WO2024240634A1 - Anti-met antibodies, antibody-drug conjugates, compositions and uses thereof - Google Patents
Anti-met antibodies, antibody-drug conjugates, compositions and uses thereof Download PDFInfo
- Publication number
- WO2024240634A1 WO2024240634A1 PCT/EP2024/063676 EP2024063676W WO2024240634A1 WO 2024240634 A1 WO2024240634 A1 WO 2024240634A1 EP 2024063676 W EP2024063676 W EP 2024063676W WO 2024240634 A1 WO2024240634 A1 WO 2024240634A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- antibody
- cancer
- antigen
- met
- seq
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/68031—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being an auristatin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6849—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a receptor, a cell surface antigen or a cell surface determinant
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2863—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against receptors for growth factors, growth regulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/33—Crossreactivity, e.g. for species or epitope, or lack of said crossreactivity
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
Definitions
- the mesenchymal-epithelial transition factor (MET or cMET) is a receptor tyrosine kinase comprising a 50 kDa a-subunit and a 145 kDa p-subunit.
- the only known ligand for MET is hepatocyte growth factor (HGF), which is also known as scatter factor.
- Binding of HGF to MET leads to receptor dimerization and autophosphorylation of p-subunit residues Y1349 and Y1356, activating downstream signaling pathways that include the phosphoinositol 3-kinase (PI3K)- protein kinase B (Akt) pathway, the signal transducer and activator of transcription factor (STAT) pathway, the mitogen-activated protein kinase (MAPK) pathway, and the nuclear factor kappa-light-chain-enhancer of activated B cells (NFKB) pathway. This ultimately leads to increased mitogenesis, cell proliferation, cell survival, and cell motility.
- PI3K phosphoinositol 3-kinase
- STAT signal transducer and activator of transcription factor
- MAPK mitogen-activated protein kinase
- NFKB nuclear factor kappa-light-chain-enhancer of activated B cells
- Dysregulation of MET or HGF activity may occur, e.g., through overexpression, gene amplification, mutation, or alternative splicing of MET, or through HGF ligand-induced autocrine/paracrine loop signaling.
- Such dysregulation plays a role in many cancers by facilitating cancer invasiveness, angiogenesis, metastasis, and tumor growth, thus leading to a more aggressive cancer phenotype and a poorer prognosis.
- MET is also known to interact with signaling pathways involving other receptors, such as EGFR, TGF-p, and HER3, and may play a role in resistance to treatments targeting those receptors.
- MET inhibitors such as anti-MET antibodies, thus may be effective in combination with other receptor inhibitors in overcoming resistant phenotypes.
- MET inhibitors include both monoclonal antibodies, which may target either MET or its ligand, HGF, and small molecule kinase inhibitors.
- Known anti- MET small molecule receptor tyrosine kinase inhibitors include tivantinib, cabozantinib, foretinib, golvatinib, and crizotinib.
- no anti-cMET antibodies have been approved for therapeutic use.
- Known antibodies targeting the cMET pathway include onartuzumab (Genentech, WO 2006/015371), ARGX- 111 (Argenx, WO 2012/059561), emibetuzumab (LY2875358; Eli Lilly, WO 2010/059654), SAIT-301 (Samsung, US 2014-0154251), telisotuzumab (ABT-700, Abbott/ Abbvie, WO 2017/201204) and Sym015 (Symphogen, WO 2016/042412).
- Bispecific antibodies targeting MET have also been described, such as the amivantamab bispecific antibody targeting EGFR and MET (JNJ-61186372, Jansseb Biotech, US 9593164).
- MET receptor is an active target in cancer treatment and an attractive target for the development of anti-MET therapeutic antibodies and antibody drug conjugates.
- the present invention relates to a novel recombinant antibody or antigen-binding portion thereof targeting MET, as well as ADCs comprising this antibody or antigen-binding portion thereof, compositions comprising this antibody or antigenbinding portion thereof, compositions comprising the ADC comprising said anti- MET antibody or antigen-binding portion thereof, the use of said antibody or antigen-binding portion thereof or ADC, and said compositions comprising anti- MET antibody or antigen-binding portion thereof or anti-MET ADC, for treatment of cancers, including cancers that express MET or rely on MET pathway activation, such as melanoma, uveal melanoma, renal cancer, thyroid cancer, mesothelioma, liver hepatocellular cancer, lung cancer including non-small cell lung cancer and small cell lung cancer, gastric cancer including stomach cancer, pancreatic cancer, colorectal cancer, esophageal cancer, cholangiocarcinoma, head and neck cancer including oral cancer, cervical and endocer
- the antibodies of the invention, ADCs comprising the antibodies of the invention and compositions comprising the antibody of the invention or ADCs of the invention are surprisingly effective on cancer cells.
- the present invention provides an anti-MET antibody or an antigen-binding portion thereof.
- the antibody or antigenbinding portion thereof competes for binding to human MET with an antibody whose H-CDR1, H-CDR2, H-CDR3, L-CDR1, L-CDR2, and L-CDR3 comprise or consist of the amino acid sequences of SEQ ID NOs: 1, 2, 3, 4, 5, and 6, respectively.
- the anti-MET antibody or antigen-binding portion thereof binds to the same epitope of human MET as an antibody whose H- CDR1, H-CDR2, H-CDR3, L-CDR1, L-CDR2, and L-CDR3 comprise or consist of the amino acid sequences of SEQ ID NOs: 1, 2, 3, 4, 5, and 6, respectively.
- the present invention provides an anti-MET antibody or antigen-binding portion thereof comprising an H-CDR1, H-CDR2, and H-CDR3 that comprise or consist of the amino acid sequences of SEQ ID NOs: 1 , 2, and 3, respectively.
- the anti-MET antibody or antigen-binding portion thereof comprises a heavy chain variable domain (VH) that is at least 90% identical in sequence to the amino acid sequence of SEQ ID NO: 7.
- the anti-MET antibody or antigen-binding portion thereof comprises a VH that comprises or consists of the amino acid sequence of SEQ ID NO: 7.
- the anti-MET antibody or antigen-binding portion thereof comprises a heavy chain (HC) that comprises or consists of the amino acid sequence of SEQ ID NO: 11 or 13.
- the anti-MET antibody or antigen-binding portion thereof comprises a L-CDR1 , L-CDR2, and L-CDR3 that comprise or consist of the amino acid sequences of SEQ ID NOs: 4, 5, and 6.
- the anti-MET antibody or antigen-binding portion thereof comprises a light chain variable domain (VL) that is at least 90% identical in sequence to the amino acid sequence of SEQ ID NO: 8.
- the anti-MET antibody or antigen-binding portion thereof comprises a VL that comprises or consists of the amino acid sequence of SEQ ID NO: 8.
- the anti-MET antibody or antigen-binding portion thereof comprises a light chain (LC) that comprises or consists of the amino acid sequence of SEQ ID NO: 12 or 14.
- the anti-MET antibody or antigen-binding portion thereof comprises a H-CDR1 , H-CDR2, H-CDR3, L-CDR1, L-CDR2, and L-CDR3 that comprise or consist of the amino acid sequences of SEQ ID NOs: 1, 2, 3, 4, 5, and 6, respectively.
- the anti-MET antibody or antigen-binding portion thereof comprises a VH that is at least 90% identical in sequence to the amino acid sequence of SEQ ID NO: 7 and a VL that is at least 90% identical in sequence to the amino acid sequence of SEQ ID NO: 8.
- the anti-MET antibody or antigen-binding portion thereof comprises a VH that comprises or consists of the amino acid sequence of SEQ ID NO: 7 and a VL that comprises or consists of the amino acid sequence of SEQ ID NO: 8.
- the anti-MET antibody or antigen-binding portion thereof comprises an HC that comprises or consists of the amino acid sequence of SEQ ID NO: 11 or 13 and an LC that comprises or consists of the amino acid sequence of SEQ ID NO: 12 or 14.
- the anti-MET antibody or antigen-binding portion thereof comprises an HC and LC comprising or consisting of the amino acid sequences of SEQ ID NOs: 11 and 12, respectively, or the HC and LC comprising or consisting of the amino acid sequences of SEQ ID NOs: 13 and 14, respectively.
- the antibody may be of isotype IgG. In certain embodiments, the antibody is of isotype subclass lgG1. In certain embodiments, the anti-MET antibody is of isotype subclass lgG2.
- the present invention also provides a multi-specific (e.g., bi-specific) binding molecule comprising the antigen-binding portion of an anti-MET antibody described herein, and the antigen-binding portion of another distinct antibody such as another anti-MET antibody or an antibody that targets a different protein.
- the bispecific binding molecule comprises an antigenbinding portion of an antibody whose H-CDR1, H-CDR2, H-CDR3, L-CDR1, L- CDR2, and L-CDR3 comprise the amino acid sequences of SEQ ID NOs: 1, 2, 3, 4, 5, and 6, respectively.
- the present disclosure provides, novel antibody-drug conjugate (ADC) compounds comprising the anti-MET antibodies of the present invention.
- ADC antibody-drug conjugate
- the ADC compounds show biological activity against cancer cells and may slow, inhibit, and/or reverse tumor growth in mammals, and/or may be useful for treating human cancer patients.
- the present disclosure more specifically relates, in some embodiments, to ADC compounds that are capable of binding and killing cancer cells.
- the ADC compounds are also capable of internalizing into a target cell after binding.
- the ADC may be represented by Ab-(L-D)p, wherein Ab is an anti-Met antibody or an antigen-binding portion thereof; D is a payload or drug or any compound to be linked to the Ab with the linker L; L is a linker that covalently attaches Ab to D; and p is an integer from 1 to 16.
- the ADC comprises an anti-Met antibody or an antigenbinding portion thereof whose H-CDR1, H-CDR2, H-CDR3, L-CDR1 , L-CDR2, and L-CDR3 comprise the amino acid sequences of SEQ ID NOs: 1 , 2, 3, 4, 5, and 6, respectively.
- L-D refers to the linker-drugs, linker-payloads, or linkercompounds.
- p is an integer from 1 to 16.
- the linker (L) may be a cleavable or non-cleavable linker.
- D refers to a drug moiety or payload and is selected from an Eg5 inhibitor, a V-ATPase inhibitor, a HSP90 inhibitor, an IAP inhibitor, an mTor inhibitor, a microtubule stabilizer, a microtubule destabilizer, an auristatin (such as Monomethyl Auristatin E or MMAE), a dolastatin, a maytansinoid, a MetAP (methionine aminopeptidase), an inhibitor of nuclear export of proteins CRM1, a DPPIV inhibitor, an inhibitor of phosphoryl transfer reactions in mitochondria, a protein synthesis inhibitor, a kinase inhibitor, a CDK2 inhibitor, a CDK9 inhibitor, a proteasome inhibitor, a kinesin inhibitor, an HDAC inhibitor, a DNA damaging agent, a DNA alkylating agent, a DNA intercalator, a DNA minor groove binder, an RNA polymerase inhibitor, an amanitin, a spliceosome inhibitor,
- the present invention also provides antibody compositions comprising an anti- MET antibody or antigen-binding portion thereof described herein.
- the antibody composition comprises an anti-MET antibody or antigen-binding portion thereof that competes for binding to human MET with an antibody whose H-CDR1, H-CDR2, H-CDR3 and L-CDR1, L-CDR2, and L-CDR3 comprise the amino acid sequences of SEQ ID NOs: 1, 2, 3, 4, 5, and 6, respectively.
- the antibody composition comprises an anti-MET antibody or antigen-binding portion thereof that binds to the same epitope of human MET as an antibody whose H-CDR1, H-CDR2, H-CDR3, L-CDR1, L-CDR2, and L-CDR3 comprise the amino acid sequences of SEQ ID NOs: 1 , 2, 3, 4, 5, and 6, respectively.
- the antibody composition comprises the anti-MET antibody or antigen-binding portion thereof comprises a H-CDR1, H-CDR2, H-CDR3, L- CDR1, L-CDR2, and L-CDR3 that comprise the amino acid sequences of SEQ ID NOs: 1, 2, 3, 4, 5, and 6, respectively.
- the antibody composition comprises an anti-MET antibody or antigen-binding portion thereof that comprises a VH and VL that are at least 90% identical in sequence to the amino acid sequences of SEQ ID NOs: 7 and 8, respectively.
- the antibody composition comprises an anti-MET antibody or antigen-binding portion thereof that comprises a VH and VL comprising the amino acid sequences of SEQ ID NOs: 7 and 8, respectively.
- the antibody composition comprises an anti-MET antibody or antigen-binding portion thereof that comprises an HC and LC comprising the amino acid sequences of SEQ ID NOs: 11 and 12, respectively, or the HC and LC comprising the amino acid sequences of SEQ ID NOs: 13 and 14, respectively.
- the antibody composition comprises an anti-MET antibody or antigen-binding portion thereof comprises an HC and LC comprising the amino acid sequences of SEQ ID NOs: 11 and 12, respectively, or the HC and LC comprising the amino acid sequences of SEQ ID NOs: 13 and 14, respectively.
- the present invention also provides ADC compositions comprising an ADC comprising an anti-MET antibody or antigen-binding portion thereof described herein.
- the ADC composition comprises an ADC wherein the anti- MET antibody or antigen-binding portion thereof comprises a H-CDR1, H-CDR2, H-CDR3, L-CDR1, L-CDR2, and L-CDR3 that comprise the amino acid sequences of SEQ ID NOs: 1 , 2, 3, 4, 5, and 6, respectively.
- the ADC composition comprises an ADC comprising an anti-MET antibody or antigenbinding portion thereof that comprises a VH and VL that are at least 90% identical in sequence to the amino acid sequences of SEQ ID NOs: 7 and 8, respectively.
- the ADC composition comprises an ADC wherein the anti- MET antibody or antigen-binding portion thereof that comprises a VH and VL comprising the amino acid sequences of SEQ ID NOs: 7 and 8, respectively.
- the ADC composition comprises an ADC wherein the anti- MET antibody or antigen-binding portion thereof that comprises an HC and LC that are at least 90% identical in sequence to the amino acid sequences of SEQ ID NOs: 11 and 12, respectively, or the HC and LC that are at least 90% identical in sequence to the amino acid sequences of SEQ ID NOs: 13 and 14, respectively.
- the present invention provides an isolated nucleic acid molecule comprising a nucleotide sequence that encodes the heavy chain or an antigen-binding portion thereof, a nucleotide sequence that encodes the light chain or an antigen-binding portion thereof, or both, of an anti-MET antibody described herein.
- the isolated nucleic acid molecule comprises a nucleotide sequence selected from the group consisting of SEQ ID NOs: 9 or 10.
- the present invention also provides a vector comprising the isolated nucleic acid molecule, wherein said vector further comprises an expression control sequence.
- the present invention also provides a host cell comprising a nucleotide sequence that encodes the heavy chain or an antigen-binding portion thereof, a nucleotide sequence that encodes the light chain or an antigen-binding portion thereof, or both, of an anti-MET antibody described herein.
- the host cell comprises a nucleotide sequence selected from the group consisting of SEQ ID NOs: 9 or 10.
- the present invention also provides a non-human transgenic animal or plant comprising a nucleotide sequence that encodes the heavy chain or an antigenbinding portion thereof, a nucleotide sequence that encodes the light chain or an antigen-binding portion thereof, or both, of an anti-MET antibody described herein, wherein said animal or plant expresses the nucleotide sequence(s).
- the animal or plant comprises a nucleotide sequence selected from the group consisting of SEQ ID NOs: 9 or 10.
- the present invention also provides a method for producing an anti-MET antibody or antigen-binding portion thereof described herein, comprising providing the above-described host cell, cultivating said host cell under conditions suitable for expression of the antibody or portion, and isolating the resulting antibody or portion. Methods of producing ADC compounds comprising the anti-MET antibodies of the present invention are also disclosed.
- the present invention also provides a method for treating a patient with a MET- mediated disorder, comprising administering to said patient an anti-MET antibody or antigen-binding portion thereof as described herein, an ADC comprising an anti- Met antibody or an antigen-binding portion thereof as described herein, an antibody composition comprising an anti-MET antibody or antigen-binding portion thereof or an ADC comprising an anti-Met antibody as described herein, or a pharmaceutical composition comprising the anti-MET antibody composition or the ADC comprising an anti-Met antibody composition.
- the present disclosure provides methods of treating a cancer (e.g., a cancer that expresses the MET antigen targeted by the antibody or antigen-binding portion of the ADC). In some embodiments, the present disclosure provides methods of reducing or slowing the expansion of a cancer cell population in a subject.
- a cancer e.g., a cancer that expresses the MET antigen targeted by the antibody or antigen-binding portion of the ADC.
- the present invention also provides a method for treating a patient having or suspected of having a cancer, comprising administering to said patient an anti- MET antibody or antigen-binding portion thereof, an ADC comprising an anti-Met antibody or an antigen-binding portion thereof as described herein, or a composition or a pharmaceutical composition comprising the anti-MET antibody or antigen-binding portion thereof or the ADC comprising an anti-Met antibody or an antigen-binding portion thereof.
- Another exemplary embodiment is a method of reducing or inhibiting the growth of a tumor in a patient, comprising administering to the subject a therapeutically effective amount of an anti-MET antibody or antigen-binding portion thereof, an ADC comprising an anti-Met antibody or an antigen-binding portion thereof, a composition or pharmaceutical composition comprising the anti-MET antibody or antigen-binding portion thereof or comprising the ADC comprising anti-MET antibody or antigen-binding portion thereof.
- Another exemplary embodiment is a method of reducing or slowing the expansion of a cancer cell population in a patient, comprising administering to the subject a therapeutically effective amount of an anti-MET antibody or antigen-binding portion thereof, an ADC comprising an anti-Met antibody or an antigen-binding portion thereof, a composition or pharmaceutical composition comprising the anti-MET antibody or antigen-binding portion thereof or comprising the ADC comprising an anti-MET antibody or antigen-binding portion thereof.
- the cancer is dependent on MET activation and/or MET expression.
- Each VH and VL is composed of three CDRs (H-CDR herein designates a CDR from the heavy chain; and L-CDR herein designates a CDR from the light chain) and four FRs, arranged from amino-terminus to carboxyl-terminus in the following order: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4.
- the assignment of amino acids to each region may be in accordance with IMGT® definitions (Lefranc et al., Dev Comp Immunol 27(1):55-77 (2003); or the definitions of Kabat, Sequences of Proteins of Immunological Interest (National Institutes of Health, Bethesda, MD (1987 and 1991)); Chothia & Lesk, J. Mol.
- antibody portion refers to one or more portions or fragments of an antibody that retain the ability to specifically bind to an antigen (e.g., human MET, or a portion thereof). It has been shown that certain portions or fragments of a full-length antibody can perform the antigen-binding function of the antibody.
- binding specificity refers to the ability of an individual antibody or antigen binding portion to preferentially react with one antigenic determinant over a different antigenic determinant. The degree of specificity indicates the extent to which an antibody or portion preferentially binds to one antigenic determinant over a different antigenic determinant.
- the term “specific,” “specifically binds,” and “binds specifically” refers to a binding reaction between an antibody or antigen-binding portion (e.g., an anti-Met antibody) and a target antigen (e.g., MET) in a heterogeneous population of proteins and other biologies.
- a “specific antibody” or a “target-specific antibody” is one that only binds the target antigen (e.g., MET), but does not bind (or exhibits minimal binding) to other antigens.
- variant refers to a nucleic acid sequence or an amino acid sequence that differs from a reference nucleic acid sequence or amino acid sequence respectively, but retains one or more biological properties of the reference sequence.
- a variant may contain one or more amino acid substitutions, deletions, and/or insertions (or corresponding substitution, deletion, and/or insertion of codons) with respect to a reference sequence. Changes in a nucleic acid variant may not alter the amino acid sequence of a peptide encoded by the reference nucleic acid sequence, or may result in amino acid substitutions, additions, deletions, fusions, and/or truncations.
- a nucleic acid variant disclosed herein encodes an identical amino acid sequence to that encoded by the unmodified nucleic acid or encodes a modified amino acid sequence that retains one or more functional properties of the unmodified amino acid sequence. Changes in the sequence of peptide variants are typically limited or conservative, so that the sequences of the unmodified peptide and the variant are closely similar overall and, in many regions, identical. In some embodiments, a peptide variant retains one or more functional properties of the unmodified peptide sequence. A variant and unmodified peptide can differ in amino acid sequence by one or more substitutions, additions, deletions in any combination.
- a peptide variant encompasses polypeptides having amino acid substitutions, deletions, and/or insertions as long as the polypeptide has at least 80%, at least 85%, at least 90%, at least 91%, at least 92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least 99% amino acid sequence identity with a reference sequence, or with a corresponding segment (e.g., a functional fragment) of a reference sequence, e.g., those variants that also retain one or more functions of the reference sequence.
- a corresponding segment e.g., a functional fragment
- the percent identity between the two sequences is a function of the number of identical positions shared by the sequences, taking into account the number of gaps, and the length of each gap, which need to be introduced for optimal alignment of the two sequences.
- amino acid identity or homology between proteins disclosed herein and variants thereof, including variants of target antigens (such as MET) and variants of antibody variable domains (including individual variant CDRs) is at least 80% to the sequences depicted herein, e.g., identities or homologies of at least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, almost 100%, or 100%.
- antibody-drug conjugate refers to one or more therapeutic compounds that is linked to one or more antibodies or antigenbinding portions.
- Ab an antibody or antigen-binding portion (e.g., an anti-Met antibody or antigen-binding portion thereof)
- L a linker moiety
- D a drug moiety
- p the number of drug moieties per antibody or antigen-binding portion.
