(I-XVI)
(I-XVIV) wherein R
2, R
4, R
5, X, Y
1, Y
2, A
1, A
2, A
3, A
4, R
8, R
8A, R
8B, R
8C and R
8D, and any groups associated therewith, are as defined anywhere herein, in particular any of the numbered paragraphs appearing hereinbefore. [00106] In an embodiment of the compounds of formula (I-I), (I-II), (I-III), (I-IV), (I-V), (I-VI), (I-VII), (I-VIII), (I-IX), (I-X), (I-XI), (I-XII), (I-XIII), (I-XIV), (I-XV), (I-XVI), (I-XVII), (I-XVIII) or (I- XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, where present: R
2 is as defined in numbered paragraph (1); R
4 is as defined in numbered paragraph (11); R
5 is as defined in numbered paragraph (16);
or R
4 and R
5 are as defined in numbered paragraph (21); X is as defined in numbered paragraph (33); Y
1 is as defined in numbered paragraph (37); Y
2 is as defined in numbered paragraph (40); A
1, A
2, A
3 and A
4 are as defined in numbered paragraph (44); and all other groups are as defined in any one of the numbered paragraphs appearing hereinbefore. [00107] In an embodiment of the compounds of formula (I-I), (I-II), (I-III), (I-IV), (I-V), (I-VI), (I-VII), (I-VIII), (I-IX), (I-X), (I-XI), (I-XII), (I-XIII), (I-XIV), (I-XV), (I-XVI), (I-XVII), (I-XVIII) or (I- XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, where present: R
2 is as defined in numbered paragraph (4); R
4 is as defined in numbered paragraph (11); R
5 is as defined in numbered paragraph (16); or R
4 and R
5 are as defined in numbered paragraph (21); X is as defined in numbered paragraph (33); Y
1 is as defined in numbered paragraph (37); Y
2 is as defined in numbered paragraph (40); A1, A2, A3 and A4 are as defined in numbered paragraph (44); and all other groups are as defined in any one of the numbered paragraphs appearing hereinbefore. [00108] In an embodiment of the compounds of formula (I-I), (I-II), (I-III), (I-IV), (I-V), (I-VI), (I-VII), (I-VIII), (I-IX), (I-X), (I-XI), (I-XII), (I-XIII), (I-XIV), (I-XV), (I-XVI), (I-XVII), (I-XVIII) or (I- XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, where present: R
2 is as defined in numbered paragraph (1); R
4 is as defined in numbered paragraph (14); R
5 is as defined in numbered paragraph (16); X is as defined in numbered paragraph (33); Y
1 is as defined in numbered paragraph (37); Y
2 is as defined in numbered paragraph (40); A1, A2, A3 and A4 are as defined in numbered paragraph (44); and all other groups are as defined in any one of the numbered paragraphs appearing hereinbefore.
[00109] In an embodiment of the compounds of formula (I-I), (I-II), (I-III), (I-IV), (I-V), (I-VI), (I-VII), (I-VIII), (I-IX), (I-X), (I-XI), (I-XII), (I-XIII), (I-XIV), (I-XV), (I-XVI), (I-XVII), (I-XVIII) or (I- XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, where present: R
2 is as defined in numbered paragraph (1); R
4 is as defined in numbered paragraph (11); R
5 is as defined in numbered paragraph (19); X is as defined in numbered paragraph (33); Y
1 is as defined in numbered paragraph (37); Y
2 is as defined in numbered paragraph (40); A
1, A
2, A
3 and A
4 are as defined in numbered paragraph (44); and all other groups are as defined in any one of the numbered paragraphs appearing hereinbefore. [00110] In an embodiment of the compounds of formula (I-I), (I-II), (I-III), (I-IV), (I-V), (I-VI), (I-VII), (I-VIII), (I-IX), (I-X), (I-XI), (I-XII), (I-XIII), (I-XIV), (I-XV), (I-XVI), (I-XVII), (I-XVIII) or (I- XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, where present: R
2 is as defined in numbered paragraph (1); R
4 and R
5 are as defined in numbered paragraph (26) or (27); R
A is as defined in numbered paragraph (28); X is as defined in numbered paragraph (33); Y
1 is as defined in numbered paragraph (37); Y
2 is as defined in numbered paragraph (40); A1, A2, A3 and A4 are as defined in numbered paragraph (44); and all other groups are as defined in any one of the numbered paragraphs appearing hereinbefore. [00111] In an embodiment of the compounds of formula (I-I), (I-II), (I-III), (I-IV), (I-V), (I-VI), (I-VII), (I-VIII), (I-IX), (I-X), (I-XI), (I-XII), (I-XIII), (I-XIV), (I-XV), (I-XVI), (I-XVII), (I-XVIII) or (I- XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, where present: R
2 is as defined in numbered paragraph (1); R
4 and R
5 are as defined in numbered paragraph (26) or (27); R
A is as defined in numbered paragraph (32); X is as defined in numbered paragraph (33);
Y
1 is as defined in numbered paragraph (37); Y
2 is as defined in numbered paragraph (40); A
1, A
2, A
3 and A
4 are as defined in numbered paragraph (44); and all other groups are as defined in any one of the numbered paragraphs appearing hereinbefore. [00112] In an embodiment of the compounds of formula (I-I), (I-II), (I-III), (I-IV), (I-V), (I-VI), (I-VII), (I-VIII), (I-IX), (I-X), (I-XI), (I-XII), (I-XIII), (I-XIV), (I-XV), (I-XVI), (I-XVII), (I-XVIII) or (I- XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, where present: R
2 is as defined in numbered paragraph (1); R
4 is as defined in numbered paragraph (11); R
5 is as defined in numbered paragraph (16); or R
4 and R
5 are as defined in numbered paragraph (21); X is as defined in numbered paragraph (36); Y
1 is as defined in numbered paragraph (37); Y
2 is as defined in numbered paragraph (40); A
1, A
2, A
3 and A
4 are as defined in numbered paragraph (44); and all other groups are as defined in any one of the numbered paragraphs appearing hereinbefore. [00113] In an embodiment of the compounds of formula (I-I), (I-II), (I-III), (I-IV), (I-V), (I-VI), (I-VII), (I-VIII), (I-IX), (I-X), (I-XI), (I-XII), (I-XIII), (I-XIV), (I-XV), (I-XVI), (I-XVII), (I-XVIII) or (I- XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, where present: R
2 is as defined in numbered paragraph (1); R
4 is as defined in numbered paragraph (11); R
5 is as defined in numbered paragraph (16); or R
4 and R
5 are as defined in numbered paragraph (21); X is as defined in numbered paragraph (33); Y
1 and Y
2 are as defined in numbered paragraph (43); A1, A2, A3 and A4 are as defined in numbered paragraph (44); and all other groups are as defined in any one of the numbered paragraphs appearing hereinbefore.
[00114] In an embodiment of the compounds of formula (I-I), (I-II), (I-III), (I-IV), (I-V), (I-VI), (I-VII), (I-VIII), (I-IX), (I-X), (I-XI), (I-XII), (I-XIII), (I-XIV), (I-XV), (I-XVI), (I-XVII), (I-XVIII) or (I- XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, where present: R
2 is as defined in numbered paragraph (1); R
4 is as defined in numbered paragraph (11); R
5 is as defined in numbered paragraph (16); or R
4 and R
5 are as defined in numbered paragraph (21); X is as defined in numbered paragraph (33); Y
1 is as defined in numbered paragraph (37); Y
2 is as defined in numbered paragraph (40); A
1, A
2, A
3 and A
4 are as defined in any one of numbered paragraphs (49)-(52), (55)-(58), (104), (109) or (111); and all other groups are as defined in any one of the numbered paragraphs appearing hereinbefore. [00115] In an embodiment of the compounds of formula (I-I), (I-II), (I-III), (I-IV), (I-V), (I-VI), (I-VII), (I-VIII), (I-IX), (I-X), (I-XI), (I-XII), (I-XIII), (I-XIV), (I-XV), (I-XVI), (I-XVII), (I-XVIII) or (I- XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, where present: R
2 is as defined in numbered paragraph (1); R
4 is as defined in numbered paragraph (11); R
5 is as defined in numbered paragraph (16); or R
4 and R
5 are as defined in numbered paragraph (21); X is as defined in numbered paragraph (33); Y
1 is as defined in numbered paragraph (37); Y
2 is as defined in numbered paragraph (40); A1, A2, A3 and A4 are as defined in numbered paragraph (44); R
7 is as defined in numbered paragraph (59); R
8A is as defined in numbered paragraph (61); R
8 is as defined in numbered paragraph (66); R
8B and R
8D are as defined in numbered paragraph (67); R
8C is as defined in numbered paragraph (72); and
all other groups are as defined in any one of the numbered paragraphs appearing hereinbefore. [00116] In an embodiment of the compounds of formula (I-I), (I-II), (I-III), (I-IV), (I-V), (I-VI), (I-VII), (I-VIII), (I-IX), (I-X), (I-XI), (I-XII), (I-XIII), (I-XIV), (I-XV), (I-XVI), (I-XVII), (I-XVIII) or (I- XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, where present: R
2 is as defined in numbered paragraph (1); R
4 is as defined in numbered paragraph (11); R
5 is as defined in numbered paragraph (16); or R
4 and R
5 are as defined in numbered paragraph (21); X is as defined in numbered paragraph (33); Y
1 is as defined in numbered paragraph (37); Y
2 is as defined in numbered paragraph (40); A
1, A
2, A
3 and A
4 are as defined in numbered paragraph (44); R
7 is as defined in numbered paragraph (59); R
8A is as defined in numbered paragraph (64); R
8 is as defined in numbered paragraph (66); R
8B and R
8D are as defined in numbered paragraph (67); R
8C is as defined in numbered paragraph (72); and all other groups are as defined in any one of the numbered paragraphs appearing hereinbefore. [00117] In an embodiment of the compounds of formula (I-I), (I-II), (I-III), (I-IV), (I-V), (I-VI), (I-VII), (I-VIII), (I-IX), (I-X), (I-XI), (I-XII), (I-XIII), (I-XIV), (I-XV), (I-XVI), (I-XVII), (I-XVIII) or (I- XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, where present: R
2 is as defined in numbered paragraph (1); R
4 is as defined in numbered paragraph (11); R
5 is as defined in numbered paragraph (16); or R
4 and R
5 are as defined in numbered paragraph (21); X is as defined in numbered paragraph (33); Y
1 is as defined in numbered paragraph (37); Y
2 is as defined in numbered paragraph (40); A1, A2, A3 and A4 are as defined in numbered paragraph (44);
R
7 is as defined in numbered paragraph (59); R
8A is as defined in numbered paragraph (65); R
8 is as defined in numbered paragraph (66); R
8B and R
8D are as defined in numbered paragraph (67); R
8C is as defined in numbered paragraph (72); and all other groups are as defined in any one of the numbered paragraphs appearing hereinbefore. [00118] In an embodiment of the compounds of formula (I-I), (I-II), (I-III), (I-IV), (I-V), (I-VI), (I-VII), (I-VIII), (I-IX), (I-X), (I-XI), (I-XII), (I-XIII), (I-XIV), (I-XV), (I-XVI), (I-XVII), (I-XVIII) or (I- XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, where present: R
2 is as defined in numbered paragraph (1); R
4 is as defined in numbered paragraph (11); R
5 is as defined in numbered paragraph (16); or R
4 and R
5 are as defined in numbered paragraph (21); X is as defined in numbered paragraph (33); Y
1 is as defined in numbered paragraph (37); Y
2 is as defined in numbered paragraph (40); A1, A2, A3 and A4 are as defined in numbered paragraph (44); R
7 is as defined in numbered paragraph (59); R
8A is as defined in numbered paragraph (61); R
8 is as defined in numbered paragraph (71); R
8C is as defined in numbered paragraph (72); and all other groups are as defined in any one of the numbered paragraphs appearing hereinbefore. [00119] In an embodiment of the compounds of formula (I-I), (I-II), (I-III), (I-IV), (I-V), (I-VI), (I-VII), (I-VIII), (I-IX), (I-X), (I-XI), (I-XII), (I-XIII), (I-XIV), (I-XV), (I-XVI), (I-XVII), (I-XVIII) or (I- XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, where present: R
2 is as defined in numbered paragraph (1); R
4 is as defined in numbered paragraph (11); R
5 is as defined in numbered paragraph (16); or R
4 and R
5 are as defined in numbered paragraph (21);
X is as defined in numbered paragraph (33); Y
1 is as defined in numbered paragraph (37); Y
2 is as defined in numbered paragraph (40); A
1, A
2, A
3 and A
4 are as defined in numbered paragraph (44); R
7 is as defined in numbered paragraph (59); R
8A is as defined in numbered paragraph (61); R
8 is as defined in numbered paragraph (66); R
8B and R
8D are as defined in numbered paragraph (67); R
8C is as defined in numbered paragraph (78); and all other groups are as defined in any one of the numbered paragraphs appearing hereinbefore. [00120] In an embodiment of the compounds of formula (I-I), (I-II), (I-III), (I-IV), (I-V), (I-VI), (I-VII), (I-VIII), (I-IX), (I-X), (I-XI), (I-XII), (I-XIII), (I-XIV), (I-XV), (I-XVI), (I-XVII), (I-XVIII) or (I- XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, where present: R
2 is as defined in numbered paragraph (1); R
4 is as defined in numbered paragraph (11); R
5 is as defined in numbered paragraph (16); or R
4 and R
5 are as defined in numbered paragraph (21); X is as defined in numbered paragraph (33); Y
1 is as defined in numbered paragraph (37); Y
2 is as defined in numbered paragraph (40); A1, A2, A3 and A4 are as defined in numbered paragraph (44); R
7 is as defined in numbered paragraph (59); R
8A is as defined in numbered paragraph (61); R
8 is as defined in numbered paragraph (66); R
8B and R
8D are as defined in numbered paragraph (67); R
8C is as defined in numbered paragraph (80); and all other groups are as defined in any one of the numbered paragraphs appearing hereinbefore.
[00121] In an embodiment of the compounds of formula (I-I), (I-II), (I-III), (I-IV), (I-V), (I-VI), (I-VII), (I-VIII), (I-IX), (I-X), (I-XI), (I-XII), (I-XIII), (I-XIV), (I-XV), (I-XVI), (I-XVII), (I-XVIII) or (I- XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, where present: R
2 is as defined in numbered paragraph (1); R
4 is as defined in numbered paragraph (11); R
5 is as defined in numbered paragraph (16); or R
4 and R
5 are as defined in numbered paragraph (21); X is as defined in numbered paragraph (33); Y
1 is as defined in numbered paragraph (37); Y
2 is as defined in numbered paragraph (40); A
1, A
2, A
3 and A
4 are as defined in numbered paragraph (44); R
7 is as defined in numbered paragraph (59); R
8A and R
8D are as defined in numbered paragraph (84); R
8 is as defined in numbered paragraph (66); R
8B is as defined in numbered paragraph (67); R
8C is as defined in numbered paragraph (72); and all other groups are as defined in any one of the numbered paragraphs appearing hereinbefore. [00122] In an embodiment of the compounds of formula (I-I), (I-II), (I-III), (I-IV), (I-V), (I-VI), (I-VII), (I-VIII), (I-IX), (I-X), (I-XI), (I-XII), (I-XIII), (I-XIV), (I-XV), (I-XVI), (I-XVII), (I-XVIII) or (I- XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, where present: R
2 is as defined in numbered paragraph (1); R
4 is as defined in numbered paragraph (11); R
5 is as defined in numbered paragraph (16); X is as defined in numbered paragraph (33); Y
1 is as defined in numbered paragraph (37); Y
2 is as defined in numbered paragraph (40); A1, A2, A3 and A4 are as defined in any one of numbered paragraphs (49), (50), (51) or (52); R
7 is as defined in numbered paragraph (59); R
8A is as defined in numbered paragraph (61);
R
8 is as defined in numbered paragraph (66); R
8B and R
8D are as defined in numbered paragraph (67); R
8C is as defined in numbered paragraph (72); R
12 is as defined in numbered paragraph (87); R
13 is as defined in numbered paragraph (92); R
14 is as defined in numbered paragraph (96); R
D is as defined in numbered paragraph (100); and all other groups are as defined in any one of the numbered paragraphs appearing hereinbefore. [00123] In an embodiment of the compounds of formula (I-I), (I-II), (I-III), (I-IV), (I-V), (I-VI), (I-VII), (I-VIII), (I-IX), (I-X), (I-XI), (I-XII), (I-XIII), (I-XIV), (I-XV), (I-XVI), (I-XVII), (I-XVIII) or (I- XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, where present: R
2 is as defined in numbered paragraph (1); R
4 is as defined in numbered paragraph (11); R
5 is as defined in numbered paragraph (16); X is as defined in numbered paragraph (33); Y
1 is as defined in numbered paragraph (37); Y
2 is as defined in numbered paragraph (40); A1, A2, A3 and A4 are as defined in any one of numbered paragraphs (55), (56), (57) or (58); R
7 is as defined in numbered paragraph (59); R
8A is as defined in numbered paragraph (61); R
8 is as defined in numbered paragraph (66); R
8B and R
8D are as defined in numbered paragraph (67); R
8C is as defined in numbered paragraph (72); R
12 is as defined in numbered paragraph (87); R
13 is as defined in numbered paragraph (92); R
14 is as defined in numbered paragraph (96); R
D is as defined in numbered paragraph (100); and all other groups are as defined in any one of the numbered paragraphs appearing hereinbefore.
[00124] In an embodiment of the compounds of formula (I-I), (I-II), (I-III), (I-IV), (I-V), (I-VI), (I-VII), (I-VIII), (I-IX), (I-X), (I-XI), (I-XII), (I-XIII), (I-XIV), (I-XV), (I-XVI), (I-XVII), (I-XVIII) or (I- XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, where present: R
2 is as defined in numbered paragraph (1); R
4 is as defined in numbered paragraph (11); R
5 is as defined in numbered paragraph (16); X is as defined in numbered paragraph (33); Y
1 is as defined in numbered paragraph (37); Y
2 is as defined in numbered paragraph (40); A
1, A
2, A
3 and A
4 are as defined in any one of numbered paragraphs (104), (109) or (111); R
7 is as defined in numbered paragraph (59); R
8A is as defined in numbered paragraph (61); R
8 is as defined in numbered paragraph (66); R
8B and R
8D are as defined in numbered paragraph (67); R
8C is as defined in numbered paragraph (72); R
12 is as defined in numbered paragraph (87); R
13 is as defined in numbered paragraph (92); R
14 is as defined in numbered paragraph (96); R
D is as defined in numbered paragraph (100); and all other groups are as defined in any one of the numbered paragraphs appearing hereinbefore. [00125] In an embodiment of the compounds of formula (I-I), (I-II), (I-III), (I-IV), (I-V), (I-VI), (I-VII), (I-VIII), (I-IX), (I-X), (I-XI), (I-XII), (I-XIII), (I-XIV), (I-XV), (I-XVI), (I-XVII), (I-XVIII) or (I- XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, where present: R
2 is as defined in numbered paragraph (3); R
4 is as defined in numbered paragraph (12); R
5 is as defined in numbered paragraph (18); or R
4 and R
5 are as defined in numbered paragraph (24); X is as defined in numbered paragraph (35); Y
1 and Y
2 are as defined in numbered paragraph (43);
A
1, A
2, A
3 and A
4 are as defined in numbered paragraph (45); R
7 is as defined in numbered paragraph (59); R
8A is as defined in numbered paragraph (63); R
8 is as defined in numbered paragraph (66); R
8B and R
8D are as defined in numbered paragraph (69); R
8C is as defined in numbered paragraph (78); R
12 is as defined in numbered paragraph (87); R
13 is as defined in numbered paragraph (92); R
14 is as defined in numbered paragraph (96); R
D is as defined in numbered paragraph (100); and all other groups are as defined in any one of the numbered paragraphs appearing hereinbefore. [00126] In an embodiment of the compounds of formula (I-I), (I-II), (I-III), (I-IV), (I-V), (I-VI), (I-VII), (I-VIII), (I-IX), (I-X), (I-XI), (I-XII), (I-XIII), (I-XIV), (I-XV), (I-XVI), (I-XVII), (I-XVIII) or (I- XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, where present: R
2 is as defined in numbered paragraph (4); R
4 is as defined in numbered paragraph (14); R
5 is as defined in numbered paragraph (19); or R
4 and R
5 are as defined in numbered paragraph (27); R
A is as defined in numbered paragraph (32); X is as defined in numbered paragraph (36); Y
1 and Y
2 are as defined in numbered paragraph (43); A1, A2, A3 and A4 are as defined in numbered paragraph (45); R
7 is as defined in numbered paragraph (59); R
8A is as defined in numbered paragraph (65); R
8 is as defined in numbered paragraph (71); R
8C is as defined in numbered paragraph (80); R
12 is as defined in numbered paragraph (88); R
13 is as defined in numbered paragraph (93);
R
14 is as defined in numbered paragraph (97); R
D is as defined in numbered paragraph (101); and all other groups are as defined in any one of the numbered paragraphs appearing hereinbefore. [00127] In an embodiment of the compounds of formula (I-I), (I-II), (I-III), (I-IV), (I-V), (I-VI), (I-VII), (I-VIII), (I-IX), (I-X), (I-XI), (I-XII), (I-XIII), (I-XIV), (I-XV), (I-XVI), (I-XVII), (I-XVIII) or (I- XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof: R
2 is as defined in numbered paragraph (5); R
4 is as defined in numbered paragraph (15); R
5 is as defined in numbered paragraph (20); X is CH; Y
1 is -CH
2-; Y
2 is -CH
2– or -CH
2-CH
2-; A1, A2, A3 and A4 are as defined in numbered paragraphs (52) or (54); R
7 is as defined in numbered paragraph (59); R
8A is as defined in numbered paragraph (65); R
8 is as defined in numbered paragraph (71); and R
8C is as defined in numbered paragraph (80). [00128] In an embodiment of the compounds of formula (I-I), (I-II), (I-III), (I-IV), (I-V), (I-VI), (I-VII), (I-VIII), (I-IX), (I-X), (I-XI), (I-XII), (I-XIII), (I-XIV), (I-XV), (I-XVI), (I-XVII), (I-XVIII) or (I- XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof: R
2 is as defined in numbered paragraph (5); R
4 is as defined in numbered paragraph (15); R
5 is hydrogen or methyl; X is CH; Y
1 is -CH2-; Y
2 is -CH2-; A1, A2, A3 and A4 are as defined in numbered paragraph (52); R
7 is as defined in numbered paragraph (59); R
8A is as defined in numbered paragraph (65);
R
8 is as defined in numbered paragraph (71); and R
8C is -CH
2-N(CH
3)
2. [00129] In an embodiment of the compounds of formula (I-I), (I-II), (I-III), (I-IV), (I-V), (I-VI), (I-VII), (I-VIII), (I-IX), (I-X), (I-XI), (I-XII), (I-XIII), (I-XIV), (I-XV), (I-XVI), (I-XVII), (I-XVIII) or (I- XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof: R
2 is OH; R
4 is OH; R
5 is hydrogen; X is CH; Y
1 is -CH
2-; Y
2 is -CH
2-; A
1, A
2, A
3 and A
4 are as defined in numbered paragraph (52); R
7 is as defined in numbered paragraph (59); R
8A is methyl; R
8 is as defined in numbered paragraph (71); and R
8C is -CH2-N(CH3)2. [00130] In an embodiment of the compounds of formula I or any appropriate sub-definition of formula I (as defined by formulae (I-I) to (I-XVIV)) or by any of the embodiments described in relation to formulae (I-I) to (I-XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, R
2 is as defined in numbered paragraph (3) above. Suitably, R
2 is as defined in numbered paragraph (4) above. More suitably, R
2 is as defined in numbered paragraph (5) above. [00131] In an embodiment of the compounds of formula I or any appropriate sub-definition of formula I (as defined by formulae (I-I) to (I-XVIV)) or by any of the embodiments described in relation to formulae (I-I) to (I-XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, R
3 is as defined in numbered paragraph (9) above. Suitably, R
3 is as defined in numbered paragraph (10) above. [00132] In an embodiment of the compounds of formula I or any appropriate sub-definition of formula I (as defined by formulae (I-I) to (I-XVIV)) or by any of the embodiments described in relation to formulae (I-I) to (I-XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, R
4 is as defined in numbered paragraph (13) above. Suitably, R
4 is as defined in numbered paragraph (14) above. More suitably, R
4 is as defined in numbered paragraph (15) above.
[00133] In an embodiment of the compounds of formula I or any appropriate sub-definition of formula I (as defined by formulae (I-I) to (I-XVIV)) or by any of the embodiments described in relation to formulae (I-I) to (I-XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, R
5 is as defined in numbered paragraph (19) above. Suitably, R
5 is as defined in numbered paragraph (20) above. [00134] In an embodiment of the compounds of formula I or any appropriate sub-definition of formula I (as defined by formulae (I-I) to (I-XVIV)) or by any of the embodiments described in relation to formulae (I-I) to (I-XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, R
4 and R
5 are as defined in numbered paragraph (25) above. Suitably, R
4 and R
5 are as defined in numbered paragraph (26) or (27) above. In both of these embodiments, R
A is as defined in numbered paragraph (30) above. [00135] In an embodiment of the compounds of formula I or any appropriate sub-definition of formula I (as defined by formulae (I-I) to (I-XVIV)) or by any of the embodiments described in relation to formulae (I-I) to (I-XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, X is as defined in numbered paragraph (35) above. Suitably, X is as defined in numbered paragraph (36) above. [00136] In an embodiment of the compounds of formula I or any appropriate sub-definition of formula I (as defined by formulae (I-I) to (I-XVIV)) or by any of the embodiments described in relation to formulae (I-I) to (I-XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, Y
1 and Y
2 are as defined in numbered paragraph (43) above. [00137] In an embodiment of the compounds of formula I or any appropriate sub-definition of formula I (as defined by formulae (I-I) to (I-XVIV)) or by any of the embodiments described in relation to formulae (I-I) to (I-XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, A1, A2, A3 and A4 are as defined in numbered paragraph (48) or (54) above. Suitably, A1, A2, A3 and A4 are as defined in numbered paragraph (52) or (58) above. Suitably, A1, A2, A3 and A4 are as defined in numbered paragraph (104), (109) or (111) above. [00138] In an embodiment of the compounds of formula I or any appropriate sub-definition of formula I (as defined by formulae (I-I) to (I-XVIV)) or by any of the embodiments described in relation to formulae (I-I) to (I-XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, R
7 is as defined in numbered paragraph (59) above. [00139] In an embodiment of the compounds of formula I or any appropriate sub-definition of formula I (as defined by formulae (I-I) to (I-XVIV)) or by any of the embodiments described in relation to formulae (I-I) to (I-XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, R
8A is as defined in numbered paragraph (64) above. Suitably, R
8A is as defined in numbered paragraph (65) above.
[00140] In an embodiment of the compounds of formula I or any appropriate sub-definition of formula I (as defined by formulae (I-I) to (I-XVIV)) or by any of the embodiments described in relation to formulae (I-I) to (I-XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, R
8 is as defined in numbered paragraph (66) above. Suitably, R
8 is as defined in numbered paragraph (71) above. [00141] In an embodiment of the compounds of formula I or any appropriate sub-definition of formula I (as defined by formulae (I-I) to (I-XVIV)) or by any of the embodiments described in relation to formulae (I-I) to (I-XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, R
8B and R
8D are as defined in numbered paragraph (69) above. Suitably, R
8B and R
8D are as defined in numbered paragraph (70) above. [00142] In an embodiment of the compounds of formula I or any appropriate sub-definition of formula I (as defined by formulae (I-I) to (I-XVIV)) or by any of the embodiments described in relation to formulae (I-I) to (I-XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, R
8C is as defined in numbered paragraph (78) above. Suitably, R
8C is as defined in numbered paragraph (79) above. More suitably, R
8C is as defined in numbered paragraph (80) above. [00143] In an embodiment of the compounds of formula I or any appropriate sub-definition of formula I (as defined by formulae (I-I) to (I-XVIV)) or by any of the embodiments described in relation to formulae (I-I) to (I-XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, R
8A and R
8D are as defined in numbered paragraph (83) above. Suitably, R
8A and R
8D are as defined in numbered paragraph (84) above. [00144] In an embodiment of the compounds of formula I or any appropriate sub-definition of formula I (as defined by formulae (I-I) to (I-XVIV)) or by any of the embodiments described in relation to formulae (I-I) to (I-XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, R
12 is as defined in numbered paragraph (87) above. Suitably, R
12 is as defined in numbered paragraph (88) above. More suitably, R
12 is as defined in numbered paragraph (89) above. [00145] In an embodiment of the compounds of formula I or any appropriate sub-definition of formula I (as defined by formulae (I-I) to (I-XVIV)) or by any of the embodiments described in relation to formulae (I-I) to (I-XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate thereof, R
13 is as defined in numbered paragraph (92) above. Suitably, R
13 is as defined in numbered paragraph (93) above. [00146] In an embodiment of the compounds of formula I or any appropriate sub-definition of formula I (as defined by formulae (I-I) to (I-XVIV)) or by any of the embodiments described in relation to formulae (I-I) to (I-XVIV), or a pharmaceutically acceptable salt, hydrate and/or solvate
thereof, R
14 is as defined in numbered paragraph (96) above. Suitably, R
14 is as defined in numbered paragraph (97) above. More suitably, R
14 is as defined in numbered paragraph (98) above. In each of these embodiments, R
D is as defined in numbered paragraph (100) above. [00147] Particular compounds of the present invention include any of the compounds exemplified in the present application, or a pharmaceutically acceptable salt or solvate thereof, and, in particular, any of the following: (E)-N-(2-(3-Chloro-4,6-dihydroxy-2-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N- methylbut-2-enamide; (E)-N-Benzyl-N-(2-(2,4-dihydroxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)but-2-enamide; (E)-N-Benzyl-N-(2-(2,4-dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)but-2- enamide; (E)-N-(2-(2,4-Dihydroxybenzoyl) isoindolin-4-yl)-4-(diethylamino)-N-((1-methyl-1H-pyrazol-4- yl)methyl)but-2-enamide; (E)-4-(3,3-Difluoropyrrolidin-1-yl)-N-(2-(2,4-dihydroxy-5-methylbenzoyl)isoindolin-4-yl)but-2- enamide; ((E)-4-(3,3-Difluoroazetidin-1-yl)-N-(2-(2,4-dihydroxy-5-methylbenzoyl)isoindolin-4-yl)but-2- enamide; N-(2-(2, 4-Dihydroxy-5-methylbenzoyl) isoindolin-4-yl) acrylamide; (E)-N-(2-(2,4-Dihydroxy-6-methylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-4- (dimethylamino)but-2-enamide; (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-4- (dimethylamino)but-2-enamide; (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-4- (dimethylamino)-N-methylbut-2-enamide; (E)-N-(2-(2,4-Dihydroxy-6-methylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-4- (dimethylamino)-N-methylbut-2-enamide; (E)-N-(2-(2,4-Dihydroxy-5-isopropylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-4- (dimethylamino)-N-methylbut-2-enamide; (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4-(pyrrolidin-1-yl)but-2-enamide; (E)-N-(2-(2, 4-Dihydroxy-5-methylbenzoyl) isoindolin-4-yl)-4-morpholinobut-2-enamide; N-(2-(3-Chloro-4,6-dihydroxy-2-methylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- methylacrylamide;
N-(2-(5-Chloro-2,4-dihydroxybenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N-methylacrylamide; (E)-N-(2-(5-(tert-Butyl)-2-hydroxy-4-methoxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N- methylbut-2-enamide; (E)-N-(2-(5-(tert-Butyl)-4-fluoro-2-hydroxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N- methylbut-2-enamide; (E)-N-(2-(4,6-Dihydroxy-2,3-dimethylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-4- (dimethylamino)-N-methylbut-2-enamide; N-(2-(2,4-Dihydroxy-5-methylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N-methylacrylamide; N-Benzyl-N-(2-(2,4-dihydroxybenzoyl)isoindolin-4-yl)acrylamide; N-Benzyl-N-(2-(2,4-dihydroxy-5-methylbenzoyl)isoindolin-4-yl)acrylamide; (E)-N-(2-(2,4-Dihydroxybenzoyl) isoindolin-4-yl)-4-(dimethyl amino)-N-phenethylbut-2-enamide; (E)-N-(2-(2-Chloro-4,6-dihydroxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-methylbut-2- enamide; (E)-4-(Dimethylamino)-N-(2-(6-hydroxy-3,3-dimethylindoline-5-carbonyl)isoindolin-4-yl)-N- methylbut-2-enamide; (E)-N-(2-(4,6-Dihydroxy-2,3-dimethylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-methylbut-2- enamide; (E)-N-(2-(5-(tert-Butyl)-2,4-dihydroxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-methylbut-2- enamide; (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-ethylbut-2- enamide; (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-(2- hydroxyethyl)but-2-enamide; (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-(pyridin-4- ylmethyl)but-2-enamide; (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-(2-(pyridin-4- yl)ethyl)but-2-enamide; (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-(2-(pyridin-2- yl)ethyl)but-2-enamide; (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-(2-(pyridin-3- yl)ethyl)but-2-enamide;
(E)-N-(2-(4,6-Dihydroxy-2,3-dimethylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-(2-(pyridin-2- yl)ethyl)but-2-enamide; E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-(2-(tetrahydro-2H- pyran-4-yl)ethyl)but-2-enamide; (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-(2- methoxyethyl)but-2-enamide; (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-methylbut-2- enamide; (E)-N-(2-(2,4-Dihydroxy-6-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-methylbut-2- enamide; (E)-N-(2-(5-Chloro-2,4-dihydroxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)but-2-enamide; (E)-N-(2-(2,4-Dihydroxy-5-methoxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)but-2-enamide; (E)-N-(2-(4,6-Dihydroxy-2,3-dimethylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)but-2-enamide; (E)-4-(Dimethylamino)-N-(2-(6-hydroxy-1H-indole-5-carbonyl)isoindolin-4-yl)but-2-enamide; (E)-N-(2-(5-Bromo-2,4-dihydroxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)but-2-enamide; (E)-N-(2-(2,4-Dihydroxy-5-(trifluoromethyl)benzoyl)isoindolin-4-yl)-4-(dimethylamino)-N- methylbut-2-enamide; (E)-N-(2-(5-Cyano-2,4-dihydroxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)but-2-enamide; (E)-N-(2-(2,4-Dihydroxy-6-(trifluoromethyl)benzoyl)isoindolin-4-yl)-4-(dimethylamino)but-2- enamide; (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)but-2-enamide; (E)-N-(2-Benzoylisoindolin-4-yl)-4-(dimethylamino)but-2-enamide; (E)-4-(Dimethylamino)-N-(2-(4-hydroxy-2-methoxybenzoyl)isoindolin-4-yl)but-2-enamide; (E)-N-(2-(2-(Difluoromethyl)-4-hydroxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)but-2-enamide; (E)-N-(2-(2-Chloro-4-hydroxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)but-2-enamide; (E)-4-(Dimethylamino)-N-(2-(6-hydroxy-1H-indazole-5-carbonyl)isoindolin-4-yl)but-2-enamide; (E)-N-(2-(2,4-Dihydroxy-6-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)but-2-enamide; (E)-N-(2-(4-(Difluoromethyl)-2-hydroxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)but-2-enamide; (E)-4-(Dimethylamino)-N-(2-(5-fluoro-2,4-dihydroxybenzoyl)isoindolin-4-yl)but-2-enamide;
(E)-4-(Dimethylamino)-N-(2-(6-hydroxybenzo[d]isoxazole-5-carbonyl)isoindolin-4-yl)but-2- enamide; (E)-N-(2-(2,4-Dihydroxy-5-isopropylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)but-2-enamide; (E)-N-(2-(4-Amino-2-hydroxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)but-2-enamide; (E)-4-(Dimethylamino)-N-(2-(5-ethyl-2,4-dihydroxybenzoyl)isoindolin-4-yl)but-2-enamide; (E)-N-(2-(2,4-Dihydroxy-5-isopropylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-methylbut-2- enamide; (E)-N-(2-(3-Chloro-4-(difluoromethoxy)-6-hydroxy-2-methylbenzoyl)isoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide; (E)-4-(Dimethylamino)-N-(2-(2-hydroxy-5-isopropylbenzoyl)isoindolin-4-yl)-N-methylbut-2- enamide; (E)-N-(2-(5-(tert-Butyl)-2-hydroxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-methylbut-2- enamide; (E)-N-(2-(4-(Difluoromethoxy)-2-hydroxy-5-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N- methylbut-2-enamide; (E)-4-(Dimethylamino)-N-(2-(4-hydroxy-2,3-dimethylbenzoyl)isoindolin-4-yl)-N-methylbut-2- enamide; (E)-N-(2-(3-(tert-Butyl)-4-hydroxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-methylbut-2- enamide; (E)-4-(Dimethylamino)-N-(2-(4-hydroxy-3-methylbenzoyl)isoindolin-4-yl)-N-methylbut-2- enamide; (E)-4-(Dimethylamino)-N-(2-(4-hydroxy-3-isopropylbenzoyl)isoindolin-4-yl)-N-methylbut-2- enamide; (E)-4-(Dimethylamino)-N-(2-(4-hydroxy-3-isopropylbenzoyl)isoindolin-4-yl)but-2-enamide; (E)-4-(Dimethylamino)-N-(2-(4-hydroxy-3-(trifluoromethyl)benzoyl)isoindolin-4-yl)-N-methylbut- 2-enamide; (E)-N-(2-(3-Chloro-4-hydroxy-2-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-methylbut- 2-enamide; N-[2-(2,4-Dihydroxy-5-methyl-benzoyl)isoindolin-4-yl]-N-[2-(4-pyridyl)ethyl]prop-2-enamide; N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-N-(2-(1-methylpyrrolidin-3- yl)ethyl)acrylamide;
N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-N-methylacrylamide; N-(2-(5-(tert-Butyl)-2,4-dihydroxybenzoyl)isoindolin-4-yl)acrylamide; N-(2-(5-(tert-Butyl)-2,4-dihydroxybenzoyl)isoindolin-4-yl)-N-methylacrylamide; N-(2-(2,4-Dihydroxy-6-methylbenzoyl)isoindolin-4-yl)acrylamide; (E)-N-[2-(2,4-Dihydroxy-5-methyl-benzoyl)isoindolin-4-yl]-4-(dimethylamino)-N-indan-2-yl-but-2- enamide; N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-N-(2-morpholinoethyl)acrylamide; N-(2-(2,4-Dihydroxy-6-methylbenzoyl)isoindolin-4-yl)-N-methylacrylamide; (E)-N-[2-(2,4-Dihydroxybenzoyl)isoindolin-4-yl]-4-(dimethylamino)but-2-enamide; (E)-4-(Dimethylamino)-N-(2-(2-hydroxy-4-methoxybenzoyl)isoindolin-4-yl)but-2-enamide; (E)-4-(Dimethylamino)-N-(2-(2-hydroxy-5-methylbenzoyl)isoindolin-4-yl)but-2-enamide; (E)-4-(Dimethylamino)-N-(2-(4-hydroxy-3-methylbenzoyl)isoindolin-4-yl)but-2-enamide; (E)-4-(Dimethylamino)-N-(2-(2-fluoro-4-hydroxybenzoyl)isoindolin-4-yl)but-2-enamide; (E)-4-(Dimethylamino)-N-(2-(4-hydroxy-2-methylbenzoyl)isoindolin-4-yl)but-2-enamide; (E)-4-(Dimethylamino)-N-(2-(5-hydroxypicolinoyl)isoindolin-4-yl)but-2-enamide; (E)-4-(Dimethylamino)-N-(2-(2-methoxybenzoyl)isoindolin-4-yl)but-2-enamide; (E)-N-(2-(2,4-Dihydroxy-5-(trifluoromethyl)benzoyl)isoindolin-4-yl)-4-(dimethylamino)but-2- enamide; (E)-N-(2-(5-(tert-Butyl)-2,4-dihydroxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)but-2-enamide; (E)-N-(2-(2,4-Dihydroxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)but-2-enamide; (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4-methoxybut-2-enamide; 1-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-3-(2-(dimethylamino)ethylidene)pyrrolidin- 2-one; N-(2-(2,4-Dihydroxy-5-methylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N-(1-methylpiperidin- 4-yl)acrylamide; (E)-N-(2-(4,6-Dihydroxy-2,3-dimethylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-ethylbut-2- enamide; N-(2-(4,6-Dihydroxy-2,3-dimethylbenzoyl)isoindolin-4-yl)-N-(2-methoxyethyl)acrylamide; N-(2-(4,6-Dihydroxy-2,3-dimethylbenzoyl)isoindolin-4-yl)-N-methylacrylamide;
(E)-N-(2-(5-(tert-Butyl)-2,4-dihydroxybenzoyl)-5-chloroisoindolin-4-yl)-4-(dimethylamino)-N- methylbut-2-enamide; (E)-N-(5-Chloro-2-(2, 4-dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4-(dimethyl amino)-N- methylbut-2-enamide; (E)-4-(Dimethylamino)-N-(2-(6-hydroxy-3,3-dimethyl-2-oxoindoline-5-carbonyl)isoindolin-4-yl)- N-methylbut-2-enamide; (E)-N-(2-(3-Chloro-4,6-dihydroxy-2-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N- ethylbut-2-enamide; (E)-N-(2-(3-Chloro-4,6-dihydroxy-2-methylbenzoyl)isoindolin-4-yl)-4-(ethyl(methyl)amino)-N- methylbut-2-enamide; (E)-N-(7-Chloro-2-(2,4-dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N- methylbut-2-enamide; (E)-N-(7-Chloro-2-(4,6-dihydroxy-2,3-dimethylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N- methylbut-2-enamide; N-(2-(3-Chloro-4,6-dihydroxy-2-methylbenzoyl)isoindolin-4-yl)-N-methyl acrylamide; (E)-N-(5-Chloro-2-(4,6-dihydroxy-2,3-dimethylbenzoyl) isoindolin-4-yl)-4-(dimethyl amino)-N- methylbut-2-enamide; (E)-N-(2-(3-Chloro-2-fluoro-4,6-dihydroxybenzoyl) isoindolin-4-yl)-4-(dimethylamino)-N- methylbut-2-enamide; (E)-N-(2-(3-Chloro-2-fluoro-4,6-dihydroxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-ethylbut- 2-enamide; N-(2-(3-Chloro-4,6-dihydroxy-2-methylbenzoyl) isoindolin-4-yl)-N-ethyl acrylamide; N-(2-(3-Chloro-2-fluoro-4,6-dihydroxybenzoyl) isoindolin-4-yl)-N-methylacrylamide; N-(2-(2,4-Dihydroxy-5-methylbenzoyl)-6-(2-(dimethylamino) ethoxy) isoindolin-4-yl)-N- methylacrylamide; (E)-4-(Dimethylamino)-N-(2-(6-hydroxy-2,3-dimethyl-1H-indole-5-carbonyl) isoindolin-4-yl)-N- methylbut-2-enamide; (E)-4-(Dimethylamino)-N-(2-(6-hydroxy-2-methyl-1H-indole-5-carbonyl) isoindolin-4-yl)-N- methylbut-2-enamide; (E)-4-(Dimethylamino)-N-(2-(6-hydroxy-3-methyl-1H-indole-5-carbonyl) isoindolin-4-yl)-N- methylbut-2-enamide;
(E)-4-(Dimethylamino)-N-(2-(4-fluoro-2-hydroxy-5-isopropylbenzoyl) isoindolin-4-yl)-N- methylbut-2-enamide; (E)-4-(Dimethylamino)-N-(2-(2-hydroxy-4-methoxy-5-methylbenzoyl)isoindolin-4-yl)-N- methylbut-2-enamide; (E)-N-(2-(3-(tert-Butyl)-2-fluoro-6-hydroxy-5-(hydroxymethyl)benzoyl)isoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide; N-(2-(3-(Aminomethyl)-5-(tert-butyl)-6-fluoro-2-hydroxybenzoyl)isoindolin-4-yl)-N- methylacrylamide; (E)-N-(2-(5-(tert-Butyl)-2-hydroxy-4-methoxybenzoyl)-5-chloroisoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide; (E)-4-(Dimethylamino)-N-(2-(2-hydroxy-5-isopropyl-4-methoxybenzoyl)isoindolin-4-yl)-N- methylbut-2-enamide; (E)-4-(Dimethylamino)-N-(2-(5-ethyl-2-hydroxy-4-methoxybenzoyl)isoindolin-4-yl)-N-methylbut- 2-enamide; (E)-N-(2-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N- methylbut-2-enamide; (E)-N-(2-(5-Chloro-2-hydroxy-4-methoxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N- methylbut-2-enamide; N-(2-(5-(tert-Butyl)-2-hydroxy-4-methoxybenzoyl)isoindolin-4-yl)-N-methylacrylamide; N-(2-(5-Ethyl-2-hydroxy-4-methoxybenzoyl)isoindolin-4-yl)-N-methylacrylamide; (E)-N-(2-(3-(tert-Butyl)-2-fluoro-6-hydroxybenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-4- (dimethylamino)-N-methylbut-2-enamide; N-(2-(3-(tert-Butyl)-2-fluoro-6-hydroxybenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- methylacrylamide; N-(2-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)isoindolin-4-yl)-N-methylacrylamide; N-(2-(5-Chloro-2-hydroxy-4-methoxybenzoyl) isoindolin-4-yl)-N-methyl acrylamide; (E)-N-(2-(5-Chloro-2-hydroxy-4-methoxybenzoyl)-5-methylisoindolin-4-yl)-4-(dimethylamino)-N- methylbut-2-enamide; (E)-N-(5-Chloro-2-(5-chloro-2-hydroxy-4-methoxybenzoyl) isoindolin-4-yl)-4-(dimethyl amino)-N- methylbut-2-enamide;
(E)-N-(5-Chloro-2-(2-hydroxy-5-isopropyl-4-methoxybenzoyl) isoindolin-4-yl)-4-(dimethyl amino)-N-methylbut-2-enamide; (E)-N-(2-(5-(tert-Butyl)-2-hydroxy-4-methoxybenzoyl)-5-methylisoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide; N-(2-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- methylacrylamide; N-(2-(4-Fluoro-2-hydroxy-5-isopropylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- methylacrylamide; N-(2-(5-Chloro-2-hydroxy-4-methoxybenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- methylacrylamide; N-(2-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)-6-methyl-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- methylacrylamide; N-(2-(6-Hydroxy-3,3-dimethyl-2-oxoindoline-5-carbonyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- methylacrylamide; N-(2-(6-Hydroxy-3,3-dimethyl-2-oxoindoline-5-carbonyl) isoindolin-4-yl)-N-methyl acrylamide; N-(6-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)-5,6,7,8-tetrahydro-2,6-naphthyridin-3-yl)-N- methylacrylamide; N-(7-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)-3-methyl-5,6,7,8-tetrahydro-1,7-naphthyridin- 2-yl)-N-methylacrylamide; N-(6-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)-2-methyl-5,6,7,8-tetrahydro-1,6-naphthyridin- 3-yl)-N-methylacrylamide; (E)-N-(2-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-4- (dimethylamino)-N-methylbut-2-enamide; (E)-4-(Dimethylamino)-N-(2-(2-hydroxy-5-isopropyl-4-methoxybenzoyl)-1,2,3,4- tetrahydroisoquinolin-7-yl)-N-methylbut-2-enamide; (E)-N-(2-(5-(tert-Butyl)-4-fluoro-2-hydroxybenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-4- (dimethylamino)-N-methylbut-2-enamide; (E)-4-(Dimethylamino)-N-(2-(4-fluoro-2-hydroxy-5-isopropylbenzoyl)-1,2,3,4- tetrahydroisoquinolin-7-yl)-N-methylbut-2-enamide; N-(2-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)isoindolin-4-yl)-N-methylacrylamide; N-(6-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)-5,6,7,8-tetrahydro-1,6-naphthyridin-3-yl) acrylamide;
(E)-N-(2-(3-Chloro-6-hydroxy-2-methylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-4- (dimethylamino)-N-methylbut-2-enamide; (E)-N-(6-(5-(tert-Butyl)-2-hydroxybenzoyl)-5,6,7,8-tetrahydro-1,6-naphthyridin-3-yl)-4- (dimethylamino)-N-methylbut-2-enamide; (E)-N-(2-(5-(tert-Butyl)-2-hydroxybenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-4-(dimethylamino)- N-methylbut-2-enamide; (E)-N-(5-Chloro-2-(4-hydroxy-3-isopropylbenzoyl) isoindolin-4-yl)-4-(dimethylamino) but-2- enamide; (E)-N-(5-Chloro-2-(4-hydroxy-3-isopropylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N- methylbut-2-enamide; N-(2-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-6-(2-(dimethylamino)ethoxy)-1,2,3,4- tetrahydroisoquinolin-7-yl)-N-methylacrylamide; N-(6-(2-(Dimethylamino)ethoxy)-2-(2-hydroxy-5-isopropyl-4-methoxybenzoyl)-1,2,3,4- tetrahydroisoquinolin-7-yl)-N-methylbut-2-ynamide; (E)-N-(5-Chloro-2-(3-chloro-6-hydroxy-4-methoxy-2-methylbenzoyl) isoindolin-4-yl)-4-(dimethyl amino)-N-methylbut-2-enamide; (E)-N-(2-(5-(tert-Butyl)-2-fluoro-4-hydroxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N- methylbut-2-enamide; (E)-N-(2-(5-(tert-Butyl)-2-hydroxy-4-methoxybenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-4- (dimethylamino)-N-methylbut-2-enamide; N-(2-(5-(tert-Butyl)-2-hydroxy-4-methoxybenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- methylacrylamide; N-(2-(3-Chloro-2-fluoro-6-hydroxy-4-methoxybenzoyl)isoindolin-4-yl)-N-methylacrylamide; N-(5-Chloro-2-(3-chloro-6-hydroxy-4-methoxy-2-methylbenzoyl) isoindolin-4-yl)-N-methyl acrylamide; (E)-4-(Dimethylamino)-N-(2-(2-hydroxy-5-isopropyl-4-methoxybenzoyl)-5-methylisoindolin-4-yl)- N-methylbut-2-enamide; N-(2-(4-Fluoro-2-hydroxy-5-isopropylbenzoyl)-6-methyl-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- methylacrylamide; N-(6-Chloro-2-(2-hydroxy-5-isopropyl-4-methoxybenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- methylacrylamide;
N-(2-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)-8-methyl-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- methylacrylamide; N-(6-(2-(Dimethylamino)ethoxy)-2-(2-hydroxy-5-isopropyl-4-methoxybenzoyl)-1,2,3,4- tetrahydroisoquinolin-7-yl)-N-methylacrylamide; N-(6-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-5,6,7,8-tetrahydro-2,6-naphthyridin-3- yl)-N-methylacrylamide; (E)-N-(7-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-5,6,7,8-tetrahydro-1,7-naphthyridin- 2-yl)-N-methylbut-2-enamide; N-(7-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-5,6,7,8-tetrahydro-1,7-naphthyridin-2- yl)-N-methylacrylamide; N-(7-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-3-methyl-5,6,7,8-tetrahydro-1,7- naphthyridin-2-yl)-N-methylacrylamide; N-(6-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-2-methyl-5,6,7,8-tetrahydro-1,6- naphthyridin-3-yl)-N-methylacrylamide; N-(2-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N-(1- methylpiperidin-4-yl) acrylamide; (E)-N-(2-(4,6-dihydroxy-2,3-dimethylbenzoyl)-5-methylisoindolin-4-yl)-4-(dimethylamino)-N- methylbut-2-enamide; (E)-N-(2-(4,6-Dihydroxy-2,3-dimethylbenzoyl)-6-methylisoindolin-4-yl)-4-(dimethylamino)-N- methylbut-2-enamide; (E)-N-(6-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-5,6,7,8-tetrahydro-1,6-naphthyridin- 3-yl)-N-methylbut-2-enamide; N-(2-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- methylacrylamide; N-(2-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N-(1- methylpyrrolidin-3-yl)acrylamide; ((E)-N-(5-Chloro-2-(2-hydroxy-5-isopropyl-4-methoxybenzoyl)isoindolin-4-yl)-4- (dimethylamino)but-2-enamide; (E)-N-(2-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)isoindolin-4-yl)-4- (dimethylamino)but-2-enamide; N-(6-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-5,6,7,8-tetrahydro-1,6-naphthyridin-3- yl)acrylamide;
(E)-N-(2-(3-Chloro-6-hydroxy-2-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-methylbut- 2-enamide; (E)-N-(7-Chloro-2-(3-chloro-6-hydroxy-4-methoxy-2-methylbenzoyl) isoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide; N-(7-Chloro-2-(3-chloro-6-hydroxy-4-methoxy-2-methylbenzoyl) isoindolin-4-yl)-N-methyl acrylamide; (E)-4-(Dimethylamino)-N-(6-(2-hydroxy-5-isopropyl-4-methoxybenzoyl)-5,6,7,8-tetrahydro-1,6- naphthyridin-3-yl)-N-methylbut-2-enamide; N-(2-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-6-(2-(dimethylamino)ethoxy)-1,2,3,4- tetrahydroisoquinolin-7-yl)-N-methylbut-2-ynamide; (E)-N-(5-Chloro-2-(3-chloro-4-hydroxy-2-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)but-2- enamide; (E)-N-(5-Chloro-2-(4-hydroxy-2,3-dimethylbenzoyl) isoindolin-4-yl)-4-(dimethylamino) but-2- enamide; (E)-4-(Dimethylamino)-N-(2-(4-hydroxy-3-isopropylbenzoyl)-3-methyl-1,2,3,4- tetrahydroisoquinolin-7-yl)-N-methylbut-2-enamide; (E)-4-(Dimethylamino)-N-(2-(4-hydroxy-3-isopropylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)- N-methylbut-2-enamide; N-(2-(4-Hydroxy-3-isopropylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N-(1-methylpiperidin-4- yl)acrylamide; (E)-4-(Dimethylamino)-N-(6-(2-hydroxy-5-isopropyl-4-methoxybenzoyl)-2-methoxy-5,6,7,8- tetrahydro-1,6-naphthyridin-3-yl)-N-methylbut-2-enamide; ((E)-4-(Dimethylamino)-N-(6-(4-hydroxy-3-isopropylbenzoyl)-2-methoxy-5,6,7,8-tetrahydro-1,6- naphthyridin-3-yl)-N-methylbut-2-enamide; (E)-N-(2-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)- N-methylbut-2-enamide; N-(6-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-5,6,7,8-tetrahydro-1,6-naphthyridin-3- yl)but-2-ynamide. [00148] The various functional groups and substituents making up the compounds of the Formula (I), or sub-formulae (I-I) to (I-XVIV), are typically chosen such that the molecular weight of the compound of the formula (I) does not exceed 700. More usually, the molecular weight of the compound will be less than 650. More preferably, the molecular weight is less than 600.
[00149] A suitable pharmaceutically acceptable salt of a compound of the invention is, for example, an acid-addition salt of a compound of the invention which is sufficiently basic, for example, an acid-addition salt with, for example, an inorganic or organic acid, for example hydrochloric, hydrobromic, sulfuric, phosphoric, trifluoroacetic, formic, citric methane sulfonate or maleic acid. In addition, a suitable pharmaceutically acceptable salt of a compound of the invention which is sufficiently acidic is an alkali metal salt, for example a sodium or potassium salt, an alkaline earth metal salt, for example a calcium or magnesium salt, an ammonium salt or a salt with an organic base which affords a pharmaceutically acceptable cation, for example a salt with methylamine, dimethylamine, trimethylamine, piperidine, morpholine or tris-(2-hydroxyethyl)amine. [00150] Compounds that have the same molecular formula but differ in the nature or sequence of bonding of their atoms or the arrangement of their atoms in space are termed “isomers”. Isomers that differ in the arrangement of their atoms in space are termed “stereoisomers”. Stereoisomers that are not mirror images of one another are termed “diastereomers” and those that are non-superimposable mirror images of each other are termed “enantiomers”. When a compound has an asymmetric centre, for example, it is bonded to four different groups, a pair of enantiomers is possible. An enantiomer can be characterized by the absolute configuration of its asymmetric centre and is described by the R- and S-sequencing rules of Cahn and Prelog, or by the manner in which the molecule rotates the plane of polarized light and designated as dextrorotatory or levorotatory (i.e., as (+) or (-)-isomers respectively). A chiral compound can exist as either individual enantiomer or as a mixture thereof. A mixture containing equal proportions of the enantiomers is called a “racemic mixture”. [00151] The compounds of this invention may possess one or more asymmetric centres; such compounds can therefore be produced as individual (R)- or (S)-stereoisomers or as mixtures thereof. Unless indicated otherwise, the description or naming of a particular compound in the specification and claims is intended to include both individual enantiomers and mixtures, racemic or otherwise, thereof. The methods for the determination of stereochemistry and the separation of stereoisomers are well-known in the art (see discussion in Chapter 4 of “Advanced Organic Chemistry”, 4th edition J. March, John Wiley and Sons, New York, 2001), for example by synthesis from optically active starting materials or by resolution of a racemic form. Some of the compounds of the invention may have geometric isomeric centres (E- and Z- isomers). [00152] It is to be understood that the present invention encompasses all optical, diastereoisomers and geometric isomers and mixtures thereof that possess activity. [00153] The present invention also encompasses compounds of the invention as defined herein which comprise one or more isotopic substitutions. For example, H may be in any isotopic form,
including
1H,
2H (D), and
3H (T); C may be in any isotopic form, including
12C,
13C, and
14C; and O may be in any isotopic form, including
16O and
18O; and the like. [00154] It is also to be understood that certain compounds of the Formula (I), or sub-formulae (I-I) to (I-XVIV), may exist in solvated as well as unsolvated forms such as, for example, hydrated forms. It is to be understood that the invention encompasses all such solvated forms that possess activity. [00155] It is also to be understood that certain compounds of the Formula (I), or sub-formulae (I-I) to (I-XVIV), may exhibit polymorphism, and that the invention encompasses all such forms that possess activity. [00156] Compounds of the Formula (I), or sub-formulae (I-I) to (I-XVIV), may exist in a number of different tautomeric forms and references to compounds of the Formula (I), or sub-formulae (I- I) to (I-XVIV), include all such forms. For the avoidance of doubt, where a compound can exist in one of several tautomeric forms, and only one is specifically described or shown, all others are nevertheless embraced by Formula (I), or sub-formulae (I-I) to (I-XVIV). Examples of tautomeric forms include keto-, enol-, and enolate-forms, as in, for example, the following tautomeric pairs: keto/enol (illustrated below), imine/enamine, amide/imino alcohol, amidine/amidine, nitroso/oxime, thioketone/enethiol, and nitro/aci-nitro.

keto enol enolate [00157] Compounds of the Formula (I), or sub-formulae (I-I) to (I-XVIV), containing an amine function may also form N-oxides. A reference herein to a compound of the Formula (I), or sub- formulae (I-I) to (I-XVIV), that contains an amine function also includes the N-oxide. Where a compound contains several amine functions, one or more than one nitrogen atom may be oxidised to form an N-oxide. Particular examples of N-oxides are the N-oxides of a tertiary amine or a nitrogen atom of a nitrogen-containing heterocycle. N-Oxides can be formed by treatment of the corresponding amine with an oxidizing agent such as hydrogen peroxide or a per-acid (e.g., a peroxycarboxylic acid), see for example Advanced Organic Chemistry, by Jerry March, 4th Edition, Wiley Interscience. More particularly, N-oxides can be made by the procedure of L. W. Deady (Syn. Comm. 1977, 7, 509-514) in which the amine compound is reacted with m- chloroperoxybenzoic acid (mCPBA), for example, in an inert solvent such as dichloromethane. [00158] The compounds of Formula (I), or sub-formulae (I-I) to (I-XVIV), may be administered in the form of a pro-drug which is broken down in the human or animal body to release a compound of the invention. A pro-drug may be used to alter the physical properties and/or the
pharmacokinetic properties of a compound of the invention. A pro-drug can be formed when the compound of the invention contains a suitable group or substituent to which a property-modifying group can be attached. Examples of pro-drugs include in vivo cleavable ester derivatives that may be formed at a carboxy group or a hydroxy group in a compound of the Formula (I), or sub- formulae (I-I) to (I-XVIV), and in-vivo cleavable amide derivatives that may be formed at a carboxy group or an amino group in a compound of the Formula (I), or sub-formulae (I-I) to (I-XVIV). [00159] Accordingly, the present invention includes those compounds of the Formula (I), or sub- formulae (I-I) to (I-XVIV), as defined hereinbefore, when made available by organic synthesis and when made available within the human or animal body by way of cleavage of a pro-drug thereof. Accordingly, the present invention includes those compounds of the Formula (I), or sub- formulae (I-I) to (I-XVIV), that are produced by organic synthetic means and also such compounds that are produced in the human or animal body by way of metabolism of a precursor compound, that is a compound of the Formula (I), or sub-formulae (I-I) to (I-XVIV), may be a synthetically-produced compound or a metabolically-produced compound. [00160] A suitable pharmaceutically acceptable pro-drug of a compound of the Formula (I), or sub-formulae (I-I) to (I-XVIV), is one that is based on reasonable medical judgement as being suitable for administration to the human or animal body without undesirable pharmacological activities and without undue toxicity. [00161] Various forms of pro-drug have been described, for example in the following documents:- a) Methods in Enzymology, Vol. 42, p. 309-396, edited by K. Widder, et al. (Academic Press, 1985); b) Design of Pro-drugs, edited by H. Bundgaard, (Elsevier, 1985); c) A Textbook of Drug Design and Development, edited by Krogsgaard-Larsen and H. Bundgaard, Chapter 5 “Design and Application of Pro-drugs”, by H. Bundgaard p. 113-191 (1991); d) H. Bundgaard, Advanced Drug Delivery Reviews, 8, 1-38 (1992); e) H. Bundgaard, et al., Journal of Pharmaceutical Sciences, 77, 285 (1988); f) N. Kakeya, et al., Chem. Pharm. Bull., 32, 692 (1984); g) T. Higuchi and V. Stella, “Pro-Drugs as Novel Delivery Systems”, A.C.S. Symposium Series, Volume 14; and h) E. Roche (editor), “Bioreversible Carriers in Drug Design”, Pergamon Press, 1987. [00162] A suitable pharmaceutically acceptable pro-drug of a compound of the Formula (I), or sub-formulae (I-I) to (I-XVIV), that possesses a carboxy group is, for example, an in vivo cleavable ester thereof. An in vivo cleavable ester of a compound of the Formula (I), or sub- formulae (I-I) to (I-XVIV), containing a carboxy group is, for example, a pharmaceutically
acceptable ester which is cleaved in the human or animal body to produce the parent acid or parent alcohol. Suitable pharmaceutically acceptable esters for carboxy include (1-6C)alkyl esters such as methyl, ethyl and tert-butyl, (1-6C)alkoxymethyl esters such as methoxymethyl esters, (1-6C)alkanoyloxymethyl esters such as pivaloyloxymethyl esters, 3-phthalidyl esters, (3- 8C)cycloalkylcarbonyloxy-(1-6C)alkyl esters such as cyclopentylcarbonyloxymethyl and 1- cyclohexylcarbonyloxyethyl esters, 2-oxo-1,3-dioxolenylmethyl esters such as 5-methyl-2-oxo- 1,3-dioxolen-4-ylmethyl esters and (1-6C)alkoxycarbonyloxy-(1-6C)alkyl esters such as methoxycarbonyloxymethyl and 1-methoxycarbonyloxyethyl esters. [00163] A suitable pharmaceutically acceptable pro-drug of a compound of the Formula (I), or sub-formulae (I-I) to (I-XVIV), that possesses a hydroxy group is, for example, an in vivo cleavable ester or ether thereof. An in vivo cleavable ester or ether of a compound of the Formula (I), or sub-formulae (I-I) to (I-XVIV), containing a hydroxy group is, for example, a pharmaceutically acceptable ester or ether which is cleaved in the human or animal body to produce the parent hydroxy compound. Suitable pharmaceutically acceptable ester forming groups for a hydroxy group include inorganic esters such as phosphate esters (including phosphoramidic cyclic esters). Further suitable pharmaceutically acceptable ester forming groups for a hydroxy group include (1-10C)alkanoyl groups such as acetyl, benzoyl, phenylacetyl and substituted benzoyl and phenylacetyl groups, (1-10C)alkoxycarbonyl groups such as ethoxycarbonyl, N,N-(1-6C)2carbamoyl, 2-dialkylaminoacetyl and 2-carboxyacetyl groups. Examples of ring substituents on the phenylacetyl and benzoyl groups include aminomethyl, N- alkylaminomethyl, N,N-dialkylaminomethyl, morpholinomethyl, piperazin-1-ylmethyl and 4-(1- 4C)alkylpiperazin-1-ylmethyl. Suitable pharmaceutically acceptable ether forming groups for a hydroxy group include ^-acyloxyalkyl groups such as acetoxymethyl and pivaloyloxymethyl groups. [00164] A suitable pharmaceutically acceptable pro-drug of a compound of the Formula (I), or sub-formulae (I-I) to (I-XVIV), that possesses a carboxy group is, for example, an in vivo cleavable amide thereof, for example an amide formed with an amine such as ammonia, a (1- 4C)alkylamine such as methylamine, a [(1-4C)alkyl]2amine such as dimethylamine, N-ethyl-N- methylamine or diethylamine, a (1-4C)alkoxy-(2-4C)alkylamine such as 2-methoxyethylamine, a phenyl-(1-4C)alkylamine such as benzylamine and amino acids such as glycine or an ester thereof. [00165] A suitable pharmaceutically acceptable pro-drug of a compound of the Formula (I), or sub-formulae (I-I) to (I-XVIV), that possesses an amino group is, for example, an in vivo cleavable amide derivative thereof. Suitable pharmaceutically acceptable amides from an amino group include, for example an amide formed with (1-10C)alkanoyl groups such as an acetyl, benzoyl, phenylacetyl and substituted benzoyl and phenylacetyl groups. Examples of ring substituents on
the phenylacetyl and benzoyl groups include aminomethyl, N-alkylaminomethyl, N,N- dialkylaminomethyl, morpholinomethyl, piperazin-1-ylmethyl and 4-(1-4C)alkyl)piperazin-1- ylmethyl. [00166] The in vivo effects of a compound of the Formula (I), or sub-formulae (I-I) to (I-XVIV), may be exerted in part by one or more metabolites that are formed within the human or animal body after administration of a compound of the Formula (I), or sub-formulae (I-I) to (I-XVIV). As stated hereinbefore, the in vivo effects of a compound of the Formula (I), or sub-formulae (I-I) to (I-XVIV), may also be exerted by way of metabolism of a precursor compound (a pro-drug). [00167] Though the present invention may relate to any compound or particular group of compounds defined herein by way of optional, preferred or suitable features or otherwise in terms of particular embodiments, the present invention may also relate to any compound or particular group of compounds that specifically excludes said optional, preferred or suitable features or particular embodiments. [00168] Suitably, the present invention excludes any individual compounds not possessing the biological activity defined herein. Synthesis [00169] The compounds of the present invention can be prepared by any suitable technique known in the art. Particular processes for the preparation of these compounds are described further in the accompanying examples. [00170] In the description of the synthetic methods described herein and in any referenced synthetic methods that are used to prepare the starting materials, it is to be understood that all proposed reaction conditions, including choice of solvent, reaction atmosphere, reaction temperature, duration of the experiment and workup procedures, can be selected by a person skilled in the art. [00171] It is understood by one skilled in the art of organic synthesis that the functionality present on various portions of the molecule must be compatible with the reagents and reaction conditions utilised. [00172] It will be appreciated that during the synthesis of the compounds of the invention in the processes defined herein, or during the synthesis of certain starting materials, it may be desirable to protect certain substituent groups to prevent their undesired reaction. The skilled chemist will appreciate when such protection is required, and how such protecting groups may be put in place, and later removed.
[00173] For examples of protecting groups see one of the many general texts on the subject, for example, ‘Protective Groups in Organic Synthesis’ by Theodora Green (publisher: John Wiley & Sons). Protecting groups may be removed by any convenient method described in the literature or known to the skilled chemist as appropriate for the removal of the protecting group in question, such methods being chosen so as to effect removal of the protecting group with the minimum disturbance of groups elsewhere in the molecule. [00174] Thus, if reactants include, for example, groups such as amino, carboxy or hydroxy it may be desirable to protect the group in some of the reactions mentioned herein. [00175] By way of example, a suitable protecting group for an amino or alkylamino group is, for example, an acyl group, for example an alkanoyl group such as acetyl, an alkoxycarbonyl group, for example a methoxycarbonyl, ethoxycarbonyl or t-butoxycarbonyl group, an arylmethoxycarbonyl group, for example benzyloxycarbonyl, or an aroyl group, for example benzoyl. The deprotection conditions for the above protecting groups necessarily vary with the choice of protecting group. Thus, for example, an acyl group such as an alkanoyl or alkoxycarbonyl group or an aroyl group may be removed by, for example, hydrolysis with a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide. Alternatively, an acyl group such as a tert-butoxycarbonyl group may be removed, for example, by treatment with a suitable acid as hydrochloric, sulfuric or phosphoric acid or trifluoroacetic acid and an arylmethoxycarbonyl group such as a benzyloxycarbonyl group may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon, or by treatment with a Lewis acid for example boron tris(trifluoroacetate). A suitable alternative protecting group for a primary amino group is, for example, a phthaloyl group which may be removed by treatment with an alkylamine, for example dimethylaminopropylamine, or with hydrazine. [00176] A suitable protecting group for a hydroxy group is, for example, an acyl group, for example an alkanoyl group such as acetyl, an aroyl group, for example benzoyl, or an arylmethyl group, for example benzyl. The deprotection conditions for the above protecting groups will necessarily vary with the choice of protecting group. Thus, for example, an acyl group such as an alkanoyl or an aroyl group may be removed, for example, by hydrolysis with a suitable base such as an alkali metal hydroxide, for example lithium, sodium hydroxide or ammonia. Alternatively, an arylmethyl group such as a benzyl group may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon. [00177] A suitable protecting group for a carboxy group is, for example, an esterifying group, for example a methyl or an ethyl group which may be removed, for example, by hydrolysis with a base such as sodium hydroxide, or for example a t-butyl group which may be removed, for example, by treatment with an acid, for example an organic acid such as trifluoroacetic acid, or
for example a benzyl group which may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon. [00178] Resins may also be used as a protecting group. [00179] The methodology employed to synthesise a compound of Formula (I), or sub-formulae (I-I) to (I-XVIV), will vary depending on the nature of R
2, R
3, R
4, R
5, X, Y
1, Y
2, A
1, A
2, A
3 and A
4 and any substituent groups or subgroups associated therewith. Suitable processes for their preparation are described further in the accompanying Examples. [00180] Once a compound of Formula (I), or sub-formulae (I-I) to (I-XVIV), has been synthesised by any one of the processes defined herein, the processes may then further comprise the additional steps of: (i) removing any protecting groups present; (ii) converting the compound Formula (I) into another compound of Formula (I); (iii) forming a pharmaceutically acceptable salt, hydrate or solvate thereof; and/or (iv) forming a prodrug thereof. [00181] An example of (ii) above is when a compound of Formula (I) is synthesised and then one or more of the groups R
2, R
3, R
4, R
5, X, Y
1, Y
2, A
1, A
2, A
3 and A
4 may be further reacted to change the nature of the group and provide an alternative compound of Formula (I). [00182] The resultant compounds of Formula (I), or sub-formulae (I-I) to (I-XVIV), can be isolated and purified using techniques well known in the art. [00183] The compounds of Formula (I), or sub-formulae (I-I) to (I-XVIV), may be synthesised by the synthetic routes shown in the Examples section below. Biological Activity [00184] The biological assays described in the Examples section herein may be used to measure the pharmacological effects of the compounds of the present invention. [00185] Although the pharmacological properties of the compounds of Formula (I) vary with structural change, as expected, the compounds of the invention were found to be active in a PMS2 in vitro assay as described in the Examples section. Pharmaceutical Compositions [00186] According to a further aspect of the invention there is provided a pharmaceutical composition which comprises a compound of the invention as defined hereinbefore, or a
pharmaceutically acceptable salt, hydrate or solvate thereof, in association with a pharmaceutically acceptable diluent or carrier. [00187] The compositions of the invention may be in a form suitable for oral use (for example as tablets, lozenges, hard or soft capsules, aqueous or oily suspensions, emulsions, dispersible powders or granules, syrups or elixirs), for topical use (for example as creams, ointments, gels, or aqueous or oily solutions or suspensions), for administration by inhalation (for example as a finely divided powder or a liquid aerosol), for administration by insufflation (for example as a finely divided powder) or for parenteral administration (for example as a sterile aqueous or oily solution for intravenous, subcutaneous, intramuscular, intraperitoneal or intramuscular dosing or as a suppository for rectal dosing). [00188] The compositions of the invention may be obtained by conventional procedures using conventional pharmaceutical excipients, well known in the art. Thus, compositions intended for oral use may contain, for example, one or more colouring, sweetening, flavouring and/or preservative agents. [00189] An effective amount of a compound of the present invention for use in therapy is an amount sufficient to treat or prevent a proliferative condition referred to herein, slow its progression and/or reduce the symptoms associated with the condition. [00190] The amount of active ingredient that is combined with one or more excipients to produce a single dosage form will necessarily vary depending upon the individual treated and the particular route of administration. For example, a formulation intended for oral administration to humans will generally contain, for example, from 0.5 mg to 0.5 g of active agent (more suitably from 0.5 to 100 mg, for example from 1 to 30 mg) compounded with an appropriate and convenient amount of excipients which may vary from about 5 to about 98 percent by weight of the total composition. [00191] The size of the dose for therapeutic or prophylactic purposes of a compound of the Formula (I) will naturally vary according to the nature and severity of the conditions, the age and sex of the animal or patient and the route of administration, according to well-known principles of medicine. [00192] In using a compound of the invention for therapeutic or prophylactic purposes it will generally be administered so that a daily dose in the range, for example, 0.1 mg/kg to 75 mg/kg body weight is received, given if required in divided doses. In general, lower doses will be administered when a parenteral route is employed. Thus, for example, for intravenous or intraperitoneal administration, a dose in the range, for example, 0.1 mg/kg to 30 mg/kg body weight will generally be used. Similarly, for administration by inhalation, a dose in the range, for example, 0.05 mg/kg to 25 mg/kg body weight will be used. Oral administration may also be
suitable, particularly in tablet form. Typically, unit dosage forms will contain about 0.5 mg to 0.5 g of a compound of this invention. Therapeutic Uses and Applications [00193] The present invention provides compounds that function as inhibitors of PMS2 activity. [00194] The compounds of Formula (I), or a pharmaceutically acceptable salt thereof, therefore, have potential therapeutic uses in a variety of disease states in which the inhibition of PMS2 activity is beneficial. [00195] The present invention therefore provides a method of treating a disease or disorder in which the inhibition PMS2 activity is beneficial in a patient in need of such treatment, said method comprising administering to said patient a therapeutically effective amount of a compound, or a pharmaceutically acceptable salt, hydrate or solvate thereof, or a pharmaceutical composition as defined herein. [00196] The present invention provides a method of inhibiting PMS2 activity, in vitro or in vivo, said method comprising contacting a cell with an effective amount of a compound or a pharmaceutically acceptable salt, hydrate or solvate thereof as defined herein, or a pharmaceutical composition as defined herein. [00197] The present invention provides a method of treating a proliferative disorder in a patient in need of such treatment, said method comprising administering to said patient a therapeutically effective amount of a compound or a pharmaceutically acceptable salt, hydrate or solvate thereof as defined herein, or a pharmaceutical composition as defined herein. [00198] The present invention provides a method of treating cancer in a patient in need of such treatment, said method comprising administering to said patient a therapeutically effective amount of a compound or a pharmaceutically acceptable salt, hydrate or solvate thereof as defined herein, or a pharmaceutical composition as defined herein. [00199] The present invention provides a compound, or a pharmaceutically acceptable salt, hydrate or solvate thereof, or a pharmaceutical composition as defined herein for use in therapy. [00200] The present invention provides a compound, or a pharmaceutically acceptable salt, hydrate or solvate thereof, or a pharmaceutical composition as defined herein for use as a medicament. [00201] The present invention provides a compound or a pharmaceutically acceptable salt, hydrate or solvate thereof as defined herein, or a pharmaceutical composition as defined herein,
for use in the treatment of a proliferative disorder. [00202] The present invention provides a compound, or a pharmaceutically acceptable salt, hydrate or solvate thereof, or a pharmaceutical composition as defined herein for use in the treatment of cancer. In a particular embodiment, the cancer is human cancer. In a particular embodiment, the cancer is human cancer, in particular oestrogen positive cancers, such as breast cancer, or androgen receptor positive cancers, such as prostate cancer. [00203] The present invention provides a compound, or a pharmaceutically acceptable salt, hydrate or solvate thereof, as defined herein, or a pharmaceutical composition as defined herein for use in the inhibition of PMS2 activity. [00204] The present invention provides a compound, or a pharmaceutically acceptable salt, hydrate or solvate thereof, as defined herein, or a pharmaceutical composition as defined herein for use in the treatment of a disease or disorder in which the inhibition of PMS2 activity is beneficial. [00205] The present invention provides a use of a compound, or a pharmaceutically acceptable salt, hydrate or solvate thereof, as defined herein, or a pharmaceutical composition as defined herein in the manufacture of a medicament for the treatment of a proliferative disorder. [00206] The present invention provides a use of a compound, or a pharmaceutically acceptable salt, hydrate or solvate thereof, as defined herein, or a pharmaceutical composition as defined herein in the manufacture of a medicament for the treatment of cancer. [00207] The present invention provides a use of a compound, or a pharmaceutically acceptable salt, hydrate or solvate thereof, as defined herein, or a pharmaceutical composition as defined herein in the manufacture of a medicament for the inhibition of PMS2 activity. [00208] The present invention provides a use of a compound, or a pharmaceutically acceptable salt, hydrate or solvate thereof, as defined herein, or a pharmaceutical composition as defined herein in the manufacture of a medicament for the treatment of a disease or disorder in which the inhibition of PMS2 activity is beneficial. [00209] The term "proliferative disorder", “proliferative condition” and “proliferative disease” are used interchangeably herein and pertain to an unwanted or uncontrolled cellular proliferation of excessive or abnormal cells which is undesired, such as, neoplastic or hyperplastic growth, whether in vitro or in vivo. [00210] In the above-outlined aspects of the invention, the proliferative disorder is suitably cancer, and the cancer is suitably a human cancer. In particular, the compounds of the present invention will be useful for the treatment of any cancer in which mis-match repair inhibition is beneficial. Any suitable cancer may be targeted (e.g., adenoid cystic carcinoma, adrenal gland
tumor, amyloidosis, anal cancer, appendix cancer, astrocytoma, ataxia-telangiectasia, Beckwith- Wiedemann Syndrome, bile duct cancer (cholangiocarcinoma), Birt-Hogg-Dubé Syndrome, bladder cancer, bone cancer, brain stem glioma, brain tumor, breast cancer, Carney Complex, central nervous system tumors, cervical cancer, colorectal cancer, Cowden Syndrome, craniopharyngioma, desmoplastic infantile ganglioglioma, ependymoma, esophageal cancer, Ewing sarcoma, eye cancer, eyelid cancer, familial adenomatous polyposis, familial GIST, familial malignant melanoma, familial non-VHL clear cell renal cell carcinoma, familial pancreatic cancer, gallbladder cancer, gastrointestinal stromal tumor – GIST, germ cell tumor, gestational trophoblastic disease, head and neck cancer, hereditary breast and ovarian cancer, hereditary diffuse gastric cancer, hereditary leiomyomatosis and renal cell cancer, hereditary mixed polyposis syndrome, hereditary pancreatitis, hereditary papillary renal carcinoma, juvenile polyposis syndrome, kidney cancer, lacrimal gland tumor, laryngeal and hypopharyngeal cancer, leukemia (acute lymphoblastic leukamia (ALL), acute myeloid leukemia (AML), B-cell prolymphocytic leukemia, hairy cell leukemia, chronic lymphocytic leukemia (CLL), chronic myeloid leukemia (CML), chronic T-cell lymphocytic leukemia, eosinophilic leukemia), Li- Fraumeni Syndrome, liver cancer, lung cancer (non-small cell lung cancer, small cell lung cancer), Lymphoma (Hodgkin, non-Hodgkin), Lynch Syndrome, mastocytosis, medulloblastoma, melanoma, meningioma, mesothelioma, multiple endocrine neoplasia Type 1 & 2, multiple myeloma, MUTYH (or MYH)-associated polyposis, myelodysplastic syndromes (MDS), nasal cavity and paranasal sinus Cancer, nasopharyngeal Cancer, neuroblastoma, neuroendocrine tumors (e.g., of the gastrointestinal tract, lung or pancreas), neurofibromatosis Type 1 & 2, nevoid basal cell carcinoma syndrome, oral and oropharyngeal cancer, osteosarcoma, ovarian / fallopian tube / peritoneal cancer, pancreatic cancer, parathyroid cancer, penile cancer, Peutz- Jeghers Syndrome, pheochromocytoma, paraganglioma, pituitary gland tumor, pleuropulmonary blastoma, prostate cancer, retinoblastoma, rhabdomyosarcoma, salivary gland cancer, sarcoma (e.g., Kaposi or soft tissue), skin cancer, small bowel cancer, stomach cancer, testicular cancer, thymoma and thymic carcinoma, thyroid cancer, tuberous sclerosis complex, uterine cancer, vaginal cancer, Von Hippel-Lindau syndrome, vulvar cancer, Waldenstrom’s macroglobulinemia, Werner syndrome, Wilms Tumor and xeroderma pigmentosum). Particular cancers of interest include haematological cancers such as lymphomas (including diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL), Burkitt lymphoma (BL) and angioimmunoblastic T-cell lymphoma (AITL)), leukaemias (including acute lymphoblastic leukaemia (ALL) and chronic myeloid leukaemia (CML)), multiple myeloma, breast cancer, non-small cell lung cancer (NSCLC), colorectal cancer, endometrial cancer, gastro-oesophageal cancer, neuroendocrine cancers, osteosarcomas, prostate cancer, pancreatic cancer, small intestine cancer, bladder cancer, rectal cancer, cholangiocarcinoma, CNS cancer, thyroid cancer, head and neck cancer, oesophageal cancer, and ovarian cancer.
[00211] The compounds of the present invention may also be used to treat triplet repeat disorders. [00212] Thus, a further aspect of the present invention provides a method of treating a triplet repeat disorder (e.g., Huntington’s disease (HD), myotonic dystrophy type 1 (DM1), fragile X syndrome type A (FRAXA), Friedreich’s ataxia (FRDA), and spinocerebellar ataxias (SCAs)) in a patient in need of such treatment, said method comprising administering to said patient a therapeutically effective amount of a compound or a pharmaceutically acceptable salt, hydrate or solvate thereof as defined herein, or a pharmaceutical composition as defined herein. [00213] According to a further aspect of the present invention, there is provided a compound, or a pharmaceutically acceptable salt, hydrate or solvate thereof, or a pharmaceutical composition as defined herein for use in the treatment of a triplet repeat disorder. In a particular embodiment, the triplet repeat disorder is selected from the group consisting of Huntington’s disease (HD), myotonic dystrophy type 1 (DM1), fragile X syndrome type A (FRAXA), Friedreich’s ataxia (FRDA), and spinocerebellar ataxias (SCAs). [00214] According to a further aspect of the present invention, there is provided the use of a compound, or a pharmaceutically acceptable salt, hydrate or solvate thereof, as defined herein, or a pharmaceutical composition as defined herein in the manufacture of a medicament for the treatment of a triplet repeat disorder. In a particular embodiment, the triplet repeat disorder is selected from the group consisting of Huntington’s disease (HD), myotonic dystrophy type 1 (DM1), fragile X syndrome type A (FRAXA), Friedreich’s ataxia (FRDA), and spinocerebellar ataxias (SCAs). Routes of Administration [00215] The compounds of the invention or pharmaceutical compositions comprising these compounds may be administered to a subject by any convenient route of administration, whether systemically, peripherally or topically (i.e., at the site of desired action). [00216] Routes of administration include, but are not limited to, oral (e.g., by ingestion); buccal; sublingual; transdermal (including, e.g., by a patch, plaster, etc.); transmucosal (including, e.g., by a patch, plaster, etc.); intranasal (e.g., by nasal spray); ocular (e.g., by eye drops); pulmonary (e.g., by inhalation or insufflation therapy using, e.g., via an aerosol, e.g., through the mouth or nose); rectal (e.g., by suppository or enema); vaginal (e.g., by pessary); parenteral, for example, by injection, including intratumoral, subcutaneous, intradermal, intramuscular, intravenous, intra-arterial, intracardiac, intrathecal, intraspinal, intracapsular, subcapsular, intraorbital, intraperitoneal, intratracheal, subcuticular, intraarticular, subarachnoid, and intrasternal; by implant of a depot or reservoir, for example,
subcutaneously or intramuscularly. Combination Therapies [00217] The compounds of the present invention may be administered as a sole therapy or may involve, in addition to a compound of the invention, conventional surgery or radiotherapy or chemotherapy or a targeted agent. Such chemotherapy or targeted agent may include one or more of the following categories: (i) Antiproliferative/antineoplastic drugs and combinations thereof, as used in medical oncology, such as, but not limited to, alkylating agents (for example cis-platin, oxaliplatin, carboplatin, cyclophosphamide, nitrogen mustard, melphalan, chlorambucil, busulphan, temozolamide and nitrosoureas); antimetabolites (for example gemcitabine and antifolates such as fluoropyrimidines like 5-fluorouracil and tegafur, raltitrexed, methotrexate, cytosine arabinoside, and hydroxyurea); antitumour antibiotics (for example anthracyclines like adriamycin, bleomycin, doxorubicin, daunomycin, epirubicin, idarubicin, mitomycin-C, dactinomycin and mithramycin); antimitotic agents (for example vinca alkaloids like vincristine, vinblastine, vindesine and vinorelbine and taxoids like taxol and taxotere and polokinase inhibitors); and topoisomerase inhibitors (for example epipodophyllotoxins like etoposide and teniposide, amsacrine, topotecan and camptothecins including irinotecan); (ii) cytostatic agents such as, but not limited to, antioestrogens (for example tamoxifen, fulvestrant, toremifene, raloxifene, droloxifene and iodoxyfene), antiandrogens (for example bicalutamide, flutamide, nilutamide and cyproterone acetate), LHRH antagonists or LHRH agonists (for example goserelin, leuprorelin and buserelin), steroid hormones, including progestogens (for example megestrol acetate) and corticosteroids (for example dexamethasone, prednisone and prednisolone), aromatase inhibitors (for example as anastrozole, letrozole, vorazole and exemestane) and inhibitors of 5 ^-reductase such as finasteride; (iii) anti-invasion agents such as, but not limited to, c-Src kinase family inhibitors 4-(6-chloro- 2,3-methylenedioxyanilino)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-tetrahydropyran-4- yloxyquinazoline (AZD0530; International Patent Application WO 01/94341), N-(2-chloro- 6-methylphenyl)-2-{6-[4-(2-hydroxyethyl)piperazin-1-yl]-2-methylpyrimidin-4- ylamino}thiazole-5-carboxamide (dasatinib, BMS-354825; J. Med. Chem., 2004, 47, 6658- 6661), bosutinib (SKI-606), and metalloproteinase inhibitors such as marimastat, inhibitors of urokinase plasminogen activator receptor function or antibodies to Heparanase;
(iv) inhibitors of growth factor function such as, but not limited to, growth factor antibodies and growth factor receptor antibodies (for example the anti-erbB2 antibody trastuzumab [Herceptin™], the anti-EGFR antibody panitumumab, the anti-erbB1 antibody cetuximab [Erbitux, C225] and any growth factor or growth factor receptor antibodies disclosed by Stern et al. (Critical reviews in oncology/haematology, 2005, Vol. 54, pp11-29); such inhibitors also include tyrosine kinase inhibitors, for example inhibitors of the epidermal growth factor family (for example EGFR family tyrosine kinase inhibitors such as N-(3- chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazolin-4-amine (gefitinib, ZD1839), N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)quinazolin-4-amine (erlotinib, OSI-774) and 6-acrylamido-N-(3-chloro-4-fluorophenyl)-7-(3-morpholinopropoxy)- quinazolin-4-amine (CI 1033), erbB2 tyrosine kinase inhibitors such as lapatinib); inhibitors of the hepatocyte growth factor family; inhibitors of the insulin growth factor family; inhibitors of the platelet-derived growth factor family such as imatinib and/or nilotinib (AMN107); inhibitors of serine/threonine kinases (for example Ras/Raf signalling inhibitors such as farnesyl transferase inhibitors, for example sorafenib (BAY 43-9006), tipifarnib (R115777) and lonafarnib (SCH66336)), inhibitors of cell signalling through MEK and/or AKT kinases, c-kit inhibitors, abl kinase inhibitors, PI3 kinase inhibitors, Plt3 kinase inhibitors, CSF-1R kinase inhibitors, IGF receptor (insulin-like growth factor) kinase inhibitors; aurora kinase inhibitors and cyclin dependent kinase inhibitors such as CDK2 and/or CDK4 inhibitors; (v) antiangiogenic agents such as, but not limited to, those which inhibit the effects of vascular endothelial growth factor, [for example the anti-vascular endothelial cell growth factor antibody bevacizumab (Avastin™) and for example, a VEGF receptor tyrosine kinase inhibitor such as vandetanib (ZD6474), vatalanib (PTK787), sunitinib (SU11248), axitinib (AG-013736) and pazopanib (GW 786034); (vi) vascular damaging agents such as, but not limited to, Combretastatin A4 and compounds disclosed in International Patent Applications WO 99/02166, WO 00/40529, WO 00/41669, WO 01/92224, WO 02/04434 and WO 02/08213; (vii) an endothelin receptor antagonist, for example zibotentan (ZD4054) or atrasentan; (viii) antisense therapies, such as, but not limited to, those directed to targets listed above, such as ISIS 2503, an anti-ras antisense; (ix) immunotherapy approaches, including for example cancer vaccines, antibody, viral (oncolytic viruses) and small molecule or cell therapy approaches to increase the immunogenicity of patient tumour cells and/or facilitate a cell mediated anti-tumour response. Such therapies could include, but are not limited to, OX40 agonists, cGAS- STING agonists, ENPP1 inhibitors, CD38 inhibitors, TBK1 inhibitors, A2a receptor
antagonists, PI3 kinase inhibitors, TLR7/8 agonists, IDO inhibitors, Arginase inhibitors, BTK inhibitors and Bromodomain inhibitors; transduction with microbial vectors of cancer antigens, direct transduction of cancer antigens into antigen presenting cells, treatment with immune cells specific for cancer antigens (e.g., CAR-T), treatment with antibodies, antibody fragments and antibody drug conjugates that enable the immune system to recognise tumour cells. [00218] Such conjoint treatment may be achieved by way of the simultaneous, sequential or separate dosing of the individual components of the treatment. Such combination products employ the compounds of this invention within the dosage range described hereinbefore and the other pharmaceutically-active agent within its approved dosage range. [00219] According to this aspect of the invention there is provided a combination for use in the treatment of a cancer (for example a cancer involving a solid tumour) comprising a compound of the invention as defined hereinbefore, or a pharmaceutically acceptable salt or solvate thereof, and an anti-tumour agent. [00220] According to this aspect of the invention there is provided a combination for use in the treatment of a proliferative condition, such as cancer (for example a cancer involving a solid tumour), comprising a compound of the invention as defined hereinbefore, or a pharmaceutically acceptable salt or solvate thereof, and any one of the anti-tumour agents listed herein above. [00221] In a further aspect of the invention there is provided a compound of the invention or a pharmaceutically acceptable salt or solvate thereof, for use in the treatment of cancer in combination with another anti-tumour agent, optionally selected from one listed herein above. [00222] In a further aspect of the invention there is provided a compound of the invention or a pharmaceutically acceptable salt or solvate thereof, for use in the treatment of cancer in combination with a tyrosine kinase inhibitor, optionally selected from one listed herein above. [00223] Herein, where the term “combination” is used it is to be understood that this refers to simultaneous, separate or sequential administration. In one aspect of the invention “combination” refers to simultaneous administration. In another aspect of the invention “combination” refers to separate administration. In a further aspect of the invention “combination” refers to sequential administration. Where the administration is sequential or separate, the delay in administering the second component should not be such as to lose the beneficial effect of the combination. [00224] According to a further aspect of the invention there is provided a pharmaceutical composition which comprises a compound of the invention, or a pharmaceutically acceptable salt or solvate thereof, in combination with an anti-tumour agent (optionally selected from one listed herein above), in association with a pharmaceutically acceptable diluent or carrier.
Combination Therapy with Immune Modulating Treatments Immune checkpoint inhibitors [00225] Immune checkpoint proteins present on immune cells and/or cancer cells [e.g., CTLA4 (also known as cytotoxic T-lymphocyte-associated protein 4 and CD152), LAG3 (also known as lymphocyte-activation gene 3 and CD223), PD1 (also known as programmed cell death protein 1 and CD279), PD-L1 (also known as programmed death-ligand 1 and CD274), TIM-3 (also known as T-cell immunoglobulin mucin-3) and TIGIT (also known as T-cell Immunoreceptor with Ig and ITIM domains)] are molecular targets that have been found to play an important role in regulating anti-tumour immune responses. Inhibitors of these immune checkpoint proteins (e.g., CTLA4, LAG3, PD1, PD-L1, TIM-3 and/or TIGIT inhibitors) promote an anti-tumour immune response that can be utilised to effectively treat certain forms of cancer. Immune stimulators [00226] Monoclonal antibodies, bispecific antibodies, recombinant ligands and small molecule therapeutics that bind to stimulatory receptors on immune cells can facilitate an effective anti- tumour response. Such receptors may be involved in cell-to-cell contact for example contact between tumour cell and immune cell or between two types of immunce cells, other receptors may bind to soluble factors that stimulate an immune response. In one such embodiment antibodies, bispecifics, recombindant proteins or small molecule therapeutics can activate stimulatory receptors, including, but not limited to, 4-1BB, OX40, cGAS-STING, CD27, CD40, and DR3 that enhance anti-tumour immunity. [00227] Modulators of antigen processing may facilitate the presentation of neoantigenic peptides on the cell surface to enhance an effective anti-tumour response. In one such embodiment inhibitors of the endoplasmic reticulum aminopeptidases ERAP1 and ERAP2 may stimulate anti-tumour immunity. [00228] In one aspect, the present invention relates to a combination comprising a compound as defined herein, or a pharmaceutically acceptable salt thereof, and an immune checkpoint inhibitor or immune stimulator as defined herein, or a pharmaceutically acceptable salt thereof, for use in the treatment of a proliferative disorder. [00229] In another aspect, the present invention relates to a use of a combination comprising a compound as defined herein, or a pharmaceutically acceptable salt thereof, and an immune checkpoint inhibitor or immune stimulator as defined herein, or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treatment of a proliferative disorder.
[00230] In another aspect, the present invention relates to a method of treating a proliferative disorder in a subject in need thereof comprising administering to said subject a combination comprising a compound as defined herein, or a pharmaceutically acceptable salt thereof, and an immune checkpoint inhibitor or immune stimulator as defined herein, or a pharmaceutically acceptable salt thereof, as defined herein. [00231] In another aspect, the present invention relates to a compound as defined herein, or a pharmaceutically acceptable salt thereof, as defined herein for use in the treatment of a proliferative disorder, wherein the compound, or a pharmaceutically acceptable salt thereof, is for simultaneous, separate or sequential administeration with an immune checkpoint inhibitor, or immune stimulator, or a pharmaceutically acceptable salt thereof. [00232] In another aspect, the present invention relates to an immune checkpoint inhibitor or immune stimulator, or a pharmaceutically acceptable salt thereof, for use in the treatment of a proliferative disorder, wherein the immune checkpoint inhibitor is for simultaneous, separate or sequential administeration with a compound as defined herein, or a pharmaceutically acceptable salt thereof, as defined herein. [00233] In another aspect, the present invention relates to a use of a compound as defined herein, or a pharmaceutically acceptable salt thereof, as defined herein in the manufacture of a medicament for treating a proliferative disorder, wherein the medicament is for simultaneous, separate or sequential administeration with an immune checkpoint inhibitor or immune stimulator, or a pharmaceutically acceptable salt thereof. [00234] In another aspect, the present invention relates to a use of an immune checkpoint inhibitor or immune stimulator, or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating a proliferative disorder, wherein the medicament is for simultaneous, separate or sequential administeration with a compound as defined herein, or a pharmaceutically acceptable salt thereof. [00235] In another aspect, the present invention relates to a method of treating a proliferative disorder comprising adminstering to a subject in need thereof a therapeutically effective amount of a compound as defined herein, or a pharmaceutically acceptable salt thereof, as defined herein and an immune checkpoint inhibitor or immune stimulator as defined herein, or a pharmaceutically acceptable salt thereof, either sequentially, separately or simultaneously. [00236] Any immune checkpoint inhibitor or immune stimulator may be used in the combination therapy defined herein. [00237] In one embodiment, the immune stimulator is selected from a 4-1BB stimulator, a OX40 stimulator, a CD27 stimulator, a CD40 stimulator, and a DR3 stimulator. In another embodiment
the immune checkpoint inhibitor is selected from a PD1-inhibitor, a PD-L1 inhibitor, a LAG3 inhibitor, CTLA-4 inhibitor, a TIM-3 inhibitor and/or a TIGIT inhibitor. In a particular embodiment, the immune checkpoint inhibitor is a PD1 or PD-L1 inhibitor. [00238] PD-1 is a cell surface receptor protein present on immune cells such as T cells. PD-1 plays an important role in down-regulating the immune system and promoting self-tolerance by suppressing T cell activation. The PD-1 protein is an immune checkpoint that guards against autoimmunity through a dual mechanism of promoting apoptosis (programmed cell death) in antigen specific T cells in lymph nodes, while simultaneously reducing apoptosis in regulatory T cells (anti-inflammatory suppressive T cells). [00239] PD-1 therefore inhibits the immune system. This prevents autoimmune diseases, but it can also prevent the immune system from killing cancer cells. [00240] PD1 binds two ligands, PD-L1 and PD-L2. PD-L1 is of particular interest as it is highly expressed in several cancers and hence the role of PD1 in cancer immune evasion is well established. Monoclonal antibodies targeting PD-1 that boost the immune system are approved or are being developed for the treatment of cancer. Many tumour cells express PD-L1, an immunosuppressive PD-1 ligand; inhibition of the interaction between PD-1 and PD-L1 can enhance T-cell responses in vitro and mediate preclinical antitumour activity. This is known as immune checkpoint blockade. [00241] Examples of drugs that target PD-1 include pembrolizumab (Keytruda) and nivolumab (Opdivo). These drugs have been shown to be effective in treating several types of cancer, including melanoma of the skin, non-small cell lung cancer, kidney cancer, bladder cancer, head and neck cancers, and Hodgkin lymphoma. They are also being studied for use against many other types of cancer. Examples of drugs in development include BMS-936559 (Bristol Myers Squibb), MGA012 (MacroGenics) and MEDI-0680 (MedImmune). [00242] Examples of drugs that inhibit PD-L1 include atezolizumab (Tecentriq), avelumab (Bavencio) and durvalumab (Imfinzi). These drugs have also been shown to be helpful in treating different types of cancer, including bladder cancer, non-small cell lung cancer, and Merkel cell skin cancer (Merkel cell carcinoma). They are also being studied for use against other types of cancer. [00243] Examples of LAG3 inhibitors include BMS-986016/Relatlimab, TSR-033, REGN3767, MGD013 (bispecific DART binding PD-1 and LAG-3), GSK2831781 and LAG525. [00244] Examples of CTLA-4 inhibitors include MDX-010/Ipilimumab, AGEN1884, and CP- 675,206/Tremelimumab.
[00245] Examples of TIM-3 inhibitors include MBG453 (Novartis), TSR-022 (Tesaro), and LY3321367 (Lilly). [00246] Examples of TIGIT inhibitors include Tiragolumab (MTIG7192A; RG6058; Genentech/Roche), AB154 (Arcus Bioscience), MK-7684 (Merck), BMS-986207 (Bristol-Myers Squibb), ASP8374 (Astellas Pharma; Potenza Therapeutics). [00247] In one embodiment, the immune checkpoint inhibitor is selected from BMS- 986016/Relatlimab, TSR-033, REGN3767, MGD013 (bispecific DART binding PD-1 and LAG-3), GSK2831781, LAG525, MDX-010/Ipilimumab, AGEN1884, and CP-675,206/Tremelimumab, pembrolizumab, nivolumab, atezolizumab, avelumab, durvalumab, MBG453, TSR-022, LY3321367, Tiragolumab (MTIG7192A; RG6058), AB154, MK-7684, BMS-986207, and/or ASP8374 or a pharmaceutically acceptable salt or solvate thereof. Combination therapy with DNA damage response modulators [00248] The compounds of the present invention are particularly suited to use in combination with agents that act as DNA damage response modulators, e.g., PARP inhibitors, ATM inhibitors and ATR inhibitors. [00249] In one aspect, the present invention relates to a combination comprising a compound as defined herein, or a pharmaceutically acceptable salt thereof, and a DNA damage response modulator (e.g., a PARP inhibitor, an ATM inhibitor and/or an ATR inhibitor), or a pharmaceutically acceptable salt thereof, for use in the treatment of a proliferative disorder. [00250] In another aspect, the present invention relates to a use of a combination comprising a compound as defined herein, or a pharmaceutically acceptable salt thereof, and a DNA damage response modulator (e.g., a PARP inhibitor, an ATM inhibitor and/or an ATR inhibitor), or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating of a proliferative disorder. [00251] In another aspect, the present invention relates to a method of treating of a proliferative disorder in a subject in need thereof comprising administering to said subject a combination comprising a compound as defined herein, or a pharmaceutically acceptable salt thereof, and a DNA damage response modulator (e.g., a PARP inhibitor, an ATM inhibitor and/or an ATR inhibitor), or a pharmaceutically acceptable salt thereof, as defined herein. [00252] In another aspect, the present invention relates to a compound as defined herein, or a pharmaceutically acceptable salt thereof, as defined herein for use in the treatment of a proliferative disorder, wherein the compound, or a pharmaceutically acceptable salt thereof, is for simultaneous, separate or sequential administeration with a DNA damage response
modulator (e.g., a PARP inhibitor, an ATM inhibitor and/or an ATR inhibitor), or a pharmaceutically acceptable salt thereof. [00253] In another aspect, the present invention relates to a use of a compound as defined herein, or a pharmaceutically acceptable salt thereof, as defined herein in the manufacture of a medicament for treating a proliferative disorder, wherein the medicament is for simultaneous, separate or sequential administeration with a DNA damage response modulator (e.g., a PARP inhibitor, an ATM inhibitor and/or an ATR inhibitor), or a pharmaceutically acceptable salt thereof. [00254] In another aspect, the present invention relates to a method of treating a proliferative disorder comprising adminstering to a subject in need thereof a therapetuically effective amount of a compound as defined herein, or a pharmaceutically acceptable salt thereof, as defined herein and a DNA damage response modulator (e.g., a PARP inhibitor, an ATM inhibitor and/or an ATR inhibitor), or a pharmaceutically acceptable salt thereof, either sequentially, separately or simultaneously. [00255] Any DNA damage response modulator (e.g., a PARP inhibitor, an ATM inhibitor and/or an ATR inhibitor) may be used in the combination therapy defined herein. EXAMPLES [00256] While specific embodiments of the invention have been described herein for the purpose of reference and illustration, various modifications will be apparent to a person skilled in the art without departing from the scope of the invention as defined by the appended claims. Abbreviations ACN Acetonitrile Boc tert-Butyloxycarbonyl Bs Broad singlet CPME Cycopentyl methyl ether DAST Diethylaminosulfur trifluoride DCM Dichloromethane Dikis Bis(triphenylphosphine)palladium(II) dichloride DIPEA N,N-Diisopropylethylamine, Hünig’s base DMA N,N-Dimethylacetamide DMAP 4-(Dimethylamino)pyridine DME Dimethyl ether DMF N,N-Dimethylformamide
DMSO Dimethylsulfoxide. EDC 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide EtOAc Ethyl acetate Et
2O Diethyl ether FA Formic acid h Hours HATU N-[(Dimethylamino)-1H-1,2,3-triazolo-[4,5-b]pyridin-1-ylmethylene]-N- methylmethanaminium hexafluorophosphate N-oxide HOBT N-Hydroxybenzotriazole HPLC High Pressure Liquid Chromatography. IPA Isopropyl alcohol LAH Lithium aluminium hydride LCMS Liquid Chromatography-Mass Spectrometry MeOH Methanol MI Molecular Ion min(s) Minutes MW Microwave NCS N-Chlorosuccinimide NMM N-Methylmorpholine NMR Nuclear Magnetic Resonance. PdCl2(PPh3)2 Bis(triphenylphosphine)palladium chloride Pd(dppf)2Cl2 [1,1'-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) (Pd(dba)2) Bis(dibenzylideneacetone)palladium RT Retention time or room temperature SFC Supercritical fluid chromatography STAB Sodium triacetoxyborohydride TFAA Trifluoroacetic anhydride TFA Trifluoroacetic acid THF Tetrahydrofuran Analytical Methods [00257] Commercially available starting materials, reagents and dry solvents were used as supplied. Flash chromatography or glass column chromatography was performed using Merck silica gel 230-400 mesh size. Flash chromatography was also performed on combi-flash RF Teledyne Isco machine. Preparative TLC was performed on Merck plates.
Liquid Chromatography-Mass Spectrometry Methods Method A [00258] Waters Acquity UPLC with binary solvent manager, PDA detector and Acquity QDA performance mass detector, column: X-Bridge BEH C18, 50 x 2.1 mm, 2.5 micron, column temperature: 35°C, auto sampler temperature: 5°C, mobile phase A : 0.1% (v/v) formic acid in water (pH = 2.70), Mobile Phase B : 0.1% formic acid (v/v) in water : acetonitrile (10:90), mobile phase gradient details: t = 0 min (97% A, 3% B) flow : 0.8 mL/min; t = 0.75 min (97% A, 3% B) flow : 0.8 mL/min; gradient to t = 2.7 min (2% A, 98% B) flow : 0.8 mL/min; gradient to t = 3 min (0% A, 100% B) flow : 1mL/min; t = 3.5 min (0% A, 100% B) flow : 1 mL/min; gradient to t = 3.51 min (97% A, 3% B) flow : 0.8 mL/min; end of run at t = 4 min (97% A, 3% B), Flow rate: 0.8 mL/min, analysis time 4 min. Mass detector parameter: ionization mode was cycled through positive and negative modes with cone voltage 10 V and 30 V and 0.8 kV capillary voltage, temperature of source and probe were 120°C and 600°C respectively. Method B [00259] Waters Acquity with PDA detector and SQ Detector, column: X-Bridge BEH C18, 50 x 2.1 mm, 2.5 micron, column temperature: 35°C, auto sampler temperature: 5°C, mobile phase A: 5 mM ammonium bicarbonate in water (pH = 7.35), mobile phase B: acetonitrile; mobile phase gradient details: t = 0 min (97% A, 3% B) flow : 0.5 mL/min; t = 0.2 min (97% A, 3% B) flow : 0.5 mL/min; gradient to t = 2.7 min (2% A, 98% B) flow: 0.5 mL/min; gradient to t = 3 min (0% A, 100% B) flow : 0.7mL/min; t = 3.5 min (0% A, 100% B) flow : 0.7 mL/min; gradient to t = 3.51 min (97% A, 3% B) flow : 0.5 mL/min; end of run at t = 4 min (97% A, 3% B), flow rate: 0.5 mL/min, analysis time 4 min. Mass detection parameter: ionization mode was cycled through positive and negative mode with cone voltage 10 V and 30 V and 3.25 kV capillary voltage, temperature of source and probe were 120°C and 400 °C respectively. Method C [00260] Waters Acquity UPLC with binary solvent manager, PDA detector and Acquity QDA performance mass detector, column: YMC Tri-art C18, 50 x 2 mm, 1.9 micron, column temperature: 35°C, auto sampler temperature: 5°C, mobile phase A : 0.1% (v/v) formic acid in water (pH = 2.70), Mobile Phase B : 0.1% formic acid (v/v) in water : acetonitrile (10:90), mobile phase gradient details: t = 0 min (97% A, 3% B) flow : 0.8 mL/min; t = 0.75 min (97% A, 3% B) flow : 0.8 mL/min; gradient to t = 2.7 min (2% A, 98% B) flow : 0.8 mL/min; gradient to t = 3 min (0% A, 100% B) flow : 1mL/min; t = 3.5 min (0% A, 100% B) flow : 1 mL/min; gradient to t = 3.51
min (97% A, 3% B) flow : 0.8 mL/min; end of run at t = 4 min (97% A, 3% B), Flow rate: 0.8 mL/min, analysis time 4 min. Mass detector parameter: ionization mode was cycled through positive and negative modes with cone voltage 10 V and 30 V and 0.8 kV capillary voltage, temperature of source and probe were 120°C and 600°C respectively. Method D [00261] Waters Acquity UPLC with quaternary solvent manager, with PDA detector and SQ detector, column: X-Bridge BEH C18, 50*2.1 mm,2.5 micron, column temperature: 35°C, auto sampler temperature: 5°C, mobile phase A : 0.1% (v/v) Formic acid in water (pH = 2.70), mobile phase B : 0.1% (v/v) formic acid in water : acetonitrile (10:90), mobile phase gradient details: t = 0 min (97% A, 3% B) flow : 0.8 mL/min; t = 0.75 min (97% A, 3% B) flow : 0.8 mL/min; gradient to t = 2.7 min (2% A, 98% B) flow : 0.8 mL/min; gradient to t = 3 min (0% A, 100% B) flow : 1mL/min; t = 3.5 min (0% A, 100% B) flow : 1 mL/min; gradient to t = 3.51 min (97% A, 3% B) flow : 0.8 mL/min; end of run at t = 4 min (97% A, 3% B), Flow rate: 0.8 mL/min, analysis time 4 min. Mass detector parameter: ESI capillary probe, ionization mode cycled through positive and negative modes with cone voltage 10 V and 30V and 0.8 kV capillary voltage, temperature of source and probe were 120°C and 400°C respectively. Method E [00262] Waters Acquity UPLC with binary solvent manager, PDA detector and Acquity QDA performance mass detector, column: Welch Xtimate C18, 50*2.1 mm, 1.8 micron, column temperature: 35°C, auto sampler temperature: 5°C, mobile phase A : 0.1% (v/v) formic acid in water (pH = 2.70), Mobile Phase B : 0.1% formic acid (v/v) in water : acetonitrile (10:90), mobile phase gradient details: t = 0 min (97% A, 3% B) flow : 0.8 mL/min; t = 0.75 min (97% A, 3% B) flow : 0.8 mL/min; gradient to t = 2.7 min (2% A, 98% B) flow : 0.8 mL/min; gradient to t = 3 min (0% A, 100% B) flow : 1mL/min; t = 3.5 min (0% A, 100% B) flow : 1 mL/min; gradient to t = 3.51 min (97% A, 3% B) flow : 0.8 mL/min; end of run at t = 4 min (97% A, 3% B), Flow rate: 0.8 mL/min, analysis time 4 min. Mass detector parameter: ionization mode was cycled through positive and negative modes with cone voltage 10 V and 30 V and 0.8 kV capillary voltage, temperature of source and probe were 120°C and 600°C respectively. Method F [00263] Waters Acquity UPLC with PDA detector and SQ Detector, column: Welch-Xtimate,C18 4.6*50mm,5 micron, column temperature: 35°C, auto sampler temperature: 5°C, mobile phase
A: 5 mM ammonium bicarbonate in water (pH = 7.35), mobile phase B: acetonitrile; mobile phase gradient details: t = 0 min (97% A, 3% B) flow : 0.5 mL/min; t = 0.2 min (97% A, 3% B) flow : 0.5 mL/min; gradient to t = 2.7 min (2% A, 98% B) flow: 0.5 mL/min; gradient to t = 3 min (0% A, 100% B) flow : 0.7mL/min; t = 3.5 min (0% A, 100% B) flow : 0.7 mL/min; gradient to t = 3.51 min (97% A, 3% B) flow : 0.5 mL/min; end of run at t = 4 min (97% A, 3% B), flow rate: 0.5 mL/min, analysis time 4 min. Mass detection parameter: ionization mode was cycled through positive and negative mode with cone voltage 10 V and 30 V and 3.25 kV capillary voltage, temperature of source and probe were 120°C and 400 °C respectively. Method G [00264] Waters Acquity UPLC with binary solvent manager, PDA detector and Acquity QDA performance mass detector, column: X-Bridge C18 2.1*50mm3.5 micron, column temperature: 35°C, auto sampler temperature: 5°C, mobile phase A: 5 mM ammonium bicarbonate in water (pH = 7.35), mobile phase B: acetonitrile; mobile phase gradient details: t = 0 min (97% A, 3% B) flow : 0.5 mL/min; t = 0.2 min (97% A, 3% B) flow : 0.5 mL/min; gradient to t = 2.7 min (2% A, 98% B) flow: 0.5 mL/min; gradient to t = 3 min (0% A, 100% B) flow : 0.7mL/min; t = 3.5 min (0% A, 100% B) flow : 0.7 mL/min; gradient to t = 3.51 min (97% A, 3% B) flow : 0.5 mL/min; end of run at t = 4 min (97% A, 3% B), flow rate: 0.5 mL/min, analysis time 4 min. Mass detector parameter: ionization mode was cycled through positive and negative modes with cone voltage 10 V and 30 V and 0.8 kV capillary voltage, temperature of source and probe were 120°C and 600°C respectively. Method H [00265] Waters 996 Photodiode Array Detector equipped with Waters Micromass ZQ detector, column: XTIMATE C185µm 4.6*150mm, Column temperature: 35°C, Auto sampler temperature: 15°C, Mobile Phase A: 5mM Ammonium Acetate and 0.1 % Formic acid (pH =3.50) in Milli Q water, Mobile Phase B: Methanol. Mobile phase gradient details: T = 0 min (90% A, 10% B); T = 7.0min (10% A, 90% B); gradient to T = 9.0 min (0% A, 100% B); gradient to T = 14.00 min (0% A, 100% B); T = 14.01 min (90% A, 10% B); end of run at T = 17 min (90% A, 10% B), Flow rate:- 1.0 mL/min, Run Time:- 17 min, UV Detection Method:- PDA. Mass parameter: Probe: ESI, Mode of Ionisation: Positive and Negative, Cone voltage :-30 and 10 V, capillary voltage:- 3.0 KV, Extractor Voltage:-2 V, Rf Lens:- 0.1 V, Temperature of source:-120°C,Temperature of Probe:- 400 °C,Cone Gas Flow:- 100 L/Hr, Desolvation Gas flow:-800 L/Hr.
Method I [00266] Waters Acquity UPLC- H Class equipped with PDA and attached with QDa detector, column: X-Bridge BEH C18, 50 x 2.1 mm, 2.5 micron, Column temperature: 30°C, Auto sampler temperature: 15°C,Mobile Phase A : 0.1 % Formic acid in Milli Q water (pH= 2.70), Mobile Phase B : 0.1%Formic acid in Milli Q water : Acetonitrile (10:90), Mobile phase gradient details: T = 0 min (97% A, 3% B) flow : 0.8 mL/min; T = 0.75 min (97% A, 3% B) flow : 0.8 mL/min; gradient to T = 2.7 min (2% A, 98% B) flow : 0.8 mL/min; gradient to T = 3 min (0% A, 100% B) flow : 1mL/min; T = 3.5 min (0% A, 100% B) flow : 1 mL/min; gradient to T= 3.51 min (97% A, 3% B) flow : 0.8 mL/min; end of run at T = 4 min (97% A, 3% B), Flow rate: 0.8 mL/min, Run Time:- 4 min. UV Detection Method: - PDA Mass parameter: Probe:-ESI, Mode of Ionisation :- positive and negative, Cone voltage :-10V and 30V, capillary voltage:- 0.8 KV, Extractor Voltage:- 1KV, Rf Lens:- 0.1,Temperature of source:-120°C,Temperature of Probe:- 600°C, Cone Gas Flow:- Default , Desolvation Gas flow:-Default. Method J [00267] Waters 996 Photodiode Array Detector equipped with Waters Micromass ZQ detector, column: XTIMATE C185µm 4.6*150mm, Column temperature: 60°C, Auto sampler temperature: 15°C, Mobile Phase A: 5mM Ammonium Acetate and 0.1 % Formic acid (pH =3.50) in Milli Q water, Mobile Phase B: Methanol. Mobile phase gradient details: T = 0 min (90% A, 10% B); T = 7.0min (10% A, 90% B); gradient to T = 9.0 min (0% A, 100% B); gradient to T = 14.00 min (0% A, 100% B); T = 14.01 min (90% A, 10% B); end of run at T = 17 min (90% A, 10% B), Flow rate:- 1.0 mL/min, Run Time:- 17 min, UV Detection Method:- PDA. Mass parameter: Probe: ESI, Mode of Ionisation: Positive and Negative, Cone voltage :-30 and 10 V, capillary voltage:- 3.0 KV, Extractor Voltage:-2 V, Rf Lens:- 0.1 V, Temperature of source:-120°C,Temperature of Probe:- 400 °C,Cone Gas Flow:- 100 L/Hr, Desolvation Gas flow:-800 L/Hr. Method K [00268] Agilent 1260 Infinity-II DAD Detector equipped with MASS detector Agilent G6125C (LC/MSD), column: XTIMATE C185µm 4.6*150mm, Column temperature: 35°C, Auto sampler temperature: 15°C, Mobile Phase A: 5mM Ammonium Acetate and 0.1 % Formic acid (pH =3.50) in Milli Q water, Mobile Phase B: Methanol. Mobile phase gradient details: T = 0 min (90% A, 10% B); T = 7.0min (10% A, 90% B); gradient to T = 9.0 min (0% A, 100% B); gradient to T = 14.00 min (0% A, 100% B); T = 14.01 min (90% A, 10% B); end of run at T = 17 min (90% A, 10% B), Flow rate:- 1.0 mL/min, Run Time:- 17 min, UV Detection Method:- DAD. Mass
parameter: Probe: MMI, Mode of Ionisation: (ESI) Positive and Negative, Fragment voltage :-30 and 70 V, capillary voltage:- 3000 V, Gas temperature of source:-325°C,Temperature of Vaporizer:- 225 °C, Gas Flow:- 12 L/min, Nebulizer:- 50 Method L [00269] Agilent 1260 Infinity-II DAD Detector equipped with MASS detector Agilent G6125C (LC/MSD), column: XTIMATE C185µm 4.6*150mm, Column temperature: 60°C, Auto sampler temperature: 15°C, Mobile Phase A: 5mM Ammonium Acetate and 0.1 % Formic acid (pH =3.50) in Milli Q water, Mobile Phase B: Methanol. Mobile phase gradient details: T = 0 min (90% A, 10% B); T = 7.0min (10% A, 90% B); gradient to T = 9.0 min (0% A, 100% B); gradient to T = 14.00 min (0% A, 100% B); T = 14.01 min (90% A, 10% B); end of run at T = 17 min (90% A, 10% B), Flow rate:- 1.0 mL/min, Run Time:- 17 min, UV Detection Method:- DAD. Mass parameter: Probe: MMI, Mode of Ionisation: (ESI) Positive and Negative, Fragment voltage :-30 and 70 V, capillary voltage:- 3000 V, Gas temperature of source:-325°C,Temperature of Vaporizer:- 225 °C, Gas Flow:- 12 L/min, Nebulizer:- 50 Method M [00270] Waters 996 Photodiode Array Detector equipped with Waters Micromass ZQ detector, column: XTIMATE C185µm 4.6*150mm, Column temperature: 35°C, Auto sampler temperature: 15°C, Mobile Phase A: 0.05% Trifluoro acetic acid (pH =3.50) in Milli Q water, Mobile Phase B: Acetonitrile. Mobile phase gradient details: T = 0 min (100% A, 00% B); T = 7.0min (50% A, 50% B); gradient to T = 9.0 min (00% A, 100% B); gradient to T = 14.00 min (0% A, 100% B); T = 14.01 min (100% A, 00% B); end of run at T = 17 min (100% A, 00% B), Flow rate:- 1.0 mL/min, Run Time:- 17 min, UV Detection Method:- PDA. Mass parameter: Probe: ESI, Mode of Ionisation: Positive and Negative, Cone voltage :-30 and 10 V, capillary voltage:- 3.0 KV, Extractor Voltage:-2 V, Rf Lens:- 0.1 V, Temperature of source:-120°C,Temperature of Probe:- 400 °C,Cone Gas Flow:- 100 L/Hr, Desolvation Gas flow:-800 L/Hr. Method N [00271] Waters Acquity UPLC- H Class equipped with PDA and attached with QDa detector, column: X-Bridge BEH C18, 50 x 2.1 mm, 2.5 micron, Column temperature: Ambient, Auto sampler temperature: 15
0C, Mobile Phase A : 2 mM ammonium acetate followed by 0.1%Formic acid in water, Mobile Phase B : 0.1% Formic Acid in Acetonitrile, Mobile phase gradient details: T = 0 min (95% A, 5% B) flow; T = 0.4 min (95% A, 5% B) ; gradient to T = 0.8 min (65% A, 35%
B) ; gradient to T = 1.20 min (45% A, 55% B) ; T = 2.5 min (0% A, 100% B) ; gradient to T= 3.30 min (0% A, 100% B) ; gradient to T= 3.31 min to end of run at T = 4 min (95% A, 5% B), Flow rate: 0.55 mL/min, Run Time: 4 min. UV Detection Method:PDA Mass parameter: Probe:ESI, Mode of Ionisation : positive and negative, Cone voltage : 10V and 30V, capillary voltage: 0.8 KV, Extractor Voltage: 1KV, Rf Lens: 0.1,Temperature of source: 120°C,Temperature of Probe: 600°C, Cone Gas Flow:- Default , Desolvation Gas flow:-Default. Method O [00272] Waters Acquity UPLC- H Class equipped with PDA and attached with QDa detector, column: X-Bridge BEH C18, 50 x 2.1 mm, 2.5 micron, Column temperature: Ambient, Auto sampler temperature: 15
0C, Mobile Phase A : 5mM Ammonium Bicarbonate in Milli Q water, Mobile Phase B : Acetonitrile, Mobile phase gradient details: T = 0 min (95% A, 5% B) flow; T = 0.4 min (95% A, 5% B) ; gradient to T = 0.8 min (65% A, 35% B) ; gradient to T = 1.20 min (45% A, 55% B) ; T = 2.5 min (0% A, 100% B) ; gradient to T= 3.30 min (0% A, 100% B) ; gradient to T= 3.31 min to end of run at T = 4 min (95% A, 5% B), Flow rate: 0.55 mL/min, Run Time:- 4 min. UV Detection Method: PDA Mass parameter: Probe:ESI, Mode of Ionisation :positive and negative, Cone voltage : 10V and 30V, capillary voltage: 0.8 KV, Extractor Voltage: 1KV, Rf Lens: 0.1,Temperature of source: 120°C,Temperature of Probe: 600°C, Cone Gas Flow:- Default , Desolvation Gas flow:-Default. Method P [00273] Waters Acquity UPLC- H Class equipped with PDA and attached with QDa detector, column: Welch Xtimate C18, 150*4.6 mm, 5 micron, Column temperature: Ambient, Auto sampler temperature: 15
0C, Mobile Phase A : 2 mM ammonium acetate followed by 0.1%Formic acid in water, Mobile Phase B : 0.1% Formic acid in Acetonitrile, Mobile phase gradient details: T = 0 min (90% A, 10% B); T = 7.0 min (10% A, 90% B); gradient to T = 9.0 min (0% A, 100% B); gradient to T = 14.00 min (0% A, 100% B); T = 14.01 min (90% A, 10% B); end of run at T = 17 min (90% A, 10% B), Flow rate: 1.00 mL/min, Run Time: 17 min. UV Detection Method: - PDA Mass parameter: Probe: ESI, Mode of Ionisation : positive and negative, Cone voltage : 10V and 30V, capillary voltage: 0.8 KV, Extractor Voltage: 1KV, Rf Lens: 0.1,Temperature of source: 120°C,Temperature of Probe: 600°C, Cone Gas Flow:- Default , Desolvation Gas flow:-Default Method Q
[00274] Waters Acquity UPLC- H Class equipped with PDA and attached with QDa detector, column: Welch Xtimate C18, 150*4.6 mm, 5 micron, Column temperature: Ambient, Auto sampler temperature: 15
0C, Mobile Phase A : 2 mM ammonium acetate followed by 0.1%Formic acid in water, Mobile Phase B : 0.1% Formic acid in Acetonitrile, Mobile phase gradient details: T = 0 min (100% A, 0% B); T = 7.0 min (50% A, 50% B); gradient to T = 9.0 min (0% A, 100% B); gradient to T = 14.00 min (0% A, 100% B); T = 14.01 min (100% A, 0% B); end of run at T = 17 min (100% A, 0% B), Flow rate: 1.00 mL/min, Run Time: 17 min. UV Detection Method: - PDA Mass parameter: Probe: ESI, Mode of Ionisation : positive and negative, Cone voltage : 10V and 30V, capillary voltage: 0.8 KV, Extractor Voltage: 1KV, Rf Lens: 0.1,Temperature of source: 120°C,Temperature of Probe: 600°C, Cone Gas Flow:- Default , Desolvation Gas flow:-Default Method R [00275] Waters Acquity UPLC- H Class equipped with PDA and attached with QDa detector, column: Welch Xtimate C18, 150*4.6 mm, 5 micron, Column temperature: Ambient, Auto sampler temperature: 15
0C, Mobile Phase A : 2 mM ammonium acetate followed by 0.1%Formic acid in water, Mobile Phase B : 0.1% Formic acid in Acetonitrile, Mobile phase gradient details: T = 0 min (50% A, 50% B); T = 7.0 min (10% A, 90% B); gradient to T = 9.0 min (0% A, 100% B); gradient to T = 14.00 min (0% A, 100% B); T = 14.01 min (50% A, 50% B); end of run at T = 17 min (50% A, 50% B), Flow rate: 1.00 mL/min, Run Time: 17 min. UV Detection Method: - PDA Mass parameter: Probe: ESI, Mode of Ionisation : positive and negative, Cone voltage : 10V and 30V, capillary voltage: 0.8 KV, Extractor Voltage: 1KV, Rf Lens: 0.1,Temperature of source: 120°C,Temperature of Probe: 600°C, Cone Gas Flow:- Default , Desolvation Gas flow:-Default. Method S [00276] Waters Acquity UPLC- H Class equipped with PDA and attached with QDa detector, column: Welch Xtimate C18, 150*4.6 mm, 5 micron, Column temperature: Ambient, Auto sampler temperature: 15
0C, Mobile Phase A : 0.05% Trifluoro Acetic Acid in Milli Q water, Mobile Phase B: Acetonitrile, Mobile phase gradient details: T = 0 min (100% A, 0% B); T = 7.0 min (50% A, 50% B); gradient to T = 9.0 min (0% A, 100% B); gradient to T = 14.00 min (0% A, 100% B); T = 14.01 min (100% A, 0% B); end of run at T = 17 min (100% A, 0% B), Flow rate: 1.00 mL/min, Run Time: 17 min. UV Detection Method: - PDA Mass parameter: Probe: ESI, Mode of Ionisation : positive and negative, Cone voltage : 10V and 30V, capillary voltage: 0.8 KV, Extractor Voltage: 1KV, Rf Lens: 0.1,Temperature of source: 120°C,Temperature of Probe: 600°C, Cone Gas Flow:- Default , Desolvation Gas flow:-Default.
Method T [00277] Waters Acquity UPLC with binary solvent manager, PDA detector and Acquity QDa detector, column: X-Bridge BEH C18, 50 x 2.1 mm, 2.5 micron, Column temperature: Ambient, Auto sampler temperature: 15
0C, Mobile Phase A : 2 mM ammonium acetate followed by 0.1%Formic acid in water, Mobile Phase B : 0.1% Formic Acid in Acetonitrile, Mobile phase gradient details: T = 0 min (95% A, 5% B) flow; T = 0.4 min (95% A, 5% B) ; gradient to T = 0.8 min (65% A, 35% B) ; gradient to T = 1.20 min (45% A, 55% B) ; T = 2.5 min (0% A, 100% B) ; gradient to T= 3.30 min (0% A, 100% B) ; gradient to T= 3.31 min to end of run at T = 4 min (95% A, 5% B), Flow rate: 0.55 mL/min, Run Time: 4 min. UV Detection Method:PDA Mass parameter: Probe:ESI, Mode of Ionisation : positive and negative, Cone voltage : 10V and 30V, capillary voltage: 0.8 KV, Extractor Voltage: 1KV, Rf Lens: 0.1,Temperature of source: 120°C,Temperature of Probe: 600°C, Cone Gas Flow:- Default , Desolvation Gas flow:-Default. Method U [00278] Waters Acquity UPLC- H Class equipped with PDA and attached with QDa detector, column: Welch Xtimate C18, 150*4.6 mm, 5 micron, Column temperature: 80
0C Auto sampler temperature: 15
0C, Phase A : 5mM Ammonium Bicarbonate in Milli Q water, Mobile Phase B : Acetonitrile,, Mobile phase gradient details: T = 0 min (90% A, 10% B); T = 7.0 min (10% A, 90% B); gradient to T = 9.0 min (0% A, 100% B); gradient to T = 14.00 min (0% A, 100% B); T = 14.01 min (90% A, 10% B); end of run at T = 17 min (90% A, 10% B), Flow rate: 1.00 mL/min, Run Time: 17 min. UV Detection Method: - PDA Mass parameter: Probe: ESI, Mode of Ionisation : positive and negative, Cone voltage : 10V and 30V, capillary voltage: 0.8 KV, Extractor Voltage: 1KV, Rf Lens: 0.1,Temperature of source: 120°C,Temperature of Probe: 600°C, Cone Gas Flow:- Default , Desolvation Gas flow:-Default. Method V [00279] Agilent 1260 Infinity-II DAD Detector equipped with MASS detector Agilent G6125C (LC/MSD), column: XTIMATE C185µm 4.6*150mm, Column temperature: 60°C, Auto sampler temperature: 15°C, Mobile Phase A : 5mM Ammonium Bicarbonate in Milli Q water, Mobile Phase B : Acetonitrile,. Mobile phase gradient details: T = 0 min (90% A, 10% B); T = 7.0min (10% A, 90% B); gradient to T = 9.0 min (0% A, 100% B); gradient to T = 14.00 min (0% A, 100% B); T = 14.01 min (90% A, 10% B); end of run at T = 17 min (90% A, 10% B), Flow rate:- 1.0 mL/min, Run Time:- 17 min, UV Detection Method:- DAD. Mass parameter: Probe: MMI, Mode of Ionisation: (ESI) Positive and Negative, Fragment voltage :-30 and 70 V, capillary voltage:-
3000 V, Gas temperature of source:-325°C,Temperature of Vaporizer:- 225 °C, Gas Flow:- 12 L/min, Nebulizer:- 50 Method W [00280] Waters Acquity UPLC- H Class equipped with PDA and attached with QDa detector, column: Welch Xtimate C18, 150*4.6 mm, 5 micron, Column temperature: Ambient Auto sampler temperature: 15
0C, Phase A : 5mM Ammonium Bicarbonate in Milli Q water, Mobile Phase B : Acetonitrile,, Mobile phase gradient details: T = 0 min (90% A, 10% B); T = 7.0 min (10% A, 90% B); gradient to T = 9.0 min (0% A, 100% B); gradient to T = 14.00 min (0% A, 100% B); T = 14.01 min (90% A, 10% B); end of run at T = 17 min (90% A, 10% B), Flow rate: 1.00 mL/min, Run Time: 17 min. UV Detection Method: - PDA Mass parameter: Probe: ESI, Mode of Ionisation : positive and negative, Cone voltage : 10V and 30V, capillary voltage: 0.8 KV, Extractor Voltage: 1KV, Rf Lens: 0.1,Temperature of source: 120°C,Temperature of Probe: 600°C, Cone Gas Flow:- Default , Desolvation Gas flow:-Default. Method X [00281] Waters Acquity UPLC- H Class equipped with PDA and attached with QDa detector, column: Welch Xtimate C18, 150*4.6 mm, 5 micron, Column temperature: Ambient, Auto sampler temperature: 15
0C, Mobile Phase A : 0.05% Trifluoro Acetic Acid in Milli Q water, Mobile Phase B: Acetonitrile, Mobile phase gradient details: T = 0 min (90% A, 10% B); T = 7.0 min (10% A, 90% B); gradient to T = 9.0 min (0% A, 100% B); gradient to T = 14.00 min (0% A, 100% B); T = 14.01 min (90% A, 10% B); end of run at T = 17 min (90% A, 10% B), Flow rate: 1.00 mL/min, Run Time: 17 min. Flow rate: 1.00 mL/min, Run Time: 17 min. UV Detection Method: - PDA Mass parameter: Probe: ESI, Mode of Ionisation : positive and negative, Cone voltage : 10V and 30V, capillary voltage: 0.8 KV, Extractor Voltage: 1KV, Rf Lens: 0.1,Temperature of source: 120°C,Temperature of Probe: 600°C, Cone Gas Flow:- Default , Desolvation Gas flow:-Default. Method Y [00282] Waters 996 Photodiode Array Detector equipped with Waters Micromass ZQ detector, column: XTIMATE C185µm 4.6*150mm, Column temperature: 35°C, Auto sampler temperature: 15°C, Mobile Phase A: 0.05% Trifluoro acetic acid (pH =3.50) in Milli Q water, Mobile Phase B: Acetonitrile. Mobile phase gradient details: T = 0 min (90% A, 10% B); T = 7.0 min (10% A, 90% B); gradient to T = 9.0 min (0% A, 100% B); gradient to T = 14.00 min (0% A, 100% B); T = 14.01 min (90% A, 10% B); end of run at T = 17 min (90% A, 10% B). Flow rate:- 1.0 mL/min, Run
Time:- 17 min, UV Detection Method:- PDA. Mass parameter: Probe: ESI, Mode of Ionisation: Positive and Negative, Cone voltage :-30 and 10 V, capillary voltage:- 3.0 KV, Extractor Voltage:- 2 V, Rf Lens:- 0.1 V, Temperature of source:-120°C,Temperature of Probe:- 400 °C,Cone Gas Flow:- 100 L/Hr, Desolvation Gas flow:-800 L/Hr. Method Z [00283] Waters Acquity UPLC- H Class equipped with PDA and attached with SQd detector, column: Welch Xtimate C18, 150*4.6 mm, 5 micron, Column temperature: Ambient, Auto sampler temperature: 15
0C, Mobile Phase A : 2 mM ammonium acetate followed by 0.1%Formic acid in water, Mobile Phase B : 0.1% Formic acid in Acetonitrile, Mobile phase gradient details: T = 0 min (90% A, 10% B); T = 7.0 min (10% A, 90% B); gradient to T = 9.0 min (0% A, 100% B); gradient to T = 14.00 min (0% A, 100% B); T = 14.01 min (90% A, 10% B); end of run at T = 17 min (90% A, 10% B), Flow rate: 1.00 mL/min, Run Time: 17 min. UV Detection Method: - PDA Mass parameter: Probe: ESI, Mode of Ionisation : positive and negative, Cone voltage : 10V and 30V, capillary voltage: 3.0 KV, Extractor Voltage: 1KV, Rf Lens: 0.1,Temperature of source: 120°C,Temperature of Probe: 600°C, Cone Gas Flow:- Default , Desolvation Gas flow:-Default Method A1 [00284] Waters Acquity UPLC- H Class equipped with PDA and attached with SQd detector, column: Welch Xtimate C18, 150*4.6 mm, 5 micron, Column temperature: Ambient, Auto sampler temperature: 15
0C, Mobile Phase A : 2 mM ammonium acetate followed by 0.1%Formic acid in water, Mobile Phase B : 0.1% Formic acid in Acetonitrile, Mobile phase gradient details: T = 0 min (100% A, 0% B); T = 7.0 min (50% A, 50% B); gradient to T = 9.0 min (0% A, 100% B); gradient to T = 14.00 min (0% A, 100% B); T = 14.01 min (100% A, 0% B); end of run at T = 17 min (100% A, 0% B), Flow rate: 1.00 mL/min, Run Time: 17 min. UV Detection Method: - PDA Mass parameter: Probe: ESI, Mode of Ionisation : positive and negative, Cone voltage : 10V and 30V, capillary voltage: 3.0 KV, Extractor Voltage: 1KV, Rf Lens: 0.1,Temperature of source: 120°C,Temperature of Probe: 600°C, Cone Gas Flow:- Default , Desolvation Gas flow:-Default Method A2 [00285] Waters Acquity UPLC equipped with PDA and attached with SQd detector, column: Welch Xtimate C18, 150*4.6 mm, 5 micron, Column temperature: Ambient, Auto sampler temperature: 15
0C, Mobile Phase A: 0.05% Trifluoro acetic acid (pH =3.50) in Milli Q water, Mobile Phase B: Acetonitrile. Mobile phase gradient details: T = 0 min (100% A, 0% B); T = 7.0
min (50% A, 50% B); gradient to T = 9.0 min (0% A, 100% B); gradient to T = 14.00 min (0% A, 100% B); T = 14.01 min (100% A, 0% B); end of run at T = 17 min (100% A, 0% B), Flow rate: 1.00 mL/min, Run Time: 17 min. UV Detection Method: - PDA Mass parameter: Probe: ESI, Mode of Ionisation : positive and negative, Cone voltage : 10V and 30V, capillary voltage: 3.0 KV, Extractor Voltage: 1KV, Rf Lens: 0.1,Temperature of source: 120°C,Temperature of Probe: 600°C, Cone Gas Flow:- Default , Desolvation Gas flow:-Default Method A3 [00286] Waters Acquity UPLC- H Class equipped with PDA and attached with QDa detector, column: Welch Xtimate C18, 150*4.6 mm, 5 micron, Column temperature: Ambient, Auto sampler temperature: 15
0C, Mobile Phase: 5mM ammonium bicarbonate in water (pH = 7.35), mobile phase B: Acetonitrile. Mobile phase gradient details: T = 0 min (100% A, 0% B); T = 7.0 min (50% A, 50% B); gradient to T = 9.0 min (0% A, 100% B); gradient to T = 14.00 min (0% A, 100% B); T = 14.01 min (100% A, 0% B); end of run at T = 17 min (100% A, 0% B), Flow rate: 1.00 mL/min, Run Time: 17 min. UV Detection Method: - PDA Mass parameter: Probe: ESI, Mode of Ionisation : positive and negative, Cone voltage : 10V and 30V, capillary voltage: 0.8 KV, Extractor Voltage: 1KV, Rf Lens: 0.1,Temperature of source: 120°C,Temperature of Probe: 600°C, Cone Gas Flow:- Default , Desolvation Gas flow:-Default. Method A4 [00287] Waters Acquity UPLC- H Class equipped with PDA and attached with SQd detector, column: X-Bridge BEH C18, 50 x 2.1 mm, 2.5 micron, Column temperature: Ambient, Auto sampler temperature: 15
0C, Mobile Phase A : 2 mM ammonium acetate followed by 0.1%Formic acid in water, Mobile Phase B : 0.1% Formic Acid in Acetonitrile, Mobile phase gradient details: T = 0 min (95% A, 5% B) flow; T = 0.4 min (95% A, 5% B) ; gradient to T = 0.8 min (65% A, 35% B) ; gradient to T = 1.20 min (45% A, 55% B) ; T = 2.5 min (0% A, 100% B) ; gradient to T= 3.30 min (0% A, 100% B) ; gradient to T= 3.31 min to end of run at T = 4 min (95% A, 5% B), Flow rate: 0.55 mL/min, Run Time: 4 min. UV Detection Method: - PDA Mass parameter: Probe: ESI, Mode of Ionisation : positive and negative, Cone voltage : 10V and 30V, capillary voltage: 3.0 KV, Extractor Voltage: 1KV, Rf Lens: 0.1,Temperature of source: 120°C,Temperature of Probe: 600°C, Cone Gas Flow:- Default , Desolvation Gas flow:-Default Method A5
[00288] Waters Acquity UPLC equipped with PDA and attached with SQd detector,column: X- Bridge BEH C18, 50 x 2.1 mm, 2.5 micron, Column temperature: Ambient, Auto sampler temperature: 150C,Mobile Phase A : 5mM ammonium bicarbonate in water (pH = 8.0), Mobile Phase B : Acetonitrile. Mobile phase gradient details: T = 0 min (95% A, 5% B) flow; T = 0.4 min (95% A, 5% B) ; gradient to T = 0.8 min (65% A, 35% B) ; gradient to T = 1.20 min (45% A, 55% B) ; T = 2.5 min (0% A, 100% B) ; gradient to T= 3.30 min (0% A, 100% B) ; gradient to T= 3.31 min to end of run at T = 4 min (95% A, 5% B), Flow rate: 0.55 mL/min, Run Time: 4 min.UV Detection Method: - PDA Mass parameter: Probe: ESI, Mode of Ionisation : positive and negative, Cone voltage : 10V and 30V, capillary voltage: 3.0 KV, Extractor Voltage: 1KV, Rf Lens: 0.1,Temperature of source: 120°C,Temperature of Probe: 600°C, Cone Gas Flow:- Default , Desolvation Gas flow:-Default LC-Method 1 [00289] UPLC-MS was performed on a Waters DAD + Waters SQD2, single quadrupole UPLC- MS spectrometer using an Acquity UPLC HSS Shield RP181.7 µm 100 x 2.1 mm (Plus guard cartridge), maintained at 40 ˚C column being initially held at 5% Acetonitrile (Far UV grade) with 0.1% (V/V) formic acid/water (High purity via PureLab Option unit) with 0.1% formic acid for 0.4 minutes, followed by a linear gradient of 5-95% within 6.4 minutes and then held at 95% for 1.2 minutes (F = 0.4 mL/min). LC-Method 2 [00290] UPLC-MS was performed on a Waters DAD + Waters SQD2, single quadrupole UPLC- MS spectrometer using an Acquity UPLC BEH Shield RP181.7 µm 100 x 2.1 mm (Plus guard cartridge), maintained at 40 ˚C column being initially held at 5% acetonitrile/water (with 10 mM ammonium bicarbonate) for 0.4 minutes, followed by a linear gradient of 5-95% within 6.4 minutes and then held at 95% for 1.2 minutes (F = 0.4 mL/min). LC-Method 3 [00291] UPLC-MS was performed on a Waters DAD + Waters SQD2, single quadrupole UPLC- MS spectrometer using an Acquity UPLC BEH C181.7um 100 x 2.1mm (Plus guard cartridge), maintained at 40°C column being initially held at 5% Acetonitrile (Far UV grade) with 0.1% (V/V) formic acid / Water (High purity via PureLab Option unit) with 0.1% formic acid for 0.4 minutes, followed by a linear gradient of 5-95% within 6.4 minutes and then held at 95% for 1.2 minutes (F = 0.4 mL/min).
LC-Method 4 [00292] UPLC-MS was performed on a Waters DAD + Waters SQD2, single quadrupole UPLC- MS spectrometer using an Acquity UPLC BEH C181.7um 100 x 2.1mm (Plus guard cartridge), maintained at 40°C column being initially held at 5% acetonitrile/water (with 0.1% v/v ammonium hydroxide) for 0.4 minutes, followed by a linear gradient of 5-95% within 6.4 minutes and then held at 95% for 1.2 minutes (F = 0.4 mL/min). Analytical HPLC Methods Method B [00293] Machine Details: - Agilent 1260 Series with PDA detector, Column temperature: 25 ˚C, Auto sampler temperature: 25 ˚C, Mobile Phase A: 0.05% Trifluoro acetic acid in Milli Q water (pH= 2.1), Mobile Phase B: Acetonitrile (100%). [00294] Mobile phase gradient details: T = 0 min (90% A, 10% B); T = 7.0min (10% A, 90% B); gradient to T = 9.0 min (0% A, 100% B); gradient to T = 14.00 min (0% A, 100% B); T = 14.01 min (90% A, 10% B); end of run at T = 17 min (90% A, 10% B), Flow rate:- 1.0 mL/min, Run Time:- 17 min, UV Detection Method:- PDA. Method C [00295] Machine Details: - Water alliance e2695 with 2998 PDA detector, Column temperature: 25 ˚C, Auto sampler temperature: 25 ˚C,Mobile Phase A: 0.1% ammonium hydroxide solution in HPLC water Mobile Phase B : Acetonitrile (100%). [00296] Mobile phase gradient details: T = 0 min (90% A, 10% B); T = 7.0min (10% A, 90% B); gradient to T = 9.0 min (0% A, 100% B); gradient to T = 14.00 min (0% A, 100% B); T = 14.01 min (90% A, 10% B); end of run at T = 17 min (90% A, 10% B), Flow rate:- 1.0 mL/min, Run Time:- 17 min, UV Detection Method:- PDA. Method D [00297] Machine Details: Shimadzu i-series LC-2050C with PDA detector, Column temperature: 25 ˚C, Auto sampler temperature: 25 ˚C, Mobile Phase A: 0.05% Trifluoro acetic acid in Milli Q water (pH= 2.1), Mobile Phase B: Acetonitrile (100%). [00298] Mobile phase gradient details: T = 0 min (90% A, 10% B); T = 7.0min (10% A, 90% B); gradient to T = 9.0 min (0% A, 100% B); gradient to T = 14.00 min (0% A, 100% B); T = 14.01
min (90% A, 10% B); end of run at T = 17 min (90% A, 10% B), Flow rate:- 1.0 mL/min, Run Time:- 17 min, UV Detection Method:- PDA NMR [00299]
1H Nuclear magnetic resonance (NMR) spectroscopy was carried out using a Bruker instrument operating at 400 MHz using the stated solvent at around room temperature unless otherwise stated. In all cases, NMR data were consistent with the proposed structures. Characteristic chemical shifts (δ) are given in parts-per-million using conventional abbreviations for designation of major peaks: e.g. s, singlet; d, doublet; t, triplet; q, quartet; dd, doublet of doublets; dt, doublet of triplets; m, multiplet; br, broad. Purification methods Preparative reverse-phase HPLC conditions [00300] Preparative HPLC purification was performed by reverse phase HPLC using a Waters Fractionlynx preparative HPLC system (2525 pump, 2996/2998 UV/VIS detector, 2767 liquid handler) or an equivalent HPLC system such as a Gilson Trilution UV directed system. The Waters 2767 liquid handler acted as both auto-sampler and fraction collector. The columns used for the preparative purification of the compounds were a Waters Sunfire OBD Phenomenex Luna Phenyl Hexyl or Waters Xbridge Phenyl at 10 µm 19 × 150 mm or Waters CSH Phenyl Hexyl, 19 × 150, 5 µm column unless otherwise stated. Appropriate focused gradients were selected based on acetonitrile and methanol solvent systems under either acidic or basic conditions. The modifiers used under acidic/basic conditions were formic acid or TFA (0.1% V/V) and ammonium bicarbonate (10 mM) respectively. The purification was controlled by Waters Fractionlynx software through monitoring at 210-400 nm and triggered a threshold collection value at 260 nm and, when using the Fractionlynx, the presence of target molecular ion as observed under API conditions. Collected fractions were analysed by LCMS (Waters Acquity systems with Waters SQD). [00301] Below is a list of methods and conditions used for preparative reverse phase HPLC purifications. Preparative HPLC Conditions Method Sunfire C1819 x 150 mm, 10 µm 5-60% ACN / H2O (10 mM NH4CO3), prep-LC-1 20 mL/min, room temperature
Luna Phenyl-Hexyl 21.2 x 150 mm, 10 µm 20-80% MeOH / H2O (0.1% prep-LC-2 FA), 20 mL/min, room temperature Sunfire C1819 x 150 mm, 10 µm 5-60% ACN / H
2O (0.1% FA), 20 prep-LC-3 mL/min, room temperature Luna Phenyl-Hexyl 21.2x150 mm, 10 µm 5-60% MeOH / H2O (0.1% prep-LC-4 FA), 20 mL/min, room temperature Xbridge Phenyl 19 x 150 mm, 10 µm 20-80% MeOH / H
2O (10 mM prep-LC-5 NH
4CO
3), 20 mL/min, room temperature Sunfire C1819 x 150 mm, 10 µm 35-45% MeOH / H2O (0.1% TFA), 20 prep-LC-6 mL/min, room temperature Xbridge Phenyl 19 x 150 mm, 10 µm 5-60% MeOH / H
2O (10 mM prep-LC-7 NH
4CO
3), 20 mL/min, room temperature Sunfire C1819 x 150 mm, 10 µm 5-60% ACN / H2O (0.1% FA), 20 prep-LC-8 mL/min, room temperature Sunfire C1819 x 150 mm, 10 µm 20-80% ACN / H2O (0.1% FA), 20 Prep-LC-9 mL/min, room temperature Luna Phenyl-Hexyl 3 x 50 mm, 3 µm 5-95% MeOH / H
2O (0.1% FA), Prep-LC-10 1.7 mL/min, room temperature Sunfire C183 x 50 mm, 3 µm 5-95% ACN / H2O (10 mM NH4CO3), 1.7 Prep-LC-11 mL/min, room temperature Sunfire C1819x150 mm, 10 µm 5-60% ACN / H2O (0.1% TFA), 20 Prep-LC-12 mL/min, room temperature Luna Phenyl-Hexyl 21.2x150 mm, 10 µm 40-100% MeOH / H2O (0.1% Prep-LC-13 FA), 20 mL/min, room temperature Chiral Supercritical Fluid Chromatography (SFC) separation protocol [00302] The enantiomeric separation of compounds was achieved by Supercritical Fluid Chromatography (SFC) using a Waters Thar Prep100 preparative SFC system (P200 CO2 pump, 2545 modifier pump, 2998 UV/VIS detector, 2767 liquid handler with Stacked Injection Module). The Waters 2767 liquid handler acted as both auto-sampler and fraction collector. Appropriate isocratic methods were selected based on methanol, ethanol or isopropanol solvent systems under un-modified or basic conditions. The standard SFC method used was modifier, CO2, 100 mL/min, 120 Bar backpressure, 40 ˚C column temperature. The modifier used under basic conditions was diethylamine (0.1% V/V). The modifier used under acidic conditions was either formic acid (0.1% V/V) or TFA (0.1% V/V). The SFC purification was controlled by Waters Fractionlynx software through monitoring at 210-400 nm and triggered at a threshold collection
value, typically 260 nm. Collected fractions were analysed by SFC (Waters/Thar SFC systems with Waters SQD). The fractions that contained the desired product were concentrated by vacuum centrifugation. [00303] Below is a list of SFC methods and conditions used to resolve enantiomers or to determine enantiomeric purity. SFC method Conditions for chiral resolution WATERS VIRIDIS 2-EP 20 x 250 mm, 5 µm 15-25% MeOH (0.1% NH
4OH) / CO
2, SFC-1 100 mL/min, 120 bar, 40° C, DAD 250 nm WATERS VIRIDIS 2-EP 20 x 250 mm, 5 µm 10-20% MeOH (0.1% NH4OH) / CO2, SFC-2 100 mL/min, 120bar, 40 ˚C, DAD 250 nm WATERS TORUS DEA 20 x 150 mm, 5 µm 20-30% MeOH (0.1% NH
4OH) / CO
2, SFC-3 100 mL/min, 120 bar, 40 ˚C, DAD 270 nm Preparation of compounds Example 1: (E)-N-(2-(3-Chloro-4,6-dihydroxy-2-methylbenzoyl)isoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide Step 1: (3-Chloro-4,6-dihydroxy-2-methylphenyl)(4-(methylamino)isoindolin-2-yl)methanone

