WO2025021997A1 - New map4k1 inhibitors - Google Patents
New map4k1 inhibitors Download PDFInfo
- Publication number
- WO2025021997A1 WO2025021997A1 PCT/EP2024/071341 EP2024071341W WO2025021997A1 WO 2025021997 A1 WO2025021997 A1 WO 2025021997A1 EP 2024071341 W EP2024071341 W EP 2024071341W WO 2025021997 A1 WO2025021997 A1 WO 2025021997A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- mixture
- salt
- hydrate
- solvate
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains three hetero rings
- C07D487/20—Spiro-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Definitions
- the present invention covers MAP4K1 inhibitor compounds of formula (I) as described and defined herein, methods of preparing said compounds, intermediate compounds useful for preparing said compounds, pharmaceutical compositions and combinations comprising said compounds, and the use of said compounds for manufacturing pharmaceutical compositions for the treatment or prophylaxis of diseases, in particular for treatment, amelioration or prevention of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, or amelioration of vaccine therapies or cell therapies, as a sole agent or in combination with other active ingredients.
- the present invention further relates to the use, respectively to the use of said compounds for manufacturing pharmaceutical compositions for the treatment or prophylaxis of protein inhibitors in benign hyperplasias, atherosclerotic disorders, sepsis, autoimmune disorders, vascular disorders, viral infections, in neurodegenerative disorders, in inflammatory disorders, in atherosclerotic disorders and in male fertility control.
- T-cell immune checkpoint such as CTLA-4, PD-1 or PD-L1 were recently shown to result in a remarkable clinical efficacy in subsets of cancer patients.
- cell surface receptors that act as negative immune regulators, several mediators of intracellular signaling have been identified that also represent potential immunoevasive mechanisms utilized by the tumor.
- MAP4K1 also known as hematopoietic progenitor kinase 1 (HPK1).
- HPK1 hematopoietic progenitor kinase 1
- MAP4K1 (GenelD11184) is a serine/threonine kinase and member of the Germinal Center Kinase family. In the adult organism MAP4K1 expression is restricted to hematopoietic cell types.
- the MAP4K1 protein consist of a N-terminal kinase domain, followed by a proline-rich domain that can interact with adaptor molecules through SH2 and SH3 domains, and a C-terminal citron homology domain of which the exact function remains to be identified.
- MAP4K1 is capable of binding to a diversity of adaptors in hematopoietic cells, including those involved in T-cell receptor (TCR), B-cell receptor (BCR) and cytokine signaling (Hu et al., Genes Dev. 1996 Sep 15;10(18):2251-64, 2.; Ling et al.,. J Biol Chem. 2001 Jun 1;276(22), Sauer et al., J Biol Chem. 2001 Nov 30;276(48):45207-16., Tsuji et al., J Exp Med. 2001 Aug 20;194(4):529-39, Boomer et al., J Cell Biochem. 2005 May 1 ;95(1):34-44).
- TCR T-cell receptor
- BCR B-cell receptor
- cytokine signaling Hu et al., Genes Dev. 1996 Sep 15;10(18):2251-64, 2.; Ling et al.,. J Biol Chem. 2001
- MAP4K1 The function of MAP4K1 has been studied in greatest detail in the context of TCR signaling.
- MAP4K1 Upon TCR stimulation, MAP4K1 is phosphorylated on tyrosine 381 (Y-381; Y-379 in mouse) (Di Bartolo et al., J Exp Med. 2007 Mar 19;204(3):681-91). Consequently, MAP4K1 is recruited to the TCR-signaling complex where it induces dissociation of this complex through its serine/threonine kinase function.
- MAP4K1 phosphorylates the SLP-76 adaptor protein at Serine-376, resulting in downregulation of AP-1 and Erk2 pathways.
- MAPK1 acts as a negative feedback on TCR-signaling (Liou et al., Immunity. 2000 Apr;12(4):399-408; Lasserre et al., J Cell Biol. 2011 Nov 28;195(5):839-53.).
- MAP4K1 can be triggered to suppress T cell function by prostaglandin E2 (PGE2), and possibly also by transforming growth factor beta (TGF-beta), factors that are commonly found in the tumor microenvironment.
- PGE2 prostaglandin E2
- TGF-beta transforming growth factor beta
- MAP4K1 activation by these mediators involves protein kinase A (PKA)-dependent phosphorylation of Serine 171 (S-171; also in mouse) (Alzabin et al., Cancer Immunol Immunother. 2010 Mar;59(3):419-29; Sawasdikosol et al., J Biol Chem. 2007 Nov 30;282(48):34693-9.).
- PKA protein kinase A
- MAP4K1 -deficient mice show an apparent normal phenotype, are fertile and exhibit normal lymphocyte development. These animals are prone to develop T-cell dependent autoimmune reactivity as indicated by development of a more severe disease score in the EAE (experimental autoimmune encephalomyelitis) model of multiple sclerosis (Shui et al., Nat Immunol. 2007 Jan;8(1):84-91). In case of the second strain, a dysregulation of immune function was observed when, at the age of approximately 6 months, MAP4K1 -deficient mice develop a spontaneous autoimmune phenotype (Alzabin et al., Cancer Immunol Immunother. 2010 Mar;59(3):419-29).
- MAP4K1-/- T-cells display hyperresponsiveness upon TCR-stimulation. These cells proliferate and secrete pro-inflammatory cytokines like IL-2 or IFNg to a significantly greater extent than their wild-type counterparts (Shui et al., Nat Immunol. 2007 Jan;8(1):84-91). Furthermore, MAP4K1-/- T-cells are resistant to PGE2-mediated suppression of T cell proliferation, suppression of IL-2 production and induction of apoptosis (Alzabin et al., Cancer Immunol Immunother. 2010 Mar;59(3):419-29).
- MAP4K1-/- mice are much more resistant to tumorigenesis by PGE2-producing Lewis lung carcinoma than wild type mice, which correlated with increased T-lymphocyte infiltration in the tumor areas.
- the crucial role of T-cells in tumor rejection was supported by experiments in which MAP4K1-/- T-cells adoptively transferred into T-cell-deficient mice were able to eradicate tumors more efficiently than wild-type T-cells (Alzabin et al., Cancer Immunol Immunother. 2010 Mar;59(3):419-29).
- MAP4K1 also regulates the stimulation and activation of dendritic cells. MAP4K1 deficient
- BMDC Bone marrow derived cells
- HPK1 HPK1 and MAP4K1 are referring to the same human protein
- Table (1) exemplifies state of the art disclosing HPK1 inhibitors with publication date between January 2021 to January 2023.
- HPK1 inhibitors see e.g. I.D. Linney, K. Neelu, Expert Opinion on Therapeutic Patents, 2021 , 31 , 893-910.
- HPK1 inhibitors see for example L. Zhou et al., Eur. J. Med. Chem. 2022, 244, 114819; see also Q. Zhu et al., J. Med. Chem. 2022, 65, 8065-8090.
- HPK1 (MAP4K1) inhibitors based on (2,2-difluorocyclopropyl)(1 ,2,4-triazolyl)methanamine substructures S-1 or S-2 or S-3 as present in the compounds of general formula (I) of the present invention have not been exemplified in the prior art.
- HPK1 (MAP4K1) inhibition activity HPK1 (MAP4K1) inhibition activity.
- WO2022147246 to have FGFR tyrosine kinase inhibitory activity.
- these compounds contain the (1 H-pyrazol-3yl)pyridine substructure S-4, according to general formula L-9 the pyrazole ring can only be a part of an indazole ring system.
- compounds of general formula L-9 have not been reported to have HPK1 (MAP4K1) inhibition activity.
- X can be a bond and Y can represent a hydrogen.
- R 1 can be a group *-A-B, where B can represent a 3-membered cycloalkyl, optionally substituted with halogen atoms, and where A can represent *-CR a H- group, where R a can be a 5-membered heteroaryl.
- the substituent R 1 together with the nitrogen to which it is attached, can represent (2,2- difluorocyclopropyl)(1 ,2,4-triazolyl)methanamine substructure (S-1 , S-2 or S-3).
- R 2 can only be a bicyclic heteroaromatic structure limited to quinoline, 1 ,5- or 1 ,6-naphthyridine, 1 H-pyrrolo[2,3- b]pyridine, 1 H-pyrazolo[3,4-b]pyridine or 3H-imidazo[4,5-b]pyridine.
- R 2 can not mean pyridine as present in the formula (I) of the present invention.
- R 4 can be Ci-3-alkyl-C3-6-cycloalkyl, optionally substituted with one or more fluorine atoms and independently optionally substituted with 5-membered heteroaryl.
- the substituent R 4 together with the nitrogen to which it is attached can form a (2,2-difluorocyclopropyl)(1 ,2,4-triazolyl)methanamine substructure (S-1 , S- 2 or S-3).
- compounds containing such a substructure are not exemplified in WO2022167627 and are not known from any prior art.
- the compound of general formula (I) of the present invention containing the unique (2,2-difluorocyclopropyl)(1 ,2,4-triazolyl)methanamine substructure (S-1 , S-2 or S-3), is shown to have multiple unexpected advantages over compounds described in the prior art. Such advantageous properties are listed below.
- the incorporation of the unique (2,2- difluorocyclopropyl)(1 ,2,4-triazolyl)methanamine substructure (S-1 , S-2 or S-3) to achieve such advantageous properties is a modification that is not obvious to the person skilled in the art.
- a further object of the present invention is to provide a compound and pharmaceutical compositions comprising this compound for prophylactic and therapeutic applications for hyperproliferative disorders, in particular for cancer, respectively tumour disorders, and conditions with dysregulated immune responses, as a sole agent or in combination with other active ingredients.
- a further object of the present invention is to provide a compound and pharmaceutical compositions comprising this compound for manufacturing pharmaceutical compositions for the treatment or prophylaxis of benign hyperplasias, atherosclerotic disorders, sepsis, autoimmune disorders, vascular disorders, viral infections, in neurodegenerative disorders, in inflammatory disorders, in atherosclerotic disorders and in male fertility control.
- the compound according to the invention inhibits the MAP4K1 protein and thereby enhances tumor immunogenicity leading to inhibition of cancer cells growth by the immune response. Accordingly, the invention provides a new compound for the therapy of human and animal disorders, in particular of cancers.
- a high aqueous solubility (Table 4: kinetic solubility in pH 6.5 aqueous phosphate buffer). It is well known to the person skilled in the art that high aqueous solubility is required to achieve desired concentration of drug substance in systemic circulation for desired pharmacological response. It is also known to the person skilled in the art that high aqueous solubility of a drug substance improves oral absorption of the drug. It is also known to the person skilled in the art that high aqueous solubility of a drug substance simplifies formulation development of the drug. For reference see e.g. E. H. Kerns, L. Di, Chapter 7 - Solubility, Editor(s): E. H. Kerns, L.
- Kerns, L. Di Drug-like Properties: Concepts, Structure Design and Methods, Academic Press, 2008, pages 137-168; see also P. Baranczewski et. al, Introduction to in vitro estimation of metabolic stability and drug interactions of new chemical entities in drug discovery and development. Pharmacol Rep. 2006, 58 (4), 453-472.
- the compound shows an unexpected unique combination of properties making it useful to treat human diseases via inhibition of hMAP4K1. While the known structurally related examples from the prior art WO2022167627A1 can have one or a small subset of the advantageous properties described above, none of them is known to have this unique combination. For example, there is no compound described in WO2022167617A1 that is a highly selective MAP4K1 inhibitor, has high selectivity against hERG cardiac ion channel and has simultaneously a moderate in vivo clearance ( ⁇ 1.5 L/h/kg) in rat.
- heteroaryl or heteroarylene groups include all possible isomeric forms thereof, e.g.: tautomers and positional isomers with respect to the point of linkage to the rest of the molecule.
- pyridinyl includes pyridin-2-yl, pyridin-3-yl and pyridin-4-yl; or the term thienyl includes thien-2-yl and thien-3-yl.
- the term “leaving group” means an atom or a group of atoms that is displaced in a chemical reaction as stable species taking with it the bonding electrons.
- a leaving group is selected from the group comprising: halide, in particular fluoride, chloride, bromide or iodide, (methylsulfonyl)oxy, [(trifluoromethyl)sulfonyl]oxy, [(nonafl uorobutyl)- sulfonyl]oxy, (phenylsulfonyl)oxy, [(4-methylphenyl)sulfonyl]oxy, [(4-bromophenyl)sulfonyl]oxy, [(4-nitrophenyl)sulfonyl]oxy, [(2-nitrophenyl)sulfonyl]oxy, [(4-isopropylphenyl)sulfonyl]oxy, [(2,4,6-triis
- the invention therefore includes one or more isotopic variant(s) of the compounds of general formula (I), particularly deuterium-containing compounds of general formula (I).
- Isotopic variant of a compound or a reagent is defined as a compound exhibiting an unnatural proportion of one or more of the isotopes that constitute such a compound.
- Isotopic variant of the compound of general formula (I) is defined as a compound of general formula (I) exhibiting an unnatural proportion of one or more of the isotopes that constitute such a compound.
- unnatural proportion means a proportion of such isotope which is higher than its natural abundance.
- the natural abundances of isotopes to be applied in this context are described in “Isotopic Compositions of the Elements 1997”, Pure Appl. Chem., 70(1), 217-235, 1998.
- isotopes include stable and radioactive isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus, sulfur, fluorine, chlorine, bromine and iodine, such as 2 H (deuterium), 3 H (tritium), 11 C, 13 C, 14 C, 15 N, 17 0, 18 0, 32 P, 33 P, 33 S, 34 S, 35 S, 36 S, 18 F, 36 CI, 82 Br, 123 l, 124 l, 125
- isotopes include stable and radioactive isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus, sulfur, fluorine, chlorine, bromine and iodine, such as 2 H (deuterium), 3 H (tritium), 11 C, 13 C, 14 C, 15 N, 17 0, 18 0, 32 P, 33 P, 33 S, 34 S, 35 S, 36 S, 18 F, 36 CI, 82 Br, 123 l, 124 l, 125
- the isotopic variant(s) of the compounds of general formula (I) preferably contain deuterium (“deuterium- containing compounds of general formula (I)”).
- Isotopic variants of the compounds of general formula (I) in which one or more radioactive isotopes, such as 3 H or 14 C, are incorporated are useful e.g. in drug and/or substrate tissue distribution studies. These isotopes are particularly preferred for the ease of their incorporation and detectability.
- Positron emitting isotopes such as 18 F or 11 C may be incorporated into a compound of general formula (I). These isotopic variants of the compounds of general formula (I) are useful for in vivo imaging applications.
- Deuterium- containing and 13 C-containing compounds of general formula (I) can be used in mass spectrometry analyses in the context of preclinical or clinical studies.
- Isotopic variants of the compounds of general formula (I) can generally be prepared by methods known to a person skilled in the art, such as those described in the schemes and/or examples herein, by substituting a reagent for an isotopic variant of said reagent, preferably for a deuterium-containing reagent.
- a reagent for an isotopic variant of said reagent preferably for a deuterium-containing reagent.
- deuterium from D2O can be incorporated either directly into the compounds or into reagents that are useful for synthesizing such compounds.
- Deuterium gas is also a useful reagent for incorporating deuterium into molecules.
- Catalytic deuteration of olefinic bonds and acetylenic bonds is a rapid route for incorporation of deuterium.
- Metal catalysts i.e. Pd, Pt, and Rh
- Pd, Pt, and Rh metal catalysts in the presence of deuterium gas can be used to directly exchange deuterium for hydrogen in functional groups containing hydrocarbons.
- a variety of deuterated reagents and synthetic building blocks are commercially available from companies such as for example C/D/N Isotopes, Quebec, Canada; Cambridge Isotope Laboratories Inc., Andover, MA, USA; and CombiPhos Catalysts, Inc., Princeton, NJ, USA.
- deuterium-containing compound of general formula (I) is defined as a compound of general formula (I), in which one or more hydrogen atom(s) is/are replaced by one or more deuterium atom(s) and in which the abundance of deuterium at each deuterated position of the compound of general formula (I) is higher than the natural abundance of deuterium, which is about 0.015%.
- the abundance of deuterium at each deuterated position of the compound of general formula (I) is higher than 10%, 20%, 30%, 40%, 50%, 60%, 70% or 80%, preferably higher than 90%, 95%, 96% or 97%, even more preferably higher than 98% or 99% at said position(s). It is understood that the abundance of deuterium at each deuterated position is independent of the abundance of deuterium at other deuterated position(s).
- the selective incorporation of one or more deuterium atom(s) into a compound of general formula (I) may alter the physicochemical properties (such as for example acidity [C. L. Perrin, et al., J. Am. Chem. Soc., 2007, 129, 4490], basicity [C. L. Perrin et al., J. Am. Chem. Soc., 2005, 127, 9641], lipophilicity [B. Testa et al., Int. J. Pharm., 1984, 19(3), 271]) and/or the metabolic profile of the molecule and may result in changes in the ratio of parent compound to metabolites or in the amounts of metabolites formed.
- physicochemical properties such as for example acidity [C. L. Perrin, et al., J. Am. Chem. Soc., 2007, 129, 4490], basicity [C. L. Perrin et al., J. Am. Chem. Soc., 2005
- Kassahun et al., WO2012/112363 are examples for this deuterium effect. Still other cases have been reported in which reduced rates of metabolism result in an increase in exposure of the drug without changing the rate of systemic clearance (e.g. Rofecoxib: F. Schneider et al., Arzneim. Forsch. I Drug. Res., 2006, 56, 295; Telaprevir: F. Maltais et al., J. Med. Chem., 2009, 52, 7993). Deuterated drugs showing this effect may have reduced dosing requirements (e.g. lower number of doses or lower dosage to achieve the desired effect) and/or may produce lower metabolite loads.
- the compound of formula (I) may have multiple potential sites of attack for metabolism.
- deuterium-containing compounds of general formula (I) having a certain pattern of one or more deuterium-hydrogen exchange(s) can be selected.
- the deuterium atom(s) of deuterium-containing compound(s) of general formula (I) is/are attached to a carbon atom and/or is/are located at those positions of the compound of general formula (I), which are sites of attack for metabolizing enzymes such as e.g. cytochrome P450.
- the present invention concerns a deuterium-containing compound of general formula (I).
- the present invention concerns a deuterium-containing compound of general formula (I) with 1 , 2, 3, 4, 5, 6, 7, 8 or 9 deuterium atoms, particularly 1 , 2, 3, 4, 5 or 6 deuterium atoms.
- the present invention concerns a deuterium-containing compound of general formula (I) having 1 , 2, 3 or 4 deuterium atoms, particularly with 1 , 2 or 3 deuterium atoms.
- the compound of the present invention contains three asymmetric centres. It is possible that one or more asymmetric carbon atoms are present in the (R) or (S) configuration, which can result in diastereomeric mixtures of up to 8 stereoisomers in variable ratios.
- Preferred compounds are those which produce the more desirable biological activity.
- Separated, pure or partially purified isomers and stereoisomers or racemic or diastereomeric mixtures of the compounds of the present invention are also included within the scope of the present invention.
- the purification and the separation of such materials can be accomplished by standard techniques known in the art.
- the optical isomers can be obtained by resolution of the racemic mixtures according to conventional processes, for example, by the formation of diastereoisomeric salts using an optically active acid or base or formation of covalent diastereomers.
- appropriate acids are tartaric, diacetyltartaric, ditoluoyltartaric and camphorsulfonic acid.
- Mixtures of diastereoisomers can be separated into their individual diastereomers on the basis of their physical and/or chemical differences by methods known in the art, for example, by chromatography or fractional crystallisation.
- the optically active bases or acids are then liberated from the separated diastereomeric salts.
- a different process for separation of optical isomers involves the use of chiral chromatography (e.g., HPLC columns using a chiral phase), with or without conventional derivatisation, optimally chosen to maximise the separation of the enantiomers.
- Suitable HPLC columns using a chiral phase are commercially available, such as those manufactured by Daicel, e.g., Chiracel OD and Chiracel OJ, for example, among many others, which are all routinely selectable.
- Enzymatic resolution with isolated enzymes or microorganisms, with or without derivatisation are also useful.
- the optically active compounds of the present invention can likewise be obtained by chiral syntheses utilizing optically active starting materials.
- the present invention includes all possible stereoisomers of the compounds of the present invention as single stereoisomers, or as any mixture of said stereoisomers, e.g. (R)- or (S)- isomers, in any ratio.
- Isolation of a single stereoisomer, e.g. a single enantiomer or a single diastereomer, of a compound of the present invention is achieved by any suitable state of the art method, such as chromatography, especially chiral chromatography, for example.
- any compound of the present invention which contains triazole as a heteroaryl group for example can exist as a 1 H tautomer, 2H-tautomer or a 4H tautomer, or even a mixture in any amount of the three tautomers, namely:
- the present invention includes all possible tautomers of the compounds of the present invention as single tautomers, or as any mixture of said tautomers, in any ratio.
- the chemical structure of compond of formula (I) described herein can also be drawn as because of potential tautomerism of the triazole ring.
- the present invention also covers useful forms of the compounds of the present invention, such as metabolites, hydrates, solvates, prodrugs, salts, in particular pharmaceutically acceptable salts, and/or co-precipitates.
- the compounds of the present invention can exist as a hydrate, or as a solvate, wherein the compounds of the present invention contain polar solvents, in particular water, methanol or ethanol for example, as structural element of the crystal lattice of the compounds. It is possible for the amount of polar solvents, in particular water, to exist in a stoichiometric or non- stoichiometric ratio.
- polar solvents in particular water
- stoichiometric solvates e.g. a hydrate, hemi-, (semi-), mono- , sesqui-, di-, tri-, tetra-, penta- etc. solvates or hydrates, respectively, are possible.
- the present invention includes all such hydrates or solvates.
- the compounds of the present invention may exist in free form, e.g. as a free base, or as a free acid, or as a zwitterion, or to exist in the form of a salt.
- Said salt may be any salt, either an organic or inorganic addition salt, particularly any pharmaceutically acceptable organic or inorganic addition salt, which is customarily used in pharmacy, or which is used, for example, for isolating or purifying the compounds of the present invention.
- pharmaceutically acceptable salt refers to an inorganic or organic acid addition salt of a compound of the present invention. For example, see S. M. Berge, et al. “Pharmaceutical Salts,” J. Pharm. Sci. 1977, 66, 1-19.
- a suitable pharmaceutically acceptable salt of the compounds of the present invention may be, for example, an acid-addition salt of a compound of the present invention bearing a nitrogen atom, in a chain or in a ring, for example, which is sufficiently basic, such as an acid-addition salt with an inorganic acid, or “mineral acid”, such as hydrochloric, hydrobromic, hydroiodic, sulfuric, sulfamic, bisulfuric, phosphoric, or nitric acid, for example, or with an organic acid, such as formic, acetic, acetoacetic, pyruvic, trifluoroacetic, propionic, butyric, hexanoic, heptanoic, undecanoic, lauric, benzoic, salicylic, 2-(4-hydroxybenzoyl)-benzoic, camphoric, cinnamic, cyclopentanepropionic, digluconic, 3-hydroxy-2-naphthoic, nico
- an alkali metal salt for example a sodium or potassium salt
- an alkaline earth metal salt for example a calcium, magnesium or strontium salt, or an aluminium or a zinc salt
- acid addition salts of the claimed compounds to be prepared by reaction of the compounds with the appropriate inorganic or organic acid via any of a number of known methods.
- alkali and alkaline earth metal salts of acidic compounds of the present invention are prepared by reacting the compounds of the present invention with the appropriate base via a variety of known methods.
- the present invention includes all possible salts of the compounds of the present invention as single salts, or as any mixture of said salts, in any ratio.
- the present invention includes all possible crystalline forms, or polymorphs, of the compounds of the present invention, either as single polymorph, or as a mixture of more than one polymorph, in any ratio.
- the present invention also includes prodrugs of the compounds according to the invention.
- prodrugs here designates compounds which themselves can be biologically active or inactive, but are converted (for example metabolically or hydrolytically) into compounds according to the invention during their residence time in the body.
- the present invention covers compounds of general formula (I), supra, in which:
- Embodiment B The compound
- Embodiment F The compound 5- ⁇ (3R)-l-[(R)-((R)-2,2-difluorocyclopropyl)(lH-l,2,4-triazol-5-yl)methyl]-5',6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[l,2-b]pyrazol]-2'-yl ⁇ -3-(trifluoromethyl)pyridin-2-amine of formula (I f) according to embodiment A, B or C or a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same.
- Embodiment I The specific stereoisomer of the compound of embodiment F, obtainable by stirring a solution of 5-[(3R)-5’,6’-dihydrospiro[pyrrolidine-3,4’-pyrrolo[1 ,2-b]pyrazol]- 2’-yl]-3-(trifluoromethyl)pyridin-2-amine hydrogen chloride (1/1) (420 mg, 1.17 mmol), [2,2-difluorocyclopropyl](1 H-1 ,2,4-triazol-5-yl)methanone (220 mg, 1.27 mmol), titanium(IV) isopropoxide (940 pL, 3.2 mmol) and DIPEA (920 pL, 5.3 mmol) in methanol (11 mL) at 60°C for 4 h adding sodium cyanoborohydride (166 mg, 2.65 mmol) and stirring the reaction mixture overnight at 60°C adding water, stirring for additional 10 min, filtering and washing the filter cake with methanol the
- eluent A water + 0.2 vol % aqueous ammonia (32%); eluent B: acetonitril;gradient: 0.00-0.50 min 10% B (150 mL/min), 0.50-6.00 min 10-50% B (150 mL/min), 6.00-6.10 min 50-100% B (150 mL/min), 6.10-8.00 min 100% B (150 mL/min), temperature: rt; DAD scan: 210-400 nm) to afford 5- ⁇ (3R)-1 -[(2,2- difluorocyclopropyl)(1 H-1 ,2,4-triazol-5-yl)methyl]-5',6'-dihydrospiro[pyrrolidine-3,4'- pyrrolo[1 ,2-b]pyrazol]-2'-yl ⁇ -3-(trifluoromethyl)pyridin-2-amine (all fractions having a molecular mass of 480 Daltons and similar retention times...
- Retention time 4.2 - 4.7 min, first/ eluting) further separating the resulting mixture by chiral HPLC method C (Instrument: PrepCon Labomatic HPLC-3; Column: Chiralpak IG 5p, 250x30; eluent A: hexane + 0.1 vol % diethylamine; eluent B: ethanol; isocratic: 70% A + 30% B; flow: 70 mL/min; temperature: 25°C; UV: 280 nm. Retention time: 8.04 - 9.75 min, first/ eluting) or a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same.
- Embodiment J The specific stereoisomer of the compound of embodiment F, obtainable by dissolving 5-[(3R)-5',6'-dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl]-3- (trifluoromethyl)pyridin-2-amine hydrogen chloride (1/1) (200 mg, 528 pmol) in DMF (3.9 mL) adding successively N,N-diisopropylethylamine (550 pL, 3.2 mmol) and then 5- ⁇ chloro[(1 R)-2,2-difluorocyclopropyl]methyl ⁇ -1 H-1 ,2,4-triazole hydrogen chloride (1/1) (diastereomer 1 and diastereomer 2) (285 mg, 1.24 mmol) dissolved in DMF (3.9 mL) stirring the reaction mixture overnight at rt concentrating the reaction mixture partitioning the residue between water and ethyl acetate
- Embodiment K A method of preparing a compound of formula (I) according to embodiment A, said method comprising the step of allowing an intermediate compound of general formula (II), a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof: in which Y represents a leaving group, to react with a compound of formula (III), a stereoisomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof: thereby giving the compound of formula (I) of claim 1: or a stereoisomer, a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same.
- Embodiment L A method of preparing a compound of formula (I) according to embodiment A, said method comprising the step of allowing an intermediate compound of general formula (Ila), a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof: in which the ketone to react with a compound of formula (III), a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof: thereby giving the compound of formula (I) of claim 1: a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof.
- Embodiment M The compound of formula (I) of embodiment A for use in the treatment or prophylaxis of a disease.
- Embodiment N A pharmaceutical composition comprising a compound of formula (I) of embodiment A and one or more pharmaceutically acceptable excipients.
- Embodiment O A pharmaceutical combination comprising:
- Embodiment Q Use of the compound of formula (I) of embodiment A for the preparation of a medicament for the treatment or prophylaxis of a disease.
- Embodiment R Use according to embodiment P or Q, wherein the disease is a neoplastic or abnormal cell proliferative disorder, such as cancer, a condition with dysregulated immune response or another disorder associated with aberrant Map4K1 signaling.
- a neoplastic or abnormal cell proliferative disorder such as cancer, a condition with dysregulated immune response or another disorder associated with aberrant Map4K1 signaling.
- the present invention covers intermediate compounds which are useful for the preparation of the compounds of general formula (I), supra.
- the present invention covers the intermediate compounds which are disclosed in the Example Section of this text, infra.
- Embodiment S The compound of general formula (II): in which Y represents a leaving group) or a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof.
- Embodiment T The compound of general formula (Il a): a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof.
- Embodiment U The compound of general formula (III): a stereoisomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof.
- the present invention covers the use of said intermediate compounds for the preparation of a compound of general formula (I) as defined supra.
- Embodiment V Use of a compound of general formula (II) a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof in which Y a leaving group, for the preparation of a compound of formula (I) of embodiment A.
- Embodiment W Use of a compound of general formula (II) a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof for the preparation of a compound of formula (I) of embodiment A.
- Embodiment X Use of a compound of general formula (III) a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof, for the preparation of a compound of formula (I) of embodiment A.
- Embodiment Y Method for controlling cancer in humans and animals by administering an antiproliferative effective amount of the compound of formula (I) of embodiment A, or of a medicament as defined in one of embodiments P, Q or R.
- the present invention covers any sub-combination within any embodiment or aspect of the present invention of intermediate compounds of general formula (I), supra.
- the compounds of general formula (I) of the present invention can be converted to any salt, preferably pharmaceutically acceptable salts, as described herein, by any method which is known to the person skilled in the art.
- any salt of a compound of general formula (I) of the present invention can be converted into the free compound, by any method which is known to the person skilled in the art.
- Compounds of general formula (I) of the present invention demonstrate a valuable pharmacological spectrum of action and pharmacokinetic profile , both of which could not have been predicted.
- Compounds of the present invention have surprisingly been found to effectively inhibit Map4K1 and it is possible therefore that said compounds be used for the treatment or prophylaxis of diseases, preferably neoplastic or abnormal cell proliferative disorder, such as cancer, a condition with dysregulated immune response or another disorder associated with aberrant Map4K1 signaling disorders in humans and animals.
- Compounds of the present invention can be utilized to inhibit, block, reduce, decrease, etc., cell proliferation and/or cell division, and/or produce apoptosis.
- This method comprises administering to a mammal in need thereof, including a human, an amount of a compound of general formula (I) of the present invention, or a pharmaceutically acceptable salt, isomer, polymorph, metabolite, hydrate, solvate or ester thereof, which is effective to treat the disorder.
- Hyperproliferative disorders include, but are not limited to, for example : psoriasis, keloids, and other hyperplasias affecting the skin, benign prostate hyperplasia (BPH), solid tumours, such as cancers of the breast, respiratory tract, brain, reproductive organs, digestive tract, urinary tract, eye, liver, skin, head and neck, thyroid, parathyroid and their distant metastases.
- BPH benign prostate hyperplasia
- solid tumours such as cancers of the breast, respiratory tract, brain, reproductive organs, digestive tract, urinary tract, eye, liver, skin, head and neck, thyroid, parathyroid and their distant metastases.
- Those disorders also include lymphomas, sarcomas, and leukaemias.
- breast cancers include, but are not limited to, invasive ductal carcinoma, invasive lobular carcinoma, ductal carcinoma in situ, and lobular carcinoma in situ.
- cancers of the respiratory tract include, but are not limited to, small-cell and non- small-cell lung carcinoma, as well as bronchial adenoma and pleuropulmonary blastoma.
- brain cancers include, but are not limited to, brain stem and hypophtalmic glioma, cerebellar and cerebral astrocytoma, medulloblastoma, ependymoma, as well as neuroectodermal and pineal tumour.
- Tumours of the male reproductive organs include, but are not limited to, prostate and testicular cancer.
- Tumours of the female reproductive organs include, but are not limited to, endometrial, cervical, ovarian, vaginal, and vulvar cancer, as well as sarcoma of the uterus.
- Tumours of the digestive tract include, but are not limited to, anal, colon, colorectal, oesophageal, gallbladder, gastric, pancreatic, rectal, small-intestine, and salivary gland cancers.
- Tumours of the urinary tract include, but are not limited to, bladder, penile, kidney, renal pelvis, ureter, urethral and human papillary renal cancers.
- Eye cancers include, but are not limited to, intraocular melanoma and retinoblastoma.
- liver cancers include, but are not limited to, hepatocellular carcinoma (liver cell carcinomas with or without fibrolamellar variant), cholangiocarcinoma (intrahepatic bile duct carcinoma), and mixed hepatocellular cholangiocarcinoma.
- Skin cancers include, but are not limited to, squamous cell carcinoma, Kaposi’s sarcoma, malignant melanoma, Merkel cell skin cancer, and non-melanoma skin cancer.
- Head-and-neck cancers include, but are not limited to, laryngeal, hypopharyngeal, nasopharyngeal, oropharyngeal cancer, lip and oral cavity cancer and squamous cell.
- Lymphomas include, but are not limited to, AIDS-related lymphoma, non-Hodgkin’s lymphoma, cutaneous T-cell lymphoma, Burkitt lymphoma, Hodgkin’s disease, and lymphoma of the central nervous system.
- Sarcomas include, but are not limited to, sarcoma of the soft tissue, osteosarcoma, malignant fibrous histiocytoma, lymphosarcoma, and rhabdomyosarcoma.
- Leukemias include, but are not limited to, acute myeloid leukemia, acute lymphoblastic leukemia, chronic lymphocytic leukemia, chronic myelogenous leukemia, and hairy cell leukemia.
- the present invention also provides methods of treating angiogenic disorders including diseases associated with excessive and/or abnormal angiogenesis.
- Inappropriate and ectopic expression of angiogenesis can be deleterious to an organism.
- a number of pathological conditions are associated with the growth of extraneous blood vessels. These include, for example, diabetic retinopathy, ischemic retinal-vein occlusion, and retinopathy of prematurity [Aiello et al., New Engl. J. Med., 1994, 331 , 1480 ; Peer et al., Lab. Invest., 1995, 72, 638], age-related macular degeneration (AMD) [Lopez et al., Invest.
- AMD age-related macular degeneration
- neovascular glaucoma neovascular glaucoma
- psoriasis retrolental fibroplasias
- angiofibroma inflammation
- RA rheumatoid arthritis
- restenosis in-stent restenosis
- vascular graft restenosis etc.
- the increased blood supply associated with cancerous and neoplastic tissue encourages growth, leading to rapid tumour enlargement and metastasis.
- the growth of new blood and lymph vessels in a tumour provides an escape route for renegade cells, encouraging metastasis and the consequence spread of the cancer.
- compounds of general formula (I) of the present invention can be utilized to treat and/or prevent any of the aforementioned angiogenesis disorders, for example by inhibiting and/or reducing blood vessel formation; by inhibiting, blocking, reducing, decreasing, etc. endothelial cell proliferation, or other types involved in angiogenesis, as well as causing cell death or apoptosis of such cell types.
- treating or “treatment” as stated throughout this document is used conventionally, for example the management or care of a subject for the purpose of combating, alleviating, reducing, relieving, improving the condition of a disease or disorder, such as a carcinoma.
- the compounds of the present invention can be used in particular in therapy and prevention, i.e. prophylaxis, of tumour growth and metastases, especially in solid tumours of all indications and stages with or without pre-treatment of the tumour growth.
- chemotherapeutic agents and/or anti-cancer agents in combination with a compound or pharmaceutical composition of the present invention will serve to:
- the compounds of general formula (I) of the present invention can also be used in combination with radiotherapy and/or surgical intervention.
- the compounds of general formula (I) of the present invention may be used to sensitize a cell to radiation, i.e. treatment of a cell with a compound of the present invention prior to radiation treatment of the cell renders the cell more susceptible to DNA damage and cell death than the cell would be in the absence of any treatment with a compound of the present invention.
- the cell is treated with at least one compound of general formula (I) of the present invention.
- the present invention also provides a method of killing a cell, wherein a cell is administered one or more compounds of the present invention in combination with conventional radiation therapy.
- the present invention also provides a method of rendering a cell more susceptible to cell death, wherein the cell is treated with one or more compounds of general formula (I) of the present invention prior to the treatment of the cell to cause or induce cell death.
- the cell is treated with at least one compound, or at least one method, or a combination thereof, in order to cause DNA damage for the purpose of inhibiting the function of the normal cell or killing the cell.
- a cell is killed by treating the cell with at least one DNA damaging agent, i.e. after treating a cell with one or more compounds of general formula (I) of the present invention to sensitize the cell to cell death, the cell is treated with at least one DNA damaging agent to kill the cell.
- DNA damaging agents useful in the present invention include, but are not limited to, chemotherapeutic agents (e.g. cis platin), ionizing radiation (X-rays, ultraviolet radiation), carcinogenic agents, and mutagenic agents.
- a cell is killed by treating the cell with at least one method to cause or induce DNA damage.
- methods include, but are not limited to, activation of a cell signalling pathway that results in DNA damage when the pathway is activated, inhibiting of a cell signalling pathway that results in DNA damage when the pathway is inhibited, and inducing a biochemical change in a cell, wherein the change results in DNA damage.
- a DNA repair pathway in a cell can be inhibited, thereby preventing the repair of DNA damage and resulting in an abnormal accumulation of DNA damage in a cell.
- a compound of general formula (I) of the present invention is administered to a cell prior to the radiation or other induction of DNA damage in the cell.
- a compound of general formula (I) of the present invention is administered to a cell concomitantly with the radiation or other induction of DNA damage in the cell.
- a compound of general formula (I) of the present invention is administered to a cell immediately after radiation or other induction of DNA damage in the cell has begun.
- the cell is in vitro. In another embodiment, the cell is in vivo.
- Compounds of the present invention can be utilized to inhibit, block, reduce, decrease, etc., Map4K1.
- This method comprises administering to a mammal in need thereof, including a human, an amount of a compound of this invention, or a pharmaceutically acceptable salt, isomer, polymorph, metabolite, hydrate, solvate or ester thereof; which is effective to treat the disorder.
- the present invention also provides methods of treating, ameliorating or preventing neoplastic disorders or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling and methods of ameliorating of vaccine therapies or cell therapies.
- Amelioration Any intervention with a goal of improvement in comparison to a situation without this interventions.
- Neoplastic disorder A disorder which causes or results in an abnormal and excessive growth of tissue.
- Abnormal cell proliferative disorder A disorder which causes or results in abnormal cell proliferation.
- Condition with dysregulated immune response A condition in which the regulation of an immune response differs from the regulation of this immune response in a healthy human.
- Vaccine therapy is a therapy which uses vaccines.
- treating or “treatment” as used in the present text is used conventionally, e.g., the management or care of a subject for the purpose of combating, alleviating, reducing, relieving, improving the condition of a disease or disorder, such as a carcinoma.
- the compounds of the present invention can be used in particular in therapy and prevention, i.e. prophylaxis, of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling and in amelioration of vaccine therapies or cell therapies.
- the present invention covers compounds of general formula (I), as described supra, or stereoisomers, tautomers, N-oxides, hydrates, solvates, and salts thereof, particularly pharmaceutically acceptable salts thereof, or mixtures of same, for use in the treatment or prophylaxis of diseases, in particular of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, and for the amelioration of vaccine therapies or cell therapies.
- diseases in particular of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, and for the amelioration of vaccine therapies or cell therapies.
- the pharmaceutical activity of the compounds according to the invention can be explained by their activity as Map4K1 inhibitor.
- the present invention covers the use of compounds of general formula (I), as described supra, or stereoisomers, tautomers, N-oxides, hydrates, solvates, and salts thereof, particularly pharmaceutically acceptable salts thereof, or mixtures of same, for the treatment or prophylaxis of diseases, in particular of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, and for the amelioration of vaccine therapies or cell therapies.
- diseases in particular of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, and for the amelioration of vaccine therapies or cell therapies.
- the present invention covers the use of a compound of formula (I), described supra, or a stereoisomer, a tautomer, an N-oxide, a hydrate, a solvate, or a salt thereof, particularly a pharmaceutically acceptable salt thereof, or a mixture of same, for the prophylaxis or treatment of diseases, in particular of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, and for the amelioration of vaccine therapies or cell therapies.
- diseases in particular of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, and for the amelioration of vaccine therapies or cell therapies.
- the present invention covers the use of compounds of general formula (I), as described supra, or stereoisomers, tautomers, N-oxides, hydrates, solvates, and salts thereof, particularly pharmaceutically acceptable salts thereof, or mixtures of same, in a method of treatment or prophylaxis of diseases, in particular of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, and for the amelioration of vaccine therapies or cell therapies.
- diseases in particular of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, and for the amelioration of vaccine therapies or cell therapies.
- the present invention covers use of a compound of general formula (I), as described supra, or stereoisomers, tautomers, N-oxides, hydrates, solvates, and salts thereof, particularly pharmaceutically acceptable salts thereof, or mixtures of same, for the preparation of a pharmaceutical composition, preferably a medicament, for the prophylaxis or treatment of diseases, in particular of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, and for the amelioration of vaccine therapies or cell therapies.
- diseases in particular of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, and for the amelioration of vaccine therapies or cell therapies.
- the present invention covers a method of treatment or prophylaxis of diseases, in particular of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, and for the amelioration of vaccine therapies or cell therapies, using an effective amount of a compound of general formula (I), as described supra, or stereoisomers, tautomers, hydrates, solvates, and salts thereof, particularly pharmaceutically acceptable salts thereof, or mixtures of same.
- diseases in particular of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, and for the amelioration of vaccine therapies or cell therapies, using an effective amount of a compound of general formula (I), as described supra, or stereoisomers, tautomers, hydrates, solvates, and salts thereof, particularly pharmaceutically acceptable salts thereof, or mixtures of same.
- the present invention covers pharmaceutical compositions, in particular a medicament, comprising a compound of general formula (I), as described supra, or a stereoisomer, a tautomer, a hydrate, a solvate, a salt thereof, particularly a pharmaceutically acceptable salt, or a mixture of same, and one or more excipients), in particular one or more pharmaceutically acceptable excipient(s).
- a medicament comprising a compound of general formula (I), as described supra, or a stereoisomer, a tautomer, a hydrate, a solvate, a salt thereof, particularly a pharmaceutically acceptable salt, or a mixture of same, and one or more excipients), in particular one or more pharmaceutically acceptable excipient(s).
- excipients in particular one or more pharmaceutically acceptable excipient(s).
- Conventional procedures for preparing such pharmaceutical compositions in appropriate dosage forms can be utilized.
- the present invention furthermore covers pharmaceutical compositions, in particular medicaments, which comprise at least one compound according to the invention, conventionally together with one or more pharmaceutically suitable excipients, and to their use for the above mentioned purposes.
- the compounds according to the invention can be administered in a suitable manner, such as, for example, via the oral, parenteral, pulmonary, nasal, sublingual, lingual, buccal, rectal, vaginal, dermal, transdermal, conjunctival, otic route or as an implant or stent.
- the compounds according to the invention for oral administration, it is possible to formulate the compounds according to the invention to dosage forms known in the art that deliver the compounds of the invention rapidly and/or in a modified manner, such as, for example, tablets (uncoated or coated tablets, for example with enteric or controlled release coatings that dissolve with a delay or are insoluble), orally- disintegrating tablets, films/wafers, films/lyophylisates, capsules (for example hard or soft gelatine capsules), sugar-coated tablets, granules, pellets, powders, emulsions, suspensions, aerosols or solutions. It is possible to incorporate the compounds according to the invention in crystalline and/or amorphised and/or dissolved form into said dosage forms.
- Parenteral administration can be effected with avoidance of an absorption step (for example intravenous, intraarterial, intracardial, intraspinal or intralumbal) or with inclusion of absorption (for example intramuscular, subcutaneous, intracutaneous, percutaneous or intraperitoneal).
- absorption step for example intravenous, intraarterial, intracardial, intraspinal or intralumbal
- absorption for example intramuscular, subcutaneous, intracutaneous, percutaneous or intraperitoneal.
- Administration forms which are suitable for parenteral administration are, inter alia, preparations for injection and infusion in the form of solutions, suspensions, emulsions, lyophylisates or sterile powders.
- Examples which are suitable for other administration routes are pharmaceutical forms for inhalation [inter alia powder inhalers, nebulizers], nasal drops, nasal solutions, nasal sprays; tablets/films/wafers/capsules for lingual, sublingual or buccal administration; suppositories; eye drops, eye ointments, eye baths, ocular inserts, ear drops, ear sprays, ear powders, ear-rinses, ear tampons; vaginal capsules, aqueous suspensions (lotions, mixturae agitandae), lipophilic suspensions, emulsions, ointments, creams, transdermal therapeutic systems (such as, for example, patches), milk, pastes, foams, dusting powders, implants or stents.
- inhalation inter alia powder inhalers, nebulizers
- nasal drops nasal solutions, nasal sprays
- tablets/films/wafers/capsules for lingual, sublingual or buccal
- the compounds according to the invention can be incorporated into the stated administration forms. This can be effected in a manner known per se by mixing with pharmaceutically suitable excipients.
- Pharmaceutically suitable excipients include, inter alia,
- fillers and carriers for example cellulose, microcrystalline cellulose (such as, for example, Avicel®), lactose, mannitol, starch, calcium phosphate (such as, for example, Di-Cafos®)),
- ointment bases for example petroleum jelly, paraffins, triglycerides, waxes, wool wax, wool wax alcohols, lanolin, hydrophilic ointment, polyethylene glycols
- ointment bases for example petroleum jelly, paraffins, triglycerides, waxes, wool wax, wool wax alcohols, lanolin, hydrophilic ointment, polyethylene glycols
- bases for suppositories for example polyethylene glycols, cacao butter, hard fat
- solvents for example water, ethanol, isopropanol, glycerol, propylene glycol, medium chain-length triglycerides fatty oils, liquid polyethylene glycols, paraffins
- surfactants for example sodium dodecyl sulfate), lecithin, phospholipids, fatty alcohols (such as, for example, Lanette®), sorbitan fatty acid esters (such as, for example, Span®), polyoxyethylene sorbitan fatty acid esters (such as, for example, Tween®), polyoxyethylene fatty acid glycerides (such as, for example, Cremophor®), polyoxethylene fatty acid esters, polyoxyethylene fatty alcohol ethers, glycerol fatty acid esters, poloxamers (such as, for example, Pluronic®), • buffers, acids and bases (for example phosphates, carbonates, citric acid, acetic acid, hydrochloric acid, sodium hydroxide solution, ammonium carbonate, trometamol, triethanolamine),
- acids and bases for example phosphates, carbonates, citric acid, acetic acid, hydrochloric acid, sodium hydroxide solution, ammonium carbonate, tro
- isotonicity agents for example glucose, sodium chloride
- adsorbents for example highly-disperse silicas
- viscosity-increasing agents for example polyvinylpyrrolidone, methylcellulose, hydroxypropylmethylcellulose, hydroxypropylcellulose, carboxymethylcellulose-sodium, starch, carbomers, polyacrylic acids (such as, for example, Carbopol®); alginates, gelatine),
- disintegrants for example modified starch, carboxymethylcellulose-sodium, sodium starch glycolate (such as, for example, Explotab®), cross- linked polyvinylpyrrolidone, croscarmellose-sodium (such as, for example, AcDiSol®)
- disintegrants for example modified starch, carboxymethylcellulose-sodium, sodium starch glycolate (such as, for example, Explotab®), cross- linked polyvinylpyrrolidone, croscarmellose-sodium (such as, for example, AcDiSol®)
- lubricants for example magnesium stearate, stearic acid, talc, highly-disperse silicas (such as, for example, Aerosil®)
- mould release agents for example magnesium stearate, stearic acid, talc, highly-disperse silicas (such as, for example, Aerosil®)
- coating materials for example sugar, shellac
- film formers for films or diffusion membranes which dissolve rapidly or in a modified manner for example polyvinylpyrrolidones (such as, for example, Kollidon®), polyvinyl alcohol, hydroxypropylmethylcellulose, hydroxypropylcellulose, ethylcellulose, hydroxypropylmethylcellulose phthalate, cellulose acetate, cellulose acetate phthalate, polyacrylates, polymethacrylates such as, for example, Eudragit®)),
- capsule materials for example gelatine, hydroxypropylmethylcellulose
- synthetic polymers for example polylactides, polyglycolides, polyacrylates, polymethacrylates (such as, for example, Eudragit®), polyvinylpyrrolidones (such as, for example, Kollidon®), polyvinyl alcohols, polyvinyl acetates, polyethylene oxides, polyethylene glycols and their copolymers and blockcopolymers),
- plasticizers for example polyethylene glycols, propylene glycol, glycerol, triacetine, triacetyl citrate, dibutyl phthalate
- stabilisers for example antioxidants such as, for example, ascorbic acid, ascorbyl palmitate, sodium ascorbate, butylhydroxyanisole, butylhydroxytoluene, propyl gallate
- antioxidants for example antioxidants such as, for example, ascorbic acid, ascorbyl palmitate, sodium ascorbate, butylhydroxyanisole, butylhydroxytoluene, propyl gallate
- preservatives for example parabens, sorbic acid, thiomersal, benzalkonium chloride, chlorhexidine acetate, sodium benzoate
- colourants for example inorganic pigments such as, for example, iron oxides, titanium dioxide
- flavourings • flavourings, sweeteners, flavour- and/or odour-masking agents.
