WO2025021712A1 - Immunogenic composition - Google Patents
Immunogenic composition Download PDFInfo
- Publication number
- WO2025021712A1 WO2025021712A1 PCT/EP2024/070618 EP2024070618W WO2025021712A1 WO 2025021712 A1 WO2025021712 A1 WO 2025021712A1 EP 2024070618 W EP2024070618 W EP 2024070618W WO 2025021712 A1 WO2025021712 A1 WO 2025021712A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- antigen
- gmma
- immunogenic composition
- paratyphi
- typhimurium
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/02—Bacterial antigens
- A61K39/025—Enterobacteriales, e.g. Enterobacter
- A61K39/0275—Salmonella
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/64—Drug-peptide, drug-protein or drug-polyamino acid conjugates, i.e. the modifying agent being a peptide, protein or polyamino acid which is covalently bonded or complexed to a therapeutically active agent
- A61K47/6415—Toxins or lectins, e.g. clostridial toxins or Pseudomonas exotoxins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/64—Drug-peptide, drug-protein or drug-polyamino acid conjugates, i.e. the modifying agent being a peptide, protein or polyamino acid which is covalently bonded or complexed to a therapeutically active agent
- A61K47/646—Drug-peptide, drug-protein or drug-polyamino acid conjugates, i.e. the modifying agent being a peptide, protein or polyamino acid which is covalently bonded or complexed to a therapeutically active agent the entire peptide or protein drug conjugate elicits an immune response, e.g. conjugate vaccines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/12—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from bacteria
- C07K16/1203—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from bacteria from Gram-negative bacteria
- C07K16/1228—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from bacteria from Gram-negative bacteria from Enterobacteriaceae (F), e.g. Citrobacter, Serratia, Proteus, Providencia, Morganella, Yersinia
- C07K16/1235—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from bacteria from Gram-negative bacteria from Enterobacteriaceae (F), e.g. Citrobacter, Serratia, Proteus, Providencia, Morganella, Yersinia from Salmonella (G)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55505—Inorganic adjuvants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/60—Medicinal preparations containing antigens or antibodies characteristics by the carrier linked to the antigen
- A61K2039/6031—Proteins
- A61K2039/6037—Bacterial toxins, e.g. diphteria toxoid [DT], tetanus toxoid [TT]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/70—Multivalent vaccine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/76—Antagonist effect on antigen, e.g. neutralization or inhibition of binding
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- the present invention relates to immunogenic compositions comprising antigens from Salmonella enterica serovar Typhimurium (S. Typhimurium), Salmonella enterica serovar Enteritidis (S. Entcritidis), and Salmonella enterica serovar Typhi (S. Typhi).
- the present invention further relates to methods and uses of compositions comprising GMMA for boosting an immune response to an S. Typhi antigen, vaccines comprising the immunogenic compositions and methods and uses of the immunogenic compositions.
- Typhoid fever is a bacterial disease caused by Salmonella enterica subspecies enterica serovar Typhi (Salmonella Typhi or S. Typhi), a human host-restricted organism [Crump, 2019]. The disease occurs globally, affecting predominantly children and young adults but is endemic in the developing countries of Africa and Asia, while in the developed countries it is reported occasionally in travellers that recently returned from endemic countries [Smith, 2016]. The exact burden of typhoid fever is said to be grossly underestimated due to difficulties in establishing its diagnosis in endemic areas. In 2017, there were estimated 10.9 million cases of typhoid fever and 116.8 thousand deaths due to Salmonella Typhi.
- MDR multidrugresistant
- Salmonella first identified in 1980 and defined as strains resistant to ampicillin, chloramphenicol and trimethoprim sulfamethoxazole.
- the emergence of resistant strains of the bacteria has been somewhat overcome with newer antimicrobials, but the challenge remains and hampers effective control of the disease [Radhakrishnan, 2018].
- XDR Extremely Drug Resistant
- S. Paratyphi A resides in the human gut and its clinical manifestations are indistinguishable from Typhoid fever.
- S. Paratyphi A is ranked second as a causative agent of enteric fever, preceded only by Salmonella enterica serovar Typhi (5. Typhi).
- Enteric fever caused by S. Paratyphi A, or Paratyphoid fever was thought to be responsible for a comparatively smaller proportion of enteric fever cases.
- S. Paratyphi A since the 1980s both the incidence and relative frequency of Paratyphoid fever have risen in Nepal, Pakistan, and Thailand.
- the populous nations of India and China have reported substantial numbers of S. Paratyphi A cases.
- the present Examples demonstrate that a trivalent vaccine comprising antigens (GMMA) from S. Typhimurium, S. Enteritidis, and S. Typhi is safe and highly immunogenic. Similarly, the present Examples demonstrate that quadrivalent vaccines comprising the trivalent vaccine and an antigen from S. Paratyphi A (GMMA or an O-antigen conjugate) are also highly immunogenic, and that no antigen interference is observed between the four antigens. Furthermore, the Examples demonstrate that GMMA (from S. Enteritidis and/or S. Typhimurium) can boost the immune response against an antigen from S. Typhi (fVi conjugated to CRM197).
- an immunogenic composition comprising:
- a method of boosting an immune response to a S. Typhi or a S. Paratyphi A antigen comprising administering a composition comprising the S. Typhi antigen or the S. Paratyphi A antigen and GMMA.
- a method of preventing infection by S. Typhi or S. Paratyphi A comprising administering an immunogenic composition comprising the S. Typhi antigen or the S. Paratyphi A antigen and GMMA, wherein the GMMA boosts the immune response to the S. Typhi antigen or the S. Paratyphi A antigen.
- an immunogenic composition comprising GMMA for use in a method of boosting an immune response to a S. Typhi or S. Paratyphi A antigen, wherein the method comprises administering an immunogenic composition comprising the S. Typhi antigen or the S. Paratyphi A antigen and GMMA.
- an immunogenic composition for use in a method of preventing infection by S. Typhi or S. Paratyphi A comprising administering an immunogenic composition comprising the S. Typhi antigen or the S. Paratyphi A antigen and GMMA, wherein the GMMA boosts the immune response to the S. Typhi or the S. Paratyphi A antigen.
- a vaccine comprising the immunogenic composition of the invention.
- a method of preventing an infection comprising administering an effective amount of the immunogenic composition or vaccine of the invention.
- an eighth aspect of the invention there is provided a use of the immunogenic composition or vaccine of the invention, for the manufacture of a medicament for use in a method of preventing an infection.
- Figure 1 iNTS-TCV vaccine induces specific serum IgG responses against the target antigens and antibodies are bactericidal in mice.
- Study design iNTS-TCV drug product; 8 mice/group; Immunization IP: days 0, 28; Bleeds: days 27, 42; Toxicology lot at time zero.
- FIG. 4 Reaction scheme for conjugation of S. Paratyphi A O-antigen to CRM197 by a random CDAP chemistry approach.
- Figure 5. Both Pan-Salmonella formulations induce specific serum IgG responses against the 4 antigens and antibodies are bactericidal in mice.
- Figures 5(a)-(d) show IgG responses at: one day before immunisation (left-hand column), 27 days after immunisation (middle column) and 42 days after immunisation (right-hand column).
- Figures 5(e)-(g) show SBA results.
- the left-hand column for each of O:2-CRM197 and ParA GMMA
- the right-hand column for each of O:2-CRM197 and ParA GMMA
- each bar is for 42 days after immunisation.
- Figures 6(a), (c), (e) and (g) show IgG responses at: one day before immunisation (left-hand column), 27 days after immunisation (middle column) and 42 days after immunisation (right-hand column).
- Figures 6(b), (d) and (f) show SBA results.
- the left-hand column is one day before immunisation and the right-hand column is 42 days after immunisation.
- each bar is for 42 days after immunisation.
- FIG. 7 Subclasses relative abundance %, calculated as subclass/subclasses total %. Top segment is IgG3, next segment is IgG2b, next segment is IgG2a and bottom segment is IgGl.
- Figures 8(a)-(d) show IgG responses at: one day before immunisation (left-hand column), 27 days after immunisation (middle column) and 42 days after immunisation (right-hand column).
- Figures 8(e)-(g) show SBA results, where the left-hand column (for each of 0:2- CRM197 and ParA GMMA) is one day before immunisation and the right-hand column (for each of O:2-CRM197 and ParA GMMA) is 42 days after immunisation.
- FIG 9 SBA on heterologous panel of mice sera elicited by monovalent components (STm or SEn GMMA) ( Figures 9(a) and (b)). SBA on heterologous panel of mice sera elicited by bivalent vaccine (STm and SEn GMMA) ( Figure 9(c)) and trivalent vaccine iNTS-TCV (STm and SEn GMMA and fVi polysaccharide from S. Typhi ( Figure 9(d)).
- FIG. 10 Quadrivalent Pan-Salmonella formulations elicit bactericidal antibodies against a broad panel of Salmonella strains. Panel includes invasive STm isolates from Africa and Southeast Asia and S. enterica serovars other than STm, SEn, ParA, Typhi.
- the term “comprising” is intended to mean including but not limited to.
- the phrase “An immunogenic composition comprising a Salmonella Typhimurium antigen” should be interpreted to mean that the immunogenic composition comprises a Salmonella Typhimurium antigen, but the immunogenic composition may comprise further components.
- the word “comprising” is replaced with the phrase “consisting of.
- the term “consisting of” is intended to be limiting.
- the phrase “An immunogenic composition consisting of a Salmonella Typhimurium antigen” should be understood to mean that the immunogenic composition has the Salmonella Typhimurium antigen and no further components.
- the word “comprising” is replaced with the phrase “consisting essentially of” .
- the term “consisting essentially of’ means that specific further components can be present, namely those not materially affecting the essential characteristics of the subject matter.
- a value refers to that value but within a reasonable degree of scientific error.
- a value is “about x” or “around x” if it is within 10%, within 5%, or within 1% of x.
- the immunogenic composition of the invention comprises a Salmonella Typhimurium (S. Typhimurium) antigen. In some aspects, the immunogenic composition of the invention comprises a Salmonella Enteritidis (S. Enteritidis) antigen. In some aspects, the immunogenic composition of the invention comprises a Salmonella Paratyphi A (S. Paratyphi A) antigen.
- S. Typhimurium, S. Enteritidis and S. Paratyphi A antigens are known to the skilled person.
- the S. Typhimurium, S. Enteritidis and S. Paratyphi A bacteria all comprise an outer membrane comprising O-antigen
- the S. Typhimurium, S. Enteritidis and/or the S. Paratyphi A antigen may comprise the O-antigen.
- O-antigen OAg and 0:2 are considered to be interchangeable.
- the outer membrane of gram-negative bacteria comprise a lipopolysaccharide. This lipopolysaccharide comprises an O-antigen, which is linked via to the core domain to a lipid A domain.
- O-antigen OAg and 0:2 ” refer to a polysaccharide made up of the O-antigen alone, or more preferably the O- antigen linked to core domain of the lipopolysaccharide.
- the S. Typhimurium antigen, the S. Enteritidis antigen and/or the S. Paratyphi A may be an O-antigen.
- a typical process for the purification of these O- antigens is based on the phenol-water method of Westphal and Jann, first described in the 1960s (Westphal and Jann (1965) Methods Carbohydr. Chem. 5:83-91), followed by detoxification of the lipopolysaccharide with acetic acid or anhydrous hydrazine.
- the O- antigen is modified to remove the lipid A.
- extraction and purification of polysaccharide can be performed by acetic acid hydrolysis as described in for example Watson et al., (1992) Infect Immun.
- the O-antigens of Salmonella serogroups A, B and D have been described and are thought to share a common backbone: - ⁇ 2-a-D-Man/?-(l - ⁇ 4)-a-L-Rha/?-(l— >3)-a-D-Gal/?-(l— >.
- the serogroup specificity of Salmonella Paratyphi A is conferred by an a-3,6- dideoxyglucose (a-D-paratose) linked (1— 3) to the mannose of the backbone.
- the a-L- rhamnose of the backbone is partially O-acetylated at C-3 (Konadu et al. (1996) Infect Immun. (7):2709-15).
- the published structures of the O-antigen from S. Paratyphi A is shown in Figure 11, including the KDO subunit and primary amine group (within a pyrophosphoethanolamine group) in the core domain.
- the O-antigen from S. Typhimurium, S. Enteritidis and/or the S. Paratyphi A may be part of a conjugate.
- conjugate refers to a molecule formed by a covalent linkage between an antigen (like O-antigen) and a carrier.
- the carrier may be a carrier protein.
- conjugation of polysaccharides to carrier proteins enhances the immunogenicity of the polysaccharides as it converts them from T- independent antigens to T-dependent antigens, thus allowing priming for immunological memory.
- Carrier proteins include bacterial toxins, such as diphtheria or tetanus toxins, or toxoids or mutants thereof.
- the carrier protein is CRM197.
- the sequence of CRM197 is provided in Figure 12 (SEQ ID NO: 1).
- the S. Paratyphi A antigen comprises S. Paratyphi A O-antigen conjugated to a carrier protein.
- the carrier protein may be diphtheria toxoid or CRM197.
- the carrier protein is CRM197.
- the O-antigen from S. Typhimurium, S. Enteritidis and/or the S. Paratyphi A may be conjugated to the carrier protein by a method that comprises introducing more than one activated site into the O-antigen.
- the O-antigen from S. Typhimurium, S. Enteritidis and/or the S. Paratyphi A comprises more than one activated site.
- activated site is intended to refer to a site or functional group on the polysaccharide that has been activated by a step in a conjugation chemistry method such that it is primed to be conjugated to a carrier protein.
- the polysaccharide is “activated” by the addition of CDAP if the addition of CDAP introduces cyanoester groups.
- the CDAP activation introduces cyanoester groups at one or more sites, and the positions of these introduced cyanoester groups would be considered to be “activated sites Once the O- antigen has been activated, it may be linked (conjugated) to a carrier protein at one or more (in some cases all) of the activated sites.
- activated sites' includes sites that have been activated and not linked to carrier protein and also sites that have been activated and are linked to a carrier protein.
- the O-antigen from S. Typhimurium, S. Enteritidis and/or the S. Paratyphi A comprises 1.5 or more, 2.0 or more, or 2.5 or more activated sites.
- the O-antigen will comprise 1.5 or more activated sites if the average number of activated sites on each O-antigen molecule in the composition is 1.5 or more.
- the S. Paratyphi A antigen is an O-antigen conjugated to a carrier protein, and the O-antigen is conjugated to the carrier protein by a method that comprises introducing more than one activated site into the S. Paratyphi O-antigen and/or the S. Paratyphi A O- antigen comprises more than one activated site.
- the O-antigen from S. Typhimurium, S. Enteritidis and/or the S. Paratyphi A may be conjugated to the carrier protein by CDAP chemistry, optionally via a linker.
- a linker is a compound that can be used to link a protein and a polysaccharide. Any suitable linker may be used in the conjugates and methods of the invention. Suitable linkers include an adipic acid dihydrazide (ADH) linker, which is a compound having the following structure: Other suitable linkers include adipic acid, glutaric acid, carbonyl, P-propionamido (WO00/10599), adipic acid bis(N-hydroxysuccinimmide), dihydrazides analogous to ADH but with different chain lengths, hexamethylenediamine (or analogous diamines with different chain lengths), nitrophenyl-ethylamine (Gever ct al. (1979) Med. Microbiol.
- ADH adipic acid dihydrazide
- haloacyl halides U.S. Pat. No. 4,057,685
- glycosidic linkages U.S. Pat. Nos. 4,673,574; 4,761,283; and 4,808,700
- 6-aminocaproic acid U.S. Pat. No. 4,459,286
- SPDP N- succinimidyl-3-(2-pyridyldithio)-propionate
- C4 to C12 moieties U.S. Pat. No. 4,663, 160
- the O-antigen from S. Typhimurium, S. Enteritidis and/or the S. Paratyphi A is/are conjugated to the carrier protein by CDAP chemistry
- the O-antigen may be conjugated to the carrier protein by a method comprising a step of activating the O- antigen by CDAP chemistry to provide an activated O-antigen.
- Activating the O-antigen by CDAP chemistry comprises mixing the O-antigen with CDAP such a way that cyanoester groups are introduced into the polysaccharide or O-antigen.
- Example 9 discloses a suitable method of activating an O-antigen by CDAP chemistry.
- Activating the O-antigen by CDAP chemistry results in an activated O-antigen.
- Activating the O-antigen by CDAP chemistry introduces cyanoester groups, and so a method comprises a step of activating an O-antigen by CDAP chemistry if the method comprises mixing the O-antigen with CDAP and the number of cyanoester groups present on the O-antigen after the step of mixing with CDAP is higher than the number of cyanoester groups present on the O-antigen prior to that step.
- the number of cyanoester groups present may be measured using the ADH quenching/TNBS colourimetric method as reported in Lees A., Vaccines (Basel), 2020; 8(4):777.
- activating the O-antigen by CDAP chemistry comprises mixing the O-antigen with CDAP at a w/w ratio of between 0.05:1 and 5:1, between 0.1:1 and 5:1, between 0.2:1 and 2:1, or around 0.3:1 (CDAP to O-antigen).
- the step of activating the O-antigen by CDAP chemistry comprising mixing the O-antigen with CDAP takes place in a salt solution, such as a solution of NaCl or KC1.
- the step of activating the O-antigen by CDAP chemistry comprising mixing the O-antigen with CDAP takes place in a solution of NaCl or KC1 at a concentration between 50 mM and IM, between 100 mM and 250 mM, between 125 mM and 200 mM, or around 150 mM.
- the pH is adjusted, optionally to a pH between 6 and 10, between 7 and 9, or between 9 and 10.
- the pH is adjusted by adding a base, such as triethylamine, sodium hydroxide, or pyridine.
- the pH is adjusted by adding between 5 % and 15%, between 8% and 12%, or around 10% (v/v) triethylamine.
- the mixture is incubated at a temperature between 18°C and 30°C, between 20°C and 28°C, room temperature, or around 25°C.
- the solution is incubated with stirring prior to conjugation of the activated O-antigen to the carrier protein.
- O-antigens that have been activated using CDAP chemistry comprise cyanoester groups (at activated sites), and these cyanoester groups may be covalently linked to hydrazide or amino groups.
- O-antigens that have been activated using CDAP chemistry may be linked directly to carrier proteins (via amino groups), or may be conjugated to a carrier protein via a linker comprising a hydrazide or amino group.
- Suitable linkers include the ADH linker described above.
- the activated O-antigen may be conjugated to the carrier protein by a method comprising reacting the activated O-antigen with hydrazide/amino groups on a carrier protein or a carrier-protein linker compound.
- the method may further comprise steps to prepare the carrier protein-linker compound.
- the linker is an ADH linker
- the method may comprise a step of preparing an ADH-carrier protein compound (such as an ADH-CRM197 compound), for example as reported in Micoli et al. Vaccine 2011, 29, (4), 712-20.
- Reacting the activated O-antigen with hydrazide/amino groups on a carrier protein or a carrier-protein linker compound may comprise mixing the carrier protein or the carrierprotein linker compound with the activated O-antigen under conditions suitable for a covalent bond to be formed between the cyanoester groups (activated sites) on the activated O-antigen and the hydrazide/amino groups on the carrier protein or the carrier protein-linker compound. For example, it may be simply a case of mixing the activated O- antigen with the carrier protein or the carrier protein-linker compound.
- Reacting the activated O-antigen with hydrazide/amino groups on the carrier protein or a carrier protein-linker compound may comprise mixing the activated O-antigen with the carrier protein or the carrier protein-linker compound at a w/w ratio of between 0.1:1 and 5:1, between 0.2:1 and 3:1, between 0.5:1 and 2:1, or around 1 : 1 (O-antigen to carrier protein or carrier protein-linker).
- the step of mixing the activated O-antigen with the carrier protein or the carrier protein-linker provides a conjugation mixture.
- mixing the activated O-antigen with the carrier protein or the carrier proteinlinker compound takes place at a pH between 8 and 11, between 9 and 10, or around 9.5.
- the pH is maintained at between 8 and 11, between 9 and 10, or around 9.5 for at least 1 hour, at least 2 hours, between 30 minutes and 10 hours, between 1 hour and 5 hours, or between 2 hours and 3 hours.
- the pH is maintained using a base, such as triethylamine, sodium hydroxide, or pyridine.
- the pH is maintained using triethylamine.
- the method may further comprise a step of adding glycine solution (to quench the cyanoester groups).
- the glycine solution is adding in a concentration of 0.5M to 5M, 0.5M to 2M, or around IM.
- the glycine solution is added to a volume of the conjugation mixture with is substantially equal to the volume of the glycine solution. A volume is substantially equal to another volume if it is within 10%.
- this further step occurs after a step of adding a glycine solution.
- the pH is adjusted to a pH between 7 and 9, or around 8.
- the incubation step comprises incubating at a temperature below 15°C, between 12°C, below 10°C, between 0°C and 10°C, or between 2°C and 8°C.
- the incubation step takes place for between 10 and 30 hours, or between 10 and 20 hours.
- the method may further comprise a chromatography step to remove any unconjugated O-antigen.
- the chromatography step comprises hydrophobic interaction chromatography or anion exchange chromatography.
- the S. Typhimurium, S. Enteritidis and/or the S. Paratyphi A antigen comprises or consists of outer membrane vesicles such as GMMA.
- OMVs include native OMVs.
- Gram-negative bacteria can spontaneously release outer membrane vesicles (OMVs) during growth due to the turgor pressure of the cell envelope, and these are native OMVs.
- OMVs are rich in immunogenic cell surface- associated, periplasmic and secreted antigens and have been used as vaccines.
- OMVs of the invention include Generalised Modules for Membrane Antigens (GMMA), native OMVs (‘NOMVs’ (see Katial et al. 2002, Infect Immun, 70: 702-707), microvesicles (MVs (see WO 02/09643)), detergent-extracted OMVs (DOMVs), mutant- derived OMVs (m-OMV), and blebs, which are outer-membrane protrusions that remain atached to bacteria prior to release as MVs (see Beveridge, 1999, J. Bacteriol. 181: 4725- 4733)).
- GMMA Generalised Modules for Membrane Antigens
- NOMVs native OMVs
- MVs microvesicles
- DOMVs detergent-extracted OMVs
- m-OMV mutant- derived OMVs
- blebs which are outer-membrane protrusions that remain atached to bacteria prior to release as MVs (see Beveridge, 1999, J
- GMMA Generalised Modules for Membrane Antigens
- NOMV native outer membrane vesicles
- the membrane structure has been modified by the deletion of genes encoding key structural components, such as tolR (leading to hyperblebbing).
- GMMA large quantities of outer membrane “bud off' (or “hyperbleb”) to provide a practical source of membrane material for vaccine production.
- GA/M4 refers to OMVs which are released spontaneously from bacteria modified to hyperbleb (such as Salmonella bacteria which are modified such that they do not comprise a gene encoding functional TolR).
- the Gram-negative bacteria from which the OMVs (such as GMMA) of the invention are purified may be one or more of the group consisting of: Salmonella enterica subspecies enterica serovar Typhimurium (Salmonella Typhimurium), Salmonella enterica subspecies enterica serovar Enteritidis (Salmonella Enteritidis), and Salmonella enterica subspecies enterica serovar Paratyphi A (Salmonella Paratyphi A).
- Suitable purification methods are known in the art, and include a variety of filtration and chromatography methods. A preferred two-step filtration purification process is described in WO 2011/036562 herein incorporated by reference.
- the S. Typhimurium, S. Enteritidis and/or the S. Paratyphi A antigen comprises or consists of GMMA, i.e. S. Typhimurium, S. Enteritidis and/or S. Paratyphi A GMMA.
- the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A GMMA may comprise modified lipid A or may be derived from S. Typhimurium, S. Enteritidis and/or S.
- a modified lipid A is a lipid A that has a different structure compared to a corresponding wild type lipid A.
- the structure of lipid A may be determined using MALDI-TOF analysis of lipid A isolated from the GMMA. For the assay, the lipid A is separated after treatment of GMMA with acetic acid and then assayed by MALDI-TOF.
- GMMA with a protein concentration of about 1 mg/mL (micro BCA calibration curve) or a cell bank suspension with an OD600 of about 3 (4 mL sample) are treated with 1% acetic acid (final concentration) for 2 or 6 hours, respectively, at 100°C to obtain a precipitate containing the lipid A.
- the precipitate is then collected, washed with water and the lipid A is extracted in chloroform / methanol 4:1.
- the final solution which contains the lipid A, is mixed 1 : 1 with Super DHB (Fluka, 50862) saturated solution (acetonitrile / water 1:1).
- Two microliters of the mixture are loaded onto the target plate and after the spot is dried at room temperature, the plate is inserted in the mass spectrometer.
- the spectra (negative reflectron mode) generally show peaks corresponding to the lipid A molecular species and contain several peaks due to fragmentation of the lipid A (i.e. loss of one or more fatty acid chains), sodium adduct (+22 m/z) and lipid A dephosphorylation (- 80 m/z).
- the species of lipid A is identified by comparison of the molecular peak mass m/z to what is expected for the sample in analysis.
- the lipid A is modified to be detoxified i.e. the modified lipid A is detoxified lipid A).
- “Detoxified” means that the lipid A is less toxic than wildtype lipid A.
- the wildtype lipid A used in the comparison is a corresponding wildtype lipid A.
- “Toxicity” or “toxic” in this context refers to the extent to which the innate immune system is activated by lipid A, particularly through the Toll-like receptor 4 pathway. Highly toxic lipid A can lead to uncontrolled inflammation, apoptosis, and in extreme cases septic shock, among other effects.
- a modified lipid A is less toxic if it is less reactogenic than a corresponding wildtype lipid A.
- lipid A that can be found in the corresponding wildtype bacterium and strain.
- lipid A that is modified relative to a “corresponding wildtype lipid in the context of S. Typhimurium GMMA is interpreted to mean a lipid A that is modified (e.g. such that it is less toxic) relative to lipid A found in wildtype S. Typhimurium.
- the modified lipid A is penta-acylated lipid A.
- the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A GMMA may be derived from S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria that comprise any suitable modification that leads to production of GMMA comprising lipid A that is less toxic than wildtype lipid A.
- HtrB, MsbB and PagP are proteins that are involved in production of lipid A in Gramnegative bacteria. Of these, MsbB and PagP are important in Salmonella. Salmonella bacteria that do not express functional versions of MsbB and/or PagP will not produce native lipid A, but rather will produce modified, detoxified lipid A.
- S. Typhimurium, S. Enteritidis and/or S. Paratyphi A GMMA may be derived from S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria that do not express functional versions of MsbB and/or PagP.
- the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A GMMA are derived from S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria that do not comprise a gene encoding a functional MsbB and/or PagP protein.
- Whether or not the bacteria from which GMMA is derived express functional versions of MsbB and/or PagP or comprise a gene encoding a functional MsbB and/or PagP protein may be determined by isolating lipid A from the GMMA and analysing its structure by MALDI-TOF as described above. If the lipid A is detoxified then the bacteria from which the GMMA is derived do not express functional versions of Msb and/or PagP or comprise a gene encoding a functional MsbB and/or PagP protein.
- the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived do not comprise a gene (such as htrB, msbB, and/or pagP) encoding a functional protein because they comprise a mutation in that gene.
- the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived do not comprise a gene encoding a functional HtrB, MsbB, and/or PagP protein.
- Paratyphi A bacteria from which the GMMA are derived comprise a gene encoding at least a portion of the HtrB, MsbB, and/or PagP protein, but either the gene is mutated such that the HtrB, MsbB, and/or PagP protein encoded is missing one or more important amino acids or a portion of the gene is deleted.
- the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived may comprise a substitution or deletion mutation in the htrB, msbB, and/or pagP gene.
- a bacteria from which the GMMA are derived may have an addition mutation in the htrB, msbB and/or PagP gene, for example an addition mutation causing a frame shift.
- the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived comprises a deletion mutation in the htrB, msbB,, and/or pagP gene.
- the htrB, msbB, and/or pagP gene comprises a deletion mutation, and at least 10%, at least 20%, at least 25%, at least 50%, or at least 75% of the htrB, msbB, and/or pagP gene is deleted.
- the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived lacks a htrB, msbB, and/or pagP gene (for example because the complete htrB, msbB, and/or pagP gene has been deleted (a tshtrB, tsmsbB, and/or tSpagP mutation)).
- the S. Paratyphi A OMVs or GMMA are derived from S. Paratyphi A where at least a part of the msbB and/or pagP gene has been replaced by a different gene.
- the S. Paratyphi A OMVs or GMMA are derived from S. Paratyphi A where at least a part of the msbB and/or pagP gene has been replaced by a tetracycline (tet) or kanamycin (kan) gene respectively.
- the S. Paratyphi A OMVs or GMMA are derived from S. Paratyphi A where at least a part of the msbB and/or pagP gene has been replaced by a tetracycline (tet) or kanamycin (kan) gene respectively.
- the S. Paratyphi A OMVs or GMMA are derived from S.
- Hyperblebbing The S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived may have been modified (for example genetically modified) to hyperbleb i.e. more quantities of outer membrane “bud off' compared to a corresponding Gram-negative bacterium that does not have the genetic mutation.
- the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived may comprise any suitable modification that leads to hyperblebbing.
- the modification is a mutation, for example the S. Typhimurium, S. Enteritidis and/or S. Paratyphi
- a bacteria from which the GMMA are derived may not comprise a gene (such as tolR) encoding a functional protein because it comprises a mutation in that gene.
- the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived do not comprise a gene encoding a functional TolR protein.
- Paratyphi A bacteria from which the GMMA are derived comprise a gene encoding at least a portion of the TolR protein, but either the gene is mutated such that the TolR protein encoded is missing one or more important amino acids or a portion of the gene is deleted.
- the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived may comprise a substitution or deletion mutation in the tolR gene.
- the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived may have an addition mutation in the tolR gene, for example an addition mutation causing a frame shift.
- Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived comprises a deletion mutation in the tolR gene.
- the tolR gene comprises a deletion mutation, and at least 10%, at least 20%, at least 25%, at least 50%, or at least 75% of the tolR gene is deleted.
- the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived lacks a tolR gene (for example because the complete tolR gene has been deleted (a AtolR mutation)).
- the S. Paratyphi A OMVs or GMMA are derived from S. Paratyphi A where at least a part of the tolR gene has been replaced by a different gene.
- the S. Paratyphi A OMVs or GMMA are derived from S. Paratyphi A where at least a part of the tolR gene has been replaced by a chloramphenicol acetyltransferase (cat) gene.
- the S. Paratyphi A OMVs or GMMA are derived from S. Paratyphi A where the tolR gene has been replaced by a chloramphenicol acetyltransferase (cat) gene.
- the S. Paratyphi A OMVs or GMMA are derived from S. Paratyphi A which is tolR::cat.
- a bacteria from which the GMMA are derived to hyperbleb may be tested using the following hyperblebbing assay.
- the user should prepare two cultures of bacterium.
- the first culture should comprise the bacterium having the genetic modification to be tested (the test culture), and the second culture should comprise an equivalent bacterium which is identical but for the genetic modification to be tested (the reference culture).
- the user should grow the test culture and the reference culture under identical conditions and determine the number of outer membrane vesicles released from the bacteria in the test culture and bacteria in the reference culture.
- the genetic modification causes the bacterium to hyperbleb.
- the level of outer membrane vesicles released may be determined by O-Antigen quantification, for example according to Example 3.
- the immunogenic composition comprises S. Typhimurium GMMA derived from S. Typhimurium strain 2192 (see e.g. De Benedetto et al, 2017, Multiple Techniques for Size Determination of Generalized Modules for Membrane Antigens from Salmonella typhimurium and Salmonella enteritidis. ACS Omega. 2017 Nov 30;2(l l):8282-8289).
- the immunogenic composition comprises S. Enteritidis GMMA derived from S. Enteritidis strain 618 (see e.g. Lanzilao L, Stefanetti G, Saul A, MacLennan CA, Micoli F, Rondini S. Strain Selection for Generation of O-Antigen-Based Glycoconjugate Vaccines against Invasive Nontyphoidal Salmonella Disease. PLoS One. 2015 Oct
- the immunogenic composition comprises S. Paratyphi A GMMA derived from S. Paratyphi A strain ED 199 (see e.g. Mylona E, Sanchez-Garrido J, Hoang Thu TN, Dongol S, Karkey A, Baker S, Shenoy AR, Frankel G. Very long O- antigen chains of Salmonella Paratyphi A inhibit inflammasome activation and pyroptotic cell death. Cell Microbiol. 2021 May;23(5):el3306).
- the S. Paratyphi A strain may be tolR::cat pagP::kan msbB::tet.
- An immunogenic composition comprises S. Typhimurium GMMA derived from S. Typhimurium strain 2192, if the strain used was based on S. Typhimurium strain 2192 even if modifications to strain 2192 have been made (for example mutation of msbB, pagP or tolR genes).
- the immunogenic composition may comprise a dose of 1 to 100 pg, 1 to 50 pg, 15 to 50 pg, 20 to 30 pg, 1 to 20 pg, 1 to 10 pg, around 25 pg, or around 5 pg of S. Paratyphi A O- antigen.
- the dose of S. Paratyphi A O-antigen in a composition may be determined by mild hydrolysis of the O-antigen in the immunogenic composition (to provide the monosaccharide Paratose) and detecting the amount of Paratose by HPAEC-PAD.
- Paratose is a monosaccharide that is present in the S. Paratyphi A O-antigen and not present in O-antigen from S. Enteritidis or S.
- S. Paratyphi A O-antigen by HPAEC-PAD is set out in Example 10. If the S. Paratyphi A O-antigen is part of a conjugate (comprising a carrier protein), then the amount of the carrier protein may vary. For example, if the ratio of carrier protein to O-antigen in the conjugate is greater than 2, then the amount of carrier protein present to achieve an O-antigen dose of 1 pg will be higher than if the ratio of carrier protein to O-antigen in the conjugate is lower than 2.
- the immunogenic composition of the invention may comprise a dose (O-antigen) of between 1 pg and 50 pg, between 2 pg and 25 pg, between 2 pg and 10 pg, between 15 pg and 25 pg, around 20 pg, or around 4 pg of the S. Typhimurium antigen or S.
- a dose O-antigen of between 1 pg and 50 pg, between 2 pg and 25 pg, between 2 pg and 10 pg, between 15 pg and 25 pg, around 20 pg, or around 4 pg of the S. Typhimurium antigen or S.
- the GMMA used in the immunogenic compositions comprises O- antigen.
- the dose of GMMA may be quantified as an O-antigen dose, i.e. if the immunogenic composition comprises a Ipg (O-antigen) dose of GMMA then the immunogenic composition comprises sufficient GMMA to provide 1 pg of the O-antigen associated with that GMMA (e.g. if the GMMA is S. Typhimurium GMMA then the immunogenic composition comprises GMMA containing a total of I pg of S. Typhimurium O-antigen).
- an immunogenic composition comprises GMMA rich in O- antigen
- the actual amount of GMMA present to achieve a dose of 1 pg (O-antigen) may be lower than the amount required if the GMMA is poor in O-antigen.
- the amount of S. Typhimurium O-antigen present in an immunogenic composition may be determined by mild hydrolysis of the O-antigen in the immunogenic composition (to provide the monosaccharide Abequose) and detecting the amount of Abequose using HPAEC-PAD. Assuming that no ‘Tree” S. Typhimurium O-antigen has been added, the amount of O- antigen in an S. Typhimurium GMMA composition will correspond to the O-antigen dose of the S. Typhimurium GMMA.
- the immunogenic composition of the invention may comprise a dose (O-antigen) of between 1 pg and 50 pg, between 2 pg and 25 pg, between 2 pg and 10 pg, between 15 pg and 25 pg, around 20 pg, or around 4 pg of S. Enteritidis antigen or S. Enteritidis GMMA.
- the amount of S. Enteritidis O-antigen present in an immunogenic composition may be determined by mild hydrolyss of the O-antigen in the immunogenic composition (to provide the monosaccharide Tyvelose) and detecting the amount of Tyvelose using HPAEC-PAD. Assuming that no ‘Tree” S. Enteritidis O-antigen has been added, the amount of O-antigen in an S. Enteritidis GMMA composition will correspond to the O- antigen dose of the S. Enteritidis GMMA.
- the immunogenic composition of the invention may comprise a dose (O-antigen) of between 1 pg and 50 pg, between 2 pg and 25 pg, between 2 pg and 10 pg, between 15 pg and 25 pg, around 20 pg, or around 4 pg of S. Paratyphi A GMMA.
- the amount S. Paratyphi A O-antigen present in an immunogenic composition may be determined by mild hydrolysis of the O-antigen in the immunogenic composition (to provide the monosaccharide Paratose) and detecting the amount of Paratose using HPAEC-PAD. Assuming that no ‘Tree” S. Paratyphi A O-antigen has been added, the amount of O- antigen in an S.
- Paratyphi A GMMA composition will correspond to the O-antigen dose of the 5.
- the O-antigen/protein ratio of the S. Paratyphi A OMVs or GMMA present in an immunogenic composition may be at least 0.2, 0.3, 0.4, 0.5 or at least 0.6, typically at least 0.4.
- the O-antigen/total protein ratio may be at most 0.8, 0.9, 1.0, or 2.0.
- the O-antigen content can be quantified by HPAEC-PAD, for example as described in Example 5.
- the protein concentration is quantified by micro-BCA, for example as described in PCT/EP2022/073501.
- the immunogenic composition of the invention may comprise a dose of 1 to 100 pg, 1 to 50 pg, 15 to 50 pg, 20 to 30 pg, 1 to 20 pg, 1 to 10 pg, around 25 pg, or around 5 pg of fVi polysaccharide.
- the dose of fVi polysaccharide in a composition may be determined by hydrolysing (by acid hydrolysis) the fVi polysaccharide in the immunogenic composition and detecting the monomer sugar of the repeating unit by HPAEC-PAD.
- a suitable method for determining the amount fVi polysaccharide by HPAEC-PAD is set out in Example 5.
- the amount of the carrier protein may vary. For example, if the ratio of carrier protein to fVi polysaccharide in the conjugate is greater than 2, then the amount of carrier protein present to achieve an fVi polysaccharide dose of Ipg will be higher than if the ratio of carrier protein to fVi polysaccharide in the conjugate is lower than 2.
- the immunogenic composition may further comprise an antigen from Salmonella Typhi (S. Typhi).
- S. Typhi Salmonella Typhi
- the antigen from S. Typhi is a Vi polysaccharide.
- Fz or “Vi polysaccharide” relates to the capsular polysaccharide of Salmonella enterica serovar Typhi purified from Citrobacter (Rondini et al., J. Infect. Dev. Ctries, 2012).
- the Vi polysaccharide is a fragmented Vi polysaccharide (fVi).
- fragmented in reference to the Vi polysaccharide refers to the Vi polysaccharide having undergone size reduction thus reducing the number of repeating units in the polysaccharide. Fragmented Vi therefore has a lower average molecular weight compared to native Vi.
- fragmented Vi may comprise 30 to 300 repeating units, compared to over 600 repeating units for native Vi.