[00304] To a stirred solution of 3-chloro-4,6-dihydroxy-2-methylbenzoic acid (Intermediate B) (0.4 g, 1.97 mmol, 1 eq.) in DMF (4 mL) were added EDC.HCl (0.56 g, 2.96 mmol, 1.5 eq.) and HOAT (0.26 g, 1.97 mmol, 1 eq.) at 0 ˚ C. N-Methylisoindolin-4-amine hydrochloride (Intermediate A) (0.43 g, 2.36 mmol, 1.2 eq.) and N-methyl morpholine (0.99 g, 9.87 mmol, 5 eq.) were added and the mixture was stirred at 0˚C for 10 mins and then at room temperature for 30 mins. The resulting mixture was poured into ice cold water (50 mL) and extracted with EtOAc (3 x 30 mL). The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The crude material was purified by chromatography on silica (product eluted in 4% MeOH in DCM) to afford the title compound as an off-white solid (0.3 g, Yield: 22.8%). [00305]
1H NMR (DMSO-d6, 400 MHz): δ 2.13 (s, 3H), 2.61 - 2.72 (dd, J= 4 Hz, 3H), 4.10 - 4.47 (m, 2H), 4.56 (s, 1H), 4.70 (s, 1H), 5.34 - 5.52 (dd, J= 4 Hz, 1H, D2O exchangeable), 6.34 - 6.40 (m, 1H), 6.44 -6.48 (m, 2H), 7.06 - 7.13 (m, 1H), 9.75 (d, J= 8 Hz, 1H, D
2O exchangeable), 10.10 (d, J= 4 Hz, 1H, D
2O exchangeable).
[00306] LCMS (Method A): 1.482 min, 1.592 min, MS: ES+ 332.8 (M+1). Step 2: (E)-N-(2-(3-Chloro-4,6-dihydroxy-2-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N- methylbut-2-enamide
[00307] Performed in 2 parallel batches, each of 0.15 g scale: To a stirred solution of (E)-4- (dimethylamino)but-2-enoic acid hydrochloride (CAS: 848133-35-7) (0.15 g, 0.90 mmol, 1.0 eq.) in DMF (1.5 mL) were added (3-chloro-4,6-dihydroxy-2-methylphenyl)(4- (methylamino)isoindolin-2-yl)methanone (Step 1) (0.15 g, 0.45 mmol, 0.5 eq.) and DCC (0.28 g, 1.35 mmol, 1.5 eq.) and the reaction mixture was heated to 150 ˚C using microwave irradiation for 15 mins. The resulting mixture was diluted with water (30 mL) and extracted with EtOAc (2 x 15 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under reduced pressure. The crude material was purified by Prep. HPLC to afford the title compound (0.04 g, Yield: 10%). [00308] High temperature
1H NMR (DMSO-d6, 400 MHz, 348K): δ 2.10 - 2.15 (s, 3H), 2.63 - 2.83 (m, 6H), 3.10 (s, 3H), 3.73 - 3.77 (m, 2H), 4.26 - 4.87 (m, 4H), 5.98 - 6.10 (m, 1H), 6.44 - 6.50 (m, 1H), 6.59 - 6.74 (m, 1H), 7.22 - 7.26 (m, 1H), 7.34 - 7.47 (m, 2H), 9.87 (d, J= 12 Hz, 1H), 10.19 - 10.21 (m, 1H). [00309] LCMS (Method A): 1.085 min, MS: ES+ 443.9 (M+1). [00310] Analytical HPLC (Method D): 4.78 min Prep HPLC purification method [00311] Chromatographic separation and isolation were conducted with Shimazu NEXERA purification system with UV detector; column Shim-Pack GIST C18 (250 mm x 20 mm x 5µm); compounds were eluted with, Mobile Phase A : 0.05% TFA in water, Mobile Phase B : Acetonitrile with a gradient of T = 0 min (85% A, 15% B); gradient to T = 21.00 min (62% A, 38% B); T = 21.01 min (2% A, 98% B); gradient to T = 23.0 min (2% A, 98% B); T= 23.01 min (85% A, 15% B); gradient to T = 26.00 min (85% A, 15% B); Flow rate= 20 mL/min; analysis time 26 min.
Example 1.1: (E)-N-Benzyl-N-(2-(2,4-dihydroxybenzoyl)isoindolin-4-yl)-4- (dimethylamino)but-2-enamide Step 1: (4-(Benzylamino)isoindolin-2-yl)(2,4-dihydroxyphenyl)methanone

[00312] The title compound was prepared from N-benzylisoindolin-4-amine hydrochloride (Intermediate E) and 2,4-dihydroxybenzoic acid (CAS 89-86-1) analogously to Example 1 Step 1. [00313]
1H NMR (DMSO-d6, 400 MHz): δ 2.73 (s, 1H), 2.89 (s, 1H), 4.31 (d, J= 20 Hz, 2H), 4.70 (t, J= 12 Hz, 4H), 6.11 - 6.54 (m, 5H), 6.96 (d, J= 8 Hz, 1H), 7.20 - 7.35 (m, 5H), 9.72 (s, 1H, D2O exchangeable), 10.56 (d, J= 36 Hz, 1H, D2O exchangeable). [00314] LCMS (Method A): 2.012 min, MS: ES+ 361 (M+1) Step 2: (E)-N-Benzyl-N-(2-(2,4-dihydroxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)but-2- enamide
[00315] The title compound was prepared from (4-(benzylamino)isoindolin-2-yl)(2,4- dihydroxyphenyl)methanone (Step 1) and (E)-4-(dimethylamino)but-2-enoic acid hydrochloride (CAS: 848133-35-7) analogously to Example 1 Step 2. [00316]
1H NMR (DMSO-d6, 400 MHz): δ 1.99 (s, 6H), 2.86 (s, br, 2H), 4.07 - 4.32 (m, 1H), 4.44 - 4.57 (m, 1H), 4.79 (s, 3H), 5.01 (d, J= 12 Hz, 1H), 5.72 (t, J= 12 Hz, 1H), 6.27 - 6.32 (m, 2H), 6.71 – 6.91 (m, 3H), 6.98 (d, J= 8 Hz, 1H), 7.11 - 7.31 (m, 6H), 9.73 (s, 1H, D
2O exchangeable), 10.34 (s, 1H, D2O exchangeable). [00317] LCMS (Method A): 1.272 min, MS: ES+ 472.2 (M+1). [00318] Analytical HPLC (Method B): 4.737 min,
Example 1.2: (E)-N-Benzyl-N-(2-(2,4-dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4- (dimethylamino)but-2-enamide Step 1: (4-(Benzylamino)isoindolin-2-yl)(2,4-dihydroxy-5-methylphenyl)methanone
[00319] The title compound was prepared from N-benzylisoindolin-4-amine hydrochloride (Intermediate E) and 2,4-dihydroxy-5-methylbenzoic acid (Intermediate F) analogously to Example 1 Step 1. [00320]
1H NMR (DMSO-d6, 400 MHz): δ 2.07 (s, 3H), 4.28 - 4.33 (m, 2H), 4.65 - 4.72 (m, 4H), 6.08 - 6.27 (m, 2H), 6.40 - 6.52 (m, 2H), 6.96 (s, 1H), 7.07 (s, 1H), 7.19 (s, 1H), 7.30 - 7.36 (m, 4H), 9.64 (d, J=8 Hz, 1H, D2O exchangeable), 10.12 - 10.22 (m, 1H, D2O exchangeable). [00321] LCMS (Method A): 2.124 min, MS: ES+ 375.1 (M+1) Step 2: (E)-N-Benzyl-N-(2-(2,4-dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino) but-2-enamide
[00322] The title compound was prepared from (4-(benzylamino)isoindolin-2-yl)(2,4-dihydroxy- 5-methylphenyl)methanone (Step 1) and (E)-4-(dimethylamino)but-2-enoic acid hydrochloride (CAS: 848133-35-7) analogously to Example 1 Step 2. [00323]
1H NMR (DMSO-d6, 400 MHz): δ 2.01 (s, 6H), 2.08 (s, 3H), 2.89 (s, br, 2H), 4.06 (d, J= 16 Hz, 1H), 4.28 (d, J= 16 Hz, 1H), 4.43 - 4.56 (m, 1H), 4.78 (s, 4H), 5.01 (d, J= 16 Hz, 1H), 5.74 (d, J= 16 Hz, 1H), 6.38 (s, 1H), 6.60 - 6.79 (m, 2H), 6.90 (d, J= 8 Hz, 2H), 7.18 - 7.34 (m, 7H), 9.65 (s, 1H, D2O exchangeable), 9.89 and 10.06 (singlets, 1H, D2O exchangeable). [00324] LCMS (Method A): 1.372 min, MS: ES+ 486.2 (M+1). [00325] Analytical HPLC (Method B): 4.93 min.
Example 1.3: (E)-N-(2-(2,4-Dihydroxybenzoyl) isoindolin-4-yl)-4-(diethylamino)-N-((1- methyl-1H-pyrazol-4-yl)methyl)but-2-enamide Step 1: (2, 4-Dihydroxyphenyl) (4-(((1-methyl-1H-pyrazol-4-yl) methyl) amino) isoindolin-2-yl) methanone
[00326] The title compound was prepared from N-((1-methyl-1H-pyrazol-4-yl) methyl) isoindolin- 4-amine (Intermediate L) and 2,4-dihydroxybenzoic acid (CAS 89-86-1) analogously to Example 1 Step 1. [00327]
1H NMR (DMSO-d6, 400 MHz): δ 3.75 (s, 3H), 4.02 - 4.13 (m, 2H), 4.61 - 4.72 (m, 4H), 5.69 (s, 1H D2O exchangeable), 6.29 - 6.33 (m, 2H), 6.46 - 6.57(m, 2H), 7.04 (d, J= 6.8 Hz, 1H), 7.24 (d, J= 8.8 Hz, 1H), 7.34 (d, J= 16.8 Hz, 1H), 7.55 (d, J= 16 Hz, 1H). 9.72 (s, 1H, D2O exchangeable), 10.5 and 10.6 (2 singlets, 1H, D2O exchangeable) [00328] LCMS (Method A): 1.468 min, MS: ES+ 365 (M+1). Step 2: (E)-N-(2-(2,4-Dihydroxybenzoyl) isoindolin-4-yl)-4-(diethylamino)-N-((1-methyl-1H- pyrazol-4-yl)methyl)but-2-enamide
[00329] The title compound was prepared from (2,4-dihydroxyphenyl) (4-(((1-methyl-1H-pyrazol- 4-yl) methyl) amino) isoindolin-2-yl) methanone (Step 1) and (E)-4-(dimethylamine) but-2-enoic acid hydrochloride (CAS: 848133-35-7) analogously to Example 1 Step 2. [00330]
1H NMR (DMSO-d6, 400 MHz, D2O exchange) δ 2.06 (s, 6H), 2.95 (s, br, 2H), 3.69 - 3.82 (m, 3H), 4.35 - 4.83 (m, 6H), 5.67 – 5.77 (m, 1H), 6.28 - 6.33 (m, 2H), 6.70 (bs, 1H), 6.98 - 7.15 (m, 3H), 7.35 - 7.51 (m, 3H).
[00331] LCMS (Method A): 0.977 min, MS: ES+ 475.1 (M+1). [00332] Analytical HPLC (Method D): 4.392 min. Prep. HPLC purification method. [00333] Chromatographic separation and isolation were conducted with Shimadzu Nexera prep with Lh-40 auto purification system. The column used was Shim-Pack GIST C18 (250 mm x 20 mm x 5 µm) and the compounds were eluted with, Mobile Phase A : 0.05% Formic acid in water, Mobile Phase B : Methanol with a gradient of T = 0.01 min (87% A, 13% B); gradient to T = 24.00 min (63% A, 37% B); T = 24.01 min (2% A, 98% B) gradient to T = 26.00 min (2% A, 98% B); T = 26.01 min (87% A, 13% B); gradient to T = 30 min (87% A, 13% B); Flow rate= 14 mL/min; analysis time 30 min. Example 2: (E)-4-(3,3-Difluoropyrrolidin-1-yl)-N-(2-(2,4-dihydroxy-5-methylbenzoyl) isoindolin-4-yl)but-2-enamide Step 1: (E)-N-(2-(2,4-bis(Methoxymethoxy)-5-methylbenzoyl)isoindolin-4-yl)-4-(3,3- difluoropyrrolidin-1-yl)but-2-enamide
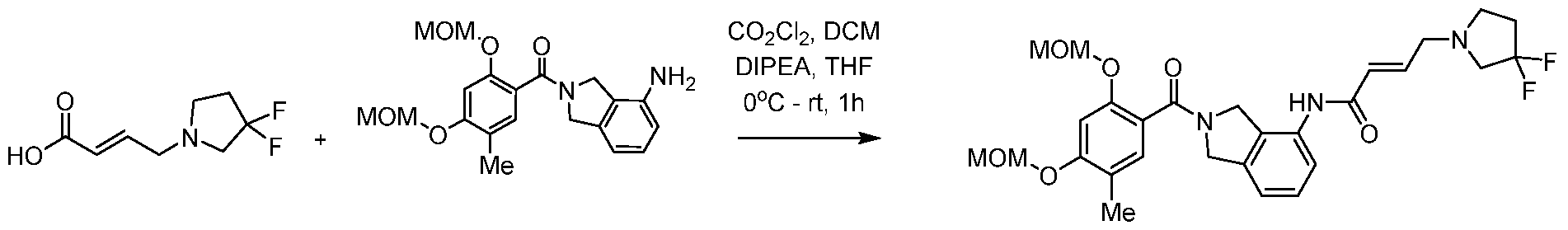
[00334] To a cooled (0 ˚C) solution of (E)-4-(3,3-difluoropyrrolidin-1-yl)but-2-enoic acid (Intermediate D) (0.35 g, 1.83 mmol, 1.0 eq.) in DCM (3.5 mL) were added CO2Cl2 (1.7 mL) and cat. DMF (2-3 drops) under nitrogen and the mixture was stirred at 0 ˚ C for 15 mins. The resulting mixture was concentrated under reduced pressure (under nitrogen), the intermediate was dissolved in THF (3.5 mL) and cooled to 0 ˚C. To this mixture was added a solution of (4- aminoisoindolin-2-yl)(2,4-bis(methoxymethoxy)-5-methylphenyl)methanone (Intermediate C) (0.2 g, 0.49 mmol, 0.3 eq.) and DIPEA (0.7 g, 5.49 mmol, 3.0 eq.) and the resulting mixture was allowed to stir at room temperature for 1 h. The mixture was poured into water (30 mL) and extracted with ethyl acetate (3 x 30 mL). The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The crude material was purified by chromatography on silica (product eluted in 3.6% MeOH in DCM) to afford the title compound (0.16 g, Yield: 31%). [00335]
1H NMR (DMSO-d6, 400 MHz): δ 2.14 (s, 3H), 2.20 - 2.34 (m, 2H), 2.67 - 2.78 (m, 2H), 2.86 - 2.98 (m, 2H), 3.24 - 3.25 (m, 1H), 3.28 - 3.31 (m, 4H), 3.42 (s, 3H), 4.59 (s, br, 2H), 4.80 (d, J= 14.8 Hz, 2H), 5.17 (s, 2H), 5.26 (s, 2H), 6.56 - 6.80 (m, 2H), 6.92 (d, J= 3.6 Hz, 1H), 7.04
(d, J= 7.6 Hz, 1H), 7.13 - 7.17 (m, 2H), 7.26 - 7.30 (q, J= 7.6 Hz, 15.6 Hz, 1H), 7.67 (t, J= 7.6 Hz, 1H). [00336] LCMS (Method A): 1.335 min, 1.420 min, 1.487 min, 1.518 min, MS: ES+ 546 (M+1). Step 2: (E)-4-(3,3-Difluoropyrrolidin-1-yl)-N-(2-(2,4-dihydroxy-5-methylbenzoyl)isoindolin-4- yl)but-2-enamide
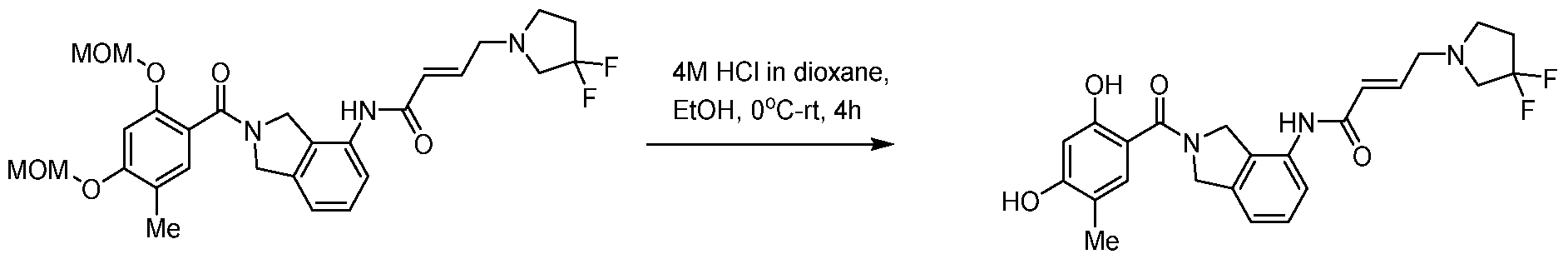
[00337] To a stirred solution of (E)-N-(2-(2,4-bis(methoxymethoxy)-5-methylbenzoyl)isoindolin- 4-yl)-4-(3,3-difluoropyrrolidin-1-yl)but-2-enamide (Step 1) (0.16 g, 0.29 mmol, 1.0 eq.) in EtOH (1.6 mL) was added dropwise 4M HCl in dioxane (0.8 mL) at 0 ˚C. The reaction mixture was stirred at room temperature for 4 h and then concentrated under reduced pressure to afford the title compound (0.045 g, Yield: 34%). [00338]
1H NMR (DMSO-d6, 400 MHz): δ 2.03 (s, 3H), 2.27 - 2.33 (m, 2H), 2.67 - 2.74 (m, 2H), 2.91 - 2.94 (m, 2H), 3.27 - 3.34 (m, 2H), 4.70 - 4.80 (m, 4H), 6.31 (s, 1H), 6.65 - 6.75 (m, 1H), 7.06 - 7.15 (m, 2H), 7.26 - 7.27 (m, 1H), 7.66 (d, J= 7.2 Hz, 1H), 9.63 - 9.72 (m, 2H, D
2O exchangeable), 9.98 - 10.17 (m, 1H, D
2O exchangeable). [00339] LCMS (Method A): 1.208 min, MS: ES+ 458 (M+1). [00340] Analytical HPLC (Method B): 4.638 min. Prep. HPLC purification method. [00341] Chromatographic separation and isolation were conducted with a Waters 2545 purification system with UV detector. The column used was SUNFIRE, C18, OBD (19 x 250)mm, 5μm and the compounds were eluted with, Mobile Phase A : 0.05% Formic acid in water, Mobile Phase B : Acetonitrile with a gradient of T = 0 min (75% A, 25% B); gradient to T = 17.00 min (65% A, 35% B); T = 17.01 min (02% A, 98% B) gradient to T = 20.00 min (02% A, 98% B); T = 20.01 min (75% A, 25% B); gradient to T = 24 min (75% A, 25% B); Flow rate= 13 mL/min; analysis time 24 min. Example 2.1: ((E)-4-(3,3-Difluoroazetidin-1-yl)-N-(2-(2,4-dihydroxy-5- methylbenzoyl)isoindolin-4-yl)but-2-enamide
Step 1: (E)-N-(2-(2,4-bis(Methoxymethoxy)-5-methylbenzoyl) isoindolin-4-yl)-4-(3,3- difluoroazetidin-1-yl) but-2-enamide
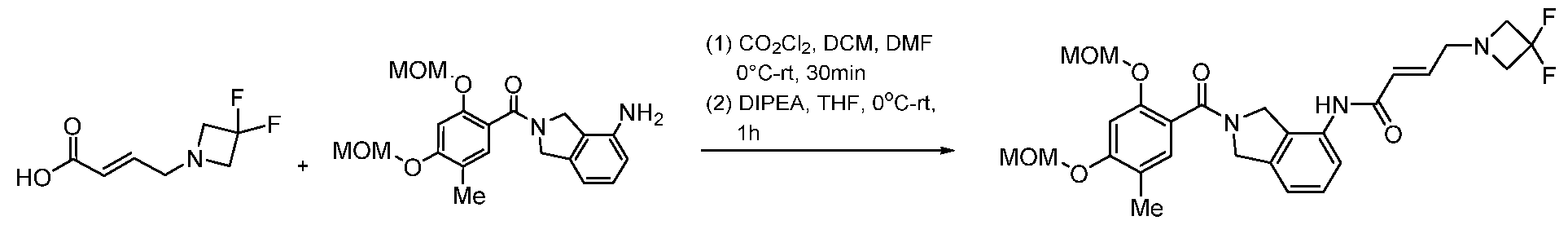
[00342] The title compound was prepared from (E)-4-(3,3-difluoroazetidin-1-yl) but-2-enoic acid (Intermediate DA) and (4-Aminoisoindolin-2-yl)(2,4-bis(methoxymethoxy)-5- methylphenyl)methanone (Intermediate C) analogously to Example 2 Step 1. The compound was used directly in Step 2. LCMS (Method A): 1.410 min, 1.437 min, 1.521 min, 1.546 min, MS: ES+ 535.1 (M+1). Step 2: ((E)-4-(3,3-Difluoroazetidin-1-yl)-N-(2-(2,4-dihydroxy-5-methylbenzoyl)isoindolin-4- yl)but-2-enamide
[00343] The title compound was prepared from (E)-N-(2-(2,4-bis(methoxymethoxy)-5- methylbenzoyl) isoindolin-4-yl)-4-(3,3-difluoroazetidin-1-yl) but-2-enamide and 4M HCl in dioxane analogously to Example 2 Step 2. [00344] High temperature
1H NMR (DMSO-d6, 400 MHz, 348K): δ 2.07 (s, 3H), 3.40 (d, J= 4 Hz, 2H), 3.63-3.69 (t, J= 12 Hz, 4H), 4.83 (d, J= 12.8 Hz, 4H), 6.35 - 6.40 (m, 2H), 6.69 - 6.74 (m, 1H), 7.11 (d, J= 8.8 Hz, 2H), 7.29 - 7.25 (t, J= 8 Hz, 1H), 7.64 (d, J= 8 Hz, 1H), 9.43 (m, 2H, D
2O exchangeable), 10.08 (s, 1H, D
2O exchangeable). [00345] LCMS (Method A): 1.208 min, MS: ES+ 444 (M+1). [00346] Analytical HPLC (Method B): 4.514 min, Prep HPLC purification method. [00347] Chromatographic separation and isolation were conducted with a Waters 2545 purification system with UV detector. The column used was SUNFIRE, C18, OBD 19 x 250 mm, 5μm and the compounds were eluted with, Mobile Phase A : 0.05% Formic acid in water, Mobile Phase B : 20% A Line in Acetonitrile with a gradient of T = 0 min (78% A, 22% B); gradient to T = 15.00 min (68% A, 32% B); T = 15.01 min (2% A, 98% B); gradient to T = 18.0 min (2% A, 98%
B); T= 18.01 min (78% A, 22% B); gradient to T = 24.00 min (78% A, 22% B); Flow rate= 19 mL/min; analysis time 24 min. Example 3: N-(2-(2, 4-Dihydroxy-5-methylbenzoyl) isoindolin-4-yl) acrylamide Step 1: (2,4-Dihydroxy-5-methylphenyl)(4-nitroisoindolin-2-yl)methanone:

[00348] The title compound was prepared from 2,4-dihydroxy-5-methylbenzoic acid (Intermediate F) and 4-nitroisoindoline hydrochloride (Intermediate C Step 1c) analogously to Example 1 Step 1. [00349]
1H NMR (DMSO-d6, 400 MHz): δ 2.04 (s, 3H), 4.90 (s, 2H), 5.19 (s, br, 2H), 6.43 (s, 1H), 7.06 (s, br, 1H), 7.62 (t, J= 8 Hz, 1H), 7.78 - 7.84 (m, 2H), 8.15 (d, J= 8.4 Hz, 1H), 9.68 (s, 1H, D2O exchangeable), 9.97 and 10.1 (singlets, br, 1H, D2O exchangeable). [00350] LCMS (Method A): 1.606 min, MS: ES+ 315.1 (M+1). Step 2: (4-Aminoisoindolin-2-yl) (2, 4-dihydroxy-5-methylphenyl) methanone
[00351] To a stirred solution of (2,4-dihydroxy-5-methylphenyl) (4-nitroisoindolin-2- yl)methanone (Step 1) (1.0 g, 3.18 mmol, 1 eq.) in EtOH (10 mL) and water (5 mL) were added NH4Cl (2.53 g, 4.78 mmol, 15.0 eq.) and Fe powder (2.14 g, 38.21 mmol, 6.0 eq.) and the reaction mixture heated to 70 ˚C for 3 h. The resulting mixture was filtered through Celite® and the filter cake washed with 10% MeOH: DCM (4 x 100 mL). The filtrate was poured in water (200 mL) and extracted with EtOAc (3 x 100 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under vacuum to afford the title compound as a brown solid (0.8 g, Yield: 94%). [00352]
1H NMR (MeOD-d6, 400 MHz): δ 2.14 (s, 3H), 4.77 (s, br, 2H), 4.92 (s, br, 2H), 6.39 (s, br, 1H), 6.63 (s, br, 2H), 7.06 - 7.14 (m, 2H). [00353] LCMS (Method A): 1.287 min, MS: ES+ 284.79 (M+1).
Step 3: N-(2-(2, 4-Dihydroxy-5-methylbenzoyl) isoindolin-4-yl) acrylamide
[00354] To a stirred solution of (4-aminoisoindolin-2-yl) (2, 4-dihydroxy-5-methylphenyl) methanone (Step 2) (0.200 g, 0.704 mmol, 1 eq.) in acetic acid (2 mL) was added dropwise acrylic anhydride (0.089 g, 0.704 mmol, 1 eq.) at 0 ˚C. The resulting mixture was allowed to warm to room temperature and stirred for 2 h. The resulting mixture was poured into ice-cold water (100 mL), neutralized using saturated solution of K
2CO
3 solution (pH ~10) and extracted in EtOAc (3 x 50 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted in 3.4% MeOH in DCM) to afford the title compound as an off-white solid (0.065 g, Yield: 27.3%). [00355] High temperature
1H NMR (DMSO-d6, 400 MHz): δ 2.07 (s, 3H), 4.82 - 4.86 (m, 4H), 5.73 - 5.76 (m, 1H), 6.25 - 6.29 (m, 1H), 6.41 (s, 1H), 6.50 - 6.57 (m, 1H), 7.12 (d, J= 6.8 Hz, 2H), 7.28 (t, J= 7.6 Hz, 1H), 7.62 (d, J= 8 Hz, 1H), 9.40 (s, 1H), 9.58 (s, 1H), 10.09 (s, 1H). [00356] LCMS (Method A): 1.398 min, MS: ES+ 338.8 (M+1). [00357] Analytical HPLC (Method B): 6.13 min. Example 4: (E)-N-(2-(2,4-Dihydroxy-6-methylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)- 4-(dimethylamino)but-2-enamide Step 1: tert-Butyl (E)-7-(4-(dimethylamino)but-2-enamido)-3,4-dihydroisoquinoline-2(1H)- carboxylate

[00358] To a stirred solution of (E)-4-(dimethylamino)but-2-enoic acid hydrochloride (CAS: 848133-35-7) (1 g, 6.02 mmol, 1 eq.) in DMF (15 mL) was added EDC.HCl (1.7 g, 9.03 mmol, 1.5 eq.) and HOAT (0.82 g, 6.02 mmol, 1 eq.) and the reaction mixture was stirred at room temperature for 30 mins. tert-Butyl 7-amino-3,4-dihydroisoquinoline-2(1H)-carboxylate (Intermediate H) (1.5 g, 6.02 mmol, 1 eq.) and NMM (1.21 g, 12.04 mmol, 2 eq.) were added and stirring continued at room temperature for 2 h. The resulting mixture diluted with water (100 mL)
and extracted with EtOAc (4 x 30 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted at 3.5% MeOH in DCM) to afford the title compound as a brown solid (3.5 g, Yield: Quantitative) [00359]
1H NMR (DMSO-d6, 400 MHz): δ 1.42 (s, 9H), 2.26 (s, 6H), 2.71 (t, J= 5.6 Hz, 2H), 2.17 (t, J= 6.0 Hz, 2H), 3.53 (t, J= 5.6 Hz, 2H), 4.45 (s, br, 2H), 6.28 (d, J= 15.2 Hz, 1H), 6.68 - 6.75 (m, 1H), 7.09 (d, J= 8.4 Hz, 1H), 7.34 - 7.38 (m, 1H), 7.50 (bs, 1H), 10.05 (s, 1H). [00360] LCMS (Method A): 1.373 min, MS: ES+ 359.9 (M+1). Step 2: (E)-7-(4-(Dimethylamino)but-2-enamido)-3,4-dihydroisoquinoline-2(1H)-carboxylate
[00361] To a cooled (0 ˚C) solution of tert-butyl (E)-7-(4-(dimethylamino)but-2-enamido)-3,4- dihydroisoquinoline-2(1H)-carboxylate (Step 1) (3.5 g, 9.74 mmol, 1 eq.) in DCM (35 mL) was added dropwise 4M HCl in dioxane (35 mL) and reaction mixture was stirred at 0 ˚C for 2 h. The resulting mixture was concentrated under vacuum and the crude material was triturated with diethyl ether (7 x 20 mL) and dried under vacuum to yield the title compound as a light yellow solid (3.0 g, Yield: quantitative). [00362]
1H NMR (DMSO-d6, 400 MHz): δ 2.74 (d, J= 4.8 Hz, 6H), 2.95 (t, J= 5.6 Hz, 2H), 3.32 (bs, 2H), 3.91 (t, J= 5.6 Hz, 2H), 4.22 (bs, 2H), 6.54 (d, J= 15.6 Hz, 1H), 6.77 - 6.85 (m, 1H), 7.17 (d, J= 8.4 Hz, 1H), 7.50 - 7.55 (m, 1H), 7.61 (s, 1H), 9.57 (bs, 2H), 10.66 (s, 1H), 11.12 (bs, 1H). HCl salt. [00363] LCMS (Method C): 4.186 min, MS: ES+ 260.2 (M+1). Step 3: (E)-N-(2-(2,4-Dihydroxy-6-methylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-4- (dimethylamino)but-2-enamide

[00364] To a stirred solution of 2,4-dihydroxy-6-methylbenzoic acid (Intermediate G) (0.3 g, 1.78 mmol, 1 eq.) in DMF (15 mL) was added EDC.HCl (0.513 g, 2.67 mmol, 1.5 eq.) and HOAT
(0.242 g, 1.78 mmol, 1 eq.) at room temperature and the mixture was stirred for 30 mins. (E)-4- (Dimethylamino)-N-(1,2,3,4-tetrahydroisoquinolin-7-yl)but-2-enamide hydrochloride (Step 2) (0.632 g, 2.14 mmol, 1.2 eq.) and NMM (0.97 mL, 8.90 mmol, 5 eq.) were added and stirring continued at room temperature for 16 h. The resulting mixture was purified directly by reverse phase purification (product eluted at 50% acetonitrile in water). The material was further triturated with MeOH (3 x 5 mL) to afford the title compound as a grey solid (0.143 g, Yield: 20%) [00365] High temperature
1H NMR (DMSO-d6, 400 MHz, 349K): δ 1.99 (s, 3H), 2.21 (s, 6H), 2.68 - 2.78 (m, 4H), 3.53 - 3.55 (m, 2H), 4.55 - 4.58 (m, 2H), 6.13 - 6.26 (m, 3H), 6.71 - 6.75 (m, 1H), 7.08 (d, J= 8.0 Hz, 1H), 7.41 (d, J= 7.2 Hz, 1H), 9.05 - 9.10 (m, 2H), 9.75 (bs, 1H). [00366] LCMS (Method A): 0.996 min, MS: ES+ 410 (M+1). [00367] Analytical HPLC (Method B): 4.357 min. Example 4.1: (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7- yl)-4-(dimethylamino)but-2-enamide

[00368] To a stirred solution of 2,4-dihydroxy-5-methylbenzoic acid (Intermediate F) (0.2 g, 1.19 mmol, 1 eq.) in DMF (5 mL) were added EDC.HCl (0.342 g, 1.78 mmol, 1.5 eq.) and HOAT (0.161 g, 1.18 mmol, 1 eq.) and the mixture was stirred at room temperature for 10 mins. (E)-4- (Dimethylamino)-N-(1,2,3,4-tetrahydroisoquinolin-7-yl)but-2-enamide (Example 4 step 2) (0.422 g, 1.42 mmol, 1.2 eq.) and NMM (0.600 g, 5.94 mmol, 5 eq.) were added and stirring continued at room temperature for 1.5 h. The resulting mixture was poured in water (10 mL) and extracted with EtOAc (3 x 10 mL). The combined organic extracts were washed with cold brine solution (2 x 10 mL), dried over Na
2SO
4 and concentrated under vacuum. The crude material was purified by Prep-HPLC followed by lyophilization of the product fractions to afford the title compound (0.160 g, Yield: 15.00%). [00369] High temperature
1H NMR (DMSO-d6, 400 MHz, 344K): δ 2.09 (s, 3H), 2.17 (s, 6H), 2.67 - 2.80 (m, 2H), 3.37 (d, J= 5.6 Hz, 2H), 3.63 - 3.66 (m, 2H), 4.60 (s, 2H), 6.23 - 6.26 (m, 1H), 6.40 (s ,1H), 6.70 - 6.77 (m, 1H), 6.80 (s, 1H), 7.08 - 7.10 (m, 1H), 7.39 - 7.45 (m, 2H), 9.30 - 9.33 (m, 2H), 9.77 (s, 1H). Formic salt. [00370] LCMS (Method A): 1.095 min, MS: ES+ 409.9 (M+1). [00371] Analytical HPLC (Method B): 4.581 min.
Prep. HPLC purification method: [00372] Chromatographic separation and isolation were conducted with flash chromatography- Selekt with uv detector; column YMC C18, 120gm, 50μm; compound eluted with, Mobile Phase A : 0.1% Formic acid in water, Mobile Phase B : Acetonitrile with a gradient of T = 0 min (100% A, 00% B); gradient to T = 25.00 min (80% A, 20% B); T = 25.01 min (00% A, 100% B) gradient to T = 30.00 min (00% A, 100% B); T = 30.01 min (100% A, 00% B); gradient to T = 35 min (100% A, 00% B); Flow rate= 70 mL/min; analysis time 35.01 min. Example 4.2: (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7- yl)-4-(dimethylamino)-N-methylbut-2-enamide Step 1: tert-Butyl (E)-7-(4-(dimethylamino)-N-methylbut-2-enamido)-3,4-dihydroisoquinoline- 2(1H)-carboxylate

[00373] Performed in 6 parallel batches, each of 0.5 g scale: To a stirred solution of (E)-4- (dimethylamino)but-2-enoic acid hydrochloride (CAS: 848133-35-7) (0.5 g, 3.01 mmol, 1.0 eq.) in DMF (0.5 mL) were added tert-butyl 7-(methylamino)-3,4-dihydroisoquinoline-2(1H)- carboxylate (Intermediate I) (0.395 g, 1.50 mmol, 0.5 eq.) and DCC (0.927 g, 4.5 mmol, 1.5 eq.) and the reaction mixture was heated to 150 ˚C using microwave irradiation for 15 mins. The resulting mixture was diluted with ice water (200 mL) and extracted with EtOAc (5 x 50 mL). The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The crude material was purified by chromatography on silica (product eluted in 4.1% MeOH in DCM) to afford the title compound (2.4 g, Yield: 35%) which was used directly in Step 2. LCMS (Method A): 1.375 min, MS ES+: 374.2 (M+1). Step 2: (E)-4-(Dimethylamino)-N-methyl-N-(1,2,3,4-tetrahydroisoquinolin-7-yl)but-2-enamide hydrochloride

[00374] The title compound was prepared from tert-butyl (E)-7-(4-(dimethylamino)-N-methylbut- 2-enamido)-3,4-dihydroisoquinoline-2(1H)-carboxylate (Step 1) and 4M HCl in dioxane analogously to Example 4 Step 2 and used directly in Step 3. [00375] LCMS (Method B): 1.55 min, MS: ES+ 273.38 (M+1). Step 3: (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-4- (dimethylamino)-N-methylbut-2-enamide
[00376] The title compound was prepared from 2,4-dihydroxy-5-methylbenzoic acid (Intermediate F) and (E)-4-(dimethylamino)-N-methyl-N-(1,2,3,4-tetrahydroisoquinolin-7-yl)but- 2-enamide hydrochloride (Step 2) analogously to Example 4 Step 3. [00377]
1H NMR (DMSO-d6, 400 MHz): δ 2.01 (s, 3H), 2.05 (s, 6H), 2.84 - 2.91 (m, 4H), 3.20 (s, 3H), 3.60 - 3.63 (m, 2H), 4.60 - 4.62 (m, 2H), 5.88 (d, J= 14.8 Hz, 1H), 6.38 (s, 1H), 6.57 - 6.64 (m, 1H), 6.85 (s, 1H), 7.05 - 7.10 (m, 2H), 7.22 (d, J= 8 Hz, 1H), 9.60 (bs, 2H). [00378] LCMS (Method A): 1.123 min, MS: ES+ 424.11 (M+1). [00379] Analytical HPLC (Method B): 4.279 min. Prep. HPLC purification method: [00380] Chromatographic separation and isolation were conducted with a Waters 2545 purification system with UV detector. The column used was SUNFIRE, C18, OBD 19 x 250 mm, 5μm and the compounds were eluted with, Mobile Phase A : 0.05% FORMIC ACID IN WATER, Mobile Phase B : Acetonitrile with a gradient of T = 0 min (90% A, 10% B); gradient to T = 17.00 min (82% A, 18% B); T = 17.01 min (02% A, 98% B) gradient to T = 19.00 min (02% A, 98% B); T = 19.01 min (90% A, 10% B); gradient to T = 24 min (90% A, 10% B); Flow rate= 15 mL/min; analysis time 24.00 min. Example 4.3: (E)-N-(2-(2,4-Dihydroxy-6-methylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7- yl)-4-(dimethylamino)-N-methylbut-2-enamide
[00381] The title compound was prepared from 2,4-dihydroxy-6-methylbenzoic acid (Intermediate G) and (E)-4-(dimethylamino)-N-methyl-N-(1,2,3,4-tetrahydroisoquinolin-7-yl)but- 2-enamide hydrochloride (Example 4.2 Step 2) analogously to Example 4 Step 3. [00382] High temperature
1H NMR (DMSO-d6, 400 MHz 348K): δ 2.00 (s, 3H), 2.07 (s, 6H), 2.86 - 2.90 (m, 4H), 3.22 (s, 2H), 3.53 (bs, 2H), 4.61 (bs, 2H), 5.91 (d, J= 15.4 Hz, 1H), 6.15 (d, J= 22.4 Hz, 2H), 6.57 - 6.64 (m, 1H), 7.04 (d, J= 8.0 Hz, 2H), 7.22 (d, J= 8.0 Hz, 1H), 9.04 (bs, 2H). [00383] LCMS (Method A): 1.025 min, MS: ES+ 424.1 (M+1). [00384] Analytical HPLC (Method B): 4.062 min Prep. HPLC purification method: [00385] Chromatographic separation and isolation were conducted with a Waters 2545 purification system with UV detector. The column used was SUNFIRE, C18, OBD (19 x 250)mm, 5μm and the compounds were eluted with, Mobile Phase A : 0.05% Formic acid in water, Mobile Phase B : Acetonitrile with a gradient of T = 3 min (93% A, 7% B); gradient to T = 16.00 min (93% A, 7% B); T = 16.01 min (2% A, 98% B) gradient to T = 19.00 min (2% A, 98% B); T = 19.01 min (93% A, 7% B); gradient to T = 25 min (93% A, 7% B); Flow rate= 19 mL/min; analysis time 25 min. Example 4.4: (E)-N-(2-(2,4-Dihydroxy-5-isopropylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7- yl)-4-(dimethylamino)-N-methylbut-2-enamide

[00386] The title compound was prepared from 2,4-dihydroxy-5-isopropylbenzoic acid (Intermediate FA) and (E)-4-(dimethylamino)-N-methyl-N-(1,2,3,4-tetrahydroisoquinolin-7-yl)but- 2-enamide hydrochloride (Example 4.2 Step 2) analogously to Example 4 Step 3.
[00387]
1H NMR (DMSO-d6, 400 MHz): δ 1.10 (d, J= 6.8 Hz, 6H), 2.06 (s, 6H), 2.85 - 2.91 (m, 4H), 3.05 - 3.08 (m, 1H), 3.20 (s, 3H), 3.37 – 3.63 (m, 2H), 4.64 (s, br, 2H), 5.85 (d, J= 13.6 Hz, 1H), 6.39 (s, 1H), 6.57 - 6.93 (m, 1H), 6.88 (s, 1H), 7.06 (d, J= 8 Hz, 1H), 7.12 (s, 1H), 7.23 (d, J= 8 Hz, 1H), 9.58 (s, 2H). [00388] LCMS (Method A): 1.337 min, MS: ES+ 452.1 (M+1). [00389] Analytical HPLC (Method D): 5.007 min. Example 5: (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4-(pyrrolidin-1- yl)but-2-enamide
[00390] The title compound was prepared from (E)-4-(pyrrolidin-1-yl) but-2-enoic acid hydrochloride (CAS: 848133-09-5) and (4-aminoisoindolin-2-yl) (2,4-dihydroxy-5- methylphenyl)methanone (Example 3, Step 2) analogously to Example 1 Step 2. [00391]
1H NMR (DMSO-d6, 400 MHz): δ 1.71 (s, 4H), 2.00 (s, 3H), 2.50 - 2.67 (m, 4H), 3.23 (d, J= 5.2 Hz, 2H), 4.81 - 4.85 (m, 4H), 6.20 - 6.22 (s, br, 1H), 6.39 (d, J= 12 Hz, 1H), 6.75 - 6.81 (m, 1H), 7.07 - 7.11 (m, 2H), 7.26 (t, J= 8 Hz, 1H), 7.68 (d, J= 8 Hz, 1H), 9.65 (s, 1H), 10.33 (bs, 2H). [00392] LCMS (Method I): 1.488 min, MS: ES+ 422.2 (M+1). [00393] Analytical HPLC (Method C): 5.02 min. Prep. HPLC purification method: [00394] Chromatographic separation and isolation were conducted with a Shimadzu Nexera purification system with UV detector. The column used was SHIMPACK GIST C18, (250 x 20)mm, 5μm and the compounds were eluted with, Mobile Phase A : 0.1% Formic acid in water, Mobile Phase B : Acetonitrile with a gradient of T = 0 min (80% A, 20% B); gradient to T = 18 min (78% A, 22% B); T = 18.01 min (02% A, 98% B); gradient to T = 20.00 min (02% A, 98% B); T = 20.01 min (80% A, 20% B) to T=22 min (80% A, 20% B); Flow rate= 20 mL/min; analysis time 22 min.
Example 5.1: (E)-N-(2-(2, 4-Dihydroxy-5-methylbenzoyl) isoindolin-4-yl)-4-morpholinobut- 2-enamide
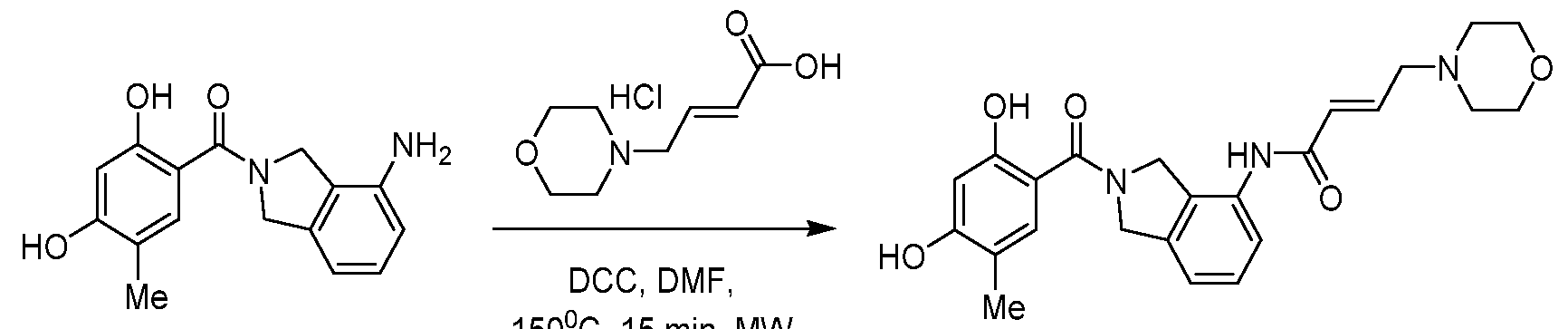
[00395] The title compound was prepared from (E)-4 -morpholino-but-2-enoic acid hydrochloride (CAS: 1419865-05-6) and (4-aminoisoindolin-2-yl) (2, 4-dihydroxy-5-methylphenyl) methanone (Example 3 Step 2) analogously to Example 1 Step 2. [00396] High Temperature
1H NMR (DMSO-d6, 400 MHz): δ 2.07 (s, 3H), 2.40 - 2.43 (m, 4H), 3.61 - 3.63 (m, 4H), 3.62 (m, 4H), 4.82 - 4.85 (m, 4H), 6.36 - 6.40 (m, 2H), 6.74 - 6.78 (m, 1H), 7.10 - 7.12 (m, 2H), 7.27 (t, J= 7.6 Hz, 1H), 7.62 (d, J= 4.6 Hz, 1H), 9.42 (bs, 1H), 9.49 (bs, 1H), 10.09 (bs, 1H). [00397] LCMS (Method A): 1.103 min, MS: ES+ 438.0 (M+1). [00398] Analytical HPLC (Method B): 4.38 min. Example 6: N-(2-(3-Chloro-4,6-dihydroxy-2-methylbenzoyl)-1,2,3,4-tetrahydroisoquinolin- 7-yl)-N-methylacrylamide Step 1: (3-Chloro-4,6-dihydroxy-2-methylphenyl)(7-(methylamino)-3,4-dihydroisoquinolin-2(1H)- yl)methanone

[00399] To a stirred solution of 3-chloro-4,6-dihydroxy-2-methylbenzoic acid (Intermediate B) (0.7 g, 3.45 mmol, 1 eq.) in DMF (7 mL) were added EDC.HCl (0.995 g, 5.18 mmol, 1.5 eq) and HOAT (0.469 g, 3.45 mmol, 1 eq) at 0 ˚C. The mixture was allowed to warm to room temperature and treated with N-methyl-1,2,3,4-tetrahydroisoquinolin-7-amine hydrochloride (Intermediate J) (0.8 g, 4.14 mmol, 1.2 eq.) and NMM (1.74 g, 17.27 mmol, 5 eq.). The reaction mixture was stirred at room temperature for 30 mins. The resulting mixture was poured into ice cold water (30 mL) and extracted with ethyl acetate (3 x 30 mL). The combined organic extracts were dried over
Na
2SO
4 and concentrated under reduced pressure. The crude material was purified by chromatography on silica (product eluting at 4% MeOH: DCM) to afford the title compound as an off-white solid (0.18 g, Yield: 15%). [00400]
1H NMR (DMSO-d6, 400 MHz): δ 2.00 - 2.08 (2 singlets, 3H), 2.57 - 2.69 (m, 5H), 3.35 (s, 1H), 3.59 - 3.96 (m, 1H), 4.17 - 4.29 (m, 1H), 4.60 – 4.70 (m, 1H), 5.49 (s, br, 1H, D
2O exchangeable), 6.11 - 6.45 (m, 3H), 6.82 - 6.88 (m, 1H), 9.65 (d, J= 12 Hz, 1H, D
2O exchangeable), 10.08 (d, J= 2 Hz, 1H, D
2O exchangeable). [00401] LCMS (Method A): 1.061 min, MS: ES+ 346.9 (M+1) [00402] Analytical HPLC (Method D): 3.727 min, Step 2: N-(2-(3-Chloro-4,6-dihydroxy-2-methylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- methylacrylamide
[00403] Performed in 2 parallel batches, each of 0.12 g scale: To a cooled (0 ˚C) stirred solution of (3-chloro-4,6-dihydroxy-2-methylphenyl)(7-(methylamino)-3,4-dihydroisoquinolin-2(1H)- yl)methanone (Step 1) (0.12 g, 0.34 mmol, 1.0 eq.) in DCM (1.2 mL) were added pyridine (0.26 g, 0.3.46 mmol, 10.0 eq.) and acrylic anhydride (0.065 g, 0.52 mmol, 1.5 eq.) and the mixture was stirred at 0 ˚C for 2 h. The resulting mixture was diluted with water (30 mL) and extracted with ethyl acetate (2 x 30 mL). The combined organic extracts were dried over Na2SO4 and reduced under vacuum. The crude material was purified by chromatography on silica (product eluted at 3.7% MeOH in DCM) to afford the title compound as white solid (0.023 g, Yield: 8.3 %). [00404]
1H NMR (DMSO-d6, 400 MHz): δ 1.99 and 2.10 (2 singlets, 3H), 2.71 - 2.74 (m, 1H), 2.80 - 2.86 (m, 1H), 3.17 - 3.24 (2 singlets, 3H), 3.38 - 3.43 (m, 1H), 3.68 - 4.02 (m, 1H), 4.31 - 4.42 (m, 1H), 4.72 - 4.85 (m, 1H), 5.59 (t, J= 12.8 Hz, 1H), 6.04 - 6.18 (m, 2H), 6.45 (d, J= 8 Hz, 1H), 7.04 - 7.09 (m, 1H), 7.18 - 7.26 (m, 2H), 9.65 - 9.71 (m, 1H, D
2O exchangeable), 10.11 (d, J= 4 Hz, 1H, D
2O exchangeable). [00405] LCMS (Method A): 1.459 min, MS ES+: 401 (M+1). [00406] Analytical HPLC (Method D): 5.471 min,
Example 6.1: N-(2-(5-Chloro-2,4-dihydroxybenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- methylacrylamide Step 1: (5-Chloro-2,4-dihydroxyphenyl)(7-(methylamino)-3,4-dihydroisoquinolin-2(1H)- yl)methanone
[00407] The title compound was prepared from 5-chloro-2,4-dihydroxybenzoic acid (Intermediate Q) and N-methyl-1,2,3,4-tetrahydroisoquinolin-7-amine hydrochloride (Intermediate J) analogously to Example 6 Step 1. This material was progressed directly to Step 2. [00408] LCMS (Method A): 1.016 min, MS: ES+ 332.9 & 334.6 (M+1 & M+3). Step 2: N-(2-(5-Chloro-2,4-dihydroxybenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- methylacrylamide
[00409] The title compound was prepared from (5-chloro-2,4-dihydroxyphenyl)(7- (methylamino)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (Step 1), acetic acid and acrylic anhydride analogously to Example 3, Step 3. [00410]
1H NMR (DMSO-d6, 400 MHz, D
2O exchange): δ 2.83 (t, J= 5.2 Hz, 2H), 3.21 (s, 3H), 3.58 (bs, 2H), 4.65 (bs, 2H), 5.56 (d, J= 11.6 Hz, 1H), 6.09 - 6.16 (m, 2H), 6.57 (s, 1H), 7.06 - 7.09 (m, 2H), 7.15 (bs, 1H), 7.24 (d, J= 8.0 Hz, 1H). [00411] LCMS (Method A): 1.423 min, MS: ES+ 387 & 389 (M+1 & M+3). [00412] Analytical HPLC (Method D): 5.538 min. Prep. HPLC purification method: [00413] Chromatographic separation and isolation were conducted with Shimadzu Nexera prep with LH-40 auto purification system. The column used was Shim-Pack GIST C18 (250 mm x 20
mm x 5 µm) and the compounds were eluted with, Mobile Phase A : 0.05% Formic acid in water, Mobile Phase B : Acetonitrile with a gradient of T = 0.01 min (90% A, 10% B); gradient to T = 17.00 min (47% A, 53% B); T = 17.01 min (2% A, 98% B) gradient to T = 19.00 min (2% A, 98% B); T = 19.01 min (90% A, 10% B); gradient to T = 22 min (90% A, 10% B); Flow rate= 22 mL/min; analysis time 22 min. Example 7.1: (E)-N-(2-(5-(tert-Butyl)-2-hydroxy-4-methoxybenzoyl)isoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide Step 1: (5-(tert-Butyl)-2-hydroxy-4-methoxyphenyl)(4-(methylamino)isoindolin-2-yl)methanone

[00414] To a stirred solution of 5-(tert-butyl)-2-hydroxy-4-methoxybenzoic acid (Intermediate N) (0.73 g, 3.25 mmol, 1.0 eq.) in DMF (15 mL) were added EDC.HCl (0.93 g, 4.88 mmol, 1.5 eq.) and HOAT (0.44 g, 3.25 mmol, 1.0 eq.) and the reaction mixture was stirred at room temperature for 30 mins. N-Methylisoindolin-4-amine hydrochloride (Intermediate A) (0.57 g, 3.90 mmol, 1.2 eq.) and NMM (1.64 g, 16.2 mmol, 5.0 eq.) were added and stirring continued for 3 h. The resulting mixture was diluted with water (100 mL) and extracted with ethyl acetate (3 x 100 mL). The combined organic extracts were dried over Na2SO4 and concentrated under vacuum. Purification of the crude material by chromatography on silica (product eluted at 3.4% MeOH: DCM) afforded the title compound as a light brown solid (0.6 g, Yield: 18.1%). [00415]
1H NMR (DMSO-d6, 400 MHz): δ 1.30 (s, 9H), 2.64 – 2.72 (m, 3H), 3.80 (s, 3H), 4.52 – 4.72 (m, 4H), 6.37 (d, J= 6.8 Hz, 1H), 6.52 (t, J= 17.6 Hz, 11.2 Hz, 2H), 7.10 (d, J= 7.6 Hz, 2H), 10.07 – 10.28 (m, 1H). [00416] LCMS (Method A): 2.463 min. MS: ES+ 355.1 (M+1). [00417] Analytical HPLC (Method B): 9.85 min. Step 2: (E)-N-(2-(5-(tert-Butyl)-2-hydroxy-4-methoxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)- N-methylbut-2-enamide
[00418] To a stirred solution (E)-4-(dimethylamino)but-2-enoic acid hydrochloride (CAS: 848133-35-7) (0.08 g, 0.48 mmol, 1.0 eq.) in DMF (1.6 mL, 20v) were added DCC (0.198 g, 0.96 mmol, 2.0 eq.) and (5-(tert-butyl)-2-hydroxy-4-methoxyphenyl)(4-(methylamino)isoindolin-2- yl)methanone (Step 1) (0.07 g, 0.19 mmol, 0.4 eq.) and the reaction mixture was heated to 150 ˚C using microwave irradiation for 15 mins. The resulting mixture was diluted with cold water (50 mL) and extracted with EtOAc (3 x 50 mL). The combined organic extracts were dried over Na2SO4 and concentrated under vacuum. Purification of the crude material by Prep. HPLC (0.05% Formic acid in water:ACN) afforded the title compound as a light brown solid (0.005 g, Yield: 2%). [00419]
1H NMR (DMSO-d6, 400 MHz): δ 1.29 (s, 9H), 2.01 (s, 6H), 2.87 (d, J= 4.8 Hz, 2H), 3.1 - 3.20 (m, 3H), 3.80 (s, 3H), 4.59 (d, J= 15.2 Hz, 1H), 4.71 - 4.89 (m, 3H), 5.75 (d, J= 14 Hz, 1H), 6.49 - 6.66 (m, 2H), 7.07 (s, 1H), 7.22 (d, J= 6.8 Hz, 1H), 7.36 - 7.44 (m, 2H), 10.32 (bs, 1H). [00420] LCMS (Method A): 1.529 min. MS: ES+ 466.1 (M+1). [00421] Analytical HPLC (Method D): 5.84 min. Prep. HPLC purification method. [00422] Chromatographic separation and isolation were conducted with an FC-01 flash purification system (Buchi model C810); column YMC-120GM C18, 50µm; compound eluted with: Mobile Phase A : 0.1% Formic acid in water, Mobile Phase B : Acetonitrile with a gradient of T = Initial (100% A, 0% B); gradient to T = 5.00 min (100% A, 0% B); T = 30.00 min (70% A, 30% B) gradient to T = 30.01 min (0% A, 100% B); T = 35.00 min (0% A, 100% B); T = 35.01 min (100% A, 0% B); T = 40.00 min (100% A, 0% B); Flow rate= 70 mL/min; analysis time 40 min. Example 7.2: (E)-N-(2-(5-(tert-Butyl)-4-fluoro-2-hydroxybenzoyl)isoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide Step 1: (5-(tert-butyl)-4-fluoro-2-hydroxyphenyl)(4-(methylamino)isoindolin-2-yl)methanone
[00423] The title compound was prepared from 5-(tert-butyl)-4-fluoro-2-hydroxybenzoic acid (Intermediate OA) and N-methylisoindolin-4-amine hydrochloride (Intermediate A) analogously to Example 7.1, Step 1. [00424]
1H NMR (DMSO-d6, 400 MHz): δ 1.23 - 1.40 (m, 9H), 2.63 and 2.72 (singlets, 3H), 4.42 - 4.72 (m, 4H), 5.37 - 5.52 (m, 1H, D
2O exchangeable), 6.37 (d, J= 10 Hz, 1H), 6.46 - 6.58 (m, 1H), 6.63 - 6.68 (dd, J= 4.4 Hz, 4.8 Hz, 1H), 7.06 - 7.15 (m, 2H), 10.29 (d, J= 17.6, 1H, D
2O exchangeable ). [00425] LCMS (Method A): 2.242 min, MS: ES+ 343.1 (M+1). [00426] Analytical HPLC (Method D): 8.260 min, Step 2: (E)-N-(2-(5-(tert-Butyl)-4-fluoro-2-hydroxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N- methylbut-2-enamide
[00427] The title compound was prepared from (E)-4-(dimethylamino)but-2-enoic acid hydrochloride (CAS: 848133-35-7) and (5-(tert-butyl)-4-fluoro-2-hydroxyphenyl)(4- (methylamino)isoindolin-2-yl)methanone (Step 1) analogously to Example 7.1, Step 2. [00428]
1H NMR (DMSO-d6, 400 MHz): δ 1.29 (d, J= 4.8 Hz, 9H), 2.04 (s, 6H), 2.91 (d, J= 4.4, 2H), 3.20 and 3.24 (singlets, 3H), 4.48 - 4.88 (m, 4H), 5.75 (t, J= 14.8 Hz, 1H), 6.57 - 6.69 (m, 2H), 7.12 (d, J= 9.6 Hz, 1H), 7.22 (d, J= 7.6 Hz, 1H), 7.34 - 7.44 (m, 2H), 10.42 (s, 1H, D
2O exchangeable). [00429] LCMS (Method A): 1.476 min, MS: ES+ 454.3 (M+1). [00430] Analytical HPLC (Method D): 4.760 min. Example 8: (E)-N-(2-(4,6-Dihydroxy-2,3-dimethylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7- yl)-4-(dimethylamino)-N-methylbut-2-enamide
Step 1: (4,6-Dihydroxy-2,3-dimethylphenyl)(7-(methylamino)-3,4-dihydroisoquinolin-2(1H)- yl)methanone

[00431] To a stirred solution of 4,6-dihydroxy-2,3-dimethylbenzoic acid (Intermediate K) (0.180 g, 0.989 mmol, 1.0 eq.) in DMF (0.9 mL) were added EDC.HCl (0.284 g, 1.483 mmol, 1.5 eq.) and HOAT (0.134 g, 0.989 mmol, 1.0 eq.) and the mixture was stirred at room temperature for 30 mins. N-Methyl-1,2,3,4-tetrahydroisoquinolin-7-amine hydrochloride (Intermediate J) (0.195 g, 0.989 mmol, 1.0 eq.) in DMF (0.9 mL) was added followed by NMM (0.49 g, 4.94 mmol, 5.0 eq.) were added and the reaction mixture was stirred at room temperature for 30 mins. The resulting mixture was diluted with ice cold water (100 mL) and extracted with ethyl acetate (3 x 25 mL). The combined organic extracts were dried over Na2SO4 and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted at 70% EtOAc in hexane) to afford the title compound (0.120 g, Yield:37%) which was used directly in Step 2 [00432] LCMS (Method A): 0.998 min, MS: ES+327 (M+1). Step 2: (E)-N-(2-(4,6-Dihydroxy-2,3-dimethylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-4- (dimethylamino)-N-methylbut-2-enamide

[00433] To a stirred solution of (E)-4-(dimethylamino)but-2-enoic acid hydrochloride (CAS:848133-35-7) (0.122g, 0.734 mmol, 1.0 eq.) and (4,6-dihydroxy-2,3-dimethylphenyl)(7- (methylamino)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (step 1) (0.120, 0.367mmol, 0.5 eq ) in DMF (1.2 mL) was added DCC (0.227 g, 1.102 mmol, 1.5 eq.) and the reaction mixture was heated to 150 ˚C using microwave irradiation for 15 mins. The resulting mixture was diluted with water (100 mL), acidified with dilute HCl and further extracted with EtOAc (3 x 30 mL). The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The crude material was purified by Prep-HPLC eluting with 0.05% formic acid in water/ACN to afford the title compound as a brown solid (0.005 g, Yield: 4.00%). [00434] High temperature
1H NMR (DMSO-d6, 400 MHz, 348.7K): δ 1.97 (s, 6H), 2.05 (s, 3H), 2.07 (s, 3H), 2.85 - 2.90 (m, 4H), 4.07 (bs, 1H), 4.39 (bs, 1H), 4.72 (bs, 2H), 5.91 (d, J= 15.6 Hz,
1H), 6.29 (s, 1H), 6.59 - 6.62 (m, 1H), 7.02 - 7.23 (m, 3H), 8.20 (s, 1H, D
2O exchangeable), 8.90 (s, br, D
2O exchangeable). [00435] LCMS (Method D): 1.431 min, MS: ES+ 438.4 (M+1). [00436] Analytical HPLC (Method D): 3.839 min. Prep. HPLC purification method [00437] Chromatographic separation and isolation were conducted with Shimadzu LC20AP with uv detector. The column used was Shim-Pack GIST C18 (250 mm x 20 mm x 5 µm) and the compounds were eluted with, Mobile Phase A : 0.05% Formic acid in water, Mobile Phase B : Acetonitrile with a gradient of T = 0.01 min (88% A, 12% B); gradient to T = 18.00 min (75% A, 25% B); T = 18.01 min (2% A, 98% B) gradient to T = 20.00 min (2% A, 98% B); T = 20.01 min (88% A, 12% B); gradient to T = 23 min (88% A, 12% B); Flow rate= 22 mL/min; analysis time 23 min. Example 9: N-(2-(2,4-Dihydroxy-5-methylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- methylacrylamide Step 1: (2,4-Dihydroxy-5-methylphenyl)(7-(methylamino)-3,4-dihydroisoquinolin-2(1H)- yl)methanone

[00438] To a stirred solution of 2,4-dihydroxy-5-methylbenzoic acid (Intermediate F) (0.5 g, 2.97 mmol, 1 eq.) in DCM: THF (10 mL) were added EDC.HCl (0.86 g, 4.46 mmol, 1.5 eq) and HOAT (0.4 g, 2.97 mmol, 1 eq) at room temperature. N-Methyl-1,2,3,4-tetrahydroisoquinolin-7-amine hydrochloride (Intermediate J) (0.58 g, 3.57 mmol, 1.2 eq) and NMM (1.5 g, 14.88 mmol, 2 eq.) were added and the reaction mixture was stirred at room temperature for 1 h. The resulting mixture was poured into water (100 mL) and extracted with DCM (2 x 100 mL). The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The crude material was purified by reverse phase chromatography (eluting product at 4% water: ACN) to afford the title compound as an off-white solid (0.4 g, Yield: 41.5%).
[00439] LCMS (Method A): 1.025 min, MS: ES+ 312.9 (M+1). This material was used directly in Step 2. Step 2: N-(2-(2,4-Dihydroxy-5-methylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- methylacrylamide

[00440] To a cooled (0 ˚C) solution of (2,4-dihydroxy-5-methylphenyl)(7-(methylamino)-3,4- dihydroisoquinolin-2(1H)-yl)methanone (0.27 g, 0.86 mmol, 1 eq.) (Step 1) in pyridine (2.7 mL) was added dropwise acrylic anhydride (0.12 mL, 0.951 mmol, 1.1 eq.) and the reaction mixture was stirred at room temperature for 1 h. The resulting mixture was neutralized with saturated citric acid solution (80 mL) and extracted with DCM (3 x 50 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under reduced pressure. The crude material was purified using Prep. HPLC (0.05% formic acid n water: ACN, MeOH, IPA) to afford the title compound as an off-white solid (0.035 g, Yield: 11.6%). [00441]
1H NMR (DMSO-d6, 400 MHz): δ 2.00 (s, 3H), 2.83 (t, J= 5.2 Hz, 2H), 3.21 (s, 3H), 3.62 (s, br, 2H), 4.64 (s, 2H), 5.56 (d, J= 11.6. Hz, 1H), 6.06 - 6.16 (m, 2H), 6.39 (s, 1H), 6.85 (s, 1H), 7.07 (t, J= 8.8 Hz, 7.65 Hz, 1H), 7.14 (s, 1H), 7.23 (d, J= 8 Hz, 1H), 9.55 (d, J= 7.2 Hz, 2H, D
2O exchangeable). [00442] LCMS (Method A): 1.461 min, MS: ES+ 367 (M+1). [00443] Analytical HPLC (Method D): 6.203 min. Example 10: N-Benzyl-N-(2-(2,4-dihydroxybenzoyl)isoindolin-4-yl)acrylamide
[00444] The title compound was prepared from (4-(benzylamino)isoindolin-2-yl)(2,4- dihydroxyphenyl)methanone (Example 1.1 Step 1), acetic acid and acrylic anhydride analogously to Example 3, Step 3.
[00445]
1H NMR (DMSO-d6, 400 MHz): δ 4.32 - 4.51 (m, 2H), 4.85 (s, 3H), 4.92 - 4.94 (m, 1H), 5.61 (d, J= 10.4 Hz, 1H), 6.20 - 6.34 (m, 3H), 6.98 (t, J= 4.0 Hz, 1H), 7.15 - 7.24 (m, 6H), 7.34 (d, J= 4.8 Hz, 2H), 9.55 (s, 1H), 10.32 (s, 1H). [00446] LCMS (Method A): 1.729 min, MS: ES+ 415.1 (M+1). [00447] Analytical HPLC (Method D): 7.278 min. Prep. HPLC purification method: [00448] Chromatographic separation and isolation were conducted with a Waters 2545 purification system with UV detector. The column used was Xtimate C18 (250 mm x 21.2 mm x 5 µm) and the compounds were eluted with, Mobile Phase A : 0.05% Formic acid in water, Mobile Phase B : Acetonitrile: MeOH: IPA (65:25:10) with a gradient of T = Initial (55% A, 45% B); gradient to T = 17.00 min (55% A, 45% B); T = 17.01 min (2% A, 98% B) gradient to T = 19.00 min (2% A, 98% B); T = 19.01 min (55% A, 45% B); gradient to T = 23 min (55% A, 45% B); gradient to T = 23 min (55% A, 45% B); Flow rate= 22 mL/min; analysis time 23 min. Example 10.1: N-Benzyl-N-(2-(2,4-dihydroxy-5-methylbenzoyl)isoindolin-4-yl)acrylamide

[00449] The title compound was prepared from (4-(benzylamino)isoindolin-2-yl)(2,4-dihydroxy- 5-methylphenyl)methanone (Example 1.2 step 1), acetic acid and acrylic anhydride analogously to Example 3, Step 3. [00450]
1H NMR (DMSO-d6, 400 MHz): δ 2.07 (s, 3H), 4.29 - 4.54 (m, 2H), 4.84 (bs, 4H), 5.61 (d, J= 9.2 Hz, 1H), 5.95 - 6.01 (m, 1H), 6.25 (dd, J= 2.0, 8.4 Hz, 1H), 6.39 (s, 1H), 6.97 - 6.99 (m, 2H), 7.17 - 7.25 (m, 5H), 7.30 - 7.34 (m, 2H), 9.45 (bs, 1H, D2O exchangeable), 9.97 (bs, 1H, D2O exchangeable). [00451] LCMS (Method A): 1.829 min, MS: ES+ 429.0 (M+1). [00452] HPLC (Method D): 7.57 min. Prep. HPLC purification method:
[00453] Chromatographic separation and isolation were conducted with a Waters 2545 purification system with a UV detector. The column used was Xtimate C18 (250 mm x 21.2 mm x 5 µm) and the compounds were eluted with, Mobile Phase A : 0.05% Formic acid in water, Mobile Phase B : 20% a line in acetonitrile + 10%THF with a gradient of T = Initial (52% A, 48% B); gradient to T = 24.00 min (52% A, 48% B); T = 24.01 min (2% A, 98% B) gradient to T = 26.00 min (2% A, 98% B); T = 26.01 min (52% A, 48% B); T = 28 min (52% A, 48% B); T = 28.01 min (52% A, 48% B); Flow rate= 20 mL/min; analysis time 28.01 min. Example 11: (E)-N-(2-(2,4-Dihydroxybenzoyl) isoindolin-4-yl)-4-(dimethyl amino)-N- phenethylbut-2-enamide Step 1: (2,4-Dihydroxyphenyl) (4-(phenethyl amino) isoindolin-2-yl) methanone

