WO2024089639A1 - Lentiviral formulations - Google Patents
Lentiviral formulations Download PDFInfo
- Publication number
- WO2024089639A1 WO2024089639A1 PCT/IB2023/060809 IB2023060809W WO2024089639A1 WO 2024089639 A1 WO2024089639 A1 WO 2024089639A1 IB 2023060809 W IB2023060809 W IB 2023060809W WO 2024089639 A1 WO2024089639 A1 WO 2024089639A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- composition
- car
- antigen binding
- sequence
- cells
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C12N15/86—Viral vectors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/0091—Purification or manufacturing processes for gene therapy compositions
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/70503—Immunoglobulin superfamily
- C07K14/7051—T-cell receptor (TcR)-CD3 complex
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2740/00—Reverse transcribing RNA viruses
- C12N2740/00011—Details
- C12N2740/10011—Retroviridae
- C12N2740/16011—Human Immunodeficiency Virus, HIV
- C12N2740/16041—Use of virus, viral particle or viral elements as a vector
- C12N2740/16043—Use of virus, viral particle or viral elements as a vector viral genome or elements thereof as genetic vector
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2740/00—Reverse transcribing RNA viruses
- C12N2740/00011—Details
- C12N2740/10011—Retroviridae
- C12N2740/16011—Human Immunodeficiency Virus, HIV
- C12N2740/16051—Methods of production or purification of viral material
Definitions
- Viruses are highly efficient at nucleic acid delivery to specific cell types, while often avoiding detection by the infected host immune system. These features make certain viruses attractive candidates as gene-delivery vehicles for use in gene therapies.
- lentiviral vectors include reconstructed viral vector systems derived from human immunodeficiency virus- 1 (HIV- 1) and are capable of introducing a gene of interest into animal and human primary cells or cell lines.
- HIV- 1 human immunodeficiency virus- 1
- Lentiviral vector-mediated gene expression can be used to achieve continuous and stable protein production, because the gene of interest has been integrated into a host cell's genome and is thus replicated upon division of the cell. Lentiviral vectors can effectively transduce non-dividing cells as well as those actively progressing through the cell cycle.
- Tissues and cells in which lentiviral vector-mediated chronic expression of a gene of interest can occur include the brain, liver, muscle cells, retina, hematopoietic stem cells, marrow mesenchymal stem cells, and macrophages, among others.
- lentiviral vectors have been hindered by several challenges, such as low titer of the viral yield and low stability of the vector. Additionally, lentiviral vectors are susceptible to inactivation during purification process which can contribute to diminished final quality and efficacy of the vector preparation, further creating another hurdle for production of large scale of purified lentiviral vector. Thus, there remains a need for formulation buffers that preserve vector stability.
- the disclosure provides, at least in part, a method for producing high titer lentiviral vectors, carrying a transgene of interest under satisfactory safety conditions.
- the disclosure also provides, at least in part, methods of purification of such lentiviral particles, e.g., from a cell culture.
- the disclosure also provides a formulation for lentiviral preparations that maintains structural integrity of the viral vector during purification, storage, and gene transfer events, e.g., ex vivo gene transfer.
- the present disclosure provides an aqueous composition
- aqueous composition comprising: a) N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid (HEPES); and b) one or both of a free positively charged amino acid (e.g., arginine, lysine, or histidine) and a free nonpolar amino acid (e.g., proline, methionine, or tryptophan).
- HEPES N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid
- the aqueous composition is substantially free of (e.g., is free of) inorganic salts.
- the aqueous composition is substantially free of (e.g., is free of) one or both of NaCl and MgCE.
- the aqueous composition comprises less than 20, 10, 5, 2, or 1 mM total of inorganic salts.
- the HEPES is at a concentration of 10-200, 10-150, 10-100, 10-50, 10-40, 10-30, 15-25, or 20 mM.
- the HEPES is at a concentration of about 20 mM.
- the aqueous composition comprises the positively charged amino acid, wherein the positively charged amino acid comprises arginine (e.g., L-arginine).
- arginine e.g., L-arginine
- the arginine is at a concentration of at least 25, 50, 75, 100, or 150 mM.
- the arginine is at a concentration of 25-50, 50-75, 75-100, 75-125, 100- 200, 125-175, or 150 mM.
- the arginine is at a concentration of about 150 mM.
- the aqueous composition comprises the nonpolar amino acid, wherein the nonpolar amino acid comprises proline (e.g., L-proline).
- proline e.g., L-proline
- the proline is at a concentration of 25-200, 50-200, 100-200, 125-175, or 150 mM. In some embodiments, the proline is at a concentration of about 150 mM.
- the aqueous composition further comprises a cryoprotectant agent, e.g., a carbohydrate, e.g., a non-reducing carbohydrate, e.g., sucrose.
- a cryoprotectant agent e.g., a carbohydrate, e.g., a non-reducing carbohydrate, e.g., sucrose.
- the aqueous composition further comprises sucrose.
- the sucrose is at a concentration of 25-200, 50-200, 100-200, 125-175, or 150 mM.
- the sucrose is at a concentration of about 150 mM.
- the aqueous composition further comprises a stabilizing agent (e.g., HSA).
- a stabilizing agent e.g., HSA
- the aqueous composition comprises HSA.
- the HSA comprises recombinant HSA (rHSA) or human-derived HSA (e.g., HSA isolated from human serum).
- rHSA recombinant HSA
- human-derived HSA e.g., HSA isolated from human serum
- the HSA is present at 0.5-3%, 0.5-2%, 0.5-1%, 1-2%, 1.5-2.5%, or 2% w/v.
- the HSA is present at about 2% w/v.
- the aqueous composition is substantially free of (e.g., is free of) HSA.
- the aqueous composition has a pH of 6.0-7.5, 6.0-7.0, 6.0-6.5, 6.5-7.0, 6.2- 6.8, 6.4-6.6, or 6.5.
- the aqueous composition has a pH of about 6.5.
- the aqueous composition is substantially free of (e.g., is free of) one, two, or three of PEG lipid, F108, and cholesterol.
- the composition comprises the positively charged amino acid, wherein the positively charged amino acid comprises L-arginine; the composition comprises the nonpolar amino acid, wherein the nonpolar amino acid comprises L-proline; the composition further comprises sucrose; and the composition is substantially free of inorganic salts.
- the L-arginine is at a concentration of 100-200 mM; the L-proline is at a concentration of 25-200 mM; and the sucrose is at a concentration of 25-200 mM.
- the HEPES formulation and/or storage buffer comprises L-arginine at a concentration of 100-200 mM, L-proline at a concentration of 25-200 mM, and sucrose at a concentration of 25-200 mM.
- the HEPES formulation and/or storage buffer comprises 20 mM HEPES, L-arginine at a concentration of 150 mM, L-proline at a concentration of 150 mM, and sucrose at a concentration of 150 mM. In some embodiments, the HEPES formulation and/or storage buffer further comprises HSA at a concentration of 2% w/v.
- the osmolality of the aqueous composition is from about 400 mOsm/kg to about 700 mOsm/kg, e.g., about 415 mOsm/kg to about 689 mOsm/kg.
- the disclosure provides a composition comprising: a lentiviral vector and an aqueous composition of any of the preceding embodiments.
- the composition comprises at least 1 x 10 6 , 1 x 10 7 , 5 x 10 7 , 1 x 10 8 , l x 10 9 , 2 x 10 9 , 3 x 10 9 , 4 x 10 9 , 5 x 10 9 , or 6 x 10 9 transducing units per milliliter (TU/mL) of the lentiviral vector.
- the lentiviral vector comprises a transgene.
- the fransgene encodes a chimeric antigen receptor (CAR).
- the CAR comprises an antigen-binding domain that binds a tumor antigen selected from the group consisting of: TSHR, CD19, CD123, CD22, CD30, CD171, CS-1, CLL-1, CD33, EGFRvIII , GD2, GD3, BCMA, Tn Ag, PSMA, R0R1, FLT3, FAP, TAG72, CD38, CD44v6, CEA, EPCAM, B7H3, KIT, IL-13Ra2, Mesothelin, IL-l lRa, PSCA, PRSS21, VEGFR2, Lewis Y, CD24, PDGFR-beta, S SEA-4, CD20, Folate receptor alpha, ERBB2 (Her2/neu), MUC1, EGFR, NCAM, Prostase, PAP, ELF2M, Ephrin B2, IGF-I receptor, CAIX, L
- the CAR comprises an antigen binding domain, a transmembrane domain, one or more primary signaling domains, and/or one or more costimulatory signaling domains.
- the one or more primary signaling domains comprises a CD3-zeta stimulatory domain.
- the one or more costimulatory signaling domains comprise an intracellular domain selected from
- a costimulatory protein selected from the group consisting of 0X40, CD27, CD28, ICAM-1, LFA-1 (CDl la/CD18), ICOS(CD278), 4-1BB (CD137), ICAM-1, GITR, BAFFR, HVEM (LIGHTR), SLAMF7, NKp80 (KLRF1), NKp44, NKp30, NKp46, CD160, CD19, CD4, CD8alpha, CD8beta, IL2R beta, IL2R gamma, IL7R alpha, ITGA4, VLA1, CD49a, ITGA4, IA4, CD49D, ITGA6, VLA-6, CD49f, ITGAD, CD1 Id, ITGAE, CD103, ITGAL, CD1 la, LFA-1, ITGAM, CD1 lb, ITGAX, CD11c, ITGB1, CD29, ITGB2, CD18, LFA-1, ITGB7, NKG2D, NKG2C
- SELPLG (CD 162), LTBR, LAT, GADS, SLP-76, PAG/Cbp, CD 19a, and
- a ligand that specifically binds with CD83 e.g., a 4-1 BB (CD137) costimulatory domain or a CD28 costimulatory domain.
- the CAR comprises a transmembrane domain, wherein:
- the transmembrane domain comprises a transmembrane domain of a protein chosen from the alpha, beta, or zeta chain of T-cell receptor, CD28, CD3 epsilon, CD45, CD4, CD5, CD8, CD9, CD16, CD22, CD33, CD37, CD64, CD80, CD86, CD134, CD137, and CD154;
- the transmembrane domain comprises a transmembrane domain of CD8
- the transmembrane domain comprises the amino acid sequence of SEQ ID NO: 6, or an amino acid sequence having at least about 90% sequence identity thereto; or
- the transmembrane domain is encoded by a nucleic acid sequence of SEQ ID NO: 17, or a nucleic acid sequence having at least about 90% sequence identity thereto.
- the composition exhibits a level of sub-visible particles of greater than or equal to 25 pm of about 50-100 particles/mL or lower.
- the composition exhibits a level of sub-visible particles of greater than or equal to 10 pm of about 1100-2000 particles/mL or lower.
- the lentiviral vector within the composition retains a hydrodynamic diameter of between about 85-200, 90-200, or 85-130 nm.
- the disclosure provides a kit comprising: a lentiviral vector and an aqueous composition of any of the preceding embodiments.
- the disclosure provides a method of making a CAR-expressing cell, the method comprising: providing a composition of any of the preceding embodiments; and contacting the composition with immune effector cells under conditions that allow for transduction of one or more of the immune effector cells, thereby making the CAR-expressing cell.
- the method is an in vitro or ex vivo method.
- the population of immune effector cells comprises one or both of T cells and NK cells.
- the composition comprises HSA, e.g., at a concentration of about 0.5% to 3% w/v.
- the HSA comprises recombinant HSA (rHSA) or human-derived HSA (e.g., HSA isolated from human serum).
- rHSA recombinant HSA
- human-derived HSA e.g., HSA isolated from human serum
- the disclosure provides a method of delivering a fransgene to a subject, the method comprising administering to the subject a composition of any of the preceding embodiments, wherein the lentiviral vector of the composition comprises the fransgene.
- the fransgene comprises a CAR, e.g., a CAR described herein.
- the CAR is a CD19 CAR, e.g., a CAR having CDRs as set out in Table 1.
- the composition further comprises a particle, e.g., a silica particle, e.g., a mesoporous particle (MSP), optionally wherein the mesoporous silica particle is a mesoporous silica rod.
- a particle e.g., a silica particle, e.g., a mesoporous particle (MSP)
- MSP mesoporous particle
- the retroviral vector is noncovalently, e.g., electrostatically, or covalently associated with the mesoporous silica particle;
- the cell activation agent is noncovalently or covalently associated with the mesoporous silica particle.
- the composition further comprises a cell activation agent.
- the cell activation agent :
- (a) comprises an agent that stimulates CD3/TCR complex and/or an agent that stimulates a costimulatory molecule and/or growth factor receptor;
- (b) is a multispecific binding molecule comprising: (i) an anti-CD3 binding domain, and (ii) a costimulatory molecule binding domain (e.g., an anti-CD2 binding domain or an anti-CD28 binding domain); and/or
- (c) is conjugated to or adsorbed on the particle, e.g., mesoporous silica particle.
- the present disclosure provides a method for manufacturing a lentiviral vector, the method comprising: a) providing a plurality of mammalian (e.g., human) cells, b) contacting the plurality of mammalian cells with: i) FectoVIR®-AAV transfection reagent, and ii) a nucleic acid encoding a therapeutic effector, e.g., a therapeutic protein (e.g., a CAR) and sufficient LTR sequence for packaging into a viral particle, and optionally nucleic acid encoding a lentiviral packaging protein, a lentiviral envelope protein, and, under conditions that allow the nucleic acid to be introduced into at least a subset of the cells; and c) culturing the cell under conditions suitable for production of the lentiviral vector.
- a therapeutic effector e.g., a therapeutic protein (e.g., a CAR) and sufficient LTR sequence for packaging into a viral particle
- the plurality of mammalian cells when the plurality of mammalian cells is in a 50L culture, it yields a number of transducing units per ml culture that is no less than 50%, 60%, 70%, or 80% the number of transducing units per ml culture in an otherwise similar 100 ml culture.
- the method yields at least IxlO 7 or 3xl0 7 or at least IxlO 8 transducing units when used under conditions described in Example 5.
- the method yields a ratio of equal to or less than 1188: 1, 953: 1, and 1800: 1 PP (physical particles): IP (infectious particles).
- the mammalian cells are 293 cells, e.g., Expi293F cells.
- the FectoVIR®-AAV is used at a concentration of 0.3 - 0.6 pl FectoVIR®- AAV / million cells, e.g., about 0.4 pl/ million cells.
- the nucleic acid is used at a concentration of 0.3 - 0.6 pg of nucleic acid / million cells, e.g., about 0.4 pg/ million cells.
- the FectoVIR®-AAV transfection reagent is complexed with the nucleic acid.
- the method further comprises admixing the FectoVIR®-AAV transfection reagent with the nucleic acid before step b).
- complexation volume of the transfection reagent and the nucleic acid is between about 1% and about 15%, e.g., about 1% and about 10% (e.g., about 5-7.5% or 7.5-10%).
- the complexation volume is 3-7%, 4-6%, or about 5%.
- the FectoVIR®-AAV transfection reagent and the nucleic acid are incubated for sufficient time to allow complexation to occur, e.g., about 10-90 minutes, e.g., 15-60, e.g., 15-30, 30-45, or 45-60 minutes.
- the present disclosure provides a method of manufacturing a lentiviral vector, the method comprising: a) culturing a plurality of mammalian (e.g., human) cells at a pH of above about 6.9 or about 6.9- 7.3, e.g., about 7.0-7. 1; b) subsequently to step a), adjusting the pH of the culture to about 6.0 - 6.8, e.g., 6.6 - 6.8, e.g., about 6.7; c) subsequently to step b), contacting the culture with a transfection reagent and DNA.
- mammalian e.g., human
- the transfection reagent comprises FectoVIR®-AAV transfection reagent.
- the DNA encodes one or more retroviral packaging protein, a retroviral envelope protein, and a therapeutic effector, e.g., a therapeutic protein (e.g., a CAR).
- a therapeutic protein e.g., a CAR
- a) comprises culturing the cells for about 2-4 days, e.g., about 3 days. In some embodiments, the method further comprises an additional step of culturing the cells between steps b) and c).
- the method further comprises an additional step of culturing the cells after step c).
- step b) comprises lowering the pH by about 0. 1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, or 1.
- the plurality of mammalian cells are inoculated at between 0. 1x10 s cells/mL - and 0.3xl0 6 cells/mL (e.g., about 0.15xl0 6 cells/mL or about 0.2xl0 6 cells/mL) in culture medium (e.g., FreeStyleTM medium) at a final volume.
- culture medium e.g., FreeStyleTM medium
- the plurality of mammalian cells are inoculated between 50 and 80 hours (e.g., about 55 hours, about 60 hours, about 65 hours, about 70 hours, about 72 hours, about 75 hours, or about 80 hours) prior to step a).
- the plurality of mammalian cells are cultured under conditions suitable to allow for cell growth and amplification to a suitable cell density at transfection (e.g., between about 1.0x10 s cells/mL and about 3.0xl0 6 cells/mL (e.g., between 1.5xl0 6 cells/mL and 2.5xl0 6 cells/mL)).
- a suitable cell density at transfection e.g., between about 1.0x10 s cells/mL and about 3.0xl0 6 cells/mL (e.g., between 1.5xl0 6 cells/mL and 2.5xl0 6 cells/mL)).
- the present disclosure provides a method of manufacturing a lentiviral vector, the method comprising: a) providing a composition comprising the lentiviral vector and at least one impurity (e.g., wherein the composition comprises a clarified cell harvest or a filtrate), and b) contacting the composition with arginine or a salt thereof.
- one or more of: i) the arginine is at a concentration of about 25-50 mM (about 50mM), 50-100 mM (e.g., about 75mM), 100-200 mM (e.g., about 150 mM), or 200-400 (e.g., about 300) mM arginine); or ii) the arginine is at a concentration sufficient to increase level of transducing units of the lentiviral vector by about 10% - 300%, about 20% - 180%, about 30% - 160%, about 50% - 150%, about 75%- 125% or about 100% compared to an otherwise similar composition, e.g., in an assay according to Example 7; iii) after step b) the composition shows a total particle concentration per ml of less than 400,000, less than 300,000, less than 200,000, or less than 100,000, as measured by micro-flow imaging, wherein optionally the particles comprise aggregated lentivirus; iv) after
- b) comprises contacting the composition with a solution comprising the arginine and a buffer, wherein optionally the buffer is PIPES, wherein optionally the PIPES is at a concentration of from about 10 mM to about 50 mM, e.g., about of 20 mM in the solution.
- the solution has a pH of about 6.0 to about 7.0, e.g., about 6.5.
- the solution further comprises a salt, wherein optionally the salt is selected from the group consisting of sodium chloride, magnesium chloride, and calcium chloride, e.g., sodium chloride.
- the salt is present in the solution at a concentration of from about 25-150 mM, e.g., about 50-100 mM, e.g., about 75 mM.
- the solution has a pH of about 6.5.
- the solution further comprises a carbohydrate, e.g., a non-reducing carbohydrate, e.g., sucrose or trehalose.
- a carbohydrate e.g., a non-reducing carbohydrate, e.g., sucrose or trehalose.
- the carbohydrate is present in the solution at a concentration of from about 1 % to about 10% by weight per volume of the solution, e.g., about 2% to about 5% by weight per volume of the solution, about 2.5% by weight per volume of the solution.
- the carbohydrate is present in the solution at a concentration of about 30- 150 mM (about 73 mM), or about 150-300 (e.g., about 220) mM.
- the solution further comprises one or both of NaCl (e.g., about 25-150 mM, e.g., about 50-100 mM, e.g., about 75 mM), and sucrose (e.g., about 30-150 mM, e.g., about 73 mM, or e.g., about 150-300 mM, e.g., about 220 mM, or about 2.5% by weight per volume) of the solution.
- NaCl e.g., about 25-150 mM, e.g., about 50-100 mM, e.g., about 75 mM
- sucrose e.g., about 30-150 mM, e.g., about 73 mM, or e.g., about 150-300 mM, e.g., about 220 mM, or about 2.5% by weight per volume
- the solution comprises 20 mM PIPES, 75 mM sodium chloride, and 2.5% sucrose by weight per volume of the solution, and wherein the solution has a pH of about 6.5. In certain embodiments, the solution comprises about 20 mM PIPES, about 75 mM sodium chloride, and about 2.5% sucrose by weight per volume of the solution, and wherein the solution has a pH of about 6.5.
- the solution comprises 20 mM PIPES, 75 mM sodium chloride, and 73 mM sucrose and wherein the solution has a pH of about 6.5. In certain embodiments, the solution comprises about 20 mM PIPES, about 75 mM sodium chloride, and about 73 mM sucrose and wherein the solution has a pH of about 6.5.
- the solution comprises 20 mM PIPES, 75 mM sodium chloride, and 220 mM sucrose and wherein the solution has a pH of about 6.5. In some embodiments, the solution comprises about 20 mM PIPES, about 75 mM sodium chloride, and about 220 mM sucrose and wherein the solution has a pH of about 6.5.
- the solution further comprises 20 mM PIPES, 75mM arginine, e.g., arginine-HCl, and wherein the solution has a pH of about 6.5. In some embodiments, the solution further comprises about 20 mM PIPES, about 75mM arginine, e.g., arginine-HCl, and wherein the solution has a pH of about 6.5.
- the osmolality of the solution is from about 270 mOsm/kg to about 330 mOsm/kg, e.g., about 275 mOsm/kg to about 300 mOsm/k, e.g., about 285 mOsm/kg.
- the method further comprises: c) performing a purification step, e.g., a filtration step, on the composition of b), thereby producing a semi -purified composition comprising the lentiviral vector.
- the method further comprises, after step c), contacting the semi-purified composition with arginine or a salt thereof.
- the arginine encapsulates the lentiviral vector.
- the arginine stabilizes the lentiviral vector.
- the impurity comprises a protein (e.g., a host cell protein), a nucleic acid (e.g., a host cell nucleic acid), a carbohydrate (e.g., a host cell carbohydrate), a lipid, an enzyme, a salt, a buffer, or any combination thereof.
- a protein e.g., a host cell protein
- nucleic acid e.g., a host cell nucleic acid
- a carbohydrate e.g., a host cell carbohydrate
- lipid e.g., an enzyme, a salt, a buffer, or any combination thereof.
- the cell density at transfection is between about 1.0x10 s cells/mL and about 3.0xl0 6 cells/mL (e.g., between 1.5xl0 6 cells/mL and 2.5xl0 6 cells/mL).
- the viability of the cells is, or is assessed to be, at least 90% (e.g., 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100%) at the time of transfection.
- the viability of the cells is measured at or around the time of transfection (e.g., within 30 minutes prior to transfection).
- the method is used for a process with two or more nucleic acids (e.g., two or more plasmids, e.g., two plasmids, three plasmids, four plasmids, or five plasmids).
- two or more nucleic acids e.g., two or more plasmids, e.g., two plasmids, three plasmids, four plasmids, or five plasmids.
- the present disclosure provides a method of manufacturing a lentiviral vector, the method comprising: a) providing a population of human cells (e.g., 293 cells); b) introducing into the cells nucleic acid encoding a retroviral packaging protein, a retroviral envelope protein, and a therapeutic effector, e.g., a therapeutic protein (e.g., a CAR), c) contacting the cells with benzonase at a time about 6-40, 10-40, 10-30, or about 20 hours after step b); and d) culturing the cells under conditions suitable for production of the lentiviral vector.
- a population of human cells e.g., 293 cells
- a therapeutic effector e.g., a therapeutic protein (e.g., a CAR)
- the present disclosure provides a method of manufacturing a lentiviral vector, the method comprising: a) providing a population of human cells (e.g., 293 cells); b) introducing into the cells nucleic acid encoding a retroviral packaging protein, a retroviral envelope protein, and a therapeutic effector, e.g., a therapeutic protein (e.g., a CAR), c) contacting the cells with benzonase; d) culturing the cells under conditions suitable for production of the lentiviral vector; e) harvesting the lentiviral vectors from cells 6-10 hours, 10-20 hours, 20-30 hours, 30-40 hours, or 40-50 hours after step c).
- a population of human cells e.g., 293 cells
- b) introducing into the cells nucleic acid encoding a retroviral packaging protein, a retroviral envelope protein, and a therapeutic effector, e.g., a therapeutic protein (e.g., a CAR
- the present disclosure provides a method of manufacturing a lentiviral vector, the method comprising: a) providing a plurality of mammalian (e.g., human) cells, wherein the plurality of cells (e.g., wherein the cell is a fibroblast cell, e.g., an embryonic kidney fibroblast cell, e.g., an Expi293F cell), wherein the cell comprises a nucleic acid (e.g., DNA) encoding one or more retroviral packaging protein, a retroviral envelope protein, and a therapeutic effector, e.g., a therapeutic protein (e.g., a CAR), b) culturing the cell under conditions suitable for production of the lentiviral vector.
- a nucleic acid e.g., DNA
- a retroviral packaging protein e.g., a retroviral envelope protein
- a therapeutic effector e.g., a therapeutic protein (e.g., a CAR)
- an aqueous composition comprising a lentiviral vector, arginine, a 1,4-piperazinediethanesulfonic acid (PIPES) buffer, and a salt.
- a lentiviral vector comprising a lentiviral vector, arginine, a 1,4-piperazinediethanesulfonic acid (PIPES) buffer, and a salt.
- the arginine in the aqueous composition is at a concentration of about 25-50 mM (about 50mM), about 50-100 mM (e.g., about 75mM), about 100-200 mM (e.g., about 150 mM), or about 200-400 (e.g., about 300) mM arginine), wherein optionally the PIPES aqueous composition is at a concentration of from about 10 mM to about 50 mM, e.g., about, e.g., 20 mM.
- the aqueous composition has a pH of about 6.0 to about 7.0, e.g., about 6.5.
- the aqueous composition further comprises a salt, wherein optionally the salt is selected from the group consisting of sodium chloride, magnesium chloride, and calcium chloride.
- the salt is sodium chloride (NaCl).
- the salt in the aqueous composition is from about 25 mM to about 150 mM, e.g., about 50mM to about 75mM.
- the aqueous composition comprises 20 mM PIPES and 75 mM sodium chloride, and wherein the aqueous composition has a pH of about 6.5.
- the aqueous composition further comprises a carbohydrate, e.g., a nonreducing carbohydrate, e.g., sucrose or trehalose.
- a carbohydrate e.g., a nonreducing carbohydrate, e.g., sucrose or trehalose.
- the carbohydrate is present in the aqueous composition at a concentration of from about 1 % to about 10% by weight per volume of the solution, e.g., about 2% to about 5% by weight per volume of the aqueous composition, about 2.5% by weight per volume of the aqueous composition.
- the carbohydrate is present in the aqueous composition at a concentration of from about 30-150 mM (about 73 mM), or about 150-300 (e.g., about 220) mM.
- the aqueous composition comprises one or both of NaCl (e.g., about 25- 150 mM, e.g., about 50-100 mM, e.g., about 75 mM), and sucrose (e.g., about 30-150 mM, e.g., about 73 mM, or e.g., about 150-300 mM, e.g., about 220 mM, or about 2.5% by weight per volume) of the aqueous composition.
- NaCl e.g., about 25- 150 mM, e.g., about 50-100 mM, e.g., about 75 mM
- sucrose e.g., about 30-150 mM, e.g., about 73 mM, or e.g., about 150-300 mM, e.g., about 220 mM, or about 2.5% by weight per volume
- the aqueous composition comprises 20 mM PIPES, 75 mM sodium chloride, and 2.5% sucrose by weight per volume of the aqueous composition and wherein the aqueous composition has a pH of about 6.5.
- the aqueous composition comprises 20 mM PIPES, 75 mM sodium chloride and 73 mM sucrose and wherein the aqueous composition has a pH of about 6.5.
- the aqueous composition comprises 20 mM PIPES, 75 mM sodium chloride and 220 mM sucrose and wherein the aqueous composition has a pH of about 6.5.
- the osmolality of the aqueous composition is from about 270 mOsm/kg to about 330 mOsm/kg, e.g., about 275 mOsm/kg to about 300 mOsm/k, e.g., about 285 mOsm/kg.
- the lentiviral vector of any preceding claims is present at a concentration of from about 3 x 10 8 TU/mL to about 5 x 10 8 TU/mL.
- the aqueous composition is free of one or more proteins selected from the group consisting of human serum albumin (HSA), recombinant human serum albumin (rHSA), bovine serum albumin (BSA), and a lipoprotein.
- HSA human serum albumin
- rHSA recombinant human serum albumin
- BSA bovine serum albumin
- lentiviral vector comprises a fransgene, e.g., a fransgene encoding a protein, e.g., a protein comprising a chimeric antigen receptor (CAR).
- a fransgene e.g., a fransgene encoding a protein, e.g., a protein comprising a chimeric antigen receptor (CAR).
- CAR chimeric antigen receptor
- the CAR comprises, in an N-terminal to C- terminal direction, an antigen binding domain, a transmembrane domain, and one or more signaling domains.
- the signaling domain comprises one or more primary signaling domains and/or one or more costimulatory signaling domains.
- one of the one or more primary signaling domains comprises a CD3-zeta stimulatory domain.
- one or more of the costimulatory signaling domains comprises an intracellular domain selected from a costimulatory protein selected from the group consisting of 0X40, CD27, CD28, ICAM-1, LFA-1 (CDlla/CD18), ICOS(CD278), 4-1BB (CD137), ICAM-1, GITR, BAFFR, HVEM (LIGHTR), SLAMF7, NKp80 (KLRF1), NKp44, NKp30, NKp46, CD160, CD19, CD4, CD8alpha, CD8beta, IL2R beta, IL2R gamma, IL7R alpha, ITGA4, VLA1, CD49a, ITGA4, IA4, CD49D, ITGA6, VLA-6, CD49f, ITGAD, CD1 Id, ITGAE, CD103, ITGAL, CD1 la, LFA-1, ITGAM, CD1 lb, ITGAX, CD11c, ITGB1, CD29, IT
- one or more of the costimulatory signaling domains comprises an intracellular domain selected from a costimulatory protein selected from the group consisting ofCD27, CD28, 4-1BB (CD137), 0X40, CD30, CD40, PD-1, ICOS, lymphocyte function-associated antigen-1 (LFA-1), CD2, CD7, LIGHT, NKG2C, B7-H3, a ligand that specifically binds with CD83, ICAM-1, GITR, BAFFR, HVEM (LIGHTR), SLAMF7, NKp80 (KLRF1), CD160, CD19, CD4, CD8alpha, CD8beta, IL2Rbeta, IL2R gamma, IL7R alpha, ITGA4, VLA1, CD49a, ITGA4, IA4, CD49D, ITGA6, VLA-6, CD49f, ITGAD, GDI Id, ITGAE, CD103, ITGAL, GDI la, LFA-1,
- the antigen binding domain is an scFv.
- the antigen binding domain binds to an antigen selected from the group consisting of CD19; CD123; CD22; CD30; CD171 ; CS-1; C- type lectin-like molecule-1, CD33; epidermal growth factor receptor variant III (EGFRvlll); ganglioside G2 (GD2); ganglioside GD3; TNF receptor family member B cell maturation (BCMA); Tn antigen ((Tn Ag) or (GalNAca-Ser/Thr)); prostate-specific membrane antigen (PSMA); Receptor tyrosine kinase-like orphan receptor 1 (R0R1); Fms-Like Tyrosine Kinase 3 (FLT3); Tumor-associated glycoprotein 72 (TAG72); CD38; CD44v6; Carcinoembryonic antigen (CEA); Epithelial cell adhesion molecule (EPCAM); B7H3 (CD276); KIT (CD1 17); Interleukin- 13 receptor sub
- the lentiviral vector comprises a second transgene, e.g., a second fransgene encoding a second protein, e.g., a second protein comprising a second chimeric antigen receptor (CAR).
- a second transgene e.g., a second fransgene encoding a second protein, e.g., a second protein comprising a second chimeric antigen receptor (CAR).
- CAR chimeric antigen receptor
- the present disclosure provides a method of manufacturing a lentiviral vector, comprising: a) providing a population of human cells (e.g., 293 cells); b) introducing into the cells nucleic acid encoding a retroviral packaging protein, a retroviral envelope protein, and a therapeutic effector, e.g., a therapeutic protein (e.g., a CAR), c) contacting the cells with benzonase at a time about 2-6 (e.g., about 3), 4-10 (e.g., about 6), 6- 40, 10-40, 10-30 (e.g., about 24), or about 20 hours after step b); and d) culturing the cells under conditions suitable for production of the lentiviral vector.
- a therapeutic protein e.g., a CAR
- Benzonase is added 6-10 hours, 10-20 hours, 20-30 hours, 30-40 hours, or 40-50 hours, before harvest of lentiviral vector from the cells.
- the present disclosure provides a method of manufacturing a lentiviral vector, comprising: a) providing a population of human cells (e.g., 293 cells); b) introducing into the cells nucleic acid encoding a retroviral packaging protein, a retroviral envelope protein, and a therapeutic effector, e.g., a therapeutic protein (e.g., a CAR), c) contacting the cells with benzonase (e.g., 3-24 hours after step b); d) culturing the cells under conditions suitable for production of the lentiviral vector; e) harvesting the lentiviral vectors from cells 6-10 hours, 10-20 hours, 20-30 hours, 30-40 hours, or 40-50 hours after step c).
- a population of human cells e.g., 293 cells
- b) introducing into the cells nucleic acid encoding a retroviral packaging protein, a retroviral envelope protein, and a therapeutic effector, e.g., a therapeutic
- benzonase is at a concentration of about 10-40 U/mL, e.g., 20-30 U/mL, e.g., about 25 U/mL.
- benzonase is at a concentration of about 3 - 60 U/mL, 3-10 U/mL, 3-7 U/mL, 4-6 U/mL, or about 5 U/mL. In one embodiment, the benzonase is at a concentration of 5-50, 5-15, 15-25, or 25-50 U/mL.
- the method further comprises, before step c), contacting the benzonase with MgC’h. e.g., at about 1-5 mM, about 1-3 mM, or about 2 mM.
- the present disclosure provides a method of manufacturing a lentiviral vector, comprising: a) providing a plurality of mammalian (e.g., human) cells, wherein the plurality of mammalian cells do not comprise SV40 large T antigen (e.g., wherein the cell is a fibroblast cell, e.g., an embryonic kidney fibroblast cell, e.g., an Expi293F cell), wherein the plurality of mammalian cells comprise a nucleic acid (e.g., DNA) encoding one or more retroviral packaging protein, a retroviral envelope protein, and a therapeutic effector, e.g., a therapeutic protein (e.g., a CAR), b) culturing the cell under conditions suitable for production of the lentiviral vector.
- a nucleic acid e.g., DNA
- a retroviral packaging protein e.g., a retroviral envelope protein
- a therapeutic effector e
- a) comprises introducing the nucleic acid into the plurality of mammalian cells.
- the method further comprises at least partially separating the lentiviral vector from the plurality of mammalian cells.
- the one or more retroviral packaging proteins comprises a lentiviral gag, a lentiviral pol, or a lentiviral rev, or any combination thereof.
- the retroviral envelope protein comprises a VSV-G.
- the present disclosure provides a preparation of lentiviral vector, the preparation comprising: a plurality of lentiviral vector that comprise: a) a lentivirus genome encoding a therapeutic effector, e.g., a therapeutic protein (e.g., a CAR), and b) an envelope enclosing the lentivirus genome (wherein optionally the envelope comprises VSV- G); wherein the preparation comprises at least 5 x IO 7 , 1 x 10 8 , 1 x 10 9 , or 1 x 10 10 , transducing units; wherein the preparation comprises less than 90% of SV40 large T antigen or less than 10 pg/ml, 1 pg/ml of nucleic acid (e.g., DNA) encoding SV40 large T antigen.
- a therapeutic effector e.g., a therapeutic protein (e.g., a CAR)
- an envelope enclosing the lentivirus genome
- the preparation comprises at least 5
- the plurality of lentiviral vectors comprises at least 1 x 10 9 , at least 2 x 10 9 , at least 5 x 10 9 , at least 1 x IO 10 , at least 2 x IO 10 , at least 5 x IO 10 , at least 1 x 10 11 , at least 2 x 10 11 , at least 5 x 10 1 , or at least 1 xlO 12 of the cells.
- the plurality of mammalian cells are in a culture volume of at least 5, at least 10, at least 20, at least 50, at least 100, at least 200, or at least 500 L.
- the plurality of mammalian cells are grown in suspension.
- the CAR comprises a CD19 CAR (e.g., a humanized CD19 CAR, e.g., as described in WO2014153270A1).
- a CD19 CAR e.g., a humanized CD19 CAR, e.g., as described in WO2014153270A1.
- the CAR comprises a dual CAR (e.g., a humanized CD19-CD22 CAR, e.g., as described in WO2016164731A2).
- a dual CAR e.g., a humanized CD19-CD22 CAR, e.g., as described in WO2016164731A2.
- the nucleic acid encoding a CAR further encodes a shRNA, e.g., as described in WO2017049166A).
- the lentiviral vector is produced in cells cultured in the absence of serum.
- the lentiviral vector is characterized by a hydrodynamic radius of 100 ⁇ 25 nm as measured by dynamic light scattering (DLS).
- DLS dynamic light scattering
- the lentiviral vector maintains the hydrodynamic radius of 100 ⁇ 25 nm within a temperature range of from 25°C to 55°C (e.g., 26°C, 27°C, 28°C, 29°C, 30°C, 31°C, 32°C, 33°C, 34°C, 35°C, 36°C, 37°C, 38°C, 39°C, 40°C, 41°C, 42°C, 43°C, 44°C, 45°C, 46°C, 47°C, 48°C, 49°C, 50°C, 51°C, 52°C, 53°C, 54°C, or 55°C).
- 25°C to 55°C e.g., 26°C, 27°C, 28°C, 29°C, 30°C, 31°C, 32°C, 33°C, 34°C, 35°C, 36°C, 37°C, 38°C, 39°C, 40°C, 41°C, 42°C, 43°C, 44°C, 45°
- the lentiviral vector is characterized by a poly dispersity of from 10% to 25% (e.g., 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19%, 20%, 21%, 22%, 23%, 24% or 25%).
- the lentiviral vector maintains the polydispersity of from 10% to 25% (e.g., 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19%, 20%, 21%, 22%, 23%, 24% or 25%) within a temperature range of from 25 °C to 55 °C (e.g., 26°C, 27°C, 28°C, 29°C, 30°C, 31°C, 32°C, 33°C, 34°C, 35°C, 36°C, 37°C, 38°C, 39°C, 40°C, 41°C, 42°C, 43°C, 44°C, 45°C, 46°C, 47°C, 48°C, 49°C, 50°C, 51°C, 52°C, 53°C, 54°C, or 55°C).
- 10% to 25% e.g., 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19%, 20%, 21%
- the lentiviral vector maintains a concentration after 3 freeze/thaw cycles of from about 70% to about 100% (e.g., about 70%, about 75%, about 80%, about 85%, about 90%, about 95%, about 97%, or about 100%) relative to the concentration of the lentiviral vector in the aqueous composition prior to the freeze/thaw cycles, wherein each of the freeze/thaw cycles comprises freezing the aqueous composition and subsequently allowing the aqueous composition to thaw at room temperature.
- the lentiviral vector maintains the concentration of from about 70% to about 100% (e.g, about 70%, about 75%, about 80%, about 85%, about 90%, about 95%, about 97%, or about 100%) after 6-10 of the freeze/thaw cycles, e.g., after 6-9 of the freeze/thaw cycles.
- the present disclosure provides an aqueous composition
- a lentiviral vector comprising a lentiviral vector, a buffer selected from the group consisting of a phosphate buffer, a sodium citrate buffer, a 2-(N- morpholino) ethanesulfonic acid (MES) buffer, a 3-morpholinopropane-l -sulfonic acid (MOPS) buffer, and a salt.
- a buffer selected from the group consisting of a phosphate buffer, a sodium citrate buffer, a 2-(N- morpholino) ethanesulfonic acid (MES) buffer, a 3-morpholinopropane-l -sulfonic acid (MOPS) buffer, and a salt.
- MES 2-(N- morpholino) ethanesulfonic acid
- MOPS 3-morpholinopropane-l -sulfonic acid
- the salt is selected from the group consisting of sodium chloride, magnesium chloride, and calcium chloride.
- the aqueous composition birther comprises a non-reducing carbohydrate selected from the group consisting of sucrose and trehalose.
- the present disclosure provides scalable processes for the production of large quantities of viral vectors (e.g., lentiviral vectors), e.g., for prophylactic, diagnostic, immunotherapeutic or therapeutic use.
- the processes may be performed using suspension cells (e.g., HEK293 cells, e.g., Expi293F cells).
- substantially all of the suspension cells do not express a large T antigen, e.g., SV40 T antigen.
- the process may be performed using a bioreactor.
- the present disclosure provides highly reproducible efficient scalable processes for the production of large quantities of viral vectors (e.g., lentiviral vectors) having one or both of a high viral titer or high viral yield.
- viral vectors e.g., lentiviral vectors
- the present disclosure provides highly reproducible efficient scalable processes for the purification of large quantities of viral vector (e.g., lentiviral) having one or both of a high viral titer or high viral yield.
- viral vector e.g., lentiviral
- compositions and methods for stabilizing viral vectors e.g., lentiviral vectors during a purification process.
- FIG. 1 is a schematic representation showing the overall study design.
- FIG. 2 is a graph showing LVV product particle size distribution (PSD; Dh) before and after incubation for 12 hours at 37°C using HEPES or XV 15 supplemented with sucrose.
- FIG. 3 is a graph showing LVV product Particle Concentration before and after incubation for 12 hours at 37°C using HEPES or XV 15 supplemented with sucrose.
- FIG. 4 is a graph showing TU titer of LVV product before and after incubation for 12 hours at 37°C using HEPES or XV15 supplemented with sucrose.
- FIG. 5 is a graph showing total p24 titer of LVV product before and after incubation for 12 hours at 37°C using HEPES or XV15 supplemented with sucrose.
- FIG. 6 is a graph showing the Zeta Potential (ZP) distribution of the LVV in HEPES and PIPES buffer.
- FIG. 7 is a graph showing the phase plot of the LVV in HEPES and PIPES buffer.
- FIG. 8 is a graph showing the relationship between pH and the size (left y-axis) or PDI (right y- axis) of the vector.
- FIG. 9 is a graph showing the relationship between pH and the zeta-potential of the vector.
- the dashed line (-30 mV) indicates potential minimum ZP for stable particle in suspension.
- FIG. 13 is a Pareto Chart for the TU titer obtained from combination 1.
- FIG. 14 is a graph showing the relationship between arginine and the TU titer obtained from combination 1.
- FIG. 15 is a graph showing the relationship between recombinant HSA (rHSA) and the TU titer obtained from combination 1.
- FIG. 16 is a graph showing the relationship between Proline and the TU titer obtained from combination 1.
- FIG. 17 is a graph showing the relationship between Lactose and the TU titer obtained from combination 1.
- FIG. 18 is a Pareto Chart for the p24 titer obtained from combination 1.
- FIG. 19 is a graph showing the relationship between Glycerol and the p24 titer obtained from combination 1.
- FIG. 20 is a graph showing the relationship between recombinant HSA (rHSA) and the p24 titer obtained from combination 1.
- FIG. 21 is a graph showing the relationship between Proline and the p24 titer obtained from combination 1.
- FIG. 22 is a graph showing the relationship between Lactose and the p24 titer obtained from combination 1.
- FIG. 23 is a Pareto Chart for the TU titer obtained from combination 2.
- FIG. 24 is a graph showing the relationship between Arginine and the TU titer obtained from combination 2.
- FIG. 25 is a graph showing the relationship between Glutamic Acid and the TU titer obtained from combination 2.
- FIG. 26 is a graph showing the relationship between recombinant HSA (HSA) and the TU titer obtained from combination 2.
- FIG. 27 is a graph showing the relationship between Arginine and the TU titer obtained from combination 2.
- FIG. 28 is a graph showing the relationship between MgC and the TU titer obtained from combination 2.
- FIG. 29 is a Pareto Chart for the p24 titer obtained from combination 2.
- FIG. 30 is a chart showing particle analysis of the factorial design of LVV product formulated in PIPES buffer. Particle concentrations greater than 10 pm are plotted against the Z-average. Dashed lines represent LVV particle size distribution (between 85 nm and 130 nm) and the number of sub-visible particles (1300) greater than 10 pm.
- FIG. 31 is a chart showing particle analysis of the factorial design of LVV product formulated in PIPES buffer. Particle concentrations greater than 25 pm are plotted against the Z-average. Dashed lines represent LVV particle size distribution (between 85 nm and 130 nm) and the number of sub-visible particles (150) greater than 25 pm.
- FIG. 32 is a graph showing TU titer for LVV product formulated with HEPES-F1 after buffer exchange (ABE) and spiked with different HSA sources using 2.0% HSA in formulation by the number of freeze thaw cycles (FT; white, OFT; striped, 3FT).
- FIG. 33 is a graph showing TU titer for LVV product formulated with HEPES-F1 after buffer exchange (ABE) and spiked with different HSA sources using 0.5% HSA in formulation by the number of freeze thaw cycles (FT; white, OFT; striped, 3FT).
- FIG. 34 is a graph showing p24 titer for LVV product formulated with HEPES-F1 after buffer exchange (ABE) and spiked with different HSA sources using 2.0% HSA in formulation by the number of freeze thaw cycles (FT; white, OFT; striped, 3FT).
- FIG. 35 is a graph showing p24 titer for LVV product formulated with HEPES-F1 after buffer exchange (ABE) and spiked with different HSA sources using 0.5% HSA in formulation by the number of freeze thaw cycles (FT; white, OFT; striped, 3FT).
- FIG. 36 is a graph showing the total number of sub-visible particles versus excipient range by freeze thaw cycles (FT; white, OFT; striped, 3FT).
- FIG. 37 is a graph showing the TU titer for three batches of LVV product formulated with HEPES-F1 after buffer exchange (ABE), HSA spike (+HSA), and three freeze-thaw cycles (+3FT).
- FIG. 38 is a graph showing p24 ELISA results for three batches of LVV product formulated with HEPES-F1 after buffer exchange (ABE), HSA spike (+HSA), and three freeze-thaw cycles (+3FT).
- FIG. 39 is a graph showing total p24 titer results for three batches of LVV product formulated with HEPES-F1 after buffer exchange (ABE), HSA spike (+HSA), and three freeze-thaw cycles (+3FT).
- FIG. 40 is a graph showing the total number of sub-visible particles greater than 10 pm observed in three batches of LVV product formulated with HEPES-F1 after buffer exchange (ABE), HSA spike (+HSA), and three freeze-thaw cycles (+3FT).
- FIG. 41 is a graph showing the total number of sub-visible particles greater than 25 pm observed in three batches of LVV product formulated with HEPES-F1 after buffer exchange (ABE), HSA spike (+HSA), and three freeze-thaw cycles (+3FT).
- FIG. 42 is a graph showing TU titer for three batches of LVV product formulated with HEPES- F2 after buffer exchange (ABE), HSA spike (+HSA), and three freeze-thaw cycles (+3FT).
- FIG. 43 is a graph showing total p24 titer results for three batches of LVV product formulated with HEPES-F2 after buffer exchange (ABE), HSA spike (+HSA), and three freeze-thaw cycles (+3FT).
- FIG. 44 is a graph showing the total number of sub-visible particles greater than 10 pm observed in three batches of LVV product formulated with HEPES-F2 after buffer exchange (ABE), HSA spike (+HSA), and three freeze-thaw cycles (+3FT).
