WO2024047247A1 - Base editing approaches for the treatment of amyotrophic lateral sclerosis - Google Patents
Base editing approaches for the treatment of amyotrophic lateral sclerosis Download PDFInfo
- Publication number
- WO2024047247A1 WO2024047247A1 PCT/EP2023/074086 EP2023074086W WO2024047247A1 WO 2024047247 A1 WO2024047247 A1 WO 2024047247A1 EP 2023074086 W EP2023074086 W EP 2023074086W WO 2024047247 A1 WO2024047247 A1 WO 2024047247A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- c9orf72
- sequence
- base
- editing
- rna
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7088—Compounds having three or more nucleosides or nucleotides
- A61K31/7105—Natural ribonucleic acids, i.e. containing only riboses attached to adenine, guanine, cytosine or uracil and having 3'-5' phosphodiester links
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/16—Hydrolases (3) acting on ester bonds (3.1)
- C12N9/22—Ribonucleases [RNase]; Deoxyribonucleases [DNase]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/20—Type of nucleic acid involving clustered regularly interspaced short palindromic repeats [CRISPR]
Definitions
- ALS Amyotrophic Lateral Sclerosis
- ALS is the most prevalent adult-onset motor neuron disease with an estimated prevalence of 8/100,000 and 1,500 new cases per year in France. ALS is caused by multiple genetic causes; the most prevalent is a repeat expansion in the C9orf72 gene ( Figure 1).
- C9orf72 repeat expansions are recognized as a major genetic cause for a related neurodegenerative disorder, Frontotemporal Dementia (FTD).
- FTD Frontotemporal Dementia
- toxic gain of function of the repeats is considered to be an important pathogenic mechanism
- significantly reduced levels of C9orf72 transcript and protein have been consistently described in ALS patients indicating that C9orf72 loss of function causes neurodegeneration (Ciura, Sorana, et al. "Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis.” Annals of neurology 74.2 (2013): 180-187). Therefore, restoring C9orf72 function represents an important therapeutic target in ALS, FTD and related disorders.
- ALS is untreatable through common pharmacological approaches.
- Current therapies for ALS include the administration of antisense oligonucleotides and gene silencing by AAV to target the RNA repeats transcribed from the C9orf72 gene (reviewed in Cappella, Marisa, et al. "Gene therapy for ALS—a perspective.” International journal of molecular sciences 20.18 (2019): 4388).
- these strategies remain either temporary and need replenishing as in the case of oligonucleotides or are still undergoing early clinical trials for gene delivery via the AAV approach. Therefore, there is a strong rationale for developing novel, definitive and universal strategies for this disease.
- CRISPR-system-based cytosine and adenine base editors can make pinpoint changes in the DNA with little or no double-strand break (DSB) generation (Rees, Holly A., and David R. Liu. “Base editing: precision chemistry on the genome and transcriptome of living cells.” Nature reviews genetics 19.12 (2016): 770-788).
- BEs The basic components of BEs are a catalytically disabled Cas9 nuclease, a guide RNA that drives the system to a specific DNA target, and a deaminase that produces C-G to T-A or A-T to G-C conversions (for CBEs and ABEs, respectively).
- Base-editing approaches allow precise DNA repair virtually in the absence of DSBs, and thus eliminate the risks of DSB-induced apoptosis, translocations and insertions or deletions of large portions of DNA (Kosicki, Michael, Kärt Tomberg, and Allan Bradley.
- the major objective of the present invention is to develop safe, effective and definitive therapeutic strategies for ALS through the innovative approach of base editing.
- the inventors have identified a unique site of RNA Polymerase II pause site in the promoter region of C9orf72. Their approach was therefore to mutate this site using the base editing technology to increase C9orf72 transcription and expression. Therefore, base editing approaches that target the RNA Polymerase II pause site should boost the expression of C9orf72 as well as likely reduce the toxicity due to repeat expression, thus doubling the therapeutic potential of the genome editing strategy.
- the polymer may be linear or branched, it may comprise modified amino acids, and it may be interrupted by non-amino acids.
- the terms also encompass an amino acid polymer that has been modified; for example, disulfide bond formation, glycosylation, lipidation, acetylation, phosphorylation, pegylation, or any other manipulation, such as conjugation with a labeling component.
- amino acid includes natural and/or unnatural or synthetic amino acids, including glycine and both the D or L optical isomers, and amino acid analogs and peptidomimetics.
- nucleic acid molecule or “polynucleotide” refers to a DNA molecule (for example, but not limited to, a cDNA or genomic DNA).
- the nucleic acid molecule can be single-stranded or double-stranded.
- isolated when referring to nucleic acid molecules or polypeptides means that the nucleic acid molecule or the polypeptide is substantially free from at least one other component with which it is associated or found together in nature.
- complementarity refers to the ability of a nucleic acid to form hydrogen bond(s) with another nucleic acid sequence by either traditional Watson-Crick base- pairing or other non-traditional types.
- a percent complementarity indicates the percentage of residues in a nucleic acid molecule which can form hydrogen bonds (e.g., Watson-Crick base pairing) with a second nucleic acid sequence (e.g., 5, 6, 7, 8, 9, 10 out of 10 being 50%, 60%, 70%, 80%, 90%, and 100% complementary).
- Perfectly complementary means that all the contiguous residues of a nucleic acid sequence will hydrogen bond with the same number of contiguous residues in a second nucleic acid sequence.
- “Substantially complementary” as used herein refers to a degree of complementarity that is at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 97%, 98%, 99%, or 100% over a region of 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 30, 35, 40, 45, 50, or more nucleotides, or refers to two nucleic acids that hybridize under stringent conditions.
- stringent conditions for hybridization refer to conditions under which a nucleic acid having complementarity to a target sequence predominantly hybridizes with the target sequence, and substantially does not hybridize to non-target sequences. Stringent conditions are generally sequence-dependent, and vary depending on a number of factors.
- hybridization or “hybridizing” refers to a process where completely or partially complementary nucleic acid strands come together under specified hybridization conditions to form a double-stranded structure or region in which the two constituent strands are joined by hydrogen bonds.
- fusion polypeptide or “fusion protein” means a protein created by joining two or more polypeptide sequences together.
- the fusion polypeptides encompassed in this invention include translation products of a chimeric gene construct that joins the nucleic acid sequences encoding a first polypeptide, e.g., an RNA-binding domain, with the nucleic acid sequence encoding a second polypeptide, e.g., an effector domain, to form a single open- reading frame.
- a “fusion polypeptide” or “fusion protein” is a recombinant protein of two or more proteins which are joined by a peptide bond or via several peptides.
- the fusion protein may also comprise a peptide linker between the two domains.
- wild type is a term of the art understood by skilled persons and means the typical form of an organism, strain, gene or characteristic as it occurs in nature as distinguished from mutant or variant forms.
- derived from refers to a process whereby a first component (e.g., a first molecule), or information from that first component, is used to isolate, derive or make a different second component (e.g., a second molecule that is different from the first).
- the comparison of sequences and determination of percent identity between two sequences can be accomplished using a mathematical algorithm, as described below.
- the percent identity between two amino acid sequences can be determined using the Needleman and Wunsch algorithm (Needleman, Saul B. & Wunsch, Christian D. (1970). "A general method applicable to the search for similarities in the amino acid sequence of two proteins". Journal of Molecular Biology.48 (3): 443–53).
- the percent identity between two nucleotide or amino acid sequences may also be determined using for example algorithms such as EMBOSS Needle (pair wise alignment; available at www.ebi.ac.uk).
- EMBOSS Needle may be used with a BLOSUM62 matrix, a “gap open penalty” of 10, a “gap extend penalty” of 0.5, a false “end gap penalty”, an “end gap open penalty” of 10 and an “end gap extend penalty” of 0.5.
- the “percent identity” is a function of the number of matching positions divided by the number of positions compared and multiplied by 100. For instance, if 6 out of 10 sequence positions are identical between the two compared sequences after alignment, then the identity is 60%.
- % identity is typically determined over the whole length of the query sequence on which the analysis is performed.
- Two molecules having the same primary amino acid sequence or nucleic acid sequence are identical irrespective of any chemical and/or biological modification.
- a first amino acid sequence having at least 90% of identity with a second amino acid sequence means that the first sequence has 90; 91; 92; 93; 94; 95; 96; 97; 98; 99 or 100% of identity with the second amino acid sequence.
- linker refers to any means, entity or moiety used to join two or more entities.
- a linker can be a covalent linker or a non-covalent linker.
- covalent linkers include covalent bonds or a linker moiety covalently attached to one or more of the proteins or domains to be linked.
- the linker can also be a non-covalent bond, e.g., an organometallic bond through a metal centre such as platinum atom.
- various functionalities can be used, such as amide groups, including carbonic acid derivatives, ethers, esters, including organic and inorganic esters, amino, urethane, urea and the like.
- the domains can be modified by oxidation, hydroxylation, substitution, reduction etc. to provide a site for coupling. Methods for conjugation are well known by persons skilled in the art and are encompassed for use in the present invention.
- Linker moieties include, but are not limited to, chemical linker moieties, or for example a peptide linker moiety (a linker sequence). It will be appreciated that modification which do not significantly decrease the function of the RNA- binding domain and effector domain are preferred.
- the “linked” as used herein refers to the attachment of two or more entities to form one entity.
- a conjugate encompasses both peptide-small molecule conjugates as well as peptide-protein/peptide conjugates.
- the term “base-editing enzyme” refers to fusion protein comprising a defective CRISPR/Cas nuclease linked to a deaminase polypeptide. The term is also known as “base- editor”.
- CBEs cytosine base-editing enzymes
- ABEs adenine base-editing enzymes
- cytosine base-editing enzymes are created by fusing the defective CRISPR/Cas nuclease to a deaminase.
- the term “deaminase” refers to an enzyme that catalyzes a deamination reaction.
- deamination refers to the removal of an amine group from one molecule.
- the deaminase is a cytidine deaminase, catalysing the hydrolytic deamination of cytidine or deoxycytidine to uracil or deoxyuracil, respectively.
- nuclease includes a protein (i.e.
- CRISPR/Cas nuclease has its general meaning in the art and refers to segments of prokaryotic DNA containing clustered regularly interspaced short palindromic repeats (CRISPR) and associated nucleases encoded by Cas genes.
- CRISPR clustered regularly interspaced short palindromic repeats
- the CRISPR/Cas loci encode RNA-guided adaptive immune systems against mobile genetic elements (viruses, transposable elements and conjugative plasmids). Three types of CRISPR systems have been identified.
- CRISPR clusters contain spacers, the sequences complementary to antecedent mobile elements. CRISPR clusters are transcribed and processed into mature CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) RNA (crRNA).
- the CRISPR/Cas nucleases Cas9 and Cpf1 belong to the type II and type V CRISPR/Cas system and have strong endonuclease activity to cut target DNA. Cas9 is guided by a mature crRNA that contains about 20 nucleotides of unique target sequence (called spacer) and a trans-activating small RNA (tracrRNA) that also serves as a guide for ribonuclease III-aided processing of pre-crRNA.
- spacer a mature crRNA that contains about 20 nucleotides of unique target sequence
- tracrRNA trans-activating small RNA
- the crRNA:tracrRNA duplex directs Cas9 to target DNA via complementary base pairing between the spacer on the crRNA and the complementary sequence (called protospacer) on the target DNA.
- Cas9 recognizes a trinucleotide (NGG for S. Pyogenes Cas9) protospacer adjacent motif (PAM) to specify the cut site (the 3 rd or the 4 th nucleotide upstream from PAM).
- NVG trinucleotide
- PAM protospacer adjacent motif
- Cas9 or “Cas9 nuclease” refers to an RNA-guided nuclease comprising a Cas9 protein, or a fragment thereof (e.g., a protein comprising an active or inactive DNA cleavage domain of Cas9, and/or the gRNA binding domain of Cas9).
- a Cas9 nuclease is also referred to sometimes as a casn1 nuclease or a CRISPR (clustered regularly interspaced short palindromic repeat)-associated nuclease.
- CRISPR is an adaptive immune system that provides protection against mobile genetic elements (viruses, transposable elements and conjugative plasmids).
- CRISPR clusters contain spacers, sequences complementary to antecedent mobile elements, and target invading nucleic acids. CRISPR clusters are transcribed and processed into CRISPR RNA (crRNA). In type II CRISPR systems correct processing of pre-crRNA requires a trans-encoded small RNA (tracrRNA), endogenous ribonuclease 3 (rnc) and a Cas9 protein. The tracrRNA serves as a guide for ribonuclease 3-aided processing of pre- crRNA. Subsequently, Cas9/crRNA/tracrRNA endonucleolytically cleaves linear or circular dsDNA target complementary to the spacer.
- tracrRNA trans-encoded small RNA
- rnc endogenous ribonuclease 3
- Cas9 protein serves as a guide for ribonuclease 3-aided processing of pre- crRNA.
- RNA single guide RNAs
- sgRNA single guide RNAs
- gNRA single guide RNAs
- Cas9 recognizes a short motif in the CRISPR repeat sequences (the PAM or protospacer adjacent motif) to help distinguish self versus non-self.
- Cas9 nuclease sequences and structures are well known to those of skill in the art (see, e.g., “Complete genome sequence of an M1 strain of Streptococcus pyogenes.” Ferretti et al., J. J., McShan W. M., Ajdic D. J., Savic D. J., Savic G., Lyon K., Primeaux C., Sezate S., Suvorov A. N., Kenton S., Lai H. S., Lin S. P., Qian Y., Jia H.
- Cas9 nucleases and sequences include Cas9 sequences from the organisms and loci disclosed in Chylinski, Rhun, and Charpentier, “The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems” (2013) RNA Biology 10:5, 726-737; the entire contents of which are incorporated herein by reference.
- Cas9 refers to Cas9 from: Corynebacterium ulcerans (NCBI Refs: NC_015683.1, NC_017317.1); Corynebacterium diphtheria (NCBI Refs: NC_016782.1, NC_016786.1); Spiroplasma syrphidicola (NCBI Ref: NC_021284.1); Prevotella intermedia (NCBI Ref: NC_017861.1); Spiroplasma taiwanense (NCBI Ref: NC_021846.1); Streptococcus iniae (NCBI Ref: NC_021314.1); Belliella baltica (NCBI Ref: NC_018010.1); Psychroflexus torquisI (NCBI Ref: NC_018721.1); Streptococcus thermophilus (NCBI Ref: YP_820832.1); Listeria innocua (NCBI Ref: NP_472073.1);
- Cas9 nuclease comprises the amino acid sequence as set forth in SEQ ID NO: 1.
- SEQ ID NO:1 Cas9 sequence MDKKYSIGLAIGTNSVGWAVITDEYKVPSKKFKVLGNTDRHSIKKNLIGALLFDSGETAEATRLKRTAR RRYTRRKNRICYLQEIFSNEMAKVDDSFFHRLEESFLVEEDKKHERHPIFGNIVDEVAYHEKYPTIYHL RKKLVDSTDKADLRLIYLALAHMIKFRGHFLIEGDLNPDNSDVDKLFIQLVQTYNQLFEENPINASGVD AKAILSARLSKSRRLENLIAQLPGEKKNGLFGNLIALSLGLTPNFKSNFDLAEDAKLQLSKDTYDDDLD NLLAQIGDQYADLFLAAKNLSDAILLSDILRVNTEITKAPLSASMIKRYDEHHQDLTLLKALVRQQQ
- nickase has its general meaning in the art and refers to an endonuclease which cleaves only a single strand of a DNA duplex. Accordingly, the term “Cas9 nickase” refers to a nickase derived from a Cas9 protein, typically by inactivating one nuclease domain of Cas9 protein.
- guide RNA molecule generally refers to an RNA molecule (or a group of RNA molecules collectively) that can bind to a Cas9 protein and target the Cas9 protein to a specific location within a target DNA.
- a guide RNA can comprise two segments: a DNA-targeting guide segment and a protein-binding segment.
- the DNA-targeting segment comprises a nucleotide sequence that is complementary to (or at least can hybridize to under stringent conditions) a target sequence.
- the protein-binding segment interacts with a CRISPR protein, such as a Cas9 or Cas9 related polypeptide. These two segments can be located in the same RNA molecule or in two or more separate RNA molecules. When the two segments are in separate RNA molecules, the molecule comprising the DNA-targeting guide segment is sometimes referred to as the CRISPR RNA (crRNA), while the molecule comprising the protein-binding segment is referred to as the trans-activating RNA (tracrRNA).
- CRISPR RNA CRISPR RNA
- tracrRNA trans-activating RNA
- target nucleic acid refers to a nucleic acid containing a target nucleic acid sequence.
- a target nucleic acid may be single-stranded or double-stranded, and often is double-stranded DNA.
- a “target nucleic acid sequence,” “target sequence” or “target region,” as used herein, means a specific sequence or the complement thereof that one wishes to bind to using the CRISPR system as disclosed herein.
- target nucleic acid strand refers to a strand of a target nucleic acid that is subject to base-pairing with a guide RNA as disclosed herein.
- each strand can be a “target nucleic acid strand” to design crRNA and guide RNAs and used to practice the method of this invention as long as there is a suitable PAM site.
- the term “mutation” has its general meaning in the art and refers to a substitution, deletion or insertion.
- substitution means that a specific amino acid residue at a specific position is removed and another amino acid residue is inserted into the same position.
- deletion means that a specific amino acid residue is removed.
- insertion means that one or more amino acid residues are inserted before or after a specific amino acid residue.
- mutagenesis refers to the introduction of mutations into a polynucleotide sequence. According to the present invention mutations are introduced into a target DNA molecule encoding for a variant domain of the antibody so as to mimic somatic hypermutation.
- variant refers to a first composition (e.g., a first molecule), that is related to a second composition (e.g., a second molecule, also termed a “parent” molecule).
- the variant molecule can be derived from, isolated from, based on or homologous to the parent molecule.
- a variant molecule can have entire sequence identity with the original parent molecule, or alternatively, can have less than 100% sequence identity with the parent molecule.
- a variant of a sequence can be a second sequence that is at least 50; 51; 52; 53; 54; 55; 56; 57; 58; 59; 60; 61; 62; 63; 64; 65; 66; 67; 68; 69; 70; 71; 72; 73; 74; 75; 76; 77; 78; 79; 80; 81; 82; 83; 84; 85; 86; 87; 88; 89; 90; 91; 92; 93; 94; 95; 96; 97; 98; 99; 100% identical in sequence compare to the original sequence.
- Sequence identity is frequently measured in terms of percentage identity (or similarity or homology); the higher the percentage, the more similar are the two sequences.
- Methods of alignment of sequences for comparison are well known in the art. Various programs and alignment algorithms are described in: Smith and Waterman, Adv. Appl. Math., 2:482, 1981; Needleman and Wunsch, J. Mol. Biol., 48:443, 1970; Pearson and Lipman, Proc. Natl. Acad. Sci. U.S.A., 85:2444, 1988; Higgins and Sharp, Gene, 73:237-244, 1988; Higgins and Sharp, CABIOS, 5:151-153, 1989; Corpet et al. Nuc.
- ALIGN compares entire sequences against one another, while LFASTA compares regions of local similarity.
- these alignment tools and their respective tutorials are available on the Internet at the NCSA Website, for instance.
- the Blast 2 sequences function can be employed using the default BLOSUM62 matrix set to default parameters, (gap existence cost of 11, and a per residue gap cost of 1).
- the alignment should be performed using the Blast 2 sequences function, employing the PAM30 matrix set to default parameters (open gap 9, extension gap 1 penalties).
- the BLAST sequence comparison system is available, for instance, from the NCBI web site; see also Altschul et al., J. Mol.
- C9orf72 has its general meaning in the art and refers to the chromosome 9 open reading frame 72 protein that is encoded by the gene C9orf72 (Gene ID: 203228).
- the human C9orf72 gene is located on the short (p) arm of chromosome 9 open reading frame 72, from base pair 27,546,546 to base pair 27,573,866 (GRCh38). Its cytogenetic location is at 9p21.2. The protein is found in many regions of the brain, in the cytoplasm of neurons as well as in presynaptic terminals.
- C9orf72 is a component of the C9orf72-SMCR8 complex that has guanine nucleotide exchange factor (GEF) activity and regulates autophagy.
- GEF guanine nucleotide exchange factor
- C9orf72 and SMCR8 probably constitute the catalytic subunits that promote the exchange of GDP to GTP, converting inactive GDP-bound RAB8A and RAB39B into their active GTP-bound form, thereby promoting autophagosome maturation.
- the C9orf72-SMCR8 complex also acts as a regulator of autophagy initiation by interacting with the ULK1/ATG1 kinase complex and modulating its protein kinase activity.
- An exemplary amino acid sequence for C9orf72 is represented by SEQ ID NO:2.
- the identified mutation of C9ORF72 is a hexanucleotide repeat expansion of the six-letter string of nucleotides GGGGCC (Bigio EH (December 2011).
- C9ORF72 the new gene on the block, causes C9FTD/ALS: new insights provided by neuropathology. Acta Neuropathologica. 122 (6): 653–5).
- the mutation is the most common identified mutation associated with frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) (Babi ⁇ Leko M, ⁇ upunski V, Kirincich J, Smilovi ⁇ D, Hortobágyi T, Hof PR, ⁇ imi ⁇ G (2019).
- FDD frontotemporal dementia
- ALS amyotrophic lateral sclerosis
- RNA Pol II pausing is a key regulatory step in transcription. RNA Pol II pauses at GC-rich regions, and almost 65% of the pause sites are cytosines (Watts, Jason A., et al. "Cis elements that mediate RNA polymerase II pausing regulate human gene expression.” The American Journal of Human Genetics 105.4 (2019): 677-688). Gene expression analysis showed that genes with more paused polymerases have lower expression levels. Therefore, mutagenesis of the pause sites might lead to a significant increase in transcription.
- pause site is located on chr9:27563356-27563366 region, is defined by the nucleotide sequence SEQ ID NO:3 (GTTCGGCTGCCGGGAAGAGGCGCGGGTAGAAGCGGGGGCTCTCCTCAGAGCT) and is depicted in Figure 1.
- ALS Amyotrophic Lateral Sclerosis
- SEQ ID NO: 29 Amyotrophic Lateral Sclerosis
- ALS has its general meaning in the art and refers to a neurodegenerative disease characterized by progressive muscular paralysis reflecting degeneration of motor neurons in the primary motor cortex, corticospinal tracts, brainstem and spinal cord. According to the present invention, ALS is caused by the C9orf72 mutation as described above.
- treatment refers to both prophylactic or preventive treatment as well as curative or disease modifying treatment, including treatment of patient at risk of contracting the disease or suspected to have contracted the disease as well as patients who are ill or have been diagnosed as suffering from a disease or medical condition, and includes suppression of clinical relapse.
- the treatment may be administered to a subject having a medical disorder or who ultimately may acquire the disorder, in order to prevent, cure, delay the onset of, reduce the severity of, or ameliorate one or more symptoms of a disorder or recurring disorder, or in order to prolong the survival of a subject beyond that expected in the absence of such treatment.
- therapeutic regimen is meant the pattern of treatment of an illness, e.g., the pattern of dosing used during therapy.
- a therapeutic regimen may include an induction regimen and a maintenance regimen.
- the phrase “induction regimen” or “induction period” refers to a therapeutic regimen (or the portion of a therapeutic regimen) that is used for the initial treatment of a disease.
- the general goal of an induction regimen is to provide a high level of drug to a patient during the initial period of a treatment regimen.
- An induction regimen may employ (in part or in whole) a "loading regimen", which may include administering a greater dose of the drug than a physician would employ during a maintenance regimen, administering a drug more frequently than a physician would administer the drug during a maintenance regimen, or both.
- maintenance regimen refers to a therapeutic regimen (or the portion of a therapeutic regimen) that is used for the maintenance of a patient during treatment of an illness, e.g., to keep the patient in remission for long periods of time (months or years).
- a maintenance regimen may employ continuous therapy (e.g., administering a drug at regular intervals, e.g., weekly, monthly, yearly, etc.) or intermittent therapy (e.g., interrupted treatment, intermittent treatment, treatment at relapse, or treatment upon achievement of a particular predetermined criteria [e.g., pain, disease manifestation, etc.]).
- the expression "therapeutically effective amount” means a sufficient amount of the active ingredient for treating or reducing the symptoms at reasonable benefit/risk ratio applicable to any medical treatment. It will be understood that the total daily usage of the compounds and compositions of the present invention will be decided by the attending physician within the scope of sound medical judgment.
- the specific therapeutically effective dose level for any particular subject will depend upon a variety of factors including the disorder being treated and the severity of the disorder; activity of the specific compound employed; the specific composition employed, the age, body weight, general health, sex and diet of the subject; the time of administration, route of administration, and rate of excretion of the specific compound employed; the duration of the treatment; drugs used in combination with the active ingredients; and like factors well known in the medical arts.
- the first object of the present invention relates to a method for restoring the expression of C9orf72 in a patient suffering from Amyotrophic Lateral Sclerosis (ALS) comprising administering to the patient a therapeutically effective amount of a gene editing platform that consists of (a) at least one base-editing enzyme and (b) at least one guide RNA molecule for guiding the base-editing enzyme to at least one target sequence in the RNA Polymerase II pause site located in the first intronic region of the gene C9orf72, thereby editing said site and subsequently increasing the expression of C9orf72.
- a gene editing platform that consists of (a) at least one base-editing enzyme and (b) at least one guide RNA molecule for guiding the base-editing enzyme to at least one target sequence in the RNA Polymerase II pause site located in the first intronic region of the gene C9orf72, thereby editing said site and subsequently increasing the expression of C9orf72.
- the gene editing platform is suitable for introducing some mutations in the RNA Polymerase II pause site. In some embodiments, the gene editing platform is particularly suitable for mutating the base C located at position Chr9:27563356-27563366- Hg18. In some embodiments, the gene editing platform is particularly suitable for introducing one C>T conversion at position Chr9:27563356-27563366-Hg18 ( Figure 1A).
- the base-editing enzyme of the present invention comprises a defective CRISPR/Cas nuclease. The sequence recognition mechanism is the same as for the non- defective CRISPR/Cas nuclease.
- the defective CRISPR/Cas nuclease of the invention comprises at least one RNA binding domain.
- the RNA binding domain interacts with a guide RNA molecule as defined hereinafter.
- the defective CRISPR/Cas nuclease of the invention is a modified version with no nuclease activity. Accordingly, the defective CRISPR/Cas nuclease specifically recognizes the guide RNA molecule and thus guides the base-editing enzyme to its target DNA sequence.
- the defective CRISPR/Cas nuclease can be modified to increase nucleic acid binding affinity and/or specificity, alter an enzymatic activity, and/or change another property of the protein.
- the nuclease domains of the protein can be modified, deleted, or inactivated.
- the protein can be truncated to remove domains that are not essential for the function of the protein.
- the protein is truncated or modified to optimize the activity of the RNA binding domain.
- the CRISPR/Cas nuclease consists of a mutant CRISPR/Cas nuclease i.e. a protein having one or more point mutations, insertions, deletions, truncations, a fusion protein, or a combination thereof.
- the mutant has the RNA-guided DNA binding activity, but lacks one or both of its nuclease active sites.
- the mutant comprises an amino acid sequence having at least 50% of identity with the wild type amino acid sequence of the CRISPR/Cas nuclease.
- CRISPR/Cas nucleases can be used in this invention.
- Non-limiting examples of suitable CRISPR/CRISPR/Cas nucleases include Cas3, Cas4, Cas5, Cas5e (or CasD), Cas6, Cas6e, Cas6f, Cas7, Cas8a1, Cas8a2, Cas8b, Cas8c, Cas9, Cas10, Cas10d, CasF, CasG, CasH, Csy1, Csy2, Csy3, Cse1 (or CasA), Cse2 (or CasB), Cse3 (or CasE), Cse4 (or CasC), Csc1, Csc2, Csa5, Csn2, Csm2, Csm3, Csm4, Csm5, Csm6, Cmr1, Cmr3, Cmr4, Cmr5, Cmr6, Csb1, Csb2, Csb3, Csx17, Csx14, Csx10, Csx16, CsaX, Csx3, Cs
- the CRISPR/Cas nuclease is derived from a type II CRISPR-Cas system. In some embodiments, the CRISPR/Cas nuclease is derived from a Cas9 protein.
- the Cas9 protein can be from Streptococcus pyogenes, Streptococcus thermophilus, Streptococcus sp., Nocardiopsis rougevillei, Streptomyces pristinaespiralis, Streptomyces viridochromogenes, Streptomyces viridochromogenes, Streptosporangium roseum, Streptosporangium roseum, Alicyclobacillus acidocaldarius, Bacillus pseudomycoides, Bacillus selenitireducens, Exiguobacterium sibiricum, Lactobacillus delbrueckii, Lactobacillus salivarius, Microscilla marina, Burkholderiales bacterium, Polaromonas naphthalenivorans, Polaromonas sp., Crocosphaera watsonii, Cyanothece sp., Microcystis aeruginosa, Synechococcus s
- the CRISPR/Cas nuclease is a mutant of a wild type CRISPR/Cas nuclease (such as Cas9) or a fragment thereof. In some embodiments, the CRISPR/Cas nuclease is a mutant Cas9 protein from S. pyogenes.
- Methods for generating a Cas9 protein (or a fragment thereof) having an inactive DNA cleavage domain are known (See, e.g., Jinek et al., Science.337:816-821(2012); Qi et al., “Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression” (2013) Cell.28; 152(5):1173-83, the entire contents of each of which are incorporated herein by reference).
- the DNA cleavage domain of Cas9 is known to include two subdomains, the HNH nuclease subdomain and the RuvC1 subdomain.
- the HNH subdomain cleaves the strand complementary to the gRNA, whereas the RuvC1 subdomain cleaves the non-complementary strand. Mutations within these subdomains can silence the nuclease activity of Cas9. For example, the mutations D10A and H841A completely inactivate the nuclease activity of S. pyogenes Cas9 (Jinek et al., Science.337:816-821(2012); Qi et al., Cell. 28; 152(5):1173-83 (2013)).
- the CRISPR/Cas nuclease of the present invention is nickase and more particularly a Cas9 nickase i.e.
- the nickase of the present invention comprises the amino acid sequence as set forth in SEQ ID NO:4 or SEQ ID NO:5. SEQ ID NO: 4> S.
- variants of dCas9 are provided which are at least about 70% identical, at least about 80% identical, at least about 90% identical, at least about 95% identical, at least about 98% identical, at least about 99% identical, at least about 99.5% identical, or at least about 99.9% to SEQ ID NO:1.
