 eq) at -70 °C, and then the mixture was stir
- reaction mixture was quenched by addition ammonium chloride (200 mL) at 0 °C and added water (200 mL) and extracted with ethyl acetate (600 mL). The combined organic layers were washed with brine, dried over sodium sulfate, filtered, and concentrated under reduced pressure to afford a residue which was purified by silica gel column chromatography (10 ⁇ 50% ethyl acetate in petroleum ether) to produce (5-chloro-2-pyridyl)-tetrahydropyran-4-yl-methanol (2.55 g, crude as a yellow oil.
- reaction mixture was quenched by addition ammonium chloride (30 mL) at 0 °C, added water (20 mL), and extracted with ethyl acetate (150 mL). The combined organic layers were washed with brine, dried over sodium sulfate, filtered, and concentrated under reduced pressure to afford a residue, which was purified by prep-HPLC (column: Phenomenex luna C18 150*25mm* 10um; mobile phase: [water (formic acid)-methanol]; 50%-80%, 30 min).
- Step 1 Preparation of tert-butyl 1-(5-chloro-2-pyridyl)-3,3,3-trifluoro-propan-1-ol
- tert-butyl 1-(5-chloro-2-pyridyl)-3,3,3-trifluoro-propan-1-ol A mixture of 3,3,3-trifluoropropanal (10 g, 1.1 eq), 2-bromo-5-chloro-pyridine (15.61 g, 1 eq) in tetrahydrofuran (150 mL) was degassed and purged with nitrogen three times, and the mixture was added n-butyllithium (2.5 M, 64.91 mL, 2 eq) at -70 °C, and then the mixture
- reaction mixture was quenched by addition ammonium chloride (200 mL) at 0 °C, added water (200 mL), and extracted with ethyl acetate (600 mL). The combined organic layers were washed with brine (400 mL), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give a residue which was purified by prep-HPLC (column: Phenomenex luna C18 (250*70mm, 10 um); mobile phase: [water (formic acid)-acetonitrile]; gradient: 20%-50%, over 22 min) to produce 1-(5-chloro-2-pyridyl)-3,3,3-trifluoro-propan-1-ol (400 mg, 2.19% yield) as a red oil.
- prep-HPLC columnumn: Phenomenex luna C18 (250*70mm, 10 um)
- mobile phase [water (formic acid)-acetonitrile]; gradient: 20%-50%, over 22 min
- Step 3 Preparation of tert-butyl 4-[1-(5-chloro-2-pyridyl)-3,3,3-trifluoro-propyl]-4- hydroxy-piperidine-1-carboxylate A mixture of tert-butyl 4-oxopiperidine-1-carboxylate (138 mg, 1 eq), 2-(1-bromo-3,3,3- trifluoro-propyl)-5-chloro-pyridine (200 mg, 1 eq) in tetrahydrofuran (1.5 mL) and then
- Step 5 Preparation of 4-[(1S)-1-(5-chloro-2-pyridyl)-3,3,3-trifluoro-propyl]piperidin-4- ol
- 4-[(1S)-1-(5-chloro-2-pyridyl)-3,3,3-trifluoro-propyl]-4-hydroxy- piperidine-1-carboxylate (80 mg, 1 eq) in dichloromethane (1 mL) was added trifluoroacetic acid (1 mL, 68.80 eq). The mixture was stirred at 25 °C for 1hour.
- Step 6 Preparation of 6-chloro-4-[4-[(1S)-1-(5-chloro-2-pyridyl)-3,3,3-trifluoro-propyl]- 4-hydroxy-1-piperidyl]-1-methyl-2-oxo-1,5-naphthyridine-3-carbonitrile
- Step 1 Preparation of 1-(5-chloropyridin-2-yl)-2,2,2-trifluoroethan-1-ol
- 5-chloropyridine-2-carbaldehyde 10 g, 1 eq
- tetrahydrofuran 200 mL
- trimethyl(trifluoromethyl)silane 12.05 g, 1.2 eq
- tetrabutylammonium fluoride solution (1 M, 5 mL, 0.071 eq) and stirred for 0.5 h at 0°C.
- the mixture was stirred at -78 °C for 0.5 h.
- the mixture was diluted with saturated water (30 mL), extracted with ethyl acetate (3 ⁇ 50 mL), washed with brine (2 ⁇ 50 mL), dried over anhydrous sodium sulfate, filtered, and concentrated.
- the resultant residue was purified by flash silica gel chromatography (ISCO®; 12 g SepaFlash® Silica Flash Column, eluent: 0-20% ethyl acetate/petroleum ether gradient @ 60 mL/min).
- reaction mixture was concentrated to afford a residue which was purified by prep-HPLC (column: Phenomenex luna C18150*25mm* 10um; mobile phase: [water (formic acid)- acetonitrile]; gradient: 40%-70%, over 30 min) to produce 6-chloro-4-[4-[(1R)-1-(5- chloro-2-pyridyl)-2,2,2-trifluoro-ethyl]-4-hydroxy-1-piperidyl]-1-methyl-2-oxo-1,5- naphthyridine-3-carbonitrile (53.9 mg, 0.1 mmol, 38% yield, 97% purity) as a yellow solid.
- Examples 92-156 The following compounds were prepared using the requisite halide and amine in an analogous fashion to the compounds above.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Disclosed herein are compounds of Formula (I), or a pharmaceutically acceptable salt thereof, which inhibit the activity of one or both of diacylglycerol kinase alpha (DGKα). These compounds are useful in the treatment of proliferative diseases (e.g., cancer) and viral infections.
Description
DGK TARGETING COMPOUNDS AND USES THEREOF RELATED APPLICATIONS This application claims priority to U.S. Provisional Application No.63/530,352, filed on August 2, 2023. The entire contents of the foregoing application are expressly incorporated herein by reference. BACKGROUND Human cancers harbor numerous genetic and epigenetic alterations, generating neoantigens potentially recognizable by the immune system (Sjoblom et al. (2006) Science 314:268–74). The adaptive immune system, comprised of T and B lymphocytes, has powerful anti-cancer potential, with a broad capacity and exquisite specificity to respond to diverse tumor antigens. Further, the immune system demonstrates considerable plasticity and a memory component. The successful harnessing of all these attributes of the adaptive immune system would make immunotherapy unique among all cancer treatment modalities. However, although an endogenous immune response to cancer is observed in preclinical models and patients, this response is ineffective, and established cancers are viewed as “self” and tolerated by the immune system. Contributing to this state of tolerance, tumors may exploit several distinct mechanisms to actively subvert anti-tumor immunity. These mechanisms include dysfunctional T-cell signaling (Mizoguchi et al. (1992) Science 258:1795–98), suppressive regulatory cells (Facciabene et al. (2012) Cancer Res.72:2162–71), and the co- opting of endogenous “immune checkpoints,” which serve to down-modulate the intensity of adaptive immune responses and protect normal tissues from collateral damage, by tumors to evade immune destruction (Topalian et al. (2012) Curr. Opin. Immunol.24:1–6; Mellman et al. (2011) Nature 480:480–89). Accordingly, new and novel anticancer agents that are safe and effective in restoring T-cell activation, lowering antigen threshold, enhancing antitumor functionality, and/or overcoming the suppressive effects of one or more endogenous immune checkpoints are needed in the art for the treatment of cancer. SUMMARY Disclosed herein are compounds that have activity as inhibitors of diacylglycerol kinase alpha (DGKα). Additionally, in embodiments, the disclosed compounds cause the degradation of DGKα.
In embodiments, compounds described herein have desirable efficacy, stability, bioavailability, therapeutic index, and toxicity values that are important to their use as pharmaceuticals. In embodiments, provided herein are compounds of having the structure of Formula (I):
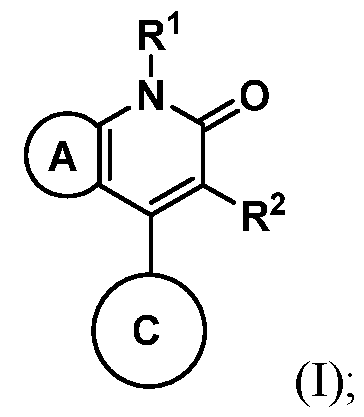 or a pharmaceutically acceptable salt thereof, wherein: Ring A is an optionally substituted phenyl or optionally substituted 5- to 6-membered monocyclic heteroaryl; Ring C is a 5- to 6-membered monocyclic heterocyclyl, an optionally substituted 8- to 15-membered bicyclic heterocyclyl,
or a pharmaceutically acceptable salt thereof, wherein: Ring A is an optionally substituted phenyl or optionally substituted 5- to 6-membered monocyclic heteroaryl; Ring C is a 5- to 6-membered monocyclic heterocyclyl, an optionally substituted 8- to 15-membered bicyclic heterocyclyl,
 Ring D is an optionally substituted phenyl or an optionally substituted 5- to 6- membered monocyclic heteroaryl; RC1 and RC2 are each independently H, C1-4alkyl, C1-4haloalkyl, optionally substituted C3-6cycloalkyl, optionally substituted 4- to 6-membered heterocyclyl, or optionally substituted phenyl; R1 is C1-4alkyl; R2 is H, halogen, SO2CF3, -N(Ra)2, NO2, SO2Ra, C≡N, C(Rb)3, CORa, CHO, CO2Ra, CON(Ra)2, or N=O; each Ra is independently H or C1-4alkyl; and Rb is halogen; provided that the compound of Formula I is not 2-chloro-4-(4-hydroxy-4-(1-(5- (trifluoromethoxy)pyridin-2-yl)propyl)piperidin-1-yl)-7-methyl-6-oxo-6,7-dihydrothieno[2,3- b]pyridine-5-carbonitrile, or a pharmaceutically acceptable or stereoisomer thereof; and
provided that (i) when Ring A is pyridinyl or thiazoyl then Ring
Ring D is an optionally substituted phenyl or an optionally substituted 5- to 6- membered monocyclic heteroaryl; RC1 and RC2 are each independently H, C1-4alkyl, C1-4haloalkyl, optionally substituted C3-6cycloalkyl, optionally substituted 4- to 6-membered heterocyclyl, or optionally substituted phenyl; R1 is C1-4alkyl; R2 is H, halogen, SO2CF3, -N(Ra)2, NO2, SO2Ra, C≡N, C(Rb)3, CORa, CHO, CO2Ra, CON(Ra)2, or N=O; each Ra is independently H or C1-4alkyl; and Rb is halogen; provided that the compound of Formula I is not 2-chloro-4-(4-hydroxy-4-(1-(5- (trifluoromethoxy)pyridin-2-yl)propyl)piperidin-1-yl)-7-methyl-6-oxo-6,7-dihydrothieno[2,3- b]pyridine-5-carbonitrile, or a pharmaceutically acceptable or stereoisomer thereof; and
provided that (i) when Ring A is pyridinyl or thiazoyl then Ring
 wherein one of RC1 and RC2 is: (a) C1-4haloalkyl, optionally substituted C3-6cycloalkyl, or optionally substituted 4- to 6-membered heterocyclyl when Ring A is pyridinyl; or (b) C1-4alkyl, C1-4haloalkyl, optionally substituted C3-6cycloalkyl, optionally substituted 4- to 6-membered heterocyclyl, or optionally substituted phenyl when Ring A is thiazoyl. Also disclosed herein are pharmaceutical compositions comprising one or more compounds disclosed herein and a pharmaceutically acceptable excipient. In embodiments, disclosed herein are methods treating cancer, comprising administering a therapeutically effective amount of one or more compounds disclosed herein, or a pharmaceutical composition thereof, to a subject in need thereof. In embodiments, disclosed herein are methods of inhibiting the activity of diacylglycerol kinase alpha (DGKα), comprising administering a therapeutically effective amount one or more compounds disclosed herein, or a pharmaceutical composition thereof, to a subject in need thereof. DETAILED DESCRIPTION Diacylglycerol kinases (DGKs) are lipid kinases that mediate the conversion of diacylglycerol to phosphatidic acid thereby terminating T-cell functions propagated through the TCR signaling pathway. Thus, DGKs serve as intracellular checkpoints, and inhibition of DGKs is expected to enhance T-cell signaling pathways and T-cell activation. Knock-out mouse models of DGKα have shown a hyper-responsive T-cell phenotype and improved anti- tumor immune activity (Zha Y et al. Nature Immunology, (2006) 12:1343; Olenchock B. A. et al., Nature (2006) 11: 1174–81). Furthermore, tumor infiltrating lymphocytes isolated from human renal cell carcinoma patients were observed to overexpress DGKα that resulted in inhibited T-cell function (Prinz, P. U. et al. J Immunology (2012) 12:5990–6000). Thus,
DGKα may be viewed as a target for cancer immunotherapy (Riese M. J. et al. Front Cell Dev Biol. (2016) 4: 108; Chen, S. S. et al. Front Cell Dev Biol. (2016) 4: 130; Avila-Flores, A. et al. Immunology and Cell Biology (2017) 95: 549-563; Noessner, E. Front Cell Dev Biol. (2017) 5: 16; Krishna, S., et al. Front Immunology (2013) 4:178; Jing, W. et al. Cancer Research (2017) 77: 5676–86). There remains a need for compounds useful as inhibitors of DGKα. Accordingly, disclosed herein are compounds that have activity as inhibitors of DGKα. In embodiments, the disclosed compounds selectively inhibit DGKα. In embodiments, the disclosed compounds are selective inhibitors for DGKα over other diacylglycerol kinases (e.g., DGKζ). In embodiments, the disclosed compounds cause the degradation of DGKα. In embodiments, the disclosed compounds cause the selective degradation of DGKα. In embodiments, the disclosed compounds cause the selective degradation of DGKα over other diacylglycerol kinases (e.g., DGKζ). The disclosed compounds can be used to treat certain diseases or disorders. In embodiments, the disclosed compounds can be used for the treatment of certain cancers (e.g., colon cancer, pancreatic cancer, breast cancer, prostate cancer, lung cancer, ovarian cancer, cervical cancer, renal cancer, head and/or neck cancer, lymphoma, lymphoma, leukemia, and melanoma). Definitions Unless otherwise defined herein, scientific, and technical terms used in this application shall have the meanings that are commonly understood by those of ordinary skill in the art. Generally, nomenclature used in connection with, and techniques of, chemistry, cell and tissue culture, molecular biology, cell and cancer biology, neurobiology, neurochemistry, virology, immunology, microbiology, pharmacology, genetics and protein and nucleic acid chemistry, described herein, are those well-known and commonly used in the art. The methods and techniques of the present disclosure are generally performed, unless otherwise indicated, according to conventional methods well-known in the art and as described in various general and more specific references that are cited and discussed throughout this specification. See, e.g., “Principles of Neural Science,” McGraw-Hill Medical, New York, N.Y. (2000); Motulsky, “Intuitive Biostatistics”, Oxford University
Press, Inc. (1995); Lodish et al. “Molecular Cell Biology, 4th ed.,” W. H. Freeman & Co., New York (2000); Griffiths et al. “Introduction to Genetic Analysis, 7th ed. ,” W. H. Freeman & Co., N.Y. (1999); and Gilbert et al. “Developmental Biology, 6th ed.,” Sinauer Associates, Inc., Sunderland, M A (2000). Chemistry terms used herein are used according to conventional usage in the art, as exemplified by “The McGraw-Hill Dictionary of Chemical Terms,” Parker S., Ed., McGraw- Hill, San Francisco, Calif. (1985). All the above, and all other publications, patents, and published patent applications referred to in this application are specifically incorporated by reference herein. In case of conflict, the present specification, including its specific definitions, will control. As used herein, the term “cell” is meant to refer to a cell that is in vitro, ex vivo, or in vivo. In embodiments, an ex vivo cell can be part of a tissue sample excised from an organism such as a mammal. In embodiments, an in vitro cell can be a cell in a cell culture. In embodiments, an in vivo cell is a cell living in an organism such as a mammal. The term “DGKα degrader” refers to an agent that targets the DGKα enzyme resulting in its degradation. The term “DGKα inhibitor” refers to an agent capable of inhibiting the enzymatic activity of diacylglycerol kinase alpha (DGKα) in T-cells resulting in enhanced T-cell stimulation. The DGKα inhibitor can be a reversible DGKα inhibitor. “A reversible DGKα inhibitor” is a compound that reversibly inhibits DGKα enzyme activity either at the catalytic site or at a non-catalytic site. The terms “patient,” “subject,” and “individual” are used interchangeably herein and refer to either a human or a non-human animal. These terms include mammals, such as humans, primates, livestock animals (including bovines, porcines, etc.), companion animals (e.g., canines, felines, etc.) and rodents (e.g., mice and rats). In embodiments, the subject is a human. As used herein, the term “selective inhibitors of DGKα” refers to a compound’s to selectively inhibit the activity of DGKα as compared to other diacylglycerol kinases (e.g., DGKζ).
As used herein, the term “selective degraders of DGKα” refers to a compound’s to selectively degrade DGKα as compared to other diacylglycerol kinases (e.g., DGKζ). “Treating” a condition or patient, and “treatment” refer to taking steps to obtain beneficial or desired results, including clinical results, via administration of a compound or composition of the present invention. Beneficial or desired results include, but are not limited to, alleviation or amelioration of one or more symptoms or conditions, diminishment of extent of disease, stabilized (i.e., not worsening) state of disease, preventing spread of disease, delay or slowing of disease progression, amelioration or palliation of the disease state, and remission (whether partial or total. “Treatment” also can mean prolonging survival as compared to expected survival if not receiving treatment. “Administering” or “administration of” a compound or a composition of the invention to a subject can be carried out using any of a variety of routes and methods known to those skilled in the art based on the directions of an attending healthcare provider. For example, a compound or pharmaceutical composition of the invention can be administered to a subject in need of such treatment by any of the following routes: intravenously, arterially, intradermally, intramuscularly, intraperitoneally, subcutaneously, ocularly, sublingually, buccally, orally (by ingestion), intranasally (by inhalation), intravaginally, intraspinally, intrathecally, intracerebrally, and transdermally (by absorption through the skin). A compound or composition of the present invention can also appropriately be introduced by rechargeable or biodegradable polymeric devices or other devices, e.g., patches and pumps, or formulations, which provide for the extended, slow, delayed, or controlled release of the compound or composition. Administering also can be performed, for example, once, a plurality of times, and/or over one or more extended periods. Appropriate methods of administering a compound or pharmaceutical composition of the invention to the subject will also depend on a variety of factors such as, for example, age, weight, gender, and physical condition of the subject, as well as the chemical and biological properties of the compound or pharmaceutical composition (e.g., solubility, absorption, bioavailability, metabolism, stability, and toxicity). In embodiments, a compound or pharmaceutical composition of present invention is administered orally, e.g., to a subject by ingestion. In embodiments, the orally administered
compound or pharmaceutical composition is in a controlled release (e.g., a delayed release, extended release, or slow release) formulation. As used herein, the phrase “conjoint administration” refers to any form of administration of two or more different therapeutic agents such that the second agent is administered while the previously administered therapeutic agent is still effective in the body (e.g., the two agents are simultaneously effective in the patient, which may include synergistic effects of the two agents). For example, the different therapeutic compounds can be administered either in the same formulation or in separate formulations, either simultaneously or sequentially. Thus, an individual who receives such treatment can benefit from a combined effect of different therapeutic agents. A “therapeutically effective amount” or a “therapeutically effective dose” of a compound or pharmaceutical composition of the invention is an amount of the drug or composition that, when administered to a subject, will have the intended therapeutic effect. The full therapeutic effect does not necessarily occur by administration of one dose and may occur only after administration of a series of doses. Thus, a therapeutically effective amount may be administered in one or more administrations. The effective amount needed for a subject will depend, for example, upon the subject’s age, weight, health, gender, and the nature and extent of the condition (e.g., cancer) being treated. The attending healthcare provider will generally determine the effective amount for a given situation according to these and other factors. The term “alkyl” refers to saturated aliphatic groups, including straight-chain alkyl groups, branched-chain alkyl groups, cycloalkyl (alicyclic) groups, alkyl-substituted cycloalkyl groups, and cycloalkyl-substituted alkyl groups. In preferred embodiments, a straight chain or branched chain alkyl has six or fewer carbon atoms in its backbone (e.g., C1–C6 for straight chains, C3–C6 for branched chains), and more preferably four or less carbons in its backbone. The terms “halo,” “halogen,” and “halogen groups,” as used herein, refer to a substituent group from Group 17 of the periodic table of the elements and includes fluoro (-F), chloro (- Cl), bromo(-Br), and iodo (-I) substituent groups. The term “heteroaryl” or “heteroaromatic” includes aromatic single ring structures, preferably 5- to 7-membered rings, more preferably 5- to 6-membered rings, whose ring structures include at least one heteroatom, preferably one to four heteroatoms, more preferably one or two heteroatoms. The term “heteroaryl” also includes polycyclic ring
systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is heteroaromatic, e.g., the other cyclic rings can be selected from cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and heterocyclyls. Heteroaryl groups include, for example, pyrrole, furan, thiophene, imidazole, oxazole, thiazole, pyrazole, pyridine, pyrazine, pyridazine, and pyrimidine, and the like. The term “heteroatom” as used herein means an atom of any element other than carbon or hydrogen. Preferred heteroatoms are nitrogen, oxygen, and sulfur. The term “heterocyclic ring” or “heterocyclyl” refers to a non-aromatic ring structure, preferably 3- to 10-membered rings, more preferably 3- to 7-membered rings, whose ring structures include at least one heteroatom, preferably one to four heteroatoms, more preferably one or two heteroatoms. The term “heterocyclic ring” also includes polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is heterocyclic, e.g., the other cyclic rings can be selected from cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and heterocyclyls. Heterocyclyl groups include, for example, diazinane, imidazolidine, piperidine, piperazine, pyrrolidine, morpholine, lactones, lactams, octahydropyrrolo[3,4-c]pyrrole, and the like. The term “substituted” refers to moieties having substituents replacing a hydrogen on one or more carbons of the backbone. It will be understood that “substitution” or “substituted with” includes the implicit proviso that such substitution is in accordance with permitted valence of the substituted atom and the substituent, and that the substitution results in a stable compound, e.g., which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, etc. As used herein, the term “substituted” is contemplated to include all permissible substituents of organic compounds. In a broad aspect, the permissible substituents include acyclic and cyclic, branched, and unbranched, carbocyclic and heterocyclic, aromatic and non-aromatic substituents of organic compounds. The permissible substituents can be one or more and the same or different for appropriate organic compounds. For purposes of this invention, the heteroatoms such as nitrogen may have hydrogen substituents and/or any permissible substituents of organic compounds described herein which satisfy the valences of the heteroatoms. Substituents can include any substituents described herein, for example, a halogen, a hydroxyl, a carbonyl (such as a carboxyl, an alkoxycarbonyl, a formyl, or an acyl), a thiocarbonyl (such as a thioester, a
thioacetate, or a thioformate), an alkoxyl, a phosphoryl, a phosphate, a phosphonate, a phosphinate, an amino, an amido, an amidine, an imine, a cyano, a nitro, an azido, a sulfhydryl, an alkylthio, a sulfate, a sulfonate, a sulfamoyl, a sulfonamido, a sulfonyl, a heterocyclyl, an aralkyl, or an aromatic or heteroaromatic moiety. It will be understood by those skilled in the art that the moieties substituted on the hydrocarbon chain can themselves be substituted, if appropriate. Certain compounds described herein may exist in various stereoisomeric forms. Stereoisomers are compounds that differ only in their spatial arrangement. When a disclosed compound is named or depicted by structure with or without indicating stereochemistry, it is understood that the name or structure encompasses all possible stereoisomers, geometric isomers, including essentially pure stereo or geometric isomers, as well as combination thereof. When the stereochemical configuration at a chiral center in a compound having one or more chiral centers is depicted by its chemical name (e.g., where the configuration is indicated in the chemical name by “R” or “S”) or structure (e.g., the configuration is indicated by “wedge” bonds), the enrichment of the indicated configuration relative to the opposite configuration is greater than 50%, 60%, 70%, 80%, 90%, 99%, or 99.9%. “Enrichment of the indicated configuration relative to the opposite configuration” is a mole percent and is determined by dividing the number of compounds with the indicated stereochemical configuration at the chiral center(s) by the total number of all the compounds with the same or opposite stereochemical configuration in a mixture. Unless otherwise indicated, all isomers are arbitrarily assigned to their relative data points. That is, while the compound is depected as either “R” or S” absolute stereochemistry, the absolute stereochemistry may be opposite to what is indicated. Enantiomers are pairs of stereoisomers whose mirror images are not superimposable, most commonly because they contain an asymmetrically substituted carbon atom that acts as a chiral center. “Enantiomer” means one of a pair of molecules that are mirror images of each other and are not superimposable. Diastereomers are stereoisomers that contain two or more asymmetrically substituted carbon atoms. “Geometric isomers” are stereoisomers that differ in the orientation of substituent atoms in relationship to a carbon-carbon double bond, to a carbocyclyl ring, or to a bridged bicyclic system.
Enantiomeric and diastereomeric mixtures can be resolved into their component enantiomers or stereoisomers by well-known methods, such as chiral-phase gas chromatography, chiral- phase high performance liquid chromatography, crystallizing the compound as a chiral salt complex, or crystallizing the compound in a chiral solvent. Enantiomers and diastereomers can also be obtained from diastereomerically- or enantiomerically-pure intermediates, reagents, and catalysts by well-known asymmetric synthetic methods. Compounds of the Invention Disclosed herein are compounds that have activity as inhibitors of DGKα. In preferred embodiments, the disclosed compounds selectively inhibit DGKα. In embodiments, the disclosed compounds are selective inhibitors of DGKα over other diacylglycerol kinases (e.g., DGKζ). In embodiments, the disclosed compounds cause the degradation of DGKα. In embodiments, the disclosed compounds cause the selective degradation of DGKα. In embodiments, the disclosed compounds cause the selective degradation of DGKα over other diacylglycerol kinases (e.g., DGKζ). In embodiments, the disclosed compounds have desirable efficacy, stability, bioavailability, therapeutic index, and toxicity values that are important to their use as pharmaceuticals. In embodiments, disclosed herein is a compound of having a structure according to Formula
wherein one of RC1 and RC2 is: (a) C1-4haloalkyl, optionally substituted C3-6cycloalkyl, or optionally substituted 4- to 6-membered heterocyclyl when Ring A is pyridinyl; or (b) C1-4alkyl, C1-4haloalkyl, optionally substituted C3-6cycloalkyl, optionally substituted 4- to 6-membered heterocyclyl, or optionally substituted phenyl when Ring A is thiazoyl. Also disclosed herein are pharmaceutical compositions comprising one or more compounds disclosed herein and a pharmaceutically acceptable excipient. In embodiments, disclosed herein are methods treating cancer, comprising administering a therapeutically effective amount of one or more compounds disclosed herein, or a pharmaceutical composition thereof, to a subject in need thereof. In embodiments, disclosed herein are methods of inhibiting the activity of diacylglycerol kinase alpha (DGKα), comprising administering a therapeutically effective amount one or more compounds disclosed herein, or a pharmaceutical composition thereof, to a subject in need thereof. DETAILED DESCRIPTION Diacylglycerol kinases (DGKs) are lipid kinases that mediate the conversion of diacylglycerol to phosphatidic acid thereby terminating T-cell functions propagated through the TCR signaling pathway. Thus, DGKs serve as intracellular checkpoints, and inhibition of DGKs is expected to enhance T-cell signaling pathways and T-cell activation. Knock-out mouse models of DGKα have shown a hyper-responsive T-cell phenotype and improved anti- tumor immune activity (Zha Y et al. Nature Immunology, (2006) 12:1343; Olenchock B. A. et al., Nature (2006) 11: 1174–81). Furthermore, tumor infiltrating lymphocytes isolated from human renal cell carcinoma patients were observed to overexpress DGKα that resulted in inhibited T-cell function (Prinz, P. U. et al. J Immunology (2012) 12:5990–6000). Thus,
DGKα may be viewed as a target for cancer immunotherapy (Riese M. J. et al. Front Cell Dev Biol. (2016) 4: 108; Chen, S. S. et al. Front Cell Dev Biol. (2016) 4: 130; Avila-Flores, A. et al. Immunology and Cell Biology (2017) 95: 549-563; Noessner, E. Front Cell Dev Biol. (2017) 5: 16; Krishna, S., et al. Front Immunology (2013) 4:178; Jing, W. et al. Cancer Research (2017) 77: 5676–86). There remains a need for compounds useful as inhibitors of DGKα. Accordingly, disclosed herein are compounds that have activity as inhibitors of DGKα. In embodiments, the disclosed compounds selectively inhibit DGKα. In embodiments, the disclosed compounds are selective inhibitors for DGKα over other diacylglycerol kinases (e.g., DGKζ). In embodiments, the disclosed compounds cause the degradation of DGKα. In embodiments, the disclosed compounds cause the selective degradation of DGKα. In embodiments, the disclosed compounds cause the selective degradation of DGKα over other diacylglycerol kinases (e.g., DGKζ). The disclosed compounds can be used to treat certain diseases or disorders. In embodiments, the disclosed compounds can be used for the treatment of certain cancers (e.g., colon cancer, pancreatic cancer, breast cancer, prostate cancer, lung cancer, ovarian cancer, cervical cancer, renal cancer, head and/or neck cancer, lymphoma, lymphoma, leukemia, and melanoma). Definitions Unless otherwise defined herein, scientific, and technical terms used in this application shall have the meanings that are commonly understood by those of ordinary skill in the art. Generally, nomenclature used in connection with, and techniques of, chemistry, cell and tissue culture, molecular biology, cell and cancer biology, neurobiology, neurochemistry, virology, immunology, microbiology, pharmacology, genetics and protein and nucleic acid chemistry, described herein, are those well-known and commonly used in the art. The methods and techniques of the present disclosure are generally performed, unless otherwise indicated, according to conventional methods well-known in the art and as described in various general and more specific references that are cited and discussed throughout this specification. See, e.g., “Principles of Neural Science,” McGraw-Hill Medical, New York, N.Y. (2000); Motulsky, “Intuitive Biostatistics”, Oxford University
Press, Inc. (1995); Lodish et al. “Molecular Cell Biology, 4th ed.,” W. H. Freeman & Co., New York (2000); Griffiths et al. “Introduction to Genetic Analysis, 7th ed. ,” W. H. Freeman & Co., N.Y. (1999); and Gilbert et al. “Developmental Biology, 6th ed.,” Sinauer Associates, Inc., Sunderland, M A (2000). Chemistry terms used herein are used according to conventional usage in the art, as exemplified by “The McGraw-Hill Dictionary of Chemical Terms,” Parker S., Ed., McGraw- Hill, San Francisco, Calif. (1985). All the above, and all other publications, patents, and published patent applications referred to in this application are specifically incorporated by reference herein. In case of conflict, the present specification, including its specific definitions, will control. As used herein, the term “cell” is meant to refer to a cell that is in vitro, ex vivo, or in vivo. In embodiments, an ex vivo cell can be part of a tissue sample excised from an organism such as a mammal. In embodiments, an in vitro cell can be a cell in a cell culture. In embodiments, an in vivo cell is a cell living in an organism such as a mammal. The term “DGKα degrader” refers to an agent that targets the DGKα enzyme resulting in its degradation. The term “DGKα inhibitor” refers to an agent capable of inhibiting the enzymatic activity of diacylglycerol kinase alpha (DGKα) in T-cells resulting in enhanced T-cell stimulation. The DGKα inhibitor can be a reversible DGKα inhibitor. “A reversible DGKα inhibitor” is a compound that reversibly inhibits DGKα enzyme activity either at the catalytic site or at a non-catalytic site. The terms “patient,” “subject,” and “individual” are used interchangeably herein and refer to either a human or a non-human animal. These terms include mammals, such as humans, primates, livestock animals (including bovines, porcines, etc.), companion animals (e.g., canines, felines, etc.) and rodents (e.g., mice and rats). In embodiments, the subject is a human. As used herein, the term “selective inhibitors of DGKα” refers to a compound’s to selectively inhibit the activity of DGKα as compared to other diacylglycerol kinases (e.g., DGKζ).
As used herein, the term “selective degraders of DGKα” refers to a compound’s to selectively degrade DGKα as compared to other diacylglycerol kinases (e.g., DGKζ). “Treating” a condition or patient, and “treatment” refer to taking steps to obtain beneficial or desired results, including clinical results, via administration of a compound or composition of the present invention. Beneficial or desired results include, but are not limited to, alleviation or amelioration of one or more symptoms or conditions, diminishment of extent of disease, stabilized (i.e., not worsening) state of disease, preventing spread of disease, delay or slowing of disease progression, amelioration or palliation of the disease state, and remission (whether partial or total. “Treatment” also can mean prolonging survival as compared to expected survival if not receiving treatment. “Administering” or “administration of” a compound or a composition of the invention to a subject can be carried out using any of a variety of routes and methods known to those skilled in the art based on the directions of an attending healthcare provider. For example, a compound or pharmaceutical composition of the invention can be administered to a subject in need of such treatment by any of the following routes: intravenously, arterially, intradermally, intramuscularly, intraperitoneally, subcutaneously, ocularly, sublingually, buccally, orally (by ingestion), intranasally (by inhalation), intravaginally, intraspinally, intrathecally, intracerebrally, and transdermally (by absorption through the skin). A compound or composition of the present invention can also appropriately be introduced by rechargeable or biodegradable polymeric devices or other devices, e.g., patches and pumps, or formulations, which provide for the extended, slow, delayed, or controlled release of the compound or composition. Administering also can be performed, for example, once, a plurality of times, and/or over one or more extended periods. Appropriate methods of administering a compound or pharmaceutical composition of the invention to the subject will also depend on a variety of factors such as, for example, age, weight, gender, and physical condition of the subject, as well as the chemical and biological properties of the compound or pharmaceutical composition (e.g., solubility, absorption, bioavailability, metabolism, stability, and toxicity). In embodiments, a compound or pharmaceutical composition of present invention is administered orally, e.g., to a subject by ingestion. In embodiments, the orally administered
compound or pharmaceutical composition is in a controlled release (e.g., a delayed release, extended release, or slow release) formulation. As used herein, the phrase “conjoint administration” refers to any form of administration of two or more different therapeutic agents such that the second agent is administered while the previously administered therapeutic agent is still effective in the body (e.g., the two agents are simultaneously effective in the patient, which may include synergistic effects of the two agents). For example, the different therapeutic compounds can be administered either in the same formulation or in separate formulations, either simultaneously or sequentially. Thus, an individual who receives such treatment can benefit from a combined effect of different therapeutic agents. A “therapeutically effective amount” or a “therapeutically effective dose” of a compound or pharmaceutical composition of the invention is an amount of the drug or composition that, when administered to a subject, will have the intended therapeutic effect. The full therapeutic effect does not necessarily occur by administration of one dose and may occur only after administration of a series of doses. Thus, a therapeutically effective amount may be administered in one or more administrations. The effective amount needed for a subject will depend, for example, upon the subject’s age, weight, health, gender, and the nature and extent of the condition (e.g., cancer) being treated. The attending healthcare provider will generally determine the effective amount for a given situation according to these and other factors. The term “alkyl” refers to saturated aliphatic groups, including straight-chain alkyl groups, branched-chain alkyl groups, cycloalkyl (alicyclic) groups, alkyl-substituted cycloalkyl groups, and cycloalkyl-substituted alkyl groups. In preferred embodiments, a straight chain or branched chain alkyl has six or fewer carbon atoms in its backbone (e.g., C1–C6 for straight chains, C3–C6 for branched chains), and more preferably four or less carbons in its backbone. The terms “halo,” “halogen,” and “halogen groups,” as used herein, refer to a substituent group from Group 17 of the periodic table of the elements and includes fluoro (-F), chloro (- Cl), bromo(-Br), and iodo (-I) substituent groups. The term “heteroaryl” or “heteroaromatic” includes aromatic single ring structures, preferably 5- to 7-membered rings, more preferably 5- to 6-membered rings, whose ring structures include at least one heteroatom, preferably one to four heteroatoms, more preferably one or two heteroatoms. The term “heteroaryl” also includes polycyclic ring
systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is heteroaromatic, e.g., the other cyclic rings can be selected from cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and heterocyclyls. Heteroaryl groups include, for example, pyrrole, furan, thiophene, imidazole, oxazole, thiazole, pyrazole, pyridine, pyrazine, pyridazine, and pyrimidine, and the like. The term “heteroatom” as used herein means an atom of any element other than carbon or hydrogen. Preferred heteroatoms are nitrogen, oxygen, and sulfur. The term “heterocyclic ring” or “heterocyclyl” refers to a non-aromatic ring structure, preferably 3- to 10-membered rings, more preferably 3- to 7-membered rings, whose ring structures include at least one heteroatom, preferably one to four heteroatoms, more preferably one or two heteroatoms. The term “heterocyclic ring” also includes polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is heterocyclic, e.g., the other cyclic rings can be selected from cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and heterocyclyls. Heterocyclyl groups include, for example, diazinane, imidazolidine, piperidine, piperazine, pyrrolidine, morpholine, lactones, lactams, octahydropyrrolo[3,4-c]pyrrole, and the like. The term “substituted” refers to moieties having substituents replacing a hydrogen on one or more carbons of the backbone. It will be understood that “substitution” or “substituted with” includes the implicit proviso that such substitution is in accordance with permitted valence of the substituted atom and the substituent, and that the substitution results in a stable compound, e.g., which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, etc. As used herein, the term “substituted” is contemplated to include all permissible substituents of organic compounds. In a broad aspect, the permissible substituents include acyclic and cyclic, branched, and unbranched, carbocyclic and heterocyclic, aromatic and non-aromatic substituents of organic compounds. The permissible substituents can be one or more and the same or different for appropriate organic compounds. For purposes of this invention, the heteroatoms such as nitrogen may have hydrogen substituents and/or any permissible substituents of organic compounds described herein which satisfy the valences of the heteroatoms. Substituents can include any substituents described herein, for example, a halogen, a hydroxyl, a carbonyl (such as a carboxyl, an alkoxycarbonyl, a formyl, or an acyl), a thiocarbonyl (such as a thioester, a
thioacetate, or a thioformate), an alkoxyl, a phosphoryl, a phosphate, a phosphonate, a phosphinate, an amino, an amido, an amidine, an imine, a cyano, a nitro, an azido, a sulfhydryl, an alkylthio, a sulfate, a sulfonate, a sulfamoyl, a sulfonamido, a sulfonyl, a heterocyclyl, an aralkyl, or an aromatic or heteroaromatic moiety. It will be understood by those skilled in the art that the moieties substituted on the hydrocarbon chain can themselves be substituted, if appropriate. Certain compounds described herein may exist in various stereoisomeric forms. Stereoisomers are compounds that differ only in their spatial arrangement. When a disclosed compound is named or depicted by structure with or without indicating stereochemistry, it is understood that the name or structure encompasses all possible stereoisomers, geometric isomers, including essentially pure stereo or geometric isomers, as well as combination thereof. When the stereochemical configuration at a chiral center in a compound having one or more chiral centers is depicted by its chemical name (e.g., where the configuration is indicated in the chemical name by “R” or “S”) or structure (e.g., the configuration is indicated by “wedge” bonds), the enrichment of the indicated configuration relative to the opposite configuration is greater than 50%, 60%, 70%, 80%, 90%, 99%, or 99.9%. “Enrichment of the indicated configuration relative to the opposite configuration” is a mole percent and is determined by dividing the number of compounds with the indicated stereochemical configuration at the chiral center(s) by the total number of all the compounds with the same or opposite stereochemical configuration in a mixture. Unless otherwise indicated, all isomers are arbitrarily assigned to their relative data points. That is, while the compound is depected as either “R” or S” absolute stereochemistry, the absolute stereochemistry may be opposite to what is indicated. Enantiomers are pairs of stereoisomers whose mirror images are not superimposable, most commonly because they contain an asymmetrically substituted carbon atom that acts as a chiral center. “Enantiomer” means one of a pair of molecules that are mirror images of each other and are not superimposable. Diastereomers are stereoisomers that contain two or more asymmetrically substituted carbon atoms. “Geometric isomers” are stereoisomers that differ in the orientation of substituent atoms in relationship to a carbon-carbon double bond, to a carbocyclyl ring, or to a bridged bicyclic system.
Enantiomeric and diastereomeric mixtures can be resolved into their component enantiomers or stereoisomers by well-known methods, such as chiral-phase gas chromatography, chiral- phase high performance liquid chromatography, crystallizing the compound as a chiral salt complex, or crystallizing the compound in a chiral solvent. Enantiomers and diastereomers can also be obtained from diastereomerically- or enantiomerically-pure intermediates, reagents, and catalysts by well-known asymmetric synthetic methods. Compounds of the Invention Disclosed herein are compounds that have activity as inhibitors of DGKα. In preferred embodiments, the disclosed compounds selectively inhibit DGKα. In embodiments, the disclosed compounds are selective inhibitors of DGKα over other diacylglycerol kinases (e.g., DGKζ). In embodiments, the disclosed compounds cause the degradation of DGKα. In embodiments, the disclosed compounds cause the selective degradation of DGKα. In embodiments, the disclosed compounds cause the selective degradation of DGKα over other diacylglycerol kinases (e.g., DGKζ). In embodiments, the disclosed compounds have desirable efficacy, stability, bioavailability, therapeutic index, and toxicity values that are important to their use as pharmaceuticals. In embodiments, disclosed herein is a compound of having a structure according to Formula
 or a pharmaceutically acceptable salt thereof, wherein: Ring A is an optionally substituted phenyl or optionally substituted 5- to 6-membered monocyclic heteroaryl;
Ring C is a 5- to 6-membered monocyclic heterocyclyl, an optionally substituted 8- to 15-membered bicyclic heterocyclyl,
or a pharmaceutically acceptable salt thereof, wherein: Ring A is an optionally substituted phenyl or optionally substituted 5- to 6-membered monocyclic heteroaryl;
Ring C is a 5- to 6-membered monocyclic heterocyclyl, an optionally substituted 8- to 15-membered bicyclic heterocyclyl,
 Ring D is an optionally substituted phenyl or an optionally substituted 5- to 6- membered monocyclic heteroaryl; RC1 and RC2 are each independently H, C1-4alkyl, C1-4haloalkyl, optionally substituted C3-6cycloalkyl, optionally substituted 4- to 6-membered heterocyclyl, or optionally substituted phenyl; R1 is C1-4alkyl; R2 is H, halogen, SO2CF3, -N(Ra)2, NO2, SO2Ra, C≡N, C(Rb)3, CORa, CHO, CO2Ra, CON(Ra)2, or N=O; each Ra is independently H or C1-4alkyl; and Rb is halogen; provided that the compound of Formula I is not 2-chloro-4-(4-hydroxy-4-(1-(5- (trifluoromethoxy)pyridin-2-yl)propyl)piperidin-1-yl)-7-methyl-6-oxo-6,7-dihydrothieno[2,3- b]pyridine-5-carbonitrile, or a pharmaceutically acceptable or stereoisomer thereof; and provided that (i) when Ring A is pyridinyl or thiazoyl then Ring
Ring D is an optionally substituted phenyl or an optionally substituted 5- to 6- membered monocyclic heteroaryl; RC1 and RC2 are each independently H, C1-4alkyl, C1-4haloalkyl, optionally substituted C3-6cycloalkyl, optionally substituted 4- to 6-membered heterocyclyl, or optionally substituted phenyl; R1 is C1-4alkyl; R2 is H, halogen, SO2CF3, -N(Ra)2, NO2, SO2Ra, C≡N, C(Rb)3, CORa, CHO, CO2Ra, CON(Ra)2, or N=O; each Ra is independently H or C1-4alkyl; and Rb is halogen; provided that the compound of Formula I is not 2-chloro-4-(4-hydroxy-4-(1-(5- (trifluoromethoxy)pyridin-2-yl)propyl)piperidin-1-yl)-7-methyl-6-oxo-6,7-dihydrothieno[2,3- b]pyridine-5-carbonitrile, or a pharmaceutically acceptable or stereoisomer thereof; and provided that (i) when Ring A is pyridinyl or thiazoyl then Ring
 wherein one of RC1 and RC2 is: (a) C1-4haloalkyl, optionally substituted C3-6cycloalkyl, or optionally substituted 4- to 6-membered heterocyclyl when Ring A is pyridinyl; or (b) C1-4alkyl, C1-4haloalkyl, optionally substituted C3-6cycloalkyl, optionally substituted 4- to 6-membered heterocyclyl, or optionally substituted phenyl when Ring A is thiazoyl. In embodiments of Formula (I), disclosed herein are compounds having the structure according to Formula (II), (III), (IV), or (V):
wherein one of RC1 and RC2 is: (a) C1-4haloalkyl, optionally substituted C3-6cycloalkyl, or optionally substituted 4- to 6-membered heterocyclyl when Ring A is pyridinyl; or (b) C1-4alkyl, C1-4haloalkyl, optionally substituted C3-6cycloalkyl, optionally substituted 4- to 6-membered heterocyclyl, or optionally substituted phenyl when Ring A is thiazoyl. In embodiments of Formula (I), disclosed herein are compounds having the structure according to Formula (II), (III), (IV), or (V):

