WO2024246270A1 - Process for the polymerization of olefins - Google Patents
Process for the polymerization of olefins Download PDFInfo
- Publication number
- WO2024246270A1 WO2024246270A1 PCT/EP2024/065010 EP2024065010W WO2024246270A1 WO 2024246270 A1 WO2024246270 A1 WO 2024246270A1 EP 2024065010 W EP2024065010 W EP 2024065010W WO 2024246270 A1 WO2024246270 A1 WO 2024246270A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- reactor
- compound
- polymerization
- mono
- process according
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F210/00—Copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F210/04—Monomers containing three or four carbon atoms
- C08F210/06—Propene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
- C08L23/02—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers not modified by chemical after-treatment
- C08L23/16—Ethene-propene or ethene-propene-diene copolymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2207/00—Properties characterising the ingredient of the composition
- C08L2207/02—Heterophasic composition
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2308/00—Chemical blending or stepwise polymerisation process with the same catalyst
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2314/00—Polymer mixtures characterised by way of preparation
- C08L2314/02—Ziegler natta catalyst
Definitions
- the present disclosure relates to a process for the preparation of polyolefins.
- it relates to a gas-phase process for preparing polyolefins, especially polypropylene, carried out in the presence of specific ester compounds.
- PPAs are polysulfone copolymers, polymeric polyamines, polyalcohols, hydroxyesters of polyalcohols, salts of alkylarylsulfonic acids, polysiloxanes, alkoxyamines, polyglycol ethers, etc.
- EP560035A1 discloses a polymerization process in which PPAs are used to eliminate or reduce the build-up of polymer particles on the walls of a gas-phase polymerization reactor. Those PPAs are capable of selectively inhibiting the polymerization on polymer particles smaller than 850 pm, the latter being considered responsible for fouling problems and polymer sheeting.
- PPAs hydroxyesters, having at least two free hydroxyl groups, obtained from carboxylic acids having at least 4 and preferably from 8 to 22 carbon atoms and from polyalcohols are mentioned.
- W02012/041811 describe the use of a method for feeding PPAs together with catalysts in the polymerization reactor. Glycerol monostearate was used as PPA in a gasphase reactor for ethylene polymerization.
- the effectiveness of PPAs may change.
- the type of polyolefin produced may play a role. For example, sticky products such as propyl ene/ethylene rubbery copolymers have higher tendency to adhere to reactor walls and create fouling. The problem may be more evident for propylene-ethylene copolymer particles with small size (less than 500 pm) and relatively high ethylene content.
- propylene-ethylene copolymer particles with small size and higher ethylene content than the average may be the result of an increased polymerization activity which also generates an excess of heat which, in turn, may render the copolymer softer and prone to stick to reactor walls. Under this situation, keeping the ethylene content on small polymer size under control may be an indication of PPA effectiveness.
- the present disclosure provides a process for the preparation of polyolefins, carried out in the presence of (i) a Ziegler-Natta catalyst, (ii) an aluminum alkyl cocatalyst, optionally (iii) an external donor, and (iv) a compound (polymerization process aid (PPA)) comprising mono or diglycerol ester derivatives of saturated C6-C12, preferably Ce-Cio monocarboxylic acids said component (iv) being used in an amount of at least 50 ppm based on the amount of polyolefin produced.
- PPA polymerization process aid
- the PPA comprises also mono or polyglycerol fatty acid esters other than mono or diglycerol ester derivatives of saturated Ce-Cio, preferably Cs- Cio monocarboxylic acids
- the amount of mono or diglycerol ester derivatives of saturated Ce-Cio, preferably Cs-Cio monocarboxylic acids is higher than 50%wt, preferably more than 55%wt and especially in the range 60-90%wt with respect to the total amount of mono or polyglycerol fatty acid esters.
- the PPA comprises one or more of mono or diglycerol ester derivatives of saturated Ce-Cio, more preferably Cs-Cio, monocarboxylic acids and mixture thereof.
- the PPA comprises, for more than 50% wt, preferably more than 55%wt and especially in the range 60-90%wt based on the total amount of fatty acid mono or polyglycerol esters, one or more of glycerol monoester or diester of octanoic acid, glycerol monoester or diester of decanoic acid, glycerol diester mixed octanoic and decanoic acid, diglycerol mono, di- or triester of octanoic acid, diglycerol mono, di- or triester of decanoic acid, diglycerol di or triester of mixed octanoic and decanoic acid and any mixture thereof.
- PPA comprises, for more than 50% wt, preferably more than 55%wt and especially in the range 60-90%wt based on the total amount of fatty acid glycerol esters, glycerol mono octanoate or a mixture comprising glycerol diester of octanoic acid, glycerol diester of mixed octanoic and decanoic acid, diglycerol mono ester of octanoic acid and diglycerol mono ester of decanoic acid and mixture thereof.
- the mono or diglycerol ester derivatives of saturated C6-C12, preferably Cs-Cio monocarboxylic can be obtained by extraction and/or treatment from natural sources such as glycerolysis of commercial edible oils into diglycerides, and are commercially available either pure or in mixtures. It is also possible that minor amounts of other components such as non-esterified mono or polyglycerol, remains in the mixture. It is also possible to synthesize substantially pure glycerol esters such as glycerol caprylate via esterification reaction as described in CN107935851B.
- Such products are widely available in the market with different purity.
- glycerol monoester of decanoic acid and glycerol monoester of octanoic acid are sold by SigmaAldrich
- glycerol diester mixed octanoic and decanoic acid is sold by Dayang Chem
- diglycerol esters of octanoic acid and decanoic acid is sold by Ality Chemical Corporation.
- a dispersing medium which is preferably selected from liquid non-aqueous solvents and in particular from liquid hydrocarbons.
- the PPA is liquid at room temperature.
- the amount of PPA introduced into the polymerization reactor is from 70 to 2000 ppm per weight, or from 150 to 1500 ppm per weight, or from 200 to 1000 ppm per weight referring to the weight of the prepared polyolefin.
- PPA of the present disclosure can be provided to the polymerization process according to different modes of operation.
- PPA can be fed to the polymerization reactor in a flow of saturated or unsaturated hydrocarbon having from 2 to 6 carbon atoms that, according to an embodiment, can be a monomer, such as propylene, or, according to another embodiment, an alkane, such as propane. A mixture of saturated and unsaturated hydrocarbons can be used as well.
- the monomer and/or the alkane can be either in liquid or gas form.
- PPA may be diluted and homogenized with a solvent at short distance from the injection point by means of customary mixing/homogenizing/dispersing systems, such as static mixers so as increase the volume and make the injection smoother and more reliable.
- PPA of the present disclosure can be introduced into the reactor in multiple ways.
- the feeding can occur directly into the reactor and/or into a line leading to the reactor and/or into a line exiting the reactor.
- PPA can be fed:
- a catalyst precontacting vessel i.e. a vessel in which the catalyst components are brought into contact with each other;
- the present disclosure provides a process for the polymerization of olefins, such as 1 -olefins, i.e. hydrocarbons having terminal double bonds, without being restricted thereto.
- Typical 1 -olefins are linear or branched 1 -alkenes having from 2 to 12 carbon atoms, in particular linear 1 -alkenes having from 2 to 10 carbon atoms such as ethylene, propylene, 1 -butene, 1 -pentene, 1 -hexene, 1 -heptene, 1 -octene, 1 -decene or branched 1- alkenes having from 2 to 10 carbon atoms such as 4-methyl-l -pentene, conjugated and nonconjugated dienes such as 1,3 -butadiene, 1,4-hexadiene or 1,7-octadiene or vinyl-aromatic compounds such as styrene or substituted styrene.
- Olefins that can be polymerized with the process of the present disclosure include those in which the double bond is part of a cyclic structure which can have one or more ring systems. Examples are cyclopentene, norbomene, tetracyclododecene or methylnorbornene or dienes such as 5-ethylidene-2-norbornene, norbornadiene or ethylnorbornadiene. It is also possible to polymerize mixtures of two or more olefins.
- the process can be used for the homopolymerization or copolymerization of ethylene or for the homopolymerization or copolymerization of propylene. Homo and/or copolymerization of propylene is preferred.
- comonomers for use in ethylene polymerization are 1-alkenes having from 3 to 8 carbon atoms such as 1 -butene, 1 -pentene, 1 -hexene and/or 1 -octene in amount of up to 20 wt.% or from 0.01 wt.% to 15 wt.%, or from 0.05 wt.% to 12 wt.% based on the total weight of the final polymer.
- comonomers for use in propylene polymerization are ethylene and/or 1-butene and/or 1-hexene in amount of up to 40 wt.% or from 0.5 wt.% to 35 wt.% based on the total weight of the final polymer.
- a particular type of propyl ene/ethylene copolymer preferably contains from 15 to 80% of ethylene, preferably from 20 to 70%wt. Such copolymers are typically rubbery and tend to adhere to the reactor walls. In particular, reactivity of fine particles is considered very important to be controlled. Since ethylene is more reactive than propylene, capability for the compound (iv) to lower the content of ethylene in the fine particles is considered also beneficial.
- the process of the present disclosure allows the preparation of any types of common olefin polymers.
- the prepared olefin polymers can be broad molecular weight olefin polymers and, particularly, multimodal olefin polymers whereby the term multimodal refers to the modality of the molecular weight distribution.
- multimodal shall include bimodal.
- Such polymers can be obtained from polymerizing olefins in a cascade of two or more polymerization reactors or in different zones of a multizone reactor under different reaction conditions.
- the "modality" indicates how many different polymerization conditions were utilized to prepare the polyolefin, independently whether this modality of the molecular weight distribution can be recognized as separated maxima in a gel permeation chromatography (GPC) curve or not.
- the olefin polymer can also have a comonomer distribution.
- the polymerization of olefins is carried out using titanium-based Ziegler-Natta- catalysts.
- Ziegler-Natta catalysts used in the process of the present disclosure comprise:
- an electron-donor compound optionally, an electron-donor compound (external donor).
- catalyst component (i) is particularly preferred when propylene is homo or copolymerized.
- the solid catalyst component (i) can be prepared by one or more steps (a) carried out at a temperature ranging from 0 to 150°C in which a Mg based compound of formula (MgCl m X2-m) LB, where m ranges from 0 to 2, n ranges from 0 to 6, X is, independently halogen, R 1 , OR 1 , -OCOR 1 or O-C(O)-OR 1 group, in which R 1 is a C1-C20 hydrocarbon group, and LB is a Lewis base, is reacted with a liquid medium comprising a Ti compound, having at least a Ti-Cl bond, in an amount such that the Ti/Mg molar ratio is greater than 3.
- a Mg based compound of formula (MgCl m X2-m) LB where m ranges from 0 to 2, n ranges from 0 to 6, X is, independently halogen, R 1 , OR 1 , -OCOR 1 or O-C(O
- the liquid medium preferably comprises a titanium compound of formula Ti(OR 1 )q- y Cly, where q is the valence of titanium, y is a number between 1 and q and R 1 is a C1-C20 hydrocarbon group.
- titanium polyhalogenated compounds such as titanium tetrahalides or halogenalcoholates.
- Preferred specific titanium compounds are TiCL and Ti(OEt)Ch.
- the liquid medium comprising the Ti compound can be a mixture of the Ti compound in another liquid diluent.
- Preferred diluents are hydrocarbons, optionally chlorinated, that are liquid at room temperature.
- the liquid medium consists of the liquid titanium compound.
- the magnesium based compound used as a starting compound in the first of the one or more steps (a) is preferably selected among adducts of formula MgCh’nR'OH, where n is a number between 0.1 and 6, and R 1 is a hydrocarbon radical having 1-18 carbon atoms.
- n ranges from 1 to 5 and more preferably from 1.5 to 4.5.
- the adduct can be suitably prepared by mixing alcohol and magnesium chloride, operating under stirring conditions at the melting temperature of the adduct (100- 130°C). Then, the adduct is mixed with an inert hydrocarbon immiscible with the adduct thereby creating an emulsion which is quickly quenched causing the solidification of the adduct in the form of spherical particles. Examples of spherical adducts prepared according to this procedure are described in US 4,399,054 and US 4,469,648.
