WO2024226890A1 - Pyrazole 3h-imidazo(4,5-b)pyridine compounds and uses thereof - Google Patents
Pyrazole 3h-imidazo(4,5-b)pyridine compounds and uses thereof Download PDFInfo
- Publication number
- WO2024226890A1 WO2024226890A1 PCT/US2024/026398 US2024026398W WO2024226890A1 WO 2024226890 A1 WO2024226890 A1 WO 2024226890A1 US 2024026398 W US2024026398 W US 2024026398W WO 2024226890 A1 WO2024226890 A1 WO 2024226890A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- compound
- mmol
- tolyl
- morpholino
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Definitions
- the present disclosure provides compounds that are phosphoinositide kinase inhibitors, in particular FYVE-type finger-containing phosphoinositide kinase (“PIKfyve”) inhibitors and are therefore useful for the treatment of central nervous system diseases. Also provided are pharmaceutical compositions containing such compounds and processes for preparing such compounds.
- PIKfyve FYVE-type finger-containing phosphoinositide kinase
- Phosphoinositide kinases catalyze the phosphory lation of phosphatidy linositol, which is a component of eukaryotic cell membranes, and related phospholipids called phosphoinositides. Phosphoinositides are involved in the regulation of diverse cellular processes, including cellular proliferation, survival, cytoskeletal organization, vesicle trafficking, glucose transport, and platelet function. Fruman et al., “Phosphoinositide Kinases,” Ann. Review. Biochem. 1998, 67, 481-507. Phosphorylated derivatives of phosphatidylinositol regulate cytoskeletal functions, membrane trafficking, and receptor signaling by recruiting protein complexes to cell and endosomal membranes.
- PIKs Phosphoinositide kinases
- FYVE-type finger-containing phosphoinositide kinase (PIKfyve; also known as phosphatidylinositol-3-phosphate 5-kinase type III or PIPKIII) is a ubiquitously expressed PIK with both lipid and protein kinase activity. In its capacity as a lipid kinase, the enzyme phosphorylates the D-5 position in endosomal phosphatidylinositol and phosphatidylinositol-3- phosphate (PI3P) to generate the corresponding 5-phosphate phospholipid analogs. Shisheva et al., Cell Biol. Int. 2008, 32(6), 591.
- PI3P is found in cell membranes with roles in protein trafficking, protein degradation, and autophagy. Nascimbeni et al., FEBSJ. 2017, 284. 1267- 1278.
- PIKfyve regulates endomembrane homeostasis and plays a role in the biogenesis of endosome carrier vesicles from early endosomes. The enlarged endosome/lysosome structure was observed in cells expressing PIKfyve dominant negative or siRNA. Ikonomov et al., J. Biol. Chem. 2001, 276(28), 26141-26147; Rutherford et al. , J. Cell Sci. 2006, 119, 3944-3957.
- Phosphorylated inositides produced by PIKfyve are localized in various cellular membranes and organelles, consistent with the various PIKfy ve functions of endolysosomal transport, endomembrane homeostasis, and biogenesis of endosome carrier vesicles (ECV)/multivesicular bodies (MVB) from early endosomes. Further, PIKfyve is required for endocytic-vacuolar pathway and nuclear migration. Thus, PIKfyve helps maintain proper morphology of the endosome and lysosome.
- FIG4 phosphoinositide 5-phosphatase
- Inhibition of PIKfyve would mimic overexpression of FIG4, thereby increasing levels of PI3P, stimulating autophagy, and improving motor neuron health.
- Numerous diseases are correlated with FIG4 deficiencies, such as deleterious FIG4 mutations or diminished FIG4 function, and are therefore suitable as target diseases for treatment with PIKfyve inhibitors, including amyotrophic lateral sclerosis (ALS), primary lateral sclerosis (PLS), Charcot-Marie-Tooth (including type 4J (CMT4J)), and Yunis- Varon syndrome.
- ALS amyotrophic lateral sclerosis
- PLS primary lateral sclerosis
- CMT4J Charcot-Marie-Tooth
- Yunis- Varon syndrome including type 4J (CMT4J)
- Exemplary diseases associated with FIG4 deficiencies are amyotrophic lateral sclerosis (ALS), primary lateral sclerosis (PLS), Charcot-Marie-Tooth (including type 4J (CMT4J)), Yunis-Varon syndrome, poly microgyri a (including polymicrogyria with seizures), temporo- occipital polymicrogyria, Pick’s disease, Parkinson's disease, Parkinson's disease with Lewy bodies, dementia with Lewy bodies.
- ALS amyotrophic lateral sclerosis
- PLS primary lateral sclerosis
- CMT4J Charcot-Marie-Tooth
- Yunis-Varon syndrome Yunis-Varon syndrome
- poly microgyri a including polymicrogyria with seizures
- temporo- occipital polymicrogyria Pick’s disease
- Parkinson's disease Parkinson's disease with Lewy bodies, dementia with Lewy bodies.
- Lewy body disease fronto-temporal dementia, diseases of neuronal nuclear inclusions of poly glutamine and intranuclear inclusion bodies, disease of Marinesco and Hirano bodies, Alzheimer’s disease, neurodegeneration, spongiform neurodegeneration, autophagy, peripheral neuropathy, leukoencephalopathy, motor neuropathy, sensory neuropathy. Bharadwaj et al., Hum. Mol. Genet. 2016, 25(4). 682-692.
- PIKfyve inhibitors are useful in a range of neurological disorders, such as tauopathies (including but not limited to Alzheimer's disease, progressive supranuclear palsy, corticobasal syndrome, frontotemporal dementias, and chronic traumatic encephalopathy), traumatic brain injury’ (TBI), cerebral ischemia, ALS, fronto-temporal dementia (FTD), Guillain-Barre Syndrome, chronic inflammatory demyelinating polyneuropathy, multiple sclerosis, CMT.
- lysosomal storage diseases including but not limited to Fabry's disorder, Gaucher's disorder, Niemann Pick C, Tay-Sachs, and Mucolipidosis type IV), as well as several types of neuropathies.
- PIKfyve inhibitors include Huntington's disease and psychiatric disorders (such as ADHD, schizophrenia, mood disorders including but not limited to major depressive disorder, bipolar disorder I, and bipolar disorder II).
- Gardiner et al. “Prevalence of carriers of intermediate and pathological polyglutamine disease-associated alleles among large population-based cohorts,"’ JAMA Neurol. 2019, 76(6), 650-656; PCT Publ. No. WO2016/210372; US Publ. No. US2018/0161335. Summary
- Embodiment 1 is a compound of Formula (I):
- W is N, CH, or CR 4 ‘;
- R is H, oxo, alkyl, alkenyl, heteroaryl, heterocyclyl, or carboxamide, each substituted with 0, 1, or 2 R x groups and each R x is oxo, hydroxyl, cyano, substituted or unsubstituted alkyd, substituted or unsubstituted cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted amino, substituted or unsubstituted ester, substituted or unsubstituted sulfonyl, substituted or unsubstituted amide, substituted or unsubstituted carboxamide, substituted or unsubstituted alkoxy, or substituted or unsubstituted alkoxyalkyl;
- R 1 is H, alkyl, cycloalkyl, heterocyclyl, amino, alkoxy, alkoxyalkyl, amide, carboxamide, or sulfonyl, each substituted with 0, 1, or 2 R y groups and each R y is substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted alkoxy, substituted or unsubstituted alkoxyalkyl, substituted or unsubstituted amino, substituted or unsubstituted ester, substituted or unsubstituted sulfonyl, substituted or unsubstituted amide, substituted or unsubstituted carboxamide, substituted or unsubstituted urea, substituted or unsubstituted carb
- R 4 and R 4 * are aryl, lieteroar l. or heterocyclyl, each substituted with 0, 1, or 2 R z groups and each R z is alkyl, cycloalkyl, haloalkyl. alkoxy, haloalkoxy, halo, or -CN, provided that only one of R 4 and R 4 * is present;
- R 5 is H, carboxyl, substituted or unsubstituted alkyl, substituted or unsubstituted alkoxy carbonyl, substituted or unsubstituted ester, or substituted or unsubstituted carboxamide;
- R 6 is absent, H, or alkyl
- R 7 and R 8 are each independently H, Ci-6 alkyl or R 7 and R 8 join together to form a fused or bridging ring system;
- — represents a single or double bond; x is 0, 1 or 2;
- R 9 is absent or oxo; and pharmaceutically acceptable salts, solvates, and prodrugs thereof; provided that when R 6 is absent, R 7 and R 8 are
- R 4 is /n-tolyl, then R is not substituted or unsubstituted piperidinyl; substituted or unsubstituted pyrrolidinyl; -CH 2 OH; -CH 2 (substituted or unsubstituted heterocyclyl); or C(O)NR*R** wherein R* and R** join to form a substituted or unsubstituted heterocyclic group.
- Embodiment 2 is the compound of embodiment 1 , wherein R is H, oxo, substituted or unsubstituted Ci-6 alkyl, substituted or unsubstituted Ci-6 alkenyl, substituted or unsubstituted monocyclic heteroaryl, substituted or unsubstituted monocyclic heterocyclyl. or R a R b NC(O)-; and each R a and R b is independently H, Ci-6 alkyL Ci-shaloalkyl, Ci-6 cycloalkyl, or CH 3 C(O)-.
- Embodiment 3 is the compound of embodiment 2, wherein each R a and R b is independently H, cyclopropyl, cyclobutyl, methyl, ethyl, CH 3 C(O)-, -CD 3 , or -CFs.
- Embodiment 4 is the compound of embodiment 1 or 2, wherein R is H, oxo, methyl, ethyl, propyl, isopropyl, isobutyl, propenyl, pyridyl, pyrimidinyl, pyrazolyl, dioxanyl, oxetanyl, morpholino, 3,6-dihydro-2H-pyranyl, or R a R b NC(O)-.
- Embodiment 5 is the compound of embodiment 1 or 2, wherein R is H.
- Embodiment 6 is the compound of any one of embodiments 1-5, wherein R x is monocyclic heteroaryl, monocyclic heterocyclyl, Ci-6 alkyl. Ci-s cycloalkyl, R a R b N, R a O-, R c , R a R b N-(Ci-6 alkyl)-, R a O-(Ci-6 alkyl)-, or R C O-(CI-6 alkyl)-; and R c is H, methyl, ethyl, HOC(O)CH 2 -, CH 3 OC(O)CH 2 -, (CH 3 ) 3 COC(O)CH 2 -, (CH 3 ) 3 COC(O)NH-, H 2 NC(O)CH 2 -,
- Embodiment 7 is the compound of any one of embodiments 1-5, wherein R x is methyl, cyclopropyl, cyclobutyl, pyridyl, pyrazolyl, morpholino, piperidinyl, piperazinyl, R a R b N, R a O-, R c , R a R b N-ethyl, R c O-ethyl-, R c O-methyl-, or R c O-isopentyl.
- R x is methyl, cyclopropyl, cyclobutyl, pyridyl, pyrazolyl, morpholino, piperidinyl, piperazinyl, R a R b N, R a O-, R c , R a R b N-ethyl, R c O-ethyl-, R c O-methyl-, or R c O-isopentyl.
- Embodiment 8 is the compound of any one of embodiments 1-7, wherein R 1 is H, Ci-6 alkyl, C i-6 cycloalkyl, monocyclic heterocyclyl, R a O-, R a O-(Ci-6 alkyd), R a R b NC(O)-, or alkyl- SO2-.
- Embodiment 9 is the compound of any one of embodiments 1-7, wherein R 1 is H, propyl, ethyl, methyl, isobutyl, cyclobutyl, cyclopropyl. azetidinyL oxetanyl. piperidinyl, tetrahydropyran, tetrahydrofuran, thietane 1,1 dioxide, hexahydropyrimidin-2-one, R a O-, R a O- ethyl, R a O-methyl, R a R b NC(O)-, CH3CH2SO2-, (CH 3 ) 2 CHSO 2 -, or cyclopropyl-SCh-.
- Embodiment 10 is the compound of any one of embodiments 1-9, wherein R y is C1-6 alkyl, C1-6 alkenyl. C1-6 haloalkyl, C1-6 cycloalkyl, monocyclic heteroaryl, monocyclic aryl, monocyclic heterocyclyl, halo, -CN, -OH, R a O-, CH3SO2-.
- R d is methyl, oxo, CHsC(O)-, CH 3 OC(O)-, CH 3 SO 2 -, -F, (CH 3 ) 3 CO-, (CD 3 ) 3 CO-, R a O-, -CN, NH 2 , (CH 3 ) 3 COC(O)-, H 2 NC(O)-, CH 3 HNC(O)-.
- Embodiment 11 is the compound of any one of embodiments 1-9, wherein R y is methyl, cyclopropyl, cyclobutyl, pyridinyl, phenyl, piperidinyl, pyrrolidinyl, oxetanyl, pyrrolidinone, imidazolidin-2-one, oxazolidin-2-one, lH-l,2,4-triazol-5-one, tetrahydropyran, dioxanyl, azetidinyl, -CN, -F,
- Embodiment 12 is the compound of any one of embodiments 1-11, wherein R 4 is H, phenyl, pyrazolyl, thiazolyl, thienyl, oxabicyclo[2.1.1]hexanyl, and norbomanyl, each substituted by 0, 1. or 2 R z wherein each R z is -CN, halo, C1-6 alkyl, C1-6 alkoxy, C1-6 haloalkyl, C1-6 cycloalkyl, deuterated alkyl, or C1-6 haloalkoxy.
- Embodiment 13 is the compound of embodiment 12, wherein R 4 is H, phenyl, pyrazolyl, thiazolyl, thienyl, oxabicyclo[2. l. l]hexanyl, or norbomanyl.
- Embodiment 14 is the compound of embodiment 12 or 13, wherein each R z is -Cl, -F, - CN, -CF 3 , -CDS, -OCF 3 , methyl, ethyl, cyclopropyl, methoxy, difluoromethoxy, or difluoromethyl.
- Embodiment 15 is the compound of any one of embodiments 1-14, wherein each R 5 is - C(O)NR a R b , -C(O)OR a , unsubstituted C1-6 alkyl, or C1-6 alkyl substituted with 1 or 2 -OR a or - NH 2 , -NH-R a , -NR a R b groups.
- Embodiment 16 is the compound of any one of embodiments 1-14, wherein each R 5 is H, hydroxymethyl, hydroxyethyl, methoxymethyl, H 2 N-methyl, CH 3 NH-methyl, or (CH 3 ) 2 N- methyl, trideuteriomethoxymethyl, -C(O)NR a R b , -C(O)OR a .
- Embodiment 17 is the compound of any one of embodiments 1-16, wherein R 6 is absent.
- Embodiment 18 is the compound of any one of embodiments 1-176, wherein R is not substituted with an R x group.
- Embodiment 19 is the compound of any one of embodiments 1-17, wherein R is substituted with 1 R x group.
- Embodiment 20 is the compound of any one of embodiments 1-17, wherein R is substituted with 2 R x groups.
- Embodiment 21 is the compound of any one of embodiments 1-20, wherein R 1 is not substituted with an R y group.
- Embodiment 22 is the compound of any one of embodiments 1-20, wherein R 1 is substituted with 1 R y group.
- Embodiment 23 is the compound of any one of embodiments 1-20, wherein R 1 is substituted with 2 R y groups.
- Embodiment 24 is the compound of any one of embodiments 10-23, wherein R y is not substituted with an R d group.
- Embodiment 25 is the compound of any one of embodiments 10-23, wherein R y is substituted with 1 R d group.
- Embodiment 26 is the compound of any one of embodiments 10-23, wherein R y is substituted with 2 R d groups.
- Embodiment 27 is the compound of any one of embodiments 1-26, wherein R 4 is not substituted with an R z group.
- Embodiment 28 is the compound of any one of embodiments 1-26, wherein R 4 is substituted with 1 R z group.
- Embodiment 29 is the compound of any one of embodiments 1-26, wherein R 4 is substituted with 2 R z groups.
- Embodiment 30 is the compound of any one of embodiments 1-29, wherein W is CH.
- Embodiment 31 is the compound of any one of embodiments 1-30, wherein R 7 and R 8 are each H.
- Embodiment 32 is the compound of any one of embodiments 1-30, wherein R 7 and R 8 join together to form a 1 or 2 carbon bridged rin g system.
- Embodiment 33 is the compound of embodiment 1, having the structure of Formula (Ic) and pharmaceutically acceptable salts, solvates, and prodrugs thereof.
- Embodiment 34 is the compound of embodiment 33, wherein R is H.
- Embodiment 35 is the compound of embodiment 33, wherein R 4 is m-tolyl or m- chlorophenyl.
- Embodiment 36 is the compound of embodiment 33, wherein R 1 is H, methyl, ethyl or isopropyl substituted with 1 R y group and R y is substituted with 1 or 2 R d groups.
- Embodiment 37 is the compound of embodiment 33, having the structure of Formula (Ic- pharmaceutically acceptable salts, solvates, and prodrugs thereof wherein n is 0, 1 or 2; and each R x is independently halo, cyano, C1-3 alkyl, C1-3 haloalkyl, C3-6 cycloalkyl, C1-3 alkoxy, or C1-3 haloalkoxy.
- Embodiment 38 is the compound of embodiment 37, wherein n is 1 or 2; and each R x is independently F, Cl, cyano, methyl, fluoromethyl, difluoromethyl, trifluoromethyl, ethyl, fluoroethyl, difluoroethyl, trifluoroethyl, cyclopropyl, methoxy, fluoromethoxy, difluoromethoxy, trifluoromethoxy, ethoxy, fluoroethoxy, difluoroethoxy, or trifluoroethoxy.
- Embodiment 39 is the compound of embodiment 37, wherein R 1 is H, methyl, ethyl or isopropyl substituted with 1 R y group and R y is substituted with 1 or 2 R d groups.
- Embodiment 40 is a compound selected from the compounds in Table 1 and pharmaceutically acceptable salts thereof or a compound selected from
- Embodiment 41 is a compound and/or a pharmaceutically acceptable salt of any one of embodiments 1-40, wherein one or more hydrogen atoms attached to carbon atoms of the compound are replaced by deuterium atoms.
- Embodiment 42 is a pharmaceutical composition comprising a compound and/or a pharmaceutically acceptable salt of any one of embodiments 1-41 and a pharmaceutically acceptable excipient.
- Embodiment 43 is a method of inhibiting PIKf ve kinase in a subject in need thereof comprising administering to the subject an effective amount of a compound of any one of embodiments 1-41, or a pharmaceutical composition of embodiment 42.
- Embodiment 44 is a method of treating a neurological disease associated with PIKfyve activity in a subject in need thereof comprising administering to the subject an effective amount of a compound of any one of embodiments 1-51, or a pharmaceutical composition of embodiment 52.
- Embodiment 45 is the method of embodiment 44, wherein the neurological disease is amyotrophic lateral sclerosis (ALS), primary' lateral sclerosis (PLS), Charcot-Marie-Tooth (CMT; including ty pe 4J (CMT4J)), and Yunis-Varon syndrome, autophagy, polymicrogyria (including polymicrogyria with seizures), temporo-occipital polymicrogyria.
- ALS amyotrophic lateral sclerosis
- PLS primary' lateral sclerosis
- CMT Charcot-Marie-Tooth
- Yunis-Varon syndrome autophagy
- polymicrogyria including polymicrogyria with seizures
- temporo-occipital polymicrogyria is the method of embodiment 44, wherein the neurological disease is amyotrophic lateral sclerosis (ALS), primary' lateral sclerosis (PLS), Charcot-Marie-Tooth (CMT; including ty
- Parkinson’s disease Parkinson’s disease with Lewy 7 bodies, dementia with Lewy bodies, Lewy' body disease, fronto-temporal dementia, diseases of neuronal nuclear inclusions of polyglutamine and intranuclear inclusion bodies, disease of Marinesco and Hirano bodies, tauopathy, Alzheimer’s disease, neurodegeneration, spongiform neurodegeneration, peripheral neuropathy, leukoencephalopathy, inclusion body disease, progressive supranuclear palsy, corticobasal syndrome, chronic traumatic encephalopathy, traumatic brain injury' (TBI), cerebral ischemia, Guillain-Barre Syndrome, chronic inflammatory' demyelinating polyneuropathy, multiple sclerosis, a lysosomal storage disease, Fabry’s disorder, Gaucher's disorder, Niemann Pick C disease, Tay-Sachs disease, and Mucolipidosis type IV, neuropathy. Huntington’s disease, a psychiatric disorder, ADHD, schizophrenia, a mood disorder, major depressive
- Embodiment 46 is the method of embodiment 44, wherein the disease is ALS, FTD, Alzheimer’s disease. Parkinson’s disease. Huntington’s disease, or CMT.
- Embodiment 47 is the method of embodiment 44, wherein the disease is ALS.
- Embodiment 48 is the method of embodiment 44, wherein the disease is a tauopathy such as Alzheimer’s disease, progressive supranuclear palsy, corticobasal syndrome, frontotemporal dementia, or chronic traumatic encephalopathy.
- a tauopathy such as Alzheimer’s disease, progressive supranuclear palsy, corticobasal syndrome, frontotemporal dementia, or chronic traumatic encephalopathy.
- Embodiment 49 is the method of embodiment 44, wherein the disease is a lysosomal storage disease such as Fabry’s disorder, Gaucher's disorder, Niemann Pick C disease, Tay- Sachs disease, or Mucolipidosis type IV.
- Fabry Fabry’s disorder
- Gaucher's disorder Gaucher's disorder
- Niemann Pick C disease Tay- Sachs disease
- Mucolipidosis type IV Mucolipidosis type IV.
- Embodiment 50 is the method of embodiment 44, wherein the disease is a psychiatric disorder such as ADHD, schizophrenia, or mood disorders such as major depressive disorder, depression, bipolar disorder I, or bipolar disorder II.
- a psychiatric disorder such as ADHD, schizophrenia, or mood disorders such as major depressive disorder, depression, bipolar disorder I, or bipolar disorder II.
- Embodiment 51 is a compound of any one of embodiments 1-41 for use as a medicament.
- Embodiment 52 is the compound of embodiment 51, wherein the compound is for use in treating a neurological disease treatable by inhibition of PIKlyve kinase.
- Embodiment 53 is the use of a compound of any one of embodiments 1-41 in the manufacture of a medicament for treating a neurological disease in a subject in which PIKl ve contributes to the pathology and/or symptoms of the disease.
- the term “about” refers to a value or composition that is within an acceptable error range for the particular value or composition as determined by one of ordinary’ skill in the art, which will depend in part on how the value or composition is measured or determined, i.e., the limitations of the measurement system.
- “about” or “approximately” can mean within one or more than one standard deviation per the practice in the art.
- “about” or “approximately” can mean a range of up to 10% (i.e., ⁇ 10%) or more depending on the limitations of the measurement system.
- about 5 mg can include any number between 4.5 mg and 5.5 mg.
- the terms can mean up to an order of magnitude or up to 5-fold of a value.
- the meaning of “about” or “approximately” should be assumed to be within an acceptable error range for that particular value or composition.
- Reference to "about” a value or parameter herein includes (and describes) embodiments that are directed to that value or parameter per se.
- B, and/or C is intended to encompass each of the following aspects: A, B, and C; A, B, or C; A or C; A or B; B or C; A and C; A and B; B and C; A (alone); B (alone); and C (alone).
- the terms “or a combination thereof’ and “or combinations thereof’ as used herein refers to any and all permutations and combinations of the listed terms preceding the term.
- “A, B, C, or combinations thereof’ is intended to include at least one of: A, B, C, AB, AC, BC, or ABC, and if order is important in a particular context, also BA, CA, CB, ACB, CBA, BCA, BAC, or CAB.
- a dash (“-”) that is not between two letters or symbols is used to indicate a point of attachment for a substituent.
- -C(O)NH2 is attached through the carbon atom.
- a dash at the front or end of a chemical group is a matter of convenience; chemical groups may be depicted with or without one or more dashes without losing their ordinary meaning.
- a wavy line or a dashed line drawn through a line in a Formula indicates a specified point of attachment of a group. Unless chemically or structurally required, no directionality or stereochemistry is indicated or implied by the order in which a chemical group is written or named.
- Cu-v indicates that the following group has from u to v carbon atoms.
- Ci-6 alkyl indicates that the alkyl group has from 1 to 6 carbon atoms.
- Alkyl means a linear saturated monovalent hydrocarbon radical of one to ten carbon atoms or a branched saturated monovalent hydrocarbon radical of three to ten carbon atoms, e.g., methyl, ethyl, propyl, 2-propyl. butyl (including all isomeric forms), pentyl (including all isomeric forms), and the like.
- Alkylene means a linear saturated divalent hydrocarbon radical of one to ten carbon atoms or a branched saturated divalent hydrocarbon radical of three to ten carbon atoms unless otherwise stated e.g., methylene, ethylene, propylene, 1 -methylpropylene, 2-methylpropylene, butylene, pentylene, and the like.
- Alkenyl means a linear unsaturated or partially unsaturated monovalent hydrocarbon radical of two to ten carbon atoms or a branched unsaturated or partially unsaturated monovalent hydrocarbon radical of three to ten carbon atoms, e.g., ethylenyl, propylenyl. 2-propylenyl, butenyl (including all isomeric forms), pentenyl (including all isomeric forms), and the like.
- C2- x alkenyl refers to an alkenyl group with from 2 to x carbon atoms.
- Alkylsulfonyl means a-SChR radical where R is alkyl as defined above, e.g., methylsulfonyl, ethyl sulfonyl, and the like.
- amino means a -NH2.
- Substituted amino means -NR*R**, where the R* and R** substituents are alkyl or substituted alkyl as defined herein.
- Alkoxy means a -OR radical where R is alky l as defined above, e g., methoxy, ethoxy, propoxy, or 2-propoxy, n-, iso-, or /m-butoxy. and the like.
- Alkoxyalkyl means a linear monovalent hydrocarbon radical of one to ten carbon atoms or a branched monovalent hydrocarbon radical of three to ten carbons substituted with an alkoxy group, (in one embodiment one or two alkoxy groups), as defined above, e.g., 2- methoxyethyl, 1-, 2-. or 3 -methoxy propyl. 2-ethoxyethyl, and the like.
- Alkoxy carbonyl means a -C(O)OR radical where R is alkyl as defined above, e.g., methoxycarbonyl, ethoxycarbonyl, and the like.
- Acyl means a -C(O)R radical where R is alkyl, haloalky 1, or cycloalkyl, e.g., acetyd, propionyl, cyclopropylcarbonyl, and the like.
- R is alky l
- the radical is also referred to herein as alkylcarbonyl.
- Aryl refers to an aromatic carbocyclic group having a single ring (e.g., monocyclic) or multiple rings (e.g., bicyclic or tricyclic) including fused systems.
- aryl has 6 to 20 ring carbon atoms (i.e., C6-20 ary l), 6 to 18 carbon ring atoms (i.e., Ce-18 ary l), 6 to 12 carbon ring atoms (i.e., C6-12 aryl) or 6 to 10 carbon ring atoms (i.e., Ce-io aryl).
- Examples of aryl groups include phenyl, naphthyl, fluorenyl and anthryl.
- Aryl does not encompass or overlap in any way with heteroaryl defined below. If one or more aryl groups are fused with a heteroary l, the resulting ring system is heteroary l. If one or more aryl groups are fused with a heterocycly 1, the resulting ring system is heterocyclyl.
- Cycloalkyl means a cyclic saturated monovalent hydrocarbon radical of three to ten carbon atoms wherein one or two carbon atoms may be replaced by an oxo group, e.g., cyclopropy 1, cyclobutyl, cyclopentyl, or cyclohexyl, and the like.
- Carboxy means -C(O)OH.
- Carboxamide means - C(O)NR*R**.
- Carboxamide means -OC(O)NR*R**.
- Halo means fluoro, chloro, bromo, or iodo; in one embodiment fluoro or chloro.
- Haloalkyl means alkyl radical as defined above, which is substituted with one or one to five halogen atoms (in one embodiment fluorine or chlorine,) including those substituted with different halogens, e.g., -CH2CI. -CF3. -CHF2. -CH2CF3, -CF2CF3, -CF(CH3)2, and the like. When the alkyl is substituted with only fluoro, it can be referred to in this disclosure as fluoroal kyl.
- Haloalkoxy means a -OR radical where R is haloalkyl as defined above e.g., -OCF3, -OCHF2, and the like.
- R is haloalkyl where the alkyl is substituted with only fluoro, it can be referred to in this disclosure as fluoroalkoxy.
- “Hydroxyalkyl” means a linear monovalent hydrocarbon radical of one to six carbon atoms or a branched monovalent hydrocarbon radical of three to six carbons substituted with one or two hydroxy groups, provided that if two hydroxy groups are present they are not both on the same carbon atom.
- Representative examples include, but are not limited to, hydroxymethyl, 2- hydroxy ethyl, 2-hydroxypropyl, 3-hydroxypropyl, l-(hydroxymethyl)-2-methylpropyl, 2- hydroxybutyl, 3-hydroxybutyl. 4-hydroxybutyl, 2,3-dihydroxypropyl, l-(hydroxymethyl)-2- hydroxyethyl, 2.3-dihydroxybutyl.
- Heterocyclyl means a saturated or unsaturated monovalent monocyclic or bi-cyclic group (fused bi-cyclic or bridged bi-cyclic or spiro compounds) of 4 to 10 ring atoms in which one or two ring atoms are heteroatom selected from N, O. and S(O) n , where n is an integer from 0 to 2, the remaining ring atoms being C. Additionally, one or two ring carbon atoms in the heterocyclyl ring can optionally be replaced by a -CO- group.
- heterocyclyl includes, but is not limited to, oxetanyl, pyrrolidino, piperidino, homopiperidino, 2- oxopyrrolidinyl, 2-oxopiperidinyl, morpholino, piperazino, tetrahydropyranyl, thiomorpholino, hexahydropyrrolo[1.2-a]pyrazin-6(2H)-one-yl, tetrahydro-lH-oxazolo[3.4-a]pyrazin-3(5H)-one- yl, 5,6,7,8-tetrahydro-[l,2,4]triazolo[4,3-a]pyrazine-yl, 3-oxa-8-azabicyclo[3.2.1]octane-yl, 6- oxa-l-azaspiro[3.3]heptanyl, 2-oxa-6-azaspiro[3.3]heptanyl, and the like.
- C x-y heterocyclyl refers to a heterocyclyl group with from x to y carbon atoms in the ring, where x and y are integers.
- Heterocyclylalkyl or “heterocycloalkyl” means a -(alkylene)-R radical where R is heterocyclyl ring as defined above e g., tetraydrofuranylmethyl, piperazinylmethyl, morpholinylethyl, and the like.
- Heterocycloamino means a saturated or unsaturated monovalent monocyclic group of 4 to 8 ring atoms in which one or two ring atoms are heteroatom selected from N, O, or S(O) n , where n is an integer from 0 to 2, the remaining ring atoms being C provided that at least one of the ring atoms is N.
- one or two ring carbon atoms in the heterocycloamino ring can optionally be replaced by a -CO- group.
- the heterocycloamino ring is unsaturated it can contain one or two ring double bonds provided that the ring is not aromatic.
- Heterocycloaminoalkyr means a -(alkylene)-R radical where R is heterocycloamino as described above.
- Heteroaryf means a monovalent monocyclic or bicyclic aromatic radical of 5 to 10 ring atoms where one or more, (in one embodiment one, two, or three), ring atoms are heteroatom selected from N, O, and S, the remaining ring atoms being carbon.
- Representative examples include, but are not limited to, pyrrolyl, thienyl, thiazolyl, imidazolyl, furanyl, indolyl, isoindolyl, oxazolyl, isoxazolyl, benzothiazolyl, benzoxazolyl, quinolinyl, isoquinolinyl, pyridinyl, pynmidinyl, pyrazinyl, pyridazinyl, triazolyl, tetrazolyl, and the like.
- Cx-yheteroaryl refers to a heteroaryl group with from x to y carbon atoms, where x and y are integers.
- Rea means -NRC(O)NR*R**.
- Triazaindene and “triazaindenyl” refers to 3f/-imidazo(4.5-6)pyridine. unless specified otherwise.
- mammal as used herein means domesticated animals (such as dogs, cats, and horses), and humans. In one embodiment, mammal is a human.
- salt or “pharmaceutically acceptable salt” refers to salts derived from a variety of organic and inorganic counter ions well known in the art.
- Pharmaceutically acceptable acid addition salts can be formed with inorganic acids and organic acids.
- Inorganic acids from which salts can be derived include, for example, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like.
- Organic acids from which salts can be derived include, for example, acetic acid, propionic acid, glycolic acid, pyruvic acid, oxalic acid, maleic acid, malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, /Moluenesul fonic acid, salicylic acid, and the like.
- Pharmaceutically acceptable base addition salts can be formed with inorganic and organic bases.
- Inorganic bases from which salts can be derived include, for example, sodium, potassium, lithium, ammonium, calcium, magnesium, iron, zinc, copper, manganese, aluminum, and the like.
- Organic bases from which salts can be derived include, for example, primary, secondary , and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, basic ion exchange resins, and the like, specifically such as isopropylamine, trimethylamine, diethylamine, triethylamine, tripropylamine, and ethanolamine.
- the pharmaceutically acceptable base addition salt is chosen from ammonium, potassium, sodium, calcium, and magnesium salts.
- heterocyclyl group optionally substituted with an alkyl group means that the al kyl may but need not be present, and the description includes situations where the heterocyclyl group is substituted with an alkyl group and situations where the heterocyclyl group is not substituted with alkyl.
- parenteral administration and “administered parenterally” as used herein means modes of administration other than enteral and topical administration, usually by injection, and includes, without limitation, intravenous, intramuscular, intraarterial, intrathecal, intracapsular, intraorbital, intracardiac, intradermal, intraperitoneal, transtracheal, subcutaneous, subcuticular, intraarticular, subcapsular. subarachnoid, intraspinal and intrastemal injection and infusion.
- phrases “pharmaceutically acceptable” is employed herein to refer to those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- phrases “pharmaceutically acceptable excipient” or “pharmaceutically acceptable carrier” as used herein means a pharmaceutically acceptable material, composition, or vehicle, such as a liquid or solid filler, diluent, excipient, solvent or encapsulating material. Each carrier must be “acceptable” in the sense of being compatible with the other ingredients of the formulation and not injurious to the patient.
- materials which can serve as pharmaceutically acceptable carriers include: (1) sugars, such as lactose, glucose and sucrose; (2) starches, such as com starch and potato starch; (3) cellulose, and its derivatives, such as sodium carboxymethyl cellulose, ethyl cellulose and cellulose acetate; (4) powdered tragacanth; (5) malt; (6) gelatin; (7) talc; (8) excipients, such as cocoa butter and suppository' waxes; (9) oils, such as peanut oil, cottonseed oil, safflower oil, sesame oil, olive oil, com oil and soybean oil; (10) glycols, such as propylene glycol; (11) polyols, such as glycerin, sorbitol, mannitol and polyethylene glycol; (12) esters, such as ethyl oleate and ethyl laurate; (13) agar; (14) buffering agents, such as magnesium hydroxide and aluminum hydro
- a “therapeutically effective amount” means the amount of a compound of Formula (I) (or any of the embodiments thereof described herein), that, when administered to a mammal for treating a disease, is sufficient to treat the disease.
- the “therapeutically effective amount” will vary depending on the compound, the disease and its severity 7 and the age, weight, etc., of the mammal to be treated.
- the compounds described herein may in some cases exist as diastereomers, enantiomers, or other stereoisomeric forms. All chiral, diastereomeric, racemic forms, as individual forms and mixtures thereof, are within the scope of this disclosure, unless the specific stereochemistry 7 or isomeric form is specifically indicated.
- Compounds of the present disclosure containing an asymmetrically substituted atom may be isolated in optically active, optically enriched, optically pure, or racemic forms. It is well known in the art how to prepare optically active forms, such as by resolution of materials. Separation of stereoisomers may be performed by chromatography or by forming diastereomers and separating by recry stallization, or chromatography, or any combination thereof.
- Stereoisomers may' also be obtained by stereoselective synthesis.
- Certain compounds of Formula (I) may exist as tautomers and/or geometric isomers. All possible tautomers and cis and trans isomers, as individual forms and mixtures thereof, are within the scope of this disclosure.
- pyrazole tautomers as shown below are equivalent structures. The depiction of one such structure is intended to encompass both structures.
- alkyl includes all the possible isomeric forms of said alkyl group albeit only a few examples are set forth. Furthermore, when the cyclic groups such as heteroaryl, heterocyclyl are substituted, they include all the positional isomers.
- the present disclosure also includes the prodrugs of compounds of Formula (I) (or any of the embodiments thereof described herein) and/or a pharmaceutically acceptable salt thereof.
- the term prodrug is intended to represent covalently bonded carriers, which are capable of releasing the active ingredient of Formula (I) (or any of the embodiments thereof described herein) when the prodrug is administered to a mammalian subject. Release of the active ingredient occurs in vivo.
- Prodrugs can be prepared by techniques known to one skilled in the art. These techniques generally modify appropriate functional groups in a given compound. These modified functional groups however regenerate original functional groups in vivo or by routine manipulation.
- Prodrugs of compounds of Formula (I) include compounds wherein a hydroxy, amino, carboxylic, or a similar group is modified.
- Examples of prodrugs include, but are not limited to esters (e.g., acetate, formate, and benzoate derivatives), carbamates (e.g., A/A imethylaminocarbonyl) of hydroxy or amino functional groups in compounds of Formula (I)), amides (e.g., trifluoroacetylamino, acetylamino, and the like), and the like.
- Prodrugs of compounds of Formula (I) (or any of the embodiments thereof described herein) and/or a pharmaceutically acceptable salt thereof are also within the scope of this disclosure.
- the present disclosure also includes polymorphic forms (amorphous as well as crystalline) and deuterated forms of compounds of Formula (I) (or any of the embodiments thereof described herein) and/or a pharmaceutically acceptable salt thereof.
- the compounds disclosed herein are used in different enriched isotopic forms, e.g., enriched in the content of 2 H, 3 H, n C, 13 C and/or 14 C.
- the compound is deuterated in at least one position.
- deuterated forms can be made by the procedure described in U.S. Patent Nos. 5,846.514 and 6,334,997.
- deuteration can improve the metabolic stability and or efficacy, thus increasing the duration of action of drugs.
- structures depicted herein are intended to include compounds which differ only in the presence of one or more isotopically enriched atoms.
- compounds having the present structures except for the replacement of a hydrogen by a deuterium or tritium, or the replacement of a carbon by 13 C- or 14 C-enriched carbon are within the scope of the present disclosure.
- the compounds of the present disclosure optionally contain unnatural proportions of atomic isotopes at one or more atoms that constitute such compounds.
- the compounds may be labeled with isotopes, such as for example, deuterium ( 2 H), tritium ( 3 H), iodine-125 ( 125 I) or carbon-14 ( 14 C).
- isotopes such as for example, deuterium ( 2 H), tritium ( 3 H), iodine-125 ( 125 I) or carbon-14 ( 14 C).
- Isotopic substitution with 2 H, n C, 13 C. 14 C, 15 C, 12 N, 13 N, 15 N, 16 N, 16 O, 17 O, 14 F, 15 F, 16 F, 17 F, 18 F, 33 S, 34 S, 35 S, 36 S, 35 CL 37 C1, 79 Br, 81 Br, and 125 I are all contemplated. All isotopic variations of the compounds of the present invention, whether radioactive or not, are encompassed within the scope of the present invention
- the compounds disclosed herein have some or all of the atoms replaced with 2 H atoms.
- the methods of synthesis for deuterium-containing compounds are known in the art and include, by way of non-limiting example only, the following synthetic methods.
- Deuterium substituted compounds are synthesized using various methods such as described in: Dean, Dennis C.; Editor. Recent Advances in the Synthesis and Applications of Radiolabeled Compounds for Drug Discovery and Development. [In: Curr., Pharm. Des., 2000; 6(10)] 2000, 110 pp; George W.; Varma, Rajender S. The Synthesis of Radiolabeled Compounds via Organometallic Intermediates, Tetrahedron, 1989, 45(21), 6601-21 ; and Evans, E. Anthony. Synthesis of radiolabeled compounds, J. Radioanal. Chem., 1981, 64(1-2), 9-32. [0119] Deuterated starting materials are readily available and are subjected to the synthetic methods described herein to provide for the synthesis of deuterium-containing compounds.
- W is N, CH, or CR 4 *
- R is H, oxo, alkyl, alkenyl, heteroaryl, heterocyclyl, or carboxamide, each substituted with 0, 1, or 2 R x groups and each R x is oxo, hydroxyl, cyano, substituted or unsubstituted alky l, substituted or unsubstituted cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted amino, substituted or unsubstituted ester, substituted or unsubstituted sulfonyl, substituted or unsubstituted amide, substituted or unsubstituted carboxamide, substituted or unsubstituted alkoxy, or substituted or unsubstituted alkoxyalkyl;
- R 1 is H, alkyl, cycloalkyl, heterocyclyl, amino, alkoxy, alkoxyalkyl, amide, carboxamide, or sulfonyl, each substituted with 0, 1, or 2 R y groups and each R y is substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted alkoxy, substituted or unsubstituted alkoxyalkyl, substituted or unsubstituted amino, substituted or unsubstituted ester, substituted or unsubstituted sulfonyl, substituted or unsubstituted amide, substituted or unsubstituted carboxamide, substituted or unsubstituted urea, substituted or unsubstituted carb
- R 4 and R 4 * are aryl, heteroaryl, or heterocyclyl, each substituted with 0, 1, or 2 R z groups and each R z is alkyl, cycloalkyl, haloalkyl, alkoxy, haloalkoxy, halo, or -CN, provided that only one of R 4 and R 4 * is present; or when R 3 and R 4 are bonded to the same carbon, then R 3 and R 4 join to form a monocyclic or bicyclic cycloalkyl;
- R 5 is H, carboxyl, substituted or unsubstituted alkyl, substituted or unsubstituted alkoxy carbonyl, substituted or unsubstituted ester, or substituted or unsubstituted carboxamide;
- R 6 is absent, H, or alkyl;
- R 7 and R 8 are each independently H, Ci-6 alkyl or R 7 and R 8 join together to form a fused or bridging ring system;
- — represents a single or double bond; x is 0, 1 or 2;
- R 9 is absent or oxo; and pharmaceutically acceptable salts, solvates, and prodrugs thereof; provided that when R 6 is absent, R 7 and R 8 are
- R 4 is m-tolyl. then R is not substituted or unsubstituted piperidinyl; substituted or unsubstituted pyrrolidinyl; -CH2OH; -CH 2 (substituted or unsubstituted heterocyclyl); or C(O)NR*R** wherein R* and R** join to form a substituted or unsubstituted heterocyclic group.
- W is CH. In some embodiments, W is N. In some embodiments, W is CR 4 *.
- R is H, oxo, substituted or unsubstituted Ci-6 alkyl, substituted or unsubstituted monocyclic heteroaryl, substituted or unsubstituted monocyclic heterocyclyl, or R a R b NC(O)-.
- R is H, oxo, methyl, ethyl, propyl, isopropyl, isobutyl, propenyl, pyridyl, pyrimidinyl, pyrazolyl, dioxanyl, oxetanyl, morpholino, 3,6-dihydro-2H- pyranyl, or R a R b NC(O)-.
- R is H and R 6 is absent.
- R is not R a R b NC(O)-.
- R x is monocyclic heteroaryl, monocyclic heterocyclyl, Ci-6 alkyl, Ci-6 cycloalkyl, R a R b N, R a O-, R c , R a R b N-(Ci-6 alkyl)-, R a O-(Ci-6 alkyl)-, or R C O-(CI-6 alkyl)-.
- R x is methyl, cyclopropyl, cyclobutyl, pyridyl, pyrazolyl, morpholino.
- R is not substituted with an R x group. In some embodiments, R is substituted with 1 R x group. In some embodiments, R is substituted with 2 R x groups.
- each R a and R b is independently H, Ci-6 alkyl, Ci-e haloalkyl, Ci-6 cycloalkyl, or CHsC(O)-.
- each R a and R b is independently H, cyclopropyl, cyclobutyl, methyl, ethyl, CH3C(O)-, -CD3, or -CF3.
- each R c is H, methyl, ethyl, HOC(O)CH2-. CHsOC(O)CH2-, (CH 3 )3COC(O)CH 2 -, (CH 3 )3COC(O)NH-, H 2 NC(O)CH 2 -, CH 3 NHC(O)CH2-, (CH 3 ) 2 NC(O)CH2-, NC-ethyl, H 2 N-, H 2 N-ethyl, CH 3 NH-ethyl, or (CH 3 ) 2 N-ethyl.
- R 1 is H, C1-6 alkyl, C1-6 cycloalkyl, monocyclic heterocyclyl, R a O-, R a O-(Ci-6 alkyl), R a R b NC(O)-, or alkyl-SCh-.
- R 1 is H, propyl, ethyl, methyl, isobutyl, cyclobutyl, cyclopropyl, azetidinyl. oxetanyl.
- R y is Ci-6 alkyl, Ci-6 alkenyl, Ci-6 haloalkyl, Ci-6 cycloalkyl, monocyclic heteroaryl, monocyclic aryl, monocyclic heterocyclyl, halo, -CN, -OH, R a O-, CH3SO2- CH 3 C(O)-, R a OC(O)-, R a R b NC(O)-, H 2 NC(O)NH-,R a R b N-, or methylallyl, each further substituted with 0, 1, or 2 R d groups. In some embodiments.
- R y is methyl, cyclopropyl, cyclobutyl, pyridinyl, phenyl, piperidinyl. pyrrolidinyl, oxetanyl. pyrrolidinone, imidazolidin-2- one, oxazolidin-2-one, lH-l,2,4-triazol-5-one, tetrahydropyran, dioxanyl, azetidinyl, -CN, -F, - CF3, -OH, R a O-, CH3SO2-, CH 3 C(O)-, HOC(O)-, R a R b NC(O)-, H 2 NC(O)NH-, R a R b N-, or methylallyl
- R 1 is not substituted with an R y group. In some embodiments. R 1 is substituted with 1 R y group. In some embodiments, R 1 is substituted with 2 R y groups.
- R d is methyl, oxo, CHsC(O)-, CHsOC(O)-, CH3SO2-, -F, (CH 3 )3CO-, (CD 3 ) 3 CO-, R a O-, -CN, -NH2, (CH 3 )3COC(O)-, H 2 NC(O)-, CH 3 HNC(O)-.
- R y is not substituted with an R d group. In some embodiments. R y is substituted with 1 R d group. In some embodiments, R y is substituted with 2 R d groups.
- R 4 is H, phenyl, pyrazolyl, thiazolyl, thienyl, oxabicyclo[2.1.1]hexanyl, and norbomanyl, each substituted by 0, 1, or 2 R z .
- R 4 is H, phenyl, pyrazolyl, thiazolyl, thienyl, oxabicyclo[2.1.1]hexanyl, or norbomanyl.
- each R z is -CN, halo, C1-6 alkyl, C1-6 alkoxy, C1-6 haloalkyl, C1-6 cycloalkyl, deuterated alkyd, or C1-6 haloalkoxy.
- R z is -Cl, -F, -CN, -CF3, -CD3, -OCF3, methyl, ethyl, cyclopropyl, methoxy, difluoromethoxy, or difluoromethyl.
- R 4 is not substituted with an R z group. In some embodiments. R 4 is substituted with 1 R z group. In some embodiments, R 4 is substituted with 2 R z groups.
- R 4 is tolyl. In some embodiments, R 4 is m-tolyl. In some embodiments, R 4 is chlorophenyl. In some embodiments, R 4 is m-chlorophenyl.
- R 5 is -C(O)NR a R b , -C(O)OR a , unsubstituted C1-6 alkyl, or C1-6 alkyl substituted with 1 or 2 OR a or alkylamino groups.
- R 5 is H, hydroxymethyl, hydroxyethyl, methoxymethyl, H 2 N-methyl, CHsNH-methyl. or (CH3) 2 N- methyl, trideuteriomethoxymethyl, -C(O)NR a R b , -C(O)OR a .
- R 6 is absent.
- R 7 and R 8 are each H. In some embodiments one of R 7 and R 8 is H. In some embodiments one of R 7 and R 8 is methyl. In some embodiments R 7 and R 8 join together to form a fused or bridging ring system. In some embodiments R 7 and R 8 join together to form a fused ring system. In some embodiments R 7 and R 8 join together to form a bridging ring system. In some embodiments when R 7 and R 8 join together to form a fused ring system the additional ring is a 4, 5, 6, or 7 membered ring. In some embodiments R 7 and R 8 join together to form a bridged ring system wherein the bridge comprises 1, 2, or 3 carbon atoms.
- R is H
- R 4 is m-tolyl or m-chlorophenyl
- R 1 is H, methyl, ethyl or isopropyl substituted with 1 R y group and R y is substituted with 1 or 2 R d groups.
- n 0, 1 or 2; and each R x is independently halo, cyano, C1-3 alky l, C1-3 haloalkyl, C3-6 cycloalkyl, C1-3 alkoxy, or C1-3 haloalkoxy.
- n is 1 or 2; and each R x is independently F, Cl, cyano, methyl, fluoromethyl, difluoromethyl, trifluoromethyl, ethyl, fluoroethyl, difluoroethyl, trifluoroethyl, cyclopropyl, methoxy, fluoromethoxy, difluoromethoxy, trifluoromethoxy, ethoxy, fluoroethoxy, difluoroethoxy, or trifluoroethoxy.
- R 1 is H, methyl, ethyl or isopropyl substituted with 1 R y group and R y is substituted with 1 or 2 R d groups.
- ring is a 5- or 6- membered heterocyclic ring comprises optionally a C, N or O in addition to the N at the fusion point for the rings, and is optionally substituted with an oxo group, an alky l group, halo, CN, or alkoxy group.
- the compound of Formula (I) is selected from:
- the compound of Formula (I) is selected from the compounds of Table 1.
- the compounds of this disclosure will be administered in a therapeutically effective amount by any of the accepted modes of administration for agents that serve similar utilities.
- Therapeutically effective amounts of compounds of Formula (I) may range from about 0.01 to about 500 mg per kg patient body weight per day, which can be administered in single or multiple doses.
- the dosage level will be about 0. 1 to about 250 mg/kg per day.
- the dosage level will be about 0.5 to about 100 mg/kg per day.
- a suitable dosage level may be about 0.01 to about 250 mg/kg per day, about 0.05 to about 100 mg/kg per day, or about 0. 1 to about 50 mg/kg per day.
- the dosage can be about 0.05 to about 0.5, about 0.5 to about 5 or about 5 to about 50 mg/kg per day.
- the compositions may be provided in the form of tablets containing about 1.0 to about 1000 milligrams of the active ingredient, particularly about 1.0, 5.0, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, 500, 600, 750, 800, 900, and 1000 milligrams of the active ingredient.
- the actual amount of the compound of this disclosure, i.e., the active ingredient will depend upon numerous factors such as the severity of the disease to be treated, the age and relative health of the subj ect, the potency of the compound being utilized, the route and form of administration, and other factors.
- compositions will be administered as pharmaceutical compositions by any one of the following routes: oral, systemic (e.g.. transdermal, intranasal or by suppository), or parenteral (e.g., intramuscular, intravenous, or subcutaneous) administration.
- routes e.g.. oral, systemic (e.g.. transdermal, intranasal or by suppository), or parenteral (e.g., intramuscular, intravenous, or subcutaneous) administration.
- parenteral e.g., intramuscular, intravenous, or subcutaneous
- compositions can take the form of tablets, pills, capsules, semisolids, powders, sustained release formulations, solutions, suspensions, elixirs, aerosols, or any other appropriate compositions.
- compositions can be formulated using one or more pharmaceutically acceptable carriers comprising excipients and auxiliaries.
- the formulation can be modified depending upon the route of administration chosen.
- the pharmaceutical compositions can also include the compounds described herein in a free base form or a pharmaceutically acceptable salt form.
- Methods for formulation of the pharmaceutical compositions can include formulating any of the compounds described herein with one or more inert, pharmaceutically acceptable excipients or carriers to form a solid, semi-solid, or liquid composition.
- Solid compositions can include, for example, powders, tablets, dispersible granules and capsules, and in some aspects, the solid compositions further contain nontoxic, auxiliary substances, for example wetting or emulsifying agents, pH buffering agents, and other pharmaceutically acceptable additives.
- the compositions described herein can be lyophilized or in powder form for reconstitution with a suitable vehicle, e.g., sterile pyrogen-free water, before use.
- the active ingredients can be entrapped in microcapsules prepared, for example, by coacervation techniques or by interfacial polymerization (e.g., hydroxymethylcellulose or gelatin microcapsules and poly-(methylmethacylate) microcapsules, respectively), in colloidal drugdelivery systems (e.g., liposomes, albumin microspheres, microemulsions, nano-particles and nanocapsules) or in macroemulsions.
- colloidal drugdelivery systems e.g., liposomes, albumin microspheres, microemulsions, nano-particles and nanocapsules
- compositions and formulations can be sterilized. Sterilization can be accomplished by filtration through sterile filtration.
- compositions described herein can be formulated for administration as an injection.
- formulations for injection can include a sterile suspension, solution, or emulsion in oily or aqueous vehicles.
- Suitable oily vehicles can include, but are not limited to, lipophilic solvents or vehicles such as fatty oils, synthetic fatty acid esters, or liposomes.
- Aqueous injection suspensions can contain substances which increase the viscosity of the suspension.
- the suspension can also contain suitable stabilizers.
- Injections can be formulated for bolus injection or continuous infusion.
- the compounds can be formulated in a unit dosage injectable form (e.g.. solution, suspension, emulsion) in association with a pharmaceutically acceptable parenteral vehicle.
- a pharmaceutically acceptable parenteral vehicle e.g.. water, saline, Ringer’s solution, dextrose solution, and 5% human serum albumin.
- Nonaqueous vehicles such as fixed oils and ethyl oleate can also be used.
- Liposomes can be used as carriers.
- the vehicle can contain minor amounts of additives such as substances that enhance isotonicity and chemical stability (e.g., buffers and preservatives).
- sustained-release preparations can also be prepared.
- sustained-release matrices can include polyesters, hydrogels (e.g., poly(2-hydroxyethyl-methacrylate), or poly(vinylalcohol)), polylactides, copolymers of L-glutamic acid and y ethyl-L-glutamate, non- degradable ethylene-vinyl acetate, degradable lactic acid-glycolic acid copolymers such as the LUPRON DEPOTM (i.e., injectable microspheres composed of lactic acid-glycolic acid copolymer and leuprolide acetate), and poly-D-(-)-3-hydroxybutyric acid.
- LUPRON DEPOTM i.e., injectable microspheres composed of lactic acid-glycolic acid copolymer and leuprolide acetate
- poly-D-(-)-3-hydroxybutyric acid i.e., injectable microspheres composed of lactic acid
- compositions described herein can be prepared for storage by mixing a compound with a pharmaceutically acceptable carrier, excipient, and/or a stabilizer.
- This formulation can be a lyophilized formulation or an aqueous solution.
- Acceptable carriers, excipients, and/or stabilizers can be nontoxic to recipients at the dosages and concentrations used.
- Acceptable carriers, excipients, and/or stabilizers can include buffers such as phosphate, citrate, and other organic acids; antioxidants including ascorbic acid and methionine; preservatives, polypeptides; proteins, such as serum albumin or gelatin; hydrophilic polymers; amino acids; monosaccharides, disaccharides, and other carbohydrates including glucose, mannose, or dextrins; chelating agents such as EDTA; sugars such as sucrose, mannitol, trehalose or sorbitol; salt-forming counter-ions such as sodium; metal complexes; and/or nonionic surfactants or polyethylene glycol.
- buffers such as phosphate, citrate, and other organic acids
- antioxidants including ascorbic acid and methionine
- preservatives polypeptides
- proteins such as serum albumin or gelatin
- hydrophilic polymers amino acids
- Compounds of the present disclosure may be used in methods of treating in combination with one or more other combination agents (e.g., one, two, or three other drugs) that are used in the prevention, treatment, control, amelioration, or reduction of risk of the diseases or conditions for which compounds of the present disclosure are useful.
- the combination of the drugs together are safer or more effective than either drug alone.
- the compound disclosed herein and the one or more combination agents have complementary activities that do not adversely affect each other.
- Such molecules can be present in combination in amounts that are effective for the purpose intended.
- Such other drug(s) may be administered, by a route and in an amount commonly used therefore, contemporaneously or sequentially with a compound of the present disclosure.
- the agents are administered together in a single pharmaceutical composition in unit dosage form.
- the pharmaceutical compositions of the present disclosure also include those that contain one or more other active ingredients, in addition to a compound of the present disclosure.
- the weight ratio of the compound of the present disclosure to the second active agent may be varied and will depend upon the effective dose of each ingredient. Generally, an effective dose of each will be used.
- combination therapy includes therapies in which the compound of the present disclosure and one or more other drugs are administered separately, and in some cases, the two or more agents are administered on different, overlapping schedules.
- the combination agent is a drug for reduction of symptoms of ALS.
- the combination agent is selected from an NAD supplement (such as nicotinamide riboside, offered under the trade names Basis® or Tru Niagen®). vitamin B12 (oral or injection), glycopyrrolate, atropine, scopolamine, baclofen, tizanidine. mexiletine. an SSRI. a benzodiazepine, Neudexta. riluzole, and edaravone, and combinations thereof.
- the compounds, pharmaceutical compositions, and methods of the present disclosure can be useful for treating a subj ect such as, but not limited to, a mammal, a human, a non-human mammal, a domesticated animal (e.g., laboratory animals, household pets, or livestock), a nondomesticated animal (e.g., wildlife), a dog, a cat, a rodent, a mouse, a hamster, a cow, a bird, a chicken, a fish, a pig, a horse, a goat, a sheep, or a rabbit.
- a subj ect such as, but not limited to, a mammal, a human, a non-human mammal, a domesticated animal (e.g., laboratory animals, household pets, or livestock), a nondomesticated animal (e.g., wildlife), a dog, a cat, a rodent, a mouse, a hamster, a cow, a bird,
- the compounds, pharmaceutical compositions, and methods described herein can be useful as a therapeutic, for example a treatment that can be administered to a subject in need thereof.
- a therapeutic effect can be obtained in a subject by reduction, suppression, remission, or eradication of a disease state, including, but not limited to, a symptom thereof.
- a therapeutic effect in a subject having a disease or condition, or pre-disposed to have or is beginning to have the disease or condition can be obtained by a reduction, a suppression, a prevention, a remission, or an eradication of the condition or disease, or pre-condition or pre-disease state.
- therapeutically effective amounts of the compounds or pharmaceutical compositions described herein can be administered to a subject in need thereof, often for treating and/or preventing a condition or progression thereof.
- a pharmaceutical composition can affect the physiol ogy of the subject, such as the immune system, inflammatory response, or other physiologic affect.
- a therapeutically effective amount can vary widely depending on the severity of the disease, the age and relative health of the subject, the potency of the compounds used, and other factors.
- Treat and/or treating can refer to any indicia of success in the treatment or amelioration of the disease or condition. Treating can include, for example, reducing, delaying or alleviating the severity of one or more symptoms of the disease or condition, or it can include reducing the frequency with which symptoms of a disease, defect, disorder, or adverse condition, and the like, are experienced by a patient. Treat can be used herein to refer to a method that results in some level of treatment or amelioration of the disease or condition and can contemplate a range of results directed to that end, including but not restricted to prevention of the condition entirely. [0161] Prevent, preventing, and the like can refer to the prevention of the disease or condition in the patient. For example, if an individual at risk of contracting a disease is treated with the methods of the present disclosure and does not later contract the disease, then the disease has been prevented, at least over a period of time, in that individual.
- a therapeutically effective amount can be the amount of a compound or pharmaceutical composition or an active component thereof sufficient to provide a beneficial effect or to otherwise reduce a detrimental non-beneficial event to the individual to whom the composition is administered.
- a therapeutically effective dose can be a dose that produces one or more desired or desirable (e.g., beneficial) effects for which it is administered, such administration occurring one or more times over a given period of time. An exact dose can depend on the purpose of the treatment and can be ascertainable by one skilled in the art using known techniques.
- the compounds or pharmaceutical compositions described herein that can be used in therapy can be formulated and dosages established in a fashion consistent with good medical practice taking into account the disorder to be treated, the condition of the individual patient, the site of delivery' of the compound or pharmaceutical composition, the method of administration and other factors known to practitioners.
- the compounds or pharmaceutical compositions can be prepared according to the description of preparation described herein.
- compositions or compounds described herein can be for administration to a subject in need thereof.
- administration of the compounds or pharmaceutical compositions can include routes of administration, non-limiting examples of administration routes include intravenous, intraarterial, subcutaneous, subdural, intramuscular, intracranial, intrastemal, intratumoral, or intraperitoneally.
- a pharmaceutical composition or compound can be administered to a subject by additional routes of administration, for example, by inhalation, oral, dermal, intranasal, or intrathecal administration.
- Pharmaceutical compositions or compounds of the present disclosure can be administered to a subject in need thereof in a first administration, and in one or more additional administrations.
- the one or more additional administrations can be administered to the subject in need thereof minutes, hours, days, weeks, or months following the first administration. Any one of the additional administrations can be administered to the subject in need thereof less than 21 days, or less than 14 days, less than 10 days, less than 7 days, less than 4 days or less than 1 day after the first administration.
- the one or more administrations can occur more than once per day, more than once per week, or more than once per month.
- the compounds or pharmaceutical compositions can be administered to the subject in need thereof in cycles of 21 days. 14 days, 10 days, 7 days, 4 days, or daily over a period of one to seven days.
- the compounds, pharmaceutical compositions, and methods provided herein can be useful for the treatment of a plurality of diseases or conditions or preventing a disease or a condition in a subject, or other therapeutic applications for subjects in need thereof.
- the disclosure relates to a method for treating a neurological disease mediated by PIKfyve activity in a subject in need thereof, comprising administering an effective amount of a compound or a pharmaceutical composition as described herein to the subject.
- the disease is associated with a FIG4 deficiency.
- the neurological disease is amyotrophic lateral sclerosis (ALS), primary lateral sclerosis (PLS), Charcot-Marie-Tooth (CMT; including type 4J (CMT4J)), and Yunis-Varon syndrome, autophagy 7 , polymicrogyria (including polymicrogyria with seizures), temporo-occipital polymicrogyria.
- ALS amyotrophic lateral sclerosis
- PLS primary lateral sclerosis
- CMT Charcot-Marie-Tooth
- Yunis-Varon syndrome autophagy 7
- polymicrogyria including polymicrogyria with seizures
- temporo-occipital polymicrogyria temporo-occipital polymicrogyria.
- Parkinson’s disease Parkinson’s disease with Lewy bodies, dementia with Lewy bodies, Lewy body disease, fronto-temporal dementia, diseases of neuronal nuclear inclusions of polyglutamine and intranuclear inclusion bodies, disease of Marinesco and Hirano bodies, tauopathy, Alzheimer's disease, neurodegeneration, spongiform neurodegeneration, peripheral neuropathy, leukoencephalopathy, motor neuropathy, sensory neuropathy, inclusion body disease, progressive supranuclear palsy, corticobasal syndrome, chronic traumatic encephalopathy, traumatic brain injury (TBI), cerebral ischemia, Guillain-Barre Syndrome, chronic inflammatory demyelinating polyneuropathy, multiple sclerosis, a lysosomal storage disease, Fabry ’s disorder, Gaucher's disorder, Niemann Pick C disease, Tay-Sachs disease, and Mucolipidosis type IV, neuropathy, Huntington's disease, a psychiatric disorder, ADHD, schizophrenia, a mood
- the neurological disease is ALS, FTD, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, or CMT. In some embodiments, the neurological disease is ALS.
- the neurological disease is a tauopathy such as Alzheimer's disease, progressive supranuclear palsy, corticobasal syndrome, frontotemporal dementia, or chronic traumatic encephalopathy.
- the neurological disease is a lysosomal storage disease such as Fabry's disorder, Gaucher's disorder, Niemann Pick C disease, Tay-Sachs disease, or Mucolipidosis type IV.
- the neurological disease is a psychiatric disorder such as ADHD, schizophrenia, or mood disorders such as major depressive disorder, depression, bipolar disorder I, or bipolar disorder II.
- the disclosure further provides any compounds disclosed herein for use in a method of treatment of the human or animal body by therapy. Therapy may be by any mechanism disclosed herein, such as inhibiting, reducing, or reducing progression of the diseases disclosed herein.
- the disclosure further provides any compound disclosed herein for prevention or treatment of any condition disclosed herein.
- the disclosure also provides any compound or pharmaceutical composition thereof disclosed herein for obtaining any clinical outcome disclosed herein for any condition disclosed herein.
- the disclosure also provides use of any compound disclosed herein in the manufacture of a medicament for preventing or treating any disease or condition disclosed herein.
- the starting materials and reagents used in preparing these compounds are either available from commercial suppliers such as Aldrich Chemical Co., (Milwaukee, Wis.), Bachem (Torrance, Calif.), or Sigma (St. Louis, Mo.) or are prepared by methods known to those skilled in the art following procedures set forth in references such as Fieser and Fieser’s Reagents for Organic Synthesis, Volumes 1-17 (John Wiley and Sons, 1991); Rodd's Chemistry of Carbon Compounds.
- the reactions described herein take place at atmospheric pressure over a temperature range from about -78 °C to about 150 °C, or from about 0 °C to about 125 °C or at about room (or ambient) temperature, e.g., about 20 °C.
- trimethoxymethane 970 mL
- TSOH-H2O 3.7 g, 0.021 mol
- reaction mixture was diluted with water (30 mL) and extracted with EtOAc (3 x 20 mL). The combined organic phases were dried over anhydrous Na2SO4. filtrated and concentrated. The resulting residue was purified by silica gel column chromatography with a gradient elution of 5% EtOAc/PE to 25% EtOAc/PE to provide ethyl l-(6-((2-methoxyethyl)amino)-4-morpholino- 5-nitropyridin-2-yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate (395 mg, 0.77 mmol) as a yellow oil.
- reaction mixture was diluted with water (30 mL) and extracted with DCM (3 x 20 mL). The combined organic phases were washed with saturated aqueous NaCl solution (2 x 20 mL), dried over anhydrous NazSO-i. filtrated and concentrated. The resulting residue was purified by slurry in MeOH to provide l-(3-(2-methoxyethyl)-7- morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-N-methyl-3-(m-tolyl)-lH-pyrazole-5-carboxamide (36 mg, 0.077 mmol) as an off-white solid.
- reaction mixture was diluted with water (30 mL) and extracted with DCM (3 x 20 mL). The combined organic phases were washed with saturated aqueous NaCl solution (2 x 20 mL), dried over anhydrous Na2SC>4, filtrated and concentrated.
- N2-(2,4-dimethoxybenzyl)-4-morpholino-6-(3-(m- tolyl)-lH- pyrazol-l-yl)pyridine-2,3-diamine 107.0 g, 0.21 mol
- trimethoxymethane 970 mL
- TSOH-H2O 3.7 g, 0.021 mol
- the reaction mixture was heated to reflux and stirred for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was concentrated directly. The resulting residual was slurry in EtOAc/PE (1/5.
- reaction mixture was diluted wi th water (20 mL) and extracted with DCM (3 x 10 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduce pressure. The resulting residue was purified by preparative TLC with an elution of 50% EtOAc/PE to provide l-((5-(3-(3-(difluoromethoxy)phenyl)-lH-pyrazol-l-yl)-7-morpholino-3H- imidazo[4,5-b]pyridin-3-yl)methyl)cyclopropanecarbonitrile (39 mg, 0.079 mmol) as a white solid.
- reaction mixture was diluted with water (20 mL).
- the aqueous solution was extracted with EtOAc (3 x 10 mL).
- the combined organic phases were washed with saturated aqueous NaCl solution (2 x 20 mL), dried over anhydrous Na2SO4, filtrated and concentrated.
- reaction mixture was stirred at rt for 3 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was quenched with water (50 mL) and extracted with DCM (3 x 100 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduce pressure. The resulting residue was used for the next step without further purification.
- reaction mixture was quenched with water (10 mL) and extracted with EtOAc (3 x 20 mL). The combined organic phases were dried over anhydrous Na2SO4, filtered, and concentrated.
- reaction mixture was stirred at that temperature for 1 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with NH4CI solution and extracted with EtOAc (3 x 20 mL). The combined organic phases were dried over anhydrous Na2SO4, filtered, and concentrated under reduce pressure.
- PIKFYVE Full length human recombinant PIKFYVE expressed in baculovirus expression system as N-termmal GST-fusion protein (265 kDa) was obtained from Cama Biosciences (Kobe, Japan).
- the kinase substrate was prepared by mixing and sonicating fluorescently-labeled phosphatidylinositol 3-phosphate (PI3P) with phospho-L-serine (PS) at a 1 : 10 ratio in 50 mM HEPES buffer pH7.5.
- kinase reactions were assembled in 384-well plates (Greiner) in a total volume of 20 mL as follows.
- Kinase protein was pre-diluted in an assay buffer comprising 25 mM HEPES, pH 7.5, 1 mM DTT, 2.5 mM MgCh, and 2.5 mM MnCh, and 0.005% Triton X-100, and dispensed into a 384-well plate (10 pL per well).
- Test compounds were serially pre-diluted in DMSO and added to the protein samples by acoustic dispensing (Labcyte Echo). The concentration of DMSO was equalized to 1% in all samples. All test compounds were tested at 12 concentrations.
- Apilimod was used as a reference compound and was tested in identical manner in each assay plate.
- Control samples (0%-inhibition, in the absence of inhibitor, DMSO only) and 100%-inhibition (in the absence of enzyme) were assembled in replicates of four and were used to calculate %-inhibition in the presence of compounds.
- the reactions were initiated by addition of 10 pL of 2x PI3P/PS substrate supplemented with ATP.
- the final concentration of enzyme was 2 nM
- the final concentration of ATP was 10 mM
- the final concentration of PI3P/PS substrate was 1 pM (PI3P).
- the kinase reactions were allowed to proceed for 3 h at room temperature.
- Terminated plates were analyzed on a microfluidic electrophoresis instrument (Caliper LabChip® 3000, Caliper Life Sciences/Perkin Elmer). The change in the relative fluorescence intensity of the PI(3)P substrate and PI(3,5)P product peaks was measured. The activity' in each test sample was determined as the product to sum ratio (PSR): P/(S+P), where P is the peak height of the product, and S is the peak height of the substrate. Percent inhibition (Pinh) was determined using the following equation:
- Pinh (PSR.0°/oinh - PSRcompound)/(PSRo%inh - PSR100%inh)* 100 in which PSRcompound is the product/sum ratio in the presence of compound.
- PSRo%inh is the product/sum ratio in the absence of compound, and the PSRioo%inh is the product/sum ratio in the absence of the enzyme.
- ICso of test compounds 50%-inhibition
- the %-inh cdata (Pinh versus compound concentration) were fitted by a four-parameter sigmoid doseresponse model using XLfit software (IDBS).
- HEK 293T Immortalized human embryonic kidney 293T
- plasmids containing TDP-43 Q331K mutation were transfected with plasmids containing TDP-43 Q331K mutation, resulting in an increase in cell death that is biologically relevant to ALS patients.
- Cell death is measured as reductions in the amount of ATP, an indicator of metabolically active cells, that is quantified by a luminescence Cell-Titer- Glo® (CTG) reagent.
- CCG luminescence Cell-Titer- Glo®
- HEK293T cells were passaged using 0.25% Trypsin-EDTA and plated at a density of 10,000 cells/well in 96- well tissue culture plates (inner 60 wells). The following day, the cells were transfected with TDP43-Q331K plasmid using Lipofectamine 3000 and treatment with compound was performed (9-point dose response curve, half-log serial dilution with lOuM as the highest concentration with 6 replicates). After 48 hours of incubation, CellTiter-Glo reagent was added, and the lysates were transferred to opaque white flat-bottom polystyrene plates. Compound induced rescue of HEK cells with mutant TDP43 related viability deficit was measured using the CellTiter-Glo assay.
- Dose-response curve graphs were generated using GraphPad Prism. ECso values for compound treatment were determined using a four-parameter variable slope dose-response curve fitted to the Hill equation. Data point(s) of high concentration compound-induced toxicity resulting in decreased cellular viability was excluded.
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Psychiatry (AREA)
- Pain & Pain Management (AREA)
- Hospice & Palliative Care (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
The present disclosure provides compounds of formula (I) that are inhibitors of PlKfyve kinases useful for the treatment of neurological diseases treatable by inhibition of PlKfyve. Also provided are pharmaceutical compositions containing such compounds, and methods of treatment using such compounds whith X.
Description
PYRAZOLE 3H-IMIDAZO(4,5-B)PYRIDINE COMPOUNDS AND USES THEREOF
Field of Invention
[0001] The present disclosure provides compounds that are phosphoinositide kinase inhibitors, in particular FYVE-type finger-containing phosphoinositide kinase (“PIKfyve”) inhibitors and are therefore useful for the treatment of central nervous system diseases. Also provided are pharmaceutical compositions containing such compounds and processes for preparing such compounds.
Background
[0002] Phosphoinositide kinases (PIKs) catalyze the phosphory lation of phosphatidy linositol, which is a component of eukaryotic cell membranes, and related phospholipids called phosphoinositides. Phosphoinositides are involved in the regulation of diverse cellular processes, including cellular proliferation, survival, cytoskeletal organization, vesicle trafficking, glucose transport, and platelet function. Fruman et al., “Phosphoinositide Kinases,” Ann. Review. Biochem. 1998, 67, 481-507. Phosphorylated derivatives of phosphatidylinositol regulate cytoskeletal functions, membrane trafficking, and receptor signaling by recruiting protein complexes to cell and endosomal membranes.
[0003] FYVE-type finger-containing phosphoinositide kinase (PIKfyve; also known as phosphatidylinositol-3-phosphate 5-kinase type III or PIPKIII) is a ubiquitously expressed PIK with both lipid and protein kinase activity. In its capacity as a lipid kinase, the enzyme phosphorylates the D-5 position in endosomal phosphatidylinositol and phosphatidylinositol-3- phosphate (PI3P) to generate the corresponding 5-phosphate phospholipid analogs. Shisheva et al., Cell Biol. Int. 2008, 32(6), 591. PI3P is found in cell membranes with roles in protein trafficking, protein degradation, and autophagy. Nascimbeni et al., FEBSJ. 2017, 284. 1267- 1278. PIKfyve regulates endomembrane homeostasis and plays a role in the biogenesis of endosome carrier vesicles from early endosomes. The enlarged endosome/lysosome structure was observed in cells expressing PIKfyve dominant negative or siRNA. Ikonomov et al., J. Biol. Chem. 2001, 276(28), 26141-26147; Rutherford et al. , J. Cell Sci. 2006, 119, 3944-3957. Inhibition of PIKfy ve activity increases levels of PI3P, stimulating autophagy and improving motor neuron health. Phosphorylated inositides produced by PIKfyve are localized in various cellular membranes and organelles, consistent with the various PIKfy ve functions of endolysosomal transport, endomembrane homeostasis, and biogenesis of endosome carrier vesicles (ECV)/multivesicular bodies (MVB) from early endosomes. Further, PIKfyve is
required for endocytic-vacuolar pathway and nuclear migration. Thus, PIKfyve helps maintain proper morphology of the endosome and lysosome.
[0004] In mammalian cells, PI3P levels are regulated by the reciprocal activities of PIKfyve and the phosphatase FIG4 phosphoinositide 5-phosphatase (FIG4). Zolov et al., “In vivo, Pikfyve generates PI(3,5)P2. which serves as both a signaling lipid and the major precursor for PI5P,” Proc. Natl. Acad. Set. USA 2012, 109(43), 17472-17477. Normally, FIG4 is localized on the cytoplasmic surface of endolysosomal vesicles in a complex. Inhibition of PIKfyve would mimic overexpression of FIG4, thereby increasing levels of PI3P, stimulating autophagy, and improving motor neuron health. Numerous diseases are correlated with FIG4 deficiencies, such as deleterious FIG4 mutations or diminished FIG4 function, and are therefore suitable as target diseases for treatment with PIKfyve inhibitors, including amyotrophic lateral sclerosis (ALS), primary lateral sclerosis (PLS), Charcot-Marie-Tooth (including type 4J (CMT4J)), and Yunis- Varon syndrome.
[0005] Exemplary diseases associated with FIG4 deficiencies are amyotrophic lateral sclerosis (ALS), primary lateral sclerosis (PLS), Charcot-Marie-Tooth (including type 4J (CMT4J)), Yunis-Varon syndrome, poly microgyri a (including polymicrogyria with seizures), temporo- occipital polymicrogyria, Pick’s disease, Parkinson's disease, Parkinson's disease with Lewy bodies, dementia with Lewy bodies. Lewy body disease, fronto-temporal dementia, diseases of neuronal nuclear inclusions of poly glutamine and intranuclear inclusion bodies, disease of Marinesco and Hirano bodies, Alzheimer’s disease, neurodegeneration, spongiform neurodegeneration, autophagy, peripheral neuropathy, leukoencephalopathy, motor neuropathy, sensory neuropathy. Bharadwaj et al., Hum. Mol. Genet. 2016, 25(4). 682-692.
[0006] PIKfyve inhibitors are useful in a range of neurological disorders, such as tauopathies (including but not limited to Alzheimer's disease, progressive supranuclear palsy, corticobasal syndrome, frontotemporal dementias, and chronic traumatic encephalopathy), traumatic brain injury’ (TBI), cerebral ischemia, ALS, fronto-temporal dementia (FTD), Guillain-Barre Syndrome, chronic inflammatory demyelinating polyneuropathy, multiple sclerosis, CMT. lysosomal storage diseases (including but not limited to Fabry's disorder, Gaucher's disorder, Niemann Pick C, Tay-Sachs, and Mucolipidosis type IV), as well as several types of neuropathies. Other therapeutic targets for intervention with PIKfyve inhibitors include Huntington's disease and psychiatric disorders (such as ADHD, schizophrenia, mood disorders including but not limited to major depressive disorder, bipolar disorder I, and bipolar disorder II). Gardiner et al., “Prevalence of carriers of intermediate and pathological polyglutamine disease-associated alleles among large population-based cohorts,"’ JAMA Neurol. 2019, 76(6), 650-656; PCT Publ. No. WO2016/210372; US Publ. No. US2018/0161335.
Summary
[0007] The present embodiments can be understood more fully by reference to the detailed description and examples, which are intended to exemplify non-limiting embodiments.
[0008] Embodiment 1 is a compound of Formula (I):
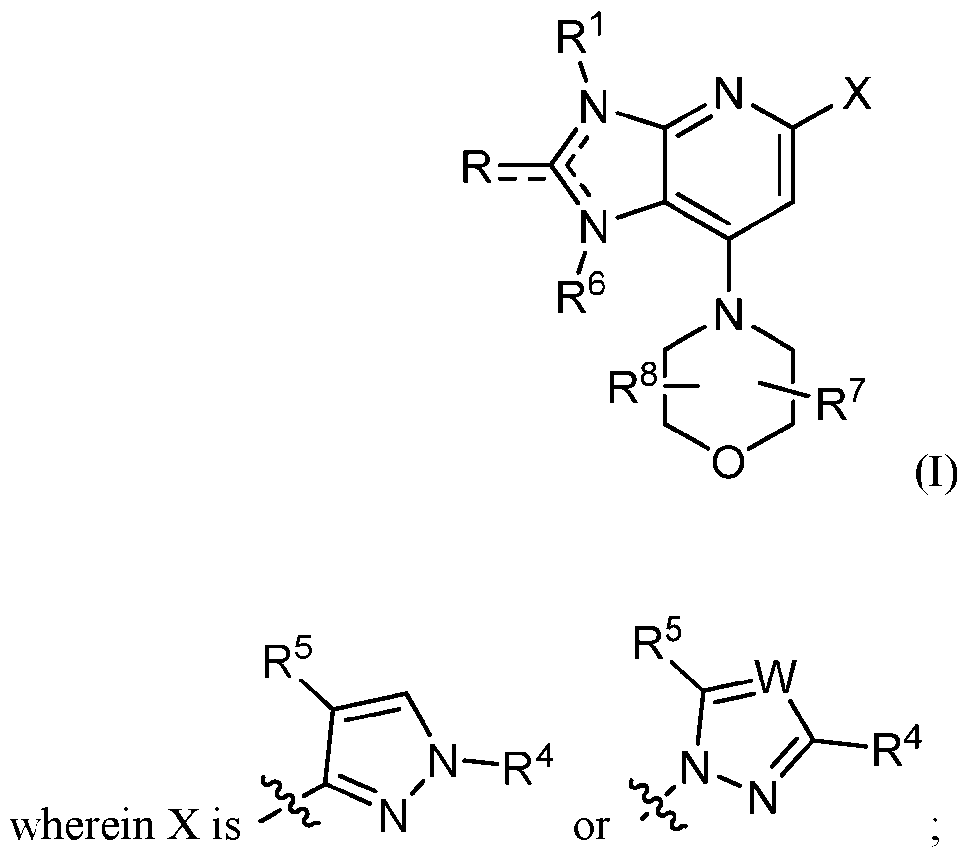
W is N, CH, or CR4‘;
R is H, oxo, alkyl, alkenyl, heteroaryl, heterocyclyl, or carboxamide, each substituted with 0, 1, or 2 Rx groups and each Rx is oxo, hydroxyl, cyano, substituted or unsubstituted alkyd, substituted or unsubstituted cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted amino, substituted or unsubstituted ester, substituted or unsubstituted sulfonyl, substituted or unsubstituted amide, substituted or unsubstituted carboxamide, substituted or unsubstituted alkoxy, or substituted or unsubstituted alkoxyalkyl;
R1 is H, alkyl, cycloalkyl, heterocyclyl, amino, alkoxy, alkoxyalkyl, amide, carboxamide, or sulfonyl, each substituted with 0, 1, or 2 Ry groups and each Ry is substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted alkoxy, substituted or unsubstituted alkoxyalkyl, substituted or unsubstituted amino, substituted or unsubstituted ester, substituted or unsubstituted sulfonyl, substituted or unsubstituted amide, substituted or unsubstituted carboxamide, substituted or unsubstituted urea, substituted or unsubstituted carbamate, halo, -CN, -OH, oxo, or methylallyl;
R4 and R4* are aryl, lieteroar l. or heterocyclyl, each substituted with 0, 1, or 2 Rz groups and each Rz is alkyl, cycloalkyl, haloalkyl. alkoxy, haloalkoxy, halo, or -CN, provided that only one of R4 and R4* is present;
R5 is H, carboxyl, substituted or unsubstituted alkyl, substituted or unsubstituted
alkoxy carbonyl, substituted or unsubstituted ester, or substituted or unsubstituted carboxamide;
R6 is absent, H, or alkyl;
R7 and R8 are each independently H, Ci-6 alkyl or R7 and R8 join together to form a fused or bridging ring system;
— represents a single or double bond; x is 0, 1 or 2;
R9 is absent or oxo; and pharmaceutically acceptable salts, solvates, and prodrugs thereof; provided that when R6 is absent, R7 and R8 are

