WO2024133292A1 - Novel ccr8 antagonists - Google Patents
Novel ccr8 antagonists Download PDFInfo
- Publication number
- WO2024133292A1 WO2024133292A1 PCT/EP2023/086683 EP2023086683W WO2024133292A1 WO 2024133292 A1 WO2024133292 A1 WO 2024133292A1 EP 2023086683 W EP2023086683 W EP 2023086683W WO 2024133292 A1 WO2024133292 A1 WO 2024133292A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- ethyl
- carboxylate
- piperidine
- naphthalene
- sulfonamido
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/22—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound oxygen atoms
- C07C311/29—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound oxygen atoms having the sulfur atom of at least one of the sulfonamide groups bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/50—Compounds containing any of the groups, X being a hetero atom, Y being any atom
- C07C311/51—Y being a hydrogen or a carbon atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/56—Nitrogen atoms
- C07D211/58—Nitrogen atoms attached in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/553—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having one nitrogen atom as the only ring hetero atom
- C07F9/576—Six-membered rings
- C07F9/59—Hydrogenated pyridine rings
Definitions
- the invention relates to naphthalene sulfonamide compounds and their medical use.
- the human CC chemokine receptor 8 (CCR8) is G protein-coupled receptor (GPCR) for which the CC chemokine ligand 1 (CCL1) is the best established endogenous agonist.
- CCR8 is expressed mainly on T-helper type 2 cells (Th2 cells) cells, which play a crucial role in adaptive immunity. Because recruitment of Th2 cells from the circulation to the site of inflammation is important for orchestrating inflammatory responses, there has been much interest in CCR8 inhibition as a therapeutic strategy.
- preclinical animal studies clearly demonstrated the involvement of CCR8 in various inflammatory diseases, including atopic dermatitis, allergic enteritis and asthma, although the data were controversial and sometimes inconclusive.
- CCR8 expression is enriched on tumor-infiltrating CD4 + Foxp3 + regulatory T cells (Tregs), compared to peripheral Tregs.
- Tregs tumor-infiltrating CD4 + Foxp3 + regulatory T cells
- These tumorresident CCR8+ Tregs are endowed with a highly immunosuppressive phenotype and hamper effective antitumor immunity, which was demonstrated for several types of cancer (breast, lung, colon and liver).
- high CCR8+ Treg numbers correlated with more advanced disease stages and poor prognosis.
- tumor-resident Tregs are targeted by various monoclonal antibodies and nanobodies, that both showed significant tumor growth inhibition and prolonged survival when evaluated in preclinical animal models, either as monotherapy or in combination with anti-PD-1 agents. Their antitumoral effect is ascribed to their ability to selectively deplete the immuno-suppressive tumorinfiltrating Treg cells.
- CCR8 small molecule CCR8 antagonists
- the number of chemotypes that are known to act as CCR8 antagonists is rather limited, and include oxazolidinone-based and naphthalene-sulfonamide-based analogues.
- SB-649701 is an oxazolidinone analogue reported by researchers from GSK [Jin etal. (2007) Bioorg Med Chem Lett 17, 1722-1725]. This compound showed low nM activity, when evaluated in a human CCR8 calcium mobilization assay, although it lacked antagonistic activity against the mouse, rat and guinea pig CCR8.
- this compound behaves as a functional human CCR8 antagonist when evaluated in various chemotaxis assays.
- SB-649701 lacked activity against other GPCRs (including various other chemokine receptors) and showed acceptable ADMET properties.
- the naphthalene sulfonamide series were reported by Jenkins et al. (2007) J Med Chem 50, 566-584 and Rummel etal. (2012) BrJ Pharmacol, 167, 1206-1217].
- This series of compounds, exemplified by NS-15 showed at least 300-fold selectivity versus other GPCRs, did not significantly inhibit P450 isozymes, and lacked significant hERG binding.
- -Ri is selected from substituents II III, or IV, wherein X, Y, Z are each independently selected from N or CH, wherein A is NH, O or S, wherein R3 is an aryl or heteroaryl group, both groups optionally substituted with a group selected from halogen, Cl-5 alkyl, Cl-5 alkoxy, cyano, haloalkyl and nitro, -R2 is
- Ri is substituent II or III
- D is S or P
- B is NH or O
- Ri is substituent IV
- D is P and B is NH
- R 4 is selected from the group consisting of a substituted or unsubstituted aliphatic, substituted or unsubstituted cycloaliphatic substituted unsubstituted heterocycloaliphatic, substituted or unsubstituted aryl and substituted or unsubstituted hetero(aryl) group, or wherein
- -Ri is selected from the group of substituents II, III or IV wherein X, Y, Z are each independently selected from N or CH, wherein A is NH, O or S, wherein R3 is an aryl or heteroaryl group, optionally substituted with a group selected from halogen, Cl-5alkyl, Cl-5 alkoxy, cyano, haloalkyl, nitro,
- -R2 is wherein D is S or P, wherein B is NH or O, wherein R 4 is selected from a substituted or unsubstituted aliphatic, substituted or unsubstituted cycloaliphatic, substituted or unsubstituted heterocycloaliphatic, substituted or unsubstituted aryl or substituted or unsubstituted hetero(aryl) group, 2.
- a compound according to any one of the statements 1 to 8, for use a medicament for use a medicament.
- CCR.8 mediated disease is selected from the group consisting of asthma, atopic dermatitis, allergic rhinitis, dermatitis, eczema, urticaria, rheumatoid arthritis, inflammatory bowel disease, psoriasis, multiple scelerosis, diabetes and cancer.
- a pharmaceutical composition comprising a compound according to any one of the statements 1 to 6, and a pharmaceutically acceptable carrier.
- -Ri is selected from substituents II or III wherein X, Y, Z are each independently selected from N or CH, wherein A is NH, O or S , wherein R3 is an aryl or heteroaryl group, both groups optionally substituted with a group selected from halogen, Cl-5 alkyl, Cl-5 alkoxy, cyano, haloalkyl and nitro,
- -R1 is selected from the group of substituents II, III or IV wherein X, Y, Z are each independently selected from N or CH, wherein A is NH, O or S, wherein R3 is an aryl or heteroaryl group, optionally substituted with a group selected from halogen, Cl-5alkyl, Cl-5 alkoxy, cyano, haloalkyl, nitro,
- -R2 is wherein D is S or P, wherein B is NH or O, wherein R4 is selected from a substituted or unsubstituted aliphatic, cycloaliphatic, heterocycloaliphatic, aryl or hetero(aryl) group,
- Reference compounds 4 and 6 were both prepared according to a previously reported procedure (Scheme 1) [Verhaegen et al. (2021) Bioorg Chem 107, 104560]. Briefly, sulfonylation of 1-bromo-naphthalene 1 afforded sulfonylchloride 2, which was subsequently reacted with ethyl 4-amino-l-piperidinecarboxylate or 4- methoxyaniline, yielding intermediates 3 and 5, respectively. Introduction of the o- toluamide moiety was achieved via a Buchwald-Hartwig amidation reaction, yielding reference compounds 4 and 6.
- Reagents and conditions (a) HSO3CI, chloroform, 0 °C; (b) amine, EtsN, DCM, rt ; (c) Pd(OAc)2, Xantphos, CS2CO3, o-toluamide, Ar, dioxane, 100 °C.
- Compound 3 was selected as a key intermediate from which easily structural variety can be introduced.
- Various /V-linked heteroaromatics were introduced on compound 3 by a typical Ullmann coupling using an appropriate azole, including an imidazole, a pyrazole and various triazoles (Scheme 2) [Zhang et al. (2015) Journal of Organic Chemistry 80, 705-710] .
- the desired target compound was isolated as the sole product.
- 4-phenyl-l,2,3-triazole and benzotriazole a mixture of the 1,4- and 2,4-disubstituted 1,2,3-triazoles (compounds 7d-e and 7f- g, respectively) was obtained.
- Reagents and conditions (a) Cui, L-proline, K2CO3, azole, Ar, DMSO, 120 °C.
- Reagents and conditions (a) Cui, DMEDA, sodium ascorbate, HzO/ethanol 3/7, 100 °C. A number of sulfur and oxygen-containing heteroaromatics (thiazole, oxazole, thiadiazole and oxadiazole) were appended the naphthalene scaffold via a carbon linkage (Scheme 4). Since the halogen-substituted heterocycles were much more readily available, when compared to the boronate containing equivalents, the bromide of key intermediate 3 was transformed into the pinacol boronate 10 using a Suzuki-Miyaura borylation with bis(pinacolato)diboron. [Seifert et al.
- Reagents and conditions (a) PdCl2(dppf), KOAc, bis(pinacolato)diboron, Ar, dioxane, 80 °C; (b) Pd(PPh 3 ) 4 , K 2 CO 3 , aromatic bromide, THF/H 2 O 4: 1, 100 °C.
- the bromide containing key intermediate 3 was first transformed into the cyano congener 12, using potassium ferrocyanide (K 4 [Fe(CN)e]) as a safe cyanide source, by palladium- catalyzed chemistry [Ren et al. (2009) Org Process Res Dev 13, 2002489] Cyclocondensation of 12 with benzhydrazide furnished the desired analogue 13 [Yeung et al. (2005) Tetrahedron 46, 3429-3432] (Scheme 5).
- K 4 [Fe(CN)e] potassium ferrocyanide
- Reagents and conditions (a) Pd(OAc) 2 , NazCCh, K 4 [Fe(CN)e], NMP, 140 °C; (b) K2CO3, benzhydrazide, n-butanol, 150 °C.
- the synthesis of the inverted sulfonamide 18 started with the coupling of 1-amino- 4-bromonaphthalene 14 with 4-methoxybenzenesulfonylchloride, yielding sulfonamide 15 (Scheme 6). Buchwald amidation of 15 with o-toluamide turned out to be cumbersome as nearly all starting material was recovered after the reaction, most probably due to poisoning of the palladium catalyst.
- Reagents and conditions (a) 4-methoxybenzene sulfonylchloride, pyridine, rt ; (b) TBAI, NaH, 4-methoxybenzylbromide, Ar, DMF, rt ; (c) Pd(OAc)2, Xantphos, CS2CO3, o-toluamide, Ar, dioxane, 100 °C; (d) TFA, DCM, rt.
- Reagents and conditions (a) 4-methoxybenzenesulfonamide, DMAP, EtsN, Mukaiyama's reagent, DCM, rt ; (b) DMAP, pyridine, BoczO, THF, 60 °C ; (c) Pd(OAc)2, Xantphos, CS2CO3, o-toluamide, Ar, dioxane, 100 °C.
- TBDPS tert-butyldiphenylsilyl Reagents and conditions: (a) NH 4 0H, Et20, acetone, 0 °C; (b) TBDPS-CI, EtsN, Ar, THF, 50 °C; (c) PPhsCl2, EtsN, Ar, Chloroform, 0 °C; (d) ethyl 4-aminopiperidine-l- carboxylate; (e) Pd(OAc)2, Xantphos, CS2CO3, o-toluamide, Ar, dioxane, 100 °C; (f) TBAF, THF, 40 °C.
- the synthesis of the phosphonoamidate analogue 32 was achieved via a different strategy (Scheme 9).
- the phosphonate moiety was introduced on the naphthalene scaffold via palladium-catalyzed chemistry using diethyl phosphite and starting from l-amino-4-bromonaphthalene 14 [Bessmertnykh et al. (2009) Chem Lett 38, 738- 739]
- the exocyclic amino group was then converted to the amide 30 by reaction with o-toluoyl chloride [Jenkins et al. cited above].
- Reagents and conditions (a) Pd2(OAc)2, PPhs, diethylphosphite, EtsN, Ar, ethanol, 80 °C; (b) o-toluoyl chloride, pyridine, 100 °C; (c) NaOH, H2O, dioxane, 100 °C; (d) 2,2'-dithiopyridine, PPF13, EtsN, ethyl 4-aminopiperidine-l-carboxylate, pyridine, 60 °C.
- Isoquinoline analog 38 was synthesized starting from 5-bromoisoquinoline 34. (Scheme 10).
- the sulfonic acid moiety was introduced using fuming sulfuric acid affording compound 35, which was isolated by precipitation from water [WO2013178362A1].
- the sulfonic acid 35 was then transformed into the sulfonamide 37 via a one pot reaction using the sulfonyl chloride as an in situ prepared intermediate [Jenkins et al. cited above].
- the amide moiety was introduced via Buchwald coupling yielding target compound 38 [Ruiz-Castello & Buchwald, cited above].
- Reagents and conditions (a) fuming sulfuric acid, 100°C; (b) thionyl chloride, DMF, 75°C; (c) DMAP, EtsN, ethyl 4-aminopiperidine-l-carboxylate, DCM, rt.; (d) Pd(OAc)2, Xantphos, CS2CO3, o-toluamide, Ar, dioxane, 100 °C.
- the basic principle of the calcium mobilization assay is to load cells with a calcium-sensitive dye, which fluoresces when bound to calcium; therefore, an increased fluorescence signal is indicative of activation of CCR8. Hence, inhibition of CCL1 induced calcium mobilization by various compounds is indicative for their CCR8 antagonistic activity.
- the pyrazole (compound 7a), the imidazole (compound 7b), the 1,2,4-triazole (compound 7c) and the 2,4- disubstituted 1,2,3-triazole analogue (compound 7e) showed potent inhibition of labeled CCL1 binding, with IC50 values in the range of 18-42 nM.
- the same analogues were also endowed with excellent activity in the functional CCR8 calcium mobilization assay (IC50 values between 0.089 and 3.2 pM), although a 5- to 76-fold loss in activity in the calcium assay is observed, when compared to the binding assay.
- the 1,4-disubstituted 1,2,3-triazole (compound 7d) was the most potent analogue within this series.
- IC50 5 nM
- This potency of compound 7d is not that surprising, since the triazole moiety has gained widespread use, as an excellent non-classical bioisoster of the trans amide bond [Kumari et al. (2020) J. Med. Chem 63, 12290- 12358].
- the potent CCR8 antagonistic activity of triazole 7d prompted us to probe the SAR around the phenyl ring (Table 2). Fusing the phenyl moiety with the triazole ring afforded the regioisomeric benzotriazole derivatives 7f and 7g. Both regioisomers are 40-fold less active in the binding assay, when compared to the phenyl-substituted triazole analogue 7d. Their decreased activity as CCR8 antagonists is more pronounced in the calcium mobilization assay, as evident from their IC50 values in the low pM range.
- the sulfur-containing heteroaromatics such as the 1,3,4-thiadiazole Ila, the 5-phenylthiazole 11b and the 2- phenylthiazole 11c do show activities in the binding assay with IC50 values of 10 nM or less, but loose quite some activity (40- to 120-fold) upon evaluation in the calcium assay.
- Table 3. CCL1 competition binding and CCR8 calcium mobilization data of compounds lla-e and 13 a ICso : the compound concentration inhibiting CCL1 AF647 binding by 50%.
- b ICso the compound concentration that inhibits CCL1 induced intracellular calcium flux by 50%.
- NMR spectra were acquired on commercial instruments (Bruker Avance 300 MHz, Bruker AMX 400 MHz or Bruker Avance 11+ 600 MHz) and chemical shifts (6) are reported in parts per million (ppm) referenced to tetramethylsilane ( 1 H), or the internal (NMR) solvent signal ( 13 C).
- High resolution mass spectra were acquired on a quadrupole orthogonal acceleration time-of-flight mass spectrometer (Synapt G2 HDMS, Waters, Milford, MA, USA). Samples were infused at 3 pL/min and spectra were obtained in positive mode with a resolution of 15 000 (FWHM) using leucine enkephalin as lock mass.
- This compound was prepared according to the general procedure using 3- phenylpyrazole (36.0 mg, 0.25 mmol, 2.5 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (13.3 mg, 26% yield).
- This compound was prepared according to the general procedure using 4- phenylimidazole (36.0 mg, 0.25 mmol, 2.5 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (21.3 mg, 41% yield).
- This compound was prepared according to the general procedure using 3-phenyl- 1,2,4-triazole (36.3 mg, 0.25 mmol, 2.5 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (31.3 mg, 62% yield).
- Both compounds were prepared according to the general procedure on a doubled scale using 4-phenyl-l,2,3-triazole (72.5 mg, 0.5 mmol, 2.5 eq.).
- the crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding title compound 7d as a solid (28.0 mg, 27% yield) and title compound 7e as a solid (29.1 mg, 28% yield).
- Both compounds were prepared according to the general procedure on a doubled scale using lH-benzo[d][l,2,3]triazole (59.6 mg, 0.5 mmol, 2.5 eq.).
- the crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding title compound 7f as a solid (13.8 mg, 14% yield) and title compound 7g as a solid (20.9 mg, 22% yield).
- This compound was prepared according to the general procedure using 2- methoxyphenyl acetylene (31 pl, 0.24 mmol, 1.2 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (54.8 mg, 51% yield).
- This compound was prepared according to the general procedure using 3- methoxyphenyl acetylene (30 pl, 0.24 mmol, 1.2 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (30.1 mg, 28% yield). 1
- This compound was prepared according to the general procedure using 4- methoxyphenyl acetylene (26 pl, 0.24 mmol, 1.2 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (71.8 mg, 67% yield).
- This compound was prepared according to the general procedure using 2- methoxyphenyl acetylene (30 pl, 0.24 mmol, 1.2 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (47.9 mg, 46% yield).
- This compound was prepared according to the general procedure using 2- methoxyphenyl acetylene (31 pl, 0.24 mmol, 1.2 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (31.4 mg, 30% yield).
- This compound was prepared according to the general procedure using 2- methoxyphenyl acetylene (30 pl, 0.24 mmol, 1.2 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (36.9 mg, 36% yield).
- This compound was prepared according to the general procedure using 2- methoxyphenyl acetylene (28 pl, 0.24 mmol, 1.2 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (31.2 mg, 30% yield).
- This compound was prepared according to the general procedure using 2- methoxyphenyl acetylene (28 pl, 0.24 mmol, 1.2 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (26.6 mg, 25% yield).
- This compound was prepared according to the general procedure using 2- methoxyphenyl acetylene (35 pl, 0.24 mmol, 1.2 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (31.8 mg, 28% yield).
- This compound was prepared according to the general procedure using 2- methoxyphenyl acetylene (39 pl, 0.24 mmol, 1.2 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (23.7 mg, 21% yield).
- the reaction tube was evacuated and backfilled with argon three times before dissolving the solids in anhydrous dioxane which was degassed using 5 freeze pump thaw cycles (20 ml).
- the reaction was allowed to stir 20h at 80 °C, after which the mixture was filtered over celite, washed with DCM and concentrated in vacuo.
- the crude residue was purified by silica gel column chromatography using petroleum ether/ethyl acetate (8/2) as the mobile phase yielding the title compound as a solid (462.2 mg, quantitative yield).
- This compound was prepared according to the general procedure using 2-bromo-5- phenyl-l,3,4-thiadiazole (36.2mg, 0.15 mmol, 1.5 eq.).
- the crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (37.3 mg, 71%).
- This compound was prepared according to the general procedure using 2-bromo-5- phenylthiazole (36.0 mg, 0.15 mmol, 1.5 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (39.3 mg, 75%).
- This compound was prepared according to the general procedure using 5-bromo-2- phenylthiazole (36.0 mg, 0.15 mmol, 1.5 eq.).
- the crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (36.5 mg, 70%).
- This compound was prepared according to the general procedure using 2-bromo-5- phenyl-l,3,4-oxadiazole (33.8 mg, 0.15 mmol, 1.5 eq.).
- the crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (29.8 mg, 59%).
- This compound was prepared according to the general procedure using 5-bromo-2- phenyloxazole (33.6 mg, 0.15 mmol, 1.5 eq.).
- the crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (32.9 mg, 65%).
- benzhydrazide (27.2 mg, 0.2 mmol, 2 eq.), ethyl-4-((4-cyanonaphthalene)-l- sulfonylamino)-piperidine-l-carboxylate 11 (38.7 mg, 0.1 mmol, 1 eq.), K2CO3 (27.6 mg, 0.2 mmol, 2 eq.) and n-butanol (0.5ml) were added and allowed to stir for 5h at 150 °C. The mixture was quenched with water and extracted three times with dichloromethane.
- N-(4- bromonaphthalen-l-yl)-4-methoxybenzenesulfonamide (15) (392.3 mg, 1 mmol, 1 eq.), TBAI (96.7 mg, 0.3 mmol, 0.3 eq.), NaH (72.0 mg, 3 mmol, 3 eq.) were added.
- the round bottom flask was evacuated and backfilled with argon three times before dissolving the solids in anhydrous DMF (10 ml). The mixture was allowed to stir for 10 minutes at 0 °C before adding 4-methoxybenzylbromide (216 pl, 1.5 mmol, 1.5 eq.).
- the reaction tube was evacuated and backfilled with argon three times. Anhydrous dioxane (0.8 ml) was added and the reaction was allowed to stir for 16h at 100 °C. The mixture was diluted with DCM (8 ml) and filtered. The filtrate was concentrated in vacuo and the crude residue was purified by silica gel column chromatography using petroleum ether/ethyl acetate (6/4) as the mobile phase yielding the title compound as a solid (367.5 mg, 81% yield).
- the reaction tube was evacuated and backfilled with argon three times. Anhydrous dioxane (0.4 ml) was added and the reaction was allowed to stir for 16h at 100 °C. The mixture was diluted with DCM (4 ml) and filtered. The filtrate was concentrated in vacuo and the crude residue was purified by silica gel column chromatography using petroleum ether/ethyl acetate (6/4) as the mobile phase yielding the title compound as a solid (152.4 mg, 80% yield).
- triphenylphosphine dichloride (249.9 mg, 0.75 mmol, 1.5 eq.) was added.
- the round bottom flask was evacuated and backfilled with argon three times.
- Chloroform (1.67 ml) was added followed by the dropwise addition of triethylamine (111 pl, 0.8 mmol, 1.6 eq.).
- the reaction mixture was allowed to stir for 20 minutes before cooling it down to 0 °C.
- the reaction tube was evacuated and backfilled with argon three times. Anhydrous dioxane (0.2 ml) was added and the reaction was allowed to stir for 16h at 100 °C. The mixture was diluted with DCM (4 ml) and filtered. The filtrate was concentrated in vacuo and the crude residue was purified by silica gel column chromatography using petroleum ether/ethyl acetate (6/4) as the mobile phase yielding the title compound as a solid (90.7 mg, 62% yield). The product was used in the next step without further purification.
- 4-bromonaphthalen-l-amine 14 (1.111 g, 5 mmol, 1 eq.), Pd(OAc)2 (22.5 mg, 0.1 mmol, 0.02 eq.) and triphenylphosphine (78.7 mg, 0.3 mmol, 0.06 eq.) were added.
- the reaction tube was evacuated and backfilled with argon three times and ethanol (10 ml), diethyl phosphite (0.78 ml, 12 mmol, 1.2 eq.) and triethylamine (1.05 ml, 7.5 mmol, 1.5 eq.) were added.
- diethyl (4- aminonaphthalen-l-yl)phosphonate 29 (1.1 g, 3.7 mmol, 1 eq.), DCM (40 ml) and 2-methylbenzoylchloride (0.724 ml, 5.55 mmol, 1.5 eq.) were added.
- the mixture was allowed to stir for 5 minutes at room temperature before addition of triethylamine (4ml).
- the reaction mixture was allowed to stir for 15h at room temperature and washed two times with HCI (IM) and water.
- the organic phase was dried over anhydrous magnesium sulfate, filtered and concentrated in vacuo.
- Silica gel column chromatography of the crude product using DCM/MeOH (95/5) as the mobile phase provided the title compound as a solid (1.294 g, 88% yield). The product was used in the next step without further purification.
- the hCCLl AF647 binding inhibition assay was performed and analyzed as described in [Liu et al. (2021) Biochem Pharmacol. 188, 114565]. Briefly, 50 pL/well of serial compound dilutions and 50 pL/well of hCCLl AF647 (2 nM final concentration) were mixed in a U-bottom 96-well plate. Then 100 pL/well of 1 x 10 5 Jurkat.hCCR8 cells, resuspended in the assay buffer, was added. After mixing samples were and incubated for 30 min at room temperature (RT) protected from light.
- RT room temperature
- MFI mean fluorescence intensity
- Human glioblastoma U87 cells stably expressing CD4 and CCR8 (U87.CD4.CCR8) were seeded at 20,000 cells per well in gelatin-coated black-walled polystyrene 96- well plates and incubated overnight at 37°C and 5% CO2. The next day, cell culture medium was replaced with 80pL/well of Calcium 6 Assay Kit loading medium (Molecular Devices), which was prepared according the manufacturer's instructions. After incubating the cells for 2h at 37°C and 5% CO2 they were transferred to the FLIPR Tetra device (Molecular Devices). Serial dilutions of compounds were added to the cells first and incubated for lOmin.
- hCCLl a fixed concentration of hCCLl (5.88nM final concentration) was added to induce a CCR8-mediated intracellular Ca 2+ release.
- All responses were normalized by dividing through a baseline determined just before the time of hCCLl addition and also background corrected by subtracting the averaged normalized response from untreated (i.e., no compound) and unstimulated (i.e., no hCCLl) cells. The percentage (%) inhibition induced by the compounds was then calculated relative to positive control samples (i.e., no compound, only hCCLl stimulation). Based on the dose-dependent % inhibition, IC50 values were calculated using non-linear curve fitting in Graphpad Prism 9.0.0.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pulmonology (AREA)
- Diabetes (AREA)
- Endocrinology (AREA)
- Biochemistry (AREA)
- Dermatology (AREA)
- Molecular Biology (AREA)
- Emergency Medicine (AREA)
- Immunology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Hydrogenated Pyridines (AREA)
- Peptides Or Proteins (AREA)
Abstract
The invention relates to novel CCR8 antagonists, based on NS-15 as lead structure, were designed and synthesized. To maintain the hydrogen bond potential, focus was on the isosteric replacements of the amide bond. In addition, isosters of the sulfonamide moiety, as well as of the naphthalene scaffold are disclosed.
Description
NOVEL CCR8 ANTAGONISTS
Field of the invention
The invention relates to naphthalene sulfonamide compounds and their medical use.
Introduction
The human CC chemokine receptor 8 (CCR8) is G protein-coupled receptor (GPCR) for which the CC chemokine ligand 1 (CCL1) is the best established endogenous agonist. CCR8 is expressed mainly on T-helper type 2 cells (Th2 cells) cells, which play a crucial role in adaptive immunity. Because recruitment of Th2 cells from the circulation to the site of inflammation is important for orchestrating inflammatory responses, there has been much interest in CCR8 inhibition as a therapeutic strategy. In addition, preclinical animal studies clearly demonstrated the involvement of CCR8 in various inflammatory diseases, including atopic dermatitis, allergic enteritis and asthma, although the data were controversial and sometimes inconclusive. More recently, it was shown that CCR8 expression is enriched on tumor-infiltrating CD4+Foxp3+ regulatory T cells (Tregs), compared to peripheral Tregs. These tumorresident CCR8+ Tregs are endowed with a highly immunosuppressive phenotype and hamper effective antitumor immunity, which was demonstrated for several types of cancer (breast, lung, colon and liver). In patients suffering from breast and pancreatic cancer, high CCR8+ Treg numbers correlated with more advanced disease stages and poor prognosis. Currently, tumor-resident Tregs are targeted by various monoclonal antibodies and nanobodies, that both showed significant tumor growth inhibition and prolonged survival when evaluated in preclinical animal models, either as monotherapy or in combination with anti-PD-1 agents. Their antitumoral effect is ascribed to their ability to selectively deplete the immuno-suppressive tumorinfiltrating Treg cells.
Altogether, these studies spurred the interest in CCR8 as a drug target and stimulated research teams to search for small molecule CCR8 antagonists. The number of chemotypes that are known to act as CCR8 antagonists is rather limited, and include oxazolidinone-based and naphthalene-sulfonamide-based analogues. SB-649701 is an oxazolidinone analogue reported by researchers from GSK [Jin etal. (2007) Bioorg Med Chem Lett 17, 1722-1725]. This compound showed low nM activity, when evaluated in a human CCR8 calcium mobilization assay, although it lacked antagonistic activity against the mouse, rat and guinea pig CCR8. Moreover, this compound behaves as a functional human CCR8 antagonist when evaluated in various chemotaxis assays. SB-649701 lacked activity against other GPCRs (including
various other chemokine receptors) and showed acceptable ADMET properties. The naphthalene sulfonamide series were reported by Jenkins et al. (2007) J Med Chem 50, 566-584 and Rummel etal. (2012) BrJ Pharmacol, 167, 1206-1217]. This series of compounds, exemplified by NS-15, showed at least 300-fold selectivity versus other GPCRs, did not significantly inhibit P450 isozymes, and lacked significant hERG binding. Unfortunately, the oral bioavailability of these compounds is low (2 %), presumably to solubility issues. An optimized compound from this series, known as ML-604086, was evaluated in a primate model of asthma [Wang et al. (2013) Thorax 68, 263-268] . Although more than 98% target inhibition by ML-604086 on circulating T-cells was shown, this did not translate into a beneficial effect on lung pathology, since CCR8 blockade did not affect mucus secretion, allergen-induced bronchoconstriction and airway hyper-responsiveness. This study suggested that CCR8 antagonism is not a viable therapeutic strategy for the treatment of asthma.
SUMMARY OF THE INVENTION
The invention is summarized in the following statements:
1. A compound with formula I
 wherein
wherein
-Q is CH,
-Ri is selected from substituents II III, or IV,
 wherein X, Y, Z are each independently selected from N or CH, wherein A is NH, O or S, wherein R3 is an aryl or heteroaryl group, both groups optionally substituted with a group selected from halogen, Cl-5 alkyl, Cl-5 alkoxy, cyano, haloalkyl and nitro,
-R2 is
wherein X, Y, Z are each independently selected from N or CH, wherein A is NH, O or S, wherein R3 is an aryl or heteroaryl group, both groups optionally substituted with a group selected from halogen, Cl-5 alkyl, Cl-5 alkoxy, cyano, haloalkyl and nitro,
-R2 is

