WO2022206888A1 - Cdk2 inhibitors and use thereof - Google Patents
Cdk2 inhibitors and use thereof Download PDFInfo
- Publication number
- WO2022206888A1 WO2022206888A1 PCT/CN2022/084355 CN2022084355W WO2022206888A1 WO 2022206888 A1 WO2022206888 A1 WO 2022206888A1 CN 2022084355 W CN2022084355 W CN 2022084355W WO 2022206888 A1 WO2022206888 A1 WO 2022206888A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- membered
- compound
- pharmaceutically acceptable
- butyl
- tert
- Prior art date
Links
- 239000003112 inhibitor Substances 0.000 title description 60
- 101150073031 cdk2 gene Proteins 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims abstract description 248
- 150000003839 salts Chemical class 0.000 claims abstract description 132
- 238000000034 method Methods 0.000 claims abstract description 56
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 52
- 108010024986 Cyclin-Dependent Kinase 2 Proteins 0.000 claims abstract description 43
- 201000010099 disease Diseases 0.000 claims abstract description 33
- 208000035475 disorder Diseases 0.000 claims abstract description 19
- 102000015792 Cyclin-Dependent Kinase 2 Human genes 0.000 claims abstract 4
- -1 pyrrolopyridinyl Chemical group 0.000 claims description 297
- 206010028980 Neoplasm Diseases 0.000 claims description 89
- 125000000623 heterocyclic group Chemical group 0.000 claims description 73
- 229910052739 hydrogen Inorganic materials 0.000 claims description 52
- 239000001257 hydrogen Substances 0.000 claims description 50
- 125000004452 carbocyclyl group Chemical group 0.000 claims description 46
- 201000011510 cancer Diseases 0.000 claims description 40
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 38
- 229910052799 carbon Inorganic materials 0.000 claims description 30
- 229910052736 halogen Inorganic materials 0.000 claims description 29
- 150000002367 halogens Chemical class 0.000 claims description 29
- 150000002431 hydrogen Chemical class 0.000 claims description 26
- 102100037858 G1/S-specific cyclin-E1 Human genes 0.000 claims description 24
- 101000738568 Homo sapiens G1/S-specific cyclin-E1 Proteins 0.000 claims description 24
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 23
- 125000000041 C6-C10 aryl group Chemical group 0.000 claims description 22
- 125000001313 C5-C10 heteroaryl group Chemical group 0.000 claims description 20
- 125000000171 (C1-C6) haloalkyl group Chemical group 0.000 claims description 19
- 125000004765 (C1-C4) haloalkyl group Chemical group 0.000 claims description 18
- 125000004076 pyridyl group Chemical group 0.000 claims description 16
- 230000002018 overexpression Effects 0.000 claims description 15
- 239000008194 pharmaceutical composition Substances 0.000 claims description 15
- 125000004429 atom Chemical group 0.000 claims description 14
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims description 13
- 230000003321 amplification Effects 0.000 claims description 13
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 13
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 claims description 13
- 238000003199 nucleic acid amplification method Methods 0.000 claims description 13
- 229910052760 oxygen Inorganic materials 0.000 claims description 13
- 125000003161 (C1-C6) alkylene group Chemical group 0.000 claims description 12
- 125000006590 (C2-C6) alkenylene group Chemical group 0.000 claims description 12
- 125000006591 (C2-C6) alkynylene group Chemical group 0.000 claims description 12
- 125000001072 heteroaryl group Chemical group 0.000 claims description 12
- 229910052717 sulfur Inorganic materials 0.000 claims description 12
- 230000002401 inhibitory effect Effects 0.000 claims description 11
- 125000003386 piperidinyl group Chemical group 0.000 claims description 11
- 125000001544 thienyl group Chemical group 0.000 claims description 11
- 108090000623 proteins and genes Proteins 0.000 claims description 10
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 9
- 125000002393 azetidinyl group Chemical group 0.000 claims description 9
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 8
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 8
- 125000004193 piperazinyl group Chemical group 0.000 claims description 8
- 125000000714 pyrimidinyl group Chemical group 0.000 claims description 8
- 125000002883 imidazolyl group Chemical group 0.000 claims description 7
- 125000000335 thiazolyl group Chemical group 0.000 claims description 7
- 125000001425 triazolyl group Chemical group 0.000 claims description 7
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 claims description 6
- 229910052801 chlorine Inorganic materials 0.000 claims description 6
- 239000003937 drug carrier Substances 0.000 claims description 6
- 229910052731 fluorine Inorganic materials 0.000 claims description 6
- 230000014509 gene expression Effects 0.000 claims description 6
- 125000003373 pyrazinyl group Chemical group 0.000 claims description 6
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 5
- 125000000842 isoxazolyl group Chemical group 0.000 claims description 5
- 125000002971 oxazolyl group Chemical group 0.000 claims description 5
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 5
- 125000003226 pyrazolyl group Chemical group 0.000 claims description 5
- 125000004853 tetrahydropyridinyl group Chemical group N1(CCCC=C1)* 0.000 claims description 5
- IISBACLAFKSPIT-UHFFFAOYSA-N bisphenol A Chemical compound C=1C=C(O)C=CC=1C(C)(C)C1=CC=C(O)C=C1 IISBACLAFKSPIT-UHFFFAOYSA-N 0.000 claims description 4
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 claims description 4
- GVNVAWHJIKLAGL-UHFFFAOYSA-N 2-(cyclohexen-1-yl)cyclohexan-1-one Chemical compound O=C1CCCCC1C1=CCCCC1 GVNVAWHJIKLAGL-UHFFFAOYSA-N 0.000 claims description 3
- 101150065749 Churc1 gene Proteins 0.000 claims description 3
- 102100038239 Protein Churchill Human genes 0.000 claims description 3
- 229910052794 bromium Inorganic materials 0.000 claims description 3
- 125000002757 morpholinyl group Chemical group 0.000 claims description 3
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims description 3
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 claims description 3
- 125000006583 (C1-C3) haloalkyl group Chemical group 0.000 claims description 2
- 125000004485 2-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])([H])C1([H])* 0.000 claims description 2
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 claims 1
- 239000000203 mixture Substances 0.000 abstract description 327
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 494
- 235000019439 ethyl acetate Nutrition 0.000 description 247
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 229
- 239000000243 solution Substances 0.000 description 168
- 230000002829 reductive effect Effects 0.000 description 157
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 142
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 122
- YMWUJEATGCHHMB-UHFFFAOYSA-N dichloromethane Substances ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 99
- 239000011541 reaction mixture Substances 0.000 description 94
- 239000007787 solid Substances 0.000 description 91
- 239000004698 Polyethylene Substances 0.000 description 80
- 239000011734 sodium Substances 0.000 description 80
- 239000003921 oil Substances 0.000 description 68
- 238000010898 silica gel chromatography Methods 0.000 description 62
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 60
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 55
- 238000003818 flash chromatography Methods 0.000 description 47
- WYURNTSHIVDZCO-UHFFFAOYSA-N tetrahydrofuran Substances C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 47
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 46
- CXNIUSPIQKWYAI-UHFFFAOYSA-N xantphos Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 CXNIUSPIQKWYAI-UHFFFAOYSA-N 0.000 description 46
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 42
- 239000012044 organic layer Substances 0.000 description 42
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical group N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 41
- 239000012267 brine Substances 0.000 description 40
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 40
- 102100036239 Cyclin-dependent kinase 2 Human genes 0.000 description 39
- 238000006243 chemical reaction Methods 0.000 description 34
- 239000000725 suspension Substances 0.000 description 33
- 238000011282 treatment Methods 0.000 description 33
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 32
- 238000002953 preparative HPLC Methods 0.000 description 31
- 239000012230 colorless oil Substances 0.000 description 29
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 28
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 28
- 229910052757 nitrogen Inorganic materials 0.000 description 28
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 26
- 239000000047 product Substances 0.000 description 26
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 25
- 239000012074 organic phase Substances 0.000 description 25
- 238000000746 purification Methods 0.000 description 25
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 24
- 125000001424 substituent group Chemical group 0.000 description 24
- NPMYYNGDMKIHFG-DTWKUNHWSA-N (1R,3S)-3-(5-amino-1-tert-butylpyrazol-3-yl)cyclopentan-1-ol Chemical compound CC(C)(C)N1N=C([C@@H](CC2)C[C@@H]2O)C=C1N NPMYYNGDMKIHFG-DTWKUNHWSA-N 0.000 description 22
- 229910000104 sodium hydride Inorganic materials 0.000 description 22
- 239000003208 petroleum Substances 0.000 description 21
- 239000000706 filtrate Substances 0.000 description 20
- 229920006395 saturated elastomer Polymers 0.000 description 20
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 18
- 206010006187 Breast cancer Diseases 0.000 description 18
- 239000003795 chemical substances by application Substances 0.000 description 18
- 208000026310 Breast neoplasm Diseases 0.000 description 17
- 102000037982 Immune checkpoint proteins Human genes 0.000 description 17
- 108091008036 Immune checkpoint proteins Proteins 0.000 description 17
- 125000003118 aryl group Chemical group 0.000 description 16
- 210000004027 cell Anatomy 0.000 description 16
- 238000005160 1H NMR spectroscopy Methods 0.000 description 14
- 125000004093 cyano group Chemical group *C#N 0.000 description 14
- 125000005842 heteroatom Chemical group 0.000 description 14
- 229940126546 immune checkpoint molecule Drugs 0.000 description 14
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 13
- 125000000217 alkyl group Chemical group 0.000 description 13
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 13
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 239000012043 crude product Substances 0.000 description 12
- 125000004484 1-methylpiperidin-4-yl group Chemical group CN1CCC(CC1)* 0.000 description 11
- 244000166102 Eucalyptus leucoxylon Species 0.000 description 11
- 235000004694 Eucalyptus leucoxylon Nutrition 0.000 description 11
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 11
- 208000009956 adenocarcinoma Diseases 0.000 description 11
- 239000012298 atmosphere Substances 0.000 description 11
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 11
- 230000000694 effects Effects 0.000 description 11
- 239000010410 layer Substances 0.000 description 11
- 239000001301 oxygen Substances 0.000 description 11
- MFYSUUPKMDJYPF-UHFFFAOYSA-N 2-[(4-methyl-2-nitrophenyl)diazenyl]-3-oxo-n-phenylbutanamide Chemical compound C=1C=CC=CC=1NC(=O)C(C(=O)C)N=NC1=CC=C(C)C=C1[N+]([O-])=O MFYSUUPKMDJYPF-UHFFFAOYSA-N 0.000 description 10
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 10
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 10
- 125000000753 cycloalkyl group Chemical group 0.000 description 10
- 238000009472 formulation Methods 0.000 description 10
- 238000004128 high performance liquid chromatography Methods 0.000 description 10