- Adducts having the desired final alcohol content can be obtained by directly using the selected amount of alcohol during the adduct preparation.
- the above mentioned adduct can be directly reacted with the Ti compound or it can be previously subjected to thermal controlled dealcoholation (80-130°C) so as to obtain an adduct in which the number of moles of alcohol is lowered and its porosity increased.
- thermal controlled dealcoholation 80-130°C
- it generally brings the number of moles of alcohol per mole of Mg to less than 3, preferably between 0.1 and 2.5.
- the reaction between the Mg based compound, in particular the MgCh-alcohol adduct and the Ti compound can be carried out by suspending the Mg based compound in large excess of cold TiCh (generally 0°C); the mixture is heated up to a temperature ranging from 60-140°C and kept at this temperature for 0.1-4 hours, preferably 0.5-2 hours. After that time, stirring is discontinued and after the settlement of the solid particles the liquid phase is removed.
- cold TiCh generally 0°C
- This reaction step (a) can be carried out one or more times under identical or different conditions.
- the temperature and duration of treatment can be changed.
- the number of steps (a) is comprised between 1 and 3.
- the electron donor compound can be added in the desired amount during one or more of the reaction steps (a) between the Mg based compound and the liquid Ti compound.
- the electron donor compound is added at least during the first step (a) of reaction between the Mg based compound and the Ti compound. In some cases such a treatment can be repeated one or two additional times.
- the electron donor compound as described in W02004/106388, can be added as a fresh reactant to the solid intermediate catalyst component obtained by the above described reaction between the adduct and the Ti compound.
- the reaction step (a) is carried out by a continuous feeding of liquid Ti compound, preferably TiCh, in an apparatus and under conditions that are described in W002/48208.
- liquid Ti compound preferably TiCh
- the Mg based compound is fed batchwise while a continuous stream of liquid Ti compound with the desired temperature profile is fed and a liquid phase containing dissolved reaction product is continuously withdrawn.
- the organic coloring agent and, optionally, the electron donor can be added at any time during the feeding of the Ti compound.
- the particles of solid catalyst component have substantially spherical morphology and average diameter ranging between 5 and 150 pm, preferably from 20 to 100 pm and more preferably from 30 to 90 pm when the polymerization process is carried out in gas-phase.
- particles having substantially spherical morphology those are meant wherein the ratio between the greater axis and the smaller axis is equal to or lower than 1.5 and preferably lower than 1.3.
- the amount of Mg preferably ranges from 8 to 30% more preferably from 10 to 25%wt with respect to the total weight of solid catalyst component.
- the amount of Ti can range from 0.5 to 5% and more preferably from 0.7 to 3% wt with respect to the total weight of solid catalyst component.
- the internal electron donor is selected from the group consisting of ethers, amines, silanes, carbamates, ketones, esters of aliphatic acids, alkyl and aryl esters of optionally substituted aromatic polycarboxylic acids, diol derivatives chosen among monoesters monocarbamates and monoesters monocarbonates or mixtures thereof.
- the internal donor is selected from alkyl and aryl esters of optionally substituted aromatic polycarboxylic acids
- preferred donors are the esters of phthalic acids.
- Preferred esters of aliphatic acids are selected from malonic, glutaric, maleic and succinic acids. Specific examples of such esters are n-butylphthalate, di-isobutylphthalate, and di- n-octylphthalate.
- the ethers can be selected from the 1,3 diethers of the formula (I): wherein R, R 1 , R n , R 111 , R IV and R v are equal to or different from each other, and are hydrogen or hydrocarbon radicals having from 1 to 18 carbon atoms; and R VI and R vn are equal to or different from each other, and have the same meaning of R-R v except that R VI and R vn cannot be hydrogen; one or more of the R-R vn groups can be linked to form a cycle.
- the 1,3-diethers in which R VI and R vn are selected from C1-C4 alkyl radicals are particularly preferred.
- ethers are the C2-C20 aliphatic ethers and in particular, cyclic ethers preferably having 3-5 carbon atoms cyclic ethers such as tetrahydrofurane, methyl-tetrahydrofurane, dioxane.
- Preferred esters are ethylacetate and methyl formiate. Among them tetrahydrofurane and ethylacetate are the most preferred.
- the final amount of electron donor compound in the solid catalyst component may range from 0.5 to 40 wt% by weight preferably in the range from 1 to 35 wt% with respect to the total weight of the solid catalyst component.
- the solid catalyst component obtained according to the present disclosure preferably shows a surface area (by B.E.T. method) generally between 20 and 500 m 2 /g and preferably between 50 and 400 m 2 /g, and a total porosity (by B.E.T. method) greater than 0.2 cm 3 /g, preferably between 0.3 and 0.6 cm 3 /g.
- the porosity (Hg method) due to pores with radius up to 10.000A generally ranges from 0.3 to 1.5 cm 3 /g, preferably from 0.45 to 1.0 cm 3 /g.
- the solid catalyst component prepared according to the process of the present disclosure is converted into catalyst for the polymerization of olefins by reacting it with alkylaluminum compounds (ii) according to known methods.
- the alkylaluminum compound (ii) is preferably chosen from the trialkyl aluminum compounds such as for example triethylaluminum, triisobutylaluminum, tri-n- butylaluminum, tri-n-hexylaluminum, tri-n-octylaluminum. It is also possible to use alkylaluminum halides, alkylaluminum hydrides or alkylaluminum sesquichlorides, such as AlEt2Cl and AhEtsCh, possibly in mixture with the above cited trialkylaluminums.
- the Al/Ti molar ratio is such that there is an excess of Al.
- the Al/Ti molar ratio may range between 50:1 and 2000: 1, preferably between 50: 1 and 500: 1.
- an external electron-donor compound (iii) can be used. It is preferably selected from silicon compounds, ethers, esters, amines, heterocyclic compounds and 2,2,6,6-tetramethylpiperidine and ketones.
- Another class of preferred external donor compounds is that of silicon compounds of formula (R6)a(R7)bSi(ORs)c, where a and b are integers from 0 to 2, c is an integer from 1 to 4 and the sum of (a+b+c) is 4; Re, R7, and Rs, are alkyl, cycloalkyl or aryl radicals with 1-18 carbon atoms optionally containing heteroatoms.
- Examples of such preferred silicon compounds are methylcyclohexyldimethoxysilane (C donor), diphenyldimethoxysilane, methyl-t- butyldimethoxysilane, dicyclopentyldimethoxysilane (D donor), diisopropyldimethoxysilane, (2-ethylpiperidinyl)t-butyldimethoxysilane, (2- ethylpiperidinyl)thexyldimethoxysilane, (3,3,3-trifluoro-n-propyl)(2- ethylpiperidinyl)dimethoxysilane, methyl(3,3,3-trifluoro-n-propyl)dimethoxysilane.
- C donor methylcyclohexyldimethoxysilane
- D donor dicyclopentyldimethoxysilane
- diisopropyldimethoxysilane (2-ethy
- the silicon compounds in which a is 0 and c is 3, R7 is a branched alkyl or cycloalkyl group, optionally containing heteroatoms, and Rs is methyl are also preferred.
- Examples of such preferred silicon compounds are cyclohexyltrimethoxysilane, t- butyltrimethoxysilane and thexyltrimethoxysilane.
- the external electron donor compound is used in such an amount to give a molar ratio organoaluminum compound to said external electron donor compound of from 0.1 : 1 to 500: 1, preferably from 1 : 1 to 300: 1, and more preferably from 3: 1 to 100: 1.
- the molar ratio of compound (iv) to alkylaluminum compound (ii) introduced into the polymerization reactor is from 0.01 to 5, preferably from 0.05 to 3.5, more preferably from 0.1 to 2.5.
- the catalyst components (i)-(iii) are preferably contacted with a liquid inert hydrocarbon solvent such as, e.g., propane, n-hexane or n-heptane, at a temperature below about 60°C and preferably from about 0 to 30°C for a time period of from about six seconds to 60 minutes.
- a liquid inert hydrocarbon solvent such as, e.g., propane, n-hexane or n-heptane
- the above catalyst components (i), (ii) and optionally (iii) are fed to a precontacting vessel, in amounts such that the weight ratio (ii/(i) is in the range of 0.1-10 and if the compound (iii) is present, the weight ratio (ii)/(iii) is preferably as defined above.
- the said components are pre-contacted at a temperature of from 10 to 20°C for 1-30 minutes.
- the precontact vessel can be either a stirred tank or a loop reactor.
- the precontacted catalyst is then fed to the prepolymerization reactor where step (ii) takes place.
- the prepolymerization step is carried out in a first reactor selected from a loop reactor or a continuously stirred tank reactor.
- the prepolymerization can be carried out either in gas-phase or in liquid-phase. Preferably it is carried out in liquid-phase.
- the liquid medium comprises liquid alpha-olefin monomer(s), optionally with the addition of an inert hydrocarbon solvent.
- Said hydrocarbon solvent can be either aromatic, such as toluene, or aliphatic, such as propane, hexane, heptane, isobutane, cyclohexane and 2,2,4-trimethylpentane.
- the amount of hydrocarbon solvent, if any, is lower than 40% by weight with respect to the total amount of alpha-olefins, preferably lower than 20% by weight.
- step (ii) is carried out in the absence of inert hydrocarbon solvents.
- the average residence time in this reactor generally ranges from 2 to 40 minutes, preferably from 10 to 25 minutes.
- the temperature is comprised between 10°C and 50°C, preferably between 20°C and 40°C. Adopting these conditions allows to obtain a pre-polymerization degree in the preferred range from 60 to 800g per gram of solid catalyst component, preferably from 150 to 500 g per gram of solid catalyst component.
- the prepolymerization step is preferably characterized by a low concentration of solid in the slurry, typically in the range from 50 g to 300 g of solid per liter of slurry.
- the process of the present disclosure can be carried out in any polymerization plant comprising one or more liquid-phase and/or gas-phase polymerization reactors.
- liquid-phase reactors are loop reactors and continuously stirred tank reactors (CSTR).
- gas-phase reactors include fluidized bed reactors, stirred bed reactors and reactors having two interconnected polymerization zones as described in EP 0782587 and EP 1012195.
- the process of the present disclosure can be carried in two or more cascade reactors, giving rise to a sequential multistage polymerization process. For instance, a fluidized bed reactor can be used to prepare a first polymer component, which is successively fed to a gas-phase reactor having two interconnected polymerization zones to prepare a second and a third polymer component.
- a first fluidized bed reactor can be used to prepare a first polymer component, which is successively fed to a second fluidized bed reactor to prepare a second polymer component and then to a third fluidized bed reactor to prepare a third polymer component. Accordingly, an olefin polymer endowed with a multi-modal molecular weight distribution can be obtained, as well as an olefin copolymer comprising two or more components having a different comonomer content.
- the polymerization process is carried out as gas-phase polymerization, i.e. by a process in which the solid polymers are obtained from a gas-phase of the monomer or the monomers.
- gas-phase polymerizations may be carried out at pressures of from 0.1 to 20 MPa, or from 0.5 to 10 MPa, or from 1.0 to 5 MPa and polymerization temperatures from 40 to 150°C or from 65 to 125°C.
- Gas-phase polymerization reactors can be, for example, horizontally or vertically stirred reactor, fluidized bed gas-phase reactors or multizone circulating reactors.
- Fluidized-bed polymerization reactors are reactors in which the polymerization takes place in a bed of polymer particles which is maintained in a fluidized state by feeding in gas at the lower end of a reactor, for example below a gas distribution grid having the function of dispensing the gas flow, and taking off the gas again at its upper end. The reactor gas is then returned to the lower end to the reactor via a recycle line equipped with a compressor and a heat exchanger.
- the circulated reactor gas is, for example, a mixture of the olefins to be polymerized, inert gases such as nitrogen and/or lower alkanes such as ethane, propane, butane, pentane or hexane and optionally a molecular weight regulator such as hydrogen.