- the present invention furthermore relates to a pharmaceutical composition which comprise at least one compound according to the invention, conventionally together with one or more pharmaceutically suitable excipient(s), and to their use according to the present invention.
- the present invention covers pharmaceutical combinations, in particular medicaments, comprising at least one compound of general formula (I) of the present invention and at least one or more further active ingredients, in particular for the treatment and/or prophylaxis of a of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, and for the amelioration of vaccine therapies or cell therapies.
- pharmaceutical combinations in particular medicaments, comprising at least one compound of general formula (I) of the present invention and at least one or more further active ingredients, in particular for the treatment and/or prophylaxis of a of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, and for the amelioration of vaccine therapies or cell therapies.
- the present invention covers a pharmaceutical combination, which comprises:
- neoplastic or abnormal cell proliferative disorders such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, and for the amelioration of vaccine therapies or cell therapies.
- a “fixed combination” in the present invention is used as known to persons skilled in the art and is defined as a combination wherein, for example, a first active ingredient, such as one or more compounds of general formula (I) of the present invention, and a further active ingredient are present together in one unit dosage or in one single entity.
- a “fixed combination” is a pharmaceutical composition wherein a first active ingredient and a further active ingredient are present in admixture for simultaneous administration, such as in a formulation.
- Another example of a “fixed combination” is a pharmaceutical combination wherein a first active ingredient and a further active ingredient are present in one unit without being in admixture.
- a non-fixed combination or “kit-of-parts” in the present invention is used as known to persons skilled in the art and is defined as a combination wherein a first active ingredient and a further active ingredient are present in more than one unit.
- a non-fixed combination or kit-of-parts is a combination wherein the first active ingredient and the further active ingredient are present separately. It is possible for the components of the non-fixed combination or kit-of- parts to be administered separately, sequentially, simultaneously, concurrently or chronologically staggered.
- the compounds of the present invention can be administered as the sole pharmaceutical agent or in combination with one or more other pharmaceutically active ingredients where the combination causes no unacceptable adverse effects.
- the present invention also covers such pharmaceutical combinations.
- the compounds of the present invention can be combined with known cancer agents.
- cancer agents examples include:
- the effective dosage of the compounds of the present invention can readily be determined for treatment of each desired indication.
- the amount of the active ingredient to be administered in the treatment of one of these conditions can vary widely according to such considerations as the particular compound and dosage unit employed, the mode of administration, the period of treatment, the age and sex of the patient treated, and the nature and extent of the condition treated.
- the compounds of the invention can further be combined with chimeric antigen receptor T cells (CAR-T cells), such as Axicabtagen-Ciloleucel or Tisagenlecleucel.
- CAR-T cells chimeric antigen receptor T cells
- the activity of CAR-T cells can be suppressed by the tumor micro environment (TME).
- the present invention covers combinations comprising one or more compounds according to the invention, or stereoisomers, tautomers, hydrates, solvates, and salts thereof, particularly pharmaceutically acceptable salts thereof, or mixtures of same, with chimeric antigen receptor T cells, (CAR-T cells), CAR-NKT cells or CAR-NK cells.
- CAR-T cells chimeric antigen receptor T cells
- CAR-NKT cells CAR-NK cells
- the chimeric antigen receptor T cells are YESCARTA® (axicabtagen ciloleucel), KYMRIAH® (tisagenlecleucel), BREYANZI® (lisocabtagene maraleucel), TECARTUS® (brecucabtqagene autoleucel), ABECMA® (idecabtagene vicleucel), or CARVYKTI® (clitacabtagene autoleucel).
- the present invention further provides the use of the compounds according to the invention for expansion of T cells including CAR-T and tumor infiltrated lymphocytes ex-vivo.
- the present invention covers compounds according to the invention, or stereoisomers, tautomers, hydrates, solvates, and salts thereof, particularly pharmaceutically acceptable salts thereof, or mixtures of same, for use in the expansion of T cells including CAR-T cells, CAR-NKT cells or CAR-NK cells and tumor infiltrated lymphocytes ex-vivo.
- the present invention also relates to the use of the compounds according to the invention for the expansion of T cells, including CAR-T cell, CAR-NKT cells or CAR-NK cells and tumor infiltrated lymphocytes, ex-vivo.
- the present invention also comprises an ex-vivo method for the expansion of T cells, including CAR-T cells, CAR-NKT cells or CAR-NK cells and tumor infiltrated lymphocytes, contacting said T cells with compounds according to the invention.
- the total amount of the active ingredient to be administered will generally range from about 0.001 mg/kg to about 200 mg/kg body weight per day, and preferably from about 0.01 mg/kg to about 20 mg/kg body weight per day.
- Clinically useful dosing schedules will range from one to three times a day dosing to once every four weeks dosing.
- drug holidays in which a patient is not dosed with a drug for a certain period of time, to be beneficial to the overall balance between pharmacological effect and tolerability. It is possible for a unit dosage to contain from about 0.5 mg to about 1500 mg of active ingredient, and can be administered one or more times per day or less than once a day.
- the average daily dosage for administration by injection will preferably be from 0.01 to 200 mg/kg of total body weight.
- the average daily rectal dosage regimen will preferably be from 0.01 to 200 mg/kg of total body weight.
- the average daily vaginal dosage regimen will preferably be from 0.01 to 200 mg/kg of total body weight.
- the average daily topical dosage regimen will preferably be from 0.1 to 200 mg administered between one to four times daily.
- the transdermal concentration will preferably be that required to maintain a daily dose of from 0.01 to 200 mg/kg.
- the average daily inhalation dosage regimen will preferably be from 0.01 to 100 mg/kg of total body weight.
- the specific initial and continuing dosage regimen for each patient will vary according to the nature and severity of the condition as determined by the attending diagnostician, the activity of the specific compound employed, the age and general condition of the patient, time of administration, route of administration, rate of excretion of the drug, drug combinations, and the like.
- the desired mode of treatment and number of doses of a compound of the present invention or a pharmaceutically acceptable salt or ester or composition thereof can be ascertained by those skilled in the art using conventional treatment tests.
- Figure 1 50% thermal ellipsoids of intermediate 15, Molecule 1.
- Figure 2 50% thermal ellipsoids of intermediate 15, Molecule 2.
- Figure 3 50% thermal ellipsoids of side product 1.
- NMR peak forms are stated as they appear in the spectra, possible higher order effects have not been considered.
- the 1 H-NMR data of selected compounds are listed in the form of 1 H-NMR peaklists. Therein, for each signal peak the 6 value in ppm is given, followed by the signal intensity, reported in round brackets. The 6 value-signal intensity pairs from different peaks are separated by commas. Therefore, a peaklist is described by the general form: 61 (intensity-i), 62 (intense), ... , 6i (intensity,), ... , 6 n (intensity,,).
- a 1 H-NMR peaklist is similar to a classical 1 H-NMR readout, and thus usually contains all the peaks listed in a classical NMR interpretation. Moreover, similar to classical 1 H- NMR printouts, peaklists can show solvent signals, signals derived from stereoisomers of the particular target compound, peaks of impurities, 13 C satellite peaks, and/or spinning sidebands.
- the peaks of stereoisomers, and/or peaks of impurities are typically displayed with a lower intensity compared to the peaks of the target compound (e.g., with a purity of >90%).
- Such stereoisomers and/or impurities may be typical for the particular manufacturing process, and therefore their peaks may help to identify a reproduction of the manufacturing process on the basis of "by-product fingerprints".
- An expert who calculates the peaks of the target compound by known methods can isolate the peaks of the target compound as required, optionally using additional intensity filters. Such an operation would be similar to peak-picking in classical 1 H-NMR interpretation.
- the compounds and intermediates produced according to the methods of the invention may require purification. Purification of organic compounds is well known to the person skilled in the art and there may be several ways of purifying the same compound. In some cases, no purification may be necessary. In some cases, the compounds may be purified by crystallization. In some cases, impurities may be stirred out using a suitable solvent. In some cases, the compounds may be purified by chromatography, particularly flash column chromatography, using for example prepacked silica gel cartridges, e.g.
- the compounds may be purified by preparative HPLC using for example a Waters autopurifier equipped with a diode array detector and/or on-line electrospray ionization mass spectrometer in combination with a suitable prepacked reverse phase column and eluents such as gradients of water and acetonitrile which may contain additives such as trifluoroacetic acid, formic acid or aqueous ammonia.
- purification methods as described above can provide those compounds of the present invention which possess a sufficiently basic or acidic functionality in the form of a salt, such as, in the case of a compound of the present invention which is sufficiently basic, a trifluoroacetate or formate salt for example, or, in the case of a compound of the present invention which is sufficiently acidic, an ammonium salt for example.
- a salt of this type can either be transformed into its free base or free acid form, respectively, by various methods known to the person skilled in the art, or be used as salts in subsequent biological assays. It is to be understood that the specific form (e.g. salt, free base etc.) of a compound of the present invention as isolated and as described herein is not necessarily the only form in which said compound can be applied to a biological assay in order to quantify the specific biological activity.
- two or more successive steps may be performed without work-up between the said steps, e.g. in a “one-pot” reaction, as it is well-known to a person skilled in the art.
- any of the described reactants and synthetic intermediates can be used in the form of single stereoisomers as well as in the form of a mixture of multiple stereoisomers.
- PG 1 represents a suitable triazole protecting group (e.g. SEM, THP or diethoxymethyl)
- Z represents a suitable leaving group (e.g. halogen, N(OMe)Me, N(OBn)Me, S-alkyl, S-aryl or S-heteroaryl).
- Starting materials a1 and b1 are commercially available or described in the literature (for racemic compound b1 and its enantiomers see: A. E. Goetz et al., Org. Process Res. Dev. 2022, 26, 683-697).
- step 2 difluorocyclopropyl carboxylic acid b1 is converted to intermediate b2 where Z is a leaving group, exemplified by but not limited to halogen, N-alkyl-N-alkoxy, N-alkyl- N-benzyloxy, thioalkyl, thioaryl or thioheteroaryl.
- Z is a leaving group, exemplified by but not limited to halogen, N-alkyl-N-alkoxy, N-alkyl- N-benzyloxy, thioalkyl, thioaryl or thioheteroaryl.
- CPTPA 1-chloro-N,N,2-trimethylprop-1-en-1-amine
- N,O- dimethylhydroxylamine or related amines can be reacted with N,O- dimethylhydroxylamine or related amines to yield N-methoxy-N-methyl amide (Weinreb-Nahm amide) or related N-alkyl-N-alkoxy amide, or N-alkyl-N-benzyloxy amide, via intermediate formation of acid chloride or using various amide coupling reagents like carbonyldiimidazole (GDI), propylphosphonic anhydride (T3P), O-(7-azabenzotriazol-1-yl)-N,N,N',N'- tetramethyluronium-hexafluorphosphat (HATU), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) and others which are well known to the person skilled in organic synthesis.
- GDI carbonyldiimidazole
- T3P propylphosphonic anhydride
- suitable reagent for e.g. L. Kurti and B. Czako, Strategic Application of Named Reactions in Organic Synthesis, Elsevier, 2005
- Fukuyama ketone synthesis see e.g. H. Tokuyama et al., Tetrahedron Lett. 1998, 39, 3189-3192
- Liebeskind-Srogl coupling see e.g.
- the resulting organometallic species can be then reacted with b2 or b3 (commercially available under the CAS-number: 36597-03-2), optionally in the presence of a transition metal catalyst, e.g. iron complex or Pd complex, to give the intermediate a3, optionally after additional hydrolysis step.
- a transition metal catalyst e.g. iron complex or Pd complex
- step 5 the carbonyl group of intermediate a4 can be reduced to give intermediate a5.
- the reduction can be done with a reagent like sodium borohydride in a solvent like ethanol at a temperature range between 0°C and room temperature.
- a reagent like sodium borohydride in a solvent like ethanol at a temperature range between 0°C and room temperature.
- other hydride reducing agents known to the person skilled in the art can be used, exemplified by lithium borohydride, lithium aluminium hydride, lithium tri-sec-butyl(hydrido)borate, optionally in the presence of an additive like TiCU or Ti(OiPr)4.
- the reduction can be performed using hydrogen gas in the presence of a catalyst, for example a transition metal catalyst.
- the reduction can be also a stereoselective reduction with non-chiral reducing agent or catalyst or a chiral reducing agent or catalyst.
- the reduction can be also an enzyme-catalyzed reduction.
- Those reduction methods are well known to the person skilled in the art and are published in the scientific literature (for asymmetric carbonyl group using chiral reagents see e.g.: E.J. Corey, L. Kurti in Enantioselective Chemical Synthesis. Methods, Logic and Practice, Elsevier, 2013; for diastereoselective carbonyl group reduction see e.g. T. Hanamoto et al., J. Org. Chem.
- step 6 the hydroxyl group of intermediate a5 can be converted to a leaving group, exemplified but not limited to Cl, Br, I, p-toluenesulfonate, methylsulfonate, triflate or acetate, to give intermediate a6 (see for e.g. Th. Netscher Recent Research Developments in Organic Chemistry 2003, 7, 71-83).
- this reaction can be carried out using stereospecific methods like Appel reaction (see e.g. Z. Wang (ed.), Comprehensive Organic Name Reactions and Reagents, John Wiley, 2009). Such methods are well known to the person skilled in the art of organic synthesis and widely exemplified in the scientific literature.
- step 7 (scheme 2) can be performed similar to step 5 starting from intermediate a3 to give intermediate a7.
- step 8 (scheme 2), the hydroxy group of intermediate a7 can be converted to a leaving group as described for step 6 to give intermediate a8.
- step 9 the protecting group of intermediate a8 can be deprotected as described for step 4 to give intermediate a6.
- PG 2 represents a suitable amine protecting group (e.g. Boc)
- G represents a functional group suitable for oxidative addition to a metal catalyst (e.g. Br, I or triflate)
- M represents a group suitable for transmetallation reaction with a metal catalyst (e.g. B(OH)2, B(OAIkyl)2,
- step 10 the intermediate of general formula c1 (prepared e.g. according to the methods and synthetic procedures described in WO2022167627 A1 can be reacted with organoelement compound d1 (e.g boronic acid derivative, exemplified for example by boronic acid CAS 1189126-37-1 or boronic acid pinacol ester CAS 947249-01-6) in a metal-catalyzed cross-coupling reaction, preferably Suzuki-Miyaura cross-coupling reaction, to give intermediate c2 (see e.g. A.
- organoelement compound d1 e.g boronic acid derivative, exemplified for example by boronic acid CAS 1189126-37-1 or boronic acid pinacol ester CAS 947249-01-6
- a metal-catalyzed cross-coupling reaction preferably Suzuki-Miyaura cross-coupling reaction
- Pd(0) catalysts like tetrakis(triphenylphosphine)palladium(0) [Pd(PPhs)4], tris(dibenzylideneacetone)di- palladium(O) [Pd2(dba)3], or by Pd(ll) catalysts like dichlorobis(triphenylphosphine)-palladium (II) [Pd(PPh 3 )2CI 2 ], palladium (II) acetate and triphenylphosphine, [1 ,1'- bis(diphenylphosphino)ferrocene] palladium dichloride as well as other catalysts and pre catalysts known to the person skilled in the art (see e.g.
- the reaction is preferably carried out in a mixture of a solvent like 1 ,2-dimethoxyethane, dioxane, DMF, THF, or isopropanol with water and in the presence of a base like potassium carbonate, sodium bicarbonate or potassium phosphate.
- a solvent like 1 ,2-dimethoxyethane, dioxane, DMF, THF, or isopropanol with water and in the presence of a base like potassium carbonate, sodium bicarbonate or potassium phosphate.
- the reaction is performed at temperatures ranging from room temperature to the boiling point of the solvent.
- the reaction is preferably completed after 1 to 36 hours.
- step 11 the amine protecting group of intermediate c2 is cleaved using procedures well known to the person skilled in the art (see e.g. T.W. Greene and P.G.M. Wuts in Protective Groups in Organic Synthesis, 4 th edition, Wiley 2006).
- the protecting group can be a Boc group and can be cleaved using a solution of hydrochloric acid in dioxane or with a solution of trifluoroacetic acid in dichloromethane at temperatures ranging from 0°C to room temperature to give intermediate c3 in a form of a salt or a free base after additional basification step.
- step 12 the spiroamine intermediate c3 can be reacted with ketone a4 under reductive amination conditions to provide the final compound of general formula c4.
- the reductive amination reaction is well known to the person skilled in the art of organic synthesis and can be performed using reducing agents like sodium cyanoborohydride, sodium triacetoxyborohydride, sodium borohydride or alfa-picoline-borane, optionally in the presence of an acid like acetic acid or para-toluenesulfonic acid or a Lewis acid like titanium (IV) tetraisopropoxide or titanium (IV) tetrachloride in a protic or aprotic organic solvent like THF, dioxane, methanol or ethanol or in water, at temperatures ranging from 0°C to the boiling point of the solvent (see e.g.
- the reductive amination reaction can be a metal-catalyzed hydrogenation reaction (see e.g. T. Irregang, R. Kempe, Chem. Rev. 2020, 120, 9583-9674).
- the reductive amination can be an asymmetric reductive amination reaction (see e.g. Y. Tian et al. , Org. Chem. Front.
- the reductive amination can be an enzyme-catalyzed reaction (see e.g. P.N. Scheller et al., ChemCatChem 2015, 7, 3239-3242).
- step 12 the spiroamine intermediate c3 can be reacted with intermediate a6 in a nucleophilic substitution reaction (N-alkylation reaction) to provide the final compound of general formula c4.
- N-alkylation reaction N-alkylation reaction
- Such reactions can proceed via SN1 or SN2 mechanism and are well known to the person skilled in the art of organic synthesis (see e.g. J. Clayden, N. Greeves, and S. Warren in Organic Chemistry, 2 nd edition, Oxford University Press 201 ).
- reaction can be performed in a solvent like DMF, THF, dioxane or toluene, in the presence of a base like sodium hydride, triethyl amine, diisopropylethylamine or pyridine at temperatures ranging from 0°C to the boiling point of the solvent.
- a base like sodium hydride, triethyl amine, diisopropylethylamine or pyridine at temperatures ranging from 0°C to the boiling point of the solvent.
- HMDS 6.5 mL, 1 .0 M in THF, 6.5 mmol
- THF 10 mL
- tert-butyl acetate 870 pL, 6.5 mmol
- 1-tert-butyl-3-methyl-3-(2- ⁇ [tert-butyl(dimethyl)silyl]oxy ⁇ ethyl)pyrrolidine-1 ,3- dicarboxylate 500 mg, 1.29 mmol
- the title compound was prepared by chiral separation of the corresponding racemic mixture.
- the title compound was prepared by chiral separation of the corresponding racemic mixture.
- Triethylamine (5.7 mL, 41 mmol) and T3P (9.6 mL, 50 % in ethyl acetate, 16 mmol) were added to 2, 2-difluorocyclopropane-1 -carboxylic acid (1.00 g, 8.19 mmol) and N-methoxymethanamine- hydrogen chloride (1/1) (879 mg, 9.01 mmol) in THF (13 mL) and stirred at rt overnight. Water (50 mL) was added and it was extracted with ethyl acetate. The organic phase was washed with brineand dried through a hydrophobic filter. The solvent was removed at a maximum of 50 °C and at a minimum of 50 mbar to yield 1 .33 g (98 % yield) of the title compound which was used without further purification in the next step.
- reaction mixture was quenched with water, extracted three times with ethyl acetate, the combined organic phases were washed with brine, filtered through a hydrophobic filter and concentrated under reduced pressure.
- the product was separated by silica gel flash chromatography (gradient 10 to 18% EtOAc in hexane) to yield 190 mg (22%) of the title compound (racemic).
- SHELXM was used for structure solution and SHELXL was used for full-matrix least-squares refinement on F 2 .
- two molecules of intermediate 15 are present. All non-hydrogen atoms were refined anisotropically. All hydrogen atoms were added in calculated positions and refined riding on their resident atoms. Hydrogens attached to N4 and C3 atoms (see Figure 1 for naming atoms) were either located in the difference Fourier map and placed manually or were refined using the riding model. The isotropic temperature factors of the hydrogen atoms were refined as 1 .2 and 1 .5 times the size of the temperature factors of the corresponding heavy atoms, respectively. The absolute stereochemistry (R-configuration) could be assigned unambiguously with a Flack Parameter of 0.05(6). The program XP was used for molecular representations. Table 3. Crystal data and structure refinement for intermediate 15.
- the reaction was stirred for 30 minutes at -75°C, thereafter the cooling bath was removed and the mixture was warmed up to room temperature.
- the reaction mixture was quenched with water and extracted 3 times with EtOAc.
- the combined organic layers were washed with water and brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure.
- the resulting crude product was purified by silica gel flash chromatography (gradient 0 to 60% EtOAc in hexane) to yield 723 mg (54 % yield) of the title compound.
- reaction mixture was stirred for 30 minutes at -75°C, thereafter the cooling bath was removed and the mixture was warmed up to rt the reaction mixturewas quenched with water and extracted 3 times with EtOAc. The combined organic layers were washed with water and brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure.
- the resulting crude product was purified by silica gel flash chromatography (gradient 0 to 30% EtOAc in hexane) to give 76.0 mg (80 % purity, 45 % yield) of the title compound as a mixture of stereoisomers.
- reaction mixture A To the stirred solution of (1 R)-2,2-difluorocyclopropane-1 -carboxylic acid (4.66, 38.17 mmol) in DCM (93 mL) was added 1 ,1'-carbonyldiimidazole (6.19 g, 38.17 mmol) and the resulting mixture was stirred for 15 minutes at room temperature and then cooled down to 0°C to give reaction mixture A.
- N-(phenylmethoxy)methanamine hydrochloride (CAS 71925-14-9, 6.026 g, 34.70 mmol) in DCM (47 mL) was added N,N-diisopropylethylamine (6.347 mL, 36.44 mmol) and the resulting mixture was stirred for 10 minutes at room temperature and then cooled down to 0°C to give reaction mixture B.
- the reaction mixtures A and B were combined at 0°C and the resulting mixture was stirred for 1 h.
- the reaction mixture was quenched with saturated aqueous ammonium chloride solution and extracted trice with DCM.
- the combined organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure to yield 8.21 g (88 % yield, 93.5% ee) of the title compound.
- the reaction mixture was concentrated, water and ethyl acetate were addd and the pH was adjusted to 7 with 1 M citric acid and saturated aqueous sodium hydrogen carbonate solution. The layers were separated and the aqueous phase was extracted twice with ethyl acetate. The combined organic phases were dried with a hydrophobic filter and concentrated. The residue was treated with ethyl acetate / toluene and concentrated again yielding 239 mg (78%) of the title compound.
- the residue was purified by HPLC (pump: Labomatic HD-5000, head HDK 280, lowpressure gradient module ND-B1000; manual injection valve: Rheodyne 3725i038; detector: Knauer Azura UVD 2.15; collector: Labomatic Labocol Vario-4000; column: Chromatorex RP C-18 10 pm, 125x30mm; eluent; gradient; UV- Detection.
- eluent A water + 0.2 vol % aqueous ammonia (32%); eluent B: acetonitril;gradient: 0.00-0.50 min 10% B (150 mL/min), 0.50-6.00 min 10-50% B (150 mL/min), 6.00-6.10 min 50- 100% B (150 mL/min), 6.10-8.00 min 100% B (150 mL/min), temperature: rt; DAD scan: 210- 400 nm) to afford 99 mg (21 % yield) of the title compound.
- Instrument Waters Autopurificationsystem; Column: Triart C18 5p, 100x30mm; eluent A: water + 0.2 vol % aqueous ammonia (32%); eluent B: methanol; gradient: 0.0-0.5 min 41% B (35-70 mL/min), 0.5-5.5 min 41-61% B; flow: 70 mL/min; temperature: 25°C; DAD scan: 210-400 nm.
- Instrument Waters Autopurificationsystem; Column: Triart C18 5p, 100x30mm; eluent A: water + 0.2 vol % aqueous ammonia (32%); eluent B: methanol; gradient: 0.0-0.5 min 41% B (35-70 mL/min), 0.5-5.5 min 41-61 % B; flow: 70 mL/min; temperature: 25°C; DAD scan: 210-400 nm.
- Instrument Waters Autopurificationsystem; Column: Triart C18 5p, 100x30mm; eluent A: water + 0.2 vol % aqueous ammonia (32%); eluent B: methanol; gradient: 0.0-0.5 min 41% B (35-70 mL/min), 0.5-5.5 min 41-61 % B; flow: 70 mL/min; temperature: 25°C; DAD scan: 210-400 nm.
- Preparative method A Instrument: Waters Autopurificationsystem; Column: Triart C18 5p, 100x30mm; eluent A: water + 0.2 vol % aqueous ammonia (32%); eluent B: methanol; gradient: 0.0-0.5 min 41% B (35-70 mL/min), 0.5-5.5 min 41-61% B; flow: 70 mL/min; temperature: 25°C; DAD scan: 210-400 nm.
- Instrument Waters Autopurificationsystem; Column: XBrigde C18 5p, 100x30mm; eluent A: water + 0.2 vol % aqueous ammonia (32%); eluent B: acetonitril; gradient: 0.0-0.5 min 15% B (35-70 mL/min), 0.5-5.5 min 15-35% B; flow: 70 mL/min; temperature: 25°C; DAD scan: 210- 400 nm.
- N,N- Diisopropylethylamine (550 pL, 3.2 mmol) and then 5- ⁇ chloro[(1R)-2,2- difluorocyclopropyl]methyl ⁇ -1 H-1,2,4-triazole hydrogen chloride (1/1) (diastereomer 1 and diastereomer 2) (285 mg, 1.24 mmol) dissolved in DMF (3.9 mL) were added successively.
- the reaction mixture was stirred overnight at rt and concentrated.
- the residue was partioned between water and ethyl acetate.
- the layers were separated and the aqueous phase was extracted with ethyl acetate.
- the combined organic phases were washed with brine, dried and concentrated obtaining 263 mg of the title compound.
- Instrument Waters Autopurificationsystem; Column: XBrigde C18 5p, 100x30mm; eluent A: water + 0.1 vol % formic acid; eluent B: methanol; gradient: 0.0-0.5 min 10% B (35-70 mL/min), 0.5-10.1 min 20-40% B; flow: 70 mL/min; temperature: rt; DAD scan: 210-400 nm.
- Instrument Waters Autopurificationsystem; Column: XBrigde C18 5p, 100x30mm; eluent A: water + 0.1 vol % formic acid; eluent B: methanol; gradient: 0.0-0.5 min 10% B (35-70 mL/min), 0.5-10.1 min 20-40% B; flow: 70 mL/min; temperature: rt; DAD scan: 210-400 nm.
- N,N- Diisopropylethylamine (5.05 mL, 29.0 mmol) and then 5- ⁇ chloro[(1 R)-2,2- difluorocyclopropyl]methyl ⁇ -1 H-1 ,2,4-triazole hydrogen chloride (1/1) (diastereomer 1 and diastereomer 2) (3.20 g, 9.74 mmol, purity 70%) dissolved in DMF (30.4 mL) were added successively.
- the reaction mixture was stirred overnight at rt and concentrated. The residue was partioned between water and ethyl acetate. The layers were separated and the aqueous phase was extracted with ethyl acetate.
- Preparative method F Instrument: Sepiatec: Prep SFClOO; Column: Chiralpak IG 5p 250x30mm; eluent A: C02; eluent B: 2-propanol; isocratic: 22%B; gradient: no; flow: 100 mL/min; temperature: 40°C; BPR: 150 bar; UV: 270 nm.
- the average value also referred to as the arithmetic mean value, represents the sum of the values obtained divided by the number of times tested
- the median value represents the middle number of the group of values when ranked in ascending or descending order. If the number of values in the data set is odd, the median is the middle value. If the number of values in the data set is even, the median is the arithmetic mean of the two middle values.
- Examples were synthesized one or more times. When synthesized more than once, data from biological assays represent average values or median values calculated utilizing data sets obtained from testing of one or more synthetic batch.
- Aqueous solubility at pH 6.5 was determined by an orientating high throughput screening method. Solubility was determined in PBS buffer pH 6.5 containing 1% DMSO. Test compounds were applied as 1mm DMSO solution. After addition of PBS buffer pH 6.5 solutions were shaken for 24 h at room temperature. Undissolved material was removed by filtration. The compound dissolved in the filtrate was quantified by HPLC-UV. The response was fitted to a one-point standard curve prepared in DMSO. Reference: Onofrey Th., Kazan, G., Barbagallo, C., Blodgett, J., Weiss, A. Millipore Corporation, Life Sciences Division, Danvers, MA USA 01923: Automated Screening of Aqueous Compound Solubility in Drug Discovery (July 31 , 2019)
- Tracer 222 from Invitrogen (catalogue no. PR9198A) was used as Alexa647-labelled ATP-competitive kinase inhibitor.
- the resulting mixture was incubated 30 min at 22°C to allow the formation of a complex between the Tracer 222, the fusion protein and Anti-GST-Tb. Subsequently the amount of this complex was evaluated by measurement of the resonance energy transfer from the Tb-cryptate to the Tracer 222. Therefore, the fluorescence emissions at 620 nm and 665 nm after excitation at 350 nm were measured in a TR-FRET reader, e.g. a Pherastar (BMG Labtechnologies, Offenburg, Germany) or a Viewlux (Perkin-Elmer). The ratio of the emissions at 665 nm and at 622 nm was taken as the measure for the amount of the complex.
- test compounds were tested on the same microtiterplate in 11 different concentrations in the range of 20 pM to 0.07 nM (20 pM, 5.7 pM, 1.6 pM, 0.47 pM, 0.13 pM, 38 nM, 11 nM, 3.1 nM, 0.9 nM, 0.25 nM and 0.07 nM, the dilution series prepared separately before the assay on the level of the lOOfold concentrated solutions in DMSO by serial dilutions, exact concentrations may vary depending pipettors used) in duplicate values for each concentration and IC50 values were calculated using Genedata ScreenerTM software.
- Flt3 inhibitory activity of compounds of the present invention at a high ATP concentration after preincubation of enzyme and test compounds was quantified employing the TR-FRET-based Flt3 activity inhibition assay as described in the following paragraphs.
- As substrate for the kinase reaction biotinylated peptide biotin-Ahx- GGEEEEYFELVKKKK (C-terminus in amide form) was used which can be purchased e.g. form the company Biosyntan (Berlin-Buch, Germany).
- nL of a 100-fold concentrated solution of the test compound in DMSO was pipetted into either a black low volume 384well microtiter plate (Greiner Bio-One, Frickenhausen, Germany), or a black 1536-well microtiter plate (Greiner Bio-one or Corning), 2 pl of a solution of Flt3 in aqueous assay buffer [25 mM HEPES pH 7.5, 10 mM MgCh, 2 mM dithiothreitol (DTT), 5 mM p-glycerolphosphate, 0.5 mM (EDTA), 0.001 % (w/v) bovine serum albumin (BSA), 0.01% (v/v) Triton X-100 (Sigma)] were added and the mixture was incubated for 15 min at 22°C to allow pre-binding of the test compounds to the enzyme before the start of the kinase reaction.
- the concentration of Flt3 was adjusted depending on the activity of the enzyme lot and was chosen appropriate to have the assay in the linear range, a typical concentration was 0.5 nM.
- TR-FRET detection reagents 167 nM streptavidine-XL665 [Cisbio Bioassays, Codolet, France] [as an alternative 333 nM streptavidine-DY648 can be used], and 1.67 nM PT66-Tb-Cryptate, an terbium-cryptate labelled anti-phospho-tyrosine antibody from Cisbio Bioassays
- aqueous EDTA-solution (66.7 mM EDTA, 0.2 % (w/v) bovine serum albumin in 50 mM HEPES/NaOH pH 7.5).
- the resulting mixture was incubated 1 h at 22°C to allow the formation of complex between the phosphorylated biotinylated peptide and the detection reagents. Subsequently the amount of phosphorylated substrate was evaluated by measurement of the resonance energy transfer from the Eu-chelate to the streptavidine-XL. Therefore, the fluorescence emissions at 620 nm and 665 nm after excitation at 350 nm was measured in a TR-FRET reader, e.g. a Pherastar FS (BMG Labtechnologies, Offenburg, Germany) or a Viewlux (Perkin-Elmer). The ratio of the emissions at 665 nm and at 622 nm was taken as the measure for the amount of phosphorylated substrate.
- TR-FRET reader e.g. a Pherastar FS (BMG Labtechnologies, Offenburg, Germany) or a Viewlux (Perkin-Elmer). The ratio of the emissions at 665 nm and at 622 nm was taken as
- test compounds were tested on the same microtiterplate in 11 different concentrations in the range of 20 pM to 0.07 nM (20 pM, 5.7 pM, 1.6 pM, 0.47 pM, 0.13 pM, 38 nM, 11 nM, 3.1 nM, 0.9 nM, 0.25 nM and 0.07 nM, the dilution series prepared separately before the assay on the level of the 100-fold concentrated solutions in DMSO by serial dilutions, exact concentrations may vary depending pipettors used) in duplicate values for each concentration and IC50 values were calculated using Genedata ScreenerTM software.
- test compound dilutions Serial dilutions of test compounds were prepared in 100% DMSO using a Precision Pipetting System (BioTek, USA). Afterwards, a 100-fold concentrated solution of the test compound (50 nL) in DMSO was transferred to microtiter test plates (384 or 1 ,536 wells, Greiner Bio-One, Germany) using either a Hummingbird liquid handler (Digilab, MA, USA) or an Echo acoustic system (Labcyte, CA, USA). Plates were sealed with adhesive foil or heat-sealed and stored at -20 °C until use.
- HTRF Homogeneous time- resolved fluorescence
- IC50 values were calculated using a four-parameter fit, with a commercial software package (Genedata Screener, Switzerland).
- Phospho-SLP-76 HTRF assay quantifies the endogenous levels of phosphorylated SLP-76, only when phosphorylated at Ser376 by using the Phospho-SLP76 HTRF kit from Cisbio (Cisbio, Condolet, France, # 63ADK076PEH).
- the assay was carried out in Jurkat E6.1 cells from American Type Culture Collection (ATCC) stably overexpressing human FLAG-tagged SLP-76 (proprietary) and frozen cell vials were generated. Cells were thawed quickly, counted and a cell suspension in assay medium (RPMI 1640, Biochrom Cat#F1275; 1% FCS Superior, Biochrom Cat#S0615 heat-inactivated; 1 % Penicillin/ Streptavidin, Biochrom Cat#A2213; 1% L-Glutamine, Gibco Cat#25030-81) with 5E+06 cells / ml was prepared.
- assay medium RPMI 1640, Biochrom Cat#F1275; 1% FCS Superior, Biochrom Cat#S0615 heat-inactivated; 1 % Penicillin/ Streptavidin, Biochrom Cat#A2213; 1% L-Glutamine, Gibco Cat#25030-81
- reaction was stopped by addition of 2 pl lysis buffer with blocking reagent (1 :25 in lysis buffer) supplied with kit. After 60 minutes for lysis at room temperature, 2 pl of detection solution from kit including the detection antibodies was dispensed to all wells. The plate was measured after 60 minutes at room temperature.
- PBMCs peripheral blood mononuclear cells
- PBMCs peripheral blood mononuclear cells
- test compounds The in vitro metabolic stability of test compounds was determined by incubating them at 1 pM in a suspension liver microsomes in 100 mM phosphate buffer, pH7.4 (NaH2PO4X H2O + Na2HPO4 x 2H2O) and at a protein concentration of 0.5 mg/mL at 37° C.
- the microsomes were activated by adding a co-factor mix containing 8 mM Glukose-6-Phosphat, 4 mM MgCI2; 0.5 mM NADP and 1 I U/ml G-6-P-Dehydrogenase in phosphate buffer, pH 7.4.
- the metabolic assay was started shortly afterwards by adding the test compound to the incubation at a final volume of 1 mL.
- Organic solvent in the incubations was limited to ⁇ 0.01 % dimethyl sulfoxide (DMSO) and ⁇ 1 % acetonitrile.
- DMSO dimethyl sulfoxide
- acetonitrile acetonitrile
- liver blood flow 1.32 L/h/kg; specific liver weight -- 38 g/kg (mouse) and 21 g/kg (human); microsomal protein content - 40 mg/g.
- the liver cells were distributed in WME containing 5% FCS to glass vials at a density of 1.0 x 10 6 vital cells/ml.
- the test compound was added to a final concentration of 1 pM.
- the hepatocyte suspensions were continuously shaken at 580 rpm and aliquots were taken at 2, 8, 16, 30, 45 and 90 min, to which equal volumes of cold methanol were immediately added. Samples were frozen at -20° C over night, after subsequently centrifuged for 15 minutes at 3000 rpm and the supernatant was analyzed with an Agilent 1290 HPLC applying MS/MS detection.
- the half-life of the test compound was determined from the concentration-time plot. From the half-life the intrinsic clearances were calculated using the 'well stirred' liver model applying the additional parameters liver blood flow, amount of liver cells in vivo and in vitro. The hepatic in vivo blood clearance (CLbiood) and the maximal oral bioavailability (F ma x) was calculated. The following parameter values were used: Liver blood flow - 4.2 L/h/kg rat, 1.32 L/h/kg human; specific liver weight - 32 g/kg (rat) and 21 g/kg (human); liver cells in vivo- 1.1 x 10 8 cells/g liver, liver cells in vitro - 1.0 x 10 6 /ml.
- Caco-2 cells (purchased from DSMZ Braunschweig, Germany) were seeded at a density of 4.5 x 10 4 cell per well on 24 well insert plates, 0.4 pm pore size, and grown for 15 days in DM EM medium supplemented with 10% fetal bovine serum, 1% GlutaMAX (100x, GIBCO), 100 U/mL penicillin, 100 pg/mL streptomycin (GIBCO) and 1 % non-essential amino acids (100 x). Cells were maintained at 37°C in a humified 5% CO2 atmosphere. Medium was changed every 2-3 day.
- TEER transepithelial electrical resistance
- Test compounds were predissolved in DMSO and added either to the apical or basolateral compartment in final concentration of 2 pM. Before and after 2 h incubation at 37°C samples were taken from both compartments. Analysis of compound content was done after precipitation with methanol by applying LC/MS/MS. Permeability (P app ) was calculated in the apical to basolateral (A - B) and basolateral to apical (B - A) directions. The apparent permeability was calculated using following equation:
- V r is the volume of medium in the receiver chamber
- P 2 is the measured peak area of the test drug in the acceptor chamber after 2 h of incubation
- t is the incubation time.
- the efflux ratio basolateral (B) to apical (A) was calculated by dividing the P app (B-A) by the Pa PP (A-B). In addition, the compound recovery was calculated.
- assay control reference compounds were analyzed in parallel.
- the inhibitory potential of the test substance towards 5 human cytochrome P450 isoforms (CYP1A2, 2C8, 2C9, 2D6, 3A4) is determined.
- CYP3A4 additionally the so-called time-dependent inhibition potential is tested.
- the test substance is preincubated in a metabolically active system for 30 minutes.
- Human liver microsomes (pool, >30 male and female donors) are used for all assays, which are incubated with individual CYP isoform-selective standard substrates (phenacetin, amodiaquine, diclofenac, dextromethorphan, midazolam).
- CYP isoform- selective inhibitors are included as positive controls (fluvoxamine for CYP1A2, montelukast for CYP2C8, sulfaphenazole for CYP2C9, fluoxetine for CYP2D6, ketoconazole for CYP3A4, and mibefradil for CYP3A4 pre-incubation).
- the incubation conditions are optimized with regard to the following parameters: protein concentration, substrate concentration, incubation time and metabolic turnover.
- the incubation medium consists of 50 mM potassium phosphate buffer (pH 7.4), 1 mM EDTA, NADPH regenerating system (1 mM NADP, 5 mM glucose 6-phosphate, glucose 6-phosphate dehydrogenase (1.5 U/mL)). Sequential dilutions and all incubations are carried out in 96-MTP plate format at 37°C in a final volume of 200 pL and under automated conditions using a Genesis Workstation (Tecan, Crailsheim). The enzymatic reaction is stopped by adding 100 pL acetonitrile including internal standard. After protein precipitation and centrifugation, the supernatants are analyzed.
- CYP1A2 The metabolites paracetamol (CYP1A2), desethylamodiaquine (CYP2C8), 4-hydroxydiclofenac (CYP2C9), dextrorphan (CYP2D6), and 1 -hydroxymidazolam (CYP3A4) are quantified using LC/MS/MS.
- the CYP-mediated enzyme activity is determined as a function of the test substance concentration and the enzyme-kinetic parameter IC50 is calculated.
- Binding of test compounds to plasma proteins has been measured by equilibrium dialysis in a 96-well format using HTDialysis equipment made of Teflon and a semipermeable membrane (regenerated cellulose, MWCO 12-14K).
- the membrane separates the plasma and buffer cavities (50 mM phosphate buffer) filled with 150 pl each.
- the test compound is added to the plasma cavity at a test concentration of 3 pM and binds to plasma proteins.
- the unbound fraction of the test compound passes the membrane and distributes on both sides until equilibrium is reached, which is usually the case after 6-8h at 37°C and 5% CO2 atmosphere.
- Relative compound concentration (peak area ratios analyte/IS) of plasma and buffer side is measured by LC/MS/MS analytics.
- BAYXXX (test item) inhibits the human Ether-a-go-go-Related Gene (hERG) potassium channel
- in vitro automated voltage clamp recordings were performed on recombinant HEK293 cells stably expressing the hERG alpha subunit (hERG cells). Following harvest, cells were transferred to the cell reservoir of a 384 channel automated patch clamp device and stored there at 20°C until usage.
- hERG cells were transferred to a 384 well patch clamp chip (pipette resistance of ⁇ 2-3 MQ) prefilled with external solution (containing in mM: 143 NaCI, 4 KOI, 2 CaCh, 1 MgCI 2 , 5 glucose, 10 HEPES; pH 7.4 (NaOH)). Underpressure was applied underneath the glass bottom of the patch clamp chip to position the hERG cells on the recording sites in the glass bottom of the chip.
- a seal enhancing solution (containing in mM: 78 NaCI, 60 NMDG, 4 KOI, 10 CaCI2, 1 MgCI2, 5 glucose, 10 HEPES; pH 7.4 (HOI)) was added to the hERG cells to facilitate the formation of stable seals between the membranes of the hERG cells and the glass next to the recording sites. Then, hERG cells were washed several times with wash solution (containing in mM: 87 NaCI, 60 NMDG, 4 KCI, 2 CaCI 2 , 1 MgCI 2 , 5 glucose, 10 HEPES; pH 7.4 (HOI)) to remove excess seal enhancing solution.
- wash solution containing in mM: 87 NaCI, 60 NMDG, 4 KCI, 2 CaCI 2 , 1 MgCI 2 , 5 glucose, 10 HEPES; pH 7.4 (HOI)
- the membrane parts of the hERG cells covering the recording sites were exposed to internal solution (containing in mM: 10 NaCI, 123 KF, 10 EGTA, 10 HEPES; pH 7.2 (KOH)) supplemented with 5-20 pM Escin, and the perforated patch configuration was established.
- the holding potential was stepwise adjusted to -80 mV, capacitance was compensated, and a series of defined voltage commands was initiated to trigger the hERG current response from the hERG cells (-80 mV for 200 ms, +20 mV for 1000 ms, -40 mV for 500 ms; repeated at a frequency of 0.1 Hz).
- a negative control i.e. wash solution supplemented with 0.3% DMSO and 0.01 % HSA
- a solution containing the test item at a final concentration of (0.1 , 1 , or 10) pM was applied for 10 min to measure eventual inhibitory effects of the test item on the hERG current (- the test item solution was produced from a 10 mM DMSO stock by using an automated pipetting device and sequential dilution).
- a positive control i.e. wash solution supplemented with 0.3% DMSO, 0.01% HSA, and 10 pM quinidine
- hERG tail current amplitudes were averaged from three consecutive current responses at the end of the negative control phase, test item phase, and positive control phase, respectively. Resulting mean hERG tail current amplitudes were normalized to the mean hERG tail current amplitude at the end of the negative control phase with nominal 0% inhibition as well as the hERG tail current amplitude at the end of the positive control phase with nominal 100% inhibition. Then, the effect of the test item was calculated as a percentage inhibition value at the test item concentration applied.
- test compounds were administered to male Wistar rats intravenously at doses of 0.3 to 0.8 mg/kg and intragastric at doses of 0.5 to 1.5 mg/kg formulated as solutions using solubilizers such as PEG400 in well-tolerated amounts.
- test compounds were given as i.v. bolus and blood samples were taken at 2 min, 8 min, 15 min, 30 min, 45 min, 1 h, 2 h, 4 h, 6 h, 8 h and 24 h after dosing.
- test compounds were given intragastric to fasted rats and blood samples were taken at 5 min, 15 min, 30 min, 45 min,
- PK parameters were calculated by noncompartmental analysis using a PK calculation software.
- t-1/2 terminal half-life (in h);
- F oral bioavailability: AUCnorm after intragastric administration divided by AUCnorm after intravenous administration (in %).
- Table 7 In vivo pharmacokinetics data for the target compound of the invention after i.v. bolus administration in Wistar rats
- Table 8 In vivo pharmacokinetics data for the target compound of the invention after oral administration in Wistar rats
- PEG400 polyethylenglycol 400
- unbound compound exposure in plasma is considered equal to the unbound compound concentration in the target tissue. Therefore, unbound plasma exposure level can be directly used to assess coverage of pharmacologically active concentrations (IC50u) that are defined in cellular or biochemical in vitro assays.
- IC50u pharmacologically active concentrations
- Table 9 In vivo exposure data for the example xx (BAY 3619082) of the invention after oral/intragastraladministration in female Balb/c AnN mice (Charles River)
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention covers MAP4K1 inhibitor compounds of formula (I) as described and defined herein, methods of preparing said compounds, intermediate compounds useful for preparing said compounds, pharmaceutical compositions and combinations comprising said compounds, and the use of said compounds for manufacturing pharmaceutical compositions for the treatment or prophylaxis of diseases, in particular for treatment, amelioration or prevention of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, or amelioration of vaccine therapies or cell therapies, as a sole agent or in combination with other active ingredients.
Description
NEW MAP4K1 INHIBITORS
The present invention covers MAP4K1 inhibitor compounds of formula (I) as described and defined herein, methods of preparing said compounds, intermediate compounds useful for preparing said compounds, pharmaceutical compositions and combinations comprising said compounds, and the use of said compounds for manufacturing pharmaceutical compositions for the treatment or prophylaxis of diseases, in particular for treatment, amelioration or prevention of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, or amelioration of vaccine therapies or cell therapies, as a sole agent or in combination with other active ingredients.
The present invention further relates to the use, respectively to the use of said compounds for manufacturing pharmaceutical compositions for the treatment or prophylaxis of protein inhibitors in benign hyperplasias, atherosclerotic disorders, sepsis, autoimmune disorders, vascular disorders, viral infections, in neurodegenerative disorders, in inflammatory disorders, in atherosclerotic disorders and in male fertility control.
This work has been conducted under a Research and Collaboration Agreement by Bayer AG and Deutsches Krebsforschungszentrum (DKFZ).
BACKGROUND
Although cancer cell commonly can be recognized by the adaptive immune system, the response generated is evidently not capable of eliminating the tumor. A major reason for this is the presence of immunosuppressive mechanisms in the tumor microenvironment. In this respect, inhibitors of T-cell immune checkpoint such as CTLA-4, PD-1 or PD-L1 were recently shown to result in a remarkable clinical efficacy in subsets of cancer patients. Besides cell surface receptors that act as negative immune regulators, several mediators of intracellular signaling have been identified that also represent potential immunoevasive mechanisms utilized by the tumor.
One of these is MAP4K1 , also known as hematopoietic progenitor kinase 1 (HPK1). MAP4K1 (GenelD11184) is a serine/threonine kinase and member of the Germinal Center Kinase family. In the adult organism MAP4K1 expression is restricted to hematopoietic cell types. The MAP4K1 protein consist of a N-terminal kinase domain, followed by a proline-rich domain that can interact with adaptor molecules through SH2 and SH3 domains, and a C-terminal citron homology domain of which the exact function remains to be identified. Through its proline-rich domain, MAP4K1 is capable of binding to a diversity of adaptors in hematopoietic cells, including those involved in T-cell receptor (TCR), B-cell receptor (BCR) and cytokine signaling (Hu et al., Genes Dev. 1996 Sep 15;10(18):2251-64, 2.; Ling et al.,. J Biol Chem. 2001 Jun
1;276(22), Sauer et al., J Biol Chem. 2001 Nov 30;276(48):45207-16., Tsuji et al., J Exp Med. 2001 Aug 20;194(4):529-39, Boomer et al., J Cell Biochem. 2005 May 1 ;95(1):34-44).
The function of MAP4K1 has been studied in greatest detail in the context of TCR signaling. Upon TCR stimulation, MAP4K1 is phosphorylated on tyrosine 381 (Y-381; Y-379 in mouse) (Di Bartolo et al., J Exp Med. 2007 Mar 19;204(3):681-91). Consequently, MAP4K1 is recruited to the TCR-signaling complex where it induces dissociation of this complex through its serine/threonine kinase function. In particular MAP4K1 phosphorylates the SLP-76 adaptor protein at Serine-376, resulting in downregulation of AP-1 and Erk2 pathways. As, such, MAPK1 acts as a negative feedback on TCR-signaling (Liou et al., Immunity. 2000 Apr;12(4):399-408; Lasserre et al., J Cell Biol. 2011 Nov 28;195(5):839-53.). Alternatively, MAP4K1 can be triggered to suppress T cell function by prostaglandin E2 (PGE2), and possibly also by transforming growth factor beta (TGF-beta), factors that are commonly found in the tumor microenvironment. Notably, MAP4K1 activation by these mediators involves protein kinase A (PKA)-dependent phosphorylation of Serine 171 (S-171; also in mouse) (Alzabin et al., Cancer Immunol Immunother. 2010 Mar;59(3):419-29; Sawasdikosol et al., J Biol Chem. 2007 Nov 30;282(48):34693-9.).
Further important insights into the function of MAP4K1 in the regulation of T cell immunity stem from in vivo and in vitro experiments respectively with MAP4K1 deficient mice produced by two laboratories and with immune cells isolated from these mice (Shui et al., Nat Immunol. 2007 Jan;8(1):84-91; Alzabin et al., Cancer Immunol Immunother. 2010 Mar;59(3):419-29).
MAP4K1 -deficient mice show an apparent normal phenotype, are fertile and exhibit normal lymphocyte development. These animals are prone to develop T-cell dependent autoimmune reactivity as indicated by development of a more severe disease score in the EAE (experimental autoimmune encephalomyelitis) model of multiple sclerosis (Shui et al., Nat Immunol. 2007 Jan;8(1):84-91). In case of the second strain, a dysregulation of immune function was observed when, at the age of approximately 6 months, MAP4K1 -deficient mice develop a spontaneous autoimmune phenotype (Alzabin et al., Cancer Immunol Immunother. 2010 Mar;59(3):419-29). In vitro studies showed that MAP4K1-/- T-cells display hyperresponsiveness upon TCR-stimulation. These cells proliferate and secrete pro-inflammatory cytokines like IL-2 or IFNg to a significantly greater extent than their wild-type counterparts (Shui et al., Nat Immunol. 2007 Jan;8(1):84-91). Furthermore, MAP4K1-/- T-cells are resistant to PGE2-mediated suppression of T cell proliferation, suppression of IL-2 production and induction of apoptosis (Alzabin et al., Cancer Immunol Immunother. 2010 Mar;59(3):419-29). In the context of tumor immunology, in vivo experiments revealed that MAP4K1-/- mice are much more resistant to tumorigenesis by PGE2-producing Lewis lung carcinoma than wild type mice, which correlated with increased T-lymphocyte infiltration in the tumor areas. The crucial role of T-cells in tumor rejection was supported by experiments in which MAP4K1-/- T-cells adoptively transferred into T-cell-deficient mice were able to eradicate tumors more efficiently
than wild-type T-cells (Alzabin et al., Cancer Immunol Immunother. 2010 Mar;59(3):419-29).
The important role of the kinase enzymatic activity was demonstrated by studies were only wild type MAP4K1 , but not the MAP4K1 kinase-dead mutant, could mediate serine-phosphorylation of the TCR-signaling complex component SLP-76 and subsequent binding of SLP-76 to the negative regulator of TCR-signaling 14-3-3-t (Shui et al., Nat Immunol. 2007 Jan;8(1):84-91).
MAP4K1 also regulates the stimulation and activation of dendritic cells. MAP4K1 deficient
Bone marrow derived cells (BMDC) express after maturation and stimulation higher level of costimulatory molecules and produce more proinflammatory cytokines. Also elimination of tumors was observed to be more efficient by MAP4K1 -/- BMDC compared to their wildtype counterparts (Alzabin et al., J Immunol. 2009 May 15;182(10):6187-94).
Prior art
The prior art discloses a multitude of HPK1 (HPK1 and MAP4K1 are referring to the same human protein) inhibitors. Table (1) exemplifies state of the art disclosing HPK1 inhibitors with publication date between January 2021 to January 2023. For the review of earlier reports on HPK1 inhibitors see e.g. I.D. Linney, K. Neelu, Expert Opinion on Therapeutic Patents, 2021 , 31 , 893-910. For additional reviews on HPK1 inhibitors see for example L. Zhou et al., Eur. J. Med. Chem. 2022, 244, 114819; see also Q. Zhu et al., J. Med. Chem. 2022, 65, 8065-8090.
Table (1) Examples of prior art describing HPK1 inhibitors (published between January 2021 and January 2023)