- variants of dCas9 are provided having amino acid sequences which are shorter, or longer than SEQ ID NO: 1 or 3, by about 5 amino acids, by about 10 amino acids, by about 15 amino acids, by about 20 amino acids, by about 25 amino acids, by about 30 amino acids, by about 40 amino acids, by about 50 amino acids, by about 75 amino acids, by about 100 amino acids or more.
- the second component of the base-editing enzyme herein disclosed comprises a non-nuclease DNA modifying enzyme that is a deaminase. Since, the targeted base of interest is a C, the deaminase is preferably a cytidine deaminase.
- the cytidine deaminase is an apolipoprotein B mRNA-editing complex (APOBEC) family deaminase. In some embodiments, the cytidine deaminase is an APOBEC1 family deaminase. In some embodiments, the cytidine deaminase is an activation-induced cytidine deaminase (AID). In some embodiments, the cytidine deaminase is an ACF1/ASE deaminase.
- APOBEC apolipoprotein B mRNA-editing complex
- the cytidine deaminase is selected from the group consisting of AID: activation induced cytidine deaminase, APOBEC1: apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 1, APOBEC3A: apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3A, APOBEC3B: apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3B, APOBEC3C: apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3C, APOBEC3D: apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3D, APOBEC3F: apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3F, APOBEC3G: apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like
- the cytidine deaminase derives from the Activation Induced cytidine Deaminase (AID).
- AID is a cytidine deaminase that can catalyze the reaction of deamination of cytosine in the context of DNA or RNA.
- AID changes a C base to U base. In dividing cells, this could lead to a C to T point mutation.
- the change of C to U could trigger cellular DNA repair pathways, mainly excision repair pathway, which will remove the mismatching U-G base-pair, and replace with a T-A, A-T, C-G, or G-C pair.
- the DNA modifying enzyme is AID* ⁇ that is an AID mutant with increased SHM activity whose Nuclear Export Signal (NES) has been removed (Hess GT, Fresard L, Han K, Lee CH, Li A, Cimprich KA, Montgomery SB, Bassik MC: Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat Methods 2016, 13(12):1036- 1042).
- the cytidine deaminase consists of a variant of the amino acid sequence as set forth in SEQ ID NO:6-16.
- the deaminase is fused to the C-terminus of the defective CRISPR/Cas nuclease.
- the defective CRISPR/Cas nuclease and the deaminase are fused via a linker.
- the linker comprises a (GGGGS)n (SEQ ID NO:17), a (G)n, an (EAAAK)n (SEQ ID NO: 18), a (GGS)n, or an SGSETPGTSESATPES (SEQ ID NO:19) motif (see, e.g., Guilinger, John P., David B. Thompson, and David R. Liu.
- suitable linker motifs and configurations include those described in Chen et al., Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev. 2013; 65(10):1357-69, the entire contents of which are incorporated herein by reference.
- the fusion protein may comprise additional features.
- localization sequences such as nuclear localization sequences (NLS), cytoplasmic localization sequences, export sequences, such as nuclear export sequences, or other localization sequences, as well as sequence tags that are useful for solubilization, purification, or detection of the fusion proteins.
- NLS nuclear localization sequences
- cytoplasmic localization sequences such as cytoplasmic localization sequences
- export sequences such as nuclear export sequences, or other localization sequences
- sequence tags that are useful for solubilization, purification, or detection of the fusion proteins.
- Suitable localization signal sequences and sequences of protein tags include, but are not limited to, biotin carboxylase carrier protein (BCCP) tags, myc-tags, calmodulin-tags, FLAG-tags, hemagglutinin (HA)-tags, polyhistidine tags, also referred to as histidine tags or His-tags, maltose binding protein (MBP)-tags, nus-tags, glutathione-S-transferase (GST)-tags, green fluorescent protein (GFP)-tags, thioredoxin-tags, S-tags, Softags (e.g., Softag 1, Softag 3), strep-tags, biotin ligase tags, FlAsH tags, V5 tags, and SBP-tags.
- BCCP biotin carboxylase carrier protein
- MBP maltose binding protein
- GST glutathione-S-transferase
- GFP green fluorescent protein
- Softags e.g., Softag
- CBE base-editing enzymes are known in the art (see e.g. Koblan, Luke W., et al. "Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction.” Nature biotechnology 36.9 (2016): 843-846) and typically include those described in Table A.
- Table A some exemplary base-editing enzymes
- the amino acid sequence of CBE-SpRY is SEQ ID NO:20.
- the guide RNA molecule of the present invention thus comprises a guide sequence for providing the targeting specificity. It includes a region that is complementary and capable of hybridization to a pre-selected target site of interest in the RNA Polymerase II pause site. According to the present disclosure, the guide RNA targets the pause site that is located on chr9:27563356-27563366 region, and more preferably the nucleotide sequence SEQ ID NO:30 (GAAGAGGCGCGGGTAGAAGC). In some embodiment, this guide sequence can comprise from about 10 nucleotides to more than about 25 nucleotides.
- the region of base pairing between the guide sequence and the corresponding target site sequence can be about 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 22, 23, 24, 25, or more than 25 nucleotides in length.
- the guide sequence is about 17-20 nucleotides in length, such as 20 nucleotides.
- a software program is used to identify candidate CRISPR target sequences on both strands of the DNA nucleic acid molecule based on desired guide sequence length and a CRISPR motif sequence (PAM) for a specified CRISPR enzyme.
- PAM CRISPR motif sequence
- Each target sequence and its corresponding PAM site/sequence are referred herein as a Cas-targeted site.
- Type II CRISPR system one of the most well characterized systems, needs only Cas 9 protein and a guide RNA complementary to a target sequence to affect target cleavage.
- target sites for Cas9 from S. pyogenes, with PAM sequences NGG may be identified by searching for 5′-Nx-NGG- 3′ both on the input sequence and on the reverse-complement of the input. Since multiple occurrences in the genome of the DNA target site may lead to nonspecific genome editing, after identifying all potential sites, the program filters out sequences based on the number of times they appear in the relevant reference genome.
- the filtering step may be based on the seed sequence.
- results are filtered based on the number of occurrences of the seed:PAM sequence in the relevant genome.
- the user may be allowed to choose the length of the seed sequence.
- the user may also be allowed to specify the number of occurrences of the seed:PAM sequence in a genome for purposes of passing the filter. The default is to screen for unique sequences. Filtration level is altered by changing both the length of the seed sequence and the number of occurrences of the sequence in the genome.
- the program may in addition or alternatively provide the sequence of a guide sequence complementary to the reported target sequence(s) by providing the reverse complement of the identified target sequence(s). Further details of methods and algorithms to optimize sequence selection can be found in U.S. application Ser. No. 61/836,080; incorporated herein by reference.
- the guide RNA targets a sequence selected from Table 1 (see EXAMPLE).
- the gene editing platform comprises a) a base-editing enzyme that is a CBE-SpRY and b) and at least one guide RNA molecule that targets one sequence selected in Table 1.
- the guide RNA molecule of the present invention can be made by various methods known in the art including cell-based expression, in vitro transcription, and chemical synthesis.
- RNA molecules of the present invention can be made with recombinant technology using a host cell system or an in vitro translation-transcription system known in the art.
- the guide RNA molecule may include one or more modifications. Such modifications may include inclusion of at least one non-naturally occurring nucleotide, or a modified nucleotide, or analogs thereof. Modified nucleotides may be modified at the ribose, phosphate, and/or base moiety. Modified nucleotides may include 2’-O-methyl analogs, 2’- deoxy analogs, or 2’-fluoro analogs.
- the nucleic acid backbone may be modified, for example, a phosphorothioate backbone may be used.
- LNA locked nucleic acids
- BNA bridged nucleic acids
- modified bases include, but are not limited to, 2-aminopurine, 5-bromo-uridine, pseudouridine, inosine, 7-methylguanosine.
- a plurality of guide RNA molecules are designed for targeting a plurality of sequences in the RNA Polymerase II pause site.
- the gene editing platform disclosed herein thus comprises 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, or 20 guide RNA molecules as disclosed herein.
- a plurality of base-editing enzyme along with a plurality of guide RNA molecules are designed for targeting a plurality of sequences in the RNA Polymerase II pause site.
- the gene editing platform disclosed herein thus comprises 2, 3 or 4 base-editing enzymes and 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, or 20 RNA molecules as disclosed herein.
- the different components of the gene editing platform of the present invention are administered to the patient through expression from one or more vectors. Examples of vectors include viral vector and non-viral vectors.
- the term “viral vector” refers to a virion or virus particle that functions as a nucleic acid delivery vehicle and which comprises a vector genome packaged within the virion or virus particle.
- the vector is a viral vector which is an adeno-associated virus (AAV), a retroviral vector, bovine papilloma virus, an adenovirus vector, a vaccinia virus, or a polyoma virus.
- AAV adeno-associated virus
- the viral vector is a AAV vector.
- AAV vector means a vector derived from an adeno- associated virus serotype, including without limitation, AAV1, AAV2, AAV3, AAV4, AAV5, AAV6, AAV7, AAV8, AAV9, and mutated forms thereof.
- AAV vectors can have one or more of the AAV wild-type genes deleted in whole or part, preferably the rep and/or cap genes, but retain functional flanking ITR sequences.
- the viral vector is a retroviral vector.
- retroviral vector refers to a vector containing structural and functional genetic elements that are primarily derived from a retrovirus.
- the retroviral vector of the present invention derives from a retrovirus selected from the group consisting of alpharetroviruses (e.g., avian leukosis virus), betaretroviruses (e.g., mouse mammary tumor virus), gammaretroviruses (e.g., murine leukemia virus), deltaretroviruses (e.g., bovine leukemia virus), epsilonretroviruses (e.g., Walley dermal sarcoma virus), lentiviruses (e.g., HIV-1, HIV-2) and spumaviruses (e.g., human spumavirus).
- alpharetroviruses e.g., avian leukosis virus
- betaretroviruses e.g., mouse mammary tumor virus
- gammaretroviruses e.g., murine leukemia virus
- deltaretroviruses e.g., bovine leukemia virus
- the retroviral vector of the present invention is a replication deficient retroviral virus particle, which can transfer a foreign imported RNA of a gene instead of the retroviral mRNA.
- the retroviral vector of the present invention is a lentiviral vector.
- the term “lentiviral vector” refers to a vector containing structural and functional genetic elements that are primarily derived from a lentivirus.
- the lentiviral vector of the present invention is selected from the group consisting of HIV-1, HIV-2, SIV, FIV, EIAV, BIV, VISNA and CAEV vectors.
- the lentiviral vector is a HIV-1 vector.
- minimum retroviral gene delivery vectors can be prepared from a vector genome, which only contains, apart from the recombinant nucleic acid molecule of the present invention, the sequences of the retroviral genome which are non-coding regions of said genome, necessary to provide recognition signals for DNA or RNA synthesis and processing.
- the retroviral vector genome comprises all the elements necessary for the nucleic import and the correct expression of the polynucleotide of interest (i.e. the base editing platform).
- elements that can be inserted in the retroviral genome of the retroviral vector of the present invention are at least one (preferably two) long terminal repeats (LTR), such as a LTR5' and a LTR3', a psi sequence involved in the retroviral genome encapsidation, and optionally at least one DNA flap comprising a cPPT and a CTS domains.
- LTR long terminal repeats
- the LTR preferably the LTR3', is deleted for the promoter and the enhancer of U3 and is replaced by a minimal promoter allowing transcription during vector production while an internal promoter is added to allow expression of the transgene.
- the vector is a Self-INactivating (SIN) vector that contains a non-functional or modified 3' Long Terminal Repeat (LTR) sequence.
- This sequence is copied to the 5' end of the vector genome during integration, resulting in the inactivation of promoter activity by both LTRs.
- a vector genome may be a replacement vector in which all the viral coding sequences between the 2 long terminal repeats (LTRs) have been replaced by the recombinant nucleic acid molecule of the present invention.
- the retroviral vector genome is devoid of functional gag, pol and/or env retroviral genes. By “functional” it is meant a gene that is correctly transcribed, and/or correctly expressed.
- the retroviral vector genome of the present invention in this embodiment contains at least one of the gag, pol and env genes that is either not transcribed or incompletely transcribed; the expression "incompletely transcribed” refers to the alteration in the transcripts gag, gag-pro or gag-pro-pol, one of these or several of these being not transcribed.
- the retroviral genome is devoid of gag, pol and/or env retroviral genes.
- the retroviral vector genome is also devoid of the coding sequences for Vif-, Vpr-, Vpu- and Nef-accessory genes (for HIV-1 retroviral vectors), or of their complete or functional genes.
- the retroviral vector of the present invention is non replicative i.e., the vector and retroviral vector genome are not able to form new particles budding from the infected host cell. This may be achieved by the absence in the retroviral genome of the gag, pol or env genes, as indicated in the above paragraph; this can also be achieved by deleting other viral coding sequence(s) and/or cis-acting genetic elements needed for particles formation.
- the present invention encompasses use of virus-like particles.
- virus-like particle or “VLP” refers to a structure resembling a virus particle but devoid of the viral genome, incapable of replication and devoid of pathogenicity.
- the particle typically comprises at least one type of structural protein from a virus. Preferably only one type of structural protein is present. Most preferably no other non-structural component of a virus is present.
- virus-like particles can be spontaneously self-assembled by viral structural proteins under appropriate conditions in vitro while excluding the genetic material and potential replication probability.
- virus-like particles with a diameter of approximately 20 to 150 nm, also have the characteristics of nanometer materials, such as large surface area, surface-accessible amino acids with reactive moieties (e.g., lysine and glutamic acid residues), inerratic spatial structure, and good biocompatibility. Therefore, assembled virus-like particles have great potential as a delivery system for specifically carrying a variety of cargos.
- one or more of the zinc finger motifs of the Gag protein is/are substituted by one or more RNA-binding domain(s).
- the RNA-binding domain is the Coat protein of the MS2 bacteriophage, of the PP7 phage or of the Q3 phage, the prophage HK022 Nun protein, the U1A protein or the hPum protein. More preferably, the RNA binding domain is the Coat protein of the MS2 bacteriophage or of the PP7 phage. Even more preferably the RNA-binding domain is the Coat protein of the MS2 bacteriophage. These embodiments are particularly suitable for packaging the mRNA encoding for the apelin polypeptide into the VLP.
- the mRNA encoding for the apelin polypeptide that is encapsuled in the virus particle of the present invention comprises at least one encapsidation sequence.
- encapsidation sequence is meant an RNA motif (sequence and three-dimensional structure) recognized specifically by an RNA-binding domain as above described.
- the encapsidation sequence is a stem-loop motif.
- the encapsidation sequence of the retroviral particle is the stem-loop motif of the RNA of the MS2 bacteriophage or of the PP7 phage such as.
- the stem-loop motif and more particularly the stem-loop motif of the RNA of the MS2 bacteriophage or that of the RNA of the PP7 phage may be used alone or repeated several times, preferably from 2 to 25 times, more preferably from 2 to 18 times, for example from 6 to 18 times.
- the present invention encompasses the use of the LentiFlash® technology that based on non-integrative lentiviral particles constructed using a bacteriophage coat protein and its cognate 19-nt stem loop, to replace the natural lentiviral Psi packaging sequence, in order to achieve active mRNA packaging into the lentiviral particles (Prel A, Caval V, Gayon R, Ravassard P, Duthoit C, Payen E, Maouche-Chretien L, Creneguy A, Nguyen TH, Martin N, Piver E, Sevrain R, Lamouroux L, Leboulch P, Deschaseaux F, Bouée P, Sensébé L, Pagès JC.
- the LentiFlash® technology that based on non-integrative lentiviral particles constructed using a bacteriophage coat protein and its cognate 19-nt stem loop, to replace the natural lentiviral Psi packaging sequence, in order to achieve active mRNA packaging into the lentivi
- the retroviral vectors of the present invention can be produced by any well-known method in the art including by transfection (s) transient (s), in stable cell lines and / or by means of helper virus. Use of stable cell lines may also be preferred for the production of the vectors (Greene, M. R. et al.
- the retroviral vector of the present invention is obtainable by a transcomplementation system (vector/packaging system) by transfecting in vitro a permissive cell (such as 293T cells) with a plasmid containing the retroviral vector genome of the present invention, and at least one other plasmid providing, in trans, the gag, pol and env sequences encoding the polypeptides GAG, POL and the envelope protein(s), or for a portion of these polypeptides sufficient to enable formation of retroviral particles.
- a transcomplementation system vector/packaging system
- permissive cells are transfected with a) transcomplementation plasmid, lacking packaging signal psi and, the plasmid is optionally deleted of accessory genes vif, nef, vpu and / or vpr, b) a second plasmid (envelope expression plasmid or pseudotyping env plasmid) comprising a gene encoding an envelope protein(s) and c) a plasmid vector comprising a recombinant genome retroviral, optionally deleted from the promoter region of the 3 'LTR or U3 enhancer sequence of the 3' LTR, including, between the LTR sequences 5 'and 3' retroviral, a psi encapsidation sequence, a nuclear export element (preferably RRE element of HIV or other retroviruses equivalent), comprising the nucleic acid molecule of the present invention and optionally a promoter and / or a nuclear import sequence (cPPT sequence eg
- the three plasmids used do not contain homologous sequence sufficient for recombination.
- Nucleic acids encoding gag, pol and env cDNA can be advantageously prepared according to conventional techniques, from viral gene sequences available in the prior art and databases.
- the trans-complementation plasmid provides a nucleic acid encoding the proteins retroviral gag and pol. These proteins are derived from a lentivirus, and most preferably, from HIV-1.
- the plasmid is devoid of encapsidation sequence, sequence coding for an envelope, accessory genes, and advantageously also lacks retroviral LTRs.
- the sequences coding for gag and pol proteins are advantageously placed under the control of a heterologous promoter, eg cellular, viral, etc.., which can be constitutive or regulated, weak or strong. It is preferably a plasmid containing a sequence transcomplement ⁇ psi-CMV-gag-pol- PolyA. This plasmid allows the expression of all the proteins necessary for the formation of empty virions, except the envelope glycoproteins.
- the plasmid transcomplementation may advantageously comprise the TAT and REV genes. Plasmid transcomplementation is advantageously devoid of vif, vpr, vpu and / or nef accessory genes.
- gag and pol genes and genes TAT and REV can also be carried by different plasmids, possibly separated. In this case, several plasmids are used transcomplementation, each encoding one or more of said proteins.
- the promoters used in the plasmid transcomplementation, the envelope plasmid and the plasmid vector respectively to promote the expression of gag and pol of the coat protein, the mRNA of the vector genome and the transgene are promoters identical or different, chosen advantageously from ubiquitous promoters or specific, for example, from viral promoters CMV, TK, RSV LTR promoter and the RNA polymerase III promoter such as U6 or H1 or promoters of helper viruses encoding env, gag and pol (i.e.
- the plasmids described above can be introduced into competent cells and viruses produced are harvested.
- the cells used may be any cell competent, particularly eukaryotic cells, in particular mammalian, eg human or animal. They can be somatic or embryonic stem or differentiated. Typically the cells include 293T cells, fibroblast cells, hepatocytes, muscle cells (skeletal, cardiac, smooth, blood vessel, etc.), nerve cells (neurons, glial cells, astrocytes) of epithelial cells, renal, ocular etc.. It may also include, insect, plant cells, yeast, or prokaryotic cells.
- the genes gag, pol and env encoded in plasmids or helper viruses can be introduced into cells by any method known in the art, suitable for cell type considered.
- the cells and the vector system are contacted in a suitable device (plate, dish, tube, pouch, etc...), for a period of time sufficient to allow the transfer of the vector system or the plasmid in the cells.
- the vector system or the plasmid is introduced into the cells by calcium phosphate precipitation, electroporation, transduction or by using one of transfection-facilitating compounds, such as lipids, polymers, liposomes and peptides, etc..
- the calcium phosphate precipitation is preferred.
- the cells are cultured in any suitable medium such as RPMI, DMEM, a specific medium to a culture in the absence of fetal calf serum, etc.
- a suitable medium such as RPMI, DMEM, a specific medium to a culture in the absence of fetal calf serum, etc.
- the retroviral vectors of the present invention may be purified from the supernatant of the cells. Purification of the retroviral vector to enhance the concentration can be accomplished by any suitable method, such as by density gradient purification (e.g., cesium chloride (CsCl)) or by chromatography techniques (e.g., column or batch chromatography).
- CsCl cesium chloride
- chromatography techniques e.g., column or batch chromatography
- the vector of the present invention can be subjected to two or three CsCl density gradient purification steps.
- the vector is desirably purified from cells infected using a method that comprises lysing cells infected with adenovirus, applying the lysate to a chromatography resin, eluting the adenovirus from the chromatography resin, and collecting a fraction containing the retroviral vector of the present invention.
- the vector of the present invention includes "control sequences", which refers collectively to promoter sequences, polyadenylation signals, transcription termination sequences, upstream regulatory domains, origins of replication, internal ribosome entry sites ("IRES"), enhancers, and the like, which collectively provide for the replication, transcription and translation of a coding sequence in a recipient cell.
- nucleic acid sequence is a "promoter" sequence, which is used herein in its ordinary sense to refer to a nucleotide region comprising a DNA regulatory sequence, wherein the regulatory sequence is derived from a gene which is capable of binding RNA polymerase and initiating transcription of a downstream (3'- direction) coding sequence.
- Transcription promoters can include "inducible promoters” (where expression of a polynucleotide sequence operably linked to the promoter is induced by an analyte, cofactor, regulatory protein, etc.), “repressible promoters” (where expression of a polynucleotide sequence operably linked to the promoter is induced by an analyte, cofactor, regulatory protein, etc.), and “constitutive promoters”.
- the different components of the gene editing platform of the present invention are provided to the population of cells through the use of an RNA-encoded system.
- the base-editing system may be provided to the population of cells through the use of a chemically modified mRNA-encoded adenine or cytidine base editor together with modified guide RNA as described in Jiang, T., Henderson, J.M., Coote, K. et al. Chemical modifications of adenine base editor mRNA and guide RNA expand its application scope. Nat Commun 11, 1979 (2020).
- engineered RNA-encoded base-editing enzymes e.g. ABE
- ABE engineered RNA-encoded base-editing enzymes
- modifications consist in uridine depleted mRNAs modified with 5-methoxyuridine: synonymous codons may be introduced to deplete uridines as much as possible without altering the coding sequence and replaced all the remaining uridines with 5-methoxyuridine.
- Said optimized base editing system exhibits higher editing efficiency at some genomic sites compared to DNA-encoded system. It is also possible to encapsulate the modified mRNA and guide RNA into lipid nanoparticle (LNP) for allowing lipid nanoparticle (LNP)-mediated delivery.
- LNP lipid nanoparticle
- the mRNA is formulated using one or more lipid-based structures that include but are not limited to liposomes, lipoplexes, or lipid nanoparticles (Paunovska, Kalina, David Loughrey, and James E. Dahlman. "Drug delivery systems for RNA therapeutics.” Nature Reviews Genetics (2022): 1-16).
- Liposomes are artificially-prepared vesicles which can primarily be composed of a lipid bilayer and can be used as a delivery vehicle for the administration of pharmaceutical formulations.
- Liposomes can be of different sizes such as, but not limited to, a multilamellar vesicle (MLV) which can be hundreds of nanometers in diameter and can contain a series of concentric bilayers separated by narrow aqueous compartments, a small unicellular vesicle (SUV) which can be smaller than 50 nm in diameter, and a large unilamellar vesicle (LUV) which can be between 50 and 500 nm in diameter.
- MLV multilamellar vesicle
- SUV small unicellular vesicle
- LUV large unilamellar vesicle
- Liposome design can include, but is not limited to, opsonins or ligands in order to improve the attachment of liposomes to unhealthy tissue or to activate events such as, but not limited to, endocytosis.
- Liposomes can contain a low or a high pH in order to improve the delivery of the pharmaceutical formulations.
- liposomes such as synthetic membrane vesicles are prepared by the methods, apparatus and devices described in US Patent Publication No. US20130177638, US20130177637, US20130177636, US20130177635, US20130177634, US20130177633, US20130183375, US20130183373 and US20130183372.
- the liposomes are formed from 1,2-dioleyloxy-N,N-dimethylaminopropane (DODMA) liposomes, DiLa2 liposomes from Marina Biotech (Bothell, Wash.), 1,2-dilinoleyloxy-3-dimethylaminopropane (DLin-DMA), 2,2-dilinoleyl-4-(2-dimethylaminoethyl)-[1,3]-dioxolane (DLin-KC2-DMA), and MC3 (as described in US20100324120) and liposomes which can deliver small molecule drugs such as, but not limited to, DOXIL® from Janssen Biotech, Inc. (Horsham, Pa.).
- DOXIL® 1,2-dioleyloxy-N,N-dimethylaminopropane
- the mRNAs can be encapsulated by the liposome and/or it can be contained in an aqueous core which can then be encapsulated by the liposome (see International Pub. Nos. WO2012031046, WO2012031043, WO2012030901 and WO2012006378 and US Patent Publication No. US20130189351, US20130195969 and US20130202684).
- the mRNA is formulated with stabilized plasmid-lipid particles (SPLP) or stabilized nucleic acid lipid particle (SNALP) that have been previously described and shown to be suitable for oligonucleotide delivery in vitro and in vivo (see Wheeler et al. Gene Therapy.
- SPLP stabilized plasmid-lipid particles
- SNALP stabilized nucleic acid lipid particle
- the vector of the present invention is combined with pharmaceutically acceptable excipients, and optionally sustained-release matrices, such as biodegradable polymers, to form pharmaceutical compositions.
- pharmaceutically acceptable excipients such as pharmaceutically acceptable polymers
- pharmaceutically acceptable carrier or excipient refers to a non-toxic solid, semi- solid or liquid filler, diluent, encapsulating material or formulation auxiliary of any type.
- Target the Pol II pause site in the C9orf72 gene using base editing A) Schematic representation of the C9orf72 gene on chromosome 9, depicting C9orf72 exons and the region containing G4C2 hexanucleotides repeats and the location of the Pol II pause site (chr9:27563356-27,563,366 – Hg18). Targets sequences of the sgRNAs are indicated with arrows and aligned with the DNA sequence that they bind to in a stranded-oriented way.
- PRO-seq or PRO-cap genome-wide methods map RNA polymerase active sites and transcription start sites, respectively, and allow to study the relationship between promoter- proximal pausing and the core promoter structure.
- PRO-seq or PRO-cap analyses performed by Watts and colleagues (Watts, Jason A., et al. "Cis elements that mediate RNA polymerase II pausing regulate human gene expression.” The American Journal of Human Genetics 105.4 (2019): 677-688) identified a Pol II pause site in the C9orf72 gene (Cheung et al., personal communication). Our base of interest is located at position Chr9:27563356-27563366-Hg18).
- RNA-mediated and RNA-mediated base editing of the RNA Pol II pause site in K562 cells A) Experimental protocol used for base editing experiments in K562 cells. K562 cells co- transfected with a BE/GFP-expressing plasmid and a sgRNA-expressing plasmid.24 hours post transfection, GFP+ cells were sorted and cultured for 3 to 5 days before DNA extraction and PCR amplification of the target region. Amplicons were subjected to Sanger sequencing. RNA extraction was performed around 7 days post transfection and first-strand cDNAs were obtained by reverse transcription, which was then followed by qPCR. B) Experimental protocol used for base editing experiments in K562 cells.
- a BE/GFP- expressing mRNA and a sgRNA were co-transfected in K562 cells.
- Cells were cultured for 3 to 5 days before DNA extraction and PCR amplification of the target region. Amplicons were subjected to Sanger sequencing. RNA extraction was performed around 7 days post transfection and first-strand cDNAs were obtained by reverse transcription, which was then followed by qPCR.
- Figure 3 A BE/GFP- expressing mRNA and a sgRNA were co-transfected in K562 cells. Cells were cultured for 3 to 5 days before DNA extraction and PCR amplification of the target region. Amplicons were subjected to Sanger sequencing. RNA extraction was performed around 7 days post transfection and first-strand cDNAs were obtained by reverse transcription, which was then followed by qPCR. Figure 3.
- RNA-mediated base editing of the RNA Pol II pause site in K562 cells Frequency of C>T conversion in the C1 base (the C9orf72 Pol II pause site) in K562 and at the C2 (bystander C), as determined by Sanger sequencing followed by EditR analysis.
- Cells were transfected with CBE-SpRY-OPT mRNA, purchased from Trilink in combination with chemically modified C9orf72_1, C9orf72_2, C9orf72_3 and C9orf72_4 sgRNAs.
- EXAMPLE 1 Methods: Cell culture Human erythroleukemia cells (K562) were grown in RPMI 1640 medium supplemented with 10% dialyzed fetal bovine serum, 100 U/mL penicillin, 100 ⁇ g/mL streptomycin and 2 mM glutamine. Cells were maintained in a humidified atmosphere with 5% CO2 at 37oC, with medium renewal every 2–3 days.
- Plasmid used in this study is the CBE-SpRY-OPT (used for in vitro mRNA production) that contains the uridine-depleted coding sequence of pCAG-CBE4max-SpRY-P2A-EGFP (RTW5133) (Addgene #139999), two copies of the 3’ untranslated region (UTR) of the HBB gene, a poly-A sequence of 96 A, and a T7 promoter followed by a G allowing efficient capping (Antoniou et al., under revision).
- sgRNA design We manually designed sgRNAs targeting the identified unique site of RNA Polymerase II pause site in the promoter region of C9orf72 (Table 1).
- oligonucleotides were annealed to create the gRNA protospacer and the duplexes were ligated into the Bbs I-digested MA128 plasmid (provided by M. Amendola, Genethon, France).
- Bbs I-digested MA128 plasmid provided by M. Amendola, Genethon, France.
- RNA-mediated base editing we used chemically modified synthetic sgRNAs harboring 2′-O- methyl analogs and 3′-phosphorothioate nonhydrolyzable linkages at the first three 5′ and 3′ nucleotides (Synthego). Table 1. gRNA target sequences.
- CBE-SpRY-OPT mRNA in vitro transcription CBE-SpRY-OPT mRNA, containing 5-methoxyuridine, capped with Cap1 analog, and subjected to silica membrane purification, was purchased from Trilink.
- Plasmid transfection 2x10 6 K562 cells per condition were transfected with 3 ug of the base editor CBE and 1 ⁇ g of sgRNA of interest (C9orf72_2, C9orf72_3 and C9orf72_4).
- C9orf72_2, C9orf72_3 and C9orf72_4 We used the Cell Line Nucleofector TM Kit V VCA-1003 for K562 cells and the U16 program of Nucleofector 2B. Cells transfected with TE buffer or with the base editor-expressing plasmid only served as negative controls.