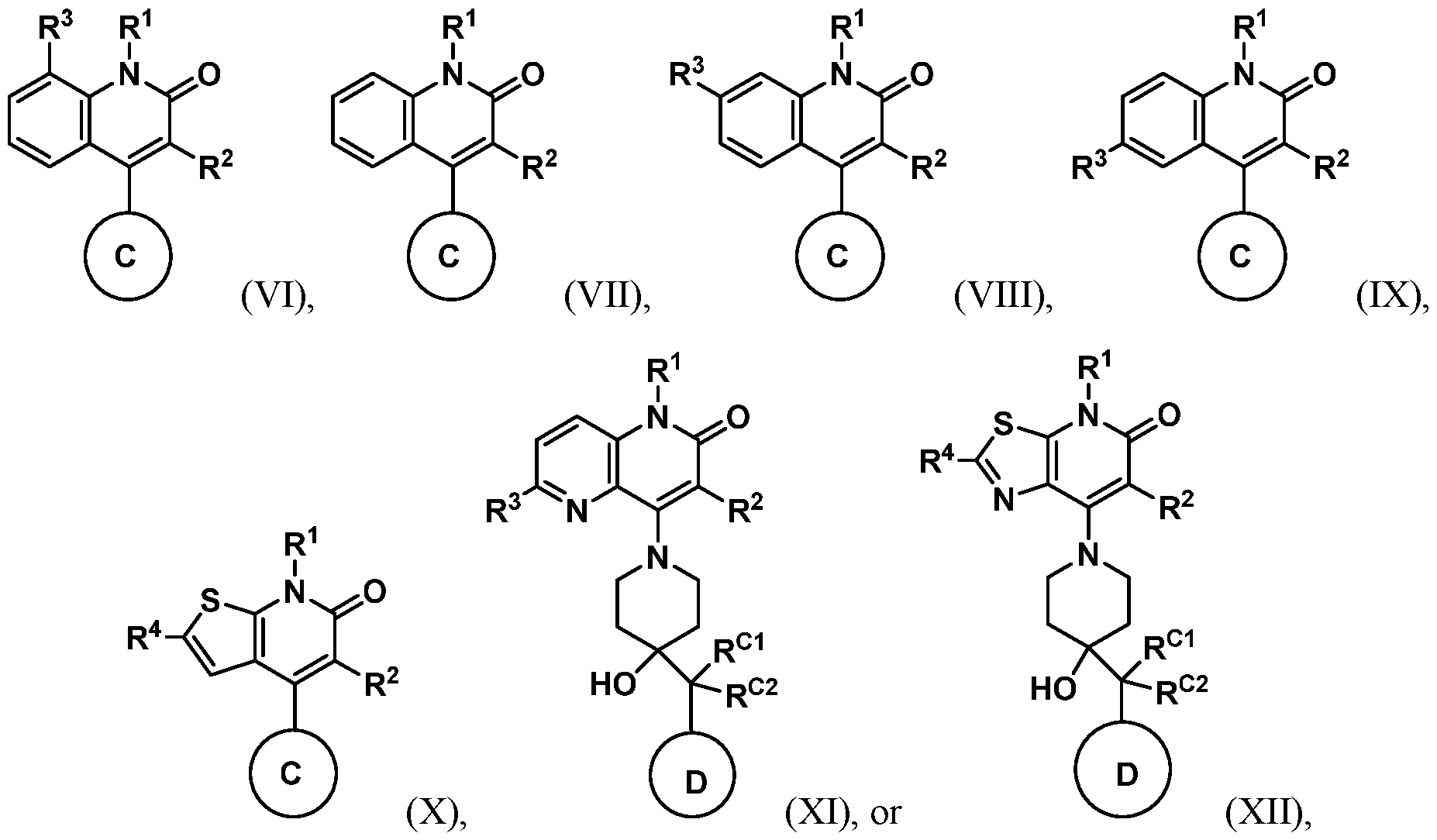












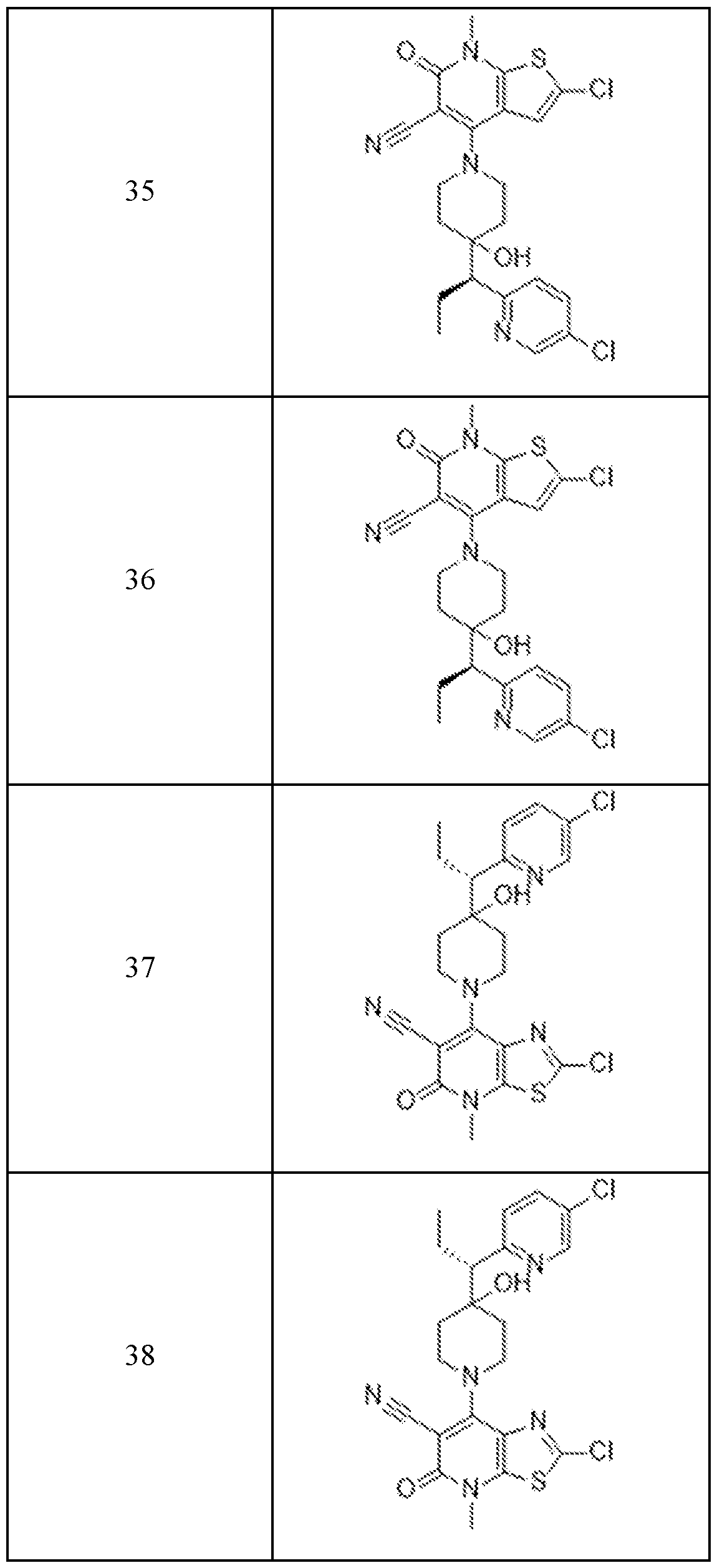
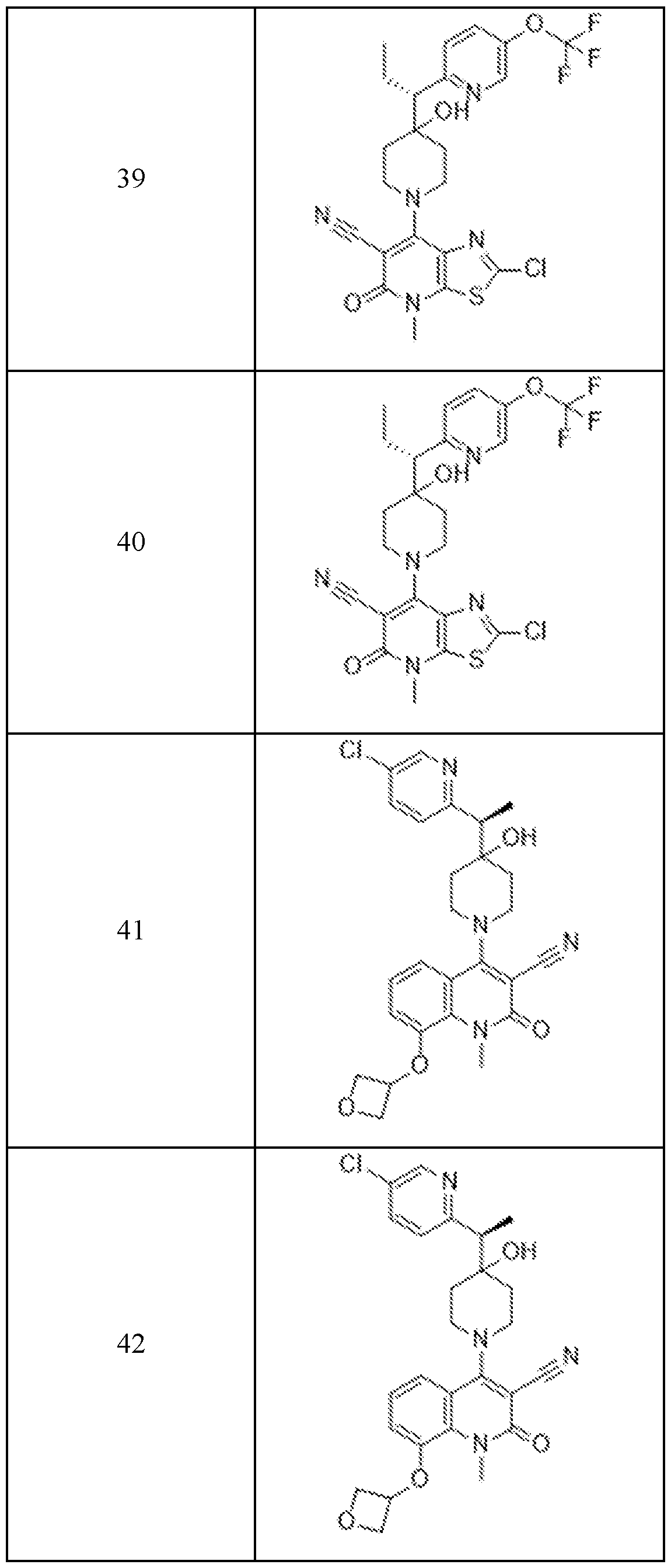