- FIG. 45 is a graph showing the total number of sub-visible particles greater than 25 pm observed in three batches of LVV product formulated with HEPES-F2 after buffer exchange (ABE), HSA spike (+HSA), and three freeze-thaw cycles (+3FT).
- FIG. 46 is a graph showing the particle size distribution by intensity for three batches of LVV product in PIPES and HEPES buffer.
- FIG. 47 is a graph showing particle concentration measured by MADLS in LVV product after one (VS) and two (VS-2SF) sterile filtrations comparing the effect of HSA formulations.
- FIG. 48 is a graph showing TU titers of LVV product after one (VS) and two (VS-2SF) sterile filtrations comparing the effect of HSA formulations.
- FIG. 49 is a graph showing the TU titers of LVV product formulated in PIPES buffer following storage at different temperatures (25°C, 4°C, -20°C and -80°C) during a period of up to 9 months.
- FIG. 50 is a graph showing the TU titers of LVV product formulated in HEPES (Fl) buffer following storage at different temperatures (25°C, 4°C, -20°C and -80°C) during a period of up to 9 months.
- FIG. 51 is a graph showing the TU titers of LVV product formulated in HEPES (F2) buffer following storage at different temperatures (25°C, 4°C, -20°C and -80°C) during a period of up to 9 months.
- This disclosure is based, at least in part, on a method for producing high titer lentiviral vectors, carrying a transgene of interest and under satisfactory safety conditions.
- the disclosure also provides, at least in part, methods of purification of such lentiviral particle, e.g., from a cell culture.
- the disclosure also provides a formulation for lentiviral preparations that maintain structural integrity of the viral vector during purification, storage, and gene transfer events ex vivo.
- “About” and “approximately” shall generally mean an acceptable degree of error for the quantity measured given the nature or precision of the measurements. Exemplary degrees of error are within 10 percent (%), within 5%, or within 2% of a given value or range of values.
- amino acid refers to naturally occurring, synthetic, and unnatural amino acids, as well as amino acid analogs and amino acid mimetics that function in a manner similar to the naturally occurring amino acids.
- Naturally occurring amino acids are those encoded by the genetic code, as well as those amino acids that are later modified, e.g., hydroxyproline, y-carboxyglutamate, and O- phosphoserine.
- Amino acid analogs refer to compounds that have the same basic chemical structure as a naturally occurring amino acid, i.e., an a-carbon that is bound to a hydrogen, a carboxyl group, an amino group, and an R group, e.g., homoserine, norleucine, methionine sulfoxide, methionine methyl sulfonium. Such analogs have modified R groups (e.g., norleucine) or modified peptide backbones, but retain the same basic chemical structure as a naturally occurring amino acid.
- Amino acid mimetics refers to chemical compounds that have a structure that is different from the general chemical structure of an amino acid, but that functions in a manner similar to a naturally occurring amino acid.
- buffer refers to a mixture of a weak acid and its conjugate base or a weak base and its conjugate acid.
- a “N-2-hydroxyethylpiperazine-N'-2- ethanesulfonic acid (HEPES) buffer refers to a mixture that includes N-2-hydroxyethylpiperazine-N'-2- ethanesulfonic acid and the N-2-hydroxyethylpiperazine-N'-2-ethanesulfonate anion. Due to the chemical equilibrium that is established between a weak acid and its conjugate base, a solution containing a buffer resists abrupt changes in pH upon the addition of small quantities of acid or base to the solution.
- inorganic salt refers to a chemical component (e.g., a component of a solution) that lacks a carbon-hydrogen bond, is composed of positively charged cations and negatively charged anions, and has no net electric charge.
- exemplary inorganic salts include sodium chloride (NaCl) and magnesium chloride (MgCE).
- the salt is present in dissociated form in an aqueous solution.
- freeze/thaw cycle refers to exposure of a liquid mixture, such as an aqueous solution or suspension, to a temperature at or less than its freezing point until the mixture is frozen, followed by thawing the mixture at a temperature greater than its freezing point.
- the freezing step can be performed, e.g., by placing the mixture in an environment in which the temperature is from about - 80 °C to about -20 °C.
- the mixture can remain frozen, e.g., for a period of one or more days, weeks, months, or years prior to thawing.
- the thawing step can be performed by exposing the mixture to conditions in which the temperature is from about 2 °C to about 8 °C, or by storing the mixture at room temperature (e.g., the ambient temperature of a laboratory, or about 25 °C). Alternatively, thawing can take place by use of a water bath (e.g., at 37°C).
- hydrodynamic radius refers to the apparent radius (Rh in nm) of a particle in a solution as inferred from the diffusional characteristics of the particle.
- the hydrodynamic radius of a viral particle is one factor that dictates the rate of diffusion of the viral particle in aqueous solution, as well as the ability of the particle to migrate in gels of macromolecules.
- the hydrodynamic radius of a viral particle is determined in part by the mass and molecular structure of each of the components of the particle, as well as its hydration state. Methods for determining the hydrodynamic radius of a viral particle are well known in the art and include the use of dynamic light scattering and size exclusion chromatography.
- non-reducing carbohydrate refers to a carbohydrate that does not exist in a state of chemical equilibrium with an aldehyde, and thus lacks the ability to be oxidized to a carboxylic acid by transition metal cations, such as silver (Ag+) and copper (Cu2+).
- exemplary nonreducing carbohydrates include, without limitation, disaccharides such as sucrose, trehalose, and palatinitol, frisaccharides such as raffinose and melezitose, as well as tetrasaccharides such as stachyose.
- Non-reducing carbohydrates additionally include monosaccharide derivatives such as sorbitol, mannitol, erythritol, and xylitol, disaccharide derivatives such as lacitol and maltitol, aldonic acids and their lactones such as gluconic acid, gluconic acid y-lactone, aldaric acids and their lactones such as ribaraic acid, arabinaric acid, and galactaric acid, uronic acids such as glucuronic acid, galaccuronic acid, and itiannuronic acid, ester derivatives such as trehalose octaacetate, sucrose octaacetate, and cellobiose octaacetate, and ether derivatives in which hydroxyl groups are O-alkylated.
- Non-reducing carbohydrates include those that have a D or L stereochemical orientation.
- osmolality refers to a measure of the osmotic pressure of dissolved solute particles in an aqueous solution.
- the solute particles include both ions as well as non-ionized molecules.
- Osmolality is expressed as the concentration of osmotically active particles (i.e., osmoles) dissolved in 1 kg of solvent (i.e., water). Osmolality is expressed herein in units of milliosmoles per 1 kg of water (mOsm/kg).
- percent by weight per volume denotes the percentage weight (in grams) of a single component relative to the total volume (in milliliters) of the mixture that contains the component. For instance, 500 mg of a component in a total volume of 8 ml is 6.25% w/v, and 500 mg of a component in a total volume of 5 ml is 10% w/v.
- polydispersity refers to the degree of homogeneity of the sizes of particles, such as lentiviral particles, within a sample.
- a higher polydispersity indicates less homogeneity and a lower poly dispersity indicates a higher level of homogeneity.
- lentiviral particles can be considered to be approaching identical sizes and are thus monodisperse.
- a lower polydispersity indicates a higher level of homogeneity.
- a formulation with 15% polydispersity has less homogeneity than a formulation with 10% poly dispersity.
- the level of homogeneity is low, the particle population can be considered to contain significantly different sizes and thus be polydisperse.
- binding domain refers to a protein, e.g., an immunoglobulin chain or fragment thereof, comprising at least one immunoglobulin variable domain sequence.
- binding domain or “antibody molecule” encompasses antibodies and antibody fragments.
- an antibody molecule is a multispecific antibody molecule, e.g., it comprises a plurality of immunoglobulin variable domain sequences, wherein a first immunoglobulin variable domain sequence of the plurality has binding specificity for a first epitope and a second immunoglobulin variable domain sequence of the plurality has binding specificity for a second epitope.
- a multispecific antibody molecule is a bispecific antibody molecule.
- a bispecific antibody has specificity for no more than two antigens.
- a bispecific antibody molecule is characterized by a first immunoglobulin variable domain sequence which has binding specificity for a first epitope and a second immunoglobulin variable domain sequence that has binding specificity for a second epitope.
- antibody heavy chain refers to the larger of the two types of polypeptide chains present in antibody molecules in their naturally occurring conformations, and which normally determines the class to which the antibody belongs.
- antibody light chain refers to the smaller of the two types of polypeptide chains present in antibody molecules in their naturally occurring conformations.
- Kappa (K) and lambda (X) light chains refer to the two major antibody light chain isotypes.
- antigen binding fragment refers to one or more portions of an antibody that retain the ability to specifically interact with e.g., by binding, steric hindrance, stabilizing/destabilizing, spatial distribution) an epitope of an antigen.
- binding fragments include, but are not limited to, single-chain Fvs (scFv), camelid antibodies, disulfide-linked Fvs (sdFv), Fab fragments, F(ab') fragments, a monovalent fragment consisting of the VL, VH, CL and CHI domains; a F(ab)2 fragment, a bivalent fragment comprising two Fab fragments linked by a disulfide bridge at the hinge region; a Fd fragment consisting of the VH and CHI domains; a Fv fragment consisting of the VL and VH domains of a single arm of an antibody; a dAb fragment (Ward et al., Nature 341:544-546, 1989), which consists of a VH domain; and an isolated complementarity determining region (CDR), or other epitope-binding fragments of an antibody.
- scFv single-chain Fvs
- sdFv camelid antibodies
- sdFv disulfide-linked
- the portion of the CAR described comprising an antibody or antibody fragment thereof may exist in a variety of forms where the antigen binding domain is expressed as part of a contiguous polypeptide chain including, for example, a single domain antibody fragment (sdAb), a single chain antibody (scFv), a humanized antibody or bispecific antibody (Harlow et al., 1999, In: Using Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory Press, NY; Harlow et al., 1989, In: Antibodies: A Laboratory Manual, Cold Spring Harbor, New York; Houston et al., 1988, Proc. Natl. Acad. Sci. USA 85:5879-5883; Bird et al., 1988, Science 242:423-426).
- sdAb single domain antibody fragment
- scFv single chain antibody
- humanized antibody or bispecific antibody Harlow et al., 1999, In: Using Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory Press, NY; Harlow et al., 1989,
- the antigen binding domain of a CAR composition comprises an antibody fragment.
- the CAR comprises an antibody fragment that comprises a scFv.
- the precise amino acid sequence boundaries of a given CDR can be determined using any of a number of well-known schemes, including those described by Rabat et al. (1991), “Sequences of Proteins of Immunological Interest,” 5th Ed. Public Health Service, National Institutes of Health, Bethesda, MD (“Rabat” numbering scheme), ALLazikani et al., (1997) JMB 273,927-948 (“Chothia” numbering scheme), or a combination thereof.
- the two domains of the Fv fragment, VL and VH are coded for by separate genes, they can be joined, using recombinant methods, by a synthetic linker that enables them to be made as a single protein chain in which the VL and VH regions pair to form monovalent molecules (known as single chain Fv (“scFv”); see, e.g. , Bird et al. , Science 242:423-426, 1988; and Huston et al. , Proc. Natl. Acad. Sci. 85:5879-5883, 1988).
- single chain Fv single chain Fv
- Such single chain antibodies are also intended to be encompassed within the term “antigen binding fragment.” These antigen binding fragments are obtained using conventional techniques known to those of skill in the art, and the fragments are screened for utility in the same manner as are intact antibodies.
- CAR Chimeric Antigen Receptor
- a “CAR” refers to a recombinant polypeptide construct comprising at least an extracellular antigen binding domain, a transmembrane domain and a cytoplasmic signaling domain (also referred to herein as “an intracellular signaling domain”) comprising a functional signaling domain derived from a stimulatory molecule as defined below.
- the domains in the CAR polypeptide construct are in the same polypeptide chain, e.g., comprise a chimeric fusion protein.
- the domains in the CAR polypeptide construct are not contiguous with each other, e.g., are in different polypeptide chains, e.g., as provided in an RCAR.
- the cytoplasmic signaling domain comprises a primary signaling domain (e.g., a primary signaling domain of CD3-zeta). In some aspects, the cytoplasmic signaling domain further comprises one or more functional signaling domains derived from at least one costimulatory molecule as defined below. In some aspects, the costimulatory molecule is chosen from 4-1BB (i.e., CD137), CD27, ICOS, and/or CD28. In some aspects, the CAR comprises a chimeric fusion protein comprising an extracellular antigen recognition domain, a transmembrane domain and an intracellular signaling domain comprising a functional signaling domain derived from a stimulatory molecule.
- 4-1BB i.e., CD137
- CD27 CD27
- ICOS ICOS
- CD28 CD28
- the CAR comprises a chimeric fusion protein comprising an extracellular antigen recognition domain, a transmembrane domain and an intracellular signaling domain comprising a functional signaling domain derived from
- the CAR comprises a chimeric fusion protein comprising an extracellular antigen recognition domain, a transmembrane domain and an intracellular signaling domain comprising a functional signaling domain derived from a co-stimulatory molecule and a functional signaling domain derived from a stimulatory molecule.
- the CAR comprises a chimeric fusion protein comprising an extracellular antigen recognition domain, a transmembrane domain and an intracellular signaling domain comprising two functional signaling domains derived from one or more co-stimulatory molecule(s) and a functional signaling domain derived from a stimulatory molecule.
- the CAR comprises a chimeric fusion protein comprising an extracellular antigen recognition domain, a transmembrane domain and an intracellular signaling domain comprising at least two functional signaling domains derived from one or more co-stimulatory molecule(s) and a functional signaling domain derived from a stimulatory molecule.
- the CAR comprises an optional leader sequence at the amino-terminus (N-term) of the CAR fusion protein.
- the CAR further comprises a leader sequence at the N-terminus of the extracellular antigen recognition domain, wherein the leader sequence is optionally cleaved from the antigen recognition domain (e.g., an scFv) during cellular processing and localization of the CAR to the cellular membrane.
- a CAR that comprises an antigen binding domain e.g., an scFv, a single domain antibody, or TCR (e.g., a TCR alpha binding domain or TCR beta binding domain)
- X CAR a tumor marker as described herein
- BCMA CAR a CAR that comprises an antigen binding domain that targets BCMA
- the CAR can be expressed in any cell, e.g., an immune effector cell as described herein (e.g., a T cell or an NK cell).
- signaling domain refers to the functional portion of a protein which acts by transmitting information within the cell to regulate cellular activity via defined signaling pathways by generating second messengers or functioning as effectors by responding to such messengers.
- stimulation molecule refers to a molecule expressed by an immune cell (e.g., T cell, NK cell, B cell) that provides the cytoplasmic signaling sequence(s) that regulate activation of the immune cell in a stimulatory way for at least some aspect of the immune cell signaling pathway.
- the signal is a primary signal that is initiated by, for instance, binding of a TCR/CD3 complex with an MHC molecule loaded with peptide, and which leads to mediation of a T cell response, including, but not limited to, proliferation, activation, differentiation, and the like.
- a primary cytoplasmic signaling sequence (also referred to as a “primary signaling domain”) that acts in a stimulatory manner may contain a signaling motif which is known as immuno-receptor tyrosine-based activation motif or ITAM.
- an ITAM containing cytoplasmic signaling sequence examples include, but are not limited to, those derived from CD3 zeta, common FcR gamma (FCER1G), Fc gamma Rlla, FcR beta (Fc Epsilon Rib), CD3 gamma, CD3 delta, CD3 epsilon, CD79a, CD79b, DAP10, and DAP12.
- the intracellular signaling domain in any one or more CARs described herein comprises an intracellular signaling sequence, e.g., a primary signaling sequence of CD3-zeta.
- the primary signaling sequence of CD3-zeta is a human sequence, or the equivalent residues from a nonhuman species, e.g., mouse, rodent, monkey, ape, and the like.
- intracellular signaling domain refers to an intracellular portion of a molecule.
- the intracellular signaling domain generates a signal that promotes an immune effector function of the CAR containing cell, e.g., a CART cell.
- immune effector function e.g., in a CART cell, include cytolytic activity and helper activity, including the secretion of cytokines.
- the intracellular signaling domain can comprise a primary intracellular signaling domain.
- Exemplary primary intracellular signaling domains include those derived from the molecules responsible for primary stimulation, or antigen dependent simulation.
- the intracellular signaling domain can comprise a costimulatory intracellular domain.
- Exemplary costimulatory intracellular signaling domains include those derived from molecules responsible for costimulatory signals, or antigen independent stimulation.
- a primary intracellular signaling domain can comprise a cytoplasmic sequence of a T cell receptor
- a costimulatory intracellular signaling domain can comprise cytoplasmic sequence from co-receptor or costimulatory molecule.
- a primary intracellular signaling domain can comprise a signaling motif which is known as an immuno-receptor tyrosine-based activation motif or ITAM.
- ITAM containing primary cytoplasmic signaling sequences include, but are not limited to, those derived from CD3 zeta, common FcR gamma (FCER1G), Fc gamma Rlla, FcR beta (Fc Epsilon Rib), CD3 gamma, CD3 delta, CD3 epsilon, CD79a, CD79b, DAP 10, and DAP 12.
- zeta or alternatively “zeta chain,” “CD3-zeta,” or “TCR-zeta” refers to CD247.
- a “zeta stimulatory domain” or alternatively a “CD3-zeta stimulatory domain” or a “TCR-zeta stimulatory domain” refers to a stimulatory domain of CD3-zeta or a variant thereof (e.g., a molecule having mutations, e.g., point mutations, fragments, insertions, or deletions).
- the cytoplasmic domain of zeta comprises residues 52 through 164 of GenBank Acc. No. BAG36664.
- zeta or alternatively “zeta chain”, “CD3-zeta” (or “CD3zeta , CD3 zeta, or CD3z) or “TCR-zeta” is defined as the protein provided as GenBank Acc. No. BAG36664.
- zeta stimulatory domain or alternatively a “CD3-zeta stimulatory domain” or a “TCR-zeta stimulatory domain” is defined as the amino acid residues from the cytoplasmic domain of the zeta chain, or functional derivatives thereof, that are sufficient to functionally transmit an initial signal necessary for T cell activation.
- the cytoplasmic domain of zeta comprises residues 52 through 164 of GenBank Acc. No. BAG36664. 1 or the equivalent residues from a non-human species, e.g., mouse, rodent, monkey, ape, and the like, that are functional orthologs thereof.
- costimulatory molecule refers to a cognate binding partner on a T cell that specifically binds with a costimulatory ligand, thereby mediating a costimulatory response by the T cell, such as, but not limited to, proliferation.
- Costimulatory molecules are cell surface molecules other than antigen receptors or their ligands that CD3 zeta, common FcR gamma (FCER1G), Fc gamma Rlla, FcR beta (Fc Epsilon Rib), CD3 gamma, CD3 delta, CD3 epsilon, CD79a, CD79b, DAP 10, and DAP 12 contribute to an efficient immune response.
- Costimulatory molecules include, but are not limited to, an MHC class I molecule, BTLA and a Toll ligand receptor, as well as 0X40, CD27, CD28, ICAM-1, LFA- 1 (CD1 la/CD18), ICOS(CD278), and 4-1BB (CD137).
- costimulatory molecules include ICAM-1, GITR, BAFFR, HVEM (LIGHTR), SLAMF7, NKp80 (KLRF1), NKp44, NKp30, NKp46, CD160, CD19, CD4, CD8alpha, CD8beta, IL2Rbeta, IL2R gamma, IL7R alpha, ITGA4, VLA1, CD49a, ITGA4, IA4, CD49D, ITGA6, VLA-6, CD49f, ITGAD, CDl ld, ITGAE, CD103, ITGAL, CDl la, LFA-1, ITGAM, CDl lb, ITGAX, CDl lc, ITGB1, CD29, ITGB2, CD18, LFA-1, ITGB7, NKG2D, NKG2C, TNFR2, TRANCE/RANKL, DNAM1 (CD226), SLAMF4 (CD244, 2B4), CD84, CD96 (Tactile), CE
- a costimulatory intracellular signaling domain can be the intracellular portion of a costimulatory molecule.
- a costimulatory molecule can be represented in the following protein families: TNF receptor proteins, Immunoglobulin-like proteins, cytokine receptors, integrins, signaling lymphocytic activation molecules (SLAM proteins), and activating NK cell receptors.
- Examples of such molecules include CD27, CD28, 4-1BB (CD137), 0X40, GITR, CD30, CD40, ICOS, BAFFR, HVEM, ICAM-1, lymphocyte function-associated antigen- 1 (LFA-1), CD2, CD7, CD287, LIGHT, NKG2C, NKG2D, SLAMF7, NKp80, NKp30, NKp44, NKp46, CD 160, B7-H3, and a ligand that specifically binds with CD83, and the like.
- the intracellular signaling domain can comprise the entire intracellular portion, or the entire native intracellular signaling domain, of the molecule from which it is derived, or a functional fragment or derivative thereof.
- CDRs complementarity -determining domains
- VL and VH Complementary -determining regions
- the CDRs are the target proteinbinding site of the antibody chains that harbors specificity for such target protein.
- CDR1-3 Three CDRs (CDR1-3, numbered sequentially from the N-terminus) in each human VL or VH, constituting about 15- 20% of the variable domains.
- the CDRs are structurally complementary to the epitope of the target protein and are thus directly responsible for the binding specificity.
- the remaining stretches of the VL or VH, the so-called framework regions exhibit less variation in amino acid sequence (Kuby, Immunology, 4th ed., Chapter 4. W.H. Freeman & Co., New York, 2000).
- the positions of the CDRs and framework regions can be determined using various well known definitions in the art, e.g., Rabat, Chothia, international ImMunoGeneTics database (IMGT) (on the worldwide web at imgt.org), and AbM (see, e.g., Johnson et al., Nucleic Acids Res., 29:205-206 (2001); Chothia and Lesk, J. Mol. Biol., 196:901-917 (1987); Chothia et a/., Nature, 342:877-883 (1989); Chothia et al., J. Mol. Biol., 227:799-817 (1992); ALLazikani et al., J. Mol.
- contaminating polynucleotide refers to a polynucleotide not derived from a lentiviral vector.
- Contaminating polynucleotides may include, e.g., non-lentiviral polynucleotides derived from a cell in which the lentiviral vector was produced, such as chromosomal mammalian DNA (e.g., human DNA) that is not included within a transgene or other component of a lentiviral vector. “Derived from” as that term is used herein, indicates a relationship between a first and a second molecule.
- first molecule generally refers to structural similarity between the first molecule and a second molecule and does not connotate or include a process or source limitation on a first molecule that is derived from a second molecule.
- first molecule that is derived from a CD3zeta molecule
- intracellular signaling domain retains sufficient CD3zeta structure such that it has the required function, namely, the ability to generate a signal under the appropriate conditions.
- the term “prevent”, “preventing,” or “prevention” of any disease or disorder refers to the prophylactic treatment of the disease or disorder; or delaying the onset or progression of the disease or disorder.
- epitopes refers to an antibody or antigen binding fragment thereof that finds and interacts (e.g., binds) with its epitope, whether that epitope is linear or conformational.
- epitope refers to a site on an antigen to which an antibody or antigen binding fragment of the disclosure specifically binds.
- Epitopes can be formed both from contiguous amino acids or noncontiguous amino acids juxtaposed by tertiary folding of a protein. Epitopes formed from contiguous amino acids are typically retained on exposure to denaturing solvents, whereas epitopes formed by tertiary folding are typically lost on treatment with denaturing solvents.
- An epitope typically includes at least 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, or 15 amino acids in a unique spatial conformation.
- Methods of determining spatial conformation of epitopes include techniques in the art, for example, x-ray crystallography and 2-dimensional nuclear magnetic resonance (see, e.g., Epitope Mapping Protocols in Methods in Molecular Biology, Vol. 66, G. E. Morris, Ed. (1996)).
- retroviral packaging protein refers to a protein derived from a retrovirus, or a variant thereof, that assists with packaging of a nucleic acid (e.g., a viral genome) into an envelope.
- exemplary retroviral packaging proteins include gag, pol, and rev, e.g., lenti viral gag, pol, and rev, e.g., the wild-type proteins or variant thereof, e.g., sequences having at least 80%, 90%, or 95% sequence identity thereto.
- one or more retroviral packaging protein is provided as a polyprotein.
- the term “retroviral envelope protein” refers to a protein derived from a retrovirus, or a variant thereof, that can be assembled into an envelope around a nucleic acid (e.g., a viral genome).
- An exemplary retroviral envelope protein is env, e.g., wild-type or a variant thereof.
- the retroviral envelope protein is a lentiviral envelope protein, e.g., wild-type or a variant thereof.
- the retroviral envelope protein is VSV-G, e.g., wild-type or variant thereof.
- the retroviral envelop protein is pseudotyped.
- the retroviral envelope protein is from a different virus than one or more of the retroviral packaging protein or LTRs of the nucleic acid to be packaged.
- the phrases “specifically binds” and “binds” refer to a binding reaction which is determinative of the presence of a particular protein in a heterogeneous population of proteins and other biological molecules that is recognized, e.g., by a ligand with particularity.
- a ligand e.g., a protein, proteoglycan, or glycosaminoglycan
- a ligand that specifically binds to a protein will bind to the protein with a KD of less than 500 nM.
- a ligand that specifically binds to a protein will bind to the protein with a KD of up to 500 nM (e.g., between 1 pM and 500 nM).
- a ligand that does not exhibit specific binding to a protein or a domain thereof will exhibit a KD of greater than 500 nM (e.g., greater than 600 nm, 700 nM, 800 nM, 900 nM, 1 pM, 1 00 pM, 500 pM, or 1 mM) for that particular protein or domain thereof.
- a variety of assay formats may be used to determine the affinity of a ligand for a specific protein. For example, solid-phase ELISA assays are routinely used to identify ligands that specifically bind a target protein.
- subject includes human and non-human animals.
- Non-human animals include all vertebrates, e.g., mammals and non-mammals, such as non-human primates, sheep, dog, cow, chickens, amphibians, and reptiles. Except when noted, the terms “patient” or “subject” are used herein interchangeably.
- RNA or polypeptide refers to a molecule (e.g., an RNA or polypeptide) that, at an effective level, can exert a therapeutic effect on a subject.
- terapéuticaally acceptable amount or “therapeutically effective dose” interchangeably refers to an amount sufficient to effect the desired result (i.e., a reduction in tumor size, inhibition of tumor growth, prevention of metastasis, inhibition or prevention of viral, bacterial, fungal or parasitic infection).
- a therapeutically acceptable amount does not induce or cause undesirable side effects.
- a therapeutically acceptable amount induces or causes side effects but only those that are acceptable by the healthcare providers in view of a patient’s condition.
- a therapeutically acceptable amount can be determined by first administering a low dose, and then incrementally increasing that dose until the desired effect is achieved.
- a “prophylactically effective dosage,” and a “therapeutically effective dosage,” can, in some embodiments, prevent the onset of, or result in a decrease in severity of, respectively, disease symptoms, including symptoms associated with cancer.
- transfection refers to the introduction of DNA into a eukaryotic cell. Transfection may be accomplished by a variety of means including but not limited to calcium phosphate- DNA co-precipitation, DEAE-dextran-mediated transfection, polybrene-mediated transfection, electroporation, microinjection, liposome fusion, lipofection, protoplast fusion, retroviral infection, and biolistics.
- the terms “treat,” “treating,” or “treatment” of any disease or disorder refers in one embodiment, to ameliorating the disease or disorder (i.e., slowing or arresting or reducing the development of the disease or at least one of the clinical symptoms thereof).
- “treat,” “treating,” or “treatment” refers to alleviating or ameliorating at least one physical parameter including those which may not be discernible by the patient.
- “treat,” “treating,” or “treatment” refers to modulating the disease or disorder, either physically, (e.g., stabilization of a discernible symptom), physiologically, (e.g., stabilization of a physical parameter), or both.
- viral titer refers to the number of infectious vector particles, or "transducing units,” that result in the transfer of a given nucleic acid sequence from the particles into a target cell.
- Viral titer can be measured by a functional assay, such as an assay described in Xiao et al., Exp. Neurobiol. 144: 1 13-124, 1997, or Fisher et al., J. Virol. 70:520-532, 1996, the disclosures of both of which are incorporated by reference in their entirety.
- viral titer can be measured by determining the quantity of viral DNA that has integrated into a host cell genome, e.g., using polymerase chain reaction (PCR) techniques known in the art.
- PCR polymerase chain reaction
- viral vector refers to a viral particle which has a capability of introducing a nucleic acid molecule into a host.
- a viral vector carrying an exogenous gene(s) is typically packaged into an infectious virus particle via virus packaging with the aid of packaging plasmids using specific cell-lines. The infectious virus particle infects a cell to achieve expression of the exogenous gene.
- a "recombinant" viral vector refers to a viral vector constructed by gene recombinant technologies.
- a recombinant viral vector can be constructed using any suitable method, such as by transducing or transfecting a packaging cell-line with a nucleic acid encoding the viral genome and subsequently isolating newly packaged viral particles.
- lentiviral vector refers to a vector derived from at least a portion of a lentivirus genome, including, for example, a self-inactivating lentiviral vector as provided in Milone et al., Mol. Ther. 17(8): 1453-1464 (2009).
- lentivirus vectors that may be used in the clinic, include but are not limited to, e.g., the LENTIVECTOR® gene delivery technology from Oxford BioMedica, the LENTIMAXTM vector system from Lentigen and the like. Nonclinical types of lentiviral vectors are also available and would be known to one skilled in the art.
- host cells can be cultured. Exemplary types of host cells, such as human cells lacking the large T antigen, are described in more detail in the section entitled “Host cells” herein. Without wishing to be bound by theory, it is believed that host cells lacking the large T antigen can lead to manufacturing advantages compared to host cells comprising the large T antigen (see, e.g., Example 1 of PCT/IB2022/053880, hereby incorporated by reference in its entirety).
- the host cells are cultured in sequentially larger vessels (e.g., bioreactors) until sufficiently large numbers of cells are produced.
- the desired nucleic acids can be introduced into the host cells.
- the nucleic acids may be introduced by transfection, e.g., using the FectoVIR®-AAV transfection reagent, e.g., as described in the section entitled “Transfection” herein.
- the transfected nucleic acids may include a viral genome to be packaged, wherein the viral genome includes a therapeutic gene of interest and sufficient LTR sequence for packaging into a viral particle.
- Additional nucleic acids that may be introduced into the host cell include plasmids that promote packaging, e.g., plasmids encoding viral gag, pol, env, and rev.
- the pH of the culture medium may be shifted downwards before transfection, e.g., from about 7. 1 to about 6.7, e.g., as described in the section herein entitled “Culture conditions and transfection conditions.”
- the cells then begin to produce lentivirus.
- a nuclease such as benzonase may be added to the culture media, e.g., as described in the section entitled “Culture media.”
- the cell culture medium is a source of contaminating nucleic acids to the final lentiviral preparation, e.g., the culture medium may contain host cell DNA from lysed host cells. Accordingly, addition of benzonase to the cell culture medium may degrade the contaminating nucleic acids, allowing for improved purification of the lentivirus.
- lentivirus can be harvested from the host cell culture to begin purification of the lentivirus.
- harvesting of lentivirus comprises separating the supernatant or cell culture media from the cell.
- the cell is not lysed before clarification.
- the cells may be lysed, and the lysate may be clarified.
- Purification of the lentivirus from the cell culture media or cell lysate typically involves several sequential purification steps. Purification steps may include filtration (e.g., ultrafiltration) and chromatography steps.
- arginine can be added during the purification process, e.g., before or after a filtration step or a chromatography step. Addition of arginine is described, e.g., in the section entitled “Purification.” Without wishing to be bound by theory, in some embodiments, the arginine stabilizes the lentiviral vectors and/or reduces their aggregation.
- the purified lentivirus can be used for a variety of applications.
- the lentivirus can be used to deliver a gene to cells ex vivo, e.g., to generate CART cells from immune effector cells from an apheresis sample.
- the lentivirus may be administered to a subject, to deliver a gene to cells of the subject in situ.
- the lentivirus may be used for in vivo CART production.
- the lentivirus is suitable for administration in a human subject, e.g., a lentivirus encoding a CAR maybe administered to a subject allowing for introduction of the CAR encoding nucleic acid into immune effector cells in the subject’s body.
- Naturally occurring lentiviruses are a genus of viruses of the Refroviridae family, characterized by a long incubation period. Lentiviruses can typically deliver a significant amount of genetic information into the DNA of the host cell.
- lentiviruses include HIV (human immunodeficiency virus; including HIV type 1, and HIV type 2), the etiologic agent of the human acquired immunodeficiency syndrome (AIDS); visna-maedi, which causes encephalitis (visna) or pneumonia (maedi) in sheep, the caprine arthritis-encephalitis virus, which causes immune deficiency, arthritis, and encephalopathy in goats; equine infectious anemia virus, which causes autoimmune hemolytic anemia, and encephalopathy in horses; feline immunodeficiency virus (FIV), which causes immune deficiency in cats; bovine immune deficiency virus (BIV), which causes lymphadenopathy, lymphocytosis, and possibly central nervous system infection in cattle; and
- viruses Diseases caused by these viruses are characterized by a long incubation period and protracted course. Usually, the viruses latently infect monocytes and macrophages, from which they spread to other cells. HIV, FIV, and SIV also readily infect T lymphocytes (i.e., T-cells).
- the lentivirus or lentiviral vector disclosed herein may include a nucleic acid, e.g., a transgene, such as a protein-encoding transgene.
- the nucleic acid may comprise a transgene, e.g., as described in the section herein entitled “Transgene”.
- the transgene may be operably linked to a promoter sequence.
- the nucleic acid may also comprise one or more (e.g., two) LTR sequences. Without wishing to be bound by theory, the LTRs may promote insertion of the transgene and promoter into a host cell genome.
- the LTR sequences may comprise wild-type lentiviral LTR sequences or variants thereof.
- the 3’ LTR may comprise a deletion that renders the virus self-inactivating after integration.
- the 5 ’ LTR may be a chimeric LTR.
- the transgene can be integrated into the chromosomal DNA of a target cell.
- Exemplary transgenes include those that encode a chimeric antigen receptor (CAR).
- the CAR may include several domains, such as an antigen binding domain, a transmembrane domain, and one or more signaling domains.
- the signaling domains may contain one or more primary signaling domains (such as a CD3-zeta stimulatory domain) and/or one or more costimulatory signaling domains (such as CD27, CD28, 4-1 BB (CD137), 0X40, GITR, CD30, CD40, ICOS, BAFFR, HVEM, ICAM-1 , lymphocyte function-associated antigen- 1 (LFA-1), CD2, CD7, CD287, LIGHT, NKG2C, NKG2D, SLAMF7, NKp80, NKp30, NKp44, NKp46, CD 160, B7-H3, or a ligand that specifically binds with CD83.
- primary signaling domains such as a CD3-zet
- the transgene may encode an antigenbinding domain (such as a scFv) that binds a particular target protein or carbohydrate.
- antigens include CD19, CD123, CD22, CD30, CD171 , CS-1, C-type lectin-like molecule-1, CD33, epidermal growth factor receptor variant III (EGFRvlll), ganglioside G2 (GD2), ganglioside GD3, TNF receptor family member B cell maturation (BCMA), Tn antigen ((Tn Ag) or (GalNAca-Ser/Thr)), prostate-specific membrane antigen (PSMA), Receptor tyrosine kinase-like orphan receptor 1 (R0R1), Fms-Like Tyrosine Kinase 3 (FLT3), Tumor- associated glycoprotein 72 (TAG 72), CD38, CD44v6, Carcinoembryonic antigen (CE)
- a lentiviral vector described herein comprises more than one transgene, e.g., a first transgene encoding a first CAR, e.g., a CD 19 CAR and a second transgene encoding a second CAR, e.g., a CD22 CAR.
- a dual CAR lentiviral vector described herein encodes two different CARs, e.g., a CD19 CAR and a CD22 CAR.
- the two CARs are part of a single open reading frame and are separated by a protease cleavage site, e.g., a self-cleavage site, e.g., a P2A site.
- the open reading frame encodes, from N-terminal to C-terminal, a first leader sequence, a first scFv (e.g., that binds CD22), optionally a first hinge domain, a first transmembrane domain, a first costimulatory domain (e.g., 4-1BB), a first primary signaling domain (e.g., CD3-zeta), a protease cleavage site (e.g., P2A), a second leader sequence, a second scFv (e.g., that binds CD19), optionally a second hinge domain, a second transmembrane domain, a second costimulatory domain (e.g., 4-1BB), and a second primary signaling domain (e.g., CD3-zeta).
- a first leader sequence e.g., binds CD22
- a first hinge domain e.g., binds CD22
- a first transmembrane domain
- first and second leader sequences have the same sequence.
- first and second hinge domains have the same sequence.
- first and second transmembrane domains have the same sequence.
- first and second costimulatory domains have the same sequence.
- first and second primary signaling domains have the same sequence.
- CAR targets Additional CARs that can be encoded by transgene described herein are provided, e.g., in the section herein entitled “CAR targets”.
- a lentiviral vector described herein encodes a siRNA or shRNA that targets a nucleic acid in an immune effector cell.
- the siRNA or shRNA may target a nucleic acid encoding a TCR and/or HLA, and/or an inhibitory molecule (e.g., PD1, PD-L1, PD-L2, CTLA4, TIM3, CEACAM (e.g., CEACAM-1, CEACAM-3 and/or CEACAM-5), LAG3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4, CD80, CD86, B7-H3 (CD276), B7-H4 (VTCN1), HVEM (TNFRSF14 or CD270), KIR, A2aR, MHC class I, MHC class II, GAL9, adenosine, and TGF beta), in a T cell.
- an inhibitory molecule e.g., PD1, PD-L1, PD-L2, CTLA4,
- siRNA and shRNAs are described, e.g., in paragraphs 649 and 650 of International Application WO2015/142675, filed March 13, 2015, which is incorporated by reference in its entirety.
- These nucleic acids can also be targeted, for example, using a CRISPER system, Zinc finger nucleases, or TALENs.
- the immune effector cell may be autologous or allogeneic to the subject to be treated.
- a lentiviral vector described herein comprises or encodes one or more inhibitor of a methylcytosine dioxygenase gene (e.g., Tetl, Tet2, or Tet3).
- a methylcytosine dioxygenase gene e.g., Tetl, Tet2, or Tet3
- Disruption of a single allele of a Tet gene e.g., a Tetl, Tet2, or Tet3 leads to decreased total levels of 5-hydroxymethylcytosine in association with enhanced proliferation, regulation of effector cytokine production and degranulation, and thereby increases CAR T cell proliferation and/or function.
- the expression and/or function of Tet2 in the cell has been reduced or eliminated.
- the inhibitor of Tetl, Tet2 and/or Tet3, is a siRNA or shRNA specific for Tetl, Tet2, Tet3, or nucleic acid encoding the siRNA or shRNA.
- the siRNA or shRNA comprises a sequence complementary to a sequence of a Tet2 mRNA, e.g., comprises a target sequence of shRNA listed in Table 4 of WO2017/049166, which application is herein incorporated by reference in its entirety, including Table 4.
- the inhibitor of Tetl, Tet2 and/or Tet3, is (1) a gene editing system targeted to one or more sites within the gene encoding Tetl, Tet2 and/or Tet3, or its regulatory elements, e.g., Tet2, or its regulatory elements; (2) nucleic acid encoding one or more components of the gene editing system; or (3) combinations thereof.
- the gene editing system is selected from the group consisting of: a CRISPR/Cas9 system, a zinc finger nuclease system, a TALEN system and a meganuclease system.
- a lentiviral vector described here comprises a transgene, e.g., a transgene encoding a chimeric antigen receptor (CAR) and further comprises a siRNA or shRNA that targets a nucleic acid in an immune effector cell.
- a transgene e.g., a transgene encoding a chimeric antigen receptor (CAR) and further comprises a siRNA or shRNA that targets a nucleic acid in an immune effector cell.
- CAR chimeric antigen receptor
- the lentiviral vectors are characterized by a hydrodynamic radius of 90- 200 nm as measured by dynamic light scattering (DLS).
- the lentiviral vectors may maintain a hydrodynamic radius of 90-200 nm within a temperature range of from 25°C to 37°C.
- the lentiviral vectors are characterized by a poly dispersity of from 10% to 25%.
- the lentiviral vectors may maintain a polydispersity of from 10% to 25% within a temperature range of from 25°C to 37°C.
- the lentiviral vectors maintain a concentration after 3, 6, or 9 freeze/thaw cycles of from about 70% to about 100% relative to the concentration of the lentiviral vector in the aqueous composition prior to the freeze/thaw cycles, wherein each of the freeze/thaw cycles includes freezing the aqueous composition and subsequently allowing the aqueous composition to thaw at room temperature.
- a lentivirus prepared, purified or stored using any of the methods or formulations disclosed herein may have lower vector copy number (VCN), e.g., at least 5%, at least 10%, at least 20%, at least 30%, at least 50%, at least 60% lower VCN compared to a lentivirus not produced, purified or stored by the methods or in formulations as described herein, e.g., when tested at MOI of 1.
- VCN vector copy number
- a packaging system can be used to package a nucleic acid, e.g., an RNA encoding a transgene into a lentiviral vector.
- the systems and methods described herein may comprise, e.g., a lentiviral packaging system comprising at least one plasmid adapted for the production of a lentiviral vector, e.g., a lentiviral vector optionally comprising a transgene.
- lentiviral components useful for the production of a lentiviral vector are known in the art. See for example Zufferey et al., 1997, Nat. Biotechnol. 15:871-875 and Dull et al, 1998, J. Virol.
- a lentiviral packaging system comprising one or more nucleic acids (e.g., plasmids), e.g., at least one, two, three, or four plasmids, wherein one plasmid encodes a retroviral envelope protein (Env plasmid), one plasmid encodes one or more retroviral packaging proteins, e.g., Gag and Pol proteins (packaging plasmid or Gag-Pol plasmid), one plasmid encodes a lentiviral Rev protein (Rev plasmid) and one or more plasmids comprising at least one transgene of interest (TOI) expression cassette.
- nucleic acids e.g., plasmids
- plasmids e.g., at least one, two, three, or four plasmids
- one plasmid encodes a retroviral envelope protein (Env plasmid)
- one plasmid encodes one or more retroviral packaging
- the lentiviral packaging system further comprises, or a method described herein comprises use of, at least one, two, three, or four plasmids. In some embodiments, the lentiviral packaging system further comprises, or a method described herein comprises use of, a fifth plasmid. In certain embodiments, a method described herein comprises transfecting five plasmids into the host cell, wherein the fifth plasmid does not encode a protein of the lentiviral vector packaging system.
- the lentiviral packaging system comprises one or more nucleic acids (e.g., plasmids), e.g., five plasmids, wherein one plasmid encodes an expression vector, one plasmid encodes a Tat (e.g., pcDNATat), one plasmid encodes a Rev protein (e.g., pHCMV- Rev), one plasmid encodes a gagpol (e.g., pHCMV-gagpol), and one plasmid encodes VSV-G (e.g., pVSVG), e.g., as described in Rout-Pitt et al., J Biol. Methods 5(2): 1-9, 2018).
- plasmids e.g., five plasmids
- one plasmid encodes an expression vector
- one plasmid encodes a Tat (e.g., pcDNATat)
- one plasmid encodes a Rev protein (e
- a plasmid may comprise a dual gene expression cassette, e.g., a bicistronic cassette, e.g., a bicistronic construct encoding two transgenes of interest.
- the first transgene of interest encodes a first CAR, e.g., a CD 19 CAR
- the second transgene of interest encodes a second CAR, e.g., a CD22 CAR.
- the retroviral packaging proteins are derived from a lentivirus, e.g., lentiviral packaging proteins, e.g., lentiviral gag and pol proteins.
- the lentiviral gag protein is a wild-type lentiviral gag protein, and in other embodiments it has one or more sequence modifications relative to the wild-type sequence.
- the lentiviral pol protein is a wild-type lentiviral pol protein, and in other embodiments it has one or more sequence modifications relative to the wild-type sequence.
- the rev protein is a wild-type rev protein, and in other embodiments it has one or more sequence modifications relative to the wild-type sequence.
- the lentiviral vector may be a pseudotyped vector, comprising a modified envelope protein, e.g., an envelope protein derived from a different virus or a chimeric envelope protein, e.g., the Env plasmid may encode a VSV-G Env protein, e.g., a wild type VSV-G protein or a modified variant.
- a modified envelope protein e.g., an envelope protein derived from a different virus or a chimeric envelope protein
- the Env plasmid may encode a VSV-G Env protein, e.g., a wild type VSV-G protein or a modified variant.
- a lentiviral vector is generated using a packaging system comprising pMDLgpRRE, pRSV-Rev and pMD.G plasmids (Dull et al., supra), but using a kanamycin resistance marker, e.g., a marker that confers resistance to both kanamycin and neomycin, e.g., neomycin phosphotransferase II instead of an ampicillin gene.
- kanamycin resistance marker e.g., a marker that confers resistance to both kanamycin and neomycin, e.g., neomycin phosphotransferase II instead of an ampicillin gene.
- a system described herein comprises a transfer vector comprising a kanamycin resistance marker, e.g., a marker that confers resistance to both kanamycin and neomycin, e.g., neomycin phosphotransferase II, e.g., instead of an ampicillin gene.
- the transfer vector comprises sequence from, e.g., a pELPS construct as disclosed in WO2017087861A or Milone et al., Mol. Ther. 17(8): 1453-1464, 2009, each of which is incorporated by reference herein in its entirety.
- the therapeutic protein is encoded on a self-inactivating transfer vector that comprises one or more of, e.g., all of, lentiviral 5’ LTR (e.g., a truncated lentiviral 5’ LTR), lentiviral 3’ LTR, cPPT, and WPRE.
- lentiviral 5’ LTR e.g., a truncated lentiviral 5’ LTR
- lentiviral 3’ LTR e.g., a truncated lentiviral 5’ LTR
- cPPT lentiviral 3’ LTR
- the transfer vector lacks one or more of, e.g., all of: a promoter active in bacteria (e.g., lacking all of a T7 promoter, a T3 promoter, and a lac promoter), M13 primer binding site (e.g., lacking both an M13 forward primer binding site and an M13 reverse primer binding site), a phage origin (e.g., fl ori), and a fluorescent protein-encoding gene (e.g., a GFP, e.g., EGFP).
- the transfer vector lacks both of a CAP binding site and lac operator.
- the transfer vector comprises pELPS construct as disclosed in WO2017087861, except that the transfer vector lacks a T7 promoter, an M13 forward primer binding site, an fl ori, a CAP binding site, an IPTG inducible promoter, a lac operator, an M13 reverse primer binding site, a T3 promoter, and EGFP wherein optionally the transfer vector encodes a therapeutic protein, e.g., a CAR.
- the transfer vector has one or more of the following properties: (a) is more stable than an otherwise similar control transfer vector, (b) results in lower cell toxicity than an otherwise similar control transfer vector, or (c) results in a lower vector copy number (VCN) when integrated into target cells, e.g., as described herein.
- the control transfer vector comprises a T7 promoter, an M13 forward primer binding site, an fl ori, a CAP binding site, an IPTG inducible promoter, a lac operator, an M13 reverse primer binding site, and a T3 promoter.
- the gene expression cassette encodes a protein, e.g., a chimeric antigen receptor (CAR). In some embodiments, the gene expression cassette encodes two proteins, e.g., a first CAR and a second CAR. Exemplary transgenes suitable for a gene expression cassette are described in the current disclosure.
- the different functions for production of a lentiviral vector are provided to a plurality of host cells, e.g., mammalian cells, e.g., HEK293 cells, e.g., Expi293F cells (e.g., plurality of Expi293F cells growing in suspension under serum-free conditions) by transfection, e.g., transient or stable transfection, of a lentiviral packaging system adapted for producing lentiviral vectors.
- host cells e.g., mammalian cells, e.g., HEK293 cells, e.g., Expi293F cells (e.g., plurality of Expi293F cells growing in suspension under serum-free conditions)
- transfection e.g., transient or stable transfection
- At least 25%, at least 30%, at least 35%, at least 40%, at least 50%, at least 60%, at least 70%, at least 80%, at least 90%, at least 95% of host cells, e.g., HEK293 cells, e.g., Expi293F cells are transfected. Methods for transfection or infection are well known by those of skill in the art. In some embodiments, at least 0.3pg, at least 0.4pg, at least 0.5pg, at least 0.6pg, at least 0.7pg, at least 0.8pg cells, at least 0.9pg, or at least 1.0 pg of lentiviral packaging system is provided per million cells for transfection.
- a transfection reagent is used for transfecting the host cells, e.g., mammalian cells, e.g., HEK293 cells, e.g., Expi293F cells.
- a transfection reagent is used.
- Transfection reagents are well known in the art and are available from commercial suppliers. Examples of transfection reagents include but are not limited to, LipofectamineTM (Invitrogen), Polifectamine, LentiTran (Origene), PEIpro® (Polyplus), FectoVIR® -AAV (Polyplus), and ProFection® (Promega).
- the transfection reagent e.g., FectoVIR® -AAV is used at a level of 0. 1 pl, 02. pl, 0.3pl, 0.4 pl, 0.5 pl, 0.6 pl, 0.7 pl, 0.8 pl, 0.9 pl, or 1.0 pl per million cells.
- the packaging system and the transfection reagent, e.g., FectoVIR® -AAV are used at ratio of about 1:0.5, 1:0.75, 1: 1, 1: 1.5, or 1:2, or any range therebetween, for transfection.
- the transfection reagent comprises a synthetic transfection reagent. Synthetic transfection reagents include those which are chemical-based and/or free of animal components.
- the transfection reagent comprises FectoVIR® -AAV transfection reagent. FectoVIR® -AAV can be obtained, e.g., from Polyplus (850 bd Sebastien Brant, 67400 Illkirch, FRANCE; 1251 Ave of the Americas; 3rd Fl, New York; NY 10020 USA). FectoVIR® -AAV is a synthetic, chemical-based, animal-free transfection reagent.
- the cells are at a density of about 0.5xl0 6 cells/mL - IxlO 7 cells/mL, 1x10 s cells/mL - 6x10 s cells/mL, 1x10 s cells/mL - 5xl0 6 cells/mL, 1.50xl0 6 cells/mL - 2.50xl0 6 cells/mL, 2.0 xlO 6 cells/mL - 3.0 xlO 6 cells/mL, 2.0 xlO 6 cells/mL - 2.5 xlO 6 cells/mL.
- the cell population has a viability of at least about 80%, 90%, or 95%.
- the PP/IP (physical particle/infectious particle) ratio is less than 500, 700, 900, 1000, 1100, 1200, 1300, 1400, 1500, 1600, 1700, 1800, 1900, 2000 after transfection.
- a suitable host cell is a eukaryotic cell, e.g., a mammalian cell.
- the mammalian cells may be genetically modified mammalian cells for expressing a virus, e.g., a lentivirus, e.g., a lentiviral vector or a lentivirus of interest.
- a number of mammalian cell lines are suitable host cells for recombinant expression of viruses.
- Mammalian host cell lines include, for example, COS, PER.C6, TM4, VERO, MDCK, BRL-3A, W138, Hep G2, MMT, MRC 5, FS4, CHO, 293T, A431, 3T3, CV-1, C3H10T1/2, Colo205, HEK293, HeLa, L cells, BHK, HL-60, FRhL-2, U937, HaK, Jurkat cells, Rat2, BaF3, 32D, FDCP-1, PC12, Mix, murine myelomas (e.g., SP2/0 and NSO) and C2C12 cells, as well as transformed primate cell lines, hybridomas, normal diploid cells, and cell strains derived from in vitro culture of primary tissue and primary explants.
- murine myelomas e.g., SP2/0 and NSO
- C2C12 cells as well as transformed primate cell lines, hybridomas, normal diploid cells, and cell strains derived from in vitro culture of
- the host cell is a HEK293 cell, including a cell derived from HEK293 cells, e.g., 293F cells, e.g., Expi293F cells.
- at least 80%, at least 85%, at least 90%, at least 90%, at least 95% of host cells in a culture express a large T antigen, e.g., a polyomaviral large T antigen, e.g., a SV40 large T antigen, e.g., a mutant SV40 large T antigen.
- at least 99%, at least 98%, at least 97%, at least 96%, at least 95% of the host cells in a culture do not express a large T cell antigen.
- the host cell is suitable for growing in suspension.
- Eukaryotic cells e.g., mammalian cells, e.g., HEK293 cells, e.g., Expi293F cells may be cultured as non-anchorage dependent cells growing freely in suspension throughout the bulk of the culture; or as anchorage-dependent cells requiring attachment to a solid substrate for their propagation (e.g., as a monolayer).
- a microcarrier system may be used to accommodate cell growth.
- the microcarrier system may comprise a suspension culture, e.g., a large-scale suspension culture.
- the suspension culture may be operated in open or closed systems, e.g., batch or fed-batch closed systems.
- nutrients are not added, and waste products are not removed through the duration of culture.
- nutrients are continuously fed into the system to prolong the growth cycle although cells, products, by products, and waste products, including toxic metabolites, are not removed.
- the culture system may be an open, e.g., a continuous system, e.g., a perfusion system or a chemostat system.
- the system may comprise one or more cell retention device.
- Cell retention devices may include, for example, microcarriers, fine mesh spin filters, hollow fibers, flat plate membrane filters, settling tubes, ultrasonic cell retention devices, and the like.
- the concentration of cells in the bioreactor is higher than the concentration of cells present the supernatant harvested from the bioreactor. In some embodiments, the concentration of cells in the bioreactor is substantially identical than the supernatant harvested from the bioreactor.
- Continuous fermentation process a defined media often is continuously added to a bioreactor while an equal amount of culture volume is removed simultaneously for product recovery.
- Continuous cultures generally maintain cells in the log phase of growth at a constant cell density.
- Continuous or semi- continuous culture methods permit the modulation of one factor or any number of factors that affect cell growth or end product concentration. For example, an approach may limit the carbon source and allow all other parameters to moderate metabolism.
- a number of factors affecting growth may be altered continuously while the cell concentration, measured by media turbidity, is kept constant.
- Continuous systems often maintain steady state growth and thus the cell growth rate often is balanced against cell loss due to media being drawn off the culture. Methods of modulating nutrients and growth factors for continuous culture processes are known and a variety of methods are known in the art.
- a culture of suspension cells comprises only cells that are in suspension.
- a culture of suspension cells may comprise a small number (e.g., less than 1%) of cells that adhere, e.g., transiently, to a surface.
- Cell culture may refer to any in vitro culture of cells. Included within this term are continuous cell lines (e.g., with an immortal phenotype), primary cell cultures, finite cell lines (e.g., non-transformed cells), and any other cell population maintained in vitro.
- a system or method described herein makes uses of packaging cells or a packaging cell line for production of a viral vector.
- the cell line may be stably transfected with elements for production of the lentiviral vector, for example retroviral packaging proteins and retroviral envelope protein.
- packaging cells typically contain one or more expression cassettes which are capable of expressing viral proteins (such as gag, pol and env) but the expression cassettes do not contain a packaging signal.
- a packaging cell may be a cell cultured in vitro.
- a packaging cell line may be utilized to create producer cell lines for production of the lentiviral particles, e.g., by providing at least one plasmid comprising at least one transgene of interest (TOI) expression cassette.
- TOI transgene of interest
- a producer cell transiently expresses a plasmid (e.g., a transfer plasmid) encoding a therapeutic effector and comprising sufficient LTR sequence to allow for packaging of RNA comprising the LTR(s) into a viral vector.
- a producer cell line stably expresses an expression cassette encoding a therapeutic effector and comprising sufficient LTR sequence to allow for packaging of RNA comprising the LTR(s) into a viral vector.
- the methods of the current disclosure may be carried out using any media suitable (e.g., supports cell growth and maintenance under the conditions of the current disclosure) for culturing eukaryotic cells, e.g., mammalian cells, e.g., HEK293 cells, e.g., Expi293F cells.
- eukaryotic cells e.g., mammalian cells, e.g., HEK293 cells, e.g., Expi293F cells.
- cell culture medium and “culture medium” (or simply “medium”) refer to a nutrient solution used for growing eukaryote cells e.g., mammalian cells, e.g., HEK293 cells, e.g., Expi293F cells, that typically provides at least one component from one or more of the following categories: (1) salts (e.g., sodium, potassium, magnesium, calcium, etc.) contributing to the osmolality of the medium; (2) an energy source, usually in the form of a carbohydrate such as glucose; (3) all essential amino acids, and usually the basic set of twenty amino acids; (4) vitamins and/or other organic compounds required at low concentrations; and (5) trace elements, where trace elements are defined as inorganic compounds that are typically required at very low concentrations, usually in the micromolar range.
- salts e.g., sodium, potassium, magnesium, calcium, etc.
- an energy source usually in the form of a carbohydrate such as glucose
- all essential amino acids and usually the basic set
- compositions of such media are known in the art (see, e.g., Mather, J. P., et al. (1999) “Culture media, animal cells, large scale production,” Encyclopedia of Bioprocess Technology: Fermentation, Biocatalysis, and Bioseparation, Vol. 2:777-785, hereby incorporated herein by reference in their entirety.)
- the nutrient solution may optionally be supplemented with one or more of the components from any of the following categories: (a) animal serum; (b) hormones and other growth factors such as, for example, insulin, transferrin, and epidermal growth factor; and (c) hydrolysates of plant, yeast, and/or tissues, including protein hydrolysates thereof.
- the culture media may comprise serum, e.g., fetal bovine serum (FBS).
- FBS fetal bovine serum
- the culture media is serum free.
- the culture media is chemically defined, e.g., medium lacking animal-derived components.
- animal-derived components are any components that are produced in an intact animal (such as, e.g., proteins isolated and purified from serum), or produced using components produced in an intact animal (such as, e.g., an amino acid made by using an enzyme isolated and purified from an animal to hydrolyze a plant source material).
- a protein which has the sequence of an animal protein i.e., has a genomic origin in an animal
- which is produced in vitro in cell culture such as, e.g., in a recombinant yeast or bacterial cells or in an established continuous eukaryote cell line, recombinant or not
- using media lacking components produced in, or isolated and purified from, an intact animal is not an “animal -derived” component.
- Chemically defined media are media in which all components have a known chemical structure. Chemically -defined medium are available from commercial suppliers, such as, for example, Sigma, ThermoFisher, Invifrogen, JRH Biosciences, and Gibco. In some embodiments, the media is FreeStyleTM 293 Expression Medium. In some embodiments, a concentrated serum may be used, e.g., medium that contains higher concentration of nutrients than is normally necessary and normally provided to a growing culture. In some embodiments, the medium may contain an amino acid(s) derived from any source or method known in the art.
- an enzyme e.g., a nuclease, e.g., an endonuclease, e.g., a recombinant endonuclease, e.g., a Benzonase® may be added in the culture media.
- a nuclease e.g., an endonuclease, e.g., a recombinant endonuclease, e.g., a Benzonase®
- an enzyme e.g., a nuclease, e.g., an endonuclease, e.g., a recombinant endonuclease, e.g., a Benzonase® may be added in the culture media.
- between 2U/mL and lOU/mL, between 10 U/mL and 20 U/mL, between 20 U/mL and 30 U/mL, between 30 U/mL and 40 U/mL, between 40 U/mL and 50 U/mL, or between 50 U/mL and 60 U/mL of Benzonase® is added.
- the Benzonase® is added after at a time about 5-40, 10-40, 10-30, 20-30, or about 20 hours or about 24 hours after transfecting the host cells, e.g., Expi293F cells.
- the benzonase is added at a concentration of 3-7 U/mL (e.g., about 5 U/mL) at 20-30 hours (e.g., about 24 hours) after transfecting the host cells. In some embodiments, the benzonase is added at a concentration of 3-7 U/mL (e.g. about 5 U/mL) at 1-5 hours (e.g., about 3 hours) after transfecting the host cells. In some embodiments, the benzonase is added at a concentration of 3-7 U/mL (e.g. about 5 U/mL) at 4-8 hours (e.g., about 6 hours) after transfecting the host cells.
- 3-7 U/mL e.g., about 5 U/mL
- 4-8 hours e.g., about 6 hours
- the benzonase is added at a concentration of 12-18 U/mL (e.g. about 15 U/mL) at 1-5 hours (e.g., about 3 hours) after transfecting the host cells. In some embodiments, the benzonase is added at a concentration of 12-18 U/mL (e.g. about 15 U/mL) at 4-8 hours (e.g., about 6 hours) after transfecting the host cells. In some embodiments, the benzonase is added at a concentration of 12-18 U/mL (e.g. about 15 U/mL) at 20-30 hours (e.g., about 24 hours) after transfecting the host cells.
- the benzonase is added at a concentration of 20-30 U/mL (e.g. about 25 U/mL) at 1-5 hours (e.g., about 3 hours) after transfecting the host cells. In some embodiments, the benzonase is added at a concentration of 20-30 U/mL (e.g. about 25 U/mL) at 4-8 hours (e.g., about 6 hours) after transfecting the host cells. In some embodiments, the benzonase is added at a concentration of 20-30 U/mL (e.g. about 25 U/mL) at 20-30 hours (e.g., about 24 hours) after transfecting the host cells.
- the benzonase is added at a concentration of 40-60 U/mL (e.g. about 50 U/mL) at 1-5 hours (e.g., about 3 hours) after transfecting the host cells. In some embodiments, the benzonase is added at a concentration of 40-60 U/mL (e.g. about 50 U/mL) at 4-8 hours (e.g., about 6 hours) after transfecting the host cells. In some embodiments, the benzonase is added at a concentration of 40-60 U/mL (e.g. about 50 U/mL) at 20-30 hours (e.g., about 24 hours) after transfecting the host cells.
- the benzonase is added at a concentration of 5 U/mL at about 3 hours after transfecting the host cells. In some embodiments, the benzonase is added at a concentration of 15 U/mL at about 3 hours after transfecting the host cells. In some embodiments, the benzonase is added at a concentration of 25 U/mL at about 3 hours after transfecting the host cells. In some embodiments, the benzonase is added at a concentration of 50 U/mL at about 3 hours after transfecting the host cells. In some embodiments, the benzonase is added at a concentration of 5 U/mL at about 6 hours after transfecting the host cells.
- the benzonase is added at a concentration of 15 U/mL at about 6 hours after transfecting the host cells. In some embodiments, the benzonase is added at a concentration of 25 U/mL at about 6 hours after transfecting the host cells. In some embodiments, the benzonase is added at a concentration of 50 U/mL at about 6 hours after transfecting the host cells. In some embodiments, the benzonase is added at a concentration of 5 U/mL at about 24 hours after transfecting the host cells. In some embodiments, the benzonase is added at a concentration of 15 U/mL at about 24 hours after transfecting the host cells.
- the benzonase is added at a concentration of 25 U/mL at about 24 hours after transfecting the host cells. In some embodiments, the benzonase is added at a concentration of 50 U/mL at about 24 hours after transfecting the host cells.
- a salt e.g., MgCL is added to the Benzonase®, e.g., in a concentration at about 1-5 mM, 1-3 mM, or about 2 mM.
- the methods disclosed herein may comprise addition of Benzonase® in production and/or purification process.
- a chemical compound may be added to the media to influence culture growth, e.g., inhibition of proliferation, induction of differentiation and induction or repression of gene expression.
- the chemical compound is sodium butyrate.
- a cell culture medium described herein comprises sodium butyrate.
- Culture conditions can include any culture conditions suitable for maintaining a cell (e.g., in a static or proliferative state).
- culture conditions can include several parameters, including without limitation, temperature, oxygen content, nutrient content (e.g., glucose content), pH (e.g., increasing or decreasing pH), agitation level (e.g., rotations per minute), gas flow rate (e.g., air, oxygen, nitrogen gas), redox potential, cell density (e.g. , optical density), cell viability and the like.
- a change in culture conditions can comprise an alteration, modification or shift of one or more culture parameters.
- a change in culture condition e.g., increasing or decreasing pH is introduced at a certain time during the culture, e.g., before transfection.
- the pH is modified, e.g., adjusted to about 6.0 - 6.8, e.g., 6.2 - 6.8, e.g., 6.4 - 6.8, e.g., 6.7- 6.75 before transfection with a lentiviral packaging system.
- the methods of the disclosure may be carried out in a small cell culture, e.g., in a laboratory scale, or in a large-scale culture, e.g., in industrial scale.
- the methods may be carried out in an appropriate culture unit, e.g., a culture flask or a bioreactor.
- the bioreactor can be of any size as long as it is useful for culturing cells, e.g., mammalian cells.
- the methods of this disclosure are highly scalable, e.g., the plurality of mammalian cells is in a scaled culture (e.g., at least 1 L, at least 2 L, at least 5 L, at least 10 L, at least 15 L, at least 20 L yields a number of transducing units per ml culture that is no less than 30%, 40%, 50%, 60%, 70%, or 80% the number of transducing units per ml culture in an otherwise similar small-scale culture, e.g., 100 ml, 200 ml, 300 ml, 400 ml, 500 ml.
- a scaled culture e.g., at least 1 L, at least 2 L, at least 5 L, at least 10 L, at least 15 L, at least 20 L yields a number of transducing units per ml culture that is no less than 30%, 40%, 50%, 60%, 70%, or 80% the number of transducing units per ml culture in an otherwise similar small-scale culture, e.g.,
- the scale culturing i.e., with culture volumes greater than 50 L
- the internal conditions of the culture unit including but not limited to pH, pOi. and temperature, are typically controlled during the culturing period.
- a production culture unit refers to the final culture unit used in the production of the polypeptide, virus, and/or any other product of interest.
- the volume of a large-scale production culture unit is generally greater than about 50 liters, and may be about 100, about 200, about 300, about 500, about 800, about 1000, about 2500, about 5000, about 8000, about 10,000, about 12,0000 L or more, or any intermediate volume.
- a suitable culture unit or production culture unit may be composed of (i.e., constructed of) any material that is suitable for holding cell cultures suspended in media under the culture conditions contemplated herein, and one that is conducive to mammalian cell, e.g., HEK293 cells, e.g., Expi293F cell growth and viability.
- suitable materials include, without limitation, glass, plastic, and/or metal.
- the material(s) do not interfere, or do not significantly or do not substantially interfere, with expression and/or stability of the desired product, e.g., the lentiviral vector.
- the cell culture process is operated in more than one distinct culture units, such as using one or more seed culture unit(s) followed by use of the production culture unit.
- the process involves transferring the propagated seed culture from one or more seed culture unit to a large production unit.
- expansion of the cells to the production culture unit and the production phase may be accomplished in one physical culture unit, e.g., the cells may be expanded to a final production scale and the process switched to production conditions.
- the spent medium is harvested at the end of culture period for down-stream processing of the lentivirus or lentiviral vector. In some embodiments, harvest may be collected after 24 hours, after 48 hours, after 72 hours, after 96 hours, or after 120 hours post-transfection.
- down-stream processing comprises purification, formulation and/or longterm storage of the lentivirus.
- the viral harvest collected at the end of culture period comprises lentivirus, at a concentration of, e.g., from about 5 x 10 6 transducing units per milliliter (TU/mL) to about 6 x 10 9 TU/mL (e.g., 5xl0 6 TU/mL, 5.5 x 10 6 TU/mL, 6 x 10 6 TU/mL , 6.5 x 10 6 TU/mL, 7 x 10 6 TU/mL, 7.5 x 10 6 TU/mL, 8 x 10 6 TU/mL, 8.5 x 10 6 TU/mL, 9 x 10 6 TU/mL, 9.5 x
- the viral harvest collected at the end of culture period comprises lentivirus, at the end of culture period.
- the viral harvest collected at the end of culture period comprises lentivirus, at a concentration of 5xl0 6 TU/mL - 6 x 10 6 TU/mL, 6 x 10 6 TU/mL - 7 x 10 6 TU/mL, 7 x 10 6 TU/mL - 8 x 10 6 TU/mL, 8 x 10 6 TU/mL - 9 x 10 6 TU/mL, 9 x 10 6 TU/mL - 1 x 10 7 TU/mL, 1 x 10 7 TU/mL - 2 x 10 7 TU/mL, 2 x 10 7 TU/mL - 3 x 10 7 TU/mL, 3 x 10 7 TU/mL - 4x 10 7 TU/mL , 4 x 10 7 TU/mL - 5 x 10 7 TU/mL, 5 x 10 7 TU/mL - 6 x 10 7 TU/mL
- the disclosure provides processes for purifying lentiviral vectors with improved efficiency, e.g., such that higher quantities of lentiviral vector are recovered.
- at least one step in the purification process comprises adding an agent, e.g., an amino acid or a salt thereof, e.g., an arginine or a salt thereof, e.g., arginine-HCl to the purification intermediate composition (an intermediate composition comprising a buffer before completion of purification) before further purification, e.g., centrifugation, filtration, or chromatography, to improve the purification process.
- filtration may refer to but are not limited to flow filtration, depth filtration, tangential flow filtration.
- chromatography may include but are not limited to Size Exclusion Chromatography, Affinity Chromatography, Hydrophobic Interaction Chromatography, Ion Exchange Chromatography.
- a lentiviral vector produced according to a method described herein has one or more of the following properties: complies with GMP guidelines, is sterile, is substantially free of contaminants, is suitable for pharmaceutical use, is suitable for administration to a human subject, or is suitable for ex vivo treatment of human cells.
- a solution or a suspension is subjected to a semi-permeable membrane (filtration) that retains larger particles e.g., viral particles, while allowing solvent and small solute molecules to pass through.
- a method described herein uses a filter to remove and exchange salts, sugars, and non-aqueous solvents, to separate free from bound species, to remove low molecular-weight material, and/or to cause the rapid change of ionic and/or pH environments.
- a filtration step may be used to increase the concentration of vectors in a solution or suspension.
- a filtration step is used to increase the concentration of a lentiviral particle in harvest.
- a method described herein makes use of a process, technique or combination of techniques comprises a filtration step (e.g., one or more of microfiltration, ultrafiltration, nanofiltration, and diafiltration) either sequentially or simultaneously.
- filtration is performed using a flat-sheet membrane or a hollow fiber.
- the filtration is performed using an average transmembrane pressure of about 0. 1 - 0.5 bar (e.g., about 0.1, 0.2, 0.3, 0.4, or 0.5 bar).
- filtration is performed using a load of 4 - 100 L/m 2 , e.g., about 4-10, 10-20, 20-30, 30-40, 40-50, 50-60, 60-70, 70-80, or 80-90.
- a filtration step is employed to exchange the various buffers used in connection with the instant disclosure, optionally in combination with chromatography or other purification steps, and optionally also to remove impurities from viral yield.
- Filtration techniques such as those described above and known in the art, can be used so as to produce lentiviral preparations that are substantially free of microorganisms and cells (e.g., mammalian cells, e.g., HEK293 cells, e.g., Expi293F cells) from which the lentiviral vector is prepared.
- microorganisms and cells e.g., mammalian cells, e.g., HEK293 cells, e.g., Expi293F cells
- lentiviral vector preparations of the disclosure may be treated with nucleases so as to produce a preparation that is substantially free of contaminating polynucleotides (e.g., non-lentiviral polynucleotides derived from the cell in which the lentiviral vector was produced, such as DNA, RNA, or other polynucleotides that are not included within the lentiviral transgene).
- contaminating polynucleotides e.g., non-lentiviral polynucleotides derived from the cell in which the lentiviral vector was produced, such as DNA, RNA, or other polynucleotides that are not included within the lentiviral transgene.
- a composition that is substantially free of a particuclar component contains less than 1%, less than 0.5%, less than 0.4%, less than 0.3%, less than 0.2%, less than 0. 1%, less than 0.01%, less than 0.001% of the component in the composition by weight.
- the component in a composition that is substantially free of a particuclar component, is not detectable, e.g., according to routine methods known in the art.
- Buffers e.g., for use in purification
- buffers e.g., an aqueous composition comprising buffering agents comprising buffering agents used for viral vector purification
- aqueous composition comprising buffering agents comprising buffering agents used for viral vector purification
- sulfonic based acid buffer e.g., 1 ,4- piperazinediethanesulfonic acid (PIPES) based buffer (PIPES buffer)
- PIPES buffer a sulfonic acid-based buffer
- a PIPES buffer may comprise, a buffering agent, e.g., PIPES at a concentration of from about 10 mM to about 50 mM, from about 15 mM to about 40mM, from about 20 mM to about 30 mM, e.g., about 20 mM.
- a buffering agent e.g., PIPES at a concentration of from about 10 mM to about 50 mM, from about 15 mM to about 40mM, from about 20 mM to about 30 mM, e.g., about 20 mM.
- a purification buffer may further comprise a salt, e.g., Sodium Chloride (NaCl), Magnesium Chloride (MgCE), or Calcium Chloride (CaCE), or any combination thereof.
- the salt may be present, e.g., at a concentration of from about 1 mM to about 1 M in the aqueous lentiviral preparation (e.g., 1 mM, 2 mM, 3 mM, 4 mM, 5 mM, 6 mM, 7 mM, 8 mM, 9 mM, 10 mM, 15 mM, 20 mM, 25 mM, 30 mM, 35 mM, 40 mM, 45 mM, 50 mM, 55 mM, 60 mM, 65 mM, 70 mM, 75 mM, 80 mM, 85 mM, 90 mM, 100 mM, 125 mM, 150 mM, 175 mM
- the concentration of salt is from about 25 mM to about 250 mM, about 50 mM to about 75 mM, about 50 mM to about 200 mM, or about 100 mM to about 150 mM (e.g., 25 mM, 30 mM, 35 mM, 40 mM, 45 mM, 50 mM, 55 mM, 60 mM, 65 mM, 70 mM, 75 mM, 80 mM, 85 mM, 90 mM, 100 mM, 125 mM, or 150 mM). In some embodiments, the concentration of salt may be 50 mM or 75 mM, as desired.
- the purification buffer may also comprise a carbohydrate, e.g., a nonreducing carbohydrate, e.g., sucrose or trehalose.
- the carbohydrate, e.g., sucrose is present at a concentration of about 30 mM to about 300 mM, from about 40 mM to about 275 mM, from about 50 mM to about 250 mM, from about 60 mM to about 240 mM, from about 70 mM to about 220 mM, from about 30 mM to 150 mm, or from about 150-300 mM.
- the purification buffer e.g., the PIPES buffer comprises sucrose at a concentration from about 50mM to about 80 mM, e.g., about 73 mM. In some embodiments, the purification buffer, e.g., the PIPES buffer comprises sucrose at a concentration of from about 200 mM to 250 mM, e.g., about 220 mM.
- a carbohydrate may be present at a concentration of, e.g., from about 1 % to about 10%, from about 2.5% to about 10%, or from about 2.5% to about 5% by weight per volume (w/v) of the aqueous lentiviral preparation during manufacturing.
- a carbohydrate such as a non-reducing carbohydrate described herein, can be present within an aqueous lentiviral preparation during manufacturing at a concentration of 1 % w/v, 1 .5% w/v, 2% w/v, 2.5% w/v, 3% w/v, 3.5% w/v, 4% w/v, 4.5% w/v, 5% w/v, 5.5% w/v, 6% w/v, 6.5% w/v, 7% w/v, 7.5% w/v, 8% w/v, 8.5% w/v, 9% w/v, 9.5% w/v, or 10% w/v.
- a carbohydrate such as a non-reducing carbohydrate described herein, can be present within an aqueous lentiviral preparation during manufacturing at a concentration of at least 1 % w/v, 1 .5% w/v, 2% w/v, 2.5% w/v, 3% w/v, 3.5% w/v, 4% w/v, 4.5% w/v, 5% w/v, 5.5% w/v, 6% w/v, 6.5% w/v, 7% w/v, 7.5% w/v, 8% w/v, 8.5% w/v, 9% w/v, 9.5% w/v, or 10% w/v.
- a carbohydrate such as a non-reducing carbohydrate described herein, can be present within an aqueous lentiviral preparation during manufacturing at a concentration of 1 % w/v - 2 % w/v, 2% w/v - 3% w/v, 3% w/v - 4% w/v, 4% w/v - 5% w/v, 5% w/v - 6% w/v, 6% w/v - 7% w/v, 7% w/v - 8% w/v, 8% w/v - 9% w/v, 9% w/v - 10% w/v.
- the buffer comprises arginine or a salt thereof, e.g., arginine-HCl.
- the agent e.g., arginine or a salt thereof, e.g., arginine monohydrochloride (arginine-HCl) is added at a concentration of about 25-50 mM (e.g., about 50mM), 50-100 mM (e.g., about 75mM), 100- 200 mM (e.g., about 150 mM), or 200-400 (e.g., about 300) mM.
- At least one of buffers e.g., PIPES buffer used for viral purification (e.g., lentiviral purification using a process disclosed herein) comprises arginine, e.g., arginine-HCl.
- the pH of the buffers used in the purification process disclosed herein is from about 5.0 to about 8.0, e.g., 6.0 to about 7.0 (e.g., 6.0, 6. 1, 6.2, 6.3, 6.4, 6.5, 6.6, 6.7, 6.8, 6.9, or 7.0), e.g., about 6.5.
- the PIPES buffer may be used as one or more of exchange buffer and/or filtration buffer.
- the ratio of concentration of PIPES, NaCl, and sucrose are different in PIPES filtration buffer and PIPES exchange buffer.
- the ratio of concentration of PIPES, NaCl, and sucrose are identical in PIPES filtration buffer and PIPES exchange buffer.
- the ratio of concentration of PIPES, NaCl, and sucrose are identical in PIPES exchange buffer and PIPES filtration buffer.
- arginine e.g., arginine-HCl is added to cell culture harvest during purification.
- arginine, e.g., arginine-HCl is added to the purification intermediate composition comprising a buffer, e.g., a PIPES buffer or PIPES buffer during purification.
- arginine, e.g., arginine-HCl is added to a PIPES buffer that does not comprise arginine.
- arginine, e.g., arginine-HCl is added to a PIPES buffer that comprises arginine.
- the agent e.g., arginine or a salt thereof, e.g., arginine monochloride (arginine-HCl) is added at a concentration of about 25-50 mM (e.g., about 50mM), 50-100 mM (e.g., about 75mM), 100- 200 mM (e.g., about 150 mM), or 200-400 (e.g., about 300) mM.
- arginine or a salt thereof e.g., arginine monochloride (arginine-HCl)
- arginine-HCl arginine-HCl
- the vector recovery e.g., the amount of transducing units of the lenti virus increases in a purification process which comprises a purification step comprising adding arginine to the purification intermediate composition by about 10% - 300%, about 20% - 180%, about 30% - 160%, about 50% - 150%, about 75%- 125% or about 100% higher relative to a purification process which does not comprise a purification step comprising adding arginine to the purification intermediate composition.
- addition of arginine decreases the process time of purification.
- the process time of the purification is improved by at least 5%, at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, or by at least 50% compared an otherwise similar purification process which does not comprise adding arginine to the purification intermediate composition.
- the purification intermediate composition after addition of arginine or a salt thereof, e.g., arginine-HCl and subsequent purification step shows a total particle concentration per ml of less than 400,000, less than 300,000, less than 200,000, or less than 100,000, as measured by micro-flow imaging.
- the micro-flow imaging does not substantially detect individual lentiviral particles (e.g., infectious viral particles), but detects larger particles comprising aggregates, e.g., aggregates of non-functional virus.
- the purification intermediate composition after addition of arginine or a salt thereof, e.g., arginine-HCl and subsequent purification step show a concentration of particles that are >10pm per ml of less than about 5,000, about 4,500, about 4,000, about 3,500, about 3,000, or about 2,500, as measured by micro-flow imaging.
- the purification intermediate composition after addition of arginine or a salt thereof, e.g., arginine-HCl and subsequent purification step show a concentration >25 pm per ml of less than about 500, about 400, about 300, or about 200, as measured by micro-flow imaging.
- the reduction of aggregates reduces blockage of filtration membrane at a given time point.
- the arginine stabilizes the lentiviral particles.
- the purified lentiviral composition comprises a lentiviral vector at a concentration of, e.g., from about 1 x 10 7 transducing units per milliliter (TU/mL) to about 7 x 10 7 TU/mL (e.g., 1 x 10 7 TU/mL, 1.5 x 10 7 TU/mL, 2 x 10 7 TU/mL, 2.5 x 10 7 TU/mL, 3 x 10 7 TU/mL, 3.5 x 10 7 TU/mL, 4x 10 7 TU/mL , 4.5 x 10 7 TU/mL, 5 x 10 7 TU/mL, 5.5 x 10 7 TU/mL, 6 x 10 7 TU/mL, 6.5 x 10 7 TU/mL, or 7
- Aqueous Compositions e.g., for lentiviral formulations and storage
- the disclosure provides a preparation, e.g., an aqueous mixture, e.g., an aqueous solution or a suspension e.g., an aqueous composition comprising a lentiviral vector disclosed herein and a buffer, e.g., a formulation buffer or a storage buffer, e.g., a HEPES buffer, e.g., a HEPES buffer comprising one or both of a free positively charged amino acid, (e.g., arginine (e.g., L-arginine), lysine, or histidine) and a free nonpolar amino acid (e.g., proline (e.g., L-proline), methionine, or tryptophan).
- a buffer e.g., a formulation buffer or a storage buffer
- a HEPES buffer e.g., a HEPES buffer comprising one or both of a free positively charged amino acid, (e.g., arginine
- lentiviral preparations comprising a formulation buffer or a storage buffer, e.g., a HEPES buffer, e.g., a HEPES buffer comprising one or both of a free positively charged amino acid, (e.g., arginine (e.g., L-arginine), lysine, or histidine) and a free nonpolar amino acid (e.g., proline (e.g., L-proline), methionine, or tryptophan) exhibit improved biological properties relative to lentiviral preparations containing a conventional lentiviral formulation buffer, such as PIPES. These improved biological characteristics include elevated resistance to aggregation across a range of temperatures herein.
- a HEPES buffer e.g., a HEPES buffer comprising one or both of a free positively charged amino acid, (e.g., arginine (e.g., L-arginine), lysine, or histidine) and a free nonpolar amino acid (e
- the HEPES buffer shows an improved transduction capacity at physiological temperatures, and greater resistance to loss of infectivity during multiple freeze/thaw cycles.
- Other buffers useful in conjunction with lentiviral preparations of the disclosure include histidine buffers, phosphate buffers, sodium citrate buffers, MES buffers, MOPS buffers, and PIPES buffers.
- Lentiviral preparations of the disclosure may optionally include a salt, such as sodium chloride, and may optionally contain a carbohydrate, such as a non-reducing carbohydrate.
- a HEPES formulation buffer and/or storage buffer may comprise, a buffering agent, e.g., HEPES at a concentration of from about 10 mM to about 200 mM, from about 10 mM to about 150, from about 10 mM to about 100 mM, from about 10 mM to about 50 mM, from about 10 mM to about 40 mM, from about 10 mM to about 30 mM, or from about 15 mM to about 25 mM, e.g., about 20 mM.
- a buffering agent e.g., HEPES at a concentration of from about 10 mM to about 200 mM, from about 10 mM to about 150, from about 10 mM to about 100 mM, from about 10 mM to about 50 mM, from about 10 mM to about 40 mM, from about 10 mM to about 30 mM, or from about 15 mM to about 25 mM, e.g., about 20
- a lentiviral vector preparation of the disclosure may comprise an amino acid or a salt thereof; such as alanine, arginine, asparagine, aspartic acid, cysteine, glutamic acid, glutamine, glycine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, proline, serine, threonine, tryptophan, tyrosine, valine or a salt thereof.
- amino acid is synonymous with “free amino acid” in this context unless clear otherwise from context (e.g., compared to reference to a polypeptide molecule comprised of a series of amino acids).
- the HEPES formulation and/or storage buffer can comprise one or both of a free positively charged amino acid (e.g., arginine, lysine, or histidine) or a salt thereof and a free nonpolar amino acid (e.g., proline, methionine, or tryptophan) or a salt thereof.
- a free positively charged amino acid e.g., arginine, lysine, or histidine
- a free nonpolar amino acid e.g., proline, methionine, or tryptophan
- the HEPES formulation and/or storage buffer comprises free arginine (e.g., L-arginine) or a salt thereof.
- the free arginine or salt thereof is at a concentration of at least 25, 50, 75, 100, or 150 mM. In some embodiments, the free arginine or salt thereof is at a concentration of about 150 mM.
- the HEPES formulation and/or storage buffer comprises free proline (e.g., L-proline) or a salt thereof.
- the free proline or salt thereof is at a concentration of 25-200, 50-200, 100-200, 125-175, or 150 mM. In some embodiments, the free proline or salt thereof is at a concentration of about 150 mM.
- the HEPES formulation and/or storage buffer comprises both free arginine and free proline, or salts thereof (e.g., arginine-HCl and proline-HCl).
- the HEPES formulation and/or storage buffer comprises less than 20, less than 10, less than 5, less than 2, or less than 1 mM total of inorganic salts. In some embodiments, the HEPES formulation and/or storage buffer comprises between about 0. 1 mM to about 20 mM total of inorganic salts. In some embodiments, the HEPES formulation and/or storage buffer is substantially free of (e.g., is free of) inorganic salts. In some embodiments, the HEPES formulation and/or storage buffer is free of inorganic salts. In some embodiments, the HEPES formulation and/or storage buffer is substantially free of (e.g., is free of) one or both of NaCl and MgCh. In some embodiments, the HEPES formulation and/or storage buffer is free of one or both of NaCl and MgCf.
- a lentiviral vector preparation of the disclosure may further contain a carbohydrate, such as a non-reducing carbohydrate as described herein, e.g., as a cryoprotectant.
- exemplary non-reducing carbohydrates include sucrose and trehalose, among others.
- a carbohydrate e.g., a non-reducing carbohydrate, e.g., sucrose
- a carbohydrate may be present at a concentration of, e.g., from about 25-200 mM, about 50-200 mM, about 100-200 mM, about 125-175 mM, or about 150 mM.
- a carbohydrate, such as a non-reducing carbohydrate described herein, e.g., sucrose can be present within an aqueous lentiviral preparation at a concentration of about 150 mM.
- the HEPES formulation and/or storage buffer further comprises a stabilizing agent.
- the HEPES formulation and/or storage buffer further comprises human serum albumin (HSA).
- HSA human serum albumin
- the HSA is human-derived HSA (e.g., HSA isolated from human serum). Human-derived HSA may obtained by isolating HSA from human serum, e.g., according to routine methods known in the art.
- the HSA is recombinant HSA (rHSA).
- the HSA is present at 0.5-3%, 0.5-2%, 0.5-1%, 1-2%, 1.5-2.5%, or 2% w/v. In some embodiments, the HSA is present at about 2% w/v.
- the HEPES formulation and/or storage buffer is substantially free of (e.g., is free of) HSA. In other embodiments, the HEPES formulation and/or storage buffer is free of HSA.
- Lentiviral vector preparations described herein may exhibit a pH, e.g., of from about 6.0 to about 7.5, e.g., 6.0-7.5, 6.0-7.0, 6.0-6.5, 6.5-7.0, 6.2-6.8, 6.4-6.6, or 6.5.
- the pH of the lentiviral vector preparation is 6.5.
- the HEPES formulation and/or storage buffer is substantially free of (e.g., is free of) one, two, or three of a PEG lipid, F108, and cholesterol. In some embodiments, the HEPES formulation and/or storage buffer is free of one, two, or three of a PEG lipid, F108, and cholesterol.
- the HEPES formulation and/or storage buffer comprises L-arginine, L- proline, sucrose, and is substantially free of inorganic salts. In some embodiments, the HEPES formulation and/or storage buffer comprises L-arginine, L-proline, sucrose, and is free of inorganic salts.
- the HEPES formulation and/or storage buffer comprises L-arginine at a concentration of 100-200 mM, L-proline at a concentration of 25-200 mM, and sucrose at a concentration of 25-200 mM.
- the HEPES formulation and/or storage buffer comprises 20 mM HEPES, L-arginine at a concentration of 150 mM, L-proline at a concentration of 150 mM, and sucrose at a concentration of 150 mM. In some embodiments, the HEPES formulation and/or storage buffer further comprises HSA at a concentration of 2% w/v.
- HEPES formulation buffer and HEPES storage buffer comprises identical composition. In some embodiments, HEPES formulation buffer and HEPES storage buffer comprises different composition.
- a lentiviral vector may be present within a lentiviral preparation of the disclosure within a range of concentrations.
- a lentiviral vector may be present within a lentiviral preparation at a concentration of, e.g., from about 1 x 1 0 7 transducing units per milliliter (TU/mL) to about 1 x 10 9 TU/mL (e.g., 1 x 10 7 TU/mL, 2 x 10 7 TU/mL, 3 x 10 7 TU/mL, 4 x 10 7 TU/mL, 5 x 10 7 TU/mL, 6 x 10 7 TU/mL, 7 x 10 7 TU/mL , 8 x 10 7 TU/mL, 9 x 10 7 TU/mL, 1 x 10 8 TU/mL, 1.5 x 10 8 TU/mL, 2 x 10 8 TU/mL, 2.5 x 10 8 TU/mL, 3 x 10 8 TU/m
- a lentiviral preparation may contain a lentiviral vector at a concentration of from about 3 x 10 8 TU/mL to about 5 x 10 8 TU/mL (e.g., 3 x 10 8 TU/mL, 3.5 x 10 8 TU/mL, 4 x 10 8 TU/mL, 4.5 x 10 8 TU/mL, or 5 x 10 8 TU/mL).
- the acqueous composition comprises cholesterol. In some embodiments, the acqueous composition comprises MgCL. In some embodiments, the acqueous composition comprises lysine. In some embodiments, the acqueous composition comprises lactose. In some embodiments, the acqueous composition comprises sorbitol. In some embodiments, the acqueous composition comprises glycerol. In some embodiments, the acqueous composition comprises PEG lipid. In some embodiments, the acqueous composition comprises F108. In some embodiments, the acqueous composition comprises glutamic acid.
- the aqueous composition e.g., an aqueous composition comprising a lentiviral vector described herein may be stored at low temperatures, e.g., at 10°C, at 6°C, at 4°C, at 0°C, at -10°C, at -20°C, at -30°C, at -40°C, at -50°C, at -60°C, at -70°C, at -80°C, or at -90°C for a period of time, e.g., for about 20 minutes, 40 minutes, 60 minutes, 1.5 hour, 2 hours, 5 hours, 10 hours, 15 hours, 20 hours, 1 day, 2 days, 3 days, 4 days, 5 days, 7 days, 10 days, 12 days, 14 days, 16 days, 18 days, 20 days, 21 days, 25 days, one month, two months, three months, four months, five months, six months, seven months, eight months, nine months, or more.
- low temperatures e.g., at 10°C, at 6°C
- the aqueous composition is stored at less than 10°C, 6°C, 4°C, 0°C, -10°C, -20°C, -30°C, -40°C, -50°C, -60°C, -70°C, -80°C, or - 90°C.
- a purified lentiviral sample stored in a HEPES storage buffer is stored at -80 °C immediately after purification in a frozen condition.
- the lentiviral preparation thus stored may be thawed prior to use and refrozen (e.g., a freeze-thaw cycle).
- a lentiviral preparation prepared and stored as disclosed herein may undergo at least 1, at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9 freeze-thaw cycles without any significant loss of stability and/or infectivity.
- the preparation displays no more than 0.5%, no more than 1%, no more than 2%, no more than 3%, no more than 4%, no more than 5%, no more than 10%, no more than 15%, no more than 20%, no more than 25%, no more than 30%, no more than 40%, no more than 50%, no more than 60%, no more than 70%, or no more than 80% loss of stability and/or infectivity compared to a lentiviral preparation that never underwent a freeze-thaw cycle.
- a lentivirus preparation as disclosed herein may be stored at a chilled condition at 4°C for a period of time, e.g., for about 20 minutes, 40 minutes, 60 minutes, 1.5 hour, 2 hours, 5 hours, 10 hours, 15 hours, 20 hours, 1 day, 2 days, 3 days, 4 days, 5 days, 7 days, 10 days, 12 days, 14 days, 16 days, 18 days, 20 days, 21 days, 25 days, one month, two months, three months, four months, five months, six months, seven months, eight months, nine months, or more.
- a lentiviral preparation as disclosed herein may be stored in a frozen condition at -80°C for a period of time, e.g., for about 20 minutes, 40 minutes, 60 minutes, 1.5 hour, 2 hours, 5 hours, 10 hours, 15 hours, 20 hours, 1 day, 2 days, 3 days, 4 days, 5 days, 7 days, 10 days, 12 days, 14 days, 16 days, 18 days, 20 days, 21 days, 25 days, one month, two months, three months, four months, five months, six months, seven months, eight months, nine months, or more.
- a lentivirus preparation stored as disclosed displays at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 92%, at least 95%, at least 98%, at least 99% or 100% infectivity compared to a lentivirus that was never frozen.
- the lentivirus preparation does not lose more than 0.5%, more than 1%, more than 2%, more than 5%, more than 7%, more than 10%, more than 15%, more than 20%, more than 25%, more than 30%, more than 40%, more than 50%, more than 60%, more than 70%, more than 80% loss of infectivity after undergoing more than 1, (e.g., 2, 3, 4, 5, 6, 7, 8, or 9) freeze-thaw cycles.
- more than 1, e.g., 2, 3, 4, 5, 6, 7, 8, or 9 freeze-thaw cycles.
- a lentivirus preparation stored as disclosed is at least 30%, at least 40%, at least 50%, at least 60%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 92%, at least 95%, at least 98%, at least 99% or 100% stable compared to a lentivirus that was never frozen.
- a lentivirus preparation is used after freezing for at least 5 hours, at least 12 hours, at least 18 hours, at least 1 days, at least 2 days, at least 3 days, at least 5 days, at least 7 days for improved vector integration.
- the disclosure further includes dried or lyophilized compositions, which are prepared by drying or lyophilizing the aqueous compositions described herein, as well as aqueous compositions that are prepared by reconstituting such dried or lyophilized compositions in a buffer described herein (or another, standard vehicle for administration).
- HEPES buffers disclosed within this section are suitable for lentiviral formulation and storage, they may also be utilized in other steps of the lentiviral manufacturing and purification process, e.g., during filtration (e.g., ultrafiltration prior to sterile filtration).
- composition comprising a lentiviral vector and an aqueous composition described herein.
- a “formulation” is synonymous with a “composition.”
- the composition is a pharmaceutical composition.
- Described herein are viral vectors to transduce immune effector cells (e.g., T cells, NK cells) that are engineered to contain one or more chimeric antigen receptors (CARs) that direct the immune effector cells to undesired cells (e.g., cancer cells). This is achieved through an antigen binding domain on the CAR that is specific for a cancer associated antigen.