- a structure of the Vi monomeric repeating unit is shown below.
- the fragmented Vi preferably no changes in the structure of the repeating unit is observed compared to native Vi. This can be confirmed byl H NMR analysis (see WO2015/068129).
- the percentage of O-acetyl groups in the fragmented Vi is preferably the same as the native Vi (i.e. about 95% O-acetylation) but may vary and decrease to about 65% O-acetylation. O-acetylation can be determined by standard measurements such asl H NMR or the Hestrin colorimetric method.
- the IVi polysaccharide In its native size, the IVi polysaccharide has an average molecular weight measured by HPLC size exclusion chromatography (HPLC-SEC) of about 165kDa.
- the fVi polysaccharide has an average molecular weight of between 10 kDa and 90 kDa, between 25 kDa and 70 kDa, between 40 kDa and 55 kDa, between 41 kDa and 49 kDa, or between 51 kDa and 55 kDa.
- the fVi polysaccharide has a target molecular weight of between 51 kDa and 55 kDa (e.g. it has been made by a method that typically generates fVi having a molecular weight within this range).
- the molecular weight of the Vi polysaccharide may be determined by HPLC-SEC.
- the average molecular weight is calculated by running the sample on a TSK gel 3000 PWXL column, (30 cm x 7.8 mm; particle size 7 pm; cod. 808021) with a TSK gel PWXL guard column (4.0 cm x 6.0 mm; particle size 12 pm; cod. 808033) (Tosoh Bioscience) using dextrans as standards (5, 25, 50, 80, 150 kDa).
- the mobile phase is 0.1 M NaCI, 0.1 M NaH2 PO4, 5% CH3 CN, pH 7.2, at the flow rate of 0.5 mL/min (isocratic method for 30 min).
- Void and bed volume calibration is performed with X-DNA (X-DNA Molecular Weight Marker III 0.12-21.2 kb; Roche) and sodium azide (NaN3; Merck), respectively.
- Fragmented Vi polysaccharide can further be separated into pools of different average molecular weight ranges. This can be achieved by methods known in the art such as anion exchange chromatography, size exclusion chromatography, and tangential flow filtration.
- fVi polysaccharide used in the present invention have certain average molecular weight (avMW) range distributions which can be further characterized in terms of polydispersity index (PDI).
- avMW average molecular weight
- PDI polydispersity index
- the polydispersity index is calculated as shown in the equation below:
- PDI Mw / Mn where Mw is the weight average molecular weight and Mn is the number average molecular weight.
- the fVi polysaccharide may have an avMW distribution characterised in that at least 80% of the pool has an avMW between 25 kDa and 70 kDa. It may have an avMW distribution characterised in that at least 50% of the pool has an avMW between 35 kDa and 60 kDa. It may have an avMW distribution characterised in that at least 30% of the pool has an avMW between 41 kDa and 55 kDa.
- Fragmentation of the Vi polysaccharide may be carried out by a number of methods known in the art such as chemical hydrolysis of the native polysaccharide, enzymatic fragmentation of the native polysaccharide, gamma irradiation of the native polysaccharide, or mechanical methods such as sonication, or high pressure homogenizer/microfluidizer/HPCDS (High pressure cell disruption system) of the native polysaccharide.
- the fragmentation method used in the present invention is selected such that it can yield fVi polysaccharide having an avMW of less than 90kDa, less than 80 kDa, less than 60 kDa, or between 40 and 55 kDa.
- the method may also be selected such that there are no alterations to the repeating units' structure.
- fragmentation is not by mechanical methods.
- fragmentation is not by alkaline hydrolysis.
- the fVi polysaccharide may be obtained by chemical hydrolysis with hydrogen peroxide. Using this method, it was found that the Vi polysaccharide could be reduced in size without altering the repeating units' structure. Also, hydrolysis with hydrogen peroxide could enable the formation of fragmented Vi having a lower average molecular weight than when using mechanical methods.
- a suitable method for fragmenting Vi polysaccharide is set out in Example 2.
- the fVi polysaccharide may be part of an fVi conjugate comprising fVi and a carrier protein.
- the carrier protein in the fVi conjugate is tetanus toxoid, CRM197, or diphtheria toxoid.
- the carrier protein is CRM197.
- the fVi polysaccharide may be conjugated to the carrier protein via any suitable conjugation chemistry.
- Conjugation of the fVi polysaccharide to the carrier protein may be via a -NH2 group, e.g., through the side chain(s) of a lysine residue(s) or arginine residue(s) in the carrier polypeptide.
- this group can react with an amine in the protein to form a conjugate by reductive amination.
- Conjugation to the carrier may also be via a -SH group, e.g., through the side chain(s) of a cysteine residue(s) in the carrier polypeptide.
- the fVi polysaccharide may be conjugated to the carrier protein via a linker molecule.
- the fVi polysaccharide will typically be activated or functionalised prior to conjugation. Activation may involve, for example, cyanylating reagents such as CDAP (l-cyano-4- dimethylamino pyridinium tetrafluoro borate).
- cyanylating reagents such as CDAP (l-cyano-4- dimethylamino pyridinium tetrafluoro borate).
- Other suitable techniques use carbodiimides, hydrazides, active esters, norborane, p-nitrobenzoic acid, N-hydroxysuccinimide, S-NHS, EDC, TSTU (see, e.g., the introduction to WO 98/42721).
- Direct conjugation to the carrier protein may comprise oxidation of the fVi polysaccharide followed by reductive amination with the protein, as described in, for example, U.S. Pat No. 4,761,283 and U.S. Pat No. 4,356,170.
- Conjugation via a linker group may be made using any known procedure, for example, the procedures described in U.S. Pat No.
- linker is attached via an anomeric carbon of the polysaccharide.
- a preferred type of linker is an adipic acid linker, which may be formed by coupling a free -NH2 group (e.g., introduced to a polysaccharide by amination) with adipic acid (using, for example, diimide activation), and then coupling a protein to the resulting saccharide-adipic acid intermediate (see, e.g., EP-B-0477508, Mol. Immunol, (1985) 22, 907-919, and EP-A-0208375).
- a similar preferred type of linker is a glutaric acid linker, which may be formed by coupling a free -NH group with glutaric acid in the same way.
- Adipic and glutaric acid linkers may also be formed by direct coupling to the polysaccharide, i.e., without prior introduction of a free group, e.g., a free -NH group, to the polysaccharide, followed by coupling a protein to the resulting saccharide - adipic/glutaric acid intermediate.
- Another preferred type of linker is a carbonyl linker, which may be formed by reaction of a free hydroxyl group of a modified polysaccharide with CDI (Bethell G.S. et al. (1979) J.
- linkers include P-propionamido (WO00/10599), nitrophenyl-ethylamine (Gever et al. (1979) Med. Microbiol. Immunol. 165, 171-288), haloacyl halides (U.S. Pat. No. 4,057,685), glycosidic linkages (U.S. Pat. Nos. 4,673,574; 4,761,283; and 4,808,700), 6-aminocaproic acid (U.S. Pat. No.
- a bifunctional linker may be used to provide a first group for coupling to an amine group in the polysaccharide (e.g., introduced to the polysaccharide by amination) and a second group for coupling to the carrier (typically for coupling to an amine in the carrier).
- the first group is capable of direct coupling to the polysaccharide, i.e., without prior introduction of a group, e.g., an amine group, to the polysaccharide.
- the fVi conjugate is obtained by or obtainable by a method (i.e. a method for preparing an fVi conjugate) comprising the steps of: a. fragmenting Vi polysaccharide to obtain a fragmented Vi (fVi) polysaccharide having an average molecular weight of between 10 kDa and 90 kDa, between 25 kDa and 70 kDa, between 40 kDa and 55 kDa, between 41 kDa and 49 kDa, or between 51 kDa and 55 kDa; b. reacting the fVi polysaccharide obtained in step a.
- step b. reacting the N-hydroxysuccinimide ester fVi derivative obtained in step b. with the carrier protein (optionally derivatised carrier protein) to produce the fVi conjugate.
- the carrier protein may be derivatised by reacting it with a carbodiimide and a linker.
- the carbodiimide is l-ethyl-3 -(3 -Dimethylaminopropyl) carbodiimide (EDAC). Any suitable linker (such as those discussed above) may be used. In some embodiments, the linker is an ADH linker.
- derivatising the carrier protein produces a derivatised carrier protein.
- the carrier protein is CRM197 and derivatising the carrier protein comprises one or more of the following steps:
- CRM 197 is an appropriate buffer, optionally MES buffer;
- the carrier protein is derivatised by a method that comprises steps (i), (ii), and (iv). In some embodiments, the carrier protein is derivatised by a method that comprises steps (i), (ii), (iii), and (iv). In some embodiments, the carrier protein is derivatised by a method that comprises steps (i), (ii), (iv), and (v). In some embodiments, the carrier protein is derivatised by a method that comprises all of steps (i) to (v) above. In some embodiments, steps (i) to (v) above are performed in the order set out above, except that steps (ii) and (iii) may be performed simultaneously.
- the fVi conjugate may be obtainable or obtained by a method comprising a step of reacting the fVi polysaccharide with a carbodiimide and N-hydroxysuccinimide at a pH of 5 to 6 to form an N-hydroxysuccinimide ester fVi derivative.
- the carbodiimide is EDC (N-3-dimethylamino propyl(-N-ethyl carbodiimide).
- reacting the fVi polysaccharide with a carbodiimide and N-hydroxysuccinimide comprises mixing the fVi with a carbodiimide such as EDC in the presence of N-hydroxysuccinimide (NHS).
- reacting the fVi polysaccharide with a carbodiimide and N- hydroxysuccinimide comprises mixing the fVi polysaccharide with NHS.
- reacting the fVi polysaccharide with a carbodiimide and N-hydroxysuccinimide comprises mixing the fVi polysaccharide with NHS such that the NHS concentration is between 0.1 M and 0.5M, or around 0.33M, and the fVi polysaccharide concentration is between 1 mg/mL and 100 mg/ml, or around 50 mg/ml.
- reacting the fVi polysaccharide with a carbodiimide and N-hydroxysuccinimide comprises mixing the fVi polysaccharide with EDC to have a molar ratio of EDC to fVi repeating unit or between 1 : 1 and 20: 1 , between 1:1 and 10:0, between 2:1 and 7:1, or around 5:1.
- mixing the fVi polysaccharide with EDC is carried out after mixing the fVi polysaccharide with NHS.
- reacting the fVi polysaccharide with a carbodiimide and N- hydroxysuccinimide comprises a step of incubating a mixture of fVi polysaccharide, NHS and EDC for at least 30 minutes, or around 1 hour at room temperature.
- reacting the N-hydroxysuccinimide ester fVi derivative with the carrier protein comprises mixing the N-hydroxysuccinimide ester fVi derivative with the carrier protein (or carrier protein derivative).
- reacting the N-hydroxysuccinimide ester fVi derivative with the carrier protein comprises mixing the N-hydroxysuccinimide ester fVi derivative with the carrier protein (or carrier protein derivative) at a ratio of between (w/w) 1:0.1 and 1:10, between 1:0.5 and 1:5, between 1:0.75 and 1:2, or around 1:1.
- mixing the N-hydroxysuccinimide ester fVi derivative with the carrier protein (or carrier protein derivative) is carried out in a buffer at a pH between 5 and 7, or around 6.
- mixing the N-hydroxysuccinimide ester fVi derivative with the carrier protein (or carrier protein derivative) is carried out in MES buffer.
- mixing the N- hydroxysuccinimide ester fVi derivative with the carrier protein (or carrier protein derivative) is carried out at a temperature between 20°C and 30°C or around room temperature, optionally with mixing.
- the method for preparing an fVi conjugate may comprise one or more of the following additional steps, after the step of reacting the N-hydroxysuccinimide ester fVi derivative with the carrier protein (or carrier protein derivative):
- the method for preparing an fVi conjugate comprises 2 or more, 3 or more, 4 or more, or all 5 of steps (i) to (v) above.
- the method comprises step (i) above.
- the method comprises steps (i) to (iii) above.
- the method comprises steps (i) to (v) above.
- the method comprises steps (i) to (iii) above in the order recited above.
- the method comprises steps (i) to (v) above in the order recited above.
- a suitable method for conjugating the fVi polysaccharide to CRM197 using ED AC chemistry via an ADH linker is set out in Example 2.
- the immunogenic compositions of the invention or used in the invention may comprise additional components, such as a pharmaceutically acceptable excipient(s), an adjuvant, and/or further antigens.
- the immunogenic composition may further comprise a pharmaceutically acceptable excipient.
- Typical ''pharmaceutically acceptable excipients include any carrier that does not itself induce the production of antibodies harmful to the individual receiving the composition.
- Suitable carriers are typically large, slowly metabolised macromolecules such as proteins, polysaccharides, polylactic acids, polyglycolic acids, polymeric amino acids, amino acid copolymers, sucrose, trehalose, lactose, and lipid aggregates (such as oil droplets or liposomes). Such carriers are well known to those of ordinary skill in the art.
- Pharmaceutically acceptable excipients may also contain diluents, such as water, saline, glycerol, etc.
- the immunogenic composition comprises phosphate buffered saline (and optionally an aluminium adjuvant as described further below).
- the immunogenic composition comprises phosphate buffered saline at a pH between 6 and 7, for example pH 6.5.
- Immunogenic compositions may be prepared as injectables, either as liquid solutions or suspensions. Solid forms suitable for solution in, or suspension in, liquid vehicles prior to injection can also be prepared (e.g. a lyophilised composition or a spray-freeze dried composition).
- the immunogenic composition may be prepared for topical administration e.g. as an ointment, cream or powder.
- the immunogenic composition may be prepared for oral administration e.g. as a tablet or capsule, as a spray, or as a syrup (optionally flavoured).
- the immunogenic composition may be prepared for pulmonary administration e.g. as an inhaler, using a fine powder or a spray.
- the composition may be prepared as a suppository or pessary.
- the immunogenic composition may be prepared for nasal, aural or ocular administration e.g. as drops.
- the immunogenic composition may be in kit form, designed such that a combined composition is reconstituted just prior to administration to a mammal.
- kits may comprise one or more antigens in liquid form and one or more lyophilised antigens.
- Immunogenic compositions may be presented in vials, or they may be presented in pre-filled syringes. The syringes may be supplied with or without needles. A syringe will include a single dose of the composition, whereas a vial may include a single dose or multiple doses.
- Immunogenic compositions of or used in the invention may be packaged in unit dose form or in multiple dose form.
- vials are preferred to pre-filled syringes.
- Effective dosage volumes can be routinely established, but a typical human dose of the composition has a volume of 0.5ml e.g. for intramuscular injection.
- composition will be sterile.
- Immunogenic compositions of or used in the invention may be isotonic with respect to humans.
- immunogenic compositions of or used in the invention may be useful as vaccines.
- Vaccines according to the invention may either be prophylactic (i.e. to prevent infection) or therapeutic (i.e. to treat infection), but will typically be prophylactic.
- Immunogenic compositions used as vaccines comprise an effective amount of antigen(s), as well as any other components, as needed.
- effective amount i.e. an immunologically effective amount
- Immunogenic compositions of the invention may include an antimicrobial, particularly when packaged in multiple dose formats.
- the immunogenic compositions of or used in the invention may comprise an adjuvant.
- Any suitable adjuvant may be used.
- the adjuvant is a mineral salt, such as an aluminium salt or a calcium salt.
- suitable mineral salts include hydroxides (e.g. oxyhydroxides), phosphates (e.g. hydroxyphosphates, orthophosphates), sulphates, etc. or mixtures of different mineral compounds, with the compounds taking any suitable form (e.g. gel, crystalline, amorphous, etc.), and with adsorption being preferred.
- the mineral containing compositions may also be formulated as a particle of metal salt.
- the immunogenic compositions of or used in the invention may comprise an aluminium adjuvant, i.e. any compound comprising Al 3+ ions.
- the aluminium adjuvant may comprise or consist of aluminium phosphate (any compound comprising Al 3+ and PO4 3 ' ions) and/or aluminium hydroxide (any compound comprising Al 3+ and OH' ions).
- the aluminium adjuvant comprises or consists of aluminium hydroxide.
- the aluminium hydroxide adjuvant may comprise or be an aluminium oxyhydroxide salt.
- the aluminium hydroxide adjuvant may comprise or be an aluminium oxyhydroxide salt that is at least partially crystalline.
- Aluminium oxyhydroxide salt which can be represented by the formula A10(0H), can be distinguished from other aluminium compounds, such as aluminium hydroxide salt (Al(0H)3), by infrared (IR) spectroscopy, in particular by the presence of an adsorption band at 1070cm' 1 and a strong shoulder at 3090-3100cm' 1 (chapter 9 of ref.
- Vaccine Design The Subunit and Adjuvant Approach (eds.
- aluminium hydroxide adjuvants will be apparent to one of skill in the art, for example ALHYDROGEL®.
- the aluminium adjuvant may comprise or consist of between 0.1 mg and 10 mg Al 3+ , between 0.1 mg and 5 mg Al 3+ , between 0.3 mg and 0.4 mg Al 3+ , or around 0.35 mg Al 3+ .
- the Examples show that the immunogenic composition of the invention demonstrate good immunogenicity. Immunogenicity can be measured according to the assays set out in Example 6.
- the immunogenic compositions of the invention may induce at least 10 3 EU/ml of anti-5.
- Enteritidis O- antigen antibodies in an immunogenicity assay comprising the following steps:
- An immunogenic composition “ induces” at least 10 3 EU/ml of anti-5.
- Typhimurium O- antigen antibodies if it is able to induce this level of antibodies when used to immunise mice. Whether or not it is able to induce this level of antibodies when used to immunise a mice may be determined by testing a sample of the immunogenic composition using the immunogenicity assay. The dose of the GMMA and the saccharide may be determined as discussed in the section entitled “dose” above.
- a suitable ELISA may involve: coating ELISA plates with S. Typhimurium or S. Enteritidis O-antigen; applying a blood sample taken from the mice at day 42 to the coated ELISA plates and then washing the plates to remove antibodies not bound to the S. Typhimurium or S.
- Enteritidis O-antigen detecting the amount of anti-5. Typhimurium or S. Enteritidis O-antigen antibodies bound to the S. Typhimurium or S. Enteritidis O-antigen on the ELISA plates using an anti-IgG antibody conjugated to a detection moiety such as alkaline phosphatase.
- the immunogenic compositions of the invention may induce at least 10 3 EU/ml of anti-fVi conjugate antibodies in an immunogenicity assay comprising the following steps:
- the dose of the GMMA and the saccharide may be determined as discussed in the section entitled “dose” above.
- a suitable ELISA may involve: coating ELISA plates with fVi polysaccharide; applying a blood sample taken from the mice at day 42 to the coated ELISA plates and then washing the plates to remove antibodies not bound to the fVi polysaccharide; and detecting the amount of anti-fVi conjugate antibodies bound to the fVi polysaccharide on the ELISA plates using an anti-IgG antibody conjugated to a detection moiety such as alkaline phosphatase.
- the immunogenic compositions of the invention may induce at least 10 3 EU/ml or 10 3 5 of S. Paratyphi A O-antigen antibodies in an immunogenicity assay comprising the following steps: (a) immunise mice at days 0 and 28 intraperitoneally with the immunogenic composition at a dose of 1 pg (O-antigen) per GMMA and 1.25 pg saccharide per saccharide conjugate; and
- the dose of the GMMA and the saccharide may be determined as discussed in the section entitled “dose” above.
- a suitable ELISA may involve: coating ELISA plates with S. Paratyphi A O-antigen; applying a blood sample taken from the mice at day 42 to the coated ELISA plates and then washing the plates to remove antibodies not bound to the S. Paratyphi A O- antigen; and detecting the amount of anti- S. Paratyphi A O-antigen antibodies bound to the S. Paratyphi A O-antigen on the ELISA plates using an anti-IgG antibody conjugated to a detection moiety such as alkaline phosphatase.
- Typhimurium O- antigen antibodies induced may be at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, or at least 98% of the level of anti-5.
- the level of antibodies in the immunogenic composition and the corresponding monovalent immunogenic composition may be determined using the “ suitable ELISA” set out above.
- a “corresponding monovalent S. Typhimurium immunogenic composition” is identical to the immunogenic composition, except that the only antigen present is the 5.
- Typhimurium antigen For example, if the immunogenic composition of the invention comprises 5 pg O- antigen from 5.
- Typhimurium O-antigen from 5. Enteritidis, an fVi polysaccharide conjugate, an aluminium adjuvant and phosphate buffered saline, the corresponding monovalent 5.
- Typhimurium immunogenic composition would comprise 5 pg O-antigen from 5.
- Enteritidis O- antigen antibodies induced may be at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, or at least 98% of the level of anti-5.
- Enteritidis immunogenic composition may be at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, or at least 98% of the level of anti-5.
- the level of antibodies in the immunogenic composition and the corresponding monovalent immunogenic composition may be determined using the “ suitable ELISA” set out above.
- a “corresponding monovalent S. Enteritidis immunogenic composition” is identical to the immunogenic composition, except that the only antigen present is the 5. Enteritidis antigen.
- the immunogenic composition of the invention comprises O- antigen from 5. Typhimurium, 5 pg O-antigen from 5. Enteritidis, an fVi polysaccharide conjugate, an aluminium adjuvant and phosphate buffered saline, the corresponding monovalent 5.
- Enteritidis immunogenic composition would comprise 5 pg O-antigen from 5. Enteritidis, an aluminium adjuvant and phosphate buffered saline.
- the level of anti-fVi conjugate antibodies induced may be at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, or at least 98% of the level of anti-fVi conjugate antibodies induced by a corresponding monovalent fVi polysaccharide immunogenic composition.
- the level of antibodies in the immunogenic composition and the corresponding monovalent immunogenic composition may be determined using the “suitable ELISA” set out above.
- a “corresponding monovalent jVi polysaccharide immunogenic composition” is identical to the immunogenic composition, except that the only antigen present is the fVi polysaccharide antigen. For example, if the immunogenic composition of the invention comprises O-antigen from 5.
- the corresponding monovalent fVi polysaccharide immunogenic composition would comprise 5 pg fVi polysaccharide, an aluminium adjuvant and phosphate buffered saline.
- the level of anti-5 In the immunogenic compositions of the invention, the level of anti-5.
- Paratyphi A O- antigen antibodies induced may be at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, or at least 98% of the level of anti-5.
- a “corresponding monovalent S. Paratyphi A immunogenic composition” is identical to the immunogenic composition, except that the only antigen present is the S. Paratyphi A antigen.
- the immunogenic composition of the invention comprises O- antigen from S. Typhimurium, 5 pg O-antigen from S. Paratyphi A, an fVi polysaccharide conjugate, an aluminium adjuvant and phosphate buffered saline, the corresponding monovalent S. Paratyphi A immunogenic composition would comprise 5 pg O-antigen from S. Paratyphi A, an aluminium adjuvant and phosphate buffered saline.
- the level of anti-5 is measured in an immunogenicity assay comprising the following steps:
- the immunogenic compositions of the invention may demonstrate cross-protection.
- the immunogenic compositions of the invention may induce antibodies against three or more, four or more, five or more, six or more, seven or more, eight or more, nine or more or all ten of the following strains:
- the immunogenic compositions of the invention may induce antibodies against:
- An immunogenic composition “induces ” antibodies against (for example) S. Typhimurium ST34 if it is able to induce these antibodies when used to immunise mice. Whether or not it is able to induce these antibodies when used to immunise a mice may be determined by testing a sample of the immunogenic composition using the cross-protection assay.
- Whether or not an immunogenic composition induces antibodies against one or more of the strains described above may be determined by carrying out a cross-protection assay. Specifically, the user may: immunise mice intraperitoneally with 500
- SBA serum bactericidal assay
- the SBA assay may be based on the assay described in Example 6, except that the user should measure the bactericidal activity against the relevant strain listed above (rather than, for example Salmonella Paratyphi A NVGH308).
- the immunogenic composition induces antibodies against one more of the strains described above, if the IC50 (serum dilution giving 50% inhibition of the ATP level) obtained in the SBA assay is above 10 2 .
- the immunogenic compositions of the invention may induce anti-5.
- Paratyphi A antibodies in each of classes IgG3, IgG2b, IgG2a, and IgGl, as determined using an antibody class assay comprising the following steps:
- Paratyphi A O-antigen antibody subtype level by ELISA at day 42.
- An immunogenic composition “induces ” anti-5.
- Paratyphi A antibodies in each of classes IgG3, IgG2b, IgG2a, and IgGl if it is able to induce these antibodies when used to immunise mice. Whether or not it is able to induce these antibodies when used to immunise a mice may be determined by testing a sample of the immunogenic composition using an ELISA assay to measure the anti-5.
- Paratyphi A O-antigen antibody subtype level as set out below.
- Paratyphi A O-antigen antibody subtype level may involve: coating ELISA plates with 5.
- Paratyphi A O-antigen may be applied a blood sample taken from the mice at day 42 to the coated ELISA plates and then washing the plates to remove antibodies not bound to the 5.
- Paratyphi A O- antigen may be detecting the amount of anti- 5.
- Paratyphi A O-antigen on the ELISA plates using an anti-IgG3 antibody conjugated to a detection moiety such as alkaline phosphatase; repeating the first three steps but using an anti-IgG2b antibody in place of the anti- IgG3 antibody, then using an anti-IgG2a antibody in place of the anti-IgG3 antibody and finally using an anti-IgGl antibody in place of the anti-IgG3 antibody.
- a detection moiety such as alkaline phosphatase
- “Tolerogenicity” or “tolerogenic” refers to the an immunogenic composition that does not induce significant negative reactions in the subject to which it is administered.
- immunogenic compositions comprising outer membrane vesicles have been known to induce fevers in patients to which they are administered.
- an immunogenic composition may be considered to be tolerogenic if it does not induce a significant fever.
- An immunogenic composition will be considered to “induce ” a fever if it induces a fever when used to immunise mice. Whether or not it induces a fever when used to immunise a mice may be determined by testing a sample of the immunogenic composition using the toxicity assay set out below.
- An immunogenic composition may be tolerogenic if it induces a temperature rise of less than 1.8°C, less than 1.7°C, less than 1.6°C, or less than 1.5°C in a toxicity assay comprising the following steps:
- An immunogenic composition may be tolerogenic if it induces a maximum temperature of 41°C or less, 40.9°C or less, or 40.8°C or less in a toxicity assay comprising the following steps:
- a method of boosting an immune response to an antigen comprising administering a composition comprising the antigen and GMMA.
- a method of preventing infection by a Salmonella enterica bacterium comprising administering an immunogenic composition comprising an antigen of the Salmonella enterica and GMMA, wherein administration of the GMMA boosts the immune response to the Salmonella enterica bacterium.
- the methods of the invention may comprise boosting an immune response to a S. Typhi or a S. Paratyphi A antigen comprising administering a composition comprising the S. Typhi antigen or the S. Paratyphi A antigen and GMMA.
- the methods of the invention may also comprise preventing infection by S. Typhi or S. Paratyphi A comprising administering an immunogenic composition comprising the S. Typhi antigen or the S. Paratyphi A antigen and GMMA, wherein the GMMA boosts the immune response to the S. Typhi antigen or the S. Paratyphi A antigen.
- an immunogenic composition of the invention comprising GMMA for use in a method of boosting an immune response to a S. Typhi or S. Paratyphi A antigen, wherein the method comprises administering an immunogenic composition comprising the S. Typhi antigen or the S. Paratyphi A antigen and GMMA.
- an immunogenic composition of the invention for use in a method of preventing infection by S. Typhi or S. Paratyphi A comprising the S. Typhi antigen or the S. Paratyphi A antigen and GMMA, wherein the GMMA boosts the immune response to the S. Typhi or the S. Paratyphi A antigen.
- the method may be a method of boosting an immune response to an S. Typhi antigen and the immunogenic composition comprises the S. Typhi antigen.
- the method may be a method of boosting an immune response to an S. Paratyphi A antigen and the immunogenic composition may comprise the S. Paratyphi A antigen.
- the method may be a method of preventing infection by S. Typhi
- the immunogenic composition may comprise the S. Typhi antigen
- the GMMA may boost the immune response to the S. Typhi antigen.
- the method may be a method of preventing infection by S. Paratyphi A
- the immunogenic composition may comprise the S. Paratyphi A antigen
- the GMMA may boost the immune response to the S. Paratyphi A antigen.
- the method or immunogenic composition for use of the invention wherein the GMMA comprises at least one are selected from the group consisting of S. Typhimurium GMMA, S. Enteritidis GMMA, and A Paratyphi A GMMA.
- the A Typhimurium GMMA may boost the immune response to the S. Typhi or S. Paratyphi A antigen.
- the S. Paratyphi A GMMA may boost the immune response to the S. Typhi antigen.
- the S. Enteritidis GMMA may boost the immune response to the S. Typhi or S. Paratyphi A antigen.
- the S. Paratyphi GMMA may boost the immune response to the S. Typhi antigen.
- a method is a method of “boosting an immune response” to a S. Typhi antigen or a S. Paratyphi A antigen, if the immune response raised to the S. Typhi or S. Paratyphi A antigen is higher when the S. Typhi antigen or a S. Paratyphi A antigen is part of the immunogenic composition comprising GMMA compared to the immune response raised when the S. Typhi antigen or the S. Paratyphi A antigen is not part of an immunogenic composition comprising GMMA. This is due to the adjuvanting effect of GMMA. In other words, when GMMA and S. Typhi antigen or S. Paratyphi A antigen are in the same composition, the GMMA increase the immune response to the S. Typhi antigen or S. Paratyphi A antigen when compared to S. Typhi antigen or S. Paratyphi A antigen on their own.
- the S. Typhi antigen and S. Paratyphi A antigen may be polysaccharides.
- the S. Typhi antigen may comprise a fVi polysaccharide, optionally wherein the fVi polysaccharide is part of an fVi conjugate comprising fVi and a carrier protein, further optionally wherein the carrier protein is CRM197.
- the S. Paratyphi A antigen may comprise S. Paratyphi A O-antigen, optionally wherein the S. Paratyphi A O- antigen is conjugated to a carrier protein, further optionally wherein the carrier protein is CRM197.
- GMMA boosts the immune response to a S. Typhi antigen or a S. Paratyphi A antigen if the immune response raised to the S. Typhi antigen or the S. Paratyphi A antigen is higher when the S. Typhi antigen or the S. Paratyphi A antigen is part of the immunogenic composition comprising the GMMA compared to the immune response raised when the S. Typhi antigen or the S. Paratyphi A antigen is not part of an immunogenic composition comprising GMMA.
- a method is a method of boosting an immune response to an S. Typhi or a S. Paratyphi A antigen or GMMA boosts the immune response to the S. Typhi antigen or the S.
- Paratyphi A antigen if the immune response raised to the S. Typhi antigen or the S. Paratyphi A antigen is at least 5 times, at least 10 times, or at least 20 times higher when the S. Typhi antigen or the S. Paratyphi A antigen is part of the immunogenic composition comprising the GMMA compared to the immune response raised when the S. Typhi antigen or the S. Paratyphi A antigen is not part of an immunogenic composition comprising GMMA.
- the immune response raised to a S. Typhi antigen or a S. Paratyphi A antigen by the immunogenic compositions of the invention, or the methods of the invention may be the number of antibodies raised as determined by ELISA 42 days after administration of the immunogenic composition comprising the S. Typhi antigen or S. Paratyphi A antigens and the GMMA at a dose of 0.78 pg of S. Typhi antigen or S. Paratyphi A antigen and 0.63 pg (O-antigen) of the GMMA.
- a suitable ELISA may involve: coating ELISA plates with S. Typhi antigen or S. Paratyphi A O-antigen; applying a blood sample taken from the mice at day 42 to the coated ELISA plates and then washing the plates to remove antibodies not bound to the S. Typhi antigen or S.
- Paratyphi A O-antigen; and detecting the amount of anti- S. Typhi antigen or S. Paratyphi A O-antigen antibodies bound to the S. Typhi antigen or S. Paratyphi A O-antigen on the ELISA plates using an anti-IgG antibody conjugated to a detection moiety such as alkaline phosphatase.
- an immunogenic composition of the invention for use in a method of preventing an infection.
- a method of preventing an infection comprising administering an effective amount of the immunogenic composition or vaccine of the invention to a subject.
- the method of preventing an infection may comprise administering an effective amount of the immunogenic composition or vaccine of the invention to a subject.
- the method of preventing an infection may be a method of preventing Salmonella infection.
- the method of preventing an infection is a method of preventing infection by S. Typhimurium, S. Entcritidis, S. Typhi and/or S. Paratyphi A.
- the term “preventing Salmonella infection” in the method/immunogenic composition for use/use of the immunogenic composition in the manufacture of a medicament of the invention comprises raising an immune response in a subject.
- the immune response may be protective and may raises antibodies, such as IgG antibodies.
- the subject of the invention is a mammal, optionally a human.
- the human may be an adult i.e. subject is 18 years old or above 18 years old.
- the human may be a child i.e. below 18 years old.
- the child may be between 12 to 72 months, preferably between 24 to 59 months, more preferably between 6 to 12 months.
- the vaccine is for prophylactic use, the child may be around 9 months.
- the human is preferably a child.
- a vaccine intended for children may also be administered to adults e.g. to assess safety, dosage, or immunogenicity.
- Salmonella enterica serovar Typhimurium wild-type (WT) strain 2192 was provided by the Salmonella Genetic Stock Center (SGSC) at the University of Calgary, Canada, which belongs to the global Salmonella reference collection A (SARA 12).
- SGSC Salmonella Genetic Stock Center
- Salmonella enterica serovar Enteritidis WT strain 618 was provided by Quotient Bioresearch Limited, UK. The strain of animal origin was isolated by the European Antimicrobial Susceptibility Surveillance in Animals (EASSA).
- EASSA European Antimicrobial Susceptibility Surveillance in Animals
- Salmonella Typhimurium olR ApagP AmsbB and Salmonella Enteritidis olR ApagP AmsbB recombinant mutants for each strain were generated as previously reported (Rossi O, Caboni M, Negrea A, Necchi F, Alfini R, Micoli F, et al. Toll-Like Receptor Activation by Generalized Modules for Membrane Antigens from Lipid A Mutants of Salmonella enterica Serovars Typhimurium and Enteritidis. Clin Vaccine Immunol. 2016;23(4):304-14.).
- GMMA derived from the Salmonella strains above (Salmonella Typhimurium GMMA (STmGMMA) and Salmonella Enteritidis GMMA (SEnGMMA)) were purified and isolated. GMMA were purified using similar methods previously reported for S. sonnei GMMA (Gerke C, Colucci AM, Giannelli C, Sanzone S, Vitali CG, Sollai L, et al. Production of a Shigella sonnei Vaccine Based on Generalized Modules for Membrane Antigens (GMMA), 1790GAHB. PLoS One. 2015;10(8):e0134478. doi:
- GMMA released into the fermentation broth were purified using two consecutive Tangential Flow Filtration (TFF) steps: a microfiltration in which the culture supernatant containing the GMMA is separated from the bacteria, and an ultrafiltration, in which the GMMA are separated from soluble proteins and nucleic acids.
- TMF Tangential Flow Filtration
- Step 1 Fragmentation Reaction and Quenching: Vi-Polysaccharide fragmentation is achieved by an Oxidation-reaction using hydrogen peroxide in the presence of iron sulphate.
- the reaction is quenched with EDTA (Ethylenediaminetetraacetic acid).
- Native Vi polysaccharide is diluted with WFI.
- a calculated volume of 10 mM FeSO4 and H2O2 is added to get a final concentration of 0.5 mM FeSO4 and 0.5% v/v H2O2 in the reaction mixture respectively.
- the reaction mixture is incubated at 15 ⁇ 5°C for 120 +/- 10 min.
- the reaction is stopped by adding equal volumes of 250 mM EDTA to get a final EDTA concentration of 10 mM and is stirred.
- Step 2 Buffer Exchange: Buffer exchange is performed by Tangential Flow Filtration (TFF) with lOOmM Sodium phosphate (pH: 7.2 ⁇ 0.2) using 30 kDa cassettes to remove residual H2O2. Fragmented Vi (fVi) polysaccharide is concentrated.
- Step 3 Stabilization of fVi polysaccharide: Post fragmentation, 30 kDa retentate is stabilized by incubating at 80 ⁇ 5° C for 120 +/- 15 min.
- Step 4 JVi Purification by Anion Exchange (Resin: Capto-Q): AA chromatography step is used to separate the desired molecular size of fVi polysaccharide (25-70 KDa). This is performed using a linear gradient elution, with Capto-Q Buffer A and Capto-Q Buffer B using a Capto-Q Resin which has the binding capacity of 13 mg of IVi/mL. The eluted fractions are collected based on the conductivity for every 1 mS/cm; i.e., from 35 to 50 mS/cm and estimating the Molecular size distribution by SEC/HPLC. The Capto-Q fractions are pooled based on the Molecular Size (kDa) distribution.
- Step 5 Desalting'. Pooled Capto-Q fractions are concentrated by Tangential Flow Filtraion (TFF) using a 10 kDa Cut-off cassette and then dia- filtered using WFI until permeate conductivity reaches ⁇ 30 pS/cm.
- TFF Tangential Flow Filtraion
- Step 6 0.2 pm Filtration of fVi polysaccharide'.
- the fVi polysaccharide is fdtered through a 0.22 pm filter.
- the purified fVi polysaccharide is stored in PETG bottles.
- Stepl Thawing of CRM197: Purified CRM197 is thawed at 2-8°C prior to buffer exchange with 100 mM MES (Morpholino Ethanesulfonic acid) buffer. Post thawing, CRM197 is filtered using 0.5 pm filter.
- MES Methyl MES
- Step 2 Buffer Exchange with 100 mM MES Buffer. Buffer exchange is performed by TFF with 100 mM MES buffer (pH 6.0 ⁇ 0.2) using 10 KDa cassette after CRM197 thawing.
- Step 3 CRM197 Derivatization'.
- the required concentration of CRM197 is diluted with 100 mM MES buffer followed by addition of calculated quantity of ADH (Adipic Acid Dihydrazide) and ED AC (l-Ethyl-3 -(3 -Dimethylaminopropyl) carbodiimide) to make a CRM : ADH : ED AC 1 : 3.5 : 0.15 w/w/w ratio.
- ADH Adipic Acid Dihydrazide
- ED AC l-Ethyl-3 -(3 -Dimethylaminopropyl) carbodiimide
- Step 4 Purification of CRM197'. Post reaction, CRM197 is purified by TFF using a 10 KDa cassette with 5 mM MES buffer.
- Step 5 Filtration ofDia-Filtered CRM197: 0.2-micron filtration is performed for dia- filtered CRM197 solution followed by storage at 2-8°C in glass bottle.
- Step 1 jVi polysaccharide drying by Rota Vapor. fVi is further concentrated by drying at 30°C using rotavapor. Concentrated fVi polysaccharide is reconstituted by using 100 mM MES buffer (pH: 6.0) in order to get a 50 mg/mL concentration.