- RNA transfection 2x10 5 K562 cells per condition were transfected with 3.0 ⁇ g of the enzyme encoding mRNA, and 3.2 ⁇ g of synthetic sgRNA (C9orf72_1, C9orf72_2, C9orf72_3 and C9orf72_4).
- C9orf72_1, C9orf72_2, C9orf72_3 and C9orf72_4 were transfected with 3.0 ⁇ g of the enzyme encoding mRNA, and 3.2 ⁇ g of synthetic sgRNA (C9orf72_1, C9orf72_2, C9orf72_3 and C9orf72_4).
- P3 Primary Cell 4D-Nucleofector X Kit S Likanza
- CA137 program Nucleofector 4D
- RNA polymerase II RNA polymerase II
- Figure 1A and B RNA polymerase II pausing is a key regulatory step in transcription. RNA Pol II pauses at GC-rich regions, and almost 65% of the pause sites are cytosines (Watts, Jason A., et al. "Cis elements that mediate RNA polymerase II pausing regulate human gene expression.” The American Journal of Human Genetics 105.4 (2019): 677-688).
- CBE-SpRY coupled with C9orf72_3 and C9orf72_4 sgRNAs showed also efficient conversion of the target base C1, but also of C2, located within the base editing window (bystander edit) ( Figure 3A).
- TIDE analysis confirmed the DSB-free nature of base editors, as we detected low frequencies of InDels in base edited samples compared to Cas9 nuclease used in combination with the C9orf72_2, C9orf72_3 or C9orf72_4 sgRNAs (Figure 3B).
- RNA-mediated base editing in K562 cells in the C9orf72 Pol II pause site To establish a clinically relevant method to deliver the base editing system in K562 cells and achieve a higher editing efficiency coupled with minimal toxicity compared to the plasmid transfection, we used a protocol based on transfection of mRNA encoding base editors and chemically modified synthetic sgRNAs (Figure 2B).
- C9orf72_1, C9orf72_2, C9orf72_3 or C9orf72_4lls were kept in culture for 3 to 5 days before DNA extraction and Sanger sequencing of the PCR- amplified target region.
- EditR analysis showed that all the sgRNA were able to modify the target base with efficiencies ranging from 9% (C9orf72_1) to 27% (C9orf72_4) with a variable efficiency of bystander C2 within the editing window, upon RNA-mediated delivery of the base editing system (Figure 5).
- EXAMPLE 2 Experimentation on patient-derived motor neurons Generation of iPSC-derived motor neurons Origin of the cells: A control iPSC line generated by the Stem Cell facility at the host institute (Institut Imagine cell line #Ctr004) was used as a starting material for all the motor neuron derivation studies. iPSC colonies were grown in mTeSR medium in vitronectin (both StemCell) coated dishes and were passaged by enzyme - free dissociation (ReleSR, StemCell) according to the manufacturer’s instructions.
- iMN differentiation To generate motor neurons, we have adapted a published protocol (Maury et al., 2015).
- N2B27 medium consists of a 1:1 ratio of advanced DMEM/F12 (Life Technologies) and Neurobasal medium (Life Technologies), 1% N2 supplement (Life Technologies), 2% B27 supplement minus vitamin A (Life Technologies), 5 mM Glutamax (Life Technologies), 5mM L- Glutamine (Sigma), 1% penicillin/streptomycin (Life Technologies) and 0,1 mM 2 - ME (Life Technologies).
- iPSCs spontaneously aggregated into embryoid bodies (EBs) in suspension in N2B27 medium.
- N2B27 medium 10 ⁇ M ROCK inhibitor Y - 27632, 3 ⁇ M CHIR99021 (TOCRIS), 0, 1 ⁇ M LDN (TOCRIS), 20 ⁇ M SB431542 (TOCRIS) and 10 ⁇ M ascorbic acid (Sigma - Aldrich).
- the medium was changed to N2B27 + 3 ⁇ M CHIR99021, 0,1 ⁇ M LDN, 20 ⁇ M 8SB431542 and 100 nM retinoic acid (Sigma - Aldrich).
- the medium was changed to N2B27 + 0, 1 ⁇ M LDN, 20 ⁇ M SB431542 , 100 nM retinoic acid and 500 nM SAG (TOCRIS).
- the medium was changed to N2B27 + 100 nM retinoic acid and 500 nM SAG.
- the medium was changed to N2B27 + 100 nM retinoic acid, 500 nM SAG and 10 ⁇ M DAPT (TOCRIS).
- the EBs are composed of motor neurons and were dissociated with 0 , 5 % Trypsin (Life Technologies). The obtained motor neurons were then seeded in 96- well plates or 4- well plates (with 10mm coverslips) previously coated with Poly-D-lysine.
- iMN testing For assessing the effect of drugs on neuronal morphology and viability, mature iPSC-derived motor neuron cultures are monitored in real time using the live cell imaging and analysis instrument Incucyte SX5 (Sartorius). A neurite detection mask has been optimized for online measurement of neurite extension and retraction. Viability is monitored live by the uptake of Propidium Iodide which is added to the cell culture medium at a concentration of 1 ⁇ g/ml. Images can be acquired and automatically analyzed every hour for the duration of the drug screening test.
- REFERENCES Throughout this application, various references describe the state of the art to which this invention pertains. The disclosures of these references are hereby incorporated by reference into the present disclosure.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Organic Chemistry (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- General Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Microbiology (AREA)
- Physics & Mathematics (AREA)
- Animal Behavior & Ethology (AREA)
- Plant Pathology (AREA)
- Epidemiology (AREA)
- Biophysics (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
The major objective of the present invention was to develop safe, effective and definitive therapeutic strategies for Amyotrophic Lateral Sclerosis through the innovative approach of base editing. In particular, the inventors have identified a unique site of RNA Polymerase II pause site in the promoter region of C9orf72. Their approach was therefore to mutate this site using the base editing technology to increase C9orf72 transcription and expression. Therefore, base editing approaches that target the RNA Polymerase II pause site should boost the expression of C9orf72 as well as likely reduce the toxicity due to repeat expression, thus doubling the therapeutic potential of the genome editing strategy. Here the inventors report the results obtained following the design of 4 single guide RNAs (sgRNAs) targeting this region that show a low predicted off-target activity. Initial results obtained in cell lines show an efficient rate of base editing by targeting the Pol II pause site of C9orf72 and C9orf72 up- regulation. Accordingly, the present invention relates to base editing approaches for the treatment of ALS.
Description
BASE EDITING APPROACHES FOR THE TREATMENT OF AMYOTROPHIC LATERAL SCLEROSIS FIELD OF THE INVENTION: The present invention is in the field of medicine, in particular neurology. BACKGROUND OF THE INVENTION: Motor neuron diseases lead to motor neuron degeneration and muscle wasting and have devastating consequences in patients. Amyotrophic Lateral Sclerosis (ALS) is the most prevalent adult-onset motor neuron disease with an estimated prevalence of 8/100,000 and 1,500 new cases per year in France. ALS is caused by multiple genetic causes; the most prevalent is a repeat expansion in the C9orf72 gene (Figure 1). Significantly, C9orf72 repeat expansions are recognized as a major genetic cause for a related neurodegenerative disorder, Frontotemporal Dementia (FTD). Although toxic gain of function of the repeats is considered to be an important pathogenic mechanism, significantly reduced levels of C9orf72 transcript and protein have been consistently described in ALS patients indicating that C9orf72 loss of function causes neurodegeneration (Ciura, Sorana, et al. "Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis." Annals of neurology 74.2 (2013): 180-187). Therefore, restoring C9orf72 function represents an important therapeutic target in ALS, FTD and related disorders. ALS is untreatable through common pharmacological approaches. Current therapies for ALS include the administration of antisense oligonucleotides and gene silencing by AAV to target the RNA repeats transcribed from the C9orf72 gene (reviewed in Cappella, Marisa, et al. "Gene therapy for ALS—a perspective." International journal of molecular sciences 20.18 (2019): 4388). However, these strategies remain either temporary and need replenishing as in the case of oligonucleotides or are still undergoing early clinical trials for gene delivery via the AAV approach. Therefore, there is a strong rationale for developing novel, definitive and universal strategies for this disease. In ALS, we have shown for the first time that there is C9orf72 haploinsufficiency in ALS patients. Results under review from our team point out that boosting C9orf72 expression can rescue motor neuron degeneration and reduce the expression of the disease-causing repeats. We have shown that C9orf72 is key factor for autophagy initiation (Sellier, Chantal, et al. "Loss of C9 ORF 72 impairs autophagy and synergizes with polyQ
Ataxin‐2 to induce motor neuron dysfunction and cell death." The EMBO journal 35.12 (2016): 1276-1297). Increasing C9orf72 expression can restore the autophagy pathway, which is mainly responsible for the degradation of the translated C9orf72 repeats. Results in motor neurons derived from iPSCs of patients with repeat expansion in C9orf72 have indeed shown that C9orf72 overexpression can reduce the toxicity due to expression of the dipeptide transcribed from the expanded repeats (Shi, Yingxiao, et al. "Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons." Nature medicine 24.3 (2018): 313-325). Importantly, combining expression of the C9orf72 hexanucleotide repeats and inactivation the C9orf72 expression leads to exacerbated phenotypic features in zebrafish and mouse models (Zhu, Qiang, et al. "Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72." Nature neuroscience 23.5 (2020): 615-624). CRISPR-system-based cytosine and adenine base editors (CBEs and ABEs) can make pinpoint changes in the DNA with little or no double-strand break (DSB) generation (Rees, Holly A., and David R. Liu. "Base editing: precision chemistry on the genome and transcriptome of living cells." Nature reviews genetics 19.12 (2018): 770-788). The basic components of BEs are a catalytically disabled Cas9 nuclease, a guide RNA that drives the system to a specific DNA target, and a deaminase that produces C-G to T-A or A-T to G-C conversions (for CBEs and ABEs, respectively). Base-editing approaches allow precise DNA repair virtually in the absence of DSBs, and thus eliminate the risks of DSB-induced apoptosis, translocations and insertions or deletions of large portions of DNA (Kosicki, Michael, Kärt Tomberg, and Allan Bradley. "Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements." Nature biotechnology 36.8 (2018): 765-771). Importantly, base editing results in homogeneous, predictable base changes as compared to the heterogeneous and unpredictable mutagenesis induced by Cas9 nuclease, thus allowing for precise edits. Due to selective base repair, this approach, while allowing the permanent genetic modification of target cells, should not pose major DNA-related side effects, and could be combined with other genetic or pharmacological interventions for ALS. SUMMARY OF THE INVENTION: The present invention is defined by the claims. In particular, the present invention relates to base editing approaches for the treatment of amyotrophic lateral sclerosis (ALS).
DETAILED DESCRIPTION OF THE INVENTION: The major objective of the present invention is to develop safe, effective and definitive therapeutic strategies for ALS through the innovative approach of base editing. In particular, the inventors have identified a unique site of RNA Polymerase II pause site in the promoter region of C9orf72. Their approach was therefore to mutate this site using the base editing technology to increase C9orf72 transcription and expression. Therefore, base editing approaches that target the RNA Polymerase II pause site should boost the expression of C9orf72 as well as likely reduce the toxicity due to repeat expression, thus doubling the therapeutic potential of the genome editing strategy. Here the inventors report the results obtained following the design of 4 single guide RNAs (sgRNAs) targeting this region that show a low predicted off-target activity, as evaluated by COSMID (Cradick, Thomas J., et al. "COSMID: a web- based tool for identifying and validating CRISPR/Cas off-target sites." Molecular Therapy- Nucleic Acids 3 (2014): e214). Initial results obtained in cell lines show an efficient rate of base editing by targeting the Pol II pause site of C9orf72 and C9orf72 up-regulation. Main definitions: As used herein, the terms “polypeptide”, “peptide” and “protein” are used interchangeably herein to refer to polymers of amino acids of any length. The polymer may be linear or branched, it may comprise modified amino acids, and it may be interrupted by non-amino acids. The terms also encompass an amino acid polymer that has been modified; for example, disulfide bond formation, glycosylation, lipidation, acetylation, phosphorylation, pegylation, or any other manipulation, such as conjugation with a labeling component. As used herein the term “amino acid” includes natural and/or unnatural or synthetic amino acids, including glycine and both the D or L optical isomers, and amino acid analogs and peptidomimetics. As used herein, the term “nucleic acid molecule” or “polynucleotide” refers to a DNA molecule (for example, but not limited to, a cDNA or genomic DNA). The nucleic acid molecule can be single-stranded or double-stranded. As used herein, the term “isolated” when referring to nucleic acid molecules or polypeptides means that the nucleic acid molecule or the polypeptide is substantially free from at least one other component with which it is associated or found together in nature.
As used herein, the term “complementarity” refers to the ability of a nucleic acid to form hydrogen bond(s) with another nucleic acid sequence by either traditional Watson-Crick base- pairing or other non-traditional types. A percent complementarity indicates the percentage of residues in a nucleic acid molecule which can form hydrogen bonds (e.g., Watson-Crick base pairing) with a second nucleic acid sequence (e.g., 5, 6, 7, 8, 9, 10 out of 10 being 50%, 60%, 70%, 80%, 90%, and 100% complementary). “Perfectly complementary” means that all the contiguous residues of a nucleic acid sequence will hydrogen bond with the same number of contiguous residues in a second nucleic acid sequence. “Substantially complementary” as used herein refers to a degree of complementarity that is at least 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 97%, 98%, 99%, or 100% over a region of 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 30, 35, 40, 45, 50, or more nucleotides, or refers to two nucleic acids that hybridize under stringent conditions. As used herein, the term “stringent conditions” for hybridization refer to conditions under which a nucleic acid having complementarity to a target sequence predominantly hybridizes with the target sequence, and substantially does not hybridize to non-target sequences. Stringent conditions are generally sequence-dependent, and vary depending on a number of factors. In general, the longer the sequence, the higher the temperature at which the sequence specifically hybridizes to its target sequence. Non-limiting examples of stringent conditions are described in detail in Tijssen (1993), Laboratory Techniques In Biochemistry And Molecular Biology- Hybridization With Nucleic Acid Probes Part I, Second Chapter “Overview of principles of hybridization and the strategy of nucleic acid probe assay”, Elsevier, N.Y. As used herein, the term “hybridization” or “hybridizing” refers to a process where completely or partially complementary nucleic acid strands come together under specified hybridization conditions to form a double-stranded structure or region in which the two constituent strands are joined by hydrogen bonds. Although hydrogen bonds typically form between adenine and thymine or uracil (A and T or U) or cytosine and guanine (C and G), other base pairs may form (e.g., Adams et al., The Biochemistry of the Nucleic Acids, 11th ed., 1992). As used herein, the term “fusion polypeptide” or “fusion protein” means a protein created by joining two or more polypeptide sequences together. The fusion polypeptides encompassed in this invention include translation products of a chimeric gene construct that joins the nucleic acid sequences encoding a first polypeptide, e.g., an RNA-binding domain, with the nucleic
acid sequence encoding a second polypeptide, e.g., an effector domain, to form a single open- reading frame. In other words, a “fusion polypeptide” or “fusion protein” is a recombinant protein of two or more proteins which are joined by a peptide bond or via several peptides. The fusion protein may also comprise a peptide linker between the two domains. As used herein the term “wild type” is a term of the art understood by skilled persons and means the typical form of an organism, strain, gene or characteristic as it occurs in nature as distinguished from mutant or variant forms. As used herein, the term “derived from” refers to a process whereby a first component (e.g., a first molecule), or information from that first component, is used to isolate, derive or make a different second component (e.g., a second molecule that is different from the first). As used herein, the “percent identity” between the two sequences is a function of the number of identical positions shared by the sequences (i.e., % identity = number of identical positions/total number of positions x 100), taking into account the number of gaps, and the length of each gap, which need to be introduced for optimal alignment of the two sequences. The comparison of sequences and determination of percent identity between two sequences can be accomplished using a mathematical algorithm, as described below. The percent identity between two amino acid sequences can be determined using the Needleman and Wunsch algorithm (Needleman, Saul B. & Wunsch, Christian D. (1970). "A general method applicable to the search for similarities in the amino acid sequence of two proteins". Journal of Molecular Biology.48 (3): 443–53). The percent identity between two nucleotide or amino acid sequences may also be determined using for example algorithms such as EMBOSS Needle (pair wise alignment; available at www.ebi.ac.uk). For example, EMBOSS Needle may be used with a BLOSUM62 matrix, a “gap open penalty” of 10, a “gap extend penalty” of 0.5, a false “end gap penalty”, an “end gap open penalty” of 10 and an “end gap extend penalty” of 0.5. In general, the “percent identity” is a function of the number of matching positions divided by the number of positions compared and multiplied by 100. For instance, if 6 out of 10 sequence positions are identical between the two compared sequences after alignment, then the identity is 60%. The % identity is typically determined over the whole length of the query sequence on which the analysis is performed. Two molecules having the same primary amino acid sequence or nucleic acid sequence are identical irrespective of any chemical and/or biological modification. According to the invention a first amino acid sequence having at least 90% of
identity with a second amino acid sequence means that the first sequence has 90; 91; 92; 93; 94; 95; 96; 97; 98; 99 or 100% of identity with the second amino acid sequence. As used herein, the term “linker” refers to any means, entity or moiety used to join two or more entities. A linker can be a covalent linker or a non-covalent linker. Examples of covalent linkers include covalent bonds or a linker moiety covalently attached to one or more of the proteins or domains to be linked. The linker can also be a non-covalent bond, e.g., an organometallic bond through a metal centre such as platinum atom. For covalent linkages, various functionalities can be used, such as amide groups, including carbonic acid derivatives, ethers, esters, including organic and inorganic esters, amino, urethane, urea and the like. To provide for linking, the domains can be modified by oxidation, hydroxylation, substitution, reduction etc. to provide a site for coupling. Methods for conjugation are well known by persons skilled in the art and are encompassed for use in the present invention. Linker moieties include, but are not limited to, chemical linker moieties, or for example a peptide linker moiety (a linker sequence). It will be appreciated that modification which do not significantly decrease the function of the RNA- binding domain and effector domain are preferred. As used herein, the “linked” as used herein refers to the attachment of two or more entities to form one entity. A conjugate encompasses both peptide-small molecule conjugates as well as peptide-protein/peptide conjugates. As used herein, the term “base-editing enzyme” refers to fusion protein comprising a defective CRISPR/Cas nuclease linked to a deaminase polypeptide. The term is also known as “base- editor”. Initially, two classes of base-editing enzymes--cytosine base-editing enzymes (CBEs) and adenine base-editing enzymes (ABEs)--were used to generate single base pair edits without double stranded breaks (Zhang, Xiaohui, et al. "Dual base editor catalyzes both cytosine and adenine base conversions in human cells." Nature biotechnology 38.7 (2020): 856-860). Typically, cytosine base-editing enzymes are created by fusing the defective CRISPR/Cas nuclease to a deaminase. As used herein, the term “deaminase” refers to an enzyme that catalyzes a deamination reaction. The term “deamination”, as used herein, refers to the removal of an amine group from one molecule. In some embodiments, the deaminase is a cytidine deaminase, catalysing the hydrolytic deamination of cytidine or deoxycytidine to uracil or deoxyuracil, respectively.
As used herein, the term “nuclease” includes a protein (i.e. an enzyme) that induces a break in a nucleic acid sequence, e.g., a single or a double strand break in a double-stranded DNA sequence. As used herein, the term “CRISPR/Cas nuclease” has its general meaning in the art and refers to segments of prokaryotic DNA containing clustered regularly interspaced short palindromic repeats (CRISPR) and associated nucleases encoded by Cas genes. In bacteria the CRISPR/Cas loci encode RNA-guided adaptive immune systems against mobile genetic elements (viruses, transposable elements and conjugative plasmids). Three types of CRISPR systems have been identified. CRISPR clusters contain spacers, the sequences complementary to antecedent mobile elements. CRISPR clusters are transcribed and processed into mature CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) RNA (crRNA). The CRISPR/Cas nucleases Cas9 and Cpf1 belong to the type II and type V CRISPR/Cas system and have strong endonuclease activity to cut target DNA. Cas9 is guided by a mature crRNA that contains about 20 nucleotides of unique target sequence (called spacer) and a trans-activating small RNA (tracrRNA) that also serves as a guide for ribonuclease III-aided processing of pre-crRNA. The crRNA:tracrRNA duplex directs Cas9 to target DNA via complementary base pairing between the spacer on the crRNA and the complementary sequence (called protospacer) on the target DNA. Cas9 recognizes a trinucleotide (NGG for S. Pyogenes Cas9) protospacer adjacent motif (PAM) to specify the cut site (the 3rd or the 4th nucleotide upstream from PAM). As used herein, the term “Cas9” or “Cas9 nuclease” refers to an RNA-guided nuclease comprising a Cas9 protein, or a fragment thereof (e.g., a protein comprising an active or inactive DNA cleavage domain of Cas9, and/or the gRNA binding domain of Cas9). A Cas9 nuclease is also referred to sometimes as a casn1 nuclease or a CRISPR (clustered regularly interspaced short palindromic repeat)-associated nuclease. CRISPR is an adaptive immune system that provides protection against mobile genetic elements (viruses, transposable elements and conjugative plasmids). CRISPR clusters contain spacers, sequences complementary to antecedent mobile elements, and target invading nucleic acids. CRISPR clusters are transcribed and processed into CRISPR RNA (crRNA). In type II CRISPR systems correct processing of pre-crRNA requires a trans-encoded small RNA (tracrRNA), endogenous ribonuclease 3 (rnc) and a Cas9 protein. The tracrRNA serves as a guide for ribonuclease 3-aided processing of pre- crRNA. Subsequently, Cas9/crRNA/tracrRNA endonucleolytically cleaves linear or circular
dsDNA target complementary to the spacer. The target strand not complementary to crRNA is first cut endonucleolytically, then trimmed 3′-5′ exonucleolytically. In nature, DNA- binding and cleavage typically requires protein and both RNAs. However, single guide RNAs (“sgRNA”, or simply “gNRA”) can be engineered so as to incorporate aspects of both the crRNA and tracrRNA into a single RNA species. See, e.g., Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J. A., Charpentier E. Science 337:816-821(2012), the entire contents of which is hereby incorporated by reference. Cas9 recognizes a short motif in the CRISPR repeat sequences (the PAM or protospacer adjacent motif) to help distinguish self versus non-self. Cas9 nuclease sequences and structures are well known to those of skill in the art (see, e.g., “Complete genome sequence of an M1 strain of Streptococcus pyogenes.” Ferretti et al., J. J., McShan W. M., Ajdic D. J., Savic D. J., Savic G., Lyon K., Primeaux C., Sezate S., Suvorov A. N., Kenton S., Lai H. S., Lin S. P., Qian Y., Jia H. G., Najar F. Z., Ren Q., Zhu H., Song L., White J., Yuan X., Clifton S. W., Roe B. A., McLaughlin R. E., Proc. Natl. Acad. Sci. U.S.A. 98:4658-4663(2001); “CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III.” Deltcheva E., Chylinski K., Sharma C. M., Gonzales K., Chao Y., Pirzada Z. A., Eckert M. R., Vogel J., Charpentier E., Nature 471:602-607(2011); and “A programmable dual- RNA-guided DNA endonuclease in adaptive bacterial immunity.” Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J. A., Charpentier E. Science 337:816-821(2012), the entire contents of each of which are incorporated herein by reference). Cas9 orthologs have been described in various species, including, but not limited to, S. pyogenes and S. thermophilus. Additional suitable Cas9 nucleases and sequences will be apparent to those of skill in the art based on this disclosure, and such Cas9 nucleases and sequences include Cas9 sequences from the organisms and loci disclosed in Chylinski, Rhun, and Charpentier, “The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems” (2013) RNA Biology 10:5, 726-737; the entire contents of which are incorporated herein by reference. In some embodiments, the term “Cas9” refers to Cas9 from: Corynebacterium ulcerans (NCBI Refs: NC_015683.1, NC_017317.1); Corynebacterium diphtheria (NCBI Refs: NC_016782.1, NC_016786.1); Spiroplasma syrphidicola (NCBI Ref: NC_021284.1); Prevotella intermedia (NCBI Ref: NC_017861.1); Spiroplasma taiwanense (NCBI Ref: NC_021846.1); Streptococcus iniae (NCBI Ref: NC_021314.1); Belliella baltica (NCBI Ref: NC_018010.1); Psychroflexus torquisI (NCBI Ref: NC_018721.1); Streptococcus thermophilus (NCBI Ref: YP_820832.1); Listeria innocua (NCBI Ref: NP_472073.1); Campylobacter jejuni (NCBI Ref: YP_002344900.1); or Neisseria. meningitidis (NCBI Ref: YP_002342100.1). Typically the Cas9 nuclease comprises the amino acid sequence as set forth in SEQ ID NO: 1.
SEQ ID NO:1: Cas9 sequence MDKKYSIGLAIGTNSVGWAVITDEYKVPSKKFKVLGNTDRHSIKKNLIGALLFDSGETAEATRLKRTAR RRYTRRKNRICYLQEIFSNEMAKVDDSFFHRLEESFLVEEDKKHERHPIFGNIVDEVAYHEKYPTIYHL RKKLVDSTDKADLRLIYLALAHMIKFRGHFLIEGDLNPDNSDVDKLFIQLVQTYNQLFEENPINASGVD AKAILSARLSKSRRLENLIAQLPGEKKNGLFGNLIALSLGLTPNFKSNFDLAEDAKLQLSKDTYDDDLD NLLAQIGDQYADLFLAAKNLSDAILLSDILRVNTEITKAPLSASMIKRYDEHHQDLTLLKALVRQQLPE KYKEIFFDQSKNGYAGYIDGGASQEEFYKFIKPILEKMDGTEELLVKLNREDLLRKQRTFDNGSIPHQI HLGELHAILRRQEDFYPFLKDNREKIEKILTFRIPYYVGPLARGNSRFAWMTRKSEETITPWNFEEVVD KGASAQSFIERMTNFDKNLPNEKVLPKHSLLYEYFTVYNELTKVKYVTEGMRKPAFLSGEQKKAIVDLL FKTNRKVTVKQLKEDYFKKIECFDSVEISGVEDRFNASLGTYHDLLKIIKDKDFLDNEENEDILEDIVL TLTLFEDREMIEERLKTYAHLFDDKVMKQLKRRRYTGWGRLSRKLINGIRDKQSGKTILDFLKSDGFAN RNFMQLIHDDSLTFKEDIQKAQVSGQGDSLHEHIANLAGSPAIKKGILQTVKVVDELVKVMGRHKPENI VIEMARENQTTQKGQKNSRERMKRIEEGIKELGSQILKEHPVENTQLQNEKLYLYYLQNGRDMYVDQEL DINRLSDYDVDHIVPQSFLKDDSIDNKVLTRSDKNRGKSDNVPSEEVVKKMKNYWRQLLNAKLITQRKF DNLTKAERGGLSELDKAGFIKRQLVETRQITKHVAQILDSRMNTKYDENDKLIREVKVITLKSKLVSDF RKDFQFYKVREINNYHHAHDAYLNAVVGTALIKKYPKLESEFVYGDYKVYDVRKMIAKSEQEIGKATAK YFFYSNIMNFFKTEITLANGEIRKRPLIETNGETGEIVWDKGRDFATVRKVLSMPQVNIVKKTEVQTGG FSKESILPKRNSDKLIARKKDWDPKKYGGFDSPTVAYSVLVVAKVEKGKSKKLKSVKELLGITIMERSS FEKNPIDFLEAKGYKEVKKDLIIKLPKYSLFELENGRKRMLASAGELQKGNELALPSKYVNFLYLASHY EKLKGSPEDNEQKQLFVEQHKHYLDEIIEQISEFSKRVILADANLDKVLSAYNKHRDKPIREQAENIIH LFTLTNLGAPAAFKYFDTTIDRKRYTSTKEVLDATLIHQSITGLYETRIDLSQLGGD As used herein, the term “defective CRISPR/Cas nuclease” refers to a CRISPR/Cas nuclease having lost at least one nuclease domain. As used herein, the term “nickase” has its general meaning in the art and refers to an endonuclease which cleaves only a single strand of a DNA duplex. Accordingly, the term “Cas9 nickase” refers to a nickase derived from a Cas9 protein, typically by inactivating one nuclease domain of Cas9 protein. As used herein, the term “guide RNA molecule” generally refers to an RNA molecule (or a group of RNA molecules collectively) that can bind to a Cas9 protein and target the Cas9 protein to a specific location within a target DNA. A guide RNA can comprise two segments: a DNA-targeting guide segment and a protein-binding segment. The DNA-targeting segment comprises a nucleotide sequence that is complementary to (or at least can hybridize to under stringent conditions) a target sequence. The protein-binding segment interacts with a CRISPR protein, such as a Cas9 or Cas9 related polypeptide. These two segments can be located in the same RNA molecule or in two or more separate RNA molecules. When the two segments are in separate RNA molecules, the molecule comprising the DNA-targeting guide segment is sometimes referred to as the CRISPR RNA (crRNA), while the molecule comprising the protein-binding segment is referred to as the trans-activating RNA (tracrRNA).