- nitrogen or propane can be used as inert gas, if appropriate in combination with further lower alkanes.
- the velocity of the reactor gas has to be sufficiently high firstly to fluidize the mixed bed of finely divided polymer present in the tube serving as polymerization zone and secondly to remove the heat of polymerization effectively.
- the polymerization can also be carried out in a condensed or super-condensed mode, in which part of the circulating reaction gas is cooled to below the dew point and returned to the reactor separately as a liquid and a gas-phase or together as a two-phase mixture in order to make additional use of the enthalpy of vaporization for cooling the reaction gas.
- Multizone circulating reactors are gas-phase reactors in which two polymerization zones are linked to one another and the polymer is passed alternately a plurality of times through these two zones.
- Such reactors are, for example, described in WO 97/04015 Al and WO 00/02929 Al and have two interconnected polymerization zones, a riser, in which the growing polymer particles flow upward under fast fluidization or transport conditions and a downcomer, in which the growing polymer particles flow in a densified form under the action of gravity.
- the polymer particles leaving the riser enter the downcomer and the polymer particles leaving the downcomer are reintroduced into the riser, thus establishing a circulation of polymer between the two polymerization zones and the polymer is passed alternately a plurality of times through these two zones.
- the gas mixture leaving the riser and entraining the polymer particles can be partially or totally prevented from entering the downcomer.
- This can for example be achieved by feeding a barrier fluid in form of a gas and/or a liquid mixture into the downcomer, in the upper part thereof according to an embodiment.
- the barrier fluid should have an appropriate composition, different from that of the gas mixture present in the riser.
- the amount of added barrier fluid can be adjusted in a way that an upward flow of gas countercurrent to the flow of the polymer particles is generated, particularly at the top thereof, acting as a barrier to the gas mixture entrained among the particles coming from the riser. In this manner it is possible to obtain two different gas composition zones in one multizone circulating reactor. Furthermore it is also possible to introduce make-up monomers, comonomers, molecular weight regulator such as hydrogen and/or inert fluids at any point of the downcomer, below the barrier feeding point according to an embodiment. Thus, it is also easily possible to create varying monomer, comonomer and hydrogen concentrations along the downcomer resulting in a further differentiation of the polymerization conditions.
- the polymerization reactor shown in Figure 1 comprises a first polymerization zone 1 (riser), wherein the polymer particles flow upward under fast fluidization conditions along the direction of the arrow A and a second polymerization zone 2 (downcomer), wherein the polymer particles flow downward under the action of gravity along the direction of the arrow B.
- the upper portion of the riser 1 is connected to a solid/gas separator 3 by the interconnection section 4.
- the separator 3 removes the major part of the unreacted monomers from the polymer particles and the polymer withdrawn from the bottom of separator 3 enters the top portion of the downcomer 2.
- the separated unreacted monomers, optionally together with polymerization diluents, such as propane, flow up to the top of separator 3 and are successively recycled to the bottom of the riser 1 via the recycle line 5.
- a mixture comprising one or more olefin monomers, hydrogen as the molecular weight regulator, propane as the polymerization diluent, is fed to the polymerization reactor via one or more lines M, which are suitably placed along the gas recycle line 5, according to the knowledge of the person skilled in art.
- the catalyst components are continuously introduced into the riser 1 via line 6.
- the produced polymer can be discharged from the reactor via a line 7, which can be placed on the lower portion of the downcomer 2 so that, due to the packed flow of densified polymer, the quantity of gas entrained with the discharged polymer is minimized.
- a control valve not shown in Figure 2
- Additional polymer discharge lines with respect to line 7 can be placed in the bottom part of the downcomer.
- the polymerization reactor further comprises a transport section 8 connecting the bottom of downcomer 2 with the lower region of the riser 1.
- the bottom of the downcomer 2 converges into a slight restriction 9.
- a control valve 10 with an adjustable opening can be placed within the restriction 9.
- the flow rate Fp of polymer continuously circulated between the downcomer 2 and the riser 1 is adjusted by the level of opening of the control valve 10.
- the control valve 10 may be a mechanical valve, such as a butterfly valve, a ball valve, etc.
- a stream of dosing gas is fed into the lower part of the downcomer 2 by means of a line 11 placed at a short distance above the restriction 9.
- the dosing gas to be introduced through line 10 can be taken from the recycle line 5.
- the flow Fp of polymer particles circulated between downcomer 2 and riser 1 can be adjusted by varying the opening of the control valve 10 at the bottom of the downcomer and/or by varying the flow rate of the dosing gas entering the downcomer via line 11.
- the flow rate of dosing gas is adjusted by means of a control valve 18, which is suitably arranged on line 11.
- the transport section 8 is designed as a bend descending from the bottom of downcomer 2 up to the lower region of the riser 1. Furthermore, a carrier gas is introduced via line 12 at the inlet of the transport section 8. The flow rate of carrier gas is adjusted by means of a control valve 13, which is suitably arranged on line 12. [0073] Also the carrier gas is taken from the gas recycle line 5. Specifically, the gas recycle stream of line 5 is first subjected to compression by means of a compressor 14 and a minor percentage of the recycle stream passes through line 12, thus entering the transport section 8 and diluting the solid phase of polymer flowing through the transport section 8.
- the major part of the recycle stream, downstream the compressor 14, is subjected to cooling in a heat exchanger 15 and successively is introduced via line 16 at the bottom of the riser 1 at a high velocity, such to ensure fast fluidization conditions in the polymer bed flowing along the riser 1.
- the carrier gas merges with the densified polymer coming from downcomer 2 at the inlet portion of transport section 8, after exiting the slits of the gas distribution grid 17.
- the gas distribution grid 17 is formed by a plurality of trays fixed to the transport section 8 in a way to form slits in the overlapping area of adjacent trays.
- a flow rate of compound (iv) is metered into the reactor at the bottom of the riser and/or at any point in the riser 1, particularly at the top of the riser (flow rate A5).
- the antistatic composition can alternatively or additionally be metered to one or more positions along the height of the downcomer via nozzles.
- the antistatic composition flow rate A2 in line 22 is metered by one or more valves 23 and then pre-dispersed either in the liquid monomer L as described above, or alternatively in a fraction of recycle gas taken from recycle line 5 via line 24.
- the carrier gas merges with the densified polymer coming from downcomer 2 at the inlet portion of transport section 8, after exiting the slits of a gas distribution grid 17.
- the top end of the distribution grid 17 is coincident with the inlet of the transport section 8 and the distribution grid 17 extends along the bending of the transport section 8 for an angle of 60°.
- the gas distribution grid 17 is formed by a plurality of trays fixed to the transport section 8 in a way to form slits in the overlapping area of adjacent trays. A detailed description of the gas distribution grid 17 can be found in WO 2012/031986.
- the polymerization reactor can be operated by properly adjusting the polymerization conditions and the monomers concentration in the riser and in the downcomer, so as to produce a wide variety of bimodal homopolymers and random copolymers.
- the gas mixture entraining the polymer particles and coming from the riser can be partially or totally prevented from entering the downcomer, so as to polymerize two different monomers compositions in the riser and the downcomer.
- This effect may be achieved by feeding a gaseous and/or a liquid barrier stream through a line placed in the upper portion of the downcomer.
- the barrier stream should have a composition different from the gas composition present inside the riser.
- the flow rate of the barrier stream can be adjusted, so that an upward flow of gas counter-current to the flow of the polymer particles is generated, particularly at the top of the downcomer, thus acting as a barrier to the gas mixture coming from the riser.
- an upward flow of gas counter-current to the flow of the polymer particles is generated, particularly at the top of the downcomer, thus acting as a barrier to the gas mixture coming from the riser.
- the different or else identical polymerization processes can also, if desired, be connected in series and thus form a polymerization cascade.
- a parallel arrangement of reactors using two or more different or identical processes is also possible.
- the gas-phase polymerization processes according to the present disclosure are carried out in the presence of an alkane having from 3 to 5 carbon atoms as polymerization diluent, e.g. in the presence of propane.
- the process of the present disclosure provides a possibility for preparing an olefin polymer by gas-phase polymerization in a polymerization reactor in which the formation of polymer agglomerates in the polymerization reactors and fluctuations in the fluid-dynamics of the reactor are prevented or considerably reduced.
- compound (iv) reduces the tendency of the olefin polymer particles to stick to the reactor walls. This does not only reduce the risk of forming chunks or wall sheeting, which mostly leads to an unavoidable shut-down of the polymerization reactor because of plugging the discharge line, but also improves the fluid-dynamics of the reactor and avoids their fluctuations.
- the level of electrostatic charges observed in a multizone circulating reactor may be sensitive to the molecular weight of the olefin polymer particles transferred into it.
- the process of the present disclosure is used in a multizone circulating reactor for producing polymer particles having a MFR2.16 at a temperature of 190°C under a load of 2.16 kg of less than 60 g/10 min.
- the antistatic composition in case of a cascade process comprising two or more fluidized-bed gas-phase reactors, can be added into the bed of each reactor or to the polymer discharge downstream each reactor or else before the first reactor.
- compound (iv) of the present disclosure does not only result in a process for the polymerization of olefins which is simple to carry out, but the polymerization process has also a good operability. That means that the tendency for forming polymer deposits on the rector wall, i.e. reactor fouling, for forming lumps and for forming fines, i.e. for forming very small sticky polyolefin particles, is reduced. Moreover, the activity of the catalyst may be improved or at least not impaired and the product properties of the prepared polyolefins are not deteriorated.
- 13C NMR spectra were acquired on a Bruker AV-600 spectrometer equipped with cryoprobe, operating at 160.91 MHz in the Fourier transform mode at 120°C.
- the peak of the SPP carbon (nomenclature according to “Monomer Sequence Distribution in Ethylene-Propylene Rubber Measured by 13C NMR. 3. Use of Reaction Probability Mode ” C. J. Carman, R. A. Harrington and C. E. Wilkes, Macromolecules, 1977, 10, 536) was used as internal reference at 29.9 ppm.
- the samples were dissolved in l,l,2,2-tetrachloroethane-d2 at 120°C with a 8 % wt/v concentration.
- Each spectrum was acquired with a 90° pulse, 15 seconds of delay between pulses and CPD to remove 1H-13C coupling. 512 transients were stored in 32K data points using a spectral window of 9000 Hz.
- Xylene Solubles [0095] Determined as follows: 2.5 g of polymer and 250 ml of xylene were introduced in a glass flask equipped with a refrigerator and a magnetical stirrer. The temperature was raised in 30 minutes up to the boiling point of the solvent. The clear solution so obtained was then kept under reflux and stirring for further 30 minutes. The closed flask was then kept in thermostatic water bath at 25° C for 30 minutes. The so formed solid was filtered on quick filtering paper. 100 ml of the filtered liquid was poured in a previously weighed aluminium container, which was heated on a heating plate under nitrogen flow, to remove the solvent by evaporation. The container was then kept in an oven at 80° C under vacuum until constant weight was obtained. The weight percentage of polymer soluble in xylene at room temperature was then calculated.
- the melting point has been measured by using a DSC instrument according to ISO 11357-3, at scanning rate of 20°C/min both in cooling and heating, on a sample of weight between 5 and 7 mg., under inert N2 flow. Instrument calibration made with Indium.
- a Ziegler-Natta catalyst system was used as the polymerization catalyst, comprising:
- Pre-polymerization [0107] The catalyst system withdrawn from the pre-contacting vessel was continuously fed to the prepolymerization loop reactor together with a liquid stream of propylene and propane. The pre-polymerization occurred at a temperature of 20°C.
- a polypropylene slurry was continuously discharged from the loop reactor and fed to the first fluidized bed reactor where propylene was polymerized using H2 as the molecular weight regulator and in the presence of propane as inert diluent.
- the polymerization was carried out according to the conditions reported in table 1.
- the polymer discharged from the first reactor was then conveyed to the second reactor where ethyl ene/propylene copolymerization was carried out.