- the present invention relates to a novel anti-MET antibody 8902 directed against human MET, or an antigen-binding portion of said antibody.
- Variable domain heavy and light chain (VH and VL) amino acid sequences of this antibody are provided in SEQ ID NOs: 7 and 8, respectively, and corresponding nucleotide sequences are provided in SEQ ID NOs: 9 and 10, respectively.
- Full-length heavy and light chain amino acid sequences (HC and LC) are available in SEQ ID NOs: 11 and 12 (lgG1 chain) and in SEQ ID NOs: 13 and 14 (lgG2 chain), respectively.
- Amino acid sequences of heavy chain CDRs (H-CDR1, H-CDR2 and H-CDR3) and light chain CDRs (L-CDR1, L-CDR-2 and L-CDR3) of 8902 antibody are shown in SEQ ID NOs: 1 , 2 and 3 and in SEQ ID NOs: 4, 5 ad 6, respectively.
- the CDR sequences were assigned in accordance with IMGT® definitions.
- an anti-MET antibody or an antigen-binding portion thereof that competes for binding to human MET with an antibody having an H-CDR1 , H-CDR2, H-CDR3, L-CDR1 , L-CDR2, and L-CDR3 that comprise or consist of the amino acid sequences of SEQ ID NOs: 1 , 2, 3, 4, 5, and 6, respectively;
- the anti-MET antibody or antigen-binding portion thereof comprises an L-CDR1 , L-CDR2, and L-CDR3 comprising or consisting of the amino acid sequences of SEQ ID NOs: 4, 5, and 6, respectively.
- the anti-MET antibody or antigen-binding portion thereof comprises a heavy chain that comprises or consists of the amino acid sequence of SEQ ID NO: 11 and a light chain that comprises or consists the amino acid sequence of SEQ ID NO: 12.
- An anti-MET antibody can also be derivatized with a chemical group such as polyethylene glycol (PEG), a methyl or ethyl group, or a carbohydrate group. These groups may be useful to improve the biological characteristics of the antibody, e.g., to increase serum half-life.
- PEG polyethylene glycol
- the antibodies of the invention may be present in a neutral form (including zwitter ionic forms) or as a positively or negatively-charged species. In some embodiments, the antibodies may be complexed with a counterion to form a pharmaceutically acceptable salt.
- salts refers to a salt which does not abrogate the biological activity and properties of the compounds of the invention, and does not cause significant irritation to a subject to which it is administered.
- examples of such salts include, but are not limited to: (a) acid addition salts formed with inorganic acids, for example, hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, nitric acid and the like; and salts formed with organic acids, for example, acetic acid, oxalic acid, tartaric acid, succinic acid, maleic acid, fumaric acid, gluconic acid, citric acid, malic acid, ascorbic acid, benzoic acid, tannic acid, palmitic acid, alginic acid, polyglutamic acid, naphthalenesulfonic acid, methanesulfonic acid, p-toluenesulfonic acid, naphthalenedisulfonic acid, polygalacturonic acid, and the like
- the antibody-drug conjugates (ADCs) described herein can contain a monovalent anionic counterion. Any suitable anionic counterion can be used. In certain embodiments, the monovalent anionic counterion is a pharmaceutically acceptable monovalent anionic counterion. In certain embodiments, the monovalent anionic counterion can be selected from bromide, chloride, iodide, acetate, trifluoroacetate, benzoate, mesylate, tosylate, triflate, formate, or the like.
- the bispecific binding molecule may comprise an anti-MET antibody or an antigen-binding portion thereof having a heavy chain variable domain comprising or consisting of the amino acid sequence of SEQ ID NO: 7 and/or a light chain variable domain comprising or consisting of the amino acid sequence of SEQ ID NO: 8.
- the bispecific binding molecule may comprise an anti-MET antibody or an antigen-binding portion thereof having a heavy chain comprising or consisting of the amino acid sequence of SEQ ID NO: 11 and/or a light chain comprising or consisting of the amino acid sequence of SEQ ID NO: 12.
- the bispecific binding molecule may be a dual variable domain antibody, i.e. , wherein the two arms of the antibody comprise two different variable domains, or may be in the form of an antibody fragment such as a bispecific Fab fragment or a bispecific scFv. This is also useful if one wants to create a divalent or polyvalent antibody on a single polypeptide chain.
- Bispecific binding molecules or polyvalent antibodies may have the binding specificity of an anti-MET antibody or antigen-binding portion thereof described herein and the binding specificity of another antibody that targets a same or a different protein, such as an immune checkpoint protein, a cancer antigen, or a cell surface molecule whose activity mediates a disease condition such as cancer.
- a bispecific binding molecule has the binding specificities of the first anti-Met antibody 8902 and a second antibody or antigen-binding portions thereof.
- a bispecific binding molecule has the binding specificities of the first anti-Met antibody 8902 and the second anti-Met antibody 9006 or antigen-binding portions thereof.
- the bispecific binding molecule comprises an antigen-binding portion of a first antibody having a heavy chain variable domain (VH) comprising the amino acid sequence of SEQ ID NO: 11 and a light chain variable domain (VL) comprising the amino acid sequence of SEQ ID NO:12 and an antigen-binding portion of a second antibody having a heavy chain variable domain (VH) comprising the amino acid sequence of SEQ ID NO:19 and a light chain variable domain (VL) comprising the amino acid sequence of SEQ ID NQ:20.
- VH heavy chain variable domain
- VL light chain variable domain
- the bispecific binding molecule comprises an antigen-binding portion of a first antibody having a heavy chain variable domain (VH) comprising the amino acid sequence of SEQ ID NO: 13 and a light chain variable domain (VL) comprising the amino acid sequence of SEQ ID NO:14 and an antigen-binding portion of a second antibody having a heavy chain variable domain (VH) comprising the amino acid sequence of SEQ ID NO:21 and a light chain variable domain (VL) comprising the amino acid sequence of SEQ ID NO:22.
- VH heavy chain variable domain
- VL light chain variable domain
- ADC Antibody-Drug Conjugates
- the antibody-drug conjugate (ADC) compounds of the present disclosure include an anti-MET antibody or antigen-binding portion thereof conjugated (i.e., covalently attached by a linker) to a drug moiety, wherein the drug moiety when not conjugated to an antibody or antigen-binding portion has a cytotoxic or cytostatic effect.
- the ADC may provide improved activity, better cytotoxic specificity, and/or reduced off-target killing as compared to the drug moiety when administered alone.
- the components of the ADC are selected to (i) retain one or more therapeutic properties exhibited by the antibody and drug moieties in isolation, (ii) maintain the specific binding properties of the antibody or antigen-binding portion; (iii) optimize drug loading and drug-to-antibody ratios; (iv) allow delivery, e.g., intracellular delivery, of the drug moiety via stable attachment to the antibody or antigen-binding portion; (v) retain ADC stability as an intact conjugate until transport or delivery to a target site; (vi) minimize aggregation of the ADC prior to or after administration; (vii) allow for the therapeutic effect, e.g., cytotoxic effect, of the drug moiety after cleavage or other release mechanism in the cellular environment; (viii) exhibit in vivo anti-cancer treatment efficacy comparable to or superior to that of the antibody and drug moieties in isolation; (ix) minimize off-target killing by the drug moiety; and/or (x) exhibit desirable pharmacokinetic and pharma
- the ADC compounds of the present disclosure may selectively deliver an effective dose of a cytotoxic or cytostatic agent to cancer cells or to tumor tissue.
- the cytotoxic and/or cytostatic activity of the ADC is dependent on target antigen expression in a cell.
- the disclosed ADCs are particularly effective at killing cancer cells expressing a target antigen while minimizing off-target killing.
- the disclosed ADCs do not exhibit a cytotoxic and/or cytostatic effect on cancer cells that do not express a target antigen.
- ADC compounds comprising an anti-Met antibody or antigen-binding portion thereof (Ab), a drug moiety (D), and a linker moiety (L) that covalently attaches Ab to D.
- the antibody or antigen-binding portion is able to bind to a tumor-associated antigen (e.g., MET), e.g., with high specificity and high affinity.
- MET tumor-associated antigen
- the antibody or antigen-binding portion is internalized into a target cell upon binding, e.g., into a degradative compartment in the cell.
- the ADCs internalize upon binding to a target cell, undergo degradation, and release drug moiety to kill cancer cells.
- the drug moiety may be released from the antibody and/or the linker moiety of the ADC by enzymatic action, hydrolysis, oxidation, or any other mechanism.
- average p refers to the average number of -L-D moieties per antibody or antigenbinding portion, also referred to as “average drug loading.”
- the anti-Met antibody or antigen-binding thereof in the antibody drug conjugates of the present disclosure is the anti-Met antibody 8902 or antigen-binding thereof.
- the invention provides an ADC comprising an anti-MET antibody or antigen-binding portion thereof selected from the group consisting of:
- an anti-MET antibody or antigen-binding portion thereof comprising at least two, three, four or five CDR sequences selected from the group consisting of H-CDR1 SEQ ID NO: 1, H-CDR2 SEQ ID NO: 2, H-CDR3 SEQ ID NO: 3, L-CDR1 SEQ ID NO: 4, L-CDR2 SEQ ID NO: 5, and L-CDR3 SEQ ID NO: 6;
- an anti-MET antibody or an antigen-binding portion thereof having an H- CDR1, H-CDR2, and H-CDR3 comprising or consisting of the amino acid sequences of SEQ ID NOs: 1, 2, and 3, respectively, and an L-CDR1, L- CDR2, and L-CDR3 comprising or consisting of the amino acid sequences of SEQ ID NOs: 4, 5, and 6, respectively;
- an anti-MET antibody or an antigen-binding portion thereof having a heavy chain variable domain comprising or consisting of the amino acid sequence of SEQ ID NO: 7;
- an anti-MET antibody or an antigen-binding portion thereof having a light chain variable domain comprising or consisting of the amino acid sequence of SEQ ID NO: 8;
- an anti-MET antibody or an antigen-binding portion thereof having a heavy chain variable domain at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to the amino acid sequence of SEQ ID NO: 7 and a light chain variable domain at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to the amino acid sequence of SEQ ID NO: 8;
- an anti-MET antibody or an antigen-binding portion thereof having a heavy chain comprising or consisting of the amino acid sequence of SEQ ID NO: 13 and a light chain comprising or consisting of the amino acid sequence of SEQ ID NO: 14;
- an anti-MET antibody or an antigen-binding portion thereof having a heavy chain at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to the amino acid sequence of SEQ ID NO: 11 and a light chain at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to the amino acid sequence of SEQ ID NO: 12; and
- the ADC comprises an anti-MET antibody or antigen-binding portion thereof comprising at least two, three, four or five CDR sequences selected from the group consisting of H-CDR1 SEQ ID NO: 1 , H-CDR2 SEQ ID NO: 2, H- CDR3 SEQ ID NO: 3, L-CDR1 SEQ ID NO: 4, L-CDR2 SEQ ID NO: 5, and L- CDR3 SEQ ID NO: 6.
- the ADC comprises an anti-MET antibody or an antigenbinding portion thereof having a heavy chain comprising or consisting of the amino acid sequence of SEQ ID NO: 11 and a light chain comprising or consisting of the amino acid sequence of SEQ ID NO: 12.
- the ADC comprises an anti-MET antibody or an antigenbinding portion thereof having a heavy chain at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to the amino acid sequence of SEQ ID NO: 11 and a light chain at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to the amino acid sequence of SEQ ID NO: 12.
- the ADC comprises a bispecific binding molecule comprising an anti-MET antibody or an antigen-binding portion thereof having an H-CDR1, H-CDR2, and H-CDR3 comprising or consisting of the amino acid sequences of SEQ ID NOs: 1 , 2, and 3, respectively, and an L-CDR1, L- CDR2, and L-CDR3 comprising or consisting of the amino acid sequences of SEQ ID NOs: 4, 5, and 6, respectively.
- the ADC comprises a bispecific binding molecule comprising an anti-MET antibody or an antigen-binding portion thereof having a heavy chain at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to the amino acid sequence of SEQ ID NO: 13 and/or a light chain at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to the amino acid sequence of SEQ ID NO: 14.
- the ADC comprises a bispecific binding molecule comprising an antigen-binding portion of a first antibody having a heavy chain variable domain (VH) comprising the amino acid sequence of SEQ ID NO:13 and a light chain variable domain (VL) comprising the amino acid sequence of SEQ ID NO:14 and an antigen-binding portion of a second antibody having a heavy chain variable domain (VH) comprising the amino acid sequence of SEQ ID NO:17 and a light chain variable domain (VL) comprising the amino acid sequence of SEQ ID NO:18.
- VH heavy chain variable domain
- VL light chain variable domain
- the ADC comprises a bispecific binding molecule has the binding specificities of a first anti-Met antibody 8902 and a second anti-Met antibody 9338 or antigen-binding portions thereof.
- the ADC comprises a bispecific binding molecule comprising an antigen-binding portion of a first antibody having a heavy chain variable domain (VH) comprising the amino acid sequence of SEQ ID NO: 11 and a light chain variable domain (VL) comprising the amino acid sequence of SEQ ID NO:12 and an antigen-binding portion of a second antibody having a heavy chain variable domain (VH) comprising the amino acid sequence of SEQ ID NO:19 and a light chain variable domain (VL) comprising the amino acid sequence of SEQ ID NQ:20.
- VH heavy chain variable domain
- VL light chain variable domain
- Whether a linker is stable extracellularly can be determined, for example, by including an ADC in plasma for a predetermined time period (e.g., 2, 4, 6, 8, 16, 24, 48, or 72 hours) and then quantifying the amount of free drug moiety present in the plasma. Stability may allow the ADC time to localize to target cancer cells and prevent the premature release of the drug moiety, which could lower the therapeutic index of the ADC by indiscriminately damaging both normal and cancer tissues.
- the linker is stable outside of a target cell and releases the drug moiety from the ADC once inside of the cell, such that the drug can bind to its target.
- an effective linker will: (i) maintain the specific binding properties of the antibody or antigen-binding portion; (ii) allow delivery, e.g., intracellular delivery, of the drug moiety via stable attachment to the antibody or antigen-binding portion; (iii) remain stable and intact until the ADC has been transported or delivered to its target site; and (iv) allow for the therapeutic effect, e.g., cytotoxic effect, of the drug moiety after cleavage or alternate release mechanism.
- Linkers may impact the physico-chemical properties of an ADC. As many cytotoxic agents are hydrophobic in nature, linking them to the antibody with an additional hydrophobic moiety may lead to aggregation. ADC aggregates are insoluble and often limit achievable drug loading onto the antibody, which can negatively affect the potency of the ADC. Protein aggregates of biologies, in general, have also been linked to increased immunogenicity. As shown below, ADCs disclosed below have low aggregation levels and desirable levels of drug loading. A linker may be "cleavable” or “non-cleavable” (Ducry and Stump (2010) Bioconjugate Chem. 21:5-13).
- Cleavable linkers are designed to release the drug moiety when subjected to certain environment factors, e.g., when internalized into the target cell, whereas non-cleavable linkers generally rely on the degradation of the antibody or antigen-binding portion itself.
- the linker MC-VC-PAB is a protease cleavable linker.
- an intermediate which is the precursor of the linker moiety, is reacted with the drug moiety or payload under appropriate conditions.
- reactive groups are used on the drug or payload (such as MMAE standing for MonoMethyl Auristatin E) and/or the intermediate or linker.
- the product of the reaction between the drug or payload and the intermediate, or the derivatized drug or payload (drug/payload plus linker) is subsequently reacted with the antibody or antigen-binding portion under conditions that facilitate conjugation of the drug and intermediate or derivatized drug/payload and antibody or antigen-binding portion.
- the intermediate or linker may first be reacted with the antibody or antigen-binding portion, or a derivatized antibody or antigen-binding portion, and then reacted with the drug or derivatized drug.
- a number of different reactions are available for covalent attachment of the drug moiety and/or linker moiety to the antibody or antigen-binding portion. This is often accomplished by reaction of one or more amino acid residues of the antibody or antigen-binding portion with techniques that are known to the skilled artisan.
- the linker moiety (L) of the ADC attaches to the antibody or antigen-binding portion through a chemically active group on one or more amino acid residues on the antibody or antigen-binding portion.
- the linker may be attached to the antibody or antigen-binding portion via a free amino, imino, hydroxyl, thiol, or carboxyl group (e.g., to the N- or C-terminus, to the epsilon amino group of one or more lysine residues, to the free carboxylic acid group of one or more glutamic acid or aspartic acid residues, or to the sulfhydryl group of one or more cysteine residues).
- the site to which the linker is attached can be a natural residue in the amino acid sequence of the antibody or antigenbinding portion, or it can be introduced into the antibody or antigen-binding portion, e.g., by DNA recombinant technology (e.g., by introducing a cysteine residue into the amino acid sequence) or by protein biochemistry (e.g., by reduction, pH adjustment, or hydrolysis).
- the conjugation is done stochastically on native antibodies.
- the number of drug moieties that can be conjugated to an antibody or antigen-binding portion is limited by the number of free cysteine residues.
- an antibody may have only one or a few cysteine thiol groups, or may have only one or a few sufficiently reactive thiol groups through which a linker may be attached.
- antibodies do not contain many free and reactive cysteine thiol groups that may be linked to a drug moiety. Indeed, most cysteine thiol residues in antibodies are involved in either interchain or intrachain disulfide bonds. Conjugation to cysteines can therefore, in some embodiments, require at least partial reduction of the antibody. Over-attachment of linker-toxin to an antibody may destabilize the antibody by reducing the cysteine residues available to form disulfide bonds.
- an optimal drug:antibody ratio should increase potency of the ADC (by increasing the number of attached drug moieties per antibody) without destabilizing the antibody or antigen-binding portion.
- an optimal ratio may be 2, 4, 6, or 8.
- an optimal ratio may be 2 or 4.
- an antibody or antigen-binding portion is exposed to reducing conditions prior to conjugation in order to generate one or more free cysteine residues.
- An antibody in some embodiments, may be reduced with a reducing agent such as dithiothreitol (DTT) or tris(2- carboxyethyl)phosphine (TCEP), under partial or total reducing conditions, to generate reactive cysteine thiol groups.
- DTT dithiothreitol
- TCEP tris(2- carboxyethyl)phosphine
- Unpaired cysteines may be generated through partial reduction with limited molar equivalents of TCEP, which can reduce the interchain disulfide bonds which link the light chain and heavy chain (one pair per H-L pairing) and the two heavy chains in the hinge region (two pairs per H-H pairing in the case of human lgG1) while leaving the intrachain disulfide bonds intact (Stefano et al. (2013) Methods Mol Biol. 1045:145-71).
- disulfide bonds within the antibodies are reduced electrochemically, e.g., by employing a working electrode that applies an alternating reducing and oxidizing voltage.
- This approach can allow for on-line coupling of disulfide bond reduction to an analytical device (e.g., an electrochemical detection device, an NMR spectrometer, or a mass spectrometer) or a chemical separation device (e.g., a liquid chromatograph (e.g., an HPLC) or an electrophoresis device (see, e.g., US 2014/0069822)).
- an analytical device e.g., an electrochemical detection device, an NMR spectrometer, or a mass spectrometer
- a chemical separation device e.g., a liquid chromatograph (e.g., an HPLC) or an electrophoresis device (see, e.g., US 2014/0069822)
- an antibody is subjected to denaturing conditions to reveal reactive nucleophilic groups on amino acid residues, such as cysteine.
- the drug loading of an ADC may be controlled in different ways, e.g., by: (i) limiting the molar excess of drug-linker intermediate or linker reagent relative to antibody; (ii) limiting the conjugation reaction time or temperature; (iii) partial or limiting reductive conditions for cysteine thiol modification; and/or (iv) engineering by recombinant techniques the amino acid sequence of the antibody such that the number and position of cysteine residues is modified for control of the number and/or position of linker-drug attachments.
- free cysteine residues are introduced into the amino acid sequence of the antibody or antigenbinding portion.
- cysteine engineered antibodies can be prepared wherein one or more amino acids of a parent antibody are replaced with a cysteine amino acid. Any form of antibody may be so engineered, i.e. mutated.
- a parent Fab antibody fragment may be engineered to form a cysteine engineered Fab referred to as a "ThioFab.”
- a parent monoclonal antibody may be engineered to form a "ThioMab.”
- a single site mutation yields a single engineered cysteine residue in a ThioFab, whereas a single site mutation yields two engineered cysteine residues in a ThioMab, due to the dimeric nature of the IgG antibody.
- the parent antibody is engineered by incorporating cystein mutations inside the heavy chain by replacing the serine at position 400 (Ell numbering) and inside the light chain by replacing the valine at position 205 (Ell numbering) (respectively, HC S400C and LC V205C) of the peptide scaffold.
- one or more free cysteine residues are already present in an antibody or antigen-binding portion, without the use of engineering, in which case the existing free cysteine residues may be used to conjugate the antibody or antigen-binding portion to a drug moiety.
- the resulting product can be a mixture of ADC compounds with a distribution of one or more drug moieties attached to each copy of the antibody or antigenbinding portion in the mixture.
- the drug loading in a mixture of ADCs resulting from a conjugation reaction ranges from 1 to 16 drug moieties attached per antibody or antigen-binding portion.
- the average number of drug moieties per antibody or antigen-binding portion may be calculated by any conventional method known in the art, e.g., by mass spectrometry (e.g., liquid chromatography-mass spectrometry (LC-MS)) and/or high-performance liquid chromatography (e.g., HIC-HPLC).