Wherein if Ri is substituent II or III, D is S or P, and B is NH or O,
Wherein if Ri is substituent IV, D is P and B is NH wherein R4 is selected from the group consisting of a substituted or unsubstituted aliphatic, substituted or unsubstituted cycloaliphatic substituted
 unsubstituted heterocycloaliphatic, substituted or unsubstituted aryl and substituted or unsubstituted hetero(aryl) group, or wherein
unsubstituted heterocycloaliphatic, substituted or unsubstituted aryl and substituted or unsubstituted hetero(aryl) group, or wherein
-Q is N
-Ri is selected from the group of substituents II, III or IV
 wherein X, Y, Z are each independently selected from N or CH, wherein A is NH, O or S, wherein R3 is an aryl or heteroaryl group, optionally substituted with a group selected from halogen, Cl-5alkyl, Cl-5 alkoxy, cyano, haloalkyl, nitro,
wherein X, Y, Z are each independently selected from N or CH, wherein A is NH, O or S, wherein R3 is an aryl or heteroaryl group, optionally substituted with a group selected from halogen, Cl-5alkyl, Cl-5 alkoxy, cyano, haloalkyl, nitro,
-R2 is
 wherein D is S or P, wherein B is NH or O, wherein R4 is selected from a substituted or unsubstituted aliphatic, substituted or unsubstituted cycloaliphatic, substituted or unsubstituted heterocycloaliphatic, substituted or unsubstituted aryl or substituted or unsubstituted hetero(aryl) group,
2. The compound according to statement 1, wherein Q is CH and Ri is substituent II with X = CH and Y and Z = N.
wherein D is S or P, wherein B is NH or O, wherein R4 is selected from a substituted or unsubstituted aliphatic, substituted or unsubstituted cycloaliphatic, substituted or unsubstituted heterocycloaliphatic, substituted or unsubstituted aryl or substituted or unsubstituted hetero(aryl) group,
2. The compound according to statement 1, wherein Q is CH and Ri is substituent II with X = CH and Y and Z = N.
3. The compound according to statement 1, wherein Q is CH, and wherein in R.2 D is S, B is O and R4 is a substituted heterocyclo aliphatic group.
4. The compound according to statement 3, wherein R4 is ethyl (4-sulfonamido)- piperidine-l-carboxylate.
5. The compound according to statement 1, selected from the group consisting of
- Ethyl 4-((4-(4-phenyl-lH-l,2,3-triazol-l-yl)naphthalene)-l-sulfonamido) - piperidine-l-carboxylate (7d),
Ethyl 4-((4-(4-(2-methylphenyl)-l H-l, 2, 3-triazol- 1-yl) naphthalene)- 1- sulfonamido)piperidine-l-carboxylate (9d),
Ethyl 4-((4-(4-(3-methylphenyl)-l H-l, 2, 3-triazol- 1-yl) naphthalene)- 1- sulfonamido)piperidine-l-carboxylate (9e),
Ethyl 4-((4-(4-(3-fluorophenyl)-l H-l, 2, 3-triazol- 1-yl) naphthalene)- 1- sulfonamido)piperidine-l-carboxylate (9g),
Ethyl 4-((4-(4-(4-fluorophenyl)-l H-l, 2, 3-triazol- 1-yl) naphthalene)- 1- sulfonamido)piperidine-l-carboxylate (9h),
- Ethyl 4-((4-(4-(3-trifluoromethylphenyl)- 1H-1, 2, 3-triazol- 1-yl) naphthalene) -l-sulfonamido)piperidine-l-carboxylate (9i),
Ethyl 4-((4-(5-phenyl-l,3,4-thiadiazol-2-yl)naphthalene)-l-sulfonamido) piperidine-l-carboxylate (Ila),
- Ethyl 4-((4-(5-phenylthiazol-2-yl)naphthalene)-l-sulfonamido) piperidine-l- carboxylate (11b), and
-Ethyl 4-(4-(2-methylbenzamido)naphthalene) sulfonimidamido) piperidine-l- carboxylate (28).
6. The compound according to statement 1, wherein Q = N.
7. The compound according to statement 6, wherein Ri is substituent IV and R2 is
 8. The compound according to statement 7, which is Ethyl 4-((5-(2- methylbenzamido)isoquinoline)-8-sulfonamido)piperidine-l-carboxylate (38).
8. The compound according to statement 7, which is Ethyl 4-((5-(2- methylbenzamido)isoquinoline)-8-sulfonamido)piperidine-l-carboxylate (38).
9. A compound according to any one of the statements 1 to 8, for use a medicament.
10. A compound according to any one of the statements 1 to 8, for use in the treatment or prevention of a CCR8 mediated disease.
11. The compound for use in the treatment or prevention according to statement 10 wherein the CCR.8 mediated disease is selected from the group consisting of asthma, atopic dermatitis, allergic rhinitis, dermatitis, eczema, urticaria, rheumatoid arthritis, inflammatory bowel disease, psoriasis, multiple scelerosis, diabetes and cancer.
12. The compound for use in the treatment or prevention according to statement 10, wherein the compound is selected from the group consisting of:
- Ethyl 4-((4-(4-phenyl-lH-l,2,3-triazol-l-yl)naphthalene)-l-sulfonamido) - piperidine-l-carboxylate (7d),
Ethyl 4-((4-(4-(2-methylphenyl)-lH-l,2,3-triazol-l-yl)naphthalene)-l- sulfonamido)piperidine-l-carboxylate (9d),
Ethyl 4-((4-(4-(3-methylphenyl)-lH-l,2,3-triazol-l-yl)naphthalene)-l- sulfonamido)piperidine-l-carboxylate (9e),
Ethyl 4-((4-(4-(3-fluorophenyl)-lH-l,2,3-triazol-l-yl)naphthalene)-l- sulfonamido)piperidine-l-carboxylate (9g),
Ethyl 4-((4-(4-(4-fluorophenyl)-lH-l,2,3-triazol-l-yl)naphthalene)-l- sulfonamido)piperidine-l-carboxylate (9h),
- Ethyl 4-((4-(4-(3-trifluoromethylphenyl)-lH-l,2,3-triazol-l-yl)naphthalene) -l-sulfonamido)piperidine-l-carboxylate (9i),
Ethyl 4-((4-(5-phenyl-l,3,4-thiadiazol-2-yl)naphthalene)-l-sulfonamido) piperidine-l-carboxylate (Ila),
- Ethyl 4-((4-(5-phenylthiazol-2-yl)naphthalene)-l-sulfonamido) piperidine-l- carboxylate (11b),
-Ethyl 4-(4-(2-methylbenzamido)naphthalene) sulfonimidamido) piperidine-l- carboxylate (28), and
- Ethyl 4-((5-(2-methylbenzamido)isoquinoline)-8-sulfonamido)piperidine-l- carboxylate (38).
13. A pharmaceutical composition comprising a compound according to any one of the statements 1 to 6, and a pharmaceutically acceptable carrier.
14. A compound with formula I
 wherein
wherein
-Q is CH,
-Ri is selected from substituents II or III
 wherein X, Y, Z are each independently selected from N or CH, wherein A is NH, O or S , wherein R3 is an aryl or heteroaryl group, both groups optionally substituted with a group selected from halogen, Cl-5 alkyl, Cl-5 alkoxy, cyano, haloalkyl and nitro,
wherein X, Y, Z are each independently selected from N or CH, wherein A is NH, O or S , wherein R3 is an aryl or heteroaryl group, both groups optionally substituted with a group selected from halogen, Cl-5 alkyl, Cl-5 alkoxy, cyano, haloalkyl and nitro,
-R2 is
 wherein D is S or P, wherein B is NH or O , wherein R4 is selected from the group consisting of a substituted or unsubstituted aliphatic, cycloaliphatic, heterocycloaliphatic, aryl and hetero(aryl) group with the proviso that when D = P or B = NH, then Ri can also be substituent IV
wherein D is S or P, wherein B is NH or O , wherein R4 is selected from the group consisting of a substituted or unsubstituted aliphatic, cycloaliphatic, heterocycloaliphatic, aryl and hetero(aryl) group with the proviso that when D = P or B = NH, then Ri can also be substituent IV
 wherein R3 is an aryl or heteroaryl group, optionally substituted with a group selected from halogen, Cl-5 alkyl, Cl-5 alkoxy, cyano, haloalkyl, nitro or wherein
wherein R3 is an aryl or heteroaryl group, optionally substituted with a group selected from halogen, Cl-5 alkyl, Cl-5 alkoxy, cyano, haloalkyl, nitro or wherein
-Q is N
-R1 is selected from the group of substituents II, III or IV wherein X, Y, Z are each independently selected from N or CH, wherein A is NH, O or S, wherein R3 is an aryl or heteroaryl group, optionally substituted with a group selected from halogen, Cl-5alkyl, Cl-5 alkoxy, cyano, haloalkyl, nitro,
-R2 is wherein D is S or P, wherein B is NH or O, wherein R4 is selected from a substituted or unsubstituted aliphatic, cycloaliphatic, heterocycloaliphatic, aryl or hetero(aryl) group,
DESCRIPTION OF THE INVENTION
Figure 1. Known CCR8 antagonists
Figure 2. Design of new CCR8 antagonists, based on NS-15
Recently, our group embarked on the expansion of the structure-activity relationship (SAR) study of the naphthalene sulfonamide series of compounds (Figure 2). Starting from lead compound NS-15, a series of analogues was prepared in which the 2- toluoylamide group was replaced by a (substituted) (hetero)aryl, a (substituted) anilino, a styryl or a (substituted) phenylacetelyne, using a variety of palladium- catalyzed cross-coupling reactions [Verhaegen et al. (2021) Bioorg Chem 107, 104560]. All compounds from this series showed a pronounced loss of CCR8 antagonism, when compared to NS-15. Homology molecular modelling, based on the X-ray structure of the closely related CCR5 receptor, revealed that the absence of a critical hydrogen bond of the amide moiety with CCR8 explains the decreased CCR8
antagonistic activity. In this work, a novel series of CCR8 antagonists, so called second-generation CCR8 antagonists, based on NS-15 as lead structure, were designed and synthesized (Figure 2). In order to maintain the hydrogen bond potential, the focus was on the isosteric replacements of the amide bond. In addition, isosters of the sulfonamide moiety, as well as of the naphthalene scaffold, were also prepared.
Chemistry
Reference compounds 4 and 6 were both prepared according to a previously reported procedure (Scheme 1) [Verhaegen et al. (2021) Bioorg Chem 107, 104560]. Briefly, sulfonylation of 1-bromo-naphthalene 1 afforded sulfonylchloride 2, which was subsequently reacted with ethyl 4-amino-l-piperidinecarboxylate or 4- methoxyaniline, yielding intermediates 3 and 5, respectively. Introduction of the o- toluamide moiety was achieved via a Buchwald-Hartwig amidation reaction, yielding reference compounds 4 and 6.
Scheme 1. Synthesis of reference compounds 4 and 6

Reagents and conditions: (a) HSO3CI, chloroform, 0 °C; (b) amine, EtsN, DCM, rt ; (c) Pd(OAc)2, Xantphos, CS2CO3, o-toluamide, Ar, dioxane, 100 °C.
Compound 3 was selected as a key intermediate from which easily structural variety can be introduced. Various /V-linked heteroaromatics were introduced on compound 3 by a typical Ullmann coupling using an appropriate azole, including an imidazole, a pyrazole and various triazoles (Scheme 2) [Zhang et al. (2015) Journal of Organic Chemistry 80, 705-710] . In most cases, the desired target compound was isolated as the sole product. However, when using 4-phenyl-l,2,3-triazole and benzotriazole, a mixture of the 1,4- and 2,4-disubstituted 1,2,3-triazoles (compounds 7d-e and 7f- g, respectively) was obtained. In contrast, the formation of the 3-substituted 1,2,3- triazole was not observed, probably due to the steric hindrance between the
naphthalene core and phenyl ring. After separation by silica gel flash chromatography, the correct assignment of the structure of both regioisomers was achieved via NMR spectroscopy. The proton on the triazole ring is observed as a singlet in the aromatic region of the proton NMR spectrum. Via a combination of HSQC, HMBC and NOESY measurements, this proton allowed the clear assignment of the peaks originating from the protons on the phenyl ring. The remaining signals in the aromatic region of the proton spectrum are therefore assigned to the naphthalene core. Only for the 1,4-disubstituted triazole 7d, NOESY cross-peaks between the protons on the naphthalene core and on the triazole ring are possible, which is not observed in the regioisomeric 2,4-disubstituted triazole 7e.
Scheme 2. Synthesis of /V-linked heteroaromatics on the naphthalene scaffold

