WO2024099898A1 - Substituted bi-and tricyclic hset inhibitors - Google Patents
Substituted bi-and tricyclic hset inhibitors Download PDFInfo
- Publication number
- WO2024099898A1 WO2024099898A1 PCT/EP2023/080663 EP2023080663W WO2024099898A1 WO 2024099898 A1 WO2024099898 A1 WO 2024099898A1 EP 2023080663 W EP2023080663 W EP 2023080663W WO 2024099898 A1 WO2024099898 A1 WO 2024099898A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mmol
- methyl
- mixture
- amino
- hplc
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains three hetero rings
- C07D487/14—Ortho-condensed systems
Definitions
- the invention relates to substituted bi- and tricycles of the general formula I, and the use of the compounds of the present invention for the treatment and/or prevention of hyperproliferative diseases and disorders such as cancer in mammals, especially humans, and pharmaceutical compositions containing such compounds.
- DNA replication followed by equal chromosome segregation, ensures the accurate transmission of the genetic information to daughter cells (Hall et al., 2003; Nigg, 2002; Zyss and Gergely, 2009).
- centrosomes act as the dominant sites for spindle pole formation (Meunier and Vernos, 2012). Centrosome duplication is also tightly controlled and occurs simultaneously with DNA replication, thereby ensuring the generation of two functional centrosomes that form the poles of the mitotic spindle (Sharp et al., 2000).
- MT microtubule
- HSET encoded by KIFC1 in humans and Kifc5a in mice
- a minus-end MT motor is of interest in cancer due to its impact on cell division (Cai et al., 2010; Goshima et al., 2005).
- centrosomes and in particular HSET, for bipolar spindle formation has attracted much attention, although the precise role of HSET in this process remains a topic for debate (Mahoney et al., 2006; Tillement et al., 2009). Recent reports have linked centrosome amplification and high HSET expression to chromosome missegregation and aneuploidy, which are hallmarks of human cancer (Marx et al., 2009).
- Centrosome amplification disrupts asymmetric cell division in neuroblastoma cells and causes tumorigenesis in a fly model (Basto et al., 2008), and supernumerary centrosomes are also found in most solid tumor types, forming markers for aggressiveness in breast, brain, prostate, cervix, kidney, and bladder cancers (Chan, 2011). Hence, it is increasingly apparent that supernumerary centrosomes are not only indicative of malignancy but may also drive malignant transformation (Ogden et al., 2013).
- centrosome clustering prevents multipolar mitosis and cell death, it prolongs mitosis and increases the frequency of chromosome missegregation as a result of merotelic kinetochore attachments (Ganem et al., 2009; Kwon et al., 2008; Yang et al., 2008). Based on previous studies, centrosome clustering may prove to be the Achilles heel of cancer cells with supernumerary centrosomes (Basto et al., 2008), and a growing body of evidence suggests that inhibition of centrosome clustering could provide a new therapeutic strategy for tumors with a high incidence of centrosome amplification (Jordan and Wilson, 2004; Ogden et al., 2012).
- HSET A key protein that is known to be crucial for centrosome clustering is HSET (Ned in flies). HSET is required by tumour cells to cluster supernumerary centrosomes (Basto et al., 2008; Kwon et al., 2008). HSET is a member of the Kinesin 14 family of MT motor proteins, which are force-generating enzymes that facilitate movement along MTs within the cell (Mountain et al., 1999) and which transport organelles, protein complexes and mRNAs along microtubules in an ATP-dependent fashion.
- HSET is a minus-end directed motor kinesin, that cross-links and slides microtubules exerting inward forces (Walczak et al., 1997; Cai et al., 2009; Rath et al., 2012). Although the precise role of HSET in cell division is not clear, previous evidence suggests that it is essential for the survival of cancer, but not normal, cells (Ganem et al., 2009; Kwon et al., 2008).
- High HSET expression levels are strongly correlated with metastasis of non-small cell lung cancer to the brain, pointing to an association between HSET, centrosome amplification, and tumorigenesis (Cai et al., 2010; Gordon et al., 2001 ; Grinberg-Rashi et al., 2009).
- Knockdown of HSET in normal retinal pigment epithelial 1 (RPE-1) cells or the breast cancer cell line MCF-7 (which does not have a high incidence of centrosome amplification) does not inhibit bipolar spindle formation, and cells undergo normal division (Kleylein-Sohn et al., 2012; Kwon et al., 2008).
- HSET depletion increases cell death and the frequency of multipolarity in cells with supernumerary centrosomes, but not in cells with a normal number of centrosomes.
- HSET depletion induces spindle multi-polarity and selectively sensitizes centrosome amplified ER- breast cancer cell lines, including triple negative breast cancer (TNBC), to cell death (Patel et al., 2018).
- TNBC triple negative breast cancer
- Depletion of HSET was identified as inducing selective cytotoxicity in centrosome amplified cancer cells (Drosopoulos et al., 2014).
- HSET overexpression has been correlated with poor prognosis and resistance to docetaxel in breast cancer (De et al., 2009; Li et al., 2015), is observed in ovarian adenocarcinoma patients (Pawar et al., 2014) and in numerous other cancer types (Pannu et al., 2015).
- NSCLC non-small cell lung carcinoma
- tumours including centrosome amplified tumours
- cytotoxic microtubule-targeted drugs e.g. taxol, eribulin
- these drugs typically show severe side effects and the emergence of drug resistance leading to early relapse.
- agents targeting kinesin motor proteins e.g. Eg5 inhibitors
- mono-polar spindles the opposite phenotype to HSET inhibition
- target all rapidly dividing cells including bone marrow cells. Consequently, they share dose-limiting toxicities with other antimitotic therapies.
- HSET inhibitor is anticipated to show reduced toxicity by selectively killing cells with centrosome amplification whereas cells with the normal number of centrosomes will remain unaffected (Ganem et al., 2009; Patel et al., 2015). These data together provide support for developing agents that selectively inhibit HSET to target centrosome-amplified tumours (Myers and Collins, 2016). Examples of small molecule HSET inhibitors have been described in the literature. AZ82 is an ADP/ATP competitive inhibitor shown to be selective against a panel of nine other kinesins including Eg5 (Wu et al., 2013).
- the compounds according to the invention are highly selective and effective inhibitors of HSET and thus the compounds of the present invention can be used for the treament of hyperproliferative diseases and disorders such as cancer.
- the invention relates to the compounds of the general formula I, wherein
- W denotes wherein 1-4 H-atoms may be replaced by D
- R 1 denotes Hal, A or OA
- R 2 denotes
- X denotes CH or N
- A denotes H, F, OH, NH2, or unbranched or branched alkyl or cycloalkyl with 1- 12 C-atoms, which may be substituted by R 4 and wherein two adjacent CH- and/or CH2-groups may form a double or triple bond and wherein one or two non-adjacent CH- and/or CH2-groups may be replaced by N-, O- and/or S- atoms and wherein 1-7 H-atoms may be replaced by D, F or Cl,
- R 3 denotes H or A
- R 4 denotes H or unbranched or branched alkyl with 1-4 C-atoms
- Hal denotes F, Cl, Br or I and physiologically acceptable salts, derivatives, solvates, prodrugs and stereoisomers thereof, including mixtures thereof in all ratios.
- a preferred embodiment of the present invention are compounds according to formula I, wherein
- R 1 , R 2 , R 3 , R 4 , X and A have the meanings as disclosed above, and physiologically acceptable salts, derivatives, solvates, prodrugs and stereoisomers thereof, including mixtures thereof in all ratios.
- a preferred embodiment of the present invention are compounds according to formula I, wherein
- R 1 , R 2 , R 3 , R 4 , X and A have the meanings as disclosed above, and physiologically acceptable salts, derivatives, solvates, prodrugs and stereoisomers thereof, including mixtures thereof in all ratios.
- a preferred embodiment of the present invention are compounds according to formula I, wherein
- R 1 denotes OA and W
- R 2 , R 3 , R 4 , X and A have the meanings as disclosed above, and physiologically acceptable salts, derivatives, solvates, prodrugs and stereoisomers thereof, including mixtures thereof in all ratios.
- a preferred embodiment of the present invention are compounds according to formula I, wherein R 1 denotes OA, wherein A denotes an unbranched or branched alkyl wherein 1-3 H- atoms may be replaced by D, F or Cl and W, R 2 , R 3 , R 4 , X and A have the meanings as disclosed above, and physiologically acceptable salts, derivatives, solvates, prodrugs and stereoisomers thereof, including mixtures thereof in all ratios.
- a preferred embodiment of the present invention are compounds according to formula I, wherein
- R 2 denotes and W, R 1 , R 3 , R 4 , X and A have the meanings as disclosed above, and physiologically acceptable salts, derivatives, solvates, prodrugs and stereoisomers thereof, including mixtures thereof in all ratios.
- a preferred embodiment of the present invention are compounds according to formula I, wherein
- R 3 denotes 5-methyloxadiazol or 2-methyltretrazol and W, R 1 , R 2 , R 4 , X and A have the meanings as disclosed above, and physiologically acceptable salts, derivatives, solvates, prodrugs and stereoisomers thereof, including mixtures thereof in all ratios.
- a preferred embodiment of the present invention are compounds according to formula I, wherein
- R 3 denotes 5-methyloxadiazol and W
- R 1 , R 2 , R 4 , X and A have the meanings as disclosed above, and physiologically acceptable salts, derivatives, solvates, prodrugs and stereoisomers thereof, including mixtures thereof in all ratios.
- a preferred embodiment of the present invention are compounds according to formula I, wherein R 4 denotes methyl and W, R 1 , R 2 , R 3 , X and A have the meanings as disclosed above, and physiologically acceptable salts, derivatives, solvates, prodrugs and stereoisomers thereof, including mixtures thereof in all ratios.
- the invention preferably relates to a compound selected from the group consisting of:
- the invention further relates to a pharmaceutical preparation comprising one or more compounds according to the present invention and/or one of its physiologically acceptable salts, derivatives, solvates, prodrugs and stereoisomers, including mixtures thereof in all ratios.
- the invention also relates to a pharmaceutical preparation according to the invention of this type, comprising further excipients and/or adjuvants.
- the invention relates to an above pharmaceutical preparation according to the invention, comprising at least one further medicament active compound.
- compositions are taken to mean, for example, salts of the compounds of the present invention, and also so-called prodrug compounds.
- Prodrug compounds are taken to mean derivatives of the compounds of the present invention which have been modified by means of, for example, alkyl or acyl groups (see also amino- and hydroxyl-protecting groups below), sugars or oligopeptides and which are rapidly cleaved or liberated in the organism to form the effective molecules.
- These also include biodegradable polymer derivatives of the compound of the present invention, as described, for example, in Int. J. Pharm. 115 (1995), 61-67.
- the compound of the present invention can be used in its final non-salt form.
- the present invention also encompasses the use of the compound of the present invention in the form of its pharmaceutically acceptable salts, which can be derived from various organic and inorganic bases by procedures known in the art.
- Pharmaceutically acceptable salt forms of the compound of the present invention are for the most part prepared by conventional methods. If the compound of the present invention contains a carboxyl group, one of its suitable salts can be formed by reacting the compound of the present invention ith a suitable base to give the corresponding base-addition salt.
- Such bases are, for example, alkali metal hydroxides, including potassium hydroxide, sodium hydroxide and lithium hydroxide; alkaline-earth metal hydroxides, such as barium hydroxide and calcium hydroxide; alkali metal alkoxides, for example potassium ethoxide and sodium propoxide; and various organic bases, such as piperidine, diethanolamine and N-methylglutamine.
- alkali metal hydroxides including potassium hydroxide, sodium hydroxide and lithium hydroxide
- alkaline-earth metal hydroxides such as barium hydroxide and calcium hydroxide
- alkali metal alkoxides for example potassium ethoxide and sodium propoxide
- organic bases such as piperidine, diethanolamine and N-methylglutamine.
- the aluminium salts of the compound of the present invetion are likewise included.
- the base salts of the compounds of the present invention include aluminium, ammonium, calcium, copper, iron(lll), iron(ll), lithium, magnesium, man- ganese(lll), manganese(ll), potassium, sodium and zinc salts, but this is not intended to represent a restriction.
- Salts of the compounds of the present invention which are derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary and tertiary amines, substituted amines, also including naturally occurring substituted amines, cyclic amines, and basic ion exchanger resins, for example arginine, betaine, caffeine, chloroprocaine, choline, N,N'-dibenzylethylenediamine (benzathine), dicyclohexylamine, diethanolamine, diethylamine, 2-diethylamino- ethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine
- the pharmaceutically acceptable base-addition salts of the compound of the present invention are formed with metals or amines, such as alkali metals and alkaline-earth metals or organic amines.
- metals are sodium, potassium, magnesium and calcium.
- Preferred organic amines are N,N’-dibenzylethylene- diamine, chloroprocaine, choline, diethanolamine, ethylenediamine, N-methyl-D- glucamine and procaine.
- the base-addition salts of the compounds of the present invention are prepared by bringing the free acid form into contact with a sufficient amount of the desired base, causing the formation of the salt in a conventional manner.
- the free acid can be regenerated by bringing the salt form into contact with an acid and isolating the free acid in a conventional manner.
- the free acid forms differ in a certain respect from the corresponding salt forms thereof with respect to certain physical properties, such as solubility in polar solvents; for the purposes of the invention, however, the salts otherwise correspond to the respective free acid forms thereof.
- the term “pharmaceutically acceptable salt” in the present connection is taken to mean an active compound which comprises the compound of the present invention in the form of one of its salts, in particular if this salt form imparts improved pharmacokinetic properties on the active compound compared with the free form of the active compound or any other salt form of the active compound used earlier.
- the pharmaceutically acceptable salt form of the active compound can also provide this active compound for the first time with a desired pharmacokinetic property which it did not have earlier and can even have a positive influence on the pharmacodynamics of this active compound with respect to its therapeutic efficacy in the body.
- Solvates of the compound of the present invention are taken to mean adductions of inert solvent molecules of the compound of the present invention which form owing to their mutual attractive force.
- Solvates are, for example, hydrates, such as monohydrates or dihydrates, or alcoholates, i.e. addition compounds with alcohols, such as, for example, with methanol or ethanol.
- Compounds of the present invention may contain one or more centres of chirality, so that all stereoisomers, enantiomers, diastereomers, etc., of the compounds of the present inventionare also claimed in the present invention.
- the invention also relates to the optically active forms (stereoisomers), the enantiomers, the racemates, the diastereomers and hydrates and solvates of these compounds.
- Compounds of the present invention according to the invention may be chiral owing to their molecular structure and may accordingly occur in various enantiomeric forms. They may therefore be in racemic or optically active form. Since the pharmaceutical efficacy of the racemates or stereoisomers of the compounds according to the invention may differ, it may be desirable to use the enantiomers. In these cases, the end product, but also even the intermediates, may be separated into enantiomeric compounds by chemical or physical measures known to the person skilled in the art or already employed as such in the synthesis.
- compositions are taken to mean, for example, salts of the compounds according to the invention and also so-called prodrug compounds.
- Prodrug compounds are taken to mean compounds of the present invention which have been modified with, for example, alkyl or acyl groups (see also amino- and hydroxyl-protecting groups below), sugars or oligopeptides and which are rapidly cleaved or liberated in the organism to form the effective compounds according to the invention. These also include biodegradable polymer derivatives of the compounds according to the invention, as described, for example, in Int. J. Pharm. 115 (1995), 61-67.
- Suitable acid-addition salts are inorganic or organic salts of all physiologically or pharmacologically acceptable acids, for example halides, in particular hydrochlorides or hydrobromides, lactates, sulfates, citrates, tartrates, maleates, fumarates, oxalates, acetates, phosphates, methylsulfonates or p-toluenesulfonates.
- Solvates of the compounds of the present invention are taken to mean adductions of inert solvent molecules onto the compounds of the present invention which form owing to their mutual attractive force.
- Solvates are, for example, hydrates, such as monohydrates or dihydrates, or alcoholates, i.e. addition compounds with alcohols, such as, for example, with methanol or ethanol.
- a compound of the present invention includes isotopelabelled forms thereof.
- An isotope-labelled form of a compound of the present invention is identical to this compound apart from the fact that one or more atoms of the compound have been replaced by an atom or atoms having an atomic mass or mass number which differs from the atomic mass or mass number of the atom which usually occurs naturally.
- isotopes which are readily commercially available, and which can be incorporated into a compound of the present invention by well-known methods include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus, fluorine and chlorine, for example 2 H, 3 H, 13 C, 14 C, 15 N, 18 O, 17 0, 31 P, 32 P, 35 S, 18 F and 36 CI, respectively.
- a compound of the present invention, a prodrug thereof or a pharmaceutically acceptable salt of either which contains one or more of the above-mentioned isotopes and/or other isotopes of other atoms is intended to be part of the present invention.
- An isotope-labelled compound of the present invention can be used in a number of beneficial ways.
- an isotope-labelled compound of the present invention into which, for example, a radioisotope, such as 3 H or 14 C, has been incorporated is suitable for medicament and/or substrate tissue distribution assays.
- radioisotopes i.e. tritium ( 3 H) and carbon-14 ( 14 C)
- 3 H tritium
- 14 C carbon-14
- Incorporation of heavier isotopes, for example deuterium ( 2 H) into a compound of the present invention has therapeutic advantages owing to the higher metabolic stability of this isotope-labelled compound. Higher metabolic stability translates directly into an increased in-vivo half-life or lower dosages, which under most circumstances would represent a preferred embodiment of the present invention.
- An isotope-labelled compound of the present invention can usually be prepared by carrying out the procedures disclosed in the synthesis schemes and the related description, in the example part and in the preparation part in the present text, replacing a non-isotope-labelled reactant with a readily available isotope-labelled reactant.
- deuterium ( 2 H) can also be incorporated into a compound of the present invention.
- the primary kinetic isotope effect is a change in the rate of a chemical reaction that results from exchange of isotopic nuclei, which in turn is caused by the change in ground state energies necessary for covalent bond formation after this isotopic exhange.
- Exchange of a heavier isotope usually results in a lowering of the ground state energy for a chemical bond and thus causes a reduction in the rate in rate-limiting bond breakage. If the bond breakage occurs in or in the vicinity of a saddle-point region along the coordinate of a multi-product reaction, the product distribution ratios can be altered substantially.
- a compound of the present invention which has multiple potential sites of attack for oxidative metabolism, for example benzylic hydrogen atoms and hydrogen atoms bonded to a nitrogen atom, is prepared as a series of analogues in which various combinations of hydrogen atoms are replaced by deuterium atoms, so that some, most or all of these hydrogen atoms have been replaced by deuterium atoms.
- Half-life determinations enable favourable and accurate determination of the extent to which the improvement in resistance to oxidative metabolism has improved. In this way, it is determined that the half-life of the parent compound can be extended by up to 100% as the result of deuterium-hydrogen exchange of this type.
- the replacement of hydrogen by deuterium in a compound of the present invention can also be used to achieve a favourable modification of the metabolite spectrum of the starting compound in order to diminish or eliminate undesired toxic metabolites.
- a toxic metabolite arises through oxidative carbonhydrogen (C-H) bond cleavage
- C-H oxidative carbonhydrogen
- the invention also relates to mixtures of the compounds of the present invention according to the invention, for example mixtures of two diastereomers, for example in the ratio 1:1, 1:2, 1 :3, 1 :4, 1:5, 1:10, 1:100 or 1:1000. These are particularly preferably mixtures of two stereoisomeric compounds. However, preference is also given to mixtures of two or more compounds of the present invention.
- the invention relates to a process for the preparation of the compounds of the present invention, characterized in that a) the base of a compound of the present invention is converted into one of its salts by treatment with an acid, or b) an acid of a compound of the present invention is converted into one of its salts by treatment with a base.
- the starting materials or starting compounds are generally known. If they are novel, they can be prepared by methods known per se. If desired, the starting materials can also be formed in situ by not isolating them from the reaction mixture, but instead immediately converting them further into the compounds of the present invention.
- the compounds of the present invention are preferably obtained by liberating them from their functional derivatives by solvolysis, in particular by hydrolysis, or by hydrogenolysis.
- Preferred starting materials for the solvolysis or hydrogenolysis are those which contain correspondingly protected amino, carboxyl and/or hydroxyl groups instead of one or more free amino, carboxyl and/or hydroxyl groups, preferably those which carry an amino-protecting group instead of an H atom which is connected to an N atom.
- Preference is furthermore given to starting materials which carry a hydroxyl-protecting group instead of the H atom of a hydroxyl group.
- Preference is also given to starting materials which carry a protected carboxyl group instead of a free carboxyl group.
- amino-protecting group is generally known and relates to groups which are suitable for protecting (blocking) an amino group against chemical reactions, but which can easily be removed after the desired chemical reaction has been carried out elsewhere in the molecule.
- Typical of such groups are, in particular, unsubstituted or substituted acyl groups, furthermore unsubstituted or substituted aryl (for example 2,4-dinitophenyl) or aralkyl groups (for example benzyl, 4- nitrobenzyl, triphenylmethyl). Since the amino-protecting groups are removed after the desired reaction or reaction sequence, their type and size are, in addition, not crucial, but preference is given to those having 1-20, in particular 1-8, C atoms.
- acyl group is to be understood in the broadest sense in connection with the present process. It encompasses acyl groups derived from aliphatic, araliphatic, aromatic or heterocyclic carboxylic acids or sulfonic acids and, in particular, alkoxycarbonyl, aryloxycarbonyl and especially aralkoxycarbonyl groups.
- acyl groups are alkanoyl, such as acteyl, propionyl, buturyl, aralkanoyl, such as phenylacetyl, aroyl, such as benzoyl or toluyl, aryoxyaklkanoyl, such as phenoxyacetyl, alkyoxycarbonyyl, such as methoxycarbonyl, ethoxycarbonyl, 2,2,2- trichloroethoxycarbonyl, BOC, 2-iodoethoxycaronyl, aralkoxycarbonyl, such as CBZ, 4-methoxybenzyloxycarbonyl or FMOC.
- Preferred acyl groups are CBZ, FMOC, benzyl and acetyl.
- acid-protecting group or “carboxyl-protecting group” is likewise generally known and relates to groups which are suitable for protecting a -COOH group against chemical reactions, but which can easily be removed after the desired chemical reaction has been carried out elsewhere in the molecule.
- esters instead of the free acids, for example of substituted and unsubstituted alkyl esters (such as methyl, ethyl, tert-butyl and substituted derivatives thereof), of substituted and unsubstituted benzyl esters or silyl esters, is typical.
- the type and size of the acid-protecting groups is not crucial, but preference is given to those having 1-20, in particular 1-10, C atoms.
- hydroxyl-protecting group is likewise generally known and relates to groups which are suitable for protecting a hydroxyl group against chemical reactions, but which can easily be removed after the desired chemical reaction has been carried out elsewhere in the molecule. Typical of such groups are the above- mentioned unsubstituted or substituted aryl, aralkyl or acyl groups, furthermore also alkyl groups. Their type and size of the hydroxyl-protecting groups is not crucial, but preference is given to those having 1-20, in particular 1-10, C atoms.
- hyrdoxyl-protecting groups are, inter alia, benzyl, p-nitrobenzoyl, p-toluenesulfonyl and acetyl, where benzyl and acetyl are preferred.
- the functional derivatives of the compounds of the present invention to be used as starting materials can be prepared by known methods of amino-acid and peptide synthesis, as described, for example, in the said standard works and patent applications.
- the compounds of the present invention are liberated from their functional derivatives, depending on the protecting group used, for example, with the aid of strong acids, advantageously using trifluoroacetic acid or perchloric acid, but also using other strong inorganic acids, such as hydrochloric acid or sulfuric acid, strong organic acids, such as trichloroacetic acid, or sulfonic acids, such as benzoyl- or p- toluenesulfonic acid.
- strong acids advantageously using trifluoroacetic acid or perchloric acid
- other strong inorganic acids such as hydrochloric acid or sulfuric acid
- strong organic acids such as trichloroacetic acid
- sulfonic acids such as benzoyl- or p- toluenesulfonic acid.
- the starting materials can optionally be reacted in the presence of an inert solvent.
- Suitable inert solvents are, for example, heptane, hexane, petroleum ether, DMSO, benzene, toluene, xylene, trichloroethylene-, 1 ,2-dichloroethane, carbon tetrachloride, chloroform or dichloromethane; alcohols, such as methanol, ethanol, isopropanol, n-propanol, n-butanol or tert-butanol; ethers, such as diethyl ether, diisopropyl ether (preferably for substitution on the indole nitrogen), tetrahydrofuran (THF) or dioxane; glycol ethers, such as ethylene glycol monomethyl or monoethyl ether, ethylene glycol dimethyl ether (diglyme); ketones, such as acetone or butanone; amides, such as acetamide, dimethylacetamide, N-methylpyrrol
- the amount of solvent is not crucial; 10 g to 500 g of solvent can preferably be added per g of the compound of the present invention to be reacted.
- an acid-binding agent for example an alkali metal or alkaline-earth metal hydroxide, carbonate or bicarbonate or other alkali or alkaline- earth metal salts of weak acids, preferably a potassium, sodium or calcium salt, or to add an organic base, such as, for example, triethylamine, dimethylamine, pyridine or quinoline, or an excess of the amine component.
- an acid-binding agent for example an alkali metal or alkaline-earth metal hydroxide, carbonate or bicarbonate or other alkali or alkaline- earth metal salts of weak acids, preferably a potassium, sodium or calcium salt
- organic base such as, for example, triethylamine, dimethylamine, pyridine or quinoline, or an excess of the amine component.
- the resultant compounds according to the invention can be separated from the corresponding solution in which they are prepared (for example by centrifugation and washing) and can be stored in another composition after separation, or they can remain directly in the preparation solution.
- the resultant compounds according to the invention can also be taken up in desired solvents for the particular use.
- the reaction duration depends on the reaction conditions selected. In general, the reaction duration is 0.5 hour to 10 days, preferably 1 to 24 hours. On use of a microwave, the reaction time can be reduced to values of 1 to 60 minutes.
- the compounds of the present invention and also the starting materials for their preparation are, in addition, prepared by known methods, as described in the literature (for example in standard works, such as Houben-Weyl, Methoden der organischen Chemie [Methods of Organic Chemistry], Georg-Thieme-Verlag, Stuttgart), for example under reaction conditions which are known and suitable for the said reactions. Use can also be made here of variants known per se, which are not described here in greater detail.
- An acid of the present invention can be converted into the associated addition salt using a base, for example by reaction of equivalent amounts of the acid and base in an inert solvent, such as ethanol, and inclusive evaporation.
- Suitable bases for this reaction are, in particular, those which give physiologically acceptable salts.
- the acid of the present invention can be converted into the corresponding metal salt, in particular alkali or alkaline-earth metal salt, using a base (for example sodium hydroxide, potassium hydroxide, sodium carbonate or potassium carbonate) or into the corresponding ammonium salt.
- Organic bases which give physiologically acceptable salts, such as, for example, ethanolamine, are also suitable for this reaction.
- a base of the present invention can be converted into the associated acid-addition salt using an acid, for example by reaction of equivalent amounts of the base and acid in an inert solvent, such as ethanol, with subsequent evaporation.
- Suitable acids for this reaction are, in particular, those which give physiologically acceptable salts.
- inorganic acids for example sulfuric acid, nitric acid, hydrohalic acids, such as hydrochloric acid or hydrobromic acid, phosphoric acids, such as orthophosphoric acid, sulfamic acid, furthermore organic acids, in particular aliphatic, alicyclic, araliphatic, aromatic or heterocyclic, mono- or polybasic carboxylic, sulfonic or sulfuric acids, for example formic acid, acetic acid, propionic acid, pivalic acid, diethylacetic acid, malonic acid, succinic acid, pimelic acid, fumaric acid, maleic acid, lactic acid, tartaric acid, malic acid, citric acid, gluconic acid, ascorbic acid, nicotinic acid, isonicotinic acid, methane- or ethanesulfonic acid, ethanedisulfonic acid, 2-hydroxysulfonic acid, benzenesulfonic acid, p-tol
- the invention therefore furthermore relates to the use of compounds according to the invention for the preparation of a medicament for the treatment and/or prophylaxis of diseases which are caused, promoted and/or propagated by HSET.
- the invention thus also relates, in particular, to a medicament comprising at least one compound according to the invention and/or one of its physiologically acceptable salts, derivatives, solvates, prodrugs and stereoisomers, including mixtures thereof in all ratios, for use in the treatment and/or prophylaxis of physiological and/or pathophysiological states.
- physiological and/or pathophysiological states which are connected to HSET.
- Physiological and/or pathophysiological states are taken to mean physiological and/or pathophysiological states which are medically relevant, such as, for example, diseases or illnesses and medical disorders, complaints, symptoms or complications and the like, in particular diseases.
- the invention furthermore relates to a medicament comprising at least one compound according to the invention and/or one of its physiologically acceptable salts, derivatives, solvates, prodrugs and stereoisomers, including mixtures thereof in all ratios, for use in the treatment and/or prophylaxis of physiological and/or pathophysiological states selected from the group consisting of hyperproliferative diseases and disorders.
- the invention further relates to a medicament comprising at least one compound according to the invention and/or one of its physiologically acceptable salts, derivatives, solvates, prodrugs and stereoisomers, including mixtures thereof in all ratios, for use in the treatment and/or prophylaxis of physiological and/or pathophysiological states selected from the group consisting of hyperproliferative and infectious diseases and disorders, wherein the hyperproliferative disease or disorder is cancer.
- the invention thus particularly preferably relates to a medicament comprising at least one compound according to the invention and/or one of its physiologically acceptable salts, derivatives, solvates, prodrugs and stereoisomers, including mixtures thereof in all ratios, wherein the cancer is selected from the group consisting of from the group consisting of acute lymphocytic leukemia, acute granulocytic leukemia, adrenal cortex cancer, bladder cancer, brain cancer, breast cancer, cervical hyperplasia, cervical cancer, chorio cancer, chronic granulocytic leukemia, chronic lymphocytic leukemia, colon cancer, endometrial ccancer, esophageal cancer, essential thrombocytosis, genitourinary carcinoma, glioma, glioblastoma, hairy cell leukemia, head and neck carcinoma, Hodgkin's disease, Kaposi's sarcoma, lung carcinoma, lymphoma, malignant carcinoid carcinoma, malignant hypercalcemia, malignant mela
- the invention further preferably relates to a medicament comprising at least one compound according to the invention and/or one of its physiologically acceptable salts, derivatives, solvates, prodrugs and stereoisomers, including mixtures thereof in all ratios, for use in the treatment and/or prophylaxis of physiological and/or pathophysiological states selected from the group consisting of hyperproliferative and infectious diseases and disorders, wherein the hyperproliferative disease or disorder is selected from the group consisting of age-related macular degeneration, Crohn's disease, cirrhosis, chronic inflammatory-related disorders, proliferative diabetic retinopathy, proliferative vitreoretinopathy, retinopathy of prematurity, granulomatosis, immune hyperproliferation associated with organ or tissue transplantation and an immunoproliferative disease or disorder selected from the group comnsisting of inflammatory bowel disease, psoriasis, rheumatoid arthritis, systemic lupus erythematosus
- the medicaments disclosed above include a corresponding use of the compounds according to the invention for the preparation of a medicament for the treatment and/or prophylaxis of the above physiological and/or pathophysiological states.
- the medicaments disclosed above include a corresponding method for the treatment and/or prophylaxis of the above physiological and/or pathophysiological states in which at least one compound according to the invention is administered to a patient in need of such a treatment.
- the compounds according to the invention preferably exhibit an advantageous biological activity which can easily be demonstrated in enzyme assays and animal experiments, as described in the examples.
- the compounds according to the invention preferably exhibit and cause an inhibiting effect, which is usually documented by IC50 values in a suitable range, preferably in the micromolar range and more preferably in the nanomolar range.
- the compounds according to the invention can be administered to humans or animals, in particular mammals, such as apes, dogs, cats, rats or mice, and can be used in the therapeutic treatment of the human or animal body and in the combating of the above-mentioned diseases. They can furthermore be used as diagnostic agents or as reagents.
- compounds according to the invention can be used for the isolation and investigation of the activity or expression of HSET.
- they are particularly suitable for use in diagnostic methods for diseases in connection with disturbed HSET activity.
- the invention therefore furthermore relates to the use of the compounds according to the invention for the isolation and investigation of the activity or expression of HSET or as binders and inhibitors of HSET.
- the compounds according to the invention can, for example, be radioactively labelled.
- radioactive labels are 3 H, 14 C, 231 l and 125 l.
- a preferred labelling method is the iodogen method (Fraker et al., 1978).
- the compounds according to the invention can be labelled by enzymes, fluorophores and chemophores.
- enzymes are alkaline phosphatase, p- galactosidase and glucose oxidase
- an example of a fluorophore is fluorescein
- an example of a chemophore is luminol
- automated detection systems for example for fluorescent colorations, are described, for example, in US 4,125,828 and US 4,207,554.
- the present invention further relates to pharmaceutical compositions containing the compounds of the present invention and their use for the treatment and/or prophylaxis of diseases and disorders where the partial or total inactivation of HSET could be beneficial.
- the compounds of the present invention can be used for the preparation of pharmaceutical preparations, in particular by non-chemical methods. In this case, they are brought into a suitable dosage form together with at least one solid, liquid and/or semi-liquid excipient or adjuvant and optionally in combination with one or more further active compound(s).
- the invention therefore furthermore relates to pharmaceutical preparations comprising at least one compound of the present invention and/or physiologically acceptable salts, derivatives, solvates and stereoisomers thereof, including mixtures thereof in all ratios.
- the invention also relates to pharmaceutical preparations which comprise further excipients and/or adjuvants, and also to pharmaceutical preparations which comprise at least one further medicament active compound.
- the invention also relates to a process for the preparation of a pharmaceutical preparation, characterised in that a compound of the present inventionand/or one of its physiologically acceptable salts, derivatives, solvates and stereoisomers, including mixtures thereof in all ratios, is brought into a suitable dosage form together with a solid, liquid or semi-liquid excipient or adjuvant and optionally with a further medicament active compound.
- the pharmaceutical preparations according to the invention can be used as medicaments in human or veterinary medicine.
- the patient or host can belong to any mammal species, for example a primate species, particularly humans; rodents, including mice, rats and hamsters; rabbits; horses, cattle, dogs, cats, etc. Animal models are of interest for experimental investigations, where they provide a model for the treatment of a human disease.
- Suitable carrier substances are organic or inorganic substances which are suitable for enteral (for example oral), parenteral or topical administration and do not react with the novel compounds, for example water, vegetable oils (such as sunflower oil or cod-liver oil), benzyl alcohols, polyethylene glycols, gelatine, carbohydrates, such as lactose or starch, magnesium stearate, talc, lanolin or vaseline. Owing to his expert knowledge, the person skilled in the art is familiar which adjuvants are suitable for the desired medicament formulation.
- solvents for example water, physiological saline solution or alcohols, such as, for example, ethanol, propanol or glycerol, sugar solutions, such as glucose or mannitol solutions, or a mixture of the said solvents, gel formers, tablet assistants and other activeingredient carriers
- lubricants for example water, physiological saline solution or alcohols, such as, for example, ethanol, propanol or glycerol
- sugar solutions such as glucose or mannitol solutions
- gel formers such as glucose or mannitol solutions
- preparations or medicaments according to the invention may comprise one or more further active compounds, for example one or more vitamins. If desired, preparations or medicaments according to the invention may comprise one or more further active compounds and/or one or more action enhancers (adjuvants).
- “pharmaceutically tolerated” relates to medicaments, precipitation reagents, excipients, adjuvants, stabilisers, solvents and other agents which facilitate the administration of the pharmaceutical preparations obtained therefrom to a mammal without undesired physiological side effects, such as, for example, nausea, dizziness, digestion problems or the like.
- the compounds according to the invention preferably have the advantage that direct use is possible and further purification steps for the removal of toxicologically unacceptable agents, such as, for example, high concentrations of organic solvents or other toxicologically unacceptable adjuvants, are thus unnecessary before use of the compounds according to the invention in pharmaceutical formulations.
- the invention particularly preferably also relates to pharmaceutical preparations comprising at least one compound according to the invention in precipitated noncrystalline, precipitated crystalline or in dissolved or suspended form, and optionally excipients and/or adjuvants and/or further pharmaceutical active compounds.
- the compounds according to the invention preferably enable the preparation of highly concentrated formulations without unfavourable, undesired aggregation of the compounds according to the invention occurring.
- ready-to-use solutions having a high active-ingredient content can be prepared with the aid of compounds according to the invention with aqueous solvents or in aqueous media.
- the compounds and/or physiologically acceptable salts and solvates thereof can also be lyophilised and the resultant lyophilisates used, for example, for the preparation of injection preparations.
- Aqueous preparations can be prepared by dissolving or suspending compounds according to the invention in an aqueous solution and optionally adding adjuvants.
- defined volumes of stock solutions comprising the said further adjuvants in defined concentration are advantageously added to a solution or suspension having a defined concentration of compounds according to the invention, and the mixture is optionally diluted with water to the pre-calculated concentration.
- the adjuvants can be added in solid form. The amounts of stock solutions and/or water which are necessary in each case can subsequently be added to the aqueous solution or suspension obtained.
- Compounds according to the invention can also advantageously be dissolved or suspended directly in a solution comprising all further adjuvants.
- solutions or suspensions comprising compounds according to the invention and having a pH of 4 to 10, preferably having a pH of 5 to 9, and an osmolality of 250 to 350 mosmol/kg can advantageously be prepared.
- the pharmaceutical preparation can thus be administered directly substantially without pain intravenously, intraarterially, intraarticularly, subcutaneously or percutaneously.
- the preparation may also be added to infusion solutions, such as, for example, glucose solution, isotonic saline solution or Ringer's solution, which may also contain further active compounds, thus also enabling relatively large amounts of active compound to be administered.
- compositions according to the invention may also comprise mixtures of a plurality of compounds according to the invention.
- the preparations according to the invention are physiologically well tolerated, easy to prepare, can be dispensed precisely and are preferably stable with respect to assay, decomposition products and aggregates throughout storage and transport and during multiple freezing and thawing processes. They can preferably be stored in a stable manner over a period of at least three months to two years at refrigerator temperature (2-8°C) and at rt (23-27 °C) and 60% relative atmospheric humidity (R.H.).
- the compounds according to the invention can be stored in a stable manner by drying and when necessary converted into a ready-to-use pharmaceutical preparation by dissolution or suspension.
- Possible drying methods are, for example, without being restricted to these examples, nitrogen-gas drying, vacuum-oven drying, lyophilisation, washing with organic solvents and subsequent air drying, liquid-bed drying, fluidised-bed drying, spray drying, roller drying, layer drying, air drying at rt and further methods.
- the term “effective amount” denotes the amount of a medicament or of a pharmaceutical active compound which causes in a tissue, system, animal or human a biological or medical response which is sought or desired, for example, by a researcher or physician.
- terapéuticaally effective amount denotes an amount which, compared with a corresponding subject who has not received this amount, has the following consequence: improved treatment, healing, prevention or elimination of a disease, syndrome, disease state, complaint, disorder or prevention of side effects or also a reduction in the progress of a disease, complaint or disorder.
- therapeutically effective amount also encompasses the amounts which are effective for increasing normal physiological function.
- the compounds according to the invention and/or physiologically acceptable salts and solvates thereof are generally used analogously to known, commercially available preparations or preparations, preferably in dosages of between 0.1 and 500 mg, in particular 5 and 300 mg, per use unit.
- the daily dose is preferably between 0.001 and 250 mg/kg, in particular 0.01 and 100 mg/kg, of body weight.
- the preparation can be administered one or more times per day, for example two, three or four times per day.
- the individual dose for a patient depends on a large number of individual factors, such as, for example, on the efficacy of the particular compound used, on the age, body weight, general state of health, sex, nutrition, on the time and method of administration, on the excretion rate, on the combination with other medicaments and on the severity and duration of the particular disease.
- a measure of the uptake of a medicament active compound in an organism is its bioavailability. If the medicament active compound is delivered to the organism intravenously in the form of an injection solution, its absolute bioavailability, i.e. the proportion of the pharmaceutical which reaches the systemic blood, i.e. the major circulation, in unchanged form, is 100%.
- the active compound In the case of oral administration of a therapeutic active compound, the active compound is generally in the form of a solid in the formulation and must therefore first be dissolved in order that it is able to overcome the entry barriers, for example the gastrointestinal tract, the oral mucous membrane, nasal membranes or the skin, in particular the stratum corneum, or can be absorbed by the body.
- Data on the pharmacokinetics, i.e. on the bioavailability can be obtained analogously to the method of J. Shaffer et al., J. Pharm. Sciences, 88 (1999), 313-318.
- medicaments of this type can be prepared by means of one of the processes generally known in the pharmaceutical art.
- Medicaments can be adapted for administration via any desired suitable route, for example by the oral (including buccal or sublingual), rectal, pulmonary, nasal, topical (including buccal, sublingual or transdermal), vaginal or parenteral (including subcutaneous, intramuscular, intravenous, intradermal and in particular intraarticular) routes.
- Medicaments of this type can be prepared by means of all processes known in the pharmaceutical art by, for example, combining the active compound with the excipient(s) or adjuvant(s).
- Parenteral administration is preferably suitable for administration of the medicaments according to the invention.
- intra-articular administration is particularly preferred.
- the compounds according to the invention are also suitable for the preparation of medicaments to be administered parenterally having slow, sustained and/or controlled release of active compound. They are thus also suitable for the preparation of delayed-release formulations, which are advantageous for the patient since administration is only necessary at relatively large time intervals.
- the medicaments adapted to parenteral administration include aqueous and nonaqueous sterile injection solutions comprising antioxidants, buffers, bacteriostatics and solutes, by means of which the formulation is rendered isotonic with the blood or synovial fluid of the recipient to be treated; as well as aqueous and non-aqueous sterile suspensions, which can comprise suspension media and thickeners.
- the formulations can be delivered in sigle-dose or multi-dose containers, for example sealed ampoules and vials, and stored in the freeze-dried (lyophilised) state, so that only the addition of the sterile carrier liquid, for example water for injection purposes, immediately before use is necessary.
- Injection solutions and suspensions prepared in accordance with the formulation can be prepared from sterile powders, granules and tablets.
- the compounds according to the invention can also be administered in the form of liposome delivery systems, such as, for example, small unilamellar vesicles, large unilamellar vesicles and multilamellar vesicles.
- Liposomes can be formed from various phospholipids, such as, for example, cholesterol, stearylamine or phosphatidylcholines.
- the compounds according to the invention can also be coupled to soluble polymers as targeted medicament excipients.
- soluble polymers can encompass polyvinylpyrrolidone, pyran copolymer, polyhydroxypropylmethacrylamidophenol, polyhydroxyethylaspartamidophenol or polyethylene oxide polylysine, substituted by palmitoyl radicals.
- the compounds according to the invention can furthermore be coupled to a class of biodegradable polymers which are suitable for achieving slow release of a medicament, for example polylactic acid, poly-epsilon-caprolactone, polyhydroxybutyric acid, polyorthoesters, polyacetals, polydihydroxypyrans, polycyanoacrylates, polylactic-co-glycolic acid, polymers, such as conjugates between dextran and methacrylates, polyphosphoesters, various polysaccharides and polyamines and poly-s-caprolactone, albumin, chitosan, collagen or modified gelatine and crosslinked or amphipathic block copolymers of hydrogels.
- biodegradable polymers which are suitable for achieving slow release of a medicament
- a medicament for example polylactic acid, poly-epsilon-caprolactone, polyhydroxybutyric acid, polyorthoesters, polyacetals, polydihydroxypyrans, polycyanoacrylates, polylactic-co-g
- Suitable for enteral administration are, in particular, tablets, dragees, capsules, syrups, juices, drops or suppositories
- suitable for topical use are ointments, creams, pastes, lotions, gels, sprays, foams, aerosols, solutions (for example solutions in alcohols, such as ethanol or isopropanol, acetonitrile, DMF, dimethylacetamide, 1,2-propanediol or mixtures thereof with one another and/or with water) or powders.
- liposomal preparations are particularly suitable for topical uses.
- the active compound in the case of formulation to give an ointment, can be employed either with a paraffinic or a water-miscible cream base. Alternatively, the active compound can be formulated to a cream with an oil-in-water cream base or a water-in-oil base.
- Medicaments adapted to transdermal administration can be delivered as independent plasters for extended, close contact with the epidermis of the recipient.
- the active compound can be supplied from the plaster by means of iontophoresis, as described in general terms in Pharmaceutical Research, 3 (6), 318 (1986).
- the medicaments according to the invention may also comprise other agents usual in the art with respect to the particular type of pharmaceutical formulation.
- the invention also relates to a set (kit) consisting of separate packs of a) an effective amount of a compound of the present invention and/or physiologically acceptable salts, derivatives, solvates, prodrugs and stereoisomers thereof, including mixtures thereof in all ratios, and b) an effective amount of a further medicament active compound.
- kit consisting of separate packs of a) an effective amount of a compound of the present invention and/or physiologically acceptable salts, derivatives, solvates, prodrugs and stereoisomers thereof, including mixtures thereof in all ratios, and b) an effective amount of a further medicament active compound.
- the set comprises suitable containers, such as boxes or cartons, individual bottles, bags or ampoules.
- the set may, for example, comprise separate ampoules each containing an effective amount of a compound of the present inventionand/or pharmaceutically acceptable salts, derivatives, solvates, prodrugs and stereoisomers thereof, including mixtures thereof in all ratios, and an effective amount of a further medicament active compound in dissolved or lyophilised form.
- the medicaments according to the invention can be used in order to provide additive or synergistic effects in certain known therapies and/or can be used in order to restore the efficacy of certain existing therapies.
- the pharmaceutical preparations according to the invention may also comprise further medicament active compounds, for example for use in the treatment of cancer, other anti-tumor medicaments.
- the pharmaceutical preparations according to the invention may also, besides the compounds according to the invention, comprise further medicament active compounds which are known to the person skilled in the art in the treatment thereof.
- methods are provided for enhancing an immune response in a host in need thereof.
- the immune response can be enhanced by reducing T cell tolerance, including by increasing IFN-y release, by decreasing regulatory T cell production or activation, or by increasing antigen-specific memory T cell production in a host.
- the method comprises administering a compound of the present invention to a host in combination or alternation with an antibody.
- the antibody is a therapeutic antibody.
- a method of enhancing efficacy of passive antibody therapy comprising administering a compound of the present invention in combination or alternation with one or more passive antibodies.
- This method can enhance the efficacy of antibody therapy for treatment of abnormal cell proliferative disorders such as cancer or can enhance the efficacy of therapy in the treatment or prevention of infectious diseases.
- the compound of the present invention can be administered in combination or alternation with antibodies such as rituximab, herceptin or erbitux, for example.
- a method of treating or preventing abnormal cell proliferation comprising administering a compound of the present invention to a host in need thereof substantially in the absence of another anticancer agent.
- a method of treating or preventing abnormal cell proliferation in a host in need thereof comprising administering a first compound of the present invention substantially in combination with a first anticancer agent to the host and subsequently administering a second compound of the present invention receptor antagonist.
- the second antagonist is administered substantially in the absence of another anti-cancer agent.
- a method of treating or preventing abnormal cell proliferation in a host in need thereof comprising administering a compound of the present invention substantially in combination with a first anticancer agent to the host and subsequently administering a second anti-cancer agent in the absence of the antagonist.
- cancer treatment disclosed here can be carried out as therapy with a compound of the present invention or in combination with an operation, irradiation or chemotherapy.
- Chemotherapy of this type can include the use of one or more active compounds of the following categories of antitumour active compounds:
- antiproliferative/antineoplastic/DNA-damaging active compounds and combinations thereof, as used in medical oncology such as alkylating active compounds (for example cis-platin, parboplatin, cyclophosphamide, nitrogen mustard, melphalan, chlorambucil, busulphan and nitrosoureas); antimetabolites (for example antifolates such as fluoropyrimidines such as 5-fluorouracil and tegafur, raltitrexed, methotrexate, cytosine arabinoside, hydroxyurea and gemcitabine); antitumour antibiotics (for example anthracyclines, such as adriamycin, bleomycin, doxorubicin, daunomycin, epirubicin, idarubicin, mitomycin-C, dactinomycin and mithramycin); antimitotic active compounds (for example vinca alkaloids, such as vincristine, vinblastine
- cytostatic active compounds such as anti-oestrogens (for example tamoxifen, toremifene, raloxifene, droloxifene and iodoxyfene), oestrogen receptor regulators (for example fulvestrant), anti-androgens (for example bicalutamide, flutamide, nilutamide and cyproterone acetate), LHRH antagonists or LHRH agonists (for example goserelin, leuprorelin and buserelin), progesterones (for example megestrol acetate), aromatase inhibitors (for example anastrozole, letrozole, vorazole and exemestane) and inhibitors of 5a-reductase, such as finasteride;
- anti-oestrogens for example tamoxifen, toremifene, raloxifene, droloxifene and iodoxyfene
- active compounds which inhibit cancer invasion including for example metalloproteinase inhibitors, like marimastat, and inhibitors of urokinase plasminogen activator receptor function;
- inhibitors of growth factor function for example growth factor antibodies, growth factor receptor antibodies, for example the anti-erbb2 antibody trastuzumab [HerceptinTM] and the anti-erbbl antibody cetuximab [C225]), farnesyl transferase inhibitors, tyrosine kinase inhibitors and serine/threonine kinase inhibitors, for example inhibitors of the epidermal growth factor family (for example EGFR family tyrosine kinase inhibitors, such as N-(3-chloro-4-fluorophenyl)-7-methoxy-6- (3- morpholinopropoxy) quinazolin-4-amine (gefitinib, AZD1839), N-(3-ethynylphenyl)- 6,7-bis (2-methoxyethoxy)quinazolin-4-amine (erlotinib, OSI-774) and 6-acrylamido- N-(3-chloro-4
- anti-angiogenic active compounds such as bevacizumab, angiostatin, endostatin, linomide, batimastat, captopril, cartilage derived inhibitor, genistein, interleukin 12, lavendustin, medroxypregesterone acetate, recombinant human platelet factor 4, tecogalan, thrombospondin, TNP-470, anti-VEGF monoclonal antibody, soluble VEGF-receptor chimaeric protein, anti-VEGF receptor antibodies, anti-PDGF receptors, inhibitors of integrins, tyrosine kinase inhibitors, serine/threonine kinase inhibitors, antisense oligonucleotides, antisense oligodexoynucleotides, siRNAs, anti-VEGF aptamers, pigment epithelium derived factor and compounds which have been published in the international patent applications WO 97/22596, WO
- vessel-destroying agents such as combretastatin A4 and compounds which have been published in the international patent applications WO 99/02166,
- antisense therapies for example those directed to the targets mentioned above, such as ISIS 2503, an anti-Ras antisense;
- gene therapy approaches including, for example, approaches for replacement of abnormal, modified genes, such as abnormal p53 or abnormal BRCA1 or BRCA2, GDEPT approaches (gene-directed enzyme pro-drug therapy), such as those which use cytosine deaminase, thymidine kinase or a bacterial nitroreductase enzyme, and approaches which increase the tolerance of a patient to chemotherapy or radiotherapy, such as multi-drug resistance therapy; and
- immunotherapy approaches including, for example, ex-vivo and in-vivo approaches for increasing the immunogenicity of tumour cells of a patient, such as transfection with cytokines, such as interleukin 2, interleukin 4 or granulocyte macrophage colony stimulating factor, approaches for decreasing T-cell anergy, approaches using transfected immune cells, such as cytokine-transfected dendritic cells, approaches for use of cytokine-transfected tumour cells and approaches for use of anti-idiotypic antibodies
- chemotherapeutic agents including foor example abarelix, aldesleukin, alemtuzumab, alitretinoin, allopurinol, altretamine, amifostine, anastrozole, arsenic trioxide, asparaginase, BCG live, bevaceizumab, bexarotene, bleomycin, bortezomib, busulfan, calusterone, camptothecin, capecitabine, carboplatin, carmustine, celecoxib, cetuximab, chlorambucil, cinacalcet, cisplatin, cladribine, cyclophosphamide, cytarabine, dacarbazine, dactinomycin, darbepoetin alfa, daunorubicin, denileukin diftitox, dexrazoxane, docetaxel, doxorubicin, dromo
- the medicaments from table 1 can preferably, but not exclusively, be combined with the compounds of the present invention.
- Vss Volume of distribution (at steady state) v/v Volume to volume
- the invention especially relates to the compounds of the following examples and physiologically acceptable salts, derivatives, solvates, prodrugs and stereoisomers thereof, including mixtures thereof in all ratios.
- Multiplicity is abbreviated as follows: s (singlet), d (doublet), t (triplet), q (quartet), sext (sextet), hept (heptet), m (multiplet), br (broad).
- HPLC/MS spectra of the products were recorded on an Agilent 1100 HPLC system (1100 high pressure gradient pump, 1100 diode array detector, wavelength: 220 nm) interfaced to an Agilent 1100 mass spectrometer detector (positive mode).
- LC-MS analyses were performed on a SHIMADZU LC-MS machine consisting of an LIFLC 20-AD system and LCMS 2020 MS detector.
- A2.1 Nitrogen gas was bubbled through a mixture of 7-bromoisoquinolin-1-ol (0.500 g, 2.23 mmol) and zinc cyanide (0.341 g, 2.90 mmol) in DMF (12.4 mL) for 15 min. Palladium tetrakis(triphenylphosphine) (0.155 g, 0.13 mmol) was added and the mixture heated at 100 °C in a sealed vial for 16 h. The reaction mixture was diluted with brine (100 mL) and extracted with ethyl acetate (100 mL). The organic layer was washed with brine (2 x 50 mL).
- Example 1.1 4-Amino-2-chloropyridine (1.96 g, 15.246 mmol) and sodium hydroxide (3.17 g, 79.279 mmol) were dissolved in dry ethanol (15.0 mL) in a microwave tube. The reaction mixture was heated to 150 °C in a microwave reactor for 7 h. The reaction mixture was cooled to room temperature, diluted with water (40 mL) and extracted with ethyl acetate. The combined organic layers were washed with brine, dried with sodium sulfate, filtered and concentrated. The residue was purified by flash-chromatography to yield 1.76 g (84%) of 2-ethoxypyridin-4-amine as a colorless solid. HPLC/MS m/z: 139.1 [M+H] + , Rt (B): 0.74 min.
- Example 1.2 A solution of 2-ethoxypyridin-4-amine (548.0 mg, 3.966 mmol) in dichloromethane (12.7 mL) was cooled to 0 °C. NBS (777.0 g, 4.366 mmol) was added at this temperature and after 5 min the reaction mixture was allowed to warm up to room temperature and was stirred for 45 min. The reaction was quenched with water (50 mL) and extracted with dichloromethane. The combined organic layers were washed with brine, dried with sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by flash-chromatography to give 789 mg (92%) of 3-bromo-2-ethoxypyridin-4-amine as a pale-brown oil. HPLC/MS m/z: 216.9/218.9 [M+H] + , Rt (B): 1.09 min.
- Example 1.3 A solution of 3-bromo-2-ethoxypyridin-4-amine (789.0 mg, 3.635 mmol) and benzoyl isothiocyanate (1.78 g, 10.907 mmol) in acetone (4.0 mL) was stirred at room temperature overnight. A yellow precipitate formed, which was filtered by suction, washed with heptane and dried to yield 1.23 g (89%) of 1- benzoyl-3-(3-bromo-2-ethoxypyridin-4-yl)thiourea as a yellow solid. HPLC/MS m/z: 379.7/381.7 [M+H] + , Rt (B): 1.09 min.
- Example 1.4 Intermediate 1.3 (1.23 g, 3.235 mmol) was dissolved in methanol (5.0 mL). Sodium hydroxide (360.0 mg, 9.001 mmol) was dissolved in water (1.5 mL) and added to the reaction mixture. The reaction mixture was refluxed for 2 h, cooled down to room temperature and treated with aqueous saturated NH4CI solution until a precipitate was formed. The precipitate was filtered off by suction, washed with water and dichloromethane and dried to afford 870 mg (97%) of (3-bromo-2- ethoxypyridin-4-yl)thiourea as a colorless solid. HPLC/MS m/z: 275.8/277.8 [M+H] + , Rt (B): 1.27 min.
- Example 1.5 Intermediate 1.4 (2.42 g, 8.763 mmol), DL-proline (302.9 mg, 2.631 mmol), cesium carbonate (5.71 g, 17.526 mmol) and copper(l) iodide (501.1 mg, 2.631 mmol) were suspended in dry DMSO (7.5 mL). The reaction mixture was heated under an argon atmosphere to 70 °C and stirred overnight, cooled to room temperature, and stirred for 2 d. The reaction mixture was filtered, and the filtrate was evaporated to dryness.
- Example 1.6 Intermediate B1 (36.6 mg, 0.144 mmol) was dissolved in DMF (1.1 mL). 1-Methylimidazole (68.5 mg, 0.834 mmol), intermediate 1.5 (43.4 mg, 0.180 mmol) and Chloro-N,N,N',N'-tetramethylformamidinium hexafluorophosphate (68.0 mg, 0.230 mmol) were added and the reaction was stirred at room temperature for 1 h, heated to 60 °C and stirred for 5 h. The reaction mixture was cooled down to room temperature and diluted with water (5 mL). A precipitate was formed, which was filtered with suction, thoroughly rinsed with water and dried.
- Example 2 3- ⁇ [3-(5-Methyl-1,2,4-oxadiazol-3-yl)phenyl]formamido ⁇ -N-[4- (propan-2-yloxy)-[1,3]thiazolo[5,4-c]pyridin-2-yl]propenamide
- Example 11 3- ⁇ [7-(5-Methyl-1 ,2,4-oxadiazol-3-yl)isoquinolin-1 -yl]amino ⁇ -N-[4- (propan-2-yloxy)-[1,3]thiazolo[5,4-c]pyridin-2-yl]propanamide
- Example 12 N-[4-(butan-2-yloxy)-[1,3]thiazolo[5,4-c]pyridin-2-yl]-3- ⁇ [7-(5- methyl-1,2,4-oxadiazol-3-yl)isoquinolin-1-yl]amino ⁇ propanamide
- Example 22 (1s,3s)-3- ⁇ [7-(5-Methyl-1,2,4-oxadiazol-3-yl)isoquinolin-1- yl]amino ⁇ -N- ⁇ 4-propoxy-[1,3]thiazolo[5,4-c]pyridin-2-yl ⁇ cyclobutane-1- carboxamide
- Example 23.1 1-Chloro-7-nitropyrrolo[1,2-a]pyrazine (190.0 mg, 0.962 mmol) was dissolved in ethanol (3 mL) and dichloromethane (3 mL), potassium hydroxide (200.0 mg, 3.565 mmol) was added and the mixture was heated to 50 °C and stirred for 1.5 h. The dark brown solution was diluted with water and extracted with ethyl acetate. The combined organic layers were washed with brine, dried with sodium sulfate, filtered, and evaporated to dryness to afford 172.5 mg (87%) of 1- ethoxy-7-nitropyrrolo[1 ,2-a]pyrazine as a red-brown solid. HPLC/MS m/z: 208.1 [M+H] + , Rt (D): 1.54 min.
- Example 23.2 1-Ethoxy-7-nitropyrrolo[1 ,2-a]pyrazine (172.5 mg, 0.833 mmol), zinc dust (272.3 mg, 4.164 mmol) and ammonium acetate (770.4 mg, 9.994 mmol) were suspended in ethanol (5.0 mL), and the mixture was heated to 80 °C and stirred for 2 min. The reaction mixture was cooled to room temperature, filtered and the filter cake was washed with ethyl acetate. The filtrate was diluted with ethyl acetate and extracted with water and 2N NaOH. The aqueous layer was extracted with ethyl acetate.
- Example 23.3 3- ⁇ [(tert-Butoxy)carbonyl]amino ⁇ propanoic acid (63.4 mg, 0.335 mmol), 1-ethoxypyrrolo[1,2-a]pyrazin-7-amine (57.7 mg, 0.305 mmol) and HATLI (140.5 mg, 0.366 mmol) were suspended in DMF (2.0 mL). N-Ethyldiisopropylamine (104.7 pl, 0.610 mmol) was added and the clear brown solution was stirred at room temperature for 1 h. The reaction mixture was diluted with 30 ml water and extracted with ethyl acetate. The combined organic layers were washed with brine, dried with sodium sulfate, filtered, and evaporated to dryness.
- Example 23.4 Intermediate 23.3 (76.5 mg, 0.220 mmol) was dissolved in 1,4- dioxane (1.0 mL) and treated with a HCI solution in 1 ,4-dioxane (4 M, 1.5 mL). The mixture was stirred at room temperature overnight. The reaction mixture was evaporated to dryness and the residue was used in the next step without further purification. Yield: 70.5 mg (100%) of 3-amino-N- ⁇ 1-ethoxypyrrolo[1,2-a]pyrazin-7- yljpropanamide dihydrochloride as a pale-orange solid. HPLC/MS m/z: 249.1 [M+H] + , Rt (D): 0.86 min.
- Example 23.5 The amide coupling reaction was performed as described for intermediate B3.1. Yield: 11 mg (13%) of N- ⁇ 1-ethoxypyrrolo[1,2-a]pyrazin-7-yl ⁇ -3- ⁇ [7-(5-methyl-1 ,2,4-oxadiazol-3-yl)isoquin- , olin-1-yl]amino ⁇ propanamide as a colorless solid. HPLC/MS m/z: 458.1 [M+H] + , Rt (D): 1.26 min.
- Example 25 11-[2-[[7-(5-Methyl-1,2,4-oxadiazol-3-yl)-1- isoquinolyl]amino]ethyl]-6-(2,2,2-trifluoroethoxy)-1,5,11- t riazatricyclo[7.4.0.02, 7]trideca-2, 4, 6, 8-tetraen-10-one
- Example 25.1 A suspension of methyl 4-chloro-1 H-pyrrolo[3,2-c]pyridine-2- carboxylate (600.00 mg, 2.8487 mmol), 2,2,2-trifluoroethanol (0.37 mL, 4.9853 mmol), cesium carbonate (1600.00 mg, 4.9107 mmol), tBuBrettPhos Pd G3 (97.36 mg, 0.1139 mmol), tBuBrettPhos (110.46 mg, 0.2279 mmol) and 4 MS (1600 mg) in toluene (4.00 mL, 0.3600 M
- Example 25.2 A solution of methyl 4-(2,2,2-trifluoroethoxy)-1 H-pyrrolo[3,2- c]pyridine-2-carboxylate (600.00 mg, 2.0569 mmol), tert-butyl-N-hydroxyethyl carbamate (0.57 mL, 3.7024 mmol), and triphenylphosphine (809.25 mg, 3.0853 mmol) in THF (5.14 mL, 0.4000 M) was cooled to 0 °C. Diisopropyl azodicarboxylate, (0.65 mL, 3.291 mmol) was added dropwise over 20 min, and the resulting mixture was warmed to RT and stirred for 18 h.
- Example 25.3 Methyl 1-[2-(tert-butoxycarbonylamino)ethyl]-4-(2,2,2- trifluoroethoxy)pyrrolo[3,2-c]pyridine-2-carboxylate (867.20 mg, 2.0777 mmol) was mixed with 4N HCI in dioxane (20.78 mL, 83.109 mmol) and 1 ,4-dioxane (20.78 mL, 0.1000 M) at RT under argon and stirred for 2 h. Volatiles were removed under reduced pressure.
- Example 25.4 The crude methyl 1-(2-aminoethyl)-4-(2,2,2- trifluoroethoxy)pyrrolo[3,2-c]pyridine-2-carboxylate hydrochloride was dissolved in EtOH (9.33 mL, 0.2100 M) and triethylamine (1.39 mL, 9.8948 mmol) and the mixture was stirred for overnight at 80 °C. The crude mixture was evaporated under reduced pressure, redissolved in DCM and water was added.
- Example 25.5 6-(2,2,2-trifluoroethoxy)-1 ,5,11-triazatricyclo[7.4.0.02,7]trideca- 2,4,6,8-tetraen-10-one (600.00 mg, 2.1036 mmol) was dissolved in DMF (30.05 mL, 0.0700 M). The obtained solution was cooled down to 0 °C in an ice bath followed by the addition of NaH (168.29 mg, 4.2073 mmol). The reaction mixture was left stirring at 0 °C for 30 min.
- /V-Boc-2-chloroethylamine (467.50 mg, 2.5244 mmol) dissolved in DMF (2.5 mL) was added dropwise to the reaction mixture and allowed to warm to RT. The reaction mixture was left stirring at RT for 2 d. 0.6 equiv. of the /V-Boc-2-chloroethylamine reagent was added to the reaction cooled to 0 °C, followed by the addition of another 1 equiv. of NaH and the reaction was left to stir overnight at RT. Water was added and the reaction was extracted with EtOAc (3x), and the combined organic layer was washed with brine, dried over MgSCL and evaporated under reduced pressure.
- Example 25.6 tert-butyl /V-[2-[10-oxo-6-(2,2,2-trifluoroethoxy)-1 ,5,11- triazatricyclo[7.4.0.02,7]trideca-2,4,6,8-tetraen-11-yl]ethyl]carbamate (52.00 mg, 0.1214 mmol) was mixed with 4N HCI in 1 ,4-dioxane (5.60 mL, 22.408 mmol) and 1 ,4-dioxane (5.60 mL, 0.1000 M) at RT under argon and stirred for 2 h.
- Example 25.7 DIPEA (0.28 mL, 1.5992 mmol) was added dropwise to a suspension of 11-(2-aminoethyl)-6-(2,2,2-trifluoroethoxy)-1 ,5,11-triazatricyclo[7.4.0.02,7]trideca- 2,4,6,8-tetraen-10-one (140.00 mg, 0.4265 mmol), 5-methyl-3-(2-oxidoisoquinolin-2- ium-7-yl)-1 ,2,4-oxadiazole (116.28 mg, 0.5117 mmol) and PyBrop (238.56 mg, 0.5117 mmol) in DCM (3.55 mL, 0.1200 M).
- the tube was sealed, and this was heated to 60 °C for 1 h in the microwave.
- the reaction was evaporated in vacuo and subjected to RP column chromatography (10-80% MeOH:water + 0.1% formic acid) afforded partially pure product.
- the pure fractions were run through a SCX-II column, released using 2N NH3 in methanol and evaporated.
- Example 26.1 DIPEA (0.03 mL, 0.1462 mmol) was added to a mixture of 3- Methyl-2H-tetrazol-5-yl)-benzoic acid (8.58 mg, 0.0420 mmol), HATLI (27.80 mg, 0.0731 mmol) and 11-(2-aminoethyl)-6-(2,2,2-trifluoroethoxy)-1 ,5,11- triazatricyclo[7.4.0.02,7]trideca-2,4,6,8-tetraen-10-one [Example 25.6] (12.00 mg, 0.0366 mmol) in DCM (0.37 mL, 0.1000 M). This was stirred for 2 h before sat. aq.
- Example 27 3-(2-Methyltetrazol-5-yl)-N-[1,1,2,2-tetradeuterio-2-[10-oxo-6- (2,2,2-trifluoroethoxy)-1,5,11-triazatricyclo[7.4.0.02,7]trideca-2(7),3,5,8-tetraen- 11-yl]ethyl]benzamide
- Example 27.1 To a stirred suspension of 6-(2,2,2-trifluoroethoxy)-1 ,5,11- triazatricyclo[7.4.0.02,7]trideca-2(7),3,5,8-tetraen-10-one (700.00 mg, 2.4542 mmol) in DMF (35.06 mL, 0.0700 M), NaH (60% dispersion in mineral oil) (215.97 mg, 5.3993 mmol) was added in portions at RT and then stirred for 30 min at 60 °C. The reaction was cooled to RT and to this solution 1 ,2-dibromoethane-d4 (1.06 mL, 12.271 mmol) was added and stirred at RT overnight.
- NaH 50% dispersion in mineral oil
- Example 27.2 To a stirred solution of sodium azide (43.32 mg, 0.6663 mmol) in DMF (4.04 mL, 0.1500 M) was added 11-(2-bromo-1 ,1 ,2,2-tetradeuterio-ethyl)-6- (2,2,2-trifluoroethoxy)-1 ,5,11-triazatricyclo[7.4.0.02,7]trideca-2(7),3,5,8-tetraen-10- one (240.00 mg, 0.6058 mmol). The reaction mixture was stirred at 80 °C overnight. Then the reaction mixture was cooled to RT and diluted with water (5 mL).
- Example 27.3 To a solution of 11-(2-azido-1 ,1 ,2,2-tetradeuterio-ethyl)-6-(2,2,2- trifluoroethoxy)-1 ,5,11-triazatricyclo[7.4.0.02, 7]trideca-2(7), 3,5, 8-tetraen-10-one (200.00 mg, 0.5582 mmol) in THF (5.14 mL, 0.0800 M) was added Triphenylphosphine (439.21 mg, 1.6745 mmol). The mixture was stirred at RT for 48 h, then H2O (1.84 mL, 0.0800 M) was added, and the reaction was heated to 60 °C and allowed to stir for 4 h.
- Example 27.4 DI PEA (0.08 mL, 0.4606 mmol) was added to a mixture of 3-(2- Methyl-2H-tetrazol-5-yl)-benzoic acid (31.80 mg, 0.1557 mmol), HATU (102.98 mg, 0.2708 mmol) and 11-(2-amino-1 ,1 ,2,2-tetradeuterio-ethyl)-6-(2,2,2-trifluoroethoxy)-
- Example 28 3-(5-Methyl-1 ,2,4-oxadiazol-3-yl)-N-[1 , 1 ,2,2-tetradeuterio-2-[10- oxo-6-(2,2,2-trifluoroethoxy)-1,5,11 -tri azatri cyclo[7.4.0.02,7]trideca-2(7), 3,5,8- tetraen-11-yl]ethyl]benzamide
- DIPEA (0.09 mL, 0.5417 mmol) was added to a mixture of 3-(5-Methyl- 1 ,2,4- oxadiazol-3-yl)benzoic acid (31.80 mg, 0.1557 mmol), HATLI (102.98 mg, 0.2708 mmol) and 11-(2-amino-1 ,1 ,2,2-tetradeuterio-ethyl)-6-(2,2,2-trifluoroethoxy)-1 ,5,11- triazatricyclo[7.4.0.02,7]trideca-2(7),3,5,8-tetraen-10-one (45.00 mg, 0.1354 mmol) in DCM (1.35 mL, 0.1000 M).
- Example 29 11-[1,1,2,2-Tetradeuterio-2-[[7-(5-methyl-1,2,4-oxadiazol-3-yl)-1- isoquinolyl]amino]ethyl]-6-(2,2,2-trifluoroethoxy)-1,5,11- t riazatricyclo[7.4.0.02, 7]trideca-2(7), 3, 5, 8-tetraen-10-one
- Example 29.1 To a dry 250 mL round-bottom flask equipped with a stirbar under N2 were added p-toluene-sulfonyl-chloride (188.76 mg, 0.9901 mmol), 4- dimethylaminopyridine (11.00 mg, 0.0900 mmol), triethylamine (0.25 mL, 1.8002 mmol), and DCM (3.00 mL, 0.1500 M).
- Example 29.2 To a stirred solution of sodium azide (39.64 mg, 0.6098 mmol) in DMF (3.70 mL, 0.1500 M) was added 11-(2-chloro-1 ,1 ,2,2-tetradeuterio-ethyl)-6- (2,2,2-trifluoroethoxy)-1 ,5,11-triazatricyclo[7.4.0.02,7]trideca-2(7),3,5,8-tetraen-10- one (195.00 mg, 0.5544 mmol). The reaction mixture was stirred at 80 °C overnight. Then the reaction mixture was cooled to RT and diluted with water (5 mL).
- Example 29.3 To a solution of 11-(2-azido-1 ,1 ,2,2-tetradeuterio-ethyl)-6-(2,2,2- trifluoroethoxy)-1 ,5,11-triazatricyclo[7.4.0.02, 7]trideca-2(7), 3,5, 8-tetraen-10-one (150.00 mg, 0.4186 mmol) in THF (5.14 mL, 0.0600 M) was added PPh 3 (329.41 mg, 1.2559 mmol). The mixture was stirred at RT for 48 h, then H2O (1.84 mL, 0.0600 M) was added, and the reaction was heated to 60 °C and allowed to stir for 4 h.
- Example 29.4 DI PEA (0.14 mL, 0.7899 mmol) was added dropwise to a suspension of 11-(2-amino-1 ,1 ,2,2-tetradeuterio-ethyl)-6-(2,2,2-trifluoroethoxy)-1 ,5,11- triazatricyclo[7.4.0.02,7]trideca-2(7),3,5,8-tetraen-10-one (70.00 mg, 0.2106 mmol), 5-methyl-3-(2-oxidoisoquinolin-2-ium-7-yl)-1 ,2,4-oxadiazole (57.44 mg, 0.2528 mmol) and PyBrop (117.84 mg, 0.2528 mmol) in DCM (1.76 mL, 0.1200 M).
- Example 30.1 Ethyl 3-amino benzoate (0.81 mL, 5 mmol) was dissolved in a mixture of water (1.00 mL) and 50% aqueous hydrofluroboric acid (1.90 mL, 30.293 mmol). Sodium nitrite (689.90 mg, 10 mmol) was dissolved in water (1.00 mL) and added dropwise. The reaction was stirred at 0 °C for 30 min, the formation of a pale pink solid was observed. The reaction was filtered and the solid was washed with Et20.
- Example 30.2 To a solution of acetamidine hydrochloride (0.39 g, 4.167 mmol) and K2CO3 (2.88 g, 20.835 mmol) in DMSO (20.25 mL) was added 3- ethoxycarbonylbenzenediazonium; tetrafluoroboron (1.10 g, 4.167 mmol) in portions. After stirring for 1.5 h at RT, KI (1.04 g, 6.2505 mmol) and iodine (1.27 g, 5.0004 mmol) were added, stirred at RT for 1.5 h. A solution of brine and Na2S20s were added to the mixture.
- Example 30.3 ethyl 3-(5-methyltetrazol-2-yl)benzoate (150.00 mg, 0.6459 mmol) was dissolved in THF (3.23 mL) and H2O (1.00 mL, 0.1500 M). LiOH (77.34 mg, 3.2294 mmol) was added, and the mixture was stirred at 50 °C for 2 h. The mixture was cooled to RT, concentrated, and diluted with 1 mL of water. The pH was adjusted to pH ⁇ 3 and the product was extracted with EtOAc (60 mL).
- Example 30.4 DI PEA (0.08 mL, 0.4606 mmol) was added to a mixture of 3-(5- methyltetrazol-2-yl) benzoic acid (27.04 mg, 0.1324 mmol), HATU (87.56 mg, 0.2303 mmol) and 11-(2-aminoethyl)-6-(2,2,2-trifluoroethoxy)-1,5,11- triazatricyclo[7.4.0.02,7]trideca-2,4,6,8-tetraen-10-one hydrochloride (42.00 mg, 0.1151 mmol) in DCM (1.15 mL, 0.1000 M). This was stirred for 2 h before sat. aq. NaHCOs was added.
- Example 31 11-[2-[[7-(2-Methyltetrazol-5-yl)-1-isoquinolyl]amino]ethyl]-6- (2,2,2-trifluoroethoxy)-1,5,11-triazatricyclo[7.4.0.02,7]trideca-2,4,6,8-tetraen-10- one
- Example 31.1 To a stirred solution of a 5-bromo-2-methyl-1H-tetrazole (75.00 mg, 0.4602 mmol), lsoquinoline-7-boronic acid (103.48 mg, 0.5982 mmol), sodium carbonate (73.16 mg, 0.6903 mmol) in a mixture of DME (1.84 mL, 0.2000 M)/water (0.46 mL, 0.2000 M) 4:1 was added Pd(PPh3)4 (53.18 mg, 0.0460 mmol) under an argon atmosphere. The solution was stirred at 90 °C for 18 h and then treated with water.
- Example 31.2 7-(2-Methyltetrazol-5-yl)isoquinoline (70.00 mg, 0.3314 mmol) was dissolved in chloroform (2.53 mL, 0.1300 M) and cooled in an ice bath. 3- Chloroperoxybenzoic acid (98.04 mg, 0.3977 mmol) was added. The reaction was stirred at r.t. overnight. K2CO3 (183.22 mg, 1.3256 mmol) was added. This was stirred for 4 h at RT before filtering through a pad of anhydr.
- Example 31.3 7-(2-Methyltetrazol-5-yl)-2-oxido-isoquinolin-2-ium (58.14 mg, 0.2559 mmol), PyBrop (119.28 mg, 0.2559 mmol) and DIPEA (0.14 mL, 0.7996 mmol) in DCM (1.08 mL, 0.1200 M) were stirred under a nitrogen atmosphere for 30 min at 40 °C in a microwave vial.
- Example 32 11-[2-[[7-(5-Methyltetrazol-2-yl)-1-isoquinolyl]amino]ethyl]-6- (2,2,2-trifluoroethoxy)-1,5,11-triazatricyclo[7.4.0.02,7]trideca-2,4,6,8-tetraen-10- one
- Example 32.1 7-Aminoisoquinoline (1000.00 mg, 6.9363 mmol) was dissolved in a mixture of 50% aqueous hydrofluroboric acid (3.47 mL, 27.647 mmol) and EtOH (2.08 mL, 3.33 M). The reaction mixture was cooled to 0 °C and tert-butyl nitrite (1.65 mL, 13.873 mmol) was added dropwise. Stirred for 1 h at RT, diethyl ether (10 mL) was added to precipitate the diazonium compound that was filtered off and washed with diethyl ether (3x5 mL).
- Example 32.2 To a solution of acetamidine hydrochloride (544.74 mg, 5.762 mmol) and potassium carbonate (3.98 g, 28.81 mmol) in DMSO (28.00 mL, 0.2100 M) was added isoquinoline-7-diazonium; tetrafluoroboron (1.40 g, 5.762 mmol) in portions. After stirring for 1 .5 h at RT LC/MS showed complete addition of the acetamidine moiety, KI (1434.75 mg, 8.643 mmol) and iodine (1754.95 mg, 6.9144 mmol) were added, stirred at RT for 1.5 h.
- Example 32.3 7-(5-Methyltetrazol-2-yl)isoquinoline (155.00 mg, 0.7338 mmol) was dissolved in CHCI3 (2.45 mL, 0.3000 M) and cooled in an ice bath. 3- Chloroperoxybenzoic acid (0.22 g, 0.8806 mmol) was added. The reaction was stirred at RT for 2 h. K2CO3 (0.41 g, 2.9353 mmol) was added, stirred for 0.5 h before filtering. The filtrate was concentrated in vacuo to give a yellow/orange powder.
- Example 33 11-[2-[[6-(5-Methyl-1,2,4-oxadiazol-3-yl)quinazolin-4- yl]amino]ethyl]-6-(2,2,2-trifluoroethoxy)-1,5,11-triazatricyclo[7.4.0.02,7]trideca- 2,4,6,8-tetraen-10-one
- Example 33.1 4-Hydroxyquinazoline-6-carbonitrile (500 mg, 2.92 mmol) and triethylamine (0.41 mL, 2.92 mmol) were heated at 80 °C in [bmim]OAc (2.9 mL). Hydroxylamine hydrochloride (406 mg, 5.84 mmol) was added, after which some foaming was observed. The reaction mixture was continued to stir at 80 °C for 30 min. The reaction mixture was cooled down to room temperature and water was added (50 mL). The precipitate was filtered off, washed with water (50 mL) and dried under reduced pressure to yield 536 mg (90%) of / ⁇ /,4-dihydroxyquinazoline-6- carboxamidine. HPLC/MS m/z: 205.1 [M+H] + , Rt (II): 0.46 min.
- Example 33.2 A suspension of /V,4-dihydroxyquinazoline-6-carboxamidine (480 mg, 2.35 mmol) and acetic anhydride (0.27 mL, 2.82 mmol) in anhydrous ACN (4.70 mL) under an argon atmosphere was heated under microwave irradiation at 180 °C for 10 min. The reaction mixture was cooled to room temperature and cold water was added (50 mL). The precipitate was filtered off, washed with water (50 mL) and diethyl ether (10 mL), and dried under reduced pressure to yield 293 mg (55%) of 6- (5-methyl-1,2,4-oxadiazol-3-yl)quinazolin-4-ol. HPLC/MS m/z: 229.0727 [M+H] + , Rt (U): 1.93 min.
- Example 33.3 6-(5-Methyl-1 ,2,4-oxadiazol-3-yl)quinazolin-4-ol (60.00 mg, 0.2629 mmol), 11-(2-aminoethyl)-6-(2,2,2-trifluoroethoxy)-1 ,5,11- triazatricyclo[7.4.0.02,7]trideca-2,4,6,8-tetraen-10-one (94.94 mg, 0.2892 mmol) and DIPEA (0.14 mL, 0.7887 mmol) were dissolved in DMF (2.63 mL, 0.1000 M) at RT under argon.
- Example 34 3-Fluoro-5-(2-methyltetrazol-5-yl)-N-[2-[10-oxo-6-(2,2,2- trifluoroethoxy)-1,5,11-triazatricyclo[7.4.0.02,7]trideca-2,4,6,8-tetraen-11- yl]ethyl] benzamide
- Example 34.1 To a stirred solution of 5-bromo-2-methyl-1 H-tetrazole (70.00 mg, 0.4295 mmol), Na 2 CO 3 (68.28 mg, 0.6443 mmol), Pd(PPh 3 ) 4 (49.63 mg, 0.0430 mmol) in a mixture of DME/water (4:1 , cone. 0.2000 M) at 60 °C was added (3- fluoro-5-methoxycarbonyl-phenyl)boronic acid (110.53 mg, 0.5584 mmol) in DME (0.36 mL, 0.2000 M) under an argon atmosphere and the reaction was heated to 90 °C for 18 h and then treated with water.
- Example 34.2 Methyl 3-fluoro-5-(2-methyltetrazol-5-yl)benzoate (40.00 mg, 0.1693 mmol), THF (0.68 mL, 0.1000 M), MeOH (0.34 mL, 0.1000 M) and H 2 O (0.68 mL, 0.1000 M) were mixed at ambient temperature. Lithium hydroxide monohydrate (35.53 mg, 0.8467 mmol) was added, and the reaction mixture was stirred for 1 h. The reaction was quenched with 2N HCI and DCM was added.
- Example 34.3 DI PEA (0.10 mL, 0.5483 mmol) was added to a mixture of 3-fluoro-5- (2-methyltetrazol-5-yl) benzoic acid (33.50 mg, 0.1508 mmol), HATU (104.24 mg, 0.2741 mmol) and 11-(2-aminoethyl)-6-(2,2,2-trifluoroethoxy)-1 ,5,11- triazatricyclo[7.4.0.02,7]trideca-2,4,6,8-tetraen-10-one (45.00 mg, 0.1371 mmol) in DCM (1.22 mL, 0.1100 M) and stirred at RT overnight before sat. aq. NaHCO 3 was added.
- Example 35.1 A solution of methyl 4-(2,2,2-trifluoroethoxy)-1 H-pyrrolo[3,2- c]pyridine-2-carboxylate (750.00 mg, 2.7352 mmol), 2-bromo- 1 ,1 ,2, 2-tetradeuterio- ethanol (0.35 mL, 4.9234 mmol), and PPh 3 (1076.14 mg, 4.1028 mmol) in THF (6.84 mL, 0.4000 M) was cooled to 0 °C. Diisopropyl azodicarboxylate (0.86 mL, 4.3764 mmol) was added dropwise over 20 min, and the resulting mixture was warmed to RT and stirred for 18 h.
- Example 35.2 To a stirred solution of sodium azide (107.68 mg, 1.6564 mmol) in DMF (11.58 mL, 0.1300 M) was added methyl 1-(2-bromo-1,1,2,2-tetradeuterio- ethyl)-4-(2,2,2-trifluoroethoxy)pyrrolo[3,2-c]pyridine-2-carboxylate (580.00 mg, 1.5058 mmol). The reaction mixture was stirred at 80 °C overnight. Then the reaction mixture was cooled to RT and diluted with water (5 mL).
- Example 35.3 To a solution of methyl 1-(2-azido-1 ,1,2,2-tetradeuterio-ethyl)-4- (2,2,2-trifluoroethoxy)pyrrolo[3,2-c]pyridine-2-carboxylate (480.00 mg, 1.3821 mmol) in THF (17.28 mL, 0.0600 M) was added PPh 3 (1087.56 mg, 4.1464 mmol). The mixture was stirred at RT for 16 h, then water (5.76 mL, 0.0600 M) was added, and the reaction was heated to 40 °C and allowed to stir for 4 h.
- Example 35.5 tert-Butyl N-[2-[12,12,13,13-tetradeuterio-10-oxo-6-(2,2,2- trifluoroethoxy)-1 ,5,11-triazatricyclo[7.4.0.02,7]trideca-2,4,6,8-tetraen-11- yl]ethyl]carbamate (240.00 mg, 0.3330 mmol) was mixed with 4N HCI in 1 ,4-dioxane (3.33 mL, 13.32 mmol) and 1 ,4-dioxane (3.33 mL, 0.1000 M) at RT under argon and stirred for 2 h.
- Example 35.6 DI PEA (0.13 mL, 0.7335 mmol) was added dropwise to a suspension of 11-(2-aminoethyl)-12,12,13,13-tetradeuterio-6-(2,2,2-trifluoroethoxy)-1 ,5,11- triazatricyclo[7.4.0.02,7]trideca-2,4,6,8-tetraen-10-one (65.00 mg, 0.1956 mmol), 5- methyl-3-(2-oxidoisoquinolin-2-ium-7-yl)-1 ,2,4-oxadiazole (53.33 mg, 0.2347 mmol) and PyBrop (109.42 mg, 0.2347 mmol) in DCM (1.63 mL, 0.1200 M ).
- Example 36 3-(2-Methyltetrazol-5-yl)-N-[2-[12,12,13,13-tetradeuterio-10-oxo-6- (2,2,2-trifluoroethoxy)-1,5,11-triazatricyclo[7.4.0.02,7]trideca-2(7),3,5,8-tetraen- 11-yl]ethyl]benzamide
- DIPEA 0.05 mL, 0.2648 mmol
- 3-(2-methyl-2H-tetrazol- 5-yl)-benzoic acid 15.55 mg, 0.0761 mmol
- HATLI 50.34 mg, 0.1324 mmol
- Example 37 8-(2-((7-(5-Methyl-1,2,4-oxadiazol-3-yl)isoquinolin-1- yQaminoJethyQ-l-propoxy ⁇ .S-dihydropyridoIS' ⁇ ' ⁇ .SJpyrrolon ⁇ -aJpyrazin- 9(6H)-one
- Example 37.1 A mixture of methyl 4-chloro-1 H-pyrrolo[3,2-c]pyridine-2-carboxylate (553 mg, 2.63 mmol, 1.0 eq.), propan-1-ol (28 pL, 3.68 mmol, 1.4 eq.), CS2CO3 (1.20 g, 3.68 mmol, 1.4 eq.), tBuBrettPhos Pd G3 (90 mg, 0.11 mmol, 4 mol%), tBuBrettPhos (102 mg, 0.21 mmol, 8 mol%) and 4 molecular sieves (1.0 g) in THF (6.56 mL, 0.4 M) was heated at 80 °C overnight in a sealed vial.
- Example 37.2 Aqueous NaOH (3N, 11.8 mL) was added to a solution of methyl 4- propoxy-1 H-pyrrolo[3,2-c]pyridine-2-carboxylate (554.00 mg, 2.37 mmol, 1.0 eq.) in EtOH (11.8 mL) and this was heated to 45 °C for 45 min before the solvent was evaporated and the crude solid triturated with Et20. The solid was filtered, washed with further Et20 and dried under vacuum to afford a mixture of NaOH and sodium 4-propoxy-1 H-pyrrolo[3,2-c]pyridine-2-carboxylate as a yellow solid which was engaged into the next step without further purification (crude mass 950 mg). HPLC/MS m/z: 221.1 [M+H] + , Rt (Y): 0.815 min.
- Example 37.3 A mixture of sodium 4-propoxy-1H-pyrrolo[3,2-c]pyridine-2- carboxylate (22 mg, 89 pmol, 1.0 eq) and HATLI (41 mg, 107 pmol, 1.2 eq.) in DCM/DMF (2:1, 300 pL) was stirred for 15 min before a solution of tert-butyl N- ⁇ 2- [(2-hydroxyethyl)amino]ethyl ⁇ carbamate (19 mg, 90 pmol, 1.05 eq.) in DCM (200 pL) was added. After 5 min, the reaction was quenched with sat. aq. NaHCCh and extracted with DCM, dried over MgSC>4, then evaporated under vacuum.
- Example 37.4 A solution of tert-butyl N-[2-[2-hydroxyethyl-(4-propoxy-1 H- pyrrolo[3,2-c]pyridine-2-carbonyl)amino]ethyl]carbamate (221.30 mg, 0.5444 mmol) and triphenylphosphine (428.39 mg, 1.6333 mmol) in THF (5.44 mL, 0.1000 M) was cooled to 0 °C. DIAD (321.58 pL, 1.6333 mmol) was added dropwise over 30 min and the resulting mixture was warmed to RT, stirred for 2 h. After the solvent was removed under reduced pressure, the residue was diluted with EtOAc and extracted with aq.
- Example 37.5 Aqueous HCI (1M) (1.63 mL, 1.63 mmol, 3.0 eq.) was added to the crude solid and this was stirred for 10 min until all the material had dissolved, then evaporated to dryness. The solid was triturated with DCM and the solvent filtered off, washed with further DCM to afford pure 2-(10-oxo-6-propoxy-1,5,11- triazatricyclo[7.4.0.02,7]trideca-2(7),3,5,8-tetraen-11-yl)ethylammonium chloride (90.4 mg, 51% yield over 2 steps) as a white solid. HPLC/MS m/z: 289.2 [M+H] + , Rt (Y): 0.547 min.
- Example 37.6 DI PEA (67.03 pL, 0.3848 mmol) was added to a mixture of 5-methyl- 3-(2-oxidoisoquinolin-2-ium-7-yl)-1,2,4-oxadiazole (27.98 mg, 0.1231 mmol), 2-(10- oxo-6-propoxy-1,5,11-triazatricyclo[7.4.0.02,7]trideca-2(7),3,5,8-tetraen-11- yl)ethylammonium chloride (25.00 mg, 0.0770 mmol) and PyBroP (43.06 mg, 0.0924 mmol) in DCM (0.51 mL, 0.1500 M) and this was heated via microwave irradiation (60 °C; 1 h).
- Example 38 3-(2-Methyl-2H-tetrazol-5-yl)-N-(2-(9-oxo-1 -propoxy-6,7- dihydropyridoIS' ⁇ ' ⁇ .SJpyrrolon ⁇ -aJpyrazin-SfSJI-Q-yQethyQbenzamide
- Example 39 1 -(Dodec-11 -yn-1 -yloxy)-8-(2-((7-(5-methyl-1 ,2,4-oxadiazol-3- yl)isoquinolin-1-yl)amino)ethyl)-7,8-dihydropyrido[3',4':4,5]pyrrolo[1,2- a]pyrazin-9(6H)-one
- Example 40 N- ⁇ -CI-chloro-g-oxo-e. -dihydropyridoIS' ⁇ ' ⁇ .SJpyrrolon ⁇ - a]pyrazin-8(9H)-yl)ethyl)-3-(5-methyl-1,2,4-oxadiazol-3-yl)benzamide
- Example 40.1 Aqueous NaOH (3 N, 21 mL) was added to a solution of methyl 4- chloro-1 H-pyrrolo[3,2-c]pyridine-2-carboxylate (1.0 g, 4.75 mmol, 1.0 eq.) in EtOH (21 mL) and this was heated to 45 °C for 1 h before the solvent was evaporated and the crude solid triturated with Et20.
- Example 40.2 DMF (9.72 mL) was added dropwise to a mixture of sodium 4-chloro- 1 H-pyrrolo[3,2-c]pyridine-2-carboxylate (1.71 g, 7.8 mmol, 1.0 eq.) and HATU (3.99 g, 10.5 mmol, 1.34 eq.) in DCM (19.6 mL) and this was stirred for 30 min before a solution of tert-butyl N- ⁇ 2-[(2-hydroxyethyl)amino]ethyl ⁇ carbamate (1.95 g, 9.53 mmol, 1.22 eq.) in DCM (19.6 mL) was added.
- Example 40.3 DIAD (3.91 mL, 19.868 mmol) was added over 40 min to a solution of tert-butyl N-[2-[(4-chloro-1H-pyrrolo[3,2-c]pyridine-2-carbonyl)-(2- hydroxyethyl)amino]ethyl]carbamate (2.54 g, 6.6226 mmol) and PPha (5.21 g, 19.868 mmol) in THF (65.30 mL, 0.1000 M) at 0 °C. After 1 h, the resulting mixture was brought back to RT and stirred for an additional 3 h. After the solvent was removed under reduced pressure, the residue was diluted with EtOAc and 10% aq.
- Example 40.4 The crude was diluted in DCM (30 mL), and TFA (30 mL) was added at 0 °C at which point the solution became red. This was stirred for 30 min at RT before the solvent was evaporated and taken back in aq. HCI (3 N). After concentration, the residue was washed several times with DCM, then reverse phase chromatography (5-25% MeOH in H2O) afforded 2-(6-chloro-10-oxo-1,5,11- triazatricyclo[7.4.0.02,7]trideca-2(7),3,5,8-tetraen-11-yl)ethylammonium chloride (1.2 g, 60%, 3.9682 mmol).
- Example 41 N-(2-(1-(3-fluoropropoxy)-9-oxo-6,7- dihydropyridoIS' ⁇ ' ⁇ .SJpyrrolon ⁇ -aJpyrazin-SfSHJ-yQethyQ-S-fS-methyl-l ⁇ - oxadiazol-3-yl)benzamide
- Example 42 N-(2-(1-(2,2-difluoroethoxy)-9-oxo-6,7- dihydropyrido[3',4':4,5]pyrrolo[1 ,2-a]pyrazin-8(9H)-yl)ethyl)-3-(5-methyl-1,2,4- oxadiazol-3-yl)benzamide
- Example 44 1 -(3-Fluoropropoxy)-8-(2-((7-(5-methyl-1 ,2,4-oxadiazol-3- yl)isoquinolin-1-yl)amino)ethyl)-7,8-dihydropyrido[3',4':4,5]pyrrolo[1,2- a]pyrazin-9(6H)-one
- Example 45.1 N-(2-(1-chloro-9-oxo-6,7-dihydropyrido[3',4':4,5]pyrrolo[1 ,2-a]pyrazin- 8(9H)-yl)ethyl)-3-(2-methyl-2H-tetrazol-5-yl)benzamide was obtained from 2-(6- chloro- 10-oxo-1 ,5,11-triazatricyclo[7.4.0.02,7]trideca-2,4,6,8-tetraen-11- yl)ethylammonium chloride and 3-(2-Methyl-2H-tetrazol-5-yl)-benzoic acid, using Example 40.5 conditions.
- Example 45.2 1-Propanol-1 ,1 ,2,2,3,3,3-d? and N-[2-(6-chloro-10-oxo-1 ,5,11- triazatricyclo[7.4.0.02,7]trideca-2,4,6,8-tetraen-11-yl)ethyl]-3-(2-methyltetrazol-5- yl)benzamide, using Example 37.1 conditions.
- Example 47 N-(2-(1-(dodec-11-yn-1-yloxy)-9-oxo-6,7- dihydropyrido[3 , ,4 , :4,5]pyrrolo[1,2-a]pyrazin-8(9H)-yl)ethyl)-3-(2-methyl-2H- tetrazol-5-yl)benzamide Obtained from 3-(2-methyl-2H-tetrazol-5-yl)-benzoic acid and 11-(2-aminoethyl)-6- dodec-11-ynoxy-1 ,5,11-triazatricyclo[7.4.0.02,7]trideca-2,4,6,8-tetraen-10-one, using Example 40.5 conditions.
- Example 48 (S)-8-(3-Amino-2-((7-(5-methyl-1,2,4-oxadiazol-3-yl)isoquinolin-1- yl)amino)propyl)-1-(2,2,2-trifluoroethoxy)-7,8- dihydropyrido[3',4':4,5]pyrrolo[1 ,2-a]pyrazin-9(6H)-one
- Example 48.1 DI PEA (1029.54 pL, 5.9109 mmol) was added to a mixture of Boc- N3-Cbz-L-2,3-diaminopropionic acid (1.00 g, 2.9554 mmol) and HATLI (1573.24 mg, 4.1376 mmol) in DCM (14.78 mL). This was heated to 70 °C for 15 min or until the mixture became a clear yellow solution. A solution of ethanolamine (231.89 pL, 3.8421 mmol) in DCM (14.78 mL) was added dropwise at RT, at which point a precipitate formed, and this was stirred at this temperature for 2 h. The mixture was washed with H2O, aq.
- Example 48.2 BF3.OEt2 (7.4 pL, 60.3 pmol, 1.0 eq.) was added dropwise to a solution of tert-butyl N-[(1S)-1-(benzyloxycarbonylaminomethyl)-2-(2- hydroxyethylamino)-2-oxo-ethyl]carbamate (23 mg, 60.3 pmol) in THF (600 pL) at 0 °C, followed immediately with BH3 THF (1M in THF) (362 pL, 0.36 mmol, 6.0 eq.) dropwise. The ice bath was removed, and the solution was stirred at RT for 2 h.
- Example 48.3 A 15 min pre-stirred yellow solution of sodium 4-(2,2,2- trifluoroethoxy)-1H-pyrrolo[3,2-c]pyridine-2-carboxylate (208.00 mg, 0.7372 mmol) and HATLI (392.43 mg, 1.0321 mmol) in DMF (3.69 mL) was added dropwise to a solution of tert-butyl N-[(1R)-1-(benzyloxycarbonylaminomethyl)-2-(2- hydroxyethylamino)ethyl]carbamate (325.05 mg, 0.8846 mmol) and DI PEA (256.81 pL, 1.4744 mmol) in DMF (3.69 mL).
- Example 48.4 DIAD (298.74 pL, 1.5172 mmol) was added dropwise to a solution of tert-butyl N-[(1S)-1-(benzyloxycarbonylaminomethyl)-2-[2-hydroxyethyl-[4-(2,2,2- trifluoroethoxy)-1H-pyrrolo[3,2-c]pyridine-2-carbonyl]amino]ethyl]carbamate (308.30 mg, 0.5057 mmol) and PPh 3 (397.96 mg, 1.5172 mmol) in THF (5.06 mL, 0.1000 M) at 0 °C. After 1 h, the ice bath was removed and stirred overnight.
- Example 48.6 DI PEA (304.48 pL, 1.7481 mmol) was added dropwise to a suspension of benzyl N-[(2S)-2-amino-3-[10-oxo-6-(2,2,2-trifluoroethoxy)-1,5,11- triazatricyclo[7.4.0.02,7]trideca-2,4,6,8-tetraen-11-yl]propyl]carbamate (229.10 mg, 0.4662 mmol), 5-methyl-3-(2-oxidoisoquinolin-2-ium-7-yl)-1,2,4-oxadiazole (127.11 mg, 0.5594 mmol) and PyBrop (260.78 mg, 0.5594 mmol) in DCM (4.66 mL, 0.1000 M).
- Example 48.7 To a solution of Pd(OAc)2 (5.6 mg, 24.9 pL mmol, 23 mol%) in DCM (543 pL) were added successively Et 3 N (13 pL, 95.3 pmol, 0.9 eq.) and EtaSiH (38 pL, 0.24 mmol, 2.2 eq.) at RT.
- Example 49.1 DI PEA (142 pL, 0.8 mmol, 4.0 eq.) was added to a mixture of 3-(2- Methyl-2H-tetrazol-5-yl)-benzoic acid (48 mg, 0.23 mmol, 1.15 eq.) and HATLI (154.7 mg, 0.41 mmol, 2.0 eq.) in DCM (1 .0 mL) and this was stirred at 40 °C for 30 min before a solution of benzyl N-[(2S)-2-amino-3-[10-oxo-6-(2,2,2-trifluoroethoxy)- 1 ,5,11-triazatricyclo[7.4.0.02,7]trideca-2(7),3,5,8-tetraen-11-yl]propyl]carbamate (100 mg, 0.2 mmol, 1.0 eq.) in DCM (1.0 mL) was added at RT.
- DI PEA 142 pL, 0.8
- Example 49.2 Et 3 SiH (50 pL, 0.3 mmol, 5.7 eq.) was added to a mixture of benzyl N-[(2S)-2-[[3-(2-methyltetrazol-5-yl)benzoyl]amino]-3-[10-oxo-6-(2,2,2- trifluoroethoxy)-1 ,5,11-triazatricyclo[7.4.0.02,7]trideca-2(7),3,5,8-tetraen-11- yl]propyl]carbamate (37 mg, 55.2 pmol, 1.0 eq.) and Pd/C (35 mg, 100% wt/wt) in EtOH (552 pL, 0.1 M) at RT.
- the mixture was passed through a SCX-2 column, eluting successively with MeOH and DCM, then 10% NH3 (2N in MeOH) in DCM to collect the expected free amine. Further purification was performed with preparative TLC (10% NH 3 (2M in MeOH) in DCM).
- Example 50 - (S)-3-(2-Methyl-2H-tetrazol-5-yl)-N-(6-(methylamino)-1-(9-oxo-1- (2,2,2-trifluoroethoxy)-6,7-dihydropyrido[3 , ,4 , :4,5]pyrrolo[1,2-a]pyrazin-8(9H)- yl)hexan-2-yl)benzamide
- Example 50.1 DIPEA (721.88 pL, 4.1445 mmol) was added to a mixture of Fmoc- Lys(Me,Boc)-OH (1.00 g, 2.0722 mmol) and HATU (1.03 g, 2.7037 mmol) in DCM (10.36 mL, 0.1000 M). This was heated to 50 °C for 15 min until the mixture became a clear yellow solution. A solution of ethanolamine (162.64 uL, 2.6939 mmol) in DCM (10.36 mL) was added dropwise at RT, at which point a precipitate formed, and this was stirred at this temperature for 2 h. The mixture was washed with aq. HCI (1M) and the solvent evaporated.
- Example 50.2 BF3'OEt2 (1.28 mL, 10.361 mmol) was added dropwise to a solution of tert-butyl N-[(5S)-5-(9H-fluoren-9-ylmethoxycarbonylamino)-6-(2- hydroxyethylamino)-6-oxo-hexyl]-N-methyl-carbamate (5.45 g, 10.361 mmol) in THF (103.61 mL) at O °C, followed immediately with BH 3 THF (1M in THF) (62.16 mL, 62.164 mmol) dropwise. The ice bath was removed, and the solution was stirred at RT overnight.
- Example 50.3 A 15 min pre-stirred yellow solution of sodium 4-(2,2,2- trifluoroethoxy)-1H-pyrrolo[3,2-c]pyridine-2-carboxylate (1.00 g, 3.5442 mmol) and HATLI (4.04 g, 10.633 mmol) in DMF (17.72 mL) was added dropwise to a solution of tert-butyl N-[(5S)-5-(9H-fluoren-9-ylmethoxycarbonylamino)-6-(2- hydroxyethylamino)hexyl]-N-methyl-carbamate (1.81 g, 3.5442 mmol) and DI PEA (1.85 mL, 10.633 mmol) in DMF (17.72 mL).
- Example 50.4 A solution of PPha (1.56 g, 6.0 mmol, 3.0 eq.) in THF (9.9 mL) was added to a solution of tert-butyl N-[(5S)-5-(9H-fluoren-9-ylmethoxycarbonylamino)-6- [2-hydroxyethyl-[4-(2,2,2-trifluoroethoxy)-1H-pyrrolo[3,2-c]pyridine-2- carbonyl]amino]hexyl]-N-methyl-carbamate (1.50 g, 2.0 mmol, 1.0 eq.) in THF (9.9 mL) at -40 °C to prevent Fmoc removal.
- Example 50.5 Piperidine (392 pL, 3.97 mmol, 2.0 eq.) was added to a solution of tert-butyl N-[(5S)-5-(9H-fluoren-9-ylmethoxycarbonylamino)-6-[10-oxo-6-(2,2,2- trifluoroethoxy)-1,5,11-triazatricyclo[7.4.0.02,7]trideca-2,4,6,8-tetraen-11-yl]hexyl]-N- methyl-carbamate (1.46 g, 2.0 mmol, 1.0 eq.) in DMF (19.9 mL, 0.1 M). After 30 min, water was added, and the solvents were evaporated.
- Example 50.6 DI PEA (60.64 pL, 0.3482 mmol) was added to a mixture of 3-(2- Methyl-2H-tetrazol-5-yl)-benzoic acid (20.44 mg, 0.1001 mmol), HATLI (66.19 mg, 0.1741 mmol) and tert-butyl N-[(5S)-5-amino-6-[10-oxo-6-(2,2,2-trifluoroethoxy)- 1 ,5,11-triazatricyclo[7.4.0.02,7]trideca-2(7),3,5,8-tetraen-11-yl]hexyl]-N-methyl- carbamate (44.70 mg, 0.0870 mmol) in DCM (870.41 uL, 0.1000 M).
- Example 50.7 tert-Butyl N-methyl-N-[(5S)-5-[[3-(2-methyltetrazol-5- yl)benzoyl]amino]-6-[10-oxo-6-(2,2,2-trifluoroethoxy)-1,5,11- triazatricyclo[7.4.0.02,7]trideca-2(7),3,5,8-tetraen-11-yl]hexyl]carbamate (46.50 mg, 0.0665 mmol) was stirred in DCM/TFA (1:1) (664.55 pL) for 30 min. The solvents were evaporated, taken back in aq. HCI (3 N) and evaporated (x2).
- Example 51 - (S)-N-(6-(methyl(prop-2-yn-1-yl)amino)-1-(9-oxo-1-(2,2,2- trifluoroethoxy)-6,7-dihydropyrido[3',4':4,5]pyrrolo[1,2-a]pyrazin-8(9H)- yl)hexan-2-yl)-3-(2-methyl-2H-tetrazol-5-yl)benzamide
- Example 52 - (S)-8-(6-(Methyl(prop-2-yn-1-yl)amino)-2-((7-(5-methyl-1,2,4- oxadiazol-3-yl)isoquinolin-1-yl)amino)hexyl)-1-(2,2,2-trifluoroethoxy)-7,8- dihydropyrido[3',4':4,5]pyrrolo[1 ,2-a]pyrazin-9(6H)-one
- Example 52.1 DI PEA (0.13 mL, 0.7302 mmol) was added dropwise to a suspension of tert-butyl N-[(5S)-5-amino-6-[10-oxo-6-(2,2,2-trifluoroethoxy)-1 ,5,11- triazatricyclo[7.4.0.02,7]trideca-2(7),3,5,8-tetraen-11-yl]hexyl]-N-methyl-carbamate (100.00 mg, 0.1947 mmol), 5-methyl-3-(2-oxidoisoquinolin-2-ium-7-yl)-1 ,2,4- oxadiazole (53.09 mg, 0.2337 mmol) and PyBrop (108.93 mg, 0.2337 mmol) in DCM (1.62 mL, 0.1200 M).
- Example 52.2 tert-Butyl N-methyl-N-[(5S)-5-[[7-(5-methyl-1 ,2,4-oxadiazol-3-yl)-1- isoquinolyl]amino]-6-[10-oxo-6-(2,2,2-trifluoroethoxy)-1 ,5,11- triazatricyclo[7.4.0.02,7]trideca-2(7),3,5,8-tetraen-11-yl]hexyl]carbamate (112.00 mg, 0.1550 mmol) was stirred in DCM/TFA (1 :1) (1.55 mL) for 30 min. The solvents were evaporated, taken back in MeOH and filtered through an SCX-2 column, washing with MeOH.
- Example 53.1 To a solution of tert-butyl (3S)-3-amino-6-[tert- butoxycarbonyl(methyl)amino]hexanoate (500.00 mg, 1.5801 mmol) and 3-(5- Methyl-1 ,2,4-oxadiazol-3-yl)benzoic acid (354.88 mg, 1.7381 mmol) in dry DMF (7.70 mL) were added T3P in DMF (3.72 mL, 3.1602 mmol) and triethylamine (0.67 mL, 4.7402 mmol). The reaction mixture was stirred at room temperature for 2 h 15 min.
- Example 53.2 To a solution of tert-butyl (3S)-6-[tert-butoxycarbonyl(methyl)amino]- 3-[[3-(5-methyl-1 ,2,4-oxadiazol-3-yl)benzoyl]amino]hexanoate (710.00 mg, 1.4127 mmol) in THF (14.13 mL) were added potassium hydroxide (792.64 mg, 14.127 mmol) and water (0.5 mL, enough to dissolve the KOH) and 8 drops of MeOH. The mixture was stirred at 50 °C for 2 h. The mixture was allowed to cool and the volatiles were removed under reduced pressure.
- Example 53.3 To a mixture of rac-(3S)-6-[tert-butoxycarbonyl(methyl)amino]-3-[[3- (5-methyl-1 ,2,4-oxadiazol-3-yl)benzoyl]amino]hexanoic acid (20.00 mg, 0.0448 mmol), 7-propoxy-1 ,3-benzothiazol-2-amine [Example 75.3] (12.13 mg, 0.0582 mmol), 1-hydroxybenzotriazole (12.10 mg, 0.0896 mmol) and 1-(3- dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (17.17 mg, 0.0896 mmol) under N2 was added dry DMF (0.22 mL).
- Example 53.4 To a flask charged with tert-butyl N-methyl-N-[(4S)-4-[[3-(5-methyl- 1 ,2,4-oxadiazol-3-yl)benzoyl]amino]-6-oxo-6-[(7-propoxy-1 ,3-benzothiazol-2- yl)amino]hexyl]carbamate (9.00 mg, 0.0141 mmol) was added 4M HCI in dioxane (1.00 mL, 4 mmol). This solution was stirred at RT for 2 h. The mixture was concentrated under reduced pressure to give a pale-yellow solid.
- Example 54.1 To a solution of tert-butyl 3-amino-6-[tert- butoxycarbonyl(methyl)amino]hexanoate (522.25 mg, 1.6504 mmol) in dry DCM (8.63 mL) were added 5-methyl-3-(2-oxidoisoquinolin-2-ium-7-yl)-1 ,2,4-oxadiazole (300 mg, 1.32 mmol), PyBroP (0.75 g, 1.6108 mmol) and DIPEA (0.86 mL, 4.9511 mmol). This mixture was stirred at room temperature under N2 for 16 h.
- Example 54.2 To a solution of tert-butyl (3S)-6-[tert-butoxycarbonyl(methyl)amino]- 3-[[7-(5-methyl-1 ,2,4-oxadiazol-3-yl)-1-isoquinolyl]amino]hexanoate (522.00 mg, 0.9931 mmol) in THF (9.93 mL) were added potassium hydroxide (557.21 mg, 9.9308 mmol) and water (0.5 mL enough to dissolve the KOH) followed by 8 drops of MeOH. The mixture was allowed to stir for 18 h at 50 °C. Additional potassium hydroxide (557.21 mg, 9.9308 mmol) was added and stirring was continued at 50 °C for 13 d.
- Example 54.3 To a mixture of (3S)-6-[tert-butoxycarbonyl(methyl)amino]-3-[[7-(5- methyl-1,2,4-oxadiazol-3-yl)-1-isoquinolyl]amino]hexanoic acid (30.00 mg, 0.0639 mmol) and 7-propoxy-1,3-benzothiazol-2-amine [Example 75.3] (13.31 mg, 0.0639 mmol) in DMF (0.32 mL) under N2 were added triethylamine (0.03 mL, 0.1917 mmol) and then 1-propanephosphonic anhydride (50% in DMF) (0.08 mL, 0.1278 mmol).
- Example 54.4 To a flask charged with tert-butyl N-methyl-N-[(4S)-4-[[7-(5-methyl- 1 ,2,4-oxadiazol-3-yl)-1-isoquinolyl]amino]-6-oxo-6-[(7-propoxy-1,3-benzothiazol-2- yl)amino]hexyl]carbamate (34.00 mg, 0.0515 mmol) was added 4M HCI in dioxane (1.03 mL, 4.1225 mmol). This solution was stirred at RT for 20 min.
- Example 56 N-[4-fluoro-7-(3-fluoropropoxy)-1,3-benzothiazol-2-yl]-3-[[7-(5- methyl-1,2,4-oxadiazol-3-yl)-1 -isoquinolyl]amino]propanamide
- Example 56.1 To a solution of 2-amino-4-fluoro-1 ,3-benzothiazol-7-ol (80.00 mg, 0.4343 mmol) in DMF (4.01 mL, 0.1100 M) stirring at RT under N2 was added potassium carbonate (120.06 mg, 0.8687 mmol). This was stirred for 30 min before the addition of 1-iodo-3-fluoropropane (0.04 mL, 0.4343 mmol). This was then stirred under N2 for 7 h. The mixture was diluted with water (2 mL), acidified to pH6 with 2N HCI solution and then MeOH (20 mL) was added.
- Example 56.2 To a mixture of 3-[[7-(5-methyl-1 ,2,4-oxadiazol-3-yl)-1- isoquinolyl]amino]propanoic acid B2 (25.00 mg, 0.0838 mmol) and 4-fluoro-7-(3- fluoropropoxy)-1 ,3-benzothiazol-2-amine (20.47 mg, 0.0838 mmol) in DMF (0.84 mL) was added triethylamine (0.04 mL, 0.2514 mmol) and then 1- propanephosphonic anhydride (50% in DMF) (0.10 mL, 0.1676 mmol). This mixture was stirred at 70 °C under N2 for 3 d. A further 2 eq.
- Example 57.1 To a solution of 2-amino-4-fluoro-1 ,3-benzothiazol-7-ol (80.00 mg, 0.4343 mmol) in DMF (4.01 mL, 0.1100 M) stirring at RT under N2 was added potassium carbonate (120.06 mg, 0.8687 mmol). This was stirred for 30 min before the addition of 1-iodo-3-fluoroethane (0.05 mL, 0.4343 mmol). This was then stirred under N2 for 25 h. The mixture was diluted with water (2 mL), acidified to pH6 with 2N HCI in water solution and then MeOH (20 mL) was added.
- Example 57.2 To a mixture of 3-[[7-(5-methyl-1 ,2,4-oxadiazol-3-yl)-1- isoquinolyl]amino]propanoic acid B2 (35.00 mg, 0.1173 mmol), and 4-fluoro-7-(2- fluoroethoxy)-1 ,3-benzothiazol-2-amine (32.42 mg, 0.1408 mmol) in DMF (0.50 mL) was added 1-propanephosphonic anhydride (0.06 mL, 0.2347 mmol). This mixture was stirred at 70 °C under N2 for 3 d.
- Example 58 3-((7-(5-Methyl-1,2,4-oxadiazol-3-yl)isoquinolin-1-yl)amino)-N-(7- propoxybenzo[d]thiazol-2-yl)propanamide
- Example 60 (1s,3s)-N-(7-methoxy-4-methylbenzo[d]thiazol-2-yl)-3-((7-(5- methyl-1, 2, 4-oxadiazol-3-yl)isoquinolin-1-yl)amino)cyclobutane-1 -carboxamide
- Example 61 (1s,3s)-N-(7-methoxybenzo[d]thiazol-2-yl)-3-((7-(5-methyl-1,2,4- oxadiazol-3-yl)isoquinolin-1 -yl)amino)cyclobutane-1 -carboxamide
- Example 62 N-(4-fluoro-7-propoxybenzo[d]thiazol-2-yl)-3-((7-(5-methyl-1,2,4- oxadiazol-3-yl)isoquinolin-1-yl)amino)propanamide
- Example 62.1 A mixture of 2-amino-4-fluorobenzo[d]thiazol-7-ol (200 mg, 1.09 mmol, 1.00 eq), potassium carbonate (599 mg, 4.33 mmol, 3.99 eq) and 1- iodopropane (222 mg, 1.30 mmol, 1.20 eq) in acetonitrile (2.00 mL) was stirred at 50 °C for 12 h. After the reaction was completed, the mixture was filtered.
- Example 62.2 A mixture of 3-((7-(5-methyl-1 ,2,4-oxadiazol-3-yl)isoquinolin-1- yl)amino)propanoic acid (60.0 mg, 201 umol, 1.00 eq), 4-fluoro-7- propoxybenzo[d]thiazol-2-amine (50.0 mg, 221 umol, 1.10 eq), propylphosphonicanhydride (256 mg, 402 umol, 239 uL, 50% purity in dimethyl formamide, 2.00 eq) in dimethyl formamide (0.500 mL) was stirred at 50 °C for 2 h.
- Example 63.1 To a solution of potassium thiocyanate (1.36 g, 14.0 mmol, 1.10 eq) in acetone (20 mL) was added a solution of acetyl chloride (1.10 g, 14.0 mmol, 0.996 mL, 1.10 eq) in acetone (10 mL) at 20 °C. The mixture was stirred at 50 °C for 0.2 h. Then to the mixture was added a solution of 2-chloro-5-methoxyaniline (2.00 g, 12.7 mmol, 1.00 eq) in acetone (20 mL). The resulting mixture was stirred at 50 °C for 0.5 h.
- Example 63.2 To a solution of 1-(2-chloro-5-methoxyphenyl)thiourea (1.50 g, 6.92 mmol, 1.00 eq) in acetic acid (15 mL) was added bromine (608 mg, 3.81 mmol, 196 uL, 0.55 eq) at 0 °C. The mixture was stirred at 20 °C for 2 h.
- Example 63.3 To a solution of 4-chloro-7-methoxybenzo[d]thiazol-2-amine (1.70 g, 7.92 mmol, 1.00 eq) in dichloromethane (10 mL) was added boron tribromide (9.92 g, 39.6 mmol, 3.82 mL, 5.00 eq) at 0 °C. The mixture was stirred at 30 °C for 12 h. Then the mixture was poured into methanol (50 mL) at 0 °C carefully. The mixture was concentrated under reduced pressure to give a residue.
- Example 63.4 To a solution of 2-amino-4-chlorobenzo[d]thiazol-7-ol (0.400 g, 1.99 mmol, 1.00 eq) in dimethyl formamide (5 mL) were added potassium carbonate (551 mg, 3.99 mmol, 2.00 eq) and 1-iodopropane (407 mg, 2.39 mmol, 234 uL, 1.20 eq). The mixture was stirred at 30 °C for 12 h. Then the mixture was filtered.
- Example 63.5 To a solution of 4-chloro-7-propoxybenzo[d]thiazol-2-amine (100 mg, 412 umol, 1.00 eq) and 3-((7-(5-methyl-1 ,2,4-oxadiazol-3-yl)isoquinolin-1- yl)amino)propanoic acid B2 (123 mg, 0.412 mmol, 1.00 eq) in dimethyl formamide (3 mL) were added 1-methylimidazole (67.7 mg, 0.824 mmol, 65.7 uL, 2.00 eq) and N,N,N,N-tetramethylchloroformamidinium hexafluorophosphate (347 mg, 1.24 mmol, 3.00 eq).
- Example 64 3-((7-(5-Methyl-1,2,4-oxadiazol-3-yl)isoquinolin-1-yl)amino)-N-(4- propoxypyrazolo[1,5-a]pyridin-2-yl)propanamide
- Example 64.1 To a solution of 2-bromopyridin-3-ol (12.0 g, 69.0 mmol, 1.00 eq) in DMF (90 mL) was added potassium carbonate (11.4 g, 82.8 mmol, 1.20 eq). The mixture was stirred at 90 °C for 1 h. To the mixture was added 1-iodopropane (21.1 g, 124 mmol, 1.80 eq). The mixture was stirred at 90 °C for 3 h. The mixture was diluted with saturated ammonium chloride aqueous solution (100 mL) and extracted with ethyl acetate (3 x 80 mL).
- Example 64.2 To a solution of acetonitrile (3.06 g, 74.5 mmol, 2.80 eq) in tetra hydrofuran (100 mL) was added n-butyllithium (2.5 M in hexane, 74.5 mmol, 29.8 mL, 2.80 eq) dropwise at -78 °C. The mixture was stirred at -78°C for 0.2 h. To the mixture was added a solution of 2-bromo-3-propoxy-pyridine (5.75 g, 26.6 mmol, 1.00 eq) in tetrahydrofuran (10 mL).
- Example 64.3 To a solution of ethyl (1E)-/V-(2,4,6- trimethylphenyl)sulfonyloxyethanimidate (10.0 g, 35.0 mmol, 1.00 eq) in dioxane (20 mL) was added perchloric acid (9.32 g, 64.9 mmol, 5.61 mL, 70% purity, 1.85 eq) at 0 °C. Then the mixture was stirred at 0 °C for 0.5 h. The solution was diluted with ice-water (30 mL) and filtered. The filter cake was extracted with dichloromethane (50 mL).
- Example 64.4 To a solution of amino 2,4,6-trimethylbenzenesulfonate (0.600 M, 15.3 mmol, 25.5 mL, 1.35 eq) in dichloromethane (15 mL) was added 2-(3- propoxypyridin-2-yl)acetonitrile (2.00 g, 11.4 mmol, 1.00 eq). The mixture was stirred at 20 °C for 2 h. The mixture was concentrated under reduced pressure to afford 1-amino-2-(cyanomethyl)-3-propoxypyridin-1-ium 2,4,6- trimethylbenzenesulfonate (5.00 g, crude) as brown oil. It was used for next step directly.
- Example 64.5 To a solution of 1-amino-2-(cyanomethyl)-3-propoxypyridin-1-ium 2,4,6-trimethylbenzenesulfonate in methanol (60 mL) was added potassium carbonate (2.82 g, 20.4 mmol, 2.00 eq). The mixture was stirred at 20 °C for 12 h. The mixture was concentrated under reduced pressure to give a residue.
- Example 64.6 To a solution of 3-((7-(5-methyl-1 ,2,4-oxadiazol-3-yl)isoquinolin-1- yl)amino)propanoic acid B2 (46.8 mg, 0.157 mmol, 1.00 eq), 4-propoxypyrazolo[1 ,5- a]pyridin-2-amine (30.0 mg, 0.157 mmol, 1.00 eq) and 1-methylimidazole (51.5 mg, 0.628 mmol, 50.0 uL, 4.00 eq) in DMF (1 mL) was added /V, /V, /V, /V- tetramethylchloroformamidinium hexafluorophosphate (132 mg, 0.471 mmol, 3.00 eq).
- Example 65 3-((7-(5-Methyl-1,2,4-oxadiazol-3-yl)isoquinolin-1-yl)amino)-N-(4- propoxypyrazolo[1,5-a]pyrazin-2-yl)propanamide
- Example 65.1 To a solution of 3-nitro-1/7-pyrazole-5-carboxylic acid (6.00 38.2 mmol, 1.00 eq) in acetonitrile (60 mL) was added 1 ,1-carbonyldiimidazole (7.43 g, 45.8 mmol, 1.20 eq). The mixture was stirred at 60 °C for 3 h. To the mixture was added 2,2-dimethoxyethanamine (4.02 g, 38.2 mmol, 1.00 eq) and the mixture was stirred at 60 °C for 12 h. The mixture was concentrated to give a residue.
- Example 65.2 A solution of / ⁇ /-(2,2-dimethoxyethyl)-3-nitro-1/7-pyrazole-5- carboxamide (6.00 g, 24.57 mmol, 1.00 eq) in hydrochloric acid (5.00 M, 20 mL) was stirred at 20 °C for 12 h. The mixture was filtered. The filter cake was concentrated under reduced pressure to afford 7-hydroxy-2-nitro-6,7-dihydropyrazolo[1 ,5- a]pyrazin-4(5/-/)-one (5.00 g, crude) as a white solid.
- Example 65.3 To a solution of 7-hydroxy-2-nitro-6,7-dihydropyrazolo[1 ,5-a]pyrazin- 4(5/-/)-one (2.00 g, 10.1 mmol, 1.00 eq) in toluene (50 mL) were added thionyl chloride (2.40 g, 20.2 mmol, 2.00 eq) and dimethyl formamide (73.8 mg, 1.01 mmol, 0.10 eq). The mixture was stirred at 125 °C for 12 h. After cooling to room temperature, the mixture was filtered.
- Example 65.4 To a solution of 2-nitropyrazolo[1 ,5-a]pyrazin-4-ol (0.800 g, 4.44 mmol, 1.00 eq) in phosphoryl trichloride (6 mL) was added dimethyl formamide (16.2 mg, 0.05 eq). The mixture was stirred at 100 °C for 12 h. The mixture was poured into water (20 mL) and the pH was adjusted to pH8 with sodium carbonate. The mixture was extracted with ethyl acetate (3 x 20 mL).
- Example 65.5 To a solution of propan-1 -ol (363 mg, 6.04 mmol, 2.00 eq) in tetra hydrofuran (20 mL) was added sodium hydride (266 mg, 6.65 mmol, 60% purity, 2.20 eq) at 0 °C. The mixture was stirred at 20 °C for 0.5 h. To the mixture was added 4-chloro-2-nitropyrazolo[1 ,5-a]pyrazine (0.600 g, 3.02 mmol, 1.00 eq). The mixture was stirred at 20 °C for 2 h.
- Example 65.6 To a solution of 2-nitro-4-propoxypyrazolo[1 ,5-a]pyrazine (300 mg, 1.35 mmol, 1.00 eq) in methanol (8 mL) and water (8 mL) were added iron powder (377 mg, 6.75 mmol, 5.00 eq) and ammonium chloride (361 mg, 6.75 mmol, 5.00 eq). The mixture was stirred at 80 °C for 3 h. The mixture was filtered. The filtrate was concentrated under reduced pressure to give a residue.
- Example 65.7 To a solution of 3-((7-(5-methyl-1 ,2,4-oxadiazol-3-yl)isoquinolin-1- yl)amino)propanoic acid B2 (15.5 mg, 52.0 umol, 1.00 eq), 4-propoxypyrazolo[1 ,5- a]pyrazin-2-amine (10 mg, 52.02 umol, 1.00 eq) and 1 -methylimidazole (12.8 mg, 156 umol, 3.00 eq) in DMF (1 mL) was added /V-(chloro(dimethylamino)methylene)- N-methylmethanaminium hexafluorophosphate(V) (29.2 mg, 104.05 umol, 2.00 eq).
- Example 66 N-(4-ethoxypyrazolo[1,5-a]pyrazin-2-yl)-3-((7-(5-methyl-1,2,4- oxadiazol-3-yl)isoquinolin-1-yl)amino)propanamide
- 4-Ethoxypyrazolo[1 ,5-a]pyrazin-2-amine (0.16 g, 898 pmol) was prepared by an analogous procedure to Example 65 using ethanol.
- 4- ethoxypyrazolo[1 ,5-a]pyrazin-2-amine (70.0 mg, 393 umol, 1.00 eq) and 3-((7-(5- methyl-1 ,2,4-oxadiazol-3-yl)isoquinolin-1-yl)amino)propanoic acid (105 mg, 354 umol, 0.900 eq) in dimethyl formamide (5.00 mL) was added propylphosphonicanhydride (500 mg, 786 umol, 467 uL, 50% in dimethyl formamide, 2.00 eq).
- Example 67.1 A mixture of 4-chloro-2-nitro-pyrazolo[1,5-a]pyrazine [Example 65] (300 mg, 1.51 mmol, 1.00 eq), cesium carbonate (985 mg, 3.02 mmol, 2.00 eq) and
- Example 67.2 A mixture of 4-(3-fluoropropoxy)-2-nitro-pyrazolo[1 ,5-a]pyrazine (170 mg, 708 umol, 1.00 eq) and palladium on activated carbon (17.0 mg, 10% purity, wet) in ethyl acetate (10.0 mL) was stirred at 25 °C for 2 h under hydrogen atmosphere (15 Psi). After the reaction was completed, the mixture was filtered and filtrate was concentrated to afford 4-(3-fluoropropoxy)pyrazolo[1 ,5-a]pyrazin-2-amine (140 mg, 666 umol, 94% yield) as yellow oil.
- Example 67.3 A mixture of 3-((7-(5-methyl-1 ,2,4-oxadiazol-3-yl)isoquinolin-1- yl)amino)propanoic acid B2 (50.0 mg, 167 umol, 1.00 eq), propylphosphonicanhydride (213 mg, 335 umol, 50% in DMF, 2.00 eq) and 4-(3- fluoropropoxy)pyrazolo[1 ,5-a]pyrazin-2-amine (38.8 mg, 184 umol, 1.10 eq) in DMF (0.500 mL) was stirred at 40 °C for 2 h.
- Example 68 N-(4-(2-fluoroethoxy)pyrazolo[1,5-a]pyrazin-2-yl)-3-((7-(5-methyl- 1 ,2,4-oxadiazol-3-yl)isoquinolin-1-yl)amino)propanamide
- Example 70 N-(4-cyclopropoxypyrazolo[1,5-a]pyrazin-2-yl)-3-((7-(5-methyl- 1 ,2,4-oxadiazol-3-yl)isoquinolin-1-yl)amino)propanamide
- Example 71.1 To a mixture of methyl 4-chloro-1/7-pyrrolo[3,2-c]pyridine-2- carboxylate (900 mg, 4.27 mmol, 1.00 eq) and cesium carbonate (2.78 g, 8.55 mmol, 2.00 eq) in /V,/V-dimethylacetamide (9.00 mL) was added iodomethane (728 mg, 5.13 mmol, 1.20 eq). The mixture was stirred at 25 °C for 2 h. After the reaction was completed, the mixture was quenched with saturated ammonium chloride aqueous solution (30 mL) and extracted with ethyl acetate (3 x 30 mL). The combined organic layers were concentrated to give a residue.
- Example 71.2 To a suspension of methyl 4-chloro-1-methyl-1/7-pyrrolo[3,2- c]pyridine-2-carboxylate (700 mg, 3.12 mmol, 1.00 eq) in acetic acid (7.00 mL) was added ammonium acetate (480 mg, 6.23 mmol, 2.00 eq). The mixture was stirred at 120 °C for 12 h. After the reaction was completed, the mixture was concentrated to give a residue. The residue was triturated with water (5 mL) at 25 °C for 0.5 h.
- Example 71.3 A mixture of methyl 4-hydroxy-1-methyl-1/7-pyrrolo[3,2-c]pyridine-2- carboxylate (700 mg, 3.39 mmol, 1.00 eq), silver carbonate (1.87 g, 6.79 mmol, 2.00 eq) and 1-iodopropane (866 mg, 5.09 mmol, 1.50 eq) in chloroform (7.00 mL) was stirred at 65 °C for 1 h. After the reaction was completed, the mixture was filtered and filtrate was concentrated to give a residue.
- Example 71.4 A mixture of methyl 1-methyl- 4-propoxy-1/7-pyrrolo[3,2-c]pyridine-2- carboxylate (480 mg, 1.93 mmol, 1.00 eq) and lithium hydroxide monohydrate (163 mg, 3.87 mmol, 2.00 eq) in a mixture solvent of tetra hydrofuran (200 uL) and water (200 uL) was stirred at 30 °C for 12 h. The mixture was concentrated to remove tetra hydrofuran and diluted with water (10 mL). The pH of the aqueous phase was adjusted to around 4 by adding hydrochloric acid (1 M). A white solid was precipitated and filtered.
- Example 71.5 A mixture of 1-methyl-4-propoxy-1/7-pyrrolo[3,2-c]pyridine-2- carboxylic acid (200 mg, 854 umol, 1.00 eq), diphenylphosphoryl azide (352 mg, 1.28 mmol, 1.50 eq) and triethylamine (259 mg, 2.56 mmol, 3.00 eq) in tert-butanol (2.00 mL) was stirred at 100 °C for 12 h.
- Example 71.6 A mixture of tert-butyl (1-methyl-4-propoxy-1/7-pyrrolo[3,2-c]pyridin-2- yl)carbamate (140 mg, 458 umol, 1.00 eq) in hydrochloric acid/ethyl acetate (4 M, 500 uL) was stirred at 25 °C for 1 h. The mixture was concentrated to afford 1- methyl-4-propoxy-pyrrolo[3,2-c]pyridin-2-amine (110 mg, crude, hydrochloride) as a white solid. HPLC/MS m/z: 206.2 [M+H] + , Rt (F): 0.75 min.
- Example 71.7 A mixture of 3-((7-(5-methyl-1 ,2,4-oxadiazol-3-yl)isoquinolin-1- yl)amino)propanoic acid B2 (85.0 mg, 285 umol, 1.00 eq), 1-methyl-4-propoxy- pyrrolo[3,2-c]pyridin-2-amine (58.5 mg, 285 umol, 1.00 eq) and propylphosphonic anhydride (181 mg, 285 umol, 169 uL, 50% in DMF, 1.00 eq) in DMF (2.00 mL) was stirred at 30 °C for 2 h.
- Example 72 (c/s)-/V-(4-methoxypyrazolo[1,5-a]pyridin-2-yl)-3-((7-(5-methyl- 1 ,2,4-oxadiazol-3-yl)isoquinolin-1-yl)amino)cyclobutanecarboxamide
- Example 72.1 To a solution of acetonitrile (769 mg, 18.7 mmol, 4.40 eq) in tetra hydrofuran (10 mL) was added n-butyllithium (2.50 M, 6.81 mL, 17.0 mmol, 4.00 eq) dropwise at -78 °C. The mixture was stirred at -78°C for 0.2 h. To the mixture was added a solution of 2-bromo-3-methoxy-pyridine (0.800 g, 4.25 mmol, 1.00 eq) in tetrahydrofuran (3 mL). The mixture was stirred at -78 °C for 1 h.
- Example 72.2 To a solution of O-(mesitylsulfonyl)hydroxylamine [Example 64.3] (0.140 M, 14.5 mL, 2.03 mmol, 1.20 eq) in dichloromethane (15 mL) was added 2- (3-methoxy-2-pyridyl)acetonitrile (0.250 g, 1.69 mmol, 1.00 eq). The mixture was stirred at 20 °C for 12 h. The mixture was concentrated to afford 1-amino-2- (cyanomethyl)-3-methoxypyridin-1-ium 2,4,6-trimethylbenzenesulfonate (0.600 g, crude) as a brown oil. It was used for next step directly. HPLC/MS m/z: 164.0 [M+H] + , Rt (G): 0.67 min.
- Example 72.3 To a solution of 1-amino-2-(cyanomethyl)-3-methoxypyridin-1-ium 2,4,6-trimethylbenzenesulfonate (0.600 g, 1.65 mmol, 1.00 eq) in methanol (10 mL) was added potassium carbonate (570 mg, 4.13 mmol, 2.50 eq). The mixture was stirred at 20 °C for 12 h. The mixture was concentrated under reduced pressure to give a residue.
- Example 72.4 To a solution of (c/s)-3-((7-(5-methyl-1,2,4-oxadiazol-3-yl)isoquinolin- 1-yl)amino)cyclobutanecarboxylic acid B3 (139 mg, 429 umol, 1.00 eq), O-(7- azabenzotriazol-1-yl)-/V,/V,/V’,/ ⁇ /-tetramethyluronium hexafluorophosphate and diisopropylethylamine (166 mg, 1.29 mmol, 3.00 eq) in dimethyl formamide (2 mL) was added 4-methoxypyrazolo[1,5-a]pyridin-2-amine (70.0 mg, 429 umol, 1.00 eq).
- Example 73.1 2-Amino-4-methyl-1 ,3-benzothiazol-7-ol (100.00 mg, 0.5548 mmol) and K2CO3 (153.36 mg, 1.1097 mmol) were mixed in anhydrous DMF (0.55 mL, 1 M ) at RT under argon. 1-lodopropane (0.05 mL, 0.5548 mmol) was added, and the reaction mixture was stirred for 24 h. The reaction mixture was mixed with water (2 mL), acidified with 1M HCI to pH 6 and diluted with MeOH (20 mL), filtered through a 2 g SCX2 column.
- Example 73.2 4-Methyl-7-propoxy-1 ,3-benzothiazol-2-amine (39.13 mg, 0.1760 mmol) and 3-[[7-(5-methyl-1 ,2,4-oxadiazol-3-yl)-1-isoquinolyl]amino]propanoic acid B2 (50.00 mg, 0.1676 mmol) were mixed in anhydrous DMF (0.34 mL, 0.5000 M ) under argon at ambient temperature. 1-Propanephosphonic anhydride (50% in DMF) (0.20 mL, 0.3352 mmol) was added, and the reaction mixture was stirred for 18 h.
- Example 75.1 To methyl 3-aminopropanoate hydrochloride (100.00 mg, 0.7164 mmol), 3-(2-methyl-2H-tetrazol-5-yl)-benzoic acid (146.29 mg, 0.7164 mmol) in DMF (4.21 mL) was added DIPEA (0.50 mL, 2.8657 mmol) followed by HATU (252.83 mg, 1.0747 mmol). The obtained yellow solution was stirred for 18 h. The reaction mixture was diluted with EtOAc (150 mL) and washed with water (120 mL). The water was extracted with fresh EtOAc (100 mL). The organics were combined and washed with aq. sat. bicarb.
- Example 75.2 To methyl 3-[[3-(2-methyltetrazol-5-yl)benzoyl]amino]propanoate (170.00 mg, 0.5876 mmol) in THF (2.90 mL) was added water (2.90 mL) followed by lithium hydroxide monohydrate (98.63 mg, 2.3506 mmol). After stirring for 45 min water (25 mL) was added and the THF removed in vacuo. The solution was acidified to pH 3 with 1M citric acid solution and extracted with EtOAc (2 x 60 mL).
- Example 75.4 To a mixture of 7-propoxy-1 ,3-benzothiazol-2-amine (15.13 mg, 0.0727 mmol), 3-[[3-(2-methyltetrazol-5-yl)benzoyl]amino]propanoic acid (20.00 mg, 0.0727 mmol), HOBt (22.25 mg, 0.1453 mmol) and 1-(3-dimethylaminopropyl)-3- ethylcarbodiimide hydrochloride (27.86 mg, 0.1453 mmol) was added under argon atmosphere DMF (0.36 mL). The resulting solution was stirred at 70 °C overnight.
- Example 76 3-((7-(5-Methyl-1,2,4-oxadiazol-3-yl)isoquinolin-1-yl)amino)-N-(7- propoxy-4-(trifluoromethyl)benzo[d]thiazol-2-yl)propanamide
- Example 76.1 3-Nitro-4-(trifluoromethyl)phenol (410.00 mg, 1.9796 mmol), potassium carbonate (300.96 mg, 2.1776 mmol) and 1-iodopropane (0.21 mL, 2.1776 mmol) were mixed in anhydrous acetone (3.96 mL, 0.5000 M) in a microwave vial under argon. The reaction mixture was heated at 100 °C for 2 h under microwave irradiation.
- Example 76.2 2-Nitro-4-propoxy-1-(trifluoromethyl)benzene (455.00 mg, 1.8259 mmol) and zinc (596.89 mg, 9.1296 mmol) were mixed under argon at 0 °C. Acetic acid (5.00 mL, 0.3700 M) was added and the reaction mixture was stirred at ambient temperature for 3 h. The reaction mixture was diluted with EtOAc (10 mL) directly loaded onto silica gel. The crude was directly purified by NP chromatography (0- 20% EtOAc in cyclohexane) to give 5-propoxy-2-(trifluoromethyl)aniline (272 mg, 68%, 1.2409 mmol) as a clear oil.
- Example 76.3 To a suspension of potassium thiocyanate (129.23 mg, 1.3298 mmol) in acetone (2.00 mL) was added dropwise at ambient temperature a solution of acetyl chloride (0.09 mL, 1.3298 mmol) in acetone (2.00 mL). The mixture was stirred for 15 min at 50 °C. Then, a solution of 5-propoxy-2-(trifluoromethyl)aniline (265.00 mg, 1.2089 mmol) in acetone (2.00 mL) was added and the reaction mixture was continued to stir for 15 min at 50 °C. The reaction mixture was continued to stir for another 15 min before being removed from the heating.
- Example 76.4 To a solution of [5-propoxy-2-(trifluoromethyl)phenyl]thiourea (220.00 mg, 0.7905 mmol) in AcOH (3.95 mL) kept at ambient temperature under argon was added a solution of bromine (0.04 mL, 0.7905 mmol) in AcOH (3.95 mL) over 15 min. The reaction mixture was continued to stir for 1 h. The reaction mixture was concentrated under reduced pressure. The crude was dissolved in MeOH filtered through an SCX2 ion exchange column.
- Example 76.5 7-Propoxy-4-(trifluoromethyl)-1 ,3-benzothiazol-2-amine B2 (27.79 mg, 0.1006 mmol) and 3-[[7-(5-methyl-1 ,2,4-oxadiazol-3-yl)-1- isoquinolyl]amino]propanoic acid (30.00 mg, 0.1006 mmol) were mixed in anhydrous DMF (0.20 mL, 0.5000 M) under argon at ambient temperature.
- 1- Propanephosphonic anhydride (50% in DMF) (0.12 mL, 0.2011 mmol) and TEA (0.04 mL, 0.3017 mmol) were added successively, and the reaction mixture was stirred for 30 min. The reaction mixture was heated to 60 °C for 1 h then at 80 °C for
- Example 77.1 To a solution of 4-fluoro-7-methoxy-1 ,3-benzothiazol-2-amine (1.00 g, 5.0449 mmol) in DCM (9.01 mL) at 0 °C was added dropwise a solution of BBra in DCM (1M) (9.23 mL, 9.2322 mmol) and the mixture was allowed to reach RT and stirred overnight. MeOH was then added at 0 °C and this was stirred for an additional 10 min after which the solvent was evaporated in vacuo.
- Example 77.2 To a mixture of 2-amino-4-fluoro-1 ,3-benzothiazol-7-ol (838.00 mg, 4.5496 mmol) and cesium carbonate (2.98 g, 9.0993 mmol) in MeCN (11.00 mL, 0.3800 M) were successively added DMF (1.00 mL) and 1 -bromopropane (454.59 uL, 5.0046 mmol). This was stirred at 65 °C for 2 h. The solvent was evaporated, the residue taken back in EtOAc and water, extracted with EtOAc, dried over MgSC>4 and evaporated.
- Example 77.3 Ethyl (2R)-3-amino-2-(benzyloxycarbonylamino)propanoate (367mg, 1.38 mmol), 5-methyl-3-(2-oxidoisoquinolin-2-ium-7-yl)-1 ,2,4-oxadiazole (345mg, 1.52 mmol), PyBroP (707 mg, 1.52 mmol), DIPEA (0.90 ml) and anhydrous DCM (2.8 mL, 0.50 M ) in a microwave vial at RT under argon. The reaction mixture was heated at 60 °C by microwave irradiation for 1 h. Volatiles were removed, and the crude was purified by RP flash chromatography (30-60% MeOH in water).
- Example 77.4 Ethyl (2R)-2-(benzyloxycarbonylamino)-3-[[7-(5-methyl-1 ,2,4- oxadiazol-3-yl)-1-isoquinolyl]amino]propanoate (200.00 mg, 0.4206 mmol) was dissolved in THF (2.10 mL) at ambient temperature. EtOH (0.42 mL) and aqueous 2M NaOH (0.42 mL, 0.8412 mmol) were added and the reaction mixture was stirred at ambient temperature for 1 h. The reaction mixture was neutralized with aq. 2M HCI and concentrated under reduced pressure.
- Example 77.5 (2R)-2-(Benzyloxycarbonylamino)-3-[[7-(5-methyl-1 ,2,4-oxadiazol-3- yl)-1-isoquinolyl]amino]propanoic acid (50.00 mg, 0.1117 mmol), 4-fluoro-7-propoxy- 1 ,3-benzothiazol-2-amine (25.28 mg, 0.1117 mmol) and TEA (0.05 mL, 0.3352 mmol) were mixed in anhydrous DMF (0.22 mL, 0.5000 M) at ambient temperature under argon.
- Example 77.6 Benzyl N-[(1 R)-2-[(4-fluoro-7-propoxy-1 ,3-benzothiazol-2-yl)amino]- 1-[[[7-(5-methyl-1,2,4-oxadiazol-3-yl)-1-isoquinolyl]amino]methyl]-2-oxo- ethyl]carbamate (50.00 mg, 0.0763 mmol) was suspended in acetic acid (1.80 mL, 0.0400 M) at room temperature under an argon atmosphere.
- Example 79.1 2-Amino-1 ,3-benzothiazol-7-ol (94.0 mg, 0.56 mmol) and K2CO3 (93.8 mg, 0.68 mmol) were dissolved in dry DMF (3.8 mL) and the obtained mixture was stirred at room temperature for 40 min. lodoethane (51 uL, 0.56 mmol) was then added at 0 °C. The mixture was stirred during 1 h at 0 °C. The ice bath was then removed, and the reaction mixture stirred at room temperature for 3 d. Water ( ⁇ 1 mL) was then added to the reaction mixture.
- Example 79.2 To a mixture of 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride [B2] (32.1 mg, 0.17 mmol), 7-ethoxy-1 ,3-benzothiazol-2-amine (19.5 mg, 0.10 mmol) in DMF (0.52 mL) was added 1 -hydroxybenzotriazole (22.6 mg, 0.17 mmol) under N2. The resulting solution was stirred at 60 °C overnight. The reaction crude purified by reverse phase column chromatography eluting with 20- 100% MeOH in water (+0.1% formic acid in both).
- Example 80 (S)-N-(7-ethoxybenzo[d]thiazol-2-yl)-2-hydroxy-3-((7-(5-methyl- 1 ,2,4-oxadiazol-3-yl)isoquinolin-1-yl)amino)propenamide
- Example 82 (R)-N-(7-ethoxybenzo[d]thiazol-2-yl)-2-hydroxy-3-((7-(5-methyl- 1 ,2,4-oxadiazol-3-yl)isoquinolin-1-yl)amino)propenamide
- Example 82.1 A1 (200.0 mg, 0.88 mmol) and methyl (2R)-3-amino-2-hydroxy- propanoate hydrochloride (150.6 mg, 0.99 mmol) were dissolved/suspended in anhydrous DCM (2.93 mL) in a microwave vial. DIPEA (0.77 mL, 4.40 mmol) was added followed by PyBroP (533.4 mg, 1.14 mmol) and the mixture stirred at room temperature for 4 d.
- Example 82.2 Methyl (2R)-2-hydroxy-3-[[7-(5-methyl-1,2,4-oxadiazol-3-yl)-1- isoquinolyl]amino]propanoate (107.5 mg, 0.33 mmol) was dissolved in anhydrous THF (0.82 mL) and water (0.82 mL). To this was added lithium hydroxide monohydrate (30.2 mg, 0.72 mmol) and the obtained mixture was stirred at RT for 21 h.
- Example 82.3 To a mixture of (2R)-2-hydroxy-3-[[7-(5-methyl-1,2,4-oxadiazol-3-yl)- 1-isoquinolyl]amino]propanoic acid (30.0 mg, 0.096 mmol), 1-(3- dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (30.5 mg, 0.16 mmol), 7- ethoxy-1,3-benzothiazol-2-amine [Example 79.1] (22.2 mg, 0.11 mmol) in anhydrous DMF (0.60 mL) was added under nitrogen atmosphere 1 -hydroxybenzotriazole (25.8 mg, 0.19 mmol).
- Example 84 N-(2,2-difluoro-3-((4-fluoro-7-propoxybenzo[d]thiazol-2-yl)amino)- 3-oxopropyl)-3-(5-methyl-1,2,4-oxadiazol-3-yl)benzamide
- Example 84.1 DI PEA (0.70 mL, 4.00 mmol) was added to a solution of ethyl 2,2- difluoro-3-aminopropanoate hydrochloride (200.0 mg, 1.00 mmol), 3-(5-methyl- 1 ,2,4-oxadiazol-3-yl)benzoic acid (204.6 mg, 1.00 mmol) and HATLI (571.7 mg, 1.50 mmol) in DMF (6.68 mL). The yellow reaction mixture was stirred at room temperature for 2 d. The reaction mixture was then diluted with EtOAc (50 mL) and washed with water (100 mL).
- Example 84.2 Lithium hydroxide (47.4 mg, 1.98 mmol) was added to a solution of ethyl 2,2-difluoro-3-[[3-(5-methyl-1,2,4-oxadiazol-3-yl)benzoyl]amino]propanoate (305.0 mg, 0.90 mmol) in THF (2.25 mL) and water (2.25 mL). The reaction mixture was left stirring at room temperature for 2 h 20 min. The reaction mixture was concentrated and the aqueous was acidified with a 1M aqueous solution of citric acid to pH 3 and extracted with EtOAc (60 mL).
- Example 84.3 2,2-Difluoro-3-[[3-(5-methyl-1 ,2,4-oxadiazol-3- yl)benzoyl]amino]propanoic acid (34.3 mg, 0.11 mmol), 4-fluoro-7-propoxy-1 ,3- benzothiazol-2-amine [Example 77.2] (24.9 mg, 0.11 mmol) and TEA (50 uL, 0.33 mmol) were mixed in anhydrous DMF (0.22 mL) at room temperature.
- Example 85 3-((7-(5-Methyl-1,2,4-oxadiazol-3-yl)isoquinolin-1-yl)amino)-N-(1- methyl-7-propoxy-1 H-benzo[d]imidazol-2-yl)propenamide
- Example 85.1 7-Propoxy-1 H-benzimidazol-2-amine [Example 86] (83.0 mg, 0.43 mmol), potassium hydroxide (43.8 mg, 0.78 mmol) and iodomethane (28 uL, 0.45 mmol) were stirred in ethanol (5.4 mL) at RT for 2 d. Potassium hydroxide (61 mg) and iodomethane (20 uL) were added to the reaction mixture and this was stirred overnight.
- Example 85.2 To a 5 mL microwave vial was added B2 (10.00 mg, 0.033 mmol), 1- methyl-7-propoxy-benzimidazol-2-amine (13.8 mg, 0.067 mmol), PyBrop (37.5 mg, 0.080 mmol) followed by anhydrous DMF (0.2 mL) and DIPEA (21 uL, 0.12 mmol). The resulting solution was stirred at RT for 2 d. The reaction was diluted with DMSO (0.3 mL) and purified by reverse phase column chromatography (Eluent: 20-100% MeOH in water (+0.1% formic acid modifier in both)).
- Example 86.1 1-lodopropane (0.35 mL, 3.57 mmol) was added to a mixture of 2- amino-3-nitrophenol (500.0 mg, 3.24 mmol) and K2CO3 (672.6 mg, 4.87 mmol) in anhydrous DMF (32.4 mL). Reaction stirred for 22 h 30 min at RT. The crude was partioned between EtOAc (50 mL) and aqueous saturated solution of sodium bicarbonate (50 mL). After phase separation, the aqueous layer was further extracted with EtOAc (50 mL).
- Example 86.2 2-Nitro-6-propoxy-aniline (280.0 mg, 1.43 mmol) and tin(ll) chloride (1367.4 mg, 7.14 mmol) were taken into EtOH (14.0 mL) in a 20 mL microwave vial. The reaction mixture was heated for 10 min at 140 °C in the microwave. Extra of tin(ll) chloride (317 mg) was added and after re-sealing the microwave vial, the reaction mixture was further heated for 5 min at 135 °C. After cooling down, the reaction mixture was poured into an aqueous saturated solution of sodium bicarbonate (75 mL). The bicarbonate phase was extracted with EtOAc (2x 60 mL).
- Example 86.3 Bromine cyanide 5M in MeCN (0.19 mL, 0.97 mmol) was added to a mixture of 3-propoxybenzene-1 ,2-diamine (135.0 mg, 0.81 mmol) in MeCN (3.9 mL) and water (0.99 mL). The reaction mixture was stirred at RT overnight. The volatiles were evaporated to dryness and the obtained crude was purified by normal phase silica column chromatography (eluent: 2-15% gradient of MeOH in DCM) to afford the 7-propoxy-1H-benzimidazol-2-amine (65 mg, 42%, 0,34 mmol). HPLC/MS m/z: 192.1 [M+H] + , Rt (Y): 0.99 min.
- Example 86.4 B2 (31.2 mg, 0.1 mmol) and 7-propoxy-1 H-benzimidazol-2-amine (20 mg, 0.1 mmol) were dissolved in anhydrous DMF (0.52 mL), followed by the addition of triethylamine (59 uL, 0.42 mmol). The reaction mixture was placed in a preheated heating block at 70 °C, 50% T3P in DMF (0.18 mL, 0.31 mmol) was added dropwise over 5 min. The reaction mixture was left stirring at 70 °C for 18 h. More T3P (92 uL) and TEA (28 uL) were added to the reaction mixture, and this was stirred further at 70 °C for 18 h.
- reaction was quenched with water (0.3 mL) and stirred for 5 min before diluting with DMSO (0.4 mL) and purified by reverse phase column chromatography (Eluent: 20-100% MeOH in water (+0.1% formic acid in both)).
- Example 87 8-(2-((7-(5-methyl-1,2,4-oxadiazol-3-yl)isoquinolin-1- yl)amino)ethyl-1,1,2,2-d4)-1-(2,2,2-trifluoroethoxy-1,1-d2)-7,8- dihydropyrido[3 , ,4 , :4,5]pyrrolo[1,2-a]pyrazin-9(6H)-one-6,6,7,7-d4
- Example 87.1 A solution of NaOH (1.04 g, 26.0 mmol, 1.0 eq.) in H2O (21.1 mL) was added to a solution of glycine-2,2-d2 (2.0 g, 26.0 mmol, 1.0 eq.) in Water (21.1 mL). Then, a solution of BOC2O (7.04 g, 31.3 mmol, 1.2 eq.) in Dioxane (44.3 mL) was added dropwise and the resulting reaction mixture was stirred at r.t. overnight. After evaporation, the residue was re-dissolved in water and extracted with diethyl.
- Example 87.2 1 ,1'-Carbonyldiimidazole (5.37 g, 33.1 mmol, 1.3 eq.) was added to a solution of 2-(tert-butoxycarbonylamino)-2,2-dideuterio-acetic acid (4.42 g, 24.9 mmol, 1 eq.) in THF (83.1 mL, 0.2 M) at RT. After 15 min, the solution was cooled to 0°C and a solution of NaBD4 (1.06 g, 25.4 mmol) in H2O (41.6 mL, 0.2 M) was added. After 1 h, HCI (1M) was added and the mixture was extracted with EtOAc.
- Example 87.3 A suspension of Methyl 4-chloro-1 H-pyrrolo[3,2-c]pyridine-2- carboxylate (1.21 g, 5.73 mmol, 1 eq.), 2,2,2-Trifluoroethanol-1 ,1-d2 (683 uL, 9.18 mmol, 1.6 eq.), 4 Molecular sieves (3.49 g), tBuBrettPhos Pd G3 (215 mg, 0.252 mmol, 4 mol%), tBuBrettPhos (125 mg, 0.250 mmol, 4 m l%) and CS2CO3 (3.49 g, 10.7 mmol, 1.9 eq.) in a mixture of toluene (8.19 mL, 0.35 M) and THF (8.19 mL, 0.35 M) was heated at 80 °C for 24 h in a sealed vial under argon atmosphere.
- Example 87.4 DIAD (2.3 mL, 11 .6 mmol, 3.2 eq.) was added over 50 min to a solution of tert-butyl N-(1 ,1 ,2,2-tetradeuterio-2-hydroxy-ethyl)carbamate (2.16 g, 13.1 mmol, 3.6 eq.), PPha (2.86 g, 10.9 mmol, 3.0 eq.) and methyl 4-(1 ,1-dideuterio-
- Example 87.5 A solution of TsCI (2.08 g, 10.9 mmol, 2 eq.) in DCM (18.2 mL) was slowly added to a solution of tert-butyl N-(1 ,1 ,2,2-tetradeuterio-2-hydroxy- ethyl)carbamate (0.90 g, 5.45 mmol, 1 eq.), DMAP (50.6 mg, 0.414 mmol, 8 mol%) and Et 3 N (2.3 mL, 16.3 mmol, 3 eq.) in DCM (36.3 mL, 0.1 M) at 0°C. The resulting solution was stirred at 0°C for 30 min and then at RT for 3h.
- Example 87.6 A solution of [2-(tert-butoxycarbonylamino)- 1 ,1 ,2, 2-tetradeuterio- ethyl] 4-methylbenzene sulfonate (1.35 g, 4.22 mmol, 1.2 eq.) in DMSO (8.7 mL) was added to 12,12,13,13-tetradeuterio-6-(1 ,1-dideuterio-2,2,2-trifluoro-ethoxy)- 1 ,5, 11 -triazatricyclo [7.4.0.02,7]trideca-2(7),3,5,8-tetraen-10-one (0.99 g, 3.39 mmol, 1 eq.), KOH (0.40 g, 7.11 mmol, 2.1 eq.), K2CO3 (1.40 g, 10.2 mmol, 3 eq.), and TBAB (0.44 g, 1.35 mmol, 0.4 eq.) in DMSO (17.37
- Example 87.7 DI PEA (96.52 uL, 0.5542 mmol) was added dropwise to a suspension of 11 -(2-amino-1 , 1 ,2,2-tetradeuterio-ethyl)-12, 12, 13, 13-tetradeuterio-6- (1 ,1-dideuterio-2,2,2-trifluoro-ethoxy)-1 ,5,11-triazatricyclo[7.4.0.02,7]trideca- 2(7),3,5,8-tetraen-10-one (50.00 mg, 0.1478 mmol), A1 (40.29 mg, 0.1773 mmol) and PyBroP (82.67 mg, 0.1773 mmol) in DOM (1.48 mL, 0.1 M).
- Example 88 3-(2-methyl-2H-tetrazol-5-yl)-N-(2-(9-oxo-1-(2,2,2-trifluoroethoxy- 1, 1-d2)-6, 7-dihydropyrido[3',4':4,5]pyrrolo[1,2-a]pyrazin-8(9H)-yl-6, 6,7,7- d4)ethyl-1,1 ,2,2-d4)benzamide
- DIPEA 164.73 uL, 0.9458 mmol
- DIPEA 164.73 uL, 0.9458 mmol
- Example 87.6 (80.00 mg, 0.2364 mmol), 3-(2-Methyl-2H-tetrazol-5-yl)-benzoic acid (55.52 mg, 0.2719 mmol) and HATU (179.80 mg, 0.4729 mmol) in DCM (1.97 mL, 0.12 M) .
- Example 89 Compound structures an IUPAC names
- Example 90 Testing compounds of the present invention for inhibitory activities against HSET - HSET ADP-Glo Assay S - 3 pM ATP
- - Buffer is 20 mM Hepes pH 7.5, 200 mM NaCI, 2 mM TCEP, 5% glycerol (5 pL aliquots at 10.2 pM)
- Buffer Stocks (filtered and stored at rt for up to 1 month)
- ECHO Protocol Create an ECHO intermediate plate by adding 24.5 pL DMSO to columns 1 & 2, and 40 pL DMSO to columns 23 & 24 of an ECHO 384PP plate
- Assay buffer (Keep on ice): HEPES pH 6.8, MgCI 2 , EGTA, Triton X-100, DTT, HPLC H 2 O
- PM buffer PIPES pH 7.0, MgCI 2 , HPLC H 2 O, Paclitaxel, mix well, store at rt
- BLANK solution is 357 pL 2XAB + 143 pL microtubule working solution 3.2.
- ATP Working solution is 1 pL 10 mM UltraPure ATP (Promega kit) + 999 pL ddH 2 O gives 10 pM ATP for a 3 pM final assay concentration, stored on ice.
- KDR Kinase Detection Reagent
- a solution of 100 g of a compound of the present invention and 5 g of disodium hydrogenphosphate in 3 L of bidistilled water is adjusted to pH 6.5 using 2 N hydrochloric acid, filtered under sterile conditions, transferred into injection vials, lyophilised under sterile conditions and sealed under sterile conditions.
- Each injection vial contains 5 mg of a compound of the present invention.
- a solution is prepared from 1 g of a compound of the present invention, 9.38 g of NaH2PC>4. 2 H2O, 28.48 g of Na2HPO4. 12 H2O and 0.1 g of benzalkonium chloride in 940 mL of bidistilled water. The pH is adjusted to 6.8, and the solution is made up to 1 L and sterilised by irradiation.
- a solution of 1 kg of a compound of the present invention in 60 L of bidistilled water is filtered under sterile conditions, transferred into ampoules, lyophilised under sterile conditions and sealed under sterile conditions. Each ampoule contains 10 mg of a compound of the present invention.
- Centrosome amplification can initiate tumorigenesis in flies. Cell 133, 1032-1042.
- ChEMBL a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 40(Database issue), D1100-D1107.
- Chromosome movement in mitosis requires microtubule anchorage at spindle poles. J. Cell Biol. 152, 425-434.
- RNA interference machinery regulates chromosome dynamics during mitosis and meiosis in fission yeast. Proc. Natl. Acad. Sci. USA 100, 193-198.
- KIFC1 is a novel potential therapeutic target for breast cancer. Cancer Biology and Therapy 16(9), pp. 1316-1322
- HSET kinesin-related protein
- Aryl acid adenylating enzymes involved in siderophore biosynthesis fluorescence polarization assay, ligand specificity, and discovery of non-nucleoside inhibitors via high- throughput screening. Biochemistry 47, 11735-11749.
- XCTK2 A kinesin-related protein that promotes mitotic spindle assembly in Xenopus laevis egg extracts. Journal of Cell Biology 136(4), pp. 859-870
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description

















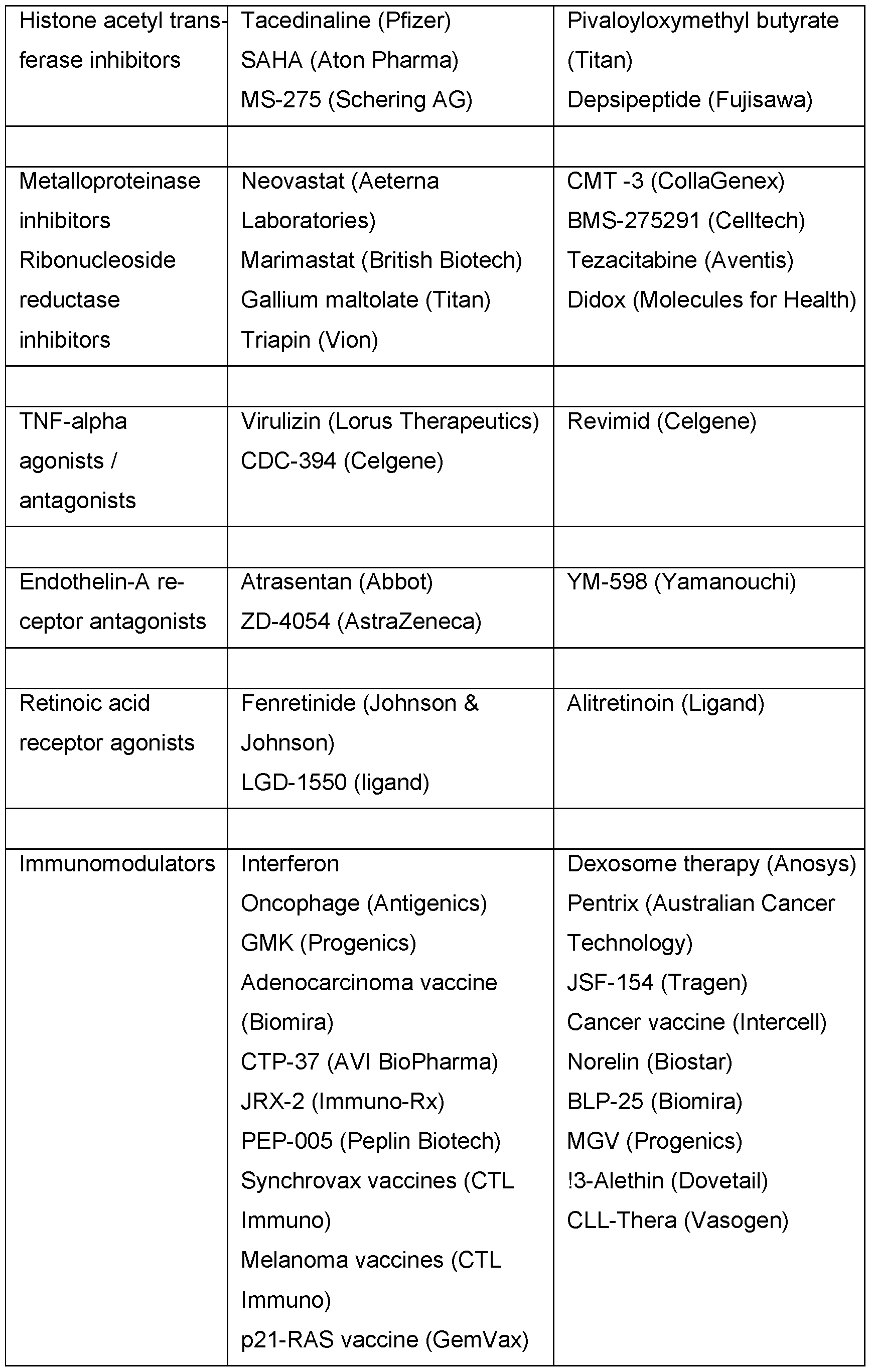
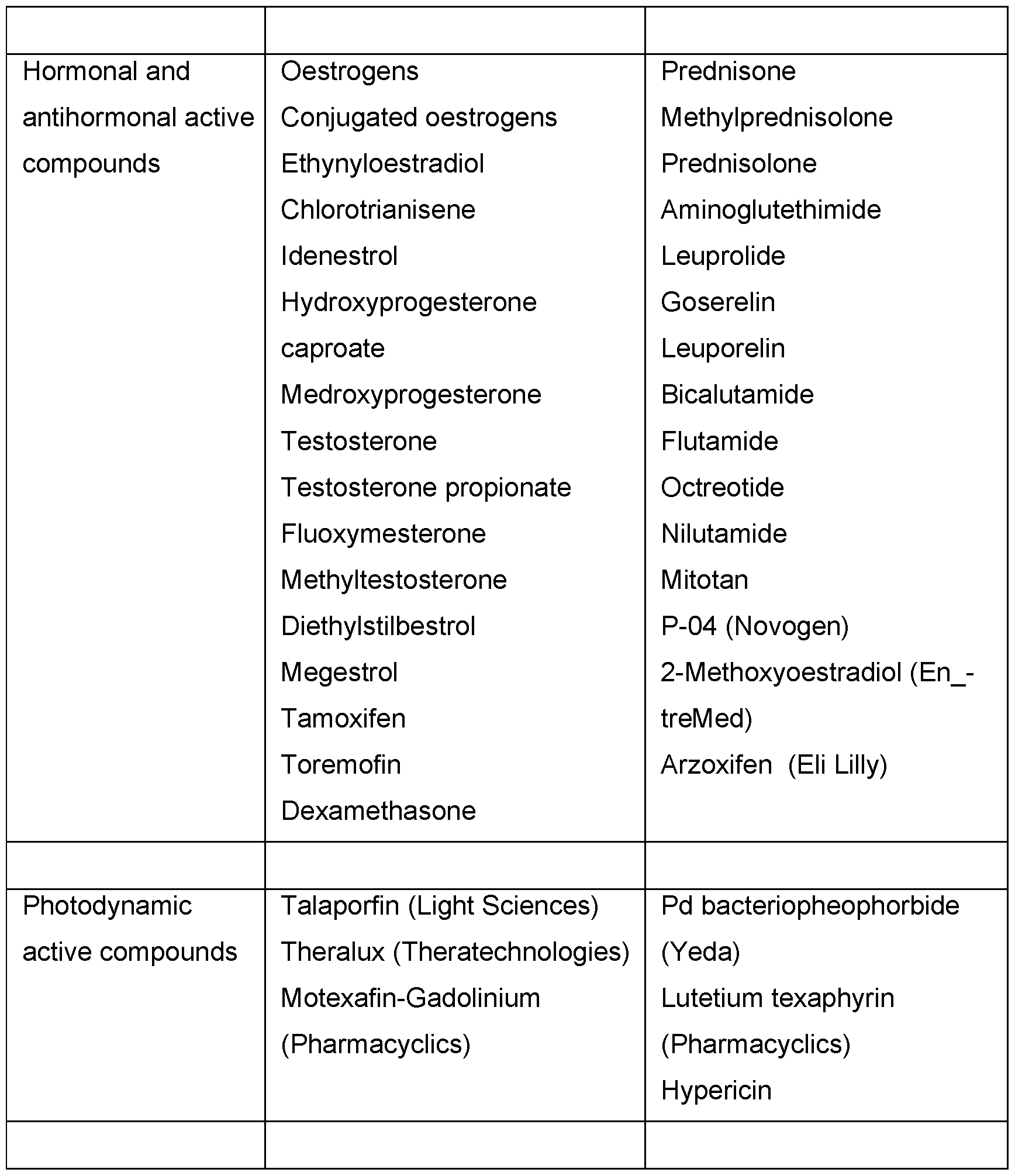



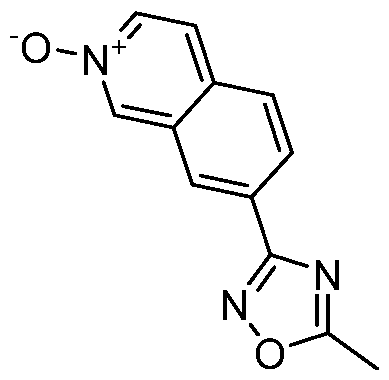

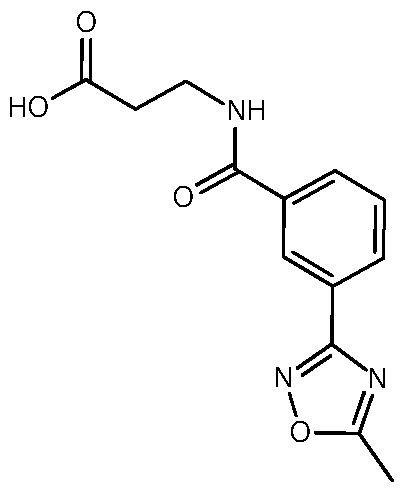

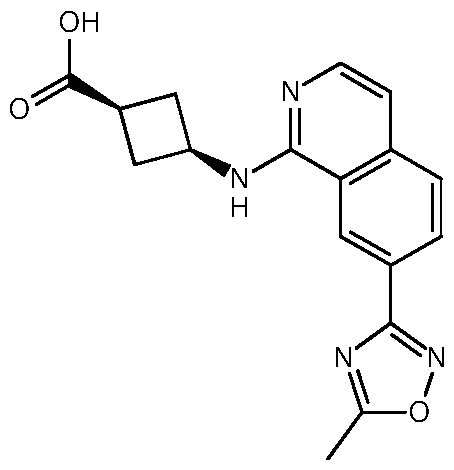

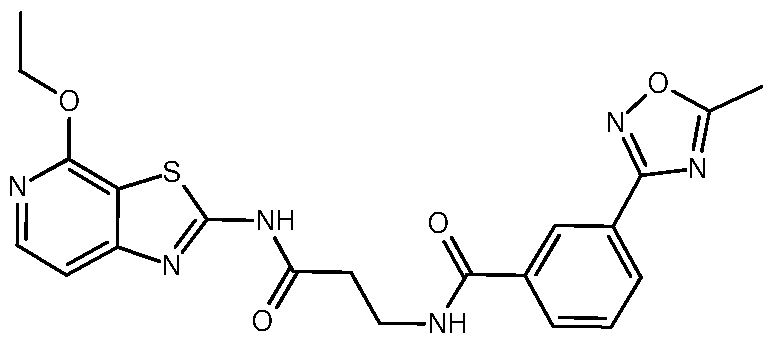




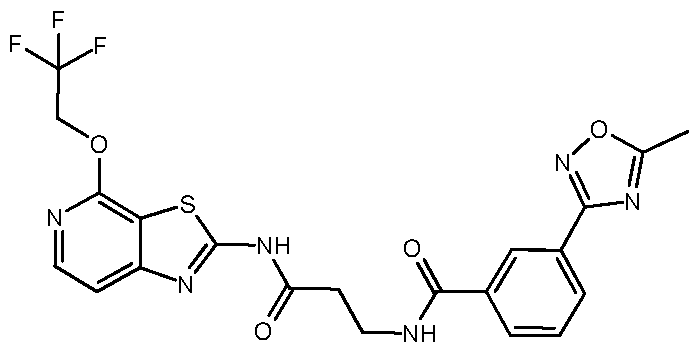

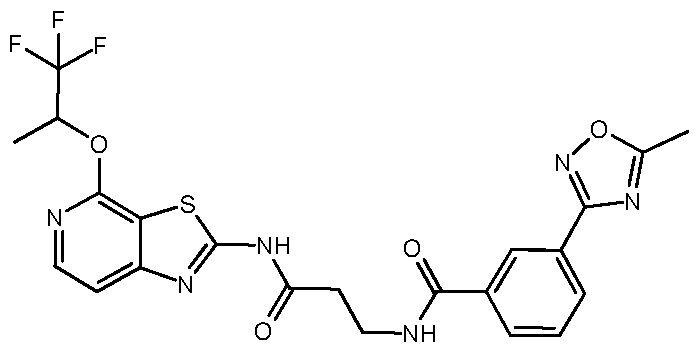
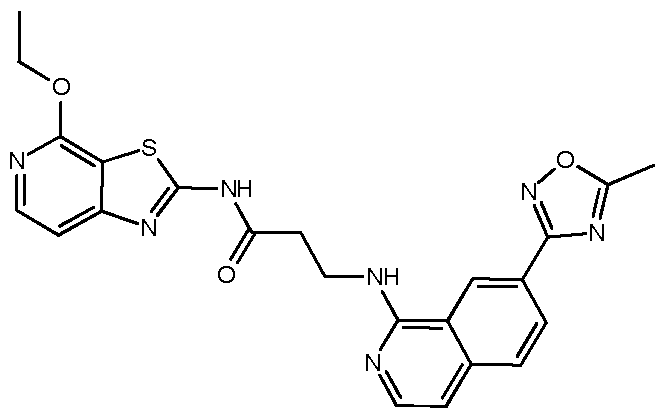






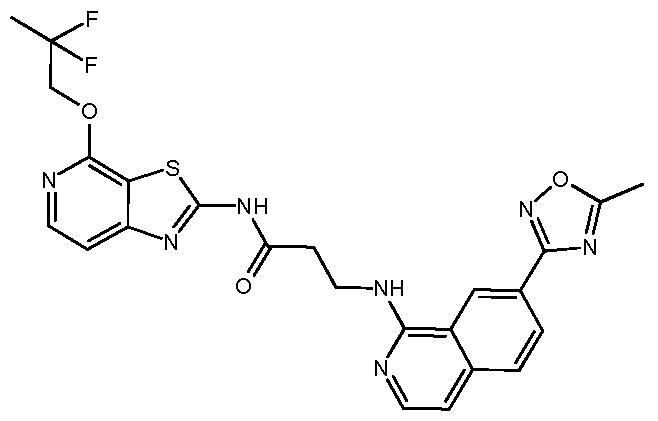
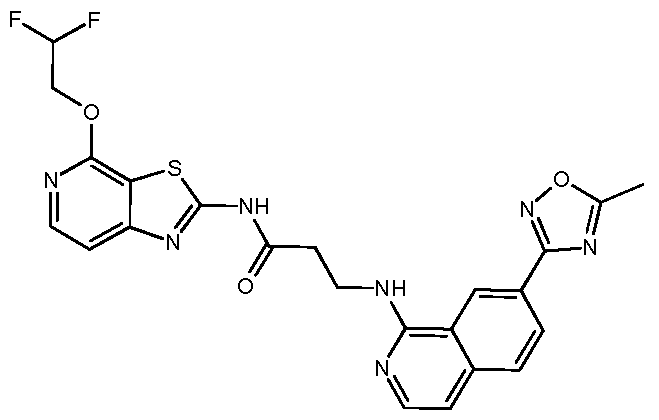


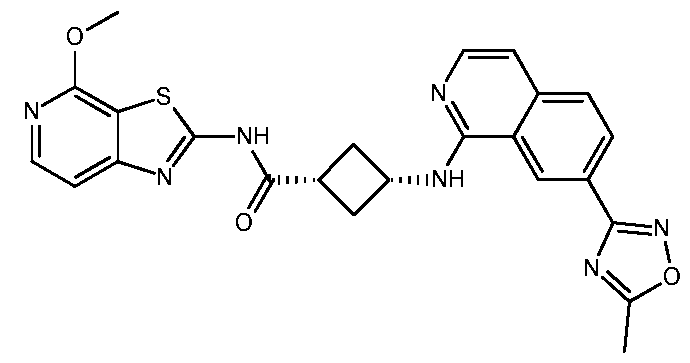




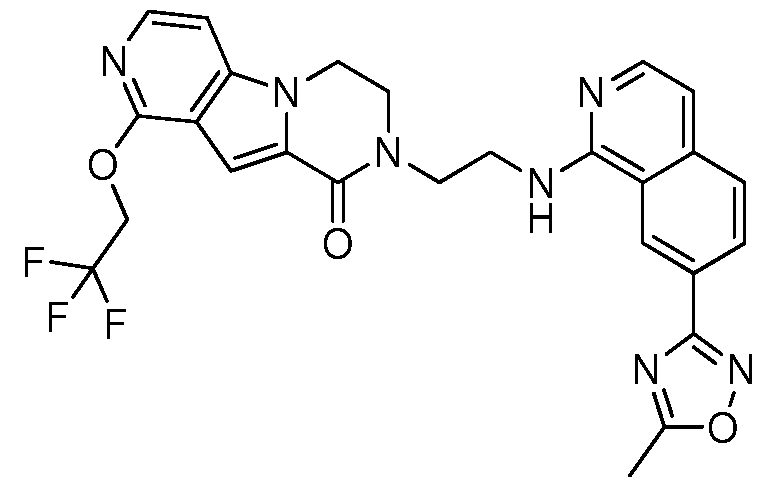





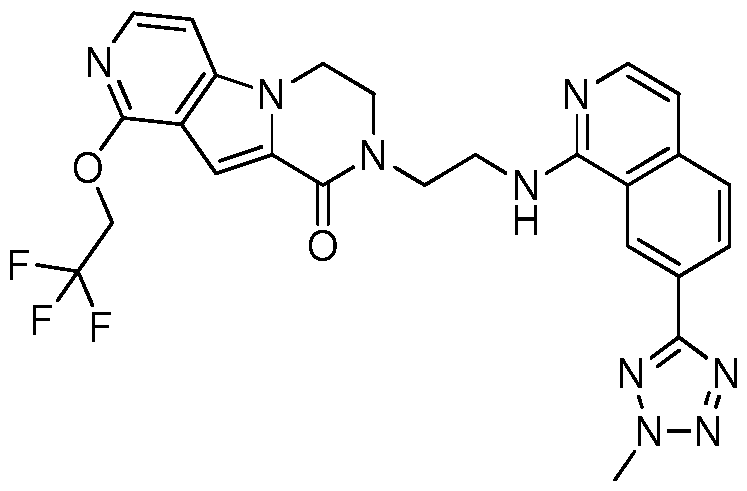
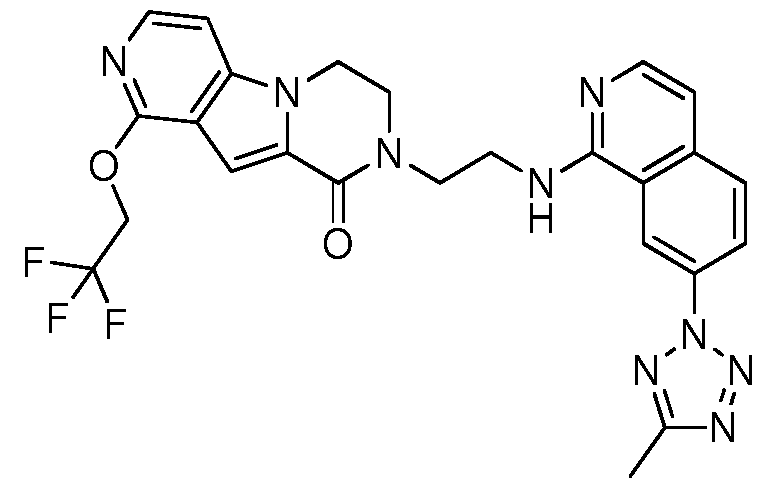






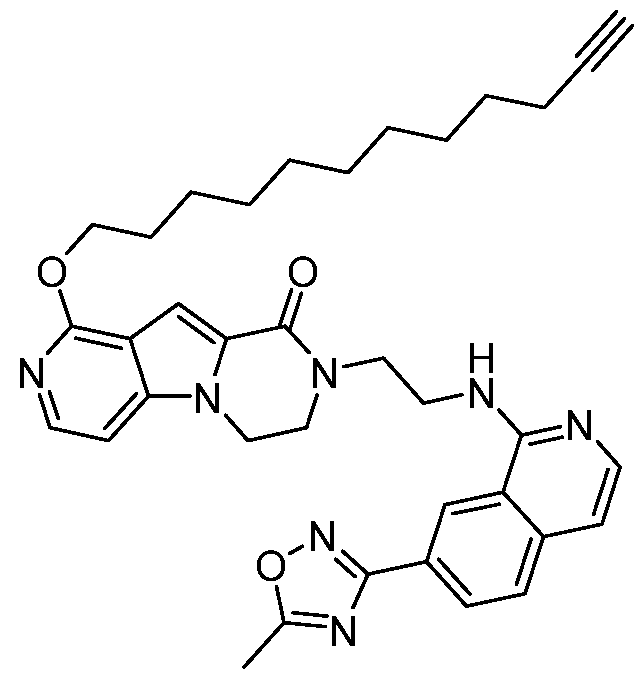





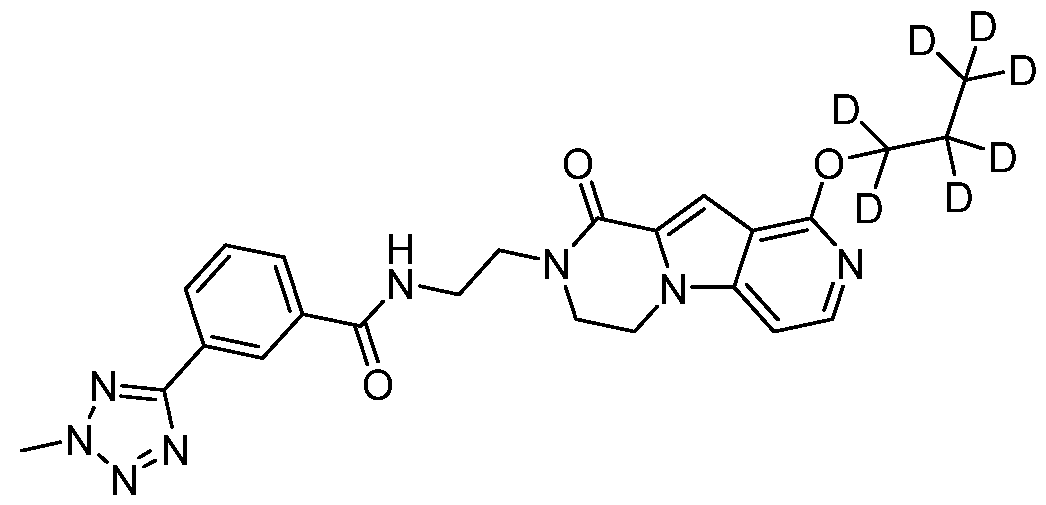




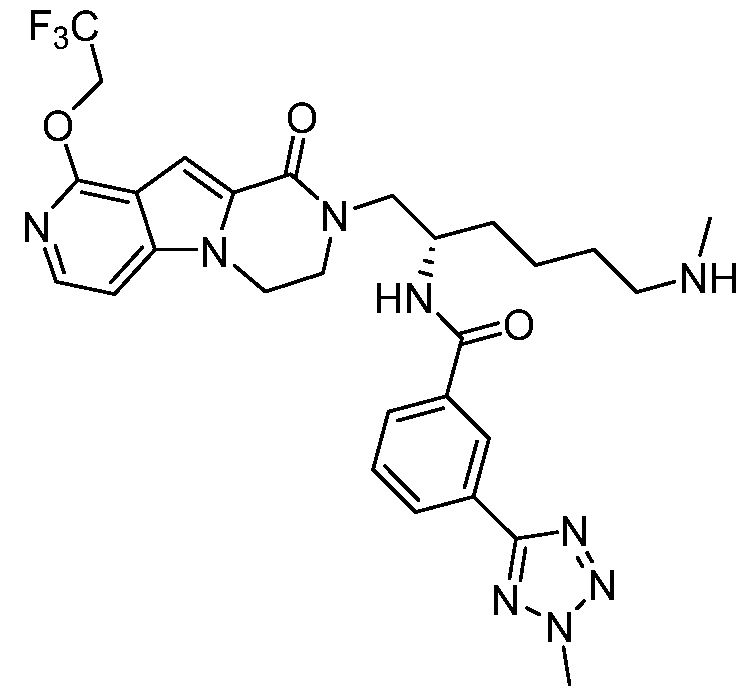

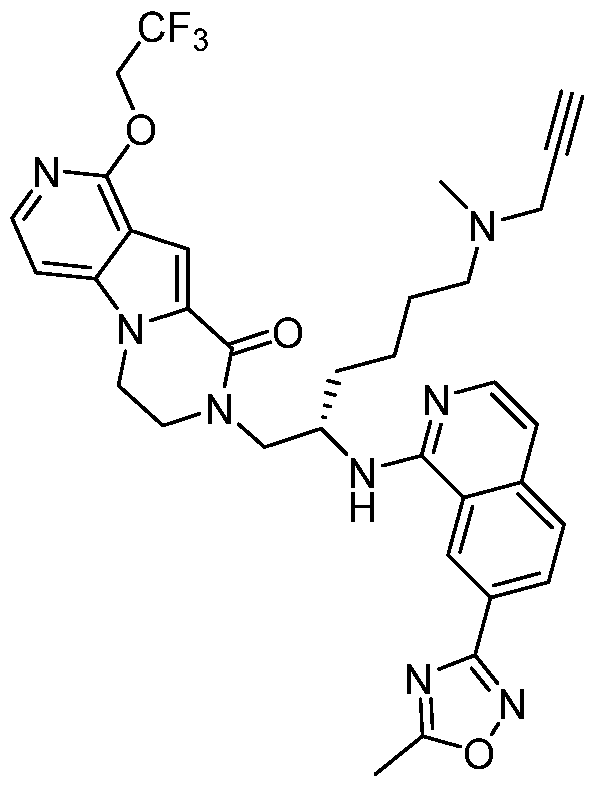




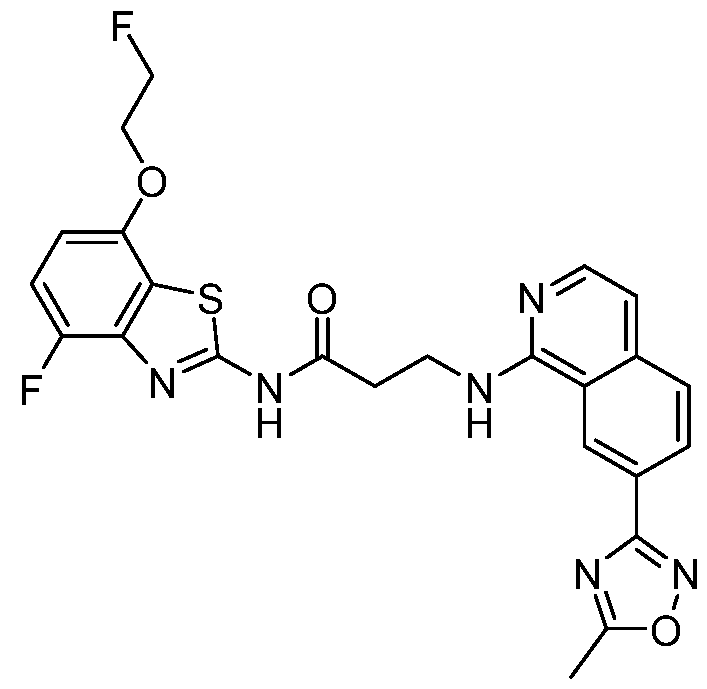



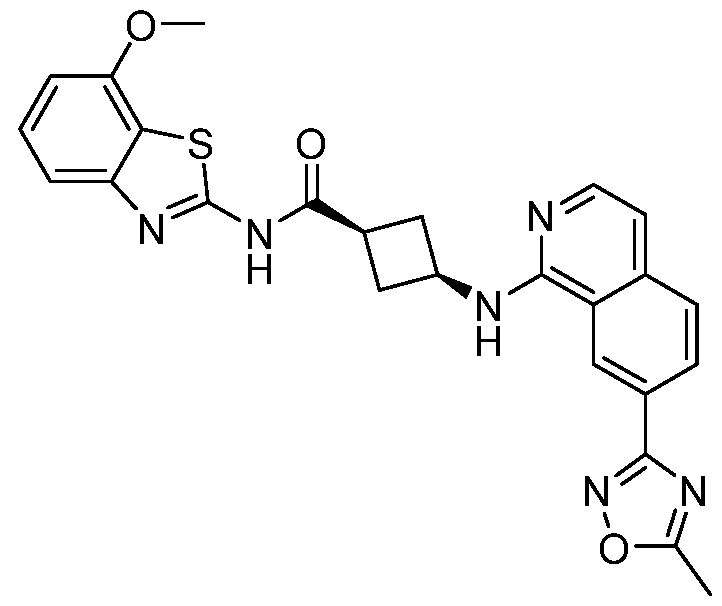

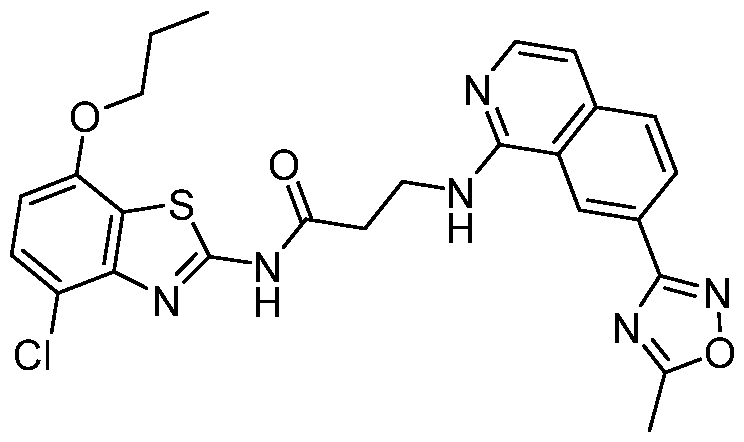


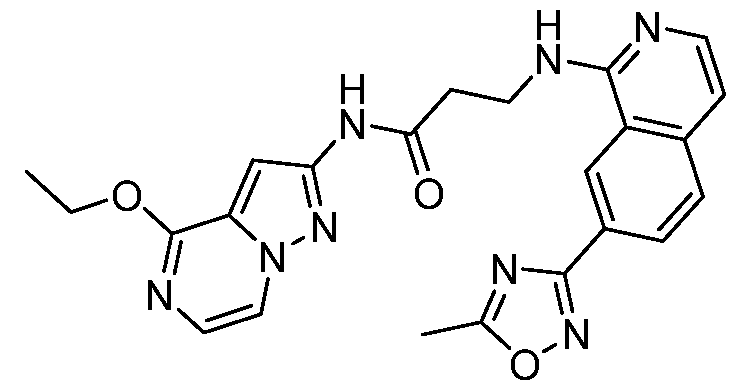











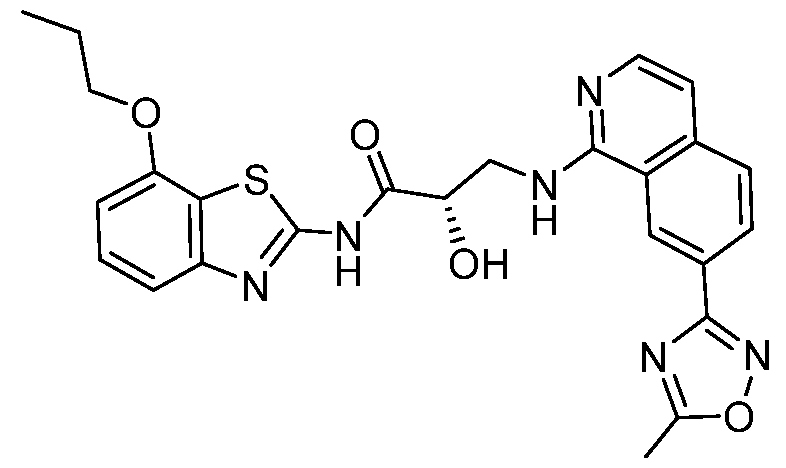


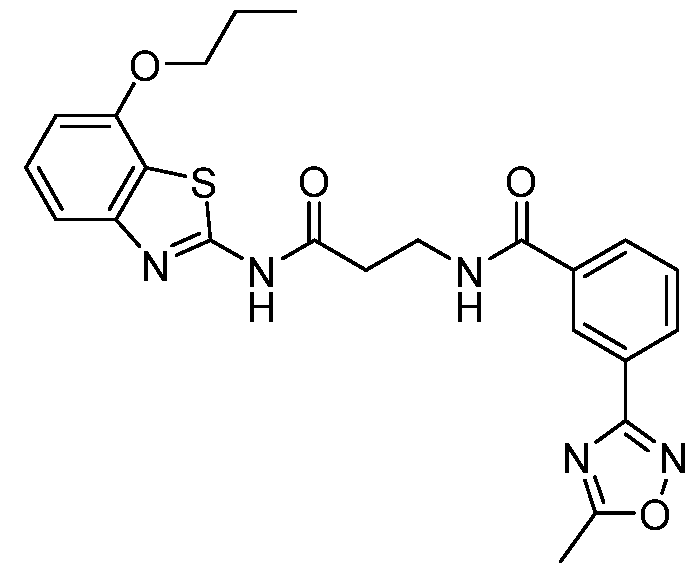


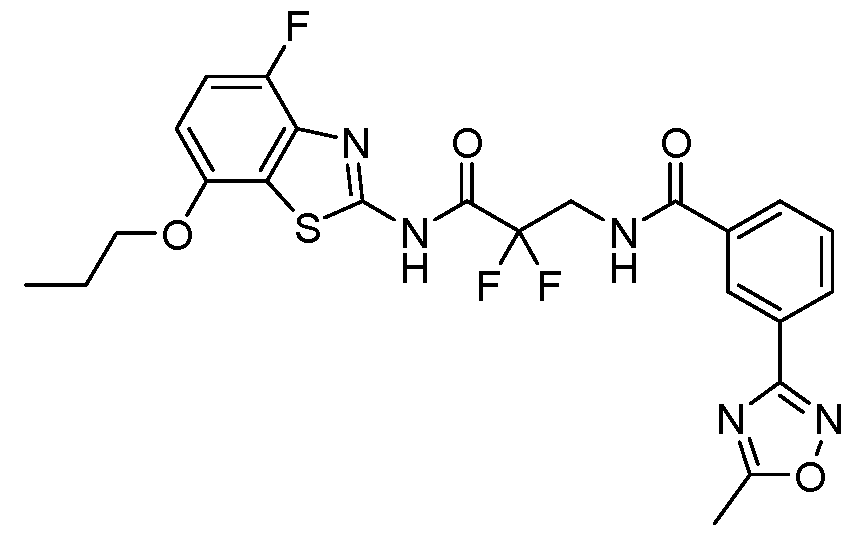









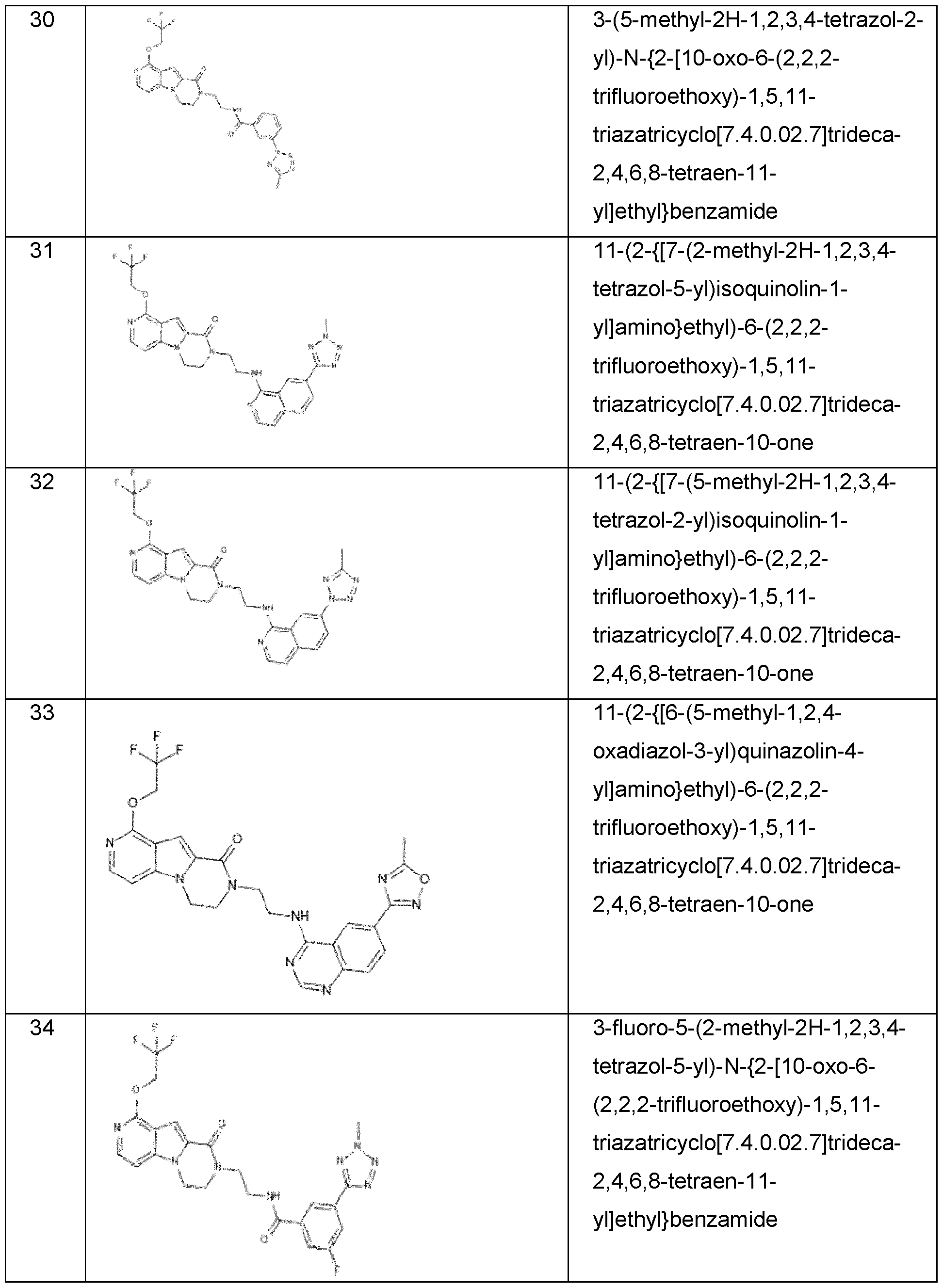

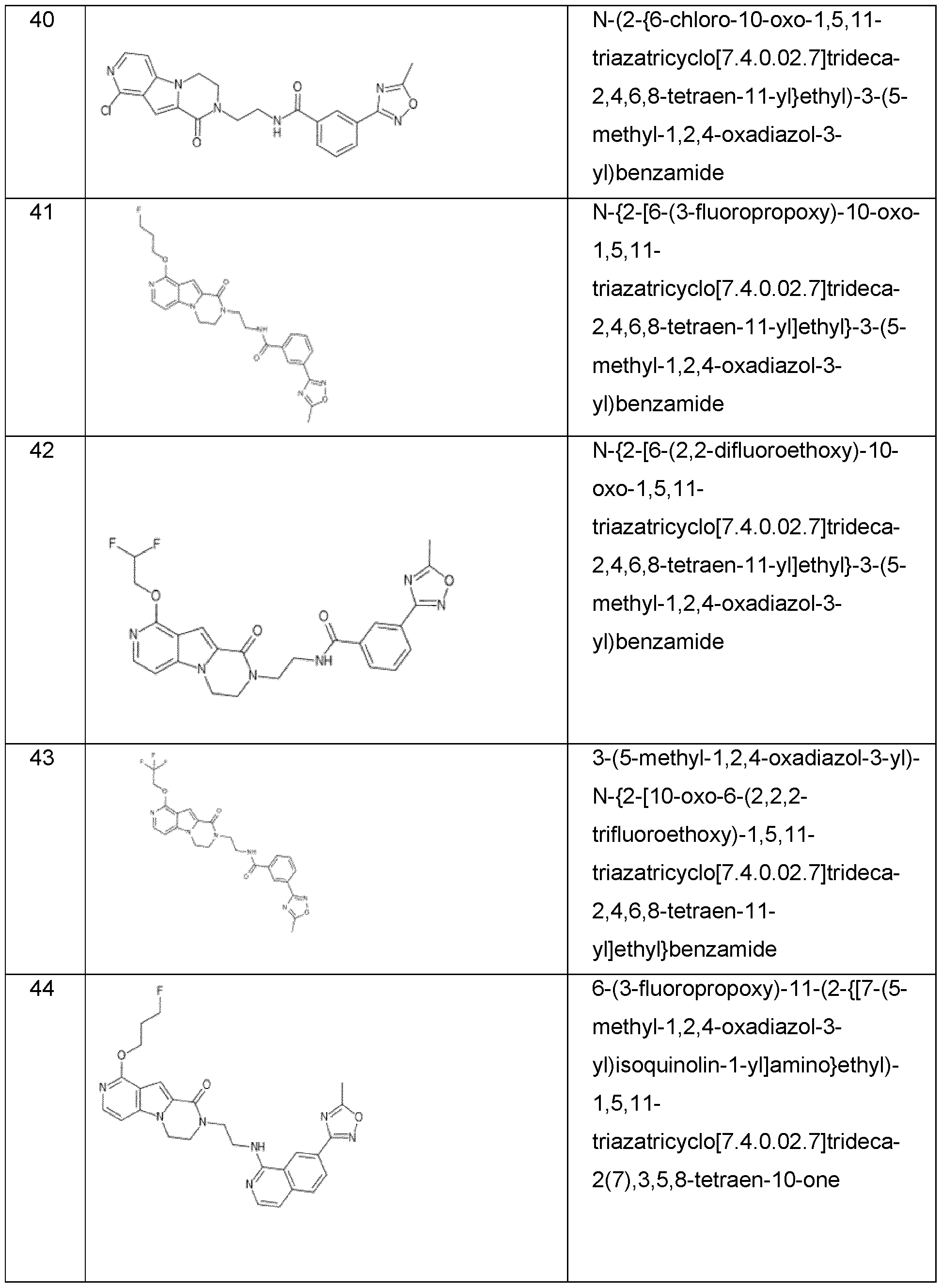











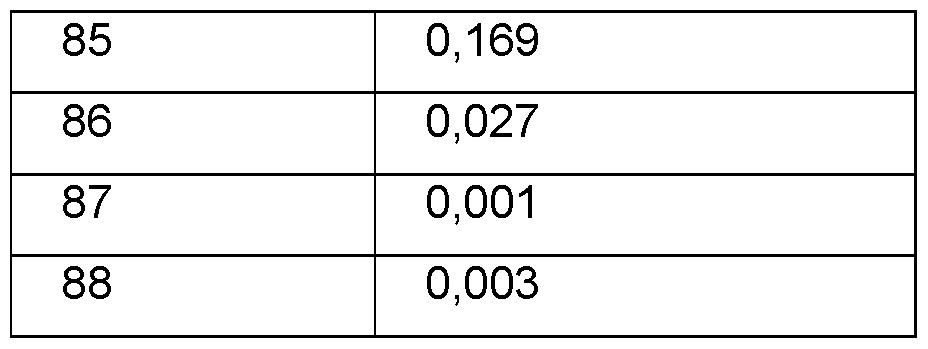
Claims

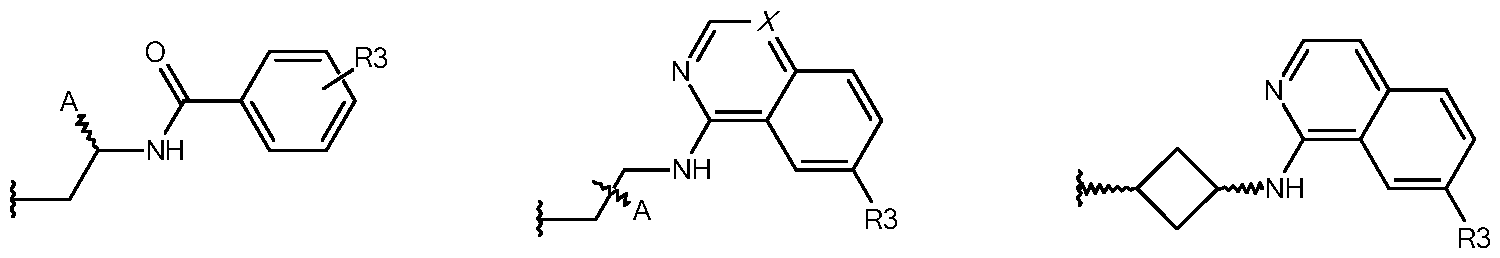










Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2023375996A AU2023375996A1 (en) | 2022-11-07 | 2023-11-03 | Substituted bi-and tricyclic hset inhibitors |
| EP23800845.2A EP4615838A1 (en) | 2022-11-07 | 2023-11-03 | Substituted bi-and tricyclic hset inhibitors |
| JP2025526291A JP2025542089A (en) | 2022-11-07 | 2023-11-03 | Substituted Bicyclic and Tricyclic HSET Inhibitors |
| CN202380077319.1A CN120265629A (en) | 2022-11-07 | 2023-11-03 | Substituted bicyclic and tricyclic HSET inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP22205800 | 2022-11-07 | ||
| EP22205800.0 | 2022-11-07 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024099898A1 true WO2024099898A1 (en) | 2024-05-16 |
Family
ID=84363857
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2023/080663 Ceased WO2024099898A1 (en) | 2022-11-07 | 2023-11-03 | Substituted bi-and tricyclic hset inhibitors |
Country Status (7)
| Country | Link |
|---|---|
| EP (1) | EP4615838A1 (en) |
| JP (1) | JP2025542089A (en) |
| CN (1) | CN120265629A (en) |
| AR (1) | AR130957A1 (en) |
| AU (1) | AU2023375996A1 (en) |
| TW (1) | TW202434603A (en) |
| WO (1) | WO2024099898A1 (en) |
Citations (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4125828A (en) | 1972-08-04 | 1978-11-14 | Med-El Inc. | Method and apparatus for automated classification and analysis of cells |
| US4207554A (en) | 1972-08-04 | 1980-06-10 | Med-El Inc. | Method and apparatus for automated classification and analysis of cells |
| WO1997022596A1 (en) | 1995-12-18 | 1997-06-26 | Zeneca Limited | Quinazoline derivatives |
| WO1997030035A1 (en) | 1996-02-13 | 1997-08-21 | Zeneca Limited | Quinazoline derivatives as vegf inhibitors |
| WO1997032856A1 (en) | 1996-03-05 | 1997-09-12 | Zeneca Limited | 4-anilinoquinazoline derivatives |
| WO1998013354A1 (en) | 1996-09-25 | 1998-04-02 | Zeneca Limited | Quinazoline derivatives and pharmaceutical compositions containing them |
| WO1999002166A1 (en) | 1997-07-08 | 1999-01-21 | Angiogene Pharmaceuticals Ltd. | Use of colchinol derivatives as vascular damaging agents |
| WO2000040529A1 (en) | 1999-01-07 | 2000-07-13 | Angiogene Pharmaceuticals Ltd. | Colchinol derivatives as vascular damaging agents |
| WO2000041669A2 (en) | 1999-01-15 | 2000-07-20 | Angiogene Pharmaceuticals Ltd. | Benzimidazole vascular damaging agents |
| WO2001092224A1 (en) | 2000-05-31 | 2001-12-06 | Astrazeneca Ab | Indole derivatives with vascular damaging activity |
| WO2002004434A1 (en) | 2000-07-07 | 2002-01-17 | Angiogene Pharmaceuticals Limited | Colchinol derivatives as vascular damaging agents |
| WO2002008213A1 (en) | 2000-07-07 | 2002-01-31 | Angiogene Pharmaceuticals Limited | Colchinol derivatives as angiogenesis inhibitors |
| WO2005070930A2 (en) * | 2004-01-23 | 2005-08-04 | Chiron Corporation | Tetrahydrocarboline compounds as anticancer agents |
| US20060084687A1 (en) * | 2004-10-19 | 2006-04-20 | Boyce Rustum S | Indole and benzimidazole derivatives |
| WO2006060737A2 (en) * | 2004-12-03 | 2006-06-08 | Takeda San Diego, Inc. | Mitotic kinesin inhibitors |
| US20070135435A1 (en) * | 2005-11-02 | 2007-06-14 | Xiangping Qian | Certain chemical entities, compositions, and methods |
| WO2009155025A1 (en) | 2008-05-30 | 2009-12-23 | Dana-Farber Cancer Institute Inc. | Methods of treating a meiotic kinesin-associated disease |
| WO2015085088A1 (en) | 2013-12-05 | 2015-06-11 | Novazoi Theranostics, Inc. | Compositions and methods for prognosis and treatment of cancer |
-
2023
- 2023-11-03 AR ARP230102952A patent/AR130957A1/en unknown
- 2023-11-03 WO PCT/EP2023/080663 patent/WO2024099898A1/en not_active Ceased
- 2023-11-03 CN CN202380077319.1A patent/CN120265629A/en active Pending
- 2023-11-03 EP EP23800845.2A patent/EP4615838A1/en active Pending
- 2023-11-03 JP JP2025526291A patent/JP2025542089A/en active Pending
- 2023-11-03 AU AU2023375996A patent/AU2023375996A1/en active Pending
- 2023-11-06 TW TW112142700A patent/TW202434603A/en unknown
Patent Citations (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4125828A (en) | 1972-08-04 | 1978-11-14 | Med-El Inc. | Method and apparatus for automated classification and analysis of cells |
| US4207554A (en) | 1972-08-04 | 1980-06-10 | Med-El Inc. | Method and apparatus for automated classification and analysis of cells |
| WO1997022596A1 (en) | 1995-12-18 | 1997-06-26 | Zeneca Limited | Quinazoline derivatives |
| WO1997030035A1 (en) | 1996-02-13 | 1997-08-21 | Zeneca Limited | Quinazoline derivatives as vegf inhibitors |
| WO1997032856A1 (en) | 1996-03-05 | 1997-09-12 | Zeneca Limited | 4-anilinoquinazoline derivatives |
| WO1998013354A1 (en) | 1996-09-25 | 1998-04-02 | Zeneca Limited | Quinazoline derivatives and pharmaceutical compositions containing them |
| WO1999002166A1 (en) | 1997-07-08 | 1999-01-21 | Angiogene Pharmaceuticals Ltd. | Use of colchinol derivatives as vascular damaging agents |
| WO2000040529A1 (en) | 1999-01-07 | 2000-07-13 | Angiogene Pharmaceuticals Ltd. | Colchinol derivatives as vascular damaging agents |
| WO2000041669A2 (en) | 1999-01-15 | 2000-07-20 | Angiogene Pharmaceuticals Ltd. | Benzimidazole vascular damaging agents |
| WO2001092224A1 (en) | 2000-05-31 | 2001-12-06 | Astrazeneca Ab | Indole derivatives with vascular damaging activity |
| WO2002004434A1 (en) | 2000-07-07 | 2002-01-17 | Angiogene Pharmaceuticals Limited | Colchinol derivatives as vascular damaging agents |
| WO2002008213A1 (en) | 2000-07-07 | 2002-01-31 | Angiogene Pharmaceuticals Limited | Colchinol derivatives as angiogenesis inhibitors |
| WO2005070930A2 (en) * | 2004-01-23 | 2005-08-04 | Chiron Corporation | Tetrahydrocarboline compounds as anticancer agents |
| US20060084687A1 (en) * | 2004-10-19 | 2006-04-20 | Boyce Rustum S | Indole and benzimidazole derivatives |
| WO2006060737A2 (en) * | 2004-12-03 | 2006-06-08 | Takeda San Diego, Inc. | Mitotic kinesin inhibitors |
| US20070135435A1 (en) * | 2005-11-02 | 2007-06-14 | Xiangping Qian | Certain chemical entities, compositions, and methods |
| WO2009155025A1 (en) | 2008-05-30 | 2009-12-23 | Dana-Farber Cancer Institute Inc. | Methods of treating a meiotic kinesin-associated disease |
| WO2015085088A1 (en) | 2013-12-05 | 2015-06-11 | Novazoi Theranostics, Inc. | Compositions and methods for prognosis and treatment of cancer |
Non-Patent Citations (70)
| Title |
|---|
| BASTO, R.BRUNK, K.VINADOGROVA, T.PEEL, N.FRANZ, A.KHODJAKOV, A.RAFF, J.W.: "Centrosome amplification can initiate tumorigenesis in flies", CELL, vol. 133, 2008, pages 1032 - 1042, XP007909980, DOI: 10.1016/j.cell.2008.05.039 |
| BENDER, A.YOUNG, D.W.JENKINS, J.L.SERRANO, M.MIKHAILOV, D.CLEMONS, P.A.DAVIES, J.W.: "Chemogenomic data analysis: prediction of small-molecule targets and the advent of biological fingerprint", COMB. CHEM. HIGH THROUGHPUT SCREEN., vol. 10, 2007, pages 719 - 731 |
| BICKERTON, G.R.PAOLINI, G.V.BESNARD, J.MURESAN, S.HOPKINS, A.L.: "Quantifying the chemical beauty of drugs", NAT. CHEM., vol. 4, 2012, pages 90 - 98 |
| BOVERI, T: "Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris", J. CELL SCI., vol. 121, 2008, pages 1 - 84 |
| CAI, S.WEAVER, L.N.EMS-MCCLUNG, S.C.WALCZAK, C.E.: "Kinesin-14 family proteins HSET/XCTK2 control spindle length by cross-linking and sliding microtubules", MOLECULAR BIOLOGY OF THE CELL, vol. 20, no. 5, 2009, pages 1348 - 1359 |
| CAI, S.WEAVER, L.N.EMS-MCCLUNG, S.C.WALCZAK, C.E.: "Proper organization of microtubule minus ends is needed for midzone stability and cytokinesis", CURR. BIOL., vol. 20, 2010, pages 880 - 885, XP027425938, DOI: 10.1016/j.cub.2010.03.067 |
| CHAN, J.Y.: "A clinical overview of centrosome amplification in human cancers", INT. J. BIOL. SCI., vol. 7, 2011, pages 1122 - 1144, XP055310877, DOI: 10.7150/ijbs.7.1122 |
| DATABASE PubChem [online] 13 November 2007 (2007-11-13), PUBCHEM: "3-methoxy-N-[3-[(6-methoxy-1,3-benzothiazol-2-yl)amino]-3-oxopropyl]benzamide", XP093120898, retrieved from NCBI Database accession no. 17226185 * |
| DATABASE PubChem [online] 13 November 2007 (2007-11-13), PUBCHEM: "N-[3-(1H-benzimidazol-2-ylamino)-3-oxopropyl]-2-methoxybenzamide", XP093120796, retrieved from NCBI Database accession no. 17409968 * |
| DATABASE PubChem [online] 13 November 2007 (2007-11-13), PUBCHEM: "N-[3-[(6-ethoxy-1,3-benzothiazol-2-yl)amino]-3-oxopropyl]-3-methylbenzamide", XP093120897, retrieved from NCBI Database accession no. 17191524 * |
| DATABASE PubChem [online] 13 November 2007 (2007-11-13), PUBCHEM: "N-[3-[(6-methoxy-1,3-benzothiazol-2-yl)amino]-3-oxopropyl]-3-methylbenzamide", XP093120896, retrieved from NCBI Database accession no. 17191471 * |
| DATABASE PubChem [online] 13 November 2007 (2007-11-13), PUBCHEM: "N-[3-[(6-methoxy-1,3-benzothiazol-2-yl)amino]-3-oxopropyl]benzamide", XP093120888, retrieved from NCBI Database accession no. 17154528 * |
| DATABASE PubChem [online] 18 October 2012 (2012-10-18), PUBCHEM: "N-[3-(1H-benzimidazol-2-ylamino)-3-oxopropyl]-4-fluorobenzamide", XP093120808, retrieved from NCBI Database accession no. 60564482 * |
| DATABASE PubChem [online] 25 May 2018 (2018-05-25), PUBCHEM: "N-[4-oxo-4-[(1-propylbenzimidazol-2-yl)amino]butan-2-yl]benzamide", XP093120809, retrieved from NCBI Database accession no. 133894627 * |
| DATABASE PubChem [online] 28 May 2009 (2009-05-28), PUBCHEM: "N-[3-oxo-3-[(1-propylbenzimidazol-2-yl)amino]propyl]benzamide", XP093120806, retrieved from NCBI Database accession no. 26186777 * |
| DATABASE PubChem [online] 30 July 2006 (2006-07-30), PUBCHEM: "N-[3-(1H-benzimidazol-2-ylamino)-3-oxopropyl]-4-tert-butylbenzamide", XP093120803, retrieved from NCBI Database accession no. 8832915 * |
| DATABASE PubChem [online] 30 July 2006 (2006-07-30), PUBCHEM: "N-[3-[(6-ethoxy-1,3-benzothiazol-2-yl)amino]-3-oxopropyl]benzamide", XP093120887, retrieved from NCBI Database accession no. 8883008 * |
| DE, S.CIPRIANO, R.JACKSON, M.W.STARK, G.R.: "Overexpression of kinesins mediates docetaxel resistance in breast cancer cells", CANCER RESEARCH, vol. 69, no. 20, 2009, pages 8035 - 8042 |
| DROSOPOULOS K.TANG C.CHAO W. C.LINARDOPOULOS S.: "APC/C is an essential regulator of centrosome clustering", NAT COMMUN, vol. 5, 2014, pages 3686 |
| FOSTER, ADV. DRUG RES., vol. 14, 1985, pages 1 - 40 |
| GANEM, N.J.COMPTON, D.A.: "The Kinl kinesin Kif2a is required for bipolar spindle assembly through a functional relationship with MCAK.", J. CELL BIOL., vol. 166, 2004, pages 473 - 478 |
| GANEM, N.J.GODINHO, S.A.PELLMAN, D.: "A mechanism linking extra centrosomes to chromosomal instability", NATURE, vol. 460, 2009, pages 278 - 282 |
| GATLIN, J.C.BLOOM, K.: "Microtubule motors in eukaryotic spindle assembly and maintenance", SEMIN. CELL DEV. BIOL., vol. 21, 2010, pages 248 - 254, XP026978623 |
| GAULTON, A.BELLIS, L.J.BENTO, A.P.CHAMBERS, J.DAVIES, M.HERSEY, A.LIGHT, Y.MCGLINCHEY, S.MICHALOVICH, D.AL-LAZIKANI, B.: "ChEMBL: a large-scale bioactivity database for drug discovery", NUCLEIC ACIDS RES., vol. 40, 2012, pages D1100 - D1107, XP055082497, DOI: 10.1093/nar/gkr777 |
| GILLETTE ET AL., BIOCHEMISTRY, vol. 33, no. 10, 1994, pages 2927 - 2937 |
| GORDON, M.B.HOWARD, L.COMPTON, D.A.: "Chromosome movement in mitosis requires microtubule anchorage at spindle poles", J. CELL BIOL., vol. 152, 2001, pages 425 - 434 |
| GOSHIMA, G.NEDELEC, F.VALE, R.D.: "Mechanisms for focusing mitotic spindle poles by minus end-directed motor proteins", J. CELL BIOL., vol. 171, 2005, pages 229 - 240 |
| GRINBERG-RASHI, H.OFEK, E.PERELMAN, M.SKARDA, J.YARON, P.HAJDUCH, M.JACOB-HIRSCH, J.AMARIGLIO, N.KRUPSKY, M.SIMANSKY, D.A. ET AL.: "The expression of three genes in primary non-small cell lung cancer is associated with metastatic spread to the brain", CLIN. CANCER RES., vol. 15, 2009, pages 1755 - 1761, XP055301482, DOI: 10.1158/1078-0432.CCR-08-2124 |
| HALL, I.M.NOMA, K.GREWAL, S.I.: "RNA interference machinery regulates chromosome dynamics during mitosis and meiosis in fission yeast", PROC. NATL. ACAD. SCI. USA, vol. 100, 2003, pages 193 - 198, XP002405201, DOI: 10.1073/pnas.232688099 |
| HANZLIK ET AL., J. ORG. CHEM., vol. 55, 1990, pages 3992 - 3997 |
| HOLDGATE, G.A.WARD, W.H.J.: "Measurements of binding thermodynamics in drug discovery", DRUG DISCOV. TODAY, vol. 10, 2005, pages 1543 - 1550, XP005133826, DOI: 10.1016/S1359-6446(05)03610-X |
| HUSZAR, D.THEOCLITOU, M.E.SKOLNIK, J.HERBST, R.: "Kinesin motor proteins as targets for cancer therapy", CANCER METASTASIS REV., vol. 28, 2009, pages 197 - 208, XP019671837 |
| INT. J. PHARM., vol. 115, 1995, pages 61 - 67 |
| J. SHAFFE ET AL., J. PHARM. SCIENCES, vol. 88, 1999, pages 313 - 318 |
| JARMAN ET AL., CARCINOGENESIS, vol. 16, no. 4, 1993, pages 683 - 688 |
| JORDAN, M.A.WILSON, L.: "Microtubules as a target for anticancer drugs", NAT. REV. CANCER, vol. 4, 2004, pages 253 - 265, XP002525599, DOI: 10.1038/NR1317 |
| KLABUNDE, T.: "Chemogenomic approaches to drug discovery: similar receptors bind similar ligands", BR. J. PHARMACOL., vol. 152, 2007, pages 5 - 7, XP071136339, DOI: 10.1038/sj.bjp.0707308 |
| KLEYLEIN-SOHN, J.PDLLINGER, B.OHMER, M.HOFMANN, F.NIGG, E.A.HEMMINGS, B.A.WARTMANN, M.: "Acentrosomal spindle organization renders cancer cells dependent on the kinesin HSET", J. CELL SCI., vol. 125, 2012, pages 5391 - 5402 |
| KRALJEVIC PAVELIC, S.SEDIC, M.BOSNJAK, H.SPAVENTI, S.PAVELIC, K.: "Metastasis: new perspectives on an old problem", MOL. CANCER, vol. 10, 2011, pages 22, XP021088452, DOI: 10.1186/1476-4598-10-22 |
| KRAMER, A.MAIER, B.BARTEK, J.: "Centrosome clustering and chromosomal (in)stability: a matter of life and death", MOL. ONCOL., vol. 5, 2011, pages 324 - 335, XP028278421, DOI: 10.1016/j.molonc.2011.05.003 |
| KWON, M.GODINHO, S.A.CHANDHOK, N.S.GANEM, N.J.AZIOUNE, A.THERY, M.PELLMAN, D.: "Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes", GENES DEV, vol. 22, 2008, pages 2189 - 2203, XP007909963, DOI: 10.1101/gad.1700908 |
| LI, Y.LU, W.CHEN, D.BOOHAKER, R. J.ZHAI, L.PADMALAYAM, I.WENNERBERG, K.XU, B.ZHANG, W.: "KIFC1 is a novel potential therapeutic target for breast cancer", CANCER BIOLOGY AND THERAPY, vol. 16, no. 9, 2015, pages 1316 - 1322 |
| LIPINSKI, C.A.LOMBARDO, F.DOMINY, B.W.FEENEY, P.J.: "Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings", ADV. DRUG DELIV. REV., vol. 46, 2001, pages 3 - 26, XP001097746, DOI: 10.1016/S0169-409X(00)00129-0 |
| LUO, L.CARSON, J.D.DHANAK, D.JACKSON, J.R.HUANG, P.S.LEE, Y.SAKOWICZ, R.COPELAND, R.A.: "Mechanism of inhibition of human KSP by monastrol: insights from kinetic analysis and the effect of ionic strength on KSP inhibition", BIOCHEMISTRY, vol. 43, 2004, pages 15258 - 15266 |
| MAHONEY, N.M.GOSHIMA, G.DOUGLASS, A.D.VALE, R.D.: "Making microtubules and mitotic spindles in cells without functional centrosomes", CURR. BIOL., vol. 16, 2006, pages 564 - 569, XP025108175, DOI: 10.1016/j.cub.2006.01.053 |
| MARX, A.HOENGER, A.MANDELKOW, E.: "Structures of kinesin motor proteins", CELL MOTIL. CYTOSKELETON, vol. 66, 2009, pages 958 - 966 |
| MAYER, T.U.KAPOOR, T.M.HAGGARTY, S.J.KING, R.W.SCHREIBER, S.L.MITCHISON, T.J.: "Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen", SCIENCE, vol. 286, 1999, pages 971 - 974, XP002166153, DOI: 10.1126/science.286.5441.971 |
| MEUNIER, S.VERNOS, !.: "Microtubule assembly during mitosis - from distinct origins to distinct functions", J. CELL SCI., vol. 125, 2012, pages 2805 - 2814 |
| MOUNTAIN, V., SIMERLY, C., HOWARD, L., ANDO, A., SCHATTEN, G., COMPTON, D.A.: "The kinesin-related protein, HSET, opposes the activity of Eg5 and cross-links microtubules in the mammalian mitotic spindle", J. CELL BIOL., vol. 147, 1999, pages 351 - 366 |
| MYERS S.COLLINS I.: "Recent findings and future directions for interpolar mitotic kinesin inhibitors in cancer therapy", FUTURE MED CHEM., vol. 8, 2016, pages 463 - 89 |
| NERES, J.WILSON, D.J.CELIA, L.BECK, B.J.ALDRICH, C.C.: "Aryl acid adenylating enzymes involved in siderophore biosynthesis: fluorescence polarization assay, ligand specificity, and discovery of non-nucleoside inhibitors via high-throughput screening", BIOCHEMISTRY, vol. 47, 2008, pages 11735 - 11749 |
| NIESEN, F.H.BERGLUND, H.VEDADI, M.: "The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability", NAT. PROTOC., vol. 2, 2007, pages 2212 - 2221, XP055391241, DOI: 10.1038/nprot.2007.321 |
| NIGG, E.A.: "Centrosome aberrations: cause or consequence of cancer progression?", NAT. REV. CANCER, vol. 2, 2002, pages 815 - 825, XP002398058, DOI: 10.1038/nrc924 |
| OGDEN, A.RIDA, P.C.ANEJA, R.: "Heading off with the herd: how cancer cells might maneuver supernumerary centrosomes for directional migration", CANCER METASTASIS REV, vol. 32, 2013, pages 269 - 287 |
| OGDEN, A.RIDA, P.C.ANEJA, R.: "Let's huddle to prevent a muddle: centrosome declustering as an attractive anticancer strategy", CELL DEATH DIFFER, vol. 19, 2012, pages 1255 - 1267 |
| PANNU, V.RIDA, P.C.G.OGDEN, A.TURAGA, R. C.DONTHAMSETTY, SBOWEN N. J.RUDD KGUPTA M. V.REID M. D.CANTUARIA G.: "HSET overexpression fuels tumor progression via centrosome clustering-independent mechanisms in breast cancer patients", ONCOTARGET, vol. 6, 2015, pages 6076 - 6091 |
| PATEL N. E.WEEKES D.DROSOPOULOS K.GAZINSKA P.NOEL E.RASHID M.MIRZA H.QUIST J.BRASO-MARISTANY F.MATHEW S.: "Integrated genomics and functional validation identifys malignant cell specific dependencies in triple negative breast cancer", NAT. COMMUN., vol. 9, 2018, pages 1044 |
| PAWAR, S.DONTHAMSETTY, S.PANNU, V.RIDA, P.OGDEN, A.BOWEN, N.OSAN, R.CANTUARIA, G.ANEJA, R.: "KIFCI, a novel putative prognostic biomarker for ovarian adenocarcinomas: Delineating protein interaction networks and signaling circuitries", JOURNAL OF OVARIAN RESEARCH, vol. 7, no. 1, 2014, pages 53, XP021186949, DOI: 10.1186/1757-2215-7-53 |
| PHARMACEUTICAL RESEARCH, vol. 3, no. 6, 1986, pages 318 |
| RATH, O.KOZIELSKI, F.: "Kinesins and cancer", NATURE REVIEWS CANCER, vol. 12, no. 8, 2012, pages 527 - 539 |
| REIDER ET AL., J. ORG. CHEM., vol. 52, 1987, pages 3326 - 3334 |
| SHARP, D.J.ROGERS, G.C.SCHOLEY, J.M.: "Microtubule motors in mitosis", NATURE, vol. 407, 2000, pages 41 - 47, XP009124176, DOI: 10.1038/35024000 |
| TILLEMENT, V.REMY, M.H.RAYNAUD-MESSINA, B.MAZZOLINI, L.HAREN, L.MERDES, A.: "Spindle assembly defects leading to the formation of a monopolar mitotic apparatus", BIOL. CELL, vol. 101, 2009, pages 1 - 11, XP071518549, DOI: 10.1042/BC20070162 |
| VICHAI, V.KIRTIKARA, K.: "Sulforhodamine B colorimetric assay for cytotoxicity screening", NAT. PROTOC., vol. 1, 2006, pages 1112 - 1116, XP055272660, DOI: 10.1038/nprot.2006.179 |
| WALCZAK, C.E.VERMA, S.MITCHISON, T.J.: "XCTK2: A kinesin-related protein that promotes mitotic spindle assembly in Xenopus laevis egg extracts", JOURNAL OF CELL BIOLOGY, vol. 136, no. 4, 1997, pages 859 - 870 |
| WATTS C. A.RICHARDS F. M.BENDER A.BOND P. J.KORB O.KERN O.RIDDICK M.OWEN P.MYERS R. M.RAFF J.: "Design, synthesis, and biological evaluation of an allosteric inhibitor of HSET that targets cancer cells with supernumerary centrosomes", CHEMISTRY & BIOLOGY, vol. 20, 2013, pages 1399 - 1410, XP093024131, DOI: 10.1016/j.chembiol.2013.09.012 |
| WU J.MIKULE K.WANG W.SU N.PETTERUTI P.GHARAHDAGHI F.CODE E.ZHU X.JACQUES K.LAI Z.: "Discovery and mechanistic study of a small molecule inhibitor for motor protein KIFC1", ACS CHEM BIOL., vol. 8, 2013, pages 2201 - 2208 |
| YANG, Z.LONCAREK, J.KHODJAKOV, A.RIEDER, C.L.: "Extra centrosomes and/or chromosomes prolong mitosis in human cells", NAT. CELL BIOL., vol. 10, 2008, pages 748 - 751 |
| ZHANG W.ZHAI L.WANG Y.BOOHAKER R. J.LU W.GUPTA V. V.PADMALAYAM I.BOSTWICK R. J.WHITE E. L.ROSS L. J.: "Discovery of a novel inhibitor of kinesin-like protein KIFC1", BIOCHEM J., vol. 473, pages 1027 - 1035, XP093024129, DOI: 10.1042/BJ20150992 |
| ZYSS, D.GERGELY, F.: "Centrosome function in cancer: guilty or innocent?", TRENDS CELL BIOL, vol. 19, 2009, pages 334 - 346, XP026281753, DOI: 10.1016/j.tcb.2009.04.001 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN120265629A (en) | 2025-07-04 |
| TW202434603A (en) | 2024-09-01 |
| JP2025542089A (en) | 2025-12-25 |
| EP4615838A1 (en) | 2025-09-17 |
| AU2023375996A1 (en) | 2025-06-19 |
| AR130957A1 (en) | 2025-02-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2718218T3 (en) | Derivatives of 4,5,6,7-tetrahydropyrazolo [1,5-a] pyrazine substituted as inhibitors of casein kinase 1 D / E | |
| ES2797252T3 (en) | Dihydronaphthyridines and related compounds useful as kinase inhibitors for the treatment of proliferative diseases | |
| TWI406864B (en) | Chemical compounds | |
| EP3841102B1 (en) | Tetrahydropyridopyrimidine derivatives as ahr modulators | |
| JP6564406B2 (en) | Imidazo-pyridazine derivatives as casein kinase 1 delta / epsilon inhibitors | |
| JP7600119B2 (en) | Thiazolopyridine derivatives as adenosine receptor antagonists. | |
| US20250049814A1 (en) | 5-azaindazole derivatives as adenosine receptor antagonists | |
| AU2023375996A1 (en) | Substituted bi-and tricyclic hset inhibitors | |
| WO2023131690A1 (en) | Substituted heterocycles as hset inhibitors | |
| KR20240128969A (en) | Heteroaromatic nitrogen oxide compounds, their preparation methods and their uses | |
| WO2024042007A1 (en) | Substituted bicycles as hset inhibitors | |
| WO2019043373A1 (en) | Substituted cyclyl-sulfonamides as modulators of hedgehog (hh) signalling pathway | |
| JP2025521723A (en) | PROTACs for targeted degradation of KAT2A and KAT2B for the treatment of cancer | |
| HK40055989A (en) | Tetrahydropyridopyrimidine derivatives as ahr modulators | |
| HK40055989B (en) | Tetrahydropyridopyrimidine derivatives as ahr modulators |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 23800845 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202380077319.1 Country of ref document: CN |
|
| ENP | Entry into the national phase |
Ref document number: 2025526291 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2025526291 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: AU2023375996 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023800845 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2023800845 Country of ref document: EP Effective date: 20250610 |
|
| ENP | Entry into the national phase |
Ref document number: 2023375996 Country of ref document: AU Date of ref document: 20231103 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 202380077319.1 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2023800845 Country of ref document: EP |