- 208000015181 infectious disease Diseases 0.000 description 10
- 239000007788 liquid Substances 0.000 description 10
- 239000000126 substance Substances 0.000 description 10
- 239000011593 sulfur Chemical group 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- 206010039491 Sarcoma Diseases 0.000 description 9
- 239000000741 silica gel Substances 0.000 description 9
- 229910002027 silica gel Inorganic materials 0.000 description 9
- 238000003756 stirring Methods 0.000 description 9
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 8
- 206010025323 Lymphomas Diseases 0.000 description 8
- 206010033128 Ovarian cancer Diseases 0.000 description 8
- 125000004432 carbon atom Chemical group C* 0.000 description 8
- 208000020816 lung neoplasm Diseases 0.000 description 8
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 8
- 229960001924 melphalan Drugs 0.000 description 8
- 239000012299 nitrogen atmosphere Substances 0.000 description 8
- 238000002360 preparation method Methods 0.000 description 8
- 125000006413 ring segment Chemical group 0.000 description 8
- 239000007858 starting material Substances 0.000 description 8
- ZIDIKYIZXMYHAW-UHFFFAOYSA-N 2-bromo-6-fluoropyridine Chemical compound FC1=CC=CC(Br)=N1 ZIDIKYIZXMYHAW-UHFFFAOYSA-N 0.000 description 7
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 7
- 108091007914 CDKs Proteins 0.000 description 7
- 229940045513 CTLA4 antagonist Drugs 0.000 description 7
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 7
- 206010061535 Ovarian neoplasm Diseases 0.000 description 7
- 239000002253 acid Substances 0.000 description 7
- 239000003814 drug Substances 0.000 description 7
- 230000005764 inhibitory process Effects 0.000 description 7
- 125000002816 methylsulfanyl group Chemical group [H]C([H])([H])S[*] 0.000 description 7
- 239000002480 mineral oil Substances 0.000 description 7
- 235000010446 mineral oil Nutrition 0.000 description 7
- 125000000636 p-nitrophenyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)[N+]([O-])=O 0.000 description 7
- 239000002904 solvent Substances 0.000 description 7
- 229960000575 trastuzumab Drugs 0.000 description 7
- NXLNNXIXOYSCMB-UHFFFAOYSA-N (4-nitrophenyl) carbonochloridate Chemical compound [O-][N+](=O)C1=CC=C(OC(Cl)=O)C=C1 NXLNNXIXOYSCMB-UHFFFAOYSA-N 0.000 description 6
- UCDBLYHPJMMFMY-UHFFFAOYSA-N 8-bromoquinoline-4-carbonitrile Chemical compound C1=CN=C2C(Br)=CC=CC2=C1C#N UCDBLYHPJMMFMY-UHFFFAOYSA-N 0.000 description 6
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 6
- 108010074708 B7-H1 Antigen Proteins 0.000 description 6
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 6
- 201000009030 Carcinoma Diseases 0.000 description 6
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 6
- 101000851370 Homo sapiens Tumor necrosis factor receptor superfamily member 9 Proteins 0.000 description 6
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 6
- 206010035226 Plasma cell myeloma Diseases 0.000 description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 6
- 102100024216 Programmed cell death 1 ligand 1 Human genes 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- 102100036856 Tumor necrosis factor receptor superfamily member 9 Human genes 0.000 description 6
- 241000700605 Viruses Species 0.000 description 6
- 239000002246 antineoplastic agent Substances 0.000 description 6
- 239000002585 base Substances 0.000 description 6
- 125000002619 bicyclic group Chemical group 0.000 description 6
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 description 6
- 229960003957 dexamethasone Drugs 0.000 description 6
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 6
- 239000002552 dosage form Substances 0.000 description 6
- 201000001441 melanoma Diseases 0.000 description 6
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 6
- 125000002950 monocyclic group Chemical group 0.000 description 6
- 239000002243 precursor Substances 0.000 description 6
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 6
- 125000006239 protecting group Chemical group 0.000 description 6
- 206010041823 squamous cell carcinoma Diseases 0.000 description 6
- 208000024891 symptom Diseases 0.000 description 6
- 239000003826 tablet Substances 0.000 description 6
- IOKGWQZQCNXXLD-UHFFFAOYSA-N tert-butyl n-(3-bromopropyl)carbamate Chemical compound CC(C)(C)OC(=O)NCCCBr IOKGWQZQCNXXLD-UHFFFAOYSA-N 0.000 description 6
- 210000004881 tumor cell Anatomy 0.000 description 6
- 206010009944 Colon cancer Diseases 0.000 description 5
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 5
- 101000801234 Homo sapiens Tumor necrosis factor receptor superfamily member 18 Proteins 0.000 description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 5
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 5
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 5
- 208000034578 Multiple myelomas Diseases 0.000 description 5
- 101710089372 Programmed cell death protein 1 Proteins 0.000 description 5
- 102100040678 Programmed cell death protein 1 Human genes 0.000 description 5
- 208000005718 Stomach Neoplasms Diseases 0.000 description 5
- 102100033728 Tumor necrosis factor receptor superfamily member 18 Human genes 0.000 description 5
- 102100022153 Tumor necrosis factor receptor superfamily member 4 Human genes 0.000 description 5
- 101710165473 Tumor necrosis factor receptor superfamily member 4 Proteins 0.000 description 5
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 5
- 229960005395 cetuximab Drugs 0.000 description 5
- 125000004122 cyclic group Chemical group 0.000 description 5
- 229960004397 cyclophosphamide Drugs 0.000 description 5
- 229960004679 doxorubicin Drugs 0.000 description 5
- 206010017758 gastric cancer Diseases 0.000 description 5
- 239000005457 ice water Substances 0.000 description 5
- 238000001990 intravenous administration Methods 0.000 description 5
- 201000005202 lung cancer Diseases 0.000 description 5
- 244000052769 pathogen Species 0.000 description 5
- 125000003367 polycyclic group Chemical group 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- 102000004196 processed proteins & peptides Human genes 0.000 description 5
- 108090000765 processed proteins & peptides Proteins 0.000 description 5
- 102000004169 proteins and genes Human genes 0.000 description 5
- 150000003254 radicals Chemical class 0.000 description 5
- 230000001105 regulatory effect Effects 0.000 description 5
- 229960004641 rituximab Drugs 0.000 description 5
- 239000007921 spray Substances 0.000 description 5
- 201000011549 stomach cancer Diseases 0.000 description 5
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 5
- 229960001796 sunitinib Drugs 0.000 description 5
- XDJCYKMWJCYQJM-UHFFFAOYSA-N tert-butyl n-(3-hydroxypropyl)carbamate Chemical compound CC(C)(C)OC(=O)NCCCO XDJCYKMWJCYQJM-UHFFFAOYSA-N 0.000 description 5
- LIYMTLVBAVHPBU-UHFFFAOYSA-N tert-butyl n-(4-hydroxybutyl)carbamate Chemical compound CC(C)(C)OC(=O)NCCCCO LIYMTLVBAVHPBU-UHFFFAOYSA-N 0.000 description 5
- 238000002560 therapeutic procedure Methods 0.000 description 5
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 4
- WEVYNIUIFUYDGI-UHFFFAOYSA-N 3-[6-[4-(trifluoromethoxy)anilino]-4-pyrimidinyl]benzamide Chemical compound NC(=O)C1=CC=CC(C=2N=CN=C(NC=3C=CC(OC(F)(F)F)=CC=3)C=2)=C1 WEVYNIUIFUYDGI-UHFFFAOYSA-N 0.000 description 4
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 description 4
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 4
- 206010005949 Bone cancer Diseases 0.000 description 4
- 208000018084 Bone neoplasm Diseases 0.000 description 4
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 4
- 229940124297 CDK 4/6 inhibitor Drugs 0.000 description 4
- 108010021064 CTLA-4 Antigen Proteins 0.000 description 4
- 102000008203 CTLA-4 Antigen Human genes 0.000 description 4
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 4
- 102000016736 Cyclin Human genes 0.000 description 4
- 108050006400 Cyclin Proteins 0.000 description 4
- 102000000577 Cyclin-Dependent Kinase Inhibitor p27 Human genes 0.000 description 4
- 108010016777 Cyclin-Dependent Kinase Inhibitor p27 Proteins 0.000 description 4
- 102100032857 Cyclin-dependent kinase 1 Human genes 0.000 description 4
- 101710106279 Cyclin-dependent kinase 1 Proteins 0.000 description 4
- 102000003903 Cyclin-dependent kinases Human genes 0.000 description 4
- 108090000266 Cyclin-dependent kinases Proteins 0.000 description 4
- 206010014733 Endometrial cancer Diseases 0.000 description 4
- 206010014759 Endometrial neoplasm Diseases 0.000 description 4
- 208000000461 Esophageal Neoplasms Diseases 0.000 description 4
- 102100034458 Hepatitis A virus cellular receptor 2 Human genes 0.000 description 4
- 241000282412 Homo Species 0.000 description 4
- 101001068133 Homo sapiens Hepatitis A virus cellular receptor 2 Proteins 0.000 description 4
- 101001137987 Homo sapiens Lymphocyte activation gene 3 protein Proteins 0.000 description 4
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 4
- 101000997835 Homo sapiens Tyrosine-protein kinase JAK1 Proteins 0.000 description 4
- 239000005511 L01XE05 - Sorafenib Substances 0.000 description 4
- 102000017578 LAG3 Human genes 0.000 description 4
- 206010024612 Lipoma Diseases 0.000 description 4
- 101100335081 Mus musculus Flt3 gene Proteins 0.000 description 4
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 4
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Substances BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 4
- 206010029260 Neuroblastoma Diseases 0.000 description 4
- 206010030155 Oesophageal carcinoma Diseases 0.000 description 4
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 4
- 206010041067 Small cell lung cancer Diseases 0.000 description 4
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 4
- 208000003721 Triple Negative Breast Neoplasms Diseases 0.000 description 4
- 102100033438 Tyrosine-protein kinase JAK1 Human genes 0.000 description 4
- 208000002495 Uterine Neoplasms Diseases 0.000 description 4
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 4
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 4
- 125000002947 alkylene group Chemical group 0.000 description 4
- 239000000427 antigen Substances 0.000 description 4
- 229960001467 bortezomib Drugs 0.000 description 4
- 210000000481 breast Anatomy 0.000 description 4
- 125000002837 carbocyclic group Chemical group 0.000 description 4
- 208000006990 cholangiocarcinoma Diseases 0.000 description 4
- 239000003246 corticosteroid Substances 0.000 description 4
- 229940127089 cytotoxic agent Drugs 0.000 description 4
- 208000005017 glioblastoma Diseases 0.000 description 4
- 230000012010 growth Effects 0.000 description 4
- 208000014829 head and neck neoplasm Diseases 0.000 description 4
- 150000002430 hydrocarbons Chemical group 0.000 description 4
- 238000001802 infusion Methods 0.000 description 4
- 229940043355 kinase inhibitor Drugs 0.000 description 4
- GOTYRUGSSMKFNF-UHFFFAOYSA-N lenalidomide Chemical compound C1C=2C(N)=CC=CC=2C(=O)N1C1CCC(=O)NC1=O GOTYRUGSSMKFNF-UHFFFAOYSA-N 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- 125000004433 nitrogen atom Chemical group N* 0.000 description 4
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 4
- 229960002621 pembrolizumab Drugs 0.000 description 4
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 description 4
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 4
- 239000008177 pharmaceutical agent Substances 0.000 description 4
- HXITXNWTGFUOAU-UHFFFAOYSA-N phenylboronic acid Chemical compound OB(O)C1=CC=CC=C1 HXITXNWTGFUOAU-UHFFFAOYSA-N 0.000 description 4
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 4
- 208000007056 sickle cell anemia Diseases 0.000 description 4
- 208000000587 small cell lung carcinoma Diseases 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- 239000012312 sodium hydride Substances 0.000 description 4