- the PPA (a mixture of commercially available 40%wt mono and 60%wt diglycerol esters of octanoic and decanoic acid diluted in hexane) was fed via propylene carrier, partly to the first and second gas-phase reactors via fluidization gas and partly to the polymer discharged from the first reactor before being introduced into the second.
- the flow rate of the PPA feed was such as to obtain in the polymer the amounts of PPA indicated in Table 2.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
Abstract
A process for the preparation of polyolefins is carried out in the presence of (i) a Ziegler- Natta catalyst, (ii) an aluminum alkyl cocatalyst, optionally (iii) an external donor, and at least 50 ppm, based on the amount of polyolefin produced, of a compound selected from mono or diglycerol ester derivatives of monocarboxylic acids.
Description
PROCESS FOR THE POLYMERIZATION OF OLEFINS
FIELD OF THE DISCLOSURE
[0001] The present disclosure relates to a process for the preparation of polyolefins. In particular, it relates to a gas-phase process for preparing polyolefins, especially polypropylene, carried out in the presence of specific ester compounds.
BACKGROUND OF THE DISCLOSURE
[0002] Polymerization process aids (PPAs) are used in processes for the polymerization of olefins to in order to reduce or eliminate wall sheeting and formation of polymer agglomerates in the polymerization reactor or in downstream equipments such as degassing and recovery vessels. In the context of olefin polymerization, PPAs are also called, depending on the specific function, antifouling agents, antistatic agents, activity inhibitors, productivity inhibitors or kinetic modifiers. PPAs may comprise compounds which have polar functional groups such as acid or ester groups, amine or amide groups or hydroxyl or ether groups. Examples of PPAs are polysulfone copolymers, polymeric polyamines, polyalcohols, hydroxyesters of polyalcohols, salts of alkylarylsulfonic acids, polysiloxanes, alkoxyamines, polyglycol ethers, etc.
[0003] EP560035A1 for example, discloses a polymerization process in which PPAs are used to eliminate or reduce the build-up of polymer particles on the walls of a gas-phase polymerization reactor. Those PPAs are capable of selectively inhibiting the polymerization on polymer particles smaller than 850 pm, the latter being considered responsible for fouling problems and polymer sheeting. Among the list of PPAs, hydroxyesters, having at least two free hydroxyl groups, obtained from carboxylic acids having at least 4 and preferably from 8 to 22 carbon atoms and from polyalcohols are mentioned. Specific examples were carried out with glycerol monostearate and sorbitan monooleate in liquid propylene polymerization in the presence of a ZN catalyst comprising a Mg compound supported Ti-based solid component including diisobutylphthalate as internal donor and a silane as external donor. No indication is given about the use of glycerol esters of fatty acids in gas-phase polymerization of propylene.
[0004] W02012/041811 describe the use of a method for feeding PPAs together with catalysts in the polymerization reactor. Glycerol monostearate was used as PPA in a gasphase reactor for ethylene polymerization.
[0005] Depending on the molecular structure, reaction conditions and reactor type, the effectiveness of PPAs may change. Also, the type of polyolefin produced may play a role. For example, sticky products such as propyl ene/ethylene rubbery copolymers have higher tendency to adhere to reactor walls and create fouling. The problem may be more evident for propylene-ethylene copolymer particles with small size (less than 500 pm) and relatively high ethylene content. One reason for that may be due to the higher reactivity of ethylene compared to propylene. In this perspective, propylene-ethylene copolymer particles with small size and higher ethylene content than the average, may be the result of an increased polymerization activity which also generates an excess of heat which, in turn, may render the copolymer softer and prone to stick to reactor walls. Under this situation, keeping the ethylene content on small polymer size under control may be an indication of PPA effectiveness.
[0006] It is thus desirable to provide compounds that are highly efficient in preventing reactor wall fouling and formation of polymer agglomerates in the polymerization reactor or in downstream equipment and, at the same time, do not overly impact on the catalyst activity, are easy to be handled and fed to the polymerization reactor while being not prone to lower the quality of the polymer product or to impact on the polymerization hardware.
SUMMARY OF THE DISCLOSURE
[0007] The present disclosure provides a process for the preparation of polyolefins, carried out in the presence of (i) a Ziegler-Natta catalyst, (ii) an aluminum alkyl cocatalyst, optionally (iii) an external donor, and (iv) a compound (polymerization process aid (PPA)) comprising mono or diglycerol ester derivatives of saturated C6-C12, preferably Ce-Cio monocarboxylic acids said component (iv) being used in an amount of at least 50 ppm based on the amount of polyolefin produced.
DETAILED DESCRIPTION OF THE DISCLOSURE
[0008] Preferably, when the PPA comprises also mono or polyglycerol fatty acid esters other than mono or diglycerol ester derivatives of saturated Ce-Cio, preferably Cs- Cio monocarboxylic acids, the amount of mono or diglycerol ester derivatives of saturated Ce-Cio, preferably Cs-Cio monocarboxylic acids, is higher than 50%wt, preferably more than 55%wt and especially in the range 60-90%wt with respect to the total amount of mono or polyglycerol fatty acid esters.
[0009] In a preferred embodiment, the PPA comprises one or more of mono or diglycerol ester derivatives of saturated Ce-Cio, more preferably Cs-Cio, monocarboxylic acids and mixture thereof. In a particularly preferred embodiment, the PPA comprises, for
more than 50% wt, preferably more than 55%wt and especially in the range 60-90%wt based on the total amount of fatty acid mono or polyglycerol esters, one or more of glycerol monoester or diester of octanoic acid, glycerol monoester or diester of decanoic acid, glycerol diester mixed octanoic and decanoic acid, diglycerol mono, di- or triester of octanoic acid, diglycerol mono, di- or triester of decanoic acid, diglycerol di or triester of mixed octanoic and decanoic acid and any mixture thereof.
[0010] In a most preferred embodiment, PPA comprises, for more than 50% wt, preferably more than 55%wt and especially in the range 60-90%wt based on the total amount of fatty acid glycerol esters, glycerol mono octanoate or a mixture comprising glycerol diester of octanoic acid, glycerol diester of mixed octanoic and decanoic acid, diglycerol mono ester of octanoic acid and diglycerol mono ester of decanoic acid and mixture thereof.
[0011] The mono or diglycerol ester derivatives of saturated C6-C12, preferably Cs-Cio monocarboxylic can be obtained by extraction and/or treatment from natural sources such as glycerolysis of commercial edible oils into diglycerides, and are commercially available either pure or in mixtures. It is also possible that minor amounts of other components such as non-esterified mono or polyglycerol, remains in the mixture. It is also possible to synthesize substantially pure glycerol esters such as glycerol caprylate via esterification reaction as described in CN107935851B.
[0012] Such products, also used in cosmetics field, are widely available in the market with different purity. As an example, glycerol monoester of decanoic acid and glycerol monoester of octanoic acid are sold by SigmaAldrich, glycerol diester mixed octanoic and decanoic acid is sold by Dayang Chem and diglycerol esters of octanoic acid and decanoic acid is sold by Ality Chemical Corporation.
[0013] In the context of the present disclosure they can be used as such or with the aid of a dispersing medium which is preferably selected from liquid non-aqueous solvents and in particular from liquid hydrocarbons.
[0014] In a preferred embodiment, the PPA is liquid at room temperature.
[0015] According to preferred embodiments of the present disclosure, the amount of PPA introduced into the polymerization reactor is from 70 to 2000 ppm per weight, or from 150 to 1500 ppm per weight, or from 200 to 1000 ppm per weight referring to the weight of the prepared polyolefin.
[0016] PPA of the present disclosure can be provided to the polymerization process according to different modes of operation.
[0017] In a method of operation, PPA can be fed to the polymerization reactor in a flow of saturated or unsaturated hydrocarbon having from 2 to 6 carbon atoms that, according to an embodiment, can be a monomer, such as propylene, or, according to another embodiment, an alkane, such as propane. A mixture of saturated and unsaturated hydrocarbons can be used as well. The monomer and/or the alkane can be either in liquid or gas form. According to an embodiment, PPA may be diluted and homogenized with a solvent at short distance from the injection point by means of customary mixing/homogenizing/dispersing systems, such as static mixers so as increase the volume and make the injection smoother and more reliable.
[0018] PPA of the present disclosure can be introduced into the reactor in multiple ways. The feeding can occur directly into the reactor and/or into a line leading to the reactor and/or into a line exiting the reactor. According to different embodiments, PPA can be fed:
• upstream or into a catalyst precontacting vessel, i.e. a vessel in which the catalyst components are brought into contact with each other;
• upstream or into a prepolymerization reactor;
• at any other point upstream a polymerization reactor;
• in case of a cascade process with one or more liquid or gas phase reactors, at any point in-between two reactors;
• suitably distributed into a reactor or in any recirculation flow thereof, for example into the bed of a fluidized bed gas phase reactor or into the bed of a fast fluidization bed or of a packed bed of -respectively - a riser or downer of a multizone circulating reactor;
• in the polymer discharge out of a polymerization reactor;
• in the unreacted monomer separation and finishing sections downstream the polymerization reactor(s).
[0019] The present disclosure provides a process for the polymerization of olefins, such as 1 -olefins, i.e. hydrocarbons having terminal double bonds, without being restricted thereto. Typical 1 -olefins are linear or branched 1 -alkenes having from 2 to 12 carbon
atoms, in particular linear 1 -alkenes having from 2 to 10 carbon atoms such as ethylene, propylene, 1 -butene, 1 -pentene, 1 -hexene, 1 -heptene, 1 -octene, 1 -decene or branched 1- alkenes having from 2 to 10 carbon atoms such as 4-methyl-l -pentene, conjugated and nonconjugated dienes such as 1,3 -butadiene, 1,4-hexadiene or 1,7-octadiene or vinyl-aromatic compounds such as styrene or substituted styrene. It is also possible to polymerize mixtures of various 1 -olefins. Olefins that can be polymerized with the process of the present disclosure include those in which the double bond is part of a cyclic structure which can have one or more ring systems. Examples are cyclopentene, norbomene, tetracyclododecene or methylnorbornene or dienes such as 5-ethylidene-2-norbornene, norbornadiene or ethylnorbornadiene. It is also possible to polymerize mixtures of two or more olefins.
[0020] According to a preferred embodiment, the process can be used for the homopolymerization or copolymerization of ethylene or for the homopolymerization or copolymerization of propylene. Homo and/or copolymerization of propylene is preferred.
[0021] According to a preferred embodiment, comonomers for use in ethylene polymerization are 1-alkenes having from 3 to 8 carbon atoms such as 1 -butene, 1 -pentene, 1 -hexene and/or 1 -octene in amount of up to 20 wt.% or from 0.01 wt.% to 15 wt.%, or from 0.05 wt.% to 12 wt.% based on the total weight of the final polymer. According to another preferred embodiment, comonomers for use in propylene polymerization are ethylene and/or 1-butene and/or 1-hexene in amount of up to 40 wt.% or from 0.5 wt.% to 35 wt.% based on the total weight of the final polymer.
[0022] A particular type of propyl ene/ethylene copolymer preferably contains from 15 to 80% of ethylene, preferably from 20 to 70%wt. Such copolymers are typically rubbery and tend to adhere to the reactor walls. In particular, reactivity of fine particles is considered very important to be controlled. Since ethylene is more reactive than propylene, capability for the compound (iv) to lower the content of ethylene in the fine particles is considered also beneficial.
[0023] The process of the present disclosure allows the preparation of any types of common olefin polymers. According to an embodiment, the prepared olefin polymers can be broad molecular weight olefin polymers and, particularly, multimodal olefin polymers whereby the term multimodal refers to the modality of the molecular weight distribution. As used in the art, and also used herein, multimodal shall include bimodal. Such polymers can be obtained from polymerizing olefins in a cascade of two or more polymerization reactors or in different zones of a multizone reactor under different reaction conditions. Thus, the "modality" indicates how many different polymerization conditions were utilized to prepare the polyolefin, independently whether this modality of the molecular weight
distribution can be recognized as separated maxima in a gel permeation chromatography (GPC) curve or not. In addition to the molecular weight distribution, the olefin polymer can also have a comonomer distribution.