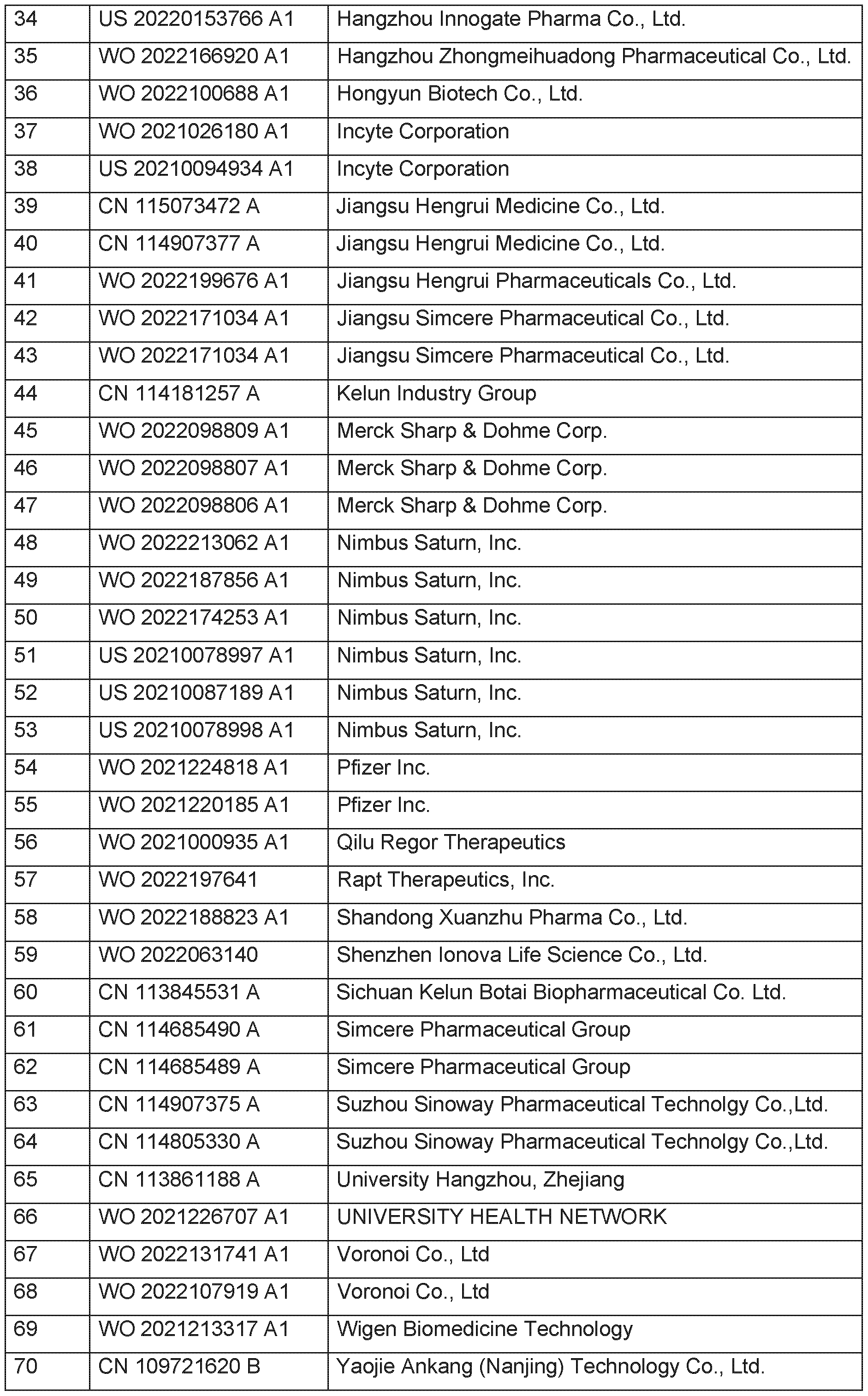

HPK1 (MAP4K1) inhibitors based on (2,2-difluorocyclopropyl)(1 ,2,4-triazolyl)methanamine substructures S-1 or S-2 or S-3 as present in the compounds of general formula (I) of the present invention have not been exemplified in the prior art.

Compounds displaying a 5-(1 H-pyrazol-3-yl)-3-(trifluoromethyl)pyridin-2-amine substructure S-
4 are known from the prior art but have not been reported to be part of a HPK1 (MAP4K1) inhibitor as described below.
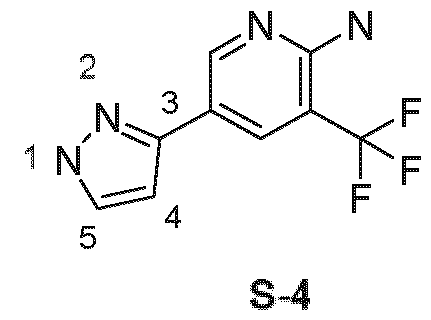
Compound L-1 and other compounds with general formula L-2 have been described in WO2014111496 as DLK (MAP3K12) inhibitors for the treatment of neurodegenerative diseases. According to the definition of the general formula L-2 the substituents R1, R5, R6and R7 can have the meaning hydrogen, substituent R4 can have the meaning CF3, however substituents R2 and R3 can not together form a cyclic system as present in formula (I) of the present invention. In addition, compounds of general formula L-2 have not been reported to have HPK1 (MAP4K1) inhibition activity. Some examples disclosed in WO2014111496 as well as further analogues corresponding to the general formula L-2 are also reported in S. Patel et al., J. Med. Chem. 2017, 60, 8083-8102.

Compounds with general formula L-3 have been described in WO2015091889 A1 to have DLK (MAP3K12) inhibitory activity. While these compounds contain the (1 H-pyrazol-3yl)pyridine substructure S-4, the definition of the general formula L-3 and the meaning of R6 does not allow
the pyrazole ring to be fused with a cycloalkyl ring or a (hetero)spirocycloalkyl ring system. In addition, compounds of general formula L-3 have not been reported to have HPK1 (MAP4K1) inhibition activity.
Similar compounds with general formula L-4 have been described in WO2018107072 to also have DLK (MAP3K12) inhibitory activity. According to the definition of general formula L-4, the meaning of X3 is a nitrogen, the meaning of X4 and X5 is a carbon and the meaning of X1 and X2 can be a carbon or a nitrogen. Thus, X1-X5 can form together a pyrazole ring. However, the definition of the general formula L-4 and the meaning of R1 does not allow the pyrazole ring to be fused with a cycloalkyl ring or a (hetero)spirocycloalkyl ring system as present in the general formula (I) of the present invention. In addition, compounds of general formula L-4 have not been reported to have HPK1 (MAP4K1) inhibition activity.

Compound L-5 and other compounds with general formula L-6 have been described in WO2014153208 as arginine methyltransferase inhibitors. A related compound L-7 has been described in WO2017136699 as arginine methyltransferase inhibitor. According to the description of the invention in WO2014153208 the meaning of general formula L-6 can match the substructure S-4, however the meaning of X, Y and Z in L-6 does not allow pyrazole ring to be fused to a cycloalkyl ring and does not allow pyrazole to be unsubstituted at position 4. In addition, compounds of general formula L-6 and compound L-7 have not been reported to have
HPK1 (MAP4K1) inhibition activity.

Compound L-8 and other examples of general formula L-9 have been described in
WO2022147246 to have FGFR tyrosine kinase inhibitory activity. Although these compounds
contain the (1 H-pyrazol-3yl)pyridine substructure S-4, according to general formula L-9 the pyrazole ring can only be a part of an indazole ring system. In addition, compounds of general formula L-9 have not been reported to have HPK1 (MAP4K1) inhibition activity.
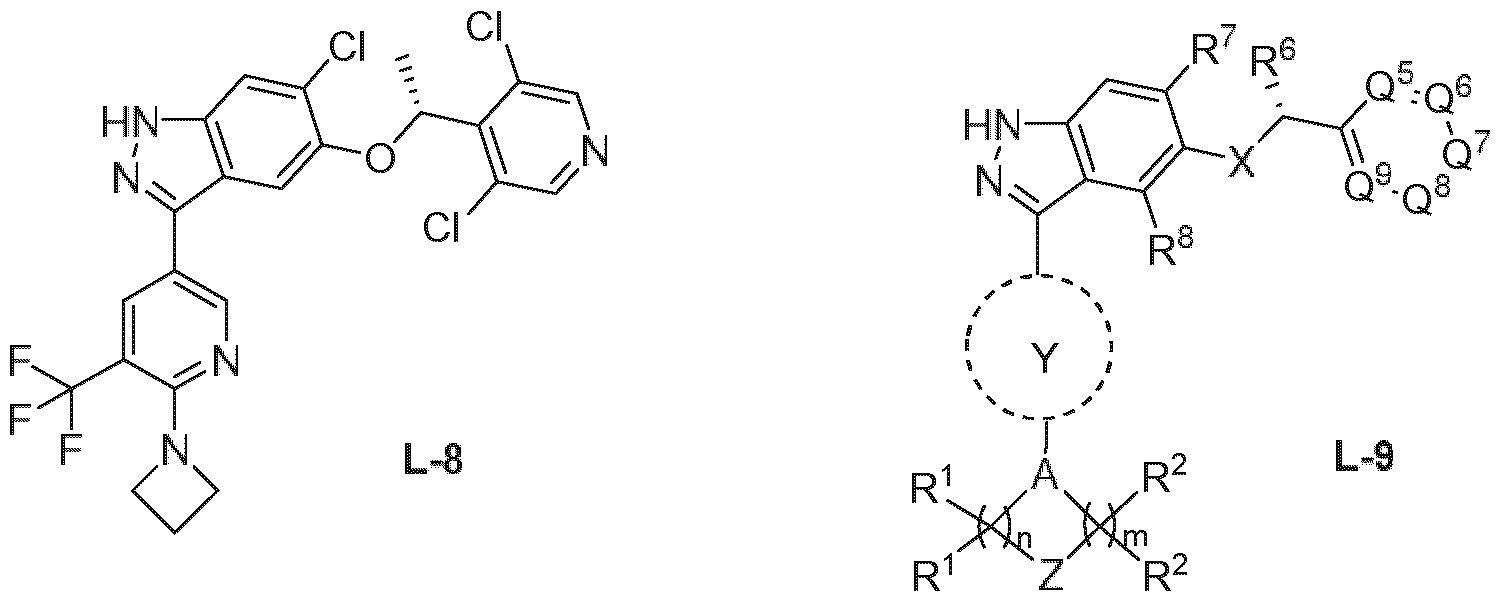
Compounds with 5',6'-dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazole] substructure S-5 as present in the general formula (I) of the present invention are known from the prior art as described below.

S-5
Compound L-10 and other compounds of general formula L-11 have been recently described in WO2021074279 as HPK1 (MAP4K1) inhibitors. According to the definition of the general formula L-11 , the meaning of X can be a bond and Y can represent a hydrogen. At the same time, the meaning of R1 can be a group *-A-B, where B can represent a 3-membered cycloalkyl, optionally substituted with halogen atoms, and where A can represent *-CRaH- group, where Ra can be a 5-membered heteroaryl. Thus, according to the definition of the general formula L-11 , the substituent R1, together with the nitrogen to which it is attached, can represent (2,2- difluorocyclopropyl)(1 ,2,4-triazolyl)methanamine substructure (S-1 , S-2 or S-3). However, such compounds are not exemplified in WO2021074279. In addition, the meaning of R2 can only be a bicyclic heteroaromatic structure limited to quinoline, 1 ,5- or 1 ,6-naphthyridine, 1 H-pyrrolo[2,3- b]pyridine, 1 H-pyrazolo[3,4-b]pyridine or 3H-imidazo[4,5-b]pyridine. Thus, R2 can not mean pyridine as present in the formula (I) of the present invention.
L-10 L-11
Compound L-12 and other compounds of general formula L-13 have been described in WO2022167627 as HPK1 (MAP4K1) inhibitors. According to the definition of the general formula L-13, the meaning of R1 can be CF3, the meaning of Q can be a carbon and the meaning of A can be a bond. At the same time, the meaning of E can be -(CH2)n- with n = 2 and the meaning of G is -(CH2)-. Thus, the substructure -E-N-G- together with the carbon to which it is attached can form a pyrrolidine ring. At the same time, the meaning of R4 can be Ci-3-alkyl-C3-6-cycloalkyl, optionally substituted with one or more fluorine atoms and independently optionally substituted with 5-membered heteroaryl. Thus, the substituent R4 together with the nitrogen to which it is attached can form a (2,2-difluorocyclopropyl)(1 ,2,4-triazolyl)methanamine substructure (S-1 , S- 2 or S-3). However, compounds containing such a substructure are not exemplified in WO2022167627 and are not known from any prior art.
At the same time, the compound of general formula (I) of the present invention, containing the unique (2,2-difluorocyclopropyl)(1 ,2,4-triazolyl)methanamine substructure (S-1 , S-2 or S-3), is shown to have multiple unexpected advantages over compounds described in the prior art. Such advantageous properties are listed below. The incorporation of the unique (2,2- difluorocyclopropyl)(1 ,2,4-triazolyl)methanamine substructure (S-1 , S-2 or S-3) to achieve such advantageous properties is a modification that is not obvious to the person skilled in the art.

Accordingly, it is an object of the present invention to provide a compound and pharmaceutical compositions comprising this compound for prophylactic and therapeutic applications for hyperproliferative disorders, in particular for cancer, respectively tumour disorders, and conditions with dysregulated immune responses, as a sole agent or in combination with other active ingredients.
A further object of the present invention is to provide a compound and pharmaceutical compositions comprising this compound for manufacturing pharmaceutical compositions for the treatment or prophylaxis of benign hyperplasias, atherosclerotic disorders, sepsis, autoimmune disorders, vascular disorders, viral infections, in neurodegenerative disorders, in inflammatory disorders, in atherosclerotic disorders and in male fertility control.
The compound according to the invention inhibits the MAP4K1 protein and thereby enhances tumor immunogenicity leading to inhibition of cancer cells growth by the immune response. Accordingly, the invention provides a new compound for the therapy of human and animal disorders, in particular of cancers.
It has now been found, and this constitutes the basis of the present invention, that the compound of the present invention shows additional surprising and advantageous properties.
This variety of unique properties makes the compound of formula (I) of the present invention most suitable as an active ingredient in a medicinal product. In particular the compound of the present invention has surprisingly been found to show or be:
1) a high aqueous solubility (Table 4: kinetic solubility in pH 6.5 aqueous phosphate buffer). It is well known to the person skilled in the art that high aqueous solubility is required to achieve desired concentration of drug substance in systemic circulation for desired pharmacological response. It is also known to the person skilled in the art that high aqueous solubility of a drug substance improves oral absorption of the drug. It is also known to the person skilled in the art that high aqueous solubility of a drug substance simplifies formulation development of the drug. For reference see e.g. E. H. Kerns, L. Di, Chapter 7 - Solubility, Editor(s): E. H. Kerns, L. Di, Drug-like Properties: Concepts, Structure Design and Methods, Academic Press, 2008, pages 56-85; see also R. J. Young, 2021. "Tactics to Improve Solubility", The Medicinal Chemist's Guide to Solving ADMET Challenges, Editor(s): P. Schnider, RSC, 2021.
2) a highly potent hMAP4K1 inhibitor (Table 5). It is obvious to the person skilled in the art that high MAP4K1 potency is advantageous for a drug substance used to treat MAP4K1- mediated diseases or disorders. High potency reduces the required concentration of the drug substance in systemic circulation required for desired pharmacological response. Thus, high potency of the drug substance towards its target of action increases the probability to achieve the desired pharmacological response as well as reduces the risk of adverse events due to interaction with other off-targets. High potency of the drug substance also reduces the required drug dose.
3) a highly selective MAP4K1 kinase inhibitor as it shows a selectivity factor of >100 against FLT3 kinase (Table 5)
4) a potent inhibitor of pSLP76 phsophorylation in a human cell line (Table 5)
5) a potent inducer of cytokine secretion in PGE2-stimulated human peripheral blood mononuclear cells (PBMCs) (Table 5, Figure 4)
6) a high in vitro metabolic stability in human liver microsomes and hepatocytes (Table 6). It is well known to the person skilled in the art that high in vitro metabolic stability in human liver microsomes and hepatocytes is required to achieve desired concentration of drug substance in systemic circulation for desired pharmacological response. It is also known to the person skilled in the art that if high stability on incubation with rat liver microsomes and hepatocytes results in a significant correlation with in vivo blood clearance in rat, the desired concentration of drug substance in systemic circulation for desired pharmacological response should be achievable in human, too. For reference see e.g. E. H. Kerns, L. Di, Chapter 11 - Metabolic Stability, Editor(s): E. H. Kerns, L. Di, Drug-like Properties: Concepts, Structure Design and Methods, Academic Press, 2008, pages 137-168; see also P. Baranczewski et. al, Introduction to in vitro estimation of metabolic stability and drug interactions of new chemical entities in drug discovery and development. Pharmacol Rep. 2006, 58 (4), 453-472.
7) moderate permeability and hint on active transport (efflux) through monolayer of Caco- 2 cells. In addition, efflux is saturated on increasing concentration (Table 6). It is well known to the person skilled in the art that moderate or high permeability and without strong efflux is a prerequisite to achieve desired concentration of drug substance in systemic circulation for desired pharmacological response following oral administration. For reference see e.g. E. H. Kerns, L. Di, Chapter 8 - Permeability, Chapter 9 - Transporters, Chapter 26 - Permeability Methods, Chapter 26 - Transporter Methods, Editor(s): E. H. Kerns, L. Di, Drug-like Properties: Concepts, Structure Design and Methods, Academic Press, 2008, pages 86-121 and 287-310.
8) no inhibition of CYP 450 metabolic enzymes in vitro at concentrations significantly above the concentration required to inhibit hMAP4K1 in a biochemical assay or to inhibit pSLP76 phosphorylation in a cellular assay (Table 6). It is well known to the person skilled in the art that correspondingly high IC50 data of the CYP inhibition assay is indicating low or very low risk for drug-drug interactions in patients. For reference see e.g. E. H. Kerns, L. Di, Chapter 15 - Cytochrome P450 Inhibition, Editor(s): E. H. Kerns, L. Di, Drug-like Properties: Concepts, Structure Design and Methods, Academic Press, 2008, pages 197-208.
9) a high selectivity against cardiac ion channels, e.g. against hERG channel (Table 6). It is known to the person skilled in the art that high selectivity against cardiac ion channels, in particular hERG channel, results in insignificantly reduced risk of cardiovascular side effects in humans. For reference see e.g. C. Bordoni, D.J. Brough, G. Davison, J.H. Hunter, J.D. Lopez-Fernandez, K. McAdam, D.C. Miller, P.A. Morese, A. Papaioannou,
M. Uguen, P. Ratcliffe, N. Sitnikov, M.J. Waring, 2021. "Cardiac Ion Channel Inhibition", The Medicinal Chemist's Guide to Solving ADMET Challenges, Patrick Schnider, RSC, 2021.
10) a moderate in vivo clearance in rat and mouse (Table 7). It is well known to the person skilled in the art that low or moderate in vivo clearance in rodents should translate to desired concentration of drug substance in systemic circulation for desired pharmacological response in humans. For reference see e.g. D.A. Smith, K. Beaumont, T.S. Maurer, L.Di, Clearance in Derug Design, J. Med. Chem. 2019, 62, 2245-2255.
11) Efficacy in EMT 6 model
Thus, the compound shows an unexpected unique combination of properties making it useful to treat human diseases via inhibition of hMAP4K1. While the known structurally related examples from the prior art WO2022167627A1 can have one or a small subset of the advantageous properties described above, none of them is known to have this unique combination. For example, there is no compound described in WO2022167617A1 that is a highly selective MAP4K1 inhibitor, has high selectivity against hERG cardiac ion channel and has simultaneously a moderate in vivo clearance (<1.5 L/h/kg) in rat.
DESCRIPTION of the INVENTION
The present invention covers the following embodiment:
Embodiment A) The compound
5-{-l-[(2,2-difluorocyclopropyl)(lH-l,2,4-triazol-5-yl)methyl]-5',6'-dihydrospiro[pyrrolidine-
3,4'-pyrrolo[l,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine of formula (I)
 or a stereoisomer, a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same.
or a stereoisomer, a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same.
DEFINITIONS
In general, and unless otherwise mentioned, the heteroaryl or heteroarylene groups include all possible isomeric forms thereof, e.g.: tautomers and positional isomers with respect to the point of linkage to the rest of the molecule. Thus, for some illustrative non-restricting examples, the
term pyridinyl includes pyridin-2-yl, pyridin-3-yl and pyridin-4-yl; or the term thienyl includes thien-2-yl and thien-3-yl.
As used herein, the term “leaving group” means an atom or a group of atoms that is displaced in a chemical reaction as stable species taking with it the bonding electrons. In particular, such a leaving group is selected from the group comprising: halide, in particular fluoride, chloride, bromide or iodide, (methylsulfonyl)oxy, [(trifluoromethyl)sulfonyl]oxy, [(nonafl uorobutyl)- sulfonyl]oxy, (phenylsulfonyl)oxy, [(4-methylphenyl)sulfonyl]oxy, [(4-bromophenyl)sulfonyl]oxy, [(4-nitrophenyl)sulfonyl]oxy, [(2-nitrophenyl)sulfonyl]oxy, [(4-isopropylphenyl)sulfonyl]oxy, [(2,4,6-triisopropylphenyl)sulfonyl]oxy, [(2,4,6-trimethylphenyl)sulfonyl]oxy, [(4-terf-butyl- phenyl)sulfonyl]oxy and [(4-methoxyphenyl)sulfonyl]oxy.
It is possible for the compounds of general formula (I) to exist as isotopic variants. The invention therefore includes one or more isotopic variant(s) of the compounds of general formula (I), particularly deuterium-containing compounds of general formula (I).
The term “Isotopic variant” of a compound or a reagent is defined as a compound exhibiting an unnatural proportion of one or more of the isotopes that constitute such a compound.
The term “Isotopic variant of the compound of general formula (I)” is defined as a compound of general formula (I) exhibiting an unnatural proportion of one or more of the isotopes that constitute such a compound.
The expression “unnatural proportion” means a proportion of such isotope which is higher than its natural abundance. The natural abundances of isotopes to be applied in this context are described in “Isotopic Compositions of the Elements 1997”, Pure Appl. Chem., 70(1), 217-235, 1998.
Examples of such isotopes include stable and radioactive isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus, sulfur, fluorine, chlorine, bromine and iodine, such as 2H (deuterium), 3H (tritium), 11C, 13C, 14C, 15N, 170, 180, 32P, 33P, 33S, 34S, 35S, 36S, 18F, 36CI, 82Br, 123l, 124l, 125|, 129| and 131l, respectively.
With respect to the treatment and/or prophylaxis of the disorders specified herein the isotopic variant(s) of the compounds of general formula (I) preferably contain deuterium (“deuterium- containing compounds of general formula (I)”). Isotopic variants of the compounds of general formula (I) in which one or more radioactive isotopes, such as 3H or 14C, are incorporated are useful e.g. in drug and/or substrate tissue distribution studies. These isotopes are particularly preferred for the ease of their incorporation and detectability. Positron emitting isotopes such as 18F or 11C may be incorporated into a compound of general formula (I). These isotopic variants of the compounds of general formula (I) are useful for in vivo imaging applications. Deuterium- containing and 13C-containing compounds of general formula (I) can be used in mass spectrometry analyses in the context of preclinical or clinical studies.
Isotopic variants of the compounds of general formula (I) can generally be prepared by methods known to a person skilled in the art, such as those described in the schemes and/or examples herein, by substituting a reagent for an isotopic variant of said reagent, preferably for a deuterium-containing reagent. Depending on the desired sites of deuteration, in some cases deuterium from D2O can be incorporated either directly into the compounds or into reagents that are useful for synthesizing such compounds. Deuterium gas is also a useful reagent for incorporating deuterium into molecules. Catalytic deuteration of olefinic bonds and acetylenic bonds is a rapid route for incorporation of deuterium. Metal catalysts (i.e. Pd, Pt, and Rh) in the presence of deuterium gas can be used to directly exchange deuterium for hydrogen in functional groups containing hydrocarbons. A variety of deuterated reagents and synthetic building blocks are commercially available from companies such as for example C/D/N Isotopes, Quebec, Canada; Cambridge Isotope Laboratories Inc., Andover, MA, USA; and CombiPhos Catalysts, Inc., Princeton, NJ, USA.
The term “deuterium-containing compound of general formula (I)” is defined as a compound of general formula (I), in which one or more hydrogen atom(s) is/are replaced by one or more deuterium atom(s) and in which the abundance of deuterium at each deuterated position of the compound of general formula (I) is higher than the natural abundance of deuterium, which is about 0.015%. Particularly, in a deuterium-containing compound of general formula (I) the abundance of deuterium at each deuterated position of the compound of general formula (I) is higher than 10%, 20%, 30%, 40%, 50%, 60%, 70% or 80%, preferably higher than 90%, 95%, 96% or 97%, even more preferably higher than 98% or 99% at said position(s). It is understood that the abundance of deuterium at each deuterated position is independent of the abundance of deuterium at other deuterated position(s).
The selective incorporation of one or more deuterium atom(s) into a compound of general formula (I) may alter the physicochemical properties (such as for example acidity [C. L. Perrin, et al., J. Am. Chem. Soc., 2007, 129, 4490], basicity [C. L. Perrin et al., J. Am. Chem. Soc., 2005, 127, 9641], lipophilicity [B. Testa et al., Int. J. Pharm., 1984, 19(3), 271]) and/or the metabolic profile of the molecule and may result in changes in the ratio of parent compound to metabolites or in the amounts of metabolites formed. Such changes may result in certain therapeutic advantages and hence may be preferred in some circumstances. Reduced rates of metabolism and metabolic switching, where the ratio of metabolites is changed, have been reported (A. E. Mutlib et al., Toxicol. Appl. Pharmacol., 2000, 169, 102). These changes in the exposure to parent drug and metabolites can have important consequences with respect to the pharmacodynamics, tolerability and efficacy of a deuterium-containing compound of general formula (I). In some cases deuterium substitution reduces or eliminates the formation of an undesired or toxic metabolite and enhances the formation of a desired metabolite (e.g. Nevirapine: A. M. Sharma et al., Chem. Res. Toxicol., 2013, 26, 410; Efavirenz: A. E. Mutlib et
al., Toxicol. Appl. Pharmacol., 2000, 169, 102). In other cases the major effect of deuteration is to reduce the rate of systemic clearance. As a result, the biological half-life of the compound is increased. The potential clinical benefits would include the ability to maintain similar systemic exposure with decreased peak levels and increased trough levels. This could result in lower side effects and enhanced efficacy, depending on the particular compound’s pharmacokinetic/ pharmacodynamic relationship. ML-337 (C. J. Wenthur et al., J. Med. Chem., 2013, 56, 5208) and Odanacatib (K. Kassahun et al., WO2012/112363) are examples for this deuterium effect. Still other cases have been reported in which reduced rates of metabolism result in an increase in exposure of the drug without changing the rate of systemic clearance (e.g. Rofecoxib: F. Schneider et al., Arzneim. Forsch. I Drug. Res., 2006, 56, 295; Telaprevir: F. Maltais et al., J. Med. Chem., 2009, 52, 7993). Deuterated drugs showing this effect may have reduced dosing requirements (e.g. lower number of doses or lower dosage to achieve the desired effect) and/or may produce lower metabolite loads.
The compound of formula (I) may have multiple potential sites of attack for metabolism. To optimize the above-described effects on physicochemical properties and metabolic profile, deuterium-containing compounds of general formula (I) having a certain pattern of one or more deuterium-hydrogen exchange(s) can be selected. Particularly, the deuterium atom(s) of deuterium-containing compound(s) of general formula (I) is/are attached to a carbon atom and/or is/are located at those positions of the compound of general formula (I), which are sites of attack for metabolizing enzymes such as e.g. cytochrome P450.
In another embodiment the present invention concerns a deuterium-containing compound of general formula (I).
In another embodiment the present invention concerns a deuterium-containing compound of general formula (I) with 1 , 2, 3, 4, 5, 6, 7, 8 or 9 deuterium atoms, particularly 1 , 2, 3, 4, 5 or 6 deuterium atoms.
In another embodiment the present invention concerns a deuterium-containing compound of general formula (I) having 1 , 2, 3 or 4 deuterium atoms, particularly with 1 , 2 or 3 deuterium atoms.
Where the plural form of the word compounds, salts, polymorphs, hydrates, solvates and the like, is used herein, this is taken to mean also a single compound, salt, polymorph, isomer, hydrate, solvate or the like.
The compound of the present invention contains three asymmetric centres. It is possible that one or more asymmetric carbon atoms are present in the (R) or (S) configuration, which can result in diastereomeric mixtures of up to 8 stereoisomers in variable ratios.
Preferred compounds are those which produce the more desirable biological activity. Separated, pure or partially purified isomers and stereoisomers or racemic or diastereomeric mixtures of the
compounds of the present invention are also included within the scope of the present invention. The purification and the separation of such materials can be accomplished by standard techniques known in the art.
The optical isomers can be obtained by resolution of the racemic mixtures according to conventional processes, for example, by the formation of diastereoisomeric salts using an optically active acid or base or formation of covalent diastereomers. Examples of appropriate acids are tartaric, diacetyltartaric, ditoluoyltartaric and camphorsulfonic acid. Mixtures of diastereoisomers can be separated into their individual diastereomers on the basis of their physical and/or chemical differences by methods known in the art, for example, by chromatography or fractional crystallisation. The optically active bases or acids are then liberated from the separated diastereomeric salts. A different process for separation of optical isomers involves the use of chiral chromatography (e.g., HPLC columns using a chiral phase), with or without conventional derivatisation, optimally chosen to maximise the separation of the enantiomers. Suitable HPLC columns using a chiral phase are commercially available, such as those manufactured by Daicel, e.g., Chiracel OD and Chiracel OJ, for example, among many others, which are all routinely selectable. Enzymatic resolution with isolated enzymes or microorganisms, with or without derivatisation, are also useful. The optically active compounds of the present invention can likewise be obtained by chiral syntheses utilizing optically active starting materials.
In order to distinguish different types of isomers from each other reference is made to IUPAC Rules Section E (Pure Appl Chem 45, 11-30, 1976).
The present invention includes all possible stereoisomers of the compounds of the present invention as single stereoisomers, or as any mixture of said stereoisomers, e.g. (R)- or (S)- isomers, in any ratio. Isolation of a single stereoisomer, e.g. a single enantiomer or a single diastereomer, of a compound of the present invention is achieved by any suitable state of the art method, such as chromatography, especially chiral chromatography, for example.
Further, it is possible for the compounds of the present invention to exist as tautomers. For example, any compound of the present invention which contains triazole as a heteroaryl group for example can exist as a 1 H tautomer, 2H-tautomer or a 4H tautomer, or even a mixture in any amount of the three tautomers, namely:

1 H tautomer 4H tautomer 2H tautomer
The present invention includes all possible tautomers of the compounds of the present invention as single tautomers, or as any mixture of said tautomers, in any ratio.
The chemical structure of compond of formula (I)
 described herein can also be drawn as
described herein can also be drawn as
 because of potential tautomerism of the triazole ring.
because of potential tautomerism of the triazole ring.
The present invention also covers useful forms of the compounds of the present invention, such as metabolites, hydrates, solvates, prodrugs, salts, in particular pharmaceutically acceptable salts, and/or co-precipitates.
The compounds of the present invention can exist as a hydrate, or as a solvate, wherein the compounds of the present invention contain polar solvents, in particular water, methanol or ethanol for example, as structural element of the crystal lattice of the compounds. It is possible for the amount of polar solvents, in particular water, to exist in a stoichiometric or non- stoichiometric ratio. In the case of stoichiometric solvates, e.g. a hydrate, hemi-, (semi-), mono- , sesqui-, di-, tri-, tetra-, penta- etc. solvates or hydrates, respectively, are possible. The present invention includes all such hydrates or solvates.
Further, it is possible for the compounds of the present invention to exist in free form, e.g. as a free base, or as a free acid, or as a zwitterion, or to exist in the form of a salt. Said salt may be any salt, either an organic or inorganic addition salt, particularly any pharmaceutically acceptable organic or inorganic addition salt, which is customarily used in pharmacy, or which is used, for example, for isolating or purifying the compounds of the present invention.
The term “pharmaceutically acceptable salt" refers to an inorganic or organic acid addition salt of a compound of the present invention. For example, see S. M. Berge, et al. “Pharmaceutical Salts,” J. Pharm. Sci. 1977, 66, 1-19.
A suitable pharmaceutically acceptable salt of the compounds of the present invention may be, for example, an acid-addition salt of a compound of the present invention bearing a nitrogen atom, in a chain or in a ring, for example, which is sufficiently basic, such as an acid-addition salt with an inorganic acid, or “mineral acid”, such as hydrochloric, hydrobromic, hydroiodic, sulfuric, sulfamic, bisulfuric, phosphoric, or nitric acid, for example, or with an organic acid, such as formic, acetic, acetoacetic, pyruvic, trifluoroacetic, propionic, butyric, hexanoic, heptanoic, undecanoic, lauric, benzoic, salicylic, 2-(4-hydroxybenzoyl)-benzoic, camphoric, cinnamic, cyclopentanepropionic, digluconic, 3-hydroxy-2-naphthoic, nicotinic, pamoic, pectinic, 3- phenylpropionic, pivalic, 2-hydroxyethanesulfonic, itaconic, trifluoromethanesulfonic, dodecylsulfuric, ethanesulfonic, benzenesulfonic, para-toluenesulfonic, methanesulfonic, 2-naphthalenesulfonic, naphthalinedisulfonic, camphorsulfonic acid, citric, tartaric, stearic, lactic, oxalic, malonic, succinic, malic, adipic, alginic, maleic, fumaric, D-gluconic, mandelic, ascorbic, glucoheptanoic, glycerophosphoric, aspartic, sulfosalicylic, or thiocyanic acid, for example.
Further, another suitably pharmaceutically acceptable salt of a compound of the present invention which is sufficiently acidic, is an alkali metal salt, for example a sodium or potassium salt, an alkaline earth metal salt, for example a calcium, magnesium or strontium salt, or an aluminium or a zinc salt, or an ammonium salt derived from ammonia or from an organic primary, secondary or tertiary amine having 1 to 20 carbon atoms, such as ethylamine, diethylamine, triethylamine, ethyldiisopropylamine, monoethanolamine, diethanolamine, triethanolamine, dicyclohexylamine, dimethylaminoethanol, diethylaminoethanol, tris(hydroxymethyl)aminomethane, procaine, dibenzylamine, /V-methylmorpholine, arginine, lysine, 1 ,2-ethylenediamine, /V-methylpiperidine, /V-methyl-glucamine, /V,/V-dimethyl-glucamine, /V-ethyl-glucamine, 1 ,6-hexanediamine, glucosamine, sarcosine, serinol, 2-amino-1 ,3- propanediol, 3-amino-1 ,2-propanediol, 4-amino-1 ,2,3-butanetriol, or a salt with a quarternary ammonium ion having 1 to 20 carbon atoms, such as tetramethylammonium, tetraethylammonium, tetra(n-propyl)ammonium, tetra(n-butyl)ammonium, /V-benzyl-/V,/V,/V- trimethylammonium, choline or benzalkonium.
Those skilled in the art will further recognise that it is possible for acid addition salts of the claimed compounds to be prepared by reaction of the compounds with the appropriate inorganic or organic acid via any of a number of known methods. Alternatively, alkali and alkaline earth metal salts of acidic compounds of the present invention are prepared by reacting the compounds of the present invention with the appropriate base via a variety of known methods.
The present invention includes all possible salts of the compounds of the present invention as single salts, or as any mixture of said salts, in any ratio.
In the present text, in particular in the Experimental Section, for the synthesis of intermediates and of examples of the present invention, when a compound is mentioned as a salt form with the corresponding base or acid, the exact stoichiometric composition of said salt form, as obtained by the respective preparation and/or purification process, is, in most cases, unknown.
Unless specified otherwise, suffixes to chemical names or structural formulae relating to salts, such as "hydrochloride", "trifluoroacetate", "sodium salt", or "x HO", "x CF3COOH", "x Na+", for example, mean a salt form, the stoichiometry of which salt form not being specified.
This applies analogously to cases in which synthesis intermediates or example compounds or salts thereof have been obtained, by the preparation and/or purification processes described, as solvates, such as hydrates, with (if defined) unknown stoichiometric composition.
Furthermore, the present invention includes all possible crystalline forms, or polymorphs, of the compounds of the present invention, either as single polymorph, or as a mixture of more than one polymorph, in any ratio.
Moreover, the present invention also includes prodrugs of the compounds according to the invention. The term “prodrugs” here designates compounds which themselves can be biologically active or inactive, but are converted (for example metabolically or hydrolytically) into compounds according to the invention during their residence time in the body.
In accordance with other embodiments, the present invention covers compounds of general formula (I), supra, in which:
Embodiment B) The compound
5-{(3R)-l-[(2,2-difluorocyclopropyl)(lH-l,2,4-triazol-5-yl)methyl]-5',6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[l,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine of formula (I b) according to embodiment A
 or a stereoisomer, a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same.
Embodiment C) The compound
or a stereoisomer, a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same.
Embodiment C) The compound
5-{(3R)-l-[((R)-2,2-difluorocyclopropyl)(lH-l,2,4-triazol-5-yl)methyl]-5',6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[l,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine of formula (I c) according to embodiment A or B
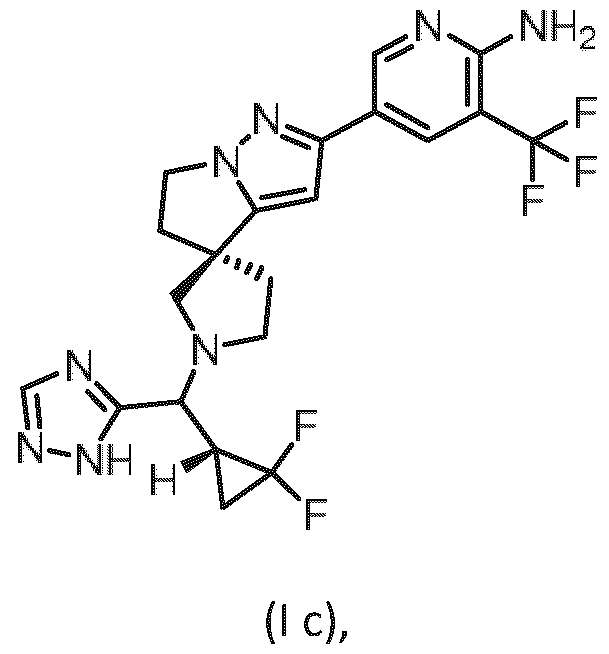 or a stereoisomer, a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same.
or a stereoisomer, a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same.
Embodiment D) The compound
5-{(3R)-l-[((S)-2,2-difluorocyclopropyl)(lH-l,2,4-triazol-5-yl)methyl]-5',6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[l,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine of formula (I d) according to embodiment A or B

(I d), or a stereoisomer, a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same. Embodiment E) The compound
5-{(3R)-l-[(S)-((R)-2,2-difluorocyclopropyl)(lH-l,2,4-triazol-5-yl)methyl]-5',6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[l,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine of formula (I e) according to embodiment A, B or C
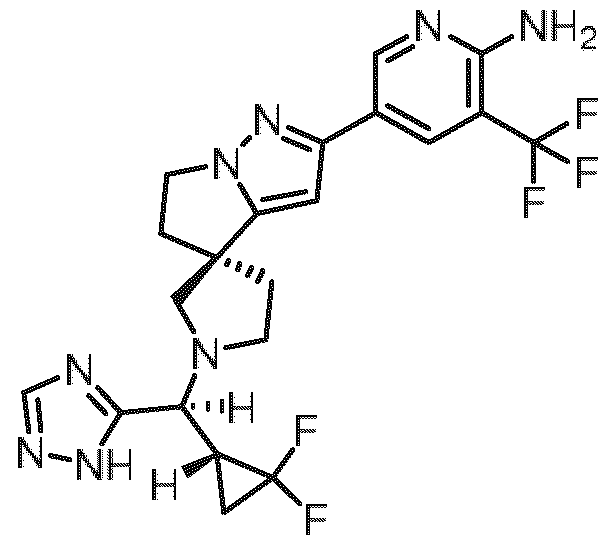
(I e), or a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same.
Embodiment F) The compound 5-{(3R)-l-[(R)-((R)-2,2-difluorocyclopropyl)(lH-l,2,4-triazol-5-yl)methyl]-5',6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[l,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine of formula (I f) according to embodiment A, B or C
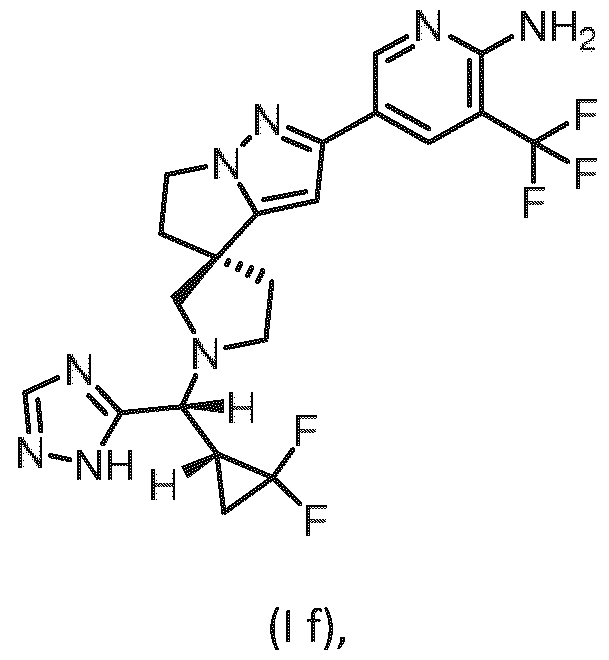 or a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same.
or a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same.
Embodiment F1) The compound
5-{(3R)-l-[(R)-[(lR)-2,2-difluorocyclopropyl](lH-l,2,4-triazol-5-yl)(2H)methyl]-5',6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[l,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine according to claim 4
 or a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same.
or a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same.
Embodiment G) The compound
5-{(3R)-l-[(S)-((S)2,2-difluorocyclopropyl)(lH-l,2,4-triazol-5-yl)methyl]-5',6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[l,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine of formula (I g) according to embodiment A, B or D