- 229960003787 sorafenib Drugs 0.000 description 4
- 150000003457 sulfones Chemical class 0.000 description 4
- 150000003462 sulfoxides Chemical class 0.000 description 4
- 230000008685 targeting Effects 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- WYWHKKSPHMUBEB-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 4
- 208000022679 triple-negative breast carcinoma Diseases 0.000 description 4
- 206010046766 uterine cancer Diseases 0.000 description 4
- 229960005486 vaccine Drugs 0.000 description 4
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 3
- HOVCGEVZLGMGSK-UHFFFAOYSA-N (3-bromo-6-chloropyridin-2-yl)methanol Chemical compound OCC1=NC(Cl)=CC=C1Br HOVCGEVZLGMGSK-UHFFFAOYSA-N 0.000 description 3
- NCCDWBWIMWQQOM-UHFFFAOYSA-N (3-methylidenecyclobutyl)methanol Chemical compound OCC1CC(=C)C1 NCCDWBWIMWQQOM-UHFFFAOYSA-N 0.000 description 3
- BFCUWOVHOAODGT-UHFFFAOYSA-N (4,6-dichloropyridin-2-yl)methanol Chemical compound OCC1=CC(Cl)=CC(Cl)=N1 BFCUWOVHOAODGT-UHFFFAOYSA-N 0.000 description 3
- LOVPHSMOAVXQIH-UHFFFAOYSA-M (4-nitrophenyl) carbonate Chemical compound [O-]C(=O)OC1=CC=C([N+]([O-])=O)C=C1 LOVPHSMOAVXQIH-UHFFFAOYSA-M 0.000 description 3
- ATSGNNIRLDMOAN-UHFFFAOYSA-N (6-bromo-5-fluoropyridin-2-yl)methanol Chemical compound OCC1=CC=C(F)C(Br)=N1 ATSGNNIRLDMOAN-UHFFFAOYSA-N 0.000 description 3
- SQMVRFXDBRYXFQ-UHFFFAOYSA-N 1-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydro-2h-pyridine Chemical compound C1N(C)CCC(B2OC(C)(C)C(C)(C)O2)=C1 SQMVRFXDBRYXFQ-UHFFFAOYSA-N 0.000 description 3
- 102000010400 1-phosphatidylinositol-3-kinase activity proteins Human genes 0.000 description 3
- HLBKNKSYLIRLPK-UHFFFAOYSA-N 2,6-dichloro-4-phenylpyridine Chemical compound ClC1=NC(Cl)=CC(C=2C=CC=CC=2)=C1 HLBKNKSYLIRLPK-UHFFFAOYSA-N 0.000 description 3
- UEJJHQNACJXSKW-UHFFFAOYSA-N 2-(2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione Chemical compound O=C1C2=CC=CC=C2C(=O)N1C1CCC(=O)NC1=O UEJJHQNACJXSKW-UHFFFAOYSA-N 0.000 description 3
- RTQWWZBSTRGEAV-PKHIMPSTSA-N 2-[[(2s)-2-[bis(carboxymethyl)amino]-3-[4-(methylcarbamoylamino)phenyl]propyl]-[2-[bis(carboxymethyl)amino]propyl]amino]acetic acid Chemical compound CNC(=O)NC1=CC=C(C[C@@H](CN(CC(C)N(CC(O)=O)CC(O)=O)CC(O)=O)N(CC(O)=O)CC(O)=O)C=C1 RTQWWZBSTRGEAV-PKHIMPSTSA-N 0.000 description 3
- QOAXYSDYEOLOSF-UHFFFAOYSA-N 2-tert-butyl-5-(3,3-dimethoxycyclopentyl)pyrazol-3-amine Chemical compound C(C)(C)(C)N1N=C(C=C1N)C1CC(CC1)(OC)OC QOAXYSDYEOLOSF-UHFFFAOYSA-N 0.000 description 3
- DFCZRLQMEMTBKM-UHFFFAOYSA-N 3,6-dibromo-2-(bromomethyl)pyridine Chemical compound BrCc1nc(Br)ccc1Br DFCZRLQMEMTBKM-UHFFFAOYSA-N 0.000 description 3
- ITIMXDXLMZTDMV-UHFFFAOYSA-N 3-(3,3-dimethoxycyclopentyl)-3-oxopropanenitrile Chemical compound COC1(CC(CC1)C(CC#N)=O)OC ITIMXDXLMZTDMV-UHFFFAOYSA-N 0.000 description 3
- NNKLICLIBKMDOY-UHFFFAOYSA-N 3-methylidenecyclobutane-1-carboxylic acid Chemical compound OC(=O)C1CC(=C)C1 NNKLICLIBKMDOY-UHFFFAOYSA-N 0.000 description 3
- NRZJXHGQGKQDDF-UHFFFAOYSA-N 4,6-dibromo-2-methylpyridin-3-ol Chemical compound CC1=NC(Br)=CC(Br)=C1O NRZJXHGQGKQDDF-UHFFFAOYSA-N 0.000 description 3
- GGOHWTJUUGWLCJ-UHFFFAOYSA-N 5-[(2,4-dimethoxyphenyl)methylamino]pentan-2-ol Chemical compound COc1ccc(CNCCCC(C)O)c(OC)c1 GGOHWTJUUGWLCJ-UHFFFAOYSA-N 0.000 description 3
- AUBAFPZVFLTWNE-UHFFFAOYSA-N 6-bromo-2-(bromomethyl)-3-fluoropyridine Chemical compound FC1=CC=C(Br)N=C1CBr AUBAFPZVFLTWNE-UHFFFAOYSA-N 0.000 description 3
- GAOUVRGJVCEHAE-UHFFFAOYSA-N 6-bromo-2-(bromomethyl)-3-methoxypyridine Chemical compound COC1=CC=C(Br)N=C1CBr GAOUVRGJVCEHAE-UHFFFAOYSA-N 0.000 description 3
- OUOXDHCCDFQUTK-UHFFFAOYSA-N 6-bromo-2-fluoro-3-iodopyridine Chemical compound BrC1=CC=C(C(=N1)F)I OUOXDHCCDFQUTK-UHFFFAOYSA-N 0.000 description 3
- NZEZVKXETZALTH-UHFFFAOYSA-N 6-bromo-2-methylpyridin-3-ol Chemical compound CC1=NC(Br)=CC=C1O NZEZVKXETZALTH-UHFFFAOYSA-N 0.000 description 3
- HUBRRMYRFLDKED-UHFFFAOYSA-N 6-bromo-3-methoxy-2-methylpyridine Chemical compound COC1=CC=C(Br)N=C1C HUBRRMYRFLDKED-UHFFFAOYSA-N 0.000 description 3
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 3
- GOLCXWYRSKYTSP-UHFFFAOYSA-N Arsenious Acid Chemical compound O1[As]2O[As]1O2 GOLCXWYRSKYTSP-UHFFFAOYSA-N 0.000 description 3
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 3
- HGWWTTCJZBSBKI-BJKOFHAPSA-N CC(C)(C)N(C([C@@H](CC1)C[C@@H]1OC(N1C=NC=C1)=O)=C1)N=C1NC1=CC=C(C=CN2CCCCNC(OC(C)(C)C)=O)C2=N1 Chemical compound CC(C)(C)N(C([C@@H](CC1)C[C@@H]1OC(N1C=NC=C1)=O)=C1)N=C1NC1=CC=C(C=CN2CCCCNC(OC(C)(C)C)=O)C2=N1 HGWWTTCJZBSBKI-BJKOFHAPSA-N 0.000 description 3
- RCEBNWANGOIEOC-UHFFFAOYSA-N CC(C)(C)[Si](C)(C)OCCCCOC1=CC=CC(Br)=N1 Chemical compound CC(C)(C)[Si](C)(C)OCCCCOC1=CC=CC(Br)=N1 RCEBNWANGOIEOC-UHFFFAOYSA-N 0.000 description 3
- JGLMVXWAHNTPRF-CMDGGOBGSA-N CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O Chemical compound CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O JGLMVXWAHNTPRF-CMDGGOBGSA-N 0.000 description 3
- 101000715943 Caenorhabditis elegans Cyclin-dependent kinase 4 homolog Proteins 0.000 description 3
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 description 3
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 3
- 208000005243 Chondrosarcoma Diseases 0.000 description 3
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 3
- 102000003909 Cyclin E Human genes 0.000 description 3
- 108090000257 Cyclin E Proteins 0.000 description 3
- 102000000578 Cyclin-Dependent Kinase Inhibitor p21 Human genes 0.000 description 3
- 108010016788 Cyclin-Dependent Kinase Inhibitor p21 Proteins 0.000 description 3
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 3
- ZBNZXTGUTAYRHI-UHFFFAOYSA-N Dasatinib Chemical compound C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1Cl ZBNZXTGUTAYRHI-UHFFFAOYSA-N 0.000 description 3
- 208000006168 Ewing Sarcoma Diseases 0.000 description 3
- BKSHXTLFNNVSGY-UHFFFAOYSA-N Fc1cnc(CCl)[nH]c1=O Chemical compound Fc1cnc(CCl)[nH]c1=O BKSHXTLFNNVSGY-UHFFFAOYSA-N 0.000 description 3
- 201000008808 Fibrosarcoma Diseases 0.000 description 3
- 206010017993 Gastrointestinal neoplasms Diseases 0.000 description 3
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 3
- 102000004457 Granulocyte-Macrophage Colony-Stimulating Factor Human genes 0.000 description 3
- 208000017604 Hodgkin disease Diseases 0.000 description 3
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 3
- 101001037256 Homo sapiens Indoleamine 2,3-dioxygenase 1 Proteins 0.000 description 3
- 101000916644 Homo sapiens Macrophage colony-stimulating factor 1 receptor Proteins 0.000 description 3
- 101000831007 Homo sapiens T-cell immunoreceptor with Ig and ITIM domains Proteins 0.000 description 3
- 241000701806 Human papillomavirus Species 0.000 description 3
- 102100040061 Indoleamine 2,3-dioxygenase 1 Human genes 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- 102000042838 JAK family Human genes 0.000 description 3
- 108091082332 JAK family Proteins 0.000 description 3
- 229940116839 Janus kinase 1 inhibitor Drugs 0.000 description 3
- 208000007766 Kaposi sarcoma Diseases 0.000 description 3
- 208000008839 Kidney Neoplasms Diseases 0.000 description 3
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 3
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 3
- 239000002067 L01XE06 - Dasatinib Substances 0.000 description 3
- 239000002144 L01XE18 - Ruxolitinib Substances 0.000 description 3
- 208000031671 Large B-Cell Diffuse Lymphoma Diseases 0.000 description 3
- 208000018142 Leiomyosarcoma Diseases 0.000 description 3
- 102100028198 Macrophage colony-stimulating factor 1 receptor Human genes 0.000 description 3
- 206010027406 Mesothelioma Diseases 0.000 description 3
- 201000003793 Myelodysplastic syndrome Diseases 0.000 description 3
- SVGIEIZXLWHQEL-UHFFFAOYSA-N N-[(2,4-dimethoxyphenyl)methyl]-4-hydroxypentanamide Chemical compound COC1=CC=C(CNC(=O)CCC(C)O)C(OC)=C1 SVGIEIZXLWHQEL-UHFFFAOYSA-N 0.000 description 3
- BXDZOYLPNAIDOC-UHFFFAOYSA-N N-[5-[(5-tert-butyl-1,3-oxazol-2-yl)methylsulfanyl]-1,3-thiazol-2-yl]-1-[2-[2-[2-[2-[2-[[2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindol-4-yl]amino]ethoxy]ethoxy]ethoxy]ethylamino]-2-oxoethyl]piperidine-4-carboxamide Chemical compound CC(C)(C)c1cnc(CSc2cnc(NC(=O)C3CCN(CC(=O)NCCOCCOCCOCCNc4cccc5C(=O)N(C6CCC(=O)NC6=O)C(=O)c45)CC3)s2)o1 BXDZOYLPNAIDOC-UHFFFAOYSA-N 0.000 description 3
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 3
- HNPJBCZRSMUZRG-UHFFFAOYSA-N NCC#CCOC1=NC(Br)=CC=C1 Chemical compound NCC#CCOC1=NC(Br)=CC=C1 HNPJBCZRSMUZRG-UHFFFAOYSA-N 0.000 description 3
- IEYBNXNUCQPICM-ZWKOTPCHSA-N NCCCCN1C2=NC(NC3=NNC([C@@H](CC4)C[C@@H]4OC(N4C=NC=C4)=O)=C3)=CC=C2C=C1 Chemical compound NCCCCN1C2=NC(NC3=NNC([C@@H](CC4)C[C@@H]4OC(N4C=NC=C4)=O)=C3)=CC=C2C=C1 IEYBNXNUCQPICM-ZWKOTPCHSA-N 0.000 description 3
- 201000004404 Neurofibroma Diseases 0.000 description 3
- YGSUPCDESGOOMI-UHFFFAOYSA-N OCC#CCOC1=NC(Br)=CC=C1 Chemical compound OCC#CCOC1=NC(Br)=CC=C1 YGSUPCDESGOOMI-UHFFFAOYSA-N 0.000 description 3
- BREFHLNCLJHGCP-UHFFFAOYSA-N OCC1CC(COC2=NC(Br)=CC=C2)C1 Chemical compound OCC1CC(COC2=NC(Br)=CC=C2)C1 BREFHLNCLJHGCP-UHFFFAOYSA-N 0.000 description 3
- 108091007960 PI3Ks Proteins 0.000 description 3
- 229930012538 Paclitaxel Natural products 0.000 description 3
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 3
- 108091000080 Phosphotransferase Proteins 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 206010060862 Prostate cancer Diseases 0.000 description 3
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 3
- 229940079156 Proteasome inhibitor Drugs 0.000 description 3
- 208000000453 Skin Neoplasms Diseases 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 102100024834 T-cell immunoreceptor with Ig and ITIM domains Human genes 0.000 description 3
- 206010043276 Teratoma Diseases 0.000 description 3
- 206010046458 Urethral neoplasms Diseases 0.000 description 3
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 3
- ADQYRFHRLXUSAZ-UHFFFAOYSA-N [N-]=[N+]=NCC1CC(COC2=NC(Br)=CC=C2)C1 Chemical compound [N-]=[N+]=NCC1CC(COC2=NC(Br)=CC=C2)C1 ADQYRFHRLXUSAZ-UHFFFAOYSA-N 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 239000000556 agonist Substances 0.000 description 3
- 125000003545 alkoxy group Chemical group 0.000 description 3
- 229960000473 altretamine Drugs 0.000 description 3
- 108091007433 antigens Proteins 0.000 description 3
- 102000036639 antigens Human genes 0.000 description 3
- 229950000971 baricitinib Drugs 0.000 description 3
- XUZMWHLSFXCVMG-UHFFFAOYSA-N baricitinib Chemical compound C1N(S(=O)(=O)CC)CC1(CC#N)N1N=CC(C=2C=3C=CNC=3N=CN=2)=C1 XUZMWHLSFXCVMG-UHFFFAOYSA-N 0.000 description 3
- PCMHACWNZMLJQH-UHFFFAOYSA-N benzyl N-[2-tert-butyl-5-(3-oxocyclopentyl)pyrazol-3-yl]carbamate Chemical compound C(C)(C)(C)N1N=C(C=C1NC(OCC1=CC=CC=C1)=O)C1CC(CC1)=O PCMHACWNZMLJQH-UHFFFAOYSA-N 0.000 description 3
- 229960000397 bevacizumab Drugs 0.000 description 3
- 229960002092 busulfan Drugs 0.000 description 3
- 229960004117 capecitabine Drugs 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 208000011892 carcinosarcoma of the corpus uteri Diseases 0.000 description 3
- 229960005243 carmustine Drugs 0.000 description 3
- 230000022131 cell cycle Effects 0.000 description 3
- 238000002512 chemotherapy Methods 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- 238000002648 combination therapy Methods 0.000 description 3
- 229960002448 dasatinib Drugs 0.000 description 3
- 206010012818 diffuse large B-cell lymphoma Diseases 0.000 description 3
- 239000002532 enzyme inhibitor Substances 0.000 description 3
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 3
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 3
- 229960001433 erlotinib Drugs 0.000 description 3
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 description 3
- 201000004101 esophageal cancer Diseases 0.000 description 3