R4 is /n-tolyl, then R is not substituted or unsubstituted piperidinyl; substituted or unsubstituted pyrrolidinyl; -CH2OH; -CH2(substituted or unsubstituted heterocyclyl); or C(O)NR*R** wherein R* and R** join to form a substituted or unsubstituted heterocyclic group.
[0009] Embodiment 2 is the compound of embodiment 1 , wherein R is H, oxo, substituted or unsubstituted Ci-6 alkyl, substituted or unsubstituted Ci-6 alkenyl, substituted or unsubstituted monocyclic heteroaryl, substituted or unsubstituted monocyclic heterocyclyl. or RaRbNC(O)-; and each Ra and Rb is independently H, Ci-6 alkyL Ci-shaloalkyl, Ci-6 cycloalkyl, or CH3C(O)-. [0010] Embodiment 3 is the compound of embodiment 2, wherein each Ra and Rb is independently H, cyclopropyl, cyclobutyl, methyl, ethyl, CH3C(O)-, -CD3, or -CFs.
[0011] Embodiment 4 is the compound of embodiment 1 or 2, wherein R is H, oxo, methyl, ethyl, propyl, isopropyl, isobutyl, propenyl, pyridyl, pyrimidinyl, pyrazolyl, dioxanyl, oxetanyl, morpholino, 3,6-dihydro-2H-pyranyl, or RaRbNC(O)-.
[0012] Embodiment 5 is the compound of embodiment 1 or 2, wherein R is H.
[0013] Embodiment 6 is the compound of any one of embodiments 1-5, wherein Rx is monocyclic heteroaryl, monocyclic heterocyclyl, Ci-6 alkyl. Ci-s cycloalkyl, RaRbN, RaO-, Rc, RaRbN-(Ci-6 alkyl)-, RaO-(Ci-6 alkyl)-, or RCO-(CI-6 alkyl)-; and Rc is H, methyl, ethyl, HOC(O)CH2-, CH3OC(O)CH2-, (CH3)3COC(O)CH2-, (CH3)3COC(O)NH-, H2NC(O)CH2-,
CH3NHC(O)CH2-, (CHS)2NC(O)CH2-, NC-ethyl, H2N-, H2N-ethyl, CH3NH-ethyl, or (CH3)2N- ethyl.
[0014] Embodiment 7 is the compound of any one of embodiments 1-5, wherein Rx is methyl, cyclopropyl, cyclobutyl, pyridyl, pyrazolyl, morpholino, piperidinyl, piperazinyl, RaRbN, RaO-, Rc, RaRbN-ethyl, RcO-ethyl-, RcO-methyl-, or RcO-isopentyl.
[0015] Embodiment 8 is the compound of any one of embodiments 1-7, wherein R1 is H, Ci-6 alkyl, C i-6 cycloalkyl, monocyclic heterocyclyl, RaO-, RaO-(Ci-6 alkyd), RaRbNC(O)-, or alkyl- SO2-.
[0016] Embodiment 9 is the compound of any one of embodiments 1-7, wherein R1 is H, propyl, ethyl, methyl, isobutyl, cyclobutyl, cyclopropyl. azetidinyL oxetanyl. piperidinyl, tetrahydropyran, tetrahydrofuran, thietane 1,1 dioxide, hexahydropyrimidin-2-one, RaO-, RaO- ethyl, RaO-methyl, RaRbNC(O)-, CH3CH2SO2-, (CH3)2CHSO2-, or cyclopropyl-SCh-.
[0017] Embodiment 10 is the compound of any one of embodiments 1-9, wherein Ry is C1-6 alkyl, C1-6 alkenyl. C1-6 haloalkyl, C1-6 cycloalkyl, monocyclic heteroaryl, monocyclic aryl, monocyclic heterocyclyl, halo, -CN, -OH, RaO-, CH3SO2-. CH?C(O)-, RaOC(O)-, RaRbNC(O)-, H2NC(O)NH-, RaRbN-, or methylallyl, each further substituted with 0, 1, or 2 Rd groups where Rd is methyl, oxo, CHsC(O)-, CH3OC(O)-, CH3SO2-, -F, (CH3)3CO-, (CD3)3CO-, RaO-, -CN, NH2, (CH3)3COC(O)-, H2NC(O)-, CH3HNC(O)-.
[0018] Embodiment 11 is the compound of any one of embodiments 1-9, wherein Ry is methyl, cyclopropyl, cyclobutyl, pyridinyl, phenyl, piperidinyl, pyrrolidinyl, oxetanyl, pyrrolidinone, imidazolidin-2-one, oxazolidin-2-one, lH-l,2,4-triazol-5-one, tetrahydropyran, dioxanyl, azetidinyl, -CN, -F,
-CF3. -OH. RaO-, CH3SO2-, CH3C(O)-, HOC(O)-. RaRbNC(O)-. H2NC(O)NH-. RaRbN-, or methylallyl.
[0019] Embodiment 12 is the compound of any one of embodiments 1-11, wherein R4 is H, phenyl, pyrazolyl, thiazolyl, thienyl, oxabicyclo[2.1.1]hexanyl, and norbomanyl, each substituted by 0, 1. or 2 Rz wherein each Rz is -CN, halo, C1-6 alkyl, C1-6 alkoxy, C1-6 haloalkyl, C1-6 cycloalkyl, deuterated alkyl, or C1-6 haloalkoxy.
[0020] Embodiment 13 is the compound of embodiment 12, wherein R4 is H, phenyl, pyrazolyl, thiazolyl, thienyl, oxabicyclo[2. l. l]hexanyl, or norbomanyl.
[0021] Embodiment 14 is the compound of embodiment 12 or 13, wherein each Rz is -Cl, -F, - CN, -CF3, -CDS, -OCF3, methyl, ethyl, cyclopropyl, methoxy, difluoromethoxy, or difluoromethyl.
[0022] Embodiment 15 is the compound of any one of embodiments 1-14, wherein each R5 is - C(O)NRaRb, -C(O)ORa, unsubstituted C1-6 alkyl, or C1-6 alkyl substituted with 1 or 2 -ORa or - NH2, -NH-Ra, -NRaRb groups.
[0023] Embodiment 16 is the compound of any one of embodiments 1-14, wherein each R5 is H, hydroxymethyl, hydroxyethyl, methoxymethyl, H2N-methyl, CH3NH-methyl, or (CH3)2N- methyl, trideuteriomethoxymethyl, -C(O)NRaRb, -C(O)ORa.
[0024] Embodiment 17 is the compound of any one of embodiments 1-16, wherein R6 is absent.
[0025] Embodiment 18 is the compound of any one of embodiments 1-176, wherein R is not substituted with an Rx group.
[0026] Embodiment 19 is the compound of any one of embodiments 1-17, wherein R is substituted with 1 Rx group.
[0027] Embodiment 20 is the compound of any one of embodiments 1-17, wherein R is substituted with 2 Rx groups.
[0028] Embodiment 21 is the compound of any one of embodiments 1-20, wherein R1 is not substituted with an Ry group.
[0029] Embodiment 22 is the compound of any one of embodiments 1-20, wherein R1 is substituted with 1 Ry group.
[0030] Embodiment 23 is the compound of any one of embodiments 1-20, wherein R1 is substituted with 2 Ry groups.
[0031] Embodiment 24 is the compound of any one of embodiments 10-23, wherein Ry is not substituted with an Rd group.
[0032] Embodiment 25 is the compound of any one of embodiments 10-23, wherein Ry is substituted with 1 Rd group.
[0033] Embodiment 26 is the compound of any one of embodiments 10-23, wherein Ry is substituted with 2 Rd groups.
[0034] Embodiment 27 is the compound of any one of embodiments 1-26, wherein R4 is not substituted with an Rz group.
[0035] Embodiment 28 is the compound of any one of embodiments 1-26, wherein R4 is substituted with 1 Rz group.
[0036] Embodiment 29 is the compound of any one of embodiments 1-26, wherein R4 is substituted with 2 Rz groups.
[0037] Embodiment 30 is the compound of any one of embodiments 1-29, wherein W is CH. [0038] Embodiment 31 is the compound of any one of embodiments 1-30, wherein R7 and R8 are each H.
[0039] Embodiment 32 is the compound of any one of embodiments 1-30, wherein R7 and R8 join together to form a 1 or 2 carbon bridged rin g system.
[0040] Embodiment 33 is the compound of embodiment 1, having the structure of Formula (Ic)
 and pharmaceutically acceptable salts, solvates, and prodrugs thereof.
and pharmaceutically acceptable salts, solvates, and prodrugs thereof.
[0041] Embodiment 34 is the compound of embodiment 33, wherein R is H.
[0042] Embodiment 35 is the compound of embodiment 33, wherein R4 is m-tolyl or m- chlorophenyl.
[0043] Embodiment 36 is the compound of embodiment 33, wherein R1 is H, methyl, ethyl or isopropyl substituted with 1 Ry group and Ry is substituted with 1 or 2 Rd groups.
[0044] Embodiment 37 is the compound of embodiment 33, having the structure of Formula (Ic-
 pharmaceutically acceptable salts, solvates, and prodrugs thereof wherein n is 0, 1 or 2; and each Rx is independently halo, cyano, C1-3 alkyl, C1-3 haloalkyl, C3-6 cycloalkyl, C1-3 alkoxy, or C1-3 haloalkoxy.
pharmaceutically acceptable salts, solvates, and prodrugs thereof wherein n is 0, 1 or 2; and each Rx is independently halo, cyano, C1-3 alkyl, C1-3 haloalkyl, C3-6 cycloalkyl, C1-3 alkoxy, or C1-3 haloalkoxy.
[0045] Embodiment 38 is the compound of embodiment 37, wherein n is 1 or 2; and each Rx is independently F, Cl, cyano, methyl, fluoromethyl, difluoromethyl, trifluoromethyl, ethyl, fluoroethyl, difluoroethyl, trifluoroethyl, cyclopropyl, methoxy, fluoromethoxy, difluoromethoxy, trifluoromethoxy, ethoxy, fluoroethoxy, difluoroethoxy, or trifluoroethoxy. [0046] Embodiment 39 is the compound of embodiment 37, wherein R1 is H, methyl, ethyl or isopropyl substituted with 1 Ry group and Ry is substituted with 1 or 2 Rd groups.
[0047] Embodiment 40 is a compound selected from the compounds in Table 1 and pharmaceutically acceptable salts thereof or a compound selected from
4-[3-(2-methoxyethyl)-5-[3-(m-tolyl)pyrazol-l-yl]imidazo[4,5-b]pyridin-7-yl]morpholine; 2-(7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-3-yl)acetonitrile;
5-({7-morpholino-5-[3-(m-tolyl)-l-pyrazolyl]-3H-l,3,4-triazainden-3-yl}methyl)-2- pyrrohdinone;
l-(2-{7-morpholino-5-[3-(m-tolyl)-l-pyrazolyl]-3H-l,3,4-triazainden-3-yl}ethyl)-2- imidazolidinone;
4-({7-morpholino-5-[3-(m-toly'l)-l-pyrazolyl]-3H-l,3,4-triazainden-3-yl}methyl)-l,3- oxazolidin-2-one;
N-methyl-3-({7-morpholino-5-[3-(m-tolyl)-l-pyrazolyl]-3H-l,3,4-triazainden-3-yl}methyl)-l- azetidinecarboxamide; l-[3-({5-[3-(m-chlorophenyl)-l-pyrazolyl]-7-morpholino-3H-l,3,4-triazainden-3-yl}methyl)-l- azetidinyl]-! -ethanone; and 3-({7-morpholino-5-[3-(m-tolyl)-l-pyrazolyl]-3H-f,3,4-triazainden-3-yl}methyl)-l- azetidinecarboxamide, and pharmaceutically acceptable salts thereof.
[0048] Embodiment 41 is a compound and/or a pharmaceutically acceptable salt of any one of embodiments 1-40, wherein one or more hydrogen atoms attached to carbon atoms of the compound are replaced by deuterium atoms.
[0049] Embodiment 42 is a pharmaceutical composition comprising a compound and/or a pharmaceutically acceptable salt of any one of embodiments 1-41 and a pharmaceutically acceptable excipient.
[0050] Embodiment 43 is a method of inhibiting PIKf ve kinase in a subject in need thereof comprising administering to the subject an effective amount of a compound of any one of embodiments 1-41, or a pharmaceutical composition of embodiment 42.
[0051] Embodiment 44 is a method of treating a neurological disease associated with PIKfyve activity in a subject in need thereof comprising administering to the subject an effective amount of a compound of any one of embodiments 1-51, or a pharmaceutical composition of embodiment 52.
[0052] Embodiment 45 is the method of embodiment 44, wherein the neurological disease is amyotrophic lateral sclerosis (ALS), primary' lateral sclerosis (PLS), Charcot-Marie-Tooth (CMT; including ty pe 4J (CMT4J)), and Yunis-Varon syndrome, autophagy, polymicrogyria (including polymicrogyria with seizures), temporo-occipital polymicrogyria. Pick’s disease, Parkinson’s disease, Parkinson’s disease with Lewy7 bodies, dementia with Lewy bodies, Lewy' body disease, fronto-temporal dementia, diseases of neuronal nuclear inclusions of polyglutamine and intranuclear inclusion bodies, disease of Marinesco and Hirano bodies, tauopathy, Alzheimer’s disease, neurodegeneration, spongiform neurodegeneration, peripheral neuropathy, leukoencephalopathy, inclusion body disease, progressive supranuclear palsy, corticobasal syndrome, chronic traumatic encephalopathy, traumatic brain injury' (TBI), cerebral ischemia, Guillain-Barre Syndrome, chronic inflammatory' demyelinating polyneuropathy, multiple sclerosis, a lysosomal storage disease, Fabry’s disorder, Gaucher's disorder, Niemann
Pick C disease, Tay-Sachs disease, and Mucolipidosis type IV, neuropathy. Huntington’s disease, a psychiatric disorder, ADHD, schizophrenia, a mood disorder, major depressive disorder, depression, bipolar disorder I, or bipolar disorder II.
[0053] Embodiment 46 is the method of embodiment 44, wherein the disease is ALS, FTD, Alzheimer’s disease. Parkinson’s disease. Huntington’s disease, or CMT.
[0054] Embodiment 47 is the method of embodiment 44, wherein the disease is ALS.
[0055] Embodiment 48 is the method of embodiment 44, wherein the disease is a tauopathy such as Alzheimer’s disease, progressive supranuclear palsy, corticobasal syndrome, frontotemporal dementia, or chronic traumatic encephalopathy.
[0056] Embodiment 49 is the method of embodiment 44, wherein the disease is a lysosomal storage disease such as Fabry’s disorder, Gaucher's disorder, Niemann Pick C disease, Tay- Sachs disease, or Mucolipidosis type IV.
[0057] Embodiment 50 is the method of embodiment 44, wherein the disease is a psychiatric disorder such as ADHD, schizophrenia, or mood disorders such as major depressive disorder, depression, bipolar disorder I, or bipolar disorder II.
[0058] Embodiment 51 is a compound of any one of embodiments 1-41 for use as a medicament.
[0059] Embodiment 52 is the compound of embodiment 51, wherein the compound is for use in treating a neurological disease treatable by inhibition of PIKlyve kinase.
[0060] Embodiment 53 is the use of a compound of any one of embodiments 1-41 in the manufacture of a medicament for treating a neurological disease in a subject in which PIKl ve contributes to the pathology and/or symptoms of the disease.
Detailed Description
[0061] Reference will now be made in detail to certain embodiments of the invention. While the invention will be described in conjunction with the described embodiments, it will be understood that such descriptions are not intended to limit the invention to those embodiments. On the contrary, the invention is intended to cover all alternatives, modifications, and equivalents, which may be included within the invention as defined by the appended claims.
[0062] The section headings used herein are for organizational purposes only and are not to be construed as limiting the desired subject matter in any way. In the event that any literature incorporated by reference contradicts any term defined in this specification, this specification controls.
[0063] Unless otherwise defined herein, scientific and technical terms used herein have the meanings that are commonly understood by those of ordinary skill in the art. In the event of any latent ambiguity, definitions provided herein take precedence over any dictionary’ or extrinsic
definition. Unless otherwise required by context, singular terms shall include pluralities and plural terms shall include the singular.
[0064] Before describing the present teachings in detail, it is to be understood that the disclosure is not limited to specific compositions or process steps, as such may vary. It should be noted that, as used in this specification and the appended claims, the singular form “a,” “an,” and “the’’ include plural references unless the context clearly dictates otherwise. Thus, for example, reference to “a surfactant” includes a plurality of surfactants and the like.
[0065] Numeric ranges are inclusive of the numbers defining the range. Measured and measurable values are understood to be approximate, taking into account significant digits and the error associated with the measurement. Also, the use of “comprise,” “comprises,” “comprising,” “contain,” “contains,” “containing,” “include,” “includes,” “included,” and “including” are not intended to be limiting. It is to be understood that both the foregoing general description and detailed description are exemplary and explanatory only and are not restrictive of the teachings.
[0066] Unless specifically noted in the above specification, embodiments in the specification that recite “comprising” various components are also contemplated as “consisting of’ or “consisting essentially of' the recited components; embodiments in the specification that recite “consisting of’ various components are also contemplated as “comprising” or “consisting essentially of’ the recited components; and embodiments in the specification that recite “consisting essentially of’ various components are also contemplated as “consisting of’ or “comprising” the recited components (this interchangeability does not apply to the use of these terms in the claims).
[0067] Unless otherwise stated, the following terms used in the specification and claims are defined for the purposes of this disclosure and have the following meanings.
[0068] As used herein, the term “about” refers to a value or composition that is within an acceptable error range for the particular value or composition as determined by one of ordinary’ skill in the art, which will depend in part on how the value or composition is measured or determined, i.e., the limitations of the measurement system. For example, “about” or “approximately” can mean within one or more than one standard deviation per the practice in the art. Alternatively, “about” or “approximately” can mean a range of up to 10% (i.e., ±10%) or more depending on the limitations of the measurement system. For example, about 5 mg can include any number between 4.5 mg and 5.5 mg. Furthermore, particularly with respect to biological systems or processes, the terms can mean up to an order of magnitude or up to 5-fold of a value. When particular values or compositions are provided in the instant disclosure, unless otherwise stated, the meaning of “about” or “approximately” should be assumed to be within an
acceptable error range for that particular value or composition. Reference to "about" a value or parameter herein includes (and describes) embodiments that are directed to that value or parameter per se.
[0069] The term “or” is used in the inclusive sense, i.e.. equivalent to “and/or,” unless the context requires otherwise.
[0070] The term “and/or” used herein is to be taken mean specific disclosure of each of the specified features or components with or without the other. For example, the term “and/or” as used in a phrase such as “A and/or B” herein is intended to include “A and B,” “A or B,” “A” (alone), and “B” (alone). Likewise, the term “and/or” as used in a phrase such as “A. B, and/or C” is intended to encompass each of the following aspects: A, B, and C; A, B, or C; A or C; A or B; B or C; A and C; A and B; B and C; A (alone); B (alone); and C (alone).The terms “or a combination thereof’ and “or combinations thereof’ as used herein refers to any and all permutations and combinations of the listed terms preceding the term. For example, “A, B, C, or combinations thereof’ is intended to include at least one of: A, B, C, AB, AC, BC, or ABC, and if order is important in a particular context, also BA, CA, CB, ACB, CBA, BCA, BAC, or CAB. Continuing with this example, expressly included are combinations that contain repeats of one or more item or term, such as BB, AAA, AAB, BBC, AAABCCCC, CBBAAA, CABABB, and so forth. The skilled artisan will understand that typically there is no limit on the number of items or terms in any combination, unless otherwise apparent from the context.
[0071] A dash (“-”) that is not between two letters or symbols is used to indicate a point of attachment for a substituent. For example, -C(O)NH2 is attached through the carbon atom. A dash at the front or end of a chemical group is a matter of convenience; chemical groups may be depicted with or without one or more dashes without losing their ordinary meaning. A wavy line or a dashed line drawn through a line in a Formula indicates a specified point of attachment of a group. Unless chemically or structurally required, no directionality or stereochemistry is indicated or implied by the order in which a chemical group is written or named.
[0072] The prefix “Cu-v” indicates that the following group has from u to v carbon atoms. For example, “Ci-6 alkyl” indicates that the alkyl group has from 1 to 6 carbon atoms.
[0073] “Alkyl” means a linear saturated monovalent hydrocarbon radical of one to ten carbon atoms or a branched saturated monovalent hydrocarbon radical of three to ten carbon atoms, e.g., methyl, ethyl, propyl, 2-propyl. butyl (including all isomeric forms), pentyl (including all isomeric forms), and the like.
[0074] “Alkylene” means a linear saturated divalent hydrocarbon radical of one to ten carbon atoms or a branched saturated divalent hydrocarbon radical of three to ten carbon atoms unless
otherwise stated e.g., methylene, ethylene, propylene, 1 -methylpropylene, 2-methylpropylene, butylene, pentylene, and the like.
[0075] “Alkenyl” means a linear unsaturated or partially unsaturated monovalent hydrocarbon radical of two to ten carbon atoms or a branched unsaturated or partially unsaturated monovalent hydrocarbon radical of three to ten carbon atoms, e.g., ethylenyl, propylenyl. 2-propylenyl, butenyl (including all isomeric forms), pentenyl (including all isomeric forms), and the like. C2- xalkenyl refers to an alkenyl group with from 2 to x carbon atoms.
[0076] “Alkylsulfonyl” means a-SChR radical where R is alkyl as defined above, e.g., methylsulfonyl, ethyl sulfonyl, and the like.
[0077] “Amino” means a -NH2. Substituted amino means -NR*R**, where the R* and R** substituents are alkyl or substituted alkyl as defined herein.
[0078] “Alkoxy” means a -OR radical where R is alky l as defined above, e g., methoxy, ethoxy, propoxy, or 2-propoxy, n-, iso-, or /m-butoxy. and the like.
[0079] “Alkoxyalkyl” means a linear monovalent hydrocarbon radical of one to ten carbon atoms or a branched monovalent hydrocarbon radical of three to ten carbons substituted with an alkoxy group, (in one embodiment one or two alkoxy groups), as defined above, e.g., 2- methoxyethyl, 1-, 2-. or 3 -methoxy propyl. 2-ethoxyethyl, and the like.
[0080] “Alkoxy carbonyl” means a -C(O)OR radical where R is alkyl as defined above, e.g., methoxycarbonyl, ethoxycarbonyl, and the like.
[0081] “Acyl” means a -C(O)R radical where R is alkyl, haloalky 1, or cycloalkyl, e.g., acetyd, propionyl, cyclopropylcarbonyl, and the like. When R is alky l, the radical is also referred to herein as alkylcarbonyl.
[0082] “Aryl” refers to an aromatic carbocyclic group having a single ring (e.g., monocyclic) or multiple rings (e.g., bicyclic or tricyclic) including fused systems. As used herein, aryl has 6 to 20 ring carbon atoms (i.e., C6-20 ary l), 6 to 18 carbon ring atoms (i.e., Ce-18 ary l), 6 to 12 carbon ring atoms (i.e., C6-12 aryl) or 6 to 10 carbon ring atoms (i.e., Ce-io aryl). Examples of aryl groups include phenyl, naphthyl, fluorenyl and anthryl. Aryl, however, does not encompass or overlap in any way with heteroaryl defined below. If one or more aryl groups are fused with a heteroary l, the resulting ring system is heteroary l. If one or more aryl groups are fused with a heterocycly 1, the resulting ring system is heterocyclyl.
[0083] “Cycloalkyl” means a cyclic saturated monovalent hydrocarbon radical of three to ten carbon atoms wherein one or two carbon atoms may be replaced by an oxo group, e.g., cyclopropy 1, cyclobutyl, cyclopentyl, or cyclohexyl, and the like.
[0084] “Carboxy ” means -C(O)OH.
[0085] “Carboxamide” means - C(O)NR*R**.
[0086] ‘Carbamate” means -OC(O)NR*R**.
[0087] ‘ ‘Halo” means fluoro, chloro, bromo, or iodo; in one embodiment fluoro or chloro. [0088] “Haloalkyl” means alkyl radical as defined above, which is substituted with one or one to five halogen atoms (in one embodiment fluorine or chlorine,) including those substituted with different halogens, e.g., -CH2CI. -CF3. -CHF2. -CH2CF3, -CF2CF3, -CF(CH3)2, and the like. When the alkyl is substituted with only fluoro, it can be referred to in this disclosure as fluoroal kyl.
[0089] “Haloalkoxy” means a -OR radical where R is haloalkyl as defined above e.g., -OCF3, -OCHF2, and the like. When R is haloalkyl where the alkyl is substituted with only fluoro, it can be referred to in this disclosure as fluoroalkoxy.
[0090] “Hydroxyalkyl” means a linear monovalent hydrocarbon radical of one to six carbon atoms or a branched monovalent hydrocarbon radical of three to six carbons substituted with one or two hydroxy groups, provided that if two hydroxy groups are present they are not both on the same carbon atom. Representative examples include, but are not limited to, hydroxymethyl, 2- hydroxy ethyl, 2-hydroxypropyl, 3-hydroxypropyl, l-(hydroxymethyl)-2-methylpropyl, 2- hydroxybutyl, 3-hydroxybutyl. 4-hydroxybutyl, 2,3-dihydroxypropyl, l-(hydroxymethyl)-2- hydroxyethyl, 2.3-dihydroxybutyl. 3,4-dihydroxybutyl and 2-(hydroxymethyl)-3-hydroxypropyl. Further examples include, but are not limited to, 2-hydroxyethyl, 2.3-dihydroxypropyl. and 1- (hydroxymethyl)-2-hydroxy ethyl.
[0091] “Heterocyclyl” means a saturated or unsaturated monovalent monocyclic or bi-cyclic group (fused bi-cyclic or bridged bi-cyclic or spiro compounds) of 4 to 10 ring atoms in which one or two ring atoms are heteroatom selected from N, O. and S(O)n, where n is an integer from 0 to 2, the remaining ring atoms being C. Additionally, one or two ring carbon atoms in the heterocyclyl ring can optionally be replaced by a -CO- group. More specifically the term heterocyclyl includes, but is not limited to, oxetanyl, pyrrolidino, piperidino, homopiperidino, 2- oxopyrrolidinyl, 2-oxopiperidinyl, morpholino, piperazino, tetrahydropyranyl, thiomorpholino, hexahydropyrrolo[1.2-a]pyrazin-6(2H)-one-yl, tetrahydro-lH-oxazolo[3.4-a]pyrazin-3(5H)-one- yl, 5,6,7,8-tetrahydro-[l,2,4]triazolo[4,3-a]pyrazine-yl, 3-oxa-8-azabicyclo[3.2.1]octane-yl, 6- oxa-l-azaspiro[3.3]heptanyl, 2-oxa-6-azaspiro[3.3]heptanyl, and the like. When the heterocyclyl ring is unsaturated it can contain one or two ring double bonds provided that the ring is not aromatic. Cx-y heterocyclyl refers to a heterocyclyl group with from x to y carbon atoms in the ring, where x and y are integers.
[0092] ‘Heterocyclylalkyl” or “heterocycloalkyl” means a -(alkylene)-R radical where R is heterocyclyl ring as defined above e g., tetraydrofuranylmethyl, piperazinylmethyl, morpholinylethyl, and the like.
[0093] “Heterocycloamino” means a saturated or unsaturated monovalent monocyclic group of 4 to 8 ring atoms in which one or two ring atoms are heteroatom selected from N, O, or S(O)n, where n is an integer from 0 to 2, the remaining ring atoms being C provided that at least one of the ring atoms is N. Additionally, one or two ring carbon atoms in the heterocycloamino ring can optionally be replaced by a -CO- group. When the heterocycloamino ring is unsaturated it can contain one or two ring double bonds provided that the ring is not aromatic.
[0094] “Heterocycloaminoalkyr means a -(alkylene)-R radical where R is heterocycloamino as described above.
[0095] “Heteroaryf’ means a monovalent monocyclic or bicyclic aromatic radical of 5 to 10 ring atoms where one or more, (in one embodiment one, two, or three), ring atoms are heteroatom selected from N, O, and S, the remaining ring atoms being carbon. Representative examples include, but are not limited to, pyrrolyl, thienyl, thiazolyl, imidazolyl, furanyl, indolyl, isoindolyl, oxazolyl, isoxazolyl, benzothiazolyl, benzoxazolyl, quinolinyl, isoquinolinyl, pyridinyl, pynmidinyl, pyrazinyl, pyridazinyl, triazolyl, tetrazolyl, and the like. Cx-yheteroaryl refers to a heteroaryl group with from x to y carbon atoms, where x and y are integers.
[0096] “Oxo” refers to a >C=O group.
[0097] “Urea” means -NRC(O)NR*R**.
[0098] “Triazaindene” and “triazaindenyl” refers to 3f/-imidazo(4.5-6)pyridine. unless specified otherwise.
[0099] “Mammal” as used herein means domesticated animals (such as dogs, cats, and horses), and humans. In one embodiment, mammal is a human.
[0100] The term “salt” or “pharmaceutically acceptable salt” refers to salts derived from a variety of organic and inorganic counter ions well known in the art. Pharmaceutically acceptable acid addition salts can be formed with inorganic acids and organic acids. Inorganic acids from which salts can be derived include, for example, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like. Organic acids from which salts can be derived include, for example, acetic acid, propionic acid, glycolic acid, pyruvic acid, oxalic acid, maleic acid, malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, /Moluenesul fonic acid, salicylic acid, and the like. Pharmaceutically acceptable base addition salts can be formed with inorganic and organic bases. Inorganic bases from which salts can be derived include, for example, sodium, potassium, lithium, ammonium, calcium, magnesium, iron, zinc, copper, manganese, aluminum, and the like. Organic bases from which salts can be derived include, for example, primary, secondary , and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, basic ion exchange resins, and the like, specifically such as
isopropylamine, trimethylamine, diethylamine, triethylamine, tripropylamine, and ethanolamine. In some embodiments, the pharmaceutically acceptable base addition salt is chosen from ammonium, potassium, sodium, calcium, and magnesium salts. It is understood that the pharmaceutically acceptable salts are non-toxic. Additional information on suitable pharmaceutically acceptable salts can be found in Remington 's Pharmaceutical Sciences, 17th ed.. Mack Publishing Company, Easton, PA, 1985, which is incorporated herein by reference. [0101] “Oxo” means an =(O) group and “carbonyl” means a >C(O) group.
[0102] “Optional” or “optionally” means that the subsequently described event or circumstance may but need not occur, and that the description includes instances where the event or circumstance occurs and instances in which it does not. For example, “heterocyclyl group optionally substituted with an alkyl group” means that the al kyl may but need not be present, and the description includes situations where the heterocyclyl group is substituted with an alkyl group and situations where the heterocyclyl group is not substituted with alkyl.
[0103] The phrases “parenteral administration” and “administered parenterally” as used herein means modes of administration other than enteral and topical administration, usually by injection, and includes, without limitation, intravenous, intramuscular, intraarterial, intrathecal, intracapsular, intraorbital, intracardiac, intradermal, intraperitoneal, transtracheal, subcutaneous, subcuticular, intraarticular, subcapsular. subarachnoid, intraspinal and intrastemal injection and infusion.
[0104] The phrase “pharmaceutically acceptable” is employed herein to refer to those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
[0105] The phrase “pharmaceutically acceptable excipient” or “pharmaceutically acceptable carrier” as used herein means a pharmaceutically acceptable material, composition, or vehicle, such as a liquid or solid filler, diluent, excipient, solvent or encapsulating material. Each carrier must be “acceptable” in the sense of being compatible with the other ingredients of the formulation and not injurious to the patient. Some examples of materials which can serve as pharmaceutically acceptable carriers include: (1) sugars, such as lactose, glucose and sucrose; (2) starches, such as com starch and potato starch; (3) cellulose, and its derivatives, such as sodium carboxymethyl cellulose, ethyl cellulose and cellulose acetate; (4) powdered tragacanth; (5) malt; (6) gelatin; (7) talc; (8) excipients, such as cocoa butter and suppository' waxes; (9) oils, such as peanut oil, cottonseed oil, safflower oil, sesame oil, olive oil, com oil and soybean oil; (10) glycols, such as propylene glycol; (11) polyols, such as glycerin, sorbitol, mannitol and
polyethylene glycol; (12) esters, such as ethyl oleate and ethyl laurate; (13) agar; (14) buffering agents, such as magnesium hydroxide and aluminum hydroxide; (15) alginic acid; (16) pyrogen- free water; (17) isotonic saline; (18) Ringer's solution; (19) ethyl alcohol; (20) phosphate buffer solutions; and (21) other non-toxic compatible substances employed in pharmaceutical formulations.
[0106] ’‘Treating” or “treatment” of a disease includes:
(1) preventing the disease, e.g., causing the clinical symptoms of the disease not to develop in a mammal that may be exposed to or predisposed to the disease but does not yet experience or display symptoms of the disease;
(2) inhibiting the disease, e.g., arresting or reducing the development of the disease or its clinical symptoms; or
(3) relieving the disease, e.g., causing regression of the disease or its clinical symptoms. [0107] A “therapeutically effective amount” means the amount of a compound of Formula (I) (or any of the embodiments thereof described herein), that, when administered to a mammal for treating a disease, is sufficient to treat the disease. The “therapeutically effective amount” will vary depending on the compound, the disease and its severity7 and the age, weight, etc., of the mammal to be treated.
[0108] The compounds described herein may in some cases exist as diastereomers, enantiomers, or other stereoisomeric forms. All chiral, diastereomeric, racemic forms, as individual forms and mixtures thereof, are within the scope of this disclosure, unless the specific stereochemistry7 or isomeric form is specifically indicated. Compounds of the present disclosure containing an asymmetrically substituted atom may be isolated in optically active, optically enriched, optically pure, or racemic forms. It is well known in the art how to prepare optically active forms, such as by resolution of materials. Separation of stereoisomers may be performed by chromatography or by forming diastereomers and separating by recry stallization, or chromatography, or any combination thereof. (Jean Jacques, Andre Collet, Samuel H. Wilen, “Enantiomers, Racemates and Resolutions”. John Wiley and Sons, Inc., 1981, herein incorporated by reference for this disclosure). Stereoisomers may' also be obtained by stereoselective synthesis.
[0109] Certain compounds of Formula (I) (or any of the embodiments thereof described herein) and/or a pharmaceutically acceptable salt thereof may exist as tautomers and/or geometric isomers. All possible tautomers and cis and trans isomers, as individual forms and mixtures thereof, are within the scope of this disclosure. For example, pyrazole tautomers as shown below are equivalent structures. The depiction of one such structure is intended to encompass both structures.