(I g), or a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same.
Embodiment H) The compound
5-{(3R)-l-[(R)-((S)-2,2-difluorocyclopropyl)(lH-l,2,4-triazol-5-yl)methyl]-5',6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[l,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine of formula (I h) according to embodiment A, B or D
 (I h), or a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same.
(I h), or a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same.
Other embodiments of the present invention cover methods of preparing compounds of general formula (I). Additionally they cover methods of preparing compounds of the present invention of general formula (I), said methods comprising the steps as described in the Experimental Section herein.
Embodiment I) The specific stereoisomer of the compound of embodiment F, obtainable by stirring a solution of 5-[(3R)-5’,6’-dihydrospiro[pyrrolidine-3,4’-pyrrolo[1 ,2-b]pyrazol]- 2’-yl]-3-(trifluoromethyl)pyridin-2-amine hydrogen chloride (1/1) (420 mg, 1.17 mmol), [2,2-difluorocyclopropyl](1 H-1 ,2,4-triazol-5-yl)methanone (220 mg, 1.27 mmol), titanium(IV) isopropoxide (940 pL, 3.2 mmol) and DIPEA (920 pL, 5.3 mmol) in methanol (11 mL) at 60°C for 4 h adding sodium cyanoborohydride (166 mg, 2.65 mmol) and stirring the reaction mixture overnight at 60°C adding water, stirring for additional 10 min, filtering and washing the filter cake with methanol the volatiles of the filtrate were removed under reduced pressure extracting the remaining aqueous phase residue twice with ethyl acetate and washing the combined ethyl acetate layers with saturated sodium chloride solution, then drying and concentrating purifying the residue by HPLC (pump: Labomatic HD-5000, head HDK 280, lowpressure gradient module ND-B1000; manual injection valve: Rheodyne 3725i038; detector: Knauer Azura UVD 2.15; collector: Labomatic Labocol Vario- 4000; column: Chromatorex RP C-18 10 pm, 125x30mm; eluent; gradient; UV- Detection. eluent A: water + 0.2 vol % aqueous ammonia (32%); eluent B: acetonitril;gradient: 0.00-0.50 min 10% B (150 mL/min), 0.50-6.00 min 10-50% B (150 mL/min), 6.00-6.10 min 50-100% B (150 mL/min), 6.10-8.00 min 100% B (150 mL/min), temperature: rt; DAD scan: 210-400 nm) to afford 5-{(3R)-1 -[(2,2- difluorocyclopropyl)(1 H-1 ,2,4-triazol-5-yl)methyl]-5',6'-dihydrospiro[pyrrolidine-3,4'- pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine (all fractions having a molecular mass of 480 Daltons and similar retention times... were combined) separating the stereoisomeric mixture of 5-{(3R)-1-[(2,2-difluorocyclopropyl)(1 H- 1 ,2,4-triazol-5-yl)methyl]-5',6'-dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'- yl}-3-(trifluoromethyl)pyridin-2-amine (95.0 mg, 198 pmol) by HPLC method A
(Instrument: Waters Autopurificationsystem; Column: Triart C18 5p, 100x30mm; eluent A: water + 0.2 vol % aqueous ammonia (32%); eluent B: methanol; gradient: 0.0-0.5 min 41 % B (35-70 mL/min), 0.5-5.5 min 41-61 % B; flow: 70 mL/min; temperature: 25°C; DAD scan: 210-400 nm. Retention time: 4.2 - 4.7 min, first/ eluting) further separating the resulting mixture by chiral HPLC method C (Instrument: PrepCon Labomatic HPLC-3; Column: Chiralpak IG 5p, 250x30; eluent A: hexane + 0.1 vol % diethylamine; eluent B: ethanol; isocratic: 70% A + 30% B; flow: 70 mL/min; temperature: 25°C; UV: 280 nm. Retention time: 8.04 - 9.75 min, first/ eluting) or a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same.
Embodiment J) The specific stereoisomer of the compound of embodiment F, obtainable by dissolving 5-[(3R)-5',6'-dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl]-3- (trifluoromethyl)pyridin-2-amine hydrogen chloride (1/1) (200 mg, 528 pmol) in DMF (3.9 mL) adding successively N,N-diisopropylethylamine (550 pL, 3.2 mmol) and then 5- {chloro[(1 R)-2,2-difluorocyclopropyl]methyl}-1 H-1 ,2,4-triazole hydrogen chloride (1/1) (diastereomer 1 and diastereomer 2) (285 mg, 1.24 mmol) dissolved in DMF (3.9 mL) stirring the reaction mixture overnight at rt concentrating the reaction mixture partitioning the residue between water and ethyl acetate separating the layers extracting the aqueous phase with ethyl acetate
- washing the combined organic phases with brine drying and concentrating separating by HPLC (Instrument: Waters Autopurificationsystem; Column: XBrigde C18 5p, 100x30mm; eluent A: water + 0.1 vol % formic acid; eluent B: methanol; gradient: 0.0-0.5 min 10% B (35-70 mL/min), 0.5-10.1 min 20-40% B; flow: 70 mL/min; temperature: rt; DAD scan: 210-400 nm. Retention time: 5.1 - 6.0 min) the first-eluting diastereomer or a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same.
Or as a second procedure
dissolving 5-[(3R)-5',6'-dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl]-3- (trifluoromethyl)pyridin-2-amine hydrogen chloride (1/1) (1.52 g, 4.14 mmol) in DMF (30.4 mL) adding successively N,N-diisopropylethylamine (5.05 mL, 29.0 mmol) and then 5- {chloro[(1 R)-2,2-difluorocyclopropyl]methyl}-1 H-1 ,2,4-triazole hydrogen chloride (1/1) (diastereomer 1 and diastereomer 2) (3.20 g, 9.74 mmol) dissolved in DMF (30.4 mL) stirring the reaction mixture overnight at rt concentrating the reaction mixture partitioning the residue between water and ethyl acetate separating the layers extracting the aqueous phase with ethyl acetate
- washing the combined organic phases with brine drying and concentrating pyrifying by chromatography (Sfaer Amino, hexane/ethyl acetate 5-100% and ethyl acetae/ethanol 0-100%) separating by HPLC (Instrument: Sepiatec: Prep SFC100; Column: Chiralpak IG 5p 250x30mm; eluent A: CO2; eluent B: 2-propanol; isocratic: 22%B; gradient: no; flow: 100 mL/min; temperature: 40°C; BPR: 150 bar; UV: 270 nm. Retention time: 7.9 -- 11.9 min) the second-eluting diastereomer or a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same.
Embodiment K) A method of preparing a compound of formula (I) according to embodiment A, said method comprising the step of allowing an intermediate compound of general formula (II), a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof:
 in which Y represents a leaving group, to react with a compound of formula (III), a stereoisomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof:
in which Y represents a leaving group, to react with a compound of formula (III), a stereoisomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof:
 thereby giving the compound of formula (I) of claim 1:
thereby giving the compound of formula (I) of claim 1:
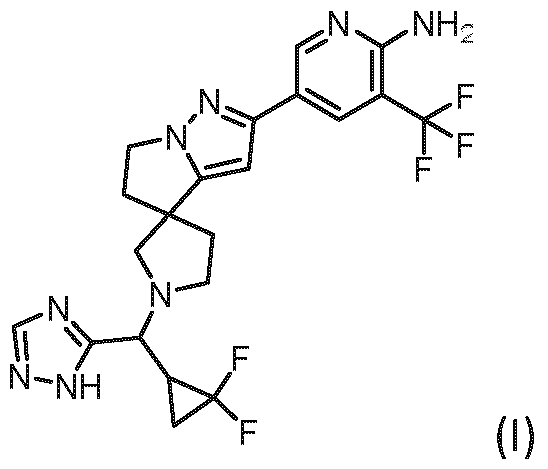 or a stereoisomer, a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same. Embodiment L) A method of preparing a compound of formula (I) according to embodiment A, said method comprising the step of allowing an intermediate compound of general formula (Ila), a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof:
or a stereoisomer, a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same. Embodiment L) A method of preparing a compound of formula (I) according to embodiment A, said method comprising the step of allowing an intermediate compound of general formula (Ila), a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof:
 in which the ketone to react with a compound of formula (III), a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof:
in which the ketone to react with a compound of formula (III), a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof:
 thereby giving the compound of formula (I) of claim 1:
thereby giving the compound of formula (I) of claim 1:
 a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof.
a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof.
Embodiment M) The compound of formula (I) of embodiment A for use in the treatment or prophylaxis of a disease.
Embodiment N) A pharmaceutical composition comprising a compound of formula (I) of embodiment A and one or more pharmaceutically acceptable excipients.
Embodiment O) A pharmaceutical combination comprising:
• one first active ingredient, in particular the compound of formula (I) embodiment A, and
• one or more further active ingredients, in particular:
1311-metuximab, 1311-chTNT, abarelix, abemaciclib, abiraterone, acalabrutinib, aclarubicin, adalimumab, ado-trastuzumab emtansine, afatinib, aflibercept, aldesleukin, alectinib, alfaferone, alemtuzumab, alendronic acid, alitretinoin, almonertinib, alpelisib, alpharadin, monosodium alpha luminol, altretamine, amifostine, aminoglutethimide, hexyl aminolevulinate, aminolevulinic acid, amrubicin, amsacrine, anastrozole, ancestim, anethole dithiolethione, anetumab ravtansine, angiotensin II, antithrombin III, apalutamide, aprepitant, arcitumomab, arglabin, arsenic trioxide, asparaginase, atezolizumab, avapritinib, avelumab, axicabtagene ciloleucel, axitinib, azacitidine, basiliximab, beclomethasone dipropionate, belantamab mafodotin, belinostat, belotecan, bendamustine, besilesomab, beta-elemene, bevacizumab, bexarotene, bicalutamide, binimetinib, bisantrene, bleomycin, blinatumomab, boanmycin hydrochloride, borofalan, bortezomib, bosutinib, budesonide, buserelin, brentuximab vedotin, brigatinib, busulfan, cabazitaxel, cabozantinib, calcitonine, calcium folinate, calcium levofolinate, capecitabine, capmatinib, capromab, carbamazepine, carboplatin, carboquone, carfilzomib, carmofur, carmustine, catumaxomab, catequentinib, celecoxib, celmoleukin, cemiplimab, ceritinib, cetuximab, chlorambucil, chlormadinone,
chlormethine, chloroxoquinoline, cidofovir, cinacalcet, cisplatin, cladribine, clodronic acid, clofarabine, cobimetinib, copanlisib, crisantaspase, crizotinib, cyclophosphamide, cyproterone, cytarabine, dacarbazine, dacomitinib, dactinomycin, daratumumab, darbepoetin alfa, dabrafenib, darolutamide, dasatinib, daunorubicin, decitabine, deferasirox, degarelix, denileukin diftitox, denosumab, depreotide, deslorelin, dianhydrogalactitol, dexrazoxane, dibrospidium chloride, dianhydrogalactitol, diclofenac, dinutuximab, docetaxel, dolasetron, doxifluridine, doxorubicin, doxorubicin + estrone, dronabinol, dupilumab, durvalumab, duvelisib, eculizumab, edrecolomab, elliptinium acetate, elotuzumab, eltrombopag, enasidenib, encorafenib, endostatin, enfortumab vedotin, enocitabine, enzalutamide, epirubicin, epitiostanol, epoetin alfa, epoetin beta, epoetin zeta, eptaplatin, eribulin, erlotinib, ensartinib, entrectinib, erdafitinib, esomeprazole, estradiol, estramustine, estrone, ethinylestradiol, etoposide, everolimus, evocalcet, exemestane, fadrozole, famotidine, fentanyl, filgrastim, flumatinib, fluoxymesterone, fluticasone, fluticasone furoate, floxuridine, fludarabine, fluorouracil, flutamide, folinate, folinic acid, formestane, forodesine, fosaprepitant, fotemustine, fruquintinib, fulvestrant, gadobutrol, gadoteridol, gadoteric acid meglumine, gadoversetamide, gadoxetic acid, gallium nitrate, ganirelix, gefitinib, gemcitabine, gemtuzumab, gendicine, gilteritinib, ginsenoside Rg3, glasdegib, glucarpidase, glutoxim, GM-CSF, goserelin, granisetron, granulocyte colony stimulating factor, hematoporphyrin, histamine dihydrochloride, histrelin, holmium-166-chitosan complex, human menopausal gonadotrophin, hydroxycarbamide, 1-125 seeds, lansoprazole, ibandronic acid, ibritumomab tiuxetan, ibrutinib, icotinib hydrochloride, idarubicin, idelalisib, iobenguane (1311), iodi ne( 13 II ) tumor necrosis factor monoclonal antibody, ifosfamide, imatinib, imiquimod, improsulfan, immunocyanin, indisetron, incadronic acid, indole-3-carbinol + epigallocatechin-3-gallate, ingenol mebutate, inotuzumab ozogamicin, interferon alfa, interferon alpha lb, interferon-alpha 2, interferon alpha-2a, interferon alfa-2b, interferon beta, interferon gamma, interleukin- 2, iobitridol, iobenguane (1231), iomeprol, ipilimumab, irinotecan, Itraconazole, isatuximab, ivosidenib, ixabepilone, ixazomib, lanreotide, lansoprazole, lapatinib, larotrectinib, lasocholine, lenalidomide, lenvatinib, lenograstim, lentinan, letrozole, leuprorelin, levamisole, levonorgestrel, levothyroxine sodium, lisuride, lobaplatin, lomustine, lonidamine, lorlatinib, lurbinectedin, luspatercept, lutetium Lu 177 dotatate, masoprocol, medroxyprogesterone, megestrol, melarsoprol, melphalan, mepitiostane,
mercaptamine, mercaptopurine, mesna, methadone, methotrexate, methoxsalen, methylaminolevulinate, methylprednisolone, methyltestosterone, metirosine, midostaurin, mifamurtide, mifepristone, miltefosine, miriplatin, mitobronitol, mitoguazone, mitolactol, mitomycin, mitotane, mitoxantrone, mogamulizumab, molgramostim, mometasone, mopidamol, morphine hydrochloride, morphine sulfate, mvasi, moxetumomab pasudotox, nabilone, nabiximols, nafarelin, naloxone + pentazocine, naltrexone, nartograstim, natalizumab, necitumumab, nedaplatin, nelarabine, neratinib, neridronic acid, netupitant/palonosetron, nivolumab, pentetreotide, nilotinib, nilutamide, nimorazole, nimotuzumab, nimustine, nintedanib, niraparib, nitracrine, nivolumab, obinutuzumab, octreotide, ofatumumab, olaparib, olaratumab, olmutinib, omacetaxine mepesuccinate, omalizumab, omeprazole, ondansetron, oprelvekin, orelabrutinib, orgotein, orilotimod, osimertinib, oxaliplatin, oxycodone, oxymethoIone, ozogamicine, p53 gene therapy, paclitaxel, padeliporfin, palbociclib, palifermin, palladium-103 seed, palonosetron, pamidronic acid, panitumumab, panobinostat, pantoprazole, pazopanib, pegaspargase, PEG-epoetin beta (methoxy PEG-epoetin beta), pembrolizumab, pemigatinib, pegfilgrastim, peginterferon alfa-2b, pembrolizumab, pemetrexed, pentazocine, pentostatin, peplomycin, Perflubutane, perfosfamide, Pertuzumab, phenoxybenzamine, picibanil, pilocarpine, pirarubicin, pixantrone, plerixafor, plicamycin, poliglusam, polatuzumab vedotin, polyestradiol phosphate, polyvinylpyrrolidone + sodium hyaluronate, polysaccharide-K, pomalidomide, ponatinib, porfimer sodium, pralatrexate, pralsetinib, prednimustine, prednisone, procarbazine, procodazole, propranolol, quinagolide, quizartinib, rabeprazole, racotumomab, radium-223 chloride, radotinib, raloxifene, raltitrexed, ramosetron, ramucirumab, ranimustine, rasburicase, razoxane, refametinib, regorafenib, relugolix, ribociclib, ripretinib, risedronic acid, rhenium-186 etidronate, rituximab, rivoceranib, rolapitant, romidepsin, romiplostim, romurtide, rucaparib, sacituzumab govitecan, samarium (153Sm) lexidronam, sargramostim, sarilumab, satumomab, secretin, selinexor, selpercatinib, selumetinib, siltuximab, sipuleucel-T, sirolimus, sizofiran, sobuzoxane, sodium glycididazole, sonidegib, sophoridine hydrochloride, sorafenib, stanozolol, streptozocin, Strontium 89, sunitinib, surufatinib, tagraxofusp, talaporfin, talazoparib, talimogene laherparepvec, tamibarotene, tamoxifen, tapentadol, tasonermin, tazemetostat, teceleukin, technetium (99mTc) nofetumomab merpentan, 99mTc-HYNIC-[Tyr3]-octreotide, tegafur, tegafur + gimeracil
+ oteracil, temoporfin, temozolomide, temsirolimus, teniposide, testosterone, tetrofosmin, thalidomide, thiotepa, thrombopoietin, thymalfasin, thyrotropin alfa, tioguanine, tirabrutinib, tisagenlecleucel, tislelizumab, tivozanib, tocilizumab, topotecan, toremifene, tositumomab, trabectedin, trametinib, tramadol, trastuzumab, trastuzumab emtansine, treosulfan, tretinoin, trifluridine + tipiracil, trilostane, triptorelin, trametinib, trofosfamide, tryptophan, tucatinib, tucidinostat, recombinant tumor necrosis a-factor of thymosine-al, ubenimex, ulipristal, umbralisib, valatinib, valrubicin, vandetanib, vapreotide, vemurafenib, venetoclax, vinblastine, vincristine, vindesine, vinflunine, vinorelbine, vismodegib, vorinostat, vorozole, yttrium-90 glass microspheres, zanubrutinib, zinostatin, zinostatin stimalamer, zoledronic acid, zorubicin.
Embodiment P) Use of the compound of formula (I) of embodiment A for the treatment or prophylaxis of a disease.
Embodiment Q) Use of the compound of formula (I) of embodiment A for the preparation of a medicament for the treatment or prophylaxis of a disease.
Embodiment R) Use according to embodiment P or Q, wherein the disease is a neoplastic or abnormal cell proliferative disorder, such as cancer, a condition with dysregulated immune response or another disorder associated with aberrant Map4K1 signaling.
In accordance with other embodiments , the present invention covers intermediate compounds which are useful for the preparation of the compounds of general formula (I), supra. The present invention covers the intermediate compounds which are disclosed in the Example Section of this text, infra.
Embodiment S) The compound of general formula (II):
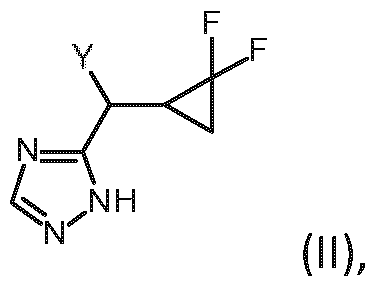 in which Y represents a leaving group) or a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof.
in which Y represents a leaving group) or a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof.
Embodiment T) The compound of general formula (Il a):
 a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof.
a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof.
Embodiment U) The compound of general formula (III):
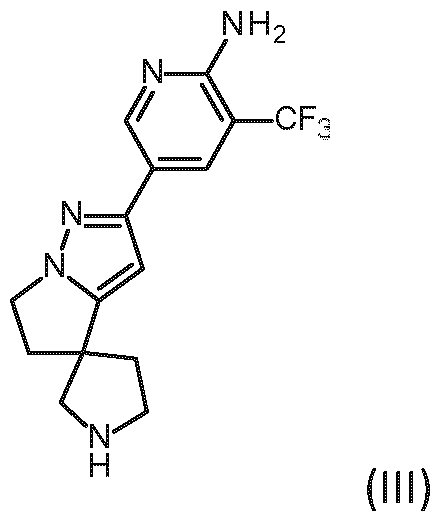 a stereoisomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof.
a stereoisomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof.
In accordance with other embodiments, the present invention covers the use of said intermediate compounds for the preparation of a compound of general formula (I) as defined supra.
Embodiment V) Use of a compound of general formula (II)
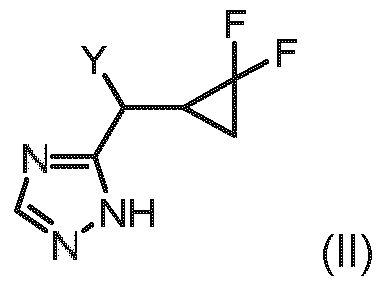 a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof in which Y a leaving group, for the preparation of a compound of formula (I) of embodiment A.
a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof in which Y a leaving group, for the preparation of a compound of formula (I) of embodiment A.
Embodiment W) Use of a compound of general formula (II)
 a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof for the preparation of a compound of formula (I) of embodiment A.
Embodiment X) Use of a compound of general formula (III)
a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof for the preparation of a compound of formula (I) of embodiment A.
Embodiment X) Use of a compound of general formula (III)
 a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof, for the preparation of a compound of formula (I) of embodiment A.
a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof, for the preparation of a compound of formula (I) of embodiment A.
Embodiment Y) Method for controlling cancer in humans and animals by administering an antiproliferative effective amount of the compound of formula (I) of embodiment A, or of a medicament as defined in one of embodiments P, Q or R.
The present invention covers any sub-combination within any embodiment or aspect of the present invention of intermediate compounds of general formula (I), supra.
The compounds of general formula (I) of the present invention can be converted to any salt, preferably pharmaceutically acceptable salts, as described herein, by any method which is known to the person skilled in the art. Similarly, any salt of a compound of general formula (I) of the present invention can be converted into the free compound, by any method which is known to the person skilled in the art.
Compounds of general formula (I) of the present invention demonstrate a valuable pharmacological spectrum of action and pharmacokinetic profile , both of which could not have been predicted. Compounds of the present invention have surprisingly been found to effectively inhibit Map4K1 and it is possible therefore that said compounds be used for the treatment or prophylaxis of diseases, preferably neoplastic or abnormal cell proliferative disorder, such as cancer, a condition with dysregulated immune response or another disorder associated with aberrant Map4K1 signaling disorders in humans and animals.
Compounds of the present invention can be utilized to inhibit, block, reduce, decrease, etc., cell proliferation and/or cell division, and/or produce apoptosis. This method comprises administering to a mammal in need thereof, including a human, an amount of a compound of general formula (I) of the present invention, or a pharmaceutically acceptable salt, isomer, polymorph, metabolite, hydrate, solvate or ester thereof, which is effective to treat the disorder.
Hyperproliferative disorders include, but are not limited to, for example : psoriasis, keloids, and other hyperplasias affecting the skin, benign prostate hyperplasia (BPH), solid tumours, such as cancers of the breast, respiratory tract, brain, reproductive organs, digestive tract, urinary
tract, eye, liver, skin, head and neck, thyroid, parathyroid and their distant metastases. Those disorders also include lymphomas, sarcomas, and leukaemias.
Examples of breast cancers include, but are not limited to, invasive ductal carcinoma, invasive lobular carcinoma, ductal carcinoma in situ, and lobular carcinoma in situ.
Examples of cancers of the respiratory tract include, but are not limited to, small-cell and non- small-cell lung carcinoma, as well as bronchial adenoma and pleuropulmonary blastoma.
Examples of brain cancers include, but are not limited to, brain stem and hypophtalmic glioma, cerebellar and cerebral astrocytoma, medulloblastoma, ependymoma, as well as neuroectodermal and pineal tumour.
Tumours of the male reproductive organs include, but are not limited to, prostate and testicular cancer.
Tumours of the female reproductive organs include, but are not limited to, endometrial, cervical, ovarian, vaginal, and vulvar cancer, as well as sarcoma of the uterus.
Tumours of the digestive tract include, but are not limited to, anal, colon, colorectal, oesophageal, gallbladder, gastric, pancreatic, rectal, small-intestine, and salivary gland cancers.
Tumours of the urinary tract include, but are not limited to, bladder, penile, kidney, renal pelvis, ureter, urethral and human papillary renal cancers.
Eye cancers include, but are not limited to, intraocular melanoma and retinoblastoma.
Examples of liver cancers include, but are not limited to, hepatocellular carcinoma (liver cell carcinomas with or without fibrolamellar variant), cholangiocarcinoma (intrahepatic bile duct carcinoma), and mixed hepatocellular cholangiocarcinoma.
Skin cancers include, but are not limited to, squamous cell carcinoma, Kaposi’s sarcoma, malignant melanoma, Merkel cell skin cancer, and non-melanoma skin cancer.
Head-and-neck cancers include, but are not limited to, laryngeal, hypopharyngeal, nasopharyngeal, oropharyngeal cancer, lip and oral cavity cancer and squamous cell.
Lymphomas include, but are not limited to, AIDS-related lymphoma, non-Hodgkin’s lymphoma, cutaneous T-cell lymphoma, Burkitt lymphoma, Hodgkin’s disease, and lymphoma of the central nervous system.
Sarcomas include, but are not limited to, sarcoma of the soft tissue, osteosarcoma, malignant fibrous histiocytoma, lymphosarcoma, and rhabdomyosarcoma.
Leukemias include, but are not limited to, acute myeloid leukemia, acute lymphoblastic leukemia, chronic lymphocytic leukemia, chronic myelogenous leukemia, and hairy cell leukemia.
The present invention also provides methods of treating angiogenic disorders including diseases associated with excessive and/or abnormal angiogenesis.
Inappropriate and ectopic expression of angiogenesis can be deleterious to an organism. A number of pathological conditions are associated with the growth of extraneous blood vessels. These include, for example, diabetic retinopathy, ischemic retinal-vein occlusion, and retinopathy of prematurity [Aiello et al., New Engl. J. Med., 1994, 331 , 1480 ; Peer et al., Lab. Invest., 1995, 72, 638], age-related macular degeneration (AMD) [Lopez et al., Invest.
Opththalmol. Vis. Sci., 1996, 37, 855], neovascular glaucoma, psoriasis, retrolental fibroplasias, angiofibroma, inflammation, rheumatoid arthritis (RA), restenosis, in-stent restenosis, vascular graft restenosis, etc. In addition, the increased blood supply associated with cancerous and neoplastic tissue, encourages growth, leading to rapid tumour enlargement and metastasis. Moreover, the growth of new blood and lymph vessels in a tumour provides an escape route for renegade cells, encouraging metastasis and the consequence spread of the cancer. Thus, compounds of general formula (I) of the present invention can be utilized to treat and/or prevent any of the aforementioned angiogenesis disorders, for example by inhibiting and/or reducing blood vessel formation; by inhibiting, blocking, reducing, decreasing, etc. endothelial cell proliferation, or other types involved in angiogenesis, as well as causing cell death or apoptosis of such cell types.
These disorders have been well characterized in humans, but also exist with a similar etiology in other mammals, and can be treated by administering pharmaceutical compositions of the present invention.
The term “treating” or “treatment” as stated throughout this document is used conventionally, for example the management or care of a subject for the purpose of combating, alleviating, reducing, relieving, improving the condition of a disease or disorder, such as a carcinoma.
The compounds of the present invention can be used in particular in therapy and prevention, i.e. prophylaxis, of tumour growth and metastases, especially in solid tumours of all indications and stages with or without pre-treatment of the tumour growth.
Generally, the use of chemotherapeutic agents and/or anti-cancer agents in combination with a compound or pharmaceutical composition of the present invention will serve to:
1. yield better efficacy in reducing the growth of a tumour or even eliminate the tumour as compared to administration of either agent alone,
2. provide for the administration of lesser amounts of the administered chemotherapeutic agents,
3. provide for a chemotherapeutic treatment that is well tolerated in the patient with fewer deleterious pharmacological complications than observed with single agent chemotherapies and certain other combined therapies,
4. provide for treating a broader spectrum of different cancer types in mammals, especially humans,
5. provide for a higher response rate among treated patients,
6. provide for a longer survival time among treated patients compared to standard chemotherapy treatments,
7. provide a longer time for tumour progression, and/or
8. yield efficacy and tolerability results at least as good as those of the agents used alone, compared to known instances where other cancer agent combinations produce antagonistic effects.
In addition, the compounds of general formula (I) of the present invention can also be used in combination with radiotherapy and/or surgical intervention.
In a further embodiment of the present invention, the compounds of general formula (I) of the present invention may be used to sensitize a cell to radiation, i.e. treatment of a cell with a compound of the present invention prior to radiation treatment of the cell renders the cell more susceptible to DNA damage and cell death than the cell would be in the absence of any treatment with a compound of the present invention. In one aspect, the cell is treated with at least one compound of general formula (I) of the present invention.
Thus, the present invention also provides a method of killing a cell, wherein a cell is administered one or more compounds of the present invention in combination with conventional radiation therapy.
The present invention also provides a method of rendering a cell more susceptible to cell death, wherein the cell is treated with one or more compounds of general formula (I) of the present invention prior to the treatment of the cell to cause or induce cell death. In one aspect, after the cell is treated with one or more compounds of general formula (I) of the present invention, the cell is treated with at least one compound, or at least one method, or a combination thereof, in order to cause DNA damage for the purpose of inhibiting the function of the normal cell or killing the cell.
In other embodiments of the present invention, a cell is killed by treating the cell with at least one DNA damaging agent, i.e. after treating a cell with one or more compounds of general formula (I) of the present invention to sensitize the cell to cell death, the cell is treated with at least one DNA damaging agent to kill the cell. DNA damaging agents useful in the present invention include, but are not limited to, chemotherapeutic agents (e.g. cis platin), ionizing radiation (X-rays, ultraviolet radiation), carcinogenic agents, and mutagenic agents.
In other embodiments, a cell is killed by treating the cell with at least one method to cause or induce DNA damage. Such methods include, but are not limited to, activation of a cell
signalling pathway that results in DNA damage when the pathway is activated, inhibiting of a cell signalling pathway that results in DNA damage when the pathway is inhibited, and inducing a biochemical change in a cell, wherein the change results in DNA damage. By way of a non-limiting example, a DNA repair pathway in a cell can be inhibited, thereby preventing the repair of DNA damage and resulting in an abnormal accumulation of DNA damage in a cell.
In one aspect of the invention, a compound of general formula (I) of the present invention is administered to a cell prior to the radiation or other induction of DNA damage in the cell. In another aspect of the invention, a compound of general formula (I) of the present invention is administered to a cell concomitantly with the radiation or other induction of DNA damage in the cell. In yet another aspect of the invention, a compound of general formula (I) of the present invention is administered to a cell immediately after radiation or other induction of DNA damage in the cell has begun.
In another aspect, the cell is in vitro. In another embodiment, the cell is in vivo.
Compounds of the present invention can be utilized to inhibit, block, reduce, decrease, etc., Map4K1. This method comprises administering to a mammal in need thereof, including a human, an amount of a compound of this invention, or a pharmaceutically acceptable salt, isomer, polymorph, metabolite, hydrate, solvate or ester thereof; which is effective to treat the disorder.
The present invention also provides methods of treating, ameliorating or preventing neoplastic disorders or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling and methods of ameliorating of vaccine therapies or cell therapies.
Amelioration: Any intervention with a goal of improvement in comparison to a situation without this interventions.
Neoplastic disorder: A disorder which causes or results in an abnormal and excessive growth of tissue.
Abnormal cell proliferative disorder: A disorder which causes or results in abnormal cell proliferation.
Condition with dysregulated immune response: A condition in which the regulation of an immune response differs from the regulation of this immune response in a healthy human.
Vaccine therapy is a therapy which uses vaccines.
These disorders have been well characterized in humans, but also exist with a similar etiology in other mammals, and can be treated by administering pharmaceutical compositions of the present invention.
The term “treating” or “treatment” as used in the present text is used conventionally, e.g., the management or care of a subject for the purpose of combating, alleviating, reducing, relieving, improving the condition of a disease or disorder, such as a carcinoma.
The compounds of the present invention can be used in particular in therapy and prevention, i.e. prophylaxis, of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling and in amelioration of vaccine therapies or cell therapies.
In accordance with a further aspect, the present invention covers compounds of general formula (I), as described supra, or stereoisomers, tautomers, N-oxides, hydrates, solvates, and salts thereof, particularly pharmaceutically acceptable salts thereof, or mixtures of same, for use in the treatment or prophylaxis of diseases, in particular of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, and for the amelioration of vaccine therapies or cell therapies.
The pharmaceutical activity of the compounds according to the invention can be explained by their activity as Map4K1 inhibitor.
In accordance with a further aspect, the present invention covers the use of compounds of general formula (I), as described supra, or stereoisomers, tautomers, N-oxides, hydrates, solvates, and salts thereof, particularly pharmaceutically acceptable salts thereof, or mixtures of same, for the treatment or prophylaxis of diseases, in particular of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, and for the amelioration of vaccine therapies or cell therapies.
In accordance with a further aspect, the present invention covers the use of a compound of formula (I), described supra, or a stereoisomer, a tautomer, an N-oxide, a hydrate, a solvate, or a salt thereof, particularly a pharmaceutically acceptable salt thereof, or a mixture of same, for the prophylaxis or treatment of diseases, in particular of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, and for the amelioration of vaccine therapies or cell therapies.
In accordance with a further aspect, the present invention covers the use of compounds of general formula (I), as described supra, or stereoisomers, tautomers, N-oxides, hydrates, solvates, and salts thereof, particularly pharmaceutically acceptable salts thereof, or mixtures of same, in a method of treatment or prophylaxis of diseases, in particular of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response,
other disorders associated with aberrant Map4K1 signaling, and for the amelioration of vaccine therapies or cell therapies.
In accordance with a further aspect, the present invention covers use of a compound of general formula (I), as described supra, or stereoisomers, tautomers, N-oxides, hydrates, solvates, and salts thereof, particularly pharmaceutically acceptable salts thereof, or mixtures of same, for the preparation of a pharmaceutical composition, preferably a medicament, for the prophylaxis or treatment of diseases, in particular of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, and for the amelioration of vaccine therapies or cell therapies.
In accordance with a further aspect, the present invention covers a method of treatment or prophylaxis of diseases, in particular of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, and for the amelioration of vaccine therapies or cell therapies, using an effective amount of a compound of general formula (I), as described supra, or stereoisomers, tautomers, hydrates, solvates, and salts thereof, particularly pharmaceutically acceptable salts thereof, or mixtures of same.
In accordance with a further aspect, the present invention covers pharmaceutical compositions, in particular a medicament, comprising a compound of general formula (I), as described supra, or a stereoisomer, a tautomer, a hydrate, a solvate, a salt thereof, particularly a pharmaceutically acceptable salt, or a mixture of same, and one or more excipients), in particular one or more pharmaceutically acceptable excipient(s). Conventional procedures for preparing such pharmaceutical compositions in appropriate dosage forms can be utilized.
The present invention furthermore covers pharmaceutical compositions, in particular medicaments, which comprise at least one compound according to the invention, conventionally together with one or more pharmaceutically suitable excipients, and to their use for the above mentioned purposes.
It is possible for the compounds according to the invention to have systemic and/or local activity. For this purpose, they can be administered in a suitable manner, such as, for example, via the oral, parenteral, pulmonary, nasal, sublingual, lingual, buccal, rectal, vaginal, dermal, transdermal, conjunctival, otic route or as an implant or stent.
For these administration routes, it is possible for the compounds according to the invention to be administered in suitable administration forms.
For oral administration, it is possible to formulate the compounds according to the invention to dosage forms known in the art that deliver the compounds of the invention rapidly and/or in a modified manner, such as, for example, tablets (uncoated or coated tablets, for example with enteric or controlled release coatings that dissolve with a delay or are insoluble), orally-
disintegrating tablets, films/wafers, films/lyophylisates, capsules (for example hard or soft gelatine capsules), sugar-coated tablets, granules, pellets, powders, emulsions, suspensions, aerosols or solutions. It is possible to incorporate the compounds according to the invention in crystalline and/or amorphised and/or dissolved form into said dosage forms.
Parenteral administration can be effected with avoidance of an absorption step (for example intravenous, intraarterial, intracardial, intraspinal or intralumbal) or with inclusion of absorption (for example intramuscular, subcutaneous, intracutaneous, percutaneous or intraperitoneal). Administration forms which are suitable for parenteral administration are, inter alia, preparations for injection and infusion in the form of solutions, suspensions, emulsions, lyophylisates or sterile powders.
Examples which are suitable for other administration routes are pharmaceutical forms for inhalation [inter alia powder inhalers, nebulizers], nasal drops, nasal solutions, nasal sprays; tablets/films/wafers/capsules for lingual, sublingual or buccal administration; suppositories; eye drops, eye ointments, eye baths, ocular inserts, ear drops, ear sprays, ear powders, ear-rinses, ear tampons; vaginal capsules, aqueous suspensions (lotions, mixturae agitandae), lipophilic suspensions, emulsions, ointments, creams, transdermal therapeutic systems (such as, for example, patches), milk, pastes, foams, dusting powders, implants or stents.
The compounds according to the invention can be incorporated into the stated administration forms. This can be effected in a manner known per se by mixing with pharmaceutically suitable excipients. Pharmaceutically suitable excipients include, inter alia,
• fillers and carriers (for example cellulose, microcrystalline cellulose (such as, for example, Avicel®), lactose, mannitol, starch, calcium phosphate (such as, for example, Di-Cafos®)),
• ointment bases (for example petroleum jelly, paraffins, triglycerides, waxes, wool wax, wool wax alcohols, lanolin, hydrophilic ointment, polyethylene glycols),
• bases for suppositories (for example polyethylene glycols, cacao butter, hard fat),
• solvents (for example water, ethanol, isopropanol, glycerol, propylene glycol, medium chain-length triglycerides fatty oils, liquid polyethylene glycols, paraffins),
• surfactants, emulsifiers, dispersants or wetters (for example sodium dodecyl sulfate), lecithin, phospholipids, fatty alcohols (such as, for example, Lanette®), sorbitan fatty acid esters (such as, for example, Span®), polyoxyethylene sorbitan fatty acid esters (such as, for example, Tween®), polyoxyethylene fatty acid glycerides (such as, for example, Cremophor®), polyoxethylene fatty acid esters, polyoxyethylene fatty alcohol ethers, glycerol fatty acid esters, poloxamers (such as, for example, Pluronic®),
• buffers, acids and bases (for example phosphates, carbonates, citric acid, acetic acid, hydrochloric acid, sodium hydroxide solution, ammonium carbonate, trometamol, triethanolamine),
• isotonicity agents (for example glucose, sodium chloride),
• adsorbents (for example highly-disperse silicas),
• viscosity-increasing agents, gel formers, thickeners and/or binders (for example polyvinylpyrrolidone, methylcellulose, hydroxypropylmethylcellulose, hydroxypropylcellulose, carboxymethylcellulose-sodium, starch, carbomers, polyacrylic acids (such as, for example, Carbopol®); alginates, gelatine),
• disintegrants (for example modified starch, carboxymethylcellulose-sodium, sodium starch glycolate (such as, for example, Explotab®), cross- linked polyvinylpyrrolidone, croscarmellose-sodium (such as, for example, AcDiSol®)),
• flow regulators, lubricants, glidants and mould release agents (for example magnesium stearate, stearic acid, talc, highly-disperse silicas (such as, for example, Aerosil®)),
• coating materials (for example sugar, shellac) and film formers for films or diffusion membranes which dissolve rapidly or in a modified manner (for example polyvinylpyrrolidones (such as, for example, Kollidon®), polyvinyl alcohol, hydroxypropylmethylcellulose, hydroxypropylcellulose, ethylcellulose, hydroxypropylmethylcellulose phthalate, cellulose acetate, cellulose acetate phthalate, polyacrylates, polymethacrylates such as, for example, Eudragit®)),
• capsule materials (for example gelatine, hydroxypropylmethylcellulose),
• synthetic polymers (for example polylactides, polyglycolides, polyacrylates, polymethacrylates (such as, for example, Eudragit®), polyvinylpyrrolidones (such as, for example, Kollidon®), polyvinyl alcohols, polyvinyl acetates, polyethylene oxides, polyethylene glycols and their copolymers and blockcopolymers),
• plasticizers (for example polyethylene glycols, propylene glycol, glycerol, triacetine, triacetyl citrate, dibutyl phthalate),
• penetration enhancers,
• stabilisers (for example antioxidants such as, for example, ascorbic acid, ascorbyl palmitate, sodium ascorbate, butylhydroxyanisole, butylhydroxytoluene, propyl gallate),
• preservatives (for example parabens, sorbic acid, thiomersal, benzalkonium chloride, chlorhexidine acetate, sodium benzoate),
• colourants (for example inorganic pigments such as, for example, iron oxides, titanium dioxide),
• flavourings, sweeteners, flavour- and/or odour-masking agents.
The present invention furthermore relates to a pharmaceutical composition which comprise at least one compound according to the invention, conventionally together with one or more pharmaceutically suitable excipient(s), and to their use according to the present invention.
In accordance with another aspect, the present invention covers pharmaceutical combinations, in particular medicaments, comprising at least one compound of general formula (I) of the present invention and at least one or more further active ingredients, in particular for the treatment and/or prophylaxis of a of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, and for the amelioration of vaccine therapies or cell therapies.
Particularly, the present invention covers a pharmaceutical combination, which comprises:
• one or more first active ingredients, in particular compounds of general formula (I) as defined supra, and
• one or more further active ingredients, in particular of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, and for the amelioration of vaccine therapies or cell therapies.
The term “combination” in the present invention is used as known to persons skilled in the art, it being possible for said combination to be a fixed combination, a non-fixed combination or a kit- of- parts.
A “fixed combination” in the present invention is used as known to persons skilled in the art and is defined as a combination wherein, for example, a first active ingredient, such as one or more compounds of general formula (I) of the present invention, and a further active ingredient are present together in one unit dosage or in one single entity. One example of a “fixed combination” is a pharmaceutical composition wherein a first active ingredient and a further active ingredient are present in admixture for simultaneous administration, such as in a formulation. Another example of a “fixed combination” is a pharmaceutical combination wherein a first active ingredient and a further active ingredient are present in one unit without being in admixture.
A non-fixed combination or “kit-of-parts” in the present invention is used as known to persons skilled in the art and is defined as a combination wherein a first active ingredient and a further active ingredient are present in more than one unit. One example of a non-fixed combination or kit-of-parts is a combination wherein the first active ingredient and the further active ingredient are present separately. It is possible for the components of the non-fixed combination or kit-of-
parts to be administered separately, sequentially, simultaneously, concurrently or chronologically staggered.
The compounds of the present invention can be administered as the sole pharmaceutical agent or in combination with one or more other pharmaceutically active ingredients where the combination causes no unacceptable adverse effects. The present invention also covers such pharmaceutical combinations. For example, the compounds of the present invention can be combined with known cancer agents.
Examples of cancer agents include:
1311-chTNT, abarelix, abemaciclib, abiraterone, acalabrutinib, aclarubicin, adalimumab, ado- trastuzumab emtansine, afatinib, aflibercept, aldesleukin, alectinib, alemtuzumab, alendronic acid, alitretinoin, alpharadin, altretamine, amifostine, aminoglutethimide, hexyl aminolevulinate, amrubicin, amsacrine, anastrozole, ancestim, anethole dithiolethione, anetumab ravtansine, angiotensin II, antithrombin III, apalutamide, aprepitant, arcitumomab, arglabin, arsenic trioxide, asparaginase, atezolizumab, avelumab, axicabtagene ciloleucel, axitinib, azacitidine, basiliximab, belotecan, bendamustine, besilesomab, belinostat, bevacizumab, bexarotene, bicalutamide, bisantrene, bleomycin, blinatumomab, bortezomib, bosutinib, buserelin, brentuximab vedotin, brigatinib, busulfan, cabazitaxel, cabozantinib, calcitonine, calcium folinate, calcium levofolinate, capecitabine, capromab, carbamazepine carboplatin, carboquone, carfilzomib, carmofur, carmustine, catumaxomab, celecoxib, celmoleukin, cemiplimab, ceritinib, cetuximab, chlorambucil, chlormadinone, chlormethine, cidofovir, cinacalcet, cisplatin, cladribine, clodronic acid, clofarabine, cobimetinib, copanlisib , crisantaspase, crizotinib, cyclophosphamide, cyproterone, cytarabine, dacarbazine, dactinomycin, daratumumab, darbepoetin alfa, dabrafenib, darolutamide, dasatinib, daunorubicin, decitabine, degarelix, denileukin diftitox, denosumab, depreotide, deslorelin, dianhydrogalactitol, dexrazoxane, dibrospidium chloride, dianhydrogalactitol, diclofenac, dinutuximab, docetaxel, dolasetron, doxifluridine, doxorubicin, doxorubicin + estrone, dronabinol, durvalumab, eculizumab, edrecolomab, elliptinium acetate, elotuzumab, eltrombopag, enasidenib, endostatin, enocitabine, enzalutamide, epirubicin, epitiostanol, epoetin alfa, epoetin beta, epoetin zeta, eptaplatin, eribulin, erlotinib, esomeprazole, estradiol, estramustine, ethinylestradiol, etoposide, everolimus, exemestane, fadrozole, fentanyl, filgrastim, fluoxymesterone, floxuridine, fludarabine, fluorouracil, flutamide, folinic acid, formestane, fosaprepitant, fotemustine, fulvestrant, gadobutrol, gadoteridol, gadoteric acid meglumine, gadoversetamide, gadoxetic acid, gallium nitrate, ganirelix, gefitinib, gemcitabine, gemtuzumab, Glucarpidase, glutoxim, GM-CSF, goserelin, granisetron, granulocyte colony stimulating factor, histamine dihydrochloride, histrelin, hydroxycarbamide, 1-125 seeds, lansoprazole, ibandronic acid, ibritumomab tiuxetan, ibrutinib, idarubicin, ifosfamide, imatinib, imiquimod, improsulfan, indisetron, incadronic acid, ingenol mebutate, inotuzumab ozogamicin,
interferon alfa, interferon beta, interferon gamma, iobitridol, iobenguane (1231), iomeprol, ipilimumab, irinotecan, Itraconazole, ixabepilone, ixazomib, lanreotide, lansoprazole, lapatinib, larotrectinib, lasocholine, lenalidomide, lenvatinib, lenograstim, lentinan, letrozole, leuprorelin, levamisole, levonorgestrel, levothyroxine sodium, lisuride, lobaplatin, lomustine, lonidamine, lutetium Lu 177 dotatate, masoprocol, medroxyprogesterone, megestrol, melarsoprol, melphalan, mepitiostane, mercaptopurine, mesna, methadone, methotrexate, methoxsalen, methylaminolevulinate, methylprednisolone, methyltestosterone, metirosine, midostaurin, mifamurtide, miltefosine, miriplatin, mitobronitol, mitoguazone, mitolactol, mitomycin, mitotane, mitoxantrone, mogamulizumab, molgramostim, mopidamol, morphine hydrochloride, morphine sulfate, mvasi, nabilone, nabiximols, nafarelin, naloxone + pentazocine, naltrexone, nartograstim, necitumumab, nedaplatin, nelarabine, neratinib, neridronic acid, netupitant/palonosetron, nivolumab, pentetreotide, nilotinib, nilutamide, nimorazole, nimotuzumab, nimustine, nintedanib, niraparib, nitracrine, nivolumab, obinutuzumab, octreotide, ofatumumab, olaparib, olaratumab, omacetaxine mepesuccinate, omeprazole, ondansetron, oprelvekin, orgotein, orilotimod, osimertinib, oxaliplatin, oxycodone, oxymethoIone, ozogamicine, p53 gene therapy, paclitaxel, palbociclib, palifermin, palladium- 103 seed, palonosetron, pamidronic acid, panitumumab, panobinostat, pantoprazole, pazopanib, pegaspargase, PEG-epoetin beta (methoxy PEG-epoetin beta), pembrolizumab, pegfilgrastim, peginterferon alfa-2b, pembrolizumab, pemetrexed, pentazocine, pentostatin, peplomycin, Perflubutane, perfosfamide, Pertuzumab, picibanil, pilocarpine, pirarubicin, pixantrone, plerixafor, plicamycin, poliglusam, polyestradiol phosphate, polyvinylpyrrolidone + sodium hyaluronate, polysaccharide-K, pomalidomide, ponatinib, porfimer sodium, pralatrexate, prednimustine, prednisone, procarbazine, procodazole, propranolol, quinagolide, rabeprazole, racotumomab, radium-223 chloride, radotinib, raloxifene, raltitrexed, ramosetron, ramucirumab, ranimustine, rasburicase, razoxane, refametinib , regorafenib, ribociclib, risedronic acid, rhenium-186 etidronate, rituximab, rolapitant, romidepsin, romiplostim, romurtide, rucaparib, samarium (153Sm) lexidronam, sargramostim, sarilumab, satumomab, secretin, siltuximab, sipuleucel-T, sizofiran, sobuzoxane, sodium glycididazole, sonidegib, sorafenib, stanozolol, streptozocin, sunitinib, talaporfin, talimogene laherparepvec, tamibarotene, tamoxifen, tapentadol, tasonermin, teceleukin, technetium (99mTc) nofetumomab merpentan, 99mTc-HYNIC-[Tyr3]-octreotide, tegafur, tegafur + gimeracil + oteracil, temoporfin, temozolomide, temsirolimus, teniposide, testosterone, tetrofosmin, thalidomide, thiotepa, thymalfasin, thyrotropin alfa, tioguanine, tisagenlecleucel, tislelizumab, tocilizumab, topotecan, toremifene, tositumomab, trabectedin, trametinib, tramadol, trastuzumab, trastuzumab emtansine, treosulfan, tretinoin, trifluridine + tipiracil, trilostane, triptorelin, trametinib, trofosfamide, thrombopoietin, tryptophan, ubenimex, valatinib, valrubicin, vandetanib, vapreotide, vemurafenib, vinblastine, vincristine, vindesine, vinflunine, vinorelbine,
vismodegib, vorinostat, vorozole, yttrium-90 glass microspheres, zinostatin, zinostatin stimalamer, zoledronic acid, zorubicin.
Based upon standard laboratory techniques known to evaluate compounds useful for the treatment of of neoplastic or abnormal cell proliferative disorders, such as cancer, conditions with dysregulated immune response, other disorders associated with aberrant Map4K1 signaling, and for the amelioration of vaccine therapies or cell therapies, by standard toxicity tests and by standard pharmacological assays for the determination of treatment of the conditions identified above in mammals, and by comparison of these results with the results of known active ingredients or medicaments that are used to treat these conditions, the effective dosage of the compounds of the present invention can readily be determined for treatment of each desired indication. The amount of the active ingredient to be administered in the treatment of one of these conditions can vary widely according to such considerations as the particular compound and dosage unit employed, the mode of administration, the period of treatment, the age and sex of the patient treated, and the nature and extent of the condition treated.
The compounds of the invention can further be combined with chimeric antigen receptor T cells (CAR-T cells), such as Axicabtagen-Ciloleucel or Tisagenlecleucel. The activity of CAR-T cells can be suppressed by the tumor micro environment (TME).
In accordance with a further aspect, the present invention covers combinations comprising one or more compounds according to the invention, or stereoisomers, tautomers, hydrates, solvates, and salts thereof, particularly pharmaceutically acceptable salts thereof, or mixtures of same, with chimeric antigen receptor T cells, (CAR-T cells), CAR-NKT cells or CAR-NK cells.
Preferably, the chimeric antigen receptor T cells (CAR-T cells) are YESCARTA® (axicabtagen ciloleucel), KYMRIAH® (tisagenlecleucel), BREYANZI® (lisocabtagene maraleucel), TECARTUS® (brecucabtqagene autoleucel), ABECMA® (idecabtagene vicleucel), or CARVYKTI® (clitacabtagene autoleucel).
The present invention further provides the use of the compounds according to the invention for expansion of T cells including CAR-T and tumor infiltrated lymphocytes ex-vivo.
In accordance with a further aspect, the present invention covers compounds according to the invention, or stereoisomers, tautomers, hydrates, solvates, and salts thereof, particularly pharmaceutically acceptable salts thereof, or mixtures of same, for use in the expansion of T cells including CAR-T cells, CAR-NKT cells or CAR-NK cells and tumor infiltrated lymphocytes ex-vivo.
Hence, the present invention also relates to the use of the compounds according to the invention for the expansion of T cells, including CAR-T cell, CAR-NKT cells or CAR-NK cells and tumor infiltrated lymphocytes, ex-vivo.
The present invention also comprises an ex-vivo method for the expansion of T cells, including CAR-T cells, CAR-NKT cells or CAR-NK cells and tumor infiltrated lymphocytes, contacting said T cells with compounds according to the invention.
The total amount of the active ingredient to be administered will generally range from about 0.001 mg/kg to about 200 mg/kg body weight per day, and preferably from about 0.01 mg/kg to about 20 mg/kg body weight per day. Clinically useful dosing schedules will range from one to three times a day dosing to once every four weeks dosing. In addition, it is possible for "drug holidays", in which a patient is not dosed with a drug for a certain period of time, to be beneficial to the overall balance between pharmacological effect and tolerability. It is possible for a unit dosage to contain from about 0.5 mg to about 1500 mg of active ingredient, and can be administered one or more times per day or less than once a day. The average daily dosage for administration by injection, including intravenous, intramuscular, subcutaneous and parenteral injections, and use of infusion techniques will preferably be from 0.01 to 200 mg/kg of total body weight. The average daily rectal dosage regimen will preferably be from 0.01 to 200 mg/kg of total body weight. The average daily vaginal dosage regimen will preferably be from 0.01 to 200 mg/kg of total body weight. The average daily topical dosage regimen will preferably be from 0.1 to 200 mg administered between one to four times daily. The transdermal concentration will preferably be that required to maintain a daily dose of from 0.01 to 200 mg/kg. The average daily inhalation dosage regimen will preferably be from 0.01 to 100 mg/kg of total body weight.
Of course the specific initial and continuing dosage regimen for each patient will vary according to the nature and severity of the condition as determined by the attending diagnostician, the activity of the specific compound employed, the age and general condition of the patient, time of administration, route of administration, rate of excretion of the drug, drug combinations, and the like. The desired mode of treatment and number of doses of a compound of the present invention or a pharmaceutically acceptable salt or ester or composition thereof can be ascertained by those skilled in the art using conventional treatment tests.
FIGURE LEGENDS
Figure 1 : 50% thermal ellipsoids of intermediate 15, Molecule 1.
Figure 2: 50% thermal ellipsoids of intermediate 15, Molecule 2.
Figure 3: 50% thermal ellipsoids of side product 1.
Figure 4: INF gamma secretion in anti-CD3 antibody/PGE2-treated hPBMCs upon incubation with the target compound of the invention.
EXPERIMENTAL SECTION
NMR peak forms are stated as they appear in the spectra, possible higher order effects have not been considered.
The 1H-NMR data of selected compounds are listed in the form of 1H-NMR peaklists. Therein, for each signal peak the 6 value in ppm is given, followed by the signal intensity, reported in round brackets. The 6 value-signal intensity pairs from different peaks are separated by commas. Therefore, a peaklist is described by the general form: 61 (intensity-i), 62 (intense), ... , 6i (intensity,), ... , 6n (intensity,,).
The intensity of a sharp signal correlates with the height (in cm) of the signal in a printed NMR spectrum. When compared with other signals, this data can be correlated to the real ratios of the signal intensities. In the case of broad signals, more than one peak, or the center of the signal along with their relative intensity, compared to the most intense signal displayed in the spectrum, are shown. A 1H-NMR peaklist is similar to a classical 1H-NMR readout, and thus usually contains all the peaks listed in a classical NMR interpretation. Moreover, similar to classical 1H- NMR printouts, peaklists can show solvent signals, signals derived from stereoisomers of the particular target compound, peaks of impurities, 13C satellite peaks, and/or spinning sidebands. The peaks of stereoisomers, and/or peaks of impurities are typically displayed with a lower intensity compared to the peaks of the target compound (e.g., with a purity of >90%). Such stereoisomers and/or impurities may be typical for the particular manufacturing process, and therefore their peaks may help to identify a reproduction of the manufacturing process on the basis of "by-product fingerprints". An expert who calculates the peaks of the target compound by known methods (MestReC, ACD simulation, or by use of empirically evaluated expectation values), can isolate the peaks of the target compound as required, optionally using additional intensity filters. Such an operation would be similar to peak-picking in classical 1H-NMR interpretation. A detailed description of the reporting of NMR data in the form of peaklists can be found in the publication "Citation of NMR Peaklist Data within Patent Applications" (cf. http://www.researchdisclosure.com/searching-disclosures, Research Disclosure Database Number 605005, 2014, 01 Aug 2014). In the peak picking routine, as described in the Research Disclosure Database Number 605005, the parameter "MinimumHeight" can be adjusted between 1 % and 4%. However, depending on the chemical structure and/or depending on the concentration of the measured compound it may be reasonable to set the parameter "MinimumHeight" <1 %.
Chemical names were generated using the ACD/Name software from ACD/Labs. In some cases generally accepted names of commercially available reagents were used in place of ACD/Name generated names.
The following table 2 lists the abbreviations used in this paragraph and in the Examples section as far as they are not explained within the text body. Other abbreviations have their meanings customary perse to the skilled person.
Abbreviations Table 2: Abbreviations