- CARs cancer associated antigen receptors
- Two classes of cancer associated antigens (tumor antigens) that can be targeted by CARs are: (1) cancer associated antigens that are expressed on the surface of cancer cells; and (2) cancer associated antigens that itself is intracellular, however, a fragment of such antigen (peptide) is presented on the surface of the cancer cells by MHC (major histocompatibility complex).
- the tumor antigen is chosen from one or more of: CD19; CD123; CD22; CD30; CD171; CS-1 (also referred to as CD2 subset 1, CRACC, SLAMF7, CD319, and 19A24); C-type lectin-like molecule-1 (CLL-1 or CLECL1); CD33; epidermal growth factor receptor variant III (EGFRvIII); ganglioside G2 (GD2); ganglioside GD3 (aNeu5Ac(2-8)aNeu5Ac(2-3)bDGalp(l- 4)bDGlcp(l-l)Cer); TNF receptor family member B cell maturation (BCMA); Tn antigen ((Tn Ag) or (GalNAca-Ser/Thr)); prostate-specific membrane antigen (PSMA); Receptor tyrosine kinase-like orphan receptor 1 (R0R1); Fms-Like Tyrosine Kinase 3 (FLT3); Tumor-associated
- a CAR described herein can comprise an antigen binding domain (e.g., antibody or antibody fragment, TCR or TCR fragment) that binds to a tumor-supporting antigen (e.g., a tumor-supporting antigen as described herein).
- the tumor-supporting antigen is an antigen present on a stromal cell or a myeloid-derived suppressor cell (MDSC).
- Stromal cells can secrete growth factors to promote cell division in the microenvironment. MDSC cells can inhibit T cell proliferation and activation.
- the CAR-expressing cells destroy the tumor-supporting cells, thereby indirectly inhibiting tumor growth or survival.
- the stromal cell antigen is chosen from one or more of: bone marrow stromal cell antigen 2 (BST2), fibroblast activation protein (FAP) and tenascin.
- BST2 bone marrow stromal cell antigen 2
- FAP fibroblast activation protein
- tenascin tenascin.
- the FAP-specific antibody is, competes for binding with, or has the same CDRs as, sibrotuzumab.
- the MDSC antigen is chosen from one or more of: CD33, CD1 lb, C14, CD15, and CD66b.
- the tumor-supporting antigen is chosen from one or more of: bone marrow stromal cell antigen 2 (BST2), fibroblast activation protein (FAP) or tenascin, CD33, CD1 lb, C14, CD15, and CD66b.
- BST2 bone marrow stromal cell antigen 2
- FAP fibroblast activation protein
- tenascin CD33, CD1 lb, C14, CD15, and CD66b.
- An non-limiting exemplary tumor antigen is CD 19.
- CARs that bind to CD 19 are known in the art. For example, those disclosed in WO2012/079000 and WO2014/153270 may be used in accordance with the present disclosure.
- Any known CD19 CAR, for example, the CD19 antigen binding domain of any known CD19 CAR, in the art can be used in accordance with the present disclosure.
- Non-limiting exemplary CD 19 CARs include CD 19 CARs described herein or an anti-CD19 CAR described in Xu et al. Blood 123.24(2014):3750-9; Kochenderfer et al. Blood 122.25(2013):4129- 39, Cruz et al.
- the antigen binding domain binds to CD 19 and has the same or a similar binding specificity as the FMC63 scFv fragment described in Nicholson et al. Mol. Immun. 34 (16-17): 1157-1165 (1997). In some embodiments, the antigen binding domain binds to CD19 and includes the scFv fragment described in Nicholson et al. Mol. Immun. 34 (16-17): 1157-1165 (1997).
- the antigen binding domain (for example, a humanized antigen binding domain) binds to CD19 and comprises a sequence from Table 3 of WO2014/153270, incorporated herein by reference.
- WO2014/153270 also describes methods of assaying the binding and efficacy of various CAR constructs.
- Humanization of murine CD 19 antibody is desired for the clinical setting, where the mousespecific residues may induce a human-anti-mouse antigen (HAMA) response in patients who receive CART19 treatment, i.e., treatment with T cells transduced with the CAR19 construct.
- HAMA human-anti-mouse antigen
- the production, characterization, and efficacy of humanized CD 19 CAR sequences is described in International Application WO2014/153270 which is herein incorporated by reference in its entirety, including Examples 1-5 (p. 115-159).
- the antigen binding domain comprises the parental murine scFv sequence of the CAR19 construct provided in WO2012/079000 (incorporated herein by reference). In some embodiments, the antigen binding domain binds CD 19 and comprises a scFv described in WO2012/079000.
- the CD 19 CAR comprises the fusion polypeptide sequence provided as SEQ ID NO: 12 in W02012/079000, which provides an scFv fragment of murine origin that specifically binds to human CD 19.
- the CD 19 CAR comprises an amino acid sequence provided as SEQ ID NO: 12 in W02012/079000.
- the CD 19 CAR comprises the amino acid sequence: diqmtqttsslsaslgdrvtiscrasqdiskylnwyqqkpdgtvklliyhtsrlhsgvpsrfsgsgtdysltisnleqediatyfcqqgntlpytfgggt kleitggggsggggsggggsevklqesgpglvapsqslsvtctvsgvslpdygvswirqpprkglewlgviwgsettyynsalksrltiikdnsksq vflkmnslqtddtaiyycakhyyyggsyamdywgqgtsvtvssttpaprpptpaptiasqplslrpeacrpaaggavhtrgl
- the CD 19 CAR comprises the amino acid sequence: eivmtqspatlslspgeratlscrasqdiskylnwyqqkpgqaprlliyhtsrlhsgiparfsgsgtdytltisslqpedfavyfcqqgntlpytfgqgt kleikggggsggggsggggsqvqlqesgpglvkpsetlsltctvsgvslpdygvswirqppgkglewigviwgsettyyqsslksrvtiskdnskn qvslklssvtaadtavyycakhyyyggsyamdywgqgtlvtvss (SEQ ID NO: 758)
- the CD 19 CAR is a humanized CD 19 CAR comprising the amino acid sequence: eivmtqspatlslspgeratlscrasqdiskylnwyqqkpgqaprlliyhtsrlhsgiparfsgsgtdytltisslqpedfavyfcqqgntlpytfgqgt kleikggggsggggsggggsqvqlqesgpglvkpsetlsltctvsgvslpdygvswirqppgkglewigviwgsettyyqsslksrvtiskdnskn qvslklssvtaadtavyycakhyyggsyamdywgqgtlvtvssttpaprpptpaptiasqplslrpeacrpaaggavht
- CD 19 CARs comprise a sequence, for example, a CDR, VH, VL, scFv, or full-CAR sequence, disclosed in Table 1 below, or a sequence having at least 80%, 85%, 90%, 95%, or 99% identity thereto.
- CD 19 CARs comprise a sequence, for example, a CDR, VH, VL, scFv, or full-CAR sequence, disclosed in Table 1 below, or a sequence having at least one amino acid substitution relative thereto, e.g., 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 ,15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, or 50 substitutions.
- the CD 19 CAR is a comprises a binding domain of the FMC63 monoclonal antibody -derived single-chain variable fragment (scFv), IgG4 hinge region, CD28 transmembrane domain, 4- IBB (CD 137) costimulatory domain, and CD3 zeta activation domain.
- the CD 19 CAR is encoded by a nucleotide sequence of Table 25, or a nucleotide sequence having at least 80%, 85%, 90%, 95%, 97%, or 100% sequence identity relative thereto.
- the CD19 CAR is encoded by a nucleotide sequence having no more than 20, 15, 10, 8, 5, 4, 3, 2, or 1 substitutions relative to a nucleotide sequence of Table 25.
- the CD 19 CAR comprises a polypeptide encoded by a nucleotide sequence of Table 25, or a nucleotide sequence having at least 80%, 85%, 90%, 95%, 97%, or 100% sequence identity relative thereto.
- the CD 19 CAR comprises a polypeptide encoded by a nucleotide sequence having no more than 20, 15, 10, 8, 5, 4, 3, 2, or 1 substitutions relative to a nucleic acid sequence of Table 25.
- the CD 19 CAR comprises a polypeptide sequence of Table 25, or a polypeptide sequence having at least 80%, 85%, 90%, 95%, 97%, or 100% sequence identity relative thereto. In some embodiments, the CD 19 CAR comprises a polypeptide sequence having no more than 20, 15, 10, 8, 5, 4, 3, 2, or 1 amino acid substitutions relative to an amino acid sequence of Table 25. In some embodiments, the CD 19 CAR comprises a heavy chain CDR1-3 and a light chain CDR1-3, of a sequence of Table 25. In some embodiments, the CD19 CAR comprises a heavy chain CDRl-3and a light chain CDR1-3, of a sequence of Table 25 according to Rabat.
- the CD 19 CAR comprises a heavy chain CDR1-3 and a light chain CDR1-3, of a sequence of Table 25 according to Chothia. In some embodiments, the CD19 CAR comprises a heavy chain CDR1-3 of a sequence of Table 25. In some embodiments, the CD19 CAR comprises a heavy chain CDR1-3 of a sequence of Table 25 according to Rabat. In some embodiments, the CD19 CAR comprises a heavy chain CDR1-3 of a sequence of Table 25 according to Chothia. In some embodiments, the CD19 CAR comprises a light chain CDR1-3 of a sequence of Table 25. In some embodiments, the CD19 CAR comprises a light chain CDR1-3 of a sequence of Table 25 according to Rabat. In some embodiments, the CD 19 CAR comprises a light chain CDR1-3 of a sequence of Table 25 according to Chothia. Table 25: Amino acid and nucleic acid sequences of an exemplary CD19 CAR
- the CD 19 CAR comprises a murine anti-CD19 single-chain variable fragment (scFv) linked to CD28 and CD3-zeta co-stimulatory domains.
- the anti- CD19 single-chain variable fragment comprises the FMC63 antibody (e.g., the antibody described in Nicholson et al., Molecular Immunology, 34(16-17): 1157-1165, 1997; the entire contents of which are incorporated herein by reference).
- the CD 19 CAR is encoded by a nucleotide sequence of Table 26, or a nucleotide sequence having at least 80%, 85%, 90%, 95%, 97%, or 100% sequence identity relative thereto.
- the CD 19 CAR is encoded by a nucleotide sequence having no more than 20, 15, 10, 8, 5, 4, 3, 2, or 1 substitutions relative to a nucleotide sequence of Table 26.
- the CD 19 CAR comprises a polypeptide encoded by a nucleotide sequence of Table 26, or a nucleotide sequence having at least 80%, 85%, 90%, 95%, 97%, or 100% sequence identity relative thereto.
- the CD 19 CAR comprises a polypeptide encoded by a nucleotide sequence having no more than 20, 15, 10, 8, 5, 4, 3, 2, or 1 substitutions relative to a nucleic acid sequence of Table 26.
- the CD 19 CAR comprises a polypeptide sequence of Table 26, or a polypeptide sequence having at least 80%, 85%, 90%, 95%, 97%, or 100% sequence identity relative thereto. In some embodiments, the CD 19 CAR comprises a polypeptide sequence having no more than 20, 15, 10, 8, 5, 4, 3, 2, or 1 amino acid substitutions relative to an amino acid sequence of Table 25. In some embodiments, the CD 19 CAR comprises a heavy chain CDR1-3 and a light chain CDR1-3, of a sequence of Table 26. In some embodiments, the CD19 CAR comprises a heavy chain CDR1-3 and a light chain CDR1-3, of a sequence of Table 26 according to Rabat.
- the CD19 CAR comprises a heavy chain CDR1-3 and a light chain CDR1-3, of a sequence of Table 26 according to Chothia. In some embodiments, the CD 19 CAR comprises a heavy chain CDR1-3 of a sequence of Table 26. In some embodiments, the CD19 CAR comprises a heavy chain CDR1-3 of a sequence of Table 26 according to Rabat. In some embodiments, the CD19 CAR comprises a heavy chain CDR1-3 of a sequence of Table 26 according to Chothia. In some embodiments, the CD19 CAR comprises a light chain CDR1-3 of a sequence of Table 26. In some embodiments, the CD19 CAR comprises a light chain CDR1-3 of a sequence of Table 26 according to Rabat. In some embodiments, the CD 19 CAR comprises a light chain CDR1-3 of a sequence of Table 26 according to Chothia.
- the CD 19 CAR comprises a murine anti-CD19 single chain variable fragment (scFv) linked to CD28 and CD3-zeta co-stimulatory domains.
- the CD 19 CAR is encoded by a nucleotide sequence of Table 27, or a nucleotide sequence having at least 80%, 85%, 90%, 95%, 97%, or 100% sequence identity relative thereto.
- the CD19 CAR is encoded by a nucleotide sequence having no more than 20, 15, 10, 8, 5, 4, 3, 2, or 1 substitutions relative to a nucleotide sequence of Table Tl .
- the CD 19 CAR comprises a polypeptide encoded by a nucleotide sequence of Table Tl, or a nucleotide sequence having at least 80%, 85%, 90%, 95%, 97%, or 100% sequence identity relative thereto.
- the CD19 CAR comprises a polypeptide encoded by a nucleotide sequence having no more than 20, 15, 10, 8, 5, 4, 3, 2, or 1 substitutions relative to a nucleic acid sequence of Table Tl .
- the CD 19 CAR comprises a polypeptide sequence of Table 27, or a polypeptide sequence having at least 80%, 85%, 90%, 95%, 97%, or 100% sequence identity relative thereto.
- the CD 19 CAR comprises a polypeptide sequence having no more than 20, 15, 10, 8, 5, 4, 3, 2, or 1 substitutions relative to an amino acid sequence of Table Tl .
- the CD 19 CAR comprises a heavy chain CDR1-3 and a light chain CDR1-3, of a sequence of Table Tl .
- the CD19 CAR comprises a heavy chain CDR1-3 and a light chain CDR1-3, of a sequence of Table Tl according to Rabat.
- the CD19 CAR comprises a heavy chain CDR1-3 and a light chain CDR1- 3, of a sequence of Table Tl according to Chothia.
- the CD19 CAR comprises a heavy chain CDR1-3 of a sequence of Table Tl .
- the CD19 CAR comprises a heavy chain CDR1-3 of a sequence of Table Tl according to Rabat. In some embodiments, the CD19 CAR comprises a heavy chain CDR1-3 of a sequence of Table 27 according to Chothia. In some embodiments, the CD19 CAR comprises a light chain CDR1-3 of a sequence of Table 27. In some embodiments, the CD19 CAR comprises a light chain CDR1-3 of a sequence of Table 27 according to Rabat. In some embodiments, the CD 19 CAR comprises a light chain CDR1-3 of a sequence of Table Tl according to Chothia.
- the CD19 CAR is encoded by a nucleotide sequence of Table 34, or a nucleotide sequence having at least 80%, 85%, 90%, 95%, 97%, or 100% sequence identity relative thereto. In some embodiments, the CD 19 CAR is encoded by a nucleotide sequence having no more than 20, 15, 10, 8, 5, 4, 3, 2, or 1 substitutions relative to a nucleotide sequence of Table 34. In some embodiments, the CD19 CAR comprises a polypeptide encoded by a nucleotide sequence of Table 34, or a nucleotide sequence having at least 80%, 85%, 90%, 95%, 97%, or 100% sequence identity relative thereto.
- the CD 19 CAR comprises a polypeptide encoded by a nucleotide sequence having no more than 20, 15, 10, 8, 5, 4, 3, 2, or 1 substitutions relative to a nucleic acid sequence of Table 34.
- the CD19 CAR comprises a polypeptide sequence of Table 34, or a polypeptide sequence having at least 80%, 85%, 90%, 95%, 97%, or 100% sequence identity relative thereto.
- the CD 19 CAR comprises a polypeptide sequence having no more than 20, 15, 10, 8, 5, 4, 3, 2, or 1 amino acid substitutions relative to an amino acid sequence of Table 34.
- the CD19 CAR comprises a heavy chain CDR1-3 and a light chain CDR1-3, of a sequence of Table 34.
- the CD 19 CAR comprises a heavy chain CDR1-3 and a light chain CDR1-3, of a sequence of Table 34 according to Rabat. In some embodiments, the CD19 CAR comprises a heavy chain CDR1-3 and a light chain CDR1-3, of a sequence of Table 34 according to Chothia. In some embodiments, the CD19 CAR comprises a heavy chain CDR1-3 of a sequence of Table 34. In some embodiments, the CD 19 CAR comprises a heavy chain CDR1-3 of a sequence of Table 34 according to Rabat. In some embodiments, the CD 19 CAR comprises a heavy chain CDR1-3 of a sequence of Table 34 according to Chothia. In some embodiments, the CD 19 CAR comprises a light chain CDR1-3 of a sequence of Table 34. In some embodiments, the CD19 CAR comprises a light chain CDR1-3 of a sequence of Table 34 according to Rabat. In some embodiments, the CD19 CAR comprises a light chain CDR1-3 of a sequence of Table 34 according to Chothia.
- the CD 19 CAR is a bispecific CAR.
- the CD 19 bispecific CAR comprises a light chain variable domain targeting CD 19 and a heavy chain variable domain targeting a different target (e.g., CD20).
- the bispecific car is an anti-CD19 and anti-CD20 CAR.
- the bispecific CAR is encoded by a nucleotide sequence of Table 35, or a nucleotide sequence having at least 80%, 85%, 90%, 95%, 97%, or 100% sequence identity relative thereto.
- the bispecific CAR is encoded by a nucleotide sequence having no more than 20, 15, 10, 8, 5, 4, 3, 2, or 1 substitutions relative to a nucleotide sequence of Table 35.
- the bispecific CAR comprises a polypeptide encoded by a nucleotide sequence of Table 35, or a nucleotide sequence having at least 80%, 85%, 90%, 95%, 97%, or 100% sequence identity relative thereto.
- the bispecific CAR comprises a polypeptide encoded by a nucleotide sequence having no more than 20, 15, 10, 8, 5, 4, 3, 2, or 1 substitutions relative to a nucleic acid sequence of Table 35.
- the bispecific CAR comprises a polypeptide sequence of Table 35, or a polypeptide sequence having at least 80%, 85%, 90%, 95%, 97%, or 100% sequence identity relative thereto. In some embodiments, the bispecific CAR comprises a polypeptide sequence having or no more than 20, 15, 10, 8, 5, 4, 3, 2, or 1 amino acid substitutions relative to an amino acid sequence of Table 35. In some embodiments, the bispecific CAR comprises a heavy chain CDR1-3 and a light chain CDR1-3, of a sequence of Table 35. In some embodiments, the bispecific CAR comprises a heavy chain CDR1-3 and a light chain CDR1-3, of a sequence of Table 35 according to Rabat.
- the bispecific CAR comprises a heavy chain CDR1-3 and a light chain CDR1-3, of a sequence of Table 35 according to Chothia. In some embodiments, the bispecific CAR comprises a heavy chain CDR1-3 of a sequence of Table 35. In some embodiments, the bispecific CAR comprises a heavy chain CDR1-3 of a sequence of Table 35 according to Rabat. In some embodiments, the bispecific CAR comprises a heavy chain CDR1-3 of a sequence of Table 35 according to Chothia. In some embodiments, the bispecific CAR comprises a light chain CDR1-3 of a sequence of Table 35. In some embodiments, the bispecific CAR comprises a light chain CDR1-3 of a sequence of Table 35 according to Rabat. In some embodiments, the bispecific CAR comprises a light chain CDR1-3 of a sequence of Table 35 according to Chothia. Table 35: Amino acid and nucleic acid sequences of an exemplary CD19 CAR
- a non-limiting exemplary tumor antigen is BCMA.
- CARs that bind to BCMA are known in the art. For example, those disclosed WO2016/014565 or WO2019/241426 can be used in accordance with the present disclosure. Any known BCMA CAR, for example, the BCMA antigen binding domain of any known BCMA CAR, in the art can be used in accordance with the present disclosure.
- the BCMA CAR comprises one or more CDRs, VH, VL, scFv, or full- length sequences of BCMA-1, BCMA-2, BCMA-3, BCMA-4, BCMA-5, BCMA-6, BCMA-7, BCMA-8, BCMA-9, BCMA-10, BCMA-11, BCMA-12, BCMA-13, BCMA-14, BCMA-15, 149362, 149363, 149364, 149365, 149366, 149367, 149368, 149369, BCMA_EBB-C1978-A4, BCMA_EBB-C1978-G1, BCMA EBB-C1979-C1, BCMA_EBB-C1978-C7, BCMA_EBB-C1978-D10, BCMA_EBB-C1979-C12, BCMA_EBB-C1980-G4, BCMA_EBB-C1980-D2, BCMA_EBB-C1978-A10, BCMA_EBB-C1978-D4, BCMA_EBB-C1978
- Exemplary antigen binding domains that bind BCMA are disclosed in W02012/0163805, WO 2017/021450, WO 2017/011804, WO 2017/025038, WO 2016/090327, WO 2016/130598, WO 2016/210293, WO 2016/090320, WO 2016/014789, WO 2016/094304, WO 2016/154055, WO 2015/166073, WO 2015/188119, WO 2015/158671, US 9,243,058, US 8,920,776, US 9,273,141, US 7,083,785, US 9,034,324, US 2007/0049735, US 2015/0284467, US 2015/0051266, US 2015/0344844, US 2016/0131655, US 2016/0297884, US 2016/0297885, US 2017/0051308, US 2017/0051252, WO 2016/020332, WO 2016/087531, WO 2016/079177, WO 2015/172800, WO 2017/008169, US 9,
- the antigen binding domain comprises a human antibody or a human antibody fragment that binds BCMA.
- the antigen binding domain comprises one or more (for example, all three) LC CDR1, LC CDR2, and LC CDR3 of a human anti-BCMA binding domain described herein (for example, in Tables 2-14), and/or one or more (for example, all three) HC CDR1, HC CDR2, and HC CDR3 of a human anti-BCMA binding domain described herein (for example, in Tables 2-14).
- the human anti-BCMA binding domain comprises a human VL described herein (for example, in Tables 2, 6, and 10) and/or a human VH described herein (for example, in Tables 2, 6, and 10).
- the antigen binding domain is a scFv comprising a VL and a VH of an amino acid sequence of Tables 2, 6, and 10.
- the antigen binding domain (for example, an scFv) comprises: a VL comprising an amino acid sequence having at least one, two or three modifications (for example, substitutions, for example, conservative substitutions) but not more than 30, 20 or 10 modifications (for example, substitutions, for example, conservative substitutions) of an amino acid sequence provided in Tables 2, 6, and 10, or a sequence with 95-99% identity with an amino acid sequence of Tables 2, 6, and 10; and/or a VH comprising an amino acid sequence having at least one, two or three modifications (for example, substitutions, for example, conservative substitutions) but not more than 30, 20 or 10 modifications (for example, substitutions, for example, conservative substitutions) of an amino acid sequence provided in Tables 2, 6, and 10, or a sequence with 95-99% identity to an amino acid sequence of Tables 2, 6, and 10.
- a VL comprising an amino acid sequence having at least one, two or three modifications (for example, substitutions, for example, conservative substitutions) but not more than 30, 20 or 10 modifications (for example, substitutions, for
- the antigen binding domain described herein includes:
- LC CDRs chosen from:
- the antigen binding domain described herein includes:
- LC CDRs from one of the following:
- the antigen binding domain described herein includes:
- LC CDRs from one of the following:
- the HC CDR1, HC CDR2, HC CDR3, LC CDR1, LC CDR2, and LC CDR3 comprise the amino acid sequences of SEQ ID NOs: 44, 45, 84, 54, 55, and 56, respectively.
- the HC CDR1, HC CDR2, HC CDR3, LC CDR1, LC CDR2, and LC CDR3 comprise the amino acid sequences of SEQ ID NOs: 44, 45, 46, 54, 55, and 56, respectively.
- the HC CDR1, HC CDR2, HC CDR3, LC CDR1, LC CDR2, and LC CDR3 comprise the amino acid sequences of SEQ ID NOs: 44, 45, 68, 54, 55, and 56, respectively.
- the HC CDR1, HC CDR2, HC CDR3, LC CDR1, LC CDR2, and LC CDR3 comprise the amino acid sequences of SEQ ID NOs: 44, 45, 76, 54, 55, and 56, respectively.
- the HC CDR1, HC CDR2, HC CDR3, LC CDR1, LC CDR2, and LC CDR3 comprise the amino acid sequences of SEQ ID NOs: 47, 48, 84, 57, 58, and 59, respectively.
- the HC CDR1, HC CDR2, HC CDR3, LC CDR1, LC CDR2, and LC CDR3 comprise the amino acid sequences of SEQ ID NOs: 47, 48, 46, 57, 58, and 59, respectively.
- the HC CDR1, HC CDR2, HC CDR3, LC CDR1, LC CDR2, and LC CDR3 comprise the amino acid sequences of SEQ ID NOs: 47, 48, 68, 57, 58, and 59, respectively. In some embodiments, the HC CDR1, HC CDR2, HC CDR3, LC CDR1, LC CDR2, and LC CDR3 comprise the amino acid sequences of SEQ ID NOs: 47, 48, 76, 57, 58, and 59, respectively.
- the HC CDR1, HC CDR2, HC CDR3, LC CDR1, LC CDR2, and LC CDR3 comprise the amino acid sequences of SEQ ID NOs: 49, 50, 85, 60, 58, and 56, respectively.
- the HC CDR1, HC CDR2, HC CDR3, LC CDR1, LC CDR2, and LC CDR3 comprise the amino acid sequences of SEQ ID NOs: 49, 50, 51, 60, 58, and 56, respectively.
- the HC CDR1, HC CDR2, HC CDR3, LC CDR1, LC CDR2, and LC CDR3 comprise the amino acid sequences of SEQ ID NOs: 49, 50, 69, 60, 58, and 56, respectively. In some embodiments, the HC CDR1, HC CDR2, HC CDR3, LC CDR1, LC CDR2, and LC CDR3 comprise the amino acid sequences of SEQ ID NOs: 49, 50, 77, 60, 58, and 56, respectively.
- a BCMA CAR comprises a sequence, for example, a CDR, VH, VL, scFv, or full-CAR sequence, disclosed in Tables 2-14, or a sequence having at least 80%, 85%, 90%, 95%, or 99% identity thereto.
- BCMA CARs may be generated using the VH and VL sequences from W02012/0163805 (the contents of which are hereby incorporated by reference in its entirety). In some embodiments, BCMA CARs may be generated using the CDRs, VHs, VLs, scFvs, or full-CAR sequences from WO2019/241426 (the contents of which are hereby incorporated by reference in its entirety).
- the BCMA CAR comprises a murine extracellular single-chain variable fragment (scFv) specific for recognizing B cell maturation antigen (BCMA) followed by a human CD8a hinge and transmembrane domain fused to the T cell cytoplasmic signaling domains of CD 137 (4- IBB) and CD3L chain, in tandem. Binding of BCMA CAR D to BCMA-expressing target cells leads to signaling initiated by CD3C and 4- IBB domains, and subsequent CAR-positive T cell activation. Antigen-specific activation of BCMA CAR D results in CAR-positive T cell proliferation, cytokine secretion, and subsequent cytolytic killing of BCMA -expressing cells.
- scFv murine extracellular single-chain variable fragment
- BCMA B cell maturation antigen
- the BCMA CAR is encoded by a nucleotide sequence of Table 28, or a nucleotide sequence having at least 80%, 85%, 90%, 95%, 97%, or 100% sequence identity relative thereto. In some embodiments, the BCMA CAR is encoded by a nucleotide sequence having no more than 20, 15, 10, 8, 5, 4, 3, 2, or 1 substitutions relative to a nucleotide sequence of Table 28. In some embodiments, the BCMA CAR comprises a polypeptide encoded by a nucleotide sequence of Table 28, or a nucleotide sequence having at least 80%, 85%, 90%, 95%, 97%, or 100% sequence identity relative thereto.
- the BCMA CAR comprises a polypeptide encoded by a nucleotide sequence having no more than 20, 15, 10, 8, 5, 4, 3, 2, or 1 substitutions relative to a nucleic acid sequence of Table 28. In some embodiments, the BCMA CAR comprises a polypeptide sequence of Table 28, or a polypeptide sequence having at least 80%, 85%, 90%, 95%, 97%, or 100% sequence identity relative thereto. In some embodiments, the BCMA CAR comprises a polypeptide sequence having no more than 20, 15, 10, 8, 5, 4, 3, 2, or 1 amino acid substitutions relative to an amino acid sequence of Table 28. In some embodiments, the BCMA CAR comprises a heavy chain CDR1-3 and a light chain CDR1-3, of a sequence of Table 28.
- the BCMA CAR comprises a heavy chain CDR1-3 and a light chain CDR1-3, of a sequence of Table 28 according to Rabat. In some embodiments, the BCMA CAR comprises a heavy chain CDR1-3 and a light chain CDR1-3, of a sequence of Table 28 according to Chothia. In some embodiments, the BCMA CAR comprises a heavy chain CDR1-3 of a sequence of Table 28. In some embodiments, the BCMA CAR comprises a heavy chain CDR1-3 of a sequence of Table 28 according to
- the BCMA CAR comprises a heavy chain CDR1-3, of a sequence of Table 28 according to Chothia. In some embodiments, the BCMA CAR comprises a light chain CDR1-3 of a sequence of Table 28. In some embodiments, the BCMA CAR comprises a light chain CDR1-3 of a sequence of Table 28 according to Rabat. In some embodiments, the BCMA CAR comprises a light chain CDR1-3 of a sequence of Table 28 according to Chothia.
- the BCMA CAR comprises two single-domain antibodies linked to a 4- 1BB costimulatory domain and a CD3-zeta signaling domain.
- the chimeric antigen receptor described herein comprises a polypeptide comprising, (a) an extracellular antigen binding domain comprising a first anti-BCMA single domain antibody (sdAb), and a second anti-BCMA sdAb.
- sdAb first anti-BCMA single domain antibody
- each of the first and second anti-BCMA antibody are independently a VhH domain.
- the first anti-BCMA sdAb comprises a CDR1, a CDR2, and a CDR3 as set forth in the VhH domain comprising the amino acid sequence of SEQ ID NO: 377, or a peptide sequence having at least 80%, 85%, 90%, 95%, 97%, or 100% sequence identity relative thereto.
- the first anti-BCMA sdAb comprises a CDR1, a CDR2, and a CDR3 as set forth in the VhH domain comprising the amino acid sequence having no more than 20, 15, 10, 8, 5, 4, 3, 2, or 1 substitutions relative to the amino acid sequence in SEQ ID NO: 377.
- the second anti-BCMA sdAb comprises a CDR1, a CDR2, and a CDR3 as set forth in the VhH domain comprising the amino acid sequence of SEQ ID NO: 381, or a peptide sequence having at least 80%, 85%, 90%, 95%, 97%, or 100% sequence identity relative thereto.
- the second anti-BCMA sdAb comprises a CDR1, a CDR2, and a CDR3 as set forth in the VhH domain comprising the amino acid sequence having no more than 20, 15, 10, 8, 5, 4, 3, 2, or 1 v substitutions relative to the amino acid sequence in SEQ ID NO: 381.
- the BCMA CAR is any BCMA CAR described in US Patent No.
- the CD 19 CAR is encoded by a nucleotide sequence of Table 29, or a nucleotide sequence having at least 80%, 85%, 90%, 95%, 97%, or 100% sequence identity relative thereto. In some embodiments, the CD19 CAR is encoded by a nucleotide sequence having no more than 20, 15, 10, 8, 5, 4, 3, 2, or 1 substitutions relative to a nucleotide sequence of Table 29.
- the CD 19 CAR comprises a polypeptide encoded by a nucleotide sequence of Table 29, or a nucleotide sequence having at least 80%, 85%, 90%, 95%, 97%, or 100% sequence identity relative thereto. In some embodiments, the CD 19 CAR comprises a polypeptide encoded by a nucleotide sequence having no more than 20, 15, 10, 8, 5, 4, 3, 2, or 1 substitutions relative to a nucleic acid sequence of Table 29. In some embodiments, the CD 19 CAR comprises a polypeptide sequence of Table 29, or a polypeptide sequence having at least 80%, 85%, 90%, 95%, 97%, or 100% sequence identity relative thereto.
- the CD 19 CAR comprises a polypeptide sequence having no more than 20, 15, 10, 8, 5, 4, 3, 2, or 1 amino acid substitutions relative to an amino acid sequence of Table 29.
- the BCMA CAR comprises a heavy chain CDR1-3 and a light chain CDR1-3, of a sequence of Table 29.
- the BCMA CAR comprises a heavy chain CDR1-3 and a light chain CDR1-3, of a sequence of Table 29 according to Rabat. In some embodiments, the BCMA CAR comprises a heavy chain CDR1-3 and a light chain CDR1-3, of a sequence of Table 29 according to Chothia. In some embodiments, the BCMA CAR comprises a heavy chain CDR1-3 of a sequence of Table 29. In some embodiments, the BCMA CAR comprises a heavy chain CDR1-3 of a sequence of Table 29 according to Rabat. In some embodiments, the BCMA CAR comprises a heavy chain CDR1-3 of a sequence of Table 29 according to Chothia. In some embodiments, the BCMA CAR comprises a light chain CDR1-3 of a sequence of Table 29. In some embodiments, the BCMA CAR comprises a light chain CDR1-3 of a sequence of Table 29 according to Rabat. In some embodiments, the BCMA CAR comprises a light chain CDR1-3 of a sequence of Table 29 according to Chothia.
- tumor antigens include CD20, CD22, EGFR, CD 123, and CLL-1.
- CD20 CARs that bind to CD20 are known in the art. For example, those disclosed in WO2018/067992 or WO2016/164731, incorporated by reference herein, can be used in accordance with the present disclosure. Any known CD20 CAR, for example, the CD20 antigen binding domain of any known CD20 CAR, in the art can be used in accordance with the present disclosure. Exemplary CD20-binding sequences or CD20 CAR sequences are disclosed in, for example, Tables 1-5 of WO2018/067992, incorporated by reference.
- the CD20 CAR comprises a CDR, variable region, scFv, or full-length sequence of a CD20 CAR disclosed in WO2018/067992 or WO2016/164731, both incorporated by reference herein.
- CD20 CARs comprise a sequence, for example, a CDR, VH, VL, scFv, or foll-CAR sequence, disclosed in Table 23 below, or a sequence having at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identity thereto.
- antigen binding domains that bind CD20 are described in WO2016/164731 and WO2018/067992, incorporated herein by reference. In some embodiments, the antigen binding domain of one or more of the CD20 antigen binding domains disclosed therein.
- Exemplary antigen binding domains that bind CD22 are described in WO2016/164731 and WO2018/067992, incorporated herein by reference.
- the antigen binding domain comprises a HC CDR1, a HC CDR2, and a HC CDR3 of any heavy chain binding domain amino acid sequences listed in Table 15. In embodiments, the antigen binding domain further comprises a LC CDR1, a LC CDR2, and a LC CDR3. In embodiments, the antigen binding domain comprises a LC CDR1, a LC CDR2, and a LC CDR3 amino acid sequences listed in Table 16.
- the antigen binding domain comprises one, two or all of LC CDR1, LC CDR2, and LC CDR3 of any light chain binding domain amino acid sequences listed in Table 16, and one, two or all of HC CDR1, HC CDR2, and HC CDR3 of any heavy chain binding domain amino acid sequences listed in Table 15.
- Exemplary antigen binding domains that bind CD 123 are described in WO 2014/130635 and WO2016/028896, incorporated herein by reference.
- the antigen binding domain comprises a sequence from Tables 1-2 of WO2014/130635, incorporated herein by reference.
- the antigen binding domain comprises a sequence from Tables 2, 6, and 9 of WO2016/028896, incorporated herein by reference.
- Exemplary antigen binding domains that bind CLL-1 are disclosed in WO2016/014535, incorporated herein by reference.
- the antigen binding domain comprises one, two three (for example, all three) heavy chain CDRs, HC CDR1, HC CDR2 and HC CDR3, from an antibody described herein (for example, an antibody described in WO2015/142675, US-2015-0283178-A1, US -2016-0046724-Al, US2014/0322212A1, US2016/0068601A1, US2016/0051651A1, US2016/0096892A1, US2014/0322275A1, or WO2015/090230, incorporated herein by reference), and/or one, two, three (for example, all three) light chain CDRs, LC CDR1, LC CDR2 and LC CDR3, from an antibody described herein (for example, an antibody described in WO2015/142675, US-2015-0283178-A1, US-2016- 0046724-A1, US2014/0322212A1, US2016/0068601A1, US2016/0051651A1,
- the antigen binding domain is an antigen binding domain described in WO2015/142675, US-2015-0283178-Al, US-2016-0046724-A1, US2014/0322212A1, US2016/0068601A1, US2016/0051651 Al, US2016/0096892A1, US2014/0322275A1, or WO2015/090230, incorporated herein by reference.
- target antigens that can be targeted using the CAR-expressing cells, include, but are not limited to, CD19, CD123, EGFRvIII, CD33, mesothelin, BCMA, and GFR ALPHA -4, among others, as described in, for example, WO2014/153270, WO 2014/130635, WO2016/028896, WO 2014/130657, WO2016/014576, WO 2015/090230, WO2016/014565, WO2016/014535, and W02016/025880, each of which is herein incorporated by reference in its entirety.
- the antigen binding domain of any of the CARs described herein comprises one, two three (for example, all three) heavy chain CDRs, HC CDR1, HC CDR2 and HC CDR3, from an antibody listed above, and/or one, two, three (for example, all three) light chain CDRs, LC CDR1, LC CDR2 and LC CDR3, from an antigen binding domain listed above.
- the antigen binding domain comprises a heavy chain variable region and/or a variable light chain region of an antibody listed or described above.
- the antigen binding domain comprises one, two three (for example, all three) heavy chain CDRs, HC CDR1, HC CDR2 and HC CDR3, from an antibody listed above, and/or one, two, three (for example, all three) light chain CDRs, LC CDR1, LC CDR2 and LC CDR3, from an antibody listed above.
- the antigen binding domain comprises a heavy chain variable region and/or a variable light chain region of an antibody listed or described above.
- CD22 CARs that bind to CD22 are known in the art. For example, those disclosed in WO2018/067992 or WO2016/164731 can be used in accordance with the present disclosure. Any known CD22 CAR, for example, the CD22 antigen binding domain of any known CD22 CAR, in the art can be used in accordance with the present disclosure.
- CD22 -binding sequences or CD22 CAR sequences are disclosed in, for example, Tables 6A, 6B, 7A, 7B, 7C, 8 A, 8B, 9A, 9B, 10A, and 10B of WO2016164731 and Tables 6-10 of WO2018067992.
- the CD22 CAR sequences comprise a CDR, variable region, scFv or full-length sequence of a CD22 CAR disclosed in WO2018067992 or WO2016164731.
- the CAR comprises an antigen binding domain that binds to CD22 (CD22 CAR).
- the antigen binding domain targets human CD22.
- the antigen binding domain includes a single chain Fv sequence as described herein.
- a human CD22 CAR is CAR22-65.
- CD22 CARs comprise a sequence, for example, a CDR, VH, VL, scFv, or full-CAR sequence, disclosed in Tables 15-16 and Table 24 below, or a sequence having at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identity thereto.
- Table 15 Heavy Chain Variable Domain CDRs of CD22 CAR (CAR22-65)
- CARs that bind to EGFR are known in the art. For example, those disclosed in WO2014/130657, incorporated by reference herein, can be used in accordance with the present disclosure. Any known EGFR CAR, for example, the EGFR antigen binding domain of any known EGFR CAR, in the art can be used in accordance with the present disclosure.
- Exemplary EGFRvIII CARs can include a CDR, a variable region, an scFv, or a full-length CAR sequence disclosed in WO2014/130657, for example, Table 2 of WO2014/130657, incorporated herein by reference.
- CARs that bind to CD123 are known in the art.
- those disclosed in WO2014/130635 or WO2016/028896 can be used in accordance with the present disclosure.
- Any known CD123 CAR for example, the CD 123 antigen binding domain of any known CD 123 CAR, in the art can be used in accordance with the present disclosure.
- the amino acid and nucleotide sequences encoding the CD 123 CAR molecules and antigen binding domains are specified in WO 2014/130635 and WO2016/028896.
- CARs that bind to CLL-1 are known in the art. For example, those disclosed in US2016/0051651A1, incorporated herein by reference. Any known CLL-1 CAR, for example, the CLL-1 antigen binding domain of any known CLL-1 CAR, in the art can be used in accordance with the present disclosure.
- the CAR comprises a CLL-1 CAR or antigen binding domain according to Table 2 of WO2016/014535, incorporated herein by reference.
- the amino acid and nucleotide sequences encoding the CLL-1 CAR molecules and antigen binding domains are specified in WO2016/014535.
- CD33 CARs that bind to CD33 are known in the art. For example, those disclosed in US2016/0096892A1 and WO2016/014576, incorporated by reference herein, can be used in accordance with the present disclosure. Any known CD33 CAR, for example, the CD33 antigen binding domain of any known CD33 CAR, in the art can be used in accordance with the present disclosure. For example, CAR33-1 to CAR33-9 disclosed in WO2016/014576 can be used in accordance with the present disclosure.
- the CAR comprises a CD33 CAR or antigen binding domain according to Table 2 or 9 of WO2016/014576, incorporated herein by reference.
- the amino acid and nucleotide sequences encoding the CD33 CAR molecules and antigen binding domains are specified in WO2016/014576.
- an antigen binding domain against CD33 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Brass et al., Clin Cancer Res 7(6): 1490-1496 (2001) (Gemtuzumab Ozogamicin, hP67.6),Caron et al., Cancer Res 52(24):6761-6767 (1992) (Lintuzumab, HuM195), Lapusan et al., Invest New Drugs 30(3): 1121-1131 (2012) (AVE9633), Aigner et al., Leukemia 27(5): 1107-1115 (2013) (AMG330, CD33 BiTE), Dutour et al., Adv hematol 2012:683065 (2012), and Pizzitola et al., Leukemia doi: 10.
- CDRs antigen binding portion
- an antigen binding domain against CD33 is an antigen binding portion, e.g., CDRs, of an antibody, antigen-binding fragment, or CAR described in WO/2017/014576.
- an antigen binding domain against GD2 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Mujoo et al., Cancer Res.
- an antigen binding domain against GD2 is an antigen binding portion of an antibody selected from mAb 14.18, 14G2a, chl4.18, hul4.18, 3F8, hu3F8, 3G6, 8B6, 60C3, 10B8, ME36.1, and 8H9, see e.g., WO2012033885, W02013040371, WO2013192294, WO2013061273, W02013123061, WO2013074916, and WO201385552.
- an antigen binding domain against GD2 is an antigen binding portion of an antibody described in US Publication No. : 20100150910 or PCT Publication No.: WO 2011160119.
- an antigen binding domain against Tn antigen is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., US8,440,798, Brooks et al., PNAS 107(22): 10056-10061 (2010), and Stone et al., Oncolmmunology l(6):863-873(2012).
- an antigen binding portion e.g., CDRs
- an antigen binding domain against PSMA is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Parker et al., Protein Expr Purif 89(2): 136-145 (2013), US 20110268656 (J591 ScFv); Frigerio et al, European J Cancer 49(9):2223-2232 (2013) (scFvD2B); WO 2006125481 (mAbs 3/A12, 3/E7 and 3/F11) and single chain antibody fragments (scFv A5 and D7).
- CDRs antigen binding portion
- an antigen binding domain against ROR1 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Hudecek et al., Clin Cancer Res 19( 12): 3153-3164 (2013); WO 2011159847; and US20130101607.
- an antigen binding domain against FLT3 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., WO2011076922, US5777084, EP0754230, US20090297529, and several commercial catalog antibodies (R&D, ebiosciences, Abeam).
- an antigen binding domain against TAG72 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Hornbach et al., Gastroenterology 113(4): 1163-1170 (1997); and Abeam ab691.
- an antigen binding domain against FAP is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Ostermann et al., Clinical Cancer Research 14:4584-4592 (2008) (FAP5), US Pat. Publication No. 2009/0304718; sibrotuzumab (see e.g., Hofheinz et al., Oncology Research and Treatment 26(1), 2003); and Tran et al., J Exp Med 210(6): 1125-1135 (2013).
- CDRs an antigen binding portion
- an antigen binding domain against CD38 is an antigen binding portion, e.g., CDRs, of daratumumab (see, e.g., Groen et al., Blood 116(21): 1261-1262 (2010); MOR202 (see, e.g., US8,263,746); or antibodies described in US8,362,211.
- an antigen binding domain against CD44v6 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Casucci et al., Blood 122(20):3461-3472 (2013).
- an antigen binding domain against CEA is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Chmielewski et al., Gastoenterology 143(4): 1095-1107 (2012).
- an antigen binding domain against EPCAM is an antigen binding portion, e.g., CDRs, of an antibody selected from MT110, EpCAM-CD3 bispecific Ab (see, e.g., clinicaltrials.gov/ct2/show/NCT00635596); Edrecolomab; 3622W94; ING-1; and adecatumumab (MT201).
- CDRs an antigen binding portion
- EpCAM-CD3 bispecific Ab see, e.g., clinicaltrials.gov/ct2/show/NCT00635596
- Edrecolomab 3622W94
- ING-1 adecatumumab
- an antigen binding domain against PRSS21 is an antigen binding portion, e.g., CDRs, of an antibody described in US Patent No.: 8,080,650.
- an antigen binding domain against B7H3 is an antigen binding portion, e.g., CDRs, of an antibody MGA271 (Macrogenics).
- an antigen binding domain against KIT is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., US7915391, US20120288506, and several commercial catalog antibodies.
- an antigen binding domain against IL-13Ra2 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., W02008/146911, W02004087758, several commercial catalog antibodies, and W02004087758.
- an antigen binding domain against CD30 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., US7090843 Bl, and EP0805871.
- an antigen binding domain against GD3 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., US7253263; US 8,207,308; US 20120276046; EP1013761; W02005035577; and US6437098.
- an antigen binding domain against CD171 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Hong et al., J hnmunother 37(2):93-104 (2014).
- an antigen binding domain against IL-1 IRa is an antigen binding portion, e.g., CDRs, of an antibody available from Abeam (cat# ab55262) or Novus Biologicals (cat# EPR5446).
- an antigen binding domain again IL-1 IRa is a peptide, see, e.g., Huang et al., Cancer Res 72(1)271-281 (2012).
- an antigen binding domain against PSCA is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Morgenroth et al., Prostate 67(10): 1121-1131 (2007) (scFv 7F5); Nejatollahi et al., J of Oncology 2013(2013), article ID 839831 (scFv C5-II); and US Pat Publication No. 20090311181.
- CDRs antigen binding portion
- an antigen binding domain against VEGFR2 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Chinnasamy et al., J Clin Invest 120(11): 3953-3968 (2010).
- an antigen binding domain against LewisY is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Kelly et al., Cancer Biother Radiopharm 23(4):411-423 (2008) (hu3S193 Ab (scFvs)); Dolezal et al., Protein Engineering 16(l):47-56 (2003) (NC10 scFv).
- CDRs antigen binding portion
- an antigen binding domain against CD24 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Maliar et al., Gastroenterology 143(5): 1375-1384 (2012).
- an antigen binding domain against PDGFR-beta is an antigen binding portion, e.g., CDRs, of an antibody Abeam ab32570.