[0110] Additionally, as used herein the term alkyl includes all the possible isomeric forms of said alkyl group albeit only a few examples are set forth. Furthermore, when the cyclic groups such as heteroaryl, heterocyclyl are substituted, they include all the positional isomers.
[0111] Pharmaceutically acceptable salts of the compounds of Formula (I) (or any of the embodiments thereof described herein) are within the scope of this disclosure. In addition, the compounds described herein include hydrates and solvates of the compounds or pharmaceutically acceptable salts thereof.
[0112] The present disclosure also includes the prodrugs of compounds of Formula (I) (or any of the embodiments thereof described herein) and/or a pharmaceutically acceptable salt thereof. The term prodrug is intended to represent covalently bonded carriers, which are capable of releasing the active ingredient of Formula (I) (or any of the embodiments thereof described herein) when the prodrug is administered to a mammalian subject. Release of the active ingredient occurs in vivo. Prodrugs can be prepared by techniques known to one skilled in the art. These techniques generally modify appropriate functional groups in a given compound. These modified functional groups however regenerate original functional groups in vivo or by routine manipulation. Prodrugs of compounds of Formula (I) (or any of the embodiments thereof described herein) include compounds wherein a hydroxy, amino, carboxylic, or a similar group is modified. Examples of prodrugs include, but are not limited to esters (e.g., acetate, formate, and benzoate derivatives), carbamates (e.g., A/A imethylaminocarbonyl) of hydroxy or amino functional groups in compounds of Formula (I)), amides (e.g., trifluoroacetylamino, acetylamino, and the like), and the like. Prodrugs of compounds of Formula (I) (or any of the embodiments thereof described herein) and/or a pharmaceutically acceptable salt thereof are also within the scope of this disclosure.
[0113] The present disclosure also includes polymorphic forms (amorphous as well as crystalline) and deuterated forms of compounds of Formula (I) (or any of the embodiments thereof described herein) and/or a pharmaceutically acceptable salt thereof.
[0114] The compounds disclosed herein, in some embodiments, are used in different enriched isotopic forms, e.g., enriched in the content of 2H, 3H, nC, 13C and/or 14C. In one particular embodiment, the compound is deuterated in at least one position. Such deuterated forms can be made by the procedure described in U.S. Patent Nos. 5,846.514 and 6,334,997. As described in U.S. Patent Nos. 5.846,514 and 6,334.997, deuteration can improve the metabolic stability and or efficacy, thus increasing the duration of action of drugs.
[0115] Unless otherwise stated, structures depicted herein are intended to include compounds which differ only in the presence of one or more isotopically enriched atoms. For example, compounds having the present structures except for the replacement of a hydrogen by a deuterium or tritium, or the replacement of a carbon by 13C- or 14C-enriched carbon are within the scope of the present disclosure.
[0116] The compounds of the present disclosure optionally contain unnatural proportions of atomic isotopes at one or more atoms that constitute such compounds. For example, the compounds may be labeled with isotopes, such as for example, deuterium (2H), tritium (3H), iodine-125 (125I) or carbon-14 (14C). Isotopic substitution with 2H, nC, 13C. 14C, 15C, 12N, 13N, 15N, 16N, 16O, 17O, 14F, 15F, 16F, 17F, 18F, 33S, 34S, 35S, 36S, 35CL 37C1, 79Br, 81Br, and 125I are all contemplated. All isotopic variations of the compounds of the present invention, whether radioactive or not, are encompassed within the scope of the present invention.
[0117] In certain embodiments, the compounds disclosed herein have some or all of the
 atoms replaced with 2H atoms. The methods of synthesis for deuterium-containing compounds are known in the art and include, by way of non-limiting example only, the following synthetic methods.
atoms replaced with 2H atoms. The methods of synthesis for deuterium-containing compounds are known in the art and include, by way of non-limiting example only, the following synthetic methods.
[0118] Deuterium substituted compounds are synthesized using various methods such as described in: Dean, Dennis C.; Editor. Recent Advances in the Synthesis and Applications of Radiolabeled Compounds for Drug Discovery and Development. [In: Curr., Pharm. Des., 2000; 6(10)] 2000, 110 pp; George W.; Varma, Rajender S. The Synthesis of Radiolabeled Compounds via Organometallic Intermediates, Tetrahedron, 1989, 45(21), 6601-21 ; and Evans, E. Anthony. Synthesis of radiolabeled compounds, J. Radioanal. Chem., 1981, 64(1-2), 9-32. [0119] Deuterated starting materials are readily available and are subjected to the synthetic methods described herein to provide for the synthesis of deuterium-containing compounds.
Large numbers of deuterium-containing reagents and building blocks are available commercially from chemical vendors, such as Aldrich Chemical Co.
[0120] In one aspect provided herein is a compound of Formula (I):
 wherein
wherein
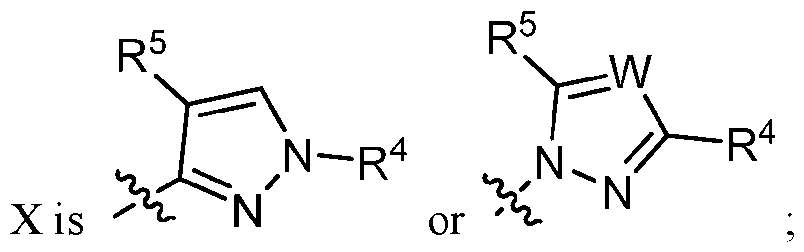
W is N, CH, or CR4*;
R is H, oxo, alkyl, alkenyl, heteroaryl, heterocyclyl, or carboxamide, each substituted with 0, 1, or 2 Rx groups and each Rx is oxo, hydroxyl, cyano, substituted or unsubstituted alky l, substituted or unsubstituted cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted amino, substituted or unsubstituted ester, substituted or unsubstituted sulfonyl, substituted or unsubstituted amide, substituted or unsubstituted carboxamide, substituted or unsubstituted alkoxy, or substituted or unsubstituted alkoxyalkyl;
R1 is H, alkyl, cycloalkyl, heterocyclyl, amino, alkoxy, alkoxyalkyl, amide, carboxamide, or sulfonyl, each substituted with 0, 1, or 2 Ry groups and each Ry is substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted alkoxy, substituted or unsubstituted alkoxyalkyl, substituted or unsubstituted amino, substituted or unsubstituted ester, substituted or unsubstituted sulfonyl, substituted or unsubstituted amide, substituted or unsubstituted carboxamide, substituted or unsubstituted urea, substituted or unsubstituted carbamate, halo, -CN, -OH, oxo, or methylallyl;
R4 and R4* are aryl, heteroaryl, or heterocyclyl, each substituted with 0, 1, or 2 Rz groups and each Rz is alkyl, cycloalkyl, haloalkyl, alkoxy, haloalkoxy, halo, or -CN, provided that only one of R4 and R4* is present; or when R3 and R4 are bonded to the same carbon, then R3 and R4 join to form a monocyclic or bicyclic cycloalkyl;
R5 is H, carboxyl, substituted or unsubstituted alkyl, substituted or unsubstituted alkoxy carbonyl, substituted or unsubstituted ester, or substituted or unsubstituted carboxamide; R6 is absent, H, or alkyl;
R7 and R8 are each independently H, Ci-6 alkyl or R7 and R8 join together to form a fused or bridging ring system;
— represents a single or double bond; x is 0, 1 or 2;
R9 is absent or oxo; and pharmaceutically acceptable salts, solvates, and prodrugs thereof;
provided that when R6 is absent, R7 and R8 are

R4 is m-tolyl. then R is not substituted or unsubstituted piperidinyl; substituted or unsubstituted pyrrolidinyl; -CH2OH; -CH2(substituted or unsubstituted heterocyclyl); or C(O)NR*R** wherein R* and R** join to form a substituted or unsubstituted heterocyclic group.
[0121] In some embodiments, W is CH. In some embodiments, W is N. In some embodiments, W is CR4*.
[0122] In some embodiments. R is H, oxo, substituted or unsubstituted Ci-6 alkyl, substituted or unsubstituted monocyclic heteroaryl, substituted or unsubstituted monocyclic heterocyclyl, or RaRbNC(O)-. In some embodiments, R is H, oxo, methyl, ethyl, propyl, isopropyl, isobutyl, propenyl, pyridyl, pyrimidinyl, pyrazolyl, dioxanyl, oxetanyl, morpholino, 3,6-dihydro-2H- pyranyl, or RaRbNC(O)-.
[0123] In some embodiments of Formula (I), R is H and R6 is absent.
[0124] In some embodiments of Formula (I), R is not RaRbNC(O)-.
[0125] In some embodiments, Rx is monocyclic heteroaryl, monocyclic heterocyclyl, Ci-6 alkyl, Ci-6 cycloalkyl, RaRbN, RaO-, Rc, RaRbN-(Ci-6 alkyl)-, RaO-(Ci-6 alkyl)-, or RCO-(CI-6 alkyl)-. In some embodiments, Rx is methyl, cyclopropyl, cyclobutyl, pyridyl, pyrazolyl, morpholino. piperidinyl, piperazinyl, RaRbN, RaO-, Rc, RaRbN-ethyl, RcO-ethy 1-, RcO-methyl-, or RCO- isopentyl.
[0126] In some embodiments, R is not substituted with an Rx group. In some embodiments, R is substituted with 1 Rx group. In some embodiments, R is substituted with 2 Rx groups.
[0127] In some embodiments, each Ra and Rbis independently H, Ci-6 alkyl, Ci-e haloalkyl, Ci-6 cycloalkyl, or CHsC(O)-. In some embodiments, each Ra and Rb is independently H, cyclopropyl, cyclobutyl, methyl, ethyl, CH3C(O)-, -CD3, or -CF3.
[0128] In some embodiments, each Rc is H, methyl, ethyl, HOC(O)CH2-. CHsOC(O)CH2-, (CH3)3COC(O)CH2-, (CH3)3COC(O)NH-, H2NC(O)CH2-, CH3NHC(O)CH2-, (CH3)2NC(O)CH2-, NC-ethyl, H2N-, H2N-ethyl, CH3NH-ethyl, or (CH3)2N-ethyl.
[0129] In some embodiments, R1 is H, C1-6 alkyl, C1-6 cycloalkyl, monocyclic heterocyclyl, RaO-, RaO-(Ci-6 alkyl), RaRbNC(O)-, or alkyl-SCh-. In some embodiments, R1 is H, propyl, ethyl, methyl, isobutyl, cyclobutyl, cyclopropyl, azetidinyl. oxetanyl. piperidinyl, tetrahydropyran, tetrahydrofuran, thietane 1 ,1 dioxide, hexahydropyrimidin-2-one, RaO-, RaO- ethyl, RaO-methyl, RaRbNC(O)-, CH3CH2SO2-, (CH3)2CHSO2-, or cyclopropyl-SO2-.
[0130] In some embodiments, Ry is Ci-6 alkyl, Ci-6 alkenyl, Ci-6 haloalkyl, Ci-6 cycloalkyl, monocyclic heteroaryl, monocyclic aryl, monocyclic heterocyclyl, halo, -CN, -OH, RaO-, CH3SO2- CH3C(O)-, RaOC(O)-, RaRbNC(O)-, H2NC(O)NH-,RaRbN-, or methylallyl, each further substituted with 0, 1, or 2 Rd groups. In some embodiments. Ry is methyl, cyclopropyl, cyclobutyl, pyridinyl, phenyl, piperidinyl. pyrrolidinyl, oxetanyl. pyrrolidinone, imidazolidin-2- one, oxazolidin-2-one, lH-l,2,4-triazol-5-one, tetrahydropyran, dioxanyl, azetidinyl, -CN, -F, - CF3, -OH, RaO-, CH3SO2-, CH3C(O)-, HOC(O)-, RaRbNC(O)-, H2NC(O)NH-, RaRbN-, or methylallyl
[0131] In some embodiments, R1 is not substituted with an Ry group. In some embodiments. R1 is substituted with 1 Ry group. In some embodiments, R1 is substituted with 2 Ry groups.
[0132] In some embodiments, Rd is methyl, oxo, CHsC(O)-, CHsOC(O)-, CH3SO2-, -F, (CH3)3CO-, (CD3)3CO-, RaO-, -CN, -NH2, (CH3)3COC(O)-, H2NC(O)-, CH3HNC(O)-.
[0133] In some embodiments, Ry is not substituted with an Rd group. In some embodiments. Ry is substituted with 1 Rd group. In some embodiments, Ry is substituted with 2 Rd groups.
[0134] In some embodiments, R4 is H, phenyl, pyrazolyl, thiazolyl, thienyl, oxabicyclo[2.1.1]hexanyl, and norbomanyl, each substituted by 0, 1, or 2 Rz. In some embodiments, R4 is H, phenyl, pyrazolyl, thiazolyl, thienyl, oxabicyclo[2.1.1]hexanyl, or norbomanyl.
[0135] In some embodiments, each Rz is -CN, halo, C1-6 alkyl, C1-6 alkoxy, C1-6 haloalkyl, C1-6 cycloalkyl, deuterated alkyd, or C1-6 haloalkoxy. In some embodiments, Rz is -Cl, -F, -CN, -CF3, -CD3, -OCF3, methyl, ethyl, cyclopropyl, methoxy, difluoromethoxy, or difluoromethyl. [0136] In some embodiments, R4 is not substituted with an Rz group. In some embodiments. R4 is substituted with 1 Rz group. In some embodiments, R4 is substituted with 2 Rz groups.
[0137] In some embodiments, R4 is tolyl. In some embodiments, R4 is m-tolyl. In some embodiments, R4 is chlorophenyl. In some embodiments, R4 is m-chlorophenyl.
[0138] In some embodiments, R5 is -C(O)NRaRb, -C(O)ORa, unsubstituted C1-6 alkyl, or C1-6 alkyl substituted with 1 or 2 ORa or alkylamino groups. In some embodiments. R5 is H, hydroxymethyl, hydroxyethyl, methoxymethyl, H2N-methyl, CHsNH-methyl. or (CH3)2N- methyl, trideuteriomethoxymethyl, -C(O)NRaRb, -C(O)ORa.
[0139] In some embodiments, R6 is absent.
[0140] In some embodiments, R7 and R8 are each H. In some embodiments one of R7 and R8 is H. In some embodiments one of R7 and R8 is methyl. In some embodiments R7 and R8join together to form a fused or bridging ring system. In some embodiments R7 and R8 join together to form a fused ring system. In some embodiments R7 and R8join together to form a bridging ring system. In some embodiments when R7 and R8join together to form a fused ring system the
additional ring is a 4, 5, 6, or 7 membered ring. In some embodiments R7 and R8join together to form a bridged ring system wherein the bridge comprises 1, 2, or 3 carbon atoms.
[0141] In some embodiments provided herein are compounds of Formula (Ic)
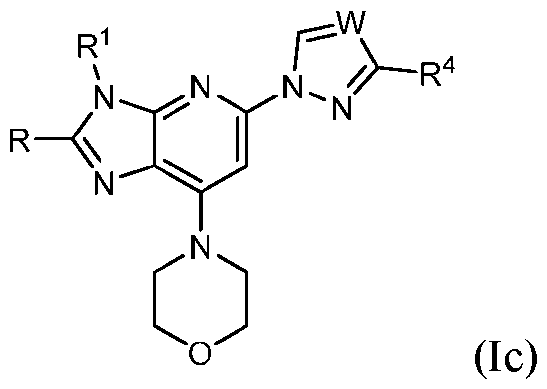 and pharmaceutically acceptable salts, solvates, and prodrugs thereof. In some embodiments of the compounds of Formula (Ic), R is H, R4 is m-tolyl or m-chlorophenyl, and R1 is H, methyl, ethyl or isopropyl substituted with 1 Ry group and Ry is substituted with 1 or 2 Rd groups. [0142] In some embodiments provided herein are compounds of Formula (Ic- 1 ):
and pharmaceutically acceptable salts, solvates, and prodrugs thereof. In some embodiments of the compounds of Formula (Ic), R is H, R4 is m-tolyl or m-chlorophenyl, and R1 is H, methyl, ethyl or isopropyl substituted with 1 Ry group and Ry is substituted with 1 or 2 Rd groups. [0142] In some embodiments provided herein are compounds of Formula (Ic- 1 ):
 and pharmaceutically acceptable salts, solvates, and prodrugs thereof, wherein n is 0, 1 or 2; and each Rx is independently halo, cyano, C1-3 alky l, C1-3 haloalkyl, C3-6 cycloalkyl, C1-3 alkoxy, or C1-3 haloalkoxy. In some embodiments of the compounds of Formula (Ic-1), n is 1 or 2; and each Rx is independently F, Cl, cyano, methyl, fluoromethyl, difluoromethyl, trifluoromethyl, ethyl, fluoroethyl, difluoroethyl, trifluoroethyl, cyclopropyl, methoxy, fluoromethoxy, difluoromethoxy, trifluoromethoxy, ethoxy, fluoroethoxy, difluoroethoxy, or trifluoroethoxy. In some embodiments of the compounds of Formula (Ic-1), R1 is H, methyl, ethyl or isopropyl substituted with 1 Ry group and Ry is substituted with 1 or 2 Rd groups.
and pharmaceutically acceptable salts, solvates, and prodrugs thereof, wherein n is 0, 1 or 2; and each Rx is independently halo, cyano, C1-3 alky l, C1-3 haloalkyl, C3-6 cycloalkyl, C1-3 alkoxy, or C1-3 haloalkoxy. In some embodiments of the compounds of Formula (Ic-1), n is 1 or 2; and each Rx is independently F, Cl, cyano, methyl, fluoromethyl, difluoromethyl, trifluoromethyl, ethyl, fluoroethyl, difluoroethyl, trifluoroethyl, cyclopropyl, methoxy, fluoromethoxy, difluoromethoxy, trifluoromethoxy, ethoxy, fluoroethoxy, difluoroethoxy, or trifluoroethoxy. In some embodiments of the compounds of Formula (Ic-1), R1 is H, methyl, ethyl or isopropyl substituted with 1 Ry group and Ry is substituted with 1 or 2 Rd groups.
[0143] In some embodiments provided herein are compounds of Formula (II):
 wherein X, R6, R7 and R8 are defined above and wherein R and R1 are defined as above, but are also joined to form a 3-, 4-, 5-, 6-, or 7 membered ring. In some
embodiments the ring is a 5- or 6- membered heterocyclic ring comprises optionally a C, N or O in addition to the N at the fusion point for the rings, and is optionally substituted with an oxo group, an alky l group, halo, CN, or alkoxy group.
wherein X, R6, R7 and R8 are defined above and wherein R and R1 are defined as above, but are also joined to form a 3-, 4-, 5-, 6-, or 7 membered ring. In some
embodiments the ring is a 5- or 6- membered heterocyclic ring comprises optionally a C, N or O in addition to the N at the fusion point for the rings, and is optionally substituted with an oxo group, an alky l group, halo, CN, or alkoxy group.
[0144] In some embodiments, the compound of Formula (I) is selected from
4-[3-(2-methoxyethyl)-5-[3-(m-tolyl)pyrazol-l-yl]imidazo[4.5-b]pyridin-7-yl]morpholine;
2-(7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-3-yl)acetonitrile;
5-({7-morpholino-5-[3-(m-tolyl)-l-pyrazolyl]-3H-l,3,4-triazainden-3-yl}methyl)-2- pyrrolidinone; l-(2- {7-morpholino-5-[3-(m-tolyl)- 1 -pyrazolyl]-3H- 1 ,3,4-triazainden-3-yl}ethyl)-2- imidazolidinone;
4-( { 7-morpholino-5 -[3-(m-tolyl)- 1 -pyrazolyl] -3H- 1 ,3,4-triazainden-3-y 1 } methyl)- 1 ,3 - oxazolidin-2-one;
N-methyl-3-({7-morpholino-5-[3-(m-tolyl)-l-pyrazolyl]-3H-l,3,4-triazainden-3-yl}methyl)-l- azetidinecarboxamide; l-[3-({5-[3-(m-chlorophenyl)-l-pyrazolyl]-7-morpholino-3H-l,3,4-triazainden-3-yl]methyl)-l- azetidinyl]-! -ethanone; and
3-({7-morpholino-5-[3-(m-tolyl)-l-pyrazolyl]-3H-l,3,4-triazainden-3-yl}methyl)-l- azetidinecarboxamide; and pharmaceutically acceptable salts thereof.
[0145] In some embodiments, the compound of Formula (I) is selected from the compounds of Table 1.
[0146] Table 1


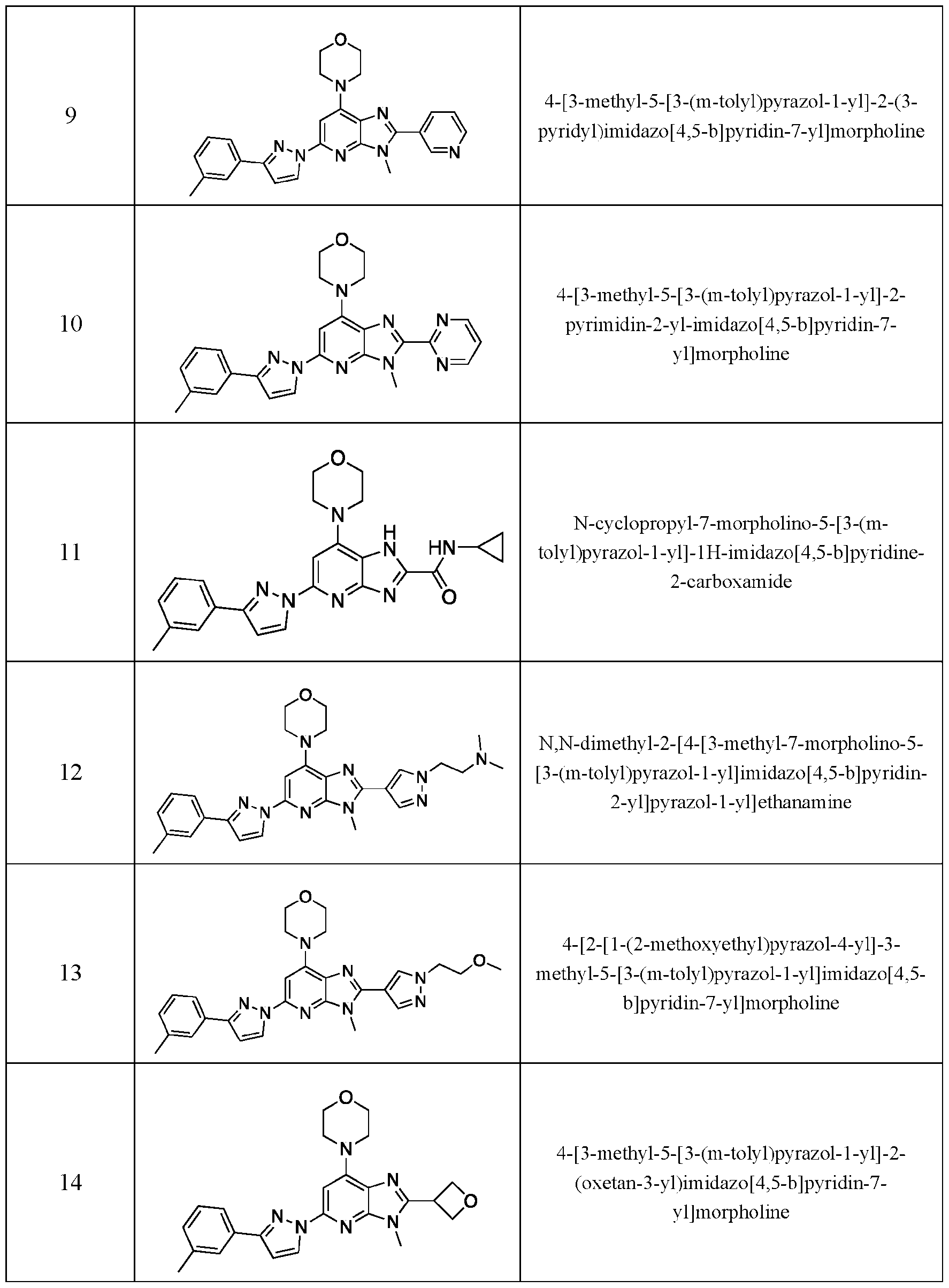















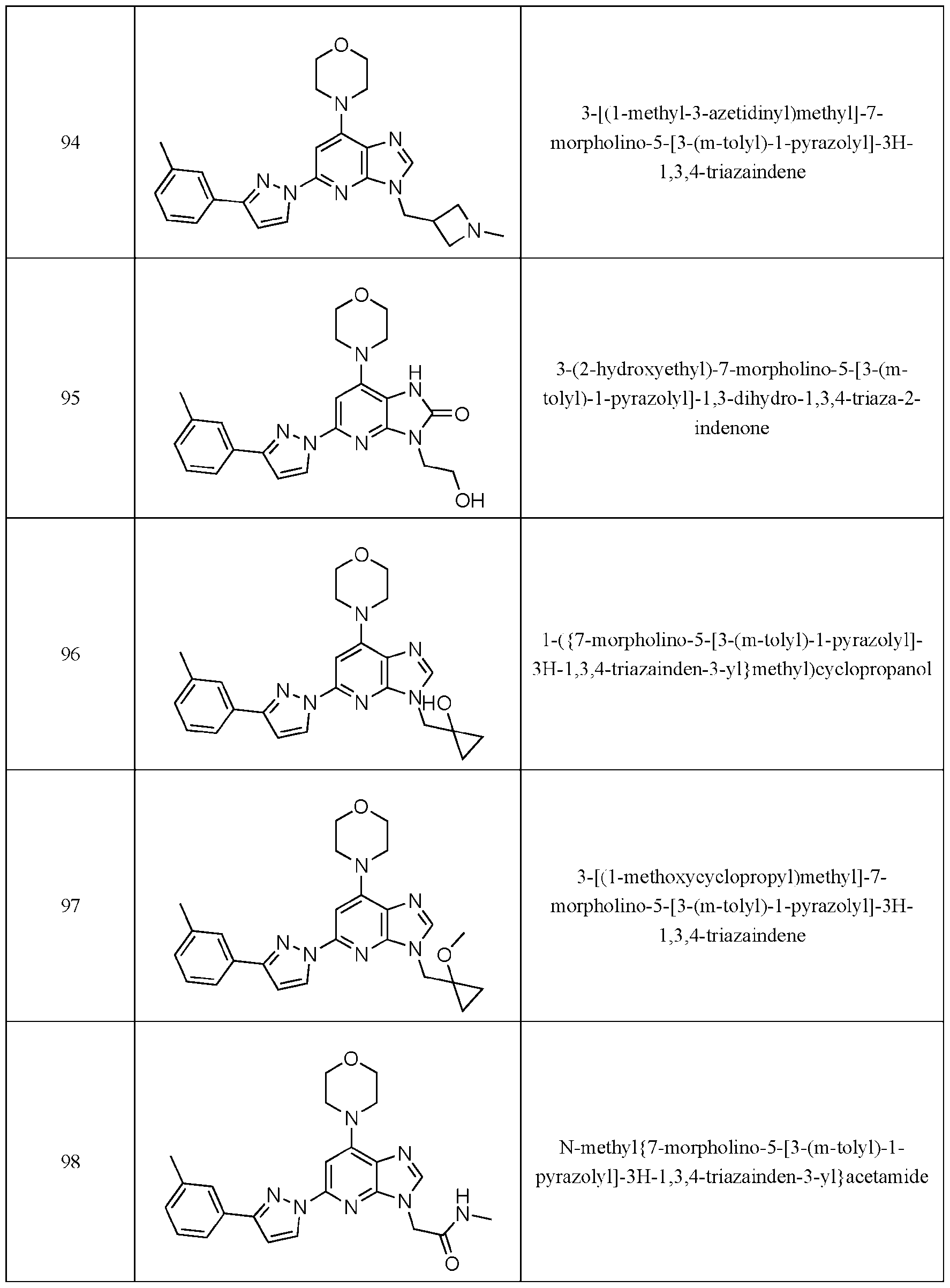

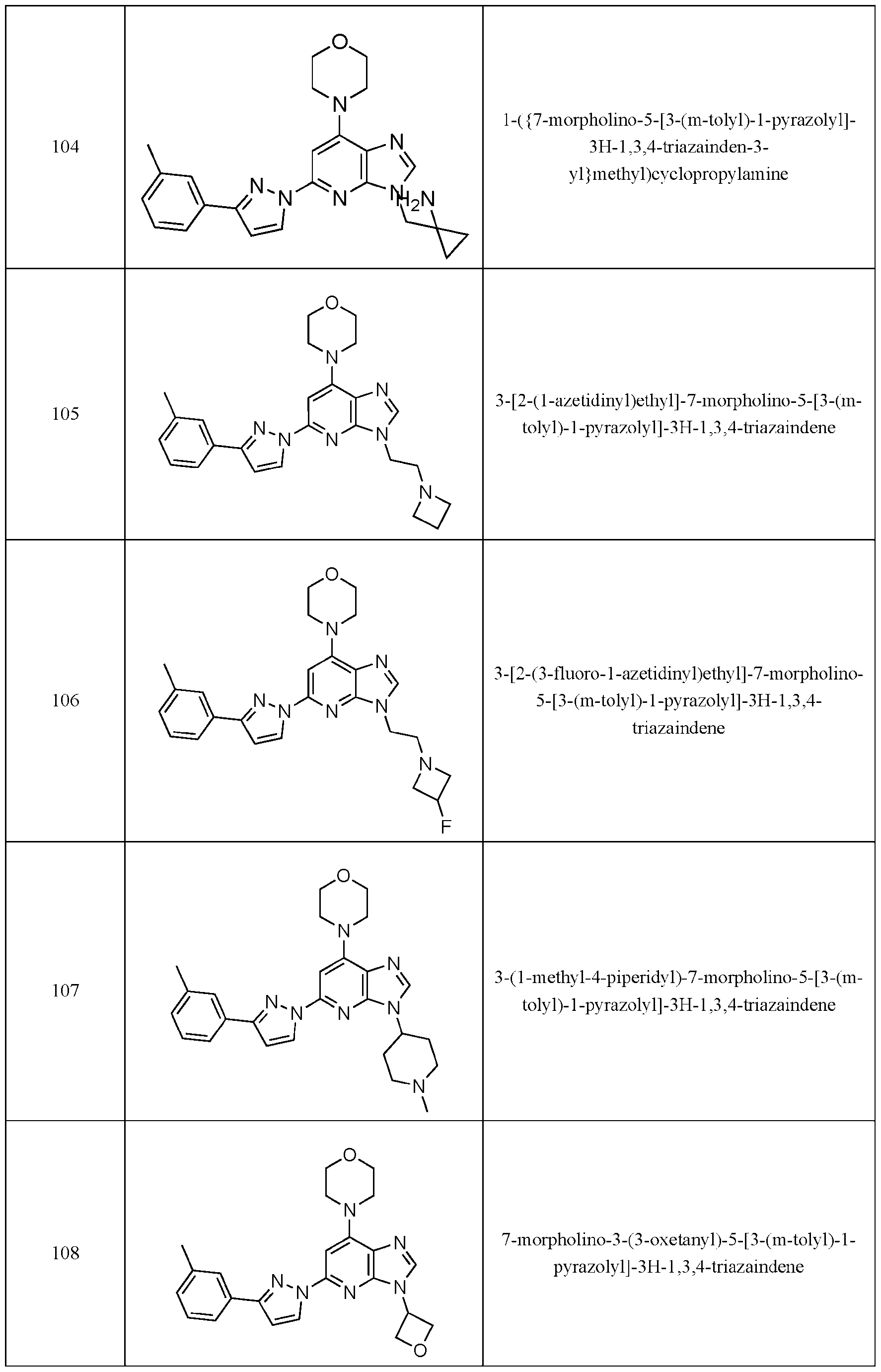






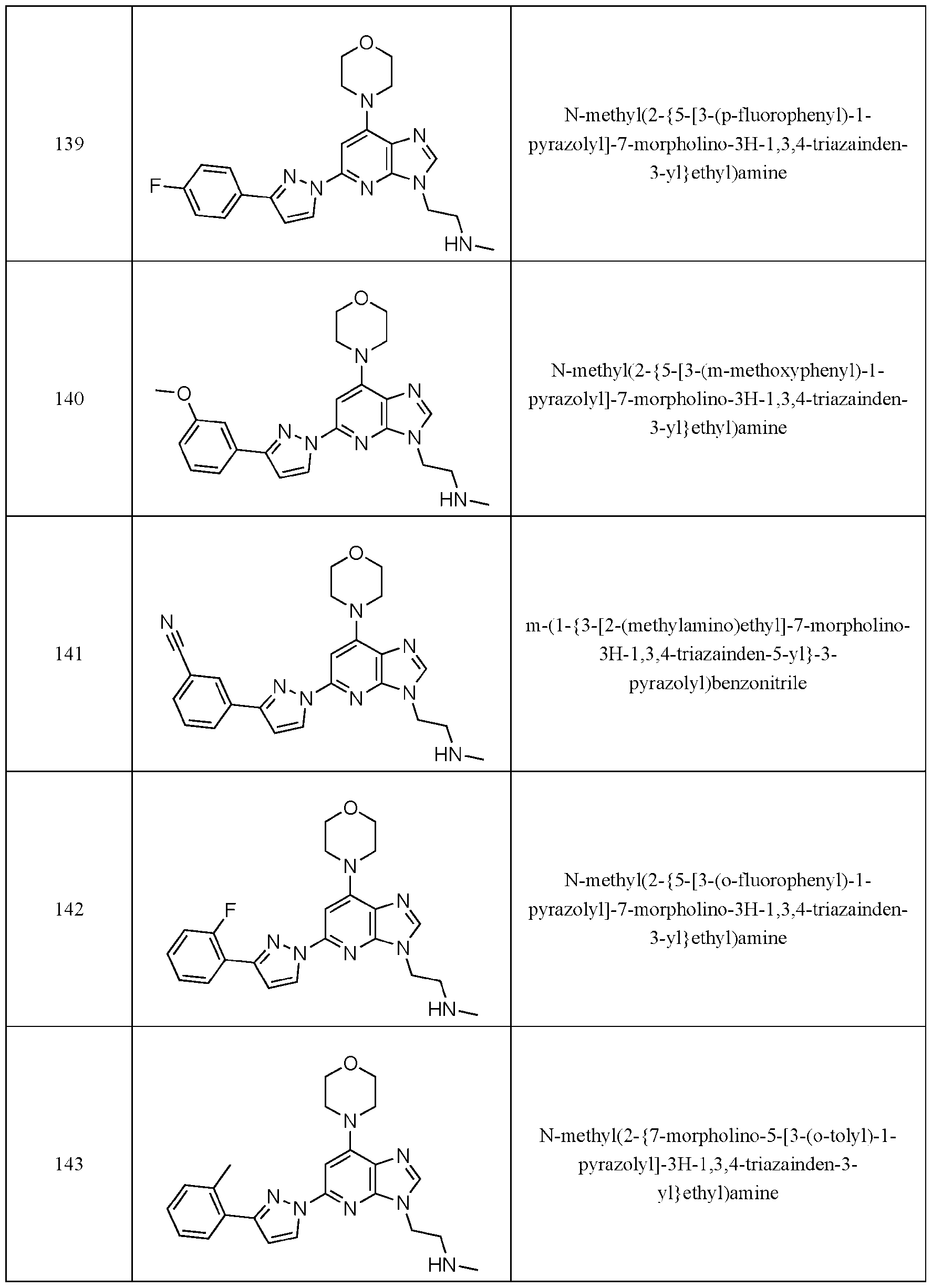







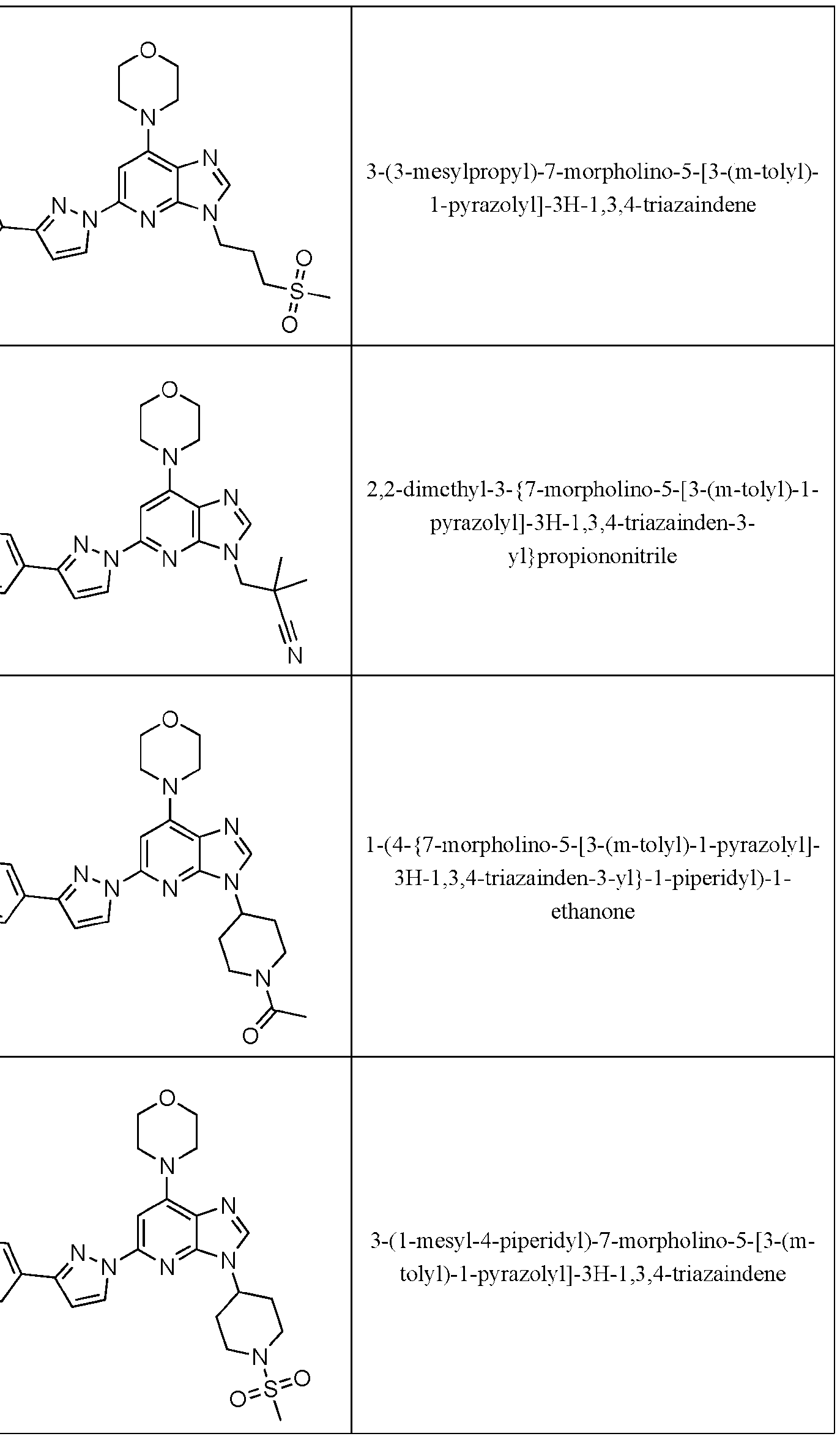
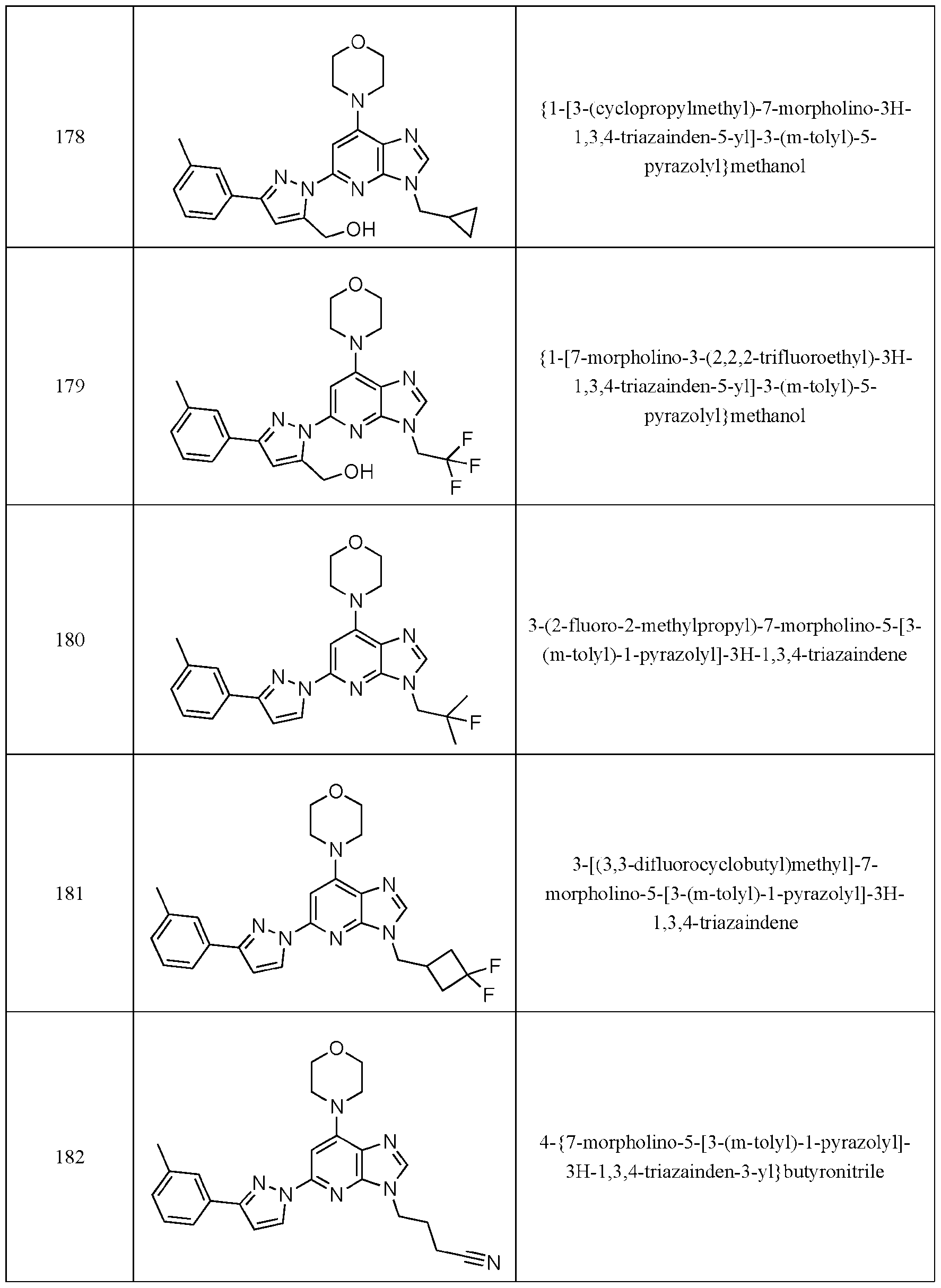





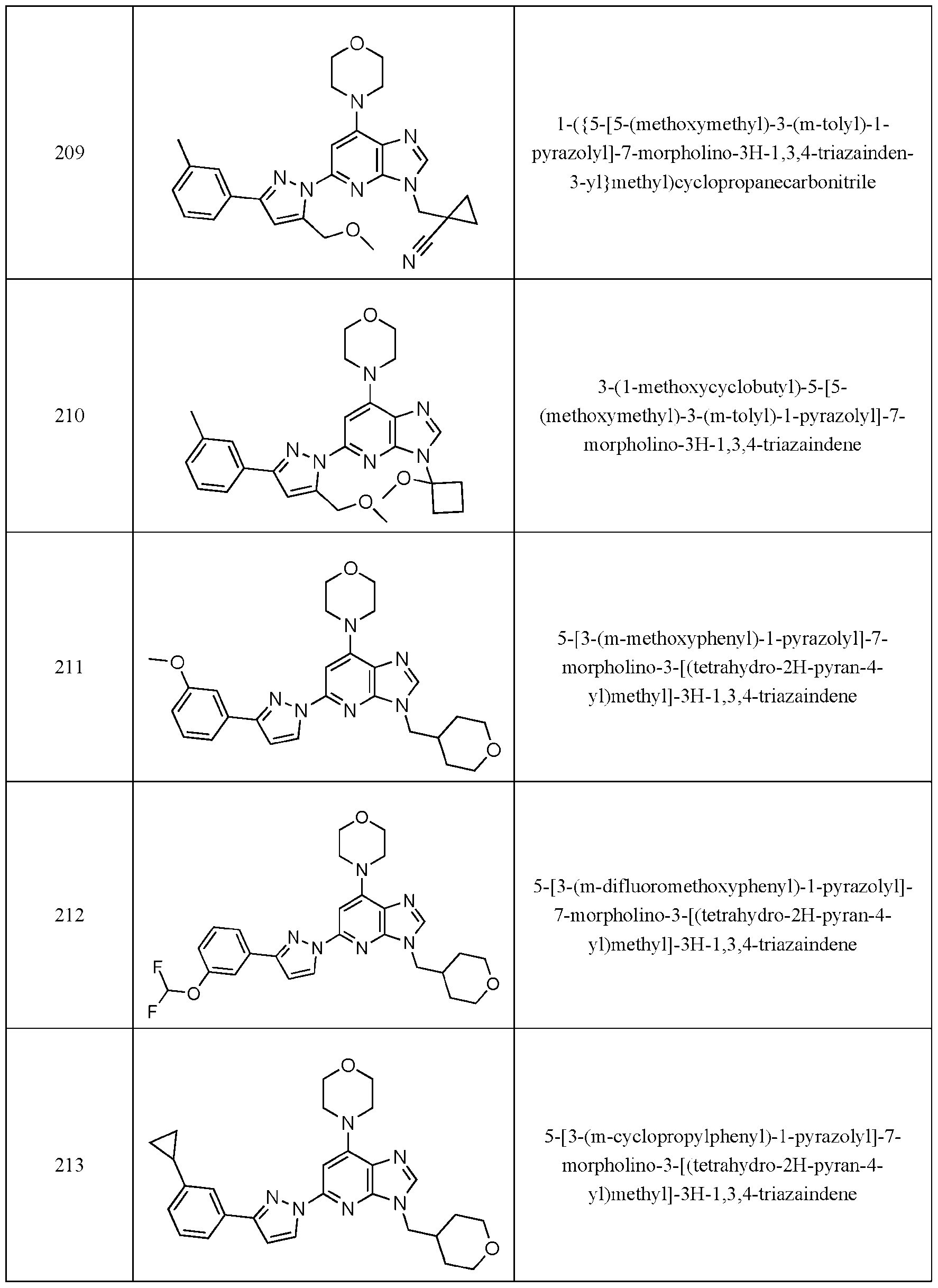

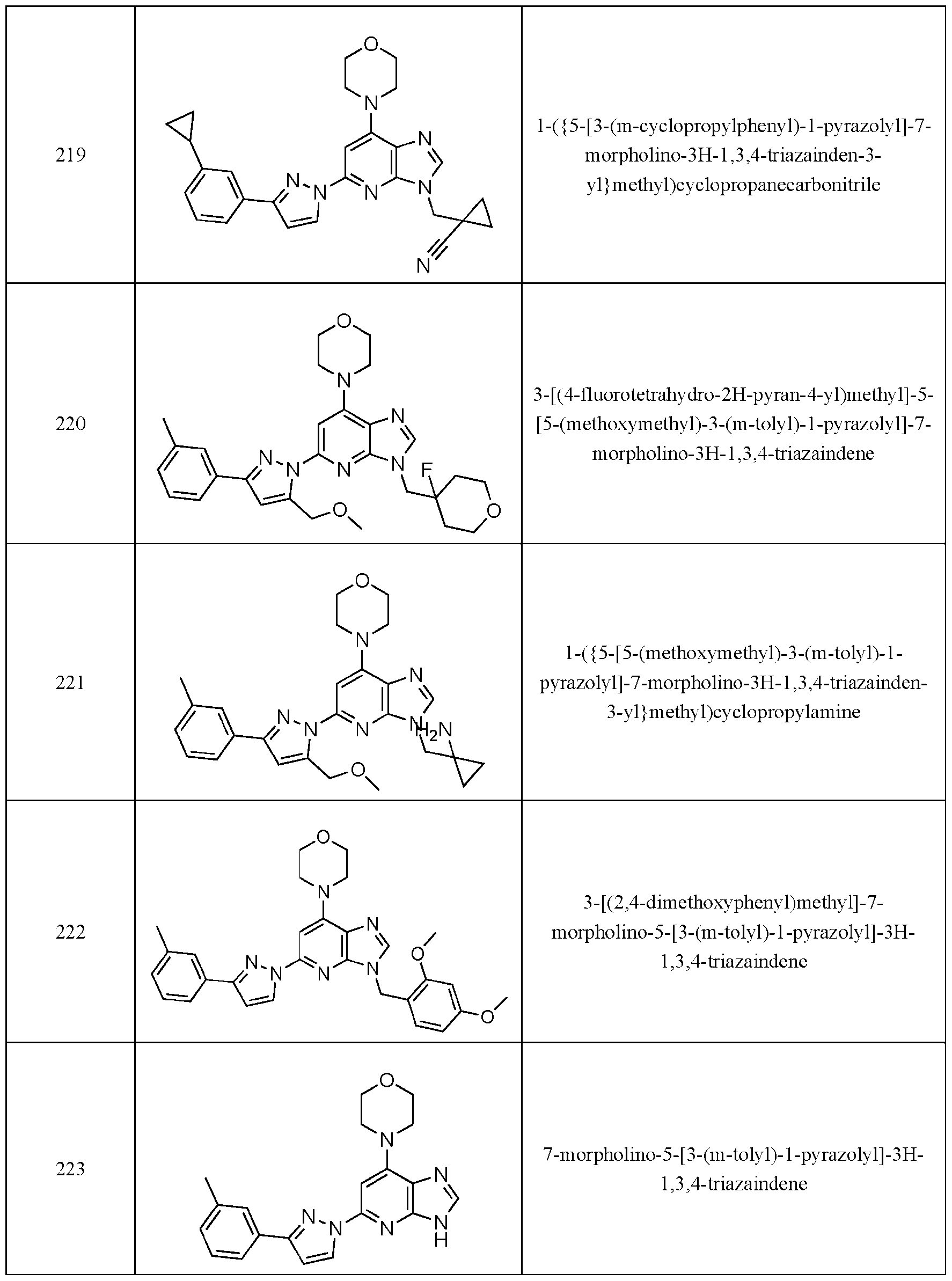













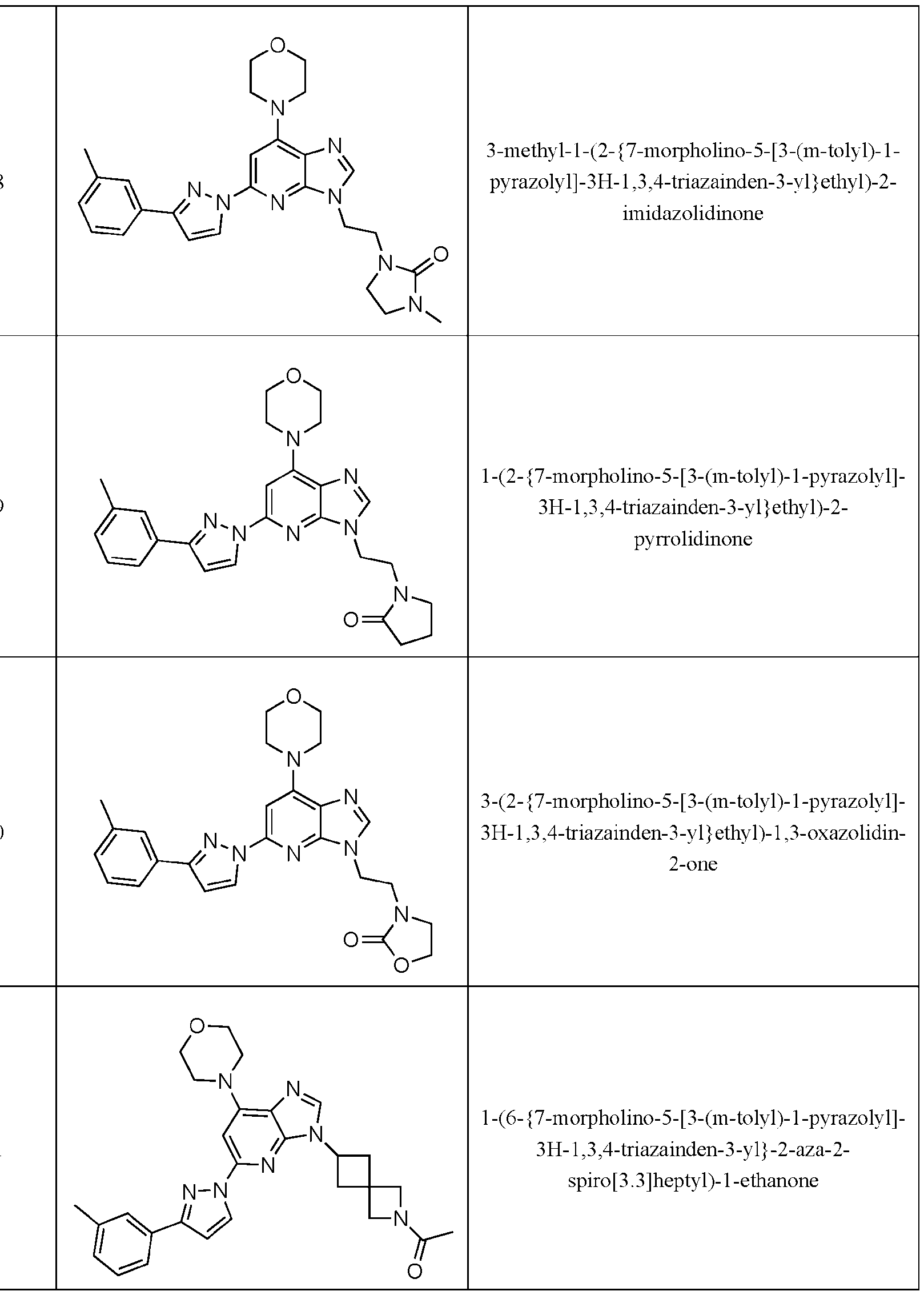






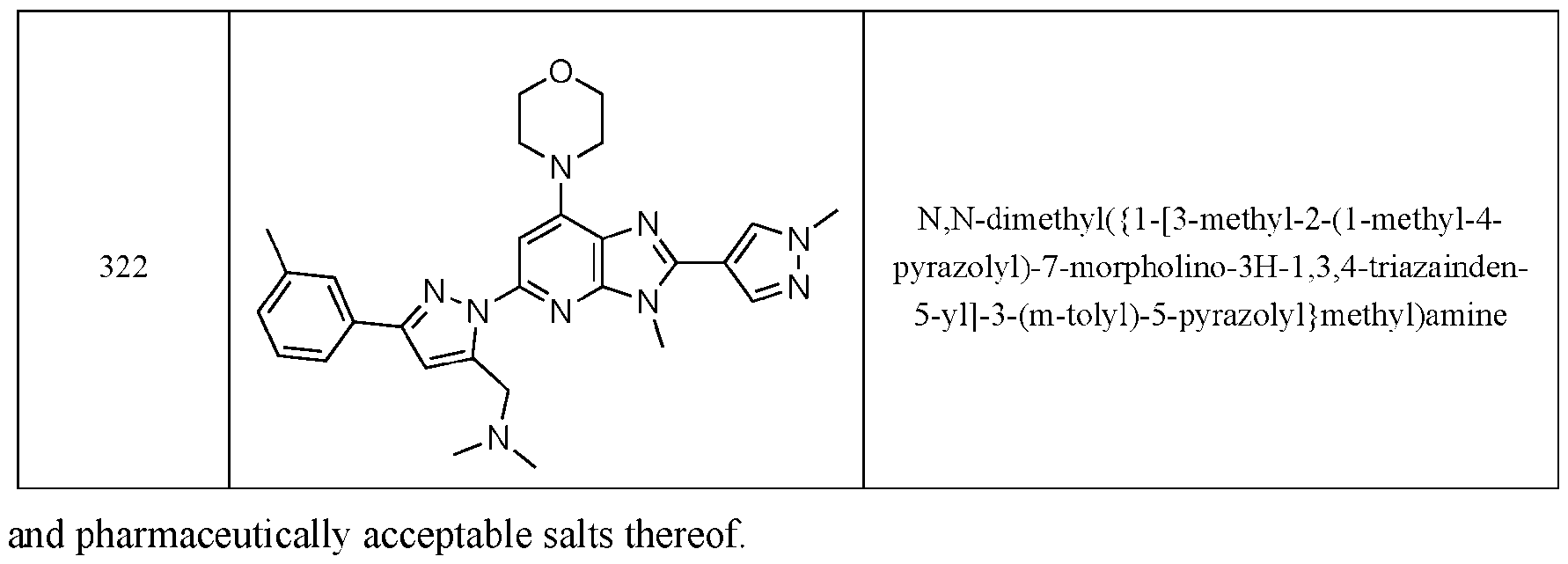
Methods of Treating, Administration, and Pharmaceutical Compositions
[0147] In general, the compounds of this disclosure will be administered in a therapeutically effective amount by any of the accepted modes of administration for agents that serve similar utilities. Therapeutically effective amounts of compounds of Formula (I) may range from about 0.01 to about 500 mg per kg patient body weight per day, which can be administered in single or multiple doses. In one embodiment, the dosage level will be about 0. 1 to about 250 mg/kg per day. In another embodiment the dosage level will be about 0.5 to about 100 mg/kg per day. A suitable dosage level may be about 0.01 to about 250 mg/kg per day, about 0.05 to about 100 mg/kg per day, or about 0. 1 to about 50 mg/kg per day. Within this range the dosage can be about 0.05 to about 0.5, about 0.5 to about 5 or about 5 to about 50 mg/kg per day. For oral administration, the compositions may be provided in the form of tablets containing about 1.0 to about 1000 milligrams of the active ingredient, particularly about 1.0, 5.0, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, 500, 600, 750, 800, 900, and 1000 milligrams of the active ingredient. The actual amount of the compound of this disclosure, i.e., the active ingredient, will depend upon numerous factors such as the severity of the disease to be treated, the age and relative health of the subj ect, the potency of the compound being utilized, the route and form of administration, and other factors.
[0148] In general, compounds of this disclosure will be administered as pharmaceutical compositions by any one of the following routes: oral, systemic (e.g.. transdermal, intranasal or by suppository), or parenteral (e.g., intramuscular, intravenous, or subcutaneous) administration. The preferred manner of administration is oral using a convenient daily dosage regimen, which can be adjusted according to the degree of affliction. Compositions can take the form of tablets, pills, capsules, semisolids, powders, sustained release formulations, solutions, suspensions, elixirs, aerosols, or any other appropriate compositions.
[0149] Pharmaceutical compositions can be formulated using one or more pharmaceutically acceptable carriers comprising excipients and auxiliaries. The formulation can be modified
depending upon the route of administration chosen. The pharmaceutical compositions can also include the compounds described herein in a free base form or a pharmaceutically acceptable salt form.
[0150] Methods for formulation of the pharmaceutical compositions can include formulating any of the compounds described herein with one or more inert, pharmaceutically acceptable excipients or carriers to form a solid, semi-solid, or liquid composition. Solid compositions can include, for example, powders, tablets, dispersible granules and capsules, and in some aspects, the solid compositions further contain nontoxic, auxiliary substances, for example wetting or emulsifying agents, pH buffering agents, and other pharmaceutically acceptable additives. Alternatively, the compositions described herein can be lyophilized or in powder form for reconstitution with a suitable vehicle, e.g., sterile pyrogen-free water, before use. The active ingredients can be entrapped in microcapsules prepared, for example, by coacervation techniques or by interfacial polymerization (e.g., hydroxymethylcellulose or gelatin microcapsules and poly-(methylmethacylate) microcapsules, respectively), in colloidal drugdelivery systems (e.g., liposomes, albumin microspheres, microemulsions, nano-particles and nanocapsules) or in macroemulsions.
[0151] The pharmaceutical compositions and formulations can be sterilized. Sterilization can be accomplished by filtration through sterile filtration.
[0152] The pharmaceutical compositions described herein can be formulated for administration as an injection. Non-limiting examples of formulations for injection can include a sterile suspension, solution, or emulsion in oily or aqueous vehicles. Suitable oily vehicles can include, but are not limited to, lipophilic solvents or vehicles such as fatty oils, synthetic fatty acid esters, or liposomes. Aqueous injection suspensions can contain substances which increase the viscosity of the suspension. The suspension can also contain suitable stabilizers. Injections can be formulated for bolus injection or continuous infusion.
[0153] For parenteral administration, the compounds can be formulated in a unit dosage injectable form (e.g.. solution, suspension, emulsion) in association with a pharmaceutically acceptable parenteral vehicle. Such vehicles can be inherently nontoxic, and non-therapeutic. A vehicle can be water, saline, Ringer’s solution, dextrose solution, and 5% human serum albumin. Nonaqueous vehicles such as fixed oils and ethyl oleate can also be used. Liposomes can be used as carriers. The vehicle can contain minor amounts of additives such as substances that enhance isotonicity and chemical stability (e.g., buffers and preservatives).
[0154] Sustained-release preparations can also be prepared. Examples of sustained-release matrices can include polyesters, hydrogels (e.g., poly(2-hydroxyethyl-methacrylate), or poly(vinylalcohol)), polylactides, copolymers of L-glutamic acid and y ethyl-L-glutamate, non-
degradable ethylene-vinyl acetate, degradable lactic acid-glycolic acid copolymers such as the LUPRON DEPO™ (i.e., injectable microspheres composed of lactic acid-glycolic acid copolymer and leuprolide acetate), and poly-D-(-)-3-hydroxybutyric acid.
[0155] Pharmaceutical formulations of the compositions described herein can be prepared for storage by mixing a compound with a pharmaceutically acceptable carrier, excipient, and/or a stabilizer. This formulation can be a lyophilized formulation or an aqueous solution. Acceptable carriers, excipients, and/or stabilizers can be nontoxic to recipients at the dosages and concentrations used. Acceptable carriers, excipients, and/or stabilizers can include buffers such as phosphate, citrate, and other organic acids; antioxidants including ascorbic acid and methionine; preservatives, polypeptides; proteins, such as serum albumin or gelatin; hydrophilic polymers; amino acids; monosaccharides, disaccharides, and other carbohydrates including glucose, mannose, or dextrins; chelating agents such as EDTA; sugars such as sucrose, mannitol, trehalose or sorbitol; salt-forming counter-ions such as sodium; metal complexes; and/or nonionic surfactants or polyethylene glycol.
[0156] Compounds of the present disclosure may be used in methods of treating in combination with one or more other combination agents (e.g., one, two, or three other drugs) that are used in the prevention, treatment, control, amelioration, or reduction of risk of the diseases or conditions for which compounds of the present disclosure are useful. In some embodiments, the combination of the drugs together are safer or more effective than either drug alone. In some embodiments the compound disclosed herein and the one or more combination agents have complementary activities that do not adversely affect each other. Such molecules can be present in combination in amounts that are effective for the purpose intended. Such other drug(s) may be administered, by a route and in an amount commonly used therefore, contemporaneously or sequentially with a compound of the present disclosure. When a compound of the present disclosure is used contemporaneously with one or more other drugs, in some embodiments, the agents are administered together in a single pharmaceutical composition in unit dosage form. Accordingly, the pharmaceutical compositions of the present disclosure also include those that contain one or more other active ingredients, in addition to a compound of the present disclosure. The weight ratio of the compound of the present disclosure to the second active agent may be varied and will depend upon the effective dose of each ingredient. Generally, an effective dose of each will be used. In some embodiments, combination therapy includes therapies in which the compound of the present disclosure and one or more other drugs are administered separately, and in some cases, the two or more agents are administered on different, overlapping schedules. It is also contemplated that when used in combination with one or more other active ingredients, the compounds of the present disclosure and the other active
ingredients may be used in lower doses than when each is used singly. In some embodiments, the combination agent is a drug for reduction of symptoms of ALS. In some embodiments, the combination agent is selected from an NAD supplement (such as nicotinamide riboside, offered under the trade names Basis® or Tru Niagen®). vitamin B12 (oral or injection), glycopyrrolate, atropine, scopolamine, baclofen, tizanidine. mexiletine. an SSRI. a benzodiazepine, Neudexta. riluzole, and edaravone, and combinations thereof.
[0157] The compounds, pharmaceutical compositions, and methods of the present disclosure can be useful for treating a subj ect such as, but not limited to, a mammal, a human, a non-human mammal, a domesticated animal (e.g., laboratory animals, household pets, or livestock), a nondomesticated animal (e.g., wildlife), a dog, a cat, a rodent, a mouse, a hamster, a cow, a bird, a chicken, a fish, a pig, a horse, a goat, a sheep, or a rabbit. In preferred embodiments, compounds, pharmaceutical compositions, and methods of the present disclosure are used for treating a human.
[0158] The compounds, pharmaceutical compositions, and methods described herein can be useful as a therapeutic, for example a treatment that can be administered to a subject in need thereof. A therapeutic effect can be obtained in a subject by reduction, suppression, remission, or eradication of a disease state, including, but not limited to, a symptom thereof. A therapeutic effect in a subject having a disease or condition, or pre-disposed to have or is beginning to have the disease or condition, can be obtained by a reduction, a suppression, a prevention, a remission, or an eradication of the condition or disease, or pre-condition or pre-disease state. [0159] In practicing the methods described herein, therapeutically effective amounts of the compounds or pharmaceutical compositions described herein can be administered to a subject in need thereof, often for treating and/or preventing a condition or progression thereof. A pharmaceutical composition can affect the physiol ogy of the subject, such as the immune system, inflammatory response, or other physiologic affect. A therapeutically effective amount can vary widely depending on the severity of the disease, the age and relative health of the subject, the potency of the compounds used, and other factors.
[0160] Treat and/or treating can refer to any indicia of success in the treatment or amelioration of the disease or condition. Treating can include, for example, reducing, delaying or alleviating the severity of one or more symptoms of the disease or condition, or it can include reducing the frequency with which symptoms of a disease, defect, disorder, or adverse condition, and the like, are experienced by a patient. Treat can be used herein to refer to a method that results in some level of treatment or amelioration of the disease or condition and can contemplate a range of results directed to that end, including but not restricted to prevention of the condition entirely.
[0161] Prevent, preventing, and the like can refer to the prevention of the disease or condition in the patient. For example, if an individual at risk of contracting a disease is treated with the methods of the present disclosure and does not later contract the disease, then the disease has been prevented, at least over a period of time, in that individual.
[0162] A therapeutically effective amount can be the amount of a compound or pharmaceutical composition or an active component thereof sufficient to provide a beneficial effect or to otherwise reduce a detrimental non-beneficial event to the individual to whom the composition is administered. A therapeutically effective dose can be a dose that produces one or more desired or desirable (e.g., beneficial) effects for which it is administered, such administration occurring one or more times over a given period of time. An exact dose can depend on the purpose of the treatment and can be ascertainable by one skilled in the art using known techniques.
[0163] The compounds or pharmaceutical compositions described herein that can be used in therapy can be formulated and dosages established in a fashion consistent with good medical practice taking into account the disorder to be treated, the condition of the individual patient, the site of delivery' of the compound or pharmaceutical composition, the method of administration and other factors known to practitioners. The compounds or pharmaceutical compositions can be prepared according to the description of preparation described herein.
[0164] One of ordinary skill in the art would understand that the amount, duration, and frequency of administration of a pharmaceutical composition or compound described herein to a subject in need thereof depends on several factors including, for example but not limited to, the health of the subject, the specific disease or condition of the patient, the grade or level of a specific disease or condition of the patient, the additional therapeutics the subject is being or has been administered, and the like.
[0165] The methods, compounds, and pharmaceutical compositions described herein can be for administration to a subject in need thereof. Often, administration of the compounds or pharmaceutical compositions can include routes of administration, non-limiting examples of administration routes include intravenous, intraarterial, subcutaneous, subdural, intramuscular, intracranial, intrastemal, intratumoral, or intraperitoneally. Additionally, a pharmaceutical composition or compound can be administered to a subject by additional routes of administration, for example, by inhalation, oral, dermal, intranasal, or intrathecal administration. [0166] Pharmaceutical compositions or compounds of the present disclosure can be administered to a subject in need thereof in a first administration, and in one or more additional administrations. The one or more additional administrations can be administered to the subject in need thereof minutes, hours, days, weeks, or months following the first administration. Any
one of the additional administrations can be administered to the subject in need thereof less than 21 days, or less than 14 days, less than 10 days, less than 7 days, less than 4 days or less than 1 day after the first administration. The one or more administrations can occur more than once per day, more than once per week, or more than once per month. The compounds or pharmaceutical compositions can be administered to the subject in need thereof in cycles of 21 days. 14 days, 10 days, 7 days, 4 days, or daily over a period of one to seven days.
[0167] The compounds, pharmaceutical compositions, and methods provided herein can be useful for the treatment of a plurality of diseases or conditions or preventing a disease or a condition in a subject, or other therapeutic applications for subjects in need thereof. In one aspect, the disclosure relates to a method for treating a neurological disease mediated by PIKfyve activity in a subject in need thereof, comprising administering an effective amount of a compound or a pharmaceutical composition as described herein to the subject. In some embodiments, the disease is associated with a FIG4 deficiency.
[0168] In some embodiments, the neurological disease is amyotrophic lateral sclerosis (ALS), primary lateral sclerosis (PLS), Charcot-Marie-Tooth (CMT; including type 4J (CMT4J)), and Yunis-Varon syndrome, autophagy7, polymicrogyria (including polymicrogyria with seizures), temporo-occipital polymicrogyria. Pick's disease, Parkinson’s disease, Parkinson’s disease with Lewy bodies, dementia with Lewy bodies, Lewy body disease, fronto-temporal dementia, diseases of neuronal nuclear inclusions of polyglutamine and intranuclear inclusion bodies, disease of Marinesco and Hirano bodies, tauopathy, Alzheimer's disease, neurodegeneration, spongiform neurodegeneration, peripheral neuropathy, leukoencephalopathy, motor neuropathy, sensory neuropathy, inclusion body disease, progressive supranuclear palsy, corticobasal syndrome, chronic traumatic encephalopathy, traumatic brain injury (TBI), cerebral ischemia, Guillain-Barre Syndrome, chronic inflammatory demyelinating polyneuropathy, multiple sclerosis, a lysosomal storage disease, Fabry ’s disorder, Gaucher's disorder, Niemann Pick C disease, Tay-Sachs disease, and Mucolipidosis type IV, neuropathy, Huntington's disease, a psychiatric disorder, ADHD, schizophrenia, a mood disorder, major depressive disorder, depression, bipolar disorder I, or bipolar disorder II.
[0169] In some embodiments, the neurological disease is ALS, FTD, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, or CMT. In some embodiments, the neurological disease is ALS.
[0170] In some embodiments, the neurological disease is a tauopathy such as Alzheimer's disease, progressive supranuclear palsy, corticobasal syndrome, frontotemporal dementia, or chronic traumatic encephalopathy.
[0171] In some embodiments, the neurological disease is a lysosomal storage disease such as Fabry's disorder, Gaucher's disorder, Niemann Pick C disease, Tay-Sachs disease, or Mucolipidosis type IV.
[0172] In some embodiments, the neurological disease is a psychiatric disorder such as ADHD, schizophrenia, or mood disorders such as major depressive disorder, depression, bipolar disorder I, or bipolar disorder II.
[0173] The disclosure further provides any compounds disclosed herein for use in a method of treatment of the human or animal body by therapy. Therapy may be by any mechanism disclosed herein, such as inhibiting, reducing, or reducing progression of the diseases disclosed herein. The disclosure further provides any compound disclosed herein for prevention or treatment of any condition disclosed herein. The disclosure also provides any compound or pharmaceutical composition thereof disclosed herein for obtaining any clinical outcome disclosed herein for any condition disclosed herein. The disclosure also provides use of any compound disclosed herein in the manufacture of a medicament for preventing or treating any disease or condition disclosed herein.
Examples
[0174] The following preparations of compounds of Formula (I) and intermediates are given to enable those skilled in the art to more clearly understand and to practice the present disclosure. They should not be considered as limiting the scope of the disclosure, but merely as being illustrative and representative thereof.
[0175] The starting materials and reagents used in preparing these compounds are either available from commercial suppliers such as Aldrich Chemical Co., (Milwaukee, Wis.), Bachem (Torrance, Calif.), or Sigma (St. Louis, Mo.) or are prepared by methods known to those skilled in the art following procedures set forth in references such as Fieser and Fieser’s Reagents for Organic Synthesis, Volumes 1-17 (John Wiley and Sons, 1991); Rodd's Chemistry of Carbon Compounds. Volumes 1-5 and Suppiementals (Elsevier Science Publishers, 1989); Organic Reactions, Volumes 1-40 (John Wiley and Sons, 1991), March’s Advanced Organic Chemistry, (John Wiley and Sons, 4th Edition) and Larock’s Comprehensive Organic Transformations (VCH Publishers Inc., 1989). These schemes are merely illustrative of some methods by which the compounds of this disclosure can be synthesized, and various modifications to these schemes can be made and will be suggested to one skilled in the art having referred to this disclosure. The starting materials and the intermediates, and the final products of the reaction may be isolated and purified if desired using conventional techniques, including but not limited to filtration,
distillation, crystallization, chromatography and the like. Such materials may be characterized using conventional means, including physical constants and spectral data.
[0176] Unless specified to the contrary, the reactions described herein take place at atmospheric pressure over a temperature range from about -78 °C to about 150 °C, or from about 0 °C to about 125 °C or at about room (or ambient) temperature, e.g., about 20 °C.
[0177] Compounds of Formula (I) and subformulae and species described herein, including those where the substituent groups as defined herein, can be prepared as illustrated and described below.
[0178] Unless otherwise noted, all reagents were used without further purification. 'H NMR spectra were obtained in CDCk DMSO-rL. or CD?OD at room temperature on a Bruker 300 MHz instrument. When more than one conformer was detected, the chemical shifts for the most abundant one is reported. Chemical shifts of 'l l NMR spectra were recorded in parts per million (ppm) on the d scale from an internal standard of residual solvent. Splitting patterns are designed as s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad. LC-MS conditions were as described below:
LCMS Column: Agilent Zorbax XDB C18 4.6x50 mm, 3.5pm a. Mobile phase: Solvent A: Water (with 0.1% formic acid); Solvent B:
MeOH b. Flow rate: 1.0 mL/min. c. Run time: 2 min gradient (20%-90% B), then 3 min @90% B, d. Temperature: 30 °C
HPLC Column: Agilent SB-C18 4.6x 150 mm, 3.5pm a. Mobile phase: Solvent A: water (with 0.02% TFA); Solvent B: MeOH b. Flow rate: 1.0 mL/min. c. Run time: 0.5 min @10% B, 9.5 min gradient (10%-90% B), then 10 min
@90% B, d. Temperature: 30 °C
Preparative LC Column: Phenomenex Luna 5u 100A, 21.2x250mm, 5pm a. Mobile phase: Solvent A: Water Solvent B: MeOH b. Flow rate: 10 mL/min, c. Run time: 1 min @20% B, 30 min gradient (20%-80% B), then 10 min
@90% B, d. Temperature: Ambient
[0179] The following abbreviations are used in the text:


[0180] Synthesis and Characterization
[0181] General procedure 1
[0182] Synthesis of Compound 1: 4-(3-methyl-2-(pyridin-2-yl)-5-(3-(m-tolyl)-lH-pyrazol- l-yl)-3H-imidazo [4,5-b] pyridin-7-yl)morpholine

[0183] Synthesis of 4-(2,6-dichloro-3-nitropyridin-4-yl)morpholme, GP1.1
[0184] To a solution of 2,4,6-trichloro-3-nitropyridine (2.0 g, 8.81 mmol) in EtOH (40 mL) at -10 °C was added morpholine (920 mg, 10.57 mmol) and a solution of EtsN (2.5 mL, 17.62 mmol) in EtOH (10 mL). The solution was stirred at -10 °C for 2 h. The completion of the reaction was monitored by TLC. The reaction mixture was concentrated directly under reduced pressure. The resulting residue was purified by silica gel column chromatography with a
gradient elution of 5% EtOAc/PE to 50% EtOAc/PE to provide 4-(2,6-dichloro-3-nitropyridin-4- yl)morpholine (1.67 g, 7.39 mmol) as a yellow solid. LC-MS (ESI+): m/z 227/229 (MH+).'HNMR (300MHZ, CDCh) <56.81 (s, 1H), 3.82-3.75 (m, 4H), 3.25-3.15 (m, 4H).
[0185] Synthesis of 6-chloro-N-methyl-4-morpholmo-3-nitropyridin-2-amine, GP1.2
[0186] To a solution of 4-(2,6-dichloro-3-nitropyridin-4-yl)morpholine (GP1.1, 500 mg, 1.78 mmol) in DMF (30 mL) at rt was added CS2CO3 (1.2 g, 3.6 mmol) and methanamine hydrochloride (122 mg, 1.80 mmol). The solution mixture was stirred at rt for 2 h. The completion of the reaction was monitored by TLC. The reaction mixture was diluted with water (60 mL) and extracted with EtOAc (3 x 40 mL). The combined organic phase was dried over anhydrous Na2SC>4, filtrated and concentrated. The resulting residue was purified by silica gel column chromatography with a gradient elution of 5% EtOAc/PE to 20% EtOAc/PE to provide 6-chloro-N-methyl-4-morpholino-3-nitropyridin-2-amine (480 mg, 1.76 mmol) as a yellow solid. LC-MS (ESI+): m/z 273/275 (MH+). 1HNMR (300MHz, CDCh) <57.94 (broad, 1H), 6.12 (s, 1H), 3.90-3.80 (m, 4H), 3.23-3.15 (m, 4H), 3.08 (d, J= 4.8 Hz, 3H).
[0187] Synthesis ofN-methyl-4-morpholino-3-nitro-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridin-2- amine, GP1.3
[0188] To a solution of 6-chloro-N-methyl-4-morpholino-3-nitropyridin-2-amine (GP1.2, 288 mg, 1.06 mmol) in DMF (17 mL) was added 3-(m-tolyl)-lH-pyrazole (251 mg, 1.59 mmol) and CS2CO3 (1.04 g, 3.18 mmol). The reaction mixture was stirred at 110 °C under N2 for 2 h. The completion of the reaction was monitored by TLC. The reaction mixture was diluted with water (30 mL) and extracted with EtOAc (3 x 30 mL). The combined organic phase was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 5% EtOAc/PE to 20% EtOAc/PE to provide N-methyl-4-morpholino-3-nitro-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridin-2- amine (300 mg, 0.76 mmol) as a yellow solid. LC-MS (ESI+): m/z 395 (MH+). 1HNMR (300MHz, CDCh) <58.55 (d, J= 2.4 Hz, 1H), 8.43 (broad, 1H), 7.75-7.70 (m, 2H), 7.34 (t, J = 7.2 Hz, 1H), 7.20 (d, J= 7.2 Hz, 1H), 6.95 (s, 1H), 6.77 (d, J= 2.7 Hz, 1H), 3.90-3.80 (m, 4H), 3.40-3.30 (m, 4H), 3.08 (d, J= 4.8 Hz, 3H), 2.44 (s. 3H).
[0189] Synthesis qfN2-methyl-4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridine-2,3- diamine, GP1.4
[0190] To a solution of N-methyl-4-morpholino-3-nitro-6-(3-(m-tolyl)-lH-pyrazol-l- yl)pyridin-2-amine (GP1.3, 300 mg, 0.76 mmol) in DCM/MeOH (10: 1, 20 mL) was added Pd/C (50 mg). The reaction mixture was stirred at rt under hydrogen atmosphere overnight. The completion of the reaction was monitored by TLC. After filtration, the filtrate was concentrated directly under reduced pressure. The resulting residue was purified by silica gel column
chromatography with a gradient elution of 10% EtOAc/PE to 50% EtOAc/PE to provide N2- methyl-4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridine-2,3-diamine (170 mg, 0.47 mmol) as a purplish solid. LC-MS (ESI+): m/z 365 (MEI+). 1HNMR (300MHz, CDCk) £8.51 (d, J= 2.4 Hz, 1H), 7.78 (s, 1H), 7.72 (d, J= 7.5 Hz, 1H), 7.32 (t, J= 7.5 Hz, 1H), 7.22 (s. 1H), 7.15 (d, J= 7.5 Hz, 1H), 6.71 (d, J= 2.4 Hz, 1H), 3.92-3.83 (m, 4H), 3.40 (brs, 2H), 3.10-3.00 (m, 7H), 2.43 (s, 3H).
[0191] Synthesis ofN-(3-amino-4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridin-2-yl)-N- methylpicolinamide, GP1.5
[0192] To a solution of N2-methyl-4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridine- 2,3-diamine (GP1.4, 44 mg, 0.12 mmol) and EtsN (24 mg, 0.24 mmol) at rt in DCM (5 mL) was added picolinoyl chloride hydrochloride (22 mg, 0. 12 mmol). The solution was stirred at rt for 4 h. The completion of the reaction was monitored by TLC. The reaction mixture was quenched with water (10 mL) and extracted with DCM (2 x 10 mL). The combined organic phase was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was used directly for the next step without further purification. LC-MS (ESI+): m/z 470 (MH+).
[0193] Synthesis of 4-(3-methyl-2-(pyridin-2-yl)-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-b]pyridin-7-yl)morphohne, Compound 1
[0194] To a solution of N-(3-amino-4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridin-2- yl)-N-methylpicolinamide (GP1.5, 40 mg, 0.085 mmol) in EtOH (5 mL) was added one drop of cone. H2SO4. The reaction mixture was stirred under reflux for 2 h. The completion of the reaction was monitored by TLC. The reaction mixture was diluted with water (20 mL) and extracted with DCM (2 x 10 mL). The combined organic phase was dried over anhydrous Na2SC>4, filtered, and concentrated under reduced pressure. The resulting residue was slurry in Et20 to provide 4-(3-methyl-2-(pyridin-2-yl)-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5- b]pyridin-7-yl)morpholine (19.5 mg, 0.043 mmol) as a purple solid. LC-MS (ESI+): m/z 452 (MH+). 1HNMR (300MHz, CDCh) 8.68 (s, 2H), 8.33 (d, J= 8.1 Hz, 1H), 7.85-7.79 (m, 3H), 7.40-7.28 (m, 3H), 7.16 (d, J= 8.1 Hz, 1H), 6.79 (s, 1H), 4.30 (s, 3H), 4.12-4.08 (m, 4H), 4.00- 3.90 (m, 4H), 2.45 (s, 3H).
[0195] General procedure 2
[0196] Synthesis of Compound 2: N-cyclopropyI-3-methyl-7-morpholino-5-(3-(m-tolyl)- lH-pyrazol-l-yl)-3H-imidazo [4,5-b] pyridine-2-carboxamide

[0197] Synthesis of methyl 3-methyl-7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidcizo[4, 5-b ]pyridine-2-carboxylate, GP2. 1
[0198] To a solution of N2-methyl-4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridine- 2,3-diamine (GP1.4, 80 mg, 0.22 mmol) in DCE (8 mL) was added DIEA (43 mg, 0.33 mmol) and methyl 2, 2-dichloro-2-methoxy acetate (46 mg, 0.26 mmol). The solution mixture was stirred at 80 °C for 4 h. The completion of the reaction was monitored by TLC. The reaction mixture was concentrated directly under reduced pressure. The resulting residue was purified by preparative TLC to provide methyl 3-methyl-7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-b]pyridine-2-carboxylate (26 mg. 0.06 mmol). LC-MS (ESI+): m/z 433 (MH+).
[0199] Synthesis of 3-methyl-7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5- b ]pyridine-2-carboxylic acid, GP2.2
[0200] To a solution of methyl 3-methyl-7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-b]pyridine-2-carboxylate (GP2.1, 26 mg. 0.06 mmol) in DCM (8 mL) was added aqueous NaOH solution (5 mg, IM, 0. 12 mL). The solution mixture was stirred at rt for 2 h. The completion of the reaction was monitored by TLC. The reaction mixture was concentrated directly under reduce pressure. The resulting residue was purified by preparative TLC to provide 3-methyl-7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridine-2- carboxylic acid (20 mg, 0.05 mmol). LC-MS (ESI+): m/z 419 (MH+).
[0201] Synthesis ofN-cyclopropyl-3-methyl-7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)- 3H-imidazo[4,5-b]pyridine-2-carboxamide, Compound 2
[0202] To a solution of 3-methyl-7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-b]pyridine-2-carboxylic acid (GP2.2, 20 mg. 0.05 mmol) in DCM (10 mL) was added cyclopropanamine (3 mg, 0.06 mmol), EDC1 (23 mg, 0. 12 mmol) and DMAP (15 mg. 0. 12 mmol). The reaction was stirred at rt for 3 h. The completion of the reaction was monitored by TLC. The reaction mixture was concentrated directly under reduced pressure. The resulting residue was purified by preparative TLC with a elution of 50% EtOAc/PE to EtOAc to provide
N-cyclopropyl-3-methyl-7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5- b]pyridine-2-carboxamide (6.6 mg, 0.014 mmol) as a yellow solid. LC-MS (ESI+): m/z 458 (MH+). ’HNMR (300MHz, CDCh) 8.65 (d, J= 2.4 Hz, 1H), 7.77-7.72 (m, 2H), 7.50 (s, 1H), 7.38-7.31 (m, 2H). 7.18 (d, J= 7.5 Hz, 1H). 6.78 (d, J = 2.4 Hz, 1H). 4.21 (s. 3H), 4.03-3.90 (m. 8H), 2.90-2.87 (m,lH), 2.44 (s, 3H), 0.91-0.80 (m, 2H), 0.80-0.70 (m, 2H).
[0203] General procedure 3
[0204] Synthesis of Compound 3: 4-(3-(3-(pyridin-2-yl)propyl)-5-(3-(m-tolyl)-lH- pyrazol- l-yl)-3H-imidazo [4,5-b] pyridin-7-yl)morpholine

[0205] Synthesis of 6-chloro-4-morpholino-3-nitro-N-( 3-(pyridin-2-yl)propyl)pyridin-2-amine, GP3.1
[0206] To a solution of 4-(2,6-dichloro-3-nitropyridin-4-yl)morpholine (GP1.1, 600 mg, 2.16 mmol) in DMF (60 mL) at rt was added CS2CO3 (1.4 g, 4.32 mmol) and 3-(pyridin-2-yl)propan- 1-amine (440 mg, 3.24 mmol). The solution mixture was stirred at rt overnight. The completion of the reaction was monitored by TLC. The reaction mixture was quenched with water (100 mL). The aqueous solution was extracted with EtOAc (3 x 50 mL). The combined organic phase was dried over anhydrous Na^SO i. fdtrated and concentrated. The resulting residue was purified by silica gel column chromatography with a gradient elution of 5% EtOAc/PE to 33% EtOAc/PE to provide 6-chloro-4-morpholino-3-nitro-N-(3-(pyridin-2-yl)propyl)pyridin-2-amine (400 mg. 1.06 mmol) as a yellow oil. LC-MS (ESI+): m/z 378/380 (MH1 ).
[0207] Synthesis of4-morpholino-3-nitro-N-(3-(pyridin-2-yl)propyl)-6-(3-(m-tolyl)-lH- pyrazol-l-yl)pyridin-2-amine, GP3.2
[0208] To a solution of 6-chloro-4-morpholino-3-nitro-N-(3-(pyridin-2-yl)propyl)pyridin-2- amine (GP3.1, 380 mg, 1.0 mmol) in DMF (20 mL) was added 3-(m-tolyl)-lH-pyrazole (190 mg, 1.2 mmol) and CS2CO3 (655 mg, 2.0 mmol). The reaction was stirred at rt for 4 h. The completion of the reaction was monitored by TLC. The reaction mixture was quenched with water (30 mL) and extracted with EtOAc (2 x 100 mL). The combined organic phase was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue
was purified by silica gel column chromatography with a gradient elution of 10% EtOAc/PE to 20% EtOAc/PE to provide 4-morpholino-3-nitro-N-(3-(pyridin-2-yl)propyl)-6-(3-(m-tolyl)-lH- pyrazol-l-yl)pyridin-2-amine (410 mg, 0.82 mmol) as a yellow oil. LC-MS (ESI+): m/z 500 (MH+). 1HNMR (300MHz, CDCh) <J8.60-8.56 (m, 1H), 8.40-8.39 (m, 2H), 7.74-7.69 (m, 2H), 7.65-7.60 (m, 1H), 7.34-7.32 (m, 1H), 7.21-7.15 (m, 3H), 6.92 (s, 1H), 6.74 (d, J = 2.1 Hz, 1H), 3.90-3.86 (m, 4H), 3.82-3.70 (m, 2H), 3.32-3.25 (m, 4H), 2.95-2.85 (m, 2H), 2.44 (s, 3H), 2.22- 2.10 (m, 2H).
[0209] Synthesis of 4-morpholino-N2-(3-(pyridin-2-yl)propyl)-6-(3-(m-tolyl)-lH-pyrazol-l- y!)pyridine-2, 3-diamine. GP3.3
[0210] To a solution of 4-morpholino-3-nitro-N-(3-(pyridin-2-yl)propyl)-6-(3-(m-tolyl)-lH- pyrazol-l-yl)pyridin-2-amine (GP3.2, 410 mg, 0.82 mmol) in DCM/MeOH (10:1, 20 mL) was added Pd/C (50 mg). The reaction mixture was stirred at rt under hydrogen atmosphere for 3 h. The completion of the reaction was monitored by TLC. After filtration, the filtrate was concentrated directly under reduced pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 2% MeOH/DCM to 7% MeOH/DCM to provide 4-morpholino-N2-(3-(pyridin-2-yl)propyl)-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridine-2,3- diamine (300 mg, 0.64 mmol) as a brown solid. LC-MS (ESI+): m/z 470 (MH+).1HNMR (300MHz, CDCh) <J8.60-8.56 (m, 1H), 8.40 (d,.7= 2.7 Hz, 1H), 7.78 (s, 1H), 7.72 (d, .7= 7.8 Hz, 1H), 7.62-7.60 (m, 1H), 7.32 (t, J= 7.8 Hz, 1H), 7.21-7.12 (m, 4H), 6.69 (d, J= 2.7 Hz, 1H), 3.90-3.86 (m, 4H), 3.60-3.55 (m, 2H), 3.40 (broad, 2H), 3.05-2.90 (m, 6H), 2.43 (s, 3H), 2.20-2.10 (m, 2H).
[0211] Synthesis of 4-(3-(3-(pyridin-2-yl)propyl)-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-h]pyridin-7-yl)morpholine, Compound 3
[0212] To a solution of 4-morpholino-N2-(3-(pyridin-2-yl)propyl)-6-(3-(m-tolyl)-lH-pyrazol- l-yl)pyridine-2, 3-diamine (GP3.3, 60 mg. 0.13 mmol) in 1.4-dioxane (5 mL) was added TsOH (2 mg) and trimethoxymethane (18 mg, 0.15 mmol). The solution mixture was stirred under reflux for 2 h. The completion of the reaction was monitored by TLC. The reaction mixture was quenched with aqueous NaHCCL solution to adjust pH to 8. The aqueous solution was extracted with EtOAc (3 x 10 mL). The combined organic phase was dried over anhydrous Na2SO4, filtrated and concentrated under reduced pressure. The resulting residue was purified by preparative TLC with a elution of 7% MeOH/DCM to provide 4-(3-(3-(pyridin-2-yl)propyl)-5- (3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (50 mg, 0.10 mmol) as a brown solid. LC-MS (ESI+): m/z 480 (MH+). 'HNMR (300 MHz, CDCh) <J8.55 (d, J= 2.7 Hz, 2H), 7.58-7.70 (m, 3H), 7.58-7.56 (m, 1H), 7.39-7.28 (m, 2H), 7.22-7.10 (m, 3H), 6.76 (d, J
= 2.7 Hz, 1H), 4.31 (t, J = 6.9 Hz, 2H), 4.08-3.98 (m, 4H), 3.97-3.87 (m, 4H), 2.85 (t, J= 7.5
Hz, 2H), 2.50-2.36 (m, 5H).
[0213] General procedure 4:
[0214] Synthesis of Compound 4: 7-morpholino-3-(3-(pyridin-2-yl)propyl)-5-(3-(m-tolyl)- lH-pyrazol-l-yl)-lH-imidazo[4,5-b]pyridin-2(3H)-one

[0215] Synthesis of7-morpholino-3-(3-(pyridm-2-yl)propyl)-5-(3-(m-tolyl)-lH-pyrazol-l-yl)- lH-imidazo[4,5-b]pyridin-2(3H)-one, Compound 4
[0216] To a solution of 4-morpholino-N2-(3-(pyridin-2-yl)propyl)-6-(3-(m-tolyl)-lH-pyrazol- l-yl)pyridine-2,3-diamine (GP3.3, 200 mg, 0.4 mmol) in THF (10 mL) was added CDI (140 mg, 0.8 mmol). The reaction mixture was stirred at 80 °C under micro wave condition for 1 h. The completion of the reaction was monitored by TLC. The reaction mixture was concentrated directly under reduced pressure. The resulting residue was slurry in Et20 to provide 7- morpholino-3-(3-(pyridin-2-yl)propyl)-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-lH-imidazo[4.5- b]pyridin-2(3H)-one (150 mg, 0.30 mmol) as a white solid. LC-MS (ESI+): m/z 496 (MH+).
1HNMR (300 MHz, CDCh) S 10.35 (s, 1H), 8.52 (s, 1H), 8.45 (d, J= 2.4 Hz, 1H), 7.77 (s, 1H), 7.66- 7.57 (m, 1H), 7.51-7.45 (m, 1H), 7.37-7.27 (m, 2H), 7.22-7.08 (m, 3H), 6.75 (d, J= 2.4 Hz. 1H), 4.09 (t, J = 7.2 Hz, 2H), 4.01-3.90 (m, 4H), 3.42- 3.30 (m. 4H), 2.93 (t, J= 7.5 Hz, 2H), 2.44 (s, 3H), 2.42-2.30 (m, 2H).
[0217] General procedure 5:
[0218] Synthesis of Compound 5: l-methyl-7-morpholino-3-(3-(pyridin-2-yl)propyl)-5-(3-
(m-tolyl)-lH-pyrazol-l-yl)-lH-imidazo[4,5-b]pyridin-2(3H)-one

Synthesis of l-methyl-7-morpholino-3-(3-(pyridin-2-yl)propyl)-5-(3-(m-tolyl)-lH-pyrazol-l-yl)- lH-imidazo[4,5-b]pyridin-2(3H)-one . Compound 5
[0219] To a solution of 7-morpholino-3-(3-(pyridin-2-yl)propyl)-5-(3-(m-tolyl)-lH-pyrazol-l- yl)-lH-imidazo[4,5-b]pyridin-2(3H)-one (Compound 4, 50 mg, 0.10 mmol) in DMF (10 mL) was added CS2CO3 (65 mg, 0.20 mmol) and CH3I (20 mg, 0.14 mmol). The reaction mixture was stirred at rt for 1 h. The completion of the reaction was monitored by TLC. The reaction mixture w as quenched with water (20 mL) and extracted with EtOAc (3 x 10 mL). The combined organic phase was dried over anhydrous Na2SOr, filtered and concentrated under reduced pressure. The resulting residue was slurry in Et20 to provide l-methyl-7-morpholino-3-(3- (pyridin-2-yl)propyl)-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-lH-imidazo[4.5-b]pyridin-2(3H)-one (30 mg, 0.30 mmol) as a white solid. LC-MS (ESI+): m/z 510 (MH+). ’HNMR (300 MHz. CDCh) <J 8.50 (d, J= 2.4 Hz, 1H), 8.43 (d, J= 2.4 Hz, 1H), 7.77 (s, 1H), 7.72 (d, J= 7.5 Hz, 1H), 7.58- 7.53 (m, 2H), 7.34 (t, J= 7.5 Hz, 1H), 7.25-7.14 (m, 2H), 7.13-7.02 (m, 1H), 6.74 (d, J= 2.4 Hz, 1H), 4.09 (t, J= 6.6 Hz, 2H), 3.98-3.80 (m, 4H), 3.67 (s, 3H). 3.21-3.10 (m, 4H). 2.92 (t, J = 7.8 Hz. 2H), 2.44 (s, 3H), 2.44-2.26 (m. 2H).
[0220] General procedure 6:
[0221] Synthesis of Compound 6: 4-(2-(pyridin-2-yl)-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-lH- imidazo [4,5-b] pyridin-7-yl)morpholine

[0222] Synthesis of 6-chloro-N-(2, 4-dimethoxybenzyl)-4-morpholino-3-nitropyridin-2-amine, GP6.1
[0223] To a solution of 4-(2.6-dichloro-3-nitropyridin-4-yl)morpholine (GP1.1, 500 mg. 2.2 mmol) in THF (20 mL) was added (2,4-dimethoxyphenyl)methanamine (251 mg, 1.59 mmol) and CS2CO3 (1.4 g, 4.4 mmol). The reaction was stirred at rt overnight. The completion of the reaction was monitored by TLC. The reaction mixture was concentrated directly under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 5% EtOAc/PE to 20% EtOAc/PE to provide 6-chloro-N-(2.4- dimethoxybenzyl)-4-morpholino-3-nitropyridin-2-amine (390 mg, 0.96 mmol) as a yellow solid. LC-MS (ESI+): m/z 409/411 (MH+). 'HNMR (300 MHz, CDCh) <J8.42-8.39 (m, 1H), 7.28-7.26
(m, 1H), 6.45-6.40 (m, 2H), 6.08 (s, 1H), 4.68-4.63 (m, 2H), 3.86 (m, 3H), 3.80-3.72 (m, 7H), 3.21-3.10 (m, 4H).
[0224] Synthesis ofN-(2,4-dimethoxybenzyl)-4-morpholino-3-nitro-6-(3-(m-tolyl)-lH-pyrazol-
1-yl)pyridin-2-amine, GP6.2
[0225] To a solution of 6-chloro-N-(2,4-dimethoxybenzyl)-4-morpholino-3-nitropyridin-2- amine (GP6.1, 390 mg, 0.96 mmol) in DMF (17 mL) was added 3-(m-tolyl)-lH-pyrazole (226 mg, 1.43 mmol), C112O (14 mg, 0.1 mmol) and CS2CO3 (935 mg, 2.87 mmol). The reaction was stirred at 110 °C under N2 for 2 h. The completion of the reaction was monitored by TLC. The reaction mixture was quenched with water (30 mL) and extracted with EtOAc (3 x 30 mL). The combined organic phase was washed with aqueous NaCl solution, dried over anhydrous Na2SO4, filtrated and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 5% EtOAc/PE to 33% EtOAc/PE to provide N-(2,4-dimethoxybenzyl)-4-morpholino-3-nitro-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridin-
2-amine (570 mg, 1.08 mmol) as ayellow solid. LC-MS (ESI+): m/z 531 (MH+). 'HNMR (300 MHz, CDCh) £8.52 (d, J= 2.4 Hz, 1H), 7.78 (s, 1H), 7.71 (d, J= 7.5 Hz, 1H), 7.33-7.29 (m, 2H), 7.20 (s, 1H), 7.14 (d, J= 7.5 Hz, 1H), 6.70 (d, J= 2.4 Hz, 1H), 6.50-6.43 (m, 2H), 4.70- 4.50 (m, 3H), 3.95-3.85 (m, 7H), 3.80 (s, 3H). 3.40 (brs, 2H). 3.04-3.95 (m, 4H), 2.44 (s. 3H).
[0226] Synthesis of4-morpholino-3-nitro-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridin-2-amine, GP6.3
[0227] To a solution of N-(2,4-dimethoxybenzyl)-4-morpholino-3-nitro-6-(3-(m-tolyl)-lH- pyrazol-l-yl)pyridin-2-amine (GP6.2, 570 mg, 1.08 mmol) in DCM (15 mL) was added TFA (5 mL). The reaction mixture was stirred at 40 °C overnight. The completion of the reaction was monitored by TLC. The reaction mixture was quenched with aqueous NaHCOs solution to adjust pH to 8. The resulting mixture was extracted with DCM/MeOH (15:1, 3 X 20 mL). The combined organic phase was concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 1% MeOH/DCM to 10% MeOH/DCM to provide 4-morpholino-3-nitro-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridin-2- amine (450 mg, 1.18 mmol) as a yellow solid. LC-MS (ESI+): m/z 381 (MH ). 1HNMR (300 MHz, CDCh) £8.47 (d, J= 2.7 Hz, 1H), 7.74 (s, 1H), 7.70 (d, J= 7.5 Hz, 1H), 7.34 (t, J= 7.5 Hz. 1H), 7.20 (d. J= 7.8 Hz. 1H), 7.01 (s. 1H), 6.76 (d. J= 2.7 Hz. 1H), 6.45 (brs, 2H), 3.90- 3.85 (m, 4H), 3.35-3.31 (m, 4H), 2.43 (s, 3H).
[0228] Synthesis of 4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridine-2,3-diamine, GP6.4 [0229] To a solution of 4-morpholino-3-nitro-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridin-2-amine (GP6.3, 450 mg. 1.18 mmol) in DCM/MeOH (10: 1, 30 mL) was added Pd/C (50 mg). The reaction mixture was stirred at rt under hydrogen atmosphere for 1.5 h. The completion of the
reaction was monitored by TLC. After filtration, the filtrate was concentrated directly. The resulting residue was purified by silica gel column chromatography with a gradient elution of 10% EtOAc/PE to 50% EtOAc/PE to provide 4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l- yl)pyridine-2,3-diamine (280 mg, 0.8 mmol) as a brown solid. LC-MS (ESI+): m/z 351 (MH+). [0230] Synthesis of N-(2-amino-4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridin-3- yl)picolinamide, GP6.5
[0231] To a solution of 4-morpholino-6-(3-(m-tolyl)-lEI-pyrazol-l-yl)pyridine-2,3-diamine (GP6.4, 50 mg, 0. 14 mmol) and EtsN (28 mg, 0.29 mmol) in DCM (5 mL) was added picolinoyl chloride hydrochloride (25 mg, 0. 14 mmol). The solution was stirred at rt overnight. The completion of the reaction was monitored by TLC. The reaction mixture was quenched with water (10 mL) and extracted with DCM (3 x 10 mL). The combined organic phase was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was used directly for the next step without further purification. LC-MS (ESI+): m/z 456 (MH+).
[0232] Synthesis of 4-(2-(pyridin-2-yl)-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-lH-imidazo[4, 5- b]pyridin-7-yl)morpholine, Compound 6
[0233] To a solution of N-(2-amino-4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridin-3- yl)picolinamide (GP6.5, 45 mg, 0.099 mmol) in EtOH (5 mL) was added cone. H2SO4 (30 mg, 0.31 mmol). The reaction mixture was stirred under reflux for 2 h. The completion of the reaction was monitored by TLC. The reaction mixture was quenched with aqueous NaHCCh solution to adjust pH to 8. The aqueous solution was extracted with DCM/MeOH (15: 1, 3 x 10 mL). The combined organic phase was dried over anhydrous Na2SC>4, filtrated and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography with a elution of EtOAc to provide 4-(2-(pyridin-2-yl)-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-lH- imidazo[4,5-b]pyridin-7-yl)morpholine (5 mg, 0.043 mmol) as a yellow solid. LC-MS (ESI+): m/z 438 (MH+). lH NMR (300 MHz, CDCh) 10.65 (s, 1H), 8.63 (s, 1H), 8.54 (d, J = 2.1 Hz, 1H), 8.32 (d, J = 7.5 Hz, 1H), 7.85-7.72 (m, 3H), 7.37-7.31 (m, 3H), 7.17 (d, J= 7.8 Hz, 1H), 6.75 (d, J= 2.4 Hz, 1H), 4.15-4.11 (m, 4H), 4.00-3.90 (m, 4H), 2.44 (s, 3H).
[0234] General procedure 7
[0235] Synthesis of Synthesis of Compound 7: 4-(3-methyl-2-(lH-pyrazol-4-yl)-5-(3-(m- tolyl)- IH-py razol- l-yl)-3H-imidazo [4,5-b] pyridin-7-yl)morpholine and
Compound 12: N,N-dimethyl-2-(4-(3-methyl-7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l- yl)-3H-imid azo [4,5-b] pyridin-2-yl)- IH-pyrazol- l-yl)ethan amine

[0236] Synthesis of 4-(3-methyl-2-( lH-pyrazol-4-yl)-5-( 3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-b]pyridin-7-yl)morpholine, Compound 7
[0237] To a solution of N2-methyl-4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridine- 2,3-diamine (GP1.4, 200 mg, 0.55 mmol) in AcOH / H2O (3:2. 25 mL) was added Cu(OAc)2 (990 mg, 5.5 mmol) and tert-butyl 4-formyl-lH-pyrazole-l -carboxylate (30 mg, 0.31 mmol). The reaction mixture was stirred at 55 °C for 2 h. The completion of the reaction was monitored by TLC. The reaction mixture was quenched with aqueous NaHCOs solution to adjust pH to 8. The aqueous solution was extracted with DCM/MeOH (15: 1, 3 x 20 mL). The combined organic phase was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 2% MeOH/DCM to 7% MeOH/DCM to provide 4-(3-methyl-2-(lH-pyrazol-4-yl)-5-(3-(m- tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (200 mg. 0.45 mmol) as a brown solid. LC-MS (ESI+): m/z 441 (MH ). 1HNMR (300 MHz, DMSO-rfc) 13.40 (s, 1H), 8.74 (s, 1H), 8.47 (s, 1H), 8.11 (s, 1H), 7.80-7.77 (m, 2H), 7.38-7.33 (m, 1H), 7.23-7.18 (m, 2H), 7.05 (s, 1H), 4.11-3.94 (m, 7H), 3.84-3.72 (m, 4H), 2.51 (s, 3H).
[0238] Synthesis of N,N-dimethyl-2-(4-( 3-methyl- 7-morpholino-5-( 3-(m-tolyl)-lH-pyrazol-l- yl)-3H-imidazo[4, 5-b ]pyridin-2-yl)-lH-pyrazol-l-yl)ethanamine. Compound 12
[0239] To a solution of 4-(3-methyl-2-(lH-pyrazol-4-yl)-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-b]pyridin-7-yl)morpholine (Compound 7, 40 mg, 0.09 mmol) in DMF (5 mL) was added NaH (9 mg, 0.23 mmol) and 2-chloro-N,N-dimethylethanamine hydrochloride (20 mg, 0. 14 mmol). The reaction mixture was stirred at rt for 4 h. The completion of the reaction was monitored by TLC. The reaction mixture was quenched with water (10 mL) and extracted with DCM/MeOH (15: 1, 3 x 10 mL). The combined organic phase was dried over anhydrous Na2SO4, filtrated and concentrated under reduced pressure. The resulting residue was purified by preparative TLC with a elution of 7% MeOH/DCM to provide N.N-dimethyl-2-(4-(3-methyl-7- morpholino-5 -(3 -(m-toly 1)- 1 H-pyrazol- 1 -yl)-3H-imidazo[4,5 -b] pyri din-2 -yl)- 1 H-py razol- 1 - yl)ethanamine (20 mg, 0.04 mmol) as a white solid. LC-MS (ESI+): m/z 512 (MH+). 1HNMR (300 MHz, CDCh) 8.65 (d, J= 2.7 Hz, 1H), 8.10 (s, 1H), 8.03 (s, 1H), 7.81 (s, 1H), 7.76 (d, J = 7.8 Hz. 1H), 7.38-7.33 (m. 2H), 7.19 (d. J= 7.8 Hz. 1H), 6.79 (d. J = 2.7 Hz. 1H), 4.36 (t, J = 6.3 Hz, 2H), 4.06-4.03 (m, 4H), 4.00-3.97 (m, 7H), 2.89 (t, J= 6.6 Hz, 2H), 2.46 (s, 3H), 2.35 (s, 6H).
[0240] General procedure 8
[0241] Synthesis of Compound 17: 4-(2-(methoxymethyl)-3-methyl-5-(3-(m-tolyl)-lH- pyrazol- l-yl)-3H-imidazo [4,5-b] pyridin-7-yl)morpholine

[0242] Synthesis of2-chloro-N-(2-(methylamino)-4-morpholmo-6-(3-(m-tolyl)-lH-pyrazol-l- yl)pyridin-3-yl)acetamide, GP8.1
[0243] To a solution of N2-methyl-4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridine- 2,3-diamine (GP1.4, 220 mg, 0.60 mmol) and EtsN (67 mg, 0.66 mmol) in THF (5 mL) was added a solution of 2-chloroacetyl chloride (82 mg. 0.72 mmol) in THF (1 mL) dropwise. The completion of the reaction was monitored by TLC. The reaction mixture was quenched with aqueous NaHCCh solution to adjust pH to 8. The aqueous solution was extracted with EtOAc (3 x 20 mL). The combined organic phase was dried over anhydrous Na2SO4, filtered, and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 10% EtOAc/PE to 33% EtOAc/PE to provide 2- chloro-N-(2-(methylamino)-4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyri din-3- yl)acetamide (230 mg, 0.52 mmol) as a yellow solid. LC-MS (ESI+): m/z 441/443 (MH ).
[0244] Synthesis of 4-(2-(methoxymethyl)-3-methyl-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-bJpyridin-7-yl)morpholine. Compound 17
[0245] To a solution of 2-chloro-N-(2-(methylamino)-4-morpholino-6-(3-(m-tolyl)-lH- pyrazol-l-yl)pyridin-3-yl)acetamide (GP8.1, 30 mg, 0.068 mmol) in MeOH (5 mL) was added one drop of cone. H2SO4. The solution mixture was stirred at 55 °C for 2 h. The completion of the reaction was monitored by TLC. The reaction mixture was quenched with aqueous NaHCCh solution to adjust pH to 8. The aqueous solution was extracted with EtOAc (3 x 20 mL). The combined organic phase was dried over anhydrous Na2SO4, filtrated and concentrated under reduced pressure. The resulting residue was purified by preparative TLC with a elution of 50% EtOAc/PE to provide 4-(2-(methoxymethyl)-3-methyl-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-b]pyridin-7-yl)morpholine as a white solid. LC-MS (ESI+): m/z 419 (MH+).
'HNMR (300 MHz, CDCh) 8.62 (s, 1H), 7.78 (s, 1H), 7.74 (d, J= 7.8 Hz, 1H), 7.36-7.31 (m, 2H), 7.17 (d, J= 7.8 Hz, 1H), 6.77 (d, J= 2.4 Hz, 1H), 4.68 (s, 2H), 4.03-4.01 (m, 4H), 3.96- 3.94 (m, 4H), 3.86 (s, 3H). 3.43 (s, 3H), 2.44 (s. 3H).
[0246] General procedure 9:
[0247] Synthesis of Compound 18: N,N-dimethyl-l-(3-methyl-7-morpholino-5-(3-(m- tolyl)- IH-py razol- l-yl)-3H-imidazo [4, 5-b] pyridin-2-yl)methanamine
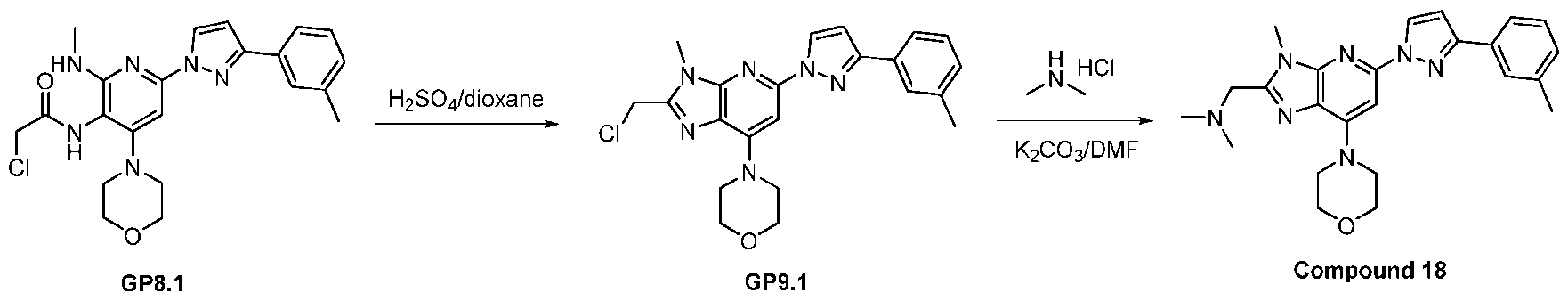
[0248] Synthesis of 4-(2-(chloromethyl)-3-methyl-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4, 5-b ]pyridin-7-yl)morpholine, GP9. 1
[0249] To a solution of 2-chloro-N-(2-(methylamino)-4-morpholino-6-(3-(m-tolyl)-lH- pyrazol-l-yl)pyridin-3-yl)acetamide (GP8.1, 150 mg, 0.34 mmol) in 1,4-dioxane (5 mL) was added one drop of cone. H2SO4. The solution mixture was stirred at 80 °C for 1 h. The completion of the reaction was monitored by TLC. The reaction mixture was quenched with aqueous NaHCCh solution to adjust pH to 8. The aqueous solution was extracted with EtOAc (3 x 30 mL). The combined organic phase was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 10% EtOAc/PE to 50% EtOAc/PE to provide 4-(2- (chloromethyl)-3-methyl-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-7- yl)morpholine (140 mg, 0.33 mmol) as a yellow7 solid. LC-MS (ESI+): m/z 423/425 (MH ). ’HNMR (300 MHz, CDCh) 8.62 (s, 1H), 7.78 (s, 1H), 7.73 (d, J= 7.5 Hz, 1H), 7.36-7.31 (m, 2H), 7.17 (d. J = 7.2 Hz. 1H), 6.77 (s, 1H), 4.79 (s, 2H), 4.10-4.00 (m, 4H), 3.96-3.90 (m, 4H), 2.44 (s, 3H).
[0250] Synthesis ofN,N-dimethyl-l-(3-methyl-7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)- 3H-imidazo[4,5-b]pyridin-2-yl)methanamine, Compound 18
[0251] To a solution of 4-(2-(chloromethyl)-3-methyl-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-b]pyridin-7-yl)morpholine (GP9.1, 140 mg, 0.33 mmol) in DMF (5 mL) was added K2CO3 (90 mg, 0.66 mmol) and dimethylamine hydrochloride (40 mg, 0.50 mmol). The solution mixture w as stirred at 80 °C for 2 h. The completion of the reaction w as monitored by TLC. The reaction mixture was diluted with water (30 mL) and extracted with EtOAc (3 x 30 mL). The combined organic phase was dried over anhydrous Na2SO4. filtered, and concentrated under reduced pressure. The resulting residue was purified by preparative TLC with by elution of 7% MeOH/DCM to provide N,N-dimethyl-l-(3-methyl-7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l- yl)-3H-imidazo[4,5-b]pyridin-2-yl)methanamine (74 mg, 0.17 mmol) as white solid. LC-MS (ESI+): m/z 432 (MH+). 1HNMR (300 MHz, CDCh) £8.62 (d. J= 2.1 Hz. 1H), 7.78 (s, 1H),
7.73 (d, J= 7.5 Hz, 1H), 7.36-7.31 (m, 2H), 7.16 (d, J= 7.2 Hz, 1H), 6.76 (d, J= 2.1 Hz, 1H), 4.02-4.00 (m, 4H), 3.96-3.94 (m, 4H), 3.87 (s, 3H), 3.68 (s, 2H), 2.44 (s, 3H), 2.32 (s, 6H).
[0252] General procedure 10:
[0253] Synthesis of Compound 11: N-cyclopropyl-7-morpholino-5-(3-(m-tolyl)-lH- pyrazol- l-yl)-3H-imidazo [4,5-b] pyridine-2-carboxamide

[0254] Synthesis of 7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazof4, 5-b pyridine- 2-carboxylic acid, GP10.1
[0255] To a solution of 4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridine-2,3-diamine (GP6.4, 106 mg, 0.30 mmol) and TEA (0.2 mL, 1.21 mmol) in MeOH (10 mL) was added methyl 2.2-di chi oro-2-methoxy acetate (106 mg. 0.60 mmol). The reaction mixture was stirred at 80 °C for 4 h. The completion of the reaction was monitored by TLC. LC-MS showed the desired acid product and the ester intermediate. The reaction mixture was concentrated directly under reduced pressure. The resulting residue was purified by preparative TLC to provide 7- morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridine-2-carboxylic acid (45 mg, 0. 11 mmol) as a yellow solid. LC-MS (ESI+): m/z 405 (MH+).
[0256] Synthesis ofN-cyclopropyl-7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-b]pyridine-2-carboxamide, Compound 11
[0257] To a solution of 7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5- b]pyridine-2-carboxylic acid (GP10.1, 30 mg. 0.07 mmol) in DCM (10 mL) was added cyclopropanamine (5 mg, 0.09 mmol), HATU (70 mg, 0.19 mmol) and DIEA (24 mg, 0.19 mmol). The reaction mixture was stirred at rt for 3 h. The completion of the reaction was monitored by TLC. The reaction mixture was concentrated directly under reduced pressure. The resulting residue was purified by preparative TLC with a elution of 7% MeOH/DCM to provide N-cyclopropyl-7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridine-2- carboxamide (2.6 mg, 0.006 mmol). LC-MS (ESI+): m/z 444 (MH+). 'H NMR (300 MHz, CDCh) 8.53 (s, 1H), 7.76 (s, 1H), 7.73 (d, J = 7.8 Hz, 1H), 7.38-7.32 (m, 3H), 7.20 (d, J= 7.5 Hz. 1H), 6.78 (d. J = 2.4 Hz. 1H), 4.04-3.90 (m. 8H), 2.95-2.90 (m. 1H), 2.45 (s, 3H), 0.99-0.90 (m, 2H), 0.77-0.70 (m, 2H).
[0258] General Procedure 11
[0259] Synthesis of Compound 23: 2-(7-morpholino-5-(3-(m- tolyl)- IH-pyrazol- l-yl)-3H- imidazo[4,5-b]pyridin-3-yl)acetonitrile

[0260] Synthesis of 4-(2,6-dichloro-3-nitropyridin-4-yl)morpholine, GP11.1
[0261] To a solution of 2,4,6-trichloro-3-nitropyridine (250.0 g, 1. 1 mol) in EtOH (4.0 L) at - 10 °C was added Et?N (222.5 g, 2.2 mol) and a solution of morpholine (95.8 g, 1.1 mol) in EtOH (1.0 L). The solution was stirred at -10 °C for 2 h. The completion of the reaction was monitored by TLC. The reaction mixture was concentrated directly under reduced pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 5% EtOAc/PE to 50% EtOAc/PE to provide 4-(2,6-dichloro-3-nitropyridin-4-yl)morpholine (180.0 g, 0.65 mol) as ayellow solid. LC-MS (ESI+): m/z 227/229 (MH+). 1HNMR (300MHz, CDCh) 6.81 (s, 1H), 3.82-3.75 (m, 4H), 3.25-3.15 (m, 4H).
[0262] Synthesis of 6-chloro-N-(2, 4-dimethoxybenzyl)-4-morpholino-3-nitropyndm-2-amine,
GP11.2
[0263] To a solution of 4-(2,6-dichloro-3-nitropyridin-4-yl)morpholine (180.0 g, 0.65 mol) in DMF (1.5 L) was added (2,4-dimethoxyphenyl)methanamine (118.9 g, 0.71 mol) and CS2CO3 (422.2 g, 1.29 mol). The reaction was stirred at 35 °C overnight. The completion of the reaction was monitored by TLC. After cool to rt. the reaction mixture was diluted with ice water (3.0 L) and a large amount of solid was precipitated. After filtration, the filter cake was slurry in EtOAc (500 rnL) and dried to provide 6-chloro-N-(2.4-dimethoxybenzyl)-4-morpholino-3-nitropyridin - 2-amine (188.0 g, 0.46 mol) as a yellow solid. LC-MS (ESI+): m/z 409/411 (MH+). 'HNMR
(300 MHz, CDCh) <J8.42-8.39 (m, 1H), 7.28-7.26 (m, 1H), 6.45-6.40 (m, 2H), 6.08 (s, 1H), 4.68-4.63 (m, 2H), 3.86 (m, 3H), 3.80-3.72 (m, 7H), 3.21-3.10 (m, 4H).
[0264] Synthesis ofN-(2,4-dimethoxybenzyl)-4-morpholino-3-nitro-6-(3-(m-tolyl)- 1H- pyrazol-l-yl)pyridin-2-amine, GPU.3
[0265] To a solution of 6-chloro-N-(2,4-dimethoxybenzyl)-4-morpholino-3-nitropyridin-2- amine (188.0 g, 0.46 mol) in DMF (1.5 L) was added 3-(m-tolyl)-lH-pyrazole (80.0 g, 0.51 mol), Cu2O (6.58 g, 0.046 mol) and CS2CO3 (449.9 g, 1.38 mol). The reaction was stirred at 110 °C under N2 for 2 h. The completion of the reaction was monitored by TLC. The reaction mixture was quenched with water (4.0 L) and extracted with EtOAc (3 x 2.0 L). The combined organic phase was washed with aqueous NaCl solution, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 5% EtOAc/PE to 33% EtOAc/PE to provide N-(2.4-dimethoxybenzyl)-4-morpholino-3-nitro-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridin-2-amine (125.6 g, 0.24 mol) as a yellow solid. LC-MS (ESI+): m/z 531 (MH+). 'HNMR (300 MHz, CDCh) 8.67-8.57 (m, 1H), 8.56 (d, J= 2.4 Hz, 1H), 7.74-7.69 (m, 2H), 7.34 (t, J= 7.5 Hz, 1H), 7.26-7.18 (m, 2H), 6.91 (s, 1H), 6.77 (d, J= 2.4 Hz, 1H), 6.50 (s, 1H), 6.43 (d, J= 8.4 Hz, 1H), 4.74 (d. J = 5.7 Hz. 2H), 3.89-3.86 (m. 7H), 3.80 (s, 3H), 3.33-3.30 (m, 4H), 2.44 (s, 3H).
[0266] Synthesis ofN2-(2,4-dimethoxybenzyl)-4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l-yl) pyridine-2, 3-diamine, GPU.4
[0267] To a solution of N-(2,4-dimethoxybenzyl)-4-morpholino-3-nitro-6-(3-(m-tolyl)-lH- pyrazol-l-yl)pyridin-2-amine (125.6 g, 0.24 mol) in THF/MeOH (900 mL/900 rnL) was added Pd/C (12.56 g, 10%W/W). The reaction mixture was purged with H2 (gas) three times. The reaction mixture was stirred at rt overnight under H2 using balloons. The completion of the reaction was monitored by TLC and LC-MS. After filtration, the filtrate was concentrated. The resulting residual was purified by silica gel column chromatography with a gradient elution of PE to 33% EtOAc/PE to provide N2-(2,4-dimethoxybenzyl)-4-morpholino -6-(3-(m-tolyl)-lH- pyrazol-l-yl)pyridine-2, 3-diamine (107.0 g, 0.21 mol) as an off-white solid. LC-MS (ESI+): m/z 501 (MH ). ’HNMR (300 MHz, CDCh) J8.57 (d, J = 2.1 Hz, 1H), 7.78 (s, 1H), 7.70 (d. J= 7.2 Hz. 1H), 7.33-7.29 (m. 2H), 7.25-7.13 (m. 2H), 6.71 (s, 1H), 6.49-6.44 (m, 2H), 4.62 (broad, 2H), 3.88-3.84 (m, 7H), 3.80 (s, 3H), 3.04-2.98 (m, 4H), 2.43 (s, 3H).
[0268] Synthesis of 4-(3-(2, 4-dimethoxybenzyl)-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo [4, 5-b ]pyridin- 7-yl)morpholine, GP11.5
[0269] To a three-neck bottle was added N2-(2,4-dimethoxybenzyl)-4-morpholino-6- (3-(m- tolyl)-lH-pyrazol-l-yl)pyridine-2.3 -diamine (107.0 g, 0.21 mol), trimethoxymethane (970 mL) and TSOH-H2O (3.7 g, 0.021 mol). The reaction mixture was heated to reflux and stirred for 2 h.
The completion of the reaction was monitored by TLC and LC-MS. The reaction mixture was concentrated directly. The resulting residual was slurry in EtOAc/PE (1/5, 1.2 L) for 30 min to provide 4-(3-(2,4-dimethoxybenzyl)-5-(3-(m-tolyl)-lH-pyrazol-l-yl) -3H-imidazo[4,5- b]pyridin-7-yl)morpholine (102.0 g, 0.20 mol) as an off-white solid. LC-MS (ESI+): m/z 511 (MH+). 1HNMR (300 MHz, CDCh) 8.64 (d, J= 2.4 Hz, 1H), 7.85 (s, 1H), 7.78 (s, 1H), 7.73 (d, J= 7.5 Hz, 1H), 7.36-7.32 (m, 3H), 7.17 (d, J= 7.5 Hz, 1H), 6.77 (d, J= 2.4 Hz, 1H), 6.46- 6.43 (m, 2H), 5.32 (s, 2H), 4.00-3.92 (m, 8H), 3.86 (s, 3H), 3.78 (s, 3H), 2.44 (s, 3H).
[0270] Synthesis of 4-(5-( 3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4, 5-b Jpyridin- 7- yl)morpholine. GP11.6
[0271] To a solution of 4-(3-(2,4-dimethoxybenzyl)-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo [4,5-b]pyridin-7-yl)morpholine (102.0 g, 0.20 mol) in DCM (200 mb) was added TFA (400 mL). The reaction mixture was stirred at rt overnight. The completion of the reaction was monitored by TLC and LC-MS. The reaction mixture was concentrated directly. The resulting residual was quenched with aqueous NaHCOs solution to adjust pH to 8 and a large amount of solid was precipitated. After filtration, the filter cake was further slurry in DCM (2.0 L) and H2O (200 mL) to provide 4-(5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-7- yljmorpholine (60.5 g. 0.167 mol) as a white solid. LC-MS (ESI+): m/z 361 (MH+). 1HNMR (300 MHz, CDCh) £8.43 (d, .7= 2.1 Hz, 1H), 7.86 (s, 1H), 7.77 (s, 1H), 7.73 (d, .7= 7.8 Hz, 1H), 7.34 (d, J= 7.5 Hz, 1H), 7.24-7.17 (m, 2H), 6.79 (d, J= 2.1 Hz, 1H), 4.07-3.04 (m, 4H), 3.97-3.94 (m, 4H), 2.44 (s. 3H).
[0272] Synthesis of 2-(7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4.5- b]pyridin-3-yl)acetonitrile, Compound 23
[0273] To a solution of 4-(5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-7- yl)morpholine (22.0 g, 0.06 mol) in DMF (300 mL) was added CS2CO3 (40.0 g, 0.12 mol) and 2- bromoacetonitrile (11.0 g, 0.09 mol). The reaction mixture was stirred at rt for 1 h. The completion of the reaction was monitored by TLC and LC-MS. The reaction mixture was diluted with H2O (500 mL) and a large amount of solid was precipitated. After filtration, the filter cake was further slurry7 in EtOAc/PE (1/5, 200 mL) to provide 2-(7-morpholino-5-(3-(m-tolyl)-lH- pyrazol-l-yl)-3H-imidazo[4.5-b]pyridin-3-yl)acetonitrile (20.8 g, 0.052 mol) as a while solid. LC-MS (ESI+): m/z 400 (MH+). 1HNMR (300 MHz, CDCh) 58.60 (d, J= 2.4 Hz, 1H), 7.87 (s, 1H), 7.77-7.72 (m, 2H), 7.38-7.32 (m, 2H), 7.18 (d, J= 7.2 Hz, 1H), 6.78 (d, J= 2.4 Hz, 1H), 5.15 (s, 2H), 4.06-4.04 (m, 4H), 3.96-3.92 (m, 4H), 2.44 (s, 3H).
[0274] General procedure 12:
[0275] Synthesis of Compound 45: Ethyl l-[3-(2-methoxyethyl)-7-morpholino-3H-l,3,4- triazainden-5-yl]-3-(m-tolyl)-5-pyrazolecarboxylate
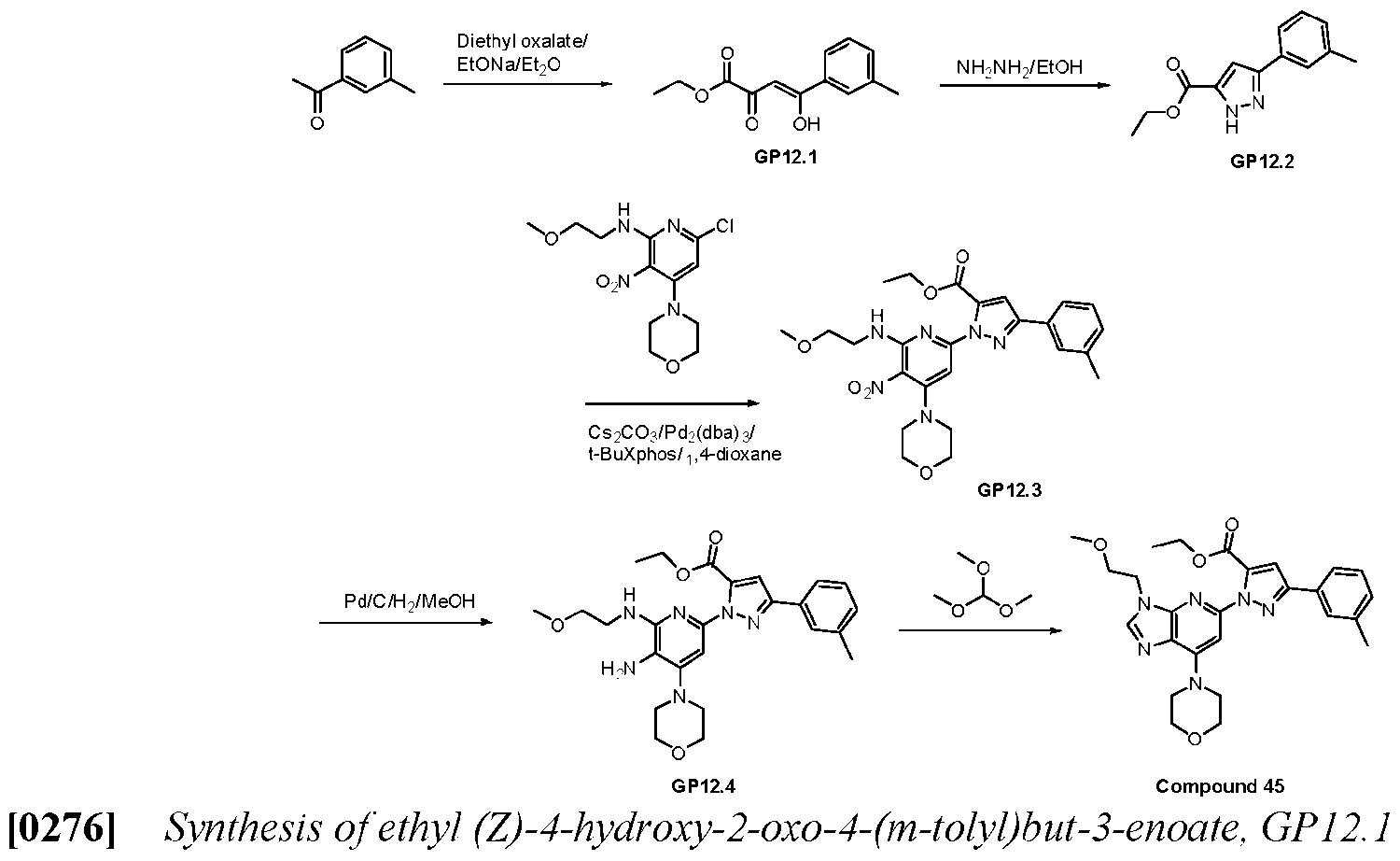
[0277] To a solution of 1 -(m-tolyl)ethanone (10 g, 75 mmol) in Et20 (400 mL) at ambient temperature was added NaOEt (51 g, 0.15 mmol) dropwise. After addition, diethyl oxalate (13.1 g, 90 mmol) was added dropwise to the above mixture. The reaction was stirred at rt for 2 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was quenched with water (200 mL) and adjusted the pH to 4 using AcOH. The aqueous solution was extracted with EtOAc (3 x 200 mL). The combined organic phases were dried over anhydrous Na2SC>4, filtrated and concentrated. The resulting residue was purified by silica gel column chromatography with a gradient elution of 5% EtOAc/PE to 30% EtOAc/PE to provide ethyl (Z)-4-hydroxy-2-oxo-4-(m-tolyl)but-3-enoate (7.6 g, 32.5 mmol). 'H NMR (300 MHz, CDCk) 5 15.34 (brs, 1H), 7.81-7.78 (m, 2H), 7.41-7.36 (m, 2H), 7.07 (s, 1H), 4.44-4.37 (m, 2H), 2.42 (s, 3H), 1.42 (t, .7= 7.5 Hz, 3H).
[0278] Synthesis of ethyl 3-(m-tolyl)-lH-pyrazole-5-carboxylate, GP12.2
[0279] A solution of ethyl (Z)-4-hydroxy-2-oxo-4-(m-tolyl)but-3-enoate (7.6 g, 32.5 mmol) and hydrazine hydrate (85%, 1.56 g, 48.75 mmol) in EtOH (50 mL) was heated to 80 °C and stirred for 2 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was concentrated directly and then quenched with water (200 mL). The aqueous solution was extracted with EtOAc (2 x 200 mL). The combined organic phases were dried over anhydrous NaiSO-i. filtrated and concentrated. The resulting residue was purified by silica gel column chromatography with a gradient elution of 5% EtOAc/PE to 30% EtOAc/PE to provide ethyl 3-(m-tolyl)-lH-pyrazole-5-carboxylate (3.5 g, 15.2 mmol). LC-MS (ESI+): m/z 231 (MH+). XH NMR (300 MHz, CDCh) 7.58-7.53 (m, 2H), 7.41-7.30 (m, 1H), 7.18 (d, J= 7.2 Hz, 1H), 7.11 (s, 1H), 4.46-4.39 (m, 2H), 2.41 (s, 3H), 1.42 (t, J= 7.2 Hz, 3H).
- I l l -
[0280] Synthesis of ethyl l-(6-((2-methoxyethyl)amino)-4-morpholino-5-nitropyridin-2-yl)-3- (m-tolyl)-lH-pyrazole-5-carboxylate, GP12.3
[0281] To a solution of 6-chloro-N-(2-methoxyethyl)-4-morpholino-3-nitropyridin-2-amine (840 mg, 2.65 mmol) in 1,4-dioxane (15 mL) was added ethyl 3-(m-tolyl)-lH-pyrazole-5- carboxylate (555 mg, 2.41 mmol). CS2CO3 (2.6 g. 7.96 mmol), t-Buxphos (451 mg, 1.06 mmol) and Pd2(dba)3 (486 mg, 0.53 mmol). The reaction mixture was purged with N2 for three times and stirred at 85 °C for 2 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was diluted with water (30 mL) and extracted with EtOAc (3 x 20 mL). The combined organic phases were dried over anhydrous Na2SO4. filtrated and concentrated. The resulting residue was purified by silica gel column chromatography with a gradient elution of 5% EtOAc/PE to 25% EtOAc/PE to provide ethyl l-(6-((2-methoxyethyl)amino)-4-morpholino- 5-nitropyridin-2-yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate (395 mg, 0.77 mmol) as a yellow oil. LC-MS (ESI+): m/z 511 (MH+). 1H NMR (300 MHz.CDCh) <78.20-8.10 (m. 1H), 7.70-7.64 (m, 2H), 7.36-7.31 (m, 1H), 7.20 (d, .7= 7.5 Hz, 1H), 7.07 (s, 1H), 6.74 (s, 1H), 4.40- 4.33 (m, 2H), 3.90-3.85 (m, 4H), 3.64-3.60 (m, 2H), 3.56-3.53 (m, 2H), 3.39 (s, 3H), 3.34-3.32 (m, 4H), 2.42 (s, 3H), 1.30 (t, J= 7.5 Hz, 3H).
[0282] Synthesis of ethyl l-(5-amino-6-((2-methoxyethyl)amino)-4-morpholinopyridin-2-yl)-3- (m-tolyl)-lH-pyrazole-5-carboxylate, GP12.4
[0283] To a solution of ethyl l-(6-((2-methoxyethyl)amino)-4-morpholino-5-nitropyridin-2- yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate (395 mg, 0.77 mmol) in MeOH (15 mL) was added Pd/C (50 mg). The mixture was purged with H2 for three times and stirred under H2 atmosphere for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was filtered and concentrated directly to provide ethyl l-(5-amino-6-((2- methoxyethyl)amino)-4-morpholinopyridin-2-yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate (355 mg, 0.74 mmol) as a brown solid. The resulting residue was used for the next step without further purification; LC-MS (ESI+): m/z 481 (MH+).
[0284] Synthesis of ethyl l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5- yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate, Compound 45
[0285] To a solution of ethyl l-(5-amino-6-((2-methoxyethyl)amino)-4-morpholinopyridin-2- yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate (300 mg, 0.63 mmol) in trimethoxymethane (15 mL) was added AcOH (46 mg, 0.75 mmol). The mixture solution was heated to 90 °C and stirred for 3 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was quenched with aqueous NaHCO? (20 mL) to pH = 9. The aqueous solution was extracted with EtOAc (3 x 20 mL). The combined organic phase was dried over anhydrous Na2SO4, filtrated and concentrated. The resulting residue was purified by silica gel column chromatography with
a gradient elution of 10% EtOAc/PE to 50% EtOAc/PE to provide ethyl l-(3-(2-methoxyethyl)- 7-morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate (250 mg, 0.51 mmol) as a white solid. LC-MS (ESI+): m/z 491 (MH+). 'HNMR (300 MHz, CDCh) 7.94 (s, 1H), 7.74 (s, 1H). 7.66 (d, J= 6.9 Hz, 1H). 7.32 (t. J= 7.2 Hz. 1H), 7.20-7.16 (m. 2H), 6.83 (s, 1H), 4.34-4.24 (m, 4H), 4.01-3.99 (m, 4H), 3.94- 3.92 (m, 4H), 3.67 (t, J= 4.8 Hz, 2H), 3.32 (s, 3H), 2.41 (s, 3H), 1.25 (t, J= 6.9 Hz, 3H).
[0286] General procedure 13:
[0287] Synthesis of Compound 46: l-[3-(2-methoxyethyl)-7-morpholino-3H- 1,3,4- triazainden-5-yl]-3-(m-tolyl)-5-pyrazolecarboxylic acid

[0288] Synthesis ofl-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-3- (m-tolyl)-lH-pyrazole-5-carhoxylic acid, Compound 46
[0289] To a solution of ethyl l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin- 5-yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate (COMPOUND 45, 230 mg, 0.47 mmol) in MeOH/thO (2:1, 15 mL) was added LiOH.lUO (197 mg, 4.7 mol). The mixture was stirred at rt overnight. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was concentrated, and the resulting residue was adjusted with IM HC1 to pH = 4. A large amount of the white solid was precipitated. After filtration, the filter cake was concentrated directly to provide l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4.5- b]pyridin-5-yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylic acid (206 mg, 0.45 mmol) as a white solid. LC-MS (ESI+): m/z 463 (MH ). 'HNMR (300 MHz, CDC13) £7.87 (s, 1H), 7.72-7.70 (m, 3H), 7.50 (s, 1H), 7.36 (t,
 7.8 Hz, 1H). 7.24-7.21 (m, 1H). 4.36 (t. J = 4.8 Hz, 2H), 4.18-4.16
7.8 Hz, 1H). 7.24-7.21 (m, 1H). 4.36 (t. J = 4.8 Hz, 2H), 4.18-4.16
(m, 4H), 3.94- 3.92 (m, 4H), 3.75 (t, J= 4.8 Hz, 2H), 3.34 (s, 3H), 2.44 (s, 3H).
[0290] General procedure 14:
[0291] Synthesis of Compound 54: {l-[3-(2-methoxyethyl)-7-morpholino-3H-l,3,4- triazainden-5-yl]-3-(m-tolyl)-5-pyrazolyl}methanol
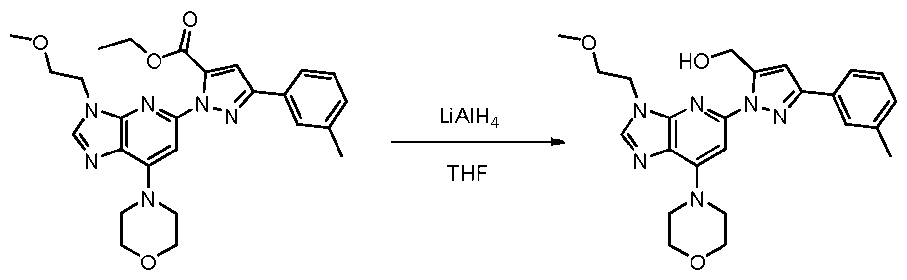 [0292] Synthesis of (l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-3- (m-tolyl)-lH-pyrazol-5-yl)methanol, Compound 54
[0292] Synthesis of (l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-3- (m-tolyl)-lH-pyrazol-5-yl)methanol, Compound 54
[0293] To a solution of ethyl l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin- 5-yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate (Compound 45, 820 mg, 1.67 mmol) in THF (10.0 mL) at 0 °C was added LiAlFL (64 mg, 1.67 mmol) in portions. The reaction mixture was allowed to warm to rt and stirred for 1 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was quenched with water (10 mL) and extracted with EtOAc (3 x 10 mL). The combined organic phases were dried over anhydrous Na2SO4, filtered and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 10% EtOAc/PE to EtOAc to provide (l-(3-(2- methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-3-(m-tolyl)-lH-pyrazol-5- yl)methanol (600 mg, 1.34 mmol) as an off-white solid. LC-MS (ESI+): m/z 449 (MH+).
1HNMR (300 MHz, CDCh) 7.88 (s, 1H), 7.72-7.67 (m, 2H), 7.35-7.32 (m, 2H), 7.16(d, J = 7.5 Hz, 1H), 6.71 (s, 1H), 6.08 (t, J= 7.2 Hz, 1H), 4.77 (d, J= 7.2 Hz, 2H), 4.34 (t, .7= 4.8 Hz, 2H), 4.08-4.06 (m, 4H), 3.95-3.92 (m, 4H), 3.73 (t, J= 4.8 Hz, 2H), 3.32(s, 3H), 2.43 (s, 3H).
[0294] General procedure 15:
[0295] Synthesis of Compound 72: N-methyl({l-[3-(2-methoxyethyl)-7-morpholino-3H- l,3,4-triazainden-5-yl]-3-(m-tolyl)-5-pyrazolyl}methyl)amine
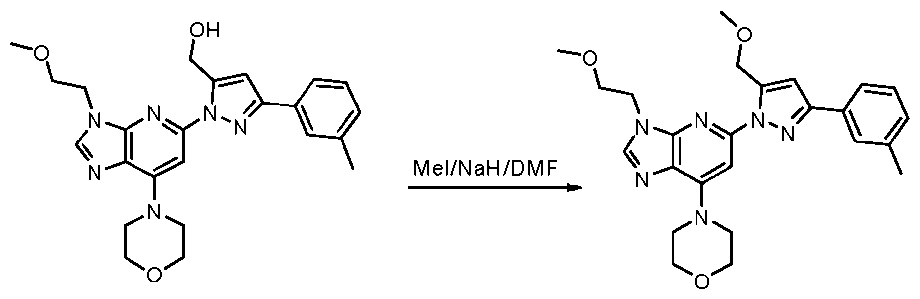
[0296] Synthesis of 4-(3-(2-methoxyethyl)-5-(5-(methoxymethyl)-3-(m-tolyl)-lH-pyrazol-l-yl)- 3H-imidazo[4,5-b]pyridin-7-yl)morpholine, Compound 72
[0297] To a solution of (l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5- yl)-3-(m-tolyl)-lH-pyrazol-5-yl)methanol (Compound 54, 50 mg, 0.11 mmol) in DMF (1.0 mL) at 0 °C was added NaH (5 mg, 60%, 0. 17 mmol) and CH3I (23.4 mg, 0. 16 mmol) in one portion. The reaction mixture was allowed to warm to rt and stirred for 2 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was quenched with water (10 rnL) and extracted with EtOAc (3 x 10 mL). The combined organic phases was dried over anhydrous Na2SO4, filtrated and concentrated under reduce pressure. The resulting residue was purified by preparative TLC with an elution of 50% EtOAc/PE to provide 4-(3-(2- methoxyethyl)-5-(5-(methoxymethyl)-3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin- 7-yl)morpholine (30 mg, 0.065 mmol) as a light yellow solid. LC-MS (ESI+): m/z 463 (MH+).
'HNMR (300 MHz, CDCh) £7.89 (s, 1H), 7.75 (s, 1H), 7.69 (d, J= 7.5 Hz, 1H), 7.31 (t, J= 7.5 Hz, 1H), 7.22 (s, 1H), 7.14 (d, J= 7.5 Hz, 1H), 6.80 (s, 1H), 5.05 (s, 2H), 4.37 (t, J= 4.8 Hz, 2H), 4.02-4.00 (m, 4H), 3.95-3.93 (m, 4H), 3.75 (t, J= 4.8 Hz, 2H), 3.49 (s, 3H). 3.34 (s, 3H), 2.42 (s, 3H).
[0298] General procedure 16:
[0299] Synthesis of Compound 55: N-methyl({l-[3-(2-methoxyethyl)-7-morpholino-3H- l,3,4-triazainden-5-yl|-3-(m-tolyl)-5-pyrazolyl]methyl)amine

[0300] Synthesis of 4-(5-(5-(chloromethyl)-3-(m-tolyl)-lH-pyrazol-l-yl)-3-(2-methoxyethyl)- 3H-imidazo[4, 5-b ]pyridin- 7-yl)morpholine, GP16. 1
[0301] To a solution of (l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5- yl)-3-(m-tolyl)-lH-pyrazol-5-yl)methanol (50 mg, 0.11 mmol) in DCM (6.0 mL) at 0 °C was added SOCh (0.5 mL). The reaction mixture was allowed to warm to rt and stirred for 1 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was concentrated directly under reduce pressure to provide crude 4-(5-(5-(chloromethyl)-3-(m-tolyl)-lH-pyrazol- l-yl)-3-(2-methoxyethyl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (47 mg, 0.10 mmol) as a yellow solid.
[0302] Synthesis of l-(l-(3-(2-methoxyethyl)~ 7-morpholino-3H-imidazo[4, 5-b ]pyridin-5-yl)-3- (m-tolyl)-lH-pyrazol-5-yl)-N-methylmethanamine, Compound 55
[0303] To a solution of crude 4-(5-(5-(chloromethyl)-3-(m-tolyl)-lH-pyrazol-l-yl)-3-(2- methoxyethyl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (47 mg, 0.10 mmol) in DMF (2 mL) was added methylamine hydrochloride (130 mg, 1.87 mmol) and CS2CO3 (940 mg, 2.88 mmol). The reaction was stirred at rt overnight. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was diluted with water (20 mL). The aqueous solution was extracted with EtOAc (3 x 20 mL). The combined organic phases were washed with saturated aqueous NaCl solution (2 x 20 mL), dried over anhydrous NazSCh, filtrated and concentrated. The resulting residue was purified by preparative TLC with an elution of 5% MeOH/DCM to provide l-(l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5- yl)-3-(m-tolyl)-lH-pyrazol-5-yl)-N-methylmethanamine (34 mg. 0.074 mmol) as an off-white solid. LC-MS (ESI+): m/z 462 (MH ). 1HNMR (300 MHz, CDCh) 7.83 (s, 1H), 7.71-7.69 (m, 2H), 7.38-7.34 (m, 2H), 7.24-7.21 (m, 1H), 6.89 (s, 1H), 4.56 (t, J= 4.8 Hz, 2H). 4.50 (s, 2H),
4.11-4.09 (m, 4H), 3.95-3.93 (m, 4H), 3.85 (t, J = 4.8 Hz, 2H), 3.26 (s, 3H), 2.66 (s, 3H), 2.44 (s, 3H).
[0304] General procedure 17:
[0305] Synthesis of Compound 56: N,N-dimethyl({l-[3-(2-methoxyethyl)-7-morpholino-
3H-l,3,4-triazainden-5-yl|-3-(m-tolyl)-5-pyrazolyl}methyl)amine

[0306] Synthesis of l-(l-(3-(2-methoxyethyl)- 7-morpholino-3H-imidazo[4, 5-b ]pyridin-5-yl)-3- (m-tolyl)-lH-pyrazol-5-yl)-N,N-dimethylmethanamine, Compound 56
[0307] To a solution of crude 4-(5-(5-(chloromethyl)-3-(m-tolyl)-lH-pyrazol-l-yl)-3-(2- methoxyethyl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (60 mg, 0.12 mmol) in DMF (3 mL) was added dimethylamine hydrochloride (20 mg, 0.24 mmol) and CS2CO3 (117 mg, 0.36 mmol). The reaction was stirred at rt for 3 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was diluted with water (20 mL). The aqueous solution was extracted with EtOAc (3 x 20 mL). The combined organic phases were washed with saturated aqueous NaCl solution (2 x 20 mL), dried over anhydrous NazSCh, filtrated and concentrated. The resulting residue was purified by preparative TLC with an elution of 5% MeOH/DCM to provide l-(l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5- yl)-3-(m-tolyl)-lH-pyrazol-5-yl)-N,N-dimethylmethanamine (9.0 mg, 0.019 mmol) as a light yellow solid. LC-MS (ESI+): m/z 476 (MH+). 1HNMR (300 MHz, CDsOD) 8.14 (s, 1H), 7.77-
7.71 (m, 2H), 7.38-7.31 (m, 2H), 7.21 (d, J= 7.5 Hz, 1H), 7.12 (s, 1H), 4.73 (s, 2H), 4.47 (t, J=
5.1 Hz. 2H), 4.02-4.00 (m. 4H), 3.92-3.90 (m. 4H), 3.83 (t, J = 5.1 Hz, 2H), 3.29 (s, 3H). 2.96 (s,
6H), 2.42 (s, 3H).
[0308] General procedure 18:
[0309] Synthesis of Compound 57: l-[3-(2-methoxyethyl)-7-morpholino-3H-l,3,4- triazainden-5-yl]-3-(m-tolyl)-5-pyrazolecarboxamide
 [0310] Synthesis of l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[ 4, 5-b ]pyridin-5-yl)-3- (m-tolyl)-lH-pyrazole-5-carboxamide, Compound 57
[0310] Synthesis of l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[ 4, 5-b ]pyridin-5-yl)-3- (m-tolyl)-lH-pyrazole-5-carboxamide, Compound 57
[0311] To a solution of l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5-yl)- 3-(m-tolyl)-lH-pyrazole-5-carboxylic acid (55 mg, 0.12 mmol) in DMAc (1 mL) was added NH4CI (13 mg. 0.24 mmol). PyBOP (93 mg, 0.18 mmol) and DIEA (47 mg, 0.36 mmol). The reaction mixture was stirred at rt for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was diluted with water (30 mL) and extracted with DCM (3 x 20 mL). The combined organic phases were washed with saturated aqueous NaCl solution (2 x 20 mL), dried over anhydrous Na2SO4, filtrated and concentrated. The resulting residue was purified by preparative TLC with an elution of 5% MeOH/DCM to provide l-(3-(2- methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-3-(m-tolyl)-lH-pyrazole-5- carboxamide (37 mg, 0.078 mmol) as an off-white solid. LC-MS (ESI+): m/z 462 (MH+).
'HNMR (300 MHz, CD3OD) 8.06 (s, 1H), 7.74 (s, 1H). 7.67 (d, J= 7.5 Hz, 1H). 7.32 (t, J= 7.5 Hz, 1H), 7.18 (d, .7= 7.5 Hz, 1H), 7.13 (s, 1H), 7.05 (s, 1H), 4.39 (t, .7= 5.1 Hz, 2H), 3.91 (s, 8H), 3.77 (t, J= 5.1 Hz, 2H), 3.32 (s, 3H), 2.41 (s, 3H).
[0312] General procedure 19:
[0313] Synthesis of Compound 58: N-methyl-l-[3-(2-methoxyethyl)-7-morpholino-3H- l,3,4-triazainden-5-yl]-3-(m-tolyl)-5-pyrazolecarboxamide
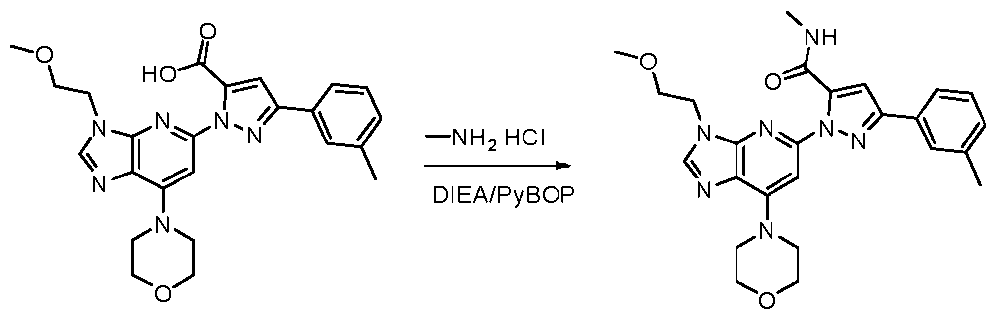
[0314] Synthesis of l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-N- methyl-3-(m-tolyl)-lH-pyrazole-5-carboxamide Compound 58
[0315] To a solution of l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5-yl)- 3-(m-tolyl)-lH-pyrazole-5-carboxylic acid (55 mg, 0.12 mmol) in DMAc (1 mL) was added methanamine hydrochloride (17 mg. 0.25 mmol), PyBOP (93 mg, 0.18 mmol) and DIEA (47 mg, 0.36 mmol). The reaction mixture was stirred at rt for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was diluted with water (30 mL) and extracted with DCM (3 x 20 mL). The combined organic phases were washed with saturated aqueous NaCl solution (2 x 20 mL), dried over anhydrous NazSO-i. filtrated and concentrated. The resulting residue was purified by slurry in MeOH to provide l-(3-(2-methoxyethyl)-7- morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-N-methyl-3-(m-tolyl)-lH-pyrazole-5-carboxamide
(36 mg, 0.077 mmol) as an off-white solid. LC-MS (ESI+): m/z 476 (MH+). 'HNMR (300 MHz, CDCh) £7.92 (s, 1H), 7.73 (s, 2H), 7.65 (d, J= 7.8 Hz, 1H), 7.32 (t, J= 7.2 Hz, 1H), 7.19-7.17 (m, 2H), 7.01 (s, 1H), 4.33 (t, J= 5.1 Hz, 2H), 4.03-4.01 (m, 4H), 3.93-3.91 (m, 4H), 3.72 (t, J= 5.1 Hz. 2H), 3.34 (s, 3H), 2.95 (d, .7 =4,8 Hz, 3H). 2.41 (s, 3H).
[0316] General procedure 20:
[0317] Synthesis of Compound 59: N,N-dimethyl-l-[3-(2-methoxyethyl)-7-morpholino- 3H-l,3?4-triazainden-5-yl]-3-(m-tolyl)-5-pyrazolecarboxamide

Compound 46 Compound 59
[0318] Synthesis of l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-N,N- dimelhyl-3-(m-lolyl)-lH-pyrazole-5-carboxcimide, Compound 59
[0319] To a solution of l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5-yl)- 3-(m-tolyl)-lH-pyrazole-5-carboxylic acid (55 mg, 0. 12 mmol) in DMAc (1 mL) was added dimethylamine hydrochloride (22 mg, 0.24 mmol), PyBOP (93 mg, 0.18 mmol) and DIEA (47 mg, 0.36 mmol). The reaction mixture was stirred at rt for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was diluted with water (30 mL) and extracted with DCM (3 x 20 mL). The combined organic phases were washed with saturated aqueous NaCl solution (2 x 20 mL), dried over anhydrous Na2SC>4, filtrated and concentrated. The resulting residue was purified by preparative TLC with an elution of 5%MeOH/DCM to provide l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-N,N-dimethyl-3- (m-tolyl)-lH-pyrazole-5-carboxamide (36 mg, 0.073 mmol) as an off-white solid. LC-MS (ESI+): m/z 490 (MH+). ’HNMR (300 MHz, CDCh) £7.87 (s, 1H), 7.74 (s, 1H), 7.67 (d, J= 7.2
Hz, 1H), 7.33-7.26 (m, 2H), 7.17(d, J= 7.2 Hz, 1H), 6.76 (s, 1H), 4.27 (t, J= 5.4 Hz, 2H), 4.03- 4.01 (m, 4H), 3.95-3.93 (m, 4H), 3.67 (t, J =5 A Hz. 2H), 3.33 (s, 3H), 3. 14 (s, 3H), 2.89 (s, 3H). 2.43 (s, 3H).
[0320] General procedure 21:
[0321] Synthesis of Compound 73: ({l-[3-(2-methoxyethyl)-7-morpholino-3H-l,3,4- triazainden-5-yl]-3-(m-tolyl)-5-pyrazolyl}methyl)amine

[0322] Synthesis of 4-(5-(5-(azidomethyl)-3-(m-tolyl)-lH-pyrazol-l-yl)-3-(2-methoxyethyl)- 3H-imidazo[4, 5-b ]pyridin- 7-yl)morpholine, GP21. 1
[0323] To a solution of 4-(5-(5-(chloromethyl)-3-(m-tolyl)-lH-pyrazol-l-yl)-3-(2- methoxyethyl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (85 mg. 0.18 mmol) in DMF (2.0 mL) at rt was added NaNs (14 mg, 0.22 mmol). The reaction mixture was stirred at rt for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with water (10 mL) and extracted with EtOAc (3 x 10 mL). The combined organic phases were dried over anhydrous Na2SO4, filtrated and concentrated. The resulting residue was used for the next step without further purification. LC-MS (ESI+): m/z 474 (MH+).
[0324] Synthesis of tert-butyl ((l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5- b ]pyridm-5-yl)-3-(m-tolyl)-lH-pyrazol-5-yl)methyl)carbamate, GP21.2
[0325] To a solution of 4-(5-(5-(azidomethyl)-3-(m-tolyl)-lH-pyrazol-l-yl)-3-(2- methoxyethyl)-3H-imidazo[4.5-b]pyridin-7-yl)morpholine (65 mg. 0.14 mmol) and BOC2O (0.5 mL) in EtOAc/EtOH (10 mL/5 mL) was added Pd/C (15 mg). The reaction mixture was purged with H2 for three times and stirred at rt overnight under H2 using balloons. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was filtered directly and the filtrate was concentrated. The resulting residual was purified by preparative TLC with a elution of 5 % MeOH/DCM to provide tert-butyl ((l-(3-(2-methoxyethyl)-7-morpholino-3H- imidazo[4,5-b]pyridin-5-yl)-3- (m-tolyl)-lH-pyrazol-5-yl)methyl)carbamate (50 mg, 0.09 mol) as a colorless oil. LC-MS (ESI+): m/z 548 (MH+).
[0326] Synthesis of ( l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4, 5-b ]pyridin-5-yl)-3- (m-tolyl)-lH-pyrazol-5-yl)methanamine hydrochloride. Compound 73
[0327] To a solution of tert-butyl ((l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5- b]pyridin-5-yl)-3-(m-tolyl)-lH-pyrazol-5-yl)methyl)carbamate (50 mg, 0.09 mmol) in
Et2O/MeOH (4.0 mL/2.0 mL) at rt was added a solution of Et2O/HCl (4.0 mL). The mixture was stirred at rt for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was concentrated directly and the resulting residue was purified by slurry in Et2O to provide (l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-3-(m- tolyl)-lH-pyrazol-5-yl)methanamine hydrochloride (40 mg, 0.089 mmol) as a white solid. LC- MS (ESI+): m/z 448 (MH ). 'HNMR (300 MHz, CDsOD) 8.80 (s, 1H), 7.77 (s, 1H), 7.71 (d, J = 7.5 Hz, 1H), 7.60 (s, 1H), 7.34 (t, J= 7.5 Hz, 1H), 7.22 (d, J= 7.5 Hz, 1H), 7.09 (s, 1H), 4.72 (s, 2H), 4.60 (t, J= 4.5 Hz, 2H), 3.95-3.93 (m, 4H), 3.85-3.83 (m, 6H), 3.33 (s. 3H), 2.42 (s,3H). [0328] General procedure 22:
[0329] Synthesis of Compound 82: 7-morpholino-3-(tetrahydro-2H-pyran-4-yl)-5-[3-(m- tolyl)- 1-py razolyl] -3H- 1,3, 4- triazaindene

[0330] Synthesis of 4-(2.6-dichloro-3-nitropyridin-4-yl)morpholine, GP22.1
[0331] To a solution of 2,4,6-trichloro-3-nitropyridine (250.0 g, 1. 1 mol) in EtOH (4.0 L) at - 10 °C was added EtsN (222.5 g, 2.2 mol) and a solution of morpholine (95.8 g, 1.1 mol) in EtOH (1.0 L). The reaction solution was stirred at -10 °C for 2 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was concentrated directly under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 5% EtOAc/PE to 50% EtOAc/PE to provide 4-(2,6-dichloro-3-nitropyridin-4- yl)morpholine (180.0 g, 0.65 mol) as a y ellow solid. LC-MS (ESI+): m/z 227/229 (MH+).
1HNMR (300MHz, CDCh) £6.81 (s, 1H), 3.82-3.75 (m, 4H), 3.25-3.15 (m, 4H).
[0332] Synthesis of 6-chloro-N-(2,4-dimethoxybenzyl)-4-morpholino-3-nitropyridin-2-amme, GP22.2
[0333] To a solution of 4-(2,6-dichloro-3-nitropyridin-4-yl)morpholine (180.0 g, 0.65 mol) in DMF (1.5 L) was added (2,4-dimethoxyphenyl)methanamine (118.9 g, 0.71 mol) and CS2CO3 (422.2 g. 1.29 mol). The reaction mixture was stirred at 35 °C overnight. After completion of the reaction as indicated by TLC analysis, the reaction mixture was cool to rt and diluted with ice water (3.0 L). A large amount of solid was precipitated. After filtration, the filter cake was slurry in EtOAc (500 mL) and dried to provide 6-chloro-N-(2,4-dimethoxybenzyl)-4-morpholino-3- nitropyridin-2-amine (188.0 g. 0.46 mol) as a yellow solid. LC-MS (ESI+): m/z 409/411 (MH+). 1HNMR (300 MHz, CDCh) 8.42-8.39 (m, 1H), 7.28-7.26 (m, 1H), 6.45-6.40 (m, 2H), 6.08 (s, 1H), 4.68-4.63 (m, 2H), 3.86 (m, 3H), 3.80-3.72 (m, 7H), 3.21-3.10 (m, 4H).
[0334] Synthesis ofN-(2.4-dimethoxybenzyl)-4-inorpholino-3-nilro-6-(3-(m-tolyl)-lH-pyrazol- l-yl)pyridin-2-amine, GP22.3
[0335] To a solution of 6-chloro-N-(2,4-dimethoxybenzyl)-4-morpholino-3-nitropyridin-2- amine (188.0 g, 0.46 mol) in DMF (1.5 L) was added 3-(m-tolyl)-lH-pyrazole (80.0 g, 0.51 mol), CU2O (6.58 g, 0.046 mol) and CS2CO3 (449.9 g, 1.38 mol). The reaction mixture was stirred at 110 °C under N2 for 2 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was quenched with water (4.0 L) and extracted with EtOAc (3 x 2.0 L). The combined organic phase was washed with aqueous NaCl solution, dried over anhydrous Na2SO4, filtrated and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 5% EtOAc/PE to 33% EtOAc/PE to provide N-(2.4-dimethoxybenzyl)-4-morpholino-3-nitro-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridine -2- amine (125.6 g, 0.24 mol) as ayellow solid. LC-MS (ESI+): m/z 531 (MH+). 1HNMR (300 MHz, CDCh) £8.67-8.57 (m, 1H), 8.56 (d, J= 2.4 Hz, 1H), 7.74-7.69 (m, 2H), 7.34 (t, J = 7.5 Hz. 1H), 7.26-7.18 (m. 2H), 6.91 (s, 1H), 6.77 (d, J = 2.4 Hz, 1H), 6.50 (s, 1H), 6.43 (d, J= 8.4 Hz, 1H), 4.74 (d, J= 5.7 Hz, 2H), 3.89-3.86 (m, 7H), 3.80 (s, 3H), 3.33-3.30 (m, 4H), 2.44 (s, 3H).
[0336] Synthesis of N2-(2, 4-dimethoxybenzyl)-4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l- yl)pyridine-2, 3-diamine, GP22.4
[0337] To a solution of N-(2,4-dimethoxybenzyl)-4-morpholino-3-nitro-6-(3-(m-tolyl)-lH- pyrazol-l -yl) pyridine-2-amine (125.6 g, 0.24 mol) in THF/MeOH (900 mL/900 mL) was added Pd/C (12.56 g, 10%W/W). The reaction mixture was purged with H2 (gas) for three times. The reaction mixture was stirred at rt overnight under H2 using balloons. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was filtered, and the filtrate was concentrated. The resulting residual was purified by silica gel column
chromatography with a gradient elution of PE to 33% EtOAc/PE to provide N2-(2,4- dimethoxybenzjd)-4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridine-2,3-diamine (107.0 g, 0.21 mol) as an off-white solid. LC-MS (ESI+): m/z 501 (MH+). ’HNMR (300 MHz. CDCh) 8 8.57 (d, 7= 2.1 Hz, 1H). 7.78 (s. 1H), 7.70 (d. 7= 7.2 Hz. 1H), 7.33-7.29 (m. 2H), 7.25-7.13 (m. 2H), 6.71 (s, 1H), 6.49-6.44 (m, 2H), 4.62 (brs, 2H), 3.88-3.84 (m, 7H), 3.80 (s, 3H), 3.04-2.98 (m, 4H), 2.43 (s, 3H).
[0338] Synthesis of 4f3-(2,4-dimethoxybenzyl)-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4, 5-b ]pyridin- 7 -yl)mor pholine, GP22.5
[0339] To a three-neck bottle was added N2-(2,4-dimethoxybenzyl)-4-morpholino-6-(3-(m- tolyl)-lH- pyrazol-l-yl)pyridine-2,3-diamine (107.0 g, 0.21 mol), trimethoxymethane (970 mL) and TSOH-H2O (3.7 g, 0.021 mol). The reaction mixture was heated to reflux and stirred for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was concentrated directly. The resulting residual was slurry in EtOAc/PE (1/5. 1.2 L) for 30 min to provide 4-(3-(2,4-dimethoxybenzyl)-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b] pyridine-7-yl)morpholine (102.0 g, 0.20 mol) as an off-white solid. LC-MS (ESI+): m/z 511 (MH+). ’HNMR (300 MHz, CDCh) 8.64 (d. J= 2.4 Hz. 1H), 7.85 (s, 1H), 7.78 (s, 1H), 7.73 (d, 7 = 7.5 Hz, 1H), 7.36-7.32 (m, 3H), 7.17 (d, J = 7.5 Hz, 1H), 6.77 (d, J = 2.4 Hz, 1H), 6.46- 6.43 (m, 2H), 5.32 (s, 2H), 4.00-3.92 (m, 8H), 3.86 (s, 3H), 3.78 (s, 3H), 2.44 (s, 3H).
[0340] Synthesis of 4-(5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-7- yl)morpholine. GP22.6
[0341] To a solution of 4-(3-(2,4-dimethoxybenzyl)-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-b] pyridin-7-yl)morpholine (102.0 g, 0.20 mol) in DCM (200 mL) was added TFA (400 mL). The reaction mixture was stirred at rt overnight. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was concentrated directly. The resulting residual was quenched with aqueous NaHCCh solution to adjust pH to 8 and a large amount of solid was precipitated. After filtration, the filter cake was further slurry in DCM (2.0 L) and H2O (200 mL) to provide 4-(5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin- 7-yl) morpholine (60.5 g, 0.167 mol) as a white solid. LC-MS (ESI+): m/z 361 (MH+). 1HNMR (300 MHz, CDCh) £8.43 (d, 7= 2.1 Hz, 1H). 7.86 (s, 1H), 7.77 (s. 1H), 7.73 (d. 7= 7.8 Hz. 1H), 7.34 (d, 7= 7.5 Hz, 1H), 7.24-7.17 (m, 2H), 6.79 (d, 7= 2.1 Hz, 1H), 4.07-3.04 (m, 4H), 3.97-3.94 (m, 4H), 2.44 (s, 3H).
[0342] Synthesis of 4-(3-(tetrahydro-2H-pyran-4-yl)-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-b]pyridin-7-yl)morpholine, Compound 82
[0343] To a solution of 4-(5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-7- yljmorpholine (247 mg, 0.69 mol) in DMF (10 mL) was added 4-iodotetrahydro-2H-pyran (290
mg, 1.37 mmol) and CS2CO3 (447 mg, 1.37 mmol). The reaction mixture was stirred at 80 °C overnight. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was concentrated directly. The resulting residual was slurry in MeOH to provide 4-(3-(tetrahydro-2H-pyran-4-yl)-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4.5- b]pyridin-7-yl)morpholine (230 mg. 0.52 mol) as a white solid. LC-MS (ESI+): m/z 445 (MH+). 'HNMR (300 MHz, CDCI3) £8.61 (d, J= 2.4 Hz, 1H), 7.84 (s, 1H), 7.78 (s, 1H), 7.73 (d, J = 7.2 Hz, 1H), 7.34-7.30 (m, 2H), 7.17 (d, J= 7.5 Hz, 1H), 6.77 (d, J= 2.1 Hz, 1H), 4.78-4.73 (m, 1H), 4.21-4.17 (m, 2H), 4.06-4.02 (m, 4H), 3.99-3.94 (m, 4H), 3.70-3.62 (m, 2H), 2.44 (s, 3H), 2.31-2.13 (m, 4H).
[0344] General procedure 23:
[0345] Synthesis of Compound 163: 3-(cyclopropylsulfonyl)-7-morpholino-5-[3-(m-tolyl)- l-pyrazolyl]-3H- 1,3, 4- triazaindene
 from GP22 Compound 163
from GP22 Compound 163
[0346] Synthesis of 4-(3-(tetrahydro-2H-pyran-4-yl)-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4, 5-b ]pyridin- 7-yl)morpholine, Compound 163
[0347] To a solution of 4-(5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-7- yl)morpholine (28 mg. 0.078 mol) in DMF (1 mL) was added cyclopropanesulfonyl chloride (32 mg, 0.233 mmol) and K2CO (54 mg, 0.39 mmol). The reaction mixture was stirred at rt for 4 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with w ater (10 mL) and extracted with EtOAc (3 x 10 mL). The combined organic phase was washed with aqueous NaCl solution, dried over anhydrous Na2SO4, filtrated and concentrated under reduce pressure. The resulting residue was purified by preparative TLC with an elution of 5% MeOH/DCM to provide 4-(3-(tetrahydro-2H-pyran-4-yl)-5-(3-(m-tolyl)- lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (13 mg, 0.028 mol) as a yellow' solid. LC-MS (ESI+): m/z 465 (MH ). ’H NMR (300 MHz. DMSO-tL) £8.63 (s, 1H), 8.43 (s. 1H), 7.86- 7.54 (m, 2H), 7.43-7.32 (m, 2H), 7.21 (d, J= 7.8 Hz, 1H), 7.09 (s, 1H), 4.08-3.94 (m, 4H), 3.89-3.76(m, 4H), 3.59-3.56 (m, 1H), 2.40(s, 3H), 1.55-1.45 (m, 2H), 1.33- 1.22 (m, 2H).
[0348] General procedure 24:
[0349] Synthesis of Compound 97: 3-[(l-methoxycyclopropyl)methyl]-7-morpholino-5-[3- (m-tolyl)-l-pyrazolyl]-3H-l,3,4-triazaindene
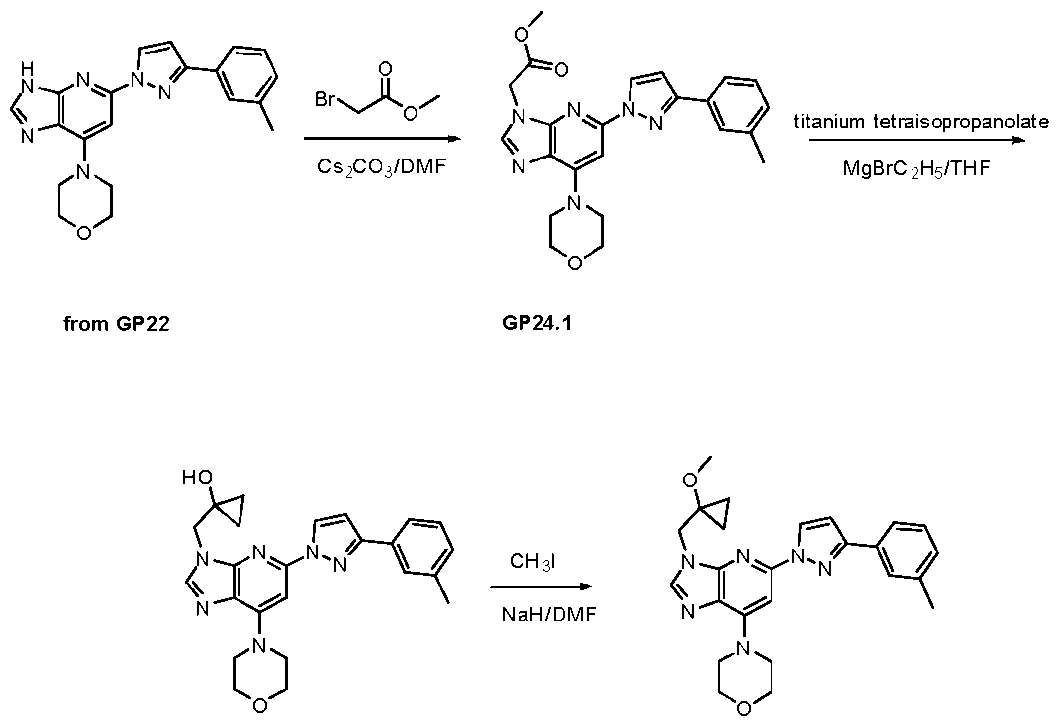
[0350] Synthesis of methyl 2-(7-morpholino-5-( 3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4.5- b ]pyridin-3-yl)acetate, GP24.1
[0351] To a solution of 4-(5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-7- yl)morpholine (400 mg, 1.11 mmol) in DMF (20.0 mL) at rt was added CS2CO3 (723 mg, 1.66 mmol) and methyl 2-bromoacetate (252 mg, 1.66 mmol). The reaction mixture was stirred at rt for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was diluted with water (20 mL) and extracted with DCM (3 x 30 mL). The combined organic phases were dried over anhydrous Na2SO4, filtrated and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 20% EtOAc/PE to 50% EtOAc/PE to provide methyl 2-(7-morpholino-5-(3- (m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-3-yl)acetate (280 mg, 0.65 mmol) as a white solid. LC-MS (ESI+): m/z 433 (MH+). 'HNMR (300 MHz, CDCh) 8.56 (d, J = 2.1Hz. 1H), 7.85 (s, 1H), 7.77 (s, 1H), 7.72 (d, J =7.5 Hz, 1H). 7.35-7.33 (m, 2H). 7. 17 (d, J =7.8 Hz. 1H), 6.75 (d, J= 2.1 Hz, 1H), 5.02 (s, 2H), 4.09-4.04 (m, 4H), 3.96-3.87 (m, 4H), 3.81 (s, 3H), 2.44 (s, 3H).
[0352] Synthesis of l-( (7-morpholino-5-( 3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4, 5- b ]pyridin-3-yl)methyl)cyclopr opanol, GP24.2
[0353] To a solution of methyl 2-(7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-b]pyridin-3-yl)acetate (100 mg, 0.23 mmol) in anhydrous THF (40.0 mL) at 0 °C under N2 atmosphere w as added titanium tetraisopropanolate (4.0 mg, 0.011 mmol). To the above solution was added a solution of CHsCH2MgBr (0.69 mL, IM. 0.69 mmol) dropwise. The mixture was stirred at that temperature for 1 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with NH4CI solution and extracted with EtOAc (3 x 30 mL). The combined organic phases was dried over anhydrous Na2SO4, filtrated and concentrated under reduce pressure. The resulting residue was purified by
preparative TLC with an elution of 10% MeOH/DCM to provide l-((7-morpholino-5-(3-(m- tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-3-yl)methyl)cyclopropanol (55 mg, 0.13 mmol) as a white solid. LC-MS (ESI+): m/z 431 (MH+). 'HNMR (300 MHz, CDCI3) £8.54 (d, J= 1.5 Hz. 1H), 7.76-7.71 (m. 3H), 7.36-7.31 (m. 2H), 7.19-7.15 (m. 1H), 6.76 (d. J= 1.5 Hz. 1H), 5.49 (s, 1H), 4.32 (s, 2H), 4.10-4.05 (m, 4H), 3.96-3.90 (m, 4H), 2.44 (s, 3H), 0.94-0.78 (m, 4H).
[0354] Synthesis of 4-(3-((l-methoxycyclopropyl)methyl)-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-b]pyridin-7-yl)morpholine, Compound 97
[0355] To a solution of l-((7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5- b]pyridin-3-yl)methyl)cyclopropanol (35 mg, 0.08 mmol) in DMF (2.0 mL) at 0 °C was added NaH (8 mg, 60%, 0.20 mmol) and a solution of CH3I (22 mg, 0.16 mmol) in DMF (0.5 mL) dropwise. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with water and extracted with DCM (3 x 10 mL). The combined organic phases were dried over anhydrous Na2SC>4, filtrated and concentrated under reduce pressure. The resulting residue was purified by preparative TLC with an elution of MeOH/DCM (1/10) to provide 4-(3-((l -methoxy cyclopropyl)methyl)-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-b]pyridin-7-yl)morpholine (15 mg, 0.034 mmol) as a white solid. LC-MS (ESI+): m/z 445 (MH+). ’HNMR (300 MHz, CDCh) 8.54 (d, J= 2.4 Hz, 1H), 7.96 (s, 1H), 7.78 (s, 1H), 7.73 (d, J= 7.5 Hz, 1H), 7.36-7.31 (m, 2H), 7.17 (d, J= 6.9 Hz, 1H), 6.75 (d,J= 2.4 Hz, 1H), 4.36 (s, 2H), 4.05-4.00 (m, 4H), 3.95-3.90 (m, 4H), 2.37 (s, 3H), 2.44(s, 3H), 0.97-0.75 (m, 4H).
[0356] General procedure 25:
[0357] Synthesis of Compound 100: 2-methyl-8-morpholino-6-[3-(m-tolyl)-l-pyrazolyl]- l,2,3,4-tetrahydro-2,4a,5,9-tetraazafluorene

Compound 100
[0358] Synthesis of N2-(2, 4-dimethoxybenzyl)-4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l- yl)pyridine-2, 3-diamine, GP25.1
[0359] To a solution of N-(2,4-dimethoxybenzyl)-4-morpholino-3-nitro-6-(3-(m-tolyl)-lH- pyrazol-l -yl) pyridine-2-amine (125.6 g, 0.24 mol) in THF/MeOH (900 mL/900 mL) was added Pd/C (12.56 g, 10%W/W). The reaction mixture was purged with H2 (gas) for three times. The reaction mixture was stirred at rt overnight under H2 using balloons. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was filtered, and the filtrate was concentrated. The resulting residual was purified by silica gel column chromatography with a gradient elution of PE to 33% EtOAc/PE to provide N2-(2,4- dimethoxybenz l)-4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l -yl)pyridine-2, 3-diamine (107.0 g, 0.21 mol) as an off-white solid. LC-MS (ESI+): m/z 501 (MH ). 'HNMR (300 MHz, CDCh) S 8.57 (d, J= 2.1 Hz, 1H), 7.78 (s, 1H), 7.70 (d, J= 7.2 Hz, 1H), 7.33-7.29 (m, 2H), 7.25-7.13 (m, 2H), 6.71 (s, 1H), 6.49-6.44 (m, 2H), 4.62 (brs, 2H), 3.88-3.84 (m, 7H), 3.80 (s, 3H), 3.04-2.98 (m, 4H), 2.43 (s, 3H).
[0360] Synthesis of 4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridine-2, 3-diamine, GP25.2
[0361] To a solution of N2-(2,4-dimethoxybenzyl)-4-morpholino-6-(3-(m-tolyl)-lH-pyrazol- 1-yl) pyridine-2, 3-diamine (25 g, 0.05 mol) in DCM (250 mL) at rt was added TFA (25 mL). The reaction mixture was stirred at rt for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with aqueous NaHCCh solution to pH ~ 8. The aqueous solution was extracted with DCM/MeOH (10/1, 3 x 200 mL). The
combined organic phases were dried over anhydrous Na2SO4, filtrated and concentrated. The resulting residue was purified by slurry in Et2O/EtOAc (100/1, 50 mL) twice to provide 4- morpholino-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridine-2,3-diamine (14.5 g, 0.04 mol) as a white solid. LC-MS (ESI+): m/z 351 (MH ). 'HNMR (300 MHz, DMSO- L) <78.33 (d. J = 2.4 Hz. 1H), 7.73-7.68 (m, 2H), 7.32 (t, J= 7.2 Hz, 1H), 7.15 (d, J= 7.2 Hz, 1H), 6.93 (s, 1H), 6.89 (d, J = 2.4 Hz, 1H), 5.70 (s, 2H), 4.31 (s, 2H), 3.83-3.78 (m, 4H), 2.93-2.86 (m, 4H), 2.38 (s, 3H).
[0362] Synthesis of tert-butyl methyl((7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo [4.5-b]pyridin-2-yl)methyl)carbamate, GP25.3
[0363] To a solution of 4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridine-2,3-diamine (100 mg, 0.29 mmol) in EtOH (3.0 mL) at rt was added Na2S2O4 (101 mg, 0.58 mmol) and tertbutyl methyl(2-oxoethyl)carbamate (55 mg, 0.34 mmol). The solution mixture was heated to 80 °C and stirred for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was diluted with water (20 mL). The aqueous solution was extracted with EtOAc (3 x 20 mL). The combined organic phases was dried over anhydrous Na2SC>4, filtrated and concentrated. The resulting residue was purified by silica gel column chromatography with a gradient elution of 10% EtOAc/PE to 50% EtOAc/PE to provide tertbutyl methyl((7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyri din-2- yl)methyl)carbamate (80 mg, 0.16 mmol) as a white solid. LC-MS (ESI+): m/z 504 (MH+).
[0364] Synthesis of tert-butyl ((3-(2-bromoethyl)-7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l- yl)-3H-imidazo[4, 5-b ]pyridin-2-yl)methyl)(methyl)carbamate, GP25.4
[0365] To a solution of tert-butyl methyl((7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4.5-b]pyridin-2-yl)methyl)carbamate (52 mg, 0.10 mmol) in DMF (2 mL) was added 1,2-di bromoethane (188 mg, 1.0 mmol) and CS2CO3 (65 mg, 0.2 mmol). The reaction was stirred at rt for 3 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was diluted with water (10 mL). The aqueous solution was extracted with EtOAc (3 x 10 mL). The combined organic phases were washed with saturated aqueous NaCl solution (2 x 20 mL), dried over anhydrous Na2SO4, filtrated and concentrated. The resulting residue was purified by preparative TLC with an elution of 33% EtOAc/PE to provide tert-butyl ((3-(2-bromoethy l)-7-morpholino-5 -(3 -(m-tolyl)- 1 H-py razol- 1 -y l)-3H-imidazo[4,5 -b]py ridin-2- yl)methyl)(methyl)carbamate (49 mg, 0.080 mmol) as a blue solid. LC-MS (ESI+): m/z 610/612 (MH+).
[0366] Synthesis of 4-(7-methyl-2-(3-(m-tolyl)-lH-pyrazol-l-yl)-6, 7,8,9-tetrahydro pyrido[3 2 4, 5 ]imidazo[ 1, 2-a ]pyrazin-4-yl)morpholine, Compound 100
[0367] To a solution of tert-butyl ((3-(2-bromoethyl)-7-morpholino-5-(3-(m-tolyl)-lH- pyrazol-l-yl)-3H-imidazo[4.5-b]pyridin-2-yl)methyl)(methyl)carbamate (49 mg. 0.080 mmol) in
DCM (5.0 mL) at rt was added TFA (0.5 mL). The reaction mixture was stirred at rt for 1 h. After completion of the reaction as indicated by TLC analysis, to the solution was added saturated Na2CCh aqueous solution (10 mL). The solution mixture was stirred at rt for another 2 h. After completion of the reaction as indicated by LC-MS analysis, the reaction mixture was extracted with MeOH/DCM (3 x 10 mL, 1 : 10). The combined organic phases was dried over anhydrous Na2SC>4, filtrated and concentrated. The resulting residue was purified by slurry in Et20 to provide 4-(7-methyl-2-(3-(m-tolyl)-lH-pyrazol-l-yl)-6, 7,8,9- tetrahydropyrido[3',2':4,5]imidazo[l,2-a]pyrazin-4-yl)morpholine (27 mg, 0.063 mmol) as a white solid. LC-MS (ESI+): m/z 430 (MH+). 'H NMR (300 MHz,CDCh) 8.58 (d, J= 2.1 Hz, 1H), 7.78 (s, 1H), 8.58 (d, J= 8.1 Hz, 1H), 7.35-7.30 (m, 2H), 7.16 (d, J= 7.5Hz, 1H), 6.75 (d, J = 2.1 Hz, 1H), 4.22 (t, J = 5.4 Hz, 2H), 3.98- 3.93 (m, 8H), 3.81 (s, 2H), 2.95 (t, J = 5.4 Hz, 2H), 2.56 (s. 3H), 2.44 (s, 3H).
[0368] General procedure 26:
[0369] Synthesis of Compound 218: l-({5-[3-(m-difluoromethoxyphenyl)-l-pyrazoIyI]-7- morpholino-3H-l,3,4-triazainden-3-yl}methyl)cyclopropanecarbonitrile
 nitropyridin-2-amine, GP26.1
nitropyridin-2-amine, GP26.1
[0371] To a solution of 6-chloro-N-(2,4-dimethoxybenzyl)-4-morpholino-3-nitropyridin-2- amine (1.0 g, 2.46 mmol) in DMF (30.0 mL) at rt was added CS2CO3 (2.0 g, 6.13 mmol) and 3- bromo-lH-pyrazole (347 mg, 2.70 mmol) in DMF (1.0 mL). The mixture was stirred at rt for 1
h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was diluted with water (20.0 mL) and extracted with EtOAc (3 x 10 rnL). The combined organic phases were washed with brine, dried over anhydrous Na2SC>4, filtrated and concentrated. The resulting residue was purified by silica gel column chromatography with a gradient elution of 20% EtOAc/PE to 50% EtOAc/PE to provide 6-(3-bromo-lH-pyrazol-l-yl)-N-(2,4- dimethoxybenzyl) -4-morpholino-3-nitropyridin-2-amine (1.23 g, 2.37 mmol) as a yellow oil. LC-MS (ESI+): m/z 519/521 (MH+).
[0372] Synthesis of 6-(3-bromo-lH-pyrazol-l-yl)-N2-(2,4-dimethoxybenzyl)-4- morpholinopyridine-2, 3-diamine, GP26.2
[0373] To a solution of 6-(3-bromo-lH-pyrazol-l-yl)-N-(2,4-dimethoxybenzyl)-4-morpholino -3-nitropyridin-2-amine (1.23 g, 2.37 mmol) in DCM/MeOH (10 mL/10 mL) was added NiCk (61 mg, 0.47 mmol) and NaBEk (180 mg, 4.74 mmol). The reaction mixture was stirred at 0 °C for 4 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with water (20 mL) and extracted with EtOAc (3 x 10 mL). The combined organic phases were dried over anhydrous Na2SO4, filtrated and concentrated. The resulting residual was purified by silica gel column chromatography with a gradient elution of 20% EtOAc/PE to 33% EtOAc/PE to provide 6-(3 -bromo- lH-pyrazol-l-yl)-N2- (2,4- dimethoxybenzyl)-4-morpholinopyridine-2.3-diamine (766 mg. 1.57 mol) as a green oil. LC-MS (ESI+): m/z 489/491 (MH+). *HNMR (300 MHz, CDCh) 8.37 (s, 1H), 7.29 (s, 1H), 7.04 (brs, 1H), 6.49-6.42 (m, 3H), 4.57 (brs, 2H), 3.96 (s, 8H), 3.80 (s, 6H).
[0374] Synthesis of 6-(3-bromo-lH-pyrazol-l-yl)-4-morpholinopyridine-2, 3-diamine, GP26.3 [0375] To a solution of 6-(3-bromo-lH-pyrazol-l-yl)-N2- (2.4-dimethoxybenzyl)-4- morpholinopyridine-2, 3-diamine (150 mg, 0.31 mmol) in DCM (10 rnL) at rt was added TFA (3 rnL). The reaction mixture was stirred at rt for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with aqueous NaHCCk solution to pH ~ 8. The aqueous solution was extracted with DCM/MeOH (10/1. 3 x 20 mL). The combined organic phases were dried over anhydrous Na2SO4, filtrated and concentrated. The resulting residual was purified by silica gel column chromatography with a gradient elution of 2% MeOH/DCM to 6.7% MeOH/DCM to provide 6-(3-bromo-lH-pyrazol-l-yl)-4- morpholinopyridine-2,3-diamine (75 mg, 0.22 mmol) as a yellow oil. LC-MS (ESI+): m/z 339/341 (MH ).
[0376] Synthesis of 4-(5-(3-bromo-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-7- yl)morpholine, GP26.4
[0377] To a solution of 6-(3-bromo-lH-pyrazol-l-yl)-4-morpholinopyridine-2.3-diamine (75 mg, 0.22 mmol) in trimethoxymethane (6.0 mL) was added TSOH-H2O (30 mg). The reaction
mixture was heated to 90 °C and stirred for 1 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was diluted with water (20 mL) and extracted with EtOAc (3 x 10 mL). The combined organic phases were dried over anhydrous Na2SO4, filtrated and concentrated. The resulting residual was purified by silica gel column chromatography with a gradient elution of 10% EtOAc/PE to 33% EtOAc/PE to provide 4-(5-(3- bromo-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (35 mg, 0.10 mmol) as a white solid. LC-MS (ESI+): m/z 349/351 (MH+).
[0378] Synthesis of l-((5-(3-bromo-lH-pyrazol-l-yl)-7-morpholino-3H-imidazo[4,5- b ]pyridin-3-yl)methyl)cyclopropanecarbonitrile, GP26.5
[0379] To a solution of 4-(5-(3-bromo-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-7- yl)morpholine (150 mg, 0.43 mmol) in DMF (6.0 mL) was added CS2CO3 (420 mg, 1.29 mmol) and l-(bromomethyl)cyclopropanecarbonitrile (136 mg, 0.86 mmol). The reaction mixture was heated to 90 °C and stirred for 1 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was diluted with water (20 mL) and extracted with EtOAc (3 x 20 mL). The combined organic phases was dried over anhydrous Na2SO4, filtrated and concentrated. The resulting residual was purified by silica gel column chromatography with a gradient elution of 10% EtOAc/PE to 50% EtOAc/PE to provide l-((5-(3-bromo-lH-pyrazol-l- yl)-7-morpholino-3H-imidazo[4,5-b]pyridin-3-yl)methyl)cyclopropanecarbonitrile (150 mg. 0.23 mmol) as a white solid. LC-MS (ESI+): m/z 428/430 (MEE).
[0380] Synthesis of l-((5-(3-(3-(difluoromethoxy)phenyl)-lH-pyrazol-l-yl)-7-morpholino-3H- imidazo[4, 5-b ]pyridin-3-yl)methyl)cyclopropanecarbonitrile, Compound 218
[0381] A suspension of l-((5-(3-bromo-lH-pyrazol-l-yl)-7-morpholino-3H-imidazo[4,5- b]pyridin-3-yl)methyl)cyclopropanecarbonitrile (70 mg, 0.16 mmol), K.2CO3 (45 mg, 0.33 mmol), Pd(PPli3)4 (23 mg, 0.032 mmol) and (3-(difluoromethoxy)phenyl)boronic acid (60 mg, 0.32 mmol) in 1 ,4-dioxane/H2O (8.0 mL, 4: 1) under N2 was heated to 100 °C and stirred for 4 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was diluted wi th water (20 mL) and extracted with DCM (3 x 10 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduce pressure. The resulting residue was purified by preparative TLC with an elution of 50% EtOAc/PE to provide l-((5-(3-(3-(difluoromethoxy)phenyl)-lH-pyrazol-l-yl)-7-morpholino-3H- imidazo[4,5-b]pyridin-3-yl)methyl)cyclopropanecarbonitrile (39 mg, 0.079 mmol) as a white solid. LC-MS (ESI+): m/z 492 (MH+). 'HNMR (300 MHz, CDCh) 8.50 (s, 1H), 7.97 (s, 1H), 7.87- 7.70 (m, 2H), 7.44 (t, J= 7.8 Hz, 1H), 7.34 (s, 1H), 7.12 (d, J= 7.8 Hz, 1H), 6.84 (s, 0.25H), 6.76 (s, 1H), 6.60 (s, 0.5H), 6.34 (s, 0.25H), 4.34 (s, 2H), 4.12-4.02 (m, 4H), 4.00- 3.92(m, 4H). 1.41 (s. 4H).
[0382] General procedure 27:
[0383] Synthesis of compound 63: 3-(2-methoxyethyl)-2-methyl-7-morpholino-5-[3-(m- tolyl)- 1-py razolyl] -3H- 1,3, 4- triazaindene
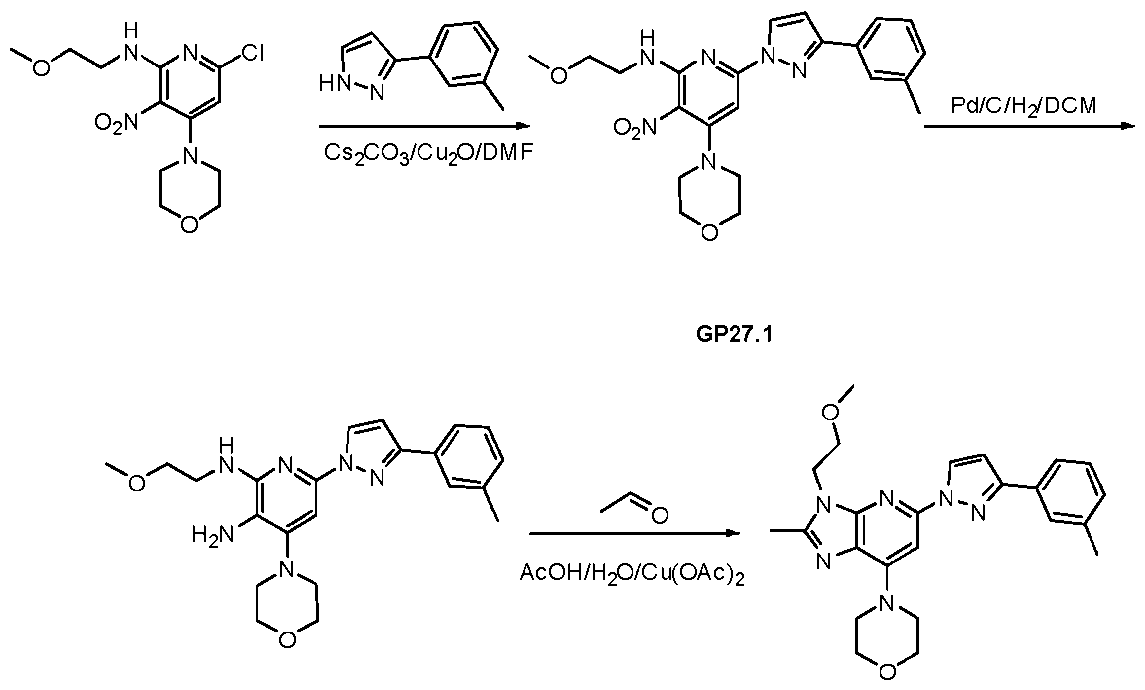
[0384] Synthesis ofN-(2-methoxyethyl)-4-morpholino-3-nitro-6-(3-(m-tolyl)-lH-pyrazol-l- yl)pyridin-2-amine, GP27. 1
[0385] To a solution of 6-chloro-N-(2-methoxyethyl)-4-morpholino-3-nitropyridin-2-amine (500 mg, 1.58 mmol) in DMF (15 mL) was added 3-(m-tolyl)-lH-pyrazole (274 mg, 1.74 mmol). Cu2O (22 mg, 0.158 mmol) and CS2CO3 (1.55 g, 4.74 mmol). The reaction mixture was heated to 1 10 °C under N2 and stirred for 2 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was quenched with water (50 mL) and extracted with EtOAc (3 x 20 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 5% EtOAc/PE to 33% EtOAc/PE to provide N-(2-methoxyethyl)-4-morpholino-3-nitro-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridin-2- amine (300 mg, 0.68 mmol) as a yellow solid. LC-MS (ESI+): m/z 439 (MEE).
[0386] Synthesis ofN2-(2-methoxyethyl)-4-morpholino-6-(3-(m-tolyl)-!H-pyrazol-l- yl) pyridine-2, 3-diamine, GP27.2
[0387] To a solution of N-(2,4-dimethoxybenzyl)-4-morpholino-3-nitro-6-(3-(m-tolyl)-lH- pyrazol-l-yl)pyridin-2-amine (300 mg, 0.68 mmol) in THF/MeOH (10 mL/10 mL) was added Pd/C (30 mg, 10%W/W). The reaction mixture was purged with H2 (gas) for three times. The reaction mixture was stirred at rt for 4 h under H2 using balloons. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was filtered and the filtrate was concentrated and the resulting residual was purified by silica gel column chromatography with a gradient elution of PE to 33% EtOAc/PE to provide N2-(2-
methoxyethyl)-4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridine-2,3-diamine (250 mg, 0.61 mmol) as an off-white solid. LC-MS (ESI+): m/z 409 (MH+).
[0388] Synthesis of 4-(3-(2-methoxyethyl)-2-methyl-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-b]pyridin-7-yl)morpholme, Compound 63
[0389] To a solution of N2-(2-methoxyethyl)-4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l- yl)pyridine-2,3-diamine (50 mg, 0.1 1 mmol) in AcOH/EhO (2: 1, 3.0 mL) at rt was added CU(OAC)2 (206 mg, 1.1 mmol) and acetaldehyde (6.0 mg, 0.14 mmol). The reaction mixture was heated to 55 °C and stirred for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with 1 M NaOH solution to pH = 9. The aqueous solution was extracted with EtOAc (3 x 20 mL). The combined organic phases were dried over anhydrous Na2SO4, filtrated and concentrated. The resulting residue was purified by preparative TLC with an elution of 50% EtOAc/PE to provide 4-(3-(2-methoxyethyl)-2-methyl- 5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (20 mg, 0.046 mmol) as a white solid. LC-MS (ESI+): m/z 433 (MH+). 1HNMR (300 MHz, CDCI3) 8.56 (d, J= 2.7 Hz, 1H), 7.78-7.71 (m, 2H), 7.35-7.31 (m, 2H), 7.16 (d, J= 7.5 Hz, 1H), 6.74 (d, J= 2.7 Hz, 1H), 4.37-4.33 (t, J= 4.8 Hz, 2H). 4.01-3.96 (m, 8H). 3.77-3.73 (m, 2H). 3.30 (s, 3H), 2.60 (s, 3H). 2.44 (s, 3H).
[0390] General procedure 28:
[0391] Synthesis of Compound 74: 8-morpholino-6-[3-(m-tolyl)-l-pyrazolyl]-3,4-dihydro- lH-2-oxa-4a,5,9-triazafluorene

