WO2024235991A1 - Rna-guided nucleases and nucleic acid targeting systems comprising such rna-guided nucleases - Google Patents
Rna-guided nucleases and nucleic acid targeting systems comprising such rna-guided nucleases Download PDFInfo
- Publication number
- WO2024235991A1 WO2024235991A1 PCT/EP2024/063269 EP2024063269W WO2024235991A1 WO 2024235991 A1 WO2024235991 A1 WO 2024235991A1 EP 2024063269 W EP2024063269 W EP 2024063269W WO 2024235991 A1 WO2024235991 A1 WO 2024235991A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- sequence
- nucleic acid
- rgn
- target
- cell
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/16—Hydrolases (3) acting on ester bonds (3.1)
- C12N9/22—Ribonucleases [RNase]; Deoxyribonucleases [DNase]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/102—Mutagenizing nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
Definitions
- the present invention relates to novel RNA-guided nucleases (RGN) and nucleic acid targeting systems comprising such.
- RGN RNA-guided nucleases
- RNA-guided nucleases such as the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-associated (Cas) proteins allow for the targeting of specific sequences by using a short RNA sequence that specifically hybridizes with a particular target sequence.
- CRISPR Clustered Regularly Interspaced Short Palindromic Repeats
- Such CRISPR systems became popular and gained multiple uses in research, diagnostics and therapeutics due to the ease of production of target-specific short RNA sequences and use of such with the same RGN protein.
- RGNs can be used to edit genomes through the introduction of a sequence-specific, double -stranded break that is either repaired and introduces a mutation or repaired by introducing a stretch of heterologous DNA.
- Inactive versions RGNs has been also widely used to target specific DNA or RNA regions and in combination with other proteins allowed to study and modulate multiple cellular processes and provide a useful tool for gene function study and modulation of their activity.
- the present invention provides novel type V RNA-guided nuclease (RGN) polypeptides, CRISPR RNAs (crRNAs), trans-activating CRISPR RNAs (tracrRNAs), gRNAs (gRNAs), nucleic acid targeting systems comprising those, nucleic acid molecules encoding the same, and vectors and host cells comprising such nucleic acid molecules.
- RGN RNA-guided nuclease
- crRNAs CRISPR RNAs
- tracrRNAs trans-activating CRISPR RNAs
- gRNAs gRNAs
- nucleic acid targeting systems comprising those, nucleic acid molecules encoding the same, and vectors and host cells comprising such nucleic acid molecules.
- nucleic acid targeting systems for binding a target nucleic acid sequence of interest, wherein the system comprises a RGN polypeptide and one or more RNA sequences targeting the nucleic acid of interest.
- methods disclosed herein are drawn to binding a target sequence of interest, and in some embodiments, cleaving or modifying the target sequence of interest.
- the target sequence of interest can be modified, for example, as a result of non-homologous end joining or homology-directed repair with an introduced donor sequence.
- Figure 1 Domain structure of EGS0023 and EGS0024 RGNs.
- Figure 4 shows the relative activity of EGS0023 with two potential tracrRNAs identified in Figure 2.
- Figure 5 shows an improvement in editing achieved with EGS0024 when using a modified tracrRNA (sgRNA v2).
- Figure 6 shows the structures of 2 version of sgRNA, one (sgRNA v2) has been truncated to remove extraneous sequence from the 5 ’ end of the tracrRNA and a shorter extended potential hairpin compared to the wild type tracrRNA (sgRNA vl).
- AAV adeno-associated virus
- a biological sample may contain whole cells and/or live cells and/or cell debris.
- the biological sample may contain (or be derived from) a "bodily fluid". Bodily fluids may be obtained from a mammal organism, for example by puncture, or other collecting or sampling procedures.
- Cas9 refers to type of an RGN that cleaves nucleic acid and is encoded by the CRISPR loci and is a part of the Type II CRISPR system.
- the Cas9 protein commonly used is from bacterial species Streptococcus pyogenes.
- the Cas9 protein may be mutated so that the nuclease activity is partly or completely inactivated.
- Casl2 refers to type of an RGN that cleaves nucleic acid and is encoded by the
- the Casl2 protein family contains many subtypes.
- the Casl2 protein may be mutated so that the nuclease activity is partly or completely inactivated.
- complement or “complementary” as used herein means a nucleic acid can mean Watson-Crick or Hoogsteen base pairing between nucleotides or nucleotide analogs of nucleic acid molecules.
- complementarity refers to a property shared between two nucleic acid sequences, such that when they are aligned antiparallel to each other, the nucleotide bases at each position will be complementary.
- CRISPR Clustered Regularly Interspaced Short Palindromic Repeats
- prokaryotic organisms such as bacteria and archaea. These sequences are derived from DNA fragments of bacteriophages that had previously infected the prokaryote. They are used to detect and destroy DNA from similar bacteriophages during subsequent infections.
- CRISPR system refers collectively to transcripts and other elements involved in the expression of or directing the activity of CRISPR-associated (“Cas") proteins, including sequences encoding a Cas protein, a tracr (trans -activating CRISPR) sequence (e.g. tracrRNA or an active partial tracrRNA), a tracr-mate sequence (containing a "direct repeat” and a tracrRNA-processed partial direct repeat in the context of an endogenous CRISPR system), a guide sequence (also referred herein to as a "spacer” in the context of an endogenous CRISPR system), or other sequences and transcripts from a CRISPR locus.
- a tracr trans -activating CRISPR
- tracr-mate sequence containing a "direct repeat” and a tracrRNA-processed partial direct repeat in the context of an endogenous CRISPR system
- guide sequence also referred herein to as a "spacer” in the context of an endogen
- an effective amount refers to an amount of a biologically active agent that is sufficient to elicit a desired biological response.
- an effective amount of a nuclease may refer to the amount of the nuclease that is sufficient to induce cleavage of a target site specifically bound and cleaved by the nuclease.
- an effective amount of a recombinase may refer to the amount of the recombinase that is sufficient to induce recombination at a target site specifically bound and recombined by the recombinase.
- an agent e.g., a nuclease, a recombinase, a hybrid protein, a fusion protein, a protein dimer, a complex of a protein (or protein dimer) and a polynucleotide, or a polynucleotide
- an agent e.g., a nuclease, a recombinase, a hybrid protein, a fusion protein, a protein dimer, a complex of a protein (or protein dimer) and a polynucleotide, or a polynucleotide
- an agent e.g., a nuclease, a recombinase, a hybrid protein, a fusion protein, a protein dimer, a complex of a protein (or protein dimer) and a polynucleotide, or a polynucleotide
- Enhancer refers to non-coding DNA sequences containing multiple activator and repressor binding sites. Enhancers range from 200 bp to 1 kb in length and may be either proximal, 5' upstream to the promoter or within the first intron of the regulated gene, or distal, in introns of neighboring genes or intergenic regions far away from the locus. Through DNA looping, active enhancers contact the promoter dependently of the core DNA binding motif promoter specificity. 4 to 5 enhancers may interact with a promoter.
- fusion protein refers to a chimeric protein created through the covalent or non-co valent joining of two or more genes, directly or indirectly, that originally coded for separate proteins.
- the translation of the fusion gene results in a single polypeptide with functional properties derived from each of the original proteins.
- gRNA also used interchangeably herein as a chimeric single guide RNA (“sgRNA”), refers to nucleic acid which is a fusion of two noncoding RNAs: a crRNA and a tracrRNA.
- gRNA is used interchangeably to refer to guide RNAs that exist as either single molecules or as a complex of two or more molecules.
- gRNAs that exist as single RNA species comprise two domains: (1) a domain that shares homology to a target nucleic acid (e.g., and directs binding of a Cas complex to the target); and (2) a domain that binds a Cas effector protein.
- an "isolated” or “purified” polypeptide, or biologically active portion thereof is substantially or essentially free from components that normally accompany or interact with the polypeptide as found in its naturally occurring environment.
- an isolated or purified polypeptide is substantially free of other cellular material, or culture medium when produced by recombinant techniques, or substantially free of chemical precursors or other chemicals when chemically synthesized.
- a protein that is substantially free of cellular material includes preparations of protein having less than 30%, 20%, 10%, 5%, or 1% (by dry weight) of contaminating protein.
- optimally culture medium represents less than 30%, 20%, 10%, 5%, or 1% (by dry weight) of chemical precursors or non-protein-of-interest chemicals.
- linker refers to a chemical group or a molecule linking two molecules or moieties, e.g., a binding domain and a cleavage domain of a nuclease. Typically, the linker is positioned between, or flanked by, two groups, molecules, or other moieties and connected to each one via a covalent bond, thus connecting the two.
- the linker is an amino acid or a plurality of amino acids (e.g., a peptide or protein).
- the linker is an organic molecule, group, polymer, or chemical moiety.
- the linker is a polypeptide of 5-100 amino acids in length, for example, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 30-35, 35-40, 40-45, 45-50, 50-60, 60-70, 70-80, 80- 90, 90-100, 100-150, or 150-200 amino acids in length. Longer or shorter linkers are also contemplated.
- modification in reference to a nucleic acid molecule refers to a change in the nucleotide sequence of the nucleic acid molecule, which can be a deletion, insertion, or substitution of one or more nucleotides, or a combination thereof.
- mutation refers to a substitution of a residue within a sequence, e.g., a nucleic acid or amino acid sequence, with another residue, or a deletion or insertion of one or more residues within a sequence. Mutations are typically described herein by identifying the original residue followed by the position of the residue within the sequence and by the identity of the newly substituted residue. Various methods for making the amino acid substitutions (mutations) provided herein are well known in the art, and are provided by, for example, Green and Sambrook, Molecular Cloning: A Laboratory Manual (4th ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. (2012)).
- nucleic acid As used herein, the terms "nucleic acid,” “nucleic acid sequence,” “nucleotide sequence,” “oligonucleotide,” and “polynucleotide” are interchangeable and refer to a polymeric form of nucleotides.
- the nucleotides may be deoxyribonucleotides (DNA), ribonucleotides (RNA), analogs thereof, or combinations thereof, and may be of any length.
- Polynucleotides may perform any function and may have any secondary and tertiary structures.
- the terms encompass known analogs of natural nucleotides and nucleotides that are modified in the base, sugar and/or phosphate moieties.
- a polynucleotide may comprise one modified nucleotide or multiple modified nucleotides. Examples of modified nucleotides include fluorinated nucleotides, methylated nucleotides, and nucleotide analogs. Nucleotide structure may be modified before or after a polymer is assembled. Following polymerization, polynucleotides may be additionally modified via, for example, conjugation with a labeling component or target binding component. A nucleotide sequence may incorporate non-nucleotide components.
- nucleic acids comprising modified backbone residues or linkages, which are synthetic, naturally occurring, and non-naturally occurring, and have similar binding properties as a reference polynucleotide (e.g., DNA or RNA).
- reference polynucleotide e.g., DNA or RNA
- analogs include, but are not limited to, phosphorothioates, phosphoramidates, methyl phosphonates, chiral-methyl phosphonates, 2-O-methyl ribonucleotides, peptide-nucleic acids (PNAs), Locked Nucleic Acid (LNATM) (Exiqon, Inc., Woburn, MA) nucleosides, glycol nucleic acid, bridged nucleic acids, and morpholino structures.
- Polynucleotide sequences are displayed herein in the conventional 5' to 3' orientation unless otherwise indicated.
- operably linked means that expression of a gene is under the control of a promoter with which it is spatially connected.
- a promoter may be positioned 5‘ (upstream) or 3‘ (downstream) of a gene under its control.
- the distance between the promoter and a gene may be approximately the same as the distance between that promoter and the gene it controls in the gene from which the promoter is derived. As is known in the art, variation in this distance may be accommodated without loss of promoter function.
- polypeptide As used herein, the terms “peptide,” “polypeptide,” and “protein” are interchangeable and refer to polymers of amino acids.
- a polypeptide may be of any length. It may be branched or linear, it may be interrupted by non-amino acids, and it may comprise modified amino acids.
- the terms may be used to refer to an amino acid polymer that has been modified through, for example, acetylation, disulfide bond formation, glycosylation, lipidation, phosphorylation, cross- linking, and/or conjugation (e.g., with a labeling component or ligand).
- Polypeptide sequences are displayed herein in the conventional N-terminal to C-terminal orientation.
- Polypeptides and polynucleotides can be made using routine techniques in the field of molecular biology (see, e.g., standard texts set forth above). Further, essentially any polypeptide or polynucleotide can be custom ordered from commercial sources.
- percentage of sequence identity means the value determined by comparing two optimally aligned sequences over a comparison window, wherein the portion of the polynucleotide sequence in the comparison window may comprise additions or deletions (i. e. , gaps) as compared to the reference sequence (which does not comprise additions or deletions) for optimal alignment of the two sequences. The percentage is calculated by determining the number of positions at which the identical nucleic acid base or amino acid residue occurs in both sequences to yield the number of matched positions, dividing the number of matched positions by the total number of positions in the window of comparison, and multiplying the result by 100 to yield the percentage of sequence identity.
- promoter means a synthetic or naturally-derived molecule which is capable of conferring, activating or enhancing expression of a nucleic acid in a cell.
- a promoter may comprise one or more specific transcriptional regulatory sequences to further enhance expression and/or to alter the spatial expression and/or temporal expression of same.
- a promoter may also comprise distal enhancer or repressor elements, which may be located as much as several thousand base pairs from the start site of transcription.
- a promoter may be derived from sources including viral, bacterial, fungal, plants, insects, and animals.
- RNA-guided endonuclease or "RGN” is used interchangeably herein and refer to a nuclease that forms a complex with (e.g., binds or associates with) one or more RNA that is not a target for cleavage.
- sequence identity or “identity” in the context of two polynucleotides or polypeptide sequences makes reference to the residues in the two sequences that are the same when aligned for maximum correspondence over a specified comparison window.
- sequence identity or “identity” in the context of two polynucleotides or polypeptide sequences makes reference to the residues in the two sequences that are the same when aligned for maximum correspondence over a specified comparison window.
- percentage of sequence identity is used in reference to proteins it is recognized that residue positions which are not identical often differ by conservative amino acid substitutions, where amino acid residues are substituted for other amino acid residues with similar chemical properties (e.g., charge or hydrophobicity) and therefore do not change the functional properties of the molecule.
- sequences differ in conservative substitutions the percent sequence identity may be adjusted upwards to correct for the conservative nature of the substitution.
- Sequences that differ by such conservative substitutions are said to have "sequence similarity" or "similarity”. Means for making this adjustment are well known to those of skill in the art. Typically this involves scoring a conservative substitution as a partial rather than a full mismatch, thereby increasing the percentage sequence identity. Thus, for example, where an identical amino acid is given a score of 1 and a non-conservative substitution is given a score of zero, a conservative substitution is given a score between zero and 1. The scoring of conservative substitutions is calculated, e.g. , as implemented in the program PC/GENE (Intelligenetics, Mountain View, California).
- spacer sequence refers to a part of gRNA nucleotide sequence or a part of a CRISPR locus that directly hybridizes with the target nucleotide sequence of interest.
- the subject may be a human or a non-human.
- the subject or patient may be undergoing other forms of treatment.
- target region refers to the region of the target gene to which the CRISPR-based system targets.
- treatment refers to a clinical intervention aimed to reverse, alleviate, delay the onset of, or inhibit the progress of a disease or disorder, or one or more symptoms thereof, as described herein.
- treatment refers to a clinical intervention aimed to reverse, alleviate, delay the onset of, or inhibit the progress of a disease or disorder, or one or more symptoms thereof, as described herein.
- treatment may be administered after one or more symptoms have developed and/or after a disease has been diagnosed.
- treatment may be administered in the absence of symptoms, e.g., to prevent or delay onset of a symptom or inhibit onset or progression of a disease.
- treatment may be administered to a susceptible individual prior to the onset of symptoms (e.g., in light of a history of symptoms and/or in light of genetic or other susceptibility factors). Treatment may also be continued after symptoms have resolved, for example, to prevent or delay their recurrence [045]
- Type II CRISPR system refers to effector system that carries out targeted DNA double-strand break in four sequential steps, using a single effector enzyme, Cas9, to cleave dsDNA.
- the Type II effector system may function in alternative contexts such as eukaryotic cells.
- the Type II effector system consists of a long pre-crRNA, which is transcribed from the spacercontaining CRISPR locus, the Cas9 protein, and a tracrRNA, which is involved in pre-crRNA processing.
- Type V CRISPR system refers to effector system that carries out targeted DNA double-strand break in four sequential steps, using a single effector enzyme, Casl2, to cleave dsDNA.
- the Type V effector system may function in alternative contexts such as eukaryotic cells.
- the Type V effector system consists of a long pre-crRNA, which is transcribed from the spacer- containing CRISPR locus, the Cast 2 protein, and, in some cases, a tracrRNA, which is involved in pre-crRNA processing.
- vector means a nucleic acid sequence containing an origin of replication.
- a vector may be a viral vector, bacteriophage, bacterial artificial chromosome or yeast artificial chromosome.
- a vector may be a DNA or RNA vector.
- a vector may be a self- replicating extrachromosomal vector, or a DNA plasmid.
- the CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) genomic locus is found in the genomes of many prokaryotes. CRISPR loci provide resistance to viruses and phages in prokaryotes. In this way, the CRISPR loci functions as a type of immune system to help defend prokaryotes against foreign invaders. In such system the response to such foreign invaders starts by cleaving the genome of invading viruses and plasmids and integrating segments (termed protospacers) of the genomic DNA into the CRISPR locus of the host organism.
- the segments that are integrated into the host genome are known as “spacers”, which mediate protection from subsequent attack by the same (or sufficiently related) virus or plasmid.
- Expression involves transcription of the CRISPR locus and subsequent enzymatic processing to produce short mature CRISPR RNAs (crRNA), each containing a single spacer sequence. Interference is induced after the CRISPR RNAs associate with Cas proteins to form effector complexes, which are then targeted to complementary protospacers in foreign genetic elements to induce nucleic acid degradation.
- Class 1 and Class 2 based upon the genes encoding the effector component.
- Class 1 systems have a multi-subunit crRNA- effector complex
- Class 2 systems have a single effector protein.
- Typical examples of Class 2 effector proteins are Cas9 and Cpfl (Cas 12a).
- Types I-VI CRISPR systems
- Class 1 systems comprise Type I, Type III and Type IV systems.
- Class 2 systems comprise Type II, Type V and Type VI systems.
- CRISPR loci include several short repeating sequences referred to as "repeats.”
- the repeats can form hairpin structures and/or the repeats can be single -stranded sequences.
- the repeats occur in clusters. Repeats frequently diverge between species. Repeats are regularly interspaced with unique intervening sequences, referred to as "spacers," resulting in a repeat-spacer-repeat locus architecture. Spacers are sequences usually identical to or homologous to foreign invader sequences (such as viral sequences).
- a spacer-repeat unit encodes a crisprRNA (crRNA).
- a crRNA refers to the mature form of the spacer-repeat unit.
- a crRNA contains a spacer sequence that is involved in targeting a target nucleic.
- crRNA has a region of complementarity to a potential DNA or RNA target sequence and in some cases, e.g., in currently characterized Type II systems, and some subtypes of Type V systems, a second region that forms base-pair hydrogen bonds with a transactivating CRISPR RNA (tracrRNA) to form a secondary structure, typically to form at least a stem structure.
- tracrRNA transactivating CRISPR RNA
- tracrRNA transactivating CRISPR RNA
- Complex formation between tracrRNA/crRNA and a Cas protein results in conformational change of the Cas protein that facilitates binding to DNA, nuclease activities of the Cas protein, and crRNA- guided sitespecific DNA cleavage by the nuclease.
- the DNA target sequence is adjacent to a cognate protospacer adjacent motif (PAM).
- PAM protospacer adjacent motif
- CRISPR locus comprises polynucleotide sequences encoding for CRISPR Associated Genes (cas) genes.
- Cas genes are involved in the biogenesis and/or the interference stages of crRNA function. Cas genes display extreme sequence diversity between different species and homologs. Some Cas proteins comprise a specific set of domain structures.
- Mature crRNAs are processed from a longer polycistronic CRISPR locus transcript, also referred to as pre-crRNA array.
- a pre-crRNA array comprises a plurality of crRNAs. The repeats in the pre-crRNA array are recognized by cas genes. Cas genes bind to the repeats and cleave the repeats. This action can liberate the plurality of crRNAs. crRNAs can be subjected to further events to produce the mature crRNA form such as trimming (e.g., with an exonuclease).
- a crRNA may comprise all, or some, of the CRISPR repeat sequences.
- Interference refers to the stage in the CRISPR system that is functionally responsible for combating infection by a foreign invader.
- CRISPR interference follows a similar mechanism to RNA interference, which results in target RNA degradation and/or destabilization.
- RNA interference results in target RNA degradation and/or destabilization.
- Currently characterized CRISPR systems perform interference of a target nucleic acid by coupling crRNAs and Cas genes, thereby forming CRISPR ribonucleoproteins (RNPs).
- crRNA of the RNP guides the RNP to foreign invader nucleic acid, (e.g. , by recognizing the foreign invader nucleic acid through hybridization).
- Hybridized target foreign invader nucleic acid- crRNA units are subjected to cleavage by Cas proteins.
- Target nucleic acid interference typically requires a protospacer adjacent motif (PAM) in a target nucleic acid.
- PAM protospacer adjacent motif
- Class 1 is characterized by multi-unit effector molecules, while class 2 contains a single effector molecule.
- Class 1 systems comprise Type I, Type III, and Type IV systems.
- Class 2 systems comprise Type II, Type V, and Type VI systems.
- Type II system is commonly represented by cas9 genes. There are two strands of RNA in Type II systems: a crRNA and a tracrRNA. The duplex formed by the tracrRNA and crRNA is recognized by, and associates with Cas9, encoded by the cas9 gene, which combines the functions of the crRNA- effector complex with target DNA cleavage. Cas9 is directed to a target nucleic acid by a sequence of the crRNA that is complementary to, and hybridizes with, a sequence in the target nucleic acid.
- nucleic acid target sequence binding involves a Casl2 protein and the crRNA, as does the nucleic acid target sequence cleavage.
- the RuvC-like nuclease domain of Cast 2 protein cleaves both strands of the nucleic acid target sequence in a sequential fashion (Swarts, et al. , Mol. Cell (2017) 66:221 -233. e4), producing 5' overhangs, which differs from the fragments generated by Cas9 protein.
- There have been multiple subtypes of Type V systems identified so far (type V-A/B/C7D/E/F/G/H/I/K/L and CRISPR-Casl2j). All of them differ by the length of Cas protein, PAM sequence and whether they require tracrRNA for its functionality.
- Type V-B is represented by Cas 12b protein.
- the Cas 12b protein cleavage activity of Type V-B systems requires hybridization of crRNA to tracrRNA to form a duplex and Cas 12a protein binds the crRNA/tracrRNA duplex in a sequence- and structure -specific manner by recognizing the stem loop and sequences adjacent to the stem loop, most notably the nucleotides 5' of the spacer sequence, which hybridizes to the nucleic acid target sequence. Substitutions that disrupt this stem-loop duplex abolish cleavage activity, whereas other substitutions that do not disrupt the stem-loop duplex do not abolish cleavage activity.
- nucleic acid target sequence binding involves Cas 12b and the crRNA/tracrRNA, as does the nucleic acid target sequence cleavage.
- the RuvC- like nuclease domain of Cas 12b cleaves one strand of the double-stranded nucleic acid target sequence
- a putative nuclease domain cleaves the other strand of the double- stranded nucleic acid target sequence in a staggered configuration, producing 5' overhangs, which is different from the blunt ends generated by Cas9 cleavage. These 5' overhangs may facilitate insertion of DNA.
- Type V crRNA and nucleic acid target sequence binding and cleavage include Cas 12a, Cas 12c, Cas 12d, and Casl2e which are similar in length to Cas 12b proteins, ranging from approximately 1000-1500 amino acids, and also require an additional RNA (either a tracRNA or a scoutRNA) besides Cas 12a.
- Still other proteins associated with Type V crRNA and nucleic acid target sequence binding and cleavage include Casl2fl, Casl2f2, Casl2f3, and Cas 12g, which are smaller in length to Casl2a proteins, ranging from approximately 300-900 amino acids, but also require a tracrRNA.
- Type VI systems include the Cas 13a protein (also known as Class 2 candidate 2 protein, or C2c2) which does not share sequence similarity with other CRISPR effector proteins (see Abudayyeh, et al, Science (2016) 353:aaf5573). Casl3a proteins have two HEPN domains and possess singlestranded RNA cleavage activity. Cas 13a proteins are similar to Cas 12a proteins in requiring a crRNA for nucleic acid target sequence binding and cleavage, but not requiring tracrRNA. [064] While many of type II systems have been identified, the discovery and characterization of CRISPR systems is ongoing.
- RNA-guided nucleases RGNs
- RNA-guided nucleases as defined by their amino acid sequences in Table 4. These systems exhibit a diversity of PAM sequences, enabling greater, and more flexible targeting space. Additionally, as the systems are derived from a variety of sources, they will be better suited for in vivo editing where pre-existing antibodies to other Cas proteins (either naturally occurring or from previous treatment) prevent effective use, and some are small enough to be effectively packaged into a single AAV vector for efficient delivery. Furthermore, Cas 12b proteins exhibit a greater specificity compared to Cas9 increasing their safety profile as a therapeutic agent (Strecker et al. Nat Commun 10, 212 (2019)).
- RNA-guided nuclease binds to a target nucleotide sequence and hybridizes with the gRNA molecule specific to the RNA-guided nuclease.
- the target sequence can then be subsequently cleaved by the RGN if the RGN polypeptide possesses nuclease activity.
- the presently disclosed RGNs can cleave nucleotides within a polynucleotide, functioning as an endonuclease.
- the disclosed RGNs can cleave nucleotides of a target nucleotide sequence within any position of a polynucleotide and thus function as both an endonuclease and exonuclease.
- the presently disclosed RGNs can be wild-type sequences derived from bacterial or archaeal species. Alternatively, the RGNs can be variants or fragments of wild-type polypeptides.
- the wildtype RGN can be modified to alter nuclease activity or alter PAM specificity, for example.
- the RGN is not naturally-occurring.
- the RGN functions as a nickase, only cleaving a single strand of the target nucleotide sequence.
- the RGNs lacks nuclease activity altogether or exhibits reduced nuclease activity and is referred to herein as nuclease-dead RGNs.
- Any method known in the art for introducing mutations into an amino acid sequence such as PCR-mediated mutagenesis and site- directed mutagenesis, can be used for generating nickases or nuclease-dead RGNs. (e.g.
- nuclease dead RGNs can be targeted to particular genomic locations to alter the expression of a desired sequence.
- the binding of a nuclease-dead RNA-guided nuclease to a target sequence results in the repression of expression of the target sequence or a gene under transcriptional control by the target sequence by interfering with the binding of RNA polymerase or transcription factors within the targeted genomic region.
- the RGN e.g.
- a nuclease- dead RGN) or its complexed gRNA further comprises an expression modulator that, upon binding to a target sequence, serves to either repress or activate the expression of the target sequence or a gene under transcriptional control by the target sequence.
- the expression modulator modulates the expression of the target sequence or regulated gene through epigenetic mechanisms.
- the nuclease-dead RGNs or a RGN with only nickase activity can be targeted to particular genomic locations to modify the sequence of a target polynucleotide through fusion to a base editing polypeptide, for example a deaminase polypeptide or active variant or fragment thereof that deaminates a nucleotide base, resulting in conversion from one nucleotide base to another.
- a base editing polypeptide for example a deaminase polypeptide or active variant or fragment thereof that deaminates a nucleotide base, resulting in conversion from one nucleotide base to another.
- the base-editing polypeptide can be fused to the RGN at its N-terminal or C-terminal end. Additionally, the base-editing polypeptide may be fused to the RGN via a peptide linker.
- a nonlimiting example of a deaminase polypeptide that is useful for such compositions and methods include cytidine deaminase or the adenosine deaminase base editor described in Gaudelli et al. (2017) Nature 551:464-471, and WO2018/027078.
- the RGN peptides of the present disclosure comprise multiple domains distributed in a recognition lobe (REC) and a nuclease lobe (NUC) for substrate recognition and cleavage.
- the REC lobe of RGN peptides provided herein consists of a RECI, and REC2 domains, where as the NUC lob consists of a WED-I, WED-II, Bridge helix domain(BH), a split Nuc domain, and a Tri-split RuvC domain.
- the RGN proteins of the present disclosure share little homology to AccCasl2b protein (in some cases less than 27%).
- the RuvC domain may comprise multiple subdomains: RuvC-I, RuvC-II and RuvC -III.
- the subdomains may be separated by other sequences on the amino acid sequence of the protein.
- RuvC domains include any polypeptides having a structural similarity and/or sequence similarity to a RuvC domain described in the art.
- the RuvC domain may share a structural similarity and/or sequence similarity to a RuvC of Cas9.
- the RuvC domain may have an amino acid sequence that share at least 80%, at least 85%, at least 90%, at least 95%, at least 99%, or 100% sequence identity with RuvC domains.
- the RuvC domain comprise RuvC-I polypeptide, RuvC-II polypeptide, and RuvC-III polypeptide.
- RuvC-I domain also include any polypeptides having a structural similarity and/or sequence similarity to a RuvC -I, II, and III domains described in the art, such as the corresponding domains of Cas9.