[00454] To a cooled (0 ˚C) solution of 2,4-dihydroxybenzoic acid (CAS 89-86-1) (0.38 g, 1.62 mmol, 1 eq.) in DMF (4.0 mL) were added HATU (0.92 g, 2.43 mmol, 1.5 eq) and DIPEA (0.45 mL, 3.24 mmol, 2.0 eq.) and the mixture was stirred stir at 0˚C for 10 mins. N-Phenethylisoindolin- 4-amine hydrochloride (Intermediate M) (0.25 g, 1.62 mmol, 1 eq.) was added and the reaction mixture was allowed to warm to room temperature and stirred for 1 h. The resulting mixture was diluted with ice cold water (50 mL) and extracted with EtOAc (3 x 50 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted at 2.0% MeOH in DCM) to afford the title compound (0.3 g, Yield: 47.6%). [00455]
1H NMR (DMSO-d6, 400 MHz): δ 2.81 - 2.89 (m, 2H), 4.59 (d, J= 10 Hz, 2H), 4.72 (s, 2H), 5.48 (d, J = 24.8 Hz, 1H), 6.29 - 6.34 (m, 2H), 6.50 - 5.59 (m, 2H), 7.09 (d, J= 6.8 Hz, 1H), 7.21 - 7.29 (m, 6H), 9.71 (s, 1H, D
2O exchangeable), 10.53 (s, 1H, D
2O exchangeable).2 protons obscured under water peak. [00456] LCMS (Method A): 2.116 min, MS: ES+ 375.1 (M+1). Step 2: (E)-N-(2-(2,4-Dihydroxybenzoyl) isoindolin-4-yl)-4-(dimethyl amino)-N-phenethylbut-2- enamide
[00457] The title compound was prepared from (E)-4-(dimethylamine) but-2-enoic acid hydrochloride (CAS: 848133-35-7 and (2, 4-dihydroxyphenyl) (4-(phenethylamine) isoindolin-2- yl) methanone (Step 1) analogously to Example 1 step 2. [00458]
1H NMR (DMSO-d6, 400 MHz): δ 2.0 (s, 6H), 2.67 - 2.87 (m, 4H), 3.70 - 3.71 (m, 1H), 3.93 - 4.02 (m, 1H), 4.71 - 4.88 (m, 2H), 4.53 - 4.67 (m, 2H), 5.66 - 5.74 (m, 1H), 6.28 - 6.34 (m, 2H), 6.65 - 6.73 (m, 1H), 7.08 - 7.36 (m, 7H), 7.38 - 7.43 (m, 2H), 9.73 (s, 1H, D2O exchangeable), 10.37 (s, 1H, D2O exchangeable). [00459] LCMS (Method A): 1.409 min, MS: ES+ 486.1 (M+1). [00460] Analytical HPLC (Method D): 5.03 min. Example 11.1: (E)-N-(2-(2-Chloro-4,6-dihydroxybenzoyl)isoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide Step 1: (2-Chloro-4,6-bis(methoxymethoxy)phenyl)(4-(methylamino)isoindolin-2-yl)methanone
[00461] The title compound was prepared from 2-chloro-4,6-bis(methoxymethoxy)benzoic acid (Intermediate P) and N-methylisoindolin-4-amine hydrochloride (Intermediate A) analogously to Example 1, Step 1. Material was used directly in Step 2 [00462] LCMS (Method A): 1.929 min.2.010 min, MS: ES+ 407.0 (M+1). Step 2: (2-Chloro-4,6-dihydroxyphenyl)(4-(methylamino)isoindolin-2-yl)
[00463] The title compound was prepared from (2-chloro-4,6-bis(methoxymethoxy)phenyl)(4- (methylamino)isoindolin-2-yl)methanone (step 1) and 4M HCl in dioxane analogously to Example 4, Step 2. [00464]
1H NMR (DMSO-d6, 400 MHz): δ 2.62 and 2.72 (2 doublets, J= 4.8 Hz, 3H), 4.23 - 4.46 (m, 2H), 4.55 (s, 1H), 4.68 (s, 1H), 5.37 - 5.52 (m, 1H, D
2O exchangeable), 6.34 - 6.40 (m, 3H), 6.46 - 6.57 (m, 1H), 7.07 - 7.14 (m, 1H), 9.97 (s, 2H, D
2O exchangeable). [00465] LCMS (Method A): 1.469 min. MS: ES+ 318.9 (M+1). [00466] Analytical HPLC (Method D): 5.105 min. Step 3: (E)-N-(2-(2-Chloro-4,6-dihydroxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N- methylbut-2-enamide
[00467] The title compound was prepared from (E)-4-(dimethylamino)but-2-enoic acid hydrochloride (CAS: 848133-35-7) and (2-chloro-4,6-dihydroxyphenyl)(4- (methylamino)isoindolin-2-yl)methanone (Step 2) analogously to Example 1, Step 2. [00468] High temperature
1H NMR (D
2O Exchange, DMSO-d6, 400 MHz, 349K): 2.09 (s, 6H), 3.09 (s, 2H), 3.22 and 3.45 (2 singlets, 3H), 4.31 - 4.63 (m, 3H), 4.83 (s, 1H), 5.80 (s, 1H), 6.31 - 6.39 (m, 2H), 6.55 - 6.70 (m, 1H), 7.17 - 7.44 (m, 3H). [00469] LCMS (Method A): 1.045 min. MS: ES+ 430.0 (M+1). [00470] Analytical HPLC (Method D): 3.73 min. Example 12: (E)-4-(Dimethylamino)-N-(2-(6-hydroxy-3,3-dimethylindoline-5- carbonyl)isoindolin-4-yl)-N-methylbut-2-enamide

[00471] To a solution 6-hydroxy-3,3-dimethylindoline-5-carboxylic acid (Intermediate R) (0.07 g, 0.33 mmol, 1 eq.) in DMF (0.7 mL) were added EDC.HCl (0.096 g, 0.50 mmol, 1.5 eq.) and HOAT
(0.045 g, 0.33 mmol, 1 eq.) and the mixture was stirred at room temperature for 15 mins. (E)-4- (Dimethylamino)-N-(isoindolin-4-yl)-N-methylbut-2-enamide hydrochloride (Intermediate S) (0.15 g, 0.50 mmol, 1.5 eq.) and NMM (0.17 g, 1.69 mmol, 5 eq.) were added and stirring continued at room temperature for 2 h. The resulting mixture was diluted with water (20 mL) causing a solid to precipitate. The solid was collected by filtration, washed with water (3 x 5 mL) and purified by preparative HPLC eluting with 0.05% HCl in water/acetonitrile. The product fractions were lyophilized to afford the title compound. (0.012 g, Yield: 6%). [00472]
1H NMR (DMSO-d6, 400 MHz, D
2O exchange): 1.28 (s, 6H), 2.62 (s, 6H), 3.20 (bs, 3H), 3.55 (bs, 2H), 3.74 - 3.93 (m, 2H), 4.60 - 4.64 (m, 1H), 4.80 - 4.87 (m, 3H), 6.04 (d, J= 15.2 Hz, 1H), 6.53 (bs, 1H), 6.71 (s, 1H), 7.18 (bs, 1H), 7.20 - 7.26 (m, 1H), 7.43 - 7.44 (m, 2H). [00473] LCMS (Method A): 1.100 min, MS: ES+ 449.1 (M+1). [00474] Analytical HPLC (Method-D): 3.580 min. Prep. HPLC purification method: [00475] Chromatographic separation and isolation were conducted with Shimadzu LC20AP with uv detector. The column used was Shim-Pack GIST C18 (250 mm x 20 mm x 5 µm) and the compounds were eluted with, Mobile Phase A : 0.05%HCL IN WATER, Mobile Phase B : Acetonitrile with a gradient of T = 0.01 min (90% A, 10% B); gradient to T = 17.00 min (74% A, 26% B); T = 17.01 min (2% A, 98% B) gradient to T = 19.00 min (2% A, 98% B); T = 19.01 min (90% A, 10% B); gradient to T = 23 min (90% A, 10% B); Flow rate= 20 mL/min; analysis time 23 min. Example 13.1: (E)-N-(2-(4,6-Dihydroxy-2,3-dimethylbenzoyl)isoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide

[00476] To a stirred solution of 4,6-dihydroxy-2,3-dimethyl-benzoic acid (Intermediate K) (150 mg, 0.41 mmol, 1.00 eq), HOBT (95 mg, 0.62 mmol, 1.5 eq), EDC (118 mg, 0.62 mmol, 1.5 eq) and (E)-4-(dimethylamino)-N-(isoindolin-4-yl)-N-methylbut-2-enamide dihydrochloride
(Intermediate V1-1) (178 mg, 0.54 mmol, 1.3 eq) in DMF (4.6 mL) was added DIPEA (0.29 mL, 1.65 mmol, 4.00 eq). The reaction mixture was stirred at room temperature for 48 h. MP carbonate resin (3.0 eq) was added and the mixture was agitated for 1 hour using a shaker. The resulting mixture was filtered through a hydrophobic frit and concentrated under reduced pressure. Purification of the crude material by preparative HPLC (Method Prep-LC-2) afforded the title compound as a formate salt (12 mg, 6.4%). [00477] ¹H NMR (400 MHz, DMSO) δ 1.93 - 2.03 (m, 12H), 2.77 - 2.89 (m, 2H), 3.08 - 3.23 (m, 3H), 4.59 - 4.88 (m, 2H), 5.66 - 5.80 (m, 1H), 6.30 - 6.32 (m, 1H), 6.60 - 6.73 (m, 1H), 7.19 - 7.23 (m, 1H), 7.28 - 7.33 (m, 2H), 7.37 - 7.46 (m, 2H), 8.21 (s, 1H). [00478] LCMS (LC-Method 1): 2.66 min, MS: ES+ 424.2 (M+1) [00479] The compounds of the following tabulated Examples (Table Ex13) were prepared analogously to Example 13.1 from the indicated intermediates. Ex. No. Name and Structure Data 13.2 ¹H NMR (400 MHz, DMSO) δ 1.30 (s, 9H), 2.00 (s, 6H), 2.81 - 2.88 (m, 2H), 3.13 - 3.21 (m, 3H), 4.57 - 4.62 (m, 1H), 4.72 - 4.85 (m, 3H), 5.75 (d, J=15.8 Hz, (E)-N-(2-(5-(tert-Butyl)-2,4- 1H), 6.39 (s, 1H), 6.65 (s, 1H), 7.03 (s, dihydroxybenzoyl)isoindolin-4-yl)-4- 1H), 7.21 (d, J=8.3 Hz, 1H), 7.41 - 7.42 (dimethylamino)-N-methylbut-2-enamide (m, 2H), 9.69 (s, 1H), 10.02 - 10.26 (m, Prepared from Intermediate V1-1 and 5- 1H). LCMS (LC-Method 1): 3.34 min, tert-butyl-2,4-dihydroxy-benzoic acid. MS: ES+ 452.2 (M+1) Purified by preparative HPLC using Method Prep-LC-7 13.3 ¹H NMR (400 MHz, DMSO) δ 0.94 - 1.09 (m, 3H), 1.99 - 2.04 (m, 9H), 2.82 - 2.89 (m, 2H), 3.45 - 3.62 (m, 1H), 3.68 - 3.87 (E)-N-(2-(2,4-Dihydroxy-5- (m, 1H), 4.53 - 4.64 (m, 1H), 4.66 - 4.73 methylbenzoyl)isoindolin-4-yl)-4- (m, 1H), 4.81 - 4.89 (m, 2H), 5.63 - 5.73 (dimethylamino)-N-ethylbut-2-enamide (m, 1H), 6.35 - 6.42 (m, 1H), 6.65 (br s, Prepared from Intermediate V1-2 and 2,4- 1H), 6.91 - 7.06 (m, 1H), 7.14 - 7.21 (m, dihydroxy-6-methylbenzoic acid and 1H), 7.32 - 7.49 (m, 2H), 8.18 (s, 1H), purified by preparative HPLC using 9.63 (br s, 1H), 10.09 (br s, 1H). Isolated Method Prep-LC-3 as formate salt. LCMS (LC-Method 1): 2.83 min, MS: ES+ 424.2 (M+1)
¹H NMR (400 MHz, DMSO) δ 2.01 (s, 9H), 2.85 - 2.87 (m, 2H), 3.47 - 3.47 (m, 1H), 3.75 - 3.76 (m, 2H), 4.55 - 4.85 (m, 5H), 6.38 (s, 1H), 6.38 (s, 1H), 6.67 (s, (E)-N-(2-(2,4-Dihydroxy-5- 1H), 6.95 - 7.03 (m, 1H), 7.24 (d, J=7.8 methylbenzoyl)isoindolin-4-yl)-4- Hz, 1H), 7.34 (s, 1H), 7.40 - 7.44 (m, (dimethylamino)-N-(2-hydroxyethyl)but-2- 1H), 8.16 (s, 1H), 9.62 (s, 1H), 10.10 (s, enamide 1H). Isolated as formate salt. LCMS Prepared from Intermediate V1-3 and 2,4- (LC-Method 1): 2.30 min, MS: ES+ dihydroxy-5-methylbenzoic acid. Purified 440.2 (M+1) by preparative HPLC using Method Prep- LC-4 ¹H NMR (400 MHz, DMSO) δ 2.01 - 2.04 (m, 9H), 2.89 (s, 2H), 4.56 - 4.64 (m, 3H), 4.80 - 4.85 (m, 2H), 4.98 - 5.15 (m, 1H), 5.78 (s, 1H), 6.40 - 6.41 (m, 1H), 6.80 (s, 1H), 7.03 (d, J=6.1 Hz, 1H), 7.13 (E)-N-(2-(2,4-Dihydroxy-5- - 7.19 (m, 1H), 7.26 - 7.39 (m, 4H), 8.24 methylbenzoyl)isoindolin-4-yl)-4- (s, 1H), 8.44 - 8.49 (m, 2H), 9.71 (s, 1H), (dimethylamino)-N-(pyridin-4- 10.13 (s, 1H). Isolated as formate salt. ylmethyl)but-2-enamide LCMS (LC-Method 1): 1.99 min, MS: Prepared from Intermediate V1-4 and 2,4- ES+ 487.2 (M+1) dihydroxy-5-methylbenzoic acid. Purified by preparative HPLC using Method Prep- LC-4 ¹H NMR (400 MHz, DMSO) δ 2.05 - 2.07 (m, 9H), 2.88 - 2.91 (m, 4H), 3.80 - 3.86 (m, 1H), 4.07 - 4.14 (m, 1H), 4.59 - 4.65 (m, 1H), 4.73 (s, 1H), 4.88 (s, 2H), 5.71 - 5.80 (m, 1H), 6.43 - 6.46 (m, 1H), 6.73 (m, 1H), 7.05 (m, 1H), 7.13 - 7.18 (m, (E)-N-(2-(2,4-Dihydroxy-5- 1H), 7.27 - 7.31 (m, 2H), 7.43 - 7.47 (m, methylbenzoyl)isoindolin-4-yl)-4- 2H), 8.31 (s, 1H), 8.47 - 8.50 (m, 2H), (dimethylamino)-N-(2-(pyridin-4- 9.69 (s, 1H), 10.16 (s, 1H). Isolated as yl)ethyl)but-2-enamide formate salt. LCMS (LC-Method 1): 1.95 min, MS: ES+ 501.2 (M+1)
Prepared from Intermediate V1-5 and 2,4- dihydroxy-5-methylbenzoic acid. Purified by preparative HPLC using Method Prep- LC-4 ¹H NMR (400 MHz, DMSO) δ 2.09 (s, 9H), 2.91 (d, J=3.3 Hz, 3H), 3.96 (s, 1H), 4.24 - 4.25 (m, 1H), 4.64 - 4.73 (m, 2H), 4.90 (s, 2H), 5.78 (m, 1H), 6.43 - 6.45 (m, 1H), 6.56 - 6.77 (m, 2H), 6.97 - 7.08 (m, 1H), 7.17 - 7.31 (m, 3H), 7.40 - 7.45 (E)-N-(2-(2,4-Dihydroxy-5- (m, 2H), 7.71 (s, 1H), 8.22 (s, 1H), 8.43 methylbenzoyl)isoindolin-4-yl)-4- - 8.50 (m, 1H), 9.63 - 9.72 (m, 1H), 10.16 (dimethylamino)-N-(2-(pyridin-2- (s, 1H). Isolated as formate salt. LCMS yl)ethyl)but-2-enamide (LC-Method 1): 2.17 min, MS: ES+ Prepared from Intermediate V1-6 and 2,4- 501.2 (M+1) dihydroxy-5-methylbenzoic acid. Purified by preparative HPLC using Method Prep- LC-4 ¹H NMR (400 MHz, DMSO) δ 2.00 - 2.02 (m, 9H) , 2.85 (m, 2H), 3.75 (m, 2H), 4.06 (s, 2H), 4.55 - 4.71 (m, 2H), 4.83 - 4.85 (m, 2H), 5.72 (m, 1H), 6.35 - 6.39 (m, 1H), 6.71 (s, 1H), 6.93 - 7.03 (m, 1H), 7.06 - 7.10 (m, 1H), 7.29 (s, 1H), (E)-N-(2-(2,4-Dihydroxy-5- 7.38 - 7.44 (m, 2H), 7.58 - 7.67 (m, 1H), methylbenzoyl)isoindolin-4-yl)-4- 8.38 - 8.42 (m, 2H), 9.65 (s, 1H), 10.10 (dimethylamino)-N-(2-(pyridin-3- (s, 1H). LCMS (LC-Method 1): 2.04 min, yl)ethyl)but-2-enamide MS: ES+ 501.4 (M+1) Prepared from Intermediate V1-7 and 2,4- dihydroxy-5-methylbenzoic acid. Purified by preparative HPLC using Method Prep- LC-4 ¹H NMR (400 MHz, DMSO) δ 1.86 - 2.04 (m, 12H), 2.75 - 2.96 (m, 3H), 3.67 - 4.17 (m, 2H), 4.29 - 4.36 (m, 1H), 4.40 - 4.59 (m, 2H), 4.69 - 4.82 (m, 1H), 5.55 - 5.71 (m, 1H), 6.21 - 6.26 (m, 1H), 6.44
(E)-N-(2-(4,6-Dihydroxy-2,3- - 6.68 (m, 1H), 7.04 - 7.14 (m, 3H), 7.17 dimethylbenzoyl)isoindolin-4-yl)-4- - 7.23 (m, 1H), 7.24 - 7.37 (m, 2H), 7.53 (dimethylamino)-N-(2-(pyridin-2- - 7.62 (m, 1H), 8.33 - 8.37 (m, 1H), 9.16 yl)ethyl)but-2-enamide - 9.20 (m, 2H). LCMS (LC-Method 1): Prepared from Intermediate V1-6 and 4,6- 2.29 min, MS: ES+ 515.3 (M+1) dihydroxy-2,3-dimethylbenzoic acid. Purified by preparative HPLC using Method SFC-1 ¹H NMR (400 MHz, DMSO) δ 1.04 - 1.23 (m, 3H), 1.53 - 1.58 (m, 6H), 1.98 - 2.02 (m, 9H), 2.82 - 2.86 (m, 2H), 3.74 - 3.79 (m, 4H), 4.56 - 4.60 (m, 1H), 4.66 (m, 1H), 4.84 - 4.88 (m, 2H), 5.64 - 5.69 (m, 1H), 6.33 - 6.35 (m, 1H), 6.61 - 6.68 (m, E)-N-(2-(2,4-Dihydroxy-5- 1H), 6.98 (s, 1H), 7.16 - 7.19 (m, 1H), methylbenzoyl)isoindolin-4-yl)-4- 7.40 - 7.44 (m, 2H), 9.88 (s, 1H). LCMS (dimethylamino)-N-(2-(tetrahydro-2H- (LC-Method 1): 2.88 min, MS: ES+ pyran-4-yl)ethyl)but-2-enamide 508.5 (M+1) Prepared from Intermediate V1-8 and 2,4- dihydroxy-5-methylbenzoic acid. Purified by preparative HPLC using Method SFC- 2 ¹H NMR (400 MHz, DMSO) δ 1.98 - 2.05 (m, 9H), 2.85 - 2.88 (m, 2H), 3.07 - 3.19 (m, 5H), 3.79 - 3.88 (m, 2H), 4.50 - 4.60 (m, 1H), 4.69 - 4.77 (m, 1H), 4.83 - 4.87 (m, 2H), 5.64 - 5.75 (m, 1H), 6.36 - 6.43 (E)-N-(2-(2,4-Dihydroxy-5- (m, 1H), 6.56 - 6.77 (m, 1H), 6.88 - 7.08 methylbenzoyl)isoindolin-4-yl)-4- (m, 1H), 7.18 - 7.25 (m, 1H), 7.31 - 7.46 (dimethylamino)-N-(2-methoxyethyl)but- (m, 2H), 9.57 - 9.74 (m, 1H), 10.08 - 2-enamide 10.21 (m, 1H). LCMS (LC-Method 1): Prepared from Intermediate V1-9 and 2,4- 2.67 min, MS: ES+ 454.2 (M+1) dihydroxy-5-methylbenzoic acid. Purified by preparative HPLC using Method Prep- LC-3
¹H NMR (400 MHz, DMSO) δ 2.01 (s, 9H), 2.85 - 2.89 (m, 2H), 4.55 - 4.60 (m, 1H), 4.65 - 4.85 (m, 3H), 5.72 - 5.78 (m, (E)-N-(2-(2,4-Dihydroxy-5- 1H), 6.37 - 6.39 (m, 1H), 6.54 - 6.68 (m, methylbenzoyl)isoindolin-4-yl)-4- 1H), 7.02 - 7.03 (m, 1H), 7.18 - 7.22 (m, (dimethylamino)-N-methylbut-2-enamide 1H), 7.39 - 7.41 (m, 2H), 8.20 (s, 1H), Prepared from Intermediate V1-1 and 2,4- 9.65 (s, 1H), 10.08 (m, 1H). NMe signal dihydroxy-5-methylbenzoic acid. Purified obscured by solvent peak. Isolated as by preparative HPLC using Method Prep- formate salt. LCMS (LC-Method 1): 2.62 LC-3 min, MS: ES+ 410.2 (M+1) ¹H NMR (400 MHz, DMSO) δ 1.90 – 2.10 (m, 9H), 2.80 - 2.87 (m, 2H), 3.17 - 3.22 (m, 3H), 4.57 - 4.68 (m, 4H), 5.72 - (E)-N-(2-(2,4-Dihydroxy-6- 5.80 (m, 1H), 6.08 - 6.13 (m, 2H), 6.64 - methylbenzoyl)isoindolin-4-yl)-4- 6.71 (m, 1H), 7.42 - 7.43 (m, 3H), 8.22 (dimethylamino)-N-methylbut-2-enamide (s, 1H), 9.51 (m, 2H). Isolated as formate Prepared from Intermediate V1-1 and 2,4- salt. LCMS (LC-Method 1): 2.37 min, dihydroxy-6-methylbenzoic acid. Purified MS: ES+ 410.2 (M+1) by preparative HPLC using Method Prep- LC-4 ¹H NMR (400 MHz, DMSO) δ 2.17 (s, 6H), 3.05 (d, J=5.7 Hz, 2H), 4.72 - 4.79 (m, 4H), 6.36 - 6.40 (m, 1H), 6.52 (s, 1H), 6.69 - 6.77 (m, 1H), 7.09 - 7.12 (m, (E)-N-(2-(5-Chloro-2,4- 1H), 7.23 - 7.28 (m, 2H), 7.67 - 7.71 (m, dihydroxybenzoyl)isoindolin-4-yl)-4- 1H), 10.46 (m, 3H). LCMS (LC-Method (dimethylamino)but-2-enamide. Prepared 1): 2.63 min, MS: ES+ 416.2 (M+1) from Intermediate V1-10 and 5-chloro- 2,4-dihydroxybenzoic acid. Purified by preparative HPLC using Method Prep-LC- 5 ¹H NMR (400 MHz, DMSO) δ 2.17 (s, 6H), 3.05 (d, J=5.1 Hz, 2H), 3.70 (s, 3H), 4.77 - 4.82 (m, 4H), 6.39 - 6.41 (m, 2H), 6.71 - 6.72 (m, 1H), 6.86 (s, 1H), 7.10 (s, 1H), 7.27 (dd, J=7.7, 7.7 Hz, 1H), 7.67
(E)-N-(2-(2,4-Dihydroxy-5- (d, J=6.8 Hz, 1H), 9.39 (s, 1H), 9.59 - methoxybenzoyl)isoindolin-4-yl)-4- 9.67 (m, 1H), 9.83 - 9.87 (m, 1H). LCMS (dimethylamino)but-2-enamide (LC-Method 1): 2.36 min, MS: ES+ Prepared from Intermediate V1-10 and 412.2 (M+1) 2,4-dihydroxy-5-methoxybenzoic acid. Purified by preparative HPLC using Method Prep-LC-5 ¹H NMR (400 MHz, DMSO) δ 1.96 (s, 3H), 2.00 - 2.03 (m, 3H), 2.14 (s, 3H), 2.19 (s, 3H), 3.00 - 3.08 (m, 2H), 4.33 - (E)-N-(2-(4,6-Dihydroxy-2,3- 4.42 (m, 1H), 4.52 - 4.57 (m, 1H), 4.73 - dimethylbenzoyl)isoindolin-4-yl)-4- 4.80 (m, 2H), 6.26 - 6.46 (m, 2H), 6.63 - (dimethylamino)but-2-enamide 6.80 (m, 1H), 6.99 - 7.17 (m, 1H), 7.22 - Prepared from Intermediate V1-10 and 7.30 (m, 1H), 7.64 - 7.71 (m, 1H), 9.22 4,6-Dihydroxy-2,3-dimethylbenzoic acid. (s, 2H), 9.47 - 9.69 (m, 1H). LCMS (LC- Purified by preparative HPLC using Method 1): 2.58 min, MS: ES+ 410.2 method SFC-3 (M+1) ¹H NMR (400 MHz, DMSO) δ 2.12 (s, 3H), 2.20 (s, 3H), 3.04 (dd, J=5.9, 41.0 Hz, 2H), 4.71 (d, J=13.0 Hz, 2H), 4.85 (E)-4-(Dimethylamino)-N-(2-(6-hydroxy- (d, J=11.8 Hz, 2H), 6.22 - 6.47 (m, 2H), 1H-indole-5-carbonyl)isoindolin-4-yl)but- 6.55 - 6.80 (m, 1H), 6.90 (s, 1H), 7.01 - 2-enamide 7.30 (m, 3H), 7.47 (d, J=11.6 Hz, 1H), Prepared from Intermediate V1-10 and 6- 7.69 (d, J=7.3 Hz, 1H), 8.18 (s, 1H), 9.49 hydroxy-1H-indole-5-carboxylic acid - 9.72 (m, 2H), 10.86 (s, 1H). Isolated as (CAS1246668-93-8). Purified by formate salt. LCMS (LC-Method 1): 2.62 preparative HPLC using Method Prep-LC- min, MS: ES+ 405.2 (M+1) 2 ¹H NMR (400 MHz, DMSO) δ 2.17 (s, 6H), 3.05 (s, 2H), 4.75 (s, 4H), 6.18 - 6.79 (m, 4H), 7.04 - 7.35 (m, 3H), 7.69 (d, J=8.4 Hz, 1H), 8.27 (s, 1H), 9.20 - (E)-N-(2-(5-Bromo-2,4- 9.90 (m, 1H), 10.00 - 11.5 (m 1H). dihydroxybenzoyl)isoindolin-4-yl)-4- Isolated as formate salt. LCMS (LC- (dimethylamino)but-2-enamide Method 1): 2.71 min, MS: ES+ 460.2 (M+1)
Prepared from Intermediate V1-10 and 5- bromo-2,4-dihydroxybenzoic acid; and purified by preparative HPLC using Method Prep-LC-2 ¹H NMR (400 MHz, DMSO) δ 2.62 - 2.70 (m, 6H), 3.38 – 3.01 (m, 3H), 3.65 - 3.82 (m, 2H), 4.51 - 4.88 (m, 3H), 5.99 - 6.10 (m, 1H), 6.45 - 6.74 (m, 3H), 7.25 - 7.27 (E)-N-(2-(2,4-Dihydroxy-5- (m, 1H), 7.34 - 7.46 (m, 2H), 9.60 - 9.70 (trifluoromethyl)benzoyl)isoindolin-4-yl)- (m, 1H), 10.80 - 10.88 (m, 2H). Isolated 4-(dimethylamino)-N-methylbut-2- as TFA salt. LCMS (LC-Method 1): 2.88 enamide min, MS: ES+ 464.2 (M+1) Prepared from Intermediate V1-1 and 2,4- dihydroxy-5-(trifluoromethyl)benzoic acid (Intermediate T). Purified by preparative HPLC using Method Prep-LC-6 ¹H NMR (400 MHz, DMSO) δ 2.23 (s, 3H, 2.27 (s, 3H), 3.01 - 3.23 (m, 3H), 4.69 - 4.87 (m, 4H), 6.35 - 6.57 (m, 1H), 6.64 (s, 1H), 6.73 - 6.85 (m, 1H), 7.10 - 7.21 (m, 1H), 7.33 (d, J=7.1 Hz, 1H), (E)-N-(2-(5-Cyano-2,4- 7.60 (s, 1H), 7.72 - 7.80 (m, 1H), 8.20 (s, dihydroxybenzoyl)isoindolin-4-yl)-4- 1H), 9.82 – 9.48 (m, 1H), 11.11 - 11.41 (dimethylamino)but-2-enamide (m, 1H). Isolated as formate salt. LCMS Prepared from Intermediate V1-10 and 5- (LC-Method 1): 2.42 min, MS: ES+ cyano-2,4-dihydroxybenzoic acid. 407.2 (M+1) Purified by preparative HPLC using Method Prep-LC-4 ¹H NMR (400 MHz, DMSO) δ 2.14 (s, 3H), 2.19 (s, 3H), 2.99 - 3.09 (m, 2H), 4.37 - 4.45 (m, 1H), 4.51 - 4.59 (m, 1H), (E)-N-(2-(2,4-Dihydroxy-6- 4.71 - 4.78 (m, 2H), 6.26 - 6.44 (m, 1H), (trifluoromethyl)benzoyl)isoindolin-4-yl)- 6.56 - 6.80 (m, 3H), 7.03 - 7.16 (m, 1H), 4-(dimethylamino)but-2-enamide 7.23 - 7.32 (m, 1H), 7.64 - 7.74 (m, 1H), Prepared from Intermediate V1-10 and 8.30 (s, 1H), 9.45 - 9.69 (m, 1H), 10.29 - 2,4-dihydroxy-6-(trifluoromethyl)benzoic 10.70 (m, 2H). Isolated as formate salt.
acid (Intermediate U) and purified using LCMS (LC-Method 2): 3.35 min, MS: Method Prep-LC-2 ES+ 450.3 (M+1) ¹H NMR (400 MHz, DMSO) δ 2.03 (s, 3H), 2.18 (s, 6H), 3.00 - 3.06 (m, 2H), 4.76 - 4.81 (m, 4H), 6.30 - 6.44 (m, 2H), 6.66 - 6.80 (m, 1H), 7.01 - 7.18 (m, 2H), 7.26 (dd, J=8.0, 8.0 Hz, 1H), 7.67 (d, (E)-N-(2-(2,4-Dihydroxy-5- J=8.1 Hz, 1H), 8.22 (s, 1H), 9.54 - 10.19 methylbenzoyl)isoindolin-4-yl)-4- (m, 3H). Isolated as formate salt. LCMS (dimethylamino)but-2-enamide (LC-Method 1): 2.57 min, MS: ES+ Prepared from Intermediate V1-10 and 396.2 (M+1) 2,4-dihydroxy-5-methylbenzoic acid. Purified by preparative HPLC using Method Prep-LC-4 ¹H NMR (400 MHz, DMSO) δ 2.14 (s, 3H), 2.19 (s, 3H), 3.00 - 3.08 (m, 2H), 4.77 (d, J=7.6 Hz, 2H), 4.87 (d, J=11.4 Hz, 2H), 6.25 - 6.47 (m, 1H), 6.64 - 6.80 (E)-N-(2-Benzoylisoindolin-4-yl)-4- (m, 1H), 7.02 - 7.18 (m, 1H), 7.23 - 7.32 (dimethylamino)but-2-enamide (m, 1H), 7.47 - 7.52 (m, 3H), 7.61 (dd, Prepared from Intermediate V1-10 and J=2.1, 5.4 Hz, 2H), 7.69 (d, J=8.3 Hz, Benzoic acid. Purified by preparative 1H), 9.50 - 9.70 (m, 1H). LCMS (LC- HPLC using Method Prep-LC-1 Method 1): 2.79 min, MS: ES+ 350.2 (M+1) ¹H NMR (400 MHz, DMSO) δ 2.15 (s, 3H), 2.20 (s, 3H), 3.00 - 3.10 (m, 2H), 3.75 (d, J=1.9 Hz, 3H), 4.53 (s, 2H), 4.77 (d, J=15.9 Hz, 2H), 6.27 - 6.46 (m, 2H), (E)-4-(Dimethylamino)-N-(2-(4-hydroxy- 6.48 - 6.51 (m, 1H), 6.64 - 6.80 (m, 1H), 2-methoxybenzoyl)isoindolin-4-yl)but-2- 7.01 - 7.16 (m, 2H), 7.23 - 7.31 (m, 1H), enamide 7.60 - 7.70 (m, 1H), 8.22 (formate, 1H), Prepared from Intermediate V1-10 and 4- 9.48 - 10.03 (m, 2H). Isolated as formate hydroxy-2-methoxybenzoic acid. Purified salt. LCMS (LC-Method 2): 2.91 min, by preparative HPLC using Method Prep- MS: ES+ 396.2 (M+1) LC-4
¹H NMR (400 MHz, DMSO) δ 2.14 (s, 3H), 2.19 (s, 3H), 2.99 - 3.09 (m, 2H), 4.60 (s, 2H), 4.84 (d, J=13.4 Hz, 2H), 6.26 - 6.45 (m, 1H), 6.64 - 6.88 (m, 1H), (E)-N-(2-(2-(Difluoromethyl)-4- 6.96 - 7.07 (m, 2H), 7.14 - 7.31 (m, 2H), hydroxybenzoyl)isoindolin-4-yl)-4- 7.48 (d, J=9.8 Hz, 1H), 7.67 - 7.73 (m, (dimethylamino)but-2-enamide 1H), 9.45 - 9.70 (m, 1H), 10.10 - 10.48 Prepared from Intermediate V1-10 and 2- (m, 1H). LCMS (LC-Method 1): 2.65 min, (difluoromethyl)-4-hydroxybenzoic acid MS: ES+ 416.2 (M+1) (CAS 2551117-27-0). Purified by preparative HPLC using Method Prep-LC- 1 ¹H NMR (400 MHz, DMSO) δ 2.27 (s, 3H), 2.34 (s, 3H), 3.17 - 3.25 (m, 2H), 4.53 (s, 2H), 4.82 (d, J=14.3 Hz, 2H), 6.30 - 6.49 (m, 1H), 6.65 - 6.81 (m, 2H), (E)-N-(2-(2-Chloro-4- 6.81 - 6.86 (m, 2H), 6.91 (t, J=2.7 Hz, hydroxybenzoyl)isoindolin-4-yl)-4- 1H), 7.04 - 7.33 (m, 2H), 7.70 (dd, J=8.6, (dimethylamino)but-2-enamide 19.5 Hz, 1H), 8.14 (formate salt, 0.3H), Prepared from Intermediate V1-10 and 2- 9.51 - 9.77 (m, 1H), 10.22 (s, 1H). chloro-4-hydroxybenzoic acid. Purified by Isolated as formate salt. LCMS (LC- preparative HPLC using Method Prep-LC- Method 2): 3.10 min, MS: ES+ 400.0 3 (M+1) N ¹H NMR (400 MHz, DMSO) δ 2.11 (s, O 3H), 2.21 (s, 3H), 2.96 - 2.99 (m, 1H), HN N N O 3.08 - 3.10 (m, 1H), 4.60 - 4.66 (m, 2H), N H OH 4.80 - 4.86 (m, 2H), 6.20 - 6.26 (m, (E)-4-(Dimethylamino)-N-(2-(6-hydroxy- 0.5H), 6.41 - 6.47 (m, 0.5H), 6.61 - 6.68 1H-indazole-5-carbonyl)isoindolin-4- (m, 0.5H), 6.73 - 6.81 (m, 0.5H), 6.93 - yl)but-2-enamide 6.94 (m, 1H), 7.01 (m, 0.5H), 7.15 - 7.18 Prepared from Intermediate V1-10 and 6- (m, 0.5H), 7.22 - 7.31 (m, 1H), 7.65 - hydroxy-1H-indazole-5-carboxylic acid 7.71 (m, 2H), 7.95 (s, 1H), 8.16 (s, 0.5H), (CAS 574758-53-5). Purified by 9.44 (s, 0.5H), 9.70 (s, 0.5H), 10.07 - preparative HPLC using Method Prep-LC- 10.18 (m, 1H), 12.69 - 12.75 (m, 1H). 4 Isolated as formate salt. LCMS (LC-
Method 1): 2.23 min, MS: ES+ 406.0 (M+1) ¹H NMR (400 MHz, DMSO) δ 2.06 (s, 3H), 2.15 (s, 3H), 2.20 (s, 3H), 3.00 - 3.09 (m, 2H), 4.36 - 4.42 (m, 1H), 4.55 - 4.61 (m, 1H), 4.74 - 4.80 (m, 2H), 6.12 (E)-N-(2-(2,4-Dihydroxy-6- (d, J=2.3 Hz, 1H), 6.20 (d, J=2.0 Hz, 1H), methylbenzoyl)isoindolin-4-yl)-4- 6.27 - 6.32 (m, 0.5H), 6.40 - 6.45 (m, (dimethylamino)but-2-enamide 0.5H), 6.64 - 6.80 (m, 1H), 7.03 (m, Prepared from Intermediate V1-10 and 0.5H), 7.15 (m, 0.5H), 7.22 - 7.30 (m, 2,4-dihydroxy-6-methylbenzoic acid. 1H), 7.67 (dd, J=5.2, 8.2 Hz, 1H), 8.17 Purified by preparative HPLC using (s, 0.6H), 9.29 - 9.33 (m, 1H), 9.47 (s, Method Prep-LC-4 0.5H), 9.50 (s, 1H), 9.67 (s, 0.5H). Isolated as formate salt. LCMS (LC- Method 1): 2.29 min, MS: ES+ 396.2 (M+1) ¹H NMR (400 MHz, DMSO) δ 2.14 (s, 3H), 2.20 (s, 3H), 3.01 (dd, J=1.4, 5.7 Hz, 1H), 3.09 (dd, J=0.9, 5.2 Hz, 1H), (E)-N-(2-(4-(Difluoromethyl)-2- 4.62 (s, 2H), 4.81 (d, J=14.9 Hz, 2H), hydroxybenzoyl)isoindolin-4-yl)-4- 6.25 - 6.46 (m, 1H), 6.64 - 6.80 (m, 1H), (dimethylamino)but-2-enamide 7.00 - 7.10 (m, 2H), 7.11 (s, 1H), 7.14 - Prepared from Intermediate V1-10 and 4- 7.18 (m, 1H), 7.23 - 7.32 (m, 1H), 7.38 (difluoromethyl)-2-hydroxybenzoic acid (d, J=7.8 Hz, 1H), 7.66 - 7.73 (m, 1H), (CAS 2551117-27-0). Purified by 8.16 (s, 0.3H), 9.49 (s, 0.5H), 9.69 (s, preparative HPLC using Method Prep-LC- 0.5H), 10.40 - 10.46 (m, 1H). Isolated as 2 formate salt. LCMS (LC-Method 1): 2.83 min, MS: ES+ 416.2 (M+1) ¹H NMR (400 MHz, DMSO) δ 2.17 (s, 6H), 3.05 (d, J=5.6 Hz, 2H), 4.77 (d, J=11.1 Hz, 4H), 6.38 - 6.40 (m, 1H), 6.50 (E)-4-(Dimethylamino)-N-(2-(5-fluoro-2,4- - 6.55 (m, 1H), 6.68 – 6.80 (m, 1H), 7.09 dihydroxybenzoyl)isoindolin-4-yl)but-2- (s, 1H), 7.12 (s, 1H), 7.27 (dd, J=7.8, 7.8 enamide Hz, 1H), 7.69 (d, J=7.6 Hz, 1H), 9.60 - Prepared from Intermediate V1-10 and 5- 9.61 (m, 1H), 10.14 - 10.24 (m, 2H). fluoro-2,4-dihydroxybenzoic acid. Purified
by preparative HPLC using Method Prep- LCMS (LC-Method 1): 2.42 min, MS: LC-1 ES+ 400.2 (M+1) ¹H NMR (400 MHz, DMSO) δ 2.17 - 2.23 (m, 6H), 3.04 (s, 2H), 3.09 (s, 2H), 4.63 - 4.85 (m, 4H), 6.26 - 6.48 (m, 1H), 6.50 (E)-4-(Dimethylamino)-N-(2-(6- - 6.59 (m, 1H), 6.67 - 6.81 (m, 1H), 7.02 hydroxybenzo[d]isoxazole-5- - 7.19 (m, 1H), 7.24 - 7.31 (m, 1H), 7.54 carbonyl)isoindolin-4-yl)but-2-enamide (s, 1H), 7.64 - 7.76 (m, 1H), 9.44 - 9.71 Prepared from Intermediate V1-10 and 6- (m, 1H), 11.01 - 11.26 (m, 1H). 1H hydroxybenzo[d]isoxazole-5-carboxylic Missing (assume NH). LCMS (LC- acid (Intermediate Z3) Purified by Method 1): 2.49 min, MS: ES+ 407.4 preparative HPLC using Method Prep-LC- (M+1) 1 ¹H NMR (400 MHz, DMSO) δ 1.13 (d, J=6.8 Hz, 6H), 2.20 (s, 6H), 2.53 (d, J=6.8 Hz, 1H), 3.03 - 3.13 (m, 2H), 4.78 (s, 4H), 6.39 (s, 2H), 6.70 - 6.71 (m, 1H), 7.05 - 7.06 (m, 3H), 7.26 - 7.27 (m, 1H), (E)-N-(2-(2,4-Dihydroxy-5- 8.18 (s, 0.5H formate), 9.61 - 9.68 (m, isopropylbenzoyl)isoindolin-4-yl)-4- 2H), 10.01 - 10.22 (m, 1H). Isolated as (dimethylamino)but-2-enamide formate salt. LCMS (LC-Method 1): 3.01 Prepared from Intermediate V1-10 and min, MS: ES+ 424.2 (M+1) 2,4-dihydroxy-5-(1-methylethyl)benzoic acid. Purified by preparative HPLC using Method Prep-LC-2 ¹H NMR (400 MHz, DMSO) δ 2.18 (s, 6H), 3.04 - 3.07 (m, 2H), 4.84 - 4.98 (m, 4H), 5.60 (s, 2H), 6.05 (d, J=2.3 Hz, 1H), (E)-N-(2-(4-Amino-2- 6.09 - 6.13 (m, 1H), 6.36 - 6.46 (m, 1H), hydroxybenzoyl)isoindolin-4-yl)-4- 6.70 - 6.78 (m, 1H), 7.09 - 7.12 (m, 1H), (dimethylamino)but-2-enamide 7.25 - 7.31 (m, 2H), 7.70 (d, J=8.1 Hz, Prepared from Intermediate V1-10 and 4- 1H), 9.59 - 9.67 (m, 1H), 11.21 - 11.31 amino-2-hydroxybenzoic acid. Purified by (m, 1H). LCMS (LC-Method 1): 2.21 preparative HPLC using Method Prep-LC- min, MS: ES+ 381.2 (M+1) 1
¹H NMR (400 MHz, DMSO) δ 1.10 (t, J=7.5 Hz, 3H), 2.12 - 2.22 (m, 6H), 2.45 (q, J=7.6 Hz, 2H), 3.01 - 3.09 (m, 2H), 4.75 - 4.83 (m, 4H), 6.26 - 6.41 (m, 2H), (E)-4-(Dimethylamino)-N-(2-(5-ethyl-2,4- 6.62 - 6.82 (m, 2H), 7.02 - 7.06 (m, 1H), dihydroxybenzoyl)isoindolin-4-yl)but-2- 7.23 - 7.31 (m, 1H), 7.59 - 7.73 (m, 1H), enamide 8.36 (s, 1H), 9.57 - 9.72 (m, 2H), 10.07 - Prepared from Intermediate V1-10 and 5- 10.21 (m, 1H). Isolated as formate salt. ethyl-2,4-dihydroxybenzoic acid. Purified LCMS (LC-Method 1): 2.81 min, MS: by preparative HPLC using Method Prep- ES+ 410.2 (M+1) LC-4 ¹H NMR (400 MHz, DMSO) δ 1.12 (6H, d, J=6.3 Hz), 1.99 - 2.02 (6H, m), 2.81 - 2.89 (2H, m), 3.20 – 3.05 (4H, m), 4.53 - 4.93 (4H, m), 5.70 - 5.79 (1H, m), 6.34 - (E)-N-(2-(2,4-Dihydroxy-5- 6.42 (1H, m), 6.54 - 6.72 (1H, m), 7.00 isopropylbenzoyl)isoindolin-4-yl)-4- (1H, s), 7.19 - 7.23 (1H, m), 7.32 - 7.46 (dimethylamino)-N-methylbut-2-enamide (2H, m), 8.31 (0.4H, s), 9.64 (1H, s), 9.95 Prepared from Intermediate V1-1 and 2,4- - 10.01 (1H, m). Isolated as formate salt. dihydroxy-5-(1-methylethyl)benzoic acid. LCMS (LC-Method 1): 3.04 min, MS: Purified by preparative HPLC using ES+ 438.2 (M+1) Method Prep-LC-2 ¹H NMR (400 MHz, DMSO) δ 2.08 - 1.99 (m, 6H), 2.24 - 2.15 (m, 3H), 2.88 - 2.82 (m, 2H), 3.20 - 3.07 (m, 3H), 4.97 - 4.37 (m, 4H), 5.80 - 5.70 (m, 1H), 6.80 - 6.53 (E)-N-(2-(3-Chloro-4-(difluoromethoxy)-6- (m, 3H), 7.27 - 7.23 (m, 2H), 7.47 - 7.39 hydroxy-2-methylbenzoyl)isoindolin-4-yl)- (m, 2H), 8.28 (s, 1H). Isolated as a 4-(dimethylamino)-N-methylbut-2- formate salt. enamide
19F NMR (376 MHz, DMSO) d -81.7 (s), Prepared from (E)-4-(dimethylamino)-N- -82.1 (s) (isoindolin-4-yl)-N-methylbut-2-enamide LCMS (LC-Method 3): 3.23 min, dihydrochloride (Intermediate V1-1) and MS:ES+ 494.3 (M+1) 3-chloro-4-(difluoromethoxy)-6-hydroxy- 2-methyl-benzoic acid (Intermediate Z4).
Purified by preparative HPLC using Method prep-LC-3 ¹H NMR (400 MHz, DMSO) δ 1.19 - 1.14 (m, 6H), 2.02 (s, 6H), 2.91 - 2.76 (m, 3H), 3.22 - 3.08 (m, 3H), 4.94 - 4.44 (m, 4H), 5.79 - 5.69 (m, 1H), 6.72 - 6.54 (m, (E)-4-(Dimethylamino)-N-(2-(2-hydroxy- 1H), 6.87 - 6.80 (m, 1H), 7.06 - 7.02 (m, 5-isopropylbenzoyl)isoindolin-4-yl)-N- 1H), 7.16 - 7.11 (m, 1H), 7.24 - 7.21 (m, methylbut-2-enamide 1H), 7.46 - 7.32 (m, 2H), 8.25 (s, 1H), Prepared from (E)-4-(dimethylamino)-N- 9.79 (s, 1H). Isolated as formate salt. (isoindolin-4-yl)-N-methylbut-2-enamide LCMS (LC-Method 3): 3.31 min, dihydrochloride (Intermediate V1-1) and MS:ES+ 422.6 (M+1) 2-hydroxy-5-isopropyl-benzoic acid CAS 31589-71-6. Purified by preparative HPLC using Method prep-LC-3 ¹H NMR (400 MHz, DMSO) δ 1.24 (d, J=5.8 Hz, 9H), 2.02 (s, 6H), 2.87 (d, J=5.8 Hz, 2H), 3.22 - 3.06 (m, 3H), 4.96 - 4.45 (m, 4H), 5.80 - 5.70 (m, 1H), 6.72 - 6.54 (m, 1H), 6.88 - 6.80 (m, 1H), 7.17 (E)-N-(2-(5-(tert-Butyl)-2- (d, J=2.7 Hz, 1H), 7.22 (d, J=8.4 Hz, 1H), hydroxybenzoyl)isoindolin-4-yl)-4- 7.46 - 7.26 (m, 3H), 8.23 (s, 0.5H), 10.16 (dimethylamino)-N-methylbut-2-enamide - 9.57 (m, 1H). Isolated as partial Prepared from (E)-4-(dimethylamino)-N- formate salt. (isoindolin-4-yl)-N-methylbut-2-enamide LCMS (LC-Method 3): 3.23 min, dihydrochloride (Intermediate V1-1) and MS:ES+ 436.6 (M+1) 5-(tert-butyl)-2-hydroxybenzoic acid CAS 16094-31-8. Purified by preparative HPLC using Method prep-LC-3 ¹H NMR (400 MHz, DMSO) δ 2.02 (d, J=4.4 Hz, 6H), 2.13 (d, J=7.4 Hz, 3H), 2.91 - 2.82 (m, 2H), 3.21 - 3.07 (m, 3H), 4.92 - 4.49 (m, 4H), 5.79 - 5.68 (m, 1H), 6.73 - 6.56 (m, 2H), 7.23 - 7.00 (m, 3H),
(E)-N-(2-(4-(Difluoromethoxy)-2-hydroxy- 7.46 - 7.32 (m, 2H), 8.29 (s, 1H), 10.70 - 5-methylbenzoyl)isoindolin-4-yl)-4- 10.05 (m, 1H). Isolated as formate salt. (dimethylamino)-N-methylbut-2-enamide
19F NMR (376 MHz, DMSO) δ -81.0 (d, Prepared from (E)-4-(dimethylamino)-N- J=19.0 Hz) (isoindolin-4-yl)-N-methylbut-2-enamide LCMS (LC-Method 3): 3.49 min, dihydrochloride (Intermediate V1-1) and MS:ES+ 460.5 (M+1) 4-(difluoromethoxy)-2-hydroxy-5- methylbenzoic acid CAS 2384115-25-5. Purified by preparative HPLC using Method prep-LC-3 ¹H NMR (400 MHz, DMSO) δ 2.15 - 2.04 (m, 12H), 2.93 - 2.88 (m, 2H), 3.26 - 3.08 (m, 3H), 4.65 - 4.23 (m, 3H), 4.96 - 4.80 (m, 1H), 5.83 - 5.71 (m, 1H), 6.77 - 6.58 (E)-4-(Dimethylamino)-N-(2-(4-hydroxy- (m, 2H), 6.97 - 6.89 (m, 1H), 7.34 - 7.22 2,3-dimethylbenzoyl)isoindolin-4-yl)-N- (m, 2H), 7.50 - 7.41 (m, 1H), 8.19 (s, methylbut-2-enamide 1H), 9.54 - 9.53 (m, 1H). Isolated as Prepared from (E)-4-(dimethylamino)-N- formate salt. (isoindolin-4-yl)-N-methylbut-2-enamide LCMS (LC-Method 3): 2.58 min, dihydrochloride (Intermediate V1-1) and MS:ES+ 408.5 (M+1) 4-hydroxy-2,3-dimethylbenzoic acid CAS 5628-59-1. Purified by preparative HPLC using Method prep-LC-3 ¹H NMR (400 MHz, DMSO) δ 1.38 - 1.32 (m, 9H), 2.02 - 1.94 (m, 6H), 2.88 - 2.77 (m, 2H), 3.21 - 3.12 (m, 3H), 4.66 - 4.57 (m, 1H), 4.94 - 4.86 (m, 3H), 5.80 - 5.67 (m, 1H), 6.72 - 6.52 (m, 1H), 6.88 - 6.79 (E)-N-(2-(3-(tert-Butyl)-4- (m, 1H), 7.23 - 7.19 (m, 1H), 7.43 - 7.32 hydroxybenzoyl)isoindolin-4-yl)-4- (m, 4H), 8.20 (s, 0.5H), 9.91 (s, 1H). (dimethylamino)-N-methylbut-2-enamide Isolated as partial formate salt. Prepared from (E)-4-(dimethylamino)-N- LCMS (LC-Method 3): 3.34 min, (isoindolin-4-yl)-N-methylbut-2-enamide MS:ES+ 436.6 (M+1) dihydrochloride (Intermediate V1-1) and 3-(tert-butyl)-4-hydroxybenzoic acid CAS 66737-88-0..
Purified by preparative HPLC using Method prep-LC-3 ¹H NMR (400 MHz, DMSO) δ 2.04 - 1.96 (m, 6H), 2.17 - 2.11 (m, 3H), 2.86 - 2.83 (m, 2H), 3.22 - 3.11 (m, 3H), 4.65 - 4.54 (m, 1H), 4.91 - 4.78 (m, 3H), 5.79 - 5.69 (E)-4-(Dimethylamino)-N-(2-(4-hydroxy- (m, 1H), 6.71 - 6.56 (m, 1H), 6.85 - 6.81 3-methylbenzoyl)isoindolin-4-yl)-N- (m, 1H), 7.45 - 7.20 (m, 5H), 8.16 (s, methylbut-2-enamide 1H), 9.88 - 9.76 (m, 1H). Isolated as Prepared from (E)-4-(dimethylamino)-N- formate salt. (isoindolin-4-yl)-N-methylbut-2-enamide LCMS (LC-Method 3): 2.62 min, dihydrochloride (Intermediate V1-1) and MS:ES+ 394.5 (M+1) 4-hydroxy-3-methylbenzoic acid CAS 499-76-3 Purified by preparative HPLC using Method prep-LC-2 ¹H NMR (400 MHz, DMSO) δ 1.21 - 1.15 (m, 6H), 2.07 - 1.92 (m, 6H), 2.87 - 2.83 (m, 2H), 3.24 - 3.10 (m, 4H), 4.66 - 4.58 (m, 1H), 4.94 - 4.78 (m, 3H), 5.78 - 5.70 (E)-4-(Dimethylamino)-N-(2-(4-hydroxy- (m, 1H), 6.72 - 6.53 (m, 1H), 6.85 - 6.81 3-isopropylbenzoyl)isoindolin-4-yl)-N- (m, 1H), 7.43 - 7.19 (m, 5H), 8.16 (s, methylbut-2-enamide 1H), 9.84 (s, 1H). Isolated as formate Prepared from (E)-4-(dimethylamino)-N- salt. (isoindolin-4-yl)-N-methylbut-2-enamide LCMS (LC-Method 3): 3.04 min, dihydrochloride (Intermediate V1-1) and MS:ES+ 422.6 (M+1) 4-hydroxy-3-isopropylbenzoic acid CAS 859034-02-9. Purified by preparative HPLC using Method prep-LC-2 ¹H NMR (400 MHz, DMSO) δ 1.23 - 1.16 (m, 6H), 2.19 - 2.13 (m, 6H), 3.08 - 3.01 (m, 2H), 3.25 - 3.19 (m, 1H), 4.85 - 4.79 (m, 4H)), 6.47 - 6.26 (m, 1H), 6.79 - 6.65 (m, 1H), 6.85 - 6.83 (m, 1H), 7.18 - 7.02 (m, 1H), 7.35 - 7.24 (m, 2H), 7.39 (s,
(E)-4-(Dimethylamino)-N-(2-(4-hydroxy- 1H), 7.72 - 7.56 (m, 1H), 8.19 (s, 0.5H), 3-isopropylbenzoyl)isoindolin-4-yl)but-2- 9.69 - 9.58 (m, 1H), 9.83 - 9.80 (m, 1H). enamide Isolated as a partial formate salt. Prepared from tert-Butyl 4- LCMS (LC-Method 3): 3.00 min, aminoisoindoline-2-carboxylate (CAS MS:ES+ 408.2 (M+1) 871013-98-8) (Intermediate V1-10) and 4- hydroxy-3-isopropylbenzoic acid CAS 859034-02-9. Purified by preparative HPLC using Method prep-LC-3 ¹H NMR (400 MHz, DMSO) δ 2.04 - 1.98 (m, 6H), 2.86 - 2.79 (m, 2H), 3.19 - 3.06 (m, 3H), 4.66 - 4.58 (m, 1H), 4.95 - 4.81 (E)-4-(Dimethylamino)-N-(2-(4-hydroxy- (m, 3H), 5.79 - 5.70 (m, 1H), 6.77 - 6.49 3-(trifluoromethyl)benzoyl)isoindolin-4- (m, 1H), 7.11 - 7.06 (m, 1H), 7.21 - 7.21 yl)-N-methylbut-2-enamide (m, 1H), 7.44 - 7.35 (m, 2H), 7.77 - 7.70 (m, 2H), 8.23 (s, 0.5H formate). Missing Prepared from (E)-4-(Dimethylamino)-N- one exchangeable proton. Isolated as a (isoindolin-4-yl)-N-methylbut-2-enamide formate salt. dihydrochloride (Intermediate V1-1) and
19F NMR (400 MHz, DMSO) -61 ppm 4-hydroxy-3-(trifluoromethyl)benzoic acid LCMS (LC-Method 3): 3.00 min, CAS: 220239-68-9. MS:ES+ 448.4 (M+1) Purified by preparative HPLC using Method Prep-LC-2 ¹H NMR (400 MHz, DMSO) δ 2.11 - 1.99 (m, 6H), 2.25 - 2.20 (m, 3H), 2.99 - 2.82 (m, 2H), 3.22 - 3.05 (m, 3H), 4.93 - 4.23 (E)-N-(2-(3-Chloro-4-hydroxy-2- (m, 4H), 5.74 (dd, J=15.1, 28.5 Hz, 1H), methylbenzoyl)isoindolin-4-yl)-4- 6.72 - 6.54 (m, 1H), 6.89 (dd, J=8.3, 15.8 (dimethylamino)-N-methylbut-2-enamide Hz, 1H), 7.09 (dd, J=8.3, 15.3 Hz, 1H), Prepared from (E)-4-(dimethylamino)-N- 7.45 - 7.18 (m, 3H), 8.37 (s, 0.5H, (isoindolin-4-yl)-N-methylbut-2-enamide formate), 10.49 (s, 1H). Isolated as a 0.5 dihydrochloride (Intermediate V1-1) and eq formate salt. 3-chloro-4-hydroxy-2-methylbenzoic acid LCMS (LC-Method 4): 2.25 min, (Example 78 Step 2) MS:ES+ 428.34 (M+1)
Purified by preparative HPLC using Method Prep-LC-3 Example 14: N-[2-(2,4-Dihydroxy-5-methyl-benzoyl)isoindolin-4-yl]-N-[2-(4- pyridyl)ethyl]prop-2-enamide
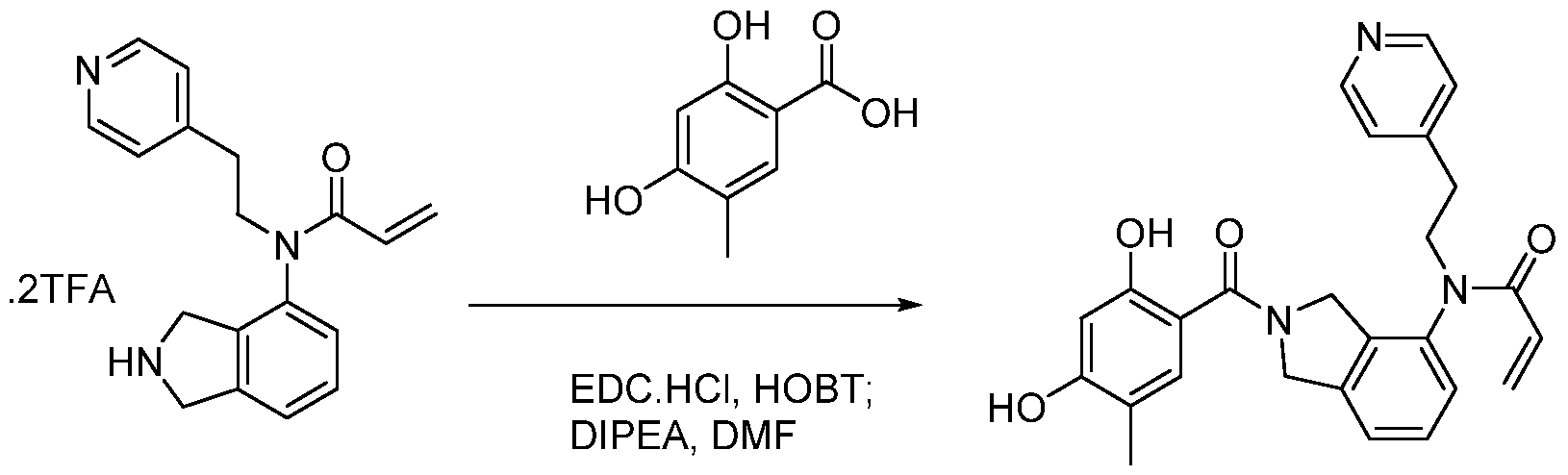
[00480] To a stirred solution of 2,4-dihydroxy-5-methyl-benzoic acid (30 mg, 0.18 mmol, 1.5 eq) in DCM (0.50 mL) was added EDC (34 mg, 0.18 mmol, 1.5 eq) and DIPEA (0.081 mL, 0.47 mmol, 4.0 eq) under an inert atmosphere. In a separate round bottom flask, a solution of N-isoindolin- 4-yl-N-[2-(4-pyridyl)ethyl]prop-2-enamide bistrifluoroacetate (Intermediate W2-1) (62 mg, 0.12 mmol, 1.0 eq) in DMF (1.0 mL) and DIPEA (0.041 mL, 0.23 mmol, 2.0 eq) were combined. This solution was added to the pre-formed ester and stirred overnight at room temperature. The resulting mixture was concentrated under reduced pressure and the crude residue was purified by chromatography on silica eluting with a gradient of 0-50% MeOH in DCM. The product was further purified by preparative HPLC (Method Prep-LC-3) to afford the title compound (1.8 mg, 3.2%). [00481] ¹H NMR (400 MHz, DMSO) δ 2.07 (3H, s), 2.79 - 2.91 (2H, m), 3.97 (2H, s), 4.69 (2H, s), 4.92 (2H, s), 5.57 (1H, d, J = 7.7 Hz), 6.02 (1H, s), 6.17 - 6.23 (1H, m), 6.40 (1H, s), 7.06 - 7.11 (2H, m), 7.18 (1H, d, J = 5.7 Hz), 7.33 - 7.43 (2H, m), 8.42 (2H, d, J = 5.3 Hz), 8.50 (1H, s), 9.31 (1H, s), 9.97 (1H, s). [00482] LCMS (LC-Method 1): 2.76 min, MS: ES+ = 444 (M+1). [00483] The compounds of the following tabulated Examples (Table Ex14) were prepared analogously to Example 14 from the appropriate acid and intermediates. Ex. No. Name and Structure Data
¹H NMR (400 MHz, DMSO) δ 1.22 - 1.29 (m, 1H), 1.45 - 1.49 (m, 2H), 1.86 (s, 1H), 1.99 - 2.02 (m, 5H), 2.17 (d, J = 1.8 Hz, 3H), 2.27 - 2.34 (m, 2H), 2.41 (d, J = 7.1 Hz, 1H), 3.48 (m, 1H), 3.67 - 3.75 (m, 1H), 4.55 (d, J= 15.4 Hz, N-(2-(2,4-Dihydroxy-5- 1H), 4.69 (d, J = 15.8 Hz, 1H), 4.84 - methylbenzoyl)isoindolin-4-yl)-N-(2-(1- 4.88 (m, 2H), 5.57 (d, J = 10.4 Hz, 1H), methylpyrrolidin-3-yl)ethyl)acrylamide 5.84 - 5.93 (m, 1H), 6.11 - 6.22 (m, Prepared from Intermediate W2-2 and 1H), 6.35 - 6.38 (m, 1H), 6.99 - 7.01 2,4-dihydroxy-5-methylbenzoic acid. (m, 1H), 7.17 (d, J = 6.8 Hz, 1H), 7.39 Purified by preparative HPLC using - 7.45 (m, 2H), 9.78 - 10.13 (m, 2H). method SFC-3 LCMS (LC-Method 1): 2.73 min, MS ES+ = 450 (M+1) ¹H NMR (400 MHz, DMSO) δ 2.03 (s, 3H), 3.13 - 3.20 (m, 3H), 4.55 - 4.87 (m, 4H), 5.56 - 6.70 (m, 4H), 6.93 - N-(2-(2,4-Dihydroxy-5- 7.43 (m, 4H), 9.68 - 10.14 (m, 2H). methylbenzoyl)isoindolin-4-yl)-N- LCMS (LC-Method 1): 3.51 min, MS methylacrylamide ES+ 353 (M+1). Prepared from Intermediate W2 and 2,4- dihydroxy-5-methylbenzoic acid. Purified by preparative HPLC using Method Prep-LC-3 ¹H NMR (400 MHz, DMSO) δ 1.30 (s, 9H), 2.54 (s, 1H), 3.19 - 3.20 (m, 2H), 3.36 - 3.43 (m, 2H), 4.59 (m, 2H), 4.83 - 4.86 (m, 4H), 5.54 (m, 1H), 6.40 (s, N-(2-(5-(tert-Butyl)-2,4- 1H), 7.04 (s, 1H), 7.22 (d, J=8.0 Hz, dihydroxybenzoyl)isoindolin-4- 1H), 7.40 - 7.42 (m, 2H). yl)acrylamide. LCMS (LC-Method 1): 4.41 min, MS: Prepared from Intermediate W2 and 5- ES+ 381.2 (M+1) (1,1-dimethylethyl)-2,4- dihydroxybenzoic acid. Purified by preparative HPLC using Method Prep- LC-9
14.4 ¹H NMR (400 MHz, DMSO) δ 1.30 - 1.33 (m, 9H), 4.81 (s, 4H), 5.75 - 5.78 (m, 1H), 6.24 - 6.29 (m, 1H), 6.39 - 6.41 (m, 2H), 7.10 (s, 2H), 7.25 - 7.32 N-(2-(5-(tert-Butyl)-2,4- (m, 1H), 7.58 - 7.61 (m, 1H), 9.77 - dihydroxybenzoyl)isoindolin-4-yl)-N- 9.80 (m, 2H), 10.03 - 10.48 (m, 1H). methylacrylamide LCMS (LC-Method 1): 4.49 min, MS: Prepared from Intermediate W2-4 and 5- ES+ 395.4 (M+1). (1,1-dimethylethyl)-2,4- dihydroxybenzoic acid. Purified by preparative HPLC using Method Prep- LC-9 14.5 ¹H NMR (400 MHz, DMSO) δ 2.05 - 2.07 (m, 3H), 4.36 - 4.43 (m, 1H), 4.55 - 4.63 (m, 1H), 4.73 - 4.79 (m, 2H), N-(2-(2,4-Dihydroxy-6- 5.68 - 5.81 (m, 1H), 6.11 - 6.13 (m, methylbenzoyl)isoindolin-4- 1H), 6.17 - 6.32 (m, 2H), 6.41 - 6.62 yl)acrylamide (m, 1H), 7.03 - 7.20 (m, 1H), 7.23 - Prepared from Intermediate W2-4 and 7.32 (m, 1H), 7.64 (d, J = 8.1 Hz, 1H), 2,4-Dihydroxy-6-methylbenzoic acid. 9.33 (s, 1H), 9.50 (d, J = 9.1 Hz, 1H), Purified by preparative HPLC using 9.61 - 9.79 (m, 1H). LCMS (LC-Method Method Prep-LC-2 1): 2.98 min, MS: ES+ 339.2 (M+1) Example 15: (E)-N-[2-(2,4-Dihydroxy-5-methyl-benzoyl)isoindolin-4-yl]-4-(dimethylamino)- N-indan-2-yl-but-2-enamide

[00484] To a cooled (0 ˚C) stirred suspension of (E)-4-(dimethylamino)but-2-enoyl chloride hydrochloride (97 mg, 0.52 mmol, 3.0 eq) in dry acetonitrile (0.58 mL) under nitrogen was added dropwise a solution of (4-((2,3-dihydro-1H-inden-2-yl)amino)isoindolin-2-yl)(2,4-dihydroxy-5- methylphenyl)methanone (Intermediate X3-1) (70 mg, 0.18 mmol, 1.0 eq) in dry acetonitrile. The reaction mixture was gradually warmed to room temperature and stirred overnight. Triethylamine
(0.073 mL, 0.52 mmol, 3.00 eq) was added and the reaction stirred for an additional 5 h. The resulting mixture was concentrated under reduced pressure and the crude residue was purified by chromatography on silica eluting with a gradient of 0-20% MeOH in DCM. The product was further purified by preparative HPLC (Method Prep-LC-2) to afford the title compound as a formate salt (1.19 mg, 1.27%). [00485] ¹H NMR (400 MHz, DMSO) δ 1.98 - 2.03 (m, 12H), 3.00 - 3.05 (m, 2H), 4.62 - 4.72 (m, 2H), 4.82 - 4.85 (m, 2H), 5.03 - 5.23 (m, 1H), 5.56 - 5.66 (m, 1H), 6.39 (s, 1H), 6.56 - 6.82 (m, 1H), 6.93 - 6.94 (m, 1H), 7.02 - 7.18 (m, 6H), 7.36 (m, 2H), 8.36 (s, 1H), 9.66 - 9.69 (m, 1H), 10.09 - 10.17 (m, 1H). LCMS (LC-Method 1): 3.46 min, MS: ES+ = 513 (M+1) Example 16: N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-N-(2- morpholinoethyl)acrylamide

[00486] To a stirred solution of (2,4-dihydroxy-5-methylphenyl)(4-((2- morpholinoethyl)amino)isoindolin-2-yl)methanone (Intermediate X3-2) (32 mg, 0.081 mmol, 1.0 eq) in pyridine (0.50 mL) was added prop-2-enoyl prop-2-enoate (0.011 mL, 0.089 mmol, 1.1 eq) and the reaction mixture was stirred under an inert atmosphere at room temperature for 1 hour. The reaction was quenched with 7N ammonia in MeOH (5 mL) and the resulting mixture concentrated under reduced pressure. Purification of the crude residue by preparative HPLC (Method Prep-LC-2) and (Method Prep-LC-7) afforded the title compound as an off-white solid (0.78 mg, 1.9%). [00487] ¹H NMR (400 MHz, DMSO) δ 2.00 (s, 3H), 2.26 - 2.26 (m, 4H), 3.72 - 3.91 (m, 4H), 4.51 - 4.57 (m, 1H), 4.87 (s, 4H), 5.58 - 5.61 (m, 1H), 5.95 - 5.98 (m, 1H), 6.18 (m, 1H), 6.32 (m, 1H), 7.01 - 7.01 (m, 1H), 7.24 - 7.27 (m, 1H), 7.37 - 7.43 (m, 2H). Exchangeable protons not observed, two protons are obscured by water and/or DMSO peaks. LCMS (LC-Method 1): 2.59 min, MS: ES+ = 452.5 (M+1) Example 16.1: N-(2-(2,4-Dihydroxy-6-methylbenzoyl)isoindolin-4-yl)-N-methylacrylamide
[00488] The title compound was prepared from ((2,4-Dihydroxy-6-methylphenyl)(4- (methylamino)isoindolin-2-yl)methanone) (Intermediate Z1) and prop-2-enoyl prop-2-enoate analogously to Example 16. The product was purified by preparative HPLC using Method Prep- LC-10. [00489] ¹H NMR (400 MHz, DMSO) δ 2.03 (d, J=21.6 Hz, 3H), 3.17 (d, J=49.2 Hz, 3H), 4.13 - 4.86 (m, 4H), 5.52 - 5.62 (m, 1H), 5.87 - 6.03 (m, 1H), 6.11 - 6.24 (m, 3H), 7.21 - 7.47 (m, 3H), 9.37 - 9.59 (m, 2H). LCMS (LC-Method 1): 3.06 min, MS: ES+ 353.2 (M+1) Example 17: (E)-N-[2-(2,4-Dihydroxybenzoyl)isoindolin-4-yl]-4-(dimethylamino)but-2- enamide

[00490] To a stirred solution of HATU (60 mg, 0.16 mmol, 1.0 eq) and DIPEA (0.11 mL, 0.63 mmol, 4.0 eq) in DMF (1.5 mL) was added to 2,4-dihydroxybenzoic acid (24 mg, 0.16 mmol, 1.0 eq). After 5 mins (E)-4-(dimethylamino)-N-(isoindolin-4-yl)but-2-enamide dihydrochloride (Intermediate V1-10) (50 mg, 0.16 mmol, 1.0 eq) was added and reaction mixture was stirred at room temperature overnight. The resulting mixture was diluted with DCM (10 mL) and washed with 10% sodium carbonate solution (1.5 mL). The organic phase was isolated using a phase separator cartridge then concentrated under reduced pressure. Purification of the resulting crude residue by preparative HPLC (Method Prep-LC-4) afforded the title compound as a formate salt (4.6 mg, 7%). [00491] ¹H NMR (400 MHz, DMSO) δ 2.19 - 2.20 (m, 6H), 3.06 - 3.09 (m, 2H), 4.80 (s, 4H), 6.29 - 6.36 (m, 2H), 6.46 (s, 1H), 6.69 - 6.80 (m, 1H), 7.08 - 7.30 (m, 3H), 7.67 - 7.71 (m, 1H), 8.16 (s, 1H, formic acid), 9.55 - 9.74 (m, 2H), 10.37 - 10.47 (m, 1H). LCMS (LC-Method 1): 3.08 min, MS: ES+ 382.2 (M+1). [00492] The compounds of the following tabulated Examples (Table Ex17) were prepared analogously to Example 17 from the indicated intermediates.
Ex. No. Name and Structure Data 17.1 ¹H NMR (400 MHz, DMSO) δ 2.13 - 2.21 (m, 6H), 3.06 – 3.03 (2H, m), 3.75 (s, 3H), 4.72 - 4.84 (m, 4H), 6.46 - 6.49 (m, 3H), 6.68 - 6.78 (m, 1H), 7.04 - 7.31 (m, (E)-4-(Dimethylamino)-N-(2-(2-hydroxy-4- 3H), 7.66 - 7.70 (m, 1H), 8.21 (s, 0.5H), methoxybenzoyl)isoindolin-4-yl)but-2- 9.53 - 9.69 (m, 1H), 10.43 - 10.49 (m, enamide 1H). Isolated as formate salt. LCMS Prepared from Intermediate V1-10 and 4- (LC-Method 2): 3.39 min, MS: ES+ methoxysalicylic acid and purified method 396.2 (M+1). using Method Prep-LC-2 17.2 ¹H NMR (400 MHz, DMSO) δ 2.14 - 2.23 (m, 9H), 3.08 – 3.02 (2H, m), 4.64 (d, J =7.8 Hz, 2H), 4.79 (d, J =13.6 Hz, 2H), 6.25 - 6.46 (m, 1H), 6.74 - 6.83 (m, 2H), (E)-4-(Dimethylamino)-N-(2-(2-hydroxy-5- 7.03 - 7.30 (m, 4H), 7.64 - 7.70 (m, 1H), methylbenzoyl)isoindolin-4-yl)but-2- 8.17 (s, 0.5H), 9.53 - 9.75 (m, 2H). enamide Isolated as formate salt. LCMS (LC- Prepared from Intermediate V1-10 and 4- Method 2): 3.40 min, MS: ES+ 380.2 methylsalicylic acid purified by preparative (M+1). HPLC using Method Prep-LC-2 17.3 ¹H NMR (400 MHz, DMSO) δ 2.15 - 2.21 (m, 9H), 3.02 - 3.09 (m, 2H), 4.80 - 4.86 (m, 4H), 6.29 - 6.86 (m, 3H), 7.04 - 7.39 (m, 4H), 7.68 - 7.71 (m, 1H), 8.16 (s, 0.5 (E)-4-(Dimethylamino)-N-(2-(4-hydroxy-3- H), 9.50 - 9.79 (m, 2H). Isolated as methylbenzoyl)isoindolin-4-yl)but-2- formate salt. LCMS (LC-Method 2): 3.12 enamide. min, MS: ES+ 380.2 (M+1). Prepared from Intermediate V1-10 and 4- hydroxy-3-methylbenzoic acid and purified using Method Prep-LC-4 17.4 ¹H NMR (400 MHz, DMSO) δ 2.14 - 2.20 (m, 6H), 3.00 - 3.08 (m, 2H), 4.64 (d, J=6.6 Hz, 2H), 4.81 (d, J = 13.1 Hz, 2H), 6.26 - 6.45 (m, 1H), 6.63 - 6.78 (m, 3H), 7.02 - 7.17 (m, 1H), 7.23 - 7.36 (m, 2H),
(E)-4-(Dimethylamino)-N-(2-(2-fluoro-4- 7.70 (t, J = 8.2 Hz, 1H), 8.20 (0.5 H, s), hydroxybenzoyl)isoindolin-4-yl)but-2- 10.1 – 10.5 (m, 1H) 9.47 - 9.69 (m, 1H). enamide Isolated as formate salt. Prepared from Intermediate V1-10 and 2- LCMS (LC-Method 1): 2.50 min, MS: fluoro-4-hydroxybenzoic acid and purified ES+ 384.2 (M+1). using Method Prep-LC-4 ¹H NMR (400 MHz, DMSO) δ 2.13 - 2.22 (m, 9H), 3.00 - 3.10 (m, 2H), 4.50 (s, 2H), 4.81 (d, J = 13.6 Hz, 2H), 6.25 - 6.46 (m, 1H), 6.62 - 6.78 (m, 3H), 6.99 - (E)-4-(Dimethylamino)-N-(2-(4-hydroxy-2- 7.16 (m, 2H), 7.22 - 7.31 (m, 1H), 7.66 - methylbenzoyl)isoindolin-4-yl)but-2- 7.69 (m, 1H), 8.15 (s, 0.5H), 9.46 - 9.69 enamide. (m, 2H). Isolated as formate salt. LCMS Prepared from Intermediate V1-10 and acid (LC-Method 2): 2.99 min, MS: ES+ 4-hydroxy-2-methylbenzoic acid and 380.0 (M+1). purified using Method Prep-LC-4. ¹H NMR (400 MHz, DMSO) δ 2.17 - 2.20 (m, 6H), 3.03 - 3.09 (m, 2H), 4.86 - 4.89 (m, 2H), 5.07 - 5.21 (m, 2H), 6.32 - 6.47 (m, 1H), 6.68 - 6.80 (m, 1H), 7.09 - 7.31 (E)-4-(Dimethylamino)-N-(2-(5- (m, 3H), 7.61 - 7.83 (m, 2H), 8.19 (d, J = hydroxypicolinoyl)isoindolin-4-yl)but-2- 2.5 Hz, 1H), 9.59 - 9.69 (m, 1H), 10.51 enamide. (s, 1H). LCMS (LC-Method 2): 2.43 min, Prepared from Intermediate V1-10 and 5- MS: ES+ 367.0 (M+1). hydroxypicolinic acid and purified using Method Prep-LC-7 ¹H NMR (400 MHz, DMSO) δ 2.61 - 2.61 (m, 3H), 3.81 (s, 3H), 4.51 - 4.53 (m, 2H), 4.77 - 4.84 (m, 2H), 6.34 - 6.55 (m, 1H), 6.63 - 6.82 (m, 1H), 7.04 - 7.16 (m, 3H), 7.20 - 7.65 (m, 4H), 8.13 (s, 0.2H), (E)-4-(Dimethylamino)-N-(2-(2- 9.65 - 9.87 (m, 1H). Some signals methoxybenzoyl)isoindolin-4-yl)but-2- obscured by solvent peaks. (formate enamide salt). LCMS (LC-Method 2): 3.46 min, MS: ES+ 380.2 (M+1).
Prepared from Intermediate V1-10 and 2- Methoxybenzoic acid and purified using Method Prep-LC-2 ¹H NMR (400 MHz, DMSO) δ 2.12 - 2.21 (m, 6H), 3.03 - 3.07 (m, 2H), 4.68 - 4.79 (m, 4H), 6.26 - 6.47 (m, 1H), 6.61 (s, 1H), 6.65 - 6.82 (m, 1H), 7.01 - 7.17 (m, 1H), 7.26 (d, J = 7.1 Hz, 1H), 7.38 (s, (E)-N-(2-(2,4-Dihydroxy-5- 1H), 7.65 - 7.70 (m, 1H), 9.47 - 9.71 (m, (trifluoromethyl)benzoyl)isoindolin-4-yl)-4- 1H), 10.69 - 11.03 (m, 2H). LCMS (LC- (dimethylamino)but-2-enamide Method 1): 2.92 min, MS: ES+ 450.2 Prepared from Intermediate V1-10 and 2,4- (M+1). dihydroxy-5-(trifluoromethyl)benzoic acid (Intermediate T) and purified using Method Prep-LC-2 ¹H NMR (400 MHz, DMSO) δ 1.31 (s, 9H), 2.14 - 2.21 (m, 6H), 3.01 - 3.09 (m, 2H), 4.75 - 4.86 (m, 4H), 6.25 - 6.42 (m, 2H), 6.66 - 6.79 (m, 1H), 7.04 - 7.31 (m, 3H), 7.50 - 7.72 (m, 1H), 9.66 - 9.73 (m, (E)-N-(2-(5-(tert-Butyl)-2,4- 2H), 10.11 - 10.30 (m, 1H). LCMS (LC- dihydroxybenzoyl)isoindolin-4-yl)-4- Method 1): 3.31 min, MS: ES+ 438.2 (dimethylamino)but-2-enamide. (M+1). Prepared from Intermediate V1-10 and acid 5-(1,1-dimethylethyl)-2,4-dihydroxybenzoic acid and purified using Method Prep-LC-4 ¹H NMR (400 MHz, DMSO) 2.14 - 2.20 (6H, m), 3.02 - 3.08 (2H, m), 4.82 - 4.87 (4H, m), 6.29 - 6.46 (1H, m), 6.66 - 6.78 (1H, m), 6.83 (2H, d, J=9.4 Hz), 7.03 - (E)-N-(2-(2,4-Dihydroxybenzoyl)isoindolin- 7.16 (1H, m), 7.23 - 7.30 (1H, m), 7.51 4-yl)-4-(dimethylamino)but-2-enamide. (2H, d, J=6.2 Hz), 7.70 (1H, d, J=7.3 Hz), Prepared from Intermediate V1-10 and acid 9.51 - 9.67 (1H, m), 9.93 (1H, s). 2,4-dihydroxybenzoic acid and purified LCMS (LC-Method 1): 2.37 min, MS: using Method Prep-LC-11 ES+ 366.2 (M+1).
Example 18: (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4-methoxybut-2- enamide
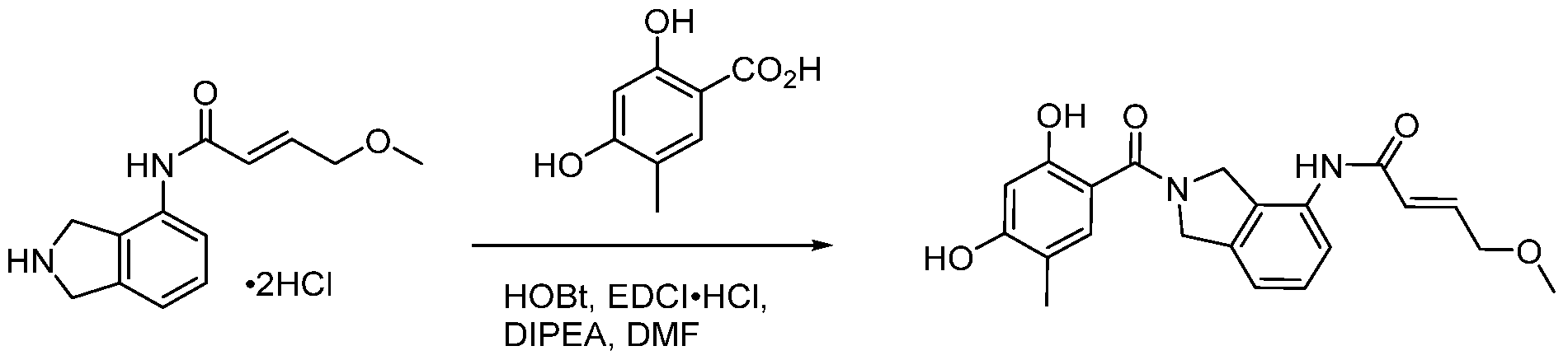
[00493] To a stirred solution of HOBT (36 mg, 0.24 mmol, 1.10 eq), EDC (45 mg, 0.24 mmol, 1.10 eq) and 2,4-dihydroxy-5-methyl-benzoic acid (36 mg, 0.22 mmol, 1.0 eq) (Intermediate F) in dry DCM (0.73 mL) was added DIPEA (0.15 mL, 0.86 mmol, 4.0 eq) and the reaction mixture stirred at room temperature for 10 mins. (E)-N-Isoindolin-4-yl)-4-methoxy-but-2-enamide dihydrochloride (Intermediate Z) (50 mg, 0.22 mmol, 1.0 eq) in DMF (0.73 mL) was added and stirring continued at room temperature for 16 h. The resulting mixture was concentrated under reduced pressure and the crude residue was purified by chromatography on silica eluting 0-20% MeOH in DCM. Further purification by preparative HPLC (Method Prep-LC-2) afforded the title compound as a formate salt (6.7 mg, 8.0%) [00494] ¹H NMR (400 MHz, DMSO) δ 2.03 (s, 3H), 4.12 (s, 2H), 4.79 (d, J=12.7 Hz, 4H), 6.36 - 6.50 (m, 2H), 6.75 - 6.83 (m, 1H), 7.04 - 7.13 (m, 2H), 7.27 (t, J=7.2 Hz, 1H), 7.69 (d, J=8.2 Hz, 1H), 8.51 (s, OH), 9.58 - 9.71 (m, 2H), 10.00 - 10.19 (m, 1H). OMe signal obscured by solvent peak. LCMS (LC-Method 1): 3.52 min, MS: ES+ 383.2 (M+1) Example 19: 1-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-3-(2- (dimethylamino)ethylidene)pyrrolidin-2-one Step 1: tert-Butyl 4-(2,4-dibromobutanamido)isoindoline-2-carboxylate

[00495] A solution of 2,4-dibromobutanoyl chloride (340 mg, 1.3 mmol, 1.2 eq) in DCM (1.0 mL) was added dropwise to a stirred mixture of tert-butyl 4-aminoisoindoline-2-carboxylate (CAS 871013-98-8) (250 mg, 1.1 mmol, 1.0 eq) and DIPEA (0.30 mL, 1.6 mmol, 1.5 eq) in DCM (4.0 mL) cooled in an ice/water bath. The reaction mixture was stirred for 4 h and then partitioned between DCM (20 mL) and water (5 mL). The organic layer was isolated using a phase separator
cartridge and concentrated under reduced pressure to afford the title compound which was used without further purification. [00496] ¹H NMR (400 MHz, DMSO) δ 1.24 - 1.29 (m, 2H), 1.45 - 1.48 (m, 9H), 3.56 - 3.71 (m, 2H), 4.54 - 4.64 (m, 4H), 4.83 - 4.91 (m, 1H), 7.16 (t, J=7.0 Hz, 1H), 7.27 - 7.33 (m, 1H), 7.54 (dd, J=8.1, 11.7 Hz, 1H), 10.10 (s, 1H). MS: ES+ 360.9, 362.8, 364.8 (M+1) Step 2: tert-Butyl 4-(3-bromo-2-oxopyrrolidin-1-yl)isoindoline-2-carboxylate

[00497] Sodium hydride (60%, 39 mg, 0.97 mmol, 1.0 eq) was added portion wise to a cooled (0 ˚C) solution of tert-butyl 4-(2,4-dibromobutanoylamino)isoindoline-2-carboxylate (Step 1) (450 mg, 0.97 mmol, 1.0 eq) in DMF (10.0 mL). The mixture was stirred at 0 ˚C for 40 mins then at room temperature for 2 h. The resulting mixture was diluted with water (5 mL) and extracted with DCM (3 x 15 mL). The combined organic extracts were dried over MgSO4 and concentrated under reduced pressure. Purification of the crude residue by chromatography on silica eluting with a gradient of 0-100% EtOAc in cyclohexane afforded the title compound (344 mg, 93%). [00498] ¹H NMR (400 MHz, DMSO) δ 1.43 - 1.47 (m, 9H), 2.32 - 2.40 (m, 1H), 2.78 - 2.87 (m, 1H), 3.77 - 3.93 (m, 2H), 4.49 - 4.55 (m, 2H), 4.60 - 4.66 (m, 2H), 4.84 - 4.89 (m, 1H), 7.27 - 7.41 (m, 3H). MS: ES+ 381.1, 382.9 (M+1) Step 3: [1-(2-tert-Butoxycarbonylisoindolin-4-yl)-2-oxo-pyrrolidin-3-yl]-triphenyl-phosphonium bromide.
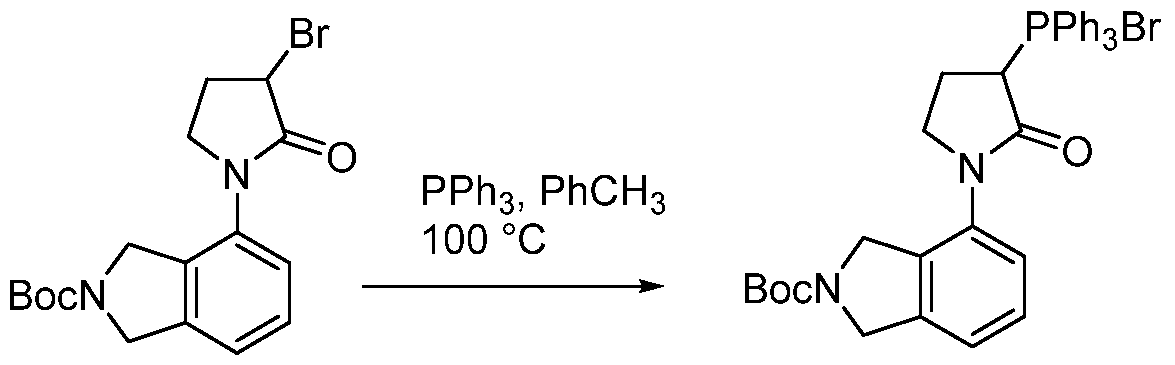
[00499] A mixture of tert-butyl 4-(3-bromo-2-oxo-pyrrolidin-1-yl)isoindoline-2-carboxylate (Step 2) (344 mg, 0.90 mmol, 1.0 eq) and triphenylphosphine (950 mg, 3.6 mmol, 4.0 eq) in toluene (3.0 mL) was heated to 100 ˚C for 4 h. EtOAc (20 mL) was added then the mixture was allowed to cool to room temperature. The precipitated solid was collected by filtration, washed with
EtOAc/toluene then dried in vacuo to afford the title compound as a purple solid which was used without further purification. (406 mg, 70%) [00500] ¹H NMR (400 MHz, DMSO) δ 1.46 - 1.48 (m, 9H), 3.58 - 3.73 (m, 2H), 3.94 - 4.13 (m, 2H), 4.22 - 4.62 (m, 4H), 5.81 - 5.97 (m, 1H), 7.12 - 7.45 (m, 6H), 7.76 - 7.96 (m, 12H). MS: ES+ 463.1 [M-Boc+1] Step 4: tert-Butyl 4-(3-(2-(dimethylamino)ethylidene)-2-oxopyrrolidin-1-yl)isoindoline-2- carboxylate

[00501] Triethylamine (0.44 mL, 3.2 mmol, 5.0 eq) was added to a mixture of [1-(2-tert- butoxycarbonylisoindolin-4-yl)-2-oxo-pyrrolidin-3-yl]-triphenyl-phosphonium bromide (Step 3) (406 mg, 0.63 mmol, 1.0 eq) and 2-(dimethylamino)acetaldehyde hydrochloride (117 mg, 0.936 mmol, 1.50 eq) in EtOH (12.0 mL) and heated to 75 ˚C for 3 h. The mixture was allowed to cool to room temperature and concentrated under reduced pressure, The residue was diluted with DCM (20 mL) and washed with water (10 mL), dried over MgSO
4 and concentrated under reduced pressure. The crude material was purified by chromatography on silica eluting with a gradient of 0-20% MeOH in DCM to afford the title compound as a purple glass. [00502] ¹H NMR (400 MHz, DMSO) δ 1.43 - 1.47 (m, 9H), 2.76 (s, 6H), 2.90 - 2.97 (m, 2H), 3.84 - 3.91 (m, 4H), 4.48 - 4.53 (m, 2H), 4.63 - 4.67 (m, 2H), 6.39 - 6.46 (m, 1H), 7.28 - 7.42 (m, 3H). MS: ES+ 372.1 (M+1) Step 5: 3-(2-(Dimethylamino)ethylidene)-1-(isoindolin-4-yl)pyrrolidin-2-one dihydrochloride
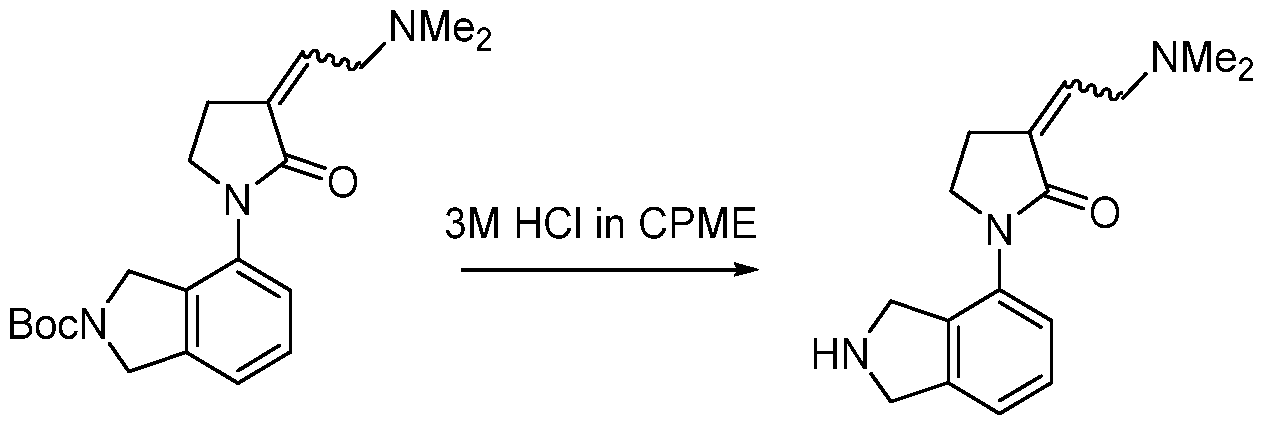
[00503] A solution of 3M HCl in CPME (2.0 mL) was added to tert-butyl 4-(3-(2- (dimethylamino)ethylidene]-2-oxo-pyrrolidin-1-yl]isoindoline-2-carboxylate (Step 4) (70 mg, 0.19
mmol, 1.0 eq) followed by a few drops of MeOH to aid dissolution. The reaction mixture was stirred at room temperature for 4 h. The resulting mixture was concentrated under reduced pressure and the crude product was used without further purification. Quantitative yield. MS: ES+ 273.2 (M+1) Step 6: 1-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-3-(2- (dimethylamino)ethylidene)pyrrolidin-2-one

[00504] DIPEA (0.16 mL, 0.94 mmol, 5.0 eq) was added to a mixture of 3-(2- (dimethylamino)ethylidene)-1-(isoindolin-4-yl)pyrrolidin-2-one dihydrochloride (Step 5) (65 mg, 0.19 mmol, 1.0 eq), 2,4-dihydroxy-5-methyl-benzoic acid (32 mg, 0.19 mmol, 1.0 eq) (Intermediate F), EDC (43 mg, 0.23 mmol, 1.2 eq) and HOBT (35 mg, 0.23 mmol, 1.2 eq) in DMF (2.0 mL). The reaction mixture was stirred for 18 h at room temperature. The resulting mixture was concentrated onto silica gel and purified by chromatography eluting with a gradient of 0-20% MeOH in DCM. The product fractions were further purified by preparative HPLC (Method Prep- LC-4) to afford the title compound (14 mg, 18%). (It was not possible to assign a cis or trans configuration to the double bond or whether the product was a cis/trans mixture). [00505] ¹H NMR (400 MHz, DMSO) δ 2.01 (s, 3H), 2.24 - 2.26 (m, 6H), 2.78 - 2.89 (m, 2H), 3.10 - 3.17 (m, 2H), 3.85 (s, 2H), 4.67 (s, 2H), 4.82 (s, 2H), 6.36 - 6.41 (m, 2H), 6.97 - 7.04 (m, 1H), 7.25 - 7.39 (m, 3H). LCMS (LC-Method 1): 2.60 min, MS: ES+ 422.2 (M+1) Example 20: N-(2-(2,4-Dihydroxy-5-methylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- (1-methylpiperidin-4-yl)acrylamide Step 1: tert-Butyl 7-[(1-methyl-4-piperidyl)amino]-3,4-dihydro-1H-isoquinoline-2-carboxylate

[00506] To a nitrogen degassed solution of tert-butyl 7-bromo-3,4-dihydro-1H-isoquinoline-2- carboxylate (CAS: 258515-65-0) (500 mg, 1.6 mmol, 1.0 eq), 1-methylpiperidin-4-amine (0.30 mL, 2.4 mmol, 1.5 eq) and sodium tert-butoxide (310 mg, 3.2 mmol, 2.00 eq) in toluene (10 mL) was added XPhos Pd G2 (130 mg, 0.16 mmol, 0.10 eq). The reaction vessel was purged with nitrogen, sealed and then stirred at 100 °C for 72 h. After cooling to room temperature, the resulting mixture was diluted with EtOAc and filtered through a Celite® pad. The filtrate was concentrated under reduced pressure and purification of the crude material by chromatography on silica eluting with 0-20% MeOH (with ammonia modifier) in DCM afforded the title compound (420 mg, 76%). [00507] ¹H NMR (400 MHz, DMSO) δ 1.28 - 1.40 (m, 2H), 1.42 (s, 9H), 1.83 (d, J=11.2 Hz, 2H), 1.99 (dd, J=9.3, 11.7 Hz, 2H), 2.16 (s, 3H), 2.59 (dd, J=5.8, 5.8 Hz, 2H), 2.70 (d, J=11.7 Hz, 2H), 3.14 - 3.09 (m, 1H), 3.48 (dd, J=5.8, 5.8 Hz, 2H), 4.36 (s, 2H), 5.20 (d, J=8.2 Hz, 1H), 6.32 (s, 1H), 6.42 (dd, J=2.1, 8.2 Hz, 1H), 6.83 (d, J=8.2 Hz, 1H). MS: ES+ 346.3 (M+1) Step 2: N-(1-Methylpiperidin-4-yl)-1,2,3,4-tetrahydroisoquinolin-7-amine

[00508] To a solution of tert-butyl 7-[(1-methyl-4-piperidyl)amino]-3,4-dihydro-1H-isoquinoline- 2-carboxylate (Step 1) (420 mg, 1.2 mmol, 1.0 eq) in DCM (3.0 mL) was added TFA (2.0 mL) and the mixture was stirred at room temperature for 1.5 h. The resulting mixture was concentrated under reduced pressure. The crude material was loaded onto an Isolute® SCX cartridge and washed with MeOH/DCM (1:1). The product was eluted in 7N methanolic ammonia/DCM and concentration of the basic fractions afforded the title compound which was used without further purification. (300 mg, Quantitative). [00509] ¹H NMR (400 MHz, DMSO) δ 1.32 - 1.42 (m, 2H), 1.86 (d, J=11.1 Hz, 2H), 2.04 (t, J=11.6 Hz, 2H), 2.20 (s, 3H), 2.70 - 2.79 (m, 4H), 3.15 - 3.22 (m, 4H), 4.01 - 4.12 (m, 2H), 5.10 - 5.35 (m, 1H), 6.23 - 6.32 (m, 1H), 6.37 - 6.51 (m, 1H), 6.77 - 6.87 (m, 1H).MS: ES+ 246.3 (M+1) Step 3: (2,4-Dihydroxy-5-methylphenyl)(7-((1-methylpiperidin-4-yl)amino)-3,4- dihydroisoquinolin-2(1H)-yl)methanone
[00510] The title compound was prepared from 2,4-dihydroxy-5-methyl-benzoic acid (Intermediate F) and N-(1-methyl-4-piperidyl)-1,2,3,4-tetrahydroisoquinolin-7-amine (Step 2) analogously to Example 13. [00511] ¹H NMR (400 MHz, DMSO) δ 0.97 (t, J=7.8 Hz, 1H), 1.31 - 1.38 (m, 2H), 1.84 (d, J=13.7 Hz, 2H), 2.01 (s, 3H), 2.10 - 2.22 (m, 5H), 2.68 - 2.75 (m, 2H), 2.96 – 3.01 (m, 2H), 3.17 - 3.19 (m, 2H), 4.47 - 4.57 (m, 1H), 5.20 (d, J=8.0 Hz, 1H), 5.75 - 5.81 (m, 1H), 6.25 - 6.33 (m, 1H), 6.38 (s, 1H), 6.40 - 6.45 (m, 1H), 6.81 - 6.84 (m, 2H), 9.50 (d, J=5.6 Hz, 1H). [00512] MS: ES+ 396.3 (M+1). Step 4: N-(2-(2,4-Dihydroxy-5-methylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N-(1- methylpiperidin-4-yl)acrylamide

[00513] The title compound was prepared from (2,4-dihydroxy-5-methyl-phenyl)-[7-[(1-methyl-4- piperidyl)amino]-3,4-dihydro-1H-isoquinolin-2-yl]methanone (Step 3) and prop-2-enoyl prop-2- enoate analogously to Example 16. The product was purified by preparative HPLC using Method Prep-LC-12. [00514] ¹H NMR (400 MHz, DMSO) δ 1.25 - 1.32 (m, 2H), 1.63 - 1.71 (m, 2H), 1.92 (t, J=11.1 Hz, 2H), 2.01 (s, 3H), 2.09 (s, 3H), 2.67 - 2.78 (m, 2H), 2.86 (t, J=5.8 Hz, 2H), 3.64 (s, 2H), 4.36 - 4.42 (m, 1H), 4.64 - 4.69 (m, 2H), 5.47 - 5.53 (m, 1H), 5.74 - 5.82 (m, 1H), 6.08 - 6.14 (m, 1H), 6.39 (s, 1H), 6.87 (s, 1H), 6.94 - 6.98 (m, 1H), 7.01 - 7.05 (m, 1H), 7.23 - 7.27 (m, 1H), 9.52 - 9.55 (m, 2H). [00515] LCMS (LC-Method 1): 2.67 min, MS: ES+ 450.48 (M+1)
Example 21: (E)-N-(2-(4,6-Dihydroxy-2,3-dimethylbenzoyl)isoindolin-4-yl)-4- (dimethylamino)-N-ethylbut-2-enamide

[00516] A solution of (E)-4-(dimethylamino)but-2-enoyl chloride (40 mg, 0.22 mmol, 1.1 eq) in acetonitrile (2.5 mL) was added dropwise to a cooled (0 ˚C) solution of 4-(4- (ethylamino)isoindoline-2-carbonyl)-5,6-dimethyl-1,3-phenylene diacetate (Intermediate X3-3) (82 mg, 0.20 mmol, 1.0 eq) and DIPEA (0.087 mL, 0.50 mmol, 2.5 eq) in acetonitrile (1.0 mL) and the mixture allowed to warm to room temperature, stirring for 16 h. Pyridine (0.50 mL) was added and the mixture was stirred for a further 24 h. Additional (E)-4-(dimethylamino)but-2-enoyl chloride (40 mg, 0.22 mmol, 1.1 eq) in acetonitrile (2.5 mL) was added and stirring continued for 16 h. The resulting mixture was diluted with DCM (20 mL) and washed with water (5.0 mL), brine (10 mL) dried over MgSO
4 and concentrated under reduced pressure. The residue was dissolved in MeOH (2.0 mL), treated dropwise with12M HCl (0.11 mL, 1.3 mmol, 10 eq) and stirred at room temperature for 48 h. The resulting mixture was concentrated under reduced pressure and purification of the crude material by preparative HPLC (Prep-LC-3) afforded the title compound as a white solid (11 mg, 20%, formate salt). [00517] ¹H NMR (400 MHz, DMSO) δ 0.91 - 1.10 (m, 3H), 1.91 - 2.10 (m, 12H), 2.82 - 2.98 (m, 2H), 4.08 - 4.90 (m, 4H), 5.60 - 5.75 (m, 1H), 6.19 - 6.34 (m, 1H), 6.52 - 6.73 (m, 1H), 7.14 - 7.23 (m, 1H), 7.29 - 7.48 (m, 2H), 9.26 (s, 1H). One exchangeable and one CH2 signals not visible. LCMS (LC-Method 1): 2.75 min. MS: ES+ 438.1 (M+1) Example 22: N-(2-(4,6-Dihydroxy-2,3-dimethylbenzoyl)isoindolin-4-yl)-N-(2- methoxyethyl)acrylamide

[00518] The title compound was prepared from 4-(4-((2-methoxyethyl)amino)isoindoline-2- carbonyl)-5,6-dimethyl-1,3-phenylene diacetate (Intermediate X3-4) and prop-2-enoyl prop-2- enoate analogously to Example 20 step 4. [00519] ¹H NMR (400 MHz, DMSO) 100 °C: 1.98 - 2.05 (m, 6H), 3.18 - 3.45 (m, 5H), 3.83 - 3.84 (m, 2H), 4.44 - 4.89 (m, 4H), 5.50 - 5.61 (m, 1H), 5.95 - 6.33 (m, 3H), 7.14 - 7.22 (m, 1H), 7.32 - 7.42 (m, 2H), 8.70 – 8.81 (m, 2H). LCMS (LC-Method 1): ES+ = 411 (M+1) Example 23: N-(2-(4,6-Dihydroxy-2,3-dimethylbenzoyl)isoindolin-4-yl)-N- methylacrylamide Step 1: 4,5-Dimethyl-6-(4-(N-methylacrylamido)isoindoline-2-carbonyl)-1,3-phenylene diacetate
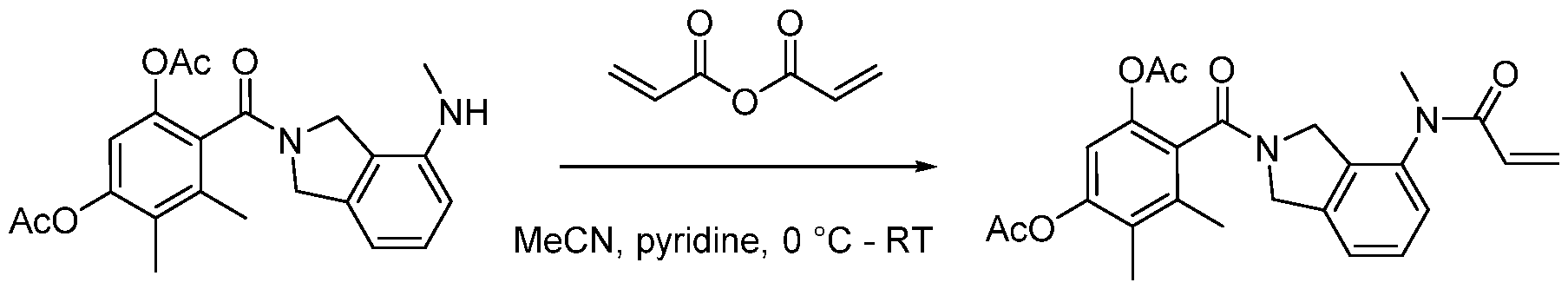
[00520] To a stirred solution of 4,5-dimethyl-6-(4-(methylamino)isoindoline-2-carbonyl)-1,3- phenylene diacetate (Intermediate Z2) (106 mg, 0.27 mmol, 1.0 eq) in pyridine (0.89 mL) under an atmosphere of nitrogen was added prop-2-enoyl prop-2-enoate (0.080 mL, 0.54 mmol, 2.0 eq) and the reaction mixture was stirred at room temperature for 3 h. The resulting mixture was concentrated under reduced pressure and purification of the crude material by chromatography on silica eluting with gradient of DCM in MeOH afforded the title compound (53 mg, 44%). MS: ES+ = 450.0 (M+1) Step 2: N-(2-(4,6-Dihydroxy-2,3-dimethylbenzoyl)isoindolin-4-yl)-N-methylacrylamide

[00521] 12M HCl (0.0070 mL, 0.084 mmol, 1.0 eq) was added to a solution of 4,5-dimethyl-6-(4- (N-methylacrylamido)isoindoline-2-carbonyl)-1,3-phenylene diacetate (Step 1) (38 mg, 0.084 mmol, 1.0 eq) in methyl alcohol (5.0 mL) and the reaction mixture was stirred at room temperature for 5 h. Additional 12M HCl (0.0070 mL, 0.084 mmol, 1.0 eq) was added and stirring continued at room temperature overnight. A further portion of 12M HCl (0.020 mL, 0.24 mmol, 2.9 eq) was
added and the mixture was stirred for an additional 2 h. The resulting mixture was concentrated under reduced pressure to afford the title compound as an off-white solid (2.7 mg, 9%). [00522] ¹H NMR (400 MHz, DMSO) δ 1.93 - 2.03 (m, 6H), 3.07 - 3.24 (m, 3H), 4.40 - 4.87 (m, 4H), 5.53 - 5.64 (m, 1H), 5.85 - 6.34 (m, 3H), 7.19 - 7.47 (m, 3H), 9.30 (s, 2H). LCMS (LC-Method 1): ES+ = 367.1 (M+1) Example 24: (E)-N-(2-(5-(tert-Butyl)-2,4-dihydroxybenzoyl)-5-chloroisoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide Step 1: tert-Butyl 5-chloro-4-(methylamino)isoindoline-2-carboxylate

[00523] To a stirred solution of tert-butyl 4-(methylamino)isoindoline-2-carboxylate (Intermediate A Step 1) (5.0 g, 20.16 mmol, 1.0 eq.) in MeCN (50 mL) was added N-chlorosuccinimide (2.68 g, 20.16 mmol, 1.0 eq.) and the reaction mixture was stirred at room temperature for 16 h. The resulting mixture was filtered and the filtrate concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted at 5.0% EtOAc in hexane) to give the title compound (1.3 g, Yield: 22.9%). tert-Butyl 4-chloro-7-(methylamino)isoindoline-2- carboxylate was also isolated as a by-product (eluted at 10.0% EtOAc in hexane) yielding (3.2 g, Yield: 56.3%). [00524]
1H NMR (DMSO-d6, 400 MHz): δ 1.45 (s, 9H), 2.96 - 2.92 (m, 3H).4.45 (d, J= 9.6 Hz, 2H), 4.81(s, 2H), 6.56 (t, J= 17.6 Hz, 1H), 7.16 (d, J= 7.6 Hz, 1H). LCMS (Method A): 2.273 min, MS: ES+ 226.8 (M-56). Step 2: 5-Chloro-N-methylisoindolin-4-amine hydrochloride
[00525] To a cooled (0 ˚C) solution of tert-butyl 5-chloro-4-(methylamino)isoindoline-2- carboxylate (Step 1) (1.3 g, 4.60 mmol, 1 eq.) in DCM (13 mL) was added dropwise 4M HCl in dioxane (6.5 mL) and the reaction mixture was stirred at room temperature for 1 h. The resulting
mixture was concentrated under vacuum and the crude material was triturated with diethyl ether (2 x 15 mL) to afford the title compound (1.1 g, Yield: Quantitative). [00526]
1H NMR (DMSO-d6, 400 MHz): δ 2.94 (s, 3H).4.37 (m, 2H), 4.719m, 2H), 6.64 (d, J= 8 Hz, 1H), 7.25 (d, J= 7.6 Hz, 1H).10.02 (m, br, 2H). LCMS (Method A): 0.796 min, MS: ES+ 182.8 (M+1). Step 3: (5-(tert-Butyl)-2, 4-dihydroxyphenyl) (5-chloro-4-(methylamino)isoindolin-2-yl)methanone

[00527] Performed in 2 parallel batches, each of 0.5 g scale: To a cooled (0 ˚C) solution of 5- (tert-butyl)-2,4-dihydroxybenzoic acid (Intermediate N, Step 1) (0.5 g, 2.38 mmol, 1 eq.) in DMF (3.5 mL) were added EDC.HCl (0.9 g, 4.76 mmol, 2.0 eq) and HOBT (0.032 g, 0.23 mmol, 0.1 eq.) and the reaction mixture was stirred at 0 ˚C for 10 mins. 5-Chloro-N-methylisoindolin-4- amine (Step 2) (0.57 g, 2.61 mmol, 1.1 eq.) and TEA (0.72 mL, 7.14 mmol, 3.0 eq.) were added at 0 ˚ C and the mixture heated to 120 ˚ C using microwave irradiation for 3 h. The resulting mixture was diluted with ice cold water (100 mL) and extracted with EtOAc (3 x 50 mL). The combined organic extracts were dried over Na2SO4 and concentrated under vacuum. Purification of the crude material by chromatography on silica (product eluted at 25.0% EtOAc in hexane) afforded the title compound (0.6 g, Yield: 33.7%). [00528]
1H NMR (DMSO-d6, 400 MHz): δ 1.45 - 1.04 (m, 9H), 2.94 (m, 3H), 4.66 - 4.59 (m, 2H), 5.04 (s, br, 2H), 5.25 (s, 1H), 6.39 (s, 1H), 6.54 (bs, 1H), 7.04 (bs, 1H), 7.17 (bs, 1H), [00529] LCMS (Method A): 2.242min, MS: ES+ 375 (M+1). HPLC (Method D): 9.458 min. Prep. HPLC purification method: [00530] Chromatographic separation and isolation were conducted with a Waters 2545 quarternary system with waters 2489 UV Detector: column Xtimate C18 (250mm x 21.2mm x 5µm); compound eluted with: Mobile Phase A : 0.05% FORMIC ACID IN WATER, Mobile Phase B : ACETONITRILE:METHANOL with a gradient of T = 0.0 min (33% A, 67% B); gradient to T = 19.00 min (33% A, 67% B); T = 19.01 min (0% A, 100% B) gradient to T = 22.00 min (0% A, 100% B); T = 22.01 min (33% A, 67% B); gradient to T = 27 min (33% A, 67% B); Flow rate= 24ml/min; analysis time 27 min.
Step 4: (E)-N-(2-(5-(tert-Butyl)-2,4-dihydroxybenzoyl)-5-chloroisoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide

[00531] To a stirred solution of (E)-4-(dimethylamine) but-2-enoic acid hydrochloride (0.14 g, 0.371 mmol, 1 eq.) in DMF (2.0 mL) were added (5-(tert-butyl)-2,4-dihydroxyphenyl)(5-chloro-4- (methylamino)isoindolin-2-yl)methanone (Step 3) (0.33 g, 0.792 mmol, 0.5 eq) and DCC (0.388 g, 1.48 mmol, 2.0 eq.) and the reaction mixture heated to 150
˚C using microwave irradiation for 15 mins. The resulting mixture was diluted with ice cold water (100 mL) and extracted with EtOAc (3 x 50 mL). The aqueous layer was lyophilized and the crude material was purified by Prep. HPLC using 0.05% FA in water: acetonitrile followed by lyophilization of the product fractions to afford the title compound as off-white solid (0.02 g, Yield: 10.3%). [00532] High Temperature
1H NMR (DMSO-d6, 400 MHz): δ 1.30 (s, 9H), 2.01 (s, 6H), 2.89 (d, J= 4.8 Hz, 2H), 3.14 (s, 3H), 4.59 - 4.84 (m, 4H), 5.70 (d, J= 13.6 Hz, 1H), 6.41 (s, 1H), 6.64 - 6.70 (m, 1H), 7.12 (s, 1H), 7.43 (d, J= 8.8 Hz, 1H), 7.56 (d, J= 8 Hz, 1H), 9.53 (s, 1H), 10.03 (bs, 1H). LCMS (Method A): 1.465 min, MS: ES+ 486.1 (M+1). HPLC (Method D): 5.605 min, Prep. HPLC purification method: [00533] Chromatographic separation and isolation were conducted with Waters 2545 quaternary system with Waters 2489 UV Detector. The column used was Xtimate C18 (250mm x 21.2mm x 5µm) and the compounds were eluted with, Mobile Phase A : 0.05% formic acid in water, Mobile Phase B : acetonitrile:methanol(50:50) with a gradient of T = 0.0 min (78% A, 22% B); gradient to T = 21.00 min (55% A, 45% B); T = 21.01 min (0% A, 100% B) gradient to T = 24.00 min (0% A, 100% B); T = 24.01 min (78% A, 22% B); gradient to T = 26.00 min (78% A, 22% B); Flow rate= 22ml/min; analysis time 26 min. Example 24.1: (E)-N-(5-Chloro-2-(2, 4-dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4- (dimethyl amino)-N-methylbut-2-enamide Step 1: (5-Chloro-4-(methylamino)isoindolin-2-yl)(2,4-dihydroxy-5-methylphenyl)methanone
[00534] The title compound was prepared from 2,4-dihydroxy-5-methylbenzoic acid (Intermediate F) and 5-chloro-N-methylisoindolin-4-amine hydrochloride (Example 24 step 2) analogously to Example 24 Step 3. [00535]
1H NMR (DMSO-d6, 400 MHz): δ 2.07 (s, 3H), 2.95 – 3.05 (m, 3H), 4.72 (s, 2H), 5.04 (m, 2H), 5.27 (m, 1H), 6.40 (s, 1H), 6.62 (d, J= 7.6 Hz, 1H), 7.13 - 7.19 (m, 2H), 9.41 (bs, 1H), 10.15 (s, 1H). HPLC (Method D): 7.525 min. LCMS (Method A): 1.752min, MS: ES+ 332.9 (M+1). Prep. HPLC purification method: [00536] Chromatographic separation and isolation were conducted with Waters 2545 quaternary system with Waters 2489 UV Detector; column Shim-Pack GIST C18 (250mm x 20mm x 5µm); compounds were eluted with: Mobile Phase A : 0.05% formic acid in water , Mobile Phase B : Acetonitrile with a gradient of T = 0 min (60% A, 40% B); gradient to T = 16.00 min (50% A, 50% B); T = 16.01 min (0% A, 100% B) gradient to T = 18.00 min (0% A, 100% B); T = 18.01 min (60% A, 40% B); gradient to T = 22.00 min (60% A, 40% B); Flow rate= 22ml/min; analysis time 22 min. Step 2: (E)-N-(5-Chloro-2-(2, 4-dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4-(dimethyl amino)- N-methylbut-2-enamide

[00537] The title compound was prepared from (E)-4-(dimethylamine) but-2-enoic acid hydrochloride and (5-chloro-4-(methylamino)isoindolin-2-yl)(2,4-dihydroxy-5- methylphenyl)methanone (Step 1) analogously to Example 24 Step 4.
[00538]
1H NMR (DMSO-d6, 400 MHz): δ 2.06 (m, 9H), 2.87 (s, 3H), 3.36 - 3.12 (m, 2H), 4.62 - 4.88 (m, 4H), 5.70 (d, J= 16 Hz, 1H), 6.40 (s, 1H), 6.60 - 6.71 (m, 1H), 7.05 (s, 1H), 7.56 (d, J= 7.6 Hz, 1H), 7.42 (d, J= 8 Hz, 1H), 9.73 (bs, 2H). LCMS (Method B): 1.534 min, MS: ES+ 444.3 (M+1). HPLC (Method D): 4.926 min, Prep. HPLC purification method: [00539] Chromatographic separation and isolation were conducted with Shimadzu LC20AP with UV detector. The column used was Shim-Pack GIST C18 (250mm x 20mm x 5µm) and the compounds were eluted with, Mobile Phase A : 0.05% formic acid in water , Mobile Phase B : acetonitrile:methanol (50:50) with a gradient of T = 0.0 min (90% A, 10% B); gradient to T = 18.00 min (58% A, 42% B); T = 18.01 min (0% A, 100% B) gradient to T = 20.00 min (0% A, 100% B); T = 20.01 min (90% A, 10% B); gradient to T = 24.00 min (90% A, 10% B); Flow rate= 21ml/min; analysis time 24 min. Example 25: (E)-4-(Dimethylamino)-N-(2-(6-hydroxy-3,3-dimethyl-2-oxoindoline-5- carbonyl)isoindolin-4-yl)-N-methylbut-2-enamide Step 1: 6-Methoxyindolin-2-one
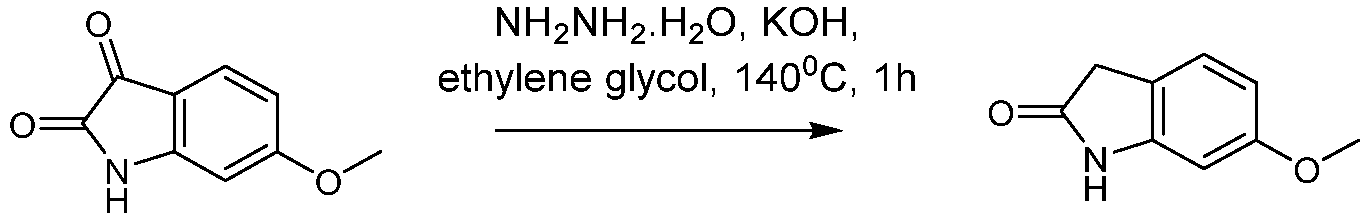
[00540] To a stirred solution of 6-methoxyindoline-2,3-dione (CAS: 52351-75-4) (10 g, 56.50 mmol, 1.0 eq.) in ethylene glycol (100 mL) were added NH
2NH
2.H
2O (5.42 g, 169.37 mmol, 3 eq.) and KOH (3.16 g, 56.42 mmol, 1 eq.) at room temperature. The reaction mixture was heated to 140 °C and stirred for 1 h. The resulting mixture was allowed to cool to room temperature, poured into ice cold water (200 mL), neutralized with sat. KHSO
4 solution and extracted into ethyl acetate (3 x 200 mL). The aqueous layer was further extracted with 15% MeOH: DCM (3 x 200 mL) and the combined organic extracts were dried over Na
2SO
4 and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted at 25% ethyl acetate in hexane) to afford the title compound (9.0 g, Yield: 98 %). [00541]
1H NMR (DMSO-d6, 400 MHz): δ 3.36 (d, J= 11.6 Hz, 2H), 3.72 (s, 3H), 6.38 - 6.39 (m, 1H), 6.47 - 6.49 (m, 1H), 7.09 (d, J= 8 Hz, 1H), 10.33 (s, 1H). LCMS (Method A): 1.066 min, MS: ES+ 163.8 Step 2: 5-Bromo-6-methoxyindolin-2-one
[00542] To a cooled (0 ˚C) solution of 6-methoxyindolin-2-one (Step 1) (9 g, 55.21 mmol, 1.0 eq.) in THF (90 mL) was added NBS (9.83 g, 55.22 mmol, 1 eq.) and the reaction mixture was stirred at room temperature for 1 h. The resulting mixture was concentrated under reduced pressure and purification of the crude material by chromatography on silica (product eluted at 30%) afforded the title compound (9 g, Yield: 67 %).
1H NMR (DMSO-d6, 400 MHz): δ 3.43 (s, 2H), 3.82 (s, 3H), 6.55 (s, 1H), 7.37 (s, 1H), 10.49 (s, 1H). LCMS (Method A): 1.371 min, MS: ES+ 241.8 & 243.8 (M & M+2). Step 3: tert-Butyl 5-bromo-6-methoxy-2-oxoindoline-1-carboxylate
[00543] To a cooled (0 ˚C) solution of 5-bromo-6-methoxyindolin-2-one (Step 2) (9 g, 37.19 mmol, 1.0 eq.) in DCM (90 mL) and DMF (18 mL) was added Boc anhydride (8.92 g, 40.91 mmol, 1.1 eq.) and DMAP (0.907 g, 7.43 mmol, 0.2 eq.) and the reaction mixture was stirred at room temperature for 2 h. The resulting mixture poured into ice cold water (1L) and extracted with EtOAc (3 x 500 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under reduced pressure. Purification of the crude material by chromatography on silica (product eluted at 10% ethyl acetate in hexane) afforded the title compound as an off-white solid. (4 g, Yield: 31 %).
1H NMR (DMSO-d6, 400 MHz): δ 1.58 (s, 9H), 3.68 (s, 2H), 3.86 (s, 3H), 7.50 (s, 1H), 7.53 (s, 1H).LCMS (Method A): 2.108 min, MS: ES+ 241.8 (M-100). Step 4: tert-Butyl 5-bromo-6-methoxy-3,3-dimethyl-2-oxoindoline-1-carboxylate
[00544] To a cooled (0 ˚C) solution of tert-butyl 5-bromo-6-methoxy-2-oxoindoline-1-carboxylate (Step 3) (4.0 g, 11.70 mmol, 1.0 eq.) in THF (40 mL) was added portionwise NaH (0.561 g, 23.37 mmol, 2 eq.) and the mixture was stirred for 30 mins. Methyl iodide (8.30 g, 58.45 mmol, 5 eq.) was added and stirring continued at 0 °C for 30 mins. The resulting mixture poured into ice cold water (100 mL) and extracted into ethyl acetate (3 x 100 mL). The combined organic extracts
were dried over Na
2SO
4 and concentrated under reduced pressure. The crude material was purified by chromatography on silica (product eluted in 10% ethyl acetate in hexane) to afford the title compound as a yellow oil (1.5 g, Yield: 35%). [00545]
1H NMR (DMSO-d6, 400 MHz): δ 1.32 (s, 6H), 1.59 (s, 9H), 3.87 (s, 3H), 7.53 (s, 1H), 7.70 (s, 1H). LCMS (Method A): 2.086 min, MS: ES+ 313.8 & 315.7 (M+1 & M+3). Step 5: 5-Bromo-6-methoxy-3,3-dimethylindolin-2-one
[00546] To a cooled (0 ˚C) solution of tert-butyl 5-bromo-6-methoxy-3,3-dimethyl-2-oxoindoline- 1-carboxylate (Step 4) (0.9 g, 2.43 mmol, 1.0 eq.) in DCM (9 mL) was added dropwise 4M HCl in dioxane (9 mL) and the reaction mixture was stirred at room temperature for 1 h. The resulting mixture was concentrated under reduced pressure and the crude material poured into ice cold water (100 mL), neutralized with sat. NaHCO
3 solution and extracted with ethyl acetate (3 x 100 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under reduced pressure. The crude material was purified by chromatography on silica (product eluted in 40% ethyl acetate in hexane) to afford the title compound (0.4 g, Yield: 61%).
1H NMR (DMSO-d6, 400 MHz): δ 1.23 (s, 6H), 3.82 (s, 3H), 6.58 (s, 1H), 7.51 (s, 1H), 10.44 (s, 1H). LCMS (Method A): 1.663 min, MS: ES+ 269.8 & 271.7 (M & M+2). Step 6: 6-Methoxy-3,3-dimethyl-2-oxoindoline-5-carboxylic acid
[00547] To a cooled (-78 ˚C) solution of 5-bromo-6-methoxy-3,3-dimethylindolin-2-one (Step 5) (0.4 g, 1.48 mmol, 1.0 eq.) in THF (4 mL) was added dropwise n-BuLi (1.6M in hexane) (1.85 mL, 2 eq.) and the reaction mixture was stirred at -78 °C for 30 mins. Dry ice was added portion wise (~8-10 pieces) over a period of 1 h and the resulting mixture was poured into ice cold NH
4Cl solution (50 mL) and extracted into ethyl acetate (3 x 50 mL). The aqueous layer was acidified with citric acid solution (pH ~5-6) and extracted with 10% MeOH: DCM solution (3 x 50 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under reduced pressure to afford the title compound which was used in the next step without further purification (0.130 g, Yield: 48%).
[00548]
1H NMR (DMSO-d6, 400 MHz): δ 1.26 (s, 6H), 3.81 (s, 3H), 6.56 (s, 1H), 7.64 (s, 1H), 10.65 (s, 1H), 12.21 (s, 1H). LCMS (Method A): 0.998 min, MS: ES+ 235.9 Step 7: 6-Hydroxy-3,3-dimethyl-2-oxoindoline-5-carboxylic acid
[00549] To a cooled (0 ˚C) solution of 6-methoxy-3,3-dimethyl-2-oxoindoline-5-carboxylic acid (Step 6) (0.13 g, 0.55 mmol, 1.0 eq.) in DCM (1.3 mL) was added dropwise BBr3 (1M in DCM) (1.1 mL, 2 eq.) and the reaction mixture was allowed to stir at room temperature for 2 h. The resulting mixture was diluted with 5% MeOH: DCM solution (50 mL), washed with ice cold water (50 mL), dried over Na
2SO
4 and concentrated under reduced pressure to afford the title compound (0.1 g, Yield: 65 %). [00550]
1H NMR (DMSO-d6, 400 MHz): δ 1.24 (s, 6H), 6.36 (s, 1H), 7.65 (s, 1H), 10.69 (s, 1H), 11.77 (s, 1H), 13.66 (s, 1H). LCMS (Method A): 1.041 min, MS: ES+ 221.9 Step 8: (E)-4-(Dimethylamino)-N-(2-(6-hydroxy-3,3-dimethyl-2-oxoindoline-5- carbonyl)isoindolin-4-yl)-N-methylbut-2-enamide
[00551] To a stirred solution of 6-hydroxy-3,3-dimethyl-2-oxoindoline-5-carboxylic acid (Step 7) (0.08 g, 0.36 mmol, 1 eq.) in DMF (0.8 mL) were added EDC.HCl (0.105 g, 0.54 mmol, 1.5 eq.) and HOAt (0.05 g, 0.37 mmol, 1.0 eq.) at room temperature and stirred for 5 mins. (E)-4- Dimethylamino)-N-(isoindolin-4-yl)-N-methylbut-2-enamide hydrochloride carboxylate (Intermediate S) (0.16 g, 0.54 mmol, 1.5 eq.) and NMM (0.182 g, 1.8 mmol, 5 eq.) were added and the reaction mixture was stirred at room temperature for 30 min. The resulting mixture was poured into ice-cold water (50 mL) and extracted with ethyl acetate (3 x 50 mL). The combined organic extracts were washed with ice cold water (2 x 50 mL), dried over Na
2SO
4 and concentrated under reduced pressure. The crude material was purified by preparative HPLC
eluting with A) 0.05% HCl in water B) ACN followed by pure fraction lyophilization to afford the title compound (0.035 g, Yield: 21%). [00552]
1H NMR (DMSO-d6, 400 MHz): δ 1.22 (s, 6H), 2.61 - 2.68 (m, 6H), 3.10 - 3.17 (m, 1H), 3.23 (s, 2H), 4.60 - 4.64 (m, 1H), 4.78 - 4.88 (m, 3H), 6.02 - 6.08 (m, 1H), 6.53 (s, 1H), 6.58 - 6.78 (m, 1H), 7.16 - 7.26 (m, 2H), 7.34 - 7.46 (m, 2H), 10.32 - 10.54 (m, 3H). LCMS (Method A): 1.054 min, MS ES+: 463.1 (M+1). HPLC (Method B): 4.287 min. Prep. HPLC purification method: [00553] Chromatographic separation and isolation were conducted with a Waters 2545 purification system with UV detector. The column used was Shim-Pack GIST C18 (250mm x 20mm x 5µm) and the compounds were eluted with, Mobile Phase A : 0.05% hydrochloric acid in water, Mobile Phase B : Acetonitrile with a gradient of T = 0 min (92% A, 8% B); gradient to T = 22.00 min (76% A, 24% B); T = 22.01 min (0% A, 100% B) gradient to T = 25.00 min (0% A, 100% B); T = 25.01 min (92% A, 8% B); gradient to T = 28 min (92% A, 8% B); Flow rate= 22ml/min; analysis time 28 min. Example 26: (E)-N-(2-(3-Chloro-4,6-dihydroxy-2-methylbenzoyl)isoindolin-4-yl)-4- (dimethylamino)-N-ethylbut-2-enamide Step 1: tert-Butyl 4-(ethylamino)isoindoline-2-carboxylate


[00554] The title compound was prepared from tert-butyl 4-bromoisoindoline-2-carboxylate (CAS:1035235-27-8) and 2M ethylamine in THF analogously to Intermediate A step 1. [00555]
1H NMR (DMSO-d6, 400 MHz): δ 1.16 (t, J= 7.2 Hz, 2H), 1.46 (d, J= 6 Hz, 9H), 3.04 - 3.11 (m, 2H), 4.39 (d, J= 5.2 Hz, 2H), 4.50 (d, J= 10.4 Hz, 2H), 5.21 - 5.24 (m, 1H), 6.41 (d, J= 8 Hz, 1H), 6.50 (t J= 5.2 Hz, 1H), 7.06 (t, J= 8 Hz, 1H). [00556] LCMS (Method A): 2.135 min, MS: ES+ 206.98 Step 2: N-Ethylisoindolin-4-amine hydrochloride
[00557] The title compound was prepared from tert-butyl 4-(ethylamino)isoindoline-2- carboxylate (Step 1) and 4M HCl in dioxane analogously to Intermediate A step 2. [00558]
1H NMR (DMSO-d6, 400 MHz): δ 1.23 (t, J= 7.2 Hz, 3H), 3.17 - 3.23 (m, 2H), 3.56 (s, 1H), 4.47 (t, J= 5.6 Hz, 2H), 4.57 (s, 2H), 7.03 (m, 2H), 7.33 (t, J= 7.6 Hz, 1H), 10.18 (s, 2H). LCMS (Method A): 0.739 min,100%, MS: ES+ 162.8 Step 3: (3-Chloro-4,6-dihydroxy-2-methylphenyl) (4-(ethylamino)isoindolin-2-yl) methanone
[00559] The title compound was prepared from 3-chloro-4,6-dihydroxy-2-methylbenzoic acid (Intermediate B) and N-ethylisoindolin-4-amine hydrochloride (Step 2) analogously to Example 1 Step 1. [00560]
1H NMR (DMSO-d6, 400 MHz): δ 1.12 (t, J= 6.8 Hz, 2H), 1.21 (t, J= 7.2 Hz, 1H), 2.17 (s, 3H), 3.04-3.16 (m, 2H), 4.22-4.51 (m, 2H), 4.62 (s, 2H), 5.02 (m, 1H), 6.42-6.58 (m, 3H), 7.04- 7.11 (m, 1H), 9.45 (bs, 1H), 9.75 (bs, 1H). LCMS (Method A): 1.627 min, MS: ES+ 347 (M+1). HPLC (Method D): 5.45 min. Step 4: (E)-N-(2-(3-Chloro-4,6-dihydroxy-2-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N- ethylbut-2-enamide
[00561] The title compound was prepared from (E)-4-(dimethylamino)but-2-enoic acid hydrochloride (CAS: 848133-35-7) and (3-chloro-4,6-dihydroxy-2-methylphenyl) (4- (ethylamino)isoindolin-2-yl) methanone (Step 3) analogously to Example 1 step 2. [00562] High temperature
1H NMR (DMSO-d6, 400 MHz): δ 0.99 (t, J= 6.8 Hz, 3H), 2.15 (s, 3H), 2.21 (s, 6H), 3.10 (bs, 2H), 3.66 (bs, 2H), 4.26 (bs, 1H), 4.48 (d, J= 14.8 Hz, 1H), 4.64 (d, J= 14 Hz, 1H), 4.88 (s, 1H), 5.79 - 5.91 (m, 1H), 6.49 (d, J= 16 Hz, 1H), 6.60 - 6.74 (m, 1H), 7.17 (t, J= 8 Hz, 1H), 7.30 - 7.45 (m, 2H), 9.52 (bs, 1H), 9.75 - 9.80 (bs, 1H). LCMS (Method A): 1.157 min, MS: ES+ 458.1 (M+1). HPLC (Method D): 4.85 min. Prep. HPLC purification method: [00563] Chromatographic separation and isolation were conducted with a Waters 2545 binary pump with Waters 2489 UV detector. The column used was Sunfire C18 OBD (250mm x 19mm 5µm) and the compounds were eluted with, Mobile Phase A : 0.05% formic acid in water, Mobile Phase B : Acetonitrile with a gradient of T = 0.01 min (88% A, 12% B); gradient to T = 18.00 min (76% A, 24% B); T = 18.01 min (0% A, 100% B) gradient to T = 21.00 min (0% A, 100% B); T = 21.01 min (88% A, 12% B); gradient to T = 25 min (88% A, 12% B); Flow rate= 14ml/min; analysis time 25 min. Example 27: (E)-N-(2-(3-Chloro-4,6-dihydroxy-2-methylbenzoyl)isoindolin-4-yl)-4- (ethyl(methyl)amino)-N-methylbut-2-enamide

[00564] The title compound was prepared from (E)-4-(ethyl(methyl)amino)but-2-enoic acid hydrochloride (CAS: 1449501-61-4) and (3-chloro-4,6-dihydroxy-2-methylphenyl)(4- (methylamino)isoindolin-2-yl)methanone (Example 1, Step 1) [00565] High temperature
1H NMR (DMSO-d6, 400 MHz, 348K): δ 0.873 (t, J = 6.8Hz, 3H), 2.05 (s, 3H), 2.16 (s, 3H), 2.256 – 2.273 (q, br, 2H), 2.98 (m, 1H), 3.11 (m, 2H), 3.24 (m, 2H), 4.20 - 4.86 (m, 4H), 5.82 (s, br, 1H), 6.47 - 6.50 (m, 1H), 6.58 – 6.72 (m, 1H), 7.20 - 7.74 (m, 3H), 9.65 (bs, 2H). [00566] LCMS (Method A): 1.119 min, MS: ES+458.0 (M+1). [00567] HPLC (Method D): 4.889 min.
Prep. HPLC purification method: [00568] Chromatographic separation and isolation were conducted with Waters 2545 quaternary system with Waters 2489 UV Detector. The column used was Shim-Pack GIST C18 (250mm x 20mm x 5µm) and the compounds were eluted with, Mobile Phase A : 0.05% formic acid in water, Mobile Phase B: acetonitrile:methanol(50:50) with a gradient of T = 0.0 min (90% A, 10% B); gradient to T = 16.00 min (70% A, 30% B); T = 16.01 min (0% A, 100% B) gradient to T = 18.00 min (0% A, 100% B); T = 18.01 min (90% A, 10% B); gradient to T = 20.00 min (90% A, 10% B); Flow rate= 24ml/min; analysis time 24 min. Example 28: (E)-N-(7-Chloro-2-(2,4-dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide Step 1: 7-Chloro-N-methylisoindolin-4-amine hydrochloride

[00569] The title compound was prepared from tert-butyl 4-chloro-7-(methylamino)isoindoline-2- carboxylate (by product of Example 24, Step 1) and 4M HCl in dioxane analogously to Example 24 Step 2. [00570]
1H NMR (DMSO-d6, 400 MHz): δ 2.72 (s, 3H), 4.36 - 4.45 (m, 4H), 5.11 (bs, 1H), 6.52 (d, J= 8.8 Hz, 1H), 7.21 (d, J= 8.8 Hz, 1H),10.08 (s, 2H). [00571] LCMS (Method A): 0.842 min, MS: ES+ 182.88 (M+1). Step 2: (4-Chloro-7-(methylamino)isoindolin-2-yl) (2,4-dihydroxy-5-methylphenyl)methanone
[00572] The title compound was prepared from 2,4-dihydroxy-5-methylbenzoic acid (Intermediate F) and 7-chloro-N-methylisoindolin-4-amine hydrochloride analogously to Example 24 Step 3.
[00573]
1H NMR (DMSO-d6, 400 MHz): δ 2.04 (s, 3H), 2.65 and 2.71 (singlets, 3H), 4.64 – 4.70 (m, 4H), 5.55 and 5.69 (singlets, 1H, D
2O exchangeable), 6.41 (s, 2H), 7.03 (s, 1H), 7.13 (s, 1H), 9.62 (s, 1H, D
2O exchangeable), 9.95 and 10.09 (singlets, 1H, D
2O exchangeable). [00574] LCMS (Method A): 1.821 min, MS: ES+ 332.9 (M+1). [00575] HPLC (Method: D): 8.19 min. Step 3: (E)-N-(7-Chloro-2-(2,4-dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N- methylbut-2-enamide
[00576] The title compound was prepared from (E)-4-(dimethylamino) but-2-enoic acid hydrochloride and (4-chloro-7-(methylamino)isoindolin-2-yl)(2,4-dihydroxy-5- methylphenyl)methanone (Step 2) analogously to Example 24, Step 4. [00577]
1H NMR (DMSO-d6, 400 MHz): δ 2.02 (s, 3H), 2.14 (s, br, 6H), 3.08 (m, 2H), 3.18 (s, 3H), 4.64 (d, J= 14.4 Hz, 1H), 4.81 (bs, 3H), 5.83 (m, 1H), 6.40 (d, J= 18.4 Hz, 1H), 6.67 - 6.97 (m, 2H), 7.30 (d, J= 7.6 Hz, 1H), 7.50 (d, J= 6.8 Hz, 1H), 9.68 (bs, 1H), 9.96 (bs, 1H). [00578] LCMS (Method A): 1.169 min, MS: ES+ 444.02 (M+1). [00579] HPLC (Method: D): 5.076 min. Prep. HPLC purification method: [00580] Chromatographic separation and isolation were conducted with a Waters 2545 binary pump with Waters 2489 UV detector with waters Acquity QDA detector; column Waters Sunfire C18 OBD (250mm x 19mm 5µm); compound eluted with, Mobile Phase A : 0.05% formic acid in water, Mobile Phase B : acetonitrile:water (80:20) with a gradient of T = 0.01 min (84% A, 16% B); gradient to T = 23.00 min (78% A, 22% B); T = 23.01 min (0% A, 100% B) gradient to T = 26.00 min (0% A, 100% B); T = 26.01 min (84% A, 16% B); gradient to T = 29 min (84% A, 16% B); Flow rate= 19ml/min; analysis time 29 min. Example 29: (E)-N-(7-Chloro-2-(4,6-dihydroxy-2,3-dimethylbenzoyl)isoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide
Step 1: (4-Chloro-7-(methylamino)isoindolin-2-yl)(4,6-dihydroxy-2,3-dimethylphenyl)- methanone

[00581] The title compound was prepared from 4,6-dihydroxy-2,3-dimethylbenzoic acid (Intermediate K) and 7-chloro-N-methylisoindolin-4-amine hydrochloride (Example 28, Step 1) analogously to Example 24 Step 3. [00582]
1H NMR (DMSO-d6, 400 MHz): δ 1.95 (d, J= 3.6 Hz, 3H), 2.01 (s, 3H), 2.60 and 2.73 (doublets, J= 4.8 Hz, 3H), 4.24 (t, J= 14 Hz, 1H), 4.38 - 4.48 (m, 1H), 4.63 - 4.68 (m, 2H), 5.52 and 5.69 (doublets, J= 4.8 Hz, 1H, D2O exchangeable), 6.31 (s, 1H), 6.38 - 6.44 (m, 1H), 7.11 - 7.15 (m, 1H), 9.22 - 9.30 (m, 2H, D2O exchangeable). [00583] LCMS (Method A): 1.685 min, MS: ES+ 346.90 (M+1). [00584] HPLC (Method D): 7.24 min. Step 2: (E)-N-(7-Chloro-2-(4,6-dihydroxy-2,3-dimethylbenzoyl)isoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide
[00585] The title compound was prepared from (E)-4-(dimethylamino) but-2-enoic acid hydrochloride and (4-chloro-7-(methylamino)isoindolin-2-yl)(4,6-dihydroxy-2,3- dimethylphenyl)methanone analogously to Example 24 Step 4. [00586] High temperature
1H NMR (DMSO-d6, 400 MHz, 349.8K): δ 1.92 - 2.04 (m, 6H), 2.08 (s, 6H), 2.93 - 3.24 (m, 6H), 4.54 - 4.84 (m, 4H), 5.86 - 5.94 (m, 1H), 6.29 – 6.34 (m, 1H), 6.59 - 6.73 (m, 1H), 7.28 (t, J= 8.4 Hz, 1H), 7.45 (t, J= 7.4 Hz, 1H), 9.01 (bs, 2H). [00587] LCMS (Method A): 1.209 min, MS: ES+ 458.02 (M+1). [00588] HPLC (Method B): 4.631 min. Prep. HPLC purification method:
[00589] Chromatographic separation and isolation were conducted with Waters 2545 quaternary system with Waters 2489 UV Detector; column Waters Sunfire C18 OBD (250mm x 19mm 5µm); compound eluted with, Mobile Phase A : 0.1% formic acid in Merck water, Mobile Phase B : Acetonitrile with a gradient of T = 0.0 min (100% A, 0% B); gradient to T = 2.00 min (90% A, 10% B); T = 19.00 min (80% A, 20% B) gradient to T = 22.00 min (80% A, 20% B); T = 22.01 min (0% A, 100% B); gradient to T = 24 min (0% A, 100% B); gradient to T = 24.01 min (100% A, 0% B); gradient to T = 26.00 min (100% A, 0% B); Flow rate= 21ml/min; analysis time 26 min. Example 30: N-(2-(3-Chloro-4,6-dihydroxy-2-methylbenzoyl)isoindolin-4-yl)-N-methyl acrylamide
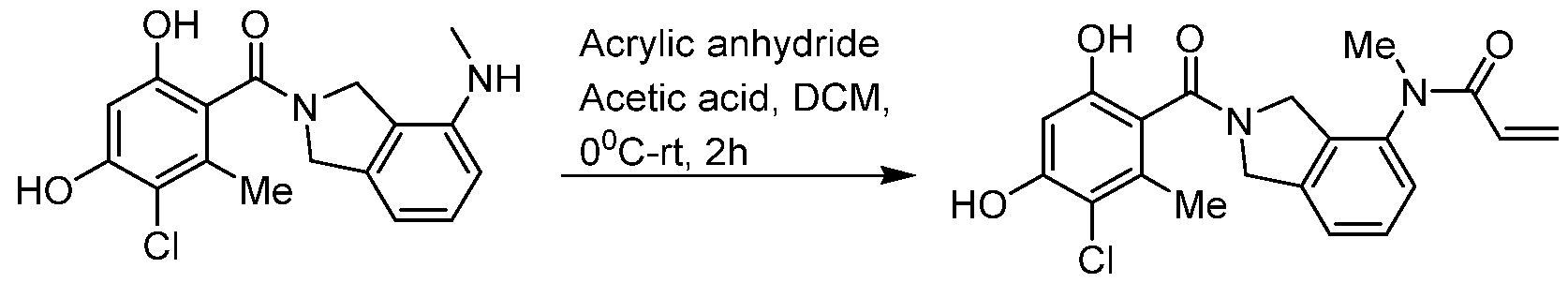
[00590] The title compound was prepared from (3-chloro-4,6-dihydroxy-2-methylphenyl)(4- (methylamino)isoindolin-2-yl)methanone (Example 1, Step 1), acetic acid and acrylic anhydride analogously to Example 3 Step 3. [00591]
1H NMR (D
2O exchange) (DMSO-d6, 400 MHz): δ 2.11 (s, 3H), 3.10 and 3.22 (singlets, 3H), 4.40 - 4.51 (m, 2H), 4.58 (t, J= 12 Hz, 1H), 4.74 - 4.88 (m, 1H), 5.62 (m, 1H), 5.86 - 5.97 (m, 1H), 6.01 - 6.24 (m, 1H), 6.47 (m, 1H), 7.24 (d, J= 7.6 Hz, 1H), 7.32 - 7.46 (m, 2H). [00592] LCMS (Method A): 1.373 min, MS ES+: 386.9 (M+1). [00593] HPLC (Method D): 6.137 min. Prep. HPLC purification method: [00594] Chromatographic separation and isolation were conducted with Waters 2545 quaternary system with Waters 2489 UV detector. The column used was Shim-Pack GIST C18 (250mm x 20mm x 5µm) and the compounds were eluted with, Mobile Phase A : 0.05% formic acid in water, Mobile Phase B: acetonitrile:methanol (50:50) with a gradient of T = 0.01 min (65% A, 35% B); gradient to T = 30.00 min (65% A, 35% B); T = 30.01 min (0% A, 100% B) gradient to T = 32.00 min (0% A, 100% B); T = 32.01 min (65% A, 35% B); gradient to T = 35 min (65% A, 35% B); Flow rate= 23ml/min; analysis time 35 min. Example 31: (E)-N-(5-Chloro-2-(4,6-dihydroxy-2,3-dimethylbenzoyl) isoindolin-4-yl)-4- (dimethyl amino)-N-methylbut-2-enamide
Step 1: (5-Chloro-4-(methylamino)isoindolin-2-yl)(4,6-dihydroxy-2,3-dimethylphenyl) methanone

[00595] Performed in 2 parallel batches, each of 0.5 g scale: To a stirred solution 4,6-dihydroxy- 2,3-dimethylbenzoic acid (Intermediate K) (0.5 g, 2.74 mmol, 1 eq) in DMF (5 mL) were added EDC.HCl (0.78 g, 4.12 mmol, 1.5 eq) and HOBT (0.037 g, 0.274 mmol, 0.1 eq) at room temperature. 5-Chloro-N-methylisoindolin-4-amine hydrochloride (Example 24 Step 2) (0.49 g, 2.74 mmol, 1.0 eq.) and TEA (1.10 mL, 8.24 mmol, 3.0 eq.) were added and the reaction mixture was heated to 120 ˚C using microwave irradiation for 3 h. The resulting mixture was poured into cold water (100 mL) and extracted with ethyl acetate (3 x 100 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under vacuum. The crude material was purified by reverse phase chromatography on silica (product eluted in 24.0% water in ACN) yielding (5-chloro-4-(methylamino)isoindolin-2-yl)(4,6-dihydroxy-2,3-dimethylphenyl) methanone as a white solid (0.5 g, Yield: 26.3%). [00596]
1H NMR (DMSO-d6, 400 MHz): δ 1.95 (d, J= 2.0 Hz, 3H), 2.00 (d, J= 3.2 Hz, 3H), 2.67 - 3.02 (m, 3H), 4.19 - 4.55 (m, 2H), 4.65 - 4.77 (m, 1H), 5.01 (d, J= 4.4 Hz, 1H), 5.27 (dd, J= 5.6 Hz, 1H), 6.30 (s, 1H), 6.65 (dd, J= 8.0 Hz, 1H), 7.17 (dd, J= 8.0 Hz, 1H), 9.27 (t, J= 4.0 Hz, 2H). [00597] LCMS (Method N): 1.635 min, MS: ES+ 347.0 (M+1). [00598] HPLC (Method H): 6.82 min Prep HPLC Purification method: [00599] Compound was purified with a Shimadzu Nexera prep with an Lh-40 auto purification system; column Shim-Pack GIST C18 (250mm x 20mm x 5µm); compound eluted with, Mobile Phase A: 0.05% formic acid in water Mobile Phase B: acetonitrile with a gradient of T = 0.01 min (65% A, 35% B); gradient to T = 20.00 min (65% A, 35% B); T = 20.01 min (0% A, 100% B); gradient to T = 23.00 min (0% A, 100% B); T = 23.01 min (65% A, 35% B); T = 26.00 min (65% A, 35% B); Flow rate= 22 ml/min; analysis time 26 min. Step 2: (E)-N-(5-Chloro-2-(4,6-dihydroxy-2,3-dimethylbenzoyl)isoindolin-4-yl)-4-(dimethyl amino)-N-methylbut-2-enamide
[00600] Performed in 4 parallel batches, each of 0.1 g scale: To a stirred solution of (E)-4- (dimethylamine) but-2-enoic acid hydrochloride (CAS: 848133-35-7) (0.1 g, 6.06 mmol, 1 eq.) in DMF (1 mL) at room temperature were added (5-chloro-4-(methylamino)isoindolin-2-yl) (4,6- dihydroxy-2,3-dimethylphenyl) methanone (Step 1) (0.104 g, 0.303 mmol, 0.5 eq) and DCC (0.185 g, 9.0 mmol, 1.5 eq.). The reaction mixture was heated to 150
˚C using microwave irradiation for 15 mins. The resulting mixture was diluted with water (100 mL) and filtered. The filtrate was lyophilized and the crude material was purified by reverse phase chromatography (product was eluted in 24% ACN in water) followed by prep. HPLC purification yielding (E)-N-(5- chloro-2-(4,6-dihydroxy-2,3-dimethylbenzoyl) isoindolin-4-yl)-4-(dimethyl amino)-N-methylbut-2- enamide as a white solid (0.020 g, Yield: 3.8%). [00601] HT
1H NMR (DMSO-d6, 400 MHz): δ 1.98 - 2.08 (m, 9H), 2.87 - 3.17 (m, 3H), 4.41 (d, J= 16.8 Hz, 1H), 4.62 (t, J= 9.6 Hz, 1H), 4.82 (d, J= 16.4 Hz, 2H), 5.72 (d, J= 15.2 Hz, 1H), 6.26 - 6.32 (m, 1H), 6.69 (s, 1H), 7.35 (d, J= 7.2 Hz, 1H), 7.53 (t, J= 7.6 Hz, 1H), 8.95 - 9.00 (m, 2H). [00602] LCMS (Method U): 6.11 min, MS: ES+ 458.20 (M+1). [00603] HPLC (Method O): 3.91 min, 4.46 min Prep. HPLC purification method [00604] Waters 2545 quaternary system with Waters 2489 UV Detector. Column YMC Triart C18 (250 x 20 mm ID), 5um; compound eluted with, Mobile Phase A 5 mM ammonium bicarbonate in water Mobile Phase B: acetonitrile with a gradient of T = 0.0 min (85% A, 15% B); gradient to T = 17.00 min (35% A, 65% B); T = 17.01 min (0% A, 100% B); gradient to T = 19.00 min (0% A, 100% B); T = 19.01 min (85% A, 15% B); T = 22.00 min (85% A, 15% B); Flow rate= 14 ml/min; analysis time 07.01. Example 32: (E)-N-(2-(3-Chloro-2-fluoro-4,6-dihydroxybenzoyl)isoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide Step 1: 3-Chloro-2-fluoro-4,6-dimethoxybenzaldehyde
[00605] To a stirred solution of 2-fluoro-4,6-dimethoxybenzaldehyde (prepared from 1-fluoro- 3,5-dimethoxybenzene (CAS: 52189-63-6) and POCl3 analogously to Intermediate B Step 1) (75.0 g, 407.23 mmol, 1 eq.) in DMF (750 mL) was added NCS (54.37 g, 407.23 mmol, 1.0 eq.); the reaction mixture was stirred at room temperature for 16 h. then filtered and dried under vacuum. The crude material was triturated with n-hexane and diethyl ether (5 x 100 mL) to yield 3-chloro-2-fluoro-4,6-dimethoxybenzaldehyde as a white solid (55g, Yield: 56.3 %). [00606]
1H NMR (DMSO-d6, 400 MHz): δ 4.00 (s, 3H), 4.05 (s, 3H), 6.78 (s, 1H), 10.16 (s, 1H). Step 2: 3-Chloro-2-fluoro-4,6-dimethoxybenzoic acid
[00607] Performed in 2 parallel batches, each of 10 g scale: To a stirred solution of 3-chloro-2- fluoro-4,6-dimethoxybenzaldehyde (Step 1) (10 g, 45.7 mmol, 1 eq.) in DMSO (50 mL) were slowly added a saturated solution of NaH
2PO
4 (38.42 g, 320.20 mmol, 7.0 eq) and NaClO
2 (28.95 g, 320.20 mmol, 7.0 eq). The reaction mixture was stirred at room temperature for 16 h. The resulting mixture was diluted with water (500 mL) and extracted with ethyl acetate (4 x 500 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum yielding 3-chloro-2-fluoro-4,6-dimethoxybenzoic acid as a white solid (15 g, yield: 51.2%). The crude material was used without further purification. [00608]
1H NMR (DMSO-d6, 400 MHz): δ 3.87 (s, 3H), 3.96 (s, 3H), 6.71 (s, 1H), 13.33 (s, 1H). Step 3: 3-Chloro-2-fluoro-4,6-dihydroxybenzoic acid
[00609] Performed in 2 parallel batches, each of 5 g scale: To a stirred solution of 3-chloro-2- fluoro-4,6-dimethoxybenzoic acid (Step 2) (5.0 g, 21.31 mmol, 1.0 eq.) in DCM (50 mL) was added BBr
3 (106.5 mL, 106.5 mmol, 5.0 eq) at 0
°C. The reaction mixture was stirred at 50
°C for 48 h. The resulting mixture was cooled to room temperature, poured into ice cold water (1000 mL) and extracted with ethyl acetate (3 x 1000 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum. The crude material was triturated with DCM and diethyl ether (3 x 20 mL) followed by high vacuum drying to yield 3-chloro-2-fluoro-4,6- dihydroxybenzoic acid as an off-white solid (6.3 g, Yield: 85.2%). [00610]
1H NMR (DMSO-d6, 400 MHz): δ 6.35 (s, 1H), 11.55 (s, 1H), 11.92 (s, 1H), 13.77 (s, 1H). Step 4: (3-Chloro-2-fluoro-4,6-dihydroxyphenyl) (4-(methylamino) isoindolin-2-yl)methanone
[00611] The title compound was prepared from 3-chloro-2-fluoro-4,6-dihydroxybenzoic acid (Step 3) and N-methylisoindolin-4-amine hydrochloride (Intermediate A) analogously to Example 1 Step 1. [00612]
1H NMR (DMSO-d6, 400 MHz): δ 2.63 - 2.72 (dd, J= 4.8 Hz, 3H), 4.36 (s, 1H), 4.54 (d, J= 20.8 Hz, 2H), 4.69 (s, 1H), 5.37 - 5.53 (dd, J= 4.8 Hz, 1H, D
2O exchangeable), 6.38 (t, J= 10.8 Hz, 1H), 6.45 - 6.58 (m, 2H), 7.07 - 7.14 (m, 1H), 10.37 (d, J= 11.2 Hz, 1H), 10.77 (s, 1H). [00613] LCMS (Method A): 1.430 min, MS: ES+ 336.9 (M+1). [00614] HPLC (Method D): 4.73 min. Step 5: (E)-N-(2-(3-Chloro-2-fluoro-4,6-dihydroxybenzoyl) isoindolin-4-yl)-4-(dimethylamino)-N- methylbut-2-enamide
[00615] To a stirred solution of (E)-4-(dimethylamino) but-2-enoic acid hydrochloride (CAS: 848133-35-7) (0.25 g, 1.50 mmol, 1.0 eq.) in DMF (2.5 mL at room temperature) were added (3- chloro-2-fluoro-4,6-dihydroxyphenyl) (4-(methylamino) isoindolin-2-yl) methanone (Step 4) (0.253 g, 0.754 mmol, 0.5 eq.) and DCC (0.623 g, 3.018 mmol, 2.0 eq.). The reaction mixture was heated to 150
˚C using microwave irradiation for 15 mins. The reaction mixture was cooled, poured into cold water (100 mL), filtered, and the filtrate concentrated under vacuum. Crude material was purified by reverse phase column chromatography followed by Prep HPLC purification. The pure fractions were lyophilized to yield (E)-N-(2-(3-chloro-2-fluoro-4,6- dihydroxybenzoyl) isoindolin-4-yl)-4-(dimethylamino)-N-methylbut-2-enamide as an off-white solid (0.05 g, Yield: 8.9%). [00616]
1H NMR (DMSO-d6, 400 MHz): δ 2.02 (s, 3H), 2.06 (s, 3H), 2.88 – 2.90 (m, 2H), 3.10 (s, 2H), 3.24 (s, 1H), 4.42 - 4.87 (m, 4H), 5.75 (t, J= 15.2 Hz, 1H), 6.45 (d, J= 26 Hz, 1H), 6.60 - 6.69 (m, 1H), 7.24 (d, J= 7.6 Hz, 1H), 7.33 (d, J= 7.6 Hz, 1H), 7.40 - 7.46 (m, 1H). [00617] LCMS (Method N): 1.110 min, MS: ES+ 448 (M+1). [00618] HPLC (Method H): 3.84 min. Prep. HPLC purification method: [00619] Waters 2545 binary pump with Waters 2489 uv detector and waters Acquity QDA detector; column Waters Sunfire C18 OBD (250mm x 19mm 5μm); compound eluted with Mobile Phase A: 0.05% formic acid in water, Mobile Phase B: Acetonitrile: MeOH : IPA (65:25:10) with a gradient of T = 0.01 min (90% A, 10% B); gradient to T = 15.00 min (75% A, 25% B); T = 17.00 min (75% A, 25% B) gradient to T = 17.01 min (0% A, 100% B); T = 20.00 min (0% A, 100% B); gradient to T = 20.01 min (90% A, 10% B); ); gradient to T = 25.00 min (90% A, 10% B); Flow rate= 15ml/min; analysis time 25 min. Example 33: (E)-N-(2-(3-Chloro-2-fluoro-4,6-dihydroxybenzoyl)isoindolin-4-yl)-4- (dimethylamino)-N-ethylbut-2-enamide Step 1: (3-Chloro-2-fluoro-4,6-dihydroxyphenyl)(4-(ethylamino)isoindolin-2-yl)methanone

[00620] The title compound was prepared from 3-chloro-2-fluoro-4,6-dihydroxybenzoic acid (Example 32 Step 3) and N-ethylisoindolin-4-amine hydrochloride (Example 26, Step 2) analogously to Example 1 Step 1. [00621]
1H NMR (DMSO-d6, 400 MHz): δ 1.09 - 1.24 (m, 3H), 3.02 - 3.12 (m, 2H), 4.38 - 4.69 (m, 4H), 5.21 - 5.32 (m, 1H, D
2O exchangeable), 6.44 - 6.57 (m, 3H), 7.07 - 7.12 (m, 1H), 10.35 (d, J= 12.8 Hz, 1H), 10.75 (s, 1H). [00622] LCMS (Method N): 1.517 min, MS: ES+ 350.9 (M+1). [00623] HPLC (Method H): 6.86 min. Step 2: (E)-N-(2-(3-Chloro-2-fluoro-4,6-dihydroxybenzoyl)isoindolin-4-yl)-4- (dimethylamino)-N-ethylbut-2-enamide
[00624] The title compound was prepared from (3-chloro-2-fluoro-4,6-dihydroxyphenyl) (4- (ethylamino) isoindolin-2-yl) methanone (Step 1) and (E)-4-(dimethylamino)but-2-enoic acid hydrochloride (CAS: 848133-35-7) analogously to Example 1 Step 2. [00625]
1H NMR (DMSO-d6, 400 MHz): δ 0.94 - 1.07 (m, 3H), 2.08 (d, J= 6.8 Hz, 6H), 2.86 (d, J= 5.2 Hz, 2H), 3.72 - 3.91 (m, 4H), 4.68 - 4.87 (m, 4H), 5.69 (t, J= 14.4 Hz, 1H), 6.49 (d, J= 6 Hz, 1H), 6.60 - 6.71 (m, 1H), 7.20 - 7.25 (m, 1H), 7.33 - 7.48 (m, 2H).
1H NMR is complex due to rotamers. [00626] LCMS (Method N): 1.166 min, MS: ES+ 462 (M+1). [00627] HPLC (Method H): 3.98 min. Prep. HPLC purification method of analysis: [00628] Chromatographic separation and isolation were conducted with Waters 2545 binary pump with waters 2489 UV detector with Waters Acquity QDA detector. The column used was Waters Sunfire C18 OBD (250mm x 19mm 5μm) and the compounds were eluted with, Mobile Phase A: 0.05% formic acid in water, Mobile Phase B: Acetonitrile: MeOH :IPA (65:25:10): Water (90:10) with a gradient of T = 0.01 min (88% A, 12% B); gradient to T = 15.00 min (70% A, 30% B); T = 19.00 min (68% A, 32% B) gradient to T = 19.01 min (0% A, 100% B); T = 22.00 min (0%
A, 100% B); gradient to T = 22.01 min (88% A, 12% B); ); gradient to T = 27.00 min (88% A, 12% B); Flow rate= 12ml/min; analysis time 27 min. Example 34: N-(2-(3-Chloro-4,6-dihydroxy-2-methylbenzoyl) isoindolin-4-yl)-N-ethyl acrylamide

[00629] The title compound was prepared from (3-chloro-4,6-dihydroxy-2-methylphenyl) (4- (ethylamino)isoindolin-2-yl) methanone (Example 26, Step 3), acetic acid and acrylic anhydride analogously to Example 3 Step 3. [00630] HT
1H NMR (DMSO-d6, 400 MHz, 349K): δ 1.01 - 1.26 (m, 3H), 1.79 - 2.16 (m, 5H), 4.51 - 4.87 (m, 4H), 5.57 - 6.20 (m, 3H), 6.50 (s, 1H), 7.16 - 7.44 (m, 3H), 9.74 (s, 2H).
1H NMR is complex due to rotamers. [00631] LCMS (Method N): 1.326 min, MS ES+: 401 (M+1). [00632] HPLC (Method H): 6.435 min. Example 35: N-(2-(3-Chloro-2-fluoro-4,6-dihydroxybenzoyl) isoindolin-4-yl)-N- methylacrylamide
[00633] The title compound was prepared from (3-chloro-2-fluoro-4,6-dihydroxyphenyl) (4- (methylamino) isoindolin-2-yl) methanone (Example 32, Step 4), acetic acid and acrylic anhydride analogously to Example 3 Step 3. [00634]
1H NMR (DMSO-d6, 400 MHz): δ 3.11 - 3.21 (m, 3H), 4.42 - 4.85 (m, 4H), 5.55 - 5.61 (m, 1H), 5.87 - 6.01 (m, 1H), 6.10 - 6.24 (m, 1H), 6.45 (d, J= 12.4 Hz, 1H), 7.24 (d, J= 8 Hz, 1H), 7.33 (d, J= 7.6 Hz, 1H), 7.43 (t, J= 7.6 Hz, 1H), 10.28 (bs, 2H).
[00635] LCMS (Method N): 1.302 min, MS: ES+ 390.9 (M+1). [00636] HPLC (Method I): 4.31 min. Prep. HPLC purification method [00637] Chromatographic separation and isolation was conducted with a Waters 2545 quaternary system with Waters 2489 UV Detector; column Shim-Pack GIST C18 (250mm x 20mm x 5μm); compound was eluted with, Mobile Phase A: 5mM ammonium acetate +0.05% formic acid in water, Mobile Phase B: acetonitrile:methanol:THF(45:45:10) with a gradient of T = 0.01 min (66% A, 34% B); gradient to T = 19.00 min (55% A, 45% B); T = 19.01 min (0% A, 100% B) gradient to T = 22.00 min (0% A, 100% B); T = 22.01 min (66% A, 34% B); gradient to T = 24.00 min (66% A, 34% B); Flow rate= 22ml/min; analysis time 24 min. Example 36: N-(2-(2,4-Dihydroxy-5-methylbenzoyl)-6-(2-(dimethylamino)ethoxy) isoindolin-4-yl)-N-methylacrylamide Step 1: 7-Bromoisoindolin-5-ol hydrochloride

[00638] BBr
3 (1M in DCM) (40 mL) was added dropwise to 4-bromo-6-methoxyisoindoline hydrochloride (CAS 1427400-90-5) (2.0 g, 7.6 mmol, 1 eq.) at room temperature. The resulting mixture was heated to 60˚C and stirred for 16 h. then cooled and concentrated under vacuum. Crude material was diluted with water (50 mL) causing a solid to precipitate. The solid was collected by filtration and dried under vacuum to yield the title compound as an off-white solid (1.35 g, Yield: 72.0%). [00639]
1H NMR (DMSO-d6, 400 MHz): δ 3.94 (s, 2H), 4.24 (s, 2H), 6.27 - 6.31 (m, 1H), 6.42 - 6.46 (m, 1H). [00640] LCMS (Method A): 0.688 min, MS: ES+ 213.9 & 215.9 (M+1 & M+3). Step 2: tert-Butyl 4-bromo-6-hydroxyisoindoline-2-carboxylate
[00641] To a stirred solution of 7-bromoisoindolin-5-ol hydrochloride (Step 1) (1.3 g, 6.1 mmol, 1 eq.) in DCM (26 mL) was added TEA (1.85 g, 18.3 mmol, 3 eq.) and the mixture stirred at room temperature for 15 mins. Boc-anhydride (1.33 g, 6.1 mmol, 1 eq.) was added and stirring continued at room temperature for 2 h. The resulting mixture was diluted with water (50 mL) and extracted with DCM (5 x 30 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under vacuum. Crude material was purified by chromatography on silica (product eluted at 45% EtOAc in hexane) to yield the title compound as an off-white solid (1.15 g, Yield: 55.0%). [00642]
1H NMR (DMSO-d6, 400 MHz): δ 1.45 (s, 9H), 4.39 (d, J= 10.4 Hz, 2H), 4.59 (d, J= 10.4 Hz, 2H), 6.73 (s, 1H), 6.87 (d, J= 1.6 Hz, 1H), 9.91 (s, 1H). [00643] LCMS (Method A): 2.024 min, MS: ES+ 257.8 (M-56). Step 3: 4-Bromo-6-(2-(dimethylamino)ethoxy)isoindoline-2-carboxylate

[00644] To a stirred solution of tert-butyl 4-bromo-6-hydroxyisoindoline-2-carboxylate (Step 2) (0.2 g, 0.63 mmol, 1 eq.) in THF (10 mL) was added KOtBu (0.353 g, 3.15 mmol, 5 eq.) and the mixture stirred at room temperature for 1 h.2-Bromo-N,N-dimethylethan-1-amine hydrobromide (CAS 2862-39-7) (0.447 g, 1.91 mmol, 3 eq.) was added and the reaction mixture heated to 70˚C for 6 h. The cooled mixture was diluted with water (100 mL) and extracted with EtOAc (4 x 30 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under vacuum. The crude material was triturated with diethyl ether (2 x 15 mL) and dried under high vacuum to yield the title compound as a yellow solid (1.05 g, Yield: 63.0%). [00645]
1H NMR (DMSO-d6, 400 MHz): δ 1.45 (s, 9H), 2.19 (s, 6H), 2.59 (d, J= 5.6 Hz, 2H), 4.03 - 4.06 (m, 2H), 4.44 (d, J= 10.4 Hz, 2H), 4.64 (d, J= 6.0 Hz, 2H), 6.96 (d, J= 6.8 Hz, 1H), 7.09 (bs, 1H). [00646] LCMS (Method A): 1.461 min, MS: ES+ 384.9 & 386.9 (M+1 & M+3).
Step 4: tert-Butyl 6-(2-(dimethylamino) ethoxy)-4-(N-methylacrylamido) isoindoline-2-carboxylate
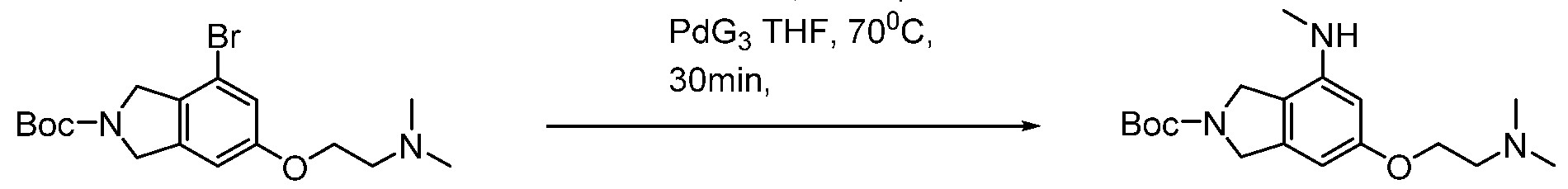
[00647] Performed in 5 parallel batches, each of 0.4 g scale: To a stirred solution of tert-butyl 4- bromo-6-(2-(dimethylamino)ethoxy)isoindoline-2-carboxylate (Step 3) (0.4 g, 1.03 mmol, 1.0 eq.) in THF (4.0 mL) was added NaOtBu (0.19 g, 2.07 mmol, 2.0 eq) at room temperature and purged with argon for 20 mins. Methylamine (2M in THF) (1 mL, 2.07 mmol, 2.0 eq) and t-BuxphosPdG3 (0.049 g, 0.062 mmol, 0.06 eq) were added and the reaction mixture was heated to 70°C temperature for 30 mins. The resulting mixture was cooled to room temperature, diluted with water (50 mL) and extracted with EtOAc (3 x 50 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted at 6% MeOH in DCM) yielding tert-butyl 6-(2- (dimethylamino) ethoxy)-4-(N-methylacrylamido) isoindoline-2-carboxylate as a yellow solid (1.2 g, Yield: 69%). [00648]
1H NMR (DMSO-d6, 400 MHz): δ 1.45 (d, J= 3.6 Hz, 9H), 2.21 (d, J= 8.8 Hz, 6H), 2.58 (t, J= 6 Hz, 2H), 2.66 (t, J= 4.4 Hz, 3H), 3.96 (t, J= 5.2 Hz, 2H), 4.29 (d, J= 10.4 Hz, 2H), 4.44 (d, J= 9.6 Hz, 2H), 5.45 (t, J= 5.2 Hz, 1H, D
2O exchangeable), 5.90 (s, 1H), 6.12 (d, J= 7.2 Hz, 1H). [00649] LCMS (Method N): 1.805 min, MS: ES+ 335.9 (M+1) Step 5: tert-Butyl 6-(2-(dimethylamino) ethoxy)-4-(N-methylacrylamido) isoindoline-2-carboxylate
[00650] To a cooled (0 ˚C) solution of tert-butyl 6-(2-(dimethylamino) ethoxy)-4-(methylamino) isoindoline-2-carboxylate (Step 4) (0.2 g, 0.59 mmol, 1.0 eq.) in DCM (2 mL) was added TEA (0.42 g, 4.17 mmol, 7.0 eq.) and the mixture allowed to stir for 10 mins. Acryloyl chloride (0.27 g, 2.98 mmol, 5.0 eq.) was added and stirring continued at 0°C for 15 mins. The resulting mixture was diluted with cold water (30 mL) and extracted with DCM (3 x 30 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum. The crude material
was triturated with n-pentane (2 x 10 mL) yielding tert-butyl 6-(2-(dimethylamino) ethoxy)-4-(N- methylacrylamido) isoindoline-2-carboxylate as yellow liquid (0.35 g, Yield: Quantitative). The crude material was used without further purification. [00651] LCMS (Method N): 1.644 min, MS ES+: 390.4 (M+1). Step 6: tert-Butyl 6-((dimethylamino ethoxy)-4-(N-methylacrylamido) isoindoline-2-carboxylate 2,2,2-trifluoroacetaldehyde

[00652] To a cooled (0 ˚C) solution of tert-butyl 6-(2-(dimethylamino) ethoxy)-4-(N- methylacrylamido) isoindoline-2-carboxylate (Step 5) (0.3 g, 0.77 mmol, 1.0 eq) in DCM (3 mL) was added TFA (1.5 mL) and the reaction mixture was stirred at room temperature for 30 mins. The resulting mixture was concentrated under reduced pressure and the crude material was triturated with n-pentane (2 x 10 mL) yielding tert-butyl 6-((dimethylamino ethoxy)-4-(N- methylacrylamido) isoindoline-2-carboxylate 2,2,2-trifluoroacetaldehyde as a brown liquid (0.45 g, Yield: quantitative) which was used directly in the next step. [00653] LCMS (Method O): 1.315 min, MS ES+: 290.1 (M+1). Step 7: N-(2-(2,4-Dihydroxy-5-methylbenzoyl)-6-(2-(dimethylamino) ethoxy) isoindolin-4-yl)-N- methylacrylamide

[00654] The title compound was prepared from 2,4-dihydroxy-5-methylbenzoic acid (Intermediate F) and tert-butyl 6-((dimethylaminoethoxy)-4-(N-methylacrylamido) isoindoline-2- carboxylate 2,2,2-trifluoro acetaldehyde (Step 6) analogously to Example 1 Step 1. [00655]
1H NMR (DMSO-d6, 400 MHz): δ 2.17 (s, 3H), 2.20 (s, 6H), 2.59 (t, J= 5.6 Hz, 2H), 3.12 - 3.19 (m, 3H), 4.04 (s, br, 2H), 4.48 (s, 1H), 4.63 (s, br, 1H), 4.79 (s, 2H), 5.59 (d, J= 6 Hz, 1H),
6.00 (d, J= 12 Hz, 1H), 6.19 (s, 1H), 6.38 (s, 1H), 6.85 (s, 1H), 6.99 (d, J= 14.8 Hz, 2H), 9.67 - 10.09 (m, 2H). [00656] LCMS (Method N): 1.564 min, MS: ES+ 439.9(M+1). [00657] HPLC (Method H): 3.49 min. Prep. HPLC purification method: [00658] Chromatographic separation and isolation were conducted with Shimadzu LC20AP purification system with UV detector; column YMC Triart C18 (250mm x 19mm x 5µm), compound was eluted with, Mobile Phase A: 5 MM Ammonium bicarbonate in water + 0.05 % NH
3 in Merck water, Mobile Phase B: Acetonitrile: MeOH: IPA (65: 25: 10) with a gradient of T = 0.01 min (60% A, 40% B); gradient to T = 20.00 min (51% A, 49% B); T = 20.01 min (0% A, 100% B) gradient to T = 22.00 min (0% A, 100% B); T = 22.01 min (60% A, 40% B); gradient to T = 26.00 min (60% A, 40% B); Flow rate= 13 ml/min; analysis time 26 min. Example 37: (E)-4-(Dimethylamino)-N-(2-(6-hydroxy-2,3-dimethyl-1H-indole-5-carbonyl) isoindolin-4-yl)-N-methylbut-2-enamide Step 1: Methyl 6-methoxy-2,3-dimethyl-1H-indole-5-carboxylate
[00659] Performed in 2 parallel batches, each of 10 g scale: Methyl 4-amino-2-methoxybenzoate (CAS: 27492-84-8) (10 g, 55.25 mmol, 1 eq), but-3-yn-2-ol (CAS: 2028-63-9) (3.87 g, 55.21 mmol, 1 eq), Ru
3(CO)
12 (0.17 g, 0.27 mmol, 0.005 eq) and aniline HCl (1.43 g, 11.04 mmol, 0.2 eq) were combined and heated to 120°C for 16 h. The resulting mixture was diluted with 10% MeOH: DCM (1000 mL) and concentrated under reduced pressure. The crude material was purified by column chromatography (product eluted at 40% ethyl acetate in hexane) to yield methyl 6-methoxy-2,3-dimethyl-1H-indole-5-carboxylate as a yellow solid (6.0 g, Yield: 31%). [00660]
1H NMR (DMSO-d6, 400 MHz): δ 2.12 (s, 3H), 2.27 (s, 3H), 3.77 (d, J= 11.2 Hz, 6H), 6.84 (s, 1H), 7.75 (s, 1H), 10.79 (s, 1H). [00661] LCMS (Method N): 2.14 min, MS ES+: 233.9 (M + 1).
Step 2: Methyl 6-hydroxy-2,3-dimethyl-1H-indole-5-carboxylic acid
[00662] The title compound was prepared from methyl 6-methoxy-2,3-dimethyl-1H-indole-5- carboxylate (Step 1) and BBr3 (1M in DCM) analogously to Example 25 Step 7. [00663]
1H NMR (DMSO-d6, 400 MHz): δ 2.11 (s, 3H), 2.24 (s, 3H), 6.64 (s, 1H), 7.84 (s, 1H), 10.72 (s, 1H), 11.21 (s, 1H), 13.19 (s, 1H). [00664] LCMS (Method N): 1.995 min, MS ES+: 203.9 (M-1). Step 3: (E)-4-(Dimethylamino)-N-(2-(6-hydroxy-2,3-dimethyl-1H-indole-5-carbonyl) isoindolin-4- yl)-N-methylbut-2-enamide
[00665] The title compound was prepared from methyl 6-hydroxy-2,3-dimethyl-1H-indole-5- carboxylic acid (Step 2) and (E)-4-(dimethylamino)-N-(isoindolin-4-yl)-N-methylbut-2-enamide hydrochloride (Intermediate S) analogously to Example 1 Step 1. [00666] HT
1H NMR (DMSO-d6, 400 MHz, 348K): δ 2.11 (s, 3H), 2.26 (s, br, 8H), 3.11 - 3.19 (m, 6H), 4.72 (s, br, 2H), 4.89 (s, 2H), 5.94 (s, 1H), 6.63 - 6.67 (m, 1H), 6.75 (s, 1H), 7.19 (d, J= 7.6 Hz, 1H), 7.29 (s, 1H), 7.35 - 7.43 (m, 2H), 9.44 (s, 1H), 10.29 (s, 1H). [00667] LCMS (Method N): 1.775 min, MS ES+: 447.0 (M+1). [00668] HPLC (Method H): 4.21 min Example 38: (E)-4-(Dimethylamino)-N-(2-(6-hydroxy-2-methyl-1H-indole-5-carbonyl) isoindolin-4-yl)-N-methylbut-2-enamide
Step 1: 4-Amino-2-methoxybenzoic acid
[00669] Performed in 2 parallel batches, each of 25 g scale: To a stirred solution of 2-methoxy- 4-nitrobenzoic acid (CAS: 2597-56-0) (25 g, 126.9 mmol, 1 eq) in MeOH: water (500 mL: 150 mL) was added Fe powder (42 g, 761.4 mmol, 6 eq) and NH
4Cl (75.4 g, 1395.9 mmol, 11 eq) and the reaction mixture heated to 80°C and for 2 h. The resulting mixture was cooled, filtered through Celite® and concentrated under reduced pressure. The crude mixture was diluted with water (600 mL), neutralized with saturated KHSO
4 solution (100 mL) and extracted with 10% MeOH in DCM (10 x 300 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under vacuum yielding 4-amino-2-methoxybenzoic acid as a brown solid (20 g, Yield: 48.0%). [00670]
1H NMR (DMSO-d6, 400 MHz): δ 3.72 (s, 3H), 5.89 (s, 2H, D2O exchangeable), 6.13 (dd, J= 1.6, 8.4 Hz, 1H), 6.19 (d, J= 1.6 Hz, 1H), 7.50 (d, J= 8.8 Hz, 1H), 11.33 (bs, 1H). [00671] LCMS (Method A): 1.204 min, MS: ES+ 167.8 (M+1) Step 2: Methyl 4-amino-2-methoxybenzoate
[00672] Performed in 2 parallel batches, each of 10 g scale: To a stirred solution of 4-amino-2- methoxybenzoic acid (Step 1) (10 g, 59.8 mmol, 1 eq.) in MeOH (200 mL) was added dropwise SOCl2 (14.25 g, 119.7 mmol, 2 eq.) at 0°C. The reaction mixture was heated to 65°C and stirred for 2 h. The resulting mixture was cooled to room temperature and concentrated under vacuum. Saturated NaHCO3 solution (500 mL) was added and the mixture extracted with EtOAc (4 x 200 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under vacuum yielding methyl 4-amino-2-methoxybenzoate as a brown solid (18 g, Yield: 83.0%). [00673]
1H NMR (DMSO-d6, 400 MHz): δ 3.64 (s, 3H), 3.70 (s, 3H), 5.94 (s, 2H, D
2O exchangeable), 6.13 (d, J= 8.4 Hz, 1H), 6.19 (s, 1H), 7.49 (d, J= 8.8 Hz, 1H). [00674] LCMS (Method A): 1.563 min, MS: ES+ 219 (M-1).
Step 3: Methyl 4-((1,1-dimethoxypropan-2-yl)amino)-2-methoxybenzoate
[00675] Performed in 2 parallel batches, each of 9 g scale: To a stirred solution of 1,1- dimethoxypropan-2-one (CAS: 6342-56-9) (11.73 g, 99.4 mmol, 2 eq) in acetic acid (270 mL) were added methyl 4-amino-2-methoxybenzoate (Step 2) (9 g, 49.7 mmol, 1 eq) and anhydrous Na
2SO
4 (70.6 g, 497 mmol, 10 eq) and the reaction mixture was stirred at room temperature for 16 h. STAB (31.6 g, 149.1 mmol, 3 eq) was added portionwise and stirring continued at room temperature for 16 h. The resulting mixture was concentrated under reduced pressure to remove acetic acid and the reaction was quenched with saturated NaHCO
3 solution (1000 mL). The mixture was extracted with EtOAc (4 x 200 mL) and the combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum. The crude material purified by chromatography on silica gel (product eluted at 20% ethyl acetate in hexane) yielding methyl 4- ((1,1-dimethoxypropan-2-yl)amino)-2-methoxybenzoate as a brown solid (18 g, Yield: 64.0%). [00676]
1H NMR (DMSO-d6, 400 MHz): δ 1.07 (d, J= 6.8 Hz, 3H), 3.35 (s, 6H), 3.65 (s, 3H), 3.68 - 3.70 (m, 1H), 3.73 (s, 3H), 4.23 (d, J= 4.4 Hz, 1H), 6.21 - 6.27 (m, 3H), 7.53 (d, J= 8.8 Hz, 1H). [00677] LCMS (Method A): 1.948 min, MS: ES+ 283.8 (M+1) Step 4: Methyl 6-methoxy-2-methyl-1H-indole-5-carboxylate
[00678] Performed in 2 parallel batches, each of 9 g scale: To a cooled (0°C) solution of methyl 4-((1,1-dimethoxypropan-2-yl)amino)-2-methoxybenzoate (9 g, 31.8 mmol, 1.0 eq.) in DCM (90 mL) was added dropwise BF
3.OEt
2 (7.68 g, 54.0 mmol, 1.7 eq.) and the mixture heated to 55°C for 4 h. The resulting mixture was diluted using ice cold water (300 mL) and extracted with DCM (4 x 100 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under reduced pressure. The crude material was purified by chromatography on silica (product
eluted at 30% ethyl acetate in hexane) yielding methyl 6-methoxy-2-methyl-1H-indole-5- carboxylate as a brown semi solid (9.0 g, Yield: 64.0%). [00679]
1H NMR (DMSO-d6, 400 MHz): δ 2.34 (s, 3H), 3.74 (s, 3H), 3.78 (s, 3H), 6.11 (s, 1H), 6.87 (s, 1H), 7.79 (s, 1H), 11.01 (s, 1H). [00680] LCMS (Method N): 1.972 min, MS: ES+ 219.9 (M+1). Step 5: Methyl 6-methoxy-2-methyl-1-tosyl-1H-indole-5-carboxylate
[00681] To a solution of methyl 6-methoxy-2-methyl-1H-indole-5-carboxylate (Step 4) (0.8 g, 3.65 mmol, 1.0 eq.) in DMF (10 mL) was added NaH (0.175 g, 7.30 mmol, 2.0 eq.) and the mixture was stirred at room temperature for 30 mins. Tosyl chloride (2.09 g, 10.9 mmol, 3.0 eq.) was added portion wise and stirring continued at room temperature for 4 h. The resulting mixture was diluted with water (50 mL) and extracted with EtOAc (3 x 30 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted at 8% EtOAc in hexane) yielding methyl 6-methoxy-2-methyl-1-tosyl-1H-indole-5-carboxylate (0.5 g, Yield: 37.0%). [00682]
1H NMR (DMSO-d6, 400 MHz): δ 2.33 (s, 3H), 2.55 (d, J= 0.8 Hz, 3H), 3.77 (s, 3H), 3.89 (s, 3H), 6.55 (s, 1H), 7.39 (d, J= 8.0 Hz, 2H), 7.68 (s, 1H), 7.77 - 7.81 (m, 3H). [00683] LCMS (Method N): 2.578 min, MS: ES+ 373.8 (M+1). Step 6: 6-Hydroxy-2-methyl-1-tosyl-1H-indole-5-carboxylic acid
[00684] The title compound was prepared from methyl 6-methoxy-2-methyl-1-tosyl-1H-indole-5- carboxylate (Step 5) and BBr3 (1M in DCM) analogously to Example 25 Step 7.
[00685]
1H NMR (DMSO-d6, 400 MHz): δ 2.32 (s, 3H), 2.51 (s, 3H), 6.39 (s, 1H), 7.17 (s, 1H), 7.38 (d, J = 8.0 Hz, 2H), 7.67 - 7.70 (m, 3H). [00686] LCMS (Method N): 2.402 min, MS: ES+ 345.7 (M+1). Step 7: 6-Hydroxy-2-methyl-1H-indole-5-carboxylic acid
[00687] Performed in 3 parallel batches, each of 0.1 g scale: To a solution of 6-hydroxy-2-methyl- 1-tosyl-1H-indole-5-carboxylic acid (Step 6) (0.1 g, 0.28 mmol, 1.0 eq.) in DMSO: water (8:2, 4 mL) was added KOH (0.325 g, 5.79 mmol, 20 eq.) and the reaction mixture heated to 130
˚C using microwave irradiation for 20 mins. The resulting mixture was diluted with water (20 mL), acidified with sat. solution of KHSO4 and extracted with EtOAc (2 x 20 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum. The crude material was purified by reverse phase chromatography (product eluted at 35% ACN in water) followed by pure fraction distillation yielding 6-hydroxy-2-methyl-1H-indole-5-carboxylic acid (0.070 g, Yield: 36.0%). [00688]
1H NMR (DMSO-d6, 400 MHz): δ 2.31 (s, 3H), 6.10 (s, 1H), 6.68 (s, 1H), 7.90 (s, 1H), 10.95 (s, 1H), 11.15 (bs, 1H), 13.31 (bs, 1H). [00689] LCMS (Method N): 1.893 min, MS: ES+ 191.9 (M+1). Step 8: (E)-4-(Dimethylamino)-N-(2-(6-hydroxy-2-methyl-1H-indole-5-carbonyl) isoindolin-4-yl)- N-methylbut-2-enamide
[00690] The title compound was prepared from 6-hydroxy-2-methyl-1H-indole-5-carboxylic acid (Step 7) and (E)-4-(dimethylamino)-N-(isoindolin-4-yl)-N-methylbut-2-enamide hydrochloride (Intermediate S) analogously to Example 1 Step 1. [00691]
1H NMR (DMSO-d6, 400 MHz): δ 2.01 (s, 6H), 2.32 (s, 3H), 2.85 (d, J= 15.2 Hz, 2H), 3.06 - 3.21 (m, 3H), 4.49 - 4.63 (m, 2H), 4.78 - 4.91 (m, 2H), 5.73 (q, J= 16.0 Hz, 1H), 6.01 (d, J= 8.0 Hz, 1H), 6.58 (s, 1H), 6.67 - 6.78 (m, 2H), 7.21 - 7.44 (m, 3H), 9.60 (s, 1H), 10.73 (s, 1H). [00692] LCMS (Method N): 1.733 min, MS: ES+ 433 (M+1). [00693] HPLC (Method H): 3.991 min Prep. HPLC purification method: [00694] Chromatographic separation and isolation was conducted with Shimadzu LC20AP system with UV detector; Column YMC Triart C18 (250 x 20 mm ID), 5um; compound eluted with: Mobile Phase A: 5 mM ammonium bicarbonate + 0.05% NH3 in water, Mobile Phase B: Acetonitrile: methanol: THF (50:50:10) with a gradient of T = 0.00 min (65% A, 35% B); gradient to T = 35.00 min (65% A, 35% B); T = 35.01 min (0% A, 100% B) gradient to T = 40.00 min (0% A, 100% B); gradient to T = 40.01 min (65% A, 35% B); gradient to T = 45.00 min (65% A, 35% B); Flow rate= 18 ml/min; analysis time 45 min. Example 39 (E)-4-(Dimethylamino)-N-(2-(6-hydroxy-3-methyl-1H-indole-5-carbonyl) isoindolin-4-yl)-N-methylbut-2-enamide Step 1: Methyl 4-(allylamino)-5-bromo-2-methoxybenzoate
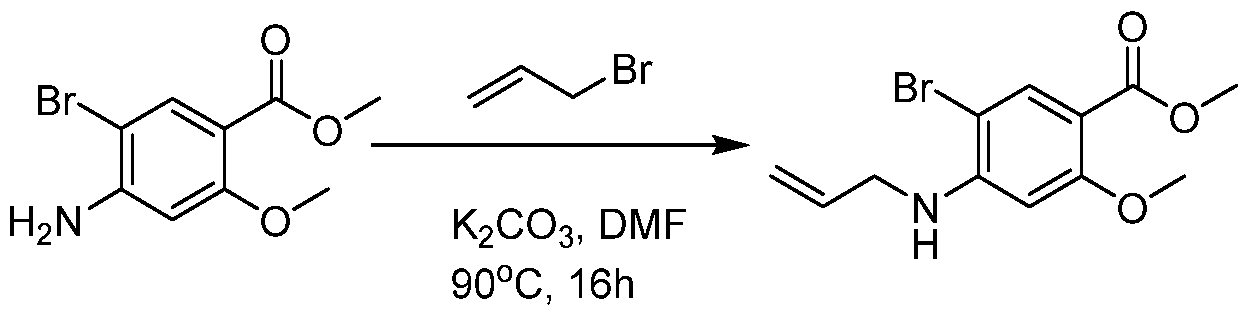
[00695] To a stirred solution of methyl 4-amino-5-bromo-2-methoxybenzoate (Intermediate R Step 2) (8 g, 30.76 mmol, 1.0 eq.) in DMF (80 mL) was added K
2CO
3 (8.5 g, 61.53 mmol, 2.0 eq.) and the mixture was stirred at room temperature for 20 mins. The mixture was cooled to 0°C, treated dropwise with allyl bromide (2.97 g, 24.61 mmol, 0.8 eq.) (CAS: 106-95-6) and heated to 80
˚C for 16 h. The resulting mixture was diluted with cold water (80 mL) and extracted with ethyl acetate (3 x 100 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica
(product eluted at 14% EtOAc in hexane) yielding methyl 4-(allylamino)-5-bromo-2- methoxybenzoate as an off-white solid (4.5 g, Yield: 49%). [00696]
1H NMR (DMSO-d6, 400 MHz): δ 3.69 (s, 3H), 3.76 (s, 3H), 3.94 (d, J= 5.2 Hz, 2H), 5.12 - 5.24 (m, 2H), 5.82 - 5.91 (m, 1H), 6.20 - 6.22 (m, 2H), 7.78 (s, 1H). [00697] LCMS (Method N): 2.272 min, MS: ES+ 301.6 (M+1). Step 2: Methyl 6-methoxy-3-methyl-1H-indole-5-carboxylate
[00698] Performed in 3 parallel batches, each of 0.5 g scale: To a stirred solution of methyl 4- (allylamino)-5-bromo-2-methoxybenzoate (Step 1) (0.5 g, 1.66 mmol, 1.0 eq.) in DMF (0.5 mL) were added K
2CO
3 (0.9 g, 6.66 mmol, 4.0 eq.), PdCl
2(Pcy
3)
2 (0.49 g, 0.66 mmol, 0.4 eq.) and P(OPh)3 (0.2 g, 0.66 mmol, 0.4 eq.) and the reaction mixture was heated to 90°C for 24 h. The resulting mixture was diluted with cold water (50 mL) and extracted with ethyl acetate (3 x 50 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted at 20% EtOAc in hexane) yielding methyl 6-methoxy-3-methyl-1H-indole-5-carboxylate as an off-white solid (0.65 g, Yield: 59%). [00699]
1H NMR (DMSO-d6, 400 MHz): δ 2.23 (s, 3H), 3.77 (s, 3H), 3.80 (s, 3H), 6.93 (s, 1H), 7.06 (s, 1H), 7.87 (s, 1H), 10.83 (s, 1H). [00700] LCMS (Method N): 2.020 min, MS: ES+ 220 (M+1). Step 3: 6-Hydroxy-3-methyl-1H-indole-5-carboxylic acid
[00701] The title compound was prepared from methyl 6-methoxy-3-methyl-1H-indole-5- carboxylate (Step 2) and BBr
3 (1M in DCM) analogously to Example 25 Step 7.The product was used directly in the next step.
[00702] LCMS (Method N): 1.873 min, MS:189.9 (M-1). Step 4: (E)-4-(Dimethylamino)-N-(2-(6-hydroxy-3-methyl-1H-indole-5-carbonyl) isoindolin-4-yl)- N-methylbut-2-enamide
[00703] The title compound was prepared from 6-hydroxy-3-methyl-1H-indole-5-carboxylic acid (Step 3) and (E)-4-(dimethylamino)-N-(isoindolin-4-yl)-N-methylbut-2-enamide hydrochloride (Intermediate S) analogously to Example 1 Step 1. [00704]
1H NMR (DMSO-d6, 400 MHz): δ 2.02 (s, 6H), 2.19 (s, 3H), 2.87 (s, 2H), 3.06 and 3.21 (singlets, 3H), 4.51 - 4.91 (m, 4H), 5.68 - 5.79 (m, 1H), 6.54 - 6.73 (m, 1H), 6.84 (d, J= 34.8 Hz, 1H), 6.95 (s, 1H), 7.21 - 7.45 (m, 4H), 9.59 (s, 1H), 10.53 (s, 1H). [00705] LCMS (Method N): 1.730 min, MS: ES+ 432.9 (M+1). [00706] HPLC (Method H): 3.94 min. Prep. HPLC purification method of analysis: [00707] Chromatographic separation and isolation were conducted with Shimadzu LC20AP purification system with a UV detector; column YMC Triart C18 (250mm x 19mm x 5µm); compound eluted with, Mobile Phase A: 0.05% Ammonium bicarbonate in water, Mobile Phase B: Acetonitrile: MeOH: THF (50: 50: 10) with a gradient of T = 0 min (55% A, 45% B); gradient to T = 28 min (55% A, 45% B); T = 28.01 min (0% A, 100% B) gradient to T = 31 min (0% A, 100% B); T = 31.01 min (55% A, 45% B); gradient to T = 33 min (55% A, 45% B); Flow rate= 7ml/min; analysis time 33 min. Example 40: (E)-4-(Dimethylamino)-N-(2-(4-fluoro-2-hydroxy-5-isopropylbenzoyl) isoindolin-4-yl)-N-methylbut-2-enamide Step 1: Methyl 5-acetyl-4-fluoro-2-hydroxybenzoate

[00708] Performed in 2 parallel batches, each of 3 g scale: To a stirred solution of anhydrous AlCl3 (7.83 g, 58.71 mmol, 3.33 eq.) in DCM (30 mL) was added dropwise acetyl chloride (4.2 mL, 58.71 mmol, 3.33 eq.) at room temperature followed by dropwise addition of methyl 4-fluoro- 2-hydroxybenzoate (3.0 g, 17.63 mmol, 1.0 eq) (CAS: 392-04-1) in DCM (30 mL). The reaction mixture was heated to 60°C and stirred for 16 h. The resulting mixture was cooled to room temperature and acidified with 1N HCl (until precipitation disappeared) and extracted with DCM (3 x 100 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under vacuum. The crude material was purified chromatography on silica (product eluting in 5% EtOAc: hexane) yielding methyl 5-acetyl-4-fluoro-2-hydroxybenzoate as an off-white solid (5.7 g, Yield: 72%). [00709]
1H NMR (DMSO-d6, 400 MHz): δ 2.50 - 2.54 (m, 3H), 3.89 (s, 3H), 6.94 (d, J= 12.8 Hz, 1H), 8.29 (d, J= 8.8 Hz, 1H), 11.30 (s, 1H). [00710] Mass: MS: ES+ 213.2 (M+1) Step 2: Methyl 5-acetyl-2-(benzyloxy)-4-fluorobenzoate
[00711] To a stirred solution of 5-acetyl-4-fluoro-2-hydroxybenzoate (Step 1) (5.7 g, 26.88 mmol, 1 eq.) in DMF (57 mL) were added K
2CO
3 (9.27 g, 67.2 mmol, 2.5 eq) followed by dropwise addition of benzyl bromide (6.9 g, 40.3 mmol, 1.5 eq) at 0°C. The reaction mixture was stirred at room temperature for 16 h. The resulting mixture was poured in ice cold water (150 mL) and extracted with DCM (2 x 100 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted in 10% EtOAc in hexane) yielding methyl 5-acetyl-2-(benzyloxy)-4- fluorobenzoate as an off-white solid (9.0 g, Yield: 70%).
[00712]
1H NMR (DMSO-d6, 400 MHz): δ 2.55 (d, J= 4.8 Hz, 3H), 3.82 (s, 3H), 5.34 (s, 2H), 7.29 - 7.37 (m, 2H), 7.43 (t, J= 7.2 Hz, 2H), 7.50 (d, J= 7.2 Hz, 2H), 8.25 (d, J= 8.8 Hz, 1H). Step 3: Methyl 2-(benzyloxy)-4-fluoro-5-(prop-1-en-2-yl) benzoate
[00713] To a cooled (0°C) solution of methyl triphenyl phosphonium bromide (21.27 g, 59.54 mmol, 2.0 eq.) in THF (90 mL) was added dropwise n-BuLi (2.5 M in hexane) (23.81 mL, 59.54 mmol, 2.0 eq) and the mixture was stirred for 1 h. Methyl 5-acetyl-2-(benzyloxy)-4-fluorobenzoate (Step 2) (9.0 g, 29.77 mmol, 1.0 eq) in THF (90 mL) was added dropwise to the reaction mixture at room temperature and stirring continued for 16 h. The resulting mixture was diluted with brine solution (150 mL) and extracted with DCM (3 x 100 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted in 4% EtOAc in hexane) yielding methyl 2-(benzyloxy)- 4-fluoro-5-(prop-1-en-2-yl) benzoate as a colourless liquid (1.2 g, Yield: 17.0%). [00714]
1H NMR (DMSO-d6, 400 MHz): δ 2.08 (s, 3H), 3.80 (s, 3H), 5.22 - 5.29 (m, 4H), 7.18 (d, J= 13.6 Hz, 1H), 7.33 (t, J = 7.2 Hz, 1H), 7.42 (t J = 7.2 Hz, 2H), 7.49 (d, J = 7.6 Hz, 2H), 7.74 (d, J= 8.8 Hz, 1H). [00715] LCMS (Method N): 2.815 min, MS: ES+ 300.8 (M+1) Step 4: Methyl 4-fluoro-2-hydroxy-5-isopropylbenzoate
[00716] To a stirred solution of methyl 2-(benzyloxy)-4-fluoro-5-(prop-1-en-2-yl) benzoate (Step 3) (1.15 g, 38.29 mmol, 1.0 eq.) in EtOH (23 mL) was added 10% Pd/C (0.575 g, 50% w/w) and the reaction mixture was stirred at room temperature under an atmosphere of H
2(g) for 16 h. The resulting mixture was filtered through Celite® and washed with 10% MeOH: DCM (250 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum yielding methyl 4-fluoro-2-hydroxy-5-isopropylbenzoate as an off-white solid (0.8 g, Yield: 98%). [00717]
1H NMR (DMSO-d6, 400 MHz): δ 1.19 (d, J= 6.8 Hz, 6H), 3.08 (m, 1H), 3.88 (s, 3H), 6.81 (d, J= 12 Hz, 1H), 7.69 (t, J= 8.8 Hz, 1H), 10.62 (s, 1H). Step 5: 4-Fluoro-2-hydroxy-5-isopropylbenzoic acid
[00718] The title compound was prepared from methyl 4-fluoro-2-hydroxy-5-isopropylbenzoate (Step 4) and KOH analogously to Example 38 Step 7. [00719]
1H NMR (DMSO-d6, 400 MHz): δ 1.19 (d, J= 6.8 Hz, 6H), 3.03 - 3.10 (m, 1H), 6.77 (d, J= 12 Hz, 1H), 7.71 (d, J= 8.8 Hz, 1H), 11.52 (bs, 1H). [00720] LCMS (Method N): 2.268 min, MS: ES+ 196.9 (M-1) Step 6: (E)-4-(Dimethylamino)-N-(2-(4-fluoro-2-hydroxy-5-isopropylbenzoyl) isoindolin-4-yl)-N- methylbut-2-enamide
[00721] The title compound was prepared from 4-fluoro-2-hydroxy-5-isopropylbenzoic acid (Step 5) and (E)-4-(dimethylamino)-N-(isoindolin-4-yl)-N-methylbut-2-enamide hydrochloride (Intermediate S) analogously to Example 1 Step 1. [00722]
1H NMR (DMSO-d6, 400 MHz): δ 1.18 (d, J= 6.8 Hz, 6H), 2.01 (s, 6H), 2.81 - 2.87 (m, 2H), 3.03 - 3.08 (m, 1H), 3.19 (s, 3H), 4.46 - 4.87 (m, 4H), 5.74 (t, J= 16 Hz, 1H), 6.57 - 6.72 (m, 2H), 7.13 - 7.23 (m, 2H), 7.33 - 7.45 (m, 2H), 10.40 (bs, 1H). [00723] LCMS (Method N): 1.872 min, MS: ES+ 440.0 (M+1).
[00724] HPLC (Method I): 6.86 min. Prep. HPLC purification method of analysis: [00725] Chromatographic separation and isolation were conducted with Shimadzu LC20AP with UV detector; column YMC Triart C18 (250 x 20 mm ID), 5um; compound eluted with: Mobile Phase A: 5 mM ammonium bicarbonate + 0.05% NH
3 in water, Mobile Phase B: Acetonitrile: methanol (50:50) with a gradient of T = 0.00 min (40% A, 60% B); gradient to T = 30.00 min (40% A, 60% B); T = 30.01 min (0% A, 100% B) gradient to T = 32.00 min (0% A, 100% B); gradient to T = 32.01 min (40% A, 60% B); gradient to T = 35.00 min (40% A, 60% B); Flow rate= 12ml/min; analysis time 35 min. Example 41: (E)-4-(Dimethylamino)-N-(2-(2-hydroxy-4-methoxy-5-methylbenzoyl) isoindolin-4-yl)-N-methylbut-2-enamide Step 1: 1-Bromo-2,4-dimethoxy-5-methylbenzene
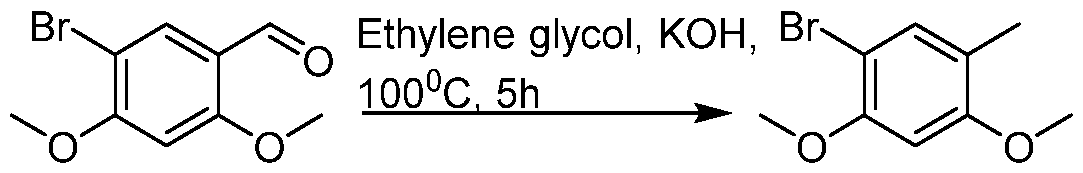
[00726] To a stirred solution of 1-bromo-2,4-dimethoxy-5-methylbenzene (CAS: 130333-46-9) (4.5 g, 18.36 mmol, 1 eq.) in ethylene glycol (10.5 mL, 187.27 mmol, 10.2 eq) was added dropwise hydrazine hydrate (2.67 g, 83.54 mmol, 4.55 eq.) followed by portion wise addition of KOH (4.12 g, 73.44 mmol, 4.0 eq) and the reaction mixture was heated to 100°C for 5 h. The cooled mixture was poured into ice cold water (100 mL), acidified with 2N HCl (20 mL) and extracted with DCM (3 x 100 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluting in 5% EtOAc: hexane) yielding 1-bromo-2,4-dimethoxy-5-methylbenzene as a light yellow solid (2.0 g, Yield: 47%). [00727]
1H NMR (DMSO-d6, 400 MHz): δ 2.05 (s, 3H), 3.84 (d, J= 8.8 Hz, 6H), 6.70 (s, 1H), 7.29 (s, 1H). [00728] Mass: m/z = 231.2 (M+1) Step 2: Methyl 2,4-dimethoxy-5-methylbenzoate
[00729] To a cooled (-78°C) solution of 1-bromo-2,4-dimethoxy-5-methylbenzene (Step 1) (1.4 g, 6.05 mmol, 1.0 eq.) in THF (14 mL) was added dropwise n-BuLi (2.5M in hexane) (4.84 mL, 12.11 mmol, 2.0 eq) and the mixture stirred at -78°C for 30 mins. Methyl chloroformate (2.8 mL) was added dropwise at -78°C and stirring continued for 30 mins. The resulting mixture was poured into ice cold water (50 mL) and extracted with ethyl acetate (3 x 50 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted in 15% EtOAc in hexane) yielding methyl 2,4-dimethoxy-5-methylbenzoate as a white solid (0.4 g, Yield: 33 %). [00730]
1H NMR (DMSO-d6, 400 MHz): δ 2.07 (s, 3H), 3.34 (s, 3H), 3.84 (s, 3H), 3.89 (s, 3H), 6.66 (s, 1H), 7.52 (s, 1H). Step 3: 2-Hydroxy-4-methoxy-5-methylbenzoate
[00731] To a cooled (0°C) solution of methyl 2,4-dimethoxy-5-methylbenzoate (Step 2) (0.4 g, 1.90 mmol, 1.0 eq.) in DCM (4 mL) was added BCl3 (7.61 mL, 7.61 mmol, 4.0 eq) and the reaction mixture was stirred for 2 h. The resulting mixture was diluted with ice cold water (30 mL) and extracted with DCM (3 x 30 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted in 5% EtOAc in hexane) yielding methyl 2-hydroxy-4-methoxy-5-methylbenzoate as a white solid (0.3 g, Yield: 80 %). [00732]
1H NMR (DMSO-d6, 400 MHz): δ 2.06 (s, 3H), 3.83 (s, 3H), 3.86 (s, 3H), 6.53 (s, 1H), 7.54 (s, 1H), 10.68 (s, 1H). [00733] LCMS (Method B): 2.07 min, MS: ES+ 181.0 (M-1) Step 4: 2-Hydroxy-4-methoxy-5-methylbenzoic acid
[00734] To a stirred solution of methyl 2,4-dimethoxy-5-methylbenzoate (Step 3) (0.3 g, 1.52 mmol, 1.0 eq.) in THF: water (3 mL) was added LiOH.H
2O (0.288 g, 6.88 mmol, 4.5 eq) at room temperature. The resulting mixture was heated to 70°C and stirred for 16 h. The resulting mixture was poured into ice cold water (30 mL), acidified with dilute HCl (5 mL) (pH ~5-6) and extracted with ethyl acetate (3 x 20 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under high vacuum yielding 2-hydroxy-4-methoxy-5-methylbenzoic acid as a white solid (0.26 g, Yield: 93%). [00735]
1H NMR (DMSO-d6, 400 MHz): δ 2.06 (s, 3H), 3.82 (s, 3H), 6.49 (s, 1H), 7.52 (s, 1H), 11.59 (bs, 1H), 13.44 (bs, 1H). [00736] LCMS (Method N): 1.938 min, MS: ES+ 181.0 (M-1) Step 5: (E)-4-(Dimethylamino)-N-(2-(2-hydroxy-4-methoxy-5-methylbenzoyl)isoindolin-4-yl)-N- methylbut-2-enamide
[00737] The title compound was prepared from 2-hydroxy-4-methoxy-5-methylbenzoic acid (Step 4) and (E)-4-(dimethylamino)-N-(isoindolin-4-yl)-N-methylbut-2-enamide hydrochloride (Intermediate S) analogously to Example 1 Step 1. [00738]
1H NMR (DMSO-d6, 400 MHz): δ 2.01 - 2.06 (m, 6H), 2.85 (s, 2H), 3.09 - 3.24 (2 singlets, 3H), 3.75 (s, 3H), 4.56 - 4.85 (m, 4H), 5.74 (t, J= 15.6 Hz, 1H), 6.44 - 6.48 (m, 1H), 6.62 - 6.70 (m, 1H), 7.03 (d, J= 36 Hz, 1H), 7.22 (d, J= 6.4 Hz, 1H), 7.35 - 7.43 (m, 2H), 9.78 (bs, 1H). [00739] LCMS (Method N): 1.66 min, MS: ES+ 424.4 (M+1). [00740] HPLC (Method H): 4.06 min. Prep. HPLC purification method of analysis: [00741] Chromatographic separation and isolation were conducted with Waters 2545 binary system with Waters 2489 UV Detector. Column Waters X-Bridge (250mm x 19mm 5μm); compound was eluted with, Mobile Phase A : 5 mM ammonium bicarbonate in Merck water +
0.05% NH
3, Mobile Phase B : acetonitrile (500mL) + methanol (500mL) + THF (100mL) with a gradient of T = 0.00 min (90% A, 10% B); gradient to T = 2.00 min (57% A, 43% B); T = 22.00 min (57% A, 43% B) gradient to T = 22.01 min (0% A, 100% B); gradient to T = 25.00 min (0% A, 100% B); gradient to T = 25.01 min (90% A, 10% B); gradient to T = 28.00 min (90% A, 10% B); Flow rate= 16ml/min; analysis time 28 min Example 42: (E)-N-(2-(3-(tert-Butyl)-2-fluoro-6-hydroxy-5- (hydroxymethyl)benzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-methylbut-2-enamide Step 1A: 2-Bromo-4-(tert-butyl)-5-fluorophenol

[00742] To a stirred solution of 2-bromo-5-fluorophenol (10 g, 52.30 mmol, 1 eq.) (CAS:147460- 41-1), and anhydrous AlCl3 (2.79 g, 20.90 mmol, 0.4 eq.) was added tert-butyl chloride (4.89 g, 52.30 mmol, 1 eq.) and the reaction mixture was stirred at room temperature for 2 h. The resulting mixture was diluted with water (200 mL) and extracted with EtOAc (3 x 250 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under vacuum. Crude material was purified by chromatography on silica (product eluted at 1% EtOAc in hexane) yielding 2-bromo-4-(tert-butyl)-5-fluorophenol as a yellow liquid (16 g, Yield: 61.9%). [00743]
1H NMR (DMSO-d6, 400 MHz): δ ppm 1.28 (s, 9H), 6.68 - 5.77 (m, 1H), 7.30 (d, J= 8.8 Hz, 1H), 10.51 (bs, 1H). [00744] LCMS (Method A): 2.47 min, MS: ES- 245, 247 (M, M+2). Step 1B: 1-Bromo-5-(tert-butyl)-4-fluoro-2-(methoxymethoxy)benzene
[00745] To a cooled (0 ˚C) solution of 2-bromo-4-(tert-butyl)-5-fluorophenol (Step 1A) (10 g, 40.46 mmol, 1 eq.) in DCM (100 mL) was added DIPEA (26.10 g, 202.33 mmol, 5 eq.) and the mixture was stirred for 15 mins. MOM-Cl (9.77 g, 121.40 mmol, 3 eq.) was added dropwise at 0
˚C and the reaction mixture was stirred at room temperature for 2 h. The resulting mixture was diluted with water (100 mL) and extracted with DCM (3 x 150 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under vacuum. Crude material was purified by chromatography on silica gel (product eluted at 1% EtOAc in hexane) yielding 1-bromo-5-(tert- butyl)-4-fluoro-2-(methoxymethoxy) benzene as an off-white solid (22 g, Yield: 76.0%). [00746]
1H NMR (DMSO-d6, 400 MHz): δ ppm 1.30 (s, 9H), 3.40 (d, J= 5.2 Hz, 3H), 5.29 (s, 2H), 7.08 (d, J= 14 Hz, 1H), 7.43 (d, J= 8.8 Hz, 1H). Step 1C: 3-(tert-Butyl)-2-fluoro-6-(methoxymethoxy)benzoic acid
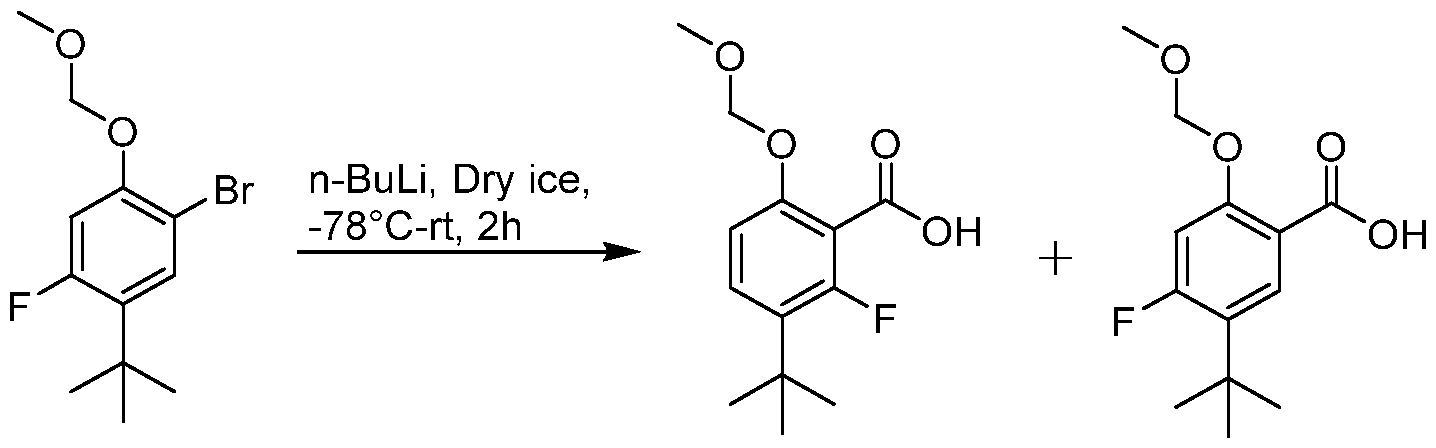
[00747] To a cooled (-78°C) solution of 1-bromo-5-(tert-butyl)-4-fluoro-2- (methoxymethoxy)benzene (Step 1B) (11 g, 37.80 mmol, 1.0 eq.) in THF (110 mL), n- BuLi was added dropwise (1.6M in hexane) (47.25 mL, 2 eq.) and the mixture was stirred for 30 mins. Dry ice (100 g) was added portion wise at -78°C and the reaction mixture was allowed to warm to room temperature stirring for 2 h. The resulting mixture was poured into sat. solution of ammonium chloride (200 mL) and extracted with EtOAc (3 x 2000 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude material was triturated with n-pentane (2 x 30 mL) to yield a mixture of 3-(tert-butyl)-2-fluoro-6- (methoxymethoxy)benzoic acid and 3-(tert-butyl)-4-fluoro-6-(methoxymethoxy)benzoic acid as a yellow liquid (20 g, Yield: 51.6%). This material was used directly in the next step. Step 1D: 3-(tert-Butyl)-2-fluoro-6-hydroxybenzoic acid