- an antigen binding domain against SSEA-4 is an antigen binding portion, e.g., CDRs, of antibody MC813 (Cell Signaling), or other commercially available antibodies.
- an antigen binding domain against CD20 is an antigen binding portion, e.g., CDRs, of the antibody Rituximab, Ofatumumab, Ocrelizumab, Veltuzumab, or GA101; or antibodies described in WO2016/ 164731.
- an antigen binding domain against Folate receptor alpha is an antigen binding portion, e.g., CDRs, of the antibody IMGN853, or an antibody described in US20120009181; US4851332, LK26: US5952484.
- an antigen binding domain against ERBB2 is an antigen binding portion, e.g., CDRs, of the antibody trastuzumab, or pertuzumab.
- an antigen binding domain against MUC1 is an antigen binding portion, e.g., CDRs, of the antibody SAR566658.
- the antigen binding domain against EGFR is antigen binding portion, e.g., CDRs, of the antibody cetuximab, panitumumab, zalutumumab, nimotuzumab, or matuzumab.
- an antigen binding domain against NCAM is an antigen binding portion, e.g., CDRs, of the antibody clone 2-2B: MAB5324 (EMD Millipore).
- an antigen binding domain against Ephrin B2 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Abengozar et al., Blood 119(19):4565-4576 (2012).
- an antigen binding domain against IGF -I receptor is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., US8344112 B2; EP2322550 Al; WO 2006/138315, or PCT/US2006/022995.
- an antigen binding domain against CAIX is an antigen binding portion, e.g., CDRs, of the antibody clone 303123 (R&D Systems).
- an antigen binding domain against LMP2 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., US7,410,640, or US20050129701.
- an antigen binding domain against gp 100 is an antigen binding portion, e.g., CDRs, of the antibody HMB45, NKIbetaB, or an antibody described in WO2013165940, or US20130295007
- an antigen binding domain against tyrosinase is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., US5843674; or US 19950504048.
- an antigen binding domain against EphA2 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Yu et al., Mol Ther 22(1): 102-111 (2014).
- an antigen binding domain against GD3 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., US7253263; US 8,207,308; US 20120276046; EP1013761 A3; 20120276046; W02005035577; or US6437098.
- an antigen binding domain against fucosyl GM1 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., US20100297138; or W02007/067992.
- an antigen binding domain against sLe is an antigen binding portion, e.g., CDRs, of the antibody G193 (for lewis Y), see Scott AM et al, Cancer Res 60: 3254-61 (2000), also as described in Neeson et al, J Immunol May 2013 190 (Meeting Abstract Supplement) 177. 10.
- an antigen binding domain against GM3 is an antigen binding portion, e.g., CDRs, of the antibody CA 2523449 (mAb 14F7).
- an antigen binding domain against HMWMAA is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Kmiecik et al., Oncoimmunology 3(l):e27185 (2014) (PMID: 24575382) (mAb9.2.27); US6528481; W02010033866; or US 20140004124.
- an antigen binding portion e.g., CDRs
- an antigen binding domain against o-acetyl-GD2 is an antigen binding portion, e.g., CDRs, of the antibody 8B6.
- an antigen binding domain against TEM1/CD248 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Marty et al., Cancer Lett 235(2):298-308 (2006); Zhao et al., J Immunol Methods 363(2):221-232 (2011).
- an antigen binding domain against CLDN6 is an antigen binding portion, e.g., CDRs, of the antibody IMAB027 (Ganymed Pharmaceuticals), see e.g., clinicaltrial.gov/show/NCT02054351.
- an antigen binding domain against TSHR is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., US8,603,466; US8,501,415; or US8,309,693.
- an antigen binding domain against GPRC5D is an antigen binding portion, e.g., CDRs, of the antibody FAB6300A (R&D Systems); or LS-A4180 (Lifespan Biosciences).
- an antigen binding domain against CD97 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., US6,846,911;de Groot et al., J Immunol 183(6):4127-4134 (2009); or an antibody from R&D:MAB3734.
- an antigen binding domain against ALK is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Mino-Kenudson et al., Clin Cancer Res 16(5): 1561-1571 (2010).
- an antigen binding domain against polysialic acid is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Nagae et al., J Biol Chem 288(47)33784-33796 (2013).
- an antigen binding domain against PLAC1 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Ghods et al., Biotechnol Appl Biochem 2013 doi: 10.1002/bab. H77.
- an antigen binding domain against GloboH is an antigen binding portion of the antibody VK9; or an antibody described in, e.g., Kudryashov V et al, Glycoconj J. 15(3):243-9 ( 1998), Lou et al., Proc Natl Acad Sci USA 111(7)2482-2487 (2014) ; MBrl: Bremer E-G et al. J Biol Chem 259: 14773-14777 (1984).
- an antigen binding domain against NY-BR-1 is an antigen binding portion, e.g., CDRs of an antibody described in, e.g., Jager et al., Appl Immunohistochem Mol Morphol 15(1):77- 83 (2007).
- an antigen binding domain against WT-1 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Dao et al., Sci Transl Med 5(176): 176ra33 (2013); or WO2012/135854.
- an antigen binding domain against MAGE-A 1 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Willemsen et al., J Immunol 174( 12): 7853-7858 (2005) (TCR-like scFv).
- an antigen binding domain against sperm protein 17 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Song et al., Target Oncol 2013 Aug 14 (PMID: 23943313); Song et al., Med Oncol 29(4)2923-2931 (2012).
- an antigen binding domain against Tie 2 is an antigen binding portion, e.g., CDRs, of the antibody AB33 (Cell Signaling Technology).
- an antigen binding domain against MAD-CT-2 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., PMID: 2450952; US7635753.
- an antigen binding domain against Fos-related antigen 1 is an antigen binding portion, e.g., CDRs, of the antibody 12F9 (Novus Biologicals).
- an antigen binding domain against MelanA/MARTl is an antigen binding portion, e.g., CDRs, of an antibody described in, EP2514766 A2; or US 7,749,719.
- an antigen binding domain against sarcoma translocation breakpoints is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Luo et al, EMBO Mol. Med. 4(6):453-46I (2012).
- an antigen binding domain against TRP-2 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Wang et al, J Exp Med. 184(6):2207-16 (1996).
- an antigen binding domain against CYP1B1 is an antigen binding portion, e.g., CDRs, of an antibody described in, e.g., Maecker et al, Blood 102 (9): 3287-3294 (2003).
- an antigen binding domain against RAGE-1 is an antigen binding portion, e.g., CDRs, of the antibody MAB5328 (EMD Millipore).
- an antigen binding domain against human telomerase reverse transcriptase is an antigen binding portion, e.g., CDRs, of the antibody cat no: LS-B95-100 (Lifespan Biosciences)
- an antigen binding domain against intestinal carboxyl esterase is an antigen binding portion, e.g., CDRs, of the antibody 4F12: cat no: LS-B6190-50 (Lifespan Biosciences).
- an antigen binding domain against mut hsp70-2 is an antigen binding portion, e.g., CDRs, of the antibody Lifespan Biosciences: monoclonal: cat no: LS-CI3326I-I00 (Lifespan Biosciences).
- an antigen binding domain against CD79a is an antigen binding portion, e.g., CDRs, of the antibody Anti-CD79a antibody [HM47/A9] (ab3121), available from Abeam; antibody CD79A Antibody #3351 available from Cell Signalling Technology; or antibody HPAO 17748 - Anti- CD79A antibody produced in rabbit, available from Sigma Aldrich.
- an antigen binding domain against CD79b is an antigen binding portion, e.g., CDRs, of the antibody polatuzumab vedotin, anti-CD79b described in Doman et al., “Therapeutic potential of an anti-CD79b antibody-drug conjugate, anti-CD79b-vc-MMAE, for the treatment of nonHodgkin lymphoma” Blood. 2009 Sep 24;114(13):2721-9. doi: 10.1182/blood-2009-02-205500.
- an antigen binding portion e.g., CDRs
- an antigen binding domain against CD72 is an antigen binding portion, e.g., CDRs, of the antibody J3-109 described in Myers, and Uckun, “An anti-CD72 immunotoxin against therapy-refractory B-lineage acute lymphoblastic leukemia.” Leuk Lymphoma. 1995 Jun;18(l-2): 119-22, or anti-CD72 (10D6.8. 1, mlgGI) described in Polson et al., “Antibody -Drug Conjugates for the Treatment of Non-Hodgkin's Lymphoma: Target and Linker-Drug Selection” Cancer Res March 15, 2009 69; 2358.
- CDRs antigen binding portion
- an antigen binding domain against LAIR1 is an antigen binding portion, e.g., CDRs, of the antibody ANT-301 LAIR1 antibody, available from ProSpec; or anti-human CD305 (LAIR1) Antibody, available from BioLegend.
- an antigen binding portion e.g., CDRs, of the antibody ANT-301 LAIR1 antibody, available from ProSpec; or anti-human CD305 (LAIR1) Antibody, available from BioLegend.
- an antigen binding domain against FC AR is an antigen binding portion, e.g., CDRs, of the antibody CD89/FCARAntibody (Catalog# 10414-H08H), available from Sino Biological Inc.
- an antigen binding domain against LILRA2 is an antigen binding portion, e.g., CDRs, of the antibody LILRA2 monoclonal antibody (M17), clone 3C7, available from Abnova, or Mouse Anti-LILRA2 antibody, Monoclonal (2D7), available from Lifespan Biosciences.
- LILRA2 monoclonal antibody M17
- clone 3C7 available from Abnova
- Mouse Anti-LILRA2 antibody Monoclonal (2D7)
- an antigen binding domain against CD300LF is an antigen binding portion, e.g., CDRs, of the antibody Mouse Anti-CMRF35-like molecule 1 antibody, Monoclonal[UP-D2], available from BioLegend, or Rat Anti-CMRF35-like molecule 1 antibody, Monoclonal [234903], available from R&D Systems.
- CDRs antigen binding portion
- an antigen binding domain against CLEC12A is an antigen binding portion, e.g., CDRs, of the antibody Bispecific T cell Engager (BiTE) scFv-antibody and ADC described in Noordhuis et al., “Targeting of CLEC12A In Acute Myeloid Leukemia by Antibody -Drug-Conjugates and Bispecific CLL-lxCD3 BiTE Antibody” 53 rd ASH Annual Meeting and Exposition, December 10-13, 2011, and MCLA-117 (Merus).
- BiTE Bispecific T cell Engager
- an antigen binding domain against BST2 is an antigen binding portion, e.g., CDRs, of the antibody Mouse Anti-CD317 antibody, Monoclonal [3H4], available from Antibodies-Online or Mouse Anti-CD317 antibody, Monoclonal[696739], available from R&D Systems.
- an antigen binding domain against EMR2 is an antigen binding portion, e.g., CDRs, of the antibody Mouse Anti-CD312 antibody, Monoclonal[LS-B8033] available from Lifespan Biosciences, or Mouse Anti-CD312 antibody, Monoclonal [494025] available from R&D Systems.
- an antigen binding domain against LY75 is an antigen binding portion, e.g., CDRs, of the antibody Mouse Anti-Lymphocyte antigen 75 antibody, Monoclonal [HD30] available from EMD Millipore or Mouse Anti-Lymphocyte antigen 75 antibody, Monoclonal [A 15797] available from Life Technologies.
- an antigen binding portion e.g., CDRs, of the antibody Mouse Anti-Lymphocyte antigen 75 antibody, Monoclonal [HD30] available from EMD Millipore or Mouse Anti-Lymphocyte antigen 75 antibody, Monoclonal [A 15797] available from Life Technologies.
- an antigen binding domain against GPC3 is an antigen binding portion, e.g., CDRs, of the antibody hGC33 described in Nakano K, Ishiguro T, Konishi H, et al. Generation of a humanized anti-glypican 3 antibody by CDR grafting and stability optimization.
- an antigen binding domain against FCRL5 is an antigen binding portion, e.g., CDRs, of the anti-FcRL5 antibody described in Elkins et al., “FcRL5 as a target of antibody-drug conjugates for the treatment of multiple myeloma” Mol Cancer Ther. 2012 Oct;l l(10):2222-32.
- an antigen binding domain against FCRL5 is an antigen binding portion, e.g., CDRs, of the anti-FcRL5 antibody described in, for example, W02001/038490, WO/2005/ 117986, W02006/039238, W02006/076691, WO2010/114940, W02010/120561, or W02014/210064.
- an antigen binding domain against IGLL1 is an antigen binding portion, e.g., CDRs, of the antibody Mouse Anti-Immunoglobulin lambda-like polypeptide 1 antibody, Monoclonal [AT 1G4] available from Lifespan Biosciences, Mouse Anti-Immunoglobulin lambda-like polypeptide 1 antibody, Monoclonal[HSLl l] available from BioLegend.
- CDRs antigen binding portion
- the antigen binding domain comprises one, two three (e.g., all three) heavy chain CDRs, HC CDR1, HC CDR2 and HC CDR3, from an antibody listed above, and/or one, two, three (e.g., all three) light chain CDRs, LC CDR1, LC CDR2 and LC CDR3, from an antibody listed above.
- the antigen binding domain comprises a heavy chain variable region and/or a variable light chain region of an antibody listed above.
- the antigen binding domain comprises a humanized antibody or an antibody fragment.
- a non-human antibody is humanized, where specific sequences or regions of the antibody are modified to increase similarity to an antibody naturally produced in a human or fragment thereof.
- the antigen binding domain is humanized.
- CARs that bind to mesothelin are known in the art. For example, those disclosed in W02015090230 and WO2017112741, for example, Tables 2, 3, 4, and 5 of WO2017112741, incorporated herein by reference, that bind human mesothelin. Any known mesothelin CAR, for example, the mesothelin antigen binding domain of any known mesothelin CAR, in the art can be used in accordance with the present disclosure.
- GFR ALPHA-4 CARs that bind to GFR ALPHA-4 are known in the art. For example, those disclosed in W02016/025880 can be used in accordance with the present disclosure. Any known GFR ALPHA-4 CAR, for example, the GFR ALPHA-4 antigen binding domain of any known GFR ALPHA -4 CAR, in the art can be used in accordance with the present disclosure.
- the amino acid and nucleotide sequences encoding the GFR ALPHA-4 CAR molecules and antigen binding domains are specified in W02016/025880.
- the antigen binding domain of the encoded CAR molecule comprises an antibody, an antibody fragment, an scFv, a Fv, a Fab, a (Fab’)2, a single domain antibody (SDAB), a VH or VL domain, a camelid VHH domain or a bi-functional (e.g. bi-specific) hybrid antibody (e.g., Lanzavecchia et al., Eur. J. Immunol. 17, 105 (1987)).
- scFvs can be prepared according to method known in the art (see, for example, Bird et al., (1988) Science 242:423-426 and Huston et al., (1988) Proc. Natl. Acad. Sci. USA 85:5879- 5883).
- ScFv molecules can be produced by linking VH and VL regions together using flexible polypeptide linkers.
- the scFv molecules comprise a linker (e.g., a Ser-Gly linker) with an optimized length and/or amino acid composition. The linker length can greatly affect how the variable regions of a scFv fold and interact.
- An scFv can comprise a linker of at least 1, at least 2, at least 3, at least 4, at least 5, at least 6, at least 7, at least 8, at least 9, at least 10, at least 11, at least 12, at least 13, at least 14, at least 15, at least 16, at least 17, at least 18, at least 19, at least 20, at least 25, at least 30, at least 35, at least 40, at least 45, at least 50, or more amino acid residues between its VL and VH regions.
- the linker sequence may comprise any naturally occurring amino acid.
- the linker sequence comprises amino acids glycine and serine.
- the linker sequence comprises sets of glycine and serine repeats such as (Gly4Ser) n , where n is a positive integer equal to or greater than 1 (SEQ ID NO:22).
- the linker can be (Gly4Ser)4 (SEQ ID NO:29) or (Gly4Ser)3 (SEQ ID NO:30). Variation in the linker length may retain or enhance activity, giving rise to superior efficacy in activity studies.
- the antigen binding domain is a T cell receptor (“TCR”), or a fragment thereof, for example, a single chain TCR (scTCR).
- TCR T cell receptor
- scTCR single chain TCR
- Methods to make such TCRs are known in the art. See, e.g., Willemsen RA et al, Gene Therapy 7: 1369-1377 (2000); Zhang T et al, Cancer Gene Ther 11: 487-496 (2004); Aggen et al, Gene Ther. 19(4):365-74 (2012) (references are incorporated herein by its entirety).
- scTCR can be engineered that contains the Va and V genes from a T cell clone linked by a linker (e.g., a flexible peptide). This approach is very useful to cancer associated target that itself is intracellular, however, a fragment of such antigen (peptide) is presented on the surface of the cancer cells by MHC.
- the encoded antigen binding domain has a binding affinity KD of 10' 4 M to IO’ 8 M.
- the encoded CAR molecule comprises an antigen binding domain that has a binding affinity KD of 10' 4 M to 10' 8 M, e.g., 10' 5 M to 10' 7 M, e.g., 10' 6 M or 10' 7 M, for the target antigen.
- the antigen binding domain has a binding affinity that is at least five-fold, at least 10-fold, at least 20-fold, at least 30-fold, at least 50-fold, at least 100-fold or at least 1,000-fold less than a reference antibody, e.g., an antibody described herein.
- the encoded antigen binding domain has a binding affinity at least 5-fold less than a reference antibody (e.g., an antibody from which the antigen binding domain is derived).
- a reference antibody e.g., an antibody from which the antigen binding domain is derived.
- antibody fragments are functional in that they provide a biological response that can include, but is not limited to, activation of an immune response, inhibition of signal -transduction origination from its target antigen, inhibition of kinase activity, and the like, as will be understood by a skilled artisan.
- the antigen binding domain of the CAR is a scFv antibody fragment that is humanized compared to the murine sequence of the scFv from which it is derived.
- the antigen binding domain of a CAR described herein is encoded by a nucleic acid molecule whose sequence has been codon optimized for expression in a mammalian cell.
- entire CAR construct is encoded by a nucleic acid molecule whose entire sequence has been codon optimized for expression in a mammalian cell.
- Codon optimization refers to the discovery that the frequency of occurrence of synonymous codons (i.e., codons that code for the same amino acid) in coding DNA is biased in different species. Such codon degeneracy allows an identical polypeptide to be encoded by a variety of nucleotide sequences.
- a variety of codon optimization methods is known in the art, and include, e.g., methods disclosed in at least US Patent Numbers 5,786,464 and 6,114,148.
- percent (%) amino acid sequence identity with respect to a nucleic acid e.g., DNA or RNA
- peptide, polypeptide can be calculated as the percentage of nucleotide or amino acid residues in a candidate sequence that are identical with the nucleotide or amino acid residues in the specific nucleotide, peptide or polypeptide sequence, after aligning the sequences and introducing gaps, if necessary, to achieve the maximum percent sequence identity, and not considering any conservative substitutions as part of the sequence identity.
- Alignment for purposes of determining percent nucleic acid amino acid sequence identity can be achieved in various ways that are within the skill in the art, for instance, using publicly available computer software such as BLAST, BLAST-2, ALIGN or MEGALIGNTM (DNASTAR) software. Those skilled in the art can determine appropriate parameters for measuring alignment, including any algorithms needed to achieve maximal alignment over the full length of the sequences being compared. Unless otherwise indicated herein, percent identity is calculated herein using BLAST.
- antigen antibody pairs are known in the art.
- Non-limiting exemplary embodiments of antigen antibody pairs and components thereof are provided herein above in the section titled Targets and below.
- the antigen binding domain is a bi- or multi- specific molecule (e.g., a multispecific antibody molecule).
- a multispecific antibody molecule is a bispecific antibody molecule.
- a bispecific antibody has specificity for no more than two antigens.
- a bispecific antibody molecule is characterized by a first immunoglobulin variable domain sequence which has binding specificity for a first epitope and a second immunoglobulin variable domain sequence that has binding specificity for a second epitope.
- the first and second epitopes are on the same antigen, e.g., the same protein (or subunit of a multimeric protein). In some embodiments the first and second epitopes overlap.
- first and second epitopes do not overlap. In some embodiments the first and second epitopes are on different antigens, e.g., different proteins (or different subunits of a multimeric protein).
- a bispecific antibody molecule comprises a heavy chain variable domain sequence and a light chain variable domain sequence which have binding specificity for a first epitope and a heavy chain variable domain sequence and a light chain variable domain sequence which have binding specificity for a second epitope. In some embodiments a bispecific antibody molecule comprises a half antibody having binding specificity for a first epitope and a half antibody having binding specificity for a second epitope.
- a bispecific antibody molecule comprises a half antibody, or fragment thereof, having binding specificity for a first epitope and a half antibody, or fragment thereof, having binding specificity for a second epitope.
- a bispecific antibody molecule comprises a scFv, or fragment thereof, have binding specificity for a first epitope and a scFv, or fragment thereof, have binding specificity for a second epitope.
- the antibody molecule is a multi-specific (e.g., a bispecific or a trispecific) antibody molecule.
- bispecific fusion proteins e.g., an expression construct containing two scFvs with a hydrophilic helical peptide linker between them and a full constant region, as described in, e.g., US5637481; minibody constructs with linked VL and VH chains further connected with peptide spacers to an antibody hinge region and CH3 region, which can be dimerized to form bispecific/multivalent molecules, as described in, e.g., US5837821; String of VH domains (or VL domains in family members) connected by peptide linkages with crosslinkable groups at the C-terminus further associated with VL domains to form a series of FVs (or scFvs), as described in, e.g., US5864019; and single chain binding polypeptides with both a VH and a V
- the VH can be upstream or downstream of the VL.
- the upstream antibody or antibody fragment e.g., scFv
- VH1 upstream of its VL
- VL2 downstream antibody or antibody fragment
- the upstream antibody or antibody fragment (e.g., scFv) is arranged with its VL (VL1) upstream of its VH (VH1) and the downstream antibody or antibody fragment (e.g., scFv) is arranged with its VH (VH2) upstream of its VL (VL2), such that the overall bispecific antibody molecule has the arrangement VL1-VH1-VH2-VL2.
- a linker is disposed between the two antibodies or antibody fragments (e.g., scFvs), e.g., between VL1 and VL2 if the construct is arranged as VH1-VL1- VL2-VH2, or between VH1 and VH2 if the construct is arranged as VL1-VH1-VH2-VL2.
- the linker may be a linker as described herein, e.g., a (Gly4-Ser) n linker, wherein n is 1, 2, 3, 4, 5, or 6, preferably 4 (SEQ ID NO: 691).
- the linker between the two scFvs should be long enough to avoid mispairing between the domains of the two scFvs.
- a linker is disposed between the VL and VH of the first scFv.
- a linker is disposed between the VL and VH of the second scFv.
- any two or more of the linkers can be the same or different.
- a bispecific CAR comprises VLs, VHs, and optionally one or more linkers in an arrangement as described herein.
- a chimeric molecule as described herein can be designed to comprise a transmembrane domain that is attached to the extracellular domain of the chimeric molecule.
- a transmembrane domain can include one or more additional amino acids adjacent to the transmembrane region, e.g., one or more amino acid associated with the extracellular region of the protein from which the transmembrane was derived (e.g., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 up to 15 amino acids of the extracellular region) and/or one or more additional amino acids associated with the intracellular region of the protein from which the transmembrane protein is derived (e.g., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 up to 15 amino acids of the intracellular region).
- one or more amino acid associated with the extracellular region of the protein from which the transmembrane was derived e.g., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 up to 15 amino acids of the extracellular region
- additional amino acids associated with the intracellular region of the protein from which the transmembrane protein is derived e.g., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 up to 15 amino acids of the intracellular region
- the transmembrane domain is one that is associated with one of the other domains of the chimeric protein (e.g., CAR) e.g., in some embodiments, the transmembrane domain may be from the same protein that the signaling domain, costimulatory domain or the hinge domain is derived from. In another aspect, the transmembrane domain is not derived from the same protein that any other domain of the chimeric protein (e.g., CAR) is derived from. In some instances, the transmembrane domain can be selected or modified by amino acid substitution to avoid binding of such domains to the transmembrane domains of the same or different surface membrane proteins, e.g., to minimize interactions with other members of the receptor complex.
- the transmembrane domain is capable of homodimerization with another CAR on the cell surface of a CAR-expressing cell.
- the amino acid sequence of the transmembrane domain may be modified or substituted so as to minimize interactions with the binding domains of the native binding partner present in the same CAR-expressing cell.
- the transmembrane domain may be derived either from a natural or from a recombinant source. Where the source is natural, the domain may be derived from any membrane -bound or transmembrane protein. In some aspects, the transmembrane domain is capable of signaling to the intracellular domain(s) whenever the CAR has bound to a target.
- a transmembrane domain may include at least the transmembrane region(s) of e.g., the alpha, beta or zeta chain of the T-cell receptor, CD28, CD27, CD3 epsilon, CD45, CD4, CD5, CD8, CD9, CD16, CD22, CD33, CD37, CD64, CD80, CD86, CD134, CD137, CD154.
- the transmembrane region(s) e.g., the alpha, beta or zeta chain of the T-cell receptor, CD28, CD27, CD3 epsilon, CD45, CD4, CD5, CD8, CD9, CD16, CD22, CD33, CD37, CD64, CD80, CD86, CD134, CD137, CD154.
- a transmembrane domain may include at least the transmembrane region(s) of, e.g., KIRDS2, 0X40, CD2, CD27, LFA-1 (CDl la, CD18), ICOS (CD278), 4-1BB (CD137), GITR, CD40, BAFFR, HVEM (LIGHTR), SLAMF7, NKp80 (KLRF1), NKp44, NKp30, NKp46, CD160, CD19, IL2R beta, IL2R gamma, IL7R a, ITGA1, VLA1, CD49a, ITGA4, IA4, CD49D, ITGA6, VLA-6, CD49f, ITGAD, CD1 Id, ITGAE, CD103, ITGAL, CD1 la, LFA-1, ITGAM, CDl lb, ITGAX, CDl lc, ITGB1, CD29, ITGB2, CD18, LFA-1, ITGB7, TNFR2,
- the transmembrane domain can be attached to the extracellular region of the CAR, e.g., the antigen binding domain of the CAR, via a hinge, e.g., a hinge from a human protein.
- the hinge can be a human Ig (immunoglobulin) hinge (e.g., an IgG4 hinge, an IgD hinge), a GS linker (e.g., a GS linker described herein), a KIR2DS2 hinge or a CD8a hinge.
- the hinge or spacer comprises (e.g., consists of) the amino acid sequence of SEQ ID N0:4.
- the transmembrane domain comprises (e.g., consists of) a transmembrane domain of SEQ ID NO: 12.
- the encoded transmembrane domain comprises an amino acid sequence of a CD8 transmembrane domain having at least one, two or three modifications but not more than 20, 10 or 5 modifications of an amino acid sequence of SEQ ID NO: 12, or a sequence with 95-99% identity to an amino acid sequence of SEQ ID NO: 12. In some embodiments, the encoded transmembrane domain comprises the sequence of SEQ ID NO: 12.
- the nucleic acid molecule encoding the CAR comprises a nucleotide sequence of a CD8 transmembrane domain, e.g., comprising the sequence of SEQ ID NO: 13, or a sequence with 95-99% identity thereof.
- the encoded antigen binding domain is connected to the transmembrane domain by a hinge region.
- the encoded hinge region comprises the amino acid sequence of a CD8 hinge, e.g., SEQ ID NO: 4; or the amino acid sequence of an IgG4 hinge, e.g., SEQ ID NO: 6, or a sequence with 95-99% identity to SEQ ID NO:4 or 6.
- the nucleic acid sequence encoding the hinge region comprises a sequence of SEQ ID NO: 5 or SEQ ID NO: 7, corresponding to a CD8 hinge or an IgG4 hinge, respectively, or a sequence with 95-99% identity to SEQ ID NO:5 or 7.
- the hinge or spacer comprises an IgG4 hinge.
- the hinge or spacer comprises a hinge of the amino acid sequence ESKYGPPCPPCPAPEFLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSQEDPEVQFNWYVDGVE VHNAKTKPREEQFNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKGLPSSIEKTISKAKGQPREPQ VYTLPPSQEEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSRLTV DKSRWQEGNVFSCSVMHEALHNHYTQKSLSLSLGKM (SEQ ID NO: 6).
- the hinge or spacer comprises a hinge encoded by a nucleotide sequence of GAGAGCAAGTACGGCCCTCCCTGCCCCCCTTGCCCTGCCCCCGAGTTCCTGGGCGGACCCAG CGTGTTCCTGTTCCCCCCCAAGCCCAAGGACACCCTGATGATCAGCCGGACCCCCGAGGTGA CCTGTGTGGTGGTGGACGTGTCCCAGGAGGACCCCGAGGTCCAGTTCAACTGGTACGTGGAC GGCGTGGAGGTGCACAACGCCAAGACCAAGCCCCGGGAGGAGCAGTTCAATAGCACCTACC GGGTGGTGTCCGTGCTGACCGTGCTGCACCAGGACTGGCTGAACGGCAAGGAATACAAGTG TAAGGTGTCCAACAAGGGCCTGCCCAGCAGCATCGAGAAAACCATCAGCAAGGCCAAGGGC CAGCCTCGGGAGCCCCAGGTGTACACCCTGCCCCCTAGCCAAGAGGAGATGACCAAGAACC AGGTGTCCCTGACCTGGTGAAGGGCTTCT
- the hinge or spacer comprises an IgD hinge.
- the hinge or spacer comprises a hinge of the amino acid sequence RWPESPKAQASSVPTAQPQAEGSLAKATTAPATTRNTGRGGEEKKKEKEKEEQEERETKTPECP SHTQPLGVYLLTPAVQDLWLRDKATFTCFVVGSDLKDAHLTWEVAGKVPTGGVEEGLLERHSN GSQSQHSRLTLPRSLWNAGTSVTCTLNHPSLPPQRLMALREPAAQAPVKLSLNLLASSDPPEAAS WLLCEVSGFSPPNILLMWLEDQREVNTSGFAPARPPPQPGSTTFWAWSVLRVPAPPSPQPATYTC VVSHEDSRTLLNASRSLEVSYVTDH (SEQ ID NO:8).
- the hinge or spacer comprises a hinge encoded by a nucleotide sequence of AGGTGGCCCGAAAGTCCCAAGGCCCAGGCATCTAGTGTTCCTACTGCACAGCCCCAGGCAG AAGGCAGCCTAGCCAAAGCTACTACTGCACCTGCCACTACGCAATACTGGCCGTGGCGG GGAGGAGAAGAAAAAGGAGAAAGAAAGAAAGAACAGGAAGAGAGGGAGACCAAGACCC CTGAATGTCCATCCCATACCCAGCCGCTGGGCGTCTATCTCTCTCTCTTGACTCCCGCAGTACAGGAC TTGTGGCTTAGAGATAAGGCCACCTTTACATGTTTCGTCGTGGGCTCTGACCTGAAGGATGC CCATTTGACTTGGGAGGTTGCCGGAAAGGTACCCACAGGGGGGGGGTTGAGGAAGGGTTGCTG GAGCCATTCCAATGGCTCTCAGAGCCAGCACTCAAGACTCACCCTTCCGAGATCCCTGTG GAACGCCGGGACCTCTGTCACATGTACTCTAAATCATCCTA
- the transmembrane domain may be recombinant, in which case it will comprise predominantly hydrophobic residues such as leucine and valine.
- a triplet of phenylalanine, tryptophan and valine can be found at each end of a recombinant transmembrane domain.
- a short oligo- or polypeptide linker may form the linkage between the transmembrane domain and the cytoplasmic region of the CAR.
- a glycine-serine doublet provides a particularly suitable linker.
- the linker comprises the amino acid sequence of GGGGSGGGGS (SEQ ID NO: 10).
- the linker is encoded by a nucleotide sequence of GGTGGCGGAGGTTCTGGAGGTGGAGGTTCC (SEQ ID NO: 11).
- the linker comprises the amino acid sequence of GGGGS (SEQ ID NO: 877).
- the linker is encoded by a nucleotide sequence of SEQ ID NO: 876.
- the hinge or spacer comprises a KIR2DS2 hinge.
- such a domain can contain, e.g., one or more of a primary signaling domain and/or a costimulatory signaling domain.
- the intracellular signaling domain comprises a sequence encoding a primary signaling domain.
- the intracellular signaling domain comprises a costimulatory signaling domain.
- the intracellular signaling domain comprises a primary signaling domain and a costimulatory signaling domain.
- the intracellular signaling sequences within the cytoplasmic portion of the CAR may be linked to each other in a random or specified order.
- a short oligo- or polypeptide linker for example, between 2 and 10 amino acids (e.g., 2, 3, 4, 5, 6, 7, 8, 9, or 10 amino acids) in length may form the linkage between intracellular signaling sequences.
- a glycine-serine doublet can be used as a suitable linker.
- a single amino acid e.g., an alanine, a glycine, can be used as a suitable linker.
- the intracellular signaling domain is designed to comprise two or more, e.g., 2, 3, 4, 5, or more, costimulatory signaling domains.
- the two or more, e.g., 2, 3, 4, 5, or more, costimulatory signaling domains are separated by a linker molecule, e.g., a linker molecule described herein.
- the intracellular signaling domain comprises two costimulatory signaling domains.
- the linker molecule is a glycine residue. In some embodiments, the linker is an alanine residue.
- a primary signaling domain regulates primary activation of the TCR complex either in a stimulatory way, or in an inhibitory way.
- Primary intracellular signaling domains that act in a stimulatory manner may contain signaling motifs which are known as immunoreceptor tyrosine-based activation motifs or ITAMs. In CARs such domains are used for the same purpose.
- ITAM containing primary intracellular signaling domains include those of CD3 zeta, common FcR gamma (FCER1G), Fc gamma Rlla, FcR beta (Fc Epsilon Rib), CD3 gamma, CD3 delta, CD3 epsilon, CD79a, CD79b, DAP 10, and DAP 12.
- a CAR comprises an intracellular signaling domain, e.g., a primary signaling domain of CD3-zeta.
- the encoded primary signaling domain comprises a Junctional signaling domain of CD3 zeta.
- the encoded CD3 zeta primary signaling domain can comprise an amino acid sequence having at least one, two or three modifications but not more than 20, 10 or 5 modifications of an amino acid sequence of SEQ ID NO: 18 or SEQ ID NO: 20, or a sequence with 95-99% identity to an amino acid sequence of SEQ ID NO: 18 or SEQ ID NO: 20.
- the encoded primary signaling domain comprises a sequence of SEQ ID NO: 18 or SEQ ID NO: 20.
- the nucleic acid sequence encoding the primary signaling domain comprises a sequence of SEQ ID NO: 19 or SEQ ID NO: 21, or a sequence with 95-99% identity thereto.
- the encoded intracellular signaling domain comprises a costimulatory signaling domain.
- the intracellular signaling domain can comprise a primary signaling domain and a costimulatory signaling domain.
- the encoded costimulatory signaling domain comprises a functional signaling domain of a protein chosen from one or more of CD27, CD28, 4-1BB (CD137), 0X40, CD30, CD40, PD-1, ICOS, lymphocyte function-associated antigen-1 (LFA-1), CD2, CD7, LIGHT, NKG2C, B7-H3, a ligand that specifically binds with CD83, ICAM-1, GITR, BAFFR, HVEM (LIGHTR), SLAMF7, NKp80 (KLRF1), CD160, CD19, CD4, CD8alpha, CD8beta, IL2Rbeta, IL2R gamma, IL7R alpha, ITGA4, VLA1, CD49a, ITGA4, IA4,
- the encoded costimulatory signaling domain comprises an amino acid sequence having at least one, two or three modifications but not more than 20, 10 or 5 modifications of an amino acid sequence of SEQ ID NO: 14 or SEQ ID NO: 16, or a sequence with 95-99% identity to an amino acid sequence of SEQ ID NO: 14 or SEQ ID NO: 16.
- the encoded costimulatory signaling domain comprises a sequence of SEQ ID NO: 14 or SEQ ID NO: 16.
- the nucleic acid sequence encoding the costimulatory signaling domain comprises a sequence of SEQ ID NO: 15 or SEQ ID NO: 17, or a sequence with 95-99% identity thereto.
- the encoded intracellular domain comprises the sequence of SEQ ID NO: 14 or SEQ ID NO: 16, and the sequence of SEQ ID NO: 18 or SEQ ID NO: 20, wherein the sequences comprising the intracellular signaling domain are expressed in the same frame and as a single polypeptide chain.
- the nucleic acid sequence encoding the intracellular signaling domain comprises a sequence of SEQ ID NO: 15 or SEQ ID NO: 17, or a sequence with 95-99% identity thereto, and a sequence of SEQ ID NO: 19 or SEQ ID NO:21, or a sequence with 95-99% identity thereto.
- the nucleic acid molecule fiirther encodes a leader sequence.
- the leader sequence comprises the sequence of SEQ ID NO: 2.
- the intracellular signaling domain is designed to comprise the signaling domain of CD3-zeta and the signaling domain of CD28. In some aspects, the intracellular signaling domain is designed to comprise the signaling domain of CD3-zeta and the signaling domain of 4-1BB. In some aspects, the signaling domain of 4-1BB is a signaling domain of SEQ ID NO: 14. In some aspects, the signaling domain of CD3-zeta is a signaling domain of SEQ ID NO: 18.
- the intracellular signaling domain is designed to comprise the signaling domain of CD3-zeta and the signaling domain of CD27.
- the signaling domain of CD27 comprises an amino acid sequence of QRRKYRSNKGESPVEPAEPCRYSCPREEEGSTIPIQEDYRKPEPACSP (SEQ ID NO: 16).
- the signaling domain of CD27 is encoded by a nucleic acid sequence of AGGAGTAAGAGGAGCAGGCTCCTGCACAGTGACTACATGAACATGACTCCCCGCCGCCCCG GGCCCACCCGCAAGCATTACCAGCCCTATGCCCCACCACGCGACTTCGCAGCCTATCGCTCC (SEQ ID NO: 17).
- the vector comprises a nucleic acid sequence that encodes a CAR, e.g., a CAR described herein, and a nucleic acid sequence that encodes an inhibitory molecule comprising: an inhibitor KIR (inhKIR) cytoplasmic domain; a transmembrane domain, e.g., a KIR transmembrane domain; and an inhibitor cytoplasmic domain, e.g., an ITIM domain, e.g., an inhKIR ITIM domain.
- KIR inhKIR
- the inhibitory molecule is a naturally occurring inhKIR, or a sequence sharing at least 50, 60, 70, 80, 85, 90, 95, or 99% homology with, or that differs by no more than 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, or 20 residues from, a naturally occurring inhKIR.
- the nucleic acid sequence that encodes an inhibitory molecule comprises: a SLAM family cytoplasmic domain; a transmembrane domain, e.g., a SLAM family transmembrane domain; and an inhibitor cytoplasmic domain, e.g., a SLAM family domain, e.g., a SLAM family ITIM domain.
- the inhibitory molecule is a naturally occurring SLAM family member, or a sequence sharing at least 50, 60, 70, 80, 85, 90, 95, or 99% homology with, or that differs by no more than 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, or 20 residues from, a naturally occurring SLAM family member.
- the vector is an in vitro transcribed vector, e.g., a vector that transcribes RNA of a nucleic acid molecule described herein.
- the nucleic acid sequence in the vector further comprises a poly(A) tail, e.g., a poly A tail.
- the nucleic acid sequence in the vector further comprises a 3’UTR, e.g., a 3’ UTR described herein, e.g., comprising at least one repeat of a 3’UTR derived from human beta-globulin.
- the nucleic acid sequence in the vector further comprises promoter, e.g., a T2A promoter.
- the vector further comprises a promoter.
- the promoter is chosen from an EF-1 promoter, a CMV IE gene promoter, an EF-1 a promoter, an ubiquitin C promoter, or a phosphoglycerate kinase (PGK) promoter.
- the promoter is an EF-1 promoter.
- the EF-1 promoter comprises a sequence of SEQ ID NO: 1.
- immune effector cells can be obtained from a unit of blood collected from a subject using any number of techniques known to the skilled artisan, such as FicollTM separation.
- cells from the circulating blood of an individual are obtained by apheresis.
- the apheresis product typically contains lymphocytes, including T cells, monocytes, granulocytes, B cells, other nucleated white blood cells, red blood cells, and platelets.
- the cells collected by apheresis may be washed to remove the plasma fraction and, optionally, to suspend the cells in a buffer or medium for subsequent processing steps.
- the cells are washed with phosphate buffered saline (PBS).
- the wash solution lacks calcium and may lack magnesium or may lack many if not all divalent cations.
- Lentiviral vectors described herein can be used, e.g., in the in vitro manufacture of CAR-T cells.
- CARTs disclosed herein can be manufactured ex vivo by any known methods in the art. For example, methods described in WO2012/079000, or W02020/047452 (both incorporated herein by reference) may be used. CARTs disclosed herein can also be manufactured in vivo by any known methods in the art. For example, methods described in WO2020/176397 (incorporated herein by reference). In vivo CART production is also described, e.g., in WO/2022/040586 (incorporated herein by reference in its entirety).
- An immune effector cell may express one CAR, or two or more CARs.
- the methods disclosed herein may manufacture immune effector cells engineered to express one or more CARs in less than 24 hours.
- the methods provided herein preserve the undifferentiated phenotype of T cells, such as naive T cells, during the manufacturing process. These CAR-expressing cells with an undifferentiated phenotype may persist longer and/or expand better in vivo after infusion.
- CART cells produced by the manufacturing methods provided herein comprise a higher percentage of stem cell memory T cells, compared to CART cells produced by the traditional manufacturing process, e.g., as measured using scRNA-seq.
- CART cells produced by the manufacturing methods provided herein comprise a higher percentage of effector T cells, compared to CART cells produced by the traditional manufacturing process, e.g., as measured using scRNA-seq.
- CART cells produced by the manufacturing methods provided herein better preserve the sternness of T cells, compared to CART cells produced by the traditional manufacturing process, e.g., as measured using scRNA-seq.
- CART cells produced by the manufacturing methods provided herein show a lower level of hypoxia, compared to CART cells produced by the traditional manufacturing process, e.g., as measured using scRNA-seq.
- CART cells produced by the manufacturing methods provided herein show a lower level of autophagy, compared to CART cells produced by the traditional manufacturing process, e.g., as measured using scRNA-seq.
- the immune effector cells are engineered to comprise a nucleic acid molecule encoding one or more CARs disclosed herein.
- the methods disclosed herein do not involve using a bead, such as Dynabeads® (for example, CD3/CD28 Dynabeads®), and do not involve a de-beading step.
- the CART cells manufactured by the methods disclosed herein may be administered to a subject with minimal ex vivo expansion, for example, less than 1 day, less than 12 hours, less than 8 hours, less than 6 hours, less than 4 hours, less than 3 hours, less than 2 hours, less than 1 hour, or no ex vivo expansion. Accordingly, the methods described herein provide a fast manufacturing process of making improved CAR-expressing cell products for use in treating a disease in a subject.
- the present disclosure provides methods of making a population of cells (for example, T cells) that express a chimeric antigen receptor (CAR) comprising: (i) contacting a population of cells (for example, T cells, for example, T cells isolated from a frozen or fresh leukapheresis product) with an agent that stimulates a CD3/TCR complex and/or an agent that stimulates a costimulatory molecule on the surface of the cells; (ii) contacting the population of cells (for example, T cells) with a nucleic acid molecule(s) (for example, a DNA or RNA molecule) encoding the CAR(s), thereby providing a population of cells (for example, T cells) comprising the nucleic acid molecule, and (iii) harvesting the population of cells (for example, T cells) for storage (for example, reformulating the population of cells in cryopreservation media) or administration, wherein: (a) step (ii) is performed together with step (i) or no
- the nucleic acid molecule in step (ii) is a DNA molecule. In some embodiments, the nucleic acid molecule in step (ii) is an RNA molecule. In some embodiments, the nucleic acid molecule in step (ii) is on a viral vector, for example, a viral vector chosen from a lentivirus vector, an adenoviral vector, or a retrovirus vector. In some embodiments, the nucleic acid molecule in step (ii) is on a non- viral vector. In some embodiments, the nucleic acid molecule in step (ii) is on a plasmid. In some embodiments, the nucleic acid molecule in step (ii) is not on any vector. In some embodiments, step (ii) comprises transducing the population of cells (for example, T cells) a viral vector(s) comprising a nucleic acid molecule encoding the CAR(s).
- the population of cells (for example, T cells) is collected from an apheresis sample (for example, a leukapheresis sample) from a subject.
- an apheresis sample for example, a leukapheresis sample
- the apheresis sample (for example, a leukapheresis sample) is collected from the subject and shipped as a frozen sample (for example, a cryopreserved sample) to a cell manufacturing facility. Then the frozen apheresis sample is thawed, and T cells (for example, CD4+ T cells and/or CD8+ T cells) are selected from the apheresis sample, for example, using a cell sorting machine (for example, a CliniMACS® Prodigy® device). The selected T cells (for example, CD4+ T cells and/or CD8+ T cells) are then seeded for CART manufacturing using the activation process described herein. In some embodiments, the selected T cells (for example, CD4+ T cells and/or CD8+ T cells) undergo one or more rounds of freeze-thaw before being seeded for CART manufacturing.
- T cells for example, CD4+ T cells and/or CD8+ T cells
- the apheresis sample (for example, a leukapheresis sample) is collected from the subject and shipped as a fresh product (for example, a product that is not frozen) to a cell manufacturing facility.
- T cells for example, CD4+ T cells and/or CD 8+ T cells
- the selected T cells are then seeded for CART manufacturing using the activation process described herein.
- the selected T cells undergo one or more rounds of freeze-thaw before being seeded for CART manufacturing.
- the apheresis sample (for example, a leukapheresis sample) is collected from the subject.
- T cells for example, CD4+ T cells and/or CD8+ T cells
- the selected T cells are then shipped as a frozen sample (for example, a cryopreserved sample) to a cell manufacturing facility.
- the selected T cells are later thawed and seeded for CART manufacturing using the activation process described herein.
- cells for example, T cells
- a vector for example, a lentiviral vector
- a CAR e.g. one or more CARs
- brief CD3 and CD28 stimulation may promote efficient transduction of self-renewing T cells.
- the activation process provided herein does not involve prolonged ex vivo expansion. Similar to the cytokine process, the activation process provided herein also preserves undifferentiated T cells during CART manufacturing.
- the population of cells is contacted with an agent that stimulates a CD3/TCR complex and/or an agent that stimulates a costimulatory molecule on the surface of the cells.
- the agent that stimulates a CD3/TCR complex is an agent that stimulates CD3.
- the agent that stimulates a costimulatory molecule is an agent that stimulates CD28, ICOS, CD27, HVEM, LIGHT, CD40, 4-1BB, 0X40, DR3, GITR, CD30, TIM1, CD2, CD226, or any combination thereof.
- the agent that stimulates a costimulatory molecule is an agent that stimulates CD28.
- the agent that stimulates a CD3/TCR complex is chosen from an antibody (for example, a single-domain antibody (for example, a heavy chain variable domain antibody), a peptibody, a Fab fragment, or a scFv), a small molecule, or a ligand (for example, a naturally-existing, recombinant, or chimeric ligand).
- the agent that stimulates a CD3/TCR complex is an antibody.
- the agent that stimulates a CD3/TCR complex is an anti-CD3 antibody.