- mass spectrometry e.g., liquid chromatography-mass spectrometry (LC-MS)
- HIC-HPLC high-performance liquid chromatography
- the average number of drug moieties per antibody or antigenbinding portion is determined by liquid chromatography-mass spectrometry (LC- MS).
- the average number of drug moieties per antibody or antigen-binding portion is from 1.5 to 3.5, from 2.5 to 4.5, from 3.5 to 5.5, from 4.5 to 6.5, from 5.5 to 7.5, from 6.5 to 8.5, or from 7.5 to 9.5. In some embodiments, the average number of drug moieties per antibody or antigen-binding portion is from 2 to 4, from 3 to 5, from 4 to 6, from 5 to 7, from 6 to 8, from 7 to 9, from 2 to 8, or from 4 to 8. In some embodiments, the average number of drug moieties per antibody or antigen-binding portion is 2, 3, 4, 5 or 6.
- ADC compounds may be identified in the mixture by mass spectroscopy and separated by, e.g., LIPLC or HPLC, e.g. hydrophobic interaction chromatography (HIC- HPLC).
- a homogeneous or nearly homogenous ADC product with a single loading value may be isolated from the conjugation mixture, e.g., by electrophoresis or chromatography.
- the present disclosure includes methods of producing the described ADCs (in Example 5 and Table 6).
- the ADCs prepared may be subjected to a purification step.
- the purification step may involve any biochemical methods known in the art for purifying proteins, or any combination of methods thereof.
- THF tangential flow filtration
- affinity chromatography affinity chromatography
- ion exchange chromatography any charge or isoelectric point-based chromatography
- mixed mode chromatography e.g., CHT (ceramic hydroxyapatite)
- hydrophobic interaction chromatography size exclusion chromatography
- dialysis filtration, selective precipitation, or any combination thereof.
- the invention provides an antibody composition comprising an anti- MET antibody or antigen-binding portion thereof of this invention.
- the invention provides an ADC composition comprising an anti- MET ADC wherein the ADC comprises an anti-MET antibody or antigen-binding portion thereof of this invention.
- the composition is an antibody composition comprising an anti-MET antibody or an antigen-binding portion thereof selected from the group consisting of:
- an anti-MET antibody or an antigen-binding portion thereof that competes for binding to human MET with an antibody having an H-CDR1 , H-CDR2, H-CDR3, L-CDR1, L-CDR2, and L-CDR3 that comprise or consist of the amino acid sequences of SEQ ID NOs: 1, 2, 3, 4, 5, and 6, respectively;
- an anti-MET antibody or an antigen-binding portion thereof that competes for binding to human MET with an antibody having a heavy chain variable domain comprising or consisting of the amino acid sequence of SEQ ID NO: 7 and a light chain variable domain comprising or consisting of the amino acid sequence of SEQ ID NO: 8; and
- the composition is an antibody composition or an ADC composition comprising an anti-MET antibody or an antigen-binding portion thereof selected from the group consisting of:
- an anti-MET antibody or an antigen-binding portion thereof having an H- CDR1 , H-CDR2, and H-CDR3 comprising or consisting of the amino acid sequences of SEQ ID NOs: 1 , 2, and 3, respectively;
- an anti-MET antibody or an antigen-binding portion thereof having an L- CDR1 , L-CDR2, and L-CDR3 comprising or consisting of the amino acid sequences of SEQ ID NOs: 4, 5, and 6, respectively;
- an anti-MET antibody or antigen-binding portion thereof comprising at least two, three, four or five CDR sequences selected from the group consisting of H-CDR1 SEQ ID NO: 1, H-CDR2 SEQ ID NO: 2, H-CDR3 SEQ ID NO: 3, L-CDR1 SEQ ID NO: 4, L-CDR2 SEQ ID NO: 5, and L-CDR3 SEQ ID NO: 6;
- an anti-MET antibody or an antigen-binding portion thereof having an H- CDR1, H-CDR2, and H-CDR3 comprising or consisting of the amino acid sequences of SEQ ID NOs: 1, 2, and 3, respectively, and an L-CDR1, L- CDR2, and L-CDR3 comprising or consisting of the amino acid sequences of SEQ ID NOs: 4, 5, and 6, respectively;
- an anti-MET antibody or an antigen-binding portion thereof having a heavy chain variable domain comprising or consisting of the amino acid sequence of SEQ ID NO: 7;
- an anti-MET antibody or an antigen-binding portion thereof having a light chain variable domain comprising or consisting of the amino acid sequence of SEQ ID NO: 8;
- an anti-MET antibody or an antigen-binding portion thereof having a heavy chain variable domain comprising or consisting of the amino acid sequence of SEQ ID NO: 7 and a light chain variable domain comprising or consisting of the amino acid sequence of SEQ ID NO: 8;
- an anti-MET antibody or an antigen-binding portion thereof having a heavy chain variable domain at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to the amino acid sequence of SEQ ID NO: 7 and a light chain variable domain at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to the amino acid sequence of SEQ ID NO: 8;
- an anti-MET antibody or an antigen-binding portion thereof having a heavy chain comprising or consisting of the amino acid sequence of SEQ ID NO: 11 and a light chain comprising or consisting of the amino acid sequence of SEQ ID NO: 12;
- an anti-MET antibody or an antigen-binding portion thereof having a heavy chain comprising or consisting of the amino acid sequence of SEQ ID NO: 13 and a light chain comprising or consisting of the amino acid sequence of SEQ ID NO: 14;
- an anti-MET antibody or an antigen-binding portion thereof having a heavy chain at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to the amino acid sequence of SEQ ID NO: 11 and a light chain at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to the amino acid sequence of SEQ ID NO: 12; and
- an anti-MET antibody or an antigen-binding portion thereof having a heavy chain at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to the amino acid sequence of SEQ ID NO: 13 and a light chain at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to the amino acid sequence of SEQ ID NO: 14.
- the antibody composition comprises an anti-MET antibody or an antigen-binding portion thereof that competes for binding to human MET with an antibody having an H-CDR1, H-CDR2, H-CDR3, L-CDR1, L-CDR2, and L- CDR3 that comprise or consist of the amino acid sequences of SEQ ID NOs: 1 , 2, 3, 4, 5, and 6, respectively.
- the antibody composition comprises an anti-MET antibody or an antigen-binding portion thereof that binds to the same epitope of human MET as an antibody having an H-CDR1, H-CDR2, H-CDR3, L-CDR1, L-CDR2, and L- CDR3 that comprise or consist of the amino acid sequences of SEQ ID NOs: 1, 2, 3, 4, 5, and 6, respectively.
- the antibody composition comprises an anti-MET antibody or an antigen-binding portion thereof that competes for binding to human MET with an antibody having a heavy chain variable domain comprising or consisting of the amino acid sequence of SEQ ID NO: 7 and a light chain variable domain comprising or consisting of the amino acid sequence of SEQ ID NO: 8.
- the antibody composition comprises an anti-MET antibody or an antigen-binding portion thereof that binds to the same epitope of human MET as an antibody having a heavy chain variable domain comprising or consisting of the amino acid sequence of SEQ ID NO: 7 and a light chain variable domain comprising or consisting of the amino acid sequence of SEQ ID NO: 8.
- the composition is an antibody composition or an ADC composition comprising an anti-MET antibody or antigen-binding portion thereof that has an H-CDR1 , H-CDR2, and H-CDR3 comprising or consisting of the amino acid sequences of SEQ ID NOs: 1, 2, and 3, respectively.
- the composition is an antibody composition or an ADC composition comprising an anti-MET antibody or antigen-binding portion thereof that has an L-CDR1 , L-CDR2, and L-CDR3 comprising or consisting of the amino acid sequences of SEQ ID NOS: 4, 5, and 6, respectively.
- the composition is an antibody composition or an ADC composition comprising an anti-MET antibody or antigen-binding portion thereof that comprises at least two, three, four or five CDR sequences selected from the group consisting of H-CDR1 SEQ ID NO: 1 , H-CDR2 SEQ ID NO: 2, H-CDR3 SEQ ID NO: 3, L-CDR1 SEQ ID NO: 4, L-CDR2 SEQ ID NO: 5, and L-CDR3 SEQ ID NO: 6.
- the composition is an antibody composition or an ADC composition comprising an anti-MET antibody or antigen-binding portion thereof that has an H-CDR1, H-CDR2, and H-CDR3 comprising or consisting of the amino acid sequences of SEQ ID NOs: 1 , 2, and 3, respectively, and an L-CDR1, L- CDR2, and L-CDR3 comprising or consisting of the amino acid sequences of SEQ ID NOS: 4, 5, and 6, respectively.
- the composition is an antibody composition or an ADC composition comprising an anti-MET antibody or antigen-binding portion thereof that has a heavy chain variable domain comprising or consisting of the amino acid sequence of SEQ ID NO: 7 and a light chain variable domain comprising or consisting of the amino acid sequence of SEQ ID NO: 8.
- the composition is an antibody composition or an ADC composition comprising an anti-MET antibody or antigen-binding portion thereof that has a heavy chain comprising or consisting of the amino acid sequence of SEQ ID NO: 11 and a light chain comprising or consisting of the amino acid sequence of SEQ ID NO: 12.
- the composition is an antibody composition or an ADC composition comprising an anti-MET antibody or antigen-binding portion thereof that has a heavy chain comprising or consisting of the amino acid sequence of SEQ ID NO: 13 and a light chain comprising or consisting of the amino acid sequence of SEQ ID NO: 14.
- the composition is an antibody composition or an ADC composition comprising an anti-MET antibody or antigen-binding portion thereof that has a heavy chain variable domain at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to the amino acid sequence of SEQ ID NO: 7 and a light chain variable domain at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to the amino acid sequence of SEQ ID NO: 8.
- the composition is an antibody composition or an ADC composition comprising an anti-MET antibody or antigen-binding portion thereof that has a heavy chain at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to the amino acid sequence of SEQ ID NO: 11 and a light chain at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to the amino acid sequence of SEQ ID NO: 12.
- the composition is an antibody composition or an ADC composition comprising an anti-MET antibody or antigen-binding portion thereof that has a heavy chain at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to the amino acid sequence of SEQ ID NO: 13 and a light chain at least 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identical to the amino acid sequence of SEQ ID NO: 14.
- the invention provides a bispecific antibody composition comprising a bispecific antibody comprising an anti-MET antibody or antigen-binding portion thereof of this invention.
- the bispecific antibody composition may comprise a bispecific binding molecule comprising the anti-MET antibody 8902.
- the bispecific antibody composition comprises an anti-MET antibody or an antigenbinding portion thereof having an H-CDR1, H-CDR2, and H-CDR3 comprising or consisting of the amino acid sequences of SEQ ID NOs: 1 , 2, and 3, respectively, and/or an L-CDR1, L-CDR2, and L-CDR3 comprising or consisting of the amino acid sequences of SEQ ID NOs: 4, 5, and 6, respectively.
- the bispecific antibody composition comprises an anti-MET antibody or an antigen-binding portion thereof having a heavy chain variable domain comprising or consisting of the amino acid sequence of SEQ ID NO: 7 and/or a light chain variable domain comprising or consisting of the amino acid sequence of SEQ ID NO: 8.
- the bispecific antibody composition comprises an anti-MET antibody or an antigen-binding portion thereof having a heavy chain comprising or consisting of the amino acid sequence of SEQ ID NO: 11 and/or a light chain comprising or consisting of the amino acid sequence of SEQ ID NO: 12.
- the bispecific antibody composition comprises an anti-MET antibody or an antigen-binding portion thereof having a heavy chain comprising or consisting of the amino acid sequence of SEQ ID NO: 13 and/or a light chain comprising or consisting of the amino acid sequence of SEQ ID NO: 14.
- the bispecific antibody composition may comprise a bispecific binding molecule having a dual variable domain antibody, i.e., wherein the two arms of the antibody comprise two different variable domains, or may be in the form of an antibody fragment such as a bispecific Fab fragment or a bispecific scFv.
- Such bispecific antibody composition may comprise bispecific binding molecules or polyvalent antibodies having the binding specificity of an anti-MET antibody or antigen-binding portion thereof described herein and the binding specificity of another antibody that targets a same or a different protein, such as an immune checkpoint protein, a cancer antigen, or a cell surface molecule whose activity mediates a disease condition such as cancer.
- the bispecific antibody composition comprises a bispecific binding molecule that has the binding specificities of the first anti-Met antibody 8902 and a second antibody or antigen-binding portions thereof. In some embodiments, the bispecific antibody composition comprises a bispecific binding molecule having the binding specificities of the first anti-Met antibody 8902 and the second anti-Met antibody 9006 or antigen-binding portions thereof. In some embodiments, the bispecific antibody composition comprises a bispecific binding molecule having the binding specificities of the first anti-Met antibody 8902 and the second anti-Met antibody 9338 or antigen-binding portions thereof.
- the present invention also provides nucleic acid molecules and sequences encoding anti-MET antibodies or antigen-binding portions thereof described herein.
- different nucleic acid molecules encode the heavy chain and light chain amino acid sequences of the anti-MET antibody or an antigen-binding portion thereof.
- the same nucleic acid molecule encodes the heavy chain and light chain amino acid sequences of the anti-MET antibody or an antigen-binding portion thereof.
- a reference to a nucleotide sequence encompasses its complement unless otherwise specified. Thus, a reference to a nucleic acid having a particular sequence should be understood to encompass its complementary strand, with its complementary sequence.
- polynucleotide as referred to herein means a polymeric form of nucleotides of at least 10 bases in length, either ribonucleotides or deoxynucleotides or a modified form of either type of nucleotide. The term includes single and double stranded forms.
- FASTA which includes, e.g., the programs FASTA2 and FASTA3, provides alignments and percent sequence identity of the regions of the best overlap between the query and search sequences (Pearson, Methods Enzymol. 183:63-98 (1990); Pearson, Methods Mol. Biol. 132:185-219 (2000); Pearson, Methods Enzymol. 266:227-258 (1996); Pearson, J. Mol. Biol. 276:71-84 (1998)). Unless otherwise specified, default parameters for a particular program or algorithm are used. For instance, percent sequence identity between nucleic acid sequences can be determined using FASTA with its default parameters (a word size of 6 and the NOPAM factor for the scoring matrix) or using Gap with its default parameters as provided in GCG Version 6.1.
- the invention provides a nucleic acid molecule comprising a nucleotide sequence of SEQ ID NOs: 9 or 10.
- the nucleic acid molecule may comprise the nucleotide sequences of SEQ ID NOs: 9 and 10.
- the nucleic acid molecules may be isolated.
- the present invention provides a vector suitable for expressing one of the chains of an antibody or antigen-binding portion thereof as described herein.
- the term “vector”, as used herein, means a nucleic acid molecule capable of transporting another nucleic acid to which it has been linked.
- the vector is a plasmid, i.e., a circular double stranded piece of DNA into which additional DNA segments may be ligated.
- the vector is a viral vector, wherein additional DNA segments may be ligated into the viral genome.
- the vectors are capable of autonomous replication in a host cell into which they are introduced (e.g., bacterial vectors having a bacterial origin of replication and episomal mammalian vectors).
- the vectors e.g., non-episomal mammalian vectors
- the vectors can be integrated into the genome of a host cell upon introduction into the host cell, and thereby are replicated along with the host genome.
- certain vectors are capable of directing the expression of genes to which they are operatively linked. Such vectors are referred to herein as “recombinant expression vectors” (or simply, “expression vectors”).
- the invention provides vectors comprising nucleic acid molecules that encode the heavy chain of an anti-MET antibody of the invention or an antigen-binding portion thereof, the light chain of an anti-MET antibody of the invention or an antigenbinding portion thereof, or both the heavy and light chains of an anti-MET antibody of the invention or an antigen-binding portion thereof.
- the invention further provides vectors comprising nucleic acid molecules encoding fusion proteins, modified antibodies, antibody fragments, and probes thereof.
- a nucleic acid molecule encoding the heavy and/or light chain of an anti-MET antibody or portion thereof can be isolated from any source that produces such an antibody or portion.
- the nucleic acid molecules are isolated from B cells that express an anti-MET antibody isolated from an animal immunized with a human MET antigen, or from an immortalized cell produced from such a B cell. Methods of isolating nucleic acids encoding an antibody are well- known in the art.
- mRNA may be isolated and used to produce cDNA for use in polymerase chain reaction (PCR) or cDNA cloning of antibody genes.
- PCR polymerase chain reaction
- a nucleic acid molecule of the invention can be synthesized rather than isolated.
- a nucleic acid molecule of the invention can comprise a nucleotide sequence encoding a VH domain from an anti-MET antibody or antigen-binding portion of the invention joined in-frame to a nucleotide sequence encoding a heavy chain constant domain from any source.
- a nucleic acid molecule of the invention can comprise a nucleotide sequence encoding a VL domain from an anti-MET antibody or antigen-binding portion of the invention joined in-frame to a nucleotide sequence encoding a light chain constant domain from any source.
- nucleic acid molecules encoding the variable domain of the heavy (VH) and/or light (VL) chains may be “converted” to full-length antibody genes.
- nucleic acid molecules encoding the VH or VL domains are converted to full-length antibody genes by insertion into an expression vector already encoding heavy chain constant (CH) or light chain constant (CL) domains, respectively, such that the VH segment is operatively linked to the CH segment(s) within the vector, and/or the VL segment is operatively linked to the CL segment within the vector.
- CH heavy chain constant
- CL light chain constant
- nucleic acid molecules encoding the VH and/or VL domains are converted into full- length antibody genes by linking, e.g., ligating, a nucleic acid molecule encoding a VH and/or VL domains to a nucleic acid molecule encoding a CH and/or CL domain using standard molecular biological techniques. Nucleic acid molecules encoding the full-length heavy and/or light chains may then be expressed from a cell into which they have been introduced and the anti-MET antibody isolated.
- the nucleic acid molecules may be used to recombinantly express large quantities of anti-MET antibodies.
- the nucleic acid molecules also may be used to produce chimeric antibodies, bispecific antibodies, single chain antibodies, immunoadhesins, diabodies, mutated antibodies and antibody derivatives, as described herein.
- a nucleic acid molecule of the invention is used as a probe or PCR primer for a specific antibody sequence.
- the nucleic acid can be used as a probe in diagnostic methods or as a PCR primer to amplify regions of DNA that could be used, inter alia, to isolate additional nucleic acid molecules encoding variable domains of anti-MET antibodies.
- the nucleic acid molecules are oligonucleotides. In some embodiments, the oligonucleotides are from highly variable domains of the heavy and light chains of the antibody of interest.
- the oligonucleotides encode all or a part of one or more of the CDRs of the anti-MET antibodies or antigen-binding portions thereof of the invention as described herein.
- the nucleic acid molecules and vectors may be used to make mutated anti-MET antibodies.
- the antibodies may be mutated in the variable domains of the heavy and/or light chains, e.g., to alter a binding property of the antibody.
- a mutation may be made in one or more of the CDR regions to increase or decrease the KD of the anti-MET antibody, to increase or decrease k O ff, or to alter the binding specificity of the antibody.
- one or more mutations are made at an amino acid residue that is known to be changed compared to the germline in a monoclonal antibody of the invention.
- the mutations may be made in a CDR region or framework region of a variable domain, or in a constant domain.
- the mutations are made in a variable domain.
- one or more mutations are made at an amino acid residue that is known to be changed compared to the germline in a CDR region or framework region of a variable domain of an antibody or antigen-binding portion thereof of the invention.
- the framework region(s) are mutated so that the resulting framework region(s) have the amino acid sequence of the corresponding germline gene.
- a mutation may be made in a framework region or constant domain to increase the half-life of the anti-MET antibody. See, e.g., PCT Publication WO 00/09560.
- a mutation in a framework region or constant domain also can be made to alter the immunogenicity of the antibody, and/or to provide a site for covalent or non-covalent binding to another molecule.
- a single antibody may have mutations in any one or more of the CDRs or framework regions of the variable domain or in the constant domain.
- the anti-MET antibodies of the invention or antigen-binding portions thereof are expressed by inserting DNAs encoding partial or full-length light and heavy chains, obtained as described above, into expression vectors such that the genes are operatively linked to necessary expression control sequences such as transcriptional and translational control sequences.
- Expression vectors include plasmids, retroviruses, adenoviruses, adeno-associated viruses (AAV), plant viruses such as cauliflower mosaic virus, tobacco mosaic virus, cosmids, YACs, EBV derived episomes, and the like.
- the antibody gene may be ligated into a vector such that transcriptional and translational control sequences within the vector serve their intended function of regulating the transcription and translation of the antibody gene.
- the expression vector and expression control sequences may be chosen to be compatible with the expression host cell used.
- the antibody light chain gene and the antibody heavy chain gene can be inserted into separate vectors. In one embodiment, both genes are inserted into the same expression vector.
- the antibody genes may be inserted into the expression vector by standard methods (e.g., ligation of complementary restriction sites on the antibody gene fragment and vector, or blunt end ligation if no restriction sites are present).
- a convenient vector is one that encodes a functionally complete human CH or CL immunoglobulin sequence, with appropriate restriction sites engineered so that any VH or VL sequence can easily be inserted and expressed, as described above.
- splicing usually occurs between the splice donor site in the inserted J region and the splice acceptor site preceding the human C domain, and also at the splice regions that occur within the human CH exons. Polyadenylation and transcription termination may occur at native chromosomal sites downstream of the coding regions.
- the recombinant expression vector also can encode a signal peptide that facilitates secretion of the antibody chain from a host cell.
- the antibody chain gene may be cloned into the vector such that the signal peptide is linked in-frame to the amino terminus of the immunoglobulin chain.
- the signal peptide can be an immunoglobulin signal peptide or a heterologous signal peptide (i.e. , a signal peptide from a non-immunoglobulin protein).
- the recombinant expression vectors of the invention may carry regulatory sequences that control the expression of the antibody chain genes in a host cell. It will be appreciated by those skilled in the art that the design of the expression vector, including the selection of regulatory sequences, may depend on such factors as the choice of the host cell to be transformed, the level of expression of protein desired, etc.
- Preferred regulatory sequences for mammalian host cell expression include viral elements that direct high levels of protein expression in mammalian cells, such as promoters and/or enhancers derived from retroviral LTRs, cytomegalovirus (CMV) (such as the CMV promoter/enhancer), Simian Virus 40 (SV40) (such as the SV40 promoter/enhancer), adenovirus, (e.g., the adenovirus major late promoter (AdMLP)), polyoma and strong mammalian promoters such as native immunoglobulin and actin promoters.
- CMV cytomegalovirus
- SV40 Simian Virus 40
- AdMLP adenovirus major late promoter
- polyoma e.g., the adenovirus major late promoter (AdMLP)
- AdMLP adenovirus major late promoter
- polyoma such as native immunoglobulin and actin promoters.
- viral regulatory elements for further description of viral regulatory elements, and sequences thereof, see e.g., US Patents 5,168,062, 4,510,245 and 4,968,615.
- Methods for expressing antibodies in plants including a description of promoters and vectors, as well as transformation of plants, are known in the art. See, e.g., US Patent 6,517,529.
- Methods of expressing polypeptides in bacterial cells or fungal cells, e.g., yeast cells are also well known in the art.
- the recombinant expression vectors of the invention may carry additional sequences, such as sequences that regulate replication of the vector in host cells (e.g., origins of replication) and selectable marker genes.
- the selectable marker gene facilitates selection of host cells into which the vector has been introduced (see e.g., US Patents 4,399,216, 4,634,665 and 5,179,017).
- the selectable marker gene confers resistance to drugs, such as G418, hygromycin or methotrexate, on a host cell into which the vector has been introduced.
- selectable marker genes include the dihydrofolate reductase (DHFR) gene (for use in dhfr-host cells with methotrexate selection/amplification), the neo gene (for G418 selection), and the glutamate synthetase gene.
- DHFR dihydrofolate reductase
- expression control sequence means polynucleotide sequences that are necessary to effect the expression and processing of coding sequences to which they are ligated.
- Expression control sequences include appropriate transcription initiation, termination, promoter and enhancer sequences; efficient RNA processing signals such as splicing and polyadenylation signals; sequences that stabilize cytoplasmic mRNA; sequences that enhance translation efficiency (i.e., Kozak consensus sequence); sequences that enhance protein stability; and when desired, sequences that enhance protein secretion.
- control sequences differs depending upon the host organism; in prokaryotes, such control sequences generally include promoter, ribosomal binding site, and transcription termination sequence; in eukaryotes, generally, such control sequences include promoters and transcription termination sequence.
- control sequences is intended to include, at a minimum, all components whose presence is essential for expression and processing, and can also include additional components whose presence is advantageous, for example, leader sequences and fusion partner sequences.
- the invention provides methods for producing a cell line that produces a human monoclonal antibody or an antigen-binding portion thereof directed against MET, comprising (a) immunizing a non-human transgenic animal with MET, a portion of MET or a cell or tissue expressing MET; (b) allowing the transgenic animal to mount an immune response to MET; (c) isolating antibody- producing cells from the transgenic animal; (d) immortalizing the antibodyproducing cells; (e) creating individual monoclonal populations of the immortalized antibody-producing cells; and (f) screening the immortalized antibody-producing cells to identify an antibody directed against MET.
- the invention provides a cell line that produces a human anti- MET antibody.
- the cell line is a hybridoma cell line.
- the hybridomas are mouse hybridomas, as described above.
- the hybridomas are produced in a non-human, non-mouse species such as rats, sheep, pigs, goats, cattle or horses.
- the hybridomas are human hybridomas.
- a transgenic animal is immunized with an MET antigen
- primary cells e.g., spleen or peripheral blood B cells
- individual cells producing antibodies specific for the desired antigen are identified.
- Polyadenylated mRNA from each individual cell is isolated and reverse transcription polymerase chain reaction (RT-PCR) is performed using sense primers that anneal to variable domain sequences, e.g., degenerate primers that recognize most or all of the FR1 regions of human heavy and light chain variable domain genes and anti-sense primers that anneal to constant or joining region sequences.
- RT-PCR reverse transcription polymerase chain reaction
- cDNAs of the heavy and light chain variable domains are then cloned and expressed in any suitable host cell, e.g., a myeloma cell, as chimeric antibodies with respective immunoglobulin constant regions, such as the heavy chain and K or A constant domains. See Babcook et al., Proc Natl Acad Sci USA 93:7843-48 (1996). Anti-MET antibodies may then be identified and isolated as described herein.
- the invention provides a method for producing an anti-MET antibody or antigenbinding portion thereof comprising the steps of synthesizing a library of human antibodies on phage, screening the library with MET or an antibody-binding portion thereof, isolating phage that bind to MET, and obtaining the antibody from the phage.
- one method for preparing the library of antibodies for use in phage display techniques comprises the steps of immunizing a non-human animal with MET or an antigenic portion thereof to create an immune response, extracting antibody-producing cells from the immunized animal; isolating RNA encoding heavy and light chains of antibodies of the invention from the extracted cells, reverse transcribing the RNA to produce cDNA, amplifying the cDNA using primers, and inserting the cDNA into a phage display vector such that antibodies are expressed on the phage.
- Recombinant anti-MET antibodies of the invention may be obtained in this way.
- Recombinant human anti-MET antibodies of the invention can be isolated by screening a recombinant combinatorial antibody library.
- the library is a scFv phage display library, generated using human VL and VH cDNAs prepared from mRNA isolated from B cells. Methods for preparing and screening such libraries are known in the art. Kits for generating phage display libraries are commercially available (e.g., the Pharmacia Recombinant Phage Antibody System, catalog no. 27-9400-01; and the Stratagene SurfZAPTM phage display kit, catalog no. 240612). There also are other methods and reagents that can be used in generating and screening antibody display libraries (see, e.g., U.S.
- a human anti-MET antibody as described herein is first used to select human heavy and light chain sequences having similar binding activity toward MET, using the epitope imprinting methods described in PCT Publication WO 93/06213, incorporated herein by reference.
- the antibody libraries used in this method are preferably scFv libraries prepared and screened as described in PCT Publication WO 92/01047, McCafferty et al., Nature 348:552-554 (1990); and Griffiths et al., EMBO J 12:725-734 (1993).
- the scFv antibody libraries preferably are screened using human MET as the antigen.
- VL and VH segments of the preferred VL/VH pair(s) can be randomly mutated, preferably within the CDR3 region of VH and/or VL, in a process analogous to the in vivo somatic mutation process responsible for affinity maturation of antibodies during a natural immune response.
- This in vitro affinity maturation can be accomplished by amplifying VH and VL domains using PCR primers complimentary to the VH CDR3 or VL CDR3, respectively, which primers have been “spiked” with a random mixture of the four nucleotide bases at certain positions such that the resultant PCR products encode VH and VL segments into which random mutations have been introduced into the VH and/or VL CDR3 regions. These randomly mutated VH and VL segments can be re-screened for binding to MET.
- nucleic acids encoding the selected antibody can be recovered from the display package (e.g., from the phage genome) and subcloned into other expression vectors by standard recombinant DNA techniques. If desired, the nucleic acid can further be manipulated to create other antibody forms of the invention, as described herein.
- the DNA encoding the antibody is cloned into a recombinant expression vector and introduced into a mammalian host cell, as described herein.
- An additional aspect of the invention relates to methods for producing the antibody compositions and antibodies and antigen-binding portions thereof of the invention.
- One embodiment of this aspect of the invention relates to a method for producing an antibody as defined herein, comprising providing a recombinant host cell capable of expressing the antibody, cultivating said host cell under conditions suitable for expression of the antibody, and isolating the resulting antibody.
- Antibodies produced by such expression in such recombinant host cells are referred to herein as “recombinant antibodies”.
- the invention also provides progeny cells of such host cells, and antibodies produced by same.
- the term “recombinant host cell” (or simply “host cell”), as used herein, means a cell into which a recombinant expression vector has been introduced.
- the invention provides host cells that may comprise, e.g., a vector according to the invention described above.
- the invention also provides host cells that comprise, e.g., a nucleotide sequence encoding the heavy chain or an antigen-binding portion thereof, a nucleotide sequence encoding the light chain or an antigenbinding portion thereof, or both, of an anti-MET antibody or antigen-binding portion thereof of the invention.
- “recombinant host cell” and “host cell” mean not only the particular subject cell but also the progeny of such a cell. Because certain modifications may occur in succeeding generations due to either mutation or environmental influences, such progeny may not, in fact, be identical to the parent cell, but are still included within the scope of the term “host cell” as used herein.
- Nucleic acid molecules encoding anti-MET antibodies and vectors comprising these nucleic acid molecules can be used for transfection of a suitable mammalian, plant, bacterial or yeast host cell. Transformation can be by any known method for introducing polynucleotides into a host cell. Methods for introduction of heterologous polynucleotides into mammalian cells are well known in the art and include dextran-mediated transfection, calcium phosphate precipitation, polybrene-mediated transfection, protoplast fusion, electroporation, encapsulation of the polynucleotide(s) in liposomes, and direct microinjection of the DNA into nuclei. In addition, nucleic acid molecules may be introduced into mammalian cells by viral vectors.
- Methods of transforming cells are well known in the art. See, e.g., US Patents 4,399,216, 4,912,040, 4,740,461, and 4,959,455.
- Methods of transforming plant cells are well known in the art, including, e.g., Agrobacterium-mediated transformation, biolistic transformation, direct injection, electroporation and viral transformation.
- Methods of transforming bacterial and yeast cells are also well known in the art.
- Mammalian cell lines available as hosts for expression are well known in the art and include many immortalized cell lines available from the American Type Culture Collection (ATCC). These include, inter alia, Chinese hamster ovary (CHO) cells, NSO cells, SP2 cells, HEK-293T cells, 293 Freestyle cells (Invitrogen), NIH-3T3 cells, HeLa cells, baby hamster kidney (BHK) cells, African green monkey kidney cells (COS), human hepatocellular carcinoma cells (e.g., Hep G2), A549 cells, and a number of other cell lines. Cell lines of particular preference are selected through determining which cell lines have high expression levels. Other cell lines that may be used are insect cell lines, such as Sf9 or Sf21 cells.
- the antibodies When recombinant expression vectors encoding antibody genes are introduced into mammalian host cells, the antibodies are produced by culturing the host cells for a period of time sufficient to allow for expression of the antibody in the host cells or, more preferably, secretion of the antibody into the culture medium in which the host cells are grown. Antibodies can be recovered from the culture medium using standard protein purification methods.
- Plant host cells include, e.g., Nicotiana, Arabidopsis, duckweed, corn, wheat, potato, etc.
- Bacterial host cells include E. coli and Streptomyces species.
- Yeast host cells include Schizosaccharomyces pombe, Saccharomyces cerevisiae and Pichia pastoris.
- the glutamine synthetase gene expression system (the GS system) is a common approach for enhancing expression under certain conditions.
- the GS system is discussed in whole or part in connection with EP patents 0216 846, 0256 055, 0 323 997 and 0 338 841.
- An antibody or antigen-binding portion thereof or antibody composition of the present invention may be produced by methods generally known in the art for production of recombinant monoclonal or polyclonal antibodies.
- any method known in the art for production of recombinant monoclonal antibodies may be used.
- the individual antibodies may be produced separately, i.e., each antibody being produced in a separate bioreactor, or the individual antibodies may be produced together in single bioreactor. If the antibody composition is produced in more than one bioreactor, the purified antibody composition can be obtained by pooling the antibodies obtained from individually purified supernatants from each bioreactor.
- Various approaches for production of a polyclonal antibody composition in multiple bioreactors, where the cell lines or antibody preparations are combined at a later point upstream or prior to or during downstream processing, are described in WO 2009/129814.
- WO 2004/061104 In the case of producing individual antibodies in a single bioreactor, this may be performed, e.g., as described in WO 2004/061104 or WO 2008/145133.
- the method described in WO 2004/061104 is based on site-specific integration of the antibody coding sequence into the genome of the individual host cells, while the method of WO 2008/145133 involves an alternative approach using random integration to produce antibodies in a single bioreactor.
- Anti-MET antibodies and antigen-binding portions thereof of the invention also can be produced transgenically through the generation of a mammal or plant that is transgenic for the immunoglobulin heavy and light chain sequences of interest and production of the antibody in a recoverable form therefrom.
- anti-MET antibodies and portions can be produced in, and recovered from, the milk of goats, cows, or other mammals. See, e.g., US patents 5,827,690, 5,756,687, 5,750,172, and 5,741 ,957.
- non-human transgenic animals that comprise human immunoglobulin loci are immunized with human MET or an immunogenic portion thereof, as described above. Methods for making antibodies in plants are described, e.g., in US patents 6,046,037 and 5,959,177.
- non-human transgenic animals or plants are produced by introducing one or more nucleic acid molecules encoding an anti-MET antibody or antigen-binding portion thereof of the invention (e.g., any of the above-described nucleic acid molecules encoding an anti-MET antibody or antigen-binding portion thereof) into the animal or plant by standard transgenic techniques. See, e.g., US Patent 6,417,429.
- the transgenic cells used for making the transgenic animal can be embryonic stem cells or somatic cells or a fertilized egg.
- the transgenic non- human organisms can be chimeric, nonchimeric heterozygotes, and nonchimeric homozygotes.
- the transgenic non-human animals have a targeted disruption and replacement by a targeting construct that encodes a heavy chain and/or a light chain of interest.
- the non-human transgenic animals or plants may comprise, e.g., a nucleotide sequence encoding the heavy chain or an antigen-binding portion thereof, a nucleotide sequence encoding the light chain or an antigen-binding portion thereof, or both, of an anti-MET antibody of the invention.
- the transgenic animals comprise and express nucleic acid molecules encoding heavy and light chains, or antigenbinding portions thereof, that specifically bind to human MET.
- the anti-MET antibodies or portions may be made in any transgenic animal.
- the non-human animals are mice, rats, sheep, pigs, goats, cattle or horses.
- the non-human transgenic animal may express said encoded polypeptides in, e.g., blood, milk, urine, saliva, tears, mucus and other bodily fluids.
- compositions comprising as an active ingredient (or as the sole active ingredient) an anti-MET antibody or antigen-binding portion thereof, an ADC comprising an anti-MET antibody or antigen-binding portion thereof, or an anti-MET antibody composition or an anti- MET antibody comprising ADC composition of the invention.
- the compositions are intended for amelioration, prevention, and/or treatment of a MET-mediated disorder (e.g., a disorder characterized by overexpression of MET) and/or cancer.
- compositions are intended for amelioration, prevention, and/or treatment of non-small cell lung cancer, gastric cancer, hepatocellular carcinoma, oesophageal cancer, colorectal cancer, kidney papillary cell cancer, glioblastoma, renal cell carcinoma, prostate cancer, and/or adrenocortical carcinoma.
- the antibodies of the invention or antigen-binding portions thereof are suitable to be administered as a formulation in association with one or more pharmaceutically acceptable excipient(s).
- excipient is used herein to describe any ingredient other than the compound(s) of the invention. The choice of excipient(s) will to a large extent depend on factors such as the particular mode of administration, the effect of the excipient on solubility and stability, and the nature of the dosage form.
- pharmaceutically acceptable excipient includes any and all solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents, and the like that are physiologically compatible.
- compositions examples include water, saline, phosphate buffered saline, dextrose, glycerol, ethanol and the like, as well as combinations thereof.
- isotonic agents for example, sugars, polyalcohols such as mannitol, sorbitol, or sodium chloride in the composition.
- additional examples of pharmaceutically acceptable substances are wetting agents or minor amounts of auxiliary substances such as wetting or emulsifying agents, preservatives or buffers, which enhance the shelf life or effectiveness of the antibody.
- compositions of the present invention and methods for their preparation will be readily apparent to those skilled in the art. Such compositions and methods for their preparation may be found, for example, in Remington’s Pharmaceutical Sciences, 19th Edition (Mack Publishing Company, 1995). Pharmaceutical compositions are preferably manufactured under GMP (good manufacturing practices) conditions.
- a pharmaceutical composition of the invention may be prepared, packaged, or sold in bulk, as a single unit dose, or as a plurality of single unit doses.
- a “unit dose” is a discrete amount of the pharmaceutical composition comprising a predetermined amount of the active ingredient.
- the amount of the active ingredient is generally equal to the dosage of the active ingredient which would be administered to a subject or a convenient fraction of such a dosage such as, for example, one-half or one-third of such a dosage.
- Any method for administering peptides, proteins or antibodies accepted in the art may suitably be employed for the antibodies and antigen-binding portions of the invention.
- parenteral administration of a pharmaceutical composition includes any route of administration characterized by physical breaching of a tissue of a subject and administration of the pharmaceutical composition through the breach in the tissue, thus generally resulting in the direct administration into the blood stream, into muscle, or into an internal organ.
- Parenteral administration thus includes, but is not limited to, administration of a pharmaceutical composition by injection of the composition, by application of the composition through a surgical incision, by application of the composition through a tissue-penetrating non-surgical wound, and the like.
- parenteral administration is contemplated to include, but is not limited to, subcutaneous, intraperitoneal, intramuscular, intrasternal, intravenous, intraarterial, intrathecal, intraventricular, intraurethral, intracranial, intrasynovial injection or infusions; and kidney dialytic infusion techniques. Regional perfusion is also contemplated. Preferred embodiments include the intravenous and the subcutaneous routes.
- Formulations of a pharmaceutical composition suitable for parenteral administration typically comprise the active ingredient combined with a pharmaceutically acceptable carrier, such as sterile water or sterile isotonic saline. Such formulations may be prepared, packaged, or sold in a form suitable for bolus administration or for continuous administration. Injectable formulations may be prepared, packaged, or sold in unit dosage form, such as in ampoules or in multi-dose containers containing a preservative. Formulations for parenteral administration include, but are not limited to, suspensions, solutions, emulsions in oily or aqueous vehicles, pastes, and the like. Such formulations may further comprise one or more additional ingredients including, but not limited to, suspending, stabilizing, or dispersing agents.
- a pharmaceutically acceptable carrier such as sterile water or sterile isotonic saline.
- Such formulations may be prepared, packaged, or sold in a form suitable for bolus administration or for continuous administration.
- injectable formulations may be prepared, packaged, or sold in unit dosage
- the active ingredient is provided in dry (i.e., powder or granular) form for reconstitution with a suitable vehicle (e.g., sterile pyrogen-free water) prior to parenteral administration of the reconstituted composition.
- a suitable vehicle e.g., sterile pyrogen-free water
- Parenteral formulations also include aqueous solutions which may contain excipients such as salts, carbohydrates and buffering agents (preferably to a pH of from 3 to 9), but, for some applications, they may be more suitably formulated as a sterile non-aqueous solution or as a dried form to be used in conjunction with a suitable vehicle such as sterile, pyrogen-free water.
- parenteral administration forms include solutions or suspensions in sterile aqueous solutions, for example, aqueous propylene glycol or dextrose solutions. Such dosage forms can be suitably buffered, if desired.
- Other parentally-administrable formulations which are useful include those which comprise the active ingredient in microcrystalline form, or in a liposomal preparation.
- Formulations for parenteral administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- sterile injectable solutions can be prepared by incorporating the anti-MET antibody or antigen-binding portion thereof or anti-MET antibody composition in the required amount in an appropriate solvent with one or a combination of ingredients enumerated above, as required, followed by filtered sterilization.
- dispersions are prepared by incorporating the active compound into a sterile vehicle that contains a basic dispersion medium and the required other ingredients from those enumerated above.
- the preferred methods of preparation are vacuum drying and freeze-drying that yields a powder of the active ingredient plus any additional desired ingredient from a previously sterile-filtered solution thereof.
- the proper fluidity of a solution can be maintained, for example, by the use of a coating such as lecithin, by the maintenance of the required particle size in the case of dispersion and by the use of surfactants.
- Prolonged absorption of injectable compositions can be brought about by including in the composition an agent that delays absorption, for example, monostearate salts and gelatin, and/or by using modified-release coatings (e.g., slow-release coatings).
- the antibodies of the invention can also be administered intranasally or by inhalation, typically in the form of a dry powder (either alone, as a mixture, or as a mixed component particle, for example, mixed with a suitable pharmaceutically acceptable excipient) from a dry powder inhaler, as an aerosol spray from a pressurised container, pump, spray, atomiser (preferably an atomiser using electrohydrodynamics to produce a fine mist), or nebuliser, with or without the use of a suitable propellant, or as nasal drops.
- a dry powder either alone, as a mixture, or as a mixed component particle, for example, mixed with a suitable pharmaceutically acceptable excipient
- atomiser preferably an atomiser using electrohydrodynamics to produce a fine mist
- nebuliser preferably an atomiser using electrohydrodynamics to produce a fine mist
- the pressurised container, pump, spray, atomizer, or nebuliser generally contains a solution or suspension of an antibody of the invention comprising, for example, a suitable agent for dispersing, solubilising, or extending release of the active, a propellant(s) as solvent.