- Step 2 Activation of jVi polysaccharide with NHS: fVi carboxylates (-COOH) are activated with EDC (N-3 -Dimethylamino propyl-N Ethyl Carbodimide) in the presence of N-hydroxysuccinimide (NHS), by forming an active ester intermediate, to increase the efficiency of conjugation with CRM197 previously activated with ADH.
- EDC N-3 -Dimethylamino propyl-N Ethyl Carbodimide
- NHS N-hydroxysuccinimide
- the dried fVi polysaccharide is re-constituted to a desired concentration (50 mg/mL) with 100 mM MES buffer (pH: 6.2 ⁇ 0.2); and activated in the presence of NHS (concentration of 0.33 M) followed by ED AC addition to have an ED AC /fVi RU molar ratio of 5 : 1.
- EDAC solution is added after addition of NHS to ensure complete dissolution. The reaction is incubated at room temperature with slow mixing for 1 h.
- Step 1 Conjugation of jVi with CRMwADI ⁇ :
- the conjugation reaction creates a covalent bond between the activated fVi and CRM197-ADH.
- the activated and derivatized reaction mixture is diluted with lOOmM MES pH: 6.0 and CRM197-ADH is added in a w/w ratio of 1 : 1 (fVi : CRM197) to reach the final fVi concentration of the activated fVi and CRM197- ADH of 5 mg/mL.
- the conjugation reaction is performed at room temperature with slow mixing until protein consumption is > 70% measured by HPLC-SEC at 280 nm absorbance.
- Step 2 Quenching and conditioning of Conjugation Reaction: The conjugation reaction is quenched by adding equal volume of Phenyl HP Buffer-B Tris 50 mM pH: 8.0. NaCl as powder is added to reach a final salt concentration of 3 M.
- Step 3 Conjugation Mixture Filtration: The fVi-CRMi97 crude conjugate is 0.65 filtered.
- Step 4 Purification of jVi-CRMi97 Crude Conjugate: Purification of the conjugate from the conditioned reaction mixture is performed through a HIC Phenyl Sepharose High Performance (HP) column. Column integrity is performed for every 5-10 cycles as per standard procedure. The column is equilibrated by using Phenyl Sepharose HP Buffer A Tris 50 mM NaCl 3M pH 8. After addition of conditioning buffer and NaCl, crude conjugate is loaded on to the column. Column washing is done using Phenyl Sepharose HP Buffer-A followed by product elution using Phenyl Sepharose HP Buffer-B Tris 50mM pH: 8. Fractions are collected and stored at 2-8°C till further usage. All the fractions from multiple runs are pooled.
- HP Phenyl Sepharose High Performance
- Step 5 Concentration and Buffer Exchange using PBS: Purified conjugate is concentrated by TFF using a 50 kDa Cut-off cassette and then buffer exchanged using PBS buffer until the permeate conductivity meets PBS buffer conductivity.
- Step 6 Pre Filtration of jVi-CRFlm Conjugate using 0.2 pm filter: fVi-CRMi97 conjugate is filtered through a 0.2 pm filter for bioburden reduction.
- Step 7 Sterile Filtration of fVi-CRMi97 Conjugate using 0.2 p cellulose acetate filter: The fVi-CRMi97 conjugate is filtered through a 0.2 pm cellulose acetate filter. The purified fVi-CRMi97 conjugate is sampled and stored at 2-8°C.
- Example 3 Formulation of a bivalent (iNTS-GMMA) vaccine against S.
- STmGMMA and SEnGMMA were separately adsorbed to aluminium hydroxide (Alum 3+ final concentration 0.7mg/mL) in phosphate buffered saline (pH 6.5), obtaining two different drug product formulations, each vialed in 3mL Type I 2 R vials.
- the bulk GMMA solutions were diluted to obtain a concentration of GMMA of 80pg/ml O-antigen based on the amount of O-Ag measured in bulk GMMAx, and a concentration of aluminium hydroxide of 0.7 mg/ml (Al 3+ ).
- the content of STmGMMA/alhydrogel drug product vial is mixed with the content of
- Quantification of the OAg in the S. Enteritidis. GMMA is performed by High- Performance Anion-Exchange Chromatography with Pulsed Amperometric Detection (HPAEC-PAD) analysis after acid hydrolysis of the sample. The quantification determines the concentration of Rhamnose, galactose, glucose and mannose present in the sample.
- HPAEC-PAD Pulsed Amperometric Detection
- the quantification of OAg is performed on the basis of the known sugar ratios present in the OAg repeating unit IxRha (Rhamnose); IxGal (Galactose); IxMan (Mannose) IxTyv (Tyvelose) and the glucose is calculated from the glucose measured in the analysis after subtraction of the glucose due to the core.
- a standard dilution series of each sugar in the range of 0.5-10 pg/mL is run in each HPAEC-PAD analysis and the peak areas are used to interpolate the pg/mL of the corresponding sugar present in the sample OAg.
- the quantification of OAg is performed on the basis of the known sugar ratios present in the OAg repeating unit IxRha (rhamnose); IxGal (galactose); lx Man (mannose); IxAbe (Abequose) and the glucose is calculated from the glucose measured in the analysis after subtraction of the glucose present in the core.
- IxRha rhamnose
- IxGal galactose
- lx Man mannose
- IxAbe Abequose
- Quantification of S. Typhimurium or S. Enteritidis OAg on GMMA adsorbed on aluminium hydroxide is performed using HPAEC-PAD after GMMA acid hydrolysis.
- HPAEC-PAD Quantification of S. Typhimurium or S. Enteritidis OAg on GMMA adsorbed on aluminium hydroxide is performed using HPAEC-PAD after GMMA acid hydrolysis.
- a standard dilution series of a solution containing fucose, rhamnose, N-acetyl glucosamine, glucose, galactose and mannose is run in each HPAEC-PAD analysis series and used as calibration curves to quantify the rhamnose, glucose, galactose and mannose in the sample.
- the procedure is the same used for quantification of S. Typhimurium and S. Enteritidis Drug Substances.
- HPAEC-PAD is performed with a Dionex ICS3000 (or 5000) equipped with a CarboPac PA10 column coupled with PA10 guard column.
- OAg The quantification of OAg is performed on the basis of the known sugar ratios present in the OAg repeating units (Rha; Glc; Gal; Man and Tyvelose (SEn) or Abequose (STm) equal to Rha) present in the formulation.
- Example 4 Adsorption of SEnGMMA and STmGMMA to aluminium hydroxide enhanced the in vivo tolerability in rabbits
- Groups of 3 New Zealand White rabbits were immunised intramuscularly with a saline control, an Alhydrogel control (a 0.5ml dose having Alhydrogel (0.7 mg/ml Al 3+ ), 20mM phosphate and 154mM NacL), or one of the following vaccines: (1) STmGMMA (20pg O- antigen in 0.5mL dose) adsorbed to Alhydrogel (STmGMMA/hydrogel), (2) SEnGMMA (20pg O-antigen in 0.5mL dose) adsorbed to Alhydrogel (SEnGMMA/hydrogel), (3) the iNTS-GMMA vaccine (20pg O-antigen in 0.5mL dose), (4) a bivalent STmGMMA and SEnGMMA vaccine at a dose of 2pg O-antigen per GMMA in 0.5mL not adsorbed to Alhydrogel. These four vaccines were made as described in Examples 1-3.
- the body temperature of the rabbits was monitored for 5 hours after vaccine administration.
- the initial temperature of each rabbit was within the range of 38.0 to 39.8°C and the temperature difference among rabbits within a group was ⁇ 1 ,0°C.
- the absolute temperature after administration did not exceed 39.2°C.
- the highest absolute temperature was 40.6°C; the temperatures of the other two rabbits in this group did not exceed 39.6°C.
- the highest temperatures measured were 39.3°C, 39.8°C and 40.2°C.
- the rabbit groups which received the Alhydrogel formulations (groups (1) to (3) above), showed an average maximum temperature rise of 1°C to 1.3°C, whereas the rabbit group (group (4) above), which received the unformulated GMMA as a 10-times lower dose, showed an average maximum temperature rise of 1 ,9°C.
- Example 5 Formulation of a trivalent (iNTS-TCV) vaccine against Salmonella Typhimurium, S. Enteritidis and 5. Typhi
- STmGMMA and SEnGMMA were sequentially adsorbed to Aluminum hydroxide phosphate buffered saline pH 6.5.
- the adsorbed STmGMMA and SEnGMMA were then mixed together, and after addition of a phosphate quencher, fVi-CRMi97 suspended in sodium phosphate buffered saline was added subsequently.
- the amount of O-Antigen (OAg) in bulk GMMA solutions was determined and then used to determine how to dilute bulk GMMA in order to obtain the correct GMMA concentrations.
- the OAg and the Vi polysaccharide amount in the final mixed drug product was also determined.
- the protocols for determining the STm GMMA and SEnGMMA OAntigen amounts are set out in Example 3. Similarly, the following paragraphs summarise the protocol used to add the correct amount of fVi-CRMi97 conjugate.
- the O-antigen identity and quantification is assessed by FAcE (Formulated Alhydrogel competitive-ELISA) assay, that is designed to detect the single antigen component for S. Enteritidis and S. Typhimurium OAg polysaccharides in the final formulation.
- the FAcE assay is a competitive-ELISA method in which serotype specific avAi-Salmonella OAg monoclonal antibodies (mAb) bind to the OAg coated on the ELISA plate and to the respective OAg of the formulated GMMA suspension present in the ELISA wells.
- the more serotype specific OAg is present in the formulated GMMA suspension, the more mAb will bind to them, and the less will be available to bind to the coated antigen.
- the ELISA signal is given by the binding of mouse mAb to the coated OAg, therefore the lowest signal is obtained at the highest GMMA concentrations and vice versa.
- Binding of the mAb is detected using an enzyme-labelled anti-mouse antibody followed by the addition of substrate solution and formation of a yellow colour detected by absorbance at 405 and 490 nm. The result is calculated as the OD difference between 405 and 490nm.
- Quantification of target antigen on test samples by FAcE assay is obtained using a reference standard curve built by sequential dilutions of Aluminium hydroxide freshly formulated serotype specific GMMA, starting from a known concentration in terms of OAg pg/mL. Test samples are assayed at different dilutions selected to fit within the linear and central part of the standard curve. OAg amount in the test samples is calculated by interpolation of absorbance readings to the standard curve fitted with a 4-parameter logistic regression analysis. As OAg quantification by FAcE is done using anti-0 Ag specific mAbs, this also confirms OAg identity. Vi identity and quantification by HPAEC-PAD in final mixed drug product iNTS-TCV
- Vi polysaccharide is hydrolysed to the monomer sugar of the repeating unit, corresponding to the chromatographic peak.
- a dilution series of Vi standard 0.16-5 pg/mL is run in each HPAEC-PAD analysis and the areas of the resulting peaks are used to build a standard curve, interpolate the peak area of the unknown sample and quantify the corresponding pg/mL.
- the standard calibration curve is prepared by acid hydrolysis; the acid hydrolysis of samples and standards is performed at the same time and with this approach it is possible to determine the concentration of polysaccharide present in the sample.
- Anti-OAg and anti-Vi antigen-specific IgG levels were measured 2 weeks after the second immunization (day 42) by ELISA as previously reported (Rondini et al, Evaluation of the immunogenicity and biological activity of the Citrobacter freundii Vi-CRM197 conjugate as a vaccine for Salmonella enterica serovar Typhi. Clin Vaccine Immunol. 2011 Mar;18(3):460-8). Briefly, 96-well round-botom MaxiSorp microtiter plates (Nunc, Roskilde, Denmark) were coated with 100 ml/well antigen overnight at 4°C. OAg purified from S.
- Paratyphi A (0:2) or S. Enteritidis (0:9) and Vi purified from C.freundii s.l. were used at 15mg/ml and 2mg/ml in carbonate or at Img/ml in phosphate buffer, respectively (Micoli et al, A scalable method for O-antigen purification applied to various Salmonella serovars. Anal Biochem. 2013 Mar 1 ;434(1): 136-45; Micoli et al, Production of a conjugate vaccine for Salmonella enterica serovar Typhi from Citrobacter Vi. Vaccine. 2012 Jan 20;30(5):853 - 61).
- ELISA units were expressed relative to a mouse antigen-specific antibody standard serum curve composed by 10 standard points and 2 blank wells (run in duplicate on each plate), with the best five-parameter fit determined by a modified Hill plot.
- One ELISA unit is defined as the reciprocal of the dilution of the standard serum that gives an absorbance value equal to 1 in this assay.
- reaction mixture containing the target bacterial cells (around 100,000 CFU/ml), BRC (50% for S. Enteritidis, 20% for S. Paratyphi A, and 5% for C.freundii s.l.), and buffer (PBS) was added to SBA plates containing HI serum dilutions and incubated for 3 h at 37°C.
- BRC 50% for S. Enteritidis, 20% for S. Paratyphi A, and 5% for C.freundii s.l.
- PBS buffer
- the plates were centrifuged for lOmin at 4,000 x g, the supernatant was discarded to remove ATP derived from dead bacteria, and live bacterial pellets resuspended in PBS were transferred to a white roundbottom 96-well plate (Greiner) and mixed 1:1 (vol/vol) with BacTiter-Glo reagent (Promega).
- the reaction mixture was incubated for 5 min at RT in an orbital shaker, and the luminescence signal was measured using a luminometer (Viktor).
- Example 12 Post-second immunization individual sera isolated from the blood samples taken from the mice immunized as described in the section entitled “Example 12 - Antibody subclasses raised by the quadrivalent Pan-Salmonella vaccines in mice” were tested to determine the isotype of the antibodies produced using a ELISA-based assay working with the same principle as described under the heading “ELISA”. The assay was repeated to determine the EU/mL on each individual sera at the dose tested using as secondary antibody antimouse-IgGl, antimouse-IgG2a, antimouse-IgG2b and antimouse-IgG3 antibodies in standard assay. Results are shown in Example 12 and expressed as calculated as subclass/subclasses total %.
- Groups of 8 CD1 mice were immunised with an iNTS-TCV vaccine produced as described in Example 5 at doses ranging from 0.01 pg to 0.63 pg (O-antigen) of each GMMA and from 0.012 pg to 0.78 pg of Vi polysaccharide (0.032-2.04 pg dose of total polysaccharide).
- the immunisations involved intraperitoneal immunisation of 0.032-2.04 pg in 500 pl at days 0 and 28. Blood samples were collected at days 27 and 42, and the antibodies raised were measured using the ELISA assay described above in Example 6. Results are shown in Figure 1.
- the iNTS-TCV vaccine candidate was well tolerated at all doses.
- the two antigen GMMA components SEnGMMA and STmGMMA
- the antigen Vi component induced an antibody dose response.
- the three antigen components (SEn OAg, STm OAg and Vi) elicited increasing SBA responses for the four lower doses on Day 42.
- mice were immunised with: the iNTS-TCV vaccine produced as described in Example 5 at 0.032, 0.13, 0.51 and 2.04 pg (total polysaccharide) dose
- SEnGMMA produced as described in Example 1 adsorbed to Alhydrogel at 0.01, 0.04, 0.16 and 0.63 pg (O-antigen) dose
- the immunisations involved intraperitoneal immunisation of the corresponding dose for each product in 500 pl at days 0 and 28.
- Blood samples were collected at days 27 and 42, and the antibodies raised were measured using the ELISA assay and SBA assay described above in Example 6. Results are shown in Figure 2.
- Overall no immune interference was observed for the iNTS GMMA components in iNTS- TCV by measurement of ELISA and SBA, but a higher anti -Vi IgG response and serum bactericidal activity of the fVi-CRMi97 component were observed when combined with GMMA in iNTS-TCV.
- a S. Paratyphi A strain ED 199 comprising tstolR tspagP LSmshB mutations was prepared using a protocol based on that described for S. Enteritidis and S. Typhimurium in Example 1, except the specific mutations used to delete tolR, pagP and msbB were tolR::cat pagP::kan msbB::tet. GMMA were isolated from that bacterium as described in Example 1.
- OAg (0:2) was conjugated to CRM 197 using a random CDAP chemistry approach either via an ADH linker or without the ADH linker.
- the random CDAP chemistry approach introduces multiple linkages between the OAg and the CRM197.
- the chemistry is described in Figure 4. Specifically, 0:2 OH groups are activated with CDAP, using 0:2 to CDAP w/w ratio of 1:0.3 in 150 mM NaCl solution. pH is adjusted to 9-10 with 10% v/v triethylamine and the solution is incubated at room temperature for 3 ⁇ 0.5 minutes on stirring.
- Activated cyanoester groups of 0:2 are covalently bound with hydrazide/amino groups of CRM197ADH/CRM197 to form O:2-CDAP-ADH-CRMi9 7 /O:2-CDAP-CRMi97.
- CRM197ADH/CRM197 is added in the equal w/w ratio (1:1) of 0:2 at a concentration of 10 mg/mL (final concentration of 0:2 and CRM197ADH/CRM197 is 5 mg/mL).
- the pH is maintained to 9.5 ⁇ 0.5 with 10% triethylamine and the solution is mixed for 2-3 hours at room temperature.
- IM glycine solution is then added to an equal volume of conjugation mixture and the pH is adjusted to 8.0 ⁇ 0.2 with 10% triethylamine; the solution is incubated at 2-8°C for 15 ⁇ 5 hours.
- the crude conjugate is then buffer exchanged and unbound and unreacted 0:2 is removed using HIC Phenyl HP resin.
- Example 10 Formulation of two quadrivalent (Pan-Salmonella) vaccines against S.
- the second (Pan-Salmonella_ParA GMMA) was similar except the S. Paratyphi A O-antigen conjugate was replaced with S. Paratyphi A GMMA (as described in Example 8).
- Concentration of phosphate buffer was optimized to obtain the highest adsorption, while quenching to ensure optimal particles size.
- Final formulation contains sufficient GMMA to provide 40 pg/ml of STm, SEn, and S. Paratyphi A O-Antigens (sPa), 50 pg/ml of Vi polysaccharide in a phosphate buffered saline matrix containing 0.7mg/mL of Aluminium Hydroxide.
- sPa O-Antigens
- the protocols for determining the amounts of STm GMMA, SEnGMMA, and Paratyphi A O-antigens are based on those set out in Example 5.
- the method used for determining the O-antigen level for S. Enteritidis, S. Typhimurium and S. Paratyphi A is as set out below.
- OAg determination for the Salmonella Enteritidis, Typhimurium and Paratyphi A Each single OAg is hydrolysed to release its relative di-deoxy monosaccharide (SEn OAg to Tyvelose; STm OAg to Abequose; SPa OAg to Paratose), corresponding to the chromatographic peak, before analysis by HPAEC-PAD.
- di-deoxy are the only sugar that differs from the others among the ones composing
- SEn, STm and SPa OAg repeating units The sample is diluted by volume (450 pL) or by weight on analytical balance, with milliQ water, in order to be within the calibration curve range for each OAg.
- 120 pL of TFA 1 M is added to vials containing standards or samples and incubated at 75°C for 1.5 hours. After hydrolysis, the vials are chilled at 2-8°C for 15 minutes in the fridge. Samples and standards are dried overnight on centrifugal evaporator at room temperature (RT) in order to remove solvent/TFA. The pellet is dissolved in in 450 pL of milliQ water. Samples and standards are filtered on AcroPrep Advance 96 Filter Plate 0.2 pm Supor 1 mL well and plates loaded on HPAEC-PAD.
- the immunisations involved intraperitoneal immunisation of 200 pl of each formulation at day 0 and day 28. Blood samples were collected at days -1, 27 and 42, and the antibodies raised were studied using the assays described in Example 6 above. The results are set out in Figure 5.
- both pan-Salmonella vaccines induced specific serum IgG responses against S. Paratyphi A O-antigen, S. Typhimurium O-antigen, S. Enteritidis O- antigen and Vi polysaccharide, and the antibodies are bactericidal in mice.
- groups of 10 CD1 mice were immunised with:
- Quadrivalent Pan-Salmonella_O:2-CRM (STm GMMA + SEn GMMA + O:2-CRMi97 + fVi-CRMw?) produced as described in Example 10 at 1.0 (pg O-antigen), 1.0 (pg O- antigen), 1.25 (pg O-antigen) and 1.25 (pg Vi polysaccharide), respectively, dose Quadrivalent Pan-Salmonella_ParAGMMA (STm GMMA + SEn GMMA + ParA GMMA + fVi-CRMw?) produced as described in Example 10 at 1.0 (pg O-antigen), 1.0 (pg O-antigen), 1.17 (pg O-antigen) and 1.25 (pg Vi polysaccharide), respectively, dose
- ParA GMMA (produced as described in Example 8) adsorbed to Alhydrogel at 1.17 pg (Vi polysaccharide) dose
- the immunisations involved intraperitoneal immunisation of the corresponding dose for each product in 200 pl at days 0 and 28. Blood samples were collected at days 27 and 42, and the antibodies raised were measured using the ELISA assay and SBA assay described above in Example 6. Results are shown in Figure 6.
- Salmonella ParAGMMA is slightly different (about 7%) from the 0:2 dose used in the Pan-Salmonella_O:2-CRM.
- Presence of iNTS GMMA in the Pan-Salmonella formulation containing 0:2-CRM had overall a positive impact on the anti-0:2 IgG response elicited: a significantly higher post 1 response was induced by the quadrivalent formulation with 0:2-CRM compared with both bivalent 0:2-CRM + fVi-CRM and monovalent 0:2-CRM, and a significantly higher post 2 response was elicited in comparison with the bivalent 0:2-CRM + fVi-CRM formulation.
- SBA titers elicited by the Pan- Salmonella formulation with 0:2-CRM were significantly higher compared to the corresponding bivalent formulation. On the contrary, no significant differences were evidenced in the anti-0:2 IgG response elicited nor in the functionality of antibodies against ParA induced by Pan- Salmonella with ParA GMMA and by the monovalent formulation ParA GMMA.
- Example 12 Antibody subclasses raised by the quadrivalent Pan-Salmonella vaccines in mice
- the immunisations involved intraperitoneal immunisation of 200pl of each formulation at day 0 and day 28. Blood samples were collected at days -1, 27 and 42. The classes of antibodies raised against the Vi antigen polysaccharide or the S. Paratyphi A 0:2 O- Antigen were determined using the assay described in Example 6 under the heading “IgG subclasses The results are set out in Figure 7. The absolute values for Figure 7 are set out in the tables below:
- Pan-Salmonella vaccines induced specific serum IgG responses against S. Paratyphi A O-antigen, S. Typhimurium O-antigen, S. Enteritidis O- antigen and fVi polysaccharide, and the antibodies are bactericidal in rabbits.
- Example 14 administration of the iNTS-TCV vaccine to human subjects
- a phase l/2a, observer-blind, randomized, dose-escalation, controlled, multi-country, two- staged, and staggered study including 9 groups will be conducted to evaluate the safety, reactogenicity, and immune response of the trivalent iNTS-TCV vaccine against invasive nontyphoidal Salmonella (iNTS) and typhoid fever when administered intramuscularly on Day 1, Day 57 and Day 169 to healthy European and African adults, compared to placebo.
- the study will be conducted overall (both Stage 1 and Stage 2) with approximately 155 healthy adult participants (18 to 50 years of age).
- the healthy European adults will be randomly assigned to 1 of the groups indicated for Stage 1.
- the healthy African adults will be randomly assigned to 1 of the groups indicated for Stage 2.
- Each group will receive 2 of the 11 study interventions at each administration, except for the Control stage 2 group which will receive 4 study interventions (a different active comparator at each administration time point together with saline).
- Each participant will receive 1 randomly selected intramuscular study intervention per arm on Day 1, Day 57, and Day 169.
- Stage 1 (Europe) will follow a 2-step staggered design, leading in with low doses of all the study interventions, in a dose-escalation manner.
- the sentinel approach will be followed for the first 10 participants each in Step 1 and Step 2, in which only 1 participant will be treated daily. This will be done to ensure maximum safety of the participants.
- Step 1 10 healthy European adults, randomized in a 2:2:1 ratio, will receive:
- Step 2 40 healthy European adults will be randomized in a 2:2:1 ratio. A staggered approach will be followed for the first 10 sentinel participants and these participants will be followed up with a safety follow-up call on the next day of administration of study intervention. The remainder of the 30 participants will receive the study intervention in a sequential (at least 60 minutes apart) manner. The participants in Step 2 will receive:
- the first 21 participants in Stage 2 will initially be recruited with administration proceeding sequentially, at least 60 minutes apart. These participants will be followed up with a safety follow-up call on the next day of administration of study intervention.
- the recruitment of the remaining 84 participants in Stage 2 will only commence if there is a positive evaluation of all safety data from these participants up to 7 days after the first administration of the study intervention.
- the study interventions will be administered in parallel in the remaining 84 participants.
- composition of 0.5 mL of the full dose of the iNTS-TCV vaccine is as follows:
- composition of 0.5 mL of the low dose of the iNTS-TCV vaccine is as follows:
- CD1 mice were immunised intraperitoneally on days 0 and 28 using 500 pL injection volume of monovalent STm GMMA (dose per mouse per injection in pg 2.5 STm OAg) or of monovalent SEn GMMA (dose per mouse per injection in pg 2.5 SEn OAg). SBA was performed on mice sera obtained on day 42.
- mice sera elicited by STm GMMA demonstrated bactericidal activity against S. Typhimurium, S. Derby and S. Dublin ( Figure 9(a)).
- Mice sera elicited by SEm GMMA demonstrated bactericidal activity against S. Enteritidis and S. Dublin ( Figure 9(b)).
- CD1 mice were immunised intraperitoneally on days 0 and 28 using 200 pL injection volume of bivalent iNTS GMMA vaccine (STm and SEn GMMA) (Dose per mouse per injection in pg 1.0 STm OAg + 1.0 SEn OAg) or trivalent iNTS-TCV GMMA vaccine (STm and SEn GMMA and S. Typhi fVi polysaccharide) (Dose per mouse per injection in pg 1.0 STm OAg + 1.0 SEn OAg + 0.125 fVi) .
- SBA was performed on mice sera obtained on day 42.
- mice sera elicited by bivalent iNTS GMMA vaccine demonstrated bactericidal activity against S. Typhimurium, S. Enteritidis, S. Derby and S. Dublin ( Figure 9(c)).
- Mice sera elicited by trivalent iNTS-TCV vaccine also demonstrated bactericidal activity against S. Typhimurium, S. Enteritidis, S. Derby and S. Dublin ( Figure 9(d)).
- New Zealand female Rabbits were immunised intramuscularly on days 0 and 28 using 500 pL inj volume of bivalent iNTS GMMA vaccine (STm and SEn GMMA) (dose per rabbit per injection in pg 20 STm OAg + 20 SEn OAg) or trivalent iNTS-TCV GMMA vaccine (STm and SEn GMMA and S. Typhi fVi polysaccharide) (dose per rabbit per injection in pg 20 STm OAg + 20 SEn OAg + 25 fVi). SBA was performed on rabbit sera obtained on day 42.
- STm and SEn GMMA bivalent iNTS GMMA vaccine
- STm and SEn GMMA trivalent iNTS-TCV GMMA vaccine
- Typhi fVi polysaccharide dose per rabbit per injection in pg 20 STm OAg + 20 SEn OAg + 25 fVi.
- SBA was performed on rabbit sera obtained on day 42.
- Paratyphi A OAg conjugate Dose per mice per injection in pg 1 STm + 1 SEn + 1.25 ParA + 1.25 fVi); and (ii) STm and SEn GMMA, 5. Typhi fVi polysaccharide and 5.
- Paratyphi A GMMA Dose per mice per injection in pg 1 STm + 1 SEn + 1.17 ParA + 1.25 fVi). SBA was performed on mice sera obtained on day 42.
- An immunogenic composition comprising:
- a method of boosting an immune response to a S. Typhi or a S. Paratyphi A antigen comprising administering a composition comprising the S. Typhi antigen or the S. Paratyphi A antigen and GMMA.
- a method of preventing infection by S. Typhi or S. Paratyphi A comprising administering an immunogenic composition comprising the S. Typhi antigen or the S. Paratyphi A antigen and GMMA, wherein the GMMA boosts the immune response to the S. Typhi antigen or the S. Paratyphi A antigen.
- An immunogenic composition comprising GMMA for use in a method of boosting an immune response to a S. Typhi or S. Paratyphi A antigen, wherein the method comprises administering an immunogenic composition comprising the S. Typhi antigen or the S. Paratyphi A antigen and GMMA.
- An immunogenic composition for use in a method of preventing infection by S. Typhi or S. Paratyphi A comprising administering an immunogenic composition comprising the S. Typhi antigen or the S. Paratyphi A antigen and GMMA, wherein the GMMA boosts the immune response to the S. Typhi or the S. Paratyphi A antigen.
- the immunogenic composition for use of embodiments 3 or 5, wherein the method is a method of preventing infection by S. Typhi, the immunogenic composition comprises the S. Typhi antigen, and the GMMA boosts the immune response to the S. Typhi antigen.
- the immunogenic composition for use of embodiments 3 or 5, wherein the method is a method of preventing infection by S. Paratyphi A, the immunogenic composition comprises the S. Paratyphi A antigen, and the GMMA boosts the immune response to the S. Paratyphi A antigen.
- GMMA comprises at least one selected from the group consisting of S. Typhimurium GMMA, A Enteritidis GMMA, and A Paratyphi A GMMA.
- immunogenic composition or method of any one of the preceding embodiments, wherein the immunogenic composition further comprises:
- the immunogenic composition comprises a dose (O-antigen) of between 1 pg and 50 pg, between 2 pg and 25 pg, between 2 pg and 10 pg, between 15 pg and 25 pg, around 20 pg, or around 4 pg of S. Enteritidis antigen or S. Enteritidis GMMA.
- a dose O-antigen of between 1 pg and 50 pg, between 2 pg and 25 pg, between 2 pg and 10 pg, between 15 pg and 25 pg, around 20 pg, or around 4 pg of S. Enteritidis antigen or S. Enteritidis GMMA.
- the S. Typhi antigen comprises a fragmented Vi (fVi) polysaccharide.
- fVi conjugate obtained by or obtainable by a method comprising the steps of: a. fragmenting Vi polysaccharide to obtain a fragmented Vi (fVi) polysaccharide having an average molecular weight of between 40 kDa and 55 kDa, between 41 kDa and 49 kDa, or between 51 kDa and 55 kDa; b. activating the fVi polysaccharide by reacting the fVi polysaccharide obtained in step a.
- step b. reacting the N-hydroxysuccinimide ester fVi derivative obtained in step b. with the carrier protein to produce the fVi conjugate.
- the immunogenic composition or method of embodiment 56 wherein the immunogenic composition comprises S. Paratyphi A O-antigen conjugated to a carrier protein.
- the carrier protein is diphtheria toxoid or CRM197.
- the immunogenic composition or method of embodiment 10 or 65, wherein the S. Paratyphi A GMMA comprise modified lipid A.
- Paratyphi A antigen if the immune response raised to the S. Typhi antigen is higher when the S. Typhi antigen or the S. Paratyphi A antigen is part of the immunogenic composition comprising GMMA compared to the immune response raised when the S. Typhi antigen or the S. Paratyphi A antigen is not part of an immunogenic composition comprising GMMA. 85.
- the immunogenic composition or method of embodiment 84 or 85 wherein a method is a method of boosting an immune response to an S. Typhi or a S. Paratyphi A antigen or GMMA boosts the immune response to the S. Typhi antigen or the S. Paratyphi A antigen, if the immune response raised to the S. Typhi antigen or the S. Paratyphi A antigen is at least 5 times, at least 10 times, or at least 20 times higher when the S. Typhi antigen or the S. Paratyphi A antigen is part of the immunogenic composition comprising the GMMA compared to the immune response raised when the S. Typhi antigen or the S. Paratyphi A antigen is not part of an immunogenic composition comprising GMMA.
- the immunogenic composition or method of any one of embodiments 84 to 86, wherein the immune response raised to the S. Typhi antigen or the S. Paratyphi A antigen is the number of antibodies raised as determined by ELISA 42 days after administration of the immunogenic composition comprising the S. Typhi antigen or S. Paratyphi A antigen and the GMMA at a dose of 0.78 pg of S. Typhi antigen or S. Paratyphi A antigen and 0.63 pg (O-antigen) of the GMMA.
- the immunogenic composition or method of embodiment 88, wherein the immunogenic composition is tolerogenic if it induces a temperature rise of less than 1.8°C, less than 1.7°C, less than 1.6°C, or less than 1.5°C in a toxicity assay comprising the following steps:
- the immunogenic composition or method of embodiment 2 or 88, wherein the immunogenic composition is tolerogenic if it induces a maximum temperature of 41 °C or less, 40.9°C or less, or 40.8°C or less in a toxicity assay comprising the following steps:
- Typhimurium O-antigen antibodies induced is at least 90%, at least 95%, or at least 98% of the level of anti-5.
- immunogenic composition or method of any one of the preceding embodiments, wherein the immunogenic composition induces antibodies against three or more, four or more, five or more, six or more, seven or more, eight or more, nine or more, or all ten of the following strains:
- immunogenic composition or method of any one of the preceding embodiments, wherein the immunogenic composition further comprises an adjuvant.
- immunogenic composition or method of any one of the preceding embodiments, wherein the immunogenic composition further comprises a pharmaceutically acceptable excipient.
- the immunogenic composition or method of embodiment 107 wherein the pharmaceutically acceptable excipient comprises phosphate buffered saline.
- the phosphate buffered saline is at a pH between 6 and 7, or around 6.5.
- a vaccine comprising the immunogenic composition of any one of embodiments 1 or 11 to 109.
- a method of preventing an infection comprising administering an effective amount of the immunogenic composition or vaccine of any one of embodiments 1 or 11 to 110 to a subject.
- the immunogenic composition or vaccine for use of embodiment 111, or the use of embodiment 113, wherein the method of preventing an infection comprises administering an effective amount of the immunogenic composition or vaccine of any one of embodiments 1 or 11 to 110 to a subject.
- the immunogenic composition or vaccine for use, method, or use of any one of embodiments 111 to 114, wherein the method of preventing an infection is a method of preventing Salmonella infection.
- the immunogenic composition or vaccine for use, method, or use of any one of embodiments 111 to 115, wherein the method of preventing an infection is a method of preventing invasive non-typeable salmonella infection.
- the immunogenic composition or vaccine for use, method, or use of any one of embodiments 111 to 116, wherein the method of preventing an infection is a method of preventing infection by S. Typhimurium, S. Enteritidis, S. Typhi and/or S. Paratyphi A.
- the immunogenic composition, immunogenic composition or vaccine for use, method, or use of any one of embodiments 10 or 65 to 117, wherein the O-antigen/protein ratio of the S. Paratyphi A GMMA is at least 0.4.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Molecular Biology (AREA)
- Veterinary Medicine (AREA)
- Organic Chemistry (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- Genetics & Genomics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Biophysics (AREA)
- Virology (AREA)
- Toxicology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Biochemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Microbiology (AREA)
- Mycology (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Peptides Or Proteins (AREA)
Abstract
The present invention relates to immunogenic compositions comprising antigens from Salmonella enterica serovar Typhimurium (S. Typhimurium), Salmonella enterica serovar Enteritidis (S. Enteritidis), and Salmonella enterica serovar Typhi (S. Typhi). The present invention further relates to methods and uses of compositions comprising GMMA for boosting an immune response to an S. Typhi antigen, vaccines comprising the immunogenic compositions and methods and uses of the immunogenic compositions.
Description
Immunogenic composition
Field of the invention
The present invention relates to immunogenic compositions comprising antigens from Salmonella enterica serovar Typhimurium (S. Typhimurium), Salmonella enterica serovar Enteritidis (S. Entcritidis), and Salmonella enterica serovar Typhi (S. Typhi). The present invention further relates to methods and uses of compositions comprising GMMA for boosting an immune response to an S. Typhi antigen, vaccines comprising the immunogenic compositions and methods and uses of the immunogenic compositions.
Background to the invention
Typhoid fever is a bacterial disease caused by Salmonella enterica subspecies enterica serovar Typhi (Salmonella Typhi or S. Typhi), a human host-restricted organism [Crump, 2019]. The disease occurs globally, affecting predominantly children and young adults but is endemic in the developing countries of Africa and Asia, while in the developed countries it is reported occasionally in travellers that recently returned from endemic countries [Smith, 2016]. The exact burden of typhoid fever is said to be grossly underestimated due to difficulties in establishing its diagnosis in endemic areas. In 2017, there were estimated 10.9 million cases of typhoid fever and 116.8 thousand deaths due to Salmonella Typhi. Similarly, the years of life lost (YLLs) attributed to typhoid fever were 8.3 million and disability-adjusted life-years (DALYs) were 8.4 million. Despite a decline in the disease burden due to improvement in water and sanitation, it still remains a significant public health problem [Global Burden of Disease, 2017]. The burden of typhoid fever is highest in school aged children and in the under-5 age group. Recent studies show that in the 5-9 years age group, the adjusted incidence of blood-culture-confirmed typhoid fever/100,000 person-years of observation with 95% confidence interval ranged from 861 (599-1203) in Malawi to 3228 (2276-4757) in Bangladesh while in the 0-4 years ago group, it was 632 (398-965) and 2625 (1764-4244) in Malawi and Bangladesh respectively [Meiring, 2021], In the absence of prompt diagnosis and treatment, intervention, typhoid fever may require hospitalization and potentially fatal complications such as Typhoid Intestinal Perforation
(TIP). In developing countries where typhoid fever is endemic, surgical intervention is often delayed thus further worsening the outcome of the disease [Contini, 2017].
Antimicrobial treatment of typhoid fever is hampered by the emergence of multidrugresistant (MDR) typhoidal Salmonella, first identified in 1980 and defined as strains resistant to ampicillin, chloramphenicol and trimethoprim sulfamethoxazole. The emergence of resistant strains of the bacteria has been somewhat overcome with newer antimicrobials, but the challenge remains and hampers effective control of the disease [Radhakrishnan, 2018]. Similarly, S. Typhi clones harboring resistance to three first-line drugs (chloramphenicol, ampicillin, and trimethoprim-sulfamethoxazole) as well as fluoroquinolones and third generation cephalosporins, which are classified as Extremely Drug Resistant (XDR) has been reported in Asia [Klemm, 2018].
S. Paratyphi A resides in the human gut and its clinical manifestations are indistinguishable from Typhoid fever. S. Paratyphi A is ranked second as a causative agent of enteric fever, preceded only by Salmonella enterica serovar Typhi (5. Typhi). Enteric fever caused by S. Paratyphi A, or Paratyphoid fever was thought to be responsible for a comparatively smaller proportion of enteric fever cases. However, since the 1980s both the incidence and relative frequency of Paratyphoid fever have risen in Nepal, Pakistan, and Thailand. Moreover, the populous nations of India and China have reported substantial numbers of S. Paratyphi A cases. Non-endemic countries like the United States report an increasing trend of Paratyphoid fever especially, amongst travelers from South Asia (Irfan et al, Ceftriaxone resistant Salmonella enterica serovar Paratyphi A identified in a case of enteric fever: first case report from Pakistan. BMC Infect Dis. 2023 Apr 26;23(1):267. doi: 10.1186/sl2879-023-08152-9. Erratum in: BMC Infect Dis. 2023 May 23;23(1):346.
PMID: 37101111; PMCID: PMC10132421).
Accordingly, there is a need for improved vaccines against Salmonella.
Summary of the invention
The present Examples demonstrate that a trivalent vaccine comprising antigens (GMMA) from S. Typhimurium, S. Enteritidis, and S. Typhi is safe and highly immunogenic. Similarly, the present Examples demonstrate that quadrivalent vaccines comprising the trivalent vaccine and an antigen from S. Paratyphi A (GMMA or an O-antigen conjugate) are also highly immunogenic, and that no antigen interference is observed between the four antigens. Furthermore, the Examples demonstrate that GMMA (from S. Enteritidis and/or S. Typhimurium) can boost the immune response against an antigen from S. Typhi (fVi conjugated to CRM197).
In a first aspect of the invention, there is provided an immunogenic composition comprising:
(a) a Salmonella enterica serovar Typhimurium (S. Typhimurium) antigen;
(b) a Salmonella enterica serovar Enteritidis (S. Enteritidis) antigen; and
(c) a Salmonella enterica serovar Typhi (S. Typhi) antigen.
In a second aspect of the invention, there is provided a method of boosting an immune response to a S. Typhi or a S. Paratyphi A antigen comprising administering a composition comprising the S. Typhi antigen or the S. Paratyphi A antigen and GMMA.
In a third aspect of the invention, there is provided a method of preventing infection by S. Typhi or S. Paratyphi A comprising administering an immunogenic composition comprising the S. Typhi antigen or the S. Paratyphi A antigen and GMMA, wherein the GMMA boosts the immune response to the S. Typhi antigen or the S. Paratyphi A antigen.
In a fourth aspect of the invention, there is provided an immunogenic composition comprising GMMA for use in a method of boosting an immune response to a S. Typhi or S. Paratyphi A antigen, wherein the method comprises administering an immunogenic composition comprising the S. Typhi antigen or the S. Paratyphi A antigen and GMMA.
In a fifth aspect of the invention, there is provided an immunogenic composition for use in a method of preventing infection by S. Typhi or S. Paratyphi A comprising administering an immunogenic composition comprising the S. Typhi antigen or the S. Paratyphi A antigen and GMMA, wherein the GMMA boosts the immune response to the S. Typhi or the S. Paratyphi A antigen.
In a sixth aspect of the invention, there is provided a vaccine comprising the immunogenic composition of the invention.
In a seventh aspect of the invention, there is provided a method of preventing an infection comprising administering an effective amount of the immunogenic composition or vaccine of the invention.
In an eighth aspect of the invention, there is provided a use of the immunogenic composition or vaccine of the invention, for the manufacture of a medicament for use in a method of preventing an infection.
Brief description of the figures
Figure 1. iNTS-TCV vaccine induces specific serum IgG responses against the target antigens and antibodies are bactericidal in mice. Study design: iNTS-TCV drug product; 8 mice/group; Immunization IP: days 0, 28; Bleeds: days 27, 42; Toxicology lot at time zero. Figure 2. Anti-STm OAg IgG response in mice of STmGMMAAlhydrogel vs iNTS- TCV2 on Days 27 (Figures 2(a)) & 42 (Figure 2(b)).
Anti-SEn OAg IgG response in mice of SEnGMMAAlhydrogel vs iNTS-TCV2 on Days 27 (Figure 2(c)) & 42 (Figure 2(d)).
Anti-Vi IgG response in mice of Vi-CRM197 vs iNTS-TCV2 on Days 27 (Figure 2(e)) & 42 (Figures 2(f)).
Figure 3. Anti-STm OAg and SEn OAg IgG antibody units detected in individual rabbit sera for bivalent iNTS-GMMA (full dose of 40 ug).
Figure 4. Reaction scheme for conjugation of S. Paratyphi A O-antigen to CRM197 by a random CDAP chemistry approach.
Figure 5. Both Pan-Salmonella formulations induce specific serum IgG responses against the 4 antigens and antibodies are bactericidal in mice.
Figures 5(a)-(d) show IgG responses at: one day before immunisation (left-hand column), 27 days after immunisation (middle column) and 42 days after immunisation (right-hand column).
Figures 5(e)-(g) show SBA results. In Figure 5(e), the left-hand column (for each of O:2-CRM197 and ParA GMMA) is one day before immunisation and the right-hand column (for each of O:2-CRM197 and ParA GMMA) is 42 days after immunisation. In Figures 5(f) and (g), each bar is for 42 days after immunisation.
Figure 6. Evaluation of immuno-interference among vaccine components in PanSalmonella formulations
Figures 6(a), (c), (e) and (g) show IgG responses at: one day before immunisation (left-hand column), 27 days after immunisation (middle column) and 42 days after immunisation (right-hand column).
Figures 6(b), (d) and (f) show SBA results. In Figure 6(b), the left-hand column is one day before immunisation and the right-hand column is 42 days after immunisation. In Figures 6(d) and (f), each bar is for 42 days after immunisation.
Figure 7. Subclasses relative abundance %, calculated as subclass/subclasses total %. Top segment is IgG3, next segment is IgG2b, next segment is IgG2a and bottom segment is IgGl.
Figure 8. Both quadrivalent Pan-Salmonella formulations induce specific serum IgG responses against the 4 antigens and antibodies are bactericidal also in rabbits.
Figures 8(a)-(d) show IgG responses at: one day before immunisation (left-hand column), 27 days after immunisation (middle column) and 42 days after immunisation (right-hand column).
Figures 8(e)-(g) show SBA results, where the left-hand column (for each of 0:2- CRM197 and ParA GMMA) is one day before immunisation and the right-hand column (for each of O:2-CRM197 and ParA GMMA) is 42 days after immunisation.
Figure 9. SBA on heterologous panel of mice sera elicited by monovalent components (STm or SEn GMMA) (Figures 9(a) and (b)).
SBA on heterologous panel of mice sera elicited by bivalent vaccine (STm and SEn GMMA) (Figure 9(c)) and trivalent vaccine iNTS-TCV (STm and SEn GMMA and fVi polysaccharide from S. Typhi (Figure 9(d)).
SBA on heterologous panel of rabbit sera elicited by bivalent vaccine (STm and SEn GMMA) (Figure 9(e)) and trivalent vaccine iNTS-TCV (STm and SEn GMMA and fVi polysaccharide from S. Typhi) (Figure 9(f)).
Figure 10. Quadrivalent Pan-Salmonella formulations elicit bactericidal antibodies against a broad panel of Salmonella strains. Panel includes invasive STm isolates from Africa and Southeast Asia and S. enterica serovars other than STm, SEn, ParA, Typhi.
Figure 11. Published structure of the O-antigen (including the core domain) from S. Paratyphi A.
Figure 12. CRM197 sequence.
General definitions
Unless defined otherwise, technical and scientific terms used herein have the same meaning as commonly understood by a person skilled in the art to which this invention belongs.
In general, the term “comprising” is intended to mean including but not limited to. For example, the phrase “An immunogenic composition comprising a Salmonella Typhimurium antigen” should be interpreted to mean that the immunogenic composition comprises a Salmonella Typhimurium antigen, but the immunogenic composition may comprise further components.
In some embodiments of the invention, the word “comprising” is replaced with the phrase “consisting of. The term “consisting of is intended to be limiting. For example, the phrase “An immunogenic composition consisting of a Salmonella Typhimurium antigen” should be understood to mean that the immunogenic composition has the Salmonella Typhimurium antigen and no further components.
In some embodiments of the invention, the word “comprising” is replaced with the phrase “consisting essentially of” . The term “consisting essentially of’ means that specific further components can be present, namely those not materially affecting the essential characteristics of the subject matter.
The term “about” or “around” when referring to a value refers to that value but within a reasonable degree of scientific error. Optionally, a value is “about x” or “around x” if it is within 10%, within 5%, or within 1% of x.
The singular forms “a”, “an ”, and “the ” include plural referents unless the content clearly dictates otherwise. Thus, for example, reference to “the GMMA” includes two or more instances or versions of such GMMA.
All publications, patents and patent applications cited herein, whether supra or infra, are hereby incorporated by reference in their entirety.
Detailed description
A Salmonella Typhimurium antigen, a Salmonella Enteritidis antigen and a Salmonella Paratyphi A antigen
In some aspects, the immunogenic composition of the invention comprises a Salmonella Typhimurium (S. Typhimurium) antigen. In some aspects, the immunogenic composition of the invention comprises a Salmonella Enteritidis (S. Enteritidis) antigen. In some aspects, the immunogenic composition of the invention comprises a Salmonella Paratyphi A (S. Paratyphi A) antigen.
O-antigen
Various S. Typhimurium, S. Enteritidis and S. Paratyphi A antigens are known to the skilled person. In particular, the S. Typhimurium, S. Enteritidis and S. Paratyphi A bacteria
all comprise an outer membrane comprising O-antigen, and the S. Typhimurium, S. Enteritidis and/or the S. Paratyphi A antigen may comprise the O-antigen.
For the purposes of the present invention, the terms O-antigen, OAg and 0:2 are considered to be interchangeable. The outer membrane of gram-negative bacteria comprise a lipopolysaccharide. This lipopolysaccharide comprises an O-antigen, which is linked via to the core domain to a lipid A domain. The terms “O-antigen” “OAg” and “0:2 ” refer to a polysaccharide made up of the O-antigen alone, or more preferably the O- antigen linked to core domain of the lipopolysaccharide.
As discussed above, the S. Typhimurium antigen, the S. Enteritidis antigen and/or the S. Paratyphi A may be an O-antigen. A typical process for the purification of these O- antigens is based on the phenol-water method of Westphal and Jann, first described in the 1960s (Westphal and Jann (1965) Methods Carbohydr. Chem. 5:83-91), followed by detoxification of the lipopolysaccharide with acetic acid or anhydrous hydrazine. The O- antigen is modified to remove the lipid A. For example, extraction and purification of polysaccharide can be performed by acetic acid hydrolysis as described in for example Watson et al., (1992) Infect Immun. 60(11):4679-86; - Konadu et al. (1996) Infect Immun. (7):2709-15; Konadu et al. (1994) Infect Immun. 62(11):5048-54; Ahmed et al. (2006) J Infect Dis. 193(4):515-21 ; Cox et al. (2011) Glycoconj J 28 :165-182; Chu ct a/. (1991) Infect Immun. 59(12):4450-58; and Micoli et al., 2012 PlosOne, 7(11): e47039.
The O-antigens of Salmonella serogroups A, B and D have been described and are thought to share a common backbone: -^■2-a-D-Man/?-(l -^4)-a-L-Rha/?-(l— >3)-a-D-Gal/?-(l— >. The serogroup specificity of Salmonella Paratyphi A is conferred by an a-3,6- dideoxyglucose (a-D-paratose) linked (1— 3) to the mannose of the backbone. The a-L- rhamnose of the backbone is partially O-acetylated at C-3 (Konadu et al. (1996) Infect Immun. (7):2709-15). The published structures of the O-antigen from S. Paratyphi A is shown in Figure 11, including the KDO subunit and primary amine group (within a pyrophosphoethanolamine group) in the core domain.
O-antigen conjugates
In embodiments where the S. Typhimurium, S. Enteritidis and/or the S. Paratyphi A antigen comprise the O-antigen, the O-antigen from S. Typhimurium, S. Enteritidis and/or the S. Paratyphi A may be part of a conjugate. The term “conjugate” refers to a molecule formed by a covalent linkage between an antigen (like O-antigen) and a carrier. The carrier may be a carrier protein. In general, conjugation of polysaccharides to carrier proteins enhances the immunogenicity of the polysaccharides as it converts them from T- independent antigens to T-dependent antigens, thus allowing priming for immunological memory.
Carrier proteins include bacterial toxins, such as diphtheria or tetanus toxins, or toxoids or mutants thereof. In some embodiments, the carrier protein is CRM197. The sequence of CRM197 is provided in Figure 12 (SEQ ID NO: 1).
In particular embodiments, the S. Paratyphi A antigen comprises S. Paratyphi A O-antigen conjugated to a carrier protein. The carrier protein may be diphtheria toxoid or CRM197. Optionally, the carrier protein is CRM197.
The O-antigen from S. Typhimurium, S. Enteritidis and/or the S. Paratyphi A may be conjugated to the carrier protein by a method that comprises introducing more than one activated site into the O-antigen. Optionally, the O-antigen from S. Typhimurium, S. Enteritidis and/or the S. Paratyphi A comprises more than one activated site.
The term “activated site” is intended to refer to a site or functional group on the polysaccharide that has been activated by a step in a conjugation chemistry method such that it is primed to be conjugated to a carrier protein. For example, if the P-antigen is conjugated by a method using CDAP chemistry, the polysaccharide is “activated” by the addition of CDAP if the addition of CDAP introduces cyanoester groups. The CDAP activation introduces cyanoester groups at one or more sites, and the positions of these introduced cyanoester groups would be considered to be “activated sites Once the O- antigen has been activated, it may be linked (conjugated) to a carrier protein at one or more
(in some cases all) of the activated sites. For the purposes of the present invention, the term “activated sites'” includes sites that have been activated and not linked to carrier protein and also sites that have been activated and are linked to a carrier protein.
Optionally, the O-antigen from S. Typhimurium, S. Enteritidis and/or the S. Paratyphi A comprises 1.5 or more, 2.0 or more, or 2.5 or more activated sites. Assuming that the O- antigen is part of a composition comprising multiple O-antigen saccharides, the O-antigen will comprise 1.5 or more activated sites if the average number of activated sites on each O-antigen molecule in the composition is 1.5 or more.
Optionally, the S. Paratyphi A antigen is an O-antigen conjugated to a carrier protein, and the O-antigen is conjugated to the carrier protein by a method that comprises introducing more than one activated site into the S. Paratyphi O-antigen and/or the S. Paratyphi A O- antigen comprises more than one activated site.
The O-antigen from S. Typhimurium, S. Enteritidis and/or the S. Paratyphi A may be conjugated to the carrier protein by CDAP chemistry, optionally via a linker.
A linker is a compound that can be used to link a protein and a polysaccharide. Any suitable linker may be used in the conjugates and methods of the invention. Suitable linkers include an adipic acid dihydrazide (ADH) linker, which is a compound having the following structure:
 Other suitable linkers include adipic acid, glutaric acid, carbonyl, P-propionamido (WO00/10599), adipic acid bis(N-hydroxysuccinimmide), dihydrazides analogous to ADH but with different chain lengths, hexamethylenediamine (or analogous diamines with different chain lengths), nitrophenyl-ethylamine (Gever ct al. (1979) Med. Microbiol. Immunol. 165, 171-288), haloacyl halides (U.S. Pat. No. 4,057,685), glycosidic linkages (U.S. Pat. Nos. 4,673,574; 4,761,283; and 4,808,700), 6-aminocaproic acid (U.S. Pat. No. 4,459,286), N- succinimidyl-3-(2-pyridyldithio)-propionate (SPDP) (U.S. Pat. No. 5,204,098), C4 to C12 moieties (U.S. Pat. No. 4,663, 160), etc.
Other suitable linkers include adipic acid, glutaric acid, carbonyl, P-propionamido (WO00/10599), adipic acid bis(N-hydroxysuccinimmide), dihydrazides analogous to ADH but with different chain lengths, hexamethylenediamine (or analogous diamines with different chain lengths), nitrophenyl-ethylamine (Gever ct al. (1979) Med. Microbiol. Immunol. 165, 171-288), haloacyl halides (U.S. Pat. No. 4,057,685), glycosidic linkages (U.S. Pat. Nos. 4,673,574; 4,761,283; and 4,808,700), 6-aminocaproic acid (U.S. Pat. No. 4,459,286), N- succinimidyl-3-(2-pyridyldithio)-propionate (SPDP) (U.S. Pat. No. 5,204,098), C4 to C12 moieties (U.S. Pat. No. 4,663, 160), etc.
In embodiments where the O-antigen from S. Typhimurium, S. Enteritidis and/or the S. Paratyphi A is/are conjugated to the carrier protein by CDAP chemistry, the O-antigen may be conjugated to the carrier protein by a method comprising a step of activating the O- antigen by CDAP chemistry to provide an activated O-antigen.
Activating the O-antigen by CDAP chemistry comprises mixing the O-antigen with CDAP such a way that cyanoester groups are introduced into the polysaccharide or O-antigen.
For example, Example 9 discloses a suitable method of activating an O-antigen by CDAP chemistry. Activating the O-antigen by CDAP chemistry results in an activated O-antigen. Activating the O-antigen by CDAP chemistry introduces cyanoester groups, and so a method comprises a step of activating an O-antigen by CDAP chemistry if the method comprises mixing the O-antigen with CDAP and the number of cyanoester groups present on the O-antigen after the step of mixing with CDAP is higher than the number of cyanoester groups present on the O-antigen prior to that step. The number of cyanoester groups present may be measured using the ADH quenching/TNBS colourimetric method as reported in Lees A., Vaccines (Basel), 2020; 8(4):777.
Optionally, activating the O-antigen by CDAP chemistry comprises mixing the O-antigen with CDAP at a w/w ratio of between 0.05:1 and 5:1, between 0.1:1 and 5:1, between 0.2:1 and 2:1, or around 0.3:1 (CDAP to O-antigen).
Optionally, the step of activating the O-antigen by CDAP chemistry comprising mixing the O-antigen with CDAP takes place in a salt solution, such as a solution of NaCl or KC1.
Optionally, the step of activating the O-antigen by CDAP chemistry comprising mixing the O-antigen with CDAP takes place in a solution of NaCl or KC1 at a concentration between 50 mM and IM, between 100 mM and 250 mM, between 125 mM and 200 mM, or around 150 mM.
Optionally, after the O-antigen has been mixed with the CDAP the pH is adjusted, optionally to a pH between 6 and 10, between 7 and 9, or between 9 and 10. Optionally, the pH is adjusted by adding a base, such as triethylamine, sodium hydroxide, or pyridine. Optionally, the pH is adjusted by adding between 5 % and 15%, between 8% and 12%, or around 10% (v/v) triethylamine. Optionally, after the O-antigen has been mixed with the CDAP, the mixture is incubated at a temperature between 18°C and 30°C, between 20°C and 28°C, room temperature, or around 25°C. Optionally, the solution is incubated with stirring prior to conjugation of the activated O-antigen to the carrier protein.
A suitable method for activating the polysaccharide by CDAP chemistry is described in Example 9.
As set out above, O-antigens that have been activated using CDAP chemistry (activated polysaccharides or activated O-antigens) comprise cyanoester groups (at activated sites), and these cyanoester groups may be covalently linked to hydrazide or amino groups. Accordingly, O-antigens that have been activated using CDAP chemistry may be linked directly to carrier proteins (via amino groups), or may be conjugated to a carrier protein via a linker comprising a hydrazide or amino group. Suitable linkers include the ADH linker described above. Accordingly, the activated O-antigen may be conjugated to the carrier protein by a method comprising reacting the activated O-antigen with hydrazide/amino groups on a carrier protein or a carrier-protein linker compound. Thus, the method may further comprise steps to prepare the carrier protein-linker compound. For example, if the linker is an ADH linker, the method may comprise a step of preparing an ADH-carrier protein compound (such as an ADH-CRM197 compound), for example as reported in Micoli et al. Vaccine 2011, 29, (4), 712-20.
Reacting the activated O-antigen with hydrazide/amino groups on a carrier protein or a carrier-protein linker compound may comprise mixing the carrier protein or the carrierprotein linker compound with the activated O-antigen under conditions suitable for a covalent bond to be formed between the cyanoester groups (activated sites) on the activated O-antigen and the hydrazide/amino groups on the carrier protein or the carrier protein-linker compound. For example, it may be simply a case of mixing the activated O- antigen with the carrier protein or the carrier protein-linker compound.
Reacting the activated O-antigen with hydrazide/amino groups on the carrier protein or a carrier protein-linker compound may comprise mixing the activated O-antigen with the carrier protein or the carrier protein-linker compound at a w/w ratio of between 0.1:1 and 5:1, between 0.2:1 and 3:1, between 0.5:1 and 2:1, or around 1 : 1 (O-antigen to carrier protein or carrier protein-linker). Optionally, the step of mixing the activated O-antigen with the carrier protein or the carrier protein-linker provides a conjugation mixture. Optionally, mixing the activated O-antigen with the carrier protein or the carrier proteinlinker compound takes place at a pH between 8 and 11, between 9 and 10, or around 9.5. Optionally, the pH is maintained at between 8 and 11, between 9 and 10, or around 9.5 for at least 1 hour, at least 2 hours, between 30 minutes and 10 hours, between 1 hour and 5 hours, or between 2 hours and 3 hours. Optionally, the pH is maintained using a base, such as triethylamine, sodium hydroxide, or pyridine. Optionally, the pH is maintained using triethylamine.
Optionally, after the activated O-antigen has been mixed and optionally after the mixture has been maintained at a pH between 8 to 11 for at least 1 hour, the method may further comprise a step of adding glycine solution (to quench the cyanoester groups). Optionally, the glycine solution is adding in a concentration of 0.5M to 5M, 0.5M to 2M, or around IM. Optionally, the glycine solution is added to a volume of the conjugation mixture with is substantially equal to the volume of the glycine solution. A volume is substantially equal to another volume if it is within 10%. Optionally, there is a further step of adjusting the pH using a base, such as triethylamine, sodium hydroxide, or pyridine. Optionally this further step occurs after a step of adding a glycine solution. Optionally, the pH is adjusted to a pH between 7 and 9, or around 8. Optionally, there is an incubation step after the
further step of adjusting the pH using a base. Optionally, the incubation step comprises incubating at a temperature below 15°C, between 12°C, below 10°C, between 0°C and 10°C, or between 2°C and 8°C. Optionally, the incubation step takes place for between 10 and 30 hours, or between 10 and 20 hours.
Once the activated O-antigen has been mixed with the carrier protein or the carrier proteinlinker compound under suitable conditions for conjugation to occur (such as those set out in the two preceding paragraphs), a conjugate will be formed. The method may further comprise a chromatography step to remove any unconjugated O-antigen. Optionally, the chromatography step comprises hydrophobic interaction chromatography or anion exchange chromatography.
A suitable method for conjugating a polysaccharide activated using CDAP to a carrier protein or a linker is described in Example 9.
Outer membrane vesicles
In some aspects, the S. Typhimurium, S. Enteritidis and/or the S. Paratyphi A antigen comprises or consists of outer membrane vesicles such as GMMA.
For the purpose of the present invention, the terms “outer membrane vesicle” or “O F” are considered to be interchangeable and to refer to any type of outer membrane vesicle. Suitable OMVs include native OMVs. Gram-negative bacteria can spontaneously release outer membrane vesicles (OMVs) during growth due to the turgor pressure of the cell envelope, and these are native OMVs. OMVs are rich in immunogenic cell surface- associated, periplasmic and secreted antigens and have been used as vaccines.
OMVs of the invention include Generalised Modules for Membrane Antigens (GMMA), native OMVs (‘NOMVs’ (see Katial et al. 2002, Infect Immun, 70: 702-707), microvesicles (MVs (see WO 02/09643)), detergent-extracted OMVs (DOMVs), mutant- derived OMVs (m-OMV), and blebs, which are outer-membrane protrusions that remain
atached to bacteria prior to release as MVs (see Beveridge, 1999, J. Bacteriol. 181: 4725- 4733)).
Generalised Modules for Membrane Antigens (GMMA) are a type of OMV. GMMA are distinct from native outer membrane vesicles (NOMV), which are released spontaneously from Gram-negative bacteria, in two crucial aspects. First, to induce GMMA formation, the membrane structure has been modified by the deletion of genes encoding key structural components, such as tolR (leading to hyperblebbing). Second, as a consequence of the genetic modification, large quantities of outer membrane “bud off' (or “hyperbleb”) to provide a practical source of membrane material for vaccine production. Accordingly, for the purpose of the present invention, the term “GA/M4” refers to OMVs which are released spontaneously from bacteria modified to hyperbleb (such as Salmonella bacteria which are modified such that they do not comprise a gene encoding functional TolR).
The Gram-negative bacteria from which the OMVs (such as GMMA) of the invention are purified may be one or more of the group consisting of: Salmonella enterica subspecies enterica serovar Typhimurium (Salmonella Typhimurium), Salmonella enterica subspecies enterica serovar Enteritidis (Salmonella Enteritidis), and Salmonella enterica subspecies enterica serovar Paratyphi A (Salmonella Paratyphi A). Suitable purification methods are known in the art, and include a variety of filtration and chromatography methods. A preferred two-step filtration purification process is described in WO 2011/036562 herein incorporated by reference.
Modification of lipid A
In some aspects, the S. Typhimurium, S. Enteritidis and/or the S. Paratyphi A antigen comprises or consists of GMMA, i.e. S. Typhimurium, S. Enteritidis and/or S. Paratyphi A GMMA. The S. Typhimurium, S. Enteritidis and/or S. Paratyphi A GMMA may comprise modified lipid A or may be derived from S. Typhimurium, S. Enteritidis and/or S.
Paratyphi A bacteria that comprise modified lipid A. A modified lipid A is a lipid A that has a different structure compared to a corresponding wild type lipid A.
The structure of lipid A may be determined using MALDI-TOF analysis of lipid A isolated from the GMMA. For the assay, the lipid A is separated after treatment of GMMA with acetic acid and then assayed by MALDI-TOF. GMMA with a protein concentration of about 1 mg/mL (micro BCA calibration curve) or a cell bank suspension with an OD600 of about 3 (4 mL sample) are treated with 1% acetic acid (final concentration) for 2 or 6 hours, respectively, at 100°C to obtain a precipitate containing the lipid A. The precipitate is then collected, washed with water and the lipid A is extracted in chloroform / methanol 4:1. The final solution, which contains the lipid A, is mixed 1 : 1 with Super DHB (Fluka, 50862) saturated solution (acetonitrile / water 1:1). Two microliters of the mixture are loaded onto the target plate and after the spot is dried at room temperature, the plate is inserted in the mass spectrometer. The spectra (negative reflectron mode) generally show peaks corresponding to the lipid A molecular species and contain several peaks due to fragmentation of the lipid A (i.e. loss of one or more fatty acid chains), sodium adduct (+22 m/z) and lipid A dephosphorylation (- 80 m/z). The species of lipid A is identified by comparison of the molecular peak mass m/z to what is expected for the sample in analysis.
Optionally, the lipid A is modified to be detoxified i.e. the modified lipid A is detoxified lipid A). “Detoxified" means that the lipid A is less toxic than wildtype lipid A. The wildtype lipid A used in the comparison is a corresponding wildtype lipid A. “Toxicity" or “toxic" in this context refers to the extent to which the innate immune system is activated by lipid A, particularly through the Toll-like receptor 4 pathway. Highly toxic lipid A can lead to uncontrolled inflammation, apoptosis, and in extreme cases septic shock, among other effects. Optionally, a modified lipid A is less toxic if it is less reactogenic than a corresponding wildtype lipid A. For example, one can determine whether a modified lipid A is less toxic by administering it to an animal such as a rabbit, and determining whether it activated more monocytes compared to a corresponding wildtype lipid A using a monocyte activation test.
 refers to lipid A that can be found in the corresponding wildtype bacterium and strain. For example, lipid A that is modified relative to a “corresponding wildtype lipid in the context of S. Typhimurium GMMA is interpreted
to mean a lipid A that is modified (e.g. such that it is less toxic) relative to lipid A found in wildtype S. Typhimurium.
refers to lipid A that can be found in the corresponding wildtype bacterium and strain. For example, lipid A that is modified relative to a “corresponding wildtype lipid in the context of S. Typhimurium GMMA is interpreted
to mean a lipid A that is modified (e.g. such that it is less toxic) relative to lipid A found in wildtype S. Typhimurium.
Optionally, the modified lipid A is penta-acylated lipid A. One may determine whether a GMMA comprises penta-acylated lipid A by determining the structure of the lipid A using MALDI-TOF analysis as set out above.
The S. Typhimurium, S. Enteritidis and/or S. Paratyphi A GMMA may be derived from S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria that comprise any suitable modification that leads to production of GMMA comprising lipid A that is less toxic than wildtype lipid A.
HtrB, MsbB and PagP are proteins that are involved in production of lipid A in Gramnegative bacteria. Of these, MsbB and PagP are important in Salmonella. Salmonella bacteria that do not express functional versions of MsbB and/or PagP will not produce native lipid A, but rather will produce modified, detoxified lipid A. Thus, the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A GMMA may be derived from S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria that do not express functional versions of MsbB and/or PagP. Optionally, the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A GMMA are derived from S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria that do not comprise a gene encoding a functional MsbB and/or PagP protein.
Whether or not the bacteria from which GMMA is derived express functional versions of MsbB and/or PagP or comprise a gene encoding a functional MsbB and/or PagP protein may be determined by isolating lipid A from the GMMA and analysing its structure by MALDI-TOF as described above. If the lipid A is detoxified then the bacteria from which the GMMA is derived do not express functional versions of Msb and/or PagP or comprise a gene encoding a functional MsbB and/or PagP protein.
Optionally, the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived do not comprise a gene (such as htrB, msbB, and/or pagP) encoding a functional protein because they comprise a mutation in that gene. Optionally,
the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived do not comprise a gene encoding a functional HtrB, MsbB, and/or PagP protein. Optionally, the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived comprise a gene encoding at least a portion of the HtrB, MsbB, and/or PagP protein, but either the gene is mutated such that the HtrB, MsbB, and/or PagP protein encoded is missing one or more important amino acids or a portion of the gene is deleted. For example, the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived may comprise a substitution or deletion mutation in the htrB, msbB, and/or pagP gene. Alternatively, the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived may have an addition mutation in the htrB, msbB and/or PagP gene, for example an addition mutation causing a frame shift. Optionally, the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived comprises a deletion mutation in the htrB, msbB,, and/or pagP gene. Optionally, the htrB, msbB, and/or pagP gene comprises a deletion mutation, and at least 10%, at least 20%, at least 25%, at least 50%, or at least 75% of the htrB, msbB, and/or pagP gene is deleted. Optionally, the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived lacks a htrB, msbB, and/or pagP gene (for example because the complete htrB, msbB, and/or pagP gene has been deleted (a tshtrB, tsmsbB, and/or tSpagP mutation)).
Optionally, the S. Paratyphi A OMVs or GMMA are derived from S. Paratyphi A where at least a part of the msbB and/or pagP gene has been replaced by a different gene.
Optionally, the S. Paratyphi A OMVs or GMMA are derived from S. Paratyphi A where at least a part of the msbB and/or pagP gene has been replaced by a tetracycline (tet) or kanamycin (kan) gene respectively. Optionally, the S. Paratyphi A OMVs or GMMA are derived from S. Paratyphi A where at least a part of the msbB and/or pagP gene has been replaced by a tetracycline (tet) or kanamycin (kan) gene respectively. Optionally, the S. Paratyphi A OMVs or GMMA are derived from S. Paratyphi A which is pagP::kan and/or refers to the gene before the
 being replaced by the gene after
being replaced by the gene after

 , gP::kan” means that the pagP gene is replaced by kanamycin. Hyperblebbing
The S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived may have been modified (for example genetically modified) to hyperbleb i.e. more quantities of outer membrane “bud off' compared to a corresponding Gram-negative bacterium that does not have the genetic mutation.
, gP::kan” means that the pagP gene is replaced by kanamycin. Hyperblebbing
The S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived may have been modified (for example genetically modified) to hyperbleb i.e. more quantities of outer membrane “bud off' compared to a corresponding Gram-negative bacterium that does not have the genetic mutation.
The S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived may comprise any suitable modification that leads to hyperblebbing.
Optionally, the modification is a mutation, for example the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived may not comprise a gene (such as tolR) encoding a functional protein because it comprises a mutation in that gene. Optionally, the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived do not comprise a gene encoding a functional TolR protein. Optionally, the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived comprise a gene encoding at least a portion of the TolR protein, but either the gene is mutated such that the TolR protein encoded is missing one or more important amino acids or a portion of the gene is deleted. For example, the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived may comprise a substitution or deletion mutation in the tolR gene. Alternatively, the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived may have an addition mutation in the tolR gene, for example an addition mutation causing a frame shift. Optionally, the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived comprises a deletion mutation in the tolR gene. Optionally, the tolR gene comprises a deletion mutation, and at least 10%, at least 20%, at least 25%, at least 50%, or at least 75% of the tolR gene is deleted. Optionally, the S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived lacks a tolR gene (for example because the complete tolR gene has been deleted (a AtolR mutation)).
Optionally, the S. Paratyphi A OMVs or GMMA are derived from S. Paratyphi A where at least a part of the tolR gene has been replaced by a different gene. Optionally, the S. Paratyphi A OMVs or GMMA are derived from S. Paratyphi A where at least a part of the tolR gene has been replaced by a chloramphenicol acetyltransferase (cat) gene. Optionally,
the S. Paratyphi A OMVs or GMMA are derived from S. Paratyphi A where the tolR gene has been replaced by a chloramphenicol acetyltransferase (cat) gene. Optionally, the S. Paratyphi A OMVs or GMMA are derived from S. Paratyphi A which is tolR::cat.
Whether or not a genetic modification causes a S. Typhimurium, S. Enteritidis and/or S. Paratyphi A bacteria from which the GMMA are derived to hyperbleb may be tested using the following hyperblebbing assay. The user should prepare two cultures of bacterium. The first culture should comprise the bacterium having the genetic modification to be tested (the test culture), and the second culture should comprise an equivalent bacterium which is identical but for the genetic modification to be tested (the reference culture). The user should grow the test culture and the reference culture under identical conditions and determine the number of outer membrane vesicles released from the bacteria in the test culture and bacteria in the reference culture. If the amount of outer membrane vesicles released in the test culture is higher than the amount of outer membrane vesicles released in the reference culture, then the genetic modification causes the bacterium to hyperbleb. The level of outer membrane vesicles released may be determined by O-Antigen quantification, for example according to Example 3.
Strain
Optionally, the immunogenic composition comprises S. Typhimurium GMMA derived from S. Typhimurium strain 2192 (see e.g. De Benedetto et al, 2017, Multiple Techniques for Size Determination of Generalized Modules for Membrane Antigens from Salmonella typhimurium and Salmonella enteritidis. ACS Omega. 2017 Nov 30;2(l l):8282-8289). Optionally, the immunogenic composition comprises S. Enteritidis GMMA derived from S. Enteritidis strain 618 (see e.g. Lanzilao L, Stefanetti G, Saul A, MacLennan CA, Micoli F, Rondini S. Strain Selection for Generation of O-Antigen-Based Glycoconjugate Vaccines against Invasive Nontyphoidal Salmonella Disease. PLoS One. 2015 Oct
7; 10(10):e0139847). Optionally, the immunogenic composition comprises S. Paratyphi A GMMA derived from S. Paratyphi A strain ED 199 (see e.g. Mylona E, Sanchez-Garrido J, Hoang Thu TN, Dongol S, Karkey A, Baker S, Shenoy AR, Frankel G. Very long O- antigen chains of Salmonella Paratyphi A inhibit inflammasome activation and pyroptotic
cell death. Cell Microbiol. 2021 May;23(5):el3306). For example, the S. Paratyphi A strain may be tolR::cat pagP::kan msbB::tet.
An immunogenic composition comprises S. Typhimurium GMMA derived from S. Typhimurium strain 2192, if the strain used was based on S. Typhimurium strain 2192 even if modifications to strain 2192 have been made (for example mutation of msbB, pagP or tolR genes).
Dose
The immunogenic composition may comprise a dose of 1 to 100 pg, 1 to 50 pg, 15 to 50 pg, 20 to 30 pg, 1 to 20 pg, 1 to 10 pg, around 25 pg, or around 5 pg of S. Paratyphi A O- antigen. The dose of S. Paratyphi A O-antigen in a composition may be determined by mild hydrolysis of the O-antigen in the immunogenic composition (to provide the monosaccharide Paratose) and detecting the amount of Paratose by HPAEC-PAD. Paratose is a monosaccharide that is present in the S. Paratyphi A O-antigen and not present in O-antigen from S. Enteritidis or S. Typhimurium. A suitable method for determining the amount of S. Paratyphi A O-antigen by HPAEC-PAD is set out in Example 10. If the S. Paratyphi A O-antigen is part of a conjugate (comprising a carrier protein), then the amount of the carrier protein may vary. For example, if the ratio of carrier protein to O-antigen in the conjugate is greater than 2, then the amount of carrier protein present to achieve an O-antigen dose of 1 pg will be higher than if the ratio of carrier protein to O-antigen in the conjugate is lower than 2.
The immunogenic composition of the invention may comprise a dose (O-antigen) of between 1 pg and 50 pg, between 2 pg and 25 pg, between 2 pg and 10 pg, between 15 pg and 25 pg, around 20 pg, or around 4 pg of the S. Typhimurium antigen or S.
Typhimurium GMMA. The GMMA used in the immunogenic compositions comprises O- antigen. The dose of GMMA may be quantified as an O-antigen dose, i.e. if the immunogenic composition comprises a Ipg (O-antigen) dose of GMMA then the immunogenic composition comprises sufficient GMMA to provide 1 pg of the O-antigen associated with that GMMA (e.g. if the GMMA is S. Typhimurium GMMA then the
immunogenic composition comprises GMMA containing a total of I pg of S. Typhimurium O-antigen). This means that if an immunogenic composition comprises GMMA rich in O- antigen, the actual amount of GMMA present to achieve a dose of 1 pg (O-antigen) may be lower than the amount required if the GMMA is poor in O-antigen. The amount of S. Typhimurium O-antigen present in an immunogenic composition may be determined by mild hydrolysis of the O-antigen in the immunogenic composition (to provide the monosaccharide Abequose) and detecting the amount of Abequose using HPAEC-PAD. Assuming that no ‘Tree” S. Typhimurium O-antigen has been added, the amount of O- antigen in an S. Typhimurium GMMA composition will correspond to the O-antigen dose of the S. Typhimurium GMMA.
The immunogenic composition of the invention may comprise a dose (O-antigen) of between 1 pg and 50 pg, between 2 pg and 25 pg, between 2 pg and 10 pg, between 15 pg and 25 pg, around 20 pg, or around 4 pg of S. Enteritidis antigen or S. Enteritidis GMMA. The amount of S. Enteritidis O-antigen present in an immunogenic composition may be determined by mild hydrolyss of the O-antigen in the immunogenic composition (to provide the monosaccharide Tyvelose) and detecting the amount of Tyvelose using HPAEC-PAD. Assuming that no ‘Tree” S. Enteritidis O-antigen has been added, the amount of O-antigen in an S. Enteritidis GMMA composition will correspond to the O- antigen dose of the S. Enteritidis GMMA.