- the RuvC domain may have an amino acid sequence that has at least 80%, at least 85%, at least 90%, at least 95%, at least 99%, or 100% sequence identity with a RuvC domain of Cas9.
- the RuvC domain of Cas9 consists of a six-stranded mixed P-sheet flanked by a-helices and two additional two-stranded antiparallel P-sheets (see e.g., Nishimasu et al. Cell, 2014).
- the RuvC domain of Cas9 shares structural similarity with the retroviral integrase superfamily members characterized by an RNase H fold, such as Escherichia coli RuvC (PDB code 1HJR, 14% identity, root-mean-square deviation (rmsd) of 3.6 A for 126 equivalent Ca atoms) and Thermus thermophilus RuvC (PDB code 4LD0, 12% identity, rmsd of 3.4 A for 131 equivalent Ca atoms).
- E. coli RuvC is a 3-layer alpha-beta sandwich containing a 5-stranded beta-sheet sandwiched between 5 alpha-helices.
- RuvC nucleases have four catalytic residues (e.g., Asp7, Glu70, Hisl43 and Aspl46 in T. thermophilus RuvC), and cleave Holliday junctions (or structurally analogous cruciform junctions) through a two-metal mechanism. Asp 10 (Ala), Glu762, His983 and Asp986 of the Cas9 RuvC domain are located at positions similar to those of the catalytic residues of T. thermophilus RuvC.
- the RGN of the present disclosure comprises a bridge helix (BH) domain.
- the bridge helix domain refers to a helix and arginine rich polypeptide.
- the bridge helix domain may be located next to anyone of the amino acid domains in the nucleic -acid guided nuclease.
- the bridge helix domain is next to a RuvC domain, e.g., next to RuvC-I, RuvC-II, or RuvC-III subdomain.
- the bridge helix domain is between a RuvC-I and RuvC-II subdomains.
- the bridge helix domain may be from 10 to 100, from 20 to 60, from 30 to 50, e.g., 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46 or 47, 48, 49, or 50 amino acids in length.
- Examples of bridge helix includes the polypeptide of amino acids 60-93 of the sequence of .S', pyogenes Cas9.
- the RGN of the present disclosure comprises a WED domain assembled from two separate regions (WED-I and WED-II).
- the WED domain may have an amino acid sequence that share at least 80%, at least 85%, at least 90%, at least 95%, at least 99%, or 100% sequence identity with a WED domain of AccCasl2b.
- the AacCasl2b WED domain is assembled from two separate regions (WED- I and WED-II) and contains a core subdomain, which adopts an oligonucleotide -binding (OBD) fold.
- the core subdomain consists of eight [3 stranded distorted [3 sheets.
- the core subdomain is flanked by two a helices and one [3-hairpin
- the RGNs may comprise one or more modifications.
- the modified RGNs may be catalytically inactive (also referred as dead).
- a catalytically inactive or dead nuclease may have reduced or no nuclease activity compared to a wildtype counterpart nuclease.
- a catalytically inactive or dead nuclease may have nickase activity.
- a catalytically inactive or dead nuclease may not have nickase activity.
- Such a catalytically inactive or dead RGN may not make either doublestrand or single-strand break on a target polynucleotide but may still bind or otherwise form complex with the target polynucleotide.
- the modifications of the RGN polypeptide may or may not cause an altered functionality. Some modifications will not result in an altered functionality include for instance codon optimization for expression into a particular host, or providing the nuclease with a particular marker. Modifications which may result in altered functionality may also include mutations, including point mutations, insertions, deletions, truncations (including split nucleases), etc., as well as chimeric RGNs (e.g., comprising domains from different orthologues or homologues) or fusion proteins.
- Fusion proteins may include, for example, fusions with heterologous domains or functional domains (e.g., localization signals, enzymes).
- functional domains e.g., localization signals, enzymes
- various different modifications may be combined (e.g., a mutated nuclease which is catalytically inactive and which further is fused to a functional domain, such as for instance to induce DNA methylation or another nucleic acid modification, such as, for example, a mutation, a deletion, an insertion, a replacement.
- the RGNs can comprise at least one nuclear localization signal (NLS) to enhance transport of the RGN to the nucleus of a cell.
- Nuclear localization signals are known in the art and generally comprise a stretch of basic amino acids (see, e.g., Lange et al., J. Biol. Chem. (2007) 282:5101-5105).
- the RGN comprises 2, 3, or more nuclear localization signals.
- the nuclear localization signal(s) can be a heterologous NLS.
- Non-limiting examples of nuclear localization signals useful for the presently disclosed RGNs are the nuclear localization signals of SV40 Large T- antigen, nucleopasmin, and c-Myc (see. e.g., Ray et al.
- the RGN comprises the NLS sequence comprising the sequence of SEQ ID NO: 27 or 28.
- the RGN may comprise one or more NLS sequences at its N-terminus, C- terminus, or both the N-terminus and C- terminus.
- the RGN may comprise two NLS sequences at the N-terminal region and four NLS sequences at the C-terminal region.
- RGNs localization signal sequences known in the art that localize polypeptides to particular subcellular location(s) can also be used to target the RGNs, including, but not limited to, plastid localization sequences, mitochondrial localization sequences, and dual-targeting signal sequences that target to both the plastid and mitochondria (see, e.g., Nassoury and Morse (2005) Biochim Biophys Acta 1743:5-19; Herrmann and Neupert (2003) IUBMB Life 55:219-225; Soil (2002) Curr Opin Plant Biol 5:529-535; Carrie and Small (2013) Biochim Biophys Acta 1833:253-259).
- the RGNs comprise at least one cell- penetrating domain that facilitates cellular uptake of the RGN.
- Cell-penetrating domains are known in the art and generally comprise stretches of positively charged amino acid residues (i.e., polycationic cell- penetrating domains), alternating polar amino acid residues and non-polar amino acid residues (i.e. amphipathic cellpenetrating domains), or hydrophobic amino acid residues (i.e.. hydrophobic cell- penetrating domains) (see, e.g., Milletti F. (2012) Drug Discov Today 17:850-860).
- a non-limiting example of a cell-penetrating domain is the trans-activating transcriptional activator (TAT) from the human immunodeficiency virus 1.
- TAT trans-activating transcriptional activator
- the nuclear localization signal, plastid localization signal, mitochondrial localization signal, dual targeting localization signal, and/or cell-penetrating domain can be located at the amino-terminus (N- terminus), the carboxyl-terminus (C-terminus), or in an internal location of the RNA-guided nuclease.
- the presently disclosed RGN polypeptides can comprise a detectable label or a purification tag.
- the detectable label or purification tag can be located at the N-terminus, the C-terminus, or an internal location of the RNA-guided nuclease, either directly or indirectly via a linker peptide.
- the RGN component of the fusion protein is a nuclease-dead RGN. In other embodiments, the RGN component of the fusion protein is a RGN with nickase activity.
- RGNs that lack nuclease activity can be used to deliver a fused polypeptide, polynucleotide, or small molecule payload to a particular genomic location.
- the RGN polypeptide or gRNA can be fused to a detectable label to allow for detection of a particular sequence.
- a nuclease-dead RGN can be fused to a detectable label (e.g., fluorescent protein) and targeted to a particular sequence associated with a disease to allow for detection of the disease-associated sequence.
- a detectable label is a molecule that can be visualized or otherwise observed.
- the detectable label may be fused to the RGN as a fusion protein (e.g., fluorescent protein) or may be a small molecule conjugated to the RGN polypeptide that can be detected visually or by other means.
- Detectable labels that can be fused to the presently disclosed RGNs as a fusion protein include any detectable protein domain, including but not limited to, a fluorescent protein or a protein domain that can be detected with a specific antibody.
- Non-limiting examples of fluorescent proteins include green fluorescent proteins (e.g., GFP, EGFP, ZsGreen) and yellow fluorescent proteins (e.g., YFP, EYFP, ZsYellow).
- RGN polypeptides can also comprise a purification tag, which is any molecule that can be utilized to isolate a protein or fused protein from a mixture (e.g., biological sample, culture medium).
- purification tags include biotin, myc, maltose binding protein (MBP), and glutathione -S- transferase (GST).
- the presently disclosed RGNs can be fused to an effector domain (a fusion protein of an RGN and an effector domain), such as a cleavage domain, a deaminase domain, or an expression modulator domain, either directly or indirectly via a linker.
- an effector domain a fusion protein of an RGN and an effector domain
- Such effector domain can be located at the N- terminus, the C-terminus, or an internal location of the RNA-guided nuclease.
- the RGN component of the fusion protein is a nuclease-dead RGN.
- RGNs that are fused to a polypeptide or domain can be separated or joined by a linker.
- a linker joins a gRNA binding domain of an RNA guided nuclease and a base-editing polypeptide, such as a deaminase.
- the RGN fusion protein comprises a cleavage domain, which is any domain that is capable of cleaving a polynucleotide (i.e.. RNA, DNA) and includes, but is not limited to, restriction endonucleases and homing endonucleases (see, e.g Linn et al. (eds.) Nucleases, Cold Spring Harbor Laboratory Press, 1993).
- the RGN fusion protein comprises a deaminase domain that deaminates a nucleotide base, resulting in conversion from one nucleotide base to another, and includes, but is not limited to, a cytidine deaminase or an adenosine deaminase base editor.
- the effector domain of the fusion protein can be an expression modulator domain, which is a domain that either serves to upregulate or downregulate transcription.
- the expression modulator domain can be an epigenetic modification domain, a transcriptional repressor domain or a transcriptional activation domain.
- the expression modulator of the RGN fusion protein comprises an epigenetic modification domain that covalently modifies DNA or histone proteins to alter histone structure and/or chromosomal structure without altering the DNA sequence, leading to changes in gene expression (i. e. , upregulation or downregulation).
- epigenetic modifications include acetylation or methylation of lysine residues, arginine methylation, serine and threonine phosphorylation, and lysine ubiquitination and sumoylation of histone proteins, and methylation and hydroxymethylation of cytosine residues in DNA.
- Non-limiting examples of epigenetic modification domains include histone acetyltransferase domains, histone deacetylase domains, histone methyltransferase domains, histone demethylase domains, DNA methyltransferase domains, and DNA demethylase domains.
- the expression modulator of the fusion protein comprises a transcriptional repressor domain, which interacts with transcriptional control elements and/or transcriptional regulatory proteins, such as RNA polymerases and transcription factors, to reduce or terminate transcription of at least one gene.
- Transcriptional repressor domains are known in the art and include, but are not limited to IKB, and Kruppel associated box (KRAB) domains.
- the expression modulator of the fusion protein comprises a transcriptional activation domain, which interacts with transcriptional control elements and/or transcriptional regulatory proteins, such as RNA polymerases and transcription factors, to increase or activate transcription of at least one gene.
- Transcriptional activation domains are known in the art and include, but are not limited to, a VP 16 activation domain and an NF AT activation domain.
- nucleic acid-targeting effector protein-gRNA complex as a whole may be associated with two or more functional domains.
- there may be two or more functional domains associated with the nucleic acid-targeting effector protein or there may be two or more functional domains associated with the gRNA (via one or more adaptor proteins), or there may be one or more functional domains associated with the nucleic acid-targeting effector protein and one or more functional domains associated with the gRNA (via one or more adaptor proteins).
- the fusion between the adaptor protein and the activator or repressor may include a linker.
- a linker For example, GlySer linkers GGGS can be used. They can be used in repeats of 3 or 6, 9 or even 12 or more, to provide suitable lengths, as required. Linkers can be used between the gRNAs and the functional domain (activator or repressor), or between the nucleic acid-targeting effector protein and the functional domain (activator or repressor). guideRNAs (gRNAs/sgRNA), tracrRNA, and crRNA
- the present disclosure provides RGNs that can bind to gRNAs.
- gRNA refers to a nucleotide sequence having sufficient complementarity with a target nucleotide sequence to hybridize with the target sequence and direct sequence -specific binding of an associated RNA- guided nuclease to the target nucleotide sequence.
- a RGN refers to a nucleotide sequence having sufficient complementarity with a target nucleotide sequence to hybridize with the target sequence and direct sequence -specific binding of an associated RNA- guided nuclease to the target nucleotide sequence.
- a RGN refers to a nucleotide sequence having sufficient complementarity with a target nucleotide sequence to hybridize with the target sequence and direct sequence -specific binding of an associated RNA- guided nuclease to the target nucleotide sequence.
- a RGN refers to a nucleotide sequence having sufficient complementarity with a target nucleotide sequence to hybrid
- a gRNA comprises a CRISPR RNA (crRNA) and a trans-activating CRISPR RNA (tracrRNA).
- Native gRNAs that comprise both a crRNA and a tracrRNA generally comprise two separate RNA molecules that hybridize to each other through the repeat sequence of the crRNA and the anti-repeat sequence of the tracrRNA.
- Native direct repeat sequences within a CRISPR array generally range in length from 28 to 37 base pairs, although the length can vary between 23 bp to 55 bp.
- Spacer sequences within a CRISPR array generally range from 28 to 34 bp in length, although the length can be between 21 bp to 72 bp.
- the spacer sequences of the present disclosure are normally between 35 and 46 nucleotides.
- Each CRISPR array generally comprises less than 60 units of the CRISPR repeat-spacer sequence.
- the CRISPRs are transcribed as part of a long transcript termed the primary CRISPR transcript, which comprises much of the CRISPR array.
- the primary CRISPR transcript is cleaved by Cas proteins to produce crRNAs or in some cases, to produce pre- crRNAs that are further processed by additional Cas proteins into mature crRNAs.
- Mature crRNAs comprise a spacer sequence and a CRISPR repeat sequence.
- maturation involves the removal of one to six or more 5', 3', or 5' and 3' nucleotides.
- these nucleotides that are removed during maturation of the pre-crRNA molecule are not necessary for generating or designing a gRNA.
- the length of the target DNA within the sequence of the gRNA with the complementary sequence is 17 to 23bp, 18 to 23bp, 19 to 23bp, more specifically 20 to 23bp, as more specifically, it may be a 21 to 23bp, but is not limited thereto.
- a CRISPR RNA comprises a spacer sequence and a CRISPR repeat sequence.
- the “spacer sequence” is the nucleotide sequence that directly hybridizes with the target nucleotide sequence of interest.
- the spacer sequence is engineered to be fully or partially complementary with the target sequence of interest.
- the spacer sequence can comprise from 8 nucleotides to 30 nucleotides, or more.
- the spacer sequence can be 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, or more nucleotides in length.
- the spacer sequence is 10 to 26 nucleotides in length, or 12 to 30 nucleotides in length.
- the spacer sequence is 30 nucleotides in length.
- a trans-activating CRISPR RNA (tracrRNA) molecule comprises a nucleotide sequence comprising a region that has sufficient complementarity to hybridize to a CRISPR repeat sequence of a crRNA, which is referred to herein as the anti-repeat region.
- the tracrRNA molecule further comprises a region with secondary structure (e.g., stem-loop) or forms secondary structure upon hybridizing with its corresponding crRNA.
- the region of the tracrRNA that is fully or partially complementary to a CRISPR repeat sequence is at the 3' end of the molecule and the 5' end of the tracrRNA comprises secondary structure.
- This region of secondary structure generally comprises several hairpin structures, including the nexus hairpin, which is found adjacent to the anti -repeat sequence. There are often hairpins at the 5' end of the tracrRNA that can vary in structure and number.
- the gRNA can be a single gRNA or a dual-gRNA system.
- a single gRNA comprises the crRNA and tracrRNA on a single molecule of RNA
- a dual-gRNA system comprises a crRNA and a tracrRNA present on two distinct RNA molecules, hybridized to one another through at least a portion of the CRISPR repeat sequence of the crRNA and at least a portion of the tracrRNA, which may be fully or partially complementary to the CRISPR repeat sequence of the crRNA.
- the crRNA and tracrRNA are separated by a linker nucleotide sequence.
- the linker nucleotide sequence is one that does not include complementary bases in order to avoid the formation of secondary structure within or comprising nucleotides of the linker nucleotide sequence.
- the linker nucleotide sequence between the crRNA and tracrRNA is at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, at least 10, at least 11, at least 12, or more nucleotides in length.
- the linker nucleotide sequence of a single gRNA is at least 4 nucleotides in length.
- the linker nucleotide sequence is the nucleotide sequence set forth in Table 5.
- the linker nucleotide sequence is at least 6 nucleotides in length.
- the linker nucleotide sequence is RAAA.
- the single gRNA or dual-gRNA can be synthesized chemically or via in vitro transcription.
- Assays for determining sequence-specific binding between a RGN and a gRNA are known in the art and include, but are not limited to, in vitro binding assays between an expressed RGN and the gRNA, which can be tagged with a detectable label (e.g., biotin) and used in a pull-down detection assay in which the gRNA:RGN complex is captured via the detectable label (e.g., with streptavidin beads).
- a control gRNA with an unrelated sequence or structure to the gRNA can be used as a negative control for non-specific binding of the RGN to RNA.
- the gRNA can be introduced into a target cell, organelle, or embryo as an RNA molecule.
- the gRNA can be transcribed in vitro or chemically synthesized.
- a nucleotide sequence encoding the gRNA is introduced into the cell, organelle, or embryo.
- the nucleotide sequence encoding the gRNA is operably linked to a promoter (e.g., an RNA polymerase III promoter).
- the promoter can be a native promoter or heterologous to the gRNA-encoding nucleotide sequence.
- the gRNA can be introduced into a target cell, organelle, or embryo as a ribonucleoprotein complex, as described herein, wherein the gRNA is bound to an RNA-guided nuclease polypeptide.
- the gRNA directs an associated RNA-guided nuclease to a particular target nucleotide sequence of interest through hybridization of the gRNA to the target nucleotide sequence.
- a target nucleotide sequence can comprise DNA, RNA, or a combination of both and can be singlestranded or double -stranded.
- a target nucleotide sequence can be genomic DNA (i.e.
- chromosomal DNA DNA
- plasmid DNA DNA
- RNA molecule e.g. , messenger RNA, ribosomal RNA, transfer RNA, micro RNA, small interfering RNA.
- the target nucleotide sequence can be bound (and in some embodiments, cleaved) by an RNA-guided nuclease in vitro or in a cell.
- the chromosomal sequence targeted by the RGN can be a nuclear, plastid or mitochondrial chromosomal sequence.
- the target nucleotide sequence is unique in the target genome.
- the present disclosure also provides methods for binding and/or modifying a target nucleotide sequence of interest.
- the methods include delivering a system comprising at least one gRNA or a polynucleotide encoding the same, and at least one fusion polypeptide comprises an RGN of the invention and a base-editing polypeptide, for example a cytidine deaminase or an adenosine deaminase, or a polynucleotide encoding the fusion polypeptide, to the target sequence or a cell, organelle, or embryo comprising the target sequence.
- methods comprise the use of a single RGN polypeptide in combination with multiple, distinct gRNAs, which can target multiple, distinct sequences within a single gene and/or multiple genes. Also encompassed herein are methods wherein multiple, distinct gRNAs are introduced in combination with multiple, distinct RGN polypeptides. These gRNAs and gRNA/RGN polypeptide systems can target multiple, distinct sequences within a single gene and/or multiple genes.
- PAM Protospacer adjacent motif
- the present disclosure also provides PAM (proto-spacer-adjacent Motif) sequences to the adjacent, target DNA sequence of the complementary chain (complementary strand) and base pair formation can be in sequence to include to, a gRNA, or a composition comprising a DNA coding for the gRNA.
- PAM proto-spacer-adjacent Motif
- the target nucleotide sequence of the RGNs is adjacent to a sequence called protospacer adjacent motif (PAM).
- a protospacer adjacent motif is generally within 1 to 30 nucleotides from the target nucleotide sequence.
- a protospacer adjacent motif can be within 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 nucleotides from the target nucleotide sequence.
- the PAM can be 5' or 3' of the target sequence depending on the RGN. In some embodiments, the PAM is 5' of the target sequence for the presently disclosed RGNs.
- the PAM is a consensus sequence of 3-4 nucleotides, but in particular embodiments, can be 2, 3, 4, 5, 6, 7, 8, 9, or more nucleotides in length.
- the RGN having or an active variant or fragment thereof binds respectively a target nucleotide sequence adjacent to a PAM sequence set forth in Table 6.
- the RGN binds to a guide sequence comprising a CRISPR repeat sequence set forth in Table 5, or an active variant or fragment thereof, and a tracrRNA sequence set forth in Table 5, or an active variant or fragment thereof.
- PAM sequence specificity for a given nuclease enzyme is affected by enzyme concentration (see, e.g. , Karvelis et al. (2015) Genome Biol 16:253), which may be modified by altering the promoter used to express the RGN, or the amount of ribonucleoprotein complex delivered to the cell, organelle, or embryo.
- the RGN can cleave the target nucleotide sequence at a specific cleavage site.
- a cleavage site is made up of the two particular nucleotides within a target nucleotide sequence between which the nucleotide sequence is cleaved by an RGN.
- the cleavage site can comprise the 1st and 2nd , 2nd and 3rd , 3rd and 4th , 4th and 5th , 5th and 6th , 7th and 8th , or 8th and 9th nucleotides from the PAM in either the 5' or 3' direction.
- the cleavage site may be over 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 nucleotides from the PAM in either the 5’ or 3’ direction. In some embodiments, the cleavage site is 4 nucleotides away from the PAM. In other embodiments, the cleavage site is at least 15 nucleotides away from the PAM.
- the cleavage site is defined based on the distance of the two nucleotides from the PAM on the positive (+) strand of the polynucleotide and the distance of the two nucleotides from the PAM on the negative (-) strand of the polynucleotide.
- the target polynucleotide of an RGN system can be any polynucleotide endogenous or exogenous to the eukaryotic cell.
- the target polynucleotide can be a polynucleotide residing in the nucleus of the eukaryotic cell.
- the target polynucleotide can be a sequence coding a gene product (e.g., a protein) or a non-coding sequence (e.g., a regions or introns).
- the target sequence is generally associated with a PAM (protospacer adjacent motif.
- the precise sequence and length requirements for the PAM differ depending on the RGN used, but PAMs are typically 2-5 base pair sequences adjacent the protospacer (that is, the target sequence).
- the target polynucleotide of a RGN/RNA complex may be a disease-associated gene or polynucleotides or a gene/ polynucleotide associated with a biological pathway.
- RGNs proteins can be complexed to an RNA (RNA/Cas complex) in order to deliver Cas in proximity with a target nucleic acid sequence.
- the RNA such as a crRNA, is a polynucleotide that site-specifically guides a Cas nuclease, or a deactivated Cas nuclease, to a target nucleic acid region.
- the binding specificity is determined jointly by the complementary region on the cognate guide and a short DNA motif (protospacer adjacent motif or PAM) juxtaposed to the complementary region.
- the spacer present in the guide specifically hybridizes to a target nucleic acid sequence and determines the location of a Cas protein's site-specific binding and nucleolytic cleavage.
- RNA/Cas complexes can be produced using methods well known in the art.
- the RNA of the complexes can be produced in vitro and RGN polypeptides can be recombinantly produced and then the RNA and RGN proteins can be complexed together using methods known in the art.
- cell lines constitutively expressing RGN proteins can be developed and can be transfected with the gRNA components, and complexes can be purified from the cells using standard purification techniques, such as but not limited to affinity, ion exchange and size exclusion chromatography. See, e.g. , Jinek M., et al, "A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity," Science (2012) 337:816-821.
- the components i.e., the gRNA and RGN-encoding polynucleotides may be provided separately to a cell, e.g., using separate constructs, or together, in a single construct, or in any combination, and complexes can be purified as above.
- guides are commonly designed to target 5' constitutively expressed exons which reduces the chances or removal of the targeted region from mRNA due to alternative splicing. Exons near the N- terminus can be targeted because frameshift mutations here will increase the likelihood of the production of a nonfunctional protein product.
- cognate guides can be designed to target exons that code for known essential protein domains.
- non-frameshift mutations such as insertions or deletions are more likely to alter protein function when they occur in protein domains that are essential for protein function.
- the target sequence should be close to the location of the desired edit. In this case, the location where the edit is desired is identified and a target sequence is selected nearby.
- the gRNA can be delivered to a cell. If the cell constitutively expresses a RGN, the RGN will then be recruited to the target site to cleave the target nucleic acid. If the cell does not express an RGN, complexes of gRNA/RGN can be delivered to the cells to make breaks in the genome, thereby triggering the repair pathways in the cells.
- Treated cells are then screened using methods well known in the art, such as using high- throughput screening techniques including, but not limited to, fluorescence-activated cell sorting (FACS)-based screening platforms, microfluidics-based screening platforms, and the like. These techniques are well known in the art. See, e.g., Wojcik et al, Int. J. Molec. Sci. (2015) 16:24918- 24945.
- the cells can then be expanded and re-transfected with additional RGN/gRNA complexes to introduce further diversity and this process can be repeated iteratively until a population with the desired properties is obtained.
- Single cell clones are sorted from the population, expanded and sequenced to recover the mutations that resulted in the desired function.
- RNA-guided nucleases comprising at least 50, 100, 150, 200, 250, 300, 350, 400, 450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950, 1000, 1050 or more contiguous amino acid residues of an amino acid sequence set forth in Table 4.
- RNA-guided nucleases provided herein can comprise at least one nuclease domain (e.g ., DNase, RNase domain) and at least one RNA recognition and/or RNA binding domain to interact with gRNAs.
- RNA-guided nucleases include, but are not limited to, DNA binding domains, helicase domains, protein-protein interaction domains, and dimerization domains.
- the RGNs provided herein can comprise at least 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% to one or more of a DNA binding domains, helicase domains, protein-protein interaction domains, and dimerization domains.
- variants or fragments While the activity of a variant or fragment may be altered compared to the polynucleotide or polypeptide of interest, the variant and fragment should retain the functionality of the polynucleotide or polypeptide of interest. For example, a variant or fragment may have increased activity, decreased activity, different spectrum of activity or any other alteration in activity when compared to the polynucleotide or polypeptide of interest.
- fragments and variants of naturally-occurring RGN polypeptides will retain sequence-specific, RNA-guided DNA-binding activity.
- fragments and variants of naturally-occurring RGN polypeptides such as those disclosed herein, will retain nuclease activity (single -stranded or double -stranded).
- a biologically active variant of an RGN polypeptide of the invention may differ by as few as 1- 15 amino acid residues, as few as 1-10, such as 6-10, as few as 5, as few as 4, as few as 3, as few as 2, or as few as 1 amino acid residue.
- the polypeptides can comprise an N- terminal or a C-terminal truncation, which can comprise at least a deletion of 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950, 1000, 1050 amino acids or more from either the N or C terminus of the polypeptide.
- gRNA Variants of gRNA, tracrRNA and CRISPR RNA repeat sequence (crRNA)
- RGN proteins can have varying sensitivity to mismatches between a spacer sequence in a gRNA and its target sequence that affects the efficiency of cleavage.
- the CRISPR RNA repeat sequence comprises a nucleotide sequence that comprises a region with sufficient complementarity to hybridize to a tracrRNA.
- the CRISPR RNA repeat sequence can comprise from 8 nucleotides to 30 nucleotides, or more.
- the CRISPR repeat sequence can be 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, or more nucleotides in length.
- the CRISPR repeat sequence is 21 nucleotides in length.
- the degree of complementarity between a CRISPR repeat sequence and its corresponding tracrRNA sequence when optimally aligned using a suitable alignment algorithm, is or more than 50%, 60%, 70%, 75%, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or more.
- the CRISPR repeat sequence comprises the nucleotide sequence set forth in Table 5, or an active variant or fragment thereof that when comprised within a gRNA, is capable of directing the sequence-specific binding of an associated RNA-guided nuclease provided herein to a target sequence of interest.
- an active CRISPR repeat sequence variant of a wild-type sequence comprises a nucleotide sequence having at least 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or more sequence identity to the nucleotide sequence set forth in Table 5.
- an active CRISPR repeat sequence fragment of a wild-type sequence comprises at least 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 contiguous nucleotides of the nucleotide sequence set forth in Table 5.
- Fragments and variants of naturally occurring CRISPR repeats will retain the ability, when part of a gRNA (comprising a tracrRNA), to bind to and guide an RNA- guided nuclease (complexed with the gRNA) to a target nucleotide sequence in a sequence -specific manner.
- Fragments and variants of naturally occurring tracrRNAs will retain the ability, when part of a gRNA (comprising a CRISPR RNA), to guide an RNA-guided nuclease (complexed with the gRNA) to a target nucleotide sequence in a sequence -specific manner.
- the present invention provides variant sequences of gRNA, wherein said gRNA has an altered linkage between tracrRNA and crRNA, and/or has hairpin mismatches removed, and/or has A:U base pairs swapped for G:C base pairs, and/or has an altered starting position of the tracrRNA, and/or has non-protein contacting regions of the sgRNA removed. Such removal also helps to minimize the design of gRNA. Swapping A:U base pairs for G:C base pairs helps to strengthen the hairpins.
- the anti-repeat region of the tracrRNA that is fully or partially complementary to the CRISPR repeat sequence comprises from 8 nucleotides to 30 nucleotides, or more.
- the region of base pairing between the tracrRNA anti-repeat sequence and the CRISPR repeat sequence can be 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, or more nucleotides in length.
- the anti-repeat region of the tracrRNA that is fully or partially complementary to a CRISPR repeat sequence is 20 nucleotides in length.
- the degree of complementarity between a CRISPR repeat sequence and its corresponding tracrRNA anti-repeat sequence when optimally aligned using a suitable alignment algorithm, is or more than 50%, 60%, 70%, 75%, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or more.
- the entire tracrRNA can comprise from 60 nucleotides to more than 140 nucleotides.
- the tracrRNA can be 60, 65, 70, 75, 80, 85, 90, 95, 100, 105, 110, 115, 120, 125, 130, 135, 140, or more nucleotides in length.
- the tracrRNA is 80 to 90 nucleotides in length, including 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, and 90 nucleotides in length.
- the tracrRNA is 85 nucleotides in length.
- the tracrRNA comprises the nucleotide sequence set forth in Table 5 or an active variant or fragment thereof that when comprised within a gRNA is capable of directing the sequence-specific binding of an associated RNA-guided nuclease provided herein to a target sequence of interest.
- an active tracrRNA sequence variant of a wild-type sequence comprises a nucleotide sequence having at least 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or more sequence identity to the nucleotide sequence set forth in Table 5.
- an active tracrRNA sequence fragment of a wild-type sequence comprises at least 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, or more contiguous nucleotides of the nucleotide sequence set forth in Table 5.
- the presently disclosed polynucleotides comprise or encode a CRISPR repeat comprising a nucleotide sequence having at least 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%,
- the presently disclosed polynucleotides can comprise or encode a tracrRNA comprising a nucleotide sequence having at least 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or greater identity to the nucleotide sequence set forth in Table 5.
- Biologically active variants of a CRISPR repeat or tracrRNA of the invention may differ by as few as 1-15 nucleotides, as few as 1-10, such as 6-10, as few as 5, as few as 4, as few as 3, as few as 2, or as few as 1 nucleotide.
- the polynucleotides can comprise a 5' or 3' truncation, which can comprise at least a deletion of 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80 nucleotides or more from either the 5' or 3' end of the polynucleotide.
- the degree of complementarity between a spacer sequence and its corresponding target sequence, when optimally aligned using a suitable alignment algorithm is or more than 80%, 85%, 90, 95%, 96%, 97%, 98%, 99%, or more.
- the spacer sequence is free of secondary structure, which can be predicted using any suitable polynucleotide folding algorithm known in the art, including but not limited to mFold (see, e.g., Zuker and Stiegler (1981) Nucleic Acids Res . 9: 133-148) and RNAfold (see, e.g., Gruber et al. (2008) Cell 106(l):23- 24).
- the present disclosure provides polynucleotides comprising the presently disclosed crRNAs, tracrRNAs, and/or gRNAs and polynucleotides comprising a nucleotide sequence encoding the presently disclosed RGNs, crRNAs, tracrRNAs, and/or gRNAs.
- Presently disclosed polynucleotides include those comprising or encoding a CRISPR repeat sequence comprising the nucleotide sequence set forth in Table 5, or an active variant or fragment thereof that when comprised within a gRNA is capable of directing the sequence -specific binding of an associated RNA- guided nuclease to a target sequence of interest.
- the disclosure also provides polynucleotides comprising or encoding a tracrRNA comprising the nucleotide sequence set forth in Table 5, or an active variant or fragment thereof that when comprised within a gRNA is capable of directing the sequence- specific binding of an associated RNA-guided nuclease to a target sequence of interest.
- Polynucleotides are also provided that encode an RGN comprising the amino acid sequence set forth in Table 4, and active fragments or variants thereof that retain the ability to bind to a target nucleotide sequence in an RNA-guided sequence specific manner.
- the expression cassette will include in the 5 '-3' direction of transcription, a transcriptional (and, in some embodiments, translational) initiation region (i.e.. a promoter), an RGN-, a transcriptional initiation region (i.e.. a promoter), crRNA-, tracrRNA -and/or gRNA- encoding polynucleotide of the invention, and a transcriptional (and in some embodiments, translational) termination region (i.e.. termination region) functional in the organism of interest.
- the promoters of the invention are capable of directing or driving expression of a coding sequence in a host cell.
- the regulatory regions e.g., promoters, transcriptional regulatory regions, and translational termination regions
- Additional regulatory signals may include, but are not limited to, transcriptional initiation start sites, operators, activators, enhancers, other regulatory elements, ribosomal binding sites, an initiation codon, and termination signals.
- the various DNA fragments may be manipulated, so as to provide for the DNA sequences in the proper orientation and, as appropriate, in the proper reading frame.
- adapters or linkers may be employed to join the DNA fragments or other manipulations may be involved to provide for convenient restriction sites, removal of superfluous DNA, removal of restriction sites, or the like.
- in vitro mutagenesis, primer repair, restriction, annealing, resubstitutions, e.g., transitions and transversions may be involved.
- a number of promoters can be used in the practice of the invention.
- the promoters can be selected based on the desired outcome.
- the nucleic acids can be combined with constitutive, inducible, growth stage -specific, cell type-specific, tissue-preferred, tissue-specific, or other promoters for expression in the organism of interest.
- the nucleotide comprises a tissue-preferred promoter.
- the nucleic acid molecules encoding a RGN, crRNA-, tracrRNA-and/or gRNA comprise a cell type-specific promoter.
- the nucleic acid sequences encoding the RGNs, crRNA-, tracrRNA-and/or gRNA can be operably linked to a promoter sequence that is recognized by a phage RNA polymerase for example, for in vitro mRNA synthesis.
- the promoter sequence can be a pol I, pol II, pol III, T7, T3, U6, CMV or SP6 promoter sequence or a variation of a T7, T3, U6, CMV or SP6 promoter sequence.
- the expressed protein and/or RNAs can be purified for use in the methods of genome modification described herein. Any Pol II promoter or terminator could express the RGN.
- the choice of a promoter depends on how strongly RGN needs to be expressed and in what tissue type.
- the RGN is expressed using is the CMV promoter.
- the gRNA can be expressed by Pol III promoters (e.g. U6 promoter).
- the polynucleotide encoding the RGN also can be linked to a polyadenylation signal (e.g., SV40 polyA signal, or sv40 polyA with rmG terminator) and/or at least one transcriptional termination sequence. Additionally, the sequence encoding the RGN also can be linked to sequence(s) encoding at least one nuclear localization signal, at least one cell- penetrating domain, and/or at least one signal peptide capable of trafficking proteins to particular subcellular locations.
- a polyadenylation signal e.g., SV40 polyA signal, or sv40 polyA with rmG terminator
- the sequence encoding the RGN also can be linked to sequence(s) encoding at least one nuclear localization signal, at least one cell- penetrating domain, and/or at least one signal peptide capable of trafficking proteins to particular subcellular locations.
- Additional regulatory signals include, but are not limited to, transcriptional initiation start sites, operators, activators, enhancers, other regulatory elements, ribosomal binding sites, an initiation codon, termination signals, and the like. See, for example, U.S. Pat. Nos. 5,039,523 and 4,853,331
- the various DNA fragments may be manipulated, so as to provide for the DNA sequences in the proper orientation and, as appropriate, in the proper reading frame.
- adapters or linkers may be employed to join the DNA fragments or other manipulations may be involved to provide for convenient restriction sites, removal of superfluous DNA, removal of restriction sites, or the like.
- in vitro mutagenesis, primer repair, restriction, annealing, resubstitutions, e.g., transitions and transversions may be involved.
- conservative variants include those sequences that, because of the degeneracy of the genetic code, encode the native amino acid sequence of the gene of interest.
- Naturally occurring allelic variants such as these can be identified with the use of well-known molecular biology techniques, as, for example, with polymerase chain reaction (PCR) and hybridization techniques as outlined below.
- Variant polynucleotides also include synthetically derived polynucleotides, such as those generated, for example, by using site-directed mutagenesis but which still encode the polypeptide or the polynucleotide of interest.
- variants of a particular polynucleotide disclosed herein will have at least 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or more sequence identity to that particular polynucleotide as determined by sequence alignment programs and parameters described elsewhere herein.
- Variants of a particular polynucleotide disclosed herein can also be evaluated by comparison of the percent sequence identity between the polypeptide encoded by a variant polynucleotide and the polypeptide encoded by the reference polynucleotide. Percent sequence identity between any two polypeptides can be calculated using sequence alignment programs and parameters described elsewhere herein.
- the percent sequence identity between the two encoded polypeptides is at least 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or more sequence identity.
- the presently disclosed polynucleotides encode an RGN polypeptide comprising an amino acid sequence having at least 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or greater identity to an amino acid sequence set forth in Table 4.
- Variant polynucleotides and proteins also encompass sequences and proteins derived from a mutagenic and recombinogenic procedure such as DNA shuffling. With such a procedure, one or more different RGN proteins disclosed herein is manipulated to create a new RGN protein possessing the desired properties. In this manner, libraries of recombinant polynucleotides are generated from a population of related sequence polynucleotides comprising sequence regions that have substantial sequence identity and can be homologously recombined in vitro or in vivo.
- sequence motifs encoding a domain of interest may be shuffled between the RGN sequences provided herein and other known RGN genes to obtain a new gene coding for a protein with an improved property of interest, such as an increased Km in the case of an enzyme.
- Strategies for such DNA shuffling are known in the art. See, for example, Stemmer (1994) Proc. Natl. Acad. Sci. USA
- the nucleic acid molecules encoding RNA-guided nucleases can be codon optimized for expression in a target cell or tissue of interest.
- Such polynucleotide coding sequence normally has its frequency of codon usage designed to mimic the frequency of preferred codon usage or transcription conditions of a particular host cell. Expression in the particular host cell or organism is enhanced as a result of the alteration of one or more codons at the nucleic acid level such that the translated amino acid sequence is not changed.
- Nucleic acid molecules can be codon optimized, either wholly or in part. Codon tables and other references providing preference information for a wide range of organisms are available in the art.
- the polynucleotide encoding the RGN, crRNA, tracrRNA, and/or sgRNA can be present in a vector or multiple vectors.
- Suitable vectors include plasmid vectors, phagemids, cosmids, artificial/mini-chromosomes, transposons, and viral vectors (e.g., lentiviral vectors, adeno-associated viral vectors, baculoviral vector).
- the vector can comprise additional expression control sequences (e.g. , enhancer sequences, Kozak sequences, polyadenylation sequences, transcriptional termination sequences), selectable marker sequences (e.g., antibiotic resistance genes), origins of replication, and the like.
- the vector can also comprise a selectable marker gene for the selection of transformed cells.
- Selectable marker genes are utilized for the selection of transformed cells or tissues.
- Marker genes include genes encoding antibiotic resistance, such as those encoding neomycin phosphotransferase II (NEO) and hygromycin phosphotransferase (HPT), as well as genes conferring resistance to herbicidal compounds, such as glufosinate ammonium, bromoxynil, imidazolinones, and 2,4- dichlorophenoxyacetate (2,4-D).
- the expression cassette or vector comprising the sequence encoding the RGN polypeptide can further comprise a sequence encoding a crRNA and/or a tracrRNA, or the crRNA and tracrRNA combined to create a gRNA.
- the sequence(s) encoding the crRNA and/or tracrRNA can be operably linked to at least one transcriptional control sequence for expression of the crRNA and/or tracrRNA in the organism or host cell of interest.
- the polynucleotide encoding the crRNA and/or tracrRNA can be operably linked to a promoter sequence that is recognized by RNA polymerase III (Pol III).
- suitable Pol III promoters include, but are not limited to, mammalian U6, U3, Hl, and 7SL RNA promoters and rice U6 and U3 promoters.
- the present disclosure provides a system for binding a target sequence of interest, wherein the system comprises at least one gRNA or a nucleotide sequence encoding the same, and at least one RGN or a nucleotide sequence encoding the same.
- the gRNA hybridizes to the target sequence of interest and also forms a complex with the RGN polypeptide, thereby directing the RGN polypeptide to bind to the target sequence.
- the RGN comprises an amino acid sequence as set forth in Table 4 or an active variant or fragment thereof.
- the gRNA comprises a CRISPR repeat sequence comprising the nucleotide sequence set forth in Table 5 or an active variant or fragment thereof.
- the gRNA comprises a tracrRNA comprising a nucleotide sequence set forth in Table 5 or an active variant or fragment thereof.
- the gRNA of the system can be a single gRNA or a dual-gRNA.
- the system comprises an RGN that is heterologous to the gRNA, wherein the RGN and gRNA are not naturally complexed in nature.
- the system for binding a target sequence of interest can be a ribonucleoprotein complex, which is at least one molecule of an RNA bound to at least one protein.
- the ribonucleoprotein complexes provided herein comprise at least one gRNA as the RNA component and an RNA-guided nuclease as the protein component.
- Such ribonucleoprotein complexes can be purified from a cell or organism that naturally expresses an RGN polypeptide and has been engineered to express a particular gRNA that is specific for a target sequence of interest.
- the ribonucleoprotein complex can be purified from a cell or organism that has been transformed with polynucleotides that encode an RGN polypeptide and a gRNA and cultured under conditions to allow for the expression of the RGN polypeptide and gRNA.
- methods are provided for making an RGN polypeptide or an RGN ribonucleoprotein complex. Such methods comprise culturing a cell comprising a nucleotide sequence encoding an RGN polypeptide, and in some embodiments a nucleotide sequence encoding a gRNA, under conditions in which the RGN polypeptide (and in some embodiments, the gRNA) is expressed.
- the RGN polypeptide or RGN ribonucleoprotein can then be purified from a lysate of the cultured cells.
- the RGN polypeptide is recombinantly produced and comprises a purification tag to aid in its purification, including but not limited to, glutathione-S-transferase (GST), chitin binding protein (CBP), maltose binding protein, thioredoxin (TRX), poly(NANP), tandem affinity purification (TAP) tag, myc, AcV5, AU1, AU5, E, ECS, E2, FLAG, HA, nus, Softag 1, Softag 3, Strep, SBP, Glu-Glu, HSV, KT3, S, SI, T7, V5, VSV-G, 6xHis, lOxHis, biotin carboxyl carrier protein (BCCP), and calmodulin.
- GST glutathione-S-transferase
- CBP chitin binding protein
- TRX thioredoxin
- TAP tandem affinity purification
- the tagged RGN polypeptide or RGN ribonucleoprotein complex is purified using immobilized metal affinity chromatography. It will be appreciated that other similar methods known in the art may be used, including other forms of chromatography or for example immunoprecipitation, either alone or in combination.
- Some methods provided herein for binding and/or cleaving a target sequence of interest involve the use of an in vitro assembled RGN ribonucleoprotein complex.
- In vitro assembly of an RGN ribonucleoprotein complex can be performed using any method known in the art in which an RGN polypeptide is contacted with a gRNA under conditions to allow for binding of the RGN polypeptide to the gRNA.
- the RGN polypeptide can be purified from a biological sample, cell lysate, or culture medium, produced via in vitro translation, or chemically synthesized.
- the gRNA can be purified from a biological sample, cell lysate, or culture medium, transcribed in vitro, or chemically synthesized.
- the RGN polypeptide and gRNA can be brought into contact in solution (e.g., buffered saline solution) to allow for in vitro assembly of the RGN ribonucleoprotein complex.
- components of the nucleic-acid targeting system are delivered using nanoscale delivery systems, such as nanoparticles.
- liposomes and other particulate delivery systems can be used.
- vectors including the components of the present methods can be packaged in liposomes prior to delivery to the subject or to cells derived therefrom. Lipid encapsulation is generally accomplished using liposomes that are able to stably bind or entrap and retain nucleic acid.
- expression constructs comprising nucleotide sequences encoding the RGNs, crRNA, tracrRNA, and/or sgRNA can be used to transform organisms of interest.
- Methods for transformation involve introducing a nucleotide construct into an organism of interest.
- the methods of the invention do not require a particular method for introducing a nucleotide construct to a host organism, only that the nucleotide construct gains access to the interior of at least one cell of the host organism.
- the host cell can be a eukaryotic or prokaryotic cell.
- the eukaryotic host cell is a plant cell, a mammalian cell, or an insect cell.
- Methods for introducing nucleotide constructs into plants and other host cells are known in the art including, but not limited to, stable transformation methods, transient transformation methods, and virus-mediated methods.
- Transformation of a host cell may be performed by infection, transfection, microinjection, electroporation, microprojection, biolistics or particle bombardment, electroporation, silica/carbon fibers, ultrasound mediated, PEG mediated, calcium phosphate co-precipitation, polycation DMSO technique, DEAE dextran procedure, and viral mediated, liposome mediated and the like.
- Viral -mediated introduction of a polynucleotide encoding an RGN, crRNA, and/or tracrRNA includes retroviral, lentiviral, adenoviral, and adeno-associated viral mediated introduction and expression, as well as the use of Caulimoviruses, Geminiviruses, and RNA plant viruses.
- Transformation may result in stable or transient incorporation of the nucleic acid into the cell.
- the cells that have been transformed may be grown into a transgenic organism, such as a plant, in accordance with conventional ways. See, for example, McCormick et al. (1986) Plant Cell Reports 5:81-84. These plants may then be grown, and either pollinated with the same transformed strain or different strains, and the resulting hybrid having constitutive expression of the desired phenotypic characteristic identified. Two or more generations may be grown to ensure that expression of the desired phenotypic characteristic is stably maintained and inherited and then seeds harvested to ensure expression of the desired phenotypic characteristic has been achieved. In this manner, the present invention provides transformed seed (also referred to as "transgenic seed") having a nucleotide construct of the invention, for example, an expression cassette of the invention, stably incorporated into their genome.
- cells that have been transformed may be introduced into an organism. These cells could have originated from the organism, wherein the cells are transformed in an ex vivo approach.
- the polynucleotides encoding the RGNs, crRNAs, and/or tracrRNAs can also be used to transform any prokaryotic species, including but not limited to, archaea and bacteria (e.g., Bacillus sp., Klebsiella sp. Streptomyces sp., Rhizobium sp., Escherichia sp., Pseudomonas sp., Salmonella sp., Shigella sp., Vibrio sp., Yersinia sp., Mycoplasma sp., Agrobacterium, Lactobacillus sp.).
- archaea and bacteria e.g., Bacillus sp., Klebsiella sp. Streptomyces sp., Rhizobium sp., Escherichia sp., Pseudomonas sp., Salmonella sp
- the polynucleotides encoding the RGNs, crRNAs, and/or tracrRNAs can be used to transform any eukaryotic species, including but not limited to animals (e.g., mammals, insects, fish, birds, and reptiles), fungi, amoeba, algae, and yeast.
- Non-viral vector delivery systems include DNA plasmids, RNA (e.g. a transcript of a vector described herein), naked nucleic acid, and nucleic acid complexed with a delivery vehicle, such as a liposome.
- Viral vector delivery systems include DNA and RNA viruses, which have either episomal or integrated genomes after delivery to the cell.
- Methods of non-viral delivery of nucleic acids include lipofection, nucleofection, microinjection, biolistics, virosomes, liposomes, immunoliposomes, polycation or lipid: nucleic acid conjugates, naked DNA, artificial virions, and agent-enhanced uptake of DNA.
- Lipofection is described in e.g., U.S. 5,049,386, 4 and lipofection reagents are sold commercially. Delivery can be to cells (e.g. in vitro or ex vivo administration) or target tissues ( e.g. in vivo administration).
- lipid: nucleic acid complexes including targeted liposomes such as immunolipid complexes, is well known to one of skill in the art (see, e.g., Gao et ah, Gene Therapy 2:710-722 (1995)).
- RNA or DNA viral based systems for the delivery of nucleic acids takes advantage of highly evolved processes for targeting a virus to specific cells in the body and trafficking the viral payload to the nucleus.
- Viral vectors can be administered directly to patients (in vivo) or they can be used to treat cells in vitro, and the modified cells may optionally be administered to patients ( ex vivo).
- Conventional viral based systems could include retroviral, lentivirus, adenoviral, adeno- associated and herpes simplex virus vectors for gene transfer. Integration in the host genome is possible with the retrovirus, lentivirus, and adeno-associated virus gene transfer methods, often resulting in long term expression of the inserted transgene. Additionally, high transduction efficiencies have been observed in many different cell types and target tissues.
- Lentiviral vectors are retroviral vectors that are able to transduce or infect non-dividing cells and typically produce high viral titers. Selection of a retroviral gene transfer system would therefore depend on the target tissue. Retroviral vectors are comprised of cis-acting long terminal repeats with packaging capacity for up to 6-10 kb of foreign sequence. The minimum cis-acting LTRs are sufficient for replication and packaging of the vectors, which are then used to integrate the therapeutic gene into the target cell to provide permanent transgene expression.
- Widely used retroviral vectors include those based upon murine leukemia virus (MuLV), gibbon ape leukemia virus (GaLV), [179] Simian Immuno deficiency vims (SIV), human immuno deficiency vims (HIV), and combinations thereof (see, e.g., Buchscher et al., J. Viral. 66:2731-2739 (1992); Johann et al., J. Viral. 66: 1635-1640 (1992); Sommnerfelt et al., Viral. 176:58-59 (1990); Wilson et al., J. Viral. 63:2374- 2378 (1989); Miller et al., 7. Viral. 65:2220-2224
- viral based systems for the delivery of nucleic acids allows targeting a vims to specific cells and trafficking the viral payload to the nucleus.
- Viral vectors can be administered directly to patients (in vivo) or they can be used to treat cells in vitro, and the modified cells may optionally be administered to patients (ex vivo).
- Conventional viral based systems could include retroviral, lentivims, adenoviral, adeno-associated and herpes simplex vims vectors for gene transfer. Integration in the host genome is possible with the retrovims, lentivims, and adeno-associated vims gene transfer methods, often resulting in long term expression of the inserted transgene. Additionally, high transduction efficiencies have been observed in many different cell types and target tissues.
- Lentiviral vectors are retroviral vectors that are able to transduce or infect non-dividing cells and typically produce high viral titers. Selection of a retroviral gene transfer system would therefore depend on the target tissue. Retroviral vectors are comprised of cis-acting long terminal repeats with packaging capacity for up to 6-10 kb of foreign sequence. The minimum cis-acting LTRs are sufficient for replication and packaging of the vectors, which are then used to integrate the therapeutic gene into the target cell to provide permanent transgene expression.
- Widely used retroviral vectors include those based upon murine leukemia vims (MuLV), gibbon ape leukemia vims (GaLV), Simian Immuno deficiency vims (SIV), human immuno deficiency vims (HIV), and combinations thereof.
- MiLV murine leukemia vims
- GaLV gibbon ape leukemia vims
- SIV Simian Immuno deficiency vims
- HAV human immuno deficiency vims
- adenoviral based systems may be used.
- Adenoviral based vectors are capable of very high transduction efficiency in many cell types and do not require cell division. With such vectors, high titer and levels of expression have been obtained. This vector can be produced in large quantities in a relatively simple system.
- Adeno-associated vims (“AAV”) vectors may also be used to transduce cells with target nucleic acids. Constmction of recombinant AAV vectors are described in a number of publications, including U.S. 5,173,414. Packaging cells are typically used to form vims particles that are capable of infecting a host cell.
- Viral vectors used in gene therapy are usually generated by producing a cell line that packages a nucleic acid vector into a viral particle.
- the vectors typically contain the minimal viral sequences required for packaging and subsequent integration into a host, other viral sequences being replaced by an expression cassette for the polynucleotide( s) to be expressed.
- the missing viral functions are typically supplied in trans by the packaging cell line.
- AAV vectors used in gene therapy typically only possess ITR sequences from the AAV genome which are required for packaging and integration into the host genome.
- Viral DNA is packaged in a cell line, which contains a helper plasmid encoding the other AAV genes, namely rep and cap, but lacking ITR sequences.
- the cell line may also be infected with adenovirus as a helper.
- the helper virus promotes replication of the AAV vector and expression of AAV genes from the helper plasmid.
- the helper plasmid is not packaged in significant amounts due to a lack of ITR sequences. Contamination with adenovirus can be reduced by, e.g., heat treatment to which adenovirus is more sensitive than AAV. Additional methods for the delivery of nucleic acids to cells are known to those skilled in the art. See, for example, US20030087817
- the disclosure provides methods of modifying a target polynucleotide in a eukaryotic cell, which may be performed in vivo, ex vivo or in vitro.
- the method comprises sampling a cell or population of cells from a human or non-human animal or plant (including microalgae) and modifying the cell or cells. Culturing may occur at any stage ex vivo. The cell or cells may even be re-introduced into the non-human animal or plant (including micro-algae).
- the present disclosure provides methods for binding, cleaving, and/or modifying a target nucleotide sequence of interest.
- the methods include delivering a system comprising at least one gRNA or a polynucleotide encoding the same, and at least one RGN polypeptide or a polynucleotide encoding the same to the target sequence or a cell, organelle, or embryo comprising the target sequence.
- the RGN comprises the amino acid sequence set forth in Table 4, or an active variant or fragment thereof.
- the gRNA comprises a CRISPR repeat sequence comprising the nucleotide sequence set forth in Table 5, or an active variant or fragment thereof.
- the gRNA comprises a tracrRNA comprising the nucleotide sequence set forth in Table 5, or an active variant or fragment thereof.
- the gRNA of the system can be a single gRNA or a dual-gRNA.
- the RGN of the system may be nuclease dead RGN, have nickase activity, or may be a fusion polypeptide.
- the fusion polypeptide comprises a base-editing polypeptide, for example a cytidine deaminase or an adenosine deaminase.
- the RGN and/or gRNA is heterologous to the cell, organelle, or embryo to which the RGN and/or gRNA (or polynucleotide(s) encoding at least one of the RGN and gRNA) are introduced.
- the method comprises delivering a polynucleotide encoding a gRNA and/or an RGN polypeptide
- the cell, organelle, or embryo can then be cultured under conditions in which the gRNA and/or RGN polypeptide are expressed.
- the method comprises contacting a target sequence with an RGN ribonucleoprotein complex.
- the RGN ribonucleoprotein complex may comprise an RGN that is nuclease dead or has nickase activity.
- the RGN of the ribonucleoprotein complex is a fusion polypeptide comprising a base-editing polypeptide.
- the method comprises introducing into a cell, organelle, or embryo comprising a target sequence an RGN ribonucleoprotein complex.
- the RGN ribonucleoprotein complex can be one that has been purified from a biological sample, recombinantly produced and subsequently purified, or in vitro- assembled as described herein.
- the method can further comprise the in vitro assembly of the complex prior to contact with the target sequence, cell, organelle, or embryo.
- a purified or in vitro assembled RGN ribonucleoprotein complex can be introduced into a cell, organelle, or embryo using any method known in the art, including, but not limited to electroporation.
- an RGN polypeptide and/or polynucleotide encoding or comprising the gRNA can be introduced into a cell, organelle, or embryo using any method known in the art (e.g., electroporation).
- the gRNA directs the RGN to bind to the target sequence in a sequence-specific manner.
- the RGN polypeptide cleaves the target sequence of interest upon binding.
- the target sequence can subsequently be modified via endogenous repair mechanisms, such as non-homologous end joining, or homology-directed repair with a provided donor polynucleotide.
- Methods to measure binding of an RGN polypeptide to a target sequence include chromatin immunoprecipitation assays, gel mobility shift assays, DNA pull-down assays, reporter assays, microplate capture and detection assays.
- methods to measure cleavage or modification of a target sequence include in vitro or in vivo cleavage assays wherein cleavage is confirmed using PCR, sequencing, or gel electrophoresis, with or without the attachment of an appropriate label (e.g., radioisotope, fluorescent substance) to the target sequence to facilitate detection of degradation products.
- an appropriate label e.g., radioisotope, fluorescent substance
- NTEXPAR nicking triggered exponential amplification reaction
- the methods involve the use of a single type of RGN complexed with more than one gRNA.
- the more than one gRNA can target different regions of a single gene or can target multiple genes.
- a double -stranded break introduced by an RGN polypeptide can be repaired by a non-homologous end-joining (NHEJ) repair process. Due to the error-prone nature of NHEJ, repair of the double -stranded break can result in a modification to the target sequence. Modification of the target sequence can result in the expression of an altered protein product or inactivation of a coding sequence.
- NHEJ non-homologous end-joining
- the donor sequence in the donor polynucleotide can be integrated into or exchanged with the target nucleotide sequence during the course of repair of the introduced double-stranded break, resulting in the introduction of the exogenous donor sequence.
- a donor polynucleotide thus comprises a donor sequence that is desired to be introduced into a target sequence of interest.
- the donor sequence alters the original target nucleotide sequence such that the newly integrated donor sequence will not be recognized and cleaved by the RGN.
- the donor polynucleotide can comprise a donor sequence flanked by compatible overhangs, allowing for direct ligation of the donor sequence to the cleaved target nucleotide sequence comprising overhangs by a non-homologous repair process during repair of the double - stranded break.
- the method can comprise introducing two RGN nickases that target identical or overlapping target sequences and cleave different strands of the polynucleotide.
- an RGN nickase that only cleaves the positive (+) strand of a double -stranded polynucleotide can be introduced along with a second RGN nickase that only cleaves the negative (-) strand of a double -stranded polynucleotide.
- a method for binding a target nucleotide sequence and detecting the target sequence, wherein the method comprises introducing into a cell, organelle, or embryo at least one gRNA or a polynucleotide encoding the same, and at least one RGN polypeptide or a polynucleotide encoding the same, expressing the gRNA and/or RGN polypeptide (if coding sequences are introduced), wherein the RGN polypeptide is a nuclease-dead RGN and further comprises a detectable label, and the method further comprises detecting the detectable label.
- the detectable label may be fused to the RGN as a fusion protein (e.g., fluorescent protein) or may be a small molecule conjugated to or incorporated within the RGN polypeptide that can be detected visually or by other means.
