WO2024118785A2 - Tlr7 agonists and antibody-drug-conjugates thereof - Google Patents
Tlr7 agonists and antibody-drug-conjugates thereof Download PDFInfo
- Publication number
- WO2024118785A2 WO2024118785A2 PCT/US2023/081614 US2023081614W WO2024118785A2 WO 2024118785 A2 WO2024118785 A2 WO 2024118785A2 US 2023081614 W US2023081614 W US 2023081614W WO 2024118785 A2 WO2024118785 A2 WO 2024118785A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- och
- compound
- heteroalkylene
- adc
- abd
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/65—Peptidic linkers, binders or spacers, e.g. peptidic enzyme-labile linkers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6839—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting material from viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6849—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a receptor, a cell surface antigen or a cell surface determinant
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6855—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from breast cancer cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6867—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from a cell of a blood cancer
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6869—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from a cell of the reproductive system: ovaria, uterus, testes, prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6889—Conjugates wherein the antibody being the modifying agent and wherein the linker, binder or spacer confers particular properties to the conjugates, e.g. peptidic enzyme-labile linkers or acid-labile linkers, providing for an acid-labile immuno conjugate wherein the drug may be released from its antibody conjugated part in an acidic, e.g. tumoural or environment
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Definitions
- TLR7 agonists and antibody-drug-conjugates thereof.
- ADCs antibody-drug-conjugates
- BACKGROUND Toll-like receptors (TLRs) are a subset of pattern recognition receptors (PRRs) and play a key role in the innate immune response. TLRs are divided into two groups depending on subcellular localization, with endosomal TLRs being of pharmaceutical interest. Of these endosomal TLRs, TLR7 has been extensively studied as a target for small molecule agonists. See, Patinote, et al., Eur. J. Med. Chem., 2020, 193:112238; U.S.
- TLR7 agonists have been reported to have antiviral and antibacterial activity, as well as activity as vaccine adjuvants and in the treatment of allergic diseases and asthma. Of interest herein, TLR7 agonists have been studied as cancer immunotherapeutics. One TLR7 agonist has been approved by the U.S. FDA, Aldara® (imiquimod) which is indicated for treatment of actinic keratosis, superficial basal cell carcinoma and external genital warts.
- ADCs combine the power of antibody specificity with the ability to site specifically target a particular type of cell or tissue with a payload.
- ADCETRIS® currentuximab vedotin
- KADCYLATM anti-trastuzumab emtansine
- TLR7 agonist payloads have been reported. See, e.g., U.S. Patent Nos.10,472,420, 10,780,180, 10,548,985, 10,722,591, 10,675,358; PCT Publication No. WO 2020/181050. However, no such ADCs have been approved for human use. [0006] Thus, there is a continuing need for TLR agonists and ADCs thereof for treatment of various diseases, including cancer and chronic hepatitis B.
- the present disclosure provides TLR7 agonists of Formula I for use in the compositions and methods provided herein: or a pharmaceutically acceptable salt thereof, wherein: R 1 is H, halo, -NHR 4 , -OR 4 , -NH-OR 4 or -R 4 ; R 2 is H, halo, or alkoxy; R 3 is -CO 2 R 23 , -CONHR 23 , -alkylene-Y, -alkylene-arylene-Y, -heteroalkylene-Y, -heteroalkylene-arylene-Y, -(hydroxy)heteroalkylene-Y, -(amino)heteroalkylene-Y, or - alkylene-PEG-Y; R 4 is alkyl optionally substituted with alkoxy or heteroalkyl; R 23 is H, alkyl or aryl; X is CH or N; Y is -OH, -Gly,
- the present disclosure provides TLR7 agonist-linkers of Formula II for use in preparation of ADCs provided herein: or a pharmaceutically acceptable salt thereof, wherein: R 1 , R 2 and X are as defined for Formula I; R 9 is a divalent group formed by removal of a terminal hydrogen (i.e., a hydrogen distal from the phenyl group to which R 9 is attached) from an R 3 group, as defined in claim 1; and L is any group or moiety that links, connects, or bonds to an antigen-binding domain ABD.
- the ADCs provided herein are useful in methods of treatment, methods of imaging, or methods of diagnosis.
- ADCs antibody-drug-conjugates
- ADCs comprising an antigen- binding domain (ABD) specific to an antigen and a Toll-like receptor 7 (TLR7) agonist
- the antigen is an HBV surface antigen (HBS sAg) and the disease is chronic Hepatitis B.
- HBS sAg HBV surface antigen
- the present disclosure provides an antibody-drug-conjugate (ADC), comprising (a) an antigen-binding domain (ABD) having binding specificity to a hepatitis B virus surface antigen (HBV sAg) and (b) a Toll-like receptor 7 (TLR7) agonist.
- the ADC further comprises a divalent linker that links the ABD to the TLR7 agonist.
- the ADC is according to Formula IV: or a pharmaceutically acceptable salt thereof, wherein: L 1 is a divalent linker; R 1 is H, halo, -NHR 4 , -OR 4 , -NH-OR 4 or -R 4 ; R 2 is H, halo, or alkoxy; R 4 is alkyl optionally substituted with alkoxy or heteroalkyl; R 9 is a divalent group formed by removal of a hydrogen from R 3 , R 3 being a group attached to the phenyl group at the position of R 9 ; R 3 is -CO 2 H, -CONHR 23 , -alkylene-Y, -alkylene-arylene-Y, -heteroalkylene-Y, - heteroalkylene-arylene-Y, -(hydroxy)heteroalkylene-Y
- the ADC comprises ABD linked to a compound of Formula III: or a pharmaceutically acceptable salt thereof, wherein: R 1 , R 2 and X are as described elsewhere for Formula I; L is any group or moiety that links to ABD; R 11 and R 12 are, independently, hydrogen, alkyl, alkoxy, alkenyl, alkynyl, aryl, arylalkyl, heteroaryl, alkylene, or heteroalkylene, wherein when R 11 is alkylene or heteroalkylene, the alkylene or heteroalkylene is further bonded to R 13 to form a 3-, 4-, 5-, 6-, 7-, or 8-membered heterocyclyl; R 14 is hydrogen, alkylene, heteroalkylene, or an amino acid side chain, wherein when R 14 is alkylene or heteroalkylene, the alkylene or heteroalkylene is further bonded to R 13 to form a 3-, 4-, 5-, 6-, 7-, or 8-membered heterocyclyl
- ABD-L 1 is linked to a compound selected from P1, P2, P6, P8, P17, P18, P19, P20, P23, P27, P29, P32, P33, P37, and P39.
- the ABD is linked to a compound selected from LP1, LP6, LP7, LP8, LP10, and LP11.
- the ADC is according to Formula V: or a pharmaceutically acceptable salt thereof, wherein: R 1 , R 2 and X are as described elsewhere for Formula I; R 10 is -alkylene-NH-, -alkylene-arylene-NH-, -heteroalkylene-NH-, -heteroalkylene- arylene-NH-, -(hydroxy)heteroalkylene-NH-, -(amino)heteroalkylene-NH-, or -alkylene- PEG-NH-;
- ABD is an antibody that contains a Q295 residue, an N297Q mutation, and/or one or more engineered LLQG (SEQ ID NO: 1), LLQGG (SEQ ID NO: 2), LLQLLQG (SEQ ID NO: 3), LLQYQG (SEQ ID NO: 4), LLQGA (SEQ ID NO: 5), LLQGSG (SEQ ID NO: 6), SLLQG (SEQ ID NO: 1)
- the ADC is according to Formula VI: or a pharmaceutically acceptable salt thereof, wherein: L 1 is a divalent linker; R 1 , R 2 , R 16 , R 11 , R 12 , R 13 , R 14 , R 15 , X, and x are as defined for Formula III; and k is an integer from one to thirty.
- the ADC comprises ABD-L 1 linked to a compound selected from P1, P2, P6, P8, P17, P18, P19, P20, P23, P27, P29, P32, P33, P37, and P39.
- k is 1, 2, 3, 4, or 5. In some embodiments, k is 2.
- the ABD comprises a heavy chain and the C-terminus of the heavy chain is conjugated to L 1 . In some embodiments, the ABD comprises two heavy chains and the C-terminus of each of the two heavy chains is conjugated to L 1 . In some embodiments, L 1 is linked to a cysteine residue of the ABD.
- the ABD is an antibody against a HBV sAg or a fragment thereof. In some embodiments, the ABD is a human antibody or a humanized antibody. In some embodiments, the ABD is IgG1 or IgG2a. In some embodiments, the ABD comprises a scFv having binding specificity to a HBV sAg.
- the ABD comprises V H chain and V L chain of an antibody against a HBV sAg.
- the ABD comprises HCDR1, HCDR2, HCDR3, LCDR1, LCDR2, and LCDR3 of an antibody against a HBV sAg.
- the ABD comprises an Fc region, wherein the Fc region comprises a modification for enhanced binding to Fc ⁇ R.
- the present disclosure provides a pharmaceutical composition comprising the ADC disclosed herein and one or more pharmaceutically acceptable carriers, excipients, or diluents.
- the present disclosure provides a method of treatment, comprising administering to a subject in need thereof an effective amount of the ADC or the pharmaceutical composition disclosed herein.
- the subject has chronic Hepatitis B.
- the subject has elevated circulating HBV DNA or HBV sAg in serum prior to administration of the ADC or the pharmaceutical composition.
- the method further comprises, before the administering, measuring circulating HBV DNA or HBV sAg in serum of the subject.
- the method further comprises, after the administering, measuring circulating HBV DNA or HBV sAg in serum of the subject to assess therapeutic efficacy of the ADC or the pharmaceutical composition.
- the step of administrating the ADC or the pharmaceutical composition is repeated. In some embodiments, the step of administrating the ADC or the pharmaceutical composition is repeated twice, three times, or more. In some embodiments, the step of administrating the ADC or the pharmaceutical composition is repeated at least at 1-week intervals, at 2-week intervals, at 3-week intervals, or at 4-week intervals. In some embodiments, the step of administrating the ADC or the pharmaceutical composition is repeated at 1-week intervals, at 2-week intervals, at 3-week intervals, or at 4-week intervals. In some embodiments, the step of administrating the ADC or the pharmaceutical composition is repeated at 1-month intervals, at 2-month intervals, or 3-month intervals.
- the ADC or pharmaceutical composition is administered by oral, intravenous, intraperitoneal, inhalation, intranasal, intramuscular, or subcutaneous administration.
- One aspect of the present disclosure provides the ADC or the pharmaceutical composition for use in treatment.
- the ADC or the pharmaceutical composition is for use in treatment of chronic Hepatitis B in a subject in need thereof.
- Another aspect of the present disclosure provides the ADC or the pharmaceutical composition for manufacture of a medicament.
- the medicament is for the treatment of chronic Hepatitis B in a subject in need thereof.
- FIG.1 shows in vitro plasma stability of anti-HER2 Ab-LP1 ADC (Example 68).
- FIG.2 shows in vitro plasma stability of anti-HER2 Ab-LP6A ADC (Example 68).
- FIG.3 shows in vitro plasma stability of anti-HER2 Ab-LP11A ADC (Example 68).
- FIG.4 shows in vitro plasma stability of anti-HER2 Ab-LP7A ADC (Example 68).
- FIG.5 shows the conjugation scheme for conjugating antibodies with linker payloads provided herein (Examples 63, 64 and 69).
- FIG.6 is a preparative SEC chromatogram of a conjugation mixture demonstrating a clean separation of ADC monomer from aggregates (“HMW”) and unconjugated linker payload (“Free drug”).
- FIG. 7 is an analytical SEC chromatogram of an SEC purified antibody-TLR7 conjugate showing a monomer purity of 99.7%.
- FIG.8 shows a LC-ESI-MS spectrum of a deglycosylated and reduced ADC sample. The calculated average DAR value was 1.84. The deconvoluted mass spectra exhibited light chain species (LC, LC1) and heavy chain species (HC, HC1, HC2, etc.). The average DAR can be calculated from the LC and HC drug-loading.
- FIG.9 shows HIC chromatograms of an antibody and its LP11A conjugate, revealing a mixture of three species: DAR2 species (51%), DAR4 species (28%) and unconjugated antibody (21%). The average DAR of this ADC is 2.1.
- FIG. 10 depicts results following a single treatment of anti-HER2 Ab-LP6A ADC (Table 3) in the N87 xenograft tumor model. Dosing was performed at Day 0.
- Regression of tumor was observed after treatment with 5 mg/kg (gray circle) of anti-HER2 Ab-LP6A ADC, while treatment with 1 mg/kg (gray square) anti-HER2 Ab-LP6A ADC resulted in tumor stasis, when compared to saline treated animals (open circle).
- Regression of N87 gastric tumors was not observed in the N87 xenograft mice treated with 5 mg/kg of isotype control Ab-LP6A ADC (Table 3) (black circle) or 0.5 mg/kg (gray triangle) anti-HER2 Ab-LP6A ADC or 0.1 mg/kg (gray diamond) anti-HER2 Ab-LP6A ADC when compared to saline treated animals (open circle).
- FIG.11 depicts results following treatment of human N87 xenograft tumors with a single dose of anti-HER2 Ab-LP6A ADC, anti-HER2 Ab-LP11A ADC or anti-HER2 Ab-LP7A ADC (Table 3). Dosing was performed at Day 0. Regression of tumor was observed after treatment with 5 mg/kg of anti-HER2 Ab-LP6A ADC (gray circle), 5 mg/kg of anti-HER2 Ab- LP11A ADC (gray square) or 5 mg/kg of anti-HER2 Ab-LP7A ADC (gray triangle), when compared to saline treated animals (open circle).
- FIG. 12 depicts results following treatment of trastuzumab-resistant HER2 medium human JIMT-1 xenograft tumors.
- Dosing was initiated at Day 0 and subsequently every 7 days for a total of 4 doses of anti-HER2 Ab-LP6A ADC (Table 3) or in combination with pertuzumab. Regression of tumor was observed after treatment with 5 mg/kg of anti-HER2 Ab-LP6A ADC in combination with 5 mg/kg pertuzumab (gray square), while treatment with 5 mg/kg of anti- HER2 Ab-LP6A ADC alone (gray circle) resulted in tumor stasis for 45 days, when compared to 5 mg/kg unconjugated mAb2 alone (open circle) treated animals.
- FIG.13 depicts results following treatment of MC38 tumors engineered to express human CD20.
- tumor bearing mice were treated with 3 total doses of anti- CD20 Ab-LP6A ADC (Table 3) with each dose separated by seven days. Tumor regression was observed in four of five mice after treatment with 5 mg/kg of anti-CD20 Ab-LP6A ADC (gray square) when compared to saline treated animals (open circle). Regression of MC38hCD20 tumors was not observed in the MC38hCD20 syngeneic mice treated with 5 mg/kg of anti-mIgG2a Ctrl Ab-LP1 ADC (black circle) (Table 3) or 5 mg/kg unconjugated anti- CD20 Ab (open square), when compared to 5 mg/kg saline treated animals (open circle).
- FIG.14 depicts HBV sAg levels measured in the chronic hepatitis B (CHB) disease mouse model after treatment with an anti-sAg mAb (mAb3), anti-sAg mAb-TLR7 agonist (mAb3+LP1 or mAb4+LP1), a TLR7 agonist (LP1), or PBS three times, two weeks apart subcutaneously (SC). (Example 51; Experiment 1).
- CHB chronic hepatitis B
- mAb3+LP1 or mAb4+LP1 anti-sAg mAb-TLR7 agonist
- LP1 TLR7 agonist
- FIG.15 depicts HBV sAg levels measured in the chronic hepatitis B (CHB) disease mouse model after treatment with an anti-sAg mAb (mAb3), anti-sAg mAb-TLR7 agonist (mAb3+LP1 or mAb4+LP1), a TLR7 agonist (LP1), or PBS three times, two weeks apart subcutaneously (SC). (Example 51; Experiment 2).
- FIG.16 depicts results following parental MC38 tumor cell rechallenge in mice having initially cleared MC38.hTAA Pos engrafted tumors (the same as used in Fig.13).
- mice On day 60 after MC38.hTAA Pos tumor cell inoculation, tumor free mice (black square) were rechallenged with parental MC38 cells without overexpression of human TAA. Compared to control na ⁇ ve mice (open circle), mice previously treated with anti-CD20 -LP6A conjugate are protected against tumor rechallenge. Data represent mean tumor volumes (mean+/-SEM) over time (post-rechallenge). [0047] FIG.
- FIG. 17 depicts results following treatment of mice having been inoculated with MC38.hTAA Pos tumor cells with 3 doses every seven days of anti-CD20-LP11A conjugate in wild type mice (closed symbols with solid lines) and in humanized IFNAR mice (open symbols with dashed lines) that lack the ability to respond to murine type I IFN. Regression of tumor was observed after treatment with 5 mg/kg of anti-CD20-LP11A conjugate (closed triangle) when compared to saline treated animals (closed circle) and isotype control antibody conjugate (closed square).
- FIG.18 depicts results following treatment of mice having been inoculated with MC38.hTAA Pos tumor cells with 3 doses every seven days of anti-CD20-LP6A conjugate with or without 5 doses every four days of anti-CD20 x anti-hCD3 bispecific antibody in mice humanized for TAA and human CD3.
- FIG.19 depicts the ring opening of the imide bond of the antibody-drug conjugates from the conjugation of the cysteine thiol with the maleimide of the linker-payload. Ring- opening of the imide bond under physiological conditions affords two regio-isomers that one is the thiol attached to the alpha carbon and the other is the thiol attached to the beta carbon to the carboxylic acid group, respectively.
- FIG. 20 is a scheme showing one possible metabolic pathway for the compound referenced herein as Q o -LP11A.
- FIG.21 is a scheme showing another possible metabolic pathway for the compound referenced herein as Q o -LP11A.
- FIG. 22 shows hepatitis B virus surface antigen (HBV sAg) levels measured in a chronic hepatitis B (CHB) disease mouse model after treatment with an anti-sAg monoclonal antibody-TLR7 agonist (mAb3+LP6A) or phosphate-buffered saline (PBS) 5 times, 1 week apart subcutaneously.
- mAb3+LP6A was effective in reducing the HBV sAg levels as compared to the PBS control.
- FIG.23 shows anti-hepatitis B virus surface antigen (HBsAG) IgG titers measured in a chronic hepatitis B (CHB) disease mouse model at day 120 (D120) post-first treatment with anti-surface antigen (sAg) monoclonal antibody-TLR7 agonist (mAb3+LP6A) or phosphate- buffered saline (PBS) 5 times, 1 week apart subcutaneously.
- sAg anti-surface antigen
- mAb3+LP6A monoclonal antibody-TLR7 agonist
- PBS phosphate- buffered saline
- Biological activity thus, encompasses therapeutic effects and pharmacokinetic behavior of such compounds, compositions and mixtures. Biological activities can be observed in in vitro systems designed to test for such activities.
- "antigen-binding domain” or “ABD” means any peptide, polypeptide, nucleic acid molecule, scaffold-type molecule, peptide display molecule, or polypeptide- containing construct that is capable of specifically binding a particular antigen of interest.
- “antigen-binding domain” includes antibodies and antigen-binding fragments of antibodies.
- proteins, polypeptides and protein fragments herein are intended to refer to the human version of the respective protein, polypeptide or protein fragment unless explicitly specified as being from a non-human species.
- the phrase “specifically binds,” or “binds specifically to,” or the like, means that an antibody or antigen-binding fragment thereof forms a complex with an antigen that is relatively stable under physiologic conditions. Specific binding can be characterized by an equilibrium dissociation constant of at least about 1x10 -8 M or less (e.g., a smaller K D denotes tighter binding). Methods for determining whether two molecules specifically bind are well known in the art and include, for example, equilibrium dialysis, surface plasmon resonance, and the like.
- Antibodies can, for example, be identified by real-time, label free bio-layer interferometry assay on an Octet® HTX biosensor, which bind specifically to a target antigen. Moreover, multi-specific antibodies that bind to one domain in the target antigen and one or more additional antigens or a bi-specific that binds to two different regions of the target antigen are nonetheless considered antibodies that “specifically bind”, as used herein. In addition to neutralizing antibodies, antibodies that bind specifically to the target antigen, but are non- neutralizing, also can be used within the scope of the present disclosure to generate antibody- drug conjugates. Such antibodies may function, for example, to deliver a payload to the cells expressing a target antigen.
- high affinity antibody refers to those mAbs having a binding affinity to a target antigen, expressed as K D , of at least 10 -8 M; preferably 10 -9 M; more preferably 10 -10 M, even more preferably 10 -11 M, even more preferably 10 -12 M, as measured by real-time, label free bio-layer interferometry assay, e.g., an Octet® HTX biosensor, or by surface plasmon resonance, e.g., BIACORETM, or by solution-affinity ELISA.
- slow off rate refers to an antibody that dissociates from a target antigen, with a rate constant of 1x10 -3 s -1 or less, preferably 1x10 -4 s -1 or less, as determined by real-time, label free bio-layer interferometry assay, e.g., an Octet® HTX biosensor, or by surface plasmon resonance, e.g., BIACORETM.
- label free bio-layer interferometry assay e.g., an Octet® HTX biosensor
- surface plasmon resonance e.g., BIACORETM.
- unrelated antigens are proteins, peptides or polypeptides that have less than 95% amino acid identity to one another.
- antibody means any antigen-binding molecule or molecular complex comprising at least one complementarity determining region (CDR) that specifically binds to or interacts with a particular antigen.
- CDR complementarity determining region
- the term “antibody” includes immunoglobulin molecules comprising four polypeptide chains, two heavy (H) chains and two light (L) chains inter-connected by disulfide bonds, as well as multimers thereof (e.g., IgM).
- Each heavy chain comprises a heavy chain variable region (abbreviated herein as HCVR or V H ) and a heavy chain constant region.
- the heavy chain constant region comprises three domains, C H 1, C H 2 and C H 3.
- Each light chain comprises a light chain variable region (abbreviated herein as LCVR or V L ) and a light chain constant region.
- the light chain constant region comprises one domain (C L 1).
- the V H and V L regions can be further subdivided into regions of hypervariability, termed complementarity determining regions (CDRs), interspersed with regions that are more conserved, termed framework regions (FR).
- CDRs complementarity determining regions
- FR framework regions
- Each V H and V L is composed of three CDRs and four FRs, arranged from amino-terminus to carboxy-terminus in the following order: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4.
- HCDR1, HCDR2, and HCDR3 Three CDRs of V H are referred to as HCDR1, HCDR2, and HCDR3, and three CDRs of V L are referred to as LCDR1, LCDR2 and LCDR3.
- antigen-binding fragment of an antibody means any naturally occurring, enzymatically obtainable, synthetic, or genetically engineered polypeptide or glycoprotein that specifically binds an antigen to form a complex.
- human antibody means antibodies having variable and constant regions derived from human germline immunoglobulin sequences.
- Human antibodies may nonetheless include amino acid residues not encoded by human germline immunoglobulin sequences (e.g., mutations introduced by random or site-specific mutagenesis in vitro or by somatic mutation in vivo), for example in the CDRs and in particular CDR3.
- human antibody as used herein, is not intended to include antibodies in which CDR sequences derived from the germline of another mammalian species, such as a mouse, have been grafted onto human framework sequences.
- the term “humanized antibody” means chimeric antibodies that contain minimal sequence derived from the non-human antibody.
- a humanized antibody is generally a human antibody (recipient antibody) in which residues from one or more CDRs are replaced by residues from one or more CDRs of a non-human antibody (donor antibody).
- the donor antibody can be any suitable non-human antibody, such as a mouse, rat, rabbit, chicken, or non-human primate antibody having a desired specificity, affinity, or biological effect.
- selected framework region residues of the recipient antibody are replaced by the corresponding framework region residues from the donor antibody.
- Humanized antibodies may also comprise residues that are not found in either the recipient antibody or the donor antibody. Such modifications may be made to further refine antibody function.
- recombinant human antibody means all human antibodies that are prepared, expressed, created or isolated by recombinant means, such as antibodies expressed using a recombinant expression vector transfected into a host cell (described further below), antibodies isolated from a recombinant, combinatorial human antibody library (described further below), antibodies isolated from an animal (e.g., a mouse) that is transgenic for human immunoglobulin genes (see, e.g., Taylor et al. (1992) Nucl. Acids Res. 20:6287-6295) or antibodies prepared, expressed, created or isolated by any other means that involves splicing of human immunoglobulin gene sequences to other DNA sequences.
- nucleic acid or fragment thereof indicates that, when optimally aligned with appropriate nucleotide insertions or deletions with another nucleic acid (or its complementary strand), there is nucleotide sequence identity in at least about 90%, and more preferably at least about 95%, 96%, 97%, 98%, or 99% of the nucleotide bases, as measured by any well-known algorithm of sequence identity, such as FASTA, BLAST, or GAP, as discussed in WO 2016/100807 or US 2016/0176953 A1, each of which are incorporated herein by reference in their entirety.
- a nucleic acid molecule having substantial identity to a reference nucleic acid molecule may, in certain instances, encode a polypeptide having the same or substantially similar amino acid sequence as the polypeptide encoded by the reference nucleic acid molecule.
- the phrase “substantial similarity” or “substantially similar” means that two peptide sequences, when optimally aligned, such as by the programs GAP or BESTFIT using default gap weights, share at least 90% sequence identity, even more preferably at least 95%, 98%, or 99% sequence identity.
- the term “surface plasmon resonance” refers to an optical phenomenon that allows for the analysis of real-time interactions by detection of alterations in protein concentrations within a biosensor matrix, for example using the BIAcoreTM system (Biacore Life Sciences division of GE Healthcare, Piscataway, N.J.).
- K D means the equilibrium dissociation constant of a particular protein-protein interaction (e.g., antibody-antigen interaction). Unless indicated otherwise, the K D values disclosed herein refer to K D values determined by surface plasmon resonance assay at 25° C.
- salts include, but are not limited to, amine salts, such as but not limited to N,N'-dibenzylethylenediamine, chloroprocaine, choline, ammonia, diethanolamine and other hydroxyalkylamines, ethylenediamine, N- methylglucamine, procaine, N-benzylphenethylamine, 1-para-chlorobenzyl-2-pyrrolidin-1'- ylmethylbenzimidazole, diethylamine and other alkylamines, piperazine and tris(hydroxymethyl)aminomethane; alkali metal salts, such as but not limited to lithium, potassium and sodium; alkali earth metal salts, such as but not limited to barium, calcium and magnesium; transition metal salts, such as but not limited to zinc; and inorganic salts, such as but not limited to, sodium hydrogen phosphate and disodium phosphate; and also including, but not limited to, salts of mineral
- the terms “treat,” “treating,” or “treatment” refer to the reduction or amelioration of the severity of at least one symptom or indication of the disease, e.g., cancer or hepatitis B infection, due to the administration of a therapeutic agent such as a disclosed antibody to a subject in need thereof.
- a therapeutic agent such as a disclosed antibody to a subject in need thereof.
- the terms include inhibition of progression of disease or of worsening of infection.
- the terms also include positive prognosis of disease, e.g., the subject may be free of infection, the subject may have reduced or no viral titers, the subject may have tumor shrinkage, upon administration of a therapeutic agent such as a disclosed antibody or antibody-drug conjugate.
- the therapeutic agent may be administered at a therapeutic dose to the subject.
- the terms “prevent,” “preventing,” or “prevention” refer to inhibition of manifestation of any symptoms or indications of a disease (e.g., cancer or hepatitis B infection) upon administration of a disclosed antibody or antibody-drug conjugate. The term includes prevention of the spread of infection in a subject exposed to the virus or at risk of having hepatitis B infection.
- the phrase “therapeutically effective amount” refers to an amount that produces the desired effect for which it is administered. The exact amount will depend on the purpose of the treatment, and will be ascertainable by one skilled in the art using known techniques (see, for example, Lloyd (1999) The Art, Science and Technology of Pharmaceutical Compounding).
- amelioration of the symptoms of a particular disorder by administration of a particular compound or pharmaceutical composition refers to any lessening, whether permanent or temporary, lasting or transient that can be attributed to or associated with administration of the compound or pharmaceutical composition.
- the IC 50 refers to an amount, concentration or dosage of a particular test compound that achieves a 50% inhibition of a maximal response in an assay that measures such response.
- moieties are specified by their conventional chemical formulae, written from left to right, they equally encompass the chemically identical moieties that would result from writing the structure from right to left, e.g., -CH 2 O- is equivalent to -OCH 2 -.
- alkyl by itself or as part of another substituent, means, unless otherwise stated, a straight (i.e., unbranched) or branched chain saturated hydrocarbon radical.
- alkylene by itself or as part of another substituent means a divalent radical derived from an alkyl.
- an alkyl (or alkylene) group will have from 1 to 24 carbon atoms, including those groups having 10 or fewer carbon atoms.
- a “lower alkyl” or “lower alkylene” is a shorter chain alkyl or alkylene group, generally having six or fewer carbon atoms.
- alkyl groups include, but are not limited to, groups such as methyl, ethyl, n-propyl, isopropyl, n- butyl, t-butyl, isobutyl, sec-butyl, homologs and isomers of, for example, n-pentyl, n-hexyl, n- heptyl, n-octyl, and the like.
- alkenyl by itself or as part of another substituent, means, unless otherwise stated, a straight (i.e., unbranched) or branched chain hydrocarbon radical having one or more carbon-carbon double bonds.
- alkenylene by itself or as part of another substituent means a divalent radical derived from an alkenyl.
- an alkenyl (or alkenylene) group will have from 1 to 24 carbon atoms, including those groups having 10 or fewer carbon atoms.
- a “lower alkenyl” or “lower alkenylene” is a shorter chain alkenyl or alkenylene group, generally having six or fewer carbon atoms.
- alkenyl groups include, but are not limited to, vinyl (i.e., ethenyl), 2-propenyl, crotyl, 2-isopentenyl, 2- (butadienyl), 2,4-pentadienyl, 3-(1,4-pentadienyl), and the higher homologs and isomers.
- alkynyl by itself or as part of another substituent, means, unless otherwise stated, a straight (i.e., unbranched) or branched chain hydrocarbon radical having one or more carbon-carbon triple bonds, which can include di- and multivalent radicals, having the number of carbon atoms designated (i.e., C 1 -C 10 means one to ten carbons).
- alkynyl groups include, but are not limited to, ethynyl, 1- and 3-propynyl, 3-butynyl, and the higher homologs and isomers.
- alkoxy alkylamino
- alkylthio thioalkoxy
- alkoxy alkylamino
- alkylthio thioalkoxy
- heteroalkyl by itself or in combination with another term, means, unless otherwise stated, a straight or branched chain hydrocarbon radical, containing at least one heteroatom in the chain selected from O, N, P, Si and S, and wherein the nitrogen and sulfur atoms may optionally be oxidized, and the nitrogen atom may have an alkyl substituent to fulfill valency and/or may optionally be quaternized.
- the heteroatom(s) O, N, P, Si and S may be placed at any interior position of the heteroalkyl group (i.e., not at the point of attachment to the rest of the molecule).
- heteroalkylene by itself or as part of another substituent means a divalent radical derived from heteroalkyl, as exemplified, but not limited by, -CH 2 -O- CH 2 -CH 2 -, -CH 2 -CH 2 -O-CH 2 -CH 2 -, -CH 2 -O-CH 2 -CH 2 -NH-CH 2 -, -CH 2 -CH 2 -S-CH 2 -CH 2 - and - CH 2 -S-CH 2 -CH 2 -NH-CH 2 -.
- cycloalkylene and “heterocycloalkylene” by themselves or as part of another substituent means a divalent radical derived from a cycloalkyl or heterocycloalkyl.
- cycloalkyl include, but are not limited to, cyclopentyl, cyclohexyl, 1-cyclohexenyl, 3-cyclohexenyl, cycloheptyl, norbornanyl, bicyclo(2.2.2)octanyl, and the like.
- heterocycloalkyl examples include, but are not limited to, 1-(1,2,5,6- tetrahydropyridyl), 1-piperidinyl, 2-piperidinyl, 3-piperidinyl, 4-morpholinyl, 3-morpholinyl, tetrahydrofuran-2-yl, tetrahydrofuran-3-yl, tetrahydrothien-2-yl, tetrahydrothien-3-yl, 1- piperazinyl, 2-piperazinyl, 1- or 2-azabicyclo(2.2.2)octanyl, and the like.
- aryl means, unless otherwise stated, a polyunsaturated, aromatic, hydrocarbon substituent which can be a single ring or multiple rings (in some embodiments from 1 to 3 rings) which are fused together or linked covalently.
- heteroaryl refers to aryl groups that contain from one to four heteroatoms selected from N, O, and S in the ring(s), wherein the nitrogen and sulfur atoms are optionally oxidized, and the nitrogen atom(s) are optionally quaternized.
- a heteroaryl group can be attached to the remainder of the molecule through a carbon or heteroatom.
- arylene and heteroarylene by themselves or as part of another substituent means a divalent radical derived from an aryl or heteroaryl.
- aryl and heteroaryl groups include phenyl, 1-naphthyl, 2-naphthyl, 4-biphenyl, 1-pyrrolyl, 2-pyrrolyl, 3-pyrrolyl, 3-pyrazolyl, 2-imidazolyl, 4-imidazolyl, pyrazinyl, 2-oxazolyl, 4-oxazolyl, 5-oxazolyl, 3-isoxazolyl, 4-isoxazolyl, 5-isoxazolyl, 2- thiazolyl, 4-thiazolyl, 5-thiazolyl, 2-furyl, 3-furyl, 2-thienyl, 3-thienyl, 2-pyridyl, 3-pyridyl, 4- pyridyl, 2-pyrimidyl, 4-pyrimidyl, 5-benzo
- heteroarylium refers to a heteroaryl group that is positively charged on one or more of the heteroatoms.
- substituent moieties for cycloalkyl, heterocycloalkyl, cycloalkenyl, and heterocycloalkenyl groups also include substituted and unsubstituted alkyl, substituted and unsubstituted alkenyl, and substituted and unsubstituted alkynyl.
- R', R", R"' and R" each in some embodiments are independently are hydrogen, substituted or unsubstituted heteroalkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted aryl (e.g., aryl substituted with 1-3 halogens), substituted or unsubstituted alkyl, alkoxy or thioalkoxy groups, or arylalkyl groups.
- each of the R groups is independently selected as are each R', R", R'" and R"" groups when more than one of these groups is present.
- R' and R" When R' and R" are attached to the same nitrogen atom, they can be combined with the nitrogen atom to form a 4-, 5-, 6-, or 7- membered ring.
- -NR'R is meant to include, but not be limited to, 1-pyrrolidinyl and 4-morpholinyl.
- alkyl is meant to include groups including carbon atoms bound to groups other than hydrogen groups, such as haloalkyl (e.g., -CF 3 and –CH 2 CF 3 ) and acyl (e.g., -C(O)CH 3 , -C(O)CF 3 , -C(O)CH 2 OCH 3 , and the like).
- haloalkyl e.g., -CF 3 and –CH 2 CF 3
- acyl e.g., -C(O)CH 3 , -C(O)CF 3 , -C(O)CH 2 OCH 3 , and the like.
- each of the R groups is independently selected as are each R', R", R'" and R"" groups when more than one of these groups is present.
- Two of the substituent moieties on adjacent atoms of an aryl or heteroaryl ring may optionally form a ring of the formula -Q'-C(O)-(CRR') q -Q''-, wherein Q' and Q'' are independently –NR-, -O-, -CRR'- or a single bond, and q is an integer of from 0 to 3.
- two of the substituent moieties on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -A-(CH 2 ) r -B-, wherein A and B are independently –CRR'-, -O-, -NR-, -S-, -S(O)-, -S(O) 2 -, -S(O) 2 NR'- or a single bond, and r is an integer of from 1 to 4.
- One of the single bonds of the new ring so formed may optionally be replaced with a double bond.
- two of the substituent moieties on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula – (CRR') s -X'-(CR''R'') d -, where s and d are independently integers of from 0 to 3, and X' is –O-, -NR'-, -S-, -S(O)-, -S(O) 2 -, or –S(O) 2 NR'-.
- the substituent moieties R, R', R" and R'" are, in some embodiments, independently selected from hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted aryl, and substituted or unsubstituted heteroaryl.
- halo by itself or as part of another substituent, means, unless otherwise stated, a fluorine, chlorine, bromine, or iodine atom. Additionally, terms such as “haloalkyl,” are meant to include monohaloalkyl and polyhaloalkyl.
- halo(C 1 - C 4 )alkyl is meant to include, but not be limited to, trifluoromethyl, 2,2,2-trifluoroethyl, 4- chlorobutyl, 3-bromopropyl, and the like.
- oxo as used herein means an oxygen atom that is double bonded to a carbon atom.
- heteroatom or “ring heteroatom” is meant to include oxygen (O), nitrogen (N), sulfur (S), phosphorus (P), and silicon (Si).
- TLR7 Agonists [0095] In one aspect, provided herein are TLR7 agonists for use in the compositions and methods provided herein.
- the TLR7 agonists are compounds of Formula I: or a pharmaceutically acceptable salt thereof, wherein: R 1 is H, halo, -NHR 4 , -OR 4 , -NH-OR 4 or -R 4 ; R 2 is H, halo, or alkoxy; R 3 is -CO 2 R 23 , -CONHR 23 , -alkylene-Y, -alkylene-arylene-Y, -heteroalkylene-Y, -heteroalkylene-arylene-Y, -(hydroxy)heteroalkylene-Y, -(amino)heteroalkylene-Y, or - alkylene-PEG-Y; R 4 is alkyl optionally substituted with alkoxy or heteroalkyl; R 23 is H, alkyl or aryl; X is CH or N; Y is -OH, -Gly, -NR 5 R 6 or -COZ; Z is -OH, alk
- the compound of the formula (I) is not a compound of the formula: [0001]
- R 5 and R 6 are selected from (i), (ii), and (iii): (i) R 5 and R 6 each H; (ii) R 5 is H and R 6 is alkyl; (iii) R 5 and R 6 , together with the N to which they are attached, form a heterocyclic ring; and R 7 and R 8 , together with the N to which they are attached, form a heterocyclic ring.
- the TLR7 agonists are selected with the proviso that R 4 is not substituted with hydroxyl.
- the TLR7 agonists are selected with the proviso that the alkylene and heteroalkylene portions of R 3 are not substituted with oxo. [0098] In some embodiments, the TLR7 agonists are selected with the proviso that the compound is not 5-(2-methoxy-4-(piperazin-1-ylmethyl)benzyl)-N4-pentyl-5H-pyrrolo[3,2- d]pyrimidine-2,4-diamine, which corresponds to P3 in Table 1; or (4-((2-amino-4- (pentylamino)-5H-pyrrolo[3,2-d]pyrimidin-5-yl)methyl)-3-methoxyphenyl)methanol, which corresponds to P1 in Table 1.
- R 1 is H. In some embodiments, R 1 is halo, -NHR 4 , -OR 4 , -NH- OR 4 or -R 4 , and is a straight chain of 6 atoms in length. In some embodiments, R 1 is halo. In some embodiments, R 1 is -NHR 4 . In some embodiments, R 1 is -OR 4 . In some embodiments, R 1 is -NH-OR 4 . In some embodiments, R 1 is -R 4 .
- R 1 is -NH-n-pentyl, -NH-O-n-butyl, -O-n-pentyl, -n-hexyl or -NH-CH 2 CH 2 -OEt. In some embodiments, R 1 is -NH- n-pentyl. In some embodiments, R 1 is -NH-O-n-butyl. In some embodiments, R 1 is -O-n- pentyl. In some embodiments, R 1 is -n-hexyl. In some embodiments, R 1 is -NH-CH 2 CH 2 -OEt. [0100] In some embodiments, R 2 is halo. In some embodiments, R 2 is alkoxy.
- R 2 is methoxy. In some embodiments, R 2 is H. [0101] In some embodiments, R 3 is -CO 2 R 23 , -CONHR 23 , , -alkylene-Y, -heteroalkylene-Y, heteroalkylene-arylene-Y, -(hydroxy)heteroalkylene-Y, -(amino)heteroalkylene-Y, or - alkylene-PEG-Y. In some embodiments, R 3 is -CONHR 23 . In some embodiments, R 3 is - alkylene-Y. In some embodiments, R 3 is -heteroalkylene-Y. In some embodiments, R 3 is - heteroalkylene-arylene-Y.
- R 3 is -(hydroxy)heteroalkylene-Y. In some embodiments, R 3 is -(amino)heteroalkylene-Y. In some embodiments, R 3 is alkylene-PEG-Y. In some embodiments, R 3 is -CONH 2 , -COOH, -CH 2 -Y, -CH 2 -O-heteroalkylene-Y, or -CH 2 -O-alkylene-Y. In some embodiments, R 3 is -CH 2 -Y, -CH 2 -O-heteroalkylene-Y, or -CH 2 - O-alkylene-Y.
- R 3 is -C(Me) 2 OH, -CO 2 H -CH 2 OCH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 CH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 OH, -CH 2 OCH 2 CH 2 OCH 2 CH 2 OH, -CH 2 OCH 2 CH 2 OCH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 OH, -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 -1-piperazinyl, -CH 2 OCH 2 NHC(O)CH 2 NH 2 , -CH 2 OCH 2 -(4-NH 2 -1-phenyl), -CH 2 OCH 2 COOH, -CH 2 OCH 2 CH 2 OCH 2 NH 2 , -CH 2 OCH 2 CH 2 CH 2 CH 2
- R 3 is -C(Me) 2 OH, -CO 2 H, -CH 2 OCH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 CH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 OH, - CH 2 OCH 2 CH 2 OCH 2 CH 2 OH, -CH 2 OCH 2 CH 2 OCH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 OH, -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 -1-piperazinyl, -CH 2 OCH 2 NHC(O)CH 2 NH 2 , -CH 2 OCH 2 -(4-NH 2 -1-phenyl), -CH 2 OCH 2 COOH, -CH 2 OCH 2 CH 2 OCH
- R 4 is n-butyl, n-pentyl, n-hexyl or ethoxyethyl. In some embodiments, R 4 is n-butyl. In some embodiments, R 4 is n-pentyl. In some embodiments, R 4 is n-hexyl. In some embodiments, R 4 is ethoxyethyl. [0103] In some embodiments, R 5 and R 6 are each independently H or alkyl, or, together with the N to which they are attached, form a piperazinyl ring. In some embodiments, R 5 and R 6 are each H. In some embodiments, R 5 is H and R 6 is alkyl.
- R 5 and R 6 together with the N to which they are attached, form 1-piperazinyl.
- Y is OH.
- Y is a divalent glycine group of the formula -NHCH 2 C(O)-.
- Y is -NR 5 R 6 .
- Y is -COZ.
- Y is -OH, -NH 2 , 1-piperazinyl, -COOH, -COOEt, -CONPr 2 or -CO-1-piperazinyl.
- Y is -OH, -NH 2 , 1-piperazinyl, -COOH or -CO-1- piperazinyl.
- Z is -OH.
- Z is alkoxy.
- Z is -NR 7 R 8 .
- Z is -OH, ethoxy, -N-n-Pr 2 or 1-piperazinyl.
- Z is -OH or 1-piperazinyl.
- R 7 and R 8 are each independently H or n-propyl, or, together with the N to which they are attached, form 1-piperazinyl.
- R 7 and R 8 together with the N to which they are attached, form 1-piperazinyl.
- the TLR7 agonist is selected from compounds P1-P39 and P41-P48 in Table 1 and pharmaceutically acceptable salts of any of these: Table 1 [0108]
- a TLR7 agonist can be a known TLR7 agonist, e.g., 852A, imiquimod, resiquimod, gardiquimod loxoribine, bropirimine, 3M-011, 3M-052, DSR-6434, DSR-29133, SC1, SZU-101, SM-360320, and SM-276001.
- TLR7 agonists are described in, for example, Chi et al., Front. Pharmacol.8:34, 31 May 2017, which is hereby incorporated by reference in its entirety. III. Synthesis of the TLR7 Agonists [0109]
- the TLR7 agonists of the disclosure can be synthesized in any suitable fashion. Non- limiting examples of synthetic schemes for the synthesis of the TLR7 agonists of the disclosure are presented herein in Schemes 1-7. [0110] Scheme 1. Synthesis of Intermediate Aa starting from Compound 1 [0111] Scheme 2. Synthesis of Intermediates Starting from Compound 5
- Scheme 3 Synthesis of payloads P1, P2, P20, P23, P27, P29, P32, P33, P37 and P39 [0114] Scheme 5. Synthesis of payloads P22, P25, P31, P35, P21, P24, P30 and P34.
- the ADC comprises ABD linked to a linker-TLR7 agonist according to Formula II: or a pharmaceutically acceptable salt thereof, wherein: R 1 , R 2 and X are as defined elsewhere for Formula I; R 9 is a divalent group formed by removal of a hydrogen from R 3 , R 3 being a group attached to the phenyl group at the position of R 9 ; and L is any group or moiety that links, connects, or bonds an antigen-binding domain (ABD), as defined elsewhere herein, with a compound of Formula I.
- R 1 , R 2 and X are as defined elsewhere for Formula I
- R 9 is a divalent group formed by removal of a hydrogen from R 3 , R 3 being a group attached to the phenyl group at the position of R 9
- L is any group or moiety that links, connects, or bonds an antigen-binding domain (ABD), as defined elsewhere herein, with a compound of Formula I.
- the compound of Formula (II) is not a compound of the formula: .
- R 9 is -alkylene-Y 1 -, -heteroalkylene-Y 1 -, -heteroalkylene-arylene-Y 1 -, -(hydroxy)heteroalkylene-Y 1 , -(amino)heteroalkylene-Y 1 , or - alkylene-PEG-Y 1 .
- R 9 is -alkylene-Y 1 -.
- R 9 is - heteroalkylene-Y 1 -.
- R 9 is -heteroalkylene-arylene-Y 1 -.
- R 9 is -(hydroxy)heteroalkylene-Y 1 . In some embodiments, R 9 is - (amino)heteroalkylene-Y 1 . In some embodiments, R 9 is -alkylene-PEG-Y 1 . In another embodiment, R 9 is -CH 2 -Y 1 -, -CH 2 -O-heteroalkylene-Y 1 -, or -CH 2 -O-alkylene-Y 1 -.
- R 9 is -C(Me) 2 O-, -CO-, -CH 2 OCH 2 CH 2 NH-, -CH 2 OCH 2 CH 2 CH 2 CH 2 NH-, - CH 2 OCH 2 CH 2 OCH 2 CH 2 O-, -CH 2 OCH 2 CH 2 OCH 2 CH 2 NH-, -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 O-, -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 NH-, -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 -1-piperazin-4-yl-, -CH 2 OCH 2 NHC(O)CH 2 NH-, -CH 2 OCH 2 -((4-NH-)-1-phenyl), -CH 2 OCH 2 COO-, -CH 2 OCH 2 CH 2 OCH 2 CO-, -CH 2 OCH 2 CH 2 OCH 2 OCH 2 CH 2 O
- Y 1 is -O-. In some embodiments, Y 1 is a divalent glycine group of the formula -NHCH 2 C(O)-. In some embodiments, Y 1 is -NR 5 -. In some embodiments, Y 1 is - COZ 1 , wherein Z 1 is -O-, -NR 7 -, -O-alkylene-, or 1-piperazin-4-yl. In some embodiments, Y 1 is -O-, -NH-, 1-piperazin-4-yl, -COO- or -CO-1-piperazin-4-yl. [0120] In some embodiments, Z 1 is -O-.
- Z 1 is -NR 7 -. In some embodiments, R 7 is H. In some embodiments, R 7 is alkyl. [0121] In some embodiments, Z 1 is 1-piperazin-4-yl. In some embodiments, Y 1 is 1- piperazin-4-yl. In some embodiments, Y 1 is -CO-1-piperazin-4-yl. [0122] In some embodiments, linkers L for use herein may be found, for example, in Antibody-Drug Conjugates and Immunotoxins, Phillips, G.
- the L group for the ADCs provided herein is sufficiently stable to exploit the circulating half-life of the antigen binding domain and, at the same time, capable of releasing its payload after antigen-mediated internalization of the ADC.
- Linker L can be cleavable or non-cleavable.
- Cleavable linkers for use as L herein include linkers that are cleaved by intracellular metabolism following internalization, e.g., cleavage via hydrolysis, reduction, or enzymatic reaction.
- Non-cleavable linkers for use as L herein include linkers that release an attached payload via lysosomal degradation of the antigen binding domain following internalization.
- Suitable L linkers include, but are not limited to, acid-labile linkers, hydrolysis-labile linkers, enzymatically cleavable linkers, reduction labile linkers, self- immolative linkers, and non-cleavable linkers.
- Suitable L linkers also include, but are not limited to, those that are or comprise peptides, carbohydrates, glucuronides, polyethylene glycol (PEG) units, hydrazones, mal-caproyl units, dipeptide units, valine-citruline units, and para-aminobenzyl (PAB) units.
- PEG polyethylene glycol
- PAB para-aminobenzyl
- PEG groups include, but are not limited to, the following [insert ChemDraw structures].
- PEG groups can have any suitable molecular weight, such as from about 60 g/mol to about 6,000 g/mol, about 60 g/mol to about 600 g/mol, about 100 g/mol to about 500 g/mol, about 300 g/mol to about 1,200 g/mol, about 200 g/mol to about 800 g/mol, about 200 g/mol to about 1,000 g/mol, about 500 g/mol to about 1,000 g/mol, about 500 g/mol to about 2,500 g/mol, or about 800 g/mol to about 2,200 g/mol.
- L linker is a cleavable linker. In other embodiments, the L linker is a non-cleavable linker.
- L linkers that can be used in the ADCs provided herein include linkers that comprise or consist of e.g., MC (6-maleimidocaproyl), MP (maleimidopropanoyl), val-cit (valine-citrulline), val-ala (valine- alanine), dipeptide site in protease-cleavable linkers, ala-phe (alanine-phenylalanine), dipeptide site in protease-cleavable linkers, PAB (p-aminobenzyloxycarbonyl), and variants and combinations thereof. Additional examples of L linkers that can be used in the ADCs provided herein are disclosed, e.g., in U.S.
- the L linkers are stable in physiological conditions.
- the L linkers are cleavable, for instance, able to release at least the payload portion in the presence of an enzyme or at a particular pH range or value.
- an L linker comprises an enzyme-cleavable moiety.
- enzyme-cleavable L linkers include, but are not limited to, peptide bonds, ester linkages, and hydrazones.
- the L linker comprises a cathepsin-cleavable linker.
- the L linker comprises a non-cleavable moiety.
- the L linker comprises one or more amino acids. Suitable amino acids include natural, non-natural, standard, non-standard, proteinogenic, non- proteinogenic, and L- or D- ⁇ -amino acids.
- the L linker comprises alanine, valine, glycine, leucine, isoleucine, methionine, tryptophan, phenylalanine, proline, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, lysine, arginine, histidine, or citrulline, a derivative thereof, or combination thereof.
- one or more side chains of the amino acids is linked to a side chain group, described below.
- the linker comprises valine and citrulline.
- the L linker comprises lysine, valine, and citrulline.
- the L linker comprises lysine, valine, and alanine. In some embodiments, the L linker comprises valine and alanine. [0128] In some embodiments, the L linker comprises a self-immolative group.
- the self- immolative group can be any such group known to those of skill in the art.
- the self-immolative group is p-aminobenzyl (PAB), or a derivative thereof.
- PAB p-aminobenzyl
- PABC p-aminobenzyloxycarbonyl
- the L group can be modified with one or more enhancement groups.
- the enhancement group can be linked to the side chain of any amino acid in L.
- amino acids for linking enhancement groups include lysine, asparagine, aspartate, glutamine, glutamate, and citrulline.
- the link to the enhancement group can be a direct bond to the amino acid side chain, or the link can be indirect via a spacer and/or reactive group.
- spacers and reactive groups include any described herein.
- the enhancement group can be any group that imparts a beneficial effect to the payload, linker payload, or ADC including, but not limited to, biological, biochemical, synthetic, solubilizing, imaging, detecting, and reactivity effects, and the like.
- the enhancement group is a hydrophilic group.
- the enhancement group is a cyclodextrin.
- the enhancement group is an alkyl sulfonic acid, heteroalkyl sulfonic acid, alkenyl sulfonic acid, heteroalkenyl sulfonic acid, heteroalkenyl taurine, heteroalkenyl phosphoric acid or phosphate, heteroalkenyl amine (e.g., quaternary amine), or heteroalkenyl sugar.
- sugars include, without limitation, monosaccharides, disaccharides, and polysaccharides. Exemplary monosaccharides include glucose, ribose, deoxyribose, xylose, arabinose, mannose, galactose, fructose, and the like.
- sugars include sugar acids such as glucuronic acid, further including conjugated forms such as glucuronides (i.e., via glucuronidation).
- exemplary disaccharides include maltose, sucrose, lactose, lactulose, trehalose, and the like.
- Exemplary polysaccharides include amylose, amylopectin, glycogen, inulin, cellulose, and the like.
- the cyclodextrin can be any cyclodextrin known to those of skill. In some embodiments, the cyclodextrin is alpha cyclodextrin, beta cyclodextrin, or gamma cyclodextrin, or mixtures thereof.
- the cyclodextrin is alpha cyclodextrin. In some embodiments, the cyclodextrin is beta cyclodextrin. In some embodiments, the cyclodextrin is gamma cyclodextrin. In some embodiments, the enhancement group is capable of improving solubility of the remainder of the ADC. In some embodiments, the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is substituted or non-substituted.
- the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is —(CH 2 ) 1-5 SO 3 H, –(CH 2 ) n –NH-(CH 2 ) 1-5 SO 3 H, –(CH 2 ) n –C(O)NH-(CH 2 ) 1-5 SO 3 H, –(CH 2 CH 2 O) m –C(O)NH-(CH 2 ) 1-5 SO 3 H, –(CH 2 ) n –N((CH 2 ) 1-5 C(O)NH(CH 2 ) 1-5 SO 3 H) 2 , –(CH 2 ) n – C(O)N((CH 2 ) 1-5 C(O)NH(CH 2 ) 1-5 SO 3 H) 2 , or —(CH 2 CH 2 O) m –C(O)N((CH 2 ) 1-5 C(O)NH(CH 2 ) 1- 5 SO 3 H) 2 ,
- the alkyl or alkenyl sulfonic acid is —(CH 2 ) 1-5 SO 3 H.
- the heteroalkyl or heteroalkenyl sulfonic acid is —(CH 2 ) n –NH-(CH 2 ) 1-5 SO 3 H, wherein n is 1, 2, 3, 4, or 5.
- the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is –(CH 2 ) n –C(O)NH-(CH 2 ) 1-5 SO 3 H, wherein n is 1, 2, 3, 4, or 5.
- the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is —(CH 2 CH 2 O) m –C(O)NH-(CH 2 ) 1-5 SO 3 H, wherein m is 1, 2, 3, 4, or 5.
- the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is — (CH 2 ) n –N((CH 2 ) 1-5 C(O)NH(CH 2 ) 1-5 SO 3 H) 2 , wherein n is 1, 2, 3, 4, or 5.
- the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is —(CH 2 ) n –C(O)N((CH 2 ) 1- 5 C(O)NH(CH 2 ) 1-5 SO 3 H) 2 , wherein n is 1, 2, 3, 4, or 5.
- the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is –(CH 2 CH 2 O) m –C(O)N((CH 2 ) 1- 5 C(O)NH(CH 2 ) 1-5 SO 3 H) 2 , wherein m is 1, 2, 3, 4, or 5.
- L comprises a maleimido (for conjugation with a thiol, e.g., cysteine, of an antigen binding domain), an N-hydroxysuccinimido ester (for conjugation with an amine, e.g., lysine, of an antigen binding domain) or cyclooctynyl group (for conjugation with an antigen binding domain using click chemistry).
- a maleimido for conjugation with a thiol, e.g., cysteine, of an antigen binding domain
- N-hydroxysuccinimido ester for conjugation with an amine, e.g., lysine, of an antigen binding domain
- cyclooctynyl group for conjugation with an antigen binding domain using click chemistry.
- the maleimido group in L reacts with a cysteine residue on an antigen binding domain to form a carbon-sulfur bond.
- L contains an N-hydroxysuccinimido ester group.
- the N-hydroxysuccinimido ester group reacts with a lysine residue on an antigen binding domain to form an amide bond.
- L contains an alkyne which can react via click chemistry with an azide, e.g., to form a click chemistry product.
- the alkyne group reacts with an azide on a modified antigen binding domain.
- L contains a functional group or moiety that is capable of undergoing a click chemistry reaction (see, e.g., click chemistry, Huisgen Proc. Chem. Soc. 1961,357-396; Wang et al. J. Am. Chem. Soc. 2003, 125(11), 3192-3193; and Agard et al. J. Am. Chem. Soc.2004, 126(46), 15046-15047).
- the reactive group is an alkyne that is capable of undergoing a 1,3- cycloaddition reaction with an azide.
- Alkynes that may be used in such embodiments include strained alkynes, e.g., those suitable for strain-promoted alkyne-azide cycloadditions (SPAAC), cycloalkynes, e.g., cyclooctynes, benzannulated alkynes, and alkynes capable of undergoing 1,3-cycloaddition reactions with alkynes in the absence of copper catalysts.
- strained alkynes e.g., those suitable for strain-promoted alkyne-azide cycloadditions (SPAAC)
- cycloalkynes e.g., cyclooctynes, benzannulated alkynes, and alkynes capable of undergoing 1,3-cycloaddition reactions with alkynes in the absence of copper catalysts.
- Alkynes that may be used in such embodiments also include, but are not limited to, dibenzoazacyclooctyne, dibenzocyclooctyne, biarylazacyclooctynone, difluorinated cyclooctyne, substituted, e.g., fluorinated alkynes, aza-cycloalkynes and bicyclo[6.1.0]nonyne.
- alkynes are useful for conjugating antibodies that have been functionalized with azido groups.
- Such functionalized antibodies include antibodies functionalized with azido-polyethylene glycol groups.
- such a functionalized antibody is derived by treating an antibody having at least one glutamine residue, e.g., heavy chain Gln295, with a compound bearing an amino group and an azide group, in the presence of the enzyme transglutaminase.
- L is selected from 2-maleimido-1-ethyl, 2-maleimidoacetyl, and 3-maleimidopropanoyl. In certain embodiments, L is selected from: , .
- L is a group selected from 2-maleimido-1-ethyl, 2- maleimidoacetyl, 3-maleimidopropanoyl, , [0136]
- the linker-TLR7 agonist is selected from those in Table 2 and pharmaceutically acceptable salts of any of these:
- the payload i.e., TLR7 agonist
- the payload can be converted to a prodrug prior to attachment to a linking group and formation of the ADC. See, e.g., WO 2020/146541.
- the payloads of Formula I can be converted to linker-TLR7 agonist of Formula III:
- L is a linker as defined elsewhere herein;
- R 1 , R 2 and X are as defined elsewhere for Formula I;
- R 11 and R 12 are, independently, hydrogen, alkyl, alkoxy, alkenyl, alkynyl, aryl, arylalkyl, heteroaryl, alkylene, or heteroalkylene, wherein when R 11 is alkylene or heteroalkylene, the alkylene or heteroalkylene is further bonded to R 13 to form a 3-, 4-, 5-, 6-, 7-, or 8-membered heterocyclyl;
- R 14 is hydrogen, alkylene, heteroalkylene, or an amino acid side chain, wherein when R 14 is alkylene or heteroalkylene, the alkylene or heteroalkylene is further bonded to R 13 to form a 3-, 4-, 5-, 6-, 7- OR 8-membered heterocyclyl;
- R 13 is hydrogen, alkyl, alkylene, or heteroalkylene, wherein when R 13 is
- the linker-TLR7 agonist has Formula III, wherein R 11 and R 12 are, independently, hydrogen, alkyl, alkoxy, alkenyl, alkynyl, aryl, arylalkyl, heteroaryl, alkylene, or heteroalkylene, wherein when R 11 is alkylene or heteroalkylene, the alkylene or heteroalkylene is further bonded to R 13 to form a 4-, 5-, or 6-membered heterocyclyl; R 14 is hydrogen, alkylene, heteroalkylene, or an amino acid side chain, wherein when R 14 is alkylene or heteroalkylene, the alkylene or heteroalkylene is further bonded to R 13 to form a 4-, 5-, or 6-membered heterocyclyl; R 13 is hydrogen, alkyl, alkylene, or heteroalkylene, wherein when R 13 is alkylene or heteroalkylene, the alkylene or heteroalkylene is further bonded to R 11 or R 14 to form a
- R 16 is the moiety of R 3 Formula I that links the phenyl ring to the oxygen atom of the alcohol.
- the TLR7 agonist used in preparing the linker-TLR7 agonist according to Formula III is P1, P2, P6, P8, P17, P18, P19, P20, P23, P27, P29, P32, P33, P37, or P39.
- V. Synthesis of the TLR7 Agonist-Linkers [0140] Scheme 8. Synthesis of Linker-payloads LP1, LP2, LP3 and LP4. [0141] Scheme 9. Synthesis of Linker-payload LP5. [0142] Scheme 10. Synthesis of Linker-payload LP6A.
- Scheme 11 Synthesis of Linker-payloads LP7A and LP10A.
- Scheme 12. Synthesis of Linker-payload LP8A. LP8A
- Scheme 13. Synthesis of Linker-payloads LP9 and LP12.
- Scheme 14. Synthesis of Linker-payload LP6A, LP6B, LP7A, LP7B, LP7C, LP7D LP10A, LP10B, LP11A, LP11B, LP11C, LP11D, LP12 and LP14
- the present disclosure provides an antibody-drug-conjugate (ADC) comprising an antigen-binding domain (ABD) (e.g., an ABD having binding specificity for a target antigen such as HBV sAg or a tumor specific antigen) and a TLR7 agonist.
- ADC antigen-binding domain
- the ADC further comprises a divalent linker that links the ABD to the TLR7 agonist.
- the ABD can bind to the TLR7 agonist with or without a linker, at any location along the ABD as long as the ABD is able to bind its target.
- the ADC is according to Formula IV: or a pharmaceutically acceptable salt thereof, wherein: L 1 is a divalent linker; R 1 is H, halo, -NHR 4 , -OR 4 , -NH-OR 4 or -R 4 ; R 2 is H, halo, or alkoxy; R 4 is alkyl optionally substituted with alkoxy or heteroalkyl; R 9 is a divalent group formed by removal of a hydrogen from R 3 , R 3 being a group attached to the phenyl group at the position of R 9 ; R 3 is -CO 2 H, -CONHR 23 , -alkylene-Y, -alkylene-arylene-Y, -heteroalkylene-Y, - heteroalkylene-arylene-Y, -(hydroxy)heteroalkylene-Y, -(amino)heteroalkylene-Y, or - alkylene-PEG-Y; R 23 is H,
- the ABC comprises one TLR7 agonist molecule conjugated to an ABD having binding specificity for a target antigen.
- the ADC comprises more than one TLR7 agonist molecules per an ABD.
- two, three, four, five or more TLR7 agonist molecules are conjugated to one ABD.
- k can be 1, 2, 3, 4, or 5.
- k is 2.
- k is 1.
- k is 4.
- k is 5 or more.
- the ADC is selected with the proviso that the ADC does not comprise 5-(2-methoxy-4-(piperazin-1-ylmethyl)benzyl)-N4-pentyl-5H-pyrrolo[3,2- d]pyrimidine-2,4-diamine or (4-((2-amino-4-(pentylamino)-5H-pyrrolo[3,2-d]pyrimidin-5- yl)methyl)-3-methoxyphenyl)methanol.
- R 1 is halo, -NHR 4 , -OR 4 , -NH-OR 4 or -R 4 , and has a straight chain of 6 atoms in length.
- R 1 is halo. In some embodiments, R 1 is - NHR 4 . In some embodiments, R 1 is -OR 4 . In some embodiments, R 1 is -NH-OR 4 . In some embodiments, R 1 is -R 4 . In some embodiments, R 1 is -NH-n-pentyl, -NH-O-n-butyl, -O-n- pentyl, -n-hexyl or -NH-CH 2 CH 2 -OEt. In some embodiments, R 1 is -NH-n-pentyl. In some embodiments, R 1 is -NH-O-n-butyl.
- R 1 is -O-n-pentyl. In some embodiments, R 1 is -n-hexyl. In some embodiments, R 1 is -NH-CH 2 CH 2 -OEt. [0156] In some embodiments, R 2 is halo. In some embodiments, R 2 is alkoxy. In some embodiments, R 2 is methoxy. In some embodiments, R 2 is H.
- R 3 is -CO 2 H, -CONHR 23 , -alkylene-Y, -heteroalkylene-Y, heteroalkylene-arylene-Y, -(hydroxy)heteroalkylene-Y, -(amino)heteroalkylene-Y, or - alkylene-PEG-Y.
- R 3 is CONHR 23 .
- R 3 is - alkylene-Y.
- R 3 is -heteroalkylene-Y.
- R 3 is - heteroalkylene-arylene-Y.
- R 3 is -(hydroxy)heteroalkylene-Y.
- R 3 is -(amino)heteroalkylene-Y. In some embodiments, R 3 is alkylene-PEG-Y. In some embodiments, R 3 is -CONH 2 , -CH 2 -Y, -CH 2 -O-heteroalkylene-Y, or -CH 2 -O-alkylene- Y. In some embodiments, R 3 is -CH 2 -Y, -CH 2 -O-heteroalkylene-Y, or -CH 2 -O-alkylene-Y.
- R 3 is -C(Me) 2 OH, -CO 2 H -CH 2 OCH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 CH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 OH, -CH 2 OCH 2 CH 2 OCH 2 CH 2 OH, -CH 2 OCH 2 CH 2 OCH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 OH, -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 -1-piperazinyl, -CH 2 OCH 2 NHC(O)CH 2 NH 2 , -CH 2 OCH 2 -(4-NH 2 -1-phenyl), -CH 2 OCH 2 COOH, -CH 2 OCH 2 CH 2 OCH 2 NH 2 , -CH 2 OCH 2 CH 2 CH 2 CH 2
- R 3 is -C(Me) 2 OH, -CO 2 H, -CH 2 OCH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 CH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 OH, - CH 2 OCH 2 CH 2 OCH 2 CH 2 OH, -CH 2 OCH 2 CH 2 OCH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 OH, -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 -1-piperazinyl, -CH 2 OCH 2 NHC(O)CH 2 NH 2 , -CH 2 OCH 2 -(4-NH 2 -1-phenyl), -CH 2 OCH 2 COOH, -CH 2 OCH 2 CH 2 OCH
- R 4 is n-butyl, n-pentyl, n-hexyl or ethoxyethyl. In some embodiments, R 4 is n-butyl. In some embodiments, R 4 is n-pentyl. In some embodiments, R 4 is n-hexyl. In some embodiments, R 4 is ethoxyethyl. [0159] In some embodiments, R 5 and R 6 are each independently H or alkyl, or, together with the N to which they are attached, form a piperazinyl ring. In some embodiments, R 5 and R 6 are each H. In some embodiments, R 5 is H and R 6 is alkyl.
- R 5 and R 6 together with the N to which they are attached, form 1-piperazinyl.
- Y is OH.
- Y is a divalent glycine group of the formula -NHCH 2 C(O)-.
- Y is -NR 5 R 6 .
- Y is -COZ.
- Y is -OH, -NH 2 , 1-piperazinyl, -COOH, -COOEt, -CONPr 2 or -CO-1-piperazinyl.
- Y is -OH, -NH 2 , 1-piperazinyl, -COOH or -CO-1- piperazinyl.
- Z is -OH.
- Z is alkoxy.
- Z is -NR 7 R 8 .
- Z is -OH, ethoxy, -N-n-Pr 2 or 1-piperazinyl.
- Z is -OH or 1-piperazinyl.
- R 7 and R 8 are each independently H or n-propyl, or, together with the N to which they are attached, form 1-piperazinyl.
- R 7 and R 8 together with the N to which they are attached, form 1-piperazinyl.
- ABD-L 1 is linked to a compound selected from P1-P43 by removal of a hydrogen from the group at the position corresponding to R 3 of the compound.
- R 9 is -alkylene-Y 1 -, -heteroalkylene-Y 1 -, -heteroalkylene-arylene-Y 1 -, -(hydroxy)heteroalkylene-Y 1 , -(amino)heteroalkylene-Y 1 , or - alkylene-PEG-Y 1 .
- R 9 is -alkylene-Y 1 -. In some embodiments, R 9 is - heteroalkylene-Y 1 -. In some embodiments, R 9 is -heteroalkylene-arylene-Y 1 -. In some embodiments, R 9 is -(hydroxy)heteroalkylene-Y 1 . In some embodiments, R 9 is - (amino)heteroalkylene-Y 1 . In some embodiments, R 9 is -alkylene-PEG-Y 1 . In some embodiments, R 9 is -CH 2 -Y 1 -, -CH 2 -O-heteroalkylene-Y 1 -, or -CH 2 -O-alkylene-Y 1 -.
- R 9 is -C(Me) 2 O-, C(O)-, -CH 2 OCH 2 CH 2 NH-, -CH 2 OCH 2 CH 2 CH 2 CH 2 NH-, - CH 2 OCH 2 CH 2 OCH 2 CH 2 O-, -CH 2 OCH 2 CH 2 OCH 2 CH 2 NH-, -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 O-, -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 NH-, -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 -1-piperazin-4-yl-, -CH 2 OCH 2 NHC(O)CH 2 NH-, -CH 2 OCH 2 -((4-NH-)-1-phenyl), -CH 2 OCH 2 COO-, -CH 2 OCH 2 CH 2 OCH 2 CO-, -CH 2 OCH 2 CH 2 CH 2 NH-, -CH 2 OCH 2
- Y 1 is -O-. In some embodiments, Y 1 is a divalent glycine group of the formula -NHCH 2 C(O)-. In some embodiments, Y 1 is -NR 5 -. In some embodiments, Y 1 is -COZ 1 , wherein Z 1 is -O-, -NR 7 -, -O-alkylene-, or 1-piperazin-4-yl. In some embodiments, Y 1 is -O-, -NH-, 1-piperazin-4-yl, -COO- or -CO-1-piperazin-4-yl. [0166] In some embodiments, Z 1 is -O-.
- Z 1 is -NR 7 -. In some embodiments, R 7 is H. In some embodiments, R 7 is alkyl. [0167] In some embodiments, Z 1 is 1-piperazin-4-yl. In some embodiments, Y 1 is 1- piperazin-4-yl. In some embodiments, Y 1 is -CO-1-piperazin-4-yl. A. L 1 Divalent Groups [0168] In some embodiments, the ADC of the present disclosure comprises a TLR7 linked to an ABD indirectly via a linker. In some embodiments, the linker is a divalent linker (L 1 ) that links the ABD to the TLR7 agonist according to Formula IV.
- the divalent linker (L 1 ) is created by the reaction between the linker (L) and the ADC for the conjugation.
- Linkers (L 1 ) for use herein may be found, for example, in Antibody-Drug Conjugates and Immunotoxins, Phillips, G. L., Ed.; Springer Verlag: New York, 2013; Antibody-Drug Conjugates, Ducry, L., Ed.; Humana Press, 2013; Antibody-Drug Conjugates, Wang, J., Shen, W.-C., and Zaro, J. L., Eds.; Springer International Publishing, 2015.
- the L 1 group for the ADCs provided herein is sufficiently stable to exploit the circulating half- life of the antigen binding domain and, at the same time, capable of releasing its payload after antigen-mediated internalization of the ADC.
- Linker L 1 can be cleavable or non-cleavable.
- Cleavable linkers for use as L 1 herein include linkers that are cleaved by intracellular metabolism following internalization, e.g., cleavage via hydrolysis, reduction, or enzymatic reaction.
- Non-cleavable linkers for use as L 1 herein include linkers that release an attached payload via lysosomal degradation of the antigen binding domain following internalization.
- Suitable L 1 linkers include, but are not limited to, acid-labile linkers, hydrolysis-labile linkers, enzymatically cleavable linkers, reduction labile linkers, self-immolative linkers, and non- cleavable linkers. Suitable L 1 linkers also include, but are not limited to, those that are or comprise peptides, carbohydrates, glucuronides, polyethylene glycol (PEG) units, hydrazones, mal-caproyl units, dipeptide units, valine-citruline units, and para-aminobenzyl (PAB) units. [0170] Any linker molecule or linker technology known in the art can be used as L 1 to create or construct an ADC provided herein.
- L 1 linker is a cleavable linker. In other embodiments, the L 1 linker is a non-cleavable linker.
- L 1 linkers that can be used in the ADCs provided herein include linkers that comprise or consist of e.g., MC (6-maleimidocaproyl), MP (maleimidopropanoyl), val-cit (valine-citrulline), val-ala (valine-alanine), dipeptide site in protease-cleavable linkers, ala-phe (alanine-phenylalanine), dipeptide site in protease-cleavable linkers, PAB (p-aminobenzyloxycarbonyl), and variants and combinations thereof.
- MC maleimidocaproyl
- MP maleimidopropanoyl
- val-cit valine-citrulline
- val-ala valine-alanine
- L 1 linkers that can be used in the ADCs provided herein are disclosed, e.g., in U.S. Pat. No. 7,754,681 and in Ducry, Bioconjugate Chem., 2010, 21:5-13, and the references cited therein.
- the L 1 linkers are stable in physiological conditions.
- the L 1 linkers are cleavable, for instance, able to release at least the payload portion in the presence of an enzyme or at a particular pH range or value.
- an L 1 linker comprises an enzyme-cleavable moiety.
- enzyme-cleavable L 1 linkers include, but are not limited to, peptide bonds, ester linkages, and hydrazones.
- the L 1 linker comprises a cathepsin-cleavable linker.
- the L 1 linker comprises a non-cleavable moiety.
- the L 1 linker comprises one or more amino acids. Suitable amino acids include natural, non-natural, standard, non-standard, proteinogenic, non- proteinogenic, and L- or D- ⁇ -amino acids.
- the L 1 linker comprises alanine, valine, glycine, leucine, isoleucine, methionine, tryptophan, phenylalanine, proline, serine, threonine, cysteine, tyrosine, asparagine, glutamine, aspartic acid, glutamic acid, lysine, arginine, histidine, or citrulline, a derivative thereof, or combination thereof.
- one or more side chains of the amino acids is linked to a side chain group, described below.
- the linker comprises valine and citrulline.
- the L 1 linker comprises lysine, valine, and citrulline.
- the L 1 linker comprises lysine, valine, and alanine. In some embodiments, the L 1 linker comprises valine and alanine. [0174] In some embodiments, the L 1 linker comprises a self-immolative group.
- the self- immolative group can be any such group known to those of skill in the art.
- the self-immolative group is p-aminobenzyl (PAB), or a derivative thereof.
- PAB p-aminobenzyl
- PABC p-aminobenzyloxycarbonyl
- the L 1 group can be modified with one or more enhancement groups.
- the enhancement group can be linked to the side chain of any amino acid in L 1 .
- amino acids for linking enhancement groups include lysine, asparagine, aspartate, glutamine, glutamate, and citrulline.
- the link to the enhancement group can be a direct bond to the amino acid side chain, or the link can be indirect via a spacer and/or reactive group.
- spacers and reactive groups include any described herein.
- the enhancement group can be any group that imparts a beneficial effect to the payload, linker payload, or ADC including, but not limited to, biological, biochemical, synthetic, solubilizing, imaging, detecting, and reactivity effects, and the like.
- the enhancement group is a hydrophilic group.
- the enhancement group is a cyclodextrin.
- the enhancement group is an alkyl, heteroalkyl, alkenyl, heteroalkenyl sulfonic acid, heteroalkenyl taurine, heteroalkenyl phosphoric acid or phosphate, heteroalkenyl amine (e.g., quaternary amine), or heteroalkenyl sugar.
- sugars include, without limitation, monosaccharides, disaccharides, and polysaccharides.
- Exemplary monosaccharides include glucose, ribose, deoxyribose, xylose, arabinose, mannose, galactose, fructose, and the like.
- sugars include sugar acids such as glucuronic acid, further including conjugated forms such as glucuronides (i.e., via glucuronidation).
- Exemplary disaccharides include maltose, sucrose, lactose, lactulose, trehalose, and the like.
- Exemplary polysaccharides include amylose, amylopectin, glycogen, inulin, cellulose, and the like.
- the cyclodextrin can be any cyclodextrin known to those of skill.
- the cyclodextrin is alpha cyclodextrin, beta cyclodextrin, or gamma cyclodextrin, or mixtures thereof.
- the cyclodextrin is alpha cyclodextrin.
- the cyclodextrin is beta cyclodextrin.
- the cyclodextrin is gamma cyclodextrin.
- the enhancement group is capable of improving solubility of the remainder of the ADC.
- the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is substituted or non-substituted.
- the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is —(CH 2 ) 1-5 SO 3 H, –(CH 2 ) n –NH-(CH 2 ) 1-5 SO 3 H, –(CH 2 ) n –C(O)NH-(CH 2 ) 1-5 SO 3 H, – (CH 2 CH 2 O) m –C(O)NH-(CH 2 ) 1-5 SO 3 H, –(CH 2 ) n –N((CH 2 ) 1-5 C(O)NH(CH 2 ) 1-5 SO 3 H) 2 , –(CH 2 ) n – C(O)N((CH 2 ) 1-5 C(O)NH(CH 2 ) 1-5 SO 3 H) 2 , or
- the alkyl or alkenyl sulfonic acid is —(CH 2 ) 1-5 SO 3 H.
- the heteroalkyl or heteroalkenyl sulfonic acid is —(CH 2 ) n –NH-(CH 2 ) 1-5 SO 3 H, wherein n is 1, 2, 3, 4, or 5.
- the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is –(CH 2 ) n –C(O)NH-(CH 2 ) 1-5 SO 3 H, wherein n is 1, 2, 3, 4, or 5.
- the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is —(CH 2 CH 2 O) m –C(O)NH-(CH 2 ) 1-5 SO 3 H, wherein m is 1, 2, 3, 4, or 5.
- the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is —(CH 2 ) n –N((CH 2 ) 1-5 C(O)NH(CH 2 ) 1-5 SO 3 H) 2 , wherein n is 1, 2, 3, 4, or 5.
- the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is —(CH 2 ) n – C(O)N((CH 2 ) 1-5 C(O)NH(CH 2 ) 1-5 SO 3 H) 2 , wherein n is 1, 2, 3, 4, or 5.
- the alkyl, heteroalkyl, alkenyl, or heteroalkenyl sulfonic acid is –(CH 2 CH 2 O) m –C(O)N((CH 2 ) 1- 5 C(O)NH(CH 2 ) 1-5 SO 3 H) 2 , wherein m is 1, 2, 3, 4, or 5.
- L 1 contains a 3-thiosuccinimido group (resulting from conjugation of a maleimido group with a thiol, e.g., cysteine, of an antigen binding domain), an amido group (resulting from conjugation of a N-hydroxysuccinimido group with an amine, e.g., lysine, of an antigen binding domain) or a triazolocyclooctyl group (resulting from conjugation of a cyclooctynyl group using click chemistry with an antigen binding domain modified to contain an azido group).
- a 3-thiosuccinimido group resulting from conjugation of a maleimido group with a thiol, e.g., cysteine, of an antigen binding domain
- an amido group resulting from conjugation of a N-hydroxysuccinimido group with an amine, e.g., lysine, of an antigen binding domain
- L 1 contains a 3-thiosuccinimido group. In such embodiments, the 3-thiosuccinimido group in L 1 results from reaction of a cysteine residue on an antigen binding domain with a maleimido group of L to form a carbon-sulfur bond.
- L 1 is derived from L containing a maleimido group. In such embodiments, the maleimido group in L reacts with a cysteine residue on an antigen binding domain to form a carbon-sulfur bond.
- L 1 is derived from L containing an N-hdroxysuccinimido ester group. In such embodiments, the N-hydroxysuccinimido ester group reacts with a lysine residue on an antigen binding domain to form an amide bond. [0180] In other embodiments, L 1 is derived from L containing an alkyne which can react via click chemistry with an azide, e.g., to form a click chemistry product. In some embodiments, the alkyne group reacts with an azide on a modified antigen binding domain.
- L contains a functional group or moiety that is capable of undergoing a click chemistry reaction (see, e.g., click chemistry, Huisgen Proc. Chem. Soc.1961,357-396; Wang et al. J. Am. Chem. Soc.2003, 125(11), 3192-3193; and Agard et al. J. Am. Chem. Soc.2004, 126(46), 15046-15047).
- the reactive group is an alkyne that is capable of undergoing a 1,3-cycloaddition reaction with an azide.
- Alkynes that may be used in such embodiments include strained alkynes, e.g., those suitable for strain-promoted alkyne-azide cycloadditions (SPAAC), cycloalkynes, e.g., cyclooctynes, benzannulated alkynes, and alkynes capable of undergoing 1,3-cycloaddition reactions with alkynes in the absence of copper catalysts.
- strained alkynes e.g., those suitable for strain-promoted alkyne-azide cycloadditions (SPAAC)
- cycloalkynes e.g., cyclooctynes, benzannulated alkynes, and alkynes capable of undergoing 1,3-cycloaddition reactions with alkynes in the absence of copper catalysts.
- Alkynes that may be used in such embodiments also include, but are not limited to, dibenzoazacyclooctyne, dibenzocyclooctyne, biarylazacyclooctynone, difluorinated cyclooctyne, substituted, e.g., fluorinated alkynes, aza-cycloalkynes and bicyclo[6.1.0]nonyne.
- alkynes are useful for conjugating antibodies that have been functionalized with azido groups.
- Such functionalized antibodies include antibodies functionalized with azido-polyethylene glycol groups.
- such a functionalized antibody is derived by treating an antibody having at least one glutamine residue, e.g., heavy chain Gln295, with a compound bearing an amino group and an azide group, in the presence of the enzyme transglutaminase.
- L 1 contains an amido group.
- the amido group in L 1 results from reaction of an N-hydroxysuccinimido ester group of L with a lysine residue on an antigen binding domain to form an amide bond.
- L 1 contains a cyclic group which results from reaction of an alkyne via click chemistry with an azide, e.g., to form a click chemistry product.
- the alkyne group reacts with an azide on a modified antigen binding domain.
- an antigen-binding domain contains an alkyne group that reacts with an azide on L. See, e.g., click chemistry, Huisgen Proc. Chem. Soc.1961,357-396; Wang et al. J. Am. Chem. Soc. 2003, 125(11), 3192-3193; and Agard et al. J. Am. Chem. Soc. 2004, 126(46), 15046-15047.
- the alkyne group is an alkyne that is capable of undergoing a 1,3-cycloaddition reaction with an azide.
- Alkynes that may be used in such embodiments include strained alkynes, e.g., those suitable for strain-promoted alkyne-azide cycloadditions (SPAAC), cycloalkynes, e.g., cyclooctynes, benzannulated alkynes, and alkynes capable of undergoing 1,3-cycloaddition reactions with alkynes in the absence of copper catalysts.
- strained alkynes e.g., those suitable for strain-promoted alkyne-azide cycloadditions (SPAAC)
- cycloalkynes e.g., cyclooctynes, benzannulated alkynes, and alkynes capable of undergoing 1,3-cycloaddition reactions with alkynes in the absence of copper catalysts.
- Alkynes that may be used in such embodiments also include, but are not limited to, dibenzoazacyclooctyne, dibenzocyclooctyne, biarylazacyclooctynone, difluorinated cyclooctyne, substituted, e.g., fluorinated alkynes, aza-cycloalkynes and bicycle[6.1.0]nonyne.
- alkynes are useful for conjugating antibodies that have been functionalized with azido groups.
- Such functionalized antibodies include antibodies functionalized with azido-polyethylene glycol groups.
- such a functionalized antibody is derived by treating an antibody having at least one glutamine residue, e.g., heavy chain Gln295, with a compound bearing an amino group and an azide group, in the presence of the enzyme transglutaminase.
- L 1 is a group derived from 2-maleimido-1-ethyl, 2- maleimidoacetyl, 3-maleimidopropanoyl, , [0184]
- L 1 is or contains a divalent group selected from:
- L 1 is or contains a group selected from , O
- L 1 is non-cleavable under physiological conditions. In some embodiments, L 1 is cleavable under physiological conditions. In some embodiments, L 1 is an acid-labile linker, a hydrolysis-labile linker, an enzymatically cleavable linker, a reduction labile linkers or a self-immolative linker. In some embodiments, L 1 is or comprises a peptide, a carbohydrate, a glucuronide, a polyethylene glycol (PEG) unit, a hydrazone, a mal-caproyl unit, a dipeptide unit, a valine-citruline unit, or a para-aminobenzyl (PAB) unit.
- PEG polyethylene glycol
- PAB para-aminobenzyl
- L 1 comprises one or more amino acids. In some embodiments, L 1 comprises a self-immolative group. In some embodiments, L 1 comprises p-aminobenzyl (PAB) or p- aminobenzyloxycarbonyl (PABC). In some embodiments, L 1 comprises a maleimido, an N- hydroxysuccinimido ester or cyclooctynyl group.
- PAB p-aminobenzyl
- PABC p- aminobenzyloxycarbonyl
- L 1 comprises a maleimido, an N- hydroxysuccinimido ester or cyclooctynyl group.
- L 1 is a group derived from 2-maleimido-1-ethyl, 2-maleimidoacetyl, 3-maleimidopropanoyl, , , O O O O N H N N H N H O O O O N N NH O , O NH 2 , O O O O O O H O O O O H N H N N H N N N N N N N N H H H H O O O O O O O O O O O O O O O OH N NH , H 2 , O
- the ADC comprises ABD linked LP1-LP15.
- B. Antigen-binding Domains (ABD) antigen-binding domains, i.e., ABD in Formula IV, for use in the ADCs provided herein include any molecule that specifically interacts with a particular antigen.
- the ABD is an antibody or antigen-binding fragment of an antibody.
- the ABD is an antibody.
- the ABD is an antibody comprising an Fc region modified to enhance binding affinity to Fc ⁇ R.
- ABD is an antibody with one or more mutations selected from F243L, R292P, Y300L, V305I, and P396L. In some embodiments, ABD is an antibody with one or more mutation selected from S239D and I332E. In some embodiments, ABD is an antibody with one or more mutations selected from S239D, I332E, and A330L. In some embodiments, ABD is an antibody with one or more mutations selected from S298A, E333A and K334A. In some embodiments, ABD is an antibody with one or more mutations selected from L234Y, L235Q, G236W, S239M, H268D, D270E, and S298A.
- ABD is an antibody with one or more mutations selected from D270E, K326D, A330M, and K334E.
- ABD is an antibody with L234Y, L235Q, G236W, S239M, H268D, D270E, and S298A in one heavy chain and D270E, K326D, A330M, and K334E in the opposing heavy chain.
- ABD is an antibody with one or more mutations selected from G236A, S239D, and I332E.
- ABD is an antibody with one or more mutations selected from M252Y, S254T, and T256E.
- ABD is an antibody with one or more mutations selected from M428L and N434S. In some embodiments, ABD is an antibody with one or more mutations selected from S267E and L328F. In some embodiments, ABD is an antibody with one or more mutations selected from N325S and L328F. [0191] In some embodiment, ABD is an antibody that comprises a glutamine residue. Antibodies comprising glutamine residues can be isolated from natural sources or engineered to comprise one or more glutamine residues. Techniques for engineering glutamine residues into an antibody polypeptide chain (glutaminyl-modified antibodies) are within the skill of the practitioners in the art. In other embodiments, ABD is an N297Q mutant antibody.
- Z is an antibody that has one or more engineered LLQG (SEQ ID NO: 1), LLQGG (SEQ ID NO: 2), LLQLLQG (SEQ ID NO: 3), LLQYQG (SEQ ID NO: 4), LLQGA (SEQ ID NO: 5), LLQGSG (SEQ ID NO: 6), SLLQG (SEQ ID NO: 7), LQG, LLQLQ (SEQ ID NO: 9), LLQLLQ (SEQ ID NO: 10), LLQGR (SEQ ID NO: 11), LLQYQGA (SEQ ID NO: 12), LQGG (SEQ ID NO: 13), LGQG (SEQ ID NO: 14) or LLQLLQGA (SEQ ID NO: 15) sites.
- LLQG SEQ ID NO: 1
- LLQGG SEQ ID NO: 2
- LLQLLQG SEQ ID NO: 3
- LLQYQG SEQ ID NO: 4
- LLQGA S
- the antibody is aglycosylated. In some embodiments, the antibody is glucosylated.
- ABD is an antibody that is a monoclonal antibody, human antibody, humanized antibody, camelised antibody, or chimeric antibody.
- ABD is an antibody of any isotype (e.g., IgG, IgE, IgM, IgD, IgA and IgY), class (e.g., IgG1, IgG2, IgG3, IgG4, IgA1 and IgA2) or subclass.
- ABD has a molecular weight of at least 500, 600, 700, 800, 900, 1000, 10000, 50000 or 100000 Daltons.
- antigen-binding domains that can be used in the ADCs provided herein include antibodies, antigen-binding fragments of antibodies, peptides that specifically interact with a particular antigen (e.g., peptibodies), receptor molecules that specifically interact with a particular antigen, proteins comprising a ligand-binding portion of a receptor that specifically binds a particular antigen, antigen-binding scaffolds (e.g., DARPins, HEAT repeat proteins, ARM repeat proteins, tetratricopeptide repeat proteins, and other scaffolds based on naturally occurring repeat proteins, etc., (see, e.g., Boersma and Pluckthun, 2011, Curr. Opin.
- DARPins e.g., DARPins, HEAT repeat proteins, ARM repeat proteins, tetratricopeptide repeat proteins, and other scaffolds based on naturally occurring repeat proteins, etc.
- ABD comprises a scFv having binding specificity to a target antigen.
- an antigen-binding domain includes polypeptides that bind a target antigen or a portion thereof with a K D of less than about 500 pM, less than about 400 pM, less than about 300 pM, less than about 200 pM, less than about 100 pM, less than about 90 pM, less than about 80 pM, less than about 70 pM, less than about 60 pM, less than about 50 pM, less than about 40 pM, less than about 30 pM, less than about 20 pM, less than about 10 pM, less than about 5 pM, less than about 4 pM, less than about 2 pM, less than about 1 pM, less than about 0.5 pM, less than about 0.2 pM, less than about 0.1 pM, or less than about 0.05 pM, as measured in a surface plasmon resonance assay.
- the framework regions (FRs) of the antibodies or antigen- binding fragment thereof for use in the ADCs provided herein may be identical to the human germline sequences, or may be naturally or artificially modified.
- An amino acid consensus sequence may be defined based on a side-by-side analysis of two or more CDRs.
- Methods and techniques for identifying CDRs within HCVR and LCVR amino acid sequences are well known in the art and can be used to identify CDRs. Exemplary conventions that can be used to identify the boundaries of CDRs include, e.g., the Kabat definition, the Chothia definition, and the AbM definition.
- the Kabat definition is based on sequence variability
- the Chothia definition is based on the location of the structural loop regions
- the AbM definition is a compromise between the Kabat and Chothia approaches. See, e.g., Kabat, "Sequences of Proteins of Immunological Interest," National Institutes of Health, Bethesda, Md. (1991); Al-Lazikani et al., J. Mol. Biol.273:927-948 (1997); and Martin et al., Proc. Natl. Acad. Sci. USA 86:9268-9272 (1989). Public databases are also available for identifying CDR sequences within an antibody.
- the antigen-binding domains for use in the ADCs provided herein may comprise or consist of antigen-binding fragments of full antibody molecules.
- Antigen-binding fragments of an antibody may be derived, e.g., from full antibody molecules using any suitable standard techniques such as proteolytic digestion or recombinant genetic engineering techniques involving the manipulation and expression of DNA encoding antibody variable and optionally constant domains.
- DNA is known and/or is readily available from, e.g., commercial sources, DNA libraries (including, e.g., phage-antibody libraries), or can be synthesized.
- the DNA may be sequenced and manipulated chemically or by using molecular biology techniques, for example, to arrange one or more variable and/or constant domains into a suitable configuration, or to introduce codons, create cysteine residues, modify, add or delete amino acids, etc.
- Non-limiting examples of antigen-binding fragments for use in the ADCs provided herein include: (i) Fab fragments; (ii) F(ab')2 fragments; (iii) Fd fragments; (iv) Fv fragments; (v) single-chain Fv (scFv) molecules; (vi) dAb fragments; and (vii) minimal recognition units consisting of the amino acid residues that mimic the hypervariable region of an antibody (e.g., an isolated complementarity determining region (CDR) such as a CDR3 peptide), or a constrained FR3-CDR3-FR4 peptide.
- CDR complementarity determining region
- an antigen-binding fragment of an antibody includes other engineered molecules, such as domain-specific antibodies, single domain antibodies, domain-deleted antibodies, chimeric antibodies, CDR-grafted antibodies, diabodies, triabodies, tetrabodies, minibodies, nanobodies (e.g., monovalent nanobodies, bivalent nanobodies, etc.), small modular immunopharmaceuticals (SMIPs), and shark variable IgNAR domains.
- an antigen-binding fragment of an antibody will comprise at least one variable domain.
- the variable domain may be of any size or amino acid composition and will generally comprise at least one CDR which is adjacent to or in frame with one or more framework sequences.
- the V H and V L domains may be situated relative to one another in any suitable arrangement.
- the variable region may be dimeric and contain V H -V H , V H -V L or V L -V L dimers.
- the antigen-binding fragment of an antibody may contain a monomeric V H or V L domain.
- an antigen-binding fragment of an antibody may contain at least one variable domain covalently linked to at least one constant domain.
- Non-limiting, exemplary configurations of variable and constant domains that may be found within an antigen-binding fragment of an antibody for use in the ADCs provided herein include: (i) V H - C H 1; (ii) V H -C H 2; (iii) V H -C H 3; (iv) V H -C H 1-C H 2; (v) V H -C H 1-C H 2-C H 3; (vi) V H -C H 2-C H 3; (vii) V H - C L ; (viii) V L -C H 1; (ix) V L -C H 2; (x) V L -C H 3; (xi) V L -C H 1-C H 2; (xii) V L -C H 1-C H 2-C H 3; (xiii) V L -C H 2- C H 3; and (xiv) V L -C L .
- variable and constant domains may be either directly linked to one another or may be linked by a full or partial hinge or linker region.
- a hinge region may consist of at least 2 (e.g., 5, 10, 15, 20, 40, 60 or more) amino acids which result in a flexible or semi-flexible linkage between adjacent variable and/or constant domains in a single polypeptide molecule.
- an antigen-binding fragment may comprise a homo-dimer or hetero-dimer (or other multimer) of any of the variable and constant domain configurations listed above in non-covalent association with one another and/or with one or more monomeric V H or V L domain (e.g., by disulfide bond(s)).
- the antigen-binding domains used in the ADCs provided herein may comprise or consist of human antibodies and/or recombinant human antibodies, or antigen-binding fragments thereof.
- the antigen-binding domains used in the ADCs provided herein may comprise or consist of recombinant human antibodies or antigen-binding fragments thereof.
- such recombinant human antibodies have variable and constant regions derived from human germline immunoglobulin sequences. In certain embodiments, however, such recombinant human antibodies are subjected to in vitro mutagenesis (or, when an animal transgenic for human Ig sequences is used, in vivo somatic mutagenesis) and thus the amino acid sequences of the V H and V L regions of the recombinant antibodies are sequences that, while derived from and related to human germline V H and V L sequences, may not naturally exist within the human antibody germline repertoire in vivo.
- the antigen-binding domains used in the ADCs provided herein also include bispecific antigen-binding molecules, such as bispecific antibodies.
- bispecific formats that can be used in the context of the present disclosure include, without limitation, e.g., scFv-based or diabody bispecific formats, IgG-scFv fusions, dual variable domain (DVD)-Ig, Quadroma, knobs-into-holes, common light chain (e.g., common light chain with knobs-into-holes, etc.), CrossMab, CrossFab, (SEED) body, leucine zipper, Duobody, IgG1/IgG2, dual acting Fab (DAF)-IgG, and Mab 2 bispecific formats (see, e.g., Klein et al.
- bispecific antigen binding molecules may comprise a first antigen-binding domain (also referred to herein as "D1"), and a second antigen-binding domain (also referred to herein as "D2").
- D1 and D2 domains of a bispecific antibody are non-competitive with one another.
- Non-competition between D1 and D2 means that, the respective monospecific antigen binding proteins from which D1 and D2 were derived do not compete with one another for binding to the target.
- Exemplary antigen-binding protein competition assays are known in the art.
- D1 and D2 bind to different (e.g., non-overlapping, or partially overlapping) epitopes on the target.
- Bispecific antigen-binding molecules may be constructed using the antigen-binding domains of two separate monospecific antibodies. For example, a collection of monoclonal monospecific antibodies may be produced using standard methods known in the art. The individual antibodies thus produced may be tested pairwise against one another for cross-competition to the target protein.
- a bispecific antigen- binding molecule can be a single multifunctional polypeptide, or it can be a multimeric complex of two or more polypeptides that are covalently or non-covalently associated with one another. Any antigen binding construct which has the ability to simultaneously bind two separate, non- identical epitopes of the target molecule is regarded as a bispecific antigen-binding molecule.
- Bispecific antigen-binding molecules may be constructed using standard molecular biological techniques (e.g., recombinant DNA and protein expression technology) as will be known to a person of skill in the art.
- bispecific antibodies are also provided wherein one arm of the bispecific antibody binds to an epitope on a first target protein, and the other arm of the bispecific antibody binds to a second epitope on a second target protein.
- bispecific formats that can be used in the context of the present disclosure include, without limitation, e.g., scFv-based or diabody bispecific formats, IgG-scFv fusions, dual variable domain (DVD)-Ig, Quadroma, knobs-into-holes, common light chain (e.g., common light chain with knobs-into-holes, etc.), CrossMab, CrossFab, (SEED)body, leucine zipper, Duobody, IgG1/IgG2, dual acting Fab (DAF)-IgG, and Mab2 bispecific formats (see, e.g., Klein et al.
- Bispecific antibodies can also be constructed using peptide/nucleic acid conjugation, e.g., wherein unnatural amino acids with orthogonal chemical reactivity are used to generate site-specific antibody-oligonucleotide conjugates which then self-assemble into multimeric complexes with defined composition, valency and geometry. (See, e.g., Kazane et al., J. Am. Chem. Soc. (Epub: Dec.4, 2012)).
- the antigen binding domains for use in the ADCs provided herein also include antibodies comprising variants of any of the HCVR, LCVR, and/or CDR amino acid sequences known in the art.
- variants include variants of any of the HCVR, LCVR, and/or CDR amino acid sequences known in the art having one or more conservative substitutions.
- the antigen binding domains include antibodies or antigen binding fragments thereof having HCVR, LCVR, and/or CDR amino acid sequences with, e.g., 10 or fewer, 8 or fewer, 6 or fewer, 4 or fewer, etc.
- the antigen binding domains include antibodies or antigen binding fragments thereof also include variants having substantial sequence identity to any of the HCVR, LCVR, and/or CDR amino acid sequences known in the art.
- residue positions which are not identical differ by conservative amino acid substitutions.
- GCG software contains programs such as GAP and BESTFIT which can be used with default parameters to determine sequence homology or sequence identity between closely related polypeptides, such as homologous polypeptides from different species of organisms or between a wild type protein and a mutein thereof. See, e.g., GCG Version 6.1. Polypeptide sequences also can be compared using FASTA using default or recommended parameters, a program in GCG Version 6.1. FASTA (e.g., FASTA2 and FASTA3) provides alignments and percent sequence identity of the regions of the best overlap between the query and search sequences (Pearson (2000) supra).
- the antigen-binding domains for use in the ADCs provided herein encompass proteins having amino acid sequences that vary from those of the described antibodies but that retain the ability to bind the target proteins.
- Such variant antigen-binding domains comprise one or more additions, deletions, or substitutions of amino acids when compared to parent sequence but exhibit biological activity that is essentially equivalent to that of the described antibodies.
- Two antigen-binding domains are considered bioequivalent if, for example, they are pharmaceutical equivalents or pharmaceutical alternatives whose rate and extent of absorption do not show a significant difference when administered at the same molar dose under similar experimental conditions, either single dose or multiple doses.
- antigen-binding domains will be considered equivalents or pharmaceutical alternatives if they are equivalent in the extent of their absorption but not in their rate of absorption and yet may be considered bioequivalent because such differences in the rate of absorption are intentional and are reflected in the labeling, are not essential to the attainment of effective body drug concentrations on, e.g., chronic use, and are considered medically insignificant for the particular drug product studied.
- two antigen-binding domains are bioequivalent if there are no clinically meaningful differences in their safety, purity, and potency.
- two antigen-binding domains are bioequivalent if a patient can be switched one or more times between the reference product and the biological product without an expected increase in the risk of adverse effects, including a clinically significant change in immunogenicity, or diminished effectiveness, as compared to continued therapy without such switching.
- two antigen-binding domains are bioequivalent if they both act by a common mechanism or mechanisms of action for the condition or conditions of use, to the extent that such mechanisms are known.
- Bioequivalence may be demonstrated by in vivo and in vitro methods.
- Bioequivalence measures include, e.g., (a) an in vivo test in humans or other mammals, in which the concentration of the antigen-binding domain or its metabolites is measured in blood, plasma, serum, or other biological fluid as a function of time; (b) an in vitro test that has been correlated with and is reasonably predictive of human in vivo bioavailability data; (c) an in vivo test in humans or other mammals in which the appropriate acute pharmacological effect of the antigen-binding domain (or its target) is measured as a function of time; and (d) in a well- controlled clinical trial that establishes safety, efficacy, or bioavailability or bioequivalence of an antigen-binding domain.
- Bioequivalent variants of antigen-binding domains for use in the ADCs provided herein may be constructed by, for example, making various substitutions of residues or sequences or deleting terminal or internal residues or sequences not needed for biological activity.
- cysteine residues not essential for biological activity can be deleted or replaced with other amino acids to prevent formation of unnecessary or incorrect intramolecular disulfide bridges upon renaturation.
- bioequivalent antigen- binding domains may include variants comprising amino acid changes which modify the glycosylation characteristics of the antigen-binding domain, e.g., mutations which eliminate or remove glycosylation.
- the antigen-binding domains for use in the ADCs provided herein bind to a human target protein but not to target protein from other species. In other embodiments, the antigen-binding domains for use in the ADCs provided herein bind to a human target protein and to a target protein from one or more non-human species.
- the antigen-binding domains for use in the ADCs provided herein may bind to a human target protein and may bind or not bind, as the case may be, to one or more of mouse, rat, guinea pig, hamster, gerbil, pig, cat, dog, rabbit, goat, sheep, cow, horse, camel, cynomologous, marmoset, rhesus or chimpanzee target protein.
- the antigen-binding domains specifically bind human target protein and cynomolgus monkey (e.g., Macaca fascicularis) target protein.
- antigen-binding domains for use herein bind human target protein but do not bind, or bind only weakly, to cynomolgus monkey target protein.
- ABD Sequences [0215] In some embodiments, the ABD comprises the heavy chain and light chain of an antibody. [0216] In some embodiments, the ABD comprises HCDR1, HCDR2, HCDR3, LCDR1, LCDR2 and LCDR3. 2. Linkage Sites [0217] The ABD can be linked to the TLR7 agonist directly or indirectly via a linker, through an attachment at a particular amino acid within the ABD.
- Exemplary amino acid attachments that can be used in the context of this embodiment of the disclosure include, e.g., lysine (see, e.g., US 5,208,020; US 2010/0129314; Hollander et al., Bioconjugate Chem., 2008, 19:358- 361; WO 2005/089808; US 5,714,586; US 2013/0101546; and US 2012/0585592), cysteine (see, e.g., US 2007/0258987; WO 2013/055993; WO 2013/055990; WO 2013/053873; WO 2013/053872; WO 2011/130598; US 2013/0101546; and US 7,750,116), selenocysteine (see, e.g., WO 2008/122039; and Hofer et al., Proc.
- lysine see, e.g., US 5,208,020; US 2010/0129314; Hollander e
- Linkers can also be conjugated to an ABD via attachment to carbohydrates (see, e.g., US 2008/0305497, WO 2014/065661, Ryan et al., Food & Agriculture Immunol., 2001, 13:127-130, and Jeger et al., Angew Chem Int Ed Engl., 2010, 49:9995-9997).
- ABD is bonded to the linker through a lysine residue.
- the antibody or antigen binding molecule is bonded to the linker through a cysteine residue, lysine residue, or glutamine residue.
- the ABD is bonded to the linker through a cysteine residue.
- a linker maleimide moiety bonds to an antibody cysteine residue.
- the ABD is bonded to the linker through a lysine residue.
- a linker N-hydroxysuccinimide moiety bonds to an antibody lysine residue to form an amide linkage.
- the ABD is bonded to the linker through a glutamine residue (see, e.g., Jeger et al., Angew Chem Int Ed Engl., 2010, 49:9995-9997 and Dennler et al., Bioconjugate Chem. 2014, 25:569-578).
- Antibodies comprising glutamine residues can be isolated from natural sources or engineered to comprise one or more glutamine residues.
- antibodies or antigen binding molecules are engineered by mutations, for example insertions or deletions to facilitate reaction via transglutaminase.
- antibodies or antigen binding molecules are engineered to remove one or more glycosylation sites.
- antibodies or antigen binding molecules are engineered to add one or more glutamine residues.
- glutamine residues are added within a TGase recognition tag, as described herein. Techniques for engineering glutamine residues into an antibody polypeptide chain (glutaminyl-modified antibodies or antigen binding molecules) are within the skill of the practitioners in the art.
- the antibody is aglycosylated.
- ABD comprises at least one glutamine residue in at least one polypeptide chain sequence.
- ABD comprises two heavy chain polypeptides, each with one Gln295 or Q295 residue.
- ABD comprises one or more glutamine residues at a site other than a heavy chain 295. Included herein are antibodies of this section bearing N297Q mutation(s) described herein. In certain embodiments, a glutamine residue is added at the heavy chain C-terminus.
- the glutamine is polypeptide engineered with a glutamine- containing tag (e.g., glutamine-containing peptide tags, Q-tags or TGase recognition tag).
- a glutamine-containing tag e.g., glutamine-containing peptide tags, Q-tags or TGase recognition tag.
- the term “TGase recognition tag” or “Q-Tag” refers to a sequence of amino acids comprising a glutamine residue that when incorporated into (e.g., appended to) a polypeptide sequence, under suitable conditions, is recognized by a transglutaminase (“TGase”) and leads to cross- linking by the TGase through a reaction between an amino acid side chain within the sequence of amino acids and a reactive group.
- the recognition tag may be a peptide sequence that is not naturally present in the polypeptide.
- the TGase recognition tag comprises at least one glutamine.
- the TGase recognition tag comprises an amino acid sequence XXQX, wherein X is any amino acid (e.g., conventional amino acid Leu, Ala, Gly, Ser, Val, Phe, Tyr, His, Arg, Asn, Glu, Asp, Cys, Gin, He, Met, Pro, Thr, Lys, or Trp or nonconventional amino acid).
- the TGase recognition tag comprises an amino acid sequence selected from the group consisting of LLQGG (SEQ ID NO: 2), LLQG (SEQ ID NO: 1), LSLSQG (SEQ ID NO: 17), GGGLLQGG (SEQ ID NO: 18), GLLQG (SEQ ID NO: 19), LLQ, GSPLAQSHGG (SEQ ID NO: 20), GLLQGGG (SEQ ID NO: 21), GLLQGG (SEQ ID NO: 22), GLLQ (SEQ ID NO: 23), LLQLLQGA (SEQ ID NO: 3), LLQGA (SEQ ID NO: 5), LLQYQGA (SEQ ID NO: 12), LLQGSG (SEQ ID NO: 6), LLQYQG (SEQ ID NO: 4), LLQLLQG (SEQ ID NO: 3), SLLQG (SEQ ID NO: 7), LLQLQ (SEQ ID NO: 9), LLQLLQ (SEQ ID NO: 2
- ABD includes an antibody heavy chain and further includes a TGase recognition tag at the C-terminus of the antibody heavy chain.
- ABD includes an antibody heavy chain and further includes a TGase recognition tag at the C- terminus of the antibody heavy chain, wherein the TGase recognition tag is the pentapeptide sequence LLQGA (SEQ ID NO: 5).
- ABD includes two antibody heavy chains and further includes a TGase recognition tag at the C-terminus of each antibody heavy chain.
- ABD includes two antibody heavy chains and further includes a TGase recognition tag at the C-terminus of each antibody heavy chain, wherein the TGase recognition tag is the pentapeptide sequence LLQGA (SEQ ID NO: 5).
- ABD can be also modified at one or more glutamine residues via transglutaminase (see, e.g., Jeger et al., Angew Chem Int Ed Engl., 2010, 49:9995-9997 and Dennler et al., Bioconjugate Chem. 2014, 25:569-578).
- one or more glutamine residues of an antibody can be coupled to a primary amine compound to provide a moiety capable of reacting with a reactive group on a linker-payload.
- the primary amine compound provides a diene or dienophile.
- the primary amine compound provides a diene or dienophile
- the linker- payload provides a complementary dienophile or diene, respectively, for conjugation via a Diels-Alder reaction.
- the primary amine compound provides an azido group.
- the primary amine compound provides an azido group
- the linker-payload provides a complementary alkyne, for conjugation via a click reaction.
- the ABD comprises a heavy chain and the heavy chain is linked to ABD directly or indirectly via a linker.
- the ABD comprises a light chain and the light chain is linked to ABD directly or indirectly via a linker.
- the ABD comprises a heavy chain and the C-terminus of the heavy chain is linked to ABD directly or indirectly via a linker.
- the ABD comprises a light chain and the C-terminus of the light chain is linked to ABD directly or indirectly via a linker.
- the ABD comprises two heavy chains and each of the two heavy chains is linked to ABD directly or indirectly via a linker. In some embodiments, the ABD comprises two light chains and each of the two light chains is linked to ABD directly or indirectly via a linker. [0227] In some embodiments, the ABD comprises two heavy chains and C-terminus of each of the two heavy chains is linked to ABD directly or indirectly via a linker. In some embodiments, the ABD comprises two light chains and C-terminus of each of the two light chains is linked to ABD directly or indirectly via a linker. 3.
- the epitope to which the antigen-binding domains bind may consist of a single contiguous sequence of 3 or more (e.g., 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 or more) amino acids of a target protein.
- the relevant epitope may consist of a plurality of non-contiguous amino acids (or amino acid sequences) of the target protein.
- the epitope is located on or near the binding domain of the target protein. In other embodiments, the epitope is located outside of the binding domain of the target protein.
- Various techniques known to persons of ordinary skill in the art can be used to determine the epitope with which the antigen-binding domains used in the ADCs provided herein interact.
- Exemplary techniques that can be used to determine an epitope or binding domain of a particular antigen-binding domain include, e.g., point mutagenesis (e.g., alanine scanning mutagenesis, arginine scanning mutagenesis, etc.), peptide blots analysis (Reineke, 2004, Methods Mol Biol 248:443-463), protease protection, and peptide cleavage analysis.
- the hydrogen/deuterium exchange method involves deuterium-labeling the protein of interest, followed by binding the antigen-binding domain to the deuterium-labeled protein. Next, the protein/antigen-binding domain complex is transferred to water to allow hydrogen- deuterium exchange to occur at all residues except for the residues protected by the antigen- binding domain (which remain deuterium-labeled).
- the target protein After dissociation of the antigen-binding domain, the target protein is subjected to protease cleavage and mass spectrometry analysis, thereby revealing the deuterium-labeled residues which correspond to the specific amino acids with which the antigen-binding domain interacts. See, e.g., Ehring (1999) Analytical Biochemistry 267(2):252-259; Engen and Smith (2001) Anal. Chem. 73:256A-265A. X-ray crystal structure analysis can also be used to identify the amino acids within a polypeptide with which an antigen-binding domain interacts. 4. Synthesis of ABDs [0230] In one embodiment, the antibodies for use in the ADCs provided herein are fully human antibodies.
- Methods for generating monoclonal antibodies, including fully human monoclonal antibodies are known in the art. Any such known methods can be used in the context of the present disclosure to make human antibodies that specifically bind to a human protein target.
- VELOCIMMUNETM technology for example, or any other similar known method for generating fully human monoclonal antibodies
- high affinity chimeric antibodies to a human protein target are initially isolated having a human variable region and a mouse constant region.
- the antibodies are characterized and selected for desirable characteristics, including affinity, ligand blocking activity, selectivity, epitope, etc.
- mouse constant regions are replaced with a desired human constant region, for example wild-type or modified IgG1 or IgG4, to generate a fully human antibody.
- Monoclonal antibodies can be generated by any techniques with which those having ordinary skill in the art will be familiar. Such methods include, but are not limited to, Epstein Barr Virus (EBV) transformation of human peripheral blood cells (e.g., containing B lymphocytes), in vitro immunization of human B-cells, fusion of spleen cells from immunized transgenic mice carrying inserted human immunoglobulin genes, isolation from human immunoglobulin V region phage libraries, or other procedures as known in the art and based on the disclosure herein.
- EBV Epstein Barr Virus
- Fully human monoclonal antibodies can be obtained from transgenic mice that have been engineered to produce specific human antibodies in response to antigenic challenge.
- Methods for obtaining fully human antibodies from transgenic mice are described, for example, by Green et al., Nature Genet. 7:13, 1994; Lonberg et al., Nature 368:856, 1994; Taylor et al., Int. Immun.6:579, 1994; U.S. Patent No. 5,877,397; Bruggemann et al., 1997 Curr. Opin. Biotechnol.8:455-58; Jakobovits et al., 1995 Ann. N. Y. Acad. Sci.764:525-35.
- human immunoglobulin transgenes may be mini-gene constructs, or transloci on yeast artificial chromosomes, which undergo B-cell-specific DNA rearrangement and hypermutation in the mouse lymphoid tissue.
- Fully human monoclonal antibodies may be obtained by immunizing the transgenic mice, which may then produce human antibodies specific for a target antigen.
- Lymphoid cells of the immunized transgenic mice can be used to produce human antibody-secreting hybridomas according to the methods described herein. Polyclonal sera containing fully human antibodies may also be obtained from the blood of the immunized animals.
- Another method for generating human antibodies of the present disclosure includes immortalizing human peripheral blood cells by EBV transformation. See, e.g., U.S. Patent No. 4,464,456. Such an immortalized B-cell line (or lymphoblastoid cell line) producing a monoclonal antibody that specifically binds to a target antigen can be identified by immunodetection methods as provided herein, for example, an ELISA, and then isolated by standard cloning techniques.
- the stability of the lymphoblastoid cell line producing an antibody against a target antigen can be improved by fusing the transformed cell line with a murine myeloma to produce a mouse-human hybrid cell line according to methods known in the art (see, e.g., Glasky et al., Hybridoma 8:377-89 (1989)).
- Still another method to generate human monoclonal antibodies is in vitro immunization, which includes priming human splenic B-cells with a target antigen, followed by fusion of primed B-cells with a heterohybrid fusion partner. See, e.g., Boerner et al., 1991 J. Immunol.147:86-95.
- a B-cell that is producing an antibody against a target antigen is selected and the light chain and heavy chain variable regions are cloned from the B-cell according to molecular biology techniques known in the art (WO 92/02551; U.S. Patent 5,627,052; Babcook et al., Proc. Natl. Acad. Sci. USA 93:7843-48 (1996)) and described herein.
- B-cells from an immunized animal may be isolated from the spleen, lymph node, or peripheral blood sample by selecting a cell that is producing an antibody that specifically binds to a target antigen.
- B-cells may also be isolated from humans, for example, from a peripheral blood sample.
- Methods for detecting single B-cells that are producing an antibody with the desired specificity are well known in the art, for example, by plaque formation, fluorescence-activated cell sorting, in vitro stimulation followed by detection of specific antibody, and the like.
- Methods for selection of specific antibody-producing B-cells include, for example, preparing a single cell suspension of B-cells in soft agar that contains a target antigen. Binding of the specific antibody produced by the B-cell to the antigen results in the formation of a complex, which may be visible as an immune-precipitate.
- the methods for obtaining antibodies of the present disclosure can also adopt various phage display technologies known in the art. See, e.g., Winter et al., 1994 Annu.
- Human or murine immunoglobulin variable region gene combinatorial libraries may be created in phage vectors that can be screened to select Ig fragments (Fab, Fv, sFv, or multimers thereof) that bind specifically to a target antigen or variant or fragment thereof. See, e.g., U.S. Patent No. 5,223,409; Huse et al., 1989 Science 246:1275-81; Sastry et al., Proc. Natl. Acad. Sci.
- a library containing a plurality of polynucleotide sequences encoding Ig variable region fragments may be inserted into the genome of a filamentous bacteriophage, such as M13 or a variant thereof, in frame with the sequence encoding a phage coat protein.
- a fusion protein may be a fusion of the coat protein with the light chain variable region domain and/or with the heavy chain variable region domain.
- immunoglobulin Fab fragments may also be displayed on a phage particle (see, e.g., U.S. Patent No.5,698,426).
- Antibody fragments fused to another protein, such as a minor coat protein can be also used to enrich phage with antigen. Then, using a random combinatorial library of rearranged heavy (V H ) and light (V L ) chains from mice immune to the target antigen (e.g., HBV sAg, tumor specific antigen), diverse libraries of antibody fragments are displayed on the surface of the phage.
- Heavy and light chain immunoglobulin cDNA expression libraries may also be prepared in lambda phage, for example, using ⁇ lmmunoZap TM (H) and ⁇ ImmunoZap TM (L) vectors (Stratagene, La Jolla, California). Briefly, mRNA is isolated from a B-cell population, and used to create heavy and light chain immunoglobulin cDNA expression libraries in the ⁇ ImmunoZap(H) and ⁇ ImmunoZap(L) vectors.
- variable regions of a gene expressing a monoclonal antibody of interest are amplified using nucleotide primers. These primers may be synthesized by one of ordinary skill in the art or may be purchased from commercially available sources.
- the specific antibody genes may be cloned by isolating and amplifying DNA or mRNA therefrom according to standard procedures as described herein.
- the antibodies produced therefrom may be sequenced and the CDRs identified and the DNA coding for the CDRs may be manipulated as described previously to generate other antibodies according to the disclosure.
- the binding agents of the present disclosure preferably modulate activity of the target antigen in the cell-based assay described herein and/or the in vivo assay described herein and/or bind to one or more of the domains described herein and/or cross-block the binding of one of the antibodies described in this application and/or are cross-blocked from binding the target antigen by one of the antibodies described in this application. Accordingly, such binding agents can be identified using the assays described herein.
- antibodies are generated by first identifying antibodies that bind to one or more of the domains provided herein and/or neutralize in the cell-based and/or in vivo assays described herein and/or cross-block the antibodies described in this application and/or are cross-blocked from binding a target antigen by one of the antibodies described in this application.
- the CDR regions from these antibodies are then used to insert into appropriate biocompatible frameworks to generate binding agents against the target antigen.
- the non-CDR portion of the binding agent may be composed of amino acids or may be a non- protein molecule.
- the assays described herein allow the characterization of binding agents.
- the binding agents of the present disclosure are antibodies as defined herein.
- CDRs complementarity determining regions
- the antigen binding proteins also may be employed in purifying a target antigenby immunoaffinity chromatography.
- human, partially human, or humanized antibodies will be suitable for many applications, particularly those involving administration of the antibody to a human subject, other types of antigen binding proteins will be suitable for certain applications.
- Non-human antibodies of the present disclosure can be, for example, derived from any antibody-producing animal, such as mouse, rat, rabbit, goat, donkey, or non-human primate (such as monkey (e.g., cynomolgus or rhesus monkey) or ape (e.g., chimpanzee)).
- An antibody from a particular species can be made by, for example, immunizing an animal of that species with the desired immunogen (e.g., HBV sAg, tumor specific antigen or using an artificial system for generating antibodies of that species (e.g., a bacterial or phage display-based system for generating antibodies of a particular species), or by converting an antibody from one species into an antibody from another species by replacing, e.g., the constant region of the antibody with a constant region from the other species, or by replacing one or more amino acid residues of the antibody so that it more closely resembles the sequence of an antibody from the other species.
- the antibody is a chimeric antibody comprising amino acid sequences derived from antibodies from two or more different species.
- Antigen binding proteins may be prepared, and screened for desired properties, by any of a number of conventional techniques. Certain of the techniques involve isolating a nucleic acid encoding a polypeptide chain (or portion thereof) of an antigen binding protein of interest (e.g., an anti-HBV sAg antibody, a tumor specific antigen), and manipulating the nucleic acid through recombinant DNA technology.
- the nucleic acid may be fused to another nucleic acid of interest, or altered (e.g., by mutagenesis or other conventional techniques) to add, delete, or substitute one or more amino acid residues, for example.
- the antigen binding proteins may be purified from cells that naturally express them (e.g., an antibody can be purified from a hybridoma that produces it), or produced in recombinant expression systems, using any technique known in the art. See, for example, Monoclonal Antibodies, Hybridomas: A New Dimension in Biological Analyses, Kennet et al. (eds.), Plenum Press, New York (1980); and Antibodies: A Laboratory Manual, Harlow and Land (eds.), Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, (1988). [0247] Any expression system known in the art can be used to make the recombinant polypeptides of the present disclosure. Expression systems are detailed comprehensively above.
- host cells are transformed with a recombinant expression vector that comprises DNA encoding a desired polypeptide.
- host cells that may be employed are prokaryotes, yeast or higher eukaryotic cells.
- Prokaryotes include gram negative or gram positive organisms, for example E. coli or Bacilli.
- Higher eukaryotic cells include insect cells and established cell lines of mammalian origin.
- suitable mammalian host cell lines include the COS-7 line of monkey kidney cells (ATCC CRL 1651) (Gluzman et al., 1981, Cell 23:175), L cells, 293 cells, C127 cells, 3T3 cells (ATCC CCL 163), Chinese hamster ovary (CHO) cells, HeLa cells, BHK (ATCC CRL 10) cell lines, and the CVI/EBNA cell line derived from the African green monkey kidney cell line CVI (ATCC CCL 70) as described by McMahan et al., 1991, EMBO J. 10: 2821.
- Appropriate cloning and expression vectors for use with bacterial, fungal, yeast, and mammalian cellular hosts are described by Pouwels et al.
- an antibody of the present disclosure may have at least one amino acid substitution, providing that the antibody retains binding specificity. Therefore, modifications to the antibody structures are encompassed within the scope of the present disclosure. These may include amino acid substitutions, which may be conservative or non- conservative that do not destroy the target binding capability of an antibody. Conservative amino acid substitutions may encompass non-naturally occurring amino acid residues, which are typically incorporated by chemical peptide synthesis rather than by synthesis in biological systems. These include peptidomimetics and other reversed or inverted forms of amino acid moieties.
- a conservative amino acid substitution may also involve a substitution of a native amino acid residue with a normative residue such that there is little or no effect on the polarity or charge of the amino acid residue at that position.
- Non-conservative substitutions may involve the exchange of a member of one class of amino acids or amino acid mimetics for a member from another class with different physical properties (e.g., size, polarity, hydrophobicity, charge). Such substituted residues may be introduced into regions of the human antibody that are homologous with non-human antibodies, or into the non-homologous regions of the molecule.
- one skilled in the art may generate test variants containing a single amino acid substitution at each desired amino acid residue.
- variants can then be screened using activity assays known to those skilled in the art. Such variants could be used to gather information about suitable variants. For example, if one discovered that a change to a particular amino acid residue resulted in destroyed, undesirably reduced, or unsuitable activity, variants with such a change may be avoided. In other words, based on information gathered from such routine experiments, one skilled in the art can readily determine the amino acids where further substitutions should be avoided either alone or in combination with other mutations. [0251] A skilled artisan will be able to determine suitable variants of the polypeptide as set forth herein using well-known techniques.
- one skilled in the art may identify suitable areas of the molecule that may be changed without destroying activity by targeting regions not believed to be important for activity. In certain embodiments, one can identify residues and portions of the molecules that are conserved among similar polypeptides. In certain embodiments, even areas that may be important for biological activity or for structure may be subject to conservative amino acid substitutions without destroying the biological activity or without adversely affecting the polypeptide structure. [0252] Additionally, one skilled in the art can review structure-function studies identifying residues in similar polypeptides that are important for activity or structure. In view of such a comparison, one can predict the importance of amino acid residues in a protein that correspond to amino acid residues which are important for activity or structure in similar proteins.
- variants of antibodies include glycosylation variants wherein the number and/or type of glycosylation site has been altered compared to the amino acid sequences of a parent polypeptide.
- variants comprise a greater or a lesser number of N-linked glycosylation sites than the native protein.
- An N-linked glycosylation site is characterized by the sequence: Asn-X-Ser or Asn-X-Thr, wherein the amino acid residue designated as X can be any amino acid residue except proline.
- the substitution of amino acid residues to create this sequence provides a potential new site for the addition of an N-linked carbohydrate chain. Alternatively, substitutions which eliminate this sequence will remove an existing N-linked carbohydrate chain. Also provided is a rearrangement of N-linked carbohydrate chains wherein one or more N-linked glycosylation sites (typically those that are naturally occurring) are eliminated and one or more new N-linked sites are created.
- Additional preferred antibody variants include cysteine variants wherein one or more cysteine residues are deleted from or substituted for another amino acid (e.g., serine) as compared to the parent amino acid sequence.
- Cysteine variants can be useful when antibodies must be refolded into a biologically active conformation such as after the isolation of insoluble inclusion bodies. Cysteine variants generally have fewer cysteine residues than the native protein, and typically have an even number to minimize interactions resulting from unpaired cysteines.
- Desired amino acid substitutions (whether conservative or non-conservative) can be determined by those skilled in the art at the time such substitutions are desired.
- preferred amino acid substitutions are those which: (1) reduce susceptibility to proteolysis, (2) reduce susceptibility to oxidation, (3) alter binding affinity for forming protein complexes, (4) alter binding affinities, and/or (4) confer or modify other physiochemical or functional properties on such polypeptides.
- single or multiple amino acid substitutions may be made in the naturally-occurring sequence (in certain embodiments, in the portion of the polypeptide outside the domain(s) forming intermolecular contacts).
- a conservative amino acid substitution typically may not substantially change the structural characteristics of the parent sequence (e.g., a replacement amino acid should not tend to break a helix that occurs in the parent sequence, or disrupt other types of secondary structure that characterizes the parent sequence).
- a replacement amino acid should not tend to break a helix that occurs in the parent sequence, or disrupt other types of secondary structure that characterizes the parent sequence.
- Examples of art-recognized polypeptide secondary and tertiary structures are described in Proteins, Structures and Molecular Principles (Creighton, Ed., W. H. Freeman and Company, New York (1984)); Introduction to Protein Structure (C. Branden and J. Tooze, eds., Garland Publishing, New York, N.Y. (1991)); and Thornton et al. Nature 354:105 (1991), which are each incorporated herein by reference.
- antibodies of the present disclosure may be chemically bonded with polymers, lipids, or other moieties.
- the binding agents may comprise at least one of the CDRs described herein incorporated into a biocompatible framework structure.
- the biocompatible framework structure comprises a polypeptide or portion thereof that is sufficient to form a conformationally stable structural support, or framework, or scaffold, which is able to display one or more sequences of amino acids that bind to an antigen (e.g., CDRs, a variable region, etc.) in a localized surface region.
- Such structures can be a naturally occurring polypeptide or polypeptide “fold” (a structural motif), or can have one or more modifications, such as additions, deletions or substitutions of amino acids, relative to a naturally occurring polypeptide or fold.
- These scaffolds can be derived from a polypeptide of any species (or of more than one species), such as a human, other mammal, other vertebrate, invertebrate, plant, bacteria or virus.
- the biocompatible framework structures are based on protein scaffolds or skeletons other than immunoglobulin domains.
- suitable binding agents include portions of these antibodies, LCDR1, LCDR2, LCDR3, HCDR1, HCDR2, and/or HCDR3.
- the non-CDR portion of the antibody may be a non-protein molecule, wherein the binding agent cross-blocks the binding of an antibody disclosed herein to a target antigen.
- the non-CDR portion of the antibody may be a non-protein molecule in which the antibody exhibits a similar binding pattern to a target antigen in a competition binding assay as that exhibited by at least one of antibodies disclosed herein.
- the non-CDR portion of the antibody may be composed of amino acids, wherein the antibody is a recombinant binding protein or a synthetic peptide, and the recombinant binding protein cross-blocks the binding of an antibody disclosed herein to a target antigen and/or neutralizes a target antigen.
- the non-CDR portion of the antibody may be composed of amino acids, wherein the antibody is a recombinant antibody, and the recombinant antibody exhibits a similar binding pattern to a target antigen in the target epitope competition binding assay (described hereinbelow) as that exhibited by at least one of the antibodies disclosed herein, and/or neutralizes the target antigen.
- an antibody comprises one or more of HCDR1, HCDR2, HCDR3, LCDR1, LCDR2 and LCDR3 as described above, it may be obtained by expression from a host cell containing DNA coding for these sequences.
- a DNA coding for each CDR sequence may be determined on the basis of the amino acid sequence of the CDR and synthesized together with any desired antibody variable region framework and constant region DNA sequences using oligonucleotide synthesis techniques, site-directed mutagenesis and polymerase chain reaction (PCR) techniques as appropriate.
- DNA coding for variable region frameworks and constant regions is widely available to those skilled in the art from genetic sequences databases such as GenBank®.
- the heavy chain and the light chain of the antibody are expressed from a single DNA construct. In some embodiments, the heavy chain and the light chain of the antibody are expressed from two or more separate DNA constructs.
- the DNA encoding an antibody of the present disclosure or fragment thereof may be propagated and expressed according to any of a variety of well-known procedures for nucleic acid excision, ligation, transformation, and transfection using any number of known expression vectors.
- expression of an antibody fragment may be preferred in a prokaryotic host, such as Escherichia coli (see, e.g., Pluckthun et al., 1989 Methods Enzymol.178:497-515).
- expression of the antibody or a fragment thereof may be preferred in a eukaryotic host cell, including yeast (e.g., Saccharomyces cerevisiae, Schizosaccharomyces pombe, and Pichia pastoris), animal cells (including mammalian cells) or plant cells.
- yeast e.g., Saccharomyces cerevisiae, Schizosaccharomyces pombe, and Pichia pastoris
- animal cells including mammalian cells
- suitable animal cells include, but are not limited to, myeloma (such as a mouse NSO line), COS, CHO, or hybridoma cells.
- plant cells include tobacco, corn, soybean, and rice cells.
- One or more replicable expression vectors containing DNA encoding an antibody variable and/or constant region may be prepared and used to transform an appropriate cell line, for example, a non-producing myeloma cell line, such as a mouse NSO line or a bacteria, such as E. coli, in which production of the antibody will occur.
- an appropriate cell line for example, a non-producing myeloma cell line, such as a mouse NSO line or a bacteria, such as E. coli, in which production of the antibody will occur.
- the DNA sequence in each vector should include appropriate regulatory sequences, particularly a promoter and leader sequence operatively linked to the variable domain sequence.
- Particular methods for producing antibodies in this way are generally well-known and routinely used. For example, basic molecular biology procedures are described by Maniatis et al.
- DNA sequencing can be performed as described in Sanger et al. (PNAS 74:5463, (1977)) and the Amersham International plc sequencing handbook, and site directed mutagenesis can be carried out according to methods known in the art (Kramer et al., Nucleic Acids Res. 12:9441, (1984); Kunkel Proc. Natl. Acad. Sci.
- the antigen-binding domains for use in the ADCs provided herein encompass proteins having amino acid sequences that vary from those of the described antibodies but that retain the ability to bind the target proteins.
- Such variant antigen-binding domains comprise one or more additions, deletions, or substitutions of amino acids when compared to parent sequence, but exhibit biological activity that is essentially equivalent to that of the described antibodies.
- Two antigen-binding domains are considered bioequivalent if, for example, they are pharmaceutical equivalents or pharmaceutical alternatives whose rate and extent of absorption do not show a significant difference when administered at the same molar dose under similar experimental conditions, either single does or multiple doses.
- antigen-binding domains will be considered equivalents or pharmaceutical alternatives if they are equivalent in the extent of their absorption but not in their rate of absorption and yet may be considered bioequivalent because such differences in the rate of absorption are intentional and are reflected in the labeling, are not essential to the attainment of effective body drug concentrations on, e.g., chronic use, and are considered medically insignificant for the particular drug product studied.
- two antigen-binding domains are bioequivalent if there are no clinically meaningful differences in their safety, purity, and potency.
- two antigen-binding domains are bioequivalent if a patient can be switched one or more times between the reference product and the biological product without an expected increase in the risk of adverse effects, including a clinically significant change in immunogenicity, or diminished effectiveness, as compared to continued therapy without such switching.
- two antigen-binding domains are bioequivalent if they both act by a common mechanism or mechanisms of action for the condition or conditions of use, to the extent that such mechanisms are known.
- Bioequivalence may be demonstrated by in vivo and in vitro methods.
- Bioequivalence measures include, e.g., (a) an in vivo test in humans or other mammals, in which the concentration of the antigen-binding domain or its metabolites is measured in blood, plasma, serum, or other biological fluid as a function of time; (b) an in vitro test that has been correlated with and is reasonably predictive of human in vivo bioavailability data; (c) an in vivo test in humans or other mammals in which the appropriate acute pharmacological effect of the antigen-binding domain (or its target) is measured as a function of time; and (d) in a well- controlled clinical trial that establishes safety, efficacy, or bioavailability or bioequivalence of an antigen-binding domain.
- Bioequivalent variants of antigen-binding domains for use in the ADCs provided herein may be constructed by, for example, making various substitutions of residues or sequences or deleting terminal or internal residues or sequences not needed for biological activity.
- cysteine residues not essential for biological activity can be deleted or replaced with other amino acids to prevent formation of unnecessary or incorrect intramolecular disulfide bridges upon renaturation.
- bioequivalent antigen- binding domains may include variants comprising amino acid changes which modify the glycosylation characteristics of the antigen-binding domain, e.g., mutations which eliminate or remove glycosylation. 6.
- the antigen-binding domains for use in the ADCs provided herein bind to a human target protein but not to target protein from other species. In other embodiments, the antigen-binding domains for use in the ADCs provided herein bind to a human target protein and to a target protein from one or more non-human species.
- the antigen-binding domains for use in the ADCs provided herein may bind to a human target protein and may bind or not bind, as the case may be, to one or more of mouse, rat, guinea pig, hamster, gerbil, pig, cat, dog, rabbit, goat, sheep, cow, horse, camel, cynomologous, marmoset, rhesus or chimpanzee target protein.
- the antigen-binding domains specifically bind human target protein and cynomolgus monkey (e.g., Macaca fascicularis) target protein.
- antigen-binding domains for use herein bind human target protein but do not bind, or bind only weakly, to cynomolgus monkey target protein. 7.
- the antigen binding domain (ABD) for use in the ADCs provided herein can have binding specificity for any antigen (target protein) deemed suitable to those of skill in the art.
- the antigen is a transmembrane molecule (e.g., receptor) or a surface protein.
- HBV Hepatitis B virus
- HBV sAg HBV surface antigen
- Current therapy with nucleoside analogs reduces HBV load in plasma but rarely is accompanied by HBV sAg loss. Thus, the nucleoside therapy needs to be given lifelong to prevent viral rebound.
- Some embodiments of the present disclosure relate to an ADC targeting an HBV antigen.
- the ADC can be used for treatment of Hepatitis B.
- the ADC comprises an ABD specific to HBV sAg, where HBV sAg could relate to a non-infectious HBV sAg particle, infectious HBV virion, or HBV sAg expressing cells.
- the ABD comprises the heavy chain and light chain of an antibody specific to HBV sAg.
- the ABD comprises the HCDR1, HCDR2, HCDR3, LCDR1, LCDR2 and LCDR3 of an antibody specific to HBV sAg .
- the ABD is an antibody specific to HBV sAg .
- the HBV antigen is HBV sAg available from Prospect Bio with Cat No. HBS-872.
- ABD against a tumor antigen Some embodiments of the present disclosure relate to an ADC targeting a tumor antigen.
- the ADC can be used for treatment of cancer.
- the ADC comprises an ABD specific to a tumor antigen.
- the antigen is expressed on a tumor.
- the binding agents interact with or bind to tumor antigens, including antigens specific for a type of tumor or antigens that are shared, overexpressed, or modified on a particular type of tumor.
- the antigen is expressed on solid tumors.
- antigens include, but are not limited to, lipoproteins; alpha1-antitrypsin; a cytotoxic T-lymphocyte associated antigen (CTLA), such as CTLA-4; vascular endothelial growth factor (VEGF); receptors for hormones or growth factors; protein A or D; fibroblast growth factor receptor 2 (FGFR2), EpCAM, GD3, FLT3, PSMA, PSCA, MUC1, MUC16, STEAP, STEAP2, CEA, TENB2, EphA receptors, EphB receptors, folate receptor, FOLRI, mesothelin, cripto, alphavbeta6, integrins, VEGF, VEGFR, EGFR, transferrin receptor, IRTA1, IRTA2, IRTA3, IRTA4, IRTA5; CD proteins such as CD2, CD3, CD4, CD5, CD6, CD8, CD11, CD14, CD19, CD20, CD21, CD22, CD25, CD26, CD28, CD30, CD
- the antigen is PRLR or HER2. In some embodiments, the antigen is HER2. In some embodiments, the antigen is human HER2. In some embodiments, the antigen is STEAP2. In some embodiments the antigen is human STEAP2. In some embodiments, the MAGE proteins are selected from MAGE-1, -2, -3, -4, - 6, and -12. In some embodiments, the GAGE proteins are selected from GAGE-1 and GAGE- 2.
- the antibody comprises a glutamine residue at one or more heavy chain positions numbered 295 in the EU numbering system. In the present disclosure, this position is referred to as glutamine 295, or as Gln295, or as Q295.
- the antibody can be engineered to comprise a glutamine residue.
- the antibody comprises one or more N297Q mutations. Techniques for modifying an antibody sequence to include a glutamine residue are within the skill of those in the art (see, e.g., Ausubel et al. Current Protoc. Mol. Biol. (John Wiley & Sons)).
- the antibody contains a Q295 residue, an N297Q mutation, or one or more engineered LLQG (SEQ ID NO: 1), LLQGG (SEQ ID NO: 2), LLQLLQG (SEQ ID NO: 3), LLQYQG (SEQ ID NO: 4), LLQGA (SEQ ID NO: 5), LLQGSG (SEQ ID NO: 6), SLLQG (SEQ ID NO: 7), LQG, LLQLQ (SEQ ID NO: 9), LLQLLQ (SEQ ID NO: 10), LLQGR (SEQ ID NO: 11), LLQYQGA (SEQ ID NO: 12), LQGG (SEQ ID NO: 13), LGQG (SEQ ID NO: 14) or LLQLLQGA (SEQ ID NO: 15) sites, a payload of Formula I where R 3 is -alkylene-Y, - alkylene-arylene-Y, -heteroalkylene-Y
- R 10 is -alkylene-NH-, -heteroalkylene-NH- or -heteroalkylene-arylene-NH-.
- R 10 is -alkylene-NH-.
- R 10 is -heteroalkylene-NH-.
- R 10 is -heteroalkylene-arylene-NH-.
- R 10 is -CH 2 -NH-, -CH 2 -O- heteroalkylene-NH-, or -CH 2 -O-alkylene-NH-.
- R 10 is - CH 2 OCH 2 CH 2 NH-, -CH 2 OCH 2 CH 2 CH 2 CH 2 NH-, -CH 2 OCH 2 CH 2 OCH 2 CH 2 NH-, -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 NH-, -CH 2 OCH 2 NHC(O)CH 2 NH-, -CH 2 OCH 2 -(4- NH-1-phenyl), -CH 2 OCH(NH-)CH 2 OH or -CH 2 NH-.
- R 10 is -CH 2 OCH 2 CH 2 NH-, -CH 2 OCH 2 CH 2 CH 2 CH 2 NH-, -CH 2 OCH 2 CH 2 OCH 2 CH 2 NH-, -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 NH-, -CH 2 OCH 2 NHC(O)CH 2 NH-, -CH 2 OCH 2 -(4- NH-1-phenyl) or -CH 2 NH-.
- the ADC provided herein has the formula ABD-P4, ABD-P5, ABD-P7, ABD-P9, ABD-P11, ABD-P12, ABD-P19, ABD-P21, ABD-P24, ABD-P30, or ABD- P34, wherein ABD is attached to the payload (i.e., TLR7 agonist) on the amino group of R 3 .
- ABD is attached to the payload (i.e., TLR7 agonist) on the amino group of R 3 .
- the ADCs provided herein for use in the compositions and methods provided herein are prepared from linker-TLR7 agonist of Formula III and have Formula VI: or a pharmaceutically acceptable salt thereof, wherein: R 1 , R 2 , R 11 , R 12 , R 13 , R 14 , R 15 , R 16 , L 1 , ABD and x are as defined elsewhere for Formula III; and k is an integer from one to thirty.
- the ADC provided herein has the formula ABD-L 1 -P1, ABD- L 1 -P2, ABD-L 1 -P6, ABD-L 1 -P8, ABD-L 1 -P17, ABD-L 1 -P18, ABD-L 1 -P19, ABD-L 1 -P20, ABD-L 1 - P23, ABD-L 1 -P27, ABD-L 1 -P29, ABD-L 1 -P32, ABD-L 1 -P33, ABD-L 1 -P37, or ABD-L 1 -P39, where ABD-L 1 is attached to the payload (i.e., TLR7 agonist).
- the payload i.e., TLR7 agonist
- ADCs comprising an ABD, linker and TLR7 agonist.
- Each component of the ADCs i.e., ABD, linker, and TLR7 agonist
- the method involves the step of partial reduction of an antigen-binding domain with tris(2-carboxyethyl)phosphine (TCEP) followed by reaction of reduced cysteine residues with maleimide functionalized linker- payload (i.e., TLR7 agonist).
- TCEP tris(2-carboxyethyl)phosphine
- an antigen-binding domain is partially reduced via addition of 1.5 - 3.0- fold molar excess of TCEP in PBS pH 7.4 and 2 mM ethylenediaminetetraacetic acid (EDTA) for 2 h at 37 °C.
- the reduced antigen-binding domain may be buffer exchanged into PBS with 1% w/v polysorbate 20.
- Linker-payloads can be added at a linker-payload / antigen-binding domain molar ratio of 5 - 10 and reacted for an additional 2 h at 25 °C in the presence of 12% v/v of dimethyl sulfoxide (DMSO).
- the mixture may be purified, e.g., via size exclusion chromatography (SEC) (AKTA pure, Superdex 200 Increase) to afford the ADCs provided herein.
- SEC size exclusion chromatography
- the reaction of ABD and the payload i.e., TLR7 agonist
- the transglutaminase enzyme is a murine transglutaminase enzyme.
- the tetrazine-linkers were designed to have two functions: (1) tetrazine-linker as a handle that has an additional chemical moiety (such as an amine) to be attached with an antibody while the tetrazine-moiety can react with a linker-payload to generate an ADC; (2) tetrazine-linker as the linker of the linker-payload that can be attached with an antibody-handle (Titas Deb, et al., Chem. Rev. 2021, 121, 12, 6850–6914; Astrid- Caroline Knall and Christian Slugovc. Chem. Soc. Rev., 2013, 42, 5131).
- the ADCs provided herein are selected from those in Table 3: VIII.
- Pharmaceutical Compositions [0290] In one aspect, the present disclosure provides a pharmaceutical composition comprising an ADC described herein a pharmaceutically acceptable carrier. In some embodiments, the pharmaceutical composition further comprises a target antigen (e.g., an HBV sAg). In some embodiments, the pharmaceutical composition comprises an immunocomplex (IC) of the ADC and the HBV sAg. [0291] The TLR7 agonists or ADCs can be formulated into suitable pharmaceutical preparations.
- the TLR7 agonists or ADCs described above are formulated into pharmaceutical compositions using techniques and procedures well known in the art (see, e.g., Ansel Introduction to Pharmaceutical Dosage Forms, Seventh Edition 1999).
- effective concentrations of one or more TLR7 agonists or ADCs or pharmaceutically acceptable salts is (are) mixed with a suitable pharmaceutical carrier.
- the concentrations of the TLR7 agonists or ADCs in the compositions are effective for delivery of an amount, upon administration, that treats, prevents, or ameliorates one or more of the symptoms and/or progression of a disease or disorder disclosed herein.
- the compositions are formulated for single dosage administration.
- the weight fraction of a TLR7 agonists or ADC is dissolved, suspended, dispersed or otherwise mixed in a selected carrier at an effective concentration such that the treated condition is relieved or ameliorated.
- Pharmaceutical carriers suitable for administration of the TLR7 agonists or ADCs provided herein include any such carriers known to those skilled in the art to be suitable for the particular mode of administration.
- the TLR7 agonist or ADC is included in the pharmaceutically acceptable carrier in an amount sufficient to exert a therapeutically useful effect in the absence of undesirable side effects on the subject treated.
- the therapeutically effective concentration may be determined empirically by testing the compounds in in vitro and in vivo systems described herein and well known to those of skill in the art, and then extrapolated therefrom for dosages for humans.
- the ADC is administered in a method to achieve a therapeutically effective concentration of the payload.
- a companion diagnostic see, e.g., Olsen D and Jorgensen J T, Front. Oncol., 2014 May 16, 4:105, doi: 10.3389/fonC.2014.00105
- a companion diagnostic see, e.g., Olsen D and Jorgensen J T, Front. Oncol., 2014 May 16, 4:105, doi: 10.3389/fonC.2014.00105
- the concentration of TLR7 agonist or ADC in the pharmaceutical composition will depend on absorption, tissue distribution, inactivation and excretion rates of the TLR7 agonist or ADC, the physicochemical characteristics of the TLR7 agonist or ADC, the dosage schedule, and amount administered as well as other factors known to those of skill in the art. For example, the amount that is delivered is sufficient to ameliorate one or more of the symptoms of a disease or disorder disclosed herein.
- the compositions may be administered at once or may be divided into a number of smaller doses to be administered at intervals of time.
- compositions may include other active compounds to obtain desired combinations of properties.
- the TLR7 agonists or ADCs provided herein, or pharmaceutically acceptable salts thereof as described herein, may also be advantageously administered for therapeutic or prophylactic purposes together with another pharmacological agent known in the general art to be of value in treating one or more of the diseases or medical conditions referred to herein. It is to be understood that such combination therapy constitutes a further aspect of the compositions and methods of treatment provided herein.
- the pharmaceutical composition can be in any form appropriate for human or veterinary medicine, including a liquid, an oil, an emulsion, a gel, a colloid, an aerosol or a solid.
- the pharmaceutical composition can be formulated for administration by any route of administration appropriate for human or veterinary medicine, including enteral and parenteral routes of administration.
- the pharmaceutical composition is formulated for intravenous, intramuscular, or subcutaneous administration.
- IX. Dosing The compounds and pharmaceutical compositions provided herein may be dosed in certain therapeutically or prophylactically effective amounts, certain time intervals, certain dosage forms, and certain dosage administration methods as described below.
- the methods provided herein encompass treating a patient regardless of subject's age, although some diseases or disorders are more common in certain age groups.
- the TLR7 agonist or ADC provided herein, or a pharmaceutically acceptable salt thereof, can be administered repeatedly if necessary, for example, until the subject experiences stable disease or regression, or until the subject experiences disease progression or unacceptable toxicity.
- the TLR7 agonist or ADC provided herein, or a pharmaceutically acceptable salt thereof can be administered once daily (QD), or divided into multiple daily doses such as twice daily (BID), three times daily (TID), and four times daily (QID).
- the administration can be continuous (i.e., daily for consecutive days or every day), intermittent, e.g., in cycles (i.e., including days, weeks, or months of rest without drug).
- the term "daily” is intended to mean that a therapeutic compound, such as the TLR7 agonist or ADC provided herein, or a pharmaceutically acceptable salt thereof, is administered once or more than once each day, for example, for a period of time.
- continuous is intended to mean that a therapeutic compound, such as the TLR7 agonist or ADC provided herein, or a pharmaceutically acceptable salt thereof, is administered daily for an uninterrupted period of at least 10 days to 52 weeks.
- intermittent or “intermittently” as used herein is intended to mean stopping and starting at either regular or irregular intervals.
- intermittent administration of the TLR7 agonist or ADC provided herein, or a pharmaceutically acceptable salt thereof is administration for one to six days per week, administration in cycles (e.g., daily administration for two to eight consecutive weeks, then a rest period with no administration for up to one week), or administration on alternate days.
- cycling as used herein is intended to mean that a therapeutic compound, such as the TLR7 agonist or ADC provided herein, or a pharmaceutically acceptable salt thereof, is administered daily or continuously but with a rest period. In some such embodiments, administration is once a day for two to six days, then a rest period with no administration for five to seven days.
- a method of treating a subject with a TLR7 agonist or ADC provided herein, or a pharmaceutically acceptable salt thereof is provided. In some embodiments, a method of treating a subject with a pharmaceutical composition comprising a TLR7 agonist or ADC provided herein, or a pharmaceutically acceptable salt thereof, is provided.
- the pharmaceutical composition comprises any of the TLR7 agonists or ADCs disclosed herein, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
- the TLR7 agonists or ADCs provided herein are useful, inter alia, for the treatment, prevention and/or amelioration of any disease or disorder associated with or mediated by expression, signaling or activity of the target protein of the antigen-binding domain.
- the ADCs provided herein are used to treat primary and/or metastatic tumors arising in the brain and meninges, oropharynx, lung and bronchial tree, gastrointestinal tract, male and female reproductive tract, muscle, bone, skin and appendages, connective tissue, spleen, immune system, blood forming cells and bone marrow, liver and urinary tract, and special sensory organs such as the eye.
- the TLR7 agonists or ADCs provided herein are used to treat one or more of the following cancers: acute myelogenous leukemia, adult T-cell leukemia, astrocytomas, bladder cancer, breast cancer, PRLR positive (PRLR+) breast cancer, cervical cancer, cholangiocarcinoma, chronic myeloid leukemia, colon cancer, colorectal cancer, endometrial cancer, esophageal cancer, gastric cancer, glioblastomata, head and neck cancer (e.g., head and neck squamous cell carcinoma (HNSCC)), Kaposi's sarcoma, kidney cancer, leiomyosarcomas, liver cancer, lung cancer (e.g., small cell lung cancer, non-small cell lung cancer (NSCLC)), lymphomas, malignant gliomas, malignant mesothelioma, melanoma, mesothelioma, malignant mesothelioma, MF
- the cancer is breast cancer. In some embodiments, the cancer is prostate cancer. [0308] In some embodiments, the subject has chronic hepatitis B. In some embodiments, the ADCs provided herein are used to treat chronic hepatitis B. [0309] In some embodiments, the subject is diagnosed to have chronic hepatitis B. In some embodiments, the subject has elevated circulating HBV DNA or HBV sAg in serum prior to administration of the ADC or the pharmaceutical composition. In some embodiments, the treatment method provided herein further comprises the step of measuring circulating HBV DNA or HBV sAg in serum of the subject before administration of the ADC or the pharmaceutical composition.
- the treatment method provided herein further comprises the step of measuring circulating HBV DNA or HBV sAg in serum of the subject to assess therapeutic efficacy of the ADC or the pharmaceutical composition after administration.
- the TLR7 agonists or ADCs may be administered as a monotherapy (i.e., as the only therapeutic agent) or in combination with one or more additional therapeutic agents (examples of which are described elsewhere herein). VII.
- the TLR7 agonists or ADCs provided herein may be co-formulated with and/or administered in combination with one or more additional therapeutically active component(s) selected from a MET antagonist (e.g., an anti-MET antibody (e.g., onartuzumab, emibetuzumab, and H4H14639D) or small molecule inhibitor of MET), an EGFR antagonist (e.g., an anti-EGFR antibody (e.g., cetuximab or panitumumab) or small molecule inhibitor of EGFR (e.g., gefitinib or erlotinib)), an antagonist of another EGFR family member such as Her2/ErbB2, ErbB3 or ErbB4 (e.g., anti-ErbB2 (e.g., trastuzumab or T-DM1 ⁇ KADCYLA® ⁇ ), anti-ErbB3 or anti-ErbB4 antibody or small molecule inhibitor of ErbB2, Er
- a PD-1 inhibitor such as an anti-PD-1 antibody can be combined with a TLR7 agonist or ADC as described herein.
- the TLR7 agonists or ADCs provided herein may also be administered and/or co- formulated in combination with antivirals, antibiotics, analgesics, corticosteroids, steroids, oxygen, antioxidants, COX inhibitors, cardioprotectants, metal chelators, IFN-gamma, and/or NSAIDs.
- the additional therapeutically active component(s) e.g., any of the agents listed above or derivatives thereof, may be administered just prior to, concurrent with, or shortly after the administration of a TLR7 agonist or ADC provided herein.
- the term “in combination” includes the use of more than one therapy (e.g., one or more prophylactic and/or therapeutic agents). However, the use of the term “in combination” does not restrict the order in which therapies (e.g., prophylactic and/or therapeutic agents) are administered to a subject with a disease or disorder.
- a first therapy (e.g., an ADC provided herein) can be administered prior to (e.g., 5 minutes, 15 minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 12 hours, 24 hours, 48 hours, 72 hours, 96 hours, 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 8 weeks, or 12 weeks before), concomitantly with, or subsequent to (e.g., 5 minutes, 15 minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 12 hours, 24 hours, 48 hours, 72 hours, 96 hours, 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 8 weeks, or 12 weeks after) the administration of a second therapy (e.g., a prophylactic or therapeutic agent) to the subject.
- a second therapy e.g., a prophylactic or therapeutic agent
- R 1 is halo, -NHR 4 , -OR 4 , -NH-OR 4 or -R 4 ;
- R 2 is halo, or alkoxy;
- R 3 is -CONHR 23 , -alkylene-Y, -alkylene-arylene-Y, -heteroalkylene-Y, - heteroalkylene-arylene-Y, -(hydroxy)heteroalkylene-Y, -(amino)heteroalkylene-Y, or - alkylene-PEG-Y;
- R 23 is H, alkyl or aryl;
- R 4 is alkyl optionally substituted with alkoxy or heteroalkyl;
- X is CH or N;
- Y is -OH, Gly, -NR 5 R 6 or -COZ;
- Z is -OH or -NR 7 R 8 ;
- R 5 and R 6 are selected from (i), (ii), and (iii): (
- Embodiment 3 The compound of embodiment 1 or 2 selected with the proviso that R 4 is not substituted with hydroxyl.
- Embodiment 4. The compound of any one of embodiments 1-3 selected with the proviso that the alkylene and heteroalkylene portions of R 3 are not substituted with oxo.
- Embodiment 7 The compound of any one of embodiments 1-6, wherein R 1 is halo.
- Embodiment 8 The compound of any one of embodiments 1-6, wherein R 1 is -NHR 4 .
- Embodiment 9. The compound of any one of embodiments 1-6, wherein R 1 is -OR 4 .
- Embodiment 10. The compound of any one of embodiments 1-6, wherein R 1 is -NH-OR 4 .
- Embodiment 11 The compound of any one of embodiments 1-6, wherein R 1 is -R 4 .
- R 3 is -C(Me) 2 OH, -CO 2 H -CH 2 OCH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 CH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 OH, -CH 2 OCH 2 CH 2 OCH 2 CH 2 OH, -CH 2 OCH 2 CH 2 OCH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 OH, -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 -1-piperazinyl, -CH 2 OCH 2 NHC(O)CH 2 NH 2 , -CH 2 OCH 2 -(4-NH 2 -1-phenyl), -CH 2 OCH 2 COOH, -CH 2 OCH 2 NHC(O)CH 2 NH 2 , -CH 2 OCH 2 -(4-
- Embodiment 33 The compound of any one of embodiments 1-22, wherein R 3 is -C(Me) 2 OH, -CO 2 H, -CH 2 OCH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 CH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 OH, - CH 2 OCH 2 CH 2 OCH 2 CH 2 OH, -CH 2 OCH 2 CH 2 OCH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 OH, -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 -1-piperazinyl, -CH 2 OCH 2 NHC(O)CH 2 NH 2 , -CH 2 OCH 2 -(4-NH 2 -1-phenyl), -CH 2 OCH 2 NHC(O)CH 2 NH 2
- Embodiment 34 The compound of any one of embodiments 1-33, wherein R 4 is n-butyl, n- pentyl, n-hexyl or ethoxyethyl.
- Embodiment 35 The compound of any one of embodiments 1-33, wherein R 4 is n-butyl.
- Embodiment 36 The compound of any one of embodiments 1-33, wherein R 4 is n-pentyl.
- Embodiment 37 The compound of any one of embodiments 1-33, wherein R 4 is n-hexyl.
- Embodiment 38 The compound of any one of embodiments 1-33, wherein R 4 is ethoxyethyl.
- Embodiment 39 The compound of any one of embodiments 1-33, wherein R 4 is ethoxyethyl.
- Embodiment 45 The compound of any one of embodiments 1-42, wherein Y is Gly.
- Embodiment 45 The compound of any one of embodiments 1-42, wherein Y is -NR 5 R 6 .
- Embodiment 46 The compound of any one of embodiments 1-42, wherein Y is -COZ.
- Embodiment 47 The compound of any one of embodiments 1-42, wherein Y is -OH, Gly, - NH 2 , 1-piperazinyl, -COOH, -COOEt, -CONPr 2 or -CO-1-piperazinyl.
- Embodiment 48 The compound of any one of embodiments 1-47, wherein Z is -OH.
- Embodiment 49 The compound of any one of embodiments 1-47, wherein Z is -OH.
- Embodiment 50 The compound of any one of embodiments 1-47, wherein Z is alkoxy.
- Embodiment 50 The compound of any one of embodiments 1-47, wherein Z is -NR 7 R 8 .
- Embodiment 51 The compound of any one of embodiments 1-47, wherein Z is -OH, ethoxy, -N-n-Pr 2 or 1-piperazinyl.
- Embodiment 52 The compound of any one of embodiments 1-47, wherein Z is -OH or 1- piperazinyl.
- Embodiment 53 The compound of any one of embodiments 1-52, wherein R 7 and R 8 are each independently H or n-propyl, or, together with the N to which they are attached, form 1- piperazinyl.
- Embodiment 54 The compound of any one of embodiments 1-52, wherein R 7 and R 8 , together with the N to which they are attached, form 1-piperazinyl.
- Embodiment 55 A compound selected from:
- Embodiment 56 A compound of Formula II: or a pharmaceutically acceptable salt thereof, wherein: R 1 , R 2 and X are as defined for Formula I in embodiment 1; R 9 is a divalent group formed by removal of a terminal hydrogen (i.e., a hydrogen distal from the phenyl group to which R 9 is attached) from an R 3 group, as defined in embodiment 1; and L is any group or moiety that links, connects, or bonds to an antigen-binding domain ABD; with the proviso that the compound is not a compound of the formula: Embodiment 57.
- R 1 , R 2 and X are as defined for Formula I in embodiment 1
- R 9 is a divalent group formed by removal of a terminal hydrogen (i.e., a hydrogen distal from the phenyl group to which R 9 is attached) from an R 3 group, as defined in embodiment 1
- L is any group or moiety that links, connects, or bonds to an antigen-binding domain ABD; with the proviso that
- R 9 is -C(Me) 2 O-, C(O)-, - CH 2 OCH 2 CH 2 NH-, -CH 2 OCH 2 CH 2 CH 2 CH 2 NH-, -CH 2 OCH 2 CH 2 OCH 2 CH 2 O-, - CH 2 OCH 2 CH 2 OCH 2 CH 2 NH-, -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 O-, -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 O-, -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 NH-, -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 -1-piperazin-4-yl-, -CH 2 OCH 2 NHC(O)CH 2 NH-, -CH 2 OCH 2 -((4-NH-)-1-phenyl), -CH 2 OCH 2 COO-, -CH 2 OCH 2 CH 2 OCH 2 CO-,
- Embodiment 66 The compound of any one of embodiments 55-65, wherein Y 1 is -O-.
- Embodiment 67 The compound of any one of embodiments 55-65, wherein Y 1 is Gly.
- Embodiment 68 The compound of any one of embodiments 55-65, wherein Y 1 is -NR 5 -.
- Embodiment 69 The compound of any one of embodiments 55-65, wherein Y 1 is -COZ 1 .
- Embodiment 70 The compound of any one of embodiments 55-65, wherein Y 1 is -O-, Glycine, -NH-, 1-piperazin-4-yl, -COO- or -CO-1-piperazin-4-yl.
- Embodiment 71 The compound of embodiment 69, wherein Z 1 is -O-.
- Embodiment 72 The compound of embodiment 69, wherein Z 1 is -NR 7 -.
- Embodiment 73 The compound of embodiment 69, wherein Z 1 is -O- or 1-piperazin-4-yl.
- Embodiment 74 The compound of embodiment 68, wherein R 5 is H.
- Embodiment 75 The compound of embodiment 68, wherein R 5 is alkyl.
- Embodiment 76 The compound of any one of embodiments 55-75, wherein L is non- cleavable under physiological conditions.
- Embodiment 77 The compound of any one of embodiments 55-75, wherein L is cleavable under physiological conditions.
- Embodiment 78 The compound of embodiment 77, wherein L is an acid-labile linker, a hydrolysis-labile linker, an enzymatically cleavable linker, a reduction labile linkers or a self- immolative linker.
- Embodiment 79 The compound of any one of embodiments 55-78, wherein L is or comprises a peptide, a carbohydrate, a glucuronide, a polyethylene glycol (PEG) unit, a hydrazone, a mal-caproyl unit, a dipeptide unit, a valine-citruline unit, or a para-aminobenzyl (PAB) unit.
- PEG polyethylene glycol
- PAB para-aminobenzyl
- Embodiment 81 The compound of any one of embodiments 55-80, wherein L comprises a self-immolative group.
- Embodiment 82 The compound of any one of embodiments 55-81, wherein L comprises p- aminobenzyl (PAB) or p-aminobenzyloxycarbonyl (PABC).
- Embodiment 83 The compound of any one of embodiments 55-82, wherein L comprises a maleimido, an N-hydroxysuccinimido ester or cyclooctynyl group.
- Embodiment 84 The compound of any one of embodiments 55-83, wherein L is a group selected from 2-maleimido-1-ethyl, 2-maleimidoacetyl, 3-maleimidopropanoyl, , OH ,
- Embodiment 85 A compound selected from:
- Embodiment 86 A compound of Formula III: or a pharmaceutically acceptable salt thereof, wherein: R 1 , R 2 and X are as defined for Formula I in embodiment 1; L is any group or moiety that links, connects, or bonds to an antigen-binding domain ABD; R 11 and R 12 are, independently, hydrogen, alkyl, alkoxy, alkenyl, alkynyl, aryl, arylalkyl, heteroaryl, alkylene, or heteroalkylene, wherein when R 11 is alkylene or heteroalkylene, the alkylene or heteroalkylene is further bonded to R 13 to form a 3-, 4-, 5-, 6-, 7-, or 8-membered heterocyclyl; R 14 is hydrogen, alkylene, heteroalkylene, or an amino acid side chain, wherein when R 14 is alkylene or heteroalkylene, the alkylene or heteroalkylene is further bonded to R 13 to form a 4-, 5-, or
- Embodiment 87 The compound of embodiment 86, wherein R 11 and R 12 are, independently, hydrogen, alkyl, alkoxy, alkenyl, alkynyl, aryl, arylalkyl, heteroaryl, alkylene, or heteroalkylene, wherein when R 11 is alkylene or heteroalkylene, the alkylene or heteroalkylene is further bonded to R 13 to form a 4-, 5-, or 6- membered heterocyclyl; R 14 is hydrogen, alkylene, heteroalkylene, or an amino acid side chain, wherein when R 14 is alkylene or heteroalkylene, the alkylene or heteroalkylene is further bonded to R 13 to form a 4-, 5-, or 6-membered heterocyclyl; R 13 is hydrogen, alkyl, alkylene, or heteroalkylene, wherein when R 13 is alkylene or heteroalkylene, the alkylene or heteroalkylene is further bonded to R 11 or R 14 to form a 4-, 5-, or 6-membere
- Embodiment 88 The compound of embodiment 86 or embodiment 87, wherein the TLR7 agonist used in preparing the compound is P1, P2, P6, P8, P17, P18, P19, P20, P23, P27, P29, P32, P33, P37, P39, P41, P42, or P43.
- Embodiment 89 An antibody-drug-conjugate (ADC), comprising the compound of any one of embodiments 1-88 or compounds of the formulae: Embodiment 90.
- ADC antibody-drug-conjugate
- R 1 , R 2 and X are as defined for Formula I in embodiment 1;
- R 10 is -alkylene-NH-, -alkylene-arylene-NH-, -heteroalkylene-NH-, -heteroalkylene- arylene-NH-, -(hydroxy)heteroalkylene-NH-, -(amino)heteroalkylene-NH-, or -alkylene-PEG- NH-;
- ABD is an antibody that contains a Q295 residue, an N297Q mutation, and/or one or more engineered LLQG (SEQ ID NO: 1), LLQGG (SEQ ID NO: 2), LLQLLQG (SEQ ID NO: 3), LLQYQG (SEQ ID NO: 4), LLQGA (SEQ ID NO: 5), LLQGSG (SEQ ID NO: 6), SLLQG (SEQ ID NO: 7), LQG, LLQ
- Embodiment 93 The ADC of embodiment 92, wherein R 10 is -alkylene-NH-, -heteroalkylene- NH-, -heteroalkylene-arylene-NH-, -(hydroxy)heteroalkylene-NH-, -(amino)heteroalkylene- NH-, or -alkylene-PEG-NH-.
- Embodiment 94 The ADC of embodiment 92 or embodiment 93, wherein R 10 is -alkylene- NH-.
- Embodiment 95 The ADC of embodiment 92 or embodiment 93, wherein R 10 is - heteroalkylene-NH-.
- Embodiment 96 The ADC of embodiment 92 or embodiment 93, wherein R 10 is - heteroalkylene-NH-.
- ADC of any one of embodiments 89-102 having the formula ABD-P4, ABD-P5, ABD-P7, ABD-P9, ABD-P11, ABD-P12, ABD-P19, ABD-P21, ABD-P24, ABD-P30, ABD-P34 or ABD-P41, wherein ABD is attached to the payload (i.e., TLR7 agonist) on the amino group of R 3 .
- Embodiment 104 Embodiment 104.
- Embodiment 105 is a divalent linker; R 1 , R 2 , R 16 , R 11 , R 12 , R 13 , R 14 , R 15 , X, and x are as defined for Formula III in embodiment 86; and k is an integer from one to thirty.
- the ADC of embodiment 104 having the formula ABD-L 1 -P1, ABD-L 1 -P2, ABD-L 1 -P6, ABD-L 1 -P8, ABD-L 1 -P17, ABD-L 1 -P18, ABD-L 1 -P19, ABD-L 1 -P20, ABD-L 1 -P23, ABD-L 1 -P27, ABD-L 1 -P29, ABD-L 1 -P32, ABD-L 1 -P33, ABD-L 1 -P37, ABD-L 1 -P39, or ABD-L 1 - P42, where ABD-L 1 is attached to the payload (i.e., TLR7 agonist) on the alcohol group of R 3 .
- the payload i.e., TLR7 agonist
- Embodiment 106 The ADC of any one of embodiments 89-105, wherein ABD has binding specificity for a transmembrane molecule (e.g., receptor) expressed on a tumor.
- Embodiment 107. A pharmaceutical composition, comprising a compound of any one of embodiments 1-88 or an ADC of any one of embodiments 89-106, and a pharmaceutically acceptable carrier.
- Embodiment 108. A method of treating or diagnosing disease, comprising administering to a subject a compound of any one of embodiments 1-88 or an ADC of any one of embodiments 89-106 or a pharmaceutical composition of embodiment 107.
- Embodiment 109. The method of embodiment 108, wherein the method treats a disease.
- An antibody-drug-conjugate comprising a. an antigen-binding domain (ABD) having binding specificity to a hepatitis B virus surface antigen (HBV sAg); and b. a Toll-like receptor 7 (TLR7) agonist.
- ADC antigen-binding domain
- HBV sAg hepatitis B virus surface antigen
- TLR7 Toll-like receptor 7
- ADC of clause 2 wherein the ADC is according to Formula IV: or a pharmaceutically acceptable salt thereof, wherein: L 1 is a divalent linker; R 1 is H, halo, -NHR 4 , -OR 4 , -NH-OR 4 or -R 4 ; R 2 is H, halo, or alkoxy; R 4 is alkyl optionally substituted with alkoxy or heteroalkyl; R 9 is a divalent group formed by removal of a hydrogen from R 3 , R 3 being a group attached to the phenyl group at the position of R 9 ; R 3 is -COOH, -CONHR 23 , -alkylene-Y, -alkylene-arylene-Y, -heteroalkylene-Y, - heteroalkylene-arylene-Y, -(hydroxy)heteroalkylene-Y, -(amino)heteroalkylene-Y, or - alkylene-PEG-Y; R 23 is H, al
- R 3 is -C(Me) 2 OH, -CO 2 H, -CH 2 OCH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 CH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 OCH 2 CH 2 OH, -CH 2 OCH 2 CH 2 OCH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 OH, -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 NH 2 , -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 -1-piperazinyl, -CH 2 OCH 2 NHC(O)CH 2 NH 2 , -CH 2 OCH 2 -(4-NH 2 -1-phenyl), -CH 2 OCH 2 COOH,-CH 2 OCH 2 COOEt, -CH 2
- Clause 33 The ADC of any one of clauses 3-32, wherein R 4 is n-butyl, n-pentyl, n-hexyl or ethoxyethyl.
- Clause 34 The ADC of clause 33, wherein R 4 is n-butyl.
- Clause 35 The ADC of clause 33, wherein R 4 is n-pentyl.
- Clause 36 The ADC of clause 33, wherein R 4 is n-hexyl.
- Clause 37 The ADC of clause 33, wherein R 4 is ethoxyethyl.
- Clause 56 The ADC of any one of clauses 3-54, wherein R 9 is -alkylene-Y 1 -, - heteroalkylene-Y 1 -, -heteroalkylene-arylene-Y 1 -, -(hydroxy)heteroalkylene-Y 1 , - (amino)heteroalkylene-Y 1 , or -alkylene-PEG-Y 1 -.
- Clause 58 The ADC of clause 56, wherein R 9 is -heteroalkylene-Y 1 -.
- Clause 59 The ADC of any one of clauses 3-54, wherein R 9 is -alkylene-Y 1 -, - heteroalkylene-Y 1 -, -heteroalkylene-arylene-Y 1 -, -(hydroxy)heteroalkylene-Y 1 , - (a
- R 9 is -C(Me) 2 O-, C(O)-, -CH 2 OCH 2 CH 2 NH-, - CH 2 OCH 2 CH 2 CH 2 CH 2 NH-, -CH 2 OCH 2 CH 2 OCH 2 CH 2 O-, -CH 2 OCH 2 CH 2 OCH 2 CH 2 NH-, -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 O-, -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 NH-, -CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 -1-piperazin-4-yl-, -CH 2 OCH 2 NHC(O)CH 2 NH-, -CH 2 OCH 2 -((4-NH-)-1-phenyl), -CH 2 OCH 2 COO-, -CH 2 OCH 2 CH 2 OCH 2 CO-, -CH 2
- Clause 65 The ADC of any one of clauses 56-64, wherein Y 1 is -O-.
- Clause 66. The ADC of any one of clauses 56-64, wherein Y 1 is Gly.
- Clause 67. The ADC of any one of clauses 56-64, wherein Y 1 is -NR 5 -.
- Clause 68. The ADC of any one of clauses 56-64, wherein Y 1 is -COZ 1 , wherein Z 1 is -O-, -NR 7 -, -O-alkylene-, or 1-piperazin-4-yl.
- L 1 is or comprises a peptide, a carbohydrate, a glucuronide, a polyethylene glycol (PEG) unit, a hydrazone, a mal-caproyl unit, a dipeptide unit, a valine-citruline unit, or a para-aminobenzyl (PAB) unit.
- PEG polyethylene glycol
- PAB para-aminobenzyl
- L 1 comprises p-aminobenzyl (PAB) or p- aminobenzyloxycarbonyl (PABC).
- PAB p-aminobenzyl
- PABC p- aminobenzyloxycarbonyl
- L 1 comprises a maleimido, an N- hydroxysuccinimido ester or cyclooctynyl group.
- Clause 85 The ADC of any one of clauses 3-76, wherein L 1 is a group derived from 2- maleimido-1-ethyl, 2-maleimidoacetyl, 3-maleimidopropanoyl, , O O O O O O N H N N H N O O H O O N N N NH O , O NH 2 , ,
- R 1 , R 2 and X are as defined for Formula I in clause 3;
- L is any group or moiety that links to ABD;
- R 11 and R 12 are, independently, hydrogen, alkyl, alkoxy, alkenyl, alkynyl, aryl, arylalkyl, heteroaryl, alkylene, or heteroalkylene, wherein when R 11 is alkylene or heteroalkylene, the alkylene or heteroalkylene is further bonded to R 13 to form a 3-, 4-, 5-, 6-, 7-, or 8-membered heterocyclyl;
- R 14 is hydrogen, alkylene, heteroalkylene, or an amino acid side chain, wherein when R 14 is alkylene or heteroalkylene, the alkylene or heteroalkylene is further bonded to R 13 to form a 3-, 4-, 5-, 6-, 7-, or 8-membered heterocyclyl;
- R 13 is hydrogen, alkyl, alkylene, or heteroalkylene, wherein
- R 11 and R 12 are, independently, hydrogen, alkyl, alkoxy, alkenyl, alkynyl, aryl, arylalkyl, heteroaryl, alkylene, or heteroalkylene, wherein when R 11 is alkylene or heteroalkylene, the alkylene or heteroalkylene is further bonded to R 13 to form a 4-, 5-, or 6-membered heterocyclyl;
- R 14 is hydrogen, alkylene, heteroalkylene, or an amino acid side chain, wherein when R 14 is alkylene or heteroalkylene, the alkylene or heteroalkylene is further bonded to R 13 to form a 4-, 5-, or 6-membered heterocyclyl;
- R 13 is hydrogen, alkyl, alkylene, or heteroalkylene, wherein when R 13 is alkylene or heteroalkylene, the alkylene or heteroalkylene is further bonded to R 11 or R 14 to form a 4-, 5-, or 6-membered
- ADC of clause 3 wherein the ADC is according to Formula V: or a pharmaceutically acceptable salt thereof, wherein: R 1 , R 2 , and X are as defined for Formula In in clause 3; R 10 is -alkylene-NH-, -alkylene-arylene-NH-, -heteroalkylene-NH-, -heteroalkylene- arylene-NH-, -(hydroxy)heteroalkylene-NH-, -(amino)heteroalkylene-NH-, or - alkylene-PEG-NH-;
- ABD is an antibody that contains a Q295 residue, an N297Q mutation, and/or one or more engineered LLQG (SEQ ID NO: 1), LLQGG (SEQ ID NO: 2), LLQLLQG (SEQ ID NO: 3), LLQYQG (SEQ ID NO: 4), LLQGA (SEQ ID NO: 5), LLQGSG (SEQ ID NO: 6),
- Clause 92 The ADC of clause 91, wherein R 10 is -alkylene-NH-, -heteroalkylene-NH-, -heteroalkylene-arylene-NH-, -(hydroxy)heteroalkylene-NH-, -(amino)heteroalkylene-NH-, or - alkylene-PEG-NH-.
- Clause 93 The ADC of clause 92, wherein R 10 is -alkylene-NH-.
- Clause 94 The ADC of clause 92, wherein R 10 is -heteroalkylene-NH-.
- Clause 95 The ADC of clause 92, wherein R 10 is -heteroalkylene-arylene-NH-.
- Clause 96 The ADC of clause 92, wherein R 10 is -(hydroxy)heteroalkylene-NH-. Clause 97. The ADC of clause 92, wherein R 10 is -(amino)heteroalkylene-NH-. Clause 98. The ADC of clause 92, wherein R 10 is -alkylene-PEG-NH-. Clause 99. The ADC of clause 91 or 92, wherein R 10 is -CH 2 -NH-, -CH 2 -O-heteroalkylene- NH-, or -CH 2 -O-alkylene-NH-. Clause 100.
- R 10 is -CH 2 OCH 2 CH 2 NH-, - CH 2 OCH 2 CH 2 CH 2 CH 2 NH-, -CH 2 OCH 2 CH 2 OCH 2 CH 2 NH-, - CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 NH-, -CH 2 OCH 2 NHC(O)CH 2 NH-, -CH 2 OCH 2 -(4-NH-1-phenyl)-, -CH 2 OCH(NH-)CH 2 OH or -CH 2 NH-.
- R 10 is -CH 2 OCH 2 CH 2 NH-, - CH 2 OCH 2 CH 2 CH 2 CH 2 NH-, -CH 2 OCH 2 CH 2 OCH 2 CH 2 NH-, - CH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 2 CH 2 NH-, -CH 2 OCH 2 NHC(O)CH 2 NH-, -CH 2 OCH 2 -(4-NH-1-phenyl)- or -CH 2 NH-.
- the ADC of clause 3 comprising ABD linked to a compound selected from P4, P5, P7, P9, P11, P12, P19, P21, P24, P30, and P34 via an amino group of R 3 .
- Clause 103 The ADC of clause 2, wherein the ADC is according to Formula VI:
- L 1 is a divalent linker
- R 1 is H, halo, -NHR 4 , -OR 4 , -NH-OR 4 or -R 4
- R 2 is H, halo, or alkoxy
- R 4 is alkyl optionally substituted with alkoxy or heteroalkyl
- R 11 and R 12 are, independently, hydrogen, alkyl, alkoxy, alkenyl, alkynyl, aryl, arylalkyl, heteroaryl, alkylene, or heteroalkylene, wherein when R 11 is alkylene or heteroalkylene, the alkylene or heteroalkylene is further bonded to R 13 to form a 3-, 4-, 5-, 6-, 7-, or 8-membered heterocyclyl
- R 14 is hydrogen, alkylene, heteroalkylene, or an amino acid side chain, wherein when R 14 is alkylene or heteroalkylene, the alkylene or heteroalkylene is further bonded to R 13
- Clause 104 The ADC of clause 103, comprising ABD-L 1 linked to a compound selected from P1, P2, P6, P8, P17, P18, P19, P20, P23, P27, P29, P32, P33, P37, and P39.
- Clause 105 The ADC of clause 103 or 104, wherein k is 1, 2, 3, 4, or 5.
- Clause 106 The ADC of clause 103, wherein k is 2.
- Clause 107 The ADC of clause 103, wherein ABD comprises a heavy chain and C-terminus of the heavy chain is conjugated to L 1 .
- Clause 118. The ADC of any one of clauses 1-114, wherein said ABD comprises three heavy chain complementarity determining regions (CDRs) (HCDR1, HCDR2 and HCDR3) contained within a heavy chain variable region (HCVR) comprising the amino acid sequence set forth in SEQ ID NO: 25, and three light chain complementarity determining regions (CDRs) (LCDR1, LCDR2 and LCDR3) contained within a light chain variable region (LCVR) comprising the amino acid sequence set forth in SEQ ID NO: 29.
- CDRs heavy chain complementarity determining regions
- LCVR light chain complementarity determining regions
- HCDR1 comprises the amino acid sequence set forth in SEQ ID NO: 26
- HCDR2 comprises the amino acid sequence set forth in SEQ ID NO: 27
- HCDR3 comprises the amino acid sequence set forth in SEQ ID NO: 28
- LCDR1 comprises the amino acid sequence set forth in SEQ ID NO: 30
- LCDR2 comprises the amino acid sequence set forth in SEQ ID NO: 31
- LCDR3 comprises the amino acid sequence set forth in SEQ ID NO: 32.
- Clause 122 The ADC of clause 118 or 119, wherein said LCVR comprises the amino acid sequence of SEQ ID NO: 29.
- Clause 123 The ADC of claim 120, wherein said LCVR is a component of a light chain comprising the amino acid sequence of SEQ ID NO: 34.
- Clause 124. The ADC of any one of clauses 118-123, wherein said ABD is a component of an antibody or antigen-binding fragment thereof. Clause 125.
- the ADC of clause 1, wherein the TLR7 agonist with a divalent linker is any one of LP1-5, LP6A-6B, LP7A-7E, LP8A-8B, LP9, LP10A-10B, LP11A-11D, and LP12-15
- a pharmaceutical composition comprising the ADC of any one of clauses 1- 117 and one or more pharmaceutically acceptable carriers, excipients, or diluents.
- Clause 127. A method of treatment, comprising administering to a subject in need thereof an effective amount of the ADC of any one of clauses 1-117 or the pharmaceutical composition of clause 118. Clause 128. The method of clause 127, wherein the subject has Hepatitis B. Clause 129.
- Clause 134 The method of clause 133, wherein the step of administrating the ADC or the pharmaceutical composition is repeated twice, three times, or more. Clause 135. The method of clause 133 or 134, wherein the step of administrating the ADC or the pharmaceutical composition is repeated at least at 1-week intervals, at 2-week intervals, at 3-week intervals, or at 4-week intervals. Clause 136. The method of clause 133 or 134, wherein the step of administrating the ADC or the pharmaceutical composition is repeated at 1-week intervals, at 2-week intervals, at 3- week intervals, or at 4-week intervals. Clause 137.
- Payload P1 (4- ⁇ [2-Amino-4-(pentylamino)-5H-pyrrolo[3,2-d]pyrimidin-5-yl]methyl ⁇ -3- methoxyphenyl)methanol (P1) [0382] Following the general procedure V starting from intermediate Aa (65 mg, 0.16 mmol), payload P1 (35 mg, 43% yield, TFA salt) was obtained as a white solid. ESI m/z: 389.3 (M + H) + .
- Payload P20 [0384] (4- ⁇ [2-Amino-4-(butoxyamino)-5H-pyrrolo[3,2-d]pyrimidin-5-yl]methyl ⁇ -3- methoxyphenyl)methanol (P20) [0385] Following the general procedure V starting from intermediate Ba (0.15 g, 0.38 mmol), payload P1 (0.13 g, 72% yield, TFA salt) was obtained as a white solid. ESI m/z: 372.5 (M + H) + .
- Payload P23 [0387] [4-( ⁇ 2-Amino-4-[(2-ethoxyethyl)amino]-5H-pyrrolo[3,2-d]pyrimidin-5-yl ⁇ methyl)-3- methoxyphenyl]methanol (P23) [0388] Following the general procedure V starting from intermediate Ca (30 mg, 75 ⁇ mol), payload P23 (15 mg, 41% yield, TFA salt) was obtained as a white solid. ESI m/z: 372.3 (M + H) + .
- Payload P32 (4- ⁇ [2-Amino-4-(butoxyamino)-5H-pyrrolo[3,2-d]pyrimidin-5- yl]methyl ⁇ phenyl)methanol (P32) [0397] Following the general procedure V starting from intermediate Bb (82 mg, 0.22 mmol), payload P32 (70 mg, 69% yield, TFA salt) was obtained as a white solid. ESI m/z: 342.4 (M + H) + .
- Payload P3 [0413] 5-( ⁇ 2-Methoxy-4-[(piperazin-1-yl)methyl]phenyl ⁇ methyl)-N 4 -pentyl-5H-pyrrolo[3,2- d]pyrimidine-2,4-diamine (P3) [0414] Following the general procedure VI starting from P1, payload P3 (0.23 g, 31% yield, TFA salt) was obtained as a white solid. ESI m/z: 438.4 (M + H) + .
- Payload P22 [0430] 4- ⁇ [2-Amino-4-(butoxyamino)-5H-pyrrolo[3,2-d]pyrimidin-5-yl]methyl ⁇ -3- methoxybenzamide (P22) [0431] Following the general procedure VII starting from Ba, payload P22 (82 mg, 28% yield, TFA salt) was obtained as a white solid. ESI m/z: 385.5 (M + H) + .
- EXAMPLE 13 [0443] Payload P21 [0444] 5- ⁇ [4-(Aminomethyl)-2-methoxyphenyl]methyl ⁇ -N 4 -butoxy-5H-pyrrolo[3,2- d]pyrimidine-2,4-diamine (P21) [0445] To a stirred suspension of lithium aluminum tetrahydride (39 mg, 1.0 mmol) in anhydrous THF (6 mL) was added dropwise a solution of compound P22 (65 mg, 0.17 mmol) in anhydrous THF (4 mL) over 10 minutes at 0 o C under nitrogen protection. The reaction mixture was stirred at 65 o C for 4 hours which was monitored by LCMS.
- EXAMPLE 16 [0452] Payload P34 [0453] 5- ⁇ [4-(Aminomethyl)phenyl]methyl ⁇ -N 4 -(2-ethoxyethyl)-5H-pyrrolo[3,2-d]pyrimidine- 2,4-diamine (P34) [0454] Following the similar procedure as Payload P30 except substituting Cc for Bc, payload P34 (0.10 g, 60% yield, TFA salt) was obtained as a white solid. ESI m/z: 341.3 (M + H) + .
- the resulting suspension was stirred at -15 to 0 o C under nitrogen protection for 10 minutes.
- To the stirred mixture were then added tetrabutylammonium iodide (TBAI) (0.05 equiv.) and benzyl halide (1.0 equiv.), and the reaction mixture was stirred at -15 to 0 o C for 30 minutes, which was monitored by LCMS.
- the resulting mixture was quenched with methanol.
- the mixture (with Boc protection or propylidene protection) was directly used for the next step without further purification.
- the mixture was neutralized with TFA (to pH 6-7) and then concentrated in vacuo.
- the residual mixture was purified by prep-HPLC (5-95% acetonitrile in aq.
- Payload P4 [0463] 5-( ⁇ 4-[(2-Aminoethoxy)methyl]-2-methoxyphenyl ⁇ methyl)-N 4 -pentyl-5H-pyrrolo[3,2- d]pyrimidine-2,4-diamine (P4) [0464] Following the general procedures VIII and IX subsequently starting from benzyl bromide P4-1 reacting with N-Boc-aminoethanol P4-2, payload P4 (13 mg, 15% yield, di-TFA salt) was obtained as a white solid.
- EXAMPLE 23 [0468] Payload P6 [0469] 2- ⁇ 2-[(4- ⁇ [2-Amino-4-(pentylamino)-5H-pyrrolo[3,2-d]pyrimidin-5-yl]methyl ⁇ -3- methoxyphenyl)methoxy]ethoxy ⁇ ethan-1-ol (P6) [0470] Following the general procedure VIII starting from benzyl bromide P4-1 reacting with diethylene glycol P6-2, payload P6 (12 mg, 23% yield, TFA salt) was obtained as an off-white solid. ESI m/z: 458.1 (M + H) + .
- Payload P44 [0475] 1-(4- ⁇ [2-amino-4-(pentylamino)-5H-pyrrolo[3,2-d]pyrimidin-5-yl]methyl ⁇ -3- methoxyphenyl)-2,5,8,11-tetraoxatridecan-13-oic acid (P44) [0476] Following the general procedure VIII starting from benzyl chloride P3-1 reacting with Hydroxy-PEG 3 -CH 2 CO 2 t Bu (CAS: 518044-31-0), payload P44 (4.6 mg, 13% yield) was obtained as a white solid (tBu group was lost during the reaction). ESI m/z: 560.3 (M + H) + .
- Payload P45 2-(2- ⁇ 2-[(4- ⁇ [2-amino-4-(pentylamino)-5H-pyrrolo[3,2-d]pyrimidin-5-yl]methyl ⁇ -3- methoxyphenyl)methoxy]ethoxy ⁇ ethoxy)acetic acid (P45) [0479] Following the general procedure VIII starting from benzyl chloride P3-1 reacting with Hydroxy-PEG 2 -CH 2 CO 2 t Bu (CAS: 149299-82-1), payload P45 (3.9 mg, 15% yield) was obtained as a white solid (tBu group was lost during the reaction). ESI m/z: 516.3 (M + H) + .
- Payload P46 [0481] 2- ⁇ 2-[(4- ⁇ [2-amino-4-(pentylamino)-5H-pyrrolo[3,2-d]pyrimidin-5-yl]methyl ⁇ -3- methoxyphenyl)methoxy]ethoxy ⁇ acetic acid (P46) [0482] Following the general procedure VIII starting from benzyl chloride P3-1 reacting with tert-butyl 2-(2-hydroxyethoxy)acetate (CAS: 287174-32-7), payload P46 (3.2 mg, 12% yield) was obtained as a white solid (tBu group was lost during the reaction). ESI m/z: 472.3 (M + H) + .
- Payload P48 2-[(4- ⁇ [2-amino-4-(pentylamino)-5H-pyrrolo[3,2-d]pyrimidin-5-yl]methyl ⁇ -3- methoxyphenyl)methoxy]ethan-1-ol (P48) [0485] Following the general procedure VIII starting from benzyl chloride P3-1 reacting with ethylene glycol (CAS: 107-21-1), payload P48 (5 mg, 10% yield, TFA salt) was obtained as an white solid. ESI m/z: 414.3 (M + H) + .
- Boc-LP3- 2 (45 mg, ESI m/z: 581.5 (M + H) + ) as a light yellow solid, which was dissolved in DCM (5 mL).
- hydrochloride in dioxane (4 N, 5 mL) and the solution was stirred at room temperature for an hour until Boc was totally removed according to LCMS.
- the volatiles were removed in vacuo and the residue was purified by reversed phase flash chromatography (0-100% acetonitrile in aq.
- linker-payload LP7B (40 mg, 36% yield) as a white solid.
- linker-payload LP8A 9.2 mg, 17% yield from P9, TFA salt
- ESI m/z 501.0 (M/2 + H) + .
- ADCs antibody drug conjugates
- TCEP tris(2-carboxyethyl)phosphine
- EDTA ethylenediaminetetraacetic acid
- Linker-payloads were added at a linker- payload / antibody molar ratio of 5 - 10 and reacted for an additional 2 h at 25 °C in the presence of 12% v/v of dimethyl sulfoxide (DMSO).
- the reaction mixtures were purified via size exclusion chromatography (SEC) (AKTA pure, Superdex 200 Increase), formulated in PBS with 5% Glycerol and stored at -80 °C.
- the protein concentration was determined via UV spectrophotometer.
- ADC monomer purity was >90% by SEC.
- the ADCs were further characterized via hydrophobic interaction chromatography (HIC), and liquid chromatography electrospray ionization mass spectrometry (LC-ESI MS) to calculate drug-antibody ratio (DAR).
- HIC hydrophobic interaction chromatography
- LC-ESI MS liquid chromatography electrospray ionization mass spectrometry
- DAR drug-antibody ratio
- EXAMPLE 64 Conjugation of TLR7 Agonist to an Antibody [0701] In a specific example shown in the conjugation scheme of FIG.5, 15 mg/mL anti- Her2 human IgG antibody in PBS was partially reduced via addition of 2.5-fold molar excess of tris(2-carboxyethyl)phosphine (TCEP) and 2 mM ethylenediaminetetraacetic acid (EDTA) for 2 h at 37 °C.
- TCEP tris(2-carboxyethyl)phosphine
- EDTA ethylenediamine
- the reduced antibody was buffer exchanged into PBS with 1% w/v polysorbate 20.
- Linker-payload was added at a linker-payload / antibody molar ratio of 6 and reacted for an additional 2 h at 25 °C in the presence of 12% v/v of dimethyl sulfoxide (DMSO).
- DMSO dimethyl sulfoxide
- the reaction mixtures were purified via size exclusion chromatography (SEC) (AKTA pure, Superdex 200 Increase), formulated in PBS with 5% Glycerol and stored at -80°C.
- the protein concentration was determined via UV spectrophotometer.
- ADC monomer purity was 99.7% by SEC.
- EXAMPLE 65 [0702] Purification method for antibody-LP11A conjugate through preparative size- exclusion chromatography (SEC).
- DTT dithiothreitol
- HIC Analytical Hydrophobic interaction Chromatography
- TLR-7 is an endosomal localized pattern recognition receptor that responds to pathogen-associated single stranded ribonucleic acid (ssRNA) and plays a key role in innate immune responses.
- TLR-7 activation induces proinflammatory cytokine and type I interferon (IFN) expression via the activation of nuclear factor kappa light chain enhancer of activated B cells (NF- ⁇ B) and interferon regulatory factor (IRF) signaling pathways.
- IFN interferon
- HEK-Blue hTLR7 cells were utilized.
- HEK-Blue TLR7 cells are a human embryonic kidney HEK293 cell line expressing human TLR7 and a secreted alkaline phosphatase (SEAP) reporter gene under the control of the interferon-beta (IFN- ⁇ ) minimal promoter fused to five NF- ⁇ B and AP-1 binding sites.
- SEAP secreted alkaline phosphatase
- a cytotoxicity assay was also performed.
- the test compounds and a reference compound, resiquimod were aliquoted at 10 concentrations with a serial of 3-fold dilution in 96 well plates. Subsequently, 50,000 cells/well were seeded into 96-well plates containing test compounds and then the plates were incubated at 37 °C, 5% CO 2 for 24 hours.
- EXAMPLE 68 [0718] To assess the plasma stability of representative mAb2 TLR7 antibody drug conjugates (ADCs) containing linker payloads provided herein, ncADCs were incubated in vitro with plasma from different species and the DAR was evaluated after incubation at 37 °C for up to 13 days.
- ADCs antibody drug conjugates
- ncADC sample (anti-HER2 Ab-LP1 ADC (comparator), anti-HER2 Ab-LP6A ADC, anti-HER2 Ab-LP11A ADC and anti-HER2 Ab-LP7A ADC) diluted in PBS buffer (Irvine Scientific, Cat#9236) was added to pooled mouse plasma (BioIVT, Cat#MSE01PLK2P2N) and IgG depleted human plasma (BiochemMed), independently, at a final concentration of 50 ⁇ g/mL, and subsequently incubated at 37 °C. A 100- ⁇ L aliquot was removed at the time 0, 24, 36, 72, 168 and 312 hours and then immediately stored frozen at -80 °C until analysis.
- Affinity capture of the ncADCs from the plasma samples was carried out on a KingFisher Apex 96 magnetic particle processor (Thermo Electron).
- biotinylated anti- human Fc antibody (Regeneron generated reagent) was immobilized on Dynabeads M280 streptavidin paramagnetic beads (Invitrogen, Cat#60210).
- Each plasma sample containing TLR7 ncADCs was mixed with 0.5 mg of the beads (Regeneron generated reagent immobilized bead) at room temperature for 2 hours in a 96 well plate.
- the beads were then washed three times with 500 ⁇ L of HBS-EP (GE healthcare, Cat#BR100188), once with 500 ⁇ L of water, and then once with 500 ⁇ L of 8% acetonitrile in water (VWR Chemicals, Cat#BDH83640.100E). Following the washes, the ncADCs were eluted by incubating the beads with 70 ⁇ L of 1% formic acid in 30% acetonitrile / 70% water for 20 minutes at room temperature. Fifty ⁇ L eluted samples were further reduced by adding 50 ⁇ L 10 mM TCEP (Sigma, Cat 646547-10X1ML) and incubated at 37 °C for 20 minutes in ThermoMixer C.
- antibodies were partially reduced via addition of 1.5 - 3.0-fold molar excess of TCEP in PBS pH 7.4 and 2 mM ethylenediaminetetraacetic acid (EDTA) for 2 hours at 37 °C.
- the reduced antibodies were buffer exchanged into PBS with 1% w/v polysorbate 20.
- Linker-payloads were added at a linker-payload / antibody molar ratio of 5 - 10 and reacted for an additional 2 hours at 25 °C in the presence of 12% v/v of dimethyl sulfoxide (DMSO).
- DMSO dimethyl sulfoxide
- reaction mixtures were purified via size exclusion chromatography (SEC) (AKTA pure, Superdex 200 Increase), formulated in PBS with 5% Glycerol and stored at -80 °C.
- SEC size exclusion chromatography
- the protein concentration was determined via UV spectrophotometer.
- ADC monomer purity was >90% by SEC.
- the ADCs were further characterized via hydrophobic interaction chromatography (HIC), and liquid chromatography electrospray ionization mass spectrometry (LC-ESI MS) to calculate drug-antibody ratio (DAR). Results are shown in Tables 6 and 7 and FIG.6, FIG.7, and FIG.8.
- Example (anti-HER2 Ab-LP1 ADC) [0728]
- 15 mg/mL of anti-PSMA antibody in PBS was partially reduced via addition of 2.5-fold molar excess of tris(2-carboxyethyl)phosphine (TCEP) and 2 mM ethylenediaminetetraacetic acid (EDTA) for 2 hours at 37 °C.
- TCEP tris(2-carboxyethyl)phosphine
- EDTA ethylenediaminetetraacetic acid
- the reduced antibody was buffer exchanged into PBS with 1% w/v polysorbate 20.
- a linker-payload LP1 was added at a linker- payload / antibody molar ratio of 6 and reacted for an additional 2 hours at 25 °C in the presence of 12% v/v of dimethyl sulfoxide (DMSO).
- DMSO dimethyl sulfoxide
- reaction mixtures were purified via size exclusion chromatography (SEC) (AKTA pure, Superdex 200 Increase), formulated in PBS with 5% Glycerol and stored at -80 °C.
- SEC size exclusion chromatography
- the protein concentration was determined via UV spectrophotometer.
- ADC monomer purity was 99.7% by SEC.
- Table 6 TLR7 antibody drug conjugates
- HEK-Blue TLR7 cells are a human embryonic kidney HEK293 cell line expressing human TLR7 and a secreted alkaline phosphatase (SEAP) reporter gene under the control of the interferon-beta (IFN- ⁇ ) minimal promoter fused to five NF- ⁇ B and AP-1 binding sites.
- SEAP secreted alkaline phosphatase
- HEK-Blue TLR7/h-antigen2 cells were also utilized.
- HEK-Blue TLR7 cells were engineered to overexpress full-length human antigen 2 via lentiviral mediated transduction and are herein referred to as HEK-Blue TLR7/h-antigen2 cells.
- HEK-Blue TLR7/h-antigen2 cells were grown for at least 2 weeks under G418 selective pressure to enrich for antigen2 positive cells and surface expression was validated via flow cytometry with mAb2.
- 40,000 cells were seeded into 96 well plates in HEK-Blue Detection media. Three-fold serial dilutions of free payloads or LPs were prepared in 100% DMSO, transferred to fresh assay media, and added to the cells at a final constant DMSO concentration of 0.2%. The last well in the plate served as a blank control containing only the assay media and 0.2% DMSO (untreated well) and was plotted as a continuation of the 3-fold serial dilution.
- SEAP activity was determined in a colorimetric assay according to manufacture instructions in which the optical density at 650 nm (OD 650 ) was measured on an Envision luminometer (PerkinElmer) and EC 50 values were determined using a four-parameter logistic equation over a 10-point dose response curve (GraphPad Prism).
- the signal to noise (S/N) was determined by taking the ratio of the highest OD 650 value on the dose response curve to the OD 650 value in the untreated wells.
- ncADCs for TLR-dependent reporter activity, HEK-Blue hTLR7 or HEK- Blue hTLR7/h-antigen2 cells were seeded in complete growth medium at 40,000 cells per well of a 96-well plate and grown overnight. Three-fold serial dilutions of ncADCs, unconjugated antibodies, or isotype control ncADCs were prepared in dilution media (Opti-Mem TM + 0.1% BSA) and added to cells for a final assay medium of 20% dilution medium + 80% growth medium (concentrations were corrected for the DAR (drug antibody ratio) and dosed based on the effective payload concentration).
- Opti-Mem TM + 0.1% BSA dilution media
- concentration concentration were corrected for the DAR (drug antibody ratio) and dosed based on the effective payload concentration.
- P9 did not increase human TLR8 within the tested dose range.
- a known TLR7 payload, P3, increased human and mouse TLR7-dependent activity with EC 50 values of 6.98 nM and 7.27 nM, respectively, and S/N values of 2.7 and 3.2 respectively.
- P3 increased human TLR8 activity with an EC 50 value of 826 nM and S/N of 4.9.
- P3 with an attached linker (LP1) increased human and mouse TLR7-dependent activity with EC 50 values of 25.7 nM and 107 nM, respectively, and S/N values of 5.8 and 2.5 respectively.
- LP1 increased human TLR8 activity with an EC 50 value of 746 ⁇ M and S/N of 2.1.
- a known TLR8 agonist increased human TLR8 activity with an EC 50 value of 4.7 nM and S/N of 5.2, but was weakly agonistic on human TLR7 and mouse TLR7 with EC 50 values > 10 uM and S/N less than or equal to 1.3.
- a known dual TLR7/8 agonist increases human TLR8 and mouse TLR7 activity with EC 50 values of 311 nM and 335 nM, respectively, and S/N values of 4.7 and 3.0 respectively. None of the tested payloads increased TLR3 or TLR9 activity within the tested dose ranges and were inactive in HEK-Blue null cells.
- P9 increased human and mouse TLR7-dependent activity.
- LPs generated from P9 (LP6A, LPA7, LP8A and LP11A) increased human TLR7 reporter activity.
- P9-derived LPs also increased mouse TLR7 reporter activity.
- P9 and associated LPs did not activate human TLR8 within the tested dose range.
- LP1 increased hTLR7 and mTLR7 activity.
- LP1 was weakly agonistic toward human TLR8. None of the test articles were active in HEK-Blue null cells within the tested dose range.
- the mAb2-TLR7 ncADCs increased human TLR7 reporter activity in HEK-Blue hTLR7/HER2 cells with EC 50 s ranging from 13.2 nM to 16.2 nM and S/N values from 2.2 to 4.9. These same ncADCs were weakly agonistic in HEK-Blue hTLR7 cells with EC 50 s > 1.0 uM and S/N values less than or equal to 1.3.
- a known anti-HER2 ncADC increased TLR7 reporter activity in HEK-Blue hTLR7/h-HER2 and HEK-Blue hTLR7 cells with EC 50 values of 9.59 nM and 465 nM, respectively, and S/N values of 6.4 and 2.9, respectively.
- the free payload of anti- HER2 Ab-LP6A ADC, anti-HER2 Ab-LP11A ADC, anti-HER2 Ab-LP7A ADC, and anti-HER2 Ab-LP8A ADC increased TLR7 activity in HEK-Blue hTLR7/h-HER2 and HEK-Blue hTLR7 cells with EC 50 values of 234nM and 153 nM, respectively and S/N values of 8.3 and 6.9, respectively.
- the known payload, P3 was agonistic in HEK-Blue hTLR7/h-HER2 and HEK- Blue hTLR7 cells with EC 50 values of 21.8 nM and 8.16 nM, respectively, and S/N values of 9.1 and 7.5, respectively.
- the known LP, LP1 increased hTLR7 activity in HEK-Blue hTLR7/h-HER2 and HEK-Blue hTLR7 cells with EC 50 values of 115 nM and 71.8 nM, respectively, and S/N values of 8.2 and 7.0, respectively.
- Non-binding isotype controls (mAb1) conjugated to TLR7 LPs were weakly cytotoxic in all tested cells with EC 50 s > 1.0 ⁇ M and S/N less than or equal to 2.0.
- EXAMPLE 71 [0740] Protocol for human PBMC TLR7 IFN- ⁇ release assay [0741] Protocol: 1.
- Protocol for human PBMC TLR7 TNF- ⁇ release assay 1. Thaw frozen human PBMC and put all cells into 40 mL assay media (RPMI1640 supplemented with 10% HI-FBS and 1% penicillin-streptomycin) in 50 mL conical tubes. 2. Spin human PBMC at 1200 rpm for 4 minutes. 3. Remove supernatant by aspiration and resuspend pellet in assay media to 1.5625 ⁇ 10 6 cells/mL. 4. Add 80 ⁇ L cells to each well (1.25 ⁇ 10 5 cells/well). 5. Incubate the plate at 37 o C 5% CO 2 incubator for 24 hours. 6.
- EXAMPLE 72 [0745] The ADCs provided herein were examined for their anti-tumor efficacy using a series of in vivo mouse models. [0746] Growth and Implantation of N87 Tumor Cells into NSGTM Mice [0747] The N87 gastric carcinoma tumor cell line was expanded in T225 flasks in RPMI1640 culture media supplemented with penicillin, streptomycin, L-glutamine, and 10% fetal bovine serum until confluent. Trypsin-EDTA (0.25%) was used to detach cells from each flask for collection.
- N87 cells were then washed twice and an aliquot of cells was collected for determining cell viability and counts using ViaStainTM, a solution containing acridine orange and propidium iodide (AOPI), in combination with the Nexcelom Cellaca MX cell counter. The remaining cells were resuspended in Matrigel Basement Membrane Matrix (50%) prepared in sterile solution and 4-5x10 6 N87 cells were implanted subcutaneously into immunodeficient Fox Chase SCID ® Beige mice across studies. In this model, a single dose (5 mg/kg) of ADCs provided herein eradicated tumors (FIG.10 and FIG.11).
- ViaStainTM a solution containing acridine orange and propidium iodide (AOPI)
- AOPI acridine orange and propidium iodide
- FIG.10 shows that tumor regression was observed after treatment with 5 mg/kg (gray circle) of anti-HER2 Ab- LP6A ADC, while treatment with 1 mg/kg (gray square) anti-HER2 Ab-LP6A ADC resulted in tumor stasis, when compared to saline treated animals (open circle).
- Regression of N87 gastric tumors was not observed in the N87 xenograft mice treated with 5 mg/kg of isotype control Ab-LP6A ADC (Table 3) (black circle), 0.5 mg/kg (gray triangle) anti-HER2 Ab-LP6A ADC, or 0.1 mg/kg (gray diamond) anti-HER2 Ab-LP6A ADC when compared to saline treated animals (open circle).
- JIMT-1 epithelial breast tumor cell line was expanded in T225 flasks in DMEM culture media supplemented with penicillin, streptomycin, L-glutamine, and 10% fetal bovine serum until confluent. Trypsin-EDTA (0.25%) was used to detach cells from each flask for collection.
- JIMT-1 cells were then washed twice and an aliquot of cells was collected for determining cell viability and counts using ViaStainTM, a solution containing acridine orange and propidium iodide (AOPI), in combination with the Nexcelom Cellaca MX cell counter. The remaining cells were resuspended in Matrigel Basement Membrane Matrix (50%) prepared in sterile solution and 2.5x10 6 JIMT-1 cells were implanted subcutaneously into immunodeficient Fox Chase SCID ® Beige mice across studies.
- ViaStainTM a solution containing acridine orange and propidium iodide (AOPI)
- AOPI propidium iodide
- ADCs provided herein were found to delay growth relative to an isotype control ADC, and when combined with a non-competing anti-HER2 antibody, pertuzumab, resulted in tumor regression, suggesting that FcR clustering can be used to enhance therapeutic efficacy (FIG.12).
- MC38 Tumor Cells Engineered to Express CD20 into C57BL/6J Mice [0751] The MC38 melanoma tumor cell line expressing a defined tumor was expanded in T225 flasks in DMEM culture media supplemented with penicillin, streptomycin, L-glutamine, sodium pyruvate, 1% HEPES, 1% non-essential amino acids and 10% fetal bovine serum until confluent. Trypsin-EDTA (0.25%) was used to detach cells from each flask for collection.
- Tumor cells were then washed twice and an aliquot of cells was collected for determining cell viability and counts using ViaStainTM, a solution containing acridine orange and propidium iodide (AOPI), in combination with the Nexcelom Cellaca MX cell counter.
- ViaStainTM a solution containing acridine orange and propidium iodide (AOPI)
- AOPI propidium iodide
- Tumor cells were resuspended in Hanks Balanced Salt Solution and 1e6 tumor cells were subcutaneously implanted into each C57BL/6J mouse.
- An anti-human CD20 ADC was shown to mediate tumor regression in this model (FIG.13).
- N87 cells were cultured in RMPI, 10% FBS, P/S/G, before implantation, cells were mixed with an equal volume of Matrigel, and 100ul of the mixture (5e6 cells) were implanted subcutaneously into the right flank of 6-8-week-old female SCID-Beige mice (Charles River).
- JIMT-1 cells were cultured in DMEM, 10% FBS, P/S/G. Before implantation, cells were mixed with an equal volume of Matrigel, and 200 uL of the mixture (2.5e6 cells) were implanted subcutaneously into the right flank of 6-8-week-old female SCID-Beige mice (Charles River).
- MC38.hTAA Pos cells were cultured in DMEM, 10% FBS, P/S/G, NaPyr, 1% HEPES, 1% NEAA.200 uL of 1e6 cells were implanted subcutaneously into the right flank of 6-8-week- old female C57BL/6J mice (The Jackson Laboratory).
- MC38 cells were cultured in DMEM, 10% FBS, P/S/G, NaPyr, 1% HEPES, 1% NEAA.200 uL of 5e5 cells were implanted subcutaneously into the left flank of 6-8-week-old female C57BL/6J mice for the rechallenge study (The Jackson Laboratory).
- MC38.hTAA Pos cells were cultured in DMEM, 10% FBS, P/S/G, NaPyr, 1% HEPES, 1% NEAA.200 uL of 1e6 cells were implanted subcutaneously into the right flank of 6-8-week- old female hIFNAR mice (Velocigene).
- MC38h.TAA Pos cells were cultured in DMEM, 10% FBS, P/S/G, NaPyr, 1% HEPES, 1% NEAA.200ul of 1e6 cells were implanted subcutaneously into the right flank of 6-8-week- old female mice having been humanized for TAA and hCD3 mice (Velocigene).
- FIG.16 depicts results following parental MC38 tumor cell rechallenge in mice having initially cleared MC38.hTAA Pos engrafted tumors (the same as used in Fig. 13).
- mice On day 60 after MC38.hTAA Pos tumor cell inoculation, tumor free mice (black square) were rechallenged with parental MC38 cells without overexpression of human TAA. Compared to control na ⁇ ve mice (open circle), mice previously treated with anti-CD20 -LP6A conjugate are protected against tumor rechallenge. Data represent mean tumor volumes (mean+/-SEM) over time (post-rechallenge). [0762] FIG.
- FIG. 17 depicts results following treatment of mice having been inoculated with MC38.hTAA Pos tumor cells with 3 doses every seven days of anti-CD20-LP11A conjugate in wild type mice (closed symbols with solid lines) and in humanized IFNAR mice (open symbols with dashed lines) that lack the ability to respond to murine type I IFN. Regression of tumor was observed after treatment with 5 mg/kg of anti-CD20-LP11A conjugate (closed triangle) when compared to saline treated animals (closed circle) and isotype control antibody conjugate (closed square).
- FIG. 18 depicts results following treatment of mice having been inoculated with MC38.hTAA Pos tumor cells with 3 doses every seven days of anti-CD20-LP6A conjugate with or without 5 doses every four days of anti-CD20 x anti-hCD3 bispecific antibody in mice humanized for TAA and human CD3.
- FIG.19 depicts the ring opening of the imide bond of the antibody-drug conjugates from the conjugation of the cysteine thiol with the maleimide of the linker-payload. Ring- opening of the imide bond under physiological conditions affords two regio-isomers that one is the thiol attached to the alpha carbon and the other is the thiol attached to the beta carbon to the carboxylic acid group, respectively.
- HBV Hepatitis B virus
- AAV Adeno-Associated Virus
- CHB chronic hepatitis B
- This AAV- HBV mouse model can be used to assess different therapeutic interventions to look at sustained HBV sAg reduction of treatment (functional cure) or complete elimination of HBV infected hepatocytes.
- mice were then treated with an anti-sAg mAb (mAb3), anti-sAg mAb-TLR7 agonist (mAb3+LP1 or mAb4+LP1), a TLR7 agonist (LP1), or PBS three times, two weeks apart subcutaneously (SC). MAb doses used are outlined in Table 12 from two independent experiments. Mice were then bled weekly or biweekly to measure sAg levels before, during and after mAb treatments. As shown in FIG. 14 and FIG.15, CHB mice treated with the anti-sAg-TLR7 ADCs displayed rapid and sustained reduction in circulating sAg levels compared to anti-sAg or TLR7 agonist treated mice.
- Table 13 shows sAg concentrations at study end for each treatment group. Mice treated with anti- sAg-TLR7 ADCs displayed lower sAg levels than anti-mAb or TLR7 agonist treated mice in both experimental studies. Furthermore, the majority of anti-sAg-TLR7 ADC-treated mice at the lower dosed arms had undetectable sAg compared to all other treatment groups even 67 days after the last treatment.
- Assay procedure Prepare 2 ⁇ LS9/compound solution: T 0 : add 199 ⁇ L 2 mg/mL liver S9 solution + 100 ⁇ L of 8 mM NADPH solution + 100 ⁇ L of 20 mM UDPGA solution + 1200 ⁇ L of ACN, vortex at 1000 rpm for 5 min, then add 1 ⁇ L of 4 mM cpd solution. T 240 : add 199 ⁇ L 2 mg/mL liver S9 solution + 100 ⁇ L of 8 mM NADPH solution + 100 ⁇ L of 20 mM UDPGA solution, prewarm the T 240 sample at 37°C for 5 min and 1 ⁇ L of 4 mM cpd solution was added.
- T 240-w/o add 199 ⁇ L 2 mg/mL liver S9 solution + 200 ⁇ L of buffer, prewarm the T 240-w/o sample at 37°C for 5 min and 1 ⁇ L of 4 mM cpd solution was added.
- T 240 min incubation add 1200 ⁇ L of ACN and then vortex at 1000 rpm for 5 min.
- Protein precipitation centrifuge quenched samples at 14000 rpm for 5 min.
- Sample preparation evaporate an aliquot of 1200 ⁇ L of the supernatant under N 2 stream until dry.
- the imide ring of the linker connecting an antibody and the payload exists as an equilibrium between an opened ring and a closed five-membered imide ring as shown in FIG.19.
- Those ADC species with an opened ring and a closed five- membered imide ring show the same or comparable activities in the research described herein.
- EXAMPLE 75 To assess whether an anti-HBV antibody conjugated with a TLR7 agonist as an antibody drug conjugate (ADC) could break B cell tolerance and elicit an antibody response to the HBV sAg protein in an AAV-HBV mouse model, male C57BL/6 mice were transduced with 1E11 viral genomes of AAV8-HBV virus intravenously (I.V.) and six weeks later measured HBV sAg levels in their serum to determine if they exhibited a CHB (chronic hepatitis B) phenotype. Mice that had HBV sAg levels ⁇ 1 ⁇ g/mL in the serum were included in the study.
- ADC antibody drug conjugate
- mice were then treated with an anti-HBV sAg mAb (mAb3) conjugated with an TLR7 agonist (LP6A) at either 44 ⁇ g or 0.44 ⁇ g per injection or given PBS five times, one week apart subcutaneously (SC).
- mAb3 The sequence of mAb3 is shown in Table 15 below.
- CHB mice treated with anti-HBV sAg-TLR7 ADC at both doses display rapid and sustained reduction in circulating HBV sAg levels until end of study (day 120).
- serum was obtained and anti-HBV sAg IgG titers were measured via ELISA to determine endogenous antibody responses.
- HBV sAg-TLR7 ADC elicited HBV sAg IgG titers above background while none of the PBS mice showed HBV sAg IgG titers which is characteristic of the AAV-HBV mouse model.
- HBV proteins One of the hallmarks of chronic hepatitis B infection is tolerogenic immune response to the HBV proteins. This is most notable for immunity to HBV sAg in which there is a minimal T cell response and no HBV sAg IgG responses despite HBV sAg being abundantly expressed.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- Cell Biology (AREA)
- Organic Chemistry (AREA)
- Oncology (AREA)
- Virology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Reproductive Health (AREA)
- Hematology (AREA)
- Biotechnology (AREA)
- Molecular Biology (AREA)
- Communicable Diseases (AREA)
- Peptides Or Proteins (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description



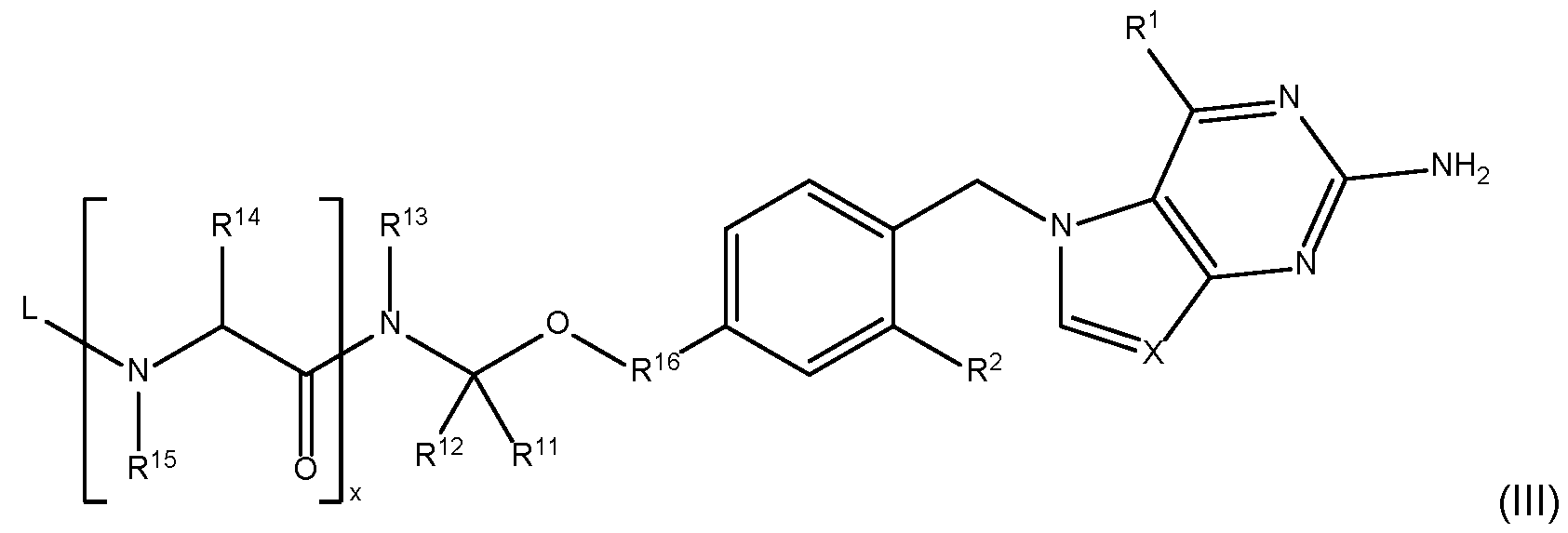






















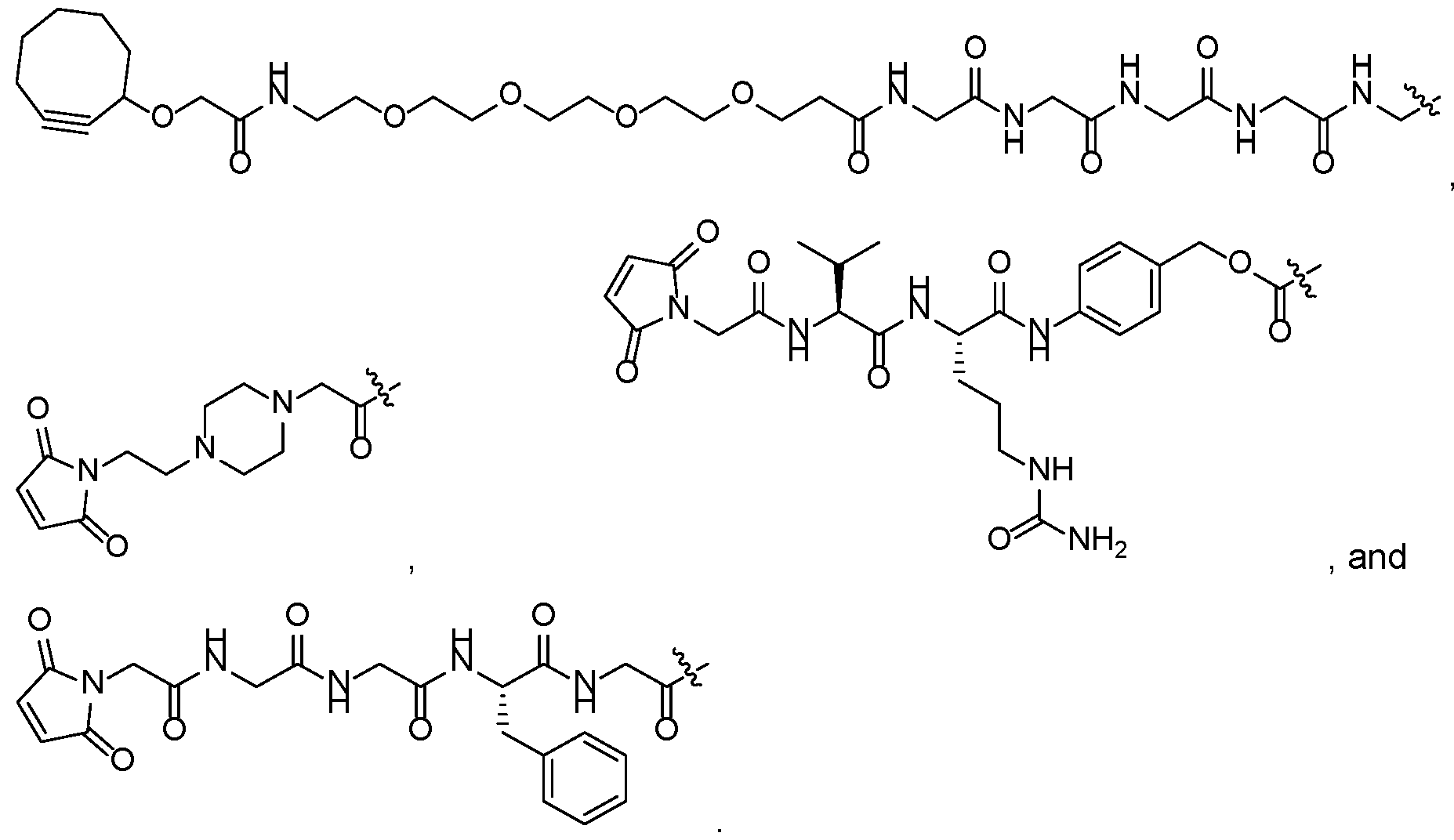













































































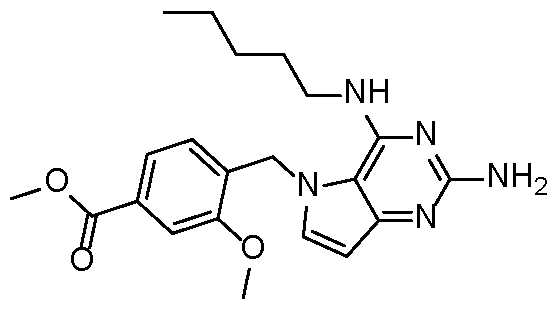





















































































































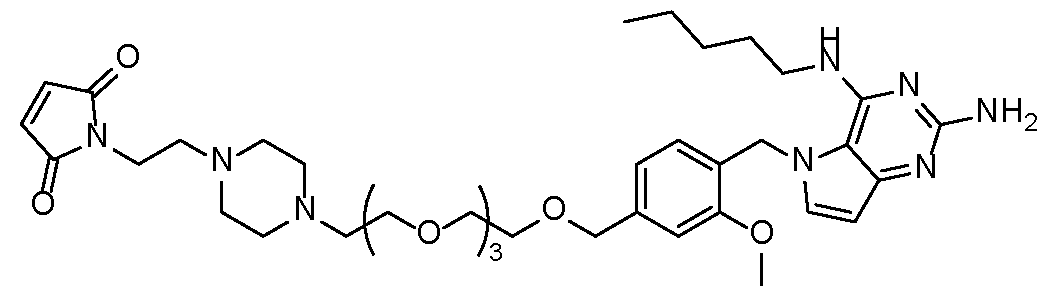

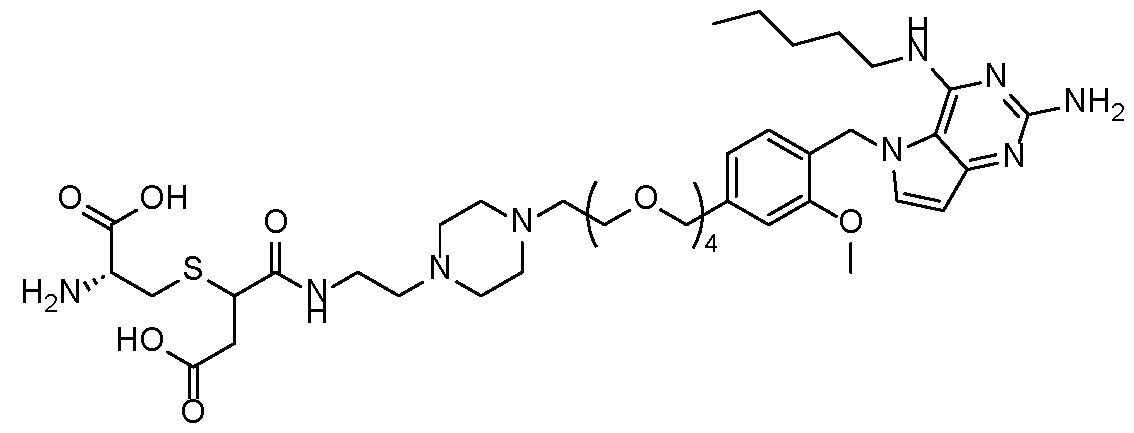





























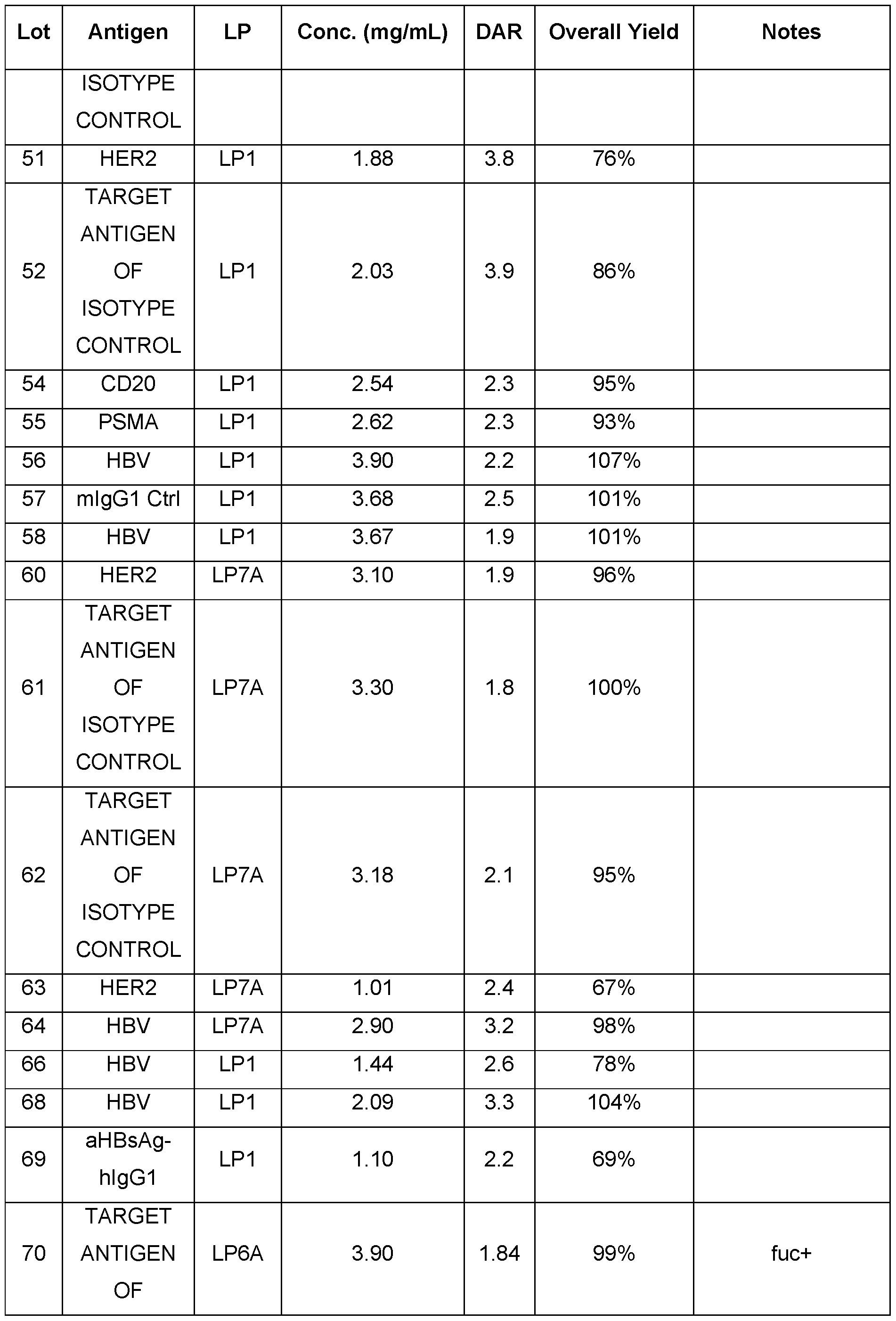






















Claims
























Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2025531228A JP2025540064A (en) | 2022-11-30 | 2023-11-29 | TLR7 agonist and its antibody-drug conjugate |
| KR1020257021135A KR20250128394A (en) | 2022-11-30 | 2023-11-29 | TLR7 agonists and antibody-drug conjugates thereof |
| AU2023403422A AU2023403422A1 (en) | 2022-11-30 | 2023-11-29 | Tlr7 agonists and antibody-drug-conjugates thereof |
| EP23836674.4A EP4626553A2 (en) | 2022-11-30 | 2023-11-29 | Tlr7 agonists and antibody-drug-conjugates thereof |
| CN202380092608.9A CN120659785A (en) | 2022-11-30 | 2023-11-29 | TLR7 agonists and antibody-drug conjugates thereof |
| MX2025006112A MX2025006112A (en) | 2022-11-30 | 2025-05-26 | Tlr7 agonists and antibody-drug-conjugates thereof |
| IL321396A IL321396A (en) | 2022-11-30 | 2025-05-29 | Tlr7 agonists and antibody-drug-conjugates thereof |
| CONC2025/0008795A CO2025008795A2 (en) | 2022-11-30 | 2025-06-27 | TLR7 agonists and antibody-drug conjugates thereof |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202263429096P | 2022-11-30 | 2022-11-30 | |
| US63/429,096 | 2022-11-30 | ||
| US202363578109P | 2023-08-22 | 2023-08-22 | |
| US63/578,109 | 2023-08-22 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2024118785A2 true WO2024118785A2 (en) | 2024-06-06 |
| WO2024118785A3 WO2024118785A3 (en) | 2024-07-04 |
Family
ID=89474538
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2023/081614 Ceased WO2024118785A2 (en) | 2022-11-30 | 2023-11-29 | Tlr7 agonists and antibody-drug-conjugates thereof |
Country Status (10)
| Country | Link |
|---|---|
| EP (1) | EP4626553A2 (en) |
| JP (1) | JP2025540064A (en) |
| KR (1) | KR20250128394A (en) |
| CN (1) | CN120659785A (en) |
| AU (1) | AU2023403422A1 (en) |
| CL (1) | CL2025001577A1 (en) |
| CO (1) | CO2025008795A2 (en) |
| IL (1) | IL321396A (en) |
| MX (1) | MX2025006112A (en) |
| WO (1) | WO2024118785A2 (en) |
Citations (42)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4464456A (en) | 1982-01-06 | 1984-08-07 | Toray Industries, Inc. | Photosensitive polymer compositions comprising polyether-ester amides |
| WO1992002551A1 (en) | 1990-08-02 | 1992-02-20 | B.R. Centre Limited | Methods for the production of proteins with a desired function |
| US5208020A (en) | 1989-10-25 | 1993-05-04 | Immunogen Inc. | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| US5223409A (en) | 1988-09-02 | 1993-06-29 | Protein Engineering Corp. | Directed evolution of novel binding proteins |
| US5698426A (en) | 1990-09-28 | 1997-12-16 | Ixsys, Incorporated | Surface expression libraries of heteromeric receptors |
| US5714586A (en) | 1995-06-07 | 1998-02-03 | American Cyanamid Company | Methods for the preparation of monomeric calicheamicin derivative/carrier conjugates |
| US5877397A (en) | 1990-08-29 | 1999-03-02 | Genpharm International Inc. | Transgenic non-human animals capable of producing heterologous antibodies of various isotypes |
| US20030138785A1 (en) | 2001-12-21 | 2003-07-24 | Stephan Kopytek | In vivo protein screen based on enzyme-assisted chemically induced dimerization ("CID") |
| WO2005089808A2 (en) | 2004-03-15 | 2005-09-29 | Wyeth | Antibody calicheamicin conjugates |
| US7087411B2 (en) | 1999-06-08 | 2006-08-08 | Regeneron Pharmaceuticals, Inc. | Fusion protein capable of binding VEGF |
| US20070258987A1 (en) | 2000-11-28 | 2007-11-08 | Seattle Genetics, Inc. | Recombinant Anti-Cd30 Antibodies and Uses Thereof |
| US20080171040A1 (en) | 2004-06-01 | 2008-07-17 | Genentech, Inc. | Antibody-drug conjugates and methods |
| WO2008122039A2 (en) | 2007-04-02 | 2008-10-09 | The Government Of The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Selenocysteine mediated hybrid antibody molecules |
| US20080305044A1 (en) | 2004-11-29 | 2008-12-11 | Seattle Genetics, Inc. | Engineered Antibodies and Immunoconjugates |
| US20080305497A1 (en) | 2007-05-23 | 2008-12-11 | Ventana Medical Systems, Inc. | Polymeric carriers for immunohistochemistry and in situ hybridization |
| US20090142354A1 (en) | 2006-12-14 | 2009-06-04 | Regeneron Pharmaceuticals, Inc. | Human antibodies to human delta like ligand 4 |
| US20100129314A1 (en) | 2008-04-30 | 2010-05-27 | Immunogen Inc. | Potent conjugates and hydrophilic linkers |
| US7750116B1 (en) | 2006-02-18 | 2010-07-06 | Seattle Genetics, Inc. | Antibody drug conjugate metabolites |
| US7754681B2 (en) | 2004-02-23 | 2010-07-13 | Seattle Genetics, Inc. | Heterocyclic self-immolative linkers and conjugates |
| US20110027286A1 (en) | 2009-07-29 | 2011-02-03 | Regeneron Pharmaceuticals, Inc. | High Affinity Human Antibodies to Human Angiopoietin-2 |
| WO2011130598A1 (en) | 2010-04-15 | 2011-10-20 | Spirogen Limited | Pyrrolobenzodiazepines and conjugates thereof |
| WO2012005982A2 (en) | 2010-07-06 | 2012-01-12 | Board Of Supervisors Of Louisiana State University And Agricultural And Mechanical College | Reporter for rna polymerase ii termination |
| WO2012059882A2 (en) | 2010-11-05 | 2012-05-10 | Rinat Neuroscience Corporation | Engineered polypeptide conjugates and methods for making thereof using transglutaminase |
| WO2012166559A1 (en) | 2011-05-27 | 2012-12-06 | Ambrx, Inc. | Compositions containing, methods involving, and uses of non-natural amino acid linked dolastatin derivatives |
| WO2013053872A1 (en) | 2011-10-14 | 2013-04-18 | Spirogen Sàrl | Synthesis method and intermediates useful in the preparation of pyrrolobenzodiazepines |
| WO2013053873A1 (en) | 2011-10-14 | 2013-04-18 | Spirogen Sàrl | Pyrrolobenzodiazepines |
| WO2013055990A1 (en) | 2011-10-14 | 2013-04-18 | Seattle Genetics, Inc. | Pyrrolobenzodiazepines and targeted conjugates |
| WO2013055993A1 (en) | 2011-10-14 | 2013-04-18 | Seattle Genetics, Inc. | Pyrrolobenzodiazepines and targeted conjugates |
| US20130101546A1 (en) | 2011-06-10 | 2013-04-25 | Mersana Therapeutics, Inc. | Protein-Polymer-Drug Conjugates |
| WO2013068874A1 (en) | 2011-11-11 | 2013-05-16 | Pfizer Inc. | Antibody-drug conjugates |
| WO2014065661A1 (en) | 2012-10-23 | 2014-05-01 | Synaffix B.V. | Modified antibody, antibody-conjugate and process for the preparation thereof |
| US20160176953A1 (en) | 2014-12-19 | 2016-06-23 | Regeneron Pharmaceuticals, Inc. | Human Antibodies to Influenza Hemagglutinin |
| US9944649B2 (en) | 2014-05-01 | 2018-04-17 | Novartis Ag | Compounds and compositions as toll-like receptor 7 agonists |
| US20180134794A1 (en) | 2016-11-16 | 2018-05-17 | Regeneron Pharmaceuticals, Inc. | Anti-met antibodies, bispecific antigen binding molecules that bind met, and methods of use thereof |
| US10472420B2 (en) | 2006-02-22 | 2019-11-12 | 3M Innovative Properties Company | Immune response modifier conjugates |
| US10548985B2 (en) | 2014-01-10 | 2020-02-04 | Birdie Biopharmaceuticals, Inc. | Compounds and compositions for treating EGFR expressing tumors |
| WO2020050406A1 (en) | 2018-09-06 | 2020-03-12 | 第一三共株式会社 | Novel cyclic dinucleotide derivative and antibody-drug conjugate thereof |
| US10675358B2 (en) | 2016-07-07 | 2020-06-09 | The Board Of Trustees Of The Leland Stanford Junior University | Antibody adjuvant conjugates |
| WO2020132658A2 (en) | 2018-12-21 | 2020-06-25 | Regeneron Pharmaceuticals, Inc. | Tubulysins and protein-tubulysin conjugates |
| WO2020146541A2 (en) | 2019-01-08 | 2020-07-16 | Regeneron Pharmaceuticals, Inc. | Traceless linkers and protein-conjugates thereof |
| US10722591B2 (en) | 2017-11-14 | 2020-07-28 | Dynavax Technologies Corporation | Cleavable conjugates of TLR7/8 agonist compounds, methods for preparation, and uses thereof |
| WO2020181050A1 (en) | 2019-03-05 | 2020-09-10 | Sperovie Biosciences, Inc. | Compounds, compositions, and methods for the treatment of disease |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4748177A (en) * | 1984-03-26 | 1988-05-31 | Warner-Lambert Company | Guanine derivatives |
| JPH1025294A (en) * | 1996-03-26 | 1998-01-27 | Akira Matsuda | Fused heterocyclic derivative, method for producing the same, and therapeutic agent for malignant tumor containing the same |
| US20120231023A1 (en) * | 2011-03-08 | 2012-09-13 | Baylor Research Institute | Novel Vaccine Adjuvants Based on Targeting Adjuvants to Antibodies Directly to Antigen-Presenting Cells |
| MA44334A (en) * | 2015-10-29 | 2018-09-05 | Novartis Ag | ANTIBODY CONJUGATES INCLUDING A TOLL-TYPE RECEPTOR AGONIST |
| WO2020252015A1 (en) * | 2019-06-10 | 2020-12-17 | Sutro Biopharma, Inc. | Immunomodulator antibody drug conjugates and uses thereof |
| EP3980423A1 (en) * | 2019-06-10 | 2022-04-13 | Sutro Biopharma, Inc. | 5h-pyrrolo[3,2-d]pyrimidine-2,4-diamino compounds and antibody conjugates thereof |
| EP4114852A1 (en) * | 2020-03-03 | 2023-01-11 | Sutro Biopharma, Inc. | Antibodies comprising site-specific glutamine tags, methods of their preparation and methods of their use |
-
2023
- 2023-11-29 KR KR1020257021135A patent/KR20250128394A/en active Pending
- 2023-11-29 WO PCT/US2023/081614 patent/WO2024118785A2/en not_active Ceased
- 2023-11-29 EP EP23836674.4A patent/EP4626553A2/en active Pending
- 2023-11-29 AU AU2023403422A patent/AU2023403422A1/en active Pending
- 2023-11-29 CN CN202380092608.9A patent/CN120659785A/en active Pending
- 2023-11-29 JP JP2025531228A patent/JP2025540064A/en active Pending
-
2025
- 2025-05-26 MX MX2025006112A patent/MX2025006112A/en unknown
- 2025-05-28 CL CL2025001577A patent/CL2025001577A1/en unknown
- 2025-05-29 IL IL321396A patent/IL321396A/en unknown
- 2025-06-27 CO CONC2025/0008795A patent/CO2025008795A2/en unknown
Patent Citations (46)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4464456A (en) | 1982-01-06 | 1984-08-07 | Toray Industries, Inc. | Photosensitive polymer compositions comprising polyether-ester amides |
| US5223409A (en) | 1988-09-02 | 1993-06-29 | Protein Engineering Corp. | Directed evolution of novel binding proteins |
| US5208020A (en) | 1989-10-25 | 1993-05-04 | Immunogen Inc. | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| WO1992002551A1 (en) | 1990-08-02 | 1992-02-20 | B.R. Centre Limited | Methods for the production of proteins with a desired function |
| US5627052A (en) | 1990-08-02 | 1997-05-06 | B.R. Centre, Ltd. | Methods for the production of proteins with a desired function |
| US5877397A (en) | 1990-08-29 | 1999-03-02 | Genpharm International Inc. | Transgenic non-human animals capable of producing heterologous antibodies of various isotypes |
| US5698426A (en) | 1990-09-28 | 1997-12-16 | Ixsys, Incorporated | Surface expression libraries of heteromeric receptors |
| US5714586A (en) | 1995-06-07 | 1998-02-03 | American Cyanamid Company | Methods for the preparation of monomeric calicheamicin derivative/carrier conjugates |
| US7087411B2 (en) | 1999-06-08 | 2006-08-08 | Regeneron Pharmaceuticals, Inc. | Fusion protein capable of binding VEGF |
| US20070258987A1 (en) | 2000-11-28 | 2007-11-08 | Seattle Genetics, Inc. | Recombinant Anti-Cd30 Antibodies and Uses Thereof |
| US20030138785A1 (en) | 2001-12-21 | 2003-07-24 | Stephan Kopytek | In vivo protein screen based on enzyme-assisted chemically induced dimerization ("CID") |
| US7754681B2 (en) | 2004-02-23 | 2010-07-13 | Seattle Genetics, Inc. | Heterocyclic self-immolative linkers and conjugates |
| WO2005089808A2 (en) | 2004-03-15 | 2005-09-29 | Wyeth | Antibody calicheamicin conjugates |
| US20080171040A1 (en) | 2004-06-01 | 2008-07-17 | Genentech, Inc. | Antibody-drug conjugates and methods |
| US20080305044A1 (en) | 2004-11-29 | 2008-12-11 | Seattle Genetics, Inc. | Engineered Antibodies and Immunoconjugates |
| US7750116B1 (en) | 2006-02-18 | 2010-07-06 | Seattle Genetics, Inc. | Antibody drug conjugate metabolites |
| US10472420B2 (en) | 2006-02-22 | 2019-11-12 | 3M Innovative Properties Company | Immune response modifier conjugates |
| US20090142354A1 (en) | 2006-12-14 | 2009-06-04 | Regeneron Pharmaceuticals, Inc. | Human antibodies to human delta like ligand 4 |
| WO2008122039A2 (en) | 2007-04-02 | 2008-10-09 | The Government Of The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Selenocysteine mediated hybrid antibody molecules |
| US20080305497A1 (en) | 2007-05-23 | 2008-12-11 | Ventana Medical Systems, Inc. | Polymeric carriers for immunohistochemistry and in situ hybridization |
| US20100129314A1 (en) | 2008-04-30 | 2010-05-27 | Immunogen Inc. | Potent conjugates and hydrophilic linkers |
| US20110027286A1 (en) | 2009-07-29 | 2011-02-03 | Regeneron Pharmaceuticals, Inc. | High Affinity Human Antibodies to Human Angiopoietin-2 |
| WO2011130598A1 (en) | 2010-04-15 | 2011-10-20 | Spirogen Limited | Pyrrolobenzodiazepines and conjugates thereof |
| WO2012005982A2 (en) | 2010-07-06 | 2012-01-12 | Board Of Supervisors Of Louisiana State University And Agricultural And Mechanical College | Reporter for rna polymerase ii termination |
| WO2012059882A2 (en) | 2010-11-05 | 2012-05-10 | Rinat Neuroscience Corporation | Engineered polypeptide conjugates and methods for making thereof using transglutaminase |
| US9676871B2 (en) | 2010-11-05 | 2017-06-13 | Pfizer Inc. | Engineered polypeptide conjugates and methods for making thereof using transglutaminase |
| WO2012166559A1 (en) | 2011-05-27 | 2012-12-06 | Ambrx, Inc. | Compositions containing, methods involving, and uses of non-natural amino acid linked dolastatin derivatives |
| US20130101546A1 (en) | 2011-06-10 | 2013-04-25 | Mersana Therapeutics, Inc. | Protein-Polymer-Drug Conjugates |
| WO2013055993A1 (en) | 2011-10-14 | 2013-04-18 | Seattle Genetics, Inc. | Pyrrolobenzodiazepines and targeted conjugates |
| WO2013055990A1 (en) | 2011-10-14 | 2013-04-18 | Seattle Genetics, Inc. | Pyrrolobenzodiazepines and targeted conjugates |
| WO2013053873A1 (en) | 2011-10-14 | 2013-04-18 | Spirogen Sàrl | Pyrrolobenzodiazepines |
| WO2013053872A1 (en) | 2011-10-14 | 2013-04-18 | Spirogen Sàrl | Synthesis method and intermediates useful in the preparation of pyrrolobenzodiazepines |
| WO2013068874A1 (en) | 2011-11-11 | 2013-05-16 | Pfizer Inc. | Antibody-drug conjugates |
| WO2014065661A1 (en) | 2012-10-23 | 2014-05-01 | Synaffix B.V. | Modified antibody, antibody-conjugate and process for the preparation thereof |
| US10780180B2 (en) | 2014-01-10 | 2020-09-22 | Birdie Biopharmaceuticals, Inc. | Compounds and compositions for immunotherapy |
| US10548985B2 (en) | 2014-01-10 | 2020-02-04 | Birdie Biopharmaceuticals, Inc. | Compounds and compositions for treating EGFR expressing tumors |
| US9944649B2 (en) | 2014-05-01 | 2018-04-17 | Novartis Ag | Compounds and compositions as toll-like receptor 7 agonists |
| US20160176953A1 (en) | 2014-12-19 | 2016-06-23 | Regeneron Pharmaceuticals, Inc. | Human Antibodies to Influenza Hemagglutinin |
| WO2016100807A2 (en) | 2014-12-19 | 2016-06-23 | Regeneron Pharmaceuticals, Inc. | Human antibodies to influenza hemagglutinin |
| US10675358B2 (en) | 2016-07-07 | 2020-06-09 | The Board Of Trustees Of The Leland Stanford Junior University | Antibody adjuvant conjugates |
| US20180134794A1 (en) | 2016-11-16 | 2018-05-17 | Regeneron Pharmaceuticals, Inc. | Anti-met antibodies, bispecific antigen binding molecules that bind met, and methods of use thereof |
| US10722591B2 (en) | 2017-11-14 | 2020-07-28 | Dynavax Technologies Corporation | Cleavable conjugates of TLR7/8 agonist compounds, methods for preparation, and uses thereof |
| WO2020050406A1 (en) | 2018-09-06 | 2020-03-12 | 第一三共株式会社 | Novel cyclic dinucleotide derivative and antibody-drug conjugate thereof |
| WO2020132658A2 (en) | 2018-12-21 | 2020-06-25 | Regeneron Pharmaceuticals, Inc. | Tubulysins and protein-tubulysin conjugates |
| WO2020146541A2 (en) | 2019-01-08 | 2020-07-16 | Regeneron Pharmaceuticals, Inc. | Traceless linkers and protein-conjugates thereof |
| WO2020181050A1 (en) | 2019-03-05 | 2020-09-10 | Sperovie Biosciences, Inc. | Compounds, compositions, and methods for the treatment of disease |
Non-Patent Citations (78)
| Title |
|---|
| "Antibodies: A Laboratory Manual,", 1988, COLD SPRING HARBOR LABORATORY PRESS |
| "Antibody-Drug Conjugates", 2015, SPRINGER INTERNATIONAL PUBLISHING |
| "Current Protocols in Molecular Biology", 1999, WILEY INTERSCIENCE |
| "Monoclonal Antibodies, Hybridomas: A New Dimension in Biological Analyses", 1980, PLENUM PRESS |
| AGARD ET AL., J. AM. CHEM. SOC., vol. 126, no. 46, 2004, pages 15046 - 15047 |
| AGARWAL ET AL., PROC. NATL. ACAD. SCI., USA, vol. 110, 2013, pages 46 - 51 |
| AL-LAZIKANI ET AL., J. MOL. BIOL., vol. 273, 1997, pages 927 - 948 |
| ALTING-MEES ET AL., , STRATEGIES IN MOLECULAR BIOLOGY, vol. 3, 1990, pages 1 - 9 |
| ALTSCHUL ET AL., NUCLEIC ACIDS RES., vol. 25, 1997, pages 3389 - 402 |
| ALTSCHUL, J. MOL. BIOL., vol. 215, 1990, pages 403 - 410 |
| ANGEW. CHEM., INT. ED, vol. 51, no. 48, 2012, pages 12000 - 12004 |
| ASTRID-CAROLINE KNALLCHRISTIAN SLUGOVC., CHEM. SOC. REV., vol. 42, 2013, pages 5131 |
| BABCOOK ET AL., PROC. NATL. ACAD. SCI. USA, vol. 93, 1996, pages 7843 - 48 |
| BIRD ET AL., SCIENCE, vol. 242, 1988, pages 423 - 426 |
| BOERNER ET AL., J. IMMUNOL., vol. 147, 1991, pages 86 - 95 |
| BOERSMAPLUCKTHUN, CURR. OPIN. BIOTECHNOL., vol. 22, 2011, pages 849 - 857 |
| BRUGGEMANN ET AL., , CURR. OPIN. BIOTECHNOL., vol. 8, 1997, pages 455 - 58 |
| BRUGGEMANN ET AL., CURR. OPIN. BIOTECHNOL., vol. 8, 1997, pages 724 - 733 |
| BURTON ET AL., ADV. IMMUNOL., vol. 57, 1994, pages 191 - 280 |
| CARRICO ET AL., NAT. CHEM. BIOL., vol. 3, 2007, pages 321 - 322 |
| CHI ET AL., FRONT. PHARMACOL., vol. 8, 31 May 2017 (2017-05-31), pages 34 |
| CHIO ET AL., METHODS MOL. BIOL, vol. 2078, 2020, pages 83 - 87 |
| CHIO ET AL., METHODS MOL. BIOL., vol. 2078, 2020, pages 83 - 87 |
| CLACKSON ET AL., NATURE, V., vol. 352, 1991, pages 624 - 628 |
| CLARK, M., IMMUNOLOGY TODAY., vol. 21, no. 8, 2000, pages 397 - 402 |
| CRAMERI ET AL., NATURE, vol. 391, 1998, pages 288 - 291 |
| DALL'ACQUA WF ET AL., METHODS, vol. 36, no. 1, 2005, pages 43 - 60 |
| DENNLER ET AL., BIOCONJUGATE CHEM, vol. 25, 2014, pages 569 - 578 |
| DUCRY, BIOCONJUGATE CHEM., vol. 21, 2010, pages 5 - 13 |
| EHRING, ANALYTICAL BIOCHEMISTRY, vol. 267, no. 2, 1999, pages 252 - 259 |
| ENGENSMITH, ANAL. CHEM., vol. 73, 2001, pages 256 |
| GLASKY ET AL., HYBRIDOMA, vol. 8, 1989, pages 377 - 89 |
| GLUZMAN ET AL., CELL, vol. 23, 1981, pages 175 |
| GREEN ET AL., NATURE GENET., vol. 7, 1994, pages 13 |
| HARRIS, R.J., JOURNAL OF CHROMATOGRAPHY, vol. 705, 1995, pages 129 - 134 |
| HOFER ET AL., PROC. NATL. ACAD. SCI., USA, vol. 105, 2008, pages 12451 - 12456 |
| HOLLANDER ET AL., BIOCONJUGATE CHEM.,, vol. 19, 2008, pages 358 - 361 |
| HOOGENBOOM ET AL., J. MOLEC. BIOL., vol. 227, 1992, pages 381 - 388 |
| HUISGEN, PROC. CHEM. SOC, 1961, pages 357 - 396 |
| HUISGEN, PROC. CHEM. SOC., 1961, pages 357 - 396 |
| HUSE ET AL., SCIENCE, vol. 246, 1989, pages 1275 - 81 |
| JAKOBOVITS ET AL., ANN. N. Y. ACAD. SCI., vol. 764, 1995, pages 525 - 35 |
| JEGER ET AL., ANGEW CHEM INT ED ENGL., vol. 49, 2010, pages 9995 - 9997 |
| JONES ET AL., NATURE, vol. 321, 1986, pages 522 - 525 |
| KANG ET AL., PROC. NATL. ACAD. SCI. USA, vol. 88, 1991, pages 4363 - 66 |
| KAZANE ET AL., J. AM. CHEM. SOC., 4 December 2012 (2012-12-04) |
| KLEIN ET AL., MABS, vol. 4, no. 6, 2012, pages 1 - 11 |
| KRAMER ET AL., NUCLEIC ACIDS RES., vol. 12, 1984, pages 9441 |
| KUNKEL ET AL., METHODS IN ENZYMOL., vol. 154, 1987, pages 367 - 82 |
| KUNKEL, PROC. NATL. ACAD. SCI. USA, vol. 82, 1985, pages 488 - 92 |
| LONBERG ET AL., NATURE, vol. 368, 1994, pages 856 |
| MANIATIS ET AL.: "Molecular Cloning, A Laboratory Manual", 1989, COLD SPRING HARBOR LABORATORY |
| MARKS ET AL., BIOLTECHNOLOGY, vol. 10, 1992, pages 779 - 783 |
| MCMAHAN ET AL., EMBO J., vol. 10, 1991, pages 2821 |
| no. 2413428-34-7 |
| NYGRENUHLEN, CURR. OPIN. IN STRUCT. BIOL., vol. 7, 1997, pages 463 - 469 |
| OLSEN DJORGENSEN J T, FRONT. ONCOL., vol. 4, 16 May 2014 (2014-05-16), pages 105 |
| PATINOTE ET AL., EUR. J. MED. CHEM., vol. 193, 2020, pages 112238 |
| PLUCKTHUN ET AL., METHODS ENZYMOL., vol. 178, 1989, pages 497 - 515 |
| PRESTA, CURR. OP. STRUCT. BIOL., vol. 2, 1992, pages 593 - 596 |
| RABUKA ET AL., NAT. PROTOCOLS, vol. 10, 2012, pages 1052 - 1067 |
| REINEKE, METHODS MOL BIOL, vol. 248, 2004, pages 443 - 463 |
| RIECHMANN ET AL., NATURE, vol. 332, 1988, pages 323 - 329 |
| RYAN ET AL., FOOD & AGRICULTURE IMMUNOL.,, vol. 13, 2001, pages 127 - 130 |
| SANGER ET AL., PNAS, vol. 74, 1977, pages 5463 |
| SASTRY ET AL., PROC. NATL. ACAD. SCI. USA, vol. 86, 1989, pages 5728 - 9272 |
| SCHLEBUSCH ET AL., HYBRIDOMA, vol. 16, 1997, pages 47 - 52 |
| TAYLOR ET AL., , INT. IMMUN., vol. 6, 1994, pages 579 |
| TAYLOR ET AL., NUCL. ACIDS RES, vol. 20, 1992, pages 6287 - 6295 |
| THOMPSON ET AL., J. MOL. BIOL., vol. 256, 1996, pages 350 - 368 |
| THORNTON ET AL., NATURE, vol. 354, 1991, pages 105 |
| TITAS DEB ET AL., CHEM. REV., vol. 121, no. 12, 2021, pages 6850 - 6914 |
| TOMER, PROTEIN SCIENCE, vol. 9, 2000, pages 487 - 496 |
| VAUGHAN ET AL., NATURE BIOTECH., vol. 16, 1998, pages 535 - 539 |
| WANG ET AL., J. AM. CHEM. SOC., vol. 125, no. 11, 2003, pages 3192 - 3193 |
| WINTER ET AL., ANNU. REV. IMMUNOL., vol. 12, 1994, pages 433 - 55 |
| YANG ET AL., J. MOL. BIOL., vol. 254, 1995, pages 392 - 403 |
| ZHANG, W. ET AL., , MOLECULAR IMMUNOLOGY., vol. 42, no. 12, 2005, pages 1445 - 1451 |
Also Published As
| Publication number | Publication date |
|---|---|
| CL2025001577A1 (en) | 2025-11-28 |
| MX2025006112A (en) | 2025-08-01 |
| CN120659785A (en) | 2025-09-16 |
| WO2024118785A3 (en) | 2024-07-04 |
| AU2023403422A1 (en) | 2025-07-17 |
| JP2025540064A (en) | 2025-12-11 |
| IL321396A (en) | 2025-08-01 |
| KR20250128394A (en) | 2025-08-27 |
| CO2025008795A2 (en) | 2025-07-17 |
| EP4626553A2 (en) | 2025-10-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7728821B2 (en) | Improved method for producing antibody-drug conjugates | |
| CA3078218C (en) | Antibody-pyrrolobenzodiazepine derivative conjugate | |
| JP7051692B2 (en) | Antibodies that specifically bind to PD-1 and their use | |
| US20210139604A1 (en) | Compositions of antibody construct-agonist conjugates and methods of use thereof | |
| JP7767411B2 (en) | Anti-FGFR2 antibodies and methods of use thereof | |
| KR102877468B1 (en) | Traceless linker and its protein-conjugate | |
| ES2401536T3 (en) | Monoclonal antibodies that bind to HGM-CSF and medicinal compositions that comprise them | |
| KR20230038738A (en) | Camptothecin analogues conjugated to glutamine residues in proteins and uses thereof | |
| EP3386536A2 (en) | Composition of antibody construct-agonist conjugates and methods of use thereof | |
| WO2018140831A2 (en) | Tumor targeting conjugates and methods of use thereof | |
| CA3042950A1 (en) | Anti-met antibodies, bispecific antigen binding molecules that bind met, and methods of use thereof | |
| EP3576782A1 (en) | Construct-peptide compositions and methods of use thereof | |
| JP2024509099A (en) | Anti-HER2 antibody-drug conjugates and uses thereof | |
| CA3166619A1 (en) | Bispecific antigen binding molecules that bind her2, and methods of use thereof | |
| WO2020100954A1 (en) | (anti-cdh6 antibody)-(pyrrolobenzodiazepine derivative) conjugate | |
| TW202019960A (en) | Immune conjugate targeting ADAM9 and method of use | |
| JP2023553808A (en) | MCL-1 inhibitor antibody-drug conjugates and methods of use | |
| CA3128519A1 (en) | Methods of treating ocular cancer using anti-met antibodies and bispecific antigen binding molecules that bind met | |
| WO2025214486A1 (en) | Anti-trop2 antibody and anti-5t4 antibody-natural killer cell conjugate and use thereof | |
| AU2023403422A1 (en) | Tlr7 agonists and antibody-drug-conjugates thereof | |
| TW202509062A (en) | Anti-b7-h3 antibodies and uses thereof | |
| TW202430557A (en) | Anti-Met antibody, antibody-drug conjugate, and preparation method and use thereof | |
| WO2026021476A1 (en) | Anti-cmet and anti-egfr multispecific antibody drug conjugates | |
| WO2025130990A1 (en) | Tumor microenvironment-activated drug conjugate and antibody-drug conjugate | |
| GB2552041A (en) | Compositions of antibody construct-agonist conjugates and methods thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 23836674 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2025/006112 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2025531228 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2025531228 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2501003562 Country of ref document: TH |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112025010835 Country of ref document: BR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 822727 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: DZ2025000858 Country of ref document: DZ Ref document number: DZP2025000858 Country of ref document: DZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11202503634U Country of ref document: SG Ref document number: 202517061569 Country of ref document: IN Ref document number: 202591695 Country of ref document: EA |
|
| WWP | Wipo information: published in national office |
Ref document number: 822727 Country of ref document: NZ Ref document number: 11202503634U Country of ref document: SG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: AU2023403422 Country of ref document: AU Ref document number: 2023836674 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2023836674 Country of ref document: EP Effective date: 20250630 |
|
| ENP | Entry into the national phase |
Ref document number: 2023403422 Country of ref document: AU Date of ref document: 20231129 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202380092608.9 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: MX/A/2025/006112 Country of ref document: MX |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020257021135 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 202380092608.9 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2023836674 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 202517061569 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 112025010835 Country of ref document: BR Kind code of ref document: A2 Effective date: 20250529 |