- the agent that stimulates a costimulatory molecule is chosen from an antibody (for example, a single-domain antibody (for example, a heavy chain variable domain antibody), a peptibody, a Fab fragment, or a scFv), a small molecule, or a ligand (for example, a naturally-existing, recombinant, or chimeric ligand).
- the agent that stimulates a costimulatory molecule is an antibody.
- the agent that stimulates a costimulatory molecule is an anti-CD28 antibody.
- the agent that stimulates a CD3/TCR complex or the agent that stimulates a costimulatory molecule does not comprise a bead.
- the agent that stimulates a CD3/TCR complex comprises an anti-CD3 antibody covalently attached to a colloidal polymeric nanomatrix.
- the agent that stimulates a costimulatory molecule comprises an anti-CD28 antibody covalently attached to a colloidal polymeric nanomatrix.
- the agent that stimulates a CD3/TCR complex and the agent that stimulates a costimulatory molecule comprise T Cell TransActTM.
- the matrix comprises or consists of a polymeric, for example, biodegradable or biocompatible inert material, for example, which is non-toxic to cells.
- the matrix is composed of hydrophilic polymer chains, which obtain maximal mobility in aqueous solution due to hydration of the chains.
- the mobile matrix may be of collagen, purified proteins, purified peptides, polysaccharides, glycosaminoglycans, or extracellular matrix compositions.
- a polysaccharide may include for example, cellulose ethers, starch, gum arabic, agarose, dextran, chitosan, hyaluronic acid, pectins, xanthan, guar gum or alginate.
- polymers may include polyesters, polyethers, poly acrylates, polyacrylamides, polyamines, polyethylene imines, polyquatemium polymers, polyphosphazenes, polyvinylalcohols, poly vinylacetates, polyvinylpyrrolidones, block copolymers, or polyurethanes.
- the mobile matrix is a polymer of dextran.
- the population of cells is contacted with a nucleic acid molecule (e.g. one or more nucleic acid molecules) encoding a CAR (e.g. one or more CARs).
- a nucleic acid molecule e.g. one or more nucleic acid molecules
- the population of cells is transduced with a DNA molecule (e.g. one or more DNA molecules) encoding a CAR (e.g. one or more CARs).
- each of the vectors containing nucleic acid molecules encoding the CAR can be added to the reaction mixture (e.g., containing a cell population) at a different multiplicity of infection (MOI).
- MOI multiplicity of infection
- MOIs for the vectors containing nucleic acid molecules which encode distinct CAR molecules may affect the final composition of the cellular population.
- different MOIs can be used to maximize the percent of preferred mono CART cells and dual CART cells, while resulting in fewer undesired mono CART cells and untransduced cells.
- contacting the population of cells with the nucleic acid molecule(s) encoding the CAR(s) occurs simultaneously with contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above.
- contacting the population of cells with the nucleic acid molecule(s) encoding the CAR(s) occurs no later than 30, 29, 28, 27, 26, 25, 24, 23, 22, 21, 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, 3, 2, 1, or 0.5 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above.
- contacting the population of cells with the nucleic acid molecule(s) encoding the CAR(s) occurs no later than 20 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above. In some embodiments, contacting the population of cells with the nucleic acid molecule(s) encoding the CAR(s) occurs no later than 19 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above.
- contacting the population of cells with the nucleic acid molecule(s) encoding the CAR(s) occurs no later than 18 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above. In some embodiments, contacting the population of cells with the nucleic acid molecule(s) encoding the CAR(s) occurs no later than 17 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above.
- contacting the population of cells with the nucleic acid molecule(s) encoding the CAR(s) occurs no later than 16 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above. In some embodiments, contacting the population of cells with the nucleic acid molecule(s) encoding the CAR(s) occurs no later than 15 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above.
- contacting the population of cells with the nucleic acid molecule(s) encoding the CAR(s) occurs no later than 14 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above. In some embodiments, contacting the population of cells with the nucleic acid molecule(s) encoding the CAR(s) occurs no later than 14 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above.
- contacting the population of cells with the nucleic acid molecule(s) encoding the CAR(s) occurs no later than 13 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above. In some embodiments, contacting the population of cells with the nucleic acid molecule(s) encoding the CAR(s) occurs no later than 12 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above.
- contacting the population of cells with the nucleic acid molecule(s) encoding the CAR(s) occurs no later than 11 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above. In some embodiments, contacting the population of cells with the nucleic acid molecule(s) encoding the CAR(s) occurs no later than 10 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above.
- contacting the population of cells with the nucleic acid molecule(s) encoding the CAR(s) occurs no later than 9 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above. In some embodiments, contacting the population of cells with the nucleic acid molecule(s) encoding the CAR(s) occurs no later than 8 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above.
- contacting the population of cells with the nucleic acid molecule(s) encoding the CAR(s) occurs no later than 7 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above. In some embodiments, contacting the population of cells with the nucleic acid molecule(s) encoding the CAR(s) occurs no later than 6 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above.
- contacting the population of cells with the nucleic acid molecule(s) encoding the CAR(s) occurs no later than 5 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above. In some embodiments, contacting the population of cells with the nucleic acid molecule(s) encoding the CAR(s) occurs no later than 4 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above.
- contacting the population of cells with the nucleic acid molecule(s) encoding the CAR(s) occurs no later than 3 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above. In some embodiments, contacting the population of cells with the nucleic acid molecule encoding the CAR(s) occurs no later than 2 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above.
- contacting the population of cells with the nucleic acid molecule(s) encoding the CAR(s) occurs no later than 1 hour after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above. In some embodiments, contacting the population of cells with the nucleic acid molecule(s) encoding the CAR(s) occurs no later than 30 minutes after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above.
- the population of cells is harvested for storage or administration.
- the population of cells is harvested for storage or administration no later than 72, 60, 48, 36, 32, 31, 30, 29, 28, 27 , 26, 25, 24, 23, 22, 21, 20, 19, or 18 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above. In some embodiments, the population of cells is harvested for storage or administration no later than 26 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above.
- the population of cells is harvested for storage or administration no later than 25 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above. In some embodiments, the population of cells is harvested for storage or administration no later than 24 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above.
- the population of cells is harvested for storage or administration no later than 23 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above. In some embodiments, the population of cells is harvested for storage or administration no later than 22 hours after the beginning of contacting the population of cells with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above.
- the population of cells is not expanded ex vivo.
- the population of cells is expanded by no more than 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 25, 30, 35, 40, 45, 50, 55, or 60%, for example, as assessed by the number of living cells, compared to the population of cells before it is contacted with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above.
- the population of cells is expanded by no more than 5%, for example, as assessed by the number of living cells, compared to the population of cells before it is contacted with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above.
- the population of cells is expanded by no more than 10%, for example, as assessed by the number of living cells, compared to the population of cells before it is contacted with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above. In some embodiments, the population of cells is expanded by no more than 15%, for example, as assessed by the number of living cells, compared to the population of cells before it is contacted with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above.
- the population of cells is expanded by no more than 20%, for example, as assessed by the number of living cells, compared to the population of cells before it is contacted with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above. In some embodiments, the population of cells is expanded by no more than 25%, for example, as assessed by the number of living cells, compared to the population of cells before it is contacted with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above.
- the population of cells is expanded by no more than 30%, for example, as assessed by the number of living cells, compared to the population of cells before it is contacted with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above. In some embodiments, the population of cells is expanded by no more than 35%, for example, as assessed by the number of living cells, compared to the population of cells before it is contacted with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above.
- the population of cells is expanded by no more than 40%, for example, as assessed by the number of living cells, compared to the population of cells before it is contacted with the agent that stimulates a CD3/TCR complex and/or the agent that stimulates a costimulatory molecule on the surface of the cells described above.
- the population of cells is expanded by no more than 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 6, 7, 8, 9, 10, 11, 12, 16, 20, 24, 36, or 48 hours, for example, as assessed by the number of living cells, compared to the population of cells before it is contacted with the one or more cytokines described above.
- the activation process is conducted in serum free cell media. In some embodiments, the activation process is conducted in cell media comprising one or more cytokines chosen from: IL-2, IL-15 (for example, hetIL-15 (IL15/sIL-15Ra)), or IL-6 (for example, IL-6/sIL-6Ra).
- cytokines chosen from: IL-2, IL-15 (for example, hetIL-15 (IL15/sIL-15Ra)), or IL-6 (for example, IL-6/sIL-6Ra).
- hetIL-15 comprises the amino acid sequence of NWVNVISDLKKIEDLIQSMHIDATLYTESDVHPSCKVTAMKCFLLELQVISLESGDASIHDTVENL IILANNSLSSNGNVTESGCKECEELEEKNIKEFLQSFVHIVQMFINTSITCPPPMSVEHADIWVKSY SLYSRERYICNSGFKRKAGTSSLTECVLNKATNVAHWTTPSLKCIRDPALVHQRPAPPSTVTTAG VTPQPESLSPSGKEPAASSPSSNNTAATTAAIVPGSQLMPSKSPSTGTTEISSHESSHGTPSQTTAK NWELTASASHQPPGVYPQG (SEQ ID NO: 309).
- hetIL-15 comprises an amino acid sequence having at least about 70, 75, 80, 85, 90, 95, or 99% identity to SEQ ID NO: 309.
- the activation process is conducted in cell media comprising a LSD1 inhibitor.
- the activation process is conducted in cell media comprising a MALT 1 inhibitor.
- the serum free cell media comprises a serum replacement.
- the serum replacement is CTSTM Immune Cell Serum Replacement (ICSR).
- the level of ICSR can be, for example, up to 5%, for example, about 1%, 2%, 3%, 4%, or 5%.
- using cell media for example, Rapid Media shown in Table 21 or Table 25, comprising ICSR, for example, 2% ICSR, may improve cell viability during a manufacture process described herein.
- the present disclosure provides methods of making a population of cells (for example, T cells) that express a chimeric antigen receptor (CAR) comprising: (a) providing an apheresis sample (for example, a fresh or cryopreserved leukapheresis sample) collected from a subject; (b) selecting T cells from the apheresis sample (for example, using negative selection, positive selection, or selection without beads); (c) seeding isolated T cells at, for example, 1 x 10 6 to 1 x 10 7 cells/mL; (d) contacting T cells with an agent that stimulates T cells, for example, an agent that stimulates a CD3/TCR complex and/or an agent that stimulates a costimulatory molecule on the surface of the cells (for example, contacting T cells with anti-CD3 and/or anti-CD28 antibody, for example, contacting T cells with TransAct); (e) contacting T cells with a nucleic acid molecule(s) (for example, a
- a population of cells for example, immune effector cells, for example, T cells or NK cells
- a population of cells made by any of the manufacturing processes described herein.
- the percentage of naive cells, for example, naive T cells, for example, CD45RA+ CD45RO- CCR7+ T cells, in the population of cells at the end of the manufacturing process (for example, at the end of the cytokine process or the activation process described herein) (1) is the same as, (2) differs, for example, by no more than 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, or 15%, from, or (3) is increased, for example, by at least 5, at least 6, at least 7, at least 8, at least 9, at least 10, at least 11, at least 12, at least 13, at least 14, at least 15, at least 16, at least 17, at least 18, at least 19, at least 20, at least 21, at least 22, at least 23, at least 24, or at least 25%, as compared to, the percentage of naive cells, for example, naive T cells, for example, CD45RA+ CD45RO- CCR7+ cells, in the population of cells at the beginning of the manufacturing process (for example, at the beginning of the cytokine process or the activation
- the population of cells at the end of the manufacturing process shows a higher percentage of naive cells, for example, naive T cells, for example, CD45RA+ CD45RO- CCR7+ T cells (for example, at least 5, at least 6, at least 7, at least 8, at least 9, at least 10, at least 11, at least 12, at least 13, at least 14, at least 15, at least 16, at least 17, at least 18, at least 19, at least 20, at least 25, at least 30, at least 35, at least 40, at least 45, or at least 50% higher), compared with cells made by an otherwise similar method which lasts, for example, more than 26 hours (for example, which lasts more than 5, more than 6, more than 7, more than 8, more than 9, more than 10, more than 11, or more than 12 days) or which involves expanding the population of cells in vitro for, for example, more than 3 days (for example, expanding the population of cells in vitro for 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, or
- the percentage of naive cells, for example, naive T cells, for example, CD45RA+ CD45RO- CCR7+ T cells, in the population of cells at the end of the manufacturing process (for example, at the end of the cytokine process or the activation process described herein) is not less than 20, less than 25, less than 30, less than 35, less than 40, less than 45, less than 50, less than 55, or less than 60%.
- the percentage of central memory cells, for example, central memory T cells, for example, CD95+ central memory T cells, in the population of cells at the end of the manufacturing process (for example, at the end of the cytokine process or the activation process described herein) (1) is the same as, (2) differs, for example, by no more than 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, or 15% from, or (3) is decreased, for example, by at least 5, at least 6, at least 7, at least 8, at least 9, at least 10, at least 11, at least 12, at least 13, at least 14, at least 15, at least 16, at least 17, at least 18, at least 19, at least 20, at least 21, at least 22, at least 23, at least 24, or at least 25%, as compared to, the percentage of central memory cells, for example, central memory T cells, for example, CD95+ central memory T cells, in the population of cells at the beginning of the manufacturing process (for example, at the beginning of the cytokine process or the activation process described herein).
- the population of cells at the end of the manufacturing process shows a lower percentage of central memory cells, for example, central memory T cells, for example, CD95+ central memory T cells (for example, at least 5, at least 6, at least 7, at least 8, at least 9, at least 10, at least 11, at least 12, at least 13, at least 14, at least 15, at least 16, at least 17, at least 18, at least 19, at least 20, at least 25, at least 30, at least 35, at least 40, at least 45, or at least 50% lower), compared with cells made by an otherwise similar method which lasts, for example, more than 26 hours (for example, which lasts more than 5, 6, 7, 8, 9, 10, 11, or 12 days) or which involves expanding the population of cells in vitro for, for example, more than 3 days (for example, expanding the population of cells in vitro for 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, or 15 days).
- central memory T cells for example, CD95+ central memory T cells (for example, at least 5, at least 6, at least 7, at least 8, at least 9, at least 10, at least 11, at least 12, at least 13, at least
- the percentage of central memory cells, for example, central memory T cells, for example, CD95+ central memory T cells, in the population of cells at the end of the manufacturing process is no more than 40, no more than 45, no more than 50, no more than 55, no more than 60, no more than 65, no more than 70, no more than 75, or no more than 80%.
- the population of cells at the end of the manufacturing process (for example, at the end of the cytokine process or the activation process described herein) after being administered in vivo, persists longer or expands at a higher level (for example, at least 20, at least 25, at least 30, at least 35, at least 40, at least 45, at least 50, at least 55, at least 60, at least 65, at least 70, at least 75, at least 80, at least 85, or at least 90% higher), compared with cells made by an otherwise similar method which lasts, for example, more than 26 hours (for example, which lasts more than 5, more than 6, more than 7, more than 8, more than 9, more than 10, more than 11, or more than 12 days) or which involves expanding the population of cells in vitro for, for example, more than 3 days (for example, expanding the population of cells in vitro for 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, or 15 days).
- a higher level for example, at least 20, at least 25, at least 30, at least 35, at least 40, at least 45, at least 50,
- the population of cells has been enriched for IL6R-expressing cells (for example, cells that are positive for IL6Ra and/or I L6 Kb) prior to the beginning of the manufacturing process (for example, prior to the beginning of the cytokine process or the activation process described herein).
- IL6R-expressing cells for example, cells that are positive for IL6Ra and/or I L6 Kb
- the population of cells comprises, for example, no less than 30, no less than 35, no less than 40, no less than 45, no less than 50, no less than 55, no less than 60, no less than 65, no less than 70, no less than 75, or no less than 80% of IL6R-expressing cells (for example, cells that are positive for IL6Ra and/or I L6 Kb) at the beginning of the manufacturing process (for example, at the beginning of the cytokine process or the activation process described herein).
- IL6R-expressing cells for example, cells that are positive for IL6Ra and/or I L6 Kb
- Lentiviral vectors described herein can be used, e.g., in the in vitro manufacture of CAR-T cells.
- cells transduced with the viral vector as described herein are expanded, e.g., by a method described herein.
- the cells are expanded in culture for a period of several hours (e.g., about 2, about 3, about 4, about 5, about 6, about 7, about 8, about 9, about 10, about 15, about 18, about 21 hours) to about 14 days (e.g., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 or 14 days).
- the cells are expanded for a period of 4 to 9 days.
- the cells are expanded for a period of 8 days or less, e.g., 7, 6 or 5 days.
- the cells are expanded in culture for 5 days, and the resulting cells are more potent than the same cells expanded in culture for 9 days under the same culture conditions. Potency can be defined, e.g., by various T cell functions, e.g. proliferation, target cell killing, cytokine production, activation, migration, or combinations thereof.
- the cells are expanded for 5 days show at least one, two, three, or four-fold increase in cells doublings upon antigen stimulation as compared to the same cells expanded in culture for 9 days under the same culture conditions.
- the cells are expanded in culture for 5 days, and the resulting cells exhibit higher proinflammatory cytokine production, e.g., IFN-y and/or GM-CSF levels, as compared to the same cells expanded in culture for 9 days under the same culture conditions.
- the cells expanded for 5 days show at least a one, two, three, four, five, ten-fold or more increase in pg/ml of proinflammatory cytokine production, e.g., IFN-y and/or GM-CSF levels, as compared to the same cells expanded in culture for 9 days under the same culture conditions.
- a washing step may be accomplished by methods known to those in the art, such as by using a semi-automated “flow-through” centrifuge (for example, the Cobe 2991 cell processor, the Baxter CytoMate, or the Haemonetics Cell Saver 5) according to the manufacturer’s instructions.
- the cells may be resuspended in a variety of biocompatible buffers, such as, for example, Ca-free, Mg-free PBS, PlasmaLyte A, or other saline solution with or without buffer.
- the undesirable components of the apheresis sample may be removed, and the cells directly resuspended in culture media.
- the in vitro methods of the application can utilize culture media conditions comprising 5% or less, for example 2%, human AB serum, and employ known culture media conditions and compositions, for example those described in Smith et al., “Ex vivo expansion of human T cells for adoptive immunotherapy using the novel Xeno-free CTS Immune Cell Serum Replacement” Clinical & Translational Immunology (2015) 4, e31; doi: 10. 1038/cti.2014.31.
- T cells are isolated from peripheral blood lymphocytes by lysing the red blood cells and depleting the monocytes, for example, by centrifugation through a PERCOLLTM gradient or by counterflow centrifugal elutriation.
- the isolated T cells may be further used in the methods described herein.
- the methods described herein can include, e.g., selection of a specific subpopulation of immune effector cells, e.g., T cells, that are a T regulatory cell-depleted population, CD25+ depleted cells, using, e.g., a negative selection technique, e.g., described herein.
- the population of T regulatory depleted cells contains less than 30%, less than 25%, less than 20%, less than 15%, less than 10%, less than 5%, less than 4%, less than 3%, less than 2%, or less than 1% of CD25+ cells.
- T regulatory cells e.g., CD25+ T cells
- T regulatory cells are removed from the population using an anti-CD25 antibody, or fragment thereof, or a CD25 -binding ligand, IL-2.
- the anti-CD25 antibody, or fragment thereof, or CD25-binding ligand is conjugated to a substrate, e.g., a bead, or is otherwise coated on a substrate, e.g., a bead.
- the anti- CD25 antibody, or fragment thereof is conjugated to a substrate as described herein.
- the T regulatory cells e.g., CD25+ T cells, are removed from the population using CD25 depletion reagent from MiltenyiTM.
- the ratio of cells to CD25 depletion reagent is 1 x 10 7 cells to 20 pL, or 1 x 10 7 cells to 15 pL, or 1 x 10 7 cells to 10 pL, or 1 x 10 7 cells to 5 pL, or 1 x 10 7 cells to 2.5 pL, or 1 x 10 7 cells to 1.25 pL.
- T regulatory cells e.g., CD25+ depletion
- greater than 500 million cells/ml is used.
- a concentration of cells of 600, 700, 800, or 900 million cells/ml is used.
- the population of immune effector cells to be depleted includes about 6 x 10 9 CD25+ T cells. In other aspects, the population of immune effector cells to be depleted include about 1 x 10 9 to lx 10 10 CD25+ T cell, and any integer value in between. In some embodiments, the resulting population T regulatory depleted cells has 2 x 10 9 T regulatory cells, e.g., CD25+ cells, or less (e.g., 1 x 10 9 , 5 x 10 8 , 1 x 10 8 , 5 x 10 7 , 1 x 10 7 , or less CD25+ cells).
- the T regulatory cells e.g., CD25+ cells
- a depletion tubing set such as, e.g., tubing 162-01.
- the CliniMAC system is run on a depletion setting such as, e.g., DEPLETION2. 1.
- decreasing the level of negative regulators of immune cells e.g., decreasing the number of unwanted immune cells, e.g., TREG cells
- decreasing the level of negative regulators of immune cells e.g., decreasing the number of unwanted immune cells, e.g., TREG cells
- methods of depleting TREG cells are known in the art. Methods of decreasing TREG cells include, but are not limited to, cyclophosphamide, anti-GITR antibody (an anti-GITR antibody described herein), CD25-depletion, and combinations thereof.
- the manufacturing methods comprise reducing the number of (e.g., depleting) TREG cells prior to manufacturing of the CAR-expressing cell.
- manufacturing methods comprise contacting the sample, e.g., the apheresis sample, with an anti-GITR antibody and/or an anti-CD25 antibody (or fragment thereof, or a CD25-binding ligand), e.g., to deplete TREG cells prior to manufacturing ofthe CAR-expressing cell (e.g., T cell, NK cell) product.
- a subject is pre-treated with one or more therapies that reduce TREG cells prior to collection of cells for CAR-expressing cell product manufacturing, thereby reducing the risk of subject relapse to CAR-expressing cell treatment.
- methods of decreasing TREG cells include, but are not limited to, administration to the subject of one or more of cyclophosphamide, anti-GITR antibody, CD25 -depletion, or a combination thereof. Administration of one or more of cyclophosphamide, anti-GITR antibody, CD25 -depletion, or a combination thereof, can occur before, during or after an infusion of the CAR-expressing cell product.
- a subject is pre-treated with cyclophosphamide prior to collection of cells for CAR-expressing cell product manufacturing, thereby reducing the risk of subject relapse to CAR- expressing cell treatment.
- a subject is pre-treated with an anti-GITR antibody prior to collection of cells for CAR-expressing cell product manufacturing, thereby reducing the risk of subject relapse to CAR-expressing cell treatment.
- the population of cells to be removed are neither the regulatory T cells or tumor cells, but cells that otherwise negatively affect the expansion and/or function of CART cells, e.g. cells expressing CD14, CD1 lb, CD33, CD15, or other markers expressed by potentially immune suppressive cells.
- such cells are envisioned to be removed concurrently with regulatory T cells and/or tumor cells, or following the depletion, or in another order.
- the methods described herein can include more than one selection step, e.g., more than one depletion step.
- Enrichment of a T cell population by negative selection can be accomplished, e.g., with a combination of antibodies directed to surface markers unique to the negatively selected cells.
- One method is cell sorting and/or selection via negative magnetic immunoadherence or flow cytometry that uses a cocktail of monoclonal antibodies directed to cell surface markers present on the cells negatively selected.
- a monoclonal antibody cocktail can include antibodies to CD14, CD20, GDI lb, CD16, HLA-DR, and CD8.
- the methods described herein can further include removing cells from the population which express a tumor antigen, e.g., a tumor antigen that does not comprise CD25, e.g., CD19, CD30, CD38, CD123, CD20, CD14 or GDI lb, to thereby provide a population of T regulatory depleted, e.g., CD25+ depleted, and tumor antigen depleted cells that are suitable for expression of a CAR, e.g., a CAR described herein.
- tumor antigen expressing cells are removed simultaneously with the T regulatory, e.g., CD25+ cells.
- an anti-CD25 antibody, or fragment thereof, and an anti-tumor antigen antibody, or fragment thereof can be attached to the same substrate, e.g., bead, which can be used to remove the cells or an anti-CD25 antibody, or fragment thereof, or the anti-tumor antigen antibody, or fragment thereof, can be attached to separate beads, a mixture of which can be used to remove the cells.
- the removal of T regulatory cells, e.g., CD25+ cells, and the removal of the tumor antigen expressing cells is sequential, and can occur, e.g., in either order.
- a check point inhibitor e.g., a check point inhibitor described herein, e.g., one or more of PD1+ cells, LAG3+ cells, and TIM3+ cells
- check point inhibitors include B7-H1, B7-1, CD160, P1H, 2B4, PD1, TIM3, CEACAM (e.g., CEACAM-1, CEACAM-3 and/or CEACAM-5), LAG3, TIGIT, CTLA-4, BTLA, and LAIR1.
- check point inhibitor expressing cells are removed simultaneously with the T regulatory, e.g., CD25+ cells.
- an anti-CD25 antibody, or fragment thereof, and an anti-check point inhibitor antibody, or fragment thereof can be attached to the same bead which can be used to remove the cells, or an anti-CD25 antibody, or fragment thereof, and the anti-check point inhibitor antibody, or fragment there, can be attached to separate beads, a mixture of which can be used to remove the cells.
- the removal of T regulatory cells, e.g., CD25+ cells, and the removal of the check point inhibitor expressing cells is sequential, and can occur, e.g., in either order.
- T cells can isolated by incubation with anti-CD3/anti-CD28 (e.g., 3x28)-conjugated beads, such as DYNABEADS® M-450 CD3/CD28 T, for a time period sufficient for positive selection of the desired T cells.
- the time period is about 30 minutes.
- the time period ranges from 30 minutes to 36 hours or longer and all integer values there between.
- the time period is at least 1, at least 2, at least 3, at least 4, at least 5, or at least 6 hours.
- the time period is 10 to 24 hours, e.g., 24 hours.
- TIL tumor infiltrating lymphocytes
- a T cell population can be selected that expresses one or more of IFN- ⁇ TNFa, IL-17A, IL-2, IL-3, IL-4, GM-CSF, IL- 10, IL- 13, granzyme B, and perforin, or other appropriate molecules, e.g., other cytokines.
- Methods for screening for cell expression can be determined, e.g., by the methods described in PCT Publication No.: WO 2013/126712.
- the concentration of cells and surface can be varied.
- it may be desirable to significantly decrease the volume in which beads and cells are mixed together e.g., increase the concentration of cells, to ensure maximum contact of cells and beads.
- a concentration of 10 billion cells/ml, 9 billion/ml, 8 billion/ml, 7 billion/ml, 6 billion/ml, or 5 billion/ml is used.
- a concentration of 1 billion cells/ml is used.
- a concentration of cells from 75, 80, 85, 90, 95, or 100 million cells/ml is used.
- concentrations of 125 or 150 million cells/ml can be used.
- Using high concentrations can result in increased cell yield, cell activation, and cell expansion.
- use of high cell concentrations allows more efficient capture of cells that may weakly express target antigens of interest, such as CD28-negative T cells, or from samples where there are many tumor cells present (e.g., leukemic blood, tumor tissue, etc.). Such populations of cells may have therapeutic value and would be desirable to obtain.
- using high concentration of cells allows more efficient selection of CD8+ T cells that normally have weaker CD28 expression.
- the concentration of cells used is 5 x 10 6 /ml. In other aspects, the concentration used can be from about 1 x 10 5 /ml to 1 x 10 6 /ml, and any integer value in between.
- the cells may be incubated on a rotator for varying lengths of time at varying speeds at either 2-10°C or at room temperature.
- T cells for stimulation can also be frozen after a washing step.
- the freeze and subsequent thaw step provide a more uniform product by removing granulocytes and to some extent monocytes in the cell population.
- the cells may be suspended in a freezing solution.
- one method involves using PBS containing 20% DMSO and 8% human serum albumin, or culture media containing 10% Dextran 40 and 5% Dextrose, 20% Human Serum Albumin and 7.5% DMSO, or 31.25% Plasmalyte-A, 31.25% Dextrose 5%, 0.45% NaCl, 10% Dextran 40 and 5% Dextrose, 20% Human Serum Albumin, and 7.5% DMSO or other suitable cell freezing media containing for example, Hespan and PlasmaLyte A, the cells then are frozen to -80°C at a rate of 1° per minute and stored in the vapor phase of a liquid nitrogen storage tank. Other methods of controlled freezing may be used as well as uncontrolled freezing immediately at -20° C or in liquid nitrogen.
- cryopreserved cells are thawed and washed as described herein and allowed to rest for one hour at room temperature prior to activation using the methods described herein.
- a blood sample or an apheresis product is taken from a generally healthy subject.
- a blood sample or an apheresis is taken from a generally healthy subject who is at risk of developing a disease, but who has not yet developed a disease, and the cells of interest are isolated and frozen for later use.
- the T cells may be expanded, frozen, and used at a later time.
- samples are collected from a patient shortly after diagnosis of a particular disease as described herein but prior to any treatments.
- the cells are isolated from a blood sample or an apheresis from a subject prior to any number of relevant treatment modalities, including but not limited to treatment with agents such as natalizumab, efalizumab, antiviral agents, chemotherapy, radiation, immunosuppressive agents, such as cyclosporin, azathioprine, methotrexate, mycophenolate, and FK506, antibodies, or other immunoablative agents such as CAMPATH, anti-CD3 antibodies, cytoxan, fludarabine, cyclosporin, FK506, rapamycin, mycophenolic acid, steroids, FR901228, and irradiation.
- agents such as natalizumab, efalizumab, antiviral agents, chemotherapy, radiation, immunosuppressive agents, such as
- T cells are obtained from a patient directly following treatment that leaves the subject with functional T cells.
- the quality of T cells obtained may be optimal or improved for their ability to expand ex vivo.
- these cells may be in a preferred state for enhanced engraftment and in vivo expansion.
- mobilization for example, mobilization with GM-CSF
- conditioning regimens can be used to create a condition in a subject wherein repopulation, recirculation, regeneration, and/or expansion of particular cell types is favored, especially during a defined window of time following therapy.
- Illustrative cell types include T cells, B cells, dendritic cells, and other cells of the immune system.
- a T cell population is diaglycerol kinase (DGK)-deficient.
- DGK-deficient cells include cells that do not express DGK RNA or protein or have reduced or inhibited DGK activity.
- DGK-deficient cells can be generated by genetic approaches, e.g., administering RNA-interfering agents, e.g., siRNA, shRNA, miRNA, to reduce or prevent DGK expression.
- RNA-interfering agents e.g., siRNA, shRNA, miRNA
- DGK-deficient cells can be generated by treatment with DGK inhibitors described herein.
- a T cell population is Ikaros-deficient.
- Ikaros-deficient cells include cells that do not express Ikaros RNA or protein, or have reduced or inhibited Ikaros activity, Ikaros-deficient cells can be generated by genetic approaches, e.g., administering RNA-interfering agents, e.g., siRNA, shRNA, miRNA, to reduce or prevent Ikaros expression. Alternatively, Ikaros-deficient cells can be generated by treatment with Ikaros inhibitors, e.g., lenalidomide.
- a T cell population is DGK -deficient and Ikaros-deficient, e.g., does not express DGK and Ikaros, or has reduced or inhibited DGK and Ikaros activity.
- DGK and Ikaros-deficient cells can be generated by any of the methods described herein.
- the NK cells are obtained from the subject.
- the NK cells are an NK cell line, e.g., NK-92 cell line (Conkwest).
- subjects may undergo leukapheresis, wherein leukocytes are collected, enriched, or depleted ex vivo to select and/or isolate the cells of interest, e.g., T cells.
- T cell isolates may be expanded by methods described herein.
- Subjects in need thereof may subsequently undergo standard treatment with high dose chemotherapy followed by peripheral blood stem cell transplantation.
- subjects receive an infusion of the expanded CAR T cells as prepared by the methods described herein.
- expanded cells are administered before or following surgery.
- a formulation described herein e.g., a formulation comprising one or more of, e.g., all of, HEPES, L-Arginine, L-Proline, and sucrose
- In vivo CART is also described, e.g., in WO/2022/040586 (incorporated herein by reference in its entirety).
- the in vivo CART method comprises: (i) administering a biomaterial and a cell recruitment factor to a site (e.g., high subcutaneous space or subcutaneous space adjacent to dermis) in the subject, and (ii) administering a lentiviral vector comprising a transgene in a formulation described herein and optionally one or both of a particle and a cell activation agent to the subject; thereby transducing cells of the subject with the transgene.
- the biomaterial and the cell recruitment factor are administered in an amount sufficient to induce lymphangiogenesis and/or recruitment of T cells to the site of administration in the subject.
- the composition comprising the lentiviral vector is administered to the site in the subject that has undergone lymphangiogenesis.
- the particle may promote local positioning of the virus which could favor transduction of locally recruited T cells and that activation could further improve transduction, prevent leakage, and transduction of unwanted cells.
- the biomaterial and the cell recruitment factor are administered prior to the administration of the composition comprising the lentiviral vector, optionally wherein: (i) the biomaterial and the cell recruitment factor are administered about 1-4 weeks, e.g., about 2 weeks, prior to the administration of the composition comprising the lentiviral vector; or (ii) the biomaterial and the cell recruitment factor are administered at least two weeks prior to the administration of the composition comprising the lentiviral vector.
- the biomaterial comprises a hydrogel; (ii) comprises a cryogel; (iii) comprises a gelatin, hyaluronic acid, collagen, alginate, laminin, chitosan, silk fibroin, agarose, polyethylene glycol), poly-vinyl alcohol, and/or hydroxyethyl methylacrylate; (iv) comprises alginate hydrogel, optionally wherein the alginate hydrogel further comprises norbomene and/or tetrazine, optionally wherein the norbomene and/or tetrazine is covalently associated with, e.g., chemically linked to, or non-covalently associated with, e.g., adsorbed on, the alginate; and/or (v) comprises pores between about 10 pm to about 300 pm, e.g., between about 50 pm to about 300 pm, in diameter, or no pores; and/or (vi) is chemically crosslinked.
- the cell recruitment factor is: (i) noncovalently associated with, e.g., adsorbed on, the biomaterial; or (ii) covalently associated with, e.g., conjugated to, the biomaterial.
- the cell recruitment factor : (i) induces lymphangiogenesis; (ii) induce growth of lymphatic endothelial cells; and/or (ii) recruits immune cells, optionally wherein the immune cells comprise T-cells and/or NK -cells.
- the cell recruitment factor is chosen from VEGF-C, IL-2, IL-7, IL- 15 (e.g., hetIL-15 (IL15/sIL-15Ra)), GM-CSF, CXCL12, CXC3L1, CCL19, CCL21, CXCL10, or CXCL11.
- the cell recruitment factor comprises VEGF-C or a functional variant thereof; IL- 15 (e.g., hetIL-15 (IL15/sIL-15Ra)) or a functional variant thereof; IL-7 or a functional variant thereof; or a combination thereof.
- the cell recruitment factor comprises VEGF-C, optionally wherein the VEGF-C: (i) comprises a mature VEGF-C peptide, optionally the minor or major mature form or a mutated variant thereof; (ii) is a monomer or dimer; and/or (iii) is present in an effective amount, optionally, in an amount of less than or about 1 mg, less than or about 10 mg, greater than or about 10 pg, greater than or about 1 pg, between about 1 pg and 1 mg, between about 10 pg and 1 mg, between about 1 pg and 10 mg, or between about 10 pg and 10 mg.
- the cell recruitment factor comprises: (i) an amino acid sequence according to SEQ ID NO: 741 of WO/2022/040586, or an amino acid sequence with at least 80%, at least 85%, at least 90%, at least 95%, or at least 99% sequence identity thereto, provided that the amino acid at position 26 of the SEQ ID NO: 741 is not Cysteine (C), optionally wherein the amino acid at position 26 of the SEQ ID NO: 741 is Alanine (A); (ii) the amino acid sequence according to SEQ ID NO: 743 of WO/2022/040586 or a sequence an amino acid sequence with at least 80%, at least 85%, at least 90%, at least 95%, or at least 99% sequence identity thereto; (iii) the amino acid sequence according to SEQ ID NO: 740 of WO/2022/040586 or an amino acid sequence with at least 80%, at least 85%, at least 90%, at least 95%, or at least 99% sequence identity thereto; (iv) the amino acid sequence according to
- the composition comprising the lentiviral vector further comprises a particle.
- the particle is a mesoporous particle, a silica particle and/or a mesoporous silica particle, optionally wherein the mesoporous silica particle is a mesoporous silica rod.
- the mesoporous silica particle comprises a surface modification, optionally wherein the surface modification comprises: (a) a -OH (hydroxyl), amine, carboxylic acid, phosphonate, halide, azide, alkyne, epoxide, sulfhydryl, polyethyleneimine, a hydrophobic moiety, or salts thereof, optionally using a Cl to C20 alkyl or ( O(CH2 CH2 ) 1-25 linker; (b) a primary, secondary, tertiary, or quaternary amine; and/or (c) a polyethyleneimine having an average molecular weight of about 1000 to 20,000 Da, about 1,200 to 15,000 Da, about 1,500 to 12,000 Da, about 2,000 Da, about 3,000 Da, about 4,000 Da, about 5,000 Da, about 6,000 Da, about 7,000 Da, about 8,000 Da, about 9,000 Da, or about 10,000 Da, as measured by gel permeation chromatography (GPC).
- GPC gel perme
- the mesoporous silica particle (i) is a trimethylammonium functionalized mesoporous silica particle, e.g., a N,N,N-trimethylpropan-l -ammonium functionalized mesoporous silica particle; (iii) comprises a plurality of pores, optionally wherein the pores are between 2-50 nm in diameter; and/or (iv) comprises a surface area of at least about 100 m2/g.
- the composition comprising the lentiviral vector further comprises a cell activation agent.
- the cell activation agent comprises an agent that stimulates CD3/TCR complex and/or an agent that stimulates a costimulatory molecule and/or growth factor receptor.
- the cell activation agent comprises a multispecific binding molecule comprising: (A) an anti-CD3 binding domain, and (B) a costimulatory molecule binding domain (e.g., an anti-CD2 binding domain or an anti-CD28 binding domain).
- the lentiviral vector is noncovalently, e.g., electrostatically, or covalently associated with the mesoporous silica particle; and/or (ii) the cell activation agent is noncovalently or covalently associated with the mesoporous silica particle.
- a formulation described herein that comprises silica particles lacks HSA.
- agents may be encoded in the vectors described herein above. Accordingly, these agents are described below in relation to the CAR-expressing cell.
- a CAR-expressing immune effector cell described herein can further express another agent, e.g., an agent which enhances the activity of a CAR-expressing cell.
- the agent can be an agent which inhibits an inhibitory molecule.
- inhibitory molecules include PD-1, PD-L1, CTLA-4, TIM-3, CEACAM (e.g., CEACAM-1, CEACAM-3 and/or CEACAM-5), LAG-3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4 and TGFR beta, e.g., as described herein.
- the agent that inhibits an inhibitory molecule comprises a first polypeptide, e.g., an inhibitory molecule, associated with a second polypeptide that provides a positive signal to the cell, e.g., an intracellular signaling domain described herein.
- the agent comprises a first polypeptide, e.g., of an inhibitory molecule such as PD-1, PD-L1, CTLA-4, TIM- 3, CEACAM (e.g., CEACAM-1, CEACAM-3 and/or CEACAM-5), LAG-3, VISTA, BTLA, TIGIT, LAIR1, CD 160, 2B4, or TGFR beta, or a fragment of any of these, and a second polypeptide which is an intracellular signaling domain described herein (e.g., comprising a costimulatory domain (e.g., 41BB, CD27 or CD28, e.g., as described herein) and/or a primary signaling domain (e.g., a CD3 zeta signaling domain described herein).
- an inhibitory molecule such as PD-1, PD-L1, CTLA-4, TIM- 3, CEACAM (e.g., CEACAM-1, CEACAM-3 and/or CEACAM-5), LAG-3, VIS
- the agent comprises a first polypeptide of PD-1 or a fragment thereof, and a second polypeptide of an intracellular signaling domain described herein (e.g., a CD28, CD27, 0X40 or 4-IBB signaling domain described herein and/or a CD3 zeta signaling domain described herein).
- a first polypeptide of PD-1 or a fragment thereof and a second polypeptide of an intracellular signaling domain described herein (e.g., a CD28, CD27, 0X40 or 4-IBB signaling domain described herein and/or a CD3 zeta signaling domain described herein).
- the CAR-expressing cell described herein can further comprise a second CAR, for example, a second CAR that includes a different antigen binding domain, for example, to the same target (for example, CD 19) or a different target (for example, a target other than CD 19, for example, a target described herein).
- a second CAR for example, a second CAR that includes a different antigen binding domain, for example, to the same target (for example, CD 19) or a different target (for example, a target other than CD 19, for example, a target described herein).
- the CAR-expressing cell described herein e.g., the CAR-expressing cell manufactured using a method described herein, comprises (i) a first nucleic acid molecule encoding a first CAR that binds to BCMA and (ii) a second nucleic acid molecule encoding a second CAR that binds to CD 19.
- the first CAR comprises an anti-BCMA binding domain, a first transmembrane domain, and a first intracellular signaling domain
- the anti-BCMA binding domain comprises a heavy chain variable region (VH) comprising a heavy chain complementary determining region 1 (HC CDR1), a heavy chain complementary determining region 2 (HC CDR2), and a heavy chain complementary determining region 3 (HC CDR3)
- VH heavy chain variable region
- VL light chain variable region
- LC CDR1 light chain complementary determining region 1
- LC CDR2 a light chain complementary determining region 2
- LC CDR3 light chain complementary determining region 3
- the HC CDR1, HC CDR2, HC CDR3, LC CDR1, LC CDR2, and LC CDR3 comprise the amino acid sequences of SEQ ID NOs: 86, 87, 88, 95, 96, and 97, respectively.
- the second CAR comprises an anti-CD19 binding domain, a second transmembrane domain, and a second intracellular signaling domain
- the anti-CD19 binding domain comprises a VH comprising a HC CDR1, a HC CDR2, and a HC CDR3, and a VL comprising a LC CDR1, a LC CDR2, and a LC CDR3, wherein the HC CDR1, HC CDR2, HC CDR3, LC CDR1, LC CDR2, and LC CDR3 comprise the amino acid sequences of SEQ ID NOs: 760, 687, 762, 763, 764, and 765, respectively.
- the VH and VL of the anti-BCMA binding domain comprise the amino acid sequences of SEQ ID NOs: 93 and 102, respectively.
- the VH and VL of the anti-CD19 binding domain comprise the amino acid sequences of SEQ ID NOs: 250A and 251 A, respectively.
- the anti-BCMA binding domain comprises the amino acid sequence of SEQ ID NO: 105.
- the anti-CD19 binding domain comprises the amino acid sequence of SEQ ID NO: 758.
- the first CAR comprises the amino acid sequence of SEQ ID NO: 107.
- the second CAR comprises the amino acid sequence of SEQ ID NO: 225.
- the CAR-expressing cell described herein e.g., the CAR-expressing cell manufactured using a method described herein, comprises (i) a first nucleic acid molecule encoding a first CAR that binds to CD22 and (ii) a second nucleic acid molecule encoding a second CAR that binds to CD 19.
- the CD22 CAR comprises a CD22 antigen binding domain, and a first transmembrane domain; a first co-stimulatory signaling domain; and/or a first primary signaling domain.
- the CD 19 CAR comprises a CD 19 antigen binding domain, and a second transmembrane domain; a second co-stimulatory signaling domain; and/or a second primary signaling domain.
- the CD22 antigen binding domain comprises one or more (e.g., all three) light chain complementarity determining region 1 (LC CDR1), light chain complementarity determining region 2 (LC CDR2), and light chain complementarity determining region 3 (LC CDR3) of a CD22 binding domain described herein, e.g., in Tables 15, 16, 30, 31, or 32; and/or one or more (e.g., all three) heavy chain complementarity determining region 1 (HC CDR1), heavy chain complementarity determining region 2 (HC CDR2), and heavy chain complementarity determining region 3 (HC CDR3) of a CD22 binding domain described herein, e.g., in Tables 15, 16, 30, 31 or 32.
- the CD22 antigen binding domain comprises a LC CDR1, LC CDR2 and LC CDR3 of a CD22 binding domain described herein, e.g., in Table 15, 16, 30, 31 or 32; and/or a HC CDR1, HC CDR2 and HC CDR3 of a CD22 binding domain described herein, e.g., in Tables 15, 16, 30, 31 or 32.
- the CD19 antigen binding domain comprises: one or more (e.g., all three) LC CDR1, LC CDR2, and LC CDR3 of a CD19 binding domain described herein, e.g., in Tables 1, 30, 31, or 32; and/or one or more (e.g., all three) HC CDR1, HC CDR2, and HC CDR3 of a CD 19 binding domain described herein, e.g., in Tables 1, 30, 31, and 32.
- the CD19 antigen binding domain comprises a LC CDR1, LC CDR2 and LC CDR3 of a CD19 binding domain described herein, e.g., in Tables 1, 30, 31, and 32; and/or a HC CDR1, HC CDR2 and HC CDR3 of a CD19 binding domain described herein, e.g., in Tables 1, 30, 31, and 32.
- the CD22 antigen binding domain (e.g., an scFv) comprises a light chain variable (VL) region of a CD22 binding domain described herein, e.g., in Tables 30 or 32; and/or a heavy chain variable (VH) region of a CD22 binding domain described herein, e.g., in Tables 30 or 32.
- the CD22 antigen binding domain comprises a VL region comprising an amino acid sequence having at least one, two or three modifications (e.g., substitutions) but not more than 30, 20 or 10 modifications (e.g., substitutions) of a CD22 VL region sequence provided in Table 30 or 32.
- the CD22 antigen binding domain comprises a VL region comprising an amino acid sequence provided in Table 30 or 32, or a sequence having at least about 80%, about 85%, about 90%, about 92%, about 95%, about 97%, about 98%, or about 99% sequence identity to any of the aforesaid sequences.
- the CD22 antigen binding domain comprises a VH region comprising an amino acid sequence having at least one, two or three modifications (e.g., substitutions) but not more than 30, 20 or 10 modifications (e.g., substitutions) of a CD22 VH region sequence provided in Table 30 or 32.
- the CD22 antigen binding domain comprises a VH region comprising the amino acid sequence of a CD22 VH region sequence provided in Table 30 or 32, or a sequence having at least about 80%, about 85%, about 90%, about 92%, about 95%, about 97%, about 98%, or about 99% sequence identity to any of the aforesaid sequences.
- the CD 19 antigen binding domain e.g., an scFv
- the CD 19 antigen binding domain comprises a VL region of a CD19 binding domain described herein, e.g., in Tables 1, 30, or 32; and/or a VH region of a CD19 binding domain described herein, e.g., in Tables 1, 30, or 32.
- the CD 19 antigen binding domain comprises a VL region comprising an amino acid sequence having at least one, two or three modifications (e.g., substitutions) but not more than 30, 20 or 10 modifications (e.g., substitutions) of a CD19 VL region sequence provided in Tables 1, 30, or 32.
- the CD 19 antigen binding domain comprises a VL region comprising the amino acid sequence of a CD 19 VL region sequence provided in Tables 1, 30, or 32, or a sequence having at least about 80%, about 85%, about 90%, about 92%, about 95%, about 97%, about 98%, or about 99% sequence identity to any of the aforesaid sequences.
- the CD 19 antigen binding domain comprises a VH region comprising an amino acid sequence having at least one, two or three modifications (e.g., substitutions) but not more than 30, 20 or 10 modifications (e.g., substitutions) of a CD19 VH region sequence provided in Tables 1, 30, or 32.