The immunogenic composition of the invention may comprise a dose (O-antigen) of between 1 pg and 50 pg, between 2 pg and 25 pg, between 2 pg and 10 pg, between 15 pg and 25 pg, around 20 pg, or around 4 pg of S. Paratyphi A GMMA. The amount S. Paratyphi A O-antigen present in an immunogenic composition may be determined by mild hydrolysis of the O-antigen in the immunogenic composition (to provide the monosaccharide Paratose) and detecting the amount of Paratose using HPAEC-PAD. Assuming that no ‘Tree” S. Paratyphi A O-antigen has been added, the amount of O- antigen in an S. Paratyphi A GMMA composition will correspond to the O-antigen dose of the 5. Paratyphi A GMMA.
The O-antigen/protein ratio of the S. Paratyphi A OMVs or GMMA present in an immunogenic composition may be at least 0.2, 0.3, 0.4, 0.5 or at least 0.6, typically at least 0.4. The O-antigen/total protein ratio may be at most 0.8, 0.9, 1.0, or 2.0. Optionally, the O-antigen content can be quantified by HPAEC-PAD, for example as described in Example 5. Optionally, the protein concentration is quantified by micro-BCA, for example as described in PCT/EP2022/073501.
The immunogenic composition of the invention may comprise a dose of 1 to 100 pg, 1 to 50 pg, 15 to 50 pg, 20 to 30 pg, 1 to 20 pg, 1 to 10 pg, around 25 pg, or around 5 pg of fVi polysaccharide. The dose of fVi polysaccharide in a composition may be determined by hydrolysing (by acid hydrolysis) the fVi polysaccharide in the immunogenic composition and detecting the monomer sugar of the repeating unit by HPAEC-PAD. A suitable method for determining the amount fVi polysaccharide by HPAEC-PAD is set out in Example 5. If the fVi polysaccharide is part of a conjugate (comprising a carrier protein), then the amount of the carrier protein may vary. For example, if the ratio of carrier protein to fVi polysaccharide in the conjugate is greater than 2, then the amount of carrier protein present to achieve an fVi polysaccharide dose of Ipg will be higher than if the ratio of carrier protein to fVi polysaccharide in the conjugate is lower than 2.
Salmonella Typhi antigen
The immunogenic composition may further comprise an antigen from Salmonella Typhi (S. Typhi). Optionally the antigen from S. Typhi is a Vi polysaccharide.
The term “Fz” or “Vi polysaccharide” relates to the capsular polysaccharide of Salmonella enterica serovar Typhi purified from Citrobacter (Rondini et al., J. Infect. Dev. Ctries, 2012).
Optionally, the Vi polysaccharide is a fragmented Vi polysaccharide (fVi). The term “fragmented” in reference to the Vi polysaccharide refers to the Vi polysaccharide having undergone size reduction thus reducing the number of repeating units in the polysaccharide. Fragmented Vi therefore has a lower average molecular weight compared
to native Vi. For example, fragmented Vi may comprise 30 to 300 repeating units, compared to over 600 repeating units for native Vi. A structure of the Vi monomeric repeating unit is shown below.
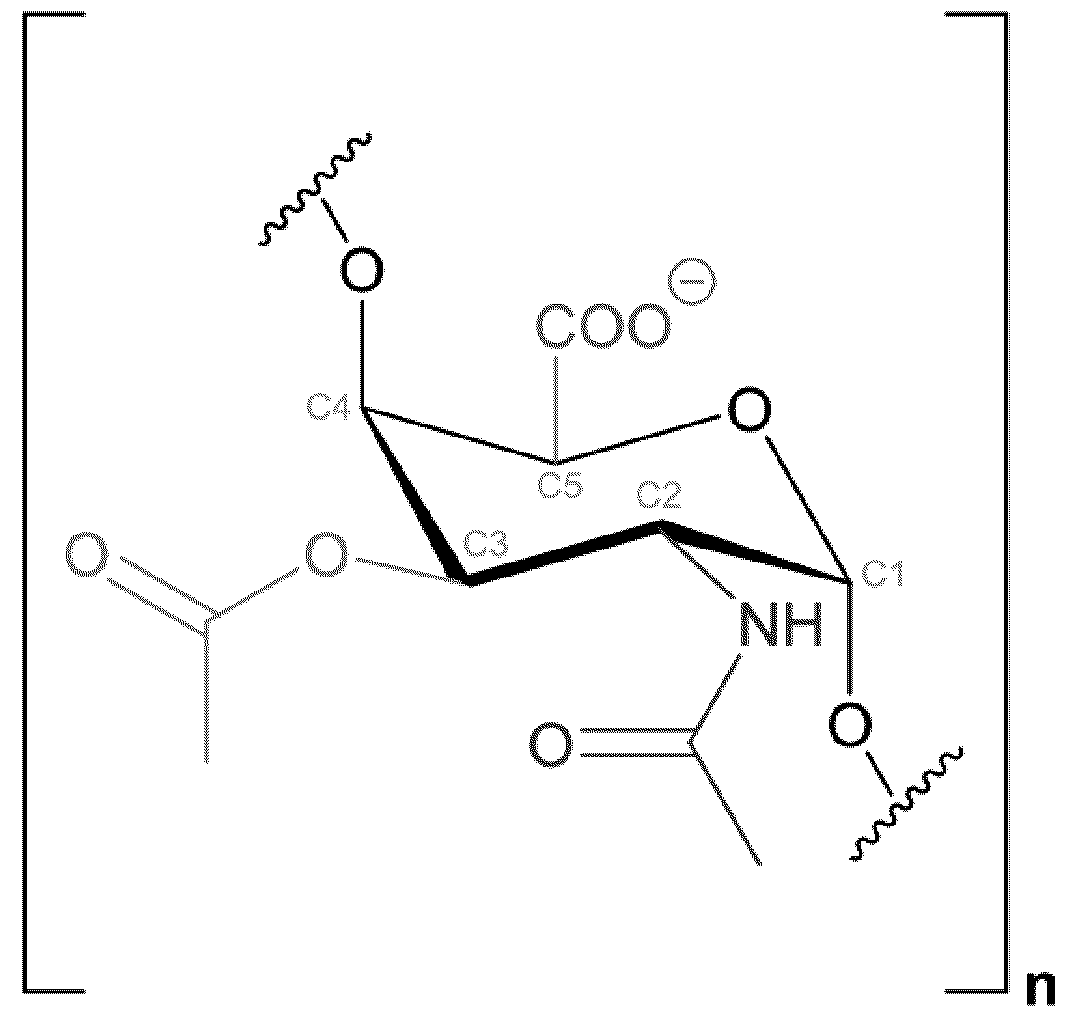
In the fragmented Vi, preferably no changes in the structure of the repeating unit is observed compared to native Vi. This can be confirmed byl H NMR analysis (see WO2015/068129). In addition, the percentage of O-acetyl groups in the fragmented Vi is preferably the same as the native Vi (i.e. about 95% O-acetylation) but may vary and decrease to about 65% O-acetylation. O-acetylation can be determined by standard measurements such asl H NMR or the Hestrin colorimetric method.
In its native size, the IVi polysaccharide has an average molecular weight measured by HPLC size exclusion chromatography (HPLC-SEC) of about 165kDa. In some embodiments, the fVi polysaccharide has an average molecular weight of between 10 kDa and 90 kDa, between 25 kDa and 70 kDa, between 40 kDa and 55 kDa, between 41 kDa and 49 kDa, or between 51 kDa and 55 kDa. Optionally, the fVi polysaccharide has a target molecular weight of between 51 kDa and 55 kDa (e.g. it has been made by a method
that typically generates fVi having a molecular weight within this range). The molecular weight of the Vi polysaccharide may be determined by HPLC-SEC.
Typically, the average molecular weight is calculated by running the sample on a TSK gel 3000 PWXL column, (30 cm x 7.8 mm; particle size 7 pm; cod. 808021) with a TSK gel PWXL guard column (4.0 cm x 6.0 mm; particle size 12 pm; cod. 808033) (Tosoh Bioscience) using dextrans as standards (5, 25, 50, 80, 150 kDa). The mobile phase is 0.1 M NaCI, 0.1 M NaH2 PO4, 5% CH3 CN, pH 7.2, at the flow rate of 0.5 mL/min (isocratic method for 30 min). Void and bed volume calibration is performed with X-DNA (X-DNA Molecular Weight Marker III 0.12-21.2 kb; Roche) and sodium azide (NaN3; Merck), respectively.
Fragmented Vi polysaccharide can further be separated into pools of different average molecular weight ranges. This can be achieved by methods known in the art such as anion exchange chromatography, size exclusion chromatography, and tangential flow filtration.
The fVi polysaccharide used in the present invention have certain average molecular weight (avMW) range distributions which can be further characterized in terms of polydispersity index (PDI).
The polydispersity index is calculated as shown in the equation below:
PDI = Mw / Mn where Mw is the weight average molecular weight and Mn is the number average molecular weight.
The narrower the molecular weight distribution, the closer the PDI value is to 1.
The fVi polysaccharide may have an avMW distribution characterised in that at least 80% of the pool has an avMW between 25 kDa and 70 kDa. It may have an avMW distribution characterised in that at least 50% of the pool has an avMW between 35 kDa and 60 kDa. It may have an avMW distribution characterised in that at least 30% of the pool has an avMW between 41 kDa and 55 kDa.
Fragmentation of the Vi polysaccharide may be carried out by a number of methods known in the art such as chemical hydrolysis of the native polysaccharide, enzymatic fragmentation of the native polysaccharide, gamma irradiation of the native polysaccharide, or mechanical methods such as sonication, or high pressure homogenizer/microfluidizer/HPCDS (High pressure cell disruption system) of the native polysaccharide. The fragmentation method used in the present invention is selected such that it can yield fVi polysaccharide having an avMW of less than 90kDa, less than 80 kDa, less than 60 kDa, or between 40 and 55 kDa. The method may also be selected such that there are no alterations to the repeating units' structure.
Preferably, fragmentation is not by mechanical methods. Preferably, fragmentation is not by alkaline hydrolysis. The fVi polysaccharide may be obtained by chemical hydrolysis with hydrogen peroxide. Using this method, it was found that the Vi polysaccharide could be reduced in size without altering the repeating units' structure. Also, hydrolysis with hydrogen peroxide could enable the formation of fragmented Vi having a lower average molecular weight than when using mechanical methods. A suitable method for fragmenting Vi polysaccharide is set out in Example 2.
The fVi polysaccharide may be part of an fVi conjugate comprising fVi and a carrier protein. Optionally, the carrier protein in the fVi conjugate is tetanus toxoid, CRM197, or diphtheria toxoid. Optionally, the carrier protein is CRM197.
The fVi polysaccharide may be conjugated to the carrier protein via any suitable conjugation chemistry.
Conjugation of the fVi polysaccharide to the carrier protein may be via a -NH2 group, e.g., through the side chain(s) of a lysine residue(s) or arginine residue(s) in the carrier polypeptide. Where the fVi polysaccharide has a free aldehyde group, this group can react with an amine in the protein to form a conjugate by reductive amination. Conjugation to the carrier may also be via a -SH group, e.g., through the side chain(s) of a cysteine residue(s) in the carrier polypeptide. Alternatively, the fVi polysaccharide may be conjugated to the carrier protein via a linker molecule.
The fVi polysaccharide will typically be activated or functionalised prior to conjugation. Activation may involve, for example, cyanylating reagents such as CDAP (l-cyano-4- dimethylamino pyridinium tetrafluoro borate). Other suitable techniques use carbodiimides, hydrazides, active esters, norborane, p-nitrobenzoic acid, N-hydroxysuccinimide, S-NHS, EDC, TSTU (see, e.g., the introduction to WO 98/42721).
Direct conjugation to the carrier protein may comprise oxidation of the fVi polysaccharide followed by reductive amination with the protein, as described in, for example, U.S. Pat No. 4,761,283 and U.S. Pat No. 4,356,170. Conjugation via a linker group may be made using any known procedure, for example, the procedures described in U.S. Pat No.
4,882,317 and U.S. Pat No. 4,695,624. Typically, the linker is attached via an anomeric carbon of the polysaccharide. A preferred type of linker is an adipic acid linker, which may be formed by coupling a free -NH2 group (e.g., introduced to a polysaccharide by amination) with adipic acid (using, for example, diimide activation), and then coupling a protein to the resulting saccharide-adipic acid intermediate (see, e.g., EP-B-0477508, Mol. Immunol, (1985) 22, 907-919, and EP-A-0208375). A similar preferred type of linker is a glutaric acid linker, which may be formed by coupling a free -NH group with glutaric acid in the same way. Adipic and glutaric acid linkers may also be formed by direct coupling to the polysaccharide, i.e., without prior introduction of a free group, e.g., a free -NH group, to the polysaccharide, followed by coupling a protein to the resulting saccharide - adipic/glutaric acid intermediate. Another preferred type of linker is a carbonyl linker, which may be formed by reaction of a free hydroxyl group of a modified polysaccharide with CDI (Bethell G.S. et al. (1979) J. Biol. Chem. 254, 2572-4 and Hearn M.T.W. (1981) J. Chromatogr. 218, 509-18); followed by reaction with a protein to form a carbamate linkage. Other linkers include P-propionamido (WO00/10599), nitrophenyl-ethylamine (Gever et al. (1979) Med. Microbiol. Immunol. 165, 171-288), haloacyl halides (U.S. Pat. No. 4,057,685), glycosidic linkages (U.S. Pat. Nos. 4,673,574; 4,761,283; and 4,808,700), 6-aminocaproic acid (U.S. Pat. No. 4,459,286), N- succinimidyl-3-(2-pyridyldithio)- propionate (SPDP) (U.S. Pat. No. 5,204,098), adipic acid dihydrazide (ADH) (U.S. Pat. No. 4,965,338), C4 to C12 moieties (U.S. Pat. No. 4,663, 160), etc. Carbodiimide condensation can also be used (W02007/000343).
A bifunctional linker may be used to provide a first group for coupling to an amine group in the polysaccharide (e.g., introduced to the polysaccharide by amination) and a second group for coupling to the carrier (typically for coupling to an amine in the carrier). Alternatively, the first group is capable of direct coupling to the polysaccharide, i.e., without prior introduction of a group, e.g., an amine group, to the polysaccharide.
Optionally, the fVi conjugate is obtained by or obtainable by a method (i.e. a method for preparing an fVi conjugate) comprising the steps of: a. fragmenting Vi polysaccharide to obtain a fragmented Vi (fVi) polysaccharide having an average molecular weight of between 10 kDa and 90 kDa, between 25 kDa and 70 kDa, between 40 kDa and 55 kDa, between 41 kDa and 49 kDa, or between 51 kDa and 55 kDa; b. reacting the fVi polysaccharide obtained in step a. with a carbodiimide and N- hydroxysuccinimide at a pH of 5 to 6 to form an N-hydroxysuccinimide ester fVi derivative; and c. reacting the N-hydroxysuccinimide ester fVi derivative obtained in step b. with the carrier protein (optionally derivatised carrier protein) to produce the fVi conjugate.
Such a method is described in more detail in WO2015068129.
The carrier protein may be derivatised by reacting it with a carbodiimide and a linker. Optionally, the carbodiimide is l-ethyl-3 -(3 -Dimethylaminopropyl) carbodiimide (EDAC). Any suitable linker (such as those discussed above) may be used. In some embodiments, the linker is an ADH linker. Optionally, derivatising the carrier protein produces a derivatised carrier protein. Optionally, the carrier protein is CRM197 and derivatising the carrier protein comprises one or more of the following steps:
(i) providing CRM 197 is an appropriate buffer, optionally MES buffer;
(ii) mixing the CRM197 with EDAC at a ratio between 1 :0.05 and 1 :0.5, between 1:0.1 and 1:0.3, or around 1:0.15 (w/w CRM197 to EDAC);
(iii) mixing the CRM197 with ADH at a ratio of between 1 : 1 and 1 :6, between 1:2 and 1:4, or around 1:3.5 (w/w CRM197 to ADH);
(iv) incubating a mixture of CRM 197 and ED AC and optionally ADH for at least 30 minutes, or between 30 minutes and 2 hours, optionally with stirring; and
(v) purifying derivatised CRM197, optionally by tangential flow filtration.
In some embodiments, the carrier protein is derivatised by a method that comprises steps (i), (ii), and (iv). In some embodiments, the carrier protein is derivatised by a method that comprises steps (i), (ii), (iii), and (iv). In some embodiments, the carrier protein is derivatised by a method that comprises steps (i), (ii), (iv), and (v). In some embodiments, the carrier protein is derivatised by a method that comprises all of steps (i) to (v) above. In some embodiments, steps (i) to (v) above are performed in the order set out above, except that steps (ii) and (iii) may be performed simultaneously.
The fVi conjugate may be obtainable or obtained by a method comprising a step of reacting the fVi polysaccharide with a carbodiimide and N-hydroxysuccinimide at a pH of 5 to 6 to form an N-hydroxysuccinimide ester fVi derivative. Optionally, the carbodiimide is EDC (N-3-dimethylamino propyl(-N-ethyl carbodiimide). Optionally, reacting the fVi polysaccharide with a carbodiimide and N-hydroxysuccinimide comprises mixing the fVi with a carbodiimide such as EDC in the presence of N-hydroxysuccinimide (NHS). Optionally, reacting the fVi polysaccharide with a carbodiimide and N- hydroxysuccinimide comprises mixing the fVi polysaccharide with NHS. Optionally, reacting the fVi polysaccharide with a carbodiimide and N-hydroxysuccinimide comprises mixing the fVi polysaccharide with NHS such that the NHS concentration is between 0.1 M and 0.5M, or around 0.33M, and the fVi polysaccharide concentration is between 1 mg/mL and 100 mg/ml, or around 50 mg/ml. Optionally, reacting the fVi polysaccharide with a carbodiimide and N-hydroxysuccinimide comprises mixing the fVi polysaccharide with EDC to have a molar ratio of EDC to fVi repeating unit or between 1 : 1 and 20: 1 , between 1:1 and 10:0, between 2:1 and 7:1, or around 5:1. Optionally, mixing the fVi polysaccharide with EDC is carried out after mixing the fVi polysaccharide with NHS. Optionally, reacting the fVi polysaccharide with a carbodiimide and N- hydroxysuccinimide comprises a step of incubating a mixture of fVi polysaccharide, NHS and EDC for at least 30 minutes, or around 1 hour at room temperature.
Optionally, reacting the N-hydroxysuccinimide ester fVi derivative with the carrier protein (optionally the derivatised carrier protein) comprises mixing the N-hydroxysuccinimide ester fVi derivative with the carrier protein (or carrier protein derivative). Optionally, reacting the N-hydroxysuccinimide ester fVi derivative with the carrier protein (optionally the derivatised carrier protein) comprises mixing the N-hydroxysuccinimide ester fVi derivative with the carrier protein (or carrier protein derivative) at a ratio of between (w/w) 1:0.1 and 1:10, between 1:0.5 and 1:5, between 1:0.75 and 1:2, or around 1:1. Optionally, mixing the N-hydroxysuccinimide ester fVi derivative with the carrier protein (or carrier protein derivative) is carried out in a buffer at a pH between 5 and 7, or around 6. Optionally, mixing the N-hydroxysuccinimide ester fVi derivative with the carrier protein (or carrier protein derivative) is carried out in MES buffer. Optionally, mixing the N- hydroxysuccinimide ester fVi derivative with the carrier protein (or carrier protein derivative) is carried out at a temperature between 20°C and 30°C or around room temperature, optionally with mixing.
The method for preparing an fVi conjugate may comprise one or more of the following additional steps, after the step of reacting the N-hydroxysuccinimide ester fVi derivative with the carrier protein (or carrier protein derivative):
(i) quenching by addition of a quencher such as Phenyl HP buffer;
(ii) fdtering the fVi conjugate;
(iii) purifying the fVi conjugate, optionally using hydrophobic interaction chromato graphy ;
(iv) concentrating the fVi conjugate, optionally by tangential flow filtration; and
(v) filtering the conjugate, optionally using one or more 0.2pm filters.
Optionally, the method for preparing an fVi conjugate comprises 2 or more, 3 or more, 4 or more, or all 5 of steps (i) to (v) above. Optionally, the method comprises step (i) above. Optionally, the method comprises steps (i) to (iii) above. Optionally, the method comprises steps (i) to (v) above. Optionally, the method comprises steps (i) to (iii) above in the order recited above. Optionally, the method comprises steps (i) to (v) above in the order recited above.
A suitable method for conjugating the fVi polysaccharide to CRM197 using ED AC chemistry via an ADH linker is set out in Example 2.
Immunogenic compositions
The immunogenic compositions of the invention or used in the invention may comprise additional components, such as a pharmaceutically acceptable excipient(s), an adjuvant, and/or further antigens.
The immunogenic composition may further comprise a pharmaceutically acceptable excipient. Typical ''pharmaceutically acceptable excipients ’ include any carrier that does not itself induce the production of antibodies harmful to the individual receiving the composition. Suitable carriers are typically large, slowly metabolised macromolecules such as proteins, polysaccharides, polylactic acids, polyglycolic acids, polymeric amino acids, amino acid copolymers, sucrose, trehalose, lactose, and lipid aggregates (such as oil droplets or liposomes). Such carriers are well known to those of ordinary skill in the art. Pharmaceutically acceptable excipients may also contain diluents, such as water, saline, glycerol, etc. Additionally, auxiliary substances, such as wetting or emulsifying agents, pH buffering substances, and the like, may be present. Sterile pyrogen-free, Tris-buffered physiologic saline is a suitable carrier particularly when using aluminium adjuvants since the phosphate in phosphate buffered saline may interfere with outer membrane vesicle binding to aluminium. However, in a particular embodiment, the immunogenic composition comprises phosphate buffered saline (and optionally an aluminium adjuvant as described further below). Optionally, the immunogenic composition comprises phosphate buffered saline at a pH between 6 and 7, for example pH 6.5.
Immunogenic compositions may be prepared as injectables, either as liquid solutions or suspensions. Solid forms suitable for solution in, or suspension in, liquid vehicles prior to injection can also be prepared (e.g. a lyophilised composition or a spray-freeze dried composition). The immunogenic composition may be prepared for topical administration e.g. as an ointment, cream or powder. The immunogenic composition may be prepared for oral administration e.g. as a tablet or capsule, as a spray, or as a syrup (optionally flavoured). The immunogenic composition may be prepared for pulmonary administration
e.g. as an inhaler, using a fine powder or a spray. The composition may be prepared as a suppository or pessary. The immunogenic composition may be prepared for nasal, aural or ocular administration e.g. as drops. The immunogenic composition may be in kit form, designed such that a combined composition is reconstituted just prior to administration to a mammal. Such kits may comprise one or more antigens in liquid form and one or more lyophilised antigens. Immunogenic compositions may be presented in vials, or they may be presented in pre-filled syringes. The syringes may be supplied with or without needles. A syringe will include a single dose of the composition, whereas a vial may include a single dose or multiple doses.
Immunogenic compositions of or used in the invention may be packaged in unit dose form or in multiple dose form. For multiple dose forms, vials are preferred to pre-filled syringes. Effective dosage volumes can be routinely established, but a typical human dose of the composition has a volume of 0.5ml e.g. for intramuscular injection.
The composition will be sterile. Immunogenic compositions of or used in the invention may be isotonic with respect to humans.
Thus, immunogenic compositions of or used in the invention may be useful as vaccines. Vaccines according to the invention may either be prophylactic (i.e. to prevent infection) or therapeutic (i.e. to treat infection), but will typically be prophylactic.
Immunogenic compositions used as vaccines comprise an effective amount of antigen(s), as well as any other components, as needed. By “ effective amount” (i.e. an immunologically effective amount), it is meant that the administration of that amount to an individual, either in a single dose or as part of a series, is effective for treatment or prevention. This amount varies depending upon the health and physical condition of the individual to be treated, age, the taxonomic group of individual to be treated (e.g. non-human primate, primate, etc.), the capacity of the individual's immune system to synthesise antibodies, the degree of protection desired, the formulation of the vaccine, the treating doctor's assessment of the medical situation, and other relevant factors.
Immunogenic compositions of the invention may include an antimicrobial, particularly when packaged in multiple dose formats.
Adjuvants
The immunogenic compositions of or used in the invention may comprise an adjuvant. Any suitable adjuvant may be used. However, in some embodiments the adjuvant is a mineral salt, such as an aluminium salt or a calcium salt. Suitable mineral salts include hydroxides (e.g. oxyhydroxides), phosphates (e.g. hydroxyphosphates, orthophosphates), sulphates, etc. or mixtures of different mineral compounds, with the compounds taking any suitable form (e.g. gel, crystalline, amorphous, etc.), and with adsorption being preferred. The mineral containing compositions may also be formulated as a particle of metal salt.
The immunogenic compositions of or used in the invention may comprise an aluminium adjuvant, i.e. any compound comprising Al3+ ions.
The aluminium adjuvant may comprise or consist of aluminium phosphate (any compound comprising Al3+ and PO43' ions) and/or aluminium hydroxide (any compound comprising Al3+ and OH' ions).
Optionally, the aluminium adjuvant comprises or consists of aluminium hydroxide. The aluminium hydroxide adjuvant may comprise or be an aluminium oxyhydroxide salt. The aluminium hydroxide adjuvant may comprise or be an aluminium oxyhydroxide salt that is at least partially crystalline. Aluminium oxyhydroxide salt, which can be represented by the formula A10(0H), can be distinguished from other aluminium compounds, such as aluminium hydroxide salt (Al(0H)3), by infrared (IR) spectroscopy, in particular by the presence of an adsorption band at 1070cm'1 and a strong shoulder at 3090-3100cm'1 (chapter 9 of ref. Vaccine Design: The Subunit and Adjuvant Approach (eds. Powell & Newman) Plenum Press 1995 (ISBN 0-306-44867-X)). The degree of crystallinity of an aluminium hydroxide adjuvant is reflected by the width of the diffraction band at 20 half height (WHH), with poorly-crystalline particles showing greater line broadening due to smaller crystallite sizes. The surface area increases as WHH increases, and adjuvants with
higher WHH values have been seen to have greater capacity for antigen adsorption. A fibrous morphology (e.g. as seen in transmission electron micrographs) is typical for aluminium hydroxide adjuvants.
Suitable examples of aluminium hydroxide adjuvants will be apparent to one of skill in the art, for example ALHYDROGEL®.
The aluminium adjuvant may comprise or consist of between 0.1 mg and 10 mg Al3+, between 0.1 mg and 5 mg Al3+, between 0.3 mg and 0.4 mg Al3+, or around 0.35 mg Al3+.
Immunogenicity
The Examples show that the immunogenic composition of the invention demonstrate good immunogenicity. Immunogenicity can be measured according to the assays set out in Example 6.
The immunogenic compositions of the invention may induce at least 103 EU/ml of anti-5. Typhimurium O-antigen antibodies and/or at least 103 EU/ml of anti-5. Enteritidis O- antigen antibodies in an immunogenicity assay comprising the following steps:
(a) immunise mice at days 0 and 28 intraperitoneally with the immunogenic composition at a dose of 1 pg (O-antigen) per GMMA and 1.25 pg saccharide per saccharide; and
(b) measure the anti-5. Typhimurium O-antigen and/or 5. Enteritidis O-antigen antibody level by ELISA at day 42.
An immunogenic composition “ induces” at least 103 EU/ml of anti-5. Typhimurium O- antigen antibodies if it is able to induce this level of antibodies when used to immunise mice. Whether or not it is able to induce this level of antibodies when used to immunise a mice may be determined by testing a sample of the immunogenic composition using the immunogenicity assay.
The dose of the GMMA and the saccharide may be determined as discussed in the section entitled “dose” above.
A suitable ELISA may involve: coating ELISA plates with S. Typhimurium or S. Enteritidis O-antigen; applying a blood sample taken from the mice at day 42 to the coated ELISA plates and then washing the plates to remove antibodies not bound to the S. Typhimurium or S.
Enteritidis O-antigen; and detecting the amount of anti-5. Typhimurium or S. Enteritidis O-antigen antibodies bound to the S. Typhimurium or S. Enteritidis O-antigen on the ELISA plates using an anti-IgG antibody conjugated to a detection moiety such as alkaline phosphatase.
The immunogenic compositions of the invention may induce at least 103 EU/ml of anti-fVi conjugate antibodies in an immunogenicity assay comprising the following steps:
(a) immunise mice at days 0 and 28 intraperitoneally with the immunogenic composition at a dose of 1 pg (O-antigen) per GMMA and 1.25 pg saccharide per saccharide conjugate; and
(b) measure the anti-fVi conjugate antibody level by ELISA at day 42.
The dose of the GMMA and the saccharide may be determined as discussed in the section entitled “dose” above.
A suitable ELISA may involve: coating ELISA plates with fVi polysaccharide; applying a blood sample taken from the mice at day 42 to the coated ELISA plates and then washing the plates to remove antibodies not bound to the fVi polysaccharide; and detecting the amount of anti-fVi conjugate antibodies bound to the fVi polysaccharide on the ELISA plates using an anti-IgG antibody conjugated to a detection moiety such as alkaline phosphatase.
The immunogenic compositions of the invention may induce at least 103 EU/ml or 103 5 of S. Paratyphi A O-antigen antibodies in an immunogenicity assay comprising the following steps:
(a) immunise mice at days 0 and 28 intraperitoneally with the immunogenic composition at a dose of 1 pg (O-antigen) per GMMA and 1.25 pg saccharide per saccharide conjugate; and
(b) measure the anti-5. Paratyphi A O-antigen antibody level by ELISA at day 42.
The dose of the GMMA and the saccharide may be determined as discussed in the section entitled “dose” above.
A suitable ELISA may involve: coating ELISA plates with S. Paratyphi A O-antigen; applying a blood sample taken from the mice at day 42 to the coated ELISA plates and then washing the plates to remove antibodies not bound to the S. Paratyphi A O- antigen; and detecting the amount of anti- S. Paratyphi A O-antigen antibodies bound to the S. Paratyphi A O-antigen on the ELISA plates using an anti-IgG antibody conjugated to a detection moiety such as alkaline phosphatase.
In the immunogenic compositions of the invention, the level of anti-5. Typhimurium O- antigen antibodies induced may be at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, or at least 98% of the level of anti-5. Typhimurium O-antigen antibodies induced by a corresponding monovalent 5. Typhimurium immunogenic composition. The level of antibodies in the immunogenic composition and the corresponding monovalent immunogenic composition may be determined using the “ suitable ELISA” set out above. A “corresponding monovalent S. Typhimurium immunogenic composition” is identical to the immunogenic composition, except that the only antigen present is the 5. Typhimurium antigen. For example, if the immunogenic composition of the invention comprises 5 pg O- antigen from 5. Typhimurium, O-antigen from 5. Enteritidis, an fVi polysaccharide conjugate, an aluminium adjuvant and phosphate buffered saline, the corresponding monovalent 5. Typhimurium immunogenic composition would comprise 5 pg O-antigen from 5. Typhimurium, an aluminium adjuvant and phosphate buffered saline.
In the immunogenic compositions of the invention, the level of anti-5. Enteritidis O- antigen antibodies induced may be at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, or at least 98% of the level of anti-5. Enteritidis O-antigen antibodies induced by a corresponding monovalent 5. Enteritidis immunogenic composition. The level of antibodies in the immunogenic composition and the corresponding monovalent immunogenic composition may be determined using the “ suitable ELISA” set out above. A “corresponding monovalent S. Enteritidis immunogenic composition” is identical to the immunogenic composition, except that the only antigen present is the 5. Enteritidis antigen. For example, if the immunogenic composition of the invention comprises O- antigen from 5. Typhimurium, 5 pg O-antigen from 5. Enteritidis, an fVi polysaccharide conjugate, an aluminium adjuvant and phosphate buffered saline, the corresponding monovalent 5. Enteritidis immunogenic composition would comprise 5 pg O-antigen from 5. Enteritidis, an aluminium adjuvant and phosphate buffered saline.
In the immunogenic compositions of the invention, the level of anti-fVi conjugate antibodies induced may be at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, or at least 98% of the level of anti-fVi conjugate antibodies induced by a corresponding monovalent fVi polysaccharide immunogenic composition. The level of antibodies in the immunogenic composition and the corresponding monovalent immunogenic composition may be determined using the “suitable ELISA” set out above. A “corresponding monovalent jVi polysaccharide immunogenic composition” is identical to the immunogenic composition, except that the only antigen present is the fVi polysaccharide antigen. For example, if the immunogenic composition of the invention comprises O-antigen from 5. Typhimurium, O-antigen from 5. Enteritidis, 5 pg fVi polysaccharide, an aluminium adjuvant and phosphate buffered saline, the corresponding monovalent fVi polysaccharide immunogenic composition would comprise 5 pg fVi polysaccharide, an aluminium adjuvant and phosphate buffered saline.
In the immunogenic compositions of the invention, the level of anti-5. Paratyphi A O- antigen antibodies induced may be at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, or at least 98% of the level of anti-5. Paratyphi A O-antigen antibodies induced by a corresponding monovalent 5. Paratyphi A immunogenic composition. The level of
antibodies in the immunogenic composition and the corresponding monovalent immunogenic composition may be determined using the “ suitable ELISA” set out above.
A “corresponding monovalent S. Paratyphi A immunogenic composition” is identical to the immunogenic composition, except that the only antigen present is the S. Paratyphi A antigen. For example, if the immunogenic composition of the invention comprises O- antigen from S. Typhimurium, 5 pg O-antigen from S. Paratyphi A, an fVi polysaccharide conjugate, an aluminium adjuvant and phosphate buffered saline, the corresponding monovalent S. Paratyphi A immunogenic composition would comprise 5 pg O-antigen from S. Paratyphi A, an aluminium adjuvant and phosphate buffered saline.
In the immunogenic compositions or methods of the invention, the level of anti-5. Typhimurium O-antigen antibodies, anti-5. Enteritidis O-antigen antibodies, anti-fVi conjugate antibodies and/or anti-5. Paratyphi A O-antigen antibodies is measured in an immunogenicity assay comprising the following steps:
(a) immunise mice at days 0 and 28 intraperitoneally with the immunogenic composition at a dose of 1 pg (O-antigen) per GMMA and 1.25 pg saccharide per saccharide conjugate; and
(b) measure the anti-5. Typhimurium, anti-5. Enteritidis, anti-fVi conjugate and/or anti-5. Paratyphi A O-antigen antibodies levels by ELISA at day 42.
Cross-protection
The immunogenic compositions of the invention may demonstrate cross-protection. For example, the immunogenic compositions of the invention may induce antibodies against three or more, four or more, five or more, six or more, seven or more, eight or more, nine or more or all ten of the following strains:
(a) 5. Typhimurium ST34 (Mather et al, New Variant of Multidrug-Resistant Salmonella enterica Serovar Typhimurium Associated with Invasive Disease in Immunocompromised Patients in Vietnam. mBio. 2018 Sep 4;9(5):e01056- 18);
(b) 5. Typhimurium 10433 3 (Van Puyvelde et al, An African Salmonella Typhimurium ST313 sublineage with extensive drug-resistance and signatures of host adaptation. Nat Commun. 2019 Sep 19; 10(l):4280);
(c) S. Typhimurium D23580 (Van Puyvelde et al, An African Salmonella Typhimurium ST313 sublineage with extensive drug-resistance and signatures of host adaptation. Nat Commun. 2019 Sep 19; 10(l):4280);
(d) S. Typhimurium ST4/74 (Hurley et al, Atypical Salmonella enterica Serovars in Murine and Human Macrophage Infection Models. Infect Imrnun. 2020 Mar 23;88(4):e00353-19)
(e) S. Typhimurium Al 30 (Okoro et al, Intracontinental spread of human invasive Salmonella Typhimurium pathovariants in sub-Saharan Africa. Nat Genet. 2012 Nov;44(l l): 1215-21);
(f) S. enterica serovar Derby (Pullinger et al, Identification of Salmonella enterica serovar Dublin-specific sequences by subtractive hybridization and analysis of their role in intestinal colonization and systemic translocation in cattle. Infect Immun. 2008 Nov;76(l l):5310-21);
(g) S. enterica serovar Dublin (Pullinger et al, Identification of Salmonella enterica serovar Dublin-specific sequences by subtractive hybridization and analysis of their role in intestinal colonization and systemic translocation in cattle. Infect Immun. 2008 Nov;76(l l):5310-21);
(h) S. Enteritidis Al 636 (Perez- Sepulveda et al, Complete Genome Sequences of African Salmonella enterica Serovar Enteritidis Clinical Isolates Associated with Bloodstream Infection. Microbiol Resour Announc. 2021 Mar 25;10(12):e01452-20);
(i) S. Enteritidis CP255 (Perez- Sepulveda et al, Complete Genome Sequences of African Salmonella enterica Serovar Enteritidis Clinical Isolates Associated with Bloodstream Infection. Microbiol Resour Announc. 2021 Mar 25;10(12):e01452-20); and
(j) S. Enteritidis D7795 (Perez- Sepulveda et al, Complete Genome Sequences of African Salmonella enterica Serovar Enteritidis Clinical Isolates Associated with Bloodstream Infection. Microbiol Resour Announc. 2021 Mar 25;10(12):e01452-20).
The immunogenic compositions of the invention may induce antibodies against:
(i) S. Enteritidis and S. Dublin;
(ii) S. Typhimurium, S. Derby and S. Dublin;
(iii) S. Typhimurium, S. Enteritidis, S. Derby and S. Dublin; or
(iv) S. Typhimurium, S. Enteritidis, S. Derby, S. Dublin and S. Paratyphi A.
An immunogenic composition “induces ” antibodies against (for example) S. Typhimurium ST34 if it is able to induce these antibodies when used to immunise mice. Whether or not it is able to induce these antibodies when used to immunise a mice may be determined by testing a sample of the immunogenic composition using the cross-protection assay.
Whether or not an immunogenic composition induces antibodies against one or more of the strains described above may be determined by carrying out a cross-protection assay. Specifically, the user may: immunise mice intraperitoneally with 500|iL of the immunogenic composition; and measure the level of bactericidal antibodies raised against the relevant strain using a serum bactericidal assay (SBA).
The SBA assay may be based on the assay described in Example 6, except that the user should measure the bactericidal activity against the relevant strain listed above (rather than, for example Salmonella Paratyphi A NVGH308).
Optionally, the immunogenic composition induces antibodies against one more of the strains described above, if the IC50 (serum dilution giving 50% inhibition of the ATP level) obtained in the SBA assay is above 102.
The immunogenic compositions of the invention may induce anti-5. Paratyphi A antibodies in each of classes IgG3, IgG2b, IgG2a, and IgGl, as determined using an antibody class assay comprising the following steps:
(a) immunise mice at days 0 and 28 intraperitoneally with the immunogenic composition at a dose of 1 pg (O-antigen) per GMMA and 1.25 pg saccharide per saccharide conjugate; and
(b) measure the anti-5. Paratyphi A O-antigen antibody subtype level by ELISA at day 42.
An immunogenic composition “induces ” anti-5. Paratyphi A antibodies in each of classes IgG3, IgG2b, IgG2a, and IgGl if it is able to induce these antibodies when used to immunise mice. Whether or not it is able to induce these antibodies when used to immunise a mice may be determined by testing a sample of the immunogenic composition using an ELISA assay to measure the anti-5. Paratyphi A O-antigen antibody subtype level as set out below.