[0024] The polymerization of olefins is carried out using titanium-based Ziegler-Natta- catalysts.
[0025] According to preferred embodiment, Ziegler-Natta catalysts used in the process of the present disclosure comprise:
(i) a solid catalyst component comprising Mg, Ti, a halogen and optionally an electron donor compound (internal donor),
(ii) an alkylaluminum compound, and
(iii) optionally, an electron-donor compound (external donor).
[0026] The presence of an internal donor in catalyst component (i) is particularly preferred when propylene is homo or copolymerized.
[0027] The solid catalyst component (i) can be prepared by one or more steps (a) carried out at a temperature ranging from 0 to 150°C in which a Mg based compound of formula (MgClmX2-m) LB, where m ranges from 0 to 2, n ranges from 0 to 6, X is, independently halogen, R1, OR1, -OCOR1 or O-C(O)-OR1 group, in which R1 is a C1-C20 hydrocarbon group, and LB is a Lewis base, is reacted with a liquid medium comprising a Ti compound, having at least a Ti-Cl bond, in an amount such that the Ti/Mg molar ratio is greater than 3.
[0028] The liquid medium preferably comprises a titanium compound of formula Ti(OR1)q-yCly, where q is the valence of titanium, y is a number between 1 and q and R1 is a C1-C20 hydrocarbon group.
[0029] Among them, particularly preferred are titanium polyhalogenated compounds such as titanium tetrahalides or halogenalcoholates. Preferred specific titanium compounds are TiCL and Ti(OEt)Ch.
[0030] The liquid medium comprising the Ti compound can be a mixture of the Ti compound in another liquid diluent. Preferred diluents are hydrocarbons, optionally chlorinated, that are liquid at room temperature. In a very preferred embodiment the liquid medium consists of the liquid titanium compound.
[0031] The magnesium based compound used as a starting compound in the first of the one or more steps (a) is preferably selected among adducts of formula MgCh’nR'OH, where n is a number between 0.1 and 6, and R1 is a hydrocarbon radical having 1-18 carbon atoms. Preferably, n ranges from 1 to 5 and more preferably from 1.5 to 4.5.
[0032] The adduct can be suitably prepared by mixing alcohol and magnesium chloride, operating under stirring conditions at the melting temperature of the adduct (100- 130°C). Then, the adduct is mixed with an inert hydrocarbon immiscible with the adduct thereby creating an emulsion which is quickly quenched causing the solidification of the adduct in the form of spherical particles. Examples of spherical adducts prepared according to this procedure are described in US 4,399,054 and US 4,469,648.
[0033] Another useable method for the spherulization is the spray cooling described for example in US 5,100,849 and US 4,829,034. Adducts having the desired final alcohol content can be obtained by directly using the selected amount of alcohol during the adduct preparation.
[0034] The above mentioned adduct can be directly reacted with the Ti compound or it can be previously subjected to thermal controlled dealcoholation (80-130°C) so as to obtain an adduct in which the number of moles of alcohol is lowered and its porosity increased. When the dealcoholation is carried out, it generally brings the number of moles of alcohol per mole of Mg to less than 3, preferably between 0.1 and 2.5.
[0035] The reaction between the Mg based compound, in particular the MgCh-alcohol adduct and the Ti compound can be carried out by suspending the Mg based compound in large excess of cold TiCh (generally 0°C); the mixture is heated up to a temperature ranging from 60-140°C and kept at this temperature for 0.1-4 hours, preferably 0.5-2 hours. After that time, stirring is discontinued and after the settlement of the solid particles the liquid phase is removed.
[0036] This reaction step (a) can be carried out one or more times under identical or different conditions. For example, the temperature and duration of treatment can be changed. In a preferred embodiment, the number of steps (a) is comprised between 1 and 3.
[0037] If used, the electron donor compound can be added in the desired amount during one or more of the reaction steps (a) between the Mg based compound and the liquid Ti compound.
[0038] Preferably the electron donor compound is added at least during the first step (a) of reaction between the Mg based compound and the Ti compound. In some cases such a treatment can be repeated one or two additional times.
[0039] In another embodiment, the electron donor compound, as described in W02004/106388, can be added as a fresh reactant to the solid intermediate catalyst component obtained by the above described reaction between the adduct and the Ti compound.
[0040] In a particular embodiment, the reaction step (a) is carried out by a continuous feeding of liquid Ti compound, preferably TiCh, in an apparatus and under conditions that are described in W002/48208. Under this embodiment the Mg based compound is fed batchwise while a continuous stream of liquid Ti compound with the desired temperature profile is fed and a liquid phase containing dissolved reaction product is continuously withdrawn. Under these basic conditions the organic coloring agent and, optionally, the electron donor can be added at any time during the feeding of the Ti compound.
[0041] The particles of solid catalyst component have substantially spherical morphology and average diameter ranging between 5 and 150 pm, preferably from 20 to 100 pm and more preferably from 30 to 90 pm when the polymerization process is carried out in gas-phase. As particles having substantially spherical morphology, those are meant wherein the ratio between the greater axis and the smaller axis is equal to or lower than 1.5 and preferably lower than 1.3.
[0042] In general the amount of Mg preferably ranges from 8 to 30% more preferably from 10 to 25%wt with respect to the total weight of solid catalyst component.
[0043] The amount of Ti can range from 0.5 to 5% and more preferably from 0.7 to 3% wt with respect to the total weight of solid catalyst component.
[0044] When used, the internal electron donor is selected from the group consisting of ethers, amines, silanes, carbamates, ketones, esters of aliphatic acids, alkyl and aryl esters of optionally substituted aromatic polycarboxylic acids, diol derivatives chosen among monoesters monocarbamates and monoesters monocarbonates or mixtures thereof.
[0045] When the internal donor is selected from alkyl and aryl esters of optionally substituted aromatic polycarboxylic acids preferred donors are the esters of phthalic acids. Preferred esters of aliphatic acids are selected from malonic, glutaric, maleic and succinic acids. Specific examples of such esters are n-butylphthalate, di-isobutylphthalate, and di- n-octylphthalate.
[0046] Preferably, the ethers can be selected from the 1,3 diethers of the formula (I):
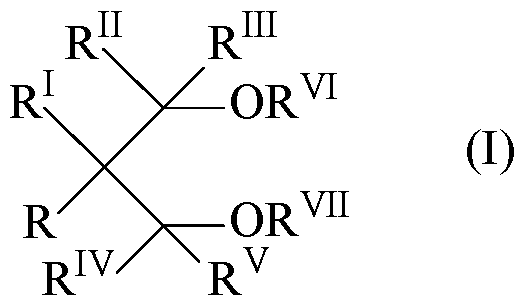 wherein R, R1, Rn, R111, RIV and Rv are equal to or different from each other, and are hydrogen or hydrocarbon radicals having from 1 to 18 carbon atoms; and RVI and Rvn are equal to or different from each other, and have the same meaning of R-Rv except that RVI and Rvn cannot be hydrogen; one or more of the R-Rvn groups can be linked to form a cycle. The 1,3-diethers in which RVI and Rvn are selected from C1-C4 alkyl radicals are particularly preferred.
wherein R, R1, Rn, R111, RIV and Rv are equal to or different from each other, and are hydrogen or hydrocarbon radicals having from 1 to 18 carbon atoms; and RVI and Rvn are equal to or different from each other, and have the same meaning of R-Rv except that RVI and Rvn cannot be hydrogen; one or more of the R-Rvn groups can be linked to form a cycle. The 1,3-diethers in which RVI and Rvn are selected from C1-C4 alkyl radicals are particularly preferred.
[0047] It is also possible to use mixtures of the above mentioned donors. Specific mixtures are those constituted by esters of succinic acids and 1,3 diethers as disclosed in WO201 1/061134.
[0048] When it is desired to increase the capability of the catalyst to distribute an olefin co-monomer within a polymer chain, such as in case of production of ethylene/a-olefin copolymers, it is preferred to choose the electron donor among monofunctional donors, chosen among ethers and C1-C4 alkyl esters of aliphatic mono carboxylic acids. Preferred ethers are the C2-C20 aliphatic ethers and in particular, cyclic ethers preferably having 3-5 carbon atoms cyclic ethers such as tetrahydrofurane, methyl-tetrahydrofurane, dioxane. Preferred esters are ethylacetate and methyl formiate. Among them tetrahydrofurane and ethylacetate are the most preferred.
[0049] In general, the final amount of electron donor compound in the solid catalyst component may range from 0.5 to 40 wt% by weight preferably in the range from 1 to 35 wt% with respect to the total weight of the solid catalyst component.
[0050] The solid catalyst component obtained according to the present disclosure preferably shows a surface area (by B.E.T. method) generally between 20 and 500 m2/g and preferably between 50 and 400 m2/g, and a total porosity (by B.E.T. method) greater than 0.2 cm3/g, preferably between 0.3 and 0.6 cm3/g. The porosity (Hg method) due to
pores with radius up to 10.000A, generally ranges from 0.3 to 1.5 cm3/g, preferably from 0.45 to 1.0 cm3/g.
[0051] The solid catalyst component prepared according to the process of the present disclosure is converted into catalyst for the polymerization of olefins by reacting it with alkylaluminum compounds (ii) according to known methods.
[0052] The alkylaluminum compound (ii) is preferably chosen from the trialkyl aluminum compounds such as for example triethylaluminum, triisobutylaluminum, tri-n- butylaluminum, tri-n-hexylaluminum, tri-n-octylaluminum. It is also possible to use alkylaluminum halides, alkylaluminum hydrides or alkylaluminum sesquichlorides, such as AlEt2Cl and AhEtsCh, possibly in mixture with the above cited trialkylaluminums.
[0053] The Al/Ti molar ratio is such that there is an excess of Al. In particular, the Al/Ti molar ratio may range between 50:1 and 2000: 1, preferably between 50: 1 and 500: 1.
[0054] Optionally, an external electron-donor compound (iii) can be used. It is preferably selected from silicon compounds, ethers, esters, amines, heterocyclic compounds and 2,2,6,6-tetramethylpiperidine and ketones. Another class of preferred external donor compounds is that of silicon compounds of formula (R6)a(R7)bSi(ORs)c, where a and b are integers from 0 to 2, c is an integer from 1 to 4 and the sum of (a+b+c) is 4; Re, R7, and Rs, are alkyl, cycloalkyl or aryl radicals with 1-18 carbon atoms optionally containing heteroatoms. Particularly preferred are the silicon compounds in which a is 1, b is 1, and c is 2, at least one of Re and R7 is selected from branched alkyl, cycloalkyl or aryl groups with 3-10 carbon atoms optionally containing heteroatoms, and Rs is a C1-C10 alkyl group, in particular methyl. Examples of such preferred silicon compounds are methylcyclohexyldimethoxysilane (C donor), diphenyldimethoxysilane, methyl-t- butyldimethoxysilane, dicyclopentyldimethoxysilane (D donor), diisopropyldimethoxysilane, (2-ethylpiperidinyl)t-butyldimethoxysilane, (2- ethylpiperidinyl)thexyldimethoxysilane, (3,3,3-trifluoro-n-propyl)(2- ethylpiperidinyl)dimethoxysilane, methyl(3,3,3-trifluoro-n-propyl)dimethoxysilane. Moreover, the silicon compounds in which a is 0 and c is 3, R7 is a branched alkyl or cycloalkyl group, optionally containing heteroatoms, and Rs is methyl are also preferred. Examples of such preferred silicon compounds are cyclohexyltrimethoxysilane, t- butyltrimethoxysilane and thexyltrimethoxysilane.
[0055] The external electron donor compound is used in such an amount to give a molar ratio organoaluminum compound to said external electron donor compound of from 0.1 : 1 to 500: 1, preferably from 1 : 1 to 300: 1, and more preferably from 3: 1 to 100: 1.
[0056] According to a preferred embodiment of the disclosure, the molar ratio of compound (iv) to alkylaluminum compound (ii) introduced into the polymerization reactor is from 0.01 to 5, preferably from 0.05 to 3.5, more preferably from 0.1 to 2.5.