As used herein, the term “target nucleic acid” or “target” refers to a nucleic acid containing a target nucleic acid sequence. A target nucleic acid may be single-stranded or double-stranded, and often is double-stranded DNA. A “target nucleic acid sequence,” “target sequence” or “target region,” as used herein, means a specific sequence or the complement thereof that one wishes to bind to using the CRISPR system as disclosed herein. As used herein, the term “target nucleic acid strand” refers to a strand of a target nucleic acid that is subject to base-pairing with a guide RNA as disclosed herein. That is, the strand of a target nucleic acid that hybridizes with the crRNA and guide sequence is referred to as the “target nucleic acid strand.” The other strand of the target nucleic acid, which is not complementary to the guide sequence, is referred to as the “non-complementary strand.” In the case of double-stranded target nucleic acid (e.g., DNA), each strand can be a “target nucleic acid strand” to design crRNA and guide RNAs and used to practice the method of this invention as long as there is a suitable PAM site. As used herein, the term “mutation” has its general meaning in the art and refers to a substitution, deletion or insertion. The term "substitution" means that a specific amino acid residue at a specific position is removed and another amino acid residue is inserted into the same position. The term "deletion" means that a specific amino acid residue is removed. The term "insertion" means that one or more amino acid residues are inserted before or after a specific amino acid residue. As used herein, the term “mutagenesis” refers to the introduction of mutations into a polynucleotide sequence. According to the present invention mutations are introduced into a target DNA molecule encoding for a variant domain of the antibody so as to mimic somatic hypermutation. As used herein, the term “variant” refers to a first composition (e.g., a first molecule), that is related to a second composition (e.g., a second molecule, also termed a “parent” molecule). The variant molecule can be derived from, isolated from, based on or homologous to the parent molecule. A variant molecule can have entire sequence identity with the original parent molecule, or alternatively, can have less than 100% sequence identity with the parent molecule. For example, a variant of a sequence can be a second sequence that is at least 50; 51; 52; 53; 54; 55; 56; 57; 58; 59; 60; 61; 62; 63; 64; 65; 66; 67; 68; 69; 70; 71; 72; 73; 74; 75; 76; 77; 78;
79; 80; 81; 82; 83; 84; 85; 86; 87; 88; 89; 90; 91; 92; 93; 94; 95; 96; 97; 98; 99; 100% identical in sequence compare to the original sequence. Sequence identity is frequently measured in terms of percentage identity (or similarity or homology); the higher the percentage, the more similar are the two sequences. Methods of alignment of sequences for comparison are well known in the art. Various programs and alignment algorithms are described in: Smith and Waterman, Adv. Appl. Math., 2:482, 1981; Needleman and Wunsch, J. Mol. Biol., 48:443, 1970; Pearson and Lipman, Proc. Natl. Acad. Sci. U.S.A., 85:2444, 1988; Higgins and Sharp, Gene, 73:237-244, 1988; Higgins and Sharp, CABIOS, 5:151-153, 1989; Corpet et al. Nuc. Acids Res., 16:10881-10890, 1988; Huang et al., Comp. Appls Biosci., 8:155-165, 1992; and Pearson et al., Meth. Mol. Biol., 24:307-31, 1994). Altschul et al., Nat. Genet., 6:119-129, 1994, presents a detailed consideration of sequence alignment methods and homology calculations. By way of example, the alignment tools ALIGN (Myers and Miller, CABIOS 4:11- 17, 1989) or LFASTA (Pearson and Lipman, 1988) may be used to perform sequence comparisons (Internet Program® 1996, W. R. Pearson and the University of Virginia, fasta20u63 version 2.0u63, release date December 1996). ALIGN compares entire sequences against one another, while LFASTA compares regions of local similarity. These alignment tools and their respective tutorials are available on the Internet at the NCSA Website, for instance. Alternatively, for comparisons of amino acid sequences of greater than about 30 amino acids, the Blast 2 sequences function can be employed using the default BLOSUM62 matrix set to default parameters, (gap existence cost of 11, and a per residue gap cost of 1). When aligning short peptides (fewer than around 30 amino acids), the alignment should be performed using the Blast 2 sequences function, employing the PAM30 matrix set to default parameters (open gap 9, extension gap 1 penalties). The BLAST sequence comparison system is available, for instance, from the NCBI web site; see also Altschul et al., J. Mol. Biol., 215:403-410, 1990; Gish. & States, Nature Genet., 3:266-272, 1993; Madden et al. Meth. Enzymol., 266:131-141, 1996; Altschul et al., Nucleic Acids Res., 25:3389-3402, 1997; and Zhang & Madden, Genome Res., 7:649-656, 1997. As used herein, the term “C9orf72” has its general meaning in the art and refers to the chromosome 9 open reading frame 72 protein that is encoded by the gene C9orf72 (Gene ID: 203228). The human C9orf72 gene is located on the short (p) arm of chromosome 9 open reading frame 72, from base pair 27,546,546 to base pair 27,573,866 (GRCh38). Its cytogenetic location is at 9p21.2. The protein is found in many regions of the brain, in the cytoplasm of neurons as well as in presynaptic terminals. C9orf72 is a component of the C9orf72-SMCR8
complex that has guanine nucleotide exchange factor (GEF) activity and regulates autophagy. In the complex, C9orf72 and SMCR8 probably constitute the catalytic subunits that promote the exchange of GDP to GTP, converting inactive GDP-bound RAB8A and RAB39B into their active GTP-bound form, thereby promoting autophagosome maturation. The C9orf72-SMCR8 complex also acts as a regulator of autophagy initiation by interacting with the ULK1/ATG1 kinase complex and modulating its protein kinase activity. An exemplary amino acid sequence for C9orf72 is represented by SEQ ID NO:2. The identified mutation of C9ORF72 is a hexanucleotide repeat expansion of the six-letter string of nucleotides GGGGCC (Bigio EH (December 2011). "C9ORF72, the new gene on the block, causes C9FTD/ALS: new insights provided by neuropathology". Acta Neuropathologica. 122 (6): 653–5). The mutation is the most common identified mutation associated with frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) (Babić Leko M, Župunski V, Kirincich J, Smilović D, Hortobágyi T, Hof PR, Šimić G (2019). "C9orf72 Hexanucleotide Repeat Expansion". Behavioural Neurology.2019: 2909168). SEQ ID NO:2 >sp|Q96LT7|CI072_HUMAN Guanine nucleotide exchange factor C9orf72 OS=Homo sapiens OX=9606 GN=C9orf72 PE=1 SV=2 MSTLCPPPSPAVAKTEIALSGKSPLLAATFAYWDNILGPRVRHIWAPKTEQVLLSDGEIT FLANHTLNGEILRNAESGAIDVKFFVLSEKGVIIVSLIFDGNWNGDRSTYGLSIILPQTE LSFYLPLHRVCVDRLTHIIRKGRIWMHKERQENVQKIILEGTERMEDQGQSIIPMLTGEV IPVMELLSSMKSHSVPEEIDIADTVLNDDDIGDSCHEGFLLNAISSHLQTCGCSVVVGSS AEKVNKIVRTLCLFLTPAERKCSRLCEAESSFKYESGLFVQGLLKDSTGSFVLPFRQVMY APYPTTHIDVDVNTVKQMPPCHEHIYNQRRYMRSELTAFWRATSEEDMAQDTIIYTDESF TPDLNIFQDVLHRDTLVKAFLDQVFQLKPGLSLRSTFLAQFLLVLHRKALTLIKYIEDDT QKGKKPFKSLRNLKIDLDLTAEGDLNIIMALAEKIKPGLHSFIFGRPFYTSVQERDVLMT F As used herein, the term “RNA Polymerase II pause site” refers to the RNA polymerase II (Pol II) pause site located in the first intronic region of the gene C9orf72 (Figure 1A and B). RNA Pol II pausing is a key regulatory step in transcription. RNA Pol II pauses at GC-rich regions, and almost 65% of the pause sites are cytosines (Watts, Jason A., et al. "Cis elements that mediate RNA polymerase II pausing regulate human gene expression." The American Journal of Human Genetics 105.4 (2019): 677-688). Gene expression analysis showed that genes with more paused polymerases have lower expression levels. Therefore, mutagenesis of the pause sites might lead to a significant increase in transcription. More particularly the pause site is located on chr9:27563356-27563366 region, is defined by the nucleotide sequence SEQ ID NO:3 (GTTCGGCTGCCGGGAAGAGGCGCGGGTAGAAGCGGGGGCTCTCCTCAGAGCT) and is depicted in Figure 1. The reverse sequence depicted in Figure 1 is defined by the
nucleotide sequence SEQ ID NO: 29 (AGCTCTGAGGAGAGCCCCCGCTTCTACCCG CGCCTCTTCCCGGCAGCCGAAC) As used herein, the term “Amyotrophic Lateral Sclerosis” or “ALS” has its general meaning in the art and refers to a neurodegenerative disease characterized by progressive muscular paralysis reflecting degeneration of motor neurons in the primary motor cortex, corticospinal tracts, brainstem and spinal cord. According to the present invention, ALS is caused by the C9orf72 mutation as described above. As used herein, the term "treatment" or "treat" refer to both prophylactic or preventive treatment as well as curative or disease modifying treatment, including treatment of patient at risk of contracting the disease or suspected to have contracted the disease as well as patients who are ill or have been diagnosed as suffering from a disease or medical condition, and includes suppression of clinical relapse. The treatment may be administered to a subject having a medical disorder or who ultimately may acquire the disorder, in order to prevent, cure, delay the onset of, reduce the severity of, or ameliorate one or more symptoms of a disorder or recurring disorder, or in order to prolong the survival of a subject beyond that expected in the absence of such treatment. By "therapeutic regimen" is meant the pattern of treatment of an illness, e.g., the pattern of dosing used during therapy. A therapeutic regimen may include an induction regimen and a maintenance regimen. The phrase "induction regimen" or "induction period" refers to a therapeutic regimen (or the portion of a therapeutic regimen) that is used for the initial treatment of a disease. The general goal of an induction regimen is to provide a high level of drug to a patient during the initial period of a treatment regimen. An induction regimen may employ (in part or in whole) a "loading regimen", which may include administering a greater dose of the drug than a physician would employ during a maintenance regimen, administering a drug more frequently than a physician would administer the drug during a maintenance regimen, or both. The phrase "maintenance regimen" or "maintenance period" refers to a therapeutic regimen (or the portion of a therapeutic regimen) that is used for the maintenance of a patient during treatment of an illness, e.g., to keep the patient in remission for long periods of time (months or years). A maintenance regimen may employ continuous therapy (e.g., administering a drug at regular intervals, e.g., weekly, monthly, yearly, etc.) or intermittent therapy (e.g., interrupted treatment, intermittent treatment, treatment at relapse, or treatment upon achievement of a particular predetermined criteria [e.g., pain, disease manifestation, etc.]).
As used herein, the expression "therapeutically effective amount" means a sufficient amount of the active ingredient for treating or reducing the symptoms at reasonable benefit/risk ratio applicable to any medical treatment. It will be understood that the total daily usage of the compounds and compositions of the present invention will be decided by the attending physician within the scope of sound medical judgment. The specific therapeutically effective dose level for any particular subject will depend upon a variety of factors including the disorder being treated and the severity of the disorder; activity of the specific compound employed; the specific composition employed, the age, body weight, general health, sex and diet of the subject; the time of administration, route of administration, and rate of excretion of the specific compound employed; the duration of the treatment; drugs used in combination with the active ingredients; and like factors well known in the medical arts. For example, it is well within the skill of the art to start doses of the compound at levels lower than those required to achieve the desired therapeutic effect and to gradually increase the dosage until the desired effect is achieved. Methods of the present invention: The first object of the present invention relates to a method for restoring the expression of C9orf72 in a patient suffering from Amyotrophic Lateral Sclerosis (ALS) comprising administering to the patient a therapeutically effective amount of a gene editing platform that consists of (a) at least one base-editing enzyme and (b) at least one guide RNA molecule for guiding the base-editing enzyme to at least one target sequence in the RNA Polymerase II pause site located in the first intronic region of the gene C9orf72, thereby editing said site and subsequently increasing the expression of C9orf72. In some embodiments, the gene editing platform is suitable for introducing some mutations in the RNA Polymerase II pause site. In some embodiments, the gene editing platform is particularly suitable for mutating the base C located at position Chr9:27563356-27563366- Hg18. In some embodiments, the gene editing platform is particularly suitable for introducing one C>T conversion at position Chr9:27563356-27563366-Hg18 (Figure 1A). In some embodiments, the base-editing enzyme of the present invention comprises a defective CRISPR/Cas nuclease. The sequence recognition mechanism is the same as for the non-
defective CRISPR/Cas nuclease. Typically, the defective CRISPR/Cas nuclease of the invention comprises at least one RNA binding domain. The RNA binding domain interacts with a guide RNA molecule as defined hereinafter. However, the defective CRISPR/Cas nuclease of the invention is a modified version with no nuclease activity. Accordingly, the defective CRISPR/Cas nuclease specifically recognizes the guide RNA molecule and thus guides the base-editing enzyme to its target DNA sequence. In some embodiments, the defective CRISPR/Cas nuclease can be modified to increase nucleic acid binding affinity and/or specificity, alter an enzymatic activity, and/or change another property of the protein. In some embodiments, the nuclease domains of the protein can be modified, deleted, or inactivated. In some embodiments, the protein can be truncated to remove domains that are not essential for the function of the protein. In some embodiments, the protein is truncated or modified to optimize the activity of the RNA binding domain. In some embodiments, the CRISPR/Cas nuclease consists of a mutant CRISPR/Cas nuclease i.e. a protein having one or more point mutations, insertions, deletions, truncations, a fusion protein, or a combination thereof. In some embodiments, the mutant has the RNA-guided DNA binding activity, but lacks one or both of its nuclease active sites. In some embodiments, the mutant comprises an amino acid sequence having at least 50% of identity with the wild type amino acid sequence of the CRISPR/Cas nuclease. Various CRISPR/Cas nucleases can be used in this invention. Non-limiting examples of suitable CRISPR/CRISPR/Cas nucleases include Cas3, Cas4, Cas5, Cas5e (or CasD), Cas6, Cas6e, Cas6f, Cas7, Cas8a1, Cas8a2, Cas8b, Cas8c, Cas9, Cas10, Cas10d, CasF, CasG, CasH, Csy1, Csy2, Csy3, Cse1 (or CasA), Cse2 (or CasB), Cse3 (or CasE), Cse4 (or CasC), Csc1, Csc2, Csa5, Csn2, Csm2, Csm3, Csm4, Csm5, Csm6, Cmr1, Cmr3, Cmr4, Cmr5, Cmr6, Csb1, Csb2, Csb3, Csx17, Csx14, Csx10, Csx16, CsaX, Csx3, Csz1, Csx15, Csf1, Csf2, Csf3, Csf4, and Cu1966. See e.g., WO2014144761 WO2014144592, WO2013176772, US20140273226, and US20140273233, the contents of which are incorporated herein by reference in their entireties. In some embodiments, the CRISPR/Cas nuclease is derived from a type II CRISPR-Cas system. In some embodiments, the CRISPR/Cas nuclease is derived from a Cas9 protein. The Cas9 protein can be from Streptococcus pyogenes, Streptococcus thermophilus, Streptococcus sp., Nocardiopsis dassonvillei, Streptomyces pristinaespiralis, Streptomyces viridochromogenes, Streptomyces viridochromogenes, Streptosporangium roseum, Streptosporangium roseum,
Alicyclobacillus acidocaldarius, Bacillus pseudomycoides, Bacillus selenitireducens, Exiguobacterium sibiricum, Lactobacillus delbrueckii, Lactobacillus salivarius, Microscilla marina, Burkholderiales bacterium, Polaromonas naphthalenivorans, Polaromonas sp., Crocosphaera watsonii, Cyanothece sp., Microcystis aeruginosa, Synechococcus sp., Acetohalobium arabaticum, Ammonifex degensii, Caldicelulosiruptor becscii, Candidatus Desulforudis, Clostridium botulinum, Clostridium difficile, Finegoldia magna, Natranaerobius thermophilus, Pelotomaculum thermopropionicum, Acidithiobacillus caldus, Acidithiobacillus ferrooxidans, Allochromatium vinosum, Marinobacter sp., Nitrosococcus halophilus, Nitrosococcus watsoni, Pseudoalteromonas haloplanktis, Ktedonobacter racemifer, Methanohalobium evestigatum, Anabaena variabilis, Nodularia spumigena, Nostoc sp., Arthrospira maxima, Arthrospira platensis, Arthrospira sp., Lyngbya sp., Microcoleus chthonoplastes, Oscillatoria sp., Petrotoga mobilis, Thermosipho africanus, or Acaryochloris marina, inter alia. In some embodiments, the CRISPR/Cas nuclease is a mutant of a wild type CRISPR/Cas nuclease (such as Cas9) or a fragment thereof. In some embodiments, the CRISPR/Cas nuclease is a mutant Cas9 protein from S. pyogenes. Methods for generating a Cas9 protein (or a fragment thereof) having an inactive DNA cleavage domain are known (See, e.g., Jinek et al., Science.337:816-821(2012); Qi et al., “Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression” (2013) Cell.28; 152(5):1173-83, the entire contents of each of which are incorporated herein by reference). For example, the DNA cleavage domain of Cas9 is known to include two subdomains, the HNH nuclease subdomain and the RuvC1 subdomain. The HNH subdomain cleaves the strand complementary to the gRNA, whereas the RuvC1 subdomain cleaves the non-complementary strand. Mutations within these subdomains can silence the nuclease activity of Cas9. For example, the mutations D10A and H841A completely inactivate the nuclease activity of S. pyogenes Cas9 (Jinek et al., Science.337:816-821(2012); Qi et al., Cell. 28; 152(5):1173-83 (2013)). In some embodiments, the CRISPR/Cas nuclease of the present invention is nickase and more particularly a Cas9 nickase i.e. the Cas9 from S. pyogenes having one mutation selected from the group consisting of D10A and H840A. In some embodiments, the nickase of the present invention comprises the amino acid sequence as set forth in SEQ ID NO:4 or SEQ ID NO:5.
SEQ ID NO: 4> S. pyogenes nCas9 Protein Sequence having the D10A mutation MDKKYSIGLAIGTNSVGWAVITDEYKVPSKKFKVLGNTDRHSIKKNLIGALLFDSGETAEATRLKRTAR RRYTRRKNRICYLQEIFSNEMAKVDDSFFHRLEESFLVEEDKKHERHPIFGNIVDEVAYHEKYPTIYHL RKKLVDSTDKADLRLIYLALAHMIKFRGHFLIEGDLNPDNSDVDKLFIQLVQTYNQLFEENPINASGVD AKAILSARLSKSRRLENLIAQLPGEKKNGLFGNLIALSLGLTPNFKSNFDLAEDAKLQLSKDTYDDDLD NLLAQIGDQYADLFLAAKNLSDAILLSDILRVNTEITKAPLSASMIKRYDEHHQDLTLLKALVRQQLPE KYKEIFFDQSKNGYAGYIDGGASQEEFYKFIKPILEKMDGTEELLVKLNREDLLRKQRTFDNGSIPHQI HLGELHAILRRQEDFYPFLKDNREKIEKILTFRIPYYVGPLARGNSRFAWMTRKSEETITPWNFEEVVD KGASAQSFIERMTNFDKNLPNEKVLPKHSLLYEYFTVYNELTKVKYVTEGMRKPAFLSGEQKKAIVDLL FKTNRKVTVKQLKEDYFKKIECFDSVEISGVEDRFNASLGTYHDLLKIIKDKDFLDNEENEDILEDIVL TLTLFEDREMIEERLKTYAHLFDDKVMKQLKRRRYTGWGRLSRKLINGIRDKQSGKTILDFLKSDGFAN RNFMQLIHDDSLTFKEDIQKAQVSGQGDSLHEHIANLAGSPAIKKGILQTVKVVDELVKVMGRHKPENI VIEMARENQTTQKGQKNSRERMKRIEEGIKELGSQILKEHPVENTQLQNEKLYLYYLQNGRDMYVDQEL DINRLSDYDVDHIVPQSFLKDDSIDNKVLTRSDKNRGKSDNVPSEEVVKKMKNYWRQLLNAKLITQRKF DNLTKAERGGLSELDKAGFIKRQLVETRQITKHVAQILDSRMNTKYDENDKLIREVKVITLKSKLVSDF RKDFQFYKVREINNYHHAHDAYLNAVVGTALIKKYPKLESEFVYGDYKVYDVRKMIAKSEQEIGKATAK YFFYSNIMNFFKTEITLANGEIRKRPLIETNGETGEIVWDKGRDFATVRKVLSMPQVNIVKKTEVQTGG FSKESILPKRNSDKLIARKKDWDPKKYGGFDSPTVAYSVLVVAKVEKGKSKKLKSVKELLGITIMERSS FEKNPIDFLEAKGYKEVKKDLIIKLPKYSLFELENGRKRMLASAGELQKGNELALPSKYVNFLYLASHY EKLKGSPEDNEQKQLFVEQHKHYLDEIIEQISEFSKRVILADANLDKVLSAYNKHRDKPIREQAENIIH LFTLTNLGAPAAFKYFDTTIDRKRYTSTKEVLDATLIHQSITGLYETRIDLSQLGGD SEQ ID NO: 5> S. pyogenes nCas9 Protein Sequence having the H840A mutation MDKKYSIGLDIGTNSVGWAVITDEYKVPSKKFKVLGNTDRHSIKKNLIGALLFDSGETAEATRLKRTAR RRYTRRKNRICYLQEIFSNEMAKVDDSFFHRLEESFLVEEDKKHERHPIFGNIVDEVAYHEKYPTIYHL RKKLVDSTDKADLRLIYLALAHMIKFRGHFLIEGDLNPDNSDVDKLFIQLVQTYNQLFEENPINASGVD AKAILSARLSKSRRLENLIAQLPGEKKNGLFGNLIALSLGLTPNFKSNFDLAEDAKLQLSKDTYDDDLD NLLAQIGDQYADLFLAAKNLSDAILLSDILRVNTEITKAPLSASMIKRYDEHHQDLTLLKALVRQQLPE KYKEIFFDQSKNGYAGYIDGGASQEEFYKFIKPILEKMDGTEELLVKLNREDLLRKQRTFDNGSIPHQI HLGELHAILRRQEDFYPFLKDNREKIEKILTFRIPYYVGPLARGNSRFAWMTRKSEETITPWNFEEVVD KGASAQSFIERMTNFDKNLPNEKVLPKHSLLYEYFTVYNELTKVKYVTEGMRKPAFLSGEQKKAIVDLL FKTNRKVTVKQLKEDYFKKIECFDSVEISGVEDRFNASLGTYHDLLKIIKDKDFLDNEENEDILEDIVL TLTLFEDREMIEERLKTYAHLFDDKVMKQLKRRRYTGWGRLSRKLINGIRDKQSGKTILDFLKSDGFAN RNFMQLIHDDSLTFKEDIQKAQVSGQGDSLHEHIANLAGSPAIKKGILQTVKVVDELVKVMGRHKPENI VIEMARENQTTQKGQKNSRERMKRIEEGIKELGSQILKEHPVENTQLQNEKLYLYYLQNGRDMYVDQEL DINRLSDYDVDAIVPQSFLKDDSIDNKVLTRSDKNRGKSDNVPSEEVVKKMKNYWRQLLNAKLITQRKF DNLTKAERGGLSELDKAGFIKRQLVETRQITKHVAQILDSRMNTKYDENDKLIREVKVITLKSKLVSDF RKDFQFYKVREINNYHHAHDAYLNAVVGTALIKKYPKLESEFVYGDYKVYDVRKMIAKSEQEIGKATAK YFFYSNIMNFFKTEITLANGEIRKRPLIETNGETGEIVWDKGRDFATVRKVLSMPQVNIVKKTEVQTGG FSKESILPKRNSDKLIARKKDWDPKKYGGFDSPTVAYSVLVVAKVEKGKSKKLKSVKELLGITIMERSS FEKNPIDFLEAKGYKEVKKDLIIKLPKYSLFELENGRKRMLASAGELQKGNELALPSKYVNFLYLASHY EKLKGSPEDNEQKQLFVEQHKHYLDEIIEQISEFSKRVILADANLDKVLSAYNKHRDKPIREQAENIIH LFTLTNLGAPAAFKYFDTTIDRKRYTSTKEVLDATLIHQSITGLYETRIDLSQLGGD In some embodiments, the Cas9 variants having mutations other than D10A or H840A are used, which e.g., result in nuclease inactivated Cas9 (dCas9). Such mutations, by way of example, include other amino acid substitutions at D10 and H840, or other substitutions within the nuclease domains of Cas9 (e.g., substitutions in the HNH nuclease subdomain and/or the RuvC1 subdomain). In some embodiments, variants of dCas9 are provided which are at least about 70% identical, at least about 80% identical, at least about 90% identical, at least about 95% identical, at least about 98% identical, at least about 99% identical, at least about 99.5%
identical, or at least about 99.9% to SEQ ID NO:1. In some embodiments, variants of dCas9 are provided having amino acid sequences which are shorter, or longer than SEQ ID NO: 1 or 3, by about 5 amino acids, by about 10 amino acids, by about 15 amino acids, by about 20 amino acids, by about 25 amino acids, by about 30 amino acids, by about 40 amino acids, by about 50 amino acids, by about 75 amino acids, by about 100 amino acids or more. According to the present invention, the second component of the base-editing enzyme herein disclosed comprises a non-nuclease DNA modifying enzyme that is a deaminase. Since, the targeted base of interest is a C, the deaminase is preferably a cytidine deaminase. In some embodiments, the cytidine deaminase is an apolipoprotein B mRNA-editing complex (APOBEC) family deaminase. In some embodiments, the cytidine deaminase is an APOBEC1 family deaminase. In some embodiments, the cytidine deaminase is an activation-induced cytidine deaminase (AID). In some embodiments, the cytidine deaminase is an ACF1/ASE deaminase. In some embodiments, the cytidine deaminase is selected from the group consisting of AID: activation induced cytidine deaminase, APOBEC1: apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 1, APOBEC3A: apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3A, APOBEC3B: apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3B, APOBEC3C: apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3C, APOBEC3D: apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3D, APOBEC3F: apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3F, APOBEC3G: apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3G, APOBEC3H: apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3H, ADA: adenosine deaminase, ADAR1: adenosine deaminase acting on RNA 1, Dnmt1: DNA (cytosine-5-)-methyltransferase 1, Dnmt3a: DNA (cytosine-5-)- methyltransferase 3 alpha, Dnmt3b: DNA (cytosine-5-)-methyltransferase 3 beta and Tet1: methylcytosine dioxygenase. In some embodiments, the cytidine deaminase derives from the Activation Induced cytidine Deaminase (AID). AID is a cytidine deaminase that can catalyze the reaction of deamination of cytosine in the context of DNA or RNA. When brought to the targeted site, AID changes a C base to U base. In dividing cells, this could lead to a C to T point mutation. Alternatively, the
change of C to U could trigger cellular DNA repair pathways, mainly excision repair pathway, which will remove the mismatching U-G base-pair, and replace with a T-A, A-T, C-G, or G-C pair. As a result, a point mutation would be generated at the target C-G site. In some embodiments, the DNA modifying enzyme is AID*Δ that is an AID mutant with increased SHM activity whose Nuclear Export Signal (NES) has been removed (Hess GT, Fresard L, Han K, Lee CH, Li A, Cimprich KA, Montgomery SB, Bassik MC: Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat Methods 2016, 13(12):1036- 1042). In some embodiments, the cytidine deaminase consists of a variant of the amino acid sequence as set forth in SEQ ID NO:6-16. SEQ ID NO:6 Human AID: MDSLLMNRRKFLYQFKNVRWAKGRRETYLCYVVKRRDSATSFSLDFGYLRNKNGCHVELLFLRYISDWD LDPGRCYRVTWFTSWSPCYDCARHVADFLRGNPNLSLRIFTARLYFCEDRKAEPEGLRRLHRAGVQIAI MTFKDYFYCWNT FVENHERTFKAWEGLHENSVRLSRQLRRILLPLYEVDDLRDAFRTLGL SEQ ID NO:7 Human APOBEC-3G MKPHFRNTVERMYRDTFSYNFYNRPILSRRNTVWLCYEVKTKGPSRPPLDAKIFRGQVYSELKYHPEMR FFHWFSKWRKLHRDQEYEVTWYISWSPCTKCTRDMATFLAEDPKVTLTIFVARLYYFWDPDYQEALRSL CQKRDGPRATMKIMNYDEFQHCWSKFVYSQRELFEPWNNLPKYYILLHIMLGEILRHSMDPPTFTFNFN NEPWVRGRHETYLCYEVERMHNDTWVLLNQRRGFLCNQAPHKHGFLEGRHAELCFLDVIPFWKLDLDQD YRVTCFTSWSPCFSCAQEMAKFISKNKHVSLCIFTARIYDDQGRCQEGLRTLAEAGAKISIMTYSEFKH CWDTFVDHQGCPFQPWDGLDEHSQDLSGRLRAILQNQEN SEQ ID NO:8 Human APOBEC-3F MKPHFRNTVERMYRDTFSYNFYNRPILSRRNTVWLCYEVKTKGPSRPRLDAKIFRGQVYSQPEHHAEMC FLSWFCGNQLPAYKCFQITWFVSWTPCPDCVAKLAEFLAEHPNVTLTISAARLYYYWERDYRRALCRLS QAGARVKIMDDEEFAYCWENFVYSEGQPFMPWYKFDDNYAFLHRTLKEILRNPMEAMYPHIFYFHFKNL RKAYGRNESWLCFTMEVVKHHSPVSWKRGVFRNQVDPETHCHAERCFLSWFCDDILSPNTNYEVTWYTS WSPCPECAGEVAEFLARHSNVNLTIFTARLYYFWDTDYQEGLRSLSQEGASVEIMGYKDFKYCWENFVY NDDEPFKPWKGLKYNFLFLDSKLQEILE SEQ ID NO:9 Human APOBEC-3B: MNPQIRNPMERMYRDTFYDNFENEPILYGRSYTWLCYEVKIKRGRSNLLWDTGVFRGQVYFKPQYHAEM CFLSWFCGNQLPAYKCFQITWFVSWTPCPDCVAKLAEFLSEHPNVTLTISAARLYYYWERDYRRALCRL SQAGARVTIMDYEEFAYCWENFVYNEGQQFMPWYKFDENYAFLHRTLKEILRYLMDPDTFTFNFNNDPL VLRRRQTYLCYEVERLDNGTWVLMDQHMGFLCNEAKNLLCGFYGRHAELRFLDLVPSLQLDPAQIYRVT WFISWSPCFSWGCAGEVRAFLQENTHVRLRIFAARIYDYDPLYKEALQMLRDAGAQVSIMTYDEFEYCW DTFVYRQGCPFQPWDGLEEHSQALSGRLRAILQNQGN SEQ ID NO:10 Human APOBEC-3C: MNPQIRNPMKAMYPGTFYFQFKNLWEANDRNETWLCFTVEGIKRRSVVSWKTGVFRNQVDSETHCHAER CFLSWFCDDILSPNTKYQVTWYTSWSPCPDCAGEVAEFLARHSNVNLTIFTARLYYFQYPCYQEGLRSL SQEGVAVEIMDYEDFKYCWENFVYNDNEPFKPWKGLKTNFRLLKRRLRESLQ SEQ ID NO:11 Human APOBEC-3A: MEASPASGPRHLMDPHIFTSNFNNGIGRHKTYLCYEVERLDNGTSVKMDQHRGFLHNQAKNLLCGFYGR HAELRFLDLVPSLQLDPAQIYRVTWFISWSPCFSWGCAGEVRAFLQENTHVRLRIFAARIYDYDPLYKE ALQMLRDAGAQVSIMTYDEFKHCWDTFVDHQGCPFQPWDGLDEHSQALSGRLRAILQNQGN
SEQ ID NO:12 Human APOBEC-3H: MALLTAETFRLQFNNKRRLRRPYYPRKALLCYQLTPQNGSTPTRGYFENKKKCHAEICFINEIKSMGLD ETQCYQVTCYLTWSPCSSCAWELVDFIKAHDHLNLGIFASRLYYHWCKPQQKGLRLLCGSQVPVEVMGF PKFADCWENFVDHEKPLSFNPYKMLEELDKNSRAIKRRLERIKIPGVRAQGRYMDILCDAEV SEQ ID NO:13 Human APOBEC-3D MNPQIRNPMERMYRDTFYDNFENEPILYGRSYTWLCYEVKIKRGRSNLLWDTGVFRGPVLPKRQSNHRQ EVYFRFENHAEMCFLSWFCGNRLPANRRFQITWFVSWNPCLPCVVKVTKFLAEHPNVTLTISAARLYYY RDRDWRWVLLRLHKAGARVKIMDYEDFAYCWENFVCNEGQPFMPWYKFDDNYASLHRTLKEILRNPMEA MYPHIFYFHFKNLLKACGRNESWLCFTMEVTKHHSAVFRKRGVFRNQVDPETHCHAERCFLSWFCDDIL SPNTNYEVTWYTSWSPCPECAGEVAEFLARHSNVNLTIFTARLCYFWDTDYQEGLCSLSQEGASVKIMG YKDFVSCWKNFVYSDDEPFKPWKGLQTNFRLLKRRLREILQ SEQ ID NO:14 Human APOBEC-1: MTSEKGPSTGDPTLRRRIEPWEFDVFYDPRELRKEACLLYEIKWGMSRKIWRSSGKNTTNHVEVNFIKK FTSERDFHPSMSCSITWFLSWSPCWECSQAIREFLSRHPGVTLVIYVARLFWHMDQQNRQGLRDLVNSG VTIQIMRASEYYHCWRNFVNYPPGDEAHWPQYPPLWMMLYALELHCIILSLPPCLKISRRWQNHLTFFR LHLQNCHYQTIPPHILLATGLIHPSVAWR SEQ ID NO:15 Human ADAT-2: MEAKAAPKPAASGACSVSAEETEKWMEEAMHMAKEALENTEVPVGCLMVYNNEVVGKGRNEVNQTKNAT RHAEMVAIDQVLDWCRQSGKSPSEVFEHTVLYVTVEPCIMCAAALRLMKIPLVVYGCQNERFGGCGSVL NIASADLPNTGR PFQCIPGYRAEEAVEMLKTFYKQENPNAPKSKVRKKECQKS SEQ ID NO:16 Human ADAT-1: MWTADEIAQLCYEHYGIRLPKKGKPEPNHEWTLLAAVVKIQSPADKACDTPDKPVQVTKEVVSMGTGTK CIGQSKMRKNGDILNDSHAEVIARRSFQRYLLHQLQLAATLKEDSIFVPGTQKGVWKLRRDLIFVFFSS HTPCGDASIIPMLEFEDQPCCPVFRNWAHNSSVEASSNLEAPGNERKCEDPDSPVTKKMRLEPGTAARE VTNGAAHHQSFGKQKSGPISPGIHSCDLTVEGLATVTRIAPGSAKVIDVYRTGAKCVPGEAGDSGKPGA AFHQVGLLRVKPGRGDRTRSMSCSDKMARWNVLGCQGALLMHLLEEPIYLSAVVIGKCPYSQEAMQRAL IGRCQNVSALPKGFGVQELKILQSDLLFEQSRSAVQAKRADSPGRLVPCGAAISWSAVPEQPLDVTANG FPQGTTKKTIGSLQARSQISKVELFRSFQKLLSRIARDKWPHSLRVQKLDTYQEYKEAASSYQEAWSTL RKQVFGSWIRNPPDYHQFK In some embodiments, the deaminase is fused to the N-terminus of the defective CRISPR/Cas nuclease. In some embodiments, the deaminase is fused to the C-terminus of the defective CRISPR/Cas nuclease. In some embodiments, the defective CRISPR/Cas nuclease and the deaminase are fused via a linker. In some embodiments, the linker comprises a (GGGGS)n (SEQ ID NO:17), a (G)n, an (EAAAK)n (SEQ ID NO: 18), a (GGS)n, or an SGSETPGTSESATPES (SEQ ID NO:19) motif (see, e.g., Guilinger, John P., David B. Thompson, and David R. Liu. "Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification." Nature biotechnology 32.6 (2014): 577-582). Additional suitable linker motifs and linker configurations will be apparent to those of skill in the art. In some embodiments, suitable linker motifs and configurations include those described in Chen et al., Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev. 2013; 65(10):1357-69, the entire contents of which are incorporated herein by reference. In some embodiments, the fusion protein may comprise additional features. Other exemplary features that may be present are localization sequences, such as nuclear localization sequences
(NLS), cytoplasmic localization sequences, export sequences, such as nuclear export sequences, or other localization sequences, as well as sequence tags that are useful for solubilization, purification, or detection of the fusion proteins. Suitable localization signal sequences and sequences of protein tags are provided herein, and include, but are not limited to, biotin carboxylase carrier protein (BCCP) tags, myc-tags, calmodulin-tags, FLAG-tags, hemagglutinin (HA)-tags, polyhistidine tags, also referred to as histidine tags or His-tags, maltose binding protein (MBP)-tags, nus-tags, glutathione-S-transferase (GST)-tags, green fluorescent protein (GFP)-tags, thioredoxin-tags, S-tags, Softags (e.g., Softag 1, Softag 3), strep-tags, biotin ligase tags, FlAsH tags, V5 tags, and SBP-tags. Additional suitable features will be apparent to those of skill in the art. Various CBE base-editing enzymes are known in the art (see e.g. Koblan, Luke W., et al. "Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction." Nature biotechnology 36.9 (2018): 843-846) and typically include those described in Table A. Table A: some exemplary base-editing enzymes
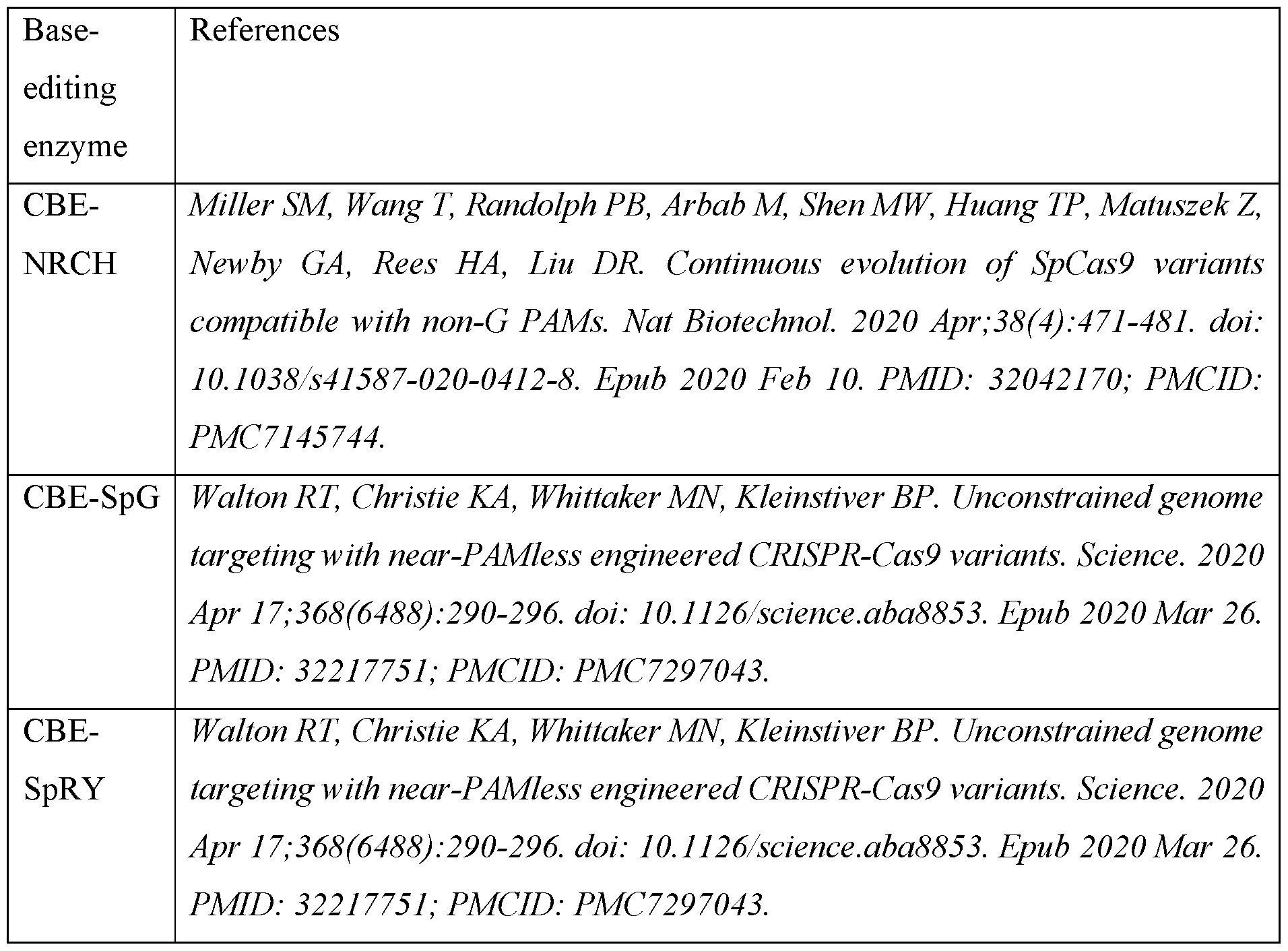 In particular, the amino acid sequence of CBE-SpRY is SEQ ID NO:20. SEQ ID NO:21> amino acid sequence of CBE-SpRY MKRTADGSEFESPKKKRKVSSETGPVAVDPTLRRRIEPHEFEVFFDPRELRKETCLLYEINWGGRHSIW RHTSQNTNKHVEVNFIEKFTTERYFCPNTRCSITWFLSWSPCGECSRAITEFLSRYPHVTLFIYIARLY HHADPRNRQGLRDLISSGVTIQIMTEQESGYCWRNFVNYSPSNEAHWPRYPHLWVRLYVLELYCIILGL PPCLNILRRKQPQLTFFTIALQSCHYQRLPPHILWATGLKSGGSSGGSSGSETPGTSESATPESSGGSS GGSDKKYSIGLAIGTNSVGWAVITDEYKVPSKKFKVLGNTDRHSIKKNLIGALLFDSGETAERTRLKRT ARRRYTRRKNRICYLQEIFSNEMAKVDDSFFHRLEESFLVEEDKKHERHPIFGNIVDEVAYHEKYPTIY HLRKKLVDSTDKADLRLIYLALAHMIKFRGHFLIEGDLNPDNSDVDKLFIQLVQTYNQLFEENPINASG VDAKAILSARLSKSRRLENLIAQLPGEKKNGLFGNLIALSLGLTPNFKSNFDLAEDAKLQLSKDTYDDD LDNLLAQIGDQYADLFLAAKNLSDAILLSDILRVNTEITKAPLSASMIKRYDEHHQDLTLLKALVRQQL PEKYKEIFFDQSKNGYAGYIDGGASQEEFYKFIKPILEKMDGTEELLVKLNREDLLRKQRTFDNGSIPH QIHLGELHAILRRQEDFYPFLKDNREKIEKILTFRIPYYVGPLARGNSRFAWMTRKSEETITPWNFEEV VDKGASAQSFIERMTNFDKNLPNEKVLPKHSLLYEYFTVYNELTKVKYVTEGMRKPAFLSGEQKKAIVD LLFKTNRKVTVKQLKEDYFKKIECFDSVEISGVEDRFNASLGTYHDLLKIIKDKDFLDNEENEDILEDI VLTLTLFEDREMIEERLKTYAHLFDDKVMKQLKRRRYTGWGRLSRKLINGIRDKQSGKTILDFLKSDGF ANRNFMQLIHDDSLTFKEDIQKAQVSGQGDSLHEHIANLAGSPAIKKGILQTVKVVDELVKVMGRHKPE NIVIEMARENQTTQKGQKNSRERMKRIEEGIKELGSQILKEHPVENTQLQNEKLYLYYLQNGRDMYVDQ ELDINRLSDYDVDHIVPQSFLKDDSIDNKVLTRSDKNRGKSDNVPSEEVVKKMKNYWRQLLNAKLITQR KFDNLTKAERGGLSELDKAGFIKRQLVETRQITKHVAQILDSRMNTKYDENDKLIREVKVITLKSKLVS DFRKDFQFYKVREINNYHHAHDAYLNAVVGTALIKKYPKLESEFVYGDYKVYDVRKMIAKSEQEIGKAT AKYFFYSNIMNFFKTEITLANGEIRKRPLIETNGETGEIVWDKGRDFATVRKVLSMPQVNIVKKTEVQT GGFSKESIRPKRNSDKLIARKKDWDPKKYGGFLWPTVAYSVLVVAKVEKGKSKKLKSVKELLGITIMER SSFEKNPIDFLEAKGYKEVKKDLIIKLPKYSLFELENGRKRMLASAKQLQKGNELALPSKYVNFLYLAS HYEKLKGSPEDNEQKQLFVEQHKHYLDEIIEQISEFSKRVILADANLDKVLSAYNKHRDKPIREQAENI IHLFTLTRLGAPRAFKYFDTTIDPKQYRSTKEVLDATLIHQSITGLYETRIDLSQLGGDSGGSGGSGGS TNLSDIIEKETGKQLVIQESILMLPEEVEEVIGNKPESDILVHTAYDESTDENVMLLTSDAPEYKPWAL VIQDSNGENKIKMLSGGSGGSGGSTNLSDIIEKETGKQLVIQESILMLPEEVEEVIGNKPESDILVHTA YDESTDENVMLLTSDAPEYKPWALVIQDSNGENKIKMLSGGSKRTADGSEFEPKKKRKVGGGGSGATNF SLLKQAGDVEENPGPMVSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKL PVPWPTLVTTLTYGVQCFSRYPDHMKQHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLVNR IELKGIDFKEDGNILGHKLEYNYNSHNVYIMADKQKNGIKVNFKIRHNIEDGSVQLADHYQQNTPIGDG PVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAAGITLGMDELYK The second component of the gene-editing platform disclosed herein consists of at least one guide RNA molecule suitable for guiding the base-editing enzyme to at least one target sequence located in the RNA Polymerase II pause site. The guide RNA molecule of the present invention thus comprises a guide sequence for providing the targeting specificity. It includes a region that is complementary and capable of hybridization to a pre-selected target site of interest in the RNA Polymerase II pause site. According to the present disclosure, the guide RNA targets the pause site that is located on chr9:27563356-27563366 region, and more preferably the nucleotide sequence SEQ ID NO:30 (GAAGAGGCGCGGGTAGAAGC). In some embodiment, this guide sequence can comprise from about 10 nucleotides to more than about 25 nucleotides. For example, the region of base pairing between the guide sequence and the corresponding target site sequence can be about 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20,
22, 23, 24, 25, or more than 25 nucleotides in length. In some embodiments, the guide sequence is about 17-20 nucleotides in length, such as 20 nucleotides. Typically, a software program is used to identify candidate CRISPR target sequences on both strands of the DNA nucleic acid molecule based on desired guide sequence length and a CRISPR motif sequence (PAM) for a specified CRISPR enzyme. One requirement for selecting a suitable target nucleic acid is that it has a 3′ PAM site/sequence. Each target sequence and its corresponding PAM site/sequence are referred herein as a Cas-targeted site. Type II CRISPR system, one of the most well characterized systems, needs only Cas 9 protein and a guide RNA complementary to a target sequence to affect target cleavage. For example, target sites for Cas9 from S. pyogenes, with PAM sequences NGG, may be identified by searching for 5′-Nx-NGG- 3′ both on the input sequence and on the reverse-complement of the input. Since multiple occurrences in the genome of the DNA target site may lead to nonspecific genome editing, after identifying all potential sites, the program filters out sequences based on the number of times they appear in the relevant reference genome. For those CRISPR enzymes for which sequence specificity is determined by a “seed” sequence, such as the 11-12 bp 5′ from the PAM sequence, including the PAM sequence itself, the filtering step may be based on the seed sequence. Thus, to avoid editing at additional genomic loci, results are filtered based on the number of occurrences of the seed:PAM sequence in the relevant genome. The user may be allowed to choose the length of the seed sequence. The user may also be allowed to specify the number of occurrences of the seed:PAM sequence in a genome for purposes of passing the filter. The default is to screen for unique sequences. Filtration level is altered by changing both the length of the seed sequence and the number of occurrences of the sequence in the genome. The program may in addition or alternatively provide the sequence of a guide sequence complementary to the reported target sequence(s) by providing the reverse complement of the identified target sequence(s). Further details of methods and algorithms to optimize sequence selection can be found in U.S. application Ser. No. 61/836,080; incorporated herein by reference. In some embodiments, the guide RNA targets a sequence selected from Table 1 (see EXAMPLE).
In some embodiments, the gene editing platform comprises a) a base-editing enzyme that is a CBE-SpRY and b) and at least one guide RNA molecule that targets one sequence selected in Table 1. The guide RNA molecule of the present invention can be made by various methods known in the art including cell-based expression, in vitro transcription, and chemical synthesis. The ability to chemically synthesize relatively long RNAs (as long as 200 mers or more) using TC- RNA chemistry (see, e.g., U.S. Pat. No.8,202,983) allows one to produce RNAs with special features that outperform those enabled by the basic four ribonucleotides (A, C, G and U). In particular, the RNA molecule of the present invention can be made with recombinant technology using a host cell system or an in vitro translation-transcription system known in the art. Details of such systems and technology can be found in e.g., WO2014144761 WO2014144592, WO2013176772, US20140273226, and US20140273233, the contents of which are incorporated herein by reference in their entireties. In some embodiments, the guide RNA molecule may include one or more modifications. Such modifications may include inclusion of at least one non-naturally occurring nucleotide, or a modified nucleotide, or analogs thereof. Modified nucleotides may be modified at the ribose, phosphate, and/or base moiety. Modified nucleotides may include 2’-O-methyl analogs, 2’- deoxy analogs, or 2’-fluoro analogs. The nucleic acid backbone may be modified, for example, a phosphorothioate backbone may be used. The use of locked nucleic acids (LNA) or bridged nucleic acids (BNA) may also be possible. Further examples of modified bases include, but are not limited to, 2-aminopurine, 5-bromo-uridine, pseudouridine, inosine, 7-methylguanosine. In some embodiments, a plurality of guide RNA molecules are designed for targeting a plurality of sequences in the RNA Polymerase II pause site. In some embodiments, the gene editing platform disclosed herein thus comprises 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, or 20 guide RNA molecules as disclosed herein. In some embodiments, a plurality of base-editing enzyme along with a plurality of guide RNA molecules are designed for targeting a plurality of sequences in the RNA Polymerase II pause site. In some embodiments, the gene editing platform disclosed herein thus comprises 2, 3 or 4 base-editing enzymes and 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, or 20 RNA molecules as disclosed herein.
In some embodiments, the different components of the gene editing platform of the present invention are administered to the patient through expression from one or more vectors. Examples of vectors include viral vector and non-viral vectors. As used herein, the term “viral vector” refers to a virion or virus particle that functions as a nucleic acid delivery vehicle and which comprises a vector genome packaged within the virion or virus particle. Typically, the vector is a viral vector which is an adeno-associated virus (AAV), a retroviral vector, bovine papilloma virus, an adenovirus vector, a vaccinia virus, or a polyoma virus. In some embodiments, the viral vector is a AAV vector. As used herein, the term "AAV vector" means a vector derived from an adeno- associated virus serotype, including without limitation, AAV1, AAV2, AAV3, AAV4, AAV5, AAV6, AAV7, AAV8, AAV9, and mutated forms thereof. AAV vectors can have one or more of the AAV wild-type genes deleted in whole or part, preferably the rep and/or cap genes, but retain functional flanking ITR sequences. In some embodiments, the viral vector is a retroviral vector. As used herein, the term “retroviral vector” refers to a vector containing structural and functional genetic elements that are primarily derived from a retrovirus. In some embodiments, the retroviral vector of the present invention derives from a retrovirus selected from the group consisting of alpharetroviruses (e.g., avian leukosis virus), betaretroviruses (e.g., mouse mammary tumor virus), gammaretroviruses (e.g., murine leukemia virus), deltaretroviruses (e.g., bovine leukemia virus), epsilonretroviruses (e.g., Walley dermal sarcoma virus), lentiviruses (e.g., HIV-1, HIV-2) and spumaviruses (e.g., human spumavirus). In some embodiments, the retroviral vector of the present invention is a replication deficient retroviral virus particle, which can transfer a foreign imported RNA of a gene instead of the retroviral mRNA.
In some embodiments, the retroviral vector of the present invention is a lentiviral vector. As used herein, the term “lentiviral vector” refers to a vector containing structural and functional genetic elements that are primarily derived from a lentivirus. In some embodiments, the lentiviral vector of the present invention is selected from the group consisting of HIV-1, HIV-2, SIV, FIV, EIAV, BIV, VISNA and CAEV vectors. In some embodiments, the lentiviral vector is a HIV-1 vector. The structure and composition of the vector genome used to prepare the retroviral vectors of the present invention are in accordance with those described in the art. Especially, minimum retroviral gene delivery vectors can be prepared from a vector genome, which only contains, apart from the recombinant nucleic acid molecule of the present invention, the sequences of the retroviral genome which are non-coding regions of said genome, necessary to provide recognition signals for DNA or RNA synthesis and processing. In some embodiment, the retroviral vector genome comprises all the elements necessary for the nucleic import and the correct expression of the polynucleotide of interest (i.e. the base editing platform). As examples of elements that can be inserted in the retroviral genome of the retroviral vector of the present invention are at least one (preferably two) long terminal repeats (LTR), such as a LTR5' and a LTR3', a psi sequence involved in the retroviral genome encapsidation, and optionally at least one DNA flap comprising a cPPT and a CTS domains. In some embodiments of the present invention, the LTR, preferably the LTR3', is deleted for the promoter and the enhancer of U3 and is replaced by a minimal promoter allowing transcription during vector production while an internal promoter is added to allow expression of the transgene. In particular, the vector is a Self-INactivating (SIN) vector that contains a non-functional or modified 3' Long Terminal Repeat (LTR) sequence. This sequence is copied to the 5' end of the vector genome during integration, resulting in the inactivation of promoter activity by both LTRs. Hence, a vector genome may be a replacement vector in which all the viral coding sequences between the 2 long terminal repeats (LTRs) have been replaced by the recombinant nucleic acid molecule of the present invention. In some embodiments, the retroviral vector genome is devoid of functional gag, pol and/or env retroviral genes. By "functional" it is meant a gene that is correctly transcribed, and/or correctly expressed. Thus, the retroviral vector genome of the present invention in this embodiment
contains at least one of the gag, pol and env genes that is either not transcribed or incompletely transcribed; the expression "incompletely transcribed" refers to the alteration in the transcripts gag, gag-pro or gag-pro-pol, one of these or several of these being not transcribed. In some embodiments, the retroviral genome is devoid of gag, pol and/or env retroviral genes. In some embodiments the retroviral vector genome is also devoid of the coding sequences for Vif-, Vpr-, Vpu- and Nef-accessory genes (for HIV-1 retroviral vectors), or of their complete or functional genes. Typically, the retroviral vector of the present invention is non replicative i.e., the vector and retroviral vector genome are not able to form new particles budding from the infected host cell. This may be achieved by the absence in the retroviral genome of the gag, pol or env genes, as indicated in the above paragraph; this can also be achieved by deleting other viral coding sequence(s) and/or cis-acting genetic elements needed for particles formation. Thus the present invention encompasses use of virus-like particles. As used herein, the term “virus-like particle” or “VLP” refers to a structure resembling a virus particle but devoid of the viral genome, incapable of replication and devoid of pathogenicity. The particle typically comprises at least one type of structural protein from a virus. Preferably only one type of structural protein is present. Most preferably no other non-structural component of a virus is present. Thus, virus-like particles can be spontaneously self-assembled by viral structural proteins under appropriate conditions in vitro while excluding the genetic material and potential replication probability. virus-like particles, with a diameter of approximately 20 to 150 nm, also have the characteristics of nanometer materials, such as large surface area, surface-accessible amino acids with reactive moieties (e.g., lysine and glutamic acid residues), inerratic spatial structure, and good biocompatibility. Therefore, assembled virus-like particles have great potential as a delivery system for specifically carrying a variety of cargos. In some embodiments, one or more of the zinc finger motifs of the Gag protein is/are substituted by one or more RNA-binding domain(s). In some embodiments, the RNA-binding domain is the Coat protein of the MS2 bacteriophage, of the PP7 phage or of the Q3 phage, the prophage HK022 Nun protein, the U1A protein or the hPum protein. More preferably, the RNA binding domain is the Coat protein of the MS2 bacteriophage or of the PP7 phage. Even more preferably the RNA-binding domain is the Coat protein of the MS2 bacteriophage. These embodiments are particularly suitable for packaging the mRNA encoding for the apelin polypeptide into the VLP.
Thus, in some embodiments, the mRNA encoding for the apelin polypeptide that is encapsuled in the virus particle of the present invention comprises at least one encapsidation sequence. By “encapsidation sequence” is meant an RNA motif (sequence and three-dimensional structure) recognized specifically by an RNA-binding domain as above described. Preferably, the encapsidation sequence is a stem-loop motif. Even more preferably, the encapsidation sequence of the retroviral particle is the stem-loop motif of the RNA of the MS2 bacteriophage or of the PP7 phage such as. The stem-loop motif and more particularly the stem-loop motif of the RNA of the MS2 bacteriophage or that of the RNA of the PP7 phage may be used alone or repeated several times, preferably from 2 to 25 times, more preferably from 2 to 18 times, for example from 6 to 18 times. In some embodiments, the present invention encompasses the use of the LentiFlash® technology that based on non-integrative lentiviral particles constructed using a bacteriophage coat protein and its cognate 19-nt stem loop, to replace the natural lentiviral Psi packaging sequence, in order to achieve active mRNA packaging into the lentiviral particles (Prel A, Caval V, Gayon R, Ravassard P, Duthoit C, Payen E, Maouche-Chretien L, Creneguy A, Nguyen TH, Martin N, Piver E, Sevrain R, Lamouroux L, Leboulch P, Deschaseaux F, Bouillé P, Sensébé L, Pagès JC. Highly efficient in vitro and in vivo delivery of functional RNAs using new versatile MS2-chimeric retrovirus-like particles. Mol Ther Methods Clin Dev.2015 Oct 21;2:15039. doi: 10.1038/mtm.2015.39. PMID: 26528487; PMCID: PMC4613645). The retroviral vectors of the present invention can be produced by any well-known method in the art including by transfection (s) transient (s), in stable cell lines and / or by means of helper virus. Use of stable cell lines may also be preferred for the production of the vectors (Greene, M. R. et al. Transduction of Human CD34 + Repopulating Cells with a Self-Inactivating Lentiviral Vector for SCID-X1 Produced at Clinical Scale by a Stable Cell Line. Hum. Gene Ther. Methods 23, 297–308 (2012).) For instance, the retroviral vector of the present invention is obtainable by a transcomplementation system (vector/packaging system) by transfecting in vitro a permissive cell (such as 293T cells) with a plasmid containing the retroviral vector genome of the present invention, and at least one other plasmid providing, in trans, the gag, pol and env sequences encoding the polypeptides GAG, POL and the envelope protein(s), or for a portion of these polypeptides sufficient to enable formation of retroviral particles. As an example, permissive cells are transfected with a) transcomplementation plasmid, lacking packaging signal psi and, the plasmid is optionally deleted of accessory genes vif, nef, vpu and / or vpr, b) a second plasmid (envelope expression plasmid or pseudotyping env plasmid) comprising a gene encoding an envelope protein(s) and c) a plasmid vector comprising a
recombinant genome retroviral, optionally deleted from the promoter region of the 3 'LTR or U3 enhancer sequence of the 3' LTR, including, between the LTR sequences 5 'and 3' retroviral, a psi encapsidation sequence, a nuclear export element (preferably RRE element of HIV or other retroviruses equivalent), comprising the nucleic acid molecule of the present invention and optionally a promoter and / or a nuclear import sequence (cPPT sequence eg CTS ) of the RNA. Advantageously, the three plasmids used do not contain homologous sequence sufficient for recombination. Nucleic acids encoding gag, pol and env cDNA can be advantageously prepared according to conventional techniques, from viral gene sequences available in the prior art and databases. The trans-complementation plasmid provides a nucleic acid encoding the proteins retroviral gag and pol. These proteins are derived from a lentivirus, and most preferably, from HIV-1. The plasmid is devoid of encapsidation sequence, sequence coding for an envelope, accessory genes, and advantageously also lacks retroviral LTRs. Therefore, the sequences coding for gag and pol proteins are advantageously placed under the control of a heterologous promoter, eg cellular, viral, etc.., which can be constitutive or regulated, weak or strong. It is preferably a plasmid containing a sequence transcomplement Δpsi-CMV-gag-pol- PolyA. This plasmid allows the expression of all the proteins necessary for the formation of empty virions, except the envelope glycoproteins. The plasmid transcomplementation may advantageously comprise the TAT and REV genes. Plasmid transcomplementation is advantageously devoid of vif, vpr, vpu and / or nef accessory genes. It is understood that the gag and pol genes and genes TAT and REV can also be carried by different plasmids, possibly separated. In this case, several plasmids are used transcomplementation, each encoding one or more of said proteins. The promoters used in the plasmid transcomplementation, the envelope plasmid and the plasmid vector respectively to promote the expression of gag and pol of the coat protein, the mRNA of the vector genome and the transgene are promoters identical or different, chosen advantageously from ubiquitous promoters or specific, for example, from viral promoters CMV, TK, RSV LTR promoter and the RNA polymerase III promoter such as U6 or H1 or promoters of helper viruses encoding env, gag and pol (i.e. adenoviral, baculoviral, herpes viruses). For the production of the retroviral vector of the present invention, the plasmids described above can be introduced into competent cells and viruses produced are harvested. The cells used may be any cell competent, particularly eukaryotic cells, in particular mammalian, eg human or animal. They can be somatic or embryonic stem or differentiated. Typically the cells include 293T cells, fibroblast cells, hepatocytes, muscle cells (skeletal, cardiac, smooth, blood vessel, etc.), nerve cells (neurons, glial cells, astrocytes) of epithelial cells, renal, ocular etc.. It may also include, insect, plant cells, yeast, or prokaryotic cells. It can
also be cells transformed by the SV40 T antigen. The genes gag, pol and env encoded in plasmids or helper viruses can be introduced into cells by any method known in the art, suitable for cell type considered. Usually, the cells and the vector system are contacted in a suitable device (plate, dish, tube, pouch, etc...), for a period of time sufficient to allow the transfer of the vector system or the plasmid in the cells. Typically, the vector system or the plasmid is introduced into the cells by calcium phosphate precipitation, electroporation, transduction or by using one of transfection-facilitating compounds, such as lipids, polymers, liposomes and peptides, etc.. The calcium phosphate precipitation is preferred. The cells are cultured in any suitable medium such as RPMI, DMEM, a specific medium to a culture in the absence of fetal calf serum, etc. Once transfected the retroviral vectors of the present invention may be purified from the supernatant of the cells. Purification of the retroviral vector to enhance the concentration can be accomplished by any suitable method, such as by density gradient purification (e.g., cesium chloride (CsCl)) or by chromatography techniques (e.g., column or batch chromatography). For example, the vector of the present invention can be subjected to two or three CsCl density gradient purification steps. The vector, is desirably purified from cells infected using a method that comprises lysing cells infected with adenovirus, applying the lysate to a chromatography resin, eluting the adenovirus from the chromatography resin, and collecting a fraction containing the retroviral vector of the present invention. In some embodiments, the vector of the present invention includes "control sequences", which refers collectively to promoter sequences, polyadenylation signals, transcription termination sequences, upstream regulatory domains, origins of replication, internal ribosome entry sites ("IRES"), enhancers, and the like, which collectively provide for the replication, transcription and translation of a coding sequence in a recipient cell. Not all of these control sequences need always be present so long as the selected coding sequence is capable of being replicated, transcribed and translated in an appropriate host cell. Another nucleic acid sequence, is a "promoter" sequence, which is used herein in its ordinary sense to refer to a nucleotide region comprising a DNA regulatory sequence, wherein the regulatory sequence is derived from a gene which is capable of binding RNA polymerase and initiating transcription of a downstream (3'- direction) coding sequence. Transcription promoters can include "inducible promoters" (where expression of a polynucleotide sequence operably linked to the promoter is induced by an analyte, cofactor, regulatory protein, etc.), "repressible promoters" (where expression of a polynucleotide sequence operably linked to the promoter is induced by an analyte, cofactor, regulatory protein, etc.), and "constitutive promoters”.