[00748] To a cooled (0°C) solution of 3-(tert-butyl)-2-fluoro-6-(methoxymethoxy)benzoic acid and 3-(tert-butyl)-4-fluoro-6-(methoxymethoxy)benzoic acid (Step 1C) (10 g, 39.02 mmol, 1 eq.)
in EtOH (100 mL) was added dropwise 4M HCl in dioxane (20 mL) and the reaction mixture was stirred at room temperature for 2 h. The resulting mixture was concentrated under vacuum and the crude material was purified by trituration with n-pentane yielding a mixture of 3-(tert-butyl)-2- fluoro-6-hydroxybenzoic acid and 3-(tert-butyl)-4-fluoro-6-hydroxybenzoic acid as an off-white gummy solid (15 g, Yield: 46.7 %). This material was used directly in the next step. [00749] LCMS (Method A): 1.888 min, 2.048 min, MS: ES- 211 (M-1). Step 1: Methyl 3-(tert-butyl)-2-fluoro-6-hydroxybenzoate
[00750] To a cooled (0°C) solution of 3-(tert-butyl)-2-fluoro-6-hydroxybenzoic acid and 3-(tert- butyl)-4-fluoro-6-hydroxybenzoic acid (Step 1D) (4 g, 18.86 mmol, 1.0 eq) in MeOH (40 mL) was added dropwise conc. H
2SO
4 (1.2 mL) and the reaction mixture was heated to 70°C for 16 h. The resulting mixture was cooled to room temperature and concentrated under vacuum. The crude material was purified by chromatography on silica (product was eluted at 5% EtOAc in hexane) yielding methyl 3-(tert-butyl)-2-fluoro-6-hydroxybenzoate as a yellow liquid (2.9 g, Yield: 68%) with methyl 3-(tert-butyl)-4-fluoro-6-hydroxybenzoate as a minor impurity. [00751]
1H NMR (DMSO-d6, 400 MHz): δ 1.28 (s, 9H), 3.81 (s, 3H), 6.67 (d, J= 8.8 Hz, 1H), 7.24 (t, J= 9.2 Hz, J= 18.8 Hz, 1H), 10.29 (s, 1H). [00752] LCMS (Method N): 2.67 min, MS: ES+ 226.9 (M+1). Step 2: Methyl 3-bromo-5-(tert-butyl)-6-fluoro-2-hydroxybenzoate
[00753] To a cooled (0 ˚C) solution of methyl 3-(tert-butyl)-2-fluoro-6-hydroxybenzoate (Step 1) (2.2 g, 9.71 mmol, 1.0 eq.) in CHCl3 (22 mL) was added dropwise a solution of Br2 in CHCl3 (0.5 mL, 9.71 mmol, 1.0 eq.) and the reaction mixture was stirred at room temperature for 30 mins. The resulting mixture was poured into saturated solution of NH
4Cl (70 mL) and extracted with ethyl acetate (3 x 50 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and
concentrated under reduced pressure. The crude material was purified by chromatography on silica (product was eluted in 1% EtOAc in hexane) yielding methyl 3-bromo-5-(tert-butyl)-6-fluoro- 2-hydroxybenzoate as a yellow liquid (2.2 g, Yield: 74%). [00754]
1H NMR (DMSO-d6, 400 MHz): δ 1.29 (s, 9H), 3.87 (s, 3H), 7.53 (d, J= 8.4 Hz, 1H), 10.61 (s, 1H). Step 3: Methyl 3-(tert-butyl)-2-fluoro-6-hydroxy-5-vinylbenzoate
[00755] To a stirred solution of methyl 3-bromo-5-(tert-butyl)-6-fluoro-2-hydroxybenzoate (Step 2) (2.2 g, 7.20 mmol, 1.0 eq.) and potassium trifluoro(vinyl)borate (1.44 g, 10.74 mmol, 1.5 eq.) in THF: water (44 mL: 10mL) were added Na
2CO
3 (5.34 g, 50.37 mmol, 7 eq.) and PdCl
2(PPh
3) (0.25 g, 0.356 mmol, 0.05 eq) at room temperature. The reaction mixture was heated to 70°C and stirred for 16 h. The resulting mixture was diluted with water (40 mL) and extracted with EtOAc (2 x 30 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted at 1% EtOAc in hexane) yielding methyl 3-(tert-butyl)-2-fluoro-6-hydroxy-5-vinylbenzoate as a white solid (1.0 g, Yield: 55 %). [00756]
1H NMR (DMSO-d6, 400 MHz): δ 1.32 (s, 9H), 3.88 (s, 3H), 5.28 (d, J= 11.6 Hz, 1H), 5.80 (d, J= 17.6 Hz, 1H), 6.89 - 6.96 (m, 1H), 7.53 (d, J= 9.2 Hz, 1H), 10.5 (s, 1H). Step 4: Methyl 3-(tert-butyl)-2-fluoro-5-formyl-6-hydroxybenzoate
[00757] To a stirred solution of potassium osmate (K
2OSO
4) (0.011 g, 0.031 mmol, 0.01 eq.) in water (4 mL) was slowly dropwise added solution of methyl 3-(tert-butyl)-2-fluoro-6-hydroxy-5- vinylbenzoate (Step 3) (0.8 g, 3.17 mmol, 1.0 eq.) in 1,4-dioxane (8 mL) and the mixture was stirred for 10 mins. NaIO
4 (1.69 g, 7.93 mmol, 2.5 eq.) was added portion wise and stirring continued at room temperature for 2 h. The resulting mixture was diluted with water (30 mL) and
extracted with EtOAc (2 x 30 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under vacuum yielding methyl 3-(tert-butyl)-2-fluoro-5-formyl-6-hydroxybenzoate as a yellow oil (0.8 g, Yield: 99%). [00758]
1H NMR (DMSO-d6, 400 MHz): δ 1.34 (s, 9H), 3.88 (s, 3H), 7.86 (d, J= 9.2 Hz, 1H), 10.13 (s, 1H), 11.36 (s, 1H). [00759] LCMS (Method N): 2.53 min, MS: ES+ 255.2 (M+1). Step 5: Methyl 3-(tert-butyl)-2-fluoro-6-hydroxy-5-(hydroxymethyl)benzoate
[00760] To a stirred solution methyl 3-(tert-butyl)-2-fluoro-5-formyl-6-hydroxybenzoate (Step 4) (0.6 g, 2.36 mmol, 1.0 eq.) in EtOH (18 mL) was added portion wise NaBH
4 (0.104 g, 2.83 mmol, 1.2 eq.) and the reaction mixture was stirred at room temperature for 2 h. The resulting mixture was acidified with 2N HCl (15 mL) and extracted with EtOAc (2 x 25 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted at 100% DCM) yielding methyl 3-(tert-butyl)- 2-fluoro-6-hydroxy-5-(hydroxymethyl)benzoate as a white solid (0.3 g, Yield: 49%). [00761]
1H NMR (DMSO-d6, 400 MHz): δ 1.31 (s, 9H), 3.87 (s, 3H), 4.49 (s, 2H), 5.26 (s, br,1H, D2O exchangeable), 7.46 (d, J= 9.6 Hz, 1H), 10.20 (s, 1H). Step 6: 3-(tert-Butyl)-2-fluoro-6-hydroxy-5-(hydroxymethyl)benzoic acid
[00762] To a stirred solution of methyl 3-(tert-butyl)-2-fluoro-6-hydroxy-5- (hydroxymethyl)benzoate (Step 5) (0.3 g, 1.17 mmol, 1.0 eq.) in MeOH:THF:H
2O (7 : 3 : 1) (3.3 mL) was added LiOH.H
2O (0.49 g, 11.71 mmol, 10 eq.) and the reaction mixture was heated to 70°C and stirred for 4 h. The resulting mixture was concentrated to remove the volatile solvents, acidified with dilute HCl (20 mL) and extracted with ethyl acetate (2 x 30 mL). The combined organic extracts were dried over Na
2SO
4, filtered and concentrated under vacuum yielding 3-
(tert-butyl)-2-fluoro-6-hydroxy-5-(hydroxymethyl)benzoic acid as a white solid (0.25 g, Yield: 88%). [00763]
1H NMR (DMSO-d6, 400 MHz): δ 1.32 (s, 9H), 4.48 (s, 2H), 5.12 (s, 1H, D
2O exchangeable), 7.51 (d, J= 8.8 Hz, 1H), 11.85 (bs,1H). Step 7: (E)-N-(2-(3-(tert-Butyl)-2-fluoro-6-hydroxy-5-(hydroxymethyl)benzoyl)isoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide
[00764] The title compound was prepared from 3-(tert-butyl)-2-fluoro-6-hydroxy-5- (hydroxymethyl)benzoic acid (Step 6) and (E)-4-(dimethylamino)-N-(isoindolin-4-yl)-N- methylbut-2-enamide hydrochloride (Intermediate S) analogously to Example 1 Step 1. [00765]
1H NMR (DMSO-d6, 400 MHz): δ 1.23 - 1.32 (m, 9H), 2.00 - 2.06 (2 singlets, 6H), 2.93 - 2.94 (m, 4H), 3.07 and 3.21 (2 singlets, 3H), 4.45 - 4.59 (m, 4H), 4.97 - 4.89 (m, 1H), 5.69 - 5.81 (m, 1H), 6.53 - 6.75 (m, 1H), 7.24 - 7.47 (m, 4H), 9.30 (bs, 1H). [00766] LCMS (Method N): 1.83 min, MS: ES+ 483.9 (M+1). [00767] HPLC (Method H): 4.34 min. Example 43 N-(2-(3-(Aminomethyl)-5-(tert-butyl)-6-fluoro-2-hydroxybenzoyl)isoindolin- 4-yl)-N-methylacrylamide Step 1: Methyl 3-(aminomethyl)-5-(tert-butyl)-6-fluoro-2-hydroxybenzoate
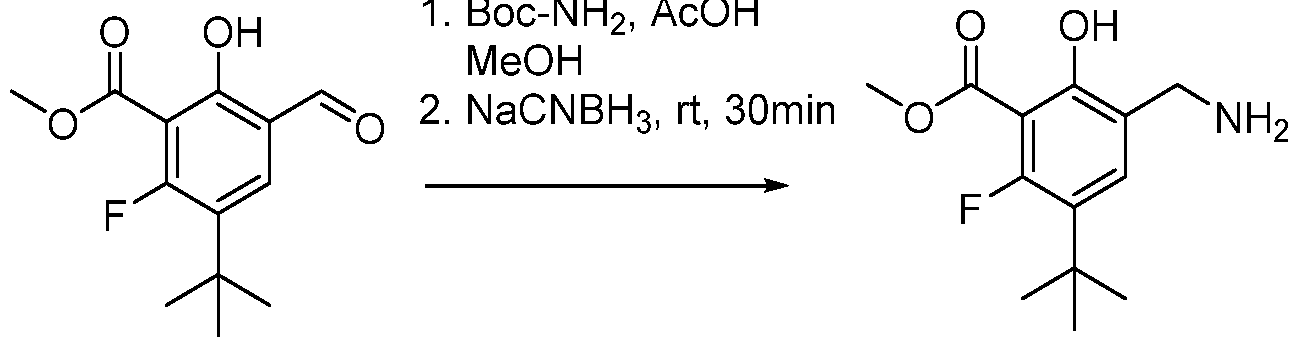
[00768] To a stirred solution of methyl 3-(tert-butyl)-2-fluoro-5-formyl-6-hydroxybenzoate (Example 42 Step 4) (0.7 g, 2.73 mmol, 1 eq) and tert-butyl carbamate (CAS: 4248-19-5) (0.16 g, 1.37 mmol, 0.5 eq) in ethylene dichloride (7.0 mL) was added acetic acid (0.99 g, 16.51 mmol, 6.0 eq.) at room temperature and the mixture was stirred for 30 mins. STAB (1.75 g, 8.25 mmol,
3.0 eq) was added portion wise and stirring continued at room temperature for 30 mins. The resulting mixture was diluted with water (20 mL) and extracted with ethyl acetate (2 x 25 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted at 9% EtOAc: hexane) yielding methyl 3-(aminomethyl)-5-(tert-butyl)-6-fluoro-2-hydroxybenzoate (0.7 g, Yield: 99%). [00769]
1H NMR (DMSO-d6, 400 MHz): δ 1.31 (s, 9H), 3.87 (s, 3H), 4.49 (s, 2H), 5.19 (bs, 1H, D
2O exchangeable), 6.15 (bs, 1H), 7.46 (d, J= 9.2 Hz, 1H), 10.17 (s, 1H). [00770] LCMS (Method N): 2.25 min, MS: ES+ 255.8 (M+1). Step 2: 3-(Aminomethyl)-5-(tert-butyl)-6-fluoro-2-hydroxybenzoic acid
[00771] To a stirred solution of methyl 3-(aminomethyl)-5-(tert-butyl)-6-fluoro-2- hydroxybenzoate (Step 1) (0.7 g, 1.96 mmol, 1.0 eq.) in MeOH (7 mL) was added KOH (1.1 g, 19.69 mmol, 10 eq.) dissolved in water (3.5 mL) at room temperature. The reaction mixture was heated to 70°C and stirred for 3 h. The resulting mixture was acidified with dilute HCl (25 mL) and extracted with ethyl acetate (2 x 30 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum yielding 3-(aminomethyl)-5-(tert-butyl)-6- fluoro-2-hydroxybenzoic acid as an off-white solid (0.45 g, Yield: 68%). [00772]
1H NMR (DMSO-d6, 400 MHz): δ 1.32 (s, 9H), 4.48 (s, 2H), 5.13 (bs, 1H, D
2O exchangeable), 7.51 (d, J= 9.2 Hz, 1H), 12.36 (bs, 2H). Step 3: N-(2-(3-(Aminomethyl)-5-(tert-butyl)-6-fluoro-2-hydroxybenzoyl)isoindolin-4-yl)-N- methylacrylamide
[00773] The title compound was prepared from 3-(aminomethyl)-5-(tert-butyl)-6-fluoro-2- hydroxybenzoic acid (Step 2) and (N-(isoindolin-4-yl)-N-methyl acrylamide (Intermediate W2) analogously to Example 1 Step 1. [00774]
1H NMR (DMSO-d6, 400 MHz): δ 1.23 - 1.32 (m, 9H), 3.09 - 3.23 (2 singlets, 3H), 4.31 - 4.60 (m, 5H), 4.80 - 4.90 (m, 2H), 5.60 - 5.63 (m, 1H), 5.87 - 6.25 (m, 2H), 7.25 - 7.47 (m, 4H), 8.73 (bs, 1H). [00775] LCMS (Method N): 2.05 min, MS: ES+ 426.8 (M+1). [00776] HPLC (Method H): 6.66 min. Prep. HPLC purification method of analysis: [00777] Chromatographic separation and isolation were conducted using a Waters 2545 binary pump with waters 2489 UV detector with waters Acquity QDA detector; Column YMC Triart C18 (250 x 20 mm ID, 5µm); compound eluted with, Mobile Phase A: 5mM ammonium bicarbonate+0.05% NH3 in Merck water, Mobile Phase B: acetonitrile with a gradient of T = 0.00 min (68% A, 32% B); gradient to T = 18.00 min (45% A, 55% B); T = 18.01 min (0% A, 100% B) gradient to T = 20.00 min (0% A, 100% B); T = 20.01 min (68% A, 32% B); gradient to T = 24.00 min (68% A, 32% B); Flow rate= 13 ml/min; analysis time 24 min. Example 44: (E)-N-(2-(5-(tert-Butyl)-2-hydroxy-4-methoxybenzoyl)-5-chloroisoindolin-4- yl)-4-(dimethylamino)-N-methylbut-2-enamide Step 1: tert-Butyl (E)-5-chloro-4-(4-(dimethylamino)-N-methylbut-2-enamido)isoindoline-2- carboxylate

[00778] The title compound was prepared from (E)-4-(dimethylamino) but-2-enoic acid (CAS: 848133-35-7) and tert-butyl 5-chloro-4-(methylamino)isoindoline-2-carboxylate (Example 24, Step 1) analogously to Example 24, Step 4. [00779]
1H NMR (DMSO-d6, 400 MHz): δ 1.45 (m, 9H), 2.24 (s, 6H), 3.12 (s, 3H), 3.17 - 3.19 (m, 2H), 4.36 - 4.66 (m, 4H), 5.77 - 5.83 (m, 1H), 6.67 - 6.74 (m, 1H), 7.43 - 7.46 (m, 1H), 7.59 - 7.61 (d, J= 8 Hz, 1H).
[00780] LCMS (Method N): 1.849 min, MS ES+: 394 (M+1). Step 2: (E)-N-(5-Chloroisoindolin-4-yl)-4-(dimethylamino)-N-methylbut-2-enamide hydrochloride
[00781] To a cooled (0°C) solution of tert-butyl (E)-5-chloro-4-(4-(dimethylamino)-N-methylbut- 2-enamido)isoindoline-2-carboxylate (0.4 g, 1.01 mmol, 1 eq) in DCM (4 mL) was added dropwise 4M HCl in dioxane (4 mL) and the reaction mixture was stirred at room temperature for 1 h. The resulting mixture was concentrated under reduced pressure and the crude material was triturated with diethyl ether (2 x 20 mL) followed by high vacuum drying to yield (E)-N-(5- chloroisoindolin-4-yl)-4-(dimethylamino)-N-methylbut-2-enamide hydrochloride (0.3 g, Yield: 89%). [00782]
1H NMR (DMSO-d6, 400 MHz): δ 2.63 (bs, 6H), 3.14 (s, 3H), 3.74 - 3.79 (m, 2H), 4.39 - 4.42 (d, J= 14 Hz, 3H), 4.57 (bs, 3H), 6.05 (d, J= 15.2 Hz, 1H), 6.71 - 6.78 (m, 1H), 7.51 (d, J= 8 Hz, 1H), 7.67 (d, J= 8.0 Hz, 1H), 10.06 (bs, 1H), 10.44 (bs, 1H), 10.65 (bs, 1H). [00783] LCMS (Method O): 1.449 min, MS ES+: 294.12 (M+1). Step 3: (E)-N-(2-(5-(tert-Butyl)-2-hydroxy-4-methoxybenzoyl)-5-chloroisoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide
[00784] The title compound was prepared from 5-(tert-butyl)-2-hydroxy-4-methoxybenzoic acid (Intermediate N) and (E)-N-(5-chloroisoindolin-4-yl)-4-(dimethylamino)-N-methylbut-2-enamide hydrochloride (Step 2) analogously to Example 1 Step 1. [00785]
1H NMR (DMSO-d6, 400 MHz): δ 1.21 (bs, 9H), 2.11 (bs, 6H), 2.89 (s, 2H), 3.05 and 3.15 (2 singlets, 3H), 3.85 (s, 3H), 4.59 - 4.63 (d, J= 16 Hz, 1H), 4.80 - 4.89 (m, 3H), 5.71 - 5.75 (m, 1H), 6.50 - 6.53 (d, J=13.2 Hz, 1H), 6.69 (m, br, 1H), 7.07 (s, br, 1H), 7.40 - 7.59 (m, 2H), 10.17 (s, 1H). [00786] LCMS (Method N): 1.996 min, MS ES+: 499.8 (M+1).
[00787] HPLC (Method H): 4.82 min. Prep. HPLC purification method: [00788] Chromatographic separation and isolation were conducted with a Waters 2545 binary pump with Waters 2489 UV detector with waters Acquity QDA detector; column Waters X-Bridge C18 (250mm x 19mm x 5µm); compound eluted with, Mobile Phase A: 0.05% ammonium bicarbonate+0.05% NH
3 in water Mobile Phase B : Acetonitrile with a gradient of T = 0.01 min (45% A, 55% B); gradient to T = 21.00 min (45% A, 55% B); T = 21.01 min (0% A, 100% B) gradient to T = 24.00 min (0% A, 100% B); T = 24.01 min (45% A, 55% B); gradient to T = 28.00 min (45% A, 55% B); Flow rate= 9ml/min; analysis time 28 min. Example 45: (E)-4-(Dimethylamino)-N-(2-(2-hydroxy-5-isopropyl-4- methoxybenzoyl)isoindolin-4-yl)-N-methylbut-2-enamide Step 1: Methyl 2,4-dihydroxy-5-isopropylbenzoate
[00789] The title compound was prepared from 2,4-dihydroxy-5-isopropylbenzoic acid (Intermediate FA) and conc. H
2SO
4 analogously to Example 42 Step 1. [00790]
1H NMR (DMSO-d6, 400 MHz): δ 1.12 (d, J = 6.8Hz, 6H), 3.06 - 3.09 (m, 1H), 3.84 (s, 3H), 6.35 (s, 1H), 7.50 (s, 1H), 10.46 (s, 1H), 10.56 (s, 1H). Step 2: Methyl 2,4-dihydroxy-5-isopropylbenzoate
[00791] To a stirred solution of methyl 2,4-dihydroxy-5-isopropylbenzoate (Step 1) (0.7 g, 3.34 mmol, 1.0 eq) in acetonitrile (7 mL) was added K
2CO
3 anhydrous (0.55 g, 3.9 mmol., 1.2 eq.) and the mixture was stirred at room temperature for 10 mins. Dimethyl sulphate (0.44 g, 3.5 mmol., 1.05 eq.) was added and stirring continued at room temperature for 16 h. The resulting mixture
was concentrated under vacuum and the crude material diluted with water (20 mL) and extracted with EtOAc (2 x 100 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under reduced pressure. The crude material was purified by chromatography on silica (product eluted in 5% EtOAc in hexane) yielding methyl 2-hydroxy-5-isopropyl-4- methoxybenzoate as an off-white solid (0.56 g, Yield: 75%). [00792]
1H NMR (DMSO-d6, 400 MHz): δ 1.13 (d, J= 6.8 Hz, 6H), 3.07 - 3.14 (m, 1H), 3.86 (d, J= 12 Hz, 6H), 6.54 (s, 1H), 7.54 (s, 1H), 10.71 (s, 1H). [00793] LCMS (Method N): 2.731 min, MS: ES+ 225.05 (M+1). Step 3: 2-Hydroxy-5-isopropyl-4-methoxybenzoic acid
[00794] To a stirred solution of methyl 2-hydroxy-5-isopropyl-4-methoxybenzoate (Step 2) (0.56 g, 2.5 mmol, 1.0 eq) in MeOH: H
2O (5.6 mL) was added KOH (1.4 g, 2.5 mmol, 10 eq) and the reaction mixture was heated to 70°C for 2 h. The resulting mixture was poured into ice cold sat. solution of KHSO
4 solution (10 mL) (pH adjusted to pH 5-6) and extracted with EtOAc (3 x 100 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under reduced pressure yielding 2-hydroxy-5-isopropyl-4-methoxybenzoic acid as a light pink solid (0.54 g, Yield: 99.5%). The crude material was used without further purification. [00795]
1H NMR (DMSO-d6, 400 MHz): δ 1.10 (d, J= 6.8 Hz, 6H), 3.05 - 3.09 (m, 1H), 3.74 (s, 3H), 6.24 (s, 1H), 7.46 (s, 1H), 12.08 (bs, 1H), 14.71 (bs, 1H). [00796] LCMS (Method N): 2.586 min, MS: ES+ 209.07 (M-1). Step 4: (E)-4-(Dimethylamino)-N-(2-(2-hydroxy-5-isopropyl-4-methoxybenzoyl)isoindolin-4-yl)- N-methylbut-2-enamide
[00797] The title compound was prepared from 2-hydroxy-5-isopropyl-4-methoxybenzoic acid (Step 3) and (E)-4-(dimethylamino)-N-(isoindolin-4-yl)-N-methylbut-2-enamide hydrochloride (Intermediate S) analogously to Example 1 Step 1. [00798]
1H NMR (DMSO-d6, 400 MHz): δ 1.12 (s, br, 6H), 2.02 (s, 6H), 2.89 (s, 2H), 3.10 (s, br, 2H), 3.20 (s, br, 2H), 3.78 (s, 3H), 5.75 (m, 1H), 6.46 - 6.56 (m, 3H), 7.05 (s, 1H), 7.22 (d, J= 8.4 Hz, 1H), 7.36 - 7.44 (m, 2H), 10.15 (s, 1H) [00799] LCMS (Method N): 1.895 min, MS ES+: 452 (M+1). [00800] HPLC (Method H): 4.53 min. Prep. HPLC purification method: [00801] Chromatographic separation and isolation were conducted with Shimadzu LC20AP with UV detector; column Waters X-Bridge (250mm x 19mm 5μm); compound was eluted with: Mobile Phase A: 0.05% ammonium bicarbonate in water, Mobile Phase B: acetonitrile:MeOH:THF[50:50:10] with a gradient of T = 0.00 min (60% A, 40% B); gradient to T = 30.00 min (60% A, 40% B); T = 30.01 min (0% A, 100% B) gradient to T = 35.00 min (0% A, 100% B); gradient to T = 35.01 min (60% A, 40% B); gradient to T = 40.00 min (60% A, 40% B); Flow rate= 9ml/min; analysis time 40min. Example 46: (E)-4-(Dimethylamino)-N-(2-(5-ethyl-2-hydroxy-4- methoxybenzoyl)isoindolin-4-yl)-N-methylbut-2-enamide Step 1: 5-Ethyl-2,4-dihydroxybenzoic acid

[00802] Performed in 2 parallel batches, each of 5 g scale: To a 250 mL autoclave, 4- ethylbenzene-1,3-diol (CAS: 2896-60-8) (5 g, 34.72 mmol, 1.0 eq) in glycerol (50 mL) was added followed by KHCO
3 (17.38 g, 173.61 mmol, 5 eq) and dry ice (50 g) at room temperature. The resulting mixture was placed under a CO
2 pressure of 15 kg/cm
2 at room temperature and heated to 150°C for 24 h. The resulting mixture was poured into ice cold water (500 mL) and extracted with EtOAc (3 x 500 mL) to remove impurities. The aqueous portion was acidified with 2N HCl (20 mL) (to pH ~5 to 6) and extracted with 10% MeOH in DCM (3 x 500 mL) and 30% IPA in
CHCl
3 (3 x 500 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under reduced pressure. The crude material was purified by reverse phase column chromatography (product eluted at 26% ACN in water). The product fractions were combined and concentrated under vacuum. The residue was triturated with n-hexane (2 x 1L) yielding 5- ethyl-2,4-dihydroxybenzoic acid as an off-white solid (6 g, Yield: 32%). [00803]
1H NMR (DMSO-d6, 400 MHz): δ 1.09 (t, J= 7.2 Hz, 14.8 Hz, 3H), 2.45 (q, J= 7.6 Hz, 14.8 Hz, 2H), 6.32 (s, 1H), 7.47 (s, 1H), 10.35 (s, 1H), 11.24 (s, 1H), 13.30 (bs, 1H). [00804] LCMS (Method N): 1.84 min, Step 2: Methyl 5-ethyl-2,4-dihydroxybenzoate
[00805] Performed in 2 parallel batches, each of 0.5 g scale: To a cooled (0°C) solution of 5- ethyl-2,4-dihydroxybenzoic acid (Step 1) (0.5 g, 2.74 mmol, 1.0 eq) in DCM (5 mL) under an atmosphere of N2 gas was added dropwise oxalyl chloride (0.349 g, 2.75 mmol, 1 eq) followed by DMF (0.1 mL, catalytic amount) and the mixture stirred at room temperature for 30 mins. The mixture was re-cooled to 0°C, MeOH (2.5 mL) was added dropwise and stirring continued at room temperature for 30 mins. The resulting mixture was poured into ice cold water (100 mL) and the pH neutralized with sat. NaHCO
3 solution. The aqueous layer was extracted with DCM (3 x 100 mL) and the combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted at 20% EtOAc in hexane) yielding methyl 5-ethyl-2,4-dihydroxybenzoate as a yellow oil (0.54 g, Yield: 50%). [00806]
1H NMR (DMSO-d6, 400 MHz): δ 1.09 (t, J= 7.6 Hz, 3H), 2.46 (q, J= 7.6 Hz, 15.2 Hz, 2H), 3.84 (s, 3H), 6.35 (s, 1H), 7.49 (s, 1H), 10.46 (s, 1H), 10.57 (s, 1H). Step 3: Methyl 5-ethyl-2-hydroxy-4-methoxybenzoate
[00807] To a stirred solution of methyl 5-ethyl-2,4-dihydroxybenzoate (Step 2) (0.54 g, 2.75 mmol, 1.0 eq) in acetonitrile (5.4 mL) were added anhydrous K
2CO
3 (0.45 g, 3.34 mmol, 1.2 eq.) and dimethyl sulphate (0.36 g, 2.89 mmol, 1.05 eq) and the reaction mixture stirred at room temperature for 16 h. The resulting mixture was poured into ice cold water (50 mL) and extracted with EtOAc (3 x 50 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica (product was eluted in 18% EtOAc in hexane) yielding methyl 5-ethyl-2-hydroxy-4- methoxybenzoate as a white solid (0.44 g, Yield: 76%). [00808]
1H NMR (DMSO-d6, 400 MHz): δ 1.09 (t, J= 7.6 Hz, 3H), 2.47 - 2.48 (m, 2H), 3.84 (s, 3H), 3.87 (s, 3H), 6.54 (s, 1H), 7.53 (s, 1H), 10.70 (s, 1H). Step 4: 5-Ethyl-2-hydroxy-4-methoxybenzoic acid
[00809] To a stirred solution of methyl 5-ethyl-2-hydroxy-4-methoxybenzoate (0.44 g, 2.10 mmol, 1.0 eq) in MeOH: H2O (2:1) (6 mL) was added KOH (1.17 g, 20.89 mmol, 10 eq) and the reaction mixture was heated to 60°C for 2 h. The resulting mixture was poured into ice cold water (50 mL) and the pH was adjusted to pH ~7 with KHSO4 solution. The mixture was extracted with EtOAc (3 x 100 mL and the combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under reduced pressure to yield 5-ethyl-2-hydroxy-4-methoxybenzoic acid as an off-white solid (0.44 g, Yield: quantitative). The crude material was used without further purification. [00810]
1H NMR (DMSO-d6, 400 MHz): δ 1.09 (t, J= 7.6 Hz, 14.8 Hz, 3H), 2.47 - 2.50 (m, 2H), 3.82 (s, 3H), 6.48 (s, 1H), 7.50 (s, 1H), 12.35 (s, 1H). [00811] LCMS (Method N): 2.18 min, MS: ES+ 195 (M-1). Step 5: (E)-4-(Dimethylamino)-N-(2-(5-ethyl-2-hydroxy-4-methoxybenzoyl)isoindolin-4-yl)-N- methylbut-2-enamide
[00812] The title compound was prepared from 5-ethyl-2-hydroxy-4-methoxybenzoic acid (Step 4) and (E)-4-(dimethylamino)-N-(isoindolin-4-yl)-N-methylbut-2-enamide hydrochloride (Intermediate S) analogously to Example 1 Step 1. [00813] HT
1H NMR (DMSO-d6, 400 MHz, 348K): δ 1.12 (t, J= 7.2 Hz, 3H), 2.05 (s, 6H), 2.48 - 2.51 (m, 2H), 2.87 (d, J= 5.6 Hz, 2H), 3.19 (s, 3H), 3.81 (s, 3H), 4.71 (bs, 2H), 4.89 (s, 2H), 5.81 - 5.84 (s, br, 1H), 6.51 (s, 1H), 6.62 - 6.69 (m, 1H), 7.11 (s, 1H), 7.19 (d, J= 7.6 Hz, 1H), 7.37 - 7.44 (m, 2H), 10.06 - 10.07 (m, 1H). [00814] LCMS (Method A): 1.853 min, MS ES+: 438 (M+1). [00815] HPLC (Method H): 4.32 min. Prep. HPLC purification method: [00816] Chromatographic separation and isolation were conducted Waters 2545 binary pump with Waters 2489 UV detector with Waters Acquity QDA detector; column Waters Sunfire C18 OBD (250mm x 19mm x 5µm); compound was eluted with: Mobile Phase A: 0.05% formic acid in water. Mobile Phase B: acetonitrile: water: THF (80:20:10) with a gradient of T = 0.01 min (80% A, 20% B); gradient to T = 36.00 min (75% A, 25% B); T = 36.01 min (0% A, 100% B) gradient to T = 39.00 min (0% A, 100% B); T = 39.01 min (80% A, 20% B); gradient to T = 46.00 min (80% A, 20% B); Flow rate= 9 ml/min; analysis time 46 min. Example 47 (E)-N-(2-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)isoindolin-4-yl)- 4-(dimethylamino)-N-methylbut-2-enamide Step 1: (6-Chloro-7-hydroxy-2,2,5-trimethyl-4H-benzo[d][1,3]dioxin-4-one

[00817] To a stirred solution 3-chloro-4,6-dihydroxy-2-methylbenzoic acid (Intermediate B) (2 g, 9.87 mmol, 1.0 eq.) in DMF (20 mL) at room temperature were added acetone (7.3 mL, 29.6 mmol, 10 eq.) and DMAP (0.12 g, 0.98 mmol, 0.1 eq.). The mixture was cooled to 0°C and treated dropwise with thionyl chloride (2.15 mL, 29.61 mmol, 3 eq.). The reaction mixture was allowed to warm to room temperature and stirred for 2 h. The resulting mixture was neutralized with sat. NaHCO3 (30 mL) and extracted with ethyl acetate (2 x 40 mL). The combined organic extracts
were dried over Na
2SO
4, filtered, and concentrated under reduced pressure. The crude material was purified by chromatography on silica (product was eluted in 100% DCM) yielding (6-chloro- 7-hydroxy-2,2,5-trimethyl-4H-benzo[d][1,3]dioxin-4-one as a white solid (1.0 g, Yield: 41%). [00818]
1H NMR (DMSO-d6, 400 MHz): δ 1.64 (s, 6H), 2.65 (s, 3H), 6.49 (s, 1H), 11.57 (s, 1H). [00819] LCMS (Method N): 2.14 min, MS: ES+ 242.78 (M+1). Step 2: 6-Chloro-7-methoxy-2,2,5-trimethyl-4H-benzo[d][1,3]dioxin-4-one

[00820] To a stirred solution of (6-chloro-7-hydroxy-2,2,5-trimethyl-4H-benzo[d][1,3]dioxin-4-one (Step 1) (1 g, 4.12 mmol, 1.0 eq.) in acetonitrile (10 mL) was added K2CO3 (0.68g, 4.94 mmol, 1.2 eq.) and the mixture was stirred at room temperature for 10 mins. Dimethyl sulphate (0.54g, 4.32 mmol, 1.05 eq.) was added and stirring continued at room temperature for 16 h. The resulting mixture was diluted with water (20 mL) and extracted with ethyl acetate (2 x 20 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude material was purified by chromatography on silica (product was eluted in 100% DCM) yielding 6-chloro-7-methoxy-2,2,5-trimethyl-4H-benzo[d][1,3]dioxin-4-one as a yellow gummy (0.8 g, Yield: 75%). [00821]
1H NMR (DMSO-d6, 400 MHz): δ 1.67 (s, 6H), 2.68 (s, 3H), 3.92 (s, 3H), 6.78 (s, 1H). Step 3: 3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoic acid
[00822] To a stirred solution of 6-chloro-7-methoxy-2,2,5-trimethyl-4H-benzo[d][1,3]dioxin-4-one (Step 2) (0.7 g, 2.73 mmol, 1.0 eq.) in THF:H2O (1: 1) (14 mL) was added LiOH.H2O (1.14 g, 27.3 mmol, 10 eq.) and the reaction mixture was stirred at room temperature for 16 h. The resulting mixture was acidified with dilute HCl and extracted with ethyl acetate (2 x 30 mL). The combined
organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum yielding 3- chloro-6-hydroxy-4-methoxy-2-methylbenzoic acid as a white solid (0.4 g, Yield: 76%). [00823]
1H NMR (DMSO-d6, 400 MHz): δ 2.40 (s, 3H), 3.83 (s, 3H), 6.52 (s, 1H), 12.35 (bs, 1H). Step 4: (E)-N-(2-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)isoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide
[00824] The title compound was prepared from 3-chloro-6-hydroxy-4-methoxy-2-methylbenzoic acid (Step 3) and (E)-4-(dimethylamino)-N-(isoindolin-4-yl)-N-methylbut-2-enamide hydrochloride (Intermediate S) analogously to Example 1 Step 1. [00825]
1H NMR (DMSO-d6, 400 MHz): δ 2.00 - 2.03 (m, 6H), 2.11 - 2.18 (m, 3H), 2.84 - 2.98 (m, 2H), 3.09 and 3.21 (2 singlets, 3H), 3.81 (s, 3H), 4.42 - 4.90 (m, 4H), 5.71 - 5.80 (m, 1H), 6.32 - 6.70 (m, 2H), 7.23 - 7.24 (m, 1H), 7.31 - 7.39 (m, 1H), 7.41 - 7.45 (m, 1H), 10.10 (bs, 1H). [00826] LCMS (Method N): 1.75 min, MS: ES+ 457.8 (M+1). [00827] HPLC (Method G): 4.14 min. Prep. HPLC purification method: [00828] Chromatographic separation and isolation were conducted using a Shimadzu LC20AP with UV detector; column Waters X-Bridge (250mm x 19mm 5µm); compound was eluted with: Mobile Phase A: 0.05% ammonium bicarbonate in water, Mobile Phase B: acetonitrile:MeOH:THF [50:50:10] with a gradient of T = 0.00 min (60% A, 40% B); gradient to T = 30.00 min (60% A, 40% B); T = 30.01 min (0% A, 100% B) gradient to T = 32.00 min (0% A, 100% B); T = 32.01 min (60% A, 40% B); gradient to T = 35.00 min (60% A, 40% B); Flow rate= 9 ml/min; analysis time 35 min. Example 48: (E)-N-(2-(5-Chloro-2-hydroxy-4-methoxybenzoyl)isoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide
[00829] To a stirred solution of 5-chloro-2-hydroxy-4-methoxybenzoic acid (CAS: 1378866-39- 7) (0.2 g, 0.98 mmol, 1 eq) in DMF (2 mL) were added EDC.HCl (0.28 g, 1.48 mmol, 1.5 eq) and HOAT (0.134 g, 0.98 mmol, 1 eq) and the mixture was stirred at room temperature for 5 mins. N- methylisoindolin-4-amine hydrochloride (Intermediate A) (0.35 g, 1.18 mmol, 1.2 eq) and NMM (0.498 g, 4.9 mmol, 5 eq) were added and the reaction mixture was stirred at room temperature for 1 h. The resulting mixture was diluted using ice cold water (100 mL) and extracted with EtOAc (3 x 50 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under vacuum. The crude material was purified using reverse phase column chromatography (product eluted at 10% ACN: water) followed by preparative HPLC purification [A: 0.05% formic acid in water B: ACN:MeOH:THF (50:50:10)] yielding (E)-N-(2-(5-chloro-2-hydroxy-4- methoxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-methylbut-2-enamide as a pale pink solid (0.020 g, Yield: 4.6%). [00830]
1H NMR (DMSO-d6, 400 MHz): δ 2.078 (s, 6H), 2.89 - 2.90 (d, J= 5.2 Hz, 2H), 3.10 and 3.20 (2 singlets, 3H), 3.84 (s, 3H), 4.54 - 4.87 (s, 4H), 5.75 (t, J= 14.0 Hz, 1H), 6.58 - 6.70 (m, 2H), 7.22 - 7.44 (m, 4H), 10.61 (bs, 1H). [00831] LCMS (Method N): 1.75 min, MS ES+: 443.88, 445.88 (M, M+2). [00832] HPLC (Method G): 4.05 min. Prep. HPLC purification method: [00833] Chromatographic separation and isolation were conducted using a Waters 2545 quaternary system with Waters 2489 UV Detector; column Waters Sunfire C18 (250mm x 19mm 5µm); compound was eluted with: Mobile Phase A: 0.05% formic acid in Merck water. Mobile Phase B: acetonitrile: MeOH: THF (50:50:10) with a gradient of T = 0.00 min (78% A, 22% B); gradient to T = 28.00 min (78% A, 22% B); T = 28.01 min (0% A, 100% B) gradient to T = 30.00 min (0% A, 100% B); T = 30.01 min (78% A, 22% B); gradient to T = 40.00 min (78% A, 22% B); Flow rate= 9 ml/min; analysis time 40 min. The compounds of the following tabulated Examples (Table Ex48) were prepared analogously to Example 48 from the indicated intermediates. Ex. No. Name and Structure Data
1H NMR (DMSO-d6, 400 MHz): δ 1.29 (s, 9H), 3.12 and 3.24 (2 singlets, 3H), 3.80 (s, 3H), 4.58 - 4.61 (m, 1H), 4.72 - 4.89 (m, 3H), 5.58 (t, J= 11.2, 1H), 5.94 - 6.00 (m, 1H), 6.10 - 6.24 (m, 1H), 6.52 N-(2-(5-(tert-Butyl)-2-hydroxy-4- (bs, 1H), 7.09 (s, 1H), 7.23 (d, J= 6.4 Hz, methoxybenzoyl)isoindolin-4-yl)-N- 1H), 7.37 - 7.44 (m, 2H), 10.19 (bs, 1H). methylacrylamide LCMS (Method N): 2.441 min, MS: ES+ Prepared from 5-(tert-butyl)-2-hydroxy-4- 409.0 (M+1). methoxybenzoic acid (Intermediate N) and HPLC (Method H): 8.68 min. N-(isoindolin-4-yl)-N-methyl acrylamide TFA salt (TFA salt of Intermediate W2) 1H NMR (DMSO-d6, 400 MHz): δ 1.05 (s, br, 3H), 2.40 - 2.41 (m, 2H), 3.11 and 3.17 (2 singlets, 3H), 3.78 (s, 3H), 4.55 – 4.74 (m, 2H), 4.85 (d, J= 19.6 Hz, 2H), N-(2-(5-Ethyl-2-hydroxy-4- 5.59 (t, J= 10.8 Hz, 1H), 5.94 - 6.01 (m, methoxybenzoyl)isoindolin-4-yl)-N- 1H), 6.11 - 6.24 (m, 1H), 6.50 (s, 1H), methylacrylamide 7.06 (d, J= 18 Hz, 1H), 7.23 - 7.25 (m, Prepared from 5-ethyl-2-hydroxy-4- 1H), 7.35 - 7.44 (m, 2H), 9.69 - 9.70 (m, methoxybenzoic acid (Example 46, step 4) 1H). and N-(isoindolin-4-yl)-N-methyl LCMS (Method N): 2.206 min, MS ES+: acrylamide TFA salt (TFA salt of 380.9 (M+1). Intermediate W2) HPLC (Method H): 7.60 min. 1H NMR (DMSO-d6, 400 MHz): δ 1.32 (s, 9H), 2.05 - 2.09 (m, 6H), 2.90 (bs, 4H), 3.10 - 3.25 (m, 3H), 3.47 (bs, 1H), 3.82 - 3.95 (m, 1H), 4.38 (bs, 1H), 4.80 (s, 1H), 5.86 - 5.97 (m, 1H), 6.62 - 6.69 (E)-N-(2-(3-(tert-butyl)-2-fluoro-6- (m, 2H), 7.06 (d, J=7.6 Hz, 1H), 7.16 - hydroxybenzoyl)-1,2,3,4- 7.23 (m, 3H), 9.73 (s, 1H) tetrahydroisoquinolin-7-yl)-4- LCMS (Method N): 1.763 min, MS: ES+ (dimethylamino)-N-methylbut-2-enamide 468.10 (M+1). Prepared from 3-(tert-butyl)-2-fluoro-6- HPLC (Method H): 4.50 min. hydroxybenzoic acid (Intermediate O) and (E)-4-(dimethylamino)-N-methyl-N- (1,2,3,4-tetrahydroisoquinolin-7-yl)but-2-
enamide hydrochloride (Example 4.2, Step 2) 1H NMR (DMSO-d6, 400 MHz): δ 1.29 - 1.32 (m, 9H), 2.81 - 2.91 (m, 2H), 3.19 and 3.26 (2 singlets, H), 3.48 (s, 1H), 3.80 - 3.98 (m, 1H), 4.39 (bs, 1H), 4.82 (s, 1H), 5.51 - 5.56 (m, 1H), 6.09 - 6.16 N-(2-(3-(tert-butyl)-2-fluoro-6- (m, 2H), 6.68 (d, J= 8.0 Hz, 1H), 6.96 hydroxybenzoyl)-1,2,3,4- (bs, 1H), 7.07 (d, J= 7.2 Hz, 1H), 7.16 - tetrahydroisoquinolin-7-yl)-N- 7.24 (m, 2H), 9.54 (bs, 1H). methylacrylamide LCMS (Method N): 2.123 min, MS: ES+ Prepared from 3-(tert-butyl)-2-fluoro-6- 411.0 (M+1). hydroxybenzoic acid (Intermediate O) and HPLC (Method H): 7.45 min. N-methyl-N-(1,2,3,4-tetrahydroisoquinolin- 7-yl) acrylamide TFA salt (Example 51 step 2) 1H NMR (DMSO-d6, 400 MHz): δ 2.17 (s, 3H), 3.09 and 3.22 (2 singlets, 3H), 3.81 (s, 3H), 4.38 - 4.95 (m, 4H), 5.50 - 5.69 (m, 1H), 5.90 - 6.12 (m, 1H), 6.16 - N-(2-(3-Chloro-6-hydroxy-4-methoxy-2- 6.25 (m, 1H), 6.50 - 6.54 (m, 1H), 7.23 - methylbenzoyl)isoindolin-4-yl)-N- 7.25 (m, 1H), 7.31 - 7.33 (m, 1H), 7.40 - methylacrylamide 7.46 (m, 1H), 10.07 (bs, 1H). Prepared from 3-chloro-6-hydroxy-4- LCMS (Method N): 1.867 min, MS: ES+ methoxy-2-methylbenzoic acid (Example 400.81 (M+1). 47 step 3) and N-(isoindolin-4-yl)-N-methyl HPLC (Method H): 6.20 min. acrylamide TFA salt (TFA salt of Intermediate W2) HT
1H NMR (DMSO-d6, 400 MHz, 348K): δ 3.20 (s, 3H), 3.86 (s, 3H), 4.68 (s, 2H), 4.86 (s, 2H), 5.57 (d, J= 9.2 Hz, 1H), 6.03 (s, br, 1H), 6.17 (d, J= 15.2 Hz, N-(2-(5-Chloro-2-hydroxy-4- 1H), 6.68 (s, 1H), 7.20 (d, J= 7.6 Hz, 1H), methoxybenzoyl) isoindolin-4-yl)-N-methyl 7.33 (s, 1H), 7.38 - 7.44 (m, 2H), 9.29 acrylamide (s, 1H).
Prepared from 5-chloro-2-hydroxy-4- LCMS (Method A): 1.986 min, MS ES+: methoxybenzoic acid (CAS: 1378866-39-7) 386.75 (M+1). and N-(isoindolin-4-yl)-N-methyl HPLC (Method H): 6.68 min. acrylamide TFA salt (TFA salt of Intermediate W2) 1H NMR (DMSO-d6, 400 MHz): δ 2.00 (s, 6H), 2.13 (d, J= 11.6 Hz, 3H), 2.84 - 2.98 (m, 2H), 3.01 and 3.13 (2 singlets, 3H), 3.82 (s, 3H), 4.48 - 4.53 (m, 1H), (E)-N-(2-(5-Chloro-2-hydroxy-4- 4.65 - 4.81 (m, 3H), 5.62 - 5.70 (m, 1H), methoxybenzoyl)-5-methylisoindolin-4-yl)- 6.60 - 6.72 (m, 2H), 7.21 - 7.34 (m, 3H), 4-(dimethylamino)-N-methylbut-2-enamide 10.65 (s, 1H). Prepared from 5-chloro-2-hydroxy-4- LCMS (Method A): 1.66 min, MS: ES+ methoxybenzoic acid (CAS: 1378866-39-7) 457.7 (M+1). and (E)-4-(dimethylamino)-N-methyl-N-(5- HPLC (Method H): 4.22 min. methylisoindolin-4-yl)but-2-enamide hydrochloride (Example 54 step 6) HT
1H NMR (DMSO-d6, 400 MHz, 348K): δ 2.04 (s, 6H), 2.88 (s, br, 2H), 3.30 and 3.86 (2 singlets, 3H), 4.62 (d, J= 15.6 Hz, 1H), 4.70 - 4.82 (m, 3H), (E)-N-(5-Chloro-2-(5-chloro-2-hydroxy-4- 5.70 (t, J= 15.2 Hz, 1H), 6.68 (d, J= 8.4 methoxybenzoyl) isoindolin-4-yl)-4- Hz, 2H), 7.23 -7.30 (m, 1H), 7.41 – 7.51 (dimethyl amino)-N-methylbut-2-enamide (m, 1H) 7.57 (t, J= 8.0 Hz, 1H), 10.20 Prepared from 5-chloro-2-hydroxy-4- (bs, 1H). methoxybenzoic acid (CAS: 1378866-39-7) LCMS (Method A): 1.690 min, MS: ES+ and (E)-N-(5-chloroisoindolin-4-yl)-4- 477.7(M+1). (dimethylamino)-N-methylbut-2-enamide HPLC (Method H): 4.23 min, hydrochloride (Example 44 step 2) HT
1H NMR (DMSO-d6, 400 MHz, 348K): δ 1.15 (d, J= 6.4 Hz, 6H), 2.03 (s, 6H), 2.16 - 2.21 (m, 1H), 2.87 (m, 2H), 3.22 - 3.17 (m, 3H), 3.80 (s, 3H), 4.62 - (E)-N-(5-Chloro-2-(2-hydroxy-5-isopropyl- 4.66 (m, 1H), 4.82 - 4.87 (m, 3H), 5.71 4-methoxybenzoyl) isoindolin-4-yl)-4- (d, J= 15.2 Hz, 1H), 6.51 (s, 1H), 6.65 - (dimethyl amino)-N-methylbut-2-enamide 6.69 (m, 1H), 7.12 (s, 1H), 7.43 (d, J=
Prepared from 2-hydroxy-5-isopropyl-4- 7.6 Hz, 1H), 7.57 (d, J= 8.0 Hz, 1H), methoxybenzoic acid (Example 45 step 3) 10.10 (s, 1H). and (E)-N-(5-chloroisoindolin-4-yl)-4- LCMS (Method A): 1.831 min, MS: ES+ (dimethylamino)-N-methylbut-2-enamide 485.9 (M+1). hydrochloride (Example 44 step 2) HPLC (Method H): 4.64 min. HT
1H NMR (DMSO-d6, 400 MHz, 348K): δ 1.32 (s, 9H), 2.02 (s, 6H), 2.16 (s, 3H), 2.85 (d, J= 5.6 Hz, 2H), 3.82 (s, 3H), 4.60 (d, J= 15.2 Hz, 1H), 4.81 (t, J= 14.8 Hz, 3H), 5.69 (d, J= 15.2 Hz, 1H), (E)-N-(2-(5-(tert-Butyl)-2-hydroxy-4- 6.53 (s, 1H), 6.67 (t, J= 6.4 Hz, 1H), 7.17 methoxybenzoyl)-5-methylisoindolin-4-yl)- (s, 1H), 7.31 (s, 2H), 10.20 (bs, 1H). 4-(dimethylamino)-N-methylbut-2-enamide LCMS (Method A): 1.899 min, MS: ES+ Prepared from 5-(tert-butyl)-2-hydroxy-4- 479.9 (M+1). methoxybenzoic acid (Intermediate N) and HPLC (Method H): 4.79 min, (E)-4-(dimethylamino)-N-methyl-N-(5- methylisoindolin-4-yl)but-2-enamide hydrochloride (Example 54 step 6) 1H NMR (DMSO-d6, 400 MHz): δ 1.10 (d, J= 6.8 Hz, 6H), 2.85 (s, 2H), 3.07 - 3.14 (m, 1H), 3.21 (s, 3H), 3.62 (s, 2H), 3.74 (s, 3H), 4.66 (s, 2H), 5.55 (d, J= 10 Hz, 1H), 6.08 - 6.16 (m, 2H), 6.46 (s, N-(2-(2-Hydroxy-5-isopropyl-4- 1H), 6.94 (s, 1H), 7.06 (d, J= 7.6 Hz, 1H), methoxybenzoyl)-1,2,3,4- 7.14 (s, 1H), 7.23 (d, J= 8 Hz, 1H), 9.88 tetrahydroisoquinolin-7-yl)-N- (s, 1H) methylacrylamide. Prepared from 2- LCMS (Method A): 2.274 min, MS: ES+ hydroxy-5-isopropyl-4-methoxybenzoic 408.85 (M+1). acid (Example 45 step 3) and N-methyl-N- HPLC (Method H): 7.89 min, (1,2,3,4-tetrahydroisoquinolin-7- yl)acrylamide TFA salt (Example 51 step 2) 1H NMR (DMSO-d6, 400 MHz): δ 1.17 (d, J= 6.4 Hz, 6H), 2.54 - 2.84 (m, 2H), 3.05 (t, J= 7.2 Hz, 1H), 3.22 (s, 3H), 3.46 (s, br, 1H), 3.84 (s, br, 1H), 4.46 (s, br, 1H), 4.76 (s, br, 1H), 5.57 (s, br, 1H), 6.15 (d, J= 15.2 Hz, 2H), 6.62 (d, J= 12
N-(2-(4-Fluoro-2-hydroxy-5- Hz, 1H), 7.07 (d, J= 5.6 Hz, 2H), 7.24 (d, isopropylbenzoyl)-1,2,3,4- J= 8 Hz, 2H), 10.10 (s, 1H). tetrahydroisoquinolin-7-yl)-N- LCMS (Method A): 2.171 min, MS: ES+ methylacrylamide. Prepared from 4-fluoro- 396.9 (M+1). 2-hydroxy-5-isopropylbenzoic acid HPLC (Method H): 7.70 min. (Example 40 step 5) and N-methyl-N- (1,2,3,4-tetrahydroisoquinolin-7- yl)acrylamide TFA salt (Example 51 step 2) 1H NMR (DMSO-d6, 400 MHz): δ 2.84 (s, 2H), 3.21 (s, 3H), 3.55 (s, br, 2H), 3.81 (s, 3H), 4.69 (s, br, 2H), 5.56 (d, J= 9.2 Hz, 1H), 6.08 - 6.16 (m, 2H), 6.61 (s, 1H), 7.07 (d, J= 8.0 Hz, 1H), 7.19 - 7.24 N-(2-(5-Chloro-2-hydroxy-4- (m, 3H), 10.35 (s, 1H). methoxybenzoyl)-1,2,3,4- LCMS (Method A): 1.948 min, MS: ES+ tetrahydroisoquinolin-7-yl)-N- 401.28 (M+1). methylacrylamide. Prepared from 5-chloro- HPLC (Method H): 6.820 min. 2-hydroxy-4-methoxybenzoic acid (CAS: 1378866-39-7) and N-methyl-N-(1,2,3,4- tetrahydroisoquinolin-7-yl) acrylamide TFA salt (Example 51 step 2) 1H NMR (DMSO-d6, 400 MHz): δ 1.14 (d, J= 6.8 Hz, 6H), 2.09 (s, 3H), 2.83 (t, J= 5.6 Hz, 2H), 3.14 – 3.18, m 4H), 3.67 (t, J = 5.6Hz, 2H), 3.79 (s, 3H), 4.64 (s, 2H), 5.50 (d, J= 10 Hz, 1H), 5.87 - 5.94 N-(2-(2-Hydroxy-5-isopropyl-4- (m, 1H), 6.15 (dd, J= 5.6 Hz, 1H), 6.49 methoxybenzoyl)-6-methyl-1,2,3,4- (s, 1H), 6.97 (s, 1H), 7.04 (s, 1H), 7.15 tetrahydroisoquinolin-7-yl)-N- (s, 1H) 9.50 (s, 1H). methylacrylamide LCMS (Method N): 2.29 min, MS: ES+ Prepared from 2-hydroxy-5-isopropyl-4- 423.2 (M+1). methoxybenzoic acid (Example 45 step 3) HPLC (Method H): 8.47 min and N-methyl-N-(6-methyl-1,2,3,4- tetrahydroisoquinolin-7-yl) acrylamide TFA salt (Example 55 step 3)
1H NMR (DMSO-d6, 400 MHz): δ 1.21 (s, 6H), 2.85 (t, J= 5.2 Hz, 2H), 3.21 (s, 3H), 3.60 (bs, 2H), 4.66 (bs, 2H), 5.56 (d, J= 10 Hz, 1H), 6.07- 6.16 (m, 2H), 6.45 (s, 1H), 7.06 - 7.09 (m, 2H), 7.16 (s, 1H), 7.24 (d, J= 8 Hz, 1H), 9.61 (s, 1H), 10.33 N-(2-(6-Hydroxy-3,3-dimethyl-2- (s, 1H). oxoindoline-5-carbonyl)-1,2,3,4- LCMS (Method N): 1.776 min, MS ES+: tetrahydroisoquinolin-7-yl)-N- 419.9 (M+1). methylacrylamide. Prepared from 6- HPLC (Method H): 5.35 min, hydroxy-3,3-dimethyl-2-oxoindoline-5- carboxylic acid (Example 25 step 7) and N- methyl-N-(1,2,3,4-tetrahydroisoquinolin-7- yl)acrylamide TFA salt (Example 51 step 2) 1H NMR (DMSO-d6, 400 MHz): δ 1.22 (s, 6H), 3.11 and 3.22 (2 singlets, 3H), 4.53 - 4.57 (m, 1H), 4.68 - 4.87 (m, 3H), 5.54 - 5.61 (m, 1H), 5.94 - 6.00 (m, 1H), 6.09 - 6.24 (m, 1H), 6.47 (s, 1H), 7.17 - N-(2-(6-Hydroxy-3,3-dimethyl-2- 7.25 (m, 2H), 7.36 - 7.44 (m, 2H), 10.03 oxoindoline-5-carbonyl) isoindolin-4-yl)-N- (bs, 1H), 10.36 (s, 1H). methyl acrylamide. Prepared from 6- LCMS (Method N): 1.777 min, MS ES+: hydroxy-3,3-dimethyl-2-oxoindoline-5- 405.9 (M+1). carboxylic acid (Example 25 step 7) and N- HPLC (Method H): 5.38 min, (isoindolin-4-yl)-N-methyl acrylamide TFA salt (TFA salt of Intermediate W2) 1H NMR (DMSO-d6, 400 MHz): δ 1.11 (d, J= 7.2 Hz, 6H), 2.86 (s, br, 2H), 3.08 - 3.14 (m, 1H), 3.27 (s, 3H), 3.64 - 3.70 (m, 2H), 3.77 (s, 3H), 4.66 - 4.71 (m, N-(6-(2-Hydroxy-5-isopropyl-4- 2H), 5.61 (dd, J= 3.2 Hz, 1H), 6.18 (t, J= methoxybenzoyl)-5,6,7,8-tetrahydro-2,6- 9.6 Hz, 2H), 6.47 (s, 1H), 6.96 (s, 1H), naphthyridin-3-yl)-N-methylacrylamide 7.28 (s, 1H), 8.30 (s, 1H), 9.80 (s, 1H). Prepared from 2-hydroxy-5-isopropyl-4- LCMS (Method N): 1.991 min, MS: ES+ methoxybenzoic acid (Example 45 step 3) 410.2 (M+1). and N-methyl-N-(5,6,7,8-tetrahydro-2,6- HPLC (Method H): 7.053 min.
naphthyridin-3-yl)acrylamide TFA salt (Example 59, step 3) 1H NMR (DMSO-d6, 400 MHz): δ 1.10 (d, J= 6.8 Hz, 6H), 2.12 (s, 3H), 2.86 (s, br, 2H), 3.07 - 3.14 (m, 4H), 3.68 (s, br, 2H), 3.77 (s, 3H), 4.59 (s, br, 2H), 5.54 N-(7-(2-Hydroxy-5-isopropyl-4- (d, J= 10 Hz, 1H), 5.73 - 5.80 (m, 1H), methoxybenzoyl)-3-methyl-5,6,7,8- 6.17 (d, J= 16.4 Hz, 1H), 6.47 (s, 1H), tetrahydro-1,7-naphthyridin-2-yl)-N- 6.96 (s, 1H), 7.65 (s, 1H), 8.60 (s, 1H). methylacrylamide. Prepared from 2- LCMS (Method N): 2.05 min, MS: ES+ hydroxy-5-isopropyl-4-methoxybenzoic 424.2 (M+1). acid (Example 45 step 3) and N-methyl-N- HPLC (Method H): 7.33 min, (3-methyl-5,6,7,8-tetrahydro-1,7- naphthyridin-2-yl) acrylamide TFA salt (Example 62 step 4) 1H NMR (DMSO-d6, 400 MHz): δ 1.11 (d, J= 6.8 Hz, 6H), 2.26 (s, 3H), 2.92 (t, J= 5.6 Hz, 2H), 3.08 - 3.14 (m, 4H), 3.71 (s, br, 2H), 3.77 (s, 3H), 4.67 (s, 2H), N-(6-(2-Hydroxy-5-isopropyl-4- 5.57 (dd, J= 1.2 Hz, 10.4 Hz, 1H), 5.85 - methoxybenzoyl)-2-methyl-5,6,7,8- 5.91 (m, 1H), 6.18 (dd, J= 2 Hz, 16.8 Hz, tetrahydro-1,6-naphthyridin-3-yl)-N- 1H), 6.47 (s, 1H), 6.96 (s, 1H), 7.58 (s, methylacrylamide. Prepared from 2- 1H), 9.79 (s, 1H). hydroxy-5-isopropyl-4-methoxybenzoic LCMS (Method A): 1.89 min, MS: ES+ acid (Example 45 step 3) and N-methyl-N- 423.9 (M+1). (2-methyl-5,6,7,8-tetrahydro-1,6- HPLC (Method H): 6.26 min, naphthyridin-3-yl)acrylamide TFA salt (Example 63 step 6) HT
1H NMR (DMSO-d6, 400 MHz, 348K): δ 2.04 (s, 6H), 2.14 (s, br, 2H), 2.75 (s, br, 1H), 2.89 (s, br, 3H), 3.23 (s, br, 3H), 3.44 (s, br, 1H), 3.72 (s, br, 1H), (E)-N-(2-(3-Chloro-6-hydroxy-4-methoxy- 3.82 (s, 3H), 4.05 (s, br, 1H), 4.34 - 4.39 2-methylbenzoyl)-1,2,3,4- (s, br, 1H), 4.78 (s, br, 1H), 5.91 (s, 1H), tetrahydroisoquinolin-7-yl)-4- 6.53 - 6.60 (m, 2H), 7.06 (d, J= 7.2 Hz, (dimethylamino)-N-methylbut-2-enamide 1H), 7.19 - 7.22 (m, 2H), 9.67 (bs, 1H).
Prepared from 3-chloro-6-hydroxy-4- LCMS (Method A): 1.661 min, MS: ES+ methoxy-2-methylbenzoic acid (Example 471.7 (M+1). 47 step 3) and (E)-4-(dimethylamino)-N- HPLC (Method H): 4.18 min. methyl-N-(1,2,3,4-tetrahydroisoquinolin-7- yl)but-2-enamide hydrochloride (Example 4.2, step 2) 1H NMR (DMSO-d6, 400 MHz): δ 1.10 (d, J= 6.8 Hz, 2H), 2.02 (s, 6H), 2.85 – 2.87 (m, 4H), 3.11 (m, 1H), 3.20 (s, 3H), 3.62 (bs, 2H), 3.77 (s, 3H), 4.64 (s, 2H), (E)-4-(Dimethylamino)-N-(2-(2-hydroxy-5- 5.87 (d, J= 13.2 Hz, 1H), 6.47 (s, 1H), isopropyl-4-methoxybenzoyl)-1,2,3,4- 6.57 - 6.64 (m, 1H), 6.95 (s, 1H), 7.06 (d, tetrahydroisoquinolin-7-yl)-N-methylbut-2- J= 8.8 Hz, 1H), 7.13 (bs, 1H), 7.23 (d, J= enamide. Prepared from 2-hydroxy-5- 8 Hz, 1H), 9.82 (s, 1H). isopropyl-4-methoxybenzoic acid (Example LCMS (Method A): 1.791 min, MS: ES+ 45 step 3) and (E)-4-(dimethylamino)-N- 465.9 (M+1). methyl-N-(1,2,3,4-tetrahydroisoquinolin-7- HPLC (Method H): 4.56 min. yl)but-2-enamide hydrochloride (Example 4.2, step 2) HT
1H NMR (DMSO-d6, 400 MHz, 348K): δ 1.32 (s, 9H), 2.07 (s, 6H), 2.86 - 2.90 (m, 4H), 3.22 (s, 3H), 3.64 (s, br, 2H), 4.65 (s, br, 2H), 5.93 (d, J= 15.6 Hz, (E)-N-(2-(5-(tert-Butyl)-4-fluoro-2- 1H), 6.58 - 6.65 (m, 2H), 7.07 (m, 3H), hydroxybenzoyl)-1,2,3,4- 7.23 (d, J= 8Hz, 1H), 9.89 (s, 1H). tetrahydroisoquinolin-7-yl)-4- LCMS (Method A): 1.817 min, MS: ES+ (dimethylamino)-N-methylbut-2-enamide 468.3 (M+1). Prepared from 5-(tert-butyl)-4-fluoro-2- HPLC (Method H): 4.62 min. hydroxybenzoic acid (Intermediate OA) and (E)-4-(dimethylamino)-N-methyl-N- (1,2,3,4-tetrahydroisoquinolin-7-yl)but-2- enamide hydrochloride (Example 4.2, step 2)
1H NMR (DMSO-d6, 400 MHz): δ 1.17 (d, J= 6.8Hz, 6H), 2.04 (s, 6H), 2.86 (s, br, 4H), 3.05 (m, 1H), 3.20 (s, br, 3H), 3.46 (s, br, 1H), 3.83 (s, br 1H), 4.45 (s, (E)-4-(Dimethylamino)-N-(2-(4-fluoro-2- br, 1H), 4.75 (s, br, 1H), 5.87 (s, br, 1H), hydroxy-5-isopropylbenzoyl)-1,2,3,4- 6.62 (d, J= 12 Hz, 2H), 7.06 - 7.24 (m, tetrahydroisoquinolin-7-yl)-N-methylbut-2- 4H), 10.15 (s, 1H). enamide. Prepared from 4-fluoro-2- LCMS (Method A): 1.749 min, MS: ES+ hydroxy-5-isopropylbenzoic acid (Example 454.3 (M+1). 40 step 5) and (E)-4-(dimethylamino)-N- HPLC (Method I): 6.948 min. methyl-N-(1,2,3,4-tetrahydroisoquinolin-7- yl)but-2-enamide hydrochloride (Example 4.2, step 2) 1H NMR (DMSO-d6, 400 MHz): δ 1.12 (s, 6H), 3.11 - 3.21 (m, 2H), 3.34 (s, 2H), 3.78 (s, 3H), 4.54 - 4.89 (m, 4H), 5.58 - 5.61 (m, 1H), 5.94 - 6.01 (m, 1H), 6.10 - N-(2-(2-Hydroxy-5-isopropyl-4- 6.24 (m, 1H), 6.49 (s, 1H), 7.07 (s, 1H), methoxybenzoyl)isoindolin-4-yl)-N- 7.23 (d, J= 6.4 Hz, 1H), 7.36 - 7.44 (m, methylacrylamide. Prepared from 2- 2H). hydroxy-5-isopropyl-4-methoxybenzoic LCMS (Method N): 2.217 min, MS: ES+ acid (Example 45 step 3) and N-(isoindolin- 395.2 (M+1). 4-yl)-N-methyl acrylamide TFA salt (TFA HPLC (Method I): 7.650 min. salt of Intermediate W2) 1H NMR (DMSO-d6, 400 MHz): δ 1.10 (d, J= 6.8 Hz, 6H), 2.87 (t, J= 1.2 Hz, 2H), 3.09 - 3.13 (m, 1H), 3.70 (s, br, 2H), 3.77 (s, 3H), 4.67 (s, br, 2H), 5.79 (d, J= N-(6-(2-Hydroxy-5-isopropyl-4- 10.4 Hz, 1H), 6.27 (d, J= 16.8 Hz, 1H), methoxybenzoyl)-5,6,7,8-tetrahydro-1,6- 6.40 - 6.46 (m, 2H), 6.96 (s, 1H), 7.93 (s, naphthyridin-3-yl) acrylamide. Prepared 1H), 8.57 (s, 1H), 9.77 (s, 1H), 10.33 (s, from 2-hydroxy-5-isopropyl-4-methoxy 1H). benzoic acid (Example 45 step 3) and N- LCMS (Method N): 1.82 min, MS: ES+ (5,6,7,8-tetrahydro-1,6-naphthyridin-3-yl) 396.2 (M+1). acrylamide (Example 72 step 2) HPLC (Method H): 5.652 min,
1H NMR (DMSO-d6, 400 MHz): δ 2.03, 2.07 and 2.13 - 2.02 (3 singlets, 9H), 2.83 - 2.94 (m, 4H), 3.18 - 3.25 (2 singlets, 3H), 3.39 - 3.42 (m, 1H), 3.70 - (E)-N-(2-(3-Chloro-6-hydroxy-2- 4.08 (m, 1H), 4.27 - 4.40 (m, 1H), 4.73 - methylbenzoyl)-1,2,3,4- 4.87 (m, 1H), 5.90 - 5.93 (m, 1H), 6.54 - tetrahydroisoquinolin-7-yl)-4- 6.66 (m, 1H), 6.74 - 6.78 (m, 1H), 6.99 - (dimethylamino)-N-methylbut-2-enamide 7.07 (m, 1H), 7.21 - 7.27 (m, 3H), 9.97 Prepared from 3-chloro-6-hydroxy-2- (s, 1H). methylbenzoic acid (Example 73 Step 3) LCMS (Method T): 1.214 min, MS: ES+ and (E)-4-(dimethylamino)-N-methyl-N- 442.0 & 443.5 (M, M+2). (1,2,3,4-tetrahydroisoquinolin-7-yl)but-2- HPLC (Method I): 5.97 min, enamide hydrochloride (Example 4.2 Step 2) 1H NMR (DMSO-d6, 400 MHz): δ 1.28 (s, 9H), 2.05 (s, 6H), 2.91 - 2.94 (m, 4H), 3.24 (s, br, 3H), 3.56 - 3.96 (m, 2H), 4.49 - 4.82 (m, 2H), 5.88 - 5.92 (m, 1H), 6.65 (s, br,1H), 6.83 (d, J= 8.0 Hz, 1H), 7.14 (E)-N-(6-(5-(tert-Butyl)-2-hydroxybenzoyl)- (s, 1H), 7.28 (dd, J= 2.4 Hz, J= 8.8 Hz, 5,6,7,8-tetrahydro-1,6-naphthyridin-3-yl)-4- 1H), 7.55 - 7.73 (m, 1H), 8.30 (s, 1H), (dimethylamino)-N-methylbut-2-enamide 9.67 (s, 1H). Prepared from 5-(tert-butyl)-2- LCMS (Method A4): 1.743 min, MS: ES+ hydroxybenzoic acid (CAS: 16094-31-8) 451.7 (M+1). and (E)-4-(dimethylamino)-N-methyl-N- HPLC (Method H): 4.26 min. (5,6,7,8-tetrahydro-1,6-naphthyridin-3- yl)but-2-enamide hydrochloride (Example 76 Step 3) 1H NMR (DMSO-d6, 400 MHz): δ 1.24 (s, 9H), 2.05 (bs, 6H), 2.84 - 2.88 (m, 4H), 3.21 (s, br, 3H), 3.46 (s, br, 1H), 3.84 (s, br, 1H), 4.45 (bs, 1H), 4.76 (s, 1H), 5.90 (s, br, 1H), 6.60 - 6.62 (m, 1H), (E)-N-(2-(5-(tert-Butyl)-2-hydroxybenzoyl)- 6.83 (d, J= 8.4 Hz, 1H), 7.07 - 7.11 (m, 1,2,3,4-tetrahydroisoquinolin-7-yl)-4- 2H), 7.22 - 7.29 (m, 3H), 9.63 (s, 1H). (dimethylamino)-N-methylbut-2-enamide LCMS (Method N): 1.751 min, MS: ES+ 450.3 (M+1).
Prepared from 5-(tert-butyl)-2-hydroxy HPLC (Method H): 4.02 min. benzoic acid (CAS: 16094-31-8) and (E)-4- (dimethylamino)-N-methyl-N-(1,2,3,4- tetrahydroisoquinolin-7-yl)but-2-enamide hydrochloride (Example 4.2 Step 2) 1H NMR (DMSO-d6, 400 MHz): δ 1.14 - 1.19 (m, 6H), 2.15 and 2.19 (2 singlets, 6H), 3.02 (s, br, 1H), 3.09 (s, br, 1H), 3.21 (m, 1H), 4.69 (s, 2H), 4.86 (d, J= (E)-N-(5-Chloro-2-(4-hydroxy-3- 8.4 Hz, 2H), 6.28 - 6.39 (m, 1H), 6.66 - isopropylbenzoyl) isoindolin-4-yl)-4- 6.84 (m, 2H), 7.20 - 7.49 (m, 4H), 9.86 (dimethylamino) but-2-enamide. Prepared (s, 1H), 9.96 (s, 1H). from (E)-N-(5-chloroisoindolin-4-yl)-4- LCMS (Method N): 1.64 min, MS: ES+ (dimethylamino)but-2-enamide 442.2 (M+1). hydrochloride (Example 70 Step 3) and 4- HPLC (Method H): 3.66 min. hydroxy-3-isopropylbenzoic acid (CAS: 859034-02-9) 1H NMR (DMSO-d6, 400 MHz): δ 1.15 - 1.23 (m, 6H), 1.98 and 2.00 (2 singlets, 6H), 2.85 (d, J= 14.0 Hz, 2H), 3.05 - 3.25 (m, 4H), 4.57 - 4.68 (m, 1H), 4.84 - 4.97 (E)-N-(5-Chloro-2-(4-hydroxy-3- (m, 3H), 5.68 (t, J= 14.4 Hz, 1H), 6.59 - isopropylbenzoyl)isoindolin-4-yl)-4- 6.85 (m, 2H), 7.30 - 7.59 (m, 4H), 9.86 (dimethylamino)-N-methylbut-2-enamide. (bs, 1H). Prepared from (E)-N-(5-Chloroisoindolin-4- LCMS (Method: N): 1.67 min, MS ES+: yl)-4-(dimethylamino)-N-methylbut-2- 456.2, 458.2 (M, M+2). enamide hydrochloride (Example 44 Step HPLC (Method: H): 3.79 min, 2) and 4-hydroxy-3-isopropylbenzoic acid (CAS: 859034-02-9). 1H NMR (DMSO, 400 MHz): δ 2.03 (d, J= 20 Hz, 1H), 2.12 (d, J= 4.8 Hz, 2H), 2.28 (s, br, 6H), 2.63 - 2.72 (m, 2H), 2.82 - 2.87 (m, 2H), 2.96 - 3.01 (m, 1H), 3.05 N-(2-(3-Chloro-6-hydroxy-4-methoxy-2- - 3.06 (m, 1H), 3.12 (s, 2H), 3.39 - 3.40 methylbenzoyl)-6-(2- (m, 2H), 3.63 - 3.66 (m, 1H), 3.79 - 3.81 (dimethylamino)ethoxy)-1,2,3,4- (m, 3H), 4.08 - 4.31 (m, 2H), 4.27 - 4.31
tetrahydroisoquinolin-7-yl)-N- (m, 1H), 4.64 - 4.78 (m, 1H), 5.46 - 5.53 methylacrylamide. Prepared from N-(6-(2- (m, 1H), 5.92 - 6.14 (m, 2H), 6.48 - 6.54 (dimethylamino)ethoxy)-1,2,3,4- (m, 1H), 6.96 - 7.00 (m, 1H), 7.18 (s, tetrahydroisoquinolin-7-yl)-N- 1H). methylacrylamide TFA salt (Example 58 LCMS (Method T): 1.642 min, MS: ES+ Step 10) and 3-chloro-6-hydroxy-4- 502.2, 504.2 (M, M+2). methoxy-2-methylbenzoic acid (Example HPLC (Method K): 5.705 min. 47 Step 3). 1H NMR (MeOD, 400 MHz): δ ppm 1.11 (d, J= 6.8 Hz, 6H), 1.73 (s, 3H), 2.20 (s, 6H), 2.59 - 2.62 (m, 2H), 2.84 - 2.85 (m, 2H), 3.04 (s, 2H), 3.08 - 3.15 (m, 1H), 3.6 - 3.7 (m, 2H), 3.77 (s, br, 3H), 4.04 - 4.11 N-(6-(2-(Dimethylamino)ethoxy)-2-(2- (m, 2H), 4.58 (bs, 2H), 6.47 (s, 1H), 6.92 hydroxy-5-isopropyl-4-methoxybenzoyl)- - 6.95 (m, 2H), 7.12 (s, 1H), 9.80 (s, 1H). 1,2,3,4-tetrahydroisoquinolin-7-yl)-N- LCMS (Method T): 1.724 min, MS: ES+ methylbut-2-ynamide. Prepared from 2- 508.3 (M+1). hydroxy-5-isopropyl-4-methoxybenzoic HPLC (Method H): 4.067 min acid (Example 45 Step 3) and N-(6-(2- (dimethylamino)ethoxy)-1,2,3,4- tetrahydroisoquinolin-7-yl)-N-methylbut-2- ynamide TFA salt (Example 77 step 9) 1H NMR (DMSO-d6, 400 MHz): δ ppm 1.98 - 2.19 (multiple singlets, 9H), 2.84 - 2.91 (m, 2H), 3.02 and 3.15 (2 singlets, 3H), 3.80 (s, 3H), 4.45 - 4.89 (m, 4H), (E)-N-(5-Chloro-2-(3-chloro-6-hydroxy-4- 5.68 (dd, J= 15.2 Hz, 1H), 6.53 (s, 1H), methoxy-2-methylbenzoyl) isoindolin-4-yl)- 6.58 - 6.78 (m, 1H), 7.36 - 7.64 (m, 2H). 4-(dimethyl amino)-N-methylbut-2- LCMS (Method U): 6.00 min, 6.291 min enamide. Prepared from 3-chloro-6- MS: ES+ 492.2 (M+1). hydroxy-4-methoxy-2-methylbenzoic acid HPLC (Method O): 3.96 min, 4.49 min. (Example 47 Step 3) and (E)-N-(5- chloroisoindolin-4-yl)-4-(dimethylamino)-N- methylbut-2-enamide hydrochloride (Example 44 step 2)
Example 49: (E)-N-(2-(5-(tert-Butyl)-2-fluoro-4-hydroxybenzoyl)isoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide Step 1: 5-(tert-Butyl)-2-fluoro-4-hydroxybenzoate

[00834] To a cooled (-5°C) solution of methyl 2-fluoro-4-hydroxybenzoate (CAS: 197507-22-5) (19.0 g, 111.67 mmol, 1.0 eq.) in DCM (285 mL) was added dropwise conc. H2SO4 (30 mL, 558.36 mmol, 5.0 eq.) followed by tert-butanol (53.0 ml, 558.36 mmol, 5.0 eq.) over 1 h. The reaction mixture was stirred at -5°C for 4 h and then at room temperature for 12 h. The resulting mixture was added to ice cold water (300 mL) and extracted with DCM (3 x 300 mL). The combined organic extracts were washed with brine solution (300 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude material was purified by chromatography eluting with a gradient of 0-100% EtOAc (product eluted at 8% ethyl acetate in hexane) yielding methyl 5-(tert-butyl)-2-fluoro-4-hydroxybenzoate as an off-white solid. (9.0 g, Yield: 35 %). [00835]
1H NMR (DMSO-d6, 400 MHz): δ 1.33 (s, 9H), 3.78 (s, 3H), 6.64 (d, J= 12.4 Hz, 1H), 7.69 (d, J= 8.0 Hz, 1H), 10.85 (s, 1H). [00836] LCMS (Method N): 2.248 min, MS: ES+ 225.01 (M-1). Step 2: 5-(tert-Butyl)-2-fluoro-4-hydroxybenzoic acid
[00837] To a stirred solution of methyl 5-(tert-butyl)-2-fluoro-4-hydroxybenzoate (Step 1) (9.5 g, 42.00 mmol, 1.0 eq.) in MeOH: water (1:1) (190 mL) was added LiOH.H
2O (17.6 g, 420.00 mmol, 10.0 eq.) and the reaction mixture was heated to 65°C for 2 h. The resulting mixture was concentrated under reduced pressure to remove MeOH and the remaining was acidified to pH 3-4 with 2N HCl (250 mL). The mixture was extracted with ethyl acetate (3 x 300 mL) and the combined organic extracts were washed with brine solution (2 x 250 mL), dried over Na
2SO
4, filtered, and concentrated under reduced pressure to yield 5-(tert-butyl)-2-fluoro-4- hydroxybenzoic acid as an off-white solid (9.0 g, Yield: Quantitative).
[00838]
1H NMR (DMSO-d6, 400 MHz): δ 1.33 (s, 9H), 6.61 (d, J= 12.4 Hz, 1H), 7.69 (d, J= 8.8 Hz, 1H), 10.71 (s, 1H), 12.65 (bs, 1H). [00839] LCMS (Method N): 1.912 min, MS: 211.01 (M-1). Step 3: (E)-N-(2-(5-(tert-Butyl)-2-fluoro-4-hydroxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N- methylbut-2-enamide
[00840] The title compound was prepared from 5-(tert-butyl)-2-fluoro-4-hydroxybenzoic acid (Step 2) and (E)-4-(dimethylamino)-N-(isoindolin-4-yl)-N-methylbut-2-enamide hydrochloride (Intermediate S) analogously to Example 1 Step 1. [00841]
1H NMR (DMSO-d6, 400 MHz): δ 1.36 (s, 9H), 2.06 (s, 6H), 2.90 (bs, 2H), 3.10 - 3.23 (m, 3H), 4.52 - 4.71 (m, 2H), 4.85 - 4.90 (m, 2H), 5.85 (bs, 1H), 6.59 - 6.68 (m, 2H), 7.20 (d, J= 8.0 Hz, 2H), 7.32 - 7.41 (m, 2H), 10.08 (s, 1H). [00842] LCMS (Method N): 1.720 min, MS: ES+ 454.03 (M+1). [00843] HPLC (Method H): 4.35 min. Prep. HPLC purification: [00844] Chromatographic separation and isolation were conducted with an FC-01 flash purification system (Buchi model C810); column REDISEP GOLD C18100g 50µm; compound eluted with: Mobile Phase A: 5 mM ammonium bicarbonate in water, Mobile Phase B: Acetonitrile with a gradient of T = initial (100% A, 0% B); gradient to T = 3.00 min (100% A, 0% B); T = 20.00 min (70% A, 30% B) gradient to T = 20.01 min (0% A, 100% B); T = 23.00 min (100% A, 100% B); gradient to T = 23.01 min (100% A, 0% B); Flow rate= 70ml/min; analysis time 23.01 min. Example 50: (E)-N-(2-(5-(tert-Butyl)-2-hydroxy-4-methoxybenzoyl)-1,2,3,4- tetrahydroisoquinolin-7-yl)-4-(dimethylamino)-N-methylbut-2-enamide Step 1: tert-Butyl (E)-7-(4-(dimethylamino)-N-methylbut-2-enamido)-3,4-dihydroisoquinoline- 2(1H)-carboxylate
[00845] To a stirred solution of (E)-4-(dimethylamino) but-2-enoic acid (CAS:848133-35-7) (0.5 g, 3.03 mmol, 1.0 eq.) in DCM (5 mL) were added tert-butyl 7-(methylamino)-3,4- dihydroisoquinoline-2(1H)-carboxylate (Intermediate I) (0.397 g, 1.15 mmol, 0.5 eq.) and pyridine (0.97 mL, 12.12 mmol, 4.0 eq.) at room temperature. After stirring for 5 mins, the mixture was cooled to 0°C and treated with POCl
3 (0.417 mL, 4.54 mmol, 1.5 eq.). The reaction mixture was stirred at room temperature for 1 h. The resulting mixture was diluted with sat. NaHCO
3 solution (50 mL) and extracted with EtOAc (3 x 30 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted at 7% MeOH: DCM) yielding tert-butyl (E)-7-(4- (dimethylamino)-N-methylbut-2-enamido)-3,4-dihydroisoquinoline-2(1H)-carboxylate as a yellow liquid (0.205 g, Yield: 28%). [00846]
1H NMR (DMSO-d6, 400 MHz): δ 1.41 (s, 9H), 2.78 (s, 2H), 3.15 (s, 6H), 3.28 (s, 3H), 3.50 - 3.64 (m, 5H), 4.12 (s, br, 2H), 4.48 (s, 2H), 6.06 (d, J=13.6 Hz, 1H), 6.59 - 6.62 (m, 1H), 7.06 - 7.12 (m, 2H), 7.23 - 7.24 (m, 1H). [00847] LCMS (Method N): 1.720 min, MS ES+: 374 (M+1). Step 2: (E)-4-(Dimethylamino)-N-methyl-N-(1,2,3,4-tetrahydroisoquinolin-7-yl) but-2-enamide hydrochloride

[00848] To a cooled (0°C) solution of tert-butyl (E)-7-(4-(dimethylamino)-N-methylbut-2- enamido)-3,4-dihydroisoquinoline-2(1H)-carboxylate (Step 1) (0.20 g, 0.548 mmol, 1.0 eq) in DCM (2.5 mL) was added dropwise 4M HCl in dioxane (1.3 mL) and the reaction mixture was stirred at room temperature for 1 h. The resulting mixture was concentrated under reduced pressure, triturated with diethyl ether (20 mL) and dried under high vacuum to yield (E)-4- (dimethylamino)-N-methyl-N-(1,2,3,4-tetrahydroisoquinolin-7-yl) but-2-enamide hydrochloride as a yellow solid (0.198 g, Yield: quantitative) which was used directly in the next step. [00849] LCMS (Method N): 0.257 min, MS ES+: 273.9 (M+1).
Step 3: (E)-N-(2-(5-(tert-Butyl)-2-hydroxy-4-methoxybenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)- 4-(dimethylamino)-N-methylbut-2-enamide

[00850] The title compound was prepared from (E)-4-(dimethylamino)-N-methyl-N-(1,2,3,4- tetrahydroisoquinolin-7-yl) but-2-enamide hydrochloride (Step 2) and 5-(tert-butyl)-2-hydroxy-4- methoxybenzoic acid (Intermediate N) analogously to Example 1 Step 1. [00851]
1H NMR (DMSO-d6, 400 MHz): δ 1.28 (s, 9H), 2.04 (s, 6H), 2.87 (bs, 4H), 3.20 (s, 3H), 3.63 - 3.67 (m, 2H), 3.79 (s, 3H), 4.65 (bs, 2H), 5.87 - 5.89 (m, 1H), 6.50 (s, 1H), 6.56 - 6.63 (m, 1H), 6.98 (s, 1H), 7.06 - 7.13 (m, 2H), 7.24 (d, J=7.2 Hz, 1H), 9.83 (s, 1H). [00852] LCMS (Method N): 1.850 min, MS: ES+ 479.9 (M+1). [00853] HPLC (Method H): 4.65 min. Prep. HPLC purification method: [00854] Chromatographic separation and isolation were conducted with an FC-01 flash purification system (Buchi model C810); column REDISEP GOLD C18100g 50µm; compound eluted with, Mobile Phase A: 5 mM ammonium bicarbonate IN WATER, Mobile Phase B: Acetonitrile with a gradient of T = 0.00 min (100% A, 00% B); gradient to T = 3.00 min (90% A, 10% B); T = 20.0 min (50% A, 50% B) gradient to T = 20.01 min (0% A, 100% B); T = 23.00 min (100% A, 0% B); gradient to T = 23.01 min (100% A, 0% B); Flow rate= 70 ml/min; analysis time 23.01 min. Example 51: N-(2-(5-(tert-Butyl)-2-hydroxy-4-methoxybenzoyl)-1,2,3,4- tetrahydroisoquinolin-7-yl)-N-methylacrylamide Step 1: tert-Butyl 7-(N-methylacrylamido)-3,4-dihydroisoquinoline-2(1H)-carboxylate

[00855] The title compound was prepared from tert-butyl 7-(methylamino)-3,4- dihydroisoquinoline-2(1H)-carboxylate (Intermediate I) and acryloyl chloride analogously to Example 36 Step 5. [00856]
1H NMR (DMSO-d6, 400 MHz): δ 1.43 (s, 9H), 2.80 (bs, 2H), 3.22 (s, 3H), 3.57 (bs, 2H), 4.51 (s, 2H), 5.55 (d, J= 8 Hz, 1H), 6.06 - 6.17 (m, 2H), 7.01 (d, J= 7.2 Hz, 1H), 7.14 (s, 1H), 7.24 (d, J= 7.6 Hz, 1H). [00857] LCMS (Method N): 2.166 min, MS ES+: 316.9 (M+1). Step 2: N-Methyl-N-(1,2,3,4-tetrahydroisoquinolin-7-yl) acrylamide TFA salt
[00858] The title compound was prepared from tert-butyl 7-(N-methylacrylamido)-3,4- dihydroisoquinoline-2(1H)-carboxylate (Step 1) and TFA analogously to Example 36 Step 6. [00859]
1H NMR (DMSO-d6, 400 MHz): δ 3.02 (s, 2H), 3.22 (s, 3H), 3.32 - 3.40 (m, 2H), 4.28 (s, 2H), 5.58 (d, J= 9.2 Hz, 1H), 6.04 (bs, 1H), 6.15 - 6.19 (m, 1H), 7.19 - 7.33 (m, 3H), 9.11 (s, 1H). [00860] LCMS (Method O): 1.485 min, MS ES+: 217.2 (M+1) Step 3: N-(2-(5-(tert-Butyl)-2-hydroxy-4-methoxybenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- methylacrylamide
[00861] The title compound was prepared from 5-(tert-butyl)-2-hydroxy-4-methoxybenzoic acid (Intermediate N) and N-methyl-N-(5,6,7,8-tetrahydro-2,6-naphthyridin-3-yl) acrylamide TFA salt (Step 2) analogously to Example 1 Step 1. [00862]
1H NMR (DMSO-d6, 400 MHz): δ 1.28 (s, 9H), 2.86 (s, 2H), 3.21 (s, 3H), 3.63 (bs, 2H), 3.79 (s, 3H), 4.67 (s, 2H), 5.56 (d, J= 8.4 Hz, 1H), 6.13 - 6.17 (m, 2H), 6.50 (s, 1H), 6.97 (s, 1H), 7.07 (d, J= 6.8 Hz, 1H), 7.16 (bs, 1H), 7.24 (d, J= 6.8 Hz, 1H), 9.82 (s, 1H). [00863] LCMS (Method N): 2.32 min, MS: ES+ 423.1 (M+1). [00864] HPLC (Method H): 8.64 min.
Example 52: N-(2-(3-Chloro-2-fluoro-6-hydroxy-4-methoxybenzoyl)isoindolin-4-yl)-N- methylacrylamide Step 1: 3-Chloro-2-fluoro-6-hydroxy-4-methoxybenzoic acid

[00865] To a cooled (0°C) solution of 3-chloro-2-fluoro-4,6-dimethoxybenzoic acid (Example 32 Step 2) (2 g, 8.50 mmol, 1 eq.) in DCM (10 mL) was added dropwise BBr3 (1M in DCM) (10 mL). The reaction mixture was allowed to warm to room temperature and stirred for 16 h. The resulting mixture was concentrated under vacuum and the crude residue was diluted with ice-cold (50 mL) causing precipitation of a solid. The solid was collected by filtration and dried under high vacuum to yield 3-chloro-2-fluoro-6-hydroxy-4-methoxybenzoic acid as an off-white solid (1.2 g, Yield: 63.0%). [00866]
1H NMR (DMSO-d6, 400 MHz): δ 3.91 (s, 3H), 6.59 (d, J= 1.6 Hz, 1H), 12.18 (bs, 2H). [00867] LCMS (Method A): 1.800 min, MS: ES- 219 (M-1). Step 2: N-(2-(3-Chloro-2-fluoro-6-hydroxy-4-methoxybenzoyl)isoindolin-4-yl)-N- methylacrylamide
[00868] The title compound was prepared from 3-chloro-2-fluoro-6-hydroxy-4-methoxybenzoic acid (Step 1) and N-(isoindolin-4-yl)-N-methyl acrylamide TFA salt (TFA salt of Intermediate W2) analogously to Example 1 Step 1. [00869]
1H NMR (DMSO-d6, 400 MHz): δ 3.10 and 3.22 (2 singlets, 3H), 3.84 (s, 3H), 4.43 - 4.54 (m, 1H), 4.62 - 4.87 (m, 3H), 5.54 - 5.61 (m, 1H), 5.87 - 6.01 (m,1H), 6.09 - 6.24 (m, 1H), 6.50 (d, J= 11.6 Hz, 1H), 7.22 - 7.26 (m, 1H), 7.32 - 7.33 (m, 1H), 7.40 - 7.46 (m, 1H), 10.57 (bs, 1H). [00870] LCMS (Method N): 1.908 min, MS: ES+ 404.8 & 406.6 (M+1 & M+2).
[00871] HPLC (Method H): 6.58 min. Prep. HPLC purification method: [00872] Chromatographic separation and isolation was conducted with Waters 2545 binary pump with Waters 2489 UV detector with waters Acquity QDA detector; column Waters X-Bridge C18 (250mm x 19mm x 5µm); compound was eluted with, Mobile Phase A : 5mM ammonium bicartbonate + 0.05% NH
3 in water, Mobile Phase B: acetonitrile:water:THF(80:20:10), gradient of T= 0.01 min (72% A, 28% B); gradient to T= 22.00 min (61% A, 39% B); T= 22.01 min (0% A, 100% B) gradient to T= 25.00 min (0% A, 100% B); gradient to T= 25.01 min (72% A, 28% B); gradient to T= 30.00 min (72% A, 28% B); Flow rate= 10 ml/min; analysis time 30.00 min Example 53: N-(5-Chloro-2-(3-chloro-6-hydroxy-4-methoxy-2-methylbenzoyl) isoindolin-4- yl)-N-methyl acrylamide Step 1: tert-Butyl 5-chloro-4-(N-methylacrylamido) isoindoline-2-carboxylate
[00873] The title compound was prepared from tert-butyl 5-chloro-4-(methylamino)isoindoline-2- carboxylate (Example 24 Step 1) and acryloyl chloride analogously to Example 36 Step 5. [00874]
1H NMR (DMSO-d6, 400 MHz): δ 1.44 (s, 9H), 3.12 (s, 3H), 4.35 (t, J= 15.6 Hz, 1H), 4.64 (d, J= 8.4 Hz, 3H), 5.60 (d, J= 10.0 Hz, 1H), 5.85 - 5.93 (m, 1H), 6.21 (dd, J= 2.0 Hz, 1H), 7.43 (t, J= 6.0 Hz, 1H), 7.58 (d, J= 8.0 Hz, 1H). [00875] LCMS (Method N): 2.261 min, MS: ES- 280.70 (M-56). Step 2: N-(5-Chloroisoindolin-4-yl)-N-methyl acrylamide TFA salt
[00876] The title compound was prepared from tert-butyl 5-chloro-4-(N-methylacrylamido) isoindoline-2-carboxylate (Step 1) and TFA analogously to Example 36 Step 6 and was used directly in the next step. [00877] LCMS (Method O): 1.27 min, MS: ES+ 236.8 (M+1). Step 3: N-(5-chloro-2-(3-chloro-6-hydroxy-4-methoxy-2-methylbenzoyl) isoindolin-4-yl)-N-methyl acrylamide
[00878] The title compound was prepared from 3-chloro-6-hydroxy-4-methoxy-2-methylbenzoic acid (Example 47 Step 3) and N-(5-chloroisoindolin-4-yl)-N-methyl acrylamide TFA salt (Step 2) analogously to Example 1 Step 1. [00879] HT
1H NMR (DMSO-d6, 400 MHz): δ 2.14 - 2.21 (m, 3H), 3.05 and 3.22 (2 singlets, 3H), 3.83 (s, 3H), 4.40 - 4.91 (m, 4H), 5.58 - 5.63 (m, 1H), 5.86 - 5.98 (m, 1H), 6.14 - 6.26 (m, 1H), 6.52 - 6.56 (m, 1H), 7.36 - 7.60 (m, 2H), 9.09 (s, 1H). [00880] LCMS (Method U): 5.926 min, MS: ES+ 435.1 (M+1). [00881] HPLC (Method O): 5.924 min. Prep. HPLC purification method: [00882] Chromatographic separation and isolation were conducted on a PHP-08-Waters 2545 quaternary system with Waters 2489 UV Detector; column Waters X-Bridge Prep C8 (250mm x 19mm 5µm); compound was eluted with, Mobile Phase A: 5mM ammonium bicarbonate in water Mobile Phase B: acetonitrile:water (80:20) with a gradient of T= 0.0 min (60% A, 40% B); gradient to T= 30.00 min (47% A, 53% B); T= 30.01 min (0% A, 100% B); gradient to T= 34.00 min (0% A, 100% B); T= 34.01 min (60% A, 40% B); gradient to T= 36.00 min (60% A, 40% B); Flow rate= 10 ml/min; analysis time 36.00 min. Example 54: (E)-4-(Dimethylamino)-N-(2-(2-hydroxy-5-isopropyl-4-methoxybenzoyl)-5- methylisoindolin-4-yl)-N-methylbut-2-enamide Step 1: 4-Bromo-3-hydroxy-5-methylisoindolin-1-one

[00883] To a cooled (-78°C) solution of LDA (2M in THF) (14.03 mL, 28.06 mmol, 1.1 eq) in THF (15mL) was added 3-bromo-4-methylbenzonitrile (CAS: 42872-74-2) (5.0 g, 25.5 mmol, 1.0 eq.) in THF (20 mL) and the mixture was stirred for 5 mins. Ethyl formate (2.77 mL, 1.35 eq.) was added dropwise and stirring continued at -78°C for 15 mins. The resulting mixture was diluted with ice cold water (100 mL), acidified with dilute HCl (50 mL) and extracted with EtOAc (3 x 200 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted in 5% MeOH in DCM) yielding 4-bromo-3-hydroxy-5-methylisoindolin-1-one as an off-white solid (2.0 g, 32% yield). [00884]
1H NMR (DMSO-d6, 400 MHz): δ 2.50 (s, 3H) 5.79 (d, J= 9.2 Hz, 1H), 6.37 (d, J= 9.2 Hz, 1H, D
2O exchangeable), 7.50 (s, br, 2H), 9.00 (s, 1H) [00885] LCMS (Method: N): 1.601 min, MS ES+: 243.62 (M+1). Step 2: 4-Bromo-5-methylisoindoline
[00886] To a stirred solution of sodium borohydride (0.66 g, 17.355 mmol, 6.0 eq.) in THF (7.0 mL) was added boron trifluoride etherate (4.4 mL, 17.35 mmol, 6.0 eq.) at room temperature and stirred for 5 mins.4-Bromo-3-hydroxy-5-methylisoindolin-1-one (Step 1) (0.7 g, 2.89 mmol, 1.0 eq) was added and the reaction mixture was heated to 70°C and stirred for 16 h. The resulting mixture was poured in 1M NaOH solution (50 mL) and extracted with EtOAc (3 x 100 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum. The crude material was triturated with hexane (20 mL) in a dry ice bath and concentrated under high vacuum to yield 4-bromo-5-methylisoindoline as a light green solid. (0.6 g, yield: 51%). The crude material was used without further purification. [00887] LCMS (Method: N): 1.508 min, MS ES+: 213.79 (M + 1). Step 3: tert-Butyl 4-bromo-5-methylisoindoline-2-carboxylate
[00888] To a solution of 4-bromo-5-methylisoindoline (Step 2)(0.6 g, 2.83 mmol, 1.0 eq.) in THF (6.0 mL) were added 1M NaOH solution in water (6.6 mL) followed by Boc anhydride (0.9 mL, 3.67 mmol, 1.3 eq.) and the reaction mixture was stirred at room temperature for 16 h. The resulting mixture poured into water (50 mL) and extracted with EtOAc (2 x 100 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted in 6% EtOAc in hexane). The pure fractions were combined and concentrated under reduced pressure to yield tert-butyl 4-bromo-5-methylisoindoline-2-carboxylate as an off-white solid (0.3 g, 34% yield). [00889]
1H NMR (DMSO-d6, 400 MHz): δ 1.45 (s, 9H), 2.34 (s, 3H), 4.51 (d, J= 9.6 Hz, 2H), 4.65 (d, J= 10.4 Hz, 2H), 7.23 - 7.27 (m, 2H) [00890] LCMS (Method: N): 2.889 min, MS ES+: 255.68 (M - 56). Step 4: tert-Butyl 5-methyl-4-(methylamino)isoindoline-2-carboxylate
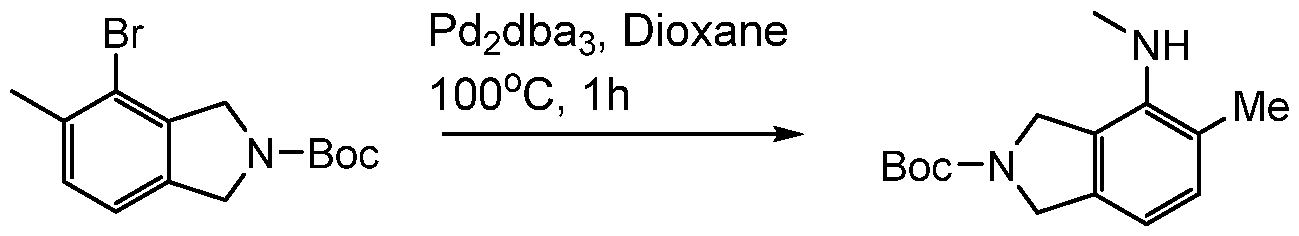
[00891] Performed in 2 parallel batches, each of 0.3 g scale: To a stirred solution of tert-butyl 4- bromo-5-methylisoindoline-2-carboxylate (Step 3) (0.3 g, 0.96 mmol, 1.0 eq.) in dioxane (3.0 mL) was added NaOtBu (0.23 g, 2.40 mmol, 2.5 eq.) and the mixture was purged with N2 gas for 15 minutes. Methyl amine (2M in THF) (0.96 mL, 2.11 mmol, 2.2 eq.), Xphos (0.02 g, 0.057 mmol, 0.06 eq.), and Pd2dba3 (0.11 g, 0.038 mmol, 0.04 eq.) were added and the reaction mixture was heated to 100°C for 1 h. The resulting mixture poured in water (30 mL) and extracted with EtOAc (2 x 20 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted in 29% EtOAc in hexane) to yield tert-butyl 5-methyl-4-(methylamino)isoindoline-2-carboxylate as an brown sticky solid (0.27 g, 64% yield) [00892]
1H NMR (DMSO-d6, 400 MHz): δ 1.45 (s, 9H), 2.07 (s, 3H), 2.85 - 2.88 (m, 3H), 4.43 (d, J= 8.4 Hz, 2H), 4.61 - 4.65 (m, 1H, D
2O exchangeable), 4.73 (s, 2H), 6.49 (dd, J= 7.6 Hz, J= 12.4 Hz, 1H), 6.89 (d, J= 7.2 Hz, 1H)
[00893] LCMS (Method: N): 1.997 min, MS ES+: 206.85 (M - 56). Step 5: tert-Butyl (E)-4-(4-(dimethylamino)-N-methylbut-2-enamido)-5-methylisoindoline-2- carboxylate
[00894] Performed in 3 parallel batches, each of 1 g scale: To a cooled (0°C) solution of (E)-4- (dimethylamino)but-2-enoic acid hydrochloride (CAS: 848133-35-7) (1.0 g, 6.037 mmol, 1.0 eq.) and tert-butyl 5-methyl-4-(methylamino)isoindoline-2-carboxylate (Step 4) (0.79 g, 3.018 mmol, 0.5 eq.) in DCM (10.0 mL) was added pyridine (1.95 mL, 24.148 mmol, 4.0 eq.) and the mixture was stirred for 5 mins. POCl
3 (0.85 mL, 9.055 mmol, 1.5 eq.) was added dropwise over 5 mins and the reaction mixture was stirred at room temperature for 1 h. The resulting mixture was concentrated under reduced pressure and purification of the crude material by reverse phase chromatography eluting with a gradient of 0-100% ACN: water (product was eluted at 25% ACN: water) followed by lyophilization of the pure fractions yielded tert-butyl (E)-4-(4-(dimethylamino)- N-methylbut-2-enamido)-5-methylisoindoline-2-carboxylate as brown sticky solid. (1.28 g, Yield: 30%). [00895]
1H NMR (DMSO-d6, 400 MHz): δ 1.44 (s, 9H), 2.13 (d, J= 4.8 Hz, 3H), 3.11 and 3.16 (2 singlets, 3H), 4.30 - 4.36 (m, 1H), 4.54 - 4.63 (m, 3H), 5.86 - 5.90 (m, 1H), 6.70 - 6.73 (m, 1H), 7.28 - 7.33 (m, 2H). [00896] LCMS (Method N): 1.698 min, MS: ES+ 374.0 (M+1) Step 6: (E)-4-(Dimethylamino)-N-methyl-N-(5-methylisoindolin-4-yl)but-2-enamide hydrochloride
[00897] The title compound was prepared from tert-butyl (E)-4-(4-(dimethylamino)-N-methylbut- 2-enamido)-5-methylisoindoline-2-carboxylate (Step 5) and 4M HCl in dioxane analogously to Example 44 Step 2 and wasused directly in the next step [00898] LCMS (Method O): 1.26 min, MS: ES+ 274.3 (M+1)
Step 7: (E)-4-(Dimethylamino)-N-(2-(2-hydroxy-5-isopropyl-4-methoxybenzoyl)-5- methylisoindolin-4-yl)-N-methylbut-2-enamide
[00899] The title compound was prepared from 2-hydroxy-5-isopropyl-4-methoxybenzoic acid (Example 45 Step 3) and (E)-4-(dimethylamino)-N-methyl-N-(5-methylisoindolin-4-yl)but-2- enamide hydrochloride (Step 6) analogously to Example 1 Step 1. [00900]
1H NMR (DMSO-d6, 400 MHz): δ 1.11 - 1.15 (m, 6H), 1.99 (s, 6H), 2.14 (s, br, 3H), 2.85 (s, br, 2H), 3.02 (s, br, 1H), 3.09 - 3.13 (m, 3H), 3.78 (s, 3H), 4.52 - 4.55 (m, 1H), 4.70 - 4.85 (m, 3H), 5.66 (t, J= 14.4 Hz, 1H), 6.48 (d, J= 9.2 Hz, 1H), 6.59 - 6.72 (m, 1H), 7.06 (s, 1H), 7.25 - 7.34 (m, 2H), 10.16 (bs, 1H). [00901] LCMS (Method A): 1.819 min, MS: ES+ 465.9 (M+1). [00902] HPLC (Method H): 4.60 min. Prep. HPLC purification method of analysis: [00903] Chromatographic separation and isolation were conducted with a Waters 2545 binary pump with Waters 2489 UV detector with waters Acquity QDA detector UV Detector purification system with UV detector; column Waters X-Bridge C18 (250mm x 19mm x 5µm); compound eluted with, Mobile Phase A: 5mM ammonium bicarbonate + 0.05% NH
3 in Merck water, Mobile Phase B: ACETONITRILE with a gradient of T = 0.01 (56% A, 44% B); gradient to T = 26.00 min (56% A, 44% B); T = 26.01 min (0% A, 100% B) gradient to T = 29.00 min (0% A, 100% B); T = 29.01 min (56% A, 44% B); gradient to T = 35.00 min (56% A, 44% B); Flow rate= 12ml/min; analysis time 35.00 min. Example 55: N-(2-(4-Fluoro-2-hydroxy-5-isopropylbenzoyl)-6-methyl-1,2,3,4- tetrahydroisoquinolin-7-yl)-N-methylacrylamide Step 1: tert-Butyl 6-methyl-7-(methylamino)-3,4-dihydroisoquinoline-2(1H)-carboxylate
[00904] To a stirred solution of tert-butyl 6-chloro-7-(methylamino)-3,4-dihydroisoquinoline- 2(1H)-carboxylate (Example 56 Step 5) (1 g, 3.36 mmol, 1 eq) in toluene (10 mL) were added methyl boronic acid (5.04 g, 84.23 mmol, 25 eq), Cs
2CO
3 (5.4 g, 16.84 mmol, 5 eq), and RuPhos PdG2 (0.39 g, 0.505 mmol, 0.15 eq) and the reaction mixture was heated to 100°C for 16 h. The resulting mixture was diluted with water (35 mL) and extracted with ethyl acetate (2 x 40 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted at 8% EtOAc: hexane) to yield tert-butyl 6-methyl-7-(methylamino)-3,4-dihydroisoquinoline-2(1H)- carboxylate as off-white solid (0.8g, Yield: 85%). [00905]
1H NMR (DMSO-d6, 400 MHz): δ 1.42 (s, 9H), 2.01 (s, 3H), 2.57 (t, J= 5.2 Hz, J= 10.08 Hz, 2H), 2.68 (d, J= 5.2 Hz, 2H), 3.48 (m, 2H), 4.38 (s, br, 2H), 4.88 (d, J= 4.8 Hz, 2H), 6.19 (s, 1H), 6.72 (s, 1H). [00906] LCMS (Method N): 2.07 min, 100%, 220nm, MS: ES+ 276.9 (M+1). Step 2: tert-Butyl 6-methyl-7-(N-methylacrylamido)-3,4-dihydroisoquinoline-2(1H)-carboxylate
[00907] The title compound was prepared from tert-butyl 6-methyl-7-(methylamino)-3,4- dihydroisoquinoline-2(1H)-carboxylate (Step 1) and acryloyl chloride analogously to Example 36 Step 5. [00908]
1H NMR (DMSO-d6, 400 MHz): δ 1.42 (s, 9H), 2.06 (s, 3H), 2.76 (t, J= 5.6 Hz, J= 11.2 Hz, 2H), 3.12 (s, 3H), 3.55 (m, 2H), 4.46 (s, 2H), 5.53 (dd, J= 2 Hz, 1H), 5.83 - 5.90 (m, 1H), 6.16 (dd, J= 2 Hz, 1H) 7.08 (s, 1H), 7.14 (s, 1H). [00909] LCMS (Method N): 2.25 min, MS: ES+330.7 (M+1). Step 3: N-Methyl-N-(6-methyl-1,2,3,4-tetrahydroisoquinolin-7-yl) acrylamide TFA salt
[00910] The title compound was prepared from tert-butyl 6-methyl-7-(N-methylacrylamido)-3,4- dihydroisoquinoline-2(1H)-carboxylate (Step 2) and TFA analogously to Example 36 Step 6. [00911]
1H NMR (DMSO-d6, 400 MHz): δ 2.09 (s, 3H), 2.98 (t, J= 6 Hz, J= 12.4 Hz, 2H), 3.12 (s, 3H), 3.35 - 3.41 (m, 2H), 4.25 (d, J= 5.2 Hz, 2H), 5.55 (dd, J= 2 Hz, 1H), 5.80 - 5.87 (m, 1H), 6.17 (dd, J= 2.4 Hz, 1H) 7.15 (s, 1H), 7.23 (s, 1H) 9.07 (s, 1H). Step 4: N-(2-(4-Fluoro-2-hydroxy-5-isopropylbenzoyl)-6-methyl-1,2,3,4-tetrahydroisoquinolin-7- yl)-N-methylacrylamide
[00912] The title compound was prepared from 4-fluoro-2-hydroxy-5-isopropylbenzoic acid (Example 40 Step 5) and N-methyl-N-(6-methyl-1,2,3,4-tetrahydroisoquinolin-7-yl) acrylamide TFA salt (Step 3) analogously to Example 1 Step 1. [00913]
1H NMR (DMSO-d6, 400 MHz): δ 1.16 (d, J= 6.8 Hz, 6H), 2.06 (s, 3H), 2.81 (s, br, 2H), 3.02 - 3.08 (m, 1H), 3.12 (s, br, 3H), 3.45 (s, 1H), 3.73 - 3.81 (m, 1H), 4.41 (s, 1H), 4.72 (s, 1H), 5.53 (d, J= 10 Hz, 1H), 5.88 (m, 1H), 6.15 (d, J= 16.4 Hz, 1H), 6.62 (d, J= 12.4 Hz, 1H), 6.98 - 7.15 (m, 3H), 10.12 (s, 1H). [00914] LCMS (Method N): 2.19 min, MS: ES+ 410.81 (M+1). [00915] HPLC (Method H): 7.93 min. Example 56: N-(6-Chloro-2-(2-hydroxy-5-isopropyl-4-methoxybenzoyl)-1,2,3,4- tetrahydroisoquinolin-7-yl)-N-methylacrylamide Step 1: 6-Chloro-7-nitro-1,2,3,4-tetrahydroisoquinoline
[00916] To a cooled (0 ˚C) solution of 6-chloro-1,2,3,4-tetrahydroisoquinoline (CAS: 33537-99- 4) (10 g, 59.52 mmol, 1 eq) in conc. H
2SO
4 (60 mL) was added dropwise a solution of KNO
3 (6.01 g, 59.52 mmol, 1 eq) in conc. H
2SO
4 (60 mL) over 10 mins and stirring continued at 0°C for 15 mins. The resulting mixture was diluted with ice cold water (500 mL) and carefully neutralized with sat. NaOH solution at 0°C. The mixture was extracted with EtOAc (3 x 500 mL) and the combined organic extracts were dried over Na
2SO
4, filtered and concentrated under vacuum. The crude material was triturated with n-hexane (50 mL) followed by high vacuum drying to yield 6-chloro-7-nitro-1,2,3,4-tetrahydroisoquinoline as yellow solid (10 g, Yield: 78%). The crude material was used in next step without purification. [00917]
1H NMR (DMSO-d6, 400 MHz): δ 2.74 (t, J= 5.6 Hz, J= 11.2 Hz, 2H), 2.90 - 2.94 (m, 2H), 3.86 (s, 2H), 7.49 (s, 1H), 7.80 (s, 1H). [00918] LCMS (Method N): 1.44 min, MS: ES+ 212.8 (M+1). Step 2: tert-Butyl 6-chloro-7-nitro-3,4-dihydroisoquinoline-2(1H)-carboxylate
[00919] To a cooled (0 ˚C) solution of 6-chloro-7-nitro-1,2,3,4-tetrahydroisoquinoline (Step 1) (10 g, 46.94 mmol, 1 eq) in DCM (100 mL) was added dropwise TEA (14.22 g, 140.84 mmol, 3 eq) and the mixture was stirred for 5 mins. Boc anhydride (20.46 g, 93.89 mmol, 2 eq) was added at 0°C and the reaction mixture was allowed to warm to room temperature and stirred for 30 mins. The resulting mixture was diluted with ice cold water (200 mL) and extracted with DCM (3 x 200 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted at 10% EtOAc: hexane) to yield tert-butyl 6-chloro-7-nitro-3,4-dihydroisoquinoline-2(1H)-carboxylate as yellow liquid (12 g, Yield: 81%). [00920]
1H NMR (DMSO-d6, 400 MHz): δ 1.43 (s, 9H), 2.86 (t, J= 5.6 Hz, J= 11.6 Hz, 2H), 3.54 - 3.59 (m, 2H), 4.57 (s, 2H), 7.62 (s, 1H), 8.01 (s, 1H). [00921] LCMS (Method N): 2.49 min, MS: ES+ 310.8 (M+1).
Step 3: tert-Butyl 7-amino-6-chloro-3,4-dihydroisoquinoline-2(1H)-carboxylate
[00922] The title compound was prepared from tert-butyl 6-chloro-7-nitro-3,4- dihydroisoquinoline-2(1H)-carboxylate (Step 2), Fe powder and NH4Cl analogously to Example 38 Step 1. [00923] LCMS (Method N): 2.31 min, MS: ES+ 182.8 (M-100). Step 4: tert-Butyl 6-chloro-7-formamido-3,4-dihydroisoquinoline-2(1H)-carboxylate
[00924] A solution of tert-butyl 7-amino-6-chloro-3,4-dihydroisoquinoline-2(1H)-carboxylate (Step 3) (0.3 g, 1.06 mmol, 1eq) in ethyl formate (3 mL) was heated to 80°C and stirred for 16 h. The resulting mixture was concentrated under vacuum yielding tert-butyl 6-chloro-7-formamido- 3,4-dihydroisoquinoline-2(1H)-carboxylate (0.3 g, Yield: 90%). The crude material was used in next step without purification. [00925] LCMS (Method N): 2.14 min, MS: ES+ 210.9 (M-100). Step 5: tert-Butyl 6-chloro-7-(methylamino)-3,4-dihydroisoquinoline-2(1H)-carboxylate