[0393] To a solution of 4-(2.6-dichloro-3-nitropyridin-4-yl)morpholine (350 mg, 1.26 mmol) in 1,4-dioxane (14.0 mL) was added morpholin-3-one (140 mg. 1.40 mmol), Pd2(dba)3 (231 mg. 0.252 mmol), CS2CO3 (819 mg, 2.52 mmol) and t-Buxphos (133 mg, 0.32 mmol). The reaction mixture was stirred at 90 °C under N2 overnight. After completion of the reaction as indicated by
TLC analysis, the reaction mixture was quenched with water (50 mL) and extracted with EtOAc (3 x 20 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SC>4, filtered, and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 5% EtOAc/PE to 20% EtOAc/PE to provide 4-(6-chloro-4-morpholino-3-nitropyridin-2-yl)morpholin-3-one (65 mg, 0.19 mmol) as a yellow solid. LC-MS (ESI+): m/z 343/345 (MH ).
[0394] Synthesis of 4-(4-morpholino-3-nitro-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridin-2- yl)morpholin- 3-one, GP28.2
[0395] To a solution of 4-(6-chloro-4-morpholino-3-nitropyridin-2-yl)morpholin-3-one (65 mg, 0.19 mmol) in DMF (2.0 mL) was added 3-(m-tolyl)-lH-pyrazole (33 mg, 0.21 mmol), CS2CO3 (155 mg, 0.48 mmol) and Cui (3.0 mg, 0.016 mmol). The solution was stirred at 110 °C for 2 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was diluted with water (20 mL) and extracted with EtOAc (3 x 10 mL). The combined organic phases were dried over anhydrous Na2SO4, filtrated and concentrated. The resulting residue was purified by silica gel column chromatography with a gradient elution of 5% EtOAc/PE to 33% EtOAc/PE to provide 4-(4-morpholino-3-nitro-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridin-2- yl)morpholin-3-one (35 mg, 0.075 mmol) as a yellow solid. LC-MS (ESI+): m/z 465 (MH ). 1HNMR (300 MHz, CD3CI) 8.47 (s, 1H), 7.74-7.69 (m, 2H), 7.63 (s, 1H), 7.35 (t, J= 7.5 Hz, 1H), 7.21 (d, J= 7.2 Hz, 1H), 6.79 (d, J= 2.7 Hz, 1H), 4.31 (s, 2H), 4.09-4.06 (m, 2H), 3.95- 3.93 (m, 2H), 3.87-3.83 (m, 4H), 3.32-3.29 (m, 4H), 2.44 (s, 3H).
[0396] Synthesis of 4-morpholino-2-( 3-(m-tolyl)-lH-pyrazol-l-yl)-8, 9-dihydro-6H- pyrido[3',2':4,5]imidazo[2, l-c][1.4]oxazine, Compound 74
[0397] To a solution of 4-(4-morpholino-3-nitro-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridin-2- yl)morpholin-3-one (35 mg, 0.075 mmol) in AcOH (1.0 mL) was added Fe powder (43 mg, 0.77 mmol). The mixture was stirred at rt for 2 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was quenched with aqueous NaHCOs solution to pH ~ 9. The mixture was extracted with EtOAc (3 x 10 mL). The combined organic phases was dried over anhydrous Na2SO4, filtrated and concentrated. The resulting residue was purified by preparative TLC with an elution of 50% EtOAc/PE to provide 4-morpholino-2-(3-(m-tolyl)-lH-pyrazol-l- yl)-8.9-dihydro-6H-pyrido[3',2':4,5]imidazo[2,l-c][l,4]oxazine (11.2 mg, 0.027 mmol) as a green solid. LC-MS (ESI+): m/z 417 (MH+). 'H NMR (300 MHz, CDCI3) £8.57 (s, 1H), 7.78- 7.68 (m, 2H), 7.35-7.28 (m, 2H), 7.17 (d, J= 7.2 Hz, 1H), 6.75 (d, J= 2.1 Hz, 1H), 4.98 (s, 2H), 4.24-4.18 (m, 4H), 3.97-3.90 (m, 8H), 2.44 (s, 3H).
[0398] General procedure 29:
[0399] Synthesis of Compound 115: 5-ethyl-l-[3-(2-methoxyethyl)-7-morpholino-3H- 1 ,3,4-triazainden-5-yl] -3-(m- tolyl)- 1H- 1 ,2,4-triazole
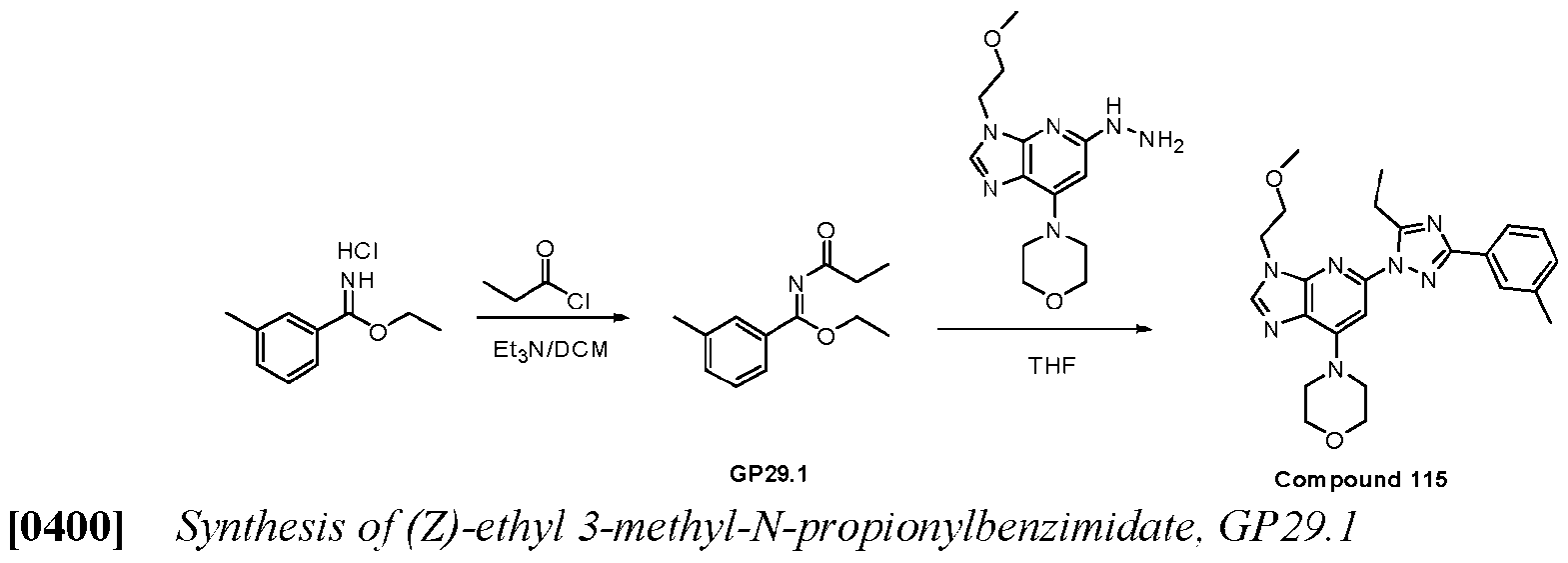
[0401] To a solution of ethyl 3-methylbenzimidate hydrochloride (100 mg. 0.50 mmol) in DCM (5.0 mL) at 0 °C was added EtaN (167 mg, 1.60 mmol) and a solution of propionyl chloride (93 mg, 1.00 mmol) in DCM (0.5 mL) dropwise. After completion of the reaction as indicated by TLC analysis, the reaction mixture was quenched with KI ISO-i solution (10%, 10 mL) and extracted with DCM (3 x 10 mL). The combined organic phases were dried over anhydrous Na2SO4, filtered, and concentrated under reduce pressure. The resulting residue was used directly for the next step without further purification.
[0402] Synthesis of 4-(5-(5-ethyl-3-(m-tolyl)-lH-l, 2, 4-triazol-l-yl)-3-(2-methoxyethyl)-3H- imidazo[4,5-b]pyridin-7-yl)morpholine, Compound 115
[0403] To a solution of 4-(5-hydrazinyl-3-(2-methoxyethyl)-3H-imidazo[4,5-b]pyridin-7- yl)morpholine (200 mg, 0.68 mmol) in anhydrous THF (5.0 mL) at 0 °C under N2 atmosphere was added EtaN (79 mg, 0.76 mmol and crude (Z)-ethyl 3-methyl-N-propionylbenzimidate (85 mg, 0.38 mmol). The mixture was stirred at rt overnight. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with water (10 mL) and extracted with EtOAc (3 x 20 mL). The combined organic phases were dried over anhydrous Na2SC>4, filtered, and concentrated under reduce pressure. The resulting residue was purified by preparative TLC with an elution of 50% EtOAc/PE to provide 4-(5-(5-ethyl-3-(m- tolyl)-lH-l,2,4-triazol-l-yl)-3-(2-methoxyethyl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (22 mg, 0.049 mmol) as a white solid. LC-MS (ES1+): m/z 448 (MH+). 1HNMR (300 MHz, CD3OD) 8.12 (s, 1H), 7.94 (s, 1H), 7.88 (d, J= 7.5 Hz, 1H), 7.35 (t, .7 =7.5 Hz, 1H), 7.26 (d, J
= 6.9 Hz, 1H), 7.10 (s, 1H), 4.44 (t, J= 5.1 Hz, 2H), 3.96-3.92 (m, 4H), 3.91- 3.89 (m, 4H), 3.78 (t, J= 5.1 Hz, 2H). 3.33 (s, 3H), 3.27-3.25 (m, 2H), 2.42 (s. 3H), 1.41 (t, J = 7.5Hz, 3H).
[0404] General procedure 30:
[0405] Synthesis of Compound 116: {l-[3-(2-methoxyethyl)-7-morpholino-3H-l,3,4- triazainden-5-yl] -3-(m-tolyl)- 1H- 1,2,4- triazol-5-yl}methanol
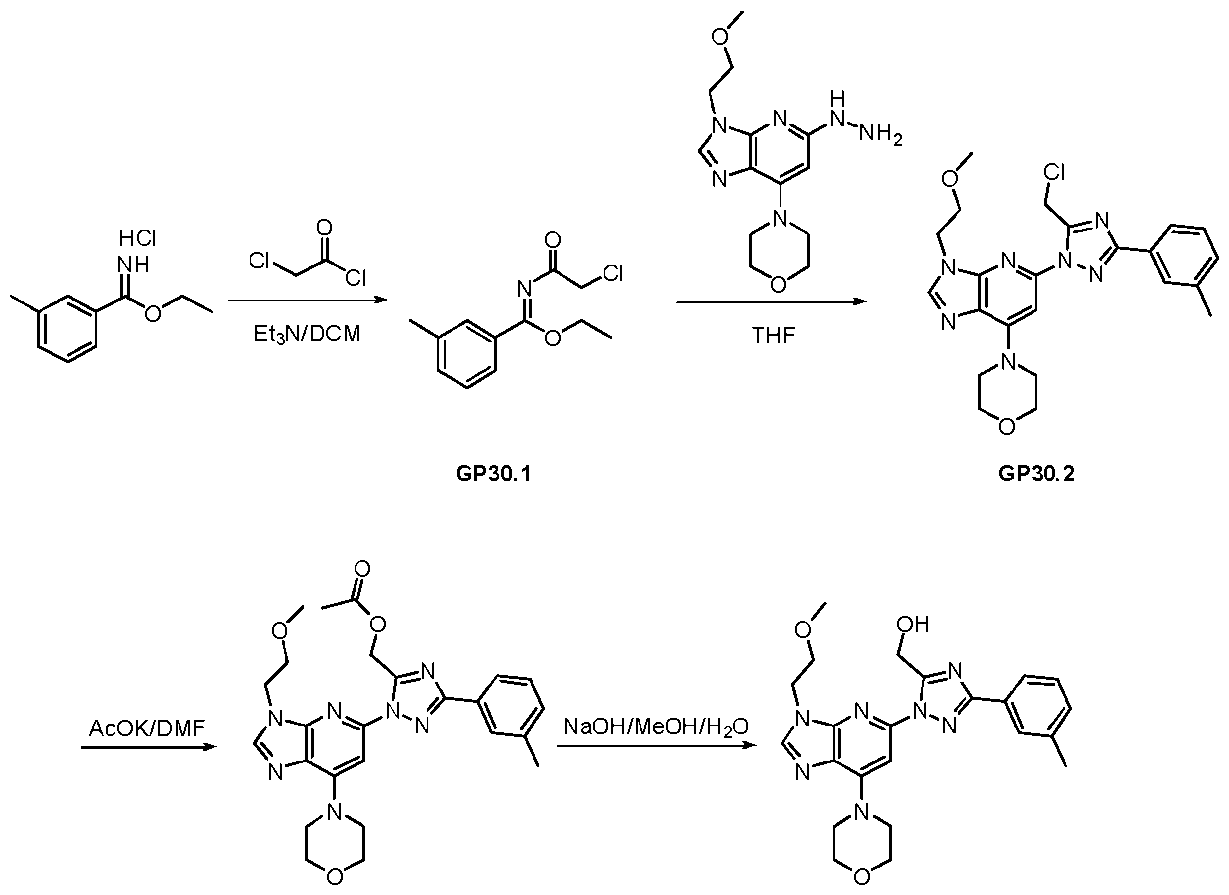
[0406] Synthesis of (Z)-ethyl N-(2-chloroacetyl)-3-methylbenzimidate, GP30. 1
[0407] To a solution of ethyl 3-methylbenzimidate hydrochloride (250 mg. 1.26 mmol) in DCM (5.0 mL) at 0 °C was added EtsN (406 mg, 4.0 mmol) and a solution of 2-chloroacetyl chloride (285 mg, 2.00 mmol) in DCM (1 mL) dropwise. After completion of the reaction as indicated by TLC analysis, the reaction mixture was quenched with KHSCh solution (10%, 10 mL) and extracted with DCM (3 x 10 mL). The combined organic phases were dried over anhydrous Na2SO4, filtered, and concentrated under reduce pressure. The resulting residue was used directly for the next step without further purification.
[0408] Synthesis of 4-(5-(5-(chloromethyl)-3-(m-tolyl)-lH-l, 2, 4-triazol-l-yl)-3-(2- methoxyethyl)-3H-imidazo[4, 5-b ]pyridin- 7-yl)morpholine, GP30.2
[0409] To a solution of 4-(5-hydrazinyl-3-(2-methoxyethyl)-3H-imidazo[4,5-b]pyridin-7- yl)morpholine (500 mg, 1.70 mmol) in anhydrous THF (15.0 mL) at 0 °C under N2 atmosphere was added Et?N (192 mg, 1.89 mmol and crude (Z)-ethyl 3-methyl-N-propionylbenzimidate (227 mg, 0.95 mmol). The mixture w as stirred at rt overnight. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with water (10 mL) and extracted with EtOAc (3 x 20 mL). The combined organic phases were dried over anhydrous Na2SC>4, filtered, and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 50% EtOAc/PE to EtOAc to provide 4-(5-(5-(chloromethyl)-3-(m-tolyl)-lH-l,2.4-triazol-l-yl)-3-(2- methoxyethyl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (200 mg, 0.43 mmol) as a white solid. LC-MS (ESI+): m/z 468/470 (MH+).
[0410] Synthesis of (l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-h]pyridin-5-yl)-3- (m-tolyl)-lH-l,2,4-triazol-5-yl)methyl acetate. GP30.3
[0411] To a solution of 4-(5-(5-(chloromethyl)-3-(m-tolyl)-lH-l,2,4-triazol-l-yl)-3-(2- methoxyethyl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (30 mg, 0.063 mmol) in DMF (2.0 mL) was added AcOK (8.0 mg, 0.077 mmol). The mixture was stirred at rt for 3 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with water (10 mL) and extracted with EtOAc (3 x 5 mL). The combined organic phases were dried over anhydrous Na2SC>4, fdtered and concentrated under reduce pressure to provide (l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-3-(m-tolyl)-lH- l,2,4-triazol-5-yl)methyl acetate (29 mg, 0.06 mmol) as a yellow oil. LC-MS (ESI+): m/z 492 (MH+).
[0412] Synthesis of(l-(3-(2-methoxyethyl)-7-morpholino-3H-imidcizo[4,5-b]pyridin-5-yl)-3- (m-tolyl)-lH-l,2,4-triazol-5-yl)methanol< Compound 116
[0413] To a solution of (l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5- yl)-3-(m-tolyl)-lH-l,2,4-triazol-5-yl)methyl acetate (29 mg, 0.06 mmol) in MeOH (5 mL) was added aqueous NaOH (2 mL, 6 M). The mixture was stirred at rt for 2h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with water (10 mL) and extracted with EtOAc (3 x 20 mL). The combined organic phases were dried over anhydrous Na2SO4, filtered, and concentrated under reduce pressure. The resulting residue was purified by preparative TLC with an elution of EtOAc to provide (l-(3-(2-methoxyethyl)-7- morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-3-(m-tolyl)-lH-l,2,4-triazol-5-yl)methanol (17 mg, 0.038 mmol) as a white solid. LC-MS (ESI+): m/z 450 (MH+). 1HNMR (300 MHz, CDCls) 5 8.02-7.99 (m, 2H), 7.91 (s. 1H), 7.36 (t, J= 7.5 Hz, 1H), 7.26-7.22 (m, 2H), 5.74 (t, J= 7.2 Hz, 1H), 5.02 (d. J = 7.2 Hz. 2H), 4.37 (t, J = 4.8Hz, 2H). 4. 12-4.09 (m, 4H). 3.96-3.92 (m, 4H). 3.73 (t, J = 4.8 Hz, 2H), 3.33 (s, 3H), 2.44 (s, 3H).
[0414] General procedure 31:
[0415] Synthesis of Compound 117: l-[3-(2-methoxyethyl)-7-morpholino-3H- 1,3,4- triazainden-5-yl]-5-(methoxymethyl)-3-(m-tolyl)-lH-l,2,4-triazole

[0416] Synthesis of 4-(3-(2-methoxyethyl)-5-(5-(methoxymethyl)-3-(m-tolyl)-lH-l,2,4-triazol- l-yl)-3H-imidazo[ 4, 5-b ]pyridin- 7-yl)morpholine, Compound 117
[0417] To a solution of 4-(5-(5-(chloromethyl)-3-(m-tolyl)-lH-l,2,4-triazol-l-yl)-3-(2- methoxyethyl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (40 mg, 0.086 mmol) in THF (1 mL) under N2 was added a solution of CHsONa (0.1 mL, 30% in MeOH). The mixture was stirred at rt overnight. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with water (10 mL) and extracted with EtOAc (3 x 20 mL). The combined organic phases were dried over anhydrous Na2SO4, filtered, and concentrated under reduce pressure. The resulting residue was purified by preparative TLC with an elution of 50% EtOAc/PE to provide 4-(3-(2-methoxyethyl)-5-(5-(methoxymethyl)-3-(m-tolyl)-lH-l,2,4- triazol-l-yl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (28 mg, 0.06 mmol) as a white solid. LC-MS (ESI+): m/z 464 (MH+). 'HNMR (300 MHz, CDCh) 8.06-8.03 (m, 2H), 7.94 (s, 1H), 7.35 (t, J= 7.5 Hz, 1H), 7.24-7.22 (m, 1H), 7.12 (s, 1H), 5.14(s, 2H), 4.39 (t, J= 4.8 Hz, 2H), 4.05-4.03 (m, 4H), 3.95-3.93(m, 4H), 3.75 (t, J= 4.8 Hz, 2H), 3.54 (s, 3H). 3.35 (s, 3H), 2.43 (s. 3H).
[0418] General procedure 32:
[0419] Synthesis of Compound 118: ({l-[3-(2-methoxyethyl)-7-inorpholino-3H-l,3,4- triazainden-5-yl]-3-(m-tolyl)-lH-l,2,4-triazol-5-yl}methyl)amine
 methoxyethyl)-3H-imidazo[4, 5-b ]pyridin- 7-yl)morpholine, GP32.1
methoxyethyl)-3H-imidazo[4, 5-b ]pyridin- 7-yl)morpholine, GP32.1
[0421] To a solution of 4-(5-(5-(chloromethyl)-3-(m-tolyl)-lH-L2.4-triazol-l-yl)-3-(2- methoxyethyl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (70 mg, 0.15 mmol) in DMF (3 mL) was added NaNs (30 mg, 0.45 mmol). The mixture was stirred at rt overnight. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with water (10 mL) and extracted with EtOAc (3 x 10 mL). The combined organic phases were dried over anhydrous Na2SO4, filtrated. The filtrate was used directly for the next step without further concentration. LC-MS (ESI+): m/z 475 (MH+).
[0422] Synthesis of tert-butyl ((l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5- b ]pyridin-5-yl)-3-(m-tolyl)-lH-l, 2, 4-triazol-5-yl)methyl)carbamate, GP30.2
[0423] To a solution of 4-(5-(5-(azidomethyl)-3-(m-tolyl)-lH-l,2,4-triazol-l-yl)-3-(2- methoxyethyl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (70 mg, 0.15 mmol) and Boc2O (0.5 mL) in EtOAc/EtOH (10 mL/5 mL) was added Pd/C (20 mg). The reaction mixture was purged with H2 for three times and stirred at rt overnight under H2 using balloons. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was filtered directly, and the filtrate was concentrated. The resulting residue was purified by silica gel column chromatography with a gradient elution of 15% EtOAc/PE to 50% EtOAc/PE to provide tertbutyl ((l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-3-(m-tolyl)-lH-
1.2.4-triazol-5-yl)methyl)carbamate (54 mg, 0.098 mol) as a white solid. LC-MS (ESI+): m/z 549 (MH ).
[0424] Synthesis of (l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-3- (m-tolyl)- 1H- 1.2.4-triazol-5-yl)methanamine hydrochloride, Compound 118
[0425] To a solution of tert-butyl ((l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5- b]pyridin-5-yl)-3-(m-tolyl)-lH-l,2,4-triazol-5-yl)methyl)carbamate (54 mg, 0.098 mol) in MeOH (4 mL) at rt was added a solution of HCl/Et2O (4.0 mL). The solution mixture was stirred at rt for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was concentrated directly and the resulting residue was purified by slurry in Et2O to provide (l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-3-(m- tolyl)-lH-l,2,4-triazol-5-yl)methanamine hydrochloride (38 mg, 0.079 mmol) as a yellow solid. LC-MS (ESI+): m/z 449 (MH1). 1HNMR (300 MHz, CD3OD) 8.60 (s, 1H), 8.06 (s, 1H). 8.00 (d, J= 8.1 Hz, 1H), 7.45 (s, 1H), 7.38-7.30 (m, 2H), 4.95 (s, 2H), 4.57 (t, J = 4.5 Hz, 2H), 3.92 (s, 8H), 3.84 (t, J= 4.5 Hz, 2H), 3.35 (s, 3H), 2.44(s, 3H).
[0426] General procedure 33:
[0427] Synthesis of Compound 119: N-methyl({l-[3-(2-methoxyethyl)-7-morpholino-3H-
1.3.4-triazainden-5-yl]-3-(m-tolyl)-lH-l,2,4-triazol-5-yl}methyI)amine
 from GP30 Compound 119
from GP30 Compound 119
[0428] Synthesis of l-(l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-3-
(m-tolyl)-lH-l.2.4-triazol-5-yl)-N-methylmethanamine, Compound 119
[0429] To a solution of 4-(5-(5-(chloromethyl)-3-(m-tolyl)-lH-l,2,4-triazol-l-yl)-3-(2- methoxyethyl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (34 mg, 0.073 mmol) in DMF (3 mL) was added methylamine hydrochloride (11 mg, 0. 146 mmol) and CS2CO3 (72 mg, 0.219 mmol). The reaction was stirred at rt overnight. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was diluted with water (20 mL). The aqueous solution was extracted with EtOAc (3 x 10 mL). The combined organic phases were washed with saturated aqueous NaCl solution (2 x 20 mL), dried over anhydrous Na2SO4, filtrated and concentrated. The resulting residue was purified by preparative TLC with an elution of EtOAc to provide l-(l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-3-(m-tolyl)- lH-L2,4-triazol-5-yl)-N-methylmethanamine (14 mg, 0.03 mmol) as an off-white solid. LC-MS (ESI+): m/z 463 (MH+). 'HNMR (300 MHz, CDCls) 8.02-8.00 (m, 2H), 7.93(s, 1H), 7.36 (t, J = 7.8 Hz. 1H), 7.26-7.25 (m. 1H), 7.19 (s, 1H), 4.39 (s, 4H), 4.06-4.02 (m, 4H), 3.95- 3.90 (m, 4H), 3.75 (t, J= 4.8 Hz, 2H), 3.33 (s, 3H). 2.53 (s, 3H). 2.44 (s. 3H).
[0430] General procedure 34:
[0431] Synthesis of Compound 120: N,N-dimethyl({l-[3-(2-methoxyethyl)-7-morpholino- 3H-l,3?4-triazainden-5-yl]-3-(m-tolyl)-lH-l,2,4-triazol-5-yl}methyl)amine
 from GP30 Compound 120
from GP30 Compound 120
[0432] Synthesis of l-(l-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-3- (m-tolyl)-lH-l, 2, 4-triazol-5-yl)-N,N-dimethylmethanamine, Compound 120
[0433] To a solution of 4-(5-(5-(chloromethyl)-3-(m-tolyl)-lH-1.2,4-triazol-l-yl)-3-(2- methoxyethyl)-3H-imidazo[4.5-b]pyridin-7-yl)morpholine (80 mg. 0.172 mmol) in DMF (3 mL) was added dimethylamine hydrochloride (29 mg, 0.34 mmol) and CS2CO3 (169 mg, 0.51 mmol). The reaction was stirred at rt overnight. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was diluted with water (20 mL). The aqueous solution was extracted with EtOAc (3 x 10 mL). The combined organic phases were washed with saturated aqueous NaCl solution (2 x 20 mL), dried over anhydrous NazSCL, filtrated and concentrated. The resulting residue was purified by silica gel column chromatography with a gradient elution of DCM to 3% MeOH/DCM to provide l-(l-(3-(2-methoxyethyl)-7- morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-3-(m-tolyl)-lH-l,2,4-triazol-5-yl)-N,N- dimethylmethanamine (60 mg, 0.13 mmol) as a yellow solid. LC-MS (ESI+): m/z 477 (MH+).
1HNMR (300 MHz, CDCh) £8.06-8.01 (m, 2H), 7.95 (s, 1H), 7.34 (t, J= 7.5 Hz, 1H), 7.24- 7.21 (m, 1H), 7.09 (s, 1H), 4.40 (t, J = 4.8 Hz, 2H), 4.19 (s, 2H), 4.04-4.00 (m, 4H), 3.94-3.90 (m, 4H), 3.74 (t, J= 4.8 Hz, 2H), 3.35 (s, 3H), 2.43 (s, 3H), 2.39 (s. 6H).
[0434] General procedure 35:
[0435] Synthesis of Compound 122: l-[3-(2-methoxyethyI)-7-morpholino-3H-l,3,4- triazainden-5-yl] -3-(m-tolyl)- 1H- 1,2,4-triazole

GP35.1 Compound 122
[0436] Synthesis ofN'-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4, 5-b ]pyridin-5-yl)-3- methylbenzimidohydrazide, GP35. 1
[0437] To a solution of ethyl 3-methylbenzimidate hydrochloride (50 mg, 0.25 mmol) in MeOH (10 mL) at rt was added EtsN (76 mg, 0.75 mmol) and 4-(5-hydrazinyl-3-(2- methoxyethyl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (87.6 mg. 0.30 mmol). The reaction mixture was stirred at rt overnight. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with water (10 mL) and extracted with EtOAc (3 x 10 mL). The combined organic phases were dried over anhydrous Na2SO4, filtrated and concentrated under reduce pressure to provide the crude product which was used directly for the next step without further purification. LC-MS (ESI+): m/z 410 (MH+).
[0438] Synthesis of 4-(3-(2-methoxyethyl)-5-(3-(m-tolyl)-lH-l, 2, 4-triazol-l -yl)-3H- imidazo[4,5-b]pyridin-7-yl)morpholine, Compound 122
[0439] A solution of crude N'-(3-(2-methoxyethyl)-7-morpholino-3H-imidazo[4.5-b]pyridin- 5-yl)-3-methylbenzimidohydrazide (30 mg, 0.07 mmol) in HCOOH (5.0 mL) was heated to reflux and stirred for 2 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was concentrated directly and the resulting residue was purified by silica gel column chromatography with an elution of 5% MeOH/DCM to provide 4-(4-morpholino-3- nitro-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyri din-2 -yl)morpholin-3-one (8.6 mg, 0.02 mmol) as an off white solid. LC-MS (ESI+): m/z 420 (MH+). 'H NMR (300 MHz, CDCk) £9.15 (s, 1H), 8.11-7.98 (m, 3H), 7.38 (t, J= 7.5 Hz, 1H), 7.37-7.35 (m, 1H), 7.22 (s, 1H), 4.43 (t, J = 4.8 Hz, 2H), 4.11-4.01 (m, 4H), 3.99-3.89 (m, 4H), 3.77 (t, J= 4.8 Hz, 2H), 3.37 (s, 3H). 2.46 (s, 3H).
[0440] General procedure 36:
[0441] Synthesis of Compound 123: N-methyl(2-{5-[3-(4-fluoro-3-tolyl)-l-pyrazolyl]-7- morpholino-3H-l,3,4-triazainden-3-yl}ethyl)amine
 [0443] To a solution of tert-butyl (2-hydroxyethyl)(methyl)carbamate (10 g, 57.1 mmol) in DCM (100 mL) at rt was added ELN (8.1 g, 80.2 mmol) and a solution of MsCl (7.2 g, 62.8 mmol) in DCM (10 mL) drop wise. The mixture was stirred at rt for 3 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was quenched with water (50 mL) and extracted with DCM (3 x 100 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduce pressure. The resulting residue was used for the next step without further purification.
[0443] To a solution of tert-butyl (2-hydroxyethyl)(methyl)carbamate (10 g, 57.1 mmol) in DCM (100 mL) at rt was added ELN (8.1 g, 80.2 mmol) and a solution of MsCl (7.2 g, 62.8 mmol) in DCM (10 mL) drop wise. The mixture was stirred at rt for 3 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was quenched with water (50 mL) and extracted with DCM (3 x 100 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduce pressure. The resulting residue was used for the next step without further purification.
[0444] Synthesis of tert-butyl (2-(5, 7-dichloro-3H-imidazo[4,5-b]pyridin-3- yl)ethyl)(methyl)carbamate, GP36.2
[0445] To a solution of 5,7-dichloro-3H-imidazo[4,5-b]pyridine (5.0 g, 26.6 mmol) in DMF (100 mL) at rt was added K2CO3 (8.4 g, 61.2 mmol) and 2-((tert-butoxycarbonyl)(methyl) amino)ethyl methanesulfonate (13.5 g, 53.2 mmol). The reaction mixture was stirred at rt overnight. After completion of the reaction as indicated by TLC analysis, the reaction mixture
was quenched with water (100 mL) and extracted with EtOAc (3 x 100 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SC>4, filtered and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 20% EtOAc/PE to 50% EtOAc to provide tert-butyl (2-(5,7-dichloro- 3H-imidazo[4.5-b]pyridin-3-yl)ethyl)(methyl) carbamate (2.0 g, 5.8 mmol) as a yellow solid. LC-MS (ESI+): m/z 345/347 (MH ). *H NMR (300 MHz, CDCh) 8.00 (s, 1H), 7.33 (s, 1H), 4.53-4.43 (m, 2H), 3.69-3.65 (m, 2H), 2.85 (s, 1.5H), 2.74 (s, 1.5H), 1.39 (s, 4.5H), 1.20 (s, 4.5H).
[0446] Synthesis of tert-butyl (2-(5-chloro-7-morpholino-3H-imidazo[4,5-bJpyridin-3- yl)ethyl)(methyl)carbamate, GP36.3
[0447] To a solution of tert-buty l (2-(5,7-dichloro-3H-imidazo[4,5-b]pyridin-3- yl)ethyl)(methyl)carbamate (2.0 g. 5.80 mmol) in DMF (100 mL) at rt was added K2CO3 (1.6 g, 11.6 mmol) and morpholine (756 mg, 8.70 mmol). The reaction mixture was stirred at 110 °C overnight. After completion of the reaction as indicated by TLC analysis, the reaction mixture was diluted with water (100 mL) and extracted with EtOAc (3 x 100 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 20% EtOAc/PE to 50% EtOAc to provide tert-butyl (2-(5-chloro-7- morpholino-3H-imidazo[4,5-b]pyridin-3-yl)ethyl)(methyl)carbamate (1.7 g, 4.3 mmol) as a white solid. LC-MS (ESI+): m/z 396/398 (MH+). 'I I NMR (300 MHz, CDCh) £7.70-7.64 (m, 1H), 6.43 (s, 1H), 4.50-4.30 (m, 2H), 3.90 (s, 8H), 3.78-3.64 (m, 2H), 2.80 (s, 1.5H). 2.62 (s, 1.5H), 1.41 (s, 4.5H), 1.26 (s, 4.5H).
[0448] Synthesis of 3-(4-fluoro-3-methylphenyl)-l-(tetrahydro-2H-pyran-2-yl)-lH-pyrazole,
GP36.4
[0449] A suspension of 3-bromo-l-(tetrahydro-2H-pyran-2-yl)-lH-pyrazole (200 mg, 0.86 mmol), (4-fluoro-3-methylphenyl)boronic acid (198 mg, 1.30 mmol), Na2COs (275 mg, 2.60 mmol) and Pd(PPh?)4 (25 mg, 0.022 mmol) in 1 ,4-dioxane/H2O (10: 1, 11 mL) under N2 was heated to reflux and stirred for 2 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was diluted with water (30.0 mL) and extracted with EtOAc (3 x 20 mL). The combined organic phases were dried over anhydrous Na2SO4. filtered, and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 5% EtOAc/PE to 20% EtOAc/PE to provide 3-(4- fluoro-3-methylphenyl)-l-(tetrahydro-2H-pyran-2-yl)-lH-pyrazole (150 mg, 0.58 mmol). LC- MS (ESI+): m/z 261 (MH ).
[0450] Synthesis of 3-(4-fluoro-3-methylphenyl)-lH-pyrazole hydrochloride, GP36.5
[0451] To a solution of 3-(4-fluoro-3-methylphenyl)-l-(tetrahydro-2H-pyran-2-yl)-lH- pyrazole (150 mg, 0.58 mmol) in DCM (5.0 mL) was added a solution of Et2O/HCl (2.0 rnL). The reaction mixture was stirred at rt for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was concentrated under reduce pressure. The resulting residue was purified by slurry in Et20 to provide 3-(4-fluoro-3-methylphenyl)-lH- pyrazole hydrochloride (100 mg, 0.47 mmol) as ayellow solid. LC-MS (ESI+): m/z 177 (MH ). [0452] Synthesis of tert-butyl (2-(5-(3-(4-fluoro-3-methylphenyl)-lH-pyrazol-l-yl)-7- morpholino-3H-imidazo[4.5-b]pyridin-3-yl)ethyl)(methyl)carbamate, GP36.6
[0453] A suspension of tert-butyl (2-(5-chloro-7-morpholino-3H-imidazo[4,5-b]pyridin-3- yl)ethyl)(methyl)carbamate (80 mg, 0.20 mmol), t-BuXphos (43 mg, 0. 10 mmol), t-BuONa (49 mg, 0.51 mmol), Pd2(dba)s (93 mg, 0.10 mmol) and 3-(4-fluoro-3-methylphenyl)-lH-pyrazole hydrochloride (51 mg, 0.24 mmol) in xylene (5 mL) under N2 was heated to 130 °C and stirred for 2 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was concentrated under reduce pressure directly. The resulting residue was purified by silica gel column chromatography with a gradient elution of 5% EtOAc/PE to 33% EtOAc/PE to provide tert-butyl (2-(5-(3-(4-fluoro-3-methylphenyl)-lH-pyrazol-l-yl)-7-morpholino-3H-imidazo[4,5- b]pyridin-3-yl)ethyl)(methyl)carbamate (110 mg, 0.21 mmol) as a colorless oil. LC-MS (ESI+): m/z 536 (MH1).
[0454] Synthesis of2-(5-(3-(4fluoro-3-methylphenyl)-lH-pyrazol-l-yl)-7-morpholino-3H- imidazo[4,5-b]pyridin-3-yl)-N-methylethanamine hydrochloride, Compound 123
[0455] To a solution of tert-butyl (2-(5-(3-(4-fluoro-3-methylphenyl)-lH-pyrazol-l-yl)-7- morpholino-3H-imidazo[4,5-b]pyridin-3-yl)ethyl)(methyl)carbamate (110 mg, 0.21 mmol) in DCM (4.0 mL) was added a solution of EtzO/HCl (1 mL). The mixture was stirred at rt for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was concentrated directly and the resulting residue was purified by slurry in Et20 to provide 2- (5-(3-(4-fluoro-3-methylphenyl)-lH-pyrazol-l-yl)-7-morpholino-3H-imidazo[4,5-b]pyri din-3- yl)-N-methylethanamine hydrochloride (65.8 mg. 0.14 mmol). LC-MS (ESI+): m/z 436 (MH+). 1H NMR (300 MHz, DMSO-cL) 8.83 (d, J= 2.1 Hz, 1H), 8.25 (s, 1H), 7.91-7.81 (m, 2H), 7.25-7.17 (m, 2H), 7.04 (d, J= 2.1 Hz, 1H), 4.70-4.65 (m, 2H), 4.00-4.95 (m, 4H), 3.85-3.80 (m, 4H), 3.50-3.45 (m, 2H), 2.62 (s, 3H), 2.33 (s, 3H).
[0456] General procedure 37:
[0457] Synthesis of Compound 124: N-methyl{2-[5-(l'-methyl-3,3'-bipyrazolyl-l-yl)-7- morpholino-3H- 1 ,3,4- triazainden-3-y 11 ethyl } amine
 [0459] A suspension of 3-bromo-l-(tetrahydro-2H-pyran-2-yl)-lH-pyrazole (300 mg, 1.29 mmol), l-methyl-3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (402 mg, 1.94 mmol), K2CO3 (534 mg, 3.87 mmol) and Pd(dppf)C12(94 mg, 0.0129 mmol) in l,4-dioxane/H2O (10: 1, 11 mL) under N2 was heated to 90 °C and stirred for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was diluted with water (20.0 mL) and extracted with EtOAc (3 x 20 mL). The combined organic phases were dried over anhydrous Na2SC>4, filtered, and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 1% MeOH/DCM to 5% MeOH/DCM to provide 1 -methyl- T-(tetrahydro-2H-pyran-2-yl)-lH,rH-3,3'-bipyrazole (280 mg, 1.22 mmol) as a brown oil. LC-MS (ESI+): m/z 233 (MH+).
[0459] A suspension of 3-bromo-l-(tetrahydro-2H-pyran-2-yl)-lH-pyrazole (300 mg, 1.29 mmol), l-methyl-3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (402 mg, 1.94 mmol), K2CO3 (534 mg, 3.87 mmol) and Pd(dppf)C12(94 mg, 0.0129 mmol) in l,4-dioxane/H2O (10: 1, 11 mL) under N2 was heated to 90 °C and stirred for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was diluted with water (20.0 mL) and extracted with EtOAc (3 x 20 mL). The combined organic phases were dried over anhydrous Na2SC>4, filtered, and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 1% MeOH/DCM to 5% MeOH/DCM to provide 1 -methyl- T-(tetrahydro-2H-pyran-2-yl)-lH,rH-3,3'-bipyrazole (280 mg, 1.22 mmol) as a brown oil. LC-MS (ESI+): m/z 233 (MH+).
[0460] Synthesis of 1 -methyl- ! H. I'H-S.S'-bipyrazole. GP37.2
[0461] To a solution of l-methyl-T-(tetrahydro-2H-pyran-2-yl)-lH,l'H-3,3'-bipyrazole (280 mg, 1.22 mmol) in DCM (5.0 mL) was added a solution of EtzO/HCl (2.0 mL). The reaction mixture was stirred at rt for 2 h. After completion of the reaction as indicated by TLC and LC- MS analysis, the reaction mixture was quenched with aqueous Na2COs solution to pH ~ 9. The mixture was extracted with EtOAc (3 x 20 mL). The combined organic phases were dried over anhydrous Na2SO4, filtered, and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 1% MeOH/DCM to 3% MeOH/DCM to provide l-methyl-lH,l'H-3,3'-bipyrazole (70 mg, 0.47 mmol) as a blue solid. LC-MS (ESI+): m/z 149 (MH+). 'HNMR (300 MHz, CDCh) 7.60 (s, 1H), 7.38 (s, 1H), 6.59 (s, 1H). 6.54 (s, 1H). 3.95 (s. 3H).
[0462] Synthesis of tert-butyl methyl(2-(5-( 1 '-methyl-lH, 1 'H-[ 3.3'-bipyrazol ]-l-yl)-7- morpholino-3H-imidazo[4, 5-b ]pyridin-3-yl)ethyl)carbamate, GP37.3
[0463] A suspension of tert-butyl (2-(5-chloro-7-morpholino-3H-imidazo[4,5-b]pyridin-3- yl)ethyl)(methyl)carbamate (50 mg, 0.13 mmol), t-BuXphos (43 mg, 0.06 mmol), t-BuONa (38 mg, 0.39 mmol), Pd2(dba)3 (55 mg, 0.06 mmol) and 1 -methyl- lH,rH-3,3'-bipyrazole (23 mg, 0. 16 mmol) in xylene (5 mL) under N2 was heated to 130 °C under microwave for 30 min. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was diluted with water (10 mL) and extracted with EtOAc (3 x 20 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SC>4, filtered, and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of DCM to 2% MeOH/DCM to provide tert-butyl methyl(2-(5-(l'-methyl- lH,TH-[3,3'-bipyrazol]-l-yl)-7-morpholino-3H-imidazo[4,5-b]pyridin-3-yl)ethyl)carbamate (43 mg, 0.085 mmol) as a colorless oil. LC-MS (ESI+): m/z 508 (MEI+).
[0464] Synthesis ofN-methyl-2-(5-( 1 '-methyl-lH, 1 'H-[3, 3 '-bipyrazol ]-l-yl)-7-morpholino-3H- imidazo[4,5-b]pyridin-3-yl)ethanamine hydrochloride, Compound 124
[0465] To a solution of tert-butyl methyl(2-(5-(T-methyl-lH,l'H-[3,3'-bipyrazol]-l-yl)-7- morpholino-3EI-imidazo[4,5-b]pyridin-3-yl)ethyl)carbamate (43 mg, 0.085 mmol) in DCM (4.0 mL) was added a solution of Et2O/HCl (1 mL). The mixture was stirred at rt for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was concentrated directly and the resulting residue was purified by slurry in Et20 to provide N- methyl-2-(5-(T-methyl-lH,TH-[3,3,-bipyrazol]-l-yl)-7-morpholino-3H-imidazo[4,5-b]pyridin- 3-yl)ethanamine hydrochloride (36.7 mg, 0.083 mmol) as a white solid. LC-MS (ESI+): m/z 408 (MH+). ’HNMR (300 MHz, DMSO-tL + D2O) <78.73 (d, J= 2.4Hz, 1H), 8.21 (s. 1H), 7.77 (s, 1H), 7.20 (s, 1H), 6.85(d, J = 2.4 Hz, 1H). 6.71 (d, J = 1.8 Hz, 1H). 4.57-4.55(m. 2H), 3.99-3.91 (m, 4H), 3.90 (s, 3H), 3.82-3.80 (m, 4H), 3.51-3.49 (m, 2H), 2.64 (s, 3H).
[0466] General procedure 38:
[0467] Synthesis of Compound 148: 3-[2-(2H3)methoxyethyl]-5-[3-(m-methoxyphenyl)-l- pyrazolyl] -7-morpholino-3H- 1 ,3, 4- triazaindene
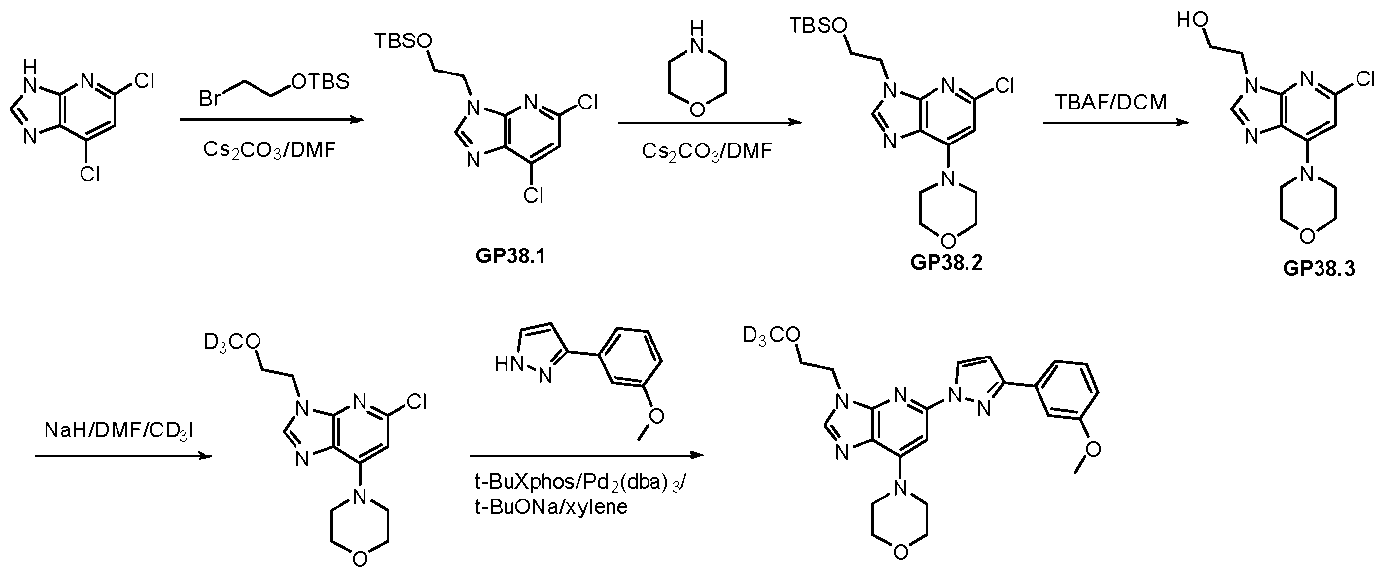 [0468] Synthesis of 3-(2-((tert-butyldimethylsilyl)oxy)ethyl)-5, 7-dichloro-3H-imidazo[4,5- b ]pyridine, GP38. 1
[0468] Synthesis of 3-(2-((tert-butyldimethylsilyl)oxy)ethyl)-5, 7-dichloro-3H-imidazo[4,5- b ]pyridine, GP38. 1
[0469] To a solution of 5,7-dichloro-3H-imidazo[4,5-b]pyridine (2.1 g, 11.2 mmol) in DMF (40.0 mL) at rt was added CS2CO3 (8.4 g, 61.2 mmol) and (2-bromoethoxy)(tert- butyl)dimethylsilane (3.2 g, 13.4 mmol). The mixture was stirred at 70 °C for 1 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was diluted with water (100 mL) and extracted with EtOAc (3 x 50 mL). The combined organic phases were washed with brine, dried over anhydrous NazSCh, fdtered, and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 10% EtOAc/PE to 30% EtOAc to provide 3-(2-((tert- butyldimethylsilyl)oxy)ethyl)-5,7-dichloro-3H-imidazo[4,5-b]pyridine (2.4 g, 6.96 mmol) as a yellow solid. LC-MS (ESI+): m/z 346/348 (MH ). ’HNMR (300 MHz, CDCh) 8.25 (s, 1H), 7.39 (s, 1H), 4.46 (t, J = 4.8 Hz, 2H), 4.02 (t, J= 4.8 Hz, 2H), 0.92 (s, 9H). 0.08 (s. 6H).
[0470] Synthesis of 4-(3-(2-((tert-butyldimethylsilyl)oxy)ethyl)-5-chloro-3H-imidazo[4, 5- b ]pyridin- 7 -yl)mor pholine, GP38.2
[0471] To a solution of 3-(2-((tert-butyldimethylsilyl)oxy)ethyl)-5,7-dichloro-3H-imidazo[4,5- b]pyridine (908 mg, 10.44 mmol) in DMF (40.0 mL) at rt was added CS2CO3 (4.5 g, 13.92 mmol) and morpholine (756 mg, 8.70 mmol). The mixture was stirred at 70 °C overnight. After completion of the reaction as indicated by TLC analysis, the reaction mixture was diluted with water (100 mL) and extracted with EtOAc (3 x 100 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 20% EtOAc/PE to 50% EtOAc to provide 4-(3-(2-((tert- butyldimethylsilyl)oxy)ethyl)-5-chloro-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (1.7 g, 4.29 mmol) as an off-white solid. LC-MS (ESI+): m/z 397/399 (MH+).
[0472] Synthesis of 2-(5-chloro-7-morpholino-3H-imidazo[4.5-b]pyridin-3-yl)ethanol. GP38.3 [0473] To a solution of 4-(3-(2-((tert-butyldimethylsilyl)oxy)ethyl)-5-chloro-3H-imidazo[4,5- b]pyridin-7-yl)morpholine (1.7 g, 4.29 mmol) in DCM (40.0 mL) at rt was added TBAF (1.12 g, 4.29 mmol). The mixture w as stirred at rt for 2 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was diluted with water (100 mL) and extracted with EtOAc (3 x 50 mL). The combined organic phase was dried over anhydrous Na2SO4, filtrated and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 20% EtOAc/PE to 50% EtOAc to provide 2-(5- chloro-7-morpholino-3H-imidazo[4.5-b]pyridin-3-yl)ethanol (1.07 g, 3.79 mmol) as an off- white solid. LC-MS (ESI+): m/z 283/285 (MH1 ).
[0474] Synthesis of 4-(5-chloro-3-(2-(methoxy-d3)ethyl)-3H-imidazo[4, 5-b ]pyridin-7- yl)morpholine, GP38.4
[0475] To a solution of 2-(5-chloro-7-morpholino-3H-imidazo[4,5-b]pyridin-3-yl)ethanol (200 mg, 0.71 mmol) in DMF (20 mL) at rt was added NaH (57 mg, 1.42 mmol) and CD3I (0.85 mmol, 123 mg). The mixture was stirred at rt overnight. After completion of the reaction as indicated by TLC analysis, the reaction mixture was diluted with water (20 mL) and extracted with EtOAc (3 x 50 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SC>4, fdtered, and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 20% EtOAc/PE to 50% EtOAc to provide 4-(5-chloro-3-(2-(methoxy-d3)ethyl)-3H-imidazo[4,5-b]pyridin-7- yl)morpholine (150 mg, 0.50 mmol) as an off white solid. LC-MS (ESI+): m/z 300/302 (MH+).
[0476] Synthesis of 4-(3-(2-(methoxy-d3)ethyl)-5-(3-(3-methoxyphenyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-b]pyridin-7-yl)morpholine, Compound 148
[0477] A suspension of 4-(5-chloro-3-(2-(methoxy-d3)ethyl)-3H-imidazo[4,5-b]pyridin-7- yl)morpholine (20 mg, 0.067 mmol), Xphos (15.9 mg, 0.03 mmol), t-BuONa (12.7 mg, 0.13 mmol), Pd2(dba)3 (30.5 mg, 0.033 mmol) and 3-(3-methoxyphenyl)-lH-pyrazole (14 mg, 0.08 mmol) in xylene (2 mL) under N2 was heated to 130 °C and stirred for 2 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was diluted with water (20 mL) and extracted with EtOAc (3 x 15 mL). The combined organic phases were dried over anhydrous Na2SO4, filtered, and concentrated under reduce pressure. The resulting residue was purified by preparative TLC with an elution of 7% MeOH/DCM to provide 4-(3-(2-(methoxy- d3)ethyl)-5-(3-(3-methoxyphenyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (16.4 mg, 0.037 mmol) as a white solid. LC-MS (ESI+): m/z 438 (MH+). 'HNMR (300 MHz, CD3OD) 8.61 (d, J = 2.7 Hz, 1H), 8.04 (s, 1H), 7.50-7.48 (m, 2H), 7.34 (t, J= 8.1 Hz, 1H), 7.29 (s. 1H), 6.93 (d, J= 8.4 Hz. 1H), 6.86 (d, J= 2.4 Hz, 1H), 4.45 (t, .7= 5.1 Hz, 2H), 3.91 (s, 8H), 3.87 (s, 3H), 3.80 (t, J = 4.8 Hz, 2H).
[0478] General procedure 39:
[0479] Synthesis of Compound 158: 5-[3-(m-difluoromethoxyphenyl)-l-pyrazolyl]-3-[2- (2H3)methoxyethyl] -7-morpholino-3H- 1,3, 4- triazaindene
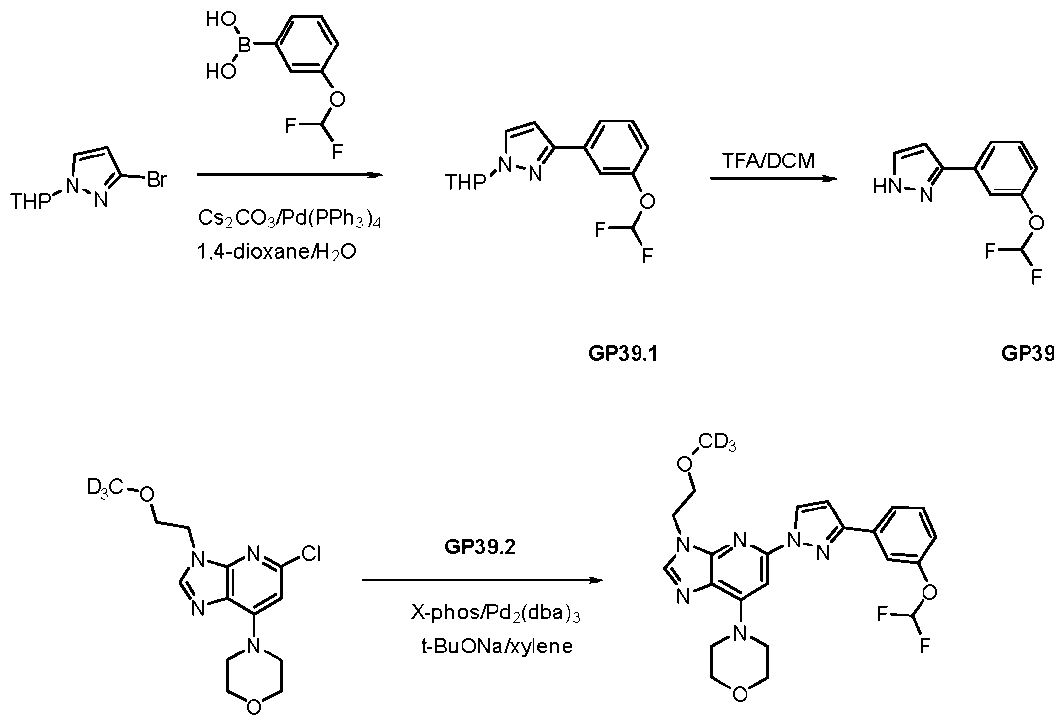
[0480] Synthesis of 3-(3-(difluoromethoxy)phenyl)-l-(tetrahydro-2H-pyran-2-yl)-lH-pyrazole.
GP39. 1
[0481] A suspension of 3-bromo-l-(tetrahydro-2H-pyran-2-yl)-lH-pyrazole (410 mg, 1.77 mmol), (3-(difluoromethoxy)phenyl)boronic acid (500 mg, 2.66 mmol), Na2COs (564 mg, 5.32 mmol) and Pd(PPhs)4 (205 mg, 0. 177 mmol) in 1 ,4-dioxane/H2O (10: 1. 11 mL) under N2 was heated to reflux for 2 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was diluted with water (20.0 mL) and extracted with EtOAc (3 x 20 mL). The combined organic phases were dried over anhydrous Na2SO4, filtered, and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 10% EtOAc/PE to 50% EtOAc/PE to provide 3-(3- (difluoromethoxy)phenyl)-l-(tetrahydro-2H-pyran-2-yl)-lH-pyrazole (390 mg, 1.33 mmol) as a yellow oil.
[0482] Synthesis of 3-(3-(difluoromethoxy)phenyl)-lH-pyrazole, GP39.2
[0483] To a solution of 3-(3-(difluoromethoxy)phenyl)-l-(tetrahydro-2H-pyran-2-yl)-lH- pyrazole (390 mg, 1.33 mmol) in DCM (5.0 mL) was added TFA (1.0 mL). The reaction mixture was stirred at rt for 2 h. After completion of the reaction as indicated by TLC and LC- MS analysis, the reaction mixture was quenched with aqueous Na2COs solution to pH ~ 9. The mixture was extracted with DCM (3 x 20 mL). The combined organic phases were dried over anhydrous Na2SO4, filtrated and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 1% MeOH/DCM to 3% MeOH/DCM to provide 3 -(3 -(difluoromethoxy) phenyl)- IH-pyrazole (130 mg, 0.62 mmol) as a yellow oil. LC-MS (ESI+): m/z 211 (MH ).
[0484] Synthesis of 5-[3-(m-difluoromethoxyphenyl)-l-pyrazolyl ]-3-[2-(2Hs)methoxyethyl ]-7- morpholino-3H-l,3,4-triazaindene, Compound 158
[0485] A suspension of 4-(5-chloro-3-(2-(methoxy-d3)ethyl)-3H-imidazo[4,5-b]pyridin-7- yl)morpholine (60 mg, 0.152 mmol), Xphos (32 mg, 0.076 mmol), t-BuONa (29 mg, 0.30 mmol), Pd2(dba)3 (69.4 mg, 0.076 mmol) and 3-(3-(difluoromethoxy)phenyl)-lH-pyrazole (47 mg, 0.23 mmol) in xylene (5 mL) under N2 was heated to 130 °C and stirred for 2 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was diluted with water (20 mL) and extracted with EtOAc (3 x 15 mL). The combined organic phases were dried over anhydrous Na2SO4, filtered, and concentrated under reduce pressure. The resulting residue was purified by preparative TLC with an elution of 50% EtOAc/PE to provide 5-[3-(m- difluoromethoxyphenyl)-l-pyrazolyl]-3-[2-(2H3)methoxyethyl]-7-morpholino-3H-l,3,4- triazaindene (12.5 mg, 0.026 mmol) as a white solid. LC-MS (ESI+): m/z 473 (MH+). 1HNMR (300 MHz, CD3OD) 8.65 (s, 1H), 8.05 (s, 1H), 7.79 (d, J= 7.5 Hz, 1H), 7.71 (s, 1H), 7.47 (t, J = 7.8 Hz. 1H), 7.32 (s, 1H), 7.15-7.13 (s, 1.3H), 6.91 (s, 1.5H), 6.66(s, 0.2H), 4.45 (t, J= 4.5 Hz, 2H), 3.92 (s, 8H), 3.81 (t, J= 4.5 Hz, 2H).
[0486] General procedure 40:
[0487] Synthesis of Compound 172: 3-(2-methoxyethyl)-7-morpholino-5-[l-(m-tolyl)-3- pyrazolyl] -3H- 1 ,3,4-triazaindene