The various aspects of the invention described in this application are illustrated by the following examples which are not meant to limit the invention in any way.
The example testing experiments described herein serve to illustrate the present invention and the invention is not limited to the examples given.
EXPERIMENTAL SECTION - GENERAL PART
All reagents, for which the synthesis is not described in the experimental part, are either commercially available, or are known compounds or may be formed from known compounds by known methods by a person skilled in the art.
The compounds and intermediates produced according to the methods of the invention may require purification. Purification of organic compounds is well known to the person skilled in the art and there may be several ways of purifying the same compound. In some cases, no purification may be necessary. In some cases, the compounds may be purified by crystallization. In some cases, impurities may be stirred out using a suitable solvent. In some cases, the compounds may be purified by chromatography, particularly flash column chromatography, using for example prepacked silica gel cartridges, e.g. Biotage SNAP cartidges KP-Sil® or KP- NH® in combination with a Biotage autopurifier system (SP4® or Isolera Four®) and eluents such as gradients of hexane/ethyl acetate or DCM/methanol. In some cases, the compounds may be purified by preparative HPLC using for example a Waters autopurifier equipped with a diode array detector and/or on-line electrospray ionization mass spectrometer in combination with a suitable prepacked reverse phase column and eluents such as gradients of water and acetonitrile which may contain additives such as trifluoroacetic acid, formic acid or aqueous ammonia.
In some cases, purification methods as described above can provide those compounds of the present invention which possess a sufficiently basic or acidic functionality in the form of a salt, such as, in the case of a compound of the present invention which is sufficiently basic, a trifluoroacetate or formate salt for example, or, in the case of a compound of the present invention which is sufficiently acidic, an ammonium salt for example. A salt of this type can either be transformed into its free base or free acid form, respectively, by various methods known to the person skilled in the art, or be used as salts in subsequent biological assays. It is to be understood that the specific form (e.g. salt, free base etc.) of a compound of the present invention as isolated and as described herein is not necessarily the only form in which said compound can be applied to a biological assay in order to quantify the specific biological activity.
EXPERIMENTAL SECTION - GENERAL SYNTHESIS
The following paragraphs outline a variety of synthetic approaches suitable to prepare compounds of the general formula (I), and intermediates useful for their synthesis.
In addition to the routes described below, also other routes may be used to synthesize the title compounds, in accordance with common general knowledge of a person skilled in the art of organic synthesis. The order of transformations exemplified in the following schemes is therefore not intended to be limiting, and suitable synthesis steps from various schemes can be combined to form additional synthesis sequences.
Further, it is possible that two or more successive steps may be performed without work-up between the said steps, e.g. in a “one-pot” reaction, as it is well-known to a person skilled in the art.
Further, it is possible that one or more steps are carried out using stereoselective methods. Such stereoselective methods are exemplified by but not limited to asymmetric carbonyl group reductions (for asymmetric carbonyl group using chiral reagents see e.g.: E.J. Corey, L. Kurti in Enantioselective Chemical Synthesis. Methods, Logic and Practice, Elsevier, 2013; for diastereoselective carbonyl group reduction see e.g. T. Hanamoto et al., J. Org. Chem. 1990, 55, 4969-4971 ; see also e.g. P.V. Ramachandran et al., J. Org. Chem. 1996, 61 , 95-99; see also e.g. P.K. Mohanta et. al., J. Am. Chem. Soc. 2005, 127, 11896-11897; for asymmetric carbonyl group reduction with dynamic kinetic resolution see e.g. X. Tan et al., Org. Lett. 2020, 22, 7230-7233) as well as asymmetric reductive aminations (see e.g. Y. Tian et al., Org. Chem. Front. 2021 , 8, 2328-2342; see also N. U. Din Reshi, 4CS Catal. 2021 , 11 , 13809-13837; see also P.N. Scheller et al., ChemCatChem 2015, 7, 3239-3242).
Further, it is possible that any of the described reactants and synthetic intermediates can be used in the form of single stereoisomers as well as in the form of a mixture of multiple stereoisomers.
The synthesis of the derivatives according to the present invention is preferably carried out according to the general synthetic sequences shown in schemes 1-3.

Scheme 1 : Route for the preparation of building block of general formula a4, wherein X = H or Br, PG1
represents a suitable triazole protecting group (e.g. SEM, THP or diethoxymethyl), Z represents a suitable leaving group (e.g. halogen, N(OMe)Me, N(OBn)Me, S-alkyl, S-aryl or S-heteroaryl). Starting materials a1 and b1 are commercially available or described in the literature (for racemic compound b1 and its enantiomers see: A. E. Goetz et al., Org. Process Res. Dev. 2022, 26, 683-697).
In step 1 (scheme 1), 1 ,2,4-triazole a1 (X=H) can be protected with a suitable protecting group, preferably [2-(trimethylsilyl)ethoxy]methyl, 2-tetrahydropyranyl, diethoxymethyl or any other suitable metalation-directing protecting group, as known to the person skilled in the art (see e.g. S. Ohta et al., Chem. Pharm. Bull. 1993, 41 , 1226-1231 ; also see T.W. Greene and P.G.M. Wuts in Protective Groups in Organic Synthesis, 4th edition, Wiley 2006), to give intermediate a2 (X=H) or any possible regioisomer thereof. Alternatively, 3-bromo-1 ,2,4-triazole a1 (X=Br) is protected with a suitable protecting group, preferably [2-(trimethylsilyl)ethoxy]methyl, 2-tetrahydropyranyl, diethoxymethyl, benzyl or any other suitable protecting group to give a2 (X=Br) or any possible regioisomer thereof.
In step 2 (scheme 1), difluorocyclopropyl carboxylic acid b1 is converted to intermediate b2 where Z is a leaving group, exemplified by but not limited to halogen, N-alkyl-N-alkoxy, N-alkyl- N-benzyloxy, thioalkyl, thioaryl or thioheteroaryl. For example, carboxylic acid b1 can be converted to the acid chloride b2 (Z = Cl) using such reagents as oxalyl chloride, thionyl chloride or 1-chloro-N,N,2-trimethylprop-1-en-1-amine (CTPA) in a solvent like THF at a temperature range between -10 °C and room temperature. Alternatively, it can be reacted with N,O- dimethylhydroxylamine or related amines to yield N-methoxy-N-methyl amide (Weinreb-Nahm amide) or related N-alkyl-N-alkoxy amide, or N-alkyl-N-benzyloxy amide, via intermediate formation of acid chloride or using various amide coupling reagents like carbonyldiimidazole (GDI), propylphosphonic anhydride (T3P), O-(7-azabenzotriazol-1-yl)-N,N,N',N'- tetramethyluronium-hexafluorphosphat (HATU), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) and others which are well known to the person skilled in organic synthesis.
In step 3 (scheme 1), intermediate a2 (X = H) can be metalated with a suitable reagent followed by nucleophilic addition to the carboxylic acid derivative b2 or nitrile b3, optionally in the presence of a metal catalyst, to give intermediate a3. Such transformations are exemplified, but not limited to Weinreb ketone synthesis (see e.g. L. Kurti and B. Czako, Strategic Application of Named Reactions in Organic Synthesis, Elsevier, 2005), Fukuyama ketone synthesis (see e.g. H. Tokuyama et al., Tetrahedron Lett. 1998, 39, 3189-3192), Liebeskind-Srogl coupling (see e.g. J.M. Villalobos, J. Am. Chem. Soc. 2007, 129, 15734-15735) and metal-catalyzed crosscouplings with acyl chlorides (see e.g. B. Scheiper et al., J. Org. Chem. 2004, 69, 3943-3949).
For example, a2 (X=H) can be treated with nBuLi, LDA or TMPMgCFLiCI, in the presence or absence of additives like TMEDA, in an organic solvent like THF at a temperature range between -78°C and room temperature. Alternatively, a2 (X=Br) can be metallated using halogen-lithium exchange reaction with such reagents as BuLi or (BuLi or converted to an organomagnesium
reagent using such reagents as Mg or iPrMgCI in an organic solvent like TH F at a temperature range between -78°C and room temperature.
Optionally, the resulting organometallic reactant can be further transmetallated to give e.g. organozinc, organocerium or organoboron reagent using a suitable transmetallation reagent, e.g. ZnCh, CeCh or B(OAIkyl)3 as well known to the person skilled in the art.
The resulting organometallic species can be then reacted with b2 or b3 (commercially available under the CAS-number: 36597-03-2), optionally in the presence of a transition metal catalyst, e.g. iron complex or Pd complex, to give the intermediate a3, optionally after additional hydrolysis step.
Alternatively, the intermediate a2 (X=H) can be directly reacted with b2 (preferably Z = Cl) in the presence of a base like triethyl amine or diisopropylethylamine (DIPEA) in an organic solvent such as THF or toluene at a temperature ranging from -78°C to room temperature to give intermediate a3.
In step 4 (scheme 1), the protecting group can be removed according to the respective standard procedures known to the person skilled in the art (see e.g. T.W. Greene and P.G.M. Wuts in Protective Groups in Organic Synthesis, 4th edition, Wiley 2006). As an example, the 2- tetrahydropyranyl group can be removed using a solution of acid like hydrochloric acid in a solvent like dioxane at a temperature range between 0°C and reflux.

 a3 a7 a8
a3 a7 a8
Scheme 2: Route for the preparation of building block of general formula a6, wherein PG1 represents a suitable triazole protecting group (e.g. SEM, THP or diethoxymethyl), Y represents a suitable leaving group (e.g. halogen, tosylate, mesylate, acetate or triflate).
In step 5 (scheme 2), the carbonyl group of intermediate a4 can be reduced to give intermediate
a5. For example, the reduction can be done with a reagent like sodium borohydride in a solvent like ethanol at a temperature range between 0°C and room temperature. Alternatively, other hydride reducing agents known to the person skilled in the art can be used, exemplified by lithium borohydride, lithium aluminium hydride, lithium tri-sec-butyl(hydrido)borate, optionally in the presence of an additive like TiCU or Ti(OiPr)4. Alternatively, the reduction can be performed using hydrogen gas in the presence of a catalyst, for example a transition metal catalyst. Optionally, the reduction can be also a stereoselective reduction with non-chiral reducing agent or catalyst or a chiral reducing agent or catalyst. Optionally, the reduction can be also an enzyme-catalyzed reduction. Those reduction methods are well known to the person skilled in the art and are published in the scientific literature (for asymmetric carbonyl group using chiral reagents see e.g.: E.J. Corey, L. Kurti in Enantioselective Chemical Synthesis. Methods, Logic and Practice, Elsevier, 2013; for diastereoselective carbonyl group reduction see e.g. T. Hanamoto et al., J. Org. Chem. 1990, 55, 4969-4971 ; see also e.g. P.V. Ramachandran et al., J. Org. Chem. 1996, 61 , 95-99; see also e.g. P.K. Mohanta et. al., J. Am. Chem. Soc. 2005, 127, 11896-11897; for asymmetric carbonyl group reduction with dynamic kinetic resolution see e.g. X. Tan et al., Org. Lett. 2020, 22, 7230-7233, for enzymatic reduction methods see e.g. T. Matsuda, R. Yamanaka, and K. Nakamura in Enzymatic Asymmetric Reduction of Carbonyl Compounds; Green Biocatalysis, 2016, R.N. Patel (Ed.)).
In step 6 (scheme 2), the hydroxyl group of intermediate a5 can be converted to a leaving group, exemplified but not limited to Cl, Br, I, p-toluenesulfonate, methylsulfonate, triflate or acetate, to give intermediate a6 (see for e.g. Th. Netscher Recent Research Developments in Organic Chemistry 2003, 7, 71-83). Optionally, this reaction can be carried out using stereospecific methods like Appel reaction (see e.g. Z. Wang (ed.), Comprehensive Organic Name Reactions and Reagents, John Wiley, 2009). Such methods are well known to the person skilled in the art of organic synthesis and widely exemplified in the scientific literature.
Alternatively, step 7 (scheme 2) can be performed similar to step 5 starting from intermediate a3 to give intermediate a7. In step 8 (scheme 2), the hydroxy group of intermediate a7 can be converted to a leaving group as described for step 6 to give intermediate a8. Finally, in step 9 the protecting group of intermediate a8 can be deprotected as described for step 4 to give intermediate a6.

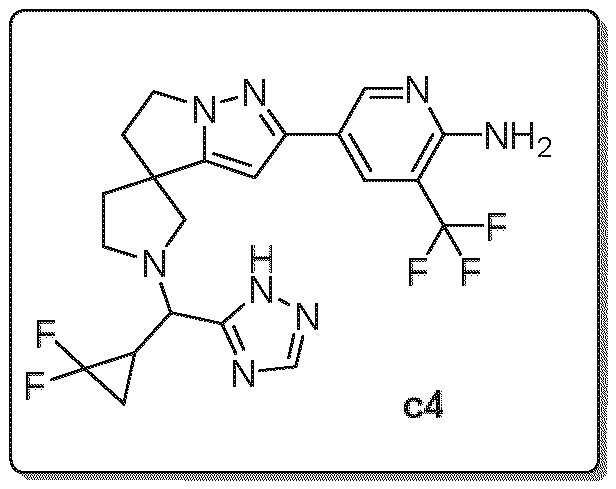
Scheme 3: Route for the preparation of the examples of the invention having general formula c4, wherein PG2 represents a suitable amine protecting group (e.g. Boc), G represents a functional group suitable for oxidative addition to a metal catalyst (e.g. Br, I or triflate), M represents a group suitable for transmetallation reaction with a metal catalyst (e.g. B(OH)2, B(OAIkyl)2, BPin (=4,4,5,5-tetramethyl - 1 ,3,2- dioxaborolane), Y represents a suitable leaving group (e.g. halogen, tosylate, mesylate, acetate or triflate).
In step 10 (scheme 3), the intermediate of general formula c1 (prepared e.g. according to the methods and synthetic procedures described in WO2022167627 A1 can be reacted with organoelement compound d1 (e.g boronic acid derivative, exemplified for example by boronic acid CAS 1189126-37-1 or boronic acid pinacol ester CAS 947249-01-6) in a metal-catalyzed cross-coupling reaction, preferably Suzuki-Miyaura cross-coupling reaction, to give intermediate c2 (see e.g. A. Zapf in Coupling of Aryl and Alkyl Halides with Organoboron Reagents (Suzuki Reaction); in Transition Metals for Organic Synthesis (2nd edition, (2004), edited by M. Beller and C. Bolm); see also e.g. X.-F. Wu et al., Angew. Chem. Int. Ed. 2010, 49, 9047-9050). Such a coupling reaction can be catalyzed, for example, by palladium catalysts, e.g. by Pd(0) catalysts like tetrakis(triphenylphosphine)palladium(0) [Pd(PPhs)4], tris(dibenzylideneacetone)di- palladium(O) [Pd2(dba)3], or by Pd(ll) catalysts like dichlorobis(triphenylphosphine)-palladium (II) [Pd(PPh3)2CI2], palladium (II) acetate and triphenylphosphine, [1 ,1'- bis(diphenylphosphino)ferrocene] palladium dichloride as well as other catalysts and pre
catalysts known to the person skilled in the art (see e.g. John Hartwig in Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books 2010 (Chapter 19: Transition Metal-Catalyzed Coupling Reactions)). The reaction is preferably carried out in a mixture of a solvent like 1 ,2-dimethoxyethane, dioxane, DMF, THF, or isopropanol with water and in the presence of a base like potassium carbonate, sodium bicarbonate or potassium phosphate. The reaction is performed at temperatures ranging from room temperature to the boiling point of the solvent. The reaction is preferably completed after 1 to 36 hours.
In step 11 (scheme 3), the amine protecting group of intermediate c2 is cleaved using procedures well known to the person skilled in the art (see e.g. T.W. Greene and P.G.M. Wuts in Protective Groups in Organic Synthesis, 4th edition, Wiley 2006). For example, the protecting group can be a Boc group and can be cleaved using a solution of hydrochloric acid in dioxane or with a solution of trifluoroacetic acid in dichloromethane at temperatures ranging from 0°C to room temperature to give intermediate c3 in a form of a salt or a free base after additional basification step.
In step 12 (scheme 3), the spiroamine intermediate c3 can be reacted with ketone a4 under reductive amination conditions to provide the final compound of general formula c4. The reductive amination reaction is well known to the person skilled in the art of organic synthesis and can be performed using reducing agents like sodium cyanoborohydride, sodium triacetoxyborohydride, sodium borohydride or alfa-picoline-borane, optionally in the presence of an acid like acetic acid or para-toluenesulfonic acid or a Lewis acid like titanium (IV) tetraisopropoxide or titanium (IV) tetrachloride in a protic or aprotic organic solvent like THF, dioxane, methanol or ethanol or in water, at temperatures ranging from 0°C to the boiling point of the solvent (see e.g. J.C. DiCesare et al., Synth. Commun. 2005, 35, 663-668 and references therein; see also e.g. S. Sato et al., Tetrahedron 2004, 60, 7899-7906 and references therein). Optionally, the reductive amination reaction can be a metal-catalyzed hydrogenation reaction (see e.g. T. Irregang, R. Kempe, Chem. Rev. 2020, 120, 9583-9674). Optionally, the reductive amination can be an asymmetric reductive amination reaction (see e.g. Y. Tian et al. , Org. Chem. Front. 2021 , 8, 2328-2342; see also N. U. Din Reshi, ACS Catal. 2021 , 11 , 13809-13837). Optionally, the reductive amination can be an enzyme-catalyzed reaction (see e.g. P.N. Scheller et al., ChemCatChem 2015, 7, 3239-3242).
Alternatively, in step 12 (scheme 3), the spiroamine intermediate c3 can be reacted with intermediate a6 in a nucleophilic substitution reaction (N-alkylation reaction) to provide the final compound of general formula c4. Such reactions can proceed via SN1 or SN2 mechanism and are well known to the person skilled in the art of organic synthesis (see e.g. J. Clayden, N. Greeves, and S. Warren in Organic Chemistry, 2nd edition, Oxford University Press 201 ). For example, such reaction can be performed in a solvent like DMF, THF, dioxane or toluene, in the presence of a base like sodium hydride, triethyl amine, diisopropylethylamine or pyridine at temperatures ranging from 0°C to the boiling point of the solvent.
EXPERIMENTAL SECTION - METHODS:
Analytical LC-MS methods:
Method 1 :
Instrument: Waters Acquity UPLCMS SingleQuad; Column: Acquity UPLC BEH C18 1.7 m, 50x2.1 mm; eluent A: water + 0.2 vol % aq. ammonia (32%), eluent B: acetonitrile; gradient: 0-
1.6 min 1-99% B, 1.6-2.0 min 99% B; flow 0.8 mL/min; temperature: 60 °C; DAD scan: 210-400 nm.
Method 2:
Instrument: Waters Acquity UPLCMS SingleQuad; Column: Acquity UPLC BEH C18 1.7 pm, 50x2.1 mm; eluent A: water + 0.1 vol % formic acid (99%), eluent B: acetonitrile; gradient: 0-1.6 min 1-99% B, 1.6-2.0 min 99% B; flow 0.8 mL/min; temperature: 60 °C; DAD scan: 210-400 nm.
Method 3:
Instrument: Waters Acquity UPLCMS SingleQuad; Column: Acquity UPLC BEH C18 1.7 pm, 50x2.1 mm; eluent A: water + 0.1 vol % formic acid (99%), eluent B: acetonitrile; gradient: 0-1.6 min 1-99% B, 1.6-2.0 min 99% B; flow 0.8 mL/min; temperature: 60 °C; ELSD.
Method 5:
Instrument: Thermo Scientific FT-MS UHPLC+: Thermo Scientific UltiMate 3000; column: Waters, HSST3, 2.1 x 75 mm, C18 1.8 pm; eluent A: water + 0.01% formic acid; eluent B: acetonitrile + 0.01% formic acid; gradient: 0.0 min 10% B - 2.5 min 95% B - 3.5 min 95% B; temperature: 50°C; flow: 0.90 mL/min; UV-detection: 210 nm/ optimum integration path 210-300 nm.
Method 6:
Instrument: Waters Acquity UPLCMS SingleQuad; Column: Acquity UPLC BEH C18 1.7 pm, 50x2.1 mm; eluent A: water + 0.1 vol % formic acid (99%), eluent B: acetonitrile; gradient: 0-
1.7 min 1-45% B, 1.7-1.72 min 45-99% B, 1.72-2.0 min 99% B; flow 0.8 mL/min; temperature: 60 °C; DAD scan: 210-400 nm.
EXPERIMENTAL SECTION - INTERMEDIATES
Intermediate 1
1-tert-butyl-3-methyl-3-(2-{[tert-butyl(dimethyl)silyl]oxy}ethyl)pyrrolidine-1 ,3-dicarboxylate (enantiomer 1 and enantiomer 2)

1-Tert-butyl 3-methyl pyrrolidine-1 , 3-dicarboxylate (9.59 g, 41.83 mmol) was dissolved in THF (96 mL) under argon atmosphere. It was cooled down to -78°C and LiHMDS (83.7 mL, 83.7 mmol, 1 M in THF) was added. It was stirred 1h at -78°C and then (2-bromoethoxy)(tert- butyl)dimethylsilane (20.01 g, 83.7 mmol) in THF (53 mL) was added dropwise. It was stirred overnight and allowed to reach rt. On an ice bath saturated ammonium chloride solution (160 mL) was added and stirred for 1h. It was extracted with ethyl acetate / THF, dried through a silicone filter and concentrated. The residue was purified by flash chromatography yielding 9.19 g (57%) of the title compound.
1H NMR (400 MHz, DMSO-cfe) 6 ppm: 0.01 (s, 6H), 0.85 (s, 9H), 1.38 (s, 9H), 1.75 - 1.97 (m, 3H), 2.14 - 2.25 (m, 1 H), 3.02 - 3.17 (m, 2H), 3.24 - 3.30 (m, 1 H), 3.46 - 3.64 (m, 5H), 3.67 - 3.82 (m, 1 H).
Intermediate 2 tert-butyl-3-(3-tert-butoxy-3-oxopropanoyl)-3-(2-{[tert-butyl(dimethyl)silyl]oxy}ethyl)pyrrolidine-
1 -carboxylate (enantiomer 1 and enantiomer 2)

□ HMDS (6.5 mL, 1 .0 M in THF, 6.5 mmol) was diluted in THF (10 mL) under argon and cooled down to -78°C then tert-butyl acetate (870 pL, 6.5 mmol) was added dropwise and stirred for 1h at -78°C. Then 1-tert-butyl-3-methyl-3-(2-{[tert-butyl(dimethyl)silyl]oxy}ethyl)pyrrolidine-1 ,3- dicarboxylate (500 mg, 1.29 mmol) dissolved in THF (10 mL) was added dropwise. The reaction mixture was allowed to warm up to rt and stirred overnight at rt. The mixture was poured into saturated ammonium chloride solution and extracted twice with ethyl acetate, dried over sodium sulfate and concentrated to afford 500 mg (82 % yield) of the title compound.
1H-NMR (400 MHz, DMSO-d6) 6 [ppm]: -0.029 (0.99), -0.011 (0.72), -0.003 (16.00), 0.009 (14.92), 0.017 (0.48), 0.057 (1.56), 0.069 (1.62), 0.079 (0.51), 0.168 (0.89), 0.840 (7.12), 1.345 (0.44), 1.381 (10.57), 1.443 (0.60), 1.987 (0.53), 2.518 (0.98), 2.522 (0.66), 3.593 (0.43), 3.655 (0.76).
Intermediate 3 tert-butyl-3-(2-{[tert-butyl(dimethyl)silyl]oxy}ethyl)-3-(5-hydroxy-1 H-pyrazol-3-yl)pyrrolidine-1- carboxylate (enantiomer 1 and enantiomer 2)

Tert-butyl-3-(3-tert-butoxy-3-oxopropanoyl)-3-(2-{[tert-butyl(dimethyl)silyl]oxy}ethyl)pyrrolidine-
1 -carboxylate (8.60 g, 18.2 mmol) and hydrazine hydrate (3.5 mL, 73 mmol) were dissolved in ethanol (180 mL) under argon and stirred for 6h at 80°C. The reaction mixture was concentrated to give 10.5 g of the title compound.
1H-NMR (400 MHz, DMSO-d6) 6 [ppm]: -0.185 (0.47), -0.149 (0.52), -0.035 (13.08), -0.030 (1.92), 0.685 (0.59), 0.831 (11.97), 0.843 (16.00), 1.039 (6.72), 1.056 (14.62), 1.074 (6.60), 1.111 (10.59), 1.228 (0.50), 1.234 (0.50), 1.272 (0.68), 1.356 (3.62), 1.367 (9.35), 1.387 (13.19), 1.853 (0.46), 1.886 (0.43), 2.081 (11.55), 3.195 (0.87), 3.243 (0.41), 3.256 (0.46), 3.269 (0.46), 3.281 (0.46), 3.415 (2.96), 3.432 (8.65), 3.449 (6.98), 3.467 (2.59), 3.692 (0.46), 3.719 (0.43), 5.203 (2.88), 5.230 (1.05), 5.253 (0.66).
Intermediate 4 tert-butyl-3-(2-hydroxyethyl)-3-(3-hydroxy-1 H-pyrazol-5-yl)pyrrolidine-1 -carboxyl ate (enantiomer 1 and enantiomer 2)
 Tert-butyl-3-(2-{[tert-butyl(dimethyl)silyl]oxy}ethyl)-3-(5-hydroxy-1 H-pyrazol-3-yl)pyrrolidine-1- carboxylate (243 mg, 590 pmol) was solubilised in methanol (1.1 mL), hydrochloric acid (440 pL, 1.76 mmol, 4.0 M in dioxane) was added and the mixture was stirred for 2h at rt under argon. Potassium carbonate (490 mg, 3.54 mmol) and di-tert-butyl dicarbonate (200 pL, 890 pmol) were added and the reaction mixture was stirred for 1h at rt under argon. The mixture was filtered and evaporated to give the title compound.
Tert-butyl-3-(2-{[tert-butyl(dimethyl)silyl]oxy}ethyl)-3-(5-hydroxy-1 H-pyrazol-3-yl)pyrrolidine-1- carboxylate (243 mg, 590 pmol) was solubilised in methanol (1.1 mL), hydrochloric acid (440 pL, 1.76 mmol, 4.0 M in dioxane) was added and the mixture was stirred for 2h at rt under argon. Potassium carbonate (490 mg, 3.54 mmol) and di-tert-butyl dicarbonate (200 pL, 890 pmol) were added and the reaction mixture was stirred for 1h at rt under argon. The mixture was filtered and evaporated to give the title compound.
Intermediate 5 tert-butyl-2'-hydroxy-5',6'-dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazole]-1 -carboxylate
(enantiomer 1 and enantiomer 2)
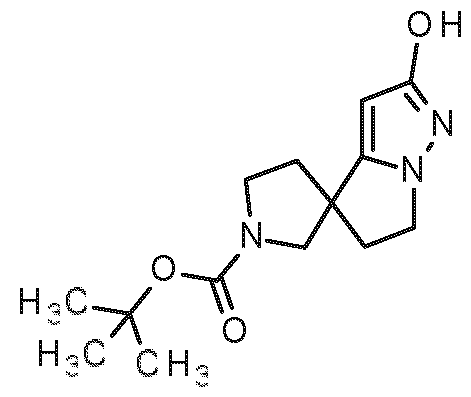
Tert-butyl-3-(2-hydroxyethyl)-3-(3-hydroxy-1 H-pyrazol-5-yl)pyrrolidine-1 -carboxylate (5.24 g, 17.6 mmol) and triphenylphosphine (11.09 g, 42.3 mmol) were solubilised in THF (82 mL) under argon, DIAD (8.0 mL) was added dropwise and the mixture was stirred for 4h at rt. The mixture was evaporated and purified by flash chromatography to give 2.83 g (95 % purity, 55 % yield) of the title compound.
1H-NMR (400 MHz, DMSO-d6) 6 [ppm]: 1.055 (0.45), 1.391 (16.00), 1.418 (13.84), 1.936 (0.55), 1.950 (0.82), 1.964 (0.84), 1.982 (0.88), 2.008 (0.64), 2.325 (0.40), 2.329 (0.55), 2.334 (0.43),
2.347 (1.09), 2.363 (1.82), 2.381 (1.13), 2.521 (1.64), 2.525 (1.12), 2.671 (0.43), 3.365 (0.83),
3.426 (0.74), 3.434 (0.63), 3.441 (0.65), 3.454 (0.50), 3.913 (1.22), 3.918 (1.44), 3.931 (2.38),
3.935 (2.82), 3.947 (1.19), 3.952 (1.45), 5.208 (10.10), 9.584 (1.93).
Intermediate 6 tert-butyl-2'-[(trifluoromethanesulfonyl)oxy]-5',6'-dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2- b]pyrazole]-1 -carboxylate (enantiomer 1 and enantiomer 2)
 Tert-butyl-2’-hydroxy-5’,6’-dihydrospiro[pyrrolidine-3,4’-pyrrolo[1 ,2-b]pyrazole]-1 -carboxylate
Tert-butyl-2’-hydroxy-5’,6’-dihydrospiro[pyrrolidine-3,4’-pyrrolo[1 ,2-b]pyrazole]-1 -carboxylate
(3.30 g, 11.8 mmol, 22% purity) was dissolved in THF (100 mL) under argon and treated with N,N-diisopropylethylamine (10 mL, 59 mmol) then N,N-bis(trifluoromethanesulfonyl)aniline (6.33 g, 17.7 mmol) was added and stirred at 60°C for 1h. The reaction mixture was concentrated and the residue was purified by flash chromatography to afford 600 mg (56 % yield) of the title compound.
1H-NMR (400 MHz, DMSO-d6) 6 [ppm]: 0.000 (12.78), 1.156 (1.57), 1.173 (5.35), 1.188 (5.10), 1.192 (2.37), 1.231 (0.58), 1.235 (0.48), 1.248 (0.83), 1.265 (0.42), 1.394 (16.00), 1.422 (12.77), 1.989 (4.21), 2.032 (0.58), 2.048 (0.79), 2.060 (0.85), 2.077 (1.07), 2.097 (0.81), 2.108 (0.44),
2.121 (0.86), 2.471 (1.67), 2.520 (1.72), 2.525 (1.11), 3.371 (0.69), 3.379 (0.88), 3.397 (0.97),
3.428 (4.90), 3.455 (0.50), 3.462 (0.81), 3.474 (0.88), 3.482 (1.12), 3.489 (0.76), 3.494 (0.94),
3.501 (0.68), 3.509 (0.66), 3.521 (0.49), 4.020 (0.97), 4.037 (0.96), 4.191 (0.42), 4.201 (1.17),
4.212 (1.51), 4.217 (1.63), 4.220 (1.58), 4.229 (1.66), 4.232 (1.87), 4.237 (1.32), 4.248 (1.33),
4.258 (0.51), 4.768 (0.45), 6.255 (2.91), 8.883 (0.91).
Intermediate 7 tert-butyl (3R)-2'-[(trifluoromethanesulfonyl)oxy]-5',6'-dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2- b]pyrazole]-1 -carboxylate (enantiomer 1 , used for the synthesis of the target compound)

The title compound was prepared by chiral separation of the corresponding racemic mixture.
Chiral separation protocol for the first-eluting enantiomer :
Preparative method:
Instrument: Sepiatec: Prep SFC100; Column: Chiralpak IG 5pm 250x30mm; eluent A: CO2; eluent B: ethanol + 0.2 vol % aqueous ammonia (32%); isocratic: 5% B; flow: 100 mL/min; temperature: 40°C; BPR: 150 bar; UV: 210 nm
Retention time preparative method : 3.8 - 4.7 min
Analytical method:
Instrument: Agilent: 1260, Aurora SFC-Modul; Column: Chiralpak IG 5pm 100x4.6mm; eluent A: CO2; eluent B: ethanol + 0.2 vol % aqueous ammonia (32%); isocratic: 5% B; flow: 4 mL/min; temperature: 37.5°C; BPR: 100 bar; UV: 210 nm
Retention time analytical method: 1 .05 min
[Q]20D : +10.4° (c = 1.00, DMSO)
Intermediate 8 tert-butyl (3S)-2'-[(trifluoromethanesulfonyl)oxy]-5',6'-dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2- b]pyrazole]-1 -carboxylate (enantiomer 2, not used further)
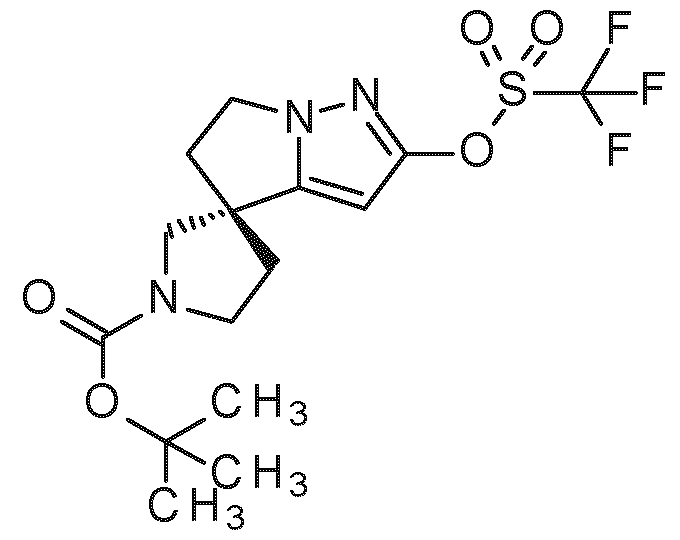
The title compound was prepared by chiral separation of the corresponding racemic mixture.
Chiral separation protocol for the second-eluting enantiomer :
Preparative method:
Instrument: Sepiatec: Prep SFC100; Column: Chiralpak IG 5pm 250x30mm; eluent A: CO2; eluent B: ethanol + 0.2 vol % aqueous ammonia (32%); isocratic: 5% B; flow: 100 mL/min; temperature: 40°C; BPR: 150 bar; UV: 210 nm
Retention time preparative method : 6.0 - 7.5 min
Analytical method:
Instrument: Agilent: 1260, Aurora SFC-Modul; Column: Chiralpak IG 5pm 100x4.6mm; eluent A: CO2; eluent B: ethanol + 0.2 vol % aqueous ammonia (32%); isocratic: 5% B; flow: 4 mL/min; temperature: 37.5°C; BPR: 100 bar; UV: 210 nm
Retention time analytical method: 1 .60 min
[a]20 D : -9.6° (c = 1.00, DMSO)
Intermediate 9 tert-butyl (3R)-2'-[6-amino-5-(trifluoromethyl)pyridin-3-yl]-5',6'-dihydrospiro[pyrrolidine-3,4'- pyrrolo[1 ,2-b]pyrazole]-1 -carboxylate

To a crude solution of 5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-3-(trifluoromethyl)pyridin- 2-amine (2.00 g, 6.94 mmol), tert-butyl (3R)-2’-[(trifluoromethanesulfonyl)oxy]-5’,6’- dihydrospiro[pyrrolidine-3,4’-pyrrolo[1 ,2-b]pyrazole]-1-carboxylate (3.00 g, 7.29 mmol) and potassium phosphate (42 mL, 0.50 M in water, 21 mmol) were added. The mixture was degassed and XPhos Pd G2 (273 mg, 347 pmol) was added and stirred at 100°C under argon for 2h. The mixture was diluted with water and then extracted twice with ethyl acetate. The combined organic layers were washed with brine, dried over sodium sulfate, filterd and concentrated. The crude was purified by column chromatography to give 3.50 g of the title compound.
LC-MS (Method 1): Rt = 1.20 min; MS (ESIpos): m/z = 425 [M+H]+
Intermediate 10
5-[(3R)-5',6'-dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl]-3-(trifluoromethyl)pyridin-
2-amine hydrogen chloride
HCI

To a mixture of tert-butyl (3R)-2’-[6-amino-5-(trifluoromethyl)pyridin-3-yl]-5’,6’- dihydrospiro[pyrrolidine-3,4’-pyrrolo[1 ,2-b]pyrazole]-1-carboxylate (2.90 g, 6.85 mmol) in dioxane (34 mL), hydrochloric acid (34 mL, 136 mmol, 4.0 M in dioxane) was added. The solution was stirred at rt overnight. The resultant mixture was concentrated in vacuo to give 3.10 g of the title compound.
LC-MS (Method 1): Rt = 0.83 min; MS (ESIpos): m/z = 324 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 6 [ppm]: 1.064 (16.00), 1.122 (0.62), 1.154 (0.46), 2.144 (0.45),
2.156 (0.50), 2.163 (1.04), 2.171 (0.76), 2.183 (0.64), 2.188 (0.62), 2.522 (0.93), 2.580 (0.52),
2.594 (0.53), 2.678 (0.45), 2.696 (0.54), 2.712 (0.48), 3.287 (0.49), 3.402 (0.44), 3.409 (0.46),
3.419 (0.60), 3.436 (0.56), 3.448 (0.54), 3.564 (1.08), 4.184 (0.64), 4.192 (0.67), 4.203 (0.95),
4.211 (0.84), 4.219 (0.68), 4.226 (0.60), 6.666 (3.69), 8.107 (1.24), 8.112 (1.27), 8.584 (1.30),
8.589 (1.26).
Intermediate 11
2, 2-difluoro-N-methoxy-N-methylcyclopropane-1 -carboxamide (enantiomer 1 and enantiomer
2)

Triethylamine (5.7 mL, 41 mmol) and T3P (9.6 mL, 50 % in ethyl acetate, 16 mmol) were added to 2, 2-difluorocyclopropane-1 -carboxylic acid (1.00 g, 8.19 mmol) and N-methoxymethanamine- hydrogen chloride (1/1) (879 mg, 9.01 mmol) in THF (13 mL) and stirred at rt overnight. Water (50 mL) was added and it was extracted with ethyl acetate. The organic phase was washed with brineand dried through a hydrophobic filter. The solvent was removed at a maximum of 50 °C and at a minimum of 50 mbar to yield 1 .33 g (98 % yield) of the title compound which was used without further purification in the next step.
LC-MS (Method 6): Rt = 1.01 min; MS (ESIpos): m/z = 166 [M+H]+
1H NMR (400 MHz, DMSO-cfe, 22°C) 6 ppm 1.85 - 1.98 (m, 2 H), 3.05 - 3.13 (m, 1 H), 3.16 (s, 3 H), 3.72 (s, 3 H).
Intermediate 12
Method 1 :
(2,2-difluorocyclopropyl)(1-{[2-(trimethylsilyl)ethoxy]methyl}-1 H-1 ,2,4-triazol-5-yl)methanone
(enantiomer 1 and enantiomer 2)
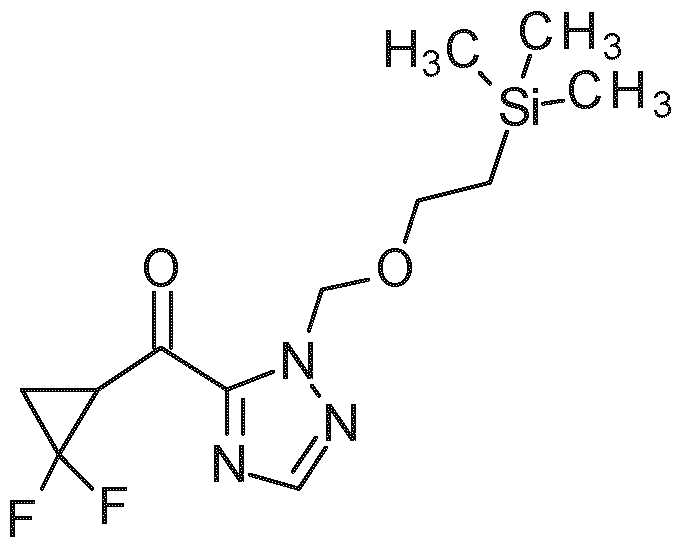
1-{[2-(Trimethylsilyl)ethoxy]methyl}-1 H-1 ,2,4-triazole (10.0 g, 50.2 mmol) was dissolved in THF (150 mL) and cooled to -78 °C. At a maximum temperature of -60 °C n-butyllithium (30 mL, 2.5 M in hexane, 75 mmol) was added dropwise and the reaction mixture was stirred for 1 h at -70
°C. At -78 °C 2, 2-difluoro-N-methoxy-N-methylcyclopropane-1 -carboxamide (8.28 g, 50.2 mmol) in THF (15 mL) was added dropwise and then the reaction mixture was allowed to reach rt. Saturated sodium hydrogen carbonate solution was added and stirred for 5 min. Ethyl acetate was added, the layers were separated and the aqueous phase was extracted once with ethyl acetate. The combined organic phases were washed with saturated sodium chloride solution, dried with a hydrophobic filter, and concentrated. The residue was purified by silica gel chromatography affording 8.48 g (56 % yield) of the title compound.
LC-MS (Method 1): Rt = 1.43 min; MS (ESIpos): m/z = 302 [M-H]’
1H NMR (400 MHz, DMSO-cfe, 22°C) 6 ppm -0.09 (s, 9 H), 0.81 (td, 2 H), 2.20 - 2.34 (m, 2 H), 3.58 (ddd, 2 H), 3.96 - 4.06 (m, 1 H), 5.72 - 5.83 (m, 2 H), 8.33 (s, 1 H).
Method 2:
(2,2-difluorocyclopropyl)(1-{[2-(trimethylsilyl)ethoxy]methyl}-1 H-1 ,2,4-triazol-5-yl)methanone
(enantiomer 1 and enantiomer 2)
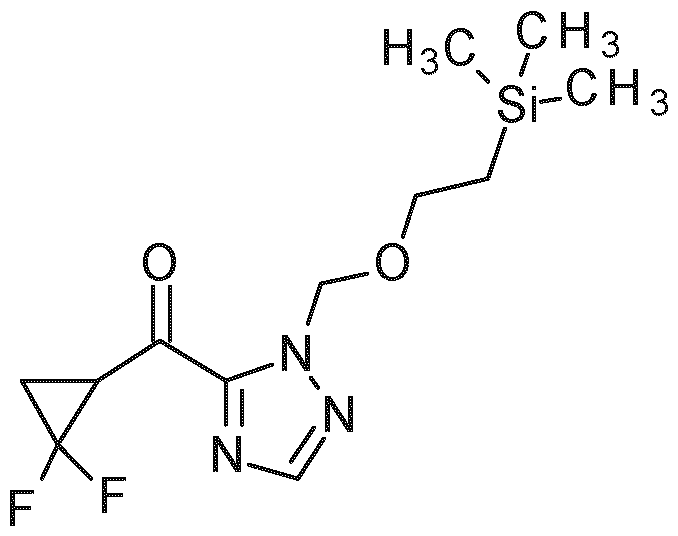
The solution of 1-{[2-(trimethylsilyl)ethoxy]methyl}-1 H-1 ,2,4-triazole (567 mg, 2.85 mmol) in THF (2.5 mL) was cooled to 0°C and the solution of 2, 2-difluorocyclopropane-1 -carbonyl chloride (400 mg, 2.85 mmol) in THF (1 mL) was added at 0°C. To the resulting mixture the solutiuon of triethylamine (1.19 mL) in THF (2.5 mL) was added dropwise at 0°C and the reaction mixture was stirred for 30 minutes at 0°C. The reaction mixture was quenched with water, extracted three times with ethyl acetate, the combined organic phases were washed with brine, filtered through a hydrophobic filter and concentrated under reduced pressure. The product was separated by silica gel flash chromatography (gradient 10 to 18% EtOAc in hexane) to yield 190 mg (22%) of the title compound (racemic).
LC-MS (Method 1): Rt = 1.39; MS (ESIpos): m/z = 304.1 [M+H]+
1H NMR (400 MHz, DMSO-cfe) 6 ppm -0.09 (m, 9 H), 0.74 - 0.86 (m, 2 H), 2.19 - 2.35 (m, 2 H), 3.53 - 3.64 (m, 2 H), 3.93 - 4.08 (m, 1 H), 5.70 - 5.83 (m, 2 H), 8.33 (s, 1 H).
Intermediate 13
Method 1 :
(2,2-difluorocyclopropyl)(1 H-1 ,2,4-triazol-5-yl)methanone (enantiomer 1 and enantiomer 2)

(2,2-Difluorocyclopropyl)(1-{[2-(trimethylsilyl)ethoxy]methyl}-1 H-1 ,2,4-triazol-5-yl)methanone
(490 mg, 1.62 mmol) was dissolved in dichloromethane (10 mL) and trifluoroacetic acid (6.2 mL, 81 mmol). The reaction mixture was stirred at 45 °C for 15 min and overnight at rt. The reaction mixture was concentrated and sodium carbonate solution (1.5 M in water) and ethyl acetate were added. The organic layer was separated, washed with saturated sodium chloride solution, dried with a hydrophobic filter, and concentrated. The aqueous phase was adjusted to pH 8 with citric acid (1 M in water), extracted with ethyl acetate, dichloromethane and ethyl acetate. The combined organic layers were dried with a hydrophobic filter and concentrated. The residues were combines and dissolved in methanol and concentrated. This prodedure was repeated several times to give 228 mg (69 % yield) of the title product.
1H NMR (400 MHz, DMSO-cfe, 22°C) 6 ppm 2.09 - 2.20 (m, 1 H), 2.21 - 2.31 (m, 1 H), 3.88 - 4.00 (m, 1 H), 8.76 (br s, 1 H), 14.93 (br s, 1 H).
Method 2:
(2,2-difluorocyclopropyl)(1 H-1 ,2,4-triazol-5-yl)methanone (enantiomer 1 and enantiomer 2)