- QRSCFNPVDJKGGB-UHFFFAOYSA-N ethyl 2-fluoro-3-oxopropanoate Chemical compound CCOC(=O)C(F)C=O QRSCFNPVDJKGGB-UHFFFAOYSA-N 0.000 description 3
- 229960005420 etoposide Drugs 0.000 description 3
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 3
- 206010016629 fibroma Diseases 0.000 description 3
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 3
- 229960002584 gefitinib Drugs 0.000 description 3
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- 125000001188 haloalkyl group Chemical group 0.000 description 3
- 201000011066 hemangioma Diseases 0.000 description 3
- 230000002489 hematologic effect Effects 0.000 description 3
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 3
- UUVWYPNAQBNQJQ-UHFFFAOYSA-N hexamethylmelamine Chemical compound CN(C)C1=NC(N(C)C)=NC(N(C)C)=N1 UUVWYPNAQBNQJQ-UHFFFAOYSA-N 0.000 description 3
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- 229960001001 ibritumomab tiuxetan Drugs 0.000 description 3
- 229960001101 ifosfamide Drugs 0.000 description 3
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 3
- YLMAHDNUQAMNNX-UHFFFAOYSA-N imatinib methanesulfonate Chemical compound CS(O)(=O)=O.C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 YLMAHDNUQAMNNX-UHFFFAOYSA-N 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 230000002519 immonomodulatory effect Effects 0.000 description 3
- 238000009169 immunotherapy Methods 0.000 description 3
- 238000007918 intramuscular administration Methods 0.000 description 3
- 238000007912 intraperitoneal administration Methods 0.000 description 3
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 3
- 229960004942 lenalidomide Drugs 0.000 description 3
- 229960003881 letrozole Drugs 0.000 description 3
- HPJKCIUCZWXJDR-UHFFFAOYSA-N letrozole Chemical compound C1=CC(C#N)=CC=C1C(N1N=CN=C1)C1=CC=C(C#N)C=C1 HPJKCIUCZWXJDR-UHFFFAOYSA-N 0.000 description 3
- 208000032839 leukemia Diseases 0.000 description 3
- 239000002502 liposome Substances 0.000 description 3
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 3
- 208000014018 liver neoplasm Diseases 0.000 description 3
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 3
- 230000002503 metabolic effect Effects 0.000 description 3
- CBVJMGDRQPEDJV-UHFFFAOYSA-N methyl 3,3-dimethoxycyclopentane-1-carboxylate Chemical compound COC(=O)C1CCC(OC)(OC)C1 CBVJMGDRQPEDJV-UHFFFAOYSA-N 0.000 description 3
- APCPBNNLVCVDPL-UHFFFAOYSA-N methyl 3-bromo-6-chloropyridine-2-carboxylate Chemical compound COC(=O)C1=NC(Cl)=CC=C1Br APCPBNNLVCVDPL-UHFFFAOYSA-N 0.000 description 3
- MHKKUZDJUGIOBC-UHFFFAOYSA-N methyl 3-hydroxypyridine-2-carboxylate Chemical compound COC(=O)C1=NC=CC=C1O MHKKUZDJUGIOBC-UHFFFAOYSA-N 0.000 description 3
- MXHPWLMQTZILLL-UHFFFAOYSA-N methyl 4,6-dichloropyridine-2-carboxylate Chemical compound COC(=O)C1=CC(Cl)=CC(Cl)=N1 MXHPWLMQTZILLL-UHFFFAOYSA-N 0.000 description 3
- UGFPNFKMYRHGBG-UHFFFAOYSA-N methyl 6-bromo-3-hydroxypyridine-2-carboxylate Chemical compound COC(=O)C1=NC(Br)=CC=C1O UGFPNFKMYRHGBG-UHFFFAOYSA-N 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 3
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 3
- 239000002674 ointment Substances 0.000 description 3
- 201000008968 osteosarcoma Diseases 0.000 description 3
- 229960001756 oxaliplatin Drugs 0.000 description 3
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 3
- 229960001592 paclitaxel Drugs 0.000 description 3
- 201000002528 pancreatic cancer Diseases 0.000 description 3
- 208000008443 pancreatic carcinoma Diseases 0.000 description 3
- 229960001972 panitumumab Drugs 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 229960002340 pentostatin Drugs 0.000 description 3
- 102000020233 phosphotransferase Human genes 0.000 description 3
- 239000006187 pill Substances 0.000 description 3
- 229920000642 polymer Polymers 0.000 description 3
- 229960004618 prednisone Drugs 0.000 description 3
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 3
- 239000003207 proteasome inhibitor Substances 0.000 description 3
- 230000000306 recurrent effect Effects 0.000 description 3
- 201000009410 rhabdomyosarcoma Diseases 0.000 description 3
- 229960000215 ruxolitinib Drugs 0.000 description 3
- HFNKQEVNSGCOJV-OAHLLOKOSA-N ruxolitinib Chemical compound C1([C@@H](CC#N)N2N=CC(=C2)C=2C=3C=CNC=3N=CN=2)CCCC1 HFNKQEVNSGCOJV-OAHLLOKOSA-N 0.000 description 3
- 230000019491 signal transduction Effects 0.000 description 3
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 3
- VMWOETMUNAQFAX-UHFFFAOYSA-N spiro[3.5]nonane Chemical compound C1CCC21CCCCC2 VMWOETMUNAQFAX-UHFFFAOYSA-N 0.000 description 3
- 239000012258 stirred mixture Substances 0.000 description 3
- 238000007920 subcutaneous administration Methods 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- 238000004808 supercritical fluid chromatography Methods 0.000 description 3
- 238000003419 tautomerization reaction Methods 0.000 description 3
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 3
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 3
- 229960003433 thalidomide Drugs 0.000 description 3
- 238000011200 topical administration Methods 0.000 description 3
- 229960005267 tositumomab Drugs 0.000 description 3
- 206010044412 transitional cell carcinoma Diseases 0.000 description 3
- 230000004614 tumor growth Effects 0.000 description 3
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 3
- 201000005290 uterine carcinosarcoma Diseases 0.000 description 3
- 229960004528 vincristine Drugs 0.000 description 3
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 3
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 3
- 239000000080 wetting agent Substances 0.000 description 3
- YPBKTZBXSBLTDK-PKNBQFBNSA-N (3e)-3-[(3-bromo-4-fluoroanilino)-nitrosomethylidene]-4-[2-(sulfamoylamino)ethylamino]-1,2,5-oxadiazole Chemical compound NS(=O)(=O)NCCNC1=NON\C1=C(N=O)/NC1=CC=C(F)C(Br)=C1 YPBKTZBXSBLTDK-PKNBQFBNSA-N 0.000 description 2
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 description 2
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 2
- HJTAZXHBEBIQQX-UHFFFAOYSA-N 1,5-bis(chloromethyl)naphthalene Chemical compound C1=CC=C2C(CCl)=CC=CC2=C1CCl HJTAZXHBEBIQQX-UHFFFAOYSA-N 0.000 description 2
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 2
- AQSRRZGQRFFFGS-UHFFFAOYSA-N 2-methylpyridin-3-ol Chemical compound CC1=NC=CC=C1O AQSRRZGQRFFFGS-UHFFFAOYSA-N 0.000 description 2
- UDJKDMSGTSDWCE-UONOGXRCSA-N 2-tert-butyl-5-[(1S,3R)-3-[tert-butyl(dimethyl)silyl]oxycyclopentyl]pyrazol-3-amine Chemical compound C(C)(C)(C)N1N=C(C=C1N)[C@@H]1C[C@@H](CC1)O[Si](C)(C)C(C)(C)C UDJKDMSGTSDWCE-UONOGXRCSA-N 0.000 description 2
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 2
- BRARRAHGNDUELT-UHFFFAOYSA-N 3-hydroxypicolinic acid Chemical compound OC(=O)C1=NC=CC=C1O BRARRAHGNDUELT-UHFFFAOYSA-N 0.000 description 2
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 2
- IDPUKCWIGUEADI-UHFFFAOYSA-N 5-[bis(2-chloroethyl)amino]uracil Chemical compound ClCCN(CCCl)C1=CNC(=O)NC1=O IDPUKCWIGUEADI-UHFFFAOYSA-N 0.000 description 2
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 2
- OZAIFHULBGXAKX-VAWYXSNFSA-N AIBN Substances N#CC(C)(C)\N=N\C(C)(C)C#N OZAIFHULBGXAKX-VAWYXSNFSA-N 0.000 description 2
- 208000036762 Acute promyelocytic leukaemia Diseases 0.000 description 2
- 208000010507 Adenocarcinoma of Lung Diseases 0.000 description 2
- 201000003076 Angiosarcoma Diseases 0.000 description 2
- 101100059333 Arabidopsis thaliana CYCA1-2 gene Proteins 0.000 description 2
- 102000004452 Arginase Human genes 0.000 description 2
- 108700024123 Arginases Proteins 0.000 description 2
- BFYIZQONLCFLEV-DAELLWKTSA-N Aromasine Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC(=C)C2=C1 BFYIZQONLCFLEV-DAELLWKTSA-N 0.000 description 2
- 108010024976 Asparaginase Proteins 0.000 description 2
- 206010003571 Astrocytoma Diseases 0.000 description 2
- 102100029822 B- and T-lymphocyte attenuator Human genes 0.000 description 2
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 description 2
- 101000840545 Bacillus thuringiensis L-isoleucine-4-hydroxylase Proteins 0.000 description 2
- 208000035143 Bacterial infection Diseases 0.000 description 2
- 206010004146 Basal cell carcinoma Diseases 0.000 description 2
- 206010004593 Bile duct cancer Diseases 0.000 description 2
- 206010005003 Bladder cancer Diseases 0.000 description 2
- 108010006654 Bleomycin Proteins 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- 208000003174 Brain Neoplasms Diseases 0.000 description 2
- 206010055113 Breast cancer metastatic Diseases 0.000 description 2
- DBWYLWFEYODFGF-WFRZBGSESA-N CC(C)(C)N1N=C([C@@H](CC2)C[C@@H]2OC(N2C=NC=C2)=O)C=C1NC1=NC(OC/C=C\CN)=CC=C1 Chemical compound CC(C)(C)N1N=C([C@@H](CC2)C[C@@H]2OC(N2C=NC=C2)=O)C=C1NC1=NC(OC/C=C\CN)=CC=C1 DBWYLWFEYODFGF-WFRZBGSESA-N 0.000 description 2
- OAXCVJUQBBGUDR-UYRGJFBYSA-N CC(C)(C)N1N=C([C@@H](CC2)C[C@@H]2OC(N2C=NC=C2)=O)C=C1NC1=NC(OC/C=C\CNC(OC(C)(C)C)=O)=CC=C1 Chemical compound CC(C)(C)N1N=C([C@@H](CC2)C[C@@H]2OC(N2C=NC=C2)=O)C=C1NC1=NC(OC/C=C\CNC(OC(C)(C)C)=O)=CC=C1 OAXCVJUQBBGUDR-UYRGJFBYSA-N 0.000 description 2
- AUQFRFVMROLBAI-XZOQPEGZSA-N CC(C)(C)N1N=C([C@@H](CC2)C[C@@H]2OC(N2C=NC=C2)=O)C=C1NC1=NC(OCCCCNC(OC(C)(C)C)=O)=CC(C#N)=C1 Chemical compound CC(C)(C)N1N=C([C@@H](CC2)C[C@@H]2OC(N2C=NC=C2)=O)C=C1NC1=NC(OCCCCNC(OC(C)(C)C)=O)=CC(C#N)=C1 AUQFRFVMROLBAI-XZOQPEGZSA-N 0.000 description 2
- APMWVKCRLYGEEJ-GHTZIAJQSA-N CC(C)(C)N1N=C([C@@H](CC2)C[C@@H]2OC(OC(C=C2)=CC=C2[N+]([O-])=O)=O)C=C1NC1=NC(COCCCN)=NC=C1 Chemical compound CC(C)(C)N1N=C([C@@H](CC2)C[C@@H]2OC(OC(C=C2)=CC=C2[N+]([O-])=O)=O)C=C1NC1=NC(COCCCN)=NC=C1 APMWVKCRLYGEEJ-GHTZIAJQSA-N 0.000 description 2
- HQBNJKJOUUBUBZ-XUZZJYLKSA-N CC(C)(C)N1N=C([C@@H](CC2)C[C@@H]2OC(OC(C=C2)=CC=C2[N+]([O-])=O)=O)C=C1NC1=NC(COCCCNC(OC(C)(C)C)=O)=NC=C1 Chemical compound CC(C)(C)N1N=C([C@@H](CC2)C[C@@H]2OC(OC(C=C2)=CC=C2[N+]([O-])=O)=O)C=C1NC1=NC(COCCCNC(OC(C)(C)C)=O)=NC=C1 HQBNJKJOUUBUBZ-XUZZJYLKSA-N 0.000 description 2
- PDQNJKMXOCIPBG-OFNKIYASSA-N CC(C)(C)N1N=C([C@H](CC2)C[C@H]2OC(N2C=NC=C2)=O)C=C1NC1=NC(COCCCNC(OC(C)(C)C)=O)=CC(Cl)=C1 Chemical compound CC(C)(C)N1N=C([C@H](CC2)C[C@H]2OC(N2C=NC=C2)=O)C=C1NC1=NC(COCCCNC(OC(C)(C)C)=O)=CC(Cl)=C1 PDQNJKMXOCIPBG-OFNKIYASSA-N 0.000 description 2
- OPLZWYORCKERSC-FNTXCZFBSA-N CC(CCCN(CC(C=CC(OC)=C1)=C1OC)C(OC(C)(C)C)=O)OC1=CC=CC(NC2=CC([C@@H](CC3)C[C@@H]3OC(N3C=NC=C3)=O)=NN2C(C)(C)C)=N1 Chemical compound CC(CCCN(CC(C=CC(OC)=C1)=C1OC)C(OC(C)(C)C)=O)OC1=CC=CC(NC2=CC([C@@H](CC3)C[C@@H]3OC(N3C=NC=C3)=O)=NN2C(C)(C)C)=N1 OPLZWYORCKERSC-FNTXCZFBSA-N 0.000 description 2
- ZGYMZDQFSAFFSA-NRRUETGQSA-N CC(CCCN)OC1=CC=CC(NC2=CC([C@@H](CC3)C[C@@H]3OC(N3C=NC=C3)=O)=NN2C(C)(C)C)=N1 Chemical compound CC(CCCN)OC1=CC=CC(NC2=CC([C@@H](CC3)C[C@@H]3OC(N3C=NC=C3)=O)=NN2C(C)(C)C)=N1 ZGYMZDQFSAFFSA-NRRUETGQSA-N 0.000 description 2
- 102100027207 CD27 antigen Human genes 0.000 description 2
- 102100038078 CD276 antigen Human genes 0.000 description 2
- 101710185679 CD276 antigen Proteins 0.000 description 2
- 101150013553 CD40 gene Proteins 0.000 description 2
- 239000004215 Carbon black (E152) Substances 0.000 description 2
- 208000017897 Carcinoma of esophagus Diseases 0.000 description 2
- ZEOWTGPWHLSLOG-UHFFFAOYSA-N Cc1ccc(cc1-c1ccc2c(n[nH]c2c1)-c1cnn(c1)C1CC1)C(=O)Nc1cccc(c1)C(F)(F)F Chemical compound Cc1ccc(cc1-c1ccc2c(n[nH]c2c1)-c1cnn(c1)C1CC1)C(=O)Nc1cccc(c1)C(F)(F)F ZEOWTGPWHLSLOG-UHFFFAOYSA-N 0.000 description 2
- 102000009410 Chemokine receptor Human genes 0.000 description 2
- 108050000299 Chemokine receptor Proteins 0.000 description 2
- JWBOIMRXGHLCPP-UHFFFAOYSA-N Chloditan Chemical compound C=1C=CC=C(Cl)C=1C(C(Cl)Cl)C1=CC=C(Cl)C=C1 JWBOIMRXGHLCPP-UHFFFAOYSA-N 0.000 description 2
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 2
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 description 2
- 102100025191 Cyclin-A2 Human genes 0.000 description 2
- 102000004127 Cytokines Human genes 0.000 description 2
- 108090000695 Cytokines Proteins 0.000 description 2
- 108010092160 Dactinomycin Proteins 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 102100031968 Ephrin type-B receptor 2 Human genes 0.000 description 2
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 2
- 208000032027 Essential Thrombocythemia Diseases 0.000 description 2
- 229940124783 FAK inhibitor Drugs 0.000 description 2
- 102100023593 Fibroblast growth factor receptor 1 Human genes 0.000 description 2
- 101710182386 Fibroblast growth factor receptor 1 Proteins 0.000 description 2
- 102100023600 Fibroblast growth factor receptor 2 Human genes 0.000 description 2
- 101710182389 Fibroblast growth factor receptor 2 Proteins 0.000 description 2
- 102100027842 Fibroblast growth factor receptor 3 Human genes 0.000 description 2
- 101710182396 Fibroblast growth factor receptor 3 Proteins 0.000 description 2
- 102100027844 Fibroblast growth factor receptor 4 Human genes 0.000 description 2
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 2
- 102100037813 Focal adhesion kinase 1 Human genes 0.000 description 2
- VWUXBMIQPBEWFH-WCCTWKNTSA-N Fulvestrant Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3[C@H](CCCCCCCCCS(=O)CCCC(F)(F)C(F)(F)F)CC2=C1 VWUXBMIQPBEWFH-WCCTWKNTSA-N 0.000 description 2
- 230000004707 G1/S transition Effects 0.000 description 2
- 241000224466 Giardia Species 0.000 description 2
- 208000032612 Glial tumor Diseases 0.000 description 2
- 206010018338 Glioma Diseases 0.000 description 2
- 108010069236 Goserelin Proteins 0.000 description 2
- 102100039619 Granulocyte colony-stimulating factor Human genes 0.000 description 2
- 208000002927 Hamartoma Diseases 0.000 description 2
- 208000001258 Hemangiosarcoma Diseases 0.000 description 2
- 208000021519 Hodgkin lymphoma Diseases 0.000 description 2
- 101000864344 Homo sapiens B- and T-lymphocyte attenuator Proteins 0.000 description 2
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 description 2
- 101000914511 Homo sapiens CD27 antigen Proteins 0.000 description 2
- 101000917134 Homo sapiens Fibroblast growth factor receptor 4 Proteins 0.000 description 2
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 description 2
- 101000997832 Homo sapiens Tyrosine-protein kinase JAK2 Proteins 0.000 description 2
- 101000666896 Homo sapiens V-type immunoglobulin domain-containing suppressor of T-cell activation Proteins 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- XQFRJNBWHJMXHO-RRKCRQDMSA-N IDUR Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(I)=C1 XQFRJNBWHJMXHO-RRKCRQDMSA-N 0.000 description 2
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 2
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 2
- 229940076838 Immune checkpoint inhibitor Drugs 0.000 description 2
- 102000037984 Inhibitory immune checkpoint proteins Human genes 0.000 description 2
- 108091008026 Inhibitory immune checkpoint proteins Proteins 0.000 description 2
- 229940122245 Janus kinase inhibitor Drugs 0.000 description 2
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 2
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 2
- 239000002118 L01XE12 - Vandetanib Substances 0.000 description 2
- 239000002137 L01XE24 - Ponatinib Substances 0.000 description 2
- 108010000817 Leuprolide Proteins 0.000 description 2
- HLFSDGLLUJUHTE-SNVBAGLBSA-N Levamisole Chemical compound C1([C@H]2CN3CCSC3=N2)=CC=CC=C1 HLFSDGLLUJUHTE-SNVBAGLBSA-N 0.000 description 2
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 2
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 2
- 208000025205 Mantle-Cell Lymphoma Diseases 0.000 description 2
- 241000712079 Measles morbillivirus Species 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- 208000003445 Mouth Neoplasms Diseases 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- 208000031888 Mycoses Diseases 0.000 description 2
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- KIIQIMJEPAVHTF-UHFFFAOYSA-N NCC1CC(COC2=NC(Br)=CC=C2)C1 Chemical compound NCC1CC(COC2=NC(Br)=CC=C2)C1 KIIQIMJEPAVHTF-UHFFFAOYSA-N 0.000 description 2
- 102000004473 OX40 Ligand Human genes 0.000 description 2
- 108010042215 OX40 Ligand Proteins 0.000 description 2
- YGACXVRLDHEXKY-WXRXAMBDSA-N O[C@H](C[C@H]1c2c(cccc2F)-c2cncn12)[C@H]1CC[C@H](O)CC1 Chemical compound O[C@H](C[C@H]1c2c(cccc2F)-c2cncn12)[C@H]1CC[C@H](O)CC1 YGACXVRLDHEXKY-WXRXAMBDSA-N 0.000 description 2
- 241000283973 Oryctolagus cuniculus Species 0.000 description 2
- 208000030852 Parasitic disease Diseases 0.000 description 2
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 2
- 102100036052 Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform Human genes 0.000 description 2
- 101710096503 Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform Proteins 0.000 description 2
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 2
- 241000288906 Primates Species 0.000 description 2
- 208000033826 Promyelocytic Acute Leukemia Diseases 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- 208000015634 Rectal Neoplasms Diseases 0.000 description 2
- 206010038389 Renal cancer Diseases 0.000 description 2
- 201000000582 Retinoblastoma Diseases 0.000 description 2
- 241000283984 Rodentia Species 0.000 description 2
- 230000018199 S phase Effects 0.000 description 2
- 101001037255 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) Indoleamine 2,3-dioxygenase Proteins 0.000 description 2
- 229940124639 Selective inhibitor Drugs 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- 101100215487 Sus scrofa ADRA2A gene Proteins 0.000 description 2
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 description 2
- 210000001744 T-lymphocyte Anatomy 0.000 description 2
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 description 2
- 208000024313 Testicular Neoplasms Diseases 0.000 description 2
- 206010057644 Testis cancer Diseases 0.000 description 2
- MUMGGOZAMZWBJJ-DYKIIFRCSA-N Testostosterone Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 MUMGGOZAMZWBJJ-DYKIIFRCSA-N 0.000 description 2
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 2
- 208000024770 Thyroid neoplasm Diseases 0.000 description 2
- 102100040653 Tryptophan 2,3-dioxygenase Human genes 0.000 description 2
- 101710136122 Tryptophan 2,3-dioxygenase Proteins 0.000 description 2
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 description 2
- 102100033444 Tyrosine-protein kinase JAK2 Human genes 0.000 description 2
- 108010079206 V-Set Domain-Containing T-Cell Activation Inhibitor 1 Proteins 0.000 description 2
- 102100038929 V-set domain-containing T-cell activation inhibitor 1 Human genes 0.000 description 2
- 102100038282 V-type immunoglobulin domain-containing suppressor of T-cell activation Human genes 0.000 description 2
- 108091008605 VEGF receptors Proteins 0.000 description 2
- 108010053099 Vascular Endothelial Growth Factor Receptor-2 Proteins 0.000 description 2
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 2
- 208000036142 Viral infection Diseases 0.000 description 2
- 208000008383 Wilms tumor Diseases 0.000 description 2
- XMYKNCNAZKMVQN-NYYWCZLTSA-N [(e)-(3-aminopyridin-2-yl)methylideneamino]thiourea Chemical compound NC(=S)N\N=C\C1=NC=CC=C1N XMYKNCNAZKMVQN-NYYWCZLTSA-N 0.000 description 2
- SQYNJZFYBCGAEI-UHFFFAOYSA-N [3-(hydroxymethyl)cyclobutyl]methanol Chemical compound OCC1CC(CO)C1 SQYNJZFYBCGAEI-UHFFFAOYSA-N 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 208000017733 acquired polycythemia vera Diseases 0.000 description 2
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 2
- 239000002671 adjuvant Substances 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 229960000548 alemtuzumab Drugs 0.000 description 2
- 229940100198 alkylating agent Drugs 0.000 description 2
- 239000002168 alkylating agent Substances 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 229960002932 anastrozole Drugs 0.000 description 2
- YBBLVLTVTVSKRW-UHFFFAOYSA-N anastrozole Chemical compound N#CC(C)(C)C1=CC(C(C)(C#N)C)=CC(CN2N=CN=C2)=C1 YBBLVLTVTVSKRW-UHFFFAOYSA-N 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 229960002594 arsenic trioxide Drugs 0.000 description 2
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 2
- 208000022362 bacterial infectious disease Diseases 0.000 description 2
- 244000052616 bacterial pathogen Species 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 2
- GEWUGXSJBKRVCE-UHFFFAOYSA-N benzyl N-[2-tert-butyl-5-(3,3-dimethoxycyclopentyl)pyrazol-3-yl]carbamate Chemical compound C(C)(C)(C)N1N=C(C=C1NC(OCC1=CC=CC=C1)=O)C1CC(CC1)(OC)OC GEWUGXSJBKRVCE-UHFFFAOYSA-N 0.000 description 2
- 208000026900 bile duct neoplasm Diseases 0.000 description 2
- 229960000074 biopharmaceutical Drugs 0.000 description 2
- 229960001561 bleomycin Drugs 0.000 description 2
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 2
- 210000001185 bone marrow Anatomy 0.000 description 2
- QYJXDIUNDMRLAO-UHFFFAOYSA-N butyl 4-methylbenzenesulfonate Chemical compound CCCCOS(=O)(=O)C1=CC=C(C)C=C1 QYJXDIUNDMRLAO-UHFFFAOYSA-N 0.000 description 2
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 229960004562 carboplatin Drugs 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical group 0.000 description 2
- 150000001735 carboxylic acids Chemical group 0.000 description 2
- 208000002458 carcinoid tumor Diseases 0.000 description 2
- 208000019065 cervical carcinoma Diseases 0.000 description 2
- 230000000973 chemotherapeutic effect Effects 0.000 description 2
- 229960004630 chlorambucil Drugs 0.000 description 2
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 2
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 2
- 229960004316 cisplatin Drugs 0.000 description 2
- 229960002436 cladribine Drugs 0.000 description 2
- 208000009060 clear cell adenocarcinoma Diseases 0.000 description 2
- 229960000928 clofarabine Drugs 0.000 description 2
- WDDPHFBMKLOVOX-AYQXTPAHSA-N clofarabine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1F WDDPHFBMKLOVOX-AYQXTPAHSA-N 0.000 description 2
- 208000029742 colonic neoplasm Diseases 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 125000001162 cycloheptenyl group Chemical group C1(=CCCCCC1)* 0.000 description 2
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 2
- 229960000684 cytarabine Drugs 0.000 description 2
- 229960003901 dacarbazine Drugs 0.000 description 2
- 229960000640 dactinomycin Drugs 0.000 description 2
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 2
- 229960000975 daunorubicin Drugs 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 229960003668 docetaxel Drugs 0.000 description 2
- NOTIQUSPUUHHEH-UXOVVSIBSA-N dromostanolone propionate Chemical compound C([C@@H]1CC2)C(=O)[C@H](C)C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H](OC(=O)CC)[C@@]2(C)CC1 NOTIQUSPUUHHEH-UXOVVSIBSA-N 0.000 description 2
- 239000006196 drop Substances 0.000 description 2
- 229950004683 drostanolone propionate Drugs 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 229950009791 durvalumab Drugs 0.000 description 2
- 238000003821 enantio-separation Methods 0.000 description 2
- 201000003914 endometrial carcinoma Diseases 0.000 description 2
- 229940088598 enzyme Drugs 0.000 description 2
- 229950006370 epacadostat Drugs 0.000 description 2
- 229960001904 epirubicin Drugs 0.000 description 2
- 229960001842 estramustine Drugs 0.000 description 2
- FRPJXPJMRWBBIH-RBRWEJTLSA-N estramustine Chemical compound ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 FRPJXPJMRWBBIH-RBRWEJTLSA-N 0.000 description 2
- 239000000328 estrogen antagonist Substances 0.000 description 2
- 229960000255 exemestane Drugs 0.000 description 2
- 239000012091 fetal bovine serum Substances 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 229960000961 floxuridine Drugs 0.000 description 2
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 description 2
- 229960000390 fludarabine Drugs 0.000 description 2
- 229960002949 fluorouracil Drugs 0.000 description 2
- IJJVMEJXYNJXOJ-UHFFFAOYSA-N fluquinconazole Chemical compound C=1C=C(Cl)C=C(Cl)C=1N1C(=O)C2=CC(F)=CC=C2N=C1N1C=NC=N1 IJJVMEJXYNJXOJ-UHFFFAOYSA-N 0.000 description 2
- 230000003325 follicular Effects 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- 238000001640 fractional crystallisation Methods 0.000 description 2
- 229960002258 fulvestrant Drugs 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 108020001507 fusion proteins Proteins 0.000 description 2
- 102000037865 fusion proteins Human genes 0.000 description 2
- 201000006585 gastric adenocarcinoma Diseases 0.000 description 2
- 230000002496 gastric effect Effects 0.000 description 2
- 229960005277 gemcitabine Drugs 0.000 description 2
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- 125000005843 halogen group Chemical group 0.000 description 2
- 208000006454 hepatitis Diseases 0.000 description 2
- 231100000283 hepatitis Toxicity 0.000 description 2
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 2
- 229940121372 histone deacetylase inhibitor Drugs 0.000 description 2
- 239000003276 histone deacetylase inhibitor Substances 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 229960000908 idarubicin Drugs 0.000 description 2
- 229960002411 imatinib Drugs 0.000 description 2
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 2
- 229960003685 imatinib mesylate Drugs 0.000 description 2
- 239000012274 immune-checkpoint protein inhibitor Substances 0.000 description 2
- 238000002649 immunization Methods 0.000 description 2
- 230000003053 immunization Effects 0.000 description 2
- 239000007943 implant Substances 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 230000002779 inactivation Effects 0.000 description 2
- 125000001041 indolyl group Chemical group 0.000 description 2
- 206010022000 influenza Diseases 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 238000010255 intramuscular injection Methods 0.000 description 2
- 239000007927 intramuscular injection Substances 0.000 description 2
- 208000024312 invasive carcinoma Diseases 0.000 description 2
- 229960004768 irinotecan Drugs 0.000 description 2
- 239000007951 isotonicity adjuster Substances 0.000 description 2
- 201000010982 kidney cancer Diseases 0.000 description 2
- 201000010260 leiomyoma Diseases 0.000 description 2
- GFIJNRVAKGFPGQ-LIJARHBVSA-N leuprolide Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 GFIJNRVAKGFPGQ-LIJARHBVSA-N 0.000 description 2
- 229960004338 leuprorelin Drugs 0.000 description 2
- 229960001614 levamisole Drugs 0.000 description 2
- 125000005647 linker group Chemical group 0.000 description 2
- KRTIYQIPSAGSBP-KLAILNCOSA-N linrodostat Chemical compound C1(CCC(CC1)C1=C2C=C(F)C=CC2=NC=C1)[C@@H](C)C(=O)NC1=CC=C(Cl)C=C1 KRTIYQIPSAGSBP-KLAILNCOSA-N 0.000 description 2
- 239000008297 liquid dosage form Substances 0.000 description 2
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 2
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 2
- 229960002247 lomustine Drugs 0.000 description 2
- 239000007937 lozenge Substances 0.000 description 2
- 201000005249 lung adenocarcinoma Diseases 0.000 description 2
- 208000026037 malignant tumor of neck Diseases 0.000 description 2
- 238000001819 mass spectrum Methods 0.000 description 2
- 229960004296 megestrol acetate Drugs 0.000 description 2
- RQZAXGRLVPAYTJ-GQFGMJRRSA-N megestrol acetate Chemical compound C1=C(C)C2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)C)[C@@]1(C)CC2 RQZAXGRLVPAYTJ-GQFGMJRRSA-N 0.000 description 2
- 206010027191 meningioma Diseases 0.000 description 2
- 229960001428 mercaptopurine Drugs 0.000 description 2
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 2
- 229960000485 methotrexate Drugs 0.000 description 2
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 2
- CFCUWKMKBJTWLW-BKHRDMLASA-N mithramycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@H](O)[C@H](O[C@@H]3O[C@H](C)[C@@H](O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@@H](O)[C@H](O)[C@@H](C)O1 CFCUWKMKBJTWLW-BKHRDMLASA-N 0.000 description 2
- 229960004857 mitomycin Drugs 0.000 description 2
- 229960000350 mitotane Drugs 0.000 description 2
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 2
- 229960001156 mitoxantrone Drugs 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- 206010028537 myelofibrosis Diseases 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- 125000001624 naphthyl group Chemical group 0.000 description 2
- 239000006199 nebulizer Substances 0.000 description 2
- 201000011682 nervous system cancer Diseases 0.000 description 2
- 229960003301 nivolumab Drugs 0.000 description 2
- 125000006574 non-aromatic ring group Chemical group 0.000 description 2
- WPHGSKGZRAQSGP-UHFFFAOYSA-N norcarane Chemical compound C1CCCC2CC21 WPHGSKGZRAQSGP-UHFFFAOYSA-N 0.000 description 2
- 201000008106 ocular cancer Diseases 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 201000010302 ovarian serous cystadenocarcinoma Diseases 0.000 description 2
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 2
- 230000036281 parasite infection Effects 0.000 description 2
- 230000001717 pathogenic effect Effects 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 230000035515 penetration Effects 0.000 description 2
- 235000019271 petrolatum Nutrition 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- RLOWWWKZYUNIDI-UHFFFAOYSA-N phosphinic chloride Chemical compound ClP=O RLOWWWKZYUNIDI-UHFFFAOYSA-N 0.000 description 2
- XKJCHHZQLQNZHY-UHFFFAOYSA-N phthalimide Chemical compound C1=CC=C2C(=O)NC(=O)C2=C1 XKJCHHZQLQNZHY-UHFFFAOYSA-N 0.000 description 2
- 229960000952 pipobroman Drugs 0.000 description 2
- NJBFOOCLYDNZJN-UHFFFAOYSA-N pipobroman Chemical compound BrCCC(=O)N1CCN(C(=O)CCBr)CC1 NJBFOOCLYDNZJN-UHFFFAOYSA-N 0.000 description 2
- 229960003171 plicamycin Drugs 0.000 description 2
- 208000037244 polycythemia vera Diseases 0.000 description 2
- 229960000688 pomalidomide Drugs 0.000 description 2
- UVSMNLNDYGZFPF-UHFFFAOYSA-N pomalidomide Chemical compound O=C1C=2C(N)=CC=CC=2C(=O)N1C1CCC(=O)NC1=O UVSMNLNDYGZFPF-UHFFFAOYSA-N 0.000 description 2
- 229960001131 ponatinib Drugs 0.000 description 2
- PHXJVRSECIGDHY-UHFFFAOYSA-N ponatinib Chemical compound C1CN(C)CCN1CC(C(=C1)C(F)(F)F)=CC=C1NC(=O)C1=CC=C(C)C(C#CC=2N3N=CC=CC3=NC=2)=C1 PHXJVRSECIGDHY-UHFFFAOYSA-N 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 150000003141 primary amines Chemical class 0.000 description 2
- 208000003476 primary myelofibrosis Diseases 0.000 description 2
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 2
- 229960000624 procarbazine Drugs 0.000 description 2
- 239000003380 propellant Substances 0.000 description 2
- 125000004289 pyrazol-3-yl group Chemical group [H]N1N=C(*)C([H])=C1[H] 0.000 description 2
- 125000002098 pyridazinyl group Chemical group 0.000 description 2
- 108091006082 receptor inhibitors Proteins 0.000 description 2
- 102000005962 receptors Human genes 0.000 description 2
- 108020003175 receptors Proteins 0.000 description 2
- 206010038038 rectal cancer Diseases 0.000 description 2
- 201000001275 rectum cancer Diseases 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 210000003491 skin Anatomy 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 239000007909 solid dosage form Substances 0.000 description 2
- 229950007213 spartalizumab Drugs 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 150000003431 steroids Chemical class 0.000 description 2
- 229960001052 streptozocin Drugs 0.000 description 2
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 2
- 238000010254 subcutaneous injection Methods 0.000 description 2
- 239000007929 subcutaneous injection Substances 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 229960001603 tamoxifen Drugs 0.000 description 2
- 238000002626 targeted therapy Methods 0.000 description 2
- 229960004964 temozolomide Drugs 0.000 description 2
- 229960001278 teniposide Drugs 0.000 description 2
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 2
- 201000003120 testicular cancer Diseases 0.000 description 2
- 229960005353 testolactone Drugs 0.000 description 2
- BPEWUONYVDABNZ-DZBHQSCQSA-N testolactone Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(OC(=O)CC4)[C@@H]4[C@@H]3CCC2=C1 BPEWUONYVDABNZ-DZBHQSCQSA-N 0.000 description 2
- 125000003831 tetrazolyl group Chemical group 0.000 description 2
- GFFXZLZWLOBBLO-ASKVSEFXSA-N tezacitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(=C/F)/[C@H](O)[C@@H](CO)O1 GFFXZLZWLOBBLO-ASKVSEFXSA-N 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- 229960001196 thiotepa Drugs 0.000 description 2
- 229960003087 tioguanine Drugs 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- 229960000303 topotecan Drugs 0.000 description 2
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 2
- 229960005026 toremifene Drugs 0.000 description 2
- XFCLJVABOIYOMF-QPLCGJKRSA-N toremifene Chemical compound C1=CC(OCCN(C)C)=CC=C1C(\C=1C=CC=CC=1)=C(\CCCl)C1=CC=CC=C1 XFCLJVABOIYOMF-QPLCGJKRSA-N 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- 238000000844 transformation Methods 0.000 description 2
- 229960005526 triapine Drugs 0.000 description 2
- PYOKUURKVVELLB-UHFFFAOYSA-N trimethyl orthoformate Chemical compound COC(OC)OC PYOKUURKVVELLB-UHFFFAOYSA-N 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 229960001055 uracil mustard Drugs 0.000 description 2
- 210000003708 urethra Anatomy 0.000 description 2
- 201000005112 urinary bladder cancer Diseases 0.000 description 2
- 229960000241 vandetanib Drugs 0.000 description 2
- UHTHHESEBZOYNR-UHFFFAOYSA-N vandetanib Chemical compound COC1=CC(C(/N=CN2)=N/C=3C(=CC(Br)=CC=3)F)=C2C=C1OCC1CCN(C)CC1 UHTHHESEBZOYNR-UHFFFAOYSA-N 0.000 description 2
- 229940099039 velcade Drugs 0.000 description 2
- 229960003048 vinblastine Drugs 0.000 description 2
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 2
- 229960002066 vinorelbine Drugs 0.000 description 2
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 description 2
- 230000009385 viral infection Effects 0.000 description 2
- 235000012431 wafers Nutrition 0.000 description 2
- 239000003643 water by type Substances 0.000 description 2
- 238000009736 wetting Methods 0.000 description 2
- BMKDZUISNHGIBY-ZETCQYMHSA-N (+)-dexrazoxane Chemical compound C([C@H](C)N1CC(=O)NC(=O)C1)N1CC(=O)NC(=O)C1 BMKDZUISNHGIBY-ZETCQYMHSA-N 0.000 description 1
- XYLPKCDRAAYATL-OAHLLOKOSA-N (11S)-7-(3,5-dimethyl-1,2-oxazol-4-yl)-11-pyridin-2-yl-9-oxa-1,3-diazatricyclo[6.3.1.04,12]dodeca-4(12),5,7-trien-2-one Chemical compound CC1=NOC(C)=C1C1=CC=C2C3=C1OC[C@H](C=1N=CC=CC=1)N3C(=O)N2 XYLPKCDRAAYATL-OAHLLOKOSA-N 0.000 description 1
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 1
- XFQNWPYGEGCIMF-HCUGAJCMSA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].[Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 XFQNWPYGEGCIMF-HCUGAJCMSA-N 0.000 description 1
- QOWBXWFYRXSBAS-UHFFFAOYSA-N (2,4-dimethoxyphenyl)methanamine Chemical compound COC1=CC=C(CN)C(OC)=C1 QOWBXWFYRXSBAS-UHFFFAOYSA-N 0.000 description 1
- FRPHIGOLOLSXAI-UHFFFAOYSA-N (2-bromo-1,3-thiazol-4-yl)methanol Chemical compound OCC1=CSC(Br)=N1 FRPHIGOLOLSXAI-UHFFFAOYSA-N 0.000 description 1
- KIUKXJAPPMFGSW-DNGZLQJQSA-N (2S,3S,4S,5R,6R)-6-[(2S,3R,4R,5S,6R)-3-Acetamido-2-[(2S,3S,4R,5R,6R)-6-[(2R,3R,4R,5S,6R)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylic acid Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 KIUKXJAPPMFGSW-DNGZLQJQSA-N 0.000 description 1
- VEEGZPWAAPPXRB-BJMVGYQFSA-N (3e)-3-(1h-imidazol-5-ylmethylidene)-1h-indol-2-one Chemical compound O=C1NC2=CC=CC=C2\C1=C/C1=CN=CN1 VEEGZPWAAPPXRB-BJMVGYQFSA-N 0.000 description 1
- ZQPDJCIXJHUERQ-QWRGUYRKSA-N (4r)-4-[3-[(1s)-1-(4-amino-3-methylpyrazolo[3,4-d]pyrimidin-1-yl)ethyl]-5-chloro-2-ethoxy-6-fluorophenyl]pyrrolidin-2-one Chemical compound CCOC1=C([C@H](C)N2C3=NC=NC(N)=C3C(C)=N2)C=C(Cl)C(F)=C1[C@@H]1CNC(=O)C1 ZQPDJCIXJHUERQ-QWRGUYRKSA-N 0.000 description 1
- KZEDPVFJLQLDIZ-UHFFFAOYSA-N (5-diphenylphosphanyl-9,9-dimethylxanthen-4-yl)-diphenylphosphane Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1.C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZEDPVFJLQLDIZ-UHFFFAOYSA-N 0.000 description 1
- CCBGOKJSSNIORL-UHFFFAOYSA-N (6-chloropyrazin-2-yl)methanol Chemical compound OCC1=CN=CC(Cl)=N1 CCBGOKJSSNIORL-UHFFFAOYSA-N 0.000 description 1
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 1
- XGQXULJHBWKUJY-LYIKAWCPSA-N (z)-but-2-enedioic acid;n-[2-(diethylamino)ethyl]-5-[(z)-(5-fluoro-2-oxo-1h-indol-3-ylidene)methyl]-2,4-dimethyl-1h-pyrrole-3-carboxamide Chemical compound OC(=O)\C=C/C(O)=O.CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C XGQXULJHBWKUJY-LYIKAWCPSA-N 0.000 description 1
- YFMFNYKEUDLDTL-UHFFFAOYSA-N 1,1,1,2,3,3,3-heptafluoropropane Chemical compound FC(F)(F)C(F)C(F)(F)F YFMFNYKEUDLDTL-UHFFFAOYSA-N 0.000 description 1
- LVGUZGTVOIAKKC-UHFFFAOYSA-N 1,1,1,2-tetrafluoroethane Chemical compound FCC(F)(F)F LVGUZGTVOIAKKC-UHFFFAOYSA-N 0.000 description 1
- WTAPZWXVSZMMDG-UHFFFAOYSA-N 1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].C=1C=CC=CC=1C=CC(=O)C=CC1=CC=CC=C1 WTAPZWXVSZMMDG-UHFFFAOYSA-N 0.000 description 1
- QCZZSANNLWPGEA-UHFFFAOYSA-N 1-(4-phenylphenyl)ethanone Chemical compound C1=CC(C(=O)C)=CC=C1C1=CC=CC=C1 QCZZSANNLWPGEA-UHFFFAOYSA-N 0.000 description 1
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 1
- WBPWDDPSYSUQJA-VQTJNVASSA-N 1-[[4-(methoxymethyl)-4-[[[(1R,2S)-2-phenylcyclopropyl]amino]methyl]piperidin-1-yl]methyl]cyclobutane-1-carboxylic acid Chemical compound COCC1(CCN(CC1)CC1(CCC1)C(=O)O)CN[C@H]1[C@@H](C1)C1=CC=CC=C1 WBPWDDPSYSUQJA-VQTJNVASSA-N 0.000 description 1
- 102100025573 1-alkyl-2-acetylglycerophosphocholine esterase Human genes 0.000 description 1
- MICMHFIQSAMEJG-UHFFFAOYSA-N 1-bromopyrrolidine-2,5-dione Chemical compound BrN1C(=O)CCC1=O.BrN1C(=O)CCC1=O MICMHFIQSAMEJG-UHFFFAOYSA-N 0.000 description 1
- DFPYXQYWILNVAU-UHFFFAOYSA-N 1-hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1.C1=CC=C2N(O)N=NC2=C1 DFPYXQYWILNVAU-UHFFFAOYSA-N 0.000 description 1
- KKVYYGGCHJGEFJ-UHFFFAOYSA-N 1-n-(4-chlorophenyl)-6-methyl-5-n-[3-(7h-purin-6-yl)pyridin-2-yl]isoquinoline-1,5-diamine Chemical compound N=1C=CC2=C(NC=3C(=CC=CN=3)C=3C=4N=CNC=4N=CN=3)C(C)=CC=C2C=1NC1=CC=C(Cl)C=C1 KKVYYGGCHJGEFJ-UHFFFAOYSA-N 0.000 description 1
- VSNHCAURESNICA-NJFSPNSNSA-N 1-oxidanylurea Chemical compound N[14C](=O)NO VSNHCAURESNICA-NJFSPNSNSA-N 0.000 description 1
- RQEUFEKYXDPUSK-UHFFFAOYSA-N 1-phenylethylamine Chemical compound CC(N)C1=CC=CC=C1 RQEUFEKYXDPUSK-UHFFFAOYSA-N 0.000 description 1
- 125000001462 1-pyrrolyl group Chemical group [*]N1C([H])=C([H])C([H])=C1[H] 0.000 description 1
- GCKMFJBGXUYNAG-UHFFFAOYSA-N 17alpha-methyltestosterone Natural products C1CC2=CC(=O)CCC2(C)C2C1C1CCC(C)(O)C1(C)CC2 GCKMFJBGXUYNAG-UHFFFAOYSA-N 0.000 description 1
- DBPWSSGDRRHUNT-CEGNMAFCSA-N 17α-hydroxyprogesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)C)(O)[C@@]1(C)CC2 DBPWSSGDRRHUNT-CEGNMAFCSA-N 0.000 description 1
- XEZNGIUYQVAUSS-UHFFFAOYSA-N 18-crown-6 Chemical compound C1COCCOCCOCCOCCOCCO1 XEZNGIUYQVAUSS-UHFFFAOYSA-N 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- KIPSRYDSZQRPEA-UHFFFAOYSA-N 2,2,2-trifluoroethanamine Chemical compound NCC(F)(F)F KIPSRYDSZQRPEA-UHFFFAOYSA-N 0.000 description 1
- LFCADBSUDWERJT-UHFFFAOYSA-N 2,6-dichloropyridine-3-carbonitrile Chemical compound ClC1=CC=C(C#N)C(Cl)=N1 LFCADBSUDWERJT-UHFFFAOYSA-N 0.000 description 1
- BTUKLHWINORBTN-UHFFFAOYSA-N 2,6-dichloropyridine-4-carbonitrile Chemical compound ClC1=CC(C#N)=CC(Cl)=N1 BTUKLHWINORBTN-UHFFFAOYSA-N 0.000 description 1
- UHPUYOIOPLNWKU-UHFFFAOYSA-N 2-(chloromethyl)-1h-pyrimidin-6-one Chemical compound OC1=CC=NC(CCl)=N1 UHPUYOIOPLNWKU-UHFFFAOYSA-N 0.000 description 1
- QXLQZLBNPTZMRK-UHFFFAOYSA-N 2-[(dimethylamino)methyl]-1-(2,4-dimethylphenyl)prop-2-en-1-one Chemical compound CN(C)CC(=C)C(=O)C1=CC=C(C)C=C1C QXLQZLBNPTZMRK-UHFFFAOYSA-N 0.000 description 1
- KTBSXLIQKWEBRB-UHFFFAOYSA-N 2-[1-[1-[3-fluoro-2-(trifluoromethyl)pyridine-4-carbonyl]piperidin-4-yl]-3-[4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)pyrazol-1-yl]azetidin-3-yl]acetonitrile Chemical compound C1=CN=C(C(F)(F)F)C(F)=C1C(=O)N1CCC(N2CC(CC#N)(C2)N2N=CC(=C2)C=2C=3C=CNC=3N=CN=2)CC1 KTBSXLIQKWEBRB-UHFFFAOYSA-N 0.000 description 1
- UHTQHHLSGVOGQR-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-4-ium-1-yl]ethanesulfonate Chemical compound OCCN1CCN(CCS(O)(=O)=O)CC1.OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 UHTQHHLSGVOGQR-UHFFFAOYSA-N 0.000 description 1
- PDGKHKMBHVFCMG-UHFFFAOYSA-N 2-[[5-(4-methylpiperazin-1-yl)pyridin-2-yl]amino]spiro[7,8-dihydropyrazino[5,6]pyrrolo[1,2-d]pyrimidine-9,1'-cyclohexane]-6-one Chemical compound C1CN(C)CCN1C(C=N1)=CC=C1NC1=NC=C(C=C2N3C4(CCCCC4)CNC2=O)C3=N1 PDGKHKMBHVFCMG-UHFFFAOYSA-N 0.000 description 1
- BVAHPPKGOOJSPU-UHFFFAOYSA-N 2-[[5-chloro-2-[(5-methyl-2-propan-2-ylpyrazol-3-yl)amino]pyridin-4-yl]amino]-n-methoxybenzamide Chemical compound CONC(=O)C1=CC=CC=C1NC1=CC(NC=2N(N=C(C)C=2)C(C)C)=NC=C1Cl BVAHPPKGOOJSPU-UHFFFAOYSA-N 0.000 description 1
- NJAHGEUFOYIGKR-UHFFFAOYSA-N 2-chloroethanimidamide Chemical compound NC(=N)CCl NJAHGEUFOYIGKR-UHFFFAOYSA-N 0.000 description 1
- ULMMVBPTWVRPSI-UHFFFAOYSA-N 2-fluoro-5-methoxy-4-[[4-[(2-methyl-3-oxo-1h-isoindol-4-yl)oxy]-5-(trifluoromethyl)pyrimidin-2-yl]amino]-n-(1-methylpiperidin-4-yl)benzamide Chemical compound FC=1C=C(NC=2N=C(OC=3C=4C(=O)N(C)CC=4C=CC=3)C(=CN=2)C(F)(F)F)C(OC)=CC=1C(=O)NC1CCN(C)CC1 ULMMVBPTWVRPSI-UHFFFAOYSA-N 0.000 description 1
- LIOLIMKSCNQPLV-UHFFFAOYSA-N 2-fluoro-n-methyl-4-[7-(quinolin-6-ylmethyl)imidazo[1,2-b][1,2,4]triazin-2-yl]benzamide Chemical compound C1=C(F)C(C(=O)NC)=CC=C1C1=NN2C(CC=3C=C4C=CC=NC4=CC=3)=CN=C2N=C1 LIOLIMKSCNQPLV-UHFFFAOYSA-N 0.000 description 1
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000389 2-pyrrolyl group Chemical group [H]N1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- NDMPLJNOPCLANR-UHFFFAOYSA-N 3,4-dihydroxy-15-(4-hydroxy-18-methoxycarbonyl-5,18-seco-ibogamin-18-yl)-16-methoxy-1-methyl-6,7-didehydro-aspidospermidine-3-carboxylic acid methyl ester Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 NDMPLJNOPCLANR-UHFFFAOYSA-N 0.000 description 1
- UCHKRHGVKYVGTC-UHFFFAOYSA-N 3,6-dibromo-2-methylpyridine Chemical compound CC1=NC(Br)=CC=C1Br UCHKRHGVKYVGTC-UHFFFAOYSA-N 0.000 description 1
- HCDMJFOHIXMBOV-UHFFFAOYSA-N 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-4,7-dihydropyrrolo[4,5]pyrido[1,2-d]pyrimidin-2-one Chemical compound C=1C2=C3N(CC)C(=O)N(C=4C(=C(OC)C=C(OC)C=4F)F)CC3=CN=C2NC=1CN1CCOCC1 HCDMJFOHIXMBOV-UHFFFAOYSA-N 0.000 description 1
- MXKLDYKORJEOPR-UHFFFAOYSA-N 3-(5-fluoro-1h-indol-3-yl)pyrrolidine-2,5-dione Chemical compound C12=CC(F)=CC=C2NC=C1C1CC(=O)NC1=O MXKLDYKORJEOPR-UHFFFAOYSA-N 0.000 description 1
- LKKMLIBUAXYLOY-UHFFFAOYSA-N 3-Amino-1-methyl-5H-pyrido[4,3-b]indole Chemical compound N1C2=CC=CC=C2C2=C1C=C(N)N=C2C LKKMLIBUAXYLOY-UHFFFAOYSA-N 0.000 description 1
- RDZPMWLHALUNCD-UHFFFAOYSA-N 3-bromo-6-chloropyridine-2-carboxylic acid Chemical compound OC(=O)C1=NC(Cl)=CC=C1Br RDZPMWLHALUNCD-UHFFFAOYSA-N 0.000 description 1
- 125000003682 3-furyl group Chemical group O1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- ZRWMAMOBIQQJSA-UHFFFAOYSA-N 3-methylidenecyclobutane-1-carbonitrile Chemical compound C=C1CC(C#N)C1 ZRWMAMOBIQQJSA-UHFFFAOYSA-N 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000001397 3-pyrrolyl group Chemical group [H]N1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 125000001541 3-thienyl group Chemical group S1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- AYYUSDKNXRPJBH-UHFFFAOYSA-N 4,6-dichloropyridine-2-carboxylic acid Chemical compound OC(=O)C1=CC(Cl)=CC(Cl)=N1 AYYUSDKNXRPJBH-UHFFFAOYSA-N 0.000 description 1
- IJEMXJANZPVITP-UHFFFAOYSA-N 4-[tert-butyl(dimethyl)silyl]oxybutan-1-ol Chemical compound CC(C)(C)[Si](C)(C)OCCCCO IJEMXJANZPVITP-UHFFFAOYSA-N 0.000 description 1
- GUBXTQINPBZVJP-UHFFFAOYSA-N 4-bromo-2,6-dichloropyridine Chemical compound ClC1=CC(Br)=CC(Cl)=N1 GUBXTQINPBZVJP-UHFFFAOYSA-N 0.000 description 1
- KDDQRKBRJSGMQE-UHFFFAOYSA-N 4-thiazolyl Chemical group [C]1=CSC=N1 KDDQRKBRJSGMQE-UHFFFAOYSA-N 0.000 description 1
- 102100022464 5'-nucleotidase Human genes 0.000 description 1
- XAUDJQYHKZQPEU-KVQBGUIXSA-N 5-aza-2'-deoxycytidine Chemical compound O=C1N=C(N)N=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 XAUDJQYHKZQPEU-KVQBGUIXSA-N 0.000 description 1
- NMUSYJAQQFHJEW-KVTDHHQDSA-N 5-azacytidine Chemical compound O=C1N=C(N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NMUSYJAQQFHJEW-KVTDHHQDSA-N 0.000 description 1
- CWDWFSXUQODZGW-UHFFFAOYSA-N 5-thiazolyl Chemical group [C]1=CN=CS1 CWDWFSXUQODZGW-UHFFFAOYSA-N 0.000 description 1
- LKXJGVGBEDEAAW-UHFFFAOYSA-N 6-bromo-1h-pyrrolo[2,3-b]pyridine Chemical compound BrC1=CC=C2C=CNC2=N1 LKXJGVGBEDEAAW-UHFFFAOYSA-N 0.000 description 1
- BFQONZCQHGIKIY-UHFFFAOYSA-N 6-bromo-3-fluoro-2-methylpyridine Chemical compound CC1=NC(Br)=CC=C1F BFQONZCQHGIKIY-UHFFFAOYSA-N 0.000 description 1
- VVIAGPKUTFNRDU-UHFFFAOYSA-N 6S-folinic acid Natural products C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-UHFFFAOYSA-N 0.000 description 1
- RHXHGRAEPCAFML-UHFFFAOYSA-N 7-cyclopentyl-n,n-dimethyl-2-[(5-piperazin-1-ylpyridin-2-yl)amino]pyrrolo[2,3-d]pyrimidine-6-carboxamide Chemical compound N1=C2N(C3CCCC3)C(C(=O)N(C)C)=CC2=CN=C1NC(N=C1)=CC=C1N1CCNCC1 RHXHGRAEPCAFML-UHFFFAOYSA-N 0.000 description 1
- SHGAZHPCJJPHSC-ZVCIMWCZSA-N 9-cis-retinoic acid Chemical compound OC(=O)/C=C(\C)/C=C/C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-ZVCIMWCZSA-N 0.000 description 1
- 101150107888 AKT2 gene Proteins 0.000 description 1
- 229940126654 ALK2 inhibitor Drugs 0.000 description 1
- 241000235389 Absidia Species 0.000 description 1
- 241000224424 Acanthamoeba sp. Species 0.000 description 1
- 206010000830 Acute leukaemia Diseases 0.000 description 1
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 1
- 206010052747 Adenocarcinoma pancreas Diseases 0.000 description 1
- 206010001233 Adenoma benign Diseases 0.000 description 1
- ZKHQWZAMYRWXGA-UHFFFAOYSA-N Adenosine triphosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)C(O)C1O ZKHQWZAMYRWXGA-UHFFFAOYSA-N 0.000 description 1
- 101150051188 Adora2a gene Proteins 0.000 description 1
- 101150078577 Adora2b gene Proteins 0.000 description 1
- 101150051155 Akt3 gene Proteins 0.000 description 1
- ITPDYQOUSLNIHG-UHFFFAOYSA-N Amiodarone hydrochloride Chemical compound [Cl-].CCCCC=1OC2=CC=CC=C2C=1C(=O)C1=CC(I)=C(OCC[NH+](CC)CC)C(I)=C1 ITPDYQOUSLNIHG-UHFFFAOYSA-N 0.000 description 1
- 102100021569 Apoptosis regulator Bcl-2 Human genes 0.000 description 1
- 208000006400 Arbovirus Encephalitis Diseases 0.000 description 1
- 102000015790 Asparaginase Human genes 0.000 description 1
- 241000228212 Aspergillus Species 0.000 description 1
- 239000012664 BCL-2-inhibitor Substances 0.000 description 1
- 108091012583 BCL2 Proteins 0.000 description 1
- 102100035080 BDNF/NT-3 growth factors receptor Human genes 0.000 description 1
- OLCWFLWEHWLBTO-HSZRJFAPSA-N BMS-214662 Chemical compound C=1C=CSC=1S(=O)(=O)N([C@@H](C1)CC=2C=CC=CC=2)CC2=CC(C#N)=CC=C2N1CC1=CN=CN1 OLCWFLWEHWLBTO-HSZRJFAPSA-N 0.000 description 1
- 229940125565 BMS-986016 Drugs 0.000 description 1
- 241000223848 Babesia microti Species 0.000 description 1
- 241000193738 Bacillus anthracis Species 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 241001235572 Balantioides coli Species 0.000 description 1
- 229940123711 Bcl2 inhibitor Drugs 0.000 description 1
- 241000228405 Blastomyces dermatitidis Species 0.000 description 1
- 206010073106 Bone giant cell tumour malignant Diseases 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 208000003508 Botulism Diseases 0.000 description 1
- 206010006143 Brain stem glioma Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 102000001805 Bromodomains Human genes 0.000 description 1
- 108050009021 Bromodomains Proteins 0.000 description 1
- 102100031151 C-C chemokine receptor type 2 Human genes 0.000 description 1
- 101710149815 C-C chemokine receptor type 2 Proteins 0.000 description 1
- 102100035875 C-C chemokine receptor type 5 Human genes 0.000 description 1
- 101710149870 C-C chemokine receptor type 5 Proteins 0.000 description 1
- FLQRQXJIQNBEBE-DTWKUNHWSA-N CC(C)(C)N(C([C@@H](CC1)C[C@@H]1O)=C1)N=C1N Chemical compound CC(C)(C)N(C([C@@H](CC1)C[C@@H]1O)=C1)N=C1N FLQRQXJIQNBEBE-DTWKUNHWSA-N 0.000 description 1
- RYYBJVVRFIAWIJ-PDGQHHTCSA-N CC(C)(C)N/N=C(/CC1)\CC1C1=NN(C(C)(C)C)C(N)=C1 Chemical compound CC(C)(C)N/N=C(/CC1)\CC1C1=NN(C(C)(C)C)C(N)=C1 RYYBJVVRFIAWIJ-PDGQHHTCSA-N 0.000 description 1
- CPNORVIVFZSSSS-UHFFFAOYSA-N CC(C)(C)N1N=C(C(CC2)CC2N)C=C1C(C(N)=N1)=CC=C1OCCCCO[Si](C)(C)C(C)(C)C Chemical compound CC(C)(C)N1N=C(C(CC2)CC2N)C=C1C(C(N)=N1)=CC=C1OCCCCO[Si](C)(C)C(C)(C)C CPNORVIVFZSSSS-UHFFFAOYSA-N 0.000 description 1
- KYVZEMHWDMGMFO-AZUAARDMSA-N CC(C)(C)N1N=C([C@@H](CC2)C[C@@H]2OC(N2C=NC=C2)=O)C=C1NC(C=C1)=NC(OCCCCN)=C1C#N Chemical compound CC(C)(C)N1N=C([C@@H](CC2)C[C@@H]2OC(N2C=NC=C2)=O)C=C1NC(C=C1)=NC(OCCCCN)=C1C#N KYVZEMHWDMGMFO-AZUAARDMSA-N 0.000 description 1
- KSNFTIPNICULHZ-FUHWJXTLSA-N CC(C)(C)N1N=C([C@@H](CC2)C[C@@H]2OC(N2C=NC=C2)=O)C=C1NC1=NC(COCCCN)=CS1 Chemical compound CC(C)(C)N1N=C([C@@H](CC2)C[C@@H]2OC(N2C=NC=C2)=O)C=C1NC1=NC(COCCCN)=CS1 KSNFTIPNICULHZ-FUHWJXTLSA-N 0.000 description 1
- FSNSKEGCDZFGLH-PZJWPPBQSA-N CC(C)(C)N1N=C([C@@H](CC2)C[C@@H]2OC(N2C=NC=C2)=O)C=C1NC1=NC(COCCCNC(OC(C)(C)C)=O)=CS1 Chemical compound CC(C)(C)N1N=C([C@@H](CC2)C[C@@H]2OC(N2C=NC=C2)=O)C=C1NC1=NC(COCCCNC(OC(C)(C)C)=O)=CS1 FSNSKEGCDZFGLH-PZJWPPBQSA-N 0.000 description 1
- MKYAIEIKTSUZRV-FCHUYYIVSA-N CC(C)(C)N1N=C([C@@H](CC2)C[C@@H]2O[Si](C(C)(C)C)(C2=CC=CC=C2)C2=CC=CC=C2)C=C1N Chemical compound CC(C)(C)N1N=C([C@@H](CC2)C[C@@H]2O[Si](C(C)(C)C)(C2=CC=CC=C2)C2=CC=CC=C2)C=C1N MKYAIEIKTSUZRV-FCHUYYIVSA-N 0.000 description 1
- NPMYYNGDMKIHFG-BDAKNGLRSA-N CC(C)(C)N1N=C([C@H](CC2)C[C@H]2O)C=C1N Chemical compound CC(C)(C)N1N=C([C@H](CC2)C[C@H]2O)C=C1N NPMYYNGDMKIHFG-BDAKNGLRSA-N 0.000 description 1
- HMEWSBGIDRGJMW-XLIONFOSSA-N CC(C)(C)N1N=C([C@H](CC2)C[C@H]2OC(N2C=NC=C2)=O)C=C1NC1=NC(COCCCN)=CC(Cl)=C1 Chemical compound CC(C)(C)N1N=C([C@H](CC2)C[C@H]2OC(N2C=NC=C2)=O)C=C1NC1=NC(COCCCN)=CC(Cl)=C1 HMEWSBGIDRGJMW-XLIONFOSSA-N 0.000 description 1
- 101100381481 Caenorhabditis elegans baz-2 gene Proteins 0.000 description 1
- 101100123850 Caenorhabditis elegans her-1 gene Proteins 0.000 description 1
- 241000222120 Candida <Saccharomycetales> Species 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- 208000005623 Carcinogenesis Diseases 0.000 description 1
- 208000009458 Carcinoma in Situ Diseases 0.000 description 1
- 101150056334 Ccne1 gene Proteins 0.000 description 1
- 206010007953 Central nervous system lymphoma Diseases 0.000 description 1
- 206010008263 Cervical dysplasia Diseases 0.000 description 1
- 229920001661 Chitosan Polymers 0.000 description 1
- 241000606161 Chlamydia Species 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 206010008631 Cholera Diseases 0.000 description 1
- 201000005262 Chondroma Diseases 0.000 description 1
- 201000009047 Chordoma Diseases 0.000 description 1
- 208000006332 Choriocarcinoma Diseases 0.000 description 1
- 241000223205 Coccidioides immitis Species 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 206010048832 Colon adenoma Diseases 0.000 description 1
- 206010010356 Congenital anomaly Diseases 0.000 description 1
- 201000002847 Cowden syndrome Diseases 0.000 description 1
- 241000709687 Coxsackievirus Species 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- 201000007336 Cryptococcosis Diseases 0.000 description 1
- 241000221204 Cryptococcus neoformans Species 0.000 description 1
- 241000295636 Cryptosporidium sp. Species 0.000 description 1
- 108010068192 Cyclin A Proteins 0.000 description 1
- 108010060273 Cyclin A2 Proteins 0.000 description 1
- 229920000858 Cyclodextrin Polymers 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 230000003350 DNA copy number gain Effects 0.000 description 1
- 239000012623 DNA damaging agent Substances 0.000 description 1
- 230000006820 DNA synthesis Effects 0.000 description 1
- 101100481408 Danio rerio tie2 gene Proteins 0.000 description 1
- 241000725619 Dengue virus Species 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 206010059866 Drug resistance Diseases 0.000 description 1
- 208000007033 Dysgerminoma Diseases 0.000 description 1
- 208000000471 Dysplastic Nevus Syndrome Diseases 0.000 description 1
- 108010093502 E2F Transcription Factors Proteins 0.000 description 1
- 102000001388 E2F Transcription Factors Human genes 0.000 description 1
- 101150029707 ERBB2 gene Proteins 0.000 description 1
- 241001115402 Ebolavirus Species 0.000 description 1
- 241001466953 Echovirus Species 0.000 description 1
- 201000009051 Embryonal Carcinoma Diseases 0.000 description 1
- 241000224432 Entamoeba histolytica Species 0.000 description 1
- 241000709661 Enterovirus Species 0.000 description 1
- 241000991587 Enterovirus C Species 0.000 description 1
- 206010014967 Ependymoma Diseases 0.000 description 1
- 108010055196 EphA2 Receptor Proteins 0.000 description 1
- 108010055191 EphA3 Receptor Proteins 0.000 description 1
- 108010055334 EphB2 Receptor Proteins 0.000 description 1
- 102100030322 Ephrin type-A receptor 1 Human genes 0.000 description 1
- 102100030340 Ephrin type-A receptor 2 Human genes 0.000 description 1
- 102100030324 Ephrin type-A receptor 3 Human genes 0.000 description 1
- 102100031983 Ephrin type-B receptor 4 Human genes 0.000 description 1
- 201000005231 Epithelioid sarcoma Diseases 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- 240000001414 Eucalyptus viminalis Species 0.000 description 1
- 108010007457 Extracellular Signal-Regulated MAP Kinases Proteins 0.000 description 1
- 102100028138 F-box/WD repeat-containing protein 7 Human genes 0.000 description 1
- 101150009958 FLT4 gene Proteins 0.000 description 1
- 201000001342 Fallopian tube cancer Diseases 0.000 description 1
- 208000013452 Fallopian tube neoplasm Diseases 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 208000007659 Fibroadenoma Diseases 0.000 description 1
- 206010053717 Fibrous histiocytoma Diseases 0.000 description 1
- 108010029961 Filgrastim Proteins 0.000 description 1
- 241000710831 Flavivirus Species 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 1
- 101710113436 GTPase KRas Proteins 0.000 description 1
- 206010051066 Gastrointestinal stromal tumour Diseases 0.000 description 1
- 208000000527 Germinoma Diseases 0.000 description 1
- 208000007569 Giant Cell Tumors Diseases 0.000 description 1
- 201000005409 Gliomatosis cerebri Diseases 0.000 description 1
- 206010018404 Glucagonoma Diseases 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- BLCLNMBMMGCOAS-URPVMXJPSA-N Goserelin Chemical compound C([C@@H](C(=O)N[C@H](COC(C)(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N1[C@@H](CCC1)C(=O)NNC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 BLCLNMBMMGCOAS-URPVMXJPSA-N 0.000 description 1
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 description 1
- 206010018691 Granuloma Diseases 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- 208000017891 HER2 positive breast carcinoma Diseases 0.000 description 1
- 229940125962 HPK1 kinase inhibitor Drugs 0.000 description 1
- 101710113864 Heat shock protein 90 Proteins 0.000 description 1
- 102100034051 Heat shock protein HSP 90-alpha Human genes 0.000 description 1
- 102000002812 Heat-Shock Proteins Human genes 0.000 description 1
- 108010004889 Heat-Shock Proteins Proteins 0.000 description 1
- 206010019629 Hepatic adenoma Diseases 0.000 description 1
- 206010073069 Hepatic cancer Diseases 0.000 description 1
- 208000005176 Hepatitis C Diseases 0.000 description 1
- 208000037262 Hepatitis delta Diseases 0.000 description 1
- 102100035108 High affinity nerve growth factor receptor Human genes 0.000 description 1
- 102000003964 Histone deacetylase Human genes 0.000 description 1
- 108090000353 Histone deacetylase Proteins 0.000 description 1
- 102100038715 Histone deacetylase 8 Human genes 0.000 description 1
- 241000228404 Histoplasma capsulatum Species 0.000 description 1
- 101000678236 Homo sapiens 5'-nucleotidase Proteins 0.000 description 1
- 101000596896 Homo sapiens BDNF/NT-3 growth factors receptor Proteins 0.000 description 1
- 101000938354 Homo sapiens Ephrin type-A receptor 1 Proteins 0.000 description 1
- 101001060231 Homo sapiens F-box/WD repeat-containing protein 7 Proteins 0.000 description 1
- 101000596894 Homo sapiens High affinity nerve growth factor receptor Proteins 0.000 description 1
- 101001032118 Homo sapiens Histone deacetylase 8 Proteins 0.000 description 1
- 101001055145 Homo sapiens Interleukin-2 receptor subunit beta Proteins 0.000 description 1
- 101000599886 Homo sapiens Isocitrate dehydrogenase [NADP], mitochondrial Proteins 0.000 description 1
- 101000868279 Homo sapiens Leukocyte surface antigen CD47 Proteins 0.000 description 1
- 101001138062 Homo sapiens Leukocyte-associated immunoglobulin-like receptor 1 Proteins 0.000 description 1
- 101001059991 Homo sapiens Mitogen-activated protein kinase kinase kinase kinase 1 Proteins 0.000 description 1
- 101000692455 Homo sapiens Platelet-derived growth factor receptor beta Proteins 0.000 description 1
- 101000932478 Homo sapiens Receptor-type tyrosine-protein kinase FLT3 Proteins 0.000 description 1
- 101000984753 Homo sapiens Serine/threonine-protein kinase B-raf Proteins 0.000 description 1
- 101000596234 Homo sapiens T-cell surface protein tactile Proteins 0.000 description 1
- 101001102797 Homo sapiens Transmembrane protein PVRIG Proteins 0.000 description 1
- 101000617285 Homo sapiens Tyrosine-protein phosphatase non-receptor type 6 Proteins 0.000 description 1
- 241000598436 Human T-cell lymphotropic virus Species 0.000 description 1
- 241000700588 Human alphaherpesvirus 1 Species 0.000 description 1
- 241000701024 Human betaherpesvirus 5 Species 0.000 description 1
- 241000701044 Human gammaherpesvirus 4 Species 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 1
- 229940043367 IDO1 inhibitor Drugs 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 208000005045 Interdigitating dendritic cell sarcoma Diseases 0.000 description 1
- 108010078049 Interferon alpha-2 Proteins 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010002350 Interleukin-2 Proteins 0.000 description 1
- 102000000588 Interleukin-2 Human genes 0.000 description 1
- 102100026879 Interleukin-2 receptor subunit beta Human genes 0.000 description 1
- 208000005016 Intestinal Neoplasms Diseases 0.000 description 1
- 102100037845 Isocitrate dehydrogenase [NADP], mitochondrial Human genes 0.000 description 1
- 241000701460 JC polyomavirus Species 0.000 description 1
- 229940121730 Janus kinase 2 inhibitor Drugs 0.000 description 1
- 102000002698 KIR Receptors Human genes 0.000 description 1
- 108010043610 KIR Receptors Proteins 0.000 description 1
- 208000002260 Keloid Diseases 0.000 description 1
- 229930194542 Keto Natural products 0.000 description 1
- 241000588748 Klebsiella Species 0.000 description 1
- 239000005536 L01XE08 - Nilotinib Substances 0.000 description 1
- 239000003798 L01XE11 - Pazopanib Substances 0.000 description 1
- 239000002145 L01XE14 - Bosutinib Substances 0.000 description 1
- 239000002138 L01XE21 - Regorafenib Substances 0.000 description 1
- 239000002176 L01XE26 - Cabozantinib Substances 0.000 description 1
- UIARLYUEJFELEN-LROUJFHJSA-N LSM-1231 Chemical compound C12=C3N4C5=CC=CC=C5C3=C3C(=O)NCC3=C2C2=CC=CC=C2N1[C@]1(C)[C@](CO)(O)C[C@H]4O1 UIARLYUEJFELEN-LROUJFHJSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 241001245510 