[00926] To a cooled (0 ˚C) solution of tert-butyl 6-chloro-7-formamido-3,4-dihydroisoquinoline- 2(1H)-carboxylate (Step 4) (0.3 g, 0.96 mmol, 1eq) in THF (3 mL) was added dropwise LAH (1M in THF) (1.93 mL, 1.93 mmol, 2eq) and the reaction mixture was stirred at room temperature for 2 h. The resulting mixture was diluted with NH4Cl solution (20 mL) and extracted with EtOAc (2 x 25 mL). The combined organic extracts were dried over Na2SO4 and concentrated under vacuum. The crude material was purified using by chromatography on silica (product eluted at
10% EtOAc: hexane) to yield tert-butyl 6-chloro-7-(methylamino)-3,4-dihydroisoquinoline-2(1H)- carboxylate as a colourless liquid (0.15 g, Yield: 52%). [00927]
1H NMR (DMSO-d6, 400 MHz): δ 1.42 (s, 9H), 2.61 (t, J= 6 Hz, J= 11.6 Hz 3H), 2.72 (d, J= 4.8 Hz, 3H), 3.49 (t, J= 5.6 Hz, J= 11.2, 2H), 4.42 (s, 2H), 5.33 (d, J= 4.8 Hz, 1H, D
2O exchangeable) 6.42 (s, 1H), 7.04 (s, 1H). [00928] LCMS (Method N): 2.59 min, 100%, 220nm, MS: ES+ 296.8 (M+1). Step 6: tert-Butyl 6-chloro-7-(N-methylacrylamido)-3,4-dihydroisoquinoline-2(1H)-carboxylate
[00929] The title compound was prepared from tert-butyl 6-chloro-7-(methylamino)-3,4- dihydroisoquinoline-2(1H)-carboxylate (Step 5) and acryloyl chloride analogously to Example 36 Step 5. [00930]
1H NMR (DMSO-d6, 400 MHz): δ 1.42 (s, 9H), 2.81 (t, J= 5.6 Hz, J= 11.2 Hz 3H), 3.13 (s, 3H), 3.55 (d, J= 5.6 Hz, 2H), 4.50 (s, br, 2H), 5.57 (dd, J= 2.4 Hz, J= 2 Hz, 1H), 5.84 - 5.91(m, 1H), 6.17 (dd, J= 2 Hz, J= 2.4 Hz, 1H), 7.38 (s, 1H), 7.46 (s, 1H). [00931] LCMS (Method N): 2.35 min, MS: ES+350.7 (M+1). Step 7: N-(6-Chloro-1,2,3,4-tetrahydroisoquinolin-7-yl)-N-methylacrylamide TFA salt
[00932] The title compound was prepared from tert-butyl 6-chloro-7-(N-methylacrylamido)-3,4- dihydroisoquinoline-2(1H)-carboxylate (Step 6) and TFA analogously to Example 36 Step 6. [00933]
1H NMR (DMSO-d6, 400 MHz): δ 3.03 (t, J= 6 Hz, J= 12 Hz, 2H), 3.35 - 3.41 (m, 2H), 4.28 (s, br, 2H), 5.60 (dd, J= 2 Hz, J= 2 Hz, 1H), 5.80 - 5.87 (m, 1H), 6.19 (dd, J= 2 Hz, J= 2 Hz, 1H), 7.44 (s, 1H), 7.58 (s, 1H), 9.09 (s, 1H). [00934] LCMS (Method O): 1.28 min, MS: ES+ 251.1 (M+1).
Step 8: N-(6-Chloro-2-(2-hydroxy-5-isopropyl-4-methoxybenzoyl)-1,2,3,4-tetrahydroisoquinolin- 7-yl)-N-methylacrylamide
[00935] The title compound was prepared from 2-hydroxy-5-isopropyl-4-methoxybenzoic acid (Example 45 Step 3) and N-(6-chloro-1,2,3,4-tetrahydroisoquinolin-7-yl)-N-methylacrylamide TFA salt (Step 7) analogously to Example 1 Step 1. [00936]
1H NMR (DMSO-d6, 400 MHz): δ 1.11 (d, J= 6.8 Hz, 6H), 2.87 (s, br, 2H), 3.13 (s, 4H), 3.61 (s, br, 2H), 3.76 (s, 3H), 4.66 (s, 2H), 5.57 (d, J= 11.2 Hz, 1H), 5.89 - 5.92 (m, 1H), 6.16 (d, J= 17.6 Hz, 1H), 6.47 (s, 1H), 6.95 (s, 1H), 7.39 (s, 1H), 7.47 (s, 1H). [00937] LCMS (Method A): 2.34 min, MS: ES+ 442.7 (M+1). [00938] HPLC (Method H): 8.68 min. Prep. HPLC purification method: [00939] Chromatographic separation and isolation were conducted using a PHP-09 Waters 2545 binary pump with Waters 2489 UV detector with Waters Acquity QDA detector; column Waters X-Bridge C18 (250mm x 19mm x 5µm); compounds eluted with: Mobile Phase A: 5mM ammonium bicarbonate +0.05% NH3 in Merck water, Mobile Phase B: ACN with a gradient of T = 0.01 min (48% A, 52% B); gradient to T = 21.00 min (48% A, 52% B); T = 21.00 min (0% A, 100% B) gradient to T = 27.00 min (0% A, 100% B); T = 27.01 min (48% A, 52% B); gradient to T = 30.00 min (48% A, 52% B); Flow rate= 13 ml/min; analysis time 30 min. Example 57 N-(2-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)-8-methyl-1,2,3,4- tetrahydroisoquinolin-7-yl)-N-methylacrylamide Step 1: N-(3-Bromo-2-methylbenzyl)-2,2-dimethoxyethan-1-amine

[00940] To a stirred solution of 3-bromo-2-methylbenzaldehyde (CAS: 83647-40-9) (10 g, 50.25 mmol, 1.0 eq.) in MeOH (100 mL) was added 2,2-dimethoxyethan-1-amine (CAS: 22483-09-6) (5.27 g, 50.25 mmol, 1 eq.) and the reaction mixture was heated to 70°C for 2 h. The mixture was cooled to 0°C and treated portion wise with NaBH
4 (1.91 g, 50.25 mmol, 1 eq.). The reaction mixture was stirred at room temperature for 4 h. The resulting mixture was concentrated under vacuum to remove MeOH, diluted using water (100 mL) and extracted with EtOAc (4 x 50 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum yielding N-(3-bromo-2-methylbenzyl)-2,2-dimethoxyethan-1-amine as a yellow liquid (13 g, Yield: 92.7%). [00941]
1H NMR (DMSO-d6, 400 MHz): δ 2.36 (s, 3H), 2.61 (d, J= 5.2 Hz, 2H), 3.25 (s, 6H), 3.73 (s, 2H), 4.41 (t, J= 5.2 Hz, 1H), 7.08 (t, J= 8.0 Hz, 1H), 7.31 (d, J= 7.6 Hz, 1H), 7.47 (d, J= 8.0 Hz, 1H). [00942] LCMS (Method N): 1.559 min, MS: ES+ 287.7 & 289.6 (M+1 & M+3). Step 2: 7-Bromo-8-methylisoquinoline
[00943] To a cooled (0 ˚ C) solution of chlorosulfonic acid (52.8 g, 452.9 mmol, 10 eq.) was added dropwise N-(3-bromo-2-methylbenzyl)-2,2-dimethoxyethan-1-amine (Step 1) (13 g, 45.29 mmol, 1 eq.) and the reaction mixture was heated to 100°C and for 2 h. The resulting mixture was carefully added to ice-cold water (1000 mL) and extracted with EtOAc (3 x 200 mL). The aqueous phase was basified with saturated NaOH solution (250 mL) (to pH14) and extracted with EtOAc (3 x 200 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under reduced pressure yielding 7-bromo-8-methylisoquinoline as a yellow solid (7 g, Yield: 69.0%). [00944]
1H NMR (DMSO-d6, 400 MHz): δ 2.85 (s, 3H), 7.78 (d, J= 8.8 Hz, 1H), 7.85 (d, J= 5.6 Hz, 1H), 7.92 (d, J= 8.8 Hz, 1H), 8.59 (d, J= 5.6 Hz, 1H), 9.56 (s, 1H). [00945] LCMS (Method N): 1.613 min, MS: ES+ 221.7 & 223.7 (M+1 & M+3). Step 3: 7-Bromo-8-methyl-1,2,3,4-tetrahydroisoquinoline
[00946] Performed in 2 parallel batches, each of 1 g scale: To a cooled (0 ˚C) solution of acetic acid (20 mL) was added portion wise NaBH
4 (0.514 g, 13.5 mmol, 3.0 eq.) and the mixture was stirred at room temperature for 1 h.7-Bromo-8-methylisoquinoline (1.0 g, 4.5 mmol, 1.0 eq.) was added and stirring continued at room temperature for 16 h. An additional 3 equivalents of NaBH
4 was added and the reaction mixture was stirred at room temperature for further 4h. The resulting mixture was basified with 2M NaOH (50 mL) and extracted with EtOAc (4 x 50 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under reduced pressure to yield 7-bromo-8-methyl-1,2,3,4-tetrahydroisoquinoline as a light-yellow solid crude (2 g, Yield: 27%). The crude material was used without further purification. [00947] LCMS (Method N): 1.563 min, MS: ES+ 225.9 & 227.8 (M+1 & M+3). Step 4: tert-Butyl 7-bromo-8-methyl-3,4-dihydroisoquinoline-2(1H)-carboxylate
[00948] To a stirred solution of 7-bromo-8-methyl-1,2,3,4-tetrahydroisoquinoline (Step 3) (2 g, 8.8 mmol, 1 eq) in DCM (40 mL) was added TEA (2.6 g, 26.4 mmol, 3 eq) and the mixture was stirred at room temperature for 15 mins. Boc anhydride was added (2.89 g, 13.27 mmol, 1.5 eq) and the reaction mixture was stirred at room temperature for 1 h. The resulting mixture was diluted with water (50 mL) and extracted with DCM (2 x 50 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted at 10% ethyl acetate in hexane) yielding tert- butyl 7-bromo-8-methyl-3,4-dihydroisoquinoline-2(1H)-carboxylate (0.850 g, Yield: 29%). [00949]
1H NMR (DMSO-d6, 400 MHz): δ 1.49 (s, 9H), 2.26 (s, 3H), 2.73 (t, J= 6.0 Hz, 2H), 3.52 (t, J= 5.6 Hz, 2H), 4.45 (s, 2H), 6.96 (d, J= 8.4 Hz, 1H), 7.42 (d, J= 8.4 Hz, 1H). [00950] LCMS (Method N): 2.890 min, MS: ES+ 269.7 (M-56). Step 5: tert-Butyl 8-methyl-7-(methylamino)-3,4-dihydroisoquinoline-2(1H)-carboxylate
[00951] The title compound was prepared from tert-butyl 7-bromo-8-methyl-3,4- dihydroisoquinoline-2(1H)-carboxylate (Step 4) and methylamine (2M in THF) analogously to Intermediate A Step 1. [00952]
1H NMR (DMSO-d6, 400 MHz): δ 1.42 (s, 9H), 1.91 (s, 3H), 2.63 (t, J= 5.6 Hz, 2H), 2.69 (d, J= 7.2 Hz, 3H), 3.49 (t, J= 5.2 Hz, 2H), 4.38 (s, 2H), 4.91 (t, J= 4.8 Hz, 1H, D2O exchangeable), 6.38 (d, J= 8.0 Hz, 1H), 6.83 (d, J= 8.0 Hz, 1H). [00953] LCMS (Method A): 1.883 min, MS: ES+ 276.9 (M+1). Step 6: tert-Butyl 8-methyl-7-(N-methylacrylamido)-3,4-dihydroisoquinoline-2(1H)-carboxylate
[00954] The title compound was prepared from tert-butyl 8-methyl-7-(methylamino)-3,4- dihydroisoquinoline-2(1H)-carboxylate (Step 5) and acryloyl chloride analogously to Example 36 Step 5. [00955]
1H NMR (DMSO-d6, 400 MHz): δ 1.44 (m, 9H), 1.97 (s, 3H), 2.80 (t, J= 5.6 Hz, 2H), 3.11 (s, 3H), 3.52 - 3.57 (m, 2H), 4.44 (s, 2H), 5.52 (dd, J= 2.0, 10 Hz, 1H), 5.85 (q, J= 10 Hz, 1H), 6.15 (dd, J= 2.0, 16.1 Hz, 1H), 7.08 (q, J= 8.0 Hz, 2H). [00956] LCMS (Method N): 2.220 min, MS: ES+ 330.8 (M+1). Step 7: N-Methyl-N-(8-methyl-1,2,3,4-tetrahydroisoquinolin-7-yl) acrylamide TFA salt
[00957] The title compound was prepared from tert-butyl 8-methyl-7-(N-methylacrylamido)-3,4- dihydroisoquinoline-2(1H)-carboxylate (Step 6) and TFA analogously to Example 36 Step 6. [00958]
1H NMR (DMSO-d6, 400 MHz): δ 1.99 (s, 3H), 3.02 (t, J= 6.0 Hz, 2H), 3.12 (s, 3H), 3.36 (bs, 2H), 4.24 (s, 2H), 5.53 (dd, J= 1.6 Hz, 12 Hz, 1H), 5.80 (dd, J= 10 Hz, 1H), 6.16 (dd, J= 2.0, 16.8 Hz, 2H), 7.19 (s 2H), 9.11 (bs, 1H). [00959] LCMS (Method B): 1.571 min, MS: ES+ 231.2 (M+1). Step 8: N-(2-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)-8-methyl-1,2,3,4-tetrahydroisoquinolin- 7-yl)-N-methylacrylamide
[00960] The title compound was prepared from 2-hydroxy-5-isopropyl-4-methoxybenzoic acid (Example 45 Step 3) and N-methyl-N-(8-methyl-1,2,3,4-tetrahydroisoquinolin-7-yl) acrylamide TFA salt (Step 7) analogously to Example 1 Step 1. [00961]
1H NMR (DMSO-d6, 400 MHz): δ 1.11 (d, J= 6.8 Hz, 6H), 1.96 (s, br, 3H), 2.86 (s, br, 2H), 3.09 (s, 3H), 3.09 – 3.16 (m, 1H), 3.60 (s, br, 2H), 3.77 (s, 3H), 4.61 (s, br, 2H), 5.52 (d, J= 10.4 Hz, 1H), 5.85 (q, J= 10 Hz, 1H), 6.15 (d, J= 16.8 Hz, 1H), 6.47 (s, 1H), 6.96 (s, 1H), 7.09 (q, J= 8.0 Hz, 2H), 9.34 (bs, 1H) [00962] LCMS (Method N): 2.270 min, MS: ES+ 422.9 (M+1). [00963] HPLC (Method H): 8.43 min, Prep. HPLC purification method of analysis: [00964] Chromatographic separation and isolation were conducted using a Waters 2545 binary pump with Waters 2489 UV detector with Waters Acquity QDA detector; column YMC Triart C18 (250 x 20 mm ID, 5μm); compounds eluted with: Mobile Phase A: 5 mM ammonium bicarbonate +0.05% NH3 in Merck water, Mobile Phase B: ACN with a gradient of T = 0.01 min (50% A, 50% B); gradient to T = 25.00 min (60% A, 40% B); T = 25.01 min (0% A, 100% B) gradient to T =
27.00 min (0% A, 100% B); T = 27.01 min (50% A, 50% B); gradient to T = 33.00 min (50% A, 50% B); Flow rate= 13 ml/min; analysis time 33 min. Example 58: N-(6-(2-(Dimethylamino)ethoxy)-2-(2-hydroxy-5-isopropyl-4- methoxybenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N-methylacrylamide Step 1: 1,2,3,4-Tetrahydroisoquinolin-6-ol hydrochloride

[00965] To a cooled (0 ˚C) solution of tert-butyl 6-hydroxy-3,4-dihydroisoquinoline-2(1H)- carboxylate (CAS: 158984-83-9) (50 g, 200.8 mmol, 1 eq.) in DCM (500 mL) was added dropwise 4M HCl in dioxane (500 mL) and the reaction mixture was stirred at room temperature for 1 h. The resulting mixture was concentrated under vacuum and the crude material was triturated with diethyl ether (500 mL) and n-pentane (500 mL) followed by high vacuum drying to yield 1,2,3,4- tetrahydroisoquinolin-6-ol hydrochloride as an off-white solid (40 g, Yield: Quantitative). [00966]
1H NMR (DMSO-d6, 400 MHz): δ 2.89 - 2.92 (m, 2H), 3.28 (s, 2H), 4.10 (s, 2H), 6.61 (s, 1H), 6.66 (d, J= 8 Hz, 1H), 7.00 (d, J= 8 Hz, 1H), 9.52 - 9.56 (m, 3H). [00967] LCMS (Method N): 0.274 min, MS: ES+ 150.0 (M+1). Step 2: 7-Nitro-1,2,3,4-tetrahydroisoquinolin-6-ol
[00968] Performed in 4 parallel batches, each of 10 g scale: To a cooled (0 ˚C) solution of 1,2,3,4-tetrahydroisoquinolin-6-ol hydrochloride (Step 1) (10 g, 53.91 mmol, 1 eq) in conc. H
2SO
4 (100 mL) was added dropwise guanidine nitrate (4.60 g, 37.70 mmol, 0.7 eq) in conc. H
2SO
4 and the reaction mixture was stirred at 0°C for 1 min. The resulting mixture was poured slowly in ice cold water (1L) and the pH adjusted to pH ~8 with saturated NaOH solution at 0°C. The aqueous layer (4L) was used directly in the next step without further purification. [00969] LCMS (Method N): 0.52 min, .65 min, MS: ES+ 194.8 (M+1).
Step 3: tert-Butyl 6-((tert-butoxycarbonyl)oxy)-7-nitro-3,4-dihydroisoquinoline-2(1H)-carboxylate

[00970] To a stirred aqueous solution of 7-nitro-1,2,3,4-tetrahydroisoquinolin-6-ol (Step 2) (40 g (quantitative, 206.18 mmol, 1 eq.) was added Boc anhydride (44.94 g, 206.14 mmol, 1 eq) and the reaction mixture was stirred at room temperature for 1 h. The resulting mixture was filtered and the bed was washed with EtOAc (2 L). The aqueous layer (4L) was extracted into EtOAc (2 x 2L) and 10% MeOH: DCM solution (3 x 1L). The combined organic extracts were dried over Na2SO4, filtered and concentrated under vacuum to yield tert-butyl 6-((tert-butoxycarbonyl)oxy)- 7-nitro-3,4-dihydroisoquinoline-2(1H)-carboxylate as a yellow oil (63 g, 158.89 mmol). The crude material was used in the next step without further purification. [00971] LCMS (Method N): 2.638 min, 2.663 min, 65.00 %, 254 nm MS: ES+ 194.2 (M-200) Step 4: tert-Butyl 6-hydroxy-7-nitro-3,4-dihydroisoquinoline-2(1H)-carboxylate

[00972] To a cooled (0 ˚C) solution of tert-butyl 6-((tert-butoxycarbonyl)oxy)-7-nitro-3,4- dihydroisoquinoline-2(1H)-carboxylate (63 g, 158.89 mmol, 1 eq.) in MeOH (630 mL) was added dropwise NaOMe (30% solution in MeOH) (252 mL) and the reaction mixture was stirred at room temperature for 30 mins. The resulting mixture was poured into ice cold water (1L) and neutralized with KHSO4 solution (pH ~7). The aqueous portion was extracted with EtOAc (1L) and DCM (3 x 1L). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted at 17% EtOAc in hexane) to yield tert-butyl 6-hydroxy-7-nitro-3,4- dihydroisoquinoline-2(1H)-carboxylate as an orange solid (16 g, Yield: 34%). [00973]
1H NMR (DMSO-d6, 400 MHz): δ 1.43 (s, 9H), 2.79 (s, 2H), 3.52 (s, 2H), 4.46 (s, 2H), 6.92 (s, 1H), 7.80 (s, 1H). Step 5: tert-Butyl 6-(2-(dimethylamino)ethoxy)-7-nitro-3,4-dihydroisoquinoline-2(1H)-carboxylate
[00974] Performed in 16 parallel batches, each of 1 g scale: To a stirred solution of tert-butyl 6- hydroxy-7-nitro-3,4-dihydroisoquinoline-2(1H)-carboxylate (Step 4) (1.0 g, 3.40 mmol, 1 eq.) in DMF (10 mL) were added 2-chloro-N,N-dimethylethan-1-amine hydrochloride (CAS: 4584-46-7) (1.47 g, 10.21 mmol, 3 eq) and Cs
2CO
3 (3.33 g, 10.21 mmol, 3 eq) and the reaction mixture was heated to 110°C for 16 h. The resulting mixture was poured into ice cold water (1 L) and extracted with EtOAc (3 x 1 L). The combined organic extracts were washed with ice cold water (3 x 500 mL), dried over Na
2SO
4, filtered, and concentrated under vacuum. The crude material was purified twice by reverse phase column chromatography (product eluted at 50% ACN in water) followed by chromatography on silica (product eluted at 20% MeOH in DCM) to yield tert-butyl 6- (2-(dimethylamino)ethoxy)-7-nitro-3,4-dihydroisoquinoline-2(1H)-carboxylate as a yellow oil (8 g, Yield: 40%). [00975]
1H NMR (DMSO-d6, 400 MHz): δ 1.43 (s, 9H), 2.22 (s, 6H), 2.66 (t, J= 5.2 Hz, 2H), 2.84 (t, J= 6.0 Hz, 2H), 3.55 (t, J= 5.2 Hz, 2H), 4.20 (t, J= 5.6 Hz, 2H), 4.48 (s, 2H), 7.20 (s, 1H), 7.77 (s, 1H). [00976] LCMS (Method N): 1.740 min MS: ES+ 365.9 (M+1) Step 6: tert-Butyl 7-amino-6-(2-(dimethylamino)ethoxy)-3,4-dihydroisoquinoline-2(1H)- carboxylate
[00977] To a solution of tert-butyl 6-(2-(dimethylamino)ethoxy)-7-nitro-3,4-dihydroisoquinoline- 2(1H)-carboxylate (Step 5) (8 g, 21.92 mmol, 1 eq.) in MeOH (80 mL) was added 10% Pd/C (4 g, 50% W/W) and the reaction mixture was stirred under an atmosphere of hydrogen for 2 h. The resulting mixture was filtered through Celite® and washed with 50% MeOH: DCM (500 mL). The combined filtrate was concentrated under reduced pressure to yield tert-butyl 7-amino-6-(2- (dimethylamino)ethoxy)-3,4-dihydroisoquinoline-2(1H)-carboxylate as a black oil (6 g, Yield: 82%). The crude material was used in the next step without purification.
[00978]
1H NMR (DMSO-d6, 400 MHz): δ 1.42 (s, 9H), 2.25 (s, 6H), 2.58 - 2.67 (m, 4H), 3.48 (t, J= 5.6 Hz, 2H), 3.98 (t, J= 5.6 Hz, 2H), 4.29 (s, 2H), 4.71 - 4.72 (m, 2H), 6.37 (s, 1H), 6.58 (s, 1H). [00979] LCMS (Method N): 1.622 min MS: ES+ 335.9 (M+1) Step 7: tert-Butyl 6-(2-(dimethylamino)ethoxy)-7-formamido-3,4-dihydroisoquinoline-2(1H)- carboxylate
[00980] The title compound was prepared from tert-butyl 7-amino-6-(2-(dimethylamino)ethoxy)- 3,4-dihydroisoquinoline-2(1H)-carboxylate (Step 6) and ethyl formate analogously to Example 56 Step 4. [00981]
1H NMR (DMSO-d6, 400 MHz): δ 1.42 (s, 9H), 2.46 (s, 6H), 2.70 - 2.71 (m, 2H), 3.00 - 3.01 (m, 2H), 3.51 - 3.52 (m, 2H), 4.13 - 4.17 (m, 2H), 4.39 (s, 2H), 7.97 (s, 1H), 8.19 (s, 2H), 8.34 (s, 1H), 9.96 (s, 1H). [00982] LCMS (Method N): 1.749 min MS: ES+ 363.9 (M+1). Step 8: tert-Butyl 6-(2-(dimethylamino)ethoxy)-7-(methylamino)-3,4-dihydroisoquinoline-2(1H)- carboxylate
[00983] The title compound was prepared from tert-butyl 6-(2-(dimethylamino)ethoxy)-7- formamido-3,4-dihydroisoquinoline-2(1H)-carboxylate (Step 7) and LAH (1M in THF) analogously to Example 56 Step 5. [00984]
1H NMR (DMSO-d6, 400 MHz): δ 1.42 (s, 9H), 2.60 (t, J= 6.0 Hz, 3H), 2.68 - 2.69 (m, 3H), 3.97 (t, J= 6.0 Hz, 2H), 4.36 (s, 2H), 4.87 - 4.88 (m, 1H), 6.22 (s, 1H), 6.58 (s, 1H). [00985] LCMS (Method N): 1.682 min MS: ES+ 349.9 (M+1) Step 9: tert-Butyl 6-(2-(dimethylamino)ethoxy)-7-(N-methylacrylamido)-3,4-dihydroisoquinoline- 2(1H)-carboxylate
[00986] The title compound was prepared from tert-butyl 6-(2-(dimethylamino)ethoxy)-7- (methylamino)-3,4-dihydroisoquinoline-2(1H)-carboxylate (Step 8) and acryloyl chloride analogously to Example 36 Step 5. This material was used directly in the next step [00987] LCMS (Method N): 1.777 min, MS: ES+ 404.0 (M+1). Step 10: N-(6-(2-(Dimethylamino)ethoxy)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- methylacrylamide TFA salt

[00988] The title compound was prepared from tert-butyl 6-(2-(dimethylamino)ethoxy)-7-(N- methylacrylamido)-3,4-dihydroisoquinoline-2(1H)-carboxylate (Step 9) and TFA analogously to Example 36 Step 6. This material was used directly in the next step [00989] LCMS (Method O): 1.12 min, MS: ES+ 304.3 (M+1). Step 11: N-(6-(2-(Dimethylamino)ethoxy)-2-(2-hydroxy-5-isopropyl-4-methoxybenzoyl)-1,2,3,4- tetrahydroisoquinolin-7-yl)-N-methylacrylamide
[00990] The title compound was prepared from 2-hydroxy-5-isopropyl-4-methoxybenzoic acid (Example 45 Step 3) and N-(6-(2-(dimethylamino)ethoxy)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- methylacrylamide TFA salt (Step 10) analogously to Example 1 Step 1. [00991]
1H NMR (MeOD, 400 MHz): δ 1.18 (d, J= 6.8 Hz, 6H), 2.34 (s, 6H), 2.77 (t, J= 5.6 Hz, 2H), 2.99 (t, J= 6.0 Hz, 2H), 3.18 – 3.23 (m, 1H), 3.25 (s, 3H), 3.78 - 3.84 (m, 2H), 3.84 (s, 3H), 4.09 - 4.21 (m, 2H), 4.70 (s, 2H), 5.52 - 5.55 (m, 1H), 6.04 - 6.11 (m, 1H), 6.20 - 6.25 (m, 1H), 6.47 (s, 1H), 6.98 (s, 1H), 7.03 - 7.06 (m, 2H),
[00992] LCMS (Method N): 1.743 min, MS: ES+ 495.9 (M+1). [00993] HPLC (Method H): 4.48 min Prep. HPLC purification method: [00994] Chromatographic separation and isolation were conducted with a Shimadzu LC20AP with UV detector; column YMC Triart C18 (250 x 20 mm ID), 5um; compound was eluted with Mobile Phase A: 5mM ammonium bicarbonate +0.05% NH
3 in Merck water Mobile Phase B: ACN:THF[90:10] with a gradient of T = 0.0 min (54% A, 46% B); gradient to T = 23.00 min (54% A, 46% B); T = 23.01 min (0% A, 100% B); gradient to T = 25.00 min (0% A, 100% B); T = 25.01 min (54% A, 46% B); T = 30.00 min (54% A, 46% B); Flow rate= 15 ml/min; analysis time 30 min. Example 59: N-(6-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-5,6,7,8-tetrahydro- 2,6-naphthyridin-3-yl)-N-methylacrylamide Step 1: tert-Butyl 7-(methylamino)-3,4-dihydro-2,6-naphthyridine-2(1H)-carboxylate
[00995] The title compound was prepared from tert-butyl 7-chloro-3,4-dihydro-2,6- naphthyridine-2(1H)-carboxylate (CAS:1060816-50-3) and methyl amine in (2M in THF) analogously to Intermediate A Step 1. [00996]
1H NMR (DMSO-d6, 400 MHz): δ 1.42 (s, 9H), 2.59 (t, J= 5.6 Hz, 2H), 2.71 (d, J= 4.8 Hz, 3H), 3.49 (d, J= 5.2 Hz, 2H), 4.37 (s, 2H), 6.20 (s, 2H), 7.79 (s, 1H). [00997] LCMS (Method N): 1.547 min, MS: ES+ 263.8 (M+1). Step 2: tert-Butyl 7-(N-methylacrylamido)-3,4-dihydro-2,6-naphthyridine-2(1H)-carboxylate
[00998] The title compound was prepared from tert-butyl 7-(methylamino)-3,4-dihydro-2,6- naphthyridine-2(1H)-carboxylate (Step 1) and acryloyl chloride analogously to Example 36 Step 5. [00999]
1H NMR (DMSO-d6, 400 MHz): δ 1.40 (s, 9H), 2.79 (t, J= 5.6 Hz, 2H), 3.27 (s, 3H), 3.59 (t, J= 4.8 Hz, 2H), 4.55 (s, 2H), 5.59 - 5.62 (m, 1H), 6.19 (m, 2H), 7.26 (s, 1H), 8.29 (s, 1H). [001000] LCMS (Method N): 1.923 min, MS ES+: 317.8 (M+1). Step 3: N-Methyl-N-(5,6,7,8-tetrahydro-2,6-naphthyridin-3-yl) acrylamide TFA salt
[001001] The title compound was prepared from tert-butyl 7-(N-methylacrylamido)-3,4- dihydro-2,6-naphthyridine-2(1H)-carboxylate (Step 2) and TFA analogously to Example 36 Step 6. [001002]
1H NMR (DMSO-d6, 400 MHz): δ 3.02 (t, J= 6 Hz, 2H), 3.30 (s, 3H), 3.43 (s, 2H), 4.32 (s, 2H), 5.65 (dd, J= 3.2 Hz, 1H), 6.18-6.24 (m, 2H), 7.31 (t, J= 6.4 Hz, 1H), 8.38 (s, 1H), 9.19 (s, 1H). [001003] LCMS (Method O): 1.24 min, MS ES+: 218.2 (M+1). Step 4: N-(6-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-5,6,7,8-tetrahydro-2,6- naphthyridin-3-yl)-N-methylacrylamide
[001004] The title compound was prepared from 3-chloro-6-hydroxy-4-methoxy-2- methylbenzoic acid (Example 47 Step 3) and N-methyl-N-(5,6,7,8-tetrahydro-2,6-naphthyridin-3- yl)acrylamide TFA salt (Step 3) analogously to Example 1 Step 1. [001005] HT
1H NMR (DMSO-d6, 400 MHz): δ 2.06 and 2.13 (2 singlets, 3H), 2.67 - 2.90 (m, 2H), 3.24 and 3.29 (2 singlets, 3H), 3.45 (m, 1H), 3.79 and 3.81 (2 singlets, 3H),3.81 – 3.86 (m, 1H), 4.42 (d, J= 3.6 Hz, 1H), 4.77 - 4.92 (m, 1H), 5.59 – 5.64 (m, 1H), 6.18 - 6.25 (m, 2H), 6.53 (d, J= 14.8 Hz, 1H), 7.12 - 7.29 (m, 1H), 8.30 (s, br, 1H), 9.60 (bs, 1H).
[001006] LCMS (Method N): 1.771 min, MS: ES+ 416.1 (M+1). [001007] HPLC (Method H): 5.90 min. Prep. HPLC purification method [001008] Chromatographic separation and isolation were conducted with a Waters 2545 binary pump with waters 2489 purification system with UV detector; column YMC Triart C18 (250mm x 20mm x 5µm); compound was eluted with: Mobile Phase A: 5 mM Ammonium bicarbonate in Merck water, Mobile Phase B: Acetonitrile with a gradient of T = 0.01 min (63% A, 37% B); gradient to T = 15 min (63% A, 37% B); T = 15.01 min (0% A, 100% B) gradient to T = 18 min (0% A, 100% B); T = 18.01 min (63% A, 37% B); gradient to T = 25 min (63% A, 37% B); Flow rate= 11ml/min; analysis time 25 min. Example 60: (E)-N-(7-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-5,6,7,8- tetrahydro-1,7-naphthyridin-2-yl)-N-methylbut-2-enamide Step 1: tert-Butyl 2-(methylamino)-5,8-dihydro-1,7-naphthyridine-7(6H)-carboxylate


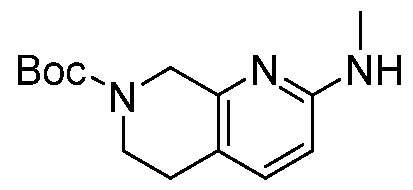
[001009] The title compound was prepared from tert-butyl 2-chloro-5,8-dihydro-1,7- naphthyridine-7(6H)-carboxylate (CAS: 1211581-54-2) and methyl amine in (2M in THF) analogously to Intermediate A Step 1. [001010]
1H NMR (DMSO-d6, 400 MHz): δ 1.42 (s, 9H), 2.55 (t, J= 5.6 Hz, 2H), 2.71 (d, J= 4.8 Hz, 3H), 3.52 (s, br, 2H), 4.29 (s, br, 2H), 6.28 (d, J= 8.4 Hz, 1H), 6.33 (s, br, 1H, D
2O exchangeable), 7.15 (d, J= 8.4 Hz, 1H) [001011] LCMS (Method: N): 1.598 min, MS ES+: 263.98 (M+1). Step 2: tert-Butyl (E)-2-(N-methylbut-2-enamido)-5,8-dihydro-1,7-naphthyridine-7(6H)- carboxylate
[001012] To a cooled (0 ˚C) solution of crotonic acid (CAS: 107-93-7) (0.45 g, 5.22 mmol, 1.0 eq.) and tert-butyl 2-(methylamino)-5,8-dihydro-1,7-naphthyridine-7(6H)-carboxylate (0.688 g, 2.61 mmol, 0.5 eq.) in DCM (4.5 mL) was added pyridine (1.68 mL, 20.90 mmol, 4.0 eq.) and the mixture stirred for 5 mins. POCl
3 (0.71 mL, 7.84 mmol, 1.5 eq) was added dropwise and the reaction mixture stirred at 0°C for 5 mins. The reaction mixture was poured into ice cold water (50 mL) and extracted with DCM (3 x 50 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum. Crude material was purified by chromatography on silica (product eluted in 43% EtOAc in hexane) to yield tert-butyl (E)-2-(N- methylbut-2-enamido)-5,8-dihydro-1,7-naphthyridine-7(6H)-carboxylate as a yellow oil (0.37 g, 43% yield). [001013]
1H NMR (DMSO-d6, 400 MHz): δ 1.43 (s, 9H), 1.75 (d, J= 6.8 Hz, 3H), 2.81 (t, J= 5.6 Hz, 2H), 3.26 (s, 3H), 3.61 (t, J= 5.6 Hz, 2H), 4.49 (s, br, 2H), 5.97 (d, J= 13.6 Hz, 1H), 6.71 - 6.80 (m, 1H), 7.16 (d, J= 8.4 Hz, 1H), 7.68 (d, J= 8.4 Hz, 1H). [001014] LCMS (Method: N): 2.050 min, MS ES+: 331.92 (M+1). Step 3: (E)-N-methyl-N-(5,6,7,8-tetrahydro-1,7-naphthyridin-2-yl)but-2-enamide TFA salt
[001015] The title compound was prepared from tert-butyl (E)-2-(N-methylbut-2-enamido)- 5,8-dihydro-1,7-naphthyridine-7(6H)-carboxylate (Step 2) and TFA analogously to Example 36 Step 6. [001016]
1H NMR (DMSO-d6, 400 MHz): δ 1.76 (d, J= 6.4 Hz, 3H), 3.02 (s, 2H), 3.28 (s, 3H), 3.43 (m, 2H), 4.29 (s, br, 2H), 5.98 (d, J= 14.8 Hz, 1H), 6.74 - 6.83 (m, 1H), 7.32 (d, J= 8.0 Hz, 1H), 7.77 (d, J= 8.4 Hz, 1H), 9.17 (s, 1H) [001017] LCMS (Method A3): 5.65 min, MS ES+: 232.2 Step 4: (E)-N-(7-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-5,6,7,8-tetrahydro-1,7- naphthyridin-2-yl)-N-methylbut-2-enamide
[001018] The title compound was prepared from 3-chloro-6-hydroxy-4-methoxy-2- methylbenzoic acid (Example 47 Step 3) and (E)-N-methyl-N-(5,6,7,8-tetrahydro-1,7- naphthyridin-2-yl)but-2-enamide TFA salt (Step 3) analogously to Example 1 Step 1. [001019] HT
1H NMR (DMSO-d6, 400 MHz, 348K): δ 1.76 (s, br, 3H), 2.06 - 2.16 (m, 3H), 2.76 - 2.81 (m, 1H), 2.91 (s, br, 1H), 3.24 - 3.31 (m, 3H), 3.48 (s, br, 1H), 3.82 (s, 3H), 3.98 - 4.03 (m, 1H), 4.31 - 4.37 (m, 1H), 4.76 - 4.81 (m, 1H), 5.93 - 6.06 (m, 1H), 6.53 (bs, 1H), 6.75 (s, br, 1H), 7.18 (d, J= 8.4 Hz, 1H), 7.67 (s, br, 1H), 9.63 (s, 1H). [001020] LCMS (Method N): 1.864 min, MS ES+: 430.1, 432.1 (M, M+2). [001021] HPLC (Method H): 6.29 min, Prep. HPLC purification method: [001022] Chromatographic separation and isolation were conducted using a Waters 2545 Binary pump with Waters 2489 UV detector with Waters Acquity QDA detector; column YMC Triart C18 (250 x 20 mm ID, 5µm); compound eluted with: Mobile Phase A: 5 mM ammonium bicarbonate + 0.05% NH
3 in Merck water. Mobile Phase B: ACN: MeOH: THF (50:50:10) with a gradient of T = 0.01 min (55% A, 45% B); gradient to T = 30.00 min (55% A, 45% B); T = 30.01min (00% A, 100% B), gradient to T = 34.00min (0% A, 100% B); T =34.01 min (55% A, 45% B); gradient to T = 40.00 min (55% A, 45% B); Flow rate= 10 ml/min; analysis time 40.00 min. Example 61: N-(7-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-5,6,7,8-tetrahydro- 1,7-naphthyridin-2-yl)-N-methylacrylamide Step 1: tert-Butyl 2-(N-methylacrylamido)-5,8-dihydro-1,7-naphthyridine-7(6H)-carboxylate
[001023] The title compound was prepared from tert-butyl 2-(methylamino)-5,8-dihydro-1,7- naphthyridine-7(6H)-carboxylate (Example 60 Step 1) and acryloyl chloride analogously to Example 36 Step 5. [001024]
1H NMR (DMSO-d6, 400 MHz): δ 1.43 (s, 9H), 2.81 (t, J= 5.6 Hz, 2H), 3.29 (s, 3H), 3.60 - 3.61 (m, 2H), 4.49 (s, 2H), 5.62 (dd, J= 9.6 Hz, J= 9.2 Hz, 1H), 6.16 - 6.24 (m, 2H), 7.20 (d, J= 8.0 Hz, 1H), 7.70 (d, J= 8.0 Hz, 1H)
[001025] LCMS (Method: N): 1.986 min, MS ES+: 317.76 (M+1). Step 2: N-Methyl-N-(5,6,7,8-tetrahydro-1,7-naphthyridin-2-yl) acrylamide TFA salt
[001026] The title compound was prepared from tert-butyl 2-(N-methylacrylamido)-5,8- dihydro-1,7-naphthyridine-7(6H)-carboxylate (Step 1) and TFA analogously to Example 36 Step 6 and used directly in the next step. [001027] LCMS (Method A3): 5.16 min, 210 nm, MS ES+: 218.2 Step 3: N-(7-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-5,6,7,8-tetrahydro-1,7- naphthyridin-2-yl)-N-methylacrylamide
[001028] The title compound was prepared from 3-chloro-6-hydroxy-4-methoxy-2- methylbenzoic acid (Example 47 Step 3) and N-methyl-N-(5,6,7,8-tetrahydro-1,7-naphthyridin-2- yl) acrylamide TFA salt (Step 2) analogously to Example 1 Step 1. [001029]
1H NMR (DMSO-d6, 400 MHz): δ 2.04 and 2.13 (2 singlets, 3H), 2.67 - 2.73 (m, 1H), 2.87 - 2.92 (m, 1H), 3.17 and 3.32 (2 singlets, 3H), 3.47 (s, br, 1H), 3.77 and 3.78 (2 singlets, 3H), 3.81 (m, 1H), 4.03 - 4.06 (m, 1H), 4.26 - 4.41 (m, 1H), 4.70 - 4.85 (m, 1H), 5.56 - 5.66 (m, 1H), 6.15 - 6.28 (m, 2H), 6.47 (d, J= 13.2 Hz, 1H), 7.19 - 7.22 (m, 1H), 7.70 (dd, 1H), 10.2 (bs, 1H). [001030] LCMS (Method: N): 1.815 min, MS ES+: 416.1, 418.1 (M, M+2). [001031] HPLC (Method: I): 5.742 min Prep. HPLC purification method: [001032] Chromatographic separation and isolation were conducted with a Waters 2545 binary pump with Waters 2489 UV detector with Waters Acquity QDA detector; column YMC Triart C18 (250 x 20 mm ID, 5µm); compound eluted with: Mobile Phase A: 05 mM ammonium bicarbonate in Merck water. Mobile Phase B: ACN with a gradient of T = 0.01 min (67% A, 33%
B); gradient to T = 21.00 min (62% A, 38% B); T = 21.01min (00% A, 100% B), gradient to T = 24.00min (0% A, 100% B); T = 24.01 min (67% A, 33% B); gradient to T = 28.00 min (67% A, 33% B); Flow rate= 11 ml/min; analysis time 28.00 min. Example 62: N-(7-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-3-methyl-5,6,7,8- tetrahydro-1,7-naphthyridin-2-yl)-N-methylacrylamide Step 1: tert-Butyl 3-bromo-2-(methylamino)-5,8-dihydro-1,7-naphthyridine-7(6H)-carboxylate

[001033] Performed in 3 parallel batches, each of 0.34 g scale: To a stirred solution of tert- butyl 2-(methylamino)-5,8-dihydro-1,7-naphthyridine-7(6H)-carboxylate (Example 60 Step 1) (0.34, 1.291 mmol, 1.0 eq) in acetonitrile (3.4 mL) was added portion wise NBS (0.229 mL, 1.291 mmol, 1.0 eq.) and the reaction mixture was stirred at room temperature for 5 mins. The resulting mixture was concentrated to remove acetonitrile and the remaining slurry poured into a saturated solution of Na2S2O3.5H2O in water (100 mL). The mixture was extracted with DCM (2 x 50 mL) and the combined organic extracts were dried over Na2SO4, filtered, and concentrated under vacuum. The crude material was triturated with n-pentane (2 x 20 mL) and dried under reduced pressure yielding tert-butyl 3-bromo-2-(methylamino)-5,8-dihydro-1,7-naphthyridine-7(6H)- carboxylate as a yellow solid (1.2 g, 92% yield). [001034]
1H NMR (DMSO-d6, 400 MHz): δ 1.42 (s, 9H) 2.56 - 2.60 (m, 2H), 2.80 (d, J= 4.4 Hz, 3H), 3.51 (t, J= 5.2 Hz, 2H), 4.30 (s, 2H), 6.22 (d, J= 4.4 Hz, 1H, D2O exchangeable), 7.55 (s, 1H) [001035] LCMS (Method: N): 2.421 min, MS ES+: 342.1 (M). Step 2: tert-Butyl 3-methyl-2-(methylamino)-5,8-dihydro-1,7-naphthyridine-7(6H)-carboxylate
[001036] Performed in 4 parallel batches, each of 0.3 g scale: To a stirred solution of tert- butyl 3-bromo-2-(methylamino)-5,8-dihydro-1,7-naphthyridine-7(6H)-carboxylate (Step 1) (0.30
g, 0.87 mmol, 1.0 eq.) in dioxane:water (9:1) at room temperature was added Cs
2CO
3 (0.85 g, 2.62 mmol, 3.0 eq.) and the mixture purged with nitrogen gas for 10 mins. Trimethyl boroxine (50% in THF) (0.32 mL, 1.31 mmol, 1.5 eq) and PdCl
2(dppf) (0.032 g, 0.043 mmol, 0.05 eq.) were added and the reaction mixture heated to 110°C for 30 mins. The resulting cooled mixture was poured into ice cold water (100mL) and extracted with EtOAc (3 x 50 mL). Combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum. Crude material was purified by chromatography on silica (product eluted in 21% EtOAc in hexane) to yield tert-butyl 3-methyl-2-(methylamino)-5,8-dihydro-1,7-naphthyridine-7(6H)-carboxylate as a yellow solid (0.75 g, 77% yield). [001037]
1H NMR (DMSO-d6, 400 MHz): δ 1.42 (s, 9H), 1.99 (s, 3H), 2.50 - 2.55 (m, 2H), 2.78 (d, J= 4.8 Hz, 3H), 3.51 (m, 2H), 4.29 (s, 2H), 5.76 - 5.83 (m, 1H, D
2O exchangeable), 6.98 (s, 1H) [001038] LCMS (Method: N): 1.628 min, MS ES+: 278.20 (M+1). Step 3: tert-Butyl 3-methyl-2-(N-methylacrylamido)-5,8-dihydro-1,7-naphthyridine-7(6H)- carboxylate
[001039] The title compound was prepared from tert-butyl 3-methyl-2-(methylamino)-5,8- dihydro-1,7-naphthyridine-7(6H)-carboxylate (Step 2) and acryloyl chloride analogously to Example 36 Step 5. [001040]
1H NMR (DMSO-d6, 400 MHz): δ 1.43 (s, 9H), 2.12 (s, 3H), 2.80 (s, br, 2H), 3.12 (s, 3H), 3.60 (s, br, 2H), 4.46 (s, 2H), 5.55 (d, J= 10.0 Hz, 1H), 5.74 - 5.80 (m, 1H), 6.18 (d, J= 16.8 Hz, 1H), 7.64 (s, 1H) [001041] LCMS (Method: N): 1.984 min, MS ES+: 332.18 (M+1). Step 4: N-Methyl-N-(3-methyl-5,6,7,8-tetrahydro-1,7-naphthyridin-2-yl) acrylamide TFA salt
[001042] The title compound was prepared from tert-butyl 3-methyl-2-(N- methylacrylamido)-5,8-dihydro-1,7-naphthyridine-7(6H)-carboxylate (Step 3) and TFA analogously to Example 36 Step 6 and used directly in the next step. [001043] LCMS (Method O): 1.10 min, 210 nm, MS ES+: 232.2 Step 5: N-(7-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-3-methyl-5,6,7,8-tetrahydro-1,7- naphthyridin-2-yl)-N-methylacrylamide
[001044] The title compound was prepared from 3-chloro-6-hydroxy-4-methoxy-2- methylbenzoic acid (Example 47 Step 3) and N-methyl-N-(3-methyl-5,6,7,8-tetrahydro-1,7- naphthyridin-2-yl) acrylamide TFA salt (Step 4) analogously to Example 1 Step 1. [001045]
1H NMR (DMSO-d6, 400 MHz, 348K): δ 2.07 - 2.14 (m, 6H), 2.76 - 2.91 (m, 2H), 3.07 and 3.15 (2 singlets, br, 3H), 3.48 (bs, 1H), 3.82 (s, br, 3H), 3.83 – 4.10 (m, br, 1H), 4.27 - 4.34 (m, 1H), 4.69 - 4.79 (m, 1H), 5.55 - 5.86 (m, 2H), 6.15 (s, br, 1H), 6.53 (s, 1H), 7.61 (s, br, 1H), 9.49 (s, 1H) [001046] LCMS (Method: N): 1.82 min, MS ES+: 430.1, 432.1 (M, M+2). [001047] HPLC (Method: H): 6.15 min Prep. HPLC purification method: [001048] Chromatographic separation and isolation were conducted on an FC-01 flash purification system (Buchi model C810); column Redisep RF C18100g 50µm; compound eluted with: Mobile Phase A: 0.05% NH3 + 5mM ammonium bicarbonate in Merck water. Mobile Phase B: ACN with a gradient of T = 0.00 min (100% A, 00% B); gradient to T = 30.00 min (70% A, 30% B); T = 30.01min (00% A, 100% B), gradient to T = 33.00min (0% A, 100% B); T = 33.01 min (100% A, 00% B); gradient to T = 36.00 min (100% A, 00% B); Flow rate= 70 ml/min; analysis time 36.00 min. Example 63: N-(6-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-2-methyl-5,6,7,8- tetrahydro-1,6-naphthyridin-3-yl)-N-methylacrylamide
Step 1: tert-Butyl 3-amino-2-bromo-7,8-dihydro-1,6-naphthyridine-6(5H)-carboxylate

[001049] To a cooled (0°C) solution of tert-butyl 3-amino-7,8-dihydro-1,6-naphthyridine- 6(5H)-carboxylate (CAS: 355819-02-2, Angene) (10 g, 40.16 mmol, 1 eq) in DMF (100 mL) was added acetic acid (1 mL) followed by portion wise addition of NBS (7.51 g, 42.19 mmol, 1.05 eq) over a period of 5 mins. The resulting mixture was diluted with EtOAc (500 mL) and washed with ice cold sodium thiosulfate solution (3 x 200 mL). The organic portion were dried over Na2SO4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted in 30% EtOAc in hexane) to yield tert-butyl 3-amino-2-bromo-7,8-dihydro- 1,6-naphthyridine-6(5H)-carboxylate as a pink solid (11 g, Yield: 83%) [001050]
1H NMR (DMSO-d6, 400 MHz): δ 1.42 (s, 9H), 2.67 (t, J= 5.6 Hz, 2H), 3.58 (t, J= 6.0 Hz, 2H), 4.39 (s, 2H), 5.33 (s, 2H, D2O exchangeable), 6.87 (s, 1H). [001051] LCMS (Method N): 2.003 min, MS: ES+ 327.6 (M, M+2). Step 2: tert-Butyl 3-amino-2-methyl-7,8-dihydro-1,6-naphthyridine-6(5H)-carboxylate
[001052] The title compound was prepared from tert-butyl 3-amino-2-bromo-7,8-dihydro- 1,6-naphthyridine-6(5H)-carboxylate (Step 1) and trimethyl boroxine (50% W/W in THF) analogously to Example 62 Step 2. [001053]
1H NMR (DMSO-d6, 400 MHz): δ 1.41 (s, 9H), 2.20 (s, 3H), 2.64 (t, J= 5.6 Hz, 2H), 3.56 (t, J= 5.2 Hz, 2H), 4.35 (s, 2H), 4.88 (s, 2H, D
2O exchangeable), 6.65 (s, 1H). [001054] LCMS (Method A): 1.535 min, MS: ES+ 264.0 (M+1). Step 3: tert-Butyl 3-formamido-2-methyl-7,8-dihydro-1,6-naphthyridine-6(5H)-carboxylate
[001055] The title compound was prepared from tert-butyl 3-amino-2-methyl-7,8-dihydro- 1,6-naphthyridine-6(5H)-carboxylate (Step 2) and ethyl formate analogously to Example 56 Step 4. [001056]
1H NMR (DMSO-d6, 400 MHz): δ 1.43 (s, 9H), 2.38 (s, 3H), 2.78 (t, J= 5.6 Hz, 2H), 3.63 (t, J= 5.6 Hz, 2H), 4.48 (s, 2H), 7.89 (s, 1H), 8.32 (s, 1H), 9.76 (s, 1H, D
2O exchangeable). [001057] LCMS (Method A): 1.547 min, MS: ES+ 219.9 (M+1). Step 4: tert-Butyl 2-methyl-3-(methylamino)-7,8-dihydro-1,6-naphthyridine-6(5H)-carboxylate
[001058] The title compound was prepared from tert-butyl 3-formamido-2-methyl-7,8- dihydro-1,6-naphthyridine-6(5H)-carboxylate (Step 3) and LAH (1M in THF) analogously to Example 56 Step 5. [001059]
1H NMR (DMSO-d6, 400 MHz): δ 1.40 (s, 9H), 2.23 (s, 3H), 2.65 - 2.69 (m, 5H), 3.57 - 3.58 (m, 2H), 4.43 (s, 2H), 5.18 (d, J= 4.8 Hz, 1H, D
2O exchangeable), 6.56 (s, 1H). [001060] LCMS (Method A): 1.593 min, MS: ES+ 278.0 (M+1). Step 5: tert-Butyl 2-methyl-3-(N-methylacrylamido)-7,8-dihydro-1,6-naphthyridine-6(5H)- carboxylate
[001061] The title compound was prepared from tert-butyl 2-methyl-3-(methylamino)-7,8- dihydro-1,6-naphthyridine-6(5H)-carboxylate (Step 4) and acryloyl chloride analogously to Example 36 Step 5. [001062]
1H NMR (DMSO-d6, 400 MHz): δ 1.44 (s, 9H), 2.26 (s, 3H), 2.86 (t, J= 6 Hz, 2H), 3.16 (s, 3H), 3.66 (t, J= 6 Hz, 2H), 4.52 (s, 2H), 5.57 (dd, J= 2 Hz, 10 Hz, 1H), 5.84 - 5.91 (m, 1H), 6.19 (dd, J= 2.4 Hz, 16.8 Hz, 1H), 7.57 (s, 1H).
[001063] LCMS (Method N): 1.762 min, MS: ES+ 331.8 (M+1). Step 6: N-Methyl-N-(2-methyl-5,6,7,8-tetrahydro-1,6-naphthyridin-3-yl)acrylamide TFA salt
[001064] The title compound was prepared from tert-butyl 2-methyl-3-(N- methylacrylamido)-7,8-dihydro-1,6-naphthyridine-6(5H)-carboxylate (Step 5) and TFA analogously to Example 36 Step 6. [001065]
1H NMR (DMSO-d6, 400 MHz): δ 2.31 (s, 3H), 3.08 (t, J= 6.0 Hz, 2H), 3.15 (s, 3H), 3.50 - 3.51 (m, 2H), 4.30 - 4.32 (m, 2H), 5.60 (dd, J= 2 Hz, 10.4 Hz, 1H), 5.76 - 5.86 (m, 1H), 6.21 (dd, J= 2 Hz, 16.8 Hz, 1H), 7.66 (s, 1H), 9.15 (s, 2H). [001066] LCMS (Method O): 1.11 min, MS: ES+ 232.2 (M+1). Step 7: N-(6-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-2-methyl-5,6,7,8-tetrahydro-1,6- naphthyridin-3-yl)-N-methylacrylamide
[001067] The title compound was prepared from 3-chloro-6-hydroxy-4-methoxy-2- methylbenzoic acid (Example 47 Step 3) and N-methyl-N-(2-methyl-5,6,7,8-tetrahydro-1,6- naphthyridin-3-yl)acrylamide TFA salt (Step 6) analogously to Example 1 Step 1. [001068] HT
1H NMR (DMSO-d6, 400 MHz, 348K): δ 2.16 (m, 3H), 2.29 (s, 3H), 2.75 - 2.95 (m, 2H), 3.11 - 3.17 (m, 3H), 3.54 (bs, 1H), 3.82 (s, 3H), 4.09 (bs, 1H), 4.38 (m, 1H), 4.78 - 4.86 (m, 1H), 5.55 - 5.57 (m, 1H), 5.91 (bs, 1H), 6.18 (d, J= 16 Hz, 1H), 6.53 (s, 1H), 7.41 - 7.60 (m, 1H), 9.69 (s, 1H). [001069] LCMS (Method A): 1.706 min, MS: ES+ 429.8 (M). [001070] HPLC (Method H): 5.200 min. Prep. HPLC purification method of analysis:
[001071] Chromatographic separation and isolation were conducted with an FC-01 flash purification system (Buchi model C810); column REDISEP RF C18100g 50µm; compound eluted with: Mobile Phase A: 5 mM ammonium bicarbonate +0.05%NH3 IN HPLC WATER, Mobile Phase B: acetonitrile with a gradient of T = 0.0 min (100% A, 0% B); gradient to T = 4.00 min (100% A, 0% B); T = 24.00 min (85% A, 15% B); gradient to T = 24.01 min (0% A, 100% B); T = 28.00 min (0% A, 100% B); T = 28.01 min (100% A, 0% B); T = 32.00 min (100% A, 0% B); Flow rate= 70 ml/min; analysis time 32 min. Example 64: N-(2-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)-1,2,3,4- tetrahydroisoquinolin-7-yl)-N-(1-methylpiperidin-4-yl) acrylamide Step 1 tert-Butyl 7-((1-methylpiperidin-4-yl) amino)-3,4-dihydroisoquinoline-2(1H)-carboxylate
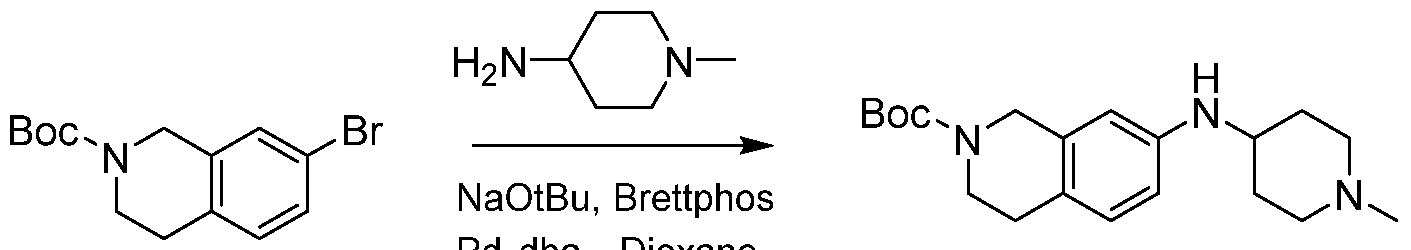
[001072] The title compound was prepared from tert-butyl 7-bromo-3,4- dihydroisoquinoline-2(1H)-carboxylate (CAS: 41838-46-4) and 1-methylpiperidin-4-amine analogously to Intermediate A Step 1. [001073]
1H NMR (DMSO-d6, 400 MHz): δ 1.42 (s, 9H), 1.85 (d, J= 11.6 Hz, 2H), 1.97- 2.02 (m, 3H), 2.16 (s, 3H), 2.58 (t, J= 5.6 Hz, 2H), 2.71 (d, J= 11.2 Hz, 2H), 3.14 (m, 2H), 3.48 (t, J= 5.6 Hz, 2H), 4.35 (s, br, 2H), 5.21 (d, J= 8.0 Hz, 1H, D2O exchangeable), 6.32 (s, 1H), 6.41 (t, J= 8.4 Hz, 1H), 6.82 (d, J= 8.0 Hz, 1H). [001074] LCMS (Method N): 1.721 min, MS: ES+ 345.90 (M+1). Step 2 tert-Butyl 7-(N-(1-methylpiperidin-4-yl) acylamido)-3,4-dihydroisoquinoline-2(1H)- carboxylate
[001075] The title compound was prepared from tert-butyl 7-((1-methylpiperidin-4-yl) amino)-3,4-dihydroisoquinoline-2(1H)-carboxylate (Step 1) and acryloyl chloride analogously to Example 36 Step 5. [001076]
1H NMR (DMSO-d6, 400 MHz): δ 1.43 (s, 9H), 1.75 (d, J= 9.6 Hz, 2H), 2.24 (s, br, 4H), 2.89 (t, J= 10.0 Hz, 2H), 2.99 (d, J= 7.2 Hz, 2H), 3.34 (m, br, 1H), 3.58 (s, br, 2H), 4.48 – 4.52 (m, 3H), 5.50 (d, J= 10.4 Hz, 1H), 5.74 - 5.88 (m, 1H), 6.22 - 6.45 (m, 1H), 6.97 (d, J= 8.0 Hz, 1H), 7.05 (s, 1H), 7.25 (d, J= 8.0 Hz, 1H), 7.47 - 7.63 (m, 1H), 7.94 (d, J= 7.6 Hz, 1H). [001077] LCMS (Method N): 1.729 min, MS: ES+ 236.8 (M+1). Step 3: N-(1-Methylpiperidin-4-yl)-N-(1,2,3,4-tetrahydroisoquinolin-7-yl) acrylamide TFA salt
[001078] The title compound was prepared from tert-butyl 7-(N-(1-methylpiperidin-4-yl) acylamido)-3,4-dihydroisoquinoline-2(1H)-carboxylate (Step 2) and TFA analogously to Example 36 Step 6 which was used directly in the next step. [001079] LCMS (Method N): 0.275 min, MS: ES+ 299.9 (M+1). Step 4: N-(2-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- (1-methylpiperidin-4-yl) acrylamide
[001080] The title compound was prepared from 2-hydroxy-5-isopropyl-4-methoxybenzoic acid (Example 45 Step 3) and N-(1-methylpiperidin-4-yl)-N-(1,2,3,4-tetrahydroisoquinolin-7-yl) acrylamide TFA salt (Step 3) analogously to Example 1 Step 1. [001081] HT
1H NMR (DMSO-d6, 400 MHz, 348K): δ 1.14 (d, J= 6.8 Hz, 6H), 1.32 – 1.40 (m, 2H), 1.70 (d, J= 11.2 Hz, 2H), 1.89 - 1.98 (m, 2H), 2.11 (s, 3H), 2.75 (d, J= 11.2 Hz, 2H), 2.91 (t, J= 5.6 Hz, 2H), 3.68 (t, J= 5.2 Hz, 2H), 3.79 (s, 3H), 4.35 – 4.42 (m, 1H), 4.69 (s, 2H),
5.46 (d, J= 12.0 Hz, 1H), 5.81 - 5.88 (m, 1H), 6.10 (d, J= 14.8 Hz, 2H), 6.50 (s, 1H), 6.94 - 7.00 (m, 3H), 7.26 (d, J= 8.0 Hz, 1H), 9.59 (s, 1H). [001082] LCMS (Method A): 1.813 min, MS: ES+ 491.98 (M+1). [001083] HPLC (Method H): 4.61 min, Prep. HPLC purification method: [001084] Chromatographic separation and isolation were conducted with a Waters 2545 quaternary system with Waters 2489 UV Detector; column Waters X-Bridge C18 (250mm x 19mm x 5µm); compound eluted with: Mobile Phase A : 5mM ammonium bicarbonate +0.05% NH
3 in Merck water Mobile Phase B : acetonitrile with a gradient of T = 0.0 min (56% A, 44% B); gradient to T = 20.00 min (40% A, 60% B); T = 20.01 min (0% A, 100% B); gradient to T = 22.00 min (0% A, 100% B); T = 22.01 min (56% A, 44% B); gradient to T = 24.00 min (56% A, 44% B); Flow rate= 12 ml/min; analysis time 24.0. Example 65: (E)-N-(2-(4,6-dihydroxy-2,3-dimethylbenzoyl)-5-methylisoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide Step 1: tert-Butyl (E)-4-(4-(dimethylamino)-N-methylbut-2-enamido)-5-methylisoindoline-2- carboxylate

[001085] To a cooled (0°C), stirred solution of tert-butyl 5-methyl-4- (methylamino)isoindoline-2-carboxylate (Example 54 Step 4) (20 mg, 0.076 mmol, 1.0 eq) in DMA (200 µL) was added a solution of (E)-4-(dimethylamino)but-2-enoyl chloride hydrochloride (CAS 1056149-69-9, 14 mg, 0.076 mmol, 1.0 eq) in acetonitrile (280 µL). The reaction mixture was stirred briefly at 0 °C then at room temperature for 16 h. The resulting mixture was concentrated under reduced pressure to afford the title compound which was used in the next step without purification. (45 mg, Yield: quantitative). MS: ES+ 374.2 (M+1) Step 2: (E)-4-(Dimethylamino)-N-methyl-N-(5-methylisoindolin-4-yl)but-2-enamide 2,2,2 trifluoroacetate
[001086] Trifluoroacetic acid (0.25 mL, 3.26 mmol, 43.5 eq) was added dropwise to a suspension of tert-butyl 4-[[(E)-4-(dimethylamino)but-2-enoyl]-methyl-amino]-5-methyl- isoindoline-2-carboxylate (Step 1) (28 mg, 0.075 mmol, 1.0 eq) in DCM (0.25 mL). The reaction mixture was stirred at room temperature for 1 h then concentrated under reduced pressure co- evaporating with toluene to afford the title compound which was used in the next step (45 mg, Yield: quantitative). MS: ES+ 274.2 (M+1) Step 3: (E)-4-(4-(4-(Dimethylamino)-N-methylbut-2-enamido)-5-methylisoindoline-2-carbonyl)- 5,6-dimethyl-1,3-phenylene diacetate

[001087] To a solution of 4,6-diacetoxy-2,3-dimethyl-benzoic acid (CAS 1081769-29-0, 49 mg, 0.183 mmol, 1.2 eq), HOBT (23.4 mg, 0.152 mmol, 1.0 eq) and EDC (88 mg, 0.458 mmol, 3.0 eq) in DCM (0.9 mL) was added DIEA (53 µL, 0.305 mmol, 2.0 eq) and the reaction mixture stirred for 10 mins. (E)-4-(Dimethylamino)-N-methyl-N-(5-methylisoindolin-4-yl)but-2-enamide- 2,2,2-trifluoroacetic acid (Step 2) (76 mg, 0.153 mmol, 1.0 eq) and DIEA (0.080 mL, 0.458 mmol, 3.0 eq) in DMF (0.9 mL) were added and reaction mixture was stirred at room temperature for 16 h. The reaction mixture was concentrated under reduced pressure. The residue was purified by chromatography on silica eluting with a gradient of 0-50% DCM in MeOH to afford the title compound which was used in next step without further purification. MS: ES+ 522.2 (M+1) Step 4: (E)-N-(2-(4,6-Dihydroxy-2,3-dimethylbenzoyl)-5-methylisoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide

[001088] 12M conc. HCl (0.19 mL, 2.29 mmol, 10.0 eq) was added to a cooled stirred solution of (E)-4-(4-(4-(dimethylamino)-N-methylbut-2-enamido)-5-methylisoindoline-2-
carbonyl)-5,6-dimethyl-1,3-phenylene diacetate (Step 3) (119 mg, 0.229 mmol, 1.0 eq) in methanol (3.6 mL). The reaction mixture was stirred at room temperature for 16 h then concentrated under reduced pressure. The residue was purified by preparative HPLC (prep-LC- 2) to afford the title compound (8.5 mg, Yield: 8.1%) as a formate salt. [001089] ¹H NMR (400 MHz, DMSO) δ 2.04 - 1.91 (m, 12H), 2.25 - 2.09 (m, 3H), 2.89 - 2.81 (m, 2H), 3.00 (s, 1H), 3.14 (s, 2H), 4.86 - 4.25 (m, 4H), 5.74 - 5.57 (m, 1H), 6.34 - 6.23 (m, 1H), 6.77 - 6.55 (m, 1H), 7.38 - 7.10 (m, 2H), 8.25 (s, 1H), 9.29 (s, 2H). Rotamers present. [001090] LCMS (LC-Method 1): 2.32 min and 2.85 min* ES+ 438.5 (M+1). * Rotamers observed, which coalesce to show a single peak at 2.78 min at 80°C. Example 66: (E)-N-(2-(4,6-Dihydroxy-2,3-dimethylbenzoyl)-6-methylisoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide Step 1: 4-Bromo-3-hydroxy-6-methylisoindolin-1-one


[001091] The title compound was prepared from 3-bromo-5-methylbenzonitrile (CAS 124289-21-0) analogously to Example 54 Step 1. [001092] ¹H NMR (400 MHz, DMSO): δ 2.39 (s, 3H), 5.81 - 5.76 (m, 1H), 6.39 - 6.31 (m, 1H), 7.44 (s, 1H), 7.63 (s, 1H), 9.02 (s, 1H). MS: ES+ 243.9 (M+1) Step 2: 4-Bromo-6-methylisoindoline hydrochloride.

[001093] To a stirred solution of 4-bromo-3-hydroxy-6-methylisoindolin-1-one (Step 1) (4673 mg, 19.3 mmol, 1.0 eq) in toluene (17.5 mL) at 55 °C was added dropwise 5M borane dimethyl sulfide complex solution (8.5 mL, 42.5 mmol, 2.2 eq) over 10-15 minutes. The reaction mixture was heated to 110 °C and stirred for 16 h. The resulting mixture was cooled to 75 °C
then quenched with 6M HCl in IPA (9 ml). The reaction was allowed to stir at 75 °C for 1 h then cooled to room temperature and allowed to stir at room temperature for an additional 30 mins. The resulting suspension was collected by filtration and washed with toluene (2 x 50 mL) and ether (50 mL) to obtain the product as a mixture with the partially reduced lactam which was subjected to another round of reduction using the same conditions afford 4-bromo-6-methyl- isoindoline hydrochloride. [001094] ¹H NMR (400 MHz, DMSO) δ 2.33 (s, 3H), 4.44 (s, 2H), 4.58 (s, 2H), 7.24 (s, 1H), 7.44 (s, 1H), 9.77 (s, 2H). MS: ES+ 213.9 (M+1) Step 3: tert-butyl 4-bromo-6-methylisoindoline-2-carboxylate.

[001095] The title compound was prepared from 4-bromo-6-methylisoindoline hydrochloride (step 2) analogously to Example 54 Step 3. [001096] ¹H NMR (400 MHz, DMSO) δ 1.46 (s, 9H), 2.31 (s, 3H), 4.50 - 4.45 (m, 2H), 4.65 (d, J=8.7 Hz, 2H), 7.18 - 7.13 (m, 1H), 7.34 (s, 1H). MS: ES+ 213.9 (M+1) des-Boc mass observed Step 4: tert-Butyl 6-methyl-4-(methylamino)isoindoline-2-carboxylate

[001097] The title compound was prepared from 4-bromo-6-methylisoindoline hydrochloride (step 2) analogously to Intermediate A Step 1. [001098] ¹H NMR (400 MHz, DMSO) δ 1.47 - 1.45 (m, 9H), 2.23 (s, 3H), 2.70 - 2.67 (m, 3H), 4.36 - 4.30 (m, 2H), 4.48 - 4.43 (m, 2H), 5.34 - 5.31 (m, 1H), 6.19 (s, 1H), 6.33 (d, J=9.5 Hz, 1H) Step 5: N,6-Dimethylisoindolin-4-amine hydrochloride
[001099] To a stirred solution of tert-butyl 6-methyl-4-(methylamino)isoindoline-2- carboxylate (step 4) (362 mg, 1.38 mmol, 1.0 eq) in methanol (3 mL) was added 3M HCl in CPME (2.3 mL, 6.90 mmol, 5.0 eq). The reaction was stirred at room temperature for 6 hours then concentrated under reduced pressure to afford the title compound (224 mg, Yield: quantitative). MS: ES+ 163 (M+1) Step 6: [5-Acetoxy-2,3-dimethyl-4-[6-methyl-4-(methylamino)isoindoline-2-carbonyl]phenyl] acetate

[001100] To a stirred solution of HOBT (232 mg, 1.52 mmol, 1.1 eq), EDC (291 mg, 1.52 mmol, 1.1 eq) and 4,6-diacetoxy-2,3-dimethylbenzoic acid (CAS 1081769-29-0, 367 mg, 1.38 mmol, 1.00 eq) in DCM (5.3 mL) was added DIEA (1.2 mL, 6.90 mmol, 5.0 eq). The reaction mixture was stirred at room temperature for 10 minutes then N,6-dimethylisoindolin-4-amine hydrochloride (Step 5) (224 mg, 1.38 mmol, 1.0 eq) in DMF (13.3 mL) was added. The reaction mixture was stirred at room temperature for 16 hours then concentrated under reduced pressure. The residue was purified by chromatography on silica eluting with a gradient of 0-20% DCM in MeOH to afford the title compound (215 mg, Yield: 38%). MS: ES+ 411 (M+1) Step 7: [5-Acetoxy-4-[4-[[(E)-4-(dimethylamino)but-2-enoyl]-methyl-amino]-6-methyl-isoindoline- 2-carbonyl]-2,3-dimethyl-phenyl] acetate

[001101] To a cooled stirred suspension of (E)-4-(dimethylamino)but-2-enoyl chloride hydrochloride (CAS 1056149-69-9, 116 mg, 0.629 mmol, 1.2 eq) in acetonitrile (1.2 mL) under nitrogen was added dropwise a solution of [5-acetoxy-2,3-dimethyl-4-[6-methyl-4- (methylamino)isoindoline-2-carbonyl]phenyl] acetate (Step 6) (215 mg, 0.524 mmol, 1.0 eq) in acetonitrile (1.2 mL). The reaction was allowed to gradually warm to room temperature and stirred for 16 hours. The resulting mixture was concentrated under reduced pressure to afford the title compound (215 mg, Yield: 78%). MS: ES+ 522.6 (M+1) Step 8: (E)-N-[2-(4,6-Dihydroxy-2,3-dimethyl-benzoyl)-6-methyl-isoindolin-4-yl]-4- (dimethylamino)-N-methyl-but-2-enamide

[001102] 12M conc. HCl (0.069 mL, 0.824 mmol, 2.0 eq) was added to a solution of [5- acetoxy-4-[4-[[(E)-4-(dimethylamino)but-2-enoyl]-methyl-amino]-6-methyl-isoindoline-2- carbonyl]-2,3-dimethyl-phenyl] acetate (Step 7) (215 mg, 0.412 mmol, 1.0 eq) in methanol (5.0 mL). The mixture was stirred at room temperature for 16 hours then concentrated under reduced pressure. The residue was purified by preparative HPLC (prep-LC-3) to afford the title compound (47 mg, 26%) as the formate salt. [001103] ¹H NMR (400 MHz, DMSO) δ 2.09 - 1.90 (m, 12H), 2.35 - 2.30 (m, 3H), 2.94 - 2.81 (m, 2H), 3.21 - 3.05 (m, 3H), 4.48 - 4.04 (m, 2H), 4.86 - 4.50 (m, 2H), 5.82 - 5.69 (m, 1H), 6.32 - 6.21 (m, 1H), 6.70 - 6.56 (m, 1H), 7.27 - 7.04 (m, 2H), 8.17 (s, 1H), 9.28 - 9.18 (m, 2H). [001104] LCMS (LC-Method 1): 2.79 min, ES+ 438.5 (M+1) Example 67: (E)-N-(6-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-5,6,7,8- tetrahydro-1,6-naphthyridin-3-yl)-N-methylbut-2-enamide Step 1: tert-Butyl 3-(methylamino)-7,8-dihydro-1,6-naphthyridine-6(5H)-carboxylate

[001105] tert-Butyl 3-bromo-7,8-dihydro-5H-1,6-naphthyridine-6-carboxylate (CAS 1184950-48-4.514 mg, 1.6 mmol, 1.0 eq) and sodium tert-butoxide (394 mg, 4.1 mmol, 2.5 eq)
were dissolved in toluene (6.5 mL) and the mixture was degassed under a stream of nitrogen for 15 minutes. 2M Methylamine in THF (4.9 mL, 9.9 mmol, 6.0 eq), tris(dibenzylideneacetone)dipalladium(0) (60 mg, 0.07 mmol, 0.04 eq) and BrettPhos (53 mg, 0.1 mmol, 0.06 eq) were added and the reaction mixture was heated at 90 °C for 18 hours. The resulting mixture was diluted with water (75 mL) and extracted with EtOAc (3 x 75mL). The combined organic extracts were washed with brine (100 mL), dried over MgSO
4 filtered and concentrated under reduced pressure. The residue was purified by chromatography on silica eluting with a gradient of 0-10% DCM in MeOH to afford the title compound (345 mg, 80%). [001106]
1H NMR (400 MHz, DMSO) δ 1.42 (s, 9H), 2.71 - 2.65 (m, 5H), 3.60 - 3.55 (m, 2H), 4.44 (s, 2H), 5.74 - 5.67 (m, 1H), 6.65 (d, J = 3.0 Hz, 1H), 7.77 (d, J = 2.5 Hz, 1H). MS:ES+ 264.1 (M+1). Step 2: (E)-N-Methyl-N-(5,6,7,8-tetrahydro-1,6-naphthyridin-3-yl)but-2-enamide;dihydrochloride

[001107] tert-Butyl 3-(methylamino)-7,8-dihydro-1,6-naphthyridine-6(5H)-carboxylate (Step 1) (280 mg, 0.9 mmol, 1.0 eq) was dissolved in DMA (2.0 mL) and cooled to 0 °C. Crotonoyl chloride (0.1 mL, 1.0 mmol, 1.2 eq) was added slowly and the reaction was stirred at 0°C for 10 minutes then at room temperature for 18 hours. The solvent was removed under reduced pressure. The crude solid was dissolved in methanol (3.0 mL) and 3M HCl in CPME (5.7 mL, 17.0 mmol, 20 eq) was added. The reaction mixture was stirred for 4 hours. The solvent was removed under reduced pressure to afford the title compound (259 mg, 100%) as a brown solid. The compound was used crude in the next step. MS:ES+ 232.2 (M+1) Step 3: (E)-N-(6-(3-chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-5,6,7,8-tetrahydro-1,6- naphthyridin-3-yl)-N-methylbut-2-enamide
[001108] 4-Methylmorpholine (0.47 mL, 4.3 mmol, 15 eq) and 3-chloro-6-hydroxy-4- methoxy-2-methylbenzoic acid (Example 47 Step 3) (77 mg, 0.36 mmol, 1.3 eq) were added to a stirred mixture of (E)-N-methyl-N-(5,6,7,8-tetrahydro-1,6-naphthyridin-3-yl)but-2-enamide dihydrochloride (Step 2) (87 mg, 0.28 mmol, 1.0 eq), EDC (218 mg, 1.1 mmol, 4.0 eq) and HOBT (52 mg, 0.34 mmol, 1.20 eq) in DCM (2.0 mL) . The mixture was stirred at room temperature for 18 hours. The resulting mixture was diluted with DCM (ca.5 mL) and water (ca.5 mL), filtered through a phase separator and the organic layer was concentrated under reduced pressure. The residue was purified by preparative HPLC (prep LC-9) to afford the title compound (13 mg, Yield: 10%) as an off-white solid. [001109] ¹H NMR (400 MHz, DMSO) δ 1.77 - 1.70 (m, 3H), 2.16 - 2.05 (m, 3H), 2.99 - 2.78 (m, 2H), 3.26 - 3.16 (m, 3H), 3.56 - 3.50 (m, 1H), 4.13 - 3.78 (m, 4H), 4.45 - 4.36 (m, 1H), 4.97 - 4.77 (m, 1H), 5.92 - 5.70 (m, 1H), 6.53 - 6.48 (m, 1H), 6.82 - 6.69 (m, 1H), 7.74 - 7.53 (m, 1H), 8.29 - 8.26 (m, 1H), 10.07 - 9.99 (m, 1H). [001110] LCMS (LC-Method 3): 3.42 min, ES+ 430.39 (M+1) Example 68: N-(2-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-1,2,3,4- tetrahydroisoquinolin-7-yl)-N-methylacrylamide Step 1: tert-Butyl 7-(methylamino)-3,4-dihydroisoquinoline-2(1H)-carboxylate

[001111] A mixture of tert-butyl 7-bromo-3,4-dihydroisoquinoline-2(1H)-carboxylate (CAS 258515-65-0, 500 mg, 1.6 mmol, 1.0 eq) and sodium tert-butoxide (385 mg, 4.0 mmol, 2.5 eq) in toluene (15.0 mL) was degassed with nitrogen for 10 mins. 2M methylamine in tetrahydrofuran (0.88 mL, 1.76 mmol, 1.1 eq), tris(dibenzylideneacetone)dipalladium(0) (59 mg, 0.064 mmol, 0.04
eq) and BrettPhos (52 mg, 0.096 mmol, 0.06 eq) were added the mixture was heated to 120 °C using microwave irradiation for 1 h. The resulting mixture was poured into water (50 mL) then extracted with EtOAc (3 × 50 mL). The combined organic extracts were washed with brine (50 mL), dried over MgSO
4 and concentrated under reduced pressure. The crude material was purified by chromatography on silica eluting with a gradient of 0 – 30% EtOAc in cyclohexane to afford the title compound as an orange oil (284 mg, 68%). [001112] ¹H NMR (400 MHz, DMSO) δ 1.42 (s, 9H), 2.60 (t, J=5.9 Hz, 2H), 2.63 (d, J=5.3 Hz, 3H), 3.49 (t, J=5.9 Hz, 2H), 4.36 (s, 2H), 5.45 - 5.41 (m, 1H), 6.28 (s, 1H), 6.40 - 6.37 (m, 1H), 6.85 (d, J=8.3 Hz, 1H). MS: ES+ 263.1 (M+1) Step 2: tert-Butyl 7-(N-methylacrylamido)-3,4-dihydroisoquinoline-2(1H)-carboxylate

[001113] To a cooled (0 °C), stirred solution of tert-butyl 7-(methylamino)-3,4- dihydroisoquinoline-2(1H)-carboxylate (Step 1) (284 mg, 1.08 mmol, 1.0 eq) in pyridine (5.0 mL) was added prop-2-enoyl prop-2-enoate (0.14 mL, 1.19 mmol, 1.1 eq). The vessel was sealed and stirred for 16 h. The reaction was quenched with 7N ammonia in methanol (2.0 mL) and concentrated under reduced pressure. The crude material was purified by chromatography on silica, eluting with a gradient of 0-50 % EtOAc in cyclohexane to afford the title compound as a yellow oil (157 mg, 46%). [001114] ¹H NMR (400 MHz, DMSO) δ 1.43 (s, 9H), 2.79 (t, J=6.2 Hz, 2H), 3.22 (s, 3H), 3.57 (t, J=5.6 Hz, 2H), 4.52 (s, 2H), 5.57 - 5.53 (m, 1H), 6.25 - 5.83 (m, 2H), 7.08 - 7.04 (m, 1H), 7.14 (d, J=2.2 Hz, 1H), 7.24 (d, J=7.9 Hz, 1H). MS: ES+ 317.0 (M+1) Step 3: N-Methyl-N-(1,2,3,4-tetrahydroisoquinolin-7-yl)acrylamide; trifluoroacetate

[001115] To a solution of tert-butyl 7-(N-methylacrylamido)-3,4-dihydroisoquinoline-2(1H)- carboxylate (Step 2) (188 mg, 0.59 mmol, 1.0 eq) in DCM (3.0 mL) was added TFA (1.0 mL, 13.1 mmol, 22 eq) and the reaction mixture was stirred under an inert atmosphere at room temperature for 1 h. The resulting mixture was concentrated under reduced pressure to give the title
compound as a yellow oil (196 mg, quant.). The material was used in the next step without further purification. [001116] MS: ES+ 217.1 (M+1) Step 4: N-(2-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7- yl)-N-methylacrylamide

[001117] To a solution of N-methyl-N-(1,2,3,4-tetrahydroisoquinolin-7-yl)acrylamide; trifluoroacetate (Step 3) (100 mg, 0.46 mmol, 1.0 eq), 3-chloro-6-hydroxy-4-methoxy-2- methylbenzoic acid (Example 47 Step 3) (120 mg, 0.55 mmol, 1.2 eq), EDCI (354 mg, 1.85 mmol, 4 eq) and HOBT (71 mg, 0.46 mmol, 1.0 eq) in DCM (3.5 mL) was added NMM (0.51 mL, 4.6 mmol, 10.0 eq) and the reaction mixture stirred under an inert atmosphere at room temperature for 16 h. The resulting mixture was diluted with DCM (20 mL) and washed with water (2 × 10 mL) and brine (10 mL). The combined organic extracts were filtered through a hydrophobic frit and the solvent was concentrated under reduced pressure. The crude material was purified by preparative HPLC using Method prep-LC-9 to afford the title compound as an off-white solid (10 mg, 5%) [001118] ¹H NMR (400 MHz, DMSO) δ 2.13 - 2.02 (m, 3H), 2.75 - 2.67 (m, 1H), 2.89 - 2.79 (m, 1H), 3.25 - 3.18 (m, 3H), 3.43 - 3.39 (m, 1H), 4.01 - 3.70 (m, 4H), 4.43 - 4.30 (m, 1H), 4.87 - 4.71 (m, 1H), 5.60 - 5.51 (m, 1H), 6.18 - 5.95 (m, 2H), 6.58 - 6.48 (m, 1H), 7.27 - 7.01 (m, 3H), 10.07 - 9.79 (m, 1H). [001119] LCMS (LC-Method 3): 3.94 min, MS:ES+ 415.18 (M+1) Example 69: N-(2-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)-1,2,3,4- tetrahydroisoquinolin-7-yl)-N-(1-methylpyrrolidin-3-yl)acrylamide Step 1: tert-Butyl 7-((1-methylpyrrolidin-3-yl)amino)-3,4-dihydroisoquinoline-2(1H)-carboxylate

[001120] Performed in 2 parallel batches, each of 0.5 g scale: To a stirred solution of tert- butyl 7-bromo-3,4-dihydroisoquinoline-2(1H)-carboxylate (0.5 g, 1.60 mmol, 1.0 eq.) (CAS: 258515-65-0) in 1,4-dioxane (5 mL) were added sodium tert-butoxide (0.384 g, 4.00 mmol, 2.5 eq.) and 1-methylpyrrolidin-3-amine (CAS 13220-27-4) at room temperature. The mixture was purged with N
2(g) for 20 mins. Brettphos (0.051 g, 0.096 mmol, 0.06 eq) and Pd
2(dba)
3 (0.058 g, 0.064 mmol, 0.04 eq) were added and the reaction mixture was heated to 100°C for 30 mins. The resulting mixture was cooled to room temperature, diluted with water (50 mL) and extracted with EtOAc (3 x 50 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluting in 15% MeOH: DCM) to yield tert-butyl 7-((1-methylpyrrolidin-3-yl)amino)-3,4- dihydroisoquinoline-2(1H)-carboxylate as a light brown liquid (1.05 g, Yield: 98 %). [001121]
1H NMR (DMSO-d6, 400 MHz): δ 1.23 - 1.35 (m, 1H), 1.42 (s, 9H), 1.50 - 1.56 (m, 1H), 2.16 - 2.20 (m, 1H), 2.22 (s, 3H), 2.25 - 2.33 (m, 1H), 2.38 – 2.44 (m, 1H), 2.56 - 2.60 (m, 2H), 2.73 (t, J= 9.2 Hz, 1H), 3.48 (t, J= 5.6 Hz, 2H), 3.83 (bs, 1H), 4.36 (s, 2H), 5.52 (d, J= 7.2 Hz, 1H, D
2O exchangeable), 6.28 (s, 1H), 6.40 (d, J= 8.0 Hz, 1H), 6.83 (d, J= 8.4 Hz, 1H). [001122] LCMS (Method N): 1.737 min, MS: ES+ 331.9 (M+1) Step 2: N-(1-Methylpyrrolidin-3-yl)-1,2,3,4-tetrahydroisoquinolin-7-amine hydrochloride
[001123] The title compound was prepared from tert-butyl 7-((1-methylpyrrolidin-3- yl)amino)-3,4-dihydroisoquinoline-2(1H)-carboxylate (Step 1) and 4M HCl in dioxane analogously to Example 4 Step 2. [001124]
1H NMR (DMSO-d6, 400 MHz): δ 1.85 - 1.95 (m, 1H), 2.80 - 2.90 (m, 5H), 3.02 - 3.07 (m, 1H), 3.28 - 3.36 (m, 4H), 3.54 (s, 2H), 4.12 - 4.22 (m, 3H), 6.45 (d, J= 7.6 Hz, 1H), 6.97 - 6.99 (m, 1H), 6.97 - 6.99 (m, 1H), 9.49 (s, 2H). [001125] LCMS (Method O): 1.10 min, MS: ES+ 232.2 (M+1)
Step 3: (2-Hydroxy-5-isopropyl-4-methoxyphenyl)(7-((1-methylpyrrolidin-3-yl)amino)-3,4- dihydroisoquinolin-2(1H)-yl)methanone
[001126] The title compound was prepared from 2-hydroxy-5-isopropyl-4-methoxybenzoic acid (Example 45 Step 3) and N-(1-methylpyrrolidin-3-yl)-1,2,3,4-tetrahydroisoquinolin-7-amine hydrochloride (Step 2) analogously to Example 9 Step 1. This material was used directly in the next step [001127] LCMS (Method N): 1.812 min, MS: ES+ 424.4 (M+1). Step 4: N-(2-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- (1-methylpyrrolidin-3-yl)acrylamide
[001128] The title compound was prepared from (2-hydroxy-5-isopropyl-4- methoxyphenyl)(7-((1-methylpyrrolidin-3-yl)amino)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (Step 3) and acrylic anhydride analogously to Example 9 Step 2. [001129] HT
1H NMR (DMSO-d6, 400 MHz): δ 1.14 (d, J= 6.8 Hz, 6H), 2.03 (m, 1H), 2.13 (s, 3H), 2.33 - 2.37 (m, 1H), 2.40 - 2.46 (m, 2H), 2.74 (t, J= 8.8 Hz, 1H), 2.86 - 2.92 (m, 3H), 3.67 - 3.75 (m, 2H), 3.79 (s, 3H), 4.66 - 4.68 (m, 2H), 4.95 (m, 1H), 5.45 - 5.48 (m, 1H), 5.82 - 5.89 (m, 1H), 6.08 - 6.13 (m, 1H), 6.51 (s, 1H), 6.98 - 7.05 (m, 3H), 7.19 - 7.27 (m, 1H), 9.56 (bs, 2H). [001130] LCMS (Method_A1): 6.79 min, 7.48 min, MS: ES+ 478.5 (M+1). [001131] HPLC (Method H): 4.36 min, 4.67 min, 4.77 min, Prep. HPLC purification method:
[001132] Chromatographic separation and isolation were conducted with Shimadzu LC20AP with UV detector; column Waters X-Bridge C18 (250mm x 19mm 5μm); compound eluted with: Mobile Phase A: 5 mM ammonium bicarbonate + 0.05% NH
3 in Merck water, Mobile Phase B: Acetonitrile with a gradient of T = 0.00 min (48% A, 52% B); gradient to T = 35.00 min (48% A, 52% B); T = 35.01 min (0% A, 100% B) gradient to T = 38.00 min (0% A, 100% B); gradient to T = 38.01 min (48% A, 52% B); gradient to T = 40.00 min (48% A, 52% B); Flow rate= 10ml/min; analysis time 40 min. Example 70: (E)-N-(5-Chloro-2-(2-hydroxy-5-isopropyl-4-methoxybenzoyl)isoindolin-4- yl)-4-(dimethylamino)but-2-enamide Step 1: tert-Butyl 4-amino-5-chloroisoindoline-2-carboxylate
[001133] To a stirred solution of tert-butyl 4-aminoisoindoline-2-carboxylate (CAS 871013- 98-8) (10.0 g, 0.042 mmol, 1.0 eq.) in ACN (150 mL) was added NCS (5.71 g, 0.042 mmol, 1.0 eq.) and the reaction mixture was stirred at room temperature for 24 h. The resulting mixture was poured in water (400 mL), filtered and the solid was washed with water (100 mL). The aqueous filtrate was extracted with EtOAc (5 x 100 mL), dried over Na
2SO
4 and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted at 12.0% EtOAc in hexane) to yield tert-butyl 4-amino-5-chloroisoindoline-2-carboxylate (1.7 g, Yield: 15%). [001134]
1H NMR (DMSO-d6, 400 MHz): δ 1.45 (d, J= 5.6 Hz, 9H), 4.46 (dd, J= 21.6 Hz, J= 11.2 Hz, 4H), 5.37 (d, J= 8.8 Hz, 2H, D2O exchangeable), 6.51 (t, J= 6.8 Hz, J= 14.0 Hz, 1H), 7.12 (d, J= 8.0 Hz, 1H) [001135] LCMS (Method A): 2.357 min, MS: ES+ 213 (M-56). Step 2: tert-Butyl (E)-5-chloro-4-(4-(dimethylamino)but-2-enamido)isoindoline-2-carboxylate
[001136] The title compound was prepared from tert-butyl 4-amino-5-chloroisoindoline-2- carboxylate (Step 1) and (2E)-4-(dimethylamino)but-2-enoic acid hydrochloride (CAS: 848133- 35-7) analogously to Example 50 Step 1; the resultant material was used directly in the next step. [001137] LCMS (Method: N): 1.723 min, MS ES+: 380.2, 382.2 (M, M+2). Step 3: (E)-N-(5-Chloroisoindolin-4-yl)-4-(dimethylamino)but-2-enamide hydrochloride
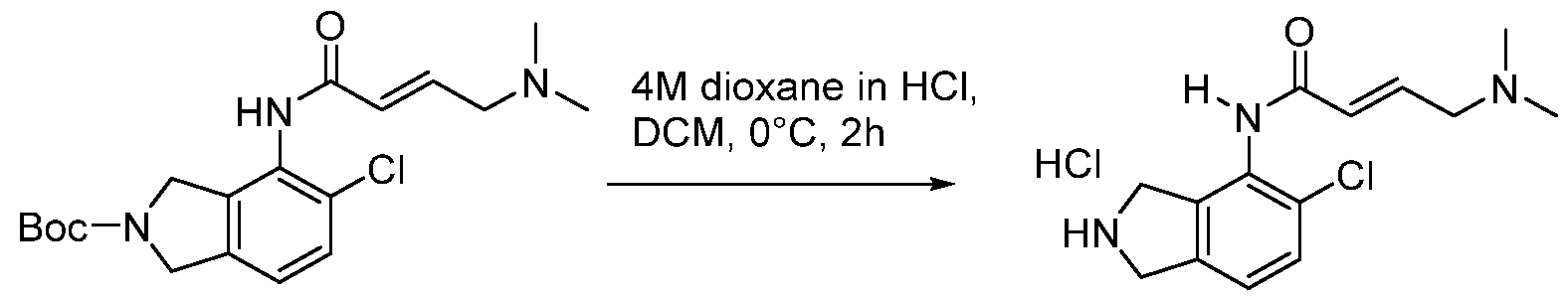
[001138] The title compound was prepared from tert-butyl (E)-5-chloro-4-(4- (dimethylamino)but-2-enamido)isoindoline-2-carboxylate (Step 2) and 4M HCl in dioxane analogously to Example 4 Step 2; the resultant material was used directly in the next step. [001139] LCMS (Method N): 0.305 min, MS: ES+ 280.2, 282.1 (M, M+2). Step 4: ((E)-N-(5-Chloro-2-(2-hydroxy-5-isopropyl-4-methoxybenzoyl)isoindolin-4-yl)-4- (dimethylamino)but-2-enamide
[001140] The title compound was prepared from 2-hydroxy-5-isopropyl-4-methoxybenzoic acid (Example 45 Step 3) and ((E)-N-(5-chloroisoindolin-4-yl)-4-(dimethylamino)but-2-enamide hydrochloride (Step 3) analogously to Example 9 Step 1. [001141]
1H NMR (DMSO-d6, 400 MHz): δ 1.11 (m, 6H), 2.14 - 2.19 (m, 6H), 3.02 - 3.13 (m, 3H), 3.76 (s, 3H), 4.65 - 4.87 (m, 4H), 6.25 - 6.48 (m, 2H), 6.67 - 6.81 (m, 1H), 7.07 - 7.49 (m, 3H), 9.88 - 9.97 (m, 1H), 10.45 (bs, 1H) [001142] LCMS (METHOD: T): 1.351 min, MS ES+: 472.02, 474.3 (M, M+2).
[001143] HPLC (METHOD: I): 7.175 min Prep. HPLC purification method: [001144] Chromatographic separation and isolation was conducted using a Shimadzu LC20AP system with UV detector; column Waters X-Bridge C18 (250mm x 19mm 5µm); compound was eluted with: Mobile Phase A: 5mM ammonium bicarbonate + 0.05% NH
3 in Merck water. Mobile Phase B: ACN:MeOH:THF (50:50:10) with a gradient of T = 0.00 min (88% A, 12% B); gradient to T = 50.00 min (88% A, 12% B); T = 50.01min (00% A, 100% B), gradient to T = 53.00min (0% A, 100% B); T = 53.01 min (88% A, 12% B); gradient to T = 60.00 min (88% A, 12% B); Flow rate= 8 ml/min; analysis time 60.00 min. Example 71: (E)-N-(2-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)isoindolin-4-yl)-4- (dimethylamino)but-2-enamide Step 1: tert-Butyl (E)-4-(4-(dimethylamino)but-2-enamido)isoindoline-2-carboxylate
[001145] The title compound was prepared from tert-butyl 4-aminoisoindoline-2- carboxylate (CAS:871013-98-8) and (E)-4-(dimethylamino)but-2-enoic acid hydrochloride (CAS: 848133-35-7) analogously to Example 1 Step 2. [001146]
1H NMR (DMSO-d6, 400 MHz): 1.46 (s, 9H), 2.69 (s, 6H), 3.81 (m, 2H), 4.59 (t, J= 11.2 Hz, 4H), 6.56 - 6.59 (m, 1H), 6.76 - 6.84 (m, 1H), 7.13 (m, 1H), 7.28 (t, J= 8.0 Hz, 1H), 7.63 (d, J= 7.6 Hz, 1H), 9.93 (d, J= 8.0 Hz, 1H). [001147] LCMS (Method N): 1.613 min, MS: ES+ 346.3 (M+1). Step 2: (E)-4-(Dimethylamino)-N-(isoindolin-4-yl)but-2-enamide hydrochloride
[001148] The title compound was prepared from tert-butyl (E)-4-(4-(dimethylamino)but-2- enamido)isoindoline-2-carboxylate (Step 1) and 4M HCl in dioxane analogously to Example 4 Step 2. [001149]
1H NMR (DMSO-d6, 400 MHz): 2.75 - 2.76 (m, 8H), 4.48 - 4.52 (m, 4H), 6.60 - 6.64 (m, 1H), 6.81 - 6.87 (m, 1H), 7.21 (d, J= 7.2 Hz, 1H), 7.36 (t, J= 7.6 Hz, 1H), 7.54 (d, J= 7.6 Hz, 1H), 10.05 (bs, 2H). [001150] LCMS (Method_A2): 3.11 min, MS: ES+ 246.1 (M+1). Step 3: (E)-N-(2-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)isoindolin-4-yl)-4- (dimethylamino)but-2-enamide
[001151] The title compound was prepared from 3-chloro-6-hydroxy-4-methoxy-2- methylbenzoic acid (Example 47 Step 3) and (E)-4-(dimethylamino)-N-(isoindolin-4-yl)but-2- enamide hydrochloride (Step 2) analogously to Example 9 Step 1. [001152] HT
1H NMR (DMSO-d6, 400 MHz, 348K): δ 2.22 (d, J= 4.0 Hz, 6H), 2.20 (s, 3H), 3.05 - 3.07 (m, 1H), 3.10 - 3.18 (m, 1H), 3.83 (s, 3H), 4.41 - 4.47 (m, 1H), 4.56 - 4.61 (m, 1H), 4.80 - 4.83 (m, 2H), 6.40 - 6.44 (m, 1H), 6.57 (s, 1H), 6.68 - 6.81 (m, 1H), 7.02 - 7.17 (m, 1H), 7.23 - 7.31 (m, 1H), 7.67 (dd, J= 8.0 Hz, 1H), 9.53 (s, 1H), 9.74 (bs, 1H). [001153] LCMS (Method N): 1.626 min, MS: ES+ 444.23 (M+1). [001154] HPLC (Method I): 5.787 min, Prep. HPLC purification method of analysis: [001155] Chromatographic separation and isolation were conducted with a Waters 2545 quaternary system with Waters 2489 UV Detector; column Waters X-Bridge Prep C18 (250mm x 19mm 5μm); compound eluted with: Mobile Phase A: 5mM ammonium bicarbonate + 0.05% ammonium hydroxide in water, Mobile Phase B: acetonitrile with a gradient of T = 0.01 (75% A, 25% B); gradient to T = 30.00 min (65% A, 35% B); T = 30.01 min (0% A, 100% B) gradient to T = 32.00 min (0% A, 100% B); T = 32.01 min (75% A, 25% B); gradient to T = 40.00 min (75% A, 25% B); gradient); Flow rate= 8.0ml/min; analysis time 40.00 min.
Example 72: N-(6-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-5,6,7,8-tetrahydro- 1,6-naphthyridin-3-yl)acrylamide Step 1: tert-Butyl 3-acrylamido-7,8-dihydro-1,6-naphthyridine-6(5H)-carboxylate
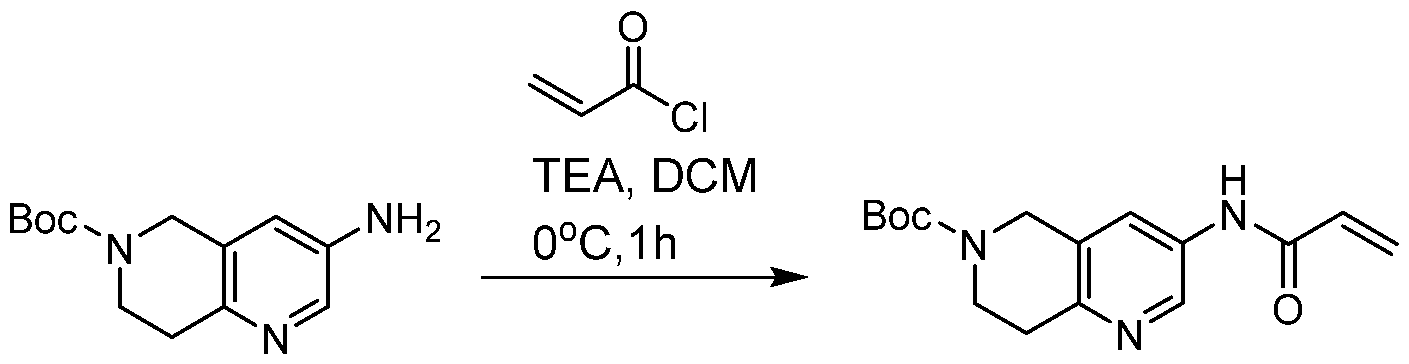
[001156] To a cooled (0 ˚C) solution of tert-butyl 3-amino-7,8-dihydro-1,6-naphthyridine- 6(5H)-carboxylate (CAS: 355819-02-2) (0.50 g, 2.00 mmol, 1.0 eq.) in DCM (5 mL) was added TEA (0.83 mL, 6.01 mmol, 3.0 eq.) and the mixture was stirred at 0°C for 15 mins. Acryloyl chloride (0.27 g, 3.008 mmol, 1.5 eq.) was dropwise and stirring continued at 0°C for approx.50 mins. The resulting mixture was diluted with water (100 mL) and extracted with DCM (3 x 30 mL). The combined organic extracts were dried over Na2SO4, filtered, and reduced under vacuum. Crude material was purified by chromatography on silica (product eluted at 5% methanol in dichloromethane) yielding tert-butyl 3-acrylamido-7,8-dihydro-1,6-naphthyridine-6(5H)- carboxylate as yellow solid (0.48 g, Yield: 89.9 %). [001157]
1H NMR (DMSO-d6, 400 MHz): δ 1.43 (s, 9H), 2.82 (t, J= 5.6 Hz, 2H), 3.65 (t, J= 6 Hz, 2H), 4.53 (s, 2H), 5.78 - 5.81 (m, 1H), 6.26 - 6.30 (m, 1H), 6.40 – 6.47 (m, 1H), 7.94 (s, 1H), 8.55 (s, 1H), 10.33 (s, 1H). [001158] LCMS (Method N): 1.676 min, MS ES+: 304.2 (M+1). Step 2: N-(5,6,7,8-Tetrahydro-1,6-naphthyridin-3-yl) acrylamide TFA salt
[001159] To a cooled (0 ˚C) solution of tert-butyl 3-acrylamido-7,8-dihydro-1,6- naphthyridine-6(5H)-carboxylate (0.47 g, 1.54 mmol, 1.0 eq) in DCM (4.7 mL) was added dropwise TFA (2.35 mL) and the reaction mixture was allowed to warm to room temperature stirring for 1 h. The resulting mixture was concentrated under vacuum. The crude material was triturated with n-pentane (3 x 15 mL) and dried under high vacuum to yield N-(5,6,7,8-tetrahydro- 1,6-naphthyridin-3-yl) acrylamide TFA salt as a yellow liquid (0.71 g, Yield: quantitative).
[001160]
1H NMR (DMSO-d6, 400 MHz): δ 3.04 (t, J= 6 Hz, 2H), 3.49 (d, J= 6.4 Hz, 2H), 4.35 (s, 2H), 5.81 - 5.84 (dd, J= 1.6 Hz, 1H), 6.27 - 6.48 (m, 2H), 8.08 (s, 1H), 8.62 (d, J= 2 Hz ,1H), 9.12 (s, 2H), 10.47 (s, 1H). [001161] LCMS (Method O): 0.98 min, MS ES+: 204.2 (M+1). Step 3: N-(6-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-5,6,7,8-tetrahydro-1,6- naphthyridin-3-yl)acrylamide
[001162] The title compound was prepared from 3-chloro-6-hydroxy-4-methoxy-2- methylbenzoic acid (Example 47 Step 3) and N-(5,6,7,8-tetrahydro-1,6-naphthyridin-3-yl) acrylamide TFA salt (Step 2) analogously to Example 9 Step 1. [001163] HT
1H NMR (DMSO-d6, 400 MHz, 348K): δ 2.15 (s, br, 3H), 2.79 - 2.91 (m, 2H), 3.52 (s, 1H), 3.82 (s, 3H), 3.96 (s, 1H), 4.39 (s, 1H), 4.81 – 4.85 (m, 1H), 5.78 (d, J= 10 Hz, 1H), 6.26 - 6.52 (m, 3H), 7.78 - 7.95 (m, 1H), 8.61 (s, 1H), 9.62 (bs, 1H), 10.10 (s, 1H) [001164] LCMS (Method N): 1.659 min, MS: ES+ 402.1 (M+1). [001165] HPLC (Method H): 4.87 min. Prep. HPLC purification method of analysis: [001166] Chromatographic separation and isolation were conducted with a Shimadzu LC20AP with UV detector; column REDISEP GOLD C18100g 50µm; compounds eluted with: Mobile Phase A: 5mM ammonium bicarbonate +0.05% NH3 in Merck water , Mobile Phase B: Acetonitrile with a gradient of T = 0.00 min (57% A, 18% B); gradient to T = 20.00 min (61% A, 39% B); T = 20.01 min (00% A, 100% B) gradient to T = 23.00 min (0% A, 100% B); T = 23.01 min (82% A, 18% B); gradient to T = 30.00 min (82% A, 18% B); Flow rate= 10 ml/min; analysis time 30 min. Example 73: (E)-N-(2-(3-Chloro-6-hydroxy-2-methylbenzoyl)isoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide Step 1: Methyl 3-chloro-6-methoxy-2-methylbenzoate
[001167] To a stirred solution of methyl 2-methoxy-6-methylbenzoate (0.65 g, 3.61 mmol, 1.0 eq) in DMF (6.5 mL) was added NCS (0.57 g, 4.33 mmol, 1.2 eq.) and the mixture stirred at room temperature for 3 days. The resulting mixture was poured into cold water (100 mL) and extracted with EtOAc (3 x 500 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under vacuum. Crude material was purified by chromatography on silica (product eluted in 10% EtOAc in hexane) to yield methyl 3-chloro-6-methoxy-2- methylbenzoate as a yellow oil (0.6 g, 77% yield). [001168]
1H NMR (CDCl3, 400 MHz): δ 2.30 (s, 3H), 3.82 (s, 3H), 3.94 (s, 3H), 6.74 (d, J= 8.8 Hz, 1H), 7.34 (d, J= 8.8 Hz, 1H) Step 2: Methyl 3-chloro-6-methoxy-2-methylbenzoate
[001169] To a stirred solution of methyl 3-chloro-6-methoxy-2-methylbenzoate (Step 1) (0.6 g, 2.79 mmol, 1.0 eq.) in DCM (14 mL) was added portion wise anhydrous AlCl3 (1.49 g, 11.18 mmol, 4.0 eq.) and the reaction mixture was heated to 45°C for 4 h. The resulting mixture was poured in dilute HCl solution (30 mL) and extracted with DCM (3 x 50 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted in 5% EtOAc in hexane) to yield methyl 3-chloro-6-methoxy-2-methylbenzoate as a yellow gummy (0.45 g, 80% yield). [001170]
1H NMR (CDCl3, 400 MHz): δ 2.61 (s, 3H), 4.01 (s, 3H), 6.83 (d, J= 8.8 Hz, 1H), 7.42 (d, J= 8.8 Hz, 1H), 10.89 (s, 1H) Step 3: 3-Chloro-6-hydroxy-2-methylbenzoic acid
[001171] A solution of methyl 3-chloro-6-methoxy-2-methylbenzoate (Step 2) (0.45 g, 2.24 mmol, 1.0 eq) in 1.5M NaOH solution (22.5 mL) was heated to 100°C for 2 h. The resulting mixture was cooled to room temperature and extracted with EtOAc (2 x 50 mL). The aqueous layer was acidified with 1N HCl solution (30 mL) and extracted with EtOAc (2 x 100 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under high vacuum yielding 3-chloro-6-hydroxy-2-methylbenzoic acid as a white solid (0.35 g, Yield: 83%). [001172]
1H NMR (DMSO-d6, 400 MHz): δ 2.23 (s, 3H), 6.74 (d, J= 8.8 Hz, 1H), 7.26 (d, J= 8.8 Hz, 1H), 10.55 (bs, 1H), 13.09 (bs, 1H) [001173] LCMS (Method N): 1.352 min, MS ES+: 184.9 (M-1) Step 4: (E)-N-(2-(3-Chloro-6-hydroxy-2-methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N- methylbut-2-enamide
[001174] The title compound was prepared from 3-chloro-6-hydroxy-2-methylbenzoic acid (Step 3) and (E)-4-(dimethylamino)-N-(isoindolin-4-yl)-N-methylbut-2-enamide hydrochloride (Intermediate S) analogously to Example 9 Step 1. [001175] HT
1H NMR (DMSO-d6, 400 MHz, 348K): δ 2.06 (s, 6H), 2.17 - 2.18 (m, 3H), 2.85 - 2.91 (m, 2H), 3.25 (s, 3H), 4.46 - 4.64 (m, 2H), 4.71 - 4.91 (m, 2H), 5.68 – 5.79 (m, 1H), 6.56 - 6.82 (m, 2H), 7.19 - 7.45 (m, 4H), 9.77 (bs, 1H) [001176] LCMS (Method V): 5.882 min, MS ES+: 428.3, 430.3 (M, M+2). [001177] HPLC (Method O): 5.882 min. Prep. HPLC purification method: [001178] Chromatographic separation and isolation were conducted Shimadzu Nexera prep with LH-40 auto purification system; column YMC Triart C18; 250 x 20 mm ID, 5µm); compounds eluted with: Mobile Phase A: 5mM ammonium bicarbonate + 0.05% NH4OH in water. Mobile Phase B: ACN with a gradient of T = 0.01 min (76% A, 24% B); gradient to T = 19.00 min (44% A, 56% B); T = 19.01min (00% A, 100% B), gradient to T = 21.00min (0% A, 100% B); T = 21.01 min (76% A, 24% B); gradient to T = 24.00 min (76% A, 24% B); Flow rate= 16 ml/min; analysis time 24.00 min.
Example 74: (E)-N-(7-Chloro-2-(3-chloro-6-hydroxy-4-methoxy-2-methylbenzoyl) isoindolin-4-yl)-4-(dimethylamino)-N-methylbut-2-enamide Step 1: tert-Butyl 4-amino-7-chloroisoindoline-2-carboxylate