[0488] Synthesis of 4-(3-(2-methoxyethyl)-5-( l-(tetrahydro-2H-pyran-2-yl)-lH-pyrazol-5-yl)- 3H-imidazo[4, 5-b ]pyridin- 7-yl)morpholine, GP40.1
[0489] A suspension of 4-(5-chloro-3-(2-methoxyethyl)-3H-imidazo[4,5-b]pyridin-7- yl)morpholine (300 mg, 1.01 mmol), K2CO3 (414 mg. 3.0 mmol), Pd2(PPh3)2C12 (73 mg. 0.1 mmol) and l-(tetrahydro-2H-pyran-2-yl)-5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH- pyrazole (423 mg, 1.52 mmol) in 1,4-dioxane (8.0 mL) under N2 was heated to 110 °C. The reaction mixture was stirred at that temperature for 2 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was quenched with water (20 mL) and extracted
wi th EtOAc (3 x 20 mL). The combined organic phases were dried over anhydrous Na2SO4, filtered and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 20% EtOAc/PE to 50% EtOAc to provide 4- (3-(2-methoxyethyl)-5-(l-(tetrahydro-2H-pyran-2-yl)-lH-pyrazol-5-yl)-3H-imidazo[4.5- b]pyridin-7-yl)morphohne (190 mg. 0.46 mmol) as a yellow oil. LC-MS (ESI+): m/z 413 (MH+).
[0490] Synthesis of 4-(3-(2-methoxyethyl)-5-(lH-pyrazol-5-yl)-3H-imidazo[4,5-b]pyridin-7- yl)morpholine, GP40.2
[0491] To a solution of 4-(3-(2-methoxyethyl)-5-(l-(tetrahydro-2H-pyran-2-yl)-lH-pyrazol-5- yl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (190 mg, 0.46 mmol) in DCM (5.0 mL) was added TFA (0.5 mL). The reaction mixture was stirred at rt overnight. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was concentrated directly to provide crude 4-(3-(2-methoxyethyl)-5-(lH-pyrazol-5-yl)-3H-imidazo[4,5-b]pyridin- 7-yl)morphohne (65 mg, 0.20 mmol) as a yellow oil. LC-MS (ESI+): m/z 327 (MH+).
[0492] Synthesis of 4-(3-(2-methoxyethyl)-5-( l-(m-tolyl)-lH-pyrazol-3-yl)-3H-imidazo[ 4, 5- b]pyridin-7-yl)morpholine hydrochloride, Compound 172
[0493] A suspension of crude 4-(3-(2-methoxyethyl)-5-(lH-pyrazol-5-yl)-3H-imidazo[4,5- b]pyridin-7-yl)morpholine (65 mg, 0.20 mmol). K2CO3 (67 mg, 0.5 mmol), Cui (4 mg. 0.02 mmol) and l-bromo-3 -methylbenzene (250 mg, 2.0 mmol) in DMF (3.0 mL) under N2 was heated to 110 °C. The reaction mixture was stirred at that temperature overnight. After completion of the reaction as indicated by TLC analysis, the reaction mixture was quenched with water (20 mL) and extracted with EtOAc (3 x 20 mL). The combined organic phases were dried over anhydrous Na2SO4, filtered, and concentrated under reduce pressure. The resulting residue w as purified by silica gel column chromatography with a gradient elution of 2% MeOH/DCM to 5% MeOH/DCM to provide impure 4-(3-(2-methoxyethyl)-5-(l-(m-tolyl)-lH- pyrazol-3-yl)-3H-imidazo[4.5-b]pyridin-7-yl)morpholine (5.0 mg, 0.012 mmol) as a white solid. To a solution of impure 4-(3-(2-methoxyethyl)-5-(l-(m-tolyl)- lH-pyrazol-3-yl)-3H- imidazo[4,5-b]pyridin-7-yl)morpholine (5.0 mg, 0.012 mmol) in DCM (2.0 mL) was added HCl/Et2O (0.5 mL). The mixture was stirred at rt for 2 h. The mixture w as concentrated directly and the resulting residue was purified by slurry in Et20 to provide 4-(3-(2-methoxyethyl)-5-(l- (m-tolyl)-lH-pyrazol-3-yl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine hydrochloride (5.0 mg, 0.012 mmol) as a white solid. LC-MS (ESI+): m/z 419 (MH+). 'H NMR (300 MHz, CDsOD) S 8.23 (d, J= 2.4 Hz, 1H), 8.08 (s, 1H), 7.69 (s, 1H), 7.62 (d, J= 8.1 Hz, 1H), 7.43- 7.33 (m, 2H), 7.17 (d, J= 7.2 Hz, 1H), 7.12 (d, J= 2.4 Hz, 1H), 4.52 (t, J= 5.1 Hz, 2H), 3.95-3.81 (m, 10H), 3.35 (s, 3H), 2.45 (s. 3H).
[0494] General procedure 41:
[0495] Synthesis of Compound 188: 7-{(lR,4R)-2-oxa-5-azabicyclo[2.2.1]hept-5-yl}-3-(2- methoxyethyl)-5-[3-(m-tolyl)-l-pyrazolyl]-3H-l, 3, 4- triazaindene
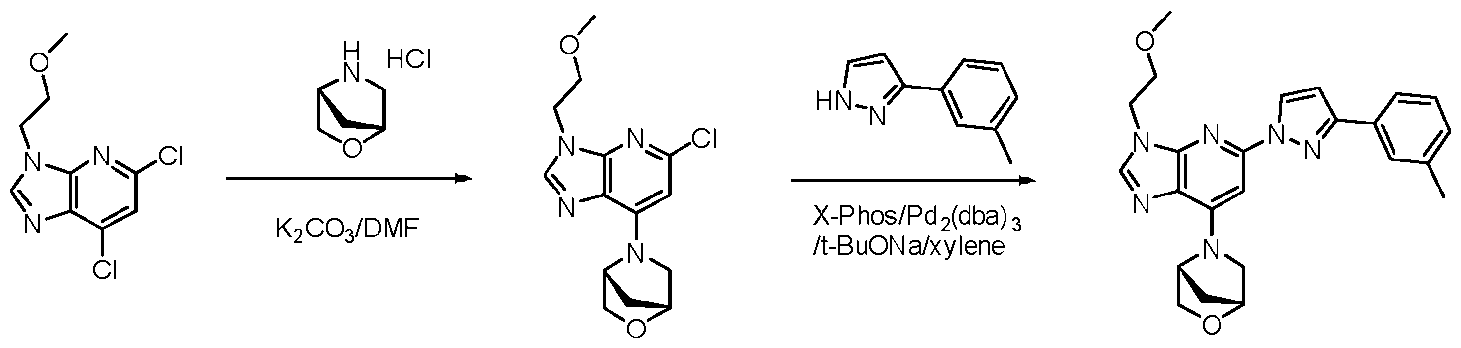
GP41.1 Compound 188
[0496] Synthesis of (lR,4R)-5-(5-chloro-3-(2-methoxyethyl)-3H-imidazo[4.5-b]pyridin-7-yl)- 2-oxa-5 -azabicyclo [2.2. 1 ]heptane. GP41. 1
[0497] To a solution of 5,7-dichloro-3-(2-methoxyethyl)-3H-imidazo[4,5-b]pyridine (200 mg, 0.81 mmol) in DMF (5.0 mL) was added K2CO3 (224 mg, 1.62 mmol) and (lR,4R)-2-oxa-5- azabicyclo[2.2.1]heptane hydrochloride (170 mg, 1.22 mmol). The reaction mixture was heated to 100 °C and stirred for 4 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was diluted with water (15 mL) and extracted with EtOAc (2 x 10 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SC>4, fdtered and concentrated to provide (lR,4R)-5-(5-chloro-3-(2-methoxyethyl)-3H-imidazo[4,5- b]pyridin-7-yl)-2-oxa-5-azabicyclo[2.2.1]heptane (59 mg, 0.19 mmol) as a colorless oil. LC-MS (ESI+): m/z 309/311 (MH+).
[0498] Synthesis of (lR,4R)-5-(3-(2-methoxyethyl)-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-b]pyridin-7-yl)-2-oxa-5-azabicyclo[2.2. l]heptane, Compound 188
[0499] A suspension of (lR,4R)-5-(5-chloro-3-(2-methoxyethyl)-3H-imidazo[4,5-b]pyridin-7- yl)-2-oxa-5-azabicyclo[2.2.1]heptane (20 mg, 0.065 mmol), t-BuONa (12.4 mg, 0.13 mmol), Pd2(dba)s (29.7 mg, 0.032 mmol), X-Phos (15.48 mg, 0.032 mmol) and 3-(m-tolyl)-lH-pyrazole (15.4 mg, 0.097 mmol) in xylene (2.0 mL) under N2 was heated to 130 °C and stirred overnight. After completion of the reaction as indicated by TLC analysis, the reaction mixture was diluted with water (20 mL) and extracted with EtOAc (3 x 10 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduce pressure. The resulting residue was purified by preparative HPLC to provide (lR,4R)-5-(3-(2- methoxyethyl)-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-7-yl)-2-oxa-5- azabicyclo[2.2.1]heptane (16.3 mg, 0.038 mmol) as a white solid. LC-MS (ESI+): m/z 431 (MH+). 1HNMR (300 MHz, CDCh) 8.58 (d, J= 2.4 Hz, 1H), 7.85 (s, 1H), 7.78 (s, 1H), 7.73 (d, J= 7.8 Hz, 1H), 7.33 (t, J= 7.8 Hz, 1H), 7.16 (d, J= 7.5 Hz, 1H), 7.03 (s, 1H), 6.74 (d, J =
2.4 Hz, 1H), 4.78 (s, 1H), 4.39 (t, J= 5.1 Hz, 2H), 3.99 (s, 2H), 3.93-3.56 (m, 5H), 3.37 (s, 3H), 2.44 (s, 3H), 2.05 (s, 2H).
[0500] General procedure 42:
[0501] Synthesis of Compound 201 and 191: 4-(2-(3-(m-tolyl)-lH-pyrazol-l-yl)-6,7,8,9- tetrahydropyrido[3',2':4,5] i id azo | l,2-a]pyrazin-4-yl)morpholine (201) and 2-mesyl-8- morpholino-6-[3-(m-tolyl)-l-pyrazolyl]-l,2,3,4-tetrahydro-2,4a,5,9-tetraazafluorene (191)

GP42.1
[0503] To a solution of N2-(2,4-dimethoxybenzyl)-4-morpholino-6-(3-(m-tolyl)-lH-pyrazol- 1-yl) pyridine-2, 3-diamine (25 g, 0.05 mol) in DCM (250 mL) at rt was added TFA (25 mL). The reaction mixture was stirred at rt for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with aqueous NaHCOs solution to pH ~ 8. The aqueous solution was extracted with DCM/MeOH (10/1, 3 x 200 mL). The combined organic phases were dried over anhydrous Na2SO4, filtrated and concentrated. The resulting residue was purified by slurry in Et2O/EtOAc (100/1, 50 mL) twice to provide 4- morpholino-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridine-2.3-diamine (14.5 g, 0.04 mol) as a white solid. LC-MS (ESI+): m/z 351 (MH+). 1HNMR (300 MHz, DMSO- e) 8.33 (d, J= 2.4 Hz, 1H), 7.73-7.68 (m, 2H), 7.32 (t, J= 7.2 Hz, 1H), 7.15 (d, J= 7.2 Hz, 1H), 6.93 (s, 1H), 6.89 (d, J = 2.4 Hz. 1H), 5.70 (s, 2H), 4.31 (s, 2H), 3.83-3.78 (m, 4H), 2.93-2.86 (m, 4H), 2.38 (s, 3H).
[0504] Synthesis of tert-butyl ((7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-I-yl)-3H- imidazo[4, 5-b ]pyridin-2-yl)methyl)carbamate. GP42.2
[0505] To a solution of 4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridine-2,3-diamine (400 mg, 1.14 mmol) in AcOH/I-bO (2: 1, 30 mL) at rt was added Cu(OAc)2 (2.06 g, 11.4 mmol) and tert-butyl (2-oxoethyl)carbamate (271 mg, 1.71 mmol). The mixture was heated to 60 °C and stirred for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with NaHCOs solution to pH ~ 9 and extracted with EtOAc (3 x 20 mL). The combined organic phases were dried over anhydrous Na2SC>4, filtrated and concentrated. The resulting residue was purified by silica gel column chromatography with a gradient elution of 20% EtOAc/PE to 50% EtOAc/PE to provide tert-butyl ((7-morpholino-5-(3- (m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-2-yl)methyl)carbamate (103 mg, 0.21 mmol) as a yellow oil. LC-MS (ESI+): m/z 490 (MH+).
[0506] Synthesis of tert-butyl ((3-(2-bromoethyl)-7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l- yl)-3H-imidazo[4, 5-b ]pyridin-2-yl)methyl) carbamate, GP42.3
[0507] To a solution of tert-butyl ((7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-b]pyridin-2-yl)methyl)carbamate (103 mg, 0.21 mmol) in DMF (3.0 mL) at rt was added CS2CO3 (137 mg, 0.42 mmol) and 1 ,2-dibromoethane (395 mg, 2.1 mmol). The mixture was stirred at rt for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was diluted with water (20.0 mL) and extracted with EtOAc (3 x 10 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4. filtrated and concentrated. The resulting residue was purified by preparative TLC with an elution of 50% EtOAc/PE to provide tert-butyl ((3-(2-bromoethyl)-7-morpholino-5-(3-(m-tolyl)-lH- pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-2-yl)methyl)carbamate (23.5 mg, 0.039 mmol) as a yellow solid. LC-MS (ESI+): m/z 596/598 (MH+).
[0508] Synthesis of 4-(2-( 3-(m-tolyl)-lH-pyrazol-l-yl)-6, 7, 8, 9-tetrahydropyrido[3 2': 4,5] imidazo[l, 2-a ]pyrazin-4-yl)morpholine, Compound 201
[0509] To a solution of tert-buty l ((3-(2-bromoethyl)-7-morpholino-5-(3-(m-tolyl)-lH- pyrazol-l-yl)-3H-imidazo[4.5-b]pyridin-2-yl)methyl)carbamate (23.5 mg, 0.039 mmol) in DCM (5.0 mL) at rt was added TFA (0.5 mL). The reaction mixture was stirred at rt for 1 h. After completion of the reaction as indicated by TLC analysis, to the solution was added saturated Na2CCh aqueous solution (10 mL). The solution mixture was stirred at rt for another 2 h. After completion of the reaction as indicated by LC-MS analysis, the reaction mixture was extracted with MeOH/DCM (3 x 10 mL, 1: 10). The combined organic phases were dried over anhydrous Na2SO4, filtrated and concentrated. The resulting residue was purified by slurry in Et20 to provide 4-(2-(3-(m-tolyl)-lH-pyrazol-l-yl)-6,7,8,9-tetrahydropyrido[3',2':4,5]imidazo[l,2- a]pyrazin-4-yl)morpholine (35 mg, 0.084 mmol) as a white solid.LC-MS (ESI+): m/z 416 (MH+). 1HNMR (300 MHz, CDCh) 8.58 (d. J= 2.4 Hz. 1H), 7.78 (s. 1H), 7.73 (d, J= 8.1 Hz.
1H), 7.35-7.30 (m, 2H), 7. 17 (d, J= 7.2Hz, 1H), 6.75 (d, J= 2.1 Hz, 1H), 4.25 (s, 2H), 4. 17 (t, J = 5.4Hz, 2H), 4.09-3.95 (m, 8H), 3.37 (t, J= 5.4 Hz, 2H), 2.44 (s, 3H).
[0510] Synthesis of 4-(7-(methylsulfonyl)-2-(3-(m-tolyl)-lH-pyrazol-l-yl)-6, 7,8,9- tetrahydropyrido[3 ', 2 4, 5 ]imidazo[l, 2-a ]pyrazin-4-yl)mor pholine, Compound 191 [0511] To a solution of 4-(2-(3-(m-tolyl)-lH-pyrazol-l-yl)-6.7.8.9-tetrahydropyrido[3',2':4,5] imidazo[l,2-a]pyrazin-4-yl)morpholine (12. 1 mg, 0.029 mmol) in DCM (1.0 mL) at rt was added EtsN (6.0 mg, 0.058 mmol) and MsCl (6.5 mg, 0.058 mmol). The mixture was stirred at rt for 0.5 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was diluted with water (5.0 mL) and extracted with MeOH/DCM (3 x 5 mL, 1 : 10). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtrated and concentrated. The resulting residue was purified by preparative TLC with an elution of 20% EtOAc/PE to provide 4-(7-(methylsulfonyl)-2-(3-(m-tolyl)-lH-pyrazol-l-yl)-6, 7,8,9- tetrahydropyrido[3',2':4,5] imidazo[l,2-a]pyrazin-4-yl)morpholine (6.0 mg, 0.0086 mmol) as a white solid. LC-MS (ESI+): m/z 494 (MH+). 1HNMR (300 MHz, CDCk) 8.57 (d, J= 2.1 Hz, 1H), 7.77 (s, 1H), 7.73 (d, J= 7.5 Hz, 1H), 7.36-7.31 (m, 2H), 7.17 (d, J= 7.2 Hz, 1H), 6.76 (d, .7= 2.1 Hz, 1H), 4.72 (s, 2H), 4. 33 (t, J= 5.1Hz, 2H). 3.98-3.95 (m, 8H). 3.87 (t. J= 5.1 Hz, 2H), 2.97 (s, 3H), 2.44 (s, 3H).
[0512] General procedure 43:
[0513] Synthesis of Compound 200: 9-morpholino-ll-[3-(m-tolyl)-l-pyrazolyl]-2,4,7,12- tetraazatricyclo[6.4.0.02,6]dodeca-l(12),6,8,10-tetraen-3-one

2-yl)methanamine hydrochloride, GP43. 1
[0515] To a solution of tert-butyl ((7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-b]pyridin-2-yl)methyl)carbamate (67 mg, 0.14 mmol) in DCM/MeOH (3.0 mL, 2: 1) was added HCl/Et2O (2 mL). The mixture was stirred at rt for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the mixture was concentrated directly and the resulting residual was purified by slurry in Et20 to provide (7-morpholino-5-(3-(m-tolyl)-lH- pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-2-yl)methanamine hydrochloride (116 mg, 0.27 mmol) as a white solid. LC-MS (ESI+): m/z 390 (MH+).
[0516] Synthesis of 4-morpholino-2-( 3-(m-tolyl)-lH-pyrazol-l-yl)-6H-imidazo[5', 1 2, 3 ] imidazo[4,5-b]pyridin-8(7H)-one, Compound 200
[0517] To a solution of (7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5- b]pyri din-2 -yl)methanamine hydrochloride (116 mg, 0.27 mmol) in CH3CN (5.0 mL) was added K2CO3 (113 mg. 0.82 mmol) and CD1 (70 mg, 0.82 mmol). The mixture was stirred at 80 °C for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was filtered, and the filtrate was concentrated. The resulting residue was purified by preparative TLC with an elution of 8% MeOH/DCM to provide 4-morpholino-2-(3-(m-tolyl)- lH-pyrazol-l-yl)-6H-imidazo[5',T:2,3]imidazo[4.5-b]pyridin-8(7H)-one (4.9 mg, 0.012 mmol) as a white solid. LC-MS (ESI+): m/z 416 (MH+). 'HWIR (300 MHz, DMSO-rfc) 8.54 (s, 1H), 7.77-7.74 (m, 2H), 7.36 (t, J= 7.2 Hz, 1H), 7.20-7.19 (m, 2H), 7.00 (s, 1H), 6.85-6.81 (m, 1H), 4.44 (d, J= 5.2 Hz, 2H), 3.95-3.91 (m, 4H), 3.85-3.80 (m, 4H), 2.39 (s. 3H).
[0518] General procedure 44:
[0519] Synthesis of Compounds 208 and 210: 3-[(l-methoxycyclopropyl)methyI]-5-[5- (methoxymethyl)-3-(m-tolyl)-l-pyrazolyl]-7-morpholino-3H-l,3,4-triazaindene (208); 3-(l- methoxycyclobutyl)-5- [5-(methoxymethyl)-3-(m-tolyl)- 1-pyrazolyl] -7-morpholino-3H- 1,3,4-triazaindene (210)
 [0520] Synthesis of ethyl l-(6-((2,4-dimethoxybenzyl)amino)-4-morpholino-5-nitropyridin-2- yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate, GP44. 1
[0520] Synthesis of ethyl l-(6-((2,4-dimethoxybenzyl)amino)-4-morpholino-5-nitropyridin-2- yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate, GP44. 1
[0521] A suspension of 6-chloro-N-(2,4-dimethoxybenzyl)-4-morpholino-3-nitropyridin-2- amine (5.7 g. 13.97 mmol), CS2CO3 (13.67 mg, 41.9 mmol), Pd2(dba)s (2.17 g, 2.79 mmol), t- BuXphos (2.38 g, 5.58 mmol) and ethyl 3-(m-tolyl)-lH-pyrazole-5-carboxylate (423 mg, 1.52 mmol) in 1,4-dioxane (100 mb) under N2 was heated to 80 °C for 2 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was diluted with water (60 mL) and EtOAc (3 x 60 mL). The combined organic phases were dried over anhydrous Na2SO4, filtrated and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 10% EtOAc/PE to 30% EtOAc/PE to provide ethyl 1- (6-((2,4-dimethoxybenzyl)amino)-4-morpholino-5-nitropyridin-2-yl)-3-(m-tolyl)-lH-pyrazole- 5-carboxylate (1.77 g, 2.94 mmol) as a yellow solid. LC-MS (ESI+): m/z 603 (MH+).
[0522] Synthesis of ethyl l-(5-amino-6-((2, 4-dimethoxybenzyl)amino)-4-morpholinopyridin-2- yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate, GP44.2
[0523] To a solution of ethyl l-(6-((2,4-dimethoxybenzyl)amino)-4-morpholino-5- nitropyridin-2-yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate (1.77 g, 2.94 mmol) in MeOH/THF (20 mL/20 mL) was added Pd/C (200 mg). The reaction mixture was purged with H2 (gas) for three times and stirred at rt overnight under H2 using balloons. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was filtered directly and the filtrate was concentrated. The resulting residue was purified by silica gel column chromatography with a gradient elution of 10% EtOAc/PE to 50% EtOAc to provide ethyl l-(5- amino-6-((2, 4-dimethoxybenzyl)amino)-4-morpholinopyri din-2 -yl)-3-(m-tolyl)-lH-pyrazole-5- carboxylate (1.38 g, 2.41 mmol) as a light yellow solid. LC-MS (ESI+): m/z 573 (MH+). 'H NMR (300 MHz, CDCh) 7.74 (s, 1H), 7.66 (d, J= 8.1 Hz, 1H), 7.34-7.31 (m, 1H), 7.25-7.22 (m, 1H), 7.17-7.13 (m, 2H), 6.87 (s, 1H), 6.46 (s, 1H), 6.43 (d, J= 8.1 Hz, 1H), 4.45 (s, 2H). 4.30-4.20 (m, 2H). 3.91-3.83 (m, 7H). 3.79 (s. 3H), 3.48 (brs. 1H), 3.03-2.95 (m. 4H), 2.41 (s, 3H), 1.23 (t, J= 7.5 Hz, 3H).
[0524] Synthesis of ethyl l-( 3-(2, 4-dimethoxybenzyl)- 7-morpholino-3H-imidazo[4, 5- b ]pyridm-5-yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate, GP44.3
[0525] To a solution of ethyl l-(5-amino-6-((2,4-dimethoxybenzyl)amino)-4- morpholinopyridin-2-yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate (1.38 g, 2.41 mmol) in trimethoxymethane (50 mL) was added AcOH (180 mg, 3.0 mmol). The reaction mixture was heated to reflux and stirred for 3 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with NaHCCh to pH = 9. The mixture was extracted with EtOAc (3 x 50 mL). The combined organic phases were dried over anhydrous
Na2SO4, filtrated and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 20% EtOAc/PE to 50% EtOAc to provide ethyl l-(3-(2,4-dimethoxybenzyl)-7-morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-3-(m- tolyl)-lH-pyrazole-5-carboxylate (1.03 g, 1.77 mmol) as a yellow solid. LC-MS (ESI+): m/z 583 (MH+). 'H NMR (300 MHz, CDCh) 81.87 (s, 1H), 7.75 (s, 1H), 7.68 (d, J= 8.4 Hz, 1H), 7.35-7.31 (m, 1H), 7.25-7.16 (m, 3H), 6.87 (s, 1H), 6.45-6.40 (m, 2H), 5.24 (s, 2H), 4.25-4.18 (m, 2H), 3.96-3.92 (m, 8H), 3.84 (s, 3H), 3.78 (s, 3H), 3.42 (s, 3H), 1.16 (t, J= 7.5 Hz. 3H). [0526] Synthesis of (l-(3-(2, 4-dimethoxybenzyl)- 7-morpholino-3H-imidazo[4, 5-b ]pyridin-5- yl)-3-(m-tolyl)-lH-pyrazol-5-yl)methanol. GP44.4
[0527] To a solution of ethyl l-(3-(2,4-dimethoxybenzyl)-7-morpholino-3H-imidazo[4,5- b]pyridin-5-yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate (620 mg, 1.06 mmol) in THF (10 mL) at 0 °C was added LiAlH4 (42 mg, 1.06 mmol) in portions. The solution mixture was allowed to warm to rt and stirred for 1 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with water (20 mL) and extracted with EtOAc (3 x 20 mL). The combined organic phases was dried over anhydrous NazSO4, filtrated and concentrated. The resulting residue was purified by silica gel column chromatography with a gradient elution of 20% EtOAc/PE to 50% EtOAc/PE to provide (l-(3-(2,4-dimethoxybenzyl)-7- morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-3-(m-tolyl) -lH-pyrazol-5-yl)methanol (470 mg, 0.87 mmol) as a white solid. LC-MS (ESI+): m/z 541 (MH+). 'HNMR (300 MHz, CDCh) 7.85 (s, 1H), 7.73-7.70 (m, 2H), 7.35-7.27 (m, 2H), 7.26-7.23 (m, 1H), 7.17 (d, J= 7.8 Hz, 1H), 6.71 (s, 1H), 6.46-6.42 (m, 2H), 6.25-6.15 (m, 1H), 5.28 (s, 2H). 4.81 (s. 2H), 4.11-4.05 (m. 4H), 3.95-3.92 (m, 4H), 3.82 (s, 3H), 3.78 (s, 3H), 2.43 (s, 3H).
[0528] Synthesis of 4-(3-(2, 4-dimethoxybenzyl)-5-(5-(methoxymethyl)-3-(m-tolyl)-lH-pyrazol- l-yl)-3H-imidazo[4, 5-b ]pyridin- 7 -yl)mor pholine, GP44.5
[0529] To a solution of (l-(3-(2.4-dimethoxybenzyl)-7-morpholino-3H-imidazo[4.5-b]pyridin- 5-yl)-3-(m-tolyl)-lH-pyrazol-5-yl)methanol (300 mg, 0.56 mmol) in DMF/THF (10 mL) at 0 °C was added NaH (20 mg, 0.83 mmol) and CH3I (200 mg, 1.34 mmol). The solution mixture was allowed to warm to rt and stirred for 2 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was quenched with water (20 mL) and extracted with EtOAc (3 x 20 mL). The combined organic phases was washed with brine, dried over anhydrous Na2SO4, filtrated and concentrated. The resulting residue was purified by silica gel column chromatography with a gradient elution of 20% EtOAc/PE to EtOAc to provide 4-(3-(2,4- dimethoxybenzyl)-5-(5-(methoxymethyl)-3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5- b]pyridin-7-yl)morpholine (250 mg. 0.45 mmol) as a yellow solid. LC-MS (ESI+): m/z 555 (MH+).
[0530] Synthesis of 4-(5-(5-(methoxymethyl)-3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[ 4, 5- b ]pyridin- 7 -yl)mor pholine, GP44.6
[0531] To a solution of 4-(3-(2,4-dimethoxybenzyl)-5-(5-(methoxymethyl)-3-(m-tolyl)-lH- pyrazol-l-yl)-3H-imidazo[4.5-b]pyridin-7-yl)morpholine (250 mg, 0.45 mmol) in DCM (5.0 mL) at 0 °C was added TFA (5.0 mL). The solution mixture was stirred at it for 4 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was quenched with aqueous NaHCOs solution and adjusted the pH to 9. The mixture was extracted with EtOAc (3 x 20 mL). The combined organic phases was dried over anhydrous Na2SC>4, filtrated and concentrated. The resulting residue was purified by silica gel column chromatography with a gradient elution of 20% EtOAc/PE to EtOAc to provide 4-(5-(5-(methoxymethyl)-3-(m-tolyl)- lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (155 mg, 0.38 mmol) as a white solid. LC-MS (ESI+): m/z 405 (MH ). 'HNMR (300 MHz, CDCh) S 12.20 (s, 1H), 7.81-7.70 (m. 3H), 7.33 (t, J= 7.5 Hz, 1H), 7.18 (d, J = 7.5 Hz, 1H), 7.07 (s, 1H), 6.85 (s, 1H). 4.74 (s. 2H), 4.09-4.00 (m, 4H), 3.95-3.81 (m, 4H), 3.45 (s, 3H), 2.41 (s, 3H).
[0532] Synthesis of 4-(3-( ( l-methoxycyclopropyl)methyl)-5-(5-(methoxymethyl)-3-(m-tolyl)- lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine, Compound 208, and 4-(3-(l- methoxycyclobutyl)-5-(5-(methoxymethyl)-3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4.5- b]pyridin-7-yl)morpholine. Compound 210
[0533] To a solution of 4-(5-(5-(methoxymethyl)-3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-b]pyridin-7-yl)morpholine (20 mg, 0.05 mmol) in DMF (3.0 mL) at 0 °C was added CS2CO3 (113 mg, 0.82 mmol) and (1 -methoxy cy cl opropyl)methyl methanesulfonate (80 mg. 0.44 mmol). The mixture was stirred at ft overnight. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was diluted with water and extracted with EtOAc (3 x 20 mL). The combined organic phases was dried over anhydrous Na2SO4, filtrated and concentrated. The resulting residue was purified by preparative TLC with an elution of 67% EtOAc/PE to provide 4-(3-(l-methoxycyclobutyl)-5-(5-(methoxymethyl)-3- (m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (Compound 210, 7.2 mg, 0.015 mmol, upper spot) and 4-(3-((l-methoxycyclopropyl)methyl)-5-(5-(methoxymethyl)- 3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (8.5 mg, 0.016 mmol, lower spot) as a white solid. Compound 210, the upper spot on the preparative TLC: LC-MS (ESI+): m/z 489 (MH ). 'HNMR (300 MHz, CDCh) £7.89 (s, 1H), 7.75 (s, 1H), 7.69(d, J= 8.1 Hz, 1H), 7.34-7.30(m, 2H), 7.14 (d, J= 8.1 Hz, 1H), 6.80 (s, 1H), 5.06 (s, 2H), 4.04-3.90 (m, 8H), 3.50 (s, 3H), 3.08 (s, 3H), 3.02-2.95 (m, 2H), 2.85-2.79 (m, 2H), 2.42 (s, 3H), 2.16-2.10 (m. 1H), 2.05-1.96(m, 1H). Compound 208, the lower spot on the preparative TLC: LC-MS (ESI+): m/z 489 (MH+). 'HNMR (300 MHz, CDCh) £8.01 (s, 1H), 7.75 (s, 1H), 7.69 (d, J= 7.8
Hz, 1H), 7.32 (t, J = 7.8 Hz, 1H), 7.25- 7.24 (m, 1H), 7.15 (d, J= 7.8 Hz, 1H), 6.80 (s, 1H), 5.02(s, 2H), 4.34 (s, 2H), 4.02-4.00 (m, 4H), 3.96-3.90 (m, 4H), 3.48 (s, 3H), 3.35 (s, 3H), 2.42 (s, 3H), 0.97-0.90 (m, 2H), 0.80-0.75 (m, 2H).
[0534] General procedure 45:
[0535] Synthesis of Compound 238 and 239: (S)-2-methyl-l-(3-methyl-7-morpholino-5- (3-(m-tolyl)-lH-pyrazol-l-yI)-3H-imidazo[4,5-b]pyridin-2-yl)propan-l-amine (238) and N- [(S)-2-methyl-l- {3-methyl-7-morpholino-5- [3-(m-tolyl)- 1-pyrazolyl] -3H- 1,3,4- triazainden- 2-yl}propyl] acetamide (239)

[0537] To a solution of 4-(2,6-dichloro-3-nitropyridin-4-yl)morpholine (2.0 g, 7. 19 mmol) in DMF (25 mL) was added Methylamine hydrochloride (488.9 mg, 7.19 mmol) and CS2CO3 (4.69 g, 14.38 mmol). The reaction was stirred at rt for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was diluted with water (50 mL). The aqueous solution was extracted with EtOAc (3 x 50 mL). The combined organic phases were dried over anhydrous Na2SC>4, filtrated and concentrated. The resulting residue was purified by silica gel column chromatography with a gradient elution of 10% EtOAc/PE to 50% EtOAc/PE to provide 6-chloro-N-methyl-4-morpholino-3-nitropyridin-2 -amine (1.3 g, 4.78 mmol) as a yellow solid. LC-MS (ESI+): m/z 273/275 (MH ). 1HNMR (300 MHz, CDCh) 8 7.92 (brs, 1H), 6.12 (s, 1H), 3.83-3.79 (m, 4H), 3.22-3.18 (m, 4H), 3.08 (d, J= 4.8 Hz, 3H).
[0538] Synthesis of N-methyl-4-morpholino-3-nitro-6-( 3-(m-tolyl)-lH-pyrazol-l-yl)pyridin-2- amine, GP45.2
[0539] To a solution of 6-chloro-N-methyl-4-morpholino-3-nitropyridin-2-arnine (1.3 g, 4.78 mmol) in DMF (44 rnL) was added CS2CO3 (4.7 g, 4.74 mmol), CU2O (70 mg, 0.47 mmol) and 3-(m-tolyl)-lH-pyrazole (1.13 g, 7.05 mmol). The reaction was stirred at 110 °C under N2 for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was diluted with water (50 mL) and extracted with EtOAc (3 x 50 mL). The combined organic phases were washed with aqueous NaCl solution, dried over anhydrous Na2SO4, filtrated and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 5% EtOAc/PE to 33% EtOAc/PE to provide N- methyl-4-morpholino-3-nitro-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridin-2-amine (1.2 g, 3.04 mmol) as a yellow solid. LC-MS (ESI+): m/z 395 (MET).
[0540] Synthesis ofN2 -methyl-4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridine-2,3- diamine, GP45.3
[0541] To a solution of N-methyl-4-morpholino-3-nitro-6-(3-(m-tolyl)-lH-pyrazol-l- yl)pyridin-2-amine (1.2 g, 3.04 mmol) in DCM/MeOH (50 mL/10 mL) was added Pd/C (120 mg, 10%W/W). The reaction mixture was purged with H2 (gas) for three times. The reaction mixture was stirred at rt for 4 h under H2 balloons. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was filtered, and the filtrate was concentrated directly. The resulting residue was purified by silica gel column chromatography with a gradient elution of PE to 33% EtOAc/PE to provide N2-methyl-4-morpholino-6-(3-(m-tolyl)-lH-pyrazol- l-yl)pyridine-2,3-diamine (1.0 g, 2.74 mmol) as an off-white solid. LC-MS (ESI+): m/z 365 (MH+). ’HNMR (300 MHz, CDCh) £8.51 (s, 1H), 7.78 (s, 1H), 7.72 (d, J= 7.8 Hz, 1H), 7.34- 7.29 (m, 1H), 7.22 (s, 1H). 7. 14 (d, J = 7.2 Hz, 1H). 6.70 (d, J = 2. 1 Hz, 1H). 3.90-3.87 (m, 4H). 3.37 (brs, 2H), 3.06-3.02 (m, 7H), 2.43 (s, 3H).
[0542] Synthesis of (S)-tert-butyl (2-methyl-l-(3-methyl-7-morpholino-5-(3-(m-tolyl)-lH- pyrazol-l-yl)-3H-imidazo[4, 5-b ]pyridin-2-yl)propyl)carbamate, GP45.4
[0543] To a solution of N2-methyl-4-morpholino-6-(3-(m-tolyl)-lH-pyrazol-l-yl)pyridine- 2,3-diamine (250 mg, 0.69 mmol) in EtOH (6.0 mL) at rt was added Na2S2C>4 (240 mg, 1.37 mmol) and (S)-tert-butyl (3 -methyl- l-oxobutan-2-yl)carbamate (207 mg, 1.03 mmol). The solution mixture was heated to 80 °C and stirred for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was concentrated. The resulting residue was diluted with water (10 rnL) and filtrated. The filter cake was purified by slurry in Et20 to provide (S)-lert-but l (2-methyl-l-(3-methyl-7-morpholino-5-(3-(m-tolyl)-lH-pyrazol- l-yl)-3H-imidazo[4,5-b]pyridin-2-yl)propyl)carbamate (130 mg, 0.24 mmol) as a white solid. LC-MS (ESI+): m/z 546 (MH+). 1HNMR (300 MHz, CD3OD) £8.67 (d, J= 1.8 Hz, 1H), 7.78 (s, 1H), 7.72 (d, J= 8.1 Hz, 1H), 7.37-7.30 (m, 2H), 7.20 (d, J= 8.4 Hz, 1H), 6.87 (d, J= 2.4
Hz, 1H), 4.05-3.95 (m, 4H), 3.94-3.90 (m, 4H), 3.88 (s, 3H), 2.44 (s, 3H), 2.38-2.36 (m, 1H), 1.50-1.40 (m, 10 H), 1.13 (d, J= 6.3 Hz, 3H), 1.13 (d, J= 6.6 Hz, 3H).
[0544] Synthesis of (S)-2-methyl-l-(3-methyl-7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)- 3H-imidazo[4, 5-b ]pyridin-2-yl)propan- 1 -amine, Compound 238
[0545] To a solution of (S)-tert-butyl (2 -methyl- 1 -(3-methyl-7-morpholino-5-(3-(m-tolyl)- 1H- pyrazol-l-yl)-3H-imidazo[4,5-b]pyridin-2-yl)propyl)carbamate (130 mg, 0.24 mmol) in DCM (5.0 mL) at 0 °C was added TFA (1.0 mL). The solution mixture was stirred at rt for 3 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was quenched with aqueous NaHCCh solution and adjusted the pH to 9. The mixture was extracted with DCM (3 x 20 mL). The combined organic phases were dried over anhydrous Na2SO4, filtrated and concentrated. The resulting residue was purified by silica gel column chromatography with a gradient elution of 1% MeOH/DCM to 2% MeOH/DCM to provide (S)-2-methyl-l-(3-methyl-7- morpholino-5 -(3 -(m-tolyl)- 1 H-py razol- 1 -yl)-3H-imidazo [4, 5-b] pyridin-2-y l)propan- 1 -amine (100 mg, 0.22 mmol) as a white solid. LC-MS (ESI+): m/z 446 (MH+). 1HNMR (300 MHz, CDCh) 8.61 (d, J = 2.1 Hz, 1H), 7.78 (s, 1H), 7.73 (d, J= 7.5 Hz, 1H), 7.39-7.25 (m, 2H), 7.16 (d, J= 6.9 Hz, 1H), 6.75 (d, J= 2.4 Hz, 1H), 4.11-3.86 (m, 9H), 3.71(s, 3H), 2.43 (s, 3H), 2.23-2.06 (m, 1H), 1.05-0.97 (m, 6H).
[0546] Synthesis of(S)-N-(2-methyl-l-(3-methyl-7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l- yl)-3H-imidazo[ 4, 5-b ]pyridin-2-yl)propyl)acetamide, Compound 239
[0547] To a solution of ((S)-2-methyl-l-(3-methyl-7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l- yl)-3H-imidazo[4,5-b]pyridin-2-yl)propan-l-amine (80 mg. 0.18 mmol) in DCM (5.0 mL) at 0 °C was added TEA (55 mg, 0.54 mmol) and acetyl chloride (28 mg, 0.36 mmol) in DCM (1 mL) dropwise. The solution mixture was stirred at rt for 1 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture w as diluted with w ater (10 mL) and extracted with DCM (3 x 20 mL). The combined organic phases were dried over anhydrous Na2SO4, filtrated and concentrated. The resulting residue was purified by slurry in hexene to provide (S)-N-(2-methyl-l-(3-methyl-7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-b]pyridin-2-yl)propyl)acetamide (60 mg, 0.12 mmol) as a white solid. LC-MS (ESI+): m/z 488 (MH+). 1HNMR (300 MHz, CDCh) £8.61 (s, 1H), 7.78 (s, 1H). 7.73 (d, J= 7.5 Hz. 1H), 7.39-7.25 (m. 2H), 7.16 (d. J= 7.2 Hz. 1H), 6.76 (d. J= 2.4 Hz. 1H), 6.20 (d. J=7.8 Hz, 1H), 5. 16 (t, J = 8.4 Hz, 1H), 4.14- 3.86 (m, 8H), 3.85 (s, 3H), 2.44 (s, 3H), 2.43-2.22 (m, 1H), 2.05 (s, 3H), 0.96 (d, J= 6.6 Hz, 3H), 0.89 (d, J= 6.6 Hz, 3H).
[0548] General procedure 46:
[0549] Synthesis of Compound 240: 3-(cyclopropylmethyl)-7-morpholino-5-[l-(m-tolyl)- 3-pyrazolyl]-3H-l, 3, 4- tri azaindene

[0551] To a solution of (E)-3-ethoxyacrylic acid (5.0 g, 43. 1 mmol) in DCM (50.0 mL) at rt was added TEA (13.0 g, 129.3 mmol), HATU (33.0 g, 86.8 mmol) and m-tolylhydrazine (13.0 g, 129.3 mmol). The solution mixture was stirred at rt for 4 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was diluted with water (10 mL) and extracted with EtOAc (3 x 50 mL). The combined organic phases was dried over anhydrous Na2SC>4, filtrated and concentrated. The resulting residue was purified by silica gel column chromatography with a gradient elution of 20% EtOAc/PE to 50% EtOAc/PE to provide (E)-3- methoxy-N'-(m-tolyl)acrylohydrazide (5.0 g, 24.3 mmol) as a yellow oil. LC-MS (ESI+): m/z 207 (MH+).
[0552] Synthesis of l-(m-tolyl)-lH-pyrazol-3-ol, GP46.2
[0553] The mixture of (E)-3-methoxy-N'-(m-tolyl)acrylohydrazide (5.0 g, 24.3 mmol) in 12 M HC1 (60 mL) w as heated to reflux for Ih. After completion of the reaction as indicated by TLC analysis, the reaction mixture was quenched with aqueous NaHCCL solution and adjusted the pH to 9. The mixture was extracted with EtOAc (3 x 20 mL). The combined organic phases w ere dried over anhydrous Na2SO4, filtered and concentrated to provide crude l-(m-tolyl)-lH- pyrazol-3-ol (5 g, 28.7 mmol) as yellow oil. LC-MS (ESI+): m/z 175 (MH+).
[0554] Synthesis of l-(m-tolyl)-lH-pyrazol-3-yl trifluoromethanesulfonate, GP46.3
[0555] To a solution of crude l-(m-tolyl)-lH-pyrazol-3-ol (5 g, 28.7 mmol) in DCM (50.0 mL) at 0 °C was added DIEA (7.4 g, 57.4 mmol) and TfzO (9.7 g, 34.44 mmol) dropwise. The solution mixture was stirred at rt for 2 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was quenched with aqueous NaHCOs solution and adjusted the pH to 9. The mixture was extracted with EtOAc (3 x 50 mL). The combined organic phases were dried over anhydrous Na2SO4, filtrated and concentrated. The resulting residue was purified by
silica gel column chromatography with a gradient elution of 5% EtOAc/PE to 20% EtOAc/PE to provide l-(m-tolyl)-lH-pyrazol-3-yl trifluoromethanesulfonate (5.0 g, 16.3 mmol) as lightyellow oil. LC-MS (ESI+): m/z 307 (MH ).
[0556] Synthesis of (l-(m-tolyl)-lH-pyrazol-3-yl)boronic acid, GP46.4
[0557] To a solution of l-(m-tolyl)-lH-pyrazol-3-yl trifluoromethanesulfonate (2.0 g, 6.5 mmol) in 1,4-dioxane (30.0 mL) was added 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi(l,3,2- dioxaborolane) (1.83 g, 7.2 mmol), Pd(PPhs)4 (376 mg, 0.32 mmol), KBr (855 mg, 7.2 mmol) and KOAc (1.9 g, 19.6 mmol). The reaction mixture was purged with N2 for three times and heated to reflux overnight. After completion of the reaction as indicated by LC-MS analysis, the reaction mixture was concentrated. The resulting residue was purified by flash column purification with an elution of 10% MeOH/EtOAc to provide crude (l-(m-tolyl)-lH-pyrazol-3- yl)boronic acid (3.0 g, 14.85 mmol). LC-MS (ESI+): m/z 203 (MH ).
[0558] Synthesis of 4-(3-(2, 4-dimethoxybenzyl)-5-( 1 -(m-tolyl)-lH-pyrazol-3-yl)-3H- imidazo[4, 5-b Jpyridin-7-yl)morpholine. GP46.5
[0559] To a solution of 4-(5-chloro-3-(2,4-dimethoxybenzyl)-3H-imidazo[4,5-b]pyridin-7- yl)morpholine (1.3 g, 3.35 mmol) in 1,4-dioxane (30.0 mL) was added (l-(m-tolyl)-lH-pyrazol- 3-yl)boronic acid (1.3 g, 5.03 mmol), Pd(PPhs)4 (380 mg, 0.34 mmol) and K2CO3 (1.4 g. 10.0 mmol). The reaction mixture was purged with N2 for three times and heated to reflux for 2 h. After completion of the reaction as indicated by LC-MS analysis, the reaction mixture was concentrated. The resulting residue was purified by silica gel column chromatography with a gradient elution of 10% EtOAc/PE to 50% EtOAc/PE to provide 4-(3-(2,4-dimethoxybenzyl)-5- (l-(m-tolyl)-lH-pyrazol-3-yl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (800 mg, 1.57 mmol). LC-MS (ESI+): m/z 511 (MH+).
[0560] Synthesis of 4-(5-(l-(m-tolyl)-lH-pyrazol-3-yl)-3H-imidazo[4,5-b]pyridin-7- yl) morpholine, GP46. 6
[0561] To a solution of 4-(3-(2,4-dimethoxybenzyl)-5-(l-(m-tolyl)-lH-pyrazol-3-yl)-3H- imidazo[4,5-b]pyridin-7-yl)morpholine (800 mg, 1.57 mmol) in DCM (20.0 mL) at 0 °C was added TFA (4 mL). The solution mixture was stirred at rt for 3 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was quenched with aqueous NaHCCh solution and adjusted the pH to 9. The mixture was extracted with DCM/MeOH (3 x 30 mL. 15: 1). The combined organic phases were dried over anhydrous Na2SC>4, filtrated and concentrated. The resulting residue was purified by silica gel column chromatography with a gradient elution of 20% EtOAc/PE to 50% EtOAc/PE to provide 4-(5-(l-(m-tolyl)-lH-pyrazol- 3-yl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (600 mg, 1.67 mmol) as a white solid. LC-MS (ESI+): m/z 361 (MH+).
[0562] Synthesis of 4-(3-(cyclopropylmethyl)-5-(l-(m-tolyl)-lH-pyrazol-3-yl)-3H-imidazo[4,5- b]pyridin-7-yl)morpholine, Compound 240
[0563] To a solution of 4-(5-(l-(m-tolyl)-lH-pyrazol-3-yl)-3H-imidazo[4,5-b]pyridin-7- yl)morpholine (50 mg, 0.14 mmol) in DMF (5.0 mL) was added CS2CO3 (50 mg. 0.14 mmol) and (bromomethyl)cyclopropane (38 mg, 0.28 mmol). The solution mixture was stirred at 80 °C for 2 h. After completion of the reaction as indicated by TLC analysis, the reaction mixture was diluted with water (20 mL) and extracted with EtOAc (3 x 20 mL). The combined organic phases were dried over anhydrous Na2SO4, filtered, and concentrated. The resulting residue was purified by preparative TLC with an elution of 50% EtOAc/PE to provide 4-(3- (cyclopropylmethyl)-5-(l-(m-tolyl)-lH-pyrazol-3-yl)-3H-imidazo[4,5-b]pyri din-7- yl)morpholine (27.6 mg, 0.067 mmol) as a white solid. LC-MS (ESI+): m/z 415 (MH+). 1EINMR (300 MHz, CDCh) 7.97 (s, 2H), 7.65 (s, 1H), 7.57(d, J= 7.2 Hz, 1H), 7.36-7.33 (m, 2H), 7.15-7.11 (m, 2H). 4.17 (d, J= 7.2 Hz, 2H). 3.97-3.90 (m, 8H). 2.46 (s. 3H), 1.41- 1.35 (m, 1H). 0.69-0.61 (m, 2H), 0.52-0.50 (m, 2H).
[0564] General procedure 47:
[0565] Synthesis of Compound 252: 3-ethyl-5-[5-(methoxymethyl)-3-(m- tolyl)- 1- pyrazolyl]-7-morpholino-2-[(l-pyrazolyl)methyl]-3H-l, 3, 4- triazaindene
 [0567] To a solution of 4-(2,6-dichloro-3-nitropyridin-4-yl)morpholine (4.6 g, 16.61 mmol) in DMF (50 rnL) at rt was added K2CO3 (6.87 g, 49.82 mmol) and ethanamine hydrochloride (2.01 g, 24.92 mmol). The reaction mixture was stirred at rt overnight. After completion of the reaction as indicated by TLC analysis, the reaction mixture was diluted with water (100 mL) and extracted with EtOAc (3 x 50 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 2% EtOAc/PE to 7% EtOAc/PE to provide 6-chloro-N-ethyl-4-morpholino-3-nitropyridin-2-amine (3.22 g, 11.25 mmol) as a yellow solid. LC-MS (ESI+): m/z 439/441 (MH+).
[0567] To a solution of 4-(2,6-dichloro-3-nitropyridin-4-yl)morpholine (4.6 g, 16.61 mmol) in DMF (50 rnL) at rt was added K2CO3 (6.87 g, 49.82 mmol) and ethanamine hydrochloride (2.01 g, 24.92 mmol). The reaction mixture was stirred at rt overnight. After completion of the reaction as indicated by TLC analysis, the reaction mixture was diluted with water (100 mL) and extracted with EtOAc (3 x 50 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 2% EtOAc/PE to 7% EtOAc/PE to provide 6-chloro-N-ethyl-4-morpholino-3-nitropyridin-2-amine (3.22 g, 11.25 mmol) as a yellow solid. LC-MS (ESI+): m/z 439/441 (MH+).
[0568] Synthesis of ethyl l-(6-(ethylamino)-4-morpholmo-5-nitropyridm-2-yl)-3-(m-tolyl)-lH- pyrazole-5-carboxylate, GP47.2
[0569] To a solution of 6-chloro-N-ethyl-4-morpholino-3-nitropyridin-2-amine (700 mg, 2.44 mmol) in 1,4-dioxane (15 mL) was added ethyl 3-(m-tolyl)-lH-pyrazole-5-carboxylate (673 mg, 2.92 mmol), CS2CO3 (2.34 g, 7.32 mmol), t-Buxphos (414 mg, 0.98 mmol) and Pd2(dba)s (447 mg, 0.49 mmol). The reaction mixture was purged with N2 for three times and stirred at 95 °C for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was diluted with water (30 mL) and extracted with EtOAc (3 x 30 mL). The combined organic phases were dried over anhydrous Na2SO-i. filtrated and concentrated. The resulting residue was purified by silica gel column chromatography with a gradient elution of 5% EtOAc/PE to 25% EtOAc/PE to provide ethyl l-(6-(ethylamino)-4-morpholino-5-nitropyridin-2- yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate (520 mg, 1.08 mmol) as ayellow solid. LC-MS (ESI+): m/z 481 (MH ).
[0570] Synthesis of ethyl l-(5-amino-6-(ethylamino)-4-morpholinopyridin-2-yl)-3-(m-tolyl)- lH-pyrazole-5-carboxylate, GP47.3
[0571] To a solution of ethyl l-(6-(ethylamino)-4-morpholino-5-nitropyridin-2-yl)-3-(m- tolyl)-lH-pyrazole-5-carboxylate (520 mg, 1.08 mmol) in DCM/MeOH (10 mL/10 mL) was added NiCh (51 mg. 0.22 mmol) and NaBEk (82 mg. 2. 16 mmol). The reaction mixture was stirred at 0 °C for 0.5 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with water (20 mL) and extracted with MeOH/DCM (3 x 20 mL, 1 : 15). The combined organic phases were dried over anhydrous Na2SO4, filtrated and concentrated. The resulting residual was purified by silica gel column chromatography with a gradient elution of 20% EtOAc/PE to 50% EtOAc/PE to provide ethyl 1- (5-amino-6-(ethylamino)-4-morpholinopyri din-2 -yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate (450 mg, 1.0 mmol) as a yellow solid. LC-MS (ESI+): m/z 451 (MH ).
[0572] Synthesis of ethyl l-(5-amino-6-(2-chloro-N-ethylacetamido)-4-morpholinopyridin-2- yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate, GP47.4
[0573] To a solution of ethyl l-(5-arrnno-6-(ethylamino)-4-morpholinopyridin-2-yl)-3-(m- tolyl)-lH-pyrazole-5-carboxylate (450 mg, 1.0 mmol) in DCM (25.0 mL) at 0 °C was added TEA (202 mg, 2.0 mmol) and 2-chloroacetyl chloride (123 mg. 1.1 mmol) in DCM (1 mL) dropwise. The solution mixture was stirred at rt for 1 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was concentrated directly. The resulting residual was purified by silica gel column chromatography with a gradient elution of 10% EtOAc/PE to 33% EtOAc/PE to provide ethyl l-(5-amino-6-(2-chloro-N-ethylacetamido)- 4-morpholinopyridin-2-yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate (350 mg, 0.66 mmol) as a yellow oil. LC-MS (ESI+): m/z 527/529 (MH+).
[0574] Synthesis of ethyl l-(2-(chloromethyl)-3-ethyl-7-morpholino-3H-imidazo[4,5- b ]pyridin-5-yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate, GP47.5
[0575] To a solution of ethyl l-(5-amino-6-(2-chloro-N-ethylacetamido)-4- morpholinopyridin-2-yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate (350 mg, 0.66 mmol) in 1,4- di oxane (15.0 mL) was added one drop of concentrated H2SO4. The solution mixture was stirred at 70 °C for 1 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was concentrated directly. The resulting residual was purified by silica gel column chromatography with a gradient elution of 10% EtOAc/PE to 33% EtOAc/PE to provide ethyl l-(2-(chloromethyl)-3-ethyl-7-morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-3-(m-tolyl)- lH-pyrazole-5-carboxylate (220 mg, 0.43 mmol) as a white solid. LC-MS (ESI+): m/z 509/511 (MH ). XH NMR (300 MHz. CDCh) 7.74 (s, 1H). 7.67 (d, J= 7.2 Hz, 1H). 7.35-7.30 (m, 1H). 7.19-7.17 (m, 2H), 6.87 (s, 1H), 4.80 (s, 2H), 4.34-4.27 (m, 4H), 3.97-3.92 (m, 4H), 3.90-3.83 (m, 4H), 2.45 (s, 3H), 1.47 (t, J= 7.5 Hz, 3H), 1.27 (t, J= 7.2 Hz, 3H).
[0576] Synthesis of ethyl l-(2-( ( lH-pyrazol-l-yl)methyl)-3-ethyl- 7 -morpholino- 3H- imidazo[4, 5-b ]pyndm-5-yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate, GP47.6
[0577] To a solution of ethyl l-(2-(chloromethyl)-3-ethyl-7-morpholino-3H-imidazo[4,5- b]pyridin-5-yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate (220 mg, 0.43 mmol) in DMF (20 mL) was added K.2CO3 (120 mg, 0.87 mmol) and IH-pyrazole (32 mg, 1.1 mmol). The reaction mixture was stirred at rt overnight. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with water (40 mL) and extracted with EtOAc (3 x 20 mL). The combined organic phases were dried over anhydrous Na2SC>4, filtrated and concentrated. The resulting residual was purified by silica gel column chromatography with a gradient elution of DCM to 3% MeOH/DCM to provide ethyl l-(2-((lH-pyrazol-l-yl)methyl)-
3-ethyl-7-morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate (160 mg, 0.30 mmol) as a yellow oil. LC-MS (ESI+): m/z 541 (MH+).
[0578] Synthesis of (l-(2-((lH-pyrazol-l-yl)methyl)-3-ethyl-7-morpholino-3H-imidazo[4,5- b]pyridin-5-yl)-3-(m-tolyl)-lH-pyrazol-5-yl)methanol, GP47.7
[0579] To a solution of ethyl l-(2-((lH-pyrazol-l-yl)methyl)-3-ethyl-7-morpholino-3H- imidazo[4,5-b]pyridin-5-yl)-3-(m-tolyl)-lH-pyrazole-5-carboxylate (160 mg, 0.30 mmol) in anhydrous THF (10 mL) at 0 °C was added LiAlF (17 mg, 0.44 mmol) in portions. The reaction mixture was wormed to rt and stirred for 1 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with water (10 mL) and extracted with DCM/MeOH (3 x 20 mL, 10:1). The combined organic phases were dried over anhydrous Na2SO4, filtrated and concentrated. The resulting residual was purified by preparative TLC with an elution of 5% MeOH/DCM to provide (l-(2-((lH-pyrazol-l- yl)methyl)-3-ethyl-7-morpholino-3H-imidazo[4,5-b]pyridin-5-yl)-3-(m-tolyl)-lH-pyrazol-5- yl)methanol (74 mg, 0.15 mmol) as a white solid. LC-MS (ESI+): m/z 499 (MH+).
[0580] Synthesis of 4-(2-( ( lH-pyrazol-l-yl)methyl)-3-ethyl-5-(5-(methoxymethyl)-3-(m-tolyl)- lH-pyrazol-l-yl)-3H-imidazo[4, 5-b ]pyridin-7-yl)morpholine, Compound 252
[0581] To a solution of (l-(2-((lH-pyrazol-l-yl)methyl)-3-ethyl-7-morpholino-3H- imidazo[4,5-b]pyridin-5-yl)-3-(m-tolyl)-lH-pyrazol-5-yl)methanol (74 mg, 0.15 mmol) in DMF/THF (6 mL, 1:5) at 0 °C was added NaH (18 mg, 60%, 0.45 mmol) and CH3I (63 mg, 0.45 mmol). The reaction mixture was wormed to 40 °C and stirred for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with water (10 mL) and extracted with EtOAc (3 x 20 mL). The combined organic phases were dried over anhydrous Na2SO4, filtered, and concentrated. The resulting residual was purified by silica gel column chromatography with a gradient elution of DCM to 3% MeOH/DCM to provide 4- (2-((lH-pyrazol-l-yl)methyl)-3-ethyl-5-(5-(methoxymethyl)-3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-b]pyridin-7-yl)morpholine (30.5 mg, 0.06 mmol) as a yellow solid. LC-MS (ESI+): m/z 513 (MH+). 1HNMR (300 MHz, CDCh) 7.78 (s, 1H), 7.73 (d, J= 7.5 Hz, 1H), 7.63-7.54 (m, 2H), 7.37-7.24 (m, 2H), 7.18 (d, J= 6.9 Hz, 1H), 6.83 (s, 1H), 6.34(s, 1H), 5.61 (s, 2H), 5.09 (s, 2H), 4.28-4.20 (m, 2H), 4.05-4.00 (m, 4H), 3.98-3.90 (m, 4H), 3.51(s, 3H), 2.44 (s, 3H), 1.19 (t, J= 7.2 Hz, 3H).
[0582] General procedure 48:
[0583] Synthesis of Compound 253: 3-ethyl-5-[5-(methoxymethyl)-3-(m- tolyl)- 1- pyrazolyl]-2-(3-methoxypropyl)-7-morpholino-3H-l,3?4-triazaindene

[0584] Synthesis of 4-(3-ethyl-5-(5-(methoxymethyl)-3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4, 5-b ]pyridin-7-yl)morpholine, GP48. 1
[0585] To a solution of 4-(5-(5-(methoxymethyl)-3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-b]pyridin-7-yl)morpholine (122 mg, 0.30 mmol) in DMF (10 mL) at rt was added
CS2CO3 (197 mg, 0.61 mmol) and bromoethane (56 mg, 0.36 mmol). The reaction mixture was stirred at rt for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with water (10 mL) and extracted with EtOAc (3 x 20 mL). The combined organic phases were dried over anhydrous Na2SO4, filtrated and concentrated. The resulting residual was purified by silica gel column chromatography with a gradient elution of 20% EtOAc/PE to 50% EtOAc/PE to provide 4-(3-ethyl-5-(5-(methoxymethyl)-3-(m-tolyl)- lH-pyrazol-l-yl)-3H-imidazo[4.5-b]pyridin-7-yl)morpholine (102 mg, 0.24 mmol) as a yellow solid. LC-MS (ESI+): m/z 433 (MH+).
[0586] Synthesis of 3-ethyl-5-(5-(methoxymethyl)-3-(m-tolyl)-lH-pyrazol-l-yl)-7-morpholino- 3H-imidazo[4, 5-b ] pyridine -2 -carb aldehyde, GP48.2
[0587] To a solution of 4-(3-ethyl-5-(5-(methoxymethyl)-3-(m-tolyl)-lH-pyrazol-l-yl)-3H- imidazo[4,5-b]pyridin-7-yl)morpholine (102 mg, 0.24 mmol) in THF (15 mL) at -78 °C under N2 was added n-BuLi (0.14 mL, 2.5M, 0.35 mmol) dropwise. After addition, the mixture was stirred at -78 °C for 1 h. To the above mixture was added a solution of DMF (19 mg, 0.26 mmol) in THF (1 mL) in one portion. The reaction mixture was stirred at that temperature for 1 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was quenched with NH4CI solution and extracted with EtOAc (3 x 20 mL). The combined organic phases were dried over anhydrous Na2SO4, filtered, and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography with a gradient elution of 20% EtOAc/PE to 50% EtOAc to provide 3-ethyl-5-(5-(methoxymethyl)-3-(m-tolyl)-lH- pyrazol-l-yl)-7-morpholino-3H-imidazo[4,5-b]pyridine-2-carbaldehyde (51 mg, 0.11 mmol) as a yellow solid. LC-MS (ESI+): m/z 461 (MH+).
[0588] Synthesis of4-(3-ethyl-5-(5-(methoxymethyl)-3-(m-tolyl)-lH-pyrazol-l-yl)-2-(3- methoxyprop-l-en-l-yl)-3H-imidazo[ 4, 5-b ]pyridin-7-yl)morpholine, GP48.3
[0589] To a suspension of 3-ethyl-5-(5-(methoxymethyl)-3-(m-tolyl)-lH-pyrazol-l-yl)-7- morpholino-3H-imidazo[4,5-b]pyridine-2-carbaldehyde (51 mg, 0.11 mmol) and bromolmethoxy ethyl)triphenylphosphorane (52 mg, 0.13 mmol) in DCM (5 mL) at rt was added a solution of NaOH (0.25 mL, 2 M) dropwise. The color of the reaction mixture was turned to a red suspension during the addition of aq. NaOH. The reaction mixture was stirred at rt for 3 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was concentrated under reduce pressure directly. The resulting residue was purified by preparative TLC with an elution of 50% EtOAc/PE to provide 4-(3 -ethyl -5 -(5 -(methoxy methyl)-
3-(m-tolyl)-lH-pyrazol-l-yl)-2-(3-methoxyprop-l-en-l-yl)-3H-imidazo[4,5-b]pyri din-7- yl)morpholine (38 mg, 0.076 mmol). LC-MS (ESI+): m/z 503 (MH+).
[0590] Synthesis of 4-(3-ethyl-5-(5-(methoxymethyl)-3-(m-tolyl)-lH-pyrazol-l-yl)-2-(3- methoxypropyl)-3H-imidazo[ 4, 5-b Jpyridin-7-yl)morpholine Compound 253
[0591] To a solution of 4-(3-ethyl-5-(5-(methoxymethyl)-3-(m-tolyl)-lH-pyrazol-l-yl)-2-(3- methoxyprop-l-en-l-yl)-3H-imidazo[4,5-b]pyridin-7-yl)morpholine (38 mg, 0.076 mmol) in DCM (10 mL) was added Pd/C (10 mg). The reaction mixture was purged with H2 for three times and stirred under H2 atmosphere for 2 h. After completion of the reaction as indicated by TLC and LC-MS analysis, the reaction mixture was filtered and concentrated directly. The resulting residue was purified by preparative TLC with an elution of 50% EtOAc/PE to provide
4-(3-ethyl-5-(5-(methoxymethyl)-3-(m-tolyl)-lH-pyrazol-l-yl)-2-(3-methoxypropyl)-3H- imidazo[4,5-b]pyridin-7-yl)morpholine (8.7 mg, 0.017 mmol) as a white solid. LC-MS (ESI+): m/z 505 (MH+). ’HNMR (300 MHz, CDCk) 7.77 (s, 1H), 7.71 (d, J= 6.6Hz, 1H), 7.34-7.29 (m, 1H), 7.18-7.14 (m, 2H), 6.81 (s, 1H), 5.09 (s, 2H), 4.24-4.19 (m, 2H), 4.02-3.94 (m, 8H), 3.54-3.50 (m, 5H), 3.38 (s. 3H), 2.92 (t, J= 7.5 Hz, 2H), 2.42 (s, 3H), 2.22-2.15 (m, 2H), 1.43 (t, J= 7.5 Hz. 3H).
[0592] Additional compounds were made by using appropriate starting material(s) in the general procedure identified in the table below.
Table 2