- the CD19 antigen binding domain comprises a VH region comprising the amino acid sequence of a CD 19 VH region sequence provided in Tables 1, 30, or 32, or a sequence having at least about 80%, about 85%, about 90%, about 92%, about 95%, about 97%, about 98%, or about 99% sequence identity to any of the aforesaid sequences.
- the CD22 antigen binding comprises an scFv comprising an amino acid sequence having at least one, two or three modifications (e.g., substitutions) but not more than 30, 20 or 10 modifications (e.g., substitutions) of a CD22 scFv sequence provided in Table 30 or 32.
- the CD22 antigen binding comprises an scFv comprising an amino acid sequence of a CD22 scFv sequence provided in Table 30 or 32, or a sequence having at least about 80%, about 85%, about 90%, about 92%, about 95%, about 97%, about 98%, or about 99% sequence identity to any of the aforesaid sequences.
- the CD19 antigen binding domain comprises an scFv comprising an amino acid sequence having at least one, two or three modifications (e.g., substitutions) but not more than 30, 20 or 10 modifications (e.g., substitutions) of a CD19 scFv sequence provided in Tables 1, 30, or 32.
- the CD 19 antigen binding domain comprises an scFv comprising the amino acid sequence of a CD 19 scFv sequence provided in Tables 1, 30, or 32, or a sequence having at least about 80%, about 85%, about 90%, about 92%, about 95%, about 97%, about 98%, or about 99% sequence identity to any of the aforesaid sequences.
- the CD22 CAR molecule and/or the CD 19 CAR molecule comprises an additional component, e.g., a signal peptide, a hinge, a transmembrane domain, a co-stimulatory signaling domain and/or a first primary signaling domain, a P2A site, and/or a linker, comprising an amino acid sequence provided in Table 33, or a sequence having at least about 70%, about 75%, about 80%, about 85%, about 90%, about 92%, about 95%, about 97%, about 98%, or about 99% sequence identity to any of the aforesaid sequences; or is encoded by a nucleotide sequence provided in Table 33, or a sequence having at least about 70%, about 75%, about 80%, about 85%, about 90%, about 92%, about 95%, about 97%, about 98%, or about 99% sequence identity to any of the aforesaid sequences.
- a signal peptide e.g., a hinge, a trans
- Exemplary nucleotide and amino acid sequences of a CAR molecule e.g., a dual CAR molecule comprising (i) a first CAR that binds to CD22 and (ii) a second CAR that binds to CD 19 disclosed herein, is provided in Table 30.
- CD22 and CD19 CDRs of a dual CAR of the disclosure are provided in T able 31.
- Table 32 provides nucleotide and amino acid sequence for CD19 and CD22 binding domains of a dual CAR or a tandem CAR disclosed herein, e.g., a dual CAR or a tandem CAR comprising (i) a first CAR that binds to CD22 and (ii) a second CAR that binds to CD 19.
- Table 33 provides nucleotide and amino acid sequences for additional CAR components, e.g., signal peptide, linkers and P2A sites, that can be used in a CAR molecule, e.g., a dual CAR molecule described herein (for example, a dual CAR molecule comprising (i) a first CAR that binds to CD22 and (ii) a second CAR that binds to CD 19).
- CAR components e.g., signal peptide, linkers and P2A sites
- the CAR-expressing immune effector cell described herein can further comprise a second CAR, e.g., a second CAR that includes a different antigen binding domain, e.g., to the same target (e.g., a target described above) or a different target.
- the second CAR includes an antigen binding domain to a target expressed on the same cancer cell type as the target of the first CAR.
- the CAR-expressing immune effector cell comprises a first CAR that targets a first antigen and includes an intracellular signaling domain having a costimulatory signaling domain but not a primary signaling domain, and a second CAR that targets a second, different, antigen and includes an intracellular signaling domain having a primary signaling domain but not a costimulatory signaling domain.
- a costimulatory signaling domain e.g., 4-1BB, CD28, CD27, or OX-40
- the CAR expressing immune effector cell comprises a first CAR that includes an antigen binding domain that targets, e.g., a target described above, a transmembrane domain and a costimulatory domain and a second CAR that targets an antigen other than antigen targeted by the first CAR (e.g., an antigen expressed on the same cancer cell type as the first target) and includes an antigen binding domain, a transmembrane domain and a primary signaling domain.
- a first CAR that includes an antigen binding domain that targets, e.g., a target described above, a transmembrane domain and a costimulatory domain
- a second CAR that targets an antigen other than antigen targeted by the first CAR e.g., an antigen expressed on the same cancer cell type as the first target
- the CAR expressing immune effector cell comprises a first CAR that includes an antigen binding domain that targets, e.g., a target described above, a transmembrane domain and a primary signaling domain and a second CAR that targets an antigen other than antigen targeted by the first CAR (e.g., an antigen expressed on the same cancer cell type as the first target) and includes an antigen binding domain to the antigen, a transmembrane domain and a costimulatory signaling domain.
- a first CAR that includes an antigen binding domain that targets, e.g., a target described above, a transmembrane domain and a primary signaling domain
- a second CAR that targets an antigen other than antigen targeted by the first CAR (e.g., an antigen expressed on the same cancer cell type as the first target) and includes an antigen binding domain to the antigen, a transmembrane domain and a costimulatory signaling domain.
- the CAR-expressing immune effector cell comprises a CAR described herein, e.g., a CAR to a target described above, and an inhibitory CAR.
- the inhibitory CAR comprises an antigen binding domain that binds an antigen found on normal cells but not cancer cells, e.g., normal cells that also express the target.
- the inhibitory CAR comprises the antigen binding domain, a transmembrane domain and an intracellular domain of an inhibitory molecule.
- the intracellular domain of the inhibitory CAR can be an intracellular domain of PD1, PD-L1, CTLA-4, TIM-3, CEACAM (e.g., CEACAM-1, CEACAM-3 and/or CEACAM- 5), LAG-3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4, or TGFRbeta.
- CEACAM e.g., CEACAM-1, CEACAM-3 and/or CEACAM- 5
- LAG-3 e.g., VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4, or TGFRbeta.
- an immune effector cell (e.g., T cell, NK cell) comprises a first CAR comprising an antigen binding domain that binds to a tumor antigen as described herein, and a second CAR comprising a PD1 extracellular domain or a fragment thereof.
- the cell further comprises an inhibitory molecule as described above.
- the second CAR in the cell is an inhibitory CAR, wherein the inhibitory CAR comprises an antigen binding domain, a transmembrane domain, and an intracellular domain of an inhibitory molecule.
- the inhibitory molecule can be chosen from one or more of: PD1, PD-L1, CTLA-4, TIM-3, LAG-3, VISTA, BTLA, TIGIT, LAIR1, CD160, 2B4, TGFR beta, CEACAM-1, CEACAM-3, and CEACAM-5.
- the second CAR molecule comprises the extracellular domain of PD1 or a fragment thereof.
- the second CAR molecule in the cell further comprises an intracellular signaling domain comprising a primary signaling domain and/or an intracellular signaling domain.
- the intracellular signaling domain in the cell comprises a primary signaling domain comprising the functional domain of CD3 zeta and a costimulatory signaling domain comprising the functional domain of 4- IBB.
- the antigen binding domain of the first CAR molecule comprises a scFv and the antigen binding domain of the second CAR molecule does not comprise a scFv.
- the antigen binding domain of the first CAR molecule comprises a scFv and the antigen binding domain of the second CAR molecule comprises a camelid VHH domain. Conformation of CARs
- the CAR-expressing cell uses a split CAR.
- the split CAR approach is described in more detail in publications WO2014/055442 and WO2014/055657.
- a split CAR system comprises a cell expressing a first CAR having a first antigen binding domain and a costimulatory domain (e.g., 41BB), and the cell also expresses a second CAR having a second antigen binding domain and an intracellular signaling domain (e.g., CD3 zeta).
- the costimulatory domain is activated, and the cell proliferates.
- the intracellular signaling domain is activated and cell-killing activity begins.
- the CAR-expressing cell is only fully activated in the presence of both antigens.
- the CAR-expressing cell described herein can further comprise a second CAR, e.g., a second CAR that includes a different antigen binding domain, e.g., to the same target or a different target (e.g., a target other than a cancer associated antigen described herein or a different cancer associated antigen described herein).
- the second CAR includes an antigen binding domain to a target expressed the same cancer cell type as the cancer associated antigen.
- the CAR-expressing cell comprises a first CAR that targets a first antigen and includes an intracellular signaling domain having a costimulatory signaling domain but not a primary signaling domain, and a second CAR that targets a second, different, antigen and includes an intracellular signaling domain having a primary signaling domain but not a costimulatory signaling domain.
- a costimulatory signaling domain e.g., 4-1BB, CD28, CD27 or OX- 40
- the CAR expressing cell comprises a first cancer associated antigen CAR that includes an antigen binding domain that binds a target antigen described herein, a transmembrane domain and a costimulatory domain and a second CAR that targets a different target antigen (e.g., an antigen expressed on that same cancer cell type as the first target antigen) and includes an antigen binding domain, a transmembrane domain and a primary signaling domain.
- a first cancer associated antigen CAR that includes an antigen binding domain that binds a target antigen described herein, a transmembrane domain and a costimulatory domain
- a second CAR that targets a different target antigen (e.g., an antigen expressed on that same cancer cell type as the first target antigen) and includes an antigen binding domain, a transmembrane domain and a primary signaling domain.
- the CAR expressing cell comprises a first CAR that includes an antigen binding domain that binds a target antigen described herein, a transmembrane domain and a primary signaling domain and a second CAR that targets an antigen other than the first target antigen (e.g., an antigen expressed on the same cancer cell type as the first target antigen) and includes an antigen binding domain to the antigen, a transmembrane domain and a costimulatory signaling domain.
- a first CAR that includes an antigen binding domain that binds a target antigen described herein, a transmembrane domain and a primary signaling domain
- a second CAR that targets an antigen other than the first target antigen e.g., an antigen expressed on the same cancer cell type as the first target antigen
- the disclosure provides a first and second CAR, wherein the antigen binding domain of one of the first CAR the second CAR does not comprise a variable light domain and a variable heavy domain.
- the antigen binding domain of one of the first CAR the second CAR is an scFv, and the other is not an scFv.
- the antigen binding domain of one of the first CAR the second CAR comprises a single VH domain, e.g., a camelid, shark, or lamprey single VH domain, or a single VH domain derived from a human or mouse sequence.
- the antigen binding domain of one of the first CAR and the second CAR comprises a nanobody.
- the antigen binding domain of one of the first CAR and the second CAR comprises a camelid VHH domain.
- various assays can be used to evaluate the activity of, for e.g., the ability to expand T cells following antigen stimulation, sustain T cell expansion in the absence of re-stimulation, and anti-cancer activities in appropriate in vitro and animal models.
- Assays to evaluate the effects of a CAR described herein are known to those of skill in the art and generally described below.
- T cells (1: 1 mixture of CD4 + and CD8 + T cells) expressing the CARs are expanded in vitro for more than 10 days followed by lysis and SDS-PAGE under reducing conditions.
- CARs containing the full length TCR-c cytoplasmic domain and the endogenous TCR-c chain are detected by western blotting using an antibody to the TCR-c chain.
- the same T cell subsets are used for SDS-PAGE analysis under non-reducing conditions to permit evaluation of covalent dimer formation.
- Sustained CAR + T cell expansion in the absence of re-stimulation can also be measured. See, e.g., Milone et al., Molecular Therapy 17(8): 1453-1464 (2009). Briefly, mean T cell volume (fl) is measured on day 8 of culture using a Coulter Multisizer III particle counter, a Nexcelom Cellometer Vision or Millipore Scepter, following stimulation with aCD3/aCD28 coated magnetic beads on day 0, and transduction with the indicated CAR on day 1.
- Animal models can also be used to measure a CART activity.
- xenograft model using human a cancer associated antigen described herein-specific CAR + T cells to treat a primary human pre-B ALL in immunodeficient mice can be used. See, e.g., Milone et al., Molecular Therapy 17(8): 1453-1464 (2009).
- Dose dependent CAR treatment response can be evaluated. See, e.g., Milone et al., Molecular Therapy 17(8): 1453-1464 (2009).
- peripheral blood is obtained 35-70 days after establishing leukemia in mice injected on day 21 with CAR T cells, an equivalent number of mock- transduced T cells, or no T cells. Mice from each group are randomly bled for determination of peripheral blood a cancer associate antigen as described herein + ALL blast counts and then killed on days 35 and 49. The remaining animals are evaluated on days 57 and 70.
- Cytotoxicity can be assessed by a standard 51 Cr-release assay. See, e.g., Milone et al., Molecular Therapy 17(8): 1453-1464 (2009
- Imaging technologies can be used to evaluate specific trafficking and proliferation of CARs in tumor-bearing animal models. Such assays have been described, for example, in Barrett et al., Human Gene Therapy 22: 1575-1586 (2011).
- the following examples describe the results of several studies directed to the development of a new lentiviral vector (LVV) formulation with improvements relative to existing formulations.
- the main quality attributes analyzed were the transduction titer (e.g., active vector particles, TU titer), particle concentration (P24 ELISA and total particles by multi-angle dynamic light scattering (DLS), particulate matter (microflow imaging (MFI)), and size distribution of the vector product.
- the formulation development activities for LVV product were divided into seven phases (Table 1-1). Experiments were performed on three different LVV constructs (humanized CD19-CAR (humanized CAR2), CD22-CAR, and CD20-CAR).
- Results were compared to controls (e.g., to the LVV in PIPES buffer).
- a general scheme of the study flow is shown in FIG. 1.
- cryoprotecting agents e.g., sucrose, lactose
- protein stabilizers e.g., salts, surfactant
- double lipid layer stabilizers e.g., polyols, membrane components, and amino acids
- the impact of different combinations of freezing rates and thawing temperatures on the LVV product was assessed to inform future handling and screening procedures.
- the effect of the freeze-thaw program on post-thaw vector recovery and functionality was investigated comparing three freezing rates (l°C/min, 5°C/min, and 10°C/min) and three thawing temperatures (5°C, 25°C, and 37°C).
- All LVVs batches were produced by transient transfection of suspension Expi293FTM cells in a single-use 50 L bioreactor. After 3 days of amplification, cells were transfected with four plasmids and FectoVIR®-AAV as transfection reagent. After 48 hours of viral production, the downstream process involves a cell removal filtration and required several steps of filtration, ultra/diafiltration, and chromatography during 3 days in order to generate the lentiviral vector in 20 mM PIPES, 75 mM NaCl, 75 mM sucrose and 75 mM L-Arginine. The product was then sterile filtered using a PES 0.2 pm filter and stored in a deep freezer (-80 °C).
- Buffer exchange was performed at 4 °C using Float-A-lyzer devices consisting of a semi permeable cellulose membrane to allow for diffusion of small molecules but retention of the sample (50 KDa cut-off (MWCO)).
- the buffer contents within the membrane were allowed to reach equilibrium with the buffer outside.
- a 10 mL volume of VS formulated in 20 mM PIPES buffer NaCl 75 mM, L-Arg 75 mM, sucrose 220 mM, pH 6.5
- the fransductive unit (TU) titer assay was based on transduction of HEK293T cells followed by isolation of the genomic DNA and quantification of the LVV WPRE element and a house-keeping gene in a duplex qPCR. After normalization and correlation to the number of cells seeded, the concentration of transducing units, (LVV particles that were able to deliver their genome into a target cell followed by integration in the host cell genome) was calculated.
- the internal method variability for the TU assay is 30% CV.
- the HIV-1 p24 enzyme-linked immunosorbent assay is a quantitative assay that measures p24 concentrations.
- the p24 antigen is a structural component of the LVV capsid and approximately 2000 p24 molecules are present per vector particle, the p24 concentration can be used as an approximation for quantifying the concentration of vector particles in a sample.
- the LVV physical particle titer based on p24 quantification may represent an overestimation as the measurement includes free p24 and defective viral particles.
- a variation of the assay to discriminate between LVV associated versus free p24 was used in certain experiments.
- the internal method variability for the p24 assay is 25% CV.
- the multi angle dynamic light scattering (MADLS) measurements were performed using a Zetasizer Ultra instrument with a 633 nm He-Ne diode-pumped laser, which was operated at an angle of 173 degrees.
- a quartz cuvette with a path length of 10 mm was loaded with 45 pL of sample. The measurements were taken at a position of 6.45 mm from the cuvette wall with an automatic signal attenuator.
- Particle size distribution (PSD) was determined in triplicate measurements after equilibration at 25 °C for 2 min. Data were expressed as mean ⁇ standard deviation (SD).
- SD standard deviation
- System suitability test was performed with 60 nm polystyrene particle standards prior to running samples.
- Particle size (hydrodynamic diameter in nm, Dh), size distribution (PSD), Z-A verage, polydispersity index (PDI), and zeta potential (ZP, mV) were measured as indicated below.
- MFI Microscopic flow imaging
- the buffer composition is critical during the lentiviral vector (LVV) manufacturing process, fill and finish (F&F) activities, and storage. After determination of a suitable pH for LVV product, a buffer system must maintain a stable pH over a long period of time. Other excipients such as salts, reducing agents, and/or stabilizers (e.g., polyols or detergents) aiming at stabilizing the LVV product can then be added.
- LVV lentiviral vector
- F&F fill and finish
- Other excipients such as salts, reducing agents, and/or stabilizers (e.g., polyols or detergents) aiming at stabilizing the LVV product can then be added.
- HEPES (20 mM) and X-VIVO 15 medium were selected as buffer systems. Both are suitable for cell culture medium during CAR-T manufacturing.
- the impact of buffer type was evaluated after incubation for 12 hours at 37 °C in presence of 220 mM sucrose.
- the vector was tested at concentrations mimicking a multiplicity of infection (MOI) of 1 as used for activated rapid manufacturing (ARM) but without T cells being present.
- MOI multiplicity of infection
- Two different vectors were used for the present buffer screening studies: humanized CD19-CAR (e.g., humanized CAR-2) and CD22-CAR.
- the LVV product stability was determined under ARM process conditions without the presence of T-cells.
- Physicochemical properties of the viral particles such as particle size (hydrodynamic diameter in nm, Dh), size distribution (PSD), polydispersity index (PDI), and zeta potential (ZP), and titers were monitored over a period of 12 hours at 37 °C in the incubator (5% CCL). Once the candidate buffer was identified, different pH values ranging between 6.0 and 7.5 were assessed.
- the LVV product PSD (Dh) and particle concentration was determined by multi-angle dynamic light scattering (MADLS) before and after incubation for 12 hours at 37 °C (FIGs. 2 - 3). Two peaks at approximately 175 nm and 415 nm before starting the incubation were found in both buffers (HEPES and X-VIVO 15 media). The first peak was associated with LVV while the second peak represented potential vector aggregates. The hydrodynamic diameter of the LVV did not change when formulated in HEPES after incubation. For LVV formulated in X-VIVO 15, the Dh dropped to approximately 120 nm and multiple additional peaks were observed. When measured in these experiments, the number distribution of the peak associated to the LVV was always above 95% and the cumulative contribution of the aggregate peaks was below 5%.
- MADLS multi-angle dynamic light scattering
- the transduction titer decreased for both buffer systems tested (FIG. 4). This effect was more pronounced for LVV formulated in X-VIVO 15 where a drop in TU of about 75% was observed after incubation (from 2.86E7 TU/mL to 7.32E6 TU/mL). In HEPES buffer, a reduction of 30% in TU titer was observed (from 3.86E6 TU/mL to 2.67E6 TU/mL).
- the viral particle titer (p24/mL) remained constant for LVV in HEPES buffer, while a reduction of 20% was found for LVV formulated in X-VIVO 15 buffer (from 9.98E10 LP/mL to 8.03E10 LP/mL) (FIG. 5).
- the incubation temperature of 37 °C is not an intended storage condition of the LVV vector and that the LVV buffer used in this Example was not fully formulated, but rather contained sucrose only.
- One potential impact of the buffer pH is on the resulting LVV ZP. Without wishing to be bound by theory, electrostatic repulsion may stabilize the vector by preventing vector aggregation. For example, particles with a ZP value above +30 mV or below -30 mV are considered stable.
- the impact of the pH buffer on physicochemical properties of the LVV was assessed.
- the LVV formulated in 20 mM HEPES (supplemented with 220 mM sucrose) and 20 mM PIPES buffer (both at pH 6.5) presented an average ZP of -28.6 mV and -15.0 mV, respectively (FIGs. 6 - 7).
- the effect of the pH on the ZP in HEPES buffer was also evaluated.
- a stock of LV vector substance (LV-VS) (humanized CD19-CAR (humanized CAR2)) was formulated in 20 mM HEPES buffer at different pH values in the range of pH 6.0 and pH 7.5. A broader pH range was not evaluated as the LVV product is intended to be used for transducing cells at a physiological pH and because extreme pH values may result in LVV degradation.
- the vector particles In 20 mM HEPES buffer supplemented with sucrose (220 mM) the vector particles have a lower ZP than in PIPES buffer (fully formulated). Without wishing to be bound by theory, it is believed that the vectors would tend to repel each other with a lesser tendency for the particles to aggregate. This trend was also confirmed in HEPES buffer at molarity up to 150 mM (data not shown).
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Genetics & Genomics (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Molecular Biology (AREA)
- Zoology (AREA)
- General Health & Medical Sciences (AREA)
- Biotechnology (AREA)
- Immunology (AREA)
- Medicinal Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- Biophysics (AREA)
- Wood Science & Technology (AREA)
- Biochemistry (AREA)
- Cell Biology (AREA)
- Gastroenterology & Hepatology (AREA)
- Toxicology (AREA)
- Veterinary Medicine (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Public Health (AREA)
- Virology (AREA)
- Physics & Mathematics (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Plant Pathology (AREA)
- Pharmacology & Pharmacy (AREA)
- Microbiology (AREA)
- Manufacturing & Machinery (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
Abstract
Description


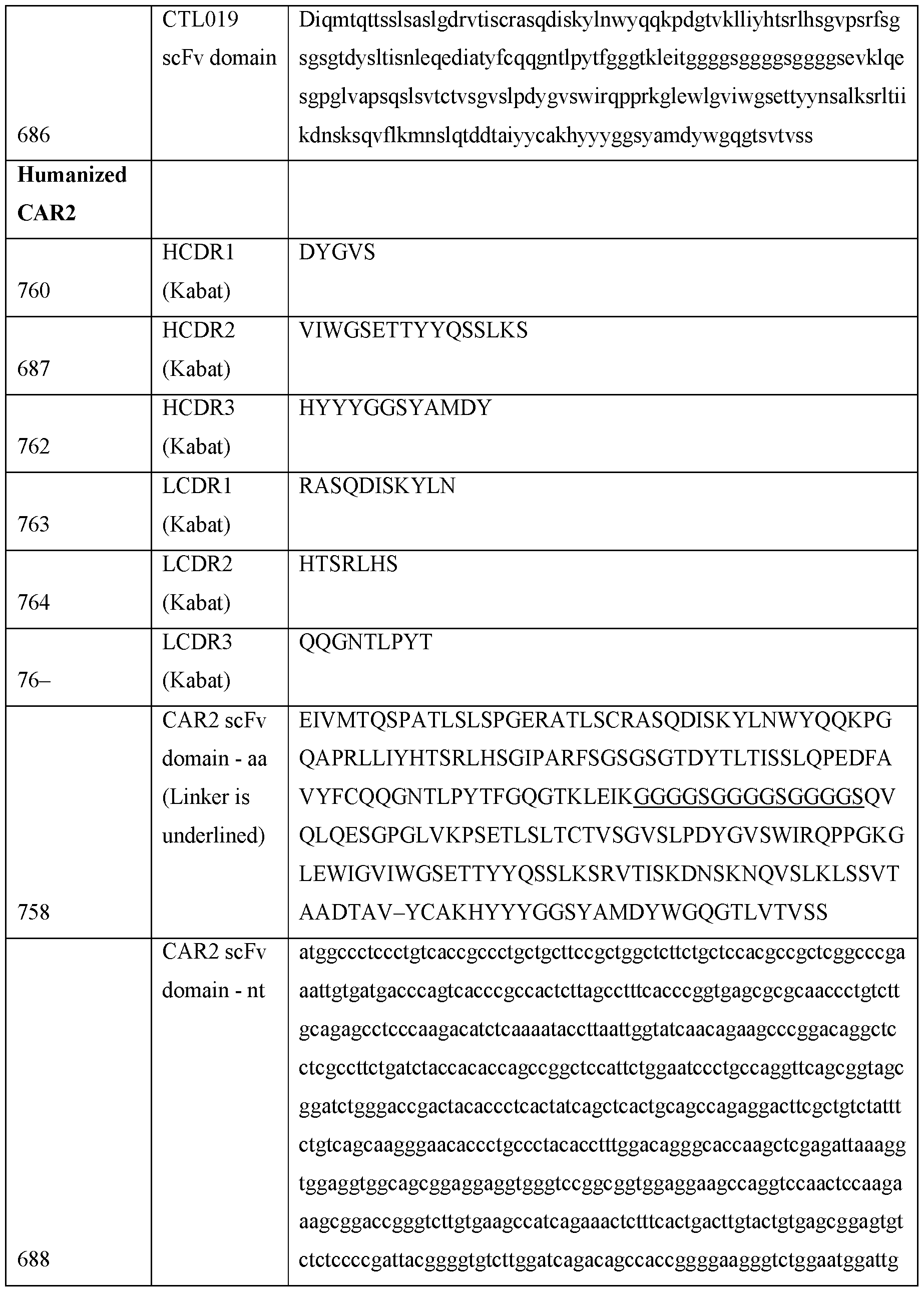















































































































































































Claims
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202380075451.9A CN120112650A (en) | 2022-10-26 | 2023-10-26 | Lentiviral preparations |
| EP23800978.1A EP4608983A1 (en) | 2022-10-26 | 2023-10-26 | Lentiviral formulations |
| AU2023369684A AU2023369684A1 (en) | 2022-10-26 | 2023-10-26 | Lentiviral formulations |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202263381107P | 2022-10-26 | 2022-10-26 | |
| US63/381,107 | 2022-10-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024089639A1 true WO2024089639A1 (en) | 2024-05-02 |
Family
ID=88695650
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2023/060809 Ceased WO2024089639A1 (en) | 2022-10-26 | 2023-10-26 | Lentiviral formulations |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP4608983A1 (en) |
| CN (1) | CN120112650A (en) |
| AU (1) | AU2023369684A1 (en) |
| WO (1) | WO2024089639A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN119709874A (en) * | 2025-02-28 | 2025-03-28 | 江苏谱新生物医药有限公司 | Preparation for lentivirus transduction and application thereof |
Citations (144)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4851332A (en) | 1985-04-01 | 1989-07-25 | Sloan-Kettering Institute For Cancer Research | Choriocarcinoma monoclonal antibodies and antibody panels |
| US5504048A (en) | 1993-06-02 | 1996-04-02 | Fina Technology, Inc. | Addition of lithium compounds to Ziegler-Natta catalysts for increased molecular weight in polyolefins |
| EP0754230A1 (en) | 1994-04-04 | 1997-01-22 | Genentech, Inc. | Agonist antibodies against the flk2/flt3 receptor and uses thereof |
| US5637481A (en) | 1993-02-01 | 1997-06-10 | Bristol-Myers Squibb Company | Expression vectors encoding bispecific fusion proteins and methods of producing biologically active bispecific fusion proteins in a mammalian cell |
| EP0805871A1 (en) | 1995-01-18 | 1997-11-12 | Roche Diagnostics GmbH | Anti-cd 30 antibodies preventing proteolytic cleavage and release of membrane-bound cd 30 antigen |
| US5777084A (en) | 1996-03-07 | 1998-07-07 | Eberhard-Karls-Universitat Tubingen | Antibody BV10A4H2 specific for human FLT3/FLK2 receptor and mybridoma |
| US5786464A (en) | 1994-09-19 | 1998-07-28 | The General Hospital Corporation | Overexpression of mammalian and viral proteins |
| US5837821A (en) | 1992-11-04 | 1998-11-17 | City Of Hope | Antibody construct |
| US5843674A (en) | 1993-11-16 | 1998-12-01 | Pola Chemical Industries Inc. | Anti-human tyrosinase monoclonal antibody |
| US5864019A (en) | 1990-06-11 | 1999-01-26 | Celltech Limited | Multivalent antigen-binding proteins |
| US5869620A (en) | 1986-09-02 | 1999-02-09 | Enzon, Inc. | Multivalent antigen-binding proteins |
| US5952484A (en) | 1994-03-08 | 1999-09-14 | Sloan-Kettering Cancer Center | Recombinant human anti-LK26 antibodies |
| EP1013761A2 (en) | 1991-09-18 | 2000-06-28 | Kyowa Hakko Kogyo Co., Ltd. | Humanized chimeric antibody directed against ganglioside GD3 |
| US6114148A (en) | 1996-09-20 | 2000-09-05 | The General Hospital Corporation | High level expression of proteins |
| WO2001038490A2 (en) | 1999-11-29 | 2001-05-31 | The Trustees Of Columbia University In The City Of New York | ISOLATION OF FIVE NOVEL GENES CODING FOR NEW Fc RECEPTORS-TYPE MELANOMA INVOLVED IN THE PATHOGENESIS OF LYMPHOMA/MELANOMA |
| US6528481B1 (en) | 1999-02-16 | 2003-03-04 | The Burnam Institute | NG2/HM proteoglycan-binding peptides that home to angiogenic vasculature and related methods |
| WO2004087758A2 (en) | 2003-03-26 | 2004-10-14 | Neopharm, Inc. | Il 13 receptor alpha 2 antibody and methods of use |
| CA2523449A1 (en) | 2003-04-23 | 2004-11-04 | Centro De Inmunologia Molecular | Recombinant antibodies and fragments which recognize n glycolil gm3 ganglioside and its use in diagnosis and treatment of tumours |
| US6846911B2 (en) | 1996-10-25 | 2005-01-25 | The United States Of America As Represented By The Department Of Health And Human Services | Methods and compositions for inhibiting inflammation and angiogenesis comprising a mammalian CD97 α subunit |
| WO2005035577A1 (en) | 2003-10-08 | 2005-04-21 | Kyowa Hakko Kogyo Co., Ltd. | Antibody composition specifically binding to ganglioside gd3 |
| US20050100543A1 (en) | 2003-07-01 | 2005-05-12 | Immunomedics, Inc. | Multivalent carriers of bi-specific antibodies |
| US20050129701A1 (en) | 2001-12-04 | 2005-06-16 | Marasco Wayne A. | Antibody to latent membrane proteins and uses thereof |
| US20050175606A1 (en) | 2001-04-11 | 2005-08-11 | Hua-Liang Huang | Cyclic single-chain trispecific antibody |
| WO2005117986A2 (en) | 2004-06-01 | 2005-12-15 | Genentech, Inc. | Antibody drug conjugates and methods |
| WO2006020258A2 (en) | 2004-07-17 | 2006-02-23 | Imclone Systems Incorporated | Novel tetravalent bispecific antibody |
| WO2006039238A2 (en) | 2004-09-30 | 2006-04-13 | The Goverment Of The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Irta2 antibodies and methods of use |
| WO2006076691A2 (en) | 2005-01-12 | 2006-07-20 | Medarex, Inc. | Irta-2 antibodies and their uses |
| US7083785B2 (en) | 1999-08-17 | 2006-08-01 | Biogen Idcc MA Inc. | Methods of treatment by administering an anti-BCMA antibody |
| US7090843B1 (en) | 2000-11-28 | 2006-08-15 | Seattle Genetics, Inc. | Recombinant anti-CD30 antibodies and uses thereof |
| WO2006125481A1 (en) | 2005-05-27 | 2006-11-30 | Universitätsklinikum Freiburg | Monoclonal antibodies and single chain antibody fragments against cell-surface prostate specific membrane antigen |
| WO2006138315A2 (en) | 2005-06-15 | 2006-12-28 | Schering Corporation | Anti-igf1r antibody formulations |
| US20070014794A1 (en) | 1995-03-01 | 2007-01-18 | Genentech, Inc. | Method for making heteromultimeric polypeptides |
| US20070049735A1 (en) | 2001-02-20 | 2007-03-01 | Zymogenetics, Inc. | Antibodies that bind both bcma and taci |
| WO2007024715A2 (en) | 2005-08-19 | 2007-03-01 | Abbott Laboratories | Dual variable domain immunoglobin and uses thereof |
| WO2007067992A2 (en) | 2005-12-08 | 2007-06-14 | Medarex, Inc. | Human monoclonal antibodies to fucosyl-gm1 and methods for using anti-fucosyl-gm1 |
| US7253263B1 (en) | 1999-09-30 | 2007-08-07 | Kyowa Hakko Kogyo Co., Ltd. | Complementarity determining region-grafted antibody against ganglioside GD3 and derivative of antibody against ganglioside GD3 |
| US7410640B2 (en) | 1998-07-22 | 2008-08-12 | Vanderbilt University | GBS toxin receptor antibodies |
| US7446190B2 (en) | 2002-05-28 | 2008-11-04 | Sloan-Kettering Institute For Cancer Research | Nucleic acids encoding chimeric T cell receptors |
| WO2008146911A1 (en) | 2007-06-01 | 2008-12-04 | Sapporo Medical University | Antibody directed against il13ra2, and diagnostic/therapeutic agent comprising the antibody |
| US20090297529A1 (en) | 2008-05-30 | 2009-12-03 | Yiwen Li | Anti-flt3 antibodies |
| US20090304718A1 (en) | 2006-01-05 | 2009-12-10 | Guenther Adolf | Antibody Molecules Specific for Fibroblast Activation Protein and Immunoconjugates Containing Them |
| US20090311181A1 (en) | 2006-03-20 | 2009-12-17 | The Regents Of The University Of California | Engineered Anti-Prostate Stem Cell Antigen (PSCA) Antibodies for Cancer Targeting |
| US7635753B2 (en) | 2007-02-19 | 2009-12-22 | Wisconsin Alumni Research Foundation | Prostate cancer and melanoma antigens |
| WO2010033866A2 (en) | 2008-09-19 | 2010-03-25 | University Of Pittsburgh-Of The Commonwealth System Of Higher Education | Monoclonal antibodies for cspg4 for the diagnosis and treatment of basal breast carcinoma |
| US20100150910A1 (en) | 2006-10-10 | 2010-06-17 | Universite De Nantes | Use of monoclonal antibodies specific to the o-acetylated form of gd2 ganglioside for treatment of certain cancers |
| US7749719B2 (en) | 1994-04-22 | 2010-07-06 | The United States Of America As Represented By The Department Of Health And Human Services | Melanoma antigens and their use in diagnostic and therapeutic methods |
| WO2010114940A1 (en) | 2009-04-01 | 2010-10-07 | Genentech, Inc. | Anti-fcrh5 antibodies and immunoconjugates and methods of use |
| WO2010120561A1 (en) | 2009-04-01 | 2010-10-21 | Genentech, Inc. | Anti-fcrh5 antibodies and immunoconjugates and methods of use |
| US7915391B2 (en) | 2006-04-24 | 2011-03-29 | Amgen Inc. | Humanized c-Kit antibody |
| EP2322550A1 (en) | 2004-12-22 | 2011-05-18 | Amgen, Inc | Compositions comprising anti-IGF-1R Antibodies and Methods for obtaining said Antibodies |
| WO2011076922A1 (en) | 2009-12-23 | 2011-06-30 | Synimmune Gmbh | Anti-flt3 antibodies and methods of using the same |
| US20110268656A1 (en) | 2009-12-02 | 2011-11-03 | David Ho | J591 minibodies and cys-diabodies for targeting human prostate specific membrane antigen (psma) and methods for their use |
| US8080650B2 (en) | 2003-06-27 | 2011-12-20 | Diadexus, Inc. | Pro104 antibody compositions and methods of use |
| WO2011160119A2 (en) | 2010-06-19 | 2011-12-22 | Memorial Sloan-Kettering Cancer Center | Anti-gd2 antibodies |
| WO2011159847A2 (en) | 2010-06-15 | 2011-12-22 | The Regents Of The University Of California | Receptor tyrosine kinase-like orphan receptor 1 (ror1) single chain fv antibody fragment conjugates and methods of use thereof |
| US20120009181A1 (en) | 2010-02-24 | 2012-01-12 | Ab Olga | Folate Receptor 1 Antibodies and Immunoconjugates and Uses Thereof |
| WO2012033885A1 (en) | 2010-09-08 | 2012-03-15 | Baylor College Of Medicine | Immunotherapy of cancer using genetically engineered gd2-specific t cells |
| WO2012079000A1 (en) | 2010-12-09 | 2012-06-14 | The Trustees Of The University Of Pennsylvania | Use of chimeric antigen receptor-modified t cells to treat cancer |
| US8207308B2 (en) | 2007-02-16 | 2012-06-26 | Sloan-Kettering Institute For Cancer Research | Anti ganglioside GD3 antibodies and uses thereof |
| US8263746B2 (en) | 2004-02-06 | 2012-09-11 | Morphosys Ag | Anti-CD38 human antibodies and uses thereof |
| WO2012135854A2 (en) | 2011-04-01 | 2012-10-04 | Memorial Sloan-Kettering Cancer Center | Antibodies to cytosolic peptides |
| EP2514766A2 (en) | 2007-03-29 | 2012-10-24 | Technion Research & Development Foundation Ltd. | Antibodies, methods and kits for diagnosing and treating melanoma |
| US8309693B2 (en) | 2001-08-23 | 2012-11-13 | Rsr Limited | Epitope regions of a thyrotrophin (TSH) receptor, uses thereof and antibodies thereto |
| US20120288506A1 (en) | 2011-05-12 | 2012-11-15 | Imclone Llc | C-kit antibodies and uses thereof |
| WO2012163805A1 (en) | 2011-05-27 | 2012-12-06 | Glaxo Group Limited | Bcma (cd269/tnfrsf17) -binding proteins |
| US8344112B2 (en) | 2007-07-31 | 2013-01-01 | Merck Sharp & Dohme Limited | IGF-1R specific antibodies useful in the detection and diagnosis of cellular proliferative disorders |
| US8362211B2 (en) | 2010-12-30 | 2013-01-29 | Takeda Pharmaceutical Company Limited | Anti-CD38 antibodies |
| US8399645B2 (en) | 2003-11-05 | 2013-03-19 | St. Jude Children's Research Hospital, Inc. | Chimeric receptors with 4-1BB stimulatory signaling domain |
| WO2013040371A2 (en) | 2011-09-16 | 2013-03-21 | Baylor College Of Medicine | Targeting the tumor microenvironment using manipulated nkt cells |
| WO2013061273A1 (en) | 2011-10-25 | 2013-05-02 | Massimo Dominici | A modified effector cell (or chimeric receptor) for treating disialoganglioside gd2 -expressing neoplasia |
| US8440798B2 (en) | 2006-10-04 | 2013-05-14 | Københavns Universitet | Generation of a cancer-specific immune response toward MUC1 and cancer specific MUC1 antibodies |
| WO2013074916A1 (en) | 2011-11-18 | 2013-05-23 | Board Of Regents, The University Of Texas System | Car+ t cells genetically modified to eliminate expression of t- cell receptor and/or hla |
| WO2013085552A1 (en) | 2011-12-08 | 2013-06-13 | Cleveland Clinic Foundation | Glenoid vault fixation |
| US8501415B2 (en) | 2002-11-26 | 2013-08-06 | B.R.A.H.M.S. Gmbh | Identification of TSH receptor autoantibodies using affinity-purified antibodies |
| WO2013123061A1 (en) | 2012-02-13 | 2013-08-22 | Seattle Children's Hospital D/B/A Seattle Children's Research Institute | Bispecific chimeric antigen receptors and therapeutic uses thereof |
| WO2013126712A1 (en) | 2012-02-22 | 2013-08-29 | The Trustees Of The University Of Pennsylvania | Compositions and methods for generating a persisting population of t cells useful for the treatment of cancer |
| US20130273055A1 (en) | 2010-11-16 | 2013-10-17 | Eric Borges | Agents and methods for treating diseases that correlate with bcma expression |
| WO2013165940A1 (en) | 2012-05-01 | 2013-11-07 | Genentech, Inc. | Anti-pmel17 antibodies and immunoconjugates |
| US8603466B2 (en) | 2006-03-29 | 2013-12-10 | King's College London | Agonist antibodies against TSHR |
| WO2013192294A1 (en) | 2012-06-20 | 2013-12-27 | Boston 3T Biotechnologies, Inc. | Cellular therapies for treating and preventing cancers and other immune system disorders |
| WO2014055442A2 (en) | 2012-10-01 | 2014-04-10 | The Trustees Of The University Of Pennsylvania | Compositions and methods for targeting stromal cells for the treatment of cancer |
| WO2014055657A1 (en) | 2012-10-05 | 2014-04-10 | The Trustees Of The University Of Pennsylvania | Use of a trans-signaling approach in chimeric antigen receptors |
| WO2014130635A1 (en) | 2013-02-20 | 2014-08-28 | Novartis Ag | Effective targeting of primary human leukemia using anti-cd123 chimeric antigen receptor engineered t cells |
| WO2014130657A1 (en) | 2013-02-20 | 2014-08-28 | The Trustees Of The University Of Pennsylvania | Treatment of cancer using humanized anti-egfrviii chimeric antigen receptor |
| WO2014153270A1 (en) | 2013-03-16 | 2014-09-25 | Novartis Ag | Treatment of cancer using humanized anti-cd19 chimeric antigen receptor |
| US8920776B2 (en) | 2002-01-22 | 2014-12-30 | Corixa Corporation | Compositions and methods for the detection diagnosis and therapy of hematological malignancies |
| WO2014210064A1 (en) | 2013-06-24 | 2014-12-31 | Genentech, Inc. | Anti-fcrh5 antibodies |
| US20150051266A1 (en) | 2012-04-11 | 2015-02-19 | The USA, as represented by the Secretary, Department of Health and Human Serivces | Chimeric antigen receptors targeting b-cell maturation antigen |
| US9034324B2 (en) | 2009-03-10 | 2015-05-19 | Biogen Idec Ma Inc. | Anti-BCMA antibodies |
| WO2015090230A1 (en) | 2013-12-19 | 2015-06-25 | Novartis Ag | Human mesothelin chimeric antigen receptors and uses thereof |
| US20150232557A1 (en) | 2012-04-20 | 2015-08-20 | Emergent Product Development Seattle Llc | Cd3 binding polypeptides |
| WO2015142675A2 (en) | 2014-03-15 | 2015-09-24 | Novartis Ag | Treatment of cancer using chimeric antigen receptor |
| US20150284467A1 (en) | 2012-11-01 | 2015-10-08 | Max-Delbrück-Centrum für Molekulare Medizin | Antibody that binds cd269 (bcma) suitable for use in the treatment of plasma cell diseases such as multiple myeloma and autoimmune diseases |
| US20150283178A1 (en) | 2014-04-07 | 2015-10-08 | Carl H. June | Treatment of cancer using anti-cd19 chimeric antigen receptor |
| WO2015158671A1 (en) | 2014-04-14 | 2015-10-22 | Cellectis | Bcma (cd269) specific chimeric antigen receptors for cancer immunotherapy |
| WO2015166073A1 (en) | 2014-04-30 | 2015-11-05 | Max-Delbrück-Centrum für Molekulare Medizin | Humanized antibodies against cd269 (bcma) |
| WO2015172800A1 (en) | 2014-05-12 | 2015-11-19 | Numab Ag | Novel multispecific molecules and novel treatment methods based on such multispecific molecules |
| US20150344844A1 (en) | 2014-02-04 | 2015-12-03 | Marc Better | Methods for producing autologous t cells useful to treat b cell malignancies and other cancers and compositions thereof |