(2,2-Difluorocyclopropyl)(1-{[2-(trimethylsilyl)ethoxy]methyl}-1 H-1 ,2,4-triazol-5-yl)methanone
(7.27 g, 24.0 mmol) was dissolved in dichloromethane (140 mL) and trifluoroacetic acid (74 mL, 960 mmol) was added. The reaction mixture was stirred at rt overnight. The reaction mixture was concentrated and saturated sodium hydrogen carbonate solution and ethyl acetate were added. The organic layer was separated and the aqueous phase was extracted three times with ethyl acetate / methanol = 8:2. The combined organic layers were washed once with brine, dried with a hydrophobic filter, and concentrated. The aqueous phase was extracted twice with ethyl acetate / methanol = 9:1 and concentrated thoroughly. The combined organic batches gave 4.05 g of a mixture of the title compound and (2,2-difluorocyclopropyl)[1-(hydroxymethyl)-1 H-1 ,2,4- triazol-5-yl]methanone.
A mixture of (2,2-difluorocyclopropyl)[1-(hydroxymethyl)-1 H-1 ,2,4-triazol-5-yl]methanone and (2,2-difluorocyclopropyl)(1 H-1 ,2,4-triazol-5-yl)methanone (2.90g) was dissolved in dioxane (64 mL) and ammonia in methanol (61 mL, 7 M, 430 mmol) were added and the reaction mixture was stirred overnight at rt. The reaction mixture was concentrated to yield 2.32 g of the title product.
LC-MS (Methode 6): Rt = 0.67 min; MS (ESIpos): m/z = 174 [M+H]+
1H NMR (400 MHz, DMSO-cfe, 22°C) 6 ppm 2.09 - 2.20 (m, 1 H), 2.21 - 2.31 (m, 1 H), 3.90 - 3.99 (m, 1 H), 8.76 (s, 1 H).
Intermediate 14
(S)-(2,2-difluorocyclopropyl)(1 H-1 ,2,4-triazol-5-yl)methanone (enantiomer 1 , not used further)

The racemic mixture of (2,2-difluorocyclopropyl)(1 H-1 ,2,4-triazol-5-yl)methanone (710 mg, 4.10 mmol) was separated by chrial HPLC to give 290 mg (99 % purity, 39 % yield) of the title compound as the first-eluting enantiomer.
Preparative method:
Instrument: Sepiatec: Prep SFC100; Column: Chiral Art Amylose-SA 5p 250x30mm; eluent A: CO2; eluent B: methanol + 0.2 vol % aqueous ammonia (32%); isocratic: 5% B; gradient: no; flow: 100 mL/min; temperature: 40°C; BPR: 150 bar; UV: 230 nm
Retention time: 3.7 - 4.15 min;
Analytical method:
Instrument: Waters Acquity UPC2 QDA; Column: Chiral Art Amylose-SA 3p 100x4.6mm; eluent A: CO2; eluent B: methanol + 0.2 vol % aqueous ammonia (32%); isocratic: 5% B; gradient: no; flow: 4 mL/min; temperature: 37.5°C; BPR: 100 bar; UV: 220 nm
Retention time: 0.85 min
[Q]20D : +45.8° (c = 1.00, methanol)
Intermediate 15
(R)-(2,2-difluorocyclopropyl)(1 H-1 ,2,4-triazol-5-yl)methanone (enantiomer 2, used for the synthesis of the target compound)
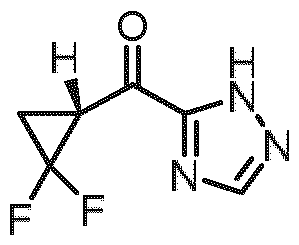
The racemic mixture of (2,2-difluorocyclopropyl)(1 H-1 ,2,4-triazol-5-yl)methanone (710 mg, 4.10 mmol) was separated by chrial HPLC to give 269 mg (99 % purity, 36 % yield) of the title compound as the second-eluting enantiomer.
Preparative method:
Instrument: Sepiatec: Prep SFC100; Column: Chiral Art Amylose-SA 5p 250x30mm; eluent A: CO2; eluent B: methanol + 0.2 vol % aqueous ammonia (32%); isocratic: 5% B; gradient: no; flow: 100 mL/min; temperature: 40°C; BPR: 150 bar; UV: 230 nm
Retention time: 4.3 - 4.85 min;
Analytical method:
Instrument: Waters Acquity UPC2 QDA; Column: Chiral Art Amylose-SA 3p 100x4.6mm; eluent A: CO2; eluent B: methanol + 0.2 vol % aqueous ammonia (32%); isocratic: 5% B; gradient: no; flow: 4 mL/min; temperature: 37.5°C; BPR: 100 bar; UV: 220 nm
Retention time: 1.03 min
[a]20D : -48.4° (c = 1.00, methanol)
The crystallographic data of intermediate 15 as well as a figure depicting the thermal ellipsoids and numbering of the structure, are shown in Table 3 and Figures 1 and 2. Colorless crystals of ntermediate 15 were obtained by slow evaporation from an ethanol solution. A single crystal was mounted on a cryoloop using a protective oil. Single-crystal X-ray diffraction data were collected at 100 K on a Rigaku MicroMax-007HF system with a Rigaku AFC-11 partial 4-axis goniometer and a Dectris Pilatus 3R 200K-A detector using Cu X-ray radiation (CuKa, = 1.54178 A). Data were integrated using the program CrysAlisPRO. SHELXM was used for structure solution and SHELXL was used for full-matrix least-squares refinement on F2. In the asymmetric unit two molecules of intermediate 15 are present. All non-hydrogen atoms were refined anisotropically. All hydrogen atoms were added in calculated positions and refined riding on their resident atoms. Hydrogens attached to N4 and C3 atoms (see Figure 1 for naming atoms) were either located in the difference Fourier map and placed manually or were refined using the riding model. The isotropic temperature factors of the hydrogen atoms were refined as 1 .2 and 1 .5 times the size of the temperature factors of the corresponding heavy atoms, respectively. The absolute stereochemistry (R-configuration) could be assigned unambiguously with a Flack Parameter of 0.05(6). The program XP was used for molecular representations.
Table 3. Crystal data and structure refinement for intermediate 15.
Identification code intermediate 15
Empirical formula 06 H5 F2 N3 O
Formula weight 173.13
Temperature 173(2) K
Wavelength 1.54178 A
Crystal system Monoclinic
Space group P21
Unit cell dimensions a = 5.02500(10) A a= 90°. b = 16.4766(2) A b= 90.7280(10)°. c = 8.65160(10) A g = 90°.
Volume 716.251(19) A3
Z 4
Density (calculated) 1.606 Mg/m3
Absorption coefficient 1.319 mm’1
F(000) 352
Crystal size 0.16 x 0.06 x 0.01 mm3
Theta range for data collection 5.112 to 68.240°.
Index ranges -6<=h<=6, -19<=k<=19, -10<=l<=10
Reflections collected 24766
Independent reflections 2489 [R(int) = 0.0889]
Completeness to theta = 67.679° 96.4 % Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 2489 / 33 / 229
Goodness-of-fit on F2 1.113
Final R indices [l>2sigma(l)] R1 = 0.0526, wR2 = 0.1132
R indices (all data) R1 = 0.0532, wR2 = 0.1137
Absolute structure parameter 0.05(6)
Extinction coefficient n/a
Largest diff. peak and hole 0.268 and -0.431 e.A’3 cf. Figure land Figure 2
Intermediate 16
Method 1 :
[(R)-2,2-difluorocyclopropyl](1 H-1,2,4-triazol-5-yl)methanol (diastereomer 1 and diastereomer
2)
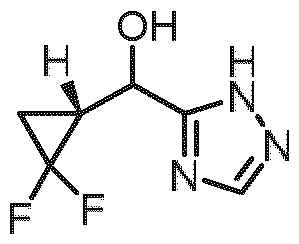
Under argon (2,2-difluorocyclopropyl)(1 H-1 ,2,4-triazol-5-yl)methanone (enantiomer 2) (260 mg, 1.50 mmol) was dissolved in ethanol (6.00 mL) and sodium tetrahydroborate (42.6 mg, 1.13 mmol) was added in portions at 0°C. The reaction mixture was stirred at rt over night. Water (6 mL) was added, the pH was adjusted to pH 7 with citric acid (1 M in water) and saturated sodium hydrogen carbonate solution, and extracted twice with ethyl acetate. The combined organic phases were dreid through a hydrophobic filter and concentrated. The residue was treated with ethyl acetate/toluene and concentrated. Afterwards the residue was treated with dioxane and concentrated to yield 220 mg (84 % yield) of the title product which was used without further purification in the next step.
LC-MS (Method 3): Rt = 0.40 min; MS (ESIpos): m/z = 174 [M-H]~
1H NMR (400 MHz, DMSO-cfe, 22°C) (mixture of diastereomers/tautomers) 6 ppm 1.10 - 1.75 (m, 4 H), 2.08 - 2.30 (m, 2 H), 4.26 - 4.35 (m, 1 H), 4.42 - 4.53 (m, 1 H), 5.64 and 5.72 (2 br d, 1 H), 6.18 and 6.28 (2 d, 1 H), 7.87 and 8.49 (2s, 1 H), 13.86 and 13.94 (2br s, 1 H).
Method 2:
(R)-(2,2-difluorocyclopropyl)(1 H-1 ,2,4-triazol-5-yl)methanone hydrochloride (intermediate 25) (226 mg, 1.078 mmol) was dissolved in EtOH (5.4 mL) and sodium borohydride (71.4 mg, 1.887 mmol) was added at rt. The reaction mixture was stirred for 1 h at rt and then concentrated under reduced pressure. The residue was suspended in EtOAc and small amount of water was added to give a clear biphasic solution. The pH of the aqueous layer was adjusted to pH = 7 with 10wt% aqueous citric acid solution and saturated aqueous NaHCOs solution and saturated with solid NaCI. The organic phase was then separated and the aqueous layer was washed with EtOAc. The combined organic layers were washed with brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure to give 120 mg (60.5% yield) of the title compound.
1H NMR (400 MHz, DMSO-cfe, 22°C) (mixture of diastereomers and tautomers) 6 ppm 1.10 - 1 .78 (m, 2 H), 2.04 - 2.28 (m, 1 H), 4.23 - 4.38 and 4.40 - 4.55 (both m, each 1 H), 5.55 - 5.77 and 6. 11 - 6.34 (both m, together 1 H), 7.86 and 8.48 (both s, together 1 H), 13.86 and 14.94 (both br s, together 2 H).
Intermediate 17
5-[chloro[(S)-2,2-difluorocyclopropyl]methyl]-1 H-1 ,2,4-triazole hydrogen chloride (1/1) (diastereomer 1 and diastereomer 2)

[(R)-2,2-Difluorocyclopropyl](1 H-1 ,2,4-triazol-5-yl)methanol (diastereomer 1 and diastereomer 2) was dissolved in dioxane (2.0 mL) and thionyl chloride (270 pL, 3.8 mmol) was added. The reaction mixture was stirred 45 min at rt and concentrated under reduced pressure. The residue was treated twice with dioxane and concentrated affording 299 mg of the title compound which was used without further purification in the next step.
LC-MS (Method 3): Rt = 0.64 and 0.67 min; MS (ESIpos): m/z = 194 [M+H]+
1H NMR (400 MHz, DMSO-cfe, 22°C) (diastereomeric mixture) 6 ppm 1.59 (dtd, 1 H), 1.71 - 1.85 (m, 2 H), 1 .88 - 1 .98 (m, 1 H), 2.64 - 2.77 (m, 2 H), 5.00 (dd, 1 H), 5.06 (dd, 1 H), 8.56 (s, 2 H).
Intermediate 18
1-(oxan-2-yl)-1 H-1 ,2,4-triazole
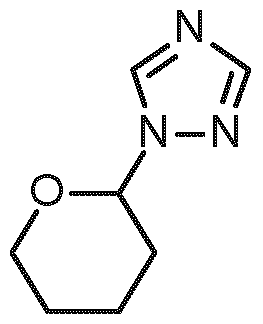
To the solution/suspension of 1 H-1 ,2,4-triazole (10.0 g, 145 mmol) in THF (73 mL) were added para-toluenesulfonic acid monohydrate (2.75 g, 14.5 mmol) and 3,4-dihydro-2H-pyran (26 mL, 290 mmol). The mixture was stirred for 2h at 70°C, then cooled to room temperature, quenched with saturated aqueous NaHCCh, diluted with water and EtOAc and extracted 3 times with EtOAc. The combined organic layers were washed with water and brine, dried over anhydrous Na2SO4, filtered over a short layer of silica gel and filter cake washed with EtOAc. The combined filtrate was concentrated under reduced pressure to yield 22.56 g (~80 % purity, 86 % yield) of the title product.
'HNMR (400 MHz, CDC13) 5 ppm 1.49 - 1.85 (m, 4 H), 1.98 - 2.16 (m, 2 H), 3.67 - 3.76 (m, 1 H), 4.03 - 4.10 (m, 1 H), 5.42 - 5.53 (m, 1 H), 7.96 (m, 1 H), 8.28 (m, 1 H).
Intermediate 19
Method 1 :
(2,2-difluorocyclopropyl)[1-(oxan-2-yl)-1 H-1 ,2,4-triazol-5-yl]methanone

1-(Oxan-2-yl)-1 H-1 ,2,4-triazole (1.00 g, ca. 80 % purity, 5.22 mmol) was dissolved in THF (12 mL) and 2.5 M n-BuLi solution in hexane (3.1 mL, 7.8 mmol) was added dropwise between - 70°C and -75°C under stirring. After 1h 2,2-difluoro-N-methoxy-N-methylcyclopropane-1- carboxamide (1.08 g, ca. 80 % purity, 5.22 mmol) in THF (4 mL) was added dropwise at -75°C. The reaction was stirred for 30 minutes at -75°C, thereafter the cooling bath was removed and the mixture was warmed up to room temperature. The reaction mixture was quenched with water and extracted 3 times with EtOAc. The combined organic layers were washed with water and brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The resulting crude product was purified by silica gel flash chromatography (gradient 0 to 60% EtOAc in hexane) to yield 723 mg (54 % yield) of the title compound.
LC-MS (Method 1): Rt = 1.04 min and 1.06 min; MS (ESIpos): m/z = 258 [M+H]+
1H-NMR (400 MHz, CHLOROFORM-d) 6 [ppm]: 1.268 (0.76), 1.285 (0.40), 1.404 (0.73), 1.545 (0.80), 1.556 (1.23), 1.575 (11.44), 1.587 (0.79) , 1.608 (1.94), 1.613 (1.86), 1.618 (1.72), 1.624 (2.06), 1.631 (2.22), 1.637 (1.98), 1.640 (1.72), 1.652 (0.95), 1.669 (0.89), 1.679 (0.91), 1.698 (1.70), 1.708 (2.22), 1.715 (1.85), 1.728 (3.53), 1.731 (3.26), 1.737 (3.64), 1.740 (3.53), 1.743 (3.13), 1.751 (3.15), 1.763 (1.91), 1.772 (1.45), 1.779 (1.04), 1.789 (0.40), 1.892 (1.65), 1.905 (1.49), 1.911 (1.73), 1.919 (2.22), 1.924 (1.71), 1.932 (2.54), 1.938 (2.98), 1.946 (2.55), 1.951 (3.57), 1.959 (2.68), 1.965 (2.79), 1.973 (1.90), 1.978 (2.48), 1.990 (1.68), 1.997 (1.14), 2.000 (1.16), 2.007 (0.61), 2.055 (1.36), 2.082 (1.03), 2.087 (1.07), 2.093 (1.42), 2.100 (1.39), 2.106 (1.80), 2.119 (1.96), 2.132 (1.13), 2.146 (0.44), 2.260 (0.47), 2.271 (0.64), 2.285 (0.64), 2.290 (0.72), 2.295 (0.98), 2.300 (1.24), 2.303 (0.98), 2.312 (1.58), 2.325 (1.46), 2.328 (2.17), 2.332 (2.41), 2.343 (2.73), 2.347 (2.04), 2.351 (1.61), 2.358 (2.82), 2.363 (3.44), 2.366 (1.69), 2.373 (1.70), 2.378 (2.66), 2.382 (2.16), 2.389 (1.27), 2.393 (1.59), 2.397 (2.02), 2.408 (1.29), 2.412 (0.96), 2.428 (0.79), 3.264 (2.23), 3.705 (1.08), 3.711 (0.87), 3.726 (1.32), 3.733 (2.79), 3.739 (1.68), 3.755 (2.23), 3.762 (2.72), 3.769 (1.25), 3.773 (5.97), 3.783 (1.27), 3.790 (0.91), 3.981 (1.32), 4.001 (1.52), 4.006 (1.87), 4.014 (1.65), 4.026 (2.08), 4.031 (1.96), 4.034 (1.97), 4.038 (2.02), 4.051 (1.44), 4.058 (2.08), 4.063 (1.72), 4.069 (0.96), 4.084 (3.27), 4.098 (1.16), 4.114 (2.09), 4.121 (1.67), 4.820 (0.56), 4.827 (0.53), 4.832 (0.68), 4.839 (0.54), 6.276 (2.25), 6.283 (2.35), 6.289 (2.39), 6.296 (2.53), 6.301 (2.45), 6.307 (2.28), 6.314 (2.43), 6.321 (2.29), 8.035 (16.00).
Method 2:
(2,2-difluorocyclopropyl)[1 -(oxan-2-yl)-1 H-1 ,2,4-triazol-5-yl]methanone

1-(Oxan-2-yl)-1 H-1 ,2,4-triazole (100 mg, 80 % purity, 522 pmol) was dissolved in THF (1.2 mL) and 2.5M n-BuLi solution in hexane (310 pL, 780 pmol) was added dropwise between -70°C and - 75°C under stirring. After 1 h N-(benzyloxy)-2,2-difluoro-N-methylcyclopropane-1-carboxamide (140 mg, 90 % purity, 522 pmol) in THF (400 pL) was added dropwise at -75°C. The reaction mixture was stirred for 30 minutes at -75°C, thereafter the cooling bath was removed and the mixture was warmed up to rt the reaction mixturewas quenched with water and extracted 3 times with EtOAc. The combined organic layers were washed with water and brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure. The resulting crude product was purified by silica gel flash chromatography (gradient 0 to 30% EtOAc in hexane) to give 76.0 mg (80 % purity, 45 % yield) of the title compound as a mixture of stereoisomers.
LC-MS (Method 1): Rt = 1 .05 min and 1.06 min; MS (ESIpos): m/z = 258 [M+H]+
1H-NMR (400 MHz, CHLOROFORM-d) 6 [ppm]: 1.557 (0.73), 1.594 (0.54), 1.609 (0.74), 1.614 (0.71), 1.625 (0.75), 1.631 (0.78), 1.637 (0.73), 1.698 (0.52), 1.708 (0.70), 1.716 (0.62), 1.729
(1.08), 1.735 (1.18), 1.740 (1.10), 1.743 (1.03), 1.752 (1.03), 1.764 (0.67), 1.772 (0.48), 1.892
(0.56), 1.905 (0.51), 1.911 (0.57), 1.918 (0.70), 1.924 (0.55), 1.932 (0.76), 1.937 (0.88), 1.946
(0.76), 1.951 (1.12), 1.959 (0.89), 1.965 (0.88), 1.974 (0.59), 1.978 (0.78), 1.991 (0.55), 2.094
(0.43), 2.101 (0.43), 2.106 (0.55), 2.120 (0.61), 2.313 (0.47), 2.328 (0.59), 2.333 (0.68), 2.343
(0.85), 2.348 (0.57), 2.351 (0.46), 2.358 (0.88), 2.363 (1.04), 2.373 (0.59), 2.378 (0.82), 2.382
(0.65), 2.389 (0.45), 2.394 (0.53), 2.398 (0.65), 2.408 (0.44), 2.636 (1.60), 2.744 (16.00), 3.267 (0.51), 3.727 (0.46), 3.734 (0.81), 3.740 (0.48), 3.755 (0.75), 3.761 (0.81), 3.784 (0.42), 3.981
(0.45), 4.001 (0.50), 4.006 (0.62), 4.014 (0.56), 4.026 (0.67), 4.034 (0.66), 4.039 (0.66), 4.051
(0.44), 4.059 (0.68), 4.063 (0.52), 4.084 (1.00), 4.114 (0.65), 4.121 (0.42), 4.723 (7.76), 6.276
(0.60), 6.283 (0.63), 6.290 (0.82), 6.296 (0.86), 6.301 (0.68), 6.308 (0.63), 6.314 (0.84), 6.321
(0.79), 7.297 (0.41), 7.305 (0.77), 7.314 (0.76), 7.321 (0.75), 7.327 (0.68), 7.332 (0.41), 7.340
(0.64), 7.343 (0.51), 7.347 (0.42), 7.354 (0.55), 7.361 (2.80), 7.362 (2.45), 7.369 (2.84), 7.375
(5.99), 7.387 (1.44), 7.414 (0.55), 8.035 (4.87).
Method 3:
(2,2-difluorocyclopropyl)[1-(oxan-2-yl)-1 H-1 ,2,4-triazol-5-yl]methanone

The solution of 1-(oxan-2-yl)-1 H-1 ,2,4-triazole (3.14 g, 80 % purity, 16.4 mmol) and DMAP (20.0 mg, 164 pmol) in DCM (10 mL) was cooled to 0°C and the solution of 2,2-difluorocyclopropane- 1 -carbonyl chloride (2.30 g, 16.4 mmol) in DCM (5 mL) was added followed by dropwise addition of triethylamine (6.9 mL, 49 mmol). The reaction mixture was stirred 1h at 0°C and then 16h at room temperature. The resulting mixture was treated with water and extracted 3 times with DCM. The combined organic layers were washed with water and brine, dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude product was purified using silica gel falsh chromatography (gradient 0 to 30% ethyl acetate in hexane) to yield 660 mg (11% yield) of the title compound as a mixture of stereoisomers.
LC-MS (Method 1): Rt = 1 .05 min and 1.06 min; MS (ESIpos): m/z = 258 [M+H]+
1H-NMR (400 MHz, CHLOROFORM-d) 6 [ppm]: 0.870 (1.02), 0.889 (2.36), 0.898 (0.71), 0.903
(0.96), 0.908 (1.15), 0.913 (0.70), 0.917 (0.78), 0.921 (1.00), 0.936 (0.98), 0.953 (0.48), 0.960
(0.43), 1.149 (1.69), 1.173 (1.55), 1.183 (0.57), 1.215 (1.04), 1.223 (4.23), 1.236 (0.61), 1.240
(0.68), 1.259 (0.99), 1.268 (0.95), 1.285 (0.46), 1.291 (0.47), 1.347 (0.47), 1.395 (0.91), 1.405
(4.47), 1.518 (4.81), 1.524 (6.66), 1.527 (7.05), 1.533 (8.43), 1.547 (14.42), 1.564 (15.27), 1.570 (16.00), 1.577 (10.79), 1.581 (12.61), 1.588 (11.20), 1.593 (9.78), 1.598 (8.75), 1.601 (8.08), 1.605 (8.36), 1.612 (7.79), 1.618 (7.01), 1.625 (9.38), 1.630 (7.16), 1.634 (7.90), 1.637 (7.36),
1.653 (4.51), 1.669 (2.91), 1.679 (1.68), 1.684 (1.88), 1.699 (2.80), 1.703 (3.32), 1.708 (5.18),
1.711 (4.98), 1.715 (5.11), 1.723 (5.89), 1.732 (8.55), 1.740 (8.04), 1.742 (7.89), 1.751 (7.05),
1.755 (7.45), 1.763 (8.41), 1.772 (6.76), 1.779 (2.42), 1.787 (3.17), 1.796 (4.72), 1.803 (2.66),
1.816 (1.86), 1.836 (2.63), 1.843 (3.35), 1.848 (3.68), 1.859 (5.21), 1.863 (4.53), 1.869 (4.49),
1.880 (4.20), 1.889 (4.04), 1.892 (4.22), 1.906 (3.03), 1.911 (2.69), 1.919 (2.85), 1.924 (2.04),
1.932 (2.76), 1.938 (3.04), 1.946 (2.66), 1.951 (3.52), 1.959 (2.70), 1.965 (2.78), 1.974 (1.80),
1.978 (2.41), 1.991 (1.65), 2.055 (0.97), 2.064 (0.67), 2.083 (1.25), 2.093 (1.88), 2.109 (2.28),
2.119 (2.23), 2.142 (0.86), 2.259 (0.68), 2.270 (0.73), 2.295 (1.04), 2.299 (1.29), 2.303 (1.04),
2.312 (1.53), 2.328 (2.12), 2.332 (2.35), 2.343 (2.80), 2.347 (1.96), 2.351 (1.48), 2.358 (2.63),
2.363 (3.25), 2.367 (1.64), 2.373 (1.62), 2.378 (2.50), 2.382 (1.96), 2.389 (1.12), 2.394 (1.55),
2.397 (1.99), 2.408 (1.27), 2.413 (0.93), 2.428 (0.83), 2.463 (0.49), 2.473 (1.94), 2.477 (1.88),
2.492 (2.84), 2.496 (2.66), 2.510 (1.43), 2.513 (1.60), 3.384 (0.82), 3.392 (0.51), 3.399 (1.61),
3.408 (1.24), 3.414 (0.97), 3.423 (1.84), 3.438 (0.97), 3.501 (3.11), 3.506 (2.16), 3.510 (3.67),
3.513 (4.51), 3.518 (2.73), 3.528 (5.44), 3.533 (3.10), 3.541 (5.66), 3.547 (3.74), 3.556 (4.59),
3.561 (1.93), 3.572 (2.85), 3.586 (2.94), 3.590 (1.79), 3.599 (1.72), 3.705 (1.15), 3.711 (0.93),
3.726 (1.52), 3.733 (2.89), 3.740 (1.81), 3.746 (1.42), 3.755 (2.39), 3.762 (4.28), 3.778 (1.52),
3.783 (1.62), 3.786 (2.08), 3.790 (1.09), 3.802 (0.83), 3.836 (0.77), 3.846 (0.76), 3.863 (4.25),
3.872 (3.36), 3.879 (3.45), 3.889 (5.55), 3.898 (2.68), 3.909 (2.84), 3.917 (2.76), 3.981 (1.39),
4.001 (1.75), 4.005 (2.08), 4.014 (3.36), 4.026 (3.71), 4.030 (3.64), 4.033 (3.41), 4.043 (3.52),
4.051 (2.38), 4.059 (3.31), 4.062 (2.92), 4.069 (2.32), 4.083 (3.04), 4.098 (1.09), 4.114 (1.96),
4.121 (1.46), 4.570 (1.05), 4.582 (1.82), 4.588 (1.23), 4.820 (4.45), 4.828 (4.22), 4.833 (5.23),
4.840 (4.11), 4.910 (0.47), 4.951 (7.13), 4.958 (6.41), 4.964 (8.68), 4.970 (5.70), 6.276 (2.08),
6.283 (2.18), 6.290 (2.35), 6.296 (2.47), 6.301 (2.28), 6.307 (2.10), 6.314 (2.41), 6.321 (2.21),
7.102 (0.44), 7.133 (0.40), 7.529 (0.42), 8.036 (15.59), 8.286 (0.41), 9.785 (1.97), 9.789 (3.66), 9.793 (2.01).
Intermediate 20
5-(2,2-difluorocyclopropane-1-carbonyl)-1 H-1 ,2,4-triazol-1-ium chloride

The solution of (2,2-difluorocyclopropyl)[1-(oxan-2-yl)-1 H-1 ,2,4-triazol-5-yl]methanone (3.52 g, 13.7 mmol) in DCM (75 mL) was cooled to 0°C and 4M solution of HCI in dioxane (17 mL, 68 mmol) was added. The reaction mixture was stirred for 0,5h, cooled with an icebath and the precipitate was collected by filtration. The resulting white solid was washed with DCM and hexene and dried under reduced pressure to yield 2.16 g (75 % yield) of the title racemic compound.
1H NMR (400 MHz, DMSO-cfe) 6 ppm 2.07 - 2.20 (m, 1 H), 2.21 - 2.32 (m, 1 H), 3.85 - 4.03 (m, 2 H), 8.77 (s, 1 H).
Intermediate 21
N-(benzyloxy)-2,2-difluoro-N-methylcyclopropane-1 -carboxamide

To the mixture of N-(benzyloxy)methanamine (400 mg, 2.92 mmol), 2,2- difluorocyclopropanecarboxylic acid (356 mg, 2.92 mmol) and N,N-diisopropylethylamine (2.5 mL, 15 mmol) in ethyl acetate (5.7 mL) was added 50 wt% solution of 1-propanephosphonic anhydride in ethyl acetate (4.3 mL, 7.3 mmol). The reaction mixture was stirred for 1h at r.t. , then
poured into brine/water and extracted 2 times with EtOAc. The combined organic layers were dried with Na2SO4, filtered and concentrated under reduced pressure to give 674 mg (90 % purity, 86 % yield) of the title compound which was used further without additional purification.
LC-MS (Method 1): Rt = 1.10 min; MS (ESIpos): m/z = 242 [M+H]+
1H-NMR (400 MHz, CHLOROFORM-d) 6 [ppm]: 1.490 (0.42), 1.518 (0.45), 1.981 (0.49), 1.995 (0.53), 2.012 (0.52), 2.027 (0.46), 3.122 (16.00), 4.710 (1.10), 4.735 (2.33), 4.777 (3.11), 4.803 (1.49), 7.124 (1.60).
Intermediate 22
2, 2-difluorocyclopropane-1 -carbonyl chloride

2, 2-Difluorocyclopropane-1 -carboxylic acid (5.00 g, 41.0 mmol) was dissolved in DCM (40 mL), DMF (0.16 mL, 2.05 mmol) and oxalyl chloride (3.93 mL, 45 mmol) was added dropwise at room temperature (gas evolution). The mixture was stirred for 2 h at room temperature and DCM was removed under reduced pressure (room temperature, 400 mbar). The residue was distilled using bulb tube distillation (50°C, 9 mbar) to give 1.79 g (31% yield) of the title compound as a colourless liquid.
1H NMR (400 MHz, CHLOROFORM-d) 6 ppm 1.94 - 2.03 (m, 1 H), 2.24 - 2.33 (m, 1 H), 2.98 - 3.07 (m, 1 H).
The following sequence of steps describes a method to get access to the chiral analogue of intermediate 12 which can be used to synthesize intermediate 15 if the (R)-enantiomer of 2,2- difluorocyclopropane-1 -carboxylic acid is used.
Intermediate 23
Method 1 :
(1 R)-N-(benzyloxy)-2,2-difluoro-N-methylcyclopropane-1 -carboxamide

To the stirred solution of (1 R)-2,2-difluorocyclopropane-1 -carboxylic acid (4.66, 38.17 mmol) in DCM (93 mL) was added 1 ,1'-carbonyldiimidazole (6.19 g, 38.17 mmol) and the resulting mixture was stirred for 15 minutes at room temperature and then cooled down to 0°C to give reaction
mixture A. To the solution of N-(phenylmethoxy)methanamine hydrochloride (CAS 71925-14-9, 6.026 g, 34.70 mmol) in DCM (47 mL) was added N,N-diisopropylethylamine (6.347 mL, 36.44 mmol) and the resulting mixture was stirred for 10 minutes at room temperature and then cooled down to 0°C to give reaction mixture B. The reaction mixtures A and B were combined at 0°C and the resulting mixture was stirred for 1 h. The reaction mixture was quenched with saturated aqueous ammonium chloride solution and extracted trice with DCM. The combined organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure to yield 8.21 g (88 % yield, 93.5% ee) of the title compound.
Analytical chiral SFC:
Instrument: Waters Acquity UPC2 QDA; column: Chiralpak IG 3 pm 100x4.6 mm; mobile phase A: carbon dioxide; mobile phase B: methanol + 0.2 vol % aqueous ammonia (32%); isocratic: 5% B; flow: 4 mL/min; temperature: 40.0°C; backpressure: 1800 psi; wavelength: UV 210nm.
Retention time: 0.74 min (first eluting, desired stereoisomer
Peak area (210 nm): 95.5% (93.5% ee)
LC-MS (Method 1): Rt = 1.09; MS (ESIpos): m/z = 242.1 [M+H]+
1H NMR (400 MHz, CHLOROFORM-d) 6 ppm 1.41 - 1.55 (m, 1 H), 1.99 (m, 1 H), 2.72 - 2.87 (m, 1 H), 3.11 (s, 3 H), 4.71 (d, 1 H), 4.78 (d, 1 H), 7.24 - 7.29 (m, 5 H).
Intermediate 24
[(R)-2,2-difluorocyclopropyl][1-(oxan-2-yl)-1 H-1 ,2,4-triazol-5-yl]methanone

1-(Oxan-2-yl)-1 H-1 ,2,4-triazole (5.865 g, 80 % purity, 30.63 mmol) was dissolved in THF (70 mL), the solution was cooled down to -75°C and 2.5 M n-BuLi solution in hexane (13.48 mL, 33.69 mmol) was added dropwise between -70°C and - 75°C under stirring. After 1 h N- (benzyloxy)-2,2-difluoro-N-methylcyclopropane-1 -carboxamide (8.21 g, 90 % purity, 30.63 mmol) in THF (23.5 mL) was added dropwise at -75°C. The reaction mixture was stirred for 30 min at -75°C, thereafter the cold reaction mixture was poured into an ice-cold stirred mixture consisting of saturated aqueous ammonium chloride solution (490 mL) and 5 wt% aqueous citric acid solution (328 mL). The the resuting mixture was checked to have pH = 3-4 and was extracted trice with ethyl acetate. The combined organic layers were washed with brine, dried
over anhydrous Na2SO4and concentrated under reduced pressure. The resulting crude product was purified by silica gel flash chromatography (gradient 0 to 15% EtOAc in hexane) to give 6.04 g (70 % purity, 54 % yield) of the title compound containing some residual amount of O-benzyl- N-methylhydroxylamine. The product was used in the next step without further purification.
LC-MS (Method 1): Rt = 1 .04 min and 1.06 min; MS (ESIpos): m/z = 258 [M+H]+
1H NMR (400 MHz, CDCI3) 6 [ppm]: 1.58 - 1.69 (m, 1 H), 1.69 - 1.80 (m, 2 H), 1.88 - 2.02 (m, 2 H), 2.05 - 2.19 (m, 1 H), 2.23 - 2.43 (m, 2 H), 3.67 - 3.81 (m, 1 H), 3.96 - 4.14 (m, 2 H), 6.22 - 6.35 (m, 1 H), 8.03 (s, 1 H).
Intermediate 25
[(R)-2,2-difluorocyclopropyl](1 H-1 ,2,4-triazol-5-yl)methanone hydrochloride

[(R)-2,2-difluorocyclopropyl][1-(oxan-2-yl)-1 H-1 ,2,4-triazol-5-yl]methanone (6.00 g, 16.327 mmol) was dissolved in DCM (84 mL) and the solution was cooled to 0°C. To the reaction mixture was added 4M HCI solution in dioxane (20.4 mL, 81.64 mmol HCI) at 0°C and the resulting mixture was stirred for 1 h. The precipitate formed was collected by filtration, washed with DCM and hexane and dried under reduced pressure to give 2.58 g (74.6% yield) of the title compound.
1H NMR (400 MHz, CDCh) 6 [ppm]: 2.09 - 2.19 (m, 1 H), 2.20 - 2.31 (m, 1 H), 3.82 - 4.05 (m, 1 H), 8.78 (s, 1 H).
Analytical chiral SFC:
Instrument: Waters Acquity UPC2 QDA; Column: Chiral Art Amylose-SA 3p 100x4.6mm; eluent A: CO2; eluent B: methanol + 0.2 vol % aqueous ammonia (32%); isocratic: 5% B; gradient: no; flow: 4 mL/min; temperature: 40°C; BPR: 100 bar; UV: 220 nm
Retention time: 1.01 min (second eluting)
Peak area (220 nm): 95.8% (92% ee).
Intermediate 26
[(1 R)-2,2-difluorocyclopropyl](1 H-1 ,2,4-triazol-5-yl)(2H)methanol
 [(1 R)-2,2-Difluorocyclopropyl](1 H-1 ,2,4-triazol-5-yl)methanone (300 mg, 1.73 mmol) was dissolved in (2H4)-methanol (5.0 mL) and cooled with an ice bath. Sodium (2H4)tetrahydroborate (54.4 mg, 1 .30 mmol) was added and the ice bath was removed after a few minutes and stirred at rt over night. The reaction mixture was concentrated, water and ethyl acetate were addd and the pH was adjusted to 7 with 1 M citric acid and saturated aqueous sodium hydrogen carbonate solution. The layers were separated and the aqueous phase was extracted twice with ethyl acetate. The combined organic phases were dried with a hydrophobic filter and concentrated. The residue was treated with ethyl acetate / toluene and concentrated again yielding 239 mg (78%) of the title compound.
[(1 R)-2,2-Difluorocyclopropyl](1 H-1 ,2,4-triazol-5-yl)methanone (300 mg, 1.73 mmol) was dissolved in (2H4)-methanol (5.0 mL) and cooled with an ice bath. Sodium (2H4)tetrahydroborate (54.4 mg, 1 .30 mmol) was added and the ice bath was removed after a few minutes and stirred at rt over night. The reaction mixture was concentrated, water and ethyl acetate were addd and the pH was adjusted to 7 with 1 M citric acid and saturated aqueous sodium hydrogen carbonate solution. The layers were separated and the aqueous phase was extracted twice with ethyl acetate. The combined organic phases were dried with a hydrophobic filter and concentrated. The residue was treated with ethyl acetate / toluene and concentrated again yielding 239 mg (78%) of the title compound.
LC-MS (Method 3): Rt = 0.39 min; MS (ESIpos): m/z = 175 [M-H]~
1H NMR (400 MHz, DMSO-cfe, 22°C) 6 ppm 1.33 - 1.76 (m, 2 H), 2.08 - 2.26 (m, 1 H), 5.2, 5.74, 6.16 and 6.24 (4 s, 1 H), 7.87 and 8.49 (2 s, 1 H), 13.86 and 13.93 (2 s, 1 H).
Intermediate 27
5-[chloro[(1S)-2,2-difluorocyclopropyl](2H)methyl]-1 H-1 ,2,4-triazole hydrogen chloride (1/1)

[(1 R)-2,2-Difluorocyclopropyl](1 H-1 ,2,4-triazol-5-yl)(2H)methanol (230 mg, 1.31 mmol) was dissolved in dioxane (3.0 mL) followed by dropwise addition of thionyl dichloride (290 pL, 3.9 mmol). The reaction mixture was stirred for 45 minutes at rt and and concentrated. The residue was dissolved twice in dioxane and concentrated agisn to obtain 410 mg of the title compound.
LC-MS (Method 3): Rt = 0.63 and 0.66 min; MS (ESIpos): m/z = 195 [M+H]+
1H NMR (400 MHz, DMSO-cfe, 22°C) 6 ppm 1.54 - 1.64, 1.71 - 1.84 (m, 1 H) and 1.87 - 1.98 (3 m, 2 H), 2.64 - 2.77 (m, 1 H), 8.56 (s, 1 H).
Target Compound
Route 1 to the target compound:
Mixture 1 of Target Compound and Sideproducts 1 , 2, and 3
5-{(3R)-1-[(2,2-difluorocyclopropyl)(1 H-1 ,2,4-triazol-5-yl)methyl]-5',6'-dihydrospiro[pyrrolidine- 3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine (diastereomer 1 , diastereomer 2, diasteromer 3, and diastereomer 4)

A solution of 5-[(3R)-5’,6’-dihydrospiro[pyrrolidine-3,4’-pyrrolo[1,2-b]pyrazol]-2’-yl]-3- (trifluoromethyl)pyridin-2-amine hydrogen chloride (1/1) (420 mg, 1.17 mmol), [2,2- difluorocyclopropyl](1H-1 ,2,4-triazol-5-yl)methanone (220 mg, 1.27 mmol), titanium(IV) isopropoxide (940 pL, 3.2 mmol) and DIPEA (920 pL, 5.3 mmol) in methanol (11 mL) was stirred at 60°C for 4 h. Sodium cyanoborohydride (166 mg, 2.65 mmol) was added and the reaction mixture was stirred overnight at 60°C. Water was added and after stirring for 10 min the mixture was filtered and the filter cake was washed with methanol. The volatiles of the filtrate were removed under reduced pressure and the remaining aqueous phase was extracted twice with ethyl acetate. The combined ethyl acetate layers were washed with saturated sodium chloride solution, dried and concentrated. The residue was purified by HPLC (pump: Labomatic HD-5000, head HDK 280, lowpressure gradient module ND-B1000; manual injection valve: Rheodyne 3725i038; detector: Knauer Azura UVD 2.15; collector: Labomatic Labocol Vario-4000; column: Chromatorex RP C-18 10 pm, 125x30mm; eluent; gradient; UV- Detection. eluent A: water + 0.2 vol % aqueous ammonia (32%); eluent B: acetonitril;gradient: 0.00-0.50 min 10% B (150 mL/min), 0.50-6.00 min 10-50% B (150 mL/min), 6.00-6.10 min 50- 100% B (150 mL/min), 6.10-8.00 min 100% B (150 mL/min), temperature: rt; DAD scan: 210- 400 nm) to afford 99 mg (21 % yield) of the title compound.
LC-MS (Method 2): Rt = 0.70 min; MS (ESIpos): m/z = 481 [M+H]+
1H-NMR (400 MHz, DMSO-d6) 6 [ppm]: 1.017 (0.44), 1.231 (1.03), 1.544 (0.96), 1.762 (0.55),
1.949 (2.02), 2.005 (2.14), 2.075 (1.57), 2.269 (0.93), 2.322 (2.27), 2.326 (2.86), 2.332 (2.28),
2.358 (0.81), 2.518 (10.42), 2.522 (7.43), 2.644 (1.50), 2.664 (3.29), 2.668 (4.04), 2.673 (2.72), 2.705 (5.56), 2.729 (2.93), 2.741 (3.12), 2.763 (2.30), 2.777 (2.29), 2.796 (1.81), 2.812 (1.08),
2.877 (3.01), 2.900 (2.76), 3.430 (0.64), 3.572 (0.79), 4.090 (5.78), 4.107 (5.26), 4.125 (1.89),
6.317 (0.95), 6.356 (1.52), 6.411 (1.00), 6.524 (13.29), 7.923 (0.61), 7.998 (10.32), 8.135 (16.00), 8.527 (1.42), 8.573 (9.07), 13.927 (1.10).
Side product 1
5-{(3R)-1-[(S)-[(1 R)-2,2-difluorocyclopropyl](1 H-1,2,4-triazol-5-yl)methyl]-5',6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine (diastereomer 1 , undesired)
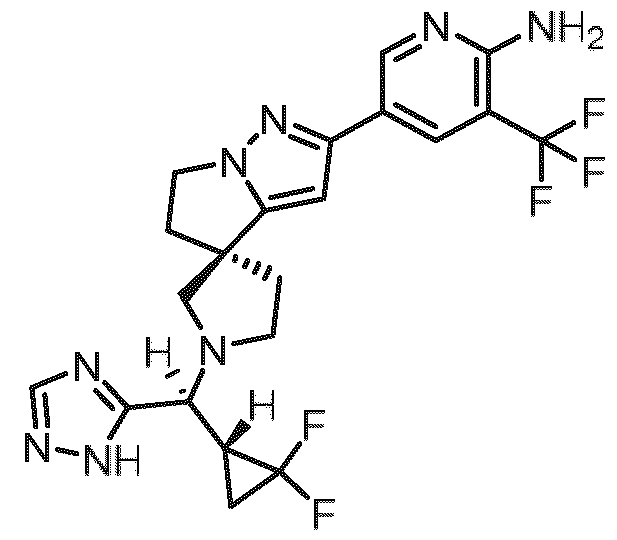
The diastereomeric mixture of 5-{(3R)-1-[(2,2-difluorocyclopropyl)(1 H-1 ,2,4-triazol-5-yl)methyl]- 5',6'-dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine (95.0 mg, 198 pmol) was separated by HPLC method A followed by chiral HPLC method B to yield 12.0 mg (95 % purity, 12 % yield) of the title compound.
Preparative method A:
Instrument: Waters Autopurificationsystem; Column: Triart C18 5p, 100x30mm; eluent A: water + 0.2 vol % aqueous ammonia (32%); eluent B: methanol; gradient: 0.0-0.5 min 41% B (35-70 mL/min), 0.5-5.5 min 41-61% B; flow: 70 mL/min; temperature: 25°C; DAD scan: 210-400 nm.
Retention time: 5.2 - 5.8 min
Analytical method A:
Analytic method: Instrument: Waters Acquity UPLCMS SingleQuad; column: Triart C18 1.7p, 50x2.1 mm; eluent A: water + 0.2 vol % aqueous ammonia (32%); eluent B: methanol; gradient: 0-1.6 min 1-99% B, 1.6-2.0 min 99% B; flow: 0.7 mL/min; temperature: 60°C; DAD scan: 210- 400 nm.
Retention time: 1.27 min
Preparative method B:
Instrument: Sepiatec: Prep SFC100; column: Chiralpak IG 5p 250x30mm; eluent A: CO2; eluent B: methanol + 0.2 vol % aqueous ammonia (32%); isocratic: 25% B; gradient: no; flow: 100 mL/min; temperature: 40°C; BPR: 150 bar; UV: 280 nm.
Retention time: 3.8 - 4.5 min
Analytical method B:
Instrument: Agilent: 1260, Aurora SFC-Modul; column: Chiralpak IG 3p 100x4.6mm; eluent A: CO2; eluent B: methanol + 0.2 vol % aqueous ammonia (32%); isocratic: 25% B; gradient: no; flow: 4 mL/min; temperature: 40.0°C; BPR: 100 bar; UV: 280 nm.
Retention time: 1.23 min
[Q]20D : +73.2° (c = 1.00, methanol)
1H NMR (400 MHz, DMSO-d6, 22°C) 6 ppm 1.03 - 1.15 (m, 1 H), 1.51 - 1.64 (m, 1 H), 1.91 - 2.07 (m, 2 H), 2.20 - 2.32 (m, 1 H), 2.53 - 2.61 (m, 1 H), 2.63 - 2.81 (m, 3 H), 2.88 (d, J=9.12 Hz, 1 H), 3.46 (br d, J=9.89 Hz, 1 H), 4.11 (t, J=6.97 Hz, 2 H), 6.42 (s, 1 H), 6.52 (s, 2 H), 8.00 (d, J=2.03 Hz, 1 H), 8.27 (br s, 1 H), 8.58 (d, J=1 .77 Hz, 1 H), 13.94 (br s, 1 H).
Side product 2
5-{(3R)-1-[(1S)-2,2-difluorocyclopropyl(1 H-1 ,2,4-triazol-5-yl)methyl]-5',6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine
(diastereomer 2, undesired)

The diastereomeric mixture of 5-{(3R)-1-[(2,2-difluorocyclopropyl)(1 H-1 ,2,4-triazol-5-yl)methyl]- 5',6'-dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine (95.0 mg, 198 pmol) was separated by HPLC method A followed by chiral HPLC method B to yield 12.0 mg (95 % purity, 12 % yield) of the title compound.
Preparative method A:
Instrument: Waters Autopurificationsystem; Column: Triart C18 5p, 100x30mm; eluent A: water + 0.2 vol % aqueous ammonia (32%); eluent B: methanol; gradient: 0.0-0.5 min 41% B (35-70 mL/min), 0.5-5.5 min 41-61 % B; flow: 70 mL/min; temperature: 25°C; DAD scan: 210-400 nm.
Retention time: 5.2 - 5.8 min
Analytical method A:
Instrument: Waters Acquity UPLCMS SingleQuad; column: Triart C18 1.7p, 50x2.1mm; eluent A: water + 0.2 vol % aqueous ammonia (32%); eluent B: methanol; gradient: 0-1.6 min 1-99% B, 1.6-2.0 min 99% B; flow: 0.7 mL/min; temperature: 60°C; DAD scan: 210-400 nm.
Retention time: 1.27 min
Preparative method B:
Instrument: Sepiatec: Prep SFC100; column: Chiralpak IG 5p 250x30mm; eluent A: CO2; eluent B: methanol + 0.2 vol % aqueous ammonia (32%); isocratic: 25% B; gradient: no; flow: 100 mL/min; temperature: 40°C; BPR: 150 bar; UV: 280 nm.
Retention time: 4.9 - 6.0 min
Analytical method B:
Instrument: Agilent: 1260, Aurora SFC-Modul; column: Chiralpak IG 3p 100x4.6mm; eluent A: CO2; eluent B: methanol + 0.2 vol % aqueous ammonia (32%); isocratic: 25% B; gradient: no; flow: 4 mL/min; temperature: 40.0°C; BPR: 100 bar; UV: 280 nm.
Retention time: 1.79 min
[CI]20D : +44.7° (c = 1.00, methanol)
1H NMR (400 MHz, DMSO-cfe, 22°C) 6 ppm 1.01 - 1.15 (m, 1 H), 1.51 - 1.62 (m, 1 H), 1.95 - 2.06 (m, 2 H), 2.22 - 2.34 (m, 1 H), 2.65 - 2.74 (m, 3 H), 2.86 - 2.96 (m, 1 H), 3.49 (br d, J=9.89 Hz, 1 H), 4.03 - 4.14 (m, 2 H), 6.40 (s, 1 H), 6.52 (s, 2 H), 8.00 (d, =2.03 Hz, 1 H), 8.37 (br s, 1 H), 8.57 (d, J=1.77 Hz, 1 H), 13.94 (br s, 1 H).
Target Compound
5-{(3R)-1-[(R)-[(1 R)-2,2-difluorocyclopropyl](1 H-1 ,2,4-triazol-5-yl) methyl]-5' ,6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine (diastereomer 3, desired)