- a suitable agent for dispersing, solubilising, or extending release of the active, a propellant(s) as solvent Prior to use in a dry powder or suspension formulation, the drug product is generally micronised to a size suitable for delivery by inhalation (typically less than 5 microns). This may be achieved by any appropriate comminuting method, such as spiral jet milling, fluid bed jet milling, supercritical fluid processing to form nanoparticles, high pressure homogenisation, or spray drying.
- Capsules, blisters and cartridges for use in an inhaler or insufflator may be formulated to contain a powder mix of the compound of the invention, a suitable powder base and a performance modifier.
- a suitable solution formulation for use in an atomiser using electrohydrodynamics to produce a fine mist may contain a suitable dose of the antibody of the invention per actuation and the actuation volume may for example vary from 1 pL to 100 pL.
- Suitable flavours, such as menthol and levomenthol, or sweeteners, such as saccharin or saccharin sodium, may be added to those formulations of the invention intended for inhaled/intranasal administration.
- Formulations for inhaled/intranasal administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed- , sustained-, pulsed-, controlled-, targeted and programmed release.
- the dosage unit is determined by means of a valve which delivers a metered amount.
- Units in accordance with the invention are typically arranged to administer a metered dose or “puff” of an antibody of the invention.
- the overall daily dose will typically be administered in a single dose or, more usually, as divided doses throughout the day.
- the antibodies and antibody portions of the invention may also be formulated for an oral route administration.
- Oral administration may involve swallowing, so that the compound enters the gastrointestinal tract, and/or buccal, lingual, or sublingual administration by which the compound enters the blood stream directly from the mouth.
- Formulations suitable for oral administration include solid, semi-solid and liquid systems such as tablets; soft or hard capsules containing multi- or nanoparticulates, liquids, or powders; lozenges (including liquid-filled); chews; gels; fast dispersing dosage forms; films; ovules; sprays; and buccal/mucoadhesive patches.
- Liquid formulations include suspensions, solutions, syrups and elixirs. Such formulations may be employed as fillers in soft or hard capsules (made, for example, from gelatin or hydroxypropylmethylcellulose) and typically comprise a carrier, for example, water, ethanol, polyethylene glycol, propylene glycol, methylcellulose, or a suitable oil, and one or more emulsifying agents and/or suspending agents. Liquid formulations may also be prepared by the reconstitution of a solid, for example, from a sachet.
- antibody compositions and antibodies and antigen-binding portions thereof of the invention is in the form of immunoconjugates, i.e., antibodies or antigen-binding portions conjugated to one or more agents such as anti-cancer agents.
- anti-cancer agents may be conjugated to the antibodies of the invention, including cytotoxic agents (e.g., conventional chemotherapy agents and other small molecule anti-cancer drugs), cytokines (in which case the conjugate may be termed an “immunocytokine”), toxins (in which case the conjugate may be termed an “immunotoxin”) and radionuclides.
- cytotoxic agents e.g., conventional chemotherapy agents and other small molecule anti-cancer drugs
- cytokines in which case the conjugate may be termed an “immunocytokine”
- toxins in which case the conjugate may be termed an “immunotoxin”
- radionuclides radionuclides.
- immunoconjugates have already been approved for clinical use. These include Zevalin® (a murine anti- CD20 antibody conjugated to 90 Y), Bexxar® (a murine anti-CD20 antibody conjugated to 131 1) and Mylotarg® (a humanized anti-CD33 antibody conjug
- immunoconjugates that have been tested in clinical trials include antibodies conjugated to, e.g., doxorubicin or a maytansinoid compound.
- Immunotoxins that have been tested in clinical trials include several antibodies conjugated to a truncated Pseudomonas exotoxin A.
- An immunocytokine comprising a humanized EpCAM antibody conjugated to IL-2 has also been tested.
- antibodies of the invention conjugated to cytotoxic agents may belong, e.g., to any of the major classes of chemotherapy drugs, including alkylating agents (e.g., carboplatin, cisplatin, oxaliplatin), antimetabolites (e.g., methotrexate, capecitabine, gemcitabine), anthracyclines (e.g., bleomycin, doxorubicin, mitomycin-C) and plant alkaloids (e.g., taxanes such as docetaxel and paclitaxel, and vinca alkaloids such as vinblastine, vincristine and vinorelbine).
- alkylating agents e.g., carboplatin, cisplatin, oxaliplatin
- antimetabolites e.g., methotrexate, capecitabine, gemcitabine
- anthracyclines e.g., bleomycin, doxorubicin, mitomycin-C
- immunoconjugates based on the antibodies of the invention may advantageously be based on highly cytotoxic agents such as calicheamicin or maytansine derivatives, or on toxins such as bacterial toxins (e.g., Pseudomonas exotoxin A, diphtheria toxin) or plant toxins (e.g., ricin).
- highly cytotoxic agents such as calicheamicin or maytansine derivatives
- toxins such as bacterial toxins (e.g., Pseudomonas exotoxin A, diphtheria toxin) or plant toxins (e.g., ricin).
- the conjugated anti-cancer agent in an immunoconjugate is generally linked to the antibody by means of a labile linker that is relatively stable in serum but which allows release of the agent when the immunoconjugate is internalized into the target cell.
- Suitable linkers include, for example, chemical linkers that are stable at neutral pH in serum but are subjected to acid hydrolysis in the mildly acidic conditions within the lysosomes subsequent to internalization, disulfide linkers that are cleaved by intracellular thiols, and peptide linkers that are stable in serum but which are subjected to enzymatic cleavage in intracellular compartments.
- the anti-MET antibodies and antigen-binding portions thereof and anti-MET compositions of the invention are used in the treatment of a MET- mediated disorder.
- the MET-mediated disorder is a condition characterized by overexpression of MET.
- the pharmaceutical composition is for use in the treatment of cancer, e.g., non-small cell lung cancer, gastric cancer, hepatocellular carcinoma, esophageal cancer, colorectal cancer, kidney papillary cell cancer, glioblastoma, adrenocortical carcinoma, renal cell carcinoma, prostate cancer, and other cancers that express or overexpress MET or rely on MET pathway activation.
- the antibodies or antibody compositions are used to treat a disorder, such as a cancer, characterized by abnormal MET overactivity.
- a disorder such as a cancer
- the abnormal overactivity stems from gene amplification, protein overexpression, a MET activating gene mutation (e.g., a point mutation or abnormal gene splicing event), or HGF overexpression.
- the anti-MET antibodies and antigen-binding portions thereof and anti-MET compositions of the invention may be used to treat a patient who is resistant to treatment with an agent targeting a different tyrosine kinase receptor.
- the patient is resistant to treatment with an ErbB kinase inhibitor.
- the ErbB kinase inhibitor targets EGFR, ErbB2, ErbB3, or ErbB4.
- the ErbB kinase inhibitor targets EGFR.
- the ErbB kinase inhibitor targets HER3.
- the ErbB kinase inhibitor may be selected from, e.g., gefitinib, erlotinib, cetuximab, pantinumumab, trastuzumab, or any combination thereof.
- agent is used herein to refer to a chemical compound, a mixture of chemical compounds, a biological molecule, an extract made from biological molecules, or a combination of two or more thereof.
- therapeutic agent or “drug” refers to an agent that is capable of modulating a biological process and/or has biological activity.
- chemotherapeutic agent or “anti-cancer agent” is used herein to refer to all agents that are effective in treating cancer (regardless of mechanism of action). Inhibition of metastasis or angiogenesis is frequently a property of a chemotherapeutic agent.
- Chemotherapeutic agents include antibodies, biological molecules, and small molecules.
- a chemotherapeutic agent may be a cytotoxic or cytostatic agent.
- cytostatic agent refers to an agent that inhibits or suppresses cell growth and/or multiplication of cells.
- cytotoxic agent refers to a substance that causes cell death primarily by interfering with a cell’s expression activity and/or functioning.
- cancer refers to the presence of cells possessing characteristics typical of cancer-causing cells, such as excessive cell growth or proliferation, uncontrolled proliferation, immortality, metastatic potential, rapid growth and proliferation rate, and/or certain morphological features. Often, cancer cells can be in the form of a tumor or mass, but such cells may exist alone within a subject, or may circulate in the blood stream as independent cells, such as leukemic or lymphoma cells.
- cancer includes all types of cancers and cancer metastases, including hematological cancers, solid tumors, sarcomas, carcinomas and other solid and non-solid tumor cancers.
- Hematological cancers may include B-cell malignancies, cancers of the blood (leukemias), cancers of plasma cells (myelomas, e.g., multiple myeloma), or cancers of the lymph nodes (lymphomas).
- B-cell malignancies include chronic lymphocytic leukemia (CLL), follicular lymphoma, mantle cell lymphoma, and diffuse large B- cell lymphoma.
- CLL chronic lymphocytic leukemia
- follicular lymphoma mantle cell lymphoma
- mantle cell lymphoma mantle cell lymphoma
- diffuse large B- cell lymphoma diffuse large B- cell lymphoma.
- Leukemias may include acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), chronic lymphocytic leukemia (CLL), chronic myelogenous leukemia (CML), chronic myelomonocytic leukemia (CMML), acute monocytic leukemia (AMoL), etc.
- ALL acute lymphoblastic leukemia
- AML acute myeloid leukemia
- CLL chronic lymphocytic leukemia
- CML chronic myelogenous leukemia
- CMML chronic myelomonocytic leukemia
- AoL acute monocytic leukemia
- Lymphomas may include Hodgkin's lymphoma, non-Hodgkin's lymphoma, etc.
- Other hematologic cancers may include myelodysplasia syndrome (MDS).
- Solid tumors may include carcinomas such as adenocarcinoma, e.g., breast cancer, pancreatic cancer, prostate cancer, colon or colorectal cancer, lung cancer, gastric cancer, cervical cancer, endometrial cancer, ovarian cancer, cholangiocarcinoma, glioma, melanoma, etc.
- carcinomas such as adenocarcinoma, e.g., breast cancer, pancreatic cancer, prostate cancer, colon or colorectal cancer, lung cancer, gastric cancer, cervical cancer, endometrial cancer, ovarian cancer, cholangiocarcinoma, glioma, melanoma, etc.
- the cancer is a melanoma, uveal melanoma, renal cancer, kidney cancer thyroid cancer, mesothelioma, liver hepatocellular cancer, lung cancer including non-small cell lung cancer and small cell lung cancer, gastric cancer including stomach cancer, pancreatic cancer, colorectal cancer, esophageal cancer, cholangiocarcinoma, head and neck cancer including oral cancer, cervical and endocervical cancer, bladder and urothelial cancer, uterine cancer, ovarian cancer, breast cancer, prostate cancer, sarcoma, testicular cancer, glioblastoma, adrenocortical cancer, brain cancer, spleen cancer, thymoma, multiple myeloma, plasma cell myeloma, leukemia, lymphoma, acute myeloid leukemia, bone marrow cancer, chronic lymphocytic leukemia, lymphoblastic leukemia including acute lymphoblastic leukemia
- the cancer is a lung cancer, pancreatic cancer or gastric cancer.
- tumor and “cancer” may be used interchangeably herein, and refer to a cellular mass of excessive cell growth or proliferation.
- tumor cell and “cancer cell” may be used interchangeably herein.
- Non-human animals include all vertebrates (e.g., mammals and non-mammals) such as any mammal.
- Nonlimiting examples of mammals include humans, chimpanzees, apes, monkeys, cattle, horses, sheep, goats, swine, rabbits, dogs, cats, rats, mice, and guinea pigs.
- Non-limiting examples of non-mammals include birds and fish.
- the patient is a human.
- a subject in need of treatment refers to a subject that would benefit biologically, medically, or in quality of life from a treatment (e.g., a treatment with any one or more of the exemplary antibody or ADC compound).
- treatment refers to any improvement of any consequence of disease, disorder, or condition, such as prolonged survival, less morbidity, and/or a lessening of side effects which result from an alternative therapeutic modality.
- treatment comprises delaying or ameliorating a disease, disorder, or condition (i.e., slowing or arresting or reducing the development of a disease or at least one of the clinical symptoms thereof).
- treatment comprises delaying, alleviating, or ameliorating at least one physical parameter of a disease, disorder, or condition, including those which may not be discernible by the patient.
- treatment comprises modulating a disease, disorder, or condition, either physically (e.g., stabilization of a discernible symptom), physiologically (e.g., stabilization of a physical parameter), or both.
- treatment comprises administration of a described antibody or ADC compound or composition to a subject, e.g., a patient, to obtain a treatment benefit enumerated herein.
- the treatment can be to cure, heal, alleviate, delay, prevent, relieve, alter, remedy, ameliorate, palliate, improve, or affect a disease, disorder, or condition (e.g., a cancer), the symptoms of a disease, disorder, or condition (e.g., a cancer), or a predisposition toward a disease, disorder, or condition (e.g., a cancer).
- a composition disclosed herein in addition to treating a subject having a disease, disorder, or condition, can also be provided prophylactically to prevent or reduce the likelihood of developing that disease, disorder, or condition.
- the term “prevent”, “preventing,” or “prevention” of a disease, disorder, or condition refers to the prophylactic treatment of the disease, disorder, or condition; or delaying the onset or progression of the disease, disorder, or condition.
- the term “therapeutically effective amount” or “therapeutically effective dose” refers to an amount of a compound described herein, e.g., an anti- MET antibody or antigen-binding portion thereof, an ADC compound comprising an anti-MET antibody or antigen-binding portion thereof or an anti-MET antibody or ADC compound composition described herein, to effect the desired therapeutic result (i.e., reduction or inhibition of an enzyme or a protein activity, amelioration of symptoms, alleviation of symptoms or conditions, delay of disease progression, a reduction in tumor size, inhibition of tumor growth, prevention of metastasis).
- a compound described herein e.g., an anti- MET antibody or antigen-binding portion thereof, an ADC compound comprising an anti-MET antibody or antigen-binding portion thereof or an anti-MET antibody or ADC compound composition described herein, to effect the desired therapeutic result (i.e., reduction or inhibition of an enzyme or a protein activity, amelioration of symptoms, alleviation of symptoms or conditions, delay of disease progression, a reduction in tumor
- a therapeutically effective amount is effective for detectable killing, reduction, and/or inhibition of the growth or spread of cancer cells, the size or number of tumors, and/or other measure of the level, stage, progression and/or severity of a cancer.
- the term also applies to a dose that will induce a particular response in target cells, e.g., a reduction, slowing, or inhibition of cell growth.
- a therapeutically effective amount can be determined by first administering a low dose, and then incrementally increasing that dose until the desired effect is achieved.
- a therapeutically effective amount of an ani-MET antibody or of an ADC comprising an anti-MET antibody may reduce the number of cancer cells, reduce tumor size, inhibit (e.g., slow or stop) tumor metastasis, inhibit (e.g., slow or stop) tumor growth, and/or relieve one or more symptoms.
- prophylactically effective amount refers to an amount of a compound disclosed herein, e.g., an anti- MET antibody or antigen-binding portion thereof, an ADC compound comprising an anti-MET antibody or antigen-binding portion thereof or an anti-MET antibody or ADC compound composition described herein, that is effective, at dosages and for periods of time necessary, to achieve the desired prophylactic result.
- a prophylactic dose is used in subjects prior to or at an earlier stage of disease, the prophylactically effective amount will be less than the therapeutically effective amount.
- a prophylactically effective amount can prevent the onset of disease symptoms, including symptoms associated with a cancer.
- the antibody compositions or antibodies or antigen-binding portions thereof of the invention may be administered alone or in combination with one or more other drugs or antibodies (or as any combination thereof).
- the pharmaceutical compositions, methods and uses of the invention thus also encompass embodiments of combinations (co-administration) with other active agents, as detailed below.
- the terms “co-administration”, “co-administered” and “in combination with,” referring to the antibody compositions and antibodies and antigen-binding portions thereof with one or more other therapeutic agents, is intended to mean, and does refer to and include the following: simultaneous administration of such combination of antibody composition I antibody I antigen-binding portion of the invention and therapeutic agent(s) to a patient in need of treatment, when such components are formulated together into a single dosage form which releases said components at substantially the same time to said patient, substantially simultaneous administration of such combination of antibody composition I antibody I antigen-binding portion of the invention and therapeutic agent(s) to a patient in need of treatment, when such components are formulated apart from each other into separate dosage forms which are taken at substantially the same time by said patient, whereupon said components are released at substantially the same time to said patient, sequential administration of such combination of antibody composition I antibody I antigen-binding portion of the invention and therapeutic agent(s) to a patient in need of treatment, when such components are formulated apart from each other into separate dosage forms which
- the antibodies or ADCs compositions of the invention, the anti-MET antibodies and antigen-binding portions thereof or ADCs of the invention may be administered without additional therapeutic treatments, i.e., as a stand-alone therapy.
- treatment with the antibodies or ADCs compositions of the invention, the anti-MET antibodies and antigen-binding portions thereof or ADCs of the invention may include at least one additional therapeutic treatment (combination therapy).
- the antibodies or ADCs compositions of the invention, the anti-MET antibodies and antigen-binding portions thereof or ADCs of the invention may be co-administered or formulated with another medication/drug for the treatment of cancer.
- the additional therapeutic treatment may comprise, e.g., an chemotherapeutic agent, an anti- neoplastic agent, an anti-angiogenic agent, a different anti-cancer antibody, a tyrosine kinase inhibitor, a MET pathway inhibitor and/or radiation therapy.
- an chemotherapeutic agent e.g., an anti- neoplastic agent, an anti-angiogenic agent, a different anti-cancer antibody, a tyrosine kinase inhibitor, a MET pathway inhibitor and/or radiation therapy.
- antibodies or ADCs compositions of the invention By combining antibodies or ADCs compositions of the invention, anti-MET antibodies and antigen-binding portions thereof or ADCs of the invention with agents known to induce terminal differentiation of cancer cells, the effect may be improved further.
- Such compounds may, for example, be selected from the group consisting of retinoic acid, trans-retinoic acids, cis-retinoic acids, phenyl butyrate, nerve growth factor, dimethyl sulfoxide, active form vitamin D3, peroxisome proliferator-activated receptor gamma, 12-O-tetradecanoylphorbol 13-acetate, hexamethylene-bis-acetamide, transforming growth factor-beta, butyric acid, cyclic AMP, and vesnarinone.
- the compound is selected from the group consisting of retinoic acid, phenylbutyrate, all-trans-retinoic acid and active form vitamin D.
- compositions of the invention comprising an antibody or ADC compositions of the invention, an anti-MET antibody and antigen-binding portions thereof or an ADC of the invention, and at least one other agent (e.g., a chemotherapeutic, anti- neoplastic, or anti-angiogenic agent) may be used as a combination treatment for simultaneous, separate or successive administration in cancer therapy.
- agent e.g., a chemotherapeutic, anti- neoplastic, or anti-angiogenic agent
- the other agent may by any agent suitable for treatment of the particular cancer in question, for example, an agent selected from the group consisting of alkylating agents, e.g., platinum derivatives such as cisplatin, carboplatin and/or oxaliplatin; plant alkoids, e.g., paclitaxel, docetaxel and/or irinotecan; antitumor antibiotics, e.g., doxorubicin (adriamycin), daunorubicin, epirubicin, idarubicin mitoxantrone, dactinomycin, bleomycin, actinomycin, luteomycin, and/or mitomycin; topoisomerase inhibitors such as topotecan; and/or antimetabolites, e.g., fluorouracil and/or other fluoropyrimidines.
- alkylating agents e.g., platinum derivatives such as cisplatin, carboplatin and/or
- an antibody or ADC compositions of the invention may be used in adjunctive therapy in connection with tyrosine kinase inhibitors.
- tyrosine kinase inhibitors synthetic, mainly quinazoline-derived, low molecular weight molecules that interact with the intracellular tyrosine kinase domain of receptors and inhibiting ligand-induced receptor phosphorylation by competing for the intracellular Mg-ATP binding site.
- Pharmaceutical articles comprising an antibody composition of the invention and at least one TKI targeting MET thus may also be used as a combination treatment for simultaneous, separate or successive administration in cancer therapy.
- the antibody or ADC compositions of the invention, the anti- MET antibody and antigen-binding portions thereof or the ADC of the invention may be administered in combination with another inhibitor of the MET pathway, which may target MET or HGF.
- the inhibitor is selected from the group consisting of, but not limited to, AMG 102, AMG 208, AMG 458, ARQ 197, AV299, BAY-853474, CGEN241, DN30, E7050, EMD 1204831, EMD 1214063, INCB28060, JNJ38877605, K252a, LY-2875358, MGCD265, MK-2461, MP-470, NK4, OA-5D5, PF-02341066, PF-04217903, PF-02341066, PHA-665752, SGX-523, SU5416, SU 11274, TAK701, XL184, XL880, cabozantinib, crizotinib
- the antibody or ADC compositions of the invention, the anti-MET antibody and antigen-binding portions thereof or the ADC of the invention may be administered in combination with an ErbB inhibitor (such as gefitinib or erlotinib) or a heat shock protein 90 (hsp90) inhibitor (such as 17-AAG).
- an ErbB inhibitor such as gefitinib or erlotinib
- hsp90 heat shock protein 90
- the antibody or ADC compositions of the invention, the anti- MET antibody and antigen-binding portions thereof or the ADC of the invention may be used in combination with other antibody therapeutics, e.g., an antibody against VEGF (e.g., Avastin®).
- the antibody or ADC compositions of the invention, the anti-MET antibody and antigen-binding portions thereof or the ADC of the invention may be used in combination with an agent known to stimulate cells of the immune system, such combination treatment leading to enhanced immune-mediated enhancement of the efficacy of the antibody compositions of the invention.
- an agent known to stimulate cells of the immune system such combination treatment leading to enhanced immune-mediated enhancement of the efficacy of the antibody compositions of the invention.
- immune-stimulating agents include recombinant interleukins (e.g., IL-21 and IL-2).
- the antibody or ADC compositions of the invention, the anti- MET antibody and antigen-binding portions thereof or the ADC of the invention may be used in a method of treatment as described above, may be for use in a treatment as described above, and/or may be for use in the manufacture of a medicament for a treatment as described above.
- the antibody compositions or ADC compositions of the invention will be administered in an effective amount for treatment of the condition in question, i.e. , at dosages and for periods of time necessary to achieve a desired result.
- a therapeutically effective amount may vary according to factors such as the particular condition being treated, the age, sex and weight of the patient, and whether the antibodies or ADCs are being administered as a stand-alone treatment or in combination with one or more additional anti-cancer treatments.
- Dosage regimens may be adjusted to provide the optimum desired response. For example, a single bolus may be administered, several divided doses may be administered over time or the dose may be proportionally reduced or increased as indicated by the exigencies of the therapeutic situation. It is especially advantageous to formulate parenteral compositions in dosage unit form for ease of administration and uniformity of dosage.
- Dosage unit form refers to physically discrete units suited as unitary dosages for the patients/subjects to be treated; each unit containing a predetermined quantity of active compound calculated to produce the desired therapeutic effect in association with the required pharmaceutical carrier.
- the specification for the dosage unit forms of the invention are generally dictated by and directly dependent on (a) the unique characteristics of the chemotherapeutic agent and the particular therapeutic or prophylactic effect to be achieved, and (b) the limitations inherent in the art of compounding such an active compound for the treatment of sensitivity in individuals.
- the dose and dosing regimen is adjusted in accordance with methods well-known in the therapeutic arts. That is, the maximum tolerable dose can be readily established, and the effective amount providing a detectable therapeutic benefit to a patient may also be determined, as can the temporal requirements for administering each agent to provide a detectable therapeutic benefit to the patient. Accordingly, while certain dose and administration regimens are exemplified herein, these examples in no way limit the dose and administration regimen that may be provided to a patient in practicing the present invention.
- dosage values may vary with the type and severity of the condition to be alleviated, and may include single or multiple doses. It is to be further understood that for any particular subject, specific dosage regimens should be adjusted over time according to the individual need and the professional judgment of the person administering or supervising the administration of the compositions, and that dosage ranges set forth herein are exemplary only and are not intended to limit the scope or practice of the embodied composition. Further, the dosage regimen with the compositions of this invention may be based on a variety of factors, including the type of disease, the age, weight, sex, medical condition of the patient, the severity of the condition, the route of administration, and the particular antibody employed. Thus, the dosage regimen can vary widely, but can be determined routinely using standard methods.
- doses may be adjusted based on pharmacokinetic or pharmacodynamic parameters, which may include clinical effects such as toxic effects and/or laboratory values.
- the present invention encompasses intra-patient dose-escalation as determined by the skilled artisan. Determining appropriate dosages and regimens are well-known in the relevant art and would be understood to be encompassed by the skilled artisan once provided the teachings disclosed herein.
- a suitable dose of an antibody composition or an ADC composition of the invention will be in the range of 0.1-100 mg/kg, such as about 0.5-50 mg/kg, e.g., about 1-20 mg/kg.
- the antibody composition may for example be administered in a dosage of at least 0.25 mg/kg, e.g., at least 0.5 mg/kg, such as at least 1 mg/kg, e.g., at least 1.5 mg/kg, such as at least 2 mg/kg, e.g., at least 3 mg/kg, such as at least 4 mg/kg, e.g., at least 5 mg/kg; and e.g., up to at most 50 mg/kg, such as up to at the most 30 mg/kg, e.g., up to at the most 20 mg/kg, such as up to at the most 15 mg/kg.
- Administration will normally be repeated at suitable intervals, e.g., once every week, once every two weeks, once every three weeks, or once every four weeks, and for as
- An effective amount for tumor therapy may be measured by its ability to stabilize disease progression and/or ameliorate symptoms in a patient, and preferably to reverse disease progression, e.g., by reducing tumor size.
- the ability of an antibody or composition of the invention to inhibit cancer may be evaluated by in vitro assays, e.g., as described in the examples, as well as in suitable animal models that are predictive of the efficacy in human tumors.
- Suitable dosage regimens will be selected in order to provide an optimum therapeutic response in each particular situation, for example, administered as a single bolus or as a continuous infusion, and with possible adjustment of the dosage as indicated by the exigencies of each case.
- the antibodies of the present invention and the ADCs of the present invention also are useful in diagnostic processes (e.g., in vitro, ex vivo).
- the antibodies and the ADCs can be used to detect and/or measure the level of MET in a sample from a patient (e.g., a tissue sample, or a body fluid sample such as an inflammatory exudate, blood, serum, bowel fluid, saliva, or urine).
- a sample from a patient e.g., a tissue sample, or a body fluid sample such as an inflammatory exudate, blood, serum, bowel fluid, saliva, or urine.
- Suitable detection and measurement methods include immunological methods such as flow cytometry, enzyme-linked immunosorbent assays (ELISA), chemiluminescence assays, radioimmunoassay, and immunohistology.
- kits e.g., diagnostic kits comprising the antibodies described herein.
- Anti-MET antibodies were obtained using the SymplexTM procedure essentially as described in WO 2005/042774. Briefly, BALB/c, C57 and C3H mice were immunized bi-weekly with human cancer cell lines over-expressing MET (HCT- 116), recombinant human MET protein (Sino Biologicals), recombinant human MET protein pre-incubated with ligand (HGF), or trypsin-digested MET.
- HCT- 116 human cancer cell lines over-expressing MET
- HGF recombinant human MET protein pre-incubated with ligand
- trypsin-digested MET trypsin-digested MET.
- Murine plasma cells obtained from spleens and inguinal lymph nodes were FACS sorted, and linkage of VH and VL coding sequences was performed on the sorted plasma cells, facilitating cognate pairing of the sequences, utilizing a two-step PCR procedure based on a one-step multiplex overlap-extension RT-PCR followed by nested PCR.
- the principle for linkage of cognate VH and VL sequences is described in detail in WO 2005/042774 and in Meijer et al., J Mol Biol 358(3):764- 72 (2006).
- VH and VL coding sequences obtained above were expressed as full-length antibodies. This involved insertion of the repertoire of VH and VL coding pairs into an expression vector and transfection into a host cell using the method described in WO 2012/059858.
- the specificity of the produced antibodies was determined by ELISA using as antigen either the extracellular domain of the MET protein or the extracellular domain of the MET protein translationally fused to a human immunoglobulin Fc domain.
- Nunc MaxiSorp plates (Cat. No. 464718) were coated with 1 pg/ml of the recombinant MET protein diluted in PBS at 4°C overnight. The plates were washed once with PBS + 0.05% Tween 20 (PBS-T) prior to blocking in 50 pl 2% Milk- PBS-T. The plates were washed once again with PBS-T, then 20 pl of 2% milk-PBS-T.
- VH and VL domains of the novel anti-MET antibody 8902 directed against human MET is provided in the present specification, as well as DNA sequences corresponding to said VH and VL.
- VH and VL amino acid sequences are provided in SEQ ID NOs: 7 and 8, respectively, and corresponding nucleotide sequences are provided in SEQ ID NOs: 9 and 10, respectively.
- Full-length heavy and light chain amino acid sequences are available in SEQ ID NOs: 11 and 12 (lgG1 chain) and in SEQ ID NOs: 13 and 14 (lgG2 chain), respectively.
- Amino acid sequences of heavy chain CDRs (H-CDR1 , H-CDR2 and H-CDR3) and light chain CDRs (L- CDR1, L-CDR-2 and L-CDR3) of 8902 antibody are shown in SEQ ID NOs: 1, 2 and 3 and in SEQ ID NOs: 4, 5 and 6, respectively.
- the CDR sequences were assigned in accordance with IMGT® definitions.
- This example describes the grouping of anti-MET antibodies into epitope bins based on paired competition patterns measured by Biolayer Interferometry (BLI). Antibodies belonging to different bins recognize different epitopes on the cMET extracellular domain (ECD).
- BBI Biolayer Interferometry
- the competition analysis of cMET antibodies was performed by BLI as a tandem assay: The human recombinant biotinylated cMET antigen was captured, and at saturating antigen binding conditions the 1st antibody was bound followed by binding of the 2nd antibody. Antibody pairs were defined as blockers or nonblockers based on a lack of response or a response above 0,1 nm, respectively.
- Tables 1 and 2 below show the competition of the anti-cMET antibodies, 9338 (HC and LC sequences provided herein in SEQ ID NOs: 19 and 20 in lgG1 format and in SEQ ID NOs: 21 and 22 in lgG2 format), 9006 (HC and LC sequences provided in SEQ ID NOs: 15 and 16 in lgG1 format and in SEQ ID NOs: 17 and 18 in lgG2 format), 8902, Telisotuzumab (HC and LC sequences provided in SEQ ID NOs: 23 and 24 in lgG1 format and in SEQ ID NOs: 25 and 26 in lgG2 format) and bispecific Amivantamab in lgG1 format (HC-1 and HC-2 sequences provided in SEQ ID NOs: 27 and 28 and LC-1 and LC-2 sequences provided in SEQ ID NOs: 29 and 30). Responses (nm) of second antibody binding captured antigenantibody complex are shown. Dark grey highlight self-blocking. The group
- Table 1 Heatmap of the tested lgG1 anti-cMET antibodies.
- Table 2 Heatmap of the tested lgG2 anti-cMET antibodies.
- the epitope is independent of isotype as confirmed by the comparable epitope binning data of lgG1 and lgG2 antibodies. All antibodies were self-competing as shown by dark gray.
- the epitope binning analysis showed that the cMET antibodies could be grouped into four distinct epitope bins highlighted in light grey and numbered bin 1-4.
- the antibodies 9338 and Amivantamab bound a similar epitope (bin 2), while the antibodies 8902 (bin 1), 9006 (bin 4) and Telisotuzumab (bin 3) each bound non-competing unique epitopes.
- Anti-human IgG (Fc) antibodies (Human antibody capture kit, Cytiva) were immobilized by amine coupling on 2 flow cells of a CM5 sensor chip (Cytiva) according to the manufacturer’s instructions (reference and active flow cell). Anti- MET antibodies were captured at 200 ng/mL on active flow cell during 60s at 10pL/min.
- Binding is measured by injection of 5 sequential injections of increasing concentrations (SCK - Single Cycle Kinetics) of human or cynomolgus MET ECD with associations of 240s and dissociation of 900s, both at 50pl/min.
- a regeneration is performed to wash the surface of anti-MET / MET complexes by injecting a solution of 3M MgCI2 during 30s at 20pl/min on both flow cell.
- Anti-cMET antibodies 9006, 8902 and 9338 in lgG1 or lgG2 format have been tested for binding to human and cynomolgus cMET-ECD transiently transfected into CHO-S cells by flow cytometry (iQue® Screener PLUS, IntelliCyt, Sartorius). Further, reference anti-cMET antibodies telisotuzumab in lgG1 or lgG2 format and amivantamab in lgG1 format have also been tested for binding to human or cynomolgus cMET in CHO-S cells. lgG1 or lgG2 isotype control antibodies and mock-transfected cells have been used as negative controls. Antibodies have been titrated in duplicates, in a 3-fold dilution from 30 pg/ml to 0.5 ng/ml.
- the different populations of transfected CHO-S cells were labeled with encoder dye VL-1 BV421 (Sartorius, 97055) or BL-1 FITC (Sartorius, 90355) in different intensities to make it possible to test more targets per well and still be able to discriminate the different cell populations.
- Antibodies were incubated on ice with CHO-S cells for 30 minutes. After washing step, a secondary AF647-fluorochrome labelled goat anti-human lgG(H+L) (A- 21445, Invitrogen) was added and cells were incubated 20 minutes on ice. After washing step, antibody binding was detected using the high-throughput flow cytometer iQue Screener PLUS (Sartorius) measuring the geometric mean (GeoMean) of AF647 signal in each well of both experiments.
- EC50 values are given in ng/ml in Table 5.
- ADCs antibody-drug conjugates
- linker-payload abbreviated by MC-VC-PAB-MMAE for maleimide caproyle (MC) valine citrulline (VC) para-aminobenzyle (PAB) monomethyl auristatine E (MMAE) (CAS:646502-53-6, Interchim) was used for the synthesis of the exemplified ADCs, wherein the linker is MC-VC-PAB and the payload is MMAE.
- Either the exemplified ADCs were synthesised using native 8902, 9006, 9338 and telizotuzumab antibodies and the conjugation to the maleimide group of the linkerpayload was carried out via naturally present interchain disulphide bonds through stochastic conjugation for an average DAR4. Or 8902, 9006, 9338 and telizotuzumab antibodies were endowed with cysteine mutations incorporated inside the heavy chain (HC S400C) and the light chain (LC V205C) of the peptide scaffold and their conjugation was performed to the maleimide group of the linkerpayload to afford an average DAR4.
- HC S400C heavy chain
- LC V205C light chain
- the conjugations on lgG1 mAb were performed in a range of 6 mg antibody.
- To the antibody was added 10mM EDTA solution in PBS 1X pH7.4 at a ratio 1/1 v/v, followed by 3-fold molar excess of 1M solution of TCEP.HCI in PBS 1X pH7.4.
- the mixture was incubated for 2 hours at +37°C. After reduction, the antibody solution was cooled down to room temperature and 6-fold molar excess of 5 mM linkerpayload solution was added to the mixture. The reaction was incubated at +4°C for 1h30.
- the conjugation was monitored by Hydrophobic Interaction Column (HIC) using TSKgel Butyl-NPR column (Tosoh Bioscience, 0014947) with mobile phase A (1.5M Ammonium Sulfate (NH4)2SO4, 25mM Potassium Phosphate dibasic (K2HPO4), adjusted at pH 7) and B (25mM Potassium Phosphate dibasic (K2HPO4), 20% Isopropanol, adjusted at pH 7).
- HIC Hydrophobic Interaction Column
- All exemplified ADCs synthesized with this method were buffer exchanged by dialysis (Thermo Fisher, 88254) in PBS 1X pH 7.4 (Sigma Life Science, P3813, 10PAK) at room temperature for 2 hours, purified by SEC column HiLoad® 26/600 Superdex® 200 prep grade with PBS 1X pH 7.4 and concentrated using Vivaspin 20, 50KD, PES (Sartorius Stedim, VS2031), filtered sterilely through 0.2pm sterile PES Filter, 25mm (Whatmann, G896-2502) and stored at +4°C. 5.1.b - Conjugation method M2
- the conjugations on lgG1 or lgG2 mAb were performed in a range of 6 mg antibody.
- To the antibody was added 10mM EDTA solution in PBS 1X pH7.4 at a ratio 1/1 v/v, followed by 6-fold molar excess of 1M solution of TCEP.HCI in PBS 1X pH7.4.
- the mixture was incubated for 2 hours at +37°C.
- All exemplified ADCs synthesized with this method were buffer exchanged by dialysis (Thermo Fisher, 88254) in PBS 1X pH 7.4 (Sigma Life Science, P3813, 10PAK) at room temperature for 2 hours, purified by SEC column HiLoad® 26/600 Superdex® 200 prep grade with PBS 1X pH 7.4 and concentrated using Vivaspin 20, 50KD, PES (Sartorius Stedim, VS2031), filtered sterilely through 0.2pm sterile PES Filter, 25mm (Whatmann, G896-2502) and stored at +4°C.
- the conjugations on lgG2 mAb were performed in a range of 13 mg antibody.
- To the antibody was added 10mM EDTA solution in PBS 1X pH7.4 at a ratio 1/1 v/v, followed by 50-fold molar excess of 1M solution of TCEP.HCI in PBS 1X pH7.4.
- the mixture was incubated for 2 hours at +37°C. After reduction, the antibody solution was cooled down to room temperature and 25-fold molar excess of 5 mM linker-payload solution was added to the mixture. The reaction was incubated at +4°C for 1h30.
- This step was carried out by adding PBS 1X pH7.4 in order to obtain only 5% solvent in the slurry and by mixing in Biorad sized disposable column for 30 minutes.
- the ADC was eluted with the IgG elution buffer and was buffer exchanged by dialysis (Thermo Fisher, 88254) in PBS 1X pH 7.4 (Sigma Life Science, P3813, 10PAK) at room temperature for 2 hours.
- All exemplified ADCs by this method were purified by SEC column HiLoad® 26/600 Superdex® 200 prep grade with PBS 1X pH 7.4, concentrated using Vivaspin 20, 50KD, PES (Sartorius Stedim, VS2031), filtered sterilely through 0.2pm sterile PES Filter, 25mm (Whatmann, G896-2502) and stored at +4°C.
- the conjugations were performed in a range of 10 mg antibody.
- To the antibody was added 10mM EDTA solution in PBS 1X pH7.4 at a ratio 1/1 v/v, followed by 10-fold molar excess of 1M solution of TCEP.HCI in PBS 1X pH7.4.
- the mixture was stirred for 2 hours at +37°C.
- After reduction, the antibody was cooled to room temperature and buffer exchanged by diafiltration using Vivaspin 20, 50KD, PES (Sartorius Stedim, VS2031).
- the conjugations were performed in a range of 10 mg antibody.
- To the monoclonal antibody was added 10mM EDTA solution in PBS 1X pH7.4 at a ratio 1/1 v/v, followed by 10-fold molar excess of 1M solution of TCEP.HCI in PBS 1X pH7.4. The mixture was stirred for 2 hours at +37°C. After reduction, the antibody was cooled to room temperature and buffer exchanged by diafiltration using Vivaspin 20, 50KD, PES (Sartorius Stedim, VS2031).
- the conjugations were performed in a range of 10 mg antibody.
- To the monoclonal antibody was added 10mM EDTA solution in PBS 1X pH7.4 at a ratio 1/1 v/v, followed by 10-fold molar excess of 1M solution of TCEP.HCI in PBS 1X pH7.4. The mixture was stirred for 2 hours at +37°C. After reduction, the antibody was cooled to room temperature and buffer exchanged by diafiltration using Vivaspin 20, 50KD, PES (Sartorius Stedim, VS2031).
- Drug-to-antibody ratio (DAR) of the exemplary ADCs was determined by liquid chromatography hyphenated with mass spectrometry (LC-MS) (80% Phase A (Water/0.1% FA), 20% Phase B (Acetonitrile/0.1%FA)).
- the ADC was either analysed in intact condition with a deglycosylation step using PNGase F enzyme (New England Biolabs®, P0705L) or following reduction with 5 mM (final concentration) of dithiothreitol DTT (Thermo Scientific, Rockford, IL, 20291).
- the ADC was loaded onto a Bioresolve RP mAb Polyphenyl, column 450A, 2.7pm, 2.1*150mm (Waters, Saint- Quentin-en-Yvelines, France, 186008946).
- a desalting step was performed for 1.5 min at 20% of B with a flow rate of 0.6 mL/min.
- Elution step was performed with a gradient from 1.5 min at 20% B to 16.5 min at 50 % B with a flow rate of 0.6 mL/min.
- a wash step was set from 16.8 min to 18.8 min at 100% B with a flow rate of 0.6 mL/min.
- mobile phase A was ultrapure water obtained with Mili-Q® system and mobile phase B was MS grade acetonitrile (Biosolve, Dieuze, France, 0001204101 BS) supplemented with 0.1% of FA (Fisher Chemical: A117-50- 50ML). Column temperature was set at +80°C.
- LC-MS analysis was performed using a Waters UPLC H-Class Bio chromatography system hyphenated with a Xevo G2 XS Q-TOF ESI mass spectrometer (Waters, Manchester, UK). Electrospray-ionization time-of-flight mass spectra of the analytes were acquired using UNI FlTM acquisition software (Waters, Manchester, UK). Then, the extracted intensity vs. m/z spectrum was deconvoluted using Maximum Entropy (MaxEntl) method of MassLynxTM software in order to determine the mass of each intact antibody species or each reduced antibody fragment depending on the treatment.
- Maximum Entropy MaxEntl
- DAR was determined from the deconvoluted spectra by summing the integrated MS (total ion current) peak area of unconjugated and conjugated given species (mAb or associated fragment). For the DAR determination, the percentage of each specie identified was calculated by intensity peak value from deconvoluted spectra. The percentage obtained, was multiplied by the number of drugs attached. The summed results produced an estimation of the final average DAR value for the full ADC*2.
- Size exclusion chromatography was performed for the quality control of each ADCs by measuring monomer percentage of the conjugate. The analysis was performed on analytical column Superdex 200 Increase 5/150 GL (GE Healthcare, 28990945) in isocratic conditions 100% PBS pH7.4 (Sigma Life Science, P3813, 10PAK), flow 0.45 ml/min for 12 minutes. The % aggregate fraction of the conjugate sample was quantified based on the peak area absorbance at 280 nm. Its calculation was based on the ratio between the high molecular weight eluent at 280 nm divided by the sum of peak area absorbance at the same wavelength of the high molecular weight and monomeric eluents multiplied by 100.
- Characterization of the exemplary ADCs was summarized in Table 6 (coupling method, LC-MS method, DAR, aggregation status after conjugation (%Agg), ADC stability (%Agg, stab w1 + 37°C) and yield.
- the average DAR values were determined using the above LC-MS methods and the percentage of aggregates was measured by size exclusion chromatography (SEC) during the quality control of the ADC and after the stability study (incubation at +37°C for 168 h in PBS buffer).
- the cell lines EBC-1, SNll-5 (both being MET amplified) and H1650 (MET nonamplified, mentioned here as H1650 3D) were cultured at 37°C in a humidified atmosphere containing 5% CO2. Cells were seeded in 96 well clear bottom plates and exposed to the anti-MET monoclonal antibodies or to the anti-MET ADCs for 120h.
- IC50s were calculated using standard four-parametric curve fitting. IC50 is defined as the compound concentration at which the CTG signal is reduced to 50% of that measured for the control. Two independent experiments were performed. IC50 data of each experiment and the arithmetic mean is shown in Tables 7 and 8 for all the antibodies and ADCs tested. For some antibodies and ADCs, the curves are shown in Fig. 2A and 2B.
- ADCs comprising lgG1 or lgG2 antibodies in those MET amplified cell lines
- all the ADCs were more potent than the payload MMAE, the ADC comprising the antibody 8902 lgG1 being one of the most potent ADCs as compared to the ADCs comprising other antibodies (IC50 of 0.025nM in EBC-1 and 0.013nM in SNU-5 for 8902 lgG1 ADC and IC50 of 0.025nM in EBC-1 and 0.015 nM in SNU-5 for 8902 lgG2 ADC) (Table 7).
- both lgG1 and lgG2 naked antibodies do not show a significant dose-dependent effect (Figure 2A and Figure 2B).
- the ADCs were less potent than the payload MMAE, the ADCs show a strong dose-dependent effect on cell viability.
- the ADCs comprising the 8902 lgG1 or lgG2 antibodies show a potent activity in this model, with an IC50 of 33.85nM and 44.2nM respectively (Table 7).
- 8902 antibody either in lgG1 or lgG2 format show a potent activity on cell viability of MET amplified cell lines.
- ADCs comprising 8902 antibody either in lgG1 or lgG2 format shows a strong effect on cell viability in either MET amplified or non-amplified cell lines.
- SNll-5 cells obtained from ATCC, were cultured in IMDM supplemented with 20% FBS and 0.5% Penicillin-Streptomycin. Cells were resuspended in RPMI without red phenol and 0.2ml containing 5x10 6 cells were subcutaneously inoculated into the right flank of female SCID mice, provided by Charles River. SNll-5 tumor cell implantation was performed 24 to 72 hours after a whole-body irradiation with a gamma-source (1.44 Gy, 60Co, BioMep, France).
- Animals were randomized based on their individual tumor volume. Randomization was performed when values reach a mean of 100-200 mm3. Animals (60/84) were randomized into ten groups of six animals each. Homogeneity between groups was tested by an analysis of variance (ANOVA). 8902 lgG2 Met antibody (15mg/kg) was injected once IV in PBS.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- Immunology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Organic Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Molecular Biology (AREA)
- Cell Biology (AREA)
- Biochemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Peptides Or Proteins (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
Description
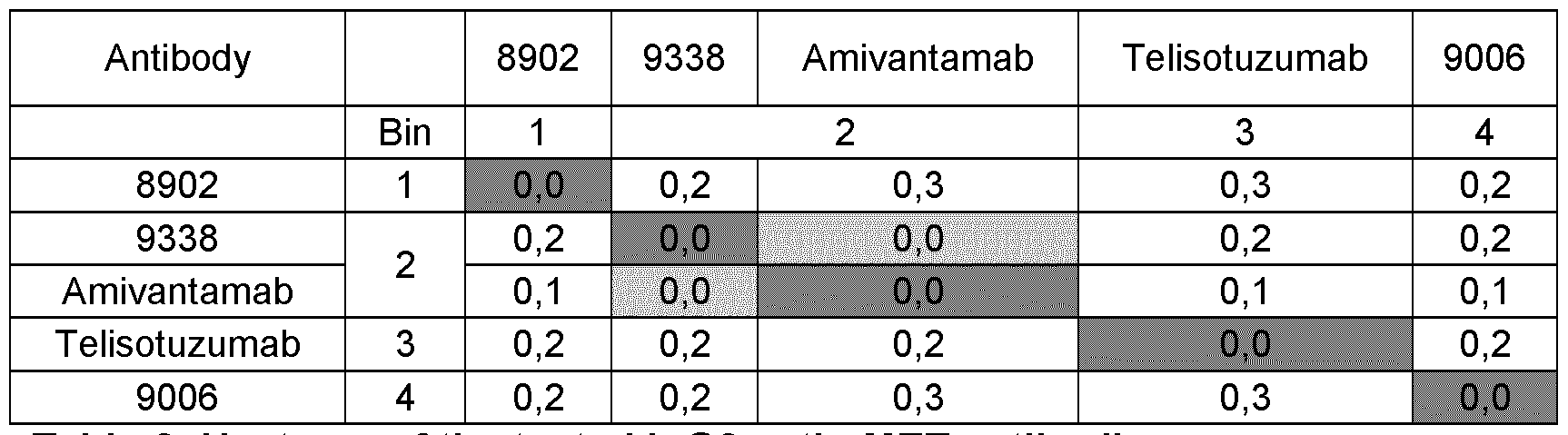



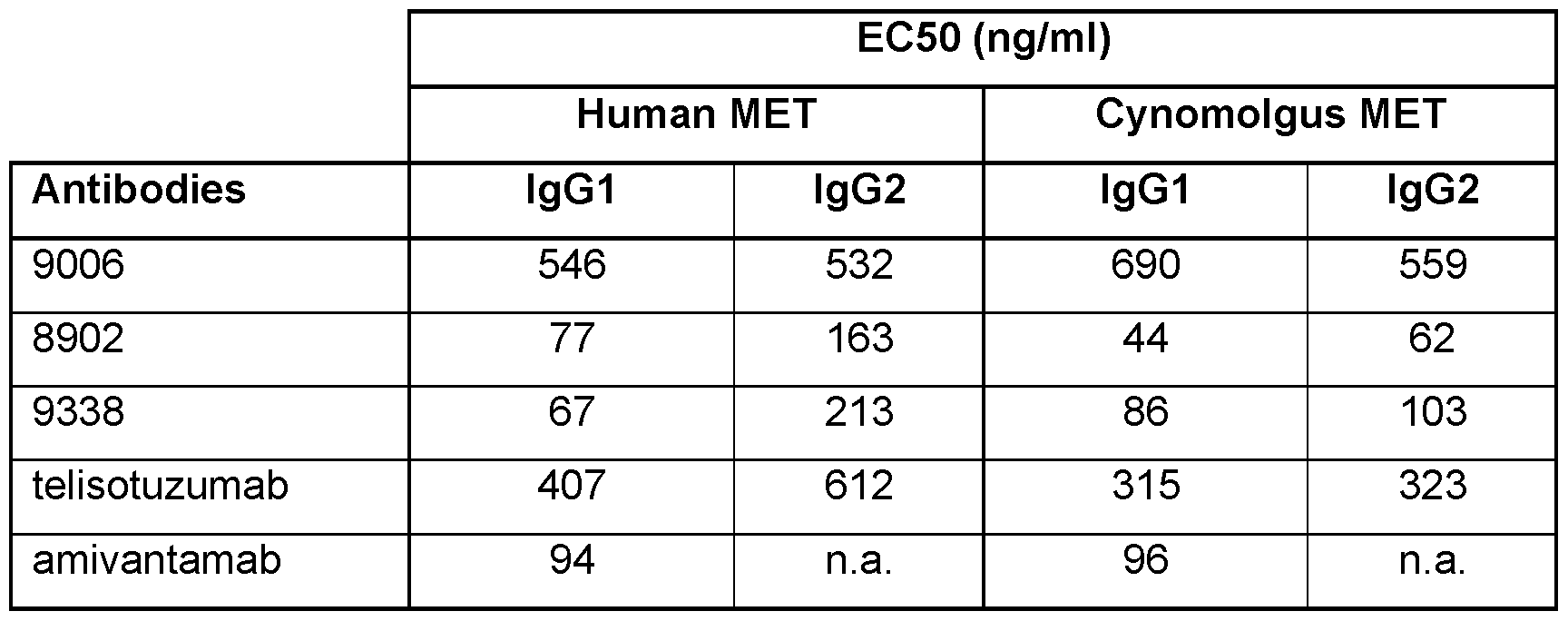
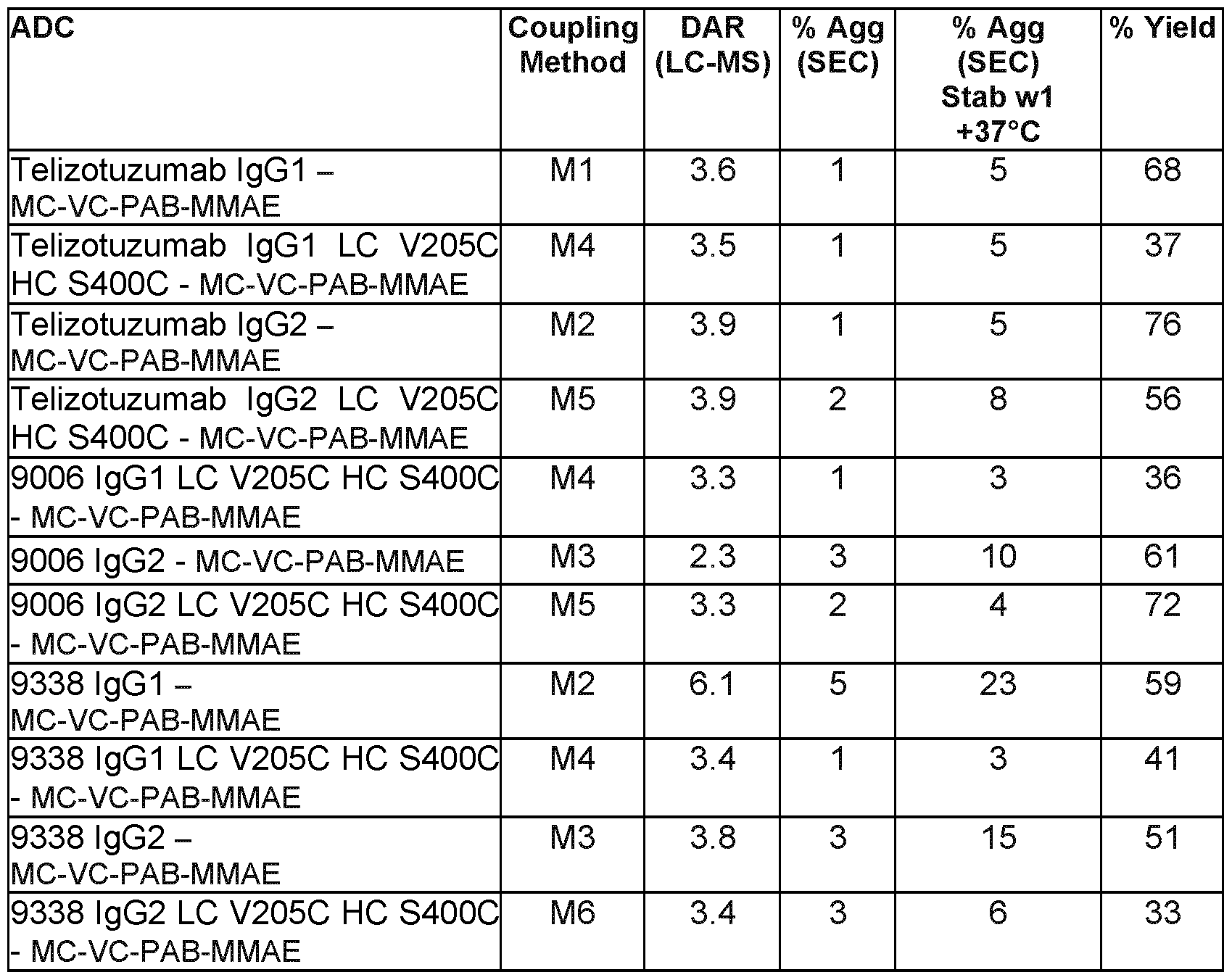

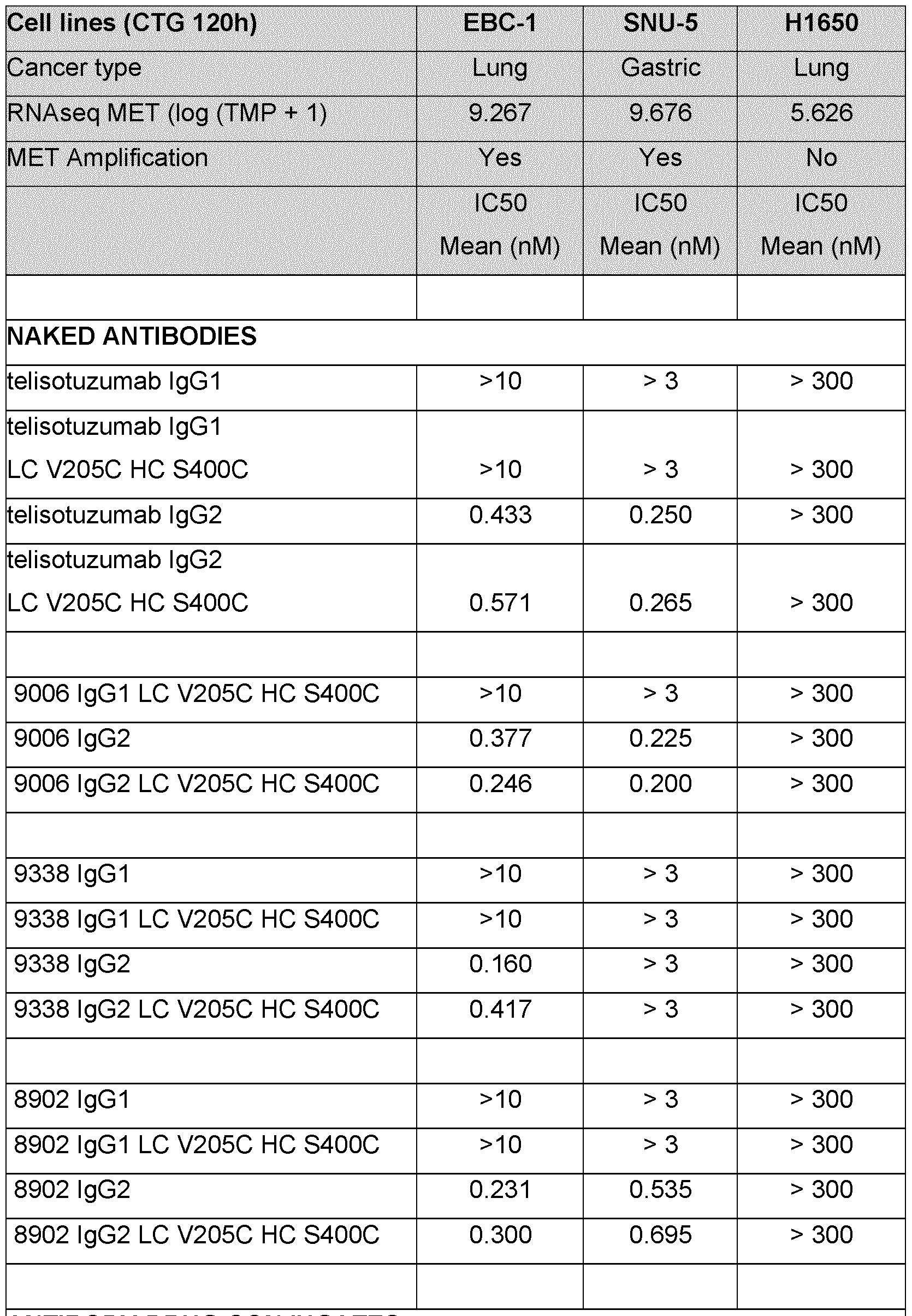
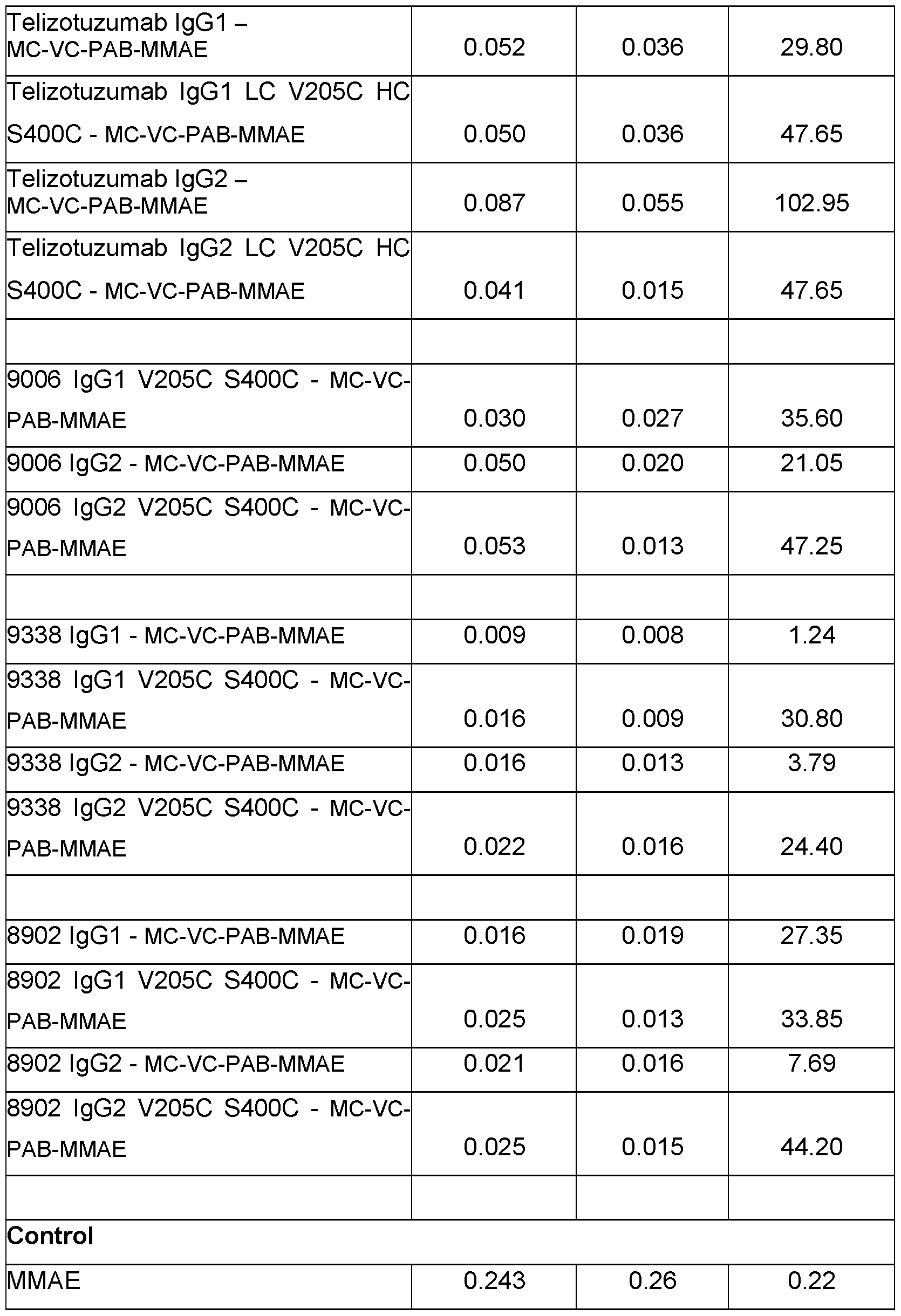
Claims
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2024277829A AU2024277829A1 (en) | 2023-05-19 | 2024-05-17 | Anti-met antibodies, antibody-drug conjugates, compositions and uses thereof |
| IL324556A IL324556A (en) | 2023-05-19 | 2025-11-10 | Anti-met antibodies, antibody-drug conjugates, compositions and uses thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP23315207.3 | 2023-05-19 | ||
| EP23315207 | 2023-05-19 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024240634A1 true WO2024240634A1 (en) | 2024-11-28 |
Family
ID=87001902
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2024/063676 Pending WO2024240634A1 (en) | 2023-05-19 | 2024-05-17 | Anti-met antibodies, antibody-drug conjugates, compositions and uses thereof |
Country Status (5)
| Country | Link |
|---|---|
| AR (1) | AR132710A1 (en) |
| AU (1) | AU2024277829A1 (en) |
| IL (1) | IL324556A (en) |
| TW (1) | TW202500584A (en) |
| WO (1) | WO2024240634A1 (en) |
Citations (45)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4399216A (en) | 1980-02-25 | 1983-08-16 | The Trustees Of Columbia University | Processes for inserting DNA into eucaryotic cells and for producing proteinaceous materials |
| US4510245A (en) | 1982-11-18 | 1985-04-09 | Chiron Corporation | Adenovirus promoter system |
| US4634665A (en) | 1980-02-25 | 1987-01-06 | The Trustees Of Columbia University In The City Of New York | Processes for inserting DNA into eucaryotic cells and for producing proteinaceous materials |
| EP0216846A1 (en) | 1985-04-01 | 1987-04-08 | Celltech Ltd | TRANSFORMED MYELOMA CELL LINE AND METHOD FOR EXPRESSING A GENE ENCODING A EUKARYOTIC POLYPEPTIDE EMPLOYING THIS LINE. |
| EP0256055A1 (en) | 1986-01-23 | 1988-02-24 | Celltech Ltd | RECOMBINANT DNA SEQUENCES, VECTORS CONTAINING THEM AND METHOD OF USING THE SAME. |
| US4740461A (en) | 1983-12-27 | 1988-04-26 | Genetics Institute, Inc. | Vectors and methods for transformation of eucaryotic cells |
| EP0323997A1 (en) | 1987-07-23 | 1989-07-19 | Celltech Limited | Recombinant dna expression vectors |
| EP0338841A1 (en) | 1988-04-18 | 1989-10-25 | Celltech Limited | Recombinant DNA methods, vectors and host cells |
| US4912040A (en) | 1986-11-14 | 1990-03-27 | Genetics Institute, Inc. | Eucaryotic expression system |
| US4959455A (en) | 1986-07-14 | 1990-09-25 | Genetics Institute, Inc. | Primate hematopoietic growth factors IL-3 and pharmaceutical compositions |
| US4968615A (en) | 1985-12-18 | 1990-11-06 | Ciba-Geigy Corporation | Deoxyribonucleic acid segment from a virus |
| WO1991017271A1 (en) | 1990-05-01 | 1991-11-14 | Affymax Technologies N.V. | Recombinant library screening methods |
| WO1992001047A1 (en) | 1990-07-10 | 1992-01-23 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| WO1992009690A2 (en) | 1990-12-03 | 1992-06-11 | Genentech, Inc. | Enrichment method for variant proteins with altered binding properties |
| WO1992015679A1 (en) | 1991-03-01 | 1992-09-17 | Protein Engineering Corporation | Improved epitode displaying phage |
| WO1992018619A1 (en) | 1991-04-10 | 1992-10-29 | The Scripps Research Institute | Heterodimeric receptor libraries using phagemids |
| WO1992020791A1 (en) | 1990-07-10 | 1992-11-26 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| US5168062A (en) | 1985-01-30 | 1992-12-01 | University Of Iowa Research Foundation | Transfer vectors and microorganisms containing human cytomegalovirus immediate-early promoter-regulatory DNA sequence |
| US5179017A (en) | 1980-02-25 | 1993-01-12 | The Trustees Of Columbia University In The City Of New York | Processes for inserting DNA into eucaryotic cells and for producing proteinaceous materials |
| WO1993001288A1 (en) | 1991-07-08 | 1993-01-21 | Deutsches Krebsforschungszentrum Stiftung des öffentlichen Rechts | Phagemide for screening antibodies |
| WO1993006213A1 (en) | 1991-09-23 | 1993-04-01 | Medical Research Council | Production of chimeric antibodies - a combinatorial approach |
| US5223409A (en) | 1988-09-02 | 1993-06-29 | Protein Engineering Corp. | Directed evolution of novel binding proteins |
| US5741957A (en) | 1989-12-01 | 1998-04-21 | Pharming B.V. | Transgenic bovine |
| US5750172A (en) | 1987-06-23 | 1998-05-12 | Pharming B.V. | Transgenic non human mammal milk |
| US5756687A (en) | 1993-03-09 | 1998-05-26 | Genzyme Transgenics Corporation | Isolation of components of interest from milk |
| US5827690A (en) | 1993-12-20 | 1998-10-27 | Genzyme Transgenics Corporatiion | Transgenic production of antibodies in milk |
| WO1998052976A1 (en) | 1997-05-21 | 1998-11-26 | Biovation Limited | Method for the production of non-immunogenic proteins |
| US5959177A (en) | 1989-10-27 | 1999-09-28 | The Scripps Research Institute | Transgenic plants expressing assembled secretory antibodies |
| WO2000009560A2 (en) | 1998-08-17 | 2000-02-24 | Abgenix, Inc. | Generation of modified molecules with increased serum half-lives |
| US6046037A (en) | 1994-12-30 | 2000-04-04 | Hiatt; Andrew C. | Method for producing immunoglobulins containing protection proteins in plants and their use |
| WO2000034317A2 (en) | 1998-12-08 | 2000-06-15 | Biovation Limited | Method for reducing immunogenicity of proteins |
| US6517529B1 (en) | 1999-11-24 | 2003-02-11 | Radius International Limited Partnership | Hemodialysis catheter |
| WO2004061104A2 (en) | 2003-01-07 | 2004-07-22 | Symphogen A/S | Method for manufacturing recombinant polyclonal proteins |
| WO2005042774A2 (en) | 2003-09-18 | 2005-05-12 | Symphogen A/S | Method for linking sequences of interest |
| WO2006015371A2 (en) | 2004-08-05 | 2006-02-09 | Genentech, Inc. | Humanized anti-cmet antagonists |
| WO2008145133A2 (en) | 2007-05-25 | 2008-12-04 | Symphogen A/S | Method for manufacturing a recombinant polyclonal protein |
| WO2009129814A1 (en) | 2008-04-23 | 2009-10-29 | Symphogen A/S | Methods for manufacturing a polyclonal protein |
| WO2010059654A1 (en) | 2008-11-21 | 2010-05-27 | Eli Lilly And Company | c-MET ANTIBODIES |
| WO2012059857A2 (en) | 2010-11-01 | 2012-05-10 | Symphogen A/S | Pan-her antibody composition |
| WO2012059561A1 (en) | 2010-11-03 | 2012-05-10 | Argen-X-Bv | Anti c-met antibodies |
| US20140069822A1 (en) | 2012-09-10 | 2014-03-13 | Antec Leyden B.V. | Electrochemical reduction of disulfide bonds in proteinaceous substances and electrochemical cell for carrying out such reduction |
| US20140154251A1 (en) | 2011-10-05 | 2014-06-05 | Samsung Electronics Co., Ltd. | Anti c-met antibody and uses thereof |
| WO2016042412A1 (en) | 2014-09-16 | 2016-03-24 | Symphogen A/S | Anti-met antibodies and compositions |
| US9593164B2 (en) | 2012-11-21 | 2017-03-14 | Janssen Biotech, Inc. | Bispecific EGFR/c-Met antibodies |
| WO2017201204A1 (en) | 2016-05-17 | 2017-11-23 | Abbvie Biotherapeutics Inc. | ANTI-cMet ANTIBODY DRUG CONJUGATES AND METHODS FOR THEIR USE |
-
2024
- 2024-05-17 AU AU2024277829A patent/AU2024277829A1/en active Pending
- 2024-05-17 AR ARP240101252A patent/AR132710A1/en unknown
- 2024-05-17 WO PCT/EP2024/063676 patent/WO2024240634A1/en active Pending
- 2024-05-17 TW TW113118374A patent/TW202500584A/en unknown
-
2025
- 2025-11-10 IL IL324556A patent/IL324556A/en unknown
Patent Citations (47)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4634665A (en) | 1980-02-25 | 1987-01-06 | The Trustees Of Columbia University In The City Of New York | Processes for inserting DNA into eucaryotic cells and for producing proteinaceous materials |
| US4399216A (en) | 1980-02-25 | 1983-08-16 | The Trustees Of Columbia University | Processes for inserting DNA into eucaryotic cells and for producing proteinaceous materials |
| US5179017A (en) | 1980-02-25 | 1993-01-12 | The Trustees Of Columbia University In The City Of New York | Processes for inserting DNA into eucaryotic cells and for producing proteinaceous materials |
| US4510245A (en) | 1982-11-18 | 1985-04-09 | Chiron Corporation | Adenovirus promoter system |
| US4740461A (en) | 1983-12-27 | 1988-04-26 | Genetics Institute, Inc. | Vectors and methods for transformation of eucaryotic cells |
| US5168062A (en) | 1985-01-30 | 1992-12-01 | University Of Iowa Research Foundation | Transfer vectors and microorganisms containing human cytomegalovirus immediate-early promoter-regulatory DNA sequence |