| WO2015188119A1 (en) | 2014-06-06 | 2015-12-10 | Bluebird Bio, Inc. | Improved t cell compositions |
| US20150368351A1 (en) | 2013-02-05 | 2015-12-24 | Engmab Ag | Method for the selection of antibodies against bcma |
| US9243058B2 (en) | 2012-12-07 | 2016-01-26 | Amgen, Inc. | BCMA antigen binding proteins |
| WO2016014576A1 (en) | 2014-07-21 | 2016-01-28 | Novartis Ag | Treatment of cancer using a cd33 chimeric antigen receptor |
| WO2016014565A2 (en) | 2014-07-21 | 2016-01-28 | Novartis Ag | Treatment of cancer using humanized anti-bcma chimeric antigen receptor |
| WO2016014535A1 (en) | 2014-07-21 | 2016-01-28 | Novartis Ag | Treatment of cancer using a cll-1 chimeric antigen receptor |
| WO2016014789A2 (en) | 2014-07-24 | 2016-01-28 | Bluebird Bio, Inc. | Bcma chimeric antigen receptors |
| WO2016020332A1 (en) | 2014-08-04 | 2016-02-11 | Engmab Ag | Bispecific antibodies against cd3epsilon and bcma |
| WO2016025880A1 (en) | 2014-08-14 | 2016-02-18 | Novartis Ag | Treatment of cancer using gfr alpha-4 chimeric antigen receptor |
| WO2016028896A1 (en) | 2014-08-19 | 2016-02-25 | Novartis Ag | Anti-cd123 chimeric antigen receptor (car) for use in cancer treatment |
| US9273141B2 (en) | 2011-05-27 | 2016-03-01 | Glaxo Group Limited | B cell maturation antigen (BCMA) binding proteins |
| US20160131655A1 (en) | 2011-04-21 | 2016-05-12 | Boehringer Ingelheim International Gmbh | Bcma-based stratification and therapy for multiple myeloma patients |
| US9340621B2 (en) | 2011-11-15 | 2016-05-17 | Boehringer Ingelheim International Gmbh | Binding molecules for BCMA and CD3 |
| WO2016079177A1 (en) | 2014-11-20 | 2016-05-26 | Engmab Ag | Bispecific antibodies against cd3epsilon and bcma |
| WO2016090320A1 (en) | 2014-12-05 | 2016-06-09 | Memorial Sloan-Kettering Cancer Center | Chimeric antigen receptors targeting b-cell maturation antigen and uses thereof |
| WO2016090327A2 (en) | 2014-12-05 | 2016-06-09 | Memorial Sloan-Kettering Cancer Center | Antibodies targeting b-cell maturation antigen and methods of use |
| WO2016087531A1 (en) | 2014-12-03 | 2016-06-09 | Engmab Ag | Bispecific antibodies against cd3epsilon and bcma for use in treatment of diseases |
| WO2016094304A2 (en) | 2014-12-12 | 2016-06-16 | Bluebird Bio, Inc. | Bcma chimeric antigen receptors |
| US20160176973A1 (en) | 2013-03-15 | 2016-06-23 | Amgen Research (Munich) Gmbh | Binding molecules for bcma and cd3 |
| WO2016130598A1 (en) | 2015-02-09 | 2016-08-18 | University Of Florida Research Foundation, Inc. | Bi-specific chimeric antigen receptor and uses thereof |
| WO2016154055A1 (en) | 2015-03-20 | 2016-09-29 | Bluebird Bio, Inc. | Vector formulations |
| US20160297884A1 (en) | 2015-04-13 | 2016-10-13 | Pfizer Inc. | Chimeric antigen receptors targeting b-cell maturation antigen |
| US20160297885A1 (en) | 2015-04-13 | 2016-10-13 | Pfizer Inc. | Therapeutic antibodies and their uses |
| WO2016164731A2 (en) | 2015-04-08 | 2016-10-13 | Novartis Ag | Cd20 therapies, cd22 therapies, and combination therapies with a cd19 chimeric antigen receptor (car) - expressing cell |
| US20160368988A1 (en) | 2015-07-10 | 2016-12-22 | Merus N.V. | Human cd3 binding antibody |
| WO2016210293A1 (en) | 2015-06-25 | 2016-12-29 | Icell Gene Therapeutics Llc | CHIMERIC ANTIGEN RECEPTORS (CARs), COMPOSITIONS AND METHODS OF USE THEREOF |
| WO2017011804A1 (en) | 2015-07-15 | 2017-01-19 | Juno Therapeutics, Inc. | Engineered cells for adoptive cell therapy |
| WO2017008169A1 (en) | 2015-07-15 | 2017-01-19 | Zymeworks Inc. | Drug-conjugated bi-specific antigen-binding constructs |
| WO2017021450A1 (en) | 2015-08-03 | 2017-02-09 | Engmab Ag | Monoclonal antibodies against bcma |
| WO2017025038A1 (en) | 2015-08-11 | 2017-02-16 | Nanjing Legend Biotech Co., Ltd. | Chimeric antigen receptors based on single-domain antibodies and methods of use thereof |
| US20170051252A1 (en) | 2014-04-25 | 2017-02-23 | Bluebird Bio, Inc. | Improved methods for manufacturing adoptive cell therapies |
| US20170051308A1 (en) | 2014-04-25 | 2017-02-23 | Bluebird Bio, Inc. | Mnd promoter chimeric antigen receptors |
| US20170051068A1 (en) | 2015-08-17 | 2017-02-23 | Janssen Pharmaceutica Nv | Anti-BCMA Antibodies, Bispecific Antigen Binding Molecules that Bind BCMA and CD3, and Uses Thereof |
| WO2017049166A1 (en) | 2015-09-17 | 2017-03-23 | Novartis Ag | Car t cell therapies with enhanced efficacy |
| WO2017087861A1 (en) | 2015-11-19 | 2017-05-26 | Novartis Ag | Buffers for stabilzation of lentiviral preparations |
| WO2017112741A1 (en) | 2015-12-22 | 2017-06-29 | Novartis Ag | Mesothelin chimeric antigen receptor (car) and antibody against pd-l1 inhibitor for combined use in anticancer therapy |
| WO2018067992A1 (en) | 2016-10-07 | 2018-04-12 | Novartis Ag | Chimeric antigen receptors for the treatment of cancer |
| WO2019241426A1 (en) | 2018-06-13 | 2019-12-19 | Novartis Ag | Bcma chimeric antigen receptors and uses thereof |
| WO2020047452A2 (en) | 2018-08-31 | 2020-03-05 | Novartis Ag | Methods of making chimeric antigen receptor-expressing cells |
| WO2020176397A1 (en) | 2019-02-25 | 2020-09-03 | Novartis Ag | Mesoporous silica particles compositions for viral delivery |
| CN109157518B (en) * | 2018-09-26 | 2021-03-12 | 厚朴生物科技(苏州)有限公司 | Lentiviral vector freeze-drying preservation method and preparation |
| CN113980917A (en) * | 2021-12-27 | 2022-01-28 | 苏州博腾生物制药有限公司 | Lentiviral lysis buffer solution and application thereof |
| WO2022040586A2 (en) | 2020-08-21 | 2022-02-24 | Novartis Ag | Compositions and methods for in vivo generation of car expressing cells |
| WO2022053880A1 (en) | 2020-09-11 | 2022-03-17 | metacluster lt, UAB | Dynamic optimization of request parameters for proxy server |
| WO2023003844A1 (en) * | 2021-07-19 | 2023-01-26 | 2Seventy Bio, Inc. | Vector manufacturing processes |
| WO2023230512A1 (en) * | 2022-05-26 | 2023-11-30 | 2Seventy Bio, Inc. | Compositions for maintaining lentiviral vector and uses thereof |
-
2023
- 2023-10-26 WO PCT/IB2023/060809 patent/WO2024089639A1/en not_active Ceased
- 2023-10-26 EP EP23800978.1A patent/EP4608983A1/en active Pending
- 2023-10-26 AU AU2023369684A patent/AU2023369684A1/en active Pending
- 2023-10-26 CN CN202380075451.9A patent/CN120112650A/en active Pending
Patent Citations (158)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4851332A (en) | 1985-04-01 | 1989-07-25 | Sloan-Kettering Institute For Cancer Research | Choriocarcinoma monoclonal antibodies and antibody panels |
| US5869620A (en) | 1986-09-02 | 1999-02-09 | Enzon, Inc. | Multivalent antigen-binding proteins |
| US5864019A (en) | 1990-06-11 | 1999-01-26 | Celltech Limited | Multivalent antigen-binding proteins |
| US6437098B1 (en) | 1991-09-18 | 2002-08-20 | Kyowa Hakko Kogyo Co., Ltd. | Human chimeric antibody specific for the ganglioside GD3 |
| EP1013761A2 (en) | 1991-09-18 | 2000-06-28 | Kyowa Hakko Kogyo Co., Ltd. | Humanized chimeric antibody directed against ganglioside GD3 |
| US5837821A (en) | 1992-11-04 | 1998-11-17 | City Of Hope | Antibody construct |
| US5637481A (en) | 1993-02-01 | 1997-06-10 | Bristol-Myers Squibb Company | Expression vectors encoding bispecific fusion proteins and methods of producing biologically active bispecific fusion proteins in a mammalian cell |
| US5504048A (en) | 1993-06-02 | 1996-04-02 | Fina Technology, Inc. | Addition of lithium compounds to Ziegler-Natta catalysts for increased molecular weight in polyolefins |
| US5843674A (en) | 1993-11-16 | 1998-12-01 | Pola Chemical Industries Inc. | Anti-human tyrosinase monoclonal antibody |
| US5952484A (en) | 1994-03-08 | 1999-09-14 | Sloan-Kettering Cancer Center | Recombinant human anti-LK26 antibodies |
| EP0754230A1 (en) | 1994-04-04 | 1997-01-22 | Genentech, Inc. | Agonist antibodies against the flk2/flt3 receptor and uses thereof |
| US7749719B2 (en) | 1994-04-22 | 2010-07-06 | The United States Of America As Represented By The Department Of Health And Human Services | Melanoma antigens and their use in diagnostic and therapeutic methods |
| US5786464A (en) | 1994-09-19 | 1998-07-28 | The General Hospital Corporation | Overexpression of mammalian and viral proteins |
| US5786464C1 (en) | 1994-09-19 | 2012-04-24 | Gen Hospital Corp | Overexpression of mammalian and viral proteins |
| EP0805871A1 (en) | 1995-01-18 | 1997-11-12 | Roche Diagnostics GmbH | Anti-cd 30 antibodies preventing proteolytic cleavage and release of membrane-bound cd 30 antigen |
| US20070014794A1 (en) | 1995-03-01 | 2007-01-18 | Genentech, Inc. | Method for making heteromultimeric polypeptides |
| US5777084A (en) | 1996-03-07 | 1998-07-07 | Eberhard-Karls-Universitat Tubingen | Antibody BV10A4H2 specific for human FLT3/FLK2 receptor and mybridoma |
| US6114148A (en) | 1996-09-20 | 2000-09-05 | The General Hospital Corporation | High level expression of proteins |
| US6114148C1 (en) | 1996-09-20 | 2012-05-01 | Gen Hospital Corp | High level expression of proteins |
| US6846911B2 (en) | 1996-10-25 | 2005-01-25 | The United States Of America As Represented By The Department Of Health And Human Services | Methods and compositions for inhibiting inflammation and angiogenesis comprising a mammalian CD97 α subunit |
| US7410640B2 (en) | 1998-07-22 | 2008-08-12 | Vanderbilt University | GBS toxin receptor antibodies |
| US6528481B1 (en) | 1999-02-16 | 2003-03-04 | The Burnam Institute | NG2/HM proteoglycan-binding peptides that home to angiogenic vasculature and related methods |
| US7083785B2 (en) | 1999-08-17 | 2006-08-01 | Biogen Idcc MA Inc. | Methods of treatment by administering an anti-BCMA antibody |
| US7253263B1 (en) | 1999-09-30 | 2007-08-07 | Kyowa Hakko Kogyo Co., Ltd. | Complementarity determining region-grafted antibody against ganglioside GD3 and derivative of antibody against ganglioside GD3 |
| WO2001038490A2 (en) | 1999-11-29 | 2001-05-31 | The Trustees Of Columbia University In The City Of New York | ISOLATION OF FIVE NOVEL GENES CODING FOR NEW Fc RECEPTORS-TYPE MELANOMA INVOLVED IN THE PATHOGENESIS OF LYMPHOMA/MELANOMA |
| US7090843B1 (en) | 2000-11-28 | 2006-08-15 | Seattle Genetics, Inc. | Recombinant anti-CD30 antibodies and uses thereof |
| US20070049735A1 (en) | 2001-02-20 | 2007-03-01 | Zymogenetics, Inc. | Antibodies that bind both bcma and taci |
| US20050175606A1 (en) | 2001-04-11 | 2005-08-11 | Hua-Liang Huang | Cyclic single-chain trispecific antibody |
| US8309693B2 (en) | 2001-08-23 | 2012-11-13 | Rsr Limited | Epitope regions of a thyrotrophin (TSH) receptor, uses thereof and antibodies thereto |
| US20050129701A1 (en) | 2001-12-04 | 2005-06-16 | Marasco Wayne A. | Antibody to latent membrane proteins and uses thereof |
| US8920776B2 (en) | 2002-01-22 | 2014-12-30 | Corixa Corporation | Compositions and methods for the detection diagnosis and therapy of hematological malignancies |
| US7446190B2 (en) | 2002-05-28 | 2008-11-04 | Sloan-Kettering Institute For Cancer Research | Nucleic acids encoding chimeric T cell receptors |
| US8501415B2 (en) | 2002-11-26 | 2013-08-06 | B.R.A.H.M.S. Gmbh | Identification of TSH receptor autoantibodies using affinity-purified antibodies |
| WO2004087758A2 (en) | 2003-03-26 | 2004-10-14 | Neopharm, Inc. | Il 13 receptor alpha 2 antibody and methods of use |
| CA2523449A1 (en) | 2003-04-23 | 2004-11-04 | Centro De Inmunologia Molecular | Recombinant antibodies and fragments which recognize n glycolil gm3 ganglioside and its use in diagnosis and treatment of tumours |
| US8080650B2 (en) | 2003-06-27 | 2011-12-20 | Diadexus, Inc. | Pro104 antibody compositions and methods of use |
| US20050100543A1 (en) | 2003-07-01 | 2005-05-12 | Immunomedics, Inc. | Multivalent carriers of bi-specific antibodies |
| WO2005035577A1 (en) | 2003-10-08 | 2005-04-21 | Kyowa Hakko Kogyo Co., Ltd. | Antibody composition specifically binding to ganglioside gd3 |
| US8399645B2 (en) | 2003-11-05 | 2013-03-19 | St. Jude Children's Research Hospital, Inc. | Chimeric receptors with 4-1BB stimulatory signaling domain |
| US8263746B2 (en) | 2004-02-06 | 2012-09-11 | Morphosys Ag | Anti-CD38 human antibodies and uses thereof |
| WO2005117986A2 (en) | 2004-06-01 | 2005-12-15 | Genentech, Inc. | Antibody drug conjugates and methods |
| WO2006020258A2 (en) | 2004-07-17 | 2006-02-23 | Imclone Systems Incorporated | Novel tetravalent bispecific antibody |
| WO2006039238A2 (en) | 2004-09-30 | 2006-04-13 | The Goverment Of The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Irta2 antibodies and methods of use |
| EP2322550A1 (en) | 2004-12-22 | 2011-05-18 | Amgen, Inc | Compositions comprising anti-IGF-1R Antibodies and Methods for obtaining said Antibodies |
| WO2006076691A2 (en) | 2005-01-12 | 2006-07-20 | Medarex, Inc. | Irta-2 antibodies and their uses |
| WO2006125481A1 (en) | 2005-05-27 | 2006-11-30 | Universitätsklinikum Freiburg | Monoclonal antibodies and single chain antibody fragments against cell-surface prostate specific membrane antigen |
| WO2006138315A2 (en) | 2005-06-15 | 2006-12-28 | Schering Corporation | Anti-igf1r antibody formulations |
| WO2007024715A2 (en) | 2005-08-19 | 2007-03-01 | Abbott Laboratories | Dual variable domain immunoglobin and uses thereof |
| WO2007067992A2 (en) | 2005-12-08 | 2007-06-14 | Medarex, Inc. | Human monoclonal antibodies to fucosyl-gm1 and methods for using anti-fucosyl-gm1 |
| US20100297138A1 (en) | 2005-12-08 | 2010-11-25 | Vistica Cynthia A | Human monoclonal antibodies to fucosyl-gm1 and methods for using anti-fucosyl-gm1 |
| US20090304718A1 (en) | 2006-01-05 | 2009-12-10 | Guenther Adolf | Antibody Molecules Specific for Fibroblast Activation Protein and Immunoconjugates Containing Them |
| US20090311181A1 (en) | 2006-03-20 | 2009-12-17 | The Regents Of The University Of California | Engineered Anti-Prostate Stem Cell Antigen (PSCA) Antibodies for Cancer Targeting |
| US8603466B2 (en) | 2006-03-29 | 2013-12-10 | King's College London | Agonist antibodies against TSHR |
| US7915391B2 (en) | 2006-04-24 | 2011-03-29 | Amgen Inc. | Humanized c-Kit antibody |
| US8440798B2 (en) | 2006-10-04 | 2013-05-14 | Københavns Universitet | Generation of a cancer-specific immune response toward MUC1 and cancer specific MUC1 antibodies |
| US20100150910A1 (en) | 2006-10-10 | 2010-06-17 | Universite De Nantes | Use of monoclonal antibodies specific to the o-acetylated form of gd2 ganglioside for treatment of certain cancers |
| US20120276046A1 (en) | 2007-02-16 | 2012-11-01 | Sloan-Kettering Institute For Cancer Research | Anti ganglioside gd3 antibodies and uses thereof |
| US8207308B2 (en) | 2007-02-16 | 2012-06-26 | Sloan-Kettering Institute For Cancer Research | Anti ganglioside GD3 antibodies and uses thereof |
| US7635753B2 (en) | 2007-02-19 | 2009-12-22 | Wisconsin Alumni Research Foundation | Prostate cancer and melanoma antigens |
| EP2514766A2 (en) | 2007-03-29 | 2012-10-24 | Technion Research & Development Foundation Ltd. | Antibodies, methods and kits for diagnosing and treating melanoma |
| WO2008146911A1 (en) | 2007-06-01 | 2008-12-04 | Sapporo Medical University | Antibody directed against il13ra2, and diagnostic/therapeutic agent comprising the antibody |
| US8344112B2 (en) | 2007-07-31 | 2013-01-01 | Merck Sharp & Dohme Limited | IGF-1R specific antibodies useful in the detection and diagnosis of cellular proliferative disorders |
| US20090297529A1 (en) | 2008-05-30 | 2009-12-03 | Yiwen Li | Anti-flt3 antibodies |
| US20140004124A1 (en) | 2008-09-19 | 2014-01-02 | University Of Pittsburgh - Of The Commonwealth System Of Higher Education | Monoclonal antibodies for cspg4 for the diagnosis and treatment of basal breast carcinoma |
| WO2010033866A2 (en) | 2008-09-19 | 2010-03-25 | University Of Pittsburgh-Of The Commonwealth System Of Higher Education | Monoclonal antibodies for cspg4 for the diagnosis and treatment of basal breast carcinoma |
| US9034324B2 (en) | 2009-03-10 | 2015-05-19 | Biogen Idec Ma Inc. | Anti-BCMA antibodies |
| WO2010114940A1 (en) | 2009-04-01 | 2010-10-07 | Genentech, Inc. | Anti-fcrh5 antibodies and immunoconjugates and methods of use |
| WO2010120561A1 (en) | 2009-04-01 | 2010-10-21 | Genentech, Inc. | Anti-fcrh5 antibodies and immunoconjugates and methods of use |
| US20110268656A1 (en) | 2009-12-02 | 2011-11-03 | David Ho | J591 minibodies and cys-diabodies for targeting human prostate specific membrane antigen (psma) and methods for their use |
| WO2011076922A1 (en) | 2009-12-23 | 2011-06-30 | Synimmune Gmbh | Anti-flt3 antibodies and methods of using the same |
| US20120009181A1 (en) | 2010-02-24 | 2012-01-12 | Ab Olga | Folate Receptor 1 Antibodies and Immunoconjugates and Uses Thereof |
| WO2011159847A2 (en) | 2010-06-15 | 2011-12-22 | The Regents Of The University Of California | Receptor tyrosine kinase-like orphan receptor 1 (ror1) single chain fv antibody fragment conjugates and methods of use thereof |
| US20130101607A1 (en) | 2010-06-15 | 2013-04-25 | Thomas J. Kipps | Receptor Tyrosine Kinase-Like Orphan Receptor 1 (ROR1) Single Chain FV Antibody Fragment Conjugates and Methods of Use Thereof |
| WO2011160119A2 (en) | 2010-06-19 | 2011-12-22 | Memorial Sloan-Kettering Cancer Center | Anti-gd2 antibodies |
| WO2012033885A1 (en) | 2010-09-08 | 2012-03-15 | Baylor College Of Medicine | Immunotherapy of cancer using genetically engineered gd2-specific t cells |
| US20130273055A1 (en) | 2010-11-16 | 2013-10-17 | Eric Borges | Agents and methods for treating diseases that correlate with bcma expression |
| WO2012079000A1 (en) | 2010-12-09 | 2012-06-14 | The Trustees Of The University Of Pennsylvania | Use of chimeric antigen receptor-modified t cells to treat cancer |
| US8362211B2 (en) | 2010-12-30 | 2013-01-29 | Takeda Pharmaceutical Company Limited | Anti-CD38 antibodies |
| WO2012135854A2 (en) | 2011-04-01 | 2012-10-04 | Memorial Sloan-Kettering Cancer Center | Antibodies to cytosolic peptides |
| US20160131655A1 (en) | 2011-04-21 | 2016-05-12 | Boehringer Ingelheim International Gmbh | Bcma-based stratification and therapy for multiple myeloma patients |
| US20120288506A1 (en) | 2011-05-12 | 2012-11-15 | Imclone Llc | C-kit antibodies and uses thereof |
| US9273141B2 (en) | 2011-05-27 | 2016-03-01 | Glaxo Group Limited | B cell maturation antigen (BCMA) binding proteins |
| WO2012163805A1 (en) | 2011-05-27 | 2012-12-06 | Glaxo Group Limited | Bcma (cd269/tnfrsf17) -binding proteins |
| WO2013040371A2 (en) | 2011-09-16 | 2013-03-21 | Baylor College Of Medicine | Targeting the tumor microenvironment using manipulated nkt cells |
| WO2013061273A1 (en) | 2011-10-25 | 2013-05-02 | Massimo Dominici | A modified effector cell (or chimeric receptor) for treating disialoganglioside gd2 -expressing neoplasia |
| US9340621B2 (en) | 2011-11-15 | 2016-05-17 | Boehringer Ingelheim International Gmbh | Binding molecules for BCMA and CD3 |
| WO2013074916A1 (en) | 2011-11-18 | 2013-05-23 | Board Of Regents, The University Of Texas System | Car+ t cells genetically modified to eliminate expression of t- cell receptor and/or hla |
| WO2013085552A1 (en) | 2011-12-08 | 2013-06-13 | Cleveland Clinic Foundation | Glenoid vault fixation |
| WO2013123061A1 (en) | 2012-02-13 | 2013-08-22 | Seattle Children's Hospital D/B/A Seattle Children's Research Institute | Bispecific chimeric antigen receptors and therapeutic uses thereof |
| WO2013126712A1 (en) | 2012-02-22 | 2013-08-29 | The Trustees Of The University Of Pennsylvania | Compositions and methods for generating a persisting population of t cells useful for the treatment of cancer |
| US20150051266A1 (en) | 2012-04-11 | 2015-02-19 | The USA, as represented by the Secretary, Department of Health and Human Serivces | Chimeric antigen receptors targeting b-cell maturation antigen |
| US20150232557A1 (en) | 2012-04-20 | 2015-08-20 | Emergent Product Development Seattle Llc | Cd3 binding polypeptides |
| US20130295007A1 (en) | 2012-05-01 | 2013-11-07 | Genentech, Inc. | Anti-pmel17 antibodies and immunoconjugates |
| WO2013165940A1 (en) | 2012-05-01 | 2013-11-07 | Genentech, Inc. | Anti-pmel17 antibodies and immunoconjugates |
| WO2013192294A1 (en) | 2012-06-20 | 2013-12-27 | Boston 3T Biotechnologies, Inc. | Cellular therapies for treating and preventing cancers and other immune system disorders |
| WO2014055442A2 (en) | 2012-10-01 | 2014-04-10 | The Trustees Of The University Of Pennsylvania | Compositions and methods for targeting stromal cells for the treatment of cancer |
| WO2014055657A1 (en) | 2012-10-05 | 2014-04-10 | The Trustees Of The University Of Pennsylvania | Use of a trans-signaling approach in chimeric antigen receptors |
| US20150284467A1 (en) | 2012-11-01 | 2015-10-08 | Max-Delbrück-Centrum für Molekulare Medizin | Antibody that binds cd269 (bcma) suitable for use in the treatment of plasma cell diseases such as multiple myeloma and autoimmune diseases |
| US9243058B2 (en) | 2012-12-07 | 2016-01-26 | Amgen, Inc. | BCMA antigen binding proteins |
| US20150368351A1 (en) | 2013-02-05 | 2015-12-24 | Engmab Ag | Method for the selection of antibodies against bcma |
| US20140322275A1 (en) | 2013-02-20 | 2014-10-30 | Jennifer Brogdon | TREATMENT OF CANCER USING HUMANIZED ANTI-EGFRvIII CHIMERIC ANTIGEN RECEPTOR |
| US20140322212A1 (en) | 2013-02-20 | 2014-10-30 | Jennifer Brogdon | Effective targeting of primary human leukemia using anti-cd123 chimeric antigen receptor engineered t cells |
| WO2014130657A1 (en) | 2013-02-20 | 2014-08-28 | The Trustees Of The University Of Pennsylvania | Treatment of cancer using humanized anti-egfrviii chimeric antigen receptor |
| WO2014130635A1 (en) | 2013-02-20 | 2014-08-28 | Novartis Ag | Effective targeting of primary human leukemia using anti-cd123 chimeric antigen receptor engineered t cells |
| US20160176973A1 (en) | 2013-03-15 | 2016-06-23 | Amgen Research (Munich) Gmbh | Binding molecules for bcma and cd3 |
| WO2014153270A1 (en) | 2013-03-16 | 2014-09-25 | Novartis Ag | Treatment of cancer using humanized anti-cd19 chimeric antigen receptor |
| WO2014210064A1 (en) | 2013-06-24 | 2014-12-31 | Genentech, Inc. | Anti-fcrh5 antibodies |
| WO2015090230A1 (en) | 2013-12-19 | 2015-06-25 | Novartis Ag | Human mesothelin chimeric antigen receptors and uses thereof |
| US20150344844A1 (en) | 2014-02-04 | 2015-12-03 | Marc Better | Methods for producing autologous t cells useful to treat b cell malignancies and other cancers and compositions thereof |
| WO2015142675A2 (en) | 2014-03-15 | 2015-09-24 | Novartis Ag | Treatment of cancer using chimeric antigen receptor |
| US20150283178A1 (en) | 2014-04-07 | 2015-10-08 | Carl H. June | Treatment of cancer using anti-cd19 chimeric antigen receptor |
| WO2015158671A1 (en) | 2014-04-14 | 2015-10-22 | Cellectis | Bcma (cd269) specific chimeric antigen receptors for cancer immunotherapy |
| US20170051308A1 (en) | 2014-04-25 | 2017-02-23 | Bluebird Bio, Inc. | Mnd promoter chimeric antigen receptors |
| US20170051252A1 (en) | 2014-04-25 | 2017-02-23 | Bluebird Bio, Inc. | Improved methods for manufacturing adoptive cell therapies |
| WO2015166073A1 (en) | 2014-04-30 | 2015-11-05 | Max-Delbrück-Centrum für Molekulare Medizin | Humanized antibodies against cd269 (bcma) |
| WO2015172800A1 (en) | 2014-05-12 | 2015-11-19 | Numab Ag | Novel multispecific molecules and novel treatment methods based on such multispecific molecules |
| WO2015188119A1 (en) | 2014-06-06 | 2015-12-10 | Bluebird Bio, Inc. | Improved t cell compositions |
| US20160096892A1 (en) | 2014-07-21 | 2016-04-07 | The Trustees Of The University Of Pennsylvania | Treatment of cancer using a cd33 chimeric antigen receptor |
| WO2016014535A1 (en) | 2014-07-21 | 2016-01-28 | Novartis Ag | Treatment of cancer using a cll-1 chimeric antigen receptor |
| US20160051651A1 (en) | 2014-07-21 | 2016-02-25 | The Trustees Of The University Of Pennsylvania | Treatment of cancer using a cll-1 chimeric antigen receptor |
| US20160046724A1 (en) | 2014-07-21 | 2016-02-18 | The Trustees Of The University Of Pennsylvania | Treatment of cancer using humanized anti-bcma chimeric antigen receptor |
| WO2016014576A1 (en) | 2014-07-21 | 2016-01-28 | Novartis Ag | Treatment of cancer using a cd33 chimeric antigen receptor |
| WO2016014565A2 (en) | 2014-07-21 | 2016-01-28 | Novartis Ag | Treatment of cancer using humanized anti-bcma chimeric antigen receptor |
| WO2016014789A2 (en) | 2014-07-24 | 2016-01-28 | Bluebird Bio, Inc. | Bcma chimeric antigen receptors |
| WO2016020332A1 (en) | 2014-08-04 | 2016-02-11 | Engmab Ag | Bispecific antibodies against cd3epsilon and bcma |
| WO2016025880A1 (en) | 2014-08-14 | 2016-02-18 | Novartis Ag | Treatment of cancer using gfr alpha-4 chimeric antigen receptor |
| US20160068601A1 (en) | 2014-08-19 | 2016-03-10 | The Trustees Of The University Of Pennsylvania | Treatment of cancer using a cd123 chimeric antigen receptor |
| WO2016028896A1 (en) | 2014-08-19 | 2016-02-25 | Novartis Ag | Anti-cd123 chimeric antigen receptor (car) for use in cancer treatment |
| WO2016079177A1 (en) | 2014-11-20 | 2016-05-26 | Engmab Ag | Bispecific antibodies against cd3epsilon and bcma |
| WO2016087531A1 (en) | 2014-12-03 | 2016-06-09 | Engmab Ag | Bispecific antibodies against cd3epsilon and bcma for use in treatment of diseases |
| WO2016090327A2 (en) | 2014-12-05 | 2016-06-09 | Memorial Sloan-Kettering Cancer Center | Antibodies targeting b-cell maturation antigen and methods of use |
| WO2016090320A1 (en) | 2014-12-05 | 2016-06-09 | Memorial Sloan-Kettering Cancer Center | Chimeric antigen receptors targeting b-cell maturation antigen and uses thereof |
| WO2016094304A2 (en) | 2014-12-12 | 2016-06-16 | Bluebird Bio, Inc. | Bcma chimeric antigen receptors |
| WO2016130598A1 (en) | 2015-02-09 | 2016-08-18 | University Of Florida Research Foundation, Inc. | Bi-specific chimeric antigen receptor and uses thereof |
| WO2016154055A1 (en) | 2015-03-20 | 2016-09-29 | Bluebird Bio, Inc. | Vector formulations |
| WO2016164731A2 (en) | 2015-04-08 | 2016-10-13 | Novartis Ag | Cd20 therapies, cd22 therapies, and combination therapies with a cd19 chimeric antigen receptor (car) - expressing cell |
| US20160297884A1 (en) | 2015-04-13 | 2016-10-13 | Pfizer Inc. | Chimeric antigen receptors targeting b-cell maturation antigen |
| US20160297885A1 (en) | 2015-04-13 | 2016-10-13 | Pfizer Inc. | Therapeutic antibodies and their uses |
| WO2016210293A1 (en) | 2015-06-25 | 2016-12-29 | Icell Gene Therapeutics Llc | CHIMERIC ANTIGEN RECEPTORS (CARs), COMPOSITIONS AND METHODS OF USE THEREOF |
| US20160368988A1 (en) | 2015-07-10 | 2016-12-22 | Merus N.V. | Human cd3 binding antibody |
| WO2017011804A1 (en) | 2015-07-15 | 2017-01-19 | Juno Therapeutics, Inc. | Engineered cells for adoptive cell therapy |
| WO2017008169A1 (en) | 2015-07-15 | 2017-01-19 | Zymeworks Inc. | Drug-conjugated bi-specific antigen-binding constructs |
| WO2017021450A1 (en) | 2015-08-03 | 2017-02-09 | Engmab Ag | Monoclonal antibodies against bcma |
| WO2017025038A1 (en) | 2015-08-11 | 2017-02-16 | Nanjing Legend Biotech Co., Ltd. | Chimeric antigen receptors based on single-domain antibodies and methods of use thereof |
| US20170051068A1 (en) | 2015-08-17 | 2017-02-23 | Janssen Pharmaceutica Nv | Anti-BCMA Antibodies, Bispecific Antigen Binding Molecules that Bind BCMA and CD3, and Uses Thereof |
| WO2017049166A1 (en) | 2015-09-17 | 2017-03-23 | Novartis Ag | Car t cell therapies with enhanced efficacy |
| WO2017087861A1 (en) | 2015-11-19 | 2017-05-26 | Novartis Ag | Buffers for stabilzation of lentiviral preparations |
| WO2017112741A1 (en) | 2015-12-22 | 2017-06-29 | Novartis Ag | Mesothelin chimeric antigen receptor (car) and antibody against pd-l1 inhibitor for combined use in anticancer therapy |
| WO2018067992A1 (en) | 2016-10-07 | 2018-04-12 | Novartis Ag | Chimeric antigen receptors for the treatment of cancer |
| WO2019241426A1 (en) | 2018-06-13 | 2019-12-19 | Novartis Ag | Bcma chimeric antigen receptors and uses thereof |
| WO2020047452A2 (en) | 2018-08-31 | 2020-03-05 | Novartis Ag | Methods of making chimeric antigen receptor-expressing cells |
| CN109157518B (en) * | 2018-09-26 | 2021-03-12 | 厚朴生物科技(苏州)有限公司 | Lentiviral vector freeze-drying preservation method and preparation |
| WO2020176397A1 (en) | 2019-02-25 | 2020-09-03 | Novartis Ag | Mesoporous silica particles compositions for viral delivery |
| WO2022040586A2 (en) | 2020-08-21 | 2022-02-24 | Novartis Ag | Compositions and methods for in vivo generation of car expressing cells |
| WO2022053880A1 (en) | 2020-09-11 | 2022-03-17 | metacluster lt, UAB | Dynamic optimization of request parameters for proxy server |
| WO2023003844A1 (en) * | 2021-07-19 | 2023-01-26 | 2Seventy Bio, Inc. | Vector manufacturing processes |
| CN113980917A (en) * | 2021-12-27 | 2022-01-28 | 苏州博腾生物制药有限公司 | Lentiviral lysis buffer solution and application thereof |
| WO2023230512A1 (en) * | 2022-05-26 | 2023-11-30 | 2Seventy Bio, Inc. | Compositions for maintaining lentiviral vector and uses thereof |
Non-Patent Citations (96)
| Title |
|---|
| "16th Annu Meet Am Soc Gen Cell Ther (ASGCT", 15 May 2013 |
| "Abstracts of 56th ASH Annual Meeting and Exposition", 6 December 2014, article "4507 Pre-Clinical Characterization of T Cell-Dependent Bispecific Antibody Anti-CD79b/CD3 As a Potential Therapy for B Cell Malignancies" |
| ABENGOZAR ET AL., BLOOD, vol. 119, no. 2014, 2012, pages 4565 - 4576 |
| AGGEN ET AL., GENE THER, vol. 19, no. 4, 2012, pages 365 - 74 |
| AIGNER ET AL., LEUKEMIA, vol. 27, no. 5, 2013, pages 1107 - 1115 |
| AL-LAZIK ET AL., JMB, vol. 273, 1997, pages 927 - 948 |
| AL-LAZIKANI ET AL., J. MOL. BIOL., vol. 273, 1997, pages 927 - 748 |
| BARRETT ET AL., HUMAN GENE THERAPY, vol. 22, 2011, pages 1575 - 1586 |
| BIRD ET AL., SCIENCE, vol. 242, 1988, pages 5879 - 5883 |
| BREMER E-G ET AL., J BIOL CHEM, vol. 259, 1984, pages 14773 - 14777 |
| BRENTJENS ET AL., BLOOD, vol. 118, no. 18, 2011, pages 4817 - 4828 |
| BROOKS ET AL., PNAS, vol. 107, no. 22, 2010, pages 10056 - 10061 |
| BROSS ET AL., CLIN CANCER RES, vol. 7, no. 6, 2001, pages 1490 - 1496 |
| CARON ET AL., CANCER RES, vol. 52, no. 24, 1992, pages 6761 - 6767 |
| CHEUNG ET AL., CANCER RES, vol. 45, no. 6, 1985, pages 2642 - 2649 |
| CHEUNG ET AL., J CLIN ONCOL, vol. 16, no. 9, 1998, pages 3053 - 3060 |
| CHEUNG ET AL., J CLIN ONCOL, vol. 5, no. 9, 1987, pages 1430 - 1440 |
| CHINNASAMY ET AL., J CLIN INVEST, vol. 120, no. 11, 2010, pages 3953 - 3968 |
| CHMIELEWSKI ET AL., GASTOENTEROLOGY, vol. 143, no. 4, 2012, pages 1095 - 1107 |
| CHOTHIA ET AL., J. MOL. BIOL., vol. 227, 1992, pages 799 - 817 |
| CHOTHIA ET AL., NATURE, vol. 342, 1989, pages 877 - 883 |
| CHOTHIALESK, J. MOL. BIOL., vol. 196, 1987, pages 901 - 917 |
| DAO ET AL., SCI TRANSL MED, vol. 5, no. 176, 2013, pages 176ra33 |
| DE GROOT ET AL., J IMMUNOL, vol. 183, no. 6, 2009, pages 4127 - 4134 |
| DOLEZAL ET AL., PROTEIN ENGINEERING, vol. 16, no. 1, 2003, pages 47 - 56 |
| DOMAN ET AL.: "Therapeutic potential of an anti-CD79b antibody-drug conjugate, anti-CD79b-vc-MMAE, for the treatment of non-Hodgkin lymphoma", BLOOD, vol. 114, no. 13, 24 July 2009 (2009-07-24), pages 2721 - 9 |
| DULL ET AL., J. VIROL, vol. 72, no. 11, 1998, pages 8463 - 8471 |
| DUTOUR ET AL., ADV HEMATOL 2012, 2012, pages 683065 |
| ELKINS ET AL.: "FcRL5 as a target of antibody-drug conjugates for the treatment of multiple myeloma", MOL CANCER THER, vol. 1 1, no. 10, October 2012 (2012-10-01), pages 2222 - 32, XP055141107, DOI: 10.1158/1535-7163.MCT-12-0087 |
| FENG ET AL.: "Glypican-3 antibodies: a new therapeutic target for liver cancer", FEES LETT, vol. 588, no. 2, 21 January 2014 (2014-01-21), pages 377 - 82, XP028669969, DOI: 10.1016/j.febslet.2013.10.002 |
| FISHER ET AL., J. VIROL., vol. 70, 1996, pages 520 - 532 |
| FRIGERIO ET AL., EUROPEAN J CANCER, vol. 49, no. 9, 2013, pages 2223 - 2232 |
| GHODS ET AL., BIOTECHNOL APPL BIOCHEM, 2013 |
| HANDGRETINGER ET AL., CANCER IMMUNOL IMMUNOTHER, vol. 35, no. 3, 1992, pages 199 - 204 |
| HOFHEINZ ET AL., ONCOLOGY RESEARCH AND TREATMENT, vol. 26, no. 1, 2003 |
| HOLLINGER ET AL., PROC NATL ACAD. SCI. U.S.A., vol. 90, pages 6444 - 6448 |
| HOMBACH ET AL., GASTROENTEROLOGY, vol. 113, no. 4, 1997, pages 1163 - 1170 |
| HONG ET AL., J IMMUNOTHER, vol. 37, no. 2, 2014, pages 93 - 104 |
| HUANG ET AL., CANCER RES, vol. 72, no. 1, 2012, pages 271 - 281 |
| HUDECEK ET AL., CLIN CANCER RES, vol. 19, no. 12, 2013, pages 3153 - 3164 |
| HUSTON ET AL., PROC. NATL. ACAD. SCI. USA, vol. 85, 1988, pages 5879 - 5883 |
| HUSTON ET AL., PROC. NATL. ACAD. SCI., vol. 85, 1988, pages 5879 - 5883 |
| JAGER ET AL., APPL IMMUNOHISTOCHEM MOL MORPHOL, vol. 15, no. 1, 2007, pages 77 - 83 |
| JOHNSON ET AL., NUCLEIC ACIDS RES., vol. 29, 2001, pages 205 - 206 |
| KELLY ET AL., CANCER BIOTHER RADIOPHARM, vol. 23, no. 4, 2008, pages 411 - 423 |
| KMIECIK ET AL., ONCOIMMUNOLOGY, vol. 3, no. 1, 2014, pages e27185 |
| KOCHENDERFER ET AL., BLOOD, vol. 116, no. 21, 2010, pages 1261 - 1262 |
| KOCHENDERFER ET AL., BLOOD, vol. 122, no. 20, 2013, pages 3461 - 3472 |
| KOCHENDERFER ET AL., BLOOD, vol. 122, no. 25, 15 May 2013 (2013-05-15), pages 4129 - 39 |
| KUDRYASHOV V ET AL., GLYCOCONJ J, vol. 15, no. 3, 1998, pages 243 - 9 |
| LANZAVECCHIA ET AL., EUR. J. IMMUNOL., vol. 17, 1987, pages 105 |
| LAPUSAN ET AL., INVEST NEW DRUGS, vol. 30, no. 3, 2012, pages 1121 - 1131 |
| LEFRANC, M.P, NUCLEIC ACIDS RES, vol. 29, 2001, pages 207 - 209 |
| LOU ET AL., PROC NATL ACAD SCI USA, vol. 111, no. 7, 2014, pages 2482 - 2487 |
| LUO ET AL., EMBO MOL. MED, vol. 4, no. 6, 2012, pages 453 - 461 |
| MACCALLUM ET AL., J. MOL. BIOL., vol. 262, 1996, pages 732 - 745 |
| MAECKER ET AL., BLOOD, vol. 102, no. 9, 2003, pages 3287 - 3294 |
| MALIAR ET AL., GASTROENTEROLOGY, vol. 143, no. 5, 2012, pages 1375 - 1384 |
| MARTIN ET AL., METHODS ENZYMOL., vol. 203, 1991, pages 121 - 153 |
| MARTIN ET AL., PROC. NATL. ACAD. SCI. USA, vol. 86, 1989, pages 9268 - 9272 |
| MARTY ET AL., CANCER LETT, vol. 235, no. 2, 2006, pages 298 - 308 |
| MATHER, J. P ET AL.: "Culture media, animal cells, large scale production", ENCYCLOPEDIA OF BIOPROCESS TECHNOLOGY: FERMENTATION, BIOCATALYSIS, AND BIOSEPARATION, vol. 2, 1999, pages 777 - 785 |
| MILONE ET AL., MOL. THER, vol. 17, no. 8, 2009, pages 1453 - 1464 |
| MILONE ET AL., MOLECULAR THERAPY, vol. 17, no. 8, 2009, pages 1453 - 1464 |
| MINO-KENUDSON ET AL., CLIN CANCER RES, vol. 16, no. 5, 2010, pages 1561 - 1571 |
| MORGENROTH ET AL., PROSTATE, vol. 67, no. 10, 2007, pages 1121 - 1131 |
| MUJOO ET AL., CANCER RES, vol. 47, no. 4, 1987, pages 1098 - 1104 |
| MYERSUCKUN: "An anti-CD72 immunotoxin against therapy-refractory B-lineage acute lymphoblastic leukemia", LEUK LYMPHOMA, vol. 18, no. 1-2, June 1995 (1995-06-01), pages 119 - 22, XP002964114 |
| NAGAE ET AL., J BIOL CHEM, vol. 288, no. 47, 2013, pages 33784 - 33796 |
| NAKANO KISHIGURO TKONISHI H ET AL.: "Generation of a humanized anti-glypican 3 antibody by CDR grafting and stability optimization", ANTICANCER DRUGS, vol. 21, no. 10, November 2010 (2010-11-01), pages 907 - 916, XP008177208, DOI: 10.1097/CAD.0b013e32833f5d68 |
| NEESON ET AL., J IMMUNOL, May 2013 (2013-05-01), pages 190 |
| NEJATOLLAHI ET AL., J OF ONCOLOGY, 2013, pages 2013 |
| NICHOLSON ET AL., MOL. IMMUN, vol. 34, no. 16-17, 1997, pages 1157 - 1165 |
| NICHOLSON ET AL., MOLECULAR IMMUNOLOGY, vol. 34, no. 16-17, 1997, pages 1157 - 1165 |
| NOORDHUIS ET AL.: "Targeting of CLEC12A In Acute Myeloid Leukemia by Antibody-Drug-Conjugates and Bispecific CLL-lxCD3 BiTE Antibody", 53RD ASH ANNUAL MEETING AND EXPOSITION, 10 December 2011 (2011-12-10) |
| OSTERMANN ET AL., CLINICAL CANCER RESEARCH, vol. 14, 2008, pages 4584 - 4592 |
| PARKER ET AL., PROTEIN EXPR PURIF, vol. 89, no. 2, 2013, pages 136 - 145 |
| PIZZITOLA ET AL., LEUKEMIA, 2014 |
| POLSON ET AL.: "Antibody-Drug Conjugates for the Treatment of Non-Hodgkin's Lymphoma: Target and Linker-Drug Selection", CANCER RES, vol. 69, 15 March 2009 (2009-03-15), pages 2358, XP055076856, DOI: 10.1158/0008-5472.CAN-08-2250 |
| ROUT-PITT ET AL., JBIOL. METHODS, vol. 5, no. 2, 2018, pages 1 - 9 |
| RUIZ ET AL., NUCLEIC ACIDS RES, vol. 28, 2000, pages 219 - 221 |
| SCOTT AM ET AL., CANCER RES, vol. 60, 2000, pages 3254 - 61 |
| SMITH ET AL.: "Ex vivo expansion of human T cells for adoptive immunotherapy using the novel Xeno-free CTS Immune Cell Serum Replacement", CLINICAL & TRANSLATIONAL IMMUNOLOGY, vol. 4, 2015, pages e31 |
| SONG ET AL., MED ONCOL, vol. 29, no. 4, 2012, pages 2923 - 2931 |
| SONG ET AL., TARGET ONCOL, 14 August 2013 (2013-08-14) |
| STONE ET AL., ONCOIMMUNOLOGY, vol. 1, no. 6, 2012, pages 863 - 873 |
| TRAN ET AL., J EXP MED, vol. 210, no. 6, 2013, pages 1125 - 1135 |
| WANG ET AL., J EXP MED., vol. 184, no. 6, 1996, pages 2207 - 16 |
| WILLEMSEN ET AL., J IMMUNOL, vol. 174, no. 12, 2005, pages 7853 - 7858 |
| WILLEMSEN RA ET AL., GENE THERAPY, vol. 7, 2000, pages 1369 - 1377 |
| XIAO ET AL., EXP. NEUROBIOL, vol. 144, 1997, pages 1 13 - 124 |
| XU ET AL., LEUK LYMPHOMA. 2013, vol. 54, no. 2, 2012, pages 255 - 260 |
| YU ET AL., MOL THER, vol. 22, no. 1, 2014, pages 102 - 111 |
| ZHANG T ET AL., CANCER GENE THER, vol. 11, 2004, pages 487 - 496 |
| ZHAO ET AL., J IMMUNOL METHODS, vol. 363, no. 2, 2011, pages 221 - 232 |
| ZUFFEREY ET AL., NAT. BIOTECHNOL., vol. 15, 1997, pages 871 - 875 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN119709874A (en) * | 2025-02-28 | 2025-03-28 | 江苏谱新生物医药有限公司 | Preparation for lentivirus transduction and application thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2023369684A1 (en) | 2025-04-17 |
| EP4608983A1 (en) | 2025-09-03 |
| CN120112650A (en) | 2025-06-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12325728B2 (en) | Methods and compositions for genetically modifying lymphocytes to express polypeptides comprising the intracellular domain of CD79A and CD79B | |
| US20220340927A1 (en) | Methods and compositions for the modification and delivery of lymphocytes | |
| US20230111159A1 (en) | Methods and compositions for the delivery of modified lymphocyte aggregates | |
| JP2018533968A (en) | Optimized lentiviral transfer vector and its use | |
| US20230044451A1 (en) | Methods and compositions for the delivery of modified lymphocytes and/or retroviral particles | |
| US20210317408A1 (en) | Methods and compositions for genetically modifying lymphocytes in blood or in enriched pbmcs | |
| US12351618B2 (en) | Engineered trimeric CD70 proteins and uses thereof | |
| JP2022546315A (en) | chimeric inhibitory receptor | |
| US20200255864A1 (en) | Methods and compositions for genetically modifying and expanding lymphocytes and regulating the activity thereof | |
| WO2020047527A2 (en) | Methods and compositions for genetically modifying lymphocytes in blood or in enriched pbmcs | |
| US20240384293A1 (en) | Viral vector production system | |
| WO2024089639A1 (en) | Lentiviral formulations | |
| WO2022187289A1 (en) | Methods and compositions for the delivery of retroviral particles | |
| US20250297282A1 (en) | Viral-binding protein and related reagents, articles, and methods of use | |
| KR20250011945A (en) | Production of virus particles | |
| JP2024510933A (en) | Methods and compositions for delivery of retroviral particles | |
| CN117043346A (en) | Methods and compositions for delivering retroviral particles |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 23800978 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: AU2023369684 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2023369684 Country of ref document: AU Date of ref document: 20231026 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2025524393 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2025524393 Country of ref document: JP Ref document number: 202380075451.9 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023800978 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11202502287Y Country of ref document: SG |
|
| WWP | Wipo information: published in national office |
Ref document number: 11202502287Y Country of ref document: SG |
|
| ENP | Entry into the national phase |
Ref document number: 2023800978 Country of ref document: EP Effective date: 20250526 |
|
| WWP | Wipo information: published in national office |
Ref document number: 202380075451.9 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2023800978 Country of ref document: EP |