[0057] The catalyst components (i)-(iii) are preferably contacted with a liquid inert hydrocarbon solvent such as, e.g., propane, n-hexane or n-heptane, at a temperature below about 60°C and preferably from about 0 to 30°C for a time period of from about six seconds to 60 minutes.
[0058] The above catalyst components (i), (ii) and optionally (iii) are fed to a precontacting vessel, in amounts such that the weight ratio (ii/(i) is in the range of 0.1-10 and if the compound (iii) is present, the weight ratio (ii)/(iii) is preferably as defined above. Preferably, the said components are pre-contacted at a temperature of from 10 to 20°C for 1-30 minutes. The precontact vessel can be either a stirred tank or a loop reactor.
[0059] In a preferred embodiment, the precontacted catalyst is then fed to the prepolymerization reactor where step (ii) takes place. The prepolymerization step is carried out in a first reactor selected from a loop reactor or a continuously stirred tank reactor. The prepolymerization can be carried out either in gas-phase or in liquid-phase. Preferably it is carried out in liquid-phase. The liquid medium comprises liquid alpha-olefin monomer(s), optionally with the addition of an inert hydrocarbon solvent. Said hydrocarbon solvent can be either aromatic, such as toluene, or aliphatic, such as propane, hexane, heptane, isobutane, cyclohexane and 2,2,4-trimethylpentane. The amount of hydrocarbon solvent, if any, is lower than 40% by weight with respect to the total amount of alpha-olefins, preferably lower than 20% by weight. Preferably, step (ii) is carried out in the absence of inert hydrocarbon solvents.
[0060] The average residence time in this reactor generally ranges from 2 to 40 minutes, preferably from 10 to 25 minutes. The temperature is comprised between 10°C and 50°C, preferably between 20°C and 40°C. Adopting these conditions allows to obtain a pre-polymerization degree in the preferred range from 60 to 800g per gram of solid catalyst component, preferably from 150 to 500 g per gram of solid catalyst component. The prepolymerization step is preferably characterized by a low concentration of solid in the slurry, typically in the range from 50 g to 300 g of solid per liter of slurry.
[0061] The process of the present disclosure can be carried out in any polymerization plant comprising one or more liquid-phase and/or gas-phase polymerization reactors. Examples of liquid-phase reactors are loop reactors and continuously stirred tank reactors (CSTR). Examples of gas-phase reactors include fluidized bed reactors, stirred bed reactors and reactors having two interconnected polymerization zones as described in EP 0782587
and EP 1012195. The process of the present disclosure can be carried in two or more cascade reactors, giving rise to a sequential multistage polymerization process. For instance, a fluidized bed reactor can be used to prepare a first polymer component, which is successively fed to a gas-phase reactor having two interconnected polymerization zones to prepare a second and a third polymer component. Or a first fluidized bed reactor can be used to prepare a first polymer component, which is successively fed to a second fluidized bed reactor to prepare a second polymer component and then to a third fluidized bed reactor to prepare a third polymer component. Accordingly, an olefin polymer endowed with a multi-modal molecular weight distribution can be obtained, as well as an olefin copolymer comprising two or more components having a different comonomer content.
[0062] In a preferred embodiment of the present disclosure, the polymerization process is carried out as gas-phase polymerization, i.e. by a process in which the solid polymers are obtained from a gas-phase of the monomer or the monomers. Such gas-phase polymerizations may be carried out at pressures of from 0.1 to 20 MPa, or from 0.5 to 10 MPa, or from 1.0 to 5 MPa and polymerization temperatures from 40 to 150°C or from 65 to 125°C.
[0063] Gas-phase polymerization reactors can be, for example, horizontally or vertically stirred reactor, fluidized bed gas-phase reactors or multizone circulating reactors.
[0064] Fluidized-bed polymerization reactors are reactors in which the polymerization takes place in a bed of polymer particles which is maintained in a fluidized state by feeding in gas at the lower end of a reactor, for example below a gas distribution grid having the function of dispensing the gas flow, and taking off the gas again at its upper end. The reactor gas is then returned to the lower end to the reactor via a recycle line equipped with a compressor and a heat exchanger. The circulated reactor gas is, for example, a mixture of the olefins to be polymerized, inert gases such as nitrogen and/or lower alkanes such as ethane, propane, butane, pentane or hexane and optionally a molecular weight regulator such as hydrogen. According to an embodiment, nitrogen or propane can be used as inert gas, if appropriate in combination with further lower alkanes. The velocity of the reactor gas has to be sufficiently high firstly to fluidize the mixed bed of finely divided polymer present in the tube serving as polymerization zone and secondly to remove the heat of polymerization effectively. The polymerization can also be carried out in a condensed or super-condensed mode, in which part of the circulating reaction gas is cooled to below the dew point and returned to the reactor separately as a liquid and a gas-phase or together as a two-phase mixture in order to make additional use of the enthalpy of vaporization for cooling the reaction gas.
[0065] Multizone circulating reactors are gas-phase reactors in which two polymerization zones are linked to one another and the polymer is passed alternately a plurality of times through these two zones. Such reactors are, for example, described in WO 97/04015 Al and WO 00/02929 Al and have two interconnected polymerization zones, a riser, in which the growing polymer particles flow upward under fast fluidization or transport conditions and a downcomer, in which the growing polymer particles flow in a densified form under the action of gravity. The polymer particles leaving the riser enter the downcomer and the polymer particles leaving the downcomer are reintroduced into the riser, thus establishing a circulation of polymer between the two polymerization zones and the polymer is passed alternately a plurality of times through these two zones. It is further also possible to operate the two polymerization zones of one multizone circulating reactor with different polymerization conditions by establishing different polymerization conditions in its riser and its downcomer. For this purpose, the gas mixture leaving the riser and entraining the polymer particles can be partially or totally prevented from entering the downcomer. This can for example be achieved by feeding a barrier fluid in form of a gas and/or a liquid mixture into the downcomer, in the upper part thereof according to an embodiment. The barrier fluid should have an appropriate composition, different from that of the gas mixture present in the riser. The amount of added barrier fluid can be adjusted in a way that an upward flow of gas countercurrent to the flow of the polymer particles is generated, particularly at the top thereof, acting as a barrier to the gas mixture entrained among the particles coming from the riser. In this manner it is possible to obtain two different gas composition zones in one multizone circulating reactor. Furthermore it is also possible to introduce make-up monomers, comonomers, molecular weight regulator such as hydrogen and/or inert fluids at any point of the downcomer, below the barrier feeding point according to an embodiment. Thus, it is also easily possible to create varying monomer, comonomer and hydrogen concentrations along the downcomer resulting in a further differentiation of the polymerization conditions.
[0066] A gas phase polymerization reactor having two interconnected polymerization zones (riser and downcomer), representing an embodiment, will now be described in detail with reference to the enclosed Figure 1, which is a diagrammatic representation and has to be considered illustrative and not limitative of the scope of the disclosure.
[0067] The polymerization reactor shown in Figure 1 comprises a first polymerization zone 1 (riser), wherein the polymer particles flow upward under fast fluidization conditions along the direction of the arrow A and a second polymerization zone 2 (downcomer), wherein the polymer particles flow downward under the action of gravity along the direction of the arrow B.
[0068] The upper portion of the riser 1 is connected to a solid/gas separator 3 by the interconnection section 4. The separator 3 removes the major part of the unreacted monomers from the polymer particles and the polymer withdrawn from the bottom of separator 3 enters the top portion of the downcomer 2. The separated unreacted monomers, optionally together with polymerization diluents, such as propane, flow up to the top of separator 3 and are successively recycled to the bottom of the riser 1 via the recycle line 5.
[0069] A mixture comprising one or more olefin monomers, hydrogen as the molecular weight regulator, propane as the polymerization diluent, is fed to the polymerization reactor via one or more lines M, which are suitably placed along the gas recycle line 5, according to the knowledge of the person skilled in art.
[0070] The catalyst components, optionally after a prepolymerization step, are continuously introduced into the riser 1 via line 6. The produced polymer can be discharged from the reactor via a line 7, which can be placed on the lower portion of the downcomer 2 so that, due to the packed flow of densified polymer, the quantity of gas entrained with the discharged polymer is minimized. By inserting a control valve (not shown in Figure 2) on the polymer discharge line 7, it becomes possible to continuously control the flow rate of polymer produced by the polymerization reactor. Additional polymer discharge lines with respect to line 7 can be placed in the bottom part of the downcomer.
[0071] The polymerization reactor further comprises a transport section 8 connecting the bottom of downcomer 2 with the lower region of the riser 1. The bottom of the downcomer 2 converges into a slight restriction 9. A control valve 10 with an adjustable opening can be placed within the restriction 9. The flow rate Fp of polymer continuously circulated between the downcomer 2 and the riser 1 is adjusted by the level of opening of the control valve 10. The control valve 10 may be a mechanical valve, such as a butterfly valve, a ball valve, etc. A stream of dosing gas is fed into the lower part of the downcomer 2 by means of a line 11 placed at a short distance above the restriction 9. The dosing gas to be introduced through line 10 can be taken from the recycle line 5. In synthesis, the flow Fp of polymer particles circulated between downcomer 2 and riser 1 can be adjusted by varying the opening of the control valve 10 at the bottom of the downcomer and/or by varying the flow rate of the dosing gas entering the downcomer via line 11. The flow rate of dosing gas is adjusted by means of a control valve 18, which is suitably arranged on line 11.
[0072] The transport section 8 is designed as a bend descending from the bottom of downcomer 2 up to the lower region of the riser 1. Furthermore, a carrier gas is introduced via line 12 at the inlet of the transport section 8. The flow rate of carrier gas is adjusted by means of a control valve 13, which is suitably arranged on line 12.
[0073] Also the carrier gas is taken from the gas recycle line 5. Specifically, the gas recycle stream of line 5 is first subjected to compression by means of a compressor 14 and a minor percentage of the recycle stream passes through line 12, thus entering the transport section 8 and diluting the solid phase of polymer flowing through the transport section 8. The major part of the recycle stream, downstream the compressor 14, is subjected to cooling in a heat exchanger 15 and successively is introduced via line 16 at the bottom of the riser 1 at a high velocity, such to ensure fast fluidization conditions in the polymer bed flowing along the riser 1.
[0074] The carrier gas merges with the densified polymer coming from downcomer 2 at the inlet portion of transport section 8, after exiting the slits of the gas distribution grid 17. In the embodiment shown in Figure 2 the top end of the distribution grid 17 is coincident with the inlet of the transport section 8 and the distribution grid 17 extends along the bending of the transport section 8 for an angle a=60°. The gas distribution grid 17 is formed by a plurality of trays fixed to the transport section 8 in a way to form slits in the overlapping area of adjacent trays.
[0075] According to an embodiment, a flow rate of compound (iv) is metered into the reactor at the bottom of the riser and/or at any point in the riser 1, particularly at the top of the riser (flow rate A5).
[0076] Alternative or additional feeding point depicted in Fig 1, are flow rate A3, line 25 metered by valve 26 into catalyst feed line 6) or into the main gas recycle line 5 (flow rate A4, line 27 metered by valve 28).
[0077] As described in WO 2011/029735, the antistatic composition can alternatively or additionally be metered to one or more positions along the height of the downcomer via nozzles. In such a case, the antistatic composition flow rate A2 in line 22 is metered by one or more valves 23 and then pre-dispersed either in the liquid monomer L as described above, or alternatively in a fraction of recycle gas taken from recycle line 5 via line 24.
[0078] The carrier gas merges with the densified polymer coming from downcomer 2 at the inlet portion of transport section 8, after exiting the slits of a gas distribution grid 17. In the embodiment shown in Figure 1 the top end of the distribution grid 17 is coincident with the inlet of the transport section 8 and the distribution grid 17 extends along the bending of the transport section 8 for an angle of 60°. The gas distribution grid 17 is formed by a plurality of trays fixed to the transport section 8 in a way to form slits in the overlapping area of adjacent trays. A detailed description of the gas distribution grid 17 can be found in WO 2012/031986.