The diastereomeric mixture of 5-{(3R)-1-[(2,2-difluorocyclopropyl)(1 H-1 ,2,4-triazol-5-yl)methyl]- 5',6'-dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine (95.0 mg, 198 pmol) was separated by HPLC method A followed by chiral HPLC method C to yield 12.0 mg (95 % purity, 12 % yield) of the title compound.
Preparative method A:
Instrument: Waters Autopurificationsystem; Column: Triart C18 5p, 100x30mm; eluent A: water + 0.2 vol % aqueous ammonia (32%); eluent B: methanol; gradient: 0.0-0.5 min 41% B (35-70 mL/min), 0.5-5.5 min 41-61 % B; flow: 70 mL/min; temperature: 25°C; DAD scan: 210-400 nm.
Retention time: 4.2 - 4.7 min
Analytical method A:
Instrument: Waters Acquity UPLCMS SingleQuad; Column: Acquity UPLC BEH C18 1.7pmm, 50x2.1mm; eluent A: water + 0.2 vol % aqueous ammonia (32%); eluent B: acetonitril; gradient:
0-1.6 min 1-99% B, 1.6-2.0 min 99% B; flow: 0.8 mL/min; temperature: 60°C; DAD scan: 210- 400 nm.
Retention time: 1.19 min
Preparative method C:
Instrument: PrepCon Labomatic HPLC-3; Column: Chiralpak IG 5p, 250x30; eluent A: hexane + 0.1 vol % diethylamine; eluent B: ethanol; isocratic: 70% A + 30% B; flow: 70 mL/min; temperature: 25°C; UV: 280 nm.
Retention time: 8.04 - 9.75 min
Analytical method C:
Instrument: Thermo Fisher UltiMate 3000; Column: Chiralpak IG 3p, 100x4.6; eluent A: hexane + 0.1 vol % diethylamine; eluent B: ethanol; isocratic: 70% A + 30% B; flow: 1.4 mL/min; temperature: 25°C; UV: 280 nm.
Retention time: 4.31 min
[Q]20D : +50.7° (c = 1.00, methanol)
1H NMR (400 MHz, DMSO-d6, 22°C) 6 ppm 1.51 - 1.63 (m, 1 H), 1.74 - 1.84 (m, 1 H), 1.95 (t, J=6.97 Hz, 2 H), 2.34 - 2.47 (m, 2 H), 2.69 - 2.80 (m, 3 H), 2.86 - 2.94 (m, 1 H), 3.60 (br d, J=10.14 Hz, 1 H), 4.03 - 4.13 (m, 2 H), 6.32 (s, 1 H), 6.52 (s, 2 H), 7.99 (d, J=2.03 Hz, 1 H), 8.32 (br s, 1 H), 8.57 (d, J=1.77 Hz, 1 H)
Side product 3
5-{(3R)-1-[(1S)-2,2-difluorocyclopropyl](1 H-1 ,2,4-triazol-5-yl)methyl]-5',6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine (diastereomer 4, undesired)

The diastereomeric mixture of 5-{(3R)-1-[(2,2-difluorocyclopropyl)(1 H-1 ,2,4-triazol-5-yl)methyl]- 5',6'-dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine (95.0 mg, 198 pmol) was separated by HPLC method A, chiral HPLC method C and method D to yield 5.5 mg (95 % purity, 6 % yield) of the title compound.
Preparative method A:
Instrument: Waters Autopurificationsystem; Column: Triart C18 5p, 100x30mm; eluent A: water + 0.2 vol % aqueous ammonia (32%); eluent B: methanol; gradient: 0.0-0.5 min 41% B (35-70 mL/min), 0.5-5.5 min 41-61% B; flow: 70 mL/min; temperature: 25°C; DAD scan: 210-400 nm.
Retention time: 4.2 - 4.7 min
Analytical method A:
Instrument: Waters Acquity UPLCMS SingleQuad; Column: Acquity UPLC BEH C18 1.7pm, 50x2.1mm; eluent A: water + 0.2 vol % aqueous ammonia (32%); eluent B: acetonitril; gradient: 0-1.6 min 1-99% B, 1.6-2.0 min 99% B; flow: 0.8 mL/min; temperature: 60°C; DAD scan: 210- 400 nm.
Retention time: 1.19 min
Preparative method C:
Instrument: PrepCon Labomatic HPLC-3; Column: Chiralpak IG 5p, 250x30; eluent A: hexane + 0.1 vol % diethylamine; eluent B: ethanol; isocratic: 70% A + 30% B; flow: 70 mL/min; temperature: 25°C; UV: 280 nm.
Retention time: 9.75 - 12.70 min
Analytical method C:
Instrument: Thermo Fisher UltiMate 3000; Column: Chiralpak IG 3p, 100x4.6; eluent A: hexane + 0.1 vol % diethylamine; eluent B: ethanol; isocratic: 70% A + 30% B; flow: 1.4 mL/min; temperature: 25°C; UV: 280 nm.
Retention time: 5.57 min
Preparative method D:
Instrument: Waters Autopurificationsystem; Column: XBrigde C18 5p, 100x30mm; eluent A: water + 0.2 vol % aqueous ammonia (32%); eluent B: acetonitril; gradient: 0.0-0.5 min 15% B (35-70 mL/min), 0.5-5.5 min 15-35% B; flow: 70 mL/min; temperature: 25°C; DAD scan: 210- 400 nm.
Retention time: 3.1 - 3.9
Analytical method D:
Instrument: Waters Acquity UPLCMS SingleQuad; Column: Acquity UPLC BEH C18 1.7p, 50x2.1mm; eluent A: water + 0.2 vol % aqueous ammonia (32%); eluent B: acetonitril; gradient: 0-1.6 min 1-99% B, 1.6-2.0 min 99% B; flow: 0.8 mL/min; temperature: 60°C; DAD scan: 210- 400 nm.
Retention time: 0.84 min
[Q]20D : +38.2° (c = 1.00, methanol)
1H NMR (400 MHz, DMSO-d6, 22°C) 6 ppm 1.46 - 1.85 (m, 2 H), 1.85 - 2.04 (m, 2 H), 2.34 - 2.45 (m, 2 H), 2.68 - 2.84 (m, 3 H), 2.89 (d, J=9.12 Hz, 1 H), 3.53 - 3.66 (m, 1 H), 4.09 (br t, J=6.59 Hz, 2 H), 6.33 - 6.39 (m, 1 H), 6.52 (s, 2 H), 7.94 and 8.53 (2 br s, 1 H), 8.00 (d, J=2.03 Hz, 1 H), 8.57 (d, J=1.52 Hz, 1 H), 13.93 and 14.03 (2 br s, 1 H).
Route 2 to the target compound:
Mixture 2 of Target Compound and Side product 1
Procedure 1
5-{(3R)-1-[(1 R)-2,2-difluorocyclopropyl(1H-1 ,2,4-triazol-5-yl)methyl]-5',6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine (diastereomer 1 and diastereomer 3)

5-[(3R)-5',6'-Dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl]-3-(trifluoromethyl)pyridin- 2-amine hydrogen chloride (1/1) (200 mg, 528 pmol) was dissolved in DMF (3.9 mL). N,N- Diisopropylethylamine (550 pL, 3.2 mmol) and then 5-{chloro[(1R)-2,2- difluorocyclopropyl]methyl}-1 H-1,2,4-triazole hydrogen chloride (1/1) (diastereomer 1 and diastereomer 2) (285 mg, 1.24 mmol) dissolved in DMF (3.9 mL) were added successively. The reaction mixture was stirred overnight at rt and concentrated. The residue was partioned between water and ethyl acetate. The layers were separated and the aqueous phase was extracted with ethyl acetate. The combined organic phases were washed with brine, dried and concentrated obtaining 263 mg of the title compound.
Target Compound
5-{(3R)-1-[(R)-[(1 R)-2,2-difluorocyclopropyl](1 H-1,2,4-triazol-5-yl)methyl]-5',6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine (diastereomer 3, desired)
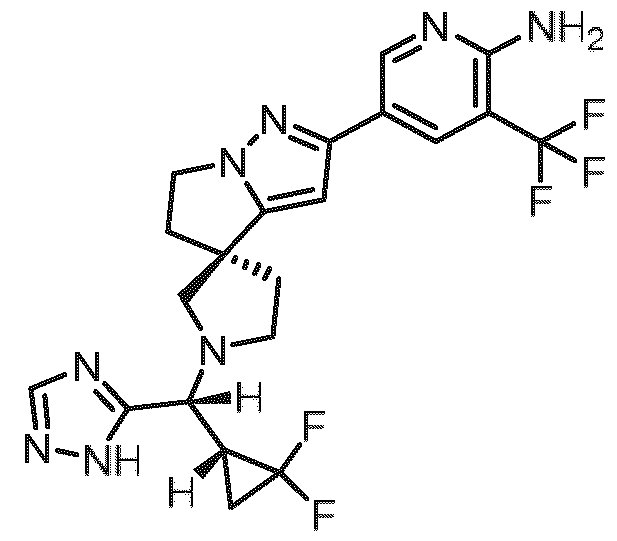
The diastereomeric mixture of 5-{(3R)-1-[(2,2-difluorocyclopropyl)(1 H-1 ,2,4-triazol-5-yl)methyl]- 5',6'-dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine (diastereomer 1 and diastereomer 3) was separated by HPLC to yield 41.0 mg (95 % purity, 15 % yield) of the title compound as the first-eluting diastereomer, the target compound.
Preparative method E:
Instrument: Waters Autopurificationsystem; Column: XBrigde C18 5p, 100x30mm; eluent A: water + 0.1 vol % formic acid; eluent B: methanol; gradient: 0.0-0.5 min 10% B (35-70 mL/min), 0.5-10.1 min 20-40% B; flow: 70 mL/min; temperature: rt; DAD scan: 210-400 nm.
Retention time: 5.1 - 6.0 min
Analytical method E:
Instrument: Waters Acquity UPLCMS SingleQuad; Column: Acquity UPLC BEH C18 1.7pm, 50x2.1 mm; eluent A: water + 0.1 vol % formic acid; eluent B: methanol; gradient: 0-1.6 min 1- 99% B, 1.6-2.0 min 99% B; flow: 0.8 mL/min; temperature: 60°C; DAD scan: 210-400 nm
Retention time: 1.08 min
[CI]20D : +48.8° (c = 1.00, methanol)
1H N MR (400 MHz, DMSO-cfe, 22°C) 6 ppm 1.45 - 1.66 (m, 1 H), 1.69 - 1.86 (m, 1 H), 1.90 - 1.99 (m, 2 H), 2.30 - 2.46 (m, 2 H), 2.69 - 2.82 (m, 3 H), 2.85 - 2.94 (m, 1 H), 3.52 - 3.68 (m, 1 H), 4.03 - 4.13 (m, 2 H), 6.32 (s, 1 H), 6.52 (s, 2 H), 7.99 (d, 1 H), 8.14 and 8.53 (2 s, 1 H), 8.57 (d, 1 H), 13.87 - 14.09 (m, 1 H).
The absolute configuration of the stereocenter at carbon C-4 of the target compound (carbon atom connected to the triazole ring, see below was elucidated by NMR based on the known (R)- configuration of stereocenters at asymmetric carbon C-5 (cyclopropyl ring) and at asymmetric carbon C-1 (spirocyclic ring system). In the target compound as well as in the side product 1 , the preferred axial alignment of the protons attached to C-4 and C-5 led to an identical trans relative orientation, the bulkier triazole moiety and the rest of difluorocyclopropane adopting the energetically favorable equatorial conformations. This preferred trans arrangement in both studied diastereomers was consistent with the observed 3J-coupling constant of 10Hz measured in both 1H-NMR spectra. The absolute configurations at C-4 were unambiguously determined by
2D-NMR, namely by homonuclear (1H,1H-NOESY) and heteronuclear Overhauser Effect experiments (1H,19F-HOESY), acquired at a broad range of temperatures, between -40°C and +25°C, in deuterated methanol. For the target compound, specific cross peaks were observed in 1H,1H-NOESY between the two diastereotopic protons in the difluorocyclopropane ring and methylene protons in the spiro-system. At the same time no correlation was observed between the geminal fluorines to the methylene protons in the spiro-system in 1H,19F-HOESY experiments at various temperatures. Relative to the known R-configuration at C-5, the absolute configuration at C-4 was henceforth assigned to be R in the target compound. The opposite correlation pattern was observed for side product 1 , i.e. , 1H,19F-HOESY cross peaks were observed between the geminal fluorines and diastereotopic protons of the spirocyclic system, while no 1H,1H-NOESY correlation was apparent from this latter to the methylene diastereotopic protons in difluorocyclopropane. This evidenced the stereoconfiguration at C-4 to be S in the side product 1.
Carbon atom numbering of the target compound as used in the NMR study.

Side 1
5-{(3R)-1-[(S)-[(1 R)-2,2-difluorocyclopropyl](1 H-1 ,2,4-triazol-5-yl)methyl]-5',6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine
(diastereomer 1 , undesired)

The diastereomeric mixture of 5-{(3R)-1-[(2,2-difluorocyclopropyl)(1 H-1 ,2,4-triazol-5-yl)methyl]- 5',6'-dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine (diastereomer 1 and diastereomer 3) was separated by HPLC to yield 41.0 mg (95 % purity, 15 % yield) of the title compound as the second-eluting diastereomer.
Preparative method E:
Instrument: Waters Autopurificationsystem; Column: XBrigde C18 5p, 100x30mm; eluent A: water + 0.1 vol % formic acid; eluent B: methanol; gradient: 0.0-0.5 min 10% B (35-70 mL/min), 0.5-10.1 min 20-40% B; flow: 70 mL/min; temperature: rt; DAD scan: 210-400 nm.
Retention time: 7.3 - 8.4 min
Analytical method E:
Instrument: Waters Acquity UPLCMS SingleQuad; Column: Acquity UPLC BEH C18 1.7p, 50x2.1mm; eluent A: water + 0.1 vol % formic acid; eluent B: methanol; gradient: 0-1.6 min 1- 99% B, 1.6-2.0 min 99% B; flow: 0.8 mL/min; temperature: 60°C; DAD scan: 210-400 nm Retention time: 1.18 min
[Q]20D : +61.2° (c = 1.00, methanol)
1H N MR (400 MHz, DMSO-cfe, 22°C) 6 ppm 0.98 - 1.16 (m, 1 H), 1.50 - 1.64 (m, 1 H), 1.93 - 2.08 (m, 2 H), 2.20 - 2.33 (m, 1 H), 2.54 - 2.62 (m, 1 H), 2.64 - 2.82 (m, 3 H), 2.88 (d, J=8.87 Hz, 1 H), 3.42 - 3.51 (m, 1 H), 4.11 (t, J=6.84 Hz, 2 H), 6.42 (s, 1 H), 6.53 (s, 2 H), 8.00 (d, J=2.03 Hz, 1 H), 8.16 (s, 1 H), 8.58 (d, J=1.52 Hz, 1 H), 13.93 (br s, 1 H).
The crystallographic data of side product 1 (5-{(3R)-1-[(S)-((R)-2,2-difluorocyclopropyl)(1 H- 1,2,4-triazol-5-yl)methyl]-5',6'-dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3- (trifluoromethyl)pyridin-2-amine - diastereomer 1 , undesired) as well as a figure depicting the thermal ellipsoids and numbering of the structure, are shown in Table 4 and Figure 3.
Colorless crystals of side product 1 were obtained by slow evaporation from an ethanol solution. A single crystal was mounted on a cryoloop using a protective oil. Single-crystal X-ray diffraction data were collected at 100 K on a Rigaku Synergy S system with a kappa goniometer and a HyPix-6000HE Hybrid Photon Counting (HPC) detector using Cu X-ray radiation (CuKa, = 1.54178 A). Data were integrated using the program CrysAlisPRO. SHELXS was used for structure solution and SHELXL was used for full-matrix least-squares refinement on F2. In the asymmetric unit one molecule of side product 1 is present. All non-hydrogen atoms were refined anisotropically. All hydrogen atoms were added in calculated positions and refined riding on their resident atoms. The isotropic temperature factors of the hydrogen atoms were refined as 1.2 and 1.5 times the size of the temperature factors of the corresponding heavy atoms, respectively. The absolute stereochemistry could be assigned unambiguously
with a Flack Parameter of -0.05 (8). The stereochemistry at C14 is R, at C19 S and at C25 R (see Figure 3 for naming atoms). The program XP was used for molecular representations.
Table 4. Crystal data and structure refinement for side product 1.
Identification code side product 1
Empirical formula C21 H21 F5 N8
Formula weight 480.46
Temperature 100(2) K
Wavelength 1.54178 A
Crystal system Orthorhombic
Space group P212121
Unit cell dimensions a = 8.9959(3) A a= 90°. b = 13.4320(4) A b= 90°. c = 18.4775(6) A g = 90°
Volume 2232.69(12) A3
Z 4
Density (calculated) 1.429 Mg/m3
Absorption coefficient 1.035 mm’1
F(000) 992
Crystal size 0.1 x 0.05 x 0.05
Theta range for data collection 4.069 to 77.134°.
Reflections collected 44983
Independent reflections 4623 [R(int) = 0.0751]
Completeness to theta = 67.679° 99.8 %
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 4623 / 0 / 307
Goodness-of-fit on F2 1.091
Final R indices [l>2sigma(l)] R1 = 0.0877, wR2 = 0.2235
R indices (all data) R1 = 0.0984, wR2 = 0.2332
Absolute structure parameter -0.05(8)
Extinction coefficient n/a
Largest diff. peak and hole 0.866 and -0.368 e.A’3 cf. Figure 3
Mixture 2 of Target Compound and Sideproduct 1
Procedure 2
5-{(3R)-1-[(1 R)-2,2-difluorocyclopropyl(1H-1 ,2,4-triazol-5-yl)methyl]-5',6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[1,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine
(diastereomer 1 and diastereomer 3)
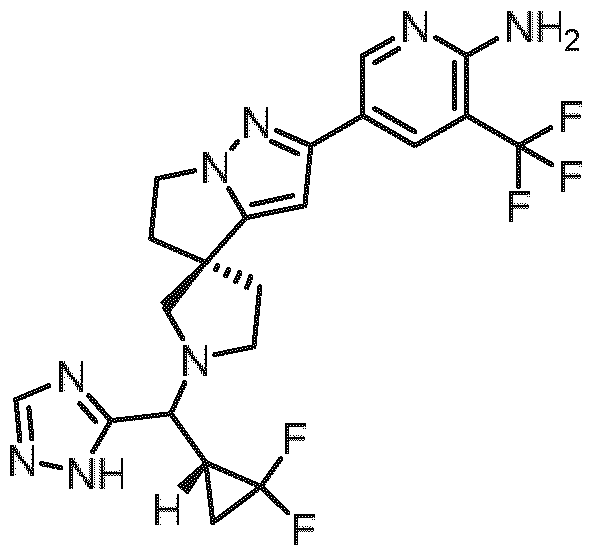
5-[(3R)-5',6'-Dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl]-3-(trifluoromethyl)pyridin- 2-amine hydrogen chloride (1/1) (1.52 g, 4.14 mmol) was dissolved in DMF (30.4 mL). N,N- Diisopropylethylamine (5.05 mL, 29.0 mmol) and then 5-{chloro[(1 R)-2,2- difluorocyclopropyl]methyl}-1 H-1 ,2,4-triazole hydrogen chloride (1/1) (diastereomer 1 and diastereomer 2) (3.20 g, 9.74 mmol, purity 70%) dissolved in DMF (30.4 mL) were added successively. The reaction mixture was stirred overnight at rt and concentrated. The residue was partioned between water and ethyl acetate. The layers were separated and the aqueous phase was extracted with ethyl acetate. The combined organic phases wered washed with brine, dried and concentrated obtaining 2.84 g. The crude material was purified by chromatography (Sfaer Amino, hexane/ethyl acetate 5-100% and ethyl acetae/ethanol 0-100%) to yield 0.83 g of the title compound.
Side product 1
5-{(3R)-1-[(S)-[(1 R)-2,2-difluorocyclopropyl](1 H-1 ,2,4-triazol-5-yl)methyl]-5' ,6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine (diastereomer 1 , undesired)
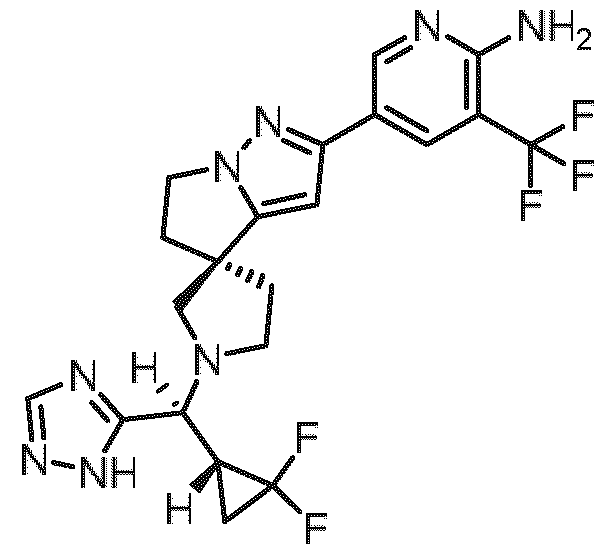
The diastereomeric mixture of 5-{(3R)-1-[(1 R)-2,2-difluorocyclopropyl(1 H-1 ,2,4-triazol-5- yl)methyl]-5',6'-dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3- (trifluoromethyl)pyridin-2-amine (diastereomer 1 and diastereomer 3) was separated by chiral HPLC method to yield 331.0 mg (98 % purity, 16 % yield) of the title compound as the first- eluting diastereomer.
Preparative method F:
Instrument: Sepiatec: Prep SFClOO; Column: Chiralpak IG 5p 250x30mm; eluent A: C02; eluent B: 2-propanol; isocratic: 22%B; gradient: no; flow: 100 mL/min; temperature: 40°C; BPR: 150 bar; UV: 270 nm.
Retention time: 4.3 - 6.4 min
Analytical method F:
Instrument: Waters Acquity UPC2 QDA; Column: Chiralpak IG 3p 100x4.6mm; eluent A: CO2; eluent B: 2-propanol + 0.4 vol % diethylamine; isocratic: 20%B; gradient: no; flow: 4 mL/min; temperature: 40.0°C; BPR:100 bar; UV: 280 nm.
Retention time: 2.38 min
[Q]20D : +70.3° (c = 1.00, methanol)
1H NMR (400 MHz, DMSO-cfe, 22°C) 5 ppm 0.93 - 1.20 (m, 1 H), 1.49 - 1.66 (m, 1 H), 1.91 - 2.07 (m, 2 H), 2.20 - 2.34 (m, 1 H), 2.53 - 2.62 (m, 1 H), 2.63 - 2.81 (m, 3 H), 2.88 (br d, 1 H), 3.38 - 3.56 (m, 1 H), 4.11 (t, 2 H), 6.42 (br s, 1 H), 6.52 (s, 2 H), 7.91 and 8.52 (2 br s, 1 H), 8.00 (d, 1 H), 8.58 (d, 1 H), 13.93 (br s, 1 H).
Target Compound
5-{(3R)-1-[(R)-[(1 R)-2,2-difluorocyclopropyl](1 H-1 ,2,4-triazol-5-yl)methyl]-5',6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine (diastereomer 3, desired)

The diastereomeric mixture of 5-{(3R)-1-[(1 R)-2,2-difluorocyclopropyl(1 H-1 ,2,4-triazol-5- yl)methyl]-5',6'-dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3-
(trifluoromethyl)pyridin-2-amine (diastereomer 1 and diastereomer 3) was separated by chiral
HPLC to yield 331 mg (98 % purity, 16 % yield) of the title compound as the second-eluting diastereomer, the target compound.
Preparative method F:
Instrument: Sepiatec: Prep SFClOO; Column: Chiralpak IG 5p 250x30mm; eluent A: CO2; eluent B: 2-propanol; isocratic: 22%B; gradient: no; flow: 100 mL/min; temperature: 40°C; BPR: 150 bar; UV: 270 nm.
Retention time: 7.9 - 11.9 min
Analytical method F:
Instrument: Waters Acquity UPC2 QDA; Column: Chiralpak IG 3p 100x4.6mm; eluent A: CO2; eluent B: 2-propanol + 0.4 vol % diethylamine; isocratic: 20%B; gradient: no; flow: 4 mL/min; temperature: 40.0°C; BPR:100 bar; UV: 280 nm.
Retention time: 5.05 min
[Q]20D : +51.9° (c = 1.00, methanol)
1H NMR (400 MHz, DMSO-cfe, 22°C) 6 ppm 1.45 - 1.69 (m, 1 H), 1.70 - 2.03 (m, 3 H), 2.29 - 2.42 (m, 1 H), 2.69 - 2.83 (m, 3 H), 2.85 - 2.94 (m, 1 H), 3.52 - 3.70 (m, 1 H), 4.02 - 4.14 (m, 2 H), 6.32 (br s, 1 H), 6.52 (s, 2 H), 7.93 and 8.53 (2 br s, 1 H), 7.99 (d, 1 H), 8.57 (d, 1 H), 13.93 and 14.03 (2 br s, 1 H).
Mixture of Deuterated Target Compound and Deuterated Sideproduct 1
5-{(3R)-1 -[(1 R)-2,2-difluorocyclopropyl(1 H-1 ,2,4-triazol-5-yl)(2H)methyl]-5',6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2- amine (deuterated diastereomer 1 and deuterated diastereomer 2)

5-[(3R)-5’,6’-Dihydrospiro[pyrrolidine-3,4’-pyrrolo[1 ,2-b]pyrazol]-2’-yl]-3-(trifluoromethyl)pyridin- 2-amine hydrogen chloride (1/1) (321 mg, 875 pmol) was dissolved in DMF (6.4 mL) and N,N- diisopropylethylamine (1.1 mL, 6.1 mmol) was added. 5-[Chloro[(1S)-2,2- difluorocyclopropyl](2H)methyl]-1 H-1 ,2,4-triazole hydrogen chloride (1/1) (402 mg, 73 % purity, 1.27 mmol) in DMF (6.4 mL) was added at rt. The reaction mixture was stirred at rt for 2 h and
5-[(3R)-5’,6’-dihydrospiro[pyrrolidine-3,4’-pyrrolo[1 ,2-b]pyrazol]-2’-yl]-3-(trifluoromethyl)pyridin- 2-amine hydrogen chloride (1/1) (0.2 equivalnets, 64.3 mg, 98 % purity, 175 pmol) was added and stirred at rt over night. Water and ethyl acetate were added, the pH of the aqueous phase was adjusted to pH 7 with saturated aqueous sodium hydrogen carbonate solution, the layers were separated and the aqueous phase was extracted twice with ethyl acetate. The combined organic layers were washed with brine, dried and concentrated. The residue was purified by
chromatography (Isolera Sfaer sillica HC 25g (dichloromethane/methanol; 1-16%; methanol premix dichloromethane/methanol 8:2) affording 311 mg (74%) of the title compound.
LC-MS (Method 1): Rt = 0.79 min; MS (ESIpos): m/z = 482 [M+H]+
1H NMR (400 MHz, DMSO-cfe, 22°C) 6 ppm 0.96 - 1.10 and 1.41 - 2.07 (m, 4 H), 2.22 - 2.40 (m, 1 H), 2.64 - 2.82 (m, 4 H), 2.85 - 2.93 (m, 1 H), 4.06 - 4.15 (m, 3 H), 6.28 - 6.46 (m, 1 H), 6.52 (s, 2 H), 7.89 - 7.96 (m, 1 H), 8.00 (t, 1 H), 8.48 - 8.55 (m, 1 H), 8.57 (s, 1 H), 13.82 - 14.11 (m, 1 H).
Deuterated Sideproduct 1
5-{(3R)-1-[(S)-[(1 R)-2,2-difluorocyclopropyl](1 H-1 ,2,4-triazol-5-yl)(2H)methyl]-5',6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine

The diastereomeric mixture of 5-{(3R)-1-[(1 R)-2,2-difluorocyclopropyl(1 H-1 ,2,4-triazol-5- yl)(2H)methyl]-5',6'-dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3- (trifluoromethyl)pyridin-2-amine (diastereomer 1 and diastereomer 2) was separated by chiral
HPLC to yield 126 mg (30 % yield) of the title compound as the first-eluting diastereomer, the deuterated sideproduct 1 .
Preparative method:
Instrument: Sepiatec: Prep SFC100; column: Chiralpak IG 5 pm 250x30 mm; mobile phase A: carbon dioxide; mobile phase B: 2-propanol; isocratic: 78% A + 22% B; flow: 100 mL/min; temperature: 40°C; backpressure: 150 bar; wavelength: 280 nm.
Retention time: 4.3 - 6.4 min
Analytical method:
Instrument: Waters Acquity UPC2 QDA; column: Chiralpak IG 3 pm 100x4.6 mm; mobile phase A: carbon dioxide; mobile phase B: 2-propanol + 0.4 vol % diethylamin; isocratic: 80% A + 20% B; flow: 4 mL/min; temperature: 40.0°C; backpressure: 100 bar; wavelength: 280 nm.
Retention time: 2.35 min
[CI]20D : +68.0° (c = 1.00, methanol)
1H NMR (400 MHz, DMSO-cfe, 22°C) 6 ppm 0.96 - 1.07 and 1.12 - 1.25 (2 m, 1 H), 1.47 - 1.68 (m, 1 H), 1.90 - 2.07 (m, 2 H), 2.21 - 2.32 (m, 1 H), 2.54 - 2.61 (m, 1 H), 2.63 - 2.69 (m, 1 H), 2.71 - 2.81 (m, 2 H), 2.85 - 2.92 (m, 1 H), 4.11 (br t, 2 H), 6.39 and 6.44 (2 br s, 1 H), 6.52 (br s, 2 H), 7.92 and 8.52 (2 br s, 1 H), 8.00 (d, 1 H), 8.57 (s, 1 H), 13.91 and 13.98 (2 br s, 1 H).
Deuterated Target Compound
5-{(3R)-1-[(R)-[(1 R)-2,2-difluorocyclopropyl](1 H-1 ,2,4-triazol-5-yl)(2H)methyl]-5',6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2-amine

The diastereomeric mixture of 5-{(3R)-1-[(1 R)-2,2-difluorocyclopropyl(1 H-1 ,2,4-triazol-5- yl)(2H)methyl]-5',6'-dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3- (trifluoromethyl)pyridin-2-amine (diastereomer 1 and diastereomer 2) was separated by chiral HPLC and purified by chromatography (Sfaer Amino 11g, ethyl acetate/ethanol 0-9%; ethanol premix ethyl acetate/ethanol 8:2). and reversed HPLC to yield 63.5 mg (15 % yield) of the title compound as the second-eluting diastereomer, the deuterated target compound.
Preparative method:
Instrument: Sepiatec: Prep SFC100; column: Chiralpak IG 5 pm 250x30 mm; mobile phase A: carbon dioxide; mobile phase B: 2-propanol; isocratic: 78% A + 22% B; flow: 100 mL/min; temperature: 40°C; backpressure: 150 bar; wavelength: 280 nm.
Retention time: 7.9 - 11.9 min
Analytical method:
Instrument: Waters Acquity UPC2 QDA; column: Chiralpak IG 3 pm 100x4.6 mm; mobile phase A: carbon dioxide; mobile phase B: 2-propanol + 0.4 vol % diethylamin; isocratic: 80% A + 20% B; flow: 4 mL/min; temperature: 40.0°C; backpressure: 100 bar; wavelength: 280 nm.
Retention time: 4.88 min
[CI]20D : +48.5° (c = 1.00, methanol)
1H NMR (400 MHz, DMSO-cfe, 22°C) 6 ppm 1.46 - 1.68 (m, 1 H), 1.69 - 2.03 (m, 3 H), 2.34 - 2.48 (m, 2 H), 2.68 - 2.82 (m, 3 H), 2.86 - 2.94 (m, 1 H), 4.08 (br t, 2 H), 6.30 and 6.34 (2 s, 1 H), 6.52 (br s, 2 H), 7.93 and 8.53 (2 s, 1 H), 7.99 (d, 1 H), 8.57 (s, 1 H), 13.93 and 14.03 (2 s, 1 H).
EXPERIMENTAL SECTION - BIOLOGICAL ASSAYS
Examples were tested in selected biological assays one or more times. When tested more than once, data are reported as either average values or as median values, wherein
• the average value, also referred to as the arithmetic mean value, represents the sum of the values obtained divided by the number of times tested, and
• the median value represents the middle number of the group of values when ranked in ascending or descending order. If the number of values in the data set is odd, the median is the middle value. If the number of values in the data set is even, the median is the arithmetic mean of the two middle values.
Examples were synthesized one or more times. When synthesized more than once, data from biological assays represent average values or median values calculated utilizing data sets obtained from testing of one or more synthetic batch.
The in vitro activity and other features of the compounds of the present invention can be demonstrated in the following assays or experiments:
Chromatographic LogD
Log D values at pH 7.5 were recorded using an indirect method for determining hydrophibicity constant by reversed-phase high performance liquid chromatography (HPLC). A homologous series of n-alkan-2-ones (C4-C16, 0.02 mol in ACN) was used for calibration. Test compounds were applied as 0.2 mmol DMSO stock solutions. The lipophilicity of compounds was then assessed by comparison to the calibration curve. Reference: Minick, D.J., Frenz, J.H., Patrick, M.A. & Brent, D.A. A comprehensive method for determining hydrophobicity constants by reversed-phase high performance liquid chromatography. Journal of Medicinal Chemistry, 31 , 1923-1933 (1988)
Solubility from DMSO stock solution in aqueous buffer pH 6.5
Aqueous solubility at pH 6.5 was determined by an orientating high throughput screening method. Solubility was determined in PBS buffer pH 6.5 containing 1% DMSO. Test compounds were applied as 1mm DMSO solution. After addition of PBS buffer pH 6.5 solutions were shaken for 24 h at room temperature. Undissolved material was removed by filtration. The compound dissolved in the filtrate was quantified by HPLC-UV. The response was fitted to a one-point standard curve prepared in DMSO. Reference: Onofrey Th., Kazan, G., Barbagallo, C., Blodgett, J., Weiss, A. Millipore Corporation, Life Sciences Division, Danvers, MA USA 01923: Automated Screening of Aqueous Compound Solubility in Drug Discovery (July 31 , 2019)
Table 4: In vitro physico-chemical profiling results for the target compound of the invention

Human MAP4K1 binding competition assay
The ability of the compounds of the present invention to inhibit the binding of an Alexa647- labelled ATP-competitive kinase inhibitor to a Glutathione-S-transferase-MAP4K1 (GST- MAP4K1) fusion protein was quantified employing the TR-FRET-based MAP4K1 binding competition assay as described in the following paragraphs.
A recombinant fusion protein of N-terminal GST and full-length human MAP4K1 , expressed by baculovirus infected SF9 insect cells and purified by Glutathione Sepharose affinity chromatography, was used as GST-MAP4K1 fusion protein. Tracer 222 from Invitrogen (catalogue no. PR9198A) was used as Alexa647-labelled ATP-competitive kinase inhibitor.
For the assay 50 nL of a 100fold concentrated solution of the test compound in DMSO was pipetted into either a black low volume 384well microtiter plate (Greiner Bio-One, Frickenhausen, Germany) or a black 1536well microtiter plate (Greiner Bio-One or Corning), 3 pl solution of Tracer 222 (25 nM => final concentration in 5 pl assay volume is 15 nM) in aqueous assay buffer [25 mM Tris/HCI pH 7.5, 10 mM MgCh, 5 mM [3-glycerolphosphate, 2.5 mM dithiothreitol, 0.5 mM ethylene glycol-bis(2-aminoethylether)-/V,/V,/V',/V'-tetraacetic acid [EGTA], 0.5 mM sodium ortho-vanadate, 0.01 % (w/v) bovine serum albumin [BSA], 0.005% (w/v) Pluronic F-127 (Sigma)] were added. Then the binding competition was started by the addition of 2 pl of a solution of the GST-MAP4K1 fusion protein (2.5 nM => final cone, in the 5 pl assay volume is 1 nM) and of Anti-GST-Tb (1.25 nM => final cone, in the 5 pl assay volume is 0.5 nM), a Lumi4®- Tb Cryptate-conjugated anti-GST-antibody from Cisbio Bioassays (France), in assay buffer.
The resulting mixture was incubated 30 min at 22°C to allow the formation of a complex between the Tracer 222, the fusion protein and Anti-GST-Tb. Subsequently the amount of this complex was evaluated by measurement of the resonance energy transfer from the Tb-cryptate to the Tracer 222. Therefore, the fluorescence emissions at 620 nm and 665 nm after excitation at 350 nm were measured in a TR-FRET reader, e.g. a Pherastar (BMG Labtechnologies, Offenburg, Germany) or a Viewlux (Perkin-Elmer). The ratio of the emissions at 665 nm and at 622 nm was taken as the measure for the amount of the complex. The data were normalised (assay reaction without inhibitor = 0 % inhibition, all other assay components but GST-MAP4K1 fusion protein = 100 % inhibition). Usually the test compounds were tested on the same microtiterplate in 11 different concentrations in the range of 20 pM to 0.07 nM (20 pM, 5.7 pM, 1.6 pM, 0.47 pM, 0.13 pM, 38 nM, 11 nM, 3.1 nM, 0.9 nM, 0.25 nM and 0.07 nM, the dilution series prepared
separately before the assay on the level of the lOOfold concentrated solutions in DMSO by serial dilutions, exact concentrations may vary depending pipettors used) in duplicate values for each concentration and IC50 values were calculated using Genedata Screener™ software.
Flt3 kinase activity inhibition assay
Flt3 inhibitory activity of compounds of the present invention at a high ATP concentration after preincubation of enzyme and test compounds was quantified employing the TR-FRET-based Flt3 activity inhibition assay as described in the following paragraphs.
A recombinant fusion protein of N-terminal GST and C-terminal fragment of human Flt3 (aa 561- end), expressed by baculovirus infected SF21 insect cells and purified by Glutathione Sepharose affinity chromatography, was used as enzyme. As substrate for the kinase reaction biotinylated peptide biotin-Ahx- GGEEEEYFELVKKKK (C-terminus in amide form) was used which can be purchased e.g. form the company Biosyntan (Berlin-Buch, Germany).
For the assay 50 nL of a 100-fold concentrated solution of the test compound in DMSO was pipetted into either a black low volume 384well microtiter plate (Greiner Bio-One, Frickenhausen, Germany), or a black 1536-well microtiter plate (Greiner Bio-one or Corning), 2 pl of a solution of Flt3 in aqueous assay buffer [25 mM HEPES pH 7.5, 10 mM MgCh, 2 mM dithiothreitol (DTT), 5 mM p-glycerolphosphate, 0.5 mM (EDTA), 0.001 % (w/v) bovine serum albumin (BSA), 0.01% (v/v) Triton X-100 (Sigma)] were added and the mixture was incubated for 15 min at 22°C to allow pre-binding of the test compounds to the enzyme before the start of the kinase reaction. Then the kinase reaction was started by the addition of 3 pl of a solution of adenosine-tri- phosphate (ATP, 16.7 pM => final cone, in the 5 pl assay volume is 1 mM) and substrate (1 .67 pM => final cone, in the 5 pl assay volume is 1 pM) in assay buffer and the resulting mixture was incubated for a reaction time of 45 min at 22°C. The concentration of Flt3 was adjusted depending on the activity of the enzyme lot and was chosen appropriate to have the assay in the linear range, a typical concentration was 0.5 nM. The reaction was stopped by the addition of 3 pl of a solution of TR-FRET detection reagents (167 nM streptavidine-XL665 [Cisbio Bioassays, Codolet, France] [as an alternative 333 nM streptavidine-DY648 can be used], and 1.67 nM PT66-Tb-Cryptate, an terbium-cryptate labelled anti-phospho-tyrosine antibody from Cisbio Bioassays) in an aqueous EDTA-solution (66.7 mM EDTA, 0.2 % (w/v) bovine serum albumin in 50 mM HEPES/NaOH pH 7.5).
The resulting mixture was incubated 1 h at 22°C to allow the formation of complex between the phosphorylated biotinylated peptide and the detection reagents. Subsequently the amount of phosphorylated substrate was evaluated by measurement of the resonance energy transfer from the Eu-chelate to the streptavidine-XL. Therefore, the fluorescence emissions at 620 nm and 665 nm after excitation at 350 nm was measured in a TR-FRET reader, e.g. a Pherastar FS
(BMG Labtechnologies, Offenburg, Germany) or a Viewlux (Perkin-Elmer). The ratio of the emissions at 665 nm and at 622 nm was taken as the measure for the amount of phosphorylated substrate. The data were normalised (enzyme reaction without inhibitor = 0 % inhibition, all other assay components but no enzyme = 100 % inhibition). Usually the test compounds were tested on the same microtiterplate in 11 different concentrations in the range of 20 pM to 0.07 nM (20 pM, 5.7 pM, 1.6 pM, 0.47 pM, 0.13 pM, 38 nM, 11 nM, 3.1 nM, 0.9 nM, 0.25 nM and 0.07 nM, the dilution series prepared separately before the assay on the level of the 100-fold concentrated solutions in DMSO by serial dilutions, exact concentrations may vary depending pipettors used) in duplicate values for each concentration and IC50 values were calculated using Genedata Screener™ software.
Jurkat Phospho-SLP-76 HTRF assay:
Preparation of test compound dilutions. Serial dilutions of test compounds were prepared in 100% DMSO using a Precision Pipetting System (BioTek, USA). Afterwards, a 100-fold concentrated solution of the test compound (50 nL) in DMSO was transferred to microtiter test plates (384 or 1 ,536 wells, Greiner Bio-One, Germany) using either a Hummingbird liquid handler (Digilab, MA, USA) or an Echo acoustic system (Labcyte, CA, USA). Plates were sealed with adhesive foil or heat-sealed and stored at -20 °C until use.
Measurement and evaluation of inhibition data, calculation of IC50 values. Homogeneous time- resolved fluorescence (HTRF) was measured with a PHERAstar reader (BMG Labtech, Ortenberg, Germany) using the HTRF module (excitation: 337 nm; emission 1 : 620 nm, emission 2: 665 nm). The ratio of the emissions at 665 and 620 nm was used as the specific signal for further evaluation. The data were normalized using the controls: DMSO = 0% inhibition, inhibition control wells were not stimulated with anti-CD3 = 100% inhibition. Compounds were tested in duplicates at up to 11 concentrations (e.g. 20 pM, 5.7 pM, 1.6 pM, 0.47 pM, 0.13 pM, 38 nM, 11 nM, 3.1 nM, 0.89 nM, 0.25 nM and 0.073 nM). IC50 values were calculated using a four-parameter fit, with a commercial software package (Genedata Screener, Switzerland).
Phospho-SLP-76 HTRF assay. This assay quantifies the endogenous levels of phosphorylated SLP-76, only when phosphorylated at Ser376 by using the Phospho-SLP76 HTRF kit from Cisbio (Cisbio, Condolet, France, # 63ADK076PEH).
The assay was carried out in Jurkat E6.1 cells from American Type Culture Collection (ATCC) stably overexpressing human FLAG-tagged SLP-76 (proprietary) and frozen cell vials were generated. Cells were thawed quickly, counted and a cell suspension in assay medium (RPMI 1640, Biochrom Cat#F1275; 1% FCS Superior, Biochrom Cat#S0615 heat-inactivated; 1 % Penicillin/ Streptavidin, Biochrom Cat#A2213; 1% L-Glutamine, Gibco Cat#25030-81) with 5E+06 cells / ml was prepared. To all wells of the test plate 3 pl of this cell suspension was
added by using a Multidrop dispenser (Thermo LabSystems) resulting in 15,000 cells / well. To all wells except inhibitor control wells, 2 pl of anti-CD3 (invitrogen Cat#16-0037-85) / anti-mouse- IgG (invitrogen Cat#31160) in assay media (final concentration in 5 pl: 1 pg / ml anti-CD3, 2 pg I ml anti-mouse-IgG) was added. In inhibitor control wells 2 pl of assay media was dispensed. After an incubation time of 30 minutes at 37°C, 5% CO2 and 95% humidity, the reaction was stopped by addition of 2 pl lysis buffer with blocking reagent (1 :25 in lysis buffer) supplied with kit. After 60 minutes for lysis at room temperature, 2 pl of detection solution from kit including the detection antibodies was dispensed to all wells. The plate was measured after 60 minutes at room temperature.
Cytokine secretion in human peripheral blood mononuclear cells (PBMCs)
To examine the effects of MAP4K1 inhibitors on cytokine secretion in the presence of MAP4K1 enhancers, human peripheral blood mononuclear cells (PBMCs) were isolated using a density gradient from buffy coats of healthy donors. The cells were activated with plate-coated anti-CD3 (24h at 4°C, using the OKT3 clone) and treated with PGE2 at 1 pM plus the MAP4K1 inhibitors. After 24h incubation, the supernatant of the culture was acquired and the IFN- levels were measured by ELISA (BD Biosciences).
Table 5: In vitro pharmacological profiling results for the target compound of the invention
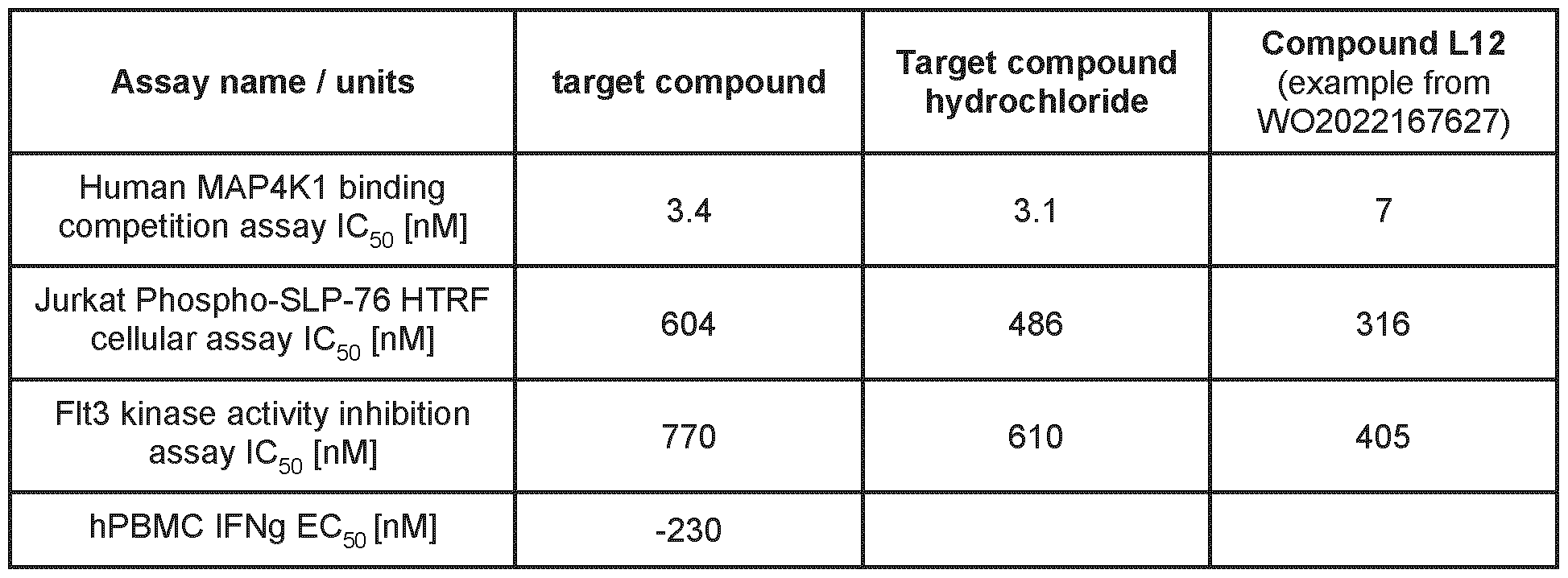 cf. Figure 4
cf. Figure 4
Determination of metabolic stability in microsomes
The in vitro metabolic stability of test compounds was determined by incubating them at 1 pM in a suspension liver microsomes in 100 mM phosphate buffer, pH7.4 (NaH2PO4X H2O + Na2HPO4 x 2H2O) and at a protein concentration of 0.5 mg/mL at 37° C. The microsomes were activated by adding a co-factor mix containing 8 mM Glukose-6-Phosphat, 4 mM MgCI2; 0.5 mM NADP and 1 I U/ml G-6-P-Dehydrogenase in phosphate buffer, pH 7.4. The metabolic assay was started shortly afterwards by adding the test compound to the incubation at a final volume of 1 mL.
Organic solvent in the incubations was limited to <0.01 % dimethyl sulfoxide (DMSO) and <1 % acetonitrile. During incubation, the microsomal suspensions were continuously shaken at 580 rpm and aliquots were taken at 2, 8, 16, 30, 45 and 60 min, to which equal volumes of cold methanol were immediately added. Samples were frozen at -20°C over night, subsequently centrifuged for 15 minutes at 3000 rpm and the supernatant was analyzed with an Agilent 1290 HPLC applying MS/MS detection.
The half-life of a test compound was determined from the concentration-time plot. From the halflife the intrinsic clearances were calculated. Together with the additional parameters liver blood flow, specific liver weight and microsomal protein content the hepatic in vivo blood clearance (CLbiood) and the maximal oral bioavailability (Fmax) were calculated using the 'well stirred' liver model. The following parameter values were used: Liver blood flow - 1.32 L/h/kg; specific liver weight -- 38 g/kg (mouse) and 21 g/kg (human); microsomal protein content - 40 mg/g.
In vitro metabolic stability in hepatocytes
The liver cells were distributed in WME containing 5% FCS to glass vials at a density of 1.0 x 106 vital cells/ml. The test compound was added to a final concentration of 1 pM. During incubation, the hepatocyte suspensions were continuously shaken at 580 rpm and aliquots were taken at 2, 8, 16, 30, 45 and 90 min, to which equal volumes of cold methanol were immediately added. Samples were frozen at -20° C over night, after subsequently centrifuged for 15 minutes at 3000 rpm and the supernatant was analyzed with an Agilent 1290 HPLC applying MS/MS detection.
The half-life of the test compound was determined from the concentration-time plot. From the half-life the intrinsic clearances were calculated using the 'well stirred' liver model applying the additional parameters liver blood flow, amount of liver cells in vivo and in vitro. The hepatic in vivo blood clearance (CLbiood) and the maximal oral bioavailability (Fmax) was calculated. The following parameter values were used: Liver blood flow - 4.2 L/h/kg rat, 1.32 L/h/kg human; specific liver weight - 32 g/kg (rat) and 21 g/kg (human); liver cells in vivo- 1.1 x 108 cells/g liver, liver cells in vitro - 1.0 x 106/ml.
Caco-2 Permeation Assay
Caco-2 cells (purchased from DSMZ Braunschweig, Germany) were seeded at a density of 4.5 x 104 cell per well on 24 well insert plates, 0.4 pm pore size, and grown for 15 days in DM EM medium supplemented with 10% fetal bovine serum, 1% GlutaMAX (100x, GIBCO), 100 U/mL penicillin, 100 pg/mL streptomycin (GIBCO) and 1 % non-essential amino acids (100 x). Cells were maintained at 37°C in a humified 5% CO2 atmosphere. Medium was changed every 2-3 day. Before running the permeation assay, the culture medium was replaced by a FCS-free
hepes-carbonate transport puffer (pH 7.2) For assessment of monolayer integrity the transepithelial electrical resistance (TEER) was measured. Test compounds were predissolved in DMSO and added either to the apical or basolateral compartment in final concentration of 2 pM. Before and after 2 h incubation at 37°C samples were taken from both compartments. Analysis of compound content was done after precipitation with methanol by applying LC/MS/MS. Permeability (Papp) was calculated in the apical to basolateral (A - B) and basolateral to apical (B - A) directions. The apparent permeability was calculated using following equation:
Papp = (Vr/P0)(1/S)(P2/t)
Where Vr is the volume of medium in the receiver chamber, Po is the measured peak area of the test drug in the donor chamber at t = 0, S the surface area of the monolayer, P2 is the measured peak area of the test drug in the acceptor chamber after 2 h of incubation, and t is the incubation time. The efflux ratio basolateral (B) to apical (A) was calculated by dividing the Papp(B-A) by the PaPP(A-B). In addition, the compound recovery was calculated. As assay control reference compounds were analyzed in parallel.
Inhibition of Cytochrome P450 (CYP) enzymes
The inhibitory potential of the test substance towards 5 human cytochrome P450 isoforms (CYP1A2, 2C8, 2C9, 2D6, 3A4) is determined. In the case of CYP3A4, additionally the so-called time-dependent inhibition potential is tested. For this purpose, the test substance is preincubated in a metabolically active system for 30 minutes. Human liver microsomes (pool, >30 male and female donors) are used for all assays, which are incubated with individual CYP isoform-selective standard substrates (phenacetin, amodiaquine, diclofenac, dextromethorphan, midazolam). The metabolism of these standard substrates is analyzed and the concentrationdependent effect of the test substance on these enzymatic reactions is quantified. Incubation batches without test substance serve as the reference. In addition, established CYP isoform- selective inhibitors are included as positive controls (fluvoxamine for CYP1A2, montelukast for CYP2C8, sulfaphenazole for CYP2C9, fluoxetine for CYP2D6, ketoconazole for CYP3A4, and mibefradil for CYP3A4 pre-incubation). The incubation conditions are optimized with regard to the following parameters: protein concentration, substrate concentration, incubation time and metabolic turnover. The incubation medium consists of 50 mM potassium phosphate buffer (pH 7.4), 1 mM EDTA, NADPH regenerating system (1 mM NADP, 5 mM glucose 6-phosphate, glucose 6-phosphate dehydrogenase (1.5 U/mL)). Sequential dilutions and all incubations are carried out in 96-MTP plate format at 37°C in a final volume of 200 pL and under automated conditions using a Genesis Workstation (Tecan, Crailsheim). The enzymatic reaction is stopped by adding 100 pL acetonitrile including internal standard. After protein precipitation and centrifugation, the supernatants are analyzed. The metabolites paracetamol (CYP1A2),
desethylamodiaquine (CYP2C8), 4-hydroxydiclofenac (CYP2C9), dextrorphan (CYP2D6), and 1 -hydroxymidazolam (CYP3A4) are quantified using LC/MS/MS.
Evaluation: The CYP-mediated enzyme activity is determined as a function of the test substance concentration and the enzyme-kinetic parameter IC50 is calculated.
Estimation of Plasma Protein Binding by Equilibrium Dialysis
Binding of test compounds to plasma proteins has been measured by equilibrium dialysis in a 96-well format using HTDialysis equipment made of Teflon and a semipermeable membrane (regenerated cellulose, MWCO 12-14K). The membrane separates the plasma and buffer cavities (50 mM phosphate buffer) filled with 150 pl each. The test compound is added to the plasma cavity at a test concentration of 3 pM and binds to plasma proteins. The unbound fraction of the test compound passes the membrane and distributes on both sides until equilibrium is reached, which is usually the case after 6-8h at 37°C and 5% CO2 atmosphere. Relative compound concentration (peak area ratios analyte/IS) of plasma and buffer side is measured by LC/MS/MS analytics. To this end both sides are matrix matched, i.e. diluted with buffer and plasma to achieve the same matrix (10% plasma) and subsequently are precipitated with a fourfold volume of methanol containing an appropriate internal standard (IS). From the quotient of buffer and plasma concentration the mean free (unbound) fraction (fu) is calculated (at least n = 3 replicates, CV%). Stability and recovery controls are included. Additionally, the test compound is dialyzed in buffer against buffer in order to estimate non-specific binding to equipment and/or membrane and to ensure equilibrium. Due to the osmotic pressure of the plasma proteins a dilution of the plasma takes place during the incubation (volume shift). The potential imprecision is addressed by inclusion of an empirical factor in the calculation of the fu. Establishment of equilibrium in bufferbuffer dialysis and stability in plasma should be at least 80%, the recovery in bufferbuffer dialysis is aimed to be >30%. A free fraction of <1 % is designated as high, between 1 and 10% as moderate and of >10% as low plasma protein.
Automated hERG Voltage clamp assay
To investigate whether BAYXXX (test item) inhibits the human Ether-a-go-go-Related Gene (hERG) potassium channel, in vitro automated voltage clamp recordings were performed on recombinant HEK293 cells stably expressing the hERG alpha subunit (hERG cells). Following harvest, cells were transferred to the cell reservoir of a 384 channel automated patch clamp device and stored there at 20°C until usage.
Upon start of the automated voltage clamp procedure, hERG cells were transferred to a 384 well patch clamp chip (pipette resistance of ~2-3 MQ) prefilled with external solution (containing in mM: 143 NaCI, 4 KOI, 2 CaCh, 1 MgCI2, 5 glucose, 10 HEPES; pH 7.4 (NaOH)). Underpressure
was applied underneath the glass bottom of the patch clamp chip to position the hERG cells on the recording sites in the glass bottom of the chip. Following successful cell catch, underpressure was stopped and a seal enhancing solution (containing in mM: 78 NaCI, 60 NMDG, 4 KOI, 10 CaCI2, 1 MgCI2, 5 glucose, 10 HEPES; pH 7.4 (HOI)) was added to the hERG cells to facilitate the formation of stable seals between the membranes of the hERG cells and the glass next to the recording sites. Then, hERG cells were washed several times with wash solution (containing in mM: 87 NaCI, 60 NMDG, 4 KCI, 2 CaCI2, 1 MgCI2, 5 glucose, 10 HEPES; pH 7.4 (HOI)) to remove excess seal enhancing solution. In the meantime, the membrane parts of the hERG cells covering the recording sites were exposed to internal solution (containing in mM: 10 NaCI, 123 KF, 10 EGTA, 10 HEPES; pH 7.2 (KOH)) supplemented with 5-20 pM Escin, and the perforated patch configuration was established. Next, the holding potential was stepwise adjusted to -80 mV, capacitance was compensated, and a series of defined voltage commands was initiated to trigger the hERG current response from the hERG cells (-80 mV for 200 ms, +20 mV for 1000 ms, -40 mV for 500 ms; repeated at a frequency of 0.1 Hz). To determine whether the test item inhibits hERG, different solutions were sequentially applied to the hERG cells: First, a negative control (i.e. wash solution supplemented with 0.3% DMSO and 0.01 % HSA) was applied for at least 3 min to investigate the basic electrophysiological properties of the hERG cells as well as define their standard current response. Next, a solution containing the test item at a final concentration of (0.1 , 1 , or 10) pM was applied for 10 min to measure eventual inhibitory effects of the test item on the hERG current (- the test item solution was produced from a 10 mM DMSO stock by using an automated pipetting device and sequential dilution). Finally, a positive control (i.e. wash solution supplemented with 0.3% DMSO, 0.01% HSA, and 10 pM quinidine) was added to the cells for ~ 3 min to block the hERG current and, thereby, define the maximum inhibition.
For data analysis, experimental results were processed using the automated patch clamp device-specific software as well as a custom-made software, and TIBCO Spotfire. For each successful recording, hERG tail current amplitudes were averaged from three consecutive current responses at the end of the negative control phase, test item phase, and positive control phase, respectively. Resulting mean hERG tail current amplitudes were normalized to the mean hERG tail current amplitude at the end of the negative control phase with nominal 0% inhibition as well as the hERG tail current amplitude at the end of the positive control phase with nominal 100% inhibition. Then, the effect of the test item was calculated as a percentage inhibition value at the test item concentration applied. Finally, percentage inhibition values from all successful recordings at a particular test item concentration were averaged and combined to construct a standard sigmoidal dose response curve, determine the half maximal inhibitory concentration (IC50) of the test item as well as extrapolate its IC2o.
Table 6: In vitro pharmacokinetics and safety pharmacology data for the target compound of the invention