[001179] To a stirred solution of tert-butyl 4-aminoisoindoline-2-carboxylate (10.0 g, 42.7 mmol, 1.0 eq.) in ACN (150 mL) was added portion wise NCS (5.71 g, 42.7 mmol, 1.0 eq) and the reaction mixture was stirred at room temperature for 24 h. The resulting mixture was diluted with water (400 mL) and filtered. The solid material was washed with water (100 mL) and dried under vacuum to yield tert-butyl 4-amino-7-chloroisoindoline-2-carboxylate as an off-white solid (5.5 g, Yield: 48.0 %). [001180]
1H NMR (DMSO-d6, 400 MHz): δ 1.46 (s, 9H), 4.42 - 4.49 (m, 4H), 5.32 (d, J= 10.4 Hz, 2H, D
2O exchangeable), 6.50 (d, J= 8.8 Hz, 1H), 6.98 (d, J= 8.4 Hz, 1H). [001181] LCMS (Method N): 2.31 min, MS: ES+ 213.1 (M-56). Step 2: tert-Butyl 4-chloro-7-(methylamino)isoindoline-2-carboxylate
[001182] To a cooled (0 ˚C) solution of NaOMe (6.01 g, 111.63 mmol, 10.0 eq.) in MeOH (45 mL) was added tert-butyl 4-amino-7-chloroisoindoline-2-carboxylate (Step 1) (3.0 g, 11.163 mmol, 1.0 eq.) followed by paraformaldehyde (0.67 g, 22.33 mmol, 2.0 eq) and the reaction mixture stirred at room temperature for 16 h. The mixture was cooled to 0°C and treated portion wise with NaBH
4 (2.12 g, 55.82 mmol, 5.0 eq) added over 20 mins. The reaction mixture was allowed to warm to room temperature and stirred for 3 h. The resulting mixture was diluted with ice cold water (200 mL) and extracted with ethyl acetate (2 x 150 mL). The combined organic extracts were washed with sat. brine solution (150mL), dried over Na2SO4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica eluting
with a gradient of 0-100% EtOAc: hexane (product was eluted at 15% EtOAc: hexane) to yield tert-butyl 4-chloro-7-(methylamino)isoindoline-2-carboxylate as light yellow solid (2.2 g, Yield: 69%). [001183]
1H NMR (DMSO-d6, 400 MHz): δ 1.46 (s, 9H), 2.68 - 2.69 (m, 3H), 4.29 - 4.50 (m, 4H), 5.57 - 5.60 (m, 1H, D
2O exchangeable), 6.42 (d, J= 8.8 Hz, 1H), 7.11 (d, J= 8.4 Hz, 1H), [001184] LCMS (Method N): 2.62 min, MS: ES+ 227.2 (M-56). Step 3: tert-Butyl (E)-4-chloro-7-(4-(dimethylamino)-N-methylbut-2-enamido) isoindoline-2- carboxylate
[001185] The title compound was prepared from tert-butyl 4-chloro-7- (methylamino)isoindoline-2-carboxylate (Step 2) and (E)-4-(dimethylamino)but-2-enoic acid hydrochloride (CAS: 848133-35-7) analogously to Example 1 Step 2. [001186]
1H NMR (DMSO-d6, 400 MHz): δ 1.44 (d, J= 6.4 Hz, 9H), 2.61 (s, 6H), 3.16 (s, 3H), 3.714 (m, 2H), 4.46 - 4.64 (m, 4H), 6.03 - 6.07 (m, 1H), 6.66 - 6.71 (m, 1H), 7.30 (d, J= 8.0 Hz, 1H), 7.50 (d, J= 8.4 Hz, 1H). [001187] LCMS (Method N): 1.802 min, MS: ES+ 394.2 (M+1). Step 4: (E)-N-(7-Chloroisoindolin-4-yl)-4-(dimethylamino)-N-methylbut-2-enamide hydrochloride
[001188] The title compound was prepared from tert-butyl (E)-4-chloro-7-(4- (dimethylamino)-N-methylbut-2-enamido) isoindoline-2-carboxylate (Step 3) and 4M HCl in dioxane analogously to Example 4 Step 2. LCMS (Method O): 1.24 min, MS: ES+ 294.2 (M+1).
Step 5: (E)-N-(7-Chloro-2-(3-chloro-6-hydroxy-4-methoxy-2-methylbenzoyl) isoindolin-4-yl)-4- (dimethylamino)-N-methylbut-2-enamide
[001189] The title compound was prepared from 3-chloro-6-hydroxy-4-methoxy-2- methylbenzoic acid (Example 47 Step 3) and (E)-N-(7-chloroisoindolin-4-yl)-4-(dimethylamino)- N-methylbut-2-enamide hydrochloride (Step 4) analogously to Example 9 Step 1. [001190] HT
1H NMR (DMSO-d6, 400 MHz, 348K): δ 2.06 (s, 6H), 2.3 (s, 3H), 2.90 - 2.95 (m, 3H), 3.25 (m, 2H), 3.81 - 3.83 (d, J= 5.2 Hz, 3H), 4.48 - 4.52 (m, 1H), 4.58 - 4.84 (m, 3H), 5.75 - 5.84 (m, 1H), 6.43 - 6.72 (m, 2H), 7.33 (d, J= 8.0 Hz, 1H), 7.49 - 7.55 (m, 1H), 10.15 (m, 1H). [001191] LCMS (Method T): 1.249 min, MS: ES+ 492.03 (M+1). [001192] HPLC (Method H): 3.83 min, Example 75: N-(7-Chloro-2-(3-chloro-6-hydroxy-4-methoxy-2-methylbenzoyl) isoindolin- 4-yl)-N-methyl acrylamide Step 1: tert-Butyl 4-chloro-7-(N-methylacrylamido) isoindoline-2-carboxylate
[001193] The title compound was prepared from tert-butyl 4-chloro-7- (methylamino)isoindoline-2-carboxylate (Example 74 Step 2) and acryloyl chloride analogously to Example 72 Step 1. [001194]
1H NMR (DMSO-d6, 400 MHz): δ 1.44 (d, J= 5.6 Hz, 9H), 3.15 (s, 3H), 4.42 (d, J= 16 Hz, 1H), 4.62 (d, J= 9.2 Hz, 3H), 5.57 (d, J= 10 Hz, 1H), 5.93 - 6.00 (m, 1H), 6.16 - 6.20 (m, 1H), 7.26 - 7.29 (m, 1H), 7.48 (d, J= 8.4 Hz, 1H). [001195] LCMS (Method N): 2.351 min, MS ES+: 281.1 (M-56).
Step 2: N-(7-Chloroisoindolin-4-yl)-N-methyl acrylamide TFA salt
[001196] The title compound was prepared from tert-butyl 4-chloro-7-(N-methylacrylamido) isoindoline-2-carboxylate (Example 74 Step 2) and TFA analogously to Example 72 Step 2. [001197]
1H NMR (DMSO-d6, 400 MHz): δ 3.16 (s, 3H), 4.39 (bs, 1H), 4.60 (s, 3H), 5.62 (d, J= 10 Hz, 1H), 5.95 - 5.99 (m, 1H), 6.21 (d, J= 17.6 Hz, 1H), 7.36 (d, J= 8.4 Hz, 1H), 7.57 (d, J= 8.4 Hz, 1H), 9.69 (s, 1H). [001198] LCMS (Method O): 1.407 min, MS ES+: 237.2 (M+1). Step 3: N-(7-Chloro-2-(3-chloro-6-hydroxy-4-methoxy-2-methylbenzoyl) isoindolin-4-yl)-N- methyl acrylamide
[001199] The title compound was prepared from 3-chloro-6-hydroxy-4-methoxy-2- methylbenzoic acid (Example 47 Step 3) and N-(7-chloroisoindolin-4-yl)-N-methyl acrylamide TFA salt (Step 2) analogously to Example 9 Step 1. [001200] HT
1H NMR (DMSO-d6, 400 MHz, 348K): δ 2.07 - 2.19 (m, 3H), 3.26 (s, 3H), 3.82 (d, J= 8.4 Hz, 3H), 4.49 - 4.92 (m, 4H), 5.55 - 5.91 (m, 1H), 5.91 - 6.25 (m, 2H), 6.50 - 6.55 (m, 1H), 7.30 - 7.34 (m, 1H), 7.49 - 7.52 (m, 1H), 10.11 (s, 1H). [001201] LCMS (Method T): 1.506 min, MS: ES+ 436.8 (M+1). [001202] HPLC (Method H): 6.59 min. Prep. HPLC purification method: [001203] Chromatographic separation and isolation were conducted with a Waters 2545 binary pump with waters 2489 purification system with UV detector; column YMC Triart C18
(250mm x 20mm x 5µm); compound eluted with: Mobile Phase A: 5 mM Ammonium bicarbonate in Merck water, Mobile Phase B: Acetonitrile with a gradient of T = 0.01 min (63% A, 37% B); gradient to T = 35 min (63% A, 37% B); T = 35.01 min (0% A, 100% B) gradient to T = 38 min (0% A, 100% B); T = 38.01 min (63% A, 37% B); gradient to T = 45 min (63% A, 37% B); Flow rate= 16ml/min; analysis time 45 min. Example 76: (E)-4-(Dimethylamino)-N-(6-(2-hydroxy-5-isopropyl-4-methoxybenzoyl)- 5,6,7,8-tetrahydro-1,6-naphthyridin-3-yl)-N-methylbut-2-enamide Step 1: tert-Butyl 3-(methylamino)-7,8-dihydro-1,6-naphthyridine-6(5H)-carboxylate

[001204] To a cooled (0 ˚C) solution of NaOMe (10.84 g, 200.68 mmol, 10.0 eq.) in MeOH (75 mL) was added tert-butyl 3-amino-7,8-dihydro-1,6-naphthyridine-6(5H)-carboxylate (CAS: 355819-02-2) (5.0 g, 20.06 mmol, 1.0 eq.) followed by paraformaldehyde (1.2 g, 40.13 mmol, 2.0 eq) and the reaction mixture stirred at room temperature for 16 h. The mixture was cooled to 0°C and treated portion wise with NaBH4 (3.81 g, 100.3 mmol, 5.0 eq). The resulting mixture was allowed to warm to room temperature and stirred for 3 h. The reaction was quenched with ice cold water (300 mL) and extracted with ethyl acetate (2 x 200 mL). The combined organic extracts were washed with brine solution (1 x 100 mL), dried over Na2SO4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica eluting with a gradient of 0-20% MeOH-DCM (product was eluted at 12% MeOH-DCM) to yield tert-butyl 3- (methylamino)-7,8-dihydro-1,6-naphthyridine-6(5H)-carboxylate (3.0 g, Yield: 57%). [001205]
1H NMR (DMSO-d6, 400 MHz): δ 1.42 (s, 9H), 2.66 - 2.70 (m, 5H), 3.59 (t, J= 5.6 Hz, J= 11.6 Hz, 2H), 4.43 (s, 2H), 5.76 (bs, 1H, D2O exchangeable), 6.65 (d, J= 1.6 Hz, 1H), 7.77 (d, J= 2.4 Hz, 1H). [001206] LCMS (Method N): 1.65 min, MS: ES+ 264.4 (M+1) Step 2: tert-Butyl (E)-3-(4-(dimethylamino)-N-methylbut-2-enamido)-7,8-dihydro-1,6- naphthyridine-6(5H)-carboxylate
[001207] The title compound was prepared from and tert-butyl 3-(methylamino)-7,8- dihydro-1,6-naphthyridine-6(5H)-carboxylate (Step 1) and (E)-4-(dimethylamino)but-2-enoic acid hydrochloride (CAS: 848133-35-7) analogously to Example 1 Step 2. [001208]
1H NMR (DMSO-d6, 400 MHz): δ 1.43 (s, 9H), 2.61 (s, 6H), 2.89 (bs, 2H), 3.16 - 3.33 (m, 3H), 3.68 (d, J= 5.2 Hz, 4H), 4.56 (s, 2H), 6.16 (bs, 1H), 6.67 - 6.74 (m, 1H), 7.68 (s, 1H), 8.33 (s, 1H). [001209] LCMS (Method N): 1.135 min, MS: ES+ 375.1 (M+1). Step 3: (E)-4-(Dimethylamino)-N-methyl-N-(5,6,7,8-tetrahydro-1,6-naphthyridin-3-yl)but-2- enamide hydrochloride
[001210] The title compound was prepared from tert-butyl (E)-3-(4-(dimethylamino)-N- methylbut-2-enamido)-7,8-dihydro-1,6-naphthyridine-6(5H)-carboxylate (Step 2) and 4M HCl in dioxane analogously to Example 4 Step 2. [001211]
1H NMR (DMSO-d6, 400 MHz): δ 2.65 - 2.66 (m, 6H), 3.24 (s, 2H), 3.32 (s, 3H), 3.46 (bs, 2H), 3.73 - 3.79 (m, 2H), 4.36 (s, 2H), 6.32 (bs, 1H), 6.71 - 6.78 (m, 1H), 7.97 (s, 1H), 8.61 (s, 1H). [001212] LCMS (Method O): 1.32 min, MS: ES+ 549.4 (Mx2 +1) Step 4: (E)-4-(Dimethylamino)-N-(6-(2-hydroxy-5-isopropyl-4-methoxybenzoyl)-5,6,7,8- tetrahydro-1,6-naphthyridin-3-yl)-N-methylbut-2-enamide
[001213] The title compound was prepared from 2-Hydroxy-5-isopropyl-4-methoxybenzoic acid (Example 45 Step 3) and (E)-4-(dimethylamino)-N-methyl-N-(5,6,7,8-tetrahydro-1,6- naphthyridin-3-yl)but-2-enamide hydrochloride (Step 3) analogously to Example 9 Step 1. [001214]
1H NMR (DMSO-d6, 400 MHz): δ 1.11 (d, J= 6.8 Hz, 6H), 2.04 (s, 6H), 2.87 - 2.95 (m, 4H), 3.08 - 3.15 (m, 1H), 3.24 (s, 3H), 3.74 - 3.77 (m, 5H), 4.70 (bs, 2H), 5.89 (bs, 1H), 6.42 - 6.47 (m, 1H), 6.62 - 6.68 (m, 1H), 6.97 (s, 1H), 7.66 (s, 1H), 8.30 (s, 1H), 9.80 (s, 1H). [001215] LCMS (Method N): 1.753 min, MS: ES+ 467.4 (M+1). [001216] HPLC (Method I): 6.15 min, Prep. HPLC purification method: [001217] Chromatographic separation and isolation were conducted with a Waters 2545 binary pump, Waters 2489 UV detector with Waters Acquity QDA detector purification system; column YMC Triart C18; 250 x 20 mm ID, 5μm); compounds eluted with: Mobile Phase A: 5mM ammonium bicarbonate + 0.05% NH3 in Merck water, Mobile Phase B: acetonitrile with a gradient of T = 0.01 (84% A, 43% B); gradient to T = 26.00 min (84% A, 44% B); T = 26.01 min (0% A, 100% B) gradient to T = 28.00 min (0% A, 100% B); T = 28.01 min (57% A, 43% B); gradient to T = 35.00 min (57% A, 43% B); gradient); Flow rate= 10ml/min; analysis time 35.00 min. Example 77: N-(2-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-6-(2- (dimethylamino)ethoxy)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N-methylbut-2-ynamide Step 1: 6-Bromo-7-nitro-1,2,3,4-tetrahydroisoquinoline

[001218] The title compound was prepared from 6-bromo-1,2,3,4-tetrahydroisoquinoline (CAS: 893566-74-0) and conc. H2SO4 /KNO3 analogously to Example 56 Step 1. [001219]
1H NMR (DMSO-d6, 400 MHz): δ 2.74 (t, J= 5.6 Hz, 2H), 2.92 (t, J= 5.2 Hz, 2H), 3.84 (s, 2H), 7.64 (s, 1H), 7.76 (s, 1H). [001220] LCMS (Method N): 1.485 min, MS: ES+ 257.0, 259.0 (M, M+2). Step 2: tert-Butyl 6-bromo-7-nitro-3,4-dihydroisoquinoline-2(1H)-carboxylate
[001221] The title compound was prepared from 6-bromo-7-nitro-1,2,3,4- tetrahydroisoquinoline (Step 1) and Boc anhydride analogously to Example 56 Step 2. [001222]
1H NMR (DMSO-d6, 400 MHz): δ 1.43 (s, 9H), 2.86 (t, J= 6.0 Hz, 2H), 3.56 (t, J= 5.6 Hz, 2H), 4.54 (s, 2H), 7.75 (s, 1H), 7.96 (s, 1H). [001223] LCMS (Method N): 2.550 min, MS: ES+ 257.0, 259.0 (M-100). Step 3: tert-Butyl 7-nitro-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,4-dihydroisoquinoline- 2(1H)-carboxylate

[001224] Performed in 2 parallel batches, each of 70 g scale: To a stirred solution of tert- butyl 6-bromo-7-nitro-3,4-dihydroisoquinoline-2(1H)-carboxylate (Step 2) (70 g, 196.08 mmol, 1 eq) in 1,4-dioxane (700 mL) were added Bis(pinacolato)diboron (74.70 g, 294.09 mmol, 1.5 eq) and KOAc (38.43 g, 392.14 mmol, 2 eq); the reaction mixture was degassed using N2 gas for 30 mins. PdCl2(dppf) (14.35 g, 19.60 mmol, 0.1 eq) was added and the reaction mixture was heated to 100°C for 2 h. The resulting mixture was filtered through Celite® and washed with EtOAc (3L). The combined organic extracts were washed with ice cold water (2L) and the aqueous layer (2L) extracted into EtOAc (2L). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted at 15% EtOAc in hexane). The resulting material was triturated with hexane (1L), filtered, and dried under vacuum to yield tert-butyl 7-nitro-6-(4,4,5,5-tetramethyl-1,3,2- dioxaborolan-2-yl)-3,4-dihydroisoquinoline-2(1H)-carboxylate as a white solid (100 g, Yield: 63%). [001225]
1H NMR (DMSO-d6, 400 MHz): δ 1.33 (s, 12H), 1.43 (s, 9H), 2.90 (t, J= 5.6 Hz, 2H), 3.57 (t, J= 5.6 Hz, 2H), 4.63 (s, 2H), 7.41 (s, 1H), 8.08 (s, 1H). Step 4: tert-Butyl 6-hydroxy-7-nitro-3,4-dihydroisoquinoline-2(1H)-carboxylate
[001226] Performed in 3 parallel batches, each of 33.4 g scale: To a cooled (0°C) solution of tert-butyl 7-nitro-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,4-dihydroisoquinoline- 2(1H)-carboxylate (Step 3) (33.4 g, 82.67 mmol, 1 eq.) in THF (334 mL) was added portion wise NaOH (6.61 g, 165.25 mmol, 2 eq) followed by dropwise addition of H2O2 (30% solution in MeOH) (39.35 mL, 1157.35 mmol, 4.2 eq). The reaction mixture was stirred at room temperature for 1 h. The resulting mixture was poured into ice cold water (2L) and extracted into EtOAc (2L). The aqueous layer was neutralized with dil. HCl solution to pH ~7 and extracted into EtOAc (3 x 1L). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted at 15% EtOAc in hexane) yielding tert-butyl 6-hydroxy-7-nitro-3,4-dihydroisoquinoline-2(1H)-carboxylate as a yellow solid (55 g, Yield: 75%). [001227]
1H NMR (DMSO-d6, 400 MHz): δ 1.41 (s, 9H), 2.77 (t, J= 6.0 Hz, 2H), 3.50 (t, J= 6.0 Hz, 2H), 4.44 (s, 2H), 6.90 (s, 1H), 7.79 (s, 1H), 10.74 (s, 1H). Step 5: tert-Butyl 6-(2-(dimethylamino)ethoxy)-7-nitro-3,4-dihydroisoquinoline-2(1H)-carboxylate

[001228] Performed in 20 parallel batches, each of 1 g scale: To a stirred solution of tert- butyl 6-hydroxy-7-nitro-3,4-dihydroisoquinoline-2(1H)-carboxylate (Step 4) (1.0 g, 3.40 mmol, 1 eq.) in DMF (10 mL) was added Cs2CO3 (3.33 g, 10.21 mmol, 3 eq); the mixture was stirred at room temperature for 1 h. The resulting mixture was cooled to 0°C, 2-chloro-N,N-dimethylethan- 1-amine hydrochloride (CAS: 4584-46-7) (1.47 g, 10.21 mmol, 3 eq) and KI (0.056 g, 0.33 mmol, 0.1 eq) were added and the reaction mixture was heated to 110°C for 16 h. The resulting mixture was diluted with EtOAc (2L) and washed with ice cold water (3 x 2L). The organic portion was dried over Na2SO4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on neutral aluminium oxide (product eluted at 40-100% EtOAc in hexane) yielding tert-butyl 6-(2-(dimethylamino)ethoxy)-7-nitro-3,4-dihydroisoquinoline-2(1H)-carboxylate as a yellow oil (9 g, Yield: 36%).
[001229]
1H NMR (DMSO-d6, 400 MHz): δ 1.43 (s, 9H), 2.21 (s, 6H), 2.63 (t, J= 5.6 Hz, 2H), 2.84 (t, J= 5.6 Hz, 2H), 3.55 (t, J= 5.6 Hz, 2H), 4.19 (t, J= 5.6 Hz, 2H), 4.48 (s, 2H), 7.20 (s, 1H), 7.77 (s, 1H). [001230] LCMS (Method N): 1.319 min, MS: ES+ 366.1 (M+1) Step 6: tert-Butyl 7-amino-6-(2-(dimethylamino)ethoxy)-3,4-dihydroisoquinoline-2(1H)- carboxylate

[001231] To a stirred solution of tert-butyl 6-(2-(dimethylamino)ethoxy)-7-nitro-3,4- dihydroisoquinoline-2(1H)-carboxylate (Step 5) (9 g, 34.65 mmol, 1 eq.) in MeOH (90 mL) was added 10% Pd/C (50% moisture) (2.7 g, 30%w/w) and the reaction mixture was stirred under an atmosphere of H2(g) at room temperature for 5 h. The resulting mixture was filtered through Celite® and the filter bed was washed with 50% MeOH: DCM (500 mL). The filtrate was concentrated under reduced pressure yielding tert-butyl 7-amino-6-(2-(dimethylamino)ethoxy)- 3,4-dihydroisoquinoline-2(1H)-carboxylate as a yellow oil (9 g, Yield: 97%). The crude material was used in the next step without further purification. [001232]
1H NMR (DMSO-d6, 400 MHz): δ 1.42 (s, 9H), 2.22 (s, 6H), 2.58 – 2.62 (m, 4H), 3.48 (t, J= 5.6 Hz, 2H), 3.97 (t, J= 5.6 Hz, 2H), 4.29 (s, 2H), 4.60 (s, 2H), 6.37 (s, 1H), 6.58 (s, 1H). [001233] LCMS (Method N): 1.159 min, MS: ES+ 336.0 (M+1) Step 7: tert-Butyl 6-(2-(dimethylamino)ethoxy)-7-(methylamino)-3,4-dihydroisoquinoline-2(1H)- carboxylate
[001234] The title compound was prepared from tert-butyl 7-amino-6-(2- (dimethylamino)ethoxy)-3,4-dihydroisoquinoline-2(1H)-carboxylate (Step 6) and paraformaldehyde analogously to Example 74 Step 2.
[001235]
1H NMR (DMSO-d6, 400 MHz): δ 1.42 (s, 9H), 2.21 (s, 6H), 2.60 (t, J= 5.6 Hz, 4H), 2.69 (d, J= 4.8 Hz, 3H), 3.49 (t, J= 5.6 Hz, 2H), 3.97 (t, J= 5.6 Hz, 2H), 4.36 (s, 2H), 4.87 – 4.88 (m, 1H), 6.22 (s, 1H), 6.59 (s, 1H). [001236] LCMS (Method N): 1.224 min MS: ES+ 350.1 (M+1) Step 8: tert-Butyl 6-(2-(dimethylamino)ethoxy)-7-(N-methylbut-2-ynamido)-3,4- dihydroisoquinoline-2(1H)-carboxylate
[001237] To a cooled (0 ˚ C) solution of but-2-ynoic acid (CAS: 590-93-2) (0.15 g, 1.78 mmol, 1 eq) and tert-butyl 6-(2-(dimethylamino)ethoxy)-7-(methylamino)-3,4-dihydroisoquinoline-2(1H)- carboxylate (Step 7) (0.31 g, 0.88 mmol, 1 eq) in DCM (1.5 mL) was added dropwise pyridine (0.56 g, 7.14 mmol, 4 eq) and the reaction mixture was stirred at 0°C for 5 mins. To this mixture was added dropwise POCl
3 (0.41 g, 2.67 mmol, 1.5 eq) and stirring continued at 0°C for 5 mins. The resulting mixture was poured into ice cold water (50 mL) and extracted with10% MeOH in DCM (3 x 50 mL). The combined organic extracts were dried over Na
2SO
4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on neutral aluminium oxide (product eluted at 10% MeOH in DCM) to give tert-butyl 6-(2- (dimethylamino)ethoxy)-7-(N-methylbut-2-ynamido)-3,4-dihydroisoquinoline-2(1H)-carboxylate (0.25 g, Yield: 59%). [001238]
1H NMR (DMSO-d6, 400 MHz): δ 1.43 (s, 9H), 1.73 (s, 3H), 2.20 (s, 6H), 2.58 - 2.63 (m, 2H), 2.78 - 2.79 (m, 2H), 3.04 (s, 3H), 3.54 - 3.55 (m, 2H), 4.00 - 4.13 (m, 2H), 4.42 (s, 2H), 6.94 (s, 1H), 7.11 (s, 1H). [001239] LCMS (Method N): 1.823 min, MS: ES+ 416.7 (M+1). Step 9: N-(6-(2-(Dimethylamino)ethoxy)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N-methylbut-2- ynamide
[001240] The title compound was prepared from tert-butyl 6-(2-(dimethylamino)ethoxy)-7- (N-methylbut-2-ynamido)-3,4-dihydroisoquinoline-2(1H)-carboxylate (Step 8) and TFA analogously to Example 72 Step 2. [001241]
1H NMR (DMSO-d6, 400 MHz): δ 1.74 (s, 3H), 2.85 - 2.86 (m, 6H), 2.99 - 3.04 (m, 2H), 3.07 (s, 3H), 3.37 (m, 1H), 3.52 - 3.56 (m, 3H), 4.20 - 4.39 (m, 6H), 7.07 (s, 1H), 7.24 (s, 1H), 9.11 - 9.15 (m, 3H), 10.05 (m, 1H). [001242] LCMS (Method A2): 3.37 min, MS: ES+ 316.6 (M+1). Step 10: N-(2-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-6-(2-(dimethylamino)ethoxy)- 1,2,3,4-tetrahydroisoquinolin-7-yl)-N-methylbut-2-ynamide
[001243] The title compound was prepared from 3-chloro-6-hydroxy-4-methoxy-2- methylbenzoic acid (Example 47 Step 3) and N-(6-(2-(dimethylamino)ethoxy)-1,2,3,4- tetrahydroisoquinolin-7-yl)-N-methylbut-2-ynamide TFA salt (Step 9) analogously to Example 9 Step 1. [001244] HT
1H NMR (DMSO, 400 MHz, 348K): δ 1.60 - 1.81 (m, 3H), 1.95 - 2.14 (m, 3H), 2.20 (s, 6H), 2.61 - 2.65 (m, 2H), 2.81 - 2.93 (m, 2H), 2.96 - 3.1 (m, 2H), 3.83 (s, 3H), 4.01 - 4.12 (m, 3H), 4.23 - 4.33 (m, 1H), 4.63 - 4.77 (m, 1H), 6.53 (s, 1H), 6.87 - 7.13 (m, 2H). [001245] LCMS (Method T): 1.691 min, MS: ES+ 514.2, 516.3 (M, M+2). [001246] HPLC (Method H): 3.618 min, 210 nm Prep. HPLC purification method: [001247] Chromatographic separation and isolation was conducted on a Waters 2545 binary pump with a Waters 2489 UV detector and a Waters Acquity QDA detector; column Waters X-Bridge C18 (250mm x 19mm x 5µm); compound was eluted with: Mobile Phase A: 5mM ammonium bicarbonate + NH3 in Merck water Mobile Phase B: ACN:THF[90:10] with a gradient of T = 0.01 min (84% A, 25% B); gradient to T = 26.00 min (84% A, 43% B); T = 26.01 min (0% A, 100%B); gradient to T = 28.00 min (0% A, 100% B); T = 28.01 min (75% A, 25% B); T = 35.00 min (75% A, 25% B); Flow rate= 8 ml/min; analysis time 35 min.
Example 78: (E)-N-(5-Chloro-2-(3-chloro-4-hydroxy-2-methylbenzoyl)isoindolin-4-yl)-4- (dimethylamino)but-2-enamide Step 1: Methyl 3-chloro-4-hydroxy-2-methylbenzoate

[001248] Performed in 2 parallel batches, each of 0.5 g scale: A stirred solution 4-bromo-2- chloro-3-methylphenol (CAS: 1799612-08-0) (0.5 g, 2.26 mmol, 1 eq) in MeOH: DMF (10 mL) was degassed with N2 gas for 10 mins and treated with TEA (0.683 g, 6.76 mol, 3 eq) and PdCl2(dppf) (0.33 g, 0.45 mmol, 0.2 eq) at room temperature. The reaction mixture was placed under a positive pressure of CO(g) (20kg/cm
2) and heated to 120°C for 16 h. After cooling to room temperature, the resulting mixture was poured into ice cold water (50 mL) and extracted with EtOAc (3 x 100 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted at 15% EtOAc in hexane) yielding methyl 3-chloro-4-hydroxy-2-methylbenzoate as a white solid (0.55 g, Yield: 61%). [001249]
1H NMR (DMSO, 400 MHz): δ 2.56 (s, 3H), 3.78 (s, 3H), 6.90 (d, J= 8.4 Hz, 1H), 7.66 (d, J= 8.4 Hz, 1H), 10.94 (s, 1H). [001250] LCMS (Method N): 1.936 min, MS: ES+ 199.1, 201.1 (M, M+2). Step 2: 3-Chloro-4-hydroxy-2-methylbenzoic acid
[001251] To a stirred solution of methyl 3-chloro-4-hydroxy-2-methylbenzoate (Step 1) (0.5 g, 2.5 mmol, 1 eq) in MeOH: H2O (1:1) (5 mL) was added KOH (2.8 g, 50 mmol, 20 eq) and the reaction mixture was heated to 60°C for 30 mins. The resulting mixture was poured into ice cold water (50 mL), neutralized with dil. HCl (2 mL) and extracted with EtOAc (3 x 50 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under vacuum to yield 3-chloro-4-hydroxy-2-methylbenzoic acid as an off-white solid (0.5 g, Yield: 98%). The crude material was used in the next step without further purification.
[001252]
1H NMR (DMSO, 400 MHz): δ 2.57 (s, 3H), 6.87 (d, J= 8.8 Hz, 1H), 7.67 (d, J= 8.8 Hz, 1H), 10.82 (s, 1H), 12.49 (s, 1H). [001253] LCMS (Method N): 1.659 min, MS: ES+ 185.0, 187.0 (M, M+2). Step 3: (E)-N-(5-Chloro-2-(3-chloro-4-hydroxy-2-methylbenzoyl)isoindolin-4-yl)-4- (dimethylamino)but-2-enamide
[001254] The title compound was prepared from 3-chloro-4-hydroxy-2-methylbenzoic acid (Step 2) and (E)-N-(5-chloroisoindolin-4-yl)-4-(dimethylamino)but-2-enamide hydrochloride (Example 70 Step 3) analogously to Example 9 Step 1. [001255]
1H NMR (DMSO, 400 MHz): δ 2.13 (s, 3H), 2.20 - 2.24 (m, 6H), 3.00 - 3.09 (m, 2H), 4.36 - 4.51 (m, 2H), 4.67 - 4.87 (m, 2H), 6.20 - 6.40 (m, 1H), 6.62 - 6.83 (m, 1H), 6.86 - 6.90 (m, 1H), 7.10 - 7.16 (m, 1H), 7.18 - 7.34 (m, 1H), 7.44 - 7.50 (m, 1H), 9.78 - 9.98 (m, 1H). [001256] LCMS (Method A5): 5.273 min, MS: ES+ 448.2, 450.1 (M, M+2). [001257] HPLC (Method K): 5.402 min, Prep. HPLC purification method: [001258] Chromatographic separation and isolation were conducted Waters 2545 binary pump with a Waters 2489 UV detector and a Waters Acquity QDA detector; column Waters X- Bridge C18 (150mm x 19mm x 5µm); compounds eluted with: Mobile Phase A: 5 mM ammonium bicarbonate + 0.05% NH
3 in Merck water Mobile Phase B: acetonitrile with a gradient of T = 0.01 min (86% A, 14% B); gradient to T = 26.00 min (79% A, 21% B); T = 26.01 min (0% A, 100% B); gradient to T = 28.00 min (0% A, 100% B); T = 28.01 min (86% A, 14% B); T = 32.00 min (86% A, 14% B) Flow rate= 17 ml/min; analysis time 32 min. Example 79: (E)-N-(5-Chloro-2-(4-hydroxy-2,3-dimethylbenzoyl) isoindolin-4-yl)-4- (dimethylamino) but-2-enamide Step 1: 4-Methoxy-2,3-dimethylbenzoic acid
[001259] The title compound was prepared from 4-methoxy-2,3-dimethylbenzaldehyde (CAS: 38998-17-3), NaClO2 and NaH2PO4 analogously to Example 32 Step 2. [001260]
1H NMR (DMSO-d6, 400 MHz): δ 2.11 (s, 3H), 2.43 (s, 3H), 3.82 (s, 3H), 6.87 (d, J= 8.8 Hz, 1H), 7.67 (d, J= 8.8 Hz, 1H), 12.44 (s, 1H). Step 2: 4-Hydroxy-2,3-dimethylbenzoic acid
[001261] The title compound was prepared from 4-methoxy-2,3-dimethylbenzoic acid (Step 1) and BBr
3 (1M in DCM) analogously to Example 32 Step 3. [001262]
1H NMR (DMSO-d6, 400 MHz): δ 2.07 (s, 3H), 2.42 (s, 3H), 6.69 (d, J= 8.4 Hz, 1H), 7.52 (d, J=8.8 Hz, 1H), 9.91 (s, 1H), 12.28 (s, 1H). [001263] LCMS (Method N): 1.601 min, MS: ES- 163 (M-3). Step 3: (E)-N-(5-Chloro-2-(4-hydroxy-2,3-dimethylbenzoyl) isoindolin-4-yl)-4-(dimethylamino) but-2-enamide
[001264] The title compound was prepared from 4-hydroxy-2,3-dimethylbenzoic acid (Step 2) (Step 2) and (E)-N-(5-chloroisoindolin-4-yl)-4-(dimethylamino)but-2-enamide hydrochloride (Example 70 Step 3) analogously to Example 9 Step 1. [001265]
1H NMR (DMSO-d6, 400 MHz): δ 2.06 - 2.11 (m, 6H), 2.20 (s, 6H), 3.00 - 3.09 (m, 2H), 4.33 - 4.47 (m, 2H), 4.66 - 4.85 (m, 2H), 6.30 - 6.40 (m, 1H), 6.61 - 6.82 (m, 2H), 6.92 - 6.96 (m, 1H), 7.16 - 7.34 (m, 1H), 7.34 - 7.50(m, 1H), 9.83 (bs, 1H) [001266] LCMS (Method T): 1.548 min MS: ES+ 428.3 (M+1).
[001267] HPLC (Method K): 5.41 min, Prep. HPLC purification method: [001268] Chromatographic separation and isolation were conducted with Waters 2545 binary pump with a Waters 2489 UV detector and a Waters Acquity QDA detector; column Waters X-Bridge C18 (250mm x 19mm x 5µm); compound was eluted with: Mobile Phase A: 5 mM ammonium bicarbonate + 0.05% NH
3 in Merck water, Mobile Phase B: acetonitrile:methanol(50:50) with a gradient of T = 0.01 min (55% A, 45% B); gradient to T = 23 min (55% A, 45% B); T = 23.01 min (0% A, 100% B) gradient to T = 26 min (0% A, 100% B); T = 26.01 min (5% A, 45% B); gradient to T = 35 min (55% A, 45% B); Flow rate= 8ml/min; analysis time 35 min. Example 80: (E)-4-(Dimethylamino)-N-(2-(4-hydroxy-3-isopropylbenzoyl)-3-methyl-1,2,3,4- tetrahydroisoquinolin-7-yl)-N-methylbut-2-enamide Step 1: 1-(4-Bromophenyl) propan-2-amine

[001269] To a stirred solution of 1-(4-bromophenyl) propan-2-one (CAS: 6186-22-7) (5.0 g, 23.4 mmol, 1.0 eq.) in titanium tetraisopropoxide (13.33 g, 2 eq) was added dropwise 7M ammonia in methanol (60 mL) and the mixture was stirred at room temperature for 16 h. The mixture was cooled to 0˚C, treated portion wise with NaBH4 (1.33 g, 35.12 mmol, 1.5 eq) and allowed to warm to room temperature stirring for 2 h. The resulting mixture was diluted with aqueous ammonia solution causing precipitation of a white solid. The solid was filtered and washed with EtOAc (200 mL). The filtrate was concentrated under reduced pressure causing further solid to precipitate out. EtOAc (200 mL) was added and the mixture was filtered. The filtrate was acidified with 2M HCl (200 mL) and the layers were separated. The pH of the aqueous layer was adjusted pH ~10 with aqueous ammonia (50 mL) and the mixture was extracted with DCM (500 mL). The organic extract was dried over Na2SO4, filtered, and concentrated under vacuum yielding 1-(4-bromophenyl) propan-2-amine as a yellow solid (2.4 g, Yield: 48%). The material was used in the next step without further purification.
[001270]
1H NMR (DMSO-d6, 400 MHz): δ 0.93 (d, J= 6.4 Hz, 3H), 2.50 (d, J= 3.6 Hz, 2H), 2.94 - 2.99 (m, 1H), 7.14 (d, J= 8.4 Hz, 2H), 7.45 (d, J= 7.6 Hz, 2H). [001271] LCMS (Method N): 1.65 min, MS ES+: 216.2 (M). Step 2: Methyl (1-(4-bromophenyl) propan-2-yl) carbamate
[001272] To a stirred solution of 1-(4-bromophenyl) propan-2-amine (Step 1) (2.4 g, 11.2 mmol, 1.0 eq.) in DCM (24 mL) was added dropwise pyridine (2.08 g, 26.35 mmol, 2.35 eq) and methyl chloro formate (1.85 g, 19.02 mmol, 1.75 eq) and the reaction mixture stirred at room temperature for 16 h. The resulting mixture was concentrated under reduced pressure. The crude material was dissolved in EtOAc (200 mL) and washed with sat. NaHCO
3 solution (200 mL), 1N HCl (50 mL), brine solution (50 mL), dried over Na
2SO
4, filtered, and concentrated under vacuum to yield methyl (1-(4-bromophenyl) propan-2-yl) carbamate as yellow solid (2.0 g, Yield: 48%). The material was used in the next step without further purification. [001273]
1H NMR (DMSO-d6, 400 MHz): δ 1.03 (d, J= 6.8 Hz, 3H), 2.56 - 2.69 (m, 2H), 3.46 (s, 3H), 3.65 - 3.68 (m, 1H), 7.10 - 7.15 (d, J= 8.4 Hz, 2H),7.45 (d, J= 8.4 Hz, 2H). Step 3: 7-Bromo-3-methyl-3,4-dihydroisoquinoline-2(1H)-carboxylate
[001274] A mixture of concentrated H2SO4 (11.13 mL) and glacial acetic acid (16.80 mL) were stirred for 15 mins then treated with methyl (1-(4-bromophenyl) propan-2-yl) carbamate (Step 2) (2.10 g, 7.71 mmol, 1.0 eq.) and paraformaldehyde (0.37 g, 12.34 mmol, 1.60 eq.). The reaction mixture was stirred at room temperature for 16 h. The resulting mixture was diluted with ice cold water (500 mL) and extracted with DCM (2 x 50 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under vacuum. The crude material was triturated with diethyl ether (2 x 50 mL) and dried under high vacuum drying to yield methyl 7- bromo-3-methyl-3,4-dihydroisoquinoline-2(1H)-carboxylate as a white solid (1.8 g, Yield: 87%).
[001275]
1H NMR (DMSO-d6, 400 MHz): δ 0.99 (d, J= 6.8 Hz, 3H), 2.58 - 2.62 (m, 1H), 2.92 - 2.97 (m, 1H), 3.64 (s, 3H), 4.27 (d, J= 17.2 Hz, 1H), 4.46 (bs, 1H), 4.69 (d, J= 17.2 Hz, 1H), 7.15 (d, J= 8.4 Hz, 1H), 7.36 - 7.39 (m, 1H), 7.46 (s, 1H). Step 4: 7-Bromo-3-methyl-1,2,3,4-tetrahydroisoquinoline
[001276] To a stirred solution of methyl 7-bromo-3-methyl-3,4-dihydroisoquinoline-2(1H)- carboxylate (Step 3) (2.0 g, 7.04 mmol, 1.0 eq.) in DCM (40 mL) was added dropwise iodo trimethyl silane (5.63 g, 28.16 mmol, 4 eq) at room temperature. The reaction mixture was heated to 50°C and stirred for 2 h. The resulting mixture was diluted with MeOH (20 mL) and stirred for 10 mins. The mixture was concentrated under vacuum and the crude material was triturated with n-pentane (2 x 20 mL) and diethyl ether (5 x 20 mL) to yield 7-bromo-3-methyl-1,2,3,4- tetrahydroisoquinoline as a yellow solid (1.8 g, Yield: quantitative). [001277]
1H NMR (DMSO-d6, 400 MHz): δ 1.34 (d, J= 6.4 Hz, 3H), 2.69 - 2.76 (m, 1H), 3.01 - 3.07 (m, 1H), 3.54 - 3.60 (m, 1H), 4.35 (s, 2H), 7.19 (d, J= 18.0 Hz,1H), 7.40 - 7.51 (m, 2H), 8.97 (bs, 1H). Step 5: 7-Bromo-3-methyl-3,4-dihydroisoquinoline-2(1H)-carboxylate
[001278] To a cooled (0 ˚C) solution of 7-bromo-3-methyl-1,2,3,4-tetrahydroisoquinoline (Step 4) (1.8 g, 7.96 mmol, 1.0 eq.) in DCM (18 mL) were added dropwise TEA (3.32 mL, 23.89 mmol, 3 eq) followed by Boc anhydride (2.6 g, 11.9 mmol, 1.5 eq). The reaction mixture was allowed to warm to room temperature and stirred for 1 h. The resulting mixture was diluted with ice cold water (50 mL) and extracted with DCM (3 x 50 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under vacuum yielding tert-butyl 7-bromo-3-methyl- 3,4-dihydroisoquinoline-2(1H)-carboxylate as a yellow liquid (1.7 g, Yield: 65%). The crude material was used in the next step without further purification. [001279]
1H NMR (DMSO-d6, 400 MHz): δ 1.43 (s, 9H), 2.50 - 2.60 (m, 1H), 2.89 - 2.95 (m, 1H), 4.22 (d, J= 18.4 Hz, 1H), 4.41 (bs, 1H), 4.63 (d, J= 17.2 Hz, 1H), 7.13 (d, J= 8.0 Hz,1H), 7.36 (d, J= 8.0 Hz, 1H), 7.43 (s, 1H).
[001280] LCMS (Method N): 2.260 min, MS ES+: 227.6 (M-100). Step 6: tert-Butyl 3-methyl-7-(methylamino)-3,4-dihydroisoquinoline-2(1H)-carboxylate
[001281] The title compound was prepared from tert-butyl 7-bromo-3-methyl-3,4- dihydroisoquinoline-2(1H)-carboxylate (Step 5) and methyl amine (40% in water) analogously to Example 36 Step 4. [001282]
1H NMR (DMSO-d6, 400 MHz): δ 0.998 (d, J = 6.4 Hz, 3H), 1.42 (s, 9H), 2.38 - 2.43 (m, 1H), 2.64 (d, J= 5.2 Hz, 3H), 2.81 - 2.86 (m, 1H), 4.10 (d, J= 5.6 Hz, 1H), 4.35 (bs, 1H), 4.50 (d, J= 16.4 Hz, 1H), 5.45 (d, J= 5.2 Hz,1H, D
2O exchangeable), 6.31 (s, 1H), 6.38 - 6.41 (m, 1H), 6.86 (d, J= 8.0 Hz, 1H). [001283] LCMS (Method N): 1.89 min, MS ES+: 221.15 (M-55). Step 7: tert-Butyl (E)-7-(4-(dimethylamino)-N-methylbut-2-enamido)-3-methyl-3,4- dihydroisoquinoline-2(1H)-carboxylate
[001284] The title compound was prepared from (E)-4-(dimethylamino) but-2-enoic acid (CAS: 848133-35-7) and tert-butyl 3-methyl-7-(methylamino)-3,4-dihydroisoquinoline-2(1H)- carboxylate (Step 6) analogously to Example 50 Step 1. The material was used in the next step without further purification. [001285] LCMS (Method N): 1.763 min, MS ES+: 388.4 (M+1). Step 8: (E)-4-(Dimethylamino)-N-methyl-N-(3-methyl-1,2,3,4-tetrahydroisoquinolin-7-yl)but-2- enamide hydrochloride
[001286] The title compound was prepared from tert-butyl (E)-7-(4-(dimethylamino)-N- methylbut-2-enamido)-3-methyl-3,4-dihydroisoquinoline-2(1H)-carboxylate (Step 7) and 4M HCl in dioxane analogously to Example 50 Step 2. [001287]
1H NMR (DMSO-d6, 400 MHz): 1.42 (d, J = 6.4 Hz, 3H), 2.65 - 2.66 (m, 6H), 2.78 - 2.90 (m, 1H), 3.04 - 3.09 (m, 1H), 3.25 (s, 3H), 3.53 - 3.67 (m, 4H), 4.29 (s, 2H), 6.12 - 6.15 (m, 1H), 6.65 - 6.72 (m, 1H), 7.20 - 7.29 (m, 3H), 9.65 (s, 1H), 9.80 (s, 1H), 10.77 (s, 1H). [001288] LCMS (Method N): 0.326 min, MS ES+: 288.22 (M+1). Step 9: (E)-4-(dimethylamino)-N-(2-(4-hydroxy-3-isopropylbenzoyl)-3-methyl-1,2,3,4- tetrahydroisoquinolin-7-yl)-N-methylbut-2-enamide
[001289] The title compound was prepared from 4-hydroxy-3-isopropylbenzoic acid and (E)-4-(dimethylamino)-N-methyl-N-(3-methyl-1,2,3,4-tetrahydroisoquinolin-7-yl)but-2-enamide hydrochloride (Step 8) analogously to Example 1 Step 1. [001290]
1H NMR (DMSO-d6, 400 MHz): δ 1.11 (d, J= 6.4 Hz, 3H), 1.17 (t, J= 6.8 Hz, 6H), 2.03 (s, 6H), 2.66 (d, J= 15.6 Hz, 1H), 2.87 - 2.89 (m, 2H), 3.08 - 3.13 (m, 1H), 3.21 (m, 4H), 4.36 (s, br, 1H), 4.59 (s, br, 1H), 4.95 (s, br, 1H), 5.85 - 5.89 (m, 1H), 6.56 - 6.63 (m, 1H), 6.84 (d, J= 8.4 Hz, 1H), 7.08 - 7.13 (m, 3H), 7.18 (s, 1H), 7.19 - 7.25 (m, 1H), 9.78 (s, 1H). [001291] LCMS (Method A4): 1.753 min, MS: ES+ 450.31 (M+1). [001292] HPLC (Method H): 3.77 min, Prep. HPLC purification method: [001293] Chromatographic separation and isolation were conducted with Waters 2545 binary pump with a Waters 2489 UV detector and a Waters Acquity QDA detector; column Waters X-Bridge Prep C8 (250mm x 19mm x 5µm); compound was eluted with: Mobile Phase A: 5mM ammonium bicarbonate +0.05% NH
3 in Merck water, Mobile Phase B: Acetonitrile with a gradient of T = 0.01 min (60% A, 40% B); gradient to T = 18.00 min (56% A, 44% B); T = 18.01 min (0%
A, 100% B) gradient to T = 21.00 min (0% A, 100% B); T = 21.01 min (60% A, 40% B); gradient to T = 27.00 min (60% A, 40% B); Flow rate= 10 ml/min; analysis time Example 81: (E)-4-(Dimethylamino)-N-(2-(4-hydroxy-3-isopropylbenzoyl)-1,2,3,4- tetrahydroisoquinolin-7-yl)-N-methylbut-2-enamide

[001294] The title compound was prepared from (E)-4-(dimethylamino)-N-methyl-N- (1,2,3,4-tetrahydroisoquinolin-7-yl)but-2-enamide 2,2,2-trifluoroacetic acid (Example 4.2 Step 2) and 4-hydroxy-3-isopropylbenzoic acid (CAS: 859034-02-9) analogously to Example 67 Step 3. [001295] ¹H NMR (400 MHz, DMSO) δ 1.16 (d, J=7.0 Hz, 6H), 2.02 (s, 6H), 2.91 - 2.85 (m, 4H), 3.24 - 3.17 (m, 4H), 3.75 - 3.69 (m, 2H), 4.68 (s, 2H), 5.89 - 5.84 (m, 1H), 6.64 - 6.56 (m, 2H), 6.83 (d, J=8.3 Hz, 1H), 7.07 (dd, J=2.2, 8.2 Hz, 1H), 7.15 (dd, J=2.0, 8.3 Hz, 1H), 7.27 - 7.21 (m, 2H), 9.88 - 9.80 (m, 1H). [001296] LCMS (LC-Method 3): 4.1min, MS:ES+ 436.4 (M+1) Example 82: N-(2-(4-Hydroxy-3-isopropylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N-(1- methylpiperidin-4-yl)acrylamide Step 1: tert-butyl 7-(N-(1-methylpiperidin-4-yl)acrylamido)-3,4-dihydroisoquinoline-2(1H)- carboxylate

[001297] To a cooled (0 ˚C) solution of tert-butyl 7-[(1-methyl-4-piperidyl)amino]-3,4- dihydro-1H-isoquinoline-2-carboxylate (Example 20 Step 1) (248 mg, 0.72 mmol, 1.0 eq) in pyridine (3.3 mL) under nitrogen was added dropwise prop-2-enoyl prop-2-enoate (0.09 mL, 0.79 mmol, 1.1 eq). The reaction was stirred at 0 °C for 10 minutes then stirred at room temperature for 16 h. The reaction was quenched with 7N methanolic ammonia (2 mL) then concentrated under vacuum to afford the title compound (331 mg, Yield: quantitative). The compound was used crude in the next step.
[001298] MS: ES+ 400.1(M+1) Step 2: N-(1-Methylpiperidin-4-yl)-N-(1,2,3,4-tetrahydroisoquinolin-7-yl)acrylamide

[001299] To a solution of tert-butyl 7-(N-(1-methylpiperidin-4-yl)acrylamido)-3,4- dihydroisoquinoline-2(1H)-carboxylate (Step 1) (331 mg, 0.83 mmol, 1.0 eq) in DCM (2.8 mL) was added TFA (2.5 mL, 33.1 mmol, 40.0 eq); the reaction mixture was stirred for 1.5 h. The resulting mixture was concentrated under vacuum to afford the title compound (285 mg, Yield: 65%). The compound was used crude in the next step. [001300] MS: ES+ 300.1 (M+1) Step 3: N-(2-(4-Hydroxy-3-isopropylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N-(1- methylpiperidin-4-yl)acrylamide

[001301] The title compound was prepared from N-(1-methylpiperidin-4-yl)-N-(1,2,3,4- tetrahydroisoquinolin-7-yl)acrylamide 2,2,2-trifluoroacetic acid (Step 2) and 4-hydroxy-3- isopropyl-benzoic acid (CAS: 859034-02-9) analogously to Example 67 Step 3. [001302] ¹H NMR (400 MHz, DMSO) δ 1.19 - 1.15 (m, 6H), 1.32 - 1.23 (m, 2H), 1.69 - 1.66 (m, 2H), 1.95 - 1.89 (m, 2H), 2.09 (s, 3H), 2.75 (d, J=11.5 Hz, 2H), 2.94 - 2.89 (m, 2H), 3.75 - 3.68 (m, 2H), 4.43 - 4.35 (m, 2H), 4.72 (s, 2H), 5.51 - 5.47 (m, 1H), 5.82 - 5.74 (m, 1H), 6.11 (dd, J=2.2, 16.9 Hz, 1H), 6.86 - 6.82 (m, 1H), 6.98 - 6.96 (m, 1H), 7.05 - 7.05 (m, 1H), 7.16 (dd, J=2.1, 8.2 Hz, 1H), 7.24 (s, 1H), 7.28 (d, J=8.0 Hz, 1H), 8.33 (s, 1H), 9.86(s,1H). Formate salt. [001303] LCMS (LC-Method 3): 4.1min, ES+ 462.7 (M+1).
Example 83: (E)-4-(Dimethylamino)-N-(6-(2-hydroxy-5-isopropyl-4-methoxybenzoyl)-2- methoxy-5,6,7,8-tetrahydro-1,6-naphthyridin-3-yl)-N-methylbut-2-enamide Step 1: tert-Butyl 3-amino-2-methoxy-7,8-dihydro-1,6-naphthyridine-6(5H)-carboxylate
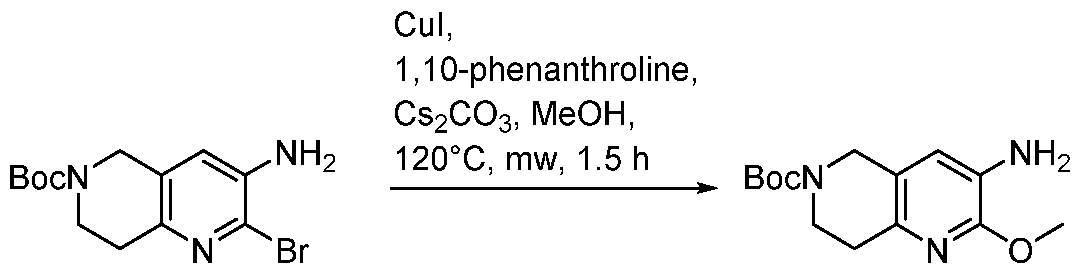
[001304] To a microwave vial charged with tert-butyl 3-amino-2-bromo-7,8-dihydro-5H-1,6- naphthyridine-6-carboxylate (Example 63 Step 1) (500 mg, 1.52 mmol, 1.0 eq), 1,10- phenanthroline (55 mg, 0.31 mmol, 0.2 eq), cesium carbonate (993 mg, 3.05 mmol, 2.0 eq) and copper(I) iodide (29 mg, 0.15 mmol, 0.10 eq) under nitrogen was added MeOH (6.0 mL, 0.15 mol, 97.2 eq). The stirred suspension was degassed with nitrogen for 5 minutes then heated to 120 °C using microwave irradiation for 2 h. The resulting mixture was filtered through Celite®, washing through with MeOH; the filtrate was concentrated under vacuum. The residue was dissolved in EtOAc (30 ml); washed with water (30 ml) and brine (30 ml). The aqueous layer was re-extracted with EtOAc (30 ml) and the combined organic extracts were dried over MgSO4, filtered and concentrated under vacuum. The crude material was purified by chromatography on silica, eluting with a gradient of 0 – 30% (10% MeOH/0.1% 7M NH3 in MeOH/DCM) and DCM to afford the title compound as a brown oil (274 mg, Yield: 64%). [001305] ¹H NMR (400 MHz, DMSO) δ 1.43 - 1.42 (m, 9H), 2.61 (t, J=5.8 Hz, 2H), 3.57 (t, J=5.9 Hz, 2H), 3.82 (s, 3H), 4.32 (s, 2H), 4.78 (s, 2H), 6.63 (s, 1H). [001306] MS: ES+ 280.0 (M+1) Step 2: tert-Butyl 3-formamido-2-methoxy-7,8-dihydro-1,6-naphthyridine-6(5H)-carboxylate
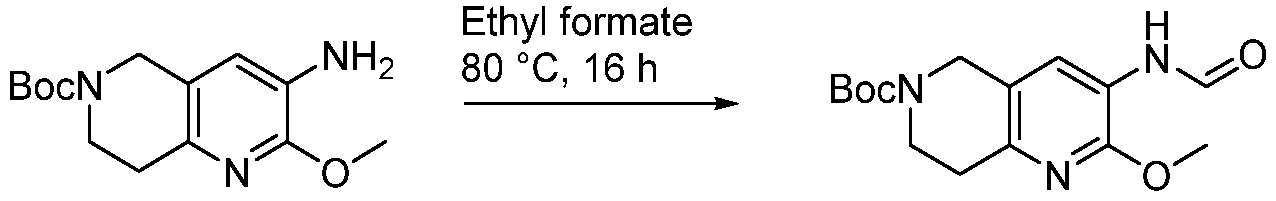
[001307] A solution of tert-butyl 3-amino-2-methoxy-7,8-dihydro-5H-1,6-naphthyridine-6- carboxylate (Step 1) (274 mg, 0.93 mmol, 1.0 eq) in ethyl formate (4.8 mL, 59.5 mmol, 63.8 eq) was stirred in a capped reaction tube and heated to 80°C for 16 h. The resulting mixture was concentrated under vacuum to afford the title compound (290 mg, Yield: quantitative). The compound was used crude in the next step.
[001308] ¹H NMR (400 MHz, DMSO): δ 1.44 - 1.42 (m, 9H), 2.72 (t, J=5.8 Hz, 2H), 3.62 (t, J=5.9 Hz, 2H), 3.91 (s, 3H), 4.42 (s, 2H), 8.23 (s, 1H), 8.32 - 8.31 (m, 1H), 9.82 (s, 1H). [001309] MS: ES+ 308.0 (M+1) Step 3: tert-Butyl 2-methoxy-3-(methylamino)-7,8-dihydro-1,6-naphthyridine-6(5H)-carboxylate

[001310] 1 M Lithium aluminum hydride (1M in THF) (4.3 mL, 4.31 mmol, 2.0 eq) was added dropwise to a cooled (0°C) solution of tert-butyl 3-formamido-2-methoxy-7,8-dihydro-1,6- naphthyridine-6(5H)-carboxylate (Step 2) (785 mg, 2.15 mmol, 1.0 eq) in dry THF (6.2 mL). The reaction was stirred at 0°C for 2 h then quenched with ice water (~20 ml) and extracted with EtOAc (3 x 20 ml). The combined organic extracts were dried over magnesium sulfate, filtered, and concentrated under vacuum. The crude material was purified by chromatography on silica, eluting with a gradient of 0 – 70% (10% MeOH:0.1% 7M methanolic ammonia in DCM) in DCM to afford the title compound as a yellow oil (477 mg, Yield: 63%). [001311] ¹H NMR (400 MHz, DMSO) δ 1.43 (s, 9H), 2.62 (t, J=6.1 Hz, 2H), 2.67 (d, J=5.4 Hz, 3H), 3.58 (t, J=5.5 Hz, 2H), 3.83 (s, 3H), 4.38 (s, 2H), 5.14 (q, J=5.1 Hz, 1H), 6.48 (s, 1H). [001312] MS: ES+ 351.1 (M+1) Steps 4 and 5: (E)-4-(Dimethylamino)-N-(2-methoxy-5,6,7,8-tetrahydro-1,6-naphthyridin-3-yl)-N- methylbut-2-enamide

[001313] To a solution of (E)-4-(dimethylamino)but-2-enoyl chloride hydrochloride (127 mg, 0.69 mmol, 1.15 eq) in acetonitrile (1.1 mL) was added tert-butyl 2-methoxy-3-(methylamino)- 7,8-dihydro-1,6-naphthyridine-6(5H)-carboxylate (Step 3) (176 mg, 0.60 mmol, 1.0 eq) in acetonitrile (5.0 mL) at 0 °C under nitrogen. The reaction mixture was stirred at 0°C for 1 h then concentrated under vacuum. The partially deprotected crude material was suspended in DCM (2 mL) and treated with TFA (2.3 mL, 30.7 mmol, 40.0 eq). After stirring at room temperature for
1 h the mixture was concentrated under vacuum to afford the title compound (530 mg, Yield: assumed quantitative). The compound was used crude in the next step. [001314] MS: ES+ 351.1 (M+1) Step 6: (E)-4-(Dimethylamino)-N-(6-(2-hydroxy-5-isopropyl-4-methoxybenzoyl)-2-methoxy- 5,6,7,8-tetrahydro-1,6-naphthyridin-3-yl)-N-methylbut-2-enamide

[001315] The title compound was prepared from 2-hydroxy-5-isopropyl-4-methoxy-benzoic acid (Example 45 Step 3) and (E)-4-(dimethylamino)-N-(2-methoxy-5,6,7,8-tetrahydro-1,6- naphthyridin-3-yl)-N-methylbut-2-enamide (Step 6) analogously to Example 67 Step 3. [001316] ¹H NMR (400 MHz, DMSO) δ 1.11 (d, J=6.9 Hz, 6H), 2.67 (s, 6H), 2.89 - 2.87 (m, 2H), 3.13 - 3.09 (m, 4H), 3.70 - 3.64 (m, 2H), 3.79 - 3.76 (m, 5H), 3.85 (s, 3H), 4.68 - 4.55 (m, 2H), 6.10 (d, J=15.3 Hz, 1H), 6.48 (s, 1H), 6.54 (s, 1H), 6.66 - 6.57 (m, 1H), 6.96 (s, 1H), 7.65 (s, 1H), 9.60 (s, 1H), 9.81 (s, 1H). Exchangeable protons at 9.81, 9.60 and 6.53 ppm. TFA salt. [001317] LCMS (LC-Method 3): 3.46 min, MS:ES+ 497.7 (M+1) [001318] The compound in the following table was prepared analogously to the Example 83 step 6 from the indicated intermediates. Ex. No. Name and Structure Data 84 ¹H NMR (400 MHz, DMSO) δ 1.17 (d, J=6.9 Hz, 6H), 2.66 (s, 6H), 2.90 (t, J=5.4 Hz, 2H), 3.12 - 3.09 (m, 3H), 3.23 - 3.17 (m, 2H), 3.75 (d, J=6.4 Hz, 3H), (E)-4-(Dimethylamino)-N-(6-(4-hydroxy-3- 3.85 (s, 3H), 4.66 - 4.63 (m, 2H), 6.10 isopropylbenzoyl)-2-methoxy-5,6,7,8- (d, J=14.9 Hz, 1H), 6.53 (s, 1H), 6.67 - tetrahydro-1,6-naphthyridin-3-yl)-N- 6.57 (m, 1H), 6.86 - 6.83 (m, 1H), 7.17 methylbut-2-enamide (dd, J=1.9, 8.9 Hz, 1H), 7.23 (d, J=2.4 Prepared from (E)-4-(dimethylamino)-N- Hz, 1H), 7.65 - 7.64 (m, 1H), 9.55 (s, (2-methoxy-5,6,7,8-tetrahydro-1,6- 1H), 9.85 (s, 1H). Exchangeable naphthyridin-3-yl)-N-methylbut-2- enamide (Example 83 Step 5) and 4-
hydroxy-3-isopropyl-benzoic acid (CAS: protons at 9.85, 9.55 and 6.52 ppm. 859034-02-9). Purified by preparative TFA salt HPLC using Method Prep-LC-12 LCMS (LC-Method 3): 3.07 min, MS:ES+ 467.5 (M+1) Example 85: (E)-N-(2-(3-chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-1,2,3,4- tetrahydroisoquinolin-7-yl)-N-methylbut-2-enamide

Step 1: (E)-N-Methyl-N-(1,2,3,4-tetrahydroisoquinolin-7-yl)but-2-enamide hydrochloride
[001319] The title compound was prepared from tert-butyl 7-(methylamino)-3,4- dihydroisoquinoline-2(1H)-carboxylate (Intermediate I) and crotonyl chloride analogously to Example 67 Step 2. [001320] MS:ES+ 231.3 (M+1) Step 2: (E)-N-(2-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-1,2,3,4- tetrahydroisoquinolin-7-yl)-N-methylbut-2-enamide

[001321] The title compound was prepared from (E)-N-methyl-N-(1,2,3,4- tetrahydroisoquinolin-7-yl)but-2-enamide hydrochloride (Step 1) and 3-chloro-6-hydroxy-4- methoxy-2-methylbenzoic acid (Example 47 Step 3) analogously to Example 67 Step 3.
[001322] ¹H NMR (400 MHz, DMSO) δ 1.73 - 1.67 (m, 3H), 2.14 - 2.07 (m, 3H), 2.91 - 2.66 (m, 2H), 3.22 - 3.14 (m, 3H), 3.44 - 3.39 (m, 1H), 4.04 - 3.68 (m, 4H), 4.43 - 4.30 (m, 1H), 4.89 - 4.72 (m, 1H), 5.84 - 5.70 (m, 1H), 6.52 - 6.48 (m, 1H), 6.76 - 6.64 (m, 1H), 7.07 - 6.98 (m, 1H), 7.27 - 7.19 (m, 2H), 9.96 (s, 1H). [001323] LCMS (LC-Method 3): 4.86 min MS:ES+ 429.3 (M+1) Example 86: N-(6-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-5,6,7,8-tetrahydro- 1,6-naphthyridin-3-yl)but-2-ynamide Step 1: tert-Butyl 3-(but-2-ynamido)-7,8-dihydro-1,6-naphthyridine-6(5H)-carboxylate

[001324] To a cooled (0 °C), stirred solution of tert-butyl 3-amino-7,8-dihydro-5H-1,6- naphthyridine-6-carboxylate (340 mg, 1.4 mmol, 1.0 eq) in DCM (3.0 mL) was added TEA (0.6 mL, 4.1 mmol, 3.0 eq) followed by gradual addition of but-2-ynoyl chloride (182 mg, 1.8 mmol, 1.3 eq) in DCM (1 mL). The reaction mixture was allowed to warm to room temperature and stirred for 12 h. The resulting mixture was diluted with DCM (25 mL) and washed with water (25 mL) and brine (25 mL). The organic layer was dried over MgSO
4, filtered, and concentrated under vacuum to afford the title compound (550 mg, 1.2 mmol, 91%) The compound was used crude in the next step. [001325] MS:ES+ 316.2 (M+1). Step 2: N-(5,6,7,8-Tetrahydro-1,6-naphthyridin-3-yl)but-2-ynamide trifluoroacetate
[001326] To a cooled (0 °C), stirred solution of tert-butyl 3-(but-2-ynoylamino)-7,8-dihydro- 5H-1,6-naphthyridine-6-carboxylate (Step 1) (550 mg, 1.2 mmol, 1.0 eq) in DCM (7.0 mL) was added trifluoroacetic acid (2.1 mL, 27mmol, 22.0 eq) and the reaction mixture was stirred at room temperature for 1 hour under a nitrogen atmosphere. The resulting mixture was concentrated under vacuum to afford the title compound (410 mg, 0.81 mmol, 65%). The compound was used crude in the next step.
[001327] MS:ES+ 316.2 (M+1). Step 3: N-(6-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-5,6,7,8-tetrahydro-1,6- naphthyridin-3-yl)but-2-ynamide

[001328] The title compound was prepared from N-(5,6,7,8-tetrahydro-1,6-naphthyridin-3- yl)but-2-ynamide trifluoroacetate (Step 2) and 3-chloro-6-hydroxy-4-methoxy-2-methylbenzoic acid (Example 47 Step 3) analogously to Example 67 Step 3. [001329] ¹H NMR (400 MHz, DMSO) δ 2.12 - 2.03 (m, 6H), 2.90 - 2.83 (m, 2H), 3.50 - 3.46 (m, 1H), 3.80 (d, J = 4.4 Hz, 3H), 3.97 - 3.90 (m, 1H), 4.38 - 4.35 (m, 1H), 4.86 - 4.73 (m, 1H), 6.52 - 6.46 (m, 1H), 7.89 - 7.68 (m, 1H), 8.54 - 8.45 (m, 1H), 10.04 - 9.90 (m, 1H), 10.81 - 10.73 (m, 1H); [001330] LCMS: 3.25 min, MS:ES+ 414.3 (M+1). Preparation of Intermediates Intermediate A N-Methylisoindolin-4-amine hydrochloride Step 1: tert-Butyl 4-(methylamino)isoindoline-2-carboxylate


[001331] Performed in 10 parallel batches, each of 1 g scale: To a stirred solution of tert- butyl 4-bromoisoindoline-2-carboxylate (CAS:1035235-27-8) (1.0 g, 3.35 mmol, 1 eq.) in 1,4- dioxane (10 mL) was added 2M methyl amine in THF (2.5 mL, 5.03 mmol, 1.5 eq) and NaOtBu (0.806 g, 8.38 mmol, 2.5 eq) at room temperature. The mixture was degassed under nitrogen for 15 mins. Brettphos (0.108 g, 0.20 mmol, 0.06 eq) and Pd
2(dba)
3 (0.122 g, 0.13 mmol, 0.04 eq)
were added and the reaction mixture was heated to 120 °C using microwave irradiation for 1 h. The resulting mixture was filtered through Celite® and the filter cake washed with 10% MeOH in DCM (100 mL). The organic filtrate was concentrated under vacuum and purification of the crude material by chromatography on silica eluting with 15% EtOAc in hexane afforded the title compound as a light yellow solid (7.0 g, Yield: 84%). [001332]
1H NMR (DMSO-d6, 400 MHz): δ 1.46 (s, 9H), 2.68 - 2.0 (m, 3H), 4.37 (d, J= 10 Hz, 2H), 4.50 (d, J= 10.4 Hz, 2H), 5.42 (dd, J= 4.8, 8.8 Hz, 1H), 6.37 (d, J= 8.0 Hz, 1H), 6.51 (t, J= 7.2 Hz, 1H), 7.09 (t, J= 8.0 Hz, 1H). [001333] LCMS (Method A): 2.012 min, MS: ES+ 192.9 (M+1). Step 2: N-Methylisoindolin-4-amine hydrochloride
[001334] A stirred solution of tert-butyl 4-(methylamino)isoindoline-2-carboxylate (step 1) (5.0 g, 20.1 mmol, 1 eq.) in DCM (50 mL) was treated dropwise with 4M HCl in dioxane (25 mL) at 0 ˚C and allowed to warm to room temperature, stirring for 2 h. The resulting mixture was concentrated under vacuum and trituration with n-pentane (3 x 10 mL) afforded the title compound as an off-white solid (5.0 g, Yield: Quantitative). [001335]
1H NMR (DMSO-d6, 400 MHz): δ ppm 2.75 (s, 3H), 4.37 - 4.44 (m, 4H), 6.64 (d, J= 7.6 Hz, 1H), 6.74 (d, J= 7.2 Hz, 1H), 7.23 (t, J= 8.0 Hz, 1H), 9.94 (bs, 2H, D
2O exchangeable). [001336] LCMS (Method A): 0.667 min, MS: ES+ 148.8 (M+1). Intermediate B 3-Chloro-4,6-dihydroxy-2-methylbenzoic acid Step 1: 2,4-Dihydroxy-6-methylbenzaldehyde
[001337] To a cooled (0 ˚C) solution of 5-methylbenzene-1,3-diol (3.0 g, 24.16 mmol, 1.0 eq.) (CAS: 504-15-4) in DMF (30 mL) was added dropwise POCl3 (7.41 g, 48.33 mmol, 2.0 eq.). The reaction mixture was allowed to warm to room temperature and stirred for 16 h. The resulting
mixture was poured into ice cold water (50 mL) and basified with 10% NaOH solution causing a suspension to form. The solid was collected by filtration and dried under vacuum. The crude material was triturated with n-pentane (2 x 20 mL) followed by high vacuum drying to afford the title compound as an off-white solid (1.8 g, 43.95%). [001338]
1H NMR (Acetone, 400 MHz): δ 2.55 (s, 3H), 6.18 (d, J= 4 Hz, 1H), 6.31 (s, 1H), 9.73 (s, 1H, D
2O exchangeable), 10.11 (d, J= 2 Hz, 1H), 12.51 (s, 1H, D
2O exchangeable). [001339] LCMS (Method A): 1.552 min, MS: ES+ 153.1 (M+1). Step 2: 3-Chloro-4,6-dihydroxy-2-methylbenzoic acid
[001340] To a cooled (0 ˚C) solution of 2,4-dihydroxy-6-methylbenzaldehyde (0.5 g, 3.28 mmol, 1.0 eq.) in THF: H
2O (2:1) were added dropwise sulfamic acid (0.63 g, 6.57 mmol, 2.0 eq.) and NaClO
2 (0.59 g, 6.57 mmol, 2.0 eq.) and the reaction mixture was stirred at 0 ˚C for 2 h. The resulting mixture was poured into water (10 mL) and extracted with ethyl acetate (3 x 15 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under reduced pressure. The crude material was triturated with n-pentane (3 x 10 mL) and dried under high vacuum to afford the title compound as a yellow solid (0.5 g, 75.32%). [001341]
1H NMR (DMSO-d6, 400 MHz): δ 2.37 (s, 3H), 6.41 (s, 1H), 10.56 (s, 1H), 10.95 (s, 1H, D
2O exchangeable), 12.98 (s, 1H, D
2O exchangeable). Intermediate C (4-Aminoisoindolin-2-yl)(2,4-bis(methoxymethoxy)-5-methylphenyl)methanone Step 1a: 4-Nitroisoindoline
[001342] Performed in 2 parallel batches, each of 25 g scale: To a cooled (0 ˚C) solution of 4-nitroisoindoline-1,3-dione (25 g, 33.8 mmol, 1.0 eq.) (CAS: 603-62-3) in THF (500 mL) was
added dropwise 1M borane THF (525 mL, 4 eq.) and the reaction mixture was heated to 70
˚C stirring for 12 h. After cooling to room temperature, the mixture was treated dropwise with MeOH (48 mL) and 6N HCl (120 mL) then heated to 60
˚C for a further hour. The resulting mixture was concentrated under vacuum and the crude material was poured into ice cold water (3 L) and extracted with EtOAc (3 x 3 L). The aqueous layer was neutralized with sat. NaHCO
3 solution and extracted with IPA: CHCl
3 (3:7, 3 x 8 L). The combined organic extracts were dried over Na
2SO
4 and concentrated under vacuum to afford the title compound as a brown solid (47 g, quantitative). [001343] LCMS (Method A): 1.93 min, MS: ES+ 165.2 Step 1b: tert-Butyl 4-nitroisoindoline-2-carboxylate
[001344] To a stirred solution of 4-nitroisoindoline (Step 1a) (45 g, 274 mmol, 1 eq) in DCM (450 mL) was added TEA (83.14 g, 823 mmol, 3 eq) and the mixture was stirred at room temperature for 15 mins. Boc-anhydride (89.72 g, 411 mmol, 1.5 eq) was added at 0 ˚C and stirring continued at room temperature for 2 h. The resulting mixture diluted with ice cold water (1000 mL) and extracted with DCM (3 x 500 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under vacuum. The crude material was purified by chromatography on silica eluting with 25% EtOAc in hexane to afford the title compound as a pale yellow solid (25 g, Yield: 34.5%). [001345]
1H NMR (DMSO-d6, 400 MHz): δ ppm 1.47 (d, J= 3.6 Hz, 9H), 4.70 (d, J= 9.6 Hz, 2H), 4.98 (d, J= 9.2 Hz, 2H), 7.61 (t, J= 7.6 Hz, 1H), 7.80 (t, J= 6.8 Hz, 1H), 8.15 (d, J= 8.0 Hz, 1H). [001346] LCMS (Method A): 2.130 min, MS: ES+ 208.8 (M-56) Step 1c: 4-Nitroisoindoline hydrochloride
[001347] To a cooled (0 ˚C) solution of tert-butyl 4-nitroisoindoline-2-carboxylate (Step 1b) (10 g, 37.87 mmol, 1 eq.) in DCM (100 mL) was added dropwise 4M HCl in dioxane (50 mL) and the mixture was stirred at room temperature for 1 h. The resulting mixture was concentrated under vacuum and the crude material was triturated with diethyl ether (4 x 100 mL) to afford the title compound as a brown solid (7.0 g, Yield: 93.9%). [001348]
1H NMR (DMSO-d6, 400 MHz): δ ppm 4.63 (s, 2H), 4.93 (s, 2H), 7.70 (t, J= 8.0 Hz, 1H), 7.88 (d, J= 7.2 Hz, 1H), 8.21 (d, J= 8.4 Hz, 1H), 10.34 (bs, 2H). [001349] LCMS (Method A): 0.410 min, MS: ES+ 164.8 (M+1). Step 1: (2, 4-Dihydroxy-5-methylphenyl) (4-nitroisoindolin-2-yl) methanone
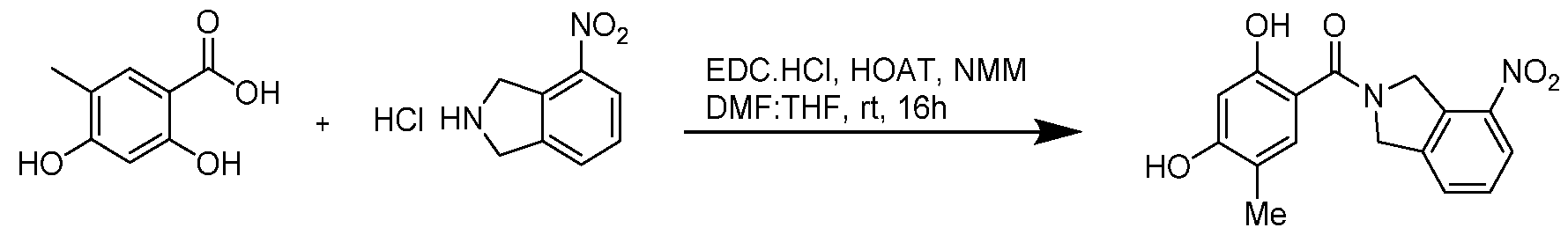
[001350] Performed in 2 parallel batches, each of 2.5 g scale: To a stirred solution of 2,4- dihydroxy-5-methylbenzoic acid (Intermediate F) (2.5 g, 14.88 mmol, 1 eq.) in DMF: THF (1:1) were added EDAC.HCl (4.35 g, 22.13 mmol, 1.5 eq.), HOAT (2.02 g, 14.88 mmol, 1.0 eq.), 4- nitroisoindoline hydrochloride (Step 1c) (3.89 g, 19.34 mmol, 1.3 eq.) and NMM (3.0 g, 29.76 mmol, 2.0 eq.) at room temperature. The reaction mixture stirred at room temperature for 16 h. The resulting mixture was poured into water (500 mL) and extracted in ethyl acetate (3 x 100 mL). The combined organic layer washed with cold brine solution (3 x 100 mL), dried over Na2SO4 and concentrated under vacuum. The crude material was purified by chromatography on silica (product was eluted at 2.7 % MeOH in DCM) followed by reverse phase column chromatography (ACN in Water) to afford the title compound as a light yellow solid (1.8 g, Yield: 33.9%). [001351]
1H NMR (DMSO-d6, 400 MHz): δ 2.04 (s, 3H), 4.89 (s, 2H), 5.19 (s, 2H), 6.44 (s, 1H), 7.03 (d, J= 16.4 Hz, 1H), 7.64 (m, 1H), 7.80 (d, J= 27.6 Hz, 1H), 8.15 (d, J= 8 Hz, 1H), 9.72 (s, 1H), 10.15 (s, 1H). [001352] LCMS (Method A): 1.621 min, MS: ES+ 314.9 (M+1). Step 2: (2, 4-bis(Methoxymethoxy)-5-methylphenyl)(4-nitroisoindolin-2-yl)methanone
[001353] To a cooled (0 ˚C) solution of (2,4-dihydroxy-5-methylphenyl) (4-nitroisoindolin-2- yl) methanone (Step 1) (1.8 g, 5.73 mmol, 1 eq.) in DCM (18 mL) was added DIPEA (4.52 g, 35.03 mmol, 10 eq.) followed by dropwise addition of MOM-Cl (1.4 g, 17.51 mmol, 5 eq.). The reaction mixture was allowed to warm to room temperature and stirred for 3 h. The resulting mixture was poured into water (100 mL) and extracted in DCM (3 x 50 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted in 58% EtOH in hexane to afford the title compound as a light yellow solid (1.0 g, Yield: 47.7%) [001354]
1H NMR (DMSO-d6, 400 MHz) : δ 2.15 (s, 3H), 3.32 (3, 3H), 3.43 (d, J= 4.8 Hz, 3H), 4.70 (s, 1H), 4.93 (s, 1H), 5.03 (s, 1H), 5.19 (t, J= 3.6 Hz, 2H), 5.20 (s, 1H), 5.29 (d, J= 7.6 Hz, 2H), 6.94 (s, 1H), 7.13 (s, 1H),7.59 - 7.67 (m, 1H), 7.75 (d, J= 7.2 Hz, 0.5H), 7.87 (d, J= 7.2 Hz, 0.5H), 8.16 (t, J=8.4 Hz, 1H). [001355] LCMS (Method A): 1.939 min, MS: ES+ 403 (M+1). Step 3: (4-Aminoisoindolin-2-yl)(2,4-bis(methoxymethoxy)-5-methylphenyl)methanone

[001356] To a stirred solution of (2,4-bis (methoxymethoxy)-5-methylphenyl)(4- nitroisoindolin-2-yl)methanone (Step 2) (1.0 g, 2.48 mmol, 1 eq.) in EtOH (10 mL) and H2O (5 mL) were added NH4Cl (1.97 g, 37.31 mmol, 15 eq.) and Fe (0.83 g, 14.92 mmol, 6.0 eq.) at room temperature. The reaction mixture heated was heated to 80 ˚C and stirred for 2 h. The resulting mixture was filtered through Celite® and the filter cake washed with 10% MeOH/DCM (4 x 100 mL). The filtrate was poured in water (200 mL) and extracted with ethyl acetate (3 x 100 mL). The combined organic extracts were dried over Na2SO4 and concentrated under vacuum to afford the title compound as a yellow solid (1.0 g, Yield: Quantitative). [001357]
1H NMR (DMSO-d6, 400 MHz): δ 2.14 (s, 3H), 3.31 (d, J= 9.2 Hz, 3H), 4.36 - 4.69 (m, 4H), 5.15 (d, J= 3.2 Hz, 2H), 5.24 (s, 2H), 6.39 - 6.53 (m, 2H), 6.89 - 7.16 (m, 4H), 7.29 (bs, 1H). One methoxylmethyl obscured under the water peak. LCMS (Method A): 1.673 min, 1.732 min, MS: ES+ 373.1 (M+1). Intermediate D (E)-4-(3,3-Difluoropyrrolidin-1-yl)but-2-enoic acid
Step 1: Methyl (E)-4-(3,3-difluoropyrrolidin-1-yl)but-2-enoate
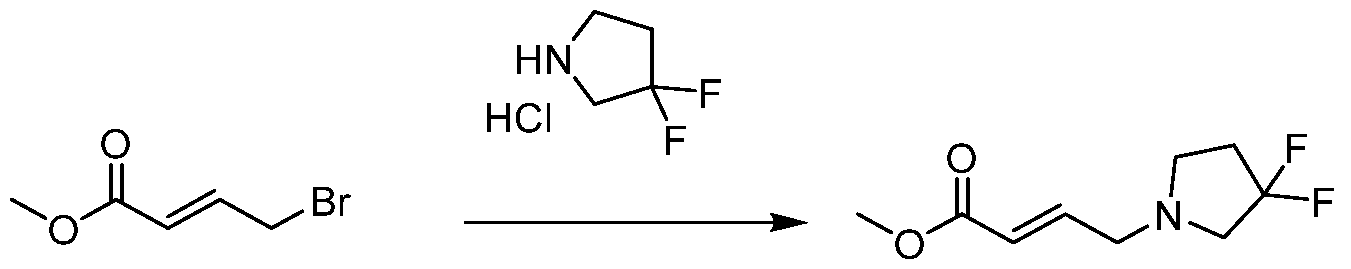
[001358] To a stirred solution of methyl (E)-4-bromobut-2-enoate (CAS: 6000-00-6) (1.0 g, 5.58 mmol, 1.0 eq.) and 3,3-difluoropyrrolidine hydrochloride (0.69 g, 5.58 mmol, 1.0 eq.) in DMF (35 mL) was added K2CO3 (2.31 g, 16.75 mmol, 3.0 eq.) at room temperature. The reaction mixture was heated to 95 ˚ C and stirred for 30 mins. The resulting mixture was diluted with EtOAc (40 mL) and washed with cold brine solution (3 x 40 mL). The organic layer was dried over Na2SO4 and concentrated under reduced pressure. The crude material was purified by chromatography on silica (product was eluted in 5% EtOAc in hexane) to afford the title compound (0.63 g, Yield: 55.3 %). [001359]
1H NMR (DMSO-d6, 400 MHz): δ 2.19 - 2.30 (m, 2H), 2.71 (t, J = 6.8 Hz, 2H), 2.86 - 2.95 (m, 2H), 3.27 (d, J= 5.2 Hz, 2H), 3.66 (s, 3H), 6.01 - 6.06 (m, 1H), 6.80 - 6.86 (m, 1H). [001360] LCMS (Method A): 0.572 min, MS: ES+ 205.8 (M+1). Step 2: (E)-4-(3,3-Difluoropyrrolidin-1-yl)but-2-enoic acid
[001361] To a stirred solution of methyl (E)-4-(3,3-difluoropyrrolidin-1-yl)but-2-enoate (Step 1) (0.6 g, 2.92 mmol, 1.0 eq.) in THF: water (1:1) (12 mL) was added LiOH.H
2O (0.61 g, 14.63 mmol, 5 eq.) and the reaction mixture stirred at room temperature for 16 h. The resulting mixture was neutralized with diluted HCl (pH ~7) and extracted with EtOAc (3 x 50 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under reduced pressure to afford the title compound (1 g, Yield: quantitative). [001362]
1H NMR (MeOD, 400 MHz): δ 2.60 - 2.71 (m, 2H), 3.63 (t, J= 7.2 Hz, 2H), 3.82 (t, J= 12 Hz, 23.6 Hz, 2H), 4.05 (d, J= 6.4 Hz, 2H), 6.26 - 6.30 (m, 1H), 6.87 - 6.95 (m, 1H). [001363] LCMS (Method A): 1.13 min, MS: ES+ 192.1 (M+1). Intermediate DA
(E)-4-(3,3-Difluoroazetidin-1-yl) but-2-enoic acid Step 1: Methyl (E)-4-(3,3-Difluoroazetidin-1-yl) but-2-enoate
[001364] The title compound was prepared from methyl (E)-4-bromobut-2-enoate (CAS: 6000-00-6) and 3,3-difluoroazetidine hydrochloride analogously to Intermediate D Step 1. [001365]
1H NMR (DMSO-d6, 400 MHz): δ 3.36 - 3.38 (m, 2H), 3.61 (s, 1H), 3.65 (t, J = 16Hz, 4H), 3.65 (s, 1H), 3.74 (s, 3H), 5.97 - 6.04 (m, 1H), 6.94 - 6.98 (m, 1H). [001366] LCMS (Method A): 0.489 min, MS: ES+ 191.7 (M+1). Step 2: (E)-4-(3,3-Difluoroazetidin-1-yl) but-2-enoic acid
[001367] The title compound was prepared from methyl(E)-4-(3,3-difluoroazetidin-1-yl) but- 2-enoate (Step 1) and LiOH.H2O analogously to Intermediate D Step 2. [001368]
1H NMR (MeOD, 400 MHz): δ 3.65 (t, J= 12.4 Hz, 4H), 4.89 (m, 2H), 5.99 (d, J= 15.6 Hz, 1H), 6.46 - 6.53 (m, 1H). [001369] LCMS (Method A): 1.22 min, MS: ES+ 178.2 (M+1). Intermediate E N-Benzylisoindolin-4-amine hydrochloride Step 1: tert-Butyl 4-(benzylamino)isoindoline-2-carboxylate

[001370] Performed in 2 parallel batches, each of 1.0 g scale: A stirred solution of tert-butyl 4-bromoisoindoline-2-carboxylate (1.0 g, 3.35 mmol, 1 eq.), phenylmethanamine (CAS: 100-46- 9) (0.431 g, 4.02 mmol, 1.2 eq), NaOtBu (0.806 g, 8.38 mmol, 2.5 eq) in THF (10 mL) was degassed with nitrogen at room temperature for 30 mins. t-BuXphos-PdG3 (0.159 g, 0.20 mmol, 0.06 eq) was added and the reaction mixture heated to 80 ˚C for 1 h. The resulting mixture was filtered through Celite® and the filter cake washed with 10% MeOH: DCM (100 mL) and the combined organic extracts concentrated under reduced vacuum. The crude material was purified by chromatography on silica (product eluted in 4% EtOAc in hexane) to afford the title compound as an off-white solid (2.0 g, Yield: 92 %). [001371]
1H NMR (DMSO-d6, 400 MHz): δ 1.47 (d, J= 8 Hz, 9H), 4.32 (t, J= 4 Hz 2H), 4.49 (t, J= 12 Hz, 4H), 6.07 - 6.13 (m, 1H, D
2O exchangeable), 6.28 (d, J= 8 Hz, 1H), 6.48 (t, J= 8 Hz, 1H), 6.95 (t, J= 8 Hz, 1H), 7.20 (t, J= 4 Hz, 1H), 7.28 - 7.36 (m, 4H). [001372] LCMS (Method A): 2.537 min, MS: ES+ 268.8(M-56). Step 2: N-Benzylisoindolin-4-amine hydrochloride
[001373] To a cooled (0 ˚C) solution of tert-butyl 4-(benzylamino)isoindoline-2-carboxylate (Step 1) (1.0 g, 3.08 mmol, 1.0 eq.) in DCM (10 mL) was added dropwise 4M HCl in dioxane (10 mL) and the reaction mixture was stirred at room temperature for 2 h. The resulting mixture was concentrated under vacuum and the crude material was triturated with n-pentane (3 x 8 mL). to afford the title compound as an off-white solid (1.0 g, Quantitative). [001374]
1H NMR (DMSO-d6, 400 MHz): δ 4.34 – 4.42 (m, 6H), 6.38 (d, J= 8 Hz, 1H), 6.57 (d, J= 8 Hz, 1H), 7.05 (t, J= 8 Hz, 1H), 7.20 (d, J= 8 Hz, 1H), 7.23 - 7.35 (m, 4H), 9.85 (s, 2H, D
2O exchangeable). [001375] LCMS (Method A): 1.236 min, MS: ES+ 225.1 (M+1). Intermediate F 2,4-Dihydroxy-5-methylbenzoic acid Step 1: 4-Methylbenzene-1, 3-diol
[001376] To a stirred solution of 2,4-dihydroxybenzaldehyde (16.5 g, 119.56 mmol, 1 eq.) in THF (500 mL) was added NaCNBH3 (11.3g, 179.93 mmol, 1.5 eq.) at room temperature. The mixture was cooled to 0
˚C, treated dropwise with 1N HCl (330 mL) and allowed to stir at room temperature for 2.5 h. The resulting mixture was poured into water (500 mL) and extracted with 10% MeOH in DCM (4 x 100 mL). The combined organic extracts were dried over Na2SO4 and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted in 4% MeOH in DCM) to afford the title compound as off-white solid (10 g, Yield: 69.6%) [001377]
1H NMR (DMSO-d6, 400 MHz): δ 1.91 (s, 3H), 6.08 - 6.10 (m, 1H), 6.25 (d, J= 2 Hz, 1H), 6.77 (d, J= 8 Hz, 1H), 8.90 (s, 1H), 9.05 (s, 1H). Step 2: 2,4-Dihydroxy-5-methylbenzoic acid
[001378] To a stirred solution of 4-methylbenzene-1, 3-diol (10.0 g, 80.64 mmol, 1 eq.) in water (100 mL) was added KHCO
3 (44.4 g, 443.55 mmol, 5.5 eq.) at room temperature. The reaction mixture was heated to 85 ˚C and purged with CO2
(g) whilst stirring for 16 h. The resulting mixture was diluted with water (50 mL), acidified with KHSO
4 (pH ~3) and extracted in 10% MeOH in DCM (3 x 100 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under vacuum. The crude material was purified by chromatography on silica (product was eluted in 3.6% MeOH in DCM) to afford the title compound as a light yellow solid (5.5 g, Yield: 40.6%). [001379]
1H NMR (DMSO-d6, 400 MHz): δ 2.03 (s, 3H), 6.25 (d, J= 2 Hz, 1H), 7.48 (s, 1H), 10.36 (s, 1H), 11.25 (bs, 1H), 13.27 (bs, 1H). [001380] LCMS (Method A): 1.196 min, MS: ES- 166.93 (M-1). Intermediate FA 2,4-Dihydroxy-5-isopropylbenzoic acid Step 1: Methyl 2, 4-dihydroxybenzoate
[001381] To a stirred solution of 2,4-dihydroxybenzoic acid (100 g, 649.35 mmol) in methanol (400 mL) was added concentrated H2SO4 (50 mL) at room temperature. The reaction mixture was heated to 100
˚C and stirred for 12 h. The resulting solution was concentrated under vacuum and poured into water (250 mL) causing a precipitate to form. The solids were collected by filtration and dried under high vacuum to afford the title compound as an off-white solid (85 g, 78%). This material was used in the next step without any further purification. [001382]
1H NMR (CDCl3, 400 MHz) δ 3.39 (s, 3H), 5.72 (s, br, 1H), 6.38 - 6.42 (m, 2H), 7.74 (d, J= 8.8 Hz, 1 H), 11.02 (s, 1H). [001383] LCMS: 4.24 min, MS: ES+ 169.19(M+1) Step 2: 2,4-Dihydroxy-5-isopropylbenzoate
[001384] To a solution of methyl 2,4-dihydroxybenzoate (Step 1) (80 g, 475.7 mmol) in DCM (1200 mL) was added anhydrous AlCl
3 (126.87 g, 951.48 mmol) followed by isopropyl bromide (351.09 g, 2854.62 mmol) in 3 equal portions (117.03 g added at 6 h intervals) at 50
˚C. The reaction mixture was stirred for a further 6 h at 50 ˚C (total time 24 h) and then concentrated under vacuum. The crude material was diluted with water (1000 mL) and extracted with EtOAc (4 x 500 mL). The combined organic extracts were dried over Na2SO4 and concentrated under vacuum. The resulting crude material was purified three times by chromatography on silica eluting with 3% EtOAc in hexane to afford the title compound as a yellow solid (25.5g, 26%). [001385]
1H NMR (DMSO, 400 MHz) δ 1.13 (d, J= 7.6Hz, 6H), 3.03 - 3.16 (m, 1H), 3.83 (s, 3H), 6.35 (s, 1H), 7.49 (s, 1H), 10.48 (s, 1H), 10.57 (s, 1H). [001386] LCMS: 2.069 min, MS: ES+ 211.11 (M+1). Step 3: 2, 4-Dihydroxy-5-isopropylbenzoic acid
[001387] To a solution of methyl 2,4-dihydroxy-5-isopropylbenzoate (Step 2) (23.0 g, 109.40 mmol) in MeOH: water (2:1, 300 mL) was added NaOH (21.88 g, 547 mmol) at room temperature and the reaction mixture was heated to 70
˚C for 4 h. After cooling to room temperature, the mixture was diluted with ice-cold water (500 mL) and acidified with 1N HCl to pH~5. The resulting precipitate was collected by filtration and dried under high vacuum to afford the title compound as a white solid (13 g, 61%) [001388]
1H NMR (DMSO-d
6, 400 MHz): δ 1.02 (d, J= 6.8Hz, 6H), 3.02 - 3.09 (m, 1H), 6.34 (s, 1H), 7.48 (s, 1H), 10.44 (s, 1H), 11.24 (s, 1H), 13.33 (s,br, 1H). [001389] LCMS: 2.044 min, MS: ES+ 197.22 (M+1). Intermediate G 2,4-Dihydroxy-6-methylbenzoic acid
[001390] To a stirred solution of 5-methylbenzene-1,3-diol (CAS: 504-15-4) (1.0 g, 8.06 mmol, 1 eq.) in ACN (20 mL) was added DBU (3.67 g, 24.1 mmol, 3 eq.) at room temperature in a hydrogenation vessel. The reaction mixture was taken to 20 bar CO
2(g) pressure and stirred at room temperature for 24 h. The resulting mixture diluted with water (50 mL), acidified with 1M HCl solution (10 mL) and extracted with ethyl acetate (3 x 40 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under vacuum to afford the title compound as a grey solid (0.8 g, Yield: 59%). [001391]
1H NMR (DMSO-d6, 400 MHz): δ 2.39 (s, 3H), 6.12 - 6.18 (m, 2H), 10.15 (s, 1H), 12.10 (bs, 1H), 13.45 (bs, 1H). [001392] LCMS (Method A): 1.093 min, MS: ES+ 166.9 (M-1). Intermediate H tert-Butyl 7-amino-3,4-dihydroisoquinoline-2(1H)-carboxylate Step 1: tert-Butyl 7-nitro-3,4-dihydroisoquinoline-2(1H)-carboxylate

[001393] To a stirred solution of 7-nitro-1,2,3,4-tetrahydroisoquinoline (CAS: 42923-79-5) (10 g, 56.1 mmol, 1 eq.) in DCM (100 mL) was added TEA (16.96 g, 168 mmol, 3 eq.) and the mixture was stirred at room temperature for 30 mins. Boc anhydride (24.45 g, 112.2 mmol, 2 eq.) was added and stirring continued at room temperature for 4 h. The resulting mixture diluted with water (50 mL) and extracted with DCM (3 x 40 mL). The combined organic extracts were dried over Na2SO4 and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted at 13% ethyl acetate in hexane) to afford the title compound as an off-white solid (14 g, Yield: 64%) [001394]
1H NMR (DMSO-d6, 400 MHz): δ 1.43 (s, 9H), 2.90 (t, J= 6.0 Hz, 2H), 3.58 (t, J= 5.6 Hz, 2H), 4.63 (bs, 2H), 7.46 (d, J= 8.4 Hz, 1H), 8.03 (dd, J= 2.4 Hz, 8.4 Hz, 1H), 8.14 (d, J= 2.0 Hz, 1H). [001395] LCMS (Method A): 2.210 min, MS: ES+ 222.8 (M-56). Step 2: tert-Butyl 7-amino-3,4-dihydroisoquinoline-2(1H)-carboxylate
[001396] The title compound was prepared from tert-butyl 7-nitro-3,4-dihydroisoquinoline- 2(1H)-carboxylate (Step 1), Fe powder and NH4Cl analogously to Intermediate C step 3. [001397]
1H NMR (DMSO-d6, 400 MHz): δ 1.42 (s, 9H), 2.57 (t, J= 6.0 Hz, 2H), 3.48 (t, J= 5.6 Hz, 2H), 4.32 (s, 2H), 4.92 (s, br, 2H, D2O exchangeable), 6.31 (s, 1H), 6.39 (dd, J= 2.4 Hz, 8.4 Hz, 1H), 6.79 (d, J= 8.4 Hz, 1H). [001398] LCMS (Method A): 1.198 min, MS: ES+ 192.9 (M-56). Intermediate I tert-Butyl 7-(methylamino)-3,4-dihydroisoquinoline-2(1H)-carboxylate
[001399] A solution of tert-butyl 7-amino-3,4-dihydroisoquinoline-2(1H)-carboxylate (Intermediate H) (5 g, 20.16 mmol, 1.0 eq.) in ethyl formate (50 mL) was heated to 80 ˚C and stirred for 2 h. The resulting mixture was concentrated under vacuum and the intermediate compound was dissolved in THF (50 mL) and cooled to 0˚C. Lithium aluminium hydride (1.0 M in THF) (40 mL, 40.32 mmol, 2 eq.) was added dropwise and the reaction mixture was stirred at 0˚C for 2 h. The resulting mixture was poured into an ice-cold saturated solution of NH
4Cl (300 mL) and extracted in DCM (3 x 50 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under vacuum to afford the title compound (3 g, Yield: 47%). [001400]
1H NMR (DMSO-d6, 400 MHz): δ 1.42 (s, 9H), 2.56 - 2.58 (m, 3H), 2.60 - 2.71 (m, 2H), 3.48 - 3.50 (m, 2H), 4.36 - 4.38 (m, 2H), 5.26 (d, J= 5.2 Hz, 1H), 6.19 - 6.27 (m, 1H), 6.34 - 6.40 (m, 1H), 6.79 - 6.86 (m, 1H). [001401] LCMS (Method A): 1.349 min, MS ES+: 262.90 (M+1). Intermediate J N-Methyl-1,2,3,4-tetrahydroisoquinolin-7-amine hydrochloride
[001402] To a cooled (0 ˚C) solution of tert-butyl-7-(methylamino)-3,4-dihydroisoquinoline- 2(1H)-carboxylate (Intermediate I) (0.2 g, 0.763 mmol, 1.0 eq.) in DCM (2 mL) was added 4M HCl in dioxane (1 mL) and the reaction mixture was stirred at room temperature for 1 h. The resulting mixture was concentrated under vacuum and the crude material was triturated with n- hexane (2 x 15 mL) to afford the title compound (0.18 g, Yield: Quantitative). [001403]
1H NMR (DMSO-d6, 400 MHz): δ 2.80 (s, 3H), 2.96 (t, J= 6.0 Hz, 2H), 3.31 (s, br, 2H), 4.24 (s, 2H), 7.06 - 7.24 (m, 3H), 9.52 (s, 2H), 10.38 (s, 1H). [001404] LCMS (Method A): 0.200 min, MS: ES+163 (M+1). Intermediate K [001405] 4,6-Dihydroxy-2,3-dimethylbenzoic acid Step 1: 4,5-Dimethyl-1,3-phenylene diacetate
[001406] Performed in 2 parallel batches, each of 30 g scale: To a stirred solution of 5,5- dimethylcyclohexane-1,3-dione (CAS: 126-81-8) (30 g, 214.5 mmol, 1.0 eq.) in acetic anhydride (340 mL) was added dropwise concentrated H2SO4 (12.4 mL, 235 mmol, 1.1 eq.) at room temperature. The reaction mixture was heated to 150 ˚C and stirred for 1 h. The resulting mixture was slowly poured into ice cold water (500 mL) and extracted with ethyl acetate (3 x 500 mL). The combined organic extracts were washed with NaHCO3 solution (3 x 500 mL), dried over Na2SO4 and concentrated under reduced pressure. The crude material was purified by chromatography on silica (product was eluted in 15% EtOAc in hexane) yielding to afford the title compound as a white solid (80 g, Yield: 84%). [001407]
1H NMR (DMSO-d6, 400 MHz): δ 2.24 (s, 3H), 2.26 (s, 3H), 2.30 (s, 3H), 2.32 (s, 3H), 6.76 (d, J = 2.4 Hz, 1H), 6.88 (d, J = 2.4 Hz, 1H). Step 2: 4, 5-Dimethylbenzene-1, 3-diol
[001408] To a solution of 4,5-dimethyl-1,3-phenylene diacetate (Step 1) (80 g, 360.3 mmol, 1.0 eq) in EtOH; H2O (1:1) (800 mL) was added NaOH (57.6 g, 1441.4 mmol., 4 eq.) and the mixture heated to 70 ˚C for 2 h. The resulting mixture was acidified with dilute HCl (pH 5) and concentrated under reduced pressure. The crude material was diluted with ethyl acetate (300 mL) and washed with brine solution (3 x 300 mL). The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The crude material was purified by chromatography on silica (product was eluted in 15% EtOAc in hexane) to afford the title compound (25 g, Yield: 55%). [001409]
1H NMR (DMSO-d6, 400 MHz): δ 1.83 (s, 3H), 2.06 (s, 3H), 6.03 (s, 1H), 6.12 (s, 1H), 8.78 (s, 1H) 8.94 (s, 1H). Step 3: 4,6-Dihydroxy-2,3-dimethylbenzaldehyde
[001410] Phosphorus oxychloride (41.5 g, 271.7 mmol, 1.5 eq.) was added dropwise to DMF (250 mL) at 0 ˚C. To this solution was added dropwise 4,5-dimethylbenzene-1,3-diol (Step 2) (25 g 181.1 mmol, 1 eq.) in DMF at 0 ˚C and the reaction mixture was stirred at room temperature for 1 h. The resulting mixture was slowly poured into ice cold water, basified using NaOH solution (pH ~10) and acidified with dilute HCl (pH 4 - 5) causing precipitation of a solid. The solid was collected by filtration and dried under high vacuum to afford the title compound as a yellow solid (15 g, Yield: 65%). [001411]
1H NMR (DMSO-d6, 400 MHz): δ 1.99 (s, 3H), 2.41 (s, 3H), 6.22 (s, 1H), 10.13 (s, 1H), 10.75 (s, 1H) 12.24 (s, 1H). [001412] LCMS (Method A): 1.467 min, MS: ES+ 166.8 (M+1) Step 4: 4,6-Dihydroxy-2,3-dimethylbenzoic acid
[001413] To a solution of 4,6-dihydroxy-2,3-dimethylbenzaldehyde (Step 3) (5 g, 30.12 mmol, 1.0 eq.) in DMSO: water (50 mL) were added dropwise NaClO2 (13.5 g, 150.6 mmol, 5 eq.) and NaH2PO4 (18.0 g, 150.6 mmol, 5 eq.) and the reaction mixture stirred at room temperature for 1 h. The resulting mixture was diluted with water (100 mL) and the pH adjusted with dilute HCl (pH 4 – 5). The mixture was extracted with ethyl acetate (3 x 80 mL) and the combined organic extracts were washed with brine solution (3 x 100 mL), dried over Na2SO4 and concentrated under reduced pressure to afford the title compound (25 g, Yield: 55%). [001414]
1H NMR (DMSO-d6, 400 MHz): δ 1.96 (s, 3H), 2.27 (s, 3H), 6.24 (s, 1H), 9.86 (s, 1H), 10.89 (s, 1H) 13.07 (bs, 1H). [001415] LCMS (Method A): 1.220 min, MS: ES+ 182.9 (M+1). Intermediate L N-((1-Methyl-1H-pyrazol-4-yl) methyl)isoindolin-4-amine
Step 1: tert-Butyl 4-(((1-methyl-1H-pyrazol-4-yl) methyl) amino) isoindoline-2-carboxylate
[001416] To a stirred solution of tert-butyl 4-aminoisoindoline-2-carboxylate (CAS:871013- 98-8) (0.5 g, 2.13 mmol, 1.0 eq.) in MeOH (5 mL) was added 1-methyl-1H-pyrazole-4- carbaldehyde (CAS: 25016-11-9) (0.47 g, 4.27 mmol, 2.0 eq.) at room temperature. After 5 min ZnCl
2 (0.87 g, 6.41 mmol, 3.0 eq) was added and stirring continued at room temperature for 16 h. NaCNBH
3 (0.67 g, 10.68 mmol, 5.0 eq.) was added and the mixture was stirred for a further hour at room temperature. The resulting mixture was poured into water (100 mL) and extracted with EtOAc (3 x 60 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted at 2.0% MeOH in DCM) to afford the title compound (0.5 g, Yield: 71.3%). [001417]
1H NMR (DMSO-d6, 400 MHz): δ 1.49 (s, 9H), 3.37 and 3.74 (singlets, 3H), 4.11 (t, J= 4.4 Hz, 2H), 4.40 (d, J= 6.0 Hz, 2H), 4.49 (d, J= 11.2 Hz, 2H), 6.44 - 6.52 (m, 2H), 7.03 (t, J= 7.6 Hz, 1H), 7.35 (d, J= 2.4 Hz, 1H), 7.57 (d, J= 2.8 Hz, 1H) [001418] LCMS (Method A): 1.924 min, MS: ES+ 229.2 (M-100). Step 2: N-((1-Methyl-1H-pyrazol-4-yl) methyl) isoindolin-4-amine
[001419] To a cooled (0 ˚C) solution of tert-butyl 4-(((1-methyl-1H-pyrazol-4-yl) methyl) amino) isoindoline-2-carboxylate (Step 1) (0.5 g, 1.52 mmol, 1 eq.) in DCM (5 mL) was added dropwise 4M HCl in dioxane (2.5 mL) and the reaction mixture stirred at room temperature for 1 h. The resulting mixture concentrated under vacuum and trituration of the crude material with n- pentane (4 x 30 mL) afforded the title compound (0.65 g, Yield: Quantitative). [001420]
1H NMR (DMSO-d6, 400 MHz): δ 3.79 and 3.81 (singlets, 3H), 4.22 (d, J= 12 Hz, 2H), 4.41 – 4,43 (m, 4H), 6.76 - 6.82 (m, 2H), 7.21 (t, J= 8.0 Hz, 1H), 7.43 (s, 1H), 7.67(s, 1H).
[001421] LCMS (Method A): 0.850 min, MS: ES+ 229.1 (M+1). Intermediate M N-Phenethylisoindolin-4-amine hydrochloride Step 1: tert-Butyl 4-(phenethylamino) isoindoline-2-carboxylate
[001422] To a stirred solution of tert-butyl 4-aminoisoindoline-2-carboxylate (CAS: 871013- 98-8) (0.8 g, 4.26 mmol, 1.0 eq.) in DMF (5 mL) were added (2-bromoethyl) benzene (CAS: 103- 63-9) (0.47 g, 8.52 mmol, 2.0 eq.) and K2CO3 (0.87 g, 12.78 mmol, 3.0 eq.) and the reaction mixture heated to 80
˚C for 16 h. The resulting mixture was diluted with ice cold water (100 mL) and extracted with EtOAc (3 x 100 mL). The combined organic extracts were dried over Na2SO4 and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted at 10.0% EtOAc in hexane) to afford the title compound (0.45 g, Yield: 38.9%). [001423]
1H NMR (DMSO-d6, 400 MHz): δ 1.99 (s, 9H), 2.86 (t, J= 8 Hz, 2H), 3.27 - 3.31 (m, 2H), 4.38 (d, J= 8.4 Hz, 2H), 4.51 (d, J= 9.6 Hz, 2H), 5.40 (t, J= 5.6 Hz, 1H), 6.48 - 6.54 (m, 2H), 7.08 (t, J= 7.2 Hz, 1H).7.22 - 7.32 (m, 5H). [001424] LCMS (Method A): 2.682 min, MS: ES+ 283 (M-56). Step 2: N-Phenethylisoindolin-4-amine hydrochloride
[001425] The title compound was prepared from tert-butyl 4-(phenethylamine) isoindoline- 2-carboxylate (Step 1) and 4M HCl in dioxane analogously to Example 4 Step 2. [001426]
1H NMR (DMSO-d6, 400 MHz): δ 2.90 (t, J= 8 Hz, 2H), 3.34 (t, J= 8 Hz, 2H), 4.36 - 4.42 (m, 4H), 6.71 (t, J= 5.6 Hz, 2H), 7.18 - 7.33 (m, 6H), 10.04 (s, br, 2H). [001427] LCMS (Method A): 1.343 min, MS: ES+ 239.1 (M+1)
Intermediate N 5-(tert-Butyl)-2-hydroxy-4-methoxybenzoic acid Step 1: 5-(tert-Butyl)-2,4-dihydroxybenzoic acid
[001428] To a cooled (0 ˚C) solution of 2,4-dihydroxybenzoic acid (5 g, 32.40 mmol, 1 eq.) in tert- butanol (24.06 g, 324 mmol, 10 eq.) and TFA (25.90 g, 227.20 mmol, 7 eq.) was added dropwise cH
2SO
4 (1.58 g, 16.20 mmol, 0.5 eq.) and the reaction mixture heated to 80
°C for 16 h. The resulting solution was cooled to room temperature and poured into ice cold water (500 mL). Hexane was added resulting in the formation of a precipitate. Solids were collected by filtration and dried under high vacuum to afford the title compound as a light pink solid (4.1 g, Yield: 93.9%). [001429]
1H NMR (DMSO-d6, 400 MHz): δ ppm 1.30 (s, 9H), 6.33 (s, 1H), 7.55 (s, 1H), 10.40 (s, 1H), 11.20 (s, 1H), 13.32 (s, 1H). [001430] LCMS (Method A): 1.730 min, MS: ES+ 209 (M-1). Step 2: 6-(tert-Butyl)-7-hydroxy-2,2-dimethyl-4H-benzo[d][1,3]dioxin-4-one
[001431] To a stirred solution of 5-(tert-butyl)-2,4-dihydroxybenzoic acid (Step 1) (4 g, 19.04 mmol, 1.0 eq.) in DME (40 mL) were added acetone (14.5 mL, 190.4 mmol, 10.0 eq.), DMAP (0.232 g, 1.90 mmol, 0.1 eq.) and SOCl
2 (6.20 mL, 85.7 mmol, 4.5 eq.) and the reaction mixture stirred at room temperature for 3 h. The resulting mixture was basified with saturated NaHCO
3 solution (200 mL) and extracted with ethyl acetate (2 x 200 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under vacuum. The crude material was purified by
chromatography on silica (product eluted in 1% MeOH: DCM) to afford the title compound as a light brown liquid (3.0 g, Yield: 73.5%). [001432] LCMS (Method A): 1.997 min. MS: ES+ 250.9 (M+1). Step 3: 6-(tert-Butyl)-7-methoxy-2,2-dimethyl-4H-benzo[d][1,3]dioxin-4-one
[001433] To a solution of 6-(tert-butyl)-7-hydroxy-2,2-dimethyl-4H-benzo[d][1,3]dioxin-4- one (Step 2) (3.0 g, 11.98 mmol, 1.0 eq.) in DMF (30 mL) was added K2CO3 (4.96 g, 35.95 mmol, 3.0 eq.) and the mixture stirred at room temperature for 15 mins. Methyl iodide (3.40 g, 23.97 mmol, 2.0 eq.) was added and the reaction mixture was heated to 50
˚C for 24 h. The resulting mixture was diluted with water (500 mL) and extracted with ethyl acetate (2 x 500 mL). The combined organic extracts were dried over Na2SO4 and concentrated under vacuum. The crude material was purified by chromatography on silica (product eluted in 2.5% EtOAc: hexane) to afford the title compound as a light yellow liquid (1.5 g, Yield: 41.67%). [001434]
1H NMR (DMSO-d6, 400 MHz): δ 1.32 (s, 9H), 1.67 (s, 6H), 3.89 (s, 3H), 6.70 (s, 1H), 7.62 (s, 1H). [001435] LCMS (Method A): 2.374 min. MS: ES+ 264.8 (M+1). Step 4: 5-(tert-Butyl)-2-hydroxy-4-methoxybenzoic acid
[001436] The title compound was prepared from 6-(tert-butyl)-7-methoxy-2,2-dimethyl-4H- benzo[d][1,3]dioxin-4-one (Step 3) and LiOH.H2O analogously to Intermediate D Step 2. [001437] LCMS (Method A): 2.088 min. MS: ES+ 224.9 (M+1). Intermediate O
3-(tert-Butyl)-2-fluoro-6-hydroxybenzoic acid Step 1: 2-Bromo-4-(tert-butyl)-5-fluorophenol
[001438] The title compound was prepared from 2-bromo-5-fluorophenol (CAS: 147460-41- 1), aluminium chloride and tert-butyl chloride analogously to Intermediate FA Step 2. [001439]
1H NMR (DMSO-d6, 400 MHz): δ 1.27 (s, 9H), 6.70 (d, J= 13.6 Hz, 1H), 7.30 (d, J= 8.8 Hz, 1H), 10.53 (s, 1H, D2O exchangeable). Step 2: 1-Bromo-5-(tert-butyl)-4-fluoro-2-(methoxymethoxy)benzene
[001440] The title compound was prepared from 2-bromo-4-(tert-butyl)-5-fluorophenol (Step 1) and MOM-Cl analogously to Intermediate C Step 2. [001441]
1H NMR (DMSO-d6, 400 MHz): δ 1.30 (s, 9H), 3.36 and 3.40 (singlets, 3H), 5.29 (s, 2H), 7.08 (d, J= 14 Hz, 1H), 7.43 (d, J= 8.8 Hz, 1H). Step 3: 3-(tert-Butyl)-2-fluoro-6-(methoxymethoxy)benzoic acid
[001442] A stirred solution 1-bromo-5-(tert-butyl)-4-fluoro-2-(methoxymethoxy)benzene (step 2) (3.5 g, 12.03 mmol, 1.0 eq.) in THF (35 mL) was cooled to -78
˚C and treated dropwise
with n-BuLi (1.6M in hexane) (15 mL, 2 eq.). After stirring at -78 ˚C for 30 mins, dry ice (1 small piece) was added and the reaction mixture allowed to warm to room temperature and stirred for 2 h. The resulting mixture was poured into cold water (30 mL), basified with 1N NaOH solution and extracted with EtOAc (3 x 50 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under reduced pressure. The crude material was triturated with n-pentane (2 x 30 mL) to afford 3-(tert-butyl)-2-fluoro-6-(methoxymethoxy)benzoic acid along with 5-(tert- butyl)-4-fluoro-2-(methoxymethoxy)benzoic acid as a minor impurity. (2.5 g, Yield: 81%). [001443]
1H NMR (DMSO-d6, 400 MHz): δ 1.31 (d, J= 2.8 Hz, 9H), 3.39 and 3.42 (singlets, 3H), 5.22 and 5.25(singlets, 2H), 6.93 - 7.04 (m, 1H), 7.28 - 7.67 (m, 1H), 12.78 (s, 1H, D
2O exchangeable). Step 4: 3-(tert-Butyl)-2-fluoro-6-hydroxybenzoic acid
[001444] The title compound was prepared from 3-(tert-butyl)-2-fluoro-6- (methoxymethoxy)benzoic acid (Step 3) and 4M HCl in dioxane analogously to Example 2 Step 2. [001445]
1H NMR (DMSO-d6, 400 MHz): δ 1.25 -1.30 (m, 9H), 6.65 - 6.78 (m, 1H), 7.25 (t, J = 9.2Hz, 0.4H), 7.73 (d, J= 9.2 Hz, 0.6H), 12.13 (s, 1H, D2O exchangeable). [001446] LCMS (Method A): 1.874 min, 2.048 min, MS: ES- 211 (M-1). Intermediate OA 5-(tert-Butyl)-4-fluoro-2-hydroxybenzoic acid Step 1: 4-(tert-Butyl)-5-fluoro-2-vinylphenol
[001447] A stirred solution of 2-bromo-4-(tert-butyl)-5-fluorophenol (Intermediate O Step 1) (5.0 g, 20.24 mmol, 1.0 eq.) and potassium trifluoro(vinyl)borate (4.06 g, 30.36 mmol, 1.5 eq.) in THF: Water (8:2) at room temperature was treated with Na
2CO
3 (14.6 g, 139.6 mmol, 6.9 eq.) and Bis(triphenylphosphine)palladium(II) dichloride (Dikis) (0.71 g, 1.021 mmol, 0.05 eq); the resulting reaction mixture was heated to 110°C and stirred for 4h. The reaction mixture was cooled, diluted with water (150 mL) and extracted using EtOAc (3 x 150 mL). The combined organic layer was dried over Na
2SO
4 and concentrated under vacuum. Crude material was purified by flash chromatography on silica (product eluted with 4% EtOAc in Hexane) yielding 4- (tert-butyl)-5-fluoro-2-vinylphenol as a yellow liquid. (3.0 g, Yield: 76.4 %). [001448]
1H NMR (DMSO-d6, 400 MHz): δ ppm 1.30 (s, 9H), 5.16 (d, J= 11.6 Hz, 1H), 5.74 (d, J= 1.2 Hz, 1H), 6.50 - 6.58 (m, 1H), 6.81 - 6.88 (m, 1H), 7.28 - 7.32 (m, 1H), 9.94s (s, 1H). [001449]
LCMS (Method N): 2.602 min, MS: ES - 192.8 (M-1). Step 2: 5-(tert-Butyl)-4-fluoro-2-hydroxybenzaldehyde
[001450] A stirred solution of potassium osmate (K
2OsO
4) (0.05 g, 0.154 mmol, 0.01 eq.) in water (30 mL) at room temperature was treated dropwise with a solution of 4-(tert-butyl)-5-fluoro- 2-vinylphenol (Step 1) (3.0 g, 15.44 mmol, 1.0 eq.) dissolved in 1,4-dioxane (30 mL). NaIO
4 (8.27 g, 38.65 mmol, 2.5 eq.) was added portion-wise to the reaction mixture and the resulting reaction mixture stirred at room temperature for 30 min; diluted with water (150 mL) and extracted using EtOAc (3 x 100 mL). The combined organic layer was dried over Na
2SO
4 and concentrated under vacuum. Crude material was purified by flash chromatography on silica (product eluted at 100% Hexane) yielding 5-(tert-butyl)-4-fluoro-2-hydroxybenzaldehyde (1.5 g, Yield: 49.5 %, 7.64 mmol). [001451]
1H NMR (DMSO-d6, 400 MHz): δ ppm 1.31 (s, 9H), 6.74 (d, J= 14 Hz, 1H), 7.63 (d, J= 9.6 Hz, 1H), 10.18 (s, 1H), 11.04 (s, 1H). Step 3: 5-(tert-Butyl)-4-fluoro-2-(methoxymethoxy)benzaldehyde
[001452] A stirred solution of 5-(tert-butyl)-4-fluoro-2-hydroxybenzaldehyde (Step 2) (1.5 g, 7.63 mmol, 1.0 eq.) in DCM (15 mL) at 0°C was treated with DIPEA (2.96 g, 22.95 mmol, 3.0 eq.) and stirred for 15 mins. MOM-Cl (1.22 g, 15.30 mmol, 2.0 eq.) was added dropwise and the resulting reaction mixture stirred at 0°C for 45 mins. The resulting reaction mixture was diluted with water (100 mL) and extracted using EtOAc (3 x 50 mL). The combined organic layer was dried over Na2SO4 and concentrated under vacuum. Crude material was purified by flash chromatography on silica (product eluted with 2% EtOAc in Hexane) yielding 5-(tert-butyl)-4- fluoro-2-(methoxymethoxy)benzaldehyde as a yellow liquid (1.7 g, Yield: 92.5%). [001453]
1H NMR (DMSO-d6, 400 MHz): δ ppm 1.32 (s, 9H), 3.43 (s, 3H), 5.37 (s, 2H), 7.12 (d, J= 14.8 Hz, 1H), 7.68 (d, J= 9.6 Hz, 1H), 10.3 (s, 1H). [001454] LCMS (Method N): 2.635 min, 210 nm MS: ES + 240.7 (M+1). Step 4: 5-(tert-Butyl)-4-fluoro-2-(methoxymethoxy)benzoic acid