In some embodiments, the different components of the gene editing platform of the present invention are provided to the population of cells through the use of an RNA-encoded system. In particular, the base-editing system may be provided to the population of cells through the use of a chemically modified mRNA-encoded adenine or cytidine base editor together with modified guide RNA as described in Jiang, T., Henderson, J.M., Coote, K. et al. Chemical modifications of adenine base editor mRNA and guide RNA expand its application scope. Nat Commun 11, 1979 (2020). In particular, engineered RNA-encoded base-editing enzymes (e.g. ABE) system are prepared by introducing various chemical modifications to both mRNA that encoded the base-editing enzyme and guide RNA. In particular said modifications consist in uridine depleted mRNAs modified with 5-methoxyuridine: synonymous codons may be introduced to deplete uridines as much as possible without altering the coding sequence and replaced all the remaining uridines with 5-methoxyuridine. Said optimized base editing system exhibits higher editing efficiency at some genomic sites compared to DNA-encoded system. It is also possible to encapsulate the modified mRNA and guide RNA into lipid nanoparticle (LNP) for allowing lipid nanoparticle (LNP)-mediated delivery. Thus, in some embodiments, the mRNA is formulated using one or more lipid-based structures that include but are not limited to liposomes, lipoplexes, or lipid nanoparticles (Paunovska, Kalina, David Loughrey, and James E. Dahlman. "Drug delivery systems for RNA therapeutics." Nature Reviews Genetics (2022): 1-16). Liposomes are artificially-prepared vesicles which can primarily be composed of a lipid bilayer and can be used as a delivery vehicle for the administration of pharmaceutical formulations. Liposomes can be of different sizes such as, but not limited to, a multilamellar vesicle (MLV) which can be hundreds of nanometers in diameter and can contain a series of concentric bilayers separated by narrow aqueous compartments, a small unicellular vesicle (SUV) which can be smaller than 50 nm in diameter, and a large unilamellar vesicle (LUV) which can be between 50 and 500 nm in diameter. Liposome design can include, but is not limited to, opsonins or ligands in order to improve the attachment of liposomes to unhealthy tissue or to activate events such as, but not limited to, endocytosis. Liposomes can contain a low or a high pH in order to improve the delivery of the pharmaceutical formulations. As a non- limiting example, liposomes such as synthetic membrane vesicles are prepared by the methods, apparatus and devices described in US Patent Publication No. US20130177638, US20130177637, US20130177636, US20130177635, US20130177634, US20130177633, US20130183375, US20130183373 and US20130183372. In some embodiments, the liposomes are formed from 1,2-dioleyloxy-N,N-dimethylaminopropane (DODMA) liposomes, DiLa2
liposomes from Marina Biotech (Bothell, Wash.), 1,2-dilinoleyloxy-3-dimethylaminopropane (DLin-DMA), 2,2-dilinoleyl-4-(2-dimethylaminoethyl)-[1,3]-dioxolane (DLin-KC2-DMA), and MC3 (as described in US20100324120) and liposomes which can deliver small molecule drugs such as, but not limited to, DOXIL® from Janssen Biotech, Inc. (Horsham, Pa.). The mRNAs can be encapsulated by the liposome and/or it can be contained in an aqueous core which can then be encapsulated by the liposome (see International Pub. Nos. WO2012031046, WO2012031043, WO2012030901 and WO2012006378 and US Patent Publication No. US20130189351, US20130195969 and US20130202684). In some embodiments, the mRNA is formulated with stabilized plasmid-lipid particles (SPLP) or stabilized nucleic acid lipid particle (SNALP) that have been previously described and shown to be suitable for oligonucleotide delivery in vitro and in vivo (see Wheeler et al. Gene Therapy. 19996:271- 281; Zhang et al. Gene Therapy.19996:1438-1447; Jeffs et al. Pharm Res.200522:362-372; Morrissey et al., Nat Biotechnol. 2005 2:1002-1007; Zimmermann et al., Nature. 2006 441:111-114; Heyes et al. J Contr Rel.2005107:276-287; Semple et al. Nature Biotech.2010 28:172-176; Judge et al. J Clin Invest. 2009119:661-673; deFougerolles Hum Gene Ther. 200819:125-132; U.S. Patent Publication No US20130122104). Typically the vector of the present invention is combined with pharmaceutically acceptable excipients, and optionally sustained-release matrices, such as biodegradable polymers, to form pharmaceutical compositions. The term "Pharmaceutically" or "pharmaceutically acceptable" refers to molecular entities and compositions that do not produce an adverse, allergic or other untoward reaction when administered to a mammal, especially a human, as appropriate. A pharmaceutically acceptable carrier or excipient refers to a non-toxic solid, semi- solid or liquid filler, diluent, encapsulating material or formulation auxiliary of any type. The invention will be further illustrated by the following figures and examples. However, these examples and figures should not be interpreted in any way as limiting the scope of the present invention. FIGURES: Figure 1. Target the Pol II pause site in the C9orf72 gene using base editing. A) Schematic representation of the C9orf72 gene on chromosome 9, depicting C9orf72 exons and the region containing G4C2 hexanucleotides repeats and the location of the Pol II pause site
(chr9:27563356-27,563,366 – Hg18). Targets sequences of the sgRNAs are indicated with arrows and aligned with the DNA sequence that they bind to in a stranded-oriented way. B) PRO-seq or PRO-cap genome-wide methods map RNA polymerase active sites and transcription start sites, respectively, and allow to study the relationship between promoter- proximal pausing and the core promoter structure. PRO-seq or PRO-cap analyses performed by Watts and colleagues (Watts, Jason A., et al. "Cis elements that mediate RNA polymerase II pausing regulate human gene expression." The American Journal of Human Genetics 105.4 (2019): 677-688) identified a Pol II pause site in the C9orf72 gene (Cheung et al., personal communication). Our base of interest is located at position Chr9:27563356-27563366-Hg18). Figure 2. Plasmid-mediated and RNA-mediated base editing of the RNA Pol II pause site in K562 cells A) Experimental protocol used for base editing experiments in K562 cells. K562 cells co- transfected with a BE/GFP-expressing plasmid and a sgRNA-expressing plasmid.24 hours post transfection, GFP+ cells were sorted and cultured for 3 to 5 days before DNA extraction and PCR amplification of the target region. Amplicons were subjected to Sanger sequencing. RNA extraction was performed around 7 days post transfection and first-strand cDNAs were obtained by reverse transcription, which was then followed by qPCR. B) Experimental protocol used for base editing experiments in K562 cells. A BE/GFP- expressing mRNA and a sgRNA were co-transfected in K562 cells. Cells were cultured for 3 to 5 days before DNA extraction and PCR amplification of the target region. Amplicons were subjected to Sanger sequencing. RNA extraction was performed around 7 days post transfection and first-strand cDNAs were obtained by reverse transcription, which was then followed by qPCR. Figure 3. C to T conversion of the RNA Pol II pause site in K562 cells using plasmid transfection A) Frequency of C>T conversion in the C1 base bases (the C9orf72 Pol II pause site) in K562 and at the C2 (bystander C), as determined by Sanger sequencing followed by EditR analysis (Kluesner, Mitchell G., et al. "EditR: a method to quantify base editing from Sanger sequencing." The CRISPR journal 1.3 (2018): 239-250). Data are expressed as mean ± standard error of the mean (SEM) (n=3 biologically independent experiments). B) Frequency of InDels, measured by TIDE analysis (Brinkman, Eva K., et al. "Easy quantitative assessment of genome editing by sequence trace decomposition." Nucleic acids
research 42.22 (2014): e168-e168), for control (CTR, non-transfected cells; cells transfected with a CBE-expressing plasmid only), base-edited samples (C9orf72_2-4) and Cas9-treated samples subjected to Sanger sequencing. Data are expressed as mean ± SEM (n=3 biologically independent experiments). **** p<0.0001, (Ordinary One-way ANOVA, multiple comparison). Figure 4. Increase of C9orf72 transcripts upon targeting of the Pol II Pause site. RT-qPCR analysis of C9orf72 mRNA levels in transfected K562 cells. C9orf72 gene expression was normalized to α-globin mRNA and expressed as fold-change, as compared to control samples (CBE, cells transfected with a CBE-expressing plasmid only). The exon that was amplified and the sgRNA used are indicated on top of each graph. Data are expressed as mean ± SEM (n=3-4 biologically independent experiments). * p≤ 0.05, ns, not significant (Ordinary One-way ANOVA, multiple comparison). Figure 5. RNA-mediated base editing of the RNA Pol II pause site in K562 cells Frequency of C>T conversion in the C1 base (the C9orf72 Pol II pause site) in K562 and at the C2 (bystander C), as determined by Sanger sequencing followed by EditR analysis. Cells were transfected with CBE-SpRY-OPT mRNA, purchased from Trilink in combination with chemically modified C9orf72_1, C9orf72_2, C9orf72_3 and C9orf72_4 sgRNAs. Data are expressed as mean ± SEM (12±[4.3] for C9orf72_1, 12±[6] for C9orf72_2, 12.6±[1] for C9orf72_3, 23±[5.8] for C9orf72_4, (n=3 biologically independent experiments). EXAMPLE: EXAMPLE 1 Methods: Cell culture Human erythroleukemia cells (K562) were grown in RPMI 1640 medium supplemented with 10% dialyzed fetal bovine serum, 100 U/mL penicillin, 100 µg/mL streptomycin and 2 mM glutamine. Cells were maintained in a humidified atmosphere with 5% CO2 at 37ºC, with medium renewal every 2–3 days. Plasmids
Plasmid used in this study is the CBE-SpRY-OPT (used for in vitro mRNA production) that contains the uridine-depleted coding sequence of pCAG-CBE4max-SpRY-P2A-EGFP (RTW5133) (Addgene #139999), two copies of the 3’ untranslated region (UTR) of the HBB gene, a poly-A sequence of 96 A, and a T7 promoter followed by a G allowing efficient capping (Antoniou et al., under revision). sgRNA design We manually designed sgRNAs targeting the identified unique site of RNA Polymerase II pause site in the promoter region of C9orf72 (Table 1). To generate the sgRNA expression plasmid, oligonucleotides were annealed to create the gRNA protospacer and the duplexes were ligated into the Bbs I-digested MA128 plasmid (provided by M. Amendola, Genethon, France). For RNA-mediated base editing, we used chemically modified synthetic sgRNAs harboring 2′-O- methyl analogs and 3′-phosphorothioate nonhydrolyzable linkages at the first three 5′ and 3′ nucleotides (Synthego). Table 1. gRNA target sequences.
In particular, the amino acid sequence of CBE-SpRY is SEQ ID NO:20. SEQ ID NO:21> amino acid sequence of CBE-SpRY MKRTADGSEFESPKKKRKVSSETGPVAVDPTLRRRIEPHEFEVFFDPRELRKETCLLYEINWGGRHSIW RHTSQNTNKHVEVNFIEKFTTERYFCPNTRCSITWFLSWSPCGECSRAITEFLSRYPHVTLFIYIARLY HHADPRNRQGLRDLISSGVTIQIMTEQESGYCWRNFVNYSPSNEAHWPRYPHLWVRLYVLELYCIILGL PPCLNILRRKQPQLTFFTIALQSCHYQRLPPHILWATGLKSGGSSGGSSGSETPGTSESATPESSGGSS GGSDKKYSIGLAIGTNSVGWAVITDEYKVPSKKFKVLGNTDRHSIKKNLIGALLFDSGETAERTRLKRT ARRRYTRRKNRICYLQEIFSNEMAKVDDSFFHRLEESFLVEEDKKHERHPIFGNIVDEVAYHEKYPTIY HLRKKLVDSTDKADLRLIYLALAHMIKFRGHFLIEGDLNPDNSDVDKLFIQLVQTYNQLFEENPINASG VDAKAILSARLSKSRRLENLIAQLPGEKKNGLFGNLIALSLGLTPNFKSNFDLAEDAKLQLSKDTYDDD LDNLLAQIGDQYADLFLAAKNLSDAILLSDILRVNTEITKAPLSASMIKRYDEHHQDLTLLKALVRQQL PEKYKEIFFDQSKNGYAGYIDGGASQEEFYKFIKPILEKMDGTEELLVKLNREDLLRKQRTFDNGSIPH QIHLGELHAILRRQEDFYPFLKDNREKIEKILTFRIPYYVGPLARGNSRFAWMTRKSEETITPWNFEEV VDKGASAQSFIERMTNFDKNLPNEKVLPKHSLLYEYFTVYNELTKVKYVTEGMRKPAFLSGEQKKAIVD LLFKTNRKVTVKQLKEDYFKKIECFDSVEISGVEDRFNASLGTYHDLLKIIKDKDFLDNEENEDILEDI VLTLTLFEDREMIEERLKTYAHLFDDKVMKQLKRRRYTGWGRLSRKLINGIRDKQSGKTILDFLKSDGF ANRNFMQLIHDDSLTFKEDIQKAQVSGQGDSLHEHIANLAGSPAIKKGILQTVKVVDELVKVMGRHKPE NIVIEMARENQTTQKGQKNSRERMKRIEEGIKELGSQILKEHPVENTQLQNEKLYLYYLQNGRDMYVDQ ELDINRLSDYDVDHIVPQSFLKDDSIDNKVLTRSDKNRGKSDNVPSEEVVKKMKNYWRQLLNAKLITQR KFDNLTKAERGGLSELDKAGFIKRQLVETRQITKHVAQILDSRMNTKYDENDKLIREVKVITLKSKLVS DFRKDFQFYKVREINNYHHAHDAYLNAVVGTALIKKYPKLESEFVYGDYKVYDVRKMIAKSEQEIGKAT AKYFFYSNIMNFFKTEITLANGEIRKRPLIETNGETGEIVWDKGRDFATVRKVLSMPQVNIVKKTEVQT GGFSKESIRPKRNSDKLIARKKDWDPKKYGGFLWPTVAYSVLVVAKVEKGKSKKLKSVKELLGITIMER SSFEKNPIDFLEAKGYKEVKKDLIIKLPKYSLFELENGRKRMLASAKQLQKGNELALPSKYVNFLYLAS HYEKLKGSPEDNEQKQLFVEQHKHYLDEIIEQISEFSKRVILADANLDKVLSAYNKHRDKPIREQAENI IHLFTLTRLGAPRAFKYFDTTIDPKQYRSTKEVLDATLIHQSITGLYETRIDLSQLGGDSGGSGGSGGS TNLSDIIEKETGKQLVIQESILMLPEEVEEVIGNKPESDILVHTAYDESTDENVMLLTSDAPEYKPWAL VIQDSNGENKIKMLSGGSGGSGGSTNLSDIIEKETGKQLVIQESILMLPEEVEEVIGNKPESDILVHTA YDESTDENVMLLTSDAPEYKPWALVIQDSNGENKIKMLSGGSKRTADGSEFEPKKKRKVGGGGSGATNF SLLKQAGDVEENPGPMVSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKL PVPWPTLVTTLTYGVQCFSRYPDHMKQHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLVNR IELKGIDFKEDGNILGHKLEYNYNSHNVYIMADKQKNGIKVNFKIRHNIEDGSVQLADHYQQNTPIGDG PVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAAGITLGMDELYK The second component of the gene-editing platform disclosed herein consists of at least one guide RNA molecule suitable for guiding the base-editing enzyme to at least one target sequence located in the RNA Polymerase II pause site. The guide RNA molecule of the present invention thus comprises a guide sequence for providing the targeting specificity. It includes a region that is complementary and capable of hybridization to a pre-selected target site of interest in the RNA Polymerase II pause site. According to the present disclosure, the guide RNA targets the pause site that is located on chr9:27563356-27563366 region, and more preferably the nucleotide sequence SEQ ID NO:30 (GAAGAGGCGCGGGTAGAAGC). In some embodiment, this guide sequence can comprise from about 10 nucleotides to more than about 25 nucleotides. For example, the region of base pairing between the guide sequence and the corresponding target site sequence can be about 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20,
22, 23, 24, 25, or more than 25 nucleotides in length. In some embodiments, the guide sequence is about 17-20 nucleotides in length, such as 20 nucleotides. Typically, a software program is used to identify candidate CRISPR target sequences on both strands of the DNA nucleic acid molecule based on desired guide sequence length and a CRISPR motif sequence (PAM) for a specified CRISPR enzyme. One requirement for selecting a suitable target nucleic acid is that it has a 3′ PAM site/sequence. Each target sequence and its corresponding PAM site/sequence are referred herein as a Cas-targeted site. Type II CRISPR system, one of the most well characterized systems, needs only Cas 9 protein and a guide RNA complementary to a target sequence to affect target cleavage. For example, target sites for Cas9 from S. pyogenes, with PAM sequences NGG, may be identified by searching for 5′-Nx-NGG- 3′ both on the input sequence and on the reverse-complement of the input. Since multiple occurrences in the genome of the DNA target site may lead to nonspecific genome editing, after identifying all potential sites, the program filters out sequences based on the number of times they appear in the relevant reference genome. For those CRISPR enzymes for which sequence specificity is determined by a “seed” sequence, such as the 11-12 bp 5′ from the PAM sequence, including the PAM sequence itself, the filtering step may be based on the seed sequence. Thus, to avoid editing at additional genomic loci, results are filtered based on the number of occurrences of the seed:PAM sequence in the relevant genome. The user may be allowed to choose the length of the seed sequence. The user may also be allowed to specify the number of occurrences of the seed:PAM sequence in a genome for purposes of passing the filter. The default is to screen for unique sequences. Filtration level is altered by changing both the length of the seed sequence and the number of occurrences of the sequence in the genome. The program may in addition or alternatively provide the sequence of a guide sequence complementary to the reported target sequence(s) by providing the reverse complement of the identified target sequence(s). Further details of methods and algorithms to optimize sequence selection can be found in U.S. application Ser. No. 61/836,080; incorporated herein by reference. In some embodiments, the guide RNA targets a sequence selected from Table 1 (see EXAMPLE).
In some embodiments, the gene editing platform comprises a) a base-editing enzyme that is a CBE-SpRY and b) and at least one guide RNA molecule that targets one sequence selected in Table 1. The guide RNA molecule of the present invention can be made by various methods known in the art including cell-based expression, in vitro transcription, and chemical synthesis. The ability to chemically synthesize relatively long RNAs (as long as 200 mers or more) using TC- RNA chemistry (see, e.g., U.S. Pat. No.8,202,983) allows one to produce RNAs with special features that outperform those enabled by the basic four ribonucleotides (A, C, G and U). In particular, the RNA molecule of the present invention can be made with recombinant technology using a host cell system or an in vitro translation-transcription system known in the art. Details of such systems and technology can be found in e.g., WO2014144761 WO2014144592, WO2013176772, US20140273226, and US20140273233, the contents of which are incorporated herein by reference in their entireties. In some embodiments, the guide RNA molecule may include one or more modifications. Such modifications may include inclusion of at least one non-naturally occurring nucleotide, or a modified nucleotide, or analogs thereof. Modified nucleotides may be modified at the ribose, phosphate, and/or base moiety. Modified nucleotides may include 2’-O-methyl analogs, 2’- deoxy analogs, or 2’-fluoro analogs. The nucleic acid backbone may be modified, for example, a phosphorothioate backbone may be used. The use of locked nucleic acids (LNA) or bridged nucleic acids (BNA) may also be possible. Further examples of modified bases include, but are not limited to, 2-aminopurine, 5-bromo-uridine, pseudouridine, inosine, 7-methylguanosine. In some embodiments, a plurality of guide RNA molecules are designed for targeting a plurality of sequences in the RNA Polymerase II pause site. In some embodiments, the gene editing platform disclosed herein thus comprises 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, or 20 guide RNA molecules as disclosed herein. In some embodiments, a plurality of base-editing enzyme along with a plurality of guide RNA molecules are designed for targeting a plurality of sequences in the RNA Polymerase II pause site. In some embodiments, the gene editing platform disclosed herein thus comprises 2, 3 or 4 base-editing enzymes and 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, or 20 RNA molecules as disclosed herein.
In some embodiments, the different components of the gene editing platform of the present invention are administered to the patient through expression from one or more vectors. Examples of vectors include viral vector and non-viral vectors. As used herein, the term “viral vector” refers to a virion or virus particle that functions as a nucleic acid delivery vehicle and which comprises a vector genome packaged within the virion or virus particle. Typically, the vector is a viral vector which is an adeno-associated virus (AAV), a retroviral vector, bovine papilloma virus, an adenovirus vector, a vaccinia virus, or a polyoma virus. In some embodiments, the viral vector is a AAV vector. As used herein, the term "AAV vector" means a vector derived from an adeno- associated virus serotype, including without limitation, AAV1, AAV2, AAV3, AAV4, AAV5, AAV6, AAV7, AAV8, AAV9, and mutated forms thereof. AAV vectors can have one or more of the AAV wild-type genes deleted in whole or part, preferably the rep and/or cap genes, but retain functional flanking ITR sequences. In some embodiments, the viral vector is a retroviral vector. As used herein, the term “retroviral vector” refers to a vector containing structural and functional genetic elements that are primarily derived from a retrovirus. In some embodiments, the retroviral vector of the present invention derives from a retrovirus selected from the group consisting of alpharetroviruses (e.g., avian leukosis virus), betaretroviruses (e.g., mouse mammary tumor virus), gammaretroviruses (e.g., murine leukemia virus), deltaretroviruses (e.g., bovine leukemia virus), epsilonretroviruses (e.g., Walley dermal sarcoma virus), lentiviruses (e.g., HIV-1, HIV-2) and spumaviruses (e.g., human spumavirus). In some embodiments, the retroviral vector of the present invention is a replication deficient retroviral virus particle, which can transfer a foreign imported RNA of a gene instead of the retroviral mRNA.
In some embodiments, the retroviral vector of the present invention is a lentiviral vector. As used herein, the term “lentiviral vector” refers to a vector containing structural and functional genetic elements that are primarily derived from a lentivirus. In some embodiments, the lentiviral vector of the present invention is selected from the group consisting of HIV-1, HIV-2, SIV, FIV, EIAV, BIV, VISNA and CAEV vectors. In some embodiments, the lentiviral vector is a HIV-1 vector. The structure and composition of the vector genome used to prepare the retroviral vectors of the present invention are in accordance with those described in the art. Especially, minimum retroviral gene delivery vectors can be prepared from a vector genome, which only contains, apart from the recombinant nucleic acid molecule of the present invention, the sequences of the retroviral genome which are non-coding regions of said genome, necessary to provide recognition signals for DNA or RNA synthesis and processing. In some embodiment, the retroviral vector genome comprises all the elements necessary for the nucleic import and the correct expression of the polynucleotide of interest (i.e. the base editing platform). As examples of elements that can be inserted in the retroviral genome of the retroviral vector of the present invention are at least one (preferably two) long terminal repeats (LTR), such as a LTR5' and a LTR3', a psi sequence involved in the retroviral genome encapsidation, and optionally at least one DNA flap comprising a cPPT and a CTS domains. In some embodiments of the present invention, the LTR, preferably the LTR3', is deleted for the promoter and the enhancer of U3 and is replaced by a minimal promoter allowing transcription during vector production while an internal promoter is added to allow expression of the transgene. In particular, the vector is a Self-INactivating (SIN) vector that contains a non-functional or modified 3' Long Terminal Repeat (LTR) sequence. This sequence is copied to the 5' end of the vector genome during integration, resulting in the inactivation of promoter activity by both LTRs. Hence, a vector genome may be a replacement vector in which all the viral coding sequences between the 2 long terminal repeats (LTRs) have been replaced by the recombinant nucleic acid molecule of the present invention. In some embodiments, the retroviral vector genome is devoid of functional gag, pol and/or env retroviral genes. By "functional" it is meant a gene that is correctly transcribed, and/or correctly expressed. Thus, the retroviral vector genome of the present invention in this embodiment
contains at least one of the gag, pol and env genes that is either not transcribed or incompletely transcribed; the expression "incompletely transcribed" refers to the alteration in the transcripts gag, gag-pro or gag-pro-pol, one of these or several of these being not transcribed. In some embodiments, the retroviral genome is devoid of gag, pol and/or env retroviral genes. In some embodiments the retroviral vector genome is also devoid of the coding sequences for Vif-, Vpr-, Vpu- and Nef-accessory genes (for HIV-1 retroviral vectors), or of their complete or functional genes. Typically, the retroviral vector of the present invention is non replicative i.e., the vector and retroviral vector genome are not able to form new particles budding from the infected host cell. This may be achieved by the absence in the retroviral genome of the gag, pol or env genes, as indicated in the above paragraph; this can also be achieved by deleting other viral coding sequence(s) and/or cis-acting genetic elements needed for particles formation. Thus the present invention encompasses use of virus-like particles. As used herein, the term “virus-like particle” or “VLP” refers to a structure resembling a virus particle but devoid of the viral genome, incapable of replication and devoid of pathogenicity. The particle typically comprises at least one type of structural protein from a virus. Preferably only one type of structural protein is present. Most preferably no other non-structural component of a virus is present. Thus, virus-like particles can be spontaneously self-assembled by viral structural proteins under appropriate conditions in vitro while excluding the genetic material and potential replication probability. virus-like particles, with a diameter of approximately 20 to 150 nm, also have the characteristics of nanometer materials, such as large surface area, surface-accessible amino acids with reactive moieties (e.g., lysine and glutamic acid residues), inerratic spatial structure, and good biocompatibility. Therefore, assembled virus-like particles have great potential as a delivery system for specifically carrying a variety of cargos. In some embodiments, one or more of the zinc finger motifs of the Gag protein is/are substituted by one or more RNA-binding domain(s). In some embodiments, the RNA-binding domain is the Coat protein of the MS2 bacteriophage, of the PP7 phage or of the Q3 phage, the prophage HK022 Nun protein, the U1A protein or the hPum protein. More preferably, the RNA binding domain is the Coat protein of the MS2 bacteriophage or of the PP7 phage. Even more preferably the RNA-binding domain is the Coat protein of the MS2 bacteriophage. These embodiments are particularly suitable for packaging the mRNA encoding for the apelin polypeptide into the VLP.
Thus, in some embodiments, the mRNA encoding for the apelin polypeptide that is encapsuled in the virus particle of the present invention comprises at least one encapsidation sequence. By “encapsidation sequence” is meant an RNA motif (sequence and three-dimensional structure) recognized specifically by an RNA-binding domain as above described. Preferably, the encapsidation sequence is a stem-loop motif. Even more preferably, the encapsidation sequence of the retroviral particle is the stem-loop motif of the RNA of the MS2 bacteriophage or of the PP7 phage such as. The stem-loop motif and more particularly the stem-loop motif of the RNA of the MS2 bacteriophage or that of the RNA of the PP7 phage may be used alone or repeated several times, preferably from 2 to 25 times, more preferably from 2 to 18 times, for example from 6 to 18 times. In some embodiments, the present invention encompasses the use of the LentiFlash® technology that based on non-integrative lentiviral particles constructed using a bacteriophage coat protein and its cognate 19-nt stem loop, to replace the natural lentiviral Psi packaging sequence, in order to achieve active mRNA packaging into the lentiviral particles (Prel A, Caval V, Gayon R, Ravassard P, Duthoit C, Payen E, Maouche-Chretien L, Creneguy A, Nguyen TH, Martin N, Piver E, Sevrain R, Lamouroux L, Leboulch P, Deschaseaux F, Bouillé P, Sensébé L, Pagès JC. Highly efficient in vitro and in vivo delivery of functional RNAs using new versatile MS2-chimeric retrovirus-like particles. Mol Ther Methods Clin Dev.2015 Oct 21;2:15039. doi: 10.1038/mtm.2015.39. PMID: 26528487; PMCID: PMC4613645). The retroviral vectors of the present invention can be produced by any well-known method in the art including by transfection (s) transient (s), in stable cell lines and / or by means of helper virus. Use of stable cell lines may also be preferred for the production of the vectors (Greene, M. R. et al. Transduction of Human CD34 + Repopulating Cells with a Self-Inactivating Lentiviral Vector for SCID-X1 Produced at Clinical Scale by a Stable Cell Line. Hum. Gene Ther. Methods 23, 297–308 (2012).) For instance, the retroviral vector of the present invention is obtainable by a transcomplementation system (vector/packaging system) by transfecting in vitro a permissive cell (such as 293T cells) with a plasmid containing the retroviral vector genome of the present invention, and at least one other plasmid providing, in trans, the gag, pol and env sequences encoding the polypeptides GAG, POL and the envelope protein(s), or for a portion of these polypeptides sufficient to enable formation of retroviral particles. As an example, permissive cells are transfected with a) transcomplementation plasmid, lacking packaging signal psi and, the plasmid is optionally deleted of accessory genes vif, nef, vpu and / or vpr, b) a second plasmid (envelope expression plasmid or pseudotyping env plasmid) comprising a gene encoding an envelope protein(s) and c) a plasmid vector comprising a
recombinant genome retroviral, optionally deleted from the promoter region of the 3 'LTR or U3 enhancer sequence of the 3' LTR, including, between the LTR sequences 5 'and 3' retroviral, a psi encapsidation sequence, a nuclear export element (preferably RRE element of HIV or other retroviruses equivalent), comprising the nucleic acid molecule of the present invention and optionally a promoter and / or a nuclear import sequence (cPPT sequence eg CTS ) of the RNA. Advantageously, the three plasmids used do not contain homologous sequence sufficient for recombination. Nucleic acids encoding gag, pol and env cDNA can be advantageously prepared according to conventional techniques, from viral gene sequences available in the prior art and databases. The trans-complementation plasmid provides a nucleic acid encoding the proteins retroviral gag and pol. These proteins are derived from a lentivirus, and most preferably, from HIV-1. The plasmid is devoid of encapsidation sequence, sequence coding for an envelope, accessory genes, and advantageously also lacks retroviral LTRs. Therefore, the sequences coding for gag and pol proteins are advantageously placed under the control of a heterologous promoter, eg cellular, viral, etc.., which can be constitutive or regulated, weak or strong. It is preferably a plasmid containing a sequence transcomplement Δpsi-CMV-gag-pol- PolyA. This plasmid allows the expression of all the proteins necessary for the formation of empty virions, except the envelope glycoproteins. The plasmid transcomplementation may advantageously comprise the TAT and REV genes. Plasmid transcomplementation is advantageously devoid of vif, vpr, vpu and / or nef accessory genes. It is understood that the gag and pol genes and genes TAT and REV can also be carried by different plasmids, possibly separated. In this case, several plasmids are used transcomplementation, each encoding one or more of said proteins. The promoters used in the plasmid transcomplementation, the envelope plasmid and the plasmid vector respectively to promote the expression of gag and pol of the coat protein, the mRNA of the vector genome and the transgene are promoters identical or different, chosen advantageously from ubiquitous promoters or specific, for example, from viral promoters CMV, TK, RSV LTR promoter and the RNA polymerase III promoter such as U6 or H1 or promoters of helper viruses encoding env, gag and pol (i.e. adenoviral, baculoviral, herpes viruses). For the production of the retroviral vector of the present invention, the plasmids described above can be introduced into competent cells and viruses produced are harvested. The cells used may be any cell competent, particularly eukaryotic cells, in particular mammalian, eg human or animal. They can be somatic or embryonic stem or differentiated. Typically the cells include 293T cells, fibroblast cells, hepatocytes, muscle cells (skeletal, cardiac, smooth, blood vessel, etc.), nerve cells (neurons, glial cells, astrocytes) of epithelial cells, renal, ocular etc.. It may also include, insect, plant cells, yeast, or prokaryotic cells. It can
also be cells transformed by the SV40 T antigen. The genes gag, pol and env encoded in plasmids or helper viruses can be introduced into cells by any method known in the art, suitable for cell type considered. Usually, the cells and the vector system are contacted in a suitable device (plate, dish, tube, pouch, etc...), for a period of time sufficient to allow the transfer of the vector system or the plasmid in the cells. Typically, the vector system or the plasmid is introduced into the cells by calcium phosphate precipitation, electroporation, transduction or by using one of transfection-facilitating compounds, such as lipids, polymers, liposomes and peptides, etc.. The calcium phosphate precipitation is preferred. The cells are cultured in any suitable medium such as RPMI, DMEM, a specific medium to a culture in the absence of fetal calf serum, etc. Once transfected the retroviral vectors of the present invention may be purified from the supernatant of the cells. Purification of the retroviral vector to enhance the concentration can be accomplished by any suitable method, such as by density gradient purification (e.g., cesium chloride (CsCl)) or by chromatography techniques (e.g., column or batch chromatography). For example, the vector of the present invention can be subjected to two or three CsCl density gradient purification steps. The vector, is desirably purified from cells infected using a method that comprises lysing cells infected with adenovirus, applying the lysate to a chromatography resin, eluting the adenovirus from the chromatography resin, and collecting a fraction containing the retroviral vector of the present invention. In some embodiments, the vector of the present invention includes "control sequences", which refers collectively to promoter sequences, polyadenylation signals, transcription termination sequences, upstream regulatory domains, origins of replication, internal ribosome entry sites ("IRES"), enhancers, and the like, which collectively provide for the replication, transcription and translation of a coding sequence in a recipient cell. Not all of these control sequences need always be present so long as the selected coding sequence is capable of being replicated, transcribed and translated in an appropriate host cell. Another nucleic acid sequence, is a "promoter" sequence, which is used herein in its ordinary sense to refer to a nucleotide region comprising a DNA regulatory sequence, wherein the regulatory sequence is derived from a gene which is capable of binding RNA polymerase and initiating transcription of a downstream (3'- direction) coding sequence. Transcription promoters can include "inducible promoters" (where expression of a polynucleotide sequence operably linked to the promoter is induced by an analyte, cofactor, regulatory protein, etc.), "repressible promoters" (where expression of a polynucleotide sequence operably linked to the promoter is induced by an analyte, cofactor, regulatory protein, etc.), and "constitutive promoters”.
In some embodiments, the different components of the gene editing platform of the present invention are provided to the population of cells through the use of an RNA-encoded system. In particular, the base-editing system may be provided to the population of cells through the use of a chemically modified mRNA-encoded adenine or cytidine base editor together with modified guide RNA as described in Jiang, T., Henderson, J.M., Coote, K. et al. Chemical modifications of adenine base editor mRNA and guide RNA expand its application scope. Nat Commun 11, 1979 (2020). In particular, engineered RNA-encoded base-editing enzymes (e.g. ABE) system are prepared by introducing various chemical modifications to both mRNA that encoded the base-editing enzyme and guide RNA. In particular said modifications consist in uridine depleted mRNAs modified with 5-methoxyuridine: synonymous codons may be introduced to deplete uridines as much as possible without altering the coding sequence and replaced all the remaining uridines with 5-methoxyuridine. Said optimized base editing system exhibits higher editing efficiency at some genomic sites compared to DNA-encoded system. It is also possible to encapsulate the modified mRNA and guide RNA into lipid nanoparticle (LNP) for allowing lipid nanoparticle (LNP)-mediated delivery. Thus, in some embodiments, the mRNA is formulated using one or more lipid-based structures that include but are not limited to liposomes, lipoplexes, or lipid nanoparticles (Paunovska, Kalina, David Loughrey, and James E. Dahlman. "Drug delivery systems for RNA therapeutics." Nature Reviews Genetics (2022): 1-16). Liposomes are artificially-prepared vesicles which can primarily be composed of a lipid bilayer and can be used as a delivery vehicle for the administration of pharmaceutical formulations. Liposomes can be of different sizes such as, but not limited to, a multilamellar vesicle (MLV) which can be hundreds of nanometers in diameter and can contain a series of concentric bilayers separated by narrow aqueous compartments, a small unicellular vesicle (SUV) which can be smaller than 50 nm in diameter, and a large unilamellar vesicle (LUV) which can be between 50 and 500 nm in diameter. Liposome design can include, but is not limited to, opsonins or ligands in order to improve the attachment of liposomes to unhealthy tissue or to activate events such as, but not limited to, endocytosis. Liposomes can contain a low or a high pH in order to improve the delivery of the pharmaceutical formulations. As a non- limiting example, liposomes such as synthetic membrane vesicles are prepared by the methods, apparatus and devices described in US Patent Publication No. US20130177638, US20130177637, US20130177636, US20130177635, US20130177634, US20130177633, US20130183375, US20130183373 and US20130183372. In some embodiments, the liposomes are formed from 1,2-dioleyloxy-N,N-dimethylaminopropane (DODMA) liposomes, DiLa2
liposomes from Marina Biotech (Bothell, Wash.), 1,2-dilinoleyloxy-3-dimethylaminopropane (DLin-DMA), 2,2-dilinoleyl-4-(2-dimethylaminoethyl)-[1,3]-dioxolane (DLin-KC2-DMA), and MC3 (as described in US20100324120) and liposomes which can deliver small molecule drugs such as, but not limited to, DOXIL® from Janssen Biotech, Inc. (Horsham, Pa.). The mRNAs can be encapsulated by the liposome and/or it can be contained in an aqueous core which can then be encapsulated by the liposome (see International Pub. Nos. WO2012031046, WO2012031043, WO2012030901 and WO2012006378 and US Patent Publication No. US20130189351, US20130195969 and US20130202684). In some embodiments, the mRNA is formulated with stabilized plasmid-lipid particles (SPLP) or stabilized nucleic acid lipid particle (SNALP) that have been previously described and shown to be suitable for oligonucleotide delivery in vitro and in vivo (see Wheeler et al. Gene Therapy. 19996:271- 281; Zhang et al. Gene Therapy.19996:1438-1447; Jeffs et al. Pharm Res.200522:362-372; Morrissey et al., Nat Biotechnol. 2005 2:1002-1007; Zimmermann et al., Nature. 2006 441:111-114; Heyes et al. J Contr Rel.2005107:276-287; Semple et al. Nature Biotech.2010 28:172-176; Judge et al. J Clin Invest. 2009119:661-673; deFougerolles Hum Gene Ther. 200819:125-132; U.S. Patent Publication No US20130122104). Typically the vector of the present invention is combined with pharmaceutically acceptable excipients, and optionally sustained-release matrices, such as biodegradable polymers, to form pharmaceutical compositions. The term "Pharmaceutically" or "pharmaceutically acceptable" refers to molecular entities and compositions that do not produce an adverse, allergic or other untoward reaction when administered to a mammal, especially a human, as appropriate. A pharmaceutically acceptable carrier or excipient refers to a non-toxic solid, semi- solid or liquid filler, diluent, encapsulating material or formulation auxiliary of any type. The invention will be further illustrated by the following figures and examples. However, these examples and figures should not be interpreted in any way as limiting the scope of the present invention. FIGURES: Figure 1. Target the Pol II pause site in the C9orf72 gene using base editing. A) Schematic representation of the C9orf72 gene on chromosome 9, depicting C9orf72 exons and the region containing G4C2 hexanucleotides repeats and the location of the Pol II pause site
(chr9:27563356-27,563,366 – Hg18). Targets sequences of the sgRNAs are indicated with arrows and aligned with the DNA sequence that they bind to in a stranded-oriented way. B) PRO-seq or PRO-cap genome-wide methods map RNA polymerase active sites and transcription start sites, respectively, and allow to study the relationship between promoter- proximal pausing and the core promoter structure. PRO-seq or PRO-cap analyses performed by Watts and colleagues (Watts, Jason A., et al. "Cis elements that mediate RNA polymerase II pausing regulate human gene expression." The American Journal of Human Genetics 105.4 (2019): 677-688) identified a Pol II pause site in the C9orf72 gene (Cheung et al., personal communication). Our base of interest is located at position Chr9:27563356-27563366-Hg18). Figure 2. Plasmid-mediated and RNA-mediated base editing of the RNA Pol II pause site in K562 cells A) Experimental protocol used for base editing experiments in K562 cells. K562 cells co- transfected with a BE/GFP-expressing plasmid and a sgRNA-expressing plasmid.24 hours post transfection, GFP+ cells were sorted and cultured for 3 to 5 days before DNA extraction and PCR amplification of the target region. Amplicons were subjected to Sanger sequencing. RNA extraction was performed around 7 days post transfection and first-strand cDNAs were obtained by reverse transcription, which was then followed by qPCR. B) Experimental protocol used for base editing experiments in K562 cells. A BE/GFP- expressing mRNA and a sgRNA were co-transfected in K562 cells. Cells were cultured for 3 to 5 days before DNA extraction and PCR amplification of the target region. Amplicons were subjected to Sanger sequencing. RNA extraction was performed around 7 days post transfection and first-strand cDNAs were obtained by reverse transcription, which was then followed by qPCR. Figure 3. C to T conversion of the RNA Pol II pause site in K562 cells using plasmid transfection A) Frequency of C>T conversion in the C1 base bases (the C9orf72 Pol II pause site) in K562 and at the C2 (bystander C), as determined by Sanger sequencing followed by EditR analysis (Kluesner, Mitchell G., et al. "EditR: a method to quantify base editing from Sanger sequencing." The CRISPR journal 1.3 (2018): 239-250). Data are expressed as mean ± standard error of the mean (SEM) (n=3 biologically independent experiments). B) Frequency of InDels, measured by TIDE analysis (Brinkman, Eva K., et al. "Easy quantitative assessment of genome editing by sequence trace decomposition." Nucleic acids
research 42.22 (2014): e168-e168), for control (CTR, non-transfected cells; cells transfected with a CBE-expressing plasmid only), base-edited samples (C9orf72_2-4) and Cas9-treated samples subjected to Sanger sequencing. Data are expressed as mean ± SEM (n=3 biologically independent experiments). **** p<0.0001, (Ordinary One-way ANOVA, multiple comparison). Figure 4. Increase of C9orf72 transcripts upon targeting of the Pol II Pause site. RT-qPCR analysis of C9orf72 mRNA levels in transfected K562 cells. C9orf72 gene expression was normalized to α-globin mRNA and expressed as fold-change, as compared to control samples (CBE, cells transfected with a CBE-expressing plasmid only). The exon that was amplified and the sgRNA used are indicated on top of each graph. Data are expressed as mean ± SEM (n=3-4 biologically independent experiments). * p≤ 0.05, ns, not significant (Ordinary One-way ANOVA, multiple comparison). Figure 5. RNA-mediated base editing of the RNA Pol II pause site in K562 cells Frequency of C>T conversion in the C1 base (the C9orf72 Pol II pause site) in K562 and at the C2 (bystander C), as determined by Sanger sequencing followed by EditR analysis. Cells were transfected with CBE-SpRY-OPT mRNA, purchased from Trilink in combination with chemically modified C9orf72_1, C9orf72_2, C9orf72_3 and C9orf72_4 sgRNAs. Data are expressed as mean ± SEM (12±[4.3] for C9orf72_1, 12±[6] for C9orf72_2, 12.6±[1] for C9orf72_3, 23±[5.8] for C9orf72_4, (n=3 biologically independent experiments). EXAMPLE: EXAMPLE 1 Methods: Cell culture Human erythroleukemia cells (K562) were grown in RPMI 1640 medium supplemented with 10% dialyzed fetal bovine serum, 100 U/mL penicillin, 100 µg/mL streptomycin and 2 mM glutamine. Cells were maintained in a humidified atmosphere with 5% CO2 at 37ºC, with medium renewal every 2–3 days. Plasmids
Plasmid used in this study is the CBE-SpRY-OPT (used for in vitro mRNA production) that contains the uridine-depleted coding sequence of pCAG-CBE4max-SpRY-P2A-EGFP (RTW5133) (Addgene #139999), two copies of the 3’ untranslated region (UTR) of the HBB gene, a poly-A sequence of 96 A, and a T7 promoter followed by a G allowing efficient capping (Antoniou et al., under revision). sgRNA design We manually designed sgRNAs targeting the identified unique site of RNA Polymerase II pause site in the promoter region of C9orf72 (Table 1). To generate the sgRNA expression plasmid, oligonucleotides were annealed to create the gRNA protospacer and the duplexes were ligated into the Bbs I-digested MA128 plasmid (provided by M. Amendola, Genethon, France). For RNA-mediated base editing, we used chemically modified synthetic sgRNAs harboring 2′-O- methyl analogs and 3′-phosphorothioate nonhydrolyzable linkages at the first three 5′ and 3′ nucleotides (Synthego). Table 1. gRNA target sequences.
 mRNA in vitro transcription CBE-SpRY-OPT mRNA, containing 5-methoxyuridine, capped with Cap1 analog, and subjected to silica membrane purification, was purchased from Trilink.
Plasmid transfection 2x106 K562 cells per condition were transfected with 3 ug of the base editor CBE and 1 µg of sgRNA of interest (C9orf72_2, C9orf72_3 and C9orf72_4). We used the Cell Line NucleofectorTM Kit V VCA-1003 for K562 cells and the U16 program of Nucleofector 2B. Cells transfected with TE buffer or with the base editor-expressing plasmid only served as negative controls. RNA transfection 2x105 K562 cells per condition were transfected with 3.0 μg of the enzyme encoding mRNA, and 3.2 µg of synthetic sgRNA (C9orf72_1, C9orf72_2, C9orf72_3 and C9orf72_4). We used the P3 Primary Cell 4D-Nucleofector X Kit S (Lonza) and the CA137 program (Nucleofector 4D). Cells transfected with TE buffer or with the enzyme-encoding mRNA only served as negative controls. Evaluation of editing efficiency Genomic DNA was extracted from control and edited cells using PURE LINK Genomic DNA Mini kit (LifeTechnologies), following manufacturers’ instructions. To evaluate base editing efficiency at sgRNA target sites, we performed PCR (using primers listed in Table 2) followed by Sanger sequencing and EditR analysis (Kluesner, Mitchell G., et al. "EditR: a method to quantify base editing from Sanger sequencing." The CRISPR journal 1.3 (2018): 239-250). Frequency of InDels was measured by TIDE analysis of Sanger sequencing data, for control and base edited samples (Brinkman, Eva K., et al. "Easy quantitative assessment of genome editing by sequence trace decomposition." Nucleic acids research 42.22 (2014): e168-e168). Table 2. Primers used to detect base editing and InDel events.
mRNA in vitro transcription CBE-SpRY-OPT mRNA, containing 5-methoxyuridine, capped with Cap1 analog, and subjected to silica membrane purification, was purchased from Trilink.
Plasmid transfection 2x106 K562 cells per condition were transfected with 3 ug of the base editor CBE and 1 µg of sgRNA of interest (C9orf72_2, C9orf72_3 and C9orf72_4). We used the Cell Line NucleofectorTM Kit V VCA-1003 for K562 cells and the U16 program of Nucleofector 2B. Cells transfected with TE buffer or with the base editor-expressing plasmid only served as negative controls. RNA transfection 2x105 K562 cells per condition were transfected with 3.0 μg of the enzyme encoding mRNA, and 3.2 µg of synthetic sgRNA (C9orf72_1, C9orf72_2, C9orf72_3 and C9orf72_4). We used the P3 Primary Cell 4D-Nucleofector X Kit S (Lonza) and the CA137 program (Nucleofector 4D). Cells transfected with TE buffer or with the enzyme-encoding mRNA only served as negative controls. Evaluation of editing efficiency Genomic DNA was extracted from control and edited cells using PURE LINK Genomic DNA Mini kit (LifeTechnologies), following manufacturers’ instructions. To evaluate base editing efficiency at sgRNA target sites, we performed PCR (using primers listed in Table 2) followed by Sanger sequencing and EditR analysis (Kluesner, Mitchell G., et al. "EditR: a method to quantify base editing from Sanger sequencing." The CRISPR journal 1.3 (2018): 239-250). Frequency of InDels was measured by TIDE analysis of Sanger sequencing data, for control and base edited samples (Brinkman, Eva K., et al. "Easy quantitative assessment of genome editing by sequence trace decomposition." Nucleic acids research 42.22 (2014): e168-e168). Table 2. Primers used to detect base editing and InDel events.
 F, forward primer; R, reverse primer. RT-qPCR
Total RNA was isolated from 106 K562 cells using TRIzol Reagent (Sigma) according to the manufacturer's protocol. RNA was treated with DNase using the DNase I kit (Invitrogen), following manufacturer’s instructions. First - strand cDNAs were obtained by reverse transcription of 1 μg of total RNA using All-in-One cDNA Synthesis SuperMix according to the manufacturer's instructions. Quantitative PCR amplification was performed using primers listed in Table 3, BlasTaqTM2X qPCR master mix (abm) and the Viia7 Real-Time PCR system (ThermoFisher Scientific), or the CFX384 Touch Real-Time PCR Detection System (Biorad). Data were analyzed transforming raw Cq values into relative quantification data using the delta delta Cq method. Table 3. Primers used for RT-qPCR.
F, forward primer; R, reverse primer. RT-qPCR
Total RNA was isolated from 106 K562 cells using TRIzol Reagent (Sigma) according to the manufacturer's protocol. RNA was treated with DNase using the DNase I kit (Invitrogen), following manufacturer’s instructions. First - strand cDNAs were obtained by reverse transcription of 1 μg of total RNA using All-in-One cDNA Synthesis SuperMix according to the manufacturer's instructions. Quantitative PCR amplification was performed using primers listed in Table 3, BlasTaqTM2X qPCR master mix (abm) and the Viia7 Real-Time PCR system (ThermoFisher Scientific), or the CFX384 Touch Real-Time PCR Detection System (Biorad). Data were analyzed transforming raw Cq values into relative quantification data using the delta delta Cq method. Table 3. Primers used for RT-qPCR.
 F = Forward primer, R = Reverse primer Fluorescence activated cell sorting (FACS) and flow cytometry analysis Cells were sorted using FACSAria II (BD Biosciences) or SH800 Cell Sorter (Sony Biotechnology). Flow cytometry analysis was performed using Gallios (Beckman coulter). Sorted and unsorted populations were cultured at a concentration of 5 × 105/ml in a RPMI K562 medium for 4/5 days before collection for DNA extraction. Data were analyzed using the FlowJo (BD Biosciences) software. Results: Plasmid-mediated base editing in K562 cells in the C9orf72 Pol II pause site To boost C9orf72 gene expression in K562 cells, we targeted the RNA polymerase II (Pol II) pause site in the first intronic region of the gene C9orf72 (Figure 1A and B). RNA Pol II pausing is a key regulatory step in transcription. RNA Pol II pauses at GC-rich regions, and almost 65% of the pause sites are cytosines (Watts, Jason A., et al. "Cis elements that mediate
RNA polymerase II pausing regulate human gene expression." The American Journal of Human Genetics 105.4 (2019): 677-688). Gene expression analysis showed that genes with more paused polymerases have lower expression levels. Therefore, mutagenesis of the pause sites might lead to a significant increase in transcription. As our base of interest is a C, we used the CBE-SpRY enzyme (allowing C to T conversions) (Walton, Russell T., et al. "Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants." Science 368.6488 (2020): 290-296), given also its PAM-less nature that allowed us to design sgRNAs by placing the base editing window within different strands and positions of the Pol II pause site (Figure 1A). The CBE-SpRY-expressing plasmid was transfected in K562 cells in combination with individual sgRNA-expressing plasmids (C9orf72_2, C9orf72_3 and C9orf72_4) targeting the Pol II site (Figure 1 and Figure 2A). 24 hours post transfection, K562 were sorted and the GFP+ cells were grown for 3 to 5 days before performing DNA extraction and Sanger sequencing of the PCR-amplified target region, followed by EditR analysis. CBE-SpRY coupled with C9orf72_2 sgRNA led to high C>T conversion with efficiencies of 47% ± [5.2], n=3) for the target base C2 (Figure 3A). CBE-SpRY coupled with C9orf72_3 and C9orf72_4 sgRNAs showed also efficient conversion of the target base C1, but also of C2, located within the base editing window (bystander edit) (Figure 3A). In fact, CBE-SpRY coupled with C9orf72_3 sgRNA resulted in a C>T conversion of 35% of the C1 and 15% of bystander C2 (25.5 ±10.5, n=3), and the C9orf72_4 sgRNA to 25% for the C1 and 18% for the bystander C2 (25 ± [5.5]; n=3) (Figure 3A). TIDE analysis confirmed the DSB-free nature of base editors, as we detected low frequencies of InDels in base edited samples compared to Cas9 nuclease used in combination with the C9orf72_2, C9orf72_3 or C9orf72_4 sgRNAs (Figure 3B). In conclusion, we were able to efficiently target the Pol II pause site and convert the target base without causing DSBs in K562 cells. Base editing-mediated C to T conversion increases C9orf72 gene transcript levels upon targeting of the Pol II pause site
C9orf72 transcript levels were significantly increased in K562 cells upon targeting of the Pol II pause site when transfection was performed with the C9orf72_2 sgRNA coupled with CBE SpRY compared to control cells (Figure 4). A similar trend was observed when using the C9orf72_3 or C9orf72_4 sgRNAs. As expected, C9orf72 transcript levels were significantly increased when using the C9orf72_2 sgRNA that in fact showed the highest percentage of C to T conversion and precision amongst the three sgRNAs tested in this study. RNA-mediated base editing in K562 cells in the C9orf72 Pol II pause site To establish a clinically relevant method to deliver the base editing system in K562 cells and achieve a higher editing efficiency coupled with minimal toxicity compared to the plasmid transfection, we used a protocol based on transfection of mRNA encoding base editors and chemically modified synthetic sgRNAs (Figure 2B). In vitro transcribed CBE-SpRY-OPT mRNAs were transfected in K562 cells in combination with chemically modified sgRNA C9orf72_1, C9orf72_2, C9orf72_3 or C9orf72_4lls were kept in culture for 3 to 5 days before DNA extraction and Sanger sequencing of the PCR- amplified target region. EditR analysis showed that all the sgRNA were able to modify the target base with efficiencies ranging from 9% (C9orf72_1) to 27% (C9orf72_4) with a variable efficiency of bystander C2 within the editing window, upon RNA-mediated delivery of the base editing system (Figure 5). EXAMPLE 2: Experimentation on patient-derived motor neurons Generation of iPSC-derived motor neurons Origin of the cells: A control iPSC line generated by the Stem Cell facility at the host institute (Institut Imagine cell line #Ctr004) was used as a starting material for all the motor neuron derivation studies. iPSC colonies were grown in mTeSR medium in vitronectin (both StemCell) coated dishes and were passaged by enzyme - free dissociation (ReleSR, StemCell) according to the manufacturer’s instructions. iMN differentiation:
To generate motor neurons, we have adapted a published protocol (Maury et al., 2015). Briefly, on day 0, iPSC colonies were dissociated to single cells using Accutase (Life Technologies) and seeded in ultra- low attachment 6- well plates (3 × 105 cells/well) or cell - repellent surface T25 flasks (3 × 105 cells/flask) in N2B27 medium. N2B27 medium consists of a 1:1 ratio of advanced DMEM/F12 (Life Technologies) and Neurobasal medium (Life Technologies), 1% N2 supplement (Life Technologies), 2% B27 supplement minus vitamin A (Life Technologies), 5 mM Glutamax (Life Technologies), 5mM L- Glutamine (Sigma), 1% penicillin/streptomycin (Life Technologies) and 0,1 mM 2 - ME (Life Technologies). In these conditions, iPSCs spontaneously aggregated into embryoid bodies (EBs) in suspension in N2B27 medium. To induce proper differentiation of these iPSCs into motor neurons, the medium was changed at specific days and different factors were added each time. On day 0, was added N2B27 medium + 10 μM ROCK inhibitor Y - 27632, 3 μM CHIR99021 (TOCRIS), 0, 1 μM LDN (TOCRIS), 20 μM SB431542 (TOCRIS) and 10 μM ascorbic acid (Sigma - Aldrich). On day 2, the medium was changed to N2B27 + 3 μM CHIR99021, 0,1 μM LDN, 20 μM 8SB431542 and 100 nM retinoic acid (Sigma - Aldrich). On day 4, the medium was changed to N2B27 + 0, 1 μM LDN, 20 μM SB431542 , 100 nM retinoic acid and 500 nM SAG (TOCRIS). On day 7, the medium was changed to N2B27 + 100 nM retinoic acid and 500 nM SAG. On day 9 and day 11, the medium was changed to N2B27 + 100 nM retinoic acid, 500 nM SAG and 10 μM DAPT (TOCRIS). On day 14, the EBs are composed of motor neurons and were dissociated with 0 , 5 % Trypsin (Life Technologies). The obtained motor neurons were then seeded in 96- well plates or 4- well plates (with 10mm coverslips) previously coated with Poly-D-lysine. iMN testing: For assessing the effect of drugs on neuronal morphology and viability, mature iPSC-derived motor neuron cultures are monitored in real time using the live cell imaging and analysis
instrument Incucyte SX5 (Sartorius). A neurite detection mask has been optimized for online measurement of neurite extension and retraction. Viability is monitored live by the uptake of Propidium Iodide which is added to the cell culture medium at a concentration of 1µg/ml. Images can be acquired and automatically analyzed every hour for the duration of the drug screening test. REFERENCES: Throughout this application, various references describe the state of the art to which this invention pertains. The disclosures of these references are hereby incorporated by reference into the present disclosure.
F = Forward primer, R = Reverse primer Fluorescence activated cell sorting (FACS) and flow cytometry analysis Cells were sorted using FACSAria II (BD Biosciences) or SH800 Cell Sorter (Sony Biotechnology). Flow cytometry analysis was performed using Gallios (Beckman coulter). Sorted and unsorted populations were cultured at a concentration of 5 × 105/ml in a RPMI K562 medium for 4/5 days before collection for DNA extraction. Data were analyzed using the FlowJo (BD Biosciences) software. Results: Plasmid-mediated base editing in K562 cells in the C9orf72 Pol II pause site To boost C9orf72 gene expression in K562 cells, we targeted the RNA polymerase II (Pol II) pause site in the first intronic region of the gene C9orf72 (Figure 1A and B). RNA Pol II pausing is a key regulatory step in transcription. RNA Pol II pauses at GC-rich regions, and almost 65% of the pause sites are cytosines (Watts, Jason A., et al. "Cis elements that mediate
RNA polymerase II pausing regulate human gene expression." The American Journal of Human Genetics 105.4 (2019): 677-688). Gene expression analysis showed that genes with more paused polymerases have lower expression levels. Therefore, mutagenesis of the pause sites might lead to a significant increase in transcription. As our base of interest is a C, we used the CBE-SpRY enzyme (allowing C to T conversions) (Walton, Russell T., et al. "Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants." Science 368.6488 (2020): 290-296), given also its PAM-less nature that allowed us to design sgRNAs by placing the base editing window within different strands and positions of the Pol II pause site (Figure 1A). The CBE-SpRY-expressing plasmid was transfected in K562 cells in combination with individual sgRNA-expressing plasmids (C9orf72_2, C9orf72_3 and C9orf72_4) targeting the Pol II site (Figure 1 and Figure 2A). 24 hours post transfection, K562 were sorted and the GFP+ cells were grown for 3 to 5 days before performing DNA extraction and Sanger sequencing of the PCR-amplified target region, followed by EditR analysis. CBE-SpRY coupled with C9orf72_2 sgRNA led to high C>T conversion with efficiencies of 47% ± [5.2], n=3) for the target base C2 (Figure 3A). CBE-SpRY coupled with C9orf72_3 and C9orf72_4 sgRNAs showed also efficient conversion of the target base C1, but also of C2, located within the base editing window (bystander edit) (Figure 3A). In fact, CBE-SpRY coupled with C9orf72_3 sgRNA resulted in a C>T conversion of 35% of the C1 and 15% of bystander C2 (25.5 ±10.5, n=3), and the C9orf72_4 sgRNA to 25% for the C1 and 18% for the bystander C2 (25 ± [5.5]; n=3) (Figure 3A). TIDE analysis confirmed the DSB-free nature of base editors, as we detected low frequencies of InDels in base edited samples compared to Cas9 nuclease used in combination with the C9orf72_2, C9orf72_3 or C9orf72_4 sgRNAs (Figure 3B). In conclusion, we were able to efficiently target the Pol II pause site and convert the target base without causing DSBs in K562 cells. Base editing-mediated C to T conversion increases C9orf72 gene transcript levels upon targeting of the Pol II pause site
C9orf72 transcript levels were significantly increased in K562 cells upon targeting of the Pol II pause site when transfection was performed with the C9orf72_2 sgRNA coupled with CBE SpRY compared to control cells (Figure 4). A similar trend was observed when using the C9orf72_3 or C9orf72_4 sgRNAs. As expected, C9orf72 transcript levels were significantly increased when using the C9orf72_2 sgRNA that in fact showed the highest percentage of C to T conversion and precision amongst the three sgRNAs tested in this study. RNA-mediated base editing in K562 cells in the C9orf72 Pol II pause site To establish a clinically relevant method to deliver the base editing system in K562 cells and achieve a higher editing efficiency coupled with minimal toxicity compared to the plasmid transfection, we used a protocol based on transfection of mRNA encoding base editors and chemically modified synthetic sgRNAs (Figure 2B). In vitro transcribed CBE-SpRY-OPT mRNAs were transfected in K562 cells in combination with chemically modified sgRNA C9orf72_1, C9orf72_2, C9orf72_3 or C9orf72_4lls were kept in culture for 3 to 5 days before DNA extraction and Sanger sequencing of the PCR- amplified target region. EditR analysis showed that all the sgRNA were able to modify the target base with efficiencies ranging from 9% (C9orf72_1) to 27% (C9orf72_4) with a variable efficiency of bystander C2 within the editing window, upon RNA-mediated delivery of the base editing system (Figure 5). EXAMPLE 2: Experimentation on patient-derived motor neurons Generation of iPSC-derived motor neurons Origin of the cells: A control iPSC line generated by the Stem Cell facility at the host institute (Institut Imagine cell line #Ctr004) was used as a starting material for all the motor neuron derivation studies. iPSC colonies were grown in mTeSR medium in vitronectin (both StemCell) coated dishes and were passaged by enzyme - free dissociation (ReleSR, StemCell) according to the manufacturer’s instructions. iMN differentiation:
To generate motor neurons, we have adapted a published protocol (Maury et al., 2015). Briefly, on day 0, iPSC colonies were dissociated to single cells using Accutase (Life Technologies) and seeded in ultra- low attachment 6- well plates (3 × 105 cells/well) or cell - repellent surface T25 flasks (3 × 105 cells/flask) in N2B27 medium. N2B27 medium consists of a 1:1 ratio of advanced DMEM/F12 (Life Technologies) and Neurobasal medium (Life Technologies), 1% N2 supplement (Life Technologies), 2% B27 supplement minus vitamin A (Life Technologies), 5 mM Glutamax (Life Technologies), 5mM L- Glutamine (Sigma), 1% penicillin/streptomycin (Life Technologies) and 0,1 mM 2 - ME (Life Technologies). In these conditions, iPSCs spontaneously aggregated into embryoid bodies (EBs) in suspension in N2B27 medium. To induce proper differentiation of these iPSCs into motor neurons, the medium was changed at specific days and different factors were added each time. On day 0, was added N2B27 medium + 10 μM ROCK inhibitor Y - 27632, 3 μM CHIR99021 (TOCRIS), 0, 1 μM LDN (TOCRIS), 20 μM SB431542 (TOCRIS) and 10 μM ascorbic acid (Sigma - Aldrich). On day 2, the medium was changed to N2B27 + 3 μM CHIR99021, 0,1 μM LDN, 20 μM 8SB431542 and 100 nM retinoic acid (Sigma - Aldrich). On day 4, the medium was changed to N2B27 + 0, 1 μM LDN, 20 μM SB431542 , 100 nM retinoic acid and 500 nM SAG (TOCRIS). On day 7, the medium was changed to N2B27 + 100 nM retinoic acid and 500 nM SAG. On day 9 and day 11, the medium was changed to N2B27 + 100 nM retinoic acid, 500 nM SAG and 10 μM DAPT (TOCRIS). On day 14, the EBs are composed of motor neurons and were dissociated with 0 , 5 % Trypsin (Life Technologies). The obtained motor neurons were then seeded in 96- well plates or 4- well plates (with 10mm coverslips) previously coated with Poly-D-lysine. iMN testing: For assessing the effect of drugs on neuronal morphology and viability, mature iPSC-derived motor neuron cultures are monitored in real time using the live cell imaging and analysis
instrument Incucyte SX5 (Sartorius). A neurite detection mask has been optimized for online measurement of neurite extension and retraction. Viability is monitored live by the uptake of Propidium Iodide which is added to the cell culture medium at a concentration of 1µg/ml. Images can be acquired and automatically analyzed every hour for the duration of the drug screening test. REFERENCES: Throughout this application, various references describe the state of the art to which this invention pertains. The disclosures of these references are hereby incorporated by reference into the present disclosure.
Claims
CLAIMS: 1. A method for restoring the expression of C9orf72 in a patient suffering from Amyotrophic Lateral Sclerosis (ALS) comprising administering to the patient a therapeutically effective amount of a gene editing platform that consists of (a) at least one base-editing enzyme and (b) at least one guide RNA molecule for guiding the base- editing enzyme to at least one target sequence in the RNA Polymerase II pause site located in the first intronic region of the gene C9orf72, thereby editing said site and subsequently increasing the expression of C9orf72. 2. The method of claim 1 wherein the gene editing platform is particularly suitable for mutating the base C located at position Chr9:27563356-27563366-Hg18. 3. The method of claim 2 wherein the gene editing platform is particularly suitable for introducing one C>T conversion at position Chr9:27563356-27563366-Hg18. 4. The method of claim 1 wherein the base-editing enzyme comprises a defective CRISPR/Cas nuclease. 5. The method of claim 4 wherein the CRISPR/Cas nuclease of the present invention is a nickase and more particularly a Cas9 nickase i.e. the Cas9 from S. pyogenes having one mutation selected from the group consisting of D10A and H840A. 6. The method of claim 5 wherein the nickase comprises the amino acid sequence as set forth in SEQ ID NO:4 or SEQ ID NO:5. 7. The method of claim 1 wherein the second component of the base-editing enzyme comprises a non-nuclease DNA modifying enzyme that is a cytidine deaminase. 8. The method of claim 7 wherein the cytidine deaminase consists of a variant of the amino acid sequence as set forth in SEQ ID NO:6-16. 9. The method of claim 1 wherein the base-editing enzyme consists of the amino acid sequence as set forth in SEQ ID NO:20 (CBE-SpYR). 10. The method of claim 1 wherein the guide RNA targets the pause site that is located on chr9:27563356-27563366 region, and more preferably the nucleotide sequence SEQ ID NO:3.
11. The method of claim 10 wherein the guide RNA targets a sequence selected from Table 1. 12. The method of claim 1 wherein the gene editing platform comprises a) a base-editing enzyme that is a CBE-SpRY and b) and at least one guide RNA molecule that targets one sequence selected in Table 1.. 13. The method of claim 1 wherein the different components of the gene editing platform of the present invention are administered to the patient through expression from one or more vectors 14. The method of claim 13 wherein the vector is a viral vector. 15. The method of claim 1 wherein the different components of the gene editing platform are administered to the patient through the use of an RNA-encoded system.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP22306302 | 2022-09-02 | ||
| EP22306302.5 | 2022-09-02 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024047247A1 true WO2024047247A1 (en) | 2024-03-07 |
Family
ID=83690167
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2023/074086 Ceased WO2024047247A1 (en) | 2022-09-02 | 2023-09-01 | Base editing approaches for the treatment of amyotrophic lateral sclerosis |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2024047247A1 (en) |
Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20100324120A1 (en) | 2009-06-10 | 2010-12-23 | Jianxin Chen | Lipid formulation |
| WO2012006378A1 (en) | 2010-07-06 | 2012-01-12 | Novartis Ag | Liposomes with lipids having an advantageous pka- value for rna delivery |
| WO2012030901A1 (en) | 2010-08-31 | 2012-03-08 | Novartis Ag | Small liposomes for delivery of immunogen-encoding rna |
| WO2012031043A1 (en) | 2010-08-31 | 2012-03-08 | Novartis Ag | Pegylated liposomes for delivery of immunogen-encoding rna |
| WO2012031046A2 (en) | 2010-08-31 | 2012-03-08 | Novartis Ag | Lipids suitable for liposomal delivery of protein-coding rna |
| US8202983B2 (en) | 2007-05-10 | 2012-06-19 | Agilent Technologies, Inc. | Thiocarbon-protecting groups for RNA synthesis |
| US20130122104A1 (en) | 2009-07-01 | 2013-05-16 | Protiva Biotherapeutics, Inc. | Novel lipid formulations for delivery of therapeutic agents to solid tumors |
| US20130177637A1 (en) | 2010-04-09 | 2013-07-11 | Pacira Pharmaceuticals, Inc. | Method for formulating large diameter synthetic membrane vesicles |
| WO2013176772A1 (en) | 2012-05-25 | 2013-11-28 | The Regents Of The University Of California | Methods and compositions for rna-directed target dna modification and for rna-directed modulation of transcription |
| WO2014144592A2 (en) | 2013-03-15 | 2014-09-18 | The General Hospital Corporation | Using truncated guide rnas (tru-grnas) to increase specificity for rna-guided genome editing |
| US20140273226A1 (en) | 2013-03-15 | 2014-09-18 | System Biosciences, Llc | Crispr/cas systems for genomic modification and gene modulation |
| US20140273233A1 (en) | 2013-03-15 | 2014-09-18 | Sigma-Aldrich Co., Llc | Crispr-based genome modification and regulation |
| WO2022070107A1 (en) * | 2020-09-30 | 2022-04-07 | Crispr Therapeutics Ag | Materials and methods for treatment of amyotrophic lateral sclerosis |
-
2023
- 2023-09-01 WO PCT/EP2023/074086 patent/WO2024047247A1/en not_active Ceased
Patent Citations (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8202983B2 (en) | 2007-05-10 | 2012-06-19 | Agilent Technologies, Inc. | Thiocarbon-protecting groups for RNA synthesis |
| US20100324120A1 (en) | 2009-06-10 | 2010-12-23 | Jianxin Chen | Lipid formulation |
| US20130122104A1 (en) | 2009-07-01 | 2013-05-16 | Protiva Biotherapeutics, Inc. | Novel lipid formulations for delivery of therapeutic agents to solid tumors |
| US20130177633A1 (en) | 2010-04-09 | 2013-07-11 | Pacira Pharmaceuticals, Inc. | Method for formulating large diameter synthetic membrane vesicles |
| US20130183375A1 (en) | 2010-04-09 | 2013-07-18 | Pacira Pharmaceuticals, Inc. | Method for formulating large diameter synthetic membrane vesicles |
| US20130183373A1 (en) | 2010-04-09 | 2013-07-18 | Pacira Pharmaceuticals, Inc. | Method for formulating large diameter synthetic membrane vesicles |
| US20130183372A1 (en) | 2010-04-09 | 2013-07-18 | Pacira Pharmaceuticals, Inc. | Method for formulating large diameter synthetic membrane vesicles |
| US20130177637A1 (en) | 2010-04-09 | 2013-07-11 | Pacira Pharmaceuticals, Inc. | Method for formulating large diameter synthetic membrane vesicles |
| US20130177636A1 (en) | 2010-04-09 | 2013-07-11 | Pacira Pharmaceuticals, Inc. | Method for formulating large diameter synthetic membrane vesicles |
| US20130177634A1 (en) | 2010-04-09 | 2013-07-11 | Pacira Pharmaceuticals, Inc. | Method for formulating large diameter synthetic membrane vesicles |
| US20130177638A1 (en) | 2010-04-09 | 2013-07-11 | Pacira Pharmaceuticals, Inc. | Method for formulating large diameter synthetic membrane vesicles |
| US20130177635A1 (en) | 2010-04-09 | 2013-07-11 | Pacira Pharmaceuticals, Inc. | Method for formulating large diameter synthetic membrane vesicles |
| WO2012006378A1 (en) | 2010-07-06 | 2012-01-12 | Novartis Ag | Liposomes with lipids having an advantageous pka- value for rna delivery |
| US20130202684A1 (en) | 2010-08-31 | 2013-08-08 | Lichtstrasse | Pegylated liposomes for delivery of immunogen encoding rna |
| WO2012031046A2 (en) | 2010-08-31 | 2012-03-08 | Novartis Ag | Lipids suitable for liposomal delivery of protein-coding rna |
| WO2012031043A1 (en) | 2010-08-31 | 2012-03-08 | Novartis Ag | Pegylated liposomes for delivery of immunogen-encoding rna |
| US20130189351A1 (en) | 2010-08-31 | 2013-07-25 | Novartis Ag | Lipids suitable for liposomal delivery of protein coding rna |
| US20130195969A1 (en) | 2010-08-31 | 2013-08-01 | Novartis Ag | Small liposomes for delivery of immunogen encoding rna |
| WO2012030901A1 (en) | 2010-08-31 | 2012-03-08 | Novartis Ag | Small liposomes for delivery of immunogen-encoding rna |
| WO2013176772A1 (en) | 2012-05-25 | 2013-11-28 | The Regents Of The University Of California | Methods and compositions for rna-directed target dna modification and for rna-directed modulation of transcription |
| WO2014144592A2 (en) | 2013-03-15 | 2014-09-18 | The General Hospital Corporation | Using truncated guide rnas (tru-grnas) to increase specificity for rna-guided genome editing |
| US20140273226A1 (en) | 2013-03-15 | 2014-09-18 | System Biosciences, Llc | Crispr/cas systems for genomic modification and gene modulation |
| WO2014144761A2 (en) | 2013-03-15 | 2014-09-18 | The General Hospital Corporation | Increasing specificity for rna-guided genome editing |
| US20140273233A1 (en) | 2013-03-15 | 2014-09-18 | Sigma-Aldrich Co., Llc | Crispr-based genome modification and regulation |
| WO2022070107A1 (en) * | 2020-09-30 | 2022-04-07 | Crispr Therapeutics Ag | Materials and methods for treatment of amyotrophic lateral sclerosis |
Non-Patent Citations (61)
| Title |
|---|
| "NCBI", Database accession no. YP_002342100.1 |
| ADAMS ET AL., THE BIOCHEMISTRY OF THE NUCLEIC ACIDS, 1992 |
| ALTSCHUL ET AL., J. MOL. BIOL., vol. 215, 1990, pages 403 - 410 |
| ALTSCHUL ET AL., NAT. GENET., vol. 6, 1994, pages 119 - 129 |
| ALTSCHUL ET AL., NUCLEIC ACIDS RES., vol. 25, 1997, pages 3389 - 3402 |
| BABIC LEKO MZUPUNSKI VKIRINCICH JSMILOVIC DHORTOBAGYI THOF PRSIMIC G: "C9orf72 Hexanucleotide Repeat Expansion", BEHAVIOURAL NEUROLOGY, 2019, pages 2909168 |
| BIGIO EH: "C90RF72, the new gene on the block, causes C9FTD/ALS: new insights provided by neuropathology", ACTA NEUROPATHOLOGICA, vol. 122, no. 6, December 2011 (2011-12-01), pages 653 - 5, XP019984648, DOI: 10.1007/s00401-011-0919-7 |
| BRINKMAN, EVA K. ET AL.: "Easy quantitative assessment of genome editing by sequence trace decomposition", NUCLEIC ACIDS RESEARCH, vol. 42, no. 22, 2014, pages e168 - e168, XP055788071, DOI: 10.1093/nar/gku936 |
| CAPPELLA, MARISA ET AL.: "Gene therapy for ALS a perspective", INTERNATIONAL JOURNAL OF MOLECULAR SCIENCES, vol. 20, no. 18, 2019, pages 4388 |
| CHEN ET AL.: "Fusion protein linkers: property, design and functionality.", ADV DRUG DELIV REV., vol. 65, no. 10, 2013, pages 1357 - 69, XP028737352, DOI: 10.1016/j.addr.2012.09.039 |
| CHYLINSKI, RHUNCHARPENTIER: "The tracrRNA and Cas9 families of type II CRLSPR-Cas immunity systems", RNA BIOLOGY, vol. 10, no. 5, 2013, pages 726 - 737, XP055116068, DOI: 10.4161/rna.24321 |
| CIURA, SORANA ET AL.: "Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis", ANNALS OF NEUROLOGY, vol. 74, no. 2, 2013, pages 180 - 187, XP071640915, DOI: 10.1002/ana.23946 |
| CORPET ET AL., NUC. ACIDS RES., vol. 16, 1988, pages 10881 - 10890 |
| CRADICK, THOMAS J. ET AL.: "COSMID: a web-based tool for identifying and validating CRISPR/Cas off-target sites", MOLECULAR THERAPY-NUCLEIC ACIDS, vol. 3, 2014, pages e214 |
| DEFOUGEROLLES, HUM GENE THER., vol. 19, 2008, pages 125 - 132 |
| DELTCHEVA E.CHYLINSKI K.SHARMA C. M.GONZALES K.CHAO Y.PIRZADA Z. A.ECKERTM. P.VOGEL J.CHARPENTIER E.: "CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III.", NATURE, vol. 471, 2011, pages 602 - 607, XP055308803, DOI: 10.1038/nature09886 |
| GISHSTATES, NATURE GENET., vol. 3, 1993, pages 266 - 272 |
| GREENE, M. R. ET AL.: "Transduction of Human CD34 + Repopulating Cells with a Self-Inactivating Lentiviral Vector for SCID-X1 Produced at Clinical Scale by a Stable Cell Line.", HUM. GENE THER. METHODS, vol. 23, 2012, pages 297 - 308, XP055503306, DOI: 10.1089/hgtb.2012.150 |
| GUILINGER, JOHN P.DAVID B. THOMPSONDAVID R. LIU: "Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification", NATURE BIOTECHNOLOGY, vol. 32, no. 6, 2014, pages 577 - 582, XP055157221, DOI: 10.1038/nbt.2909 |
| HESS GTFRESARD LHAN KLEE CHLI ACINPRICH KAMONTGOMERY SBBASSIK MC: "Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells", NAT METHODS, vol. 13, no. 12, 2016, pages 1036 - 1042, XP055832741, DOI: 10.1038/nmeth.4038 |
| HEYES ET AL., J CONTR REL., vol. 107, 2005, pages 276 - 287 |
| HIGGINSSHARP, CABIOS, vol. 4, 1989, pages 151 - 153 |
| HIGGINSSHARP, GENE, vol. 73, 1988, pages 237 - 244 |
| HUANG ET AL., COMP. APPLS BIOSCI., vol. 8, 1992, pages 155 - 16 |
| J. J., MCSHAN W. M.AJDIC D. J.SAVIC D. J.SAVIC G.LYON K.PRIMEAUX C.SEZATE S.SUVOROV A. N.KENTON S.LAI H. S.: "Complete genome sequence of an M1 strain of Streptococcus pyogenes", PROC. NATL. ACAD. SCI. U.S.A., vol. 98, 2001, pages 4658 - 4663 |
| JEFFS ET AL., PHARM RES., vol. 22, 2005, pages 362 - 372 |
| JIANG, T.HENDERSON, J.M.COOTE, K. ET AL.: "Chemical modifications of adenine base editor mRNA and guide RNA expand its application scope", NAT COMMUN, vol. 11, 2020, pages 1979, XP055905003, DOI: 10.1038/s41467-020-15892-8 |
| JINEK ET AL., SCIENCE, vol. 337, 2012, pages 16 - 821 |
| JINEK M.CHYLINSKI K.FONFARA 1.HAUER M.DOUDNA J. A.CHARPENTIER E.: "A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity", SCIENCE, vol. 337, 2012, pages 816 - 821, XP055229606, DOI: 10.1126/science.1225829 |
| JUDGE ET AL., J CLIN INVEST., vol. 119, 2009, pages 661 - 673 |
| KLUESNER, MITCHELL G. ET AL.: "EditR: a method to quantify base editing from Sanger sequencing", THE CRISPR JOURNAL 1, vol. 3, 2018, pages 239 - 250, XP055715954, DOI: 10.1089/crispr.2018.0014 |
| KLUESNER, MITCHELL G. ET AL.: "EditR: a method to quantify base editing from Sanger sequencing", THE CRISPR JOURNAL, vol. 1, no. 3, 2018, pages 239 - 250, XP055715954, DOI: 10.1089/crispr.2018.0014 |
| KOBLAN, LUKE W.: "Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction", NATURE BIOTECHNOLOGY, vol. 36, no. 9, 2018, pages 843 - 846, XP036929657, DOI: 10.1038/nbt.4172 |
| KOSICKI, MICHAEL, KART TOMBERG, ALLAN BRADLEY: "Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements", NATURE BIOTECHNOLOGY, vol. 36, no. 8, 2018, pages 765 - 771, XP036929645, DOI: 10.1038/nbt.4192 |
| MADDEN ET AL., METH. ENZYMOL., vol. 266, 1996, pages 131 - 141 |
| MICCIO ANNARITA ET AL: "Novel genome-editing-based approaches to treat motor neuron diseases: Promises and challenges", MOLECULAR THERAPY, vol. 30, no. 1, 1 January 2022 (2022-01-01), US, pages 47 - 53, XP093015223, ISSN: 1525-0016, DOI: 10.1016/j.ymthe.2021.04.003 * |
| MINGAJ X ET AL: "P.0208 Base editing therapeutic strategies for motor neuron diseases", EUROPEAN NEUROPSYCHOPHARMACOLOGY, ELSEVIER SIENCE PUBLISHERS BV , AMSTERDAM, NL, vol. 53, 1 December 2021 (2021-12-01), XP086908044, ISSN: 0924-977X, [retrieved on 20211230], DOI: 10.1016/J.EURONEURO.2021.10.201 * |
| MORRISSEY ET AL., NAT BIOTECHNOL., vol. 2, 2005, pages 1002 - 1007 |
| NEEDLEMAN, SAUL B.WUNSCH, CHRISTIAN D.: "A general method applicable to the search for similarities in the amino acid sequence of two proteins", JOURNAL OF MOLECULAR BIOLOGY, vol. 48, no. 3, 1970, pages 443 - 53, XP024011703, DOI: 10.1016/0022-2836(70)90057-4 |
| NEEDLEMANWUNSCH, J. MOL. BIOL., vol. 48, 1970, pages 443 |
| PAUNOVSKA, KALINADAVID LOUGHREYJAMES E. DAHLMAN: "Drug delivery systems for RNA therapeutics", NATURE REVIEWS GENETICS, 2022, pages 1 - 16 |
| PEARSON ET AL., METH. MOL. BIOL., vol. 24, 1994, pages 307 - 31 |
| PEARSONLIPMAN, PROC. NATL. ACAD. SCI. U.S.A., vol. 85, 1988, pages 2444 |
| PREL ACAVAL IGAYON RRAVASSARD PDUTHOIT CPAYEN EMCIOUCHE-CHRETIEN LCRENEGUY ANGUYEN THMARTIN A: "Highly efficient in vitro and in vivo delivery o RNAs using new versatile MS2-chimeric retrovirus-like particles", MOL THER METHODS CLIN DEV., vol. 2, 21 October 2015 (2015-10-21), pages 15039, XP002768084, DOI: 10.1038/mtm.2015.39 |
| QI ET AL., CELL, vol. 152, no. 5, 2013, pages 1173 - 83 |
| QI ET AL.: "Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression", CELL, vol. 152, no. 5, 2013, pages 1173 - 83, XP055346792, DOI: 10.1016/j.cell.2013.02.022 |
| REES, HOLLY A.DAVID R. LIU: "Base editing: precision chemistry on the genome and transcriptome of living cells", NATURE REVIEWS GENETICS, vol. 19, no. 12, 2018, pages 770 - 788, XP036637441, DOI: 10.1038/s41576-018-0068-0 |
| SELLIER, CHANTAL: "Loss of C9 ORF 72 impairs autophagy and synergizes with polyQ Ataxin - 2 to induce motor neuron dysfunction and cell death", THE EMBO JOURNAL, vol. 35, no. 12, 2016, pages 1276 - 1297 |
| SEMPLE ET AL., NATURE BIOTECH., vol. 28, 2010, pages 172 - 176 |
| SHI, YINGXIAO ET AL.: "Haploinsufficiency leads to neurodegeneration in C90RF72 ALS/FTD human induced motor neurons", NATURE MEDICINE, vol. 24, no. 3, 2018, pages 313 - 325, XP055973357, DOI: 10.1038/nm.4490 |
| SMITHWATERMAN, ADV. APPL. MATH., vol. 2, 1981, pages 482 |
| TIJSSEN: "Laboratory Techniques In Biochemistry And Molecular Biology-Hybridization With Nucleic Acid Probes Part I", 1993, ELSEVIER, article "Overview of principles of hybridization and the strategy of nucleic acid probe assay" |
| WALTON, RUSSELL Σ ET AL.: "Unconstrained genome targeting with near-PAM less engineered CRISPR-Cas9 variants", SCIENCE, vol. 368, no. 6488, 2020, pages 290 - 296 |
| WATTS, JASON A. ET AL.: "Cis elements that mediate RNA polymerase II pausing regulate human gene expression", THE AMERICAN JOURNAL OF HUMAN GENETICS, vol. 105, no. 4, 2019, pages 677 - 688, XP085849312, DOI: 10.1016/j.ajhg.2019.08.003 |
| WHEELER ET AL., GENE THERAPY., vol. 6, 1999, pages 1438 - 1447 |
| YINGXIAO SHI ET AL: "Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons", NATURE MEDICINE, vol. 24, no. 3, 5 February 2018 (2018-02-05), New York, pages 313 - 325, XP055497485, ISSN: 1078-8956, DOI: 10.1038/nm.4490 * |
| YUN YEOMIN ET AL: "CRISPR/Cas9-Mediated Gene Correction to Understand ALS", INTERNATIONAL JOURNAL OF MOLECULAR SCIENCES, vol. 21, no. 11, 27 May 2020 (2020-05-27), pages 3801, XP093015226, DOI: 10.3390/ijms21113801 * |
| ZHANG, XIAOHUI ET AL.: "Dual base editor catalyzes both cytosine and adenine base conversions in human cells", NATURE BIOTECHNOLOGY, vol. 38, no. 7, 2020, pages 856 - 860, XP037187540, DOI: 10.1038/s41587-020-0527-y |
| ZHANGMADDEN, GENOME RES., vol. 7, 1997, pages 49 - 656 |
| ZHU, QIANG ET AL.: "Reduced C9ORF72function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72", NATURE NEUROSCIENCE, vol. 23, no. 5, 2020, pages 615 - 624, XP037099667, DOI: 10.1038/s41593-020-0619-5 |
| ZIMMERMANN ET AL., NATURE, vol. 441, 2006, pages 111 - 114 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20240093193A1 (en) | Dead guides for crispr transcription factors | |
| US11624078B2 (en) | Protected guide RNAS (pgRNAS) | |
| JP2020508685A (en) | RNA targeting of mutations by suppressor tRNA and deaminase | |
| JP2016521555A5 (en) | ||
| US11492614B2 (en) | Stem loop RNA mediated transport of mitochondria genome editing molecules (endonucleases) into the mitochondria | |
| US20240052371A1 (en) | Programmable transposases and uses thereof | |
| EP4150081A1 (en) | Base editing approaches for the treatment of betahemoglobinopathies | |
| WO2024047247A1 (en) | Base editing approaches for the treatment of amyotrophic lateral sclerosis | |
| CN114072518B (en) | Methods and compositions for treating thalassemia or sickle cell disease | |
| JP2024529400A (en) | Hepatitis B virus (HBV) knockout | |
| CN117279671A (en) | Strategies for knocking in C3 safe harbors | |
| JP2023553701A (en) | Therapeutic LAMA2 Payload for the Treatment of Congenital Muscular Dystrophy | |
| Rathbone | Nonviral Approaches for Delivery of CRISPR-Cas9 Into Hepatocytes for Treatment of Inherited Metabolic Disease | |
| Chauhan et al. | CRISPR/Cas systems in therapeutics: Transforming gene editing into medical solutions | |
| WO2023212657A2 (en) | Enhancement of safety and precision for crispr-cas induced gene editing by variants of dna polymerase using cas-plus variants | |
| CN120112634A (en) | Knock-in strategy for B2M safe harbor sites | |
| WO2025096936A2 (en) | Use of prime editing in correcting mutations in cdkl5 | |
| US20220064635A1 (en) | Crispr compositions and methods for promoting gene editing of adenosine deaminase 2 (ada2) | |
| HK40115053A (en) | Novel omni crispr nuclease | |
| Nguyen | CRISPR/Cas9-mediated Homology Directed Repair disease modeling and lentiviral gene therapy for CDKL5 deficiency disorder | |
| WO2025043140A1 (en) | Methods and compositions for modifying expression of a mutant transforming growth factor beta induced (tgfbi) allele | |
| AU2024240582A1 (en) | Methods for creating regulatory t cells (tregs) using genome engineering | |
| HK40111655A (en) | Novel omni 115, 124, 127, 144-149, 159, 218, 237, 248, 251-253 and 259 crispr nucleases | |
| CN118786212A (en) | SARM1 expression reduction for cell therapy | |
| HK40109572A (en) | Novel omni 117, 140, 150-158, 160-165, 167-177, 180-188, 191-198, 200, 201, 203, 205-209, 211-217, 219, 220, 222, 223, 226, 227, 229, 231-236, 238-245, 247, 250, 254, 256, 257, 260 and 262 crispr nucleases |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 23764895 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 23764895 Country of ref document: EP Kind code of ref document: A1 |