- the methods comprise introducing into a cell, organelle, or embryo at least one gRNA or a polynucleotide encoding the same, and at least one RGN polypeptide or a polynucleotide encoding the same, expressing the gRNA and/or RGN polypeptide (if coding sequences are introduced), wherein the RGN polypeptide is a nuclease-dead RGN.
- the nuclease-dead RGN is a fusion protein comprising an expression modulator domain (i.e., epigenetic modification domain, transcriptional activation domain or a transcriptional repressor domain) as described herein.
- an expression modulator domain i.e., epigenetic modification domain, transcriptional activation domain or a transcriptional repressor domain
- the invention provides kits containing any one or more of the elements disclosed in the above methods and compositions.
- the kit comprises a vector system and instructions for using the kit.
- the vector system comprises (a) a first regulatory element operably linked to a tracr mate sequence and one or more insertion sites for inserting a guide sequence upstream of the tracr mate sequence, wherein when expressed, the guide sequence directs sequence-specific binding of a CRISPR complex to a target sequence in a eukaryotic cell, wherein the CRISPR complex comprises a CRIS PR enzyme complexed with (1) the guide sequence that is hybridized to the target sequence, and (2) the tracr mate sequence that is hybridized to the tracr sequence; and/or (b) a second regulatory element operably linked to an enzyme coding sequence encoding said CRISPR enzyme comprising a nuclear localization sequence.
- Elements may be provided individually or in combinations, and may be provided in any suitable container, such as a vial, a bottle
- kits includes instructions in one or more languages.
- a kit comprises one or more reagents for use in a process utilizing one or more of the elements described herein.
- Reagents may be provided in any suitable container.
- a kit may provide one or more reaction or storage buffers.
- Reagents may be provided in a form that is usable in a particular assay, or in a form that requires addition of one or more other components before use ( e.g. in concentrate or lyophilized form).
- a buffer can be any buffer, including but not limited to a sodium carbonate buffer, a sodium bicarbonate buffer, a borate buffer, a Tris buffer, a MOPS buffer, a HEPES buffer, and combinations thereof.
- the buffer is alkaline.
- the buffer has a pH from 7 to 10.
- the kit comprises one or more oligonucleotides corresponding to a guide sequence for insertion into a vector so as to operably link the guide sequence and a regulatory element.
- the kit comprises a homologous recombination template polynucleotide.
- the invention provides methods for using one or more elements of a CRISPR system.
- the CRISPR complex of the invention provides an effective means for modifying a target polynucleotide.
- the CRISPR complex of the invention has a wide variety of utility including modifying (e.g., deleting, inserting, translocating, inactivating, activating) a target polynucleotide in a multiplicity of cell types.
- An exemplary CRISPR complex comprises a CRISPR enzyme complexed with a guide sequence hybridized to a target sequence within the target polynucleotide.
- cells and organisms comprising a target sequence of interest that has been modified using a process or the system based an RGN, crRNA, and/or tracrRNA as described herein. Also are provided cells and organisms comprising the system for binding a target sequence of interest comprising an RGN, crRNA, and/or tracrRNA as described herein
- the RGN comprises the amino acid sequence as disclosed above, or an active variant or fragment thereof.
- the gRNA comprises a CRISPR repeat sequence comprising the nucleotide sequence as disclosed above, or an active variant or fragment thereof.
- the gRNA comprises a tracrRNA comprising the nucleotide sequence as disclosed above, or an active variant or fragment thereof.
- the gRNA of the system can be a single gRNA or a dual-gRNA.
- the modified cells can be eukaryotic (e.g., mammalian, plant, insect cell) or prokaryotic.
- organelles and embryos comprising at least one nucleotide sequence that has been modified by a process utilizing an RGN, crRNA, and/or tracrRNA as described herein.
- the genetically modified cells, organisms, organelles, and embryos can be heterozygous or homozygous for the modified nucleotide sequence.
- the chromosomal modification of the cell, organism, organelle, or embryo can result in altered expression (up-regulation or down-regulation), inactivation, or the expression of an altered protein product or an integrated sequence.
- the genetically modified cell, organism, organelle, or embryo is referred to as a “knock out”.
- the knock out phenotype can be the result of a deletion mutation (i.e.. deletion of at least one nucleotide), an insertion mutation (i.e.. insertion of at least one nucleotide), or a nonsense mutation (/. e. , substitution of at least one nucleotide such that a stop codon is introduced).
- the chromosomal modification of a cell, organism, organelle, or embryo can produce a “knock in”, which results from the chromosomal integration of a nucleotide sequence that encodes a protein.
- the coding sequence is integrated into the chromosome such that the chromosomal sequence encoding the wild-type protein is inactivated, but the exogenously introduced protein is expressed.
- the chromosomal modification results in the production of a variant protein product.
- the expressed variant protein product can have at least one amino acid substitution and/or the addition or deletion of at least one amino acid.
- the variant protein product encoded by the altered chromosomal sequence can exhibit modified characteristics or activities when compared to the wild-type protein, including but not limited to altered enzymatic activity or substrate specificity.
- the chromosomal modification can result in an altered expression pattern of a protein.
- chromosomal alterations in the regulatory regions controlling the expression of a protein product can result in the overexpression or downregulation of the protein product or an altered tissue or temporal expression pattern.
- the composition of the present invention may be in a pharmaceutical composition.
- the pharmaceutical composition may comprise 1 ng to 10 mg of DNA encoding the CRISPR/Cas- based system or CRISPR/Cas-based system protein component, i.e., the fusion protein.
- the pharmaceutical composition may comprise 1 ng to 10 mg of the DNA of the modified lentiviral vector.
- the pharmaceutical composition may comprise 1 ng to 10 mg of the DNA of the modified AAV vector and a nucleotide sequence encoding the site-specific nuclease.
- the pharmaceutical compositions according to the present invention can be formulated according to the mode of administration to be used. In cases where pharmaceutical compositions are injectable pharmaceutical compositions, they are sterile, pyrogen free and particulate free.
- An isotonic formulation is preferably used.
- additives for isotonicity may include sodium chloride, dextrose, mannitol, sorbitol and lactose.
- isotonic solutions such as phosphate buffered saline are preferred.
- Stabilizers include gelatin and albumin.
- a vasoconstriction agent is added to the formulation.
- the composition may further comprise a pharmaceutically acceptable excipient.
- the pharmaceutically acceptable excipient may be functional molecules as vehicles, adjuvants, carriers, or diluents.
- the pharmaceutically acceptable excipient may be a transfection facilitating agent, which may include surface active agents, such as immune-stimulating complexes (ISCOMS), Freunds incomplete adjuvant, LPS analog including monophosphoryl lipid A, muramyl peptides, quinone analogs, vesicles such as squalene and squalene, hyaluronic acid, lipids, liposomes, calcium ions, viral proteins, polyanions, polycations, or nanoparticles, or other known transfection facilitating agents.
- ISCOMS immune-stimulating complexes
- LPS analog including monophosphoryl lipid A, muramyl peptides, quinone analogs, vesicles such as squalene and squalene, hyaluronic acid,
- the transfection facilitating agent can be a polyanion, polycation, including poly-L- glutamate (LGS), or lipid.
- the transfection facilitating agent is poly-L-glutamate, and more preferably, the poly- L-glutamate is present in the composition for genome editing in skeletal muscle or cardiac muscle at a concentration less than 6 mg/ml.
- the transfection facilitating agent may also include surface active agents such as immune -stimulating complexes (ISCOMS), Freunds incomplete adjuvant, LPS analog including monophosphoryl lipid A, muramyl peptides, quinone analogs and vesicles such as squalene and squalene, and hyaluronic acid may also be used administered in conjunction with the genetic construct.
- surface active agents such as immune -stimulating complexes (ISCOMS), Freunds incomplete adjuvant, LPS analog including monophosphoryl lipid A, muramyl peptides, quinone analogs and vesicles such as squalene and squalene, and hyaluronic acid may also be used administered in conjunction with the genetic construct.
- the DNA vector encoding the composition may also include a transfection facilitating agent such as lipids, liposomes, including lecithin liposomes or other liposomes known in the art, as a DNA-liposome mixture (see for example W09324640), calcium ions, viral proteins, polyanions, polycations, or nanoparticles, or other known transfection facilitating agents.
- the transfection facilitating agent is a polyanion, polycation, including poly-L- glutamate (LGS), or lipid.
- LGS poly-L- glutamate
- Metagenomic samples were searched for open reading frames (ORFs) and those that were predicted to be genes were selected.
- a hidden Markov model (HMM) was used to compare the putative genes to profdes of known Cas proteins.
- the identified Cas genes were grouped into operons, and the operon type was determined based on the presence of known signature genes.
- the CRISPR arrays were identified based on the presence of regularly spaced repeats. The subtype of each CRISPR array was predicted using machine learning. Cas operons were linked to CRISPR arrays if they were less than 10 kilobases apart.
- Identified repeat-arrays were then used to identify anti-repeat locations flanking the putative effector protein within 3500 nt.
- Flanking regions around putative anti-repeat locations were extracted and the repeat-anti-repeat hybrid structures were predicted and used to identify putative crRNAs, tracrRNAs, and sgRNAs (Chyou, et al (2019). RNA biology, 16(4), 423-434).
- Example 2 Determination of PAM requirements for each RGN through Bacterial PAM Depletion
- PAM requirements for each RGN were determined using a bacterial PAM depletion assay essentially adapted from Kleinstiver et al. (2015) Nature 523:481-485 and Zetsche et al. (2015) Cell 163:759-771. Briefly, two plasmid libraries (C2 and T2) were generated in a pUC18 backbone (ampR), with each containing a distinct 23bp protospacer (target) sequence flanked by 8 random nucleotides (i.e., the PAM region). The target sequence and flanking PAM region of library T2 and library C2 for each RGN are set forth in Table 14.
- the libraries were separately electroporated into T7 Express E. coli (NEB) cells harboring pET28b expression vectors containing the minimal CRISPR operon with the repeat spacer array modified to contain three copies of the intended library target sequence at the average spacer length of the CRISPR repeat in C2 or T2.
- Sufficient library plasmid was used in the transformation reaction to obtain > 10 A 8 cfu.
- the modified minimal CRISPR operon in the pET28b backbone were under the control of T7 promoters.
- the transformation reaction was allowed to recover for 1 hr after which it was diluted into LB media containing carbenicillin and kanamycin and grown overnight.
- the PAM and protospacer regions of uncleaved plasmids were PCR-amplified and prepared for sequencing following published protocols (16s-metagenomic library prep guide 15044223B, Illumina, San Diego, CA). Deep sequencing (55bp paired end reads) was performed on a NextSeq (Illumina). Typically, 1-4M reads were obtained per amplicon. PAM regions were extracted, counted, and normalized to total reads for each sample. PAMs that lead to plasmid cleavage were identified by being underrepresented when compared to controls (i.e., when the library is transformed into E. coli containing the RGN but lacking an appropriate sgRNA).
- an enrichment value was computed for each kmer as the difference between the library size-normalized read counts in the control sample and in the targeting sample. This value was rounded to the nearest integer for positive numbers and set to zero for negative numbers. Enrichment values were then summed across all kmers to yield a position frequency matrix, which was represented visually as a sequence logo using the command line utility weblogo. Those RGNs with consistency among the most enriched kmers — sequence logo information content > 0.2 when including the top 100 enriched kmers — and with qualitatively consistent PAMs across plasmid libraries (T2 and C2) were deemed to have bonafide PAMs.
- the final PAM for these RGNs were obtained by summing counts across both plasmid libraries, normalizing counts, computing kmer enrichment values, summing across kmers to yield a position frequency matrix, then visually representing the PAM as a sequence logo using the command line utility weblogo.
- RNAseq pipeline was written to detect the expressed small non coding RNA transcripts (Fig 2-3). Manual curation of RNAs was performed using secondary structure prediction by NUPACK, an RNA folding software to identify the boundaries of the expressed putative tracrRNA and processed crRNA. gRNA designs were then made by linking the expressed boundaries between the putative tracrRNA and the crRNA with a RAAA linker sequence.
- the RGN was codon optimized for human expression and cloned into expression cassettes with a Nterm SV40 NLS, and a Cterm Nucleoplasmin NLS under control of a CMV promoter for mammalian expression.
- Tag sequences are set forth in Table 15.
- the gRNA expression constructs encoding a single gRNA each under the control of a human RNA polymerase III U6 promoter were produced and introduced into an expression vector containing GFP under control of a CMV promoter. Guides were design to targeted regions of selected genes with the appropriate PAM for the system.
- the constructs described were introduced into mammalian cells.
- HEK293T cells Sigma
- DMEM Dulbecco’s modified Eagle medium
- Sibco 10% (vol/vol) fetal bovine serum
- Penicillin- Streptomycin Gibco
- the coding sequence of the identified RGN is codon-optimized for expression in mammalian cells and introduced into the expression cassette, which produces a fusion protein that includes a NLS tag at its N-terminal end operably linked to a codon optimized deaminase sequences (identified and developed in house) at its C-terminal end.
- the deaminase is operably linked to a flexible amino acid linker at their C-terminal end, and the amino acid linker is operably linked to the RNA guided nuclease at its C-terminal end, that has been mutated to have an inactive RuvC domain (That is, it has been mutated into RGN that is catalytically dead).
- RNA-guided DNA binding polypeptide is operably linked to a flexible amino acid linker at their C-terminal end, and the amino acid linker is operably linked to a uracil protecting peptide.
- the uracil protecting peptide is operably linked to a flexible amino acid linker at their C-terminal end, and the amino acid linker is operably linked to a second NLS at its C-terminal end.
- Each of these expression cassettes is introduced into a vector capable of driving the expression of the fusion protein in mammalian cells.
- a vector capable of expressing guide RNA to target the deaminase-RGN-UPP fusion protein to the determined genomic location was also produced.
- These guide RNAs can guide the deaminase-RGN-UPP fusion protein to the target genome sequence for base editing.
- liposome transfection vectors capable of expressing the deaminase-RGN-UPP fusion protein and guide RNAs are transfected into HEK293T cells.
- the day before transfection the cells are distributed in a 24-well plate of growth medium (DMEM + 10% fetal bovine serum + 1% penicillin/streptomycin) at 1.3* 10 5 cells/well.
- DMEM + 10% fetal bovine serum + 1% penicillin/streptomycin 1.3* 10 5 cells/well.
- Lipofectamine® 3000 reagent Thermo Fisher Scientific
- genomic DNA is harvested from the transfected cells, and the DNA is sequenced and analyzed for the presence of targeted cytosine base editing mutations using CRISPResso2 (Clement K, et al Nat Biotechnol. 2019 Mar; 37(3):224-226. doi: 10.1038/s41587-019-0032-3. PubMed PMID: 30809026).
- Improvements to the editing ability of novel RGNs can be accomplished by changing the sgRNA scaffolds by altering the tracrRNA and crRNA linkage, removing hairpin mismatches, strengthening hairpins by swapping A:U base pairs for G:C base pairs, altering the starting position of the tracrRNA, removing non-protein contacting regions of the sgRNA to minimize the design.
- novel sgRNA designs were developed and tested for genomic editing (Table 12).
- Figure 4 shows the relative activity of EGS0023 with two potential tracrRNA.
- Figure 5 shows an improvement in editing achieved with EGS0024 when using a modified tracrRNA (sgRNA v2) that has been truncated to remove extraneous sequence from the 5 ’ end of the tracrRNA and shorten one of the extended potential hairpins compared to the wild type tracrRNA (sgRNA vl) ( Figure 6).
- sgRNA v2 modified tracrRNA
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Biomedical Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Organic Chemistry (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Plant Pathology (AREA)
- Physics & Mathematics (AREA)
- Biophysics (AREA)
- Crystallography & Structural Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Enzymes And Modification Thereof (AREA)
Abstract
The present invention provides RNA-guided nuclease proteins and nucleic acid targeting system comprising such for cleaving and/or modifying the target nucleotide of interest.
Description
RNA-GUIDED NUCLEASES AND NUCLEIC ACID TARGETING SYSTEMS COMPRISING SUCH RNA-GUIDED NUCLEASES
[001] The present invention relates to novel RNA-guided nucleases (RGN) and nucleic acid targeting systems comprising such.
BACKGROUND
[002] Targeted genome editing or modification has been undergoing many changes in the past years since the discovery of novel technologies and systems. First systems relied on meganucleases, zinc finger fusion proteins or Transcription activator-like effector nucleases (TALENs), requiring the generation of chimeric nucleases with engineered, sequence- specific DNA-binding domains specific for each particular target sequence. RNA-guided nucleases (RGNs), such as the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-associated (Cas) proteins allow for the targeting of specific sequences by using a short RNA sequence that specifically hybridizes with a particular target sequence. Such CRISPR systems became popular and gained multiple uses in research, diagnostics and therapeutics due to the ease of production of target-specific short RNA sequences and use of such with the same RGN protein. Such RGNs can be used to edit genomes through the introduction of a sequence-specific, double -stranded break that is either repaired and introduces a mutation or repaired by introducing a stretch of heterologous DNA. Inactive versions RGNs has been also widely used to target specific DNA or RNA regions and in combination with other proteins allowed to study and modulate multiple cellular processes and provide a useful tool for gene function study and modulation of their activity.
SUMMARY OF THE INVENTION
[003] The present invention provides novel type V RNA-guided nuclease (RGN) polypeptides, CRISPR RNAs (crRNAs), trans-activating CRISPR RNAs (tracrRNAs), gRNAs (gRNAs), nucleic acid targeting systems comprising those, nucleic acid molecules encoding the same, and vectors and host cells comprising such nucleic acid molecules.
[004] Also provided are nucleic acid targeting systems for binding a target nucleic acid sequence of interest, wherein the system comprises a RGN polypeptide and one or more RNA sequences targeting the nucleic acid of interest.
[005] Thus, methods disclosed herein are drawn to binding a target sequence of interest, and in some embodiments, cleaving or modifying the target sequence of interest. The target sequence of interest can be modified, for example, as a result of non-homologous end joining or homology-directed repair with an introduced donor sequence.
BRIEF DESCRIPTION OF THE DRAWINGS
[006] The present invention is described below by reference to the following figures.
[007] Figure 1. Domain structure of EGS0023 and EGS0024 RGNs.
[008] Figure 2. Small RNAseq data showing boundaries of tracrRNA and crRNA expression for EGS0023 [009] Figure 3. Small RNAseq data showing boundaries of tracrRNA and crRNA expression for
EGS0024
[010] Figure 4 shows the relative activity of EGS0023 with two potential tracrRNAs identified in Figure 2.
[Oil] Figure 5 shows an improvement in editing achieved with EGS0024 when using a modified tracrRNA (sgRNA v2).
[012] Figure 6 shows the structures of 2 version of sgRNA, one (sgRNA v2) has been truncated to remove extraneous sequence from the 5 ’ end of the tracrRNA and a shorter extended potential hairpin compared to the wild type tracrRNA (sgRNA vl).
DETAILED DESCRIPTION OF THE INVENTION Definitions
[013] Table 1. Abbreviations used throughout the specification

[014] Table 2. Amino acids abbreviations

[015] Table 3. Nucleotide Code abbreviations

[016] The following definitions are used throughout the description.
[017] The term "adeno-associated virus" or "AAV" as used interchangeably herein refers to a small virus belonging to the genus Dependovirus of the Parvoviridae family that infects humans and some other primate species. AAV is not currently known to cause disease and consequently the virus causes a very mild immune response.
[018] As used herein, a "biological sample" may contain whole cells and/or live cells and/or cell debris. The biological sample may contain (or be derived from) a "bodily fluid". Bodily fluids may be obtained from a mammal organism, for example by puncture, or other collecting or sampling procedures.
[019] The term "Cas9" refers to type of an RGN that cleaves nucleic acid and is encoded by the CRISPR loci and is a part of the Type II CRISPR system. The Cas9 protein commonly used is from bacterial species Streptococcus pyogenes. The Cas9 protein may be mutated so that the nuclease activity is partly or completely inactivated. [020] The term "Casl2" refers to type of an RGN that cleaves nucleic acid and is encoded by the
CRISPR loci and is a part of the Type V CRISPR system. The Casl2 protein family contains many
subtypes. The Casl2 protein may be mutated so that the nuclease activity is partly or completely inactivated.
[021] The term "complement" or "complementary" as used herein means a nucleic acid can mean Watson-Crick or Hoogsteen base pairing between nucleotides or nucleotide analogs of nucleic acid molecules. The term "complementarity" refers to a property shared between two nucleic acid sequences, such that when they are aligned antiparallel to each other, the nucleotide bases at each position will be complementary.
[022] The term "CRISPR" (Clustered Regularly Interspaced Short Palindromic Repeats) refers to a family of DNA sequences found in the genomes of prokaryotic organisms such as bacteria and archaea. These sequences are derived from DNA fragments of bacteriophages that had previously infected the prokaryote. They are used to detect and destroy DNA from similar bacteriophages during subsequent infections.
[023] The term "CRISPR system" refers collectively to transcripts and other elements involved in the expression of or directing the activity of CRISPR-associated ("Cas") proteins, including sequences encoding a Cas protein, a tracr (trans -activating CRISPR) sequence (e.g. tracrRNA or an active partial tracrRNA), a tracr-mate sequence (containing a "direct repeat" and a tracrRNA-processed partial direct repeat in the context of an endogenous CRISPR system), a guide sequence (also referred herein to as a "spacer" in the context of an endogenous CRISPR system), or other sequences and transcripts from a CRISPR locus.
[024] The term "effective amount," as used herein, refers to an amount of a biologically active agent that is sufficient to elicit a desired biological response. For example, in some embodiments, an effective amount of a nuclease may refer to the amount of the nuclease that is sufficient to induce cleavage of a target site specifically bound and cleaved by the nuclease. In some embodiments, an effective amount of a recombinase may refer to the amount of the recombinase that is sufficient to induce recombination at a target site specifically bound and recombined by the recombinase. As will be appreciated by the skilled artisan, the effective amount of an agent, e.g., a nuclease, a recombinase, a hybrid protein, a fusion protein, a protein dimer, a complex of a protein (or protein dimer) and a polynucleotide, or a polynucleotide, may vary depending on various factors as, for example, on the desired biological response, the specific allele, genome, target site, cell, or tissue being targeted, and the agent being used.
[025] The term "enhancer" as used herein refers to non-coding DNA sequences containing multiple activator and repressor binding sites. Enhancers range from 200 bp to 1 kb in length and may be either proximal, 5' upstream to the promoter or within the first intron of the regulated gene, or distal, in introns of neighboring genes or intergenic regions far away from the locus. Through DNA looping, active enhancers contact the promoter dependently of the core DNA binding motif promoter specificity. 4 to 5 enhancers may interact with a promoter.
[026] As used herein, the term "fusion protein" refers to a chimeric protein created through the covalent or non-co valent joining of two or more genes, directly or indirectly, that originally coded for separate proteins. In some embodiments, the translation of the fusion gene results in a single polypeptide with functional properties derived from each of the original proteins.
[027] The term "gRNA", also used interchangeably herein as a chimeric single guide RNA ("sgRNA"), refers to nucleic acid which is a fusion of two noncoding RNAs: a crRNA and a tracrRNA. "gRNA" is used interchangeably to refer to guide RNAs that exist as either single molecules or as a complex of two or more molecules. Typically, gRNAs that exist as single RNA species comprise two domains: (1) a domain that shares homology to a target nucleic acid (e.g., and directs binding of a Cas complex to the target); and (2) a domain that binds a Cas effector protein. [028] An "isolated" or "purified" polypeptide, or biologically active portion thereof, is substantially or essentially free from components that normally accompany or interact with the polypeptide as found in its naturally occurring environment. Thus, an isolated or purified polypeptide is substantially free of other cellular material, or culture medium when produced by recombinant techniques, or substantially free of chemical precursors or other chemicals when chemically synthesized. A protein that is substantially free of cellular material includes preparations of protein having less than 30%, 20%, 10%, 5%, or 1% (by dry weight) of contaminating protein. When a protein or biologically active portion thereof is recombinantly produced, optimally culture medium represents less than 30%, 20%, 10%, 5%, or 1% (by dry weight) of chemical precursors or non-protein-of-interest chemicals.
[029] The term "linker," as used herein, refers to a chemical group or a molecule linking two molecules or moieties, e.g., a binding domain and a cleavage domain of a nuclease. Typically, the linker is positioned between, or flanked by, two groups, molecules, or other moieties and connected to each one via a covalent bond, thus connecting the two. In some embodiments, the linker is an amino acid or a plurality of amino acids (e.g., a peptide or protein). In some embodiments, the linker is an organic molecule, group, polymer, or chemical moiety. In some embodiments, the linker is a polypeptide of 5-100 amino acids in length, for example, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 30-35, 35-40, 40-45, 45-50, 50-60, 60-70, 70-80, 80- 90, 90-100, 100-150, or 150-200 amino acids in length. Longer or shorter linkers are also contemplated.
[030] The term "modification" in reference to a nucleic acid molecule refers to a change in the nucleotide sequence of the nucleic acid molecule, which can be a deletion, insertion, or substitution of one or more nucleotides, or a combination thereof.
[031] The term "mutation," as used herein, refers to a substitution of a residue within a sequence, e.g., a nucleic acid or amino acid sequence, with another residue, or a deletion or insertion of one or more residues within a sequence. Mutations are typically described herein by identifying the original residue followed by the position of the residue within the sequence and by the identity of the newly substituted residue. Various methods for making the amino acid substitutions (mutations) provided
herein are well known in the art, and are provided by, for example, Green and Sambrook, Molecular Cloning: A Laboratory Manual (4th ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. (2012)).
[032] As used herein, the terms "nucleic acid," "nucleic acid sequence," "nucleotide sequence," "oligonucleotide," and "polynucleotide" are interchangeable and refer to a polymeric form of nucleotides. The nucleotides may be deoxyribonucleotides (DNA), ribonucleotides (RNA), analogs thereof, or combinations thereof, and may be of any length. Polynucleotides may perform any function and may have any secondary and tertiary structures. The terms encompass known analogs of natural nucleotides and nucleotides that are modified in the base, sugar and/or phosphate moieties. Analogs of a particular nucleotide have the same base-pairing specificity (e.g., an analog of A base pairs with T). A polynucleotide may comprise one modified nucleotide or multiple modified nucleotides. Examples of modified nucleotides include fluorinated nucleotides, methylated nucleotides, and nucleotide analogs. Nucleotide structure may be modified before or after a polymer is assembled. Following polymerization, polynucleotides may be additionally modified via, for example, conjugation with a labeling component or target binding component. A nucleotide sequence may incorporate non-nucleotide components. The terms also encompass nucleic acids comprising modified backbone residues or linkages, which are synthetic, naturally occurring, and non-naturally occurring, and have similar binding properties as a reference polynucleotide (e.g., DNA or RNA). Examples of such analogs include, but are not limited to, phosphorothioates, phosphoramidates, methyl phosphonates, chiral-methyl phosphonates, 2-O-methyl ribonucleotides, peptide-nucleic acids (PNAs), Locked Nucleic Acid (LNA™) (Exiqon, Inc., Woburn, MA) nucleosides, glycol nucleic acid, bridged nucleic acids, and morpholino structures. Polynucleotide sequences are displayed herein in the conventional 5' to 3' orientation unless otherwise indicated.
[033] The term "operably linked" as used herein means that expression of a gene is under the control of a promoter with which it is spatially connected. A promoter may be positioned 5‘ (upstream) or 3‘ (downstream) of a gene under its control. The distance between the promoter and a gene may be approximately the same as the distance between that promoter and the gene it controls in the gene from which the promoter is derived. As is known in the art, variation in this distance may be accommodated without loss of promoter function.
[034] The term "optional" or "optionally" means that the subsequent described event, circumstance or substituent may or may not occur, and that the description includes instances where the event or circumstance occurs and instances where it does not.
[035] As used herein, the terms "peptide," "polypeptide," and "protein" are interchangeable and refer to polymers of amino acids. A polypeptide may be of any length. It may be branched or linear, it may be interrupted by non-amino acids, and it may comprise modified amino acids. The terms may be used to refer to an amino acid polymer that has been modified through, for example, acetylation, disulfide bond formation, glycosylation, lipidation, phosphorylation, cross- linking, and/or
conjugation (e.g., with a labeling component or ligand). Polypeptide sequences are displayed herein in the conventional N-terminal to C-terminal orientation. Polypeptides and polynucleotides can be made using routine techniques in the field of molecular biology (see, e.g., standard texts set forth above). Further, essentially any polypeptide or polynucleotide can be custom ordered from commercial sources.
[036] As used herein, "percentage of sequence identity" means the value determined by comparing two optimally aligned sequences over a comparison window, wherein the portion of the polynucleotide sequence in the comparison window may comprise additions or deletions (i. e. , gaps) as compared to the reference sequence (which does not comprise additions or deletions) for optimal alignment of the two sequences. The percentage is calculated by determining the number of positions at which the identical nucleic acid base or amino acid residue occurs in both sequences to yield the number of matched positions, dividing the number of matched positions by the total number of positions in the window of comparison, and multiplying the result by 100 to yield the percentage of sequence identity.
[037] The term "promoter" as used herein means a synthetic or naturally-derived molecule which is capable of conferring, activating or enhancing expression of a nucleic acid in a cell. A promoter may comprise one or more specific transcriptional regulatory sequences to further enhance expression and/or to alter the spatial expression and/or temporal expression of same. A promoter may also comprise distal enhancer or repressor elements, which may be located as much as several thousand base pairs from the start site of transcription. A promoter may be derived from sources including viral, bacterial, fungal, plants, insects, and animals.