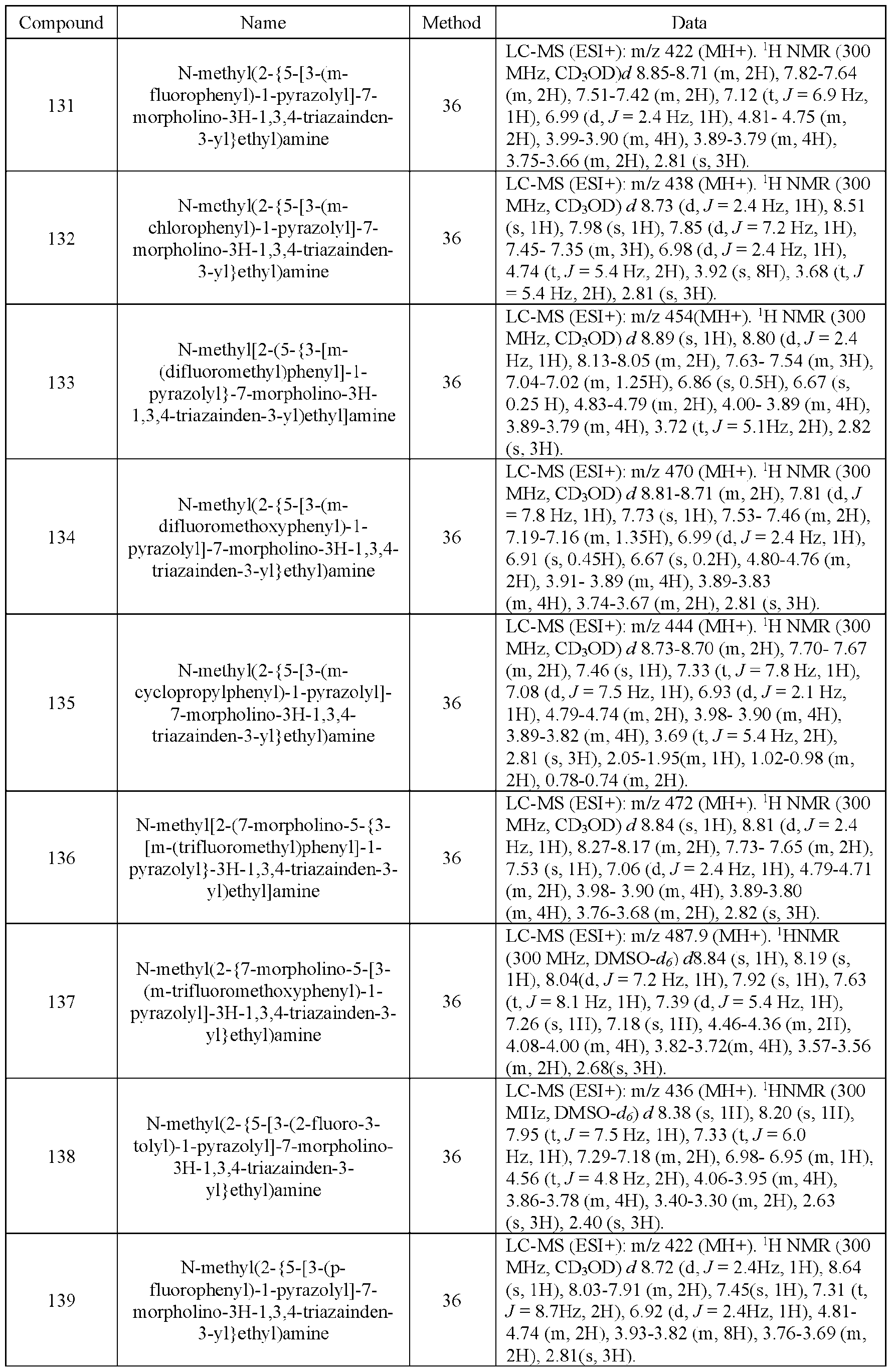



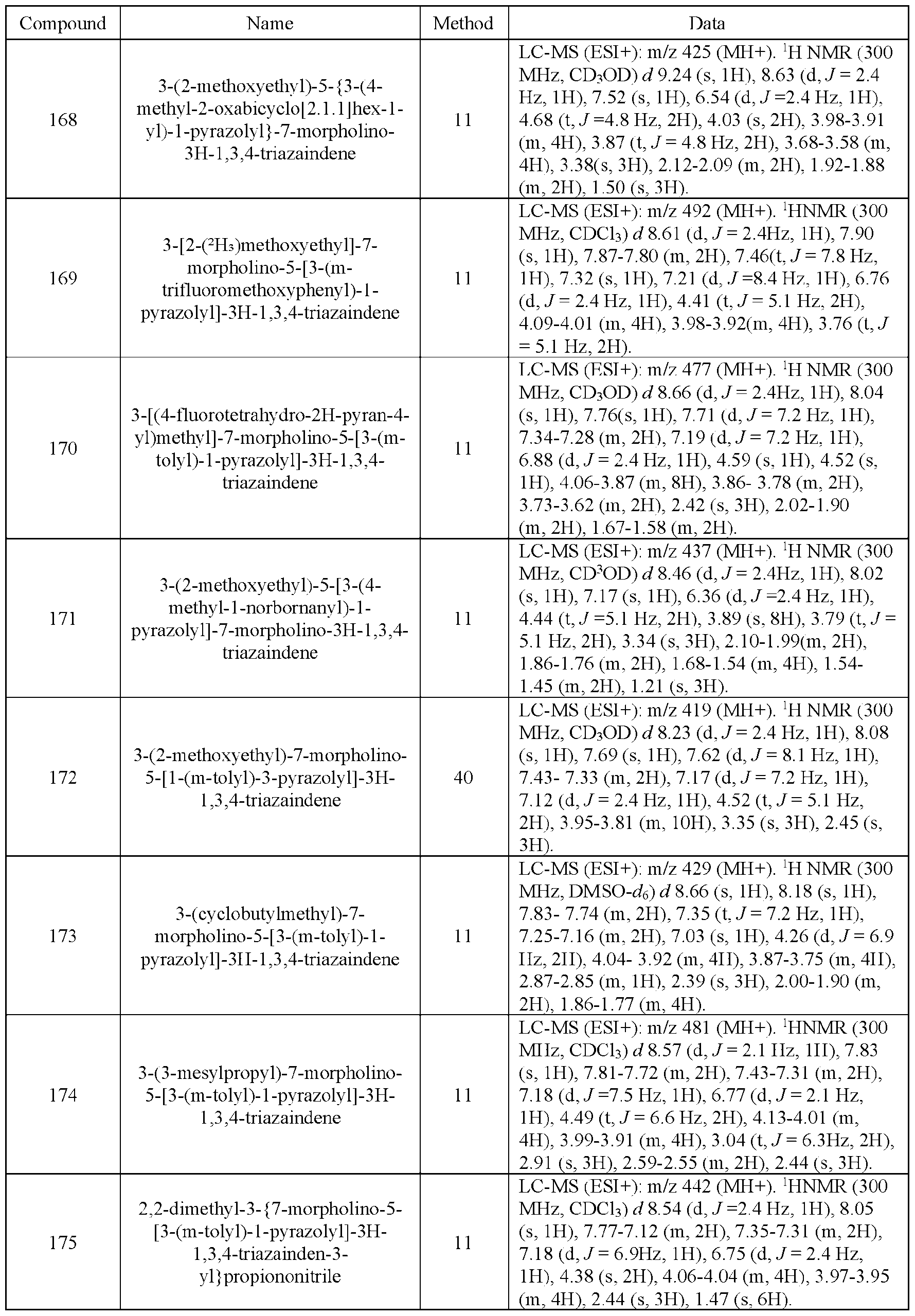

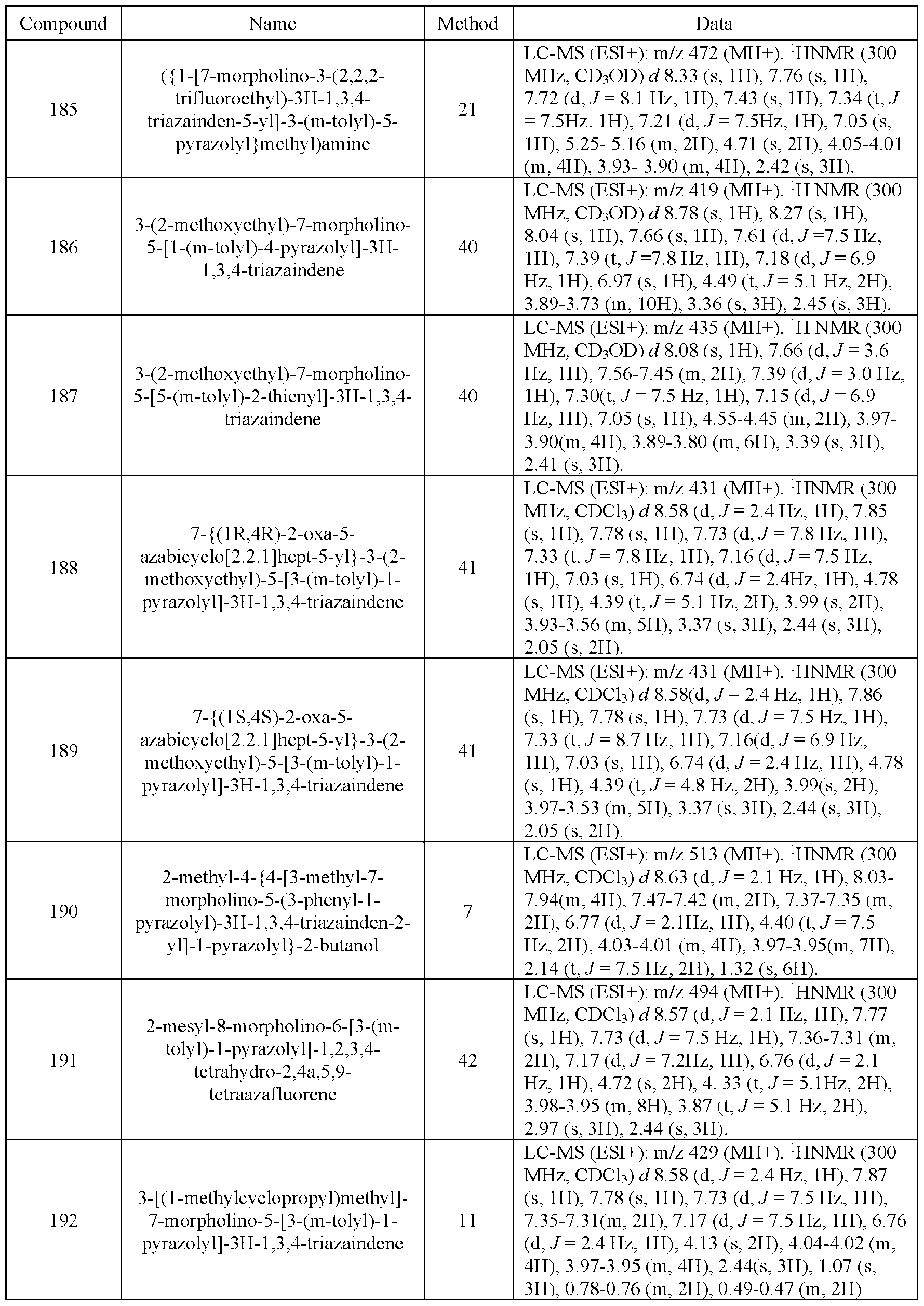

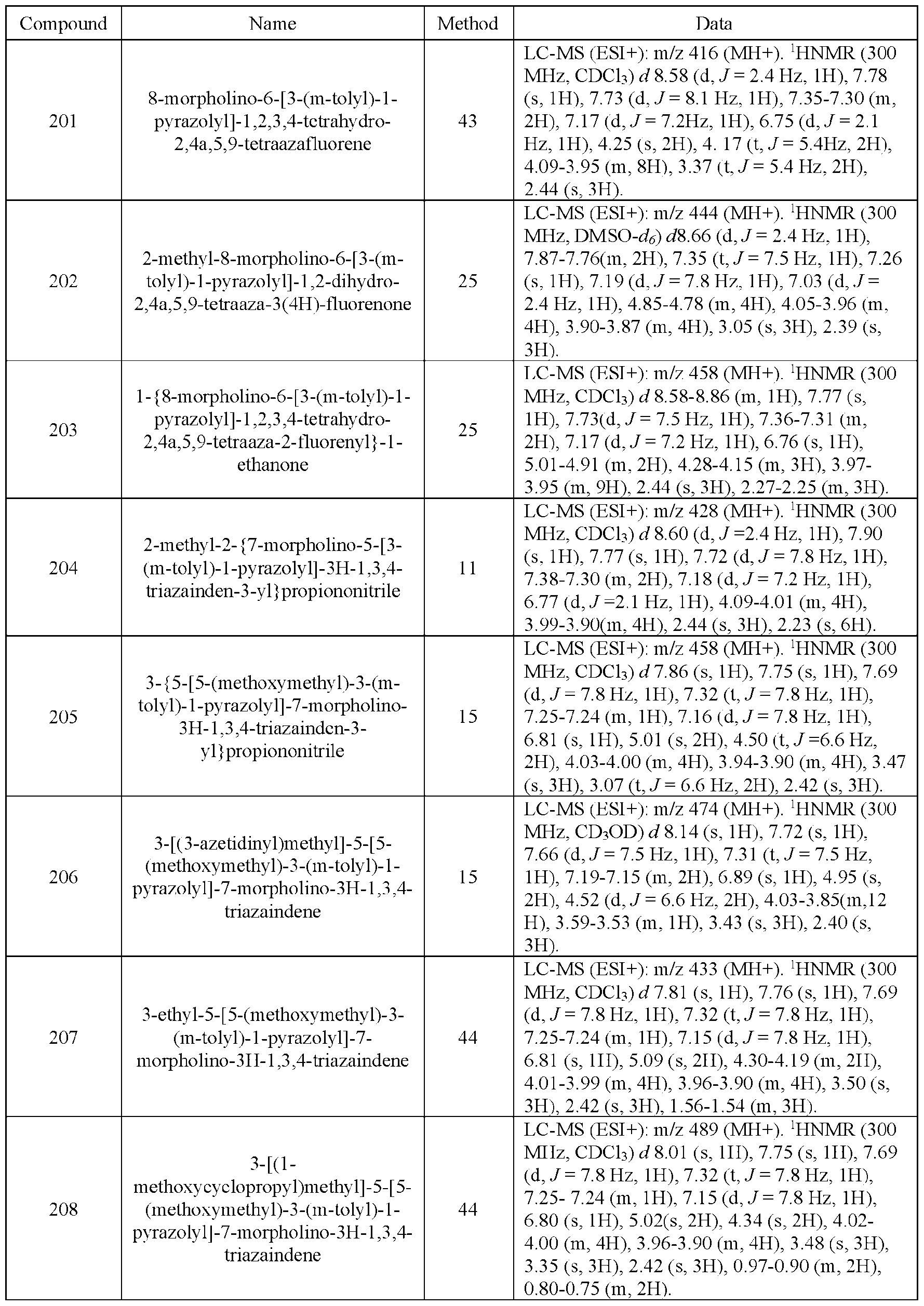


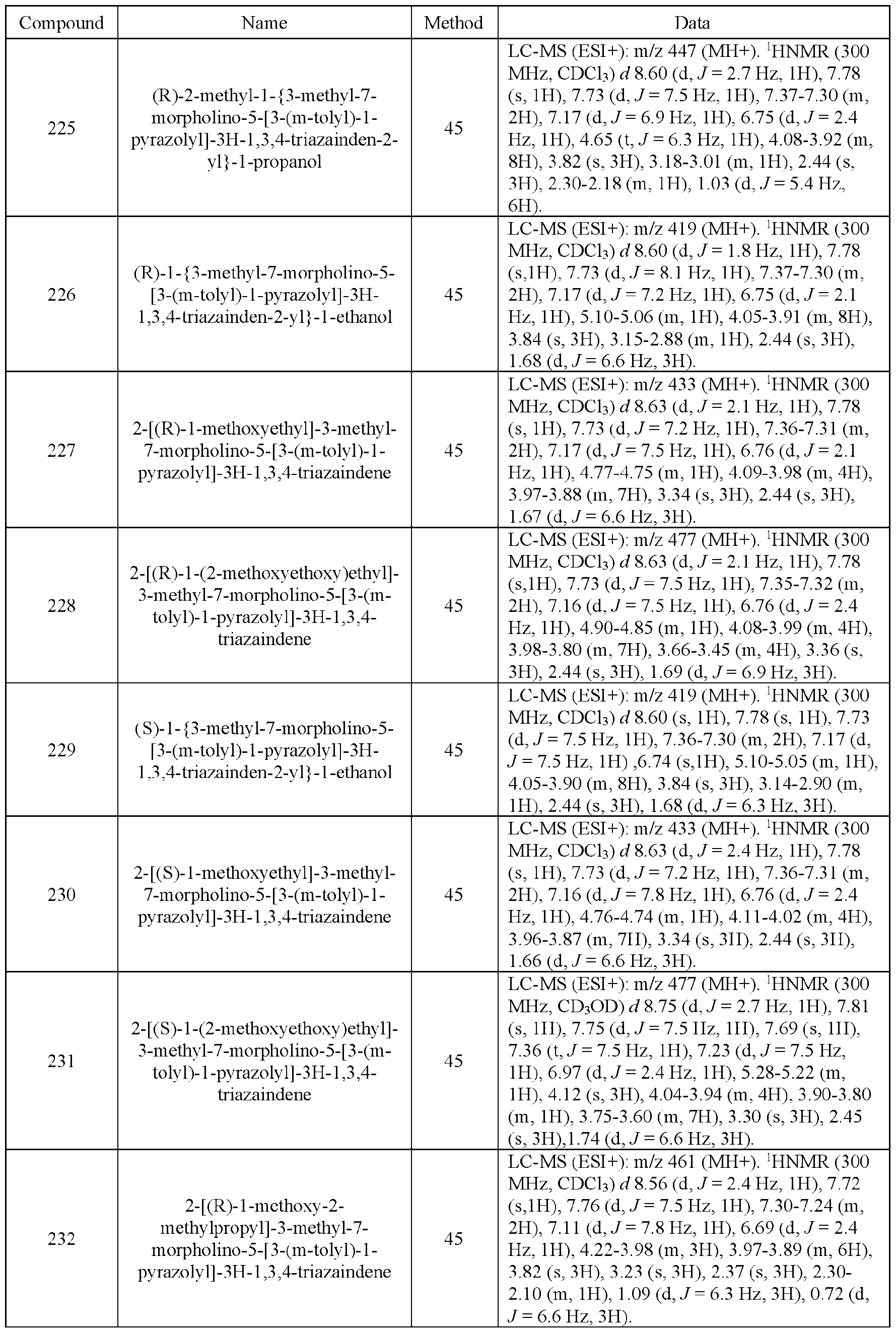



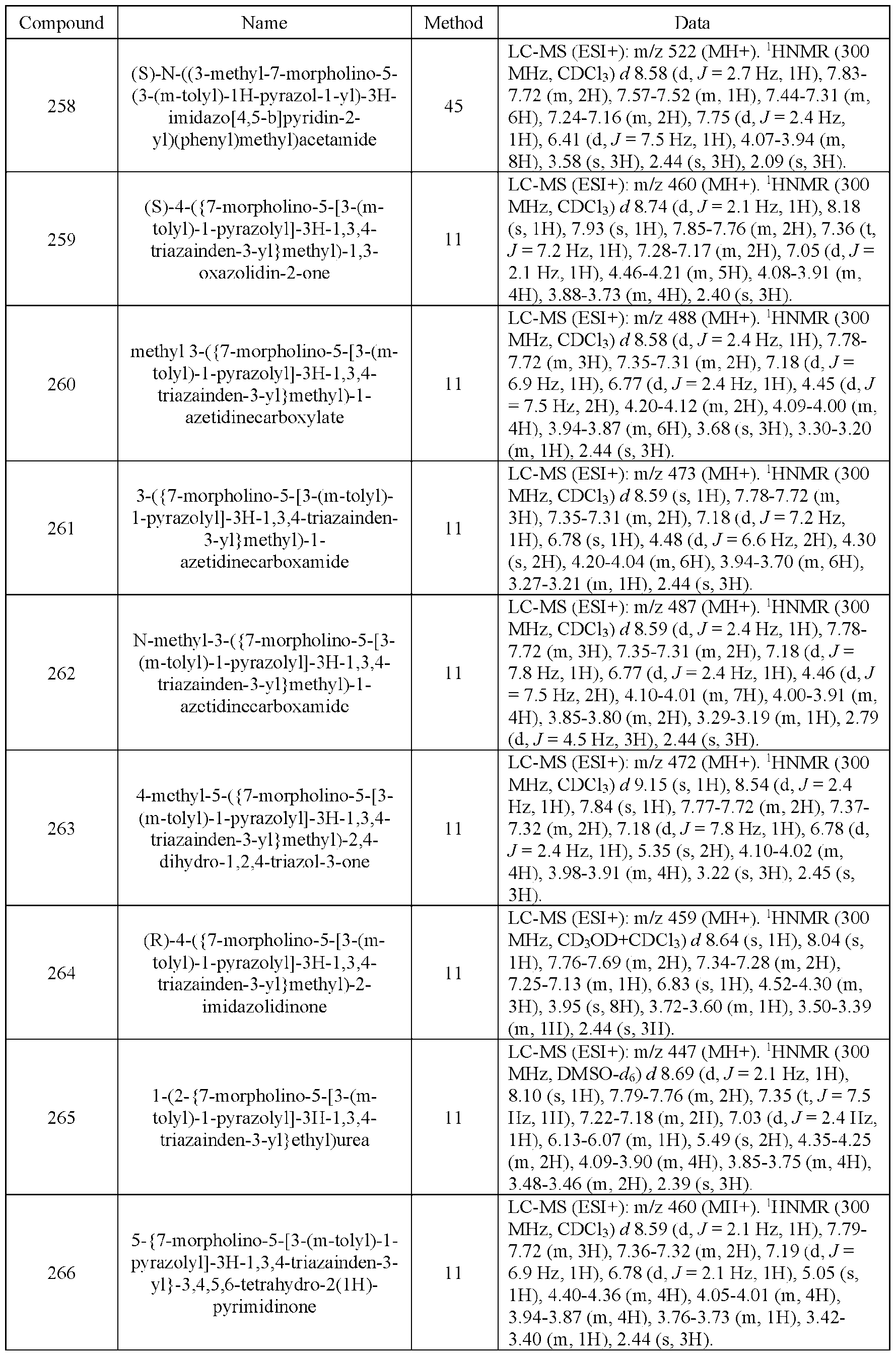
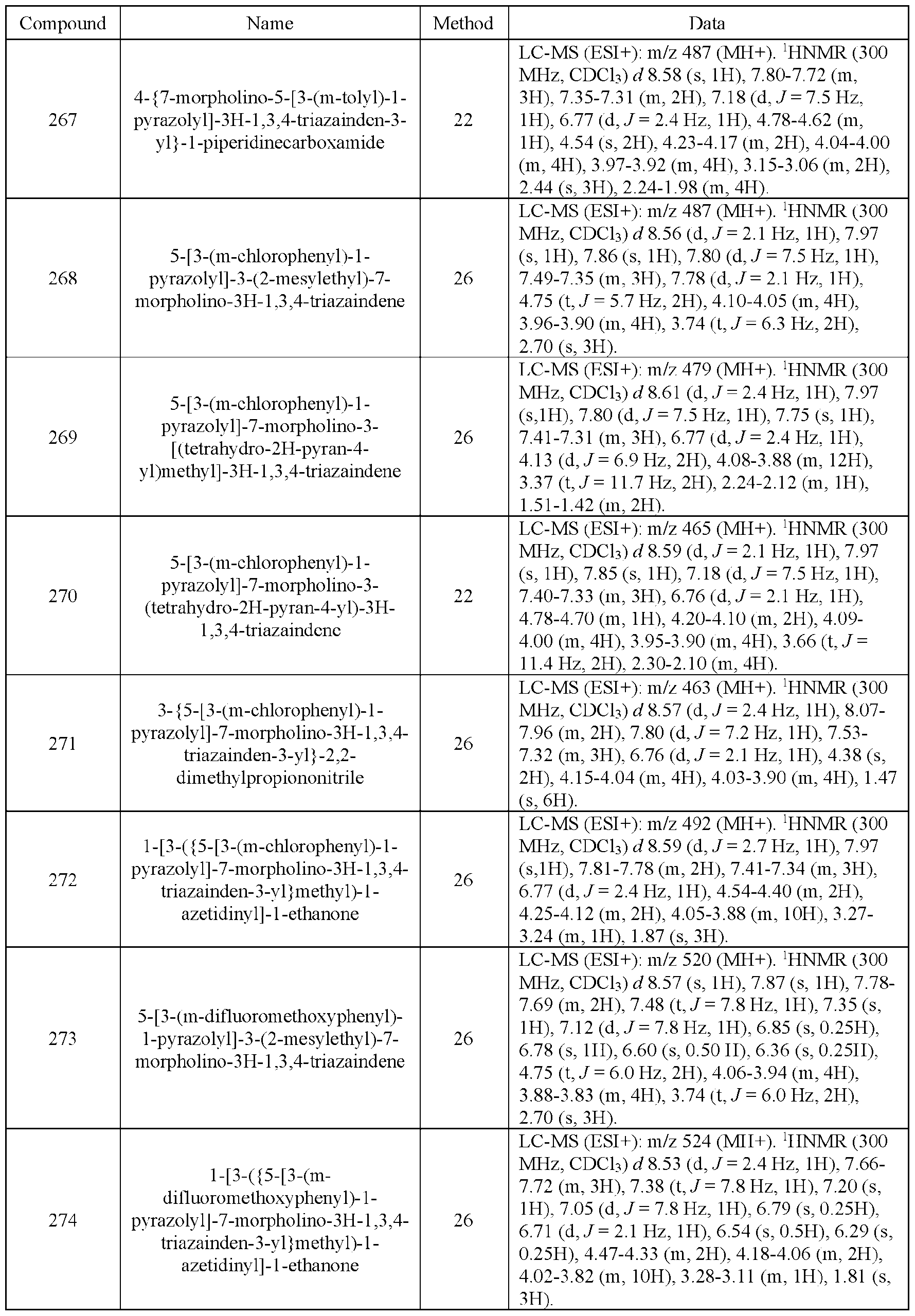
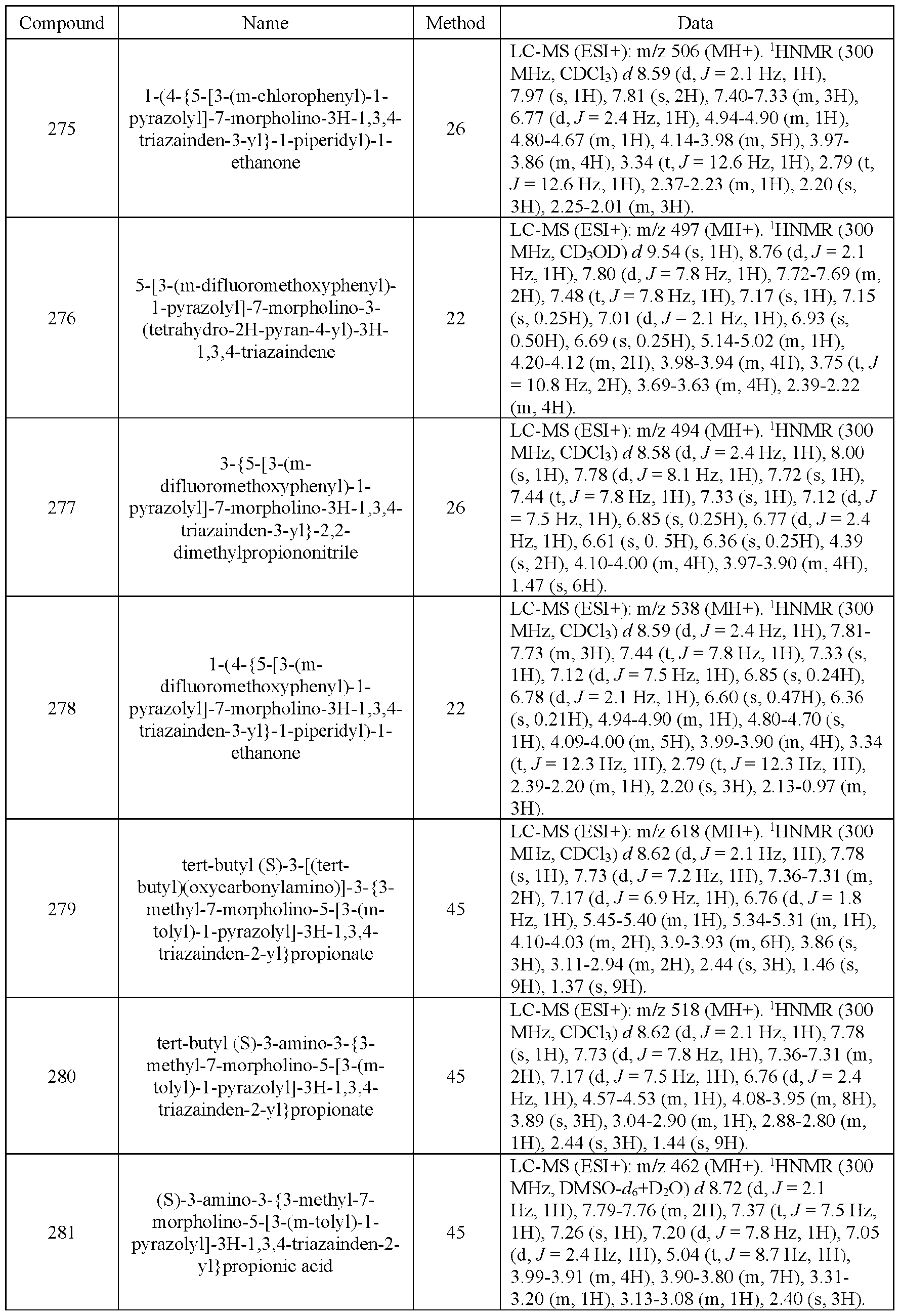






Biological Example 1: Inhibition of PIKfyve
[0593] Full length human recombinant PIKFYVE expressed in baculovirus expression system as N-termmal GST-fusion protein (265 kDa) was obtained from Cama Biosciences (Kobe, Japan). The kinase substrate was prepared by mixing and sonicating fluorescently-labeled phosphatidylinositol 3-phosphate (PI3P) with phospho-L-serine (PS) at a 1 : 10 ratio in 50 mM HEPES buffer pH7.5.
[0594] The kinase reactions were assembled in 384-well plates (Greiner) in a total volume of 20 mL as follows. Kinase protein was pre-diluted in an assay buffer comprising 25 mM HEPES, pH 7.5, 1 mM DTT, 2.5 mM MgCh, and 2.5 mM MnCh, and 0.005% Triton X-100, and dispensed into a 384-well plate (10 pL per well). Test compounds were serially pre-diluted in DMSO and added to the protein samples by acoustic dispensing (Labcyte Echo). The concentration of DMSO was equalized to 1% in all samples. All test compounds were tested at 12 concentrations. Apilimod was used as a reference compound and was tested in identical manner in each assay plate. Control samples (0%-inhibition, in the absence of inhibitor, DMSO only) and 100%-inhibition (in the absence of enzyme) were assembled in replicates of four and were used to calculate %-inhibition in the presence of compounds. The reactions were initiated by addition of 10 pL of 2x PI3P/PS substrate supplemented with ATP. The final concentration of enzyme was 2 nM, the final concentration of ATP was 10 mM, and the final concentration of PI3P/PS substrate was 1 pM (PI3P). The kinase reactions were allowed to proceed for 3 h at room temperature. Following incubation, the reactions were quenched by addition of 50 mL of termination buffer (100 mM HEPES, pH 7.5, 0.01% Triton X-100, 20 mM EDTA). Terminated
plates were analyzed on a microfluidic electrophoresis instrument (Caliper LabChip® 3000, Caliper Life Sciences/Perkin Elmer). The change in the relative fluorescence intensity of the PI(3)P substrate and PI(3,5)P product peaks was measured. The activity' in each test sample was determined as the product to sum ratio (PSR): P/(S+P), where P is the peak height of the product, and S is the peak height of the substrate. Percent inhibition (Pinh) was determined using the following equation:
Pinh = (PSR.0°/oinh - PSRcompound)/(PSRo%inh - PSR100%inh)* 100 in which PSRcompound is the product/sum ratio in the presence of compound. PSRo%inh is the product/sum ratio in the absence of compound, and the PSRioo%inh is the product/sum ratio in the absence of the enzyme. To determine the ICso of test compounds (50%-inhibition) the %-inh cdata (Pinh versus compound concentration) were fitted by a four-parameter sigmoid doseresponse model using XLfit software (IDBS).
[0595] The IC50 values for certain compounds of the disclosure are provided in the table below.
Table 4
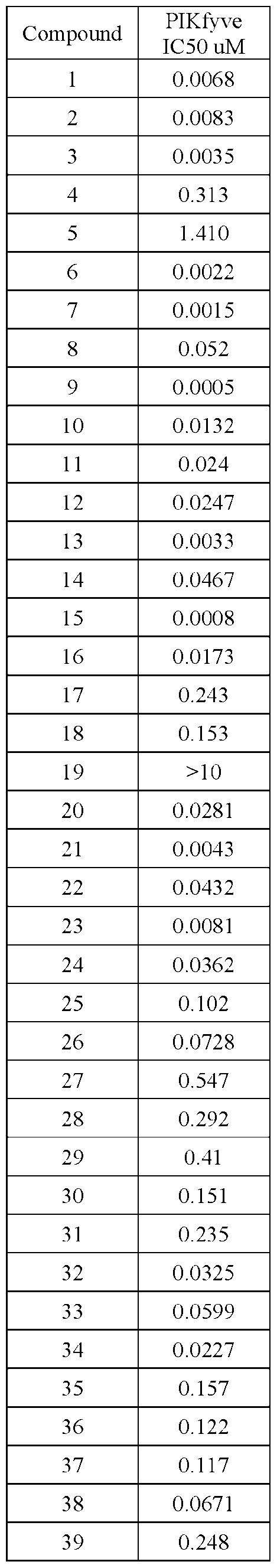
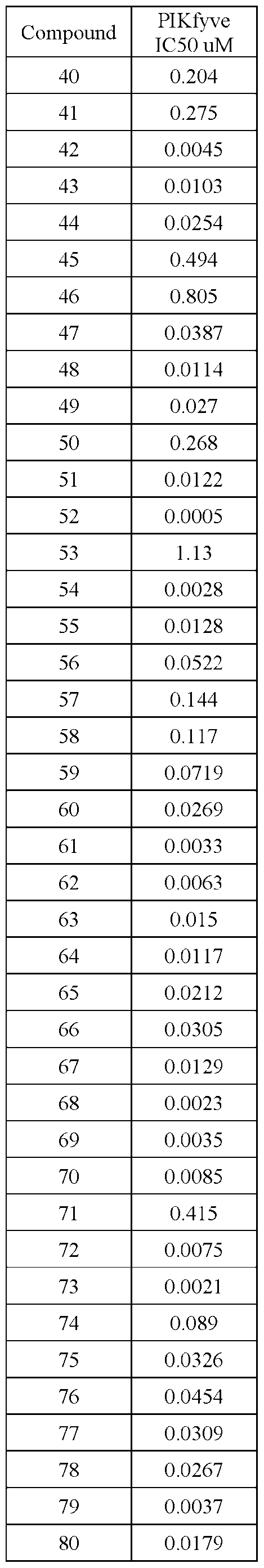
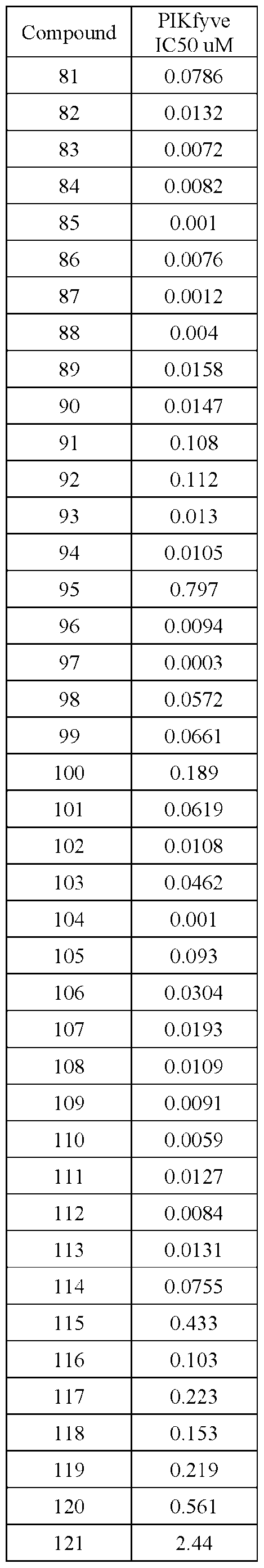




 Biological Example 2: Cell Data
Biological Example 2: Cell Data
[0596] HEK/TDP Survival assay:
[0597] Assay Description
[0598] Immortalized human embryonic kidney 293T (HEK 293T) were transfected with plasmids containing TDP-43 Q331K mutation, resulting in an increase in cell death that is biologically relevant to ALS patients. Cell death is measured as reductions in the amount of ATP, an indicator of metabolically active cells, that is quantified by a luminescence Cell-Titer- Glo® (CTG) reagent. Compounds are evaluated in this model for changes in CTG compared to no treatment group. Increased signal indicates improved survival (rescue) and decreased signal indicates decreased survival.
[0599] Reagent

[0600] Assay Procedure
[0601] HEK293T cells were passaged using 0.25% Trypsin-EDTA and plated at a density of 10,000 cells/well in 96- well tissue culture plates (inner 60 wells). The following day, the cells were transfected with TDP43-Q331K plasmid using Lipofectamine 3000 and treatment with compound was performed (9-point dose response curve, half-log serial dilution with lOuM as the highest concentration with 6 replicates). After 48 hours of incubation, CellTiter-Glo reagent was added, and the lysates were transferred to opaque white flat-bottom polystyrene plates.
Compound induced rescue of HEK cells with mutant TDP43 related viability deficit was measured using the CellTiter-Glo assay.
[0602] Analytical Method
[0603] CellTiter-Glo Luminescent Cell Viability Assay (Promega, G9243) was used to quantify ATP, an indicator of metabolically active cells. The luminescence signal was detected using the PerkinElmer EnVision. Measurements are recorded as relative luminescence units and normalized to negative control of HEK 293T cells without TDP43 transfection.
[0604] Data Analysis
[0605] Dose-response curve graphs were generated using GraphPad Prism. ECso values for compound treatment were determined using a four-parameter variable slope dose-response curve fitted to the Hill equation. Data point(s) of high concentration compound-induced toxicity resulting in decreased cellular viability was excluded.
Table 5
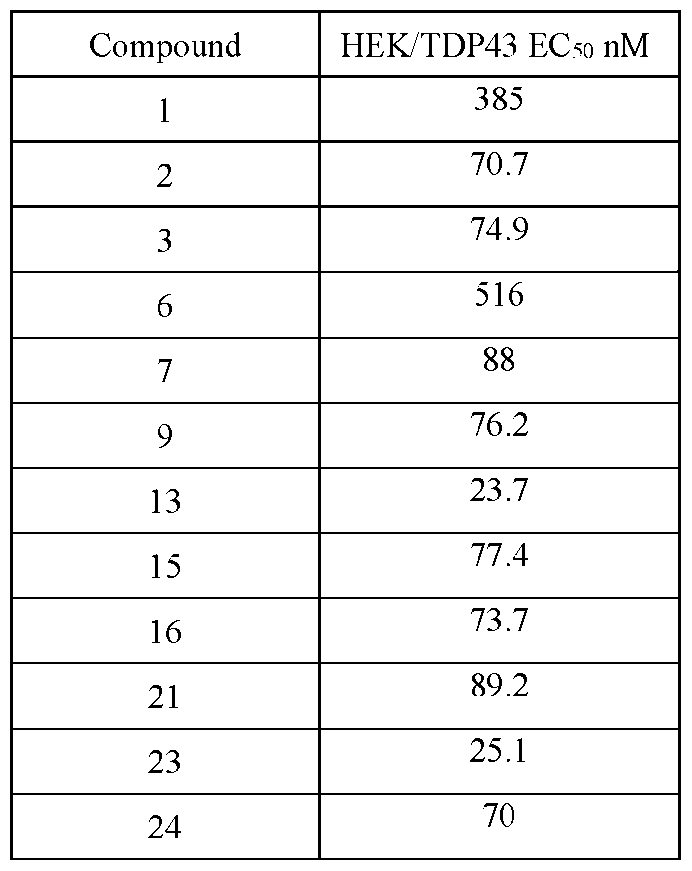
[0606] The foregoing disclosure has been described in some detail by way of illustration and example, for purposes of clarity and understanding. Therefore, it is to be understood that the above description is intended to be illustrative and not restrictive. The scope of the disclosure should, therefore, be determined not with reference to the above description, but should instead be determined with reference to the following appended claims, along with the full scope of equivalents to which such claims are entitled.
Claims
What is claimed is:
1. A compound of F ormula (I) :

W is N, CH, or CR4*;
R is H, oxo, alkyl, alkenyl, heteroaryl, heterocyclyl, or carboxamide, each substituted with 0, 1. or 2 Rx groups and each Rx is oxo. hydroxyl, cyano, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted amino, substituted or unsubstituted ester, substituted or unsubstituted sulfonyl, substituted or unsubstituted amide, substituted or unsubstituted carboxamide, substituted or unsubstituted alkoxy, or substituted or unsubstituted alkoxy alkyl;
R1 is H, alkyl, cycloalkyl, heterocyclyl, amino, alkoxy, alkoxyalkyl, amide, carboxamide, or sulfonyl, each substituted with 0, 1 , or 2 Ry groups and each Ry is substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted or unsubstituted alkoxy, substituted or unsubstituted alkoxyalkyl, substituted or unsubstituted amino, substituted or unsubstituted ester, substituted or unsubstituted sulfonyl, substituted or unsubstituted amide, substituted or unsubstituted carboxamide, substituted or unsubstituted urea, substituted or unsubstituted carbamate, halo, -CN, -OH, oxo, or methylallyl;
R4 and R4* are and, heteroary l, or heterocyclyl, each substituted with 0, 1, or 2 Rz groups and each Rz is alkyl, cycloalkyl, haloalkyl. alkoxy, haloalkoxy, halo, or -CN, provided that only one of R4 and R4* is present;
R5 is H, carboxyl, substituted or unsubstituted alkyl, substituted or unsubstituted
alkoxy carbonyl, substituted or unsubstituted ester, or substituted or unsubstituted carboxamide;
R6 is absent, H, or alkyl;
R7 and R8 are each independently H, Ci-6 alkyl or R7 and R8join together to form a fused or bridging ring system;
— represents a single or double bond; x is 0, 1 or 2;
R9 is absent or oxo; and pharmaceutically acceptable salts, solvates, and prodrugs thereof; provided that when R6 is absent, R7 and R8 are

H and R4 is m-tolyl, then R is not substituted or unsubstituted piperidinyl; substituted or unsubstituted pyrrolidinyl; -CH2OH; -CH2(substituted or unsubstituted heterocyclyl); or C(O)NR*R** wherein R* and R** join to form a substituted or unsubstituted heterocyclic group.
2. The compound of claim 1, wherein R is H, oxo, substituted or unsubstituted Ci-6 alkyl, substituted or unsubstituted Ci-6 alkenyl, substituted or unsubstituted monocyclic heteroaryl, substituted or unsubstituted monocyclic heterocyclyl, or RaRbNC(O)-; and each Ra and Rb is independently H, Ci-6 alkyl, Ci-ehaloalkyl, Ci-6 cycloalkyl, or CH3C(O)-.
The compound of claim 2, wherein each Ra and Rb is independently H, cyclopropyl, cyclobutyl, methyl, ethyl, CH3C(O)-, -CD3, or -CF3.
The compound of claim 1 or 2, wherein R is H, oxo, methyl, ethyl, propyl, isopropyl, isobutyl, propenyl, pyridyl, pyrimidinyl, pyrazolyl, dioxanyl, oxetanyl, morpholino, 3,6- dihydro-2H-pyranyl, or RaRbNC(O)-.
The compound of claim 1 or 2, wherein R is H.
The compound of any one of claims 1-5, wherein Rx is monocyclic heteroaryl, monocyclic heterocyclyl, Ci-6 alkyl, Ci-6 cycloalkyl, RaRbN, RaO-, Rc, RaRbN-(Ci-6 alkyl)-, RaO-(Ci-6 alkyl)-, or RCO-(CI-6 alkyl)-; and Rc is H, methyl, ethyl, HOC(O)CH2-, CH3OC(O)CH2-. (CH3)3COC(O)CH2-. (CH3)3COC(O)NH-. H2NC(O)CH2-,
CH3NHC(O)CH2-, (CH3)2NC(O)CH2-, NC-ethyl, H2N-, H2N-ethyl, CH3NH-ethyl, or (CH3)2N-ethyl.
7. The compound of any one of claims 1 -5. wherein Rx is methyl, cyclopropyl, cyclobutyl, pyridyl, pyrazolyl, morpholino, piperidinyl, piperazinyl, RaRbN, RaO-, Rc, RaRbN-ethyl, RcO-ethyl-, RcO-methyl-, or RcO-isopenty 1.
8. The compound of any one of claims 1-7. wherein R1 is H, Ci-6 alkyl, Ci-6 cycloalkyl, monocyclic heterocyclyl. RaO-, RaO-(Ci-6 alkyd), RaRbNC(O)-, or alkyl-SCh-
9. The compound of any one of claims 1-7, wherein R1 is H, propyl, ethyl, methyl, isobutyl, cyclobuty l, cyclopropyl, azetidinyl, oxetanyl, piperidinyl, tetrahydropyran, tetrahydrofuran, thietane 1,1 dioxide, hexahydropyrimidin-2-one, RaO-, RaO-ethyl, RaO- methyl. RaRbNC(O)-, CH3CH2SO2-, (CHs CHSCh-, or cyclopropyl-SCh-.
10. The compound of any one of claims 1-9, wherein Ry is C1-6 alky 1, C1-6 alkenyl, C1-6 haloalkyl, C 1-6 cycloalkyl, monocyclic heteroary l, monocy clic ary l, monocy clic heterocyclyl, halo, -CN, -OH, RaO-, CH3SO2-, CH3C(O)-, RaOC(O)-, RaRbNC(O)-, H2NC(O)NH-, RaRbN-_ or methylallyl, each further substituted with 0, 1, or 2 Rd groups where Rd is methyl, oxo, CH3C(O)-, CH3OC(O)-, CH3SO2-, -F, (CH3)3CO-, (CD3)3CO-, RaO-, -CN, NH2, (CH3)3COC(O)-, H2NC(O)-, CH3HNC(O)-.
11. The compound of any one of claims 1-9, wherein Ry is methy l, cyclopropyl, cyclobuty l, pyridinyl, phenyl, piperidinyl, pyrrolidinyl, oxetanyl, pyrrolidinone, imidazolidin-2-one, oxazolidin-2-one, lH-l,2,4-triazol-5-one, tetrahydropyran, dioxanyl, azetidinyl, -CN, -F. -CF3, -OH, RaO-, CH3SO2-. CH3C(O)-, HOC(O)-, RaRbNC(O)-, H2NC(O)NH-, RaRbN-, or methylallyl.
12. The compound of any one of claims 1-11, wherein R4 is H, phenyl, pyrazoly l, thiazolyl, thienyl, oxabi cyclo[2. 1.1 ]hexanyl, and norbomanyl. each substituted by 0, 1, or 2 Rz wherein each Rz is -CN, halo, C1-6 alkyd, C1-6 alkoxy, C1-6 haloalkyl, C1-6 cycloalkyl, deuterated alkyd, or C1-6 haloalkoxy.
13. The compound of claim 12, wherein R4 is H, phenyl, pyrazolyl, thiazolyl, thienyl, oxabicyclo[2.1. l]hexanyl, or norbomanyl.
14. The compound of claim 12 or 13, wherein each Rz is -Cl, -F, -CN, -CF3, -CD3, -OCF3, methyl, ethyl, cyclopropyl, methoxy, difluoromethoxy, or difluoromethyl.
15. The compound of any one of claims 1-14, wherein each R5 is -C(O)NRaRb. -C(O)ORa, unsubstituted C1-6 alkyl, or C1-6 alkyl substituted with 1 or 2 -ORa or -NH2, -NH-Ra, - NRaRb groups.
16. The compound of any one of claims 1-14, wherein each R5 is H, hydroxymethyl, hydroxyethyl, methoxymethyl. H2N-methyl. CH3NH-methyl, or (CH3)2N-methyl, trideuteriomethoxymethyL -C(O)NRaRb, -C(O)ORa.
17. The compound of any one of claims 1-16, wherein R6 is absent.
18. The compound of any one of claims 1-17, wherein R is not substituted with an Rx group.
19. The compound of any one of claims 1-17, wherein R is substituted with 1 Rx group.
20. The compound of any one of claims 1-17, wherein R is substituted with 2 Rx groups.
21. The compound of any one of claims 1-20. wherein R1 is not substituted with an Ry group.
22. The compound of any one of claims 1-20, wherein R1 is substituted with 1 Ry group.
23. The compound of any one of claims 1-20. wherein R1 is substituted with 2 Ry groups.
24. The compound of any one of claims 10-23, wherein Ry is not substituted with an Rd group.
25. The compound of any one of claims 10-23, wherein Ry is substituted with 1 Rd group.
26. The compound of any one of claims 10-23, wherein Ry is substituted with 2 Rd groups.
27. The compound of any one of claims 1-26, wherein R4 is not substituted with an Rz group.
28. The compound of any one of claims 1-26, wherein R4 is substituted with 1 Rz group.
29. The compound of any one of claims 1-26, wherein R4 is substituted with 2 Rz groups.
30. The compound of any one of claims 1-29, wherein W is CH.
31. The compound of any one of claims 1-30, wherein R7 and R8 are each H.
32. The compound of any one of claims 1-30, wherein R7 and R8join together to form a 1 or 2 carbon bridged ring system.
33. The compound of claim 1, having the structure of Formula (Ic)
 and pharmaceutically acceptable salts, solvates, and prodrugs thereof.
and pharmaceutically acceptable salts, solvates, and prodrugs thereof.
34. The compound of claim 33, wherein R is H.
35. The compound of claim 33, wherein R4 is m-tolyl or m-chlorophenyl.
36. The compound of claim 33, wherein R1 is H, methyl, ethyl or isopropyl substituted with
1 Ry group and Ry is substituted with 1 or 2 Rd groups.
37. The compound of claim 33, having the structure of Formula (Ic-1)
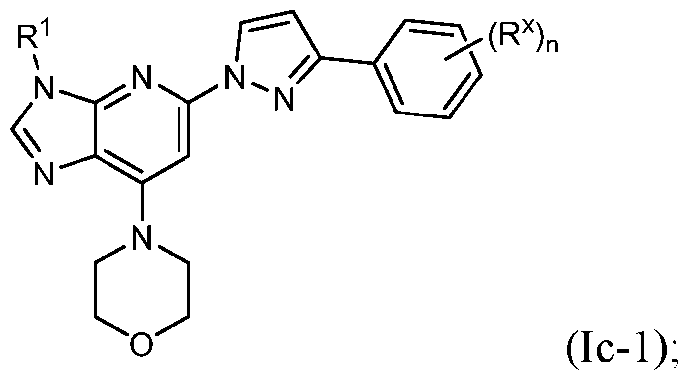 pharmaceutically acceptable salts, solvates, and prodrugs thereof wherein n is 0, 1 or 2; and each Rx is independently halo, cyano, Ci-3 alkyl. Ci-3 haloalkyl, Cs-6 cycloalkyl, Ci-3 alkoxy, or C1-3 haloalkoxy.
pharmaceutically acceptable salts, solvates, and prodrugs thereof wherein n is 0, 1 or 2; and each Rx is independently halo, cyano, Ci-3 alkyl. Ci-3 haloalkyl, Cs-6 cycloalkyl, Ci-3 alkoxy, or C1-3 haloalkoxy.
38. The compound of claim 37, wherein n is 1 or 2; and each Rx is independently F, Cl, cyano, methyl, fluoromethyl, difluoromethyl, trifluoromethyl, ethyl, fluoroethyl, difluoroethyl, trifluoroethyl, cyclopropyl, methoxy, fluoromethoxy, difluoromethoxy, trifluoromethoxy, ethoxy, fluoroethoxy, difluoroethoxy, or trifluoroethoxy.
39. The compound of claim 37, wherein R1 is H, methyl, ethyl or isopropyl substituted with 1 Ry group and Ry is substituted with 1 or 2 Rd groups.
40. A compound selected from the compounds in Table 1 and pharmaceutically acceptable salts thereof or a compound selected from
4-[3-(2-methoxyethyl)-5-[3-(m-tolyl)pyrazol-l-yl]imidazo[4,5-b]pyridin-7- yl] morpholine;
2-(7-morpholino-5-(3-(m-tolyl)-lH-pyrazol-l-yl)-3H-imidazo[4,5-b]pyri din-3- yl)acetonitrile;
5-({7-morpholino-5-[3-(m-tolyl)-l-pyrazolyl]-3H-l,3,4-triazainden-3-yl}methyl)-2- pyrrolidinone;
1 -(2- { 7 -morpholino-5-[3-(m-tolyl)- 1 -py razolyl] -3H- 1 ,3,4-triazainden-3-y 1 } ethyl)-2- imidazolidinone;
4-( { 7 -morpholino-5- [3 -(m-toly 1)- 1 -pyrazoly 1] -3H- 1 ,3 ,4-triazainden-3-y 1 } methyl)- 1,3- oxazolidin-2-one;
N-methyl-3-({7-morpholino-5-[3-(m-tolyl)-l-pyrazolyl]-3H-l,3,4-triazainden-3- yl } methyl)- 1 -azetidinecarboxamide; l-[3-({5-[3-(m-chlorophenyl)-l-pyrazolyl]-7-morpholino-3H-l,3,4-triazainden-3- yl } methyl )- 1 -azetidiny 1J - 1 -ethanone; and
3-({7-morpholino-5-[3-(m-tolyl)-l-pyrazolyl]-3H-l,3,4-triazainden-3-yl)methyl)-l- azetidinecarboxamide, and pharmaceutically acceptable salts thereof.
41. A compound and/or a pharmaceutically acceptable salt of any one of claims 1-40, wherein one or more hydrogen atoms attached to carbon atoms of the compound are replaced by deuterium atoms.
42. A pharmaceutical composition comprising a compound and/or a pharmaceutically acceptable salt of any one of claims 1-41 and a pharmaceutically acceptable excipient.
43. A method of inhibiting PIKfyve kinase in a subject in need thereof comprising administering to the subject an effective amount of a compound of any one of claims 1- 41, or a pharmaceutical composition of claim 42.
44. A method of treating a neurological disease associated with PIKfy ve activity in a subject in need thereof comprising administering to the subject an effective amount of a compound of any one of claims 1-41, or a pharmaceutical composition of claim 42.
45. The method of claim 44, wherein the neurological disease is amyotrophic lateral sclerosis (ALS), primary lateral sclerosis (PLS), Charcot-Marie-Tooth (CMT; including type 4J (CMT4J)), and Yunis-Varon syndrome, autophagy, polymicrogyria (including polymicrogyria with seizures), temporo-occipital polymicrogyria, Pick’s disease, Parkinson’s disease, Parkinson’s disease with Lewy bodies, dementia with Lewy bodies, Lewy body disease, fronto-temporal dementia, diseases of neuronal nuclear inclusions of polyglutamine and intranuclear inclusion bodies, disease of Marinesco and Hirano bodies, tauopathy, Alzheimer’s disease, neurodegeneration, spongiform neurodegeneration, peripheral neuropathy, leukoencephalopathy, inclusion body disease, progressive supranuclear palsy, corticobasal syndrome, chronic traumatic encephalopathy, traumatic brain injury (TBI), cerebral ischemia, Guillain-Barre Syndrome, chronic inflammatory demyelinating polyneuropathy, multiple sclerosis, a lysosomal storage disease, Fabry’s disorder, Gaucher’s disorder, Niemann Pick C disease, Tay-Sachs disease, and Mucolipidosis type IV, neuropathy, Huntington’s disease, a psychiatric disorder, ADHD, schizophrenia, a mood disorder, major depressive disorder, depression, bipolar disorder I, or bipolar disorder II.
46. The method of claim 44, wherein the disease is ALS, FTD, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, or CMT.
47. The method of claim 44, wherein the disease is ALS.
48. The method of claim 44, wherein the disease is a tauopathy such as Alzheimer’s disease, progressive supranuclear palsy, corticobasal syndrome, frontotemporal dementia, or chronic traumatic encephalopathy.
49. The method of claim 44, wherein the disease is a lysosomal storage disease such as Fabry’s disorder, Gaucher's disorder, Niemann Pick C disease, Tay-Sachs disease, or Mucolipidosis type IV.
50. The method of claim 44, wherein the disease is a psychiatric disorder such as ADHD, schizophrenia, or mood disorders such as major depressive disorder, depression, bipolar disorder I, or bipolar disorder II.
51. A compound of any one of claims 1-41 for use as a medicament.
52. The compound of claim 51, wherein the compound is for use in treating a neurological disease treatable by inhibition of PIKIyve kinase.
53. Use of a compound of any one of claims 1-41 in the manufacture of a medicament for treating a neurological disease in a subject in which PI K Fx x e contributes to the pathology' and/or symptoms of the disease.


Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202363498536P | 2023-04-27 | 2023-04-27 | |
| US63/498,536 | 2023-04-27 | ||
| US202463571744P | 2024-03-29 | 2024-03-29 | |
| US63/571,744 | 2024-03-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024226890A1 true WO2024226890A1 (en) | 2024-10-31 |
Family
ID=91334802
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/026398 Pending WO2024226890A1 (en) | 2023-04-27 | 2024-04-26 | Pyrazole 3h-imidazo(4,5-b)pyridine compounds and uses thereof |
| PCT/US2024/026401 Pending WO2024226892A1 (en) | 2023-04-27 | 2024-04-26 | Hydrazone 3h-imidazo(4,5-b)pyridine compounds and uses thereof |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2024/026401 Pending WO2024226892A1 (en) | 2023-04-27 | 2024-04-26 | Hydrazone 3h-imidazo(4,5-b)pyridine compounds and uses thereof |
Country Status (2)
| Country | Link |
|---|---|
| TW (2) | TW202448438A (en) |
| WO (2) | WO2024226890A1 (en) |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5846514A (en) | 1994-03-25 | 1998-12-08 | Isotechnika, Inc. | Enhancement of the efficacy of nifedipine by deuteration |
| US6334997B1 (en) | 1994-03-25 | 2002-01-01 | Isotechnika, Inc. | Method of using deuterated calcium channel blockers |
| WO2016210372A2 (en) | 2015-06-25 | 2016-12-29 | University Of Southern California | Methods to treat neurological diseases |
| WO2023058003A1 (en) * | 2021-10-07 | 2023-04-13 | Tme Therapeutics Llc | Novel inhibitors of pikfyve and methods using same |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2021252895A2 (en) * | 2020-06-11 | 2021-12-16 | Yumanity Therapeutics, Inc. | Compositions and methods for the treatment and prevention of neurological disorders |
-
2024
- 2024-04-26 WO PCT/US2024/026398 patent/WO2024226890A1/en active Pending
- 2024-04-26 TW TW113115774A patent/TW202448438A/en unknown
- 2024-04-26 TW TW113115772A patent/TW202506672A/en unknown
- 2024-04-26 WO PCT/US2024/026401 patent/WO2024226892A1/en active Pending
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5846514A (en) | 1994-03-25 | 1998-12-08 | Isotechnika, Inc. | Enhancement of the efficacy of nifedipine by deuteration |
| US6334997B1 (en) | 1994-03-25 | 2002-01-01 | Isotechnika, Inc. | Method of using deuterated calcium channel blockers |
| WO2016210372A2 (en) | 2015-06-25 | 2016-12-29 | University Of Southern California | Methods to treat neurological diseases |
| US20180161335A1 (en) | 2015-06-25 | 2018-06-14 | University Of Southern California | Methods to treat neurological diseases |
| WO2023058003A1 (en) * | 2021-10-07 | 2023-04-13 | Tme Therapeutics Llc | Novel inhibitors of pikfyve and methods using same |
Non-Patent Citations (15)
| Title |
|---|
| "Curr., Pharm. Des.", vol. 6, 2000, article "Recent Advances in the Synthesis and Applications of Radiolabeled Compounds for Drug Discovery and Development", pages: 110 |
| "Fieser and Fieser's Reagents for Organic Synthesis", vol. 1-40, 1991, JOHN WILEY AND SONS |
| "Larock's Comprehensive Organic Transformations", vol. 1-5, 1989, ELSEVIER SCIENCE PUBLISHERS |
| "March's Advanced Organic Chemistry", JOHN WILEY AND SONS |
| "Remington's Pharmaceutical Sciences", 1985, MACK PUBLISHING COMPANY |
| BHARADWAJ ET AL., HUM. MOL. GENET., vol. 25, no. 4, 2016, pages 682 - 692 |
| EVANS, E. ANTHONY: "Synthesis of radiolabeled compounds", J. RADIOANAL. CHEM., vol. 64, no. 1-2, 1981, pages 9 - 32 |
| FRUMAN ET AL.: "Phosphoinositide Kinases", ANN. REVIEW. BIOCHEM., vol. 67, 1998, pages 481 - 507, XP009006080, DOI: 10.1146/annurev.biochem.67.1.481 |
| GARDINER ET AL.: "Prevalence of carriers of intermediate and pathological polyglutamine disease-associated alleles among large population-based cohorts", JAMA NEUROL., vol. 76, no. 6, 2019, pages 650 - 656 |
| GEORGE W.VARMA, RAJENDER S.: "The Synthesis of Radiolabeled Compounds via Organometallic Intermediates", TETRAHEDRON, vol. 45, no. 21, 1989, pages 6601 - 21 |
| IKONOMOV ET AL., J. BIOL. CHEM., vol. 276, no. 28, 2001, pages 26141 - 26147 |
| NASCIMBENI ET AL., FEBS J., vol. 284, 2017, pages 1267 - 1278 |
| RUTHERFORD ET AL., J. CELL SCI., vol. 119, 2006, pages 3944 - 3957 |
| SHISHEVA ET AL., CELL BIOL. INT., vol. 32, no. 6, 2008, pages 591 |
| ZOLOV ET AL.: "In vivo, Pikfyve generates PI(3,5)P2, which serves as both a signaling lipid and the major precursor for PI5P", PROC. NATL. ACAD. SCI. USA, vol. 109, no. 43, 2012, pages 17472 - 17477, XP055350575, DOI: 10.1073/pnas.1203106109 |
Also Published As
| Publication number | Publication date |
|---|---|
| TW202448438A (en) | 2024-12-16 |
| TW202506672A (en) | 2025-02-16 |
| WO2024226892A1 (en) | 2024-10-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR102583317B1 (en) | Pyrazole derivatives as MALT1 inhibitors | |
| US10723725B2 (en) | Aminopyridine derivatives as TAM family kinase inhibitors | |
| KR102319857B1 (en) | P2x7 modulators | |
| KR102376354B1 (en) | 6-(5-hydroxy-1h-pyrazol-1-yl)nicotinamide derivatives and their use as phd inhibitors | |
| JP7744350B2 (en) | Substituted pyrazolo-pyrimidines and uses thereof | |
| AU2015299173A1 (en) | 2-(morpholin-4-yl)-l,7-naphthyridines | |
| EP3177619A1 (en) | 2-(morpholin-4-yl)-1,7-naphthyridines | |
| CA2952307A1 (en) | 3-amino-1,5,6,7-tetrahydro-4h-indol-4-ones | |
| WO2021183439A1 (en) | Substituted furo[3,2-d]pyrimidines and uses thereof | |
| US11795172B2 (en) | Substituted imidazo[1,2-b]pyridazines and [1,2,4]triazolo[4,3-b]pyridazines as CaMKII inhibitors | |
| JP2025520511A (en) | Kinase modulators and methods of use thereof | |
| CA3253414A1 (en) | Compounds and methods useful for stabilizing phenylalanine hydroxylase mutations | |
| EP4069699A1 (en) | Fused tricyclic heterocyclic compounds as inhibitors of pikfyve kinase useful for the treatment of neurological diseases | |
| WO2024226890A1 (en) | Pyrazole 3h-imidazo(4,5-b)pyridine compounds and uses thereof | |
| WO2022256299A1 (en) | 7-morpholino-5-(3-phenyl-1 h-pyrazol-1 -yl)-furo[3,2-b]pyridine derivatives and similar compounds as pikfyve kinase inhibitors for the treatment of e.g. amyotrophic lateral sclerosis (als) | |
| TW202313619A (en) | Fused bicyclic heterocyclic compounds and uses thereof | |
| HK40075899A (en) | Substituted pyrazolo-pyrimidines and uses thereof | |
| EA045333B1 (en) | PYRAZOLE DERIVATIVES AS MALT1 INHIBITORS | |
| HK1262908A1 (en) | Aromatic sulfonamide derivatives | |
| HK1262908B (en) | Aromatic sulfonamide derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24730109 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |