WO2023016528A1 - Class of benzomorpholine compounds, and preparation method therefor and use thereof - Google Patents
Class of benzomorpholine compounds, and preparation method therefor and use thereof Download PDFInfo
- Publication number
- WO2023016528A1 WO2023016528A1 PCT/CN2022/111884 CN2022111884W WO2023016528A1 WO 2023016528 A1 WO2023016528 A1 WO 2023016528A1 CN 2022111884 W CN2022111884 W CN 2022111884W WO 2023016528 A1 WO2023016528 A1 WO 2023016528A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- reaction solution
- added
- stirred
- esi calculated
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/538—1,4-Oxazines, e.g. morpholine ortho- or peri-condensed with carbocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D267/00—Heterocyclic compounds containing rings of more than six members having one nitrogen atom and one oxygen atom as the only ring hetero atoms
- C07D267/02—Seven-membered rings
- C07D267/08—Seven-membered rings having the hetero atoms in positions 1 and 4
- C07D267/12—Seven-membered rings having the hetero atoms in positions 1 and 4 condensed with carbocyclic rings or ring systems
- C07D267/14—Seven-membered rings having the hetero atoms in positions 1 and 4 condensed with carbocyclic rings or ring systems condensed with one six-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/04—Ortho-condensed systems
Definitions
- the invention relates to a class of benzomorpholine compounds, a preparation method and application thereof. It specifically relates to the compound represented by formula (I) and its pharmaceutically acceptable salt.
- Tumor immunotherapy is a therapeutic area that has attracted much attention in recent years.
- the main mechanism is to enhance the anti-tumor ability of the immune microenvironment by mobilizing the body's immune system.
- monoclonal antibody drugs for tumor immunotherapy such as Keytruda and OPDIVO, have been used for the treatment of various cancers such as non-small cell lung cancer and melanoma.
- RORs Retinoic acid-related orphan receptors
- RORs Retinoic acid-related orphan receptors
- RORs belong to the nuclear receptor superfamily and are a member of intracellular transcription factors, which can regulate a variety of physiological processes, including reproductive development, metabolism, and immune system regulation.
- ROR has three family members: ROR- ⁇ , - ⁇ and - ⁇ , which are encoded by RORA, RORB and RORC genes, respectively.
- ROR ⁇ includes two subtypes, ROR ⁇ 1 and ROR ⁇ t (ROR ⁇ 2).
- ROR ⁇ 1 is expressed in various tissues and organs such as thymus, muscle, pancreas, prostate, and liver, while the short-chain subtype ROR ⁇ t of ROR ⁇ is mainly distributed in the thymus and promotes the differentiation of initial T cells into Th17 and Tc17 cells.
- Th17 and Tc17 cells promote inflammatory and autoimmune responses by secreting IL-17, IL-22, GM-CSF and other cytokines and inflammatory factors.
- IL-17 can promote the recruitment and infiltration of CTLs and NK cells in the tumor microenvironment, and enhance the anti-tumor effect of effector T cells.
- ROR ⁇ agonists currently have no drugs approved for marketing, and Lycera Corp’s
- ROR ⁇ agonist LYC-55716 monotherapy for the treatment of advanced solid tumors is in phase II clinical research, and it is combined with PD-1 monoclonal antibody pembrolizumab in the treatment of advanced non-small cell lung cancer The treatment is in a phase 1b clinical study.
- this field still needs candidate compounds with better activity and better pharmacokinetic parameters to advance to clinical trials to meet the therapeutic needs.
- the present invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof,
- L 1 and L 2 are independently selected from single bonds and -NH-;
- T 1 is selected from N and C (R 12 );
- T 2 is selected from N and C (R 15 );
- T 3 when When it is a single bond, T 3 is CR t3 or N;
- T 3 is C
- T4 is CH or N
- n 0, 1 or 2;
- n 0, 1 or 2;
- R 11 , R 12 , R 13 , R 14 and R 15 are independently selected from H, F, Cl, Br, I, -OH, -NH 2 , -CN, C 1-3 alkyl, C 1-3 Alkoxy and C 1-3 alkylamino, wherein said C 1-3 alkyl, C 1-3 alkoxy and C 1-3 alkylamino are independently optionally substituted by 1, 2 or 3 R a ;
- R 13 and R 14 and the carbon atoms attached to them are linked together so that the structural unit selected from
- T 5 and T 6 are independently selected from N and CH;
- R 2 is selected from C 1-3 alkyl, wherein said C 1-3 alkyl is optionally substituted by 1, 2 or 3 R b ;
- R 31 and R 32 are independently selected from H, F, Cl, Br, I, -OH, -NH 2 , C 1-3 alkyl and C 1-3 alkoxy, wherein the C 1-3 alkoxy and C 1-3 alkoxy are independently optionally substituted by 1, 2 or 3 R c ;
- R t3 is selected from H, F, Cl, Br, I, -OH, -NH 2 , C 1-3 alkyl and C 1-3 alkoxy, wherein the C 1-3 alkyl and C 1-3 Alkoxy groups are independently optionally substituted by 1, 2 or 3 R d ;
- R a is independently selected from F, Cl, Br, I, -OH, -NH 2 and C 1-3 alkyl, wherein the C 1-3 alkyl is optionally substituted by 1, 2 or 3 R;
- R b are independently selected from F, Cl, Br, I, -OH, -NH 2 , -CN, -COOH and C 1-3 alkyl;
- R c are independently selected from F, Cl, Br, I, -OH and -NH 2 ;
- R d are independently selected from F, Cl, Br, I, -OH and -NH 2 ;
- R are each independently selected from F, Cl, Br, I, -OH and -NH 2 .
- the present invention provides a compound of formula (II) or a pharmaceutically acceptable salt thereof,
- L 1 and L 2 are independently selected from single bonds and -NH-;
- T 1 is selected from N and C (R 12 );
- T 2 is selected from N and C (R 15 );
- T 3 is CH or N
- T 3 is C
- T4 is CH or N
- n 0, 1 or 2;
- n 0, 1 or 2;
- R 11 , R 12 , R 13 , R 14 and R 15 are independently selected from H, F, Cl, Br, I, OH, NH 2 , CN, C 1-3 alkyl, C 1-3 alkoxy and C 1-3 alkylamino, wherein the C 1-3 alkyl, C 1-3 alkoxy and C 1-3 alkylamino are optionally substituted by 1, 2 or 3 R a ;
- R 13 and R 14 and the carbon atoms attached to them are linked together such that selected from
- T 5 and T 6 are independently selected from N and CH;
- R 2 is selected from C 1-3 alkyl optionally substituted by 1, 2 or 3 R b ;
- R a is independently selected from F, Cl, Br, I, OH, NH 2 and C 1-3 alkyl, wherein the C 1-3 alkyl is optionally substituted by 1, 2 or 3 R;
- R b are independently selected from F, Cl, Br, I, OH, NH 2 , CN, COOH and C 1-3 alkyl;
- R are each independently selected from F, Cl, Br, I, OH and NH 2 .
- T 1 is selected from C(R 12 ), and other variables are as defined in the present invention.
- T1 is selected from N, and other variables are as defined in the present invention.
- T 2 is selected from C(R 15 ), and other variables are as defined in the present invention.
- T 2 is selected from N, and other variables are as defined in the present invention.
- R a is independently selected from F, Cl, Br, I, -OH, -NH 2 and -CH 3 , and other variables are as defined in the present invention.
- R a are independently selected from F, and other variables are as defined in the present invention.
- R b are independently selected from -COOH, and other variables are as defined in the present invention.
- R 11 , R 12 , R 13 , R 14 and R 15 are independently selected from H, F, Cl, Br, I, -OH, -NH 2 , -CN, -CH 3 , -O-CH 3 and -NH-CH 3 , wherein said -CH 3 , -O-CH 3 and -NH-CH 3 are independently optionally substituted by 1, 2 or 3 R a , R a and Other variables are as defined herein.
- R 11 , R 12 , R 13 , R 14 and R 15 are selected from H, F, Cl, Br, I, -OH, -NH 2 , -CN, -CH 3 , -CF 3 , -O-CH 3 , -O-CH 2 F, -O-CHF 2 , -O-CF 3 and Other variables are as defined herein.
- R 11 is selected from H, and other variables are as defined in the present invention.
- R 12 is selected from H and F, and other variables are as defined in the present invention.
- R 13 is selected from H, and other variables are as defined in the present invention.
- R 14 is selected from H, F, Cl, -CH 3 and -OCH 3 , wherein the -O-CH 3 is optionally substituted by 1, 2 or 3 R a , R a and Other variables are as defined herein.
- R 14 is selected from H, F, Cl, -CH 3 , -CF 3 and -OCF 3 , and other variables are as defined in the present invention.
- R 15 is selected from H, F, Cl and -OCH 3 , wherein the -CH 3 is optionally substituted by 1, 2 or 3 R a , and R a and other variables are as described in the present invention definition.
- R 15 is selected from H, F, Cl, -OCH 3 , -OCF 3 and -OCHF 2 , and other variables are as defined in the present invention.
- R 2 is selected from -CH 2 -CH 3 , wherein the -CH 2 -CH 3 is optionally substituted by 1, 2 or 3 R b , and R b and other variables are as described herein invention defined.
- R 31 and R 32 are independently selected from H, F, Cl, Br, I, -OH, -NH 2 , -CH 3 and -O-CH 3 , wherein -CH 3 and -O-CH 3 are each independently optionally substituted by 1, 2 or 3 R c , R c and other variables are as defined in the present invention.
- R 31 and R 32 are independently selected from H, F, Cl, Br, I, -OH, -NH 2 , -CH 3 , -CF 3 , -O-CH 3 , - O- CH2F , -O- CHF2 and -O- CF3 , other variables are as defined in the present invention.
- R t3 is selected from H, F, Cl, Br, I, -OH, -NH 2 , -CH 3 and -O-CH 3 , and other variables are as defined in the present invention.
- R t3 is selected from H, F, -OH and -O-CH 3 , and other variables are as defined in the present invention.
- T 1 is selected from N and C (R 12 );
- T 2 is selected from N and C (R 15 );
- n, L 1 , L 2 , R 11 , R 12 , R 13 , R 14 , R 15 , R 2 , R 31 , R 32 and R t3 are as defined in the present invention.
- T 1 is selected from N and C (R 12 );
- T 2 is selected from N and C (R 15 );
- Carbon atoms with "*" are chiral carbon atoms and exist as (R) or (S) single enantiomer or enrichment of one enantiomer.
- T 1 is selected from N and C (R 12 );
- T 2 is selected from N and C (R 15 );
- R 11 , R 12 , R 13 , R 14 , R 15 , R 2 , R 31 , R 32 and R t3 are as defined in the present invention.
- T 1 is selected from N and C (R 12 );
- T 2 is selected from N and C (R 15 );
- R 11 , R 12 , R 13 , R 14 , R 15 , R 2 , R 31 , R 32 and R t3 are as defined in the present invention.
- Carbon atoms with "*" are chiral carbon atoms and exist as (R) or (S) single enantiomer or enrichment of one enantiomer.
- n, L 1 , L 2 , T 1 , T 2 , R 11 , R 12 , R 13 , R 14 , R 15 and R 2 are as defined in the present invention.
- Carbon atoms with "*" are chiral carbon atoms and exist as (R) or (S) single enantiomer or enrichment of one enantiomer.
- the present invention also provides a compound of the following formula or a pharmaceutically acceptable salt thereof,
- the compound of the present invention has remarkable in vitro activity and good pharmacokinetic properties.
- pharmaceutically acceptable refers to those compounds, materials, compositions and/or dosage forms, which are suitable for use in contact with human and animal tissues within the scope of sound medical judgment , without undue toxicity, irritation, allergic reaction or other problems or complications, commensurate with a reasonable benefit/risk ratio.
- pharmaceutically acceptable salt refers to a salt of a compound of the present invention, which is prepared from a compound having a specific substituent found in the present invention and a relatively non-toxic acid or base.
- base addition salts can be obtained by contacting such compounds with a sufficient amount of base, either neat solution or in a suitable inert solvent.
- Pharmaceutically acceptable base addition salts include sodium, potassium, calcium, ammonium, organic amine or magnesium salts or similar salts.
- acid addition salts can be obtained by contacting such compounds with a sufficient amount of the acid, either neat solution or in a suitable inert solvent.
- Examples of pharmaceutically acceptable acid addition salts include salts of inorganic acids including, for example, hydrochloric acid, hydrobromic acid, nitric acid, carbonic acid, bicarbonate, phosphoric acid, monohydrogenphosphate, dihydrogenphosphate, sulfuric acid, Hydrogen sulfate, hydroiodic acid, phosphorous acid, etc.; and organic acid salts, such as acetic acid, propionic acid, isobutyric acid, maleic acid, malonic acid, benzoic acid, succinic acid, suberic acid, Fumaric acid, lactic acid, mandelic acid, phthalic acid, benzenesulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid and methanesulfonic acid and similar acids; also salts of amino acids such as arginine and the like , and salts of organic acids such as glucuronic acid. Certain specific compounds of the present invention contain basic and acidic functional groups and can thus be converted into either
- the pharmaceutically acceptable salts of the present invention can be synthesized from the parent compound containing acid groups or bases by conventional chemical methods.
- such salts are prepared by reacting the free acid or base form of these compounds with a stoichiometric amount of the appropriate base or acid in water or an organic solvent or a mixture of both.
- the compounds of the invention may exist in particular geometric or stereoisomeric forms.
- the present invention contemplates all such compounds, including cis and trans isomers, (-)- and (+)-enantiomers, (R)- and (S)-enantiomers, diastereomers isomers, (D)-isomers, (L)-isomers, and their racemic and other mixtures, such as enantiomerically or diastereomerically enriched mixtures, all of which are subject to the present within the scope of the invention.
- Additional asymmetric carbon atoms may be present in substituents such as alkyl groups. All such isomers, as well as mixtures thereof, are included within the scope of the present invention.
- enantiomer or “optical isomer” refer to stereoisomers that are mirror images of each other.
- cis-trans isomers or “geometric isomers” arise from the inability to rotate freely due to the double bond or the single bond of the carbon atoms forming the ring.
- diastereoisomer refers to stereoisomers whose molecules have two or more chiral centers and which are not mirror images of the molecules.
- keys with wedge-shaped solid lines and dotted wedge keys Indicates the absolute configuration of a stereocenter, with a straight solid-line bond and straight dashed keys Indicates the relative configuration of the stereocenter, with a wavy line Indicates wedge-shaped solid-line bond or dotted wedge key or with tilde Indicates a straight solid line key and straight dashed keys
- the following formula (A) means that the compound exists as a single isomer of formula (A-1) or formula (A-2) or as two isomers of formula (A-1) and formula (A-2).
- the following formula (B) means that the compound exists in the form of a single isomer of formula (B-1) or formula (B-2) or in the form of both formula (B-1) and formula (B-2) It exists as a mixture of isomers.
- the following formula (C) represents that the compound exists in the form of a single isomer of formula (C-1) or formula (C-2) or in the form of two isomers of formula (C-1) and formula (C-2). It exists in the form of a mixture.
- tautomer or “tautomeric form” means that isomers with different functional groups are in dynamic equilibrium at room temperature and are rapidly interconvertible. If tautomerism is possible (eg, in solution), then chemical equilibrium of the tautomers can be achieved.
- proton tautomers also called prototropic tautomers
- prototropic tautomers include interconversions via migration of a proton, such as keto-enol isomerization and imine-ene Amine isomerization.
- Valence isomers (valence tautomers) involve interconversions by recombination of some bonding electrons.
- keto-enol tautomerization is the interconversion between two tautomers of pentane-2,4-dione and 4-hydroxypent-3-en-2-one.
- the terms “enriched in an isomer”, “enriched in an isomer”, “enriched in an enantiomer” or “enantiomerically enriched” refer to one of the isomers or enantiomers
- the content of the enantiomer is less than 100%, and the content of the isomer or enantiomer is greater than or equal to 60%, or greater than or equal to 70%, or greater than or equal to 80%, or greater than or equal to 90%, or greater than or equal to 95%, or Greater than or equal to 96%, or greater than or equal to 97%, or greater than or equal to 98%, or greater than or equal to 99%, or greater than or equal to 99.5%, or greater than or equal to 99.6%, or greater than or equal to 99.7%, or greater than or equal to 99.8%, or greater than or equal to 99.9%.
- the terms “isomer excess” or “enantiomeric excess” refer to the difference between the relative percentages of two isomers or two enantiomers. For example, if the content of one isomer or enantiomer is 90% and the other isomer or enantiomer is 10%, then the isomer or enantiomeric excess (ee value) is 80% .
- Optically active (R)- and (S)-isomers as well as D and L-isomers can be prepared by chiral synthesis or chiral reagents or other conventional techniques. If one enantiomer of a compound of the invention is desired, it can be prepared by asymmetric synthesis or derivatization with chiral auxiliary agents, wherein the resulting diastereomeric mixture is separated and the auxiliary group is cleaved to provide pure desired enantiomer.
- a diastereoisomeric salt is formed with an appropriate optically active acid or base, and then a diastereomeric salt is formed by a conventional method known in the art. Diastereomeric resolution is performed and the pure enantiomers are recovered. Furthermore, the separation of enantiomers and diastereomers is usually accomplished by the use of chromatography using chiral stationary phases, optionally in combination with chemical derivatization methods (e.g. amines to amino groups formate).
- the compounds of the present invention may contain unnatural proportions of atomic isotopes at one or more of the atoms that constitute the compounds.
- compounds may be labeled with radioactive isotopes such as tritium ( 3 H), iodine-125 ( 125 I) or C-14 ( 14 C).
- radioactive isotopes such as tritium ( 3 H), iodine-125 ( 125 I) or C-14 ( 14 C).
- heavy hydrogen can be used to replace hydrogen to form deuterated drugs.
- the bond formed by deuterium and carbon is stronger than the bond formed by ordinary hydrogen and carbon.
- deuterated drugs can reduce toxic side effects and increase drug stability. , enhance the efficacy, prolong the biological half-life of drugs and other advantages. All changes in isotopic composition of the compounds of the invention, whether radioactive or not, are included within the scope of the invention.
- substituted means that any one or more hydrogen atoms on a specified atom are replaced by a substituent, which may include deuterium and hydrogen variants, as long as the valence of the specified atom is normal and the substituted compound is stable.
- any variable eg, R
- its definition is independent at each occurrence.
- said group may optionally be substituted with up to two R, with independent options for each occurrence of R.
- substituents and/or variations thereof are permissible only if such combinations result in stable compounds.
- linking group When the number of a linking group is 0, such as -(CRR) 0 -, it means that the linking group is a single bond.
- substituent When a substituent is vacant, it means that the substituent does not exist. For example, when X in A-X is vacant, it means that the structure is actually A. When the enumerated substituent does not indicate which atom it is connected to the substituted group, this substituent can be bonded through any atom, for example, pyridyl as a substituent can be connected to any atom on the pyridine ring. The carbon atom is attached to the group being substituted.
- linking group listed does not indicate its linking direction
- its linking direction is arbitrary, for example,
- the connecting group L in the middle is -MW-, at this time -MW- can connect ring A and ring B in the same direction as the reading order from left to right to form It can also be formed by connecting loop A and loop B in the opposite direction to the reading order from left to right
- any one or more sites of the group can be linked to other groups through chemical bonds.
- the chemical bonds that the site connects with other groups can use straight solid line bonds Straight dotted key or tilde express.
- the straight-shaped solid-line bond in -OCH3 indicates that it is connected to other groups through the oxygen atom in the group;
- the straight dotted line bond in indicates that the two ends of the nitrogen atom in the group are connected to other groups;
- the wavy lines in indicate that the 1 and 2 carbon atoms in the phenyl group are connected to other groups.
- the number of atoms in a ring is generally defined as the number of ring members, eg, "5-7 membered ring” means a “ring” with 5-7 atoms arranged around it.
- C 1-3 alkyl is used to denote a straight or branched chain saturated hydrocarbon group consisting of 1 to 3 carbon atoms.
- the C 1-3 alkyl group includes C 1-2 and C 2-3 alkyl groups, etc.; it can be monovalent (such as methyl), divalent (such as methylene) or multivalent (such as methine) .
- Examples of C 1-3 alkyl include, but are not limited to, methyl (Me), ethyl (Et), propyl (including n-propyl and isopropyl), and the like.
- C 1-3 alkoxy denotes those alkyl groups containing 1 to 3 carbon atoms attached to the rest of the molecule through an oxygen atom.
- the C 1-3 alkoxy group includes C 1-2 , C 2-3 , C 3 and C 2 alkoxy groups and the like.
- Examples of C 1-3 alkoxy include, but are not limited to, methoxy, ethoxy, propoxy (including n-propoxy and isopropoxy), and the like.
- C 1-3 alkylamino denotes those alkyl groups containing 1 to 3 carbon atoms attached to the rest of the molecule through an amino group.
- the C 1-3 alkylamino group includes C 1-2 , C 3 and C 2 alkylamino groups and the like.
- Examples of C 1-3 alkylamino include, but are not limited to, -NHCH 3 , -N(CH 3 ) 2 , -NHCH 2 CH 3 , -N(CH 3 )CH 2 CH 3 , -NHCH 2 CH 2 CH 3 , -NHCH 2 CH 2 CH 3 , -NHCH 2 (CH 3 ) 2 etc.
- C n-n+m or C n -C n+m includes any specific instance of n to n+m carbons, for example C 1-12 includes C 1 , C 2 , C 3 , C 4 , C 5 , C 6 , C 7 , C 8 , C 9 , C 10 , C 11 , and C 12 , also including any range from n to n+m, for example, C 1-12 includes C 1-3 , C 1-6 , C 1-9 , C 3-6 , C 3-9 , C 3-12 , C 6-9 , C 6-12 , and C 9-12 etc.; similarly, n to n +m means that the number of atoms on the ring is n to n+m, for example, a 3-12-membered ring includes a 3-membered ring, a 4-membered ring, a 5-membered ring, a 6-membered ring, a 7-membered ring, an 8-membere
- leaving group refers to a functional group or atom that can be replaced by another functional group or atom through a substitution reaction (eg, a nucleophilic substitution reaction).
- a substitution reaction eg, a nucleophilic substitution reaction
- representative leaving groups include triflate; chlorine, bromine, iodine; sulfonate groups such as mesylate, tosylate, brosylate, tosylate esters, etc.; acyloxy groups such as acetoxy, trifluoroacetoxy, and the like.
- protecting group includes, but is not limited to, "amino protecting group", “hydroxyl protecting group” or “mercapto protecting group”.
- amino protecting group refers to a protecting group suitable for preventing side reactions at the amino nitrogen position.
- Representative amino protecting groups include, but are not limited to: formyl; acyl, such as alkanoyl (such as acetyl, trichloroacetyl or trifluoroacetyl); alkoxycarbonyl, such as tert-butoxycarbonyl (Boc) ; arylmethoxycarbonyl, such as benzyloxycarbonyl (Cbz) and 9-fluorenylmethyloxycarbonyl (Fmoc); arylmethyl, such as benzyl (Bn), trityl (Tr), 1,1-di -(4'-methoxyphenyl)methyl; silyl groups such as trimethylsilyl (TMS) and tert-butyldimethylsilyl (TBS) and the like.
- acyl such as alkanoyl (such as acetyl, trichloroacetyl or trifluoroacetyl); alkoxycarbonyl, such as
- hydroxyl protecting group refers to a protecting group suitable for preventing side reactions of the hydroxy group.
- Representative hydroxy protecting groups include, but are not limited to: alkyl, such as methyl, ethyl, and tert-butyl; acyl, such as alkanoyl (such as acetyl); arylmethyl, such as benzyl (Bn), p-formyl Oxybenzyl (PMB), 9-fluorenylmethyl (Fm) and diphenylmethyl (diphenylmethyl, DPM); silyl groups such as trimethylsilyl (TMS) and tert-butyl Dimethylsilyl (TBS) and the like.
- alkyl such as methyl, ethyl, and tert-butyl
- acyl such as alkanoyl (such as acetyl)
- arylmethyl such as benzyl (Bn), p-formyl Oxybenzyl (PMB), 9
- the compounds of the present invention can be prepared by a variety of synthetic methods well known to those skilled in the art, including the specific embodiments listed below, the embodiments formed by combining them with other chemical synthesis methods, and the methods well known to those skilled in the art Equivalent alternatives, preferred embodiments include but are not limited to the examples of the present invention.
- the structure of the compounds of the present invention can be confirmed by conventional methods known to those skilled in the art. If the present invention involves the absolute configuration of the compound, the absolute configuration can be confirmed by conventional technical means in the art. For example, in single crystal X-ray diffraction (SXRD), the cultured single crystal is collected with a Bruker D8 venture diffractometer to collect diffraction intensity data, the light source is CuK ⁇ radiation, and the scanning method is: After collecting relevant data, the absolute configuration can be confirmed by further analyzing the crystal structure by direct method (Shelxs97).
- SXRD single crystal X-ray diffraction
- aq stands for water
- HATU O-(7-azabenzotriazol-1-yl)-N,N,N'.N'-tetramethyluronium hexafluorophosphate
- EDCI represents N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride
- m-CPBA 3-chloroperoxybenzoic acid
- eq represents equivalent, equivalent
- CDI represents Carbonyldiimidazole
- DCM stands for dichloromethane
- PE stands for petroleum ether
- DIAD stands for diisopropyl azodicarboxylate
- DMF stands for N,N-dimethylformamide
- DMSO stands for dimethylsulfoxide
- EtOAc stands for ethyl acetate EtOH stands for ethanol
- MeOH stands for methanol
- CBz stands for benzyl
- reaction solution was concentrated under reduced pressure, the residue was dissolved in ethyl acetate (200mL), washed with water (200mL x 1) and saturated brine (200mL x 1) successively, the organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was decompressed After concentration, the residue was separated and purified by silica gel column chromatography (4/1 petroleum ether/ethyl acetate) to obtain compound 1-6.
- compound 1-8 (800mg, 0.634mmol) was dissolved in dry 1,4-dioxane (10mL), and diboronic acid pinacol ester (440mg, 1.73mmol), potassium acetate (463mg, 4.72mmol) and [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II) dichloromethane complex (129mg, 0.157mmol), the reaction solution was stirred at 90°C for 12 hours , water (40 mL) was added to the reaction solution, and extracted with ethyl acetate (40 mL x 1).
- compound 2-5 (250mg, 0.450mmol) was dissolved in acetone (4mL) and water (2mL), sodium periodate (356mg, 1.67mmol) and ammonium acetate (69.4mg, 0.900mmol) were added, The reaction solution was stirred at 50°C for 12 hours, adjusted to pH ⁇ 3 with 1M hydrochloric acid aqueous solution, and extracted with ethyl acetate (20 mL x 1). The organic phase was washed with saturated brine (20 mL x 1), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to obtain compound 2-6. MS-ESI calculated [M+Na] + 496, found 496.
- reaction solution was concentrated under reduced pressure, water (20 mL) was added, the pH value was adjusted to 4 with hydrochloric acid (1M), and then extracted with ethyl acetate (20 mL ⁇ 2), the combined organic phases were washed with saturated brine (50 mL ⁇ 1), and then It was dried over anhydrous sodium sulfate, filtered, concentrated under reduced pressure, and the residue was separated and purified by high performance liquid chromatography (neutral, ammonium bicarbonate system) to obtain compound 3.
- reaction solution was diluted with water (100mL) and extracted with ethyl acetate (200mL x 2), the organic phase was washed with saturated brine (500mL x 1), dried over anhydrous sodium sulfate, filtered, the filtrate was concentrated under reduced pressure, and the residue was passed through Compound 7-2 was obtained by separation and purification by silica gel column chromatography (4/1 petroleum ether/ethyl acetate).
- reaction solution was concentrated under reduced pressure, the residue was dissolved in water (5 mL), the pH was adjusted to 4 with hydrochloric acid (1 M), extracted with ethyl acetate (5 mL x 3), the organic phases were combined, and the organic phase was washed with saturated aqueous sodium chloride ( 10mL x 1), dried over anhydrous sodium sulfate, filtered, the filtrate was concentrated under reduced pressure, and the residue was separated by high performance liquid chromatography (neutral, ammonium bicarbonate system) to obtain compound 15.
- compound 2-5 (185mg, 0.391mmol) was dissolved in anhydrous dichloromethane (10mL), compound 16-4 (123mg, 0.469mmol), pyridine N-oxide (112mg, 1.17mmol) were added , pyridine (92.8mg, 1.17mmol) and copper acetate (142mg, 0.782mmol), the reaction solution was stirred at 36°C for 36 hours under an oxygen (15Psi) atmosphere, the reaction solution was concentrated under reduced pressure, and the residue was separated and purified by silica gel column chromatography (3/1 petroleum ether/ethyl acetate), to obtain compound 16-5. MS-ESI calculated [M+H] + 689, found 689.
- compound 18-1 (100mg, 0.526mmol) and compound 14-4 (155mg, 0.526mmol) were added to anhydrous 1,4-dioxane (4mL) and water (1mL), and then potassium acetate was added (103mg, 1.05mmol) and [1,1′-bis(diphenylphosphino)ferrocene]palladium(II) dichloride (38.5mg, 0.053mmol), the reaction solution was stirred at 90°C for 12 hours, and Water (20 mL) was added to the reaction solution, extracted with ethyl acetate (20 mL x 1), the organic phase was washed with saturated brine (20 mL x 1), dried over anhydrous sodium sulfate, filtered, concentrated under reduced pressure, and the residue was subjected to silica gel column chromatography Compound 18-2 was obtained by separation and purification (2/1 petroleum ether/ethyl acetate).
- compound 20-3 (150mg, 0.541mmol) was dissolved in ethyl acetate (5mL), then ethyl acetate hydrochloride (4M, 5mL, 20.0mmol) was added, and the reaction solution was stirred at 25°C for 2 hours.
- MS-ESI calculated value [M+H] + 178, found value 178.
- Zinc dust (1.81 g, 27.7 mmol) and 1,2-dibromoethane (307 mg, 1.63 mmol) were added to N,N-dimethylformamide (20 mL).
- the reaction solution was stirred and reacted at 70° C. for 10 minutes.
- the reaction solution was then cooled to 20°C.
- Chlorotrimethylsilane (177mg, 1.63mmol) was added dropwise to the reaction solution, and then the reaction solution was stirred at 20°C for 50 minutes.
- Zinc powder (1.30 g, 19.9 mmol) and 1,2-dibromoethane (220 mg, 1.17 mmol) were added to N,N-dimethylformamide (20 mL). The reaction solution was stirred and reacted at 70° C. for 10 minutes. The reaction solution was then cooled to 20°C. Chlorotrimethylsilane (127mg, 1.17mmol) was added dropwise to the reaction solution, and then the reaction solution was stirred at 20°C for 50 minutes.
- reaction solution was filtered through diatomaceous earth, the filter residue was washed with ethyl acetate (30mL), the filtrate was washed with saturated ammonium chloride aqueous solution (30mL ⁇ 1), and the organic phase was sequentially washed with water (30mL ⁇ 3) and saturated brine (30mL ⁇ 1). After washing, drying with anhydrous sodium sulfate, filtration, the filtrate was concentrated under reduced pressure, and the residue was separated and purified by silica gel column chromatography (4/1 petroleum ether/ethyl acetate) to obtain compound 31-3.
- Zinc dust (845 mg, 12.5 mmol) and 1,2-dibromoethane (165 mg, 0.879 mmol) were added to N,N-dimethylformamide (10 mL).
- the reaction solution was stirred and reacted at 70° C. for 10 minutes.
- the reaction solution was then cooled to 20°C.
- Chlorotrimethylsilane (95.5mg, 0.879mmol) was added dropwise to the reaction solution, and then the reaction solution was stirred at 20°C for 50 minutes.
- a solution of 28-1 (3.11 g, 10.9 mmol) in N,N-dimethylformamide (6 mL) was added to the reaction solution, and the reaction solution was stirred and reacted at 40°C for 1 hour under the protection of nitrogen.
- the N, N-di Methylformamide (24 mL) was added to the reaction solution.
- the reaction solution was stirred and reacted at 60° C. for 10 hours under the protection of nitrogen.
- the reaction solution was filtered through diatomaceous earth to remove solid residues, the filtrate was treated with saturated ammonium chloride (50mL x 2) and ethyl acetate (50mL x 2), and the organic phase was combined with water (50mL x 3), saturated brine (50mL x 1) washed, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure.
- the residue was separated and purified by silica gel column chromatography to obtain compound 32-2.
- reaction solution was quenched with 10% citric acid aqueous solution (30mL), extracted with ethyl acetate (30mL ⁇ 3), the combined organic phase was washed with saturated brine (100mL ⁇ 1), dried over anhydrous sodium sulfate, filtered, and the filtrate was decompressed concentrate. The residue was separated and purified by thin layer chromatography (2:1 petroleum ether/ethyl acetate) to obtain compound 38-2.
- reaction solution was quenched with saturated aqueous ammonium chloride (20 mL), extracted with ethyl acetate (20 mL ⁇ 3), the combined organic phases were washed with saturated brine (50 mL ⁇ 1), dried over anhydrous sodium sulfate, filtered, and the filtrate was decompressed concentrate. The residue was separated and purified by thin layer chromatography (4:1 petroleum ether/ethyl acetate) to obtain compound 39-1.
- ROR ⁇ ligand-binding domain LBD
- TR-FRET time-resolved fluorescence energy transfer
- the compound to be tested was diluted in DMSO and further diluted in assay buffer (50mM Tris pH 7.0, 50mM KCl, 1mM Na-EDTA, 0.1mM DTT, 0.01% BSA) (4-fold dilution, 10 concentrations, highest concentration 5000 nM), the final DMSO concentration was 1%.
- the hROR ⁇ -LBD protein was diluted in assay buffer to give a final hROR ⁇ -LBD concentration of 15 nM in 384-well plates.
- a stock solution of biotin-SRC1 polypeptide Biotin-SPSSHSSLTERHKILHRLLQEGSP
- SA-eu (1 nM final concentration) and SA-APC 50 nM final concentration
- the final assay mixture was incubated overnight at 4°C, equilibrated at room temperature for 1 hour, and centrifuged at 1000 rpm for 1 minute. Fluorescence readings were detected on the Envision microplate detector, and the logarithmic curve of the ratio of the fluorescence signal of emission wavelength 665nM/615nM to the compound concentration was drawn by GraphPad Prism software, and the 50% effective concentration (EC 50 ) and 50% inhibition of the compound were calculated Concentration ( IC50 ). The maximum response (Emax) was the upper peak peak of the signal determined by GraphPad Prism fit.
- the compound of the present invention has obvious agonistic or inverse agonistic activity on the in vitro activity of ROR ⁇ .
- Mouse CD4+T cell isolation kit (Mouse CD4+T cell isolation kit) (Stemcell)
- Non-essential amino acids (Gibco)
- the CD3 antibody was diluted to 5 ⁇ g/mL in DPBS, added to a 96-well U-bottom plate, 50 ⁇ L of liquid per well, and coated overnight at 4°C.
- C57BL/6 mouse spleen in culture medium (RPMI 1640+10% fetal bovine serum+1% penicillin+streptomycin+1% non-essential amino acid+0.05mM ⁇ -mercaptoethanol), pass through a 70 ⁇ m filter to prepare a single cell suspension , centrifuge at 300g for 3min. Red blood cell lysate was added to lyse at room temperature for 3 min. CD4 + cells were isolated using the Mouse CD4 + T cell isolation kit.
- CD4 + cells obtained above at a density of 5*10 5 /mL into the coated wells , 200 ⁇ L cell suspension per well; then add CD28 antibody (3 ⁇ g/mL), TGF ⁇ (3ng/mL), IL-6 (30ng/mL), IL-23 (10ng/mL), IL-1 ⁇ (10ng/mL ), IFN ⁇ antibody (10 ⁇ g/mL) and IL-4 antibody (10 ⁇ g/mL); then the compound of the present invention was added to the well, and cultured at 37° C. under 5% CO 2 for 3 days.
- the U-bottom plate was centrifuged at 300 g for 3 min, the supernatant was discarded, and washed twice with staining buffer.
- the IL-17A antibody was diluted 1:200 in permeabilization buffer, 50 ⁇ L of dye solution was added to each well, stained at room temperature for 30 min, and then washed twice with staining buffer. Finally, the cells were resuspended with 150 ⁇ L staining buffer, and the proportion of Th17 cells was detected by flow cytometry.
- Table 3 The compounds of the present invention promote the ability of CD4 + cells to differentiate into Th17 cells to measure results
- Compound test concentration 1 ⁇ M.
- the compounds of the present invention can obviously promote the differentiation of CD4 + cells into Th17 cells.
- the pharmacokinetic characteristics of the compounds were tested in rodents after intravenous injection and oral administration according to the standard protocol.
- the mice were given a single intravenous injection (IV) and oral administration (PO).
- the solvent for intravenous injection is a mixed solvent made up of 5% dimethyl sulfoxide, 30% PEG400, and 65% 10% hydroxypropyl ⁇ -cyclodextrin.
- the oral vehicle is a mixed vehicle made of 0.5% hypromellose and 0.2% Tween.
- the project used four female Balb/c mice, two mice were administered intravenously, the dose was 0.5mg/kg, and the collection 0h (before administration) and after administration were 0.0833, 0.25, 0.5, 1, Plasma samples at 2, 4, 8, and 24 hours were administered orally to the other two mice at a dose of 1 mg/kg, collected at 0 h (before administration) and at 0.25, 0.5, 1, 2, and 4 hours after administration , 8, 24h plasma samples, collect whole blood samples within 24 hours, centrifuge at 3000g for 15 minutes, separate supernatant to obtain plasma samples, add 4 times volume of acetonitrile solution containing internal standard to precipitate protein, centrifuge to take supernatant and add equal volume The water was then centrifuged to take the supernatant sample, and the blood drug concentration was quantitatively analyzed by LC-MS/MS analysis method, and the pharmacokinetic parameters were calculated, such as peak concentration (C max ), clearance rate (CL), half-life (T 1 / 2 ), tissue distribution (V
- the compounds of this invention have good pharmacokinetic properties, including good oral bioavailability, oral exposure, half-life and clearance rate.
- the purpose of this experiment is to study the evaluation of the compound of the present invention on the MC38 mouse colon cancer xenograft tumor model in vivo.
- the culture medium is 1640 medium containing 10% fetal bovine serum, and the culture conditions are 37°C and 5% carbon dioxide.
- the subculture ratio was 1:2 ⁇ 1:3, and subcultured 2 ⁇ 3 times a week.
- 0.1 mL (2 ⁇ 10 5 ) cells were inoculated subcutaneously on the right back of each mouse. On the same day, animals were randomized into groups based on body weight.
- the experimental vehicle was 5% DMSO/95% (20% hydroxypropyl beta cyclodextrin).
- the test substance was dissolved in a solvent, prepared into a uniform solution with a certain concentration, and stored at 4°C.
- the experimental index is to investigate whether tumor growth is inhibited, delayed or cured.
- Tumor diameters were measured twice a week with vernier calipers.
- T/C Relative tumor proliferation rate
- PD-1 monoclonal antibody BioXcell. PD-1 monoclonal antibody was administered on the 7th day after grouping, and compound 32 was administered on the day of grouping.
- the combination of the compound of the present invention and PD-1 monoclonal antibody has an excellent tumor-inhibiting effect on the transplanted tumor model of MC38 mouse colon cancer.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description
本申请主张如下优先权:This application claims the following priority:
CN202110933021.9,2021年08月13日。CN202110933021.9, August 13, 2021.
本发明涉及一类苯并吗啉类化合物及其制备方法和用途。具体涉及式(Ⅰ)所示化合物及其药学上可接受的盐。The invention relates to a class of benzomorpholine compounds, a preparation method and application thereof. It specifically relates to the compound represented by formula (I) and its pharmaceutically acceptable salt.
肿瘤免疫疗法是近年来备受关注的治疗领域,主要机理是通过调动机体的免疫系统,增强免疫微环境的抗肿瘤能力。目前已有肿瘤免疫治疗的单抗药物如Keytruda和OPDIVO等用于非小细胞肺癌和黑色素瘤等多种癌症的治疗。Tumor immunotherapy is a therapeutic area that has attracted much attention in recent years. The main mechanism is to enhance the anti-tumor ability of the immune microenvironment by mobilizing the body's immune system. Currently, monoclonal antibody drugs for tumor immunotherapy, such as Keytruda and OPDIVO, have been used for the treatment of various cancers such as non-small cell lung cancer and melanoma.
维甲酸相关孤儿受体(RAR-related orphan receptor,RORs)属于核受体超家族,是细胞内转录因子的一员,能够调控多种生理过程,包括生殖发育、新陈代谢、免疫系统调节等。ROR有三个家族成员:ROR-α、-β和-γ,分别由RORA、RORB、RORC基因编码。RORγ又包括RORγ1和RORγt(RORγ2)两种亚型。RORγ1在胸腺、肌肉、胰腺、前列腺和肝脏等多种组织和器官中表达,而RORγ的短链亚型RORγt主要分布于胸腺,促进初始T细胞向Th17和Tc17细胞分化。Th17和Tc17细胞通过分泌IL-17,IL-22,GM-CSF等细胞因子和炎症因子,促进炎症反应和自身免疫应答。IL-17可以促进肿瘤微环境中CTLs和NK细胞的招募和浸润,提升效应T细胞的抗肿瘤作用。Retinoic acid-related orphan receptors (RAR-related orphan receptors, RORs) belong to the nuclear receptor superfamily and are a member of intracellular transcription factors, which can regulate a variety of physiological processes, including reproductive development, metabolism, and immune system regulation. ROR has three family members: ROR-α, -β and -γ, which are encoded by RORA, RORB and RORC genes, respectively. RORγ includes two subtypes, RORγ1 and RORγt (RORγ2). RORγ1 is expressed in various tissues and organs such as thymus, muscle, pancreas, prostate, and liver, while the short-chain subtype RORγt of RORγ is mainly distributed in the thymus and promotes the differentiation of initial T cells into Th17 and Tc17 cells. Th17 and Tc17 cells promote inflammatory and autoimmune responses by secreting IL-17, IL-22, GM-CSF and other cytokines and inflammatory factors. IL-17 can promote the recruitment and infiltration of CTLs and NK cells in the tumor microenvironment, and enhance the anti-tumor effect of effector T cells.
RORγ激动剂目前没有药物获批上市,Lycera Corp公司的RORγ激动剂LYC-55716单药用于晚期实体瘤的治疗处于临床二期研究,与PD-1单抗pembrolizumab联用治疗晚期非小细胞肺癌的治疗处于临床1b期研究。面对巨大的未满足市场,该领域仍然需要活性更好,药代动力学参数更优的候选化合物推进临床试验,以满足治疗需求。RORγ agonists currently have no drugs approved for marketing, and Lycera Corp’s RORγ agonist LYC-55716 monotherapy for the treatment of advanced solid tumors is in phase II clinical research, and it is combined with PD-1 monoclonal antibody pembrolizumab in the treatment of advanced non-small cell lung cancer The treatment is in a phase 1b clinical study. In the face of a huge unmet market, this field still needs candidate compounds with better activity and better pharmacokinetic parameters to advance to clinical trials to meet the therapeutic needs.
发明内容Contents of the invention
本发明提供了式(Ⅰ)化合物或其药学上可接受的盐,The present invention provides a compound of formula (I) or a pharmaceutically acceptable salt thereof,

其中,in,
![]()
![]()
L 1和L 2分别独立地选自单键和-NH-; L 1 and L 2 are independently selected from single bonds and -NH-;
T 1选自N和C(R 12); T 1 is selected from N and C (R 12 );
T 2选自N和C(R 15); T 2 is selected from N and C (R 15 );
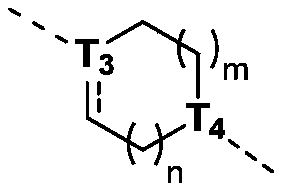
当
![]()
![]()
当
![]()
![]()
T 4为CH或N; T4 is CH or N;
m为0、1或2;m is 0, 1 or 2;
n为0、1或2;n is 0, 1 or 2;
R 11、R 12、R 13、R 14和R 15分别独立地选自H、F、Cl、Br、I、-OH、-NH 2、-CN、C 1-3烷基、C 1-3烷氧基和C 1-3烷氨基,其中所述C 1-3烷基、C 1-3烷氧基和C 1-3烷氨基分别独立地任选被1、2或3个R a取代; R 11 , R 12 , R 13 , R 14 and R 15 are independently selected from H, F, Cl, Br, I, -OH, -NH 2 , -CN, C 1-3 alkyl, C 1-3 Alkoxy and C 1-3 alkylamino, wherein said C 1-3 alkyl, C 1-3 alkoxy and C 1-3 alkylamino are independently optionally substituted by 1, 2 or 3 R a ;
或者,R
13和R
14及其所接的碳原子连接在一起,使结构单元


T 5和T 6分别独立地选自N和CH; T 5 and T 6 are independently selected from N and CH;
R 2选自C 1-3烷基,其中所述C 1-3烷基任选被1、2或3个R b取代; R 2 is selected from C 1-3 alkyl, wherein said C 1-3 alkyl is optionally substituted by 1, 2 or 3 R b ;
R 31和R 32分别独立地选自H、F、Cl、Br、I、-OH、-NH 2、C 1-3烷基和C 1-3烷氧基,其中所述C 1-3烷基和C 1-3烷氧基分别独立地任选被1、2或3个R c取代; R 31 and R 32 are independently selected from H, F, Cl, Br, I, -OH, -NH 2 , C 1-3 alkyl and C 1-3 alkoxy, wherein the C 1-3 alkoxy and C 1-3 alkoxy are independently optionally substituted by 1, 2 or 3 R c ;
R t3选自H、F、Cl、Br、I、-OH、-NH 2、C 1-3烷基和C 1-3烷氧基,其中所述C 1-3烷基和C 1-3烷氧基分别独立地任选被1、2或3个R d取代; R t3 is selected from H, F, Cl, Br, I, -OH, -NH 2 , C 1-3 alkyl and C 1-3 alkoxy, wherein the C 1-3 alkyl and C 1-3 Alkoxy groups are independently optionally substituted by 1, 2 or 3 R d ;
R a分别独立地选自F、Cl、Br、I、-OH、-NH 2和C 1-3烷基,其中所述C 1-3烷基任选被1、2或3个R取代; R a is independently selected from F, Cl, Br, I, -OH, -NH 2 and C 1-3 alkyl, wherein the C 1-3 alkyl is optionally substituted by 1, 2 or 3 R;
R b分别独立地选自F、Cl、Br、I、-OH、-NH 2、-CN、-COOH和C 1-3烷基; R b are independently selected from F, Cl, Br, I, -OH, -NH 2 , -CN, -COOH and C 1-3 alkyl;
R c分别独立地选自F、Cl、Br、I、-OH和-NH 2; R c are independently selected from F, Cl, Br, I, -OH and -NH 2 ;
R d分别独立地选自F、Cl、Br、I、-OH和-NH 2; R d are independently selected from F, Cl, Br, I, -OH and -NH 2 ;
R分别独立地选自F、Cl、Br、I、-OH和-NH 2。 R are each independently selected from F, Cl, Br, I, -OH and -NH 2 .
本发明提供了式(Ⅱ)化合物或其药学上可接受的盐,The present invention provides a compound of formula (II) or a pharmaceutically acceptable salt thereof,

其中,in,
![]()
![]()
L 1和L 2分别独立地选自单键和-NH-; L 1 and L 2 are independently selected from single bonds and -NH-;
T 1选自N和C(R 12); T 1 is selected from N and C (R 12 );
T 2选自N和C(R 15); T 2 is selected from N and C (R 15 );
当
![]()
![]()
当
![]()
![]()
T 4为CH或N; T4 is CH or N;
m为0、1或2;m is 0, 1 or 2;
n为0、1或2;n is 0, 1 or 2;
R 11、R 12、R 13、R 14和R 15分别独立地选自H、F、Cl、Br、I、OH、NH 2、CN、C 1-3烷基、C 1-3烷氧基和C 1-3烷氨基,其中所述C 1-3烷基、C 1-3烷氧基和C 1-3烷氨基任选被1、2或3个R a取代; R 11 , R 12 , R 13 , R 14 and R 15 are independently selected from H, F, Cl, Br, I, OH, NH 2 , CN, C 1-3 alkyl, C 1-3 alkoxy and C 1-3 alkylamino, wherein the C 1-3 alkyl, C 1-3 alkoxy and C 1-3 alkylamino are optionally substituted by 1, 2 or 3 R a ;
或者,R
13和R
14及其所接的碳原子连接一起,使


T 5和T 6分别独立地选自N和CH; T 5 and T 6 are independently selected from N and CH;
R 2选自任选被1、2或3个R b取代的C 1-3烷基; R 2 is selected from C 1-3 alkyl optionally substituted by 1, 2 or 3 R b ;
R a分别独立地选自F、Cl、Br、I、OH、NH 2和C 1-3烷基,其中所述C 1-3烷基任选被1、2或3个R取代; R a is independently selected from F, Cl, Br, I, OH, NH 2 and C 1-3 alkyl, wherein the C 1-3 alkyl is optionally substituted by 1, 2 or 3 R;
R b分别独立地选自F、Cl、Br、I、OH、NH 2、CN、COOH和C 1-3烷基; R b are independently selected from F, Cl, Br, I, OH, NH 2 , CN, COOH and C 1-3 alkyl;
R分别独立地选自F、Cl、Br、I、OH和NH 2。 R are each independently selected from F, Cl, Br, I, OH and NH 2 .
本发明的一些方案中,上述T 1选自C(R 12),其他变量如本发明所定义。 In some solutions of the present invention, the above T 1 is selected from C(R 12 ), and other variables are as defined in the present invention.
本发明的一些方案中,上述T 1选自N,其他变量如本发明所定义。 In some solutions of the present invention, the above T1 is selected from N, and other variables are as defined in the present invention.
本发明的一些方案中,上述T 2选自C(R 15),其他变量如本发明所定义。 In some solutions of the present invention, the above T 2 is selected from C(R 15 ), and other variables are as defined in the present invention.
本发明的一些方案中,上述T 2选自N,其他变量如本发明所定义。 In some solutions of the present invention, the above T 2 is selected from N, and other variables are as defined in the present invention.
本发明的一些方案中,上述R a分别独立地选自F、Cl、Br、I、-OH、-NH 2和-CH 3,其他变量如本发明所定义。 In some solutions of the present invention, the above R a is independently selected from F, Cl, Br, I, -OH, -NH 2 and -CH 3 , and other variables are as defined in the present invention.
本发明的一些方案中,上述R a分别独立地选自F,其他变量如本发明所定义。 In some solutions of the present invention, the above R a are independently selected from F, and other variables are as defined in the present invention.
本发明的一些方案中,上述R b分别独立地选自-COOH,其他变量如本发明所定义。 In some solutions of the present invention, the above-mentioned R b are independently selected from -COOH, and other variables are as defined in the present invention.
本发明的一些方案中,上述R 11、R 12、R 13、R 14和R 15分别独立地选自H、F、Cl、Br、I、-OH、-NH 2、-CN、-CH 3、-O-CH 3和-NH-CH 3,其中所述-CH 3、-O-CH 3和-NH-CH 3分别独立地任选被1、2或3个R a取代,R a及其他变量如本发明所定义。 In some solutions of the present invention, the above-mentioned R 11 , R 12 , R 13 , R 14 and R 15 are independently selected from H, F, Cl, Br, I, -OH, -NH 2 , -CN, -CH 3 , -O-CH 3 and -NH-CH 3 , wherein said -CH 3 , -O-CH 3 and -NH-CH 3 are independently optionally substituted by 1, 2 or 3 R a , R a and Other variables are as defined herein.
本发明的一些方案中,上述R
11、R
12、R
13、R
14和R
15选自H、F、Cl、Br、I、-OH、-NH
2、-CN、-CH
3、-CF
3、-O-CH
3、-O-CH
2F、-O-CHF
2、-O-CF
3和
![]()
![]()
本发明的一些方案中,上述R 11选自H,其他变量如本发明所定义。 In some solutions of the present invention, the above-mentioned R 11 is selected from H, and other variables are as defined in the present invention.
本发明的一些方案中,上述R 12选自H和F,其他变量如本发明所定义。 In some solutions of the present invention, the above-mentioned R 12 is selected from H and F, and other variables are as defined in the present invention.
本发明的一些方案中,上述R 13选自H,其他变量如本发明所定义。 In some solutions of the present invention, the above-mentioned R 13 is selected from H, and other variables are as defined in the present invention.
本发明的一些方案中,上述R 14选自H、F、Cl、-CH 3和-OCH 3,其中所述-O-CH 3任选被1、2或3个R a取代,R a及其他变量如本发明所定义。 In some schemes of the present invention, the above-mentioned R 14 is selected from H, F, Cl, -CH 3 and -OCH 3 , wherein the -O-CH 3 is optionally substituted by 1, 2 or 3 R a , R a and Other variables are as defined herein.
本发明的一些方案中,上述R 14选自H、F、Cl、-CH 3、-CF 3和-OCF 3,其他变量如本发明所定义。 In some solutions of the present invention, the above R 14 is selected from H, F, Cl, -CH 3 , -CF 3 and -OCF 3 , and other variables are as defined in the present invention.
本发明的一些方案中,上述R 15选自H、F、Cl和-OCH 3,其中所述-CH 3任选被1、2或3个R a取代,R a及其他变量如本发明所定义。 In some schemes of the present invention, the above-mentioned R 15 is selected from H, F, Cl and -OCH 3 , wherein the -CH 3 is optionally substituted by 1, 2 or 3 R a , and R a and other variables are as described in the present invention definition.
本发明的一些方案中,上述R 15选自H、F、Cl、-OCH 3、-OCF 3和-OCHF 2,其他变量如本发明所定义。 In some solutions of the present invention, the above R 15 is selected from H, F, Cl, -OCH 3 , -OCF 3 and -OCHF 2 , and other variables are as defined in the present invention.
本发明的一些方案中,上述R 2选自选自-CH 2-CH 3,其中所述-CH 2-CH 3任选被1、2或3个R b取代,R b及其他变量如本发明所定义。 In some schemes of the present invention, the above-mentioned R 2 is selected from -CH 2 -CH 3 , wherein the -CH 2 -CH 3 is optionally substituted by 1, 2 or 3 R b , and R b and other variables are as described herein invention defined.
本发明的一些方案中,上述R
2选自
![]()
![]()
本发明的一些方案中,上述R 31和R 32分别独立地选自H、F、Cl、Br、I、-OH、-NH 2、-CH 3和-O-CH 3,其中所述-CH 3和-O-CH 3分别独立地任选被1、2或3个R c取代,R c及其他变量如本发明所定义。 In some solutions of the present invention, the above-mentioned R 31 and R 32 are independently selected from H, F, Cl, Br, I, -OH, -NH 2 , -CH 3 and -O-CH 3 , wherein -CH 3 and -O-CH 3 are each independently optionally substituted by 1, 2 or 3 R c , R c and other variables are as defined in the present invention.
本发明的一些方案中,上述R 31和R 32分别独立地选自H、F、Cl、Br、I、-OH、-NH 2、-CH 3、-CF 3、-O-CH 3、-O-CH 2F、-O-CHF 2和-O-CF 3,其他变量如本发明所定义。 In some solutions of the present invention, the above-mentioned R 31 and R 32 are independently selected from H, F, Cl, Br, I, -OH, -NH 2 , -CH 3 , -CF 3 , -O-CH 3 , - O- CH2F , -O- CHF2 and -O- CF3 , other variables are as defined in the present invention.
本发明的一些方案中,上述R t3选自H、F、Cl、Br、I、-OH、-NH 2、-CH 3和-O-CH 3,其他变量如本发明所定义。 In some solutions of the present invention, the above-mentioned R t3 is selected from H, F, Cl, Br, I, -OH, -NH 2 , -CH 3 and -O-CH 3 , and other variables are as defined in the present invention.
本发明的一些方案中,上述R t3选自H、F、-OH和-O-CH 3,其他变量如本发明所定义。 In some solutions of the present invention, the above-mentioned R t3 is selected from H, F, -OH and -O-CH 3 , and other variables are as defined in the present invention.
本发明的一些方案中,上述结构单元



本发明的一些方案中,上述结构单元元



本发明的一些方案中,上述结构单元


本发明还有一些方案是由上述各变量任意组合而来。Some schemes of the present invention are formed by any combination of the above-mentioned variables.
本发明的一些方案中,上述化合物或其药学上可接受的盐,其中,化合物选自In some schemes of the present invention, the above compound or a pharmaceutically acceptable salt thereof, wherein the compound is selected from

其中,in,
T 1选自N和C(R 12); T 1 is selected from N and C (R 12 );
T 2选自N和C(R 15); T 2 is selected from N and C (R 15 );
m、n、L 1、L 2、R 11、R 12、R 13、R 14、R 15、R 2、R 31、R 32和R t3如本发明所定义。 m, n, L 1 , L 2 , R 11 , R 12 , R 13 , R 14 , R 15 , R 2 , R 31 , R 32 and R t3 are as defined in the present invention.
本发明的一些方案中,上述化合物或其药学上可接受的盐,其中,化合物选自In some schemes of the present invention, the above compound or a pharmaceutically acceptable salt thereof, wherein the compound is selected from

其中,in,
T 1选自N和C(R 12); T 1 is selected from N and C (R 12 );
T 2选自N和C(R 15); T 2 is selected from N and C (R 15 );
m、n、L 1、L 2、R 11、R 12、R 13、R 14、R 15、R 2、R 31、R 32和R t3如本发明所定义; m, n, L 1 , L 2 , R 11 , R 12 , R 13 , R 14 , R 15 , R 2 , R 31 , R 32 and R t3 are as defined in the present invention;
带“*”碳原子为手性碳原子,以(R)或(S)单一对映体形式或富含一种对映体形式存在。Carbon atoms with "*" are chiral carbon atoms and exist as (R) or (S) single enantiomer or enrichment of one enantiomer.
本发明的一些方案中,上述化合物或其药学上可接受的盐,其中,化合物选自In some schemes of the present invention, the above compound or a pharmaceutically acceptable salt thereof, wherein the compound is selected from

其中,in,
T 1选自N和C(R 12); T 1 is selected from N and C (R 12 );
T 2选自N和C(R 15); T 2 is selected from N and C (R 15 );
R 11、R 12、R 13、R 14、R 15、R 2、R 31、R 32和R t3如本发明所定义。 R 11 , R 12 , R 13 , R 14 , R 15 , R 2 , R 31 , R 32 and R t3 are as defined in the present invention.
本发明的一些方案中,上述化合物或其药学上可接受的盐,其中,化合物选自In some schemes of the present invention, the above compound or a pharmaceutically acceptable salt thereof, wherein the compound is selected from

其中,in,
T 1选自N和C(R 12); T 1 is selected from N and C (R 12 );
T 2选自N和C(R 15); T 2 is selected from N and C (R 15 );
R 11、R 12、R 13、R 14、R 15、R 2、R 31、R 32和R t3如本发明所定义; R 11 , R 12 , R 13 , R 14 , R 15 , R 2 , R 31 , R 32 and R t3 are as defined in the present invention;
带“*”碳原子为手性碳原子,以(R)或(S)单一对映体形式或富含一种对映体形式存在。Carbon atoms with "*" are chiral carbon atoms and exist as (R) or (S) single enantiomer or enrichment of one enantiomer.
本发明的一些方案中,上述化合物或其药学上可接受的盐,其中,化合物选自In some schemes of the present invention, the above compound or a pharmaceutically acceptable salt thereof, wherein the compound is selected from


其中,in,
m、n、L 1、L 2、T 1、T 2、R 11、R 12、R 13、R 14、R 15和R 2如本发明所定义。 m, n, L 1 , L 2 , T 1 , T 2 , R 11 , R 12 , R 13 , R 14 , R 15 and R 2 are as defined in the present invention.
本发明的一些方案中,上述化合物或其药学上可接受的盐,其中,化合物选自In some schemes of the present invention, the above compound or a pharmaceutically acceptable salt thereof, wherein the compound is selected from

其中,in,
m、n、L 1、L 2、T 1、T 2、R 11、R 12、R 13、R 14、R 15和R 2如本发明所定义; m, n, L 1 , L 2 , T 1 , T 2 , R 11 , R 12 , R 13 , R 14 , R 15 and R 2 are as defined in the present invention;
带“*”碳原子为手性碳原子,以(R)或(S)单一对映体形式或富含一种对映体形式存在。Carbon atoms with "*" are chiral carbon atoms and exist as (R) or (S) single enantiomer or enrichment of one enantiomer.
本发明还提供了下式化合物或其药学上可接受的盐,The present invention also provides a compound of the following formula or a pharmaceutically acceptable salt thereof,



本发明的一些方案中,上述化合物或其药学上可接受的盐,其中,化合物选自In some schemes of the present invention, the above compound or a pharmaceutically acceptable salt thereof, wherein the compound is selected from





技术效果technical effect
作为一类具有RORγ激动活性或反向激动活性的苯并环类化合物,本发明的化合物体外活性显著,并且具有良好的药代动力学性质。As a class of benzocyclic compounds with RORγ agonistic activity or inverse agonistic activity, the compound of the present invention has remarkable in vitro activity and good pharmacokinetic properties.
定义和说明Definition and Description
除非另有说明,本文所用的下列术语和短语旨在具有下列含义。一个特定的术语或短语在没有特别定义的情况下不应该被认为是不确定的或不清楚的,而应该按照普通的含义去理解。当本文中出现商品名时,意在指代其对应的商品或其活性成分。Unless otherwise stated, the following terms and phrases used herein are intended to have the following meanings. A specific term or phrase should not be considered indeterminate or unclear if it is not specifically defined, but should be understood according to its ordinary meaning. When a trade name appears herein, it is intended to refer to its corresponding trade name or its active ingredient.
这里所采用的术语“药学上可接受的”,是针对那些化合物、材料、组合物和/或剂型而言,它们在可靠的医学判断的范围之内,适用于与人类和动物的组织接触使用,而没有过多的毒性、刺激性、过敏性反应或其它问题或并发症,与合理的利益/风险比相称。The term "pharmaceutically acceptable" as used herein refers to those compounds, materials, compositions and/or dosage forms, which are suitable for use in contact with human and animal tissues within the scope of sound medical judgment , without undue toxicity, irritation, allergic reaction or other problems or complications, commensurate with a reasonable benefit/risk ratio.
术语“药学上可接受的盐”是指本发明化合物的盐,由本发明发现的具有特定取代基的化合物与相对无毒的酸或碱制备。当本发明的化合物中含有相对酸性的功能团时,可以通过在纯的溶液或合适的惰性溶剂中用足够量的碱与这类化合物接触的方式获得碱加成盐。药学上可接受的碱加成盐包括钠、钾、钙、铵、有机胺或镁盐或类似的盐。当本发明的化合物中含有相对碱性的官能团时,可以通过在纯的溶液或合适的惰性溶剂中用足够量的酸与这类化合物接触的方式获得酸加成盐。药学上可接受的酸加成盐的实例包括无机酸盐,所述无机酸包括例如盐酸、氢溴酸、硝酸、碳酸,碳酸氢根,磷酸、磷酸一氢根、磷酸二氢根、硫酸、硫酸氢根、氢碘酸、亚磷酸等;以及有机酸盐,所述有机酸包括如乙酸、丙酸、异丁酸、马来酸、 丙二酸、苯甲酸、琥珀酸、辛二酸、反丁烯二酸、乳酸、扁桃酸、邻苯二甲酸、苯磺酸、对甲苯磺酸、柠檬酸、酒石酸和甲磺酸等类似的酸;还包括氨基酸(如精氨酸等)的盐,以及如葡糖醛酸等有机酸的盐。本发明的某些特定的化合物含有碱性和酸性的官能团,从而可以被转换成任一碱或酸加成盐。The term "pharmaceutically acceptable salt" refers to a salt of a compound of the present invention, which is prepared from a compound having a specific substituent found in the present invention and a relatively non-toxic acid or base. When compounds of the present invention contain relatively acidic functional groups, base addition salts can be obtained by contacting such compounds with a sufficient amount of base, either neat solution or in a suitable inert solvent. Pharmaceutically acceptable base addition salts include sodium, potassium, calcium, ammonium, organic amine or magnesium salts or similar salts. When compounds of the present invention contain relatively basic functionalities, acid addition salts can be obtained by contacting such compounds with a sufficient amount of the acid, either neat solution or in a suitable inert solvent. Examples of pharmaceutically acceptable acid addition salts include salts of inorganic acids including, for example, hydrochloric acid, hydrobromic acid, nitric acid, carbonic acid, bicarbonate, phosphoric acid, monohydrogenphosphate, dihydrogenphosphate, sulfuric acid, Hydrogen sulfate, hydroiodic acid, phosphorous acid, etc.; and organic acid salts, such as acetic acid, propionic acid, isobutyric acid, maleic acid, malonic acid, benzoic acid, succinic acid, suberic acid, Fumaric acid, lactic acid, mandelic acid, phthalic acid, benzenesulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid and methanesulfonic acid and similar acids; also salts of amino acids such as arginine and the like , and salts of organic acids such as glucuronic acid. Certain specific compounds of the present invention contain basic and acidic functional groups and can thus be converted into either base or acid addition salts.
本发明的药学上可接受的盐可由含有酸根或碱基的母体化合物通过常规化学方法合成。一般情况下,这样的盐的制备方法是:在水或有机溶剂或两者的混合物中,经由游离酸或碱形式的这些化合物与化学计量的适当的碱或酸反应来制备。The pharmaceutically acceptable salts of the present invention can be synthesized from the parent compound containing acid groups or bases by conventional chemical methods. In general, such salts are prepared by reacting the free acid or base form of these compounds with a stoichiometric amount of the appropriate base or acid in water or an organic solvent or a mixture of both.
本发明的化合物可以存在特定的几何或立体异构体形式。本发明设想所有的这类化合物,包括顺式和反式异构体、(-)-和(+)-对映体、(R)-和(S)-对映体、非对映异构体、(D)-异构体、(L)-异构体,及其外消旋混合物和其他混合物,例如对映异构体或非对映体富集的混合物,所有这些混合物都属于本发明的范围之内。烷基等取代基中可存在另外的不对称碳原子。所有这些异构体以及它们的混合物,均包括在本发明的范围之内。The compounds of the invention may exist in particular geometric or stereoisomeric forms. The present invention contemplates all such compounds, including cis and trans isomers, (-)- and (+)-enantiomers, (R)- and (S)-enantiomers, diastereomers isomers, (D)-isomers, (L)-isomers, and their racemic and other mixtures, such as enantiomerically or diastereomerically enriched mixtures, all of which are subject to the present within the scope of the invention. Additional asymmetric carbon atoms may be present in substituents such as alkyl groups. All such isomers, as well as mixtures thereof, are included within the scope of the present invention.
除非另有说明,术语“对映异构体”或者“旋光异构体”是指互为镜像关系的立体异构体。Unless otherwise stated, the terms "enantiomer" or "optical isomer" refer to stereoisomers that are mirror images of each other.
除非另有说明,术语“顺反异构体”或者“几何异构体”系由因双键或者成环碳原子单键不能自由旋转而引起。Unless otherwise stated, the terms "cis-trans isomers" or "geometric isomers" arise from the inability to rotate freely due to the double bond or the single bond of the carbon atoms forming the ring.
除非另有说明,术语“非对映异构体”是指分子具有两个或多个手性中心,并且分子间为非镜像的关系的立体异构体。Unless otherwise indicated, the term "diastereoisomer" refers to stereoisomers whose molecules have two or more chiral centers and which are not mirror images of the molecules.
除非另有说明,“(+)”表示右旋,“(-)”表示左旋,“(±)”表示外消旋。Unless otherwise specified, "(+)" means dextrorotation, "(-)" means levorotation, and "(±)" means racemization.
除非另有说明,用楔形实线键
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
除非另有说明,当化合物中存在双键结构,如碳碳双键、碳氮双键和氮氮双键,且双键上的各个原子均连接有两个不同的取代基时(包含氮原子的双键中,氮原子上的一对孤对电子视为其连接的一个取代基),如果该化合物中双键上的原子与其取代基之间用波浪线
![]()
![]()

除非另有说明,术语“互变异构体”或“互变异构体形式”是指在室温下,不同官能团异构体处于动态平衡,并能很快的相互转化。若互变异构体是可能的(如在溶液中),则可以达到互变异构体的化学平衡。例如,质子互变异构体(proton tautomer)(也称质子转移互变异构体(prototropic tautomer))包括通过质子迁移来进行的互相转化,如酮-烯醇异构化和亚胺-烯胺异构化。价键异构体(valence tautomer)包括一些成键电子的重组来进行的相互转化。其中酮-烯醇互变异构化的具体实例是戊烷-2,4-二酮与4-羟基戊-3-烯-2-酮两个互变异构体之间的互变。Unless otherwise stated, the term "tautomer" or "tautomeric form" means that isomers with different functional groups are in dynamic equilibrium at room temperature and are rapidly interconvertible. If tautomerism is possible (eg, in solution), then chemical equilibrium of the tautomers can be achieved. For example, proton tautomers (also called prototropic tautomers) include interconversions via migration of a proton, such as keto-enol isomerization and imine-ene Amine isomerization. Valence isomers (valence tautomers) involve interconversions by recombination of some bonding electrons. A specific example of keto-enol tautomerization is the interconversion between two tautomers of pentane-2,4-dione and 4-hydroxypent-3-en-2-one.
除非另有说明,术语“富含一种异构体”、“异构体富集”、“富含一种对映体”或者“对映体富集”指其中一种异构体或对映体的含量小于100%,并且,该异构体或对映体的含量大于等于60%,或者大于等于70%,或者大于等于80%,或者大于等于90%,或者大于等于95%,或者大于等于96%,或者大于等于97%,或者大于等于98%,或者大于等于99%,或者大于等于99.5%,或者大于等于99.6%,或者大于等于99.7%,或者大于等于99.8%,或者大于等于99.9%。Unless otherwise stated, the terms "enriched in an isomer", "enriched in an isomer", "enriched in an enantiomer" or "enantiomerically enriched" refer to one of the isomers or enantiomers The content of the enantiomer is less than 100%, and the content of the isomer or enantiomer is greater than or equal to 60%, or greater than or equal to 70%, or greater than or equal to 80%, or greater than or equal to 90%, or greater than or equal to 95%, or Greater than or equal to 96%, or greater than or equal to 97%, or greater than or equal to 98%, or greater than or equal to 99%, or greater than or equal to 99.5%, or greater than or equal to 99.6%, or greater than or equal to 99.7%, or greater than or equal to 99.8%, or greater than or equal to 99.9%.
除非另有说明,术语“异构体过量”或“对映体过量”指两种异构体或两种对映体相对百分数之间的差值。例如,其中一种异构体或对映体的含量为90%,另一种异构体或对映体的含量为10%,则异构体或对映体过量(ee值)为80%。Unless otherwise stated, the terms "isomer excess" or "enantiomeric excess" refer to the difference between the relative percentages of two isomers or two enantiomers. For example, if the content of one isomer or enantiomer is 90% and the other isomer or enantiomer is 10%, then the isomer or enantiomeric excess (ee value) is 80% .
可以通过的手性合成或手性试剂或者其他常规技术制备光学活性的(R)-和(S)-异构体以及D和L异构体。如果想得到本发明某化合物的一种对映体,可以通过不对称合成或者具有手性助剂的衍生作用来制备,其中将所得非对映体混合物分离,并且辅助基团裂开以提供纯的所需对映异构体。或者,当分子中含有碱性官能团(如氨基)或酸性官能团(如羧基)时,与适当的光学活性的酸或碱形成非对映异构体的盐,然后通过本领域所公知的常规方法进行非对映异构体拆分,然后回收得到纯的对映体。此外,对映异构体和非对映异构体的分离通常是通过使用色谱法完成的,所述色谱法采用手性固定相,并任选地与化学衍生法相结合(例如由胺生成氨基甲酸盐)。Optically active (R)- and (S)-isomers as well as D and L-isomers can be prepared by chiral synthesis or chiral reagents or other conventional techniques. If one enantiomer of a compound of the invention is desired, it can be prepared by asymmetric synthesis or derivatization with chiral auxiliary agents, wherein the resulting diastereomeric mixture is separated and the auxiliary group is cleaved to provide pure desired enantiomer. Alternatively, when the molecule contains a basic functional group (such as an amino group) or an acidic functional group (such as a carboxyl group), a diastereoisomeric salt is formed with an appropriate optically active acid or base, and then a diastereomeric salt is formed by a conventional method known in the art. Diastereomeric resolution is performed and the pure enantiomers are recovered. Furthermore, the separation of enantiomers and diastereomers is usually accomplished by the use of chromatography using chiral stationary phases, optionally in combination with chemical derivatization methods (e.g. amines to amino groups formate).
本发明的化合物可以在一个或多个构成该化合物的原子上包含非天然比例的原子同位素。例如,可用放射性同位素标记化合物,比如氚( 3H),碘-125( 125I)或C-14( 14C)。又例如,可用重氢取代氢形成氘 代药物,氘与碳构成的键比普通氢与碳构成的键更坚固,相比于未氘化药物,氘代药物有降低毒副作用、增加药物稳定性、增强疗效、延长药物生物半衰期等优势。本发明的化合物的所有同位素组成的变换,无论放射性与否,都包括在本发明的范围之内。 The compounds of the present invention may contain unnatural proportions of atomic isotopes at one or more of the atoms that constitute the compounds. For example, compounds may be labeled with radioactive isotopes such as tritium ( 3 H), iodine-125 ( 125 I) or C-14 ( 14 C). For another example, heavy hydrogen can be used to replace hydrogen to form deuterated drugs. The bond formed by deuterium and carbon is stronger than the bond formed by ordinary hydrogen and carbon. Compared with non-deuterated drugs, deuterated drugs can reduce toxic side effects and increase drug stability. , enhance the efficacy, prolong the biological half-life of drugs and other advantages. All changes in isotopic composition of the compounds of the invention, whether radioactive or not, are included within the scope of the invention.
术语“任选”或“任选地”指的是随后描述的事件或状况可能但不是必需出现的,并且该描述包括其中所述事件或状况发生的情况以及所述事件或状况不发生的情况。The term "optional" or "optionally" means that the subsequently described event or circumstance can but need not occur, and that the description includes instances where said event or circumstance occurs and instances where said event or circumstance does not occur .
术语“被取代的”是指特定原子上的任意一个或多个氢原子被取代基取代,取代基可以包括重氢和氢的变体,只要特定原子的价态是正常的并且取代后的化合物是稳定的。当取代基为氧(即=O)时,意味着两个氢原子被取代。氧取代不会发生在芳香基上。The term "substituted" means that any one or more hydrogen atoms on a specified atom are replaced by a substituent, which may include deuterium and hydrogen variants, as long as the valence of the specified atom is normal and the substituted compound is stable. When a substituent is oxygen (ie =0), it means that two hydrogen atoms are replaced. Oxygen substitution does not occur on aromatic groups.
术语“任选被取代的”是指可以被取代,也可以不被取代,除非另有规定,取代基的种类和数目在化学上可以实现的基础上可以是任意的。The term "optionally substituted" means that it may or may not be substituted, and unless otherwise specified, the type and number of substituents may be arbitrary on a chemically realizable basis.
当任何变量(例如R)在化合物的组成或结构中出现一次以上时,其在每一种情况下的定义都是独立的。因此,例如,如果一个基团被0-2个R所取代,则所述基团可以任选地至多被两个R所取代,并且每种情况下的R都有独立的选项。此外,取代基和/或其变体的组合只有在这样的组合会产生稳定的化合物的情况下才是被允许的。When any variable (eg, R) occurs more than once in the composition or structure of a compound, its definition is independent at each occurrence. Thus, for example, if a group is substituted with 0-2 R, said group may optionally be substituted with up to two R, with independent options for each occurrence of R. Also, combinations of substituents and/or variations thereof are permissible only if such combinations result in stable compounds.
当一个连接基团的数量为0时,比如-(CRR) 0-,表示该连接基团为单键。 When the number of a linking group is 0, such as -(CRR) 0 -, it means that the linking group is a single bond.
当其中一个变量选自单键时,表示其连接的两个基团直接相连,比如A-L-Z中L代表单键时表示该结构实际上是A-Z。When one of the variables is selected from a single bond, it means that the two groups connected are directly connected. For example, when L in A-L-Z represents a single bond, it means that the structure is actually A-Z.
当一个取代基为空缺时,表示该取代基是不存在的,比如A-X中X为空缺时表示该结构实际上是A。当所列举的取代基中没有指明其通过哪一个原子连接到被取代的基团上时,这种取代基可以通过其任何原子相键合,例如,吡啶基作为取代基可以通过吡啶环上任意一个碳原子连接到被取代的基团上。When a substituent is vacant, it means that the substituent does not exist. For example, when X in A-X is vacant, it means that the structure is actually A. When the enumerated substituent does not indicate which atom it is connected to the substituted group, this substituent can be bonded through any atom, for example, pyridyl as a substituent can be connected to any atom on the pyridine ring. The carbon atom is attached to the group being substituted.
当所列举的连接基团没有指明其连接方向,其连接方向是任意的,例如,



除非另有规定,当某一基团具有一个或多个可连接位点时,该基团的任意一个或多个位点可以通过化学键与其他基团相连。所述位点与其他基团连接的化学键可以用直形实线键
![]()
![]()
![]()
![]()

![]()
![]()
![]()
![]()
除非另有规定,环上原子的数目通常被定义为环的元数,例如,“5-7元环”是指环绕排列5-7个原子的“环”。Unless otherwise specified, the number of atoms in a ring is generally defined as the number of ring members, eg, "5-7 membered ring" means a "ring" with 5-7 atoms arranged around it.
除非另有规定,术语“C 1-3烷基”用于表示直链或支链的由1至3个碳原子组成的饱和碳氢基团。所述C 1-3烷基包括C 1-2和C 2-3烷基等;其可以是一价(如甲基)、二价(如亚甲基)或者多价(如次甲基)。C 1-3烷基的实例包括但不限于甲基(Me)、乙基(Et)、丙基(包括n-丙基和异丙基)等。 Unless otherwise specified, the term "C 1-3 alkyl" is used to denote a straight or branched chain saturated hydrocarbon group consisting of 1 to 3 carbon atoms. The C 1-3 alkyl group includes C 1-2 and C 2-3 alkyl groups, etc.; it can be monovalent (such as methyl), divalent (such as methylene) or multivalent (such as methine) . Examples of C 1-3 alkyl include, but are not limited to, methyl (Me), ethyl (Et), propyl (including n-propyl and isopropyl), and the like.
除非另有规定,术语“C 1-3烷氧基”表示通过一个氧原子连接到分子的其余部分的那些包含1至3个碳原子的烷基基团。所述C 1-3烷氧基包括C 1-2、C 2-3、C 3和C 2烷氧基等。C 1-3烷氧基的实例包括但不限于甲氧基、乙氧基、丙氧基(包括正丙氧基和异丙氧基)等。 Unless otherwise specified, the term "C 1-3 alkoxy" denotes those alkyl groups containing 1 to 3 carbon atoms attached to the rest of the molecule through an oxygen atom. The C 1-3 alkoxy group includes C 1-2 , C 2-3 , C 3 and C 2 alkoxy groups and the like. Examples of C 1-3 alkoxy include, but are not limited to, methoxy, ethoxy, propoxy (including n-propoxy and isopropoxy), and the like.
除非另有规定,术语“C 1-3烷氨基”表示通过氨基连接到分子的其余部分的那些包含1至3个碳原子的烷基基团。所述C 1-3烷氨基包括C 1-2、C 3和C 2烷氨基等。C 1-3烷氨基的实例包括但不限于-NHCH 3、-N(CH 3) 2、-NHCH 2CH 3、-N(CH 3)CH 2CH 3、-NHCH 2CH 2CH 3、-NHCH 2(CH 3) 2等。 Unless otherwise specified, the term "C 1-3 alkylamino" denotes those alkyl groups containing 1 to 3 carbon atoms attached to the rest of the molecule through an amino group. The C 1-3 alkylamino group includes C 1-2 , C 3 and C 2 alkylamino groups and the like. Examples of C 1-3 alkylamino include, but are not limited to, -NHCH 3 , -N(CH 3 ) 2 , -NHCH 2 CH 3 , -N(CH 3 )CH 2 CH 3 , -NHCH 2 CH 2 CH 3 , -NHCH 2 CH 2 CH 3 , - NHCH 2 (CH 3 ) 2 etc.
除非另有规定,C n-n+m或C n-C n+m包括n至n+m个碳的任何一种具体情况,例如C 1-12包括C 1、C 2、C 3、C 4、C 5、C 6、C 7、C 8、C 9、C 10、C 11、和C 12,也包括n至n+m中的任何一个范围,例如C 1-12包括C 1-3、C 1-6、C 1-9、C 3-6、C 3-9、C 3-12、C 6-9、C 6-12、和C 9-12等;同理,n元至n+m元表示环上原子数为n至n+m个,例如3-12元环包括3元环、4元环、5元环、6元环、7元环、8元环、9元环、10元环、11元环、和12元环,也包括n至n+m中的任何一个范围,例如3-12元环包括3-6元环、3-9元环、5-6元环、5-7元环、6-7元环、6-8元环、和6-10元环等。 Unless otherwise specified, C n-n+m or C n -C n+m includes any specific instance of n to n+m carbons, for example C 1-12 includes C 1 , C 2 , C 3 , C 4 , C 5 , C 6 , C 7 , C 8 , C 9 , C 10 , C 11 , and C 12 , also including any range from n to n+m, for example, C 1-12 includes C 1-3 , C 1-6 , C 1-9 , C 3-6 , C 3-9 , C 3-12 , C 6-9 , C 6-12 , and C 9-12 etc.; similarly, n to n +m means that the number of atoms on the ring is n to n+m, for example, a 3-12-membered ring includes a 3-membered ring, a 4-membered ring, a 5-membered ring, a 6-membered ring, a 7-membered ring, an 8-membered ring, and a 9-membered ring , 10-membered rings, 11-membered rings, and 12-membered rings, also including any range from n to n+m, for example, 3-12-membered rings include 3-6-membered rings, 3-9-membered rings, 5-6-membered rings ring, 5-7-membered ring, 6-7-membered ring, 6-8-membered ring, and 6-10-membered ring, etc.
术语“离去基团”是指可以被另一种官能团或原子通过取代反应(例如亲核取代反应)所取代的官能团或原子。例如,代表性的离去基团包括三氟甲磺酸酯;氯、溴、碘;磺酸酯基,如甲磺酸酯、甲苯磺酸酯、对溴苯磺酸酯、对甲苯磺酸酯等;酰氧基,如乙酰氧基、三氟乙酰氧基等等。The term "leaving group" refers to a functional group or atom that can be replaced by another functional group or atom through a substitution reaction (eg, a nucleophilic substitution reaction). For example, representative leaving groups include triflate; chlorine, bromine, iodine; sulfonate groups such as mesylate, tosylate, brosylate, tosylate esters, etc.; acyloxy groups such as acetoxy, trifluoroacetoxy, and the like.
术语“保护基”包括但不限于“氨基保护基”、“羟基保护基”或“巯基保护基”。术语“氨基保护基”是指适合用于阻止氨基氮位上副反应的保护基团。代表性的氨基保护基包括但不限于:甲酰基;酰基,例如链烷酰基(如乙酰基、三氯乙酰基或三氟乙酰基);烷氧基羰基,如叔丁氧基羰基(Boc);芳基甲氧羰基,如苄氧羰基(Cbz)和9-芴甲氧羰基(Fmoc);芳基甲基,如苄基(Bn)、三苯甲基(Tr)、1,1-二-(4'-甲氧基苯基)甲基;甲硅烷基,如三甲基甲硅烷基(TMS)和叔丁基二甲基甲硅烷基(TBS)等等。术语“羟基保护基”是指适合用于阻止羟基副反应的保护基。代表性羟基保护基包括但不限于:烷基,如甲基、乙基和叔丁基; 酰基,例如链烷酰基(如乙酰基);芳基甲基,如苄基(Bn),对甲氧基苄基(PMB)、9-芴基甲基(Fm)和二苯基甲基(二苯甲基,DPM);甲硅烷基,如三甲基甲硅烷基(TMS)和叔丁基二甲基甲硅烷基(TBS)等等。The term "protecting group" includes, but is not limited to, "amino protecting group", "hydroxyl protecting group" or "mercapto protecting group". The term "amino protecting group" refers to a protecting group suitable for preventing side reactions at the amino nitrogen position. Representative amino protecting groups include, but are not limited to: formyl; acyl, such as alkanoyl (such as acetyl, trichloroacetyl or trifluoroacetyl); alkoxycarbonyl, such as tert-butoxycarbonyl (Boc) ; arylmethoxycarbonyl, such as benzyloxycarbonyl (Cbz) and 9-fluorenylmethyloxycarbonyl (Fmoc); arylmethyl, such as benzyl (Bn), trityl (Tr), 1,1-di -(4'-methoxyphenyl)methyl; silyl groups such as trimethylsilyl (TMS) and tert-butyldimethylsilyl (TBS) and the like. The term "hydroxyl protecting group" refers to a protecting group suitable for preventing side reactions of the hydroxy group. Representative hydroxy protecting groups include, but are not limited to: alkyl, such as methyl, ethyl, and tert-butyl; acyl, such as alkanoyl (such as acetyl); arylmethyl, such as benzyl (Bn), p-formyl Oxybenzyl (PMB), 9-fluorenylmethyl (Fm) and diphenylmethyl (diphenylmethyl, DPM); silyl groups such as trimethylsilyl (TMS) and tert-butyl Dimethylsilyl (TBS) and the like.
本发明的化合物可以通过本领域技术人员所熟知的多种合成方法来制备,包括下面列举的具体实施方式、其与其他化学合成方法的结合所形成的实施方式以及本领域技术上人员所熟知的等同替换方式,优选的实施方式包括但不限于本发明的实施例。The compounds of the present invention can be prepared by a variety of synthetic methods well known to those skilled in the art, including the specific embodiments listed below, the embodiments formed by combining them with other chemical synthesis methods, and the methods well known to those skilled in the art Equivalent alternatives, preferred embodiments include but are not limited to the examples of the present invention.
本发明的化合物可以通过本领域技术人员所熟知的常规方法来确认结构,如果本发明涉及化合物的绝对构型,则该绝对构型可以通过本领域常规技术手段予以确证。例如单晶X射线衍射法(SXRD),把培养出的单晶用Bruker D8 venture衍射仪收集衍射强度数据,光源为CuKα辐射,扫描方式:
![]()
![]()
本发明所使用的溶剂可经市售获得。本发明采用下述缩略词:aq代表水;HATU代表O-(7-氮杂苯并三唑-1-基)-N,N,N'.N'-四甲基脲六氟磷酸盐;EDCI代表N-(3-二甲基氨基丙基)-N'-乙基碳二亚胺盐酸盐;m-CPBA代表3-氯过氧苯甲酸;eq代表当量、等量;CDI代表羰基二咪唑;DCM代表二氯甲烷;PE代表石油醚;DIAD代表偶氮二羧酸二异丙酯;DMF代表N,N-二甲基甲酰胺;DMSO代表二甲亚砜;EtOAc代表乙酸乙酯;EtOH代表乙醇;MeOH代表甲醇;CBz代表苄氧羰基,是一种胺保护基团;BOC代表叔丁氧羰基是一种胺保护基团;HOAc代表乙酸;NaCNBH 3代表氰基硼氢化钠;r.t.代表室温;O/N代表过夜;THF代表四氢呋喃;Boc 2O代表二-叔丁基二碳酸酯;TFA代表三氟乙酸;DIPEA代表二异丙基乙基胺;SOCl 2代表氯化亚砜;CS 2代表二硫化碳;TsOH代表对甲苯磺酸;NFSI代表N-氟-N-(苯磺酰基)苯磺酰胺;NCS代表1-氯吡咯烷-2,5-二酮;n-Bu 4NF代表氟化四丁基铵;iPrOH代表2-丙醇;mp代表熔点;LDA代表二异丙基胺基锂。 The solvent used in the present invention is commercially available. The following abbreviations are used in the present invention: aq stands for water; HATU stands for O-(7-azabenzotriazol-1-yl)-N,N,N'.N'-tetramethyluronium hexafluorophosphate ; EDCI represents N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride; m-CPBA represents 3-chloroperoxybenzoic acid; eq represents equivalent, equivalent; CDI represents Carbonyldiimidazole; DCM stands for dichloromethane; PE stands for petroleum ether; DIAD stands for diisopropyl azodicarboxylate; DMF stands for N,N-dimethylformamide; DMSO stands for dimethylsulfoxide; EtOAc stands for ethyl acetate EtOH stands for ethanol; MeOH stands for methanol; CBz stands for benzyloxycarbonyl, an amine protecting group; BOC stands for tert-butoxycarbonyl, an amine protecting group; HOAc stands for acetic acid; NaCNBH3 stands for sodium cyanoborohydride ; rt stands for room temperature; O /N stands for overnight; THF stands for tetrahydrofuran; Boc2O stands for di-tert-butyldicarbonate; TFA stands for trifluoroacetic acid; DIPEA stands for diisopropylethylamine; Sulfone; CS 2 represents carbon disulfide; TsOH represents p-toluenesulfonic acid; NFSI represents N-fluoro-N-(benzenesulfonyl)benzenesulfonamide; NCS represents 1-chloropyrrolidine-2,5-dione; n-Bu 4 NF stands for tetrabutylammonium fluoride; iPrOH stands for 2-propanol; mp stands for melting point; LDA stands for lithium diisopropylamide.
化合物依据本领域常规命名原则或者使用
![]()
![]()
下面通过实施例对本发明进行详细描述,但并不意味着对本发明任何不利限制。本文已经详细地描述了本发明,其中也公开了其具体实施例方式,对本领域的技术人员而言,在不脱离本发明精神和范围的情况下针对本发明具体实施方式进行各种变化和改进将是显而易见的。The present invention will be described in detail through examples below, but it does not imply any unfavorable limitation to the present invention. The present invention has been described in detail herein, and its specific embodiments are also disclosed. For those skilled in the art, various changes and improvements can be made to the specific embodiments of the present invention without departing from the spirit and scope of the present invention. will be obvious.
实施例1Example 1
合成路线:synthetic route:

第一步first step
将化合物1-1(50.0g,0.384mol)溶于甲醇(500mL)中,向其中加入浓盐酸(1mL),反应液在70℃下搅拌12小时。反应液冷至室温后向其中加入固体碳酸氢钠(3.00g),室温下搅拌0.5小时后过滤,滤液浓缩得到化合物1-2。 1H NMR(400MHz,CDCl 3)δ4.28-4.25(m,1H),3.81(s,3H),3.70(s,3H),2.93(br s,1H),2.53-2.46(m,2H),2.22-2.17(m,1H),1.99-1.91(m,1H)。 Compound 1-1 (50.0 g, 0.384 mol) was dissolved in methanol (500 mL), concentrated hydrochloric acid (1 mL) was added thereto, and the reaction solution was stirred at 70° C. for 12 hours. After the reaction solution was cooled to room temperature, solid sodium bicarbonate (3.00 g) was added thereto, stirred at room temperature for 0.5 hours, filtered, and the filtrate was concentrated to obtain compound 1-2. 1 H NMR (400MHz, CDCl 3 )δ4.28-4.25(m,1H),3.81(s,3H),3.70(s,3H),2.93(br s,1H),2.53-2.46(m,2H) ,2.22-2.17(m,1H),1.99-1.91(m,1H).
第二步second step
将化合物1-2(33.8g,0.192mol),化合物1-3(50.2g,0.230mol)和三苯基膦(60.4g,0.230mol)溶于二氯甲烷(300mL)中,反应液冷至0℃,向其中缓慢滴加偶氮二甲酸二异丙酯(46.6g,0.230mol)的二氯甲烷(50mL)溶液,滴完后反应液在0℃下搅拌0.5小时,然后在20℃下搅拌12小时,减压浓缩除去溶剂,粗产物经过柱层析法分离用二氯甲烷为洗脱剂以除去三苯基氧膦,洗脱液浓缩后所得剩余物溶于乙酸乙酯(500mL),再依次用氢氧化钠水溶液(1M,500mL x 4)和饱和食盐水(500mL x 1)洗涤,有机相用无水硫 酸钠干燥,过滤,滤液减压浓缩得到化合物1-4。 1H NMR(400MHz,CDCl 3)δ8.01(d,J=2.4Hz,1H),7.61(dd,J=8.8,2.4Hz,1H),6.84(d,J=8.8Hz,1H),4.94-4.91(m,1H),3.79(s,3H),3.71(s,3H),2.71-2.61(m,2H),2.43-2.30(m,2H)。MS-ESI计算值[M+Na] +398和400,实测值398和400。 Compound 1-2 (33.8g, 0.192mol), compound 1-3 (50.2g, 0.230mol) and triphenylphosphine (60.4g, 0.230mol) were dissolved in dichloromethane (300mL), and the reaction solution was cooled to 0°C, slowly dropwise add diisopropyl azodicarboxylate (46.6g, 0.230mol) in dichloromethane (50mL) solution, after the drop, the reaction solution was stirred at 0°C for 0.5 hours, then heated at 20°C Stir for 12 hours, concentrate under reduced pressure to remove the solvent, the crude product is separated by column chromatography using dichloromethane as the eluent to remove triphenylphosphine oxide, and the residue obtained after the eluent is concentrated is dissolved in ethyl acetate (500mL) , and then washed successively with aqueous sodium hydroxide solution (1M, 500mL x 4) and saturated brine (500mL x 1), the organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain compound 1-4. 1 H NMR (400MHz, CDCl 3 ) δ8.01 (d, J=2.4Hz, 1H), 7.61 (dd, J=8.8, 2.4Hz, 1H), 6.84 (d, J=8.8Hz, 1H), 4.94 -4.91(m,1H),3.79(s,3H),3.71(s,3H),2.71-2.61(m,2H),2.43-2.30(m,2H). MS-ESI calculated [M+Na] + 398 and 400, found 398 and 400.
第三步third step
将化合物1-4(35.8g,0.952mol)溶于醋酸(284mL)中,再加入铁粉(26.6g,0.476mol)。60℃条件下,搅拌反应2小时。将反应液趁热用硅藻土过滤,滤渣用乙酸乙酯(500mL)洗涤,滤液依次用水(500mL x 3)和饱和食盐水(500mL x 1)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩得到化合物1-5。 1H NMR(400MHz,CDCl 3)δ8.50(br s,1H),7.03-7.00(m,1H),6.89(d,J=2.4Hz,1H),6.77(d,J=8.4Hz,1H),4.55(dd,J=8.4,4.4Hz,1H),3.63(s,3H),2.54-2.50(m,2H),2.32-2.24(m,1H),2.18-2.09(m,1H)。MS-ESI计算值[M+H] +314和316,实测值314和316。 Compound 1-4 (35.8g, 0.952mol) was dissolved in acetic acid (284mL), and iron powder (26.6g, 0.476mol) was added. Under the condition of 60°C, the reaction was stirred for 2 hours. The reaction solution was filtered with diatomaceous earth while it was hot, the filter residue was washed with ethyl acetate (500mL), the filtrate was washed with water (500mL x 3) and saturated brine (500mL x 1) successively, the organic phase was dried over anhydrous sodium sulfate, and filtered , the filtrate was concentrated under reduced pressure to obtain compound 1-5. 1 H NMR (400MHz, CDCl 3 ) δ8.50(br s, 1H), 7.03-7.00(m, 1H), 6.89(d, J=2.4Hz, 1H), 6.77(d, J=8.4Hz, 1H ), 4.55 (dd, J = 8.4, 4.4 Hz, 1H), 3.63 (s, 3H), 2.54-2.50 (m, 2H), 2.32-2.24 (m, 1H), 2.18-2.09 (m, 1H). MS-ESI calculated [M+H] + 314 and 316, found 314 and 316.
第四步the fourth step
将化合物1-5(20.0g,63.7mmol)溶于四氢呋喃(200mL)中,在0℃下向其中缓慢滴加硼烷二甲硫醚络合物(10M,15.9mL,0.159mol),滴加完后反应液在50℃下搅拌1小时。将反应液冷至0℃用甲醇(130mL)淬灭,再在60℃下搅拌1小时。反应液减压浓缩,剩余物用乙酸乙酯(200mL)溶解,再依次用水(200mL x 1)和饱和食盐水(200mL x 1)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩后剩余物经过硅胶柱层析法(4/1石油醚/乙酸乙酯)分离纯化得到化合物1-6。 1H NMR(400MHz,CDCl 3)δ6.76-6.72(m,2H),6.66-6.64(m,1H),4.14-4.09(m,1H),3.82(br s,1H),3.72(s,3H),3.41-3.37(m,1H),3.17-3.12(m,1H),2.66-2.52(m,2H),2.01-1.96(m,2H)。MS-ESI计算值[M+H] +300和302,实测值300和302。 Compound 1-5 (20.0g, 63.7mmol) was dissolved in tetrahydrofuran (200mL), and borane dimethyl sulfide complex (10M, 15.9mL, 0.159mol) was slowly added dropwise at 0°C, and After completion, the reaction solution was stirred at 50° C. for 1 hour. The reaction solution was cooled to 0°C and quenched with methanol (130 mL), then stirred at 60°C for 1 hour. The reaction solution was concentrated under reduced pressure, the residue was dissolved in ethyl acetate (200mL), washed with water (200mL x 1) and saturated brine (200mL x 1) successively, the organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was decompressed After concentration, the residue was separated and purified by silica gel column chromatography (4/1 petroleum ether/ethyl acetate) to obtain compound 1-6. 1 H NMR (400MHz, CDCl 3 )δ6.76-6.72(m,2H),6.66-6.64(m,1H),4.14-4.09(m,1H),3.82(br s,1H),3.72(s, 3H), 3.41-3.37(m, 1H), 3.17-3.12(m, 1H), 2.66-2.52(m, 2H), 2.01-1.96(m, 2H). MS-ESI calculated [M+H] + 300 and 302, found 300 and 302.
第五步the fifth step
将化合物1-6(19.0g,63.3mmol)和化合物1-7(18.6g,76.0mmol)溶于吡啶(110mL)中,反应液在60℃下搅拌反应24小时。反应液减压浓缩后剩余物经过硅胶柱层析法(5/1石油醚/乙酸乙酯)分离纯化得到化合物1-8。 1H NMR(400MHz,CDCl 3)δ8.00-7.99(m,2H),7.88-7.85(m,2H),7.68-7.65(m,1H),7.20(dd,J=8.8,2.4Hz,1H),6.71(d,J=8.8Hz,1H),4.30(dd,J=14.4,2.4Hz,1H),3.69(s,3H),3.52-3.48(m,1H),3.20-3.14(m,1H),2.51-2.39(m,2H),1.94-1.79(m,2H)。MS-ESI计算值[M+H] +508和510,实测值508和510。 Compound 1-6 (19.0g, 63.3mmol) and compound 1-7 (18.6g, 76.0mmol) were dissolved in pyridine (110mL), and the reaction solution was stirred at 60°C for 24 hours. After the reaction solution was concentrated under reduced pressure, the residue was separated and purified by silica gel column chromatography (5/1 petroleum ether/ethyl acetate) to obtain compound 1-8. 1 H NMR (400MHz, CDCl 3 ) δ8.00-7.99 (m, 2H), 7.88-7.85 (m, 2H), 7.68-7.65 (m, 1H), 7.20 (dd, J=8.8, 2.4Hz, 1H ),6.71(d,J=8.8Hz,1H),4.30(dd,J=14.4,2.4Hz,1H),3.69(s,3H),3.52-3.48(m,1H),3.20-3.14(m, 1H), 2.51-2.39 (m, 2H), 1.94-1.79 (m, 2H). MS-ESI calculated [M+H] + 508 and 510, found 508 and 510.
第六步step six
将化合物1-8(800mg,1.57mmol),1-9(584mg,1.89mmol),碳酸钾(435mg,3.15mmol)和1,1-双(二苯基膦)二茂铁氯化钯(115mg,0.157mmol)加入到1,4-二氧六环(10mL)和水(1mL)中。反应液在氮气保护下100℃搅拌反应3小时。向反应液中加入水(20mL),用乙酸乙酯(30mL x 2)萃取。合并有机相,用饱和食盐水洗涤(20mL x 1),无水硫酸钠干燥,过滤,滤液减压浓缩。剩余物用硅胶柱色谱法分离纯化(3/1石油醚/乙酸乙酯)得到化合物1-10。 1H NMR(400MHz,CDCl 3)δ7.96(s,1H),7.85-7.81(m,3H),7.65-7.63(m,1H),7.10-7.07(m,1H),6.75(d,J=8.0Hz,1H),6.15-6.14(m,1H),4.33-4.32(m,1H),4.30-4.29(m,2H), 3.68(s,3H),3.57-3.49(m,3H),3.23-3.17(m,1H),2.48-2.42(m,2H),2.32-2.31(m,2H),1.89-1.82(m,2H),1.51(s,9H)。MS-ESI计算值[M-56+H] +555,实测值555。 Compound 1-8 (800mg, 1.57mmol), 1-9 (584mg, 1.89mmol), potassium carbonate (435mg, 3.15mmol) and 1,1-bis(diphenylphosphine)ferrocenepalladium chloride (115mg , 0.157 mmol) was added to 1,4-dioxane (10 mL) and water (1 mL). The reaction solution was stirred and reacted at 100° C. for 3 hours under the protection of nitrogen. Water (20 mL) was added to the reaction solution, and extracted with ethyl acetate (30 mL x 2). The organic phases were combined, washed with saturated brine (20 mL x 1), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was separated and purified by silica gel column chromatography (3/1 petroleum ether/ethyl acetate) to obtain compound 1-10. 1 H NMR (400MHz, CDCl 3 )δ7.96(s,1H),7.85-7.81(m,3H),7.65-7.63(m,1H),7.10-7.07(m,1H),6.75(d,J =8.0Hz,1H),6.15-6.14(m,1H),4.33-4.32(m,1H),4.30-4.29(m,2H), 3.68(s,3H),3.57-3.49(m,3H), 3.23-3.17 (m, 1H), 2.48-2.42 (m, 2H), 2.32-2.31 (m, 2H), 1.89-1.82 (m, 2H), 1.51 (s, 9H). MS-ESI calculated value [M-56+H] + 555, found value 555.
第七步step seven
将化合物1-10(900mg,1.47mmol)溶于甲醇(20mL)中,加入钯碳(100mg,10%纯度)。反应液在氢气(15Psi)保护下25℃搅拌反应12小时。反应液过滤,滤液减压浓缩。剩余物为化合物1-11。 1H NMR(400MHz,CDCl 3)δ7.90(s,1H),7.83-7.81(m,2H),7.69-7.68(m,1H),7.64-7.60(m,1H),6.96-6.94(m,1H),6.74(d,J=8.0Hz,1H),4.34-4.33(m,1H),4.32-4.30(m,2H),3.67(s,3H),3.49-3.47(m,1H),3.19-3.18(m,1H),2.66-2.65(m,2H),2.47-2.44(m,2H),2.00-1.76(m,4H),1.64-1.59(m,3H),1.48(s,9H)。MS-ESI计算值[M-56+H] +557,实测值557。 Compound 1-10 (900 mg, 1.47 mmol) was dissolved in methanol (20 mL), and palladium on carbon (100 mg, 10% purity) was added. The reaction solution was stirred and reacted at 25° C. for 12 hours under the protection of hydrogen gas (15 Psi). The reaction solution was filtered, and the filtrate was concentrated under reduced pressure. The residue was compound 1-11. 1 H NMR (400MHz, CDCl 3 )δ7.90(s,1H),7.83-7.81(m,2H),7.69-7.68(m,1H),7.64-7.60(m,1H),6.96-6.94(m ,1H),6.74(d,J=8.0Hz,1H),4.34-4.33(m,1H),4.32-4.30(m,2H),3.67(s,3H),3.49-3.47(m,1H), 3.19-3.18(m,1H),2.66-2.65(m,2H),2.47-2.44(m,2H),2.00-1.76(m,4H),1.64-1.59(m,3H),1.48(s,9H ). MS-ESI calculated value [M-56+H] + 557, found value 557.
第八步eighth step
将化合物1-11(850mg,1.39mmol)溶于甲醇(5mL)中,加入盐酸甲醇(4M,5mL,20.0mmol)。反应液在25℃搅拌反应12小时。向反应液加入饱和碳酸氢钠水溶液(30mL),用乙酸乙酯(10mL x 3)萃取。合并有机相,用饱和食盐水洗涤(20mL x 1),无水硫酸钠干燥,过滤,滤液减压浓缩。剩余物为化合物1-12。 1H NMR(400MHz,CDCl 3)δ7.90(s,1H),7.83-7.81(m,2H),7.68-7.60(m,2H),6.94-6.91(m,1H),6.72(d,J=8.0Hz,1H),4.34-4.29(m,1H),3.68(s,3H),3.54-3.52(m,1H),3.23-3.19(m,3H),2.72-2.66(m,4H),2.46-2.44(m,2H),1.89-1.62(m,5H)。MS-ESI计算值[M+H] +513,实测值513。 Compound 1-11 (850 mg, 1.39 mmol) was dissolved in methanol (5 mL), and methanol hydrochloride (4M, 5 mL, 20.0 mmol) was added. The reaction solution was stirred and reacted at 25° C. for 12 hours. Saturated aqueous sodium bicarbonate (30 mL) was added to the reaction solution, and extracted with ethyl acetate (10 mL x 3). The organic phases were combined, washed with saturated brine (20 mL x 1), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The remainder was compound 1-12. 1 H NMR (400MHz, CDCl 3 )δ7.90(s,1H),7.83-7.81(m,2H),7.68-7.60(m,2H),6.94-6.91(m,1H),6.72(d,J =8.0Hz,1H),4.34-4.29(m,1H),3.68(s,3H),3.54-3.52(m,1H),3.23-3.19(m,3H),2.72-2.66(m,4H), 2.46-2.44 (m, 2H), 1.89-1.62 (m, 5H). MS-ESI calculated [M+H] + 513, found 513.
第九步Ninth step
将化合物1-12(300mg,0.585mmol),1-13(107mg,0.702mmol),醋酸铜(213mg,1.17mmol),吡啶(139mg,1.76mmol)和氧化吡啶(167mg,1.76mmol)加入到二氯甲烷(10mL)中。反应液在氧气保护下40℃搅拌反应12小时。向反应液中加入水(20mL),用二氯甲烷(30mL x 3)萃取。合并有机相,用饱和食盐水洗涤(30mL x 1),无水硫酸钠干燥,过滤,滤液减压浓缩。剩余物用硅胶柱色谱法分离纯化(3/1石油醚/乙酸乙酯)得到化合物1-14。MS-ESI计算值[M+H] +619,实测值619。 The compound 1-12 (300mg, 0.585mmol), 1-13 (107mg, 0.702mmol), copper acetate (213mg, 1.17mmol), pyridine (139mg, 1.76mmol) and pyridine oxide (167mg, 1.76mmol) were added to two Chloromethane (10 mL). The reaction solution was stirred and reacted at 40° C. for 12 hours under the protection of oxygen. Water (20 mL) was added to the reaction liquid, and extracted with dichloromethane (30 mL x 3). The organic phases were combined, washed with saturated brine (30 mL x 1), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was separated and purified by silica gel column chromatography (3/1 petroleum ether/ethyl acetate) to obtain compound 1-14. MS-ESI calculated [M+H] + 619, found 619.
第十步tenth step
将化合物1-14(40.0mg,0.0539mmol)和一水合氢氧化锂(4.5mg,0.108mmol)加入到四氢呋喃(3mL)和水(1mL)中。反应液在氮气保护下20℃搅拌反应12小时。反应液减压浓缩。剩余物经高效液相色谱法分离(中性,碳酸氢铵体系)纯化得到化合物1。 1H NMR(400MHz,CD 3OD)δ8.00-7.99(m,1H),7.98-7.97(m,1H),7.76-7.72(m,3H),7.04-6.90(m,5H),6.75(d,J=8.0Hz,1H),4.44-4.39(m,1H),3.89(s,3H),3.50-3.42(m,3H),3.25-3.21(m,1H),3.00-2.98(m,1H),2.70-2.68(m,1H),2.55-2.36(m,3H),2.02-1.89(m,4H),1.79-1.76(m,1H),1.63-1.61(m,1H)。MS-ESI计算值[M+H] +605,实测值605。 Compound 1-14 (40.0 mg, 0.0539 mmol) and lithium hydroxide monohydrate (4.5 mg, 0.108 mmol) were added to tetrahydrofuran (3 mL) and water (1 mL). The reaction solution was stirred and reacted at 20° C. for 12 hours under the protection of nitrogen. The reaction solution was concentrated under reduced pressure. The residue was separated and purified by high performance liquid chromatography (neutral, ammonium bicarbonate system) to obtain compound 1. 1 H NMR (400MHz, CD 3 OD) δ8.00-7.99 (m, 1H), 7.98-7.97 (m, 1H), 7.76-7.72 (m, 3H), 7.04-6.90 (m, 5H), 6.75 ( d,J=8.0Hz,1H),4.44-4.39(m,1H),3.89(s,3H),3.50-3.42(m,3H),3.25-3.21(m,1H),3.00-2.98(m, 1H), 2.70-2.68(m, 1H), 2.55-2.36(m, 3H), 2.02-1.89(m, 4H), 1.79-1.76(m, 1H), 1.63-1.61(m, 1H). MS-ESI calculated value [M+H] + 605, found value 605.
实施例2Example 2
合成路线:synthetic route:

第一步first step
将化合物2-1(800mg,4.21mmol),1-9(1.30g,4.21mmol),磷酸钾(1.79g,8.42mmol)和1,1-双(二苯基膦)二茂铁氯化钯(308mg,0.421mmol)加入到1,4-二氧六环(10mL)和水(1mL)中。反应液在氮气保护下80℃搅拌反应12小时。向反应液中加入水(10mL),用乙酸乙酯(20mL x 2)萃取。合并有机相,用饱和食盐水洗涤(20mL x 1),无水硫酸钠干燥,过滤,滤液减压浓缩。剩余物用硅胶柱色谱法分离纯化(1/1石油醚/乙酸乙酯)得到化合物2-2。 1H NMR(400MHz,CDCl 3)δ7.05-6.99(m,1H),6.50-6.43(m,2H),5.91-5.90(m,1H),4.10(s,2H),3.62-3.59(m,2H),2.33-2.32(m,2H),1.48(s,9H)。MS-ESI计算值[M-56+H] +237,实测值237。 Compound 2-1 (800mg, 4.21mmol), 1-9 (1.30g, 4.21mmol), potassium phosphate (1.79g, 8.42mmol) and 1,1-bis(diphenylphosphine)ferrocenepalladium chloride (308 mg, 0.421 mmol) was added to 1,4-dioxane (10 mL) and water (1 mL). The reaction solution was stirred and reacted at 80° C. for 12 hours under the protection of nitrogen. Water (10 mL) was added to the reaction solution, and extracted with ethyl acetate (20 mL x 2). The organic phases were combined, washed with saturated brine (20 mL x 1), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was separated and purified by silica gel column chromatography (1/1 petroleum ether/ethyl acetate) to obtain compound 2-2. 1 H NMR (400MHz, CDCl 3 )δ7.05-6.99(m,1H),6.50-6.43(m,2H),5.91-5.90(m,1H),4.10(s,2H),3.62-3.59(m ,2H), 2.33-2.32(m,2H), 1.48(s,9H). MS-ESI calculated value [M-56+H] + 237, found value 237.
第二步second step
将化合物2-2(900mg,3.08mmol)溶于甲醇(20mL)中,加入钯碳(100mg,10%纯度)。反应液在氢气(15Psi)保护下25℃搅拌反应12小时。反应液过滤,滤液减压浓缩。剩余物为化合物2-3。 1H NMR(400MHz,CDCl 3)δ7.00-6.94(m,1H),6.49-6.42(m,2H),4.17-4.06(m,2H),3.87-3.83(m,2H),3.06-3.03(m,1H),2.79-2.64(m,2H),1.79-1.75(m,2H),1.48(s,9H)。MS-ESI计算值[M-56+H] +239,实测值239。 Compound 2-2 (900 mg, 3.08 mmol) was dissolved in methanol (20 mL), and palladium on carbon (100 mg, 10% purity) was added. The reaction solution was stirred and reacted at 25° C. for 12 hours under the protection of hydrogen gas (15 Psi). The reaction solution was filtered, and the filtrate was concentrated under reduced pressure. The residue was compound 2-3. 1 H NMR (400MHz, CDCl 3 )δ7.00-6.94(m,1H),6.49-6.42(m,2H),4.17-4.06(m,2H),3.87-3.83(m,2H),3.06-3.03 (m,1H), 2.79-2.64(m,2H), 1.79-1.75(m,2H), 1.48(s,9H). MS-ESI calculated value [M-56+H] + 239, found value 239.
第三步third step
将化合物2-3(200mg,0.679mmol)溶于盐酸(12M,4.66mL,56.0mmol)中,在-5℃下滴加亚硝酸钠 (117mg,1.70mmol)的水(2mL)的溶液,在该温度下搅拌反应30分钟。然后向反应液中加入氯化亚铜(202mg,2.04mmol),反应液在25℃下搅拌反应12小时。向反应液加入水(10mL),用二氯甲烷(20mL x3)萃取。合并有机相,用饱和食盐水洗涤(20mL x 1),无水硫酸钠干燥,过滤,滤液减压浓缩。剩余物用制备薄层层析法分离纯化(10/1二氯甲烷/甲醇)得到化合物2-4。MS-ESI计算值[M+H] +214,实测值214。 Compound 2-3 (200mg, 0.679mmol) was dissolved in hydrochloric acid (12M, 4.66mL, 56.0mmol), and a solution of sodium nitrite (117mg, 1.70mmol) in water (2mL) was added dropwise at -5°C. The reaction was stirred at this temperature for 30 minutes. Then cuprous chloride (202mg, 2.04mmol) was added to the reaction solution, and the reaction solution was stirred and reacted at 25°C for 12 hours. Water (10 mL) was added to the reaction solution, and extracted with dichloromethane (20 mL x 3). The organic phases were combined, washed with saturated brine (20 mL x 1), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was separated and purified by preparative thin layer chromatography (10/1 dichloromethane/methanol) to obtain compound 2-4. MS-ESI calculated value [M+H] + 214, found value 214.
第四步the fourth step
氮气保护下,将化合物1-8(800mg,0.634mmol)溶于干燥1,4-二氧六环(10mL)中,加入联硼酸频那醇酯(440mg,1.73mmol),乙酸钾(463mg,4.72mmol)和[1,1′-双(二苯基膦)二茂铁]二氯化钯(II)二氯甲烷络合物(129mg,0.157mmol),反应液在90℃下搅拌12小时,向反应液中加入水(40mL),用乙酸乙酯(40mL x 1)萃取。饱和食盐水洗涤(40mL x 1),无水硫酸钠干燥,过滤,减压浓缩,剩余物用硅胶柱色谱法分离纯化(3/1石油醚/乙酸乙酯)得到化合物2-5。MS-ESI计算值[M+Na] +578,实测值578。 Under nitrogen protection, compound 1-8 (800mg, 0.634mmol) was dissolved in dry 1,4-dioxane (10mL), and diboronic acid pinacol ester (440mg, 1.73mmol), potassium acetate (463mg, 4.72mmol) and [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II) dichloromethane complex (129mg, 0.157mmol), the reaction solution was stirred at 90°C for 12 hours , water (40 mL) was added to the reaction solution, and extracted with ethyl acetate (40 mL x 1). Washed with saturated brine (40mL x 1), dried over anhydrous sodium sulfate, filtered, concentrated under reduced pressure, and the residue was separated and purified by silica gel column chromatography (3/1 petroleum ether/ethyl acetate) to obtain compound 2-5. MS-ESI calculated [M+Na] + 578, found 578.
第五步the fifth step
氮气保护下,将化合物2-5(250mg,0.450mmol)溶于丙酮(4mL)和水(2mL)中,加入高碘酸钠(356mg,1.67mmol)和乙酸铵(69.4mg,0.900mmol),反应液在50℃下搅拌12小时,反应液用1M盐酸水溶液调节pH<3,用乙酸乙酯(20mL x 1)萃取。有机相用饱和食盐水洗涤(20mL x 1),无水硫酸钠干燥,过滤,减压浓缩得到化合物2-6。MS-ESI计算值[M+Na] +496,实测值496。 Under nitrogen protection, compound 2-5 (250mg, 0.450mmol) was dissolved in acetone (4mL) and water (2mL), sodium periodate (356mg, 1.67mmol) and ammonium acetate (69.4mg, 0.900mmol) were added, The reaction solution was stirred at 50°C for 12 hours, adjusted to pH<3 with 1M hydrochloric acid aqueous solution, and extracted with ethyl acetate (20 mL x 1). The organic phase was washed with saturated brine (20 mL x 1), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to obtain compound 2-6. MS-ESI calculated [M+Na] + 496, found 496.
第六步step six
将化合物2-4(70.0mg,0.289mmol),2-6(137mg,0.289mmol),醋酸铜(105mg,0.578mmol),吡啶(68.6mg,0.867mmol)和氧化吡啶(82.4mg,0.867mmol)加入到二氯甲烷(5mL)中。反应液在氧气下40℃搅拌反应12小时。向反应液中加入水(10mL),用二氯甲烷(10mL x 3)萃取。合并有机相,用饱和食盐水洗涤(10mL x 1),无水硫酸钠干燥,过滤,滤液减压浓缩。剩余物用制备薄层层析法分离纯化(3/1石油醚/乙酸乙酯)得到化合物2-7。MS-ESI计算值[M+H] +641,实测值641。 Compound 2-4 (70.0mg, 0.289mmol), 2-6 (137mg, 0.289mmol), copper acetate (105mg, 0.578mmol), pyridine (68.6mg, 0.867mmol) and pyridine oxide (82.4mg, 0.867mmol) Add to dichloromethane (5 mL). The reaction solution was stirred and reacted at 40° C. under oxygen for 12 hours. Water (10 mL) was added to the reaction liquid, and extracted with dichloromethane (10 mL x 3). The organic phases were combined, washed with saturated brine (10 mL x 1), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was separated and purified by preparative thin layer chromatography (3/1 petroleum ether/ethyl acetate) to obtain compound 2-7. MS-ESI calculated value [M+H] + 641, found value 641.
第七步step seven
将化合物2-7(90.0mg,0.115mmol)和一水合氢氧化锂(9.7mg,0.230mmol)加入到四氢呋喃(2mL)和水(1mL)中。反应液在氮气保护下20℃搅拌反应12小时。反应液减压浓缩。剩余物经高效液相色谱法分离(中性,碳酸氢铵体系)纯化得到化合物2。 1H NMR(400MHz,CD 3OD)δ8.00-7.97(m,1H),7.94-7.91(m,1H),7.87-7.85(m,1H),7.75-7.73(m,1H),7.41-7.39(m,1H),7.28-7.24(m,2H),7.10-7.08(m,1H),6.81-6.80(m,1H),6.74-6.72(m,1H),4.42-4.37(m,1H),3.71-3.68(m,2H),3.44-3.40(m,2H),3.22-3.16(m,2H),2.76-2.73(m,1H),2.38-2.34(m,2H),2.08-2.04(m,1H),1.91-1.77(m,5H)。MS-ESI计算值[M+H] +627,实测值627。 Compound 2-7 (90.0 mg, 0.115 mmol) and lithium hydroxide monohydrate (9.7 mg, 0.230 mmol) were added to tetrahydrofuran (2 mL) and water (1 mL). The reaction solution was stirred and reacted at 20° C. for 12 hours under the protection of nitrogen. The reaction solution was concentrated under reduced pressure. The residue was separated and purified by high performance liquid chromatography (neutral, ammonium bicarbonate system) to obtain compound 2. 1 H NMR (400MHz, CD 3 OD) δ8.00-7.97(m,1H),7.94-7.91(m,1H),7.87-7.85(m,1H),7.75-7.73(m,1H),7.41- 7.39(m,1H),7.28-7.24(m,2H),7.10-7.08(m,1H),6.81-6.80(m,1H),6.74-6.72(m,1H),4.42-4.37(m,1H ),3.71-3.68(m,2H),3.44-3.40(m,2H),3.22-3.16(m,2H),2.76-2.73(m,1H),2.38-2.34(m,2H),2.08-2.04 (m,1H), 1.91-1.77(m,5H). MS-ESI calculated [M+H] + 627, found 627.
实施例3Example 3
合成路线:synthetic route:

第一步first step
将化合物3-1(400mg,1.56mmol),化合物3-2(291mg,1.56mmol),甲烷磺酸(2-环己基膦基-3,6-二甲氧基-2,4,6-三异丙基-1,1-联苯)(2-氨基-1,1-联苯-2-基)钯(Ⅱ)(141mg,0.156mmol)和碳酸铯(1.27g,3.90mmol)溶于N,N-二甲基甲酰胺(20mL)中,反应液在氮气保护下升温至90℃并搅拌10小时。反应液通过硅藻土过滤,滤渣用乙酸乙酯(50mL)洗涤,滤液依次用水(50mL x 3)和饱和食盐水(50mL x 1)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩后剩余物经过硅胶柱层析法(10/1石油醚/乙酸乙酯)分离纯化得到化合物3-3。MS-ESI计算值[M-56+H] +259,实测值259。 Compound 3-1 (400mg, 1.56mmol), compound 3-2 (291mg, 1.56mmol), methanesulfonic acid (2-cyclohexylphosphino-3,6-dimethoxy-2,4,6-tri Isopropyl-1,1-biphenyl)(2-amino-1,1-biphenyl-2-yl)palladium (II) (141mg, 0.156mmol) and cesium carbonate (1.27g, 3.90mmol) were dissolved in N , N-dimethylformamide (20mL), the reaction solution was heated to 90°C under nitrogen protection and stirred for 10 hours. The reaction solution was filtered through diatomaceous earth, the filter residue was washed with ethyl acetate (50 mL), the filtrate was washed with water (50 mL x 3) and saturated brine (50 mL x 1) successively, the organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was reduced to After concentrated under reduced pressure, the residue was separated and purified by silica gel column chromatography (10/1 petroleum ether/ethyl acetate) to obtain compound 3-3. MS-ESI calculated value [M-56+H] + 259, found value 259.
第二步second step
将化合物3-3(300mg,0.953mmol)溶于乙酸乙酯(5mL)中,在30℃下加入盐酸乙酸乙酯溶液(4M,5mL,20.0mmol),反应液在30℃下搅拌反应2小时。反应液过滤,滤饼用石油醚(50mL)洗涤后干燥。干燥后的滤饼溶于甲醇(5mL)中,并向其中加入固体碳酸氢钠(100mg),所得混合物在室温下搅拌0.5小时,再向其中加入二氯甲烷(50mL),过滤,滤液浓缩后得到化合物3-4。MS-ESI计算值[M+H] +215,实测值215。 Dissolve compound 3-3 (300mg, 0.953mmol) in ethyl acetate (5mL), add hydrochloric acid ethyl acetate solution (4M, 5mL, 20.0mmol) at 30°C, and stir the reaction solution at 30°C for 2 hours . The reaction solution was filtered, and the filter cake was washed with petroleum ether (50 mL) and dried. The dried filter cake was dissolved in methanol (5 mL), and solid sodium bicarbonate (100 mg) was added thereto, and the resulting mixture was stirred at room temperature for 0.5 hours, then dichloromethane (50 mL) was added thereto, filtered, and the filtrate was concentrated Compound 3-4 was obtained. MS-ESI calculated value [M+H] + 215, found value 215.
第三步third step
将化合物1-8(250mg,0.492mmol),化合物3-4(127mg,0.592mmol),甲烷磺酸(2-环己基膦基-3,6-二甲氧基-2,4,6-三异丙基-1,1-联苯)(2-氨基-1,1-联苯-2-基)钯(Ⅱ)(44.6mg,49.2umol)和碳酸铯(401mg,1.23 mmol)溶于N,N-二甲基甲酰胺(10mL)中,反应液在氮气保护下升温至90℃并搅拌12小时。反应液通过硅藻土过滤,滤渣用乙酸乙酯(30mL)洗涤,滤液依次用水(30mL x 3)和饱和食盐水(30mL x 1)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩后剩余物经过薄层层析法(4/1石油醚/乙酸乙酯)分离纯化得到化合物3-5。MS-ESI计算值[M+H] +642,实测值642。 Compound 1-8 (250mg, 0.492mmol), compound 3-4 (127mg, 0.592mmol), methanesulfonic acid (2-cyclohexylphosphino-3,6-dimethoxy-2,4,6-tri Isopropyl-1,1-biphenyl)(2-amino-1,1-biphenyl-2-yl)palladium (Ⅱ) (44.6mg, 49.2umol) and cesium carbonate (401mg, 1.23mmol) were dissolved in N , N-dimethylformamide (10 mL), the reaction solution was heated to 90 ° C under nitrogen protection and stirred for 12 hours. The reaction solution was filtered through diatomaceous earth, the filter residue was washed with ethyl acetate (30 mL), the filtrate was washed with water (30 mL x 3) and saturated brine (30 mL x 1) successively, the organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was reduced to After concentration under reduced pressure, the residue was separated and purified by thin layer chromatography (4/1 petroleum ether/ethyl acetate) to obtain compound 3-5. MS-ESI calculated value [M+H] + 642, found value 642.
第四步the fourth step
将氢氧化钠(50mg,1.24mmol)加入化合物3-5(265mg,0.413mmol)的四氢呋喃(3mL)和水(6mL)溶液中,反应液在50℃下搅拌10小时。反应液减压浓缩,加水(20mL),用盐酸(1M)调节pH值到4,再用乙酸乙酯萃取(20mL×2),有机相合并后用饱和食盐水洗涤(50mL×1),再用无水硫酸钠干燥,过滤,减压浓缩,剩余物经过高效液相色谱法(中性,碳酸氢铵体系)分离纯化得到化合物3。 1H NMR(400MHz,CD 3OD)δ8.00-7.93(m,3H),7.77-7.73(m,1H),7.18-7.15(m,1H),7.09-7.02(m,2H),6.86-6.80(m,1H),6.71-6.69(m,1H),6.46-6.42(m,1H),4.51-4.50(m,1H),4.41-4.37(m,1H),3.53-3.47(m,2H),3.37-3.35(m,1H),3.30-3.20(m,3H),2.47-2.30(m,3H),2.09-2.03(m,1H),1.88-1.73(m,2H)。MS-ESI计算值[M+H] +628,实测值628。 Sodium hydroxide (50mg, 1.24mmol) was added to a solution of compound 3-5 (265mg, 0.413mmol) in tetrahydrofuran (3mL) and water (6mL), and the reaction solution was stirred at 50°C for 10 hours. The reaction solution was concentrated under reduced pressure, water (20 mL) was added, the pH value was adjusted to 4 with hydrochloric acid (1M), and then extracted with ethyl acetate (20 mL×2), the combined organic phases were washed with saturated brine (50 mL×1), and then It was dried over anhydrous sodium sulfate, filtered, concentrated under reduced pressure, and the residue was separated and purified by high performance liquid chromatography (neutral, ammonium bicarbonate system) to obtain compound 3. 1 H NMR (400MHz, CD 3 OD) δ8.00-7.93 (m, 3H), 7.77-7.73 (m, 1H), 7.18-7.15 (m, 1H), 7.09-7.02 (m, 2H), 6.86- 6.80(m,1H),6.71-6.69(m,1H),6.46-6.42(m,1H),4.51-4.50(m,1H),4.41-4.37(m,1H),3.53-3.47(m,2H ), 3.37-3.35(m,1H), 3.30-3.20(m,3H), 2.47-2.30(m,3H), 2.09-2.03(m,1H), 1.88-1.73(m,2H). MS-ESI calculated value [M+H] + 628, found value 628.
实施例4Example 4
合成路线:synthetic route:

第一步first step
参照实施例3第一步得到化合物4-1。MS-ESI计算值[M-56+H] +558,实测值558。 Referring to the first step of Example 3, compound 4-1 was obtained. MS-ESI calculated value [M-56+H] + 558, found value 558.
第二步second step
将化合物4-1(275mg,0.448mmol)溶于乙酸乙酯(5mL)中,加入盐酸乙酸乙酯溶液(4M,5mL,20.0mmol),反应液在30℃下搅拌反应1小时。反应液过滤,滤饼用石油醚(50mL)洗涤后干燥。干燥后的滤饼 悬浮于饱和碳酸氢钠水溶液(30mL)中,并用乙酸乙酯萃取(30mL x 2),合并后的有机相用饱和食盐水(50mL x 1)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩得到化合物4-2。MS-ESI计算值[M+H] +514,实测值514。 Compound 4-1 (275mg, 0.448mmol) was dissolved in ethyl acetate (5mL), ethyl acetate hydrochloride solution (4M, 5mL, 20.0mmol) was added, and the reaction solution was stirred at 30°C for 1 hour. The reaction solution was filtered, and the filter cake was washed with petroleum ether (50 mL) and dried. The dried filter cake was suspended in saturated aqueous sodium bicarbonate (30 mL), and extracted with ethyl acetate (30 mL x 2), the combined organic phase was washed with saturated brine (50 mL x 1), dried over anhydrous sodium sulfate, After filtration, the filtrate was concentrated under reduced pressure to obtain compound 4-2. MS-ESI calculated value [M+H] + 514, found value 514.
第三步third step
将化合物4-2(100mg,0.195mmol),化合物3-1(54.9mg,0.214mmol),三(二亚苄基丙酮)二钯(0)(17.8mg,0.0195mmol),4,5-双(二苯基膦)-9,9-二甲基氧杂蒽(22.5mg,0.0389mmol)和碳酸铯(127mg,0.389mmol)溶于N,N-二甲基甲酰胺(3mL)中,反应液在氮气保护下升温至100℃并搅拌10小时。反应液通过硅藻土过滤,滤渣用乙酸乙酯(50mL)洗涤,滤液依次用水(50mL x 3)和饱和食盐水(50mL x 1)洗涤,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩后剩余物经过薄层层析法(4/1石油醚/乙酸乙酯)分离纯化得到化合物4-3。MS-ESI计算值[M+H] +642,实测值642。 Compound 4-2 (100 mg, 0.195 mmol), compound 3-1 (54.9 mg, 0.214 mmol), tris(dibenzylideneacetone) dipalladium (0) (17.8 mg, 0.0195 mmol), 4,5-bis (Diphenylphosphine)-9,9-dimethylxanthene (22.5mg, 0.0389mmol) and cesium carbonate (127mg, 0.389mmol) were dissolved in N,N-dimethylformamide (3mL), reacted The liquid was heated to 100° C. under nitrogen protection and stirred for 10 hours. The reaction solution was filtered through diatomaceous earth, the filter residue was washed with ethyl acetate (50 mL), the filtrate was washed with water (50 mL x 3) and saturated brine (50 mL x 1) successively, the organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was reduced to After concentration under reduced pressure, the residue was separated and purified by thin layer chromatography (4/1 petroleum ether/ethyl acetate) to obtain compound 4-3. MS-ESI calculated value [M+H] + 642, found value 642.
第四步the fourth step
参照实施例3第四步得到化合物4。 1H NMR(400MHz,CD 3OD)δ8.02-7.99(m,1H),7.94-7.88(m,2H),7.77-7.73(m,1H),7.22-7.19(m,2H),7.07-7.03(m,2H),6.66-6.64(m,1H),6.57-6.54(m,1H),4.40-4.36(m,1H),4.11-4.05(m,1H),3.73-3.65(m,1H),3.54-3.51(m,1H),3.46-3.40(m,1H),3.38-3.34(m,1H),3.27-3.18(m,2H),2.44-2.29(m,3H),1.98-1.90(m,1H),1.88-1.81(m,1H),1.79-1.72(m,1H)。MS-ESI计算值[M+H] +628,实测值628。 Referring to the fourth step of Example 3, compound 4 was obtained. 1 H NMR (400MHz, CD 3 OD) δ8.02-7.99 (m, 1H), 7.94-7.88 (m, 2H), 7.77-7.73 (m, 1H), 7.22-7.19 (m, 2H), 7.07- 7.03(m,2H),6.66-6.64(m,1H),6.57-6.54(m,1H),4.40-4.36(m,1H),4.11-4.05(m,1H),3.73-3.65(m,1H ),3.54-3.51(m,1H),3.46-3.40(m,1H),3.38-3.34(m,1H),3.27-3.18(m,2H),2.44-2.29(m,3H),1.98-1.90 (m,1H), 1.88-1.81(m,1H), 1.79-1.72(m,1H). MS-ESI calculated value [M+H] + 628, found value 628.
实施例5Example 5
合成路线:synthetic route:

第一步first step
将化合物5-1(500mg,3.29mmol),化合物3-2(643mg,3.45mmol),醋酸铜(1.20g,6.58mmol),吡啶(781mg,9.87mmol)和氧化吡啶(939mg,9.87mmol)溶于二氯甲烷(15mL)中,反应液在氧气下30℃搅拌12小时。反应液用水(100mL)稀释后用二氯甲烷(50mL x 3)萃取,合并有机相后用饱和食盐水(100mL x1)洗涤,再用无水硫酸钠干燥,过滤,滤液减压浓缩后剩余物经过硅胶柱层析法(5/1石油醚/乙酸乙酯)分离纯化得到化合物5-2。MS-ESI计算值[M-56+H] +237,实测值237。 Compound 5-1 (500mg, 3.29mmol), compound 3-2 (643mg, 3.45mmol), copper acetate (1.20g, 6.58mmol), pyridine (781mg, 9.87mmol) and pyridine oxide (939mg, 9.87mmol) were dissolved In dichloromethane (15 mL), the reaction solution was stirred at 30°C for 12 hours under oxygen. The reaction solution was diluted with water (100mL) and extracted with dichloromethane (50mL x 3). The combined organic phases were washed with saturated brine (100mL x1), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain the residue Compound 5-2 was obtained by separation and purification by silica gel column chromatography (5/1 petroleum ether/ethyl acetate). MS-ESI calculated value [M-56+H] + 237, found value 237.
第二步second step
将化合物5-2(260mg,0.889mmol)溶于乙酸乙酯(5mL)中,加入盐酸乙酸乙酯溶液(4M,5mL,20.0mmol),反应液在30℃下搅拌反应1小时。反应液减压浓缩后剩余物溶于饱和碳酸氢钠水溶液(30mL)中,并用二氯甲烷/甲醇萃取(10/1,50mL x 2),合并有机相后用饱和食盐水(50mL x 1)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩得到化合物5-3。MS-ESI计算值[M+H] +193,实测值193。 Compound 5-2 (260mg, 0.889mmol) was dissolved in ethyl acetate (5mL), ethyl acetate hydrochloride solution (4M, 5mL, 20.0mmol) was added, and the reaction solution was stirred at 30°C for 1 hour. After the reaction solution was concentrated under reduced pressure, the residue was dissolved in saturated aqueous sodium bicarbonate (30mL), extracted with dichloromethane/methanol (10/1, 50mL x 2), and the combined organic phases were washed with saturated brine (50mL x 1) Wash, dry over anhydrous sodium sulfate, filter, and concentrate the filtrate under reduced pressure to obtain compound 5-3. MS-ESI calculated value [M+H] + 193, found value 193.
第三步third step
参照实施例3第一步得到化合物5-4。MS-ESI计算值[M+H] +620,实测值620。 Referring to the first step of Example 3, compound 5-4 was obtained. MS-ESI calculated value [M+H] + 620, found value 620.
第四步the fourth step
参照实施例3第四步得到化合物5。 1H NMR(400MHz,CD 3OD)δ8.01-7.98(m,1H),7.95-7.93(m,2H),7.77-7.72(m,1H),7.02(d,J=1.6Hz,1H),6.89-6.85(m,2H),6.78-6.76(m,1H),6.72-6.69(m,2H),6.46-6.43(m,1H),4.40-4.36(m,1H),4.27-4.23(m,1H),3.85(s,3H),3.63-3.61(m,1H),3.48-3.44(m,1H),3.39-3.37(m,1H),3.26-3.24(m,1H),3.23-3.15(m,2H),2.47-2.31(m,3H),2.09-2.03(m,1H),1.87-1.74(m,2H)。MS-ESI计算值[M+H] +606,实测值606。 Referring to the fourth step of Example 3, compound 5 was obtained. 1 H NMR (400MHz, CD 3 OD) δ8.01-7.98 (m, 1H), 7.95-7.93 (m, 2H), 7.77-7.72 (m, 1H), 7.02 (d, J = 1.6Hz, 1H) ,6.89-6.85(m,2H),6.78-6.76(m,1H),6.72-6.69(m,2H),6.46-6.43(m,1H),4.40-4.36(m,1H),4.27-4.23( m,1H),3.85(s,3H),3.63-3.61(m,1H),3.48-3.44(m,1H),3.39-3.37(m,1H),3.26-3.24(m,1H),3.23- 3.15 (m, 2H), 2.47-2.31 (m, 3H), 2.09-2.03 (m, 1H), 1.87-1.74 (m, 2H). MS-ESI calculated value [M+H] + 606, found value 606.
实施例6Example 6
合成路线:synthetic route:

第一步first step
参照实施例3第一步得到化合物6-2。MS-ESI计算值[M+H] +620,实测值620。 Referring to the first step of Example 3, compound 6-2 was obtained. MS-ESI calculated value [M+H] + 620, found value 620.
第二步second step
参照实施例3第四步得到化合物6。 1H NMR(400MHz,CD 3OD)δ8.02-8.00(m,1H),7.95-7.90(m,2H),7.78-7.74(m,1H),7.21-7.19(m,1H),6.96-6.91(m,1H),6.90-6.84(m,3H),6.67-6.64(m,1H),6.57-6.54(m,1H),4.40-4.36(m,1H),4.09-4.02(m,1H),3.84(d,J=10.0Hz,3H),3.69-3.55(m,1H),3.45-3.43(m,1H),3.31-3.28(m,2H),3.24-3.18(m,2H),2.42-2.31(m,3H),1.94-1.75(m,3H)。MS-ESI计算值[M+H] +606,实测值606。 Referring to the fourth step of Example 3, compound 6 was obtained. 1 H NMR (400MHz, CD 3 OD) δ8.02-8.00 (m, 1H), 7.95-7.90 (m, 2H), 7.78-7.74 (m, 1H), 7.21-7.19 (m, 1H), 6.96- 6.91(m,1H),6.90-6.84(m,3H),6.67-6.64(m,1H),6.57-6.54(m,1H),4.40-4.36(m,1H),4.09-4.02(m,1H ),3.84(d,J=10.0Hz,3H),3.69-3.55(m,1H),3.45-3.43(m,1H),3.31-3.28(m,2H),3.24-3.18(m,2H), 2.42-2.31(m,3H),1.94-1.75(m,3H). MS-ESI calculated value [M+H] + 606, found value 606.
实施例7Example 7
合成路线:synthetic route:

第一步first step
将化合物1-8(10.1g,19.9mmol),化合物7-1(9.38g,39.7mmol),锌粉(3.90g,59.6mmol),四甲基乙二胺(6.93g,59.6mmol),正辛酸钠(3.3g,19.9mmol),正辛醇(5.18g,39.7mmol),(2-二环己基膦-2,4,6-三异丙基-1,1-联苯)[2-(2-氨基-1,1-联苯)]甲磺酸钯(841mg,0.994mmol)和氯化钠(3.48g,59.6mmol)溶于水(100mL)中,反应液在氮气保护下升温至60℃搅拌12小时。反应液用水(100mL)稀释后用乙酸乙酯(200mL x 2)萃取,有机相用饱和食盐水(500mL x 1)洗涤,再用无水硫酸钠干燥,过滤,滤液减压浓缩后剩余物经过硅胶柱层析法(4/1石油醚/乙酸乙酯)分离纯化得到化合物7-2。 1H NMR(400MHz,CDCl 3)δ 7.94(s,1H),7.87-7.84(m,2H),7.71-7.63(m,2H),7.11-7.08(m,1H),6.83-6.81(m,1H),4.37-4.29(m,3H),3.96-3.92(m,2H),3.74-3.70(m,4H),3.53-3.51(m,1H),3.24-3.18(m,1H),2.50-2.40(m,2H),1.96-1.81(m,2H),1.50(s,9H)。MS-ESI计算值[M-56+H] +529,实测值529。 Compound 1-8 (10.1g, 19.9mmol), compound 7-1 (9.38g, 39.7mmol), zinc powder (3.90g, 59.6mmol), tetramethylethylenediamine (6.93g, 59.6mmol), n Sodium octanoate (3.3g, 19.9mmol), n-octanol (5.18g, 39.7mmol), (2-dicyclohexylphosphine-2,4,6-triisopropyl-1,1-biphenyl)[2- (2-Amino-1,1-biphenyl)]palladium methanesulfonate (841mg, 0.994mmol) and sodium chloride (3.48g, 59.6mmol) were dissolved in water (100mL), and the reaction solution was heated to Stir at 60°C for 12 hours. The reaction solution was diluted with water (100mL) and extracted with ethyl acetate (200mL x 2), the organic phase was washed with saturated brine (500mL x 1), dried over anhydrous sodium sulfate, filtered, the filtrate was concentrated under reduced pressure, and the residue was passed through Compound 7-2 was obtained by separation and purification by silica gel column chromatography (4/1 petroleum ether/ethyl acetate). 1 H NMR (400MHz, CDCl 3 )δ 7.94(s,1H),7.87-7.84(m,2H),7.71-7.63(m,2H),7.11-7.08(m,1H),6.83-6.81(m, 1H),4.37-4.29(m,3H),3.96-3.92(m,2H),3.74-3.70(m,4H),3.53-3.51(m,1H),3.24-3.18(m,1H),2.50- 2.40(m,2H),1.96-1.81(m,2H),1.50(s,9H). MS-ESI calculated value [M-56+H] + 529, found value 529.
第二步second step
参照实施例5第二步得到化合物7-3。MS-ESI计算值[M+H] +485,实测值485。 Referring to the second step of Example 5, compound 7-3 was obtained. MS-ESI calculated [M+H] + 485, found 485.
第三步third step
参照实施例3第一步得到化合物7-5。MS-ESI计算值[M+H] +659,实测值659。 Referring to the first step of Example 3, compound 7-5 was obtained. MS-ESI calculated [M+H] + 659, found 659.
第四步the fourth step
参照实施例3第四步得到化合物7。 1H NMR(400MHz,CD 3OD)δ8.01-7.99(m,1H),7.95-7.93(m,2H),7.85(s,1H),7.76-7.72(m,1H),7.25-7.17(m,3H),7.11-7.06(m,1H),6.83-6.81(m,1H),4.47-4.42(m,3H),4.24-4.17(m,2H),3.95(s,3H),3.85-3.75(m,1H),3.49-3.44(m,1H),3.28-3.22(m,1H),2.48-2.34(m,2H),1.92-1.77(m,2H)。MS-ESI计算值[M+H] +645,实测值645。 Referring to the fourth step of Example 3, compound 7 was obtained. 1 H NMR (400MHz, CD 3 OD) δ8.01-7.99 (m, 1H), 7.95-7.93 (m, 2H), 7.85 (s, 1H), 7.76-7.72 (m, 1H), 7.25-7.17 ( m,3H),7.11-7.06(m,1H),6.83-6.81(m,1H),4.47-4.42(m,3H),4.24-4.17(m,2H),3.95(s,3H),3.85- 3.75 (m, 1H), 3.49-3.44 (m, 1H), 3.28-3.22 (m, 1H), 2.48-2.34 (m, 2H), 1.92-1.77 (m, 2H). MS-ESI calculated [M+H] + 645, found 645.
实施例8Example 8
合成路线:synthetic route:

第一步first step
参照实施例3第一步得到化合物8-2。MS-ESI计算值[M+H] +612,实测值612。 Referring to the first step of Example 3, compound 8-2 was obtained. MS-ESI calculated value [M+H] + 612, found value 612.
第二步second step
参照实施例3第四步得到化合物8。 1H NMR(400MHz,CD 3OD)δ8.80-8.78(m,1H),8.58-8.56(m,1H),8.00-7.98(m,1H),7.92-7.90(m,2H),7.89-7.87(m,1H),7.71-7.65(m,2H),7.55-7.53(m,1H),7.48-7.45(m,1H),7.23-7.20(m,1H),6.86-6.84(m,1H),6.78-6.76(m,1H),4.67-4.63(m,2H),4.44-4.40(m,1H),4.16-4.12(m,2H),4.05-3.98(m,1H),3.51-3.49(m,1H),3.31-3.25(m,1H),2.45-2.39(m,2H),1.95-1.78(m,2H)。MS-ESI 计算值[M+H] +598,实测值598。 Referring to the fourth step of Example 3, compound 8 was obtained. 1 H NMR (400MHz, CD 3 OD) δ8.80-8.78(m,1H),8.58-8.56(m,1H),8.00-7.98(m,1H),7.92-7.90(m,2H),7.89- 7.87(m,1H),7.71-7.65(m,2H),7.55-7.53(m,1H),7.48-7.45(m,1H),7.23-7.20(m,1H),6.86-6.84(m,1H ),6.78-6.76(m,1H),4.67-4.63(m,2H),4.44-4.40(m,1H),4.16-4.12(m,2H),4.05-3.98(m,1H),3.51-3.49 (m,1H), 3.31-3.25(m,1H), 2.45-2.39(m,2H), 1.95-1.78(m,2H). MS-ESI calculated value [M+H] + 598, found value 598.
实施例9Example 9
合成路线:synthetic route:

第一步first step
参照实施例3第一步得到化合物9-2。MS-ESI计算值[M+H] +641,实测值641。 Referring to the first step of Example 3, compound 9-2 was obtained. MS-ESI calculated value [M+H] + 641, found value 641.
第二步second step
参照实施例3第四步得到化合物9。 1H NMR(400MHz,CD 3OD)δ8.21-8.19(m,1H),8.02-7.98(m,2H),7.91-7.87(m,2H),7.77-7.75(m,1H),7.70-7.66(m,1H),7.52-7.50(m,1H),7.40-7.30(m,3H),7.21-7.18(m,1H),6.83-6.81(m,1H),4.72-4.69(m,2H),4.46-4.42(m,1H),4.31-4.25(m,2H),3.96(s,3H),3.91-3.84(m,1H),3.53-3.48(m,1H),3.30-3.24(m,1H),2.46-2.31(m,2H),1.95-1.78(m,2H)。MS-ESI计算值[M+H] +627,实测值627。 Referring to the fourth step of Example 3, compound 9 was obtained. 1 H NMR (400MHz, CD 3 OD) δ8.21-8.19 (m, 1H), 8.02-7.98 (m, 2H), 7.91-7.87 (m, 2H), 7.77-7.75 (m, 1H), 7.70- 7.66(m,1H),7.52-7.50(m,1H),7.40-7.30(m,3H),7.21-7.18(m,1H),6.83-6.81(m,1H),4.72-4.69(m,2H ),4.46-4.42(m,1H),4.31-4.25(m,2H),3.96(s,3H),3.91-3.84(m,1H),3.53-3.48(m,1H),3.30-3.24(m ,1H), 2.46-2.31(m,2H), 1.95-1.78(m,2H). MS-ESI calculated [M+H] + 627, found 627.
实施例10Example 10
合成路线:synthetic route:

第一步first step
参照实施例3第一步得到化合物10-2。MS-ESI计算值[M+H] +595,实测值595。 Referring to the first step of Example 3, compound 10-2 was obtained. MS-ESI calculated [M+H] + 595, found 595.
第二步second step
参照实施例3第四步得到化合物10。 1H NMR(400MHz,CD 3OD)δ8.03-8.01(m,1H),7.94-7.92(m,1H),7.85-7.83(m,2H),7.75-7.71(m,1H),7.27-7.25(m,1H),7.23-7.19(m,1H),7.17-7.14(m,1H),6.83-6.79(m,2H),6.73-6.70(m,1H),4.50-4.42(m,3H),3.99-3.94(m,2H),3.88-3.81(m,1H),3.52-3.47(m,1H),3.29-3.23(m,1H),2.44-2.26(m,2H),1.93-1.80(m,2H)。MS-ESI计算值[M+H] +581,实测值581。 Referring to the fourth step of Example 3, compound 10 was obtained. 1 H NMR (400MHz, CD 3 OD) δ8.03-8.01(m,1H),7.94-7.92(m,1H),7.85-7.83(m,2H),7.75-7.71(m,1H),7.27- 7.25(m,1H),7.23-7.19(m,1H),7.17-7.14(m,1H),6.83-6.79(m,2H),6.73-6.70(m,1H),4.50-4.42(m,3H ),3.99-3.94(m,2H),3.88-3.81(m,1H),3.52-3.47(m,1H),3.29-3.23(m,1H),2.44-2.26(m,2H),1.93-1.80 (m,2H). MS-ESI calculated value [M+H] + 581, found value 581.
实施例11Example 11
合成路线:synthetic route:

第一步first step
参照实施例3第一步得到化合物11-2。MS-ESI计算值[M+H] +629,实测值629。 Referring to the first step of Example 3, compound 11-2 was obtained. MS-ESI calculated [M+H] + 629, found 629.
第二步second step
参照实施例3第四步得到化合物11。 1H NMR(400MHz,CD 3OD)δ8.07-8.05(m,1H),7.94-7.92(m,1H),7.86-7.84(m,2H),7.79-7.75(m,1H),7.22-7.20(m,2H),7.17-7.14(m,1H),6.83-6.81(m,1H),6.76-6.71(m,1H),4.91-4.89(m,2H),4.49-4.45(m,1H),4.41-4.36(m,2H),3.76-3.70(m,1H),3.50-3.46(m,1H),3.28-3.22(m,1H),2.36-2.20(m,2H),1.92-1.77(m,2H)。MS-ESI计算值[M+H] +615,实测值615。 Referring to the fourth step of Example 3, compound 11 was obtained. 1 H NMR (400MHz, CD 3 OD) δ8.07-8.05(m,1H),7.94-7.92(m,1H),7.86-7.84(m,2H),7.79-7.75(m,1H),7.22- 7.20(m,2H),7.17-7.14(m,1H),6.83-6.81(m,1H),6.76-6.71(m,1H),4.91-4.89(m,2H),4.49-4.45(m,1H ),4.41-4.36(m,2H),3.76-3.70(m,1H),3.50-3.46(m,1H),3.28-3.22(m,1H),2.36-2.20(m,2H),1.92-1.77 (m,2H). MS-ESI calculated [M+H] + 615, found 615.
实施例12Example 12
合成路线:synthetic route:
第一步first step
参照实施例3第一步得到化合物12-2。MS-ESI计算值[M+H] +609,实测值609。 Referring to the first step of Example 3, compound 12-2 was obtained. MS-ESI calculated value [M+H] + 609, found value 609.
第二步second step
参照实施例3第四步得到化合物12。 1H NMR(400MHz,CD 3OD)δ8.02-8.00(m,1H),7.95-7.91(m,2H),7.87(s,1H),7.75-7.71(m,1H),7.19-7.16(m,1H),7.11-7.09(m,1H),7.01-6.99(m,1H),6.83-6.81(m,1H),6.77-6.73(m,1H),4.74-4.69(m,2H),4.46-4.41(m,1H),4.24-4.20(m,2H),3.78-3.72(m,1H),3.51-3.47(m,1H),3.30-3.24(m,1H),2.44-2.41(m,2H),2.38(s,3H),1.93-1.77(m,2H)。MS-ESI计算值[M+H] +595,实测值595。 Referring to the fourth step of Example 3, compound 12 was obtained. 1 H NMR (400MHz, CD 3 OD) δ8.02-8.00(m,1H),7.95-7.91(m,2H),7.87(s,1H),7.75-7.71(m,1H),7.19-7.16( m,1H),7.11-7.09(m,1H),7.01-6.99(m,1H),6.83-6.81(m,1H),6.77-6.73(m,1H),4.74-4.69(m,2H), 4.46-4.41(m,1H),4.24-4.20(m,2H),3.78-3.72(m,1H),3.51-3.47(m,1H),3.30-3.24(m,1H),2.44-2.41(m ,2H), 2.38(s,3H), 1.93-1.77(m,2H). MS-ESI calculated [M+H] + 595, found 595.
实施例13Example 13
合成路线:synthetic route:

第一步first step
参照实施例3第一步得到化合物13-2。MS-ESI计算值[M+H] +596,实测值596。 Referring to the first step of Example 3, compound 13-2 was obtained. MS-ESI calculated [M+H] + 596, found 596.
第二步second step
参照实施例3第四步得到化合物13。 1H NMR(400MHz,CD 3OD)δ8.08-8.06(m,1H),8.01-7.99(m,1H),7.95-7.93(m,1H),7.86-7.84(m,2H),7.76-7.72(m,1H),7.62-7.60(m,1H),7.17-7.15(m,1H),6.85-6.83(m,1H),6.78-6.74(m,1H),4.68-4.64(m,2H),4.45-4.41(m,1H),4.21-4.16(m,2H),3.91-3.84(m,1H),3.50-3.45(m,1H),3.29-3.23(m,1H),2.47-2.35(m,2H),1.94-1.75(m,2H)。MS-ESI计算值[M+H] +582,实测值582。 Referring to the fourth step of Example 3, compound 13 was obtained. 1 H NMR (400MHz, CD 3 OD) δ8.08-8.06 (m, 1H), 8.01-7.99 (m, 1H), 7.95-7.93 (m, 1H), 7.86-7.84 (m, 2H), 7.76- 7.72(m,1H),7.62-7.60(m,1H),7.17-7.15(m,1H),6.85-6.83(m,1H),6.78-6.74(m,1H),4.68-4.64(m,2H ),4.45-4.41(m,1H),4.21-4.16(m,2H),3.91-3.84(m,1H),3.50-3.45(m,1H),3.29-3.23(m,1H),2.47-2.35 (m,2H), 1.94-1.75(m,2H). MS-ESI calculated [M+H] + 582, found 582.
实施例14Example 14
合成路线:synthetic route:

第一步first step
将化合物14-1(10g,54.0mmol)溶于无水四氢呋喃(100mL)中,将溶液冷至-78℃,在氮气保护下缓慢滴加双(三甲基硅基)氨基锂(1M,64.8mL),反应液在-78℃下搅拌0.5小时,接着在-78℃下缓慢滴加化合物14-2(21.2g,59.4mmol)的无水四氢呋喃(80mL)溶液,反应液在0℃下搅拌2小时。反应液用水(300mL)淬灭后用乙酸乙酯(500mL×1)萃取,有机相用饱和食盐水(500mL×1)洗涤,再用无水硫酸钠干燥,过滤,滤液减压浓缩后剩余物经过硅胶柱层析法(10/1石油醚/乙酸乙酯)分离纯化得到化合物14-3。 1H NMR(400MHz,CDCl 3)δ5.78-5.73(m,1H),4.27-4.21(m,4H),1.50(s,9H)。 Compound 14-1 (10g, 54.0mmol) was dissolved in anhydrous tetrahydrofuran (100mL), the solution was cooled to -78°C, and lithium bis(trimethylsilyl)amide (1M, 64.8 mL), the reaction solution was stirred at -78°C for 0.5 hours, then a solution of compound 14-2 (21.2g, 59.4mmol) in anhydrous tetrahydrofuran (80mL) was slowly added dropwise at -78°C, and the reaction solution was stirred at 0°C 2 hours. The reaction solution was quenched with water (300mL) and extracted with ethyl acetate (500mL×1), the organic phase was washed with saturated brine (500mL×1), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain the residue Compound 14-3 was obtained by separation and purification through silica gel column chromatography (10/1 petroleum ether/ethyl acetate). 1 H NMR (400 MHz, CDCl 3 ) δ 5.78-5.73 (m, 1H), 4.27-4.21 (m, 4H), 1.50 (s, 9H).
第二步second step
将化合物14-3(9.5g,29.9mmol),双联频哪醇硼酸酯(9.12g,35.9mmol),醋酸钾(8.82g,89.8mmol),1,1-双(二苯基膦)二茂铁氯化钯(2.19g,2.99mmol)和1,1-双(二苯基膦)二茂铁(1.66g,2.99mmol)溶于1,4-二氧六环(100mL)中,反应液在氮气保护下升温至80℃搅拌12小时。反应液通过硅藻土过滤,滤液减压浓缩后剩余物经过硅胶柱层析法(10/1石油醚/乙酸乙酯)分离纯化得到化合物14-4。 1H NMR(400MHz,CDCl 3)δ6.50-6.45(m,1H),4.23-4.17(m,4H),1.49(s,9H),1.29(s,12H)。 Compound 14-3 (9.5g, 29.9mmol), bis-pinacol borate (9.12g, 35.9mmol), potassium acetate (8.82g, 89.8mmol), 1,1-bis(diphenylphosphine) Ferrocenepalladium chloride (2.19g, 2.99mmol) and 1,1-bis(diphenylphosphino)ferrocene (1.66g, 2.99mmol) were dissolved in 1,4-dioxane (100mL), The reaction solution was heated to 80° C. and stirred for 12 hours under the protection of nitrogen. The reaction solution was filtered through diatomaceous earth, the filtrate was concentrated under reduced pressure, and the residue was separated and purified by silica gel column chromatography (10/1 petroleum ether/ethyl acetate) to obtain compound 14-4. 1 H NMR (400 MHz, CDCl 3 ) δ 6.50-6.45 (m, 1H), 4.23-4.17 (m, 4H), 1.49 (s, 9H), 1.29 (s, 12H).
第三步third step
将化合物14-5(1.00g,4.88mmol),化合物14-4(2.00g,6.78mmol),碳酸钾(1.35g,9.75mmol)和1,1-双(二苯基膦)二茂铁氯化钯(357mg,0.488mmol)溶于乙二醇二甲醚(30mL)和水(5mL)中,反应液在氮气保护下升温至90℃搅拌10小时。反应液通过硅藻土过滤,滤液减压浓缩后剩余物经过硅胶柱层析法(10/1石油醚/乙酸乙酯)分离纯化得到化合物14-6。 1H NMR(400MHz,CDCl 3)δ7.25-7.19(m,1H),6.76-6.70(m,2H),6.13-6.08(m,1H),4.54-4.44(m,2H),4.35-4.27(m,2H),3.86(d,J=9.6Hz,3H),1.52(d,J=2.0Hz,9H)。MS-ESI计算值[M-56+H] +238,实测值238。 Compound 14-5 (1.00g, 4.88mmol), compound 14-4 (2.00g, 6.78mmol), potassium carbonate (1.35g, 9.75mmol) and 1,1-bis(diphenylphosphine) ferrocene chloride Palladium chloride (357mg, 0.488mmol) was dissolved in ethylene glycol dimethyl ether (30mL) and water (5mL), and the reaction solution was heated to 90°C under nitrogen protection and stirred for 10 hours. The reaction solution was filtered through diatomaceous earth, the filtrate was concentrated under reduced pressure, and the residue was separated and purified by silica gel column chromatography (10/1 petroleum ether/ethyl acetate) to obtain compound 14-6. 1 H NMR (400MHz, CDCl 3 )δ7.25-7.19(m,1H),6.76-6.70(m,2H),6.13-6.08(m,1H),4.54-4.44(m,2H),4.35-4.27 (m, 2H), 3.86 (d, J=9.6Hz, 3H), 1.52 (d, J=2.0Hz, 9H). MS-ESI calculated value [M-56+H] + 238, found value 238.
第四步the fourth step
将化合物14-6(850mg,2.90mmol)溶于甲醇(10mL)中,在氮气保护下加入湿钯碳(100mg,10%),反应液在氢气保护下20℃搅拌12小时。反应液通过硅藻土过滤,滤液减压浓缩后得到化合物14-7。MS-ESI计算值[M-56+H] +240,实测值240。 Compound 14-6 (850mg, 2.90mmol) was dissolved in methanol (10mL), wet palladium carbon (100mg, 10%) was added under the protection of nitrogen, and the reaction solution was stirred at 20°C for 12 hours under the protection of hydrogen. The reaction solution was filtered through diatomaceous earth, and the filtrate was concentrated under reduced pressure to obtain compound 14-7. MS-ESI calculated value [M-56+H] + 240, measured value 240.
第五步the fifth step
将化合物14-7(860mg,2.91mmol)溶于乙酸乙酯(5mL)中,向反应液中加入盐酸乙酸乙酯(4M,5mL,20.0mmol),反应液在20℃下搅拌2小时。反应液减压浓缩,剩余物用饱和碳酸氢钠水溶液(50mL)溶解,用乙酸乙酯萃取(50mL x 3),合并有机相,有机相用饱和氯化钠水溶液洗涤(100mL x 1),无水硫酸钠干燥,过滤,滤液减压浓缩得到粗品化合物14-8。MS-ESI计算值[M+H] +196,实测值196。 Compound 14-7 (860mg, 2.91mmol) was dissolved in ethyl acetate (5mL), ethyl acetate hydrochloride (4M, 5mL, 20.0mmol) was added to the reaction solution, and the reaction solution was stirred at 20°C for 2 hours. The reaction solution was concentrated under reduced pressure, the residue was dissolved in saturated aqueous sodium bicarbonate (50 mL), extracted with ethyl acetate (50 mL x 3), the organic phases were combined, and the organic phase was washed with saturated aqueous sodium chloride (100 mL x 1). Dry over sodium sulfate, filter, and concentrate the filtrate under reduced pressure to obtain crude compound 14-8. MS-ESI calculated value [M+H] + 196, found value 196.
第六步step six
将化合物1-8(150mg,0.295mmol),化合物14-8(69.1mg,0.354mmol),碳酸铯(240mg,0.738mmol)和甲烷磺酸(2-叔丁基膦基-2,4 6-三异丙基-1,1-联苯基)(2-氨基-1,1-联苯-2-基)钯(Ⅱ)(23.4mg,29.5umol)溶于N,N-二甲基甲酰胺(5mL)中,反应液在90℃氮气保护下搅拌反应10小时,减压浓缩除去溶剂,粗产物经过薄层层析法分离(5/1石油醚/乙酸乙酯)得到化合物14-9。MS-ESI计算值[M+H] +623实测值623。 Compound 1-8 (150 mg, 0.295 mmol), compound 14-8 (69.1 mg, 0.354 mmol), cesium carbonate (240 mg, 0.738 mmol) and methanesulfonic acid (2-tert-butylphosphino-2,4 6- Triisopropyl-1,1-biphenyl)(2-amino-1,1-biphenyl-2-yl)palladium (II) (23.4mg, 29.5umol) dissolved in N,N-dimethylformaldehyde Amide (5 mL), the reaction solution was stirred at 90°C under nitrogen protection for 10 hours, concentrated under reduced pressure to remove the solvent, and the crude product was separated by thin layer chromatography (5/1 petroleum ether/ethyl acetate) to obtain compound 14-9 . MS-ESI calculated [M+H] + 623 found 623.
第七步step seven
将化合物14-9(44.0mg,56.0umol)溶于四氢呋喃(1.5mL)和水(3mL)中,加入氢氧化钠(6.72mg,0.168mmol),50℃条件下搅拌反应10小时。减压浓缩,剩余物用水(20mL)溶解,用盐酸(1M)调节pH值到4,用乙酸乙酯萃取(20mL x 3),合并有机相,有机相用饱和氯化钠水溶液洗涤(50mL x 1),无水硫 酸钠干燥,过滤,滤液减压浓缩后剩余物经过高效液相色谱法分离(中性,碳酸氢铵体系)(中性,碳酸氢铵体系)得到化合物14。 1H NMR(400MHz,CD 3OD)δ8.00-7.98(m,1H),7.94-7.92(m,2H),7.77-7.75(m,1H),7.25-7.20(m,1H),7.02-7.00(m,1H),6.84-6.81(m,1H),6.71-6.67(m,2H),6.43-6.41(m,1H),4.40-4.36(m,1H),4.08-4.00(m,1H),3.85(s,3H),3.45-3.39(m,5H),3.38-3.23(m,1H),2.54-2.40(m,1H),2.37-2.32(m,2H),2.27-2.18(m,1H),1.90-1.80(m,1H),1.80-1.73(m,1H)。MS-ESI计算值[M+H] +609,实测值609。 Compound 14-9 (44.0mg, 56.0umol) was dissolved in tetrahydrofuran (1.5mL) and water (3mL), sodium hydroxide (6.72mg, 0.168mmol) was added, and the reaction was stirred at 50°C for 10 hours. Concentrate under reduced pressure, dissolve the residue in water (20mL), adjust the pH value to 4 with hydrochloric acid (1M), extract with ethyl acetate (20mL x 3), combine the organic phases, and wash the organic phase with saturated aqueous sodium chloride (50mL x 1), dried over anhydrous sodium sulfate, filtered, the filtrate was concentrated under reduced pressure, and the residue was separated by high performance liquid chromatography (neutral, ammonium bicarbonate system) (neutral, ammonium bicarbonate system) to obtain compound 14. 1 H NMR (400MHz, CD 3 OD) δ8.00-7.98(m,1H),7.94-7.92(m,2H),7.77-7.75(m,1H),7.25-7.20(m,1H),7.02- 7.00(m,1H),6.84-6.81(m,1H),6.71-6.67(m,2H),6.43-6.41(m,1H),4.40-4.36(m,1H),4.08-4.00(m,1H ),3.85(s,3H),3.45-3.39(m,5H),3.38-3.23(m,1H),2.54-2.40(m,1H),2.37-2.32(m,2H),2.27-2.18(m ,1H), 1.90-1.80(m,1H), 1.80-1.73(m,1H). MS-ESI calculated value [M+H] + 609, found value 609.
实施例15Example 15
合成路线:synthetic route:

第一步first step
将化合物15-1(90.2mg,0.433mmol),化合物7-3(200mg,0.413mmol),碳酸铯(336mg,1.03mmol)和甲磺酸(2-环己基膦基-3,6-二甲氧基-2,4,6-三异丙基-1,1-联苯)(2-氨基-1,1-联苯-2-基)钯(Ⅱ)(374mg,0.413mmol)溶于N,N-二甲基甲酰胺(3mL)中,氮气置换三次,反应液在90℃氮气保护下搅拌反应2小时,反应液过滤,滤液减压浓缩除去溶剂,粗产物经过薄层层析法分离(10/1石油醚/乙酸乙酯)得到化合物15-2。MS-ESI计算值[M+H] +612实测值612。 Compound 15-1 (90.2mg, 0.433mmol), compound 7-3 (200mg, 0.413mmol), cesium carbonate (336mg, 1.03mmol) and methanesulfonic acid (2-cyclohexylphosphino-3,6-dimethyl Oxygen-2,4,6-triisopropyl-1,1-biphenyl)(2-amino-1,1-biphenyl-2-yl)palladium (Ⅱ) (374mg, 0.413mmol) dissolved in N , N-dimethylformamide (3mL), nitrogen replacement three times, the reaction solution was stirred and reacted at 90°C under nitrogen protection for 2 hours, the reaction solution was filtered, the filtrate was concentrated under reduced pressure to remove the solvent, and the crude product was separated by thin-layer chromatography. (10/1 petroleum ether/ethyl acetate) to obtain compound 15-2. MS-ESI calculated [M+H] + 612 found 612.
第二步second step
将化合物15-2(5.06mg,53.5umol)溶于四氢呋喃(2mL)和水(4mL)中,加入氢氧化钠(6.4mg,0.161mmol),反应液在50℃条件下搅拌反应10小时。反应液减压浓缩,剩余物用水(5mL)溶解,用盐酸(1M)调节pH值到4,用乙酸乙酯萃取(5mL x 3),合并有机相,有机相用饱和氯化钠水溶液洗涤(10mL x 1),无水硫酸钠干燥,过滤,滤液减压浓缩后剩余物经过高效液相色谱法分离(中性,碳酸氢铵体系)得到化合物15。 1H NMR(400MHz,CD 3OD)δ9.19-9.13(m,1H),8.35-8.34(m,1H),7.98-7.94(m,2H),7.90-7.84(m,3H),7.70-7.66(m,1H),7.61-7.54(m,2H),7.21-7.19(m,1H),6.93-6.89(m,1H),6.84-6.82(m,1H),4.67-4.63(m,2H),4.42-4.39(m,1H),4.15-4.11(m,2H),4.05-3.95(m,1H),3.48-3.44(m,2H),2.42-2.36(m,2H), 1.89-1.84(m,1H),1.81-1.76(m,1H)。MS-ESI计算值[M+H] +598,实测值598。 Compound 15-2 (5.06mg, 53.5umol) was dissolved in tetrahydrofuran (2mL) and water (4mL), sodium hydroxide (6.4mg, 0.161mmol) was added, and the reaction solution was stirred at 50°C for 10 hours. The reaction solution was concentrated under reduced pressure, the residue was dissolved in water (5 mL), the pH was adjusted to 4 with hydrochloric acid (1 M), extracted with ethyl acetate (5 mL x 3), the organic phases were combined, and the organic phase was washed with saturated aqueous sodium chloride ( 10mL x 1), dried over anhydrous sodium sulfate, filtered, the filtrate was concentrated under reduced pressure, and the residue was separated by high performance liquid chromatography (neutral, ammonium bicarbonate system) to obtain compound 15. 1 H NMR (400MHz, CD 3 OD) δ9.19-9.13 (m, 1H), 8.35-8.34 (m, 1H), 7.98-7.94 (m, 2H), 7.90-7.84 (m, 3H), 7.70- 7.66(m,1H),7.61-7.54(m,2H),7.21-7.19(m,1H),6.93-6.89(m,1H),6.84-6.82(m,1H),4.67-4.63(m,2H ),4.42-4.39(m,1H),4.15-4.11(m,2H),4.05-3.95(m,1H),3.48-3.44(m,2H),2.42-2.36(m,2H), 1.89-1.84 (m,1H), 1.81-1.76(m,1H). MS-ESI calculated value [M+H] + 598, found value 598.
实施例16Example 16
合成路线:synthetic route:

第一步first step
氮气保护下将化合物16-1(1.00g,3.26mmol)和化合物16-2(1.01g,3.26mmol)加入乙二醇二甲醚(8mL)和水(2mL)中,再加入碳酸钾(1.35g,9.79mmol)和[1,1′-双(二苯基膦)二茂铁]二氯化钯(II)(239mg,0.326mmol),反应液在90℃下搅拌12小时,向反应液中加入水(40mL),用乙酸乙酯(40mL x 1)萃取,有机相用饱和食盐水洗涤(40mL x 1),无水硫酸钠干燥,过滤,减压浓缩,剩余物用硅胶柱色谱法分离纯化(10/1石油醚/乙酸乙酯)得到化合物16-3。MS-ESI计算值[M-56+H] +306,实测值306。 Under nitrogen protection, compound 16-1 (1.00g, 3.26mmol) and compound 16-2 (1.01g, 3.26mmol) were added to ethylene glycol dimethyl ether (8mL) and water (2mL), and potassium carbonate (1.35 g, 9.79mmol) and [1,1′-bis(diphenylphosphino)ferrocene]palladium(II) dichloride (239mg, 0.326mmol), the reaction solution was stirred at 90°C for 12 hours, and added to the reaction solution Water (40mL) was added to the solution, extracted with ethyl acetate (40mL x 1), the organic phase was washed with saturated brine (40mL x 1), dried over anhydrous sodium sulfate, filtered, concentrated under reduced pressure, and the residue was subjected to silica gel column chromatography Separation and purification (10/1 petroleum ether/ethyl acetate) gave compound 16-3. MS-ESI calculated value [M-56+H] + 306, found value 306.
第二步second step
参照实施例5第二步得到化合物16-4。MS-ESI计算值[M+H] +262,实测值262。 Referring to the second step of Example 5, compound 16-4 was obtained. MS-ESI calculated value [M+H] + 262, found value 262.
第三步third step
氮气保护下,将化合物2-5(185mg,0.391mmol)溶于无水二氯甲烷(10mL)中,加入化合物16-4(123mg,0.469mmol),吡啶N-氧化物(112mg,1.17mmol),吡啶(92.8mg,1.17mmol)和乙酸铜(142mg,0.782mmol),反应液在氧气(15Psi)氛围下36℃下搅拌36小时,反应液减压浓缩,剩余物用硅胶柱色谱法分离纯化(3/1石油醚/乙酸乙酯),得到化合物16-5。MS-ESI计算值[M+H] +689,实测值689。 Under nitrogen protection, compound 2-5 (185mg, 0.391mmol) was dissolved in anhydrous dichloromethane (10mL), compound 16-4 (123mg, 0.469mmol), pyridine N-oxide (112mg, 1.17mmol) were added , pyridine (92.8mg, 1.17mmol) and copper acetate (142mg, 0.782mmol), the reaction solution was stirred at 36°C for 36 hours under an oxygen (15Psi) atmosphere, the reaction solution was concentrated under reduced pressure, and the residue was separated and purified by silica gel column chromatography (3/1 petroleum ether/ethyl acetate), to obtain compound 16-5. MS-ESI calculated [M+H] + 689, found 689.
第四步the fourth step
参照实施例1第十步得到化合物16。 1H NMR(400MHz,CD 3OD)δ8.01-7.95(m,3H),7.80-7.76(m,1H),7.74-7.71(m,2H),7.51-7.47(m,2H),6.91-6.88(m,1H),6.78(d,J=9.2Hz,1H),5.80-5.72(m,1H),4.45-4.40m,1H),3.82-3.81(m,2H),3.47-3.45(m,2H),3.39-3.36(m,1H),3.28-3.22(m,1H),2.68-2.64(m,1H),2.49-2.35(m,3H),1.93-1.73(m,2H)。MS-ESI计算值[M+H] +675,实测值675。 Referring to the tenth step of Example 1, compound 16 was obtained. 1 H NMR (400MHz, CD 3 OD) δ8.01-7.95 (m, 3H), 7.80-7.76 (m, 1H), 7.74-7.71 (m, 2H), 7.51-7.47 (m, 2H), 6.91- 6.88(m,1H),6.78(d,J=9.2Hz,1H),5.80-5.72(m,1H),4.45-4.40m,1H),3.82-3.81(m,2H),3.47-3.45(m ,2H), 3.39-3.36(m,1H), 3.28-3.22(m,1H), 2.68-2.64(m,1H), 2.49-2.35(m,3H), 1.93-1.73(m,2H). MS-ESI calculated [M+H] +675 , found 675.
实施例17Example 17
合成路线:synthetic route:

第一步first step
氮气保护下将化合物17-1(1.00g,4.43mmol)和化合物16-2(1.44g,4.66mmol)加入无水1,4-二氧六环(16mL)和水(4mL)中,再加入磷酸钾(1.88g,8.87mmol)和[1,1′-双(二苯基膦)二茂铁]二氯化钯(II)(324mg,0.443mmol),反应液在90℃下搅拌12小时,向反应液中加入水(80mL),用乙酸乙酯(100mL x1)萃取,有机相用饱和食盐水洗涤(100mL x 1),无水硫酸钠干燥,过滤,减压浓缩,剩余物用硅胶柱色谱法分离纯化(5/1石油醚/乙酸乙酯)得到化合物17-2。MS-ESI计算值[M-56+H] +317,实测值317。 Under nitrogen protection, compound 17-1 (1.00g, 4.43mmol) and compound 16-2 (1.44g, 4.66mmol) were added to anhydrous 1,4-dioxane (16mL) and water (4mL), and then added Potassium phosphate (1.88g, 8.87mmol) and [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium (II) (324mg, 0.443mmol), the reaction solution was stirred at 90°C for 12 hours , water (80 mL) was added to the reaction solution, extracted with ethyl acetate (100 mL x 1), the organic phase was washed with saturated brine (100 mL x 1), dried over anhydrous sodium sulfate, filtered, concentrated under reduced pressure, and the residue was washed with silica gel Separation and purification by column chromatography (5/1 petroleum ether/ethyl acetate) gave compound 17-2. MS-ESI calculated value [M-56+H] + 317, found value 317.
第二步second step
氮气保护下将化合物17-2(1.10g,2.95mmol)溶于无水乙醇(10mL)中,加入10%钯碳(1.10g),反应液在氢气(50Psi)氛围下60℃下搅拌12小时,将反应液过滤除去钯碳,母液浓缩得到化合物17-3。MS-ESI计算值[M-56+H] +289,实测值289。 Dissolve compound 17-2 (1.10g, 2.95mmol) in absolute ethanol (10mL) under nitrogen protection, add 10% palladium carbon (1.10g), and stir the reaction solution at 60°C for 12 hours under hydrogen (50Psi) atmosphere , the reaction solution was filtered to remove palladium carbon, and the mother liquor was concentrated to obtain compound 17-3. MS-ESI calculated value [M-56+H] + 289, found value 289.
第三步third step
氮气保护下将化合物17-3(100mg,0.290mmol)溶于浓盐酸(2mL)中,-5℃再滴加溶于水(2mL)的氯化亚铜(35.9mg,0.363mmol)和亚硝酸钠(52.1mg,0.755mmol)溶液,反应液在25℃下搅拌12小时,用10%氢氧化钠水溶液调节pH>7,有大量固体析出,过滤,滤液用乙酸乙酯(20mL x 1)萃取,无水硫酸钠干燥,过滤,减压浓缩,剩余物用薄层层析法分离纯化(10/1二氯甲烷/甲醇)得到化合物17-4。MS-ESI计算值[M+H] +264,实测值264。 Compound 17-3 (100mg, 0.290mmol) was dissolved in concentrated hydrochloric acid (2mL) under nitrogen protection, and cuprous chloride (35.9mg, 0.363mmol) and nitrous acid dissolved in water (2mL) were added dropwise at -5°C Sodium (52.1mg, 0.755mmol) solution, the reaction solution was stirred at 25°C for 12 hours, adjusted to pH>7 with 10% aqueous sodium hydroxide solution, a large amount of solid precipitated, filtered, and the filtrate was extracted with ethyl acetate (20mL x 1) , dried over anhydrous sodium sulfate, filtered, concentrated under reduced pressure, and the residue was separated and purified by thin-layer chromatography (10/1 dichloromethane/methanol) to obtain compound 17-4. MS-ESI calculated [M+H] + 264, found 264.
第四步the fourth step
参照实施例16第三步得到化合物17-5。MS-ESI计算值[M+H] +691,实测值691。 Referring to the third step of Example 16, compound 17-5 was obtained. MS-ESI calculated [M+H] + 691, found 691.
第五步the fifth step
参照实施例1第十步得到化合物17。 1H NMR(400MHz,CD 3OD)δ8.02-7.97(m,2H),7.93(s,1H),7.82-7.80(m,1H),7.72(d,J=8.0Hz,1H),7.67(d,J=8.0Hz,1H),7.49-7.45(br s,1H),7.43-7.39(m,1H),6.89-6.87(m,1H),6.77-6.75(m,1H),4.65-4.61(m,1H),4.43-4.40(m,1H),3.73-3.70(m,2H),3.40-3.37(m,1H),3.29-3.25(m,1H),3.01-2.93(m,2H),2.79-2.73(m,2H),2.60-2.40(m,2H),1.88-1.80(m,2H),1.71-1.68(m,2H)。MS-ESI计算值[M+H] +678,实测值678。 Referring to the tenth step of Example 1, compound 17 was obtained. 1 H NMR (400MHz, CD 3 OD) δ8.02-7.97 (m, 2H), 7.93 (s, 1H), 7.82-7.80 (m, 1H), 7.72 (d, J = 8.0Hz, 1H), 7.67 (d,J=8.0Hz,1H),7.49-7.45(br s,1H),7.43-7.39(m,1H),6.89-6.87(m,1H),6.77-6.75(m,1H),4.65- 4.61(m,1H),4.43-4.40(m,1H),3.73-3.70(m,2H),3.40-3.37(m,1H),3.29-3.25(m,1H),3.01-2.93(m,2H ), 2.79-2.73(m,2H), 2.60-2.40(m,2H), 1.88-1.80(m,2H), 1.71-1.68(m,2H). MS-ESI calculated [M+H] + 678, found 678.
实施例18Example 18
合成路线:synthetic route:

第一步first step
氮气保护下将化合物18-1(100mg,0.526mmol)和化合物14-4(155mg,0.526mmol)加入无水1,4-二氧六环(4mL)和水(1mL)中,再加入乙酸钾(103mg,1.05mmol)和[1,1′-双(二苯基膦)二茂铁]二氯化钯(II)(38.5mg,0.053mmol),反应液在90℃下搅拌12小时,向反应液中加入水(20mL),用乙酸乙酯(20mL x1)萃取,有机相用饱和食盐水洗涤(20mL x 1),无水硫酸钠干燥,过滤,减压浓缩,剩余物用硅胶柱色谱法分离纯化(2/1石油醚/乙酸乙酯)得到化合物18-2。 1H NMR(400MHz,CDCl 3)δ7.06-6.97(m,1H),6.53-6.42(m,2H),5.96-5.92(m,1H),4.40-4.25(m,4H),4.08-3.92(m,2H),1.50-1.49(m,9H)。 Under nitrogen protection, compound 18-1 (100mg, 0.526mmol) and compound 14-4 (155mg, 0.526mmol) were added to anhydrous 1,4-dioxane (4mL) and water (1mL), and then potassium acetate was added (103mg, 1.05mmol) and [1,1′-bis(diphenylphosphino)ferrocene]palladium(II) dichloride (38.5mg, 0.053mmol), the reaction solution was stirred at 90°C for 12 hours, and Water (20 mL) was added to the reaction solution, extracted with ethyl acetate (20 mL x 1), the organic phase was washed with saturated brine (20 mL x 1), dried over anhydrous sodium sulfate, filtered, concentrated under reduced pressure, and the residue was subjected to silica gel column chromatography Compound 18-2 was obtained by separation and purification (2/1 petroleum ether/ethyl acetate). 1 H NMR (400MHz, CDCl 3 )δ7.06-6.97(m,1H),6.53-6.42(m,2H),5.96-5.92(m,1H),4.40-4.25(m,4H),4.08-3.92 (m,2H),1.50-1.49(m,9H).
第二步second step
氮气保护下将化合物18-2(398mg,1.43mmol)溶于乙酸乙酯(10mL)中,加入10%钯碳(40.0mg),反应液在氢气(15PsiI)氛围下50℃下搅拌12小时,将反应液过滤除去钯碳,母液浓缩,剩余物用硅胶柱色谱法分离纯化(2/1石油醚/乙酸乙酯),得到化合物18-3。MS-ESI计算值[M-56+H] +225,实测值225。 Compound 18-2 (398mg, 1.43mmol) was dissolved in ethyl acetate (10mL) under nitrogen protection, 10% palladium carbon (40.0mg) was added, and the reaction solution was stirred at 50°C for 12 hours under a hydrogen (15PsiI) atmosphere. The reaction solution was filtered to remove palladium carbon, the mother liquor was concentrated, and the residue was separated and purified by silica gel column chromatography (2/1 petroleum ether/ethyl acetate) to obtain compound 18-3. MS-ESI calculated value [M-56+H] + 225, found value 225.
第三步third step
氮气保护下将化合物18-3(180mg,0.642mmol)溶于浓盐酸(3mL)中,-5℃再滴加溶于水(2mL)的亚硝酸钠溶液(115mg,1.67mmol),反应液在-5℃下搅拌0.5小时,在加入氯化亚铜(318mg,3.21mmol),反应液在25℃下搅拌1.5小时,用10%氢氧化钠水溶液调节pH>7,有大量固体析出,过滤,滤液用二氯甲烷/甲醇=10/1(20mL x 4)萃取,合并有机相,无水硫酸钠干燥,过滤,减压浓缩,剩余物用薄层层析法分离纯化(10/1二氯甲烷/甲醇)得到化合物18-4。MS-ESI计算值[M+H] +200,实测值200。 Compound 18-3 (180mg, 0.642mmol) was dissolved in concentrated hydrochloric acid (3mL) under the protection of nitrogen, and sodium nitrite solution (115mg, 1.67mmol) dissolved in water (2mL) was added dropwise at -5°C. Stirring at -5°C for 0.5 hours, adding cuprous chloride (318 mg, 3.21 mmol), and stirring the reaction solution at 25°C for 1.5 hours, adjusting pH>7 with 10% aqueous sodium hydroxide solution, a large amount of solids precipitated, filtered, The filtrate was extracted with dichloromethane/methanol=10/1 (20mL x 4), the organic phases were combined, dried over anhydrous sodium sulfate, filtered, concentrated under reduced pressure, and the residue was separated and purified by thin-layer chromatography (10/1 dichloro methane/methanol) to give compound 18-4. MS-ESI calculated value [M+H] + 200, measured value 200.
第四步the fourth step
参照实施例16第三步得到化合物18-5。MS-ESI计算值[M+H] +627,实测值627。 Referring to the third step of Example 16, compound 18-5 was obtained. MS-ESI calculated [M+H] + 627, found 627.
第五步the fifth step
参照实施例1第十步得到化合物18。 1H NMR(400MHz,CD 3OD)δ8.00-7.98(m,1H),7.94-7.93(m,2H),7.77-7.73(m,1H),7.30-7.24(m,2H),7.13-7.08(m,1H),7.04-7.02(m,1H),6.69(d,J=8.8Hz,1H),6.46-6.43(m,1H),4.40-4.36(m,1H),4.24-4.15(m,1H),3.55-3.39(m,5H),3.26-3.19(m,1H),2.55-2.34(m,4H),1.84-1.73(m,2H)。MS-ESI计算值[M+H] +614,实测值614。 Referring to the tenth step of Example 1, compound 18 was obtained. 1 H NMR (400MHz, CD 3 OD) δ8.00-7.98(m,1H),7.94-7.93(m,2H),7.77-7.73(m,1H),7.30-7.24(m,2H),7.13- 7.08(m,1H),7.04-7.02(m,1H),6.69(d,J=8.8Hz,1H),6.46-6.43(m,1H),4.40-4.36(m,1H),4.24-4.15( m, 1H), 3.55-3.39 (m, 5H), 3.26-3.19 (m, 1H), 2.55-2.34 (m, 4H), 1.84-1.73 (m, 2H). MS-ESI calculated [M+H] + 614, found 614.
实施例19Example 19
合成路线:synthetic route:

第一步first step
参照实施例17第一步得到化合物19-1。MS-ESI计算值[M-56+H] +541,实测值541。 Referring to the first step of Example 17, compound 19-1 was obtained. MS-ESI calculated value [M-56+H] + 541, found value 541.
第二步second step
氮气保护下将化合物19-1(1.10g,1.84mmol)溶于乙酸乙酯(10mL)中,加入10%钯碳(100mg),反应液在氢气(15Psi)氛围下50℃下搅拌12小时,将反应液过滤除去钯碳,母液浓缩得到化合物19-2。MS-ESI计算值[M-56+H] +543,实测值543。 Compound 19-1 (1.10 g, 1.84 mmol) was dissolved in ethyl acetate (10 mL) under nitrogen protection, 10% palladium on carbon (100 mg) was added, and the reaction solution was stirred at 50° C. for 12 hours under a hydrogen (15 Psi) atmosphere. The reaction solution was filtered to remove palladium carbon, and the mother liquor was concentrated to obtain compound 19-2. MS-ESI calculated value [M-56+H] + 543, found value 543.
第三步third step
氮气保护下,将化合物19-2(1.10g,1.84mmol)溶于乙酸乙酯(5mL)中,再加入盐酸乙酸乙酯(4M,5mL,20.0mmol),反应液在25℃下搅拌2小时,将反应液旋干,向反应液中加入20mL饱和碳酸氢钠水溶液,用乙酸乙酯(20mL x 1)萃取,饱和食盐水(20mL x 1)洗涤有机相,无水硫酸钠干燥,过滤,减压浓缩得到化合物19-3。MS-ESI计算值[M+H] +499,实测值499。 Under nitrogen protection, compound 19-2 (1.10g, 1.84mmol) was dissolved in ethyl acetate (5mL), then ethyl acetate hydrochloride (4M, 5mL, 20.0mmol) was added, and the reaction solution was stirred at 25°C for 2 hours , spin the reaction solution to dryness, add 20mL saturated aqueous sodium bicarbonate solution to the reaction solution, extract with ethyl acetate (20mL x 1), wash the organic phase with saturated brine (20mL x 1), dry over anhydrous sodium sulfate, filter, Concentration under reduced pressure gave compound 19-3. MS-ESI calculated value [M+H] + 499, found value 499.
第四步the fourth step
参照实施例16第三步得到化合物19-4。MS-ESI计算值[M+H] +605,实测值605。 Referring to the third step of Example 16, compound 19-4 was obtained. MS-ESI calculated value [M+H] + 605, found value 605.
第五步the fifth step
参照实施例1第十步得到化合物19。 1H NMR(400MHz,CD 3OD)δ7.98(d,J=8.0Hz,1H),7.93-7.91m,1H), 7.83(s,1H),7.79-7.78(m,1H),7.69(q,J=8.0Hz,1H),7.07(d,J=8.2Hz,1H),6.94-6.91(m,1H),6.88-6.82(m,3H),6.78-6.75(m,1H),4.43-4.39(m,1H),3.82(d,J=1.2Hz,3H),3.61-3.53(m,2H),3.46-3.37(m,4H),3.26-3.19(m,1H),2.46-2.33(m,3H),2.05-1.98(m,1H),1.91-1.72(m,2H)。MS-ESI计算值[M+H] +591,实测值591。 Referring to the tenth step of Example 1, compound 19 was obtained. 1 H NMR (400MHz, CD 3 OD) δ7.98(d, J=8.0Hz, 1H), 7.93-7.91m, 1H), 7.83(s, 1H), 7.79-7.78(m, 1H), 7.69( q,J=8.0Hz,1H),7.07(d,J=8.2Hz,1H),6.94-6.91(m,1H),6.88-6.82(m,3H),6.78-6.75(m,1H),4.43 -4.39(m,1H),3.82(d,J=1.2Hz,3H),3.61-3.53(m,2H),3.46-3.37(m,4H),3.26-3.19(m,1H),2.46-2.33 (m,3H), 2.05-1.98(m,1H), 1.91-1.72(m,2H). MS-ESI calculated [M+H] + 591, found 591.
实施例20Example 20
合成路线:synthetic route:

第一步first step
参照实施例18第一步得到化合物20-2。MS-ESI计算值[M-56+H] +220,实测值220。 Referring to the first step of Example 18, compound 20-2 was obtained. MS-ESI calculated value [M-56+H] + 220, measured value 220.
第二步second step
参照实施例18第二步得到化合物20-3。MS-ESI计算值[M-56+H] +222,实测值222。 Referring to the second step of Example 18, compound 20-3 was obtained. MS-ESI calculated value [M-56+H] + 222, found value 222.
第三步third step
氮气保护下,将化合物20-3(150mg,0.541mmol)溶于乙酸乙酯(5mL)中,再加入盐酸乙酸乙酯(4M,5mL,20.0mmol),反应液在25℃下搅拌2小时,将反应液旋干,向剩余物中加入20mL饱和碳酸钠水溶液,用混合溶剂(二氯甲烷:甲醇=5:1)(20mL x 3)萃取,饱和食盐水(20mL x 1)洗涤有机相,无水硫酸钠干燥,过滤,减压浓缩得到化合物20-4。MS-ESI计算值[M+H] +178,实测值178。 Under nitrogen protection, compound 20-3 (150mg, 0.541mmol) was dissolved in ethyl acetate (5mL), then ethyl acetate hydrochloride (4M, 5mL, 20.0mmol) was added, and the reaction solution was stirred at 25°C for 2 hours. The reaction solution was spin-dried, and 20 mL of saturated aqueous sodium carbonate solution was added to the residue, extracted with a mixed solvent (dichloromethane:methanol=5:1) (20 mL x 3), and the organic phase was washed with saturated brine (20 mL x 1). Dry over anhydrous sodium sulfate, filter, and concentrate under reduced pressure to obtain compound 20-4. MS-ESI calculated value [M+H] + 178, found value 178.
第四步the fourth step
参照实施例16第三步得到化合物20-5。MS-ESI计算值[M+H] +605,实测值605。 Referring to the third step of Example 16, compound 20-5 was obtained. MS-ESI calculated value [M+H] + 605, found value 605.
第五步the fifth step
参照实施例1第十步得到化合物20。 1H NMR(400MHz,CD 3OD)δ8.00(d,J=8.0Hz,1H),7.96-7.95(m,2H),7.78-7.74(m,1H),7.25-7.20(m,2H),7.03-6.98(m,2H),6.94-6.90(m,1H),6.70(d,J=9.2Hz,1H),6.45-6.42(m,1H),4.41-4.37(m,1H),3.88(s,3H),3.85-3.83(m,1H),3.66-3.61(m,1H),3.42-3.36(m,3H),3.27-3.21(m,2H),2.47-2.35(m,3H),2.23-2.12(m,1H),1.91-1.73(m,2H)。MS-ESI计算值[M+H] +591,实测值591。 Referring to the tenth step of Example 1, compound 20 was obtained. 1 H NMR (400MHz, CD 3 OD) δ8.00 (d, J = 8.0Hz, 1H), 7.96-7.95 (m, 2H), 7.78-7.74 (m, 1H), 7.25-7.20 (m, 2H) ,7.03-6.98(m,2H),6.94-6.90(m,1H),6.70(d,J=9.2Hz,1H),6.45-6.42(m,1H),4.41-4.37(m,1H),3.88 (s,3H),3.85-3.83(m,1H),3.66-3.61(m,1H),3.42-3.36(m,3H),3.27-3.21(m,2H),2.47-2.35(m,3H) ,2.23-2.12(m,1H),1.91-1.73(m,2H). MS-ESI calculated [M+H] + 591, found 591.
实施例21Example 21
合成路线:synthetic route:

第一步first step
将化合物7-3(110mg,0.196mmol),21-1(60.2mg,0.196mmol),碳酸铯(128mg,0.393mmol),4,5-双二苯基膦-9,9-二甲基氧杂蒽(11.4mg,0.0196mmol)和三(二亚苄基丙酮)二钯(9.0mg,9.82nmol)加入到N,N-二甲基甲酰胺(3mL)中。反应液在氮气保护下100℃搅拌反应12小时。向反应液中加入水(10mL),用乙酸乙酯(20mL x 3)萃取。合并有机相,用饱和食盐水洗涤(20mL x 3),无水硫酸钠干燥,过滤,滤液减压浓缩。剩余物用制备薄层层析法分离纯化(3/1石油醚/乙酸乙酯)得到化合物21-2。MS-ESI计算值[M+H] +663,实测值663。 Compound 7-3 (110mg, 0.196mmol), 21-1 (60.2mg, 0.196mmol), cesium carbonate (128mg, 0.393mmol), 4,5-bisdiphenylphosphine-9,9-dimethyloxy Xanthene (11.4 mg, 0.0196 mmol) and tris(dibenzylideneacetone)dipalladium (9.0 mg, 9.82 nmol) were added to N,N-dimethylformamide (3 mL). The reaction solution was stirred and reacted at 100° C. for 12 hours under the protection of nitrogen. Water (10 mL) was added to the reaction liquid, and extracted with ethyl acetate (20 mL x 3). The organic phases were combined, washed with saturated brine (20 mL x 3), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was separated and purified by preparative thin layer chromatography (3/1 petroleum ether/ethyl acetate) to obtain compound 21-2. MS-ESI calculated [M+H] + 663, found 663.
第二步second step
将化合物21-2(50.0mg,57.5nmol)和一水合氢氧化锂(4.8mg,11.5nmol)加入到四氢呋喃(2mL)和水(0.5mL)中。反应液在氮气保护下20℃搅拌反应12小时。反应液减压浓缩。剩余物经高效液相色谱法分离(中性,碳酸氢铵体系)纯化得到化合物21。 1H NMR(400MHz,CD 3OD)δ8.01-7.98(m,1H),7.94-7.92(m,2H),7.87-7.85(m,1H),7.76-7.73(m,1H),7.58-7.56(m,2H),7.23-7.20(m,1H),7.11-7.09(m,1H),6.81(d,J=8.8Hz,1H),4.65-4.61(m,2H),4.44-4.40(m,1H),4.29-4.25(m,2H),3.87-3.85(m,1H),3.48-3.46(m,1H),3.27-3.21(m,1H),2.41-2.35(m,2H),1.87-1.79(m,2H)。MS-ESI计算值[M+H] +649,实测值649。 Compound 21-2 (50.0 mg, 57.5 nmol) and lithium hydroxide monohydrate (4.8 mg, 11.5 nmol) were added to tetrahydrofuran (2 mL) and water (0.5 mL). The reaction solution was stirred and reacted at 20° C. for 12 hours under the protection of nitrogen. The reaction solution was concentrated under reduced pressure. The residue was separated and purified by high performance liquid chromatography (neutral, ammonium bicarbonate system) to obtain compound 21. 1 H NMR (400MHz, CD 3 OD) δ8.01-7.98(m,1H),7.94-7.92(m,2H),7.87-7.85(m,1H),7.76-7.73(m,1H),7.58- 7.56(m,2H),7.23-7.20(m,1H),7.11-7.09(m,1H),6.81(d,J=8.8Hz,1H),4.65-4.61(m,2H),4.44-4.40( m,1H),4.29-4.25(m,2H),3.87-3.85(m,1H),3.48-3.46(m,1H),3.27-3.21(m,1H),2.41-2.35(m,2H), 1.87-1.79 (m, 2H). MS-ESI calculated [M+H] + 649, found 649.
实施例22Example 22
合成路线:synthetic route:

第一步first step
参照实施例21第一步得到化合物22-1。MS-ESI计算值[M+H] +591,实测值591。 Referring to the first step of Example 21, compound 22-1 was obtained. MS-ESI calculated [M+H] + 591, found 591.
第二步second step
参照实施例21第二步得到化合物22。 1H NMR(400MHz,CD 3OD)δ7.98-7.96(m,1H),7.90-7.88(m,1H),7.85-7.84(m,1H),7.76-7.75(m,1H),7.70-7.68(m,1H),7.10-7.09(m,1H),6.89-6.78(m,4H),6.59-6.58(m,1H),4.42-4.41(m,1H),4.38-4.30(m,2H),3.81-3.78(m,6H),3.48-3.46(m,1H),3.27-3.21(m,1H),2.41-2.35(m,2H),1.86-1.77(m,2H)。MS-ESI计算值[M+H] +577,实测值577。 Referring to the second step of Example 21, Compound 22 was obtained. 1 H NMR (400MHz, CD 3 OD) δ7.98-7.96(m,1H),7.90-7.88(m,1H),7.85-7.84(m,1H),7.76-7.75(m,1H),7.70- 7.68(m,1H),7.10-7.09(m,1H),6.89-6.78(m,4H),6.59-6.58(m,1H),4.42-4.41(m,1H),4.38-4.30(m,2H ), 3.81-3.78(m,6H), 3.48-3.46(m,1H), 3.27-3.21(m,1H), 2.41-2.35(m,2H), 1.86-1.77(m,2H). MS-ESI calculated [M+H] + 577, found 577.
实施例23Example 23
合成路线:synthetic route:

第一步first step
参照实施例21第一步得到化合物23-1。MS-ESI计算值[M+H] +613,实测值613。 Referring to the first step of Example 21, compound 23-1 was obtained. MS-ESI calculated [M+H] + 613, found 613.
第二步second step
参照实施例21第二步得到化合物23。 1H NMR(400MHz,CD 3OD)δ8.01-7.99(m,1H),7.91-7.89(m,1H),7.85-7.84(m,1H),7.82-7.81(m,1H),7.74-7.71(m,1H),7.15-7.12(m,1H),7.05-7.03(m,1H),6.98-6.95(m,1H),6.80(d,J=8.8Hz,1H),6.71-6.70(m,1H),4.73-4.71(m,2H),4.43-4.39(m,1H),4.23-4.21(m,2H),3.79-3.76(m,1H),3.50-3.47(m,1H),3.28-3.22(m,1H),2.41-2.34(m,2H),1.87-1.78(m,2H)。MS-ESI计算值[M+H] +599,实测值599。 Referring to the second step of Example 21, compound 23 was obtained. 1 H NMR (400MHz, CD 3 OD) δ8.01-7.99(m,1H),7.91-7.89(m,1H),7.85-7.84(m,1H),7.82-7.81(m,1H),7.74- 7.71(m,1H),7.15-7.12(m,1H),7.05-7.03(m,1H),6.98-6.95(m,1H),6.80(d,J=8.8Hz,1H),6.71-6.70( m,1H),4.73-4.71(m,2H),4.43-4.39(m,1H),4.23-4.21(m,2H),3.79-3.76(m,1H),3.50-3.47(m,1H), 3.28-3.22 (m, 1H), 2.41-2.34 (m, 2H), 1.87-1.78 (m, 2H). MS-ESI calculated value [M+H] + 599, found value 599.
实施例24Example 24
合成路线:synthetic route:

第一步first step
将化合物24-1(100mg,0.393mmol)溶于乙腈(5mL)中,加入碳酸钾(109mg,0.786mmol)和碘甲烷(112mg,0.786mmol)。反应液在80℃搅拌反应12小时。向反应液加入水(10mL),用二氯甲烷(10mL x 3)萃取。合并有机相,用饱和食盐水洗涤(10mL x 1),无水硫酸钠干燥,过滤,滤液减压浓缩。剩余物用硅胶柱色谱法分离纯化(5/1石油醚/乙酸乙酯)得到化合物24-2。 1H NMR(400MHz,CDCl 3)δ7.31-7.29(m,1H),7.14(d,J=8.8Hz,1H),6.74(d,J=8.8Hz,1H),3.94(s,3H)。 Compound 24-1 (100 mg, 0.393 mmol) was dissolved in acetonitrile (5 mL), potassium carbonate (109 mg, 0.786 mmol) and iodomethane (112 mg, 0.786 mmol) were added. The reaction solution was stirred and reacted at 80° C. for 12 hours. Water (10 mL) was added to the reaction solution, and extracted with dichloromethane (10 mL x 3). The organic phases were combined, washed with saturated brine (10 mL x 1), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was separated and purified by silica gel column chromatography (5/1 petroleum ether/ethyl acetate) to obtain compound 24-2. 1 H NMR (400MHz, CDCl 3 ) δ7.31-7.29 (m, 1H), 7.14 (d, J = 8.8Hz, 1H), 6.74 (d, J = 8.8Hz, 1H), 3.94 (s, 3H) .
第二步second step
参照实施例21第一步得到化合物24-3。MS-ESI计算值[M+H] +625,实测值625。 Referring to the first step of Example 21, compound 24-3 was obtained. MS-ESI calculated [M+H] + 625, found 625.
第三步third step
参照实施例21第二步得到化合物24。 1H NMR(400MHz,CD 3OD)δ8.00-7.99(m,1H),7.98-7.96(m,1H),7.84-7.80(m,2H),7.70-7.68(m,1H),7.12-7.11(m,1H),6.87-6.83(m,2H),6.80(d,J=8.8Hz,1H),6.75-6.73(m,1H),4.71-4.67(m,2H),4.43-4.39(m,1H),4.21-4.16(m,2H),3.74(m,3H),3.70-3.68(m,1H),3.49-3.47(m,1H),3.25-3.21(m,1H),2.42-2.36(m,2H),1.86-1.73(m,2H)。MS-ESI计算值[M+H] +611,实测值611。 Referring to the second step of Example 21, compound 24 was obtained. 1 H NMR (400MHz, CD 3 OD) δ8.00-7.99 (m, 1H), 7.98-7.96 (m, 1H), 7.84-7.80 (m, 2H), 7.70-7.68 (m, 1H), 7.12- 7.11(m,1H),6.87-6.83(m,2H),6.80(d,J=8.8Hz,1H),6.75-6.73(m,1H),4.71-4.67(m,2H),4.43-4.39( m,1H),4.21-4.16(m,2H),3.74(m,3H),3.70-3.68(m,1H),3.49-3.47(m,1H),3.25-3.21(m,1H),2.42- 2.36(m,2H),1.86-1.73(m,2H). MS-ESI calculated value [M+H] + 611, found value 611.
实施例25Example 25
合成路线:synthetic route:

第一步first step
将化合物24-1(500mg,1.97mmol)溶于N,N-二甲基甲酰胺(4.5mL)和水(0.5mL)中,加入25-1(899mg,5.90mmol)和碳酸铯(1.28g,3.93mmol)。反应液在100℃搅拌反应12小时。向反应液加入水(10mL),用乙酸乙酯(10mL x 3)萃取。合并有机相,用饱和食盐水洗涤(10mL x 1),无水硫酸钠干燥,过滤,滤液减压浓缩。剩余物用硅胶柱色谱法分离纯化(5/1石油醚/乙酸乙酯)得到化合物25-2。 1H NMR(400MHz,CDCl 3)δ7.37-7.28(m,2H),7.05(d,J=8.8Hz,1H),6.72(s,0.25H),6.54(s,0.5H),6.36(s,0.25H)。 Compound 24-1 (500mg, 1.97mmol) was dissolved in N,N-dimethylformamide (4.5mL) and water (0.5mL), and 25-1 (899mg, 5.90mmol) and cesium carbonate (1.28g ,3.93mmol). The reaction solution was stirred and reacted at 100° C. for 12 hours. Water (10 mL) was added to the reaction solution, and extracted with ethyl acetate (10 mL x 3). The organic phases were combined, washed with saturated brine (10 mL x 1), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was separated and purified by silica gel column chromatography (5/1 petroleum ether/ethyl acetate) to obtain compound 25-2. 1 H NMR (400MHz, CDCl 3 ) δ7.37-7.28 (m, 2H), 7.05 (d, J=8.8Hz, 1H), 6.72 (s, 0.25H), 6.54 (s, 0.5H), 6.36 ( s,0.25H).
第二步second step
将化合物7-3(150mg,0.310mmol),25-2(113mg,0.372mmol),碳酸铯(202mg,0.619mmol),4,5-双二苯基膦-9,9-二甲基氧杂蒽(17.9mg,31.0nmol)和三(二亚苄基丙酮)二钯(14.2mg,15.5nmol)加入到1,4-二氧六环(3mL)中。反应液在氮气保护下80℃搅拌反应12小时。向反应液中加入水(10mL),用乙酸乙酯(10mL x 3)萃取。合并有机相,用饱和食盐水洗涤(10mL x 3),无水硫酸钠干燥,过滤,滤液减压浓缩。剩余物用制备薄层层析法分离纯化(4/1石油醚/乙酸乙酯)得到化合物25-3。MS-ESI计算值[M+H] +661,实测值661。 Compound 7-3 (150mg, 0.310mmol), 25-2 (113mg, 0.372mmol), cesium carbonate (202mg, 0.619mmol), 4,5-bisdiphenylphosphine-9,9-dimethyloxa Anthracene (17.9 mg, 31.0 nmol) and tris(dibenzylideneacetone)dipalladium (14.2 mg, 15.5 nmol) were added to 1,4-dioxane (3 mL). The reaction solution was stirred and reacted at 80° C. for 12 hours under the protection of nitrogen. Water (10 mL) was added to the reaction solution, and extracted with ethyl acetate (10 mL x 3). The organic phases were combined, washed with saturated brine (10 mL x 3), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was separated and purified by preparative thin layer chromatography (4/1 petroleum ether/ethyl acetate) to obtain compound 25-3. MS-ESI calculated [M+H] + 661, found 661.
第三步third step
参照实施例21第二步得到化合物25。 1H NMR(400MHz,CD 3OD)δ8.00-7.98(m,1H),7.93-7.91(m,1H),7.85-7.84(m,2H),7.74-7.70(m,1H),7.15-7.11(m,2H),7.01-6.99(m,1H),6.84(s,0.25H),6.80(d,J=8.0Hz,1H),6.74-6.70(m,1H),6.66(s,0.5H),6.47(s,0.25H),4.78-4.73(m,2H),4.44-4.40(m,1H),4.29-4.24(m,2H),3.73-3.69(m,1H),3.48-3.43(m,1H),3.26-3.20(m,1H),2.39-2.31(m,2H),1.89-1.77(m,2H)。MS-ESI计算值[M+H] +647,实测值647。 Referring to the second step of Example 21, Compound 25 was obtained. 1 H NMR (400MHz, CD 3 OD) δ8.00-7.98(m,1H),7.93-7.91(m,1H),7.85-7.84(m,2H),7.74-7.70(m,1H),7.15- 7.11(m,2H),7.01-6.99(m,1H),6.84(s,0.25H),6.80(d,J=8.0Hz,1H),6.74-6.70(m,1H),6.66(s,0.5 H),6.47(s,0.25H),4.78-4.73(m,2H),4.44-4.40(m,1H),4.29-4.24(m,2H),3.73-3.69(m,1H),3.48-3.43 (m,1H), 3.26-3.20(m,1H), 2.39-2.31(m,2H), 1.89-1.77(m,2H). MS-ESI calculated [M+H] + 647, found 647.
实施例26Example 26
合成路线:synthetic route:

第一步first step
将化合物26-1(500mg,2.54mmol)溶于无水四氢呋喃(5mL)中,在-78℃下滴加正丁基锂(2.5M正庚烷溶液,1.02mL,2.54mmol),在该温度下搅拌反应30分钟。然后向反应液中滴加碘(646mg,2.54mmol)溶于无水四氢呋喃(5mL)中,反应液在20℃下搅拌反应12小时。向反应液加入水(10mL),用乙酸乙酯(10mL x 3)萃取。合并有机相,用饱和食盐水洗涤(10mL x 1),无水硫酸钠干燥,过滤,滤液减压浓缩。剩余物用硅胶柱色谱法分离纯化(5/1石油醚/乙酸乙酯)得到化合物26-2。 1H NMR(400MHz,CD 3OD)δ7.52(d,J=8.0Hz,1H),7.48-7.44(m,1H),7.28(d,J=8.0Hz,1H)。 Compound 26-1 (500mg, 2.54mmol) was dissolved in anhydrous tetrahydrofuran (5mL), and n-butyllithium (2.5M n-heptane solution, 1.02mL, 2.54mmol) was added dropwise at -78°C. The reaction was stirred for 30 minutes. Then iodine (646mg, 2.54mmol) was dissolved in anhydrous tetrahydrofuran (5mL) dropwise to the reaction solution, and the reaction solution was stirred at 20°C for 12 hours. Water (10 mL) was added to the reaction solution, and extracted with ethyl acetate (10 mL x 3). The organic phases were combined, washed with saturated brine (10 mL x 1), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was separated and purified by silica gel column chromatography (5/1 petroleum ether/ethyl acetate) to obtain compound 26-2. 1 H NMR (400MHz, CD 3 OD) δ 7.52 (d, J=8.0Hz, 1H), 7.48-7.44 (m, 1H), 7.28 (d, J=8.0Hz, 1H).
第二步second step
参照实施例21第一步得到化合物26-3。MS-ESI计算值[M+H] +679,实测值679。 Referring to the first step of Example 21, compound 26-3 was obtained. MS-ESI calculated [M+H] + 679, found 679.
第三步third step
参照实施例21第二步得到化合物26。 1H NMR(400MHz,CD 3OD)δ8.01-7.99(m,1H),7.92-7.90(m,1H),7.83-7.82(m,2H),7.74-7.70(m,1H),7.23-7.21(m,1H),7.15-7.10(m,2H),6.81(d,J=8.0Hz,1H),6.76-6.72(m,1H),4.78-4.74(m,2H),4.44-4.40(m,1H),4.29-4.24(m,2H),3.79-3.71(m,1H),3.50-3.44(m,1H),3.26-3.20 (m,1H),2.44-2.29(m,2H),1.92-1.74(m,2H)。MS-ESI计算值[M+H] +665,实测值665。 Referring to the second step of Example 21, compound 26 was obtained. 1 H NMR (400MHz, CD 3 OD) δ8.01-7.99(m,1H),7.92-7.90(m,1H),7.83-7.82(m,2H),7.74-7.70(m,1H),7.23- 7.21(m,1H),7.15-7.10(m,2H),6.81(d,J=8.0Hz,1H),6.76-6.72(m,1H),4.78-4.74(m,2H),4.44-4.40( m,1H),4.29-4.24(m,2H),3.79-3.71(m,1H),3.50-3.44(m,1H),3.26-3.20(m,1H),2.44-2.29(m,2H), 1.92-1.74 (m, 2H). MS-ESI calculated [M+H] +665 , found 665.
实施例27Example 27
合成路线:synthetic route:
第一步first step
将化合物5-1(1.29g,8.47mmol)溶于异丙醇(10mL)中,在25℃下加入碘化镍(79.4mg,0.254mmol),反式-2-氨基-1-环己醇盐酸盐(38.5mg,0.254mmol)和二(三甲基硅)氨基钠(1M四氢呋喃溶液,8.47mL,8.47mmol)。在该温度下搅拌反应30分钟。然后向反应液中加入7-1(1.00g,4.24mmol),反应液在80℃下搅拌反应12小时。向反应液加入水(30mL),用二氯甲烷(40mL x 2)萃取。合并有机相,用饱和食盐水洗涤(30mL x 1),无水硫酸钠干燥,过滤,滤液减压浓缩。剩余物用硅胶柱色谱法分离纯化(5/1石油醚/乙酸乙酯)得到化合物27-1。MS-ESI计算值[M-56+H] +208,实测值208。 Compound 5-1 (1.29g, 8.47mmol) was dissolved in isopropanol (10mL), and nickel iodide (79.4mg, 0.254mmol), trans-2-amino-1-cyclohexanol was added at 25°C Hydrochloride (38.5mg, 0.254mmol) and sodium bis(trimethylsilyl)amide (1M solution in tetrahydrofuran, 8.47mL, 8.47mmol). The reaction was stirred at this temperature for 30 minutes. Then 7-1 (1.00g, 4.24mmol) was added to the reaction solution, and the reaction solution was stirred and reacted at 80°C for 12 hours. Water (30 mL) was added to the reaction solution, and extracted with dichloromethane (40 mL x 2). The organic phases were combined, washed with saturated brine (30 mL x 1), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was separated and purified by silica gel column chromatography (5/1 petroleum ether/ethyl acetate) to obtain compound 27-1. MS-ESI calculated value [M-56+H] + 208, measured value 208.
第二步second step
将化合物27-1(200mg,0.147mmol)溶于甲醇(2mL)中,加入盐酸甲醇(4M,1mL,4.00mmol)。反应液在25℃搅拌反应2小时。向反应液加入饱和碳酸氢钠水溶液(20mL),用二氯甲烷(30mL x 3)萃取。合并有机相,用饱和食盐水洗涤(20mL x 1),无水硫酸钠干燥,过滤,滤液减压浓缩。剩余物用制备薄层层析法分离纯化(10/1二氯甲烷/甲醇)得到化合物27-2。MS-ESI计算值[M+H] +164,实测值164。 Compound 27-1 (200 mg, 0.147 mmol) was dissolved in methanol (2 mL), and methanol hydrochloride (4M, 1 mL, 4.00 mmol) was added. The reaction solution was stirred and reacted at 25° C. for 2 hours. Saturated aqueous sodium bicarbonate (20 mL) was added to the reaction solution, and extracted with dichloromethane (30 mL x 3). The organic phases were combined, washed with saturated brine (20 mL x 1), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was separated and purified by preparative thin layer chromatography (10/1 dichloromethane/methanol) to obtain compound 27-2. MS-ESI calculated value [M+H] + 164, found value 164.
第三步third step
将化合物27-2(30.0mg,0.143mmol),2-5(91.5mg,0.158mmol),醋酸铜(52.0mg,0.286mmol),吡啶(34.0 mg,0.430mmol)和氧化吡啶(40.9mg,0.430mmol)加入到二氯甲烷(5mL)中。反应液在氧气保护下40℃搅拌反应12小时。向反应液中加入水(20mL),用二氯甲烷(20mL x 3)萃取。合并有机相,用饱和食盐水洗涤(20mL x 1),无水硫酸钠干燥,过滤,滤液减压浓缩。剩余物用制备薄层层析法分离纯化(3/1石油醚/乙酸乙酯)得到化合物27-3。MS-ESI计算值[M+H] +591,实测值591。 Compound 27-2 (30.0 mg, 0.143 mmol), 2-5 (91.5 mg, 0.158 mmol), copper acetate (52.0 mg, 0.286 mmol), pyridine (34.0 mg, 0.430 mmol) and pyridine oxide (40.9 mg, 0.430 mmol) was added to dichloromethane (5 mL). The reaction solution was stirred and reacted at 40° C. for 12 hours under the protection of oxygen. Water (20 mL) was added to the reaction liquid, and extracted with dichloromethane (20 mL x 3). The organic phases were combined, washed with saturated brine (20 mL x 1), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was separated and purified by preparative thin layer chromatography (3/1 petroleum ether/ethyl acetate) to obtain compound 27-3. MS-ESI calculated [M+H] + 591, found 591.
第四步the fourth step
将化合物27-3(80.0mg,0.108mmol)和一水合氢氧化锂(9.0mg,0.216mmol)加入到四氢呋喃(2mL)和水(0.5mL)中。反应液在氮气保护下20℃搅拌反应12小时。反应液减压浓缩。剩余物经高效液相色谱法分离(中性,碳酸氢铵体系)纯化得到化合物27。 1H NMR(400MHz,CD 3OD)δ7.99-7.90(m,3H),7.76-7.74(m,1H),7.27-7.22(m,2H),6.99-6.94(m,3H),6.71-6.69(m,1H),6.40-6.38(m,1H),4.39-4.35(m,1H),4.28-4.24(m,3H),3.83(s,3H),3.76-3.74(m,2H),3.35-3.34(m,1H),3.24-3.20(m,1H),2.40-2.34(m,2H),1.83-1.73(m,2H)。MS-ESI计算值[M+H] +577,实测值577。 Compound 27-3 (80.0 mg, 0.108 mmol) and lithium hydroxide monohydrate (9.0 mg, 0.216 mmol) were added to tetrahydrofuran (2 mL) and water (0.5 mL). The reaction solution was stirred and reacted at 20° C. for 12 hours under the protection of nitrogen. The reaction solution was concentrated under reduced pressure. The residue was separated and purified by high performance liquid chromatography (neutral, ammonium bicarbonate system) to obtain compound 27. 1 H NMR (400MHz, CD 3 OD) δ7.99-7.90(m,3H),7.76-7.74(m,1H),7.27-7.22(m,2H),6.99-6.94(m,3H),6.71- 6.69(m,1H),6.40-6.38(m,1H),4.39-4.35(m,1H),4.28-4.24(m,3H),3.83(s,3H),3.76-3.74(m,2H), 3.35-3.34 (m, 1H), 3.24-3.20 (m, 1H), 2.40-2.34 (m, 2H), 1.83-1.73 (m, 2H). MS-ESI calculated [M+H] + 577, found 577.
实施例28Example 28
合成路线:synthetic route:

第一步first step
将锌粉(1.81g,27.7mmol)和1,2-二溴乙烷(307mg,1.63mmol)加入到N,N-二甲基甲酰胺(20mL)中。反应液在70℃搅拌反应10分钟。然后反应液冷却到20℃。三甲基氯硅烷(177mg,1.63mmol)滴加到反 应液中,然后反应液20℃搅拌反应50分钟。16-1(5.00g,16.3mmol),三(二亚苄基丙酮)二钯(299mg,0.326mmol),三(2-呋喃基)膦(151mg,0.653mmol)加入到反应液中。反应液在氮气保护下40℃搅拌反应1小时。28-1(6.93g,24.5mmol)溶于N,N-二甲基甲酰胺(40mL)加入到反应液中,反应液在氮气保护下60℃搅拌反应10小时。向反应液中加入水(100mL),用乙酸乙酯(100mL x 2)萃取。合并有机相,用饱和食盐水洗涤(100mL x 3),无水硫酸钠干燥,过滤,滤液减压浓缩。剩余物用硅胶柱色谱法分离纯化(5/1石油醚/乙酸乙酯)得到化合物28-2。 1H NMR(400MHz,CDCl 3)δ7.62-7.58(m,2H),7.33-7.29(m,1H),4.60-4.57(m,2H),4.40-4.34(m,1H),4.24-4.20(m,2H),1.48(s,9H)。MS-ESI计算值[M-56+H] +280,实测值280。 Zinc dust (1.81 g, 27.7 mmol) and 1,2-dibromoethane (307 mg, 1.63 mmol) were added to N,N-dimethylformamide (20 mL). The reaction solution was stirred and reacted at 70° C. for 10 minutes. The reaction solution was then cooled to 20°C. Chlorotrimethylsilane (177mg, 1.63mmol) was added dropwise to the reaction solution, and then the reaction solution was stirred at 20°C for 50 minutes. 16-1 (5.00g, 16.3mmol), tris(dibenzylideneacetone)dipalladium (299mg, 0.326mmol), tris(2-furyl)phosphine (151mg, 0.653mmol) were added to the reaction solution. The reaction solution was stirred and reacted at 40° C. for 1 hour under the protection of nitrogen. 28-1 (6.93g, 24.5mmol) was dissolved in N,N-dimethylformamide (40mL) and added to the reaction solution, and the reaction solution was stirred and reacted at 60°C for 10 hours under the protection of nitrogen. Water (100 mL) was added to the reaction solution, and extracted with ethyl acetate (100 mL x 2). The organic phases were combined, washed with saturated brine (100 mL x 3), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was separated and purified by silica gel column chromatography (5/1 petroleum ether/ethyl acetate) to obtain compound 28-2. 1 H NMR (400MHz, CDCl 3 )δ7.62-7.58(m,2H),7.33-7.29(m,1H),4.60-4.57(m,2H),4.40-4.34(m,1H),4.24-4.20 (m,2H), 1.48(s,9H). MS-ESI calculated value [M-56+H] + 280, measured value 280.
第二步second step
将化合物28-2(1.10g,3.28mmol)溶于甲醇(10mL)中,加入盐酸甲醇(4M,10mL,40.0mmol)。反应液在20℃搅拌反应1小时。反应液减压浓缩。剩余物为化合物28-3。MS-ESI计算值[M+H] +236,实测值236。 Compound 28-2 (1.10 g, 3.28 mmol) was dissolved in methanol (10 mL), and methanol hydrochloride (4M, 10 mL, 40.0 mmol) was added. The reaction solution was stirred and reacted at 20° C. for 1 hour. The reaction solution was concentrated under reduced pressure. The residue was compound 28-3. MS-ESI calculated [M+H] + 236, found 236.
第三步third step
将化合物1-8(200mg,0.393mmol),28-3(133mg,0.393mmol),碳酸铯(385mg,1.18mmol)和甲烷磺酸(2-二环己基膦)-3,6-二甲氧基-2,4,6-三异丙基-1,1-联苯)(2-氨基-1,1-联苯-2-基)钯(II)(35.7mg,0.0394mmol)加入到1,4-二氧六环(5mL)中。反应液在氮气保护下90℃搅拌反应12小时。向反应液中加入水(10mL),用乙酸乙酯(10mL x 3)萃取。合并有机相,用饱和食盐水洗涤(10mL x 1),无水硫酸钠干燥,过滤,滤液减压浓缩。剩余物用制备薄层层析法分离纯化(3/1石油醚/乙酸乙酯)得到化合物28-4。MS-ESI计算值[M+H] +663,实测值663。 Compound 1-8 (200mg, 0.393mmol), 28-3 (133mg, 0.393mmol), cesium carbonate (385mg, 1.18mmol) and methanesulfonic acid (2-dicyclohexylphosphine)-3,6-dimethoxy Base-2,4,6-triisopropyl-1,1-biphenyl)(2-amino-1,1-biphenyl-2-yl)palladium (II) (35.7mg, 0.0394mmol) was added to 1 , 4-dioxane (5 mL). The reaction solution was stirred and reacted at 90° C. for 12 hours under the protection of nitrogen. Water (10 mL) was added to the reaction solution, and extracted with ethyl acetate (10 mL x 3). The organic phases were combined, washed with saturated brine (10 mL x 1), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was separated and purified by preparative thin layer chromatography (3/1 petroleum ether/ethyl acetate) to obtain compound 28-4. MS-ESI calculated [M+H] + 663, found 663.
第四步the fourth step
将化合物28-4(200mg,0.130mmol)和一水合氢氧化锂(11.0mg,0.261mmol)加入到四氢呋喃(2mL)和水(0.5mL)中。反应液在氮气保护下20℃搅拌反应12小时。反应液减压浓缩。剩余物经高效液相色谱法分离(中性,碳酸氢铵体系)纯化得到化合物28。 1H NMR(400MHz,CD 3OD)δ8.00-7.94(m,2H),7.87(s,1H),7.76-7.68(m,3H),7.45-7.43(m,1H),6.96-6.95(m,1H),6.70(d,J=8.8Hz,1H),6.37-6.35(m,1H),4.67-4.61(m,1H),4.42-4.34(m,3H),4.32-4.28(m,2H),3.35-3.34(m,1H),3.24-3.18(m,1H),2.41-2.32(m,2H),1.85-1.74(m,2H)。MS-ESI计算值[M+H] +649,实测值649。 Compound 28-4 (200 mg, 0.130 mmol) and lithium hydroxide monohydrate (11.0 mg, 0.261 mmol) were added to tetrahydrofuran (2 mL) and water (0.5 mL). The reaction solution was stirred and reacted at 20° C. for 12 hours under the protection of nitrogen. The reaction solution was concentrated under reduced pressure. The residue was separated and purified by high performance liquid chromatography (neutral, ammonium bicarbonate system) to obtain compound 28. 1 H NMR (400MHz, CD 3 OD) δ8.00-7.94 (m, 2H), 7.87 (s, 1H), 7.76-7.68 (m, 3H), 7.45-7.43 (m, 1H), 6.96-6.95 ( m,1H),6.70(d,J=8.8Hz,1H),6.37-6.35(m,1H),4.67-4.61(m,1H),4.42-4.34(m,3H),4.32-4.28(m, 2H), 3.35-3.34(m, 1H), 3.24-3.18(m, 1H), 2.41-2.32(m, 2H), 1.85-1.74(m, 2H). MS-ESI calculated [M+H] + 649, found 649.
实施例29Example 29
合成路线:synthetic route:

第一步first step
将锌粉(1.30g,19.9mmol)和1,2-二溴乙烷(220mg,1.17mmol)加入到N,N-二甲基甲酰胺(20mL)中。反应液在70℃搅拌反应10分钟。然后反应液冷却到20℃。三甲基氯硅烷(127mg,1.17mmol)滴加到反应液中,然后反应液20℃搅拌反应50分钟。3-1(3.00g,11.7mmol),三(二亚苄基丙酮)二钯(214mg,0.234mmol),三(2-呋喃基)膦(109mg,0.468mmol)加入到反应液中。反应液在氮气保护下40℃搅拌反应1小时。28-1(4.97g,17.6mmol)溶于N,N-二甲基甲酰胺(30mL)加入到反应液中,反应液在氮气保护下60℃搅拌反应10小时。向反应液中加入水(100mL),用乙酸乙酯(100mL x 2)萃取。合并有机相,用饱和食盐水洗涤(100mL x 3),无水硫酸钠干燥,过滤,滤液减压浓缩。剩余物用硅胶柱色谱法分离纯化(10/1石油醚/乙酸乙酯)得到化合物29-1。 1H NMR(400MHz,CDCl 3)δ7.17-7.16(m,2H),7.02-6.98(m,1H),4.65-4.62(m,3H),4.49-4.46(m,2H),1.44(s,9H)。MS-ESI计算值[M-56+H] +230,实测值230。 Zinc powder (1.30 g, 19.9 mmol) and 1,2-dibromoethane (220 mg, 1.17 mmol) were added to N,N-dimethylformamide (20 mL). The reaction solution was stirred and reacted at 70° C. for 10 minutes. The reaction solution was then cooled to 20°C. Chlorotrimethylsilane (127mg, 1.17mmol) was added dropwise to the reaction solution, and then the reaction solution was stirred at 20°C for 50 minutes. 3-1 (3.00g, 11.7mmol), tris(dibenzylideneacetone)dipalladium (214mg, 0.234mmol), tris(2-furyl)phosphine (109mg, 0.468mmol) were added to the reaction solution. The reaction solution was stirred and reacted at 40° C. for 1 hour under the protection of nitrogen. 28-1 (4.97g, 17.6mmol) was dissolved in N,N-dimethylformamide (30mL) and added to the reaction solution, and the reaction solution was stirred and reacted at 60°C for 10 hours under the protection of nitrogen. Water (100 mL) was added to the reaction solution, and extracted with ethyl acetate (100 mL x 2). The organic phases were combined, washed with saturated brine (100 mL x 3), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was separated and purified by silica gel column chromatography (10/1 petroleum ether/ethyl acetate) to obtain compound 29-1. 1 H NMR (400MHz, CDCl 3 )δ7.17-7.16(m,2H),7.02-6.98(m,1H),4.65-4.62(m,3H),4.49-4.46(m,2H),1.44(s ,9H). MS-ESI calculated value [M-56+H] + 230, found value 230.
第二步second step
将化合物29-1(1.30g,4.55mmol)溶于甲醇(10mL)中,加入盐酸甲醇(4M,10mL,40.0mmol)。反应液在20℃搅拌反应1小时。反应液减压浓缩。剩余物为化合物29-2。MS-ESI计算值[M+H] +186,实测值186。 Compound 29-1 (1.30 g, 4.55 mmol) was dissolved in methanol (10 mL), and methanol hydrochloride (4M, 10 mL, 40.0 mmol) was added. The reaction solution was stirred and reacted at 20° C. for 1 hour. The reaction solution was concentrated under reduced pressure. The residue was compound 29-2. MS-ESI calculated value [M+H] + 186, found value 186.
第三步third step
将化合物1-8(300mg,0.590mmol),29-2(159mg,0.590mmol),碳酸铯(577mg,1.77mmol),4,5-双二苯基 膦-9,9-二甲基氧杂蒽(34.2mg,0.0590mmol)和三(二亚苄基丙酮)二钯(27.0mg,29.5nmol)加入到1,4-二氧六环(5mL)中。反应液在氮气保护下90℃搅拌反应12小时。向反应液中加入水(10mL),用乙酸乙酯(10mL x 3)萃取。合并有机相,用饱和食盐水洗涤(10mL x 1),无水硫酸钠干燥,过滤,滤液减压浓缩。剩余物用制备薄层层析法分离纯化(3/1石油醚/乙酸乙酯)得到化合物29-3。MS-ESI计算值[M+H] +613,实测值613。 Compound 1-8 (300mg, 0.590mmol), 29-2 (159mg, 0.590mmol), cesium carbonate (577mg, 1.77mmol), 4,5-bisdiphenylphosphine-9,9-dimethyloxa Anthracene (34.2 mg, 0.0590 mmol) and tris(dibenzylideneacetone)dipalladium (27.0 mg, 29.5 nmol) were added to 1,4-dioxane (5 mL). The reaction solution was stirred and reacted at 90° C. for 12 hours under the protection of nitrogen. Water (10 mL) was added to the reaction solution, and extracted with ethyl acetate (10 mL x 3). The organic phases were combined, washed with saturated brine (10 mL x 1), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was separated and purified by preparative thin layer chromatography (3/1 petroleum ether/ethyl acetate) to obtain compound 29-3. MS-ESI calculated [M+H] + 613, found 613.
第四步the fourth step
将化合物29-3(250mg,56.7nmol)和一水合氢氧化锂(4.8mg,0.113mmol)加入到四氢呋喃(5mL)和水(1mL)中。反应液在氮气保护下20℃搅拌反应12小时。反应液减压浓缩。剩余物经高效液相色谱法分离(中性,碳酸氢铵体系)纯化得到化合物29。 1H NMR(400MHz,CD 3OD)δ8.04-8.02(m,1H),7.94-7.92(m,1H),7.85(s,1H),7.81-7.79(m,1H),7.27-7.24(m,2H),7.09-7.04(m,1H),6.68(d,J=8.8Hz,1H),6.40-6.39(m,1H),6.37-6.36(m,1H),4.43-4.39(m,4H),3.92-3.89(m,2H),3.35-3.34(m,1H),3.22-3.16(m,1H),2.28-2.18(m,2H),1.82-1.75(m,2H)。MS-ESI计算值[M+H] +599,实测值599。 Compound 29-3 (250 mg, 56.7 nmol) and lithium hydroxide monohydrate (4.8 mg, 0.113 mmol) were added to tetrahydrofuran (5 mL) and water (1 mL). The reaction solution was stirred and reacted at 20° C. for 12 hours under the protection of nitrogen. The reaction solution was concentrated under reduced pressure. The residue was separated and purified by high performance liquid chromatography (neutral, ammonium bicarbonate system) to obtain compound 29. 1 H NMR (400MHz, CD 3 OD) δ8.04-8.02 (m, 1H), 7.94-7.92 (m, 1H), 7.85 (s, 1H), 7.81-7.79 (m, 1H), 7.27-7.24 ( m,2H),7.09-7.04(m,1H),6.68(d,J=8.8Hz,1H),6.40-6.39(m,1H),6.37-6.36(m,1H),4.43-4.39(m, 4H), 3.92-3.89(m, 2H), 3.35-3.34(m, 1H), 3.22-3.16(m, 1H), 2.28-2.18(m, 2H), 1.82-1.75(m, 2H). MS-ESI calculated value [M+H] + 599, found value 599.
实施例30Example 30
合成路线:synthetic route:

第一步first step
参照实施例29第一步得到化合物30-1。MS-ESI计算值[M-56+H] +296,实测值296。 Referring to the first step of Example 29, compound 30-1 was obtained. MS-ESI calculated value [M-56+H] + 296, found value 296.
第二步second step
参照实施例29第二步得到化合物30-2。MS-ESI计算值[M+H] +252,实测值252。 Referring to the second step of Example 29, compound 30-2 was obtained. MS-ESI calculated value [M+H] + 252, found value 252.
第三步third step
参照实施例29第三步得到化合物30-3。MS-ESI计算值[M+H] +679,实测值679。 Referring to the third step of Example 29, compound 30-3 was obtained. MS-ESI calculated [M+H] + 679, found 679.
第四步the fourth step
参照实施例29第四步得到化合物30。 1H NMR(400MHz,CD 3OD)δ8.03-8.01(m,1H),7.92-7.91(m,1H),7.85(s,1H),7.79-7.77(m,1H),7.45-7.43(m,1H),7.35-7.30(m,2H),7.01-7.00(m,1H),6.68(d,J=8.8Hz,1H),6.40-6.38(m,1H),4.48-4.39(m,4H),3.88-3.82(m,2H),3.35-3.34(m,1H),3.21-3.15(m,1H),2.29-2.20(m,2H),1.81-1.75(m,2H)。MS-ESI计算值[M+H] +665,实测值665。 Referring to the fourth step of Example 29, compound 30 was obtained. 1 H NMR (400MHz, CD 3 OD) δ8.03-8.01 (m, 1H), 7.92-7.91 (m, 1H), 7.85 (s, 1H), 7.79-7.77 (m, 1H), 7.45-7.43 ( m,1H),7.35-7.30(m,2H),7.01-7.00(m,1H),6.68(d,J=8.8Hz,1H),6.40-6.38(m,1H),4.48-4.39(m, 4H), 3.88-3.82(m, 2H), 3.35-3.34(m, 1H), 3.21-3.15(m, 1H), 2.29-2.20(m, 2H), 1.81-1.75(m, 2H). MS-ESI calculated [M+H] +665 , found 665.
实施例31Example 31
合成路线:synthetic route:

第一步first step
在2℃下,将亚硝酸钠(397mg,5.75mmol)的水(2.5mL)溶液加入化合物31-1(1g,5.23mmol)的浓盐酸(4.5mL)和水(24.5mL)的悬浊液中,在2℃下搅拌30分钟后于0℃向其中加入碘化钾(8.68g,52.3mmol)的水(5mL)溶液,反应液在15℃搅拌4小时。将反应液倒入冰中,用乙酸乙酯(50mL×2)萃取,合并有机相后依次用饱和碳酸氢钠水溶液(100mL×1)和水(100mL×1)洗涤,再用无水硫酸钠干燥,过滤,滤液减压浓缩后剩余物经过硅胶柱层析法(6/1石油醚/乙酸乙酯)分离纯化得到化合物31-2。 1H NMR(400MHz,CDCl 3)δ7.44-7.40(m,1H),7.31-7.29(m,1H),7.02-7.00(m,1H),3.96(s,3H)。 At 2°C, a solution of sodium nitrite (397mg, 5.75mmol) in water (2.5mL) was added to a suspension of compound 31-1 (1g, 5.23mmol) in concentrated hydrochloric acid (4.5mL) and water (24.5mL) After stirring at 2°C for 30 minutes, a solution of potassium iodide (8.68 g, 52.3 mmol) in water (5 mL) was added thereto at 0°C, and the reaction solution was stirred at 15°C for 4 hours. The reaction solution was poured into ice, extracted with ethyl acetate (50mL×2), and the combined organic phases were washed successively with saturated aqueous sodium bicarbonate (100mL×1) and water (100mL×1), and then washed with anhydrous sodium sulfate After drying and filtering, the filtrate was concentrated under reduced pressure and the residue was separated and purified by silica gel column chromatography (6/1 petroleum ether/ethyl acetate) to obtain compound 31-2. 1 H NMR (400 MHz, CDCl 3 ) δ 7.44-7.40 (m, 1H), 7.31-7.29 (m, 1H), 7.02-7.00 (m, 1H), 3.96 (s, 3H).
第二步second step
将锌粉(486mg,7.43mmol)和1,2-二溴乙烷(99.0mg,0.524mmol)溶于N,N-二甲基甲酰胺(6.6mL)中,升温至70℃搅拌10分钟,降温至20℃后向其中滴加三甲基氯硅烷(57mg,0.524mmol),继续在20℃下搅拌50分钟,然后向其中加入化合物28-1(1.86g,6.56mmol)的N,N-二甲基甲酰胺(4mL)溶液,升温至40℃搅拌1小时,最后向其中快速加入化合物31-2(1.32g,4.37mmol),三(二亚苄基丙酮)二钯(80mg,87.4umol)和三(2-呋喃基)膦(41mg,0.175mmol)的N,N-二甲基甲酰胺(16mL)溶液,反应液在氮气保护下升温至60℃搅拌10小时。反应液通过硅藻土过滤,滤渣用乙酸乙酯(30mL)洗涤,滤液用饱和氯化铵水溶液(30mL×1)洗涤,有机相依次用水(30mL×3)和饱和食盐水(30mL×1)洗涤,再用无水硫酸钠干燥,过滤,滤液减压浓缩后剩余物经过硅胶柱层析法(4/1石油醚/乙酸乙酯)分离纯化得到化合物31-3。 1H NMR(400MHz,CDCl 3)δ7.37-7.33(m,1H),7.27-7.25(m,1H),7.15-7.13(m,1H),4.38-4.35(m,2H),4.23-4.13(m,3H),3.95(s,3H),1.51(s,9H)。MS-ESI计算值[M-56+H] +276,实测值276。 Dissolve zinc powder (486mg, 7.43mmol) and 1,2-dibromoethane (99.0mg, 0.524mmol) in N,N-dimethylformamide (6.6mL), heat up to 70°C and stir for 10 minutes, After cooling down to 20°C, trimethylchlorosilane (57mg, 0.524mmol) was added dropwise, and stirring was continued at 20°C for 50 minutes, then compound 28-1 (1.86g, 6.56mmol) was added in N,N- Dimethylformamide (4mL) solution was heated to 40°C and stirred for 1 hour, and finally compound 31-2 (1.32g, 4.37mmol) and tris(dibenzylideneacetone)dipalladium (80mg, 87.4umol) were added rapidly thereto ) and tris(2-furyl)phosphine (41mg, 0.175mmol) in N,N-dimethylformamide (16mL) solution, the reaction solution was heated to 60°C under nitrogen protection and stirred for 10 hours. The reaction solution was filtered through diatomaceous earth, the filter residue was washed with ethyl acetate (30mL), the filtrate was washed with saturated ammonium chloride aqueous solution (30mL×1), and the organic phase was sequentially washed with water (30mL×3) and saturated brine (30mL×1). After washing, drying with anhydrous sodium sulfate, filtration, the filtrate was concentrated under reduced pressure, and the residue was separated and purified by silica gel column chromatography (4/1 petroleum ether/ethyl acetate) to obtain compound 31-3. 1 H NMR (400MHz, CDCl 3 )δ7.37-7.33(m,1H),7.27-7.25(m,1H),7.15-7.13(m,1H),4.38-4.35(m,2H),4.23-4.13 (m,3H), 3.95(s,3H), 1.51(s,9H). MS-ESI calculated value [M-56+H] + 276, found value 276.
第三步third step
参照实施例5第二步得到化合物31-4。MS-ESI计算值[M+H] +232,实测值232。 Referring to the second step of Example 5, compound 31-4 was obtained. MS-ESI calculated [M+H] + 232, found 232.
第四步the fourth step
参照实施例3第一步得到化合物31-5。MS-ESI计算值[M+H] +659,实测值659。 Referring to the first step of Example 3, compound 31-5 was obtained. MS-ESI calculated [M+H] + 659, found 659.
第五步the fifth step
参照实施例1第十步得到化合物31。 1H NMR(400MHz,CD 3OD)δ8.03-8.01(m,1H),7.99-7.95(m,2H),7.80-7.76(m,1H),7.44-7.40(m,1H),7.30-7.26(m,2H),6.94-6.93(m,1H),6.77-6.75(m,1H),6.40-6.37(m,1H),4.48-4.37(m,2H),4.22-4.20(m,4H),3.63(s,3H),3.42-3.39(m,1H),3.29-3.24(m,1H),2.49-2.35(m,2H),1.91-1.78(m,2H)。MS-ESI计算值[M+H] +645,实测值645。 Referring to the tenth step of Example 1, compound 31 was obtained. 1 H NMR (400MHz, CD 3 OD) δ8.03-8.01 (m, 1H), 7.99-7.95 (m, 2H), 7.80-7.76 (m, 1H), 7.44-7.40 (m, 1H), 7.30- 7.26(m,2H),6.94-6.93(m,1H),6.77-6.75(m,1H),6.40-6.37(m,1H),4.48-4.37(m,2H),4.22-4.20(m,4H ), 3.63(s,3H), 3.42-3.39(m,1H), 3.29-3.24(m,1H), 2.49-2.35(m,2H), 1.91-1.78(m,2H). MS-ESI calculated [M+H] + 645, found 645.
实施例32Example 32
合成路线:synthetic route:

第一步first step
将锌粉(845mg,12.5mmol)和1,2-二溴乙烷(165mg,0.879mmol)加入到N,N-二甲基甲酰胺(10mL)中。反应液在70℃搅拌反应10分钟。然后反应液冷却到20℃。三甲基氯硅烷(95.5mg,0.879mmol)滴加到反应液中,然后反应液20℃搅拌反应50分钟。将28-1(3.11g,10.9mmol)的N,N-二甲基甲酰胺(6mL)溶液加入到反应液中,反应液在氮气保护下40℃搅拌反应1小时。将32-1(2.00g,7.33mmol),三(二亚苄基丙酮)二钯(134mg,0.146mmol),三(2-呋喃基)膦(68.1mg,0.293mmol)的N,N-二甲基甲酰胺(24mL)加入到反应液中。反应液在氮气保护下60℃搅拌反应10小时。反应液经硅藻土过滤除去固体残渣,滤液用饱和氯化铵(50mL x 2)和乙酸乙酯(50mL x 2)分层处理,合并有机相用水(50mL x 3),饱和食盐水(50mL x 1)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩。剩余物用硅胶柱色谱法分离纯化得到化合物32-2。 1H NMR(400MHz,CDCl 3)δ7.32-7.31(m,2H),7.15-7.11(m,1H),4.55-4.46(m,3H),4.31-4.27(m,2H),1.48(s,9H)。MS-ESI计算值[M-56+H] +246,实测值246。 Zinc dust (845 mg, 12.5 mmol) and 1,2-dibromoethane (165 mg, 0.879 mmol) were added to N,N-dimethylformamide (10 mL). The reaction solution was stirred and reacted at 70° C. for 10 minutes. The reaction solution was then cooled to 20°C. Chlorotrimethylsilane (95.5mg, 0.879mmol) was added dropwise to the reaction solution, and then the reaction solution was stirred at 20°C for 50 minutes. A solution of 28-1 (3.11 g, 10.9 mmol) in N,N-dimethylformamide (6 mL) was added to the reaction solution, and the reaction solution was stirred and reacted at 40°C for 1 hour under the protection of nitrogen. The N, N-di Methylformamide (24 mL) was added to the reaction solution. The reaction solution was stirred and reacted at 60° C. for 10 hours under the protection of nitrogen. The reaction solution was filtered through diatomaceous earth to remove solid residues, the filtrate was treated with saturated ammonium chloride (50mL x 2) and ethyl acetate (50mL x 2), and the organic phase was combined with water (50mL x 3), saturated brine (50mL x 1) washed, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was separated and purified by silica gel column chromatography to obtain compound 32-2. 1 H NMR (400MHz, CDCl 3 )δ7.32-7.31(m,2H),7.15-7.11(m,1H),4.55-4.46(m,3H),4.31-4.27(m,2H),1.48(s ,9H). MS-ESI calculated value [M-56+H] + 246, found value 246.
第二步second step
将化合物32-2(400mg,1.30mmol)溶于乙酸乙酯(5mL)中,加入盐酸乙酸乙酯(4M,5mL,20.0mmol)。反应液在19℃搅拌反应1小时。反应液减压浓缩除去溶剂,剩余物用甲醇(10mL)溶解后加入碳酸氢钠(500mg),混合物在室温下搅拌30分钟后过滤除去固体碳酸氢钠,滤液减压浓缩得到化合物32-3。MS-ESI计算值[M+H] +202,实测值202。 Compound 32-2 (400 mg, 1.30 mmol) was dissolved in ethyl acetate (5 mL), and ethyl acetate hydrochloride (4M, 5 mL, 20.0 mmol) was added. The reaction solution was stirred and reacted at 19° C. for 1 hour. The reaction solution was concentrated under reduced pressure to remove the solvent. The residue was dissolved in methanol (10 mL) and sodium bicarbonate (500 mg) was added. The mixture was stirred at room temperature for 30 minutes and then filtered to remove solid sodium bicarbonate. The filtrate was concentrated under reduced pressure to obtain compound 32-3. MS-ESI calculated value [M+H] + 202, found value 202.
第三步third step
将化合物1-8(200mg,0.393mmol),32-3(120mg,0.593mmol),碳酸铯(321mg,0.984mmol)和甲烷磺酸(2-二环己基膦)-3,6-二甲氧基-2,4,6-三异丙基-1,1-联苯)(2-氨基-1,1-联苯-2-基)钯(II)(35.7mg,0.0394mmol)加入到N,N-二甲基甲酰胺(6mL)中。反应液在氮气保护下90℃搅拌反应10小时。反应液经硅藻土过滤,滤液减压浓缩。剩余物用制备薄层层析法分离纯化得到化合物32-4。MS-ESI计算值[M+H] +629,实测值629。 Compound 1-8 (200mg, 0.393mmol), 32-3 (120mg, 0.593mmol), cesium carbonate (321mg, 0.984mmol) and methanesulfonic acid (2-dicyclohexylphosphine)-3,6-dimethoxy Base-2,4,6-triisopropyl-1,1-biphenyl)(2-amino-1,1-biphenyl-2-yl)palladium (II) (35.7mg, 0.0394mmol) was added to N , in N-dimethylformamide (6 mL). The reaction solution was stirred and reacted at 90° C. for 10 hours under the protection of nitrogen. The reaction solution was filtered through diatomaceous earth, and the filtrate was concentrated under reduced pressure. The residue was separated and purified by preparative thin layer chromatography to obtain compound 32-4. MS-ESI calculated [M+H] + 629, found 629.
第四步the fourth step
将化合物32-4(138mg,0.219mmol)和氢氧化钠(27.0mg,0.675mmol)加入到四氢呋喃(2mL)和水(4mL)中。反应液在氮气保护下50℃搅拌反应10小时。反应液减压浓缩。剩余物用水(20mL)溶解,用盐酸(1M)调节pH到4,混合物用乙酸乙酯(20mL x 3)萃取,合并有机相用饱和食盐水(50mL x 1)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩。粗产物经高效液相色谱法分离(中性,碳酸氢铵体系)纯化得到化合物32。 1H NMR(400MHz,CD 3OD)δ8.01-7.99(m,1H),7.94-7.90(m,2H),7.71-7.68(m,1H),7.40-7.38(m,2H),7.25-7.23(m,1H),7.02(d,J=2.4Hz,1H),6.73-6.71(m,1H),6.43-6.41(m,1H),4.61-4.59(m,1H),4.50-4.47(m,2H),4.41-4.37(m,1H),4.05-4.03(m,2H),3.35-3.34(m,1H),3.25-3.18(m,1H),2.40-2.34(m,2H),1.85-1.76(m,2H)。MS-ESI计算值[M+H] +615,实测值615。 Compound 32-4 (138 mg, 0.219 mmol) and sodium hydroxide (27.0 mg, 0.675 mmol) were added to tetrahydrofuran (2 mL) and water (4 mL). The reaction solution was stirred and reacted at 50° C. for 10 hours under the protection of nitrogen. The reaction solution was concentrated under reduced pressure. The residue was dissolved in water (20 mL), adjusted to pH 4 with hydrochloric acid (1 M), the mixture was extracted with ethyl acetate (20 mL x 3), the combined organic phases were washed with saturated brine (50 mL x 1), dried over anhydrous sodium sulfate, Filter and concentrate the filtrate under reduced pressure. The crude product was separated and purified by high performance liquid chromatography (neutral, ammonium bicarbonate system) to obtain compound 32. 1 H NMR (400MHz, CD 3 OD) δ8.01-7.99 (m, 1H), 7.94-7.90 (m, 2H), 7.71-7.68 (m, 1H), 7.40-7.38 (m, 2H), 7.25- 7.23(m,1H),7.02(d,J=2.4Hz,1H),6.73-6.71(m,1H),6.43-6.41(m,1H),4.61-4.59(m,1H),4.50-4.47( m,2H),4.41-4.37(m,1H),4.05-4.03(m,2H),3.35-3.34(m,1H),3.25-3.18(m,1H),2.40-2.34(m,2H), 1.85-1.76 (m, 2H). MS-ESI calculated [M+H] + 615, found 615.
实施例33Example 33
合成路线:synthetic route:

第一步first step
将化合物1-6(6.00g,20.0mmol),二碳酸二叔丁酯(4.80g,22.0mmol),4-二甲氨基吡啶(244mg,2.00mmol)和N,N-二异丙基乙胺(2.84g,22.0mmol)溶于二氯甲烷(100mL),反应液在40℃搅拌10小时。再向反应液中补加二碳酸二叔丁酯(4.00g,18.3mmol),反应液在40℃下搅拌10小时。向反应液中加入二氯甲烷(100mL),用盐酸(1M,100mL×1),饱和食盐水(100mL×1)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩,粗产物经过柱层析法(4:1石油醚/乙酸乙酯)分离纯化得到化合物33-1。 1H NMR(400MHz,CD 3OD)δ7.91(s,1H),7.00-6.98(m,1H),6.67-6.64(m,1H),4.08-4.01(m,2H),3.63(s,3H),3.22-3.21(m,1H),2.52-2.47(m,2H),1.90-1.86(m,2H),1.48(s,9H)。MS-ESI计算值[M-100+H] +300和301,实测值300和301。 Compound 1-6 (6.00g, 20.0mmol), di-tert-butyl dicarbonate (4.80g, 22.0mmol), 4-dimethylaminopyridine (244mg, 2.00mmol) and N,N-diisopropylethylamine (2.84g, 22.0mmol) was dissolved in dichloromethane (100mL), and the reaction solution was stirred at 40°C for 10 hours. Further di-tert-butyl dicarbonate (4.00 g, 18.3 mmol) was added to the reaction solution, and the reaction solution was stirred at 40° C. for 10 hours. Dichloromethane (100mL) was added to the reaction solution, washed with hydrochloric acid (1M, 100mL×1), saturated brine (100mL×1), dried over anhydrous sodium sulfate, filtered, the filtrate was concentrated under reduced pressure, and the crude product was passed through the column layer Compound 33-1 was isolated and purified by analytical method (4:1 petroleum ether/ethyl acetate). 1 H NMR (400MHz, CD 3 OD) δ7.91(s,1H),7.00-6.98(m,1H),6.67-6.64(m,1H),4.08-4.01(m,2H),3.63(s, 3H), 3.22-3.21(m, 1H), 2.52-2.47(m, 2H), 1.90-1.86(m, 2H), 1.48(s, 9H). MS-ESI calculated value [M-100+H] + 300 and 301, found value 300 and 301.
第二步second step
将化合物32-3(860mg,3.61mmol)和化合物33-1(1.44g,3.61mmol)溶于N,N-二甲基甲酰胺(20mL)中,氮气保护下加入碳酸铯(3.52g,10.8mmol)和甲烷磺酸(2-二环己基膦)-3,6-二甲氧基-2,4,6-三异丙基-1,1-联苯)(2-氨基-1,1-联苯-2-基)钯(II)(327mg,0.361mmol)。反应液在95℃下搅拌12小时。向反应液中加入水(90mL),用乙酸乙酯(90mL×3)萃取,合并有机相用饱和食盐水(90mL×1)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩,粗产物经薄层层析法(2:1石油醚/乙酸乙酯)分离纯化得到化合物33-2。MS-ESI计算值[M+H] +521,实测值521。 Compound 32-3 (860mg, 3.61mmol) and compound 33-1 (1.44g, 3.61mmol) were dissolved in N,N-dimethylformamide (20mL), and cesium carbonate (3.52g, 10.8 mmol) and methanesulfonic acid (2-dicyclohexylphosphine)-3,6-dimethoxy-2,4,6-triisopropyl-1,1-biphenyl) (2-amino-1,1 -biphenyl-2-yl)palladium(II) (327 mg, 0.361 mmol). The reaction solution was stirred at 95°C for 12 hours. Water (90 mL) was added to the reaction solution, extracted with ethyl acetate (90 mL×3), the combined organic phases were washed with saturated brine (90 mL×1), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain the crude product Compound 33-2 was obtained by separation and purification by thin layer chromatography (2:1 petroleum ether/ethyl acetate). MS-ESI calculated value [M+H] + 521, found value 521.
第三步third step
将化合物33-2(800mg,1.53mmol)溶于乙酸乙酯(2mL),向其中加入氯化氢乙酸乙酯溶液(4mol/L,3.84mL),反应液在25℃搅拌1小时。反应液减压浓缩得到化合物33-3的盐酸盐。MS-ESI计算值[M+H] +421,实 测值421。 Compound 33-2 (800mg, 1.53mmol) was dissolved in ethyl acetate (2mL), hydrogen chloride ethyl acetate solution (4mol/L, 3.84mL) was added thereto, and the reaction solution was stirred at 25°C for 1 hour. The reaction solution was concentrated under reduced pressure to obtain the hydrochloride of compound 33-3. MS-ESI calculated value [M+H] + 421, found value 421.
第四步the fourth step
将化合物33-3(161mg,0.326mmol)和化合物33-4(74.5mg,0.391mmol)溶于二氯甲烷(4mL),0℃下加入N,N-二异丙基乙胺(84.2mg,0.652mmol),反应液在25℃下搅拌12小时。向反应液中加入水(20mL),用乙酸乙酯(20mL×3)萃取,合并有机相用饱和食盐水(20mL×1)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩,粗产物经薄层层析法(2:1石油醚/乙酸乙酯)分离纯化得到化合物33-5。MS-ESI计算值[M+H] +575,实测值575。 Compound 33-3 (161 mg, 0.326 mmol) and compound 33-4 (74.5 mg, 0.391 mmol) were dissolved in dichloromethane (4 mL), and N,N-diisopropylethylamine (84.2 mg, 0.652mmol), and the reaction solution was stirred at 25°C for 12 hours. Water (20mL) was added to the reaction solution, extracted with ethyl acetate (20mL×3), the combined organic phases were washed with saturated brine (20mL×1), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain the crude product Compound 33-5 was obtained by separation and purification by thin layer chromatography (2:1 petroleum ether/ethyl acetate). MS-ESI calculated [M+H] + 575, found 575.
第五步the fifth step
将化合物33-5(23.0mg,40.0μmol)溶于四氢呋喃(1mL)和水(1mL),向反应液中加入氢氧化钠(3.20mg,79.9μmol)。反应液在50℃下搅拌12小时。将反应液用盐酸(1mol/mL)调节pH至7,用乙酸乙酯(20mL×3)萃取,无水硫酸钠干燥,过滤,滤液减压浓缩,粗产物经过制备高效液相色谱法分离(中性,碳酸氢铵体系)得到化合物33。 1H NMR(400MHz,CD 3OD)δ7.51-7.45(m,3H),7.42-7.39(m,3H),7.25-7.23(m,1H),7.04-7.03(m,1H),6.72-6.70(m,1H),6.41-6.38(m,1H),4.60-4.59(m,2H),4.48-4.47(m,2H),4.30-4.28(m,1H),4.02-4.01(m,2H),3.19-3.13(m,1H),2.39-2.30(m,5H),1.85-1.74(m,2H)。MS-ESI计算值[M+H] +561,实测值561。 Compound 33-5 (23.0 mg, 40.0 μmol) was dissolved in tetrahydrofuran (1 mL) and water (1 mL), and sodium hydroxide (3.20 mg, 79.9 μmol) was added to the reaction solution. The reaction solution was stirred at 50°C for 12 hours. The reaction solution was adjusted to pH 7 with hydrochloric acid (1mol/mL), extracted with ethyl acetate (20mL×3), dried over anhydrous sodium sulfate, filtered, the filtrate was concentrated under reduced pressure, and the crude product was separated by preparative high performance liquid chromatography ( Neutral, ammonium bicarbonate system) to obtain compound 33. 1 H NMR (400MHz, CD 3 OD) δ7.51-7.45 (m, 3H), 7.42-7.39 (m, 3H), 7.25-7.23 (m, 1H), 7.04-7.03 (m, 1H), 6.72- 6.70(m,1H),6.41-6.38(m,1H),4.60-4.59(m,2H),4.48-4.47(m,2H),4.30-4.28(m,1H),4.02-4.01(m,2H ), 3.19-3.13 (m, 1H), 2.39-2.30 (m, 5H), 1.85-1.74 (m, 2H). MS-ESI calculated value [M+H] + 561, found value 561.
实施例34Example 34
合成路线:synthetic route:

第一步first step
将化合物33-3(200mg,0.405mmol)和化合物34-1(109mg,0.526mmol)溶于吡啶(3mL),反应液在60℃下搅拌12小时。将反应液减压浓缩,粗产物经薄层层析法(2:1石油醚/乙酸乙酯)分离纯化得到化合物34-2。MS-ESI计算值[M+H] +591,实测值591。 Compound 33-3 (200 mg, 0.405 mmol) and compound 34-1 (109 mg, 0.526 mmol) were dissolved in pyridine (3 mL), and the reaction solution was stirred at 60° C. for 12 hours. The reaction solution was concentrated under reduced pressure, and the crude product was separated and purified by thin layer chromatography (2:1 petroleum ether/ethyl acetate) to obtain compound 34-2. MS-ESI calculated [M+H] + 591, found 591.
第二步second step
将化合物34-2(20.0mg,33.8μmol)溶于四氢呋喃(1mL)和水(1mL),向反应液中加入氢氧化钠(2.70 mg,67.6μmol)。反应液在55℃下搅拌3小时。将反应液减压浓缩,粗产物经过制备高效液相色谱法分离(中性,碳酸氢铵体系)得到化合物34。 1H NMR(400MHz,CD 3OD)δ7.33-7.32(m,1H),7.29-7.27(m,2H),7.13-7.12(m,1H),7.12-7.11(m,1H),7.11-7.10(m,1H),7.00-6.94(m,2H),6.61-6.59(m,1H),6.31-6.30(m,1H),4.48-4.47(m,1H),4.39-4.36(m,2H),4.23-4.19(m,1H),3.92-3.88(m,2H),3.58(s,3H),3.16-3.15(m,1H),3.06-3.00(m,1H),2.25-2.20(m,2H),1.70-1.65(m,2H)。MS-ESI计算值[M+H] +577,实测值577。 Compound 34-2 (20.0 mg, 33.8 μmol) was dissolved in tetrahydrofuran (1 mL) and water (1 mL), and sodium hydroxide (2.70 mg, 67.6 μmol) was added to the reaction solution. The reaction solution was stirred at 55°C for 3 hours. The reaction solution was concentrated under reduced pressure, and the crude product was separated by preparative high performance liquid chromatography (neutral, ammonium bicarbonate system) to obtain compound 34. 1 H NMR (400MHz, CD 3 OD) δ7.33-7.32(m,1H),7.29-7.27(m,2H),7.13-7.12(m,1H),7.12-7.11(m,1H),7.11- 7.10(m,1H),7.00-6.94(m,2H),6.61-6.59(m,1H),6.31-6.30(m,1H),4.48-4.47(m,1H),4.39-4.36(m,2H ),4.23-4.19(m,1H),3.92-3.88(m,2H),3.58(s,3H),3.16-3.15(m,1H),3.06-3.00(m,1H),2.25-2.20(m ,2H), 1.70-1.65(m,2H). MS-ESI calculated [M+H] + 577, found 577.
实施例35Example 35
合成路线:synthetic route:

第一步first step
将化合物33-3(200mg,0.405mmol)和化合物35-1(118mg,0.526mmol)溶于吡啶(3mL),反应液在60℃下搅拌12小时。将反应液减压浓缩,粗产物经薄层层析法(2:1石油醚/乙酸乙酯)分离纯化得到化合物35-2。MS-ESI计算值[M+H] +609,实测值609。 Compound 33-3 (200 mg, 0.405 mmol) and compound 35-1 (118 mg, 0.526 mmol) were dissolved in pyridine (3 mL), and the reaction solution was stirred at 60° C. for 12 hours. The reaction solution was concentrated under reduced pressure, and the crude product was separated and purified by thin layer chromatography (2:1 petroleum ether/ethyl acetate) to obtain compound 35-2. MS-ESI calculated value [M+H] + 609, found value 609.
第二步second step
将化合物35-2(25mg,41.0μmol)溶于四氢呋喃(1mL)和水(1mL),向反应液中加入氢氧化钠(3.28mg,82.0μmol)。反应液在55℃下搅拌3小时。将反应液减压浓缩,粗产物经过制备高效液相色谱法分离(中性,碳酸氢铵体系)得到化合物35。 1H NMR(400MHz,CD 3OD)δ7.40-7.38(m,3H),7.30-7.29(m,2H),7.25-7.23(m,1H),7.08-7.07(m,1H),6.74-6.72(m,1H),6.44-6.42(m,1H),4.58-4.57(m,1H),4.52-4.48(m,2H),4.39-4.38(m,1H),4.05-4.01(m,2H),3.70(s,3H),3.17-3.16(m,1H),3.14-3.11(m,1H),2.31-2.23(m,2H),1.82-1.78(m,2H)。MS-ESI计算值[M+H] +595,实测值595。 Compound 35-2 (25 mg, 41.0 μmol) was dissolved in tetrahydrofuran (1 mL) and water (1 mL), and sodium hydroxide (3.28 mg, 82.0 μmol) was added to the reaction solution. The reaction solution was stirred at 55°C for 3 hours. The reaction solution was concentrated under reduced pressure, and the crude product was separated by preparative high performance liquid chromatography (neutral, ammonium bicarbonate system) to obtain compound 35. 1 H NMR (400MHz, CD 3 OD) δ7.40-7.38(m,3H),7.30-7.29(m,2H),7.25-7.23(m,1H),7.08-7.07(m,1H),6.74- 6.72(m,1H),6.44-6.42(m,1H),4.58-4.57(m,1H),4.52-4.48(m,2H),4.39-4.38(m,1H),4.05-4.01(m,2H ), 3.70(s,3H), 3.17-3.16(m,1H), 3.14-3.11(m,1H), 2.31-2.23(m,2H), 1.82-1.78(m,2H). MS-ESI calculated [M+H] + 595, found 595.
实施例36Example 36
合成路线:synthetic route:

第一步first step
将化合物33-3(200mg,0.405mmol)和化合物36-1(111mg,0.526mmol)溶于吡啶(3mL),反应液在60℃下搅拌12小时。将反应液减压浓缩,粗产物经薄层层析法(2:1石油醚/乙酸乙酯)分离纯化得到化合物36-2。MS-ESI计算值[M+H] +595,实测值595。 Compound 33-3 (200 mg, 0.405 mmol) and compound 36-1 (111 mg, 0.526 mmol) were dissolved in pyridine (3 mL), and the reaction solution was stirred at 60° C. for 12 hours. The reaction solution was concentrated under reduced pressure, and the crude product was separated and purified by thin layer chromatography (2:1 petroleum ether/ethyl acetate) to obtain compound 36-2. MS-ESI calculated [M+H] + 595, found 595.
第二步second step
将化合物36-2(23mg,38.6μmol)溶于四氢呋喃(1mL)和水(1mL),向反应液中加入氢氧化钠(3.09mg,77.2μmol)。反应液在50℃下搅拌12小时。将反应液减压浓缩,粗产物经过制备高效液相色谱法分离(中性,碳酸氢铵体系)得到化合物36。 1H NMR(400MHz,CD 3OD)δ7.57-7.53(m,3H),7.43-7.42(m,1H),7.29-7.27(m,2H),7.13-7.09(m,1H),6.89-6.88(m,1H),6.62-6.60(m,1H),6.31-6.28(m,1H),4.50-4.49(m,2H),4.39-4.37(m,2H),4.23-4.22(m,1H),3.94-3.92(m,2H),3.08-3.05(m,1H),2.24-2.21(m,2H),1.72-1.68(m,2H)。MS-ESI计算值[M+H] +581,实测值581。 Compound 36-2 (23 mg, 38.6 μmol) was dissolved in tetrahydrofuran (1 mL) and water (1 mL), and sodium hydroxide (3.09 mg, 77.2 μmol) was added to the reaction solution. The reaction solution was stirred at 50°C for 12 hours. The reaction solution was concentrated under reduced pressure, and the crude product was separated by preparative high performance liquid chromatography (neutral, ammonium bicarbonate system) to obtain compound 36. 1 H NMR (400MHz, CD 3 OD) δ7.57-7.53 (m, 3H), 7.43-7.42 (m, 1H), 7.29-7.27 (m, 2H), 7.13-7.09 (m, 1H), 6.89- 6.88(m,1H),6.62-6.60(m,1H),6.31-6.28(m,1H),4.50-4.49(m,2H),4.39-4.37(m,2H),4.23-4.22(m,1H ), 3.94-3.92(m,2H), 3.08-3.05(m,1H), 2.24-2.21(m,2H), 1.72-1.68(m,2H). MS-ESI calculated value [M+H] + 581, found value 581.
实施例37Example 37
合成路线:synthetic route:

第一步first step
将化合物37-1(5.00g,31.4mmol)溶于盐酸(30mL,12M),-5℃下滴加亚硝酸钠(2.28g,33.0mmol)的水(12mL)溶液,反应液在-5℃下搅拌1小时。向已通二氧化硫气至饱和的醋酸溶液(90mL)中加入氯化亚铜(156mg,1.57mmol)和氯化铜(2.32g,17.3mmol),-5℃下将反应液缓慢滴加入二氧化硫的醋酸液中,反应液在25℃搅拌1.5小时。将反应液滴加入水(100mL)中,用二氯甲烷(80mL×3)萃取,合并有机相用饱和食盐水(80mL×1)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩得到化合物37-2。 1H NMR(400MHz,CD 3OD)δ7.94-7.92(m,1H),7.82-7.81(m,1H),7.70-7.66(m,1H),7.56-7.55(m,1H),6.82-6.59(m,1H)。 Compound 37-1 (5.00g, 31.4mmol) was dissolved in hydrochloric acid (30mL, 12M), and a solution of sodium nitrite (2.28g, 33.0mmol) in water (12mL) was added dropwise at -5°C, and the reaction solution was maintained at -5°C Stir for 1 hour. Add cuprous chloride (156mg, 1.57mmol) and cupric chloride (2.32g, 17.3mmol) to the acetic acid solution (90mL) that has been saturated with sulfur dioxide gas, and slowly add the reaction solution to the acetic acid solution of sulfur dioxide dropwise at -5°C The reaction solution was stirred at 25°C for 1.5 hours. The reaction solution was added dropwise into water (100 mL), extracted with dichloromethane (80 mL×3), the combined organic phase was washed with saturated brine (80 mL×1), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain the compound 37-2. 1 H NMR (400MHz, CD 3 OD) δ7.94-7.92(m,1H),7.82-7.81(m,1H),7.70-7.66(m,1H),7.56-7.55(m,1H),6.82- 6.59 (m, 1H).
第二步second step
将化合物33-3(200mg,0.405mmol)和化合物37-2(295mg,1.21mmol)溶于二氯甲烷(5mL),0℃下加入N,N-二异丙基乙胺(105mg,0.809mmol),反应液在25℃下搅拌3小时。向反应液减压浓缩,粗产物经薄层层析法(2:1石油醚/乙酸乙酯)分离纯化得到化合物37-3。MS-ESI计算值[M+H] +627,实测值627。 Compound 33-3 (200mg, 0.405mmol) and compound 37-2 (295mg, 1.21mmol) were dissolved in dichloromethane (5mL), and N,N-diisopropylethylamine (105mg, 0.809mmol) was added at 0°C ), and the reaction solution was stirred at 25°C for 3 hours. The reaction solution was concentrated under reduced pressure, and the crude product was separated and purified by thin layer chromatography (2:1 petroleum ether/ethyl acetate) to obtain compound 37-3. MS-ESI calculated [M+H] + 627, found 627.
第三步third step
将化合物37-3(15mg,23.9μmol)溶于四氢呋喃(1mL)和水(1mL),向反应液中加入氢氧化钠(1.91mg,47.8μmol)。反应液在55℃下搅拌3小时。将反应液减压浓缩,粗产物经过制备高效液相色谱法分离(中性,碳酸氢铵体系)得到化合物37。 1H NMR(400MHz,CD 3OD)δ7.57-7.55(m,2H),7.42-7.38(m,4H),7.23-7.22(m,1H),7.05-7.04(m,1H),6.82-6.64(m,2H),6.44-6.43(m,1H),4.61-4.60(m,1H),4.51-4.47(m,2H),4.36-4.32(m,1H),4.06-4.04(m,2H),3.37-3.36(m,1H),3.19-3.16(m,1H),2.41-2.37(m,2H),1.86-1.84(m,1H),1.82-1.73(m,1H)。MS-ESI计算值[M+H] +613,实测值613。 Compound 37-3 (15 mg, 23.9 μmol) was dissolved in tetrahydrofuran (1 mL) and water (1 mL), and sodium hydroxide (1.91 mg, 47.8 μmol) was added to the reaction solution. The reaction solution was stirred at 55°C for 3 hours. The reaction solution was concentrated under reduced pressure, and the crude product was separated by preparative high performance liquid chromatography (neutral, ammonium bicarbonate system) to obtain compound 37. 1 H NMR (400MHz, CD 3 OD) δ7.57-7.55(m,2H),7.42-7.38(m,4H),7.23-7.22(m,1H),7.05-7.04(m,1H),6.82- 6.64(m,2H),6.44-6.43(m,1H),4.61-4.60(m,1H),4.51-4.47(m,2H),4.36-4.32(m,1H),4.06-4.04(m,2H ), 3.37-3.36(m,1H), 3.19-3.16(m,1H), 2.41-2.37(m,2H), 1.86-1.84(m,1H), 1.82-1.73(m,1H). MS-ESI calculated [M+H] + 613, found 613.
实施例38Example 38
合成路线:synthetic route:

第一步first step
将化合物31-2(530mg,1.76mmol)溶于无水四氢呋喃(5mL)中,向溶液中滴加异丙基氯化镁(0.995mL,1.99mmol),反应液在16℃下搅拌1小时。将化合物38-1(200mg,1.17mmol)溶于无水四氢呋喃(2mL)中,在0℃下滴加到上述反应液中,反应液在16℃下搅拌10小时。反应液用10%柠檬酸水溶液(30mL)淬灭,用乙酸乙酯(30mL×3)萃取,合并有机相用饱和食盐水(100mL×1)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩。剩余物用薄层层析法(2:1石油醚/乙酸乙酯)分离纯化得到化合物38-2。 1H NMR(400MHz,CDCl 3)δ7.35-7.33(m,1H),7.21-7.19(m,1H),7.07(s,1H),4.29-4.27(m,2H),4.08-4.05(m,2H),3.88(s,3H),1.37(s,9H)。MS-ESI计算值[M-56+H] +292,实测值292。 Compound 31-2 (530 mg, 1.76 mmol) was dissolved in anhydrous tetrahydrofuran (5 mL), isopropylmagnesium chloride (0.995 mL, 1.99 mmol) was added dropwise to the solution, and the reaction solution was stirred at 16°C for 1 hour. Compound 38-1 (200mg, 1.17mmol) was dissolved in anhydrous tetrahydrofuran (2mL), added dropwise to the above reaction solution at 0°C, and the reaction solution was stirred at 16°C for 10 hours. The reaction solution was quenched with 10% citric acid aqueous solution (30mL), extracted with ethyl acetate (30mL×3), the combined organic phase was washed with saturated brine (100mL×1), dried over anhydrous sodium sulfate, filtered, and the filtrate was decompressed concentrate. The residue was separated and purified by thin layer chromatography (2:1 petroleum ether/ethyl acetate) to obtain compound 38-2. 1 H NMR (400MHz, CDCl 3 )δ7.35-7.33(m,1H),7.21-7.19(m,1H),7.07(s,1H),4.29-4.27(m,2H),4.08-4.05(m ,2H), 3.88(s,3H), 1.37(s,9H). MS-ESI calculated value [M-56+H] + 292, found value 292.
第二步second step
将化合物38-2(100mg,0.288mmol)溶于乙酸乙酯(3mL)中,向其中加入氯化氢乙酸乙酯(4M,3mL, 12.0mmol)。反应液在17℃搅拌2小时。反应液减压浓缩得到化合物38-3的盐酸盐。MS-ESI计算值[M+H] +248,实测值248。 Compound 38-2 (100 mg, 0.288 mmol) was dissolved in ethyl acetate (3 mL), and ethyl hydrogen chloride (4M, 3 mL, 12.0 mmol) was added thereto. The reaction solution was stirred at 17°C for 2 hours. The reaction solution was concentrated under reduced pressure to obtain the hydrochloride of compound 38-3. MS-ESI calculated [M+H] + 248, found 248.
第三步third step
将化合物1-8(100mg,0.197mmol),38-3的盐酸盐(82mg,0.332mmol),碳酸铯(192mg,0.590mmol)和甲烷磺酸(2-二环己基膦)-3,6-二甲氧基-2,4,6-三异丙基-1,1-联苯)(2-氨基-1,1-联苯-2-基)钯(II)(17.8mg,19.7umol)溶于N,N-二甲基甲酰胺(5mL)中,反应液在氮气保护下90℃搅拌反应12小时。反应液经硅藻土过滤,滤液减压浓缩。剩余物用薄层层析法(2:1石油醚/乙酸乙酯)分离纯化得到化合物38-4。MS-ESI计算值[M+H] +675,实测值675。 Compound 1-8 (100mg, 0.197mmol), 38-3 hydrochloride (82mg, 0.332mmol), cesium carbonate (192mg, 0.590mmol) and methanesulfonic acid (2-dicyclohexylphosphine)-3,6 -Dimethoxy-2,4,6-triisopropyl-1,1-biphenyl)(2-amino-1,1-biphenyl-2-yl)palladium(II) (17.8mg, 19.7umol ) was dissolved in N,N-dimethylformamide (5 mL), and the reaction solution was stirred and reacted at 90°C for 12 hours under the protection of nitrogen. The reaction solution was filtered through diatomaceous earth, and the filtrate was concentrated under reduced pressure. The residue was separated and purified by thin layer chromatography (2:1 petroleum ether/ethyl acetate) to obtain compound 38-4. MS-ESI calculated [M+H] +675 , found 675.
第四步the fourth step
将化合物38-4(70.0mg,0.104mmol)溶于四氢呋喃(2mL)和水(4mL)中,向其中加入氢氧化钠(12.5mg,0.311mmol),反应液在50℃下搅拌10小时。反应液减压浓缩,剩余物用水(20mL)稀释,用盐酸(1M)调节pH到4,混合物用乙酸乙酯(20mL×3)萃取,合并有机相,用饱和食盐水(50mL×1)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩。粗产物经高效液相色谱法(中性,碳酸氢铵体系)分离纯化得到化合物38。 1H NMR(400MHz,CD 3OD)δ8.00-7.92(m,3H),7.78-7.74(m,1H),7.58(d,J=8.0Hz,1H),7.31-7.27(m,2H),7.00(d,J=2.4Hz,1H),6.72(d,J=8.8Hz,1H),6.40(dd,J=8.8,2.8Hz,1H),4.41-4.37(m,1H),4.24(d,J=8.4Hz,2H),4.13(d,J=8.4Hz,2H),3.88(s,3H),3.25-3.19(m,2H),2.42-2.34(m,2H),1.88-1.73(m,2H)。MS-ESI计算值[M+H] +661,实测值661。 Compound 38-4 (70.0mg, 0.104mmol) was dissolved in tetrahydrofuran (2mL) and water (4mL), sodium hydroxide (12.5mg, 0.311mmol) was added thereto, and the reaction solution was stirred at 50°C for 10 hours. The reaction solution was concentrated under reduced pressure, the residue was diluted with water (20mL), adjusted to pH 4 with hydrochloric acid (1M), the mixture was extracted with ethyl acetate (20mL×3), the organic phases were combined, and washed with saturated brine (50mL×1) , dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The crude product was separated and purified by high performance liquid chromatography (neutral, ammonium bicarbonate system) to obtain compound 38. 1 H NMR (400MHz, CD 3 OD) δ8.00-7.92(m, 3H), 7.78-7.74(m, 1H), 7.58(d, J=8.0Hz, 1H), 7.31-7.27(m, 2H) ,7.00(d,J=2.4Hz,1H),6.72(d,J=8.8Hz,1H),6.40(dd,J=8.8,2.8Hz,1H),4.41-4.37(m,1H),4.24( d,J=8.4Hz,2H),4.13(d,J=8.4Hz,2H),3.88(s,3H),3.25-3.19(m,2H),2.42-2.34(m,2H),1.88-1.73 (m,2H). MS-ESI calculated [M+H] + 661, found 661.
实施例39Example 39
合成路线:synthetic route:

第一步first step
将化合物38-2(100mg,0.288mmol)溶于无水四氢呋喃(5mL)中,冷至0℃,向溶液中加入钠氢(16.1mg,0.403mmol,60%),反应液在0℃下搅拌1小时。将碘甲烷(960mg,6.76mmol)加入0℃的反应液中,反应 液在17℃下搅拌10小时。反应液用饱和氯化铵水溶液(20mL)淬灭,用乙酸乙酯(20mL×3)萃取,合并有机相用饱和食盐水(50mL×1)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩。剩余物用薄层层析法(4:1石油醚/乙酸乙酯)分离纯化得到化合物39-1。 1H NMR(400MHz,CDCl 3)δ7.22(d,J=8.0Hz,1H),7.17(d,J=8.0Hz,1H),7.06(s,1H),4.24-4.22(m,2H),4.15-4.13(m,2H),3.83(s,3H),3.01(s,3H),1.38(s,9H)。MS-ESI计算值[M-56+H] +306,实测值306。 Compound 38-2 (100mg, 0.288mmol) was dissolved in anhydrous tetrahydrofuran (5mL), cooled to 0°C, sodium hydrogen (16.1mg, 0.403mmol, 60%) was added to the solution, and the reaction solution was stirred at 0°C 1 hour. Iodomethane (960mg, 6.76mmol) was added into the reaction solution at 0°C, and the reaction solution was stirred at 17°C for 10 hours. The reaction solution was quenched with saturated aqueous ammonium chloride (20 mL), extracted with ethyl acetate (20 mL×3), the combined organic phases were washed with saturated brine (50 mL×1), dried over anhydrous sodium sulfate, filtered, and the filtrate was decompressed concentrate. The residue was separated and purified by thin layer chromatography (4:1 petroleum ether/ethyl acetate) to obtain compound 39-1. 1 H NMR (400MHz, CDCl 3 ) δ7.22(d, J=8.0Hz, 1H), 7.17(d, J=8.0Hz, 1H), 7.06(s, 1H), 4.24-4.22(m, 2H) ,4.15-4.13(m,2H),3.83(s,3H),3.01(s,3H),1.38(s,9H). MS-ESI calculated value [M-56+H] + 306, found value 306.
第二步second step
将化合物39-1(120mg,0.332mmol)溶于乙酸乙酯(3mL)中,向其中加入氯化氢乙酸乙酯(4M,6mL,24.0mmol)。反应液在17℃搅拌1小时。反应液减压浓缩得到化合物39-2的盐酸盐。MS-ESI计算值[M+H] +262,实测值262。 Compound 39-1 (120 mg, 0.332 mmol) was dissolved in ethyl acetate (3 mL), and ethyl hydrogen chloride acetate (4M, 6 mL, 24.0 mmol) was added thereto. The reaction was stirred at 17°C for 1 hour. The reaction solution was concentrated under reduced pressure to obtain the hydrochloride salt of compound 39-2. MS-ESI calculated [M+H] + 262, found 262.
第三步third step
将化合物1-8(180mg,0.354mmol),39-2的盐酸盐(114mg,0.383mmol),碳酸铯(346mg,1.06mmol)和甲烷磺酸(2-二环己基膦)-3,6-二甲氧基-2,4,6-三异丙基-1,1-联苯)(2-氨基-1,1-联苯-2-基)钯(II)(32.1mg,35.4umol)溶于无水N,N-二甲基甲酰胺(6mL)中,反应液在氮气保护下90℃搅拌反应12小时。反应液经硅藻土过滤,滤液减压浓缩。剩余物用薄层层析法(2:1石油醚/乙酸乙酯)分离纯化得到化合物39-3。MS-ESI计算值[M+H] +689,实测值689。 Compound 1-8 (180mg, 0.354mmol), hydrochloride (114mg, 0.383mmol) of 39-2, cesium carbonate (346mg, 1.06mmol) and methanesulfonic acid (2-dicyclohexylphosphine)-3,6 -Dimethoxy-2,4,6-triisopropyl-1,1-biphenyl)(2-amino-1,1-biphenyl-2-yl)palladium(II) (32.1mg, 35.4umol ) was dissolved in anhydrous N,N-dimethylformamide (6 mL), and the reaction solution was stirred and reacted at 90°C for 12 hours under nitrogen protection. The reaction solution was filtered through diatomaceous earth, and the filtrate was concentrated under reduced pressure. The residue was separated and purified by thin layer chromatography (2:1 petroleum ether/ethyl acetate) to obtain compound 39-3. MS-ESI calculated [M+H] + 689, found 689.
第四步the fourth step
将化合物39-3(110mg,0.160mmol)溶于四氢呋喃(2mL)和水(4mL)中,向其中加入氢氧化钠(19.2mg,0.479mmol),反应液在50℃下搅拌10小时。反应液减压浓缩,剩余物用水(20mL)稀释,用盐酸(1M)调节pH到4,混合物用乙酸乙酯(20mL×3)萃取,合并有机相,用饱和食盐水(50mL×1)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩。粗产物经高效液相色谱法分离(中性,碳酸氢铵体系)纯化得到化合物39。 1H NMR(400MHz,CD 3OD)δ7.99-7.91(m,3H),7.78-7.74(m,1H),7.54(d,J=7.6Hz,1H),7.34-7.31(m,2H),7.03(d,J=2.8Hz,1H),6.72(d,J=8.8Hz,1H),6.43-6.40(m,1H),4.40-4.36(m,1H),4.26(d,J=9.2Hz,2H),4.13-4.10(m,2H),3.95(s,3H),3.37-3.35(m,1H),3.24-3.18(m,1H),3.17(s,3H),2.41-2.33(m,2H),1.89-1.73(m,2H)。MS-ESI计算值[M+H] +675,实测值675。 Compound 39-3 (110 mg, 0.160 mmol) was dissolved in tetrahydrofuran (2 mL) and water (4 mL), sodium hydroxide (19.2 mg, 0.479 mmol) was added thereto, and the reaction solution was stirred at 50° C. for 10 hours. The reaction solution was concentrated under reduced pressure, the residue was diluted with water (20mL), adjusted to pH 4 with hydrochloric acid (1M), the mixture was extracted with ethyl acetate (20mL×3), the organic phases were combined, and washed with saturated brine (50mL×1) , dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The crude product was separated and purified by high performance liquid chromatography (neutral, ammonium bicarbonate system) to obtain compound 39. 1 H NMR (400MHz, CD 3 OD) δ7.99-7.91(m, 3H), 7.78-7.74(m, 1H), 7.54(d, J=7.6Hz, 1H), 7.34-7.31(m, 2H) ,7.03(d,J=2.8Hz,1H),6.72(d,J=8.8Hz,1H),6.43-6.40(m,1H),4.40-4.36(m,1H),4.26(d,J=9.2 Hz,2H),4.13-4.10(m,2H),3.95(s,3H),3.37-3.35(m,1H),3.24-3.18(m,1H),3.17(s,3H),2.41-2.33( m,2H), 1.89-1.73(m,2H). MS-ESI calculated [M+H] +675 , found 675.
实施例40Example 40
合成路线:synthetic route:

第一步first step
将化合物38-4(200mg,0.296mmol)溶于无水二氯甲烷(8mL)中,冷至0℃,向溶液中加入二乙氨基三氟化硫(96.0mg,0.596mmol),反应液在18℃下搅拌10小时。反应液冷至0℃并用饱和碳酸氢钠水溶液(50mL)淬灭,用二氯甲烷(30mL×2)萃取,合并有机相用无水硫酸钠干燥,过滤,滤液减压浓缩。剩余物用薄层层析法(2:1石油醚/乙酸乙酯)分离纯化得到化合物40-1。MS-ESI计算值[M+H] +677,实测值677。 Compound 38-4 (200mg, 0.296mmol) was dissolved in anhydrous dichloromethane (8mL), cooled to 0°C, diethylaminosulfur trifluoride (96.0mg, 0.596mmol) was added to the solution, and the reaction solution was Stir at 18°C for 10 hours. The reaction solution was cooled to 0°C and quenched with saturated aqueous sodium bicarbonate (50 mL), extracted with dichloromethane (30 mL×2), combined organic phases were dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was separated and purified by thin layer chromatography (2:1 petroleum ether/ethyl acetate) to obtain compound 40-1. MS-ESI calculated [M+H] + 677, found 677.
第二步second step
将化合物40-1(220mg,0.325mmol)溶于四氢呋喃(2mL)和水(4mL)中,向其中加入氢氧化钠(39.0mg,0.975mmol),反应液在50℃下搅拌2小时。反应液减压浓缩,剩余物用水(20mL)稀释,用盐酸(1M)调节pH到4,混合物用乙酸乙酯(20mL×3)萃取,合并有机相,用饱和食盐水(50mL×1)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩。粗产物经高效液相色谱法分离(中性,碳酸氢铵体系)纯化得到化合物40。 1H NMR(400MHz,CD 3OD)δ8.01-7.93(m,3H),7.79-7.75(m,1H),7.63(d,J=7.6Hz,1H),7.35-7.32(m,2H),7.01(d,J=2.4Hz,1H),6.75(d,J=8.8Hz,1H),6.42(dd,J=8.8,2.8Hz,1H),4.41-4.37(m,3H),4.33(s,2H),3.87(s,3H),3.41-3.35(m,1H),3.27-3.20(m,1H),2.42-2.34(m,2H),1.89-1.72(m,2H)。MS-ESI计算值[M+H] +663,实测值663。 Compound 40-1 (220mg, 0.325mmol) was dissolved in tetrahydrofuran (2mL) and water (4mL), sodium hydroxide (39.0mg, 0.975mmol) was added thereto, and the reaction solution was stirred at 50°C for 2 hours. The reaction solution was concentrated under reduced pressure, the residue was diluted with water (20mL), adjusted to pH 4 with hydrochloric acid (1M), the mixture was extracted with ethyl acetate (20mL×3), the organic phases were combined, and washed with saturated brine (50mL×1) , dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The crude product was separated and purified by high performance liquid chromatography (neutral, ammonium bicarbonate system) to obtain compound 40. 1 H NMR (400MHz, CD 3 OD) δ8.01-7.93 (m, 3H), 7.79-7.75 (m, 1H), 7.63 (d, J=7.6Hz, 1H), 7.35-7.32 (m, 2H) ,7.01(d,J=2.4Hz,1H),6.75(d,J=8.8Hz,1H),6.42(dd,J=8.8,2.8Hz,1H),4.41-4.37(m,3H),4.33( s,2H), 3.87(s,3H), 3.41-3.35(m,1H), 3.27-3.20(m,1H), 2.42-2.34(m,2H), 1.89-1.72(m,2H). MS-ESI calculated [M+H] + 663, found 663.
生物学评价:Biological Evaluation:
实验例1:本发明化合物对RORγ体外活性的测定Experimental example 1: Determination of the in vitro activity of the compound of the present invention on RORγ
1.1实验材料及仪器见表1。1.1 The experimental materials and instruments are shown in Table 1.
表1实验材料及仪器Table 1 Experimental materials and instruments


1.2实验步骤1.2 Experimental steps
采用RORγ配体结合结构域(LBD)时间分辨荧光能量共振转移(TR-FRET)筛选本发明化合物对RORγ活性的调节。RORγ ligand-binding domain (LBD) time-resolved fluorescence energy transfer (TR-FRET) was used to screen the modulation of RORγ activity by the compounds of the present invention.
将待测化合物稀释在DMSO中,并进一步稀释在分析缓冲液(50mM Tris pH 7.0,50mM KCl,1mM Na-EDTA,0.1mM DTT,0.01%BSA)中(4倍稀释,10个浓度,最高浓度5000nM),最终DMSO浓度1%。将hRORγ-LBD蛋白稀释在分析缓冲液中,得到在384孔板中15nM的hRORγ-LBD最终浓度。在分析缓冲液中制备biotin-SRC1多肽(Biotin-SPSSHSSLTERHKILHRLLQEGSP)储液,并加入到各孔中(终浓度200nM)。将SA-eu(终浓度1nM)和SA-APC(终浓度50nM)的溶液也加入各孔中。The compound to be tested was diluted in DMSO and further diluted in assay buffer (50mM Tris pH 7.0, 50mM KCl, 1mM Na-EDTA, 0.1mM DTT, 0.01% BSA) (4-fold dilution, 10 concentrations, highest concentration 5000 nM), the final DMSO concentration was 1%. The hRORγ-LBD protein was diluted in assay buffer to give a final hRORγ-LBD concentration of 15 nM in 384-well plates. A stock solution of biotin-SRC1 polypeptide (Biotin-SPSSHSSLTERHKILHRLLQEGSP) was prepared in assay buffer and added to each well (final concentration 200 nM). Solutions of SA-eu (1 nM final concentration) and SA-APC (50 nM final concentration) were also added to each well.
将最终分析混合物在4℃下孵育过夜,室温平衡1小时,1000rpm离心1分钟。在Envision微孔板检测仪上检测荧光读数,通过GraphPad Prism软件绘制发射波长665nM/615nM的荧光信号的比值与化合物浓度的对数曲线,计算化合物的50%有效浓度(EC 50)和50%抑制浓度(IC 50)。最大应答(Emax)为通过GraphPad Prism拟合确定的信号的上限峰值。 The final assay mixture was incubated overnight at 4°C, equilibrated at room temperature for 1 hour, and centrifuged at 1000 rpm for 1 minute. Fluorescence readings were detected on the Envision microplate detector, and the logarithmic curve of the ratio of the fluorescence signal of emission wavelength 665nM/615nM to the compound concentration was drawn by GraphPad Prism software, and the 50% effective concentration (EC 50 ) and 50% inhibition of the compound were calculated Concentration ( IC50 ). The maximum response (Emax) was the upper peak peak of the signal determined by GraphPad Prism fit.
1.3实验结果见表2。1.3 The experimental results are shown in Table 2.
表2本发明化合物对RORγ体外活性的测定结果Table 2 The assay results of the compounds of the present invention to the in vitro activity of RORγ
“/”表示不适用。"/" indicates not applicable.
结论:本发明化合物对RORγ体外活性具有明显的激动作用或反向激动活性。Conclusion: the compound of the present invention has obvious agonistic or inverse agonistic activity on the in vitro activity of RORγ.
实验例2.本发明化合物促进CD4 +细胞分化为Th17细胞的能力测定 Experimental example 2. Determination of the ability of the compounds of the present invention to promote the differentiation of CD4 + cells into Th17 cells
2.1实验材料及仪器2.1 Experimental materials and instruments
1.C57BL/6小鼠脾脏1. C57BL/6 mouse spleen
2. 70μm滤网(BD)2. 70μm filter (BD)
3.红细胞裂解液(Absin)3. Red blood cell lysate (Absin)
4.Mouse CD4+T cell isolation kit(小鼠CD4+T细胞分选试剂盒)(Stemcell)4. Mouse CD4+T cell isolation kit (Mouse CD4+T cell isolation kit) (Stemcell)
5.CD3抗体(BD)5. CD3 antibody (BD)
6.CD28抗体(BD)6. CD28 antibody (BD)
7. 96孔U底板(Corning)7. 96-well U-base plate (Corning)
8.DPBS(Corning)8. DPBS (Corning)
9.TGFβ(R&D)9. TGFβ (R&D)
10.IL-6(R&D)10. IL-6 (R&D)
11.IL-1β(BioLegend)11. IL-1β (BioLegend)
12.IL-23(BioLegend)12. IL-23 (BioLegend)
13.RPMI 1640培养液(Gibco)13. RPMI 1640 culture medium (Gibco)
14.青/链霉素(HyClone)14. Penicillin/Streptomycin (HyClone)
15.胎牛血清(HyClone)15. Fetal bovine serum (HyClone)
16.非必需氨基酸(Gibco)16. Non-essential amino acids (Gibco)
17.β-巯基乙醇(Sigma)17. β-Mercaptoethanol (Sigma)
18.IFNγ抗体(BD)18. IFNγ antibody (BD)
19.IL-4抗体(BD)19. IL-4 antibody (BD)
20.PMA(sigma)20. PMA (sigma)
21.Ionomycin(离子霉素)(Invitrogen)21. Ionomycin (Ionomycin) (Invitrogen)
22.高尔基体抑制剂(BD)22. Golgi inhibitors (BD)
23.Staining buffer(染色缓冲液)(Biolegend)23. Staining buffer (Biolegend)
24.Fixation buffer(固定缓冲液)(Biolegend)24. Fixation buffer (Biolegend)
25.Permeabilization buffer(透化缓冲液)(Biolegend)25. Permeabilization buffer (Biolegend)
26.LIVE/DEAD stain kit(染色试剂盒)(Invitrogen)26. LIVE/DEAD stain kit (staining kit) (Invitrogen)
27.CD4抗体(Biolegend)27. CD4 antibody (Biolegend)
28.IL-17A抗体(Biolegend)28. IL-17A antibody (Biolegend)
29.细胞计数仪(Beckman)29. Cell counter (Beckman)
30.离心机(Eppendorf)30. Centrifuge (Eppendorf)
31.流式细胞仪(BD)31. Flow Cytometry (BD)
32.CO 2培养箱(Thermo) 32. CO2 incubator (Thermo)
2.2实验步骤2.2 Experimental steps
将CD3抗体于DPBS中稀释至5μg/mL,加入96孔U底板中,每孔50μL液体,于4℃包被过夜。The CD3 antibody was diluted to 5 μg/mL in DPBS, added to a 96-well U-bottom plate, 50 μL of liquid per well, and coated overnight at 4°C.
在培养液(RPMI 1640+10%胎牛血清+1%青链霉素+1%非必需氨基酸+0.05mMβ-巯基乙醇)中研磨C57BL/6小鼠脾脏,过70μm滤网制备单细胞悬液,300g离心3min。加入红细胞裂解液室温裂解3 min。使用Mouse CD4 +T细胞分离试剂盒分离CD4 +细胞。将前一天包被的96孔U底板取出,吸去包被的液体,并用DPBS洗两次,再将上面得到的CD4 +细胞以5*10 5/mL的密度接种至包被过的孔中,每孔200μL细胞悬液;再加入CD28抗体(3μg/mL)、TGFβ(3ng/mL)、IL-6(30ng/mL)、IL-23(10ng/mL)、IL-1β(10ng/mL)、IFNγ抗体(10μg/mL)和IL-4抗体(10μg/mL);再在孔中加入本发明化合物,于37℃,5%CO 2下培养3天。 Grind C57BL/6 mouse spleen in culture medium (RPMI 1640+10% fetal bovine serum+1% penicillin+streptomycin+1% non-essential amino acid+0.05mM β-mercaptoethanol), pass through a 70μm filter to prepare a single cell suspension , centrifuge at 300g for 3min. Red blood cell lysate was added to lyse at room temperature for 3 min. CD4 + cells were isolated using the Mouse CD4 + T cell isolation kit. Take out the 96-well U-bottom plate coated the day before, suck off the coated liquid, wash twice with DPBS, and inoculate the CD4 + cells obtained above at a density of 5*10 5 /mL into the coated wells , 200μL cell suspension per well; then add CD28 antibody (3μg/mL), TGFβ (3ng/mL), IL-6 (30ng/mL), IL-23 (10ng/mL), IL-1β (10ng/mL ), IFNγ antibody (10 μg/mL) and IL-4 antibody (10 μg/mL); then the compound of the present invention was added to the well, and cultured at 37° C. under 5% CO 2 for 3 days.
在每个孔中加入500ng/mL的PMA和离子霉素,以及高尔基体抑制剂(1:1000),37℃,5%CO 2下刺激4小时。 Add 500ng/mL of PMA and ionomycin, and Golgi inhibitor (1:1000) to each well, and stimulate for 4 hours at 37°C and 5% CO 2 .
刺激结束后将U底板300g离心3min,倒掉上清,用染色缓冲液洗两次。将CD4抗体和LIVE/DEAD染液分别按照1:200和1:1000于染色缓冲液中稀释,每孔加入50μL染液,4℃染色30min,之后用染色缓冲液洗涤细胞两次。每孔加入100μL固定缓冲液,室温固定20min,用透化缓冲液洗两次。将IL-17A抗体按照1:200于透化缓冲液中稀释,每孔加入50μL染液,室温染色30min,之后用染色缓冲液洗两次。最后用150μL染色缓冲液重悬细胞,使用流式细胞仪检测Th17细胞比例。After the stimulation, the U-bottom plate was centrifuged at 300 g for 3 min, the supernatant was discarded, and washed twice with staining buffer. Dilute CD4 antibody and LIVE/DEAD staining solution in staining buffer at 1:200 and 1:1000 respectively, add 50 μL of staining solution to each well, stain at 4°C for 30 min, and then wash cells twice with staining buffer. Add 100 μL of fixation buffer to each well, fix at room temperature for 20 min, and wash twice with permeabilization buffer. The IL-17A antibody was diluted 1:200 in permeabilization buffer, 50 μL of dye solution was added to each well, stained at room temperature for 30 min, and then washed twice with staining buffer. Finally, the cells were resuspended with 150 μL staining buffer, and the proportion of Th17 cells was detected by flow cytometry.
2.3实验结果见表3。2.3 The experimental results are shown in Table 3.
表3本发明化合物促进CD4 +细胞分化为Th17细胞的能力测定结果 Table 3 The compounds of the present invention promote the ability of CD4 + cells to differentiate into Th17 cells to measure results
化合物测试浓度:1μM。Compound test concentration: 1 μM.
结论:本发明化合物可以明显地促进CD4 +细胞分化为Th17细胞。 Conclusion: the compounds of the present invention can obviously promote the differentiation of CD4 + cells into Th17 cells.
实验例3:本发明化合物药代动力学评价Experimental example 3: Pharmacokinetic evaluation of the compound of the present invention
3.1实验目的:测试化合物在Balb/c小鼠体内的药代动力学3.1 Purpose of the experiment: to test the pharmacokinetics of compounds in Balb/c mice
3.2实验材料:3.2 Experimental materials:
Balb/c小鼠(雌性,7~9周龄,上海斯莱克)Balb/c mice (female, 7-9 weeks old, Shanghai Slack)
实验操作:Experimental operation:
以标准方案测试化合物静脉注射及口服给药后的啮齿类动物药代特征,实验中给予小鼠单次静脉注射(IV)及口服给药(PO)。静注溶媒为5%二甲基亚砜、30%的PEG400、65%的10%羟丙基β环糊精配成的混合溶媒。口服溶媒为0.5%羟丙甲纤维素和0.2%吐温配成的混合溶媒。该项目使用四只雌性Balb/c小鼠,两只小鼠进行静脉注射给药,给药剂量为0.5mg/kg,收集0h(给药前)和给药后0.0833,0.25,0.5,1,2,4,8,24h的血浆样品,另外两只小鼠口服灌胃给药,给药剂量为1mg/kg,收集0h(给药前)和给药后0.25,0.5,1,2,4,8,24h的血浆样品,收集24小时内的全血样品,3000g离心15分钟,分离上清得血浆样品,加入4倍体积含内标的乙腈溶液沉淀蛋白,离心取上清液加入等倍体积的水再离心取上清进样,以LC-MS/MS分析方法定量分析血药浓度,并计算药代参数,如达峰浓度(C max),清除率(CL),半衰期(T 1/2),组织分布(Vdss),药时曲线下面积(AUC 0-last),生物利用度(F)。 The pharmacokinetic characteristics of the compounds were tested in rodents after intravenous injection and oral administration according to the standard protocol. In the experiment, the mice were given a single intravenous injection (IV) and oral administration (PO). The solvent for intravenous injection is a mixed solvent made up of 5% dimethyl sulfoxide, 30% PEG400, and 65% 10% hydroxypropyl β-cyclodextrin. The oral vehicle is a mixed vehicle made of 0.5% hypromellose and 0.2% Tween. The project used four female Balb/c mice, two mice were administered intravenously, the dose was 0.5mg/kg, and the collection 0h (before administration) and after administration were 0.0833, 0.25, 0.5, 1, Plasma samples at 2, 4, 8, and 24 hours were administered orally to the other two mice at a dose of 1 mg/kg, collected at 0 h (before administration) and at 0.25, 0.5, 1, 2, and 4 hours after administration , 8, 24h plasma samples, collect whole blood samples within 24 hours, centrifuge at 3000g for 15 minutes, separate supernatant to obtain plasma samples, add 4 times volume of acetonitrile solution containing internal standard to precipitate protein, centrifuge to take supernatant and add equal volume The water was then centrifuged to take the supernatant sample, and the blood drug concentration was quantitatively analyzed by LC-MS/MS analysis method, and the pharmacokinetic parameters were calculated, such as peak concentration (C max ), clearance rate (CL), half-life (T 1 / 2 ), tissue distribution (Vdss), area under the drug-time curve (AUC 0-last ), bioavailability (F).
3.3实验结果见表4。3.3 The experimental results are shown in Table 4.
表4本发明化合物的药代动力学测试结果The pharmacokinetic test result of the compound of the present invention in table 4

结论:本发明化合物具有良好的药代动力学性质,包括良好的口服生物利用度,口服暴露量,半衰期和清除率等。Conclusion: The compounds of this invention have good pharmacokinetic properties, including good oral bioavailability, oral exposure, half-life and clearance rate.
实验例4:本发明化合物对MC38小鼠结肠癌移植瘤模型的体内药效学研究Experimental Example 4: In vivo pharmacodynamic study of the compound of the present invention on MC38 mouse colon cancer xenograft model
4.1实验目的:4.1 Purpose of the experiment:
本实验的目的是研究本发明化合物对MC38小鼠结肠癌移植瘤模型体内药效进行评估。The purpose of this experiment is to study the evaluation of the compound of the present invention on the MC38 mouse colon cancer xenograft tumor model in vivo.
4.2实验动物:4.2 Experimental animals:
种属:小鼠Species: Mouse
品系:C57BL/6小鼠Strain: C57BL/6 mice
周龄及体重:7周龄,体重18-23克Week age and weight: 7 weeks old, weight 18-23 grams
性别:雌性Gender: female
供应商:上海斯莱克实验动物有限公司Supplier: Shanghai Slack Experimental Animal Co., Ltd.
4.3实验方法与步骤4.3 Experimental methods and steps
4.3.1细胞培养4.3.1 Cell culture
名称:MC38(小鼠结肠癌细胞)Name: MC38 (Mouse Colon Cancer Cell)
来源:和元生物技术(上海)有限公司。由辉源生物科技(上海)有限公司保种维持传代。Source: Heyuan Biotechnology (Shanghai) Co., Ltd. The species was maintained and passed on by Huiyuan Biotechnology (Shanghai) Co., Ltd.
细胞培养:培养液为含有10%胎牛血清的1640培养基,培养条件为37℃,5%二氧化碳。传代比例为1:2~1:3,每周传代2~3次。Cell culture: the culture medium is 1640 medium containing 10% fetal bovine serum, and the culture conditions are 37°C and 5% carbon dioxide. The subculture ratio was 1:2~1:3, and subcultured 2~3 times a week.
4.3.2肿瘤细胞接种4.3.2 Tumor cell inoculation
将0.1mL(2×10 5个)细胞皮下接种于每只小鼠的右后背。同日将动物根据体重随机分组。 0.1 mL (2×10 5 ) cells were inoculated subcutaneously on the right back of each mouse. On the same day, animals were randomized into groups based on body weight.
4.3.3受试物的配制4.3.3 Preparation of test substances
实验用溶媒为5%DMSO/95%(20%羟丙基β环糊精)。受试物用溶媒溶解,配制成一定浓度均一溶液,于4℃保存。The experimental vehicle was 5% DMSO/95% (20% hydroxypropyl beta cyclodextrin). The test substance was dissolved in a solvent, prepared into a uniform solution with a certain concentration, and stored at 4°C.
4.3.4肿瘤测量和实验指标4.3.4 Tumor measurements and experimental indicators
实验指标是考察肿瘤生长是否被抑制、延缓或治愈。每周两次用游标卡尺测量肿瘤直径。肿瘤体积的计算公式为:V=0.5a×b 2,a和b分别表示肿瘤的长径和短径。 The experimental index is to investigate whether tumor growth is inhibited, delayed or cured. Tumor diameters were measured twice a week with vernier calipers. The calculation formula of tumor volume is: V=0.5a×b 2 , where a and b represent the long diameter and short diameter of the tumor respectively.
化合物的抑瘤疗效用相对肿瘤增殖率T/C(%)评价。相对肿瘤增殖率T/C(%):计算公式如下:T/C%=T RTV/C RTV×100%(T RTV:治疗组RTV;C RTV:阴性对照组RTV)。根据肿瘤测量的结果计算出相对肿瘤体积(relative tumor volume,RTV),计算公式为RTV=V t/V 0,其中V 0是分组给药时(即d 0)测量所得平均肿瘤体积,V t为某一次测量时的平均肿瘤体积,T RTV与C RTV取同一天数据。 The antitumor efficacy of compounds was evaluated by relative tumor proliferation rate T/C (%). Relative tumor proliferation rate T/C (%): the calculation formula is as follows: T/C%=T RTV /C RTV ×100% (T RTV : RTV of the treatment group; C RTV : RTV of the negative control group). According to the results of tumor measurement, the relative tumor volume (relative tumor volume, RTV) was calculated, and the calculation formula was RTV=V t /V 0 , where V 0 was the average tumor volume measured during group administration (that is, d 0 ), V t is the average tumor volume at a certain measurement, T RTV and C RTV take the data of the same day.
4.4实验结果见表5。4.4 The experimental results are shown in Table 5.
表5本发明化合物对MC38小鼠结肠癌移植瘤模型的抑瘤药效评价(基于给药后第31天肿瘤体积计算得出)Table 5 Evaluation of antitumor efficacy of compounds of the present invention on MC38 mouse colon cancer transplanted tumor model (calculated based on the tumor volume on the 31st day after administration)

PD-1单抗来源:BioXcell。PD-1单抗于分组后第7天开始给药,化合物32于分组当天开始给药。Source of PD-1 monoclonal antibody: BioXcell. PD-1 monoclonal antibody was administered on the 7th day after grouping, and compound 32 was administered on the day of grouping.
结论:本发明化合物与PD-1单抗联用对MC38小鼠结肠癌移植瘤模型具有优异的抑瘤效果。Conclusion: The combination of the compound of the present invention and PD-1 monoclonal antibody has an excellent tumor-inhibiting effect on the transplanted tumor model of MC38 mouse colon cancer.
Claims (17)





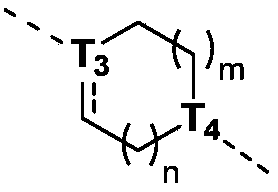



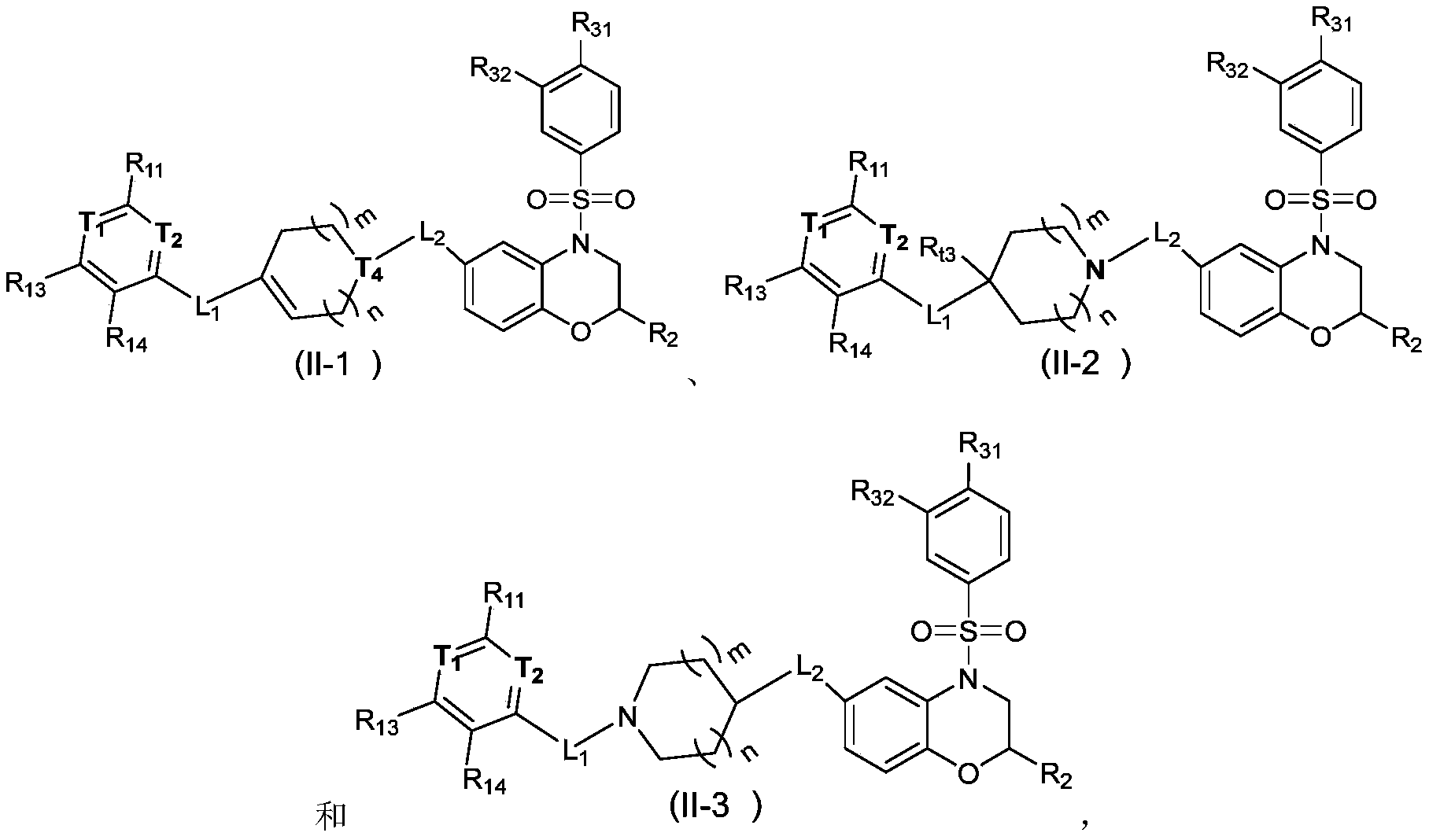







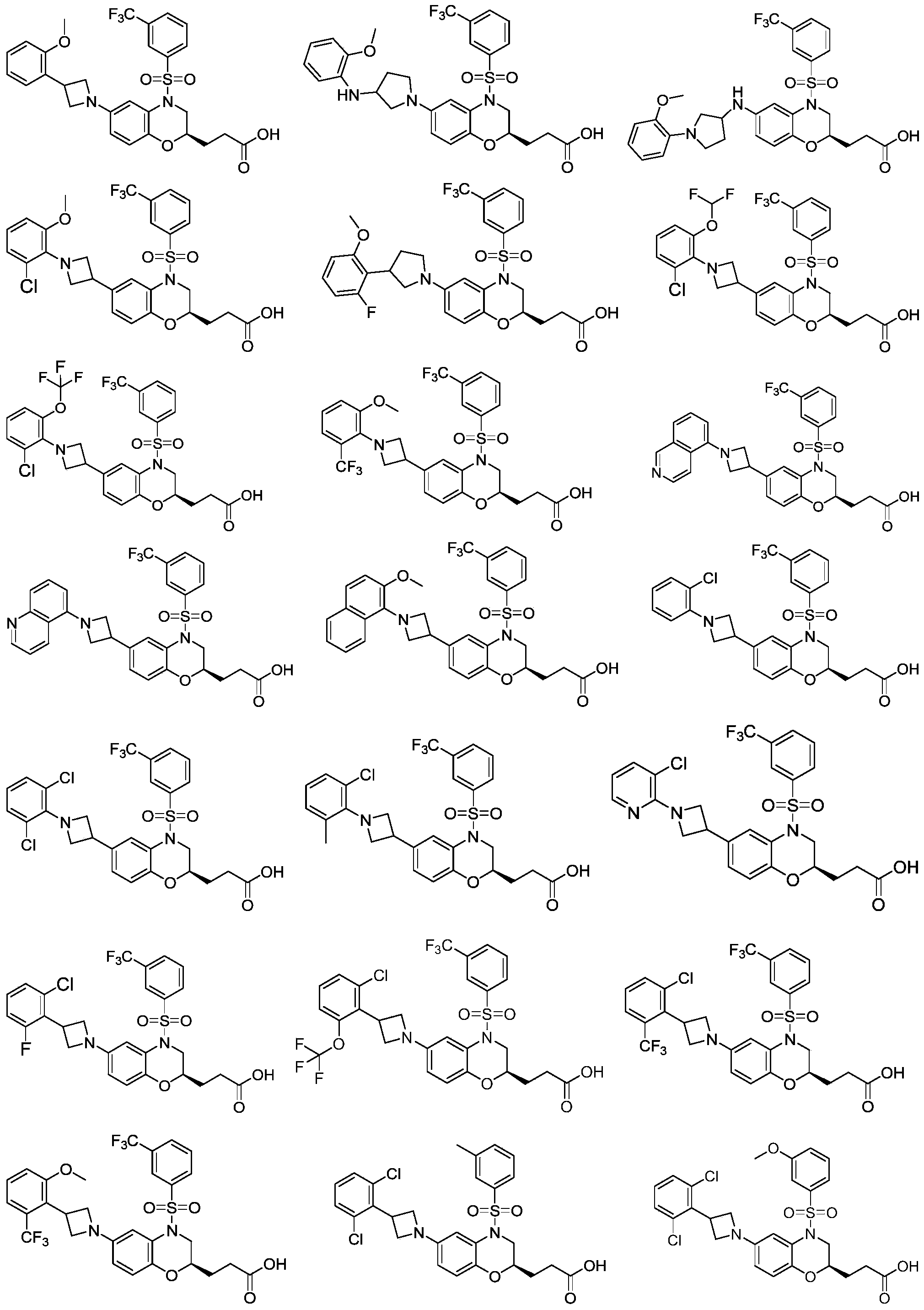

Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202110933021 | 2021-08-13 | ||
| CN202110933021.9 | 2021-08-13 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023016528A1 true WO2023016528A1 (en) | 2023-02-16 |
Family
ID=85199849
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2022/111884 Ceased WO2023016528A1 (en) | 2021-08-13 | 2022-08-11 | Class of benzomorpholine compounds, and preparation method therefor and use thereof |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2023016528A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023232870A1 (en) | 2022-05-31 | 2023-12-07 | Immunic Ag | Rorg/rorgt modulators for the treatment of virus infections like covid-19 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015171610A2 (en) * | 2014-05-05 | 2015-11-12 | Lycera Corporation | Tetrahydroquinoline sulfonamide and related compounds for use as agonists of rory and the treatment of disease |
| WO2016179343A1 (en) * | 2015-05-05 | 2016-11-10 | Lycera Corporation | DIHYDRO-2H-BENZO[b][1,4]OXAZINE SULFONAMIDE AND RELATED COMPOUNDS FOR USE AS AGONISTS OF RORy AND THE TREATMENT OF DISEASE |
| CN106132422A (en) * | 2014-02-27 | 2016-11-16 | 莱斯拉公司 | Adoptive cell therapy & related treatments using agonists of retinoic acid receptor-related orphan receptor gamma |
| CN107980042A (en) * | 2015-06-11 | 2018-05-01 | 莱斯拉公司 | As ROR gamma agonists and for treating aryl dihydro -2H- benzos [b] [1,4] oxazines sulfonamide and the related compound of disease |
| CN111499591A (en) * | 2019-01-30 | 2020-08-07 | 山东轩竹医药科技有限公司 | ROR gamma modulators |
-
2022
- 2022-08-11 WO PCT/CN2022/111884 patent/WO2023016528A1/en not_active Ceased
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106132422A (en) * | 2014-02-27 | 2016-11-16 | 莱斯拉公司 | Adoptive cell therapy & related treatments using agonists of retinoic acid receptor-related orphan receptor gamma |
| WO2015171610A2 (en) * | 2014-05-05 | 2015-11-12 | Lycera Corporation | Tetrahydroquinoline sulfonamide and related compounds for use as agonists of rory and the treatment of disease |
| WO2016179343A1 (en) * | 2015-05-05 | 2016-11-10 | Lycera Corporation | DIHYDRO-2H-BENZO[b][1,4]OXAZINE SULFONAMIDE AND RELATED COMPOUNDS FOR USE AS AGONISTS OF RORy AND THE TREATMENT OF DISEASE |
| CN107980042A (en) * | 2015-06-11 | 2018-05-01 | 莱斯拉公司 | As ROR gamma agonists and for treating aryl dihydro -2H- benzos [b] [1,4] oxazines sulfonamide and the related compound of disease |
| CN111499591A (en) * | 2019-01-30 | 2020-08-07 | 山东轩竹医药科技有限公司 | ROR gamma modulators |
Non-Patent Citations (1)
| Title |
|---|
| OLSSON, ROINE I. ET AL.: "Benzoxazepines Achieve Potent Suppression of IL-17 Release in Human T-Helper 17 (T H 17) Cells through an Induced-Fit Binding Mode to the Nuclear Receptor RORγ", CHEMMEDCHEM, vol. 11, no. 2, 10 November 2016 (2016-11-10), XP055782047, DOI: 10.1002/cmdc.201500432 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023232870A1 (en) | 2022-05-31 | 2023-12-07 | Immunic Ag | Rorg/rorgt modulators for the treatment of virus infections like covid-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2022166890A1 (en) | Substituted pyridazine phenol derivatives | |
| WO2020052649A1 (en) | Cyclopropylamine compound as lsd1 inhibitor and use thereof | |
| JP2025520714A (en) | Heterocyclic substituted pyrimidopyran compounds and uses thereof | |
| US11905276B2 (en) | Bicyclic compound that acts as CRBN protein regulator | |
| WO2020052647A1 (en) | Spiro-heterocyclic compound acting as lsd1 inhibitor and use thereof | |
| JP2022515755A (en) | Compounds for retinal diseases | |
| TWI834127B (en) | Thiophene compounds and application thereof | |
| CN103827057A (en) | Radiolabeled amino acids for diagnostic imaging | |
| CN116157405A (en) | Cathepsin C Small Molecule Inhibitors and Their Medical Uses | |
| TWI807697B (en) | Furan fused ring substituted glutarimide compounds | |
| EP4116291A1 (en) | Synthesis of novel ep4 antagonist and use in cancers and inflammations | |
| WO2023016528A1 (en) | Class of benzomorpholine compounds, and preparation method therefor and use thereof | |
| TWI808786B (en) | Benzoxazinone derivatives | |
| WO2020156564A1 (en) | Vinylpyridine carboxamide compound as pd-l1 immunomodulator | |
| WO2020156568A1 (en) | Fluorovinylbenzamide compound as pd-l1 immunomodulator | |
| WO2023016527A1 (en) | Class of benzoxazine spiro compounds and preparation method therefor | |
| WO2023016530A1 (en) | 3,4-dihydro-2h-benzo[b][1,4]oxazine compounds and preparation method therefor | |
| WO2022057836A1 (en) | Benzourea ring derivative, and preparation method therefor and use thereof | |
| WO2021175223A1 (en) | Benzo 2-azaspiro[4.4]nonane compound and use thereof | |
| CN114644635B (en) | Triazole tricyclic derivatives, preparation method and application thereof | |
| JP7296017B2 (en) | Compounds containing benzosultam | |
| CN114096525A (en) | Biaryl compounds, pharmaceutical compositions containing them, processes for their preparation and their use | |
| WO2022100614A1 (en) | Benzourea ring derivative, and preparation method therefor and use thereof | |
| CN113825750A (en) | Glucoside derivative as SGLT1 inhibitor and application thereof | |
| EA049085B1 (en) | BENZOXAZINONE DERIVATIVES |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22855508 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 22855508 Country of ref document: EP Kind code of ref document: A1 |
|
| 32PN | Ep: public notification in the ep bulletin as address of the adressee cannot be established |
Free format text: NOTING OF LOSS OF RIGHTS PURSUANT TO RULE 112(1) EPC (EPO FORM 1205A DATED 19.07.2024) |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 22855508 Country of ref document: EP Kind code of ref document: A1 |