[038] The term "RNA-guided endonuclease" or "RGN" is used interchangeably herein and refer to a nuclease that forms a complex with (e.g., binds or associates with) one or more RNA that is not a target for cleavage.
[039] As used herein, "sequence identity" or "identity" in the context of two polynucleotides or polypeptide sequences makes reference to the residues in the two sequences that are the same when aligned for maximum correspondence over a specified comparison window. When percentage of sequence identity is used in reference to proteins it is recognized that residue positions which are not identical often differ by conservative amino acid substitutions, where amino acid residues are substituted for other amino acid residues with similar chemical properties (e.g., charge or hydrophobicity) and therefore do not change the functional properties of the molecule. When sequences differ in conservative substitutions, the percent sequence identity may be adjusted upwards to correct for the conservative nature of the substitution.
[040] Sequences that differ by such conservative substitutions are said to have "sequence similarity" or "similarity". Means for making this adjustment are well known to those of skill in the art. Typically this involves scoring a conservative substitution as a partial rather than a full mismatch, thereby increasing the percentage sequence identity. Thus, for example, where an identical amino acid is
given a score of 1 and a non-conservative substitution is given a score of zero, a conservative substitution is given a score between zero and 1. The scoring of conservative substitutions is calculated, e.g. , as implemented in the program PC/GENE (Intelligenetics, Mountain View, California).
[041] As used herein the term "spacer sequence" or "spacer" refers to a part of gRNA nucleotide sequence or a part of a CRISPR locus that directly hybridizes with the target nucleotide sequence of interest.
[042] The term "subject" and "patient" as used herein interchangeably refers to any vertebrate, including, but not limited to, a mammal {e.g., cow, pig, camel, llama, horse, goat, rabbit, sheep, hamsters, guinea pig, cat, dog, rat, and mouse, a non-human primate (for example, a monkey, such as a cynomolgus or rhesus monkey, chimpanzee, etc.) and a human). In some embodiments, the subject may be a human or a non-human. The subject or patient may be undergoing other forms of treatment. [043] The term "target region", "target sequence" or "protospacer" as used interchangeably herein refers to the region of the target gene to which the CRISPR-based system targets.
[044] The terms "treatment," "treat," and "treating," refer to a clinical intervention aimed to reverse, alleviate, delay the onset of, or inhibit the progress of a disease or disorder, or one or more symptoms thereof, as described herein. As used herein, the terms "treatment," "treat," and "treating" refer to a clinical intervention aimed to reverse, alleviate, delay the onset of, or inhibit the progress of a disease or disorder, or one or more symptoms thereof, as described herein. In some embodiments, treatment may be administered after one or more symptoms have developed and/or after a disease has been diagnosed. In other embodiments, treatment may be administered in the absence of symptoms, e.g., to prevent or delay onset of a symptom or inhibit onset or progression of a disease. For example, treatment may be administered to a susceptible individual prior to the onset of symptoms (e.g., in light of a history of symptoms and/or in light of genetic or other susceptibility factors). Treatment may also be continued after symptoms have resolved, for example, to prevent or delay their recurrence [045] The term "Type II CRISPR system" refers to effector system that carries out targeted DNA double-strand break in four sequential steps, using a single effector enzyme, Cas9, to cleave dsDNA. Compared to the Type I and Type III effector systems, which require multiple distinct effectors acting as a complex, the Type II effector system may function in alternative contexts such as eukaryotic cells. The Type II effector system consists of a long pre-crRNA, which is transcribed from the spacercontaining CRISPR locus, the Cas9 protein, and a tracrRNA, which is involved in pre-crRNA processing.
[046] The term "Type V CRISPR system" refers to effector system that carries out targeted DNA double-strand break in four sequential steps, using a single effector enzyme, Casl2, to cleave dsDNA. Compared to the Type I and Type III effector systems, which require multiple distinct effectors acting as a complex, the Type V effector system may function in alternative contexts such as eukaryotic cells. The Type V effector system consists of a long pre-crRNA, which is transcribed from the spacer-
containing CRISPR locus, the Cast 2 protein, and, in some cases, a tracrRNA, which is involved in pre-crRNA processing.
[047] The term "vector" as used herein means a nucleic acid sequence containing an origin of replication. A vector may be a viral vector, bacteriophage, bacterial artificial chromosome or yeast artificial chromosome. A vector may be a DNA or RNA vector. A vector may be a self- replicating extrachromosomal vector, or a DNA plasmid.
[048] Unless otherwise defined herein, scientific and technical terms used in connection with the present disclosure shall have the meanings that are commonly understood by those of ordinary skill in the art.
CRISPR systems
[049] The CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) genomic locus is found in the genomes of many prokaryotes. CRISPR loci provide resistance to viruses and phages in prokaryotes. In this way, the CRISPR loci functions as a type of immune system to help defend prokaryotes against foreign invaders. In such system the response to such foreign invaders starts by cleaving the genome of invading viruses and plasmids and integrating segments (termed protospacers) of the genomic DNA into the CRISPR locus of the host organism. The segments that are integrated into the host genome are known as “spacers”, which mediate protection from subsequent attack by the same (or sufficiently related) virus or plasmid. Expression involves transcription of the CRISPR locus and subsequent enzymatic processing to produce short mature CRISPR RNAs (crRNA), each containing a single spacer sequence. Interference is induced after the CRISPR RNAs associate with Cas proteins to form effector complexes, which are then targeted to complementary protospacers in foreign genetic elements to induce nucleic acid degradation.
[050] Currently, two classes of CRISPR systems have been described, Class 1 and Class 2, based upon the genes encoding the effector component. Class 1 systems have a multi-subunit crRNA- effector complex, whereas Class 2 systems have a single effector protein. Typical examples of Class 2 effector proteins are Cas9 and Cpfl (Cas 12a).
[051] To date six types (Types I-VI) of CRISPR systems have been described (for an overview see Makarova et al., Nature Reviews Microbiology (2015) 13: 1-15). Class 1 systems comprise Type I, Type III and Type IV systems. Class 2 systems comprise Type II, Type V and Type VI systems.
[052] CRISPR loci include several short repeating sequences referred to as "repeats." The repeats can form hairpin structures and/or the repeats can be single -stranded sequences. The repeats occur in clusters. Repeats frequently diverge between species. Repeats are regularly interspaced with unique intervening sequences, referred to as "spacers," resulting in a repeat-spacer-repeat locus architecture. Spacers are sequences usually identical to or homologous to foreign invader sequences (such as viral sequences).
[053] In some cases, a spacer-repeat unit encodes a crisprRNA (crRNA). A crRNA refers to the mature form of the spacer-repeat unit. A crRNA contains a spacer sequence that is involved in targeting a target nucleic. crRNA has a region of complementarity to a potential DNA or RNA target sequence and in some cases, e.g., in currently characterized Type II systems, and some subtypes of Type V systems, a second region that forms base-pair hydrogen bonds with a transactivating CRISPR RNA (tracrRNA) to form a secondary structure, typically to form at least a stem structure. Complex formation between tracrRNA/crRNA and a Cas protein results in conformational change of the Cas protein that facilitates binding to DNA, nuclease activities of the Cas protein, and crRNA- guided sitespecific DNA cleavage by the nuclease. For a Cas protein/tracrRNA/crRNA complex to cleave a DNA target sequence, the DNA target sequence is adjacent to a cognate protospacer adjacent motif (PAM).
[054] Usually, CRISPR locus comprises polynucleotide sequences encoding for CRISPR Associated Genes (cas) genes. Cas genes are involved in the biogenesis and/or the interference stages of crRNA function. Cas genes display extreme sequence diversity between different species and homologs. Some Cas proteins comprise a specific set of domain structures.
[055] Mature crRNAs are processed from a longer polycistronic CRISPR locus transcript, also referred to as pre-crRNA array. A pre-crRNA array comprises a plurality of crRNAs. The repeats in the pre-crRNA array are recognized by cas genes. Cas genes bind to the repeats and cleave the repeats. This action can liberate the plurality of crRNAs. crRNAs can be subjected to further events to produce the mature crRNA form such as trimming (e.g., with an exonuclease). A crRNA may comprise all, or some, of the CRISPR repeat sequences.
[056] Interference refers to the stage in the CRISPR system that is functionally responsible for combating infection by a foreign invader. CRISPR interference follows a similar mechanism to RNA interference, which results in target RNA degradation and/or destabilization. Currently characterized CRISPR systems perform interference of a target nucleic acid by coupling crRNAs and Cas genes, thereby forming CRISPR ribonucleoproteins (RNPs). crRNA of the RNP guides the RNP to foreign invader nucleic acid, (e.g. , by recognizing the foreign invader nucleic acid through hybridization). Hybridized target foreign invader nucleic acid- crRNA units are subjected to cleavage by Cas proteins. Target nucleic acid interference typically requires a protospacer adjacent motif (PAM) in a target nucleic acid.
[057] Currently CRISPR-Cas systems are divided into two main classes based on their effector molecules: class 1 and class 2. Class 1 is characterized by multi-unit effector molecules, while class 2 contains a single effector molecule. Class 1 systems comprise Type I, Type III, and Type IV systems. Class 2 systems comprise Type II, Type V, and Type VI systems.
[058] Type II system is commonly represented by cas9 genes. There are two strands of RNA in Type II systems: a crRNA and a tracrRNA. The duplex formed by the tracrRNA and crRNA is recognized
by, and associates with Cas9, encoded by the cas9 gene, which combines the functions of the crRNA- effector complex with target DNA cleavage. Cas9 is directed to a target nucleic acid by a sequence of the crRNA that is complementary to, and hybridizes with, a sequence in the target nucleic acid.
[059] In Type V systems, nucleic acid target sequence binding involves a Casl2 protein and the crRNA, as does the nucleic acid target sequence cleavage. In Type V systems, the RuvC-like nuclease domain of Cast 2 protein cleaves both strands of the nucleic acid target sequence in a sequential fashion (Swarts, et al. , Mol. Cell (2017) 66:221 -233. e4), producing 5' overhangs, which differs from the fragments generated by Cas9 protein. There have been multiple subtypes of Type V systems identified so far (type V-A/B/C7D/E/F/G/H/I/K/L and CRISPR-Casl2j). All of them differ by the length of Cas protein, PAM sequence and whether they require tracrRNA for its functionality.
[060] Type V-B is represented by Cas 12b protein. The Cas 12b protein cleavage activity of Type V-B systems requires hybridization of crRNA to tracrRNA to form a duplex and Cas 12a protein binds the crRNA/tracrRNA duplex in a sequence- and structure -specific manner by recognizing the stem loop and sequences adjacent to the stem loop, most notably the nucleotides 5' of the spacer sequence, which hybridizes to the nucleic acid target sequence. Substitutions that disrupt this stem-loop duplex abolish cleavage activity, whereas other substitutions that do not disrupt the stem-loop duplex do not abolish cleavage activity.
[061] In Type V-B systems, nucleic acid target sequence binding involves Cas 12b and the crRNA/tracrRNA, as does the nucleic acid target sequence cleavage. In Type V systems, the RuvC- like nuclease domain of Cas 12b cleaves one strand of the double-stranded nucleic acid target sequence, and a putative nuclease domain cleaves the other strand of the double- stranded nucleic acid target sequence in a staggered configuration, producing 5' overhangs, which is different from the blunt ends generated by Cas9 cleavage. These 5' overhangs may facilitate insertion of DNA.
[062] Other proteins associated with Type V crRNA and nucleic acid target sequence binding and cleavage include Cas 12a, Cas 12c, Cas 12d, and Casl2e which are similar in length to Cas 12b proteins, ranging from approximately 1000-1500 amino acids, and also require an additional RNA (either a tracRNA or a scoutRNA) besides Cas 12a. Still other proteins associated with Type V crRNA and nucleic acid target sequence binding and cleavage include Casl2fl, Casl2f2, Casl2f3, and Cas 12g, which are smaller in length to Casl2a proteins, ranging from approximately 300-900 amino acids, but also require a tracrRNA.
[063] Type VI systems include the Cas 13a protein (also known as Class 2 candidate 2 protein, or C2c2) which does not share sequence similarity with other CRISPR effector proteins (see Abudayyeh, et al, Science (2016) 353:aaf5573). Casl3a proteins have two HEPN domains and possess singlestranded RNA cleavage activity. Cas 13a proteins are similar to Cas 12a proteins in requiring a crRNA for nucleic acid target sequence binding and cleavage, but not requiring tracrRNA.
[064] While many of type II systems have been identified, the discovery and characterization of CRISPR systems is ongoing.
RNA-guided nucleases (RGNs)
[065] The present disclosure provides novel RNA-guided nucleases (RGN) as defined by their amino acid sequences in Table 4. These systems exhibit a diversity of PAM sequences, enabling greater, and more flexible targeting space. Additionally, as the systems are derived from a variety of sources, they will be better suited for in vivo editing where pre-existing antibodies to other Cas proteins (either naturally occurring or from previous treatment) prevent effective use, and some are small enough to be effectively packaged into a single AAV vector for efficient delivery. Furthermore, Cas 12b proteins exhibit a greater specificity compared to Cas9 increasing their safety profile as a therapeutic agent (Strecker et al. Nat Commun 10, 212 (2019)).
[066] Table 4. RGN proteins of the present disclosure

[067] An RNA-guided nuclease provided herein binds to a target nucleotide sequence and hybridizes with the gRNA molecule specific to the RNA-guided nuclease. The target sequence can then be subsequently cleaved by the RGN if the RGN polypeptide possesses nuclease activity. The presently disclosed RGNs can cleave nucleotides within a polynucleotide, functioning as an endonuclease. In some embodiments, the disclosed RGNs can cleave nucleotides of a target nucleotide sequence within any position of a polynucleotide and thus function as both an endonuclease and exonuclease.
[068] The presently disclosed RGNs can be wild-type sequences derived from bacterial or archaeal species. Alternatively, the RGNs can be variants or fragments of wild-type polypeptides. The wildtype RGN can be modified to alter nuclease activity or alter PAM specificity, for example. In some embodiments, the RGN is not naturally-occurring. In certain embodiments, the RGN functions as a nickase, only cleaving a single strand of the target nucleotide sequence.
[069] In other embodiments, the RGNs lacks nuclease activity altogether or exhibits reduced nuclease activity and is referred to herein as nuclease-dead RGNs. Any method known in the art for introducing mutations into an amino acid sequence, such as PCR-mediated mutagenesis and site- directed mutagenesis, can be used for generating nickases or nuclease-dead RGNs. (e.g.
US2014/0068797 and US9,790,490).
[070] Alternatively, nuclease dead RGNs can be targeted to particular genomic locations to alter the expression of a desired sequence. In some embodiments, the binding of a nuclease-dead RNA-guided nuclease to a target sequence results in the repression of expression of the target sequence or a gene under transcriptional control by the target sequence by interfering with the binding of RNA polymerase or transcription factors within the targeted genomic region. In other embodiments, the RGN (e.g. , a nuclease- dead RGN) or its complexed gRNA further comprises an expression modulator that, upon binding to a target sequence, serves to either repress or activate the expression of the target sequence or a gene under transcriptional control by the target sequence. In some of these embodiments, the expression modulator modulates the expression of the target sequence or regulated gene through epigenetic mechanisms.
[071] In other embodiments, the nuclease-dead RGNs or a RGN with only nickase activity can be targeted to particular genomic locations to modify the sequence of a target polynucleotide through fusion to a base editing polypeptide, for example a deaminase polypeptide or active variant or fragment thereof that deaminates a nucleotide base, resulting in conversion from one nucleotide base to another. The base-editing polypeptide can be fused to the RGN at its N-terminal or C-terminal end. Additionally, the base-editing polypeptide may be fused to the RGN via a peptide linker. A nonlimiting example of a deaminase polypeptide that is useful for such compositions and methods include cytidine deaminase or the adenosine deaminase base editor described in Gaudelli et al. (2017) Nature 551:464-471, and WO2018/027078.
Structural elements of the RGN peptides
[072] The RGN peptides of the present disclosure comprise multiple domains distributed in a recognition lobe (REC) and a nuclease lobe (NUC) for substrate recognition and cleavage. In some embodiments the REC lobe of RGN peptides provided herein consists of a RECI, and REC2 domains, where as the NUC lob consists of a WED-I, WED-II, Bridge helix domain(BH), a split Nuc domain, and a Tri-split RuvC domain. While having a similar domain organization to AccCasl2b, the RGN proteins of the present disclosure share little homology to AccCasl2b protein (in some cases less than 27%).
RuvC domain
[073] The RuvC domain may comprise multiple subdomains: RuvC-I, RuvC-II and RuvC -III. The subdomains may be separated by other sequences on the amino acid sequence of the protein.
[074] Examples of RuvC domains include any polypeptides having a structural similarity and/or sequence similarity to a RuvC domain described in the art. For example, the RuvC domain may share a structural similarity and/or sequence similarity to a RuvC of Cas9. In some examples, the RuvC domain may have an amino acid sequence that share at least 80%, at least 85%, at least 90%, at least 95%, at least 99%, or 100% sequence identity with RuvC domains.
[075] In some examples, the RuvC domain comprise RuvC-I polypeptide, RuvC-II polypeptide, and RuvC-III polypeptide. Examples of the RuvC-I domain also include any polypeptides having a structural similarity and/or sequence similarity to a RuvC -I, II, and III domains described in the art, such as the corresponding domains of Cas9. The RuvC domain may have an amino acid sequence that has at least 80%, at least 85%, at least 90%, at least 95%, at least 99%, or 100% sequence identity with a RuvC domain of Cas9.
[076] the RuvC domain of Cas9 consists of a six-stranded mixed P-sheet flanked by a-helices and two additional two-stranded antiparallel P-sheets (see e.g., Nishimasu et al. Cell, 2014). The RuvC domain of Cas9 shares structural similarity with the retroviral integrase superfamily members characterized by an RNase H fold, such as Escherichia coli RuvC (PDB code 1HJR, 14% identity, root-mean-square deviation (rmsd) of 3.6 A for 126 equivalent Ca atoms) and Thermus thermophilus RuvC (PDB code 4LD0, 12% identity, rmsd of 3.4 A for 131 equivalent Ca atoms). E. coli RuvC is a 3-layer alpha-beta sandwich containing a 5-stranded beta-sheet sandwiched between 5 alpha-helices. RuvC nucleases have four catalytic residues (e.g., Asp7, Glu70, Hisl43 and Aspl46 in T. thermophilus RuvC), and cleave Holliday junctions (or structurally analogous cruciform junctions) through a two-metal mechanism. Asp 10 (Ala), Glu762, His983 and Asp986 of the Cas9 RuvC domain are located at positions similar to those of the catalytic residues of T. thermophilus RuvC.
Bridge helix
[077] The RGN of the present disclosure comprises a bridge helix (BH) domain. The bridge helix domain refers to a helix and arginine rich polypeptide. The bridge helix domain may be located next to anyone of the amino acid domains in the nucleic -acid guided nuclease. In one embodiment, the bridge helix domain is next to a RuvC domain, e.g., next to RuvC-I, RuvC-II, or RuvC-III subdomain. In one example, the bridge helix domain is between a RuvC-I and RuvC-II subdomains.
[078] The bridge helix domain may be from 10 to 100, from 20 to 60, from 30 to 50, e.g., 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46 or 47, 48, 49, or 50 amino acids in length. Examples of bridge helix includes the polypeptide of amino acids 60-93 of the sequence of .S', pyogenes Cas9.
WED Domain
[079] The RGN of the present disclosure comprises a WED domain assembled from two separate regions (WED-I and WED-II). The WED domain may have an amino acid sequence that share at least 80%, at least 85%, at least 90%, at least 95%, at least 99%, or 100% sequence identity with a WED domain of AccCasl2b. The AacCasl2b WED domain is assembled from two separate regions (WED- I and WED-II) and contains a core subdomain, which adopts an oligonucleotide -binding (OBD) fold. The core subdomain consists of eight [3 stranded distorted [3 sheets. The core subdomain is flanked by two a helices and one [3-hairpin
Modified RGN peptides
[080] The RGNs may comprise one or more modifications. The modified RGNs may be catalytically inactive (also referred as dead). A catalytically inactive or dead nuclease may have reduced or no nuclease activity compared to a wildtype counterpart nuclease. In some cases, a catalytically inactive or dead nuclease may have nickase activity. In some cases, a catalytically inactive or dead nuclease may not have nickase activity. Such a catalytically inactive or dead RGN may not make either doublestrand or single-strand break on a target polynucleotide but may still bind or otherwise form complex with the target polynucleotide.
[081] In one embodiment, the modifications of the RGN polypeptide may or may not cause an altered functionality. Some modifications will not result in an altered functionality include for instance codon optimization for expression into a particular host, or providing the nuclease with a particular marker. Modifications which may result in altered functionality may also include mutations, including point mutations, insertions, deletions, truncations (including split nucleases), etc., as well as chimeric RGNs (e.g., comprising domains from different orthologues or homologues) or fusion proteins.
[082] Fusion proteins may include, for example, fusions with heterologous domains or functional domains (e.g., localization signals, enzymes). In an embodiment, various different modifications may be combined (e.g., a mutated nuclease which is catalytically inactive and which further is fused to a functional domain, such as for instance to induce DNA methylation or another nucleic acid modification, such as, for example, a mutation, a deletion, an insertion, a replacement.
Localization signal sequences
[083] The RGNs can comprise at least one nuclear localization signal (NLS) to enhance transport of the RGN to the nucleus of a cell. Nuclear localization signals are known in the art and generally comprise a stretch of basic amino acids (see, e.g., Lange et al., J. Biol. Chem. (2007) 282:5101-5105). In embodiments, the RGN comprises 2, 3, or more nuclear localization signals. The nuclear localization signal(s) can be a heterologous NLS. Non-limiting examples of nuclear localization signals useful for the presently disclosed RGNs are the nuclear localization signals of SV40 Large T- antigen, nucleopasmin, and c-Myc (see. e.g., Ray et al. (2015) Bioconjug Chem 26(6): 1004-7). In particular embodiments, the RGN comprises the NLS sequence comprising the sequence of SEQ ID NO: 27 or 28. The RGN may comprise one or more NLS sequences at its N-terminus, C- terminus, or both the N-terminus and C- terminus. For example, the RGN may comprise two NLS sequences at the N-terminal region and four NLS sequences at the C-terminal region.
[084] Other localization signal sequences known in the art that localize polypeptides to particular subcellular location(s) can also be used to target the RGNs, including, but not limited to, plastid localization sequences, mitochondrial localization sequences, and dual-targeting signal sequences that target to both the plastid and mitochondria (see, e.g., Nassoury and Morse (2005) Biochim Biophys
Acta 1743:5-19; Herrmann and Neupert (2003) IUBMB Life 55:219-225; Soil (2002) Curr Opin Plant Biol 5:529-535; Carrie and Small (2013) Biochim Biophys Acta 1833:253-259).
[085] In certain embodiments, the RGNs comprise at least one cell- penetrating domain that facilitates cellular uptake of the RGN. Cell-penetrating domains are known in the art and generally comprise stretches of positively charged amino acid residues (i.e., polycationic cell- penetrating domains), alternating polar amino acid residues and non-polar amino acid residues (i.e. amphipathic cellpenetrating domains), or hydrophobic amino acid residues (i.e.. hydrophobic cell- penetrating domains) (see, e.g., Milletti F. (2012) Drug Discov Today 17:850-860). A non-limiting example of a cell-penetrating domain is the trans-activating transcriptional activator (TAT) from the human immunodeficiency virus 1.
[086] The nuclear localization signal, plastid localization signal, mitochondrial localization signal, dual targeting localization signal, and/or cell-penetrating domain can be located at the amino-terminus (N- terminus), the carboxyl-terminus (C-terminus), or in an internal location of the RNA-guided nuclease.
Additional tags and labels
[087] The presently disclosed RGN polypeptides can comprise a detectable label or a purification tag. The detectable label or purification tag can be located at the N-terminus, the C-terminus, or an internal location of the RNA-guided nuclease, either directly or indirectly via a linker peptide. In some of these embodiments, the RGN component of the fusion protein is a nuclease-dead RGN. In other embodiments, the RGN component of the fusion protein is a RGN with nickase activity.
[088] RGNs that lack nuclease activity can be used to deliver a fused polypeptide, polynucleotide, or small molecule payload to a particular genomic location. In some of these embodiments, the RGN polypeptide or gRNA can be fused to a detectable label to allow for detection of a particular sequence. As a non-limiting example, a nuclease-dead RGN can be fused to a detectable label (e.g., fluorescent protein) and targeted to a particular sequence associated with a disease to allow for detection of the disease-associated sequence.
[089] A detectable label is a molecule that can be visualized or otherwise observed. The detectable label may be fused to the RGN as a fusion protein (e.g., fluorescent protein) or may be a small molecule conjugated to the RGN polypeptide that can be detected visually or by other means. Detectable labels that can be fused to the presently disclosed RGNs as a fusion protein include any detectable protein domain, including but not limited to, a fluorescent protein or a protein domain that can be detected with a specific antibody. Non-limiting examples of fluorescent proteins include green fluorescent proteins (e.g., GFP, EGFP, ZsGreen) and yellow fluorescent proteins (e.g., YFP, EYFP, ZsYellow). Non-limiting examples of small molecule detectable labels include radioactive labels, such as 3H and 35 S.
[090] RGN polypeptides can also comprise a purification tag, which is any molecule that can be utilized to isolate a protein or fused protein from a mixture (e.g., biological sample, culture medium). Non-limiting examples of purification tags include biotin, myc, maltose binding protein (MBP), and glutathione -S- transferase (GST).
Fusion proteins comprising the RGNs
[091] The presently disclosed RGNs can be fused to an effector domain (a fusion protein of an RGN and an effector domain), such as a cleavage domain, a deaminase domain, or an expression modulator domain, either directly or indirectly via a linker. Such effector domain can be located at the N- terminus, the C-terminus, or an internal location of the RNA-guided nuclease. In some embodiments, the RGN component of the fusion protein is a nuclease-dead RGN.
[092] RGNs that are fused to a polypeptide or domain can be separated or joined by a linker. In some embodiments, a linker joins a gRNA binding domain of an RNA guided nuclease and a base-editing polypeptide, such as a deaminase.
[093] In some embodiments, the RGN fusion protein comprises a cleavage domain, which is any domain that is capable of cleaving a polynucleotide (i.e.. RNA, DNA) and includes, but is not limited to, restriction endonucleases and homing endonucleases (see, e.g Linn et al. (eds.) Nucleases, Cold Spring Harbor Laboratory Press, 1993).
[094] In some embodiments, the RGN fusion protein comprises a deaminase domain that deaminates a nucleotide base, resulting in conversion from one nucleotide base to another, and includes, but is not limited to, a cytidine deaminase or an adenosine deaminase base editor.
[095] In some embodiments, the effector domain of the fusion protein can be an expression modulator domain, which is a domain that either serves to upregulate or downregulate transcription. The expression modulator domain can be an epigenetic modification domain, a transcriptional repressor domain or a transcriptional activation domain.
[096] In some of these embodiments, the expression modulator of the RGN fusion protein comprises an epigenetic modification domain that covalently modifies DNA or histone proteins to alter histone structure and/or chromosomal structure without altering the DNA sequence, leading to changes in gene expression (i. e. , upregulation or downregulation). Non-limiting examples of epigenetic modifications include acetylation or methylation of lysine residues, arginine methylation, serine and threonine phosphorylation, and lysine ubiquitination and sumoylation of histone proteins, and methylation and hydroxymethylation of cytosine residues in DNA. Non-limiting examples of epigenetic modification domains include histone acetyltransferase domains, histone deacetylase domains, histone methyltransferase domains, histone demethylase domains, DNA methyltransferase domains, and DNA demethylase domains.
[097] In other embodiments, the expression modulator of the fusion protein comprises a transcriptional repressor domain, which interacts with transcriptional control elements and/or transcriptional regulatory proteins, such as RNA polymerases and transcription factors, to reduce or terminate transcription of at least one gene. Transcriptional repressor domains are known in the art and include, but are not limited to IKB, and Kruppel associated box (KRAB) domains.
[098] In yet other embodiments, the expression modulator of the fusion protein comprises a transcriptional activation domain, which interacts with transcriptional control elements and/or transcriptional regulatory proteins, such as RNA polymerases and transcription factors, to increase or activate transcription of at least one gene. Transcriptional activation domains are known in the art and include, but are not limited to, a VP 16 activation domain and an NF AT activation domain.
[099] It is also envisaged that the nucleic acid-targeting effector protein-gRNA complex as a whole may be associated with two or more functional domains. For example, there may be two or more functional domains associated with the nucleic acid-targeting effector protein, or there may be two or more functional domains associated with the gRNA (via one or more adaptor proteins), or there may be one or more functional domains associated with the nucleic acid-targeting effector protein and one or more functional domains associated with the gRNA (via one or more adaptor proteins).