| EP0216846A1 (en) | 1985-04-01 | 1987-04-08 | Celltech Ltd | TRANSFORMED MYELOMA CELL LINE AND METHOD FOR EXPRESSING A GENE ENCODING A EUKARYOTIC POLYPEPTIDE EMPLOYING THIS LINE. |
| US4968615A (en) | 1985-12-18 | 1990-11-06 | Ciba-Geigy Corporation | Deoxyribonucleic acid segment from a virus |
| EP0256055A1 (en) | 1986-01-23 | 1988-02-24 | Celltech Ltd | RECOMBINANT DNA SEQUENCES, VECTORS CONTAINING THEM AND METHOD OF USING THE SAME. |
| US4959455A (en) | 1986-07-14 | 1990-09-25 | Genetics Institute, Inc. | Primate hematopoietic growth factors IL-3 and pharmaceutical compositions |
| US4912040A (en) | 1986-11-14 | 1990-03-27 | Genetics Institute, Inc. | Eucaryotic expression system |
| US5750172A (en) | 1987-06-23 | 1998-05-12 | Pharming B.V. | Transgenic non human mammal milk |
| EP0323997A1 (en) | 1987-07-23 | 1989-07-19 | Celltech Limited | Recombinant dna expression vectors |
| EP0338841A1 (en) | 1988-04-18 | 1989-10-25 | Celltech Limited | Recombinant DNA methods, vectors and host cells |
| US5223409A (en) | 1988-09-02 | 1993-06-29 | Protein Engineering Corp. | Directed evolution of novel binding proteins |
| US6417429B1 (en) | 1989-10-27 | 2002-07-09 | The Scripps Research Institute | Transgenic plants expressing assembled secretory antibodies |
| US5959177A (en) | 1989-10-27 | 1999-09-28 | The Scripps Research Institute | Transgenic plants expressing assembled secretory antibodies |
| US5741957A (en) | 1989-12-01 | 1998-04-21 | Pharming B.V. | Transgenic bovine |
| WO1991017271A1 (en) | 1990-05-01 | 1991-11-14 | Affymax Technologies N.V. | Recombinant library screening methods |
| WO1992020791A1 (en) | 1990-07-10 | 1992-11-26 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| WO1992001047A1 (en) | 1990-07-10 | 1992-01-23 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| WO1992009690A2 (en) | 1990-12-03 | 1992-06-11 | Genentech, Inc. | Enrichment method for variant proteins with altered binding properties |
| WO1992015679A1 (en) | 1991-03-01 | 1992-09-17 | Protein Engineering Corporation | Improved epitode displaying phage |
| WO1992018619A1 (en) | 1991-04-10 | 1992-10-29 | The Scripps Research Institute | Heterodimeric receptor libraries using phagemids |
| WO1993001288A1 (en) | 1991-07-08 | 1993-01-21 | Deutsches Krebsforschungszentrum Stiftung des öffentlichen Rechts | Phagemide for screening antibodies |
| WO1993006213A1 (en) | 1991-09-23 | 1993-04-01 | Medical Research Council | Production of chimeric antibodies - a combinatorial approach |
| US5756687A (en) | 1993-03-09 | 1998-05-26 | Genzyme Transgenics Corporation | Isolation of components of interest from milk |
| US5827690A (en) | 1993-12-20 | 1998-10-27 | Genzyme Transgenics Corporatiion | Transgenic production of antibodies in milk |
| US6046037A (en) | 1994-12-30 | 2000-04-04 | Hiatt; Andrew C. | Method for producing immunoglobulins containing protection proteins in plants and their use |
| WO1998052976A1 (en) | 1997-05-21 | 1998-11-26 | Biovation Limited | Method for the production of non-immunogenic proteins |
| WO2000009560A2 (en) | 1998-08-17 | 2000-02-24 | Abgenix, Inc. | Generation of modified molecules with increased serum half-lives |
| WO2000034317A2 (en) | 1998-12-08 | 2000-06-15 | Biovation Limited | Method for reducing immunogenicity of proteins |
| US6517529B1 (en) | 1999-11-24 | 2003-02-11 | Radius International Limited Partnership | Hemodialysis catheter |
| WO2004061104A2 (en) | 2003-01-07 | 2004-07-22 | Symphogen A/S | Method for manufacturing recombinant polyclonal proteins |
| WO2005042774A2 (en) | 2003-09-18 | 2005-05-12 | Symphogen A/S | Method for linking sequences of interest |
| WO2006015371A2 (en) | 2004-08-05 | 2006-02-09 | Genentech, Inc. | Humanized anti-cmet antagonists |
| WO2008145133A2 (en) | 2007-05-25 | 2008-12-04 | Symphogen A/S | Method for manufacturing a recombinant polyclonal protein |
| WO2009129814A1 (en) | 2008-04-23 | 2009-10-29 | Symphogen A/S | Methods for manufacturing a polyclonal protein |
| WO2010059654A1 (en) | 2008-11-21 | 2010-05-27 | Eli Lilly And Company | c-MET ANTIBODIES |
| WO2012059858A1 (en) | 2010-11-01 | 2012-05-10 | Symphogen A/S | Anti-her3 antibodies and compositions |
| WO2012059857A2 (en) | 2010-11-01 | 2012-05-10 | Symphogen A/S | Pan-her antibody composition |
| WO2012059561A1 (en) | 2010-11-03 | 2012-05-10 | Argen-X-Bv | Anti c-met antibodies |
| US20140154251A1 (en) | 2011-10-05 | 2014-06-05 | Samsung Electronics Co., Ltd. | Anti c-met antibody and uses thereof |
| US20140069822A1 (en) | 2012-09-10 | 2014-03-13 | Antec Leyden B.V. | Electrochemical reduction of disulfide bonds in proteinaceous substances and electrochemical cell for carrying out such reduction |
| US9593164B2 (en) | 2012-11-21 | 2017-03-14 | Janssen Biotech, Inc. | Bispecific EGFR/c-Met antibodies |
| WO2016042412A1 (en) | 2014-09-16 | 2016-03-24 | Symphogen A/S | Anti-met antibodies and compositions |
| WO2017201204A1 (en) | 2016-05-17 | 2017-11-23 | Abbvie Biotherapeutics Inc. | ANTI-cMet ANTIBODY DRUG CONJUGATES AND METHODS FOR THEIR USE |
Non-Patent Citations (45)
| Title |
|---|
| "Proteins, Structures and Molecular Principles", 1984, W. H. FREEMAN AND COMPANY |
| "Remington's Pharmaceutical Sciences", 1995, MACK PUBLISHING COMPANY |
| AB ET AL., MOL CANCER THER, vol. 14, 2015, pages 1605 - 13 |
| ALTSCHUL ET AL., J. MOL. BIOL., vol. 215, 1990, pages 403 - 410 |
| ALTSCHUL ET AL., NUCLEIC ACIDS RES., vol. 25, 1997, pages 3389 - 402 |
| BABCOOK ET AL., PROC NATL ACAD SCI USA, vol. 93, 1996, pages 7843 - 48 |
| BARBAS ET AL., PROC NATL ACAD SCI USA, vol. 88, 1991, pages 7978 - 7982 |
| BERGE ET AL.: "Pharmaceutical Salts", J. PHARMACEUTICAL SCIENCES, vol. 66, no. 1, 1977, XP002675560, DOI: 10.1002/jps.2600660104 |
| BIRD ET AL., SCIENCE, vol. 242, 1988, pages 423 - 426 |
| CHOTHIALESK: "Sequences of Proteins of Immunological Interest", vol. 196, 1987, NATIONAL INSTITUTES OF HEALTH, pages: 901 - 917 |
| CLACKSON ET AL., NATURE, vol. 352, 1991, pages 624 - 628 |
| DUCRYSTUMP, BIOCONJUGATE CHEM, vol. 21, 2010, pages 5 - 13 |
| GARRAD ET AL., BIOLTECHNOLOGY, vol. 9, 1991, pages 1373 - 1377 |
| GONNET ET AL., SCIENCE, vol. 256, 1992, pages 1443 - 45 |
| GRAM ET AL., PROC NATL ACAD SCI USA, vol. 89, 1992, pages 3576 - 3580 |
| GRIFFITHS ET AL., EMBO J, vol. 12, 1993, pages 725 - 734 |
| HAWKINS ET AL., J MOL BIOL, vol. 226, 1992, pages 889 - 896 |
| HAY ET AL., HUM ANTIBOD HYBRIDOMAS, vol. 3, 1992, pages 81 - 85 |
| HAYNES ET AL.: "Commentary: Occurrence of Pharmaceutically Acceptable Anions and Cations in the Cambridge Structural Database", J. PHARMACEUTICAL SCIENCES, vol. 94, no. 10, 2005, XP002593272 |
| HOGAN ET AL.: "Manipulating the Mouse Embryo: A Laboratory Manual 2", 1999, COLD SPRING HARBOR PRESS |
| HOLLIGER ET AL., PROC. NATL. ACAD. SCI. USA, vol. 90, pages 6444 - 6448 |
| HOOGENBOOM ET AL., NUC ACID RES, vol. 19, 1991, pages 4133 - 4137 |
| HUSE ET AL., SCIENCE, vol. 246, 1989, pages 1275 - 1281 |
| JACKSON ET AL.: "Mouse Genetics and Transgenics: A Practical Approach", 2000, OXFORD UNIVERSITY PRESS |
| JIEYI WANG ET AL: "Anti-c-Met monoclonal antibody ABT-700 breaks oncogene addiction in tumors with MET amplification", BMC CANCER, vol. 16, no. 1, 16 February 2016 (2016-02-16), XP055394355, DOI: 10.1186/s12885-016-2138-z * |
| JUNUTULA JR ET AL., NAT BIOTECHNOL, vol. 26, 2008, pages 925 - 932 |
| KIPRIYANOV ET AL., HUMAN ANTIBODIES AND HYBRIDOMAS, vol. 6, 1995, pages 93 - 101 |
| KIPRIYANOV ET AL., MOL. IMMUNOL., vol. 31, 1994, pages 1047 - 1058 |
| LEFRANC ET AL., DEV COMP IMMUNOL, vol. 27, no. 1, 2003, pages 55 - 77 |
| LYONS ET AL., PROTEIN ENG., vol. 3, 1990, pages 703 - 708 |
| MARTIN ET AL., EMBO J., vol. 13, 1994, pages 5303 - 9 |
| MCCAFFERTY ET AL., NATURE, vol. 348, 1990, pages 552 - 554 |
| MEIJER ET AL., J MOL BIOL, vol. 358, no. 3, 2006, pages 764 - 72 |
| PEARSON, J. MOL. BIOL., vol. 276, 1998, pages 71 - 84 |
| PEARSON, METHODS ENZYMOL., vol. 183, 1990, pages 63 - 98 |
| PEARSON, METHODS ENZYMOL., vol. 266, 1996, pages 227 - 258 |
| PEARSON, METHODS MOL. BIOL., vol. 132, 2000, pages 185 - 219 |
| PEARSON, METHODS MOL. BIOL., vol. 243, 1994, pages 307 - 31 |
| PROTEIN ENG, vol. 10, 1997, pages 949 - 57 |
| ROHRER, CHIMICA OGGI/CHEMISTRY TODAY, vol. 27, no. 5, 2009, pages 56 - 60 |
| SCHRAMA ET AL., NATURE REVIEWS/DRUG DISCOVERY, vol. 5, 2006, pages 147 - 159 |
| STEFANO ET AL., METHODS MOL BIOL, vol. 1045, 2013, pages 145 - 71 |
| TRAUNECKER ET AL., EMBO J, vol. 10, 1991, pages 3655 - 3659 |
| TRAUNECKER ET AL., INT. J. CANCER, vol. 7, 1992, pages 51 - 52 |
| WU ET AL., NATURE BIOTECHNOLOGY, vol. 23, no. 9, 2005, pages 1137 - 1146 |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2024277829A9 (en) | 2025-12-11 |
| AR132710A1 (en) | 2025-07-23 |
| IL324556A (en) | 2026-01-01 |
| AU2024277829A1 (en) | 2025-12-04 |
| TW202500584A (en) | 2025-01-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7118117B2 (en) | Selective method for producing antibody-drug conjugates | |
| CN114502592B (en) | Antibodies targeting CLDN18.2 and preparation methods and applications thereof | |
| US20220002428A1 (en) | Binding molecules to the human ox40 receptor | |
| TWI751981B (en) | Anti-pd-1 antibodies and compositions | |
| JP6810687B2 (en) | Combination therapy with FAP and DR5-specific bispecific antibodies and chemotherapeutic agents | |
| KR102453226B1 (en) | Anti-axl antibodies | |
| KR102408356B1 (en) | Anti-axl antibodies | |
| EP3194444B1 (en) | Anti-met antibodies and compositions | |
| TWI726936B (en) | Binding molecules specific for asct2 and uses thereof | |
| CN109311997B (en) | Anti-AXL antagonistic antibody | |
| US12415861B2 (en) | Anti C-MET antibodies | |
| TW201731531A (en) | Site specific HER2 antibody drug conjugates | |
| JP2021531825A (en) | Antibodies specific for folic acid receptor alpha | |
| EP4142793A1 (en) | Antibodies specific to abcb5 and uses thereof | |
| JP2025134713A (en) | PD1 and VEGFR2 dual binding agents | |
| TW202246323A (en) | Antibodies against claudin-6 and uses thereof | |
| WO2022222992A1 (en) | Antibodies binding trop2 and uses thereof | |
| IL323270A (en) | Anti-ptk7 antibody and uses thereof | |
| CA3091307A1 (en) | Csf1r binding agents | |
| WO2024240634A1 (en) | Anti-met antibodies, antibody-drug conjugates, compositions and uses thereof | |
| KR20260016936A (en) | Anti-MET antibodies, antibody-drug conjugates, compositions and uses thereof | |
| US20260042850A1 (en) | Anti c-met antibodies | |
| WO2026007196A1 (en) | Antibody specifically binding to fgfr2b and drug conjugate thereof | |
| HK40062721A (en) | Anti-axl antibodies | |
| HK40057042A (en) | Anti-axl antibodies |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24728922 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 324556 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: AU2024277829 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2024277829 Country of ref document: AU Date of ref document: 20240517 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020257042254 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2025135544 Country of ref document: RU Ref document number: 2024728922 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWP | Wipo information: published in national office |
Ref document number: 324556 Country of ref document: IL |