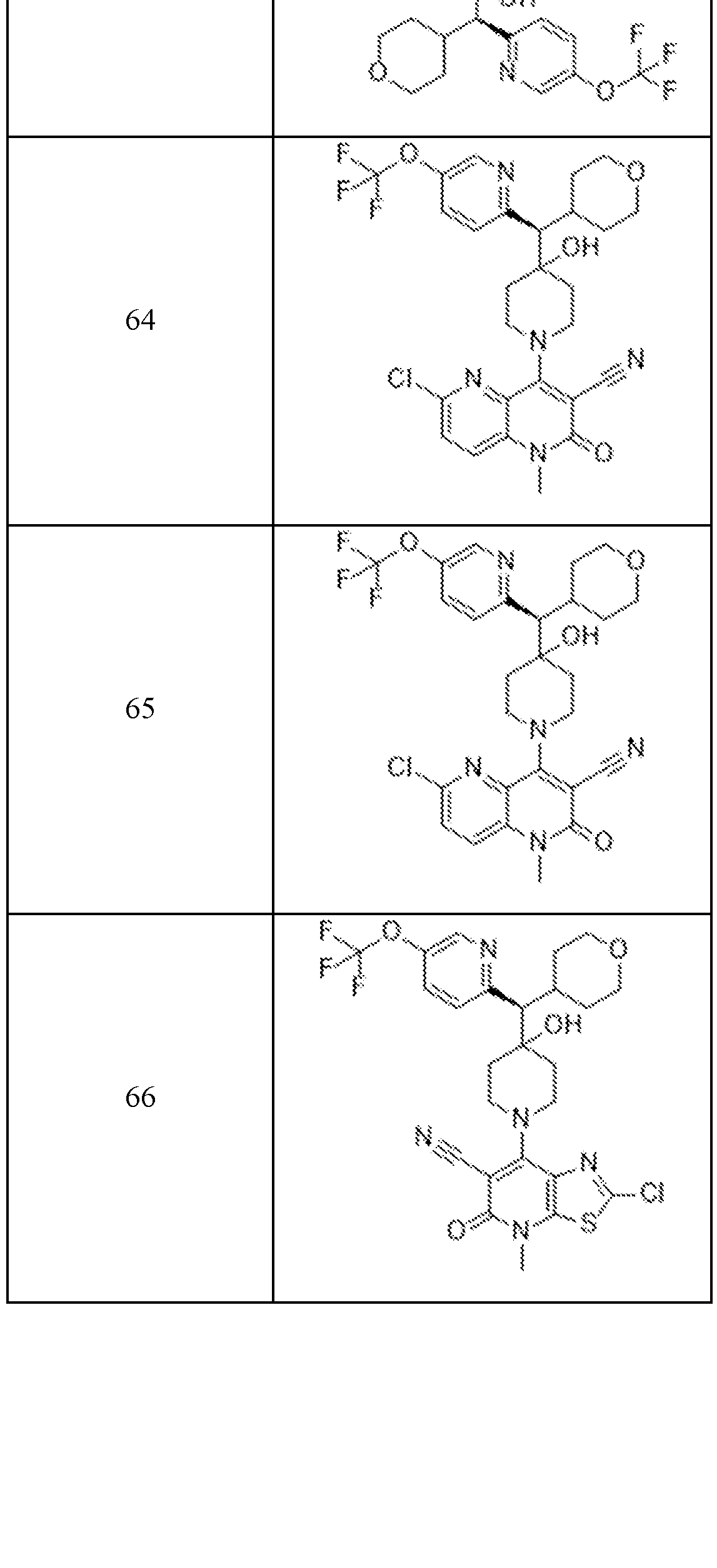
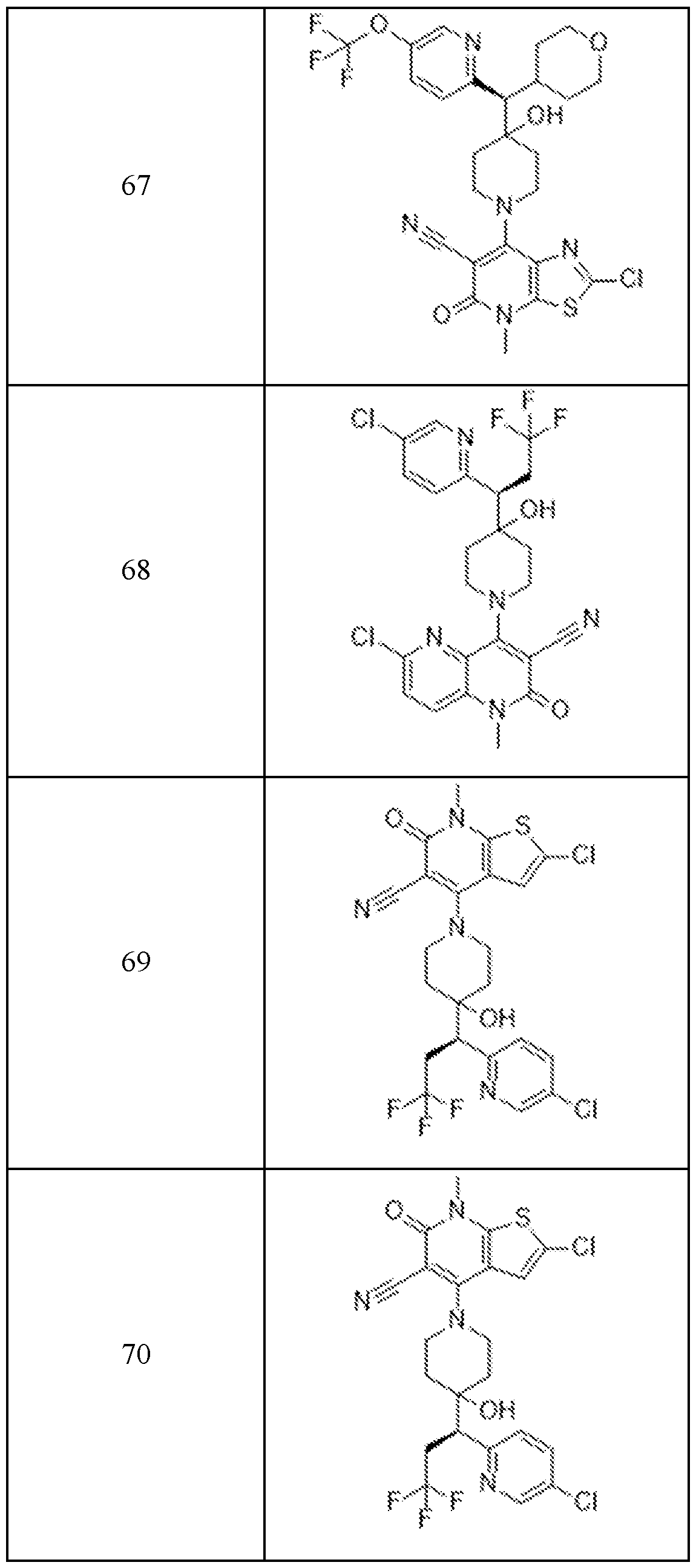



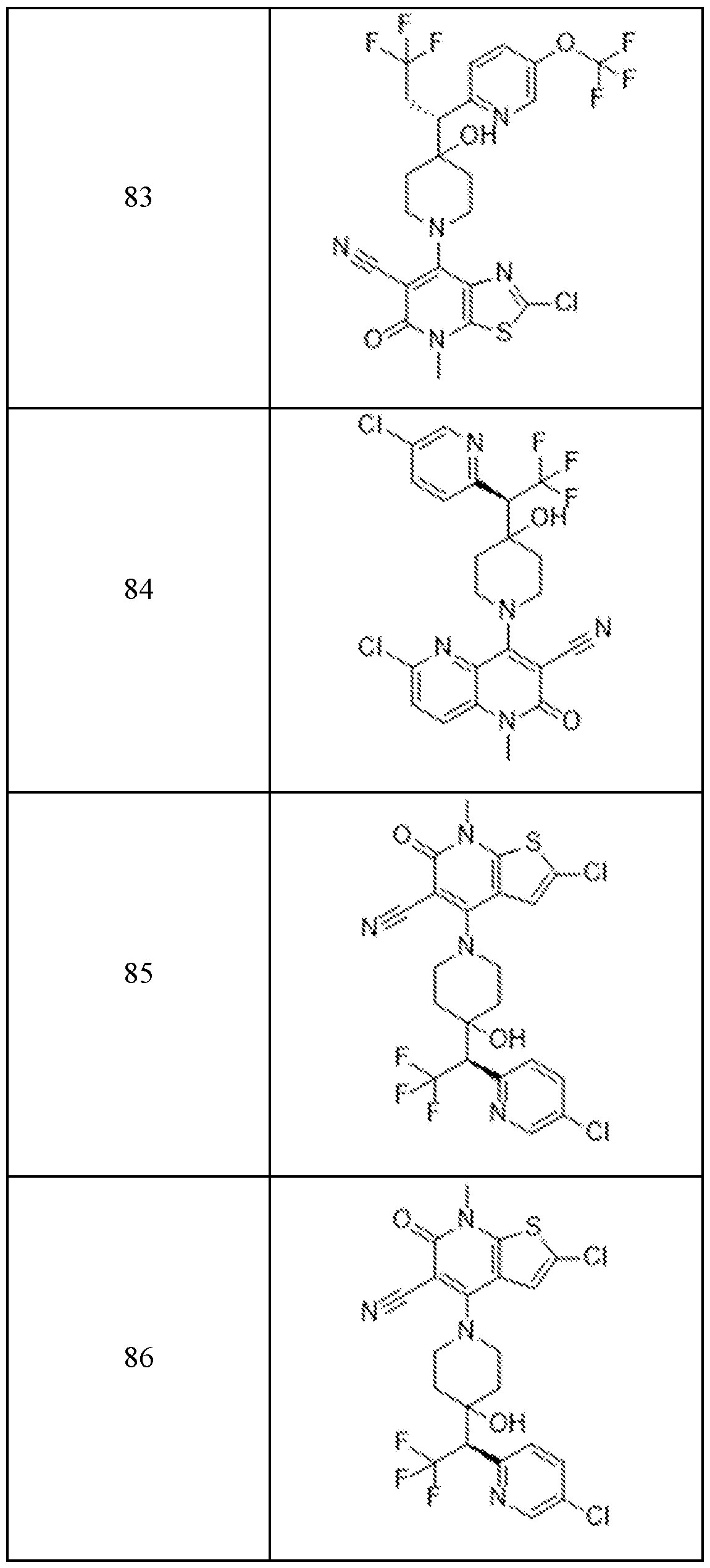
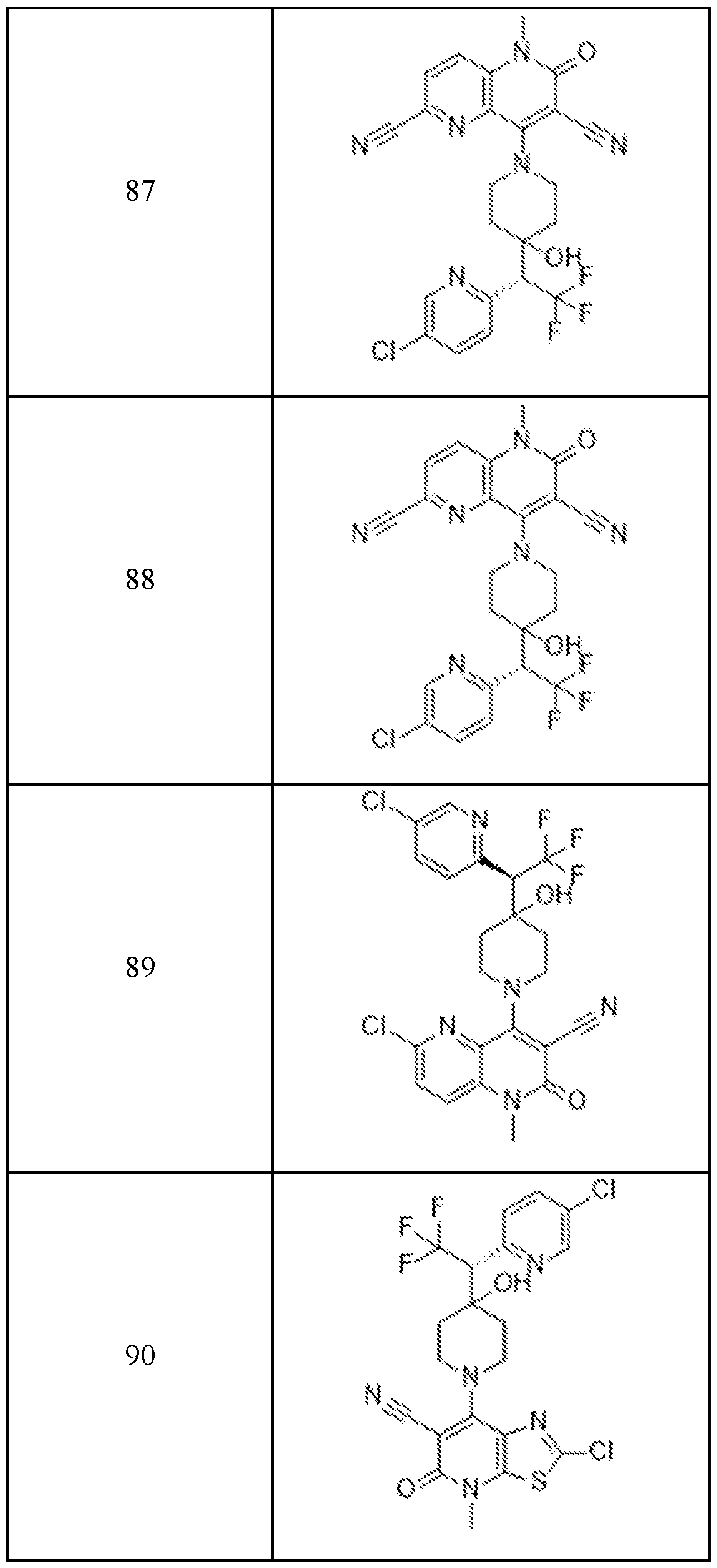
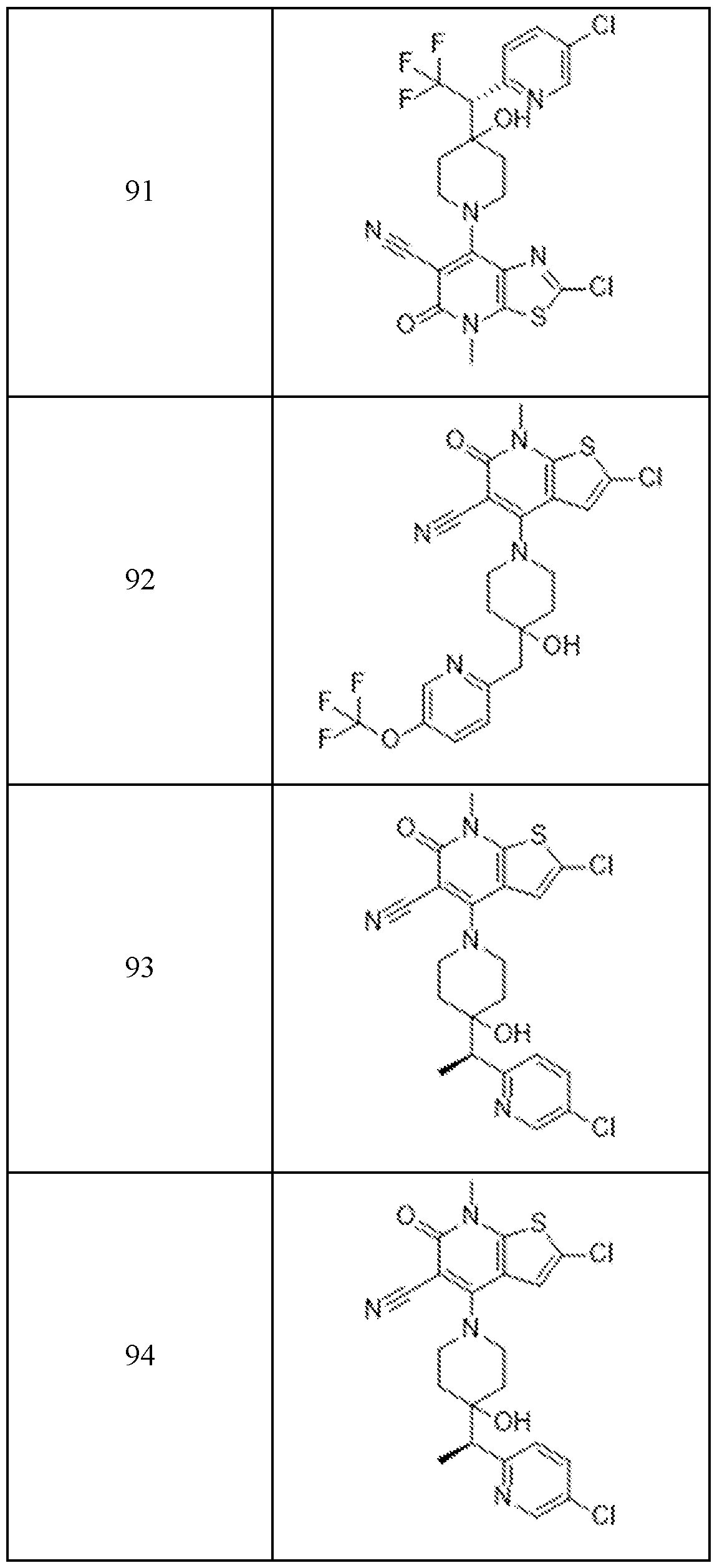

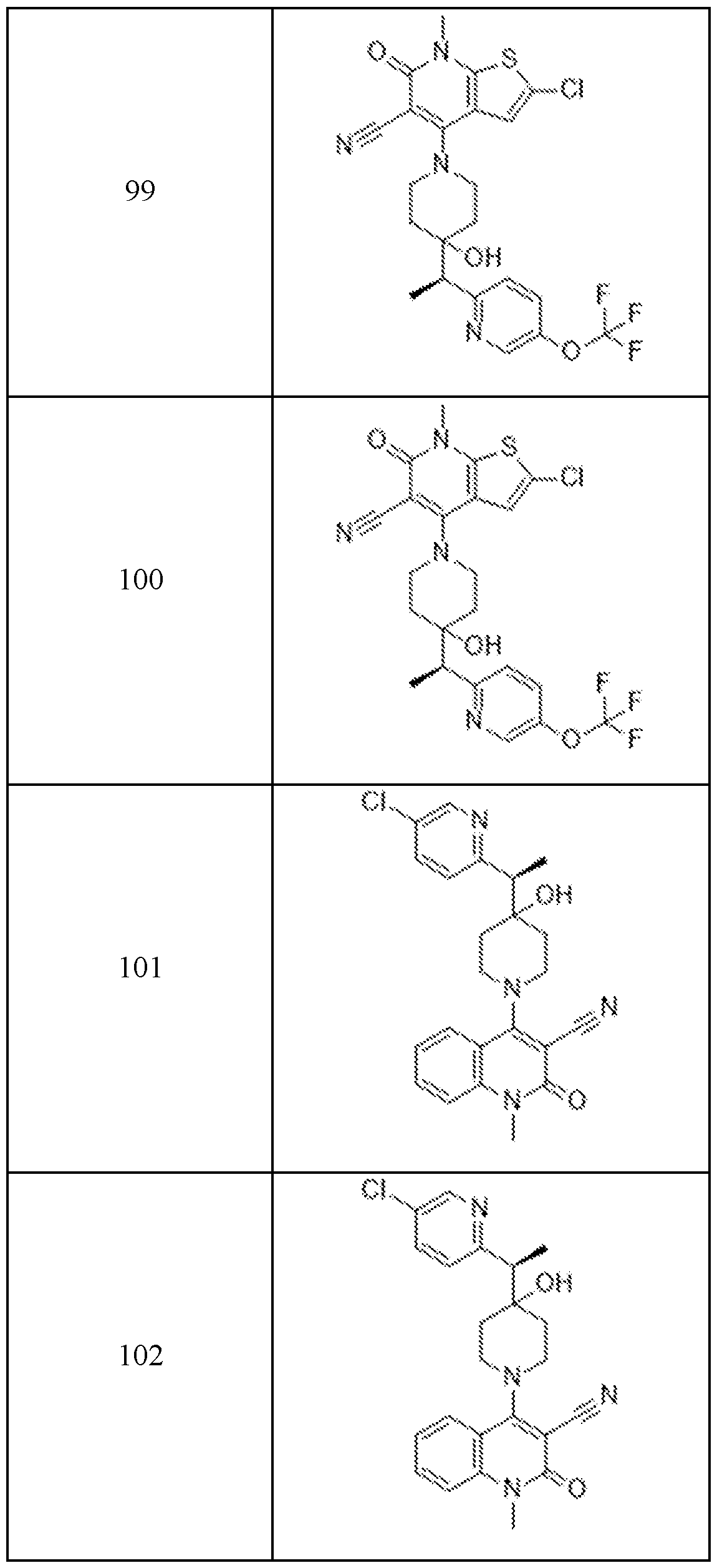



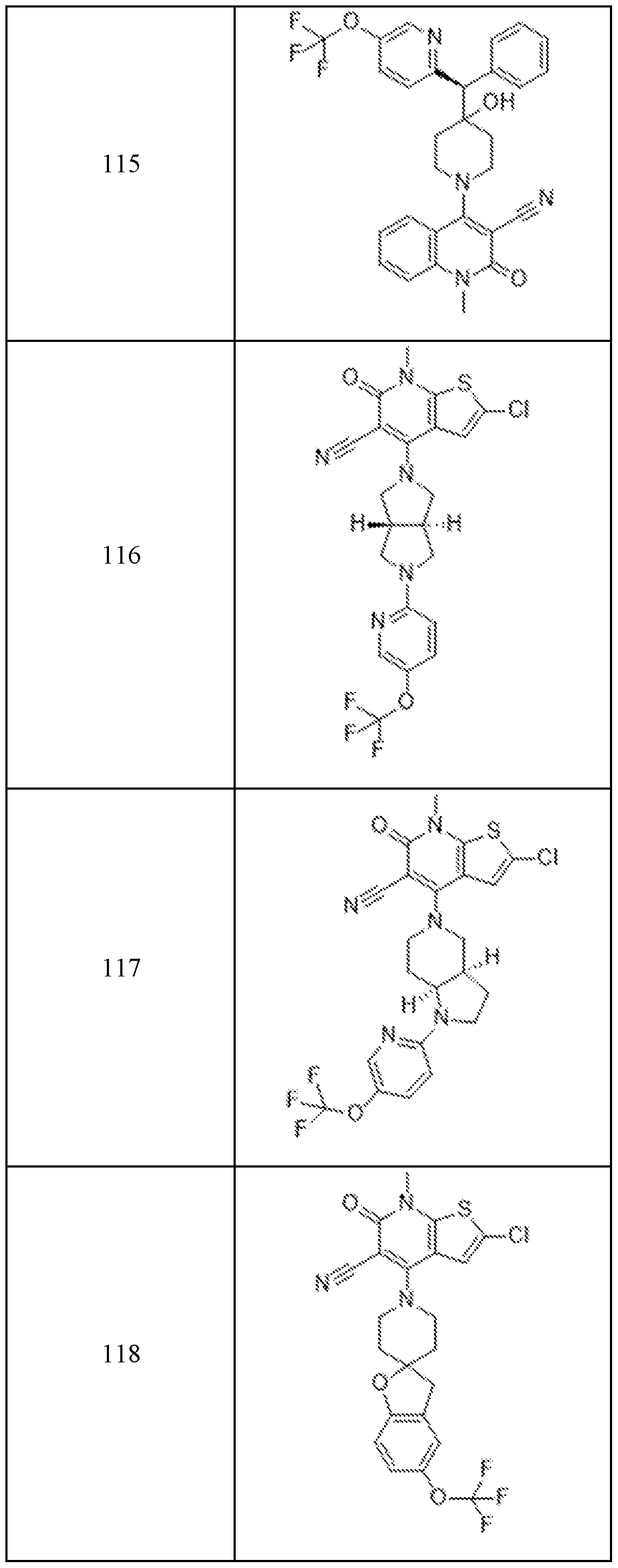
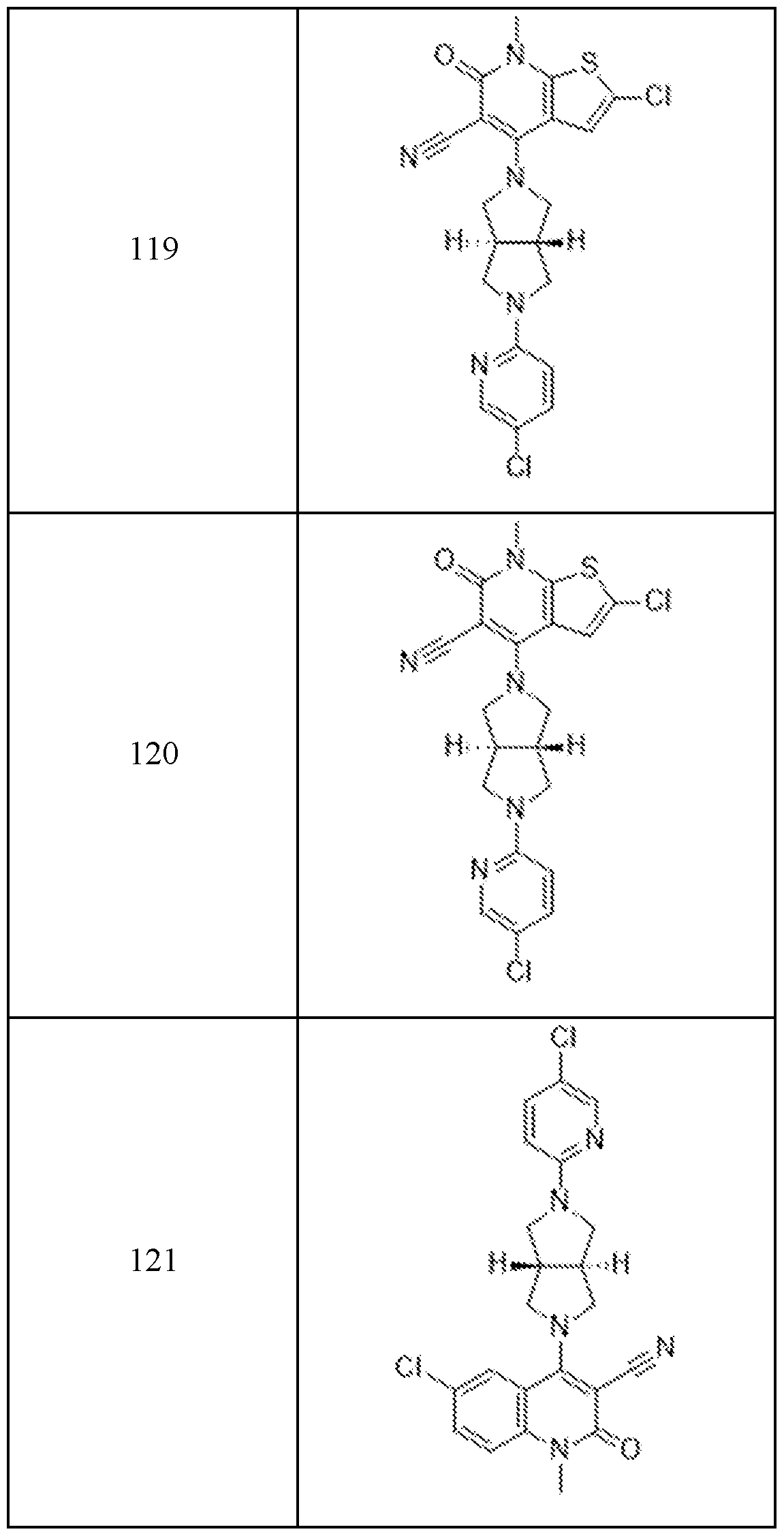



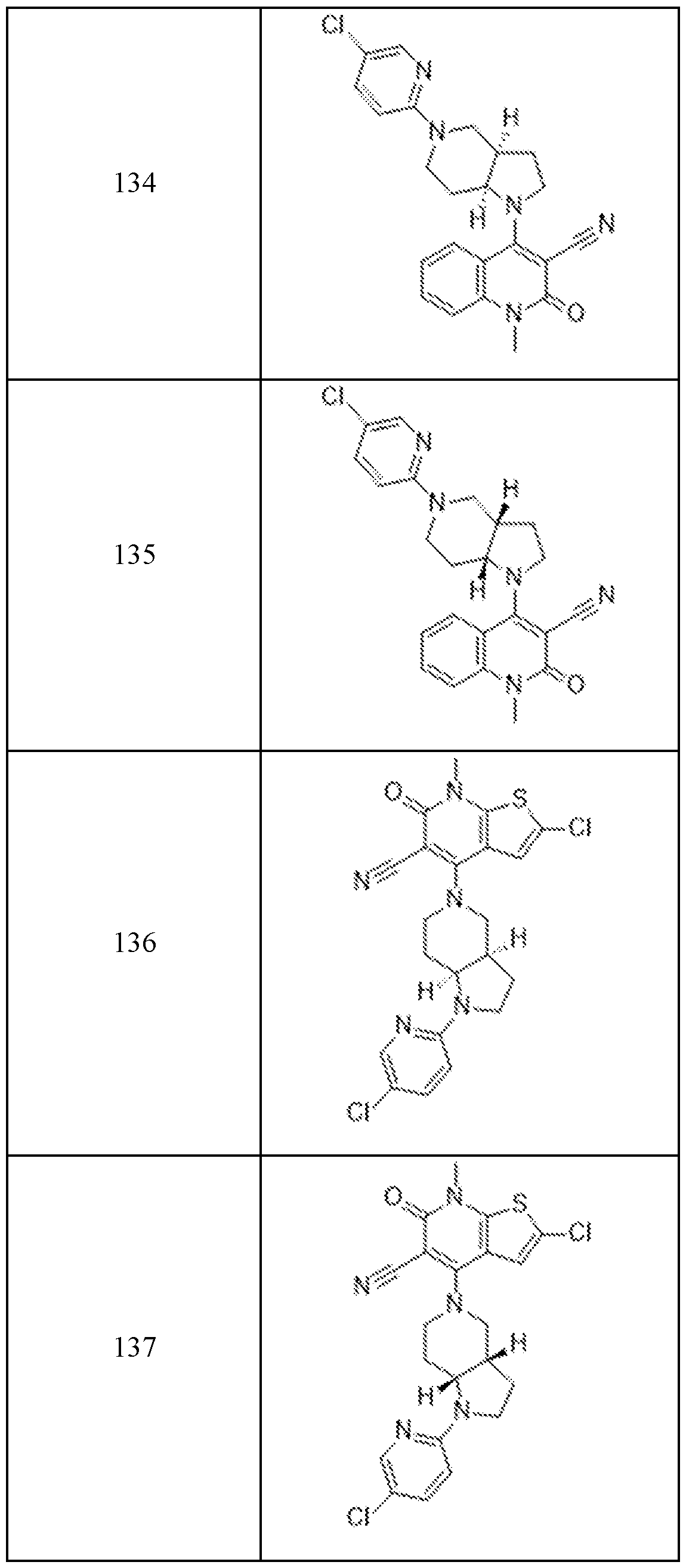


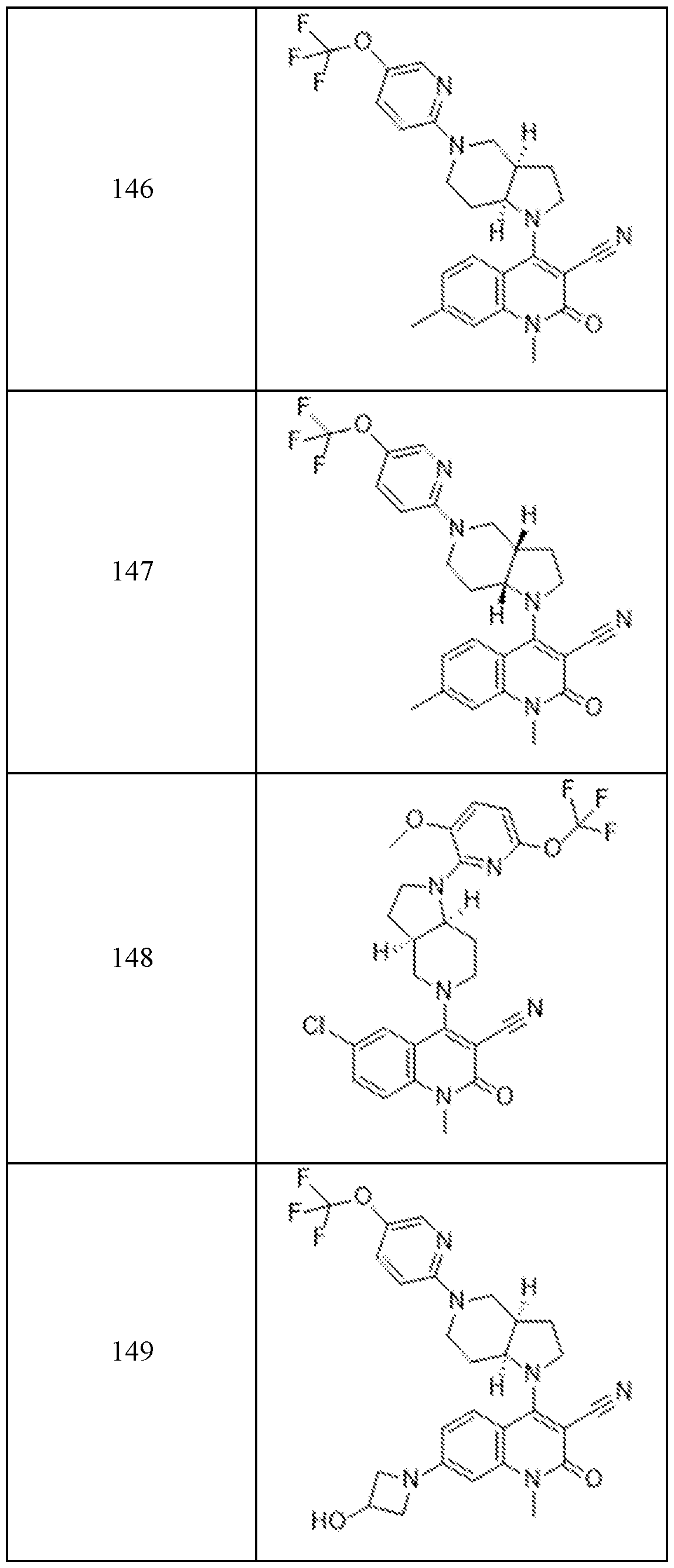
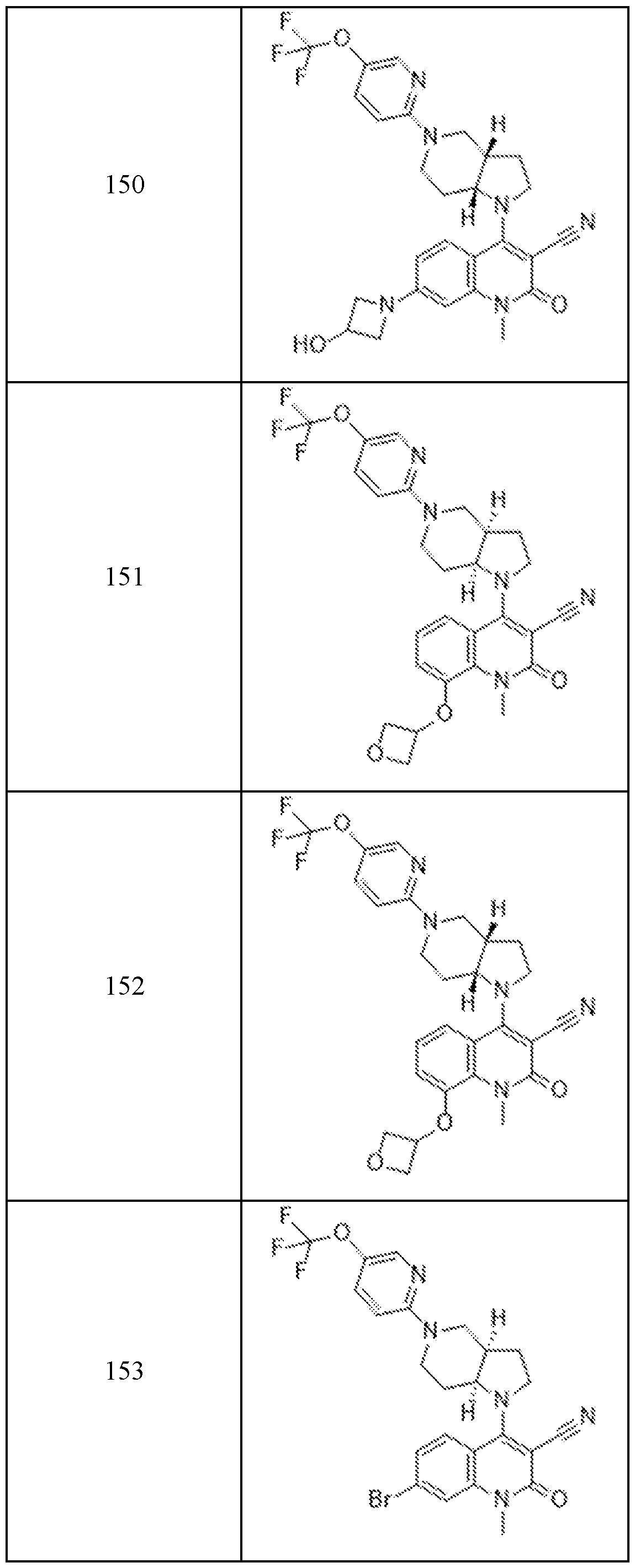
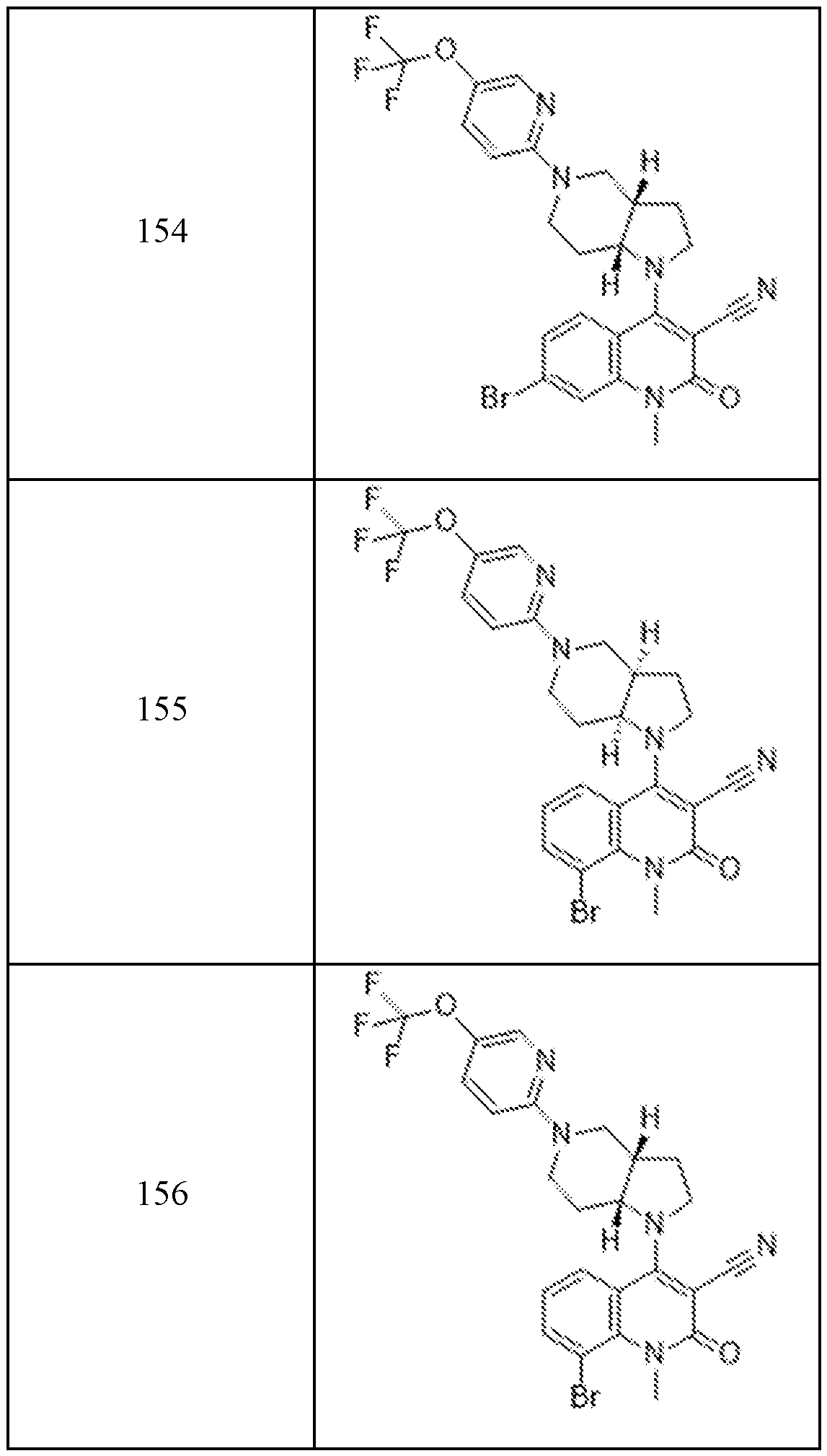


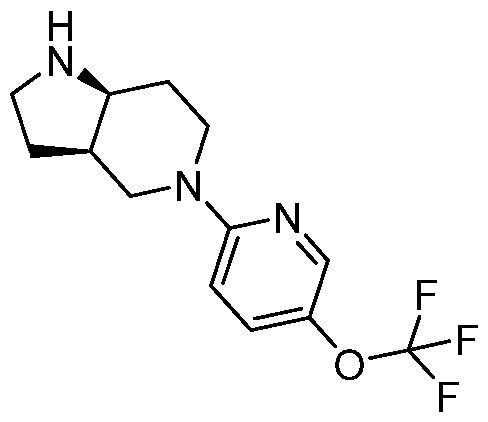



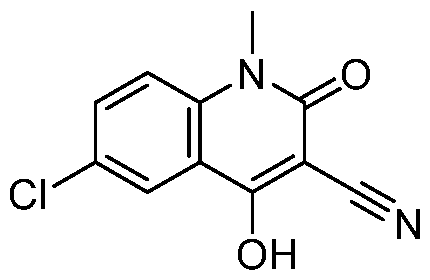
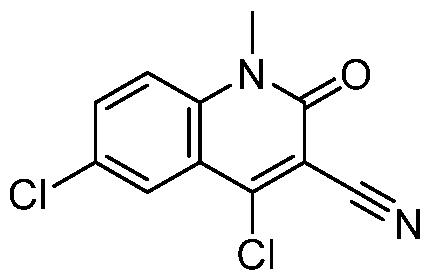

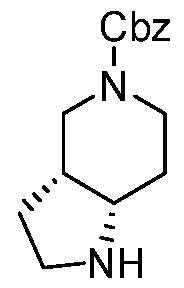


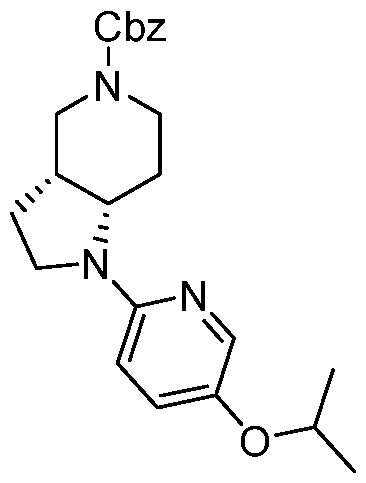






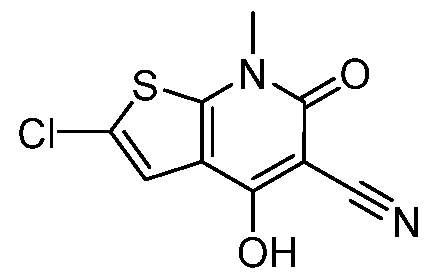


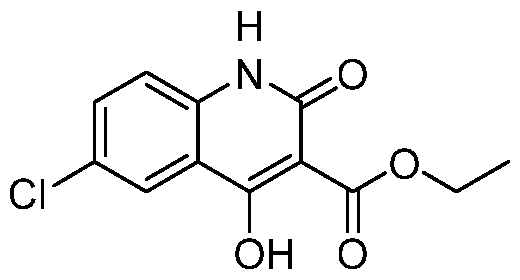





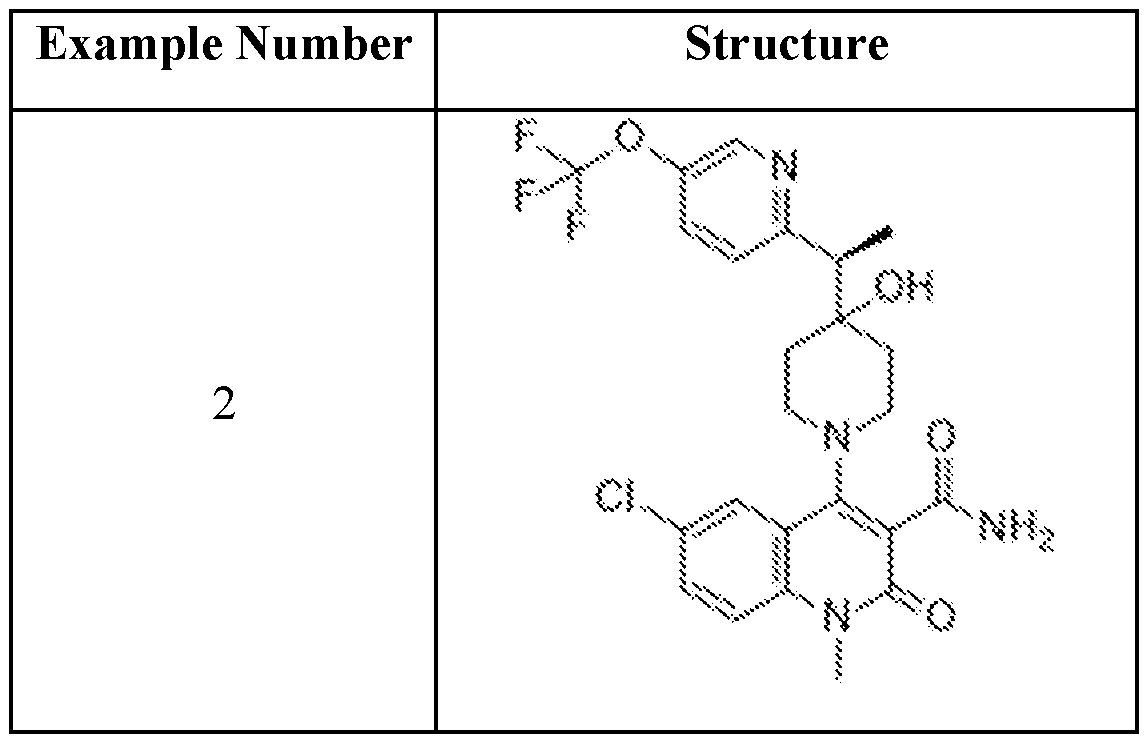
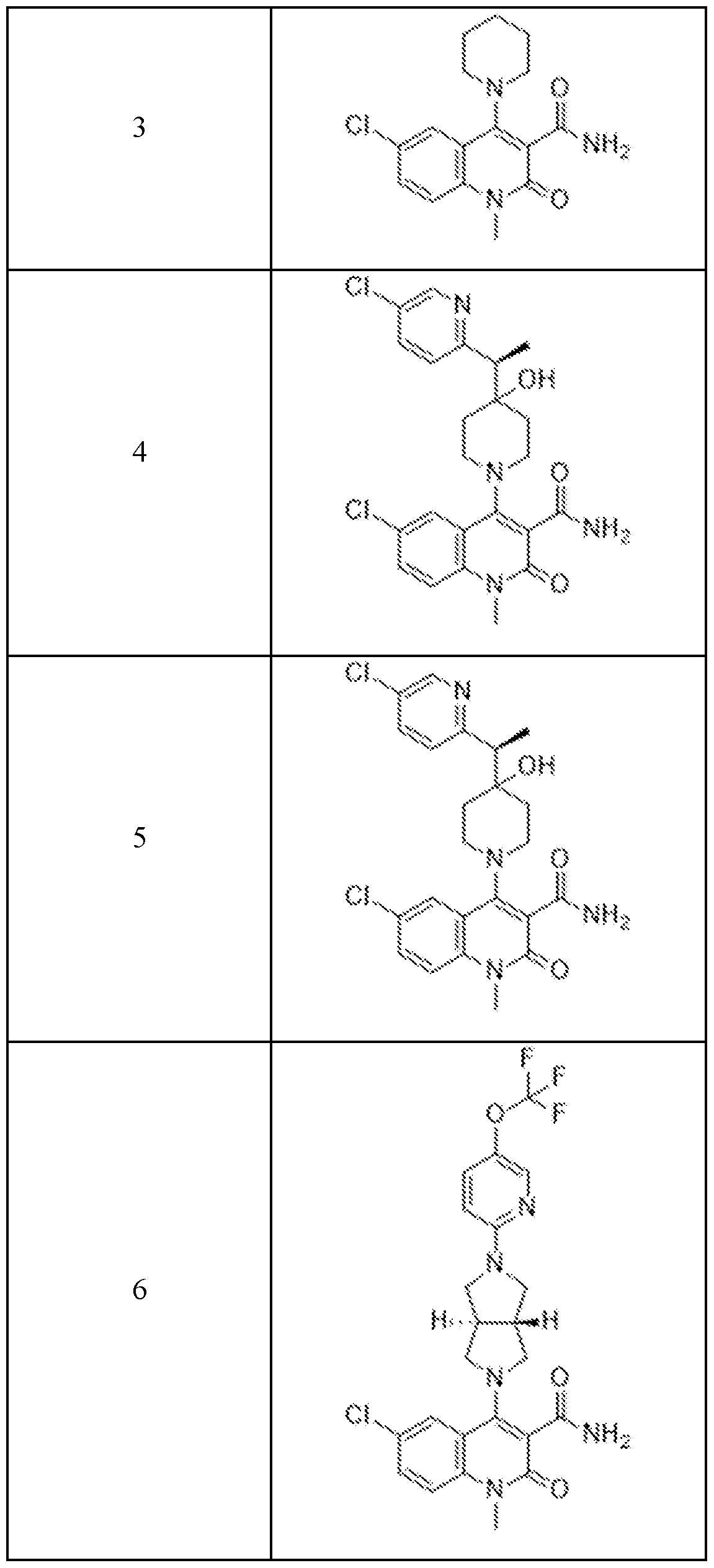
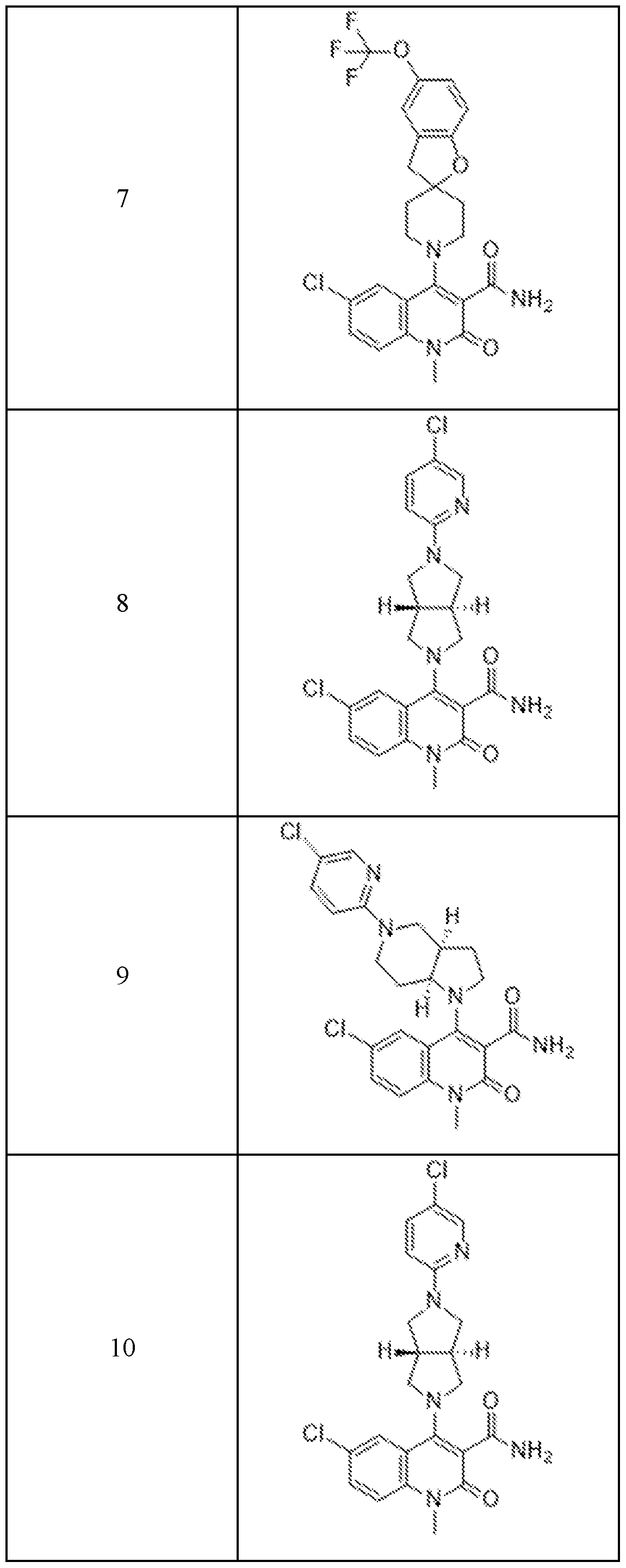












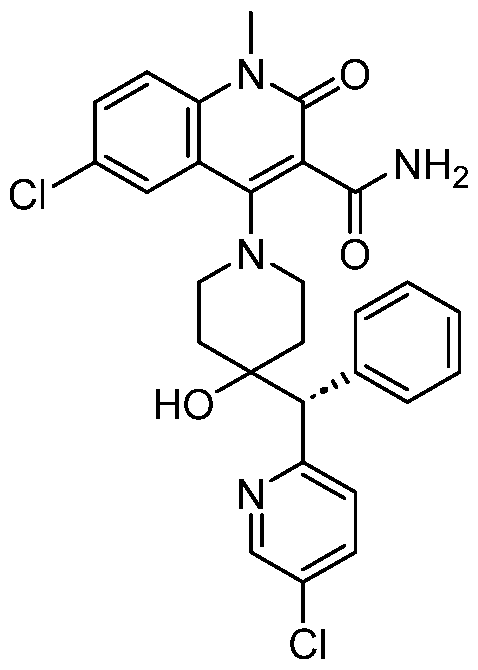


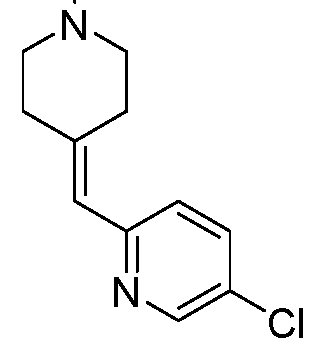

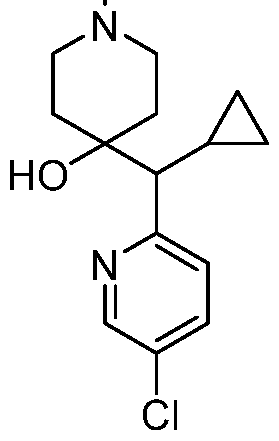











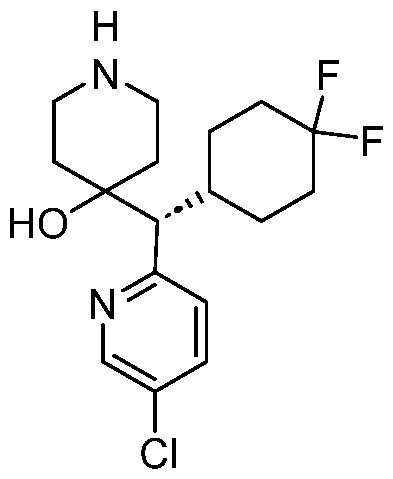

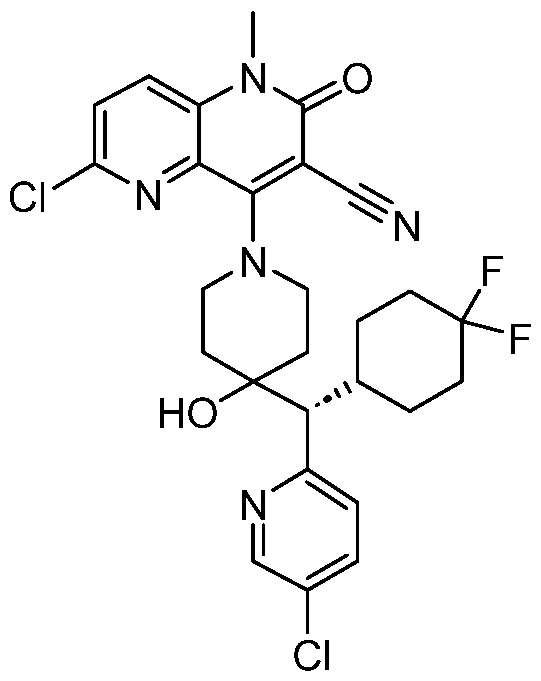






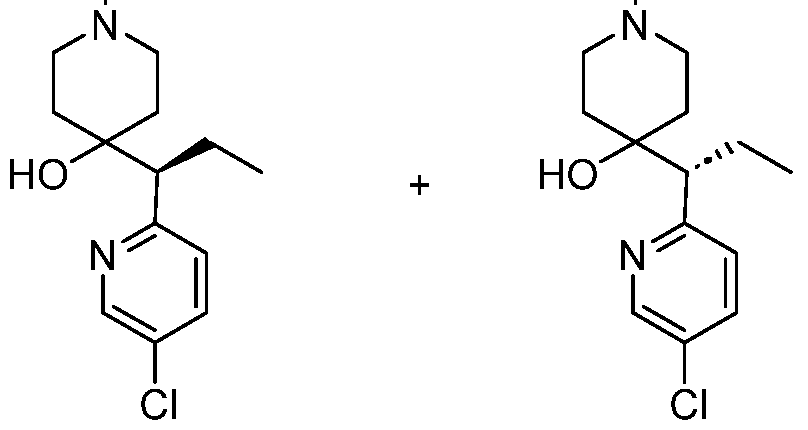

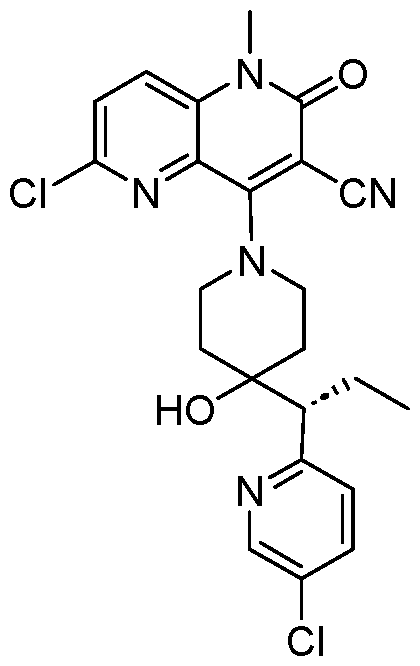



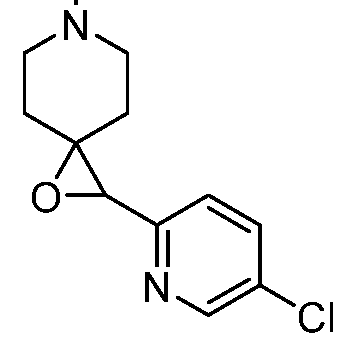


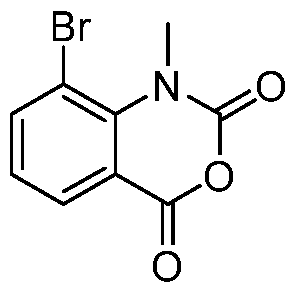


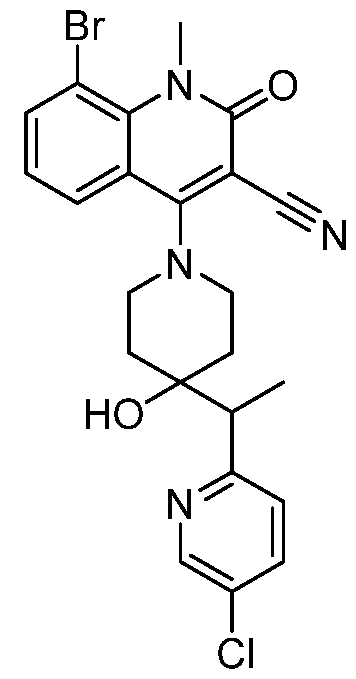

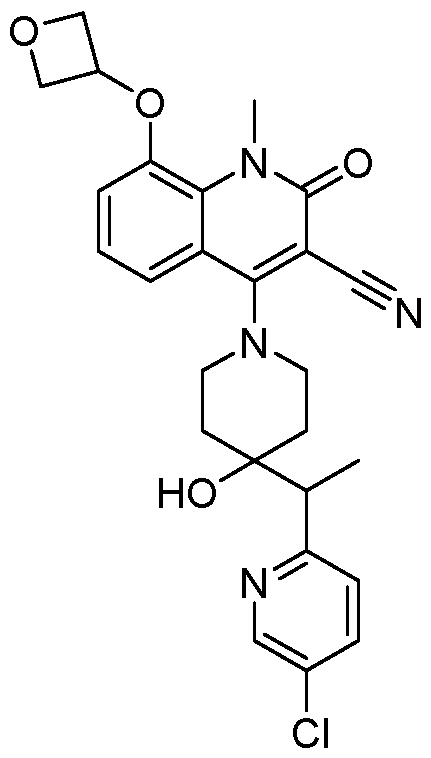

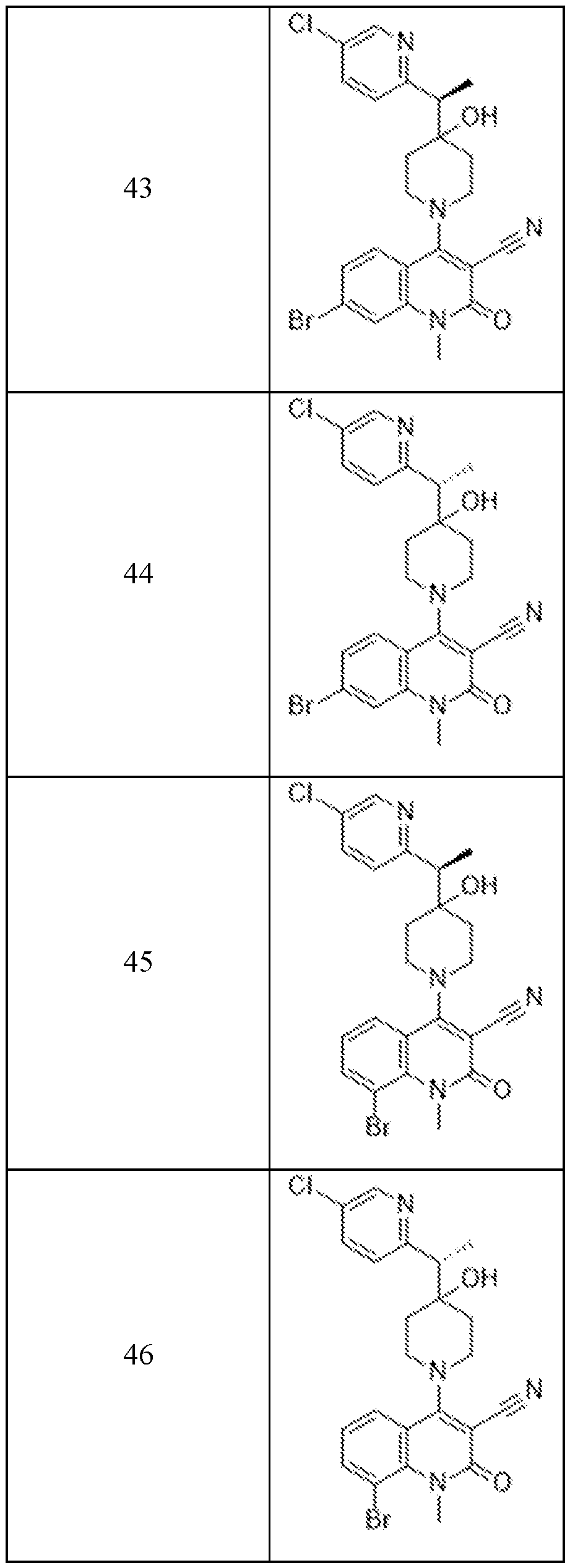
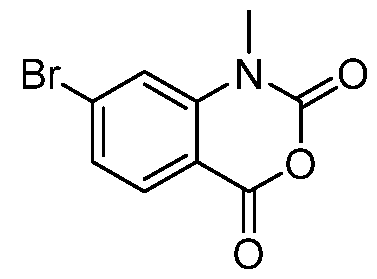







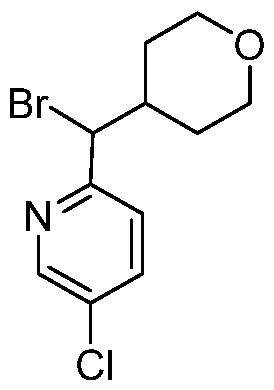



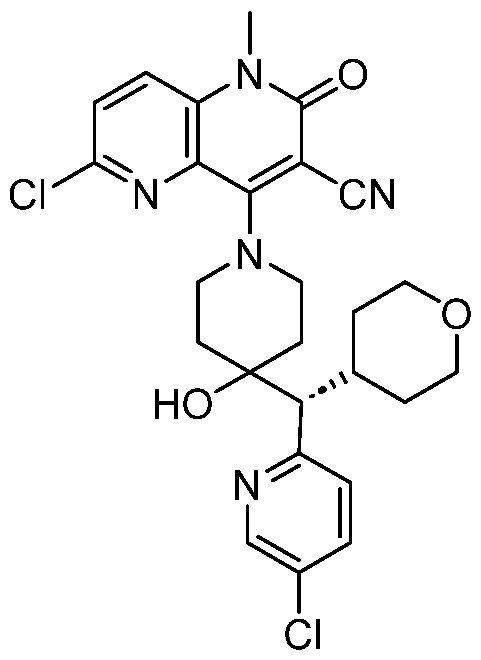
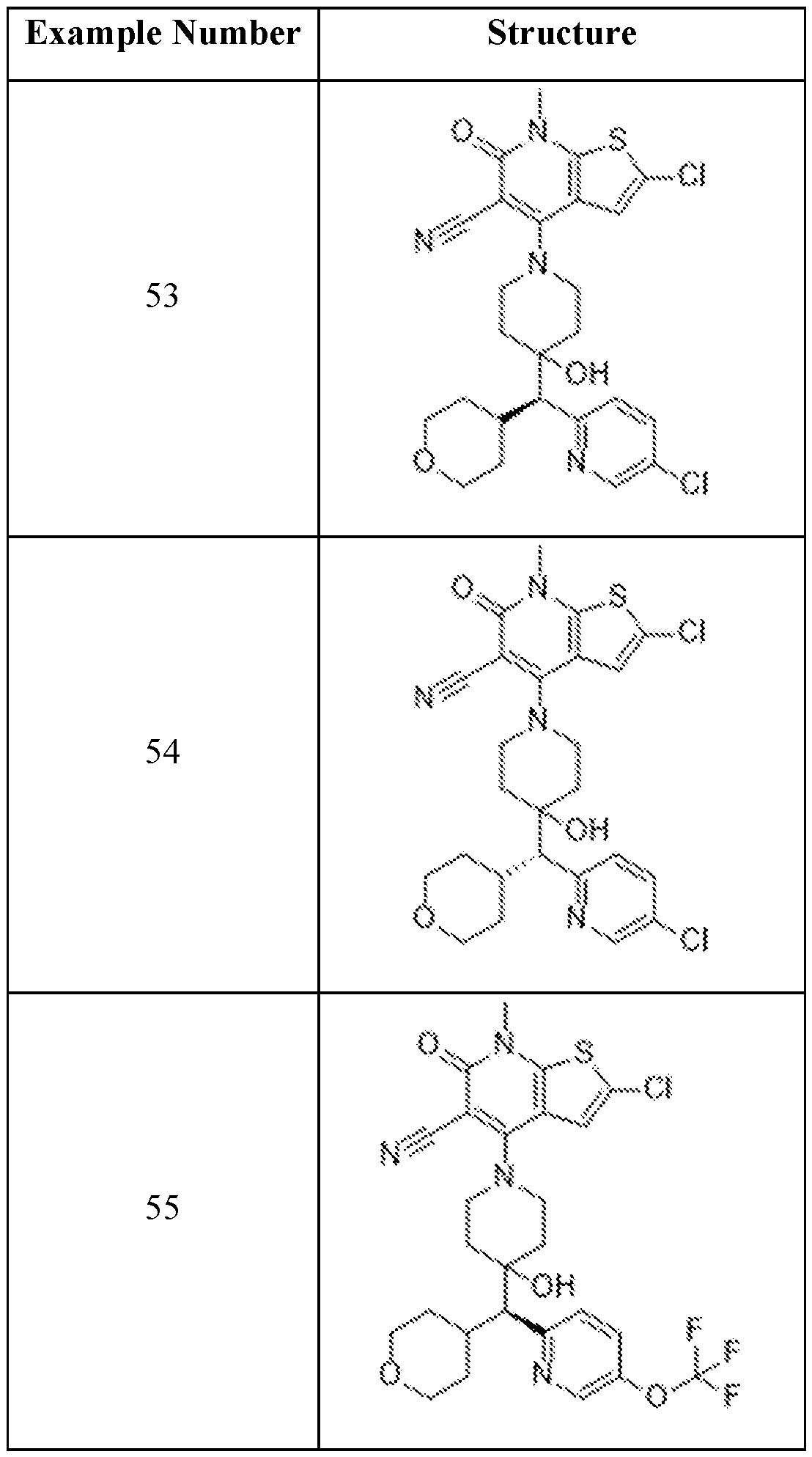

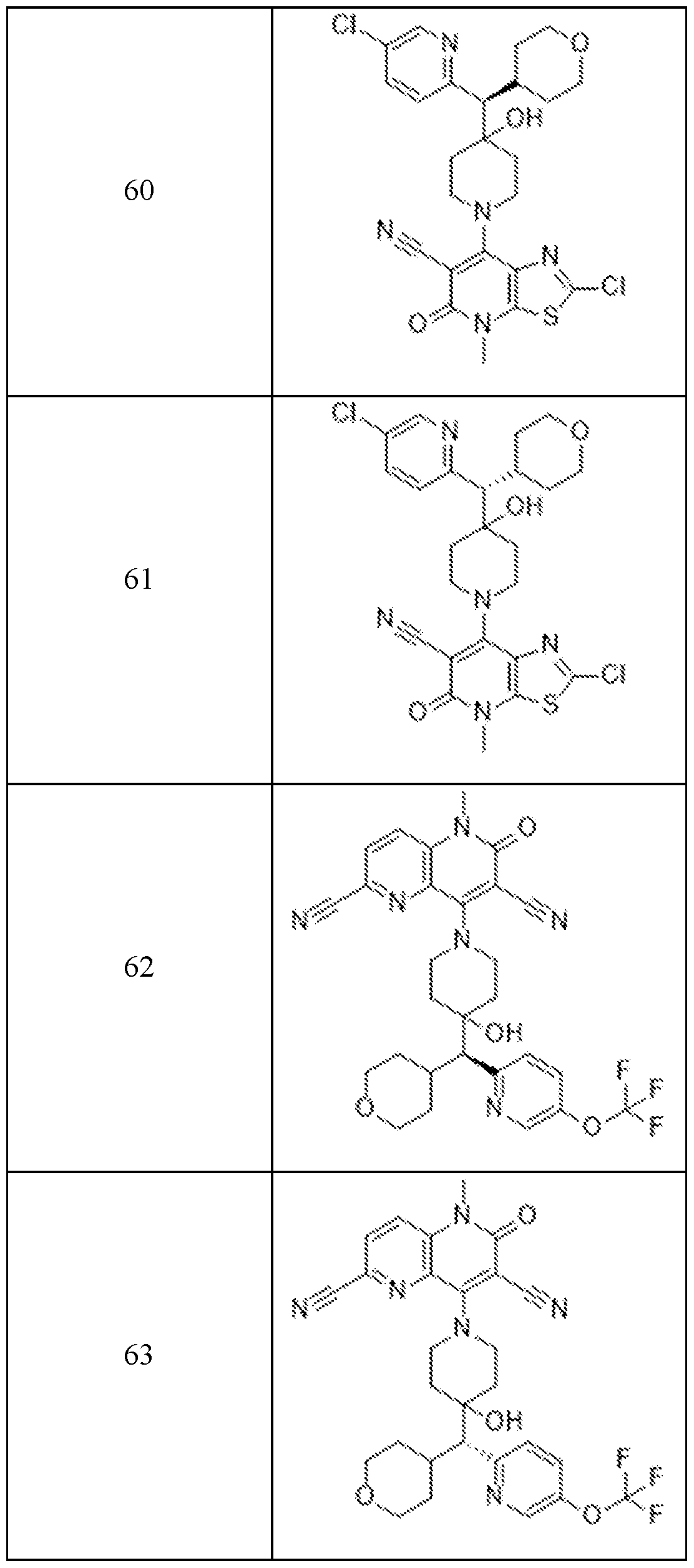








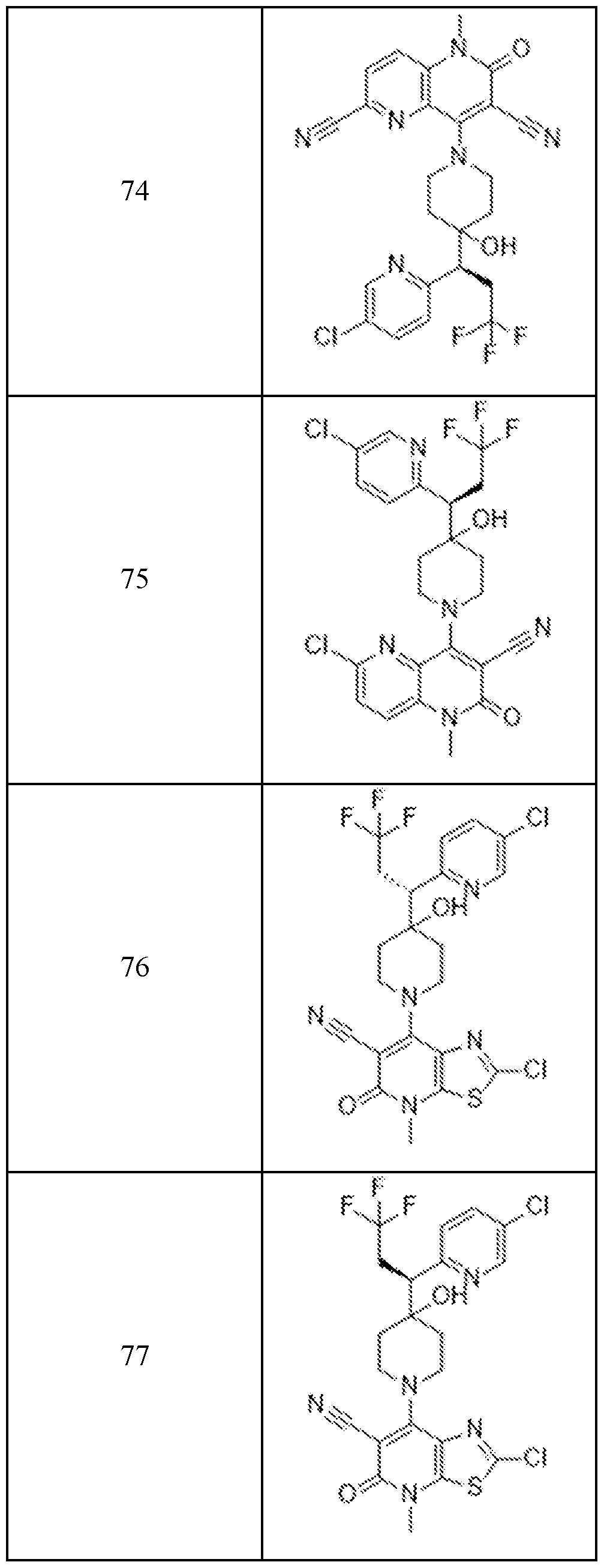
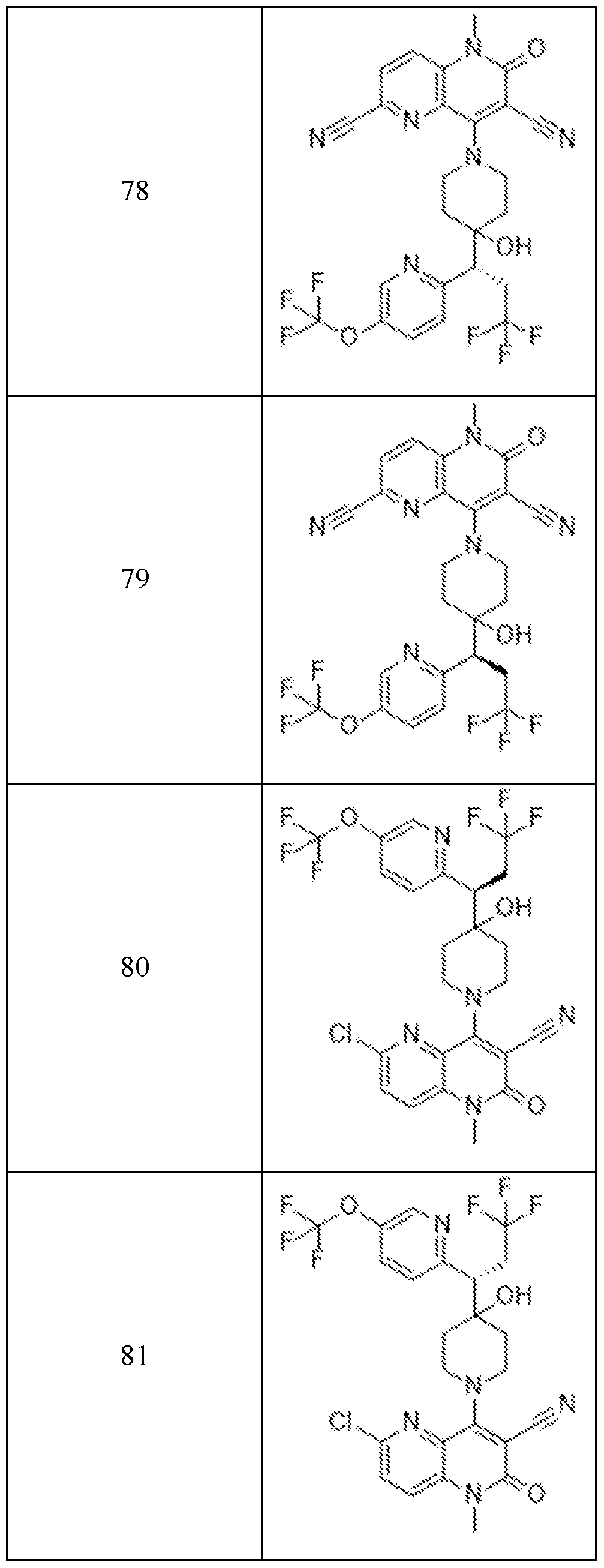
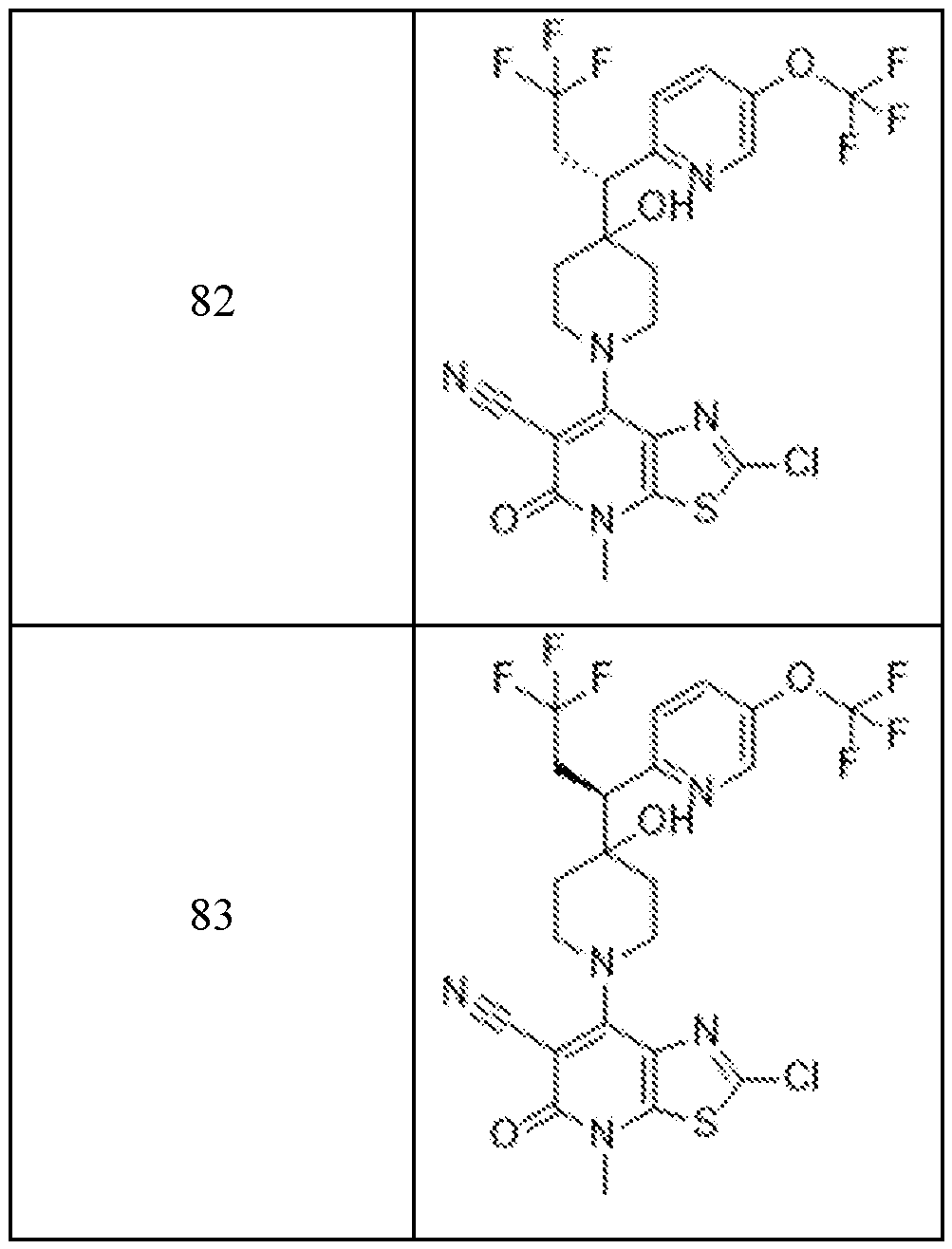
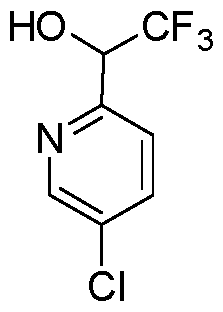







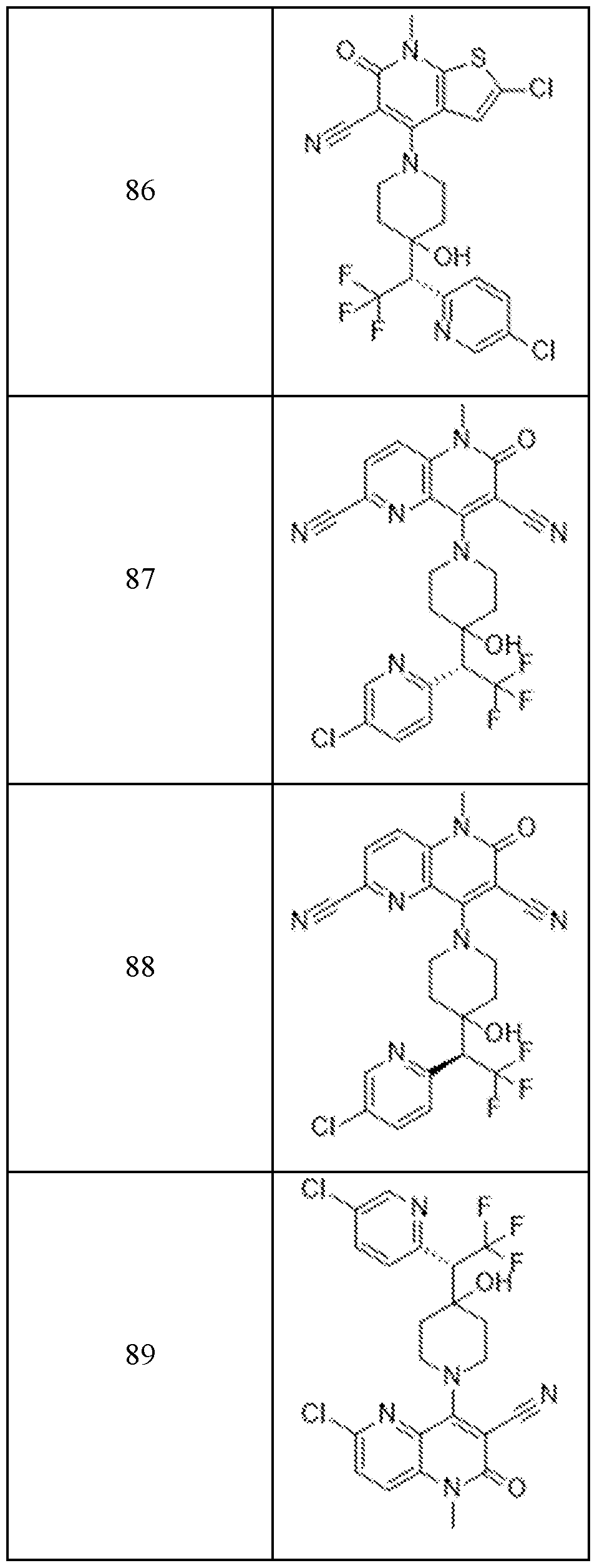
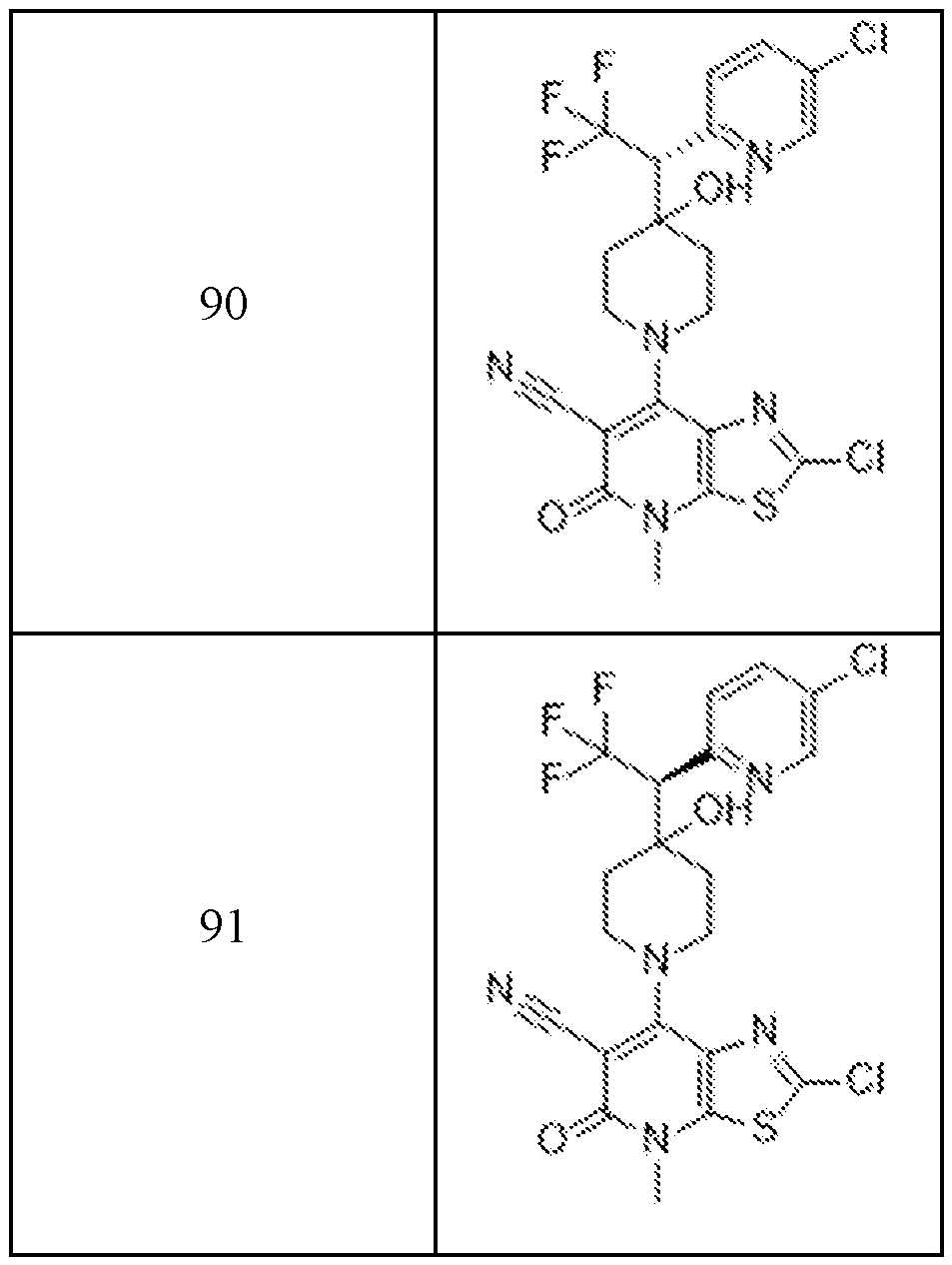
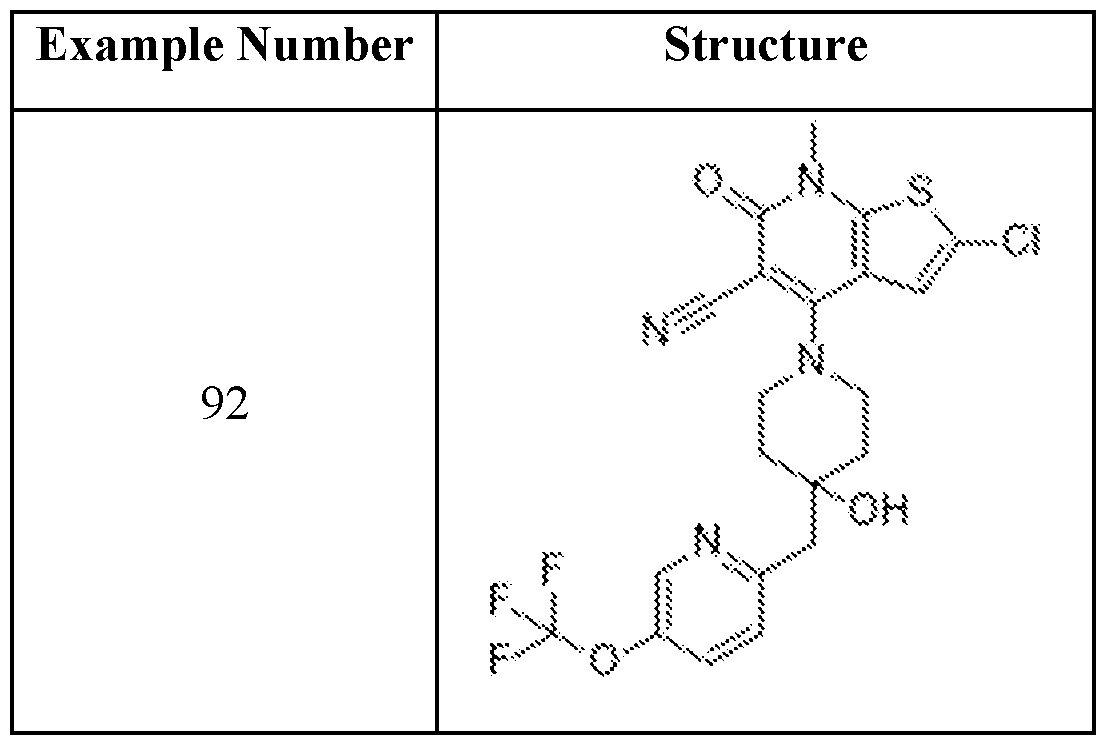
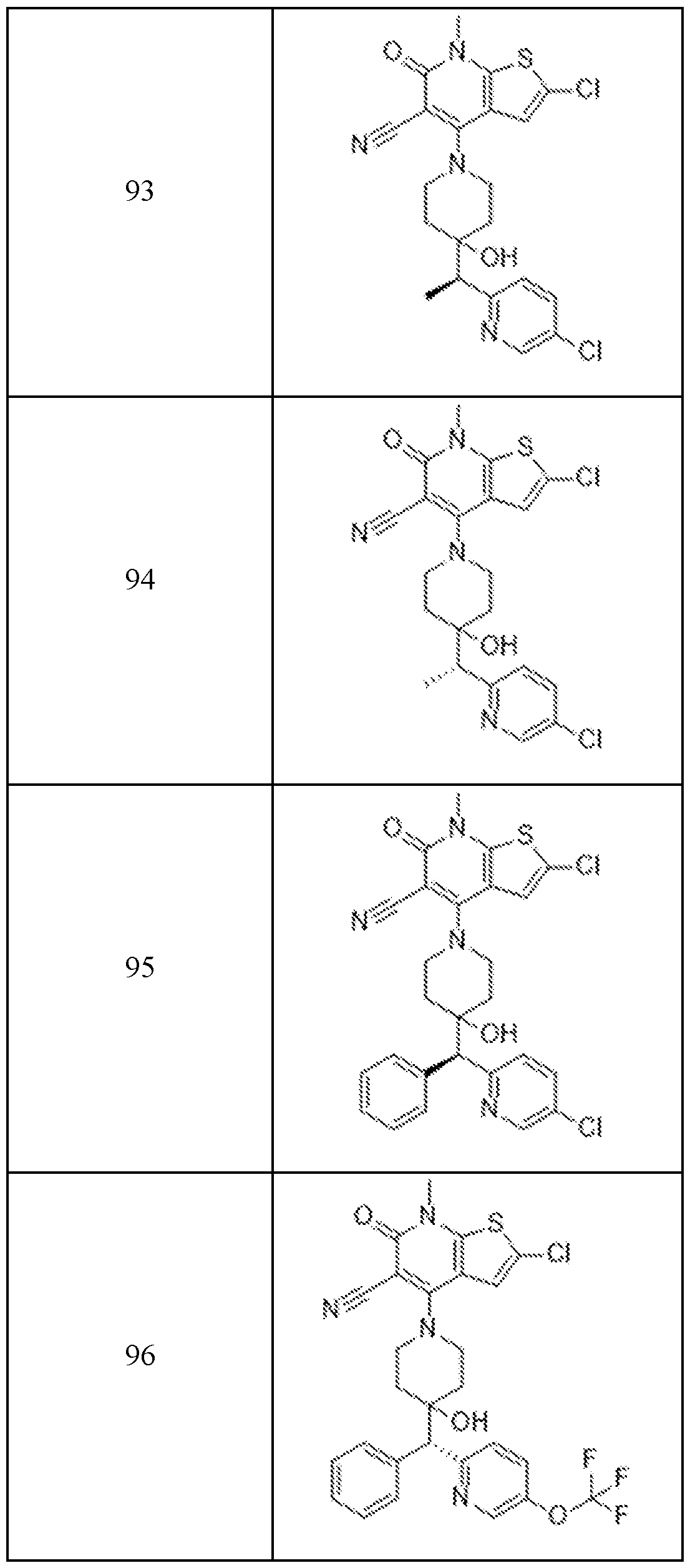


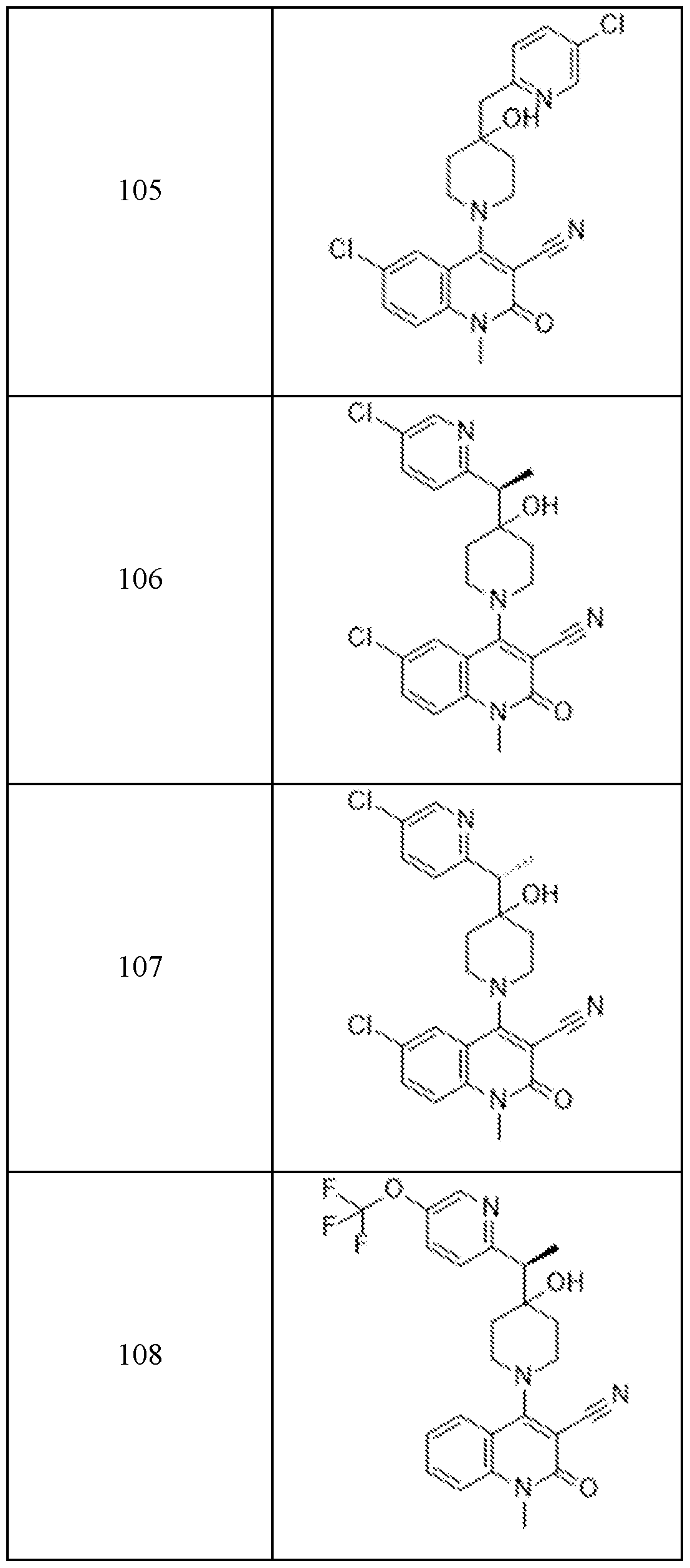







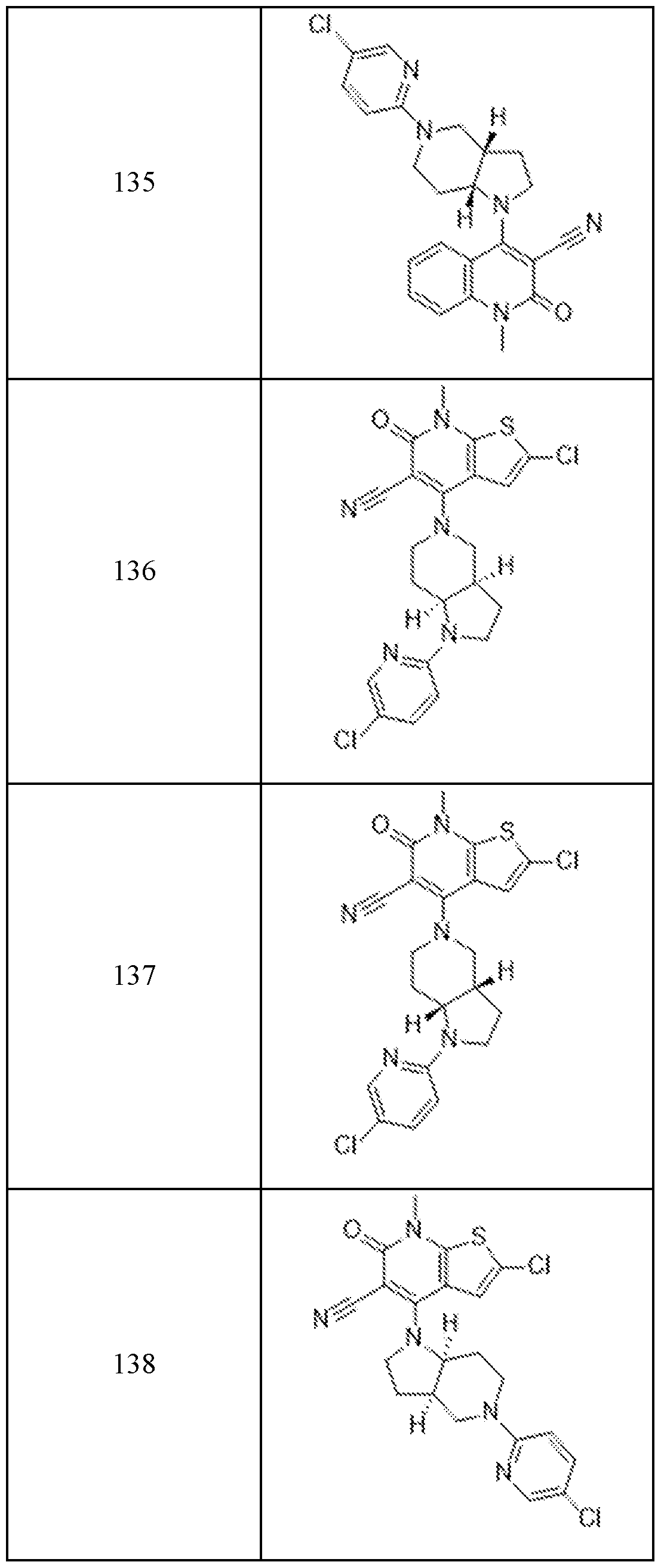
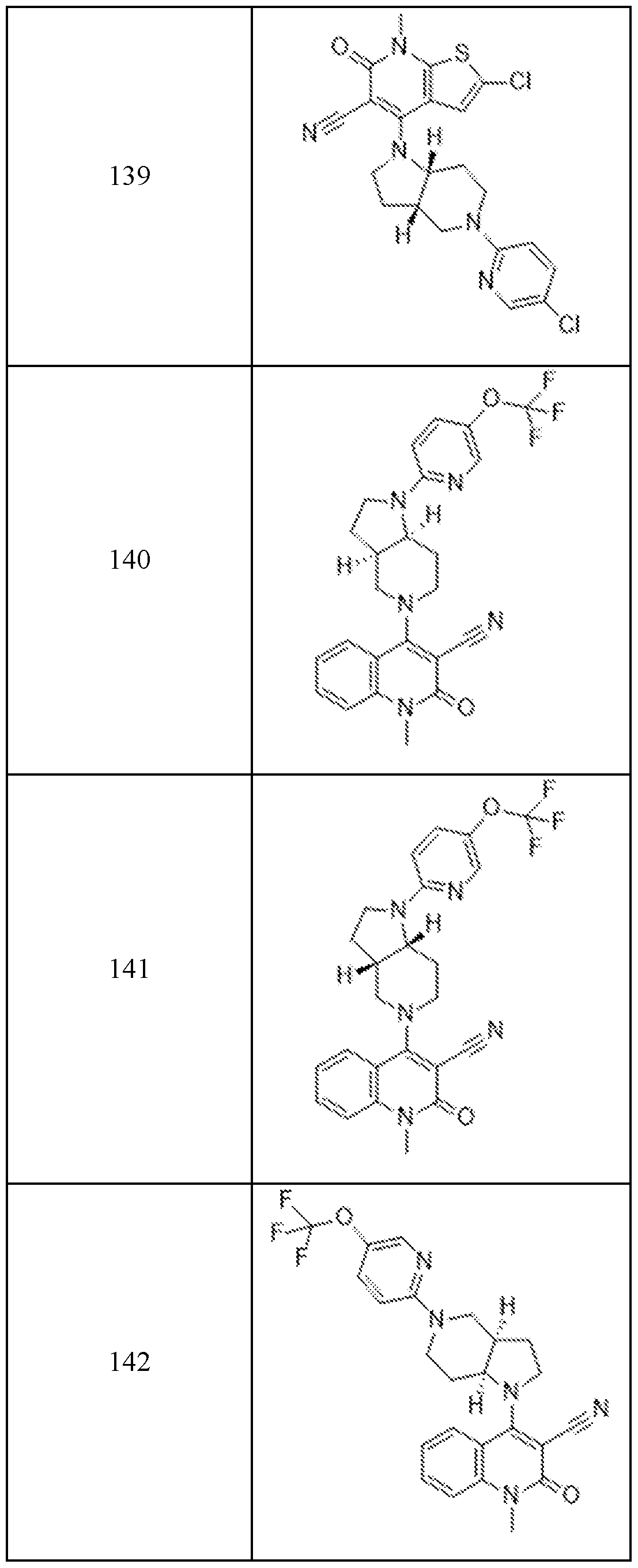

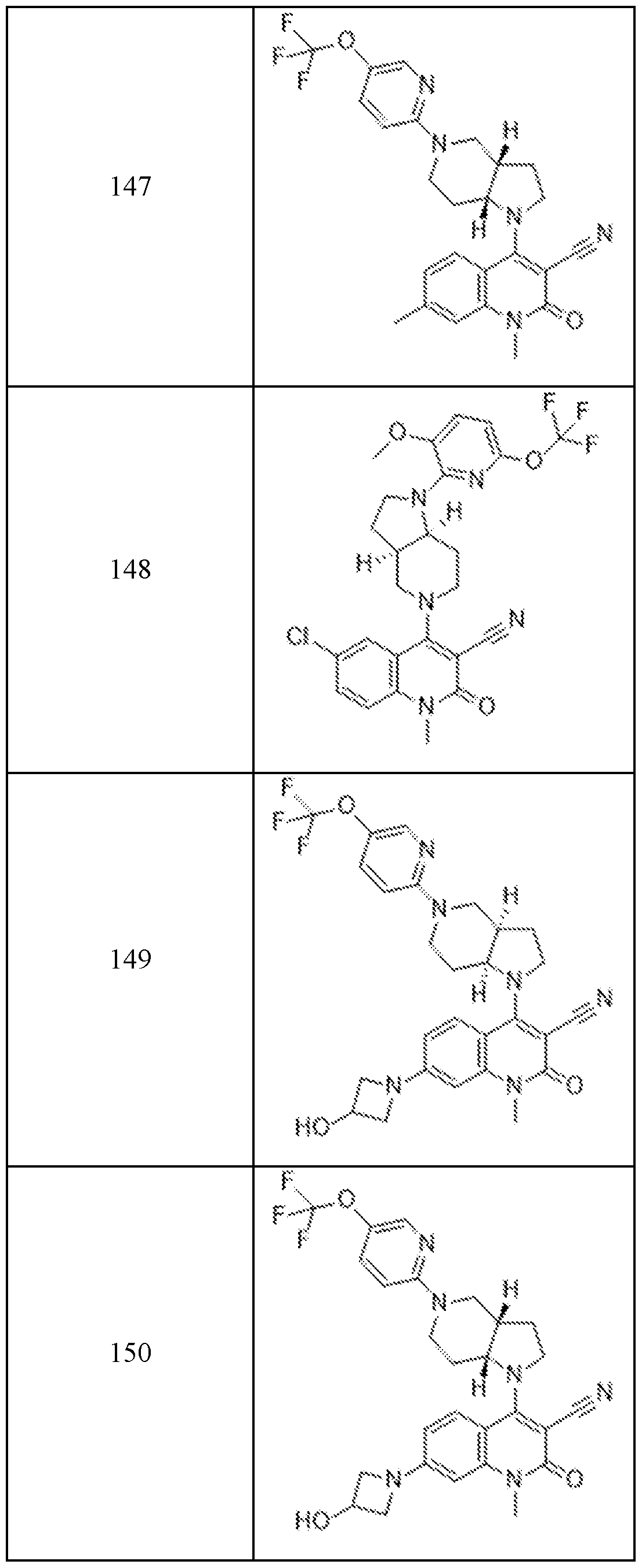

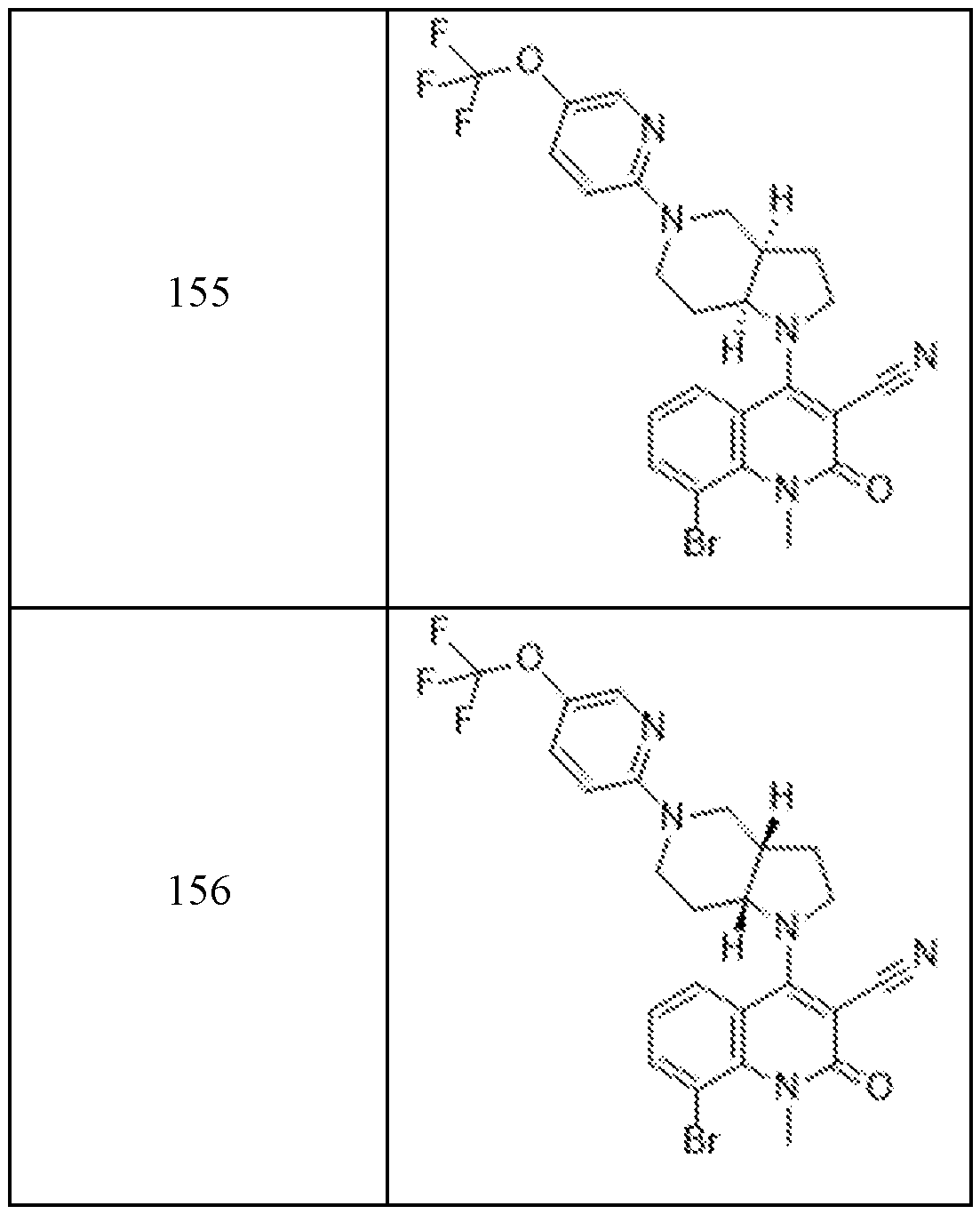



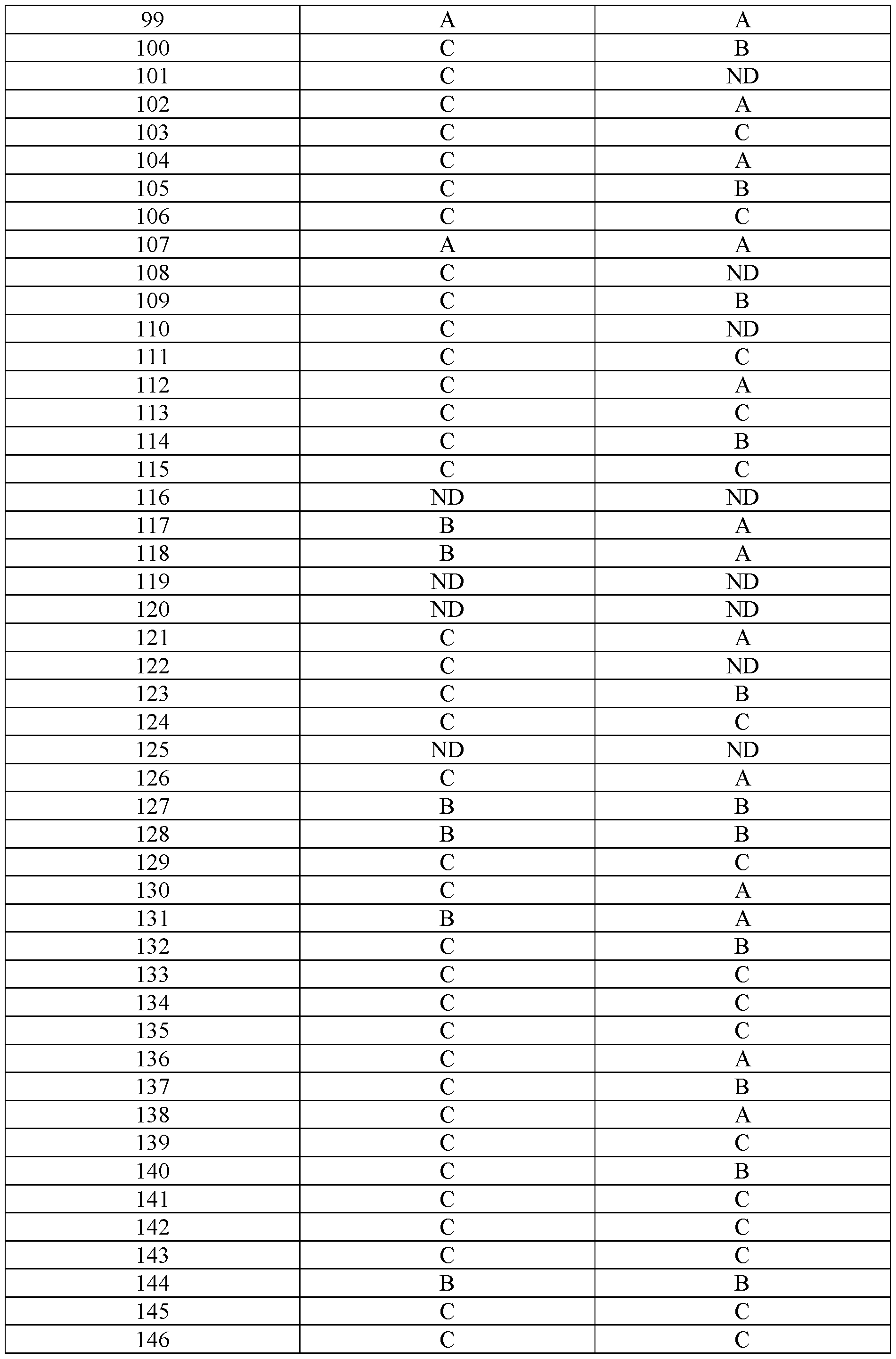

Claims
CLAIMS 1. A compound having the structure of Formula (I): (I) or a pharmaceutically acceptable salt thereof, wherein: Ring A is an optionally substituted phenyl or optionally substituted 5- to 6-membered monocyclic heteroaryl; Ring C is a 5- to 6-membered monocyclic heterocyclyl, an optionally substituted 8- to 15-membered bicyclic heterocyclyl,
 Ring D is an optionally substituted phenyl or an optionally substituted 5- to 6- membered monocyclic heteroaryl; RC1 and RC2 are each independently H, C1-4alkyl, C1-4haloalkyl, optionally substituted C3-6cycloalkyl, optionally substituted 4- to 6-membered heterocyclyl, or optionally substituted phenyl; R1 is C1-4alkyl; R2 is H, halogen, SO2CF3, -N(Ra)2, NO2, SO2Ra, C≡N, C(Rb)3, CORa, CHO, CO2Ra, CON(Ra)2, or N=O; each Ra is independently H or C1-4alkyl; and Rb is halogen; provided that the compound of Formula I is not 2-chloro-4-(4-hydroxy-4-(1-(5- (trifluoromethoxy)pyridin-2-yl)propyl)piperidin-1-yl)-7-methyl-6-oxo-6,7-dihydrothieno[2,3- b]pyridine-5-carbonitrile, or a pharmaceutically acceptable or stereoisomer thereof; and provided that
(i) when Ring A is pyridinyl or thiazoyl then Ring
Ring D is an optionally substituted phenyl or an optionally substituted 5- to 6- membered monocyclic heteroaryl; RC1 and RC2 are each independently H, C1-4alkyl, C1-4haloalkyl, optionally substituted C3-6cycloalkyl, optionally substituted 4- to 6-membered heterocyclyl, or optionally substituted phenyl; R1 is C1-4alkyl; R2 is H, halogen, SO2CF3, -N(Ra)2, NO2, SO2Ra, C≡N, C(Rb)3, CORa, CHO, CO2Ra, CON(Ra)2, or N=O; each Ra is independently H or C1-4alkyl; and Rb is halogen; provided that the compound of Formula I is not 2-chloro-4-(4-hydroxy-4-(1-(5- (trifluoromethoxy)pyridin-2-yl)propyl)piperidin-1-yl)-7-methyl-6-oxo-6,7-dihydrothieno[2,3- b]pyridine-5-carbonitrile, or a pharmaceutically acceptable or stereoisomer thereof; and provided that
(i) when Ring A is pyridinyl or thiazoyl then Ring
 wherein one of RC1 and RC2 is: (a) C1-4haloalkyl, optionally substituted C3-6cycloalkyl, or optionally substituted 4- to 6-membered heterocyclyl when Ring A is pyridinyl; or (b) C1-4alkyl, C1-4haloalkyl, optionally substituted C3-6cycloalkyl, optionally substituted 4- to 6-membered heterocyclyl, or optionally substituted phenyl when Ring A is thiazoyl. 2. The compound of claim 1, wherein the compound is of the formula (II), (III), (IV), or
wherein one of RC1 and RC2 is: (a) C1-4haloalkyl, optionally substituted C3-6cycloalkyl, or optionally substituted 4- to 6-membered heterocyclyl when Ring A is pyridinyl; or (b) C1-4alkyl, C1-4haloalkyl, optionally substituted C3-6cycloalkyl, optionally substituted 4- to 6-membered heterocyclyl, or optionally substituted phenyl when Ring A is thiazoyl. 2. The compound of claim 1, wherein the compound is of the formula (II), (III), (IV), or
 or a pharmaceutically acceptable salt thereof, wherein R3 and R4 are each independently selected from halogen, C1-4alkyl, haloC1-4alkyl, C1- 4alkoxy, haloC1-4alkoxy , C≡N, N=O, SO2Rc, -NRcRd, CORc, CO2Rc, CONRcRd, -NRcCORd,
-ORe, 5- to 6- membered heteroaryl, and 4- to 6- membered heterocyclyl, wherein said heteroaryl and heterocyclyl are each optionally substituted; Rc and Rd are each independently C1-4alkyl or haloC1-4alkyl; Re is C1-4alkyl, phenyl, 5- to 6-membered heteroaryl, or 4- to 6- membered heterocyclyl, each of which is optionally substituted; and n and p are each independently 0, 1,
or a pharmaceutically acceptable salt thereof, wherein R3 and R4 are each independently selected from halogen, C1-4alkyl, haloC1-4alkyl, C1- 4alkoxy, haloC1-4alkoxy , C≡N, N=O, SO2Rc, -NRcRd, CORc, CO2Rc, CONRcRd, -NRcCORd,
-ORe, 5- to 6- membered heteroaryl, and 4- to 6- membered heterocyclyl, wherein said heteroaryl and heterocyclyl are each optionally substituted; Rc and Rd are each independently C1-4alkyl or haloC1-4alkyl; Re is C1-4alkyl, phenyl, 5- to 6-membered heteroaryl, or 4- to 6- membered heterocyclyl, each of which is optionally substituted; and n and p are each independently 0, 1,
2, or 3.
3. The compound of claim 1 or 2, wherein, n is 0 or 1.
4. The compound of any one of claims 1 to 3, wherein, p is 0 or 1.
5. The compound of any one of claims 1 to 4, wherein the compound is of the formula (VI) (VII), (VIII), (IX), (X), (XI), or (XII):
 or a pharmaceutically acceptable salt thereof.
or a pharmaceutically acceptable salt thereof.
6. The compound of any one of claims 1 to 5, wherein R2 is CON(Ra)2 or C≡N.
7. The compound of any one of claims 1 to 6, wherein R2 is CONH2 or C≡N.
8. The compound of any one of claims 1 to 7, wherein R1 is CH3.
9. The compound of any one of claims 1 to 8, wherein R3 and R4 are each independently C≡N, -ORe, 5- or 6- membered heteroaryl, or 4- to 6- membered heterocyclyl, wherein said heteroaryl and heterocyclyl are optionally substituted with 1 to 3 R5 groups selected from halogen, C1-4alkyl, haloC1-4alkyl, C1-4alkoxy, haloC1-4alkoxy, C≡N, N=O, SO2Rc, -NRcRd, CORc, CO2Rc, CONRcRd, -NRcCORd, and OH.
10. The compound of any one of claims 1 to 9, wherein R3 is selected from C≡N, halogen, C1-4alkyl, 4- to 6- membered heterocyclyl, and -ORe, wherein said heterocyclyl is optionally substituted with 1 to 3 R5 groups selected from halogen, C1-4alkyl, haloC1-4alkyl, C1-4alkoxy, haloC1-4alkoxy , C≡N, N=O, SO2Rc, -NRcRd, CORc, CO2Rc, CONRcRd, - NRcCORd, and OH.
11. The compound of any one of claims 1 to 10, wherein Re is 4- to 5-membered heterocyclyl or C1-4alkyl optionally substituted by C3-4cycloalkyl, wherein said cycloalkyl is optionally substituted by OH.
12. The compound of any one of claims 1 to 11, wherein Re is oxetanyl, tetrahydrofuranyl
 .
.
13. The compound of any one of claims 1 to 12, wherein R3 is selected from C≡N, halogen, C1-4alkyl, azetidinyl, and -ORe, wherein said azetidinyl is optionally substituted with 1 to 3 R5 groups selected from halogen, C1-4alkyl, halo C1-4alkyl, C1-4alkoxy, halo C1-4alkoxy, C≡N, N=O, SO2Rc, -NRcRd, CORc, CO2Rc, CONRcRd, -NRcCORd, and OH.
14. The compound of any one of claims 9 to 13, wherein R5 is OH.
15. The compound of any one of claims 1 to 14, wherein R3 is selected from C≡N, Cl, Br,

16. The compound of any one of claims 1 to 9, wherein R4 is halo.
17. The compound of any one of claims 1 to 9 and 16, wherein R4 is chloro.
18. The compound of any one of claims 1 to 17, wherein ring C is a 6-membered monocyclic heterocyclyl, a substituted 5,5-, 5,6-, or 6,5-membered bicyclic heterocyclyl ring, a 10- to 15-membered spirocyclic heterocyclyl,
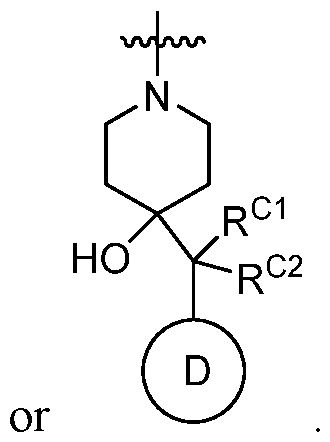 .
.
19. The compound of any one of claims 1 to 18, wherein ring C is piperdinyl, octahydropyrrolo[3,4-c]pyrrolyl, octahydro-1H-pyrrolo[3,2-c]pyridinyl, 3H- spiro[benzofuran-2,4'-piperidinyl], 3H-spiro[furo[2,3-b]pyridine-2,4'-piperidinyl], 5,7- dihydrospiro[cyclopenta[b]pyridine-6,4'-piperidinyl], or 1,3-dihydrospiro[indene-2,4'- piperidinyl].
20. The compound of any one of claims 1 to 19, wherein ring C is substituted with a phenyl or 6-membered heteroaryl, wherein said phenyl and heteroaryl are each optionally substituted with 1 to 3 R6 groups selected from halogen, C1-4alkyl, halo C1-4alkyl, C1-4alkoxy, halo C1-4alkoxy, C≡N, N=O, SO2Rc, -NRcRd, -CORc, -CO2Rc, -CONRcR’’, -NRcCORd, and OH.
21. The compound of any one of claims 1 to 20, wherein ring C is substituted with a phenyl or pyridyl, wherein said phenyl and pyridyl are each optionally substituted with 1 to 3 R6 groups selected from halogen, C1-4alkyl, halo C1-4alkyl, C1-4alkoxy, halo C1-4alkoxy, C≡N, N=O, SO2Rc, -NRcRd, -CORc, -CO2Rc, -CONRcR’’, -NRcCORd, and OH.
22. The compound of any one of claims 1 to 18, wherein ring
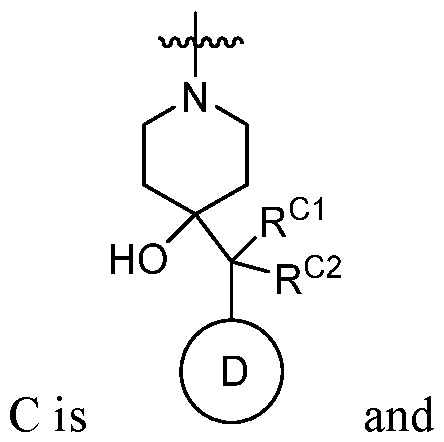 Ring D is pyridinyl optionally substituted by 1 to 3 R6 groups selected from halogen, C1-4alkyl, haloC1-4alkyl, C1-4alkoxy, halo C1-4alkoxy, C≡N, N=O, SO2Rc, -NRcRd, -CORc, -CO2Rc, - CONRcR’’, -NRcCORd, and OH.
Ring D is pyridinyl optionally substituted by 1 to 3 R6 groups selected from halogen, C1-4alkyl, haloC1-4alkyl, C1-4alkoxy, halo C1-4alkoxy, C≡N, N=O, SO2Rc, -NRcRd, -CORc, -CO2Rc, - CONRcR’’, -NRcCORd, and OH.
23. The compound of any one of claims 1 to 18 or 22, wherein RC1 and RC2 are each independently H, -CH3, -CF3, -CH2CH3, -CH2CF3, cyclopropyl, cyclohexyl, tetrahydropyranyl, or phenyl, wherein the cyclohexyl is optionally substituted by 1 to 2 halo.
24. The compound of any one of claims 19 to 23, wherein R6 is selected from halo, C1- 4alkyl, haloC1-4alkoxy, and C1-4alkoxy.
25. The compound of any one of claims 1 to 24, wherein ring C is selected from
 , ,
, ,
 , , , , ,
, , , , ,
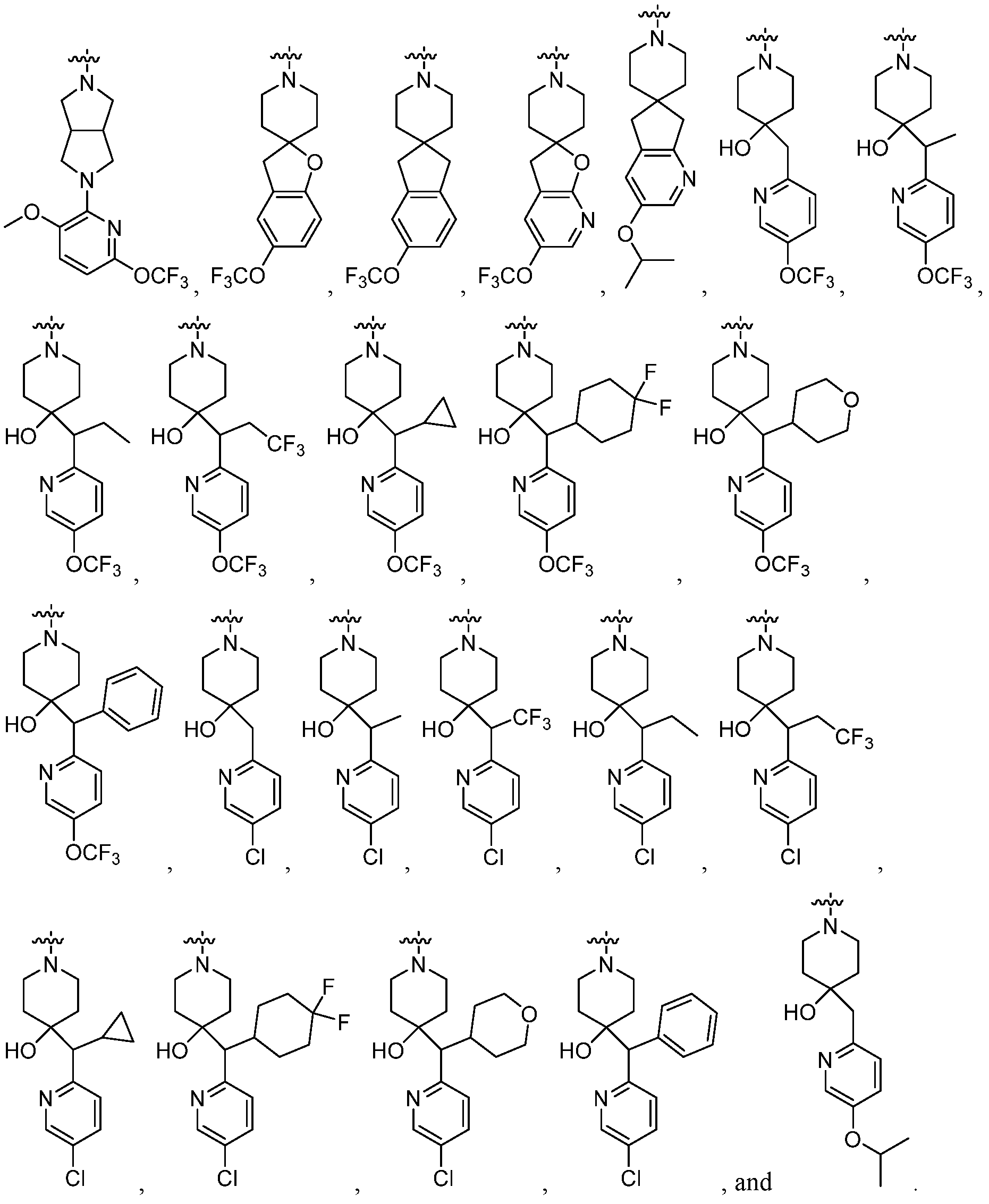
26. The compound of claim 1, wherein the compound is one of:








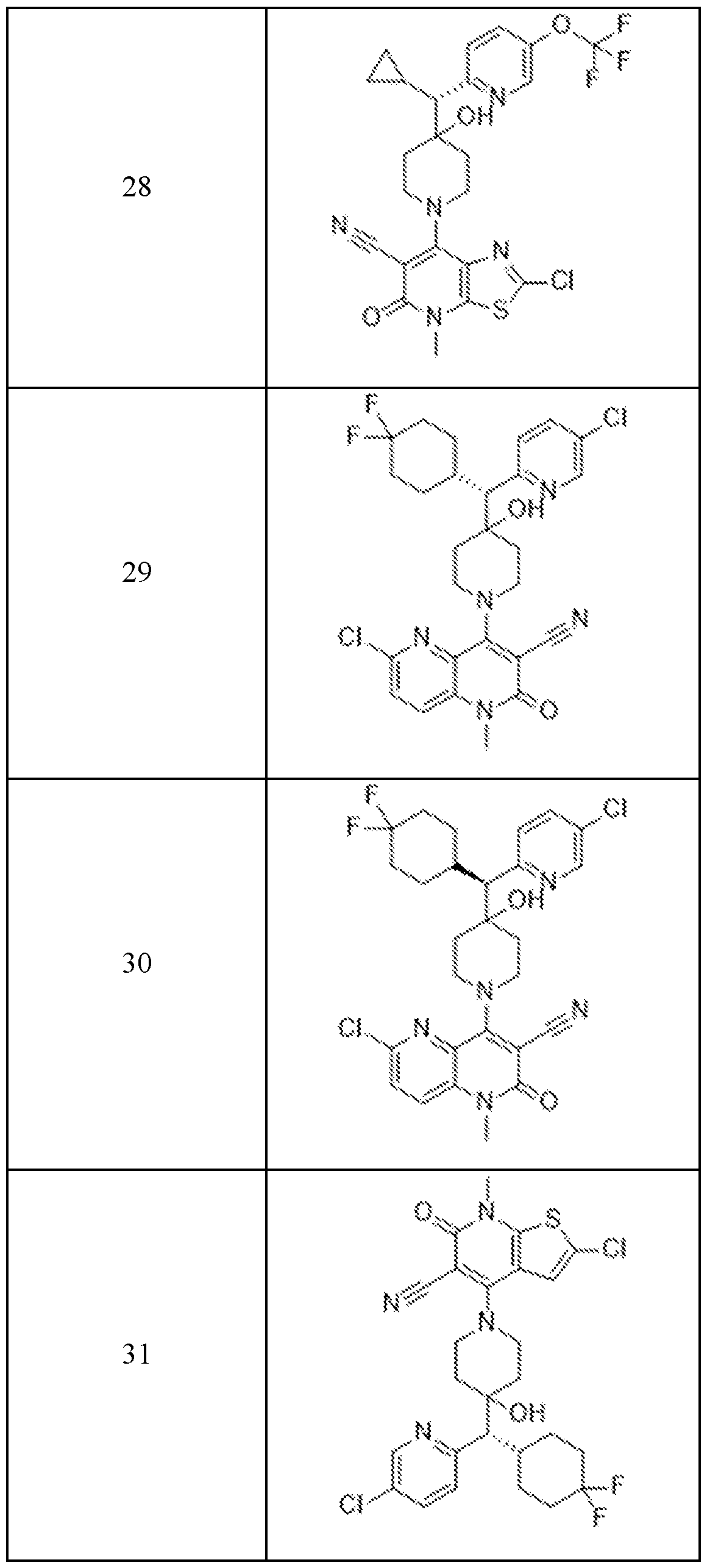
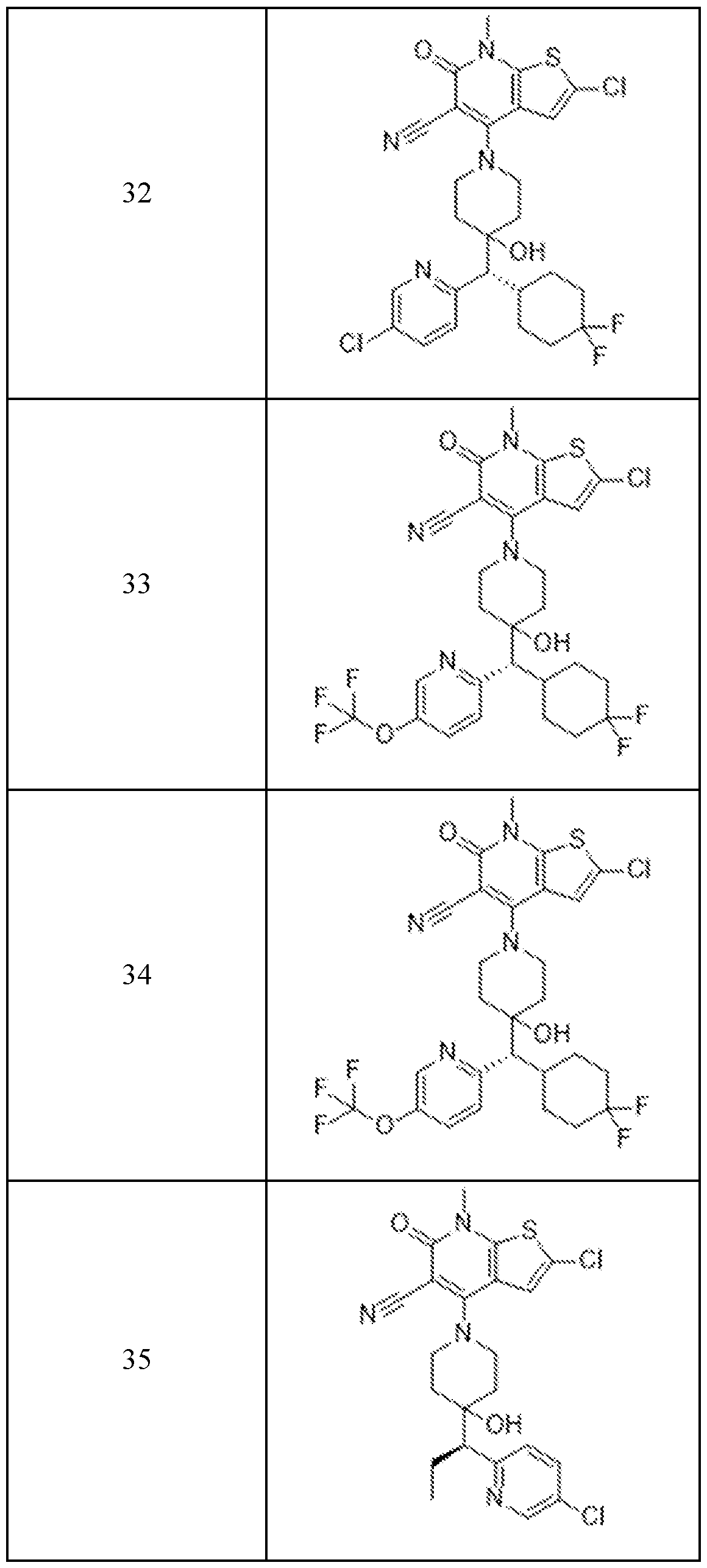
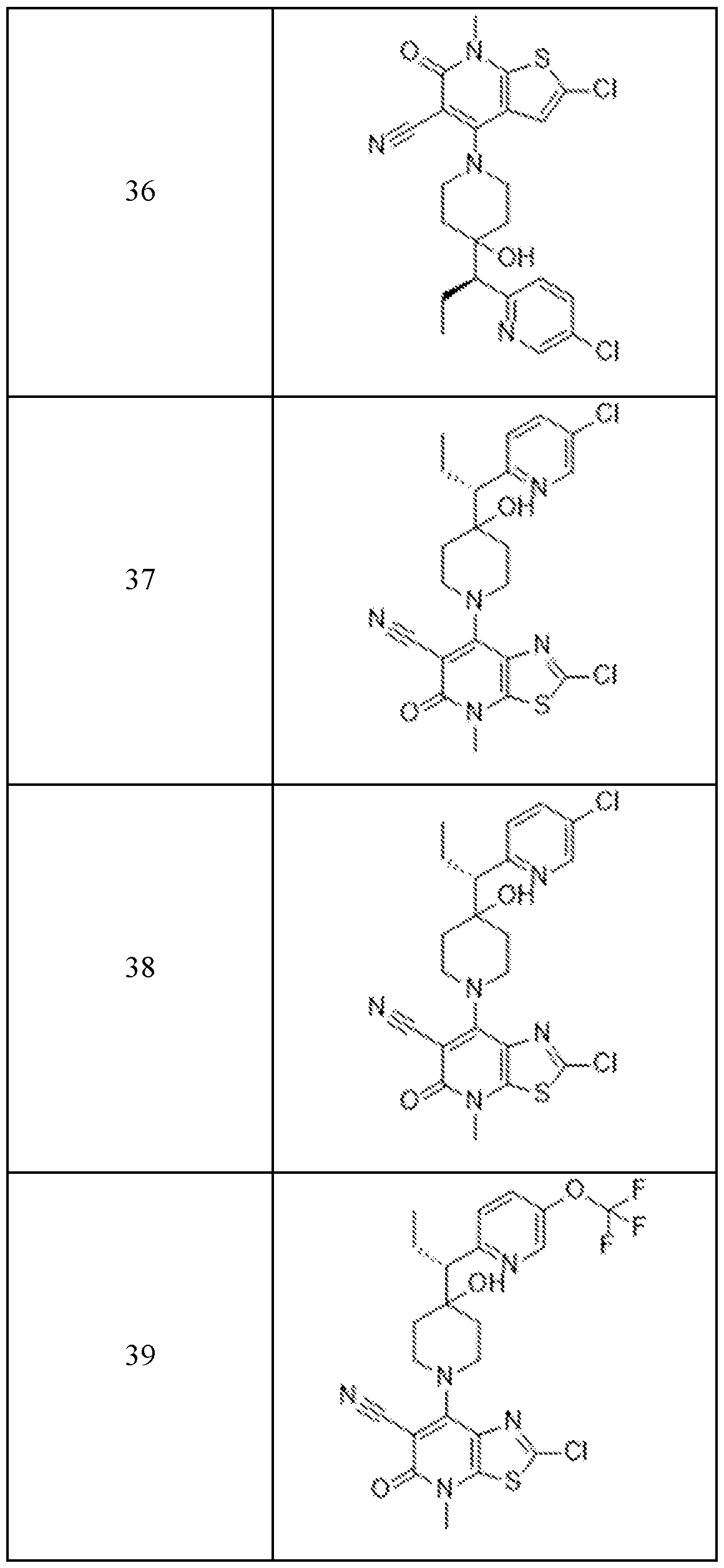

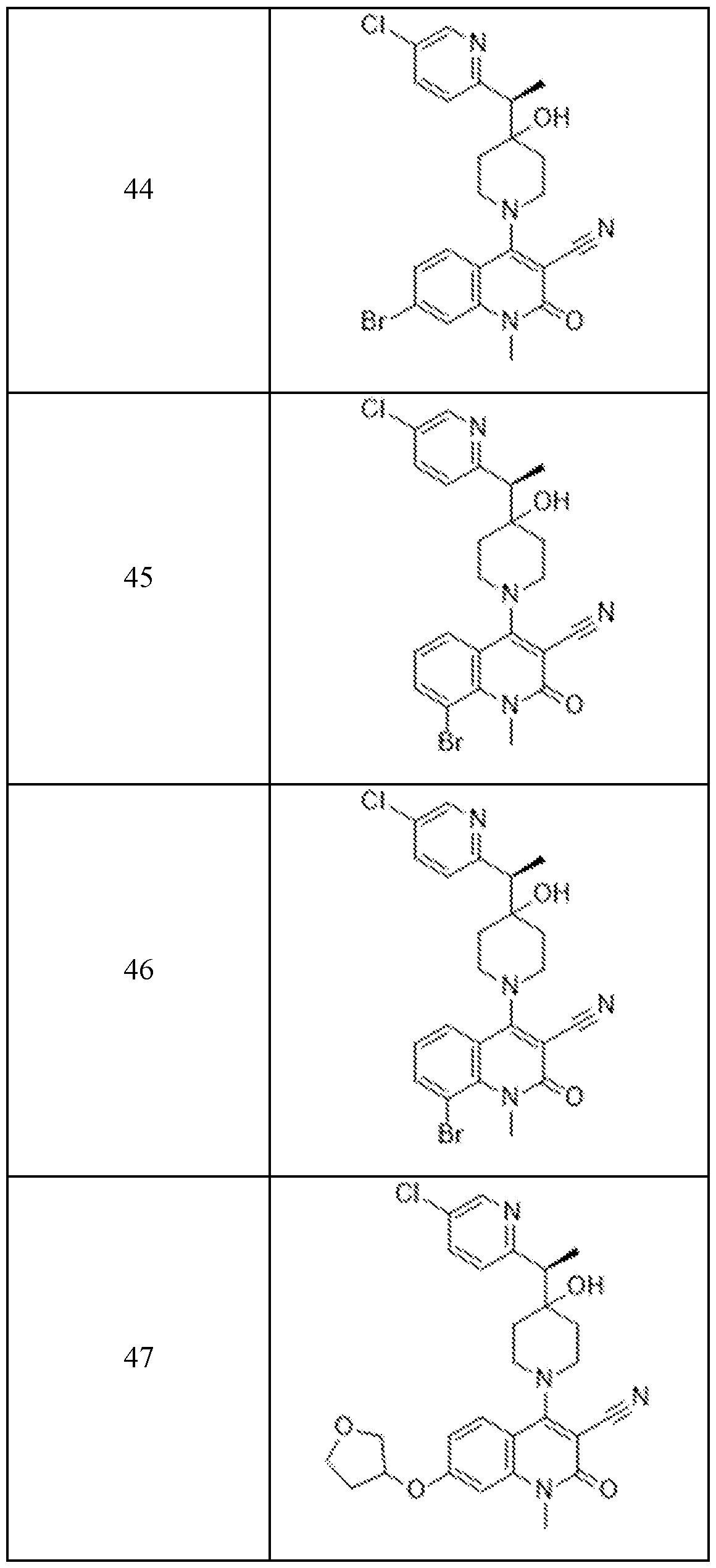
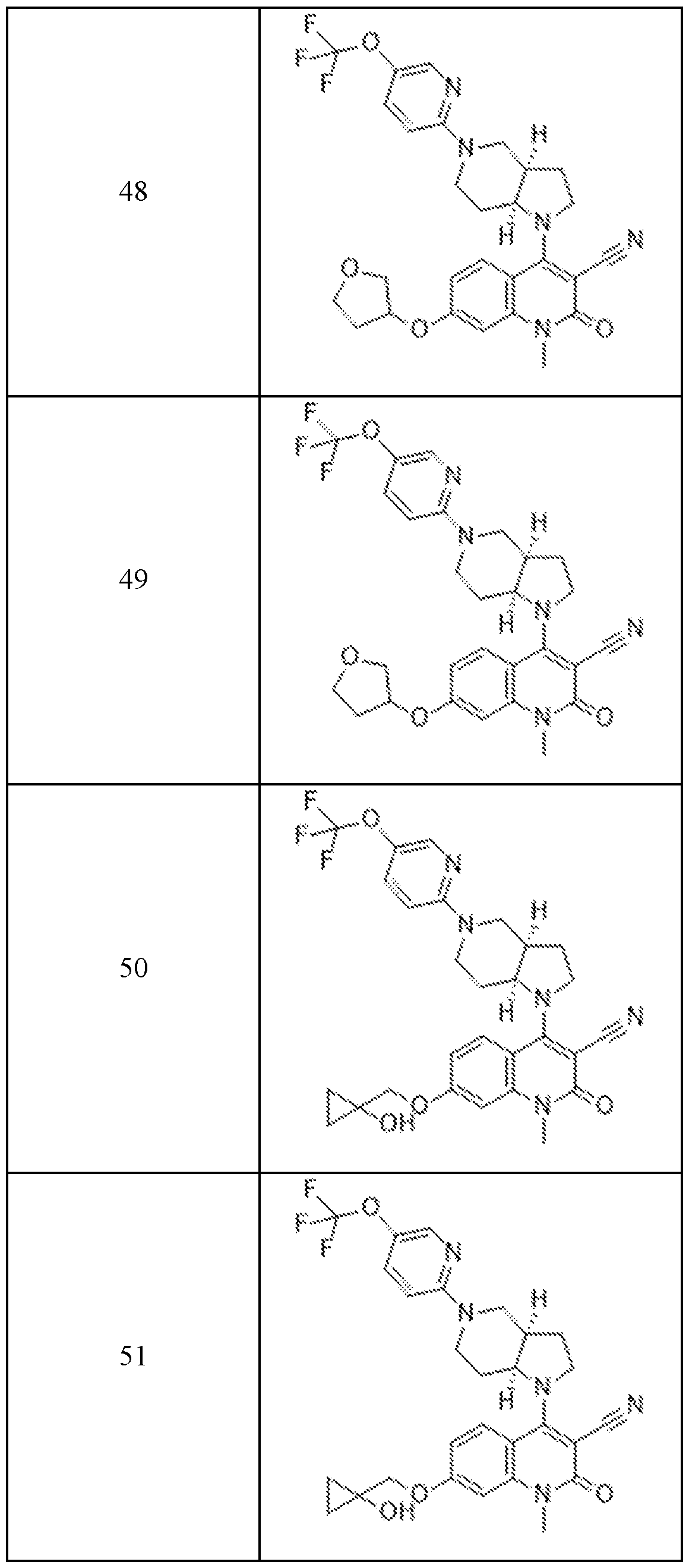







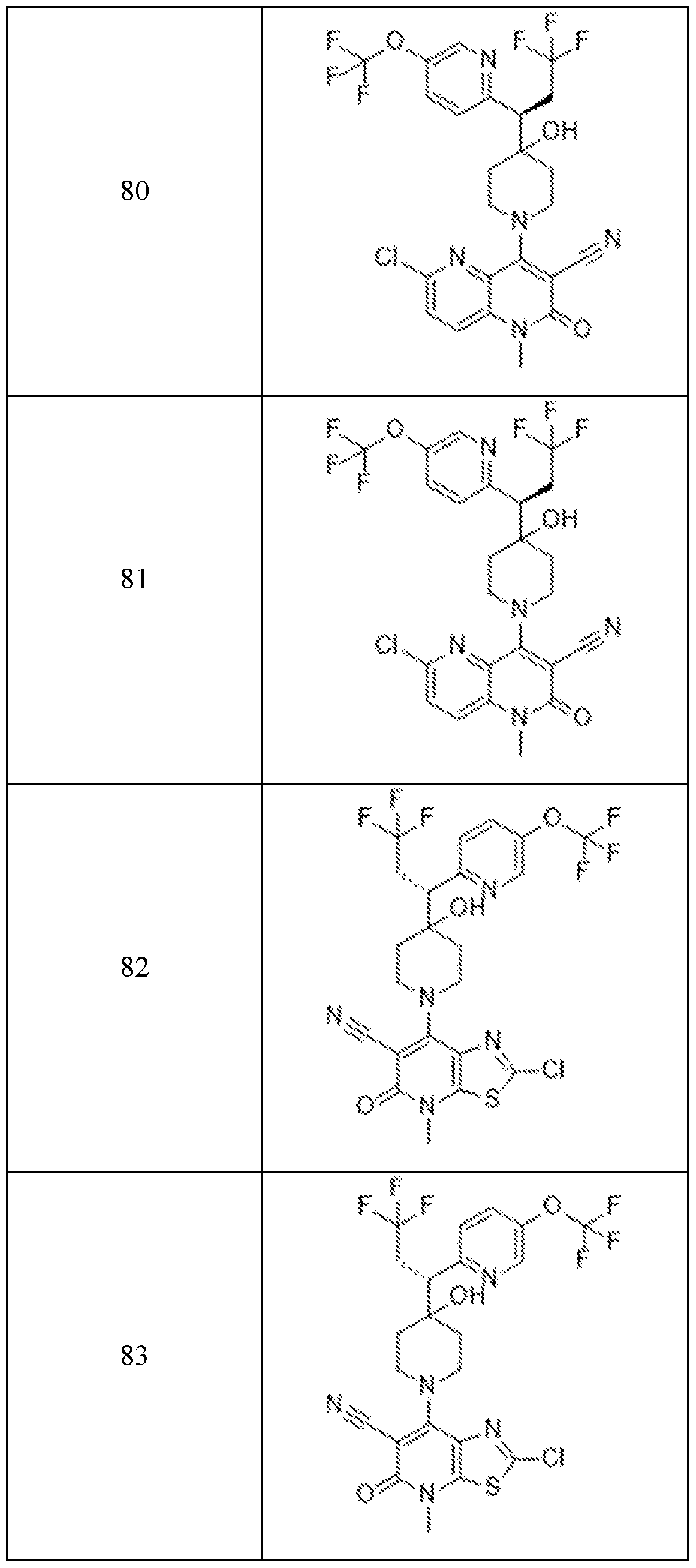





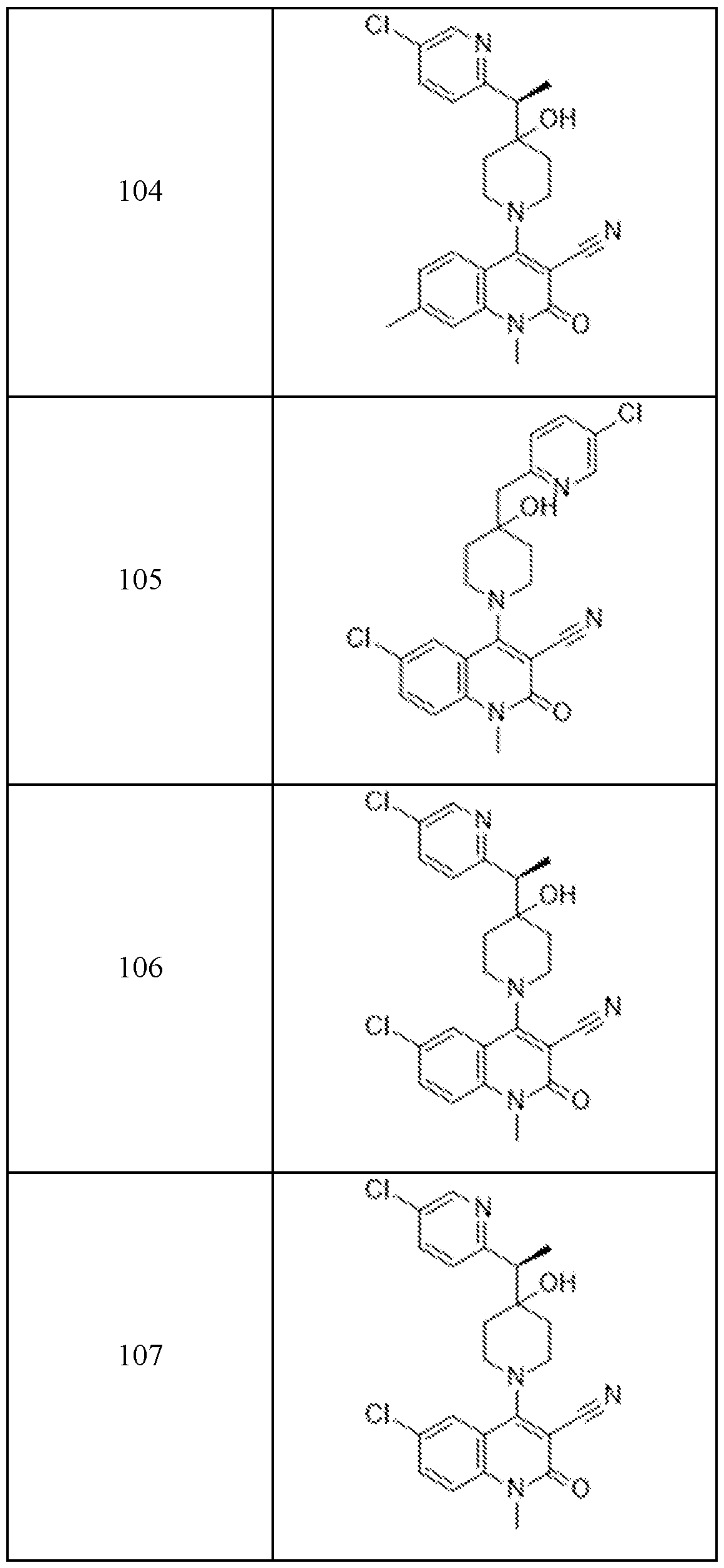






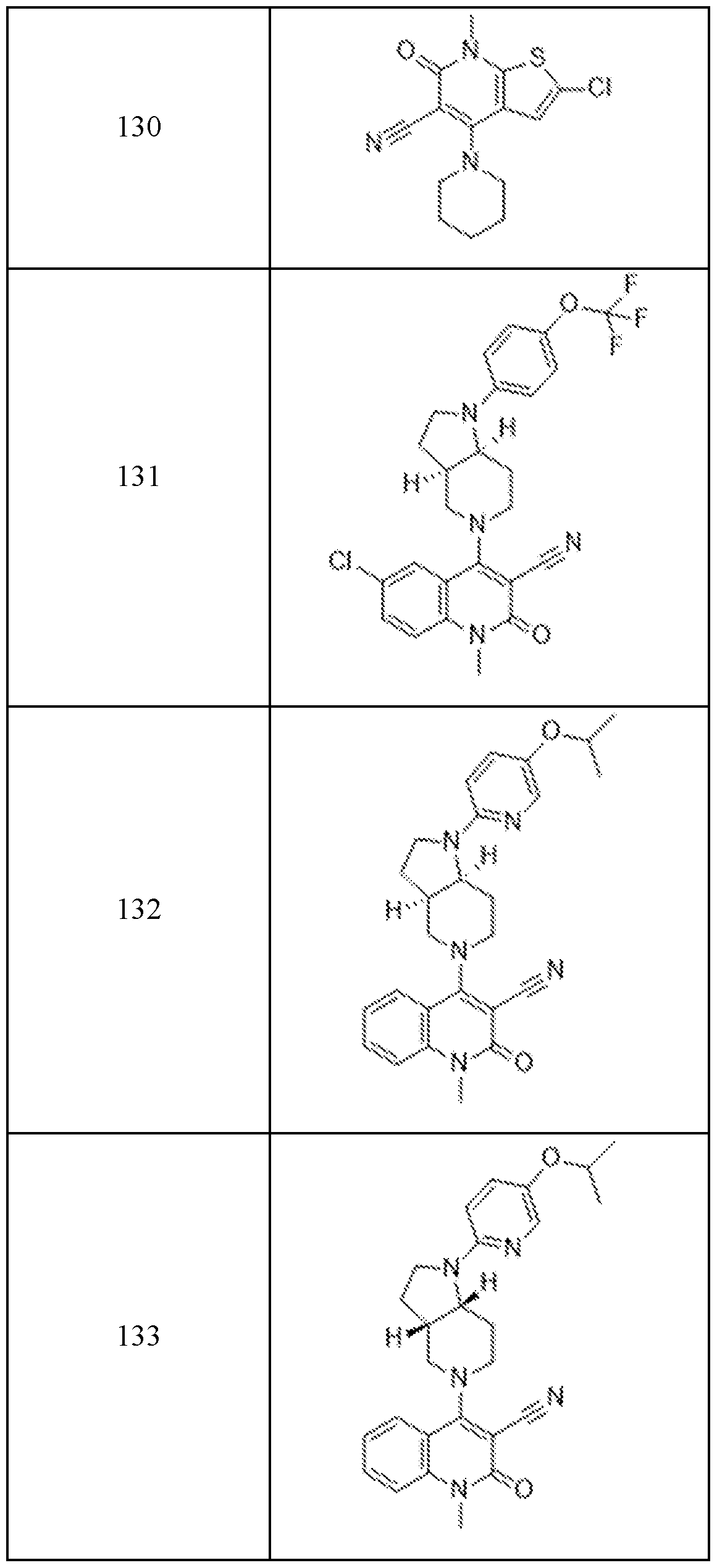






27. A pharmaceutical composition comprising a compound any one of claims 1-26 and a pharmaceutically acceptable excipient.
28. The pharmaceutical composition of claim 27, further comprising one or more additional anti-cancer agents.
29. The pharmaceutical composition of claim 28, wherein at least one of the additional anti-cancer agents is an immune checkpoint inhibitor.
30. A method of treating cancer, comprising administering an effective amount of the compound of any one of claims 1-26, or the pharmaceutical composition of any one of claims 27-30 to a subject.
31. The method of claim 30, wherein the cancer is selected from colon cancer, pancreatic cancer, breast cancer, prostate cancer, lung cancer, ovarian cancer, cervical cancer, renal cancer, head and/or neck cancer, lymphoma, lymphoma, leukemia, and melanoma.
32. A method of inhibiting the activity of diacylglycerol kinase alpha (DGKα), comprising administering a therapeutically effective amount of the compound of any one of claims 1-26, or the pharmaceutical composition of any one of claims 27-30 to a subject.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363530352P | 2023-08-02 | 2023-08-02 | |
| US63/530,352 | 2023-08-02 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2025030002A2 true WO2025030002A2 (en) | 2025-02-06 |
| WO2025030002A3 WO2025030002A3 (en) | 2025-03-27 |
Family
ID=92931924
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/040518 Pending WO2025030002A2 (en) | 2023-08-02 | 2024-08-01 | Dgk targeting compounds and uses thereof |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025030002A2 (en) |
Citations (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006029879A2 (en) | 2004-09-17 | 2006-03-23 | F.Hoffmann-La Roche Ag | Anti-ox40l antibodies |
| WO2006105021A2 (en) | 2005-03-25 | 2006-10-05 | Tolerrx, Inc. | Gitr binding molecules and uses therefor |
| WO2006122150A1 (en) | 2005-05-10 | 2006-11-16 | Incyte Corporation | Modulators of indoleamine 2,3-dioxygenase and methods of using the same |
| WO2007005874A2 (en) | 2005-07-01 | 2007-01-11 | Medarex, Inc. | Human monoclonal antibodies to programmed death ligand 1 (pd-l1) |
| WO2007075598A2 (en) | 2005-12-20 | 2007-07-05 | Incyte Corporation | N-hydroxyamidinoheterocycles as modulators of indoleamine 2,3-dioxygenase |
| WO2008036653A2 (en) | 2006-09-19 | 2008-03-27 | Incyte Corporation | N-hydroxyamidinoheterocycles as modulators of indoleamine 2,3-dioxygenase |
| WO2008036642A2 (en) | 2006-09-19 | 2008-03-27 | Incyte Corporation | N-hydroxyamidinoheterocycles as modulators of indoleamine 2,3-dioxygenase |
| WO2008132601A1 (en) | 2007-04-30 | 2008-11-06 | Immutep | Cytotoxic anti-lag-3 monoclonal antibody and its use in the treatment or prevention of organ transplant rejection and autoimmune disease |
| WO2009009116A2 (en) | 2007-07-12 | 2009-01-15 | Tolerx, Inc. | Combination therapies employing gitr binding molecules |
| WO2009044273A2 (en) | 2007-10-05 | 2009-04-09 | Immutep | Use of recombinant lag-3 or the derivatives thereof for eliciting monocyte immune response |
| WO2009073620A2 (en) | 2007-11-30 | 2009-06-11 | Newlink Genetics | Ido inhibitors |
| WO2009156652A1 (en) | 2008-05-29 | 2009-12-30 | Saint-Gobain Centre De Recherches Et D'etudes Europeen | Cellular structure containing aluminium titanate |
| WO2010019570A2 (en) | 2008-08-11 | 2010-02-18 | Medarex, Inc. | Human antibodies that bind lymphocyte activation gene-3 (lag-3), and uses thereof |
| WO2010077634A1 (en) | 2008-12-09 | 2010-07-08 | Genentech, Inc. | Anti-pd-l1 antibodies and their use to enhance t-cell function |
| WO2011028683A1 (en) | 2009-09-03 | 2011-03-10 | Schering Corporation | Anti-gitr antibodies |
| WO2011056652A1 (en) | 2009-10-28 | 2011-05-12 | Newlink Genetics | Imidazole derivatives as ido inhibitors |
| WO2011070024A1 (en) | 2009-12-10 | 2011-06-16 | F. Hoffmann-La Roche Ag | Antibodies binding preferentially human csf1r extracellular domain 4 and their use |
| WO2011107553A1 (en) | 2010-03-05 | 2011-09-09 | F. Hoffmann-La Roche Ag | Antibodies against human csf-1r and uses thereof |
| WO2011109400A2 (en) | 2010-03-04 | 2011-09-09 | Macrogenics,Inc. | Antibodies reactive with b7-h3, immunologically active fragments thereof and uses thereof |
| WO2011131407A1 (en) | 2010-03-05 | 2011-10-27 | F. Hoffmann-La Roche Ag | Antibodies against human csf-1r and uses thereof |
| WO2011140249A2 (en) | 2010-05-04 | 2011-11-10 | Five Prime Therapeutics, Inc. | Antibodies that bind csf1r |
| WO2012032433A1 (en) | 2010-09-09 | 2012-03-15 | Pfizer Inc. | 4-1bb binding molecules |
| WO2012142237A1 (en) | 2011-04-15 | 2012-10-18 | Newlink Geneticks Corporation | Fused imidazole derivatives useful as ido inhibitors |
| WO2012145493A1 (en) | 2011-04-20 | 2012-10-26 | Amplimmune, Inc. | Antibodies and other molecules that bind b7-h1 and pd-1 |
| WO2013079174A1 (en) | 2011-11-28 | 2013-06-06 | Merck Patent Gmbh | Anti-pd-l1 antibodies and uses thereof |
| WO2013087699A1 (en) | 2011-12-15 | 2013-06-20 | F. Hoffmann-La Roche Ag | Antibodies against human csf-1r and uses thereof |
| WO2013119716A1 (en) | 2012-02-06 | 2013-08-15 | Genentech, Inc. | Compositions and methods for using csf1r inhibitors |
| WO2013132044A1 (en) | 2012-03-08 | 2013-09-12 | F. Hoffmann-La Roche Ag | Combination therapy of antibodies against human csf-1r and uses thereof |
| WO2013169264A1 (en) | 2012-05-11 | 2013-11-14 | Five Prime Therapeutics, Inc. | Methods of treating conditions with antibodies that bind colony stimulating factor 1 receptor (csf1r) |
| WO2014008218A1 (en) | 2012-07-02 | 2014-01-09 | Bristol-Myers Squibb Company | Optimization of antibodies that bind lymphocyte activation gene-3 (lag-3), and uses thereof |
| WO2014036357A1 (en) | 2012-08-31 | 2014-03-06 | Five Prime Therapeutics, Inc. | Methods of treating conditions with antibodies that bind colony stimulating factor 1 receptor (csf1r) |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BE636867A (en) * | 1963-08-30 | |||
| DK111387A (en) * | 1986-03-05 | 1987-09-06 | Otsuka Pharma Co Ltd | CARBOSTYRIL DERIVATIVES AND SALTS THEREOF, MEDICINE CONTAINING SUCH DERIVATIVES AND PROCEDURES FOR THE PREPARATION OF THE DERIVATIVES |
| TW200418829A (en) * | 2003-02-14 | 2004-10-01 | Avanir Pharmaceutics | Inhibitors of macrophage migration inhibitory factor and methods for identifying the same |
| GB0605689D0 (en) * | 2006-03-21 | 2006-05-03 | Novartis Ag | Organic compounds |
| US11866430B2 (en) * | 2018-06-27 | 2024-01-09 | Bristol-Myers Squibb Company | Naphthyridinone compounds useful as T cell activators |
| CN115210225A (en) * | 2019-11-28 | 2022-10-18 | 拜耳公司 | Substituted aminoquinolones as immuno-activated DGKALPHA inhibitors |
| MX2022006466A (en) * | 2019-11-28 | 2022-08-17 | Bayer Ag | Substituted aminoquinolones as dgkalpha inhibitors for immune activation. |
| CA3163107A1 (en) * | 2019-11-28 | 2021-06-03 | Bayer Aktiengesellschaft | Substituted aminoquinolones as dgkalpha inhibitors for immune activation |
| AU2020414688A1 (en) * | 2019-12-23 | 2022-08-18 | Bristol-Myers Squibb Company | Substituted quinolinonyl piperazine compounds useful as T cell activators |
| JP7677976B2 (en) * | 2019-12-23 | 2025-05-15 | ブリストル-マイヤーズ スクイブ カンパニー | Substituted Heteroaryl Compounds Useful as T Cell Activators - Patent application |
| AR120823A1 (en) * | 2019-12-23 | 2022-03-23 | Bristol Myers Squibb Co | SUBSTITUTED BICYCLIC COMPOUNDS USEFUL AS T-CELL ACTIVATORS |
| CA3228862A1 (en) * | 2021-08-03 | 2023-02-09 | Beigene, Ltd. | Pyrazolopyridinone compounds |
| WO2023150186A1 (en) * | 2022-02-01 | 2023-08-10 | Arvinas Operations, Inc. | Dgk targeting compounds and uses thereof |
-
2024
- 2024-08-01 WO PCT/US2024/040518 patent/WO2025030002A2/en active Pending
Patent Citations (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006029879A2 (en) | 2004-09-17 | 2006-03-23 | F.Hoffmann-La Roche Ag | Anti-ox40l antibodies |
| WO2006105021A2 (en) | 2005-03-25 | 2006-10-05 | Tolerrx, Inc. | Gitr binding molecules and uses therefor |
| WO2006122150A1 (en) | 2005-05-10 | 2006-11-16 | Incyte Corporation | Modulators of indoleamine 2,3-dioxygenase and methods of using the same |
| WO2007005874A2 (en) | 2005-07-01 | 2007-01-11 | Medarex, Inc. | Human monoclonal antibodies to programmed death ligand 1 (pd-l1) |
| WO2007075598A2 (en) | 2005-12-20 | 2007-07-05 | Incyte Corporation | N-hydroxyamidinoheterocycles as modulators of indoleamine 2,3-dioxygenase |
| WO2008036653A2 (en) | 2006-09-19 | 2008-03-27 | Incyte Corporation | N-hydroxyamidinoheterocycles as modulators of indoleamine 2,3-dioxygenase |
| WO2008036642A2 (en) | 2006-09-19 | 2008-03-27 | Incyte Corporation | N-hydroxyamidinoheterocycles as modulators of indoleamine 2,3-dioxygenase |
| WO2008132601A1 (en) | 2007-04-30 | 2008-11-06 | Immutep | Cytotoxic anti-lag-3 monoclonal antibody and its use in the treatment or prevention of organ transplant rejection and autoimmune disease |
| WO2009009116A2 (en) | 2007-07-12 | 2009-01-15 | Tolerx, Inc. | Combination therapies employing gitr binding molecules |
| WO2009044273A2 (en) | 2007-10-05 | 2009-04-09 | Immutep | Use of recombinant lag-3 or the derivatives thereof for eliciting monocyte immune response |
| WO2009073620A2 (en) | 2007-11-30 | 2009-06-11 | Newlink Genetics | Ido inhibitors |
| WO2009156652A1 (en) | 2008-05-29 | 2009-12-30 | Saint-Gobain Centre De Recherches Et D'etudes Europeen | Cellular structure containing aluminium titanate |
| WO2010019570A2 (en) | 2008-08-11 | 2010-02-18 | Medarex, Inc. | Human antibodies that bind lymphocyte activation gene-3 (lag-3), and uses thereof |
| WO2010077634A1 (en) | 2008-12-09 | 2010-07-08 | Genentech, Inc. | Anti-pd-l1 antibodies and their use to enhance t-cell function |
| WO2011028683A1 (en) | 2009-09-03 | 2011-03-10 | Schering Corporation | Anti-gitr antibodies |
| WO2011056652A1 (en) | 2009-10-28 | 2011-05-12 | Newlink Genetics | Imidazole derivatives as ido inhibitors |
| WO2011070024A1 (en) | 2009-12-10 | 2011-06-16 | F. Hoffmann-La Roche Ag | Antibodies binding preferentially human csf1r extracellular domain 4 and their use |
| WO2011109400A2 (en) | 2010-03-04 | 2011-09-09 | Macrogenics,Inc. | Antibodies reactive with b7-h3, immunologically active fragments thereof and uses thereof |
| WO2011107553A1 (en) | 2010-03-05 | 2011-09-09 | F. Hoffmann-La Roche Ag | Antibodies against human csf-1r and uses thereof |
| WO2011131407A1 (en) | 2010-03-05 | 2011-10-27 | F. Hoffmann-La Roche Ag | Antibodies against human csf-1r and uses thereof |
| WO2011140249A2 (en) | 2010-05-04 | 2011-11-10 | Five Prime Therapeutics, Inc. | Antibodies that bind csf1r |
| WO2012032433A1 (en) | 2010-09-09 | 2012-03-15 | Pfizer Inc. | 4-1bb binding molecules |
| WO2012142237A1 (en) | 2011-04-15 | 2012-10-18 | Newlink Geneticks Corporation | Fused imidazole derivatives useful as ido inhibitors |
| WO2012145493A1 (en) | 2011-04-20 | 2012-10-26 | Amplimmune, Inc. | Antibodies and other molecules that bind b7-h1 and pd-1 |
| WO2013079174A1 (en) | 2011-11-28 | 2013-06-06 | Merck Patent Gmbh | Anti-pd-l1 antibodies and uses thereof |
| WO2013087699A1 (en) | 2011-12-15 | 2013-06-20 | F. Hoffmann-La Roche Ag | Antibodies against human csf-1r and uses thereof |
| WO2013119716A1 (en) | 2012-02-06 | 2013-08-15 | Genentech, Inc. | Compositions and methods for using csf1r inhibitors |
| WO2013132044A1 (en) | 2012-03-08 | 2013-09-12 | F. Hoffmann-La Roche Ag | Combination therapy of antibodies against human csf-1r and uses thereof |
| WO2013169264A1 (en) | 2012-05-11 | 2013-11-14 | Five Prime Therapeutics, Inc. | Methods of treating conditions with antibodies that bind colony stimulating factor 1 receptor (csf1r) |
| WO2014008218A1 (en) | 2012-07-02 | 2014-01-09 | Bristol-Myers Squibb Company | Optimization of antibodies that bind lymphocyte activation gene-3 (lag-3), and uses thereof |
| WO2014036357A1 (en) | 2012-08-31 | 2014-03-06 | Five Prime Therapeutics, Inc. | Methods of treating conditions with antibodies that bind colony stimulating factor 1 receptor (csf1r) |
Non-Patent Citations (21)
| Title |
|---|
| AVILA-FLORES, A. ET AL., IMMUNOLOGY AND CELL BIOLOGY, vol. 95, 2017, pages 549 - 563 |
| CHEN, S. S. ET AL., FRONT CELL DEV BIOL., vol. 4, 2016, pages 130 |
| FACCIABENE ET AL., CANCER RES., vol. 72, 2012, pages 2162 - 71 |
| GOLDSTEIN ET AL., CLIN. CANCER RES., vol. 1, 1995, pages 1311 - 18 |
| GRIFFITHS ET AL.: "Introduction to Genetic Analysis", 1999, W. H. FREEMAN & CO. |
| JING, W. ET AL., CANCER RESEARCH, vol. 77, 2017, pages 5676 - 86 |
| KOHL ET AL., NAT. MED., vol. 1, no. 8, 1995, pages 792 - 97 |
| KRISHNA, S. ET AL., FRONT IMMUNOLOGY, vol. 4, 2013, pages 178 |
| MELLMAN ET AL., NATURE, vol. 480, 2011, pages 480 - 89 |
| MIZOGUCHI ET AL., SCIENCE, vol. 258, 1992, pages 1795 - 98 |
| NOESSNER, E., FRONT CELL DEV BIOL., vol. 5, 2017, pages 16 |
| OLENCHOCK B. A. ET AL., NATURE, vol. 11, 2006, pages 1174 - 81 |
| PRINZ, P. U. ET AL., J IMMUNOLOGY, vol. 12, 2012, pages 5990 - 6000 |
| SAUSVILLE CURR. MED. CHEM. ANTI-CANC. AGENTS, vol. 3, 2003, pages 47 - 56 |
| SEKULIC ET AL., CANCER RES., vol. 60, 2000, pages 3504 - 13 |
| SJ OBLOM ET AL., SCIENCE, vol. 314, 2006, pages 268 - 74 |
| THE MCGRAW-HILL |
| TOPALIAN ET AL., CURR. OPIN. IMMUNOL., vol. 24, 2012, pages 1 - 6 |
| VLAHOS ET AL., J. BIOL., vol. 269, 1994, pages 5241 - 48 |
| WUTSGREENE: "Greene's Protective Groups in Organic Synthesis", 2007, WILEY AND SONS |
| ZHA Y ET AL., NATURE IMMUNOLOGY, vol. 12, 2006, pages 1343 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2025030002A3 (en) | 2025-03-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES3013568T3 (en) | Substituted heteroaryl compounds useful as t cell activators | |
| CN114728950B (en) | Compounds that can act as inhibitors of HELIOS proteins | |
| ES2925450T3 (en) | Isofuranone compounds useful as HPK1 inhibitors | |
| ES3008848T3 (en) | Substituted bicyclic piperidine derivatives useful as t cell activators | |
| ES2998500T3 (en) | Substituted quinolinonyl piperazine compounds useful as t cell activators | |
| CN114846007B (en) | Substituted quinazoline compounds useful as T cell activators | |
| BR112020017421A2 (en) | IMIDAZOPYRIMIDINES AND TRIAZOLOPYRIMIDINES AS A2A / A2B INHIBITORS | |
| WO2023150186A1 (en) | Dgk targeting compounds and uses thereof | |
| WO2021258010A1 (en) | Oxime compounds useful as t cell activators | |
| EP3676279A1 (en) | Cyclic dinucleotides as anticancer agents | |
| EP3697801A1 (en) | Cyclic dinucleotides as anticancer agents | |
| WO2025030002A2 (en) | Dgk targeting compounds and uses thereof | |
| WO2025064197A1 (en) | Substituted azetidinyl oxoisoindolinyl piperidine-2,6-dione compounds | |
| EP3762397A1 (en) | Cyclic dinucleotides as anticancer agents | |
| TW202321237A (en) | Map4k1 inhibitors | |
| CN121487946A (en) | Spirocyclic substituted oxo-isoindolinyl piperidine-2, 6-dione compounds | |
| TW202321238A (en) | Map4k1 inhibitors | |
| WO2025226767A1 (en) | Substituted 3-(5-(6-aminopyridin-2-yl)-4-fluoro-1-oxoisoindolin-2-yl)piperidine-2,6-dione compounds for use in the treatment of cancer | |
| EA048583B1 (en) | PYRIDINYL-SUBSTITUTED OXOISOINDOLINE COMPOUNDS FOR THE TREATMENT OF MALIGNANT TUMOR | |
| EA047609B1 (en) | SUBSTITUTED HETEROARYL COMPOUNDS USEFUL AS T-CELL ACTIVATORS | |
| TW202525802A (en) | Substituted phenyl oxooxazolyl piperidine dione compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24782660 Country of ref document: EP Kind code of ref document: A2 |