A suitable ELISA to measure the anti-5. Paratyphi A O-antigen antibody subtype level may involve: coating ELISA plates with 5. Paratyphi A O-antigen; applying a blood sample taken from the mice at day 42 to the coated ELISA plates and then washing the plates to remove antibodies not bound to the 5. Paratyphi A O- antigen; and detecting the amount of anti- 5. Paratyphi A O-antigen IgG3 antibodies bound to the 5. Paratyphi A O-antigen on the ELISA plates using an anti-IgG3 antibody conjugated to a detection moiety such as alkaline phosphatase; repeating the first three steps but using an anti-IgG2b antibody in place of the anti- IgG3 antibody, then using an anti-IgG2a antibody in place of the anti-IgG3 antibody and finally using an anti-IgGl antibody in place of the anti-IgG3 antibody.
Tolerogenicity
“Tolerogenicity” or “tolerogenic” refers to the an immunogenic composition that does not induce significant negative reactions in the subject to which it is administered. In particular, immunogenic compositions comprising outer membrane vesicles have been known to induce fevers in patients to which they are administered. Optionally, therefore, an immunogenic composition may be considered to be tolerogenic if it does not induce a significant fever.
An immunogenic composition will be considered to “induce ” a fever if it induces a fever when used to immunise mice. Whether or not it induces a fever when used to immunise a
mice may be determined by testing a sample of the immunogenic composition using the toxicity assay set out below.
An immunogenic composition may be tolerogenic if it induces a temperature rise of less than 1.8°C, less than 1.7°C, less than 1.6°C, or less than 1.5°C in a toxicity assay comprising the following steps:
(a) measure the initial temperature of the rabbits;
(b) administer the immunogenic composition at a dose of 20 pg (O-antigen) per GMMA and 25 pg saccharide per saccharide conjugate to rabbits;
(c) monitor the temperature of the rabbits for 5 hours; and
(d) record the maximum temperature of the rabbits, wherein the temperature rise is calculated as equal to the maximum temperature of the rabbits minus the initial temperature of the rabbits.
An immunogenic composition may be tolerogenic if it induces a maximum temperature of 41°C or less, 40.9°C or less, or 40.8°C or less in a toxicity assay comprising the following steps:
(a) administer the immunogenic composition at a dose of 20 pg (O-antigen) per GMMA and 25 pg per saccharide to rabbits;
(b) monitor the temperature of the rabbits for 5 hours; and
(c) record the maximum temperature of the rabbits.
Methods of boosting an immune response
In a further aspect of the invention, there is provided a method of boosting an immune response to an antigen comprising administering a composition comprising the antigen and GMMA. There is also provided a method of preventing infection by a Salmonella enterica bacterium comprising administering an immunogenic composition comprising an antigen of the Salmonella enterica and GMMA, wherein administration of the GMMA boosts the immune response to the Salmonella enterica bacterium.
For example, the methods of the invention may comprise boosting an immune response to a S. Typhi or a S. Paratyphi A antigen comprising administering a composition comprising
the S. Typhi antigen or the S. Paratyphi A antigen and GMMA. The methods of the invention may also comprise preventing infection by S. Typhi or S. Paratyphi A comprising administering an immunogenic composition comprising the S. Typhi antigen or the S. Paratyphi A antigen and GMMA, wherein the GMMA boosts the immune response to the S. Typhi antigen or the S. Paratyphi A antigen.
There is also provided an immunogenic composition of the invention comprising GMMA for use in a method of boosting an immune response to a S. Typhi or S. Paratyphi A antigen, wherein the method comprises administering an immunogenic composition comprising the S. Typhi antigen or the S. Paratyphi A antigen and GMMA. There is also provided an immunogenic composition of the invention for use in a method of preventing infection by S. Typhi or S. Paratyphi A comprising the S. Typhi antigen or the S. Paratyphi A antigen and GMMA, wherein the GMMA boosts the immune response to the S. Typhi or the S. Paratyphi A antigen.
In the methods or immunogenic compositions for use of the invention, the method may be a method of boosting an immune response to an S. Typhi antigen and the immunogenic composition comprises the S. Typhi antigen. In the methods or immunogenic compositions for use of the invention, the method may be a method of boosting an immune response to an S. Paratyphi A antigen and the immunogenic composition may comprise the S. Paratyphi A antigen.
In the methods or immunogenic compositions for use of the invention, the method may be a method of preventing infection by S. Typhi, the immunogenic composition may comprise the S. Typhi antigen, and the GMMA may boost the immune response to the S. Typhi antigen. In the methods or immunogenic compositions for use of the invention, the method may be a method of preventing infection by S. Paratyphi A, the immunogenic composition may comprise the S. Paratyphi A antigen, and the GMMA may boost the immune response to the S. Paratyphi A antigen.
The method or immunogenic composition for use of the invention, wherein the GMMA comprises at least one are selected from the group consisting of S. Typhimurium GMMA, S. Enteritidis GMMA, and A Paratyphi A GMMA. The A Typhimurium GMMA may
boost the immune response to the S. Typhi or S. Paratyphi A antigen. The S. Paratyphi A GMMA may boost the immune response to the S. Typhi antigen. The S. Enteritidis GMMA may boost the immune response to the S. Typhi or S. Paratyphi A antigen. The S. Paratyphi GMMA may boost the immune response to the S. Typhi antigen.
A method is a method of “boosting an immune response” to a S. Typhi antigen or a S. Paratyphi A antigen, if the immune response raised to the S. Typhi or S. Paratyphi A antigen is higher when the S. Typhi antigen or a S. Paratyphi A antigen is part of the immunogenic composition comprising GMMA compared to the immune response raised when the S. Typhi antigen or the S. Paratyphi A antigen is not part of an immunogenic composition comprising GMMA. This is due to the adjuvanting effect of GMMA. In other words, when GMMA and S. Typhi antigen or S. Paratyphi A antigen are in the same composition, the GMMA increase the immune response to the S. Typhi antigen or S. Paratyphi A antigen when compared to S. Typhi antigen or S. Paratyphi A antigen on their own.
For the purposes of boosting an immune response to a S. Typhi antigen or a S. Paratyphi A antigen, the S. Typhi antigen and S. Paratyphi A antigen may be polysaccharides. Optionally, the S. Typhi antigen may comprise a fVi polysaccharide, optionally wherein the fVi polysaccharide is part of an fVi conjugate comprising fVi and a carrier protein, further optionally wherein the carrier protein is CRM197. Optionally, the S. Paratyphi A antigen may comprise S. Paratyphi A O-antigen, optionally wherein the S. Paratyphi A O- antigen is conjugated to a carrier protein, further optionally wherein the carrier protein is CRM197.
GMMA boosts the immune response to a S. Typhi antigen or a S. Paratyphi A antigen if the immune response raised to the S. Typhi antigen or the S. Paratyphi A antigen is higher when the S. Typhi antigen or the S. Paratyphi A antigen is part of the immunogenic composition comprising the GMMA compared to the immune response raised when the S. Typhi antigen or the S. Paratyphi A antigen is not part of an immunogenic composition comprising GMMA.
A method is a method of boosting an immune response to an S. Typhi or a S. Paratyphi A antigen or GMMA boosts the immune response to the S. Typhi antigen or the S. Paratyphi A antigen, if the immune response raised to the S. Typhi antigen or the S. Paratyphi A antigen is at least 5 times, at least 10 times, or at least 20 times higher when the S. Typhi antigen or the S. Paratyphi A antigen is part of the immunogenic composition comprising the GMMA compared to the immune response raised when the S. Typhi antigen or the S. Paratyphi A antigen is not part of an immunogenic composition comprising GMMA.
The immune response raised to a S. Typhi antigen or a S. Paratyphi A antigen by the immunogenic compositions of the invention, or the methods of the invention, may be the number of antibodies raised as determined by ELISA 42 days after administration of the immunogenic composition comprising the S. Typhi antigen or S. Paratyphi A antigens and the GMMA at a dose of 0.78 pg of S. Typhi antigen or S. Paratyphi A antigen and 0.63 pg (O-antigen) of the GMMA.
A suitable ELISA may involve: coating ELISA plates with S. Typhi antigen or S. Paratyphi A O-antigen; applying a blood sample taken from the mice at day 42 to the coated ELISA plates and then washing the plates to remove antibodies not bound to the S. Typhi antigen or S.
Paratyphi A O-antigen; and detecting the amount of anti- S. Typhi antigen or S. Paratyphi A O-antigen antibodies bound to the S. Typhi antigen or S. Paratyphi A O-antigen on the ELISA plates using an anti-IgG antibody conjugated to a detection moiety such as alkaline phosphatase.
Medical uses and methods of treatment
In a further aspect of the invention, there is provided an immunogenic composition of the invention for use in a method of preventing an infection. In a further aspect of the invention, there is provided a method of preventing an infection comprising administering an effective amount of the immunogenic composition or vaccine of the invention to a subject. In a further aspect of the invention, there is provided a use of the immunogenic composition or vaccine of the invention for the manufacture of a medicament for use in a
method of preventing an infection. The method of preventing an infection may comprise administering an effective amount of the immunogenic composition or vaccine of the invention to a subject.
The method of preventing an infection may be a method of preventing Salmonella infection. Optionally the method of preventing an infection is a method of preventing infection by S. Typhimurium, S. Entcritidis, S. Typhi and/or S. Paratyphi A.
The term “preventing Salmonella infection" in the method/immunogenic composition for use/use of the immunogenic composition in the manufacture of a medicament of the invention comprises raising an immune response in a subject. The immune response may be protective and may raises antibodies, such as IgG antibodies.
The subject of the invention is a mammal, optionally a human. Where the vaccine is for prophylactic use, the human may be an adult i.e. subject is 18 years old or above 18 years old. Where the vaccine is for prophylactic use, the human may be a child i.e. below 18 years old. Where the vaccine is for prophylactic use, the child may be between 12 to 72 months, preferably between 24 to 59 months, more preferably between 6 to 12 months. Where the vaccine is for prophylactic use, the child may be around 9 months.
Where the vaccine is for therapeutic use, the human is preferably a child.
A vaccine intended for children may also be administered to adults e.g. to assess safety, dosage, or immunogenicity.
Examples
Example 1 - Production of S. Typhimurium and S. Enteritidis GMMA
Salmonella enterica serovar Typhimurium wild-type (WT) strain 2192 was provided by the Salmonella Genetic Stock Center (SGSC) at the University of Calgary, Canada, which belongs to the global Salmonella reference collection A (SARA 12).
Salmonella enterica serovar Enteritidis WT strain 618 was provided by Quotient Bioresearch Limited, UK. The strain of animal origin was isolated by the European Antimicrobial Susceptibility Surveillance in Animals (EASSA).
From the Salmonella strains above, Salmonella Typhimurium olR ApagP AmsbB and Salmonella Enteritidis olR ApagP AmsbB recombinant mutants for each strain were generated as previously reported (Rossi O, Caboni M, Negrea A, Necchi F, Alfini R, Micoli F, et al. Toll-Like Receptor Activation by Generalized Modules for Membrane Antigens from Lipid A Mutants of Salmonella enterica Serovars Typhimurium and Enteritidis. Clin Vaccine Immunol. 2016;23(4):304-14.).
GMMA derived from the Salmonella strains above (Salmonella Typhimurium GMMA (STmGMMA) and Salmonella Enteritidis GMMA (SEnGMMA)) were purified and isolated. GMMA were purified using similar methods previously reported for S. sonnei GMMA (Gerke C, Colucci AM, Giannelli C, Sanzone S, Vitali CG, Sollai L, et al. Production of a Shigella sonnei Vaccine Based on Generalized Modules for Membrane Antigens (GMMA), 1790GAHB. PLoS One. 2015;10(8):e0134478. doi:
10.1371/journal.pone.0134478 [ doi] ;PONE-D-15-08654). Briefly, GMMA released into the fermentation broth were purified using two consecutive Tangential Flow Filtration (TFF) steps: a microfiltration in which the culture supernatant containing the GMMA is separated from the bacteria, and an ultrafiltration, in which the GMMA are separated from soluble proteins and nucleic acids.
Example 2 - Production of fVi-CRMi97 conjugate for immunisation against S. Typhi
The following protocol was used to produce a bivalent composition comprising an S. Paratyphi A OAg-CRMi97 conjugate made as described above and a conjugate of fragmented Vi polysaccharide from S. Typhi (fVi) conjugated to CRM197.
1) Fragmentation of Vi Polysaccharide:
Step 1: Fragmentation Reaction and Quenching: Vi-Polysaccharide fragmentation is achieved by an Oxidation-reaction using hydrogen peroxide in the presence of iron sulphate. The reaction is quenched with EDTA (Ethylenediaminetetraacetic acid). Native Vi polysaccharide is diluted with WFI. A calculated volume of 10 mM FeSO4 and H2O2 is added to get a final concentration of 0.5 mM FeSO4 and 0.5% v/v H2O2 in the reaction mixture respectively. The reaction mixture is incubated at 15±5°C for 120 +/- 10 min. The reaction is stopped by adding equal volumes of 250 mM EDTA to get a final EDTA concentration of 10 mM and is stirred.
Step 2: Buffer Exchange: Buffer exchange is performed by Tangential Flow Filtration (TFF) with lOOmM Sodium phosphate (pH: 7.2 ± 0.2) using 30 kDa cassettes to remove residual H2O2. Fragmented Vi (fVi) polysaccharide is concentrated.
Step 3: Stabilization of fVi polysaccharide: Post fragmentation, 30 kDa retentate is stabilized by incubating at 80±5° C for 120 +/- 15 min.
Step 4: JVi Purification by Anion Exchange (Resin: Capto-Q): AA chromatography step is used to separate the desired molecular size of fVi polysaccharide (25-70 KDa). This is performed using a linear gradient elution, with Capto-Q Buffer A and Capto-Q Buffer B using a Capto-Q Resin which has the binding capacity of 13 mg of IVi/mL. The eluted fractions are collected based on the conductivity for every 1 mS/cm; i.e., from 35 to 50 mS/cm and estimating the Molecular size distribution by SEC/HPLC. The Capto-Q fractions are pooled based on the Molecular Size (kDa) distribution.
Step 5: Desalting'. Pooled Capto-Q fractions are concentrated by Tangential Flow Filtraion (TFF) using a 10 kDa Cut-off cassette and then dia- filtered using WFI until permeate conductivity reaches < 30 pS/cm.
Step 6: 0.2 pm Filtration of fVi polysaccharide'. The fVi polysaccharide is fdtered through a 0.22 pm filter. The purified fVi polysaccharide is stored in PETG bottles.
2) CRM197 Derivatization:
Stepl: Thawing of CRM197: Purified CRM197 is thawed at 2-8°C prior to buffer exchange with 100 mM MES (Morpholino Ethanesulfonic acid) buffer. Post thawing, CRM197 is filtered using 0.5 pm filter.
Step 2: Buffer Exchange with 100 mM MES Buffer. Buffer exchange is performed by TFF with 100 mM MES buffer (pH 6.0±0.2) using 10 KDa cassette after CRM197 thawing.
Step 3: CRM197 Derivatization'. The required concentration of CRM197 is diluted with 100 mM MES buffer followed by addition of calculated quantity of ADH (Adipic Acid Dihydrazide) and ED AC (l-Ethyl-3 -(3 -Dimethylaminopropyl) carbodiimide) to make a CRM : ADH : ED AC 1 : 3.5 : 0.15 w/w/w ratio. After addition of ADH and ED AC, the CRM 197 the reaction mixture is incubated at room temperature under mixing conditions for 60 +/- 15 min. At the end of the reaction an equal volume of 5 mM MES buffer (pH 7.0±0.2) is added.
Step 4: Purification of CRM197'. Post reaction, CRM197 is purified by TFF using a 10 KDa cassette with 5 mM MES buffer.
Step 5: Filtration ofDia-Filtered CRM197: 0.2-micron filtration is performed for dia- filtered CRM197 solution followed by storage at 2-8°C in glass bottle.
3) Conjugation of Fragmented Vi Polysaccharide with Derivatized CRM197:
Activation of fVi polysaccharide:
Step 1: jVi polysaccharide drying by Rota Vapor. fVi is further concentrated by drying at 30°C using rotavapor. Concentrated fVi polysaccharide is reconstituted by using 100 mM MES buffer (pH: 6.0) in order to get a 50 mg/mL concentration.
Step 2: Activation of jVi polysaccharide with NHS: fVi carboxylates (-COOH) are activated with EDC (N-3 -Dimethylamino propyl-N Ethyl Carbodimide) in the presence of N-hydroxysuccinimide (NHS), by forming an active ester intermediate, to increase the efficiency of conjugation with CRM197 previously activated with ADH. The dried fVi polysaccharide is re-constituted to a desired concentration (50 mg/mL) with 100 mM MES buffer (pH: 6.2±0.2); and activated in the presence of NHS (concentration of 0.33 M) followed by ED AC addition to have an ED AC /fVi RU molar ratio of 5 : 1. EDAC solution is added after addition of NHS to ensure complete dissolution. The reaction is incubated at room temperature with slow mixing for 1 h.
Conjugation:
Step 1: Conjugation of jVi with CRMwADI {: The conjugation reaction creates a covalent bond between the activated fVi and CRM197-ADH. The activated and derivatized reaction mixture is diluted with lOOmM MES pH: 6.0 and CRM197-ADH is added in a w/w ratio of 1 : 1 (fVi : CRM197) to reach the final fVi concentration of the activated fVi and CRM197- ADH of 5 mg/mL. The conjugation reaction is performed at room temperature with slow mixing until protein consumption is > 70% measured by HPLC-SEC at 280 nm absorbance.
Step 2: Quenching and conditioning of Conjugation Reaction: The conjugation reaction is quenched by adding equal volume of Phenyl HP Buffer-B Tris 50 mM pH: 8.0. NaCl as powder is added to reach a final salt concentration of 3 M.
Step 3: Conjugation Mixture Filtration: The fVi-CRMi97 crude conjugate is 0.65 filtered.
Step 4: Purification of jVi-CRMi97 Crude Conjugate: Purification of the conjugate from the conditioned reaction mixture is performed through a HIC Phenyl Sepharose High Performance (HP) column. Column integrity is performed for every 5-10 cycles as per
standard procedure. The column is equilibrated by using Phenyl Sepharose HP Buffer A Tris 50 mM NaCl 3M pH 8. After addition of conditioning buffer and NaCl, crude conjugate is loaded on to the column. Column washing is done using Phenyl Sepharose HP Buffer-A followed by product elution using Phenyl Sepharose HP Buffer-B Tris 50mM pH: 8. Fractions are collected and stored at 2-8°C till further usage. All the fractions from multiple runs are pooled.
Step 5: Concentration and Buffer Exchange using PBS: Purified conjugate is concentrated by TFF using a 50 kDa Cut-off cassette and then buffer exchanged using PBS buffer until the permeate conductivity meets PBS buffer conductivity.
Step 6: Pre Filtration of jVi-CRFlm Conjugate using 0.2 pm filter: fVi-CRMi97 conjugate is filtered through a 0.2 pm filter for bioburden reduction.
Step 7: Sterile Filtration of fVi-CRMi97 Conjugate using 0.2 p cellulose acetate filter: The fVi-CRMi97 conjugate is filtered through a 0.2 pm cellulose acetate filter. The purified fVi-CRMi97 conjugate is sampled and stored at 2-8°C.
Example 3 - Formulation of a bivalent (iNTS-GMMA) vaccine against S.
Typhimurium and S. Enteritidis adsorbed to an aluminium hydroxide adjuvant
STmGMMA and SEnGMMA were separately adsorbed to aluminium hydroxide (Alum3+ final concentration 0.7mg/mL) in phosphate buffered saline (pH 6.5), obtaining two different drug product formulations, each vialed in 3mL Type I 2 R vials. The bulk GMMA solutions were diluted to obtain a concentration of GMMA of 80pg/ml O-antigen based on the amount of O-Ag measured in bulk GMMAx, and a concentration of aluminium hydroxide of 0.7 mg/ml (Al3+). Before administration, the content of STmGMMA/alhydrogel drug product vial is mixed with the content of
SEnGMMA/ Alhydrogel vial to obtain the iNTS-GMMA vaccine, with a final concentration of 40 pg/ml of each O-Antigen adsorbed on Alum Hydroxide (final concentration 0.7mg/mL) in phosphate buffered saline to form the final mixed drug product.
The OAg amount in the final mixed drug product was also determined. The protocols for the assays used to assess the OAg amounts are set out in the following paragraphs.
Salmonella Enteritidis O-antigen quantification on bulk GMMA or final mixed drug product formulation by HPAEC-PAD:
Quantification of the OAg in the S. Enteritidis. GMMA is performed by High- Performance Anion-Exchange Chromatography with Pulsed Amperometric Detection (HPAEC-PAD) analysis after acid hydrolysis of the sample. The quantification determines the concentration of Rhamnose, galactose, glucose and mannose present in the sample.
The quantification of OAg is performed on the basis of the known sugar ratios present in the OAg repeating unit IxRha (Rhamnose); IxGal (Galactose); IxMan (Mannose) IxTyv (Tyvelose) and the glucose is calculated from the glucose measured in the analysis after subtraction of the glucose due to the core.
A standard dilution series of each sugar in the range of 0.5-10 pg/mL is run in each HPAEC-PAD analysis and the peak areas are used to interpolate the pg/mL of the corresponding sugar present in the sample OAg.
450 pL each of the dilutions of the standard and sample are treated in parallel with 150 pL of 8 M TFA for 4 hours at 100°C. Samples are then chilled at 2°C to 8°C for approximately 30 minutes, dried in a centrifugal evaporator, resuspended in 450 pL of water, filtered and analysed. HPAEC-PAD is performed with a Dionex ICS3000 (or 5000) equipped with a CarboPac PA10 column coupled with PA10 guard column. Separation is performed at 25°C using the following conditions:
• 20 min, 18 mM NaOH, flow rate 1 mL/min (separation step)
• 10 min 28 mM NaOH, Sodium acetate 100 mM, flow rate 1 mL/min (column washing step)
• 20 min 18 mM NaOH, flow rate 1 mL/min (column equilibration step) The effluent is monitored using an electrochemical detector.
Salmonella Typhimurium O-antigen quantification on bulk GMMA by HPAEC-PAD Quantification of the OAg in the S. Typhimurium GMMA is performed using High- Performance Anion-Exchange Chromatography with Pulsed Amperometric Detection (HPAEC-PAD) after GMMA acid hydrolysis. A standard dilution series of a solution containing rhamnose, galactose, glucose and mannose is run in each HPAEC-PAD analysis series and used as calibration curve to quantify the sugars present in the sample.
The quantification of OAg is performed on the basis of the known sugar ratios present in the OAg repeating unit IxRha (rhamnose); IxGal (galactose); lx Man (mannose); IxAbe (Abequose) and the glucose is calculated from the glucose measured in the analysis after subtraction of the glucose present in the core.
OAg quantification in final mixed drug product (SEn-GMMA /STm GMMA) by HPAEC- PAD
Quantification of S. Typhimurium or S. Enteritidis OAg on GMMA adsorbed on aluminium hydroxide is performed using HPAEC-PAD after GMMA acid hydrolysis. A standard dilution series of a solution containing fucose, rhamnose, N-acetyl glucosamine, glucose, galactose and mannose is run in each HPAEC-PAD analysis series and used as calibration curves to quantify the rhamnose, glucose, galactose and mannose in the sample. The procedure is the same used for quantification of S. Typhimurium and S. Enteritidis Drug Substances.
To 450 pL of each dilution of the standard and of the sample 150 pL of 8 M TFA are added. All vials are heated in parallel at 100°C for 4 hours. Samples are then chilled at 2°C to 8°C for approximately 30 minutes, dried in a centrifugal evaporator, resuspended in 450 pL of water, filtered and analysed. HPAEC-PAD is performed with a Dionex ICS3000 (or 5000) equipped with a CarboPac PA10 column coupled with PA10 guard column.
Separation is performed at 25°C using the following conditions:
• 20 min, 18 mM NaOH, flow rate 1 mL/min (separation step)
• 10 min 28 mM NaOH, 100 mM Sodium Acetate flow rate 1 mL/min (column washing step)
• 20 min 18 mM NaOH, flow rate 1 mL/min (column equilibration step) The effluent is monitored using an electrochemical detector.
The quantification of OAg is performed on the basis of the known sugar ratios present in the OAg repeating units (Rha; Glc; Gal; Man and Tyvelose (SEn) or Abequose (STm) equal to Rha) present in the formulation. Rhamnose, glucose, galactose and mannose are quantified as nmol/mL directly from the analysis and the OAg quantity is then calculated considering the molecular weight of each sugar and the content in the repeating unit (OAg =600.61 *Rha + 162.15 *Glctotai - 162.15*Galtotai + 162.15 *Man).
Example 4 - Adsorption of SEnGMMA and STmGMMA to aluminium hydroxide enhanced the in vivo tolerability in rabbits
The impact of the adsorption of SEnGMMA and STmGMMA to aluminium hydroxide was investigated, and it was determined that adsorption to aluminium hydroxide reduced the pyrogenicity of the GMMA.
Groups of 3 New Zealand White rabbits were immunised intramuscularly with a saline control, an Alhydrogel control (a 0.5ml dose having Alhydrogel (0.7 mg/ml Al3+), 20mM phosphate and 154mM NacL), or one of the following vaccines: (1) STmGMMA (20pg O- antigen in 0.5mL dose) adsorbed to Alhydrogel (STmGMMA/hydrogel), (2) SEnGMMA (20pg O-antigen in 0.5mL dose) adsorbed to Alhydrogel (SEnGMMA/hydrogel), (3) the iNTS-GMMA vaccine (20pg O-antigen in 0.5mL dose), (4) a bivalent STmGMMA and SEnGMMA vaccine at a dose of 2pg O-antigen per GMMA in 0.5mL not adsorbed to Alhydrogel. These four vaccines were made as described in Examples 1-3.
The body temperature of the rabbits was monitored for 5 hours after vaccine administration.
The initial temperature of each rabbit was within the range of 38.0 to 39.8°C and the temperature difference among rabbits within a group was <1 ,0°C. For rabbits treated with Alhydrogel control or saline, the absolute temperature after administration did not exceed 39.2°C. In one rabbit treated with STmGMMA/ Alhydrogel, the highest absolute temperature was 40.6°C; the temperatures of the other two rabbits in this group did not exceed 39.6°C. In the 3 rabbits treated with SEnGMMA/ Alhydrogel, the highest temperatures measured were 39.3°C, 39.8°C and 40.2°C. In the 2-component iNTS- GMMA vaccine group (group (3) above), the highest temperature measured did not exceed 40.4°C and the maximum temperature rise in the individual rabbits was 0.9°C, 1.2°C and 1.5°C. Whereas for rabbits treated with mixture of unadsorbed STmGMMA and SEnGMMA (group (4) described above), the maximum temperature measured was 41.3°C and similar in the three rabbits. The peak temperature was reached in all rabbits during the time period of five hours (typically by 180-270 minutes). The rabbit groups, which received the Alhydrogel formulations (groups (1) to (3) above), showed an average maximum temperature rise of 1°C to 1.3°C, whereas the rabbit group (group (4) above), which received the unformulated GMMA as a 10-times lower dose, showed an average maximum temperature rise of 1 ,9°C. The group, which received the unformulated GMMA, peaked earlier (typically by 120-180 minutes), had a steeper and higher rise and a steeper and quicker decrease.
Example 5 - Formulation of a trivalent (iNTS-TCV) vaccine against Salmonella Typhimurium, S. Enteritidis and 5. Typhi
STmGMMA and SEnGMMA were sequentially adsorbed to Aluminum hydroxide phosphate buffered saline pH 6.5. The adsorbed STmGMMA and SEnGMMA were then mixed together, and after addition of a phosphate quencher, fVi-CRMi97 suspended in sodium phosphate buffered saline was added subsequently.
In order to make solutions of the correct concentrations of STmGMMA and SEnGMMA as indicated above, the amount of O-Antigen (OAg) in bulk GMMA solutions was determined and then used to determine how to dilute bulk GMMA in order to obtain the correct GMMA concentrations. The OAg and the Vi polysaccharide amount in the final
mixed drug product was also determined. The protocols for determining the STm GMMA and SEnGMMA OAntigen amounts are set out in Example 3. Similarly, the following paragraphs summarise the protocol used to add the correct amount of fVi-CRMi97 conjugate.
STm and SEn OAg identity and quantification by FAcE in final mixed drug product iNTS- TCV
The O-antigen identity and quantification is assessed by FAcE (Formulated Alhydrogel competitive-ELISA) assay, that is designed to detect the single antigen component for S. Enteritidis and S. Typhimurium OAg polysaccharides in the final formulation. The FAcE assay is a competitive-ELISA method in which serotype specific avAi-Salmonella OAg monoclonal antibodies (mAb) bind to the OAg coated on the ELISA plate and to the respective OAg of the formulated GMMA suspension present in the ELISA wells. The more serotype specific OAg is present in the formulated GMMA suspension, the more mAb will bind to them, and the less will be available to bind to the coated antigen. The ELISA signal is given by the binding of mouse mAb to the coated OAg, therefore the lowest signal is obtained at the highest GMMA concentrations and vice versa. Binding of the mAb is detected using an enzyme-labelled anti-mouse antibody followed by the addition of substrate solution and formation of a yellow colour detected by absorbance at 405 and 490 nm. The result is calculated as the OD difference between 405 and 490nm. Quantification of target antigen on test samples by FAcE assay is obtained using a reference standard curve built by sequential dilutions of Aluminium hydroxide freshly formulated serotype specific GMMA, starting from a known concentration in terms of OAg pg/mL. Test samples are assayed at different dilutions selected to fit within the linear and central part of the standard curve. OAg amount in the test samples is calculated by interpolation of absorbance readings to the standard curve fitted with a 4-parameter logistic regression analysis. As OAg quantification by FAcE is done using anti-0 Ag specific mAbs, this also confirms OAg identity.
Vi identity and quantification by HPAEC-PAD in final mixed drug product iNTS-TCV
Identity and quantification of the Vi polysaccharide was performed by HPAEC-PAD analysis after acid hydrolysis of the sample using the following protocol.
Vi polysaccharide is hydrolysed to the monomer sugar of the repeating unit, corresponding to the chromatographic peak. A dilution series of Vi standard 0.16-5 pg/mL is run in each HPAEC-PAD analysis and the areas of the resulting peaks are used to build a standard curve, interpolate the peak area of the unknown sample and quantify the corresponding pg/mL.
The standard calibration curve is prepared by acid hydrolysis; the acid hydrolysis of samples and standards is performed at the same time and with this approach it is possible to determine the concentration of polysaccharide present in the sample.
1000 pL of a TFA/HC1 mixture (1:6.7 v/v) are added to 300 pL of each dilution of the standard curve and of the sample. All vials are heated in parallel at 80°C for 4.5 hours (final concentrations: HC1 8M TFA 10%). Samples are then chilled at 2°C to 8°C for approximately 15 minutes, dried for at least 1.5 hours under nitrogen flux and finally in a centrifugal evaporator, resuspended in 300 pL of water, filtered and analysed. HPAEC- PAD is performed with a Dionex ICS3000 (or 5000) equipped with a CarboPac PAI column coupled with PAI guard column. Separation is performed at 25°C using the following condition: 15 min, 400 mM NaOH, flow rate 1.5 mL/min (no washing step needed between injections). The effluent is monitored using an electrochemical detector.
Example 6 - immunogenicity assays
ELISA
Assessment of anti-Vi and anti-0 Ag specific total IgG by ELISA. Anti-OAg and anti-Vi antigen-specific IgG levels were measured 2 weeks after the second immunization (day 42) by ELISA as previously reported (Rondini et al, Evaluation of the immunogenicity and biological activity of the Citrobacter freundii Vi-CRM197 conjugate as a vaccine for Salmonella enterica serovar Typhi. Clin Vaccine Immunol. 2011 Mar;18(3):460-8).
Briefly, 96-well round-botom MaxiSorp microtiter plates (Nunc, Roskilde, Denmark) were coated with 100 ml/well antigen overnight at 4°C. OAg purified from S. Paratyphi A (0:2) or S. Enteritidis (0:9) and Vi purified from C.freundii s.l. were used at 15mg/ml and 2mg/ml in carbonate or at Img/ml in phosphate buffer, respectively (Micoli et al, A scalable method for O-antigen purification applied to various Salmonella serovars. Anal Biochem. 2013 Mar 1 ;434(1): 136-45; Micoli et al, Production of a conjugate vaccine for Salmonella enterica serovar Typhi from Citrobacter Vi. Vaccine. 2012 Jan 20;30(5):853 - 61). Plates were blocked with PBS plus 5% fat-free milk (Sigma) for 1 h at room temperature (RT) and then washed 3 times with PBS plus 0.05% Tween 20 (PBS-T). Serum samples were diluted 1:100 and 1:4,000 in PBS-T supplemented with 0.1% BSA (diluent buffer), and both dilutions were assayed in triplicate. After incubation for 2 h at RT, plates were washed three times with PBS-T and incubated at 25°C for 1 h with antimouse goat IgG-alkaline phosphatase (Sigma), diluted 1:6,000, 1:8,800, and 1:2,600 (for Vi, 0:2, or 0:9, respectively) in diluent buffer. After washing three times with PBS-T, plates were developed by adding the alkaline phosphatase substrate (SIGMAFAST N2770; Sigma) and read at 405 nm and 490 nm using an ELx 800 reader (BioTek). ELISA units were expressed relative to a mouse antigen-specific antibody standard serum curve composed by 10 standard points and 2 blank wells (run in duplicate on each plate), with the best five-parameter fit determined by a modified Hill plot. One ELISA unit is defined as the reciprocal of the dilution of the standard serum that gives an absorbance value equal to 1 in this assay.
SBA
Assessment of serum bactericidal activity by SBA. Individual mouse sera collected at day 42 were heat inactivated (HI) at 56°C for 30 min prior to being tested in a serum bactericidal assay based on luminescent readout against Salmonella Paratyphi A NVGH308, Salmonella Enteritidis CMCC3014, and Vi-positive Citrobacter freundii sensu lato strain 3056 (Necchi et al, Development of a high-throughput method to evaluate serum bactericidal activity using bacterial ATP measurement as survival readout. PLoS One. 2017 Feb 13; 12(2):e0172163 ; Necchi et al Setup of luminescence-based serum bactericidal assay against Salmonella Paratyphi A. J Immunol Methods. 2018 Oct;461:l 17-121). SBA was performed in 96-well round-bottom sterile plates (Coming). Dilutions of HI test sera
were incubated for 3 h in the presence of exogenous complement (baby rabbit complement [BRC]) and bacteria as previously described (Necchi et al, Development of a high- throughput method to evaluate serum bactericidal activity using bacterial ATP measurement as survival readout. PLoS One. 2017 Feb 13;12(2):e0172163). Briefly, an adequate volume of reaction mixture containing the target bacterial cells (around 100,000 CFU/ml), BRC (50% for S. Enteritidis, 20% for S. Paratyphi A, and 5% for C.freundii s.l.), and buffer (PBS) was added to SBA plates containing HI serum dilutions and incubated for 3 h at 37°C. At the end of the incubation, the plates were centrifuged for lOmin at 4,000 x g, the supernatant was discarded to remove ATP derived from dead bacteria, and live bacterial pellets resuspended in PBS were transferred to a white roundbottom 96-well plate (Greiner) and mixed 1:1 (vol/vol) with BacTiter-Glo reagent (Promega). The reaction mixture was incubated for 5 min at RT in an orbital shaker, and the luminescence signal was measured using a luminometer (Viktor). A 4-parameter nonlinear regression was applied to raw luminescence for all the serum dilutions tested, as previously described (Rossi et al, Intra-Laboratory Evaluation of Luminescence Based High-Throughput Serum Bactericidal Assay (L-SBA) to Determine Bactericidal Activity of Human Sera against Shigella. High Throughput. 2020 Jun 8;9(2):14). The SBA titer is reported as IC50, defined as serum dilutions giving 50% inhibition of the ATP level in the negative-control well. Titers below the minimum measurable level of luminescence were arbitrarily given an IC50 of 50, representing half of the first dilution of sera tested (i.e., 100). GraphPad Prism 7 software (GraphPad Software) was used for fitting and IC50 determination.
IgG subclasses
Post-second immunization individual sera isolated from the blood samples taken from the mice immunized as described in the section entitled “Example 12 - Antibody subclasses raised by the quadrivalent Pan-Salmonella vaccines in mice” were tested to determine the isotype of the antibodies produced using a ELISA-based assay working with the same principle as described under the heading “ELISA”. The assay was repeated to determine the EU/mL on each individual sera at the dose tested using as secondary antibody antimouse-IgGl, antimouse-IgG2a, antimouse-IgG2b and antimouse-IgG3 antibodies in
standard assay. Results are shown in Example 12 and expressed as calculated as subclass/subclasses total %.
Example 7 - Immunogenicity of the trivalent (iNTS-TCV) vaccine in mice
Groups of 8 CD1 mice were immunised with an iNTS-TCV vaccine produced as described in Example 5 at doses ranging from 0.01 pg to 0.63 pg (O-antigen) of each GMMA and from 0.012 pg to 0.78 pg of Vi polysaccharide (0.032-2.04 pg dose of total polysaccharide). The immunisations involved intraperitoneal immunisation of 0.032-2.04 pg in 500 pl at days 0 and 28. Blood samples were collected at days 27 and 42, and the antibodies raised were measured using the ELISA assay described above in Example 6. Results are shown in Figure 1.
The iNTS-TCV vaccine candidate was well tolerated at all doses. The two antigen GMMA components (SEnGMMA and STmGMMA) and the antigen Vi component induced an antibody dose response. The three antigen components (SEn OAg, STm OAg and Vi) elicited increasing SBA responses for the four lower doses on Day 42.
In a second experiment, groups of 8 CD1 mice were immunised with: the iNTS-TCV vaccine produced as described in Example 5 at 0.032, 0.13, 0.51 and 2.04 pg (total polysaccharide) dose
SEnGMMA (produced as described in Example 1) adsorbed to Alhydrogel at 0.01, 0.04, 0.16 and 0.63 pg (O-antigen) dose
STmGMMA (produced as described in Example 1) adsorbed to Alhydrogel at 0.01, 0.04, 0.16 and 0.63 pg (O-antigen) dose
!Vi-CRMi97 conjugate (produced as described in Example 2) at 0.012, 0.05, 0.19 and 0.78 pg (Vi polysaccharide) dose.
The immunisations involved intraperitoneal immunisation of the corresponding dose for each product in 500 pl at days 0 and 28. Blood samples were collected at days 27 and 42, and the antibodies raised were measured using the ELISA assay and SBA assay described above in Example 6. Results are shown in Figure 2.
Overall no immune interference was observed for the iNTS GMMA components in iNTS- TCV by measurement of ELISA and SBA, but a higher anti -Vi IgG response and serum bactericidal activity of the fVi-CRMi97 component were observed when combined with GMMA in iNTS-TCV.