[100] The fusion between the adaptor protein and the activator or repressor may include a linker. For example, GlySer linkers GGGS can be used. They can be used in repeats of 3 or 6, 9 or even 12 or more, to provide suitable lengths, as required. Linkers can be used between the gRNAs and the functional domain (activator or repressor), or between the nucleic acid-targeting effector protein and the functional domain (activator or repressor). guideRNAs (gRNAs/sgRNA), tracrRNA, and crRNA
[101] The present disclosure provides RGNs that can bind to gRNAs. The term “gRNA” refers to a nucleotide sequence having sufficient complementarity with a target nucleotide sequence to hybridize with the target sequence and direct sequence -specific binding of an associated RNA- guided nuclease to the target nucleotide sequence. Thus, a RGN’s respective gRNA is one or more RNA molecules (generally, one or two), that can bind to the RGN and guide the RGN to bind to a particular target nucleotide sequence, and in those instances wherein the RGN has nickase or nuclease activity, also cleave the target nucleotide sequence.
[102] In general, a gRNA comprises a CRISPR RNA (crRNA) and a trans-activating CRISPR RNA (tracrRNA). Native gRNAs that comprise both a crRNA and a tracrRNA generally comprise two separate RNA molecules that hybridize to each other through the repeat sequence of the crRNA and the anti-repeat sequence of the tracrRNA. Native direct repeat sequences within a CRISPR array generally range in length from 28 to 37 base pairs, although the length can vary between 23 bp to 55 bp. Spacer sequences within a CRISPR array generally range from 28 to 34 bp in length, although the
length can be between 21 bp to 72 bp. Specifically, the spacer sequences of the present disclosure are normally between 35 and 46 nucleotides. Each CRISPR array generally comprises less than 60 units of the CRISPR repeat-spacer sequence. The CRISPRs are transcribed as part of a long transcript termed the primary CRISPR transcript, which comprises much of the CRISPR array. The primary CRISPR transcript is cleaved by Cas proteins to produce crRNAs or in some cases, to produce pre- crRNAs that are further processed by additional Cas proteins into mature crRNAs. Mature crRNAs comprise a spacer sequence and a CRISPR repeat sequence. In some embodiments in which pre- crRNAs are processed into mature (or processed) crRNAs, maturation involves the removal of one to six or more 5', 3', or 5' and 3' nucleotides. For the purposes of genome editing or targeting a particular target nucleotide sequence of interest, these nucleotides that are removed during maturation of the pre-crRNA molecule are not necessary for generating or designing a gRNA.
[103] More specifically, the length of the target DNA within the sequence of the gRNA with the complementary sequence is 17 to 23bp, 18 to 23bp, 19 to 23bp, more specifically 20 to 23bp, as more specifically, it may be a 21 to 23bp, but is not limited thereto.
[104] A CRISPR RNA (crRNA) comprises a spacer sequence and a CRISPR repeat sequence. The “spacer sequence” is the nucleotide sequence that directly hybridizes with the target nucleotide sequence of interest. The spacer sequence is engineered to be fully or partially complementary with the target sequence of interest. In various embodiments, the spacer sequence can comprise from 8 nucleotides to 30 nucleotides, or more. For example, the spacer sequence can be 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, or more nucleotides in length. In some embodiments, the spacer sequence is 10 to 26 nucleotides in length, or 12 to 30 nucleotides in length. In particular embodiments, the spacer sequence is 30 nucleotides in length.
[105] A trans-activating CRISPR RNA (tracrRNA) molecule comprises a nucleotide sequence comprising a region that has sufficient complementarity to hybridize to a CRISPR repeat sequence of a crRNA, which is referred to herein as the anti-repeat region. In some embodiments, the tracrRNA molecule further comprises a region with secondary structure (e.g., stem-loop) or forms secondary structure upon hybridizing with its corresponding crRNA. In particular embodiments, the region of the tracrRNA that is fully or partially complementary to a CRISPR repeat sequence is at the 3' end of the molecule and the 5' end of the tracrRNA comprises secondary structure. This region of secondary structure generally comprises several hairpin structures, including the nexus hairpin, which is found adjacent to the anti -repeat sequence. There are often hairpins at the 5' end of the tracrRNA that can vary in structure and number.
[106] Table 5. RGNs and their corresponding consensus repeat sequence, tracrRNA, crRNA, sgRNA, and alternative sgRNA

[107] The gRNA can be a single gRNA or a dual-gRNA system. A single gRNA comprises the crRNA and tracrRNA on a single molecule of RNA, whereas a dual-gRNA system comprises a crRNA and a tracrRNA present on two distinct RNA molecules, hybridized to one another through at least a portion of the CRISPR repeat sequence of the crRNA and at least a portion of the tracrRNA, which may be fully or partially complementary to the CRISPR repeat sequence of the crRNA. In some of those embodiments wherein the gRNA is a single gRNA, the crRNA and tracrRNA are separated by a linker nucleotide sequence. In general, the linker nucleotide sequence is one that does not include complementary bases in order to avoid the formation of secondary structure within or comprising nucleotides of the linker nucleotide sequence. In some embodiments, the linker nucleotide sequence between the crRNA and tracrRNA is at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, at least 10, at least 11, at least 12, or more nucleotides in length. In particular embodiments, the linker nucleotide sequence of a single gRNA is at least 4 nucleotides in length. In certain embodiments, the linker nucleotide sequence is the nucleotide sequence set forth in Table 5. In other embodiments, the linker nucleotide sequence is at least 6 nucleotides in length. In certain embodiments, the linker nucleotide sequence is RAAA.
[108] The single gRNA or dual-gRNA can be synthesized chemically or via in vitro transcription. Assays for determining sequence-specific binding between a RGN and a gRNA are known in the art and include, but are not limited to, in vitro binding assays between an expressed RGN and the gRNA, which can be tagged with a detectable label (e.g., biotin) and used in a pull-down detection assay in which the gRNA:RGN complex is captured via the detectable label (e.g., with streptavidin beads). A control gRNA with an unrelated sequence or structure to the gRNA can be used as a negative control for non-specific binding of the RGN to RNA.
[109] In certain embodiments, the gRNA can be introduced into a target cell, organelle, or embryo as an RNA molecule. The gRNA can be transcribed in vitro or chemically synthesized. In other embodiments, a nucleotide sequence encoding the gRNA is introduced into the cell, organelle, or embryo. In some of these embodiments, the nucleotide sequence encoding the gRNA is operably linked to a promoter (e.g., an RNA polymerase III promoter). The promoter can be a native promoter or heterologous to the gRNA-encoding nucleotide sequence.
[HO] In various embodiments, the gRNA can be introduced into a target cell, organelle, or embryo as a ribonucleoprotein complex, as described herein, wherein the gRNA is bound to an RNA-guided
nuclease polypeptide. The gRNA directs an associated RNA-guided nuclease to a particular target nucleotide sequence of interest through hybridization of the gRNA to the target nucleotide sequence. A target nucleotide sequence can comprise DNA, RNA, or a combination of both and can be singlestranded or double -stranded. A target nucleotide sequence can be genomic DNA (i.e. , chromosomal DNA), plasmid DNA, or an RNA molecule ( e.g. , messenger RNA, ribosomal RNA, transfer RNA, micro RNA, small interfering RNA). The target nucleotide sequence can be bound (and in some embodiments, cleaved) by an RNA-guided nuclease in vitro or in a cell. The chromosomal sequence targeted by the RGN can be a nuclear, plastid or mitochondrial chromosomal sequence. In some embodiments, the target nucleotide sequence is unique in the target genome.
Multiple gRNA molecules
[Hl] The present disclosure also provides methods for binding and/or modifying a target nucleotide sequence of interest. The methods include delivering a system comprising at least one gRNA or a polynucleotide encoding the same, and at least one fusion polypeptide comprises an RGN of the invention and a base-editing polypeptide, for example a cytidine deaminase or an adenosine deaminase, or a polynucleotide encoding the fusion polypeptide, to the target sequence or a cell, organelle, or embryo comprising the target sequence.
[112] One of ordinary skill in the art will appreciate that any of the presently disclosed methods can be used to target a single target sequence or multiple target sequences. Thus, methods comprise the use of a single RGN polypeptide in combination with multiple, distinct gRNAs, which can target multiple, distinct sequences within a single gene and/or multiple genes. Also encompassed herein are methods wherein multiple, distinct gRNAs are introduced in combination with multiple, distinct RGN polypeptides. These gRNAs and gRNA/RGN polypeptide systems can target multiple, distinct sequences within a single gene and/or multiple genes.
Protospacer adjacent motif (PAM) sequences
[113] The present disclosure also provides PAM (proto-spacer-adjacent Motif) sequences to the adjacent, target DNA sequence of the complementary chain (complementary strand) and base pair formation can be in sequence to include to, a gRNA, or a composition comprising a DNA coding for the gRNA.
[114] In the context of the RGNs disclosed herein, the target nucleotide sequence of the RGNs is adjacent to a sequence called protospacer adjacent motif (PAM). A protospacer adjacent motif is generally within 1 to 30 nucleotides from the target nucleotide sequence. A protospacer adjacent motif can be within 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 nucleotides from the target nucleotide sequence. The PAM can be 5' or 3' of the target sequence depending on the RGN. In some embodiments, the PAM is 5' of the target sequence for the presently disclosed RGNs. Generally, the PAM is a consensus sequence of
3-4 nucleotides, but in particular embodiments, can be 2, 3, 4, 5, 6, 7, 8, 9, or more nucleotides in length.
[115] Table 6. PAM sequences for the RGNs

[116] In particular embodiments, the RGN having or an active variant or fragment thereof binds respectively a target nucleotide sequence adjacent to a PAM sequence set forth in Table 6. In some embodiments, the RGN binds to a guide sequence comprising a CRISPR repeat sequence set forth in Table 5, or an active variant or fragment thereof, and a tracrRNA sequence set forth in Table 5, or an active variant or fragment thereof.
[117] It is well-known in the art that PAM sequence specificity for a given nuclease enzyme is affected by enzyme concentration (see, e.g. , Karvelis et al. (2015) Genome Biol 16:253), which may be modified by altering the promoter used to express the RGN, or the amount of ribonucleoprotein complex delivered to the cell, organelle, or embryo.
[118] Upon recognizing its corresponding PAM sequence, the RGN can cleave the target nucleotide sequence at a specific cleavage site. As used herein, a cleavage site is made up of the two particular nucleotides within a target nucleotide sequence between which the nucleotide sequence is cleaved by an RGN. The cleavage site can comprise the 1st and 2nd , 2nd and 3rd , 3rd and 4th , 4th and 5th , 5th and 6th , 7th and 8th , or 8th and 9th nucleotides from the PAM in either the 5' or 3' direction. In some embodiments, the cleavage site may be over 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 nucleotides from the PAM in either the 5’ or 3’ direction. In some embodiments, the cleavage site is 4 nucleotides away from the PAM. In other embodiments, the cleavage site is at least 15 nucleotides away from the PAM. As RGNs can cleave a target nucleotide sequence resulting in staggered ends, in some embodiments, the cleavage site is defined based on the distance of the two nucleotides from the PAM on the positive (+) strand of the polynucleotide and the distance of the two nucleotides from the PAM on the negative (-) strand of the polynucleotide.
Target nucleotide sequence
[119] The target polynucleotide of an RGN system can be any polynucleotide endogenous or exogenous to the eukaryotic cell. For example, the target polynucleotide can be a polynucleotide residing in the nucleus of the eukaryotic cell. The target polynucleotide can be a sequence coding a gene product (e.g., a protein) or a non-coding sequence (e.g., a regions or introns). The target sequence is generally associated with a PAM (protospacer adjacent motif. The precise sequence and length requirements for the PAM differ depending on the RGN used, but PAMs are typically 2-5 base pair sequences adjacent the protospacer (that is, the target sequence).
[120] The target polynucleotide of a RGN/RNA complex may be a disease-associated gene or polynucleotides or a gene/ polynucleotide associated with a biological pathway.
RGNs complexes with RNA
[121] RGNs proteins can be complexed to an RNA (RNA/Cas complex) in order to deliver Cas in proximity with a target nucleic acid sequence. The RNA, such as a crRNA, is a polynucleotide that site-specifically guides a Cas nuclease, or a deactivated Cas nuclease, to a target nucleic acid region. The binding specificity is determined jointly by the complementary region on the cognate guide and a short DNA motif (protospacer adjacent motif or PAM) juxtaposed to the complementary region. The spacer present in the guide specifically hybridizes to a target nucleic acid sequence and determines the location of a Cas protein's site-specific binding and nucleolytic cleavage.
[122] RNA/Cas complexes can be produced using methods well known in the art. For example, the RNA of the complexes can be produced in vitro and RGN polypeptides can be recombinantly produced and then the RNA and RGN proteins can be complexed together using methods known in the art. Additionally, cell lines constitutively expressing RGN proteins can be developed and can be transfected with the gRNA components, and complexes can be purified from the cells using standard purification techniques, such as but not limited to affinity, ion exchange and size exclusion chromatography. See, e.g. , Jinek M., et al, "A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity," Science (2012) 337:816-821.
[123] Alternatively, the components, i.e., the gRNA and RGN-encoding polynucleotides may be provided separately to a cell, e.g., using separate constructs, or together, in a single construct, or in any combination, and complexes can be purified as above.
[124] Methods of designing particular guides, such as for use in the complexes, are known. See, e.g. , Briner et al., "GRNA Functional Modules Direct Cas9 Activity and Orthogonality," Molecular Cell (2014) 56:333-339. To do so, the genomic sequence for the gene to be targeted is first identified. The exact region of the selected gene to target will depend on the specific application. For example, in order to activate or repress a target gene using, for example, Cas activators or repressors, gRNA/RGN complexes can be targeted to the promoter driving expression of the gene of interest. For genetic knockouts, guides are commonly designed to target 5' constitutively expressed exons which reduces the chances or removal of the targeted region from mRNA due to alternative splicing. Exons near the N- terminus can be targeted because frameshift mutations here will increase the likelihood of the production of a nonfunctional protein product. Alternatively, cognate guides can be designed to target exons that code for known essential protein domains. In this regard, non-frameshift mutations such as insertions or deletions are more likely to alter protein function when they occur in protein domains that are essential for protein function. For gene editing using HDR, the target sequence should be
close to the location of the desired edit. In this case, the location where the edit is desired is identified and a target sequence is selected nearby.
[125] The gRNA can be delivered to a cell. If the cell constitutively expresses a RGN, the RGN will then be recruited to the target site to cleave the target nucleic acid. If the cell does not express an RGN, complexes of gRNA/RGN can be delivered to the cells to make breaks in the genome, thereby triggering the repair pathways in the cells.
[126] Treated cells are then screened using methods well known in the art, such as using high- throughput screening techniques including, but not limited to, fluorescence-activated cell sorting (FACS)-based screening platforms, microfluidics-based screening platforms, and the like. These techniques are well known in the art. See, e.g., Wojcik et al, Int. J. Molec. Sci. (2015) 16:24918- 24945. The cells can then be expanded and re-transfected with additional RGN/gRNA complexes to introduce further diversity and this process can be repeated iteratively until a population with the desired properties is obtained. Single cell clones are sorted from the population, expanded and sequenced to recover the mutations that resulted in the desired function.
Variants of RGNs
[127] The present disclosure provides RGNs comprising at least 50, 100, 150, 200, 250, 300, 350, 400, 450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950, 1000, 1050 or more contiguous amino acid residues of an amino acid sequence set forth in Table 4. RNA-guided nucleases provided herein can comprise at least one nuclease domain ( e.g ., DNase, RNase domain) and at least one RNA recognition and/or RNA binding domain to interact with gRNAs. Further domains that can be found in RNA-guided nucleases provided herein include, but are not limited to, DNA binding domains, helicase domains, protein-protein interaction domains, and dimerization domains. In specific embodiments, the RGNs provided herein can comprise at least 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% to one or more of a DNA binding domains, helicase domains, protein-protein interaction domains, and dimerization domains.
[128] While the activity of a variant or fragment may be altered compared to the polynucleotide or polypeptide of interest, the variant and fragment should retain the functionality of the polynucleotide or polypeptide of interest. For example, a variant or fragment may have increased activity, decreased activity, different spectrum of activity or any other alteration in activity when compared to the polynucleotide or polypeptide of interest.
[129] Fragments and variants of naturally-occurring RGN polypeptides, such as those disclosed herein, will retain sequence-specific, RNA-guided DNA-binding activity. In particular embodiments, fragments and variants of naturally-occurring RGN polypeptides, such as those disclosed herein, will retain nuclease activity (single -stranded or double -stranded).
[130] A biologically active variant of an RGN polypeptide of the invention may differ by as few as 1- 15 amino acid residues, as few as 1-10, such as 6-10, as few as 5, as few as 4, as few as 3, as few as 2, or as few as 1 amino acid residue. In specific embodiments, the polypeptides can comprise an N- terminal or a C-terminal truncation, which can comprise at least a deletion of 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950, 1000, 1050 amino acids or more from either the N or C terminus of the polypeptide.
Variants of gRNA, tracrRNA and CRISPR RNA repeat sequence (crRNA)
[131] RGN proteins can have varying sensitivity to mismatches between a spacer sequence in a gRNA and its target sequence that affects the efficiency of cleavage. The CRISPR RNA repeat sequence comprises a nucleotide sequence that comprises a region with sufficient complementarity to hybridize to a tracrRNA. In various embodiments, the CRISPR RNA repeat sequence can comprise from 8 nucleotides to 30 nucleotides, or more. For example, the CRISPR repeat sequence can be 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, or more nucleotides in length. In some embodiments, the CRISPR repeat sequence is 21 nucleotides in length. In some embodiments, the degree of complementarity between a CRISPR repeat sequence and its corresponding tracrRNA sequence, when optimally aligned using a suitable alignment algorithm, is or more than 50%, 60%, 70%, 75%, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or more. In particular embodiments, the CRISPR repeat sequence comprises the nucleotide sequence set forth in Table 5, or an active variant or fragment thereof that when comprised within a gRNA, is capable of directing the sequence-specific binding of an associated RNA-guided nuclease provided herein to a target sequence of interest. In certain embodiments, an active CRISPR repeat sequence variant of a wild-type sequence comprises a nucleotide sequence having at least 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or more sequence identity to the nucleotide sequence set forth in Table 5. In certain embodiments, an active CRISPR repeat sequence fragment of a wild-type sequence comprises at least 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 contiguous nucleotides of the nucleotide sequence set forth in Table 5.
[132] Fragments and variants of naturally occurring CRISPR repeats, such as those disclosed herein, will retain the ability, when part of a gRNA (comprising a tracrRNA), to bind to and guide an RNA- guided nuclease (complexed with the gRNA) to a target nucleotide sequence in a sequence -specific manner.
[133] Fragments and variants of naturally occurring tracrRNAs, such as those disclosed herein, will retain the ability, when part of a gRNA (comprising a CRISPR RNA), to guide an RNA-guided nuclease (complexed with the gRNA) to a target nucleotide sequence in a sequence -specific manner.
[134] In a particular embodiment the present invention provides variant sequences of gRNA, wherein said gRNA has an altered linkage between tracrRNA and crRNA, and/or has hairpin mismatches removed, and/or has A:U base pairs swapped for G:C base pairs, and/or has an altered starting position of the tracrRNA, and/or has non-protein contacting regions of the sgRNA removed. Such removal also helps to minimize the design of gRNA. Swapping A:U base pairs for G:C base pairs helps to strengthen the hairpins.
[135] In various embodiments, the anti-repeat region of the tracrRNA that is fully or partially complementary to the CRISPR repeat sequence comprises from 8 nucleotides to 30 nucleotides, or more. For example, the region of base pairing between the tracrRNA anti-repeat sequence and the CRISPR repeat sequence can be 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, or more nucleotides in length. In particular embodiments, the anti-repeat region of the tracrRNA that is fully or partially complementary to a CRISPR repeat sequence is 20 nucleotides in length. In some embodiments, the degree of complementarity between a CRISPR repeat sequence and its corresponding tracrRNA anti-repeat sequence, when optimally aligned using a suitable alignment algorithm, is or more than 50%, 60%, 70%, 75%, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or more.
[136] In various embodiments, the entire tracrRNA can comprise from 60 nucleotides to more than 140 nucleotides. For example, the tracrRNA can be 60, 65, 70, 75, 80, 85, 90, 95, 100, 105, 110, 115, 120, 125, 130, 135, 140, or more nucleotides in length. In particular embodiments, the tracrRNA is 80 to 90 nucleotides in length, including 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, and 90 nucleotides in length. In certain embodiments, the tracrRNA is 85 nucleotides in length.
[137] In particular embodiments, the tracrRNA comprises the nucleotide sequence set forth in Table 5 or an active variant or fragment thereof that when comprised within a gRNA is capable of directing the sequence-specific binding of an associated RNA-guided nuclease provided herein to a target sequence of interest. In certain embodiments, an active tracrRNA sequence variant of a wild-type sequence comprises a nucleotide sequence having at least 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or more sequence identity to the nucleotide sequence set forth in Table 5. In certain embodiments, an active tracrRNA sequence fragment of a wild-type sequence comprises at least 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, or more contiguous nucleotides of the nucleotide sequence set forth in Table 5.
[138] In certain embodiments, the presently disclosed polynucleotides comprise or encode a CRISPR repeat comprising a nucleotide sequence having at least 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%,
80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%,
97%, 98%, 99%, or greater identity to the nucleotide sequence set forth in Table 5.
[139] The presently disclosed polynucleotides can comprise or encode a tracrRNA comprising a nucleotide sequence having at least 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or greater identity to the nucleotide sequence set forth in Table 5.
[140] Biologically active variants of a CRISPR repeat or tracrRNA of the invention may differ by as few as 1-15 nucleotides, as few as 1-10, such as 6-10, as few as 5, as few as 4, as few as 3, as few as 2, or as few as 1 nucleotide. In specific embodiments, the polynucleotides can comprise a 5' or 3' truncation, which can comprise at least a deletion of 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80 nucleotides or more from either the 5' or 3' end of the polynucleotide.
Variants of spacer sequences
[141] In some embodiments, the degree of complementarity between a spacer sequence and its corresponding target sequence, when optimally aligned using a suitable alignment algorithm, is or more than 80%, 85%, 90, 95%, 96%, 97%, 98%, 99%, or more. In particular embodiments, the spacer sequence is free of secondary structure, which can be predicted using any suitable polynucleotide folding algorithm known in the art, including but not limited to mFold (see, e.g., Zuker and Stiegler (1981) Nucleic Acids Res . 9: 133-148) and RNAfold (see, e.g., Gruber et al. (2008) Cell 106(l):23- 24).
Nucleotides encoding RNA-guided nucleases, crRNA, and/or tracrRNA
[142] The present disclosure provides polynucleotides comprising the presently disclosed crRNAs, tracrRNAs, and/or gRNAs and polynucleotides comprising a nucleotide sequence encoding the presently disclosed RGNs, crRNAs, tracrRNAs, and/or gRNAs. Presently disclosed polynucleotides include those comprising or encoding a CRISPR repeat sequence comprising the nucleotide sequence set forth in Table 5, or an active variant or fragment thereof that when comprised within a gRNA is capable of directing the sequence -specific binding of an associated RNA- guided nuclease to a target sequence of interest.
[143] The disclosure also provides polynucleotides comprising or encoding a tracrRNA comprising the nucleotide sequence set forth in Table 5, or an active variant or fragment thereof that when comprised within a gRNA is capable of directing the sequence- specific binding of an associated RNA-guided nuclease to a target sequence of interest. Polynucleotides are also provided that encode an RGN comprising the amino acid sequence set forth in Table 4, and active fragments or variants thereof that retain the ability to bind to a target nucleotide sequence in an RNA-guided sequence specific manner.
[144] The expression cassette will include in the 5 '-3' direction of transcription, a transcriptional (and, in some embodiments, translational) initiation region (i.e.. a promoter), an RGN-, a transcriptional initiation region (i.e.. a promoter), crRNA-, tracrRNA -and/or gRNA- encoding polynucleotide of the
invention, and a transcriptional (and in some embodiments, translational) termination region (i.e.. termination region) functional in the organism of interest. The promoters of the invention are capable of directing or driving expression of a coding sequence in a host cell. The regulatory regions (e.g., promoters, transcriptional regulatory regions, and translational termination regions) may be endogenous or heterologous to the host cell or to each other.
[145] Additional regulatory signals may include, but are not limited to, transcriptional initiation start sites, operators, activators, enhancers, other regulatory elements, ribosomal binding sites, an initiation codon, and termination signals.
[146] In preparing the expression cassette, the various DNA fragments may be manipulated, so as to provide for the DNA sequences in the proper orientation and, as appropriate, in the proper reading frame. Toward this end, adapters or linkers may be employed to join the DNA fragments or other manipulations may be involved to provide for convenient restriction sites, removal of superfluous DNA, removal of restriction sites, or the like. For this purpose, in vitro mutagenesis, primer repair, restriction, annealing, resubstitutions, e.g., transitions and transversions, may be involved.
[147] A number of promoters can be used in the practice of the invention. The promoters can be selected based on the desired outcome. The nucleic acids can be combined with constitutive, inducible, growth stage -specific, cell type-specific, tissue-preferred, tissue-specific, or other promoters for expression in the organism of interest.
[148] In some embodiments, the nucleotide comprises a tissue-preferred promoter. In some embodiments, the nucleic acid molecules encoding a RGN, crRNA-, tracrRNA-and/or gRNA comprise a cell type-specific promoter.
[149] The nucleic acid sequences encoding the RGNs, crRNA-, tracrRNA-and/or gRNA can be operably linked to a promoter sequence that is recognized by a phage RNA polymerase for example, for in vitro mRNA synthesis. For example, the promoter sequence can be a pol I, pol II, pol III, T7, T3, U6, CMV or SP6 promoter sequence or a variation of a T7, T3, U6, CMV or SP6 promoter sequence. In such embodiments, the expressed protein and/or RNAs can be purified for use in the methods of genome modification described herein. Any Pol II promoter or terminator could express the RGN. The choice of a promoter depends on how strongly RGN needs to be expressed and in what tissue type. In a preferred embodiment the RGN is expressed using is the CMV promoter. The gRNA can be expressed by Pol III promoters (e.g. U6 promoter).
[150] In certain embodiments, the polynucleotide encoding the RGN also can be linked to a polyadenylation signal (e.g., SV40 polyA signal, or sv40 polyA with rmG terminator) and/or at least one transcriptional termination sequence. Additionally, the sequence encoding the RGN also can be linked to sequence(s) encoding at least one nuclear localization signal, at least one cell- penetrating
domain, and/or at least one signal peptide capable of trafficking proteins to particular subcellular locations.
[151] Additional regulatory signals include, but are not limited to, transcriptional initiation start sites, operators, activators, enhancers, other regulatory elements, ribosomal binding sites, an initiation codon, termination signals, and the like. See, for example, U.S. Pat. Nos. 5,039,523 and 4,853,331
[152] In preparing the expression cassette, the various DNA fragments may be manipulated, so as to provide for the DNA sequences in the proper orientation and, as appropriate, in the proper reading frame. Toward this end, adapters or linkers may be employed to join the DNA fragments or other manipulations may be involved to provide for convenient restriction sites, removal of superfluous DNA, removal of restriction sites, or the like. For this purpose, in vitro mutagenesis, primer repair, restriction, annealing, resubstitutions, e.g., transitions and transversions, may be involved.
Variants of polynucleotides
[153] For polynucleotides, conservative variants include those sequences that, because of the degeneracy of the genetic code, encode the native amino acid sequence of the gene of interest. Naturally occurring allelic variants such as these can be identified with the use of well-known molecular biology techniques, as, for example, with polymerase chain reaction (PCR) and hybridization techniques as outlined below. Variant polynucleotides also include synthetically derived polynucleotides, such as those generated, for example, by using site-directed mutagenesis but which still encode the polypeptide or the polynucleotide of interest. Generally, variants of a particular polynucleotide disclosed herein will have at least 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or more sequence identity to that particular polynucleotide as determined by sequence alignment programs and parameters described elsewhere herein.
[154] Variants of a particular polynucleotide disclosed herein (i.e., the reference polynucleotide) can also be evaluated by comparison of the percent sequence identity between the polypeptide encoded by a variant polynucleotide and the polypeptide encoded by the reference polynucleotide. Percent sequence identity between any two polypeptides can be calculated using sequence alignment programs and parameters described elsewhere herein. Where any given pair of polynucleotides disclosed herein is evaluated by comparison of the percent sequence identity shared by the two polypeptides they encode, the percent sequence identity between the two encoded polypeptides is at least 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or more sequence identity.
[155] In particular embodiments, the presently disclosed polynucleotides encode an RGN polypeptide comprising an amino acid sequence having at least 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%,
80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or greater identity to an amino acid sequence set forth in Table 4.
[156] Variant polynucleotides and proteins also encompass sequences and proteins derived from a mutagenic and recombinogenic procedure such as DNA shuffling. With such a procedure, one or more different RGN proteins disclosed herein is manipulated to create a new RGN protein possessing the desired properties. In this manner, libraries of recombinant polynucleotides are generated from a population of related sequence polynucleotides comprising sequence regions that have substantial sequence identity and can be homologously recombined in vitro or in vivo. For example, using this approach, sequence motifs encoding a domain of interest may be shuffled between the RGN sequences provided herein and other known RGN genes to obtain a new gene coding for a protein with an improved property of interest, such as an increased Km in the case of an enzyme. Strategies for such DNA shuffling are known in the art. See, for example, Stemmer (1994) Proc. Natl. Acad. Sci. USA
Codon-optimized sequences
[157] The nucleic acid molecules encoding RNA-guided nucleases can be codon optimized for expression in a target cell or tissue of interest. Such polynucleotide coding sequence normally has its frequency of codon usage designed to mimic the frequency of preferred codon usage or transcription conditions of a particular host cell. Expression in the particular host cell or organism is enhanced as a result of the alteration of one or more codons at the nucleic acid level such that the translated amino acid sequence is not changed. Nucleic acid molecules can be codon optimized, either wholly or in part. Codon tables and other references providing preference information for a wide range of organisms are available in the art.