[0079] Depending on the olefin (co)polymer to be produced, the polymerization reactor can be operated by properly adjusting the polymerization conditions and the monomers concentration in the riser and in the downcomer, so as to produce a wide variety of bimodal homopolymers and random copolymers. To this purpose, the gas mixture entraining the polymer particles and coming from the riser can be partially or totally prevented from entering the downcomer, so as to polymerize two different monomers compositions in the riser and the downcomer. This effect may be achieved by feeding a gaseous and/or a liquid barrier stream through a line placed in the upper portion of the downcomer. The barrier stream should have a composition different from the gas composition present inside the riser. The flow rate of the barrier stream can be adjusted, so that an upward flow of gas counter-current to the flow of the polymer particles is generated, particularly at the top of the downcomer, thus acting as a barrier to the gas mixture coming from the riser. For further details regarding this barrier effect at the top of the downcomer, reference is made to the disclosure of EP 1012195 Al.
[0080] The different or else identical polymerization processes can also, if desired, be connected in series and thus form a polymerization cascade. A parallel arrangement of reactors using two or more different or identical processes is also possible.
[0081] According to an embodiment, the gas-phase polymerization processes according to the present disclosure are carried out in the presence of an alkane having from 3 to 5 carbon atoms as polymerization diluent, e.g. in the presence of propane.
[0082] The process of the present disclosure provides a possibility for preparing an olefin polymer by gas-phase polymerization in a polymerization reactor in which the formation of polymer agglomerates in the polymerization reactors and fluctuations in the fluid-dynamics of the reactor are prevented or considerably reduced. In particular, compound (iv) reduces the tendency of the olefin polymer particles to stick to the reactor walls. This does not only reduce the risk of forming chunks or wall sheeting, which mostly leads to an unavoidable shut-down of the polymerization reactor because of plugging the discharge line, but also improves the fluid-dynamics of the reactor and avoids their fluctuations.
[0083] In case of a multizone circulating reactor if, for example, polymer particles have a tendency to adhere to the walls of the riser and coverage of the riser wall is continuously built-up, the polymer particle layer or a part of the layer can drop at a certain time. As a result, the amount of transportable polymer particles in the riser increases immediately, more polymer is transported to the downcomer and the polymer particle level within the downcomer rises very fast. By the increase of the number of polymer particles within the riser however not only the density of the reactor content in the riser changes temporarily
but also the fluid-dynamics of the reaction mixture fluctuate. Moreover, the variation of the polymer particle level within the downcomer also influences the fluid-dynamics within the whole multizone circulating reactor.
[0084] The level of electrostatic charges observed in a multizone circulating reactor may be sensitive to the molecular weight of the olefin polymer particles transferred into it. According to a preferred embodiment, the process of the present disclosure is used in a multizone circulating reactor for producing polymer particles having a MFR2.16 at a temperature of 190°C under a load of 2.16 kg of less than 60 g/10 min.
[0085] According to a further embodiment, in case of a cascade process comprising two or more fluidized-bed gas-phase reactors, the antistatic composition can be added into the bed of each reactor or to the polymer discharge downstream each reactor or else before the first reactor.
[0086] Using compound (iv) of the present disclosure does not only result in a process for the polymerization of olefins which is simple to carry out, but the polymerization process has also a good operability. That means that the tendency for forming polymer deposits on the rector wall, i.e. reactor fouling, for forming lumps and for forming fines, i.e. for forming very small sticky polyolefin particles, is reduced. Moreover, the activity of the catalyst may be improved or at least not impaired and the product properties of the prepared polyolefins are not deteriorated.
EXAMPLES
[0087] The following examples are given to illustrate the present disclosure without any limiting purpose.
Test Methods
Melt flow rate (MFR “L”)
[0088] Determined according to ISO 1133 (230°C, 2.16 Kg)
Ethylene content in copolymers
[0089] 13C NMR of propylene/ethylene copolymers
[0090] 13C NMR spectra were acquired on a Bruker AV-600 spectrometer equipped with cryoprobe, operating at 160.91 MHz in the Fourier transform mode at 120°C.
[0091] The peak of the SPP carbon (nomenclature according to “Monomer Sequence Distribution in Ethylene-Propylene Rubber Measured by 13C NMR. 3. Use of Reaction Probability Mode ” C. J. Carman, R. A. Harrington and C. E. Wilkes, Macromolecules, 1977, 10, 536) was used as internal reference at 29.9 ppm. The samples were dissolved in l,l,2,2-tetrachloroethane-d2 at 120°C with a 8 % wt/v concentration. Each spectrum was acquired with a 90° pulse, 15 seconds of delay between pulses and CPD to remove 1H-13C coupling. 512 transients were stored in 32K data points using a spectral window of 9000 Hz.
[0092] The assignments of the spectra, the evaluation of triad distribution and the composition were made according to Kakugo (“Carbon- 13 NMR determination of monomer sequence distribution in ethyl ene-propylene copolymers prepared with 5- titanium trichloride- diethylaluminum chloride” M. Kakugo, Y. Naito, K. Mizunuma and T. Miyatake, Macromolecules 1982, 15, 4, 1150-1152) using the following equations:
PPP = 100 Tpp/S PPE = 100 Tp5/S EPE = 100 T55/S
PEP = 100 Spp/S PEE= 100 SP5/S EEE = 100 (0.25 Sy5+0.5 S55)/S
S = Tpp + TP5 + T55 + SPP + SpS + 0.25 Sy5 + 0.5 S55
[0093] The molar percentage of ethylene content was evaluated using the following equation:
E% mol = 100 * [PEP+PEE+EEE]
[0094] The weight percentage of ethylene content was evaluated using the following equation:
100 * E% mol * MWE
E% wt. = >
E% mol * MWE + P% mol * MWp where P% mol is the molar percentage of propylene content, while MWE and MWp are the molecular weights of ethylene and propylene, respectively.
Xylene Solubles (XS)
[0095] Determined as follows: 2.5 g of polymer and 250 ml of xylene were introduced in a glass flask equipped with a refrigerator and a magnetical stirrer. The temperature was raised in 30 minutes up to the boiling point of the solvent. The clear solution so obtained was then kept under reflux and stirring for further 30 minutes. The closed flask was then kept in thermostatic water bath at 25° C for 30 minutes. The so formed solid was filtered on quick filtering paper. 100 ml of the filtered liquid was poured in a previously weighed aluminium container, which was heated on a heating plate under nitrogen flow, to remove the solvent by evaporation. The container was then kept in an oven at 80° C under vacuum until constant weight was obtained. The weight percentage of polymer soluble in xylene at room temperature was then calculated.
[0096] Melting point and crystallization point
[0097] The melting point has been measured by using a DSC instrument according to ISO 11357-3, at scanning rate of 20°C/min both in cooling and heating, on a sample of weight between 5 and 7 mg., under inert N2 flow. Instrument calibration made with Indium.
[0098] EXAMPLES
[0099] Comparative example 1-2 and inventive examples 3-4
A 4— liter steel autoclave equipped with a stirrer, pressure gauge, thermometer, catalyst feeding system, monomer feeding lines and thermostating jacket, was purged with nitrogen flow at 70°C for one hour. Then, at 30°C under propylene flow, were charged 0.1 g of PPA dissolved in hexane, 75 ml of anhydrous hexane containing 0.25 g of AlEts, about 10 mg of solid catalyst component prepared according to example 5 of EP728769 and cyclohexylmethyl dimethoxysilane as the external donor in amount to have an Aluminum/ donor molar ratio of 10. The autoclave was closed and subsequently 1.25NL of hydrogen were added. Then, under stirring, 1.2 kg of liquid propylene were fed. The temperature was raised to 70°C in ten minutes and the polymerization was carried out at this temperature for two hours. At the end of the polymerization, the non-reacted propylene was removed; the polymer was recovered and dried in an oven at 80°C. Results are reported in Table 1.
Table 1

C2 PPA= Glycerol monostearate
Ex.3 PPA= 40%wt mono and 60%wt diglycerol esters of octanoic and decanoic acid
EX.4 PPA= Glycerol monocaprylate
[0100] Example 5
[0101] The polymerization was carried out in continuous by means of a process setup comprising:
- 1.5 liter vessel for the pre-contact of the catalyst components;
- a loop prepolymerization reactor having a volume of 80 liters;
- a sequence of two serially connected fluidised bed reactors, each having a volume of 1.5m3.
[0102] Precontact
[0103] A Ziegler-Natta catalyst system was used as the polymerization catalyst, comprising:
- a solid catalyst component prepared according to example Cl of WO2012/139897; triethylaluminium (TEAL) as the cocatalyst ; dicyclopentyldimethoxysilane (DCPMS) as the external donor.
[0104] The above solid catalyst components is fed to the pre-contacting vessel, the weight ratio TEAL/solid catalyst was 4, the weight ratio TEAL/DCPMS was 5.
[0105] The above components were pre-contacted at a temperature of 12°C for 10 minutes.
[0106] Pre-polymerization
[0107] The catalyst system withdrawn from the pre-contacting vessel was continuously fed to the prepolymerization loop reactor together with a liquid stream of propylene and propane. The pre-polymerization occurred at a temperature of 20°C.
[0108] Polymerization
[0109] A polypropylene slurry was continuously discharged from the loop reactor and fed to the first fluidized bed reactor where propylene was polymerized using H2 as the molecular weight regulator and in the presence of propane as inert diluent. The polymerization was carried out according to the conditions reported in table 1. The polymer discharged from the first reactor was then conveyed to the second reactor where ethyl ene/propylene copolymerization was carried out.
[0110] The PPA (a mixture of commercially available 40%wt mono and 60%wt diglycerol esters of octanoic and decanoic acid diluted in hexane) was fed via propylene carrier, partly to the first and second gas-phase reactors via fluidization gas and partly to the polymer discharged from the first reactor before being introduced into the second. The flow rate of the PPA feed was such as to obtain in the polymer the amounts of PPA indicated in Table 2.
[0111] Comparative Example 6
[0112] Using the same reactor set-up and conditions of Example 5 the same type of product was prepared with the difference that molten glycerol monostearate was used as PPA.
[0113] Looking at table 2 it can be seen that the PPA of example 5 is much more efficient in controlling the reactivity of the fine (less than 500pm) particles (as evidenced by the lower C2 ratio) with respect to PPA used in C6. This means that polymerization run of example 5 has a much lower tendency to generate fouling occurring from particles sticking to reactor walls.
Table 2

Claims
1. A process for the polymerization of olefins, carried out in the presence of (i) a Ziegler-Natta catalyst, (ii) an aluminum alkyl cocatalyst, optionally (iii) an external donor, and (iv) a compound (polymerization process aid (PPA)) comprising mono or diglycerol ester derivatives of saturated Ce-Cn, preferably Ce-Cio, monocarboxylic acids said component (iv) being used in an amount of at least 50 ppm based on the amount of polyolefin produced.
2. The process according to claim 1, wherein the process is carried out in gasphase.
3. The process according to any of the preceding claims, in which the solid catalyst component (i) contains an internal donor.
4. The process according to claim 3 wherein the internal donor is selected from the group consisting of ethers, amines, silanes, carbamates, ketones, esters of aliphatic acids, alkyl and aryl esters of optionally substituted aromatic polycarboxylic acids, diol derivatives chosen among monoesters monocarbamates and monoesters monocarbonates or mixtures thereof.
5. The process according to any of the preceding claims, wherein external electron-donor compound (iii) is present and selected from silicon compounds of formula (Re)a(R7)bSi(OR8)c, where a and b are integers from 0 to 2, c is an integer from 1 to 4 and the sum of (a+b+c) is 4; Re, R7, and Rs, are alkyl, cycloalkyl or aryl radicals with 1-18 carbon atoms optionally containing heteroatoms.
6. The process according to any of the preceding claims, wherein the compound (iv) further includes mono or polyglycerol fatty acid esters other than mono or diglycerol ester derivatives of saturated Ce-Cio, preferably Cs-Cio monocarboxylic acids, the amount of mono or diglycerol ester derivatives of saturated Ce-Cio, preferably Cs-Cio monocarboxylic acids, being higher than 50%wt, preferably more than 55%wt and especially in the range 60-90%wt with respect to the total amount of mono or polyglycerol fatty acid esters.
7. The process according to any of the preceding claims, wherein the compound (iv) comprises one or more of mono or diglycerol ester derivatives of saturated Ce- C10, more preferably Cs-Cio, monocarboxylic acids and mixture thereof.
8. The process according to any of preceding claims, wherein compound (iv) comprises, for more than 50% wt, preferably more than 55%wt and especially in the range 60-90%wt based on the total amount of fatty acid mono or polyglycerol esters, one or more of glycerol monoester or diester of octanoic acid, glycerol monoester or diester of decanoic acid, glycerol diester mixed octanoic and decanoic acid, diglycerol mono, di- or triester of octanoic acid, diglycerol mono, di- or triester of decanoic acid, diglycerol di or triester of mixed octanoic and decanoic acid and any mixture thereof.
9. The process according to claim 8, wherein compound (iv) comprises, for more than 50% wt, preferably more than 55%wt and especially in the range 60-90%wt based on the total amount of fatty acid glycerol esters, glycerol mono octanoate or a mixture comprising glycerol diester of octanoic acid, glycerol diester of mixed octanoic and decanoic acid, diglycerol mono ester of octanoic acid and diglycerol mono ester of decanoic acid and mixture thereof.
10. The process according to any of the preceding claims in which compound (iv) is introduced into the polymerization reactor in an amount ranging from 70 to 2000 ppm per weight, preferably from 150 to 1500 ppm per weight, more preferably from 200 to 1000 ppm per weight referring to the weight of the prepared polyolefin.
11. The process according to any of the preceding claims in which compound (iv) is fed to the polymerization reactor in a flow of saturated or unsaturated hydrocarbon having from 2 to 6 carbon atoms preferably propylene and/or propane.
12. The process according to any of the preceding claims in which the molar ratio of compound (iv) to alkylaluminum compound (ii) introduced into the polymerization reactor is from 0.01 to 5, preferably from 0.05 to 3.5, more preferably from 0.1 to 2.5.
13. The process according to any of the preceding claims in which compound (iv) is fed directly into the reactor and/or into a line leading to the reactor and/or into a line exiting the reactor.
14. The process according to any of the preceding claims in which compound (iv) is fed upstream the polymerization reactor.
15. The process according to claim 2 in which the gas-phase reactor is fluidized bed reactor and/or multizone circulating reactor.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202480028830.7A CN121039175A (en) | 2023-05-31 | 2024-05-31 | Olefin polymerization process |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP23176252 | 2023-05-31 | ||
| EP23176252.7 | 2023-05-31 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024246270A1 true WO2024246270A1 (en) | 2024-12-05 |
Family
ID=86646723
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2024/065010 Pending WO2024246270A1 (en) | 2023-05-31 | 2024-05-31 | Process for the polymerization of olefins |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN121039175A (en) |
| WO (1) | WO2024246270A1 (en) |
Citations (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4399054A (en) | 1978-08-22 | 1983-08-16 | Montedison S.P.A. | Catalyst components and catalysts for the polymerization of alpha-olefins |
| US4469648A (en) | 1978-06-13 | 1984-09-04 | Montedison S.P.A. | Process for preparing spheroidally shaped products, solid at room temperature |
| US4829034A (en) | 1986-06-09 | 1989-05-09 | Neste Oy | Procedure for manufacturing catalyst components for polymerizing olefines |
| US5100849A (en) | 1989-10-02 | 1992-03-31 | Chisso Corporation | Process for producing a catalyst for olefin polymerization |
| EP0560035A1 (en) | 1992-01-31 | 1993-09-15 | Montell Technology Company bv | Process for the gas-phase polymerization of alpha-olefins |
| EP0728769A1 (en) | 1995-02-21 | 1996-08-28 | Montell North America Inc. | Components and catalysts for the polymerization of olefins |
| WO1997004015A1 (en) | 1995-07-20 | 1997-02-06 | Montell Technology Company B.V. | Process and apparatus for the gas-phase polymerization of alpha-olefins |
| WO2000002929A1 (en) | 1998-07-08 | 2000-01-20 | Montell Technology Company B.V. | Process and apparatus for the gas-phase polymerisation |
| WO2002048208A1 (en) | 2000-12-15 | 2002-06-20 | Basell Poliolefine Italia S.P.A. | Continuous process for the preparation of solid catalyst components for the polymerisation of alpha-olefins |
| WO2004106388A2 (en) | 2003-05-29 | 2004-12-09 | Basell Poliolefine Italia S.R.L. | Process for the preparation of a catalyst component and components therefrom obtained |
| WO2005030815A1 (en) * | 2003-09-23 | 2005-04-07 | Dow Global Technologies Inc. | Self limiting catalyst composition and propylene polymerization process |
| WO2011029735A1 (en) | 2009-09-11 | 2011-03-17 | Basell Poliolefine Italia S.R.L. | Process for the gas-phase polymerization of olefins |
| WO2011061134A1 (en) | 2009-11-19 | 2011-05-26 | Basell Poliolefine Italia S.R.L. | Process for the preparation of impact resistant propylene polymer compositions |
| WO2012031986A1 (en) | 2010-09-09 | 2012-03-15 | Basell Poliolefine Italia S.R.L. | Process and apparatus for the gas-phase polymerization of olefins |
| WO2012041811A1 (en) | 2010-09-28 | 2012-04-05 | Basell Polyolefine Gmbh | Method for feeding an antistatic compound to a polymerization reactor |
| WO2012139897A1 (en) | 2011-04-12 | 2012-10-18 | Basell Poliolefine Italia S.R.L. | Catalyst components for the polymerization of olefins |
| CN107935851A (en) | 2017-12-04 | 2018-04-20 | 广州星业科技股份有限公司 | A kind of Capmul MCM C8 and preparation method thereof |
| CN109535287A (en) * | 2018-12-06 | 2019-03-29 | 锦州英诺威科技服务有限公司 | A kind of external electron donor of propylene polymerization catalyst, propylene polymerization catalyst system and the preparation method and application thereof |
| CN109679001A (en) * | 2018-12-29 | 2019-04-26 | 锦州英诺威科技服务有限公司 | It is a kind of to kill agent living for polypropylene production |
-
2024
- 2024-05-31 WO PCT/EP2024/065010 patent/WO2024246270A1/en active Pending
- 2024-05-31 CN CN202480028830.7A patent/CN121039175A/en active Pending
Patent Citations (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4469648A (en) | 1978-06-13 | 1984-09-04 | Montedison S.P.A. | Process for preparing spheroidally shaped products, solid at room temperature |
| US4399054A (en) | 1978-08-22 | 1983-08-16 | Montedison S.P.A. | Catalyst components and catalysts for the polymerization of alpha-olefins |
| US4829034A (en) | 1986-06-09 | 1989-05-09 | Neste Oy | Procedure for manufacturing catalyst components for polymerizing olefines |
| US5100849A (en) | 1989-10-02 | 1992-03-31 | Chisso Corporation | Process for producing a catalyst for olefin polymerization |
| EP0560035A1 (en) | 1992-01-31 | 1993-09-15 | Montell Technology Company bv | Process for the gas-phase polymerization of alpha-olefins |
| EP0728769A1 (en) | 1995-02-21 | 1996-08-28 | Montell North America Inc. | Components and catalysts for the polymerization of olefins |
| WO1997004015A1 (en) | 1995-07-20 | 1997-02-06 | Montell Technology Company B.V. | Process and apparatus for the gas-phase polymerization of alpha-olefins |
| EP0782587A1 (en) | 1995-07-20 | 1997-07-09 | Montell Technology Company bv | Process and apparatus for the gas-phase polymerization of alpha-olefins |
| WO2000002929A1 (en) | 1998-07-08 | 2000-01-20 | Montell Technology Company B.V. | Process and apparatus for the gas-phase polymerisation |
| EP1012195A1 (en) | 1998-07-08 | 2000-06-28 | Montell Technology Company bv | Process and apparatus for the gas-phase polymerisation |
| WO2002048208A1 (en) | 2000-12-15 | 2002-06-20 | Basell Poliolefine Italia S.P.A. | Continuous process for the preparation of solid catalyst components for the polymerisation of alpha-olefins |
| WO2004106388A2 (en) | 2003-05-29 | 2004-12-09 | Basell Poliolefine Italia S.R.L. | Process for the preparation of a catalyst component and components therefrom obtained |
| WO2005030815A1 (en) * | 2003-09-23 | 2005-04-07 | Dow Global Technologies Inc. | Self limiting catalyst composition and propylene polymerization process |
| WO2011029735A1 (en) | 2009-09-11 | 2011-03-17 | Basell Poliolefine Italia S.R.L. | Process for the gas-phase polymerization of olefins |
| WO2011061134A1 (en) | 2009-11-19 | 2011-05-26 | Basell Poliolefine Italia S.R.L. | Process for the preparation of impact resistant propylene polymer compositions |
| WO2012031986A1 (en) | 2010-09-09 | 2012-03-15 | Basell Poliolefine Italia S.R.L. | Process and apparatus for the gas-phase polymerization of olefins |
| WO2012041811A1 (en) | 2010-09-28 | 2012-04-05 | Basell Polyolefine Gmbh | Method for feeding an antistatic compound to a polymerization reactor |
| WO2012139897A1 (en) | 2011-04-12 | 2012-10-18 | Basell Poliolefine Italia S.R.L. | Catalyst components for the polymerization of olefins |
| CN107935851A (en) | 2017-12-04 | 2018-04-20 | 广州星业科技股份有限公司 | A kind of Capmul MCM C8 and preparation method thereof |
| CN109535287A (en) * | 2018-12-06 | 2019-03-29 | 锦州英诺威科技服务有限公司 | A kind of external electron donor of propylene polymerization catalyst, propylene polymerization catalyst system and the preparation method and application thereof |
| CN109679001A (en) * | 2018-12-29 | 2019-04-26 | 锦州英诺威科技服务有限公司 | It is a kind of to kill agent living for polypropylene production |
Non-Patent Citations (2)
| Title |
|---|
| C. J. CARMANR. A. HARRINGTONC. E. WILKES, MACROMOLECULES, vol. 10, 1977, pages 536 |
| M. KAKUGOY. NAITOK. MIZUNUMAT. MIYATAKE, MACROMOLECULES, vol. 15, no. 4, 1982, pages 1150 - 1152 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN121039175A (en) | 2025-11-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101627058B (en) | Gas Phase Propylene Polymerization Using Staged Addition of Alkyl Aluminum | |
| US6900281B2 (en) | Gas phase olefin polymerizations using dual donor catalyst systems | |
| KR101359287B1 (en) | Process for the (co)polymerization of ethylene | |
| US7652108B2 (en) | Process for the gas-phase polymerization olefins | |
| RU2554093C2 (en) | Controlling h2 distribution in horizontal stirred bed reactor | |
| CN110392700B (en) | Process for the gas-phase polymerization of olefins | |
| US9469703B2 (en) | Process for the gas-phase polymerization of olefins | |
| KR101501074B1 (en) | Process for the polymerization of olefins | |
| EP3394110B1 (en) | Olefin polymerization process in the presence of antistatic composition | |
| US12435167B2 (en) | Processes for producing polyolefins and impact copolymers with broad molecular weight distribution and high stiffness | |
| JP5501384B2 (en) | Enlarging the molecular weight distribution of polyolefin materials produced in a horizontal stirred gas phase reactor | |
| WO2024246270A1 (en) | Process for the polymerization of olefins | |
| WO2024246273A1 (en) | Process for the polymerization of olefins | |
| KR20260015870A (en) | Olefin polymerization method | |
| WO2021150377A1 (en) | Methods for producing bimodal polyolefins and impact copolymers |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24731276 Country of ref document: EP Kind code of ref document: A1 |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112025024339 Country of ref document: BR |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2024731276 Country of ref document: EP |