In a third experiment, groups of 3 New Zealand white rabbits were immunised with the bivalent iNTS-GMMA vaccine described in Example 3. The immunisations involved intramuscular immunisation of 40 pg (O-Antigen) total GMMA at days 0 and 28. Blood samples were collected at days 21, 28 and 42, and the antibodies raised were measured using the ELISA assay described above in Example 6. Results are shown in Figure 3. The vaccine was well tolerated, and all rabbits immunized with the 2-component iNTS- GMMA candidate vaccine responded with high anti-SEn OAg and anti-STm OAg serum IgG.
Example 8 - Production of GMMA from S. Paratyphi A
A S. Paratyphi A strain ED 199 comprising tstolR tspagP LSmshB mutations was prepared using a protocol based on that described for S. Enteritidis and S. Typhimurium in Example 1, except the specific mutations used to delete tolR, pagP and msbB were tolR::cat pagP::kan msbB::tet. GMMA were isolated from that bacterium as described in Example 1.
Example 9 - Production of a conjugate of S. Paratyphi A O-Antigen and CRM197
Conjugating OAg to CRM197 using CDAP chemistry (with or without an ADH linker)
Paratyphi A OAg (0:2) was conjugated to CRM 197 using a random CDAP chemistry approach either via an ADH linker or without the ADH linker. Again, the random CDAP chemistry approach introduces multiple linkages between the OAg and the CRM197. The chemistry is described in Figure 4.
Specifically, 0:2 OH groups are activated with CDAP, using 0:2 to CDAP w/w ratio of 1:0.3 in 150 mM NaCl solution. pH is adjusted to 9-10 with 10% v/v triethylamine and the solution is incubated at room temperature for 3 ± 0.5 minutes on stirring.
Activated cyanoester groups of 0:2 are covalently bound with hydrazide/amino groups of CRM197ADH/CRM197 to form O:2-CDAP-ADH-CRMi97/O:2-CDAP-CRMi97.
CRM197ADH/CRM197 is added in the equal w/w ratio (1:1) of 0:2 at a concentration of 10 mg/mL (final concentration of 0:2 and CRM197ADH/CRM197 is 5 mg/mL). The pH is maintained to 9.5± 0.5 with 10% triethylamine and the solution is mixed for 2-3 hours at room temperature. IM glycine solution is then added to an equal volume of conjugation mixture and the pH is adjusted to 8.0 ± 0.2 with 10% triethylamine; the solution is incubated at 2-8°C for 15±5 hours. The crude conjugate is then buffer exchanged and unbound and unreacted 0:2 is removed using HIC Phenyl HP resin.
A stability study based on the study method described in Example 3 (but measuring free OAg from samples taken at 0 days, 8 days, 14 days and 28 days) was performed on the OAg-CRMi97 conjugate produced using CDAP chemistry with or without an ADH linker. The results (percentage free OAg) are shown in the table below.

Significantly less free OAg was seen in the stability study compared to OAg-CRMi97 conjugates made using the chemistries described in Examples 2-5.
Example 10 - Formulation of two quadrivalent (Pan-Salmonella) vaccines against S.
Typhimurium, S. Enteritidis, S. Typhi, and S. Paratyphi for preclinical studies
Two quadrivalent vaccines were formulated. The first (Pan-Salmonella_O:2-CRMi97) contained GMMA from S. Enteritidis and S. Typhimurium (as described in Example 1), S. Paratyphi A O-antigen conjugated to CRM197 (O:2-CRMi97 as set out in Example 9) and on fVi-CRMi97 conjugate (as set out in Example 2). The second (Pan-Salmonella_ParA GMMA) was similar except the S. Paratyphi A O-antigen conjugate was replaced with S. Paratyphi A GMMA (as described in Example 8).
STmGMMA, SEnGMMA were adsorbed to Alhydrogel. After mixing (1-2 hour), the S. Paratyphi GMMA (or the O:2-CRMi9?) are added. Subsequently, after a quenching step with Phosphate and osmolality adjustment with sodium chloride, the fVi-CRMi97 is added.
Concentration of phosphate buffer was optimized to obtain the highest adsorption, while quenching to ensure optimal particles size.
Final formulation contains sufficient GMMA to provide 40 pg/ml of STm, SEn, and S. Paratyphi A O-Antigens (sPa), 50 pg/ml of Vi polysaccharide in a phosphate buffered saline matrix containing 0.7mg/mL of Aluminium Hydroxide.
The protocols for determining the amounts of STm GMMA, SEnGMMA, and Paratyphi A O-antigens are based on those set out in Example 5. The method used for determining the O-antigen level for S. Enteritidis, S. Typhimurium and S. Paratyphi A is as set out below.
OAg determination for the Salmonella Enteritidis, Typhimurium and Paratyphi A Each single OAg is hydrolysed to release its relative di-deoxy monosaccharide (SEn OAg to Tyvelose; STm OAg to Abequose; SPa OAg to Paratose), corresponding to the chromatographic peak, before analysis by HPAEC-PAD.
Indeed, di-deoxy are the only sugar that differs from the others among the ones composing
SEn, STm and SPa OAg repeating units (RU).
The sample is diluted by volume (450 pL) or by weight on analytical balance, with milliQ water, in order to be within the calibration curve range for each OAg. 120 pL of TFA 1 M is added to vials containing standards or samples and incubated at 75°C for 1.5 hours. After hydrolysis, the vials are chilled at 2-8°C for 15 minutes in the fridge. Samples and standards are dried overnight on centrifugal evaporator at room temperature (RT) in order to remove solvent/TFA. The pellet is dissolved in in 450 pL of milliQ water. Samples and standards are filtered on AcroPrep Advance 96 Filter Plate 0.2 pm Supor 1 mL well and plates loaded on HPAEC-PAD.
Example 11 - Immunogenicity of the quadrivalent pan-Salmonella vaccines in mice
Groups of 10 CD1 mice were immunised with one of the Pan-Salmonella vaccines described in Example 10 at the following doses:
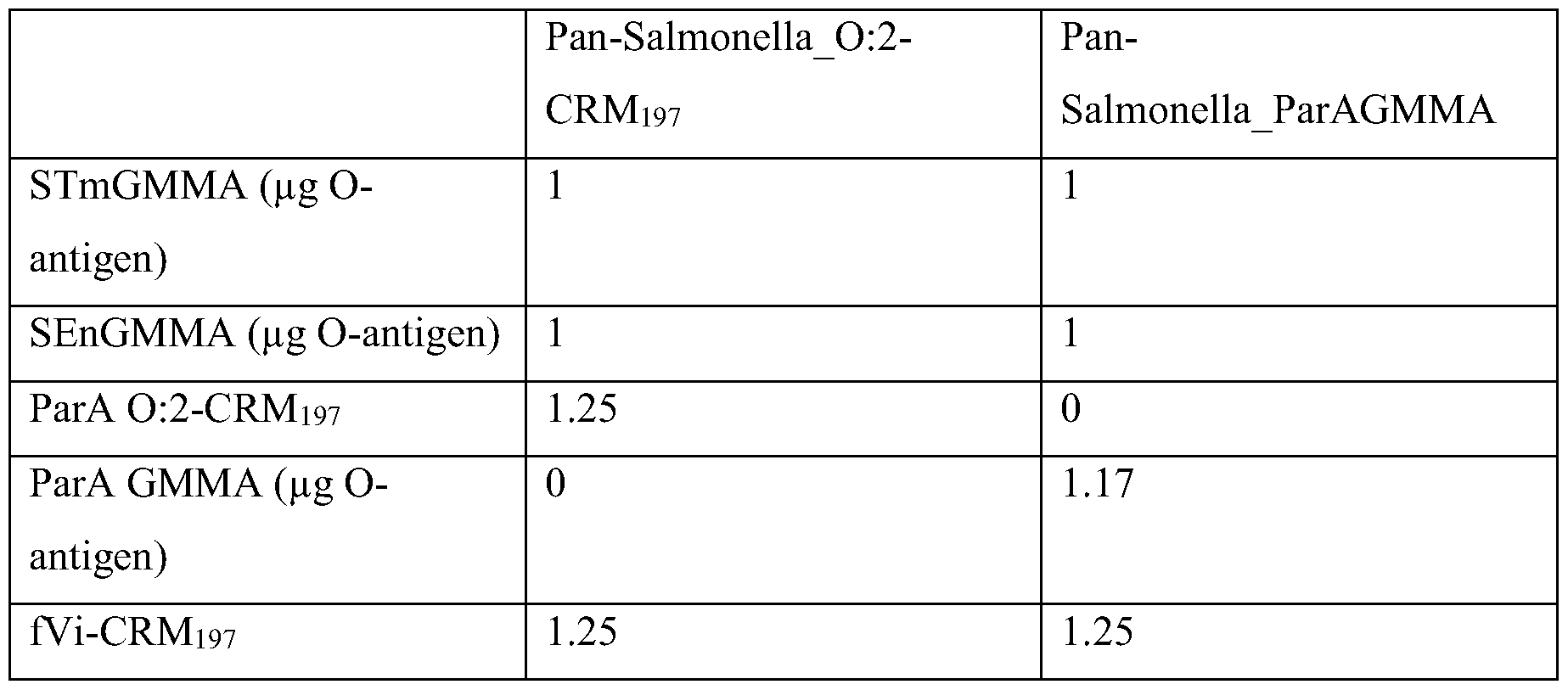
The immunisations involved intraperitoneal immunisation of 200 pl of each formulation at day 0 and day 28. Blood samples were collected at days -1, 27 and 42, and the antibodies raised were studied using the assays described in Example 6 above. The results are set out in Figure 5. In summary, both pan-Salmonella vaccines induced specific serum IgG responses against S. Paratyphi A O-antigen, S. Typhimurium O-antigen, S. Enteritidis O- antigen and Vi polysaccharide, and the antibodies are bactericidal in mice.
In a second experiment, groups of 10 CD1 mice were immunised with:
Quadrivalent Pan-Salmonella_O:2-CRM (STm GMMA + SEn GMMA + O:2-CRMi97 + fVi-CRMw?) produced as described in Example 10 at 1.0 (pg O-antigen), 1.0 (pg O- antigen), 1.25 (pg O-antigen) and 1.25 (pg Vi polysaccharide), respectively, dose Quadrivalent Pan-Salmonella_ParAGMMA (STm GMMA + SEn GMMA + ParA GMMA + fVi-CRMw?) produced as described in Example 10 at 1.0 (pg O-antigen), 1.0 (pg O-antigen), 1.17 (pg O-antigen) and 1.25 (pg Vi polysaccharide), respectively, dose
- Trivalent iNTS-TCV (STm GMMA + SEn GMMA + fVi-CRMi97) produced as described in Example 5 at 1.0 (pg O-antigen), 1.0 (pg O-antigen), and 1.25 (pg Vi polysaccharide), respectively, dose
Bivalent O:2-CRMi97 + fVi-CRMi97 produced as described in Example 3 at 1.25 (pg O-antigen), and 1.25 (pg Vi polysaccharide), respectively, dose
O:2-CRMi97 conjugate (produced as described in Example 9) at 1.25 pg (Vi polysaccharide) dose
ParA GMMA (produced as described in Example 8) adsorbed to Alhydrogel at 1.17 pg (Vi polysaccharide) dose
!Vi-CRMi97 conjugate (produced as described in Example 2) at 1.25 pg (Vi polysaccharide) dose.
The immunisations involved intraperitoneal immunisation of the corresponding dose for each product in 200 pl at days 0 and 28. Blood samples were collected at days 27 and 42, and the antibodies raised were measured using the ELISA assay and SBA assay described above in Example 6. Results are shown in Figure 6.
No negative immune-interference was detected from combining iNTS-TCV with ParA components in the anti-SEn and anti-STm functional antibody responses induced. However, a significantly higher anti-SEn IgG response was elicited by Pan-Salmonella formulation containing O:2-CRM both at days 27 and 42, and also by Pan-Salmonella formulation containing ParA GMMA only at day 27, compared with trivalent iNTS-TCV. Additionally, a significantly higher anti-STm IgG response was elicited by Pan- Salmonella formulation containing O:2-CRM only at day 27 compared with trivalent iNTS-TCV. It is
also important to consider that the 0:2-0 Ag dose used in the Pan-
Salmonella ParAGMMA is slightly different (about 7%) from the 0:2 dose used in the Pan-Salmonella_O:2-CRM.
Presence of iNTS GMMA in the Pan-Salmonella formulation containing 0:2-CRM had overall a positive impact on the anti-0:2 IgG response elicited: a significantly higher post 1 response was induced by the quadrivalent formulation with 0:2-CRM compared with both bivalent 0:2-CRM + fVi-CRM and monovalent 0:2-CRM, and a significantly higher post 2 response was elicited in comparison with the bivalent 0:2-CRM + fVi-CRM formulation. Also SBA titers elicited by the Pan- Salmonella formulation with 0:2-CRM were significantly higher compared to the corresponding bivalent formulation. On the contrary, no significant differences were evidenced in the anti-0:2 IgG response elicited nor in the functionality of antibodies against ParA induced by Pan- Salmonella with ParA GMMA and by the monovalent formulation ParA GMMA.
Example 12 - Antibody subclasses raised by the quadrivalent Pan-Salmonella vaccines in mice
Groups of 10 CD1 mice were immunised with one of the Pan-Salmonella vaccines described in Example 10 at the following doses:

Additional groups of mice were immunised with the following comparators:
• iNTS-TCV (as described in Example 5) with a dose of 1 pg (O-Antigen) STmGMMA, 1 pg (O-antigen) SEnGMMA and 1.25 pg fVi-CRMi97
• a bivalent composition comprising fVi-CRMi97 as described in Example 2 (1.25 pg) and the ParA O:2-CRMi97 conjugate described in Example 9 (1.25pg)
• a bivalent composition comprising fVi-CRMi97 as described in Example 2 (1.25 pg) and ParA GMMA as described in Example 8 (1.17pg O-Ag) • ParA O:2-CRMi97 conjugate described in Example 9 (1.25pg)
• ParA GMMA as described in Example 8 (1.17pg O-Ag)
• fVi-CRMi97 as described in Example 2 (1 ,25pg)
The immunisations involved intraperitoneal immunisation of 200pl of each formulation at day 0 and day 28. Blood samples were collected at days -1, 27 and 42. The classes of antibodies raised against the Vi antigen polysaccharide or the S. Paratyphi A 0:2 O- Antigen were determined using the assay described in Example 6 under the heading “IgG subclasses The results are set out in Figure 7. The absolute values for Figure 7 are set out in the tables below:
Antibody titres against S. Paratyphi A 0:2 O-Antigen
 Antibody titres against Vi antigen polysaccharide
Antibody titres against Vi antigen polysaccharide
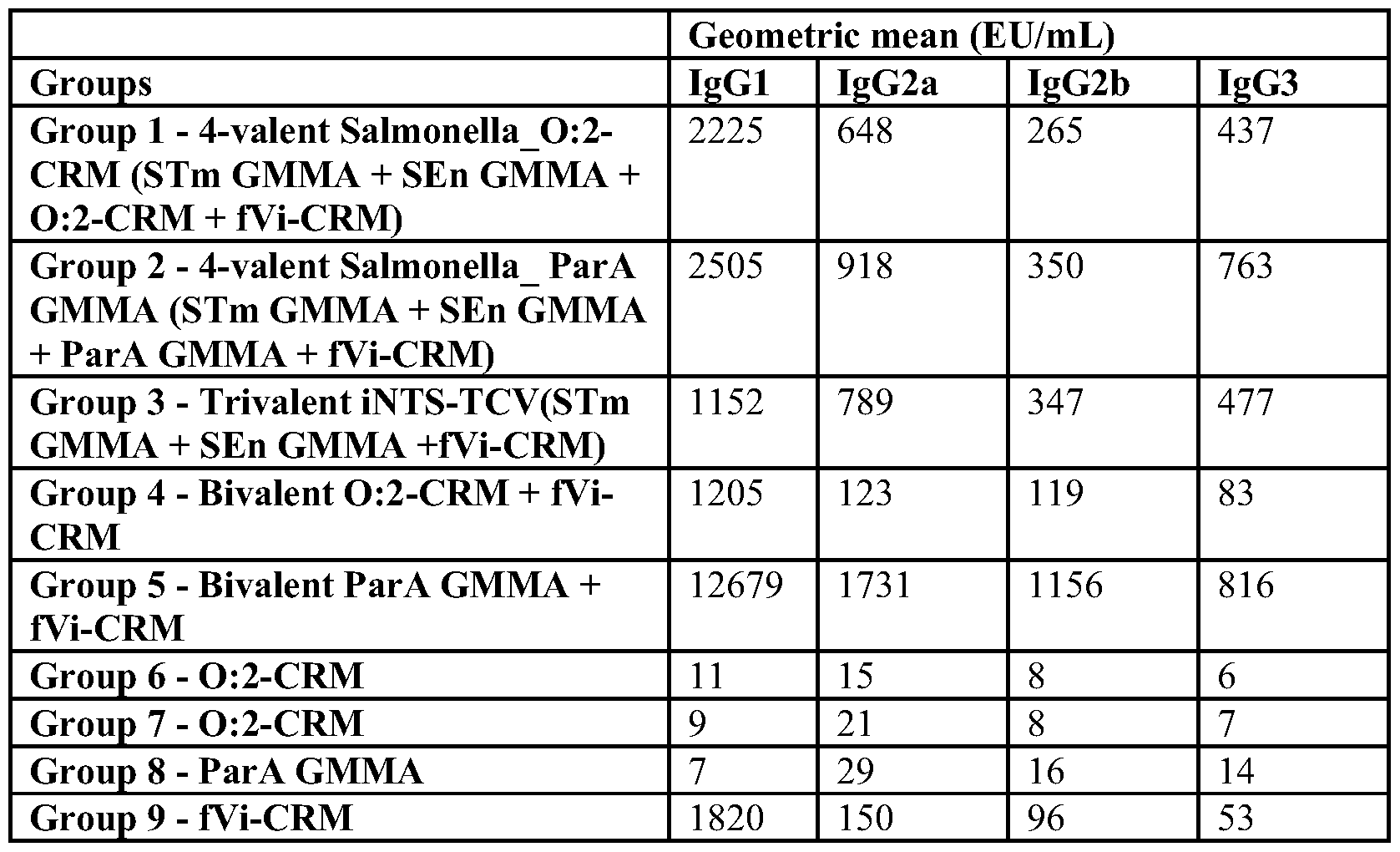
Example 13 - Immunogenicity of the quadrivalent Pan-Salmonella vaccines in rabbits
Groups of 8 New Zealand white rabbits were immunised with one of the Pan-Salmonella vaccines described in Example 10 at the following doses:
 The immunisations involved intramuscular immunisation of 500 pl of each formulation at day 0 and day 28. Blood samples were collected at days -1, 27 and 42, and the antibodies
raised were studied using the assays described in Example 6 above. The results are set out in Figure 8. In summary, both Pan-Salmonella vaccines induced specific serum IgG responses against S. Paratyphi A O-antigen, S. Typhimurium O-antigen, S. Enteritidis O- antigen and fVi polysaccharide, and the antibodies are bactericidal in rabbits.
The immunisations involved intramuscular immunisation of 500 pl of each formulation at day 0 and day 28. Blood samples were collected at days -1, 27 and 42, and the antibodies
raised were studied using the assays described in Example 6 above. The results are set out in Figure 8. In summary, both Pan-Salmonella vaccines induced specific serum IgG responses against S. Paratyphi A O-antigen, S. Typhimurium O-antigen, S. Enteritidis O- antigen and fVi polysaccharide, and the antibodies are bactericidal in rabbits.
Example 14 - administration of the iNTS-TCV vaccine to human subjects
A phase l/2a, observer-blind, randomized, dose-escalation, controlled, multi-country, two- staged, and staggered study including 9 groups will be conducted to evaluate the safety, reactogenicity, and immune response of the trivalent iNTS-TCV vaccine against invasive nontyphoidal Salmonella (iNTS) and typhoid fever when administered intramuscularly on Day 1, Day 57 and Day 169 to healthy European and African adults, compared to placebo. The study will be conducted overall (both Stage 1 and Stage 2) with approximately 155 healthy adult participants (18 to 50 years of age). The healthy European adults will be randomly assigned to 1 of the groups indicated for Stage 1. The healthy African adults will be randomly assigned to 1 of the groups indicated for Stage 2. Each group will receive 2 of the 11 study interventions at each administration, except for the Control stage 2 group which will receive 4 study interventions (a different active comparator at each administration time point together with saline).
Each participant will receive 1 randomly selected intramuscular study intervention per arm on Day 1, Day 57, and Day 169.
Stage 1
Stage 1 (Europe) will follow a 2-step staggered design, leading in with low doses of all the study interventions, in a dose-escalation manner. The sentinel approach will be followed for the first 10 participants each in Step 1 and Step 2, in which only 1 participant will be treated daily. This will be done to ensure maximum safety of the participants.
In Step 1, 10 healthy European adults, randomized in a 2:2:1 ratio, will receive:
• The low dose of the candidate iNTS-TCV vaccine and concomitant saline to be administered in different arms, or
• Separate administration of low doses of iNTS-GMMA and TCV (containing fVi-CRMi97 and S. Paratyphi A O-antigen-CRMi97 conjugates) vaccines in different arms, or
• Placebo and saline in different arms.
In Step 2, 40 healthy European adults will be randomized in a 2:2:1 ratio. A staggered approach will be followed for the first 10 sentinel participants and these participants will be followed up with a safety follow-up call on the next day of administration of study intervention. The remainder of the 30 participants will receive the study intervention in a sequential (at least 60 minutes apart) manner. The participants in Step 2 will receive:
• The full dose of the candidate iNTS-TCV vaccine and concomitant saline to be administered in different arms, or
• Separate administration of full doses of iNTS-GMMA and TCV vaccines in different arms, or
• Placebo and saline in different arms.
In Stage 2 (Africa), a total of 105 healthy African adults, randomized in a 3:3:1 ratio, will receive:
• The full dose of the candidate iNTS-TCV vaccine and concomitant saline to be administered in different arms, or
• Separate administration of full doses of iNTS-GMMA and TCV vaccines in different arms, or
• MenACWY (Menveo) and saline for the first administration, TdaP (Boostrix) and saline for the second administration and Typhoid Vi polysaccharide vaccine (Typhim Vi) and saline for the third administration in different arms. This is the Control stage 2 Group.
The first 21 participants in Stage 2 will initially be recruited with administration proceeding sequentially, at least 60 minutes apart. These participants will be followed up with a safety follow-up call on the next day of administration of study intervention. The recruitment of the remaining 84 participants in Stage 2 will only commence if there is a positive evaluation of all safety data from these participants up to 7 days after the first
administration of the study intervention. The study interventions will be administered in parallel in the remaining 84 participants.
The composition of 0.5 mL of the full dose of the iNTS-TCV vaccine is as follows:

The composition of 0.5 mL of the low dose of the iNTS-TCV vaccine is as follows:
 Example 15 - Antibodies induced by GMMA-based vaccine candidates demonstrate cross-protection
Example 15 - Antibodies induced by GMMA-based vaccine candidates demonstrate cross-protection
CD1 mice were immunised intraperitoneally on days 0 and 28 using 500 pL injection volume of monovalent STm GMMA (dose per mouse per injection in pg 2.5 STm OAg) or
of monovalent SEn GMMA (dose per mouse per injection in pg 2.5 SEn OAg). SBA was performed on mice sera obtained on day 42.
Mice sera elicited by STm GMMA demonstrated bactericidal activity against S. Typhimurium, S. Derby and S. Dublin (Figure 9(a)). Mice sera elicited by SEm GMMA demonstrated bactericidal activity against S. Enteritidis and S. Dublin (Figure 9(b)).
CD1 mice were immunised intraperitoneally on days 0 and 28 using 200 pL injection volume of bivalent iNTS GMMA vaccine (STm and SEn GMMA) (Dose per mouse per injection in pg 1.0 STm OAg + 1.0 SEn OAg) or trivalent iNTS-TCV GMMA vaccine (STm and SEn GMMA and S. Typhi fVi polysaccharide) (Dose per mouse per injection in pg 1.0 STm OAg + 1.0 SEn OAg + 0.125 fVi) . SBA was performed on mice sera obtained on day 42.
Mice sera elicited by bivalent iNTS GMMA vaccine demonstrated bactericidal activity against S. Typhimurium, S. Enteritidis, S. Derby and S. Dublin (Figure 9(c)). Mice sera elicited by trivalent iNTS-TCV vaccine also demonstrated bactericidal activity against S. Typhimurium, S. Enteritidis, S. Derby and S. Dublin (Figure 9(d)).
New Zealand female Rabbits were immunised intramuscularly on days 0 and 28 using 500 pL inj volume of bivalent iNTS GMMA vaccine (STm and SEn GMMA) (dose per rabbit per injection in pg 20 STm OAg + 20 SEn OAg) or trivalent iNTS-TCV GMMA vaccine (STm and SEn GMMA and S. Typhi fVi polysaccharide) (dose per rabbit per injection in pg 20 STm OAg + 20 SEn OAg + 25 fVi). SBA was performed on rabbit sera obtained on day 42.
Rabbit sera elicited by bivalent iNTS GMMA vaccine demonstrated bactericidal activity against S. Typhimurium, S. Enteritidis, S. Derby, S. Dublin and S. Paratyphi A (Figure 9(e)). Mice sera elicited by trivalent iNTS-TCV vaccine also demonstrated bactericidal activity against S. Typhimurium, S. Enteritidis, S. Derby, S. Dublin and S. Paratyphi A (Figure 9(f)).
CD1 mice were immunised intraperitoneally on days 0 and 28 using two quadrivalent PanSalmonella vaccines: (i) STm and SEn GMMA, S. Typhi fVi polysaccharide and S.
Paratyphi A OAg conjugate (Dose per mice per injection in pg 1 STm + 1 SEn + 1.25 ParA + 1.25 fVi); and (ii) STm and SEn GMMA, 5. Typhi fVi polysaccharide and 5. Paratyphi A GMMA (Dose per mice per injection in pg 1 STm + 1 SEn + 1.17 ParA + 1.25 fVi). SBA was performed on mice sera obtained on day 42.
Mice sera elicited by: (i) STm and SEn GMMA, S. Typhi fVi polysaccharide and S.
Paratyphi A OAg conjugate; and (ii) STm and SEn GMMA, S. Typhi fVi polysaccharide and S. Paratyphi A GMMA both demonstrated bactericidal activity against S.
Typhimurium, S. Enteritidis, S. Derby, S. Dublin and S. Paratyphi A (Figure 10).
1
Embodiments of the invention
1. An immunogenic composition comprising:
(a) a Salmonella enterica serovar Typhimurium (S. Typhimurium) antigen;
(b) a Salmonella enterica serovar Enteritidis (S. Enteritidis) antigen; and
(c) a Salmonella enterica serovar Typhi (S. Typhi) antigen.
2. A method of boosting an immune response to a S. Typhi or a S. Paratyphi A antigen comprising administering a composition comprising the S. Typhi antigen or the S. Paratyphi A antigen and GMMA.
3. A method of preventing infection by S. Typhi or S. Paratyphi A comprising administering an immunogenic composition comprising the S. Typhi antigen or the S. Paratyphi A antigen and GMMA, wherein the GMMA boosts the immune response to the S. Typhi antigen or the S. Paratyphi A antigen.
4. An immunogenic composition comprising GMMA for use in a method of boosting an immune response to a S. Typhi or S. Paratyphi A antigen, wherein the method comprises administering an immunogenic composition comprising the S. Typhi antigen or the S. Paratyphi A antigen and GMMA.
5. An immunogenic composition for use in a method of preventing infection by S. Typhi or S. Paratyphi A comprising administering an immunogenic composition comprising the S. Typhi antigen or the S. Paratyphi A antigen and GMMA, wherein the GMMA boosts the immune response to the S. Typhi or the S. Paratyphi A antigen.
6. The method or immunogenic composition for use of embodiments 2 or 4, wherein the method is a method of boosting an immune response to an S. Typhi antigen and the immunogenic composition comprises the S. Typhi antigen.
7. The method or immunogenic composition for use of embodiments 2 or 4, wherein the method is a method of boosting an immune response to an S. Paratyphi A antigen and the immunogenic composition comprises the S. Paratyphi A antigen.
8. The method or immunogenic composition for use of embodiments 3 or 5, wherein the method is a method of preventing infection by S. Typhi, the immunogenic composition comprises the S. Typhi antigen, and the GMMA boosts the immune response to the S. Typhi antigen.
9. The method or immunogenic composition for use of embodiments 3 or 5, wherein the method is a method of preventing infection by S. Paratyphi A, the immunogenic composition comprises the S. Paratyphi A antigen, and the GMMA boosts the immune response to the S. Paratyphi A antigen.
10. The method or immunogenic composition for use of any one of embodiments 2 to 9, wherein the GMMA comprises at least one selected from the group consisting of S. Typhimurium GMMA, A Enteritidis GMMA, and A Paratyphi A GMMA.
11. The immunogenic composition or method of any one of the preceding embodiments, wherein the immunogenic composition further comprises:
(d) a Salmonella enterica serovar Paratyphi A (S. Paratyphi A) antigen.
12. The immunogenic composition or method of embodiment 1 or 11, wherein the S. Typhimurium antigen comprises S. Typhimurium O-antigen.
13. The immunogenic composition or method of any one of embodiments 1, 11 or 12, wherein the S. Typhimurium antigen comprises or consists of outer membrane vesicles from S. Typhimurium.
14. The immunogenic composition or method of any one of embodiments 1 or 11 to 13, wherein the S. Typhimurium antigen comprises or consists of S. Typhimurium GMMA.
15. The immunogenic composition or method of embodiment 10 or 14, wherein the S. Typhimurium GMMA comprise modified lipid A.
16. The immunogenic composition or method of embodiment 15, wherein the modified lipid A is detoxified lipid A.
17. The immunogenic composition or method of embodiment 15 or 16, wherein the modified lipid A is penta-acylated lipid A.
18. The immunogenic composition or method of any one of embodiments 15 to 17, wherein the S. Typhimurium GMMA are derived from S. Typhimurium that does not comprise a gene encoding a functional MsbB protein.
19. The immunogenic composition or method of any one of embodiments 15 to 18, wherein the S. Typhimurium GMMA are derived from S. Typhimurium that is AmsbB.
20. The immunogenic composition or method of any of embodiments 10 or 14 to 19, wherein the S. Typhimurium GMMA are derived from S. Typhimurium that does not comprise a gene encoding a functional PagP protein.
21. The immunogenic composition or method of any one of embodiments 10 or 14 to 20, wherein the S. Typhimurium GMMA are derived from S. Typhimurium that is ApagP.
22. The immunogenic composition or method of any of embodiments 10 or 14 to 21, wherein the S. Typhimurium GMMA are derived from S. Typhimurium that does not comprise a gene encoding a functional TolR protein.
23. The immunogenic composition or method of any of embodiments 10 or 14 to 22, wherein the S. Typhimurium GMMA are derived from S. Typhimurium strain 2192.
24. The immunogenic composition or method of any one of embodiments 10 or 14 to 23, wherein the S. Typhimurium GMMA are derived from S. Typhimurium that is AtolR.
25. The immunogenic composition or method of any one of embodiments 1 or 10 to 24, wherein the immunogenic composition comprises a dose (O-antigen) of between 1 pg and 50 pg, between 2 pg and 25 pg, between 2 pg and 10 pg, between 15 pg and 25 pg, around 20 pg, or around 4 pg of the S. Typhimurium antigen or S. Typhimurium GMMA.
26. The immunogenic composition or method of any one of embodiments 10 or 14 to 25, wherein the S. Typhimurium GMMA boosts the immune response to the S. Typhi or S. Paratyphi A antigen.
27. The immunogenic composition or method of any one of embodiments 1 or 11 to 26, wherein the S. Enteritidis antigen comprises the S. Enteritidis O-antigen.
28. The immunogenic composition or method of any one of embodiments 1 or 11 to 24, wherein the S. Enteritidis antigen comprises or consists of outer membrane vesicles from S. Enteritidis.
29. The immunogenic composition or method of any one of embodiments 1 or 11 to 28, wherein the S. Enteritidis antigen comprises or consists of S. Enteritidis GMMA.
30. The immunogenic composition or method of embodiment 10 or 29, wherein the S. Enteritidis GMMA comprise modified lipid A.
31. The immunogenic composition or method of embodiment 30, wherein the modified lipid A is detoxified lipid A.
32. The immunogenic composition or method of embodiment 30 or 31, wherein the modified lipid A is penta-acylated lipid A.
33. The immunogenic composition or method of any one of embodiments 10 or 29 to 32, wherein the S. Enteritidis GMMA are derived from S. Enteritidis that does not comprise a gene encoding a functional MsbB protein.
34. The immunogenic composition or method of any one of embodiments 10 or 29 to 33, wherein the S. Enteritidis GMMA are derived from S. Enteritidis that is AmsbB.
35. The immunogenic composition or method of any of embodiments 10 or 29 to 34, wherein the S. Enteritidis GMMA are derived from S. Enteritidis that does not comprise a gene encoding a functional PagP protein.
36. The immunogenic composition or method of any one of embodiments 10 or 29 to 35, wherein the S. Enteritidis GMMA are derived from S. Enteritidis that is ApagP.
37. The immunogenic composition or method of any of embodiments 10 or 29 to 36, wherein the S. Enteritidis GMMA are derived from S. Enteritidis that does not comprise a gene encoding a functional TolR protein.
38. The immunogenic composition or method of any of embodiments 10 or 29 to 37, wherein the S. Enteritidis GMMA are derived from S. Enteritidis strain 618.
39. The immunogenic composition or method of any one of embodiments 10 or 29 to 38, wherein the S. Enteritidis GMMA are derived from S. Enteritidis that is AtolR.
40. The immunogenic composition or method of any one of embodiments 1 or 10 to 39, wherein the immunogenic composition comprises a dose (O-antigen) of between 1 pg and 50 pg, between 2 pg and 25 pg, between 2 pg and 10 pg, between 15 pg and 25 pg, around 20 pg, or around 4 pg of S. Enteritidis antigen or S. Enteritidis GMMA.
41. The immunogenic composition or method of any one of embodiments 10 or 29 to 40, wherein the S. Enteritidis GMMA boosts the immune response to the S. Typhi or S. Paratyphi A antigen.
42. The immunogenic composition or method of any one of embodiments 1 to 6, 8, or 10 to 41, wherein the S. Typhi antigen comprises a Vi polysaccharide.
43. The immunogenic composition or method of any one of embodiments 1 to 6, 8, or 10 to 42, wherein the S. Typhi antigen comprise a fragmented Vi (fVi) polysaccharide.
44. The immunogenic composition or method of embodiment 43, wherein the fVi polysaccharide has an average molecular weight of between 40 kDa and 55 kDa, between 41 kDa and 49 kDa, or between 51 kDa and 55 kDa.
45. The immunogenic composition or method of embodiment 43 or 44, wherein the fVi polysaccharide has an average molecular weight of between 51 kDa and 55 kDa.
46. The immunogenic composition or method of any one of embodiments 43 to 45, wherein the fVi polysaccharide is part of an fVi conjugate comprising fVi and a carrier protein.
47. The immunogenic composition or method of embodiment 46, wherein the carrier protein is CRM 197 or diphtheria toxoid.
48. The immunogenic composition or method of embodiment 47, wherein the carrier protein is CRM 197.
49. The immunogenic composition or method of any one of embodiments 46 to 48, wherein the fVi polysaccharide is conjugated to the carrier protein by carbodiimide chemistry, optionally via a linker.
50. The immunogenic composition or method of any one of embodiments 46 to 49, wherein the fVi conjugate is obtained by or obtainable by a method comprising the steps of: a. fragmenting Vi polysaccharide to obtain a fragmented Vi (fVi) polysaccharide having an average molecular weight of between 40 kDa and 55 kDa, between 41 kDa and 49 kDa, or between 51 kDa and 55 kDa;
b. activating the fVi polysaccharide by reacting the fVi polysaccharide obtained in step a. with a carbodiimide and N-hydroxysuccinimide at a pH of 5 to 6 to form an N-hydroxysuccinimide ester fVi derivative; and c. reacting the N-hydroxysuccinimide ester fVi derivative obtained in step b. with the carrier protein to produce the fVi conjugate.
51. The immunogenic composition or method of any one of embodiments 46 to 50, wherein the carbodiimide is EDC.
52. The immunogenic composition or method of embodiment 50 or 51, wherein the carrier protein is derivatised by reacting it with a carbodiimide and a linker.
53. The immunogenic composition or method of embodiment 52, wherein the linker is an adipic acid dihydrazide (ADH) linker.
54. The immunogenic composition or method of embodiment 52 or 53, wherein the carbodiimide is l-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDAC) or the carbodiimide chemistry is EDAC chemistry.
55. The immunogenic composition or method of any one of embodiments 1 to 6, 8, or 10 to 54, wherein the immunogenic composition comprises a dose of 1 to 100 pg, 1 to 50 pg, 15 to 50 pg, 20 to 30 pg, 1 to 20 pg, 1 to 10 pg, around 25 pg, or around 5 pg of fVi polysaccharide.
56. The immunogenic composition or method of any one of embodiments 2 to 50, wherein the S. Paratyphi A antigen comprises S. Paratyphi A O-antigen.
57. The immunogenic composition or method of embodiment 56, wherein the immunogenic composition comprises S. Paratyphi A O-antigen conjugated to a carrier protein.
58. The immunogenic composition or method of embodiment 57, wherein the carrier protein is diphtheria toxoid or CRM197.
59. The immunogenic composition or method of embodiment 58, wherein the carrier protein is CRM 197.
60. The immunogenic composition or method of any one of embodiments 57 to 59, wherein the S. Paratyphi A O-antigen is conjugated to the carrier protein by a method that comprises introducing more than activated site into the S. Paratyphi A O-antigen and/or the S. paratyphi A O-antigen comprises more than one activated site.
61. The immunogenic composition or method of any one of embodiments 57 to 60, wherein the S. Paratyphi A O-antigen is conjugated to the carrier protein by CDAP chemistry, optionally via a linker.
62. The immunogenic composition or method of embodiment 61, wherein the S. Paratyphi A O-antigen is conjugated to the carrier protein via a linker.
63. The immunogenic composition or method of embodiment 62, wherein the linker is adipic acid dihydrazide (ADH).
64. The immunogenic composition or method of any one of embodiments 56 to 63, wherein the immunogenic composition comprises a dose of 1 to 100 pg, 1 to 50 pg, 15 to 50 pg, 20 to 30 pg, 1 to 20 pg, 1 to 10 pg, around 25 pg, or around 5 pg of S. Paratyphi A O-antigen.
65. The immunogenic composition or method of any one of embodiments 2 to 64, wherein the S. Paratyphi A antigen comprises S. Paratyphi A GMMA.
66. The immunogenic composition or method of embodiment 10 or 65, wherein the S. Paratyphi A GMMA comprise modified lipid A.
67. The immunogenic composition or method of embodiment 66, wherein the modified lipid A is detoxified lipid A.
68. The immunogenic composition or method of embodiment 66 or 67, wherein the modified lipid A is penta-acylated lipid A.
69. The immunogenic composition or method of any one of embodiments 10 or 65 to 68, wherein the S. Paratyphi A GMMA are derived from S. Paratyphi A that does not comprise a gene encoding a functional MsbB protein.
70. The immunogenic composition or method of any one of embodiments 10 or 65 to 69, wherein the S. Paratyphi A GMMA are derived from S. Paratyphi A that is AmsbB.
71. The immunogenic composition or method of embodiment 69 or 70, wherein at least part of the msbB gene has been replaced with at least a portion of a tetracycline (tet) gene.
72. The immunogenic composition of embodiment 69, 70 or 71, wherein the GMMA are derived from S. Paratyphi A which is msbB::tet.
73. The immunogenic composition or method of any of embodiments 10 or 65 to 72, wherein the S. Paratyphi A GMMA are derived from S. Paratyphi A that does not comprise a gene encoding a functional PagP protein.
74. The immunogenic composition or method of any one of embodiments 10 or 65 to 73, wherein the S. Paratyphi A GMMA are derived S. Paratyphi A that is ApagP.
75. The immunogenic composition of embodiment 73 or 74, wherein at least part of the pagP gene has been replaced with at least a portion of a kanamycin (kan) gene.
76. The immunogenic composition of any one of embodiments 73 to 75, wherein the r GMMA are derived from S. Paratyphi A which is pagP::kan.
77. The immunogenic composition or method of any of embodiments 10 or 65 to 76, wherein the S. Paratyphi A GMMA are derived from S. Paratyphi A that does not comprise a gene encoding a functional TolR protein.
78. The immunogenic composition or method of any of embodiments 10 or 65 to 77, wherein the S. Paratyphi A GMMA are derived from S. Paratyphi A strain ED 199.
79. The immunogenic composition or method of any one of embodiments 10 or 65 to 78, wherein the S. Paratyphi A GMMA are derived from S. Paratyphi A that is AtolR.
80. The immunogenic composition or method of any one of embodiments 10 or 65 to 79, wherein the immunogenic composition comprises a dose (O-antigen) of between 1 pg and 50 pg, between 2 pg and 25 pg, between 2 pg and 10 pg, between 15 pg and 25 pg, around 20 pg, or around 4 pg of S. Paratyphi A GMMA.
81. The immunogenic composition of any one of embodiments 77, 79 or 80, wherein at least part of the tolR gene has been replaced with at least a portion of a chloramphenicol acetyltransferase (cat) gene.
82. The immunogenic composition of any one of embodiments 77 or 79 to 81, wherein the GMMA are derived from S. Paratyphi A which is tolR:: cat.
83. The immunogenic composition or method of any one of embodiments 10 or 65 to 82, wherein the S. Paratyphi A GMMA boosts the immune response to the S. Typhi antigen.
84. The immunogenic composition or method of any one of embodiments 2 to 83, wherein a method is a method of boosting an immune response to a S. Typhi antigen or a S.
Paratyphi A antigen, if the immune response raised to the S. Typhi antigen is higher when the S. Typhi antigen or the S. Paratyphi A antigen is part of the immunogenic composition comprising GMMA compared to the immune response raised when the S. Typhi antigen or the S. Paratyphi A antigen is not part of an immunogenic composition comprising GMMA.
85. The immunogenic composition or method of any one of embodiments 2 to 84, wherein GMMA boosts the immune response to the S. Typhi antigen or the S. Paratyphi A antigen, if the immune response raised to the S. Typhi antigen or the S. Paratyphi A antigen is higher when the S. Typhi antigen or the S. Paratyphi A antigen is part of the immunogenic composition comprising the GMMA compared to the immune response raised when the S. Typhi antigen or the S. Paratyphi A antigen is not part of an immunogenic composition comprising GMMA.
86. The immunogenic composition or method of embodiment 84 or 85, wherein a method is a method of boosting an immune response to an S. Typhi or a S. Paratyphi A antigen or GMMA boosts the immune response to the S. Typhi antigen or the S. Paratyphi A antigen, if the immune response raised to the S. Typhi antigen or the S. Paratyphi A antigen is at least 5 times, at least 10 times, or at least 20 times higher when the S. Typhi antigen or the S. Paratyphi A antigen is part of the immunogenic composition comprising the GMMA compared to the immune response raised when the S. Typhi antigen or the S. Paratyphi A antigen is not part of an immunogenic composition comprising GMMA.
87. The immunogenic composition or method of any one of embodiments 84 to 86, wherein the immune response raised to the S. Typhi antigen or the S. Paratyphi A antigen is the number of antibodies raised as determined by ELISA 42 days after administration of the immunogenic composition comprising the S. Typhi antigen or S. Paratyphi A antigen and the GMMA at a dose of 0.78 pg of S. Typhi antigen or S. Paratyphi A antigen and 0.63 pg (O-antigen) of the GMMA.
88. The immunogenic composition or method of any one of the preceding embodiments, wherein the immunogenic composition is tolerogenic.
89. The immunogenic composition or method of embodiment 88, wherein the immunogenic composition is tolerogenic if it induces a temperature rise of less than 1.8°C, less than 1.7°C, less than 1.6°C, or less than 1.5°C in a toxicity assay comprising the following steps:
(a) measure the initial temperature of the rabbits;
(b) administer the immunogenic composition at a dose of 20 pg (O-antigen) per GMMA and 25 pg saccharide per saccharide conjugate to rabbits;
(c) monitor the temperature of the rabbits for 5 hours; and
(d) record the maximum temperature of the rabbits, wherein the temperature rise is calculated as equal to the maximum temperature of the rabbits minus the initial temperature of the rabbits.
90. The immunogenic composition or method of embodiment 2 or 88, wherein the immunogenic composition is tolerogenic if it induces a maximum temperature of 41 °C or less, 40.9°C or less, or 40.8°C or less in a toxicity assay comprising the following steps:
(a) administer the immunogenic composition at a dose of 20 pg (O-antigen) per GMMA and 25 pg per saccharide to rabbits;
(b) monitor the temperature of the rabbits for 5 hours; and
(c) record the maximum temperature of the rabbits.
91. The immunogenic composition or method of any one of the preceding embodiments, wherein the immunogenic composition induces at least 103 EU/ml of anti -S. Typhimurium O-antigen antibodies and/or at least 103 EU/ml of anti-5. Enteritidis O-antigen antibodies in an immunogenicity assay comprising the following steps:
(a) immunise mice at days 0 and 28 intraperitoneally with the immunogenic composition at a dose of 1 pg (O-antigen) per GMMA and 1.25 pg saccharide per saccharide; and
(b) measure the anti-5. Typhimurium O-antigen and/or 5. Enteritidis O-antigen antibody level by ELISA at day 42.
92. The immunogenic composition or method of any one of the preceding embodiments, wherein the immunogenic composition induces at least 103 EU/ml of anti-fVi conjugate antibodies in an immunogenicity assay comprising the following steps:
(a) immunise mice at days 0 and 28 intraperitoneally with the immunogenic composition at a dose of 1 pg (O-antigen) per GMMA and 1.25 pg saccharide per saccharide conjugate; and
(b) measure the anti-fVi conjugate antibody level by ELISA at day 42.
93. The immunogenic composition or method of any one of the preceding embodiments, wherein the immunogenic composition induces at least 103 EU/ml of S. Paratyphi A O- antigen antibodies in an immunogenicity assay comprising the following steps:
(a) immunise mice at days 0 and 28 intraperitoneally with the immunogenic composition at a dose of 1 pg (O-antigen) per GMMA and 1.25 pg saccharide per saccharide conjugate; and
(b) measure the anti-5. Paratyphi A O-antigen antibody level by ELISA at day 42.
94. The immunogenic composition or method of any one of the preceding embodiments, wherein the immunogenic composition induces at least 103 5 EU/ml of S. Paratyphi A O- antigen antibodies in an immunogenicity assay comprising the following steps:
(a) immunise mice at days 0 and 28 intraperitoneally with the immunogenic composition at a dose of 1 pg (O-antigen) per GMMA and 1.25 pg saccharide per saccharide conjugate; and
(b) measure the anti-5. Paratyphi A O-antigen antibody level by ELISA at day 42.
95. The immunogenic composition or method of any one of embodiments 1 or 10 to 94, wherein the level of anti-5. Typhimurium O-antigen antibodies induced is at least 90%, at least 95%, or at least 98% of the level of anti-5. Typhimurium O-antigen antibodies induced by a corresponding monovalent 5. Typhimurium immunogenic composition.
96. The immunogenic composition or method of any one of embodiments 1 or 10 to 95, wherein the level of anti-5. Enteritidis O-antigen antibodies induced is at least 90%, at least 95%, or at least 98% of the level of anti-5. Enteritidis O-antigen antibodies induced by a corresponding monovalent 5. Enteritidis immunogenic composition.
97. The immunogenic composition or method of any one of embodiments 1 to 6, 8, or 10 to 96, wherein the level of anti-fVi conjugate antibodies induced is at least 90%, at least 95%, or at least 98% of the level of anti-fVi conjugate antibodies induced by a corresponding monovalent fVi capsular polysaccharide immunogenic composition.
98. The immunogenic composition or method of any one of embodiments 2 to 5, 7, or 11 to 97, wherein the level of anti-5. Paratyphi A O-antigen antibodies induced is at least 90%, at least 95%, or at least 98% of the level of anti-5. Paratyphi A O-antigen antibodies induced by a corresponding monovalent 5. Paratyphi A immunogenic composition.
99. The immunogenic composition or method of any one of embodiments 95 to 98, wherein the level of anti-5. Typhimurium O-antigen antibodies, anti-5. Enteritidis O- antigen antibodies, anti-fVi conjugate antibodies and/or anti-5. Paratyphi A O-antigen antibodies is measured in an immunogenicity assay comprising the following steps:
(a) immunise mice at days 0 and 28 intraperitoneally with the immunogenic composition at a dose of 1 pg (O-antigen) per GMMA and 1.25 pg saccharide per saccharide conjugate; and
(b) measure the anti-5. Typhimurium, anti-5. Enteritidis, anti-fVi conjugate and/or anti-5. Paratyphi A O-antigen antibodies levels by ELISA at day 42.
100. The immunogenic composition or method of any one of the preceding embodiments, wherein the immunogenic composition induces antibodies against three or more, four or more, five or more, six or more, seven or more, eight or more, nine or more, or all ten of the following strains:
(a) 5. Typhimurium ST34;
(b) 5. Typhimurium 10433 3;
(c) 5. Typhimurium D23580;
(d) 5. Typhimurium ST4/74;
(e) 5. Typhimurium A 130;
(f) 5. enterica serovar Derby;
(g) 5. enterica serovar Dublin;
(h) 5. Enteritidis Al 636;
(i) 5. Enteritidis CP255; and
(j) 5. Enteritidis D7795.
101. The immunogenic composition or method of any one of embodiments 2 to 5, 7, or 11 to 100, wherein the immunogenic composition induces anti-5. Paratyphi A O-antigen
antibodies in each of classes IgG3, IgG2b, IgG2a, and IgGl, as determined using an antibody class assay comprising the following steps:
(a) immunise mice at days 0 and 28 intraperitoneally with the immunogenic composition at a dose of 1 pg (O-antigen) per GMMA and 1.25 pg saccharide per saccharide conjugate; and
(b) measure the anti-5. Paratyphi A O-antigen antibody subtype level by ELISA at day 42.
102. The immunogenic composition or method of any one of the preceding embodiments, wherein the immunogenic composition further comprises an adjuvant.
103. The immunogenic composition or method of embodiment 102, wherein the adjuvant is an aluminium adjuvant.
104. The immunogenic composition or method of embodiment 102 or 103, wherein the adjuvant comprises aluminium hydroxide and/or aluminium phosphate.
105. The immunogenic composition or method of embodiment 104, wherein the adjuvant comprises aluminium hydroxide.
106. The immunogenic composition or method of any one of embodiments 103 to 105, wherein the adjuvant comprises between 0.1 mg and 10 mg Al3+, between 0.1 mg and 5 mg Al3+, between 0.3 mg and 0.4 mg Al3+, or around 0.35 mg Al3+.
107. The immunogenic composition or method of any one of the preceding embodiments, wherein the immunogenic composition further comprises a pharmaceutically acceptable excipient.
108. The immunogenic composition or method of embodiment 107, wherein the pharmaceutically acceptable excipient comprises phosphate buffered saline.
109. The immunogenic composition or method of embodiment 108, wherein the phosphate buffered saline is at a pH between 6 and 7, or around 6.5.
110. A vaccine comprising the immunogenic composition of any one of embodiments 1 or 11 to 109.
111. The immunogenic composition or vaccine of any one of embodiments 1 or 11 to 110, for use in a method of preventing an infection.
112. A method of preventing an infection comprising administering an effective amount of the immunogenic composition or vaccine of any one of embodiments 1 or 11 to 110 to a subject.
113. Use of the immunogenic composition or vaccine of any one of embodiments 1 or 11 to 110, for the manufacture of a medicament for use in a method of preventing an infection.
114. The immunogenic composition or vaccine for use of embodiment 111, or the use of embodiment 113, wherein the method of preventing an infection comprises administering an effective amount of the immunogenic composition or vaccine of any one of embodiments 1 or 11 to 110 to a subject.
115. The immunogenic composition or vaccine for use, method, or use of any one of embodiments 111 to 114, wherein the method of preventing an infection is a method of preventing Salmonella infection.
116. The immunogenic composition or vaccine for use, method, or use of any one of embodiments 111 to 115, wherein the method of preventing an infection is a method of preventing invasive non-typeable salmonella infection.
117. The immunogenic composition or vaccine for use, method, or use of any one of
embodiments 111 to 116, wherein the method of preventing an infection is a method of preventing infection by S. Typhimurium, S. Enteritidis, S. Typhi and/or S. Paratyphi A.
118. The immunogenic composition, immunogenic composition or vaccine for use, method, or use of any one of embodiments 10 or 65 to 117, wherein the O-antigen/protein ratio of the S. Paratyphi A GMMA is at least 0.4.
Claims
1. An immunogenic composition comprising:
(a) a Salmonella enterica serovar Typhimurium (5. Typhimurium) antigen, wherein the S. Typhimurium antigen comprises or consists of outer membrane vesicles from S. Typhimurium;
(b) a Salmonella enterica serovar Enteritidis (S. Enteritidis) antigen, wherein the S. Enteritidis antigen comprises or consists of outer membrane vesicles from S. Enteritidis; and
(c) a Salmonella enterica serovar Typhi (S. Typhi) antigen, wherein the S. Typhi antigen comprises a Vi polysaccharide.
2. A method of boosting an immune response to a S. Typhi or a S. Paratyphi A antigen comprising administering a composition comprising the S. Typhi antigen or the S.
Paratyphi A antigen and GMMA.
3. A method of preventing infection by S. Typhi or S. Paratyphi A comprising administering an immunogenic composition comprising the S. Typhi antigen or the S. Paratyphi A antigen and GMMA, wherein the GMMA boosts the immune response to the S. Typhi antigen or the S. Paratyphi A antigen.
4. An immunogenic composition comprising GMMA for use in a method of boosting an immune response to a S. Typhi or S. Paratyphi A antigen, wherein the method comprises administering an immunogenic composition comprising the S. Typhi antigen or the 5. Paratyphi A antigen and GMMA.
5. An immunogenic composition for use in a method of preventing infection by S. Typhi or S. Paratyphi A comprising administering an immunogenic composition comprising the S. Typhi antigen or the S. Paratyphi A antigen and GMMA, wherein the GMMA boosts the immune response to the S. Typhi or the S. Paratyphi A antigen.
6. The method or immunogenic composition for use of any one of claims 2 to 5, wherein the GMMA comprises at least one selected from the group consisting of S. Typhimurium GMMA, A Enteritidis GMMA, and A Paratyphi A GMMA.
7. The immunogenic composition or method of any one of the preceding claims, wherein the immunogenic composition further comprises:
(d) a Salmonella enterica serovar Paratyphi A (A Paratyphi A) antigen.
8. The immunogenic composition or method of claim 1 or 7, wherein the A Typhimurium antigen comprises or consists of A Typhimurium GMMA.
9. The immunogenic composition or method of claim 6 or 8, wherein the A Typhimurium GMMA comprise modified lipid A, optionally wherein the modified lipid A is detoxified lipid A.
10. The immunogenic composition or method of any one of claims 1 or 7 to 9, wherein the A Enteritidis antigen comprises or consists of A Enteritidis GMMA.
11. The immunogenic composition or method of claim 6 or 10, wherein the A Enteritidis GMMA comprise modified lipid A, optionally wherein the modified lipid A is detoxified lipid A.
12. The immunogenic composition or method of any one of the preceding claims, wherein the A Typhi antigen comprise a fragmented Vi (fVi) polysaccharide.
13. The immunogenic composition or method of claim 12, wherein the fVi polysaccharide is part of an fVi conjugate comprising fVi and a carrier protein.
14. The immunogenic composition or method of claim 13, wherein the carrier protein is CRM197 or diphtheria toxoid.
15. The immunogenic composition or method of any one of claims 2 to 14, wherein the S. Paratyphi A antigen comprises S. Paratyphi A O-antigen.
16. The immunogenic composition or method of any one of claims 2 to 15, wherein the S. Paratyphi A antigen comprises S. Paratyphi A GMMA.
17. The immunogenic composition or method of claim 6 or 16, wherein the S. Paratyphi A GMMA comprise modified lipid A.
18. The immunogenic composition or method of claim 17, wherein the modified lipid A is detoxified lipid A.
19. The immunogenic composition or method of any one of the preceding claims, wherein the immunogenic composition further comprises an adjuvant.
20. The immunogenic composition or method of claim 19, wherein the adjuvant is an aluminium adjuvant.
21. A method of preventing an infection comprising administering an effective amount of the immunogenic composition of any one of claims 1 or 7 to 20 to a subject.
22. Use of the immunogenic composition of any one of claims 1 or 7 to 20, for the manufacture of a medicament for use in a method of preventing an infection.
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB2311231.1 | 2023-07-21 | ||
| GBGB2311231.1A GB202311231D0 (en) | 2023-07-21 | 2023-07-21 | Immunigenic composition |
| IN202311049311 | 2023-07-21 | ||
| GB2311233.7 | 2023-07-21 | ||
| GBGB2311233.7A GB202311233D0 (en) | 2023-07-21 | 2023-07-21 | Immunogenic composition |
| IN202311049311 | 2023-07-21 | ||
| GBGB2318238.9A GB202318238D0 (en) | 2023-11-29 | 2023-11-29 | Vaccine |
| GB2318238.9 | 2023-11-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2025021712A1 true WO2025021712A1 (en) | 2025-01-30 |
Family
ID=92043412
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2024/070615 Pending WO2025021710A1 (en) | 2023-07-21 | 2024-07-19 | Immunogenic composition |
| PCT/EP2024/070618 Pending WO2025021712A1 (en) | 2023-07-21 | 2024-07-19 | Immunogenic composition |
| PCT/EP2024/070606 Pending WO2025021704A1 (en) | 2023-07-21 | 2024-07-19 | Vaccine |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2024/070615 Pending WO2025021710A1 (en) | 2023-07-21 | 2024-07-19 | Immunogenic composition |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2024/070606 Pending WO2025021704A1 (en) | 2023-07-21 | 2024-07-19 | Vaccine |
Country Status (1)
| Country | Link |
|---|---|
| WO (3) | WO2025021710A1 (en) |
Citations (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US160A (en) | 1837-04-17 | Process of mabrtji actubind white lead | ||
| US4663A (en) | 1846-07-28 | Smut-machibte | ||
| US4057685A (en) | 1972-02-02 | 1977-11-08 | Abbott Laboratories | Chemically modified endotoxin immunizing agent |
| US4356170A (en) | 1981-05-27 | 1982-10-26 | Canadian Patents & Development Ltd. | Immunogenic polysaccharide-protein conjugates |
| US4459286A (en) | 1983-01-31 | 1984-07-10 | Merck & Co., Inc. | Coupled H. influenzae type B vaccine |
| EP0208375A2 (en) | 1985-07-05 | 1987-01-14 | SCLAVO S.p.A. | Glycoproteinic conjugates having trivalent immunogenic activity |
| US4673574A (en) | 1981-08-31 | 1987-06-16 | Anderson Porter W | Immunogenic conjugates |
| US4695624A (en) | 1984-05-10 | 1987-09-22 | Merck & Co., Inc. | Covalently-modified polyanionic bacterial polysaccharides, stable covalent conjugates of such polysaccharides and immunogenic proteins with bigeneric spacers, and methods of preparing such polysaccharides and conjugates and of confirming covalency |
| US4761283A (en) | 1983-07-05 | 1988-08-02 | The University Of Rochester | Immunogenic conjugates |
| US4808700A (en) | 1984-07-09 | 1989-02-28 | Praxis Biologics, Inc. | Immunogenic conjugates of non-toxic E. coli LT-B enterotoxin subunit and capsular polymers |
| US4882317A (en) | 1984-05-10 | 1989-11-21 | Merck & Co., Inc. | Covalently-modified bacterial polysaccharides, stable covalent conjugates of such polysaccharides and immunogenic proteins with bigeneric spacers and methods of preparing such polysaccharides and conjugataes and of confirming covalency |
| US4965338A (en) | 1988-08-18 | 1990-10-23 | General Electric Company | PBT with improved tracking resistance |
| US5204098A (en) | 1988-02-16 | 1993-04-20 | The United States Of America As Represented By The Department Of Health And Human Services | Polysaccharide-protein conjugates |
| EP0477508B1 (en) | 1990-09-28 | 1995-07-12 | American Cyanamid Company | Improved oligosaccharide conjugate vaccines |
| WO1998042721A1 (en) | 1997-03-24 | 1998-10-01 | Andrew Lees | Uronium salt conjugate vaccines |
| WO2000010599A2 (en) | 1998-08-19 | 2000-03-02 | North American Vaccine, Inc. | IMMUNOGENIC β-PROPIONAMIDO-LINKED POLYSACCHARIDE PROTEIN CONJUGATE USEFUL AS A VACCINE PRODUCED USING AN N-ACRYLOYLATED POLYSACCHARIDE |
| WO2002009643A2 (en) | 2000-07-27 | 2002-02-07 | Children's Hospital & Research Center At Oakland | Vaccines for broad spectrum protection against diseases caused by neisseria meningitidis |
| WO2007000343A2 (en) | 2005-06-27 | 2007-01-04 | Glaxosmithkline Biologicals S.A. | Process for manufacturing vaccines |
| WO2011036562A1 (en) | 2009-09-28 | 2011-03-31 | Novartis Vaccines Institute For Global Health Srl | Purification of bacterial vesicles |
| US20130129776A1 (en) * | 2011-11-07 | 2013-05-23 | University Of Maryland, Baltimore | Broad Spectrum Vaccine Against Typhoidal and Non-typhoidal Salmonella Disease |
| WO2015068129A1 (en) | 2013-11-08 | 2015-05-14 | Novartis Ag | Salmonella conjugate vaccines |
| WO2021044436A2 (en) * | 2019-09-03 | 2021-03-11 | Serum Institute Of India Private Limited | Immunogenic compositions against enteric diseases and methods for its preparation thereof |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9358284B2 (en) | 2011-09-14 | 2016-06-07 | Glaxosmithkline Biologicals Sa | Methods for making saccharide-protein glycoconjugates |
| MX2019006105A (en) * | 2016-11-25 | 2019-08-21 | Glaxosmithkline Biologicals Sa | nOMV-ANTIGEN CONJUGATES AND USE THEREOF. |
| BR112019019440A2 (en) * | 2017-03-31 | 2020-04-14 | Indian Council Medical Res | vaccine against typhoid fever based on outer membrane vesicles of two different strains of typhoid salmonella species |
| US11260119B2 (en) * | 2018-08-24 | 2022-03-01 | Pfizer Inc. | Escherichia coli compositions and methods thereof |
-
2024
- 2024-07-19 WO PCT/EP2024/070615 patent/WO2025021710A1/en active Pending
- 2024-07-19 WO PCT/EP2024/070618 patent/WO2025021712A1/en active Pending
- 2024-07-19 WO PCT/EP2024/070606 patent/WO2025021704A1/en active Pending
Patent Citations (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4663A (en) | 1846-07-28 | Smut-machibte | ||
| US160A (en) | 1837-04-17 | Process of mabrtji actubind white lead | ||
| US4057685A (en) | 1972-02-02 | 1977-11-08 | Abbott Laboratories | Chemically modified endotoxin immunizing agent |
| US4356170A (en) | 1981-05-27 | 1982-10-26 | Canadian Patents & Development Ltd. | Immunogenic polysaccharide-protein conjugates |
| US4673574A (en) | 1981-08-31 | 1987-06-16 | Anderson Porter W | Immunogenic conjugates |
| US4459286A (en) | 1983-01-31 | 1984-07-10 | Merck & Co., Inc. | Coupled H. influenzae type B vaccine |
| US4761283A (en) | 1983-07-05 | 1988-08-02 | The University Of Rochester | Immunogenic conjugates |
| US4882317A (en) | 1984-05-10 | 1989-11-21 | Merck & Co., Inc. | Covalently-modified bacterial polysaccharides, stable covalent conjugates of such polysaccharides and immunogenic proteins with bigeneric spacers and methods of preparing such polysaccharides and conjugataes and of confirming covalency |
| US4695624A (en) | 1984-05-10 | 1987-09-22 | Merck & Co., Inc. | Covalently-modified polyanionic bacterial polysaccharides, stable covalent conjugates of such polysaccharides and immunogenic proteins with bigeneric spacers, and methods of preparing such polysaccharides and conjugates and of confirming covalency |
| US4808700A (en) | 1984-07-09 | 1989-02-28 | Praxis Biologics, Inc. | Immunogenic conjugates of non-toxic E. coli LT-B enterotoxin subunit and capsular polymers |
| EP0208375A2 (en) | 1985-07-05 | 1987-01-14 | SCLAVO S.p.A. | Glycoproteinic conjugates having trivalent immunogenic activity |
| US5204098A (en) | 1988-02-16 | 1993-04-20 | The United States Of America As Represented By The Department Of Health And Human Services | Polysaccharide-protein conjugates |
| US4965338A (en) | 1988-08-18 | 1990-10-23 | General Electric Company | PBT with improved tracking resistance |
| EP0477508B1 (en) | 1990-09-28 | 1995-07-12 | American Cyanamid Company | Improved oligosaccharide conjugate vaccines |
| WO1998042721A1 (en) | 1997-03-24 | 1998-10-01 | Andrew Lees | Uronium salt conjugate vaccines |
| WO2000010599A2 (en) | 1998-08-19 | 2000-03-02 | North American Vaccine, Inc. | IMMUNOGENIC β-PROPIONAMIDO-LINKED POLYSACCHARIDE PROTEIN CONJUGATE USEFUL AS A VACCINE PRODUCED USING AN N-ACRYLOYLATED POLYSACCHARIDE |
| WO2002009643A2 (en) | 2000-07-27 | 2002-02-07 | Children's Hospital & Research Center At Oakland | Vaccines for broad spectrum protection against diseases caused by neisseria meningitidis |
| WO2007000343A2 (en) | 2005-06-27 | 2007-01-04 | Glaxosmithkline Biologicals S.A. | Process for manufacturing vaccines |
| WO2011036562A1 (en) | 2009-09-28 | 2011-03-31 | Novartis Vaccines Institute For Global Health Srl | Purification of bacterial vesicles |
| US20130129776A1 (en) * | 2011-11-07 | 2013-05-23 | University Of Maryland, Baltimore | Broad Spectrum Vaccine Against Typhoidal and Non-typhoidal Salmonella Disease |
| WO2015068129A1 (en) | 2013-11-08 | 2015-05-14 | Novartis Ag | Salmonella conjugate vaccines |
| WO2021044436A2 (en) * | 2019-09-03 | 2021-03-11 | Serum Institute Of India Private Limited | Immunogenic compositions against enteric diseases and methods for its preparation thereof |
Non-Patent Citations (39)
| Title |
|---|
| AHMED ET AL., J INFECT DIS., vol. 193, no. 4, 2006, pages 515 - 21 |
| BETHELL G.S. ET AL., J. BIOL. CHEM., vol. 254, 1979, pages 2572 - 4 |
| BEVERIDGE, J. BACTERIOL., vol. 181, 1999, pages 4725 - 4733 |
| BMC INFECT DIS., vol. 23, no. 1, 23 May 2023 (2023-05-23), pages 346 |
| CHU ET AL., INFECT IMMUN., vol. 59, no. 12, 1991, pages 4450 - 58 |
| COX ET AL., GLYCOCONJ J, vol. 28, 2011, pages 165 - 182 |
| DE BENEDETTO ET AL.: "Multiple Techniques for Size Determination of Generalized Modules for Membrane Antigens from Salmonella typhimurium and Salmonella enteritidis", ACS OMEGA., vol. 2, no. 11, 30 November 2017 (2017-11-30), pages 8282 - 8289 |
| GASPERINI G ET AL: "Salmonella Paratyphi A Outer Membrane Vesicles Displaying Vi Polysaccharide as a Multivalent Vaccine against Enteric Fever", INFECTION AND IMMUNITY, vol. 89, no. 4, 1 January 2021 (2021-01-01), US, pages 1 - 9, XP093193963, ISSN: 0019-9567, DOI: 10.1128/IAI.00699-20 * |
| GERKE CCOLUCCI AM,GIANNELLI CSANZONE SVITALI CGSOLLAI L ET AL.: "Production of a Shigella sonnei Vaccine Based on Generalized Modules for Membrane Antigens (GMMA), 1790GAHB", PLOS ONE, vol. 10, no. 8, 2015, pages 0134478 |
| GEVER ET AL., MED. MICROBIOL. IMMUNOL., vol. 165, 1979, pages 171 - 288 |
| HEARN M.T.W., J. CHROMATOGR., vol. 218, 1981, pages 509 - 18 |
| HURLEY ET AL.: "Atypical Salmonella enterica Serovars in Murine and Human Macrophage Infection Models", INFECT IMMUN., vol. 88, no. 4, 23 March 2020 (2020-03-23), pages 00353 - 19 |
| IRFAN ET AL.: ", Ceftriaxone resistant Salmonella enterica serovar Paratyphi A identified in a case of enteric fever: first case report from Pakistan", BMC INFECT DIS., vol. 23, no. 1, 26 April 2023 (2023-04-26), pages 267, XP021317749, DOI: 10.1186/s12879-023-08152-9 |
| KATIAL ET AL., INFECT IMMUN, vol. 70, 2002, pages 702 - 707 |
| KONADU ET AL., INFECT IMMUN., vol. 62, no. 11, 1994, pages 5048 - 54 |
| KONADU ET AL., INFECT IMMUN., vol. 7, 1996, pages 2709 - 15 |
| LANZILAO LSTEFANETTI GSAUL AMACLENNAN CAMICOLI FRONDINI S.: "Strain Selection for Generation of O-Antigen-Based Glycoconjugate Vaccines against Invasive Nontyphoidal Salmonella Disease", PLOS ONE., vol. 10, no. 10, 7 October 2015 (2015-10-07), pages 0139847 |
| MATHER ET AL.: ", New Variant of Multidrug-Resistant Salmonella enterica Serovar Typhimurium Associated with Invasive Disease in Immunocompromised Patients in Vietnam", MBIO, vol. 9, no. 5, 4 September 2018 (2018-09-04), pages 01056 - 18 |
| MICOLI ET AL., PLOSONE, vol. 7, no. 11, 2012, pages 47039 |
| MICOLI ET AL., VACCINE, vol. 29, no. 4, 2011, pages 712 - 20 |
| MICOLI ET AL.: "A scalable method for O-antigen purification applied to various Salmonella serovars", ANAL BIOCHEM., vol. 434, no. 1, 1 March 2013 (2013-03-01), pages 136 - 45, XP055466360, DOI: 10.1016/j.ab.2012.10.038 |
| MICOLI ET AL.: "Production of a conjugate vaccine for Salmonella enterica serovar Typhi from Citrobacter Vi", VACCINE, vol. 30, no. 5, 20 January 2012 (2012-01-20), pages 853 - 61, XP028436164, DOI: 10.1016/j.vaccine.2011.11.108 |
| MICOLI FRANCESCA ET AL: "Comparative immunogenicity and efficacy of equivalent outer membrane vesicle and glycoconjugate vaccines against nontyphoidal Salmonella", PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES, vol. 115, no. 41, 27 September 2018 (2018-09-27), pages 10428 - 10433, XP093213287, ISSN: 0027-8424, DOI: 10.1073/pnas.1807655115 * |
| MICOLI FRANCESCA ET AL: "GMMA Is a Versatile Platform to Design Effective Multivalent Combination Vaccines", VACCINES, vol. 8, no. 3, 17 September 2020 (2020-09-17), CH, pages 540, XP093213312, ISSN: 2076-393X, DOI: 10.3390/vaccines8030540 * |
| MOL. IMMUNOL, vol. 22, 1985, pages 907 - 919 |
| MYLONA ESANCHEZ-GARRIDO JHOANG THU TNDONGOL SKARKEY ABAKER SSHENOY ARFRANKEL G.: "Very long O-antigen chains of Salmonella Paratyphi A inhibit inflammasome activation and pyroptotic cell death", CELL MICROBIOL., vol. 23, no. 5, May 2021 (2021-05-01), pages 13306 |
| NECCHI ET AL.: "Development of a high-throughput method to evaluate serum bactericidal activity using bacterial ATP measurement as survival readout", PLOS ONE, vol. 12, no. 2, 13 February 2017 (2017-02-13), pages 0172163 |
| NECCHI ET AL.: "Development of a high-throughput method to evaluate serum bactericidal activity using bacterial ATP measurement as survival readout.", PLOS ONE, vol. 12, no. 2, 13 February 2017 (2017-02-13), pages 0172163 |
| NECCHI ET AL.: "Setup of luminescence-based serum bactericidal assay against Salmonella Paratyphi A", J IMMUNOL METHODS., vol. 461, October 2018 (2018-10-01), pages 117 - 121 |
| OKORO ET AL.: "Intracontinental spread of human invasive Salmonella Typhimurium pathovariants in sub-Saharan Africa", NAT GENET., vol. 44, no. 11, November 2012 (2012-11-01), pages 1215 - 21 |
| PEREZ-SEPULVEDA ET AL.: "Complete Genome Sequences of African Salmonella enterica Serovar Enteritidis Clinical Isolates Associated with Bloodstream Infection", MICROBIOL RESOUR ANNOUNC., vol. 10, no. 12, 25 March 2021 (2021-03-25), pages 01452 - 20 |
| PULLINGER ET AL.: "Identification of Salmonella enterica serovar Dublin-specific sequences by subtractive hybridization and analysis of their role in intestinal colonization and systemic translocation in cattle", INFECT IMMUN., vol. 76, no. 11, November 2008 (2008-11-01), pages 5310 - 21 |
| RONDINI ET AL., J. INFECT. DEV. CTRIES, 2012 |
| RONDINI ET AL.: "Evaluation of the immunogenicity and biological activity of the Citrobacter freundii Vi-CRM197 conjugate as a vaccine for Salmonella enterica serovar Typhi", CLIN VACCINE IMMUNOL., vol. 18, no. 3, March 2011 (2011-03-01), pages 460 - 8 |
| ROSSI OCABONI MNEGREA ANECCHI FALFINI RMICOLI F ET AL.: "Toll-Like Receptor Activation by Generalized Modules for Membrane Antigens from Lipid A Mutants of Salmonella enterica Serovars Typhimurium and Enteritidis", CLIN VACCINE IMMUNOL., vol. 23, no. 4, 2016, pages 304 - 14, XP055506434 |
| ROSSI: "Intra-Laboratory Evaluation of Luminescence Based High-Throughput Serum Bactericidal Assay (L-SBA) to Determine Bactericidal Activity of Human Sera against Shigella", HIGH THROUGHPUT., vol. 9, no. 2, 8 June 2020 (2020-06-08), pages 14 |
| VAN PUYVELDE ET AL.: "An African Salmonella Typhimurium ST313 sublineage with extensive drug-resistance and signatures of host adaptation", NAT COMMUN., vol. 10, no. 1, 19 September 2019 (2019-09-19), pages 4280 |
| WATSON ET AL., INFECT IMMUN., vol. 60, no. 11, 1992, pages 4679 - 86 |
| WESTPHALJANN, METHODS CARBOHYDR. CHEM., vol. 5, 1965, pages 83 - 91 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2025021704A1 (en) | 2025-01-30 |
| WO2025021710A1 (en) | 2025-01-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7268125B2 (en) | Immunogenic compositions comprising conjugated capsular saccharide antigens, kits comprising same and uses thereof | |
| TWI815860B (en) | Multivalent pneumococcal polysaccharide-protein conjugate composition | |
| CN108430500B (en) | Methods and compositions for immunoprotection against enteropathogenic E.coli | |
| EP2056871B1 (en) | Protein matrix vaccines and methods of making and administering such vaccines | |
| KR101897317B1 (en) | Immunogenic Composition | |
| JP2022068303A (en) | Polyvalent pneumococcal polysaccharide-protein complex composition | |
| TW202112392A (en) | Immunogenic compositions comprising conjugated capsular saccharide antigens and uses thereof | |
| CN112741901B (en) | Vaccine containing streptococcus pneumoniae capsular polysaccharide type 5 and preparation method thereof | |
| WO2014118201A1 (en) | Multivalent glycoconjugate vaccines | |
| CZ20013378A3 (en) | Vaccine | |
| JP2013504588A (en) | Protein matrix vaccine with high immunogenicity | |
| KR20140045416A (en) | Protein matrix vaccine compositions including polycations | |
| US8795680B2 (en) | Methods for conjugation of oligosaccharides or polysaccharides to protein carriers through oxime linkages via 3-deoxy-D-manno-octulsonic acid | |
| US11097000B2 (en) | Klebsiella pneumoniae capsule polysaccharide vaccines | |
| US12521443B2 (en) | Conjugate production | |
| JPH07503238A (en) | Detoxified LPS-cholera toxin conjugate vaccine for cholera prevention | |
| Wu et al. | Investigation of nontypeable Haemophilus influenzae outer membrane protein P6 as a new carrier for lipooligosaccharide conjugate vaccines | |
| WO2025021712A1 (en) | Immunogenic composition | |
| CN110652585A (en) | Polysaccharide-protein conjugate immune preparation and application thereof | |
| HK1249403A1 (en) | Protein matrix vaccines and methods of making and administering such vac-cines | |
| 안소정 | Development of efficient Vi polysaccharide conjugate vaccine and its vaccination strategy for prevention of typhoid fever | |
| EA044044B1 (en) | IMMUNOGENIC COMPOSITIONS CONTAINING POLYPEPTIDE-ANTIGEN CONJUGATES WITH NON-NATURAL AMINO ACIDS | |
| WO2009033269A1 (en) | Novel polysaccharide immunogens from alloiococcus otitidis and synthesis of a glycoconjugate vaccine thereof | |
| HK1136220A (en) | Protein matrix vaccines and methods of making and administering such vaccines |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24748312 Country of ref document: EP Kind code of ref document: A1 |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112026001220 Country of ref document: BR |