Reagents and conditions: (a) Cui, L-proline, K2CO3, azole, Ar, DMSO, 120 °C.
Since the number of commercially available 4-phenyl-lH-l,2,3-triazoles is very limited, other l,4-disubstituted-l,2,3-triazoles 9a-j were prepared via a one-pot, three component reaction using key intermediate 3, sodium azide and a commercially available phenylacetylene, yielding target compounds 9a-j (Scheme 3) [Potratz et al. (2012) Beilstein J. Org. Chem 8, 683-692].
Scheme 3. Synthesis of l,4-disubstituted-l,2,3-triazoles 9a-j

Reagents and conditions: (a) Cui, DMEDA, sodium ascorbate, HzO/ethanol 3/7, 100 °C.
A number of sulfur and oxygen-containing heteroaromatics (thiazole, oxazole, thiadiazole and oxadiazole) were appended the naphthalene scaffold via a carbon linkage (Scheme 4). Since the halogen-substituted heterocycles were much more readily available, when compared to the boronate containing equivalents, the bromide of key intermediate 3 was transformed into the pinacol boronate 10 using a Suzuki-Miyaura borylation with bis(pinacolato)diboron. [Seifert et al. (2017) Angew. Chem. Int. Ed. 56, 7351] The subsequent Suzuki-Miyaura cross-coupling with a series of bromine substituted heteroaromatics yielded target compounds 11a- e [Suzuki (2011) Angewandte Chemie 50, 6723-6737].
Scheme 4. Introduction of heteroaromatics on the naphthalene scaffold

Reagents and conditions: (a) PdCl2(dppf), KOAc, bis(pinacolato)diboron, Ar, dioxane, 80 °C; (b) Pd(PPh3)4, K2CO3, aromatic bromide, THF/H2O 4: 1, 100 °C.
To have access to the 3,5-disubstituted 1,2,4-triazole analogue 13, the bromide containing key intermediate 3 was first transformed into the cyano congener 12, using potassium ferrocyanide (K4[Fe(CN)e]) as a safe cyanide source, by palladium- catalyzed chemistry [Ren et al. (2009) Org Process Res Dev 13, 2002489] Cyclocondensation of 12 with benzhydrazide furnished the desired analogue 13 [Yeung et al. (2005) Tetrahedron 46, 3429-3432] (Scheme 5).
Scheme 5. Synthesis of 3,5-disubstituted 1,2,4-triazole analogue 13

Reagents and conditions: (a) Pd(OAc)2, NazCCh, K4[Fe(CN)e], NMP, 140 °C; (b) K2CO3, benzhydrazide, n-butanol, 150 °C.
The synthesis of the inverted sulfonamide 18 started with the coupling of 1-amino- 4-bromonaphthalene 14 with 4-methoxybenzenesulfonylchloride, yielding sulfonamide 15 (Scheme 6). Buchwald amidation of 15 with o-toluamide turned out to be cumbersome as nearly all starting material was recovered after the reaction, most probably due to poisoning of the palladium catalyst. Therefore, the acidic proton of the sulfonamide moiety was protected with a p-methoxybenzyl (PMB) group furnishing compound 16 [Yoshioka et al. (2017J Bioorg Med Chem 25, 3461-3470]. The subsequent introduction of the amide moiety proceeded smoothly yielding compound 17. [Ruiz-Castillo & Buchwald (2016) Chem Rev 116, 12564-12649] Acidic cleavage of the protecting group using trifluoroacetic acid (TFA) afforded the desired inverted sulfonamide 18 [W02007067995].
Scheme 6. Synthesis of inverted sulfonamide 18

PMB = p-methoxybenzyl
Reagents and conditions: (a) 4-methoxybenzene sulfonylchloride, pyridine, rt ; (b) TBAI, NaH, 4-methoxybenzylbromide, Ar, DMF, rt ; (c) Pd(OAc)2, Xantphos, CS2CO3, o-toluamide, Ar, dioxane, 100 °C; (d) TFA, DCM, rt.
For the synthesis of the acylsulfonamide congener 22, initial attempts focused on the Buchwald Hartwig amidation of either 4-bromo-l-naphthoic acid 19 or the unprotected acylsulfonamide 20 with o-toluamide. Both approaches failed in the formation of the desired product and therefore an alternative strategy was needed. Protection of the nitrogen of the sulfonamide moiety as a PMB group was not a viable strategy, since the final removal of this protection group was problematic.
Therefore, 4-bromo-l-naphthoic acid 19 was reacted with 4- methoxybenzenesulfonamide using the Mukaiyama reagent as the coupling agent [Chen & Luo (2019) Tetrahedron Lett. 60, 268-271] followed by reaction with di-tert- butyl decarbonate, [Liu et al. (2018) Org Lett 20, 7771-7774] furnishing intermediate 21. This allowed the smooth introduction of the o-toluamide moiety in the subsequent step [Ruiz-Castillo & Buchwald, cited above], with concomitant cleavage of the Boc group, yielding target compound 22.
Scheme 7. Synthesis of the acylsulfonamide 22

Reagents and conditions: (a) 4-methoxybenzenesulfonamide, DMAP, EtsN, Mukaiyama's reagent, DCM, rt ; (b) DMAP, pyridine, BoczO, THF, 60 °C ; (c) Pd(OAc)2, Xantphos, CS2CO3, o-toluamide, Ar, dioxane, 100 °C.
Initial attempts to synthesize the sulfonimidamide isoster 28 followed a previously reported method [Davies et al. (2017) Angew. Chem. Int. Ed. 56, 14937-14941], which is based on a one-pot, three-component procedure for the preparation of differentially substituted sulfonimidamides. Briefly, reaction of /V-sulfinyltritylamine with the organolithium species derived from bromonaphthalene, followed by chlorination of the sulfinamide intermediate with tert-butylhypochlorite and reaction with the 4-amino-l-piperidinecarboxylate, did not allow to isolate the desired compound. Therefore, an alternative strategy was established (Scheme 8) [ Chen & Gibson (2015) RSC Adv 5, 4171]. Reaction of 4-bromo-l-naphthalenesulfonyl chloride 2 with an aqueous ammonia solution yielded the corresponding sulfonamide derivative 23. Protection of the nitrogen with a tert-butyldiphenylsilyl (TBDPS) group, allowed the in situ conversion of one oxygen atom of the sulfonamide moiety into a chlorine using triphenylphosphine dichloride. Nucleophilic substitution with ethyl 4- amino-piperidine-l-carboxylate, yielded the protected sulfonimidamide 26. Amidation via Buchwald coupling, followed by deprotection of the silyl group using tetrabutyl ammonium fluoride, resulted in the desired sulfonimidamide 28.
Scheme 8. Synthesis of sulfonimidamide analogue 28

TBDPS = tert-butyldiphenylsilyl
Reagents and conditions: (a) NH40H, Et20, acetone, 0 °C; (b) TBDPS-CI, EtsN, Ar, THF, 50 °C; (c) PPhsCl2, EtsN, Ar, Chloroform, 0 °C; (d) ethyl 4-aminopiperidine-l- carboxylate; (e) Pd(OAc)2, Xantphos, CS2CO3, o-toluamide, Ar, dioxane, 100 °C; (f) TBAF, THF, 40 °C.
The synthesis of the phosphonoamidate analogue 32 was achieved via a different strategy (Scheme 9). The phosphonate moiety was introduced on the naphthalene scaffold via palladium-catalyzed chemistry using diethyl phosphite and starting from l-amino-4-bromonaphthalene 14 [Bessmertnykh et al. (2009) Chem Lett 38, 738- 739] The exocyclic amino group was then converted to the amide 30 by reaction with o-toluoyl chloride [Jenkins et al. cited above]. Selective alkaline hydrolysis of one of the phosphono ethyl esters yielded compound 31 [van Overtveldt et al. (2015) Org Biomol Chem 13, 5260-5264] Reaction of phosphonate 31 with ethyl 4- aminopiperidine-l-carboxylate, using triphenylphosphine and 2,2'-dithiopyridine as coupling reagents yielded compound 32 [Lentini et al. (2019) ACS Med Chem Lett 10, 1284-1289]. Attempts to cleave off the second ethylester moiety were unsuccessful. Treatment of 32 with lithium hydroxide in methanol at 75°C only allowed to recover the starting material. Treatment with a Lewis acid, such as trimethylsilyl bromide at room temperature, resulted in concomitant cleavage of the phosphonoamidate bond yielding the fully deprotected phosphonate 33. Unfortunately, and in sharp contrast to the synthesis of compound 35, coupling of 33 with ethyl 4-aminopiperidine-l-carboxylate, using triphenylphosphine and 2,2'- dithiopyridine as coupling reagents, did not lead to the formation of the desired phosphonoamidate.
Scheme 9. Synthesis of phosphonoamidate analogue 32

Reagents and conditions: (a) Pd2(OAc)2, PPhs, diethylphosphite, EtsN, Ar, ethanol, 80 °C; (b) o-toluoyl chloride, pyridine, 100 °C; (c) NaOH, H2O, dioxane, 100 °C; (d) 2,2'-dithiopyridine, PPF13, EtsN, ethyl 4-aminopiperidine-l-carboxylate, pyridine, 60 °C.
Isoquinoline analog 38 was synthesized starting from 5-bromoisoquinoline 34. (Scheme 10). The sulfonic acid moiety was introduced using fuming sulfuric acid affording compound 35, which was isolated by precipitation from water [WO2013178362A1]. The sulfonic acid 35 was then transformed into the sulfonamide 37 via a one pot reaction using the sulfonyl chloride as an in situ prepared intermediate [Jenkins et al. cited above]. Finally, the amide moiety was introduced via Buchwald coupling yielding target compound 38 [Ruiz-Castello & Buchwald, cited above].
Scheme 10. Synthesis of isoquinoline analogue 38

Reagents and conditions: (a) fuming sulfuric acid, 100°C; (b) thionyl chloride, DMF, 75°C; (c) DMAP, EtsN, ethyl 4-aminopiperidine-l-carboxylate, DCM, rt.; (d) Pd(OAc)2, Xantphos, CS2CO3, o-toluamide, Ar, dioxane, 100 °C.
Biological evaluation and structure-activity relationship studies
All compounds were investigated in two types of CCR8-related assays (Tables 1-4). In a first type of experiments, the compounds were evaluated in a competition binding assay, in which increasing concentrations of the compounds compete with a fixed concentration of fluorescently labelled CCL1. It allows to determine the concentration of unlabelled compounds that is required to inhibit 50% of the binding of the labelled ligand (ICso)- Since this type of assay does not allow to distinguish CCR8 agonists from antagonists, a second, functional cell-based assay was implemented throughout the study. Binding of the natural chemokine ligand CCL1 to human CCR8 leads to a ligand-dependent release of calcium stores from the endoplasmic reticulum into the cytosol. The basic principle of the calcium mobilization assay is to load cells with a calcium-sensitive dye, which fluoresces when bound to calcium; therefore, an increased fluorescence signal is indicative of activation of CCR8. Hence, inhibition of CCL1 induced calcium mobilization by various compounds is indicative for their CCR8 antagonistic activity.
Structure-activity relationship study of the amide moiety
It has been shown before that the amide moiety is essential for CCR8 antagonism, since removal of the carbonyl group (yielding amine derivatives) [Jenkins et al. cite
above] or connecting the phenyl ring to the naphthalene core via various carboncarbon bonds [Verhaegen et al, cited above] afforded compounds displaying strongly reduced CCR.8 antagonism.
In order to retain the geometry and the hydrogen bond accepting/donating properties of the amide group, the focus in this work was redirected toward its bio-isosteric replacement. Initially, a number of nitrogen-containing five-membered heteroaromatics, linked via one of their nitrogen atoms to the naphthalene scaffold, was prepared and evaluated (Table 1). The parent amide lead structure (compound 4), included as reference compound, displayed excellent CCR.8 affinity and CCR8 antagonism, in perfect agreement with previous reports. The pyrazole (compound 7a), the imidazole (compound 7b), the 1,2,4-triazole (compound 7c) and the 2,4- disubstituted 1,2,3-triazole analogue (compound 7e) showed potent inhibition of labeled CCL1 binding, with IC50 values in the range of 18-42 nM. The same analogues were also endowed with excellent activity in the functional CCR8 calcium mobilization assay (IC50 values between 0.089 and 3.2 pM), although a 5- to 76-fold loss in activity in the calcium assay is observed, when compared to the binding assay.
The 1,4-disubstituted 1,2,3-triazole (compound 7d) was the most potent analogue within this series. When compared to the parent amide analogue 4, the triazole was equally active in the CCR8 binding assay (IC50 = 5 nM), and 10-fold less active in the calcium mobilization assay. This potency of compound 7d is not that surprising, since the triazole moiety has gained widespread use, as an excellent non-classical bioisoster of the trans amide bond [Kumari et al. (2020) J. Med. Chem 63, 12290- 12358].
Table 1. CCL1 competition binding and CCR8 calcium mobilization data of compounds 4 and 7a-e


 aICso : the compound concentration inhibiting CCL1AF647 binding by 50%. bICso : the compound concentration that inhibits CCL1 induced intracellular calcium flux by 50%.
aICso : the compound concentration inhibiting CCL1AF647 binding by 50%. bICso : the compound concentration that inhibits CCL1 induced intracellular calcium flux by 50%.
The potent CCR8 antagonistic activity of triazole 7d prompted us to probe the SAR around the phenyl ring (Table 2). Fusing the phenyl moiety with the triazole ring afforded the regioisomeric benzotriazole derivatives 7f and 7g. Both regioisomers are 40-fold less active in the binding assay, when compared to the phenyl-substituted triazole analogue 7d. Their decreased activity as CCR8 antagonists is more pronounced in the calcium mobilization assay, as evident from their IC50 values in the low pM range.
In order to probe the optimal substitution pattern, electron-donating (methyl and methoxy) as well as electron-withdrawing (fluorine and trifluoromethyl) substituents were introduced at various positions of the phenyl ring. The methoxy-substituted analogues (compounds 9a-c) are only 4-fold less active in the competition binding assay when compared to the unsubstituted analogue 7d (IC50 values in the 20 nM range versus 5 nM for compound 7d), whereas the methyl, fluoro and trifluoromethyl substituted analogues (compounds 9d-j) displayed excellent activity with IC50 values of less than 10 nM in the binding assay. Overall, compounds 9a-j are less active in the calcium mobilization assay than in the CCL1 competition binding assay, with the 2-methylphenyl analogue 9d being the most potent within this series with an IC50 value of 18 nM.
Table 2. CCL1 competition binding and CCR8 calcium mobilization data of compounds 7f-g and 9a-j


 aICso : the compound concentration inhibiting CCL1AF647 binding by 5O%.bICso : the compound concentration that inhibits CCL1 induced intracellular calcium flux by 50%.
aICso : the compound concentration inhibiting CCL1AF647 binding by 5O%.bICso : the compound concentration that inhibits CCL1 induced intracellular calcium flux by 50%.
Besides 1,2,3-triazole, other five-membered heterocycles have also been applied as amide bond surrogates [Kumari et al. cited above]. Therefore, the amide bond of compound 4 was replaced by a number of other heteroaromatics that are linked via a carbon-carbon bond to the naphthalene core (Table 3). The 1,2,4-triazole derivative 13 was the least active, as evident from an IC50 value of 0.297 pM in the binding and 5.901 pM in the calcium mobilization assay. The sulfur-containing heteroaromatics, such as the 1,3,4-thiadiazole Ila, the 5-phenylthiazole 11b and the 2- phenylthiazole 11c do show activities in the binding assay with IC50 values of 10 nM or less, but loose quite some activity (40- to 120-fold) upon evaluation in the calcium assay. The 1,3,4-oxadiazole lid and the 2-phenyloxazole lie are less active than their corresponding sulfur-containing analogues Ila and 11c, respectively, in the binding, as well as in the calcium assay.
Table 3. CCL1 competition binding and CCR8 calcium mobilization data of compounds lla-e and 13

 aICso : the compound concentration inhibiting CCL1AF647 binding by 50%. bICso : the compound concentration that inhibits CCL1 induced intracellular calcium flux by 50%.
aICso : the compound concentration inhibiting CCL1AF647 binding by 50%. bICso : the compound concentration that inhibits CCL1 induced intracellular calcium flux by 50%.
Structure-activity relationship study of the sulfonamide moiety
The piperidinyl substituent on the sulfonamide linkage has been optimized before, although the sulfonamide group itself has only received little attention. It has, however, been shown that the presence of an amide or an amine (instead of the sulfonamide) led to a complete loss of CCR8 antagonism [Jenkins et al. cited above]. In this study, we embarked on the synthesis of additional isosters. Because of synthetic feasibility, the newly synthesized sulfonamide analogues 18 and 22 carried a 4-methoxyphenyl residue (rather than the 4-amino-piperidinyl side chain). Therefore, a new reference compound (compound 5) was included to allow for comparison of the biological data and to study the influence of variation of the sulfonamide part, keeping the rest of the molecule intact. The CCR8 antagonistic data of compound 6 are in agreement with literature values [Jenkins et al. cited above]. The inverted sulfonamide (compound 18), as well as the acylsulfonamide (compound 22) and the phosphonoamidate (compound 32) are completey devoid of CCR8
affinity, with IC50 values exceeding 30 pM. Sulfonimidamides, which are the monoaza counterparts of sulfonamides, have been explored to a limited extent in medicinal chemistry [Chinthakindi et al. (2017) Angew Chem Int Ed Engl. 56, 4100-4109]. The sulfonimidamide analogue 30 is equally active as the parent sulfonamide congener 4.
Table 4. CCL1 competition binding and CCR.8 calcium mobilization data of compounds 6, 18, 22,

 aICso : the compound concentration inhibiting CCL1AF647 binding by 50%. bICso : the compound concentration that inhibits CCL1 induced intracellular calcium flux by 50%.
aICso : the compound concentration inhibiting CCL1AF647 binding by 50%. bICso : the compound concentration that inhibits CCL1 induced intracellular calcium flux by 50%.
ND : not determined.
Structure-activity relationship study of the naphthalene scaffold
Finally, the importance of the naphthalene scaffold for CCR.8 antagonism was probed by the synthesis of isoquinoline analogue 38. Side-by-side comparison of the naphthalene (compound 4) and isoquinoline (compound 38) congeners demonstrated similar levels of activity in the binding as well as in the calcium assay (Table 5).
Table 5. CCL1 competition binding and CCR.8 calcium mobilization data of compounds 4 and 38

Examples
Experimental section
Chemicals received from commercial sources (Sigma-Aldrich, Acros Organics, J&K Scientific, Alfa Aesar, Fluorochem or TCI Chemicals) were used without further purification. Dry reaction solvents were purchased from commercial sources. Thin- layer chromatography (TLC) was performed on silica gel 0.20 mm 60 with fluorescent indicator UV254 (pre-coated aluminium sheets) from Merck. For column chromatography 60-200 mesh silica gel 60 (Acros) was used as stationary phase. NMR spectra were acquired on commercial instruments (Bruker Avance 300 MHz, Bruker AMX 400 MHz or Bruker Avance 11+ 600 MHz) and chemical shifts (6) are reported in parts per million (ppm) referenced to tetramethylsilane (1H), or the internal (NMR) solvent signal (13C). High resolution mass spectra were acquired on a quadrupole orthogonal acceleration time-of-flight mass spectrometer (Synapt G2 HDMS, Waters, Milford, MA, USA). Samples were infused at 3 pL/min and spectra were obtained in positive mode with a resolution of 15 000 (FWHM) using leucine enkephalin as lock mass.
4-Bromonaphthalene-l-sulfonyl chloride (2)
In a round-bottom flask equipped with a magnetic stirring bar, 1-bromonaphthalene (700 pl, 5 mmol, 1 eq.) was dissolved in chloroform (10 ml) and allowed to stir at 0 °C. In another round-bottom flask chloroform (5 ml) was cooled down to 0 °C, after which chlorosulfonic acid (1000 pl, 15 mmol, 3 eq.) was added. To the solution of 1- bromonaphthalene was added the solution of chlorosulfonic acid in chloroform dropwise under ice bath and stirred for 4h at 0 °C. The mixture was poured slowly into ice water (50 mL) and extracted three times with dichloromethane. The combined organic layers were dried over anhydrous calcium chloride, filtered and concentrated in vacuo to give the title compound as a solid (1147 mg, 75% yield), which was used without further purification for the next step.
XH NMR (400 MHz, CDCI3) 6 (ppm) 8.82 (d, J = 8.5 Hz, 1H), 8.47 (d, J = 8.4 Hz, 1H), 8.21 (d, J = 8.1 Hz, 1H), 7.95 (d, J = 8.1 Hz, 1H), 7.87 (t, J = 7.6 Hz, 1H), 7.81 (t, J = 7.6 Hz, 1H).
Ethyl 4-((4-bromonaphthalene)-l-sulfonamido)piperidine-l-carboxylate (3)
In a round-bottom flask equipped with a magnetic stirring bar, 4-bromo-l- naphthalene sulfonyl chloride (2) (1.141 g, 3.734 mmol, 1 eq.), dichloromethane (90 ml), triethylamine (1.04 ml, 7.468 mmol, 2 eq.) and ethyl-4-aminopiperidine-l- carboxylate (769 pl, 4.481 mmol, 1.2 eq.) were added and allowed to stir for 17h at
room temperature. The mixture was quenched with water (60 ml) and extracted three times with dichloromethane. The combined organic layers were dried over anhydrous magnesium sulfate, filtered and concentrated in vacuo. Silica gel column chromatography of the crude product using petroleum ether/ethyl acetate (in a ratio of 6/4) as the mobile phase provided the title compound as a solid (92% yield, 1522 mg).
XH NMR (400 MHz, CDCI3): 6 (ppm) 8.62-8.60 (m, 1H), 8.43-8.40 (m, 1H), 8.14 (d, J = 7.9 Hz, 1H), 7.89 (d, J = 7.9 Hz, 1H), 7.74-7.72 (m, 2H), 4.62 (broad, 1H), 4.06 (q, J = 7.0 Hz, 2H), 3.90-3.87 (m, 2H), 3.30-3.28 (m, 1H), 2.79-2.73 (m, 2H), 1.66- 1.63 (m, 2H), 1.28-1.18 (m, 2H), 1.20 (t, J = 7.1 Hz, 3H). 13C (101MHz, CDCI3): 6 (ppm) 155.26, 135.60, 132.65, 129.93, 129.39, 129.10, 129.01, 128.55, 128.53, 128.33, 124.87, 61.48, 50.93, 42.22, 32.64, 14.61. Exact mass (HRMS, ESI) calculated for CisHziBrNzC S (M + H)+: 441.04785, found 441.0468
Ethyl 4-((4-(2-methylbenzamido)naphthalene)-l-sulfonamido)piperidine- 1-carboxylate (4)
To an oven-dried screw-capped reaction tube equipped with a magnetic stirring bar, Pd(OAc)2 (2.2 mg, 0.01 mmol, 0.1 eq.), Xantphos (8.7 mg, 0.015 mmol, 0.15 eq.), CS2CO3 (45.6 mg, 0.14 mmol, 1.4 eq.), o-toluamide (16.2 mg, 0.12 mmol, 1.2 eq.) and ethyl-4-(4-bromonaphthalene-l-sulfonylamino)-piperidine- 1-carboxylate (3) (44.1 mg, 0.1 mmol, 1 eq.) were added. The reaction tube was evacuated and backfilled with argon three times. Anhydrous dioxane (0.1 ml) was added and the reaction was allowed to stir for 16h at 100 °C. The mixture was diluted with DCM (1 ml) and filtered. The filtrate was concentrated in vacuo and the crude residue was purified by silica gel column chromatography using petroleum ether/ethyl acetate (4/6) as the mobile phase yielding the title compound as a solid (90% yield, 45 mg). XH NMR (400 MHz, CDCI3): 6 (ppm) 8.69 (d, J = 1H), 8.44 (d, J = 7.4 Hz, 1H), 8.36 (d, J = 8.0 Hz, 1H), 8.11 (s, 1H), 7.95 (d, J = 8.3 Hz, 1Hz), 7.74-7.63 (m, 3H), 7.48- 7.44 (m, 1H), 7.37-7.33 (m, 2H), 4.56 (d, J = 7.6 Hz, 1H), 4.07 (q, J = 7.0 Hz, 1H), 3.91-3.87 (m, 2H), 3.35-3.26 (m, 1H), 2.83-2.72 (m, 2H), 2.60 (s, 3H), 1.73-1.62 (m, 2H), 1.31-1.18 (m, 2H), 1.21 (t, J = 7.1 Hz, 3H).
4-Bromo-N-(4-methoxyphenyl)naphthalene-l-sulfonamide (5)
In a round-bottom flask equipped with a magnetic stirring bar, 4-bromo-l- naphthalene sulfonyl chloride 2 (301 mg, 0.987 mmol, 1 eq.), dichloromethane (20 ml), triethylamine (0.27 ml, 1.973 mmol, 2 eq.) and p-anisidine (146 mg, 1.184 mmol, 1.2 eq.) were added and allowed to stir for 17h at room temperature. The mixture was quenched with water (18 ml) and extracted three times with dichloromethtane. The combined organic layers were dried over anhydrous magnesium sulfate, filtered and concentrated in vacuo. Silica gel column chromatography of the crude product using petroleum ether/ethyl acetate (in a ratio
of 6/4) as the mobile phase provided the title compound as a solid (56% yield, 217.6 mg).
XH NMR (400 MHz, CDCI3) 6 8.70 (m, J = 1.9 Hz, 1H), 8.40 (m, J = 2.4 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.75 (d, J = 8.0 Hz, 1H), 7.72 (m, J = 2.0 Hz, 2H), 6.78 (m, J = 3.0 Hz, 2H), 6.64 (m, J = 3.0 Hz, 2H), 6.56 (s, 1H), 3.70 (s, 3H). 13C NMR (101 MHz, CDCI3) 6 158.32, 133.91, 132.48, 130.43, 130.09, 129.21, 129.18, 128.58, 128.50, 128.24, 128.05, 126.08, 124.75, 114.40, 55.38.
/V-(4-(/V-(4-Methoxyphenyl)sulfamoyl)naphthalen-l-yl)-2- methylbenzamide (6)
To an oven-dried screw-capped reaction tube equipped with a magnetic stirring bar, Pd(OAc)2 (11.2 mg, 0.05 mmol, 0.1 eq.), Xantphos (43.4 mg, 0.075 mmol, 0.15 eq.), CS2CO3 (228.1 mg, 0.7 mmol, 1.4 eq.), o-toluamide (81.1 mg, 0.6 mmol, 1.2 eq.) and 4-bromo-N-(4-methoxyphenyl)naphthalene-l-sulfonamide 5 (196.1 mg, 0.5 mmol, 1 eq.) were added. The reaction tube was evacuated and backfilled with argon three times. Anhydrous dioxane (0.5 ml) was added and the reaction was allowed to stir for 16 h at 100 °C. The mixture was diluted with DCM (5 ml) and filtered. The filtrate was concentrated in vacuo and the crude residue was purified by silica gel column chromatography using petroleum ether/ethyl acetate (6/4) as the mobile phase yielding the title compound as a solid (quantitative yield, 224 mg).
XH NMR (400 MHz, CDCI3) 6 8.74 (m, J = 4.6 Hz, 1H), 8.30 (d, J = 7.4 Hz, 1H), 8.15 (d, J = 8.3 Hz, 1H), 8.09 (s, 1H), 7.93 (m, J = 4.3 Hz, 1H), 7.66 (m, J = 3.2 Hz, 3H), 7.44 (m, J = 4.1 Hz, 1H), 7.33 (m, J = 7.3 Hz, 2H), 6.83 (m, J = 3.0 Hz, 2H), 6.66 (m, J = 3.0 Hz, 2H), 6.52 (s, 1H), 3.71 (s, 3H), 2.57 (s, 3H). 13C NMR (101 MHz, CDCI3) 6 158.15, 137.74, 137.02, 135.66, 131.71, 131.38, 130.96, 130.30, 129.20, 128.40, 128.38, 127.17, 126.64, 126.22, 125.75, 125.47, 120.75, 116.74, 114.43, 55.39, 20.03.
Synthesis of /V-linked heteroaromatic naphthalene sulfonamide analogues
General procedure
To an oven-dried screw-capped reaction tube equipped with a magnetic stirring bar, Cui (3.8 mg, 0.02 mmol, 0.2 eq.), ethyl-4-(4-bromonaphthalene-l-sulfonylamino)- piperidine-l-carboxylate (3) (44.1 mg, 0.1 mmol, 1 eq.), L-proline (4.6 mg, 0.04 mmol, 0.4 eq.), K2CO3 (55 mg, 0.4 mmol, 4 eq.) and phenylazole (0.25 mmol, 2.5 eq.) were added. The reaction tube was evacuated and backfilled with argon three times. Anhydrous DMSO (0.2 ml) was added and the reaction was allowed to stir for 18h at 120 °C. The mixture was quenched with brine and extracted three times with dichloromethane. The combined organic layers were dried over anhydrous magnesium sulfate, filtered and concentrated in vacuo. Silica gel column chromatography of the crude product provided the desired compound. The following compounds were made according to this procedure.
Ethyl 4-((4-(3-phenyl-lH-pyrazol-l-yl)naphthalene)-l-sulfonamido) piperidine-l-carboxylate (7a)
This compound was prepared according to the general procedure using 3- phenylpyrazole (36.0 mg, 0.25 mmol, 2.5 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (13.3 mg, 26% yield).
XH NMR (400 MHz, CDCI3) 6 8.71 (d, J = 8.6 Hz, 1H), 8.40 (d, J = 7.8 Hz, 1H), 8.20 (d, J = 8.4 Hz, 1H), 7.94 (d, J = 7.3 Hz, 2H), 7.88 (d, J = 2.4 Hz, 1H), 7.74 (t, J =
7.4 Hz, 1H), 7.67 (m, 2H), 7.46 (t, J = 7.4 Hz, 2H), 7.38 (t, J = 7.3 Hz, 1H), 6.92 (d, J = 2.3 Hz, 1H), 4.97 (d, J = 7.6 Hz, 1H), 4.07 (q, J = 7.1 Hz, 2H), 3.92 (d, J = 7.8 Hz, 2H), 3.30 (m, 1H), 2.78 (t, J = 11.9 Hz, 2H), 1.70 (d, J = 10.4 Hz, 2H), 1.29 (m, 2H), 1.21 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCI3) 6 155.27, 153.90, 142.00, 135.60, 133.36, 132.63, 129.55, 129.32, 129.25, 128.97, 128.79, 128.42, 128.04, 125.96, 124.98, 124.58, 121.17, 105.02, 61.48, 51.04, 42.27, 32.83, 14.61.
Ethyl 4-((4-(4-phenyl-lH-imidazol-l-yl)naphthalene)-l-sulfonamido) piperidine-l-carboxylate (7b)
This compound was prepared according to the general procedure using 4- phenylimidazole (36.0 mg, 0.25 mmol, 2.5 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (21.3 mg, 41% yield).
XH NMR (400 MHz, CDCI3) 6 8.76 (d, J = 8.6 Hz, 1H), 8.39 (d, J = 7.7 Hz, 1H), 7.88 (m, 4H), 7.76 (t, J = 7.5 Hz, 1H), 7.68 (t, J = 7.5 Hz, 1H), 7.60 (m, 2H), 7.43 (t, J =
7.5 Hz, 2H), 7.31 (t, J = 7.2 Hz, 1H), 5.36 (d, J = 7.6 Hz, 1H), 4.07 (q, J = 6.9 Hz, 2H), 3.93 (d, J = 10.8 Hz, 2H), 3.39 (m, 1H), 2.81 (m, 2H), 1.73 (d, J = 10.2 Hz, 2H), 1.31 (m, 2H), 1.21 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCI3) 6 155.28, 142.98, 138.70, 138.51, 136.92, 133.18, 130.04, 129.32, 129.14, 128.85, 128.82, 128.61, 127.54, 125.13, 125.06, 123.75, 121.93, 117.18, 61.54, 51.15, 42.30,
32.81, 14.62.
Ethyl 4-((4-(3-phenyl-lH-l,2,4-triazol-l-yl)naphthalene)-l-sulfonamido) piperidine-l-carboxylate (7c)
This compound was prepared according to the general procedure using 3-phenyl- 1,2,4-triazole (36.3 mg, 0.25 mmol, 2.5 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (31.3 mg, 62% yield).
NMR (400 MHz, CDCI3) 6 8.78 (d, J = 8.4 Hz, 1H), 8.56 (s, 1H), 8.41 (d, J = 7.7 Hz, 1H), 8.23 (d, J = 6.2 Hz, 2H), 8.04 (d, J = 8.2 Hz, 1H), 7.71 (m, 3H), 7.49 (m, 3H), 5.69 (d, J = 5.8 Hz, 1H), 4.07 (m, 2H), 3.91 (m, 2H), 3.33 (m, 1H), 2.79 (t, J = 10.9 Hz, 2H), 1.70 (d, J = 11.0 Hz, 2H), 1.26 (m, 5H). 13C NMR (101 MHz, CDCI3) 6
163.81, 155.31, 145.83, 138.14, 137.50, 130.10, 129.99, 129.38, 129.31, 129.22,
128.81, 128.77, 128.65, 126.69, 124.95, 124.10, 121.75, 61.55, 51.08, 42.30, 32.76, 14.62.
Ethyl 4-((4-(4-phenyl-lH-l,2,3-triazol-l-yl)naphthalene)-l-sulfonamido) piperidine-l-carboxylate (7d) and ethyl 4-((4-(4-phenyl-2H-l,2,3-triazol-2- yl)naphthalene)-l-sulfonamido)piperidine-l-carboxylate (7e)
Both compounds were prepared according to the general procedure on a doubled scale using 4-phenyl-l,2,3-triazole (72.5 mg, 0.5 mmol, 2.5 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding title compound 7d as a solid (28.0 mg, 27% yield) and title compound 7e as a solid (29.1 mg, 28% yield).
Compound 7d: NMR (300 MHz, DMSO) 6 9.22 (s, 1H), 8.84 (d, J = 8.5 Hz, 1H), 8.37 (d, J = 7.7 Hz, 2H), 7.90 (m, J = 12.5 Hz, 6H), 7.53 (t, J = 7.3 Hz, 2H), 7.42 (t, J = 6.8 Hz, 1H), 3.98 (q, J = 6.8 Hz, 2H), 3.74 (d, J = 13.1 Hz, 2H), 2.82 (t, J = 11.2 Hz, 2H), 1.56 (d, J = 11.3 Hz, 2H), 1.27 (m, 2H), 1.13 (t, J = 6.9 Hz, 3H). 13C NMR (75 MHz, DMSO) 6 154.42, 146.86, 138.59, 137.16, 130.09, 129.01, 128.79, 128.75, 128.65, 128.35, 127.89, 125.47, 125.21, 124.39, 123.45, 122.66, 60.60, 49.99, 41.82, 32.19, 14.50.
Compound 7e: NMR (400 MHz, CDCI3) 6 8.75 (d, J = 8.4 Hz, 1H), 8.59 (d, J = 8.4 Hz, 1H), 8.44 (d, J = 8.0 Hz, 1H), 8.27 (s, 1H), 7.96 (m, 3H), 7.73 (m, 2H), 7.51 (t, J = 7.4 Hz, 2H), 7.44 (t, J = 7.3 Hz, 1H), 5.16 (d, J = 7.6 Hz, 1H), 4.07 (q, J = 7.1 Hz, 2H), 3.89 (m, 2H), 3.32 (m, 1H), 2.78 (t, J = 11.9 Hz, 2H), 1.68 (d, J = 10.6 Hz, 2H), 1.30 (m, 2H), 1.20 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCI3) 6 155.27, 149.88, 140.94, 136.17, 133.57, 129.43, 129.39, 129.32, 129.12, 129.02, 128.93, 128.17, 127.95, 126.30, 125.34, 124.57, 120.77, 61.49, 51.07, 42.27, 32.76, 14.60.
Ethyl 4-((4-(2H-benzo[d][l,2,3]triazol-2-yl)naphthalene)-l-sulfonamido) piperidine-l-carboxylate (7f) and ethyl 4-((4-(lH-benzo[d][l,2,3]triazol-l- yl)naphthalene)-l-sulfonamido)piperidine-l-carboxylate (7g)
Both compounds were prepared according to the general procedure on a doubled scale using lH-benzo[d][l,2,3]triazole (59.6 mg, 0.5 mmol, 2.5 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding title compound 7f as a solid (13.8 mg, 14% yield) and title compound 7g as a solid (20.9 mg, 22% yield).
Compound 7f:
 NMR (300 MHz, CDCI3) 6 8.79 (d, J = 8.6 Hz, 1H), 8.50 (d, J = 7.9 Hz, 1H), 8.44 (d, J = 8.4 Hz, 1H), 8.07 (m, 3H), 7.76 (m, 2H), 7.56 (m, 2H), 5.01 (d, J = 7.6 Hz, 1H), 4.09 (q, J = 6.8 Hz, 2H), 3.94 (d, J = 11.3 Hz, 2H), 3.37 (m, 1H), 2.80 (t, J = 11.9 Hz, 2H), 1.72 (d, J = 12.0 Hz, 2H), 1.25 (m, 5H). 13C NMR (101 MHz, CDCI3) 6 155.25, 145.35, 141.57, 137.28, 129.27, 129.15, 128.75, 128.60, 128.40, 127.90, 125.08, 124.59, 122.37, 118.58, 61.49, 51.19, 42.29, 32.83, 14.60.
NMR (300 MHz, CDCI3) 6 8.79 (d, J = 8.6 Hz, 1H), 8.50 (d, J = 7.9 Hz, 1H), 8.44 (d, J = 8.4 Hz, 1H), 8.07 (m, 3H), 7.76 (m, 2H), 7.56 (m, 2H), 5.01 (d, J = 7.6 Hz, 1H), 4.09 (q, J = 6.8 Hz, 2H), 3.94 (d, J = 11.3 Hz, 2H), 3.37 (m, 1H), 2.80 (t, J = 11.9 Hz, 2H), 1.72 (d, J = 12.0 Hz, 2H), 1.25 (m, 5H). 13C NMR (101 MHz, CDCI3) 6 155.25, 145.35, 141.57, 137.28, 129.27, 129.15, 128.75, 128.60, 128.40, 127.90, 125.08, 124.59, 122.37, 118.58, 61.49, 51.19, 42.29, 32.83, 14.60.
Compound
 8.81 (d, J = 8.7 Hz, 1H), 8.51 (d, J =
8.81 (d, J = 8.7 Hz, 1H), 8.51 (d, J =
7.8 Hz, 1H), 8.25 (d, J = 7.7 Hz, 1H), 7.76 (d, J = 7.9 Hz, 2H), 7.57 (m, 4H), 7.33 (d, J = 7.9 Hz, 1H), 5.29 (d, J = 7.5 Hz, 1H), 4.08 (q, J = 7.0 Hz, 2H), 3.95 (d, J =
11.8 Hz, 2H), 3.41 (m, 1H), 2.83 (t, J = 11.7 Hz, 2H), 1.76 (d, J = 10.3 Hz, 2H), 1.36 (m, 2H), 1.21 (m, 3H). 13C NMR (101 MHz, CDCI3) 6 155.29, 145.85, 137.75,
137.51, 134.51, 129.83, 129.41, 129.33, 128.86, 128.55, 124.99, 124.87, 124.24, 122.87, 120.47, 110.13, 61.53, 51.23, 42.31, 32.89, 14.61.
Synthesis of l,4-disubstituted-l,2,3-triazoles
General procedure
To a screw-capped reaction tube equipped with a magnetic stirring bar, sodium azide (26.0 mg, 0.4 mmol, 2 eq.), sodium ascorbate (4.0 mg, 0.02 mmol, 0.1 eq.), N,N'- dimethylethylenediamine (4 pl, 0.04 mmol, 0.2 eq.), Cui (3.8 mg, 0.02 mmol, 0.1 eq.), ethyl-4-(4-bromonaphthalene-l-sulfonylamino)-piperidine-l-carboxylate (3) (88.3 mg, 0.2 mmol, 1 eq.) and alkyn (0.24 mmol, 1.2 eq.) were dissolved in an ethanol/water mixture (1 mL, 7:3). The mixture was allowed to stir for 18h at 100 °C. The cooled mixture was poured into 50 mL water and the aqueous solution was extracted three times with 50 mL DCM. The combined organic layers were dried over anhydrous magnesium sulfate, filtered and concentrated in vacuo. Silica gel column chromatography of the crude product provided the desired compound. The following compounds were made according to this procedure.
Ethyl 4-((4-(4-(2-methoxyphenyl)-lH-l,2,3-triazol-l-yl)naphthalene)-l- sulfonamido) piperidine-l-carboxylate (9a)
This compound was prepared according to the general procedure using 2- methoxyphenyl acetylene (31 pl, 0.24 mmol, 1.2 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (54.8 mg, 51% yield).
XH NMR (400 MHz, CDCI3) 6 8.79 (d, J = 8.6 Hz, 1H), 8.50 (m, 2H), 8.43 (d, J = 7.8 Hz, 1H), 7.85 (d, J = 8.5 Hz, 1H), 7.72 (m, 2H), 7.65 (t, J = 7.7 Hz, 1H), 7.39 (m, 1H), 7.15 (t, J = 7.6 Hz, 1H), 7.04 (d, J = 8.2 Hz, 1H), 5.71 (d, J = 7.6 Hz, 1H), 4.07 (q, J = 7.0 Hz, 1H), 3.95 (m, 5H), 3.35 (m, 1H), 2.80 (t, J = 11.8 Hz, 2H), 1.71 (m, 2H), 1.34 (m, 2H), 1.20 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCI3) 6 155.84, 155.30, 143.60, 138.38, 137.68, 129.54, 129.52, 129.23, 129.09, 128.65, 128.63, 127.84, 125.70, 124.91, 124.03, 121.99, 121.19, 118.56, 110.91, 61.52, 55.46, 51.07, 42.29, 32.73, 14.60.
Ethyl 4-((4-(4-(3-methoxyphenyl)-lH-l,2,3-triazol-l-yl)naphthalene)-l- sulfonamido)piperidine-l-carboxylate (9b)
This compound was prepared according to the general procedure using 3- methoxyphenyl acetylene (30 pl, 0.24 mmol, 1.2 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (30.1 mg, 28% yield).
1
XH NMR (400 MHz, CDCI3) 6 8.76 (d, J = 8.6 Hz, 1H), 8.42 (d, J = 7.8 Hz, 1H), 8.23 (s, 1H), 7.83 (d, J = 8.4 Hz, 1H), 7.71 (m, 3H), 7.58 (t, J = 1.9 Hz, 1H), 7.49 (d, J =
7.6 Hz, 1H), 7.39 (t, J = 7.9 Hz, 1H), 6.96 (dd, J = 1.9, 8.2 Hz, 1H), 5.43 (d, J = 7.7 Hz, 1H), 4.07 (q, J = 7.1 Hz, 2H), 3.91 (m, 5H), 3.35 (m, 1H), 2.79 (t, J = 11.7 Hz, 2H), 1.71 (m, 2H), 1.32 (m, 2H), 1.20 (t, J = 7.1 Hz, 1H). 13C NMR (101 MHz, CDCI3) 6 160.19, 155.28, 148.12, 138.12, 137.88, 131.05, 130.14, 129.37, 129.35, 129.07, 128.79, 128.61, 124.92, 123.85, 122.57, 121.98, 118.33, 114.80, 111.08, 61.53, 55.44, 51.14, 42.28, 32.78, 14.60.
Ethyl 4-((4-(4-(4-methoxyphenyl)-lH-l,2,3-triazol-l-yl)naphthalene)-l- sulfonamido)piperidine-l-carboxylate (9c)
This compound was prepared according to the general procedure using 4- methoxyphenyl acetylene (26 pl, 0.24 mmol, 1.2 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (71.8 mg, 67% yield).
NMR (400 MHz, CDCI3) 6 8.75 (d, J = 8.6 Hz, 1H), 8.43 (d, J = 7.8 Hz, 1H), 8.13 (s, 1H), 7.88 (m, 3H), 7.78 (t, J = 7.6 Hz, 1H), 7.70 (m, 2H), 7.03 (d, J = 8.6 Hz, 1H), 5.03 (d, J = 7.6 Hz, 1H), 4.07 (q, J = 7.1 Hz, 2H), 3.88 (m, 5H), 3.35 (m, 1H), 2.79 (t, J = 11.8 Hz, 2H), 1.72 (d, J = 10.4 Hz, 2H), 1.31 (m, 2H), 1.21 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCI3) 6 160.07, 155.25, 148.17, 138.32, 137.70, 129.41, 129.09, 128.76, 128.70, 127.31, 124.83, 123.99, 122.40, 121.92, 121.46, 114.49, 61.51, 55.39, 51.22, 42.29, 32.87, 14.61.
Ethyl 4-((4-(4-(2-methylphenyl)-lH-l,2,3-triazol-l-yl)naphthalene)-l- sulfonamido)piperidine-l-carboxylate (9d)
This compound was prepared according to the general procedure using 2- methoxyphenyl acetylene (30 pl, 0.24 mmol, 1.2 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (47.9 mg, 46% yield).
XH NMR (400 MHz, CDCI3) 6 8.78 (d, J = 8.6 Hz, 1H), 8.43 (d, J = 7.8 Hz, 1H), 8.14 (s, 1H), 7.94 (m, 1H), 7.86 (d, J = 8.4 Hz, 1H), 7.75 (m, 2H), 7.68 (t, J = 7.4 Hz, 1H), 7.34 (m, 3H), 5.58 (d, J = 7.7 Hz, 1H), 4.06 (q, J = 7.1 Hz, 2H), 3.91 (d, J =
11.6 Hz, 2H), 3.35 (m, J = 4.6 Hz, 1H), 2.79 (t, J = 11.8 Hz, 2H), 2.58 (s, 3H), 1.70 (m, 2H), 1.32 (m, 2H), 1.20 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCI3) 6 155.28, 147.48, 138.12, 137.86, 135.66, 131.12, 129.38, 129.33, 129.10, 129.05, 128.77, 128.70, 128.61, 126.34, 124.98, 124.43, 123.82, 122.02, 61.53, 51.10, 42.27, 32.73, 21.57, 14.60.
Ethyl 4-((4-(4-(3-methylphenyl)-lH-l,2,3-triazol-l-yl)naphthalene)-l- sulfonamido)piperidine-l-carboxylate (9e)
This compound was prepared according to the general procedure using 2- methoxyphenyl acetylene (31 pl, 0.24 mmol, 1.2 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum
ether/ethyl acetate (6/4), yielding the title compound as a solid (31.4 mg, 30% yield).
XH NMR (400 MHz, CDCI3) 6 8.77 (d, J = 8.6 Hz, 1H), 8.42 (d, J = 7.8 Hz, 1H), 8.22 (s, 1H), 7.84 (m, 2H), 7.71 (m, 4H), 7.38 (t, J = 7.6 Hz, 1H), 7.23 (d, J = 7.6 Hz, 1H), 5.42 (d, J = 7.7 Hz, 1H), 4.07 (q, J = 7.1 Hz, 2H), 3.92 (d, J = 10.8 Hz, 2H), 3.35 (m, 1H), 2.79 (t, J = 11.7 Hz, 2H), 2.44 (s, 3H), 1.70 (d, J = 10.2 Hz, 2H), 1.32 (m, 2H), 1.20 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCI3) 6 155.30, 148.38, 138.83, 138.19, 137.85, 129.62, 129.57, 129.36, 129.08, 128.99, 128.78, 128.63, 126.66, 124.93, 123.87, 123.08, 122.36, 121.98, 61.55, 51.14, 42.29, 32.78, 21.49, 14.62.
Ethyl 4-((4-(4-(4-methylphenyl)-lH-l,2,3-triazol-l-yl)naphthalene)-l- sulfonamido)piperidine-l-carboxylate (9f)
This compound was prepared according to the general procedure using 2- methoxyphenyl acetylene (30 pl, 0.24 mmol, 1.2 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (36.9 mg, 36% yield).
XH NMR (400 MHz, CDCI3) 6 8.76 (d, J = 8.6 Hz, 1H), 8.43 (d, J = 7.8 Hz, 1H), 8.18 (s, 1H), 7.85 (d, J = 7.7 Hz, 3H), 7.72 (m, 3H), 7.30 (d, J = 7.8 Hz, 2H), 6.75 (s, 1H), 5.19 (d, J = 7.7 Hz, 1H), 4.07 (q, J = 7.0 Hz, 2H), 3.92 (d, J = 8.9 Hz, 1H), 3.35 (m, 1H), 2.79 (t, J = 11.6 Hz, 2H), 2.42 (s, 3H), 1.71 (d, J = 13.6 Hz, 2H), 1.32 (qd, J = 11.6, 4.1 Hz, 2H), 1.21 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCI3) 6 155.25, 150.83, 148.36, 138.74, 138.26, 137.76, 129.76, 129.39, 129.08, 128.76, 128.67, 128.21, 126.92, 125.87, 124.86, 123.95, 121.95 (d, J = 2.0 Hz), 114.14, 61.51, 55.78, 51.19, 42.28, 32.83, 14.61.
Ethyl 4-((4-(4-(3-fluorophenyl)-lH-l,2,3-triazol-l-yl)naphthalene)-l- sulfonamido)piperidine-l-carboxylate (9g)
This compound was prepared according to the general procedure using 2- methoxyphenyl acetylene (28 pl, 0.24 mmol, 1.2 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (31.2 mg, 30% yield).
NMR (400 MHz, CDCI3) 6 8.77 (d, J = 8.6 Hz, 1H), 8.43 (d, J = 7.8 Hz, 1H), 8.25 (s, 1H), 7.82 (d, J = 8.4 Hz, 1H), 7.72 (m, 5H), 7.46 (dt, J = 6.0, 7.9 Hz, 1H), 7.11 (dt, J = 2.1, 8.4 Hz, 1H), 5.33 (d, J = 7.7 Hz, 1H), 4.07 (q, J = 7.0 Hz, 2H), 3.92 (d, J = 11.7 Hz, 2H), 3.36 (m, 1H), 2.79 (t, J = 11.5 Hz, 2H), 1.71 (m, 2H), 1.32 (m, 2H), 1.21 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCI3) 6 163.26 (d, J = 246.3 Hz), 155.28, 147.20 (d, J = 2.8 Hz), 138.02 (d, J = 3.1 Hz), 131.93 (d, J = 8.3 Hz), 130.73 (d, J = 8.3 Hz), 129.43, 129.33, 129.09, 128.87, 128.59, 124.95, 123.77, 122.75, 122.02, 121.58 (d, J = 2.8 Hz), 115.63 (d, J = 21.2 Hz), 112.96 (d, J = 23.2 Hz), 61.54, 51.19, 42.28, 32.82, 14.62.
Ethyl 4-((4-(4-(4-fluorophenyl)-lH-l,2,3-triazol-l-yl)naphthalene)-l- sulfonamido)piperidine-l-carboxylate (9h)
This compound was prepared according to the general procedure using 2- methoxyphenyl acetylene (28 pl, 0.24 mmol, 1.2 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (26.6 mg, 25% yield).
XH NMR (400 MHz, CDCI3) 6 8.76 (d, J = 8.6 Hz, 1H), 8.43 (d, J = 7.8 Hz, 1H), 8.20 (s, 1H), 7.95 (m, 2H), 7.83 (d, J = 8.3 Hz, 1H), 7.76 (m, 1H), 7.69 (m, 2H), 7.19 (t, J = 8.7 Hz, 2H), 5.29 (d, J = 7.6 Hz, 1H), 4.07 (q, J = 7.1 Hz, 2H), 3.92 (d, J =
11.7 Hz, 2H), 3.35 (m, 1H), 2.79 (t, J = 11.7 Hz, 2H), 1.71 (m, 2H), 1.32 (m, 2H), 1.21 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCI3) 6 163.02 (d, J = 248.4 Hz), 155.29, 147.41, 138.10, 137.95, 129.43, 129.35, 129.09, 128.83, 128.62, 127.77 (d, J = 8.4 Hz), 126.00 (d, J = 3.1 Hz), 124.93, 123.83, 122.10, 121.98, 116.14 (d, J = 21.8 Hz), 61.55, 51.19, 42.29, 32.81, 14.61.
Ethyl 4-((4-(4-(3-trifluoromethylphenyl)-lH-l,2,3-triazol-l-yl)naphtha- lene) -l-sulfonamido)piperidine-l-carboxylate (91)
This compound was prepared according to the general procedure using 2- methoxyphenyl acetylene (35 pl, 0.24 mmol, 1.2 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (31.8 mg, 28% yield).
NMR (400 MHz, CDCI3) 6 8.77 (d, J = 8.6 Hz, 1H), 8.43 (d, J = 7.7 Hz, 1H), 8.34 (s, 1H), 8.20 (m, 2H), 7.72 (m, 6H), 5.49 (d, J = 33.8 Hz, 1H), 4.07 (q, J = 6.7 Hz, 2H), 3.91 (d, J = 11.0 Hz, 2H), 3.35 (m, 1H), 2.79 (m, 2H), 1.70 (d, J = 11.2 Hz, 2H), 1.31 (m, 2H), 1.21 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCI3) 6 155.29, 146.95, 138.13, 137.92, 131.54 (q, J = 32.5 Hz), 130.67, 129.66, 129.46, 129.30, 129.12, 129.08, 128.92, 128.55, 125.34 (q, J = 3.9 Hz), 124.99, 123.97 (q, J =
272.7 Hz), 123.71, 122.93 (q, J = 2.2 Hz), 122.79 (q, J = 3.7 Hz), 122.06, 61.56, 51.17, 42.28, 32.78, 14.60.
Ethyl 4-((4-(4-(4-trifluoromethylphenyl)-lH-l,2,3-triazol-l-yl)naphtha- lene) -l-sulfonamido)piperidine-l-carboxylate (9j)
This compound was prepared according to the general procedure using 2- methoxyphenyl acetylene (39 pl, 0.24 mmol, 1.2 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (23.7 mg, 21% yield).
XH NMR (400 MHz, CDCI3) 6 8.77 (d, J = 8.6 Hz, 1H), 8.45 (d, J = 7.8 Hz, 1H), 8.31 (s, 1H), 8.10 (d, J = 8.1 Hz, 2H), 7.77 (m, 6H), 5.08 (d, J = 7.6 Hz, 1H), 4.08 (q, J = 7.1 Hz, 2H), 3.93 (d, J = 12.7 Hz, 2H), 3.37 (m, 1H), 2.80 (t, J = 11.9 Hz, 2H), 1.72 (d, J = 10.6 Hz, 2H), 1.32 (m, 2H), 1.21 (q, J = 4.7 Hz, 3H). 13C NMR (101 MHz, CDCI3) 6 155.26, 146.93, 138.13, 137.96, 133.24, 130.63 (q, J = 31.5 Hz), 129.53,
129.34, 129.11, 128.94, 128.60, 126.14, 126.11 (q, J = 3.4 Hz), 124.95, 124.03 (q, J = 272.2 Hz), 123.75, 123.06, 122.06, 61.54, 51.26, 42.29, 32.87, 14.62.
Ethyl 4-((4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)naphthalene)-l- sulfonamido)piperidine-l-carboxylate (10)
To an oven-dried screw-capped reaction tube equipped with a magnetic stirring bar, PdCl2(dppf) (109.8 mg, 0.15 mmol, 0.15 eq.), KOAc (196.3 mg, 2 mmol, 2 eq.), bispinacolato diboron (380.9 mg, 1.5 mmol, 1.5 eq.) and ethyl-4-(4- bromonaphthalene-l-sulfonylamino)-piperidine-l-carboxylate 3 (441.3 mg, 1 mmol, 1 eq.) were added. The reaction tube was evacuated and backfilled with argon three times before dissolving the solids in anhydrous dioxane which was degassed using 5 freeze pump thaw cycles (20 ml). The reaction was allowed to stir 20h at 80 °C, after which the mixture was filtered over celite, washed with DCM and concentrated in vacuo. The crude residue was purified by silica gel column chromatography using petroleum ether/ethyl acetate (8/2) as the mobile phase yielding the title compound as a solid (462.2 mg, quantitative yield).
XH NMR (400 MHz, CDCI3) 6 8.86 (m, 1H), 8.60 (m, 1H), 8.26 (d, J = 7.3 Hz, 1H), 8.10 (d, J = 7.3 Hz, 1H), 7.65 (m, 2H), 4.69 (d, J = 7.3 Hz, 1H), 4.05 (q, J = 7.1 Hz, 2H), 3.84 (d, J = 10.0 Hz, 2H), 3.24 (m, 1H), 2.72 (t, J = 11.8 Hz, 2H), 1.58 (d, J = 14.5 Hz, 2H), 1.45 (s, 12H), 1.25 (m, 2H), 1.19 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCI3) 6 155.27, 137.94, 137.72, 133.45, 129.60, 128.10, 127.88, 127.57, 127.20, 124.22, 84.50, 61.46, 50.93, 42.23, 32.63, 24.97, 14.59.
Synthesis of C-linked five-membered heteroaromatics
General procedure
To a screw-capped reaction tube equipped with a magnetic stirring bar, Pd(PPh3)4 (11.6 mg, 0.01 mmol, 0.1 eq.), K2CO3 (27.6 mg, 0.2 mmol, 2 eq.), ethyl 4-((4- (4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)naphthalene)-l- sulfonamido)piperidine-l-carboxylate (9) (48.8 mg, 0.1 mmol, 1 eq.), brominated heterocycle (0.15 mmol, 1.5 eq.), THF (0.8 ml) and water (0.2 ml) were added and allowed to stir for 17h at 100 °C. The mixture was diluted with water (1 ml) and extracted three times with dichloromethane. The combined organic layers were dried over anhydrous magnesium sulfate, filtered and concentrated in vacuo. Silica gel column chromatography of the crude product provided the desired compound. The following compounds were made according to this procedure.
Ethyl 4-((4-(5-phenyl-l,3,4-thiadiazol-2-yl)naphthalene)-l-sulfonamido) piperidine-l-carboxylate (Ila)
This compound was prepared according to the general procedure using 2-bromo-5- phenyl-l,3,4-thiadiazole (36.2mg, 0.15 mmol, 1.5 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (37.3 mg, 71%).
XH NMR (400 MHz, CDCI3) 6 8.81 (d, J = 9.2 Hz, 1H), 8.73 (d, J = 7.8 Hz, 1H), 8.39 (d, J = 7.6 Hz, 1H), 8.08 (m, 2H), 7.91 (d, J = 7.6 Hz, 1H), 7.75 (m, 2H), 7.56 (m, 3H), 4.89 (d, J = 7.7 Hz, 1H), 4.07 (q, J = 7.1 Hz, 2H), 3.91 (d, J = 7.0 Hz, 2H), 3.33 (m, 1H), 2.78 (t, J = 11.7 Hz, 2H), 1.69 (m, 2H), 1.28 (m, 2H), 1.20 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCI3) 6 169.91, 165.59, 155.24, 138.10, 132.71, 131.77, 131.66, 129.68, 129.40, 128.99, 128.63, 128.52, 128.32, 128.17, 127.77, 127.10, 124.61, 61.47, 51.19, 42.28, 32.86, 14.61.
Ethyl 4-((4-(5-phenylthiazol-2-yl)naphthalene)-l-sulfonamido)piperidine- 1-carboxylate (11b)
This compound was prepared according to the general procedure using 2-bromo-5- phenylthiazole (36.0 mg, 0.15 mmol, 1.5 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (39.3 mg, 75%).
NMR (400 MHz, CDCI3) 6 8.96 (m, 1H), 8.72 (m, 1H), 8.36 (d, J = 7.7 Hz, 1H), 8.23 (s, 1H), 7.89 (d, J = 7.7 Hz, 1H), 7.70 (m, 4H), 7.47 (m, 2H), 7.40 (m, 1H), 5.18 (d, J = 7.7 Hz, 1H), 4.06 (q, J = 7.1 Hz, 2H), 3.88 (d, J = 5.6 Hz, 2H), 3.32 (m, 1H), 2.77 (t, J = 11.6 Hz, 2H), 1.67 (m, 2H), 1.24 (m, 2H), 1.20 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCI3) 6 164.62, 155.25, 141.53, 139.46, 136.93, 136.36, 131.55, 130.82, 129.30, 128.86, 128.78, 128.65, 128.54, 128.09, 127.33, 126.95, 126.59, 124.50, 61.46, 51.02, 42.25, 32.75, 24.84, 14.60.
Ethyl 4-((4-(2-phenylthiazol-5-yl)naphthalene)-l-sulfonamido)piperidine- 1-carboxylate (11c)
This compound was prepared according to the general procedure using 5-bromo-2- phenylthiazole (36.0 mg, 0.15 mmol, 1.5 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (36.5 mg, 70%).
XH NMR (400 MHz, CDCI3) 6 8.74 (d, J = 8.4 Hz, 1H), 8.33 (m, 2H), 8.03 (m, 2H), 8.00 (s, 1H), 7.72 (m, 1H), 7.66 (m, 2H), 7.49 (m, 3H), 5.28 (d, J = 7.6 Hz, 1H), 4.07 (q, J = 7.1 Hz, 2H), 3.90 (d, J = 11.4 Hz, 2H), 3.37 (m, 1H), 2.81 (t, J = 11.7 Hz, 2H), 1.71 (d, J = 10.7 Hz, 2H), 1.32 (m, 2H), 1.20 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCI3) 6 169.56, 155.27, 143.54, 136.20, 135.07, 134.63, 133.23, 132.76, 130.53, 129.15, 128.63, 128.59, 128.55, 127.72, 126.85, 126.66, 126.55, 124.82, 61.47, 51.00, 42.25, 32.75, 14.61.
Ethyl 4-((4-(5-phenyl-l,3,4-oxadiazol-2-yl)naphthalene)-l-sulfonamido) piperidine-l-carboxylate (lid)
This compound was prepared according to the general procedure using 2-bromo-5- phenyl-l,3,4-oxadiazole (33.8 mg, 0.15 mmol, 1.5 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (29.8 mg, 59%).
XH NMR (400 MHz, CDCI3) 6 9.47 (m 1H), 8.74 (m, 1H), 8.44 (d, J = 7.8 Hz, 1H), 8.33 (d, J = 7.8 Hz, 1H), 8.22 (dd, J = 7.7, 1.6 Hz, 2H), 7.82 (m, 2H), 7.60 (m, 3H), 4.77 (d, J = I.! Hz, 1H), 4.06 (q, J = 7.1 Hz, 2H), 3.89 (m, 2H), 3.34 (m, 1H), 2.76 (t, J = 12.0 Hz, 2H), 1.66 (m, 2H), 1.29 (m, 2H), 1.19 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCI3) 6 164.91, 163.44, 155.23, 139.05, 132.31, 131.06, 129.29, 129.08, 128.97, 128.56, 128.13, 127.58, 127.24, 126.44, 126.06, 124.64, 123.43, 61.49, 51.29, 42.28, 32.82, 24.87, 14.60.
Ethyl 4-((4-(2-phenyloxazol-5-yl)naphthalene)-l-sulfonamido)piperidine- 1-carboxylate (lie)
This compound was prepared according to the general procedure using 5-bromo-2- phenyloxazole (33.6 mg, 0.15 mmol, 1.5 eq.). The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of petroleum ether/ethyl acetate (6/4), yielding the title compound as a solid (32.9 mg, 65%).
NMR (400 MHz, CDCI3) 6 8.77 (m, 1H), 8.53 (m, 1H), 8.40 (d, J = 7.8 Hz, 1H), 8.19 (m, 2H), 7.92 (d, J = 7.8 Hz, 1H), 7.75 (m, 2H), 7.69 (s, 1H), 7.55 (q, 3H), 5.25 (d, J = 7.7 Hz, 1H), 4.08 (q, J = 7.1 Hz, 2H), 3.90 (d, J = 10.7 Hz, 2H), 3.37 (m, 1H), 2.81 (t, J = 11.8 Hz, 2H), 1.69 (d, J = 10.8 Hz, 2H), 1.31 (m, 2H), 1.21 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCI3) 6 162.51, 155.27, 148.96, 136.05, 131.16, 130.98, 130.78, 129.01, 128.89, 128.85, 128.68, 128.57, 127.94, 126.95, 126.60, 126.11, 124.97, 124.25, 61.48, 51.03, 42.25, 32.71, 14.60.
Ethyl 4-((4-cyanonaphthalene)-l-sulfonamido)piperidine-l-carboxylate (12)
To a screw-capped reaction tube equipped with a magnetic stirring bar, NazCCh (159 mg, 1.5 mmol, 1 eq.), Pd(OAc)2 (1.4 mg, 0.006 mmol, 0.4%), NMP (2ml) and isopropanol (0.2 ml) were added and allowed to stir for 5 minutes at room temperature. To this solution, ethyl-4-(4-bromonaphthalene-l-sulfonylamino)- piperidine-l-carboxylate (3) (662 mg, 1.5 mmol, 1 eq.), NMP (2 ml) and K4[Fe(CN)6].3H2O (253.4 mg, 0.6 mmol, 0.4 eq.) were added and allowed to stir for 4h at 140 °C. The mixture was diluted with water and extracted three times with dichloromethane. The combined organic layers were dried over anhydrous magnesium sulfate, filtered and concentrated in vacuo. Silica gel column
chromatography of the crude product using petroleum ether/ethyl acetate (6/4) as the mobile phase provided the title compound as a solid (148.2 mg, 25% yield).
XH NMR (400 MHz, CDCI3) 6 8.76 (d, J = 8.3 Hz, 1H), 8.36 (m, 2H), 8.00 (d, J = 7.6 Hz, 1H), 7.80 (m, 2H), 5.74 (d, J = 7.7 Hz, 1H), 4.06 (q, J = 6.9 Hz, 2H), 3.90 (m, 2H), 3.33 (m, 1H), 2.78 (t, J = 11.7 Hz, 2H), 1.65 (d, J = 10.7 Hz, 2H), 1.31 (m, 2H), 1.20 (q, J = 4.7 Hz, 3H). 13C NMR (101 MHz, CDCI3) 6 155.29, 140.84, 133.11, 130.96, 129.73, 129.48, 127.75, 127.51, 126.38, 125.25, 116.56, 115.84, 61.59, 51.17, 42.26, 32.67, 14.60.
Ethyl 4-((4-(5-phenyl-/VH-l,2,4-triazol-3-yl)naphthalene)-l-sulfonamido) piperidine-l-carboxylate (13)
To a screw-capped reaction tube equipped with a magnetic stirring bar, benzhydrazide (27.2 mg, 0.2 mmol, 2 eq.), ethyl-4-((4-cyanonaphthalene)-l- sulfonylamino)-piperidine-l-carboxylate 11 (38.7 mg, 0.1 mmol, 1 eq.), K2CO3 (27.6 mg, 0.2 mmol, 2 eq.) and n-butanol (0.5ml) were added and allowed to stir for 5h at 150 °C. The mixture was quenched with water and extracted three times with dichloromethane. The combined organic layers were dried over anhydrous magnesium sulfate, filtered and concentrated in vacuo. Silica gel column chromatography of the crude product using petroleum ether/ethyl acetate (6/4) as the mobile phase provided the title compound as a solid (40.2 mg, 79% yield).
XH NMR (400 MHz, CDCI3) 6 8.74 (d, J = 7.7 Hz, 1H), 8.42 (d, J = 8.4 Hz, 1H), 8.36 (d, J = 7.6 Hz, 1H), 8.02 (d, J = 7.6 Hz, 1H), 7.84 (m, 3H), 7.52 (m, 2H), 5.06 (d, J = 7.7 Hz, 1H), 4.09 (q, J = 7.0 Hz, 2H), 3.94 (d, J = 9.3 Hz, 2H), 3.37 (m, 1H), 2.79 (t, J = 12.0 Hz, 2H), 1.68 (d, J = 11.9 Hz, 2H), 1.30 (m, 2H), 1.22 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCI3) 6 155.23, 140.74, 133.16, 130.89, 129.85,
129.50, 128.86, 128.74, 128.67, 127.76, 127.55, 127.36, 127.25, 126.49, 125.11,
116.51, 116.01, 61.54, 51.32, 42.25, 32.82, 14.60.
/V-(4-Bromonaphthalen-l-yl)-4-methoxybenzenesulfonamide (15)
In a round bottom flask equipped with a stirring bar 4-bromonaphthalen-l-amine 14 (444 mg, 2 mmol, 1 eq.) was dissolved in pyridine (10 ml) and cooled down to 0 °C. To the cooled down solution 4-methoxybenzene sulfonylchloride (1033 mg, 5 mmol, 2.5 eq.) was added and the mixture was allowed to stir for 16h at room temperature. The solvent was evaporated in vacuo and the residu was dissolved in ethyl acetate (10 ml) and washed twice with a saturated HCI solution, twice with water and twice with brine. The organic phase was dried over anhydrous magnesium sulfate, filtered and concentrated in vacuo. Recrystallization of the crude product in a water/ethanol mixture provided the title compound as a solid (548.6 mg, 74% yield).
XH NMR (400 MHz, CDCI3) 6 8.20 (d, J = 8.5 Hz, 1H), 7.89 (d, J = 8.4 Hz, 1H), 7.67 (m, 3H), 7.56 (t, J = 7.6 Hz, 1H), 7.48 (t, J = 7.6 Hz, 1H), 7.22 (d, J = 8.0 Hz, 1H), 7.13 (s, 1H), 6.82 (d, J = 8.7 Hz, 2H), 3.78 (s, 3H). 13C NMR (101 MHz, CDCI3) 6
163.20, 132.47, 131.52, 130.49, 130.15, 129.50, 129.42, 127.77, 127.71, 127.42,
123.14, 122.19, 121.51, 114.19, 55.57.
/V-(4-Bromonaphthalen-l-yl)-4-methoxy-/V-(4-methoxybenzyl) benzenesulfonamide (16)
To an oven dried round bottom flask equipped with a stirring bar, N-(4- bromonaphthalen-l-yl)-4-methoxybenzenesulfonamide (15) (392.3 mg, 1 mmol, 1 eq.), TBAI (96.7 mg, 0.3 mmol, 0.3 eq.), NaH (72.0 mg, 3 mmol, 3 eq.) were added. The round bottom flask was evacuated and backfilled with argon three times before dissolving the solids in anhydrous DMF (10 ml). The mixture was allowed to stir for 10 minutes at 0 °C before adding 4-methoxybenzylbromide (216 pl, 1.5 mmol, 1.5 eq.). The mixture was allowed to stir for Ih at room temperature and cooled down to 0 °C before quenching the reaction with saturated ammonium chloride. The product was extracted with ethyl acetate and washed with water and brine. The organic phase was dried over anhydrous magnesium sulfate, filtered and concentrated in vacuo. Silica gel column chromatography of the crude product using petroleum ether/ethyl acetate (8/2) as the mobile phase provided the title compound as a solid (435.9 mg, 85% yield).
XH NMR (300 MHz, CDCI3) 6 8.15 (d, J = 8.4 Hz, IH), 7.98 (d, J = 8.5 Hz, IH), 7.65 (m, 2H), 7.55 (m, 2H), 7.43 (m, IH), 7.00 (d, J = 8.7 Hz, 2H), 6.94 (d, J = 8.9 Hz, 2H), 6.68 (d, J = 8.0 Hz, IH), 6.63 (d, J = 8.7 Hz, 2H), 4.91 (d, J = 13.8 Hz, IH), 4.58 (d, J = 13.7 Hz, IH), 3.88 (s, 3H), 3.68 (s, 3H). 13C NMR (101 MHz, CDCI3) 6 163.10, 159.21, 135.93, 134.16, 132.83, 130.52, 130.33, 130.10, 128.88, 127.76, 127.51, 127.29, 127.19, 127.11, 124.82, 123.62, 114.13, 113.65, 55.75, 55.67, 55.14.
/V-(4-((4-Methoxy-/V-(4-methoxybenzyl)phenyl)sulfonamido)naphthalen- l-yl)-2-methylbenzamide (17)
To an oven-dried screw-capped reaction tube equipped with a magnetic stirring bar, Pd(OAc)2 (17.6 mg, 0.08 mmol, 0.1 eq.), Xantphos (69.6 mg, 0.12 mmol, 0.15 eq.), CS2CO3 (364.8 mg, 1.12 mmol, 1.4 eq.), o-toluamide (129.6 mg, 0.96 mmol, 1.2 eq.) and N-(4-bromonaphthalen-l-yl)-4-methoxy-N-(4-methoxybenzyl) benzenesulfonamide (16) (409.6 mg, 0.8 mmol, 1 eq.) were added. The reaction tube was evacuated and backfilled with argon three times. Anhydrous dioxane (0.8 ml) was added and the reaction was allowed to stir for 16h at 100 °C. The mixture was diluted with DCM (8 ml) and filtered. The filtrate was concentrated in vacuo and the crude residue was purified by silica gel column chromatography using petroleum ether/ethyl acetate (6/4) as the mobile phase yielding the title compound as a solid (367.5 mg, 81% yield).
XH NMR (400 MHz, CDCI3) 6 8.13 (m, 2H), 7.89 (s, 1H), 7.73 (d, J = 7.4 Hz, 1H), 7.68 (d, J = 8.5 Hz, 2H), 7.58 (m, 1H), 7.44 (m, 3H), 7.32 (d, J = 7.4 Hz, 2H), 7.06 (d, J = 8.2 Hz, 2H), 6.98 (d, J = 8.7 Hz, 2H), 6.86 (d, J = 6.7 Hz, 1H), 6.64 (d, J = 8.4 Hz, 2H), 5.02 (d, J = 13.6 Hz, 1H), 4.49 (d, J = 13.7 Hz, 1H), 3.90 (s, 3H), 3.68 (s, 3H), 2.56 (s, 3H). 13C NMR (101 MHz, CDCI3) 6 163.04, 159.13, 136.71, 133.80, 133.29, 132.87, 131.51, 130.54, 130.34, 130.14, 127.71, 126.83, 126.67, 126.57, 126.32, 126.12, 125.58, 119.66, 114.13, 113.61, 55.92, 55.66, 55.14, 20.00.
/V-(4-((4-Methoxyphenyl)sulfonamido)naphthalen-l-yl)-2-methyl- benzamide (18)
In a round bottom flask equipped with a magnetic stirring bar, /V-(4-((4-methoxy-/V- (4-methoxybenzyl)phenyl)sulfonamido)naphthalen-l-yl)-2-methylbenzamide (17) (56.7 mg, 0.1 mmol, 1 eq.) was dissolved in DCM/TFA (1/1, 0.6ml) and stirred for 2h at room temperature. The reaction was quenched with water and the product was filtered of yielding the title compound as a white solid (41.7 mg, 93% yield).
 10.30 (s, 1H), 10.10 (s, 1H), 8.06 (m, 2H), 7.66 (d, J = 8.5 Hz, 2H), 7.54 (m, 4H), 7.40 (d, J = 7.1 Hz, 1H), 7.33 (d, J = 6.4 Hz, 2H), 7.18 (d, J = 7.6 Hz, 1H), 7.04 (d, J = 8.6 Hz, 2H), 3.79 (s, 3H), 2.46 (s, 3H). 13C NMR (101 MHz, DMSO) 6 169.23, 162.81, 137.40, 135.85, 132.70, 132.20, 131.03, 131.00, 130.66, 130.13, 129.87, 129.44, 127.83, 126.65, 126.51, 126.13, 124.10, 123.75, 123.49, 123.34, 114.78, 56.09, 19.97.
10.30 (s, 1H), 10.10 (s, 1H), 8.06 (m, 2H), 7.66 (d, J = 8.5 Hz, 2H), 7.54 (m, 4H), 7.40 (d, J = 7.1 Hz, 1H), 7.33 (d, J = 6.4 Hz, 2H), 7.18 (d, J = 7.6 Hz, 1H), 7.04 (d, J = 8.6 Hz, 2H), 3.79 (s, 3H), 2.46 (s, 3H). 13C NMR (101 MHz, DMSO) 6 169.23, 162.81, 137.40, 135.85, 132.70, 132.20, 131.03, 131.00, 130.66, 130.13, 129.87, 129.44, 127.83, 126.65, 126.51, 126.13, 124.10, 123.75, 123.49, 123.34, 114.78, 56.09, 19.97.
4-Bromo-/V-((4-methoxyphenyl)sulfonyl)-l-naphthamide (20)
To a round bottom flask equipped with a magnetic stirring bar, DMAP (7.2 mg, 0.06 mmol, 0.05 eq.), Mukaiyama's reagent (367.9 mg, 1.44 mmol, 1.2 eq.), 4- bromonaphthalene-l-carboxylic acid 19 (301.2 mg, 1.2 mmol, 1 eq.), 4- methoxybenzenesulfonamide (449.2 mg, 2.4 mmol, 2 eq.) and DCM (4 ml) were added and stirred for 5 minutes before adding trimethylamine (0.5 ml, 3.6 mmol, 3 eq.) dropwise. The reaction was allowed to stir for Ih at room temperature. The solvent was evaporated in vacuo and the residu was redissolved in ethyl acetate and washed with HCI (IM). The organic phase was dried over anhydrous magnesium sulfate, filtered and concentrated in vacuo. Silica gel column chromatography of the crude product using petroleum ether/ethyl acetate (6/4) as the mobile phase provided the title compound as a solid (471.6 mg, 94% yield).
XH NMR (400 MHz, CDCI3) 6 8.83 (b, IH), 8.25 (d, J = 8.4 Hz, IH), 8.14 (d, J = 8.5 Hz, IH), 8.09 (d, J = 8.9 Hz, 2H), 7.71 (d, J = 7.7 Hz, IH), 7.61 (t, J = 7.4 Hz, IH), 7.53 (t, J = 7.4 Hz, IH), 7.45 (d, J = 7.7 Hz, IH), 7.01 (d, J = 8.9 Hz, 2H), 3.89 (s, 3H). 13C NMR (101 MHz, CDCI3) 6 164.14, 132.18, 130.98, 130.93, 130.17, 129.65, 128.63, 128.22, 128.09, 127.73, 126.39, 125.31, 114.23, 55.75.
tert-Butyl (4-bromo-l-naphthoyl)((4-methoxyphenyl)sulfonyl)carbamate
(21)
To a screw-capped reaction tube equipped with a magnetic stirring bar, 4-bromo-/V- ((4-methoxyphenyl)sulfonyl)-l-naphthamide 20 (336.2 mg, 0.8 mmol, 1 eq.), DMAP (29.3 mg, 0.24 mmol, 0.3 eq.), THF (8 ml), pyridine (0.64 ml, 8 mmol, 10 eq.) and tert-Boc anhydride (0.92 ml, 4 mmol, 5 eq.) were added and allowed to stir for 4h at 60 °C. The reaction was quenched with water and extracted three times with DCM. The combined organic layers were dried over anhydrous magnesium sulfate, filtered and concentrated in vacuo. Silica gel column chromatography of the crude product using petroleum ether/ethyl acetate (8/2) as the mobile phase provided the title compound as a solid (250.0 mg, 60% yield).
NMR (400 MHz, CDCI3) 6 8.22 (d, J = 8.5 Hz, 1H), 7.78 (d, J = 7.6 Hz, 1H), 7.57 (m, 1H), 7.49 (m, 4H), 7.36 (d, J = 7.6 Hz, 1H), 6.63 (d, J = 8.9 Hz, 2H), 3.74 (s, 3H), 1.66 (s, 9H). 13C NMR (101 MHz, CDCI3) 6 168.29, 162.43, 132.62, 131.60, 130.98, 130.44, 128.95, 128.73, 127.66, 127.62, 127.51, 126.23, 125.54, 125.02, 113.43, 87.24, 55.46, 28.05.
/V-((4-Methoxyphenyl)sulfonyl)-4-(2-methylbenzamido)-l-naphthamide
(22)
To an oven-dried screw-capped reaction tube equipped with a magnetic stirring bar, Pd(OAc)2 (9.0 mg, 0.04 mmol, 0.1 eq.), Xantphos (34.7 mg, 0.06 mmol, 0.15 eq.), CS2CO3 (182.5 mg, 0.56 mmol, 1.4 eq.), o-toluamide (64.9 mg, 0.48 mmol, 1.2 eq.) and tert-butyl (4-bromo-l-naphthoyl)((4-methoxyphenyl)sulfonyl)carbamate (21) (208.2 mg, 0.4 mmol, 1 eq.) were added. The reaction tube was evacuated and backfilled with argon three times. Anhydrous dioxane (0.4 ml) was added and the reaction was allowed to stir for 16h at 100 °C. The mixture was diluted with DCM (4 ml) and filtered. The filtrate was concentrated in vacuo and the crude residue was purified by silica gel column chromatography using petroleum ether/ethyl acetate (6/4) as the mobile phase yielding the title compound as a solid (152.4 mg, 80% yield).
XH NMR (400 MHz, DMSO) 6 12.64 (b, 1H), 10.53 (s, 1H), 8.18 (d, J = 8.6 Hz, 1H), 8.01 (m, 3H), 7.79 (m, 2H), 7.62 (m, 3H), 7.43 (m, 1H), 7.35 (m, 2H), 7.21 (d, J = 8.5 Hz, 2H), 3.90 (s, 3H), 2.48 (s, 3H).
4-Bromonaphthalene-l-sulfonamide (23)
To a round bottom flask equipped with a magnetic stirring bar, 4-bromonaphthalene- 1-sulfonylchloride 2 (0.9247g, 3.0 mmol, 1 eq.), diethyl ether (6.9 ml), acetone (2.3 ml) was added and cooled down to 0 °C. Aqueous ammonia (50%, 537 pl, 12.9 mmol, 4.3 eq.) was added dropwise and the reaction was allowed to stir for Ih at 0
°C. The mixture was neutralized with HCI (IM), diluted with water and extracted two times with ethylacetate. The combined organic layers were dried over anhydrous magnesium sulfate, filtered and concentrated in vacuo. Silica gel column chromatography of the crude product using petroleum ether/ethyl acetate (6/4) as the mobile phase provided the title compound as a solid (591.3 mg, 68% yield).
XH NMR (400 MHz, DMSO) 6 8.71 (m, 1H), 8.32 (m, 1H), 8.05 (q, 2H), 7.83 (m, 2H), 7.79 (s, 2H).
4-Bromo-/V-(tert-butyldiphenylsilyl)naphthalene-l-sulfonamide (24)
To an oven-dried screw-capped reaction tube equipped with a magnetic stirring bar, 4-bromonaphthalene-l-sulfonamide 23 (572.3 mg, 2 mmol, 1 eq.) was added. The reaction tube was evacuated and backfilled with argon three times. Anhydrous THF (2.33 ml) and triethylamine (1.11 ml, 8 mmol, 4 eq.) was added and the reaction was allowed to stir for 5 minutes at room temperature. tert-Butyldiphenylsilyl chloride (1.04 ml, 4 mmol, 2 eq.) was added and the reaction was allowed to stir 24h at 50 °C. Silica gel column chromatography of the crude product using petroleum ether/ethyl acetate (8/2) as the mobile phase provided the title compound as a solid (0.905 g, 86% yield).
NMR (400 MHz, CDCI3) 6 8.64 (m, 1H), 8.36 (m, 1H), 7.71 (m, 2H), 7.42 (m, 6H), 7.32 (d, J = 8.0 Hz, 1H), 7.24 (t, J = 7.6 Hz, 4H), 7.15 (d, J = 8.0 Hz, 1H), 5.09 (s, 1H), 0.95 (s, 9H). 13C NMR (101 MHz, CDCI3) 6 137.44, 136.04, 132.16, 130.19, 130.12, 128.79, 128.59, 128.41, 128.36, 128.34, 128.31, 127.87, 127.58, 124.78, 27.13, 18.52.
Ethyl 4-((4-bromonaphthalen-l-yl)(/Vz-(tert-butyldiphenylsilyl)) sulfon- imidamido)piperidine-l-carboxylate (26)
In an oven dried round bottom flask equipped with a stirring bar, triphenylphosphine dichloride (249.9 mg, 0.75 mmol, 1.5 eq.) was added. The round bottom flask was evacuated and backfilled with argon three times. Chloroform (1.67 ml) was added followed by the dropwise addition of triethylamine (111 pl, 0.8 mmol, 1.6 eq.). The reaction mixture was allowed to stir for 20 minutes before cooling it down to 0 °C. In a second oven dried round bottom flask, 4-bromo-/V-(tert-butyldiphenylsilyl) naphthalene-l-sulfonamide 24 (254.2 mg, 0.5 mmol, 1 eq.) was added. The round bottom flask was evacuated and backfilled with argon three times. The solid was dissolved in a minimal amount of dry chloroform and added to the triphenylphosphine dichloride solution at 0 °C. The reaction mixture was allowed to stir 20 minutes at 0 °C after which ethyl 4-aminopiperidine-l-carboxylate (257 pl, 1.5 mmol, 3 eq.) was added. The reaction mixture was allowed to stir for 2h at 0 °C. The solvent was evaporated in vacuo and resuspended in cold diethylether. The suspension was
filtered and concentrated in vacuo. Silica gel column chromatography of the crude product using petroleum ether/ethyl acetate (6/4) as the mobile phase provided the title compound as a solid (204.2 mg, 60% yield).
XH NMR (400 MHz, CDCI3) 6 8.81 (d, J = 8.4 Hz, 1H), 8.33 (d, J = 8.6 Hz, 1H), 8.09 (d, J = 8.0 Hz, 1H), 7.76 (d, J = 8.0 Hz, 1H), 7.69 (m, 2H), 7.64 (m, 3H), 7.57 (m, 1H), 7.34 (m, 2H), 7.28 (m, 3H), 4.20 (d, J = 7.1 Hz, 1H), 4.02 (q, J = 7.1 Hz, 2H), 3.75 (m, 1H), 3.61 (m, 1H), 3.12 (m, 1H), 2.66 (t, J = 11.5 Hz, 1H), 2.50 (t, J = 11.5 Hz, 1H), 1.56 (m, 2H), 1.17 (t, J = 7.1 Hz, 3H), 1.07 (s, 9H). 13C NMR (101 MHz, CDCI3) 6 155.23, 139.54, 135.68, 135.60, 132.59, 129.18, 129.08, 128.46, 128.41, 128.27, 128.12, 127.79, 127.38, 125.73, 61.34, 50.84, 42.22, 32.85, 32.11, 27.16, 19.52, 14.63.
Ethyl 4-((4-(2-methylbenzamido)naphthalene)(/V'-(tert-butyldiphenyl- silyl)) sulfonimidamido)piperidine-l-carboxylate (27)
To an oven-dried screw-capped reaction tube equipped with a magnetic stirring bar, Pd(OAc)2 (4.5 mg, 0.02 mmol, 0.1 eq.), Xantphos (17.4 mg, 0.03 mmol, 0.15 eq.), CS2CO3 (91.2 mg, 0.28 mmol, 1.4 eq.), o-toluamide (32.4 mg, 0.24 mmol, 1.2 eq.) and ethyl 4-((4-bromonaphthalen-l-yl)(/V'-(tert-butyldiphenylsilyl)) sulfonimidamido)piperidine-l-carboxylate 26 (135.8 mg, 0.2 mmol, 1 eq.) were added. The reaction tube was evacuated and backfilled with argon three times. Anhydrous dioxane (0.2 ml) was added and the reaction was allowed to stir for 16h at 100 °C. The mixture was diluted with DCM (4 ml) and filtered. The filtrate was concentrated in vacuo and the crude residue was purified by silica gel column chromatography using petroleum ether/ethyl acetate (6/4) as the mobile phase yielding the title compound as a solid (90.7 mg, 62% yield). The product was used in the next step without further purification.
 8.90 (m, 1H), 8.39 (d, J = 8.2 Hz, 1H), 8.31 (b, 1H), 8.05 (s, 1H), 7.89 (m, 1H), 7.76 (m, 2H), 7.70 (m, 2H), 7.63 (d, J = 7.4 Hz, 1H), 7.58 (m, 2H), 7.44 (t, J = 7.0 Hz, 1H), 7.32 (m, 8H), 4.18 (d, J = 7.0 Hz, 1H), 4.01 (q, J = 7.1 Hz, 2H), 3.69 (m, 1H), 3.60 (m, 1H), 3.11 (m, 1H), 2.60 (s, 3H), 2.56 (m, 2H), 1.53 (m, 2H), 1.26 (m, 2H), 1.16 (t, J = 7.1 Hz, 3H), 1.09 (s, 9H).
8.90 (m, 1H), 8.39 (d, J = 8.2 Hz, 1H), 8.31 (b, 1H), 8.05 (s, 1H), 7.89 (m, 1H), 7.76 (m, 2H), 7.70 (m, 2H), 7.63 (d, J = 7.4 Hz, 1H), 7.58 (m, 2H), 7.44 (t, J = 7.0 Hz, 1H), 7.32 (m, 8H), 4.18 (d, J = 7.0 Hz, 1H), 4.01 (q, J = 7.1 Hz, 2H), 3.69 (m, 1H), 3.60 (m, 1H), 3.11 (m, 1H), 2.60 (s, 3H), 2.56 (m, 2H), 1.53 (m, 2H), 1.26 (m, 2H), 1.16 (t, J = 7.1 Hz, 3H), 1.09 (s, 9H).
Ethyl 4-(4-(2-methylbenzamido)naphthalene)sulfonimidamido)piperidine- 1-carboxylate (28)
To an oven-dried screw-capped reaction tube equipped with a magnetic stirring bar, ethyl 4-((4-(2-methylbenzamido)naphthalene)(/V'-(tert-butyldiphenylsilyl)) sulfon- imidamido) piperidine-l-carboxylate 27 (73.3 mg, 0.1 mmol, 1 eq.) was added and dissolved in dry THF (2 ml). To this solution, a solution of TBAF in dry THF (IM, 0.1 ml, 1 eq.) was added dropwise. The reaction mixture was allowed to stir for 10 minutes at room temperature and for 2h at 40 °C. The reaction mixture was quenched with ammonium chloride (2M) and extracted two times with DCM. The
combined organic layers were washed with brine, dried over anhydrous magnesium sulfate, filtered and concentrated in vacuo. Silica gel column chromatography of the crude product using ethyl acetate as the mobile phase provided the title compound as a solid (37.6 mg, 76% yield).
XH NMR (400 MHz, CDCI3) 6 8.96 (d, J = 8.5 Hz, 1H), 8.38 (m, 2H), 8.18 (s, 1H), 7.94 (d, J = 8.3 Hz, 1H), 7.67 (m, 3H), 7.45 (t, J = 7.1 Hz, 1H), 7.35 (m, 2H), 4.06 (q, J = 7.0 Hz, 2H), 3.96 (m, 1H), 3.78 (m, 1H), 3.31 (m, 1H), 2.83 (t, J = 11.8 Hz, 1H), 2.68 (t, J = 11.1 Hz, 1H), 2.60 (s, 3H), 1.92 (d, J = 11.0 Hz, 1H), 1.36 (m, 2H), 1.20 (t, J = 7.1 Hz, 3H), 1.06 (m, 1H). 13C NMR (101 MHz, CDCI3) 6 168.73, 155.24, 137.48, 136.80, 135.90, 133.18, 131.49, 130.72, 129.79, 128.85, 127.96, 127.25, 126.94, 126.84, 126.04, 125.47, 121.54, 117.93, 61.36, 51.19, 42.44, 42.24, 33.40, 32.29, 20.03, 14.60.
Diethyl (4-aminonaphthalen-l-yl)phosphonate (29)
To an oven-dried screw-capped reaction tube equipped with a magnetic stirring bar, 4-bromonaphthalen-l-amine 14 (1.111 g, 5 mmol, 1 eq.), Pd(OAc)2 (22.5 mg, 0.1 mmol, 0.02 eq.) and triphenylphosphine (78.7 mg, 0.3 mmol, 0.06 eq.) were added. The reaction tube was evacuated and backfilled with argon three times and ethanol (10 ml), diethyl phosphite (0.78 ml, 12 mmol, 1.2 eq.) and triethylamine (1.05 ml, 7.5 mmol, 1.5 eq.) were added. The reaction was allowed to stir for 48h at 80 °C. The crude product was concentrated in vacuo and silica gel column chromatography of the crude product using DCM/MeOH (95/5) as the mobile phase provided the title compound as a solid (1.199 g, 80% yield).
NMR (400 MHz, CDCI3) 6 8.46 (d, J = 8.5 Hz, 1H), 8.07 (m, 1H), 7.84 (d, J = 8.4 Hz, 1H), 7.57 (m, 1H), 7.49 (m, 1H), 6.77 (m, 1H), 4.64 (b, 2H), 4.15 (m, 2H), 4.03 (m, 2H), 1.28 (t, J = 7.1 Hz, 6H). In accordance with literature32.
Diethyl (4-(2-methylbenzamido)naphthalen-l-yl)phosphonate (30)
To a round bottom flask equipped with a magnetic stirring bar, diethyl (4- aminonaphthalen-l-yl)phosphonate 29 (1.1 g, 3.7 mmol, 1 eq.), DCM (40 ml) and 2-methylbenzoylchloride (0.724 ml, 5.55 mmol, 1.5 eq.) were added. The mixture was allowed to stir for 5 minutes at room temperature before addition of triethylamine (4ml). The reaction mixture was allowed to stir for 15h at room temperature and washed two times with HCI (IM) and water. The organic phase was dried over anhydrous magnesium sulfate, filtered and concentrated in vacuo. Silica gel column chromatography of the crude product using DCM/MeOH (95/5) as the mobile phase provided the title compound as a solid (1.294 g, 88% yield). The product was used in the next step without further purification.
NMR (400 MHz, CDCI3) 6 8.60 (d, J = 8.2 Hz, 1H), 8.25 (m, 2H), 7.93 (d, J = 8.4 Hz, 1H), 7.60 (m, 4H), 7.42 (m, 1H), 7.33 (m, 2H), 4.10 (m, 4H), 2.58 (s, 3H), 1.30 (t, J = 7.0 Hz, 6H).
Ethyl hydrogen (4-(2-methylbenzamido)naphthalen-l-yl)phosphonate (31)
To a screw-capped reaction tube equipped with a magnetic stirring bar, diethyl (4- (2-methylbenzamido)naphthalen-l-yl)phosphonate 30 (1.5 g, 3.8 mmol, leq.), dioxane (22.5 ml) and NaOH (aq., 1.5M, 38 ml) were added. The reaction mixture was allowed to stir for 4h at 100 °C, cooled down and washed with DCM. The aqueous phase was acidified to pH = 1 using HCI (37%) and extracted three times with DCM. The combined organic layers were washed with brine, dried over anhydrous magnesium sulfate, filtered and concentrated in vacuo to provide the title compound as a solid (833.4 mg, 60% yield). The product was used in the next step without further purification.
NMR (400 MHz, DMSO) 6 10.54 (s, 1H), 8.58 (m, 1H), 8.22 (m, 1H), 8.11 (m, 1H), 7.86 (m, 1H), 7.66 (m, 3H), 7.44 (m, 1H), 7.36 (m, 2H), 3.92 (m, J = 5.9 Hz, 2H), 2.49 (s, 3H), 1.17 (t, J = 7.0 Hz, 3H).
Ethyl 4-((ethoxy(4-(2-methylbenzamido)naphthalen-l-yl)phosphoryl) amino)piperidine-l-carboxylate (32)
To a screw-capped reaction tube equipped with a magnetic stirring bar, ethyl hydrogen (4-(2-methylbenzamido)naphthalen-l-yl)phosphonate 31 (280.8 mg, 0.76 mol, 1 eq.), ethyl 4-aminopiperidine-l-carboxylate (248 pl, 1.44 mmol, 1.9 eq.), pyridine (1.9 ml) and triethylamine (1.12 ml) were added and allowed to stir for 20 minutes at room temperature. To a round bottom flask equipped with a magnetic stirring bar, 2,2'-dithiopyridine (1.22 g, 5.54 mmol, 7.3 eq.), triphenylphosphine (1.04 g, 3.96 mmol, 5.2 eq.) and pyridine (1.9 ml) were added and allowed to stir for 20 minutes at room temperature. The solution from the round bottom flask was added to the solution in the screw capped reaction tube and allowed to stir for 18h at 60 °C. The solvent was evaporated in vacuo and the residu was redissolved in ethyl acetate and filtered over celite. The crude product was concentrated in vacuo and silica gel column chromatography of the crude product using DCM/MeOH (95/5) as the mobile phase provided the title compound as a solid (312.6 mg, 79% yield). XH NMR (400 MHz, CDCI3) 6 8.70 (m, 1H), 8.41 (s, 1H), 8.15 (m, 2H), 7.94 (d, J = 7.4 Hz, 1H), 7.60 (m, 3H), 7.42 (m, 1H), 7.32 (m, 2H), 4.11 (m, 4H), 3.94 (m, 2H), 3.21 (m, 1H), 2.87 (t, J = 9.1 Hz, 1H), 2.75 (m, 1H), 2.58 (s, 3H), 1.80 (m, 2H), 1.27 (m, 8H). 13C NMR (101 MHz, CDCI3) 6 168.64, 155.37, 136.85, 136.00, 134.14, 133.70 (d, J = 12.5 Hz), 131.50, 130.65, 127.45, 127.30, 126.83, 126.74, 126.62, 126.06, 124.81, 123.12, 121.03, 118.21, 61.07 (d, J = 5.4 Hz), 48.25, 42.65, 34.70, 20.04 (d, J = 11.7 Hz), 16.45, 14.65 (d, J = 9.4 Hz).
5-Bromoisoquinoline-8-sulfonic acid (35)
To a screw-capped reaction tube equipped with a magnetic stirring bar, 5- bromoisoquinoline 34 (2.08 g, 10 mmol, leq.) and fuming sulfuric acid (40ml) were added. The reaction mixture was allowed to stir for 4h at 100 °C, cooled down and slowly poured over ice. The precipitate was filtered off, washed with water, a little amount of methanol and dried in vacuo providing the title compound as a solid (2.6 g, 90% yield). The product was used in the next step without further purification.
XH NMR (400 MHz, DMSO) 6 10.31 (s, 1H), 8.79 (d, J = 6.4 Hz, 1H), 8.39 (m, 2H), 8.08 (d, J = 7.8 Hz, 1H), 5.07 (b, 1H).
Ethyl 4-((5-bromoisoquinoline)-8-sulfonamido)piperidine-l-carboxylate (37)
To a screw-capped reaction tube equipped with a magnetic stirring bar, 5- bromoisoquinoline-8-sulfonic acid 35 (1.44 g, 5 mmol, leq.), thionyl chloride (15 ml) and DMF (0.1 ml) were added. The reaction mixture was allowed to stir for 4h at 75 °C. The mixture was concentrated in vacuo to remove the excess thionyl chloride and DMF. The residue was dissolved in DCM (50 ml) after which DMAP (122.2 mg, 1 mmol, 0.2 eq.), DCM (50 ml) and EtsN (2 ml) were added. To this mixture, ethyl 4- aminopiperidine-l-carboxylate (3.5 ml, 20 mmol, 4 eq.) was added dropwise. The reaction mixture was allowed to stir for 24h at room temperature and washed with water. The organic phase was dried over anhydrous magnesium sulfate, filtered and concentrated in vacuo. Silica gel column chromatography of the crude product using DCM/MeOH (95/5) as the mobile phase provided the title compound as a solid (458 mg, 21% yield). The product was used in the next step without further purification. XH NMR (400 MHz, CDCI3) 6 10.09 (d, J = 0.8 Hz, 1H), 8.82 (d, J = 5.9 Hz, 1H), 8.19 (m, 2H), 8.08 (d, J = 7.9 Hz, 1H), 5.63 (d, J = 7.7 Hz, 1H), 4.06 (q, J = 7.1 Hz, 2H), 3.93 (m, 2H), 3.36 (m, 1H), 2.77 (t, J = 11.5 Hz, 2H), 1.70 (m, 2H), 1.30 (m, 2H), 1.19 (t, J = 7.1 Hz, 3H).
Ethyl 4-((5-(2-methylbenzamido)isoquinoline)-8-sulfonamido)piperidine- 1-carboxylate (38)
To an oven-dried screw-capped reaction tube equipped with a magnetic stirring bar, Pd(OAc)2 (4.5 mg, 0.02 mmol, 0.1 eq.), Xantphos (17.4 mg, 0.03 mmol, 0.15 eq.), CS2CO3 (91.2 mg, 0.28 mmol, 1.4 eq.), o-toluamide (32.4 mg, 0.24 mmol, 1.2 eq.) and ethyl 4-((5-bromoisoquinoline)-8-sulfonamido)piperidine-l-carboxylate 37 (88.5 mg, 0.2 mmol, 1 eq.) were added. The reaction tube was evacuated and backfilled with argon three times. Anhydrous dioxane (0.2 ml) was added and the reaction was allowed to stir for 16h at 100 °C. The mixture was diluted with DCM (4 ml) and filtered. The filtrate was concentrated in vacuo and the crude residue was
purified by silica gel column chromatography using DCM/MeOH (95/5) as the mobile phase yielding the title compound as a solid (25.5 mg, 25% yield).
XH NMR (400 MHz, DMSO) 6 10.73 (b, 1H), 10.04 (d, J = 0.7 Hz, 1H), 8.72 (d, J = 5.9 Hz, 1H), 8.28 (m, 2H), 8.18 (m, 1H), 7.68 (m, 1H), 7.46 (m, m 1H), 7.37 (m, 2H), 3.98 (q, J = 7.1 Hz, 2H), 3.73 (d, J = 13.4 Hz, 2H), 3.28 (m, 1H), 2.79 (m, 2H), 2.49 (m, 2H), 1.53 (m, 2H), 1.23 (m, 2H), 1.13 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, DMSO) 6 169.35, 154.92, 149.62, 143.99, 138.22, 136.78, 136.10, 134.11, 131.42, 131.12, 130.56, 130.36, 128.12, 126.18, 123.69, 123.59, 116.93, 61.10, 50.40, 42.32, 32.66, 19.95, 15.00.
CCR8 competition binding assay
The hCCLlAF647 binding inhibition assay was performed and analyzed as described in [Liu et al. (2021) Biochem Pharmacol. 188, 114565]. Briefly, 50 pL/well of serial compound dilutions and 50 pL/well of hCCLlAF647 (2 nM final concentration) were mixed in a U-bottom 96-well plate. Then 100 pL/well of 1 x 105 Jurkat.hCCR8 cells, resuspended in the assay buffer, was added. After mixing samples were and incubated for 30 min at room temperature (RT) protected from light. Subsequently, cells were washed twice in assay buffer, fixed in 1% paraformaldehyde in DPBS, and immediately analyzed. The mean fluorescence intensity (MFI) value for each sample was used to calculate the percentage of hCCLlAF647 binding inhibition according to the formula: [1 - ((MFIX -MFINC) I (MFIPC - MFINC))] X 100, whereby MFIX is the MFI of the compound-treated sample, MFINC is the MFI of the negative control (i.e., autofluorescence of untreated and unlabeled cells) and MFIPC is the MFI of the positive control (i.e., cells exposed to hCCLlAF647 only). The inhibitory concentration 50 (IC50, the compound concentration that inhibits hCCLlAF647 binding by 50%) was calculated for each compound using non-linear regression analysis in GraphPad Prism 9.0.0.
CCR8 calcium mobilization assay
Human glioblastoma U87 cells stably expressing CD4 and CCR8 (U87.CD4.CCR8) were seeded at 20,000 cells per well in gelatin-coated black-walled polystyrene 96- well plates and incubated overnight at 37°C and 5% CO2. The next day, cell culture medium was replaced with 80pL/well of Calcium 6 Assay Kit loading medium (Molecular Devices), which was prepared according the manufacturer's instructions. After incubating the cells for 2h at 37°C and 5% CO2 they were transferred to the FLIPR Tetra device (Molecular Devices). Serial dilutions of compounds were added to the cells first and incubated for lOmin. Thereafter, a fixed concentration of hCCLl (5.88nM final concentration) was added to induce a CCR8-mediated intracellular Ca2+ release. All responses were normalized by dividing through a baseline determined just before the time of hCCLl addition and also background corrected by subtracting
the averaged normalized response from untreated (i.e., no compound) and unstimulated (i.e., no hCCLl) cells. The percentage (%) inhibition induced by the compounds was then calculated relative to positive control samples (i.e., no compound, only hCCLl stimulation). Based on the dose-dependent % inhibition, IC50 values were calculated using non-linear curve fitting in Graphpad Prism 9.0.0.
Claims
1. A compound with formula I
 wherein
wherein
-Q is CH,
-Ri is selected from substituents II III, or IV,
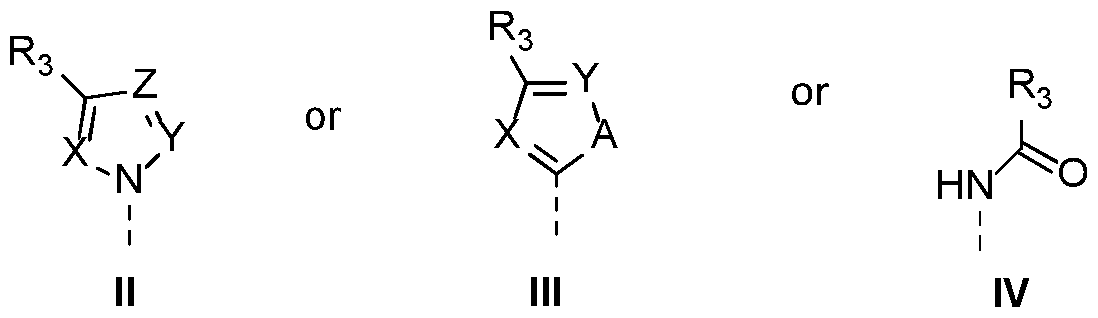 wherein X, Y, Z are each independently selected from N or CH, wherein A is NH, O or S, wherein R3 is an aryl or heteroaryl group, both groups optionally substituted with a group selected from halogen, Cl-5 alkyl, Cl-5 alkoxy, cyano, haloalkyl and nitro,
wherein X, Y, Z are each independently selected from N or CH, wherein A is NH, O or S, wherein R3 is an aryl or heteroaryl group, both groups optionally substituted with a group selected from halogen, Cl-5 alkyl, Cl-5 alkoxy, cyano, haloalkyl and nitro,
-R2 is

Wherein if Ri is substituent II or III, D is S or P, and B is NH or O, Wherein if Ri is substituent IV, D is P and B is NH, wherein R4 is selected from the group consisting of a substituted or unsubstituted aliphatic, substituted or unsubstituted cycloaliphatic, substituted or unsubstituted heterocycloaliphatic, substituted or unsubstituted aryl and substituted or unsubstituted hetero(aryl) group.
or wherein
-Q is N
-Ri is selected from the group of substituents II, III or IV
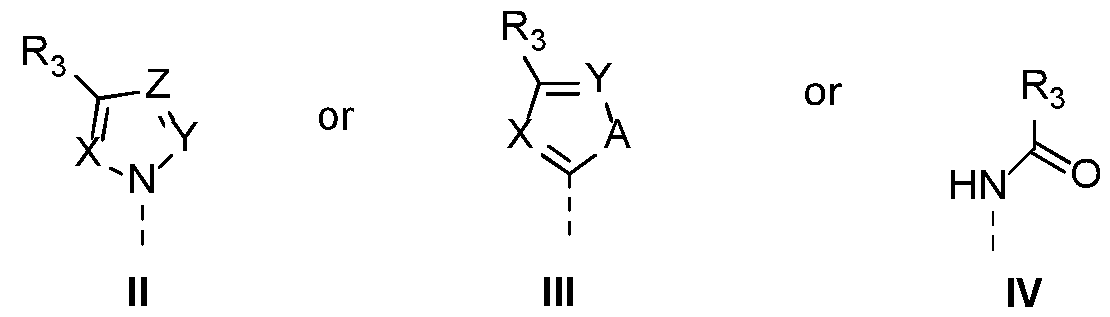 wherein X, Y, Z are each independently selected from N or CH, wherein A is NH, O or S, wherein R3 is an aryl or heteroaryl group, optionally substituted with a group selected from halogen, Cl-5alkyl, Cl-5 alkoxy, cyano, haloalkyl, nitro,
wherein X, Y, Z are each independently selected from N or CH, wherein A is NH, O or S, wherein R3 is an aryl or heteroaryl group, optionally substituted with a group selected from halogen, Cl-5alkyl, Cl-5 alkoxy, cyano, haloalkyl, nitro,
-R2 is
 wherein D is S or P, wherein B is NH or O, wherein R4 is selected from a substituted or unsubstituted aliphatic, substituted or unsubstituted cycloaliphatic, substituted or unsubstituted heterocycloaliphatic, substituted or unsubstituted aryl or substituted or unsubstituted hetero(aryl) group.
wherein D is S or P, wherein B is NH or O, wherein R4 is selected from a substituted or unsubstituted aliphatic, substituted or unsubstituted cycloaliphatic, substituted or unsubstituted heterocycloaliphatic, substituted or unsubstituted aryl or substituted or unsubstituted hetero(aryl) group.
2. The compound according to claim 1, wherein Q is CH and Ri is substituent II with X = CH and Y and Z = N.
3. The compound according to claim 1, wherein Q is CH, and wherein in R2 D is S, B is O and R4 is a substituted heterocyclo aliphatic group.
4. The compound according to claim 3, wherein R4 is ethyl (4-sulfonamido)- piperidine-l-carboxylate.
5. The compound according to claim 1, selected from the group consisting of
- Ethyl 4-((4-(4-phenyl-lH-l,2,3-triazol-l-yl)naphthalene)-l-sulfonamido) - piperidine-l-carboxylate (7d),
Ethyl 4-((4-(4-(2-methylphenyl)-l H-l, 2, 3-triazol- 1-yl) naphthalene)- 1- sulfonamido)piperidine-l-carboxylate (9d),
Ethyl 4-((4-(4-(3-methylphenyl)-l H-l, 2, 3-triazol- 1-yl) naphthalene)- 1- sulfonamido)piperidine-l-carboxylate (9e),
Ethyl 4-((4-(4-(3-fluorophenyl)-l H-l, 2, 3-triazol- 1-yl) naphthalene)- 1- sulfonamido)piperidine-l-carboxylate (9g),
Ethyl 4-((4-(4-(4-fluorophenyl)-l H-l, 2, 3-triazol- 1-yl) naphthalene)- 1- sulfonamido)piperidine-l-carboxylate (9h),
- Ethyl 4-((4-(4-(3-trifluoromethylphenyl)-lH-l, 2, 3-triazol- 1-yl) naphthalene) -l-sulfonamido)piperidine-l-carboxylate (9i),
Ethyl 4-((4-(5-phenyl-l,3,4-thiadiazol-2-yl)naphthalene)-l-sulfonamido) piperidine-l-carboxylate (Ila),
- Ethyl 4-((4-(5-phenylthiazol-2-yl)naphthalene)-l-sulfonamido) piperidine-l- carboxylate (11b), and
-Ethyl 4-(4-(2-methylbenzamido)naphthalene) sulfonimidamido) piperidine-l- carboxylate (28).
6. The compound according to claim 1, wherein Q = N.
7. The compound according to claim 6, wherein Ri is substituent IV and R.2 is

8. The compound according to claim 7, which is Ethyl 4-((5-(2- methylbenzamido)isoquinoline)-8-sulfonamido)piperidine-l-carboxylate (38).
9. A compound according to any one of the claims 1 to 8, for use a medicament.
10. A compound according to any one of the claims 1 to 8, for use in the treatment or prevention of a CCR8 mediated disease.
11. The compound for use in the treatment or prevention according to claim 10 wherein the CCR8 mediated disease is selected from the group consisting of asthma, atopic dermatitis, allergic rhinitis, dermatitis, eczema, urticaria,
rheumatoid arthritis, inflammatory bowel disease, psoriasis, multiple scelerosis, diabetes and cancer.
12. The compound for use in the treatment or prevention according to claim 10, wherein the compound is selected from the group consisting of:
- Ethyl 4-((4-(4-phenyl-lH-l,2,3-triazol-l-yl)naphthalene)-l-sulfonamido) - piperidine-l-carboxylate (7d),
Ethyl 4-((4-(4-(2-methylphenyl)-lH-l,2,3-triazol-l-yl)naphthalene)-l- sulfonamido)piperidine-l-carboxylate (9d),
Ethyl 4-((4-(4-(3-methylphenyl)-lH-l,2,3-triazol-l-yl)naphthalene)-l- sulfonamido)piperidine-l-carboxylate (9e),
Ethyl 4-((4-(4-(3-fluorophenyl)-lH-l,2,3-triazol-l-yl)naphthalene)-l- sulfonamido)piperidine-l-carboxylate (9g),
Ethyl 4-((4-(4-(4-fluorophenyl)-lH-l,2,3-triazol-l-yl)naphthalene)-l- sulfonamido)piperidine-l-carboxylate (9h),
- Ethyl 4-((4-(4-(3-trifluoromethylphenyl)-lH-l,2,3-triazol-l-yl)naphthalene) -l-sulfonamido)piperidine-l-carboxylate (9i),
Ethyl 4-((4-(5-phenyl-l,3,4-thiadiazol-2-yl)naphthalene)-l-sulfonamido) piperidine-l-carboxylate (Ila),
- Ethyl 4-((4-(5-phenylthiazol-2-yl)naphthalene)-l-sulfonamido) piperidine-l- carboxylate (11b),
-Ethyl 4-(4-(2-methylbenzamido)naphthalene) sulfonimidamido) piperidine-l- carboxylate (28), and
- Ethyl 4-((5-(2-methylbenzamido)isoquinoline)-8-sulfonamido)piperidine-l- carboxylate (38).
13. A pharmaceutical composition comprising a compound according to any one of the claims 1 to 8, and a pharmaceutically acceptable carrier.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP22214497.4 | 2022-12-19 | ||
| EP22214497 | 2022-12-19 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2024133292A1 true WO2024133292A1 (en) | 2024-06-27 |
| WO2024133292A9 WO2024133292A9 (en) | 2024-08-15 |
Family
ID=84537986
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2023/086683 Ceased WO2024133292A1 (en) | 2022-12-19 | 2023-12-19 | Novel ccr8 antagonists |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2024133292A1 (en) |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004058709A1 (en) * | 2002-12-23 | 2004-07-15 | Millennium Pharmaceuticals, Inc. | Ccr8 inhibitors |
| WO2004058736A1 (en) * | 2002-12-23 | 2004-07-15 | Millennium Pharmaceuticals, Inc. | Ccr8 inhibitors |
| US20050085518A1 (en) * | 2002-12-23 | 2005-04-21 | Millennium Pharmaceuticals, Inc. | Aryl sulfonamides useful as inhibitors of chemokine receptor activity |
| WO2007067995A2 (en) | 2005-12-09 | 2007-06-14 | Kalypsys, Inc. | Inhibitors of histone deacetylase for the treatment of disease |
| WO2013131010A2 (en) * | 2012-03-02 | 2013-09-06 | Icahn School Of Medicine At Mount Sinai | Function of chemokine receptor ccr8 in melanoma metastasis |
| WO2013178362A1 (en) | 2012-05-31 | 2013-12-05 | Phenex Pharmaceuticals Ag | Carboxamide or sulfonamide substituted thiazoles and related derivatives as modulators for the orphan nuclear receptor ror[gamma] |
| WO2022000443A1 (en) * | 2020-07-03 | 2022-01-06 | Nanjing Immunophage Biotech Co., Ltd. | Methods and compositions for targeting tregs using ccr8 inhibitors |
-
2023
- 2023-12-19 WO PCT/EP2023/086683 patent/WO2024133292A1/en not_active Ceased
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004058709A1 (en) * | 2002-12-23 | 2004-07-15 | Millennium Pharmaceuticals, Inc. | Ccr8 inhibitors |
| WO2004058736A1 (en) * | 2002-12-23 | 2004-07-15 | Millennium Pharmaceuticals, Inc. | Ccr8 inhibitors |
| US20050085518A1 (en) * | 2002-12-23 | 2005-04-21 | Millennium Pharmaceuticals, Inc. | Aryl sulfonamides useful as inhibitors of chemokine receptor activity |
| WO2007067995A2 (en) | 2005-12-09 | 2007-06-14 | Kalypsys, Inc. | Inhibitors of histone deacetylase for the treatment of disease |
| WO2013131010A2 (en) * | 2012-03-02 | 2013-09-06 | Icahn School Of Medicine At Mount Sinai | Function of chemokine receptor ccr8 in melanoma metastasis |
| WO2013178362A1 (en) | 2012-05-31 | 2013-12-05 | Phenex Pharmaceuticals Ag | Carboxamide or sulfonamide substituted thiazoles and related derivatives as modulators for the orphan nuclear receptor ror[gamma] |
| WO2022000443A1 (en) * | 2020-07-03 | 2022-01-06 | Nanjing Immunophage Biotech Co., Ltd. | Methods and compositions for targeting tregs using ccr8 inhibitors |
Non-Patent Citations (26)
| Title |
|---|
| BESSMERTNYKH ET AL., CHEM LETT, vol. 38, 2009, pages 738 - 739 |
| CHENGIBSON, RSC ADV, vol. 5, 2015, pages 4171 |
| CHENLUO, TETRAHEDRON LETT, vol. 60, 2019, pages 268 - 271 |
| CHINTHAKINDI ET AL., ANGEW CHEM INT ED ENGL, vol. 56, 2017, pages 4100 - 4109 |
| JENKINS ET AL., J MED CHEM, vol. 50, 2007, pages 566 - 584 |
| JENKINS TRACY J. ET AL: "Design, Synthesis, and Evaluation of Naphthalene-Sulfonamide Antagonists of Human CCR8", vol. 50, no. 3, 8 February 2007 (2007-02-08), US, pages 566 - 584, XP055883728, ISSN: 0022-2623, Retrieved from the Internet <URL:https://pubs.acs.org/doi/pdf/10.1021/jm061118e> DOI: 10.1021/jm061118e * |
| JIN ET AL., BIOORG MED CHEM LETT, vol. 17, 2007, pages 1722 - 1725 |
| KUMARI ET AL., J. MED. CHEM, vol. 63, 2020, pages 12290 - 12358 |
| LENTINI ET AL., ACS MED CHEM LETT, vol. 10, 2019, pages 1284 - 1289 |
| LIU ET AL., BIOCHEM PHARMACOL, vol. 188, 2021, pages 114565 |
| LIU ET AL., ORG LETT, vol. 20, 2018, pages 7771 - 7774 |
| PC RUMMEL ET AL: "Molecular requirements for inhibition of the chemokine receptor CCR8 - probe-dependent allosteric interactions", BRITISH JOURNAL OF PHARMACOLOGY, WILEY-BLACKWELL, UK, vol. 167, no. 6, 19 October 2012 (2012-10-19), pages 1206 - 1217, XP071038587, ISSN: 0007-1188, DOI: 10.1111/J.1476-5381.2012.02076.X * |
| POTRATZ, BEILSTEIN J. ORG. CHEM, vol. 8, 2012, pages 683 - 692 |
| REN ET AL., ORG PROCESS RES DEV, vol. 13, 2009, pages 2002489 |
| RUIZ-CASTILLOBUCHWALD, CHEM REV, vol. 116, 2016, pages 12564 - 12649 |
| RUMMEL ET AL., BRJ PHARMACOL, vol. 167, 2012, pages 1206 - 1217 |
| SEIFERT ET AL., ANGEW. CHEM. INT. ED., vol. 56, 2017, pages 14937 - 14941 |
| SUZUKI, ANGEWANDTE CHEMIE, vol. 50, 2011, pages 6723 - 6737 |
| VAN OVERTVELDT ET AL., ORG BIOMOL CHEM, vol. 13, 2015, pages 5260 - 5264 |
| VERHAEGEN ET AL., BIOORG CHEM, vol. 107, 2021, pages 104560 |
| VERHAEGEN YENTHEL ET AL: "Palladium-catalyzed cross-coupling reactions on a bromo-naphthalene scaffold in the search for novel human CC chemokine receptor 8 (CCR8) antagonists", BIOORGANIC CHEMISTRY, ACADEMIC PRESS INC., NEW YORK, NY, US, vol. 107, 16 December 2020 (2020-12-16), pages 104560, XP086485248, ISSN: 0045-2068, [retrieved on 20201216], DOI: 10.1016/J.BIOORG.2020.104560 * |
| WANG ET AL., THORAX, vol. 68, 2013, pages 263 - 268 |
| WANG M ET AL: "Synthesis of new carbon-11 labeled naphthalene-sulfonamides for PET imaging of human CCR8", APPLIED RADIATION AND ISOTOPES, ELSEVIER, OXFORD, GB, vol. 66, no. 10, 1 October 2008 (2008-10-01), pages 1406 - 1413, XP023785013, ISSN: 0969-8043, [retrieved on 20080320], DOI: 10.1016/J.APRADISO.2008.03.010 * |
| YEUNG ET AL., TETRAHEDRON, vol. 46, 2005, pages 3429 - 3432 |
| YOSHIOKA ET AL., BIOORG MED CHEM, vol. 25, 2017, pages 3461 - 3470 |
| ZHANG ET AL., JOURNAL OF ORGANIC CHEMISTRY, vol. 80, 2015, pages 705 - 710 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2024133292A9 (en) | 2024-08-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2008296974B2 (en) | Soluble guanylate cyclase activators | |
| CA2985542C (en) | Triazole agonists of the apj receptor | |
| CN101678013B (en) | Piperidine/piperazine derivatives | |
| Nishida et al. | Discovery, synthesis, and biological evaluation of novel pyrrole derivatives as highly selective potassium-competitive acid blockers | |
| AU2020286381B2 (en) | Imidazolo derivatives, compositions and methods as orexin antagonists | |
| US20190284173A1 (en) | Triazole phenyl compounds as agonists of the apj receptor | |
| WO2013083991A1 (en) | Novel compounds and their use in therapy | |
| CN102311448B (en) | Thienopyrimidinone DPP-IV Inhibitors | |
| JP2025512453A (en) | KIF18A inhibitors and their uses | |
| BRPI0915876B1 (en) | AZO COMPOUND, PHARMACEUTICAL COMPOSITION AND USE OF THE COMPOUND FOR NEUTROPATHIC PAIN TREATMENT | |
| AU2015226679B2 (en) | Piperidine derivatives as orexin receptor antagonist | |
| KR20130046436A (en) | Cyclic n,n'-diarylthioureas and n,n'-diarylureas as androgen receptor antagonists, anti-cancer agent, method for producing and using same | |
| CA3207590A1 (en) | Pyridopyrimidinone derivative, preparation method therefor, and use thereof | |
| EP3194403A1 (en) | Pyrrolopyrimidine derivatives as nr2b nmda receptor antagonists | |
| JP2005526024A (en) | Compounds for inhibiting insulin secretion and methods related thereto | |
| AU2023249574A1 (en) | At2r agonist | |
| EA037974B1 (en) | Compound having mutant idh inhibitory activity, preparation method and use thereof | |
| EP4037768B1 (en) | Novel substituted 6,7-dihydro-5h-benzo[7]annulene compounds, processes for their preparation and therapeutic uses thereof | |
| WO2008118319A2 (en) | Mineralocorticoid receptor modulators | |
| WO2024133292A1 (en) | Novel ccr8 antagonists | |
| CN116322692A (en) | Autotaxin inhibitor compounds | |
| CA2703593A1 (en) | 4,4-disubstituted piperidines | |
| EP3704122A1 (en) | Fused triazole agonists of the apj receptor | |
| WO2019213006A1 (en) | Substituted pyrimidinones as agonists of the apj receptor | |
| CN101918398A (en) | 4,4-Disubstituted piperidines as renin inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 23833813 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 23833813 Country of ref document: EP Kind code of ref document: A1 |