WO2025024848A1 - Methods for treating type 1 diabetes - Google Patents
Methods for treating type 1 diabetes Download PDFInfo
- Publication number
- WO2025024848A1 WO2025024848A1 PCT/US2024/040020 US2024040020W WO2025024848A1 WO 2025024848 A1 WO2025024848 A1 WO 2025024848A1 US 2024040020 W US2024040020 W US 2024040020W WO 2025024848 A1 WO2025024848 A1 WO 2025024848A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- teplizumab
- day
- course
- dose
- subject
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2809—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against the T-cell receptor (TcR)-CD3 complex
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/545—Medicinal preparations containing antigens or antibodies characterised by the dose, timing or administration schedule
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/52—Constant or Fc region; Isotype
Definitions
- Type 1 diabetes is caused by the autoimmune destruction of insulin producing beta cells in the islets of Langerhans, leading to dependence on exogeneous insulin injections for survival.
- T ID Approximately 1.6 million Americans have T ID. It is one of the most common childhood diseases.
- T1D Despite improvements in care, most affected individuals with T1D are not able to consistently achieve desired glycemic targets.
- T1D there are persisting concerns for increased risk of both morbidity and mortality.
- Two recent studies noted loss of 17.7 life-years for children diagnosed before age 10, and loss of 11 and 13 life-years respectively for Scottish men and women diagnosed as adults.
- T1D treatment methods and compositions Two recent studies noted loss of 17.7 life-years for children diagnosed before age 10, and loss of 11 and 13 life-years respectively for Scottish men and women diagnosed as adults.
- the present disclosure relates to methods of treating T1D with teplizumab, e.g., by slowing the loss of beta-cell function.
- the T1D to be treated may be Stage 3 or clinical T1D.
- the patient has new or recent onset T1D, e.g., the patient has been diagnosed with Stage 3 or clinical T1D within six weeks prior to teplizumab treatment.
- the subject has at least 20% of beta-cell function prior to teplizumab treatment.
- the patient has a peak C-peptide level of at least 0.2 pmol/mL during a mixed meal tolerance test (MMTT), for example, a two-hour MMTT, prior to teplizumab treatment.
- MMTT mixed meal tolerance test
- the patient is a child or an adolescent, e.g., about 8 to 17 (inclusive) years of age.
- the method comprises administering to a subject in need thereof a first 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 9500 pg/m 2 , wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during an MMTT (e.g., a two-hour MMTT) prior to administration of the first 12-day course of teplizumab; and administering to the subject a second 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 9500 pg/m 2 , wherein the first and the second 12-day courses of teplizumab are administered at an interval of at least about 3 months, or at about a 6-month to about a 12-month interval.
- MMTT e.g., a two-hour MMTT
- the method comprises administering to a subject in need thereof a first 12-day course of teplizumab at a total dose of from about 9000 to about 14000 pg/m 2 , wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during an MMTT (e.g., a two-hour MMTT) prior to administration of the 12-day course of teplizumab; and administering to the subject a second 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 14000 pg/m 2 , wherein the first and the second 12-day courses of teplizumab are administered at an interval of at least about 3 months, or at about a 6-month to about a 12-month interval.
- MMTT e.g., a two-hour MMTT
- teplizumab for use in a method of treating T1D, the method comprising administering to a subject in need thereof a first 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 9500 pg/m 2 , wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during an MMTT (e.g., a two-hour MMTT) prior to administration of the first 12-day course of teplizumab; and administering to the subject a second 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 9500 pg/m 2 , wherein the first and the second 12-day courses of teplizumab are administered at an interval of at least 3 months, or at about a 6-month to about a 12-month interval.
- MMTT e.g., a two-hour MMTT
- teplizumab for use in a method of treating T1D, the method comprising administering to a subject in need thereof a first 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 14000 pg/m 2 , wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during an MMTT (e.g., a two-hour MMTT) prior to administration of the first 12-day course of teplizumab; and administering to the subject a second 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 14000 pg/m 2 , wherein the first and the second 12-day courses of teplizumab are administered at an interval of at least 3 months, or at about a 6-month to about a 12-month interval.
- MMTT e.g., a two-hour MMTT
- the 12-day course comprises a first dose of 106 pg/m 2 teplizumab on day 1, a second dose of 425 pg/m 2 teplizumab on day 2, and one dose of 850 pg/m 2 teplizumab on each of days 3-12, and wherein the total teplizumab dose is approximately 9031 pg/m 2 .
- the method further comprises administering to the subject a third or more 12-day course of teplizumab, each course at a total dose of more than about 9000 pg/m 2 .
- the third or more 12-day course of teplizumab comprises a first dose of 106 pg/m 2 teplizumab on day 1, a second dose of 425 pg/m 2 teplizumab on day 2, and one dose of 850 pg/m 2 teplizumab on each of days 3-12, and the total teplizumab dose of each course is approximately 9031 pg/m 2 .
- the third or more 12-day course of teplizumab is administered at an interval of at least 3 months, or at about a 6-month to about a 24-month interval.
- the method comprises determining, after administration of each 12-day course, a baseline level of TIGIT+KLRG1+CD8+ T-cells and/or a baseline level of PD-1+CD8+ T-cells with respect to all CD3+ T-cells, monitoring the level of TIGIT+KLRG1+CD8+CD3+ T-cells and/or the level of PD-1+CD8+CD3+ T-cells, and administering an additional 12-day course of teplizumab when the level of the TIGIT+KLRG1+CD8+CD3+ T-cells and/or PD-1+CD8+CD3+ T-cells returns to the baseline level.
- the baseline level of the TIGIT+KLRG1+CD8+ T-cells and/or the baseline level of the PD-1+CD8+ T-cells is less than about 5% of all CD3+ T-cells.
- the determining of TIGIT+KLRG1+CD8+CD3+ T-cells and/or the PD- 1+CD8+CD3+ T-cells is by flow cytometry.
- the monitoring of TIGIT+KLRG1+CD8+CD3+ T-cells and/or PD-1+CD8+CD3+ T-cells is by flow cytometry.
- the determining of TIGIT+KLRG1+CD8+CD3+ T-cells and/or PD- 1+CD8+CD3+ T-cells is about 1-6 months, about 2-5 months, or about 3 months after the administration of each 12-day course. In some embodiments, if the subject has more than about 10% TIGIT+KLRG1+CD8+ T-cells and/or more than about 10% PD-1+CD8+ T-cells in all CD3+ T-cells, subsequent monitoring is annual.
- subsequent monitoring is about every 3-6 months.
- each dose of teplizumab is administered parenterally (e.g., by intravenous infusion).
- the subject may be pre-mediated with (1) a nonsteroidal anti- inflammatory drug (NSAID) or acetaminophen, (2) an antihistamine, and/or (3) an antiemetic before each dose in the first three, four, five, six, or seven days of each course.
- NSAID nonsteroidal anti- inflammatory drug
- acetaminophen acetaminophen
- an antihistamine an antiemetic
- the subject has a peak C-peptide level ranging from 0.2 pmol/mL to 0.7 pmol/mL during an MMTT (e.g., a two-hour MMTT). In some embodiments, the subject in need thereof has a peak C-peptide level of at least 0.7 pmol/mL during an MMTT (e.g., a two-hour MMTT).
- the method comprises assessing the area under the timeconcentration curve (AUC) of C-peptide following an MMTT at 78 weeks or at 18 months.
- the subject administered with teplizumab has a higher mean C-peptide value compared with a control administered with placebo.
- a “control administered with placebo” is a control subject who receives no teplizumab (i.e., no dose of teplizumab).
- the administering of teplizumab to the subject results in a 40% to 80%, or more than 80%, higher mean C-peptide value compared with subjects receiving placebo.
- the subject administered with teplizumab maintains or reduces baseline HbAlc levels, and/or maintains or increases Time in Range (TIR) with less insulin use, than a subject administered with placebo.
- TIR Time in Range
- the subject administered with teplizumab has a reduced HbAlc level compared to the pre-treatment level (i.e., before any treatment with teplizumab).
- the administering of teplizumab to the subject results in a reduction of insulin dosage by 10% to 30%, or more than 30%, compared to subjects receiving placebo.
- the administering of teplizumab to the subject results in a reduction of insulin dosage by at least 0.1 U/kg/day or in maintenance of insulin dosage.
- the administering of teplizumab to the subject results in a reduction of HbAlc baseline by 0.1 to 1 point, or more than 1 point, compared to subjects receiving placebo.
- the administering of teplizumab to the subject results in an increase of 3 to 10%, or more than 10% in TIR (%) for glycemia as assessed using a glucose monitoring system.
- the administering of teplizumab to the subject results in fewer Level 3 hypoglycemic episodes (e.g., at least 1, 2, or 3 fewer per year) than control subjects receiving placebo.
- aspects of the disclosure relate to a method of maintaining or increasing C-peptide value and/or slowing loss of beta-cell function in a subject having T1D, e.g., Stage 3 or clinical T1D.
- the method comprises administering to the subject a first 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 9500 pg/m 2 , wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during an MMTT prior to administration of the first 12-day course of teplizumab; and administering to the subject a second 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 9500 pg/m 2 , wherein the first and the second 12-day courses of teplizumab are administered at about a 6-month to about a 12-month interval, and wherein the administration of teplizumab
- the method comprises administering to the subject a first 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 14000 pg/m 2 , wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during an MMTT (e.g., a two-hour MMTT) prior to administration of the first 12-day course of teplizumab; and administering to the subject a second 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 14000 pg/m 2 , wherein the first and the second 12-day courses of teplizumab are administered at an interval of at least about 3 months, or at about a 6-month or to about a 12-month interval, and wherein the administration of teplizumab results in a 40% to 80%, or a more than 80%, increase in mean C-peptide value compared to subjects receiving placebo
- teplizumab for use in a method of maintaining or increasing C-peptide value and/or slowing loss of beta-cell function in a subject having T1D, e.g., Stage 3 or clinical T1D, the method comprising administering to the subject a first 12- day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 9500 pg/m 2 , wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during an MMTT (e.g., a two-hour MMTT) prior to administration of the first 12-day course of teplizumab; and administering to the subject a second 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 9500 pg/m 2 , wherein the first and the second 12-day courses of teplizumab are administered at an interval of at least
- teplizumab for use in a method of maintaining or increasing C-peptide value and/or slowing loss of beta-cell function in a subject having T1D, e.g., Stage 3 or clinical T1D, the method comprising administering to the subject a first 12- day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 14000 pg/m 2 , wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during an MMTT (e.g., a two-hour MMTT) prior to administration of the first 12-day course of teplizumab; and administering to the subject a second 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 14000 pg/m 2 , wherein the first and the second 12-day courses of teplizumab are administered at an interval of at least
- the 12-day course comprises a first dose of 106 pg/m 2 teplizumab on day 1, a second dose of 425 pg/m 2 teplizumab on day 2, and one dose of 850 pg/m 2 on each of days 3-12, and wherein the total dose is approximately 9031 pg/m 2 .
- the method further comprises administering to the subject in need thereof a third or more 12-day course of teplizumab, each course at a total dose of more than about 9000 pg/m 2 .
- the third or more 12-day course of teplizumab comprises a first dose of 106 pg/m 2 teplizumab on day 1, a second dose of 425 pg/m 2 teplizumab on day 2, and one dose of 850 pg/m 2 on each of days 3-12, and the total dose of each course is approximately 9031 pg/m 2 .
- the third or more 12-day course of teplizumab is administered at about a 6-month to about a 24-month interval.
- the method comprises determining, after administration of each 12-day course, a baseline of a level of TIGIT+KLRG1+CD8+ T-cells and/or a baseline of a level of PD-1+CD8+ T-cells with respect to all CD3+ T-cells, monitoring the level of TIGIT+KLRG1+CD8+CD3+ T-cells and/or the level of PD-1+CD8+CD3+ T-cells; and administering an additional 12-day course of teplizumab when the level of the TIGIT+KLRG1+CD8+CD3+ T-cells and/or PD-1+CD8+CD3+ T-cells returns to the baseline level.
- the baseline level of the TIGIT+KLRG1+CD8+ T-cells and/or the baseline level of the PD-1+CD8+ T-cells is less than about 5% of all CD3+ T-cells.
- the determining of TIGIT+KLRG1+CD8+CD3+ T-cells and/or the PD- 1+CD8+CD3+ T-cells is by flow cytometry.
- the monitoring of TIGIT+KLRG1+CD8+CD3+ T-cells and/or the PD-1+CD8+CD3+ T-cells is by flow cytometry.
- the determining of TIGIT+KLRG1+CD8+CD3+ T-cells and/or the PD-1+CD8+CD3+ T-cells is about 1-6 months, about 2-5 months, or about 3 months after the administration of each 12-day course. In some embodiments, if the subject has more than about 10% TIGIT+KLRG1+CD8+ T-cells and/or more than about 10% PD- 1+CD8+ T-cells in all CD3+ T-cells, subsequent monitoring is annual.
- subsequent monitoring is about every 3-6 months.
- each dose of teplizumab is administered parenterally. In some embodiments, each dose of teplizumab is administered by intravenous infusion. The patient may be pre-mediated as described herein.
- the subject has a peak C-peptide level ranging from 0.2 pmol/mL to 0.7 pmol/mL during an MMTT (e.g., a two-hour MMTT). In some embodiments, the subject has a peak C-peptide level of at least 0.7 pmol/mL during an MMTT (e.g., a two-hour MMTT).
- the method comprises assessing the AUC of C-peptide following an MMTT (e.g., a four-hour MMTT) at 78 weeks or 18 months.
- an MMTT e.g., a four-hour MMTT
- the subject administered with teplizumab maintains or reduces baseline HbAlc levels, and/or maintains or increases TIR with less insulin use, than a subject administered with placebo.
- the administering of teplizumab to the subject results in a reduction of insulin dosage of 10% to 30%, or more than 30%, compared to subjects receiving placebo.
- the administering of teplizumab to the subject results in a reduction of insulin dosage by at least 0.1 U/kg/day or in maintenance of insulin dosage.
- the administering of teplizumab to the subject results in a reduction of HbAlc baseline by 0.1 to 1 point, or more than 1 point, compared to subjects receiving placebo.
- the administering of teplizumab to the subject results in an increase of 3 to 10%, or more than 10%, TIR (%) for glycemia as assessed using a glucose monitoring system.
- Slowing the loss of beta-cell function includes preserving beta-cells, slowing the loss or destruction of beta-cells, and/or preserving beta-cell function (e.g., production of insulin).
- the administering of teplizumab results in slower loss of beta-cell function for at least about 18 months or 78 weeks.
- Figure 25 Diagram of the study design according to one embodiment.
- Figure 26 Modified Dosing Schedule for Participants Affected by COVID-19 Pandemic Restrictions according to one embodiment.
- Figure 27 Graph showing insulin use at different timepoints (baseline, week 12, week 26, week 39, week 52, week 65 and week 78). Figure 27 shows that insulin use was numerically lower in patients treated with teplizumab.
- Figure 28 Graph showing the percentage of subjects who met HblAc ⁇ 6.5% and insulin daily dose ⁇ 0.25 unit/kg/days at different timepoints (baseline, week 12, week 26, week 39, week 52, week 65 and week 78). Figure 28 shows that more teplizumab patients met insulin discontinuation criteria of HblAc ⁇ 6.5% and insulin daily dose ⁇ 0.25 unit/kg/days.
- Figure 29 Graph showing the HbAlc levels at different timepoints (baseline, week 12, week 26, week 39, week 52, week 65 and week 78). Figure 29 shows that HbAlc target levels were achieved in both treatment groups.
- Figure 30 Graph showing the percentage time in range at different timepoints (baseline, week 12, week 26, week 39, week 52, week 65 and week 78).
- Figure 30 shows that patients treated with teplizumab spent more Time in Range (TIR).
- Figures 31A-31D Graphs showing the efficacy endpoints.
- Figure 31A shows stimulated C-peptide levels area under the curve (ln(AUC+l) over time, data are presented as least-squares mean (95% CI).
- Figure 31B shows the proportion of patients with time in range (>70%).
- Figure 31C shows average daily insulin dose ⁇ 0.25 units/kg/d over time, data are presented as least-squares mean (95% CI).
- Figure 31D shows the proportion of patients meeting criteria for clinical remission (HbAlc ⁇ 6.5% and insulin daily dose ⁇ 0.25 units/kg/d).
- HbAlc ⁇ 6.5% and insulin daily dose ⁇ 0.25 units/kg/d.
- Figure 32 Graph showing the subgroup analysis of C-peptide AUC- ITT Population (AUC, area under the curve; CI, confidence interval).
- Figure 33 Graph showing the proportion of patients with peak C-peptide >0.2 pmol/mL over time. ***P ⁇ 0.001. Error bars indicate 95% confidence intervals. Percentages are based on the number of non-missing observations in each treatment group. Estimates are obtained from a generalized linear model for repeated measures using logit link function that includes treatment, visit, age group at randomization, and baseline peak C-peptide as fixed effects, and a treatment by visit interaction term.
- Figures 34 A-34B Graphs showing Hb Ale levels.
- Figure 34A shows Hb Ale level over time
- Figure 34B shows the proportion of Patients with HbAlc ⁇ 7% Over Time.
- MMRM mixed effect model for repeated measures. Error bars indicate 95% confidence interval. Baseline is defined as the most recent value collected prior to the first dose of study drug. Estimates are based on an, mixed effect model for repeated measures (MMRM model) with treatment group, visit, age group at randomization, screening peak C- peptide category, and a treatment by visit interaction term as fixed effects.
- Figure 35 Graph showing the proportion of patients with insulin dose ⁇ 0.25 units/kg/day by study visit. Error bars indicate 95% CI.
- Figure 36 Graph showing key clinical outcome assessment domains at Week 78.
- DTSQ Diabetes Treatment Satisfaction Questionnaire
- MMRM mixed effect model for repeated measures
- HFS Hypoglycemic Fear Survey
- PedsQL Pediatric Quality of Life Inventory.
- Estimates and p-value are based on an MMRM model with treatment group, visit, age group at randomization, screening peak C-peptide category, baseline score, and a treatment by visit interaction term as fixed effects.
- Least-squares mean difference teplizumab - placebo.
- Minimal clinically important difference scores in PedsQL were 5.27 for child/teen and 4.54 for parents.
- HFS scores range from 0 to 4 where a lower score indicates a better outcome. Signs have been adjusted for graphical representation.
- DTSQ scores range from 0 to 48 where a higher score indicates a better outcome.
- Figure 37 Plot Emax model: predicted C-peptide change vs AUC, Year 2.
- the Protege study was conducted in newly diagnosed (Stage 3) T1D patients and tested 3 teplizumab dosing regimens (full 14-day [about 9,030 g/m 2 cumulative dose], one-third of the 14-day regimen [1/3], and a 6-day curtailed [first 6 days of the full 14-day regimen]).
- T1D usually develops in childhood and adolescence; however, it can also present in adulthood as late as the 5th and 6th decades of life, although much less frequently (Atkinson 2014, Bluestone 2010, Streisand 2014). In addition to being more prone to some short- and long-term complications, there are differences in the clinical course and response to immune therapies between children/young adults and older adults. In the days or weeks before initial diagnosis, children and adolescents often suffer from severe diabetes symptoms, including polydipsia, polyuria, and weight loss, which could result in a clinical presentation of DKA and shock which requires hospitalization (Atkinson 2014, Bluestone 2010, Streisand 2014, Mittermayer 2017). Children and young adults with new-onset T1D usually have an immediate need for exogenous insulin.
- aspects of the disclosure relate to methods of treating T1D in subjects in need thereof.
- Provided herein are methods that preserve [3-cell function and improve clinical management of T1D in children compared with the natural course of disease and current standard of care including exogenous insulin therapy.
- the preservation of [3-cell function is anticipated to translate to clinical and/or metabolic benefits consistent with improved ability to maintain glycemic control and short- and/or long-term outcomes.
- the method comprises diagnosing patients 8 to 17 years of age with T1D, administering to the patients within 6 weeks of diagnosis a first course of daily doses of teplizumab for 12 days, and a second course of daily doses of teplizumab for 12 days, wherein the first and second courses are separated by a 6-month interval.
- the method further comprises assessing the AUC of C-peptide following an MMTT) at 78 weeks (18 months or 1.5 years), and/or evaluating clinical endpoints such insulin use, HbAlc levels, and hypoglycemic episodes.
- kits for treating clinical T1D comprising administering to a subject in need thereof a 12-day course of teplizumab at a total dose of more than about 9000 pg/m 2 , wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during an MMTT prior to administration of the 12-day course of teplizumab.
- a C-peptide level indicates that the subject is still producing insulin.
- the administration of the 12-day course of teplizumab can substantially protect P-cells, prevent P cell death over time, and/or significantly reduce the extent of P-cell death over time.
- the administration of the 12-day course of teplizumab can lessen or prevent reduction of pancreatic insulin production capacity. In some embodiments, the administration of a 12-day course of teplizumab can decrease or eliminate the need for insulin use. In some embodiments, the method comprises administering two 12-day courses separated by 6 or 12 months.
- each dose is administered parenterally. In some embodiments, each dose is administered by intravenous infusion.
- the administrating step results in reduction by at least 10% of insulin use as compared to subjects treated with placebo.
- the administration of teplizumab can decrease the amount of insulin needed to maintain or decrease HbAlc in the subject.
- the articles “a” and “an” refer to one or more than one, e.g., to at least one, of the grammatical object of the article.
- the use of the words “a” or “an” when used in conjunction with the term “comprising” herein may mean “one,” but it is also consistent with the meaning of “one or more,” “at least one,” and “one or more than one.”
- “about” and “approximately” generally mean an acceptable degree of error for the quantity measured given the nature or precision of the measurements.
- Exemplary degrees of error are within 20 percent (%), typically, within 10%, and more typically, within 5% of a given range of values.
- the term “substantially” means more than 50%, preferably more than 80%, and most preferably more than 90% or 95%.
- compositions, methods, and respective component(s) thereof are used in reference to compositions, methods, and respective component(s) thereof, that are present in a given embodiment, yet open to the inclusion of unspecified elements.
- the term “consisting essentially of’ refers to those elements required for a given embodiment. The term permits the presence of additional elements that do not materially affect the basic and novel or functional characteristic(s) of that embodiment of the disclosure.
- compositions, methods, and respective components thereof as described herein, which are exclusive of any element not recited in that description of the embodiment.
- antibody herein is used in the broadest sense and encompasses various antibody structures, including but not limited to monoclonal antibodies, polyclonal antibodies, multispecific antibodies (e.g., bispecific antibodies), and antibody fragments so long as they exhibit the desired antigen-binding activity.
- An “antibody fragment” refers to a molecule other than an intact antibody that comprises a portion of an intact antibody that binds the antigen to which the intact antibody binds. Examples of antibody fragments include but are not limited to Fv, Fab, Fab', Fab'-SH, F(ab')2; diabodies; linear antibodies; single-chain antibody molecules (e.g., scFv); and multispecific antibodies formed from antibody fragments.
- onset of disease with reference to T1D refers to a patient meeting the criteria established for diagnosis of T1D by the American Diabetes Association (see Mayfield et al., Am Fam Physician (2006) 58: 1355-62).
- a “protocol” includes dosing schedules and dosing regimens.
- the protocols herein are methods of use and include therapeutic protocols.
- a “dosing regimen,” “dosage regimen,” or “course of treatment” may include administration of several doses of a therapeutic agent over 1 to 20 days.
- the terms “treat,” “treatment” and “treating” refer to the amelioration of one or more symptoms associated with T1D that results from the administration of one or more CD3 binding molecules. In some embodiments, such terms refer to a reduction in a human's average number of hypoglycemic episodes. In other embodiments, such terms refer to the maintenance of a reference level of C-peptide in the peripheral blood.
- the effective amount reduces one or more T1D symptoms by at least 5%, by at least 10%, by at least 20%, by at least 25%, by at least 30%, by at least 35%, by at least 40%, by at least 45%, by at least 50%, by at least 55%, by at least 60%, by at least 65%, by at least 70%, by at least 75%, by at least 80%, by at least 85%, by at least 90%, or by at least 95%.
- anti-CD3 antibody and “an antibody that binds to CD3” refer to an antibody or antibody fragment that is capable of binding cluster of d ferentiation 3 (CD3) with sufficient affinity such that the antibody is useful as a prophylactic, diagnostic and/or therapeutic agent in targeting CD3.
- the extent of binding of an anti- CD3 antibody to an unrelated, non-CD3 protein is less than about 10% of the binding of the antibody to CD3 as measured, e.g., by a radioimmunoassay (RIA).
- RIA radioimmunoassay
- the anti-CD3 antibody can be ChAglyCD3 (otelixizumab).
- Otelixizumab is a humanized Fc nonbinding anti-CD3, which was evaluated initially in phase 2 studies by the Belgian Diabetes Registry (BDR) and then developed by Tolerx, which then partnered with GSK to conduct the phase 3 DEFEND new onset T1D trials (NCT00678886, NCT01123083, NCT00763451).
- Otelixizumab is administered IV with infusions over 8 days. See, e.g., Wiczling et al., J Clin Pharmacol. (2010) 50(5):494-506; Keymeulen et al., N Engl J Med.
- the anti-CD3 antibody can be visilizumab (also called HuM291; Nuvion).
- Visilizumab is a humanized anti-CD3 monoclonal antibody characterized by a mutated IgG2 isotype, lack of binding to Fey receptors, and the ability to induce apoptosis selectively in activated T cells. It was evaluated in patients in graft-versus- host disease (NCT00720629; NCT00032279) and in ulcerative colitis (NCT00267306) and Crohn’s Disease (NCT00267709). See, e.g., Sandborn et al., Gut (2010) 59 (11): 1485-92, incorporated herein by reference.
- the anti-CD3 antibody can be teplizumab.
- Teplizumab also known as hOKT3yl(Ala-Ala) (containing an alanine at positions 234 and 235) is an anti-CD3 antibody that has been engineered to alter the function of the T lymphocytes that mediate the destruction of the insulin-producing P-cells of the islets of the pancreas.
- Teplizumab binds to an epitope of the CD3s chain expressed on mature T-cells and by doing so changes their function.
- Circulating T-cells are transiently reduced following teplizumab treatment, in a process that may include margination and depletion (Long 2017, Sherry 2011).
- teplizumab appears to both increase the number and function of regulatory T-cells (Tregs) (Ablamunits 2010, Bisikirska 2005, Long 2017, Waldron-Lynch 2012). More recent studies indicate that teplizumab induces immunologic “exhaustion” in a subset of effector CD8+ T-cells, perhaps making them more susceptible to regulation or deletion (Long 2016, Long 2017).
- teplizumab not only exerts a “suppressive” effect on p cell immune destructive processes but rather is an immune “modulator” favoring a rebalancing of effector and regulatory arms involved with T1D autoimmunity and supporting the notion that teplizumab may have the ability to contribute to the re-introduction of P cell self-tolerance (Lebastchi 2013).
- teplizumab Sequences and compositions of teplizumab are disclosed in U.S. Patent Nos. 6,491,916; 8,663,634; and 9,056,906, each incorporated herein by reference in its entirety.
- the molecular weight of teplizumab is approximately 150 KD.
- the full sequences of the light and heavy chains are set forth below.
- Bolded portions are the complementaritydetermining regions (CDR).
- compositions comprise an effective amount of an anti-CD3 antibody, and a pharmaceutically acceptable carrier.
- pharmaceutically acceptable means approved by a regulatory agency of the Federal or a state government or listed in the U.S. Pharmacopeia or other generally recognized pharmacopeia for use in animals, and more particularly in humans.
- carrier refers to a diluent, adjuvant (e.g., Freund's adjuvant (complete and incomplete)), excipient, or vehicle with which the therapeutic is administered.
- Such pharmaceutical carriers can be sterile liquids, such as water and oils, including those of petroleum, animal, vegetable or synthetic origin, such as peanut oil, soybean oil, mineral oil, sesame oil and the like. Water is a preferred carrier when the pharmaceutical composition is administered intravenously. Saline solutions and aqueous dextrose and glycerol solutions can also be employed as liquid carriers, particularly for injectable solutions.
- Suitable pharmaceutical excipients include starch, glucose, lactose, sucrose, gelatin, malt, rice, flour, chalk, silica gel, sodium stearate, glycerol monostearate, talc, sodium chloride, dried skim milk, glycerol, propylene, glycol, water, ethanol and the like (see, for example, Handbook of Pharmaceutical Excipients, Arthur H. Kibbe (ed., 2000, which is incorporated by reference herein in its entirety), Am. Pharmaceutical Association, Washington, D.C).
- compositions can also contain minor amounts of wetting or emulsifying agents, or pH buffering agents.
- These compositions can take the form of solutions, suspensions, emulsion, tablets, pills, capsules, powders, sustained release formulations and the like.
- Oral formulation can include standard carriers such as pharmaceutical grades of mannitol, lactose, starch, magnesium stearate, sodium saccharine, cellulose, magnesium carbonate, etc. Examples of suitable pharmaceutical carriers are described in “Remington's Pharmaceutical Sciences” by E. W. Martin.
- Such compositions contain a therapeutically effective amount of a therapeutic agent preferably in purified form, together with a suitable amount of carrier so as to provide the form for proper administration to the patient.
- the formulation should suit the mode of administration.
- the pharmaceutical compositions are sterile and in suitable form for administration to a subject, preferably an animal subject, more preferably a mammalian subject, and most preferably a human subject.
- the pharmaceutical compositions may be desirable to administer the pharmaceutical compositions locally to the area in need of treatment; this may be achieved by, for example, and not by way of limitation, local infusion, by injection, or by means of an implant, said implant being of a porous, non-porous, or gelatinous material, including membranes, such as sialastic membranes, or fibers.
- an implant being of a porous, non-porous, or gelatinous material, including membranes, such as sialastic membranes, or fibers.
- care must be taken to use materials to which the anti-CD3 antibody does not absorb.
- the composition can be delivered in a vesicle, in particular a liposome (see Langer, Science (1990) 249: 1527-33; Treat et al., in Liposomes in the Therapy of Infectious Disease and Cancer, Lopez-Berestein and Fidler (eds.), Liss, New York, pp. 353-365 (1989); Lopez-Berestein, ibid., pp. 317-327; see generally ibid.).
- a liposome see Langer, Science (1990) 249: 1527-33; Treat et al., in Liposomes in the Therapy of Infectious Disease and Cancer, Lopez-Berestein and Fidler (eds.), Liss, New York, pp. 353-365 (1989); Lopez-Berestein, ibid., pp. 317-327; see generally ibid.).
- the composition can be delivered in a controlled release or sustained release system.
- a pump may be used to achieve controlled or sustained release (see Langer, supra, Sefton, 1987, CRC Crit. Ref. Biomed. Eng. 14:20; Buchwald et al., 1980, Surgery 88:507; Saudek et al., N Engl J Med. (1989) 321 :574).
- polymeric materials can be used to achieve controlled or sustained release of the antibodies of the disclosure or fragments thereof (see, e.g., Medical Applications of Controlled Release, Langer and Wise (eds.), CRC Pres., Boca Raton, Fla.
- polymers used in sustained release formulations include, but are not limited to, polyphydroxy ethyl methacrylate), poly(methyl methacrylate), poly(acrylic acid), poly(ethylene- co-vinyl acetate), poly(methacrylic acid), polyglycolides (PLG), polyanhydrides, poly(N- vinyl pyrrolidone), poly(vinyl alcohol), polyacrylamide, poly(ethylene glycol), polylactides (PLA), poly(lactide-co-glycolides) (PLGA), and poly orthoesters.
- the polymer used in a sustained release formulation is inert, free of leachable impurities, stable on storage, sterile, and biodegradable.
- a controlled or sustained release system can be placed in proximity of the therapeutic target, i.e., the lungs, thus requiring only a fraction of the systemic dose (see, e.g., Goodson, in Medical Applications of Controlled Release, supra, vol. 2, pp. 115-138 (1984)).
- a pharmaceutical composition can be formulated to be compatible with its intended route of administration.
- routes of administration include, but are not limited to, parenteral, e.g., intravenous, intradermal, subcutaneous, oral, intranasal (e.g., inhalation), transdermal (topical), transmucosal, and rectal administration.
- the composition is formulated in accordance with routine procedures as a pharmaceutical composition adapted for intravenous, subcutaneous, intramuscular, oral, intranasal or topical administration to human beings.
- a pharmaceutical composition is formulated in accordance with routine procedures for subcutaneous administration to human beings.
- compositions for intravenous administration are solutions in sterile isotonic aqueous buffer.
- the composition may also include a solubilizing agent and a local anesthetic such as lignocaine to ease pain at the site of the injection.
- compositions may be formulated for parenteral administration by injection, e.g., by bolus injection or continuous infusion.
- Formulations for injection may be presented in unit dosage form, e.g., in ampoules or in multi-dose containers, with an added preservative.
- the compositions may take such forms as suspensions, solutions or emulsions in oily or aqueous vehicles, and may contain formulary agents such as suspending, stabilizing and/or dispersing agents.
- the active ingredient may be in powder form for constitution with a suitable vehicle, e.g., sterile pyrogen-free water, before use.
- the disclosure provides dosage forms that permit administration of the anti-CD3 antibody continuously over a period of hours or days (e.g., associated with a pump or other device for such delivery), for example, over a period of 1 hour, 2 hours, 3 hours, 4 hours, 6 hours, 8 hours, 10 hours, 12 hours, 16 hours, 20 hours, 24 hours, 30 hours, 36 hours, 4 days, 5 days, 7 days, 10 days or 12 days.
- a period of hours or days e.g., associated with a pump or other device for such delivery
- the disclosure provides dosage forms that permit administration of a continuously increasing dose, for example, increasing from 106 pg/m 2 /day to 850 pg/m 2 /day or 211 pg/m 2 /day to 840 pg/m 2 /day over a period of 24 hours, 30 hours, 36 hours, 4 days, 5 days, 7 days, 10 days or 12 days.
- compositions can be formulated as neutral or salt forms.
- Pharmaceutically acceptable salts include those formed with anions such as those derived from hydrochloric, phosphoric, acetic, oxalic, tartaric acids, etc., and those formed with cations such as those derived from sodium, potassium, ammonium, calcium, ferric hydroxides, isopropylamine, triethylamine, 2-ethylamino ethanol, histidine, procaine, etc.
- the ingredients of the compositions disclosed herein are supplied either separately or mixed together in unit dosage form, for example, as a dry lyophilized powder or water free concentrate in a hermetically sealed container such as an ampoule or sachet indicating the quantity of active agent.
- a hermetically sealed container such as an ampoule or sachet indicating the quantity of active agent.
- the composition is to be administered by infusion, it can be dispensed with an infusion bottle containing sterile pharmaceutical grade water or saline.
- an ampoule of sterile water for injection or saline can be provided so that the ingredients may be mixed prior to administration.
- the disclosure provides that the anti-CD3 antibodies, or pharmaceutical compositions thereof, can be packaged in a hermetically sealed container such as an ampoule or sachet indicating the quantity of the agent.
- the anti-CD3 antibody, or pharmaceutical compositions thereof is supplied as a dry sterilized lyophilized powder or water free concentrate in a hermetically sealed container and can be reconstituted, e.g., with water or saline to the appropriate concentration for administration to a subject.
- the anti-CD3 antibody, or pharmaceutical compositions thereof is supplied as a dry sterile lyophilized powder in a hermetically sealed container at a unit dosage of at least 5 mg, more preferably at least 10 mg, at least 15 mg, at least 25 mg, at least 35 mg, at least 45 mg, at least 50 mg, at least 75 mg, or at least 100 mg.
- the lyophilized agents, or pharmaceutical compositions herein should be stored at between 2 °C and 8 °C in its original container and the therapeutic agents, or pharmaceutical compositions of the disclosure should be administered within 1 week, preferably within 5 days, within 72 hours, within 48 hours, within 24 hours, within 12 hours, within 6 hours, within 5 hours, within 3 hours, or within 1 hour after being reconstituted.
- the pharmaceutical composition is supplied in liquid form in a hermetically sealed container indicating the quantity and concentration of the agent.
- the liquid form of the administered composition is supplied in a hermetically sealed container at least 0.25 mg/ml, more preferably at least 0.5 mg/ml, at least 1 mg/ml, at least 2.5 mg/ml, at least 5 mg/ml, at least 8 mg/ml, at least 10 mg/ml, at least 15 mg/ml, at least 25 mg/ml, at least 50 mg/ml, at least 75 mg/ml or at least 100 mg/ml.
- the liquid form should be stored at between 2 °C and 8 °C in its original container.
- the disclosure provides that the composition of the disclosure is packaged in a hermetically sealed container such as an ampoule or sachet indicating the quantity of the anti-CD3 antibody.
- compositions may, if desired, be presented in a pack or dispenser device that may contain one or more unit dosage forms containing the active ingredient.
- the pack may, for example, comprise metal or plastic foil, such as a blister pack.
- the amount of the composition of the disclosure which is effective in the treatment of one or more symptoms associated with T1D can be determined by standard clinical techniques.
- the precise dose to be employed in the formulation can also depend on the route of administration and the seriousness of the condition, and should be decided according to the judgment of the practitioner and each patient's circumstances. Effective doses may be extrapolated from dose-response curves derived from in vitro or animal model test systems.
- the present disclosure encompasses administration of antihuman CD3 antibodies such as teplizumab to patients 8 through 17 years old 6 weeks from T1D diagnosis having a peak C-peptide level of >0.2 pmol/mL during an MMTT.
- the peak C-peptide level at screening rages from 0.2 pmol/mL (inclusive) to 0.7 pmol/mL (inclusive).
- T1D diagnosis is according to the American Diabetes Association (ADA) criteria. As defined by the American Diabetes Association (ADA) for the clinical diagnosis of diabetes, the individual must meet one of the following 4 criteria:
- FPG fasting plasma glucose
- the test should be performed as described by the World Health Organization (WHO), using a glucose load containing the equivalent of 75 g anhydrous glucose dissolved in water.
- WHO World Health Organization
- HbAlc hemoglobin A1C
- NGSP National Glycohemoglobin Standardization Program
- DCCT Diabetes Control and Complications Trial
- ADA suggests that plasma blood glucose rather than HbAlC should be used to diagnose the acute onset of T1D in individuals with symptoms of hyperglycemia.
- ADA a patient with classic symptoms, measurement of plasma glucose is sufficient to diagnose clinical diabetes (symptoms of hyperglycemia or hyperglycemic crisis plus a random plasma glucose >200 mg/dL [11.1 mmol/L]).
- knowing the plasma glucose level is critical because, in addition to confirming that symptoms are due to diabetes, it will inform management decisions.
- Some providers may also want to know the HbAlC to determine how long a patient has had hyperglycemia.
- T1D previously called “insulin-dependent diabetes” or “juvenile-onset diabetes,” accounts for 5-10% of diabetes and is due to cellular-mediated autoimmune destruction of the pancreatic P-cells.
- Autoimmune markers include islet cell autoantibodies and autoantibodies to GAD (GAD65), insulin, the tyrosine phosphatases IA-2 and IA-2 P, and ZnT8. T1D is defined by the presence of one or more of these autoimmune markers.
- CGM continuous glucose monitoring system
- the patient diagnosed with clinical T1D has a positive result on testing for at least one of the following T ID-related autoantibodies: Glutamic acid decarboxylase 65 (GAD65) autoantibodies, Islet antigen 2 (IA-2) autoantibodies, Zinc transporter 8 (ZnT8) autoantibodies, Islet cell cytoplasmic autoantibodies (ICA), or Insulin autoantibodies (if testing obtained within the first 14 days of insulin treatment).
- Glutamic acid decarboxylase 65 Glutamic acid decarboxylase 65 (GAD65) autoantibodies, Islet antigen 2 (IA-2) autoantibodies, Zinc transporter 8 (ZnT8) autoantibodies, Islet cell cytoplasmic autoantibodies (ICA), or Insulin autoantibodies (if testing obtained within the first 14 days of insulin treatment).
- Glutamic acid decarboxylase 65 Glutamic acid decarboxylase 65 (GAD65) autoantibodies
- Islet antigen 2 IA-2
- the methods provided herein prevent or delay the need for administration of insulin to the patients.
- [3-cell function prior to, during, and after therapy may be assessed by methods described herein or by any method known to one of ordinary skill in the art.
- DCCT Diabetes Control and Complications Trial
- HbAl and HbAlc percentage glycosylated hemoglobin
- characterization of daily insulin needs, C-peptide levels/response, hypoglycemic episodes, and/or FPIR may be used as markers of [3-cell function or to establish a therapeutic index (see Keymeulen et al., N Engl J Med.
- FPIR is calculated as the sum of insulin values at 1 and 3 minutes post IGTT, which are performed according to Islet Cell Antibody Register User's Study protocols (see, e.g., Bingley et al., Diabetes (1996) 45: 1720-8 and McCulloch et al., Diabetes Care (1993) 16:911-5).
- the effective amount comprises a 12-day course of subcutaneous intravenous (IV) infusion of the anti-CD3 antibody such as teplizumab at 106- 850 micrograms/meter squared (pg/m 2 ).
- IV subcutaneous intravenous
- the total dosage over the duration of the regimen is about 14000 pg/m 2 , 13500 pg/m 2 , 13000 pg/m 2 , 12500 pg/m 2 , 12000 pg/m 2 , 11500 pg/m 2 , 11000 pg/m 2 , 10500 pg/m 2 , 10000 pg/m 2 , 9500 pg/m 2 , 9000 pg/m 2 , 8000 pg/m 2 , 7000 pg/m 2 , 6000 pg/m 2 , and may be less than 5000 pg/m 2 , 4000 pg/m 2 , 3000 pg/m 2 , 2000 pg/m 2 , or 1000 pg/m 2 .
- the total dosage over the duration of the regimen is from about 9030 pg/m 2 to about 14000 pg/m 2 , about 9030 pg/m 2 to about 13500 pg/m 2 , about 9000 pg/m 2 to about 13000 pg/m 2 , about 9000 pg/m 2 to about 12500 pg/m 2 , about 9000 pg/m 2 to about 12000 pg/m 2 , about 9000 pg/m 2 to about 11500 pg/m 2 , about 9000 pg/m 2 to about 11000 pg/m 2 , about 9000 pg/m 2 to about 10500 pg/m 2 , about 9000 pg/m 2 to about 10000 pg/m 2 , about 9000 pg/m 2 to about 9000 pg/m 2 to about 9500 pg/m 2 .
- the total dosage over the duration of the regimen is from about 9030 pg/m 2 to about 14000 pg/m 2 , about 9030 pg/m 2 to about 13500 pg/m 2 , about 9030 pg/m 2 to about 13000 pg/m 2 , about 9030 pg/m 2 to about 12500 pg/m 2 , about 9030 pg/m 2 to about 12000 pg/m 2 , about 9030 pg/m 2 to about 11500 pg/m 2 , from about 9030 pg/m 2 to about 11000 pg/m 2 , about 9030 pg/m 2 to about 10500 pg/m 2 , about 9030pg/m 2 to about 10000 pg/m 2 , about 9030 pg/m 2 to about 9500 pg/m 2 .
- the effective amount comprises a 12-day course IV infusion of teplizumab at a first dose of 106 pg/m 2 teplizumab on day 1, a second dose of 425 pg/m 2 teplizumab on day 2, and one dose of 850 pg/m 2 on each of days 3-12.
- the effective amount comprises a 12-day course IV infusion of teplizumab at a first dose of 211 pg/m 2 teplizumab on day 1, a second dose of 423 pg/m 2 teplizumab on day 2, and one dose of 840 pg/m 2 on each of days 3-12.
- the effective amount comprises a 12-day course IV infusion of teplizumab at a first dose of approximately 100 pg/m 2 teplizumab on day 1, a second dose of approximately 400 pg/m 2 teplizumab on day 2, a third dose of approximately 850 pg/m 2 on day 3, and approximately 1,200 pg/m 2 on each of days 4-12.
- the effective amount comprises a 12-day course IV infusion of teplizumab at a first dose of approximately 100 pg/m 2 teplizumab on day 1, a second dose of approximately 400 pg/m 2 teplizumab on day 2, a third dose of approximately 850 pg/m 2 on day 3, and approximately 1,300 pg/m 2 on each of days 4-12.
- the effective amount comprises a 12-day course IV infusion of teplizumab at a first dose of approximately 100 pg/m 2 teplizumab on day 1, a second dose of approximately 400 pg/m 2 teplizumab on day 2, a third dose of approximately 850 pg/m 2 on day 3, and approximately 1,400 pg/m 2 on each of days 4-12.
- the effective amount comprises a 12-day course IV infusion of teplizumab at a first dose of approximately 200 pg/m 2 teplizumab on day 1, a second dose of approximately 400 pg/m 2 teplizumab on day 2, a third dose of approximately 850 pg/m 2 on day 3, and approximately 1,200 pg/m 2 on each of days 4-12.
- the effective amount comprises a 12-day course IV infusion of teplizumab at a first dose of approximately 200 pg/m 2 teplizumab on day 1, a second dose of approximately 400 pg/m 2 teplizumab on day 2, a third dose of approximately 850 pg/m 2 on day 3, and approximately 1,300 pg/m 2 on each of days 4-12.
- the effective amount comprises a 12-day course IV infusion of teplizumab at a first dose of approximately 200 pg/m 2 teplizumab on day 1, a second dose of approximately 400 pg/m 2 teplizumab on day 2, a third dose of approximately 850 pg/m 2 on day 3, and approximately 1,400 pg/m 2 on each of days 4-12.
- a dosing regimen comprising two or more courses of dosing with an anti-CD3 antibody such as teplizumab comprising a first course of dosing at week 1 and second course of dosing at week 26.
- an anti-CD3 antibody such as teplizumab
- teplizumab is administered via IV infusion in two courses, with the first course starting on Day 1 (Week 1) and the second course on approximately Day 182 (Week 26), each course of treatment including daily infusions for 12 days, with a cumulative teplizumab dose of 9000 pg/m 2 for each course of treatment.
- teplizumab is administered via IV infusion in two courses, with the first course starting on Day 1 (Week 1) and the second course on approximately Day 182 (Week 26), each course of treatment including daily infusions for 12 days, with a cumulative teplizumab dose of 9500 pg/m 2 for each course of treatment.
- teplizumab is administered via IV infusion in two courses, with the first course starting on Day 1 (Week 1) and the second course on approximately Day 182 (Week 26), each course of treatment including daily infusions for 12 days, with a cumulative teplizumab dose of 10000 pg/m 2 for each course of treatment.
- teplizumab is administered via IV infusion in two courses, with the first course starting on Day 1 (Week 1) and the second course on approximately Day 182 (Week 26), each course of treatment including daily infusions for 12 days, with a cumulative teplizumab dose of 10500 pg/m 2 for each course of treatment.
- teplizumab is administered via IV infusion in two courses, with the first course starting on Day 1 (Week 1) and the second course on approximately Day 182 (Week 26), each course of treatment including daily infusions for 12 days, with a cumulative teplizumab dose of 11000 pg/m 2 for each course.
- teplizumab is administered via IV infusion in two courses, with the first course starting on Day 1 (Week 1) and the second course on approximately Day 182 (Week 26), each course of treatment including daily infusions for 12 days, with a cumulative teplizumab dose of 11500 pg/m 2 for each course of treatment.
- teplizumab is administered via IV infusion in two courses, with the first course starting on Day 1 (Week 1) and the second course on approximately Day 182 (Week 26), each course of treatment including daily infusions for 12 days, with a cumulative teplizumab dose of 12000 pg/m 2 for each course of treatment.
- teplizumab is administered via IV infusion in two courses, with the first course starting on Day 1 (Week 1) and the second course on approximately Day 182 (Week 26), each course of treatment including daily infusions for 12 days, with a cumulative teplizumab dose of 12500 pg/m 2 for each course of treatment.
- teplizumab is administered via IV infusion in two courses, with the first course starting on Day 1 (Week 1) and the second course on approximately Day 182 (Week 26), each course of treatment including daily infusions for 12 days, with a cumulative teplizumab dose of 13000 pg/m 2 for each course of treatment.
- teplizumab is administered via IV infusion in two courses, with the first course starting on Day 1 (Week 1) and the second course on approximately Day 182 (Week 26), each course of treatment including daily infusions for 12 days, with a cumulative teplizumab dose of 13500 pg/m 2 for each course of treatment.
- teplizumab is administered via IV infusion in two courses, with the first course starting on Day 1 (Week 1) and the second course on approximately Day 182 (Week 26), each course of treatment including daily infusions for 12 days, with a cumulative teplizumab dose of 14000 pg/m 2 for each course of treatment.
- the 12 days course has a 2-day ramp-up phase and a 10- day fixed-, maximal dosing period.
- 106 pg/m 2 teplizumab is administered on day 1425 pg/m 2 teplizumab is administered on day 2, and 850 pg/m 2 teplizumab is administered on each of days 3-12.
- the course of dosing can be repeated at 2-month, 4-month, 5- month, 6-month, 8-month, 9-month, 10-month, 12-month, 15-month, 18-month, 24-month, 30-month, or 36-month intervals.
- efficacy of the treatment with the anti-CD3 antibody such as teplizumab is determined as described herein, or as is known in the art, at 2 months, 4 months, 5 month, 6 months, 9 months, 12 months, 15 months, 18 months, 24 months, 30 months, or 36 months subsequent to the previous treatment.
- a subject is administered one or more doses, preferably 12 daily doses, of the anti-CD3 antibody such as teplizumab at about 5-1200 pg/m 2 , preferably, 106-850 pg/m 2 to treat, or slow the progression of or ameliorate one or more symptoms of T1D.
- the anti-CD3 antibody such as teplizumab at about 5-1200 pg/m 2 , preferably, 106-850 pg/m 2 to treat, or slow the progression of or ameliorate one or more symptoms of T1D.
- the subject is administered a treatment regimen comprising two courses of daily doses of an effective amount of the anti-CD3 antibody such as teplizumab, wherein the course of treatment is administered over 2 days, 3 days, 4 days, 5 days, 6 days, 7 days, 8 days, 9 days, 10 days, 11 days or 12 days.
- the treatment regimen comprises administering doses of the effective amount every day, every 2nd day, every 3rd day or every 4th day.
- a subject is administered a treatment regimen comprising one or more doses of a prophylactically effective amount of the anti-CD3 antibody such as teplizumab, wherein the prophylactically effective amount is 200 pg/kg/day, 175 pg/kg/day, 150 pg/kg/day, 125 pg/kg/day, 100 pg/kg/day, 95 pg/kg/day, 90 pg/kg/day, 85 pg/kg/day, 80 pg/kg/day, 75 pg/kg/day, 70 pg/kg/day, 65 pg/kg/day, 60 pg/kg/day, 55 pg/kg/day, 50 pg/kg/day, 45 pg/kg/day, 40 pg/kg/day, 35 pg/kg/day, 30 pg/kg/day, 26 pg/kg/day, 25 pg/kg/day, 20 p
- the total dosage over the duration of the regimen is preferably a total of less than about 14000 pg/m 2 , 13500 pg/m 2 , 13000 pg/m 2 , 12500 pg/m 2 , 12000 pg/m 2 , 11500 pg/m 2 , 11000 pg/m 2 , 10500 pg/m 2 , 10000 pg/m 2 , 9500 pg/m 2 , 9000 pg/m 2 , 8000 pg/m 2 , 7000 pg/m 2 , 6000 pg/m 2 , and may be less than 5000 pg/m 2 , 4000 pg/m 2 , 3000 pg/m 2 , 2000 pg/m 2 , or 1000 pg/m 2 .
- the daily dosage administered in the regimen is from about 100 pg/m 2 to about 200 pg/m 2 , about 100 pg/m 2 to about 500 pg/m 2 , about 100 pg/m 2 to about 1000 pg/m 2 , or about 500 pg/m 2 to about 1000 pg/m 2 .
- a subject is administered a treatment regimen comprising one or more doses of an effective amount of the anti-CD3 antibody such as teplizumab, wherein the effective amount is increased by, e.g., 0.01 pg/kg, 0.02 pg/kg, 0.04 pg/kg, 0.05 pg/kg, 0.06 pg/kg, 0.08 pg/kg, 0.1 pg/kg, 0.2 pg/kg, 0.25 pg/kg, 0.5 pg/kg, 0.75 pg/kg, 1 pg/kg, 1.5 pg/kg, 2 pg/kg, 4 pg/kg, 5 pg/kg, 10 pg/kg, 15 pg/kg, 20 pg/kg, 25 pg/kg, 30 pg/kg, 35 pg/kg, 40 pg/kg, 45 pg/kg, 50 pg/kg, 55 pg/kg, 60 pg/kg, 0.01
- a subject is intramuscularly administered one or more doses of a 200 pg/kg or less, preferably 175 pg/kg or less, 150 pg/kg or less, 125 pg/kg or less, 100 pg/kg or less, 95 pg/kg or less, 90 pg/kg or less, 85 pg/kg or less, 80 pg/kg or less, 75 pg/kg or less, 70 pg/kg or less, 65 pg/kg or less, 60 pg/kg or less, 55 pg/kg or less, 50 pg/kg or less, 45 pg/kg or less, 40 pg/kg or less, 35 pg/kg or less, 30 pg/kg or less, 25 pg/kg or less, 20 pg/kg or less, 15 pg/kg or less, 10 pg/kg or less, 5 pg/kg or less, 2.5 pg/kg or less,
- a subject is subcutaneously administered one or more doses of a 200 pg/kg or less, preferably 175 pg/kg or less, 150 pg/kg or less, 125 pg/kg or less, 100 pg/kg or less, 95 pg/kg or less, 90 pg/kg or less, 85 pg/kg or less, 80 pg/kg or less, 75 pg/kg or less, 70 pg/kg or less, 65 pg/kg or less, 60 pg/kg or less, 55 pg/kg or less, 50 pg/kg or less, 45 pg/kg or less, 40 pg/kg or less, 35 pg/kg or less, 30 pg/kg or less, 25 pg/kg or less, 20 pg/kg or less, 15 pg/kg or less, 10 pg/kg or less, 5 pg/kg or less, 2.5 pg/kg or less, 2
- the intravenous dose of 100 pg/kg or less, 95 pg/kg or less, 90 pg/kg or less, 85 pg/kg or less, 80 pg/kg or less, 75 pg/kg or less, 70 pg/kg or less, 65 pg/kg or less, 60 pg/kg or less, 55 pg/kg or less, 50 pg/kg or less, 45 pg/kg or less, 40 pg/kg or less, 35 pg/kg or less, 30 pg/kg or less, 25 pg/kg or less, 20 pg/kg or less, 15 pg/kg or less, 10 pg/kg or less, 5 pg/kg or less, 2.5 pg/kg or less, 2 pg/kg or less, 1.5 pg/kg or less, 1 pg/kg or less, 0.5 pg/kg or less, or 0.2 pg/kg or less of the anti-CD3 antibody such
- the dose on day 1 of the regimen is 100-250 pg/m 2 /day, preferably 106 pg/m 2 /day and escalates to the daily dose as recited immediately above by day 2, and 3.
- the subject is administered a dose of approximately 106 pg/m 2 /day, on day 2 approximately 425 pg/m 2 /day, and on subsequent days of the regimen (e.g., days 3- 12) 850 pg/m 2 /day.
- the subject on day 1, is administered a dose of approximately 211 pg/m 2 /day, on day 2 approximately 423 pg/m 2 /day, on day 3 and subsequent days of the regimen (e.g., days 3-12) approximately 840 pg/m 2 /day.
- the subject in need thereof is administered an effective amount of pain reliever (such as a nonsteroidal anti-inflammatory drug (NS AID), acetaminophen), an antihistamine, an antiemetic or a combination thereof at least on each of days 1-5 of the course of IV infusion (for example, the 12-days course of IV infusion).
- the pain reliever such as a nonsteroidal anti-inflammatory drug (NSAID), acetaminophen
- an antihistamine, an antiemetic or a combination thereof can be administered on each of days 1-5, 1-6, 1-7, 1-8, 1-9, 1-10, 1-11, 1-12 or during the duration of the treatment with the anti-CD3 antibodies.
- the pain reliever (such as a nonsteroidal anti-inflammatory drug (NSAID), acetaminophen), an antihistamine, an antiemetic or a combination thereof is administered on each day about 30 min prior to the IV infusion.
- the NSAID, acetaminophen, antihistamine, anti emetic or combination thereof is administered orally.
- the NSAID, acetaminophen, antihistamine, antiemetic or combination thereof is administered intravenously.
- antipyretics, antihistamines and/or antiemetics are administered to the subject in need thereof to mitigate cytokine release syndrome.
- the liver enzymes are monitored and treatment with the anti-CD3 antibodies is discontinued or paused in subjects developing elevated ALT or AST more than 5 times the upper limit of normal.
- a set fraction of the doses for the 106 pg/m 2 /day to 850 pg/m 2 /day regimen described above is administered in escalating doses.
- the speed and duration of the infusion is designed to minimize the level of free anti-CD3 antibody such as teplizumab in the subject after administration.
- the level of free anti-CD3 antibody such as teplizumab should not exceed 200 ng/ml free antibody.
- the infusion is designed to achieve a combined T-cell receptor coating and modulation of at least 50%, 60%, 70%, 80%, 90%, 95% or of 100%.
- the anti-CD3 antibody such as teplizumab is administered chronically to treat, or slow the progression, or ameliorate one or more symptoms of T1D.
- a low dose of the anti-CD3 antibody such as teplizumab is administered once a month, twice a month, three times per month, once a week or even more frequently either as an alternative to the 6 to 14-day dosage regimen discussed above or after administration of such a regimen to enhance or maintain its effect.
- Such a low dose may be anywhere from 1 pg/m 2 to 100 pg/m 2 , such as approximately 5 pg/m 2 , 10 pg/m 2 , 15 pg/m 2 , 20 pg/m 2 , 25 pg/m 2 , 30 pg/m 2 , 35 pg/m 2 , 40 pg/m 2 , 45 pg/m 2 , or 50 pg/m 2 .
- the subject may be re-dosed at some time subsequent to administration of the two course anti-CD3 antibody such as teplizumab dosing regimen, for example, based upon one or more physiological or biomarker parameters or may be done as a matter of course.
- Such redosing may be administered and/or the need for such redosing evaluated 2 months, 4 months, 6 months, 8 months, 9 months, 1 year, 15 months, 18 months, 2 years, 30 months or 3 years after administration of a dosing regimen and may include administering a course of treatment every 6 months, 9 months, 1 year, 15 months, 18 months,
- the level (or relative amounts) of phenotypically exhausted T-cells such as
- the baseline level of TIGIT+KLRG1+CD8+CD3+ T-cells and/or PD- 1+CD8+CD3+ T-cells is less than 5% of all CD3+ T-cells. In some embodiments, the determining of TIGIT+, KLRG1+, or PD-1+ phenotypically-exhausted CD8+ T-cells is about
- the monitoring can be annual. In some embodiments, if the subject has less than about 10% TIGIT+KLRG1+CD8+ T-cells and/or PD-1+CD8+ T-cells in all CD3+ T-cells, the monitoring can be every about 3-6 months.
- the re-dosing comprises administering additional (e.g., second, third, or beyond) 12-day course(s) of teplizumab each at a total dose of more than about 9000 pg/m 2 as described herein.
- the additional 12-day course of teplizumab comprises a first dose of 106 pg/m 2 teplizumab on day 1, a second dose of 425 pg/m 2 teplizumab on day 2, and one dose of 850 pg/m 2 on each of days 3-12, and wherein the total dose is approximately 9031 pg/m 2 .
- the additional 12-day course of teplizumab comprises a first dose of 211 pg/m 2 teplizumab on day 1, a second dose of 423 pg/m 2 teplizumab on day 2, and one dose of 840 pg/m 2 on each of days 3-12, and wherein the total dose is approximately 9034 pg/m 2 .
- the additional (e.g., second, third, or beyond) 12-day course of anti-CD3 antibody such as teplizumab
- the anti-CD3 antibody such as teplizumab is administered to achieve, or maintain a level of glycosylated hemoglobin (HbAl or HbAlc) less than 8%, less than 7.5%, less than 7%, less than 6.5%, less than 6%, less than 5.5% or 5% or less.
- HbAl or HbAlc glycosylated hemoglobin
- patients have a HbAl or HbAlc level of less than 8%, less than 7.5%, less than 7%, less than 6.5%, less than 6%, or, more preferably, from 4%-6% (preferably measured in the absence of other treatment for diabetes, such as administration of exogenous insulin).
- Such patients preferably have retained at least 95%, 90%, 80%, 70%, 60%, 50%, 40% 30% or 20% of beta-cell function prior to initiation of treatment.
- the administration of the anti-CD3 antibodies prevents damage, thereby slowing progression of the disease and reducing the need for insulin administration.
- the methods of treatment provided herein result in a level of HbAl or HbAlc is 7% or less, 6.5% or less, 6% or less, 5.5% or less, or 5% or less 6 months, 9 months, 12 months, 15 months, 18 months, or 24 months after the previous treatment.
- the administration of the anti-CD3 antibodies according to the methods provided herein decreases the average level of HbAl or HbAlc in the patient by about 5%, about 10%, about 15%, about 20%, about 25%, about 30%, about 35%, about 40%, about 45%, about 50%, about 55%, about 60%, about 65% or about 70% as compared to pre-treatment levels at 6 months, 9 months, 12 months, 15 months, 18 months, or 24 months after the previous treatment.
- the administration of the anti-CD3 antibodies according to the methods provided herein results in an average level of HbAl or HbAlc in the patient that only increases by about 0.5%, about 1%, about 2.5%, about 5%, about 10%, about 15%, about 20%, about 25%, about 30%, about 35%, about 40%, about 45%, or about 50% as compared to pre-treatment levels at 6 months, 9 months, 12 months, 15 months, 18 months, or 24 months after the previous treatment.
- administering slows the loss of P-cells and/or preserves P cell function (as evidenced by e.g., higher C-peptide levels, less episodes of hypo- or hyper- glycemia, increased time in range (of glycemia), lower insulin use, or other assessment method known in the art) over 12 months, 13 months, 14 months, 15 months, 16 months, 17 months, 18 months, 19 months, 20 months, 21 months, 22 months, 2 months, 24 month or more in children and adolescents 8-17 years old who have been diagnosed with T1D in the previous 6 weeks.
- administration of the anti-CD3 antibodies slows the loss of P cells and/or preserves P cell function over 18 months (78 weeks) in children and adolescents 8-17 years old who have been diagnosed with T1D within 6 weeks prior to the administration of the first dose of the anti-CD3 antibodies.
- administration of the anti-CD3 antibodies slows the loss of P cells and/or preserves P cell function as measured by C-peptide levels over 18 months (78 weeks) in children and adolescents 8-17 years old who have been diagnosed with T1D within 6 weeks prior to the administration of the first dose of the anti-CD3 antibodies.
- administration of the anti-CD3 antibodies such as teplizumab, in 8-17 years old subjects who have been diagnosed with T1D within 6 weeks prior to the administration of the first dose of the anti-CD3 antibodies, lowers insulin use by 0.1 U/kg/day or more while achieving similar target glycemic control (HbAlc) in the subjects treated with the anti-CD3 antibodies as compared to subjects receiving placebo.
- administration of the anti-CD3 antibodies such as teplizumab, in 8-17 years old subjects who have been diagnosed with T1D within 6 weeks prior to the administration of the first dose of the anti-CD3 antibodies, improved TIR with lower insulin use by 0.1 U/kg/day or more than subjects receiving placebo.
- the administration of the anti-CD3 antibodies results in a reduction of HBAlc baseline by from 0.1 to 1 point, or more than 1 point, compared to subjects receiving placebo.
- the administration of the anti-CD3 antibodies, such as teplizumab results in a reduction of insulin use by from 10% to 15%, 15% to 20%, 20% to 25%, 25% to 30%, 10% to 20%, 20% to 30%, 10% to 30%, or more than 30%, compared to subjects receiving placebo.
- the administration of the anti-CD3 antibodies, such as teplizumab, to the subject in need thereof results in a reduction of insulin dosage by at least 0.1 U/kg/day or in maintenance of insulin dosage.
- administration of the anti-CD3 antibodies in 8-17 years old subjects who have been diagnosed with T1D within 6 weeks prior to the administration of the first dose of the anti-CD3 antibodies results in from 40% to 80%, or more than 80%, higher mean C-peptide value compared with a subject receiving placebo at 78 weeks.
- the administration of the anti-CD3 antibodies, such as teplizumab results in from 40% to 45%, 45% to 50%, 50% to 55%, 55% to 60%, 60% to 65%, 65% to 70%, 70% to 75%, 75% to 80%, or more than 80%, higher mean C- peptide value compared with a subject receiving placebo at 78 weeks.
- the administration of the anti-CD3 antibodies results in 40%, 45%, 50%. 55%. 60%, 65%, 70%, 75%, 80%, 85%, 90%, or more than 90%, higher mean C-peptide value compared with a subject receiving placebo at 78 weeks.
- administration of the anti-CD3 antibodies results in an increase of from 3% to 10%, or more than 10%, time in range for glycemia as assessed using a glucose monitoring system.
- the administration of the first dose of the anti-CD3 antibodies results in an increase of 3% to 6%, 6% to 10%, 3% to 4%, 4% to 5%, 5% to 6%, 6% to 7%, 7% to 8%, 8% to 9%, 9% to 10%, or more than 10% time in range for glycemia as assessed using a glucose monitoring system.
- Some embodiments relate to Teplizumab for use in a method of treating clinical type 1 diabetes (T1D), comprising administering to a subject in need thereof a 12-day course of the teplizumab at a total dose of more than about 9000 pg/m 2 .
- the total dose is between about 9000 and about 9500 pg/m 2 . In some embodiments, the total dose is between about 9000 and about 14000 pg/m 2 .
- the 12-day course comprises a first dose of 106 pg/m 2 teplizumab on day 1, a second dose of 425 pg/m 2 teplizumab on day 2, and one dose of 850 pg/m 2 on each of days 3-12, and wherein the total dose is approximately 9031 pg/m 2 .
- the 12-day course comprises a first dose of 211 pg/m 2 teplizumab on day 1, a second dose of 423 pg/m 2 teplizumab on day 2, and one dose of 840 pg/m 2 on each of days 3-12, and wherein the total dose is approximately 9034 pg/m 2 .
- the method can include administering a first and a second 12-day courses of teplizumab.
- the first and the second 12-day courses are administered at about 1-6 months, about 2-5 months or about 3 months interval.
- the method can include administering to the subject in need thereof a third or more 12-day course of teplizumab, each course at a total dose of more than about 9000 pg/m 2 .
- the third or more 12-day course of teplizumab comprises a first dose of 106 pg/m 2 teplizumab on day 1, a second dose of 425 pg/m 2 teplizumab on day 2, and one dose of 850 pg/m 2 on each of days 3-12, and wherein the total dose of each course is approximately 9031 pg/m 2 .
- the third or more 12-day course of teplizumab comprises a first dose of 211 pg/m 2 teplizumab on day 1, a second dose of 423 pg/m 2 teplizumab on day 2, and one dose of 840 pg/m 2 on each of days 3-12, and wherein the total dose of each course is approximately 9034 pg/m 2 .
- the third or more 12-day course of teplizumab is administered at about a 12 month to about a 24-month interval.
- the method can further include determining, after the administration of each 12-day course, a baseline of a level of TIGIT+KLRG1+CD8+ T-cells or PD-1+CD8+ T-cells with respect to all CD3+ T-cells, monitoring the level of the TIGIT+KLRG1+CD8+CD3+ T-cells, and administering an additional 12-day course of teplizumab when the level of the TIGIT+KLRG1+CD8+CD3+ T-cells or PD-1+CD8+CD3+ T-cells returns to the baseline level.
- the determining of TIGIT+KLRG1+CD8+CD3+ T-cells or PD-1+CD8+CD3+ T-cells is by flow cytometry. In some embodiments, the monitoring of TIGIT+KLRG1+CD8+CD3+ T-cells or PD- 1+CD8+CD3+ T-cells is by flow cytometry. In some embodiments, the determining of TIGIT+KLRG1+CD8+CD3+ T-cells or PD-1+CD8+CD3+ T-cells is about 1-6 months, about 2-5 months, or about 3 months after the administration of each 12-day course.
- subsequent monitoring is annual. In some embodiments, if the subject has less than about 10% TIGIT+KLRG1+CD8+ T-cells, subsequent monitoring is about every 3-6 months. In some embodiments, if the subject has less than about 10% PD-1+CD8+ T-cells in all CD3+ T-cells, subsequent monitoring is about every 3-6 months.
- the subject in need thereof has been diagnosed with T1D within 6 weeks prior to the administrating step.
- the administrating step results in reduction by at least 10% of insulin use, HbAlc levels, hypoglycemic episodes, or combinations thereof as compared to pre-treatment levels.
- each dose is administered parenterally.
- each dose is administered by intravenous infusion.
- the subject is about 8 to 17 years old.
- the subject has a peak C-peptide level of >0.2 pmol/mL during an MMTT.
- the subject in need thereof has a peak C-peptide level of from 0.2 pmol/mL to 0.7 pmol/mL during an MMTT.
- the subject receiving teplizumab has a higher mean C- peptide value compared with a control receiving placebo.
- the method further includes assessing the AUC of C-peptide following an MMTT at 78 weeks.
- the administration of teplizumab results in the maintenance of higher C-peptide levels than when the patient is administered a placebo.
- the subject has at least 20% of [3-cell function prior to the administration of the first dose of the first 12-day course of teplizumab.
- the reduction of insulin use, HbAlc levels, hypoglycemic episodes, or combinations thereof, is over a period of 12 months or more.
- Some aspects relate to a method of treating clinical T1D, comprising administering to a subject in need thereof a 12-day course of teplizumab at a total dose of more than about 9000 pg/m 2 . Some aspects relate to teplizumab for use in a method of treating clinical T1D, comprising administering to a subject in need thereof a 12-day course of the teplizumab at a total dose of more than about 9000 pg/m 2 .
- a method of treating clinical T1D comprising administering to a subject in need thereof a 12-day course of teplizumab at a total dose of from about 9000 to about 9500 pg/m 2 . In some embodiments, a method of treating clinical T1D is provided comprising administering to a subject in need thereof a 12-day course of teplizumab at a total dose of from about 9000 to about 14000 pg/m 2 .
- the anti-CD3 antibody such as teplizumab, otelixizumab, or foralumab
- the anti-CD3 antibody is administered by infusion in medical facilities or in outpatient infusion centers.
- the anti-CD3 antibody such as teplizumab, otelixizumab, or foralumab
- Home infusion therapy involves the administration of the therapeutic agents, for example, anti-CD3 antibody using intravenous, or subcutaneous routes, in the patient's home rather than in a physician's office or hospital.
- Infusion therapies in the home can be administered by a home health care worker or by a patient himself.
- a health care worker having some training in the operation of infusion equipment and the administration of anti-CD3 antibody can provide the patient with self-administration training and all the necessary equipment and/or supplies needed for the administration.
- Non-limiting, exemplary embodiments of the present disclosure are set forth below.
- a method of treating type 1 diabetes comprising: administering to a subject in need thereof a first 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 9500 pg/m 2 , wherein the subject in need thereof has a peak C-peptide level of at least 0.2 pmol/mL during a mixed meal tolerance test (MMTT) prior to administration of the first 12-day course of teplizumab; and administering to the subject in need thereof a second 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 9500 pg/m 2 , wherein the first and the second 12-day courses of teplizumab are administered at about a 6 month or to about a 12 month interval.
- T1D type 1 diabetes
- a method of treating type 1 diabetes comprising: administering to a subject in need thereof a first 12-day course of teplizumab at a total dose of from about 9000 to about 14000 pg/m 2 , wherein the subject in need thereof has a peak C-peptide level of at least 0.2 pmol/mL during a mixed meal tolerance test (MMTT) prior to administration of the 12-day course of teplizumab; and administering to the subject in need thereof a second 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 14000 pg/m 2 , wherein the first and the second 12-day courses of teplizumab are administered at about a 6 month or to about a 12 month interval.
- MMTT mixed meal tolerance test
- the 12-day course comprises a first dose of 106 pg/m 2 teplizumab on day 1, a second dose of 425 pg/m 2 teplizumab on day 2, and one dose of 850 pg/m 2 on each of days 3-12, and wherein the total dose is approximately 9031 pg/m 2 .
- the third or more 12-day course of teplizumab comprises a first dose of 106 pg/m 2 teplizumab on day 1, a second dose of 425 pg/m 2 teplizumab on day 2, and one dose of 850 pg/m 2 on each of days 3-12, and wherein the total dose of each course is approximately 9031 pg/m 2 .
- any one of embodiments 1-8 comprising: determining, after administration of each 12-day course, a baseline of a level of TIGIT+KLRG1+CD8+ cells and/or a baseline of a level of PD-1+CD8+ cells with respect to all CD3+ T cells; monitoring the level of TIGIT+KLRG1+CD8+CD3+ T-cells and/or the level of PD- 1+ CD8+ CD3+ T cells; and administering an additional 12-day course of teplizumab when the level of the TIGIT+KLRG1+CD8+CD3+ T-cells and/or PD-1+CD8+ CD3+ T cells returns to the baseline level.
- TIGIT+KLRG1+CD8+ cells and/or the baseline level of the PD-1+CD8+ is less than about 5% of all CD3+ cells.
- TIGIT+KLRG1+CD8+CD3+ T-cells and/or the PD-1+CD8+ CD3+ T-cells is about 1-6 months, about 2-5 months, or about 3 months after the administration of each 12-day course.
- a method of increasing mean C-peptide value in a subject having type 1 diabetes comprising: administering to the subject a first 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 9500 pg/m 2 , wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during a mixed meal tolerance test (MMTT) prior to administration of the first 12-day course of teplizumab; and administering to the subject a second 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 9500 pg/m 2 , wherein the first and the second 12-day courses of teplizumab are administered at about a 6 month or to about a 12 month interval, and wherein the administration of teplizumab results in a 40% to 80%, or a more than 80%, increase in mean C-peptide value compared to subjects receiving placebo.
- MMTT
- a method of increasing mean C-peptide value in a subject having type 1 diabetes comprising: administering to the subject a first 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 14000 pg/m 2 , wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during a mixed meal tolerance test (MMTT) prior to administration of the first 12-day course of teplizumab; and administering to the subject a second 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 14000 pg/m 2 , wherein the first and the second 12-day courses of teplizumab are administered at about a 6 month or to about a 12 month interval, and wherein the administration of teplizumab results in a 40% to 80%, or a more than 80%, increase in mean C-peptide value compared to subjects receiving placebo.
- MMTT
- Teplizumab for use in a method of treating type 1 diabetes (T1D), comprising administering to a subject in need thereof a first 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 9500 pg/m 2 , wherein the subject in need thereof has a peak C-peptide level of at least 0.2 pmol/mL during a mixed meal tolerance test (MMTT) prior to administration of the first 12-day course of teplizumab; and administering to the subject in need thereof a second 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 9500 pg/m 2 , wherein the first and the second 12-day courses of teplizumab are administered at about a 6 month or to about a 12 month interval.
- T1D type 1 diabetes
- Teplizumab for use in a method of treating type 1 diabetes (T1D), comprising administering to a subject in need thereof a first 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 14000 pg/m 2 , wherein the subject in need thereof has a peak C-peptide level of at least 0.2 pmol/mL during a mixed meal tolerance test (MMTT) prior to administration of the first 12-day course of teplizumab; and administering to the subject in need thereof a second 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 14000 pg/m 2 , wherein the first and the second 12-day courses of teplizumab are administered at about a 6 month or to about a 12 month interval.
- T1D type 1 diabetes
- Teplizumab for use in a method of increasing mean C-peptide value in a subject having type 1 diabetes, the method comprising: administering to the subject a first 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 9500 pg/m 2 , wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during a mixed meal tolerance test (MMTT) prior to administration of the first 12-day course of teplizumab; and administering to the subject a second 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 9500 pg/m 2 , wherein the first and the second 12-day courses of teplizumab are administered at about a 6 month or to about a 12 month interval, and wherein the administration of teplizumab results in a 40% to 80%, or a more than 80%, increase in mean C-peptide
- Teplizumab for use in a method of increasing mean C-peptide value in a subject having type 1 diabetes, the method comprising: administering to the subject a first 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 14000 pg/m 2 , wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during a mixed meal tolerance test (MMTT) prior to administration of the first 12-day course of teplizumab; and administering to the subject a second 12-day course of teplizumab at a total dose of from about 9000 pg/m 2 to about 14000 pg/m 2 , wherein the first and the second 12-day courses of teplizumab are administered at about a 6 month or to about a 12 month interval, and wherein the administration of teplizumab results in a 40% to 80%, or a more than 80%, increase in mean C-peptide
- Teplizumab is a 150 kD monoclonal antibody that binds the CD3-s epitope of the T cell receptor (TCR) complex.
- TCR T cell receptor
- the primary mechanism of action of the antibody involves binding the CD3 antigen target on T cells.
- a population pharmacokinetic (PK) model that describes teplizumab concentrations following IV administration was developed.
- Teplizumab PK was described by a Quasi-Steady-State (QSS) approximation of the Target- Mediated Drug Disposition (TMDD) model.
- QSS Quasi-Steady-State
- TMDD Target- Mediated Drug Disposition
- the Herold regimen is a 14-day course of teplizumab consisting of daily intravenous (IV) infusions (over at least 30 minutes) of 51 pg/m 2 , 103 pg/m 2 , 207 pg/m 2 , and 413 pg/m 2 on Study Days 1-4, respectively, and an infusion of 826 pg/m 2 on each of Study Days 5-14.
- the total dose for a 14-day course is approximately 9034 pg/m 2 .
- BSA body surface area
- this dosing schedule delivers approximately 17 mg of teplizumab.
- the maximum amount of drug delivered at steady-state was designed to provide coating of 50% to 80% of the available CD3 on T-cells, with no large excesses of free, unbound drug (projected to be ⁇ 200 ng/mL at steady-state).
- the new Regimen l is a 12-day course of teplizumab consisting of daily IV infusion (over at least 30 minutes) of 211 pg/m 2 and 423 pg/m 2 on Study Days 1 and 2, respectively, and an infusion of 840 pg/m 2 on each of Study Days 3-12.
- the total dose for a 12-day course is approximately 9034 pg/m 2 .
- the new Regimen 2 is a 12-day course of teplizumab consisting of daily IV infusion (over at least 30 minutes) of 106 pg/m 2 and 425 pg/m 2 on Study Days 1 and 2, respectively, and an infusion of 850 pg/m 2 on each of Study Days 3-12.
- the total dose for a 12-day course is approximately 9031 pg/m 2 .
- NONMEM software Version 7.4.1 (ICON Development Solutions).
- Computer resources included personal computers with Intel® processors, Windows 7 Professional or later operating system and Intel® Visual Fortran Professional Compiler (Version 11.0).
- Graphical and all other statistical analyses, including evaluation of NONMEM outputs were performed using R version 3.4.4 for Windows (R proj ect, r-proj ect.org) . Results
- Figures 17-24 show concentration profiles comparing Herold Regimen and Regimen 2 for a longer time period and Tables 2 and 3 summarized Cmax and AUC from 0 to 42 days in the simulations. Figures show that by day 42 concentrations are very low, so values for AUC0-42 are essentially the same as for AUCinfinity.
- Table 2 illustrates mean and standard deviation of predicted maximum concentrations (ng/mL) over 1000 simulated subjects using Protege Model 205.
- Table 3 illustrates mean and standard deviation of predicted AUC from 0 to 42 days (ng/mL*day) over 1000 simulated subjects using Protege Model 205.
- BSA-proportional dosing provides homogeneous exposure levels for adult and pediatric subjects with different body size measures.
- Example 2 A Phase 3, Randomized, Double-Blind, Multinational, Placebo-Controlled Study to Evaluate Efficacy and Safety of Teplizumab (PRV-031), a Humanized, FcR Non-Binding, anti-CD3 Monoclonal Antibody, in Children and Adolescents with Newly Diagnosed Type 1 Diabetes (T1D)
- Teplizumab (also known as PRV-031, hOKT3yl [Ala-Ala], and MGA031) is a humanized 150-kilodalton monoclonal antibody (mAb) that binds to the CD3-s epitope of the T cell receptor.
- mAb monoclonal antibody
- Teplizumab was developed when preclinical studies demonstrated that targeting T cells (the cells that are instrumental in initiating and coordinating the autoimmune process responsible for type 1 diabetes [T1D] mellitus) via this mechanism altered diabetes immunopathogenesis and prevented and reversed disease in relevant animal models. The goal of this study is to evaluate teplizumab in children and adolescents very recently diagnosed with T1D. Teplizumab holds the promise to be the first disease modifying therapy available to improve both the medical management and overall outlook in those who suffer the most devastating short- and long-term consequences of this disease.
- teplizumab is safe, well-tolerated, and effective in slowing the loss of P-cells and maintaining a clinically relevant level of P-cell function in children and adolescents newly diagnosed with T1D while improving key aspects of T1D clinical management over an 18-month period.
- the primary endpoint was:
- AUC area under the time-concentration curve (AUC) of C-peptide after a 4-hour (4h) mixed meal tolerance test (MMTT), a measure of endogenous insulin production and P cell function, at Week 78.
- Exogenous insulin use defined as a daily average in units per kilogram per day (U/kg/day), at Week 78
- TIR expressed as a daily average of the percentage of time in a 24-hour day a participant’s blood glucose (BG) is >70 but ⁇ 180 mg/dL (>3.9 to ⁇ 10.0 mmol/L), assessed using continuous glucose monitoring (CGM), at Week 78 • Clinically important hypoglycemic episodes: defined as the total number of episodes of a BG reading of ⁇ 54 mg/dL (3.0 mmol/L) and/or episodes of severe cognitive impairment requiring external assistance for recovery, from randomization through Week 78
- TEAEs treatment-emergent adverse events
- AESIs adverse events of special interest
- SAEs serious adverse events
- Incidence of treatment-emergent infections of special interest including but not limited to tuberculosis, an infection requiring IV antimicrobial treatment or hospitalization, Epstein-Barr virus (EBV) and cytomegalovirus (CMV) infection, or significant viremia (i.e., DNA-based polymerase chain reaction viral load >10,000 copies per mL or 10 6 cells), and herpes zoster
- DKA diabetic ketoacidosis
- Peak C-peptide level at screening within the range of 0.2 (inclusion criterion) to 0.7 pmol/mL (inclusive) versus >0.7 pmol/mL
- the total study duration for each participant was up to 84 weeks. This includes a screening period of up to 6 weeks and a post-randomization period of 78 weeks.
- the treatment period includes two 12-day treatment courses separated by 6 or 12 months and a post-treatment observation period of approximately 52 weeks or 26 weeks, respectively. The final visit took place at Week 78.
- each participant received the first dose of the study drug in the first 12-day treatment course, as shown in the table below.
- each participant received the first dose of the second 12-day course.
- the study drugs (teplizumab or placebo) were administered via IV infusion at the study site or other qualified facility by study-approved personnel.
- the doses of study drug were calculated based on the participant’s body surface area (BSA) measured on the first day of each treatment course. No dose adjustment is permitted.
- BSA body surface area
- MMTT In order to quantitate endogenous [3-cell function, participants underwent standardized provocative metabolic testing for C-peptide (a 1 : 1 by-product of insulin production). Participants consumed a fixed amount of a beverage with known amounts of carbohydrates, fats, and protein. Following consumption, BG, insulin, and C-peptide levels were measured over time. A 2h MMTT was conducted at screening, and 4h MMTTs was conducted at randomization and Weeks 26, 52, and 78 for key endpoint assessments.
- HbAlc This is the percent of red blood cells (measured as hemoglobin) that has become non-enzymatic glycated proportional to blood glucose levels. This indicates, on average, approximately a 3-month average of blood glucose values. It is a key clinical target in the management of T1D.
- Insulin use As an average over 7 days of data collected before each specified visit to quantify exogenously injected insulin.
- hypoglycemia Clinically important and potentially life-threating hypoglycemia is the result of insulin therapy and more likely to occur in patients who are attempting to achieve glycemic control goals. This study asks participants to record information regarding BG levels of ⁇ 70mg/dL (3.9 mmol/L) and/or events that are consistent with hypoglycemia. A particular focus is on clinically significant hypoglycemic events that are defined as a reliable glucose reading of ⁇ 54 mg/dL (3.0 mmol/L) and/or severe cognitive impairment and/or physical status requiring external assistance for recovery.
- Glucose Monitoring Intermittent glucose monitoring (e.g., spot-check or fingerstick) performed by participants or care givers multiple times a day as a necessary part of glycemic management to gauge insulin dosing and assist in diet and activity. All participants are to bring in their glucometers at all visits for review. In addition to data regarding glycemic control, at specified times during the study, participants report their daily before-meal and before-bedtime BG readings and have glucose levels assessed for 2-week intervals using CGM.
- spot-check or fingerstick multiple times a day as a necessary part of glycemic management to gauge insulin dosing and assist in diet and activity. All participants are to bring in their glucometers at all visits for review.
- participants report their daily before-meal and before-bedtime BG readings and have glucose levels assessed for 2-week intervals using CGM.
- Quality of Life Questionnaires Surveys is used to assess the general health and wellbeing of participants and the effects of teplizumab, such as the PedsQL Diabetes Module, HFS, DTSQ, and parent-reported PedsQL Family Impact Module.
- Teplizumab concentrations are analyzed in blood samples collected at specified time points throughout the study.
- Anti- teplizumab antibodies are determined, including those that are neutralizing antibodies (NAbs).
- the analysis population was all randomized participants who receive any amount of study drug, referred to as the intent-to-treat (ITT) population.
- ITT intent-to-treat
- participants were analyzed in the treatment group corresponding to the treatment to which they were randomized, regardless of what treatment they actually received.
- the analysis population consisted of all randomized study participants receiving at least one dose of study drug, referred to as the safety population. For this population, participants were analyzed in the treatment group corresponding to the treatment they actually received, regardless of the treatment to which they were randomized. [0234] For PK and immunogenicity, the analysis population was all participants in the safety population who have provided at least one evaluable sample.
- the primary endpoint is the difference between treatment groups in C-peptide ln(AUC+l) at Week 78 using the ITT population. C-peptide were measured in a 4h MMTT. Analysis of covariance (ANCOVA) was used to assess the treatment difference in C-peptide at Week 78. Missing data from patients who drop out before the end of the study was imputed based on those patients who similarly discontinue treatment in the same treatment arm but have measurement taken at scheduled visits. The model included treatment (teplizumab or placebo), age, and peak C-peptide at baseline as covariates.
- ANCOVA Analysis of covariance
- Sensitivity analysis was performed using a tipping point approach. The same imputation and model were used as in the primary analysis, but a tipping point that changes the C-peptide conclusion at 18 months was sought. Repeated measures analysis may also be used to assess sensitivity.
- ANCOVA was used to assess treatment differences in insulin use, HbAlc, and the percentage of time participants’ BG levels are within the target range of >70 to ⁇ 180 mg/dL (>3.9 to ⁇ 10.0 mmol/L) at Week 78.
- Average total exogenous insulin use (in U/kg/day) was self-recorded in an eDiary for 7 days prior to study visits and at randomization. Data from at least 5 of these 7 days was used for analysis. The model included age, baseline insulin use, peak C-peptide at baseline and treatment group as covariates.
- the model used to assess HbAlc will include age, baseline HbAlc, treatment group, and baseline peak C-peptide as covariates.
- Time in range for blood glucose was defined as the average percentage of time in range for 10 to 14 days post each study visit.
- the model included baseline time in range, treatment group, age and baseline peak C-peptide.
- the rate (number of events/ exposure time) of clinically important hypoglycemic episodes at Week 78 was compared between groups. Data was collected from intermittent glucose monitoring, continuous glucose monitors, participant eDiary and CRFs. A clinically important episode is defined as a reliable BG value of ⁇ 54 mg/dL (3.0 mmol/L) and/or a hypoglycemia event requiring external assistance, (such as seizure, syncope, severe confusion with or without a confirmatory low BG reading).
- the event rate of clinically important hypoglycemic episodes per study participant was assessed using a negative binomial model to allow for the potential for overdispersion in case episodes for hypoglycemia within participant groups were correlated.
- the model to assess clinically significant hypoglycemia included age, treatment group, and baseline peak C-peptide as covariates.
- Peak C-peptide level at screening within the range of 0.2 (inclusion criterion) to 0.7 pmol/mL (inclusive) versus >0.7 pmol/mL
- Age at randomization within the range of 8 to 12 years (inclusive) versus >12 to 17 years.
- Study duration The total study duration for each participant was up to 84 weeks. This includes a screening period of up to 6 weeks and a post-randomization period of 78 weeks. The post-randomization period includes two 12-day treatment courses separated by 6 months and a post-treatment observation period of approximately 52 weeks. The final visit took place at Week 78.
- the treatment courses were separated by an interval of approximately 12 months, and the post-treatment observation period is approximately 26 weeks. The final visit also took place at Week 78.
- the 18-month time point for the primary and key secondary clinical endpoints provide key data needed for the acceptance of teplizumab as a T1D disease modifying therapy into regular medical practice and is consistent with existing guidelines for endpoints recommended by the EMA and FDA.
- Data from T1D natural history studies and interventional trials show that P-cell loss in those with T1D can be quite variable, especially within the weeks to months following diagnosis.
- This study is enrolling participants in close proximity to T1D diagnosis (i.e. within 6 weeks) who are younger, there may be the added complexity of the consideration of the Honeymoon phenomenon (or spontaneous, transient partial remission) - that may last up to ⁇ 1 year in the study population (Abdul- Rasoul 2006).
- the 18-month timing of the primary and key secondary clinical endpoints allows for a substantial amount of the inherent, natural metabolic variability due to different trajectories of P-cell loss and/or transiently enhanced P-cell function due to the Honeymoon phenomenon to be minimized - so that the true effect on teplizumab on P-cell function and clinical parameters can be differentiated from chance.
- teplizumab will continue to be considered acceptable for its integration into care plans for children and adolescents newly diagnosed with T1D.
- a placebo control was used to establish the frequency and magnitude of changes in clinical, safety, metabolic and exploratory endpoints that may occur in the absence of active treatment. Randomization with stratification was used to minimize bias in the assignment of participants to treatment groups, to increase the likelihood that known and unknown participant attributes (e.g., demographic and baseline characteristics) are evenly balanced across treatment groups, and to enhance the validity of statistical comparisons across treatment groups. Blinded treatment was used to reduce potential bias during data collection and evaluation of all study endpoints.
- Participant is male or female.
- Participant is 8 to 17 years of age, inclusive, at the time of randomization/initiation of study drug administration.
- Participant is able to be randomized and initiate study drug within 6 weeks (42 days) of the formal T1D diagnosis according to the ADA criteria.
- Participant has a peak stimulated C-peptide of >0.2 pmol/mL from a 2-hour mixed meal tolerance test (2h MMTT) at screening. (Note: This screening 2h MMTT must occur only after 6 days following diagnosis to allow for reduction of metabolic instability.)
- Participant has a positive result on testing for at least one of the following T ID- related autoantibodies before randomization: o Glutamic acid decarboxylase 65 (GAD65) autoantibodies o Islet antigen 2 (IA-2) autoantibodies o Zinc transporter 8 (ZnT8) autoantibodies o Islet cell cytoplasmic autoantibodies (ICA) or o Insulin autoantibodies (if testing obtained within the first 14 days of insulin treatment)
- Glutamic acid decarboxylase 65 Glutamic acid decarboxylase 65 (GAD65) autoantibodies
- IA-2 Islet antigen 2
- ZnT8 Zinc transporter 8
- ICA Islet cell cytoplasmic autoantibodies
- Insulin autoantibodies if testing obtained within the first 14 days of insulin treatment
- Female participants of childbearing potential must have a negative result on highly sensitive serum (P human chorionic gonadotropin [P-HCG]) at screening. • Participants who have reached puberty must agree to adhere to the following contraceptive requirements. (Note: In countries with legislation for the age of sexual activity, the participant must comply with the local age limit regarding the use of contraception.)
- o Females with childbearing potential (defined as premenopausal females who are capable of becoming pregnant, i.e., having reached menarche or having reached Tanner stage 3 breast development even without menarche) or who gain childbearing potential during the study must practice abstinence or use 2 forms of contraception (including oral, transdermal, injectable, or implanted contraceptives, intrauterine device, female condom, diaphragm with spermicide, cervical cap, use of a condom by the sexual partner, or a sterile sexual partner) continuously from 30 days before the first dose of study drug through the end of the study.
- spermatogenesis a males who have reached puberty (i.e., spermatogenesis) with partners of childbearing potential must use barrier contraception in addition to having their partners use another method of contraception from 1 week before each study agent dose through 120 days (a complete spermatogenesis cycle) after receiving the last dose in each treatment course.
- participant Prior to receiving study drug, participant must be up to date with and/or agree to receive routine age-appropriate immunizations and comply with the guidelines for immunosuppressed individuals and those with chronic disease (diabetes mellitus) according to current local, regional and/or country-specific guidelines.
- Participant and/or appropriate legal guardian must sign an informed consent form (ICF) and/or assent according to local, regional and/or country-specific guidance for study participation.
- ICF informed consent form
- Participant has known allergies, severe reaction, intolerance, hypersensitivity, or anaphylaxis to human, humanized, or murine monoclonal antibodies, teplizumab or any of its components or its excipients. • Participant has been an active participant in a therapeutic drug, invasive medical device, or vaccine clinical trial within 12 weeks before the first dose of study drug or has received an investigational treatment with the potential for T1D disease modification.
- Participant has significant renal, cardiac, vascular, pulmonary, gastrointestinal, neurologic, hematologic, rheumatologic, oncologic, psychiatric disease, or immune deficiency.
- Participant has any autoimmune disease other than T1D (e.g., rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, multiple sclerosis, systemic lupus erythematous), with the exception of clinically stable thyroid or celiac disease.
- autoimmune disease e.g., rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, multiple sclerosis, systemic lupus erythematous
- Participant has an active infection (including a positive SARS-CoV-2 test) and/or fever >38.5°C (101.3°F) within the 48 hours prior to randomization, is prone to infections, or has chronic, recurrent or opportunistic infectious disease, including but not limited to renal, respiratory or skin infections, Pneumocystis carinii. aspergillosis, latent or active granulomatous infection, histoplasmosis, or coccidioidomycosis.
- active infection including a positive SARS-CoV-2 test
- fever >38.5°C (101.3°F) within the 48 hours prior to randomization, is prone to infections, or has chronic, recurrent or opportunistic infectious disease, including but not limited to renal, respiratory or skin infections, Pneumocystis carinii. aspergillosis, latent or active granulomatous infection, histoplasmosis, or coccidioidomycosis.
- Participant has a history of or serologic evidence at screening of current or past infection with human immunodeficiency virus (HIV), hepatitis B virus (HBV), or hepatitis C virus (HCV).
- HAV human immunodeficiency virus
- HBV hepatitis B virus
- HCV hepatitis C virus
- Participant has any of the following in regard to tuberculosis (TB): o A history of latent or active TB o Signs and/or symptoms of TB o Recent close contact with a person with known or suspected active TB, unless appropriate isoniazid prophylaxis for tuberculosis was given o A history of a chest X-ray consistent with active TB or old, inactive TB, o A history of a positive purified protein derivative skin test result (>10 mm induration); or o At screening is positive or repeatedly indeterminate with an approved interferongamma release assay (IGRA; e.g., QuantiFERON-TB test) o If required by local, regional or national regulations, a recent (within 3 months) chest X-ray or one conducted at screening read by a qualified radiologist consistent with current, active TB or old, inactive TB • At screening, participant has a clinically active infection with EBV, including but not limited to infectious mononucleosis,
- participant has a clinically active infection with CMV or a CMV viral load 10,000 copies per mL or per 10 6 lymphocytes (Verkryse 2006).
- Participant has a diagnosis of significant liver disease or at screening alanine aminotransferase (ALT) and/or aspartate aminotransferase (AST) >2X or total bilirubin (TBili) of >1.5X of the age- and sex-specific upper limit of normal (ULN) according to the central laboratory. (Note: Participants with Gilbert’s syndrome may be allowed to enroll upon approval by the Medical Monitor.)
- ALT alanine aminotransferase
- AST aspartate aminotransferase
- Tili total bilirubin
- o Live vaccines e.g., varicella, measles, mumps, rubella, cold-attenuated intranasal influenza vaccine, and smallpox
- o Non-infectious e.g., recombinant, inactivated or otherwise “non-live” vaccines: Within 2 weeks before through 6 weeks after each dosing course.
- a female who is pregnant has a positive P-HCG blood test at screening or a positive urine P-HCG test prior to initiation of study drug, wishes to become pregnant, is planning on donating eggs (ova, oocytes), and/or is lactating with the intent to provide her own breast milk to a baby during the entire study.
- Participants were randomly assigned to 1 of 2 treatment groups in a 2: 1 ratio for participants in the teplizumab group and the placebo group, respectively.
- the randomization was balanced by using randomly permuted blocks and will be stratified by peak C-peptide level at screening (0.2 to 0.7 pmol/mL inclusive, versus >0.7 pmol/mL) and age at randomization (8 to 12 years inclusive, versus >12 to 17 years).
- T1D diagnosis is according to ADA criteria.
- the patient diagnosed with T1D has a positive result on testing for at least one of the following TID-related autoantibodies: Glutamic acid decarboxylase 65 (GAD65) autoantibodies, Islet antigen 2 (IA-2) autoantibodies, Zinc transporter 8 (ZnT8) autoantibodies Islet cell cytoplasmic autoantibodies (ICA) or Insulin autoantibodies (if testing obtained within the first 14 days of insulin treatment).
- Glutamic acid decarboxylase 65 Glutamic acid decarboxylase 65 (GAD65) autoantibodies, Islet antigen 2 (IA-2) autoantibodies, Zinc transporter 8 (ZnT8) autoantibodies Islet cell cytoplasmic autoantibodies (ICA) or Insulin autoantibodies (if testing obtained within the first 14 days of insulin treatment).
- Teplizumab and placebo are prepared according to the Pharmacy Manual provided to the site.
- Two (2) mL of study drug should be drawn from the study drug vial and slowly diluted in 18 mL of 0.9% sodium chloride solution for injection by gentle mixing. The resulting 20 mL of 1 : 10 dilution is used as the initial study drug solution, which contains either placebo or teplizumab at a concentration of 100 pg/mL. This initial drug solution should then be added to 25 mL of a 0.9% sodium chloride solution. Finally, this resulting preparation should be gently mixed before administration to the participant.
- Vascular access This study requires two courses of intravenous infusions and blood draws over 12 days. It is recognized that intravenous access (for infusions and blood draws for laboratory sampling) in the pediatric population that is the focus of this study may pose a challenge. Children have smaller veins than adults, veins that may be more challenging to insert catheters and they may have a significant resistance to catheter placement and/or phlebotomy.
- Study participants received oral premedication consisting of an NSAID drug (tablet or liquid) and a locally approved antihistamine (tablet or liquid), according to local availability and practice standard, for at least the first 5 days of each study drug course and can receive one or both of these for the rest of the study drug doses if the Investigator judges it to be appropriate for the tolerability of infusions.
- the premedication should be given at least 30 minutes prior to initiation of the study drug infusion. If an NSAID is contraindicated, oral acetaminophen (tablet or liquid) may be given.
- Teplizumab for intravenous administration may only be prepared with 0.9% sodium chloride. No solutions other than 0.9% sodium chloride may be running through the same intravenous line when teplizumab is being administered. If the same intravenous line must be used for infusion of other drugs or solutions, the line should be flushed with 0.9% sodium chloride solution before and after infusion of teplizumab.
- Study drug was administered IV over a minimum of 30 minutes according to standard practices.
- participant During the infusions and for an additional 60 minutes following the infusions, participants are to have vital signs (ie, BP, RR, and HR) assessed every 15 min and be monitored for signs or symptoms of infusion reactions. These include but are not limited to fever, chills, headache, nausea, vomiting, infusion-site pain, anaphylaxis, wheezing, dyspnea, urticaria, and hypotension. If there are signs or symptoms of infusion reaction in a participant during the 60-minute post-infusion observation period, the participant should be observed for an additional 60 minutes or until the reaction resolves, whichever is longer.
- vital signs ie, BP, RR, and HR
- the glycemic goal should be attempted through proper glycemic monitoring, administration of exogenous insulin, and monitoring of activity level and diet.
- Exogenous insulin may include rapid, intermediate, and/or long-acting insulins, administered intermittently or via the use of a personal insulin pump.
- Blood glucose levels should be measured at least 4 times a day, including before meals and before bedtime.
- NS AID e.g., ibuprofen
- an antihistamine e.g., diphenhydramine
- Administration of the study drug should be performed according to the Pharmacy Manual. Study drug should be planned to be administered intravenously over 30 minutes according to standard practices, but it may be slowed if there are signs or symptoms of intolerance.
- the patient can proceed with the next infusion as described above at least 30 minutes following administration of prophylactic NSAID (acetaminophen if NSAID is contraindicated) and antihistamine. Close monitoring is to occur during the infusions and for 60 minutes following the infusions for any signs or symptoms of intolerability or infusion reactions.
- prophylactic NSAID acetaminophen if NSAID is contraindicated
- antihistamine acetaminophen if NSAID is contraindicated
- the visit window for these study visits are ⁇ 4 days from the target visit day. During these visits, participants return to the site for their scheduled visit and have clinical and/or laboratory assessments conducted. Of note at Week 12, a CGM sensor is applied and the participant is to be given instructions on CGM monitoring care and use.
- the visit window for these study visits is ⁇ 3 days from the target visit day.
- the visit window for Weeks 30, 34, 39, and 52 study visits is ⁇ 4 days from the target visit day.
- the visit window for Week 65 visit is ⁇ 7 days.
- a 4h MMTT is conducted.
- the visit window for this study visit is ⁇ 7 days from the target visit day. During this visit the 4h MMTT is conducted.
- a 2h MMTT is performed at screening to determine study eligibility (based on peak C-peptide level).
- a 4h MMTTs was performed at randomization and at Weeks 26, 52, and 78 to obtain 4h C-peptide AUCs and other data.
- a 4h MMTT was used at and postrandomization as it has been shown to be more precise and reliable in assessing the MMTT- induced C-peptide AUC than the 2h MMTT (Boyle 2015, Rigby 2013, Rigby 2016).
- the 2h-MMTT was used at screening as it is sufficient to capture the peak C- peptide level needed for study entry. Samples from these assessments were assessed for C-peptide, serum glucose, and insulin.
- Samples were stored for potential future evaluations including but not limited to proinsulin levels.
- the measurements of C-peptide and glucose in serum samples were done.
- MMTTs were to take place in the morning between approximately 7:00 a.m. and 10:00 a.m. after an overnight fast with strict guidance on insulin use.
- the 2- hour MMTT takes approximately 130 minutes to perform, and the 4-hour MMTT takes approximately 250 minutes.
- HbAlc was assessed as a blood test at select study visits.
- Patient’s daily insulin use was documented by the participant in an eDiary at select times for 7 days prior to randomization and at about Weeks 12, 26, 39, 52, 65 and 78 visits.
- the patient records all short-, intermediate- and long-acting insulin administered as intermittent injections or use with an “insulin pump” during this time. Insulin use data were not recorded on the day before or the day of the study visit. If a patient forgets to record insulin use on one or more days before a visit, they should continue to record insulin use for up to 72 hours post-dose to obtain up to 7 days of data. Every effort should be made to collect a total of 7 days of insulin use data for all the aforementioned visits except Week 78 (final visit), as patients return the eDiary at the final visit.
- BG levels were usually measured by a fingerstick glucometer at least 4 times a day, including before each meal and at bedtime.
- participants are offered a study-supplied glucometer and glucometer strips, but participants were permitted to use their own glucometers if they choose. Each participant was instructed to bring their glucometer (or glucometers if they use more than one, e.g., at home and in school) to each visit for review.
- Continuous glucose monitors record interstitial glucose levels (which closely approximate blood glucose values) at regular intervals, e.g. every 5-15 minutes depending on device.
- CGM assessments were conducted to provide key secondary clinical and exploratory endpoint data to address if and how teplizumab affects glycemic control, such as glucose excursions, time in select glucose ranges, and average daily glucose values (Steck 2014, Helminen 2016, Danne 2017).
- a recent international consensus statement on CGM monitoring supported the use of percentages of time in ranges (target, hypoglycemia, and hyperglycemia) and measurement of glycemic variability as key diabetes control metrics in clinical trials (Danne 2017).
- CGM are used to assess glycemic control approximately 7 times throughout the study: after the completion of treatment courses at randomization and Week 26; after the visit at Weeks 12, 39, 52, and 65; and before the visit at Week 78.
- CGM sensors are placed by qualified study staff, and education and training on CGM use and care are given. Sensors remain in place for up to 2 weeks. If during that 2-week period a sensor comes off, it can be replaced by the participant, a knowledgeable family member/guardian, or a qualified medical professional.
- CGM sensors were placed on participants after the study drug administration has completed for Course 1 and Course 2 and other clinical and laboratory assessments have been made on the days specified in the Schedule of Events tables. At the Weeks 12, 39, 52, and 65 visits, the sensor was placed on participants after all clinical and laboratory assessments and the MMTT have completed.
- Study CGM readings are not intended for medical management of participant’s diabetes but can be under the supervision of a participant’s health care team. Of note, routine use of the personal CGM under guidance of a participant’s regular healthcare provider is permitted.
- Spot-check and CGM blood glucose assessments are anticipated to include but are not be limited to mean BG, glycemic variability (BG standard deviation [SD]), maximum and minimum BG values over time and incidence and/or percent time with BG >70 but ⁇ 180 mg/dL (>3.9 but ⁇ 10.0 mmol/L, Level 1 (>180 but ⁇ 250 mg/dL (>10 but ⁇ 13.9 mmol/L)) and Level 2 HYPERglycemia (>250 mg/dL (>13.9 mmol/L)) and Level 1 ( ⁇ 70 but >54 mg/dL ( ⁇ 3.9 but >3.0 mmol/L)) and Level 2 ( ⁇ 54 mg/dL ( ⁇ 3.0 mmol/L)) HYPOglycemia (Seaquist 2013, International Hypoglycaemia Study Group [IHSG] 2017, Agiostratidou 2017).
- BG standard deviation [SD] glycemic variability
- SD glycemic variability
- Venous blood samples were collected for measurement of serum concentrations of teplizumab, anti-teplizumab antibodies and neutralizing antibody (NAb) according to the Schedule of Events. Additionally, samples should also be collected at the Early Termination Visit for subjects who discontinue study treatment early and from subjects who experience an AE suspected to be related to immunogenicity (e.g., infusion reactions, injection site reactions or hypersensitivity).
- samples should also be collected at the Early Termination Visit for subjects who discontinue study treatment early and from subjects who experience an AE suspected to be related to immunogenicity (e.g., infusion reactions, injection site reactions or hypersensitivity).
- Venous blood samples were collected. Samples collected for analyses of teplizumab serum concentration and antibody to teplizumab may additionally be used to evaluate safety or efficacy aspects that address concerns arising during or after the study period for further characterization of immunogenicity or for the evaluation of relevant biomarkers.
- Serum samples were analyzed using validated, specific, and sensitive immunoassay methods.
- Nonlinear mixed effects modeling (with NONMEM software) was used to analyze the serum concentration-time data of teplizumab to obtain the primary PK parameters, clearance (CL) and volume of distribution.
- the PK profiles was used, along with other available data, to develop a population PK model while including the effect of major covariates (e.g., sex, ethnicity, race, antibody) on CL and volume of distribution.
- the starting model is a previously developed model for teplizumab. All PK parameters are presented by listings and descriptive summary statistics including, arithmetic mean, geometric mean (AUC, Cmax and their derived parameters), median, range, standard deviation and coefficient of variation. The data of this study may be pooled with data from other studies.
- Immune assessments may include but are not limited to monitoring T cell profiles and B cell profiles by flow cytometry.
- serum levels of circulating pro- and antiinflammatory cytokines and other soluble factors that can impact T1D progression may be assessed.
- cytokine levels due to cytokine release or lymphocyte modulation
- blood samples were collected at select timepoints before and after study drug administration. Samples were processed and stored. It is anticipated that analyses will be conducted only at select intervals that may include after a critical number of participants have completed the Week 52 visit or other key timepoints.
- cytokines that may be evaluated are interleukin (IL) 2 (IL-2), IL-4, IL-5, IL-6, IL-8, IL-10, interferon-gamma and tumor necrosis factor-alpha.
- serum assessments including those of hormones or other metabolically active substances (e.g., glucagon, incretins, lipokines, adiponectin, or cholesterol) that may have an effect on metabolic control, may be performed.
- Approaches to evaluations may include antibody-based multiplex panels, modified-aptamer binding technology, or other platforms.
- teplizumab may have an effect on antibody subclasses in general. As such, it will be important to evaluate effects of teplizumab on T1D autoantibodies presence and titer in the context of assessment of total antibody subclasses.
- Qualitative and quantitative assessments of antibody subclasses e.g., IgG, IgA, and IgM, may indicate a change in the type of T- helper cell responses to TID-associated autoantigens.
- the disease progression has been proposed to be involved with alterations in “(3-cell stress,” specifically due to attempts at enhanced endogenous insulin production in residual P- cells that may overwhelm intracellular processes and direct or bystander injurious inflammatory meditators. It proposed that measurement of specific P-cell products may be markers of this process and that P-cell recovery due to immune interventions may result in a decrease of these markers. In this study, exploratory evaluation of two such markers, proinsulin-to-C-peptide ratios and serum levels of circulating methylated-insulin DNA, are to be evaluated. Samples that can be used for these analyses were collected, processed and stored.
- the HLA system is a grouping of genes that encode the major histocompatibility complex (MHC) proteins in humans.
- MHC haplotype gives insight to interactions of distinct types of lymphocytes that may cause T1D and may help to identify those who are at risk of developing T1D (Roark 2014).
- the MHC haplotype of participants was determined. The results of genotype analyses can be used to correlate with disease progression, therapeutic responses and identify subgroups that respond preferentially to teplizumab.
- RNA or other genetic matter may also provide insights into the mechanism of teplizumab’s effect on T1D, the immune system or individuals who preferentially respond to teplizumab.
- One example is the methylated insulin-DNA assessment anticipated, but also may include studies on the transcript/transcriptome, microarrays, or whole genome assessments. Samples that can be used for these analyses were collected, processed and/or stored.
- cytokine release syndrome In previous Phase 3 studies of teplizumab, ⁇ 6% of participants with T1D experienced cytokine release syndrome after receiving teplizumab. Symptoms of cytokine release syndrome included but were not limited to rash, headache, nausea, vomiting, chills/rigor, and fever. Most of the symptoms occurred during the first few days of treatment and are mild or moderate in severity. Cytokine release syndrome was time-limited and appeared to dissipate regardless of whether teplizumab dosing was interrupted.
- Supplemental therapy is intended for symptom management.
- the same premedications can be used as follows: • Locally approved NSAIDs should be given in compliance with appropriate age restrictions and practice standards, such as ibuprofen (tablet or liquid), diclofenac, naproxen, meloxicam, or tenoxicam.
- Acetaminophen may be added or used instead of a NS AID if the latter cannot be used. Continuation of NSAID dosing should be considered if necessary.
- Antihistamines may be continued.
- Antihistamines e.g., IV diphenhydramine
- Acetaminophen can be combined with antihistamines and can be repeated every 4 to 6 hours; hydroxyzine can be used during the day to avoid sedation.
- NSAIDs with higher potency e.g., ketorolac.
- Ondansetron for managing GI symptoms such as nausea and vomiting.
- cytokine release syndrome is associated with anaphylaxis or angioedema with or without requiring hemodynamic support (i.e., epinephrine and/or blood pressure medications) or mechanical ventilation, study drug should be permanently discontinued.
- ALT and/or AST >3X ULN but ⁇ 5X ULN
- Total Bilirubin >2X ULN but ⁇ 3X ULN
- platelet count >50,000 but ⁇ 100,000
- hemoglobin >8.5 g/dL but ⁇ 10 g/dL
- Glucocorticoids e.g., prednisone 1-1.5 mg/kg/d given twice daily
- Glucocorticoids should be reserved for intractable symptoms or Grade 3 or higher events that cannot be relieved with the above medications.
- glucocorticoids may interfere with the mechanism of action of teplizumab and therefore should be given for as short a duration as possible. If the investigator feels that glucocorticoids are necessary to resolve intractable signs or symptoms, they should follow applicable standard of care recommendations and treatment guidelines and inform the medical monitor without any delays. Blood glucose levels should be monitored carefully during glucocorticoid administration.
- HDL High density lipoprotein
- LDL Low density lipoprotein
- Type 1 diabetes antibodies (anti-insulin, anti-GAD-65, ICA, anti-ZnT8, anti-IA2)
- Baseline was defined as the most recent value collected prior to the first dose of teplizumab or placebo. Both groups achieved target HbAlc and the proportion of patients with glucose TIR was not statistically significantly different between groups (Table 6B); a greater proportion of teplizumab-treated patients achieved the glucose TIR target of >70% of the time across all study visits, but the difference to placebo was not statistically significant (Figure 31B). Throughout the study, TBR ⁇ 4% was not different between the two groups 12 (teplizumab 2.48%, placebo 2.8% at Week 78). Mean time above range was similar between groups (teplizumab 14-29%, placebo 16-33%; Table 8), with teplizumab ⁇ 25%12, 13 until Week 52 and placebo until Week 26.
- HbAlc was similar in both treatment groups, confirming the achievement of treat- to-target goal by patients. Patients receiving placebo needed more insulin to maintain or decrease HbAlc levels than patients treated with teplizumab.
- Figure 27 is a graph showing insulin use at different timepoint (baseline, week 12, week 26, week 39, week 52, week 65 and week 78). Figure 27 shows that insulin use was numerically lower in patients treated with teplizumab.
- Figure 28 is a graph showing the percentage of subjects who met Hbl Ac ⁇ 6.5% and insulin daily dose ⁇ 0.25 unit/kg/days at different timepoint (baseline, week 12, week 26, week 39, week 52, week 65 and week 78). Figure 28 shows that more teplizumab patients met insulin discontinuation criteria HblAc ⁇ 6.5% and insulin daily dose ⁇ 0.25 unit/kg/days.
- Figure 29 is graph showing the HbAlc levels at different timepoint (baseline, week 12, week 26, week 39, week 52, week 65 and week 78). Figure 29 shows that HbAlc target levels were achieved in both treatment groups.
- Figure 30 is graph showing the percentage time in range at different timepoint (baseline, week 12, week 26, week 39, week 52, week 65 and week 78).
- Figure 30 shows that patients treated with teplizumab spent more Time in Range (TIR).
- assisted technology e.g., continuous glucose monitoring, insulin pumps
- teplizumab and placebo groups likely contributed to similar rates of clinically-important hypoglycemic events in both teplizumab and placebo groups.
- Diabetes Prevention Trial-Type 1 Diabetes Study Group Effects of insulin in relatives of patients with type 1 diabetes mellitus. N Engl J Med 2002;346: 1685-91.
Landscapes
- Health & Medical Sciences (AREA)
- Immunology (AREA)
- Chemical & Material Sciences (AREA)
- Diabetes (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Emergency Medicine (AREA)
- Biochemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Obesity (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Provided herein are methods of treating type 1 diabetes (T1D). Such methods can include administering to a subject in need thereof, at an interval of about 3 months to about 12 months, a first 12-day course of teplizumab at a total dose of more than about 9000 μg/m2 and a second 12-day course of teplizumab at a total dose of more than about 9000 μg/m2.
Description
METHODS FOR TREATING TYPE 1 DIABETES
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] This application claims priority from U.S. Provisional Application 63/515,949, filed July 27, 2023, and U.S. Provisional Application 63/590,883, filed October 17, 2023, the contents of both of which are incorporated herein by reference in their entirety.
SEQUENCE LISTING
[0002] This application includes a sequence listing submitted electronically herewith. Said electronic file, created July 23, 2024, is named 122548WO061.xml and has a size of 3,380 bytes. The contents of said file are incorporated herein by reference in their entirety.
BACKGROUND
[0003] Type 1 diabetes (T1D) is caused by the autoimmune destruction of insulin producing beta cells in the islets of Langerhans, leading to dependence on exogeneous insulin injections for survival. Approximately 1.6 million Americans have T ID. It is one of the most common childhood diseases. Despite improvements in care, most affected individuals with T1D are not able to consistently achieve desired glycemic targets. For individuals with T1D, there are persisting concerns for increased risk of both morbidity and mortality. Two recent studies noted loss of 17.7 life-years for children diagnosed before age 10, and loss of 11 and 13 life-years respectively for Scottish men and women diagnosed as adults. Thus, a need exists for improved T1D treatment methods and compositions.
SUMMARY
[0004] The present disclosure relates to methods of treating T1D with teplizumab, e.g., by slowing the loss of beta-cell function. The T1D to be treated may be Stage 3 or clinical T1D. In some embodiments, the patient has new or recent onset T1D, e.g., the patient has been diagnosed with Stage 3 or clinical T1D within six weeks prior to teplizumab treatment. In some embodiments, the subject has at least 20% of beta-cell function prior to teplizumab treatment. In some embodiments, the patient has a peak C-peptide level of at least 0.2 pmol/mL during a mixed meal tolerance test (MMTT), for example, a two-hour MMTT, prior
to teplizumab treatment. In some embodiments, the patient is a child or an adolescent, e.g., about 8 to 17 (inclusive) years of age.
[0005] In some embodiments, the method comprises administering to a subject in need thereof a first 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 9500 pg/m2, wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during an MMTT (e.g., a two-hour MMTT) prior to administration of the first 12-day course of teplizumab; and administering to the subject a second 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 9500 pg/m2, wherein the first and the second 12-day courses of teplizumab are administered at an interval of at least about 3 months, or at about a 6-month to about a 12-month interval.
[0006] In some embodiments, the method comprises administering to a subject in need thereof a first 12-day course of teplizumab at a total dose of from about 9000 to about 14000 pg/m2, wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during an MMTT (e.g., a two-hour MMTT) prior to administration of the 12-day course of teplizumab; and administering to the subject a second 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 14000 pg/m2, wherein the first and the second 12-day courses of teplizumab are administered at an interval of at least about 3 months, or at about a 6-month to about a 12-month interval.
[0007] Aspects of the disclosure relate to teplizumab for use in a method of treating T1D, the method comprising administering to a subject in need thereof a first 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 9500 pg/m2, wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during an MMTT (e.g., a two-hour MMTT) prior to administration of the first 12-day course of teplizumab; and administering to the subject a second 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 9500 pg/m2, wherein the first and the second 12-day courses of teplizumab are administered at an interval of at least 3 months, or at about a 6-month to about a 12-month interval.
[0008] Aspects of the disclosure relate to teplizumab for use in a method of treating T1D, the method comprising administering to a subject in need thereof a first 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 14000 pg/m2, wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during an MMTT (e.g., a two-hour MMTT) prior to administration of the first 12-day course of teplizumab; and administering to the subject a second 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 14000 pg/m2, wherein the first and the second 12-day courses of teplizumab are
administered at an interval of at least 3 months, or at about a 6-month to about a 12-month interval.
[0009] In some embodiments, the 12-day course comprises a first dose of 106 pg/m2 teplizumab on day 1, a second dose of 425 pg/m2 teplizumab on day 2, and one dose of 850 pg/m2 teplizumab on each of days 3-12, and wherein the total teplizumab dose is approximately 9031 pg/m2.
[0010] In some embodiments, the method further comprises administering to the subject a third or more 12-day course of teplizumab, each course at a total dose of more than about 9000 pg/m2. In some embodiments, the third or more 12-day course of teplizumab comprises a first dose of 106 pg/m2 teplizumab on day 1, a second dose of 425 pg/m2 teplizumab on day 2, and one dose of 850 pg/m2 teplizumab on each of days 3-12, and the total teplizumab dose of each course is approximately 9031 pg/m2. In some embodiments, the third or more 12-day course of teplizumab is administered at an interval of at least 3 months, or at about a 6-month to about a 24-month interval.
[0011] In some embodiments, the method comprises determining, after administration of each 12-day course, a baseline level of TIGIT+KLRG1+CD8+ T-cells and/or a baseline level of PD-1+CD8+ T-cells with respect to all CD3+ T-cells, monitoring the level of TIGIT+KLRG1+CD8+CD3+ T-cells and/or the level of PD-1+CD8+CD3+ T-cells, and administering an additional 12-day course of teplizumab when the level of the TIGIT+KLRG1+CD8+CD3+ T-cells and/or PD-1+CD8+CD3+ T-cells returns to the baseline level. In some embodiments, the baseline level of the TIGIT+KLRG1+CD8+ T-cells and/or the baseline level of the PD-1+CD8+ T-cells is less than about 5% of all CD3+ T-cells. In some embodiments, the determining of TIGIT+KLRG1+CD8+CD3+ T-cells and/or the PD- 1+CD8+CD3+ T-cells is by flow cytometry. In some embodiments, the monitoring of TIGIT+KLRG1+CD8+CD3+ T-cells and/or PD-1+CD8+CD3+ T-cells is by flow cytometry. In some embodiments, the determining of TIGIT+KLRG1+CD8+CD3+ T-cells and/or PD- 1+CD8+CD3+ T-cells is about 1-6 months, about 2-5 months, or about 3 months after the administration of each 12-day course. In some embodiments, if the subject has more than about 10% TIGIT+KLRG1+CD8+ T-cells and/or more than about 10% PD-1+CD8+ T-cells in all CD3+ T-cells, subsequent monitoring is annual. In some embodiments, if the subject has less than about 10% TIGIT+KLRG1+CD8+ T-cells and/or less than about 10% PD- 1+CD8+ T-cells in all CD3+ T-cells, subsequent monitoring is about every 3-6 months.
[0012] In some embodiments, each dose of teplizumab is administered parenterally (e.g., by intravenous infusion). The subject may be pre-mediated with (1) a nonsteroidal anti-
inflammatory drug (NSAID) or acetaminophen, (2) an antihistamine, and/or (3) an antiemetic before each dose in the first three, four, five, six, or seven days of each course.
[0013] In some embodiments, the subject has a peak C-peptide level ranging from 0.2 pmol/mL to 0.7 pmol/mL during an MMTT (e.g., a two-hour MMTT). In some embodiments, the subject in need thereof has a peak C-peptide level of at least 0.7 pmol/mL during an MMTT (e.g., a two-hour MMTT).
[0014] In some embodiments, the method comprises assessing the area under the timeconcentration curve (AUC) of C-peptide following an MMTT at 78 weeks or at 18 months. [0015] In some embodiments, the subject administered with teplizumab has a higher mean C-peptide value compared with a control administered with placebo. By “administered with teplizumab” is meant that the subject has been given one or more courses of teplizumab. A “control administered with placebo” is a control subject who receives no teplizumab (i.e., no dose of teplizumab).
[0016] In some embodiments, the administering of teplizumab to the subject results in a 40% to 80%, or more than 80%, higher mean C-peptide value compared with subjects receiving placebo.
[0017] In some embodiments, the subject administered with teplizumab maintains or reduces baseline HbAlc levels, and/or maintains or increases Time in Range (TIR) with less insulin use, than a subject administered with placebo. For example, the subject administered with teplizumab has a reduced HbAlc level compared to the pre-treatment level (i.e., before any treatment with teplizumab).
[0018] In some embodiments, the administering of teplizumab to the subject results in a reduction of insulin dosage by 10% to 30%, or more than 30%, compared to subjects receiving placebo.
[0019] In some embodiments, the administering of teplizumab to the subject results in a reduction of insulin dosage by at least 0.1 U/kg/day or in maintenance of insulin dosage. [0020] In some embodiments, the administering of teplizumab to the subject results in a reduction of HbAlc baseline by 0.1 to 1 point, or more than 1 point, compared to subjects receiving placebo.
[0021] In some embodiments, the administering of teplizumab to the subject results in an increase of 3 to 10%, or more than 10% in TIR (%) for glycemia as assessed using a glucose monitoring system.
[0022] In some embodiments, the administering of teplizumab to the subject results in fewer Level 3 hypoglycemic episodes (e.g., at least 1, 2, or 3 fewer per year) than control subjects receiving placebo.
[0023] Aspects of the disclosure relate to a method of maintaining or increasing C-peptide value and/or slowing loss of beta-cell function in a subject having T1D, e.g., Stage 3 or clinical T1D. In some embodiments, the method comprises administering to the subject a first 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 9500 pg/m2, wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during an MMTT prior to administration of the first 12-day course of teplizumab; and administering to the subject a second 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 9500 pg/m2, wherein the first and the second 12-day courses of teplizumab are administered at about a 6-month to about a 12-month interval, and wherein the administration of teplizumab results in a 40% to 80%, or a more than 80%, increase in mean C-peptide value compared to subjects receiving placebo. In some embodiments, the method comprises administering to the subject a first 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 14000 pg/m2, wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during an MMTT (e.g., a two-hour MMTT) prior to administration of the first 12-day course of teplizumab; and administering to the subject a second 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 14000 pg/m2, wherein the first and the second 12-day courses of teplizumab are administered at an interval of at least about 3 months, or at about a 6-month or to about a 12-month interval, and wherein the administration of teplizumab results in a 40% to 80%, or a more than 80%, increase in mean C-peptide value compared to subjects receiving placebo.
[0024] Aspects of the disclosure relate to teplizumab for use in a method of maintaining or increasing C-peptide value and/or slowing loss of beta-cell function in a subject having T1D, e.g., Stage 3 or clinical T1D, the method comprising administering to the subject a first 12- day course of teplizumab at a total dose of from about 9000 pg/m2 to about 9500 pg/m2, wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during an MMTT (e.g., a two-hour MMTT) prior to administration of the first 12-day course of teplizumab; and administering to the subject a second 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 9500 pg/m2, wherein the first and the second 12-day courses of teplizumab are administered at an interval of at least about 3 months, or at about a 6-month to about a 12-month interval, and wherein the administration of teplizumab results in a 40% to
80%, or a more than 80%, increase in mean C-peptide value compared to subjects receiving placebo.
[0025] Aspects of the disclosure relate to teplizumab for use in a method of maintaining or increasing C-peptide value and/or slowing loss of beta-cell function in a subject having T1D, e.g., Stage 3 or clinical T1D, the method comprising administering to the subject a first 12- day course of teplizumab at a total dose of from about 9000 pg/m2 to about 14000 pg/m2, wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during an MMTT (e.g., a two-hour MMTT) prior to administration of the first 12-day course of teplizumab; and administering to the subject a second 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 14000 pg/m2, wherein the first and the second 12-day courses of teplizumab are administered at an interval of at least 3 months, or at about a 6-month to about a 12-month interval, and wherein the administration of teplizumab results in a 40% to 80%, or a more than 80%, increase in mean C-peptide value compared to subjects receiving placebo.
[0026] In some embodiments, the 12-day course comprises a first dose of 106 pg/m2 teplizumab on day 1, a second dose of 425 pg/m2 teplizumab on day 2, and one dose of 850 pg/m2 on each of days 3-12, and wherein the total dose is approximately 9031 pg/m2.
[0027] In some embodiments, the method further comprises administering to the subject in need thereof a third or more 12-day course of teplizumab, each course at a total dose of more than about 9000 pg/m2. In some embodiments, the third or more 12-day course of teplizumab comprises a first dose of 106 pg/m2 teplizumab on day 1, a second dose of 425 pg/m2 teplizumab on day 2, and one dose of 850 pg/m2 on each of days 3-12, and the total dose of each course is approximately 9031 pg/m2. In some embodiments, the third or more 12-day course of teplizumab is administered at about a 6-month to about a 24-month interval. [0028] In some embodiments, the method comprises determining, after administration of each 12-day course, a baseline of a level of TIGIT+KLRG1+CD8+ T-cells and/or a baseline of a level of PD-1+CD8+ T-cells with respect to all CD3+ T-cells, monitoring the level of TIGIT+KLRG1+CD8+CD3+ T-cells and/or the level of PD-1+CD8+CD3+ T-cells; and administering an additional 12-day course of teplizumab when the level of the TIGIT+KLRG1+CD8+CD3+ T-cells and/or PD-1+CD8+CD3+ T-cells returns to the baseline level. In some embodiments, the baseline level of the TIGIT+KLRG1+CD8+ T-cells and/or the baseline level of the PD-1+CD8+ T-cells is less than about 5% of all CD3+ T-cells. In some embodiments, the determining of TIGIT+KLRG1+CD8+CD3+ T-cells and/or the PD- 1+CD8+CD3+ T-cells is by flow cytometry. In some embodiments, the monitoring of
TIGIT+KLRG1+CD8+CD3+ T-cells and/or the PD-1+CD8+CD3+ T-cells is by flow cytometry. In some embodiments, the determining of TIGIT+KLRG1+CD8+CD3+ T-cells and/or the PD-1+CD8+CD3+ T-cells is about 1-6 months, about 2-5 months, or about 3 months after the administration of each 12-day course. In some embodiments, if the subject has more than about 10% TIGIT+KLRG1+CD8+ T-cells and/or more than about 10% PD- 1+CD8+ T-cells in all CD3+ T-cells, subsequent monitoring is annual. In some embodiments, if the subject has less than about 10% TIGIT+KLRG1+CD8+ T-cells and/or less than about 10% PD-1+CD8+ T-cells in all CD3+ T-cells, subsequent monitoring is about every 3-6 months.
[0029] In some embodiments, each dose of teplizumab is administered parenterally. In some embodiments, each dose of teplizumab is administered by intravenous infusion. The patient may be pre-mediated as described herein.
[0030] In some embodiments, the subject has a peak C-peptide level ranging from 0.2 pmol/mL to 0.7 pmol/mL during an MMTT (e.g., a two-hour MMTT). In some embodiments, the subject has a peak C-peptide level of at least 0.7 pmol/mL during an MMTT (e.g., a two-hour MMTT).
[0031] In some embodiments, the method comprises assessing the AUC of C-peptide following an MMTT (e.g., a four-hour MMTT) at 78 weeks or 18 months.
[0032] In some embodiments, the subject administered with teplizumab maintains or reduces baseline HbAlc levels, and/or maintains or increases TIR with less insulin use, than a subject administered with placebo.
[0033] In some embodiments, the administering of teplizumab to the subject results in a reduction of insulin dosage of 10% to 30%, or more than 30%, compared to subjects receiving placebo.
[0034] In some embodiments, the administering of teplizumab to the subject results in a reduction of insulin dosage by at least 0.1 U/kg/day or in maintenance of insulin dosage. [0035] In some embodiments, the administering of teplizumab to the subject results in a reduction of HbAlc baseline by 0.1 to 1 point, or more than 1 point, compared to subjects receiving placebo.
[0036] In some embodiments, the administering of teplizumab to the subject results in an increase of 3 to 10%, or more than 10%, TIR (%) for glycemia as assessed using a glucose monitoring system.
[0037] Slowing the loss of beta-cell function includes preserving beta-cells, slowing the loss or destruction of beta-cells, and/or preserving beta-cell function (e.g., production of
insulin). In some embodiments, the administering of teplizumab results in slower loss of beta-cell function for at least about 18 months or 78 weeks.
BRIEF DESCRIPTION OF THE DRAWINGS
[0038] Figure 1: Simulated Concentrations for Three Dosing Regimens: Population Predictions for a Typical Male Patient with WT = 60 kg, Age = 18 years, BSA=1.67 m2, and no Detected AD As.
[0039] Figure 2: Comparison of Concentrations for Dosing Regimens 1 and 2: Modelbased Simulations for a Typical Male Patients with WT = 60 kg, Age = 18 years, BSA=1.67 m2, and no Detected AD As.
[0040] Figure 3: Comparison of Concentrations for Herold Dosing Regimen and Dosing Regimen 1 : Model-based Simulations for a Typical Male Patients with WT = 60 kg, Age = 18 years, BSA=1.67 m2, and no Detected ADAs.
[0041] Figure 4: Comparison of Concentrations on the last dosing day for Herold Dosing Regimen and Dosing Regimen 1 : Model-based Simulations for a Typical Male Patients with WT = 60 kg, Age = 18 years, BSA=1.67 m2, and no Detected ADAs.
[0042] Figure 5: Simulated Concentrations For Three Dosing Regimens: Population Predictions for a Typical Male Patient with WT = 60 kg, Age = 18 years, BSA=1.67 m2, and High Level of Detected ADAs.
[0043] Figure 6: Comparison of Concentrations for Dosing Regimens 1 and 2: Modelbased Simulations for Typical Male Patient with WT = 60 kg, Age = 18 years, B SAM.67 m2, and High Level of Detected ADAs.
[0044] Figure 7: Comparison of Concentrations for Herold Dosing Regimen and Dosing Regimen 1 : Model -based Simulations for Typical Male Patients with WT = 60 kg, Age = 18 years, BSA=1.67 m2, and High Level of Detected ADAs.
[0045] Figure 8: Comparison of Concentrations on the last dosing day for Herold Dosing Regimen and Dosing Regimen 1 : Model-based Simulations for Typical Male Patients with WT = 60 kg, Age = 18 years, B SA .67 m2, and High Level of Detected ADAs.
[0046] Figure 9: Simulated Concentrations For Three Dosing Regimens: Population Predictions for a Typical Male Patient with WT = 45 kg, Age = 13 years, BSAM.33 m2, and no Detected ADAs.
[0047] Figure 10: Comparison of Concentrations for Dosing Regimens 1 and 2: Modelbased Simulations for Typical Male Patients with WT = 45 kg, Age = 13 years, BSAM.33 m2, and no Detected ADAs.
[0048] Figure 11: Comparison of Concentrations for Herold Dosing Regimen and Dosing Regimen 1 : Model -based Simulations for Typical Male Patients with WT = 45 kg, Age = 13 years, BSA=1.33 m2, and no Detected AD As.
[0049] Figure 12: Comparison of Concentrations on the last dosing day for Herold Dosing Regimen and Dosing Regimen 1 : Model-based Simulations for Typical Male Patients with WT = 45 kg, Age = 13 years, BSA=1.33 m2, and no Detected ADAs.
[0050] Figure 13: Simulated Concentrations For Three Dosing Regimens: Population Predictions for a Typical Male Patient with WT = 45 kg, Age = 13 years, BSA=1.33 m2, and High Level of Detected ADAs.
[0051] Figure 14: Comparison of Concentrations for Dosing Regimens 1 and 2: Modelbased Simulations for Typical Male Patients with WT = 45 kg, Age = 13 years, BSA=1.33 m2, and High Level of Detected ADAs.
[0052] Figure 15: Comparison of Concentrations for Herold Dosing Regimen and Dosing Regimen 1 : Model -based Simulations for Typical Male Patients with WT = 45 kg, Age = 13 years, BSA=1.33 m2, and High Level of Detected ADAs.
[0053] Figure 16: Comparison of Concentrations on the last dosing day for Herold Dosing Regimen and Dosing Regimen 1 : Model-based Simulations for Typical Male Patients with WT = 45 kg, Age = 13 years, BSA=1.33 m2, and High Level of Detected ADAs.
[0054] Figure 17: Comparison of Concentrations for Herold Regimen and Dosing Regimen 2: Model-based Simulations for Male Patients with WT = 60 kg, Age = 18 years, BSA=1.67 m2, and no Detected ADAs (42 days).
[0055] Figure 18: Comparison of Median Concentrations for Herold Regimen and Dosing Regimen 2: Model-based Simulations for Male Patients with WT = 60 kg, Age = 18 years, BSA=1.67 m2, and no Detected ADAs (35 days).
[0056] Figure 19: Comparison of Concentrations for Herold Regimen and Dosing Regimen 2: Model-based Simulations for Male Patients with WT = 60 kg, Age = 18 years, BSA=1.67 m2, and High Level of Detected ADAs (42 days)
[0057] Figure 20: Comparison of Median Concentrations for Herold Regimen and Dosing Regimen 2: Model-based Simulations for Male Patients with WT = 60 kg, Age = 18 years, BSA=1.67 m2, and High Level of Detected ADAs (35 days).
[0058] Figure 21: Comparison of Concentrations for Herold Regimen and Dosing Regimen 2: Model -based Simulations for Male Patients with WT = 45 kg, Age = 13 years, BSA=1.33 m2, and no Detected ADAs (42 days).
[0059] Figure 22: Comparison of Median Concentrations for Herold Regimen and Dosing
Regimen 2: Model -based Simulations for Male Patients with WT = 45 kg, Age = 13 years, BSA=1.33 m2, and no Detected AD As (35 days).
[0060] Figure 23: Comparison of Concentrations for Herold Regimen and Dosing Regimen 2: Model -based Simulations for Male Patients with WT = 45 kg, Age = 13 years, BSA=1.33 m2, and High Level of Detected AD As (42 days).
[0061] Figure 24: Comparison of Median Concentrations for Herold Regimen and Dosing Regimen 2: Model -based Simulations for Male Patients with WT = 45 kg, Age = 13 years, BSA=1.33 m2, and High Level of Detected AD As (35 days).
[0062] Figure 25: Diagram of the study design according to one embodiment.
[0063] Figure 26: Modified Dosing Schedule for Participants Affected by COVID-19 Pandemic Restrictions according to one embodiment.
[0064] Figure 27: Graph showing insulin use at different timepoints (baseline, week 12, week 26, week 39, week 52, week 65 and week 78). Figure 27 shows that insulin use was numerically lower in patients treated with teplizumab.
[0065] Figure 28: Graph showing the percentage of subjects who met HblAc <6.5% and insulin daily dose <0.25 unit/kg/days at different timepoints (baseline, week 12, week 26, week 39, week 52, week 65 and week 78). Figure 28 shows that more teplizumab patients met insulin discontinuation criteria of HblAc <6.5% and insulin daily dose <0.25 unit/kg/days.
[0066] Figure 29: Graph showing the HbAlc levels at different timepoints (baseline, week 12, week 26, week 39, week 52, week 65 and week 78). Figure 29 shows that HbAlc target levels were achieved in both treatment groups.
[0067] Figure 30: Graph showing the percentage time in range at different timepoints (baseline, week 12, week 26, week 39, week 52, week 65 and week 78). Figure 30 shows that patients treated with teplizumab spent more Time in Range (TIR).
[0068] Figures 31A-31D: Graphs showing the efficacy endpoints. Figure 31A shows stimulated C-peptide levels area under the curve (ln(AUC+l) over time, data are presented as least-squares mean (95% CI). Figure 31B shows the proportion of patients with time in range (>70%). Figure 31C shows average daily insulin dose <0.25 units/kg/d over time, data are presented as least-squares mean (95% CI). Figure 31D shows the proportion of patients meeting criteria for clinical remission (HbAlc <6.5% and insulin daily dose <0.25 units/kg/d). Estimates are obtained from a mixed model of repeated measures model with treatment group, visit, and age group at randomization baseline values as fixed effects, and a
treatment by visit interaction term. AUC, area under the curve; CGM, continuous glucose monitoring; CI = confidence interval.
[0069] Figure 32: Graph showing the subgroup analysis of C-peptide AUC- ITT Population (AUC, area under the curve; CI, confidence interval).
[0070] Figure 33: Graph showing the proportion of patients with peak C-peptide >0.2 pmol/mL over time. ***P<0.001. Error bars indicate 95% confidence intervals. Percentages are based on the number of non-missing observations in each treatment group. Estimates are obtained from a generalized linear model for repeated measures using logit link function that includes treatment, visit, age group at randomization, and baseline peak C-peptide as fixed effects, and a treatment by visit interaction term.
[0071] Figures 34 A-34B: Graphs showing Hb Ale levels. Figure 34A shows Hb Ale level over time, and Figure 34B shows the proportion of Patients with HbAlc <7% Over Time. MMRM, mixed effect model for repeated measures. Error bars indicate 95% confidence interval. Baseline is defined as the most recent value collected prior to the first dose of study drug. Estimates are based on an, mixed effect model for repeated measures (MMRM model) with treatment group, visit, age group at randomization, screening peak C- peptide category, and a treatment by visit interaction term as fixed effects.
[0072] Figure 35: Graph showing the proportion of patients with insulin dose <0.25 units/kg/day by study visit. Error bars indicate 95% CI.
[0073] Figure 36: Graph showing key clinical outcome assessment domains at Week 78. DTSQ, Diabetes Treatment Satisfaction Questionnaire; MMRM, mixed effect model for repeated measures; HFS, Hypoglycemic Fear Survey; PedsQL, Pediatric Quality of Life Inventory. Estimates and p-value are based on an MMRM model with treatment group, visit, age group at randomization, screening peak C-peptide category, baseline score, and a treatment by visit interaction term as fixed effects. Least-squares mean difference = teplizumab - placebo. PedsQL scores Scores range from 0 to 100 where a higher score indicates a better outcome. Minimal clinically important difference scores in PedsQL were 5.27 for child/teen and 4.54 for parents. HFS scores range from 0 to 4 where a lower score indicates a better outcome. Signs have been adjusted for graphical representation. DTSQ scores range from 0 to 48 where a higher score indicates a better outcome.
[0074] Figure 37: Plot Emax model: predicted C-peptide change vs AUC, Year 2. The Protege study was conducted in newly diagnosed (Stage 3) T1D patients and tested 3 teplizumab dosing regimens (full 14-day [about 9,030 g/m2 cumulative dose], one-third of the 14-day regimen [1/3], and a 6-day curtailed [first 6 days of the full 14-day regimen]).
DETAILED DESCRIPTION
[0075] T1D usually develops in childhood and adolescence; however, it can also present in adulthood as late as the 5th and 6th decades of life, although much less frequently (Atkinson 2014, Bluestone 2010, Streisand 2014). In addition to being more prone to some short- and long-term complications, there are differences in the clinical course and response to immune therapies between children/young adults and older adults. In the days or weeks before initial diagnosis, children and adolescents often suffer from severe diabetes symptoms, including polydipsia, polyuria, and weight loss, which could result in a clinical presentation of DKA and shock which requires hospitalization (Atkinson 2014, Bluestone 2010, Streisand 2014, Mittermayer 2017). Children and young adults with new-onset T1D usually have an immediate need for exogenous insulin.
[0076] This sharply contrasts with the experience of adults who develop T1D who often have months or years of non-specific symptoms or present asymptomatically from routine glycemic screening. These individuals can often be managed for prolonged periods of time (months or years) with diet or oral hypoglycemic agents before a demonstrable insulin need. More definitive studies have shown a different rate of decline of P cells according to age (Greenbaum 2012; Ludvigsson 2013). Following decades of study, the Diabetes TrialNet network has concluded that “age is the most important factor impacting the rate of decline of C-peptide post diagnosis” in that a significantly more rapid rate of decline occurs in children and adolescents compared to younger and older adults with new-onset disease. This more rapid decline appears to be due to a much more virulent and aggressive autoimmune process in children compared to adults, ostensibly supporting that there are important differences in T1D immuno-pathoetiology in younger versus older individuals (Greenbaum 2012, Campbell-Thompson 2016). Due to these fundamental differences, it is reasonable to expect that adults and children may respond differently to an immune-based disease modifying therapy. In other words, one treatment may be very effective in children but not effective at all in adults and vice versa (Rigby 2014).
[0077] Children and adolescents are those at highest risk of developing disease and suffer most substantially from short- and long-term morbidity and mortality, and thus this group has the most to benefit from a disease modifying therapy (Wherrett 2015). This has recently been reinforced by a large study showing that those diagnosed with T1D in childhood and adolescence have a 4-6-fold increase in lifetime mortality risk, including seven times the risk of mortality from cardiovascular disease, compared to counterparts without T1D. This mortality risk is in sharp contrast to individuals diagnosed with T1D in adulthood, who have
a ~3-fold higher risk from all-cause and cardiovascular disease-related mortality compared to their otherwise healthy peers (Rawshani 2017, Rawshani 2018). Recent reports indicate that those with T1D have a life expectancy -11-13 years less than otherwise healthy-age matched individuals (Lind 2014, Huo 2016). While it is a goal in T1D research to reduce the morbidity and mortality for all with T1D, it is apparent that the most urgent need is for those who develop T1D in childhood and adolescence.
[0078] There is therefore a need to develop a therapy for children who can most likely benefit from it.
[0079] Aspects of the disclosure relate to methods of treating T1D in subjects in need thereof. Provided herein are methods that preserve [3-cell function and improve clinical management of T1D in children compared with the natural course of disease and current standard of care including exogenous insulin therapy. The preservation of [3-cell function is anticipated to translate to clinical and/or metabolic benefits consistent with improved ability to maintain glycemic control and short- and/or long-term outcomes. In some embodiments, the method comprises diagnosing patients 8 to 17 years of age with T1D, administering to the patients within 6 weeks of diagnosis a first course of daily doses of teplizumab for 12 days, and a second course of daily doses of teplizumab for 12 days, wherein the first and second courses are separated by a 6-month interval. In some embodiments, the method further comprises assessing the AUC of C-peptide following an MMTT) at 78 weeks (18 months or 1.5 years), and/or evaluating clinical endpoints such insulin use, HbAlc levels, and hypoglycemic episodes.
[0080] Provided herein are methods of treating clinical T1D, comprising administering to a subject in need thereof a 12-day course of teplizumab at a total dose of more than about 9000 pg/m2, wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during an MMTT prior to administration of the 12-day course of teplizumab. Such a C-peptide level indicates that the subject is still producing insulin. In some embodiments, the administration of the 12-day course of teplizumab can substantially protect P-cells, prevent P cell death over time, and/or significantly reduce the extent of P-cell death over time. In some embodiments, the administration of the 12-day course of teplizumab can lessen or prevent reduction of pancreatic insulin production capacity. In some embodiments, the administration of a 12-day course of teplizumab can decrease or eliminate the need for insulin use. In some embodiments, the method comprises administering two 12-day courses separated by 6 or 12 months.
[0081] In some embodiments, the subject has been diagnosed with T1D within 6 weeks
prior to the administrating step. In some embodiments, the subject is about 8 to 17 years old. [0082] In some embodiments, each dose is administered parenterally. In some embodiments, each dose is administered by intravenous infusion.
[0083] In some embodiments, the administrating step results in reduction by at least 10% of insulin use as compared to subjects treated with placebo.
[0084] In some embodiments, the administration of teplizumab can decrease the amount of insulin needed to maintain or decrease HbAlc in the subject.
I. Definitions
[0085] Certain terms are defined herein below. Additional definitions are provided throughout the application.
[0086] As used herein, the articles “a” and “an” refer to one or more than one, e.g., to at least one, of the grammatical object of the article. The use of the words “a” or “an” when used in conjunction with the term “comprising” herein may mean “one,” but it is also consistent with the meaning of “one or more,” “at least one,” and “one or more than one.” [0087] As used herein, “about” and “approximately” generally mean an acceptable degree of error for the quantity measured given the nature or precision of the measurements.
Exemplary degrees of error are within 20 percent (%), typically, within 10%, and more typically, within 5% of a given range of values. The term “substantially” means more than 50%, preferably more than 80%, and most preferably more than 90% or 95%.
[0088] As used herein the term “comprising” or “comprises” is used in reference to compositions, methods, and respective component(s) thereof, that are present in a given embodiment, yet open to the inclusion of unspecified elements.
[0089] As used herein the term “consisting essentially of’ refers to those elements required for a given embodiment. The term permits the presence of additional elements that do not materially affect the basic and novel or functional characteristic(s) of that embodiment of the disclosure.
[0090] The term “consisting of’ refers to compositions, methods, and respective components thereof as described herein, which are exclusive of any element not recited in that description of the embodiment.
[0091] The term “antibody” herein is used in the broadest sense and encompasses various antibody structures, including but not limited to monoclonal antibodies, polyclonal antibodies, multispecific antibodies (e.g., bispecific antibodies), and antibody fragments so long as they exhibit the desired antigen-binding activity.
[0092] An “antibody fragment” refers to a molecule other than an intact antibody that comprises a portion of an intact antibody that binds the antigen to which the intact antibody binds. Examples of antibody fragments include but are not limited to Fv, Fab, Fab', Fab'-SH, F(ab')2; diabodies; linear antibodies; single-chain antibody molecules (e.g., scFv); and multispecific antibodies formed from antibody fragments.
[0093] As used herein, the term “onset” of disease with reference to T1D refers to a patient meeting the criteria established for diagnosis of T1D by the American Diabetes Association (see Mayfield et al., Am Fam Physician (2006) 58: 1355-62).
[0094] As used herein, a “protocol” includes dosing schedules and dosing regimens. The protocols herein are methods of use and include therapeutic protocols. A “dosing regimen,” “dosage regimen,” or “course of treatment” may include administration of several doses of a therapeutic agent over 1 to 20 days.
[0095] As used herein, the terms “subject” and “patient” are used interchangeably. As used herein, the terms “subject” and “subjects” refer to an animal, preferably a mammal including a non-primate (e.g., a cow, pig, horse, cat, dog, rat, and mouse) and a primate (e.g., a monkey or a human), and more preferably a human. In some embodiments, the patient population comprises children. In some embodiments, the patient population comprises children newly diagnosed with T1D. In some embodiments, the patient population is treated within 6 weeks of the T1D diagnosis. In some embodiments, the patient population comprises children who are positive for at least one T ID-associated autoantibody and have a peak stimulated C-peptide of >0.2 pmol/mL at screening.
[0096] As used herein, the term “children” (and variations thereof) includes those being around 8 to 17 years of age.
[0097] As used herein, the term “effective amount” refers to that amount of teplizumab sufficient to result in the delay or prevention of the development, recurrence or onset of one or more symptoms of T1D.
[0098] As used herein, the terms “treat,” “treatment” and “treating” refer to the amelioration of one or more symptoms associated with T1D that results from the administration of one or more CD3 binding molecules. In some embodiments, such terms refer to a reduction in a human's average number of hypoglycemic episodes. In other embodiments, such terms refer to the maintenance of a reference level of C-peptide in the peripheral blood.
[0099] In some embodiments, the effective amount reduces one or more T1D symptoms by at least 5%, by at least 10%, by at least 20%, by at least 25%, by at least 30%, by at least
35%, by at least 40%, by at least 45%, by at least 50%, by at least 55%, by at least 60%, by at least 65%, by at least 70%, by at least 75%, by at least 80%, by at least 85%, by at least 90%, or by at least 95%.
[0100] Various aspects of the disclosure are described in further detail below. Additional definitions are set out throughout the specification.
II. Anti-CD3 Antibodies and Pharmaceutical Compositions
[0101] The terms “anti-CD3 antibody” and “an antibody that binds to CD3” refer to an antibody or antibody fragment that is capable of binding cluster of d ferentiation 3 (CD3) with sufficient affinity such that the antibody is useful as a prophylactic, diagnostic and/or therapeutic agent in targeting CD3. In some embodiments, the extent of binding of an anti- CD3 antibody to an unrelated, non-CD3 protein is less than about 10% of the binding of the antibody to CD3 as measured, e.g., by a radioimmunoassay (RIA). In some embodiments, an antibody that binds to CD3 has a dissociation constant (Kd) of < 1 pM, < 100 nM, < 10 nM, < 1 nM, < 0.1 nM, < 0.01 nM, or < 0.001 nM (e.g., 10'8 M or less, e.g. from 10'8 M to 10'13 M, e.g., from 10'9 M to 10'13 M). In some embodiments, an anti-CD3 antibody binds to an epitope of CD3 that is conserved among CD3 from different species.
[0102] In some embodiments, the anti-CD3 antibody can be ChAglyCD3 (otelixizumab). Otelixizumab is a humanized Fc nonbinding anti-CD3, which was evaluated initially in phase 2 studies by the Belgian Diabetes Registry (BDR) and then developed by Tolerx, which then partnered with GSK to conduct the phase 3 DEFEND new onset T1D trials (NCT00678886, NCT01123083, NCT00763451). Otelixizumab is administered IV with infusions over 8 days. See, e.g., Wiczling et al., J Clin Pharmacol. (2010) 50(5):494-506; Keymeulen et al., N Engl J Med. (2005) 352:2598-608; Keymeulen et al., Diabetologia. (2010) 53:614-23; Hagopian et al., Diabetes (2013) 62:3901-8; Aronson et al., Diabetes Care (2014) 37:2746- 54; Ambery et al., Diabet Med. (2014) 31 :399-402; Bolt et al., Eur J Immunol. (1993) 23(2):403-l 1; Vlasakakis et al., Br J Clin Pharmacol. (2019) 85:704-714; Guglielmi et al, Expert Opinion on Biological Therapy (2016) 16(6):841-6; Keymeulen et al., N Engl J Med. (2005) 352(25):2598-608; Keymeulen et al., Blood 2Q\G) 115(6): 1145-55.; Sprangers et al., Immunotherapy (2011) 3(11): 1303-16; Daifotis et al., Clinical Immunology (2013) 149:268- 78; all incorporated herein by reference.
[0103] In some embodiments, the anti-CD3 antibody can be visilizumab (also called HuM291; Nuvion). Visilizumab is a humanized anti-CD3 monoclonal antibody characterized by a mutated IgG2 isotype, lack of binding to Fey receptors, and the ability to
induce apoptosis selectively in activated T cells. It was evaluated in patients in graft-versus- host disease (NCT00720629; NCT00032279) and in ulcerative colitis (NCT00267306) and Crohn’s Disease (NCT00267709). See, e.g., Sandborn et al., Gut (2010) 59 (11): 1485-92, incorporated herein by reference.
III. Teplizumab
[0104] In some embodiments, the anti-CD3 antibody can be teplizumab. Teplizumab, also known as hOKT3yl(Ala-Ala) (containing an alanine at positions 234 and 235) is an anti-CD3 antibody that has been engineered to alter the function of the T lymphocytes that mediate the destruction of the insulin-producing P-cells of the islets of the pancreas. Teplizumab binds to an epitope of the CD3s chain expressed on mature T-cells and by doing so changes their function. Circulating T-cells (and other lymphocytes) are transiently reduced following teplizumab treatment, in a process that may include margination and depletion (Long 2017, Sherry 2011). In addition to reduced effector function of T-cells, teplizumab appears to both increase the number and function of regulatory T-cells (Tregs) (Ablamunits 2010, Bisikirska 2005, Long 2017, Waldron-Lynch 2012). More recent studies indicate that teplizumab induces immunologic “exhaustion” in a subset of effector CD8+ T-cells, perhaps making them more susceptible to regulation or deletion (Long 2016, Long 2017). Taken together, these mechanistic data suggest that teplizumab not only exerts a “suppressive” effect on p cell immune destructive processes but rather is an immune “modulator” favoring a rebalancing of effector and regulatory arms involved with T1D autoimmunity and supporting the notion that teplizumab may have the ability to contribute to the re-introduction of P cell self-tolerance (Lebastchi 2013).
[0105] Sequences and compositions of teplizumab are disclosed in U.S. Patent Nos. 6,491,916; 8,663,634; and 9,056,906, each incorporated herein by reference in its entirety. The molecular weight of teplizumab is approximately 150 KD. The full sequences of the light and heavy chains are set forth below. Bolded portions are the complementaritydetermining regions (CDR).
Teplizumab Light Chain (SEQ ID NO: 1):
DIQMTQSPSSLSASVGDRVTITCSASSSVSYMNWYQQTPGKAPKRWIYDTSKLASGV PSRFSGSGSGTDYTFTISSLQPEDIATYYCQQWSSNPFTFGQGTKLQITRTVAAPSVFI FPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTY SLS STLTLSKAD YEKHKVYACEVTHQGLS SPVTKSFNRGEC
Teplizumab Heavy Chain (SEQ ID NO: 2):
QVQLVQSGGGVVQPGRSLRLSCKASGYTFTRYTMHWVRQAPGKGLEWIGYINPSR GYTNYNQKVKDRFTISRDNSKNTAFLQMDSLRPEDTGVYFCARYYDDHYCLDYW GQGTPVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTS GVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDK THTCPPCPAPEAAGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYV DGVEVHNAI<TI<PREEQYNSTYRVVSVLTVLHQDWLNGI<EYI<CI<VSNI<ALPAPIEI< TISKAKGQPREPQVYTLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNY KTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
[0106] In some embodiments, provided herein, is a pharmaceutical composition. Such compositions comprise an effective amount of an anti-CD3 antibody, and a pharmaceutically acceptable carrier. In some embodiments, the term “pharmaceutically acceptable” means approved by a regulatory agency of the Federal or a state government or listed in the U.S. Pharmacopeia or other generally recognized pharmacopeia for use in animals, and more particularly in humans. The term “carrier” refers to a diluent, adjuvant (e.g., Freund's adjuvant (complete and incomplete)), excipient, or vehicle with which the therapeutic is administered. Such pharmaceutical carriers can be sterile liquids, such as water and oils, including those of petroleum, animal, vegetable or synthetic origin, such as peanut oil, soybean oil, mineral oil, sesame oil and the like. Water is a preferred carrier when the pharmaceutical composition is administered intravenously. Saline solutions and aqueous dextrose and glycerol solutions can also be employed as liquid carriers, particularly for injectable solutions. Suitable pharmaceutical excipients include starch, glucose, lactose, sucrose, gelatin, malt, rice, flour, chalk, silica gel, sodium stearate, glycerol monostearate, talc, sodium chloride, dried skim milk, glycerol, propylene, glycol, water, ethanol and the like (see, for example, Handbook of Pharmaceutical Excipients, Arthur H. Kibbe (ed., 2000, which is incorporated by reference herein in its entirety), Am. Pharmaceutical Association, Washington, D.C).
[0107] The composition, if desired, can also contain minor amounts of wetting or emulsifying agents, or pH buffering agents. These compositions can take the form of solutions, suspensions, emulsion, tablets, pills, capsules, powders, sustained release formulations and the like. Oral formulation can include standard carriers such as pharmaceutical grades of mannitol, lactose, starch, magnesium stearate, sodium saccharine, cellulose, magnesium carbonate, etc. Examples of suitable pharmaceutical carriers are described in “Remington's Pharmaceutical Sciences” by E. W. Martin. Such compositions contain a therapeutically effective amount of a therapeutic agent preferably in purified form,
together with a suitable amount of carrier so as to provide the form for proper administration to the patient. The formulation should suit the mode of administration. In some embodiments, the pharmaceutical compositions are sterile and in suitable form for administration to a subject, preferably an animal subject, more preferably a mammalian subject, and most preferably a human subject.
[0108] In some embodiments, it may be desirable to administer the pharmaceutical compositions locally to the area in need of treatment; this may be achieved by, for example, and not by way of limitation, local infusion, by injection, or by means of an implant, said implant being of a porous, non-porous, or gelatinous material, including membranes, such as sialastic membranes, or fibers. Preferably, when administering the anti-CD3 antibody, care must be taken to use materials to which the anti-CD3 antibody does not absorb.
[0109] In some embodiments, the composition can be delivered in a vesicle, in particular a liposome (see Langer, Science (1990) 249: 1527-33; Treat et al., in Liposomes in the Therapy of Infectious Disease and Cancer, Lopez-Berestein and Fidler (eds.), Liss, New York, pp. 353-365 (1989); Lopez-Berestein, ibid., pp. 317-327; see generally ibid.).
[0110] In some embodiments, the composition can be delivered in a controlled release or sustained release system. In some embodiments, a pump may be used to achieve controlled or sustained release (see Langer, supra, Sefton, 1987, CRC Crit. Ref. Biomed. Eng. 14:20; Buchwald et al., 1980, Surgery 88:507; Saudek et al., N Engl J Med. (1989) 321 :574). In some embodiments, polymeric materials can be used to achieve controlled or sustained release of the antibodies of the disclosure or fragments thereof (see, e.g., Medical Applications of Controlled Release, Langer and Wise (eds.), CRC Pres., Boca Raton, Fla. (1974); Controlled Drug Bioavailability, Drug Product Design and Performance, Smolen and Ball (eds.), Wiley, New York (1984); Ranger and Peppas, J Macromol Sci Rev Macromol Chem. (1983) 23:61; see also Levy et al., Science (1985) 228: 190; During et al., Ann Neurol. (1989) 25:351; Howard et al., JNeurosurg. (1989) 71 :105; U.S. Pat. No. 5,679,377; U.S. Pat. No. 5,916,597; U.S. Pat. No. 5,912,015; U.S. Pat. No. 5,989,463; U.S. Pat. No. 5,128,326; PCT Publication No. WO 99/15154; and PCT Publication No. WO 99/20253. Examples of polymers used in sustained release formulations include, but are not limited to, polyphydroxy ethyl methacrylate), poly(methyl methacrylate), poly(acrylic acid), poly(ethylene- co-vinyl acetate), poly(methacrylic acid), polyglycolides (PLG), polyanhydrides, poly(N- vinyl pyrrolidone), poly(vinyl alcohol), polyacrylamide, poly(ethylene glycol), polylactides (PLA), poly(lactide-co-glycolides) (PLGA), and poly orthoesters. In some embodiments, the polymer used in a sustained release formulation is inert, free of leachable impurities, stable
on storage, sterile, and biodegradable. In some embodiments, a controlled or sustained release system can be placed in proximity of the therapeutic target, i.e., the lungs, thus requiring only a fraction of the systemic dose (see, e.g., Goodson, in Medical Applications of Controlled Release, supra, vol. 2, pp. 115-138 (1984)).
[OHl] Controlled release systems are discussed in the review by Langer (1990, Science 249: 1527-1533). Any technique known to one of skill in the art can be used to produce sustained release formulations comprising one or more antibodies of the disclosure or fragments thereof. See, e.g., U.S. Pat. No. 4,526,938; PCT Publication No. WO 91/05548; PCT Publication No. WO 96/20698; Ning et al., Radiotherapy & Oncology (1996) 39: 179- 189; Song et al., PDA Journal of Pharmaceutical Science & Technology (1995) 50:372-97; Cleek et al., Pro Int’l Symp Control Rel Bioact Mater. (1997) 24:853-4; and Lam et al., Proc Int’l Symp Control Rel Bioact Mater. (1997) 24:759-60, each of which is incorporated herein by reference in its entirety.
[0112] A pharmaceutical composition can be formulated to be compatible with its intended route of administration. Examples of routes of administration include, but are not limited to, parenteral, e.g., intravenous, intradermal, subcutaneous, oral, intranasal (e.g., inhalation), transdermal (topical), transmucosal, and rectal administration. In some embodiments, the composition is formulated in accordance with routine procedures as a pharmaceutical composition adapted for intravenous, subcutaneous, intramuscular, oral, intranasal or topical administration to human beings. In some embodiments, a pharmaceutical composition is formulated in accordance with routine procedures for subcutaneous administration to human beings. Typically, compositions for intravenous administration are solutions in sterile isotonic aqueous buffer. Where necessary, the composition may also include a solubilizing agent and a local anesthetic such as lignocaine to ease pain at the site of the injection.
[0113] The compositions may be formulated for parenteral administration by injection, e.g., by bolus injection or continuous infusion. Formulations for injection may be presented in unit dosage form, e.g., in ampoules or in multi-dose containers, with an added preservative. The compositions may take such forms as suspensions, solutions or emulsions in oily or aqueous vehicles, and may contain formulary agents such as suspending, stabilizing and/or dispersing agents. Alternatively, the active ingredient may be in powder form for constitution with a suitable vehicle, e.g., sterile pyrogen-free water, before use.
[0114] In some embodiments, the disclosure provides dosage forms that permit administration of the anti-CD3 antibody continuously over a period of hours or days (e.g., associated with a pump or other device for such delivery), for example, over a period of 1
hour, 2 hours, 3 hours, 4 hours, 6 hours, 8 hours, 10 hours, 12 hours, 16 hours, 20 hours, 24 hours, 30 hours, 36 hours, 4 days, 5 days, 7 days, 10 days or 12 days. In some embodiments, the disclosure provides dosage forms that permit administration of a continuously increasing dose, for example, increasing from 106 pg/m2/day to 850 pg/m2/day or 211 pg/m2/day to 840 pg/m2/day over a period of 24 hours, 30 hours, 36 hours, 4 days, 5 days, 7 days, 10 days or 12 days.
[0115] The compositions can be formulated as neutral or salt forms. Pharmaceutically acceptable salts include those formed with anions such as those derived from hydrochloric, phosphoric, acetic, oxalic, tartaric acids, etc., and those formed with cations such as those derived from sodium, potassium, ammonium, calcium, ferric hydroxides, isopropylamine, triethylamine, 2-ethylamino ethanol, histidine, procaine, etc.
[0116] Generally, the ingredients of the compositions disclosed herein are supplied either separately or mixed together in unit dosage form, for example, as a dry lyophilized powder or water free concentrate in a hermetically sealed container such as an ampoule or sachet indicating the quantity of active agent. Where the composition is to be administered by infusion, it can be dispensed with an infusion bottle containing sterile pharmaceutical grade water or saline. Where the composition is administered by injection, an ampoule of sterile water for injection or saline can be provided so that the ingredients may be mixed prior to administration.
[0117] In particular, the disclosure provides that the anti-CD3 antibodies, or pharmaceutical compositions thereof, can be packaged in a hermetically sealed container such as an ampoule or sachet indicating the quantity of the agent. In some embodiments, the anti-CD3 antibody, or pharmaceutical compositions thereof is supplied as a dry sterilized lyophilized powder or water free concentrate in a hermetically sealed container and can be reconstituted, e.g., with water or saline to the appropriate concentration for administration to a subject. Preferably, the anti-CD3 antibody, or pharmaceutical compositions thereof is supplied as a dry sterile lyophilized powder in a hermetically sealed container at a unit dosage of at least 5 mg, more preferably at least 10 mg, at least 15 mg, at least 25 mg, at least 35 mg, at least 45 mg, at least 50 mg, at least 75 mg, or at least 100 mg. The lyophilized agents, or pharmaceutical compositions herein should be stored at between 2 °C and 8 °C in its original container and the therapeutic agents, or pharmaceutical compositions of the disclosure should be administered within 1 week, preferably within 5 days, within 72 hours, within 48 hours, within 24 hours, within 12 hours, within 6 hours, within 5 hours, within 3 hours, or within 1 hour after being reconstituted. In some embodiments, the pharmaceutical
composition is supplied in liquid form in a hermetically sealed container indicating the quantity and concentration of the agent. Preferably, the liquid form of the administered composition is supplied in a hermetically sealed container at least 0.25 mg/ml, more preferably at least 0.5 mg/ml, at least 1 mg/ml, at least 2.5 mg/ml, at least 5 mg/ml, at least 8 mg/ml, at least 10 mg/ml, at least 15 mg/ml, at least 25 mg/ml, at least 50 mg/ml, at least 75 mg/ml or at least 100 mg/ml. The liquid form should be stored at between 2 °C and 8 °C in its original container.
[0118] In some embodiments, the disclosure provides that the composition of the disclosure is packaged in a hermetically sealed container such as an ampoule or sachet indicating the quantity of the anti-CD3 antibody.
[0119] The compositions may, if desired, be presented in a pack or dispenser device that may contain one or more unit dosage forms containing the active ingredient. The pack may, for example, comprise metal or plastic foil, such as a blister pack.
[0120] The amount of the composition of the disclosure which is effective in the treatment of one or more symptoms associated with T1D can be determined by standard clinical techniques. The precise dose to be employed in the formulation can also depend on the route of administration and the seriousness of the condition, and should be decided according to the judgment of the practitioner and each patient's circumstances. Effective doses may be extrapolated from dose-response curves derived from in vitro or animal model test systems.
IV. Methods and Use
[0121] In some embodiments, the present disclosure encompasses administration of antihuman CD3 antibodies such as teplizumab to patients 8 through 17 years old 6 weeks from T1D diagnosis having a peak C-peptide level of >0.2 pmol/mL during an MMTT. In some embodiments, the peak C-peptide level at screening rages from 0.2 pmol/mL (inclusive) to 0.7 pmol/mL (inclusive).
[0122] In some embodiments, T1D diagnosis is according to the American Diabetes Association (ADA) criteria. As defined by the American Diabetes Association (ADA) for the clinical diagnosis of diabetes, the individual must meet one of the following 4 criteria:
• A fasting plasma glucose (FPG) of >126 mg/dL (7.0 mmol/L). Fasting is defined as no caloric intake for at least 8 hours.
• A 2-hour plasma glucose (PG) of >200 mg/dL (11.1 mmol/L) during an oral glucose tolerance test (OGTT). The test should be performed as described by the World
Health Organization (WHO), using a glucose load containing the equivalent of 75 g anhydrous glucose dissolved in water.
• A hemoglobin A1C (HbAlc) of >6.5% (48 mmol/mol). The test should be performed in a laboratory using a method that is National Glycohemoglobin Standardization Program (NGSP) certified and standardized to the Diabetes Control and Complications Trial (DCCT) assay.
• In a patient with classic symptoms of hyperglycemia or hyperglycemic crisis, a random PG of >200 mg/dL (11.1 mmol/L).
[0123] For the diagnosis of clinical T1D, the ADA suggests that plasma blood glucose rather than HbAlC should be used to diagnose the acute onset of T1D in individuals with symptoms of hyperglycemia.
[0124] According to ADA, a patient with classic symptoms, measurement of plasma glucose is sufficient to diagnose clinical diabetes (symptoms of hyperglycemia or hyperglycemic crisis plus a random plasma glucose >200 mg/dL [11.1 mmol/L]). In these cases, knowing the plasma glucose level is critical because, in addition to confirming that symptoms are due to diabetes, it will inform management decisions. Some providers may also want to know the HbAlC to determine how long a patient has had hyperglycemia. In addition, T1D, previously called “insulin-dependent diabetes” or “juvenile-onset diabetes,” accounts for 5-10% of diabetes and is due to cellular-mediated autoimmune destruction of the pancreatic P-cells. Autoimmune markers include islet cell autoantibodies and autoantibodies to GAD (GAD65), insulin, the tyrosine phosphatases IA-2 and IA-2 P, and ZnT8. T1D is defined by the presence of one or more of these autoimmune markers.
[0125] In some embodiments, the diagnosis of T1D is made with the use of a continuous glucose monitoring system (CGM) revealing high sensor average glucose levels (>=110 mg/dL), or high variability of glycemia (CV >=15), or less time in range (>=10% of the time above 140 mg/dL).
[0126] In some embodiments, the patient diagnosed with clinical T1D has a positive result on testing for at least one of the following T ID-related autoantibodies: Glutamic acid decarboxylase 65 (GAD65) autoantibodies, Islet antigen 2 (IA-2) autoantibodies, Zinc transporter 8 (ZnT8) autoantibodies, Islet cell cytoplasmic autoantibodies (ICA), or Insulin autoantibodies (if testing obtained within the first 14 days of insulin treatment). In some embodiments, the presence of the autoantibodies is detected by ELISA, electrochemiluminescence (ECL), radioassay (see, e.g., Yu et al., J Clin Endocrinol Metab .
(1996) 81 :4264-7), agglutination PCR (Tsai et al, ACS Central Science (2016) 2(3):139-47) or by any other method for immuno-specific detection of antibodies described herein or as known to one of ordinary skill in the art.
[0127] It is recognized that P-cells continue to be lost following T1D diagnosis. To maximize the effect of [3-cell preservation in patients with a recoverable level of endogenous insulin production, the patients to be treated are within 6 weeks from T1D diagnosis and have a peak C-peptide level of >0.2 pmol/mL during an MMTT.
[0128] In some embodiments, the methods provided herein prevent or delay the need for administration of insulin to the patients.
[0129] [3-cell function prior to, during, and after therapy may be assessed by methods described herein or by any method known to one of ordinary skill in the art. For example, the Diabetes Control and Complications Trial (DCCT) research group has established the monitoring of percentage glycosylated hemoglobin (HbAl and HbAlc) as the standard for evaluation of blood glucose control (DCCT, N Engl J Med. (1993) 329:977-86). Alternatively, characterization of daily insulin needs, C-peptide levels/response, hypoglycemic episodes, and/or FPIR may be used as markers of [3-cell function or to establish a therapeutic index (see Keymeulen et al., N Engl J Med. (2005) 352:2598-2608; Herold et al., Diabetes (2005) 54: 1763-9; U.S. Pat. Appl. Pub. No. 2004/0038867 Al; and Greenbaum et al., Diabetes (2001) 50:470-476, respectively). For example, FPIR is calculated as the sum of insulin values at 1 and 3 minutes post IGTT, which are performed according to Islet Cell Antibody Register User's Study protocols (see, e.g., Bingley et al., Diabetes (1996) 45: 1720-8 and McCulloch et al., Diabetes Care (1993) 16:911-5).
[0130] In some embodiments, the effective amount comprises a 12-day course of subcutaneous intravenous (IV) infusion of the anti-CD3 antibody such as teplizumab at 106- 850 micrograms/meter squared (pg/m2). In some embodiments, the total dosage over the duration of the regimen is about 14000 pg/m2, 13500 pg/m2, 13000 pg/m2, 12500 pg/m2, 12000 pg/m2, 11500 pg/m2, 11000 pg/m2, 10500 pg/m2, 10000 pg/m2, 9500 pg/m2, 9000 pg/m2, 8000 pg/m2, 7000 pg/m2, 6000 pg/m2, and may be less than 5000 pg/m2, 4000 pg/m2, 3000 pg/m2, 2000 pg/m2, or 1000 pg/m2. In some embodiments, the total dosage over the duration of the regimen is from about 9030 pg/m2 to about 14000 pg/m2, about 9030 pg/m2 to about 13500 pg/m2, about 9000 pg/m2 to about 13000 pg/m2, about 9000 pg/m2 to about 12500 pg/m2, about 9000 pg/m2 to about 12000 pg/m2, about 9000 pg/m2 to about 11500 pg/m2, about 9000 pg/m2 to about 11000 pg/m2, about 9000 pg/m2 to about 10500 pg/m2, about 9000 pg/m2 to about 10000 pg/m2, about 9000 pg/m2 to about 9500 pg/m2. In some
embodiments, the total dosage over the duration of the regimen is from about 9030 pg/m2 to about 14000 pg/m2, about 9030 pg/m2 to about 13500 pg/m2, about 9030 pg/m2 to about 13000 pg/m2, about 9030 pg/m2 to about 12500 pg/m2, about 9030 pg/m2 to about 12000 pg/m2, about 9030 pg/m2 to about 11500 pg/m2, from about 9030 pg/m2 to about 11000 pg/m2, about 9030 pg/m2 to about 10500 pg/m2, about 9030pg/m2 to about 10000 pg/m2, about 9030 pg/m2 to about 9500 pg/m2.
[0131] Without being bound by the theory, cumulative doses above about 9,000 pg/m2 of teplizumab are expected to have comparable efficacy in terms of C-peptide preservation as shown for about 9,000 pg/m2. That is because the exposure/response curve surprisingly reaches a plateau above which increasing doses do not result in increased efficacy. The evaluation of C-peptide preservation was performed utilizing the Protege study data. Model- predicted teplizumab AUCs versus change from baseline in C-peptide were plotted and an Emax analysis was performed. These data demonstrate that an Emax model describes the relationship between teplizumab exposure and change in C-peptide at 2 years. As shown in Figure 37, at teplizumab AUC levels greater than about 1500 ng*hr/mL (below the lowest AUC predicted for the about 9,000 pg/m2 dose, of 1789 ng*hr/mL) no additional improvement in C-peptide with increased teplizumab exposure was observed. Therefore, these data suggest that doses above about 9000 pg/m2 of teplizumab would have comparable efficacy in terms of C-peptide preservation as shown for about 9000 pg/m2.
[0132] In some embodiments, the effective amount comprises a 12-day course IV infusion of teplizumab at a first dose of 106 pg/m2 teplizumab on day 1, a second dose of 425 pg/m2 teplizumab on day 2, and one dose of 850 pg/m2 on each of days 3-12. In some embodiments, the effective amount comprises a 12-day course IV infusion of teplizumab at a first dose of 211 pg/m2 teplizumab on day 1, a second dose of 423 pg/m2 teplizumab on day 2, and one dose of 840 pg/m2 on each of days 3-12. In some embodiments, the effective amount comprises a 12-day course IV infusion of teplizumab at a first dose of approximately 100 pg/m2 teplizumab on day 1, a second dose of approximately 400 pg/m2 teplizumab on day 2, a third dose of approximately 850 pg/m2 on day 3, and approximately 1,200 pg/m2 on each of days 4-12. In some embodiments, the effective amount comprises a 12-day course IV infusion of teplizumab at a first dose of approximately 100 pg/m2 teplizumab on day 1, a second dose of approximately 400 pg/m2 teplizumab on day 2, a third dose of approximately 850 pg/m2 on day 3, and approximately 1,300 pg/m2 on each of days 4-12. In some embodiments, the effective amount comprises a 12-day course IV infusion of teplizumab at a first dose of approximately 100 pg/m2 teplizumab on day 1, a second dose of approximately
400 pg/m2 teplizumab on day 2, a third dose of approximately 850 pg/m2 on day 3, and approximately 1,400 pg/m2 on each of days 4-12. In some embodiments, the effective amount comprises a 12-day course IV infusion of teplizumab at a first dose of approximately 200 pg/m2 teplizumab on day 1, a second dose of approximately 400 pg/m2 teplizumab on day 2, a third dose of approximately 850 pg/m2 on day 3, and approximately 1,200 pg/m2 on each of days 4-12. In some embodiments, the effective amount comprises a 12-day course IV infusion of teplizumab at a first dose of approximately 200 pg/m2 teplizumab on day 1, a second dose of approximately 400 pg/m2 teplizumab on day 2, a third dose of approximately 850 pg/m2 on day 3, and approximately 1,300 pg/m2 on each of days 4-12. In some embodiments, the effective amount comprises a 12-day course IV infusion of teplizumab at a first dose of approximately 200 pg/m2 teplizumab on day 1, a second dose of approximately 400 pg/m2 teplizumab on day 2, a third dose of approximately 850 pg/m2 on day 3, and approximately 1,400 pg/m2 on each of days 4-12.
[0133] Provided herein is a dosing regimen comprising two or more courses of dosing with an anti-CD3 antibody such as teplizumab comprising a first course of dosing at week 1 and second course of dosing at week 26. In some embodiments, teplizumab is administered via IV infusion in two courses, with the first course starting on Day 1 (Week 1) and the second course on approximately Day 182 (Week 26), each course of treatment including daily infusions for 12 days, with a cumulative teplizumab dose of 9000 pg/m2 for each course of treatment. In some embodiments, teplizumab is administered via IV infusion in two courses, with the first course starting on Day 1 (Week 1) and the second course on approximately Day 182 (Week 26), each course of treatment including daily infusions for 12 days, with a cumulative teplizumab dose of 9500 pg/m2 for each course of treatment. In some embodiments, teplizumab is administered via IV infusion in two courses, with the first course starting on Day 1 (Week 1) and the second course on approximately Day 182 (Week 26), each course of treatment including daily infusions for 12 days, with a cumulative teplizumab dose of 10000 pg/m2 for each course of treatment. In some embodiments, teplizumab is administered via IV infusion in two courses, with the first course starting on Day 1 (Week 1) and the second course on approximately Day 182 (Week 26), each course of treatment including daily infusions for 12 days, with a cumulative teplizumab dose of 10500 pg/m2 for each course of treatment. In some embodiments, teplizumab is administered via IV infusion in two courses, with the first course starting on Day 1 (Week 1) and the second course on approximately Day 182 (Week 26), each course of treatment including daily infusions for 12 days, with a cumulative teplizumab dose of 11000 pg/m2 for each course. In some
embodiments, teplizumab is administered via IV infusion in two courses, with the first course starting on Day 1 (Week 1) and the second course on approximately Day 182 (Week 26), each course of treatment including daily infusions for 12 days, with a cumulative teplizumab dose of 11500 pg/m2 for each course of treatment. In some embodiments, teplizumab is administered via IV infusion in two courses, with the first course starting on Day 1 (Week 1) and the second course on approximately Day 182 (Week 26), each course of treatment including daily infusions for 12 days, with a cumulative teplizumab dose of 12000 pg/m2 for each course of treatment. In some embodiments, teplizumab is administered via IV infusion in two courses, with the first course starting on Day 1 (Week 1) and the second course on approximately Day 182 (Week 26), each course of treatment including daily infusions for 12 days, with a cumulative teplizumab dose of 12500 pg/m2 for each course of treatment. In some embodiments, teplizumab is administered via IV infusion in two courses, with the first course starting on Day 1 (Week 1) and the second course on approximately Day 182 (Week 26), each course of treatment including daily infusions for 12 days, with a cumulative teplizumab dose of 13000 pg/m2 for each course of treatment. In some embodiments, teplizumab is administered via IV infusion in two courses, with the first course starting on Day 1 (Week 1) and the second course on approximately Day 182 (Week 26), each course of treatment including daily infusions for 12 days, with a cumulative teplizumab dose of 13500 pg/m2 for each course of treatment. In some embodiments, teplizumab is administered via IV infusion in two courses, with the first course starting on Day 1 (Week 1) and the second course on approximately Day 182 (Week 26), each course of treatment including daily infusions for 12 days, with a cumulative teplizumab dose of 14000 pg/m2 for each course of treatment. In some embodiments, the 12 days course has a 2-day ramp-up phase and a 10- day fixed-, maximal dosing period. In some embodiments, 106 pg/m2 teplizumab is administered on day 1425 pg/m2 teplizumab is administered on day 2, and 850 pg/m2 teplizumab is administered on each of days 3-12.
[0134] In other embodiments, the course of dosing can be repeated at 2-month, 4-month, 5- month, 6-month, 8-month, 9-month, 10-month, 12-month, 15-month, 18-month, 24-month, 30-month, or 36-month intervals. In some embodiments, efficacy of the treatment with the anti-CD3 antibody such as teplizumab is determined as described herein, or as is known in the art, at 2 months, 4 months, 5 month, 6 months, 9 months, 12 months, 15 months, 18 months, 24 months, 30 months, or 36 months subsequent to the previous treatment.
[0135] In some embodiments, a subject is administered one or more doses, preferably 12 daily doses, of the anti-CD3 antibody such as teplizumab at about 5-1200 pg/m2, preferably,
106-850 pg/m2 to treat, or slow the progression of or ameliorate one or more symptoms of T1D.
[0136] In some embodiments, the subject is administered a treatment regimen comprising two courses of daily doses of an effective amount of the anti-CD3 antibody such as teplizumab, wherein the course of treatment is administered over 2 days, 3 days, 4 days, 5 days, 6 days, 7 days, 8 days, 9 days, 10 days, 11 days or 12 days. In some embodiments, the treatment regimen comprises administering doses of the effective amount every day, every 2nd day, every 3rd day or every 4th day.
[0137] In some embodiments, a subject is administered a treatment regimen comprising one or more doses of a prophylactically effective amount of the anti-CD3 antibody such as teplizumab, wherein the prophylactically effective amount is 200 pg/kg/day, 175 pg/kg/day, 150 pg/kg/day, 125 pg/kg/day, 100 pg/kg/day, 95 pg/kg/day, 90 pg/kg/day, 85 pg/kg/day, 80 pg/kg/day, 75 pg/kg/day, 70 pg/kg/day, 65 pg/kg/day, 60 pg/kg/day, 55 pg/kg/day, 50 pg/kg/day, 45 pg/kg/day, 40 pg/kg/day, 35 pg/kg/day, 30 pg/kg/day, 26 pg/kg/day, 25 pg/kg/day, 20 pg/kg/day, 15 pg/kg/day, 13 pg/kg/day, 10 pg/kg/day, 6.5 pg/kg/day, 5 pg/kg/day, 3.2 pg/kg/day, 3 pg/kg/day, 2.5 pg/kg/day, 2 pg/kg/day, 1.6 pg/kg/day, 1.5 pg/kg/day, 1 pg/kg/day, 0.5 pg/kg/day, 0.25 pg/kg/day, 0.1 pg/kg/day, or 0.05 pg/kg/day; and/or wherein the prophylactically effective amount is 1200 pg/m2/day, 1150 pg/m2/day, 1100 pg/m2/day, 1050 pg/m2/day, 1000 pg/m2/day, 950 pg/m2/day, 900 pg/m2/day, 850 pg/m2/day, 800 pg/m2/day, 750 pg/m2/day, 700 pg/m2/day, 650 pg/m2/day, 600 pg/m2/day, 550 pg/m2/day, 500 pg/m2/day, 450 pg/m2/day, 400 pg/m2/day, 350 pg/m2/day, 300 pg/m2/day, 250 pg/m2/day, 200 pg/m2/day, 150 pg/m2/day, 100 pg/m2/day, 50 pg/m2/day, 40 pg/m2/day, 30 pg/m2/day, 20 pg/m2/day, 15 pg/m2/day, 10 pg/m2/day, or 5 pg/m2/day.
[0138] In some embodiments, the intravenous dose of 1200 pg/m2 or less, 1150 pg/m2 or less, 1100 pg/m2 or less, 1050 pg/m2 or less, 1000 pg/m2 or less, 950 pg/m2 or less, 900 pg/m2 or less, 850 pg/m2 or less, 800 pg/m2 or less, 750 pg/m2 or less, 700 pg/m2 or less, 650 pg/m2 or less, 600 pg/m2 or less, 550 pg/m2 or less, 500 pg/m2 or less, 450 pg/m2 or less, 400 pg/m2 or less, 350 pg/m2 or less, 300 pg/m2 or less, 250 pg/m2 or less, 200 pg/m2 or less, 150 pg/m2 or less, 100 pg/m2 or less, 50 pg/m2 or less, 40 pg/m2 or less, 30 pg/m2 or less, 20 pg/m2 or less, 15 pg/m2 or less, 10 pg/m2 or less, or 5 pg/m2 or less of the anti-CD3 antibody such as teplizumab is administered over about 24 hours, about 22 hours, about 20 hours, about 18 hours, about 16 hours, about 14 hours, about 12 hours, about 10 hours, about 8 hours, about 6 hours, about 4 hours, about 2 hours, about 1.5 hours, about 1 hour, about 50 minutes, about 40 minutes, about 30 minutes, about 20 minutes, about 10 minutes, about 5 minutes, about 2
minutes, about 1 minute, about 30 seconds or about 10 seconds to prevent, treat or ameliorate one or more symptoms of type 1 diabetes. The total dosage over the duration of the regimen is preferably a total of less than about 14000 pg/m2, 13500 pg/m2, 13000 pg/m2, 12500 pg/m2, 12000 pg/m2, 11500 pg/m2, 11000 pg/m2, 10500 pg/m2, 10000 pg/m2, 9500 pg/m2, 9000 pg/m2, 8000 pg/m2, 7000 pg/m2, 6000 pg/m2, and may be less than 5000 pg/m2, 4000 pg/m2, 3000 pg/m2, 2000 pg/m2, or 1000 pg/m2. In some embodiments, the daily dosage administered in the regimen is from about 100 pg/m2to about 200 pg/m2, about 100 pg/m2 to about 500 pg/m2, about 100 pg/m2 to about 1000 pg/m2, or about 500 pg/m2 to about 1000 pg/m2.
[0139] In some embodiments, the dose escalates over the first three, first 1/4 of the doses (e.g., over the first 3 days of a 12-day regimen of one dose per day) of the treatment regimen until the daily effective amount of the anti-CD3 antibody such as teplizumab is achieved. In some embodiments, a subject is administered a treatment regimen comprising one or more doses of an effective amount of the anti-CD3 antibody such as teplizumab, wherein the effective amount is increased by, e.g., 0.01 pg/kg, 0.02 pg/kg, 0.04 pg/kg, 0.05 pg/kg, 0.06 pg/kg, 0.08 pg/kg, 0.1 pg/kg, 0.2 pg/kg, 0.25 pg/kg, 0.5 pg/kg, 0.75 pg/kg, 1 pg/kg, 1.5 pg/kg, 2 pg/kg, 4 pg/kg, 5 pg/kg, 10 pg/kg, 15 pg/kg, 20 pg/kg, 25 pg/kg, 30 pg/kg, 35 pg/kg, 40 pg/kg, 45 pg/kg, 50 pg/kg, 55 pg/kg, 60 pg/kg, 65 pg/kg, 70 pg/kg, 75 pg/kg, 80 pg/kg, 85 pg/kg, 90 pg/kg, 95 pg/kg, 100 pg/kg, or 125 pg/kg each day; or increased by, e.g., 100 pg/m2, 150 pg/m2, 200 pg/m2, 250 pg/m2, 300 pg/m2, 350 pg/m2, 400 pg/m2, 450 pg/m2, 500 pg/m2, 550 pg/m2, 600 pg/m2, or 650 pg/m2, each day as treatment progresses. In some embodiments, a subject is administered a treatment regimen comprising one or more doses of an effective amount of the anti-CD3 antibody such as teplizumab, wherein the effective amount is increased by a factor of 1.25, a factor of 1.5, a factor of 2, a factor of 2.25, a factor of 2.5, or a factor of 5 until the daily effective amount of the anti-CD3 antibody such as teplizumab is achieved.
[0140] In some embodiments, a subject is intramuscularly administered one or more doses of a 200 pg/kg or less, preferably 175 pg/kg or less, 150 pg/kg or less, 125 pg/kg or less, 100 pg/kg or less, 95 pg/kg or less, 90 pg/kg or less, 85 pg/kg or less, 80 pg/kg or less, 75 pg/kg or less, 70 pg/kg or less, 65 pg/kg or less, 60 pg/kg or less, 55 pg/kg or less, 50 pg/kg or less, 45 pg/kg or less, 40 pg/kg or less, 35 pg/kg or less, 30 pg/kg or less, 25 pg/kg or less, 20 pg/kg or less, 15 pg/kg or less, 10 pg/kg or less, 5 pg/kg or less, 2.5 pg/kg or less, 2 pg/kg or less, 1.5 pg/kg or less, 1 pg/kg or less, 0.5 pg/kg or less, or 0.2 pg/kg or less of the anti-CD3 antibody such as teplizumab to treat or ameliorate one or more symptoms of T1D.
[0141] In some embodiments, a subject is subcutaneously administered one or more doses of a 200 pg/kg or less, preferably 175 pg/kg or less, 150 pg/kg or less, 125 pg/kg or less, 100 pg/kg or less, 95 pg/kg or less, 90 pg/kg or less, 85 pg/kg or less, 80 pg/kg or less, 75 pg/kg or less, 70 pg/kg or less, 65 pg/kg or less, 60 pg/kg or less, 55 pg/kg or less, 50 pg/kg or less, 45 pg/kg or less, 40 pg/kg or less, 35 pg/kg or less, 30 pg/kg or less, 25 pg/kg or less, 20 pg/kg or less, 15 pg/kg or less, 10 pg/kg or less, 5 pg/kg or less, 2.5 pg/kg or less, 2 pg/kg or less, 1.5 pg/kg or less, 1 pg/kg or less, 0.5 pg/kg or less, or 0.2 pg/kg or less of the anti-CD3 antibody such as teplizumab to treat or ameliorate one or more symptoms of T1D.
[0142] In some embodiments, a subject is intravenously administered one or more doses of a 100 pg/kg or less, preferably 95 pg/kg or less, 90 pg/kg or less, 85 pg/kg or less, 80 pg/kg or less, 75 pg/kg or less, 70 pg/kg or less, 65 pg/kg or less, 60 pg/kg or less, 55 pg/kg or less, 50 pg/kg or less, 45 pg/kg or less, 40 pg/kg or less, 35 pg/kg or less, 30 pg/kg or less, 25 pg/kg or less, 20 pg/kg or less, 15 pg/kg or less, 10 pg/kg or less, 5 pg/kg or less, 2.5 pg/kg or less, 2 pg/kg or less, 1.5 pg/kg or less, 1 pg/kg or less, 0.5 pg/kg or less, or 0.2 pg/kg or less of the anti-CD3 antibody such as teplizumab to treat or ameliorate one or more symptoms of T1D. In some embodiments, the intravenous dose of 100 pg/kg or less, 95 pg/kg or less, 90 pg/kg or less, 85 pg/kg or less, 80 pg/kg or less, 75 pg/kg or less, 70 pg/kg or less, 65 pg/kg or less, 60 pg/kg or less, 55 pg/kg or less, 50 pg/kg or less, 45 pg/kg or less, 40 pg/kg or less, 35 pg/kg or less, 30 pg/kg or less, 25 pg/kg or less, 20 pg/kg or less, 15 pg/kg or less, 10 pg/kg or less, 5 pg/kg or less, 2.5 pg/kg or less, 2 pg/kg or less, 1.5 pg/kg or less, 1 pg/kg or less, 0.5 pg/kg or less, or 0.2 pg/kg or less of the anti-CD3 antibody such as teplizumab, is administered over about 6 hours, about 4 hours, about 2 hours, about 1.5 hours, about 1 hour, about 50 minutes, about 40 minutes, about 30 minutes, about 20 minutes, about 10 minutes, about 5 minutes, about 2 minutes, about 1 minute, about 30 seconds or about 10 seconds to treat or ameliorate one or more symptoms of T1D.
[0143] In some embodiments, a subject is orally administered one or more doses of a 100 pg/kg or less, preferably 95 pg/kg or less, 90 pg/kg or less, 85 pg/kg or less, 80 pg/kg or less, 75 pg/kg or less, 70 pg/kg or less, 65 pg/kg or less, 60 pg/kg or less, 55 pg/kg or less, 50 pg/kg or less, 45 pg/kg or less, 40 pg/kg or less, 35 pg/kg or less, 30 pg/kg or less, 25 pg/kg or less, 20 pg/kg or less, 15 pg/kg or less, 10 pg/kg or less, 5 pg/kg or less, 2.5 pg/kg or less, 2 pg/kg or less, 1.5 pg/kg or less, 1 pg/kg or less, 0.5 pg/kg or less, or 0.2 pg/kg or less of the anti-CD3 antibody such as teplizumab to treat or ameliorate one or more symptoms of T1D. In some embodiments, the oral dose of 100 pg/kg or less, 95 pg/kg or less, 90 pg/kg or less, 85 pg/kg or less, 80 pg/kg or less, 75 pg/kg or less, 70 pg/kg or less, 65 pg/kg or less, 60
.g/kg or less, 55 gg/kg or less, 50 gg/kg or less, 45 gg/kg or less, 40 gg/kg or less, 35 gg/kg or less, 30 gg/kg or less, 25 gg/kg or less, 20 gg/kg or less, 15 gg/kg or less, 10 gg/kg or less, 5 gg/kg or less, 2.5 gg/kg or less, 2 gg/kg or less, 1.5 gg/kg or less, 1 gg/kg or less, 0.5 gg/kg or less, or 0.2 gg/kg or less of the anti-CD3 antibody such as teplizumab is administered over about 6 hours, about 4 hours, about 2 hours, about 1.5 hours, about 1 hour, about 50 minutes, about 40 minutes, about 30 minutes, about 20 minutes, about 10 minutes, about 5 minutes, about 2 minutes, about 1 minute, about 30 seconds or about 10 seconds to treat or ameliorate one or more symptoms of T1D.
[0144] In some embodiments in which escalating doses are administered for the first days of the dosing regimen, the dose on day 1 of the regimen is 100-250 pg/m2/day, preferably 106 pg/m2/day and escalates to the daily dose as recited immediately above by day 2, and 3. For example, on day 1, the subject is administered a dose of approximately 106 pg/m2/day, on day 2 approximately 425 pg/m2/day, and on subsequent days of the regimen (e.g., days 3- 12) 850 pg/m2/day. In some embodiments, on day 1, the subject is administered a dose of approximately 211 pg/m2/day, on day 2 approximately 423 pg/m2/day, on day 3 and subsequent days of the regimen (e.g., days 3-12) approximately 840 pg/m2/day.
[0145] In some embodiments, to reduce the possibility of cytokine release and other adverse effects, the first 1, 2, or 3 doses or all the doses in the regimen are administered more slowly by intravenous administration. For example, a dose of 106 pg/m2/day may be administered over about 5 minutes, about 15 minutes, about 30 minutes, about 45 minutes, about 1 hour, about 2 hours, about 4 hours, about 6 hours, about 8 hours, about 10 hours, about 12 hours, about 14 hours, about 16 hours, about 18 hours, about 20 hours, and about 22 hours. In some embodiments, the dose is administered by slow infusion over a period of, e.g., 20 to 24 hours. In some embodiments, the dose is infused in a pump, preferably increasing the concentration of antibody administered as the infusion progresses.
[0146] In some embodiments, the subject in need thereof is administered an effective amount of pain reliever (such as a nonsteroidal anti-inflammatory drug (NS AID), acetaminophen), an antihistamine, an antiemetic or a combination thereof at least on each of days 1-5 of the course of IV infusion (for example, the 12-days course of IV infusion). In some embodiments, the pain reliever (such as a nonsteroidal anti-inflammatory drug (NSAID), acetaminophen), an antihistamine, an antiemetic or a combination thereof can be administered on each of days 1-5, 1-6, 1-7, 1-8, 1-9, 1-10, 1-11, 1-12 or during the duration of the treatment with the anti-CD3 antibodies. In some embodiments, the pain reliever (such as a nonsteroidal anti-inflammatory drug (NSAID), acetaminophen), an antihistamine, an
antiemetic or a combination thereof is administered on each day about 30 min prior to the IV infusion. In some embodiments, the NSAID, acetaminophen, antihistamine, anti emetic or combination thereof is administered orally. In some embodiments, the NSAID, acetaminophen, antihistamine, antiemetic or combination thereof is administered intravenously. In some embodiments, antipyretics, antihistamines and/or antiemetics are administered to the subject in need thereof to mitigate cytokine release syndrome. In some embodiments, the liver enzymes are monitored and treatment with the anti-CD3 antibodies is discontinued or paused in subjects developing elevated ALT or AST more than 5 times the upper limit of normal.
[0147] In some embodiments, a set fraction of the doses for the 106 pg/m2/day to 850 pg/m2/day regimen described above is administered in escalating doses.
[0148] In some embodiments, the anti-CD3 antibody such as teplizumab is not administered by daily doses over a number of days, but is rather administered by infusion in an uninterrupted manner over 4 hours, 6 hours, 8 hours, 10 hours, 12 hours, 15 hours, 18 hours, 20 hours, 24 hours, 30 hours or 36 hours. The infusion may be constant or may start out at a lower dosage for, for example, the first 1, 2, 3, 5, 6, or 8 hours of the infusion and then increase to a higher dosage thereafter. Over the course of the infusion, the patient receives a dose equal to the amount administered in the 5 to 20-day regimens set forth above. For example, a dose of approximately 150 pg/m2, 200 pg/m2, 250 pg/m2, 500 pg/m2, 750 pg/m2, 1000 pg/m2, 1500 pg/m2, 2000 pg/m2, 3000 pg/m2, 4000 pg/m2, 5000 pg/m2, 6000 pg/m2, 7000 pg/m2, 8000 pg/m2, 9000 pg/m2, 9500 pg/m2, 10000 pg/m2, 10500 pg/m2, 11000 pg/m2, 11500 pg/m2, 12000 pg/m2, 12500 pg/m2, 13000 pg/m2, 13500 pg/m2 or 14000 pg/m2 can be administered. In particular, the speed and duration of the infusion is designed to minimize the level of free anti-CD3 antibody such as teplizumab in the subject after administration. In some embodiments, the level of free anti-CD3 antibody such as teplizumab should not exceed 200 ng/ml free antibody. In addition, the infusion is designed to achieve a combined T-cell receptor coating and modulation of at least 50%, 60%, 70%, 80%, 90%, 95% or of 100%.
[0149] In some embodiments, the anti-CD3 antibody such as teplizumab is administered chronically to treat, or slow the progression, or ameliorate one or more symptoms of T1D. For example, in some embodiments, a low dose of the anti-CD3 antibody such as teplizumab is administered once a month, twice a month, three times per month, once a week or even more frequently either as an alternative to the 6 to 14-day dosage regimen discussed above or after administration of such a regimen to enhance or maintain its effect. Such a low dose may
be anywhere from 1 pg/m2 to 100 pg/m2, such as approximately 5 pg/m2, 10 pg/m2, 15 pg/m2, 20 pg/m2, 25 pg/m2, 30 pg/m2, 35 pg/m2, 40 pg/m2, 45 pg/m2, or 50 pg/m2.
[0150] In some embodiments, the subject may be re-dosed at some time subsequent to administration of the two course anti-CD3 antibody such as teplizumab dosing regimen, for example, based upon one or more physiological or biomarker parameters or may be done as a matter of course. Such redosing may be administered and/or the need for such redosing evaluated 2 months, 4 months, 6 months, 8 months, 9 months, 1 year, 15 months, 18 months, 2 years, 30 months or 3 years after administration of a dosing regimen and may include administering a course of treatment every 6 months, 9 months, 1 year, 15 months, 18 months,
2 years, 30 months or 3 years indefinitely.
[0151] In some embodiments, before and/or after (e.g., at 1-6 month interval, or 2-5 month interval, or about 3 month interval) the administration of a 12-day course of teplizumab, the level (or relative amounts) of phenotypically exhausted T-cells, such as
TIGIT+KLRG1+CD8+ T-cells or PD-1+CD8+ T-cells with respect to all CD3+ T-cells is determined, for example by flow cytometry. In some embodiments, the level of the TIGIT+KLRG1+CD8+CD3+ T-cells can be monitored for example by flow cytometry. In some embodiments, an additional 12-day course of anti-CD3 antibody, such as teplizumab, is administered when the level of the TIGIT+KLRG1+CD8+CD3+ T-cells and/or PD- 1+CD8+CD3+ T-cells corresponds to (e.g., returns to) the baseline level. In some embodiments, the baseline level of TIGIT+KLRG1+CD8+CD3+ T-cells and/or PD- 1+CD8+CD3+ T-cells is less than 5% of all CD3+ T-cells. In some embodiments, the determining of TIGIT+, KLRG1+, or PD-1+ phenotypically-exhausted CD8+ T-cells is about
3 months (or about 1-6 months) after the administration of the second 12-day course. In some embodiments, if the subject has more than about 10% TIGIT+KLRG1+CD8+ T-cells and/or PD-1+CD8+ T-cells in all CD3+ T-cells, the monitoring can be annual. In some embodiments, if the subject has less than about 10% TIGIT+KLRG1+CD8+ T-cells and/or PD-1+CD8+ T-cells in all CD3+ T-cells, the monitoring can be every about 3-6 months. [0152] In some embodiments, the re-dosing comprises administering additional (e.g., second, third, or beyond) 12-day course(s) of teplizumab each at a total dose of more than about 9000 pg/m2 as described herein. In some embodiments, the additional 12-day course of teplizumab comprises a first dose of 106 pg/m2 teplizumab on day 1, a second dose of 425 pg/m2 teplizumab on day 2, and one dose of 850 pg/m2 on each of days 3-12, and wherein the total dose is approximately 9031 pg/m2. In other embodiments the additional 12-day course of teplizumab comprises a first dose of 211 pg/m2 teplizumab on day 1, a second dose
of 423 pg/m2 teplizumab on day 2, and one dose of 840 pg/m2 on each of days 3-12, and wherein the total dose is approximately 9034 pg/m2.
[0153] In some embodiments, the additional (e.g., second, third, or beyond) 12-day course of anti-CD3 antibody, such as teplizumab, can be administered about 12 months to about 24 months after the administering of the prior 12-day course, for example 12, 13, 14, 15, 16, 17, 19, 20, 21, 22, 23 or 24 months.
[0154] In some embodiments, the anti-CD3 antibody such as teplizumab is administered to achieve, or maintain a level of glycosylated hemoglobin (HbAl or HbAlc) less than 8%, less than 7.5%, less than 7%, less than 6.5%, less than 6%, less than 5.5% or 5% or less. At the initiation of treatment, patients have a HbAl or HbAlc level of less than 8%, less than 7.5%, less than 7%, less than 6.5%, less than 6%, or, more preferably, from 4%-6% (preferably measured in the absence of other treatment for diabetes, such as administration of exogenous insulin). Such patients preferably have retained at least 95%, 90%, 80%, 70%, 60%, 50%, 40% 30% or 20% of beta-cell function prior to initiation of treatment. In some embodiments, the administration of the anti-CD3 antibodies prevents damage, thereby slowing progression of the disease and reducing the need for insulin administration. In some embodiments, the methods of treatment provided herein result in a level of HbAl or HbAlc is 7% or less, 6.5% or less, 6% or less, 5.5% or less, or 5% or less 6 months, 9 months, 12 months, 15 months, 18 months, or 24 months after the previous treatment. In some embodiments, the administration of the anti-CD3 antibodies according to the methods provided herein decreases the average level of HbAl or HbAlc in the patient by about 5%, about 10%, about 15%, about 20%, about 25%, about 30%, about 35%, about 40%, about 45%, about 50%, about 55%, about 60%, about 65% or about 70% as compared to pre-treatment levels at 6 months, 9 months, 12 months, 15 months, 18 months, or 24 months after the previous treatment. In some embodiments, the administration of the anti-CD3 antibodies according to the methods provided herein results in an average level of HbAl or HbAlc in the patient that only increases by about 0.5%, about 1%, about 2.5%, about 5%, about 10%, about 15%, about 20%, about 25%, about 30%, about 35%, about 40%, about 45%, or about 50% as compared to pre-treatment levels at 6 months, 9 months, 12 months, 15 months, 18 months, or 24 months after the previous treatment.
[0155] In some embodiments, administration of the anti-CD3 antibodies, such as teplizumab according to the methods provided herein, slows the loss of P-cells and/or preserves P cell function (as evidenced by e.g., higher C-peptide levels, less episodes of
hypo- or hyper- glycemia, increased time in range (of glycemia), lower insulin use, or other assessment method known in the art) over 12 months, 13 months, 14 months, 15 months, 16 months, 17 months, 18 months, 19 months, 20 months, 21 months, 22 months, 2 months, 24 month or more in children and adolescents 8-17 years old who have been diagnosed with T1D in the previous 6 weeks. In some embodiments, administration of the anti-CD3 antibodies, such as teplizumab according to the methods provided herein, slows the loss of P cells and/or preserves P cell function over 18 months (78 weeks) in children and adolescents 8-17 years old who have been diagnosed with T1D within 6 weeks prior to the administration of the first dose of the anti-CD3 antibodies. In some embodiments, administration of the anti-CD3 antibodies, such as teplizumab according to the methods provided herein, slows the loss of P cells and/or preserves P cell function as measured by C-peptide levels over 18 months (78 weeks) in children and adolescents 8-17 years old who have been diagnosed with T1D within 6 weeks prior to the administration of the first dose of the anti-CD3 antibodies. [0156] In some embodiments, administration of the anti-CD3 antibodies, such as teplizumab, in 8-17 years old subjects who have been diagnosed with T1D within 6 weeks prior to the administration of the first dose of the anti-CD3 antibodies, lowers insulin use by 0.1 U/kg/day or more while achieving similar target glycemic control (HbAlc) in the subjects treated with the anti-CD3 antibodies as compared to subjects receiving placebo. In some embodiments, administration of the anti-CD3 antibodies, such as teplizumab, in 8-17 years old subjects who have been diagnosed with T1D within 6 weeks prior to the administration of the first dose of the anti-CD3 antibodies, improved TIR with lower insulin use by 0.1 U/kg/day or more than subjects receiving placebo. In some embodiments, the administration of the anti-CD3 antibodies, such as teplizumab, results in a reduction of HBAlc baseline by from 0.1 to 1 point, or more than 1 point, compared to subjects receiving placebo. In some embodiments, the administration of the anti-CD3 antibodies, such as teplizumab, results in a reduction of insulin use by from 10% to 15%, 15% to 20%, 20% to 25%, 25% to 30%, 10% to 20%, 20% to 30%, 10% to 30%, or more than 30%, compared to subjects receiving placebo.
[0157] In some embodiments, the administration of the anti-CD3 antibodies, such as teplizumab, to the subject in need thereof results in a reduction of insulin dosage by at least 0.1 U/kg/day or in maintenance of insulin dosage.
[0158] In some embodiments, administration of the anti-CD3 antibodies, such as teplizumab, in 8-17 years old subjects who have been diagnosed with T1D within 6 weeks prior to the administration of the first dose of the anti-CD3 antibodies results in from 40% to
80%, or more than 80%, higher mean C-peptide value compared with a subject receiving placebo at 78 weeks. In some embodiments, the administration of the anti-CD3 antibodies, such as teplizumab, results in from 40% to 45%, 45% to 50%, 50% to 55%, 55% to 60%, 60% to 65%, 65% to 70%, 70% to 75%, 75% to 80%, or more than 80%, higher mean C- peptide value compared with a subject receiving placebo at 78 weeks. For example, the administration of the anti-CD3 antibodies, such as teplizumab, results in 40%, 45%, 50%. 55%. 60%, 65%, 70%, 75%, 80%, 85%, 90%, or more than 90%, higher mean C-peptide value compared with a subject receiving placebo at 78 weeks.
[0159] In some embodiments, administration of the anti-CD3 antibodies, such as teplizumab, in 8-17 years old subjects who have been diagnosed with T1D within 6 weeks prior to the administration of the first dose of the anti-CD3 antibodies results in an increase of from 3% to 10%, or more than 10%, time in range for glycemia as assessed using a glucose monitoring system. For example, the administration of the first dose of the anti-CD3 antibodies results in an increase of 3% to 6%, 6% to 10%, 3% to 4%, 4% to 5%, 5% to 6%, 6% to 7%, 7% to 8%, 8% to 9%, 9% to 10%, or more than 10% time in range for glycemia as assessed using a glucose monitoring system.
[0160] Some embodiments relate to Teplizumab for use in a method of treating clinical type 1 diabetes (T1D), comprising administering to a subject in need thereof a 12-day course of the teplizumab at a total dose of more than about 9000 pg/m2.
[0161] In some embodiments, the total dose is between about 9000 and about 9500 pg/m2. In some embodiments, the total dose is between about 9000 and about 14000 pg/m2.
[0162] In some embodiments, the 12-day course comprises a first dose of 106 pg/m2 teplizumab on day 1, a second dose of 425 pg/m2 teplizumab on day 2, and one dose of 850 pg/m2 on each of days 3-12, and wherein the total dose is approximately 9031 pg/m2.
[0163] In some embodiments, the 12-day course comprises a first dose of 211 pg/m2 teplizumab on day 1, a second dose of 423 pg/m2 teplizumab on day 2, and one dose of 840 pg/m2 on each of days 3-12, and wherein the total dose is approximately 9034 pg/m2.
[0164] In some embodiments, the method can include administering a first and a second 12-day courses of teplizumab. In some embodiments, the first and the second 12-day courses are administered at about 1-6 months, about 2-5 months or about 3 months interval.
[0165] In some embodiments, the method can include administering to the subject in need thereof a third or more 12-day course of teplizumab, each course at a total dose of more than about 9000 pg/m2.
[0166] In some embodiments, the third or more 12-day course of teplizumab comprises a
first dose of 106 pg/m2 teplizumab on day 1, a second dose of 425 pg/m2 teplizumab on day 2, and one dose of 850 pg/m2 on each of days 3-12, and wherein the total dose of each course is approximately 9031 pg/m2.
[0167] In some embodiments, the third or more 12-day course of teplizumab comprises a first dose of 211 pg/m2 teplizumab on day 1, a second dose of 423 pg/m2 teplizumab on day 2, and one dose of 840 pg/m2 on each of days 3-12, and wherein the total dose of each course is approximately 9034 pg/m2.
[0168] In some embodiments, the third or more 12-day course of teplizumab is administered at about a 12 month to about a 24-month interval.
[0169] In some embodiments, the method can further include determining, after the administration of each 12-day course, a baseline of a level of TIGIT+KLRG1+CD8+ T-cells or PD-1+CD8+ T-cells with respect to all CD3+ T-cells, monitoring the level of the TIGIT+KLRG1+CD8+CD3+ T-cells, and administering an additional 12-day course of teplizumab when the level of the TIGIT+KLRG1+CD8+CD3+ T-cells or PD-1+CD8+CD3+ T-cells returns to the baseline level. In some embodiments, the determining of TIGIT+KLRG1+CD8+CD3+ T-cells or PD-1+CD8+CD3+ T-cells is by flow cytometry. In some embodiments, the monitoring of TIGIT+KLRG1+CD8+CD3+ T-cells or PD- 1+CD8+CD3+ T-cells is by flow cytometry. In some embodiments, the determining of TIGIT+KLRG1+CD8+CD3+ T-cells or PD-1+CD8+CD3+ T-cells is about 1-6 months, about 2-5 months, or about 3 months after the administration of each 12-day course. In some embodiments, if the subject has more than about 10% TIGIT+KLRG1+CD8+ T-cells or PD- 1+CD8+ T-cells in all CD3+ T-cells, subsequent monitoring is annual. In some embodiments, if the subject has less than about 10% TIGIT+KLRG1+CD8+ T-cells, subsequent monitoring is about every 3-6 months. In some embodiments, if the subject has less than about 10% PD-1+CD8+ T-cells in all CD3+ T-cells, subsequent monitoring is about every 3-6 months.
[0170] In some embodiments, the subject in need thereof has been diagnosed with T1D within 6 weeks prior to the administrating step.
[0171] In some embodiments, the administrating step results in reduction by at least 10% of insulin use, HbAlc levels, hypoglycemic episodes, or combinations thereof as compared to pre-treatment levels.
[0172] In some embodiments, each dose is administered parenterally.
[0173] In some embodiments, each dose is administered by intravenous infusion. [0174] In some embodiments, the subject is about 8 to 17 years old.
[0175] In some embodiments, the subject has a peak C-peptide level of >0.2 pmol/mL during an MMTT. In some embodiments, the subject in need thereof has a peak C-peptide level of from 0.2 pmol/mL to 0.7 pmol/mL during an MMTT.
[0176] In some embodiments, the subject receiving teplizumab has a higher mean C- peptide value compared with a control receiving placebo.
[0177] In some embodiments, the method further includes assessing the AUC of C-peptide following an MMTT at 78 weeks. In some embodiments, the administration of teplizumab results in the maintenance of higher C-peptide levels than when the patient is administered a placebo.
[0178] In some embodiments, the subject has at least 20% of [3-cell function prior to the administration of the first dose of the first 12-day course of teplizumab.
[0179] In some embodiments, the reduction of insulin use, HbAlc levels, hypoglycemic episodes, or combinations thereof, is over a period of 12 months or more.
[0180] Some aspects relate to a method of treating clinical T1D, comprising administering to a subject in need thereof a 12-day course of teplizumab at a total dose of more than about 9000 pg/m2. Some aspects relate to teplizumab for use in a method of treating clinical T1D, comprising administering to a subject in need thereof a 12-day course of the teplizumab at a total dose of more than about 9000 pg/m2.
[0181] In some embodiments, a method of treating clinical T1D is provided comprising administering to a subject in need thereof a 12-day course of teplizumab at a total dose of from about 9000 to about 9500 pg/m2. In some embodiments, a method of treating clinical T1D is provided comprising administering to a subject in need thereof a 12-day course of teplizumab at a total dose of from about 9000 to about 14000 pg/m2.
[0182] In some embodiments, the anti-CD3 antibody such as teplizumab, otelixizumab, or foralumab, is administered by infusion in medical facilities or in outpatient infusion centers. Yet in other embodiments, the anti-CD3 antibody such as teplizumab, otelixizumab, or foralumab, is administered by infusion in a home setting. Home infusion therapy involves the administration of the therapeutic agents, for example, anti-CD3 antibody using intravenous, or subcutaneous routes, in the patient's home rather than in a physician's office or hospital. Infusion therapies in the home can be administered by a home health care worker or by a patient himself. In some embodiments, a health care worker having some training in the operation of infusion equipment and the administration of anti-CD3 antibody can provide the patient with self-administration training and all the necessary equipment and/or supplies needed for the administration.
V. Exemplary Embodiments
[0183] Non-limiting, exemplary embodiments of the present disclosure are set forth below.
1. A method of treating type 1 diabetes (T1D), comprising: administering to a subject in need thereof a first 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 9500 pg/m2, wherein the subject in need thereof has a peak C-peptide level of at least 0.2 pmol/mL during a mixed meal tolerance test (MMTT) prior to administration of the first 12-day course of teplizumab; and administering to the subject in need thereof a second 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 9500 pg/m2, wherein the first and the second 12-day courses of teplizumab are administered at about a 6 month or to about a 12 month interval.
2. A method of treating type 1 diabetes (T1D), comprising: administering to a subject in need thereof a first 12-day course of teplizumab at a total dose of from about 9000 to about 14000 pg/m2, wherein the subject in need thereof has a peak C-peptide level of at least 0.2 pmol/mL during a mixed meal tolerance test (MMTT) prior to administration of the 12-day course of teplizumab; and administering to the subject in need thereof a second 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 14000 pg/m2, wherein the first and the second 12-day courses of teplizumab are administered at about a 6 month or to about a 12 month interval.
3. The method of embodiment 1, wherein the 12-day course comprises a first dose of 106 pg/m2 teplizumab on day 1, a second dose of 425 pg/m2 teplizumab on day 2, and one dose of 850 pg/m2 on each of days 3-12, and wherein the total dose is approximately 9031 pg/m2.
4. The method of any one of embodiments 1-3, wherein the subject in need thereof is about 8 to 17 years old.
5. The method of any one of embodiments 1-4, wherein the subject in need thereof has been diagnosed with T1D within 6 weeks prior to the administration of the first 12-day course of teplizumab .
6. The method of embodiment 1 or 2, further comprising administering to the subject in need thereof a third or more 12-day course of teplizumab, each course at a total dose of more than about 9000 pg/m2.
7. The method of embodiment 6, wherein the third or more 12-day course of teplizumab comprises a first dose of 106 pg/m2 teplizumab on day 1, a second dose of 425 pg/m2
teplizumab on day 2, and one dose of 850 pg/m2 on each of days 3-12, and wherein the total dose of each course is approximately 9031 pg/m2.
8. The method of embodiment 7, wherein the third or more 12-day course of teplizumab is administered at about a 6 month to about a 24 month interval.
9. The method of any one of embodiments 1-8, comprising: determining, after administration of each 12-day course, a baseline of a level of TIGIT+KLRG1+CD8+ cells and/or a baseline of a level of PD-1+CD8+ cells with respect to all CD3+ T cells; monitoring the level of TIGIT+KLRG1+CD8+CD3+ T-cells and/or the level of PD- 1+ CD8+ CD3+ T cells; and administering an additional 12-day course of teplizumab when the level of the TIGIT+KLRG1+CD8+CD3+ T-cells and/or PD-1+CD8+ CD3+ T cells returns to the baseline level.
10. The method of embodiment 9 wherein the baseline level of the
TIGIT+KLRG1+CD8+ cells and/or the baseline level of the PD-1+CD8+ is less than about 5% of all CD3+ cells.
11. The method of embodiment 9, wherein the determining of TIGIT+KLRG1+CD8+CD3+ T-cells and/or the PD-1+CD8+ CD3+ T-cells is by flow cytometry.
12. The method of embodiment 9, wherein the monitoring of TIGIT+KLRG1+CD8+CD3+ T-cells and/or the PD-1+CD8+ CD3+ T-cells is by flow cytometry.
13. The method of embodiment 9, wherein the determining of TIGIT+KLRG1+CD8+CD3+ T-cells and/or the PD-1+CD8+ CD3+ T-cells is about 1-6 months, about 2-5 months, or about 3 months after the administration of each 12-day course.
14. The method of embodiment 9, wherein if the subject has more than about 10% TIGIT+KLRG1+CD8+ T-cells and/or more than about 10% PD-1+CD8+ in all CD3+ T cells, subsequent monitoring is annual.
15. The method of embodiment 9, wherein if the subject has less than about 10% TIGIT+KLRG1+CD8+ T-cells and/or less than about 10% PD-1+CD8+ in all CD3+ T cells, subsequent monitoring is every about 3-6 months.
16. The method of any one of embodiments 1-15, wherein each dose of teplizumab is administered parenterally.
17. The method of any one of embodiments 1-16, wherein each dose of teplizumab is
administered by intravenous infusion.
18. The method of any one of embodiments 1-17, wherein the subject in need thereof has a peak C-peptide level ranging from 0.2 pmol/mL and 0.7 pmol/mL during a mixed meal tolerance test (MMTT).
19. The method of any one of embodiments 1-18, wherein the subject in need thereof has a peak C-peptide level of at least 0.7 pmol/mL during a mixed meal tolerance test (MMTT).
20. The method of any one of embodiments 1-19, comprising assessing the area under the time-concentration curve (AUC) of C-peptide following a mixed meal tolerance test (MMTT), at 78 weeks.
21. The method of any one of embodiments 1-20, wherein the subject in need thereof administered with teplizumab has a higher mean C-peptide value compared with a control administered with placebo.
22. The method of any one of embodiments 1-21, wherein the administering of teplizumab to the subject in need thereof results in a 40% to 80%, or more than 80%, higher mean C-peptide value compared with a subject receiving placebo at 78 weeks.
23. The method of any one of embodiments 1-22, wherein the subject in need thereof administered with teplizumab maintains or reduces baseline HbAlc levels, and/or maintains or increases Time in Range with less insulin use, than subjects administered with placebo.
24. The method of any one of embodiments 1-23, wherein the subject in need thereof has at least 20% of beta-cell function prior the administration of the first dose.
25. The method of any one of embodiments 1-24, wherein the administering of teplizumab to the subject in need thereof results in a reduction of insulin dosage of 10% to 30%, or more than 30%, compared to subjects receiving placebo.
26. The method of any one of embodiments 1-25, wherein the administering of teplizumab to the subject in need thereof results in a reduction of insulin dosage by at least 0.1 U/kg/day or in maintenance of insulin dosage.
27. The method of any one of embodiments 1-26, wherein the administering of teplizumab to the subject in need thereof results in a reduction of HbAlc baseline by 0.1 to 1 point, or more than 1 point, compared to subjects receiving placebo.
28. The method of any one of embodiments 1-27, wherein the administering of teplizumab to the subject in need thereof results in an increase of 3 to 10%, or more than 10%, time in range (%) for glycemia as assessed using a glucose monitoring system.
29. A method of increasing mean C-peptide value in a subject having type 1 diabetes, the method comprising:
administering to the subject a first 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 9500 pg/m2, wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during a mixed meal tolerance test (MMTT) prior to administration of the first 12-day course of teplizumab; and administering to the subject a second 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 9500 pg/m2, wherein the first and the second 12-day courses of teplizumab are administered at about a 6 month or to about a 12 month interval, and wherein the administration of teplizumab results in a 40% to 80%, or a more than 80%, increase in mean C-peptide value compared to subjects receiving placebo.
30. A method of increasing mean C-peptide value in a subject having type 1 diabetes, the method comprising: administering to the subject a first 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 14000 pg/m2, wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during a mixed meal tolerance test (MMTT) prior to administration of the first 12-day course of teplizumab; and administering to the subject a second 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 14000 pg/m2, wherein the first and the second 12-day courses of teplizumab are administered at about a 6 month or to about a 12 month interval, and wherein the administration of teplizumab results in a 40% to 80%, or a more than 80%, increase in mean C-peptide value compared to subjects receiving placebo.
31. Teplizumab for use in a method of treating type 1 diabetes (T1D), comprising administering to a subject in need thereof a first 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 9500 pg/m2, wherein the subject in need thereof has a peak C-peptide level of at least 0.2 pmol/mL during a mixed meal tolerance test (MMTT) prior to administration of the first 12-day course of teplizumab; and administering to the subject in need thereof a second 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 9500 pg/m2, wherein the first and the second 12-day courses of teplizumab are administered at about a 6 month or to about a 12 month interval.
32. Teplizumab for use in a method of treating type 1 diabetes (T1D), comprising administering to a subject in need thereof a first 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 14000 pg/m2, wherein the subject in need thereof has a peak C-peptide level of at least 0.2 pmol/mL during a mixed meal tolerance test (MMTT)
prior to administration of the first 12-day course of teplizumab; and administering to the subject in need thereof a second 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 14000 pg/m2, wherein the first and the second 12-day courses of teplizumab are administered at about a 6 month or to about a 12 month interval.
33. Teplizumab for use in a method of increasing mean C-peptide value in a subject having type 1 diabetes, the method comprising: administering to the subject a first 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 9500 pg/m2, wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during a mixed meal tolerance test (MMTT) prior to administration of the first 12-day course of teplizumab; and administering to the subject a second 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 9500 pg/m2, wherein the first and the second 12-day courses of teplizumab are administered at about a 6 month or to about a 12 month interval, and wherein the administration of teplizumab results in a 40% to 80%, or a more than 80%, increase in mean C-peptide value compared to subjects receiving placebo.
34. Teplizumab for use in a method of increasing mean C-peptide value in a subject having type 1 diabetes, the method comprising: administering to the subject a first 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 14000 pg/m2, wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during a mixed meal tolerance test (MMTT) prior to administration of the first 12-day course of teplizumab; and administering to the subject a second 12-day course of teplizumab at a total dose of from about 9000 pg/m2 to about 14000 pg/m2, wherein the first and the second 12-day courses of teplizumab are administered at about a 6 month or to about a 12 month interval, and wherein the administration of teplizumab results in a 40% to 80%, or a more than 80%, increase in mean C-peptide value compared to subjects receiving placebo.
EXAMPLES
Example 1. Teplizumab Population Pharmacokinetic Simulations
Introduction
[0184] Teplizumab is a 150 kD monoclonal antibody that binds the CD3-s epitope of the T cell receptor (TCR) complex. The primary mechanism of action of the antibody involves
binding the CD3 antigen target on T cells. A population pharmacokinetic (PK) model that describes teplizumab concentrations following IV administration was developed.
Teplizumab PK was described by a Quasi-Steady-State (QSS) approximation of the Target- Mediated Drug Disposition (TMDD) model. The aim of this investigation was to use the model to simulate and compare concentration-time profiles of teplizumab following several dosing regimens of interest.
Objectives
[0185] The objectives of the analysis were:
• To apply the previously developed population PK model to simulate the following three dosing regimens:
- “Herold Dosing Regimen”: Day 1 : 51 pg/m2; Day 2: 103 pg/m2; Day 3: 207 pg/m2;
Day 4: 413 pg/m2; Days 5-14: 826 pg/m2;
- Regimen 1 : Day 1 : 211 pg/m2; Day 2: 423 pg/m2; Days 3-12: 840 pg/m2;
- Regimen 2: Day 1 : 106 pg/m2; Day 2: 425 pg/m2; Day 3-12: 850 pg/m2.
• To illustrate and compare concentration-time courses of teplizumab for the 3 dosing regimens listed above.
Subjects and Methods
Dosing Regimens
[0186] The Herold regimen is a 14-day course of teplizumab consisting of daily intravenous (IV) infusions (over at least 30 minutes) of 51 pg/m2, 103 pg/m2, 207 pg/m2, and 413 pg/m2 on Study Days 1-4, respectively, and an infusion of 826 pg/m2 on each of Study Days 5-14. The total dose for a 14-day course is approximately 9034 pg/m2. For subjects with body surface area (BSA) of 1.92 m2, this dosing schedule delivers approximately 17 mg of teplizumab. The maximum amount of drug delivered at steady-state was designed to provide coating of 50% to 80% of the available CD3 on T-cells, with no large excesses of free, unbound drug (projected to be < 200 ng/mL at steady-state).
[0187] The new Regimen l is a 12-day course of teplizumab consisting of daily IV infusion (over at least 30 minutes) of 211 pg/m2 and 423 pg/m2 on Study Days 1 and 2, respectively, and an infusion of 840 pg/m2 on each of Study Days 3-12. The total dose for a 12-day course is approximately 9034 pg/m2.
[0188] The new Regimen 2 is a 12-day course of teplizumab consisting of daily IV infusion (over at least 30 minutes) of 106 pg/m2 and 425 pg/m2 on Study Days 1 and 2, respectively, and an infusion of 850 pg/m2 on each of Study Days 3-12. The total dose for a 12-day course
is approximately 9031 pg/m2.
[0189] As evident, the same total dose is to be delivered by all three regimens, but in Regimens 1 and 2, delivery is over 12 days rather than 14 days of the original Herold regimen.
Simulations
[0190] The final model of the previous analysis was used for simulations. Concentrationtime courses were simulated for 40 days (Day 0 to Day 40), with 10 time points each day. The model included the study effect as patients from Protege Encore study were found to have higher clearance and central volume than patients from Protege study. Therefore, the simulations were conducted separately for these 2 studies. Covariate values of four typical patients were used for simulations, specifically:
• Adult patients with no detected anti-drug antibodies [ADA]: 18 years old, 60 kg males with BSA of 1.67 m2;
• Adult patients with high level of AD As: 18 years old, 60 kg males with BSA of 1.67 m2;
• Pediatric patients with no detected ADA: 13 years old, 45 kg males with BSA of 1.33 m2;
• Pediatric patients with high level of ADAs: 13 years old, 45 kg males with BSA of 1.33 m2.
[0191] For each of these patients, population predictions of concentrations over time were computed for each of 3 dosing regimens, and were then compared graphically. Then, the parameters of 1000 similar patients were simulated using the model-estimated interindividual variability, and individual concentration-time courses were computed using the model. Median and 90% prediction intervals of simulated concentrations at each time point were computed for each regimen, and were compared graphically. In addition, mean and standard deviations of the simulated values at 1 day after the last dose were computed and compared.
Software
[0192] The simulations were conducted using the NONMEM software, Version 7.4.1 (ICON Development Solutions). Computer resources included personal computers with Intel® processors, Windows 7 Professional or later operating system and Intel® Visual Fortran Professional Compiler (Version 11.0). Graphical and all other statistical analyses, including evaluation of NONMEM outputs were performed using R version 3.4.4 for Windows (R proj ect, r-proj ect.org) .
Results
[0193] The results of the simulations for typical adult patients with no detected AD As are shown in Figure 1. Concentrations in the Protege study were predicted to be higher than in the Encore study for all dosing regimens. The concentrations in Dosing Regimens 1 and 2 were nearly indistinguishable except for minor differences during the first two days of dosing. During the first 12 days of dosing, concentrations in the Herold dosing regimen were lower compared to Dosing regimens 1 and 2, but they were nearly identical following the last dose (on Day 14 for the Herold regimen, and Day 12 for Regimens 1 and 2). The simulations that included inter-individual variability (Figures 2-4, Table 1) confirmed these observations. Table 1 illustrates mean and standard deviation of predicted concentrations (ng/mL) over 1000 simulated subjects from Protege study.
Table 1. Teplizumab Concentration Predictions: Ctrough 1 day After the Last Dose

[0194] The results of the simulations for typical adult patients with high level of detected AD As are shown in Figures 5-8. As expected, overall teplizumab levels are much lower for subjects with very high immunogenic response, but the conclusions about differences between the three investigated dosing regimens still hold.
[0195] The results of the simulations for typical pediatric patients are shown in Figures 9- 16. They are very similar to those for adult patients, indicating the BSA-proportional dosing
provides similar exposure for pediatric and adult populations.
[0196] Figures 17-24 show concentration profiles comparing Herold Regimen and Regimen 2 for a longer time period and Tables 2 and 3 summarized Cmax and AUC from 0 to 42 days in the simulations. Figures show that by day 42 concentrations are very low, so values for AUC0-42 are essentially the same as for AUCinfinity. Table 2 illustrates mean and standard deviation of predicted maximum concentrations (ng/mL) over 1000 simulated subjects using Protege Model 205. Table 3 illustrates mean and standard deviation of predicted AUC from 0 to 42 days (ng/mL*day) over 1000 simulated subjects using Protege Model 205.
Table 2. Teplizumab Concentration Predictions: Cmax

Table 3. Teplizumab Concentration Predictions: AUC0-42


Conclusions
[0197] The simulations indicated that:
• Predicted concentrations of teplizumab are nearly identical for 2 suggested dosing regimens (Regimen 1 and Regimen 2) except for the first day of dosing;
• Predicted concentrations of teplizumab increase faster during dosing for Regimens 1 and 2 compared to Herold regimen, but they are nearly identical for all regimens at the last day of dosing;
• Predicted concentrations of teplizumab at 1 day after the last dose are nearly identical for all 3 regimens;
• BSA-proportional dosing provides homogeneous exposure levels for adult and pediatric subjects with different body size measures.
Example 2. A Phase 3, Randomized, Double-Blind, Multinational, Placebo-Controlled Study to Evaluate Efficacy and Safety of Teplizumab (PRV-031), a Humanized, FcR Non-Binding, anti-CD3 Monoclonal Antibody, in Children and Adolescents with Newly Diagnosed Type 1 Diabetes (T1D)
[0198] Teplizumab (also known as PRV-031, hOKT3yl [Ala-Ala], and MGA031) is a humanized 150-kilodalton monoclonal antibody (mAb) that binds to the CD3-s epitope of the T cell receptor. Teplizumab was developed when preclinical studies demonstrated that targeting T cells (the cells that are instrumental in initiating and coordinating the autoimmune process responsible for type 1 diabetes [T1D] mellitus) via this mechanism altered diabetes immunopathogenesis and prevented and reversed disease in relevant animal models. The goal of this study is to evaluate teplizumab in children and adolescents very recently diagnosed with T1D. Teplizumab holds the promise to be the first disease modifying therapy available to improve both the medical management and overall outlook in those who suffer the most devastating short- and long-term consequences of this disease.
Hypothesis
[0199] The hypothesis of this study is that teplizumab is safe, well-tolerated, and effective
in slowing the loss of P-cells and maintaining a clinically relevant level of P-cell function in children and adolescents newly diagnosed with T1D while improving key aspects of T1D clinical management over an 18-month period.
Objectives
[0200] The primary objective was:
• To determine whether two courses of teplizumab slow the loss of P cells and preserve P cell function over 18 months (78 weeks) in children and adolescents 8-17 years old who have been diagnosed with T1D in the previous 6 weeks.
[0201] The secondary objectives were:
• To evaluate participant improvements in key clinical parameters of diabetes management, including insulin use, glycemic control (including hemoglobin Ale [HbAlc] and time in glycemic target range [TIR]), and clinically important hypoglycemic episodes
• To determine the safety and tolerability of two courses of teplizumab, administered intravenously (IV)
• To evaluate the pharmacokinetics (PK) and immunogenicity of two courses of IV teplizumab
[0202] The exploratory objectives were:
• To assess P-cell function and TID-focused clinical parameters
• To evaluate immunologic, endocrinologic, molecular, and genetic markers
Endpoints
[0203] The primary endpoint was:
• The area under the time-concentration curve (AUC) of C-peptide after a 4-hour (4h) mixed meal tolerance test (MMTT), a measure of endogenous insulin production and P cell function, at Week 78.
[0204] The secondary endpoints were as follows:
A. Key Clinical Endpoints:
• Exogenous insulin use: defined as a daily average in units per kilogram per day (U/kg/day), at Week 78
• HbAlc levels: expressed in % and mmol/mol, at Week 78
• TIR: expressed as a daily average of the percentage of time in a 24-hour day a participant’s blood glucose (BG) is >70 but <180 mg/dL (>3.9 to <10.0 mmol/L), assessed using continuous glucose monitoring (CGM), at Week 78
• Clinically important hypoglycemic episodes: defined as the total number of episodes of a BG reading of <54 mg/dL (3.0 mmol/L) and/or episodes of severe cognitive impairment requiring external assistance for recovery, from randomization through Week 78
B. Safety Endpoints:
• Incidence of treatment-emergent adverse events (TEAEs), adverse events of special interest (AESIs), and serious adverse events (SAEs)
• Incidence of treatment-emergent infections of special interest, including but not limited to tuberculosis, an infection requiring IV antimicrobial treatment or hospitalization, Epstein-Barr virus (EBV) and cytomegalovirus (CMV) infection, or significant viremia (i.e., DNA-based polymerase chain reaction viral load >10,000 copies per mL or 106 cells), and herpes zoster
• Incidence and severity of immediate or delayed study drug infusion-related reactions, such as hypersensitivity reactions, pain requiring interruption or discontinuation of infusions, cytokine release syndrome, and serum sickness
C. PK and Immunogenicity Endpoints:
• Teplizumab serum concentrations
• Incidence and titers of anti-teplizumab antibodies after treatment courses
3. The exploratory endpoints were as follows:
A. Assessments of P cell function and health throughout the study:
• 4h MMTT C-peptide AUC
• Participants with the recognized clinically significant stimulated peak C-peptide of >0.2 pmol/mL during 4h and 2-hour (2h) MMTTs
• Proinsulin-to-C-peptide ratios, a measure of P cell endoplasmic reticulum stress and dysfunction
B. TID-focused Clinical Endpoints during the study unless otherwise noted:
• Exogenous insulin use (in U/kg/day)
• Hb Ale levels
• Participants with poor glycemic control, defined as HbAlc of >9%
• The number of participants who do not require exogenous insulin because they are able to achieve local, regional, or national age-based glycemic management goals for HbAlc and/or routine blood glucose levels
• Evaluations of glycemic control based on BG values obtained from intermittent (i.e., spot-check, fingerstick) glucometer readings
• Evaluations of glycemic control based on BG values obtained from CGM readings, including but not limited to TIR; time in hyperglycemia and hypoglycemia ranges; daily, daytime, and nighttime average BG levels and estimated Elb Ale; and glycemic variability
• Clinically important hypoglycemic episodes from randomization through Week 39 and from Week 39 through Week 78
• Incidence of “typical” hypoglycemia, defined as BG levels >54 mg/dL (3.0 mmol/L) but <70mg/dL (3.9 mmol/L) and/or non-severe clinical episodes
• Incidence of diabetic ketoacidosis (DKA) requiring medical attention, defined as a hyperglycemic episode with serum or urine ketones elevated beyond upper limit of normal (ULN) along with serum bicarbonate <15 mmol/L or blood pH <7.3, or both, and resulting in outpatient, emergency room visit or hospitalization
• Patient-reported outcomes measured by instruments, such as Quality of Life Inventory™ (PedsQL) Diabetes Module, the Hypoglycemia Fear Scale (HFS), and the Diabetes Treatment Satisfaction Questionnaire (DTSQ)
• Impact on family life, measured by the parent-reported PedsQL Family Impact questionnaire
C. Composite Clinical Endpoints:
• Participants with both HbAlc in the American Diabetic Association (ADA) target range (i.e., <7.5%) and exogenous insulin dose in specific ranges (<0.25, 0.25 to <0.50, 0.50 to <0.75, 0.75 to <1.0, 1.0 to <1.25, and >1.25 U/k/d)
• Participants with both HbAlc of <6.5% and <7.0% and exogenous insulin dose of <0.5 U/kg/day or 0.25 U/kg/day
D. Immunologic and Endocrinologic Endpoints during the study:
• Phenotypic and functional characterizations of WBC populations, including T cells, B cells, and natural killer (NK) cells
• Serum proinflammatory and regulatory cytokine profiles and other immune mediators
• Number, type, and titer of T1D autoantibodies
• Antibody subclass levels
• Evidence of recent infection with CVB
• Levels of circulating hormones (e.g., glucagon, incretins, adiponectin) and other factors (e.g., lipokines, cholesterol, triglycerides) associated with the course of T1D pathophysiology
E. Molecular and Genetic Endpoints during the study:
• Circulating methylated- and unmethylated-insulin DNA levels as assessments of P cell stress and damage
• Gene expression and transcriptome analyses
• Association of human leukocyte antigen (HLA) type with clinical, metabolic and immune assessments
Overview of Study Design
[0205] This is a Phase 3, randomized, double-blind, placebo-controlled, multinational, multicenter study. Approximately 300 participants are enrolled and randomly assigned at a ratio of 2: 1 to either the teplizumab group (N=200) or the placebo group (N=100). A diagram of the study design is provided in Figure 25.
[0206] To minimize bias in treatment assignment, potential confounders, and enhance the validity of statistical analysis, participants were randomized at a 2: 1 ratio using randomly permuted blocks and stratification based on the following criteria:
• Peak C-peptide level at screening: within the range of 0.2 (inclusion criterion) to 0.7 pmol/mL (inclusive) versus >0.7 pmol/mL
• Age at randomization: within the range of 8 to 12 years (inclusive) versus >12 to 17 years
[0207] Modified dosing schedule for participants affected by COVID-19 pandemic restrictions:
[0208] To address COVID-19-imposed force majeure, those participants who were unable to receive the second treatment course approximately 6 months after randomization (scheduled for the Day 182 [Week 26] visit) due to COVID 19-related restrictions initiated the second course at the Day 364 (Week 52) visit instead, approximately 12 months after randomization.
[0209] The modified dosing schedule is illustrated in Figure 26. These participants underwent study procedures and assessments according to the Modified Dosing Schedule of Events.
[0210] References throughout the protocol to Day 364 (or Week 52) as the start of the second course of treatment is only applicable to this group of participants.
[0211] Teplizumab or matching placebo were administered via IV infusion in two courses, with the first course starting on Day 1 (Week 1) and the second course approximately 6 or 12 months later at Day 182 (Week 26). Each course of treatment included daily infusions for 12 days.
[0212] The total study duration for each participant was up to 84 weeks. This includes a screening period of up to 6 weeks and a post-randomization period of 78 weeks. The treatment period includes two 12-day treatment courses separated by 6 or 12 months and a post-treatment observation period of approximately 52 weeks or 26 weeks, respectively. The final visit took place at Week 78.
[0213] For participants under the modified dosing schedule, the treatment courses were separated by an interval of approximately 12 months, and the post-treatment observation period is approximately 26 weeks. The final visit also took place at Week 78.
Study Population
[0214] This study enrolled male and female participants 8 to 17 years of age with new- onset T1D who were able to be randomized and initiate study treatment within 6 weeks of their diagnosis. To be eligible for randomization, participants must be positive for at least one T ID-associated autoantibody and have a peak stimulated C-peptide of >0.2 pmol/mL at screening. They must also meet all of the specific inclusion criteria and none of the exclusion criteria.
Dosage and Administration
[0215] On the day of randomization (Day 1), each participant received the first dose of the study drug in the first 12-day treatment course, as shown in the table below. On approximately Day 182, each participant received the first dose of the second 12-day course. The study drugs (teplizumab or placebo) were administered via IV infusion at the study site or other qualified facility by study-approved personnel. The doses of study drug were calculated based on the participant’s body surface area (BSA) measured on the first day of each treatment course. No dose adjustment is permitted.
Modified Dosing Schedule
[0216] Participants who were unable to receive the second 12-day course due to COVID-19 pandemic restrictions were given the second course on approximately Day 364 (Week 52 visit). [0217] The study drug infusion on each treatment day should be administered within 20 to 28 hours after the previous dose. For example, if the dose on Day 1 is given at noon, the dose on Day 2 should be given within the interval of 8 am and 4 pm.
[0218] Dosing of the study drug (teplizumab or placebo) is based on the BSA using the height and weight obtained at this visit and the Mosteller formula (BSA=square root [height (cm) x weight (kg)/3600]).
Description of Interventions
Table 4

Key Evaluations
[0219] MMTT : In order to quantitate endogenous [3-cell function, participants underwent standardized provocative metabolic testing for C-peptide (a 1 : 1 by-product of insulin production). Participants consumed a fixed amount of a beverage with known amounts of carbohydrates, fats, and protein. Following consumption, BG, insulin, and C-peptide levels were measured over time. A 2h MMTT was conducted at screening, and 4h MMTTs was conducted at randomization and Weeks 26, 52, and 78 for key endpoint assessments.
[0220] HbAlc: This is the percent of red blood cells (measured as hemoglobin) that has become non-enzymatic glycated proportional to blood glucose levels. This indicates, on average, approximately a 3-month average of blood glucose values. It is a key clinical target in the management of T1D.
[0221] Insulin use: As an average over 7 days of data collected before each specified visit to quantify exogenously injected insulin.
[0222] Hypoglycemia: Clinically important and potentially life-threating hypoglycemia is the result of insulin therapy and more likely to occur in patients who are attempting to achieve glycemic control goals. This study asks participants to record information regarding BG levels of <70mg/dL (3.9 mmol/L) and/or events that are consistent with hypoglycemia. A particular focus is on clinically significant hypoglycemic events that are defined as a reliable glucose reading of <54 mg/dL (3.0 mmol/L) and/or severe cognitive impairment and/or physical status requiring external assistance for recovery.
[0223] Glucose Monitoring: Intermittent glucose monitoring (e.g., spot-check or fingerstick) performed by participants or care givers multiple times a day as a necessary part
of glycemic management to gauge insulin dosing and assist in diet and activity. All participants are to bring in their glucometers at all visits for review. In addition to data regarding glycemic control, at specified times during the study, participants report their daily before-meal and before-bedtime BG readings and have glucose levels assessed for 2-week intervals using CGM.
[0224] Quality of Life Questionnaires: Surveys is used to assess the general health and wellbeing of participants and the effects of teplizumab, such as the PedsQL Diabetes Module, HFS, DTSQ, and parent-reported PedsQL Family Impact Module.
[0225] Pharmacokinetic and Immunogenicity Evaluations: Teplizumab concentrations are analyzed in blood samples collected at specified time points throughout the study. Anti- teplizumab antibodies are determined, including those that are neutralizing antibodies (NAbs).
Safety Evaluation
[0226] Adverse events, serious adverse events, and adverse events of special interest. The relatedness and severity were evaluated. Severity will be graded according to the Common Terminology Criteria for Adverse Events (CTCAE), version 5.0.
[0227] Infusion-related reactions
[0228] Severe infections
[0229] Clinical laboratory tests
[0230] Vital signs and physical examination
Statistical Methods
General Considerations
[0231] All statistical inferences were based on 2-sided tests with an a-level of 0.05. All data were summarized by study drug group. Categorical variables were summarized by number and percent of individuals falling within each category. Continuous variables were summarized by mean, median, standard deviation, minimum and maximum. Unless otherwise noted, baseline values were defined as the most recent value collected prior to the first dose of study drug.
[0232] For efficacy endpoints, the analysis population was all randomized participants who receive any amount of study drug, referred to as the intent-to-treat (ITT) population. For this population, participants were analyzed in the treatment group corresponding to the treatment to which they were randomized, regardless of what treatment they actually received.
[0233] For the safety endpoints, the analysis population consisted of all randomized study participants receiving at least one dose of study drug, referred to as the safety population. For
this population, participants were analyzed in the treatment group corresponding to the treatment they actually received, regardless of the treatment to which they were randomized. [0234] For PK and immunogenicity, the analysis population was all participants in the safety population who have provided at least one evaluable sample.
Sample Size Determination
[0235] The study sample size was calculated based on the desired clinically relevant effects and the results from placebo-treated participants in previous teplizumab studies. Since C-peptide AUC is skewed right, the data was transformed using ln(AUC+l) for analysis. C- peptide data at 18 months from prior studies in children and adolescents who entered the studies with a stimulated C-peptide AUC of >0.2 pmol/mL were limited. Estimates ranged from approximately 0.22 nmol/L to 0.32 nmol/L with a standard deviation between 0.18 and 0.22. Using an estimate of 0.25 nmol/L, the transformation to geometric mean in the placebo group is exp(0.25)-l) = 0.28. This study was designed to show a difference of at least a 40% in C-peptide response between teplizumab and placebo. In geometric means this translates to a value of (1.4*0.28) = 0.392. Consequently, approximately 300 participants were planned for enrollment, assuming 2-sided a=0.05, 90% power, 2: 1 randomization, and a 10% dropout rate.
Efficacy Analyses
[0236] The primary endpoint is the difference between treatment groups in C-peptide ln(AUC+l) at Week 78 using the ITT population. C-peptide were measured in a 4h MMTT. Analysis of covariance (ANCOVA) was used to assess the treatment difference in C-peptide at Week 78. Missing data from patients who drop out before the end of the study was imputed based on those patients who similarly discontinue treatment in the same treatment arm but have measurement taken at scheduled visits. The model included treatment (teplizumab or placebo), age, and peak C-peptide at baseline as covariates.
[0237] Sensitivity analysis was performed using a tipping point approach. The same imputation and model were used as in the primary analysis, but a tipping point that changes the C-peptide conclusion at 18 months was sought. Repeated measures analysis may also be used to assess sensitivity.
[0238] The secondary clinical endpoints were assessed using the Hochberg step-up method for addressing multiplicity if primary endpoint reaches p < 0.05.
[0239] Like the primary endpoint, ANCOVA was used to assess treatment differences in insulin use, HbAlc, and the percentage of time participants’ BG levels are within the target range of >70 to <180 mg/dL (>3.9 to <10.0 mmol/L) at Week 78.
[0240] Average total exogenous insulin use (in U/kg/day) was self-recorded in an eDiary for 7 days prior to study visits and at randomization. Data from at least 5 of these 7 days was used for analysis. The model included age, baseline insulin use, peak C-peptide at baseline and treatment group as covariates.
[0241] The model used to assess HbAlc will include age, baseline HbAlc, treatment group, and baseline peak C-peptide as covariates.
[0242] Time in range for blood glucose was defined as the average percentage of time in range for 10 to 14 days post each study visit. The model included baseline time in range, treatment group, age and baseline peak C-peptide.
[0243] The rate (number of events/ exposure time) of clinically important hypoglycemic episodes at Week 78 was compared between groups. Data was collected from intermittent glucose monitoring, continuous glucose monitors, participant eDiary and CRFs. A clinically important episode is defined as a reliable BG value of <54 mg/dL (3.0 mmol/L) and/or a hypoglycemia event requiring external assistance, (such as seizure, syncope, severe confusion with or without a confirmatory low BG reading). The event rate of clinically important hypoglycemic episodes per study participant was assessed using a negative binomial model to allow for the potential for overdispersion in case episodes for hypoglycemia within participant groups were correlated. The model to assess clinically significant hypoglycemia included age, treatment group, and baseline peak C-peptide as covariates.
Other Analyses
[0244] Other analyses, including those for safety, PK, and immunogenicity, and exploratory endpoints were conducted using established and accepted statistical approaches. [0245] Additional safety and efficacy analyses were performed on the subgroup of participants who received the second treatment course starting at the Week 52 visit because of the CO VID-19 pandemic restrictions.
[0246] The study focused on individuals who have a significant amount of P cell functional capacity. It is recognized that P-cells continue to be lost following T1D diagnosis. To maximize the effect of [3-cell preservation in patients with a recoverable level endogenous insulin production, this study recruited participants within 6 weeks from T1D diagnosis and a peak C-peptide level of >0.2 pmol/mL during a mixed meal tolerance test (MMTT). The value of 0.2 pmol/mL was chosen as it is a key and accepted threshold of C-peptide correlated with clinically important lower rates of T ID-associated short- and long-term complications (Lachin 2014, Palmer 2001, Palmer 2009).
Randomization and stratification
[0247] To minimize bias in treatment assignment, potential confounders, and enhance the validity of statistical analysis, participants were randomized at a 2: 1 ratio using randomly permuted blocks and stratification based on the following criteria:
Peak C-peptide level at screening: within the range of 0.2 (inclusion criterion) to 0.7 pmol/mL (inclusive) versus >0.7 pmol/mL
[0248] Age at randomization: within the range of 8 to 12 years (inclusive) versus >12 to 17 years.
[0249] Study duration: The total study duration for each participant was up to 84 weeks. This includes a screening period of up to 6 weeks and a post-randomization period of 78 weeks. The post-randomization period includes two 12-day treatment courses separated by 6 months and a post-treatment observation period of approximately 52 weeks. The final visit took place at Week 78.
[0250] For participants under the modified dosing schedule, the treatment courses were separated by an interval of approximately 12 months, and the post-treatment observation period is approximately 26 weeks. The final visit also took place at Week 78.
[0251] The overall study length and timepoints for key assessments were chosen due to the natural course of remaining [3-cell loss following the diagnosis of T1D and study goals to demonstrate durability of effect and to confirm post-treatment safety profiles of teplizumab. At the time of diagnosis there can be substantial [3-cell reserves, often estimated at 10-20% but in some cases over 40% of normal [3-cell mass (Matveyenko 2008, Campbell-Thompson 2016). At T1D diagnosis, the majority of this reserve appears to be functionally impaired due to metabolic or immunologic (i.e., cytokine induced) stress. With exogenous insulin treatment and correction of pH, electrolyte and fluid disturbances (i.e., DKA) that are often present at diagnosis, some [3-cell function may return for days, weeks or many months. This observation is often referred to as the “Honeymoon period” where insulin requirements can be substantially reduced and at times independence from exogenous insulin can be achieved. These effects are transient and over time, usually within a year from diagnosis, inevitably full insulin replacement is required due to autoimmune elimination of these remaining P-cells. Due to the known individual variability in the natural history of [3-cell loss, the effect of disease modifying therapies intended to preserve [3-cell function is difficult to distinguish from the Honeymoon period effects during the first 12 months of T1D diagnosis.
[0252] The 18-month time point for the primary and key secondary clinical endpoints provide key data needed for the acceptance of teplizumab as a T1D disease modifying
therapy into regular medical practice and is consistent with existing guidelines for endpoints recommended by the EMA and FDA. Data from T1D natural history studies and interventional trials show that P-cell loss in those with T1D can be quite variable, especially within the weeks to months following diagnosis. As this study is enrolling participants in close proximity to T1D diagnosis (i.e. within 6 weeks) who are younger, there may be the added complexity of the consideration of the Honeymoon phenomenon (or spontaneous, transient partial remission) - that may last up to ~1 year in the study population (Abdul- Rasoul 2006). The 18-month timing of the primary and key secondary clinical endpoints allows for a substantial amount of the inherent, natural metabolic variability due to different trajectories of P-cell loss and/or transiently enhanced P-cell function due to the Honeymoon phenomenon to be minimized - so that the true effect on teplizumab on P-cell function and clinical parameters can be differentiated from chance.
[0253] Other key assessments were done at randomization, Week 26 (6 months) and Week 52 (12 months) to better understand the natural history of P-cell decline and the effect of teplizumab in this specific study population.
[0254] In addition, the primary and key clinical endpoints were assessed approximately 1 year after the last dose of study drug administration (except participants who received the modified dosing schedule). The length of effect is recognized as an important property of an intermittent disease modifying therapy for T1D. A 12-month off-therapy period whilst maintaining positive metabolic and clinical effects can, at this time, be considered a reasonable time frame to substantiate an assertion of a metabolically and clinically relevant durable benefit.
[0255] Throughout this study, participants were assessed regularly via in-person interviews, physical exams, self-reports, and laboratory examinations. Assessments occur daily during the two 12-day treatment courses and regularly between courses and the posttreatment follow up period. The on- and off-therapy observation times in this study are well within, if not significantly beyond, the periods traditionally used to assess for safety and side effects for immune therapies approved for other autoimmune conditions, including those for pediatric indications. In doses and regimens similar to that being used in this study, teplizumab has overall been well tolerated with minimal side effects and no signals of significant short- or long-term adverse effects. It is anticipated that with additional, confirmatory data from this study, the side-effect profile of teplizumab will continue to be considered acceptable for its integration into care plans for children and adolescents newly diagnosed with T1D.
[0256] A placebo control was used to establish the frequency and magnitude of changes in clinical, safety, metabolic and exploratory endpoints that may occur in the absence of active treatment. Randomization with stratification was used to minimize bias in the assignment of participants to treatment groups, to increase the likelihood that known and unknown participant attributes (e.g., demographic and baseline characteristics) are evenly balanced across treatment groups, and to enhance the validity of statistical comparisons across treatment groups. Blinded treatment was used to reduce potential bias during data collection and evaluation of all study endpoints.
Study population
Inclusion Criteria
[0257] Each potential participant must satisfy all of the following criteria to be enrolled in the study:
• Participant is male or female.
• Participant is 8 to 17 years of age, inclusive, at the time of randomization/initiation of study drug administration.
• Participant has received a diagnosis of T1D according to ADA criteria.
• Participant is able to be randomized and initiate study drug within 6 weeks (42 days) of the formal T1D diagnosis according to the ADA criteria.
• Participant has a peak stimulated C-peptide of >0.2 pmol/mL from a 2-hour mixed meal tolerance test (2h MMTT) at screening. (Note: This screening 2h MMTT must occur only after 6 days following diagnosis to allow for reduction of metabolic instability.)
• Participant has a positive result on testing for at least one of the following T ID- related autoantibodies before randomization: o Glutamic acid decarboxylase 65 (GAD65) autoantibodies o Islet antigen 2 (IA-2) autoantibodies o Zinc transporter 8 (ZnT8) autoantibodies o Islet cell cytoplasmic autoantibodies (ICA) or o Insulin autoantibodies (if testing obtained within the first 14 days of insulin treatment)
• Female participants of childbearing potential must have a negative result on highly sensitive serum (P human chorionic gonadotropin [P-HCG]) at screening.
• Participants who have reached puberty must agree to adhere to the following contraceptive requirements. (Note: In countries with legislation for the age of sexual activity, the participant must comply with the local age limit regarding the use of contraception.) o Females with childbearing potential (defined as premenopausal females who are capable of becoming pregnant, i.e., having reached menarche or having reached Tanner stage 3 breast development even without menarche) or who gain childbearing potential during the study must practice abstinence or use 2 forms of contraception (including oral, transdermal, injectable, or implanted contraceptives, intrauterine device, female condom, diaphragm with spermicide, cervical cap, use of a condom by the sexual partner, or a sterile sexual partner) continuously from 30 days before the first dose of study drug through the end of the study. o Males who have reached puberty (i.e., spermatogenesis) with partners of childbearing potential must use barrier contraception in addition to having their partners use another method of contraception from 1 week before each study agent dose through 120 days (a complete spermatogenesis cycle) after receiving the last dose in each treatment course.
• Prior to receiving study drug, participant must be up to date with and/or agree to receive routine age-appropriate immunizations and comply with the guidelines for immunosuppressed individuals and those with chronic disease (diabetes mellitus) according to current local, regional and/or country-specific guidelines.
• Participant agrees not to receive other forms of experimental treatment during the study, particularly agents that may be immune modulatory in nature and/or stimulate pancreatic P-cell regeneration or insulin secretion
• Participant and/or appropriate legal guardian must sign an informed consent form (ICF) and/or assent according to local, regional and/or country-specific guidance for study participation.
Exclusion Criteria
[0258] Any potential participant who meets any of the following criteria were excluded from participating in the study: Participant has known allergies, severe reaction, intolerance, hypersensitivity, or anaphylaxis to human, humanized, or murine monoclonal antibodies, teplizumab or any of its components or its excipients.
• Participant has been an active participant in a therapeutic drug, invasive medical device, or vaccine clinical trial within 12 weeks before the first dose of study drug or has received an investigational treatment with the potential for T1D disease modification.
• Participant has significant renal, cardiac, vascular, pulmonary, gastrointestinal, neurologic, hematologic, rheumatologic, oncologic, psychiatric disease, or immune deficiency.
• Participant has any autoimmune disease other than T1D (e.g., rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, multiple sclerosis, systemic lupus erythematous), with the exception of clinically stable thyroid or celiac disease.
• Participant has an active infection (including a positive SARS-CoV-2 test) and/or fever >38.5°C (101.3°F) within the 48 hours prior to randomization, is prone to infections, or has chronic, recurrent or opportunistic infectious disease, including but not limited to renal, respiratory or skin infections, Pneumocystis carinii. aspergillosis, latent or active granulomatous infection, histoplasmosis, or coccidioidomycosis.
• Participant has a history of or serologic evidence at screening of current or past infection with human immunodeficiency virus (HIV), hepatitis B virus (HBV), or hepatitis C virus (HCV).
• Participant has any of the following in regard to tuberculosis (TB): o A history of latent or active TB o Signs and/or symptoms of TB o Recent close contact with a person with known or suspected active TB, unless appropriate isoniazid prophylaxis for tuberculosis was given o A history of a chest X-ray consistent with active TB or old, inactive TB, o A history of a positive purified protein derivative skin test result (>10 mm induration); or o At screening is positive or repeatedly indeterminate with an approved interferongamma release assay (IGRA; e.g., QuantiFERON-TB test) o If required by local, regional or national regulations, a recent (within 3 months) chest X-ray or one conducted at screening read by a qualified radiologist consistent with current, active TB or old, inactive TB
• At screening, participant has a clinically active infection with EBV, including but not limited to infectious mononucleosis, or an EBV viral load >10,000 copies per mL or per 106 lymphocytes obtained at study screening (Rosenzweig 2010).
• At screening, participant has a clinically active infection with CMV or a CMV viral load 10,000 copies per mL or per 106 lymphocytes (Verkryse 2006).
• Participant has a diagnosis of significant liver disease or at screening alanine aminotransferase (ALT) and/or aspartate aminotransferase (AST) >2X or total bilirubin (TBili) of >1.5X of the age- and sex-specific upper limit of normal (ULN) according to the central laboratory. (Note: Participants with Gilbert’s syndrome may be allowed to enroll upon approval by the Medical Monitor.)
• An individual has any of the following hematologic parameters, confirmed by repeat tests, within 10 days before randomization/first dose of study drug: o Lymphocyte count: <1000/pL o Neutrophil count: <1000/pL o Platelet count: <100,000 platelets/pL o Hemoglobin: <10 g/dL
It should be noted that that specific hematologic, oncologic or other systemic conditions that might otherwise result in exclusion and/or is heretofore unrecognized should be considered in individuals who have one or more blood cell counts below or above the normal ranges
• Current or prior (within 30 days before screening) treatment that is known to cause a significant, ongoing change in the course of T1D or immunologic status, including high-dose inhaled, extensive topical, or systemic glucocorticoids. (Note: Short courses, i.e., approximately 2 weeks or less, of corticosteroids for transient conditions are allowed.)
• Current or prior (within 30 days before screening) use of drugs other than insulin to treat hyperglycemia (e.g., metformin, sulfonylureas, glinides, thiazolidinediones, exenatide, liraglutide, dipeptidyl peptidase-4 [DPP-IV] inhibitors, or amylin).
• Current or prior (within 30 days before screening) use of any medication known to significantly influence glucose tolerance (e.g., atypical antipsychotics, diphenylhydantoin, niacin).
• Current or planned highly restrictive dietary regimen(s) intended for T1D management, such as very-low or ultra-low carbohydrate diets.
• Recent or planned vaccinations as follows: o Live vaccines (e.g., varicella, measles, mumps, rubella, cold-attenuated intranasal influenza vaccine, and smallpox): Within the 8 weeks before randomization and initiation of study drug or planned/required administration through Week 52 or Week 78 (if Modified-Dosing Schedule is followed) of the study. o Non-infectious (e.g., recombinant, inactivated or otherwise “non-live”) vaccines: Within 2 weeks before through 6 weeks after each dosing course.
• A female who is pregnant, has a positive P-HCG blood test at screening or a positive urine P-HCG test prior to initiation of study drug, wishes to become pregnant, is planning on donating eggs (ova, oocytes), and/or is lactating with the intent to provide her own breast milk to a baby during the entire study.
• A male who is planning to father a child or donate sperm from 1 week before each study agent dose through 120 days (a complete spermatogenesis cycle) after receiving the last dose in each treatment course.
• An individual who has a history of alcohol, drug, or chemical abuse within 12 months prior to study screening.
• An individual who has a medical, psychological or social condition that, in the opinion of the Principal Investigator, would interfere with safe and proper completion of the trial.
• An individual who is an employee of the Investigator or study site, with direct involvement in the proposed study or other studies under the direction of that Investigator or study site, as well as family members of the employees or the Investigator.
Although there is not a specific exclusion based on weight, due to country, regional and/or local difference in blood draw limits for participants in a research study there may be weight-based criteria for participation depending on the location of the participant. Investigators are to confirm that potential participants meet any specific weight-based criteria due to any of these location-specific blood volume limitations.
Treatment allocation
[0259] Procedures for Randomization and Stratification
[0260] Participants were randomly assigned to 1 of 2 treatment groups in a 2: 1 ratio for participants in the teplizumab group and the placebo group, respectively. The randomization was balanced by using randomly permuted blocks and will be stratified by peak C-peptide
level at screening (0.2 to 0.7 pmol/mL inclusive, versus >0.7 pmol/mL) and age at randomization (8 to 12 years inclusive, versus >12 to 17 years).
[0261] This was a double-blind study. Blinding was maintained for all study participants throughout the study.
[0262] In some embodiments, T1D diagnosis is according to ADA criteria. In some embodiments, the patient diagnosed with T1D has a positive result on testing for at least one of the following TID-related autoantibodies: Glutamic acid decarboxylase 65 (GAD65) autoantibodies, Islet antigen 2 (IA-2) autoantibodies, Zinc transporter 8 (ZnT8) autoantibodies Islet cell cytoplasmic autoantibodies (ICA) or Insulin autoantibodies (if testing obtained within the first 14 days of insulin treatment).
[0263] Study drug preparation: At the beginning of each 12-day course of study drug administration, the participant’s current BSA is calculated using the Mosteller formula, BSA=square root [height (cm) x weight (kg) / 3600], using the height and weight obtained on that day.
[0264] Teplizumab and placebo are prepared according to the Pharmacy Manual provided to the site.
[0265] Two (2) mL of study drug should be drawn from the study drug vial and slowly diluted in 18 mL of 0.9% sodium chloride solution for injection by gentle mixing. The resulting 20 mL of 1 : 10 dilution is used as the initial study drug solution, which contains either placebo or teplizumab at a concentration of 100 pg/mL. This initial drug solution should then be added to 25 mL of a 0.9% sodium chloride solution. Finally, this resulting preparation should be gently mixed before administration to the participant.
[0266] Vascular access: This study requires two courses of intravenous infusions and blood draws over 12 days. It is recognized that intravenous access (for infusions and blood draws for laboratory sampling) in the pediatric population that is the focus of this study may pose a challenge. Children have smaller veins than adults, veins that may be more challenging to insert catheters and they may have a significant resistance to catheter placement and/or phlebotomy.
[0267] In recognition of the above, in addition to the use of “traditional” intravenous peripheral catheters, this study permits the use of temporary, intermediate term approaches for vascular access. Specifically, “midlines” or peripherally inserted central catheter (PICC) lines may be used for study drug infusions and blood draws (if appropriate according to the properties of the access line and local, regional or national guidance).
Premedication and study drug infusion:
[0268] Study participants received oral premedication consisting of an NSAID drug (tablet or liquid) and a locally approved antihistamine (tablet or liquid), according to local availability and practice standard, for at least the first 5 days of each study drug course and can receive one or both of these for the rest of the study drug doses if the Investigator judges it to be appropriate for the tolerability of infusions. The premedication should be given at least 30 minutes prior to initiation of the study drug infusion. If an NSAID is contraindicated, oral acetaminophen (tablet or liquid) may be given.
[0269] Study drug (teplizumab or placebo) was prepared and supplied by designated personnel or pharmacist equivalent according to the Pharmacy Manual.
[0270] The Pharmacy Manual should be followed to ensure that the IV drug delivery devices used are made of materials compatible with the study drug.
[0271] Teplizumab for intravenous administration may only be prepared with 0.9% sodium chloride. No solutions other than 0.9% sodium chloride may be running through the same intravenous line when teplizumab is being administered. If the same intravenous line must be used for infusion of other drugs or solutions, the line should be flushed with 0.9% sodium chloride solution before and after infusion of teplizumab.
[0272] Study drug was administered IV over a minimum of 30 minutes according to standard practices.
[0273] When the infusion solution has been completely administered, infuse an additional volume of saline equal to the volume contained in the infusion tubing at the same constant rate to ensure that all study drug has been cleared from the infusion tubing. The starting and ending times for the infusion must be recorded.
[0274] During the infusions and for an additional 60 minutes following the infusions, participants are to have vital signs (ie, BP, RR, and HR) assessed every 15 min and be monitored for signs or symptoms of infusion reactions. These include but are not limited to fever, chills, headache, nausea, vomiting, infusion-site pain, anaphylaxis, wheezing, dyspnea, urticaria, and hypotension. If there are signs or symptoms of infusion reaction in a participant during the 60-minute post-infusion observation period, the participant should be observed for an additional 60 minutes or until the reaction resolves, whichever is longer.
Diabetes Management and Insulin use:
[0275] All enrolled participants, with assistance of their health-care providers, should receive intensive diabetes management of their T1D using approved therapies according to the recommendations of American Diabetes Association (ADA) or local, regional, or national
recommendations to achieve glucose levels that appear to decrease some of the short-term and long-term sequelae of T1D. Currently the glycemic targets by the ADA are focused at management strategies to achieve a HbAlc level of <7.5% (58 mmol/mol) for individuals 17 years old and younger, and <7.0% (53 mmol/mol) 18 years and older while minimizing severe or frequent hypoglycemic events.
[0276] The glycemic goal should be attempted through proper glycemic monitoring, administration of exogenous insulin, and monitoring of activity level and diet. Exogenous insulin may include rapid, intermediate, and/or long-acting insulins, administered intermittently or via the use of a personal insulin pump. Blood glucose levels should be measured at least 4 times a day, including before meals and before bedtime.
[0277] Insulin use, including the type of products, dosages, and dosing schedules, is expected to change during the course of the study. As part of routine T1D clinical care, if the caring physician judges it to be clinically appropriate, a participant’s insulin dose may be increased, reduced, or even discontinued.
[0278] If participants are not meeting the glycemic goals, the study team should contact the participant’s primary clinical care team about possible adjustments in the insulin regimen, referral to a registered dietitian, or other approaches that may improve the glucose control.
Insulin Discontinuation
[0279] If a participant has achieved a HbAlc level of <6.5% with insulin use of <0.25 U/kg/day, insulin therapy can be discontinued. The participant’s blood glucose and HbAlc levels should continue to be monitored per protocol, and urine ketones should be monitored once a day. During routine blood glucose monitoring, if the participant’s blood glucose level exceeds 200 mg/dL (11.1 mmol/L) and/or urine ketone is moderate or greater, the participant should consult with their primary physician and/or the clinical site staff for further evaluation. If the fasting blood glucose exceeds 126 mg/dL (7 mmol/L) or HbAlc exceeds 6.5%, as documented by repeat testing, the resumption of insulin therapy should be considered.
Randomization, Treatments and Monitoring
Study Visit Week 1
[0280] Patient receives premedication of an NS AID (e.g., ibuprofen) (acetaminophen if NSAID is contraindicated) and an antihistamine (e.g., diphenhydramine) for at least the first 5 days of the treatment course, unless contraindicated by drug allergy or sensitivity. After at least 30 minutes following the premedication administration, the infusion of study drug can begin. Administration of the study drug should be performed according to the Pharmacy Manual. Study drug should be planned to be administered intravenously over 30 minutes according to
standard practices, but it may be slowed if there are signs or symptoms of intolerance. When the infusion solution has been completely administered, an additional volume of saline equal to the volume contained in the infusion tubing, at the same constant rate is to be infused to ensure that all study drug has been cleared from the infusion tubing. The starting and ending times for the infusion are to be recorded.
Day 2-12: Continued Treatment Course 1 Infusions
[0281] If there are no clinical or laboratory concerns, the patient can proceed with the next infusion as described above at least 30 minutes following administration of prophylactic NSAID (acetaminophen if NSAID is contraindicated) and antihistamine. Close monitoring is to occur during the infusions and for 60 minutes following the infusions for any signs or symptoms of intolerability or infusion reactions.
Dav 2-11
[0282] On Days 2-11, the patient is then able to leave the facility and return the following day for the next study drug infusion.
Days 12
[0283] The Day 12 following the completion of the final infusion for this course and at least a 30-minute observation a continuous Glucose Monitoring (CGM) sensor is applied and the participant is to be given instructions on CGM monitoring care and use.
Study Visits Weeks 4, 8, 12, and 20
[0284] The visit window for these study visits are ±4 days from the target visit day. During these visits, participants return to the site for their scheduled visit and have clinical and/or laboratory assessments conducted. Of note at Week 12, a CGM sensor is applied and the participant is to be given instructions on CGM monitoring care and use.
[0285] At the Week 20 visit, give participant instructions for Week 26 4h MMTT including overnight fast and pre-MMTT insulin dosing.
Study Visit Week 26: 4h MMTT and Treatment Course 2 (or Week 52, if Modified-dosing schedule followed)
[0286] The visit window for these study visits is ±3 days from the target visit day.
Davs 182-193
[0287] Day 182 clinical and laboratory assessments (including a 4h MMTT) and for initiation of the second course of study drug administration.
[0288] Of specific note, height and weight are to be obtained at this visit and used for the BSA based dosing calculation for course 2. Following the guidance as with study drug course 1, the patient is to be premedicated with an oral NSAID (acetaminophen if NSAID is
contraindicated) and anti-histamine at least 30 minutes before the first 5 study drug infusions is started (and on an as needed basis with subsequent infusions), administration of study drug should be performed according to the Pharmacy Manual, and an additional volume of saline equal to the volume contained in the infusion tubing is to be infused. During the infusions and for an additional 60 minutes following the infusions participants are to be monitored for signs or symptoms of infusion reactions.
[0289] On certain days, a blood draw is obtained for teplizumab serum levels within 30 minutes before study agent infusion.
Davs 183-192 (Dav 2-11 of Course 2 dosing)
[0290] On Days 183-192, the participant may leave the facility and return the following day for the next study drug infusion.
Dav 193 (Dav 12 of Course 2 dosing)
[0291] Following the completion of the final infusion for this course and at least a 30-minute observation, a CGM sensor is applied and the patient is to be given instructions on CGM monitoring care and use.
Study Visits Week 30, 34, 39, 52 and 65
[0292] The visit window for Weeks 30, 34, 39, and 52 study visits is ± 4 days from the target visit day. The visit window for Week 65 visit is ±7 days. At the Week 52 visit, a 4h MMTT is conducted.
[0293] At the end of the Week 39, 52, and 65 visits, a CGM sensor is to be applied and additional training and instruction updates on CGM care and use is given as needed.
Study Visits Week 39 and Week 65
[0294] Give patient instructions for Week 52 and Week 78, respectively, 4h MMTT including overnight fast and pre-MMTT insulin dosing. At the Week 65 visit, dispense to patient CGM equipment for home application to start around Week 76.
Study Visit Week 78
[0295] The visit window for this study visit is ± 7 days from the target visit day. During this visit the 4h MMTT is conducted.
Efficacy Evaluations
Mixed Meal Tolerance Tests
[0296] A 2h MMTT is performed at screening to determine study eligibility (based on peak C-peptide level). A 4h MMTTs was performed at randomization and at Weeks 26, 52, and 78 to obtain 4h C-peptide AUCs and other data. A 4h MMTT was used at and postrandomization as it has been shown to be more precise and reliable in assessing the MMTT-
induced C-peptide AUC than the 2h MMTT (Boyle 2015, Rigby 2013, Rigby 2016). Alternatively, the 2h-MMTT was used at screening as it is sufficient to capture the peak C- peptide level needed for study entry. Samples from these assessments were assessed for C-peptide, serum glucose, and insulin. Samples were stored for potential future evaluations including but not limited to proinsulin levels. The measurements of C-peptide and glucose in serum samples were done. MMTTs were to take place in the morning between approximately 7:00 a.m. and 10:00 a.m. after an overnight fast with strict guidance on insulin use. The 2- hour MMTT takes approximately 130 minutes to perform, and the 4-hour MMTT takes approximately 250 minutes.
Hemoglobin Ale
[0297] HbAlc was assessed as a blood test at select study visits.
Insulin Use
[0298] Patient’s daily insulin use was documented by the participant in an eDiary at select times for 7 days prior to randomization and at about Weeks 12, 26, 39, 52, 65 and 78 visits. The patient records all short-, intermediate- and long-acting insulin administered as intermittent injections or use with an “insulin pump” during this time. Insulin use data were not recorded on the day before or the day of the study visit. If a patient forgets to record insulin use on one or more days before a visit, they should continue to record insulin use for up to 72 hours post-dose to obtain up to 7 days of data. Every effort should be made to collect a total of 7 days of insulin use data for all the aforementioned visits except Week 78 (final visit), as patients return the eDiary at the final visit.
Episodes of Hypoglycemia
[0299] Clinically important and other non-severe and non-serious hypoglycemic episodes were recorded throughout the study by participants and through evaluation of glucometer readings.
Glucose Monitoring
(1) Intermittent Glucose Monitoring (Fingerstick)
[0300] Blood glucose levels outside of MMTT and CGM were recorded and analyzed as an endpoint at various times. As part of routine care, BG levels were usually measured by a fingerstick glucometer at least 4 times a day, including before each meal and at bedtime. At screening, participants are offered a study-supplied glucometer and glucometer strips, but participants were permitted to use their own glucometers if they choose. Each participant was instructed to bring their glucometer (or glucometers if they use more than one, e.g., at home and in school) to each visit for review. In addition, approximately 7 times throughout the
study, participants record their BG levels before breakfast, lunch, and dinner and at bedtime for 7 consecutive days in their study eDiary prior to the randomization visit and the Weeks 12, 26, 39, 52, 65, and 78 visits. Like the recording of the insulin use data, BG data on the day before and the day of the study visit were not recorded. If a participant forgets to record fingerstick glucose measurements before a visit, they should do so for 72 hours immediately after the visit. Every effort should be made to collect a total of 7 days of BG data for all the aforementioned visits except Week 78 (final visit), as participants return the eDiary at the final visit.
(2) Continuous Glucose Monitoring
[0301] “ Continuous” glucose monitors record interstitial glucose levels (which closely approximate blood glucose values) at regular intervals, e.g. every 5-15 minutes depending on device. Increasingly clinical studies are supporting that such measurements and their assessments provides valuable and unique insights to glycemic control in diabetes. In this study, CGM assessments were conducted to provide key secondary clinical and exploratory endpoint data to address if and how teplizumab affects glycemic control, such as glucose excursions, time in select glucose ranges, and average daily glucose values (Steck 2014, Helminen 2016, Danne 2017). A recent international consensus statement on CGM monitoring supported the use of percentages of time in ranges (target, hypoglycemia, and hyperglycemia) and measurement of glycemic variability as key diabetes control metrics in clinical trials (Danne 2017).
[0302] CGM are used to assess glycemic control approximately 7 times throughout the study: after the completion of treatment courses at randomization and Week 26; after the visit at Weeks 12, 39, 52, and 65; and before the visit at Week 78. CGM sensors are placed by qualified study staff, and education and training on CGM use and care are given. Sensors remain in place for up to 2 weeks. If during that 2-week period a sensor comes off, it can be replaced by the participant, a knowledgeable family member/guardian, or a qualified medical professional.
[0303] To reduce any confounding factors of glucose measurements during study drug infusions, CGM sensors were placed on participants after the study drug administration has completed for Course 1 and Course 2 and other clinical and laboratory assessments have been made on the days specified in the Schedule of Events tables. At the Weeks 12, 39, 52, and 65 visits, the sensor was placed on participants after all clinical and laboratory assessments and the MMTT have completed.
[0304] Study CGM readings are not intended for medical management of participant’s
diabetes but can be under the supervision of a participant’s health care team. Of note, routine use of the personal CGM under guidance of a participant’s regular healthcare provider is permitted.
[0305] Spot-check and CGM blood glucose assessments are anticipated to include but are not be limited to mean BG, glycemic variability (BG standard deviation [SD]), maximum and minimum BG values over time and incidence and/or percent time with BG >70 but <180 mg/dL (>3.9 but <10.0 mmol/L, Level 1 (>180 but <250 mg/dL (>10 but <13.9 mmol/L)) and Level 2 HYPERglycemia (>250 mg/dL (>13.9 mmol/L)) and Level 1 (<70 but >54 mg/dL (<3.9 but >3.0 mmol/L)) and Level 2 (<54 mg/dL (<3.0 mmol/L)) HYPOglycemia (Seaquist 2013, International Hypoglycaemia Study Group [IHSG] 2017, Agiostratidou 2017).
Pharmacokinetics and Immunogenicity
Evaluations
[0306] Venous blood samples were collected for measurement of serum concentrations of teplizumab, anti-teplizumab antibodies and neutralizing antibody (NAb) according to the Schedule of Events. Additionally, samples should also be collected at the Early Termination Visit for subjects who discontinue study treatment early and from subjects who experience an AE suspected to be related to immunogenicity (e.g., infusion reactions, injection site reactions or hypersensitivity).
[0307] Venous blood samples were collected. Samples collected for analyses of teplizumab serum concentration and antibody to teplizumab may additionally be used to evaluate safety or efficacy aspects that address concerns arising during or after the study period for further characterization of immunogenicity or for the evaluation of relevant biomarkers.
Analytical Procedures
[0308] Serum samples were analyzed using validated, specific, and sensitive immunoassay methods.
Pharmacokinetic Parameters
[0309] Nonlinear mixed effects modeling (with NONMEM software) was used to analyze the serum concentration-time data of teplizumab to obtain the primary PK parameters, clearance (CL) and volume of distribution. The PK profiles was used, along with other available data, to develop a population PK model while including the effect of major covariates (e.g., sex, ethnicity, race, antibody) on CL and volume of distribution. The starting model is a previously developed model for teplizumab. All PK parameters are presented by listings and descriptive summary statistics including, arithmetic mean, geometric mean (AUC, Cmax and their derived parameters), median, range, standard deviation and coefficient
of variation. The data of this study may be pooled with data from other studies.
Exploratory and Other Assessments
Immune and Serologic Assessments
[0310] Immune assessments may include but are not limited to monitoring T cell profiles and B cell profiles by flow cytometry. In addition, serum levels of circulating pro- and antiinflammatory cytokines and other soluble factors that can impact T1D progression may be assessed.
[0311] To investigate if and how teplizumab induces changes in lymphocyte subpopulations and expression of markers indicative of functional state (i.e., activation and exhaustion), flow cytometry is anticipated to be performed as part of an exploratory endpoint. Quantitative subpopulation analysis that includes assessment of CD4+ T cells, CD8+ T cells, B cells and NK cells (ie, Quantitative Lymphocyte Subset Panel or TBNK panel) was conducted on fresh samples. In addition, samples for PBMC evaluation were collected at the indicated timepoints, processed and stored for future “deep” phenotypic analysis, antigenspecific analyses (i.e., tetramer) and also functional responses to antigen-specific and nonspecific stimuli. Some of these analyses may take place by traditional flow cytometry, mass cytometry (CyTOF) or other technology.
[0312] To determine if and how teplizumab affects cytokine levels due to cytokine release or lymphocyte modulation, blood samples were collected at select timepoints before and after study drug administration. Samples were processed and stored. It is anticipated that analyses will be conducted only at select intervals that may include after a critical number of participants have completed the Week 52 visit or other key timepoints. Examples of cytokines that may be evaluated are interleukin (IL) 2 (IL-2), IL-4, IL-5, IL-6, IL-8, IL-10, interferon-gamma and tumor necrosis factor-alpha. In addition, other serum assessments, including those of hormones or other metabolically active substances (e.g., glucagon, incretins, lipokines, adiponectin, or cholesterol) that may have an effect on metabolic control, may be performed. Approaches to evaluations may include antibody-based multiplex panels, modified-aptamer binding technology, or other platforms.
[0313] Key markers for the presence of the autoimmune processes directed against pancreatic islets include assessing the presence and titers of anti-GAD65, anti-insulin, anti- IA-2, anti-ZnT8, and anti-ICA autoantibodies. Detection of these autoantibody combinations has proven to be an accurate predictor of T1D in several natural history studies. Therapies that impact the progression of T1D may change the presence or shift titers of autoantibodies. In addition, teplizumab may have an effect on antibody subclasses in general. As such, it will
be important to evaluate effects of teplizumab on T1D autoantibodies presence and titer in the context of assessment of total antibody subclasses. Qualitative and quantitative assessments of antibody subclasses, e.g., IgG, IgA, and IgM, may indicate a change in the type of T- helper cell responses to TID-associated autoantigens.
[0314] Recent data suggest that infection with CVB may be a trigger that breaks selftolerance in those predisposed to T1D and heralds destruction of P-cells and eventually T1D. Improved understand regarding the relationship of CVB infection in those newly diagnosed with T1D may assist in developing novels therapies to treat and/or prevent T1D. Blood samples obtained for exploratory analysis can be used for serologic, molecular and other assessments of CVB infection.
Assessment of p cell Stress
[0315] The disease progression has been proposed to be involved with alterations in “(3-cell stress,” specifically due to attempts at enhanced endogenous insulin production in residual P- cells that may overwhelm intracellular processes and direct or bystander injurious inflammatory meditators. It proposed that measurement of specific P-cell products may be markers of this process and that P-cell recovery due to immune interventions may result in a decrease of these markers. In this study, exploratory evaluation of two such markers, proinsulin-to-C-peptide ratios and serum levels of circulating methylated-insulin DNA, are to be evaluated. Samples that can be used for these analyses were collected, processed and stored.
Pharmacodynamic Substudy
[0316] In order to evaluate the pharmacodynamic (PD) effects of teplizumab, namely its CD3 receptor occupancy and modulation, a substudy was conducted.
Pharmacogenomic (DNA) Evaluations
[0317] The HLA system is a grouping of genes that encode the major histocompatibility complex (MHC) proteins in humans. An individual’s MHC haplotype gives insight to interactions of distinct types of lymphocytes that may cause T1D and may help to identify those who are at risk of developing T1D (Roark 2014). The MHC haplotype of participants was determined. The results of genotype analyses can be used to correlate with disease progression, therapeutic responses and identify subgroups that respond preferentially to teplizumab.
[0318] Pharmacogenomic studies on DNA, RNA or other genetic matter may also provide insights into the mechanism of teplizumab’s effect on T1D, the immune system or individuals who preferentially respond to teplizumab. One example is the methylated insulin-DNA
assessment anticipated, but also may include studies on the transcript/transcriptome, microarrays, or whole genome assessments. Samples that can be used for these analyses were collected, processed and/or stored.
[0319]
Quality of Life Assessment
[0320] Individuals with T1D may outwardly look healthy, but the management of their disease is a daily task for the rest of their life that requires multiple assessments and treatments per day along with close monitoring of diet, health status and exercise. As such there is a daily significant burden on those with T1D and their families (Monaghan 2015, Mittermayer 2017). In addition, those with even the most aggressively managed T1D are at risk for significant short and long-term morbidity and mortality. Therefore, it is realized that there are not just medical sequelae for those with T1D, but significant emotional, personal, familial and mental burdens as well (Monaghan 2015). Understanding if and how therapies may impact these other “quality of life” measures is an increasingly recognized aspect of beneficial effects of therapies that may alter the disease course in T1D. This study has participants complete questionnaires such as the PedsQL Diabetes Module, HFS, DTSQ, and PedsQL Family Impact Module at time points indicated in the Schedule of Events tables (Driscoll 2016, Trancone 2016, Bradley 2009, Gonder-Frederick 2011, Varni 2018).
Management of Cytokine Release Syndrome
[0321] In previous Phase 3 studies of teplizumab, ~6% of participants with T1D experienced cytokine release syndrome after receiving teplizumab. Symptoms of cytokine release syndrome included but were not limited to rash, headache, nausea, vomiting, chills/rigor, and fever. Most of the symptoms occurred during the first few days of treatment and are mild or moderate in severity. Cytokine release syndrome was time-limited and appeared to dissipate regardless of whether teplizumab dosing was interrupted.
[0322] The possibility exists that participants in this study may experience cytokine release syndrome despite premedications and may require supplemental therapy and/or modification of study drug administration. Recommendations for the management of these symptoms are as follows.
Supplemental Therapy
[0323] Supplemental therapy is intended for symptom management. The same premedications can be used as follows:
• Locally approved NSAIDs should be given in compliance with appropriate age restrictions and practice standards, such as ibuprofen (tablet or liquid), diclofenac, naproxen, meloxicam, or tenoxicam.
• Acetaminophen may be added or used instead of a NS AID if the latter cannot be used. Continuation of NSAID dosing should be considered if necessary.
• Antihistamines may be continued.
For more severe or prolonged symptoms, the following may be administered as consistent with local practice standard:
• Antihistamines, e.g., IV diphenhydramine
• Acetaminophen can be combined with antihistamines and can be repeated every 4 to 6 hours; hydroxyzine can be used during the day to avoid sedation.
• NSAIDs with higher potency, e.g., ketorolac.
• Acetaminophen with codeine or meperidine for myalgias, chills and rigors.
• Ondansetron for managing GI symptoms such as nausea and vomiting.
• Saline boluses to help with hemodynamic support
Modification of Study Drug Administration
[0324] If the cytokine release syndrome is associated with anaphylaxis or angioedema with or without requiring hemodynamic support (i.e., epinephrine and/or blood pressure medications) or mechanical ventilation, study drug should be permanently discontinued.
[0325] Symptoms alone generally do not lead to study drug modification. However, if the cytokine release syndrome is associated with the following laboratory abnormalities, the drug should be withheld: o Study drug should be permanently discontinued: ALT and/or AST >5X ULN, Total Bilirubin >3X ULN. ALT and/or AST >3X ULN AND Total Bilirubin >2X ULN (Hy’s Law criteria); platelet count <50,000/pL, neutrophil count <500 cells/pL, hemoglobin of <8.5 g/dL (laboratory test(s) should be confirmed on 2 consecutive dosing days. o Study drug may be interrupted temporarily: ALT and/or AST >3X ULN but <5X ULN, Total Bilirubin >2X ULN but <3X ULN, platelet count >50,000 but <100,000, hemoglobin >8.5 g/dL but <10 g/dL (laboratory tests may be repeated on the same day). If the repeated test normalizes, that day’s dose may be given. If the value is still in the above range or worsens, or if the test cannot be repeated on the same day, that day’s dose should be withheld.
■ If the above events resolve within 2 days, study drug dosing may be resumed according to the original schedule.
■ If the above events do not resolve after 2 consecutive days of interruption, the Medical Monitor must be consulted regarding continuation of study drug dosing.
[0326] Glucocorticoids (e.g., prednisone 1-1.5 mg/kg/d given twice daily) should be reserved for intractable symptoms or Grade 3 or higher events that cannot be relieved with the above medications. There is some evidence that glucocorticoids may interfere with the mechanism of action of teplizumab and therefore should be given for as short a duration as possible. If the investigator feels that glucocorticoids are necessary to resolve intractable signs or symptoms, they should follow applicable standard of care recommendations and treatment guidelines and inform the medical monitor without any delays. Blood glucose levels should be monitored carefully during glucocorticoid administration.
Laboratory tests
Hematology Panel
• WBC with differential
• Hemoglobin
• Hematocrit
• Platelet count
Serum Chemistry Panel with Liver Function Tests
• Sodium
• Potassium
• Chloride
• Bicarbonate
• Blood urea nitrogen (BUN)
• Creatinine
• Glucose
• Calcium
• Phosphate
• Albumin
• Total protein
Liver Function Tests
• Total bilirubin (TBili)
• Direct bilirubin (DBili)
• Aspartate aminotransferase (AST)
• Alanine aminotransferase (ALT)
• Alkaline phosphatase (ALP)
Quantitative Lymphocyte Subset (TBNK) Panel
• CD4+ T cells
• CD8+ T cells
• B cells
• NK cells
Quantitative Immunoglobulin Panel
• IgA
• IgG
. IgM
Lipid Panel (fasting)
• Total cholesterol
• High density lipoprotein (HDL)
• Low density lipoprotein (LDL)
• Triglycerides
Coagulation Panel (only at screening)
• Prothrombin time (PT)
• Partial thromboplastin time (PTT)
• International normalized ratio (INR)
Urinalysis
. pH
• Specific gravity
• Protein
• Glucose
• Ketones
• Bilirubin
• Nitrites
• Leukocyte esterase
• Blood cells/hemoglobin
Other Tests
• Serum pregnancy testing (only at screening for females of childbearing potential)
• HIV antibody serology (only at screening)
• HBV antibody serology /antigen panel (only at screening)
• HCV antibody serology (only at screening)
• VZV antibody serology
• CMV serology
• EBV serology
• CMV DNA PCR
• EBV DNA PCR
• MHC haplotype (only at Week 1, ie, randomization)
• Interferon-gamma Release Assay (IGRA) Tuberculosis testing
• HbAlc
Diabetes-related laboratory tests:
• Type 1 diabetes antibodies (anti-insulin, anti-GAD-65, ICA, anti-ZnT8, anti-IA2)
• Connecting peptide (C-peptide)
• Insulin
• Proinsulin
RESULTS
[0327] The demographics and baseline characteristics of the patients is shown at Table 5 below.
Table 5. Demographic and Clinical Characteristics of PROTECT Patients at Baseline.


[0328] The results show that the study met the primary endpoint: teplizumab treatment resulted in a significant difference in the mean change from baseline in C-peptide AUC levels compared with placebo at Week 78. Following teplizumab administration, mean C-peptide AUCs were maintained until Week 26, before declining at Weeks 52 and 78 (Figure 31A). By contrast, mean C-peptide AUCs decreased at all time points in patients receiving placebo. A prespecified exploratory analysis showed the proportion of patients with clinically meaningful peak C-peptide level >0.2 pmol/mL was greater at Week 78 with teplizumab compared to placebo (94.9% [95% CI 89.5, 97.6] versus 79.2% [95% CI 67.7, 87.4] (Figures 32 and 33). The data show that teplizumab preserves beta cell function as measured by C- peptide levels. Patients receiving teplizumab have, at week 78, a 59.3% higher mean C- peptide value compared with a control receiving placebo.
Table 6A
 [0329] The preservation of C-peptide is accompanied with positive numerical trends in insulin use and time in range (TIR) in favor of teplizumab while secondary endpoints were not statistically significant.
[0329] The preservation of C-peptide is accompanied with positive numerical trends in insulin use and time in range (TIR) in favor of teplizumab while secondary endpoints were not statistically significant.
[0330] Secondary endpoints were assessed to understand the impact of teplizumab treatment on clinical parameters. Consistent with treat-to-target glycemic management, mean HbAlc was rapidly controlled and did not differ between groups through Week 78 (Table 6B, Figures 34A-34B). The difference in TIR between the teplizumab and placebo groups was 4.71% (95% CI -1.72, 11.15) (Table 6B); the proportion of patients achieving TIR>70% is shown in Figure 31B.
Table 6B

*Prespecified additional analysis of insulin use over the entire study indicated a difference of 0.141 U/kg/day at Week 78 (95% CI, -0.230, -0.051) (MMRM, based on observed data). #Estimated rate ratio = Teplizumab / Placebo
[0331] At Week 78, mean daily insulin dose in patients treated with teplizumab and placebo were 0.45 units/kg/d and 0.6 units/kg/d, respectively (difference=0.13 (95% CI -0.28, 0.02;
P=0.085, ANCOVA) (Table 6B) and the insulin dose was lower at all time points after Week 12 when assessed with prespecified analysis of mixed model of repeated measures (MMRM) (Figure 31C).
[0332] In exploratory analyses, the proportion of patients meeting the prespecified definition of clinical remission was greater with teplizumab compared with placebo at every post-baseline time point (Figure 31D). The results are also consistent with a previously used definition of remission based on insulin dose alone (<0.25 U/kg/d) (Figure 35). Rates of clinically important hypoglycemic events were similar between groups. When differentiated by severity, the rate of Level 3 events was lower in the teplizumab group (rate ratio 0.29 [95% CI 0.13, 0.62], Table 7).
Table 7: PROTECT Efficacy Sensitivity Analysis of Clinically Important Hypoglycemic Events - eDiary and eCRFs
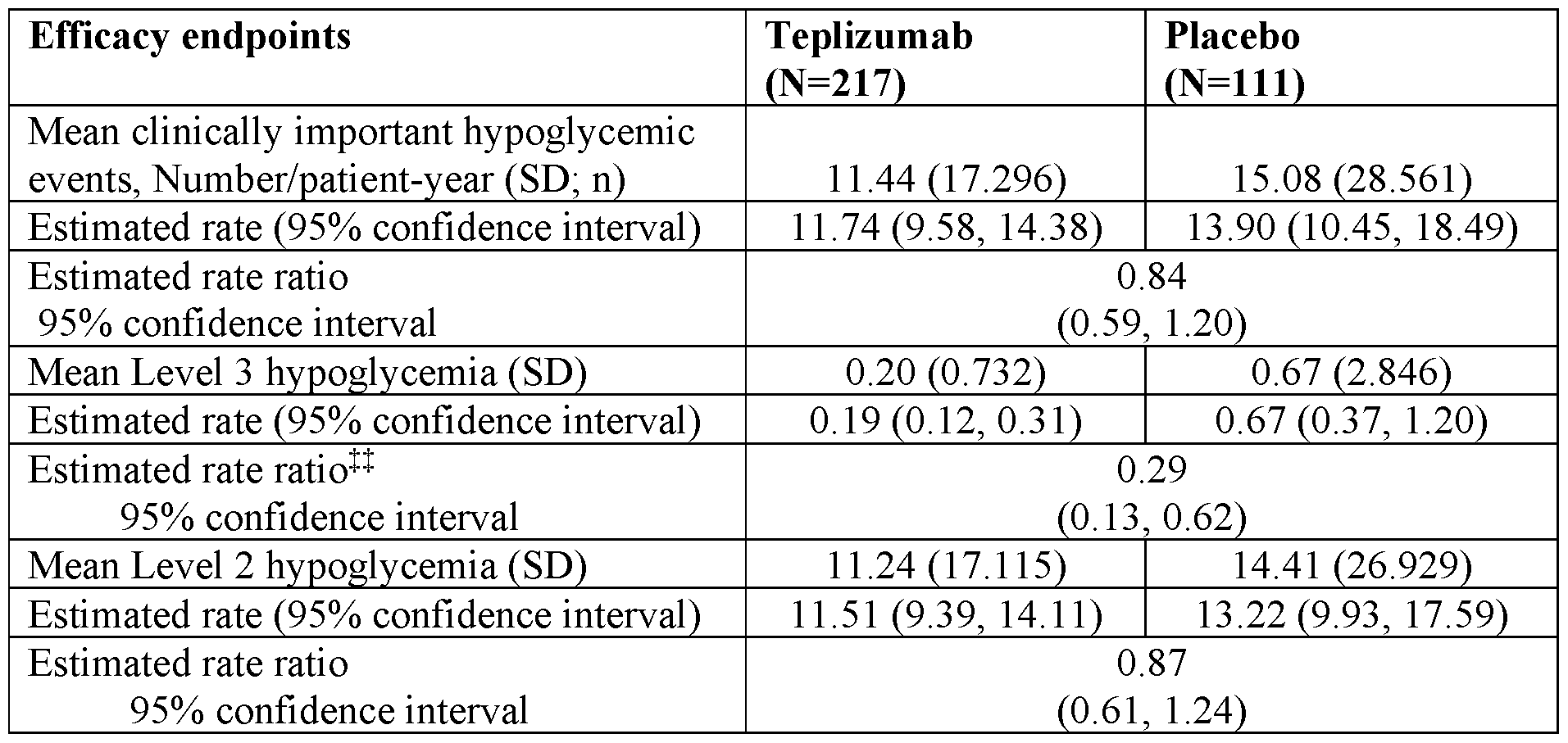
*To account for potential underreporting of clinically important hypoglycemic events in the eDiary, a sensitivity analysis was conducted post hoc that included hypoglycemic adverse events Grade 2 and above reported on the adverse events eCRFs.
[0333] Similar trends were observed in the prespecified per-protocol analysis (Table 8).
Table 8: PROTECT Efficacy Supportive Assessments - Per-Protocol Population


[0334] Baseline was defined as the most recent value collected prior to the first dose of teplizumab or placebo. Both groups achieved target HbAlc and the proportion of patients with glucose TIR was not statistically significantly different between groups (Table 6B); a greater proportion of teplizumab-treated patients achieved the glucose TIR target of >70% of the time across all study visits, but the difference to placebo was not statistically significant (Figure 31B). Throughout the study, TBR <4% was not different between the two groups 12 (teplizumab 2.48%, placebo 2.8% at Week 78). Mean time above range was similar between
groups (teplizumab 14-29%, placebo 16-33%; Table 8), with teplizumab <25%12, 13 until Week 52 and placebo until Week 26.
[0335] Analyses of COA measures (Figure 36) showed that teens in the teplizumab group experienced less decline in perceived diabetes control at Week 78 (LS mean difference 1.1 [95% CI 0.0, 2.1]) which was supported by parents who indicated greater satisfaction with their teen’s treatment at Week 78 (DTSQ-parent version; LS mean difference 2.4 [95% CI 0.3, 4.5]).
[0336] C-peptide preservation was supported by:
• A numerically lower insulin use (in U/kg/day) in patients treated with teplizumab while achieving similar target glycemic control (HbAlc) in both treatment groups (teplizumab group and placebo group)
• Patients treated with teplizumab spent numerically more Time in Range (i.e., target glucose range)
[0337] The mean insulin use (in U/kg/day) of the patients treated with teplizumab at week 78 was the same as the mean insulin use of the patients before being administered teplizumab (baseline). In contrast, the mean insulin use of the patients receiving the placebo increased from baseline to week 78.
[0338] Of note, two patients in the teplizumab group were able to stop using insulin at week 78. No patients in the placebo group were able to stop using insulin at week 78.
[0339] HbAlc was similar in both treatment groups, confirming the achievement of treat- to-target goal by patients. Patients receiving placebo needed more insulin to maintain or decrease HbAlc levels than patients treated with teplizumab.
[0340] Patients treated with teplizumab showed an improvement in time in range compared to patients receiving placebo.
[0341] Figure 27 is a graph showing insulin use at different timepoint (baseline, week 12, week 26, week 39, week 52, week 65 and week 78). Figure 27 shows that insulin use was numerically lower in patients treated with teplizumab.
[0342] The table 9 below shows that more teplizumab patients discontinued insulin.
Table 9


[0343] Figure 28 is a graph showing the percentage of subjects who met Hbl Ac <6.5% and insulin daily dose <0.25 unit/kg/days at different timepoint (baseline, week 12, week 26, week 39, week 52, week 65 and week 78). Figure 28 shows that more teplizumab patients met insulin discontinuation criteria HblAc <6.5% and insulin daily dose <0.25 unit/kg/days.
[0344] Figure 29 is graph showing the HbAlc levels at different timepoint (baseline, week 12, week 26, week 39, week 52, week 65 and week 78). Figure 29 shows that HbAlc target levels were achieved in both treatment groups.
[0345] Figure 30 is graph showing the percentage time in range at different timepoint (baseline, week 12, week 26, week 39, week 52, week 65 and week 78). Figure 30 shows that patients treated with teplizumab spent more Time in Range (TIR).
[0346] The use of assisted technology (e.g., continuous glucose monitoring, insulin pumps) likely contributed to similar rates of clinically-important hypoglycemic events in both teplizumab and placebo groups.
[0347] The safety analysis did not reveal new safety signals.
Table 10


[0348] Patients treated with teplizumab (n=217) had significantly greater stimulated C- peptide levels compared with placebo (n=l 11) at Week 78 (difference in least-square means 0.13 [95% CI 0.09, 0.17; PO.OOl]), with 95% (95% CI 89.5, 97.6) maintaining clinically meaningful peak C-peptide levels >0.2 pmol/mL versus 79% (95% CI 67.7, 87.4) with placebo (P<0.001). Teplizumab-treated patients used lower insulin doses to meet glycemic goals, with greater glucose time in range, less Grade 3 hypoglycemia, and higher frequency of predefined clinical remission. Adverse events were limited to the period of drug administration and were transient, self-limited, and consistent with prior experience.
[0349] In conclusion, treatment with teplizumab in patients (8 to 17 years) with newly diagnosed Stage 3 T1D led to a statistically significant better beta-cell preservation as shown by maintaining higher C-peptide levels at 78 weeks compared to patients receiving placebo with numerically better Time in Range and less insulin use.
[0350] Modifications and variations of the described methods and compositions of the present disclosure will be apparent to those skilled in the art without departing from the scope and spirit of the disclosure. Although the disclosure has been described in connection with specific embodiments, it should be understood that the disclosure as claimed should not be unduly limited to such specific embodiments. Indeed, various modifications of the described modes for carrying out the disclosure are intended and understood by those skilled in the relevant field in which this disclosure resides to be within the scope of the disclosure as represented by the following claims.
INCORPORATION BY REFERENCE
[0351] All patents and publications mentioned in this specification are herein incorporated by reference to the same extent as if each independent patent and publication was specifically and individually indicated to be incorporated by reference.
REFERENCES
1. Abdul -Rasoul M, Habib H, Al-Khouly M. “The honeymoon phase” in children with type 1 diabetes mellitus: frequency, duration, and influential factors. Pediatr Diabetes. 2006;7(2): 101-107.
2. Ablamunits V, Bisikirska B, Herold KC. Acquisition of regulatory function by human CD8+ T cells treated with anti-CD3 antibody requires TNF. Eur J Immunol.
2010;40(10):2891-2901.
3. Agiostratidou G, Anhalt H, Ball D, et al. Standardizing clinically meaningful outcome measures beyond HbAlc for type 1 diabetes: a consensus report of the American Association of Clinical Endocrinologists, the American Association of Diabetes Educators, the American Diabetes Association, the Endocrine Society, JDRF International, The Leona M. and Harry B. Helmsley Charitable Trust, the Pediatric Endocrine Society, and the T1D Exchange. Diabetes Care. 2017;40(12): 1622-1630.
4. Akirav EM, Kushner JA, Herold KC. P cell mass and type 1 diabetes: Going, going, gone? Diabetes. 2008;57:2883-2888.
5. American Diabetes Association. 12. Children and Adolescents: Standards of medical Care in Diabetes-2018. Diabetes Care. 2018;41(Suppl. 1):S 126-S 136.
6. Atkinson MA. Type 1 diabetes. Lancet. 2014;383(9911):69-82
7. Battelino T, Alexander CM, Amiel SA, et al. Continuous glucose monitoring and metrics for clinical trials: an international consensus statement. The lancet Diabetes & endocrinology 2023;11 :42-57.
8. Bisikirska B, Colgan J, Luban J, Bluestone, JA, Herold KC. TCR stimulation with modified anti-CD3 mAb expands CD8+ T cell population and induces CD8+CD25+ Tregs. J Clin Invest. 2005; 115(10):2904-2913.
9. Bluestone JA, Herold K, and Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature. 2010;464(7293): 1293-1300.
10. Boyle KD, Keyes-Elstein L, Ehlers MR, et al. Two- and four-hour tests differ in capture of C-peptide responses to a mixed meal in type 1 diabetes. Diabetes Care. 2016;39:e76-78.
11. Bradley C, Loewenthal K, Woodcock A, et al. Development of the diabetes treatment satisfaction questionnaire (DTSQ) for teenagers and parents: the DTSQ-Teen and the DTSQ- Parent. Diabetologia. 2009;52: (Suppl 1) S397, Abstract 1013.
12. Campbell-Thompson M, Fu A, Kaddis JS, et al. Insulitis and P-cell mass in the natural history of type 1 diabetes. Diabetes. 2016;65:719-731.
13. Danne T, Nimri R, Battelino T, et al. International consensus on use of continuous glucose monitoring. Diabetes Care 2017;40: 1631-1640.
14. Diabetes Control and Complications Trial Research Group. Hypoglycemia in the Diabetes Control and Complications Trial. Diabetes. 1997;46(2): 271-286.
15. Diabetes Prevention Trial-Type 1 Diabetes Study Group. Effects of insulin in relatives of patients with type 1 diabetes mellitus. N Engl J Med 2002;346: 1685-91.
16. DiMeglio LA, Acerini CL, Codner E, et al. ISP AD Clinical Practice Consensus Guidelines 2018: Glycemic control targets and glucose monitoring for children, adolescents, and young adults with diabetes. Pediatr Diabetes. 2018 Oct;19 Suppl 27: 105-114.
17. Driscoll KA, Raymond J, Naranjo D, et al. Fear of hypoglycemia in children and adolescents and their parents with type 1 diabetes. Curr Diab Rep. 2016;16(8):77.
18. European Medicines Agency. Ethical Considerations for Clinical Trials on Medicinal Products Conducted with the Paediatric Population. 2008. https://ec.europa.eu/health/sites/health/files/files/eudralex/vol- 10/ethical_considerations_en.pdf. Accessed September 27, 2018.
19. Feutren G, Papoz L, Assan R, et al. Cyclosporin increases the rate and length of remissions in insulin-dependent diabetes of recent onset. Results of a multicentre doubleblind trial. Lancet (London, England) 1986;2: 119-24.
20. Fonolleda M, Murillo M, Vazquez F, et al. Remission phase in paediatric type 1 diabetes: new understanding and emerging biomarkers. Horm Res Paediatr. 2017;88(5):307- 315.
21. Gitelman SE, Gottlieb PA, Rigby MR, et al. Antithymocyte globulin treatment for patients with recent-onset type 1 diabetes: 12-month results of a randomised, placebo- controlled, phase 2 trial. Lancet Diabetes Endocrinol. 2013 ; 1 (4):306- 16.
22. Gonder-Frederick L, Nyer M, Shepard JA. Assessing fear of hypoglycemia in children with Type 1 diabetes and their parents. Diabetes Manag (Lond). 2011 ; 1(6): 627-639.
23. Greenbaum CJ, Beam CA, Boulware D et al. Fall in C peptide during first 2 years from diagnosis: evidence of at least two distinct phases from composite type 1 diabetes TrialNet data. Diabetes. 2012;61 :2066-2073.
24. Hagopian W, Ferry RJ Jr, Sherry N et al, Protege Trial Investigators. Teplizumab preserves C-peptide in recent-onset type 1 diabetes: two-year results from the randomized, placebo-controlled Protege trial. Diabetes. 2013;62(l l):3901-8. doi: 10.2337/dbl3-0236. Epub 2013 Jun 25.
25. Helminen O, Pokka T, Tossavainen P, et al. Continuous glucose monitoring and HbAlc in the evaluation of glucose metabolism in children at high risk for type 1 diabetes mellitus. Diabetes Res Clin Pract. 2016;120:89-96.
26. Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, Gitelman SE, Harlan DM, Xu D, Zivin RA, Bluestone JA. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002 May 30;346(22): 1692-1698.
27. Herold KC, Gitelman SE, Umest M et al. A single course of anti-CD3 monoclonal antibody hOKT3yl(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes. 2005;54: 1-7.
28. Herold KC, Gitelman SE, Ehlers MR, et al. Teplizumab (anti-CD3 mAb) treatment preserves C-peptide responses in patients with new-onset type 1 diabetes in a randomized controlled trial: metabolic and immunologic features at baseline identify a subgroup of responders. Diabetes. 2013(a);62(l l):3766-3774.
29. Herold KC, Gitelman SE, Willi SM, et al. Teplizumab treatment may improve C- peptide responses in participants with type 1 diabetes after the new-onset period: a randomised controlled trial. Diabetologia. 2013(b);56(2):391-400.
30. Herold KC, Bundy BN, Long SA, et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N Engl J Med. 2019 Jun 9. doi:
10.1056/NEJMoal 902226. [Epub ahead of print]
31. Hilliard ME, Lawrence JM, Modi AC, et al. Identification of minimal clinically important difference scores of the PedsQL in children, adolescents, and young adults with type 1 and type 2 diabetes. Diabetes care 2013;36: 1891-7.
32. Huo L, Harding JL, Peeters A, et al. Life expectancy of type 1 diabetic patients during 1997-2010: a national Australian registry -based cohort study. Diabetologia. 2016;59(6): 1177- 85.
33. International Hypoglycaemia Study Group. Glucose Concentrations of Less Than 3.0 mmol/L (54 mg/dL) Should Be Reported in Clinical Trials: A Joint Position Statement of the American Diabetes Association and the European Association for the Study of Diabetes.
Diabetes Care. 2016;dcl62215.
34. Joint Commission. High Alert Medications and Patient Safety. Sentinel Event Alert. Issue 11. November 19,1999. https://www.j0intc0mmissi0n.0rg/assets/l/l8/SEA_l l.pdf Accessed August 21, 2018.
35. Karvonen M, Viik-Kajander M, Moltchanova E, et al. Incidence of childhood type 1 diabetes worldwide. Diabetes Mondiale (DiaMond) Project Group. Diabetes Care. 2000;23(10): 1516-1526.
36. Kuhtreiber WM, Washer SL, Hsu E, et al. Low levels of C-peptide have clinical significance for established Type 1 diabetes. Diabet Med. 2015 Oct;32(10): 1346-53.
37. Lachin JM, McGee P, Palmer JP; DCCT/EDIC Research Group. Impact of C-peptide preservation on metabolic and clinical outcomes in the Diabetes Control and Complications Trial. Diabetes. 2014;63(2):739-748.
38. Laitinen OH, Honkanen H, Pakkanen O, et al. Coxsackievirus Bl is associated with induction of P-cell autoimmunity that portends type 1 diabetes. Diabetes. 2014;63(2):446- 455.
39. Lebastchi J, Deng S, Lebastchi AH, et al. Immune therapy and P-cell death in type 1 diabetes. Diabetes. 2013;62(5): 1676-1680.
40. Lin A, Northam EA, Werther GA, Cameron FJ. Risk factors for decline in IQ in youth with type 1 diabetes over the 12 years from diagnosis/illness onset. Diabetes Care. 2015;38:236-242.
41. Lind M, Svensson AM, Kosiborod M, et al. Glycemic control and excess mortality in type 1 diabetes. N Engl J Med. 2014;371 :1972-1982.
42. Long SA, Thorpe J, DeBerg HA, et al. Partial exhaustion of CD8 T cells and clinical response to teplizumab in new-onset type 1 diabetes. Sci. Immunol. 2016; 1 (5): 1 -23.
43. Long SA, Thorpe J, Herold KC, et al. Remodeling T cell compartments during anti- CD3 immunotherapy of type 1 diabetes. Cell Immunol. 2017 Sep;319:3-9.
44. Ludvigsson J, Carlsson A, Deli A, et al. Decline of C-peptide during the first year after diagnosis of Type 1 diabetes in children and adolescents. Diabetes Res Clin Pract. 2013;100(2):203-209.
45. Masharani UB, Becker J. Teplizumab therapy for type 1 diabetes. Expert Opin Biol Ther. 2010; 10(3): 459-465.
46. Matveyneko AV and Butler PC. Relationship between p cell mass and diabetes onset. Diabetes Obes Metab. 2008;10:23-31.
47. Mittermayer F, Caveney E, De Oliveira C, et al. Addressing Unmet Medical Needs in Type 1 Diabetes: A Review of Drugs Under Development. Curr Diabetes Rev.
2017; 13(3):300-314.
48. Monaghan M, Helgeson V, Wiebe D. Type 1 diabetes in young adulthood. Curr Diabetes Rev. 2015; 11(4):239-250.
49. Mortensen HB, Hougaard P, Swift P, et al. New definition for the partial remission period in children and adolescents with type 1 diabetes. Diabetes Care. 2009;32:1384-1390.
50. National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE). https://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm. Updated March 1, 2018. Accessed September 27, 2018.
51. Orban T, Bundy B, Becker DJ, et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet 2011;378(9789):412-419.
52. Palmer JP, Fleming GA, Greenbaum CJ, et al. C-peptide is the appropriate outcome measure for type 1 diabetes clinical trials to preserve beta-cell function: report of an ADA workshop, 21-22 October 2001. Diabetes. 2004;53(l):250-264.
53. Palmer JP. C-peptide in the natural history of type 1 diabetes. Diabetes Metab Res Rev. 2009;25(4):325-328.
54. Rawshani A, Rawshani A, Franzen S, et al. Mortality and Cardiovascular Disease in Type 1 and Type 2 Diabetes. N Engl J Med. 2017;376(15): 1407-1418.
55. Rawshani A, Sattar N, Franzen S, et al. Excess mortality and cardiovascular disease in young adults with type 1 diabetes in relation to age at onset: a nationwide, register-based cohort study. Lancet. 2018;392(10146):477-486.
56. Rigby MR, DiMeglio LA, Rendell MS, et al. Targeting of memory T cells with alefacept in new-onset type Idiabetes (TIDAL study): 12-month results of a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Diabetes Endocrinol. 2013;l(4):284- 294.
57. Rigby MR, Ehlers MR. Targeted Immune Interventions for Type 1 Diabetes: Not as Easy as it Looks! Curr Opin Endocrinol Diabetes Obes. 2014;21(4): 271-278.
58. Rigby MR, Harris KM, Pinckney A, et al. Alefacept provides sustained clinical and immunological effects in new-onset type 1 diabetes patients. J Clin Invest. 2015; 125(8):3285- 3296.
59. Roark CL, Anderson KM, Simon LJ, et al. Multiple HLA epitopes contribute to type 1 diabetes susceptibility. Diabetes. 2014;63(l):323-331.
60. Rosenzweig M, Rosenthal CA, Torres VM, Vaickus L. Development of a quantitative assay to measure EBV viral load in patients with autoimmune type 1 diabetes and healthy subjects. J Virol Methods. 2010; 164(1-2): 111-115.
61. Seaquist ER, Anderson J, Childs B, et al. Hypoglycemia and diabetes: a report of a workgroup of the American Diabetes Association and the Endocrine Society. Diabetes Care. 2013;36(5): 1384-1395.
62. Secrest AM, Becker DJ, Kelsey SF, LaPorte RE, and Orchard TJ. Characterising sudden death and dead-in-bed syndrome in Type 1 diabetes: analysis from 2 childhood-onset Type 1 diabetes registries. Diabet Med. 2011;28(3): 293-300.
63. Sherry N, Hagopian W, Ludvigsson J, et al., Protege Trial Investigators. Teplizumab for treatment of type 1 diabetes (Protege study): 1-year results from a randomised, placebo- controlled trial. Lancet. 2011;378(9790):487-97.
64. Sorensen JS, Johannesen J, Pociot F, et al. Residual P-cell function 3-6 years after onset of type 1 diabetes reduces risk of severe hypoglycemia in children and adolescents. Diabetes Care. 2013;36:3454-3459.
65. Sosenko JM, Skyler JS, Herold KC, et al. The metabolic progression to type 1 diabetes as indicated by serial oral glucose tolerance testing in the Diabetes Prevention Trialtype 1. Diabetes. 2012;61(6): 1331-7.
66. Steck AK, Dong F, Taki I, et al. Early hyperglycemia detected by continuous glucose monitoring in children at risk for type 1 diabetes. Diabetes Care. 2014;37:2031-2033.
67. Steffes MW, Sibley S, Jackson M, Thomas W. Beta-cell function and the development of diabetes-related complications in diabetes control and complications trial. Diabetes Care. 2003;26:832-836.
68. Streisand R. Young children with type 1 diabetes: challenges, research, and future directions. Curr Diab Rep. 2014;14(9):520. ppl-16.
69. Trancone A, Bonfanti R, lafusco D, et al. Evaluating the experience of children with type 1 diabetes and their parents taking part in an artificial pancreas clinical trial over multiple days in a diabetes camp setting. Diabetes Care. 2016;39:2158-2164.
70. Varni JW, Delamater AM, Hood KK, et al. PedsQL 3.2 Diabetes Module for children, adolescents, and young adults: reliability and validity in type 1 diabetes. Diabetes Care. 2018; dcl72707.
71. Verkruyse LA, Storch GA, Devine SM, et al. Once daily ganciclovir as initial preemptive therapy delayed until threshold CMV load > or =10000 copies/ml: a safe and effective strategy for allogeneic stem cell transplant patients. Bone Marrow Transplant. 2006;37(l):51-6.
72. Waldron-Lynch F, Henegariu O, Deng S, et al. Teplizumab induces human gut-tropic regulatory cells in humanized mice and patients. Sci Transl Med. 2012;4(118): 118ral2.
73. Wherrett DK, Chiang JL, Delamater AM, et al. Defining pathways for development of disease-modifying therapies in children with type 1 diabetes: a consensus report. Diabetes Care. 2015;38(10): 1975-85.
74. Ziegler AG and Nepom GT. Prediction and pathogenesis in type 1 diabetes. Immunity. 2010;32:468-478.
Claims
1. A method of treating type 1 diabetes (T1D), comprising administering to a subject in need thereof two 12-day courses of teplizumab at an interval of at least about 3 months, or of about 6 months, about 26 weeks, about 12 months, or about 52 weeks, each course having a total teplizumab dose of from about 9000 pg/m2 to about 14000 pg/m2, wherein the subject has a peak C-peptide level of at least 0.2 pmol/mL during a mixed meal tolerance test (MMTT), optionally a two-hour MMTT, prior to administration of the first 12-day course of teplizumab.
2. The method of claim 1, wherein the total teplizumab dose of each 12-day course is from about 9000 pg/m2 to about 9500 pg/m2.
3. The method of claim 2, wherein each 12-day course comprises a first dose of 106 pg/m2 teplizumab on day 1, a second dose of 425 pg/m2 teplizumab on day 2, and one dose of 850 pg/m2 teplizumab on each of days 3-12, and wherein the total dose of each course is approximately 9031 pg/m2.
4. The method of any one of claims 1-3, wherein the subject is about 8 to 17 years old.
5. The method of any one of claims 1-4, wherein the subject has been diagnosed with T1D within 6 weeks prior to administration of the first 12-day course of teplizumab.
6. The method of any one of claims 1-5, wherein the subject has at least 20% of beta-cell function prior to administration of the first course.
7. The method of any one of claims 1-6, further comprising administering to the subject in need thereof one or more additional 12-day courses of teplizumab, each additional course at a total dose of more than about 9000 pg/m2.
8. The method of claim 7, wherein each additional 12-day course of teplizumab comprises a first dose of 106 pg/m2 teplizumab on day 1, a second dose of 425 pg/m2 teplizumab on day 2, and one dose of 850 pg/m2 teplizumab on each of days 3-12, and
wherein the total dose of each additional course is approximately 9031 pg/m2.
9. The method of claim 8, wherein each additional 12-day course of teplizumab is administered at about 6 months or about 26 weeks, to about 24 months or about 104 weeks, after the preceding course.
10. The method of any one of claims 1-9, further comprising: determining, after administration of each 12-day course, a baseline level of TIGIT+KLRG1+CD8+ T-cells and/or a baseline level of PD-1+CD8+ T-cells with respect to all CD3+ T-cells; monitoring the level of TIGIT+KLRG1+CD8+CD3+ T-cells and/or the level of PD- 1+CD8+CD3+ T-cells; and administering an additional 12-day course of teplizumab when the level of the TIGIT+KLRG1+CD8+CD3+ T-cells and/or PD-1+CD8+CD3+ T-cells returns to the baseline level.
11. The method of claim 10, wherein the baseline level of the TIGIT+KLRG1+CD8+ T- cells and/or the baseline level of the PD-1+CD8+ T-cells is less than about 5% of all CD3+ T- cells.
12. The method of claim 10 or 11, wherein the determining is by flow cytometry.
13. The method of any one of claims 10-12, wherein the determining of TIGIT+KLRG1+CD8+CD3+ T-cells and/or PD-1+CD8+CD3+ T-cells is about 1-6 months, about 2-5 months, or about 3 months after the administration of each 12-day course.
14. The method of any one of claims 10-13, wherein if the subject has more than about 10% TIGIT+KLRG1+CD8+ T-cells and/or more than about 10% PD-1+CD8+ T-cells in all CD3+ T-cells, subsequent monitoring is annual.
15. The method of any one of claims 10-13, wherein if the subject has less than about 10% TIGIT+KLRG1+CD8+ T-cells and/or less than about 10% PD-1+CD8+ T-cells in all CD3+ T-cells, subsequent monitoring is about every 3-6 months.
16. The method of any one of claims 1-15, wherein each dose of teplizumab is administered parenterally.
17. The method of any one of claims 1-16, wherein each dose of teplizumab is administered by intravenous infusion, optionally wherein the subject is pre-medicated with (1) a nonsteroidal anti-inflammatory drug or acetaminophen, (2) an antihistamine, and/or (3) an antiemetic before each dose in the first three, four, five, six, or seven days of each course.
18. The method of any one of claims 1-17, wherein the subject in need thereof has a peak C-peptide level that ranges from 0.2 pmol/mL to 0.7 pmol/mL, or is at least 0.7 pmol/mL, during an MMTT, optionally a two-hour MMTT, prior to administration of the first 12-day course of teplizumab.
19. The method of any one of claims 1-18, wherein the subject in need thereof administered with teplizumab maintains a peak C-peptide level of at least 0.2 pmol/mL for about 78 weeks after the first course; has a higher C-peptide level, or has a 40% to 80%, or more than 80%, higher C- peptide level, at about 78 weeks after the first course, than a control subject not receiving teplizumab; maintains or exceeds, for about 26 weeks after the first course, a pre-treatment level of area under the time-concentration curve (AUC) of C-peptide during an MMTT, optionally a four-hour MMTT; and/or has a higher AUC of C-peptide at about 78 weeks after the first course than a control subject not receiving teplizumab.
20. The method of any one of claims 1-19, wherein the subject in need thereof administered with teplizumab maintains the pre-treatment HbAlc level; or has a reduced HbAlc level relative to the pre-treatment level or to a control subject not receiving teplizumab, optionally wherein the HbAlc level is reduced by 0.1 to 1 point, or more than 1 point, further optionally wherein the HbAlc level is reduced to 6.5% or less.
21. The method of any one of claims 1-20, wherein the subject in need thereof administered with teplizumab has an increased time in range (TIR) relative to a control
subject not receiving teplizumab, optionally wherein the TIR is increased by 3 to 10%, or by more than 10%.
22. The method of any one of claims 1-21, wherein the subject in need thereof administered with teplizumab maintains the pre-treatment insulin dosage; or is in need of a reduced insulin dosage relative to the pre-treatment dosage or to a control subject not receiving teplizumab, optionally wherein the insulin use is reduced by 10-30% or by more than 30%, further optionally wherein the insulin dosage is reduced by at least 0.1 units/kg/day or to no more than 0.25 units/kg/day.
23. The method of any one of claims 1-22, wherein the subject in need thereof administered with teplizumab has an HbAlc level of < 6.5% and is in need of an insulin dosage of <0.25 units/kg/day; and/or has fewer Level 3 hypoglycemic episodes than a control subject not receiving teplizumab.
24. A method of slowing loss of beta-cell function for at least about six months or at least about 26 weeks in a subject with type 1 diabetes (T1D), the method comprising administering to the subject in need thereof two 12-day courses of teplizumab at an interval of at least about three months, or of about 6 or about 12 months, each course having a total dose of from about 9000 pg/m2 to about 9500 pg/m2, wherein the subject is about 8 to 17 years of age and (i) has been diagnosed with T1D within six weeks prior to administration of the first course and/or (ii) has a peak C-peptide level of at least 0.2 pmol/mL during a mixed meal tolerance test (MMTT), optionally a two-hour MMTT, prior to administration of the first course.
25. The method of claim 24, wherein each 12-day course comprises a first dose of 106 pg/m2 teplizumab on day 1, a second dose of 425 pg/m2 teplizumab on day 2, and one dose of 850 pg/m2 teplizumab on each of days 3-12, and wherein the total dose of each course is approximately 9031 pg/m2.
26. The method of claim 24 or 25, wherein each dose of teplizumab is administered by intravenous infusion, optionally wherein the subject is pre-medicated with (1) a nonsteroidal anti-inflammatory drug or acetaminophen, (2) an antihistamine, and/or (3) an antiemetic before each dose in the first three, four, five, six, or seven days of each course.
27. The method of any one of claims 24-26, wherein the administering of teplizumab results in slower loss of beta-cell function for at least about 18 months or 78 weeks.
28. The method of any one of claims 24-27, wherein the administering of teplizumab results in: maintenance of a pre-treatment C-peptide level or in a higher C-peptide level than a control subject not receiving teplizumab, optionally 40% to 80%, or more than 80%, higher; lower required insulin dosage than a control subject not receiving teplizumab; greater time in range than a control subject not receiving teplizumab; and/or fewer Level 3 hypoglycemic episodes than a control subject not receiving teplizumab.
29. Teplizumab for use in a method of any one of claims 1-28.
30. Use of teplizumab in the manufacture of a medicament for use in a method of any one of claims 1-28.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363515949P | 2023-07-27 | 2023-07-27 | |
| US63/515,949 | 2023-07-27 | ||
| US202363590883P | 2023-10-17 | 2023-10-17 | |
| US63/590,883 | 2023-10-17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025024848A1 true WO2025024848A1 (en) | 2025-01-30 |
Family
ID=92457137
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/040020 Pending WO2025024848A1 (en) | 2023-07-27 | 2024-07-29 | Methods for treating type 1 diabetes |
Country Status (2)
| Country | Link |
|---|---|
| TW (1) | TW202521153A (en) |
| WO (1) | WO2025024848A1 (en) |
Citations (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4526938A (en) | 1982-04-22 | 1985-07-02 | Imperial Chemical Industries Plc | Continuous release formulations |
| WO1991005548A1 (en) | 1989-10-10 | 1991-05-02 | Pitman-Moore, Inc. | Sustained release composition for macromolecular proteins |
| US5128326A (en) | 1984-12-06 | 1992-07-07 | Biomatrix, Inc. | Drug delivery systems based on hyaluronans derivatives thereof and their salts and methods of producing same |
| WO1996020698A2 (en) | 1995-01-05 | 1996-07-11 | The Board Of Regents Acting For And On Behalf Of The University Of Michigan | Surface-modified nanoparticles and method of making and using same |
| US5679377A (en) | 1989-11-06 | 1997-10-21 | Alkermes Controlled Therapeutics, Inc. | Protein microspheres and methods of using them |
| WO1999015154A1 (en) | 1997-09-24 | 1999-04-01 | Alkermes Controlled Therapeutics, Inc. | Methods for fabricating polymer-based controlled release preparations |
| WO1999020253A1 (en) | 1997-10-23 | 1999-04-29 | Bioglan Therapeutics Ab | Encapsulation method |
| US5912015A (en) | 1992-03-12 | 1999-06-15 | Alkermes Controlled Therapeutics, Inc. | Modulated release from biocompatible polymers |
| US5916597A (en) | 1995-08-31 | 1999-06-29 | Alkermes Controlled Therapeutics, Inc. | Composition and method using solid-phase particles for sustained in vivo release of a biologically active agent |
| US6491916B1 (en) | 1994-06-01 | 2002-12-10 | Tolerance Therapeutics, Inc. | Methods and materials for modulation of the immunosuppresive activity and toxicity of monoclonal antibodies |
| US20040038867A1 (en) | 2002-06-13 | 2004-02-26 | Still James Gordon | Methods of reducing hypoglycemic episodes in the treatment of diabetes mellitus |
| US8663634B2 (en) | 2005-07-11 | 2014-03-04 | Macrogenics, Inc. | Methods for the treatment of autoimmune disorders using immunosuppressive monoclonal antibodies with reduced toxicity |
| US9056906B2 (en) | 2006-06-14 | 2015-06-16 | Macrogenics, Inc. | Methods for the treatment of autoimmune disorders using immunosuppressive monoclonal antibodies with reduced toxicity |
| WO2022251253A1 (en) * | 2021-05-24 | 2022-12-01 | Provention Bio, Inc. | Methods for treating type 1 diabetes |
| WO2023039295A1 (en) * | 2021-09-13 | 2023-03-16 | Provention Bio, Inc. | Methods and compositions comprising anti-cd3 antibodies and dyrk1a inhibitors for treating diabetes |
| WO2023230476A1 (en) * | 2022-05-24 | 2023-11-30 | Provention Bio, Inc. | Methods and compositions for preventing or delaying type 1 diabetes |
| WO2024182767A1 (en) * | 2023-03-01 | 2024-09-06 | Provention Bio, Inc. | Methods and compositions for treating type 1 diabetes comprising teplizumab and verapamil |
| WO2024206739A1 (en) * | 2023-03-30 | 2024-10-03 | Provention Bio, Inc. | Methods for reducing exogenous insulin use |
-
2024
- 2024-07-29 TW TW113128151A patent/TW202521153A/en unknown
- 2024-07-29 WO PCT/US2024/040020 patent/WO2025024848A1/en active Pending
Patent Citations (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4526938A (en) | 1982-04-22 | 1985-07-02 | Imperial Chemical Industries Plc | Continuous release formulations |
| US5128326A (en) | 1984-12-06 | 1992-07-07 | Biomatrix, Inc. | Drug delivery systems based on hyaluronans derivatives thereof and their salts and methods of producing same |
| WO1991005548A1 (en) | 1989-10-10 | 1991-05-02 | Pitman-Moore, Inc. | Sustained release composition for macromolecular proteins |
| US5679377A (en) | 1989-11-06 | 1997-10-21 | Alkermes Controlled Therapeutics, Inc. | Protein microspheres and methods of using them |
| US5912015A (en) | 1992-03-12 | 1999-06-15 | Alkermes Controlled Therapeutics, Inc. | Modulated release from biocompatible polymers |
| US6491916B1 (en) | 1994-06-01 | 2002-12-10 | Tolerance Therapeutics, Inc. | Methods and materials for modulation of the immunosuppresive activity and toxicity of monoclonal antibodies |
| WO1996020698A2 (en) | 1995-01-05 | 1996-07-11 | The Board Of Regents Acting For And On Behalf Of The University Of Michigan | Surface-modified nanoparticles and method of making and using same |
| US5916597A (en) | 1995-08-31 | 1999-06-29 | Alkermes Controlled Therapeutics, Inc. | Composition and method using solid-phase particles for sustained in vivo release of a biologically active agent |
| US5989463A (en) | 1997-09-24 | 1999-11-23 | Alkermes Controlled Therapeutics, Inc. | Methods for fabricating polymer-based controlled release devices |
| WO1999015154A1 (en) | 1997-09-24 | 1999-04-01 | Alkermes Controlled Therapeutics, Inc. | Methods for fabricating polymer-based controlled release preparations |
| WO1999020253A1 (en) | 1997-10-23 | 1999-04-29 | Bioglan Therapeutics Ab | Encapsulation method |
| US20040038867A1 (en) | 2002-06-13 | 2004-02-26 | Still James Gordon | Methods of reducing hypoglycemic episodes in the treatment of diabetes mellitus |
| US8663634B2 (en) | 2005-07-11 | 2014-03-04 | Macrogenics, Inc. | Methods for the treatment of autoimmune disorders using immunosuppressive monoclonal antibodies with reduced toxicity |
| US9056906B2 (en) | 2006-06-14 | 2015-06-16 | Macrogenics, Inc. | Methods for the treatment of autoimmune disorders using immunosuppressive monoclonal antibodies with reduced toxicity |
| WO2022251253A1 (en) * | 2021-05-24 | 2022-12-01 | Provention Bio, Inc. | Methods for treating type 1 diabetes |
| WO2023039295A1 (en) * | 2021-09-13 | 2023-03-16 | Provention Bio, Inc. | Methods and compositions comprising anti-cd3 antibodies and dyrk1a inhibitors for treating diabetes |
| WO2023230476A1 (en) * | 2022-05-24 | 2023-11-30 | Provention Bio, Inc. | Methods and compositions for preventing or delaying type 1 diabetes |
| WO2024182767A1 (en) * | 2023-03-01 | 2024-09-06 | Provention Bio, Inc. | Methods and compositions for treating type 1 diabetes comprising teplizumab and verapamil |
| WO2024206739A1 (en) * | 2023-03-30 | 2024-10-03 | Provention Bio, Inc. | Methods for reducing exogenous insulin use |
Non-Patent Citations (108)
| Title |
|---|
| "Children and Adolescents: Standards of medical Care in Diabetes-2018", DIABETES CARE, vol. 41, 2018, pages S126 - S136 |
| "Controlled Drug Bioavailability, Drug Product Design and Performance", 1984, WILEY |
| ABDUL-RASOUL MHABIB HAL-KHOULY M: "The honeymoon phase'' in children with type 1 diabetes mellitus: frequency, duration, and influential factors", PEDIATR DIABETES., vol. 7, no. 2, 2006, pages 101 - 107 |
| ABLAMUNITS VBISIKIRSKA BHEROLD KC: "Acquisition of regulatory function by human CD8+ T cells treated with anti-CD3 antibody requires TNF", EUR J IMMUNOL., vol. 40, no. 10, 2010, pages 2891 - 2901, XP055700969, DOI: 10.1002/eji.201040485 |
| AGIOSTRATIDOU GANHALT HBALL D ET AL.: "Standardizing clinically meaningful outcome measures beyond HbAlc for type 1 diabetes: a consensus report of the American Association of Clinical Endocrinologists", DIABETES CARE, vol. 40, no. 12, 2017, pages 1622 - 1630 |
| AKIRAV EMKUSHNER JAHEROLD KC: "3 cell mass and type 1 diabetes: Going, going, gone?", DIABETES, vol. 57, 2008, pages 2883 - 2888 |
| AMBERY ET AL., DIABET MED., vol. 31, 2014, pages 399 - 402 |
| ATKINSON MA: "Type 1 diabetes", LANCET, vol. 383, no. 9911, 2014, pages 69 - 82 |
| BATTELINO TALEXANDER CMAMIEL SA ET AL.: "Continuous glucose monitoring and metrics for clinical trials: an international consensus statement", THE LANCET DIABETES & ENDOCRINOLOGY, vol. 11, 2023, pages 42 - 57 |
| BINGLEY ET AL., DIABETES, vol. 45, 1996, pages 1720 - 8 |
| BISIKIRSKA BCOLGAN JLUBAN JBLUESTONE, JAHEROLD KC: "TCR stimulation with modified anti-CD3 mAb expands CD8+ T cell population and induces CD8+CD25+ Tregs", J CLIN INVEST., vol. 115, no. 10, 2005, pages 2904 - 2913, XP002559116, DOI: 10.1172/JCI23961 |
| BLUESTONE JAHEROLD KEISENBARTH G: "Genetics, pathogenesis and clinical interventions in type 1 diabetes", NATURE, vol. 464, no. 7293, 2010, pages 1293 - 1300, XP037202955, DOI: 10.1038/nature08933 |
| BOLT ET AL., EUR J IMMUNOL., vol. 23, no. 2, 1993, pages 403 - 11 |
| BOYLE KDKEYES-ELSTEIN LEHLERS MR ET AL.: "Two- and four-hour tests differ in capture of C-peptide responses to a mixed meal in type 1 diabetes", DIABETES CARE, vol. 39, 2016, pages e76 - 78 |
| BRADLEY CLOEWENTHAL KWOODCOCK A ET AL.: "Development of the diabetes treatment satisfaction questionnaire (DTSQ) for teenagers and parents: the DTSQ-Teen and the DTSQ-Parent", DIABETOLOGIA, vol. 52, 2009, pages S397 |
| CAMPBELL-THOMPSON MFU AKADDIS JS ET AL.: "Insulitis and β-cell mass in the natural history of type 1 diabetes", DIABETES, vol. 65, 2016, pages 719 - 731 |
| CLEEK ET AL., PRO INT'L SYMP CONTROL REL BIOACT MATER., vol. 24, 1997, pages 853 - 4 |
| DAIFOTIS ET AL., CLINICAL IMMUNOLOGY, vol. 149, 2013, pages 268 - 78 |
| DANNE TNIMRI RBATTELINO T ET AL.: "International consensus on use of continuous glucose monitoring", DIABETES CARE, vol. 40, 2017, pages 1631 - 1640 |
| DIABETES CONTROL AND COMPLICATIONS TRIAL RESEARCH GROUP: "Hypoglycemia in the Diabetes Control and Complications Trial", DIABETES, vol. 46, no. 2, 1997, pages 271 - 286 |
| DIABETES PREVENTION TRIAL-TYPE 1 DIABETES STUDY GROUP: "Effects of insulin in relatives of patients with type 1 diabetes mellitus", N ENGL J MED, vol. 346, 2002, pages 1685 - 91 |
| DIMEGLIO LAACERINI CLCODNER E ET AL.: "Clinical Practice Consensus Guidelines 2018: Glycemic control targets and glucose monitoring for children, adolescents, and young adults with diabetes", PEDIATR DIABETES, vol. 19, October 2018 (2018-10-01), pages 105 - 114 |
| DRISCOLL KARAYMOND JNARANJO D ET AL.: "Fear of hypoglycemia in children and adolescents and their parents with type 1 diabetes", CURR DIAB REP, vol. 16, no. 8, 2016, pages 77, XP036009386, DOI: 10.1007/s11892-016-0762-2 |
| DURING ET AL., ANN NEUROL., vol. 25, 1989, pages 351 |
| EUROPEAN MEDICINES AGENCY, ETHICAL CONSIDERATIONS FOR CLINICAL TRIALS ON MEDICINAL PRODUCTS CONDUCTED WITH THE PAEDIATRIC POPULATION, 27 September 2018 (2018-09-27), Retrieved from the Internet <URL:https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-10/ethical_considerations_en.pdf.> |
| FEUTREN GPAPOZ LASSAN R ET AL.: "Cyclosporin increases the rate and length of remissions in insulin-dependent diabetes of recent onset. Results of a multicentre double-blind trial", LANCET, vol. 2, 1986, pages 119 - 24 |
| FONOLLEDA MMURILLO MVAZQUEZ F ET AL.: "Remission phase in paediatric type 1 diabetes: new understanding and emerging biomarkers", HORM RES PAEDIATR, vol. 88, no. 5, 2017, pages 307 - 315 |
| GITELMAN SEGOTTLIEB PARIGBY MR ET AL.: "Antithymocyte globulin treatment for patients with recent-onset type 1 diabetes: 12-month results of a randomised, placebo-controlled, phase 2 trial", LANCET DIABETES ENDOCRINOL, vol. 1, no. 4, 2013, pages 306 - 16 |
| GONDER-FREDERICK LNYER MSHEPARD JA: "Assessing fear of hypoglycemia in children with Type 1 diabetes and their parents", DIABETES MANAG, vol. 1, no. 6, 2011, pages 627 - 639 |
| GOODSON: "Medical Applications of Controlled Release", vol. 2, 1984, CRC PRES., pages: 115 - 138 |
| GREENBAUM CJBEAM CABOULWARE D ET AL.: "Fall in C peptide during first 2 years from diagnosis: evidence of at least two distinct phases from composite type 1 diabetes TrialNet data", DIABETES, vol. 61, 2012, pages 2066 - 2073 |
| GREENBAUM ET AL., DIABETES, vol. 50, 2001, pages 470 - 476 |
| GUGLIELMI ET AL., EXPERT OPINION ON BIOLOGICAL THERAPY, vol. 16, no. 6, 2016, pages 841 - 6 |
| HAGOPIAN WFERRY RJ JRSHERRY N ET AL.: "Protégé Trial Investigators. Teplizumab preserves C-peptide in recent-onset type 1 diabetes: two-year results from the randomized, placebo-controlled Protégé trial", DIABETES, vol. 62, no. 11, 25 June 2013 (2013-06-25), pages 3901 - 8 |
| HELMINEN OPOKKA TTOSSAVAINEN P ET AL.: "Continuous glucose monitoring and HbAlc in the evaluation of glucose metabolism in children at high risk for type 1 diabetes mellitus", DIABETES RES CLIN PRACT., vol. 120, 2016, pages 89 - 96, XP029748953, DOI: 10.1016/j.diabres.2016.07.027 |
| HEROLD KCBUNDY BNLONG SA ET AL.: "An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes", N ENGL J MED, 9 June 2019 (2019-06-09) |
| HEROLD KCGITELMAN SEEHLERS MR ET AL.: "Teplizumab (anti-CD3 mAb) treatment preserves C-peptide responses in patients with new-onset type 1 diabetes in a randomized controlled trial: metabolic and immunologic features at baseline identify a subgroup of responders", DIABETES, vol. 62, no. 11, 2013, pages 3766 - 3774 |
| HEROLD KCGITELMAN SEUMEST M ET AL.: "A single course of anti-CD3 monoclonal antibody hOKT3 1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes", DIABETES, vol. 54, 2005, pages 1763 - 7 |
| HEROLD KCGITELMAN SEWILLI SM ET AL.: "Teplizumab treatment may improve C-peptide responses in participants with type 1 diabetes after the new-onset period: a randomised controlled trial", DIABETOLOGIA, vol. 56, no. 2, 2013, pages 391 - 400, XP035158754, DOI: 10.1007/s00125-012-2753-4 |
| HEROLD KCHAGOPIAN WAUGER JAPOUMIAN-RUIZ ETAYLOR LDONALDSON DGITELMAN SEHARLAN DMXU DZIVIN RA: "Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus", N ENGL J MED., vol. 346, no. 22, 30 May 2002 (2002-05-30), pages 1692 - 1698, XP002255982, DOI: 10.1056/NEJMoa012864 |
| HEROLD KEVAN C. ET AL: "Teplizumab: A Disease-Modifying Therapy for Type 1 Diabetes That Preserves [beta]-Cell Function", DIABETES CARE, vol. 46, no. 10, 22 August 2023 (2023-08-22), US, pages 1848 - 1856, XP093219421, ISSN: 0149-5992, Retrieved from the Internet <URL:https://diabetesjournals.org/care/article-pdf/46/10/1848/734787/dc230675.pdf> DOI: 10.2337/dc23-0675 * |
| HILLIARD MELAWRENCE JMMODI AC ET AL.: "Identification of minimal clinically important difference scores of the PedsQL in children, adolescents, and young adults with type 1 and type 2 diabetes", DIABETES CARE, vol. 36, 2013, pages 1891 - 7 |
| HOWARD ET AL., J NEUROSURG., vol. 71, 1989, pages 105 |
| HUO LHARDING JLPEETERS A ET AL.: "Life expectancy of type 1 diabetic patients during 1997-2010: a national Australian registry-based cohort study", DIABETOLOGIA, vol. 59, no. 6, 2016, pages 1177 - 85, XP035804527, DOI: 10.1007/s00125-015-3857-4 |
| INTERNATIONAL HYPOGLYCAEMIA STUDY GROUP: "Glucose Concentrations of Less Than 3.0 mmol/L (54 mg/dL) Should Be Reported in Clinical Trials: A Joint Position Statement of the American Diabetes Association and the European Association for the Study of Diabetes", DIABETES CARE, 2016, pages dc162215 |
| JOINT COMMISSION: "High Alert Medications and Patient Safety", SENTINEL EVENT ALERT, 19 November 1999 (1999-11-19), Retrieved from the Internet <URL:https://www.jointcommission.org/assets/1/18/SEA_11.pdf> |
| KARVONEN MVIIK-KAJANDER MMOLTCHANOVA E ET AL.: "Incidence of childhood type 1 diabetes worldwide. Diabetes Mondiale (DiaMond) Project Group", DIABETES CARE, vol. 23, no. 10, 2000, pages 1516 - 1526 |
| KEYMEULEN ET AL., BLOOD, vol. 115, no. 6, 2010, pages 1145 - 55 |
| KEYMEULEN ET AL., DIABETOLOGIA., vol. 53, 2010, pages 614 - 23 |
| KEYMEULEN ET AL., N ENGL J MED., vol. 352, 2005, pages 2598 - 2608 |
| KEYMEULEN ET AL., N ENGLJ MED., vol. 352, no. 25, 2005, pages 2598 - 608 |
| KUHTREIBER WMWASHER SLHSU E ET AL.: "Low levels of C-peptide have clinical significance for established Type 1 diabetes", DIABET MED, vol. 32, no. 10, October 2015 (2015-10-01), pages 1346 - 53, XP071687763, DOI: 10.1111/dme.12850 |
| LACHIN JMMCGEE PPALMER JP: "Impact of C-peptide preservation on metabolic and clinical outcomes in the Diabetes Control and Complications Trial", DIABETES, vol. 63, no. 2, 2014, pages 739 - 748 |
| LAITINEN OHHONKANEN HPAKKANEN O ET AL.: "Coxsackievirus B1 is associated with induction of β-cell autoimmunity that portends type 1 diabetes", DIABETES, vol. 63, no. 2, 2014, pages 446 - 455 |
| LAM ET AL., PROC INT'L SYMP CONTROL REL BIOACT MATER., vol. 24, 1997, pages 759 - 60 |
| LANGER, SCIENCE, vol. 249, 1990, pages 1527 - 1533 |
| LEBASTCHI JDENG SLEBASTCHI AH ET AL.: "Immune therapy and β-cell death in type 1 diabetes", DIABETES, vol. 62, no. 5, 2013, pages 1676 - 1680, XP055197604, DOI: 10.2337/db12-1207 |
| LEVY ET AL., SCIENCE, vol. 228, 1985, pages 190 |
| LIN ANORTHAM EAWERTHER GACAMERON FJ: "Risk factors for decline in IQ in youth with type 1 diabetes over the 12 years from diagnosis/illness onset", DIABETES CARE, vol. 38, 2015, pages 236 - 242 |
| LIND MSVENSSON AMKOSIBOROD M ET AL.: "Glycemic control and excess mortality in type 1 diabetes", N ENGL J MED., vol. 371, 2014, pages 1972 - 1982 |
| LONG SATHORPE JDEBERG HA ET AL.: "Partial exhaustion of CD8 T cells and clinical response to teplizumab in new-onset type 1 diabetes", SCI. IMMUNOL, vol. 1, no. 5, 2016, pages 1 - 23, XP055760444, DOI: 10.1126/sciimmunol.aai7793 |
| LONG SATHORPE JHEROLD KC ET AL.: "Remodeling T cell compartments during anti-CD3 immunotherapy of type 1 diabetes", CELL IMMUNOL., vol. 319, September 2017 (2017-09-01), pages 3 - 9, XP085189944, DOI: 10.1016/j.cellimm.2017.07.007 |
| LUDVIGSSON JCARLSSON ADELI A ET AL.: "Decline of C-peptide during the first year after diagnosis of Type 1 diabetes in children and adolescents", DIABETES RES CLIN PRACT, vol. 100, no. 2, 2013, pages 203 - 209 |
| MASHARANI UBBECKER J: "Teplizumab therapy for type 1 diabetes", EXPERT OPIN BIOL THER, vol. 10, no. 3, 2010, pages 459 - 465, XP093110493, DOI: 10.1517/14712591003598843 |
| MATVEYNEKO AVBUTLER PC: "Relationship between (3 cell mass and diabetes onset", DIABETES OBES METAB, vol. 10, 2008, pages 23 - 31, XP072208533, DOI: 10.1111/j.1463-1326.2008.00939.x |
| MAYFIELD ET AL., AM FAM PHYSICIAN, vol. 58, 2006, pages 1355 - 62 |
| MCCULLOCH ET AL., DIABETES CARE, vol. 16, 1993, pages 911 - 5 |
| MITTERMAYER FCAVENEY EDE OLIVEIRA C ET AL.: "Addressing Unmet Medical Needs in Type 1 Diabetes: A Review of Drugs Under Development", CURR DIABETES REV, vol. 13, no. 3, 2017, pages 300 - 314 |
| MONAGHAN MHELGESON VWIEBE D: "Type 1 diabetes in young adulthood", CURR DIABETES REV, vol. 11, no. 4, 2015, pages 239 - 250 |
| MORTENSEN HBHOUGAARD PSWIFT P ET AL.: "New definition for the partial remission period in children and adolescents with type 1 diabetes", DIABETES CARE, vol. 32, 2009, pages 1384 - 1390, XP055095828, DOI: 10.2337/dc08-1987 |
| N ENGL J MED., vol. 329, 1993, pages 977 - 86 |
| NATIONAL CANCER INSTITUTE, COMMON TERMINOLOGY CRITERIA FOR ADVERSE EVENTS (CTCAE, 1 March 2018 (2018-03-01), Retrieved from the Internet <URL:https:Hctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm.> |
| NING ET AL., RADIOTHERAPY & ONCOLOGY, vol. 39, 1996, pages 179 - 189 |
| ORBAN TBUNDY BBECKER DJ ET AL.: "Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial", LANCET, vol. 378, no. 9789, 2011, pages 412 - 419, XP055242611, DOI: 10.1016/S0140-6736(11)60886-6 |
| PALMER JP: "C-peptide in the natural history of type 1 diabetes", DIABETES METAB RES REV, vol. 25, no. 4, 2009, pages 325 - 328 |
| PALMER JPFLEMING GAGREENBAUM CJ ET AL.: "C-peptide is the appropriate outcome measure for type 1 diabetes clinical trials to preserve beta-cell function: report of an ADA workshop, 21-22 October 2001", DIABETES, vol. 53, no. 1, 2004, pages 250 - 264 |
| RAMOS ELEANOR L. ET AL: "Teplizumab and [beta]-Cell Function in Newly Diagnosed Type 1 Diabetes", THE NEW ENGLAND JOURNAL OF MEDICINE, vol. 389, no. 23, 18 October 2023 (2023-10-18), US, pages 2151 - 2161, XP093219063, ISSN: 0028-4793, DOI: 10.1056/NEJMoa2308743 * |
| RANGERPEPPAS, J MACROMOLSCI REV MACROMOL CHEM., vol. 23, 1983, pages 61 |
| RAWSHANI ARAWSHANI AFRANZÉN S ET AL.: "Mortality and Cardiovascular Disease in Type 1 and Type 2 Diabetes", N ENGL J MED, vol. 376, no. 15, 2017, pages 1407 - 1418 |
| RAWSHANI ASATTAR NFRANZÉN S ET AL.: "Excess mortality and cardiovascular disease in young adults with type 1 diabetes in relation to age at onset: a nationwide, register-based cohort study", LANCET, vol. 392, no. 10146, 2018, pages 477 - 486, XP085439431, DOI: 10.1016/S0140-6736(18)31506-X |
| RIGBY MRDIMEGLIO LARENDELL MS ET AL.: "Targeting of memory T cells with alefacept in new-onset type 1diabetes (TIDAL study): 12-month results of a randomised, double-blind, placebo-controlled phase 2 trial", LANCET DIABETES ENDOCRINOL, vol. 1, no. 4, 2013, pages 284 - 294 |
| RIGBY MREHLERS MR: "Targeted Immune Interventions for Type 1 Diabetes: Not as Easy as it Looks!", CURR OPIN ENDOCRINOL DIABETES OBES, vol. 21, no. 4, 2014, pages 271 - 278 |
| RIGBY MRHARRIS KMPINCKNEY A ET AL.: "Alefacept provides sustained clinical and immunological effects in new-onset type 1 diabetes patients", J CLIN INVEST, vol. 125, no. 8, 2015, pages 3285 - 3296 |
| ROARK CLANDERSON KMSIMON LJ ET AL.: "Multiple HLA epitopes contribute to type 1 diabetes susceptibility", DIABETES, vol. 63, no. 1, 2014, pages 323 - 331 |
| ROSENZWEIG MROSENTHAL CATORRES VMVAICKUS L: "Development of a quantitative assay to measure EBV viral load in patients with autoimmune type 1 diabetes and healthy subjects", J VIROL METHODS, vol. 164, no. 1-2, 2010, pages 111 - 115, XP026925469, DOI: 10.1016/j.jviromet.2009.11.018 |
| SANDBORN ET AL., GUT, vol. 59, no. 11, 2010, pages 1485 - 92 |
| SAUDEK ET AL., N ENGL J MED., vol. 321, 1989, pages 574 - 365 |
| SEAQUIST ERANDERSON JCHILDS B ET AL.: "Hypoglycemia and diabetes: a report of a workgroup of the American Diabetes Association and the Endocrine Society", DIABETES CARE, vol. 36, no. 5, 2013, pages 1384 - 1395 |
| SECREST AMBECKER DJKELSEY SFLAPORTE REORCHARD TJ: "Characterising sudden death and dead-in-bed syndrome in Type 1 diabetes: analysis from 2 childhood-onset Type 1 diabetes registries", DIABET MED, vol. 28, no. 3, 2011, pages 293 - 300 |
| SEFTON: "CRC Crit. Ref. Biomed. Eng.", vol. 14, 1987, pages: 20 |
| SHERRY NHAGOPIAN WLUDVIGSSON J ET AL.: "Protégé Trial Investigators. Teplizumab for treatment of type 1 diabetes (Protégé study): 1-year results from a randomised, placebo-controlled trial", LANCET, vol. 378, no. 9790, 2011, pages 487 - 97, XP055095801, DOI: 10.1016/S0140-6736(11)60931-8 |
| SONG ET AL., PDA JOURNAL OF PHARMACEUTICALSCIENCE & TECHNOLOGY, vol. 50, 1995, pages 372 - 97 |
| SORENSEN JSJOHANNESEN JPOCIOT F ET AL.: "Residual β-cell function 3-6 years after onset of type 1 diabetes reduces risk of severe hypoglycemia in children and adolescents", DIABETES CARE, vol. 36, 2013, pages 3454 - 3459 |
| SOSENKO JMSKYLER JSHEROLD KC ET AL.: "The metabolic progression to type 1 diabetes as indicated by serial oral glucose tolerance testing in the Diabetes Prevention Trial-type 1", DIABETES, vol. 61, no. 6, 2012, pages 1331 - 7 |
| SPRANGERS ET AL., IMMUNOTHERAPY, vol. 3, no. 11, 2011, pages 1303 - 16 |
| STECK AKDONG FTAKI I ET AL.: "Early hyperglycemia detected by continuous glucose monitoring in children at risk for type 1 diabetes", DIABETES CARE, vol. 37, 2014, pages 2031 - 2033 |
| STEFFES MWSIBLEY SJACKSON MTHOMAS W: "Beta-cell function and the development of diabetes-related complications in diabetes control and complications trial", DIABETES CARE, vol. 26, 2003, pages 832 - 836 |
| STREISAND R: "Young children with type 1 diabetes: challenges, research, and future directions", CURR DIAB REP, vol. 14, no. 9, 2014, pages 1 - 16, XP035379828, DOI: 10.1007/s11892-014-0520-2 |
| TRANCONE ABONFANTI RIAFUSCO D ET AL.: "Evaluating the experience of children with type 1 diabetes and their parents taking part in an artificial pancreas clinical trial over multiple days in a diabetes camp setting", DIABETES CARE, vol. 39, 2016, pages 2158 - 2164 |
| TSAI ET AL., ACS CENTRAL SCIENCE, vol. 2, no. 3, 2016, pages 139 - 47 |
| VARNI JWDELAMATER AMHOOD KK ET AL.: "PedsQL 3.2 Diabetes Module for children, adolescents, and young adults: reliability and validity in type 1 diabetes", DIABETES CARE, 2018, pages dc172707 |
| VERKRUYSE LASTORCH GADEVINE SM ET AL.: "Once daily ganciclovir as initial pre-emptive therapy delayed until threshold CMV load > or =10000 copies/ml: a safe and effective strategy for allogeneic stem cell transplant", BONE MARROW TRANSPLANT, vol. 37, no. 1, 2006, pages 51 - 6, XP037755930, DOI: 10.1038/sj.bmt.1705213 |
| VLASAKAKIS ET AL., BR J CLIN PHARMACOL., vol. 85, 2019, pages 704 - 714 |
| WALDRON-LYNCH FHENEGARIU ODENG S ET AL.: "Teplizumab induces human gut-tropic regulatory cells in humanized mice and patients", SCI TRANSL MED, vol. 4, no. 118, 2012, pages 118 - 12 |
| WHERRETT DKCHIANG JLDELAMATER AM ET AL.: "Defining pathways for development of disease-modifying therapies in children with type 1 diabetes: a consensus report", DIABETES CARE., vol. 38, no. 10, 2015, pages 1975 - 85 |
| WICZLING ET AL., J CLIN PHARMACOL., vol. 50, no. 5, 2010, pages 494 - 506 |
| YU ET AL., J CLIN ENDOCRINOL METAB., vol. 81, 1996, pages 4264 - 7 |
| ZIEGLER AGNEPOM GT: "Prediction and pathogenesis in type 1 diabetes", IMMUNITY, vol. 32, 2010, pages 468 - 478 |
Also Published As
| Publication number | Publication date |
|---|---|
| TW202521153A (en) | 2025-06-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20220380465A1 (en) | Methods for treating type 1 diabetes | |
| Herold et al. | An anti-CD3 antibody, teplizumab, in relatives at risk for type 1 diabetes | |
| US20250304688A1 (en) | Methods and Compositions Comprising Anti-CD3 Antibodies and DYRK1A Inhibitors for Treating Diabetes | |
| US20220372146A1 (en) | Methods for treating post infectious autoimmune diabetes | |
| Pirenne et al. | How to avoid the problem of erythrocyte alloimmunization in sickle cell disease | |
| US20230013752A1 (en) | Methods for delaying onset of type 1 diabetes | |
| WO2024182767A1 (en) | Methods and compositions for treating type 1 diabetes comprising teplizumab and verapamil | |
| IL323543A (en) | Methods for reducing exogenous insulin use | |
| Floch et al. | Eleven years of alloimmunization in 6496 patients with sickle cell disease in France who received transfusion | |
| WO2025024848A1 (en) | Methods for treating type 1 diabetes | |
| AU2023275531C1 (en) | Methods and compositions for preventing or delaying type 1 diabetes | |
| US20230382993A1 (en) | Methods and compositions for preventing or delaying type 1 diabetes | |
| WO2024079046A1 (en) | Anti-cd2 antibodies for type 1 diabetes | |
| Mathieu et al. | Toward Disease-modifying therapies in type 1 diabetes: focus on Teplizumab | |
| CN117940162A (en) | Methods and compositions comprising anti-CD 3 antibodies and DYRK1A inhibitors for the treatment of diabetes | |
| CN118591558A (en) | Methods and compositions for preventing or delaying type 1 diabetes |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24758100 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 326147 Country of ref document: IL |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112026001868 Country of ref document: BR |