In vivo pharmacokinetics study in rats
For in vivo pharmacokinetic experiments test compounds were administered to male Wistar rats intravenously at doses of 0.3 to 0.8 mg/kg and intragastric at doses of 0.5 to 1.5 mg/kg formulated as solutions using solubilizers such as PEG400 in well-tolerated amounts.
For pharmacokinetics after intravenous administration test compounds were given as i.v. bolus and blood samples were taken at 2 min, 8 min, 15 min, 30 min, 45 min, 1 h, 2 h, 4 h, 6 h, 8 h and 24 h after dosing. For pharmacokinetics following oral administration test compounds were given intragastric to fasted rats and blood samples were taken at 5 min, 15 min, 30 min, 45 min,
1 h, 2 h, 4 h, 6 h, 8 h and 24 h after dosing. Blood was collected into Lithium-Heparin tubes (Monovetten®, Sarstedt) and centrifuged for 15 min at 3000 rpm. An aliquot of 100 pL from the supernatant (plasma) was taken and precipitated by addition of 400 pL cold acetonitrile and frozen at -20°C over night. Samples were subsequently thawed and centrifuged at 3000 rpm, 4°C for 20 minutes. Aliquots of the supernatants were taken for analytical testing using an Agilent
1290 HPLC-system applying MS/MS detection. PK parameters were calculated by noncompartmental analysis using a PK calculation software.
PK parameters derived from concentration-time profiles after i.v.: CLpiasma: Total plasma clearance of test compound (in L/kg/h); CLbiood: Total blood clearance of test compound: CLpiasma*Cp/Cb (in L/kg/h) with Cp/Cb being the ratio of concentrations in plasma and blood. PK parameters calculated from concentration time profiles after i.g.: Cmax: Maximal plasma concentration (in mg/L); Cmax, norm: Cmax divided by the administered dose (in kg/L); Tmax: Time point at which Cmax was observed (in h). Parameters calculated from both, i.v. and i.g. concentration-time profiles: AUCnorm: Area under the concentration-time curve from t = 0 h to infinity (extrapolated) divided by the administered dose (in kg*h/L); AUC(0-tiast)norm: Area under the concentration-time curve from t = 0 h to the last time point for which plasma concentrations could be measured divided by the administered dose (in kg*h/L); t-1/2: terminal half-life (in h); F: oral bioavailability: AUCnorm after intragastric administration divided by AUCnorm after intravenous administration (in %).
Table 7: In vivo pharmacokinetics data for the target compound of the invention after i.v. bolus administration in Wistar rats
 Table 8: In vivo pharmacokinetics data for the target compound of the invention after oral administration in Wistar rats
Table 8: In vivo pharmacokinetics data for the target compound of the invention after oral administration in Wistar rats
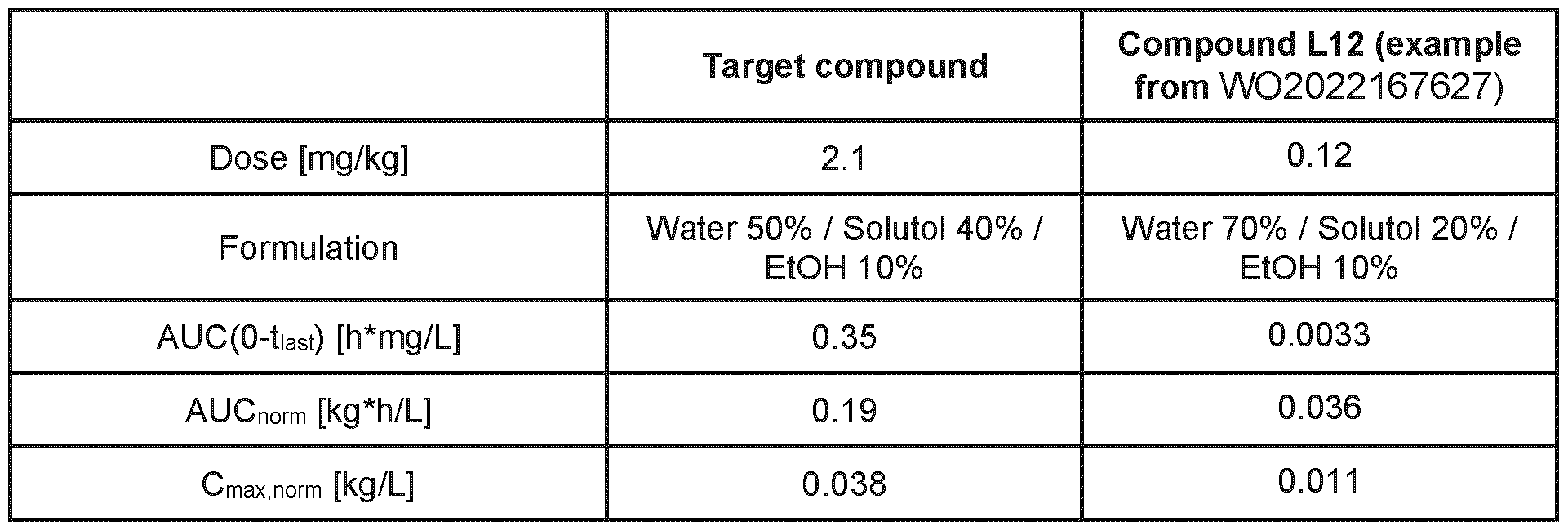

High dose exposure studies in mice
To assess in vivo compound exposure upon application of a high dose that is potentially pharmacologically active, test substance was administered orally (intragastrically) to Balb/c AnN mice (Charles River) at a dose of 100 mg/kg solubilized in a vehicle containing 60% polyethylenglycol 400 (PEG400), 10% ethanol and 30% water. 1 , 3, 7, 24 hours after compound administration to mice blood samples were taken (n = 2 mice each timepoint). Blood samples were precipitated 1 :5 (v:v) in ice-cold acetonitrile. After thawing and mixing, samples were centrifuged at 2000 rpm for 20 minutes at 4°C. Resulting supernatants are the blood plasma fraction. Plasma was collected and analysed by LC-MS/MS (AB Sciex, Framingham, MA, USA) for compound concentration. Exposure was determined by using a calibration curve generated from plasma samples of untreated mice with ex vivo added fixed compound content. To define amount of substance that is free available in plasma the measured value were further corrected for plasma protein binding. The plasma protein binding was determined using equilibrium dialysis by HT Dialysis (Gales Ferry, USA), in which a semi-permeable membrane separates 2 compartments, one containing the plasma and one containing buffer. Defined amount of compound was added to the plasma compartment and incubated under moderate agitation for 7 hours at 37°C (5% CO2, 99% humidity). Plasma samples were transferred into a deep-well plate, precipitated with 400 pl ice cold methanol and frozen over night at -20 °C. After thawing and mixing, the samples were centrifuged at 3000 rpm for 10 minutes. The supernatants, containing non-protein bound (= unbound) compound, were transferred into a 96-well plate for measurement of compound concentration by LC-MS/MS. The unbound fraction (fu) is determined calculating the ratio of unbound concentration to total concentration in plasma.
According to the free drug hypothesis (Ref. Smith DA, Di L, Kerns EH. The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat Rev Drug Discov 2010;9(12):929-39 doi 10.1038/nrd3287), the unbound compound exposure in plasma is considered equal to the unbound compound concentration in the target tissue. Therefore, unbound plasma exposure level can be directly used to assess coverage of pharmacologically active concentrations (IC50u) that are defined in cellular or biochemical in vitro assays.
Table 9: In vivo exposure data for the example xx (BAY 3619082) of the invention after oral/intragastraladministration in female Balb/c AnN mice (Charles River)


Claims
1. A compound
5-{-l-[(2,2-difluorocyclopropyl)(lH-l,2,4-triazol-5-yl)methyl]-5',6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[l,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2- amine of formula (I)
 or a stereoisomer, a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same. 2. The compound of claim 1, wherein said compound is
or a stereoisomer, a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same. 2. The compound of claim 1, wherein said compound is
5-{(3R)-l-[(2,2-difluorocyclopropyl)(lH-l,2,4-triazol-5-yl)methyl]-5',6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[l,
2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2- amine of formula (I b)
 or a stereoisomer, a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same.
or a stereoisomer, a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same.
3. The compound of claim 1 or 2, wherein said compound is
5-{(3R)-l-[((R)-2,2-difluorocyclopropyl)(lH-l,2,4-triazol-5-yl)methyl]-5',6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[l,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2- amine of formula (I c)

(I c), or a stereoisomer, a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same.
4. The compound of any one of claims 1 to 3, wherein said compound is
5-{(3R)-l-[(R)-((R)-2,2-difluorocyclopropyl)(lH-l,2,4-triazol-5-yl)methyl]-5',6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[l,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2- amine of formula (I f)
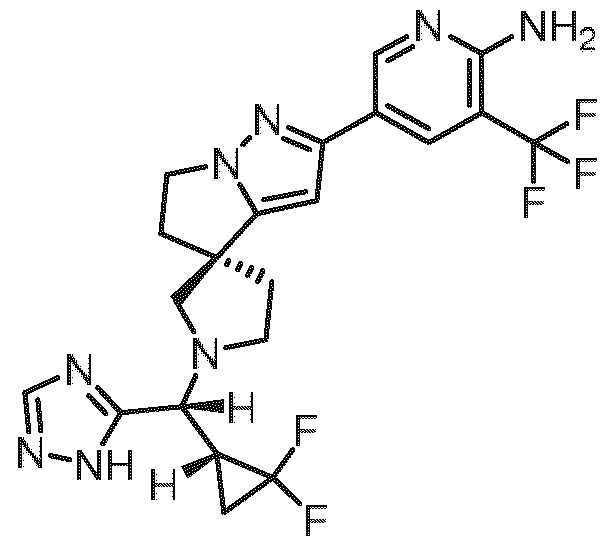
(I f), or a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same. 5. The compound of claim 4, wherein said compound is
5-{(3R)-1-[(R)-[(1 R)-2,2-difluorocyclopropyl](1 H-1,2,4-triazol-5-yl)(2H)methyl]-5',6'- dihydrospiro[pyrrolidine-3,4'-pyrrolo[1 ,2-b]pyrazol]-2'-yl}-3-(trifluoromethyl)pyridin-2- amine of formula (1f)

6. A method of preparing a compound of formula (I) according to claim 1, said method comprising the step of allowing an intermediate compound of general formula (II), a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof:
 in which Y represents a leaving group, to react with a compound of formula (III), a stereoisomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof:
in which Y represents a leaving group, to react with a compound of formula (III), a stereoisomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof:
 thereby giving the compound of formula (I) of claim 1:
thereby giving the compound of formula (I) of claim 1:
 or a stereoisomer, a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same.
or a stereoisomer, a tautomer, a hydrate, a solvate, or a salt thereof, or a mixture of same.
7. A method of preparing a compound of formula (I) according to claim 1, said method comprising the step of allowing an intermediate compound of general formula (Ila), a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof:
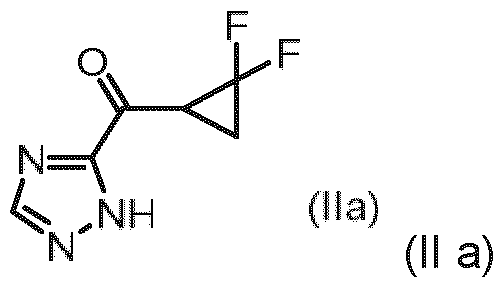 to react with a compound of formula (III), a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof:
to react with a compound of formula (III), a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof:
 thereby giving the compound of formula (I) of claim 1:
thereby giving the compound of formula (I) of claim 1:
 a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof.
a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof.
8. A compound according to any one of claims 1 to 5 for use in the treatment or prophylaxis of a disease.
9. A compound according to any one of claims 1 to 5 for use in treatment or prophylaxis of a neoplastic or abnormal cell proliferative disorder, such as cancer, a condition with ill
dysregulated immune response, or another disorder associated with aberrant Map4Kl signaling.
10. The compound for use of claim 9, wherein said neoplastic or abnormal cell proliferative disorder is cancer.
11. A pharmaceutical composition comprising a compound according to any one of claims 1 to 5 and one or more pharmaceutically acceptable excipients.
12. A pharmaceutical combination comprising:
• one first active ingredient being a compound according to any one of claims 1 to 5, and
• one or more further active ingredient(s), in particular:
1311-metuximab, 1311-chTNT, abarelix, abemaciclib, abiraterone, acalabrutinib, aclarubicin, adalimumab, ado-trastuzumab emtansine, afatinib, aflibercept, aldesleukin, alectinib, alfaferone, alemtuzumab, alendronic acid, alitretinoin, almonertinib, alpelisib, alpharadin, monosodium alpha luminol, altretamine, amifostine, aminoglutethimide, hexyl aminolevulinate, aminolevulinic acid, amrubicin, amsacrine, anastrozole, ancestim, anethole dithiolethione, anetumab ravtansine, angiotensin II, antithrombin III, apalutamide, aprepitant, arcitumomab, arglabin, arsenic trioxide, asparaginase, atezolizumab, avapritinib, avelumab, axicabtagene ciloleucel, axitinib, azacitidine, basiliximab, beclomethasone dipropionate, belantamab mafodotin, belinostat, belotecan, bendamustine, besilesomab, beta-elemene, bevacizumab, bexarotene, bicalutamide, binimetinib, bisantrene, bleomycin, blinatumomab, boanmycin hydrochloride, borofalan, bortezomib, bosutinib, budesonide, buserelin, brentuximab vedotin, brigatinib, busulfan, cabazitaxel, cabozantinib, calcitonine, calcium folinate, calcium levofolinate, capecitabine, capmatinib, capromab, carbamazepine, carboplatin, carboquone, carfilzomib, carmofur, carmustine, catumaxomab, catequentinib, celecoxib, celmoleukin, cemiplimab, ceritinib, cetuximab, chlorambucil, chlormadinone, chlormethine, chloroxoquinoline, cidofovir, cinacalcet, cisplatin, cladribine, clodronic acid, clofarabine, cobimetinib, copanlisib, crisantaspase, crizotinib, cyclophosphamide, cyproterone, cytarabine, dacarbazine, dacomitinib, dactinomycin, daratumumab, darbepoetin alfa, dabrafenib, darolutamide, dasatinib, daunorubicin, decitabine, deferasirox, degarelix, denileukin diftitox, denosumab, depreotide, deslorelin, dianhydrogalactitol, dexrazoxane, dibrospidium chloride, dianhydrogalactitol,
diclofenac, dinutuximab, docetaxel, dolasetron, doxifluridine, doxorubicin, doxorubicin + estrone, dronabinol, dupilumab, durvalumab, duvelisib, eculizumab, edrecolomab, elliptinium acetate, elotuzumab, eltrombopag, enasidenib, encorafenib, endostatin, enfortumab vedotin, enocitabine, enzalutamide, epirubicin, epitiostanol, epoetin alfa, epoetin beta, epoetin zeta, eptaplatin, eribulin, erlotinib, ensartinib, entrectinib, erdafitinib, esomeprazole, estradiol, estramustine, estrone, ethinylestradiol, etoposide, everolimus, evocalcet, exemestane, fadrozole, famotidine, fentanyl, filgrastim, flumatinib, fluoxymesterone, fluticasone, fluticasone furoate, floxuridine, fludarabine, fluorouracil, flutamide, folinate, folinic acid, formestane, forodesine, fosaprepitant, fotemustine, fruquintinib, fulvestrant, gadobutrol, gadoteridol, gadoteric acid meglumine, gadoversetamide, gadoxetic acid, gallium nitrate, ganirelix, gefitinib, gemcitabine, gemtuzumab, gendicine, gilteritinib, ginsenoside Rg3, glasdegib, glucarpidase, glutoxim, GM-CSF, goserelin, granisetron, granulocyte colony stimulating factor, hematoporphyrin, histamine dihydrochloride, histrelin, holmium-166-chitosan complex, human menopausal gonadotrophin, hydroxycarbamide, 1-125 seeds, lansoprazole, ibandronic acid, ibritumomab tiuxetan, ibrutinib, icotinib hydrochloride, idarubicin, idelalisib, iobenguane (1311), iodi ne( 13 II ) tumor necrosis factor monoclonal antibody, ifosfamide, imatinib, imiquimod, improsulfan, immunocyanin, indisetron, incadronic acid, indole-3-carbinol + epigallocatechin-3-gallate, ingenol mebutate, inotuzumab ozogamicin, interferon alfa, interferon alpha lb, interferon-alpha 2, interferon alpha-2a, interferon alfa-2b, interferon beta, interferon gamma, interleukin- 2, iobitridol, iobenguane (1231), iomeprol, ipilimumab, irinotecan, Itraconazole, isatuximab, ivosidenib, ixabepilone, ixazomib, lanreotide, lansoprazole, lapatinib, larotrectinib, lasocholine, lenalidomide, lenvatinib, lenograstim, lentinan, letrozole, leuprorelin, levamisole, levonorgestrel, levothyroxine sodium, lisuride, lobaplatin, lomustine, lonidamine, lorlatinib, lurbinectedin, luspatercept, lutetium Lu 177 dotatate, masoprocol, medroxyprogesterone, megestrol, melarsoprol, melphalan, mepitiostane, mercaptamine, mercaptopurine, mesna, methadone, methotrexate, methoxsalen, methylaminolevulinate, methylprednisolone, methyltestosterone, metirosine, midostaurin, mifamurtide, mifepristone, miltefosine, miriplatin, mitobronitol, mitoguazone, mitolactol, mitomycin, mitotane, mitoxantrone, mogamulizumab, molgramostim, mometasone, mopidamol, morphine hydrochloride, morphine sulfate, mvasi, moxetumomab pasudotox, nabilone, nabiximols, nafarelin, naloxone +
pentazocine, naltrexone, nartograstim, natalizumab, necitumumab, nedaplatin, nelarabine, neratinib, neridronic acid, netupitant/palonosetron, nivolumab, pentetreotide, nilotinib, nilutamide, nimorazole, nimotuzumab, nimustine, nintedanib, niraparib, nitracrine, nivolumab, obinutuzumab, octreotide, ofatumumab, olaparib, olaratumab, olmutinib, omacetaxine mepesuccinate, omalizumab, omeprazole, ondansetron, oprelvekin, orelabrutinib, orgotein, orilotimod, osimertinib, oxaliplatin, oxycodone, oxymethoIone, ozogamicine, p53 gene therapy, paclitaxel, padeliporfin, palbociclib, palifermin, palladium-103 seed, palonosetron, pamidronic acid, panitumumab, panobinostat, pantoprazole, pazopanib, pegaspargase, PEG-epoetin beta (methoxy PEG-epoetin beta), pembrolizumab, pemigatinib, pegfilgrastim, peginterferon alfa-2b, pembrolizumab, pemetrexed, pentazocine, pentostatin, peplomycin, Perflubutane, perfosfamide, Pertuzumab, phenoxybenzamine, picibanil, pilocarpine, pirarubicin, pixantrone, plerixafor, plicamycin, poliglusam, polatuzumab vedotin, polyestradiol phosphate, polyvinylpyrrolidone + sodium hyaluronate, polysaccharide-K, pomalidomide, ponatinib, porfimer sodium, pralatrexate, pralsetinib, prednimustine, prednisone, procarbazine, procodazole, propranolol, quinagolide, quizartinib, rabeprazole, racotumomab, radium-223 chloride, radotinib, raloxifene, raltitrexed, ramosetron, ramucirumab, ranimustine, rasburicase, razoxane, refametinib, regorafenib, relugolix, ribociclib, ripretinib, risedronic acid, rhenium-186 etidronate, rituximab, rivoceranib, rolapitant, romidepsin, romiplostim, romurtide, rucaparib, sacituzumab govitecan, samarium (153Sm) lexidronam, sargramostim, sarilumab, satumomab, secretin, selinexor, selpercatinib, selumetinib, siltuximab, sipuleucel-T, sirolimus, sizofiran, sobuzoxane, sodium glycididazole, sonidegib, sophoridine hydrochloride, sorafenib, stanozolol, streptozocin, Strontium 89, sunitinib, surufatinib, tagraxofusp, talaporfin, talazoparib, talimogene laherparepvec, tamibarotene, tamoxifen, tapentadol, tasonermin, tazemetostat, teceleukin, technetium (99mTc) nofetumomab merpentan, 99mTc-HYNIC-[Tyr3]-octreotide, tegafur, tegafur + gimeracil + oteracil, temoporfin, temozolomide, temsirolimus, teniposide, testosterone, tetrofosmin, thalidomide, thiotepa, thrombopoietin, thymalfasin, thyrotropin alfa, tioguanine, tirabrutinib, tisagenlecleucel, tislelizumab, tivozanib, tocilizumab, topotecan, toremifene, tositumomab, trabectedin, trametinib, tramadol, trastuzumab, trastuzumab emtansine, treosulfan, tretinoin, trifluridine + tipiracil, trilostane, triptorelin, trametinib, trofosfamide, tryptophan, tucatinib, tucidinostat, recombinant
tumor necrosis a-factor of thymosine-al, ubenimex, ulipristal, umbralisib, valatinib, valrubicin, vandetanib, vapreotide, vemurafenib, venetoclax, vinblastine, vincristine, vindesine, vinflunine, vinorelbine, vismodegib, vorinostat, vorozole, yttrium-90 glass microspheres, zanubrutinib, zinostatin, zinostatin stimalamer, zoledronic acid, zorubicin.
13. Use of a compound according to any one of claims I to 5 for the treatment or prophylaxis of a disease.
14. Use of a compound according to any one of claims 1 to 5 for the preparation of a medicament for the treatment or prophylaxis of a disease.
15. Use according to claim 13 or 14 wherein the disease is a neoplastic or abnormal cell proliferative disorder, such as cancer, a condition with dysregulated immune response or another disorder associated with aberrant Map4Kl signaling.
16. A compound of general formula (II):
 in which Y represents a leaving group, or a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof.
in which Y represents a leaving group, or a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof.
17. A compound of general formula (II a):
 a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof.
a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof.
18. A compound of general formula (III):
 a stereoisomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof.
a stereoisomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof.
19. Use of a compound of general formula (II)
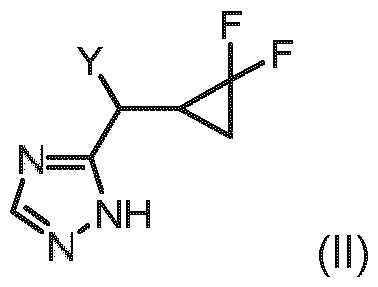 a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof in which Y a leaving group, for the preparation of a compound of formula (I) of claim 1.
a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof in which Y a leaving group, for the preparation of a compound of formula (I) of claim 1.
20. Use of a compound of general formula (II)
 a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof for the preparation of a compound of formula (I) of claim 1.
a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof for the preparation of a compound of formula (I) of claim 1.
21. Use of a compound of general formula (III)
 a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof, for the preparation of a compound of formula (I) of claim 1.
a stereoisomer, a tautomer, a salt, in particular the hydrochloride, a solvate, a hydrate or a mixture thereof, for the preparation of a compound of formula (I) of claim 1.
22. Method for controlling cancer in humans and animals by administering an antiproliferative effective amount of a compound according to any one of claims 1 to 5, or of a pharmaceutical composition according to claim 11 or 12.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP23188157 | 2023-07-27 | ||
| EP23188157.4 | 2023-07-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025021997A1 true WO2025021997A1 (en) | 2025-01-30 |
Family
ID=87517409
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2024/071341 Pending WO2025021997A1 (en) | 2023-07-27 | 2024-07-26 | New map4k1 inhibitors |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2025021997A1 (en) |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012112363A1 (en) | 2011-02-14 | 2012-08-23 | Merck Sharp & Dohme Corp. | Cathepsin cysteine protease inhibitors |
| WO2014111496A1 (en) | 2013-01-18 | 2014-07-24 | F. Hoffmann-La Roche Ag | 3-substituted pyrazoles and use as dlk inhibitors |
| WO2014153208A1 (en) | 2013-03-14 | 2014-09-25 | Epizyme, Inc. | Arginine methyltransferase inhibitors and uses thereof |
| WO2015091889A1 (en) | 2013-12-20 | 2015-06-25 | F. Hoffmann-La Roche Ag | Pyrazole derivatives and uses thereof as inhibitors of dlk |
| WO2017136699A1 (en) | 2016-02-05 | 2017-08-10 | Epizyme, Inc | Arginine methyltransferase inhibitors and uses thereof |
| WO2018107072A1 (en) | 2016-12-08 | 2018-06-14 | Board Of Regents, The University Of Texas System | Bicyclo[1.1.1]pentane inhibitors of dual leucine zipper (dlk) kinase for the treatment of disease |
| WO2021074279A1 (en) | 2019-10-16 | 2021-04-22 | Bayer Aktiengesellschaft | Spiro-fused tricyclic map4k1 inhibitors |
| WO2022147246A1 (en) | 2020-12-30 | 2022-07-07 | Tyra Biosciences, Inc. | Indazole compounds as kinase inhibitors |
| WO2022167627A1 (en) | 2021-02-05 | 2022-08-11 | Bayer Aktiengesellschaft | Map4k1 inhibitors |
| WO2022167617A1 (en) | 2021-02-05 | 2022-08-11 | Synhelix | Improved thermocycled multistep reactions device |
-
2024
- 2024-07-26 WO PCT/EP2024/071341 patent/WO2025021997A1/en active Pending
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012112363A1 (en) | 2011-02-14 | 2012-08-23 | Merck Sharp & Dohme Corp. | Cathepsin cysteine protease inhibitors |
| WO2014111496A1 (en) | 2013-01-18 | 2014-07-24 | F. Hoffmann-La Roche Ag | 3-substituted pyrazoles and use as dlk inhibitors |
| WO2014153208A1 (en) | 2013-03-14 | 2014-09-25 | Epizyme, Inc. | Arginine methyltransferase inhibitors and uses thereof |
| WO2015091889A1 (en) | 2013-12-20 | 2015-06-25 | F. Hoffmann-La Roche Ag | Pyrazole derivatives and uses thereof as inhibitors of dlk |
| WO2017136699A1 (en) | 2016-02-05 | 2017-08-10 | Epizyme, Inc | Arginine methyltransferase inhibitors and uses thereof |
| WO2018107072A1 (en) | 2016-12-08 | 2018-06-14 | Board Of Regents, The University Of Texas System | Bicyclo[1.1.1]pentane inhibitors of dual leucine zipper (dlk) kinase for the treatment of disease |
| WO2021074279A1 (en) | 2019-10-16 | 2021-04-22 | Bayer Aktiengesellschaft | Spiro-fused tricyclic map4k1 inhibitors |
| WO2022147246A1 (en) | 2020-12-30 | 2022-07-07 | Tyra Biosciences, Inc. | Indazole compounds as kinase inhibitors |
| WO2022167627A1 (en) | 2021-02-05 | 2022-08-11 | Bayer Aktiengesellschaft | Map4k1 inhibitors |
| WO2022167617A1 (en) | 2021-02-05 | 2022-08-11 | Synhelix | Improved thermocycled multistep reactions device |
Non-Patent Citations (61)
| Title |
|---|
| "Citation of NMR Peaklist Data within Patent Applications", RESEARCH DISCLOSURE, no. 605005, 1 August 2014 (2014-08-01), Retrieved from the Internet <URL:http://www.researchdisclosure.com/searching-disclosures> |
| "Comprehensive Organic Name Reactions and Reagents", 2009, JOHN WILEY |
| "Isotopic Compositions of the Elements 1997", PURE APPL. CHEM., vol. 70, no. 1, 1998, pages 217 - 235 |
| "IUPAC Rules Section E", PURE APPL CHEM, vol. 45, 1976, pages 11 - 30 |
| A. E. GOETZ ET AL., ORG. PROCESS RES. DEV., vol. 26, 2022, pages 683 - 697 |
| A. E. MUTLIB ET AL., TOXICOL. APPL. PHARMACOL., vol. 169, 2000, pages 102 |
| A. M. SHARMA ET AL., CHEM. RES. TOXICOL., vol. 26, 2013, pages 410 |
| A. ZAPF: "Transition Metals for Organic Synthesis", article "Coupling of Aryl and Alkyl Halides with Organoboron Reagents (Suzuki Reaction" |
| AIELLO ET AL., NEW ENGL. J. MED., vol. 331, 1994, pages 1480 |
| ALZABIN ET AL., CANCER IMMUNOL IMMUNOTHER, vol. 59, no. 3, March 2010 (2010-03-01), pages 419 - 29 |
| ALZABIN ET AL., J IMMUNOL., vol. 182, no. 10, 15 May 2009 (2009-05-15), pages 6187 - 94 |
| B. SCHEIPER ET AL., J. ORG. CHEM., vol. 69, 2004, pages 3943 - 3949 |
| B. TESTA ET AL., INT. J. PHARM., vol. 19, no. 3, 1984, pages 271 |
| BOOMER ET AL., J CELL BIOCHEM., vol. 95, no. 1, 1 May 2005 (2005-05-01), pages 34 - 44 |
| C. BORDONI, D.J. BROUGH, G. DAVISON, J.H. HUNTER, J.D. LOPEZ-FERNANDEZ, K. MCADAM, D.C. MILLER, P.A. MORESE, A. PAPAIOANNOU, M. UG: "The Medicinal Chemist's Guide to Solving ADMET Challenges", 2021, RSC, article "Cardiac Ion Channel Inhibition" |
| C. J. WENTHUR ET AL., J. MED. CHEM., vol. 56, 2013, pages 5208 |
| C. L. PERRIN ET AL., J. AM. CHEM. SOC., vol. 127, 2005, pages 11896 - 11897 |
| C. L. PERRIN ET AL., J. AM. CHEM. SOC., vol. 129, 2007, pages 15734 - 15735 |
| D.A. SMITHK. BEAUMONTT.S. MAURERL.DI: "Clearance in Derug Design", J. MED. CHEM., vol. 62, 2019, pages 2245 - 2255 |
| DI BARTOLO ET AL., J EXP MED., vol. 204, no. 3, 19 March 2007 (2007-03-19), pages 681 - 91 |
| E. H. KERNSL. DI: "Drug-like Properties: Concepts, Structure Design and Methods", 2008, ACADEMIC PRESS, article "Chapter 8 - Permeability, Chapter 9 - Transporters, Chapter 26 - Permeability Methods, Chapter 26 - Transporter Methods", pages: 197 - 121,287-310 |
| E.J. COREYL. KÜRTI: "Enantioselective Chemical Synthesis. Methods, Logic and Practice", 2013, ELSEVIER, article "Enantioselective Chemical Synthesis" |
| F. MALTAIS ET AL., J. MED. CHEM., vol. 52, 2009, pages 7993 |
| F. SCHNEIDER ET AL., ARZNEIM. FORSCH. / DRUG. RES., vol. 56, 2006, pages 295 |
| H. TOKUYAMA ET AL., TETRAHEDRON LETT., vol. 39, 1998, pages 3189 - 3192 |
| HU ET AL., GENES DEV, vol. 10, no. 18, 15 September 1996 (1996-09-15), pages 2251 - 64 |
| I.D. LINNEYK. NEELU, EXPERT OPINION ON THERAPEUTIC PATENTS, vol. 31, 2021, pages 893 - 910 |
| J. CLAYDENN. GREEVESS. WARREN: "Organic Chemistry", 2012, OXFORD UNIVERSITY PRESS |
| J.C. DICESARE ET AL., SYNTH. COMMUN., vol. 35, 2005, pages 663 - 668 |
| JOHN HARTWIG: "Organotransition Metal Chemistry: From Bonding to Catalysis", 2010, UNIVERSITY SCIENCE BOOKS, article "Transition Metal-Catalyzed Coupling Reactions" |
| L. ZHOU ET AL., EUR. J. MED. CHEM., vol. 244, 2022, pages 114819 |
| LASSERRE ET AL., J CELL BIOL., vol. 195, no. 5, 28 November 2011 (2011-11-28), pages 839 - 53 |
| LING ET AL., J BIOL CHEM., vol. 276, no. 22, 1 June 2001 (2001-06-01) |
| LIOU ET AL., IMMUNITY, vol. 12, no. 4, April 2000 (2000-04-01), pages 399 - 408 |
| LOPEZ ET AL., INVEST. OPTHTHALMOL. VIS. SCI., vol. 37, 1996, pages 855 |
| MINICK, D.J., FRENZ, J.H., PATRICK, M.A., BRENT, D.A.: "A comprehensive method for determining hydrophobicity constants by reversed-phase high performance liquid chromatography", JOURNAL OF MEDICINAL CHEMISTRY, vol. 31, 1988, pages 1923 - 1933, XP001179439, DOI: 10.1021/jm00118a010 |
| N. U. DIN RESHI, ACS CATAL., vol. 11, 2021, pages 13809 - 13837 |
| ONOFREY TH.KAZAN, G.BARBAGALLO, C.BLODGETT, J.WEISS, A.: "Automated Screening of Aqueous Compound Solubility in Drug Discovery", 31 July 2019, MILLIPORE CORPORATION |
| P. BARANCZEWSKI: "Introduction to in vitro estimation of metabolic stability and drug interactions of new chemical entities in drug discovery and development", PHARMACOL REP, vol. 58, no. 4, 2006, pages 453 - 472 |
| P.N. SCHELLER ET AL., CHEMCATCHEM, vol. 7, 2015, pages 3239 - 3242 |
| P.V. RAMACHANDRAN ET AL., J. ORG. CHEM., vol. 61, 1996, pages 95 - 99 |
| PEER ET AL., LAB. INVEST., vol. 72, 1995, pages 638 |
| Q. ZHU ET AL., J. MED. CHEM., vol. 65, 2022, pages 8065 - 8090 |
| RESEARCH DISCLOSURE, no. 605005 |
| S. M. BERGE ET AL.: "Pharmaceutical Salts", J. PHARM. SCI., vol. 66, 1977, pages 1 - 19, XP002675560, DOI: 10.1002/jps.2600660104 |
| S. OHTA ET AL., CHEM. PHARM. BULL., vol. 41, 1993, pages 1226 - 1231 |
| S. PATEL ET AL., J. MED. CHEM., vol. 60, 2017, pages 8083 - 8102 |
| S. SATO ET AL., TETRAHEDRON, vol. 60, 2004, pages 7899 - 7906 |
| SAUER ET AL., J BIOL CHEM., vol. 276, no. 48, 30 November 2001 (2001-11-30), pages 45207 - 16 |
| SAWASDIKOSOL ET AL., J BIOL CHEM., vol. 282, no. 48, 30 November 2007 (2007-11-30), pages 34693 - 9 |
| SHUI ET AL., NAT IMMUNOL, vol. 8, no. 1, January 2007 (2007-01-01), pages 84 - 91 |
| SMITH DADI LKERNS EH: "The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery", NAT REV DRUG DISCOV, vol. 9, no. 12, 2010, pages 929 - 39, XP055438001 |
| T. HANAMOTO ET AL., J. ORG. CHEM., vol. 55, 1990, pages 4969 - 4971 |
| T. IRREGANGR. KEMPE, CHEM. REV., vol. 120, 2020, pages 9583 - 9674 |
| T. MATSUDAR. YAMANAKAK. NAKAMURA: "Enzymatic Asymmetric Reduction of Carbonyl Compounds", GREEN BIOCATALYSIS, 2016 |
| T.W. GREENEP.G.M. WUTS: "Protective Groups in Organic Synthesis", 2006, WILEY |
| TH. NETSCHER: "Recent Research Developments in Organic Chemistry", vol. 7, 2003, pages: 71 - 83 |
| TSUJI ET AL., J EXP MED., vol. 194, no. 4, 20 August 2001 (2001-08-20), pages 529 - 39 |
| X. TAN ET AL., ORG. LETT., vol. 22, 2020, pages 7230 - 7233 |
| X.-F. WU ET AL., ANGEW. CHEM. INT. ED., vol. 49, 2010, pages 9047 - 9050 |
| Y. TIAN ET AL., ORG. CHEM. FRONT., vol. 8, 2021, pages 2328 - 2342 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP4139286B1 (en) | Substituted aminothiazoles as dgkzeta inhibitors for immune activation | |
| WO2018024602A1 (en) | 2,7-diazaspiro[4.4]nonanes | |
| WO2017207387A1 (en) | Spiro condensed azetidine derivatives as inhibitors of the menin-mml1 interaction | |
| TWI810220B (en) | Substituted macrocyclic indole derivatives | |
| WO2021105116A1 (en) | Substituted aminoquinolones as dgkalpha inhibitors for immune activation | |
| TW201840549A (en) | 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamide | |
| WO2025016899A1 (en) | Spirocyclic compounds for the treatment of cancer | |
| EP3710456B1 (en) | Macrocyclic indole derivatives | |
| US11319324B2 (en) | Pyrazolo-pyrrolo-pyrimidine-dione derivatives as P2X3 inhibitors | |
| EP3638670B1 (en) | Substituted pyrrolopyridine-derivatives as map4k1 modulators for the treatment of cancer diseases | |
| EP3774769A1 (en) | 4-(3-amino-6-fluoro-1h-indazol-5-yl)-1,2,6-trimethyl-1,4-dihydropyridine-3,5-dic arbonitrile compounds for treating hyperproliferative disorders | |
| WO2018228920A1 (en) | Substituted pyrrolopyridine-derivatives | |
| CA3070013A1 (en) | Substituted pyrrolopyridine-derivatives | |
| TW202126655A (en) | [1,2,4]triazolo[1,5-c]quinazolin-5-amines | |
| CA3074381A1 (en) | Substituted pyrrolopyridine-derivatives | |
| AU2021231312A1 (en) | Imidazotriazines acting on cancer via inhibition of CDK12 | |
| AU2020399184A1 (en) | Pyrazolotriazines | |
| EP4114837A1 (en) | Pyrazolopyrazines acting on cancers via inhibition of cdk12 | |
| WO2024003259A1 (en) | Tead inhibitors | |
| AU2023207765A1 (en) | Bicyclic triazine derivatives for the treatment of cancer | |
| WO2025021997A1 (en) | New map4k1 inhibitors | |
| WO2017207534A1 (en) | Substituted heteroarylbenzimidazole compounds | |
| WO2020048831A1 (en) | 5-aryl-3,9-diazaspiro[5.5]undecan-2-one compounds | |
| WO2020048828A1 (en) | 5-heteroaryl-3,9-diazaspiro[5.5]undecane compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24745773 Country of ref document: EP Kind code of ref document: A1 |