Vectors
[158] The polynucleotide encoding the RGN, crRNA, tracrRNA, and/or sgRNA can be present in a vector or multiple vectors. Suitable vectors include plasmid vectors, phagemids, cosmids, artificial/mini-chromosomes, transposons, and viral vectors (e.g., lentiviral vectors, adeno-associated viral vectors, baculoviral vector). The vector can comprise additional expression control sequences (e.g. , enhancer sequences, Kozak sequences, polyadenylation sequences, transcriptional termination sequences), selectable marker sequences (e.g., antibiotic resistance genes), origins of replication, and the like. Additional information can be found in "Current Protocols in Molecular Biology" Ausubel et al, John Wiley & Sons, New York, 2003 or "Molecular Cloning: A Laboratory Manual" Sambrook & Russell, Cold Spring Harbor Press, Cold Spring Harbor, N.Y., 3rd edition, 2001.
[159] The vector can also comprise a selectable marker gene for the selection of transformed cells. Selectable marker genes are utilized for the selection of transformed cells or tissues. Marker genes include genes encoding antibiotic resistance, such as those encoding neomycin phosphotransferase II
(NEO) and hygromycin phosphotransferase (HPT), as well as genes conferring resistance to herbicidal compounds, such as glufosinate ammonium, bromoxynil, imidazolinones, and 2,4- dichlorophenoxyacetate (2,4-D).
[160] In some embodiments, the expression cassette or vector comprising the sequence encoding the RGN polypeptide can further comprise a sequence encoding a crRNA and/or a tracrRNA, or the crRNA and tracrRNA combined to create a gRNA. The sequence(s) encoding the crRNA and/or tracrRNA can be operably linked to at least one transcriptional control sequence for expression of the crRNA and/or tracrRNA in the organism or host cell of interest. For example, the polynucleotide encoding the crRNA and/or tracrRNA can be operably linked to a promoter sequence that is recognized by RNA polymerase III (Pol III). Examples of suitable Pol III promoters include, but are not limited to, mammalian U6, U3, Hl, and 7SL RNA promoters and rice U6 and U3 promoters.
Systems for binding to the target nucleotide sequence of interest
[161] The present disclosure provides a system for binding a target sequence of interest, wherein the system comprises at least one gRNA or a nucleotide sequence encoding the same, and at least one RGN or a nucleotide sequence encoding the same. The gRNA hybridizes to the target sequence of interest and also forms a complex with the RGN polypeptide, thereby directing the RGN polypeptide to bind to the target sequence. In some of these embodiments, the RGN comprises an amino acid sequence as set forth in Table 4 or an active variant or fragment thereof. In various embodiments, the gRNA comprises a CRISPR repeat sequence comprising the nucleotide sequence set forth in Table 5 or an active variant or fragment thereof. In particular embodiments, the gRNA comprises a tracrRNA comprising a nucleotide sequence set forth in Table 5 or an active variant or fragment thereof. The gRNA of the system can be a single gRNA or a dual-gRNA. In particular embodiments, the system comprises an RGN that is heterologous to the gRNA, wherein the RGN and gRNA are not naturally complexed in nature.
[162] The system for binding a target sequence of interest provided herein can be a ribonucleoprotein complex, which is at least one molecule of an RNA bound to at least one protein. The ribonucleoprotein complexes provided herein comprise at least one gRNA as the RNA component and an RNA-guided nuclease as the protein component. Such ribonucleoprotein complexes can be purified from a cell or organism that naturally expresses an RGN polypeptide and has been engineered to express a particular gRNA that is specific for a target sequence of interest.
[163] Alternatively, the ribonucleoprotein complex can be purified from a cell or organism that has been transformed with polynucleotides that encode an RGN polypeptide and a gRNA and cultured under conditions to allow for the expression of the RGN polypeptide and gRNA. Thus, methods are provided for making an RGN polypeptide or an RGN ribonucleoprotein complex. Such methods comprise culturing a cell comprising a nucleotide sequence encoding an RGN polypeptide, and in
some embodiments a nucleotide sequence encoding a gRNA, under conditions in which the RGN polypeptide (and in some embodiments, the gRNA) is expressed. The RGN polypeptide or RGN ribonucleoprotein can then be purified from a lysate of the cultured cells.
[164] Methods for purifying an RGN polypeptide or RGN ribonucleoprotein complex from a lysate of a biological sample are known in the art (e.g., size exclusion and/or affinity chromatography, 2D- PAGE, HPLC, reversed-phase chromatography, immunoprecipitation). In particular methods, the RGN polypeptide is recombinantly produced and comprises a purification tag to aid in its purification, including but not limited to, glutathione-S-transferase (GST), chitin binding protein (CBP), maltose binding protein, thioredoxin (TRX), poly(NANP), tandem affinity purification (TAP) tag, myc, AcV5, AU1, AU5, E, ECS, E2, FLAG, HA, nus, Softag 1, Softag 3, Strep, SBP, Glu-Glu, HSV, KT3, S, SI, T7, V5, VSV-G, 6xHis, lOxHis, biotin carboxyl carrier protein (BCCP), and calmodulin.
[165] Generally, the tagged RGN polypeptide or RGN ribonucleoprotein complex is purified using immobilized metal affinity chromatography. It will be appreciated that other similar methods known in the art may be used, including other forms of chromatography or for example immunoprecipitation, either alone or in combination.
[166] Some methods provided herein for binding and/or cleaving a target sequence of interest involve the use of an in vitro assembled RGN ribonucleoprotein complex. In vitro assembly of an RGN ribonucleoprotein complex can be performed using any method known in the art in which an RGN polypeptide is contacted with a gRNA under conditions to allow for binding of the RGN polypeptide to the gRNA. The RGN polypeptide can be purified from a biological sample, cell lysate, or culture medium, produced via in vitro translation, or chemically synthesized. The gRNA can be purified from a biological sample, cell lysate, or culture medium, transcribed in vitro, or chemically synthesized. The RGN polypeptide and gRNA can be brought into contact in solution (e.g., buffered saline solution) to allow for in vitro assembly of the RGN ribonucleoprotein complex.
Delivery of the components to the target cells
[167] In some aspects, components of the nucleic-acid targeting system are delivered using nanoscale delivery systems, such as nanoparticles. Additionally, liposomes and other particulate delivery systems can be used. For example, vectors including the components of the present methods can be packaged in liposomes prior to delivery to the subject or to cells derived therefrom. Lipid encapsulation is generally accomplished using liposomes that are able to stably bind or entrap and retain nucleic acid.
[168] As indicated, expression constructs comprising nucleotide sequences encoding the RGNs, crRNA, tracrRNA, and/or sgRNA can be used to transform organisms of interest. Methods for transformation involve introducing a nucleotide construct into an organism of interest.
[169] The methods of the invention do not require a particular method for introducing a nucleotide construct to a host organism, only that the nucleotide construct gains access to the interior of at least one cell of the host organism. The host cell can be a eukaryotic or prokaryotic cell. In particular embodiments, the eukaryotic host cell is a plant cell, a mammalian cell, or an insect cell. Methods for introducing nucleotide constructs into plants and other host cells are known in the art including, but not limited to, stable transformation methods, transient transformation methods, and virus-mediated methods.
[170] It is recognized that other exogenous or endogenous nucleic acid sequences or DNA fragments may also be incorporated into the host cell. Transformation of a host cell may be performed by infection, transfection, microinjection, electroporation, microprojection, biolistics or particle bombardment, electroporation, silica/carbon fibers, ultrasound mediated, PEG mediated, calcium phosphate co-precipitation, polycation DMSO technique, DEAE dextran procedure, and viral mediated, liposome mediated and the like. Viral -mediated introduction of a polynucleotide encoding an RGN, crRNA, and/or tracrRNA includes retroviral, lentiviral, adenoviral, and adeno-associated viral mediated introduction and expression, as well as the use of Caulimoviruses, Geminiviruses, and RNA plant viruses.
[171] Transformation may result in stable or transient incorporation of the nucleic acid into the cell.
[172] The cells that have been transformed may be grown into a transgenic organism, such as a plant, in accordance with conventional ways. See, for example, McCormick et al. (1986) Plant Cell Reports 5:81-84. These plants may then be grown, and either pollinated with the same transformed strain or different strains, and the resulting hybrid having constitutive expression of the desired phenotypic characteristic identified. Two or more generations may be grown to ensure that expression of the desired phenotypic characteristic is stably maintained and inherited and then seeds harvested to ensure expression of the desired phenotypic characteristic has been achieved. In this manner, the present invention provides transformed seed (also referred to as "transgenic seed") having a nucleotide construct of the invention, for example, an expression cassette of the invention, stably incorporated into their genome.
[173] Alternatively, cells that have been transformed may be introduced into an organism. These cells could have originated from the organism, wherein the cells are transformed in an ex vivo approach.
[174] The polynucleotides encoding the RGNs, crRNAs, and/or tracrRNAs can also be used to transform any prokaryotic species, including but not limited to, archaea and bacteria (e.g., Bacillus sp., Klebsiella sp. Streptomyces sp., Rhizobium sp., Escherichia sp., Pseudomonas sp., Salmonella sp., Shigella sp., Vibrio sp., Yersinia sp., Mycoplasma sp., Agrobacterium, Lactobacillus sp.).
[175] The polynucleotides encoding the RGNs, crRNAs, and/or tracrRNAs can be used to transform any eukaryotic species, including but not limited to animals (e.g., mammals, insects, fish, birds, and reptiles), fungi, amoeba, algae, and yeast.
[176] Conventional viral and non-viral based gene transfer methods can be used to introduce nucleic acids in mammalian cells or target tissues. Such methods can be used to administer nucleic acids encoding components of a CRISPR system to cells in culture, or in a host organism. Non-viral vector delivery systems include DNA plasmids, RNA (e.g. a transcript of a vector described herein), naked nucleic acid, and nucleic acid complexed with a delivery vehicle, such as a liposome. Viral vector delivery systems include DNA and RNA viruses, which have either episomal or integrated genomes after delivery to the cell. Methods of non-viral delivery of nucleic acids include lipofection, nucleofection, microinjection, biolistics, virosomes, liposomes, immunoliposomes, polycation or lipid: nucleic acid conjugates, naked DNA, artificial virions, and agent-enhanced uptake of DNA. Lipofection is described in e.g., U.S. 5,049,386, 4 and lipofection reagents are sold commercially. Delivery can be to cells (e.g. in vitro or ex vivo administration) or target tissues ( e.g. in vivo administration). The preparation of lipid: nucleic acid complexes, including targeted liposomes such as immunolipid complexes, is well known to one of skill in the art (see, e.g., Gao et ah, Gene Therapy 2:710-722 (1995)).
[177] The use of RNA or DNA viral based systems for the delivery of nucleic acids takes advantage of highly evolved processes for targeting a virus to specific cells in the body and trafficking the viral payload to the nucleus. Viral vectors can be administered directly to patients (in vivo) or they can be used to treat cells in vitro, and the modified cells may optionally be administered to patients ( ex vivo). Conventional viral based systems could include retroviral, lentivirus, adenoviral, adeno- associated and herpes simplex virus vectors for gene transfer. Integration in the host genome is possible with the retrovirus, lentivirus, and adeno-associated virus gene transfer methods, often resulting in long term expression of the inserted transgene. Additionally, high transduction efficiencies have been observed in many different cell types and target tissues.
[178] The tropism of a retrovirus can be altered by incorporating foreign envelope proteins, expanding the potential target population of target cells. Lentiviral vectors are retroviral vectors that are able to transduce or infect non-dividing cells and typically produce high viral titers. Selection of a retroviral gene transfer system would therefore depend on the target tissue. Retroviral vectors are comprised of cis-acting long terminal repeats with packaging capacity for up to 6-10 kb of foreign sequence. The minimum cis-acting LTRs are sufficient for replication and packaging of the vectors, which are then used to integrate the therapeutic gene into the target cell to provide permanent transgene expression. Widely used retroviral vectors include those based upon murine leukemia virus (MuLV), gibbon ape leukemia virus (GaLV),
[179] Simian Immuno deficiency vims (SIV), human immuno deficiency vims (HIV), and combinations thereof (see, e.g., Buchscher et al., J. Viral. 66:2731-2739 (1992); Johann et al., J. Viral. 66: 1635-1640 (1992); Sommnerfelt et al., Viral. 176:58-59 (1990); Wilson et al., J. Viral. 63:2374- 2378 (1989); Miller et al., 7. Viral. 65:2220-2224
Viral delivery for therapeutic applications
[180] The use viral based systems for the delivery of nucleic acids allows targeting a vims to specific cells and trafficking the viral payload to the nucleus. Viral vectors can be administered directly to patients (in vivo) or they can be used to treat cells in vitro, and the modified cells may optionally be administered to patients (ex vivo). Conventional viral based systems could include retroviral, lentivims, adenoviral, adeno-associated and herpes simplex vims vectors for gene transfer. Integration in the host genome is possible with the retrovims, lentivims, and adeno-associated vims gene transfer methods, often resulting in long term expression of the inserted transgene. Additionally, high transduction efficiencies have been observed in many different cell types and target tissues.
[181] The tropism of a retrovims can be altered by incorporating foreign envelope proteins, expanding the potential target population of target cells. Lentiviral vectors are retroviral vectors that are able to transduce or infect non-dividing cells and typically produce high viral titers. Selection of a retroviral gene transfer system would therefore depend on the target tissue. Retroviral vectors are comprised of cis-acting long terminal repeats with packaging capacity for up to 6-10 kb of foreign sequence. The minimum cis-acting LTRs are sufficient for replication and packaging of the vectors, which are then used to integrate the therapeutic gene into the target cell to provide permanent transgene expression. Widely used retroviral vectors include those based upon murine leukemia vims (MuLV), gibbon ape leukemia vims (GaLV), Simian Immuno deficiency vims (SIV), human immuno deficiency vims (HIV), and combinations thereof.
Transient expression and gene therapy applications
[182] In applications where transient expression is preferred, adenoviral based systems may be used. Adenoviral based vectors are capable of very high transduction efficiency in many cell types and do not require cell division. With such vectors, high titer and levels of expression have been obtained. This vector can be produced in large quantities in a relatively simple system. Adeno-associated vims ("AAV") vectors may also be used to transduce cells with target nucleic acids. Constmction of recombinant AAV vectors are described in a number of publications, including U.S. 5,173,414. Packaging cells are typically used to form vims particles that are capable of infecting a host cell.
[183] Viral vectors used in gene therapy are usually generated by producing a cell line that packages a nucleic acid vector into a viral particle. The vectors typically contain the minimal viral sequences required for packaging and subsequent integration into a host, other viral sequences being replaced by an expression cassette for the polynucleotide( s) to be expressed. The missing viral functions are
typically supplied in trans by the packaging cell line. For example, AAV vectors used in gene therapy typically only possess ITR sequences from the AAV genome which are required for packaging and integration into the host genome. Viral DNA is packaged in a cell line, which contains a helper plasmid encoding the other AAV genes, namely rep and cap, but lacking ITR sequences.
[184] The cell line may also be infected with adenovirus as a helper. The helper virus promotes replication of the AAV vector and expression of AAV genes from the helper plasmid. The helper plasmid is not packaged in significant amounts due to a lack of ITR sequences. Contamination with adenovirus can be reduced by, e.g., heat treatment to which adenovirus is more sensitive than AAV. Additional methods for the delivery of nucleic acids to cells are known to those skilled in the art. See, for example, US20030087817
Methods of modifying target nucleotide sequence
[185] In one aspect, the disclosure provides methods of modifying a target polynucleotide in a eukaryotic cell, which may be performed in vivo, ex vivo or in vitro. In some embodiments, the method comprises sampling a cell or population of cells from a human or non-human animal or plant (including microalgae) and modifying the cell or cells. Culturing may occur at any stage ex vivo. The cell or cells may even be re-introduced into the non-human animal or plant (including micro-algae).
[186] The present disclosure provides methods for binding, cleaving, and/or modifying a target nucleotide sequence of interest. The methods include delivering a system comprising at least one gRNA or a polynucleotide encoding the same, and at least one RGN polypeptide or a polynucleotide encoding the same to the target sequence or a cell, organelle, or embryo comprising the target sequence. In some of these embodiments, the RGN comprises the amino acid sequence set forth in Table 4, or an active variant or fragment thereof. In various embodiments, the gRNA comprises a CRISPR repeat sequence comprising the nucleotide sequence set forth in Table 5, or an active variant or fragment thereof. In particular embodiments, the gRNA comprises a tracrRNA comprising the nucleotide sequence set forth in Table 5, or an active variant or fragment thereof. The gRNA of the system can be a single gRNA or a dual-gRNA. The RGN of the system may be nuclease dead RGN, have nickase activity, or may be a fusion polypeptide. In some embodiments, the fusion polypeptide comprises a base-editing polypeptide, for example a cytidine deaminase or an adenosine deaminase. In particular embodiments, the RGN and/or gRNA is heterologous to the cell, organelle, or embryo to which the RGN and/or gRNA (or polynucleotide(s) encoding at least one of the RGN and gRNA) are introduced.
[187] In those embodiments wherein the method comprises delivering a polynucleotide encoding a gRNA and/or an RGN polypeptide, the cell, organelle, or embryo can then be cultured under conditions in which the gRNA and/or RGN polypeptide are expressed. In various embodiments, the method comprises contacting a target sequence with an RGN ribonucleoprotein complex. The RGN
ribonucleoprotein complex may comprise an RGN that is nuclease dead or has nickase activity. In some embodiments, the RGN of the ribonucleoprotein complex is a fusion polypeptide comprising a base-editing polypeptide. In certain embodiments, the method comprises introducing into a cell, organelle, or embryo comprising a target sequence an RGN ribonucleoprotein complex. The RGN ribonucleoprotein complex can be one that has been purified from a biological sample, recombinantly produced and subsequently purified, or in vitro- assembled as described herein. In those embodiments wherein the RGN ribonucleoprotein complex that is contacted with the target sequence or a cell organelle, or embryo has been assembled in vitro, the method can further comprise the in vitro assembly of the complex prior to contact with the target sequence, cell, organelle, or embryo.
[188] A purified or in vitro assembled RGN ribonucleoprotein complex can be introduced into a cell, organelle, or embryo using any method known in the art, including, but not limited to electroporation.
[189] Alternatively, an RGN polypeptide and/or polynucleotide encoding or comprising the gRNA can be introduced into a cell, organelle, or embryo using any method known in the art (e.g., electroporation).
[190] Upon delivery to or contact with the target sequence or cell, organelle, or embryo comprising the target sequence, the gRNA directs the RGN to bind to the target sequence in a sequence-specific manner. In those embodiments wherein the RGN has nuclease activity, the RGN polypeptide cleaves the target sequence of interest upon binding. The target sequence can subsequently be modified via endogenous repair mechanisms, such as non-homologous end joining, or homology-directed repair with a provided donor polynucleotide.
[191] Methods to measure binding of an RGN polypeptide to a target sequence are known in the art and include chromatin immunoprecipitation assays, gel mobility shift assays, DNA pull-down assays, reporter assays, microplate capture and detection assays. Likewise, methods to measure cleavage or modification of a target sequence are known in the art and include in vitro or in vivo cleavage assays wherein cleavage is confirmed using PCR, sequencing, or gel electrophoresis, with or without the attachment of an appropriate label (e.g., radioisotope, fluorescent substance) to the target sequence to facilitate detection of degradation products. Alternatively, the nicking triggered exponential amplification reaction (NTEXPAR) assay can be used (see, e.g., Zhang et al. (2016) Chem. Sci.
7:4951-4957). In vivo cleavage can be evaluated using the Surveyor assay (Guschin et al. (2010) Methods Mol Biol 649:247-256).
[192] In some embodiments, the methods involve the use of a single type of RGN complexed with more than one gRNA. The more than one gRNA can target different regions of a single gene or can target multiple genes.
[193] In those embodiments wherein a donor polynucleotide is not provided, a double -stranded break introduced by an RGN polypeptide can be repaired by a non-homologous end-joining (NHEJ) repair
process. Due to the error-prone nature of NHEJ, repair of the double -stranded break can result in a modification to the target sequence. Modification of the target sequence can result in the expression of an altered protein product or inactivation of a coding sequence.
[194] In those embodiments wherein a donor polynucleotide is present, the donor sequence in the donor polynucleotide can be integrated into or exchanged with the target nucleotide sequence during the course of repair of the introduced double-stranded break, resulting in the introduction of the exogenous donor sequence. A donor polynucleotide thus comprises a donor sequence that is desired to be introduced into a target sequence of interest. In some embodiments, the donor sequence alters the original target nucleotide sequence such that the newly integrated donor sequence will not be recognized and cleaved by the RGN. Integration of the donor sequence can be enhanced by the inclusion within the donor polynucleotide of flanking sequences that have substantial sequence identity with the sequences flanking the target nucleotide sequence, allowing for a homology-directed repair process. In those embodiments wherein the RGN polypeptide introduces double -stranded staggered breaks, the donor polynucleotide can comprise a donor sequence flanked by compatible overhangs, allowing for direct ligation of the donor sequence to the cleaved target nucleotide sequence comprising overhangs by a non-homologous repair process during repair of the double - stranded break.
[195] In those embodiments wherein the method involves the use of an RGN that is a nickase (i.e.. is only able to cleave a single strand of a double-stranded polynucleotide), the method can comprise introducing two RGN nickases that target identical or overlapping target sequences and cleave different strands of the polynucleotide. For example, an RGN nickase that only cleaves the positive (+) strand of a double -stranded polynucleotide can be introduced along with a second RGN nickase that only cleaves the negative (-) strand of a double -stranded polynucleotide.
[196] In various embodiments, a method is provided for binding a target nucleotide sequence and detecting the target sequence, wherein the method comprises introducing into a cell, organelle, or embryo at least one gRNA or a polynucleotide encoding the same, and at least one RGN polypeptide or a polynucleotide encoding the same, expressing the gRNA and/or RGN polypeptide (if coding sequences are introduced), wherein the RGN polypeptide is a nuclease-dead RGN and further comprises a detectable label, and the method further comprises detecting the detectable label. The detectable label may be fused to the RGN as a fusion protein (e.g., fluorescent protein) or may be a small molecule conjugated to or incorporated within the RGN polypeptide that can be detected visually or by other means.
Methods of modulating gene expression
[197] Also provided herein are methods for modulating the expression of a target sequence or a gene of interest under the regulation of a target sequence. The methods comprise introducing into a cell,
organelle, or embryo at least one gRNA or a polynucleotide encoding the same, and at least one RGN polypeptide or a polynucleotide encoding the same, expressing the gRNA and/or RGN polypeptide (if coding sequences are introduced), wherein the RGN polypeptide is a nuclease-dead RGN. In some of these embodiments, the nuclease-dead RGN is a fusion protein comprising an expression modulator domain (i.e., epigenetic modification domain, transcriptional activation domain or a transcriptional repressor domain) as described herein.
Kits
[198] In one aspect, the invention provides kits containing any one or more of the elements disclosed in the above methods and compositions. In some embodiments, the kit comprises a vector system and instructions for using the kit. In some embodiments, the vector system comprises (a) a first regulatory element operably linked to a tracr mate sequence and one or more insertion sites for inserting a guide sequence upstream of the tracr mate sequence, wherein when expressed, the guide sequence directs sequence-specific binding of a CRISPR complex to a target sequence in a eukaryotic cell, wherein the CRISPR complex comprises a CRIS PR enzyme complexed with (1) the guide sequence that is hybridized to the target sequence, and (2) the tracr mate sequence that is hybridized to the tracr sequence; and/or (b) a second regulatory element operably linked to an enzyme coding sequence encoding said CRISPR enzyme comprising a nuclear localization sequence. Elements may be provided individually or in combinations, and may be provided in any suitable container, such as a vial, a bottle, or a tube.
[199] In some embodiments, the kit includes instructions in one or more languages. In some embodiments, a kit comprises one or more reagents for use in a process utilizing one or more of the elements described herein. Reagents may be provided in any suitable container. For example, a kit may provide one or more reaction or storage buffers. Reagents may be provided in a form that is usable in a particular assay, or in a form that requires addition of one or more other components before use ( e.g. in concentrate or lyophilized form). A buffer can be any buffer, including but not limited to a sodium carbonate buffer, a sodium bicarbonate buffer, a borate buffer, a Tris buffer, a MOPS buffer, a HEPES buffer, and combinations thereof. In some embodiments, the buffer is alkaline. In some embodiments, the buffer has a pH from 7 to 10.
[200] In some embodiments, the kit comprises one or more oligonucleotides corresponding to a guide sequence for insertion into a vector so as to operably link the guide sequence and a regulatory element. In some embodiments, the kit comprises a homologous recombination template polynucleotide. In one aspect, the invention provides methods for using one or more elements of a CRISPR system. The CRISPR complex of the invention provides an effective means for modifying a target polynucleotide. The CRISPR complex of the invention has a wide variety of utility including modifying (e.g., deleting, inserting, translocating, inactivating, activating) a target polynucleotide in a multiplicity of cell types. As such the CRISPR complex of the invention has a broad spectrum of
applications in, e.g., gene therapy, drug screening, disease diagnosis, and prognosis. An exemplary CRISPR complex comprises a CRISPR enzyme complexed with a guide sequence hybridized to a target sequence within the target polynucleotide.
Cells comprising the RGN systems
[201] Provided herein are cells and organisms comprising a target sequence of interest that has been modified using a process or the system based an RGN, crRNA, and/or tracrRNA as described herein. Also are provided cells and organisms comprising the system for binding a target sequence of interest comprising an RGN, crRNA, and/or tracrRNA as described herein
[202] In some of these embodiments, the RGN comprises the amino acid sequence as disclosed above, or an active variant or fragment thereof. In various embodiments, the gRNA comprises a CRISPR repeat sequence comprising the nucleotide sequence as disclosed above, or an active variant or fragment thereof. In particular embodiments, the gRNA comprises a tracrRNA comprising the nucleotide sequence as disclosed above, or an active variant or fragment thereof. The gRNA of the system can be a single gRNA or a dual-gRNA. The modified cells can be eukaryotic (e.g., mammalian, plant, insect cell) or prokaryotic. Also provided are organelles and embryos comprising at least one nucleotide sequence that has been modified by a process utilizing an RGN, crRNA, and/or tracrRNA as described herein. The genetically modified cells, organisms, organelles, and embryos can be heterozygous or homozygous for the modified nucleotide sequence.
[203] The chromosomal modification of the cell, organism, organelle, or embryo can result in altered expression (up-regulation or down-regulation), inactivation, or the expression of an altered protein product or an integrated sequence. In those instances wherein the chromosomal modification results in either the inactivation of a gene or the expression of a non-fiinctional protein product, the genetically modified cell, organism, organelle, or embryo is referred to as a “knock out”. The knock out phenotype can be the result of a deletion mutation (i.e.. deletion of at least one nucleotide), an insertion mutation (i.e.. insertion of at least one nucleotide), or a nonsense mutation (/. e. , substitution of at least one nucleotide such that a stop codon is introduced).
[204] Alternatively, the chromosomal modification of a cell, organism, organelle, or embryo can produce a “knock in”, which results from the chromosomal integration of a nucleotide sequence that encodes a protein. In some of these embodiments, the coding sequence is integrated into the chromosome such that the chromosomal sequence encoding the wild-type protein is inactivated, but the exogenously introduced protein is expressed.
[205] In other embodiments, the chromosomal modification results in the production of a variant protein product. The expressed variant protein product can have at least one amino acid substitution and/or the addition or deletion of at least one amino acid. The variant protein product encoded by the
altered chromosomal sequence can exhibit modified characteristics or activities when compared to the wild-type protein, including but not limited to altered enzymatic activity or substrate specificity.
[206] In yet other embodiments, the chromosomal modification can result in an altered expression pattern of a protein. As a non-limiting example, chromosomal alterations in the regulatory regions controlling the expression of a protein product can result in the overexpression or downregulation of the protein product or an altered tissue or temporal expression pattern.
Pharmaceutical compositions
[207] The composition of the present invention may be in a pharmaceutical composition. The pharmaceutical composition may comprise 1 ng to 10 mg of DNA encoding the CRISPR/Cas- based system or CRISPR/Cas-based system protein component, i.e., the fusion protein. The pharmaceutical composition may comprise 1 ng to 10 mg of the DNA of the modified lentiviral vector. The pharmaceutical composition may comprise 1 ng to 10 mg of the DNA of the modified AAV vector and a nucleotide sequence encoding the site-specific nuclease. The pharmaceutical compositions according to the present invention can be formulated according to the mode of administration to be used. In cases where pharmaceutical compositions are injectable pharmaceutical compositions, they are sterile, pyrogen free and particulate free. An isotonic formulation is preferably used. Generally, additives for isotonicity may include sodium chloride, dextrose, mannitol, sorbitol and lactose. In some cases, isotonic solutions such as phosphate buffered saline are preferred. Stabilizers include gelatin and albumin. In some embodiments, a vasoconstriction agent is added to the formulation.
[208] The composition may further comprise a pharmaceutically acceptable excipient. The pharmaceutically acceptable excipient may be functional molecules as vehicles, adjuvants, carriers, or diluents. The pharmaceutically acceptable excipient may be a transfection facilitating agent, which may include surface active agents, such as immune-stimulating complexes (ISCOMS), Freunds incomplete adjuvant, LPS analog including monophosphoryl lipid A, muramyl peptides, quinone analogs, vesicles such as squalene and squalene, hyaluronic acid, lipids, liposomes, calcium ions, viral proteins, polyanions, polycations, or nanoparticles, or other known transfection facilitating agents.
[209] The transfection facilitating agent can be a polyanion, polycation, including poly-L- glutamate (LGS), or lipid. The transfection facilitating agent is poly-L-glutamate, and more preferably, the poly- L-glutamate is present in the composition for genome editing in skeletal muscle or cardiac muscle at a concentration less than 6 mg/ml. The transfection facilitating agent may also include surface active agents such as immune -stimulating complexes (ISCOMS), Freunds incomplete adjuvant, LPS analog including monophosphoryl lipid A, muramyl peptides, quinone analogs and vesicles such as squalene and squalene, and hyaluronic acid may also be used administered in conjunction with the genetic construct. In some embodiments, the DNA vector encoding the composition may also include a transfection facilitating agent such as lipids, liposomes, including lecithin liposomes or other
liposomes known in the art, as a DNA-liposome mixture (see for example W09324640), calcium ions, viral proteins, polyanions, polycations, or nanoparticles, or other known transfection facilitating agents. Preferably, the transfection facilitating agent is a polyanion, polycation, including poly-L- glutamate (LGS), or lipid. [210] The sequences included in the present invention are shown in Tables 7-13.
[211] Table 7. RGN sequences
 [212] Table 8. Consensus repeats sequences
[212] Table 8. Consensus repeats sequences
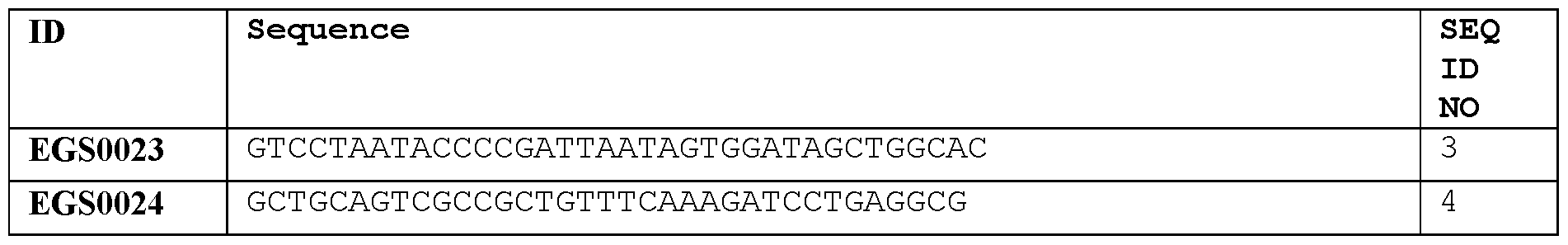
[213] Table 9. tracrRNA sequences

[214] Table 10. crRNA sequences

[215] Table 11. sgRNA sequences
 [216] Table 12. Alternative sgRNA sequences
[216] Table 12. Alternative sgRNA sequences


[217] Table 13. NLS sequences

EXAMPLES
Example 1. Identification of RNA-Guided Nuclease, crRNA, and tracrRNA
[218] Metagenomic samples were searched for open reading frames (ORFs) and those that were predicted to be genes were selected. A hidden Markov model (HMM) was used to compare the putative genes to profdes of known Cas proteins. The identified Cas genes were grouped into operons, and the operon type was determined based on the presence of known signature genes. For each genome, the CRISPR arrays were identified based on the presence of regularly spaced repeats. The subtype of each CRISPR array was predicted using machine learning. Cas operons were linked to CRISPR arrays if they were less than 10 kilobases apart. [219] Identified repeat-arrays were then used to identify anti-repeat locations flanking the putative effector protein within 3500 nt. Flanking regions around putative anti-repeat locations were extracted and the repeat-anti-repeat hybrid structures were predicted and used to identify putative crRNAs, tracrRNAs, and sgRNAs (Chyou, et al (2019). RNA biology, 16(4), 423-434).
Example 2: Determination of PAM requirements for each RGN through Bacterial PAM Depletion
[220] PAM requirements for each RGN were determined using a bacterial PAM depletion assay essentially adapted from Kleinstiver et al. (2015) Nature 523:481-485 and Zetsche et al. (2015) Cell 163:759-771. Briefly, two plasmid libraries (C2 and T2) were generated in a pUC18 backbone (ampR), with each containing a distinct 23bp protospacer (target) sequence flanked by 8 random nucleotides (i.e., the PAM region). The target sequence and flanking PAM region of library T2 and library C2 for each RGN are set forth in Table 14.
[221] Table 14. The target sequence and flanking PAM region
[222] The libraries were separately electroporated into T7 Express E. coli (NEB) cells harboring pET28b expression vectors containing the minimal CRISPR operon with the repeat spacer array modified to contain three copies of the intended library target sequence at the average spacer length of the CRISPR repeat in C2 or T2. Sufficient library plasmid was used in the transformation reaction to obtain > 10A8 cfu. The modified minimal CRISPR operon in the pET28b backbone were under the control of T7 promoters. The transformation reaction was allowed to recover for 1 hr after which it was diluted into LB media containing carbenicillin and kanamycin and grown overnight. The following day the mixture was diluted into self-inducing Overnight Express™ Instant TB Medium (Millipore Sigma) to allow expression of the RGN and sgRNA, and grown for an additional 4h at 37C and then shifted to 30C for an additional 16h after which the cells were spun down and plasmid DNA was isolated with a Mini -prep kit (Qiagen, Germantown, MD). In the presence of the appropriate crRNA, plasmids containing a PAM that is recognizable by the RGN will be cleaved resulting in their removal from the population. Plasmids containing PAMs that are not recognizable by the RGN, or that are transformed into bacteria not containing an appropriate sgRNA, will survive and replicate.
The PAM and protospacer regions of uncleaved plasmids were PCR-amplified and prepared for sequencing following published protocols (16s-metagenomic library prep guide 15044223B, Illumina, San Diego, CA). Deep sequencing (55bp paired end reads) was performed on a NextSeq (Illumina). Typically, 1-4M reads were obtained per amplicon. PAM regions were extracted, counted, and normalized to total reads for each sample. PAMs that lead to plasmid cleavage were identified by being underrepresented when compared to controls (i.e., when the library is transformed into E. coli containing the RGN but lacking an appropriate sgRNA). To identify the PAM requirements for a novel RGN, an enrichment value was computed for each kmer as the difference between the library size-normalized read counts in the control sample and in the targeting sample. This value was rounded to the nearest integer for positive numbers and set to zero for negative numbers. Enrichment values were then summed across all kmers to yield a position frequency matrix, which was represented visually as a sequence logo using the command line utility weblogo. Those RGNs with consistency
among the most enriched kmers — sequence logo information content > 0.2 when including the top 100 enriched kmers — and with qualitatively consistent PAMs across plasmid libraries (T2 and C2) were deemed to have bonafide PAMs. The final PAM for these RGNs were obtained by summing counts across both plasmid libraries, normalizing counts, computing kmer enrichment values, summing across kmers to yield a position frequency matrix, then visually representing the PAM as a sequence logo using the command line utility weblogo.
Example 3: crRNA and tracrRNA identification
[223] Systems with an identifiable PAM were grown without the PAM library to mid-log phase, pelleted, and flash frozen. RNA was isolated from the pellets using a mirVANA miRNA Isolation Kit (Life Technologies, Carlsbad, CA), and sequencing libraries were prepared from the isolated RNA using an NEBNext Small RNA Library Prep kit (NEB, Beverly, MA). The library prep was fractionated on a 6% polyacrylamide gel to capture the RNA species less than 200nt to detect crRNAs and tracrRNAs, respectively. Deep sequencing (75 bp paired-end) was performed on a Next Seq 500 (High Output kit). Reads were quality trimmed using Cutadapt and mapped to reference genomes using Bowtie2. A custom RNAseq pipeline was written to detect the expressed small non coding RNA transcripts (Fig 2-3). Manual curation of RNAs was performed using secondary structure prediction by NUPACK, an RNA folding software to identify the boundaries of the expressed putative tracrRNA and processed crRNA. gRNA designs were then made by linking the expressed boundaries between the putative tracrRNA and the crRNA with a RAAA linker sequence.
Example 4: Demonstration of gene editing activity on endogenous targets in mammalian cells
[224] The RGN was codon optimized for human expression and cloned into expression cassettes with a Nterm SV40 NLS, and a Cterm Nucleoplasmin NLS under control of a CMV promoter for mammalian expression. Tag sequences are set forth in Table 15.
[225] Table 15. Tag sequences
[226] . The gRNA expression constructs encoding a single gRNA each under the control of a human RNA polymerase III U6 promoter were produced and introduced into an expression vector containing GFP under control of a CMV promoter. Guides were design to targeted regions of selected genes with the appropriate PAM for the system. The constructs described were introduced into mammalian cells. One day prior to transfection, HEK293T cells (Sigma) were plated in 24-well dishes in Dulbecco’s modified Eagle medium (DMEM) plus 10% (vol/vol) fetal bovine serum (Gibco) and 1% Penicillin- Streptomycin (Gibco). The next day when the cells were at 50-60% confluency, 500 ng of a RGN
expression plasmid plus 500 ng of a single gRNA expression plasmid were co-transfected using 1.5 uL of Lipofectamine 3000 (Thermo Scientific) per well, following the manufacturer’s instructions. After 48 hours of growth, total genomic DNA was harvested using a genomic DNA isolation kit (Machery-Nagel) according to the manufacturer’s instructions.
[227] The total genomic DNA was then analyzed to determine the rate of editing in the targeted gene. Oligonucleotides were produced to be used for PCR amplification and subsequent analysis of the amplified genomic target site. All PCR reactions were performed using 10 uL of 2X Master Mix Platinum SuperFi DNA polymerase (Thermo Scientific) in a 20 uL reaction including 0.5 uM of each primer specific for each guide using a program of: 98°C, 1 min; 35 cycles of [98°C, 10 sec; 65°C, 15 sec; 72°C, 30 sec]; 72°C, 5 min; 12°C, forever. Primers for PCR#2 include Nextera Read 1 and Read 2 Transposase Adapter overhang sequences for Illumina sequencing.
[228] Following the PCR amplification, DNA was cleaned using a PCR cleanup kit (Zymo) according to the manufacturer’s instructions and eluted in water. Products containing the Illumina overhang sequences underwent library preparation following the Illumina 16S Metagenomic Sequencing Library protocol. Deep sequencing was performed on an Illumina NextSeq platform. Typically, 200,000 of 150 bp paired-end reads (2 x 100,000 reads) are generated per amplicon. The reads were analyzed using CRISPResso (Pinello, et al. 2016 Nature Biotech, 34:695-697) to calculate the rates of editing. Output alignments were hand-curated to confirm insertion and deletion sites as well as identify microhomology sites at the recombination sites. The overall rates of editing for actively edited samples are shown in Table 16.
[229] Table 16. Active Eukaryotic Editing

Example 5: Demonstration of base editing activity on endogenous targets in mammalian cells
[230] The coding sequence of the identified RGN is codon-optimized for expression in mammalian cells and introduced into the expression cassette, which produces a fusion protein that includes a NLS tag at its N-terminal end operably linked to a codon optimized deaminase sequences (identified and developed in house) at its C-terminal end. The deaminase is operably linked to a flexible amino acid linker at their C-terminal end, and the amino acid linker is operably linked to the RNA guided nuclease at its C-terminal end, that has been mutated to have an inactive RuvC domain (That is, it has been
mutated into RGN that is catalytically dead). The RNA-guided DNA binding polypeptide is operably linked to a flexible amino acid linker at their C-terminal end, and the amino acid linker is operably linked to a uracil protecting peptide. The uracil protecting peptide is operably linked to a flexible amino acid linker at their C-terminal end, and the amino acid linker is operably linked to a second NLS at its C-terminal end. Each of these expression cassettes is introduced into a vector capable of driving the expression of the fusion protein in mammalian cells. A vector capable of expressing guide RNA to target the deaminase-RGN-UPP fusion protein to the determined genomic location was also produced. These guide RNAs can guide the deaminase-RGN-UPP fusion protein to the target genome sequence for base editing.
[231] Using liposome transfection, vectors capable of expressing the deaminase-RGN-UPP fusion protein and guide RNAs are transfected into HEK293T cells. For liposome transfection, the day before transfection, the cells are distributed in a 24-well plate of growth medium (DMEM + 10% fetal bovine serum + 1% penicillin/streptomycin) at 1.3* 10 5 cells/well. According to the manufacturer's instructions, use Lipofectamine® 3000 reagent (Thermo Fisher Scientific) to transfect 500 ng deaminase-RGN fusion expression vector and 500 ng guide RNA expression vector. 48-72 hours after liposome transfection, genomic DNA is harvested from the transfected cells, and the DNA is sequenced and analyzed for the presence of targeted cytosine base editing mutations using CRISPResso2 (Clement K, et al Nat Biotechnol. 2019 Mar; 37(3):224-226. doi: 10.1038/s41587-019-0032-3. PubMed PMID: 30809026).
Example 6. Improved gRNA designs
[232] Improvements to the editing ability of novel RGNs can be accomplished by changing the sgRNA scaffolds by altering the tracrRNA and crRNA linkage, removing hairpin mismatches, strengthening hairpins by swapping A:U base pairs for G:C base pairs, altering the starting position of the tracrRNA, removing non-protein contacting regions of the sgRNA to minimize the design. To this end novel sgRNA designs were developed and tested for genomic editing (Table 12). Figure 4 shows the relative activity of EGS0023 with two potential tracrRNA. Figure 5 shows an improvement in editing achieved with EGS0024 when using a modified tracrRNA (sgRNA v2) that has been truncated to remove extraneous sequence from the 5 ’ end of the tracrRNA and shorten one of the extended potential hairpins compared to the wild type tracrRNA (sgRNA vl) (Figure 6).
Claims
1. A nucleic acid targeting system, comprising a) a polypeptide comprising an RNA-guided nuclease (RGN) protein comprising an amino acid sequence that is at least 80% identical to the amino acid sequence of SEQ ID NO: 1 or 2; and b) an RNA molecule binding to RGN protein (gRNA) and targeting a nucleic acid sequence of interest, said gRNA comprising in 5’ to 3’ orientation: i. a first region that binds to the RGN; and ii. a second region comprising a spacer sequence complimentary to the target nucleic acid sequence.
2. The nucleic acid targeting system of claim 1, wherein the target nucleic acid is a DNA.
3. The nucleic acid targeting system of claim 1, wherein the first region that binds to the RGN comprises the sequence of SEQ ID NO: 3 or 4 respectively or a truncated sequence thereof.
4. The nucleic acid targeting system of claim 1, wherein the target nucleic acid sequence is adjacent to a PAM sequence.
5. The nucleic acid targeting system of claim 4, wherein the PAM sequence is on the non-target strand of the target nucleic acid.
6. The nucleic acid targeting system of claim 1, wherein said RGN polypeptide is a nickase or nuclease dead.
7. The nucleic acid targeting system of claim 1, wherein said RGN polypeptide is fused to a nuclear localization signal (NLS).
8. The nucleic acid targeting system of claim 1, wherein said RGN polypeptide is fused to a heterologous polypeptide.
9. The nucleic acid targeting system of claim 1, wherein the target nucleic acid sequence is within a eukaryotic cell.
10. The nucleic acid targeting system of claim 1, wherein said RGN polypeptide is nuclease dead, and wherein the RGN polypeptide is operably linked to a base -editing polypeptide.
11. One or more isolated polynucleotides encoding the nucleic acid targeting system of any one of claims 1-10.
12. The isolated polynucleotides of claim 11, wherein the polynucleotide sequences encoding of the nucleic acid targeting system have been codon optimized for optimal expression in a target cell or organism.
13. One or more vectors comprising one or more isolated polynucleotides of claim 11.
14. The one or more vectors of claim 13, wherein said vector is a lentiviral or an AAV vector.
15. A vector comprising polynucleotides encoding said nucleic acid targeting system of any one of claims 1-10.
16. The vector of claim 15, wherein said vector is a lentiviral or an AAV vector.
17. A cell comprising the nucleic acid targeting system of any one of claims 1-10, a polynucleotide of claim 11, or a vector of claim 15.
18. A cell according to claim 17, wherein said cell is an eucaryotic cell.
19. A composition comprising the DNA targeting system of any one of claims 1-10, one or more polynucleotides of claim 9, or one or more vectors of claim 10 or 12.
20. A method for binding to a target DNA sequence comprising contacting the DNA targeting system according to any one of claims 1-10, with said target DNA sequence or a cell comprising the target DNA sequence.
21. A method for cleaving and/or modifying a target nucleic acid sequence, comprising
1) contacting the target nucleic acid sequence with a nucleic acid targeting system of any one of claims 1-10; and
2) incubating said nucleic acid targeting system with the target nucleic acid for the time and under conditions sufficient for the cleaving and/or modification to occur.
22. The method of claim 21, wherein said target nucleic acid sequence is a DNA.
23. The method of claim 21, wherein said modified target DNA sequence comprises insertion of heterologous nucleic acid sequence into the target DNA sequence.
24. The method of claim 21, wherein said modified target DNA sequence comprises deletion of at least one nucleotide from the target DNA sequence
25. The method of claim 21, wherein said modified target DNA sequence comprises mutation of at least one nucleotide in the target DNA sequence
26. The method of claim 21, wherein the target nucleic acid sequence is within a cell.
27. The method of claim 26, wherein the cell is a eukaryotic cell.
28. The method of claim 26, further comprising culturing the cell under conditions sufficient for expression of the RGN polypeptide and selecting a cell comprising said modified target nucleic acid sequence.
29. A pharmaceutical composition comprising the nucleic acid targeting system of any one of claims 1-10, one or more polynucleotides of claim 11, or one or more vectors of claim 13 or 15.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363502250P | 2023-05-15 | 2023-05-15 | |
| US63/502,250 | 2023-05-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024235991A1 true WO2024235991A1 (en) | 2024-11-21 |
Family
ID=91186852
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2024/063269 Pending WO2024235991A1 (en) | 2023-05-15 | 2024-05-14 | Rna-guided nucleases and nucleic acid targeting systems comprising such rna-guided nucleases |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2024235991A1 (en) |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4853331A (en) | 1985-08-16 | 1989-08-01 | Mycogen Corporation | Cloning and expression of Bacillus thuringiensis toxin gene toxic to beetles of the order Coleoptera |
| US5039523A (en) | 1988-10-27 | 1991-08-13 | Mycogen Corporation | Novel Bacillus thuringiensis isolate denoted B.t. PS81F, active against lepidopteran pests, and a gene encoding a lepidopteran-active toxin |
| US5173414A (en) | 1990-10-30 | 1992-12-22 | Applied Immune Sciences, Inc. | Production of recombinant adeno-associated virus vectors |
| WO1993024640A2 (en) | 1992-06-04 | 1993-12-09 | The Regents Of The University Of California | Methods and compositions for in vivo gene therapy |
| US20030087817A1 (en) | 1999-01-12 | 2003-05-08 | Sangamo Biosciences, Inc. | Regulation of endogenous gene expression in cells using zinc finger proteins |
| US20140068797A1 (en) | 2012-05-25 | 2014-03-06 | University Of Vienna | Methods and compositions for rna-directed target dna modification and for rna-directed modulation of transcription |
| US9790490B2 (en) | 2015-06-18 | 2017-10-17 | The Broad Institute Inc. | CRISPR enzymes and systems |
| WO2018027078A1 (en) | 2016-08-03 | 2018-02-08 | President And Fellows Of Harard College | Adenosine nucleobase editors and uses thereof |
| WO2020139783A2 (en) * | 2018-12-27 | 2020-07-02 | Lifeedit, Inc. | Polypeptides useful for gene editing and methods of use |
| WO2023028444A1 (en) * | 2021-08-27 | 2023-03-02 | Mammoth Biosciences, Inc. | Effector proteins and methods of use |
-
2024
- 2024-05-14 WO PCT/EP2024/063269 patent/WO2024235991A1/en active Pending
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4853331A (en) | 1985-08-16 | 1989-08-01 | Mycogen Corporation | Cloning and expression of Bacillus thuringiensis toxin gene toxic to beetles of the order Coleoptera |
| US5039523A (en) | 1988-10-27 | 1991-08-13 | Mycogen Corporation | Novel Bacillus thuringiensis isolate denoted B.t. PS81F, active against lepidopteran pests, and a gene encoding a lepidopteran-active toxin |
| US5173414A (en) | 1990-10-30 | 1992-12-22 | Applied Immune Sciences, Inc. | Production of recombinant adeno-associated virus vectors |
| WO1993024640A2 (en) | 1992-06-04 | 1993-12-09 | The Regents Of The University Of California | Methods and compositions for in vivo gene therapy |
| US20030087817A1 (en) | 1999-01-12 | 2003-05-08 | Sangamo Biosciences, Inc. | Regulation of endogenous gene expression in cells using zinc finger proteins |
| US20140068797A1 (en) | 2012-05-25 | 2014-03-06 | University Of Vienna | Methods and compositions for rna-directed target dna modification and for rna-directed modulation of transcription |
| US9790490B2 (en) | 2015-06-18 | 2017-10-17 | The Broad Institute Inc. | CRISPR enzymes and systems |
| WO2018027078A1 (en) | 2016-08-03 | 2018-02-08 | President And Fellows Of Harard College | Adenosine nucleobase editors and uses thereof |
| WO2020139783A2 (en) * | 2018-12-27 | 2020-07-02 | Lifeedit, Inc. | Polypeptides useful for gene editing and methods of use |
| WO2023028444A1 (en) * | 2021-08-27 | 2023-03-02 | Mammoth Biosciences, Inc. | Effector proteins and methods of use |
Non-Patent Citations (38)
| Title |
|---|
| "Molecular Cloning: A Laboratory Manual", 2001, COLD SPRING HARBOR PRESS |
| "Nucleases", 1993, COLD SPRING HARBOR LABORATORY PRESS |
| ABUDAYYEH ET AL., SCIENCE, vol. 353, 2016, pages 5573 |
| AUSUBEL ET AL.: "Current Protocols in Molecular Biology", 2003, JOHN WILEY & SONS |
| BRINER ET AL.: "GRNA Functional Modules Direct Cas9 Activity and Orthogonality", MOLECULAR CELL, vol. 56, 2014, pages 333 - 339 |
| BUCHSCHER ET AL., J. VIRAL., vol. 66, 1992, pages 1635 - 1640 |
| CARRIESMALL, BIOCHIM BIOPHYS ACTA, vol. 1833, 2013, pages 253 - 259 |
| CHYOU ET AL., RNA BIOLOGY, vol. 16, no. 4, 2019, pages 423 - 434 |
| CLEMENT K ET AL., NAT BIOTECHNOL., vol. 37, no. 3, March 2019 (2019-03-01), pages 224 - 226 |
| GAO, GENE THERAPY, vol. 2, 1995, pages 710 - 722 |
| GAUDELLI ET AL., NATURE, vol. 551, 2017, pages 464 - 471 |
| GRUBER ET AL., CELL, vol. 106, no. 1, 2008, pages 23 - 24 |
| GUSCHIN ET AL., METHODS MOL BIOL, vol. 649, 2010, pages 247 - 256 |
| HERRMANNNEUPERT, IUBMB LIFE, vol. 55, 2003, pages 219 - 225 |
| JINEK M ET AL.: "A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity", SCIENCE, vol. 337, 2012, pages 816 - 821, XP055229606, DOI: 10.1126/science.1225829 |
| KARVELIS ET AL., GENOME BIOL, vol. 16, 2015, pages 253 |
| KLEINSTIVER ET AL., NATURE, vol. 523, 2015, pages 481 - 485 |
| LANGE ET AL., J. BIOL. CHEM., vol. 282, 2007, pages 5101 - 5105 |
| MAKAROVA ET AL., NATURE REVIEWS MICROBIOLOGY, vol. 13, 2015, pages 1 - 15 |
| MCCORMICK ET AL., PLANT CELL REPORTS, vol. 5, 1986, pages 81 - 84 |
| MILLER ET AL., VIRAL, vol. 65, pages 2220 - 2224 |
| MILLETTI F, DRUG DISCOV TODAY, vol. 17, 2012, pages 850 - 860 |
| NASSOURYMORSE, BIOCHIM BIOPHYS ACTA, vol. 1743, 2005, pages 5 - 19 |
| NISHIMASU ET AL., CELL, 2014 |
| PINELLO ET AL., NATURE BIOTECH, vol. 34, 2016, pages 695 - 697 |
| RAY ET AL., BIOCONJUG CHEM, vol. 26, no. 6, 2015, pages 1004 - 7 |
| SOIL, CURR OPIN PLANT BIOL, vol. 5, 2002, pages 529 - 535 |
| SOMMNERFELT ET AL., VIRAL., vol. 176, 1990, pages 58 - 59 |
| STEMMER, PROC. NATL. ACAD. SCI., 1994 |
| STRECKER ET AL., NAT COMMUN, vol. 10, 2019, pages 212 |
| SWARTS ET AL., MOL. CELL, vol. 66, 2017, pages 221 - 233 |
| WANG MENG ET AL: "Diverse Class 2 CRISPR Effectors as Active Nucleases with Expanded Targeting Capabilities", THE CRISPR JOURNAL, vol. 7, no. 2, 1 April 2024 (2024-04-01), pages 120 - 130, XP093179106, ISSN: 2573-1599, DOI: 10.1089/crispr.2023.0058 * |
| WANG: "Supplemental Table 2 Coding sequence of proteins tested in Eukaryotic cells - Diverse Class 2 CRISPR Effectors as Active Nucleases with Expanded Targeting Capabilities", THE CRISPR JOURNAL, 16 April 2024 (2024-04-16), XP093179214, Retrieved from the Internet <URL:https://www.liebertpub.com/doi/suppl/10.1089/crispr.2023.0058/suppl_file/suppl_tables1-s4.zip> * |
| WILSON ET AL., J. VIRAL, vol. 63, 1989, pages 2374 - 2378 |
| WOJCIK ET AL., INT. J. MOLEC. SCI., vol. 16, 2015, pages 24918 - 24945 |
| ZETSCHE ET AL., CELL, vol. 163, 2015, pages 759 - 771 |
| ZHANG ET AL., CHEM. SCI., vol. 7, 2016, pages 4951 - 4957 |
| ZUKERSTIEGLER, NUCLEIC ACIDS RES., vol. 9, 1981, pages 133 - 148 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11555181B2 (en) | Engineered cascade components and cascade complexes | |
| AU2019282149B2 (en) | RNA-guided nucleases and active fragments and variants thereof and methods of use | |
| US10633642B2 (en) | Engineered CRISPR-Cas9 nucleases | |
| US10519454B2 (en) | Genome editing using Campylobacter jejuni CRISPR/CAS system-derived RGEN | |
| DK2784162T3 (en) | Design of systems, methods and optimized control manipulations for sequence manipulation | |
| EP4013864A1 (en) | Rna-guided nucleases and active fragments and variants thereof and methods of use | |
| CA3128876A1 (en) | Methods of editing a disease-associated gene using adenosine deaminase base editors, including for the treatment of genetic disease | |
| KR20190005801A (en) | Target Specific CRISPR variants | |
| KR20190082318A (en) | CRISPR / CPF1 system and method | |
| KR20160034901A (en) | Optimized crispr-cas double nickase systems, methods and compositions for sequence manipulation | |
| KR20160030187A (en) | Delivery and use of the crispr-cas systems, vectors and compositions for hepatic targeting and therapy | |
| KR20160044457A (en) | Delivery, engineering and optimization of tandem guide systems, methods and compositions for sequence manipulation | |
| CN114786733A (en) | Efficient DNA base editor for targeted genome modification mediated by RNA-aptamer recruitment and uses thereof | |
| EP4085133A1 (en) | Rna-guided nucleases and active fragments and variants thereof and methods of use | |
| JP2024501892A (en) | Novel nucleic acid-guided nuclease | |
| WO2024235991A1 (en) | Rna-guided nucleases and nucleic acid targeting systems comprising such rna-guided nucleases | |
| WO2024042165A2 (en) | Novel rna-guided nucleases and nucleic acid targeting systems comprising such rna-guided nucleases | |
| WO2024042168A1 (en) | Novel rna-guided nucleases and nucleic acid targeting systems comprising such rna-guided nucleases | |
| WO2024038168A1 (en) | Novel rna-guided nucleases and nucleic acid targeting systems comprising such | |
| WO2025003358A2 (en) | Novel nucleic acid targeting systems comprising rna-guided nucleases | |
| CA2957441C (en) | Genome editing using campylobacter jejuni crispr/cas system-derived rgen | |
| JP2026504733A (en) | Guide RNAs targeting the TRAC gene and methods of use | |
| EP4634384A1 (en) | Guide rnas that target trac gene and methods of use | |
| HK1240617A1 (en) | Genome editing using campylobacter jejuni crispr/cas system-derived rgen | |
| HK1240617B (en) | Genome editing using campylobacter jejuni crispr/cas system-derived rgen |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24727208 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |