WO2023170493A1 - Drug delivery using a parenteral pharmaceutical composition - Google Patents
Drug delivery using a parenteral pharmaceutical composition Download PDFInfo
- Publication number
- WO2023170493A1 WO2023170493A1 PCT/IB2023/051515 IB2023051515W WO2023170493A1 WO 2023170493 A1 WO2023170493 A1 WO 2023170493A1 IB 2023051515 W IB2023051515 W IB 2023051515W WO 2023170493 A1 WO2023170493 A1 WO 2023170493A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- exemplary
- range
- water
- glatiramer acetate
- sodium alginate
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/02—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/32—Macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. carbomers, poly(meth)acrylates, or polyvinyl pyrrolidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/34—Macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyesters, polyamino acids, polysiloxanes, polyphosphazines, copolymers of polyalkylene glycol or poloxamers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/19—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles lyophilised, i.e. freeze-dried, solutions or dispersions
Definitions
- the present disclosure generally relates to a pharmaceutical composition for drug delivery, and more particularly, relates to a method of producing a drug-delivery- pharmaceutical composition for reducing symptoms of elapsing remitting multiple sclerosis (RRMS).
- RRMS remitting multiple sclerosis
- MS Multiple sclerosis
- RRMS relapsing-remitting multiple sclerosis
- SPMS secondary progressive multiple sclerosis
- PPMS primary progressive multiple sclerosis
- PRMS progressive-relapsing multiple sclerosis
- Different methods are used to reduce symptoms of MS. These methods include medication, therapies, such as physiotherapy, and self-management techniques.
- the medication used for reducing symptoms of MS includes parenteral medications and oral medications.
- Glatiramer acetate (GA) is one of effective drugs for reducing symptoms of MS. clinical tests revealed that oral administration of GA can inhibit functionality of GA due to acidic media of digestive system. However, parenteral administration of GA may cause initial burst release of high dosage of GA in body.
- Ehud Marom et al. presented a patent on “Depot systems comprising glatiramer or pharmacologically acceptable salt thereof’ (US 8377885 B2).
- Ehud Marom et al. synthesized a composition including a therapeutically effective amount of a pharmaceutically acceptable glatiramer acetate. Glatiramer acetate was administered using a depot method.
- Nadav et al. presented a patent on “Process for preparing microparticles containing glatiramer acetate” (WO 2018/042423 Al). Nadav et al. produced microparticles containing glatiramer acetate having low levels of residual organic solvents.
- the aforementioned methods suffer from toxicity due to organic solvents and possibility of initial burst release of glatiramer acetate.
- an exemplary parenteral pharmaceutical composition for reducing symptoms of elapsing remitting multiple sclerosis (RRMS).
- an exemplary parenteral pharmaceutical composition may include a plurality of powder particles dispersed in a phosphate-buffered saline (PBS) solution.
- PBS phosphate-buffered saline
- each powder particle of an exemplary plurality of powder particles may include a first water-based portion, an oilbased portion, and a second water-based portion.
- an exemplary first water-based portion may include glatiramer acetate (GA), sodium alginate with a weight ratio of an exemplary sodium alginate to an exemplary glatiramer acetate in a range of 0.1: 1 to 0.4: 4 (sodium alginate: glatiramer acetate), polyvinyl alcohol (PVA) with a weight ratio of PVA to an exemplary sodium alginate in a range of 0.05: 0.005 to 0.02: 0.2 (PVA: sodium alginate), and polysorbate 80 with a weight ratio of an exemplary polysorbate 80 to an exemplary sodium alginate in a range of 1: 3 to 5: 15 (polysorbate 80: sodium alginate).
- G glatiramer acetate
- sodium alginate with a weight ratio of an exemplary sodium alginate to an exemplary glatiramer acetate in a range of 0.1: 1 to 0.4: 4
- PVA polyvinyl alcohol
- PVA polysorbate
- an exemplary oil-based portion may include a-Tocopherol with a weight ratio of an exemplary a-Tocopherol to an exemplary sodium alginate in a range of 0: 1 to 0.3: 3 (a-Tocopherol: sodium alginate) and poly(lactic-co-glycolic acid) (PLGA) with a weight ratio of PLGA to an exemplary sodium alginate in a range of 0: 5 to 3: 30 (PLGA: sodium alginate).
- a-Tocopherol sodium alginate
- PLGA poly(lactic-co-glycolic acid)
- an exemplary second aqueous portion may include glatiramer acetate with a weight ratio of an exemplary glatiramer acetate to an exemplary sodium alginate in a range of 0.1: 1 to 1.1: 5 (glatiramer acetate: sodium alginate), chitosan with a weight ratio of an exemplary chitosan to an exemplary sodium alginate in a range of 0.05 : 1.5 to 0.1 : 20 (chitosan: sodium alginate), and cremophor EL with a weight ratio of an exemplary cremophor EL to an exemplary sodium alginate in a range of 0:1 to 0.5:5 (cremophor EL: sodium alginate).
- an exemplary parenteral pharmaceutical composition may include an interconnected matrix of an exemplary first waterbased portion, an exemplary oil-based portion, and an exemplary second water-based portion.
- a weight ratio of an exemplary PBS solution to an exemplary plurality of exemplary powder particles may be in a range of 1: 2 to 8: 9 (an exemplary PBS solution: an exemplary plurality of the powder particles).
- an exemplary parenteral pharmaceutical composition for reducing symptoms of elapsing remitting multiple sclerosis (RRMS).
- an exemplary parenteral pharmaceutical composition may include a plurality of powder particles dispersed in a phosphate-buffered saline (PBS) solution.
- PBS phosphate-buffered saline
- each powder particle of an exemplary plurality of powder particles may include a first water-based portion, an oilbased portion, and a second water-based portion.
- an exemplary first water-based portion may include glatiramer acetate, sodium alginate, polyvinyl alcohol (PVA), and polysorbate 80.
- an exemplary oil-based portion may include a-Tocopherol and poly(lactic-co-glycolic acid) (PLGA).
- an exemplary second water-based portion may include glatiramer acetate, chitosan, and cremophor EL.
- an exemplary plurality of powder particles of an exemplary pharmaceutical composition may include glatiramer acetate with a weight percent to a total amount of an exemplary parenteral pharmaceutical composition in a range of 4 wt. % to 80 wt. %.
- an exemplary PLGA may include at least one of an acid terminated PLGA with a molecular weight in a range of 7000 g/mol to 17000 g/mol, an ester terminated PLGA with a molecular weight in a range of 7000 g/mol to 17000 g/mol, an acid terminated PLGA with a molecular weight in a range of 24000 g/mol to 38000 g/mol, and combinations thereof.
- a pH value of an exemplary PBS solution may be in a range of 6.0 to 8.0.
- an exemplary parenteral pharmaceutical composition may include a releasing rate of an exemplary glatiramer acetate in a range of 20 % to 90 % of a total amount of an exemplary glatiramer acetate within 30 days when an exemplary parenteral pharmaceutical composition may be injected intramuscularly into a patient’s body.
- each particle of an exemplary plurality of exemplary powder particles may have an average particle size in a range of 5 pm to 75 pm.
- an exemplary plurality of powder particles of an exemplary parenteral pharmaceutical composition may be stored at a temperature in a range of 15 °C to 30 °C with a relative humidity of maximum 70%.
- an exemplary method may include forming a water/oil/water emulsion, forming a plurality of powder particles of an exemplary water/oil/water emulsion by drying an exemplary water/oil/water emulsion using a freeze dryer, forming a parenteral pharmaceutical composition, and injecting a predetermined amount of an exemplary formed parenteral pharmaceutical composition intramuscularly to a patient suffering from RRMS.
- forming an exemplary water/oil/water emulsion may include forming an internal aqueous phase by mixing sodium alginate, glatiramer acetate, polyvinyl alcohol (PVA), and polysorbate 80 together, forming an organic phase by mixing an exemplary internal aqueous phase, a-Tocopherol, and poly(lactic-co-glycolic acid) (PLGA) together, forming a plurality of elementary particles by mixing a normal saline solution, PVA, an exemplary organic phase, and an exemplary internal aqueous phase together, and forming an external aqueous phase connected to an outer surface of an exemplary plurality of elementary particles by mixing an exemplary plurality of elementary particles, glatiramer acetate, chitosan, and cremophor EL together.
- PVA polyvinyl alcohol
- PLGA poly(lactic-co-glycolic acid)
- forming an exemplary parenteral pharmaceutical composition may include mixing an exemplary plurality of powder particles of an exemplary water/oil/water emulsion with a phosphate-buffered saline (PBS) solution with a weight ratio of an exemplary plurality of exemplary powder particles of an exemplary water/oil/water emulsion to an exemplary PBS solution in a range of 1: 3 to 7: 9 (an exemplary plurality of the powder particles of an exemplary water/oil/water emulsion: an exemplary PBS solution).
- PBS phosphate-buffered saline
- forming an exemplary water/oil/water emulsion may further include washing an exemplary plurality of powder particles of an exemplary water/oil/water emulsion by mixing an exemplary plurality of powder particles of an exemplary water/oil/water emulsion with a PBS solution with a weight ratio of an exemplary plurality of powder particles of an exemplary water/oil/water emulsion to an exemplary PBS solution in a range of 1 : 3 to 7: 9 (an exemplary plurality of powder particles of an exemplary water/oil/water emulsion: the PBS solution).
- washing an exemplary plurality of powder particles of an exemplary water/oil/water emulsion may include mixing an exemplary plurality of powder particles of an exemplary water/oil/water emulsion with an exemplary PBS solution with a pH value in a range of 6 to 8.
- forming an exemplary internal aqueous phase may include mixing glatiramer acetate, sodium alginate at a relative weight ratio to an exemplary glatiramer acetate in a range of 0.1: 5 to 1:20 (sodium alginate: glatiramer acetate), polyvinyl alcohol (PVA) at a relative weight ratio to an exemplary glatiramer acetate in a range of 0.01: 0.1 to 0.03: 0.5 (PVA: glatiramer acetate), and polysorbate 80 with a weight ratio of an exemplary polysorbate 80 to glatiramer acetate in a range of 1: 3 to 3: 5 (polysorbate 80: glatiramer acetate) together using a mixer with a stirring speed in a range of 500 rpm to 5000 rpm.
- PVA polyvinyl alcohol
- mixing sodium alginate, glatiramer acetate, polyvinyl alcohol (PVA), and polysorbate 80 together may include mixing sodium alginate, glatiramer acetate, polyvinyl alcohol (PVA), and polysorbate 80 together using a mixer with a stirring speed in a range of 500 rpm to 5000 rpm for a time period in a range of 10 minutes to 100 minutes.
- forming an exemplary organic phase may include mixing an exemplary internal aqueous phase, a-Tocopherol with a weight ratio of the a-Tocopherol to glatiramer acetate in a range of 0:2 to 1 :5 (a-Tocopherol: glatiramer acetate), and PLGA with a weight ratio of the PLGA to glatiramer acetate in a range of 1: 5 to 5: 50 (PLGA: glatiramer acetate) together using an ultrasonic device with an ultrasonic power in a range of 200 W/cm 2 to 1000 W/cm 2 .
- mixing an exemplary internal aqueous phase, a- Tocopherol, and PLGA together may include mixing an exemplary internal aqueous phase, a- Tocopherol, and PLGA together using an ultrasonic device for a time period in a range of 2 minutes to 60 minutes.
- forming an exemplary external aqueous phase by mixing an exemplary plurality of elementary particles, glatiramer acetate, chitosan, and cremophor EL together may be done using an ultrasonic device with an ultrasonic power in a range of 200 W/cm 2 to 1000 W/cm 2 .
- forming an exemplary external aqueous phase by mixing an exemplary plurality of elementary particles, glatiramer acetate, chitosan, and cremophor EL together may be done using an ultrasonic device for a time period in a range of 10 minutes to 100 minutes.
- drying an exemplary water/oil/water emulsion may include freezing an exemplary water/oil/water emulsion at a temperature in a range of -60 °C to -10 °C for a time period in a range of 1 hour to 10 hours and drying an exemplary water/oil/water emulsion.
- drying an exemplary water/oil/water emulsion may include initial drying an exemplary water/oil/water emulsion at a temperature in a range of -5 °C to -40 °C for a time period in a range of 10 hours to 40 hours and secondary drying an exemplary water/oil/water emulsion at a temperature in a range of 10 °C to 30 °C for a time period in a range of 5 hours to 20 hours.
- FIG. 1A illustrates a flowchart of a method of reducing symptoms of elapsing remitting multiple sclerosis (RRMS), consistent with one or more exemplary embodiments of the present disclosure
- FIG. IB illustrates a flowchart of a method of forming an exemplary water/oil/water emulsion, consistent with one or more exemplary embodiments of the present disclosure
- FIG. 2 illustrates a transmission electron microscopy (TEM) image of an exemplary prepared powder particles of an exemplary pharmaceutical composition, consistent with one or more exemplary embodiments of the present disclosure
- FIG. 3 illustrates a scanning electron microscopy (SEM) image of G-Mix22 sample before freeze drying, consistent with one or more exemplary embodiments of the present disclosure
- FIG. 4 illustrates a SEM image of G-Mix22 sample after freeze drying, consistent with one or more exemplary embodiments of the present disclosure
- FIG. 5 illustrates a SEM image of particles based on PLGA, consistent with one or more exemplary embodiments of the present disclosure
- FIG. 6 illustrates a SEM image of particles based on GA, chitosan, and sodium alginate (GA-CH-A1), consistent with one or more exemplary embodiments of the present disclosure
- FIG. 7 illustrates a TEM image of GA-CH-A1 particles, consistent with one or more exemplary embodiments of the present disclosure
- FIG. 8 illustrates a particle size distribution diagram of an exemplary plurality of powder particles of an exemplary pharmaceutical composition, consistent with one or more exemplary embodiments of the present disclosure
- FIG. 9 illustrates GA-release profiles of GMIX22, GMIX21, GC9, and GPU samples after 30 days, consistent with one or more exemplary embodiments of the present disclosure
- FIG. 10 illustrates a chart of comparative studies of mean body weights in four study groups of non-EAE, EAE-vehicle, EAE-GA, and EAE/GA/Depot mice over 28 days, consistent with one or more exemplary embodiments of the present disclosure
- FIG. 11 illustrates a chart of comparative studies of mean clinical scores of non-EAE, EAE-vehicle, EAE-GA, and EAE/GA/Depot mice over 28 days, consistent with one or more exemplary embodiments of the present disclosure
- FIG. 12A illustrates an Hematoxylin and Eosin (H&E) stained pathological image of spinal cord in an exemplary non- experimental autoimmune encephalomyelitis (EAE) group, consistent with one or more exemplary embodiments of the present disclosure
- FIG. 12B illustrates an H&E stained pathological image of spinal cord in an exemplary EAE group, consistent with one or more exemplary embodiments of the present disclosure.
- FIG. 12C illustrates an H&E stained pathological image of spinal cord in an exemplary EAEGA group, consistent with one or more exemplary embodiments of the present disclosure
- FIG. 12D illustrates an H&E stained pathological image of spinal cord in an exemplary EAEGA/Depot group, consistent with one or more exemplary embodiments of the present disclosure
- FIG. 13A illustrates an H&E stained pathological image of brain in an exemplary non- EAE group, consistent with one or more exemplary embodiments of the present disclosure
- FIG. 13B illustrates an H&E stained pathological image of brain in an exemplary EAE group, consistent with one or more exemplary embodiments of the present disclosure
- FIG. 13C illustrates an H&E stained pathological image of brain in an exemplary EAE- GA group, consistent with one or more exemplary embodiments of the present disclosure
- FIG. 13D Illustrates an H&E stained pathological image of brain in an exemplary EAEGA/Depot group, consistent with one or more exemplary embodiments of the present disclosure
- FIG. 14A illustrates an H&E stained pathological image of skin in an exemplary non- EAE group, consistent with one or more exemplary embodiments of the present disclosure
- FIG. 14B illustrates an H&E stained pathological image of skin in an exemplary EAE- GA group, consistent with one or more exemplary embodiments of the present disclosure
- FIG. 14C illustrates an H&E stained pathological image of skin in an exemplary EAEGA/Depot group, consistent with one or more exemplary embodiments of the present disclosure
- FIG. 15A illustrates a Luxol fast blue (H&E) stained pathological image captured from first of spinal cord of an exemplary EAE group for demyelination comparison, consistent with one or more exemplary embodiments of the present disclosure
- FIG. 15B illustrates a Luxol fast blue (LFB) stained pathological image captured from end of spinal cord of an exemplary EAE group for demyelination comparison, consistent with one or more exemplary embodiments of the present disclosure
- FIG. 15C illustrates a LFB stained pathological image captured from first of spinal cord of an exemplary GA group for demyelination comparison, consistent with one or more exemplary embodiments of the present disclosure
- FIG. 15D illustrates a LFB stained pathological image captured from end of spinal cord of an exemplary EAE-GA group for demyelination comparison, consistent with one or more exemplary embodiments of the present disclosure
- FIG. 15E illustrates a LFB stained pathological image captured from end of spinal cord of an exemplary EAE-GA/Depot group for demyelination comparison, consistent with one or more exemplary embodiments of the present disclosure.
- an exemplary drug may include glatiramer acetate (GA).
- GA may be used for decreasing symptoms of relapsing remitting multiple sclerosis (RRMS). Patients using GA may have symptoms of at least one of vasodilatation, chest pain, dyspnoea, palpitations, tachycardia due to immediate post-injection reaction (IPIR), and combinations thereof. Increasing receiving dosage of GA may increase exemplary symptoms which may discourage patients from continuing treatments.
- an exemplary parental pharmaceutical composition may include a plurality of powder particles and a phosphate-buffered saline (PBS) solution.
- PBS phosphate-buffered saline
- an exemplary plurality of powder particles may be dispersed inside an exemplary PBS solution before injection.
- an exemplary plurality of powder particles may be stable in an exemplary PBS solution for a time of maximum 7 days.
- an exemplary dispersion of an exemplary plurality of powder particles and an exemplary PBS solution may be injected to a body at a time of maximum 7 days after dispersing an exemplary plurality of powder particles in an exemplary PBS solution.
- each particle of an exemplary plurality of the powder particles may have an average particle size in a range of 5 pm to 75 pm.
- a pH value of an exemplary PBS solution may be in a range of 6.0 to 8.0.
- an exemplary pharmaceutical composition may include a weight ratio of an exemplary PBS solution to an exemplary plurality of powder particles in a range of 1: 2 to 8: 9 (the PBS solution: the plurality of the powder particles).
- an exemplary pharmaceutical composition may be injected intramuscularly (IM) into a body of a patient.
- IM intramuscularly
- an exemplary dispersion of an exemplary plurality of powder particles in an exemplary PBS solution may transform into a gel when being introduced into a body of a patient.
- an exemplary dispersion of an exemplary plurality of powder particles in an exemplary PBS solution may transform into a gel due to a change of at least one of pH, temperature, salt content, and combinations thereof in a target tissue being injected therein.
- an exemplary plurality of powder particles may include a first water-based portion, an oil-based portion, and a second water-based portion.
- an exemplary first water-based portion and an exemplary oil-based portion may form a plurality of elementary particles.
- an exemplary second water-based portion may form a hydrogen layer over each elementary particle of a plurality of elementary particles.
- an exemplary first water-based portion, an exemplary oil-based portion, and an exemplary second water-based portion may form a plurality of hydrogel -particle structures.
- each hydrogel-particle structure of an exemplary plurality of hydrogel-particle structures may include a hydrogel phase, a non-hydrogel phase, and combinations thereof.
- an exemplary first water based-portion and an exemplary second water-based portion may form an exemplary hydrogel phase in each hydrogel-particle structure of an exemplary plurality of hydrogel -particle structures.
- each hydrogel-particle structure of an exemplary plurality of hydrogel-particle structures may form an interconnected matrix of an exemplary hydrogel phase and an exemplary non-hydrogel phase.
- an exemplary first water-based portion may include at least one of glatiramer acetate (GA), sodium alginate, polyvinyl alcohol (PVA), polyoxyethylene (80) sorbitan monooleate (polysorbate 80), and combinations thereof.
- an exemplary first water-based portion may include sodium alginate with a weight ratio of an exemplary sodium alginate to GA in a range of 0.1 : 1 to 0.4: 4 (sodium alginate: GA).
- an exemplary first water-based portion may include PVA with a weight ratio of PVA to an exemplary sodium alginate in a range of 0.05: 0.005 to 0.02: 0.2 (PVA: sodium alginate).
- an exemplary first water-based portion may include polysorbate 80 with a weight ratio of an exemplary polysorbate 80 to an exemplary sodium alginate in a range of 1: 3 to 5: 15 (polysorbate 80: sodium alginate).
- an exemplary oil-based portion may include at least one of a-Tocopherol, poly(lactic-co-glycolic acid) (PLGA), and combinations thereof.
- an exemplary oil-based portion may include a-Tocopherol with a weight ratio of an exemplary a-Tocopherol to an exemplary sodium alginate in a range of 0: 1 to 0.3: 3 (a-Tocopherol: sodium alginate).
- an exemplary oilbased portion may include PLGA with a weight ratio of PLGA to an exemplary sodium alginate in a range of 0: 5 to 3: 30 (PLGA: sodium alginate).
- an exemplary PLGA may include at least one of an acid-terminated PLGA with a molecular weight in a range of 7000 g/mol to 17000 g/mol, an ester-terminated PLGA with a molecular weight in a range of 7000 g/mol to 17000 g/mol, an acid-terminated PLGA with a molecular weight in a range of 24000 g/mol to 38000 g/mol, and combinations thereof.
- an exemplary second water-based portion may include at least one of GA, chitosan, cremophor EL, and combinations thereof.
- an exemplary second water-based portion may include GA with a weight ratio of an exemplary GA to an exemplary sodium alginate in a range of 0.1: 1 to 1.1: 5 (GA: sodium alginate).
- an exemplary second water-based portion may include chitosan with a weight ratio of an exemplary chitosan to an exemplary sodium alginate in a range of 0.15: 1.5 to 0.1: 20 (chitosan: sodium alginate).
- an exemplary second water-based portion may include cremophor EL with a weight ratio of an exemplary cremophor EL to an exemplary sodium alginate in a range of 0: 1 to 0.5: 5 (cremophor EL: sodium alginate).
- an exemplary plurality of powder particles of an exemplary pharmaceutical composition may include glatiramer acetate with a weight percent in a range of 4 wt. % to 80 wt. % of a total amount of an exemplary parenteral pharmaceutical composition.
- an exemplary pharmaceutical composition may form a plurality of particle -hydrogel structures in an exemplary PBS solution.
- each particle-hydrogel structure of an exemplary plurality of hydrogel-particle structures may have a spherical structure.
- each particle -hydrogel structure of an exemplary plurality of hydrogel-particle structures may have an average diameter in a range of 5 pm to 75 pm.
- each particle-hydrogel structure of an exemplary plurality of hydrogel -particle structures may include a particle (nonhydrogel) portion and a hydrogel portion.
- each particle-hydrogel structure of an exemplary plurality of hydrogel-particle structures may include an interconnected matrix between an exemplary particle portion and an exemplary hydrogel portion.
- exemplary connections between an exemplary particle portion and an exemplary hydrogel portion may include at least one of a chemical connection, a physical connection, and combinations thereof.
- exemplary connections between an exemplary particle portion and an exemplary hydrogel portion may decrease releasing GA from an exemplary plurality of hydrogel-particle structures.
- releasing GA from an exemplary plurality of hydrogel-particle structures may include at least one of diffusion from an exemplary hydrogel portion, degradation of an exemplary hydrogel portion, diffusion from an exemplary particle portion, and combinations thereof.
- an exemplary plurality of powder particles of an exemplary parental pharmaceutical composition may be stored at a temperature in a range of 15 °C to 30 °C.
- an exemplary plurality of powder particles of an exemplary parenteral pharmaceutical composition may remain active at a temperature in a range of 15 °C to 30 °C.
- an exemplary parenteral pharmaceutical composition may include a releasing rate of GA in a range of 20 % to 90 % of a total amount of an exemplary GA within 30 days in body after introducing an exemplary parenteral pharmaceutical composition into an exemplary patient’s body.
- GA may be released into a body of a patient when an exemplary parenteral pharmaceutical composition may be injected intramuscularly into an exemplary patient’s body.
- an exemplary parenteral pharmaceutical composition may have a releasing rate of GA in a range of 20 % to 90 % of a total amount of an exemplary GA within 30 days when an exemplary parenteral pharmaceutical composition may be injected intramuscularly into a patient’s body.
- FIG. 1A illustrates a flowchart of a method 100 of reducing symptoms of elapsing remitting multiple sclerosis (RRMS), consistent with one or more exemplary embodiments of the present disclosure.
- method 100 may include a step 102 of forming a water/oil/water emulsion, a step 104 of forming a plurality of powder particles of the water/oil/water emulsion, a step 106 of forming a parenteral pharmaceutical composition, and a step 108 of injecting a predetermined amount of the formed parenteral pharmaceutical composition intramuscularly to a patient suffering from RRMS.
- step 102 of forming a water/oil/water emulsion may be illustrated in FIG. IB.
- FIG. IB illustrates a flowchart of a method 110 of forming an exemplary water/oil/water emulsion, consistent with one or more exemplary embodiments of the present disclosure.
- method 110 may include a step 112 of forming an internal aqueous phase, a step 114 of forming an organic phase, a step 116 of forming a plurality of elementary particles by mixing a normal saline solution, PVA, the organic phase, and the internal aqueous phase together, and a step 118 of forming an external aqueous phase connected to an outer surface of the plurality of elementary particles.
- step 112 of forming an internal aqueous phase may include mixing polyvinyl alcohol (PVA), polyoxyethylene (80) sorbitan monooleate (polysorbate 80), GA, and sodium alginate in a mixer for a time period in a range of 10 minutes to 100 minutes.
- PVA, polysorbate 80, GA, and sodium alginate may be mixed in a mixer with a stirring speed in a range of 500 rpm to 5000 rpm.
- PVA, polysorbate 80, GA, and sodium alginate may be mixed at a temperature in a range of 10 °C to 40 °C.
- an exemplary internal aqueous phase may include PVA with a weight ratio of an exemplary PVA to an exemplary sodium alginate in a range of 0.05: 0.005 to 0.02: 0.2 (PVA: sodium alginate).
- an exemplary internal aqueous phase may include GA with a weight ratio of sodium alginate to GA in a range of 0.1: 5 to 1:20 (sodium alginate: GA).
- an exemplary internal aqueous phase may include polysorbate 80 with a weight ratio of polysorbate 80 to sodium alginate in a range of 1: 3 to 5: 15 (polysorbate 80: sodium alginate).
- step 114 of forming an organic phase may include adding a-Tocopherol and (lactic-co-glycolic acid) (PLGA) to an exemplary internal aqueous solution.
- a-Tocopherol and (lactic-co-glycolic acid) (PLGA) may be added to an exemplary internal aqueous solution.
- an exemplary a-Tocopherol, PLGA, and an exemplary internal aqueous solution may be mixed using an ultrasonic device with an ultrasonic power in a range of 200 W/cm 2 to 1000 W/cm 2 .
- an exemplary a-Tocopherol, PLGA, and an exemplary internal aqueous solution may be mixed for a time period in a range of 2 minutes to 60 minutes.
- an exemplary organic phase may include an exemplary a-Tocopherol with a weight ratio of an exemplary a-Tocopherol to an exemplary sodium alginate in a range of 0: 1 to 0.3: 3 (a- Tocopherol: sodium alginate).
- an exemplary organic phase may include an exemplary PLGA to an exemplary sodium alginate with a weight ratio in a range of 0: 5 to 3: 30 (PLGA: sodium alginate).
- an exemplary PLGA may include at least one of an acid terminated PLGA with a molecular weight in a range of 7000 g/mol to 17000 g/mol, an ester terminated PLGA with a molecular weight in a range of 7000 g/mol to 17000 g/mol, an acid terminated PLGA with a molecular weight in a range of 24000 g/mol to 38000 g/mol, and combinations thereof.
- step 116 of forming a plurality of elementary particles may include adding a normal saline solution and PVA into an exemplary mixer containing an exemplary organic phase and an exemplary internal aqueous phase.
- an exemplary normal saline solution, an exemplary PVA, an exemplary organic phase, and an exemplary internal aqueous phase may be mixed together using an exemplary mixer with a stirring speed in a range of 500 rpm to 5000 rpm.
- an exemplary normal saline solution, an exemplary PVA, an exemplary organic phase, and an exemplary internal aqueous phase may be mixed together for a timed period in a range of 30 minutes to 10 hours.
- an exemplary normal saline solution, an exemplary PVA, an exemplary organic phase, and an exemplary internal aqueous phase may be mixed at a temperature in a range of 15 °C to 40 °C.
- an exemplary normal saline solution may have a concentration in a range of 0.45 mol/L to 0.9 mol/L.
- an exemplary plurality of elementary particles may be suspended inside an exemplary normal saline solution and PVA.
- each respective elementary particle may include an exemplary organic phase forming an interconnected matrix with an exemplary internal aqueous phase.
- an exemplary suspension of an exemplary plurality of elementary particles in an exemplary normal saline solution and PVA may include an exemplary PVA with a weight ratio to sodium alginate in a range of 0.05: 0.005 to 0.02: 0.2 (PVA: sodium alginate).
- step 118 of forming an external aqueous phase may include adding GA, chitosan, and cremophor EL into an exemplary plurality of elementary particles suspended in an exemplary normal saline solution and PVA.
- an exemplary plurality of elementary particles may be mixed with an exemplary GA, an exemplary chitosan, and an exemplary cremophor EL together using an ultrasonic device with an ultrasonic power in a range of 200 W/cm 2 to 1000 W/cm 2
- an exemplary plurality of elementary particles may be mixed with an exemplary GA, an exemplary chitosan, and an exemplary cremophor EL together for a time period in a range of 10 minutes to 100 minutes.
- an exemplary plurality of elementary particles may be mixed with an exemplary GA, an exemplary chitosan, and an exemplary cremophor EL together using an exemplary ultrasonic device for more than one cycle.
- an exemplary plurality of elementary particles may be mixed with an exemplary GA, an exemplary chitosan, and an exemplary cremophor EL together using an exemplary ultrasonic device for two cycles.
- an exemplary plurality of hydrogel-particle structures may be formed according to a coacervation process.
- an exemplary coacervation process may include forming an exemplary plurality of elementary particles in an exemplary normal saline solution and PVA.
- a water/oil/water emulsion may be formed by adding an exemplary external aqueous phase into an exemplary suspension of elementary particles in an exemplary normal saline solution and PVA.
- an exemplary external aqueous phase may form a liquid polymer around an exemplary plurality of elementary particles.
- an exemplary liquid polymer may turn into a hydrogel.
- an exemplary hydrogel may solidify after freeze drying an exemplary plurality of hydrogel -particle structures.
- an exemplary external aqueous phase may include GA with a weight ratio of an exemplary GA to an exemplary sodium alginate in a range of 0.1: 1 to 1.1: 5 (GA: sodium alginate).
- an exemplary external aqueous phase may include chitosan with a weight ratio of an exemplary chitosan to an exemplary sodium alginate in a range of 0.15: 1.5 to 0.1: 20 (chitosan: sodium alginate).
- an exemplary external aqueous phase may include cremophor EL with a weight ratio of an exemplary cremophor EL to an exemplary sodium alginate in a range of 0: 1 to 0.5: 5 (cremophor EL: sodium alginate).
- step 104 of forming a plurality of powder particles of an exemplary water/oil/water emulsion may include washing an exemplary water/oil/water emulsion.
- an exemplary plurality of powder particles dispersed in an exemplary water/oil/water emulsion may be mixed with a phosphate-buffered saline (PBS) solution in a mixer with a stirring speed in a range of 500 rpm to 500 rpm.
- PBS phosphate-buffered saline
- an exemplary plurality of powder particles dispersed in an exemplary water/oil/water emulsion may be mixed with an exemplary PBS solution for a time period in a range of 1 minute to 20 minutes.
- washing an exemplary water/oil/water emulsion may remove unreacted GA from an exemplary water/oil/water emulsion.
- an exemplary PBS solution may have a pH value in a range of 6 to 8.
- an exemplary PBS solution may be mixed with an exemplary water/oil/water emulsion with a weight ratio of an exemplary water/oil/water emulsion to an exemplary PBS solution in a range of 1: 3 to 7: 9 (water/oil/water emulsion: PBS).
- forming an exemplary plurality of powder particles of an exemplary water/oil/water emulsion may include freezing an exemplary water/oil/water emulsion and drying an exemplary water/oil/water emulsion.
- an exemplary plurality of powder particles of an exemplary water/oil/water emulsion may be formed utilizing a freeze dryer.
- forming an exemplary plurality of powder particles of an exemplary water/oil/water emulsion may include freezing an exemplary water/oil/water emulsion at a temperature in a range of -60 °C to -10 °C.
- an exemplary water/oil/water emulsion may be frozen for a time period in a range of 1 hour to 10 hours.
- drying an exemplary water/oil/water emulsion may include initial drying an exemplary water/oil/water emulsion and secondary drying an exemplary water/oil/water emulsion.
- initial drying an exemplary water/oil/water emulsion may include drying an exemplary frozen water/oil/water emulsion at a temperature in a range of -5 °C to -40 °C.
- initial drying an exemplary water/oil/water emulsion may include drying an exemplary frozen water/oil/water emulsion for a time period in a range of 10 hours to 40 hours.
- secondary drying an exemplary water/oil/water emulsion may include drying an exemplary frozen water/oil/water emulsion at a temperature in a range of 10 °C to 30 °C.
- secondary drying an exemplary water/oil/water emulsion may include drying an exemplary frozen dispersed powder particles in an exemplary water/oil/water emulsion for a time period in a range of 5 hours to 20 hours.
- drying an exemplary plurality of powder particles dispersed in an exemplary water/oil/water emulsion may be performed under a vacuum pressure in a range of 0.01 mbar to 0.1 mbar.
- an exemplary plurality of powder particles may be stored at a temperature in a range of 15 °C to 30 °C.
- an exemplary plurality of powder particles may be stable (active) after a time period of maximum 2 years.
- each particle of an exemplary plurality of powder particles may have an average particle size in a range of 5 pm to 75 pm.
- step 106 of forming a parenteral pharmaceutical composition may include mixing an exemplary plurality of powder particles with a phosphate-buffered saline (PBS) solution.
- PBS phosphate-buffered saline
- mixing an exemplary plurality of powder particles with an exemplary PBS solution may form a suspension of an exemplary plurality of powder particles in an exemplary PBS solution.
- an exemplary plurality of powder particles may be mixed with an exemplary PBS solution in a mixer with a stirring speed in a range of 500 rpm to 5000 rpm.
- an exemplary plurality of powder particles may be mixed with an exemplary PBS solution for a time period in a range of 30 minutes to 10 hours.
- an exemplary PBS solution may have a pH value in a range of 6 to 8.
- an exemplary plurality of powder particles may be mixed with an exemplary PBS solution with a weight ratio of an exemplary plurality of powder particles to an exemplary PBS solution in a range of 2: 1 to 9: 8 (an exemplary plurality of powder particles: PBS solution).
- an exemplary suspension of an exemplary plurality of powder particles may be stable in an exemplary PBS solution for a time of maximum 7 days.
- an exemplary suspension of an exemplary plurality of powder particles in an exemplary PBS solution may be stable after a time period of maximum 7 days.
- step 108 of injecting a predetermined amount of an exemplary formed parenteral pharmaceutical composition may include injecting intramuscularly a predetermined amount of an exemplary formed parenteral pharmaceutical composition to a patient suffering from RRMS.
- an exemplary patient suffering from RRMS may be at least a human, an animal, and combinations thereof.
- an exemplary parenteral pharmaceutical composition may be injected intramuscularly to a mouse.
- an exemplary plurality of powder particles of an exemplary pharmaceutical composition may include GA with a weight percent to a total amount of an exemplary parenteral pharmaceutical composition in a range of 4 wt. % to 80 wt. %.
- injecting an exemplary formed parenteral pharmaceutical composition intramuscularly to a mouse may include injecting an amount of an exemplary formed parenteral pharmaceutical composition in a range of 15 mg/kg to 200 mg/kg of an exemplary pharmaceutical composition every month to an exemplary mouse suffering from RRMS.
- an exemplary parenteral pharmaceutical composition may include a releasing rate of an exemplary GA in a range of 20 % to 90 % of a total amount of an exemplary GA within 30 days when an exemplary parenteral pharmaceutical composition may be injected intramuscularly into a patient’s body.
- ionic interaction between negatively charged carboxyl groups of exemplary polymers and positive charge of lysine group of GA may enhance binding capacity of GA to an exemplary plurality of powder particles.
- an exemplary polymer may include synthetic and natural polymers.
- enhancing binding capacity of GA to an exemplary plurality of powder particles may decrease release rate of GA.
- an exemplary parenteral pharmaceutical composition may transform to a hydrogel when an exemplary parenteral pharmaceutical composition may be injected intramuscularly into a patient’s body.
- an exemplary parenteral pharmaceutical composition may transform from an exemplary suspension of an exemplary plurality of powder particles in an exemplary PBS solution to a hydrogel due to change in pH and exposure to high concentration of salt in body tissues.
- forming an exemplary gel may be due to gel formation of at least one of alginate, chitosan, and combinations thereof.
- Example 1 Synthesizing a plurality of powder particles of a pharmaceutical composition for reducing RRMS symptoms
- a method similar to method 100 and method 110 may was used for synthesizing powder particles of a pharmaceutical composition for reducing RRMS symptoms.
- an internal aqueous phase, an external aqueous phase, and an organic phase were mixed together.
- a water/oil/water (W/O/W) emulsion was produced by mixing an exemplary internal aqueous phase, an exemplary external aqueous phase, and an exemplary organic phase.
- the internal aqueous phase and an exemplary external aqueous phase include glatiramer acetate, vitamin E succinate, polyvinyl alcohol (MW 30-70k), and polysorbate 80.
- An exemplary organic phase includes methylene chloride and ethanol.
- Biodegradable polymers including sodium alginate 1% was dissolved in an exemplary internal aqueous phase.
- Chitosan aqueous solution 2 % was added to an exemplary external aqueous phase, and poly (D, L,- lactide-co -glycolide) (PLGA) was dispersed in an exemplary organic phase in different proportions.
- PLGA poly (D, L,- lactide-co -glycolide)
- glatiramer acetate (GA) and sodium alginate were dissolved with a weight ratio of 1:3 (GA: sodium alginate) in an aqueous solution containing PVA 70 kDa 80 (1-2% w/w) and polysorbate 80 and added to an exemplary organic phase containing a-Tocopherol.
- PLGA in a ratio of 1: 1: 1 to 1:2:1 (PLGA 502: PLGA 503: PLGA H503) was added to an exemplary mixture and was homogenized for 20 minutes in 4 cycles of 5 minutes utilizing an ultrasonic probe with 100% power.
- addition of 0.9% normal saline solution containing 35 kDa PVA with a concentration of 1-2% w/w, temperature change and reduction of stirrer speed were used to form the primary nucleus of particles by a coacervation method.
- the aqueous phase containing GA, chitosan and cremophor EL were added to an exemplary above particle system and homogenized again in 2 cycles of 5 minutes at 100% power utilizing an exemplary ultrasonic probe.
- two methods of freeze drying and spray drying have been used to dry the particles.
- an exemplary external aqueous phase contains 30 ml of 0.9% sodium chloride containing 1.5% PVP with a molecular weight of 70 kDa and GA.
- the internal aqueous phase is prepared with a concentration of 2 mg/ml of GA and polysorbate 80-cremophor EL in a final ratio of 2% by weight.
- an exemplary organic phase To prepare an exemplary organic phase, firstly, PLGA506, PLGA503, and PLGA502h were dispersed in methylene chloride at a ratio of 1:2:1 and at the same time during sonication, a-Tocopherol was added to an exemplary organic phase at a final ratio of 1.5% (w/w). For homogenization and stability of double emulsions in final stage, an ultrasonic probe with a frequency of 100 Hz and a power of 90% was used until the size of exemplary particles after drying according to diameter of exemplary emulsified particles in an exemplary external aqueous phase were in a range of 10 pm to 55 pm.
- Table 1 illustrates formulations of exemplary produced emulsions for synthesizing exemplary powder particles of an exemplary pharmaceutical composition, consistent with one or more exemplary embodiments of the present disclosure. [0076] Table 1. Formulations of exemplary produced emulsions for synthesizing exemplary powder particles of an exemplary pharmaceutical composition.
- the prepared primary emulsion of an exemplary internal aqueous phase and an exemplary organic phase were added to an exemplary external aqueous phase.
- An exemplary mixture of an exemplary internal aqueous phase, an exemplary organic phase, and an exemplary external aqueous phase were mixed using a mixer.
- the prepared powder particles were washed using a phosphate buffer saline (PBS) solution with a pH of 7.4 to remove unreacted drugs.
- PBS phosphate buffer saline
- the method of drying was chosen based on the results obtained from drug loading and the surface morphology of microparticles and their non-spherical shape, and according to the surface pores and their effect on the way of drug release.
- Particles had a lot of surface pores due to high speed of drying of the spray drying method. Surface pores of particles can increase the speed of GA release with zero degree kinetics, which is not an appropriate method in controlling the release of GA for 30 days.
- freeze drying is optimized by setting the freeze stage up to a maximum of 7 hours and slowing down the primary drying to the range of 20 hours to 35 hours and continuing the drying to the secondary stage up to 10 hours to 15 hours, a suitable spherical shape was created. More specifically, the W/O/W emulsion was first frozen at -43 °C for 3 hours. Drying was followed by primary drying at -20 °C for 24 hours, and secondary drying at 20 °C for 15 hours.
- the second part of the formulation is the saline phosphate buffer solution with a pH in a range of 5 to 7 to form a sol solution before intramuscular injection.
- the PBS solution intended to disperse the particles. After injecting the sol solution due to the change in pH and exposure to high concentration of salt, the sol solution coverts to gel.
- FIG. 2 illustrates a transmission electron microscopy (TEM) image 200 of an exemplary prepared powder particles of an exemplary pharmaceutical composition, consistent with one or more exemplary embodiments of the present disclosure.
- TEM transmission electron microscopy
- Exemplary powder particles produced by a method similar to method 100 and method 110 had spherical, smooth, and non-aggregated micrographs under an optical microscope. To observe surfaces and morphological properties of exemplary powder particles more clearly, both conventional PLGA microparticles and exemplary produced powder particles of an exemplary pharmaceutical composition were observed using an SEM device.
- FIG. 3 illustrates a scanning electron microscopy (SEM) image 300 of G-Mix22 sample before freeze drying, consistent with one or more exemplary embodiments of the present disclosure.
- FIG. 4 illustrates a SEM image 400 of G-Mix22 sample after freeze drying, consistent with one or more exemplary embodiments of the present disclosure.
- FIG. 3 illustrates a scanning electron microscopy (SEM) image 300 of G-Mix22 sample before freeze drying, consistent with one or more exemplary embodiments of the present disclosure.
- FIG. 4 illustrates a SEM image 400 of G-Mix22 sample after freeze drying, consistent with one or more exemplary embodiments of the present disclosure.
- FIG. 5 illustrates a SEM image 500 of particles based on PLGA, consistent with one or more exemplary embodiments of the present disclosure.
- FIG. 6 illustrates a SEM image 600 of particles based on GA, chitosan, and sodium alginate (GA-CH-A1), consistent with one or more exemplary embodiments of the present disclosure.
- exemplary powder particles of an exemplary pharmaceutical composition based on GA-CH-A1 have spherical configuration with a smooth surface.
- FIG. 7 illustrates a TEM image 700 of GA-CH-A1 particles, consistent with one or more exemplary embodiments of the present disclosure.
- FIG. 8 illustrates a particle size distribution diagram 800 of exemplary powder particles of an exemplary pharmaceutical composition, consistent with one or more exemplary embodiments of the present disclosure.
- Particle size distribution diagram (802) and particle size distribution diagram (804) represent two tests for exemplary powder particles of an exemplary pharmaceutical composition.
- Example 2 Glatiramer acetate loading and entrapment efficiency (EE%) measurements
- GA glatiramer acetate
- EE% measurements of glatiramer acetate (GA) were analyzed. 4.8 mg of a GA stock solution was dissolved in 10 ml deionized water to form a GA solution with a concentration of 400 pg/ml. The GA had Assay about 87% and water content -5.5%. Then 7 ml of borate buffer 0.1 M (reaction buffer) was added into the GA solution.
- borate buffer 0.1 M reaction buffer
- Example 3 In-vitro glatiramer acetate release study from the produced powder particles of the pharmaceutical composition
- Example 1 For analyzing releasing parameters of exemplary prepared powder particles of an exemplary pharmaceutical composition prepared in Example 1 hereinabove, 50 mg of freeze dried powder particles was removed from an exemplary product sample and was added to a glass vial containing 20 ml of a PBS buffer solution with a pH of 7.4 and sodium azide 0.05%. Then 0.5 ml of the samples were collected at 0, 3 days, 6 days, 12 days, 18 days, 24 days, and 30 days. The samples were centrifuged at 10000 rpm for 5 minutes. 50 pl of supernatant was diluted with 0.5 ml of a borate buffer solution with a concentration of 0.1 M.
- FIG. 9 illustrates GA-release profile 900 of GMIX22 (902), GMIX21 (904), GC9 (906), and GPU (908) samples after 30 days, consistent with one or more exemplary embodiments of the present disclosure.
- results of GA release from GA-CH-AL hydrogel microparticles suggested that GA/polymer ratio, polymer type, hydrophobic counter ion (a-Tocopherol), and polymer charge and polymer molecular weight play roles in the GA release pattern. Suitable peptide binding was required for sufficient GA loading. However, cross linkage of a-Tocopherol’s diacid and polyamine molecules of polymer would prevent initial burst release and slowed down the release pattern in the first 1-12 days. Multi-point ionic interaction between negative charge carboxyl groups of polymers and positive charge of GA lysine directly influenced binding capacity and release rate.
- Example 4 Studying efficiency of the prepared parental pharmaceutical composition in experimental autoimmune encephalomyelitis (EAE) animal model
- EAE Experimental autoimmune encephalomyelitis
- MS multiples sclerosis
- EAE was induced by subcutaneous (sc.) injection of Myelin oligodendrocyte glycoprotein (MOG) emulsion followed by intraperitoneal injection (ip.) of Bordetella Pertussis Toxin (PTX) on day 0 and 48 hours post MOG administration.
- EAE may be assessed by daily clinical scoring of mice for 14 days post immunization according to a scale (0-5). Normal mice were sored as 0 while 1 was for limb tail, or hind limb weakness, 2 was for the limp tail and hind limb weakness, 3 was for the partial limb paralysis, 4 stand for complete hind limb paralysis and 5 was for the moribund state during 14 days.
- the Mice were kept in a specific pathogen free room with a temperature of 25+2 °C, relative humidity (RH) of 50%, 12 hours light, and 12 hours dark with free access to food and water. Anesthesia was not essential, however, immunization was done under isoflurane anesthesia. Immunization was performed as follows. Antigen (MOG35-55) was first emulsified in complete Freund’s adjuvant. The prepared emulsion was injected at maximum of 3 sites, 0.1 mL/site (0.3 mL/mouse total). PTX was freshly diluted with cold PBS and was kept in an ice bath for 2 hours before injection.
- RH relative humidity
- PTX was injected at a dose of 200 ng/mouse intraperitoneal (I.P.) on day 0 and day 1, under sterile conditions using a biosafety cabinet to induce autoimmune encephalitis.
- Typical EAE beginning is 9 days to 14 days after immunization, with the peak of disease 3 days to 5 days after EAE start for each mouse. The peak maintains 1 day to 3 days, followed by partial recovery.
- These four groups include a group A, a group B, a group c, and a group D.
- the group A include 12 participants of negative control-induction of encephalomyelitis by EAE method and by injection of carrier on days 0-9.
- the group B include 12 participants of positive control- injectable GA (commercial product) with a dose of 2 mg subcutaneously on days 0 and 1.
- the group C includes 12 participants receiving GA-DEPOT test samples with a dose of 10 mg intramuscularly on day 0 and 1 in two divided doses.
- the group D includes 6 negative control samples- healthy- no injection.
- mice Due to chronically paralysis of mice during this period, water and food were given to them individually to keep them alive and special care was taken to reduce their mortality.
- Mouse stress prior to EAE development greatly decreases EAE intensity, thus minimizing mouse sensitivity and stress was achieved by optimizing lab environmental conditions such as light, sounds, temperature and humidity, appropriate mouse handling, and suitable S.C. injection for successful EAE induction.
- Table 2 illustrates dosing regimens of an exemplary produced powder particles of an exemplary pharmaceutical composition in the EAE model, consistent with one or more exemplary embodiments of the present disclosure.
- Table 2 Groups and dosing regimens in the EAE model.
- mice were killed and selected organs were extracted and fixed for pathological studies. Data were analyzed by Anova test followed by Dunnetts’ post-Hoc test, and the Friedman test followed by Dunn’s multiple comparison test (p ⁇ 0.05 was considered as significant).
- EAE model mean clinical score and average body weight of mice have been investigated. Clinical signs of EAE and average body weight after treatment with GA and GA/DEPOT were investigated. Mice were typically monitored for 4 weeks, during which mice were remained chronically paralyzed. Their weight and clinical symptoms were reported daily. Mean body weights were examined daily for 28 days post immunization with MOG.
- FIG. 10 illustrates a chart 1000 of comparative studies of mean body weights in four study groups of non-EAE (1002), EAE-vehicle (1004), EAE-GA (1006), and EAE/GA/Depot (1008) mice over 28 days, consistent with one or more exemplary embodiments of the present disclosure.
- p ⁇ 0.05 is shown by *
- p ⁇ 0.01 is shown by **
- p ⁇ 0.001 is shown by ***.
- FIG. 11 illustrates a chart 1100 of comparative studies of mean clinical scores of non-EAE (1102), EAE-vehicle (1104), EAE-GA (1106), and EAE/GA/Depot (1108) mice over 28 days, consistent with one or more exemplary embodiments of the present disclosure.
- p ⁇ 0.05 is shown by *
- p ⁇ 0.01 is shown by **
- p ⁇ 0.001 is shown by ***.
- GA-CH-AL hydrogel microparticles completely prevent EAE development compared to 91% inhibition seen in GA commercial drug treated group.
- GA-CH-AL hydrogel microparticles significantly prevented body weight loss and ameliorated clinical symptoms in comparison to the negative control (non-EAE) group.
- mice enrolled in the EAE model were euthanized at the end of 28 days. Demyelination and inflammatory reactions in the brain and spinal cord tissues were also examined by LFB (Luxol fast blue) staining and H&E (Hematoxylin and Eosin) staining according to well established protocols. Liver, kidney, and skin samples from the mice injection site were also collected in all groups for toxicological and pathological studies by H&E staining.
- FIG. 12A illustrates an H&E stained pathological image 120 of spinal cord in an exemplary non-EAE group, consistent with one or more exemplary embodiments of the present disclosure.
- FIG. 12B illustrates an H&E stained pathological image 122 of spinal cord in an exemplary EAE group, consistent with one or more exemplary embodiments of the present disclosure.
- FIG. 12C illustrates an H&E stained pathological image 124 of spinal cord in an exemplary EAEGA group, consistent with one or more exemplary embodiments of the present disclosure.
- FIG. 12D illustrates an H&E stained pathological image 126 of spinal cord in an exemplary EAEGA/Depot group, consistent with one or more exemplary embodiments of the present disclosure.
- FIG. 12A illustrates an H&E stained pathological image 120 of spinal cord in an exemplary non-EAE group, consistent with one or more exemplary embodiments of the present disclosure.
- FIG. 12B illustrates an H&E stained pathological image 122 of spinal cord in an exemplary EAE group, consistent with one or more
- FIG. 13A illustrates an H&E stained pathological image 130 of brain in an exemplary non-EAE group, consistent with one or more exemplary embodiments of the present disclosure.
- FIG. 13B illustrates an H&E stained pathological image 132 of brain in an exemplary EAE group, consistent with one or more exemplary embodiments of the present disclosure.
- FIG. 13C illustrates an H&E stained pathological image 134 of brain in an exemplary EAE-GA group, consistent with one or more exemplary embodiments of the present disclosure.
- FIG. 13D illustrates an H&E stained pathological image 136 of brain in an exemplary EAEGA/Depot group, consistent with one or more exemplary embodiments of the present disclosure. As it is observed in FIG. 12A-D and FIG.
- FIG. 14A illustrates an H&E stained pathological image 140 of skin in an exemplary non-EAE group, consistent with one or more exemplary embodiments of the present disclosure.
- FIG. 14B illustrates an H&E stained pathological image 142 of skin in an exemplary EAE-GA group, consistent with one or more exemplary embodiments of the present disclosure.
- FIG. 14C illustrates an H&E stained pathological image 144 of skin in an exemplary EAE-GA/Depot group, consistent with one or more exemplary embodiments of the present disclosure.
- Infiltrations of hypodermic inflammatory cells and granulomatous are common adverse effects of GA conventional parenteral formulation which plays a key role in poor patient compliance who are receiving GA conventional parenteral formulation (FIG. 14B).
- FIG. 15A illustrates a Luxol fast blue (H&E) stained pathological image 150 captured from first of spinal cord of an exemplary EAE group for demyelination comparison, consistent with one or more exemplary embodiments of the present disclosure.
- H&E Luxol fast blue
- FIG. 15B illustrates a LFB stained pathological image 152 captured from end of spinal cord of an exemplary EAE group for demyelination comparison, consistent with one or more exemplary embodiments of the present disclosure.
- FIG. 15C illustrates a LFB stained pathological image 154 captured from first of spinal cord of an exemplary GA group for demyelination comparison, consistent with one or more exemplary embodiments of the present disclosure.
- FIG. 15D illustrates a LFB stained pathological image 156 captured from end of spinal cord of an exemplary EAE-GA group for demyelination comparison, consistent with one or more exemplary embodiments of the present disclosure.
- FIG. 15B illustrates a LFB stained pathological image 152 captured from end of spinal cord of an exemplary EAE group for demyelination comparison, consistent with one or more exemplary embodiments of the present disclosure.
- FIG. 15C illustrates a LFB stained pathological image 154 captured from first of spinal cord of an exemplary GA group for demyelination comparison,
- FIG. 15E illustrates a LFB stained pathological image 158 captured from end of spinal cord of an exemplary EAE- GA/Depot group for demyelination comparison, consistent with one or more exemplary embodiments of the present disclosure
- FIG. 15 illustrate demyelination in spinal cord cross sectional samples in EAE-group, GA-group, and GA/Depot groups.
- FIG. 15A-D moderate to severe demyelination in superior, medial, and lateral segments of the white region in the first and end parts of spinal cords are detectable.
- no sign of demyelination is observed in GA/Depot groups spinal cord samples (FIG. 15E).
- a parental pharmaceutical composition may be formed and used here for reducing symptoms of elapsing remitting multiple sclerosis (RRMS).
- the parental pharmaceutical composition may release glatiramer acetate when injected intramuscularly to a patient.
- the parental pharmaceutical composition may be prescribed by a doctor.
- the parental pharmaceutical composition may be administered at hospitals and clinics.
- the parental pharmaceutical composition may be administered at home with a doctor’s prescription for reducing symptoms of RRMS.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Epidemiology (AREA)
- Inorganic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Neurology (AREA)
- General Chemical & Material Sciences (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Dermatology (AREA)
- Organic Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
Abstract
A parenteral pharmaceutical composition for reducing symptoms of elapsing remitting multiple sclerosis (RRMS) is disclosed. The parenteral pharmaceutical composition includes a first water-based portion, an oil-based portion, and a second water-based portion. The first water-based portion includes glatiramer acetate, sodium alginate, polyvinyl alcohol (PVA), and polysorbate 80. The oil-based portion includes α-Tocopherol and poly(lactic-co-glycolic acid) (PLGA). The second water-based portion includes glatiramer acetate, chitosan, and cremophor EL. The parenteral pharmaceutical composition includes an interconnected matrix of the first water-based portion, the oil-based portion, and the second water-based portion.
Description
DRUG DELIVERY USING A PARENTERAL PHARMACEUTICAL COMPOSITION
TECHNICAL FIELD
[0001] The present disclosure generally relates to a pharmaceutical composition for drug delivery, and more particularly, relates to a method of producing a drug-delivery- pharmaceutical composition for reducing symptoms of elapsing remitting multiple sclerosis (RRMS).
BACKGROUND ART
[0002] Multiple sclerosis (MS) is a potentially disabling disease of central nervous system. MS is an autoimmune disease that immune system of body attacks to protective sheath covering nerve fibers. MS is categorized into five different groups of benign multiple sclerosis, relapsing-remitting multiple sclerosis (RRMS), secondary progressive multiple sclerosis (SPMS), primary progressive multiple sclerosis (PPMS), and progressive-relapsing multiple sclerosis (PRMS). MS symptoms may vary from numbness or weakness in one or more limbs, tingling, electric-shock sensations, blurry vision, fatigue, etc. MS can deteriorate quality of life and put economic burden on families and governments.
[0003] Different methods are used to reduce symptoms of MS. These methods include medication, therapies, such as physiotherapy, and self-management techniques. The medication used for reducing symptoms of MS includes parenteral medications and oral medications. Glatiramer acetate (GA) is one of effective drugs for reducing symptoms of MS. clinical tests revealed that oral administration of GA can inhibit functionality of GA due to acidic media of digestive system. However, parenteral administration of GA may cause initial burst release of high dosage of GA in body.
[0004] There are many methods and materials used for reducing symptoms of MS. For example, Ehud Marom et al. presented a patent on “Depot systems comprising glatiramer or pharmacologically acceptable salt thereof’ (US 8377885 B2). Ehud Marom et al. synthesized a composition including a therapeutically effective amount of a pharmaceutically acceptable glatiramer acetate. Glatiramer acetate was administered using a depot method. Nadav et al. presented a patent on “Process for preparing microparticles containing glatiramer acetate” (WO 2018/042423 Al). Nadav et al. produced microparticles containing glatiramer acetate having
low levels of residual organic solvents. However, the aforementioned methods suffer from toxicity due to organic solvents and possibility of initial burst release of glatiramer acetate.
[0005] There is, therefore, a need for a biocompatible and effective pharmaceutical composition for releasing glatiramer acetate with a controlled releasing rate into body. There is further a need for a cost effective method to produce a pharmaceutical composition with a controlled releasing rate of glatiramer acetate.
SUMMARY OF THE DISCLOSURE
[0006] This summary is intended to provide an overview of the subject matter of this patent, and is not intended to identify essential elements or key elements of the subject matter, nor is it intended to be used to determine the scope of the claimed implementations. The proper scope of this patent may be ascertained from the claims set forth below in view of the detailed description below and the drawings.
[0007] According to one or more exemplary embodiments, the present disclosure is directed to a parenteral pharmaceutical composition for reducing symptoms of elapsing remitting multiple sclerosis (RRMS). In an exemplary embodiment, an exemplary parenteral pharmaceutical composition may include a plurality of powder particles dispersed in a phosphate-buffered saline (PBS) solution. In an exemplary embodiment, each powder particle of an exemplary plurality of powder particles may include a first water-based portion, an oilbased portion, and a second water-based portion. In an exemplary embodiment, an exemplary first water-based portion may include glatiramer acetate (GA), sodium alginate with a weight ratio of an exemplary sodium alginate to an exemplary glatiramer acetate in a range of 0.1: 1 to 0.4: 4 (sodium alginate: glatiramer acetate), polyvinyl alcohol (PVA) with a weight ratio of PVA to an exemplary sodium alginate in a range of 0.05: 0.005 to 0.02: 0.2 (PVA: sodium alginate), and polysorbate 80 with a weight ratio of an exemplary polysorbate 80 to an exemplary sodium alginate in a range of 1: 3 to 5: 15 (polysorbate 80: sodium alginate). In an exemplary embodiment, an exemplary oil-based portion may include a-Tocopherol with a weight ratio of an exemplary a-Tocopherol to an exemplary sodium alginate in a range of 0: 1 to 0.3: 3 (a-Tocopherol: sodium alginate) and poly(lactic-co-glycolic acid) (PLGA) with a weight ratio of PLGA to an exemplary sodium alginate in a range of 0: 5 to 3: 30 (PLGA: sodium alginate). In an exemplary embodiment, an exemplary second aqueous portion may include glatiramer acetate with a weight ratio of an exemplary glatiramer acetate to an
exemplary sodium alginate in a range of 0.1: 1 to 1.1: 5 (glatiramer acetate: sodium alginate), chitosan with a weight ratio of an exemplary chitosan to an exemplary sodium alginate in a range of 0.05 : 1.5 to 0.1 : 20 (chitosan: sodium alginate), and cremophor EL with a weight ratio of an exemplary cremophor EL to an exemplary sodium alginate in a range of 0:1 to 0.5:5 (cremophor EL: sodium alginate). In an exemplary embodiment, an exemplary parenteral pharmaceutical composition may include an interconnected matrix of an exemplary first waterbased portion, an exemplary oil-based portion, and an exemplary second water-based portion. In an exemplary embodiment, a weight ratio of an exemplary PBS solution to an exemplary plurality of exemplary powder particles may be in a range of 1: 2 to 8: 9 (an exemplary PBS solution: an exemplary plurality of the powder particles).
[0008] According to one or more exemplary embodiments, the present disclosure is directed to a parenteral pharmaceutical composition for reducing symptoms of elapsing remitting multiple sclerosis (RRMS). In an exemplary embodiment, an exemplary parenteral pharmaceutical composition may include a plurality of powder particles dispersed in a phosphate-buffered saline (PBS) solution. In an exemplary embodiment, each powder particle of an exemplary plurality of powder particles may include a first water-based portion, an oilbased portion, and a second water-based portion. In an exemplary embodiment, an exemplary first water-based portion may include glatiramer acetate, sodium alginate, polyvinyl alcohol (PVA), and polysorbate 80. In an exemplary embodiment, an exemplary oil-based portion may include a-Tocopherol and poly(lactic-co-glycolic acid) (PLGA).In an exemplary embodiment, an exemplary second water-based portion may include glatiramer acetate, chitosan, and cremophor EL.
[0009] In an exemplary embodiment, an exemplary plurality of powder particles of an exemplary pharmaceutical composition may include glatiramer acetate with a weight percent to a total amount of an exemplary parenteral pharmaceutical composition in a range of 4 wt. % to 80 wt. %.
[0010] In an exemplary embodiment, an exemplary PLGA may include at least one of an acid terminated PLGA with a molecular weight in a range of 7000 g/mol to 17000 g/mol, an ester terminated PLGA with a molecular weight in a range of 7000 g/mol to 17000 g/mol, an acid terminated PLGA with a molecular weight in a range of 24000 g/mol to 38000 g/mol, and combinations thereof.
[0011] In an exemplary embodiment, a pH value of an exemplary PBS solution may be in a range of 6.0 to 8.0. In an exemplary embodiment, an exemplary parenteral pharmaceutical composition may include a releasing rate of an exemplary glatiramer acetate in a range of 20 % to 90 % of a total amount of an exemplary glatiramer acetate within 30 days when an exemplary parenteral pharmaceutical composition may be injected intramuscularly into a patient’s body.
[0012] In an exemplary embodiment, each particle of an exemplary plurality of exemplary powder particles may have an average particle size in a range of 5 pm to 75 pm. In an exemplary embodiment, an exemplary plurality of powder particles of an exemplary parenteral pharmaceutical composition may be stored at a temperature in a range of 15 °C to 30 °C with a relative humidity of maximum 70%.
[0013] According to one or more exemplary embodiments, the present disclosure is directed to a method for reducing symptoms of elapsing remitting multiple sclerosis (RRMS). In an exemplary embodiment, an exemplary method may include forming a water/oil/water emulsion, forming a plurality of powder particles of an exemplary water/oil/water emulsion by drying an exemplary water/oil/water emulsion using a freeze dryer, forming a parenteral pharmaceutical composition, and injecting a predetermined amount of an exemplary formed parenteral pharmaceutical composition intramuscularly to a patient suffering from RRMS. In an exemplary embodiment, forming an exemplary water/oil/water emulsion may include forming an internal aqueous phase by mixing sodium alginate, glatiramer acetate, polyvinyl alcohol (PVA), and polysorbate 80 together, forming an organic phase by mixing an exemplary internal aqueous phase, a-Tocopherol, and poly(lactic-co-glycolic acid) (PLGA) together, forming a plurality of elementary particles by mixing a normal saline solution, PVA, an exemplary organic phase, and an exemplary internal aqueous phase together, and forming an external aqueous phase connected to an outer surface of an exemplary plurality of elementary particles by mixing an exemplary plurality of elementary particles, glatiramer acetate, chitosan, and cremophor EL together. In an exemplary embodiment, forming an exemplary parenteral pharmaceutical composition may include mixing an exemplary plurality of powder particles of an exemplary water/oil/water emulsion with a phosphate-buffered saline (PBS) solution with a weight ratio of an exemplary plurality of exemplary powder particles of an exemplary water/oil/water emulsion to an exemplary PBS solution in a range of 1: 3 to 7: 9 (an exemplary
plurality of the powder particles of an exemplary water/oil/water emulsion: an exemplary PBS solution).
[0014] In an exemplary embodiment, forming an exemplary water/oil/water emulsion may further include washing an exemplary plurality of powder particles of an exemplary water/oil/water emulsion by mixing an exemplary plurality of powder particles of an exemplary water/oil/water emulsion with a PBS solution with a weight ratio of an exemplary plurality of powder particles of an exemplary water/oil/water emulsion to an exemplary PBS solution in a range of 1 : 3 to 7: 9 (an exemplary plurality of powder particles of an exemplary water/oil/water emulsion: the PBS solution).
[0015] In an exemplary embodiment, washing an exemplary plurality of powder particles of an exemplary water/oil/water emulsion may include mixing an exemplary plurality of powder particles of an exemplary water/oil/water emulsion with an exemplary PBS solution with a pH value in a range of 6 to 8.
[0016] In an exemplary embodiment, forming an exemplary internal aqueous phase may include mixing glatiramer acetate, sodium alginate at a relative weight ratio to an exemplary glatiramer acetate in a range of 0.1: 5 to 1:20 (sodium alginate: glatiramer acetate), polyvinyl alcohol (PVA) at a relative weight ratio to an exemplary glatiramer acetate in a range of 0.01: 0.1 to 0.03: 0.5 (PVA: glatiramer acetate), and polysorbate 80 with a weight ratio of an exemplary polysorbate 80 to glatiramer acetate in a range of 1: 3 to 3: 5 (polysorbate 80: glatiramer acetate) together using a mixer with a stirring speed in a range of 500 rpm to 5000 rpm.
[0017] In an exemplary embodiment, mixing sodium alginate, glatiramer acetate, polyvinyl alcohol (PVA), and polysorbate 80 together may include mixing sodium alginate, glatiramer acetate, polyvinyl alcohol (PVA), and polysorbate 80 together using a mixer with a stirring speed in a range of 500 rpm to 5000 rpm for a time period in a range of 10 minutes to 100 minutes.
[0018] In an exemplary embodiment, forming an exemplary organic phase may include mixing an exemplary internal aqueous phase, a-Tocopherol with a weight ratio of the a-Tocopherol to glatiramer acetate in a range of 0:2 to 1 :5 (a-Tocopherol: glatiramer acetate), and PLGA with a weight ratio of the PLGA to glatiramer acetate in a range of 1: 5 to 5: 50 (PLGA: glatiramer acetate) together using an ultrasonic device with an ultrasonic power in a range of 200 W/cm2 to 1000 W/cm2.
[0019] In an exemplary embodiment, mixing an exemplary internal aqueous phase, a- Tocopherol, and PLGA together may include mixing an exemplary internal aqueous phase, a- Tocopherol, and PLGA together using an ultrasonic device for a time period in a range of 2 minutes to 60 minutes.
[0020] In an exemplary embodiment, forming an exemplary external aqueous phase by mixing an exemplary plurality of elementary particles, glatiramer acetate, chitosan, and cremophor EL together may be done using an ultrasonic device with an ultrasonic power in a range of 200 W/cm2 to 1000 W/cm2.
[0021] In an exemplary embodiment, forming an exemplary external aqueous phase by mixing an exemplary plurality of elementary particles, glatiramer acetate, chitosan, and cremophor EL together may be done using an ultrasonic device for a time period in a range of 10 minutes to 100 minutes.
[0022] In an exemplary embodiment, drying an exemplary water/oil/water emulsion may include freezing an exemplary water/oil/water emulsion at a temperature in a range of -60 °C to -10 °C for a time period in a range of 1 hour to 10 hours and drying an exemplary water/oil/water emulsion. In an exemplary embodiment, drying an exemplary water/oil/water emulsion may include initial drying an exemplary water/oil/water emulsion at a temperature in a range of -5 °C to -40 °C for a time period in a range of 10 hours to 40 hours and secondary drying an exemplary water/oil/water emulsion at a temperature in a range of 10 °C to 30 °C for a time period in a range of 5 hours to 20 hours.
BRIEF DESCRIPTION OF THE DRAWINGS
[0023] The drawing figures depict one or more implementations in accord with the present teachings, by way of example only, not by way of limitation. In the figures, like reference numerals refer to the same or similar elements.
[0024] FIG. 1A illustrates a flowchart of a method of reducing symptoms of elapsing remitting multiple sclerosis (RRMS), consistent with one or more exemplary embodiments of the present disclosure;
[0025] FIG. IB illustrates a flowchart of a method of forming an exemplary water/oil/water emulsion, consistent with one or more exemplary embodiments of the present disclosure;
[0026] FIG. 2 illustrates a transmission electron microscopy (TEM) image of an exemplary prepared powder particles of an exemplary pharmaceutical composition, consistent with one or more exemplary embodiments of the present disclosure;
[0027] FIG. 3 illustrates a scanning electron microscopy (SEM) image of G-Mix22 sample before freeze drying, consistent with one or more exemplary embodiments of the present disclosure;
[0028] FIG. 4 illustrates a SEM image of G-Mix22 sample after freeze drying, consistent with one or more exemplary embodiments of the present disclosure;
[0029] FIG. 5 illustrates a SEM image of particles based on PLGA, consistent with one or more exemplary embodiments of the present disclosure;
[0030] FIG. 6 illustrates a SEM image of particles based on GA, chitosan, and sodium alginate (GA-CH-A1), consistent with one or more exemplary embodiments of the present disclosure;
[0031] FIG. 7 illustrates a TEM image of GA-CH-A1 particles, consistent with one or more exemplary embodiments of the present disclosure;
[0032] FIG. 8 illustrates a particle size distribution diagram of an exemplary plurality of powder particles of an exemplary pharmaceutical composition, consistent with one or more exemplary embodiments of the present disclosure;
[0033] FIG. 9 illustrates GA-release profiles of GMIX22, GMIX21, GC9, and GPU samples after 30 days, consistent with one or more exemplary embodiments of the present disclosure;
[0034] FIG. 10 illustrates a chart of comparative studies of mean body weights in four study groups of non-EAE, EAE-vehicle, EAE-GA, and EAE/GA/Depot mice over 28 days, consistent with one or more exemplary embodiments of the present disclosure;
[0035] FIG. 11 illustrates a chart of comparative studies of mean clinical scores of non-EAE, EAE-vehicle, EAE-GA, and EAE/GA/Depot mice over 28 days, consistent with one or more exemplary embodiments of the present disclosure;
[0036] FIG. 12A illustrates an Hematoxylin and Eosin (H&E) stained pathological image of spinal cord in an exemplary non- experimental autoimmune encephalomyelitis (EAE) group, consistent with one or more exemplary embodiments of the present disclosure;
[0037] FIG. 12B illustrates an H&E stained pathological image of spinal cord in an exemplary EAE group, consistent with one or more exemplary embodiments of the present disclosure.;
[0038] FIG. 12C illustrates an H&E stained pathological image of spinal cord in an exemplary EAEGA group, consistent with one or more exemplary embodiments of the present disclosure;
[0039] FIG. 12D illustrates an H&E stained pathological image of spinal cord in an exemplary EAEGA/Depot group, consistent with one or more exemplary embodiments of the present disclosure;
[0040] FIG. 13A illustrates an H&E stained pathological image of brain in an exemplary non- EAE group, consistent with one or more exemplary embodiments of the present disclosure;
[0041] FIG. 13B illustrates an H&E stained pathological image of brain in an exemplary EAE group, consistent with one or more exemplary embodiments of the present disclosure;
[0042] FIG. 13C illustrates an H&E stained pathological image of brain in an exemplary EAE- GA group, consistent with one or more exemplary embodiments of the present disclosure;
[0043] FIG. 13D Illustrates an H&E stained pathological image of brain in an exemplary EAEGA/Depot group, consistent with one or more exemplary embodiments of the present disclosure;
[0044] FIG. 14A illustrates an H&E stained pathological image of skin in an exemplary non- EAE group, consistent with one or more exemplary embodiments of the present disclosure;
[0045] FIG. 14B illustrates an H&E stained pathological image of skin in an exemplary EAE- GA group, consistent with one or more exemplary embodiments of the present disclosure;
[0046] FIG. 14C illustrates an H&E stained pathological image of skin in an exemplary EAEGA/Depot group, consistent with one or more exemplary embodiments of the present disclosure;
[0047] FIG. 15A illustrates a Luxol fast blue (H&E) stained pathological image captured from first of spinal cord of an exemplary EAE group for demyelination comparison, consistent with one or more exemplary embodiments of the present disclosure;
[0048] FIG. 15B illustrates a Luxol fast blue (LFB) stained pathological image captured from end of spinal cord of an exemplary EAE group for demyelination comparison, consistent with one or more exemplary embodiments of the present disclosure;
[0049] FIG. 15C illustrates a LFB stained pathological image captured from first of spinal cord of an exemplary GA group for demyelination comparison, consistent with one or more exemplary embodiments of the present disclosure;
[0050] FIG. 15D illustrates a LFB stained pathological image captured from end of spinal cord of an exemplary EAE-GA group for demyelination comparison, consistent with one or more exemplary embodiments of the present disclosure; and
[0051] FIG. 15E illustrates a LFB stained pathological image captured from end of spinal cord of an exemplary EAE-GA/Depot group for demyelination comparison, consistent with one or more exemplary embodiments of the present disclosure.
DESCRIPTION OF EMBODIMENTS
[0053] In the following detailed description, numerous specific details are set forth by way of examples in order to provide a thorough understanding of the relevant teachings. However, it should be apparent that the present teachings may be practiced without such details. In other instances, well known methods, procedures, components, and/or circuitry have been described at a relatively high-level, without detail, in order to avoid unnecessarily obscuring aspects of the present teachings.
[0054] The novel features which are believed to be characteristic of the present disclosure, as to its structure, organization, use and method of operation, together with further objectives and advantages thereof, will be better understood from the following discussion. In the following detailed description, numerous specific details are set forth by way of examples in order to provide a thorough understanding of the relevant teachings. However, it should be apparent that the present teachings may be practiced without such details. In other instances, well known methods, procedures, components, and/or circuitry have been described at a relatively high- level, without detail, in order to avoid unnecessarily obscuring aspects of the present teachings. The following detailed description is presented to enable a person skilled in the art to make and use the methods and devices disclosed in exemplary embodiments of the present disclosure. For purposes of explanation, specific nomenclature is set forth to provide a thorough understanding of the present disclosure. However, it will be apparent to one skilled in the art that these specific details are not required to practice the disclosed exemplary embodiments. Descriptions of specific exemplary embodiments are provided only as representative examples. Various modifications to the exemplary implementations will be readily apparent to one skilled in the art, and the general principles defined herein may be applied to other implementations and applications without departing from the scope of the present disclosure. The present disclosure is not intended to be limited to the implementations shown, but is to be accorded the widest possible scope consistent with the principles and features disclosed herein.
[0055] The present disclosure is directed to exemplary embodiments of a method for controlled drug release into a body using a parental pharmaceutical composition and a method to produce an exemplary parental pharmaceutical composition. In an exemplary embodiment, an exemplary drug may include glatiramer acetate (GA). In an exemplary embodiment, GA may be used for decreasing symptoms of relapsing remitting multiple sclerosis (RRMS). Patients using GA may have symptoms of at least one of vasodilatation, chest pain, dyspnoea,
palpitations, tachycardia due to immediate post-injection reaction (IPIR), and combinations thereof. Increasing receiving dosage of GA may increase exemplary symptoms which may discourage patients from continuing treatments.
[0056] In an exemplary embodiment, an exemplary parental pharmaceutical composition may include a plurality of powder particles and a phosphate-buffered saline (PBS) solution. In an exemplary embodiment, an exemplary plurality of powder particles may be dispersed inside an exemplary PBS solution before injection. In an exemplary embodiment, an exemplary plurality of powder particles may be stable in an exemplary PBS solution for a time of maximum 7 days. In an exemplary embodiment, an exemplary dispersion of an exemplary plurality of powder particles and an exemplary PBS solution may be injected to a body at a time of maximum 7 days after dispersing an exemplary plurality of powder particles in an exemplary PBS solution. In an exemplary embodiment, each particle of an exemplary plurality of the powder particles may have an average particle size in a range of 5 pm to 75 pm. In an exemplary embodiment, a pH value of an exemplary PBS solution may be in a range of 6.0 to 8.0. In an exemplary embodiment, an exemplary pharmaceutical composition may include a weight ratio of an exemplary PBS solution to an exemplary plurality of powder particles in a range of 1: 2 to 8: 9 (the PBS solution: the plurality of the powder particles). In an exemplary embodiment, an exemplary pharmaceutical composition may be injected intramuscularly (IM) into a body of a patient. In an exemplary embodiment, an exemplary dispersion of an exemplary plurality of powder particles in an exemplary PBS solution may transform into a gel when being introduced into a body of a patient. In an exemplary embodiment, an exemplary dispersion of an exemplary plurality of powder particles in an exemplary PBS solution may transform into a gel due to a change of at least one of pH, temperature, salt content, and combinations thereof in a target tissue being injected therein.
[0057] In an exemplary embodiment, an exemplary plurality of powder particles may include a first water-based portion, an oil-based portion, and a second water-based portion. In an exemplary embodiment, an exemplary first water-based portion and an exemplary oil-based portion may form a plurality of elementary particles. In an exemplary embodiment, an exemplary second water-based portion may form a hydrogen layer over each elementary particle of a plurality of elementary particles. In an exemplary embodiment, an exemplary first water-based portion, an exemplary oil-based portion, and an exemplary second water-based portion may form a plurality of hydrogel -particle structures. In an exemplary embodiment, each
hydrogel-particle structure of an exemplary plurality of hydrogel-particle structures may include a hydrogel phase, a non-hydrogel phase, and combinations thereof. In an exemplary embodiment, an exemplary first water based-portion and an exemplary second water-based portion may form an exemplary hydrogel phase in each hydrogel-particle structure of an exemplary plurality of hydrogel -particle structures. In an exemplary embodiment, each hydrogel-particle structure of an exemplary plurality of hydrogel-particle structures may form an interconnected matrix of an exemplary hydrogel phase and an exemplary non-hydrogel phase.
[0058] In an exemplary embodiment, an exemplary first water-based portion may include at least one of glatiramer acetate (GA), sodium alginate, polyvinyl alcohol (PVA), polyoxyethylene (80) sorbitan monooleate (polysorbate 80), and combinations thereof. In an exemplary embodiment, an exemplary first water-based portion may include sodium alginate with a weight ratio of an exemplary sodium alginate to GA in a range of 0.1 : 1 to 0.4: 4 (sodium alginate: GA). In an exemplary embodiment, an exemplary first water-based portion may include PVA with a weight ratio of PVA to an exemplary sodium alginate in a range of 0.05: 0.005 to 0.02: 0.2 (PVA: sodium alginate). In an exemplary embodiment, an exemplary first water-based portion may include polysorbate 80 with a weight ratio of an exemplary polysorbate 80 to an exemplary sodium alginate in a range of 1: 3 to 5: 15 (polysorbate 80: sodium alginate).
[0059] In an exemplary embodiment, an exemplary oil-based portion may include at least one of a-Tocopherol, poly(lactic-co-glycolic acid) (PLGA), and combinations thereof. In an exemplary embodiment, an exemplary oil-based portion may include a-Tocopherol with a weight ratio of an exemplary a-Tocopherol to an exemplary sodium alginate in a range of 0: 1 to 0.3: 3 (a-Tocopherol: sodium alginate). In an exemplary embodiment, an exemplary oilbased portion may include PLGA with a weight ratio of PLGA to an exemplary sodium alginate in a range of 0: 5 to 3: 30 (PLGA: sodium alginate). In an exemplary embodiment, an exemplary PLGA may include at least one of an acid-terminated PLGA with a molecular weight in a range of 7000 g/mol to 17000 g/mol, an ester-terminated PLGA with a molecular weight in a range of 7000 g/mol to 17000 g/mol, an acid-terminated PLGA with a molecular weight in a range of 24000 g/mol to 38000 g/mol, and combinations thereof.
[0060] In an exemplary embodiment, an exemplary second water-based portion may include at least one of GA, chitosan, cremophor EL, and combinations thereof. In an exemplary
embodiment, an exemplary second water-based portion may include GA with a weight ratio of an exemplary GA to an exemplary sodium alginate in a range of 0.1: 1 to 1.1: 5 (GA: sodium alginate). In an exemplary embodiment, an exemplary second water-based portion may include chitosan with a weight ratio of an exemplary chitosan to an exemplary sodium alginate in a range of 0.15: 1.5 to 0.1: 20 (chitosan: sodium alginate). In an exemplary embodiment, an exemplary second water-based portion may include cremophor EL with a weight ratio of an exemplary cremophor EL to an exemplary sodium alginate in a range of 0: 1 to 0.5: 5 (cremophor EL: sodium alginate). In an exemplary embodiment, an exemplary plurality of powder particles of an exemplary pharmaceutical composition may include glatiramer acetate with a weight percent in a range of 4 wt. % to 80 wt. % of a total amount of an exemplary parenteral pharmaceutical composition.
[0061] In an exemplary embodiment, an exemplary pharmaceutical composition may form a plurality of particle -hydrogel structures in an exemplary PBS solution. In an exemplary embodiment, each particle-hydrogel structure of an exemplary plurality of hydrogel-particle structures may have a spherical structure. In an exemplary embodiment, each particle -hydrogel structure of an exemplary plurality of hydrogel-particle structures may have an average diameter in a range of 5 pm to 75 pm. In an exemplary embodiment, each particle-hydrogel structure of an exemplary plurality of hydrogel -particle structures may include a particle (nonhydrogel) portion and a hydrogel portion. In an exemplary embodiment, each particle-hydrogel structure of an exemplary plurality of hydrogel-particle structures may include an interconnected matrix between an exemplary particle portion and an exemplary hydrogel portion. In an exemplary embodiment, exemplary connections between an exemplary particle portion and an exemplary hydrogel portion may include at least one of a chemical connection, a physical connection, and combinations thereof.
[0062] In an exemplary embodiment, exemplary connections between an exemplary particle portion and an exemplary hydrogel portion may decrease releasing GA from an exemplary plurality of hydrogel-particle structures. In an exemplary embodiment, releasing GA from an exemplary plurality of hydrogel-particle structures may include at least one of diffusion from an exemplary hydrogel portion, degradation of an exemplary hydrogel portion, diffusion from an exemplary particle portion, and combinations thereof. In an exemplary embodiment, an exemplary plurality of powder particles of an exemplary parental pharmaceutical composition may be stored at a temperature in a range of 15 °C to 30 °C. In an exemplary embodiment, an
exemplary plurality of powder particles of an exemplary parenteral pharmaceutical composition may remain active at a temperature in a range of 15 °C to 30 °C. In an exemplary embodiment, an exemplary parenteral pharmaceutical composition may include a releasing rate of GA in a range of 20 % to 90 % of a total amount of an exemplary GA within 30 days in body after introducing an exemplary parenteral pharmaceutical composition into an exemplary patient’s body. In an exemplary embodiment, GA may be released into a body of a patient when an exemplary parenteral pharmaceutical composition may be injected intramuscularly into an exemplary patient’s body. In an exemplary embodiment, an exemplary parenteral pharmaceutical composition may have a releasing rate of GA in a range of 20 % to 90 % of a total amount of an exemplary GA within 30 days when an exemplary parenteral pharmaceutical composition may be injected intramuscularly into a patient’s body.
[0063] FIG. 1A illustrates a flowchart of a method 100 of reducing symptoms of elapsing remitting multiple sclerosis (RRMS), consistent with one or more exemplary embodiments of the present disclosure. In an exemplary embodiment, method 100 may include a step 102 of forming a water/oil/water emulsion, a step 104 of forming a plurality of powder particles of the water/oil/water emulsion, a step 106 of forming a parenteral pharmaceutical composition, and a step 108 of injecting a predetermined amount of the formed parenteral pharmaceutical composition intramuscularly to a patient suffering from RRMS.
[0064] In further detail with respect to step 102, step 102 of forming a water/oil/water emulsion may be illustrated in FIG. IB. FIG. IB illustrates a flowchart of a method 110 of forming an exemplary water/oil/water emulsion, consistent with one or more exemplary embodiments of the present disclosure. In an exemplary embodiment, method 110 may include a step 112 of forming an internal aqueous phase, a step 114 of forming an organic phase, a step 116 of forming a plurality of elementary particles by mixing a normal saline solution, PVA, the organic phase, and the internal aqueous phase together, and a step 118 of forming an external aqueous phase connected to an outer surface of the plurality of elementary particles.
[0065] In further detail with respect to step 112, step 112 of forming an internal aqueous phase may include mixing polyvinyl alcohol (PVA), polyoxyethylene (80) sorbitan monooleate (polysorbate 80), GA, and sodium alginate in a mixer for a time period in a range of 10 minutes to 100 minutes. In an exemplary embodiment, PVA, polysorbate 80, GA, and sodium alginate may be mixed in a mixer with a stirring speed in a range of 500 rpm to 5000 rpm. In an exemplary embodiment, PVA, polysorbate 80, GA, and sodium alginate may be mixed at a
temperature in a range of 10 °C to 40 °C. In an exemplary embodiment, an exemplary internal aqueous phase may include PVA with a weight ratio of an exemplary PVA to an exemplary sodium alginate in a range of 0.05: 0.005 to 0.02: 0.2 (PVA: sodium alginate). In an exemplary embodiment, an exemplary internal aqueous phase may include GA with a weight ratio of sodium alginate to GA in a range of 0.1: 5 to 1:20 (sodium alginate: GA). In an exemplary embodiment, an exemplary internal aqueous phase may include polysorbate 80 with a weight ratio of polysorbate 80 to sodium alginate in a range of 1: 3 to 5: 15 (polysorbate 80: sodium alginate).
[0066] In further detail with respect to step 114, step 114 of forming an organic phase may include adding a-Tocopherol and (lactic-co-glycolic acid) (PLGA) to an exemplary internal aqueous solution. In an exemplary embodiment, an exemplary a-Tocopherol, PLGA, and an exemplary internal aqueous solution may be mixed using an ultrasonic device with an ultrasonic power in a range of 200 W/cm2 to 1000 W/cm2. In an exemplary embodiment, an exemplary a-Tocopherol, PLGA, and an exemplary internal aqueous solution may be mixed for a time period in a range of 2 minutes to 60 minutes. In an exemplary embodiment, an exemplary organic phase may include an exemplary a-Tocopherol with a weight ratio of an exemplary a-Tocopherol to an exemplary sodium alginate in a range of 0: 1 to 0.3: 3 (a- Tocopherol: sodium alginate). In an exemplary embodiment, an exemplary organic phase may include an exemplary PLGA to an exemplary sodium alginate with a weight ratio in a range of 0: 5 to 3: 30 (PLGA: sodium alginate). In an exemplary embodiment, an exemplary PLGA may include at least one of an acid terminated PLGA with a molecular weight in a range of 7000 g/mol to 17000 g/mol, an ester terminated PLGA with a molecular weight in a range of 7000 g/mol to 17000 g/mol, an acid terminated PLGA with a molecular weight in a range of 24000 g/mol to 38000 g/mol, and combinations thereof.
[0067] In further detail with respect to step 116, step 116 of forming a plurality of elementary particles may include adding a normal saline solution and PVA into an exemplary mixer containing an exemplary organic phase and an exemplary internal aqueous phase. In an exemplary embodiment, an exemplary normal saline solution, an exemplary PVA, an exemplary organic phase, and an exemplary internal aqueous phase may be mixed together using an exemplary mixer with a stirring speed in a range of 500 rpm to 5000 rpm. In an exemplary embodiment, an exemplary normal saline solution, an exemplary PVA, an exemplary organic phase, and an exemplary internal aqueous phase may be mixed together for
a timed period in a range of 30 minutes to 10 hours. In an exemplary embodiment, an exemplary normal saline solution, an exemplary PVA, an exemplary organic phase, and an exemplary internal aqueous phase may be mixed at a temperature in a range of 15 °C to 40 °C. In an exemplary embodiment, an exemplary normal saline solution may have a concentration in a range of 0.45 mol/L to 0.9 mol/L. In an exemplary embodiment, an exemplary plurality of elementary particles may be suspended inside an exemplary normal saline solution and PVA. In an exemplary embodiment, each respective elementary particle may include an exemplary organic phase forming an interconnected matrix with an exemplary internal aqueous phase. In an exemplary embodiment, an exemplary suspension of an exemplary plurality of elementary particles in an exemplary normal saline solution and PVA may include an exemplary PVA with a weight ratio to sodium alginate in a range of 0.05: 0.005 to 0.02: 0.2 (PVA: sodium alginate). [0068] In further detail with respect to step 118, step 118 of forming an external aqueous phase may include adding GA, chitosan, and cremophor EL into an exemplary plurality of elementary particles suspended in an exemplary normal saline solution and PVA. In an exemplary embodiment, an exemplary plurality of elementary particles may be mixed with an exemplary GA, an exemplary chitosan, and an exemplary cremophor EL together using an ultrasonic device with an ultrasonic power in a range of 200 W/cm2 to 1000 W/cm2 In an exemplary embodiment, an exemplary plurality of elementary particles may be mixed with an exemplary GA, an exemplary chitosan, and an exemplary cremophor EL together for a time period in a range of 10 minutes to 100 minutes. In an exemplary embodiment, an exemplary plurality of elementary particles may be mixed with an exemplary GA, an exemplary chitosan, and an exemplary cremophor EL together using an exemplary ultrasonic device for more than one cycle. In an exemplary embodiment, an exemplary plurality of elementary particles may be mixed with an exemplary GA, an exemplary chitosan, and an exemplary cremophor EL together using an exemplary ultrasonic device for two cycles. In an exemplary embodiment, an exemplary plurality of hydrogel-particle structures may be formed according to a coacervation process. In an exemplary embodiment, an exemplary coacervation process may include forming an exemplary plurality of elementary particles in an exemplary normal saline solution and PVA. In exemplary embodiment, a water/oil/water emulsion may be formed by adding an exemplary external aqueous phase into an exemplary suspension of elementary particles in an exemplary normal saline solution and PVA. In an exemplary embodiment, an exemplary external aqueous phase may form a liquid polymer around an exemplary plurality of elementary
particles. In an exemplary embodiment, an exemplary liquid polymer may turn into a hydrogel. In an exemplary embodiment, an exemplary hydrogel may solidify after freeze drying an exemplary plurality of hydrogel -particle structures.
[0069] In an exemplary embodiment, an exemplary external aqueous phase may include GA with a weight ratio of an exemplary GA to an exemplary sodium alginate in a range of 0.1: 1 to 1.1: 5 (GA: sodium alginate). In an exemplary embodiment, an exemplary external aqueous phase may include chitosan with a weight ratio of an exemplary chitosan to an exemplary sodium alginate in a range of 0.15: 1.5 to 0.1: 20 (chitosan: sodium alginate). In an exemplary embodiment, an exemplary external aqueous phase may include cremophor EL with a weight ratio of an exemplary cremophor EL to an exemplary sodium alginate in a range of 0: 1 to 0.5: 5 (cremophor EL: sodium alginate).
[0070] Referring back to FIG. 1A, in further detail with respect to step 104, step 104 of forming a plurality of powder particles of an exemplary water/oil/water emulsion may include washing an exemplary water/oil/water emulsion. In an exemplary embodiment, an exemplary plurality of powder particles dispersed in an exemplary water/oil/water emulsion may be mixed with a phosphate-buffered saline (PBS) solution in a mixer with a stirring speed in a range of 500 rpm to 500 rpm. In an exemplary embodiment, an exemplary plurality of powder particles dispersed in an exemplary water/oil/water emulsion may be mixed with an exemplary PBS solution for a time period in a range of 1 minute to 20 minutes. In an exemplary embodiment, washing an exemplary water/oil/water emulsion may remove unreacted GA from an exemplary water/oil/water emulsion. In an exemplary embodiment, an exemplary PBS solution may have a pH value in a range of 6 to 8. In an exemplary embodiment, an exemplary PBS solution may be mixed with an exemplary water/oil/water emulsion with a weight ratio of an exemplary water/oil/water emulsion to an exemplary PBS solution in a range of 1: 3 to 7: 9 (water/oil/water emulsion: PBS).
[0071] In an exemplary embodiment, forming an exemplary plurality of powder particles of an exemplary water/oil/water emulsion may include freezing an exemplary water/oil/water emulsion and drying an exemplary water/oil/water emulsion. In an exemplary embodiment, an exemplary plurality of powder particles of an exemplary water/oil/water emulsion may be formed utilizing a freeze dryer. In an exemplary embodiment, forming an exemplary plurality of powder particles of an exemplary water/oil/water emulsion may include freezing an exemplary water/oil/water emulsion at a temperature in a range of -60 °C to -10 °C. In an
exemplary embodiment, an exemplary water/oil/water emulsion may be frozen for a time period in a range of 1 hour to 10 hours. In an exemplary embodiment, drying an exemplary water/oil/water emulsion may include initial drying an exemplary water/oil/water emulsion and secondary drying an exemplary water/oil/water emulsion. In an exemplary embodiment, initial drying an exemplary water/oil/water emulsion may include drying an exemplary frozen water/oil/water emulsion at a temperature in a range of -5 °C to -40 °C. In an exemplary embodiment, initial drying an exemplary water/oil/water emulsion may include drying an exemplary frozen water/oil/water emulsion for a time period in a range of 10 hours to 40 hours. In an exemplary embodiment, secondary drying an exemplary water/oil/water emulsion may include drying an exemplary frozen water/oil/water emulsion at a temperature in a range of 10 °C to 30 °C. In an exemplary embodiment, secondary drying an exemplary water/oil/water emulsion may include drying an exemplary frozen dispersed powder particles in an exemplary water/oil/water emulsion for a time period in a range of 5 hours to 20 hours. In an exemplary embodiment, drying an exemplary plurality of powder particles dispersed in an exemplary water/oil/water emulsion may be performed under a vacuum pressure in a range of 0.01 mbar to 0.1 mbar. In an exemplary embodiment, an exemplary plurality of powder particles may be stored at a temperature in a range of 15 °C to 30 °C. In an exemplary embodiment, an exemplary plurality of powder particles may be stable (active) after a time period of maximum 2 years. In an exemplary embodiment, each particle of an exemplary plurality of powder particles may have an average particle size in a range of 5 pm to 75 pm.
[0072] In further detail with respect to step 106, step 106 of forming a parenteral pharmaceutical composition may include mixing an exemplary plurality of powder particles with a phosphate-buffered saline (PBS) solution. In an exemplary embodiment, mixing an exemplary plurality of powder particles with an exemplary PBS solution may form a suspension of an exemplary plurality of powder particles in an exemplary PBS solution. In an exemplary embodiment, an exemplary plurality of powder particles may be mixed with an exemplary PBS solution in a mixer with a stirring speed in a range of 500 rpm to 5000 rpm. In an exemplary embodiment, an exemplary plurality of powder particles may be mixed with an exemplary PBS solution for a time period in a range of 30 minutes to 10 hours. In an exemplary embodiment, an exemplary PBS solution may have a pH value in a range of 6 to 8. In an exemplary embodiment, an exemplary plurality of powder particles may be mixed with an exemplary PBS solution with a weight ratio of an exemplary plurality of powder particles to
an exemplary PBS solution in a range of 2: 1 to 9: 8 (an exemplary plurality of powder particles: PBS solution). In an exemplary embodiment, an exemplary suspension of an exemplary plurality of powder particles may be stable in an exemplary PBS solution for a time of maximum 7 days. In an exemplary embodiment, an exemplary suspension of an exemplary plurality of powder particles in an exemplary PBS solution may be stable after a time period of maximum 7 days.
[0073] In further detail with respect to step 108, step 108 of injecting a predetermined amount of an exemplary formed parenteral pharmaceutical composition may include injecting intramuscularly a predetermined amount of an exemplary formed parenteral pharmaceutical composition to a patient suffering from RRMS. In an exemplary embodiment, an exemplary patient suffering from RRMS may be at least a human, an animal, and combinations thereof. In an exemplary embodiment, an exemplary parenteral pharmaceutical composition may be injected intramuscularly to a mouse. In an exemplary embodiment, an exemplary plurality of powder particles of an exemplary pharmaceutical composition may include GA with a weight percent to a total amount of an exemplary parenteral pharmaceutical composition in a range of 4 wt. % to 80 wt. %. In an exemplary embodiment, injecting an exemplary formed parenteral pharmaceutical composition intramuscularly to a mouse may include injecting an amount of an exemplary formed parenteral pharmaceutical composition in a range of 15 mg/kg to 200 mg/kg of an exemplary pharmaceutical composition every month to an exemplary mouse suffering from RRMS. In an exemplary embodiment, an exemplary parenteral pharmaceutical composition may include a releasing rate of an exemplary GA in a range of 20 % to 90 % of a total amount of an exemplary GA within 30 days when an exemplary parenteral pharmaceutical composition may be injected intramuscularly into a patient’s body. In an exemplary embodiment, ionic interaction between negatively charged carboxyl groups of exemplary polymers and positive charge of lysine group of GA may enhance binding capacity of GA to an exemplary plurality of powder particles. In an exemplary embodiment, an exemplary polymer may include synthetic and natural polymers. In an exemplary embodiment, enhancing binding capacity of GA to an exemplary plurality of powder particles may decrease release rate of GA. In an exemplary embodiment, an exemplary parenteral pharmaceutical composition may transform to a hydrogel when an exemplary parenteral pharmaceutical composition may be injected intramuscularly into a patient’s body. In an exemplary embodiment, an exemplary parenteral pharmaceutical composition may transform from an exemplary suspension of an
exemplary plurality of powder particles in an exemplary PBS solution to a hydrogel due to change in pH and exposure to high concentration of salt in body tissues. In an exemplary embodiment, forming an exemplary gel may be due to gel formation of at least one of alginate, chitosan, and combinations thereof.
[0074] Example 1: Synthesizing a plurality of powder particles of a pharmaceutical composition for reducing RRMS symptoms
[0075] A method similar to method 100 and method 110 may was used for synthesizing powder particles of a pharmaceutical composition for reducing RRMS symptoms. For synthesizing an exemplary plurality of powder particles of an exemplary pharmaceutical composition, an internal aqueous phase, an external aqueous phase, and an organic phase were mixed together. A water/oil/water (W/O/W) emulsion was produced by mixing an exemplary internal aqueous phase, an exemplary external aqueous phase, and an exemplary organic phase. The internal aqueous phase and an exemplary external aqueous phase include glatiramer acetate, vitamin E succinate, polyvinyl alcohol (MW 30-70k), and polysorbate 80. An exemplary organic phase includes methylene chloride and ethanol. Biodegradable polymers including sodium alginate 1% was dissolved in an exemplary internal aqueous phase. Chitosan aqueous solution 2 % was added to an exemplary external aqueous phase, and poly (D, L,- lactide-co -glycolide) (PLGA) was dispersed in an exemplary organic phase in different proportions. First, glatiramer acetate (GA) and sodium alginate were dissolved with a weight ratio of 1:3 (GA: sodium alginate) in an aqueous solution containing PVA 70 kDa 80 (1-2% w/w) and polysorbate 80 and added to an exemplary organic phase containing a-Tocopherol. PLGA in a ratio of 1: 1: 1 to 1:2:1 (PLGA 502: PLGA 503: PLGA H503) was added to an exemplary mixture and was homogenized for 20 minutes in 4 cycles of 5 minutes utilizing an ultrasonic probe with 100% power. In a second step, addition of 0.9% normal saline solution containing 35 kDa PVA with a concentration of 1-2% w/w, temperature change and reduction of stirrer speed were used to form the primary nucleus of particles by a coacervation method. In the next step, the aqueous phase containing GA, chitosan and cremophor EL were added to an exemplary above particle system and homogenized again in 2 cycles of 5 minutes at 100% power utilizing an exemplary ultrasonic probe. After washing the particles, two methods of freeze drying and spray drying have been used to dry the particles. For example, in the GPU formulation, an exemplary external aqueous phase contains 30 ml of 0.9% sodium chloride containing 1.5% PVP with a molecular weight of 70 kDa and GA. Then, the internal aqueous
phase is prepared with a concentration of 2 mg/ml of GA and polysorbate 80-cremophor EL in a final ratio of 2% by weight. To prepare an exemplary organic phase, firstly, PLGA506, PLGA503, and PLGA502h were dispersed in methylene chloride at a ratio of 1:2:1 and at the same time during sonication, a-Tocopherol was added to an exemplary organic phase at a final ratio of 1.5% (w/w). For homogenization and stability of double emulsions in final stage, an ultrasonic probe with a frequency of 100 Hz and a power of 90% was used until the size of exemplary particles after drying according to diameter of exemplary emulsified particles in an exemplary external aqueous phase were in a range of 10 pm to 55 pm. The GA entrapment efficiency of exemplary produced powder particles showed that powder particles containing a combination of neutral PLGA, 1% sodium alginate (1%), 0.5% PVA 35k, a-Tocopherol, and 1% chitosan was significantly higher in entrapment efficiency (EE%). Table 1 illustrates formulations of exemplary produced emulsions for synthesizing exemplary powder particles of an exemplary pharmaceutical composition, consistent with one or more exemplary embodiments of the present disclosure. [0076] Table 1. Formulations of exemplary produced emulsions for synthesizing exemplary powder particles of an exemplary pharmaceutical composition.
Product code GAI-5 GC6-10 GP11-15 Gmixl6-30
Ingredients:
Aqueous Phase
Glatiramer 10-40 mg 10-40 mg 10-40 mg 10-40 mg
3CC131C
Oil phase
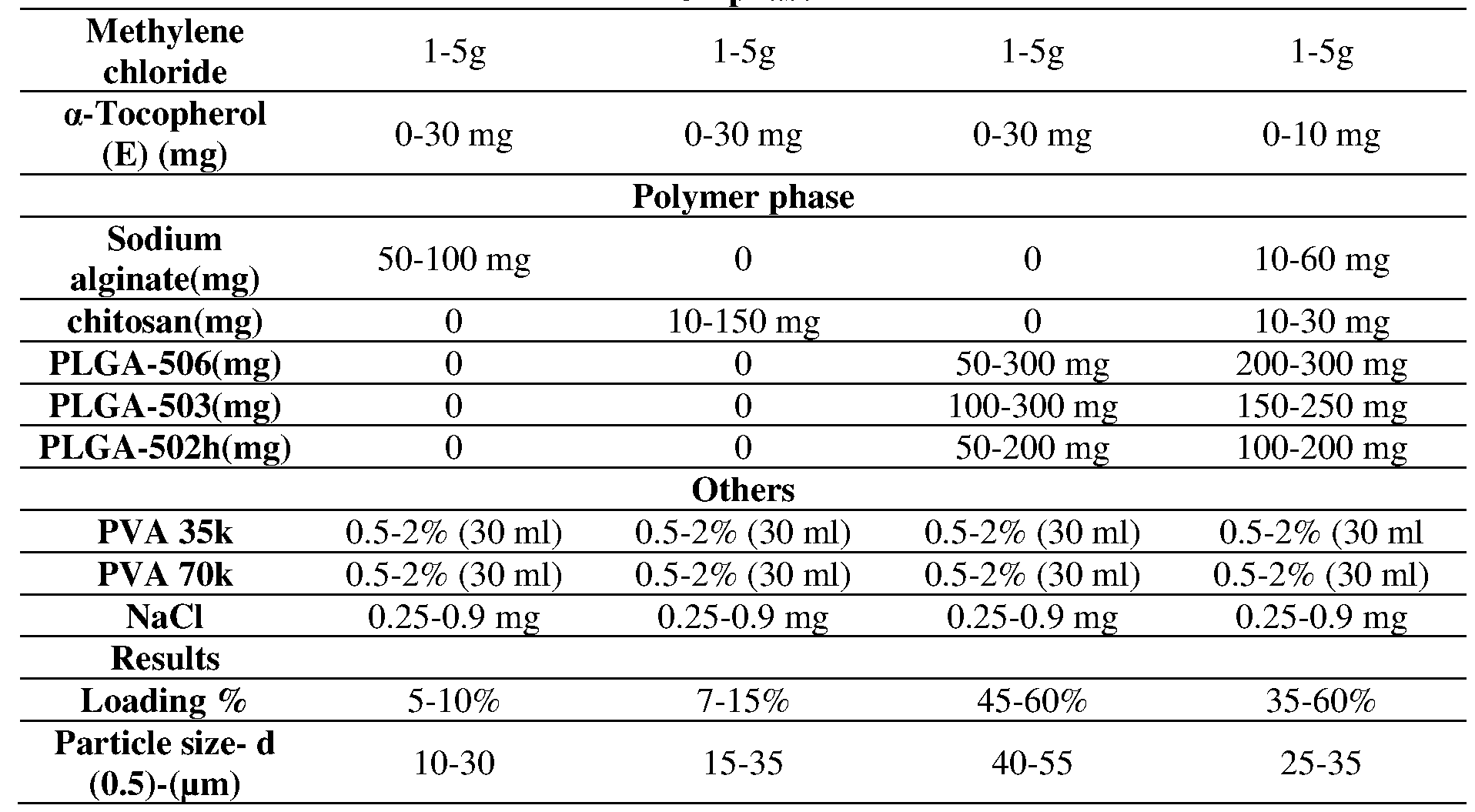 Encapsulation efficiency % 15-65% 6U-/U/o 65-85% 75-95%
Encapsulation efficiency % 15-65% 6U-/U/o 65-85% 75-95%
(EE%)
Sustained . , . , ,, ,,
, ... No No Yes Yes release profile

[0077] The prepared primary emulsion of an exemplary internal aqueous phase and an exemplary organic phase were added to an exemplary external aqueous phase. An exemplary mixture of an exemplary internal aqueous phase, an exemplary organic phase, and an exemplary external aqueous phase were mixed using a mixer. After washing the particles, the process of freeze drying or spray drying has been done. The prepared powder particles were washed using a phosphate buffer saline (PBS) solution with a pH of 7.4 to remove unreacted drugs. The method of drying was chosen based on the results obtained from drug loading and the surface morphology of microparticles and their non-spherical shape, and according to the surface pores and their effect on the way of drug release. Particles had a lot of surface pores due to high speed of drying of the spray drying method. Surface pores of particles can increase the speed of GA release with zero degree kinetics, which is not an appropriate method in controlling the release of GA for 30 days. However, in the conditions where freeze drying is optimized by setting the freeze stage up to a maximum of 7 hours and slowing down the primary drying to the range of 20 hours to 35 hours and continuing the drying to the secondary stage up to 10 hours to 15 hours, a suitable spherical shape was created. More specifically, the W/O/W emulsion was first frozen at -43 °C for 3 hours. Drying was followed by primary drying at -20 °C for 24 hours, and secondary drying at 20 °C for 15 hours. All lyophilized samples were kept at a temperature in a range of 2°C to 8 °C and were reconstituted before use. Freeze drying is known as a suitable method for the synthesis and preparation of microparticles due to control of the surface porosity of particles, the drug release kinetics of the first degree, and the control of the release within 30 days. The second part of the formulation is the saline phosphate buffer solution with a pH in a range of 5 to 7 to form a sol solution before intramuscular injection. The PBS solution intended to disperse the particles. After injecting the sol solution due to the change in pH and exposure to high concentration of salt, the sol solution coverts to gel. The gelling drug delivery system containing glatiramer acetate was used to release GA in a body.
[0078] Quantitative and qualitative evaluation of microparticles in the form of morphological examination, particle size and drug release studies were carried out within 30 days. For analyzing the powder particles of the pharmaceutical composition, the formed particles were fixed overnight with glutaraldehyde 3% in the PBS solution with a pH of 7. After dehydration and resin embedding, a thin layer with a thickness of 80 nm of SKOV3 was prepared. The prepared thin layer of SKOV3 was stained with uranyl acetate and lead citrate. FIG. 2 illustrates a transmission electron microscopy (TEM) image 200 of an exemplary prepared powder particles of an exemplary pharmaceutical composition, consistent with one or more exemplary embodiments of the present disclosure.
[0079] Exemplary powder particles produced by a method similar to method 100 and method 110 had spherical, smooth, and non-aggregated micrographs under an optical microscope. To observe surfaces and morphological properties of exemplary powder particles more clearly, both conventional PLGA microparticles and exemplary produced powder particles of an exemplary pharmaceutical composition were observed using an SEM device. FIG. 3 illustrates a scanning electron microscopy (SEM) image 300 of G-Mix22 sample before freeze drying, consistent with one or more exemplary embodiments of the present disclosure. FIG. 4 illustrates a SEM image 400 of G-Mix22 sample after freeze drying, consistent with one or more exemplary embodiments of the present disclosure. FIG. 5 illustrates a SEM image 500 of particles based on PLGA, consistent with one or more exemplary embodiments of the present disclosure. FIG. 6 illustrates a SEM image 600 of particles based on GA, chitosan, and sodium alginate (GA-CH-A1), consistent with one or more exemplary embodiments of the present disclosure. As shown in FIG. 6, exemplary powder particles of an exemplary pharmaceutical composition based on GA-CH-A1 have spherical configuration with a smooth surface. FIG. 7 illustrates a TEM image 700 of GA-CH-A1 particles, consistent with one or more exemplary embodiments of the present disclosure. FIG. 8 illustrates a particle size distribution diagram 800 of exemplary powder particles of an exemplary pharmaceutical composition, consistent with one or more exemplary embodiments of the present disclosure. Particle size distribution diagram (802) and particle size distribution diagram (804) represent two tests for exemplary powder particles of an exemplary pharmaceutical composition.
[0080] Example 2: Glatiramer acetate loading and entrapment efficiency (EE%) measurements
[0081] In this example, loading and EE% measurements of glatiramer acetate (GA) were analyzed. 4.8 mg of a GA stock solution was dissolved in 10 ml deionized water to form a GA solution with a concentration of 400 pg/ml. The GA had Assay about 87% and water content -5.5%. Then 7 ml of borate buffer 0.1 M (reaction buffer) was added into the GA solution. Standard calibration curve of GA solutions including 200 pg/ml, 100 pg/ml, 150 pg/ml, 50 pg/ml, and 20 pg/ml of GA solutions were prepared by serial dilution from stock solution 400 pg/ml in triplicate. GA assay was performed by UV-VIS spectroscopy in k=420 nm.
[0082] Example 3: In-vitro glatiramer acetate release study from the produced powder particles of the pharmaceutical composition
[0083] For analyzing releasing parameters of exemplary prepared powder particles of an exemplary pharmaceutical composition prepared in Example 1 hereinabove, 50 mg of freeze dried powder particles was removed from an exemplary product sample and was added to a glass vial containing 20 ml of a PBS buffer solution with a pH of 7.4 and sodium azide 0.05%. Then 0.5 ml of the samples were collected at 0, 3 days, 6 days, 12 days, 18 days, 24 days, and 30 days. The samples were centrifuged at 10000 rpm for 5 minutes. 50 pl of supernatant was diluted with 0.5 ml of a borate buffer solution with a concentration of 0.1 M. 50 pl trinitrobenzenesulfonic acid (TNBSA) (0.25%) was finally added to the mixture. After 30 minutes of incubation at room temperature, adsorption of the samples at 420 nm was investigated. The cumulative amount of GA release during 30 days was measured according to the calibration curve. FIG. 9 illustrates GA-release profile 900 of GMIX22 (902), GMIX21 (904), GC9 (906), and GPU (908) samples after 30 days, consistent with one or more exemplary embodiments of the present disclosure.
[0084] Results of GA release from GA-CH-AL hydrogel microparticles suggested that GA/polymer ratio, polymer type, hydrophobic counter ion (a-Tocopherol), and polymer charge and polymer molecular weight play roles in the GA release pattern. Suitable peptide binding was required for sufficient GA loading. However, cross linkage of a-Tocopherol’s diacid and polyamine molecules of polymer would prevent initial burst release and slowed down the release pattern in the first 1-12 days. Multi-point ionic interaction between negative charge carboxyl groups of polymers and positive charge of GA lysine directly influenced binding capacity and release rate. Meanwhile, lower polymer molecular weight and optimized GA/polymer ratio would provide good binding capacity and better control in initial burst release, and more reasonable release rate. As it is shown in FIG. 9, formulations containing
different PLGA types and %PVA, with or without the existence of counter ion, are distinguished in their retardants properties, the maximum cumulative amount of GA release during the 30 days and GA% release during 1-12 days were measured. Optical density calculations (y) was performed according to the following calibration equation 1 in which x is GA concentration and R2 is a regression coefficient: y = 0.0156X + 0.00099, R2 = 0.9956 (1)
[0085] Example 4: Studying efficiency of the prepared parental pharmaceutical composition in experimental autoimmune encephalomyelitis (EAE) animal model
[0086] Experimental autoimmune encephalomyelitis (EAE) is an inflammatory autoimmune demyelinating disease that can be induced in laboratory animals. Therefore, EAE would be assumed as a standard animal model for research related to autoimmune diseases such as multiples sclerosis (MS). EAE model may be applied to investigate capability of synthesized GA hydrogel microparticles, prepared in Example 1 hereinabove, as preventive care in MS management. Forty two female C57BL/6 mice were randomly enrolled in 4 groups of 6-12 weeks old and up to 25 g weight. EAE was induced by subcutaneous (sc.) injection of Myelin oligodendrocyte glycoprotein (MOG) emulsion followed by intraperitoneal injection (ip.) of Bordetella Pertussis Toxin (PTX) on day 0 and 48 hours post MOG administration. EAE may be assessed by daily clinical scoring of mice for 14 days post immunization according to a scale (0-5). Normal mice were sored as 0 while 1 was for limb tail, or hind limb weakness, 2 was for the limp tail and hind limb weakness, 3 was for the partial limb paralysis, 4 stand for complete hind limb paralysis and 5 was for the moribund state during 14 days. The Mice were kept in a specific pathogen free room with a temperature of 25+2 °C, relative humidity (RH) of 50%, 12 hours light, and 12 hours dark with free access to food and water. Anesthesia was not essential, however, immunization was done under isoflurane anesthesia. Immunization was performed as follows. Antigen (MOG35-55) was first emulsified in complete Freund’s adjuvant. The prepared emulsion was injected at maximum of 3 sites, 0.1 mL/site (0.3 mL/mouse total). PTX was freshly diluted with cold PBS and was kept in an ice bath for 2 hours before injection. PTX was injected at a dose of 200 ng/mouse intraperitoneal (I.P.) on day 0 and day 1, under sterile conditions using a biosafety cabinet to induce autoimmune encephalitis. Typical EAE beginning is 9 days to 14 days after immunization, with the peak of disease 3 days to 5 days after EAE start for each mouse. The peak maintains 1 day to 3 days, followed by partial recovery.
[0087] These four groups include a group A, a group B, a group c, and a group D. the group A include 12 participants of negative control-induction of encephalomyelitis by EAE method and by injection of carrier on days 0-9. The group B include 12 participants of positive control- injectable GA (commercial product) with a dose of 2 mg subcutaneously on days 0 and 1. The group C includes 12 participants receiving GA-DEPOT test samples with a dose of 10 mg intramuscularly on day 0 and 1 in two divided doses. The group D includes 6 negative control samples- healthy- no injection.
[0088] Due to chronically paralysis of mice during this period, water and food were given to them individually to keep them alive and special care was taken to reduce their mortality. Mouse stress prior to EAE development greatly decreases EAE intensity, thus minimizing mouse sensitivity and stress was achieved by optimizing lab environmental conditions such as light, sounds, temperature and humidity, appropriate mouse handling, and suitable S.C. injection for successful EAE induction. Table 2 illustrates dosing regimens of an exemplary produced powder particles of an exemplary pharmaceutical composition in the EAE model, consistent with one or more exemplary embodiments of the present disclosure.
[0089] Table 2. Groups and dosing regimens in the EAE model.
Dose Rout Concentration T . . . , Mg/mice/da .. .
Group Test N , . T . mL/mice/day timing e (mg/mL) y (day) S

1 PBS S‘C N/A 0.2 N/A °’9
............................ _ 1

2 hydrogel I-M 0.2 0 and 1
 microparticles
microparticles
Commercial
3 Product of GA 1 ] M 20 0 2 5 0 and 1
(positive 2 control)
Non-EAE
4 (negative 6 N/A 0 0.2 N/A 0 and 1 control)
[0090] Mean body weights were examined daily for 28 days post immunization with MOG. On day 28, mice were killed and selected organs were extracted and fixed for pathological studies. Data were analyzed by Anova test followed by Dunnetts’ post-Hoc test, and the Friedman test followed by Dunn’s multiple comparison test (p< 0.05 was considered as significant).
[0091] In the EAE model, mean clinical score and average body weight of mice have been investigated. Clinical signs of EAE and average body weight after treatment with GA and GA/DEPOT were investigated. Mice were typically monitored for 4 weeks, during which mice were remained chronically paralyzed. Their weight and clinical symptoms were reported daily. Mean body weights were examined daily for 28 days post immunization with MOG. On day 28, mice were killed and selected organs were extracted and fixed for pathological studies. Data were analyzed by Anova test followed by Dunnetts’ post-Hoc test, and the Friedman test followed by Dunn’s multiple comparison test (p< 0.05 was considered as significant). FIG. 10 illustrates a chart 1000 of comparative studies of mean body weights in four study groups of non-EAE (1002), EAE-vehicle (1004), EAE-GA (1006), and EAE/GA/Depot (1008) mice over 28 days, consistent with one or more exemplary embodiments of the present disclosure. In FIG. 10, p <0.05 is shown by *, p <0.01 is shown by ** and p <0.001 is shown by ***. FIG. 11 illustrates a chart 1100 of comparative studies of mean clinical scores of non-EAE (1102), EAE-vehicle (1104), EAE-GA (1106), and EAE/GA/Depot (1108) mice over 28 days, consistent with one or more exemplary embodiments of the present disclosure. In FIG. 11, p <0.05 is shown by *, p <0.01 is shown by ** and p <0.001 is shown by ***. As it is shown in FIG. 10 and FIG. 11 throughout the 14 days, GA-CH-AL hydrogel microparticles completely prevent EAE development compared to 91% inhibition seen in GA commercial drug treated group. GA-CH-AL hydrogel microparticles significantly prevented body weight loss and ameliorated clinical symptoms in comparison to the negative control (non-EAE) group. Parameters including mean clinical score, disease duration, and disease onset also indicated that GA-CH-AL hydrogel microparticles significantly suppressed EAE development in comparison to negative control which received vehicle. Thus, GA-CH-AL hydrogel microparticles are better than the conventional formulation when given at the same dosage level.
[0092] Stability of the synthesized product at different times after storage at room temperature (25+2 and 60% RH) has been determined and compared with the refrigerated commercial product. The results showed that the GMIX22 product is stable in non-refrigerated conditions. Table 3 illustrates stability of the synthesized product at different times after storage at room temperature, consistent with one or more exemplary embodiments of the present disclosure.
[0093] Table 3. Stability of the synthesized product at different times after storage at room temperature.
Results (n=12)
Test
Time Time Time Time
Time (0) (Monthl) (Month3) (Month6) (Monthl2)

[0094] Example 5: Histopathological studies on EAE animal model
[0095] Mice enrolled in the EAE model were euthanized at the end of 28 days. Demyelination and inflammatory reactions in the brain and spinal cord tissues were also examined by LFB (Luxol fast blue) staining and H&E (Hematoxylin and Eosin) staining according to well established protocols. Liver, kidney, and skin samples from the mice injection site were also collected in all groups for toxicological and pathological studies by H&E staining.
[0096] FIG. 12A illustrates an H&E stained pathological image 120 of spinal cord in an exemplary non-EAE group, consistent with one or more exemplary embodiments of the present disclosure. FIG. 12B illustrates an H&E stained pathological image 122 of spinal cord in an exemplary EAE group, consistent with one or more exemplary embodiments of the present disclosure. FIG. 12C illustrates an H&E stained pathological image 124 of spinal cord in an exemplary EAEGA group, consistent with one or more exemplary embodiments of the present disclosure. FIG. 12D illustrates an H&E stained pathological image 126 of spinal cord in an exemplary EAEGA/Depot group, consistent with one or more exemplary embodiments of the present disclosure. FIG. 13A illustrates an H&E stained pathological image 130 of brain in an exemplary non-EAE group, consistent with one or more exemplary embodiments of the present disclosure. FIG. 13B illustrates an H&E stained pathological image 132 of brain in an exemplary EAE group, consistent with one or more exemplary embodiments of the present disclosure. FIG. 13C illustrates an H&E stained pathological image 134 of brain in an exemplary EAE-GA group, consistent with one or more exemplary embodiments of the present disclosure. FIG. 13D illustrates an H&E stained pathological image 136 of brain in an exemplary EAEGA/Depot group, consistent with one or more exemplary embodiments of the present disclosure. As it is observed in FIG. 12A-D and FIG. 13A-D, H&E stained brain and spinal cord show the presence of mononuclear inflammatory and inflammatory cells in the white part of the spinal cord in EAE-induced mice. This phenomenon confirms the validity of the animal model. The outstanding point is that the EAE-GA/Depot group, brain, and spinal cord show mild to moderate presence of mononuclear inflammatory and inflammatory cells in
the white part of spinal cord in GA treated mice. However, brain and spinal cord tissue in GA/Depot group were comparable to the negative control (non-EAE) group. This observation confirms GA hydrogel microparticles’ efficacy in controlling symptoms of the disease in an exemplary EAE animal model. FIG. 14A illustrates an H&E stained pathological image 140 of skin in an exemplary non-EAE group, consistent with one or more exemplary embodiments of the present disclosure. FIG. 14B illustrates an H&E stained pathological image 142 of skin in an exemplary EAE-GA group, consistent with one or more exemplary embodiments of the present disclosure. FIG. 14C illustrates an H&E stained pathological image 144 of skin in an exemplary EAE-GA/Depot group, consistent with one or more exemplary embodiments of the present disclosure. Infiltrations of hypodermic inflammatory cells and granulomatous are common adverse effects of GA conventional parenteral formulation which plays a key role in poor patient compliance who are receiving GA conventional parenteral formulation (FIG. 14B). GA is an acidic medicine whose burst release in subcutaneous areas might cause irritation, necrosis, and granulomatous in the skin at the injection site. This adverse effect may be omitted in the groups which were treated with GA/Depot hydrogel microparticles (FIG. 14C). The injection site when using exemplary hydrogel-particles is comparable to EAE-group which received no GA injection. FIG. 15A illustrates a Luxol fast blue (H&E) stained pathological image 150 captured from first of spinal cord of an exemplary EAE group for demyelination comparison, consistent with one or more exemplary embodiments of the present disclosure. FIG. 15B illustrates a LFB stained pathological image 152 captured from end of spinal cord of an exemplary EAE group for demyelination comparison, consistent with one or more exemplary embodiments of the present disclosure. FIG. 15C illustrates a LFB stained pathological image 154 captured from first of spinal cord of an exemplary GA group for demyelination comparison, consistent with one or more exemplary embodiments of the present disclosure. FIG. 15D illustrates a LFB stained pathological image 156 captured from end of spinal cord of an exemplary EAE-GA group for demyelination comparison, consistent with one or more exemplary embodiments of the present disclosure. FIG. 15E illustrates a LFB stained pathological image 158 captured from end of spinal cord of an exemplary EAE- GA/Depot group for demyelination comparison, consistent with one or more exemplary embodiments of the present disclosure FIG. 15 illustrate demyelination in spinal cord cross sectional samples in EAE-group, GA-group, and GA/Depot groups. As it is shown in FIG. 15A-D, moderate to severe demyelination in superior, medial, and lateral segments of the white
region in the first and end parts of spinal cords are detectable. However, no sign of demyelination is observed in GA/Depot groups spinal cord samples (FIG. 15E).
[0097] Industrial Applicability
[0098] A parental pharmaceutical composition may be formed and used here for reducing symptoms of elapsing remitting multiple sclerosis (RRMS). The parental pharmaceutical composition may release glatiramer acetate when injected intramuscularly to a patient. The parental pharmaceutical composition may be prescribed by a doctor. The parental pharmaceutical composition may be administered at hospitals and clinics. Moreover, the parental pharmaceutical composition may be administered at home with a doctor’s prescription for reducing symptoms of RRMS.
[0099] While the foregoing has described what are considered to be the best mode and/or other examples, it is understood that various modifications may be made therein and that the subject matter disclosed herein may be implemented in various forms and examples, and that the teachings may be applied in numerous applications, only some of which have been described herein. It is intended by the following claims to claim any and all applications, modifications and variations that fall within the true scope of the present teachings.
[00100] Unless otherwise stated, all measurements, values, ratings, positions, magnitudes, sizes, and other specifications that are set forth in this specification, including in the claims that follow, are approximate, not exact. They are intended to have a reasonable range that is consistent with the functions to which they relate and with what is customary in the art to which they pertain.
[00101] The scope of protection is limited solely by the claims that now follow. That scope is intended and should be interpreted to be as broad as is consistent with the ordinary meaning of the language that is used in the claims when interpreted in light of this specification and the prosecution history that follows and to encompass all structural and functional equivalents. Notwithstanding, none of the claims are intended to embrace subject matter that fails to satisfy the requirement of Sections 101, 102, or 103 of the Patent Act, nor should they be interpreted in such a way. Any unintended embracement of such subject matter is hereby disclaimed.
[00102] Except as stated immediately above, nothing that has been stated or illustrated is intended or should be interpreted to cause a dedication of any component, step, feature,
object, benefit, advantage, or equivalent to the public, regardless of whether it is or is not recited in the claims.
[00103] It will be understood that the terms and expressions used herein have the ordinary meaning as is accorded to such terms and expressions with respect to their corresponding respective areas of inquiry and study except where specific meanings have otherwise been set forth herein. Relational terms such as first and second and the like may be used solely to distinguish one entity or action from another without necessarily requiring or implying any actual such relationship or order between such entities or actions. The terms “comprises,” “comprising,” or any other variation thereof, are intended to cover a nonexclusive inclusion, such that a process, method, article, or apparatus that comprises a list of elements does not include only those elements but may include other elements not expressly listed or inherent to such process, method, article, or apparatus. An element proceeded by “a” or “an” does not, without further constraints, preclude the existence of additional identical elements in the process, method, article, or apparatus that comprises the element.
[00104] The Abstract of the Disclosure is provided to allow the reader to quickly ascertain the nature of the technical disclosure. It is submitted with the understanding that it will not be used to interpret or limit the scope or meaning of the claims. In addition, in the foregoing Detailed Description, it can be seen that various features are grouped together in various implementations. This is for purposes of streamlining the disclosure, and is not to be interpreted as reflecting an intention that the claimed implementations require more features than are expressly recited in each claim. Rather, as the following claims reflect, inventive subject matter lies in less than all features of a single disclosed implementation. Thus, the following claims are hereby incorporated into the Detailed Description, with each claim standing on its own as a separately claimed subject matter.
[00105] While various implementations have been described, the description is intended to be exemplary, rather than limiting and it will be apparent to those of ordinary skill in the art that many more implementations and implementations are possible that are within the scope of the implementations. Although many possible combinations of features are shown in the accompanying figures and discussed in this detailed description, many other combinations of the disclosed features are possible. Any feature of any implementation may be used in combination with or substituted for any other feature or element in any other implementation unless specifically restricted. Therefore, it will be understood that any of the features shown
and/or discussed in the present disclosure may be implemented together in any suitable combination. Accordingly, the implementations are not to be restricted except in light of the attached claims and their equivalents. Also, various modifications and changes may be made within the scope of the attached claims.
Claims
1. A parenteral pharmaceutical composition for reducing symptoms of elapsing remitting multiple sclerosis (RRMS), the parenteral pharmaceutical composition comprising a plurality of powder particles dispersed in a phosphate -buffered saline (PBS) solution, each powder particle of the plurality of powder particles comprising: a first water-based portion, comprising: glatiramer acetate; sodium alginate with a weight ratio of the sodium alginate to the glatiramer acetate in a range of 0.1: 1 to 0.4: 4 (sodium alginate: glatiramer acetate); polyvinyl alcohol (PVA) with a weight ratio of PVA to the sodium alginate in a range of 0.05: 0.005 to 0.02: 0.2 (PVA: sodium alginate); and polysorbate 80 with a weight ratio of the polysorbate 80 to the sodium alginate in a range of 1: 3 to 5: 15 (polysorbate 80: sodium alginate); an oil-based portion, comprising: a-Tocopherol with a weight ratio of the a-Tocopherol to the sodium alginate in a range of 0: 1 to 0.3: 3 (a-Tocopherol: sodium alginate); and poly(lactic-co-glycolic acid) (PLGA) with a weight ratio of PLGA to the sodium alginate in a range of 0: 5 to 3: 30 (PLGA: sodium alginate); and a second water-based portion, comprising: glatiramer acetate with a weight ratio of the glatiramer acetate to the sodium alginate in a range of 0.1: 1 to 1.1: 5 (glatiramer acetate: sodium alginate); chitosan with a weight ratio of the chitosan to the sodium alginate in a range of 0.15: 1.5 to 0.1: 20(chitosan: sodium alginate); and
cremophor EL with a weight ratio of the cremophor EL to the sodium alginate in a range of 0: 1 to 0.5: 5 (cremophor EL: sodium alginate), wherein the parenteral pharmaceutical composition comprises an interconnected matrix of the first water-based portion, the oil-based portion, and the second water-based portion, wherein a weight ratio of the PBS solution to the plurality of the powder particles is in a range of 1: 2 to 8: 9 (the PBS solution: the plurality of the powder particles).
2. A parenteral pharmaceutical composition for reducing symptoms of elapsing remitting multiple sclerosis (RRMS), the parenteral pharmaceutical composition comprising a plurality of powder particles dispersed in a phosphate -buffered saline (PBS) solution, each powder particle of the plurality of powder particles comprising: a first water-based portion, comprising: glatiramer acetate; sodium alginate; polyvinyl alcohol (PVA); and polysorbate 80; an oil-based portion, comprising: a-Tocopherol; and poly(lactic-co-glycolic acid) (PLGA); and a second water-based portion, comprising: glatiramer acetate; chitosan; and cremophor EL.
3. The parenteral pharmaceutical composition of claim 2, wherein the plurality of powder particles of the pharmaceutical composition comprises glatiramer acetate with a weight percent to a total amount of the parenteral pharmaceutical composition in a range of 4 wt. % to 80 wt. %.
4. The parenteral pharmaceutical composition of claim 2, wherein the PLGA comprises at least one of an acid-terminated PLGA with a molecular weight in a range of 7000 g/mol to 17000 g/mol, an ester- terminated PLGA with a molecular weight in a range of 7000 g/mol to 17000 g/mol, an acid-terminated PLGA with a molecular weight in a range of 24000 g/mol to 38000 g/mol, and combinations thereof.
5. The parenteral pharmaceutical composition of claim 2, wherein a pH value of the PBS solution is in a range of 6.0 to 8.0.
6. The parenteral pharmaceutical composition of claim 2, wherein the parenteral pharmaceutical composition comprises a releasing rate of the glatiramer acetate in a range of 20 % to 90 % of a total amount of the glatiramer acetate within 30 days when the parenteral pharmaceutical composition being injected intramuscularly into a patient’s body.
7. The parenteral pharmaceutical composition of claim 2, wherein each particle of the plurality of the powder particles has an average particle size in a range of 5 pm to 75 pm.
8. The parenteral pharmaceutical composition of claim 2, wherein the plurality of powder particles of the parenteral pharmaceutical composition is stored at a temperature in a range of 15 °C to 30 °C with a relative humidity of maximum 70%.
9. A method for reducing symptoms of elapsing remitting multiple sclerosis (RRMS), comprising: forming a water/oil/water emulsion, comprising: forming an internal aqueous phase by mixing sodium alginate, glatiramer acetate, polyvinyl alcohol (PVA), and polysorbate 80 together; forming an organic phase by mixing the internal aqueous phase, a- Tocopherol, and poly(lactic-co-glycolic acid) (PLGA) together; forming a plurality of elementary particles by mixing a normal saline solution, PVA, the organic phase, and the internal aqueous phase together; and forming an external aqueous phase connected to an outer surface of the plurality of elementary particles by mixing the plurality of elementary particles, glatiramer acetate, chitosan, and cremophor EL together; forming a plurality of powder particles of the water/oil/water emulsion by drying the water/oil/water emulsion using a freeze dryer; forming a parenteral pharmaceutical composition by mixing the plurality of powder particles of the water/oil/water emulsion with a phosphate-buffered saline (PBS) solution with a weight ratio of the plurality of the powder particles of the water/oil/water emulsion to the PBS solution in a range of 1: 3 to 7: 9 (the plurality of the powder particles of the water/oil/water emulsion: the PBS solution); and injecting a predetermined amount of the formed parenteral pharmaceutical composition intramuscularly to a patient suffering from RRMS.
10. The method of claim 9, wherein forming the water/oil/water emulsion further comprises washing the water/oil/water emulsion by mixing the water/oil/water emulsion with a PBS solution with a weight ratio of the water/oil/water emulsion to the PBS solution in a range of 1: 3 to 7: 9 (the water/oil/water emulsion: the PBS solution).
11. The method of claim 10, wherein washing the plurality of powder particles of the water/oil/water emulsion comprises mixing the plurality of powder particles of the water/oil/water emulsion with the PBS solution with a pH value in a range of 6 to 8.
12. The method of claim 9, wherein forming the internal aqueous phase comprises mixing glatiramer acetate, sodium alginate at a relative weight ratio to the glatiramer acetate in a range of 0.1: 5 to 1:20 (sodium alginate: glatiramer acetate), polyvinyl alcohol (PVA) at a relative weight ratio to the glatiramer acetate in a range of 0.01: 0.1 to 0.03: 0.5 (PVA: glatiramer acetate), and polysorbate 80 at a relative weight ratio to glatiramer acetate in a range of 1: 3 to 3: 5 (polysorbate 80: glatiramer acetate) together using a mixer with a stirring speed in a range of 500 rpm to 5000 rpm.
13. The method of claim 12, wherein mixing sodium alginate, glatiramer acetate, polyvinyl alcohol (PVA), and polysorbate 80 together comprises mixing sodium alginate, glatiramer acetate, polyvinyl alcohol (PVA), and polysorbate 80 together using a mixer with a stirring speed in a range of 500 rpm to 5000 rpm for a time period in a range of 10 minutes to 100 minutes.
14. The method of claim 9, wherein forming the organic phase comprises mixing the internal aqueous phase, a-Tocopherol with a weight ratio of the a-Tocopherol to glatiramer acetate in a range of 0:2 to 1:5 (a-Tocopherol: glatiramer acetate), and PLGA with a weight ratio of the PLGA to glatiramer acetate in a range of 1: 5 to 5: 50 (PLGA: glatiramer acetate) together using an ultrasonic device with an ultrasonic power in a range of 200 W/cm2 to 1000 W/cm2.
15. The method of claim 14, wherein mixing the internal aqueous phase, a-Tocopherol, and PLGA together comprises mixing the internal aqueous phase, a-Tocopherol, and PLGA together using an ultrasonic device for a time period in a range of 2 minutes to 60 minutes.
16. The method of claim 9, wherein forming the external aqueous phase by mixing the plurality of elementary particles, glatiramer acetate, chitosan, and cremophor EL together is done using an ultrasonic device with an ultrasonic power in a range of 200 W/cm2 to 1000 W/cm2.
17. The method of claim 9, wherein forming the external aqueous phase by mixing the plurality of elementary particles, glatiramer acetate, chitosan, and cremophor EL together is done using an ultrasonic device for a time period in a range of 10 minutes to 100 minutes.
18. The method of claim 9, wherein drying the water/oil/water emulsion comprises: freezing the water/oil/water emulsion at a temperature in a range of -60°C to -
10 °C for a time period in a range of 1 hour to 10 hours; and drying the water/oil/water emulsion, comprising:
initial drying the water/oil/water emulsion at a temperature in a range of -5 °C to -40 °C for a time period in a range of 10 hours to 40 hours; and secondary drying the water/oil/water emulsion at a temperature in a range of 10°C to 30 °C for a time period in a range of 5 hours to 20 hours.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202263317088P | 2022-03-07 | 2022-03-07 | |
| US63/317,088 | 2022-03-07 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023170493A1 true WO2023170493A1 (en) | 2023-09-14 |
Family
ID=87936203
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2023/051515 Ceased WO2023170493A1 (en) | 2022-03-07 | 2023-02-20 | Drug delivery using a parenteral pharmaceutical composition |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2023170493A1 (en) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011080733A1 (en) * | 2010-01-04 | 2011-07-07 | Mapi Pharma Limited | Depot systems comprising glatiramer or a pharmacologically acceptable salt thereof |
| WO2018042415A1 (en) * | 2016-08-31 | 2018-03-08 | Mapi Pharma Ltd | Depot systems comprising glatiramer acetate |
| WO2018042423A1 (en) * | 2016-08-28 | 2018-03-08 | Mapi Pharma Ltd. | Process for preparing microparticles containing glatiramer acetate |
-
2023
- 2023-02-20 WO PCT/IB2023/051515 patent/WO2023170493A1/en not_active Ceased
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011080733A1 (en) * | 2010-01-04 | 2011-07-07 | Mapi Pharma Limited | Depot systems comprising glatiramer or a pharmacologically acceptable salt thereof |
| WO2018042423A1 (en) * | 2016-08-28 | 2018-03-08 | Mapi Pharma Ltd. | Process for preparing microparticles containing glatiramer acetate |
| WO2018042415A1 (en) * | 2016-08-31 | 2018-03-08 | Mapi Pharma Ltd | Depot systems comprising glatiramer acetate |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Zhang et al. | Long-acting hydrogel/microsphere composite sequentially releases dexmedetomidine and bupivacaine for prolonged synergistic analgesia | |
| EP3536333B1 (en) | Depot system comprising glatiramer acetate | |
| US8790695B2 (en) | Medicinal carriers, and preparation method and uses thereof | |
| AU2016372554B2 (en) | Sustained release cyclosporine-loaded microparticles | |
| JP2004529934A (en) | Compositions comprising anti-microtubule agents and polypeptides or polysaccharides, and uses of those compositions for the preparation of a medicament for treating inflammatory conditions | |
| KR20100062990A (en) | Injectable polymer-lipid blend for localized drug delivery | |
| Martinez-Sancho et al. | Poly (D, L-lactide-co-glycolide) microspheres for long-term intravitreal delivery of aciclovir: influence of fatty and non-fatty additives | |
| KR101208568B1 (en) | Nano capsule, medicine and food comprising the same | |
| WO2023170493A1 (en) | Drug delivery using a parenteral pharmaceutical composition | |
| Colombo et al. | Effect of excipient composition on the biocompatibility of bupivacaine‐containing microparticles at the sciatic nerve | |
| Sapra et al. | Development of mucoadhesive microspheres for intranasal delivery of fluconazole as an alternative treatment of cryptococcal meningitis infection in patients with acquired immunodeficiency | |
| CN112933225A (en) | Preparation of lipid-coated calcium carbonate carrier loaded with Ce6, and preparation method and application thereof | |
| CN116211799B (en) | A local anesthetic compound suspension | |
| CN116196266B (en) | A local anesthetic low molecular weight gel, liposome and preparation method thereof | |
| Guo et al. | Catalase-encapsulated matrix metalloproteinase-9 responsive nanogels for modulation of inflammatory response and treatment of neutrophilic asthma | |
| CN105997889A (en) | Amifostine slow-release microspheres for subcutaneous injection and preparation method thereof | |
| Xu et al. | Sustained intraocular pressure-lowering effect and biocompatibility of a single subconjunctival administration of hydrogel-encapsulated nano-brinzolamide | |
| Wang et al. | The impact of dexmedetomidine-loaded nano-microsphere combined with percutaneous acupoint electrical stimulation on the postoperative cognitive function of elderly patients with hip fracture | |
| Tanwar et al. | In Vitro Investigation of Controlled Release of Ciprofloxacin and Its β‐Cyclodextrin Inclusion Complex from Gelatin Grafted Poly (vinyl alcohol)(GPVA) Nanoparticles | |
| Sharma et al. | Enhancing therapeutic efficacy of fingolimod via intranasal delivery in an ethidium bromide-induced model of multiple sclerosis | |
| KR102047207B1 (en) | Drug delivery systems for treatment or prevention of inflammatory diseases or pain and methods for manufacturing the same | |
| Jiang et al. | Local anesthetics effects of lidocaine-loaded chitosan nanoparticles in a rat model: an in vitro and in vivo study | |
| CN107982214A (en) | Enrofloxacin solid lipid nano suspension for animals and preparation method thereof | |
| CN112451475B (en) | Long-acting sustained-release gel for treating cavernous pulmonary tuberculosis | |
| TWI681781B (en) | Hydrophilic microsphere made from pharmaceutical excipients for embolization therapy |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 23766207 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 23766207 Country of ref document: EP Kind code of ref document: A1 |