[001455] A stirred solution of 5-(tert-butyl)-4-fluoro-2-(methoxymethoxy)benzaldehyde (Step 3) (1.7 g, 7.075 mmol, 1.0 eq.) in THF : tert-butanol (17 mL) at 0°C was treated dropwise with NaH2PO4 (5.0 g, 42.45 mmol, 6.0 eq) and NaClO2 (1.91 g, 21.22 mmol, 3.0 eq) solution dissolved in water (17 mL).2-Methyl 2-butene (7.43 g, 106.1 mmol, 15.0 eq.) was then added dropwise at 0°C and the reaction mixture allowed to warm to room temperature over 30 min. The reaction mixture was poured into cold water (100 mL) and extracted in EtOAc (3 x 50 mL). The combined organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure yielding 5-(tert-butyl)-4-fluoro-2-(methoxymethoxy)benzoic acid as an off-white solid (1.7 g, Yield: 93.8%). This material was used directly in the next step.
[001456]
1H NMR (DMSO-d6, 400 MHz): δ ppm 1.32 (s, 9H), 3.39 (s, 3H), 5.25 (s, 2H), 7.02 (d, J= 14.4 Hz, 2H), 7.66 (d, J= 10 Hz, 1H), 12.69 (bs, 1H). Step 5: 5-(tert-Butyl)-4-fluoro-2-hydroxybenzoic acid
[001457] A stirred solution of 5-(tert-butyl)-4-fluoro-2-(methoxymethoxy)benzoic acid (Step 5) (1.1 g, 4.29 mmol, 1.0 eq.) in DCM (11 mL) at 0
°C was treated dropwise with 4M HCl in dioxane (5.5 mL); the resulting reaction mixture was stirred at room temperature for 30 min and concentrated under vacuum yielding 5-(tert-butyl)-4-fluoro-2-hydroxybenzoic acid as a yellow semi solid (1.0 g, Yield: quantitative). This material was used without further purification. [001458]
1H NMR (DMSO-d6, 400 MHz): δ ppm 1.31 (m, 9H), 6.78 (d, J= 14 Hz, 1H), 7.73 (d, J= 9.2 Hz, 1H), 11.39 (bs, 1H), 14.04 (bs, 1H). LCMS (Method N): 2.433 min, MS: ES - 210.9 (M-1). Intermediate P 2-Chloro-4,6-bis(methoxymethoxy)benzoic acid Step 1: 2-Chloro-4,6-dimethoxybenzaldehyde
[001459] The title compound was prepared from 1-chloro-3,5-dimethoxybenzene (CAS: 7051-16-3) and POCl3 analogously to Intermediate B Step 1. [001460]
1H NMR (DMSO-d6, 400 MHz): δ 3.89 (s, 3H), 3.90 )s, 3H), 6.69 - 6.74 (dd, J= 2.4 Hz, 2H), 10.25 (s, 1H). [001461] LCMS (Method A): 1.568 min. MS: ES+ 200.8 (M+1).
Step 2: 2-Chloro-4,6-dihydroxybenzaldehyde
[001462] To a cooled (0 ˚C) solution of 2-chloro-4,6-dimethoxybenzaldehyde (Step 1) (8 g, 39.87 mmol, 1.0 eq.) in DCM (80 mL) was added dropwise BBr3 (1M in DCM) (199.38 mL, 199.38 mmol, 5.0 eq.) and the reaction mixture was stirred at room temperature for 16 h. The resulting mixture was diluted with ice cold water (200 mL) and extracted with ethyl acetate (4 x 200 mL). The combined organic extracts were dried over Na2SO4 and concentrated under vacuum. The crude material was purified by trituration with n-pentane (3 x 30 mL) followed by high vacuum drying to afford the title compound as a red solid (5.0 g, Yield: 72.9%). [001463]
1H NMR (DMSO-d6, 400 MHz): δ 6.27 (s, 1H), 6.52 (s, 1H), 10.07 (s, 1H), 11.35 (s, 1H, D
2O exchangeable), 12.11 (s, 1H, D
2O exchangeable). [001464] LCMS (Method A): 1.565 min. MS: ES- 170.9 (M-1). Step 3: 2-Chloro-4,6-bis(methoxymethoxy)benzaldehyde
[001465] The title compound was prepared from 2-chloro-4,6-dihydroxybenzaldehyde (Step 2) and MOM-Cl analogously to Intermediate C Step 2. [001466] LCMS (Method A): 1.739 min. MS: ES+ 260.8 (M+1). Step 4: 2-Chloro-4,6-bis(methoxymethoxy)benzoic acid
[001467] Performed in 2 parallel batches, each of 2 g scale: To a stirred solution of 2-chloro- 4,6-bis(methoxymethoxy)benzaldehyde (Step 3) (2 g, 7.67 mmol, 1.0 eq.) in THF: t-BuOH (40 mL) were added a saturated solution of NaClO2 (in minimal water) (2.41 g, 26.85 mmol, 3.5 eq.) followed by NaH2PO4 (5.89 g, 49.1 mmol, 6.4 eq.) and the mixture stirred at room temperature
for 5 mins. To this mixture was added dropwise 2-methyl-2-butene (7.51 g, 107.4 mmol, 14 eq.) and stirring continued at room temperature for 1 h. The resulting mixture was diluted with water (200 mL) and extracted with DCM (3 x 100 mL). The combined organic extracts were dried over Na
2SO
4 and concentrated under vacuum to afford the title compound as a light brown liquid (4.0 g, Yield: 78.5%). The crude material was used in the next step without further purification. Intermediate Q 5-Chloro-2,4-dihydroxybenzoic acid Step 1: Methyl 5-chloro-2,4-dihydroxybenzoate
[001468] To a cooled (0 ˚C) solution of methyl 2,4-dihydroxybenzoate (Intermediate FA Step 1) (1.0 g, 5.95 mmol, 1 eq.) in DCM (10 mL) was added SO
2Cl
2 (0.45 mL) and the reaction mixture was allowed to warm to room temperature stirring for 16 h. The resulting solution was concentrated under vacuum and the crude material was diluted with water (40 mL) and extracted with ethyl acetate (4 x 40 mL). The combined organic extracts were washed with saturated NaHCO
3 solution (2 x 40 mL), dried over Na
2SO
4 and concentrated under vacuum. Purification of the crude material by chromatography on silica eluting with 4% EtOAc in hexane afforded the title compound (0.73 g, Yield: 60.6%). [001469]
1H NMR (DMSO-d6, 400 MHz): δ ppm 3.84 (s, 3H), 6.52 (s, 1H), 7.69 (s, 1H), 10.57 (s, 1H, D
2O exchangeable), 11.31 (s, 1H, D
2O exchangeable). [001470] LCMS (Method A): 1.771 min, MS: ES+ 201.2 (M-1). Step 2: 5-Chloro-2,4-dihydroxybenzoic acid
[001471] To a stirred solution of methyl 5-chloro-2,4-dihydroxybenzoate (Step 1) (1.5 g, 7.40 mmol, 1 eq.) in MeOH: water (30 mL) was added NaOH (1.49 g, 37.10 mmol, 5 eq.) and the reaction mixture heated to 80 ˚C for 4 h. The resulting mixture was diluted with water (20 mL), acidified with 1N HCl solution (10 mL) and extracted with EtOAc (3 x 30 mL). The combined
organic extracts were dried over Na
2SO
4 and concentrated under vacuum to afford the title compound as a brown solid (1.2 g, Yield: 86.0%). The crude material was used in the next step without further purification. [001472] LCMS (Method A): 1.128 min, MS: ES+ 186.8 (M-1). Intermediate R 6-Hydroxy-3,3-dimethylindoline-5-carboxylic acid Step 1: 4-Acetamido-5-bromo-2-methoxybenzoate

[001473] To methyl 4-acetamido-2-methoxybenzoate (CAS: 4093-29-2) (15 g, 67.2 mmol, 1.0 eq.) in EDC (450 mL) were added Pd(OAc)2 (1.5 g, 67.2 mmol, 0.1 eq.), Cu(OAc)2 (26 g, 134.5 mmol, 2 eq.) and CuBr2 (29.9 g, 134.5 mmol, 2.0 eq.) under nitrogen and the reaction mixture was heated to 90˚C for 72 h. The resulting mixture was diluted with ice cold water (1000 mL) and filtered and washed with DCM (500 mL). The aqueous layer was extracted with DCM (2 x 200 mL) and the combined organic extracts were dried over Na2SO4 and concentrated under vacuum. The crude material was purified by chromatography (product eluting at 1% methanol in DCM) to afford the title compound (15 g, Yield: 73.0%). [001474]
1H NMR (DMSO-d6, 400 MHz): δ 2.15 (s, 3H), 3.78 (s, 3H), 3,79 (s, 3H), 7.67 (s, 1H), 7.89 (s, 1H), 9.50 (s, 1H). [001475] LCMS (Method A): 1.431 min, MS: ES+ 301.7 & 303.6 (M+1 & M+3). Step 2: 4-Amino-5-bromo-2-methoxybenzoic acid
[001476] To a stirred solution of methyl 4-acetamido-5-bromo-2-methoxybenzoate (Step 1) (15 g, 49.61 mmol, 1.0 eq.) in MeOH: H
2O (1:1) (600 mL) was added aq. NaOH (19.86 g, 496.61 mmol, 10 eq.) and the reaction mixture heated to 80
˚C for 4 h. The resulting mixture was concentrated under reduced pressure and the crude residue was acidified with 2N HCl (pH ~ 2- 3) causing precipitation of a solid. The solid was collected by filtration, washed with water (3 x
500 mL) and triturated with n-pentane (2 x 100 mL) to afford the title compound (11 g, Yield: 90.0 %). [001477]
1H NMR (DMSO-d6, 400 MHz): δ 3.72 (s, 3H), 6.06 (s, 2H), 6.45 (s, 1H), 7.74 (s, 1H), 11.91 (s, 1H, D
2O exchangeable). [001478] LCMS (Method A): 1.489 min, MS: ES+ 245.7 & 247.9 (M+1 & M+3). Step 3: Methyl 4-amino-5-bromo-2-methoxybenzoate
[001479] To a stirred solution of methyl 4-amino-5-bromo-2-methoxybenzoic acid (Step 2) (10 g, 40.61 mmol, 1.0 eq.) in MeOH (100 mL) were dropwise added SOCl
2 (20 mL) and DMF (cat.) (0.1 mL) at 0 ˚ C. The reaction mixture was heated to 70 ˚ C and stirred for 16 h. The resulting mixture was concentrated under reduced pressure and diluted with cold water (1 L). The precipitated solid was collected by filtration, washed water (3 x 150 mL) and triturated with diethyl ether (3 x 100 mL) to afford the title compound (7 g, Yield: 66.0 %). [001480]
1H NMR (DMSO-d6, 400 MHz, D
2O exchange): δ 3.68 (s, 3H), 3.72 (s, 3H), 6.13 (bs, 2H), 6.46 (s, 1H), 7.74 (s, 1H). [001481] LCMS (Method A): 1.496 min, MS: ES+ 259.7 & 261.7 (M+1 & M+3). Step 4: Methyl 5-bromo-2-methoxy-4-((2-methylallyl)amino)benzoate
[001482] To a stirred solution of methyl 4-amino-5-bromo-2-methoxybenzoate (Step 3) (7 g, 26.9 mmol, 1.0 eq.) in DMF (70 mL) was added CS2CO3 (43.7 g, 134.6 mmol, 5.0 eq.) followed by dropwise addition of 3-bromo-2-methylprop-1-ene (CAS: 1458-98-6) (10.90 g, 80.7 mmol, 3.0 eq.) at room temperature. The reaction mixture was heated to 80 ˚C and stirred for 16 h. The resulting mixture was diluted with water (150 mL) causing a solid to precipitate. The solid was collected by filtration, washed with water (3 x 150 mL) and purified by chromatography on silica (product eluted in 10% ethyl acetate in hexane) to afford the title compound as an off-white solid (3.1 g, Yield: 36.0%).
[001483]
1H NMR (DMSO-d6, 400 MHz): δ 1.70 (s, 3H), 3.71 (s, 3H), 3.75 (s, 3H), 3.85 (d, J= 6.0 Hz, 2H), 4.88 (bs, 2H), 6.16 (s, 1H), 6.34 (t, J= 6.0 Hz, 1H, D
2O exchangeable), 7.78 (s, 1H). [001484] LCMS (Method A): 2.086 min, MS: ES+ 313.8 & 315.7 (M+1 & M+3). Step 5: Methyl 6-methoxy-3,3-dimethylindoline-5-carboxylate
[001485] To a stirred solution of methyl 5-bromo-2-methoxy-4-((2- methylallyl)amino)benzoate (Step 4) (3.1 g, 9.87 mmol, 1.0 eq.) in DMF (30 mL) were added sodium formate (0.67 g, 9.87 mmol, 1.0 eq.), TEA (2.49 g, 24.68 mmol, 2.5 eq.), Pd(OAc)2 (0.44 g, 1.97 mmol, 0.2 eq.) and tetrabutylammonium chloride (CAS: 1112-67-0) (2.74 g, 9.87 mmol, 1.0 eq.) at room temperature. The reaction mixture was heated to 80
˚C and stirred for 20 h. Additional tetrabutylammonium chloride (1.0 eq.), sodium formate (1.0 eq.) and TEA (2.5 eq.) were added and stirring continued at 80 ˚C for a further 6 h. The resulting mixture was diluted with water (50 mL) and extracted with ethyl acetate (5 x 50 mL). The combined organic extracts were washed with brine solution (3 x 100 mL), dried over Na2SO4 and concentrated under reduced pressure. The crude material was purified by chromatography on silica (product eluted in 17% ethyl acetate in hexane) to afford the title compound (0.5 g, Yield: 21.0%). [001486]
1H NMR (DMSO-d6, 400 MHz): δ 1.20 (s, 6H), 3.27 (d, J= 1.2 Hz, 2H), 3.66 (s, 3H), 3.71 (s, 3H), 6.13 (s, 1H), 6.37 (bs, 1H, D2O exchangeable), 7.36 (s, 1H). [001487] LCMS (Method A): 1.586 min, MS: ES+ 236 (M+1). Step 6: Methyl 6-methoxy-3,3-dimethylindoline-5-carboxylate
[001488] To a cooled (0 ˚C) solution of methyl 6-methoxy-3,3-dimethylindoline-5- carboxylate (Step 5) (0.5 g, 2.10 mmol, 1.0 eq.) in DCM (5 mL) was added dropwise 1M BBr
3 in DCM (2.1 mL, 2.10 mmol, 1.0 eq.) and the reaction mixture was stirred at 0 ˚C for 1 h. The
resulting mixture was diluted with ice cold water (10 mL) basified with saturated NaHCO
3 solution (pH~ 7-8). The mixture was extracted with DCM (3 x 10 mL) and the combined organic extracts were dried over Na
2SO
4 and concentrated under reduced pressure. The crude material was purified by chromatography on silica (product eluted in 5% ethyl acetate in hexane) to afford the title compound (0.140 g, Yield: 29.0%). [001489]
1H NMR (DMSO-d6, 400 MHz): δ 1.20 (s, 6H), 3.28 (s, 2H), 3.80 (s, 3H), 5.89 (s, 1H), 6.70 (bs, 1H, D
2O exchangeable), 7.29 (s, 1H), 11.08 (s, 1H, D
2O exchangeable). [001490] LCMS (Method A): 1.955 min, MS: ES+ 221.9 (M+1). Step 7: 6-Hydroxy-3,3-dimethylindoline-5-carboxylic acid
[001491] The title compound was prepared from methyl 6-methoxy-3,3-dimethylindoline-5- carboxylate (Step 6) and aq. NaOH (0.76 g, 19.09 mmol, 3.0 eq.) analogously to Intermediate R Step 2. [001492]
1H NMR (DMSO-d6, 400 MHz): δ 1.20 (s, 6H), 3.27 (s, 2H), 5.86 (s, 1H), 6.57 (bs, 1H, D
2O exchangeable), 7.27 (s, 1H), 11.75 (bs, 1H, D
2O exchangeable), 12.78 (bs, 1H, D
2O exchangeable ). [001493] LCMS (Method A): 1.436 min, MS: ES+ 208 (M+1). Intermediate S (E)-4-Dimethylamino)-N-(isoindolin-4-yl)-N-methylbut-2-enamide carboxylate. HCl Step 1: tert-Butyl (E)-4-(4-(dimethylamino)-N-methylbut-2-enamido)isoindoline-2-carboxylate
[001494] The title compound was prepared from (E)-4-(dimethylamino)but-2-enoic acid hydrochloride (CAS: 848133-35-7) and tert-butyl 4-(methylamino)isoindoline-2-carboxylate (Intermediate A Step 1) analogously to Example 1 Step 2. This material was used directly in the next step [001495] LCMS (Method A): 1.571 min, MS ES+: 360.27 (M+1). Step 2: (E)-4-(Dimethylamino)-N-(isoindolin-4-yl)-N-methylbut-2-enamide carboxylate. HCl
[001496] The title compound was prepared from tert-butyl (E)-4-(4-(dimethylamino)-N- methylbut-2-enamido)isoindoline-2-carboxylate (Step 1) and 4M HCl in dioxane analogously to Example 4 Step 2. [001497]
1H NMR (DMSO-d6, 400 MHz): 2.66 (s, br, 6H), 3.21 (s, 3H), 3.68 - 3.80 (m, 2H), 4.30 - 4.37 (m, 1H), 4.50 - 4.58 (m, 3H), 6.07 (d, J= 14.8 Hz, 1H), 6.69 - 6.77 (m, 1H), 7.33 (d, J= 7.2 Hz, 1H), 7.47 - 7.52 (m, 2H), 9.87 (bs, 1H, D2O exchangeable), 10.34 (bs, 1H, D2O exchangeable), 10.55 (bs, 1H, D2O exchangeable ). [001498] LCMS (Method D): 1.58 min, MS: ES+ 260.4 (M+1). Intermediate T 2,4-Dihydroxy-5-(trifluoromethyl)benzoic acid Step 1: Benzyl 2,4-bis(benzyloxy)benzoate

[001499] To a stirred solution of 2,4-dihydroxybenzoic acid (5.0 g, 32 mmol, 1.0 eq) in acetonitrile (45 mL) was added potassium carbonate (21 g, 0.16 mol, 4.9 eq) followed by dropwise addition of benzyl bromide (12 mL, 0.10 mol, 3.1 eq) at room temperature. The reaction mixture was heated to 85 ˚C for 16 hours under an atmosphere of nitrogen and then allowed to
cool to room temperature. The resulting mixture was poured into water (300 mL) causing a solid to precipitate. The solids were collected by filtration, washed with water (2 x 150 mL) and dried under high vacuum overnight to afford the title compound (13 g, 94%). [001500] ¹H NMR (400 MHz, DMSO) δ 5.19 (s, 2H), 5.20 (s, 2H), 5.27 (s, 2H), 6.72 (dd, J=2.3, 8.7 Hz, 1H), 6.88 (d, J=2.4 Hz, 1H), 7.29 - 7.49 (m, 15H), 7.78 (d, J=9.2 Hz, 1H). [001501] MS: ES+ 425 (M+1) Step 2: Benzyl 4-(benzyloxy)-2-hydroxy-5-iodobenzoate

[001502] Iodine monochloride (1M in DCM, 34 mL, 34 mmol, 1.1 eq) was added dropwise to a stirred solution of benzyl 2,4-dibenzyloxybenzoate (Step 1) (13 g, 31 mmol, 1.0 eq) in DCM (250 mL), under a nitrogen atmosphere and stirred at room temperature for 2 h. The resulting solution was washed with saturated aqueous sodium hydrogen carbonate solution (3 x 150 mL), aqueous sodium thiosulphate (150 mL), water (3 x 150 mL) and saturated aqueous sodium chloride solution (150 mL). The organics were dried over anhydrous magnesium sulphate and concentrated under reduced pressure to obtain crude product which was triturated with DCM/methanol and collected by filtration to afford the title compound (9.6 g, 68%). [001503] ¹H NMR (400 MHz, DMSO) δ 5.27 (s, 2H), 5.36 - 5.38 (m, 2H), 6.73 (s, 1H), 7.35 - 7.51 (m, 10H), 8.11 (s, 1H), 10.73 - 10.75 (m, 1H). Step 3: Benzyl 2,4-bis(benzyloxy)-5-iodobenzoate

[001504] To a stirred suspension of benzyl 4-benzyloxy-2-hydroxy-5-iodo-benzoate (Step 2) (9.6 g, 21 mmol, 1.0 eq) and potassium carbonate (12 g, 84 mmol, 4.0 eq) in acetone (50 mL) at room temperature under nitrogen was added dropwise benzyl bromide (5.0 mL, 42 mmol, 2.0 eq). The reaction mixture was heated to 75 ˚C for 5 hours, then cooled to room temperature and poured into water (250 mL). The resulting mixture was extracted with DCM (250 mL). The
organic phase was washed with water (2 × 150 mL), brine (150 mL), dried over magnesium sulfate and concentrated under reduced pressure. The crude material was azeotroped with toluene (3 × 50 mL) and ether (3 × 50 mL) to afford the title compound (10 g, 89%). [001505] ¹H NMR (400 MHz, DMSO) δ 5.25 - 5.27 (m, 4H), 5.32 - 5.33 (m, 2H), 7.02 (s, 1H), 7.31 - 7.53 (m, 15H), 8.12 (s, 1H). Step 4: Benzyl 2,4-bis(benzyloxy)-5-(trifluoromethyl)benzoate
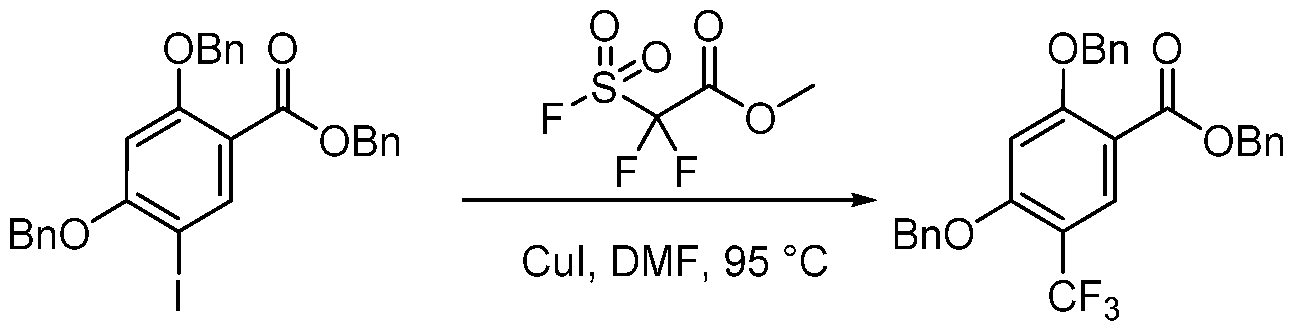
[001506] To a degassed suspension of benzyl 2,4-dibenzyloxy-5-iodo-benzoate (Step 3) (2.0 g, 3.6 mmol, 1.0 eq) and copper(I) iodide (3.5 mg, 18 mmol, 5.0 eq) in DMF (8.4 mL) was added dropwise methyl-2,2-difluoro-2-(fluorosulfonyl)acetate (2.3 mL, 18 mmol, 5.0 eq) under an atmosphere of nitrogen. The reaction mixture was placed in a preheated block at 95 ˚C, stirred for 16 hours then cooled to room temperature. The reaction mixture was diluted with EtOAc (80 mL), filtered through a pad of Celite® and the filter cake was washed with EtOAc (80 mL). The filtrate was washed with saturated aqueous ammonium chloride (80 mL), saturated aqueous sodium hydrogen carbonate solution (2 × 80 mL), water (3 × 80 mL) and brine (80 mL). The organic phase was dried over magnesium sulfate and concentrated under reduced pressure . The residue was purified by chromatography on silica, eluting with 0-15% EtOAc in cyclohexane to afford the title compound (1.0 g, 57%). [001507] ¹H NMR (400 MHz, DMSO) δ 5.29 (s, 2H), 5.34 (s, 2H), 5.39 - 5.42 (m, 2H), 7.21 (s, 1H), 7.32 - 7.49 (m, 15H), 8.01 - 8.02 (m, 1H). Step 5: 2,4-Dihydroxy-5-(trifluoromethyl)benzoic acid

[001508] To a degassed solution of benzyl 2,4-dibenzyloxy-5-(trifluoromethyl)benzoate (step 4) (1.0 g, 2.0 mmol, 1.0 eq) in ethanol (21 mL) and methanol (7.0 mL) was added palladium on carbon (5.0%, 860 mg, 0.41 mmol, 0.20 eq). The reaction mixture was stirred under an
atmosphere of hydrogen at room temperature for 16 h. The palladium was removed by filtration using a Whatmann funnel and the filter cake was washed with DCM/ethanol (1:1). The filtrate was concentrated under reduced pressure to afford the title compound (480 mg, 90%). [001509] ¹H NMR (400 MHz, DMSO) δ 6.49 (s, 1H), 7.90 (s, 1H), 11.48 (s, 1H). MS: ES- 221 (M-1) Intermediate U 2,4-Dihydroxy-6-(trifluoromethyl)benzoic acid Step 1: 2-Iodo-4,6-dimethoxybenzaldehyde
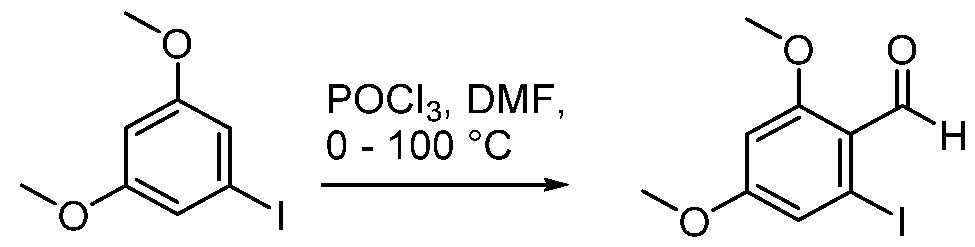
[001510] To a cooled (0 ˚C) solution of 1-iodo-3,5-dimethoxy-benzene (2.5 g, 9.5 mmol, 1.0 eq) in DMF (16 mL) was added dropwise phosphorus(V) oxychloride (2.6 mL, 28 mmol, 3.0 eq) and the mixture was stirred for 15 mins. The mixture was warmed to room temperature over 15 mins then heated to 100 ˚ C for 2.5 hours. After cooling to room temperature, the reaction mixture was poured onto ice (400 mL), stirred for 1.5 h and filtered. The residue was purified by chromatography on silica eluting with 0-12% methanol in DCM to afford the title compound (1.2 g, 43%). [001511] ¹H NMR (400 MHz, DMSO) δ 3.87 (s, 3H), 3.89 (s, 3H), 6.76 (d, J=2.3 Hz, 1H), 7.18 (d, J=2.1 Hz, 1H), 10.04 (s, 1H). MS: ES+ 293 (M+1) Step 2: 2-Iodo-4,6-dimethoxybenzoic acid
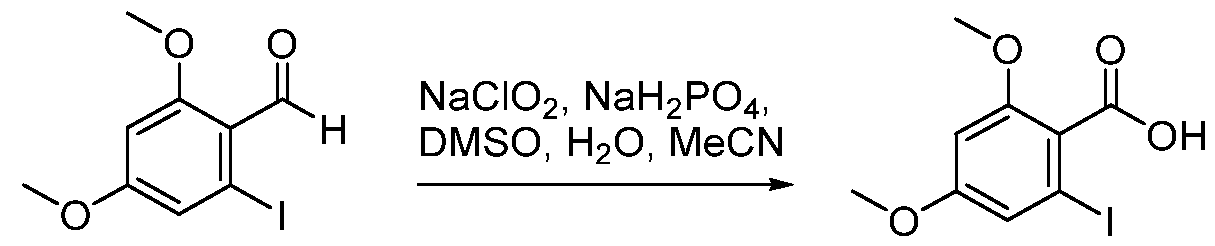
[001512] To a cooled (0 ˚C) solution of 2-iodo-4,6-dimethoxy-benzaldehyde (Step 1) (1.2 g, 4.1 mmol, 1.00 eq) in DMSO (23 mL) and acetonitrile (23 mL) was added a solution of sodium phosphate monobasic (1.2 g, 10 mmol, 2.5 eq) and sodium chlorite (80%, 1.2 g, 10 mmol, 2.5 eq) in water (23 mL). The reaction mixture was stirred at 0 ˚C for 30 minutes then at room temperature for 1 hour. The resulting mixture was diluted with water (200 mL) and acidified to pH 1 with 2N HCl solution. The mixture was extracted with cyclohexane/EtOAc (1:1) (3 × 200 mL) and the combined organic extracts were washed with brine (200 mL), dried over magnesium
sulfate and concentrated under reduced pressure to afford the title compound (1.2 g, 98%) as an off-white solid. [001513] ¹H NMR (400 MHz, DMSO) δ 3.76 (s, 3H), 3.78 (s, 3H), 6.64 (d, J=2.2 Hz, 1H), 6.95 (d, J=2.1 Hz, 1H), 13.01 (s, 1H). MS: ES+ 309 (M+1) Step 3: 2,4-Dihydroxy-6-iodobenzoic acid

[001514] To a cooled (0 ˚C) solution of 2-iodo-4,6-dimethoxy-benzoic acid (step 2) (1.0 g, 3.3 mmol, 1.0 eq) in DCM (23 mL) under and an atmosphere of nitrogen was added boron tribromide (1M in DCM, 16 mL, 16 mmol, 5.0 eq) portion wise over 5 mins. The reaction mixture was stirred at 0 ˚C for 1.5 hours then at room temperature for 16 h. The reaction was quenched with saturated aqueous sodium hydrogen carbonate solution (50 mL) and acidified with 2M aqueous HCl solution. The resulting mixture was extracted with EtOAc (3 × 50 mL) and the combined organic extracts were washed with brine (50 mL), dried over magnesium sulfate and concentrated under reduced pressure. The residue was purified by chromatography on silica eluting with a gradient of 0-40% EtOAc and cyclohexane to afford the title compound (450 mg, 50%). [001515] ¹H NMR (400 MHz, DMSO) δ 6.30 (d, J=2.1 Hz, 1H), 6.77 (d, J=2.1 Hz, 1H), 9.93 (s, 1H). MS: ES- 279 (M-1) Step 4: Benzyl 2,4-bis(benzyloxy)-6-iodobenzoate

[001516] Benzyl bromide (0.59 mL, 5.0 mmol, 3.1 eq) was added dropwise at room temperature to a stirred suspension of 4-dihydroxy-6-iodo-benzoic acid (Step 3) (450 mg, 1.6 mmol, 1.0 eq) and potassium carbonate (1.1 g, 7.8 mmol, 4.9 eq) in acetonitrile (2.5 mL) and the mixture was heated to 85 ˚C for 16 h under an atmosphere of nitrogen. The resulting mixture was cooled to room temperature and poured into water (300 mL). The solution was extracted with EtOAc (3 x 100 mL) and the combined organic extracts were washed with brine (100 mL), dried over magnesium sulfate and concentrated under reduced pressure. The crude material
was purified by chromatography on silica, eluting with 0-20% EtOAc in cyclohexane to afford the title compound as a colourless oil. (580 mg, 65%) [001517] ¹H NMR (400 MHz, DMSO) δ 5.15 (d, J=5.3 Hz, 4H), 5.28 (s, 2H), 6.89 (d, J=2.1 Hz, 1H), 7.11 (d, J=2.1 Hz, 1H), 7.31 - 7.44 (m, 15H). MS: ES+ 551 (M+1) Step 5: Benzyl 4-(benzyloxy)-2-hydroxy-6-(trifluoromethyl)benzoate

[001518] To a degassed suspension of benzyl 2,4-dibenzyloxy-6-iodo-benzoate (Step 4) (580 mg, 1.0 mmol, 1.0 eq) and copper(I) iodide (1.0 g, 5.2 mmol, 5.0 eq) in DMF (2.5 mL) was added dropwise methyl 2,2-difluoro-2-(fluorosulfonyl)acetate (0.67 mL, 5.2 mmol, 5.0 eq) under an atmosphere of nitrogenand the mixture was heated to 95 ˚C and for 16 h. The resulting mixture was cooled to room temperature then diluted with EtOAc (80 mL) and filtered through a pad of Celite®. The filter cake was further washed with EtOAc (80 mL) and the combined filtrate was washed with saturated aqueous ammonium chloride (80 mL), saturated aqueous sodium hydrogen carbonate solution (2 x 80 mL), water (3 x 80 mL) and brine (80 mL). The organic phase was dried over magnesium sulfate and concentrated under reduced pressure. The residue was purified by chromatography on silica eluting with a gradient of 0-25% EtOAc in cyclohexane to afford the title compound (340 mg, 81%). [001519] ¹H NMR (400 MHz, DMSO) δ 5.25 (d, J=14.6 Hz, 4H), 6.98 (d, J=2.2 Hz, 1H), 7.19 (d, J=2.1 Hz, 1H), 7.31 - 7.48 (m, 10H). MS: ES- 402 (M-1) Step 6: 2,4-Dihydroxy-6-(trifluoromethyl)benzoic acid

[001520] To solution of benzyl 4-benzyloxy-2-hydroxy-6-(trifluoromethyl)benzoate (Step 5) (340 mg, 0.85 mmol, 1.0 eq) in ethanol (6.0 mL) under nitrogen was added EtOAc (6.0 mL) and methanol (2.0 mL) followed by palladium on carbon (10%, 180 mg, 0.17 mmol, 0.20 eq). The reaction mixture was evacuated and placed under an atmosphere of hydrogen stirring at room temperature for 16 h. The palladium was removed by filtration and the filtrate concentrated under reduced pressure. The residue was re-dissolved in EtOAc (6.0 mL) and methanol (2.0 mL),
degassed with nitrogen and treated with Palladium on carbon (10%, 180 mg, 0.17 mmol, 0.20 eq). The reaction mixture was again evacuated and placed under an atmosphere of hydrogen and stirred at room temperature for a further 16 h. The palladium was removed by filtration and the filtrate concentrated under reduced pressure to yield the title compound (145 mg, 78%). [001521] ¹H NMR (400 MHz, DMSO) δ 6.53 (d, J=1.9 Hz, 1H), 6.57 (d, J=1.9 Hz, 1H), 10.24 (s, 1H), 10.41 - 10.66 (m, 1H), 12.71 - 13.47 (m, 1H). MS: ES- 221 (M-1) Intermediate V1-1 (E)-4-(Dimethylamino)-N-(isoindolin-4-yl)-N-methylbut-2-enamide dihydrochloride
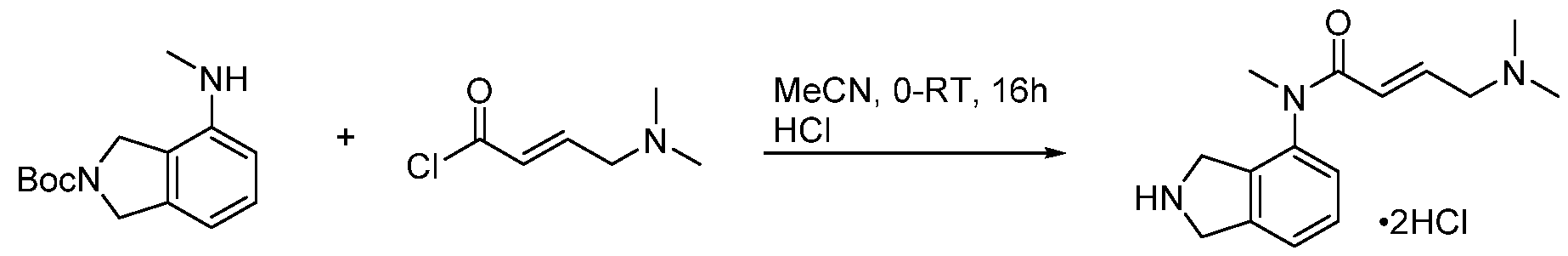
[001522] A solution of tert-butyl 4-(methylamino)isoindoline-2-carboxylate (Intermediate A Step 1) (1.2 g, 4.7 mmol, 1.0 eq) in acetonitrile (40 mL) was added dropwise to a cooled (0 ˚C) solution of (E)-4-(dimethylamino)but-2-enoyl chloride (860 mg, 4.7 mmol, 1.0 eq) in acetonitrile (10 mL). The mixture was allowed to warm gradually to room temperature stirring for 16 h. The resulting mixture was concentrated under reduced pressure and the residue suspended in 4M HCl in dioxane (40 mL). After stirring for 3 h, the mixture was concentrated under reduced pressure. The residue was triturated with IPA, isolated by filtration, washed with further IPA and dried under high vacuum. The mother liquors from the trituration were concentrated under reduced pressure, dissolved in IPA (10 mL) then added dropwise to Et
2O (100 mL) with rapid stirring. The precipitated solid was isolated by filtration, dried under high vacuum and combined with the previous batch to afford the title compound which was used without further purification (1.3 g, 87%). [001523] MS: ES+ 260.2 (M+1) [001524] The following intermediates were prepared analogously to Intermediate V1-1 by replacing tert-butyl 4-(methylamino)isoindoline-2-carboxylate (Intermediate A Step 1) with the indicated intermediate reagent. Int. Intermediate reagent Product Name and Structure Data V1-2 tert-Butyl 4- MS: ES+ (ethylamino)isoindoline-2- 274 carboxylate (Intermediate (M+1) Y
4-9)
E)-4-(Dimethylamino)-N-ethyl-N- (isoindolin-4-yl)but-2-enamide dihydrochloride V1-3 tert-Butyl 4-(2- MS: ES+ hydroxyethylamino)isoindoli 290 ne-2-carboxylate (M+1) (Intermediate Y4-10) (E)-4-(Dimethylamino)-N-(2- hydroxyethyl)-N-(isoindolin-4-yl)but- 2-enamide dihydrochloride V1-4 tert-Butyl 4-(4- MS: ES+ pyridylmethylamino)isoindoli 337 ne-2-carboxylate (M+1) (Intermediate Y4-1) (E)-4-(Dimethylamino)-N-(isoindolin- 4-yl)-N-(pyridin-4-ylmethyl)but-2- enamide trihydrochloride V1-5 tert-Butyl 4-[2-(4- MS: ES+ pyridyl)ethylamino]isoindolin 351 e-2-carboxylate (M+1) (Intermediate Y4-2) (E)-4-(Dimethylamino)-N-(isoindolin- 4-yl)-N-(2-(pyridin-4-yl)ethyl)but-2- enamide trifluoroacetate V1-6 tert-Butyl 4-((2-(pyridin-2- MS: ES+ yl)ethyl)amino)isoindoline-2- 351 carboxylate (Intermediate (M+1) Y
4-11) (E)-4-(Dimethylamino)-N-(isoindolin- 4-yl)-N-(2-(pyridin-2-yl)ethyl)but-2- enamide trifluoroacetate V1-7 tert-Butyl 4-((2-(pyridin-3- MS: ES+ yl)ethyl)amino)isoindoline-2- 351 carboxylate (M+1) (Intermediate Y4-3)
(E)-4-(Dimethylamino)-N-(isoindolin- 4-yl)-N-(2-(pyridin-3-yl)ethyl)but-2- enamide V1-8 tert-Butyl 4-((2-(tetrahydro- MS: ES+ 2H-pyran-4- 358 yl)ethyl)amino)isoindoline-2- (M+1) carboxylate (Intermediate Y4-4) (E)-4-(Dimethylamino)-N-(isoindolin- 4-yl)-N-(2-(tetrahydro-2H-pyran-4- yl)ethyl)but-2-enamide dihydrochloride V1-9 tert-Butyl 4-((2- MS: ES+ methoxyethyl)amino)isoindo 304 line-2-carboxylate (M+1) (Intermediate Y4-6) (E)-4-(Dimethylamino)-N-(isoindolin- 4-yl)-N-(2-methoxyethyl)but-2- enamide V1-10 tert-Butyl 4- MS: ES+ aminoisoindoline-2- 246 carboxylate (CAS 871013- (M+1) 9
8-8) (E)-4-(Dimethylamino)-N-(isoindolin- 4-yl)but-2-enamide dihydrochloride Intermediate W2 N-(Isoindolin-4-yl)-N-methylacrylamide Step 1: tert-Butyl 4-(N-methylacrylamido)isoindoline-2-carboxylate
[001525] To a cooled (0 ˚C) solution of tert-butyl 4-(methylamino)isoindoline-2-carboxylate (Intermediate A Step 1) (0.25 g, 1.0 mmol, 1.0 eq) in acetonitrile (11 mL) was added dropwise acryloyl chloride (0.082 mL, 1.0 mmol, 1.0 eq) and the reaction mixture was stirred under an inert atmosphere at 0 ˚ C for 1 h. The reaction was quenched with saturated aqueous sodium hydrogen carbonate (20 mL) and the mixture was extracted with EtOAc (3 x 20 mL). The combined organic
extracts were washed with water (20 mL), brine (20 mL), dried over MgSO
4 and concentrated under reduced pressure to afford the title compound (0.29 g, 95%). [001526] ¹H NMR (400 MHz, DMSO) δ 1.46 (d, J=3.3 Hz, 9H), 3.18 (s, 3H), 4.35 (d, J=14.1 Hz, 1H), 4.50 - 4.70 (m, 3H), 5.57 (d, J=10.4 Hz, 1H), 5.95 (dd, J=10.6, 16.6 Hz, 1H), 6.19 (dd, J=2.1, 16.6 Hz, 1H), 7.21 (d, J=6.7 Hz, 1H), 7.36 - 7.48 (m, 2H). MS: ES+ 247.1 (M-tBu) Step 2: N-(Isoindolin-4-yl)-N-methylacrylamide
[001527] To a solution of tert-butyl 4-(N-methylacrylamido)isoindoline-2-carboxylate (Step 1) (0.29 g, 0.95 mmol, 1.0 eq) in DCM (1.0 mL) was added TFA (0.73 mL, 9.5 mmol, 10.0 eq) and the reaction mixture was stirred under an inert atmosphere at room temperature for 2.5 h. The resulting mixture was concentrated under reduced pressure and the residue was loaded onto an Isolute® SCX-2 cartridge. The column was washed with DCM/MeOH (1:1) and the product was eluted with 7N NH
3 in MeOH/DCM (1:1). The methanolic ammonia product fractions were concentrated under reduced pressure to afford the title compound (0.12 g, 61%). MS: ES+ 203.1 (M+1). [001528] The following tabulated Intermediates were prepared analogously to Intermediate W2 from the indicated intermediate reagents. Int. Name and Structure Data W2-1 MS: ES+ 294 (M+1) N-(Isoindolin-4-yl)-N-(2-(pyridin-4-yl)ethyl)acrylamide bis(2,2,2-trifluoroacetate) Prepared from tert-butyl 4-((2-(pyridin-4- yl)ethyl)amino)isoindoline-2-carboxylate (Intermediate Y4-2) (Step 1) followed by TFA deprotection (Step 2)
W2-2 MS: ES+ 300 (M+1) N-(Isoindolin-4-yl)-N-(2-(1-methylpyrrolidin-3- yl)ethyl)acrylamide bis(2,2,2-trifluoroacetate) Prepared from tert-butyl 4-((2-(1-methylpyrrolidin-3- yl)ethyl)amino)isoindoline-2-carboxylate (Intermediate Y4-5)(Step 1) followed by TFA deprotection (Step 2) W2-4 ¹H NMR (400 MHz, DMSO) δ 4.43 - 4.56 (m, 4H), 5.81 (dd, J=1.9, 10.2 Hz, 1H), N-(Isoindolin-4-yl)acrylamide hydrochloride. 6.29 (dd, J=1.9, 17.0 Prepared using tert-butyl 4-aminoisoindoline-2- Hz, 1H), 6.49 - 6.58 carboxylate (CAS 871013-98-8) and HCl. (m, 1H), 7.22 (d, J=7.4 Hz, 1H), 7.37 (dd, J=7.7, 7.7 Hz, 1H), 7.50 (d, J=8.0 Hz, 1H), 9.66 - 9.66 (m, 2H), 10.17 (s, 1H). MS: ES+ 189.1 (M+1) Intermediate X3-2 (2,4-Dihydroxy-5-methylphenyl)(4-((2-morpholinoethyl)amino)isoindolin-2-yl)methanone Step 1: N-(2-Morpholinoethyl)isoindolin-4-amine

[001529] To a stirred solution of tert-butyl 4-((2-morpholinoethyl)amino)isoindoline-2- carboxylate (Intermediate Y4-8) (470 mg, 1.4 mmol, 1.0 eq) in DCM (5.0 mL) was added TFA (2.9 mL, 37 mmol, 28 eq) and the reaction mixture was stirred at room temperature for 1.5 h. The
resulting mixture was concentrated under reduced pressure and the residue was loaded onto an Isolute® SCX-2 cartridge. The column was washed with DCM/MeOH (1:1) and the product eluted with 7N NH
3 in MeOH/ DCM (1:1). The methanolic ammonia product fractions were concentrated under reduced pressure to afford the title compound which was used without further purification (350 mg, quantitative yield). [001530] ¹H NMR (400 MHz, CDCl¬3) δ 2.05 - 2.08 (m, 4H), 2.47 (m, 4H), 2.66 (m, 2H), 3.21 (m, 2H), 3.71 (m, 4H), 4.09 (s, 1H), 4.14 (m, 2H), 4.27 (m, 2H), 6.48 (d, J=7.9 Hz, 1H), 6.66 (d, J=7.4 Hz, 1H), 7.14 (m, 1H). MS: ES+ 248 (M+1) Step 2: (2,4-Dihydroxy-5-methylphenyl)(4-((2-morpholinoethyl)amino)isoindolin-2-yl)methanone

[001531] To a stirred solution of 2,4-dihydroxy-5-methyl-benzoic acid (20 mg, 0.12 mmol, 1.0 eq) in DCM (2.0 mL) was added EDC (57 mg, 0.30 mmol, 2.5 eq) and the mixture was stirred at room temperature for 1.5 h. N-(2-Morpholinoethyl)isoindolin-4-amine (Step 1) (29 mg, 0.12 mmol, 1.0 eq) and DIPEA (0.12 mL, 0.69 mmol, 5.8 eq) were added and stirring continued at room temperature for 2 h. The resulting mixture was concentrated under reduced pressure and purification of the crude residue by chromatography on silica eluting with a gradient of 0-100% EtOAc in cyclohexane followed by 0-25% EtOH in EtOAc afforded the title compound (32 mg, 19%). MS: ES+ 398 (M+1). [001532] The following tabulated Intermediates were prepared analogously to Intermediate X3-2 Step 1 and Step 2 using the indicated intermediate reagents. Int. Name and Structure Data X3-1 MS: ES+ 401 (M+1). (4-((2,3-Dihydro-1H-inden-2-yl)amino)isoindolin-2- yl)(2,4-dihydroxy-5-methylphenyl)methanone
Prepared using tert-butyl 4-(((2,3-dihydro-1H-inden-2- yl)methyl)amino)isoindoline-2-carboxylate (Intermediate Y4-7) and 2,4-dihydroxy-5- methylbenzoic acid (Intermediate F) X3-3 ¹H NMR (400 MHz, CDCl
3) δ 1.21-1.33 (m, 3H), 2.07-2.36 (m, 12H), 3.13 - 3.46 (m, 3H), 4.16 4-(4-(Ethylamino)isoindoline-2-carbonyl)-5,6-dimethyl- - 5.03 (m, 4H), 6.48 - 1,3-phenylene diacetate 6.71 (m, 2H), 6.85 - 6.89 Prepared using tert-butyl 4-(ethylamino)isoindoline-2- (m, 1H), 7.14 - 7.25 (m, carboxylate (Intermediate Y4-9) and 4,6-diacetoxy-2,3- 1H). dimethylbenzoic acid (CAS: 1081769-29-0) MS: ES+ 411.1 (M+1). X3-4 MS: ES+ 441 (M+1). 4-(4-((2-Methoxyethyl)amino)isoindoline-2-carbonyl)- 5,6-dimethyl-1,3-phenylene diacetate Prepared using tert-butyl 4-((2- methoxyethyl)amino)isoindoline-2-carboxylate (Intermediate Y4-6) and 4,6-diacetoxy-2,3- dimethylbenzoic acid (CAS: 1081769-29-0) Intermediate Y4-1 tert-Butyl 4-((pyridin-4-ylmethyl)amino)isoindoline-2-carboxylate
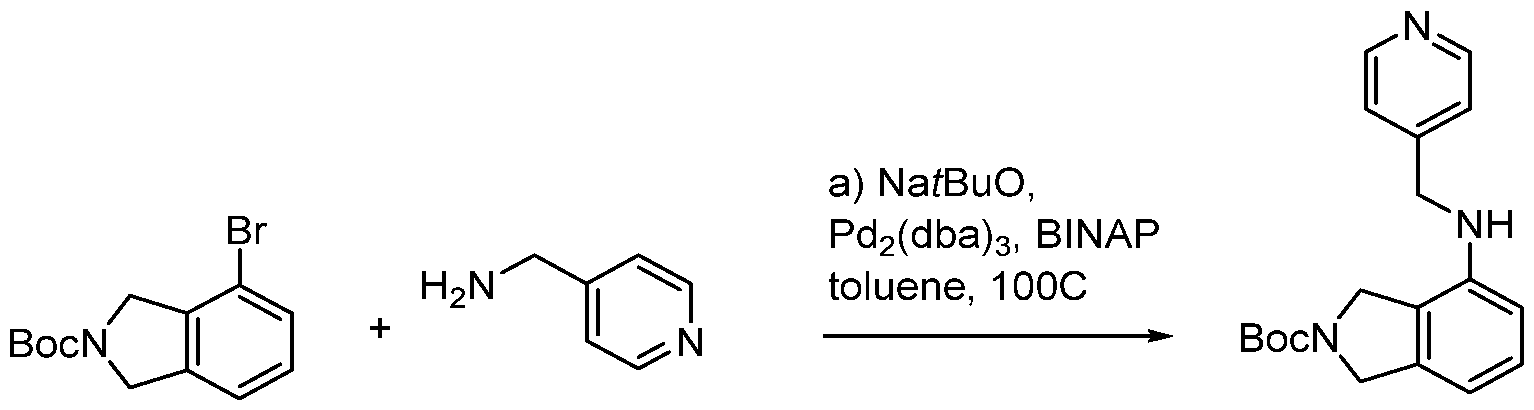
[001533] To a sealed reaction tube under nitrogen charged with tert-butyl 4- bromoisoindoline-2-carboxylate (350 mg, 1.2 mmol, 1.0 eq), 4-(aminomethyl)pyridine (0.13 mL, 1.3 mmol, 1.1 eq), sodium tert-butoxide (160 mg, 1.6 mmol, 1.4 eq), 2,2'-bis(diphenylphosphino)- 1,1'-binaphthalene (220 mg, 0.35 mmol, 0.3 eq) and tris(dibenzylideneacetone)dipalladium(0)
(110 mg, 0.12 mmol, 0.1 eq) was added dry degassed toluene (7.0 mL). The mixture was heated to 100 ˚C and stirred under nitrogen for 16 h. The resulting mixture was cooled and diluted with EtOAc (50 ml) then washed with water (50 ml). The organic phase was dried over MgSO
4 and concentrated under reduced pressure. Purification by chromatography on silica eluting with 0- 100% EtOAc in cyclohexane afforded the title compound (190 mg, 49%) as a yellow solid. [001534] ¹H NMR (400 MHz, DMSO) δ 1.46 - 1.49 (m, 9H), 4.35 - 4.38 (m, 2H), 4.47 - 4.55 (m, 4H), 6.14 - 6.23 (m, 2H), 6.51 (t, J=6.8 Hz, 1H), 6.96 (t, J=7.7 Hz, 1H), 7.34 (d, J=4.5 Hz, 2H), 8.47 (d, J=6.0 Hz, 2H). MS: ES+ 326 (M+1) [001535] The following tabulated Intermediates were prepared analogously to tert-butyl 4- ((pyridin-4-ylmethyl)amino)isoindoline-2-carboxylate (Intermediate Y4-1) from tert-butyl 4- bromoisoindoline-2-carboxylate and the appropriate commercially available amines. Int. Name and Structure Data Y4-2 ¹H NMR (400 MHz, DMSO) δ 1.45 - 1.48 (m, 9H), 2.88 (t, J=7.2 Hz, 2H), 3.33 - 3.38 (m, 2H), 4.37 (m, 2H), 4.51 (m, 2H), 5.43 (t, J=5.3 Hz, 1H), 6.49 - 6.56 (m, 2H), 7.08 (t, J=7.7 Hz, 1H), 7.29 (d, J=5.8 Hz, 2H), 8.47 (d, J=5.1 Hz, 2H). tert-Butyl 4-((2-(pyridin-4- MS: ES+ 340 (M+1) yl)ethyl)amino)isoindoline-2- carboxylate Prepared using 2-(pyridin-4- yl)ethan-1-amine Y4-3 ¹H NMR (400 MHz, CDCl
3) δ 1.50 - 1.52 (m, 9H), 2.94 (q, J=7.0 Hz, 2H), 3.33 - 3.38 (m, 1H), 3.44 - 3.52 (m, 2H), 4.35 - 4.43 (m, 2H), 4.61 - 4.68 (m, 2H), 6.53 - 6.69 (m, 2H), 7.16 - 7.25 (m, 2H), 7.50 - 7.54 (m, 1H), 8.47 - 8.52 tert-Butyl 4-((2-(pyridin-3- (m, 2H). yl)ethyl)amino)isoindoline-2- MS: ES+ 340 (M+1) carboxylate Prepared using 2-(pyridin-3- yl)ethan-1-amine Y4-4 ¹H NMR (400 MHz, CDCl
3) δ 1.33 - 1.39 (m, 2H), 1.51 - 1.53 (m, 9H), 1.58 - 1.68 (m, 5H), 3.21 (t, J=5.8 Hz, 3H), 3.35 - 3.44 (m, 2H), 3.94 - 4.00 (m, 2H), 4.43 - 4.50 (m, 2H), 4.61 -
tert-Butyl 4-((2-(tetrahydro-2H- 4.68 (m, 2H), 6.50 (dd, J=3.6, 8.0 Hz, 1H), pyran-4- 6.62 (dd, J=7.3, 23.1 Hz, 1H), 7.14 - 7.20 (m, yl)ethyl)amino)isoindoline-2- 1H). MS: ES+ 347 (M+1) carboxylate Prepared using 2-(tetrahydro- 2H-pyran-4-yl)ethan-1-amine Y4-5 ¹H NMR (400 MHz, CDCl
3) δ 1.45 - 1.49 (m, 1H), 1.51 - 1.53 (m, 9H), 1.69 - 1.79 (m, 2H), 2.02 - 2.19 (m, 2H), 2.23 - 2.33 (m, 1H), 2.34 - 2.36 (m, 3H), 2.43 - 2.51 (m, 1H), 2.57 - 2.66 tert-Butyl 4-((2-(1- (m, 1H), 2.73 - 2.80 (m, 1H), 3.13 - 3.20 (m, methylpyrrolidin-3- 2H), 3.33 - 3.35 (m, 1H), 4.41 - 4.49 (m, 2H), yl)ethyl)amino)isoindoline-2- 4.61 - 4.68 (m, 2H), 6.48 - 6.52 (m, 1H), 6.56 - carboxylate 6.65 (m, 1H), 7.13 - 7.19 (m, 1H). Prepared using 2-(1- MS: ES+ 346 (M+1) methylpyrrolidin-3-yl)ethan-1- amine Y4-6 ¹H NMR (400 MHz, DMSO) δ , 1.45 - 1.48 (m, 9H), 3.21 - 3.27 (m, 2H), 3.28 (s, 3H), 3.46 - 3.51 (m, 2H), 4.38 - 4.41 (m, 2H), 4.48 - 4.53 tert-Butyl 4-((2- (m, 2H), 5.22 - 5.28 (m, 1H), 6.45 - 6.55 (m, methoxyethyl)amino)isoindoline- 2H), 7.04 - 7.10 (m, 1H). 2-carboxylate MS: ES+ 293 (M+1) Prepared using 2- Methoxyethylamine Y4-7 ¹H NMR (400 MHz, DMSO) δ 1.45 (s, 9H), 2.86 - 2.92 (m, 2H), 3.28 - 3.29 (m, 2H), 4.29 (dd, J=6.5, 13.2 Hz, 1H), 4.38 (dd, J=1.1, 6.1 Hz, 2H), 4.51 (d, J=11.8 Hz, 2H), 5.45 (dd, J=6.7, 13.7 Hz, 1H), 6.53 - 6.60 (m, 2H), 7.09 tert-Butyl 4-(((2,3-dihydro-1H- - 7.17 (m, 3H), 7.20 - 7.24 (m, 2H). inden-2- MS: ES+ 351 (M+1) yl)methyl)amino)isoindoline-2- carboxylate Prepared using (2,3-dihydro-1H- inden-2-yl)methanamine
Y4-8 ¹H NMR (400 MHz, DMSO) δ 1.52 (s, 9H), 2.62 - 2.69 (m, 2H), 3.17 - 3.23 (m, 2H), 3.49 (s, 4H), 3.68 - 3.74 (m, 4H), 4.06 - 4.16 (m, 1H), 4.45 - 4.48 (m, 2H), 4.62 - 4.69 (m, 2H), 6.47 - 6.53 (m, 1H), 6.58 - 6.68 (m, 1H), 7.14 - tert-Butyl 4-((2- 7.20 (m, 1H). MS: ES+ 348 (M+1) morpholinoethyl)amino)isoindoli ne-2-carboxylate Prepared using 2-morpholino- ethan-1-amine Y4-11 ¹H NMR (400 MHz, DMSO) δ 1.45 - 1.48 (m, 9H), 3.01 (t, J=7.4 Hz, 2H), 3.42 (t, J=7.3 Hz, 2H), 4.37 (m, 2H), 4.51 (m, , 2H), 5.44 (s, 1H), 6.49 - 6.56 (m, 2H), 7.09 (t, J=7.7 Hz, 1H), 7.20 - 7.26 (m, 1H), 7.29 - 7.33 (m, 1H), 7.69 - tert-Butyl 4-((2-(pyridin-2- 7.74 (m, 1H), 8.51 - 8.53 (m, 1H). yl)ethyl)amino)isoindoline-2- MS: ES+ 340 (M+1) carboxylate Prepared using 2-(pyridin-2- yl)ethan-1-amine Intermediate Y4-9 tert-Butyl 4-(ethylamino)isoindoline-2-carboxylate

[001536] To a stirred solution of tert-butyl 4-aminoisoindoline-2-carboxylate (600 mg, 2.6 mmol, 1.0 eq) and bromoethane (0.19 mL, 2.6 mmol, 1.0 eq) in acetonitrile (6.4 mL) was added DIPEA (0.45 mL, 2.6 mmol, 1.0 eq) and the reaction mixture was heated to 80 ˚C for 44 h. The resulting mixture was cooled to room temperature and diluted with EtOAc (50 mL). The mixture was washed with water (50 mL) dried over MgSO4 and concentrated under reduced pressure. Purification of the crude residue by chromatography on silica eluting with a gradient of 0-25% EtOAc in cyclohexane afforded the title compound as an off-white solid. (230 mg, 34%). [001537] ¹H NMR (400 MHz, CDCl3) δ 1.24 - 1.32 (m, 3H), 1.51 - 1.53 (m, 9H), 3.17 - 3.26 (m, 2H), 4.43 - 4.50 (m, 2H), 4.61 - 4.68 (m, 2H), 6.49 - 6.53 (m, 1H), 6.57 - 6.66 (m, 1H), 7.14 - 7.20 (m, 1H). MS: ES+ 263 (M+1)
Intermediate Y4-10 tert-Butyl 4-(2-hydroxyethylamino)isoindoline-2-carboxylate

[001538] The title compound was prepared from tert-butyl 4-aminoisoindoline-2- carboxylate and 2-bromoethanol analogously to Intermediate Y4-9. [001539] ¹H NMR (400 MHz, DMSO) δ 1.45 - 1.48 (m, 9H), 3.11 - 3.17 (m, 2H), 3.56 (t, J = 6.0 Hz, 2H), 4.39 (d, J=7.2 Hz, 2H), 4.51 (d, J = 11.3 Hz, 2H), 4.68 - 4.68 (m, 1H), 5.12 - 5.14 (m, 1H), 6.45 (d, J = 7.9 Hz, 1H), 6.52 (t, J = 7.0 Hz, 1H), 7.07 (t, J = 7.7 Hz, 1H). MS: ES+ 277 (M+1) Intermediate Z (E)-N-(Isoindolin-4-yl)-4-methoxybut-2-enamide

Step 1: tert-Butyl (E)-4-(4-methoxybut-2-enamido)isoindoline-2-carboxylate [001540] To a solution of (E)-4-methoxybut-2-enoic acid (55 mg, 0.47 mmol, 1.10 eq), HATU (180 mg, 0.47 mmol, 1.10 eq) and DIPEA (0.22 mL, 1.29 mmol, 3.00 eq) in DMF (4.3 mL) was added tert-butyl 4-aminoisoindoline-2-carboxylate (100 mg, 0.43 mmol, 1.0 eq) and the reaction mixture was stirred at room temperature overnight. The resulting mixture was concentrated under reduced pressure and purification of the crude residue by chromatography on silica eluting with a gradient of 0-100% EtOAc in cyclohexane afforded the title compound (42 mg, 29%). [001541] ¹H NMR (400 MHz, DMSO) δ 1.45 - 1.48 (m, 9H), 3.39 (s, 3H), 4.10 - 4.13 (m, 2H), 4.56 - 4.61 (m, 4H), 6.43 - 6.49 (m, 1H), 6.77 - 6.84 (m, 1H), 7.07 - 7.12 (m, 1H), 7.24 - 7.29 (m, 1H), 7.65 - 7.70 (m, 1H), 9.60 - 9.64 (m, 1H). MS: 333 (M+1) Step 2: (E)-N-(isoIndolin-4-yl)-4-methoxybut-2-enamide
[001542] To a solution of tert-butyl (E)-4-(4-methoxybut-2-enamido)isoindoline-2- carboxylate (Step 1) (70 mg, 0.21 mmol, 1.0 eq) in DCM (1 mL) was added TFA (0.081 mL, 1.0 mmol, 5.0 eq) and the reaction mixture was stirred at room temperature overnight. The resulting mixture was concentrated under reduced pressure. The residue was dissolved in DCM and loaded onto an Isolute® SCX cartridge. The column was washed with DCM/MeOH (1:1) and the product eluted with 1N methanolic ammonia. The methanolic ammonia product fractions were concentrated under reduced pressure to afford the title compound which was used without further purification. (50 mg, quantitative yield). [001543] ¹H NMR (400 MHz, CDCl
3) δ 3.38 (s, 3H), 4.06 - 4.12 (m, 2H), 4.41 (s, 2H), 4.47 (s, 2H), 6.24 - 6.50 (m, 1H), 6.92 - 7.04 (m, 2H), 7.17 - 7.25 (m, 1H), 7.56 - 7.72 (m, 1H), 8.65 - 8.75 (m, 1H). MS: 233 (M+1) Intermediate Z1 (2,4-Dihydroxy-6-methylphenyl)(4-(methylamino)isoindolin-2-yl)methanone

[001544] To a solution of 2,4-dihydroxy-6-methylbenzoic acid (263 mg, 1.56 mmol, 1.00 eq) in DCM (14 mL) was added EDC (899 mg, 4.69 mmol, 3.00 eq) and the mixture was stirred at room temperature for 1.5 h. N-Methylisoindolin-4-amine (278 mg, 1.88 mmol, 1.20 eq) in DMF (4.00 mL) was added and stirring continued at room temperature for 1.5 h. DIPEA (1.9 mL, 10.9 mmol, 7.00 eq) was added and the mixture was stirred at room temperature for 24 h followed by heating to 60 ˚C for 3 h. After cooling to room temperature, the mixture was allowed to stand for 72 h. The resulting mixture was concentrated under reduced pressure and purification by chromatography on silica eluting with 0-100% EtOAc in cyclohexane followed by 0-25% MeOH in EtOAc afforded the title compound (75 mg, 0.251 mmol, 16%). [001545] MS: ES+ 299 (M+1) [001546] The following compound was prepared analogously to Intermediate Z1 using the indicated intermediates. Int. Name and Structure Data
Z2 MS: ES+ 396 (M+1). 4,5-Dimethyl-6-(4-(methylamino)isoindoline-2- carbonyl)-1,3-phenylene diacetate Prepared using N-methylisoindolin-4-amine and 4,6-diacetoxy-2,3-dimethylbenzoic acid (CAS: 1081769-29-0) Intermediate Z3 6-Hydroxybenzo[d]isoxazole-5-carboxylic acid

[001547] To a stirred solution of methyl 5-formyl-2,4-dihydroxy-benzoate (CAS 1889953- 48-3) (650 mg, 2.7 mmol, 1.0 eq) and sodium azide (259 mg, 3.98 mmol, 1.5 eq) in acetonitrile (5 mL) was added dropwise over 10 minutes a solution of trifluoromethanesulfonic acid (1.4 mL, 15.9 mmol, 6.0 eq) in acetonitrile (5 mL). The reaction mixture was allowed to stir for 10 minutes until no more effervescence was observed. The resulting mixture was concentrated under reduced pressure then diluted with saturated sodium bicarbonate aq. (25 mL) and extracted with EtOAc (3 x 25 mL). The aqueous layer was treated with 2M NaOH (25 mL) and heated to 100 °C for 4 h. After cooling to room temperature, the mixture was acidified to pH 1 with HCl aq. resulting in precipitate formation. The solid was collected by filtration and purified by C18 reverse phase chromatography eluting with a gradient of 5 to 55% acetonitrile in water (with 0.1% formic acid modifier) to afford the title compound as a cream solid (26 mg, 5.5%). [001548] MS: ES+ 178 (M-1) Intermediate Z4 3-Chloro-4-(difluoromethoxy)-6-hydroxy-2-methyl-benzoic acid Step 1: 6-Chloro-7-hydroxy-2,2,5-trimethyl-4H-benzo[d][1,3]dioxin-4-one
[001549] To a cooled (0 °C) solution of 3-chloro-4,6-dihydroxy-2-methyl-benzoic acid (Intermediate B) (1 g, 4.9 mmol, 1.0 eq) and 4-(dimethylamino)pyridine (60 mg, 0.5 mmol, 0.1 eq) in DME (10 mL) was added acetone (3.6 mL, 49.4 mmol, 10 eq) and thionyl chloride (1.6 mL, 22.2 mmol, 4.5 eq) sequentially. The cooling bath was removed after 10 mins and the reaction mixture was stirred at room temperature for 3 h under an inert atmosphere. The reaction was quenched by the addition of saturated aqueous sodium hydrogen carbonate until no gas evolution was observed. The resulting mixture was extracted with EtOAc (3 × 60 mL). The combined organic extracts were dried over MgSO4 and concentrated under reduced pressure. The crude material was purified by chromatography on silica gel, eluting with a gradient of 0-10 % MeOH in DCM to afford the title compound as an off-white solid (493 mg, 41%). [001550] ¹H NMR (400 MHz, DMSO) d 1.64 (s, 6H), 2.65 (s, 3H), 6.48 (s, 1H), 11.55 (s, 1H). MS: ES- 241 (M-1) Step 2: 6-Chloro-7-(difluoromethoxy)-2,2,5-trimethyl-4H-benzo[d][1,3]dioxin-4-one

[001551] To a cooled (0 °C) solution of 6-chloro-7-hydroxy-2,2,5-trimethyl-4H- benzo[d][1,3]dioxin-4-one (Step 1) (250 mg, 1.0 mmol, 1.0 eq) and potassium hydroxide (578 mg, 10.3 mmol, 10.0 eq) in acetonitrile (1.6 mL) and water (1.6 mL) was stirred for 10 minutes under an inert atmosphere. Diethyl (bromodifluoromethyl)phosphonate (0.26 mL, 1.4 mmol, 1.4 eq) was added dropwise and the cooling bath was removed. The reaction mixture was stirred for 2 h under an inert atmosphere. The reaction was diluted with EtOAc (30 mL). The phases were separated, and the aqueous phase extracted with EtOAc (2 × 15 mL). The combined organic extracts were dried over MgSO4 and concentrated under reduced pressure. The crude material
was purified by chromatography on silica, eluting with a gradient of 0-10 % MeOH in DCM to afford the title compound as an off-white solid (166 mg, 55%). [001552] ¹H NMR (400 MHz, DMSO) δ 1.69 (s, 6H), 2.72 (d, J=0.4 Hz, 3H), 7.03 (s, 1H), 7.49 (t, J=72.1 Hz, 1H). Step 3: 3-Chloro-4-(difluoromethoxy)-6-hydroxy-2-methyl-benzoic acid

[001553] To a solution of 6-chloro-7-(difluoromethoxy)-2,2,5-trimethyl-4H- benzo[d][1,3]dioxin-4-one (Step 2) (166 mg, 0.6 mmol, 1.0 eq) in THF (7.5 mL) was added 1M lithium hydroxide (7.5 mL, 7.5 mmol, 13.2 eq). The reaction mixture was stirred at room temperature for 16 h. The reaction mixture was acidified with 2N HCl and extracted with EtOAc (3 × 25 mL). The combined organic extracts were washed with brine (20 mL), dried over MgSO
4 and concentrated under reduced pressure to give the title compound as an off-white solid (135 mg, 94%). [001554] ¹H NMR (400 MHz, DMSO) d 2.31 (s, 3H), 6.72 (s, 1H), 7.25 (t, J=72.7 Hz, 1H). Exchangeable protons not visible. MS: ES+ 251 (M+1). Biological assays Fluorescence polarisation assay for PMS2 [001555] Test compounds, as 10 mM DMSO stocks, were dispensed into a Black Fluotrac 200384 well medium binding plate (Greiner Bio-One, item number 781076) using a Labcyte Echo acoustic liquid handler. For single point screening, test compounds were added to wells in columns 1-22 whilst DMSO was added to wells in columns 23 and 24 in order to normalise the plate. For potency determination, serial dilutions of test compounds were added to wells in columns 3-22 and DMSO volume was normalised across the plate. [001556] 20 μL of a 2 x solution (20 nM) of recombinant N-terminal PMS2 (residues 1-365) in assay buffer (25 mM HEPES, pH 7.5, 250 mM NaCl, 10 mM MgCl2, 0.01% Triton X-100, 5 mM Dithiothreitol) was added to all wells in columns 2-23 for potency determination or columns 1-23 for single point screening. 20 μL assay buffer was added to all wells in columns 1 and 24
(column 24 only for single point screening) using a MultiDrop Combi (ThermoFisher). Plates were centrifuged for 1 minute at 250 xg and were incubated at room temperature for 30 minutes prior to the addition of 20 μL of 2 x (20 nM) of 5-((5-(4-((2-(2,4-dihydroxy-5- isopropylbenzoyl)isoindolin5-yl)methyl)piperazin-1-yl)pentyl)carbamoyl)-2-(6-(dimethylamino)-3- (dimethyliminio)-3H-xanthen-9-yl)benzoate (referred to hereinafter as “probe compound”) in assay buffer (prepared from a 100 μM DMSO stock) with a MultiDrop Combi (ThermoFisher). The final concentration of N-terminal PMS2 was 10 nM and the final concentration of probe compound was 5 nM. [001557] Compound plates were centrifuged for 1 minute at 250 xg for 1 minute and were incubated at room temperature for 1 hour before being read on a PheraStar FSX (fitted with 384- well aperture spoon and 540590590 FP optic module). The gain and focus were adjusted before each plate was read so that the polarisation of a no enzyme control (column 24) was equal to 35 mP. Data were normalised against the no inhibitor controls (column 23) and no enzyme controls (column 24). [001558] Data obtained in this assay is shown in Table A1 shown below Table A1 * >1 µM ** 0.1 – 1 µM *** less than 0.1 µM Example Full Name PMS2 IC50 (E)-N-(2-(3-Chloro-4,6-dihydroxy-2-methylbenzoyl)isoindolin-4-yl)- 1 *** 4-(dimethylamino)-N-methylbut-2-enamide (E)-N-Benzyl-N-(2-(2,4-dihydroxybenzoyl)isoindolin-4-yl)-4- 1.1 ** (dimethylamino)but-2-enamide (E)-N-Benzyl-N-(2-(2,4-dihydroxy-5-methylbenzoyl)isoindolin-4-yl)- 1.2 *** 4-(dimethylamino)but-2-enamide (E)-N-(2-(2,4-Dihydroxybenzoyl) isoindolin-4-yl)-4-(diethylamino)- 1.3 ** N-((1-methyl-1H-pyrazol-4-yl)methyl)but-2-enamide (E)-4-(3,3-Difluoropyrrolidin-1-yl)-N-(2-(2,4-dihydroxy-5- 2 ** methylbenzoyl)isoindolin-4-yl)but-2-enamide
((E)-4-(3,3-Difluoroazetidin-1-yl)-N-(2-(2,4-dihydroxy-5- * methylbenzoyl)isoindolin-4-yl)but-2-enamide N-(2-(2, 4-Dihydroxy-5-methylbenzoyl) isoindolin-4-yl) acrylamide ** (E)-N-(2-(2,4-Dihydroxy-6-methylbenzoyl)-1,2,3,4- ** tetrahydroisoquinolin-7-yl)-4-(dimethylamino)but-2-enamide (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)-1,2,3,4- ** tetrahydroisoquinolin-7-yl)-4-(dimethylamino)but-2-enamide (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)-1,2,3,4- tetrahydroisoquinolin-7-yl)-4-(dimethylamino)-N-methylbut-2- *** enamide (E)-N-(2-(2,4-Dihydroxy-6-methylbenzoyl)-1,2,3,4- tetrahydroisoquinolin-7-yl)-4-(dimethylamino)-N-methylbut-2- *** enamide (E)-N-(2-(2,4-Dihydroxy-5-isopropylbenzoyl)-1,2,3,4- tetrahydroisoquinolin-7-yl)-4-(dimethylamino)-N-methylbut-2- *** enamide (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4- ** (pyrrolidin-1-yl)but-2-enamide (E)-N-(2-(2, 4-Dihydroxy-5-methylbenzoyl) isoindolin-4-yl)-4- ** morpholinobut-2-enamide N-(2-(3-Chloro-4,6-dihydroxy-2-methylbenzoyl)-1,2,3,4- *** tetrahydroisoquinolin-7-yl)-N-methylacrylamide N-(2-(5-Chloro-2,4-dihydroxybenzoyl)-1,2,3,4- *** tetrahydroisoquinolin-7-yl)-N-methylacrylamide (E)-N-(2-(5-(tert-Butyl)-2-hydroxy-4-methoxybenzoyl)isoindolin-4- * yl)-4-(dimethylamino)-N-methylbut-2-enamide (E)-N-(2-(5-(tert-Butyl)-4-fluoro-2-hydroxybenzoyl)isoindolin-4-yl)- * 4-(dimethylamino)-N-methylbut-2-enamide
(E)-N-(2-(4,6-Dihydroxy-2,3-dimethylbenzoyl)-1,2,3,4- tetrahydroisoquinolin-7-yl)-4-(dimethylamino)-N-methylbut-2- *** enamide N-(2-(2,4-Dihydroxy-5-methylbenzoyl)-1,2,3,4- *** tetrahydroisoquinolin-7-yl)-N-methylacrylamide N-Benzyl-N-(2-(2,4-dihydroxybenzoyl)isoindolin-4-yl)acrylamide * N-Benzyl-N-(2-(2,4-dihydroxy-5-methylbenzoyl)isoindolin-4- ** yl)acrylamide (E)-N-(2-(2,4-Dihydroxybenzoyl) isoindolin-4-yl)-4-(dimethyl *** amino)-N-phenethylbut-2-enamide (E)-N-(2-(2-Chloro-4,6-dihydroxybenzoyl)isoindolin-4-yl)-4- *** (dimethylamino)-N-methylbut-2-enamide (E)-4-(Dimethylamino)-N-(2-(6-hydroxy-3,3-dimethylindoline-5- * carbonyl)isoindolin-4-yl)-N-methylbut-2-enamide (E)-N-(2-(4,6-Dihydroxy-2,3-dimethylbenzoyl)isoindolin-4-yl)-4- *** (dimethylamino)-N-methylbut-2-enamide (E)-N-(2-(5-(tert-Butyl)-2,4-dihydroxybenzoyl)isoindolin-4-yl)-4- *** (dimethylamino)-N-methylbut-2-enamide (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4- *** (dimethylamino)-N-ethylbut-2-enamide (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4- *** (dimethylamino)-N-(2-hydroxyethyl)but-2-enamide (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4- *** (dimethylamino)-N-(pyridin-4-ylmethyl)but-2-enamide (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4- *** (dimethylamino)-N-(2-(pyridin-4-yl)ethyl)but-2-enamide (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4- *** (dimethylamino)-N-(2-(pyridin-2-yl)ethyl)but-2-enamide (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4- *** (dimethylamino)-N-(2-(pyridin-3-yl)ethyl)but-2-enamide
(E)-N-(2-(4,6-Dihydroxy-2,3-dimethylbenzoyl)isoindolin-4-yl)-4- *** (dimethylamino)-N-(2-(pyridin-2-yl)ethyl)but-2-enamide E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4- (dimethylamino)-N-(2-(tetrahydro-2H-pyran-4-yl)ethyl)but-2- *** enamide (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4- *** (dimethylamino)-N-(2-methoxyethyl)but-2-enamide (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4- *** (dimethylamino)-N-methylbut-2-enamide (E)-N-(2-(2,4-Dihydroxy-6-methylbenzoyl)isoindolin-4-yl)-4- *** (dimethylamino)-N-methylbut-2-enamide (E)-N-(2-(5-Chloro-2,4-dihydroxybenzoyl)isoindolin-4-yl)-4- *** (dimethylamino)but-2-enamide (E)-N-(2-(2,4-Dihydroxy-5-methoxybenzoyl)isoindolin-4-yl)-4- ** (dimethylamino)but-2-enamide (E)-N-(2-(4,6-Dihydroxy-2,3-dimethylbenzoyl)isoindolin-4-yl)-4- *** (dimethylamino)but-2-enamide (E)-4-(Dimethylamino)-N-(2-(6-hydroxy-1H-indole-5- ** carbonyl)isoindolin-4-yl)but-2-enamide (E)-N-(2-(5-Bromo-2,4-dihydroxybenzoyl)isoindolin-4-yl)-4- *** (dimethylamino)but-2-enamide (E)-N-(2-(2,4-Dihydroxy-5-(trifluoromethyl)benzoyl)isoindolin-4-yl)- ** 4-(dimethylamino)-N-methylbut-2-enamide (E)-N-(2-(5-Cyano-2,4-dihydroxybenzoyl)isoindolin-4-yl)-4- ** (dimethylamino)but-2-enamide (E)-N-(2-(2,4-Dihydroxy-6-(trifluoromethyl)benzoyl)isoindolin-4-yl)- *** 4-(dimethylamino)but-2-enamide (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4- *** (dimethylamino)but-2-enamide (E)-N-(2-Benzoylisoindolin-4-yl)-4-(dimethylamino)but-2-enamide *
(E)-4-(Dimethylamino)-N-(2-(4-hydroxy-2- * methoxybenzoyl)isoindolin-4-yl)but-2-enamide (E)-N-(2-(2-(Difluoromethyl)-4-hydroxybenzoyl)isoindolin-4-yl)-4- * (dimethylamino)but-2-enamide (E)-N-(2-(2-Chloro-4-hydroxybenzoyl)isoindolin-4-yl)-4- * (dimethylamino)but-2-enamide (E)-4-(Dimethylamino)-N-(2-(6-hydroxy-1H-indazole-5- * carbonyl)isoindolin-4-yl)but-2-enamide (E)-N-(2-(2,4-Dihydroxy-6-methylbenzoyl)isoindolin-4-yl)-4- *** (dimethylamino)but-2-enamide (E)-N-(2-(4-(Difluoromethyl)-2-hydroxybenzoyl)isoindolin-4-yl)-4- * (dimethylamino)but-2-enamide (E)-4-(Dimethylamino)-N-(2-(5-fluoro-2,4- ** dihydroxybenzoyl)isoindolin-4-yl)but-2-enamide (E)-4-(Dimethylamino)-N-(2-(6-hydroxybenzo[d]isoxazole-5- * carbonyl)isoindolin-4-yl)but-2-enamide (E)-N-(2-(2,4-Dihydroxy-5-isopropylbenzoyl)isoindolin-4-yl)-4- *** (dimethylamino)but-2-enamide (E)-N-(2-(4-Amino-2-hydroxybenzoyl)isoindolin-4-yl)-4- * (dimethylamino)but-2-enamide (E)-4-(Dimethylamino)-N-(2-(5-ethyl-2,4- *** dihydroxybenzoyl)isoindolin-4-yl)but-2-enamide (E)-N-(2-(2,4-Dihydroxy-5-isopropylbenzoyl)isoindolin-4-yl)-4- *** (dimethylamino)-N-methylbut-2-enamide (E)-N-(2-(3-Chloro-4-(difluoromethoxy)-6-hydroxy-2- methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-methylbut-2- ** enamide (E)-4-(Dimethylamino)-N-(2-(2-hydroxy-5- * isopropylbenzoyl)isoindolin-4-yl)-N-methylbut-2-enamide
(E)-N-(2-(5-(tert-Butyl)-2-hydroxybenzoyl)isoindolin-4-yl)-4- * (dimethylamino)-N-methylbut-2-enamide (E)-N-(2-(4-(Difluoromethoxy)-2-hydroxy-5- methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-methylbut-2- * enamide (E)-4-(Dimethylamino)-N-(2-(4-hydroxy-2,3- ** dimethylbenzoyl)isoindolin-4-yl)-N-methylbut-2-enamide (E)-N-(2-(3-(tert-Butyl)-4-hydroxybenzoyl)isoindolin-4-yl)-4- ** (dimethylamino)-N-methylbut-2-enamide (E)-4-(Dimethylamino)-N-(2-(4-hydroxy-3- * methylbenzoyl)isoindolin-4-yl)-N-methylbut-2-enamide (E)-4-(Dimethylamino)-N-(2-(4-hydroxy-3- * isopropylbenzoyl)isoindolin-4-yl)-N-methylbut-2-enamide (E)-4-(Dimethylamino)-N-(2-(4-hydroxy-3- * isopropylbenzoyl)isoindolin-4-yl)but-2-enamide (E)-4-(Dimethylamino)-N-(2-(4-hydroxy-3- * (trifluoromethyl)benzoyl)isoindolin-4-yl)-N-methylbut-2-enamide (E)-N-(2-(3-Chloro-4-hydroxy-2-methylbenzoyl)isoindolin-4-yl)-4- ** (dimethylamino)-N-methylbut-2-enamide N-[2-(2,4-Dihydroxy-5-methyl-benzoyl)isoindolin-4-yl]-N-[2-(4- ** pyridyl)ethyl]prop-2-enamide N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-N-(2-(1- ** methylpyrrolidin-3-yl)ethyl)acrylamide N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-N- ** methylacrylamide N-(2-(5-(tert-Butyl)-2,4-dihydroxybenzoyl)isoindolin-4-yl)acrylamide *** N-(2-(5-(tert-Butyl)-2,4-dihydroxybenzoyl)isoindolin-4-yl)-N- *** methylacrylamide N-(2-(2,4-Dihydroxy-6-methylbenzoyl)isoindolin-4-yl)acrylamide **
(E)-N-[2-(2,4-Dihydroxy-5-methyl-benzoyl)isoindolin-4-yl]-4- *** (dimethylamino)-N-indan-2-yl-but-2-enamide N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-N-(2- *** morpholinoethyl)acrylamide N-(2-(2,4-Dihydroxy-6-methylbenzoyl)isoindolin-4-yl)-N- ** methylacrylamide (E)-N-[2-(2,4-Dihydroxybenzoyl)isoindolin-4-yl]-4- ** (dimethylamino)but-2-enamide (E)-4-(Dimethylamino)-N-(2-(2-hydroxy-4- * methoxybenzoyl)isoindolin-4-yl)but-2-enamide (E)-4-(Dimethylamino)-N-(2-(2-hydroxy-5- * methylbenzoyl)isoindolin-4-yl)but-2-enamide (E)-4-(Dimethylamino)-N-(2-(4-hydroxy-3- * methylbenzoyl)isoindolin-4-yl)but-2-enamide (E)-4-(Dimethylamino)-N-(2-(2-fluoro-4-hydroxybenzoyl)isoindolin- * 4-yl)but-2-enamide (E)-4-(Dimethylamino)-N-(2-(4-hydroxy-2- * methylbenzoyl)isoindolin-4-yl)but-2-enamide (E)-4-(Dimethylamino)-N-(2-(5-hydroxypicolinoyl)isoindolin-4- * yl)but-2-enamide (E)-4-(Dimethylamino)-N-(2-(2-methoxybenzoyl)isoindolin-4-yl)but- * 2-enamide (E)-N-(2-(2,4-Dihydroxy-5-(trifluoromethyl)benzoyl)isoindolin-4-yl)- *** 4-(dimethylamino)but-2-enamide (E)-N-(2-(5-(tert-Butyl)-2,4-dihydroxybenzoyl)isoindolin-4-yl)-4- *** (dimethylamino)but-2-enamide (E)-N-(2-(2,4-Dihydroxybenzoyl)isoindolin-4-yl)-4- * (dimethylamino)but-2-enamide (E)-N-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-4- ** methoxybut-2-enamide
1-(2-(2,4-Dihydroxy-5-methylbenzoyl)isoindolin-4-yl)-3-(2- * (dimethylamino)ethylidene)pyrrolidin-2-one N-(2-(2,4-Dihydroxy-5-methylbenzoyl)-1,2,3,4- *** tetrahydroisoquinolin-7-yl)-N-(1-methylpiperidin-4-yl)acrylamide (E)-N-(2-(4,6-Dihydroxy-2,3-dimethylbenzoyl)isoindolin-4-yl)-4- *** (dimethylamino)-N-ethylbut-2-enamide N-(2-(4,6-Dihydroxy-2,3-dimethylbenzoyl)isoindolin-4-yl)-N-(2- *** methoxyethyl)acrylamide N-(2-(4,6-Dihydroxy-2,3-dimethylbenzoyl)isoindolin-4-yl)-N- *** methylacrylamide (E)-N-(2-(5-(tert-Butyl)-2,4-dihydroxybenzoyl)-5-chloroisoindolin-4- *** yl)-4-(dimethylamino)-N-methylbut-2-enamide (E)-N-(5-Chloro-2-(2, 4-dihydroxy-5-methylbenzoyl)isoindolin-4-yl)- *** 4-(dimethyl amino)-N-methylbut-2-enamide (E)-4-(Dimethylamino)-N-(2-(6-hydroxy-3,3-dimethyl-2-oxoindoline- ** 5-carbonyl)isoindolin-4-yl)-N-methylbut-2-enamide (E)-N-(2-(3-Chloro-4,6-dihydroxy-2-methylbenzoyl)isoindolin-4-yl)- *** 4-(dimethylamino)-N-ethylbut-2-enamide (E)-N-(2-(3-Chloro-4,6-dihydroxy-2-methylbenzoyl)isoindolin-4-yl)- *** 4-(ethyl(methyl)amino)-N-methylbut-2-enamide (E)-N-(7-Chloro-2-(2,4-dihydroxy-5-methylbenzoyl)isoindolin-4-yl)- *** 4-(dimethylamino)-N-methylbut-2-enamide (E)-N-(7-Chloro-2-(4,6-dihydroxy-2,3-dimethylbenzoyl)isoindolin-4- *** yl)-4-(dimethylamino)-N-methylbut-2-enamide N-(2-(3-Chloro-4,6-dihydroxy-2-methylbenzoyl)isoindolin-4-yl)-N- *** methyl acrylamide (E)-N-(5-Chloro-2-(4,6-dihydroxy-2,3-dimethylbenzoyl) isoindolin- *** 4-yl)-4-(dimethyl amino)-N-methylbut-2-enamide (E)-N-(2-(3-Chloro-2-fluoro-4,6-dihydroxybenzoyl) isoindolin-4-yl)- *** 4-(dimethylamino)-N-methylbut-2-enamide
(E)-N-(2-(3-Chloro-2-fluoro-4,6-dihydroxybenzoyl)isoindolin-4-yl)- *** 4-(dimethylamino)-N-ethylbut-2-enamide N-(2-(3-Chloro-4,6-dihydroxy-2-methylbenzoyl) isoindolin-4-yl)-N- *** ethyl acrylamide N-(2-(3-Chloro-2-fluoro-4,6-dihydroxybenzoyl) isoindolin-4-yl)-N- *** methylacrylamide N-(2-(2,4-Dihydroxy-5-methylbenzoyl)-6-(2-(dimethylamino) *** ethoxy) isoindolin-4-yl)-N-methylacrylamide (E)-4-(Dimethylamino)-N-(2-(6-hydroxy-2,3-dimethyl-1H-indole-5- ** carbonyl) isoindolin-4-yl)-N-methylbut-2-enamide (E)-4-(Dimethylamino)-N-(2-(6-hydroxy-2-methyl-1H-indole-5- * carbonyl) isoindolin-4-yl)-N-methylbut-2-enamide (E)-4-(Dimethylamino)-N-(2-(6-hydroxy-3-methyl-1H-indole-5- ** carbonyl) isoindolin-4-yl)-N-methylbut-2-enamide (E)-4-(Dimethylamino)-N-(2-(4-fluoro-2-hydroxy-5- * isopropylbenzoyl) isoindolin-4-yl)-N-methylbut-2-enamide (E)-4-(Dimethylamino)-N-(2-(2-hydroxy-4-methoxy-5- * methylbenzoyl)isoindolin-4-yl)-N-methylbut-2-enamide (E)-N-(2-(3-(tert-Butyl)-2-fluoro-6-hydroxy-5- (hydroxymethyl)benzoyl)isoindolin-4-yl)-4-(dimethylamino)-N- * methylbut-2-enamide N-(2-(3-(Aminomethyl)-5-(tert-butyl)-6-fluoro-2- * hydroxybenzoyl)isoindolin-4-yl)-N-methylacrylamide (E)-N-(2-(5-(tert-Butyl)-2-hydroxy-4-methoxybenzoyl)-5- ** chloroisoindolin-4-yl)-4-(dimethylamino)-N-methylbut-2-enamide (E)-4-(Dimethylamino)-N-(2-(2-hydroxy-5-isopropyl-4- ** methoxybenzoyl)isoindolin-4-yl)-N-methylbut-2-enamide (E)-4-(Dimethylamino)-N-(2-(5-ethyl-2-hydroxy-4- * methoxybenzoyl)isoindolin-4-yl)-N-methylbut-2-enamide
(E)-N-(2-(3-Chloro-6-hydroxy-4-methoxy-2- methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)-N-methylbut-2- *** enamide (E)-N-(2-(5-Chloro-2-hydroxy-4-methoxybenzoyl)isoindolin-4-yl)-4- * (dimethylamino)-N-methylbut-2-enamide N-(2-(5-(tert-Butyl)-2-hydroxy-4-methoxybenzoyl)isoindolin-4-yl)-N- * methylacrylamide N-(2-(5-Ethyl-2-hydroxy-4-methoxybenzoyl)isoindolin-4-yl)-N- * methylacrylamide (E)-N-(2-(3-(tert-Butyl)-2-fluoro-6-hydroxybenzoyl)-1,2,3,4- tetrahydroisoquinolin-7-yl)-4-(dimethylamino)-N-methylbut-2- ** enamide N-(2-(3-(tert-Butyl)-2-fluoro-6-hydroxybenzoyl)-1,2,3,4- ** tetrahydroisoquinolin-7-yl)-N-methylacrylamide N-(2-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)isoindolin-4- ** yl)-N-methylacrylamide N-(2-(5-Chloro-2-hydroxy-4-methoxybenzoyl)isoindolin-4-yl)-N- * methyl acrylamide (E)-N-(2-(5-Chloro-2-hydroxy-4-methoxybenzoyl)-5- * methylisoindolin-4-yl)-4-(dimethylamino)-N-methylbut-2-enamide (E)-N-(5-Chloro-2-(5-chloro-2-hydroxy-4-methoxybenzoyl) ** isoindolin-4-yl)-4-(dimethyl amino)-N-methylbut-2-enamide (E)-N-(5-Chloro-2-(2-hydroxy-5-isopropyl-4-methoxybenzoyl) ** isoindolin-4-yl)-4-(dimethyl amino)-N-methylbut-2-enamide (E)-N-(2-(5-(tert-Butyl)-2-hydroxy-4-methoxybenzoyl)-5- ** methylisoindolin-4-yl)-4-(dimethylamino)-N-methylbut-2-enamide N-(2-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)-1,2,3,4- ** tetrahydroisoquinolin-7-yl)-N-methylacrylamide N-(2-(4-Fluoro-2-hydroxy-5-isopropylbenzoyl)-1,2,3,4- * tetrahydroisoquinolin-7-yl)-N-methylacrylamide
N-(2-(5-Chloro-2-hydroxy-4-methoxybenzoyl)-1,2,3,4- ** tetrahydroisoquinolin-7-yl)-N-methylacrylamide N-(2-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)-6-methyl-1,2,3,4- ** tetrahydroisoquinolin-7-yl)-N-methylacrylamide N-(2-(6-Hydroxy-3,3-dimethyl-2-oxoindoline-5-carbonyl)-1,2,3,4- ** tetrahydroisoquinolin-7-yl)-N-methylacrylamide N-(2-(6-Hydroxy-3,3-dimethyl-2-oxoindoline-5-carbonyl) isoindolin- * 4-yl)-N-methyl acrylamide N-(6-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)-5,6,7,8- ** tetrahydro-2,6-naphthyridin-3-yl)-N-methylacrylamide N-(7-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)-3-methyl-5,6,7,8- *** tetrahydro-1,7-naphthyridin-2-yl)-N-methylacrylamide N-(6-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)-2-methyl-5,6,7,8- ** tetrahydro-1,6-naphthyridin-3-yl)-N-methylacrylamide (E)-N-(2-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-1,2,3,4- tetrahydroisoquinolin-7-yl)-4-(dimethylamino)-N-methylbut-2- ** enamide (E)-4-(Dimethylamino)-N-(2-(2-hydroxy-5-isopropyl-4- methoxybenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N-methylbut- ** 2-enamide (E)-N-(2-(5-(tert-Butyl)-4-fluoro-2-hydroxybenzoyl)-1,2,3,4- tetrahydroisoquinolin-7-yl)-4-(dimethylamino)-N-methylbut-2- * enamide (E)-4-(Dimethylamino)-N-(2-(4-fluoro-2-hydroxy-5- isopropylbenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N-methylbut- * 2-enamide N-(2-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)isoindolin-4-yl)-N- * methylacrylamide N-(6-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)-5,6,7,8- * tetrahydro-1,6-naphthyridin-3-yl) acrylamide
(E)-N-(2-(3-Chloro-6-hydroxy-2-methylbenzoyl)-1,2,3,4- tetrahydroisoquinolin-7-yl)-4-(dimethylamino)-N-methylbut-2- * enamide (E)-N-(6-(5-(tert-Butyl)-2-hydroxybenzoyl)-5,6,7,8-tetrahydro-1,6- * naphthyridin-3-yl)-4-(dimethylamino)-N-methylbut-2-enamide (E)-N-(2-(5-(tert-Butyl)-2-hydroxybenzoyl)-1,2,3,4- tetrahydroisoquinolin-7-yl)-4-(dimethylamino)-N-methylbut-2- * enamide (E)-N-(5-Chloro-2-(4-hydroxy-3-isopropylbenzoyl)isoindolin-4-yl)-4- ** (dimethylamino) but-2-enamide (E)-N-(5-Chloro-2-(4-hydroxy-3-isopropylbenzoyl)isoindolin-4-yl)-4- ** (dimethylamino)-N-methylbut-2-enamide N-(2-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-6-(2- (dimethylamino)ethoxy)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- *** methylacrylamide N-(6-(2-(Dimethylamino)ethoxy)-2-(2-hydroxy-5-isopropyl-4- methoxybenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N-methylbut- * 2-ynamide (E)-N-(5-Chloro-2-(3-chloro-6-hydroxy-4-methoxy-2- methylbenzoyl) isoindolin-4-yl)-4-(dimethyl amino)-N-methylbut-2- * enamide (E)-N-(2-(5-(tert-Butyl)-2-fluoro-4-hydroxybenzoyl)isoindolin-4-yl)- * 4-(dimethylamino)-N-methylbut-2-enamide (E)-N-(2-(5-(tert-Butyl)-2-hydroxy-4-methoxybenzoyl)-1,2,3,4- tetrahydroisoquinolin-7-yl)-4-(dimethylamino)-N-methylbut-2- ** enamide N-(2-(5-(tert-Butyl)-2-hydroxy-4-methoxybenzoyl)-1,2,3,4- ** tetrahydroisoquinolin-7-yl)-N-methylacrylamide N-(2-(3-Chloro-2-fluoro-6-hydroxy-4-methoxybenzoyl)isoindolin-4- * yl)-N-methylacrylamide
N-(5-Chloro-2-(3-chloro-6-hydroxy-4-methoxy-2-methylbenzoyl) * isoindolin-4-yl)-N-methyl acrylamide (E)-4-(Dimethylamino)-N-(2-(2-hydroxy-5-isopropyl-4- ** methoxybenzoyl)-5-methylisoindolin-4-yl)-N-methylbut-2-enamide N-(2-(4-Fluoro-2-hydroxy-5-isopropylbenzoyl)-6-methyl-1,2,3,4- * tetrahydroisoquinolin-7-yl)-N-methylacrylamide N-(6-Chloro-2-(2-hydroxy-5-isopropyl-4-methoxybenzoyl)-1,2,3,4- ** tetrahydroisoquinolin-7-yl)-N-methylacrylamide N-(2-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)-8-methyl-1,2,3,4- * tetrahydroisoquinolin-7-yl)-N-methylacrylamide N-(6-(2-(Dimethylamino)ethoxy)-2-(2-hydroxy-5-isopropyl-4- methoxybenzoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- *** methylacrylamide N-(6-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-5,6,7,8- *** tetrahydro-2,6-naphthyridin-3-yl)-N-methylacrylamide (E)-N-(7-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-5,6,7,8- * tetrahydro-1,7-naphthyridin-2-yl)-N-methylbut-2-enamide N-(7-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-5,6,7,8- *** tetrahydro-1,7-naphthyridin-2-yl)-N-methylacrylamide N-(7-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-3-methyl- *** 5,6,7,8-tetrahydro-1,7-naphthyridin-2-yl)-N-methylacrylamide N-(6-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-2-methyl- ** 5,6,7,8-tetrahydro-1,6-naphthyridin-3-yl)-N-methylacrylamide N-(2-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)-1,2,3,4- *** tetrahydroisoquinolin-7-yl)-N-(1-methylpiperidin-4-yl) acrylamide (E)-N-(2-(4,6-dihydroxy-2,3-dimethylbenzoyl)-5-methylisoindolin-4- *** yl)-4-(dimethylamino)-N-methylbut-2-enamide (E)-N-(2-(4,6-Dihydroxy-2,3-dimethylbenzoyl)-6-methylisoindolin-4- *** yl)-4-(dimethylamino)-N-methylbut-2-enamide
(E)-N-(6-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-5,6,7,8- * tetrahydro-1,6-naphthyridin-3-yl)-N-methylbut-2-enamide N-(2-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-1,2,3,4- *** tetrahydroisoquinolin-7-yl)-N-methylacrylamide N-(2-(2-Hydroxy-5-isopropyl-4-methoxybenzoyl)-1,2,3,4- *** tetrahydroisoquinolin-7-yl)-N-(1-methylpyrrolidin-3-yl)acrylamide ((E)-N-(5-Chloro-2-(2-hydroxy-5-isopropyl-4- ** methoxybenzoyl)isoindolin-4-yl)-4-(dimethylamino)but-2-enamide (E)-N-(2-(3-Chloro-6-hydroxy-4-methoxy-2- *** methylbenzoyl)isoindolin-4-yl)-4-(dimethylamino)but-2-enamide N-(6-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-5,6,7,8- * tetrahydro-1,6-naphthyridin-3-yl)acrylamide (E)-N-(2-(3-Chloro-6-hydroxy-2-methylbenzoyl)isoindolin-4-yl)-4- * (dimethylamino)-N-methylbut-2-enamide (E)-N-(7-Chloro-2-(3-chloro-6-hydroxy-4-methoxy-2- methylbenzoyl) isoindolin-4-yl)-4-(dimethylamino)-N-methylbut-2- ** enamide N-(7-Chloro-2-(3-chloro-6-hydroxy-4-methoxy-2-methylbenzoyl) ** isoindolin-4-yl)-N-methyl acrylamide (E)-4-(Dimethylamino)-N-(6-(2-hydroxy-5-isopropyl-4- methoxybenzoyl)-5,6,7,8-tetrahydro-1,6-naphthyridin-3-yl)-N- * methylbut-2-enamide N-(2-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-6-(2- (dimethylamino)ethoxy)-1,2,3,4-tetrahydroisoquinolin-7-yl)-N- * methylbut-2-ynamide (E)-N-(5-Chloro-2-(3-chloro-4-hydroxy-2-methylbenzoyl)isoindolin- ** 4-yl)-4-(dimethylamino)but-2-enamide (E)-N-(5-Chloro-2-(4-hydroxy-2,3-dimethylbenzoyl) isoindolin-4-yl)- ** 4-(dimethylamino) but-2-enamide
(E)-4-(Dimethylamino)-N-(2-(4-hydroxy-3-isopropylbenzoyl)-3- ** methyl-1,2,3,4-tetrahydroisoquinolin-7-yl)-N-methylbut-2-enamide (E)-4-(Dimethylamino)-N-(2-(4-hydroxy-3-isopropylbenzoyl)- ** 1,2,3,4-tetrahydroisoquinolin-7-yl)-N-methylbut-2-enamide N-(2-(4-Hydroxy-3-isopropylbenzoyl)-1,2,3,4-tetrahydroisoquinolin- *** 7-yl)-N-(1-methylpiperidin-4-yl)acrylamide (E)-4-(Dimethylamino)-N-(6-(2-hydroxy-5-isopropyl-4- methoxybenzoyl)-2-methoxy-5,6,7,8-tetrahydro-1,6-naphthyridin-3- ** yl)-N-methylbut-2-enamide ((E)-4-(Dimethylamino)-N-(6-(4-hydroxy-3-isopropylbenzoyl)-2- methoxy-5,6,7,8-tetrahydro-1,6-naphthyridin-3-yl)-N-methylbut-2- ** enamide (E)-N-(2-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-1,2,3,4- * tetrahydroisoquinolin-7-yl)-N-methylbut-2-enamide N-(6-(3-Chloro-6-hydroxy-4-methoxy-2-methylbenzoyl)-5,6,7,8- * tetrahydro-1,6-naphthyridin-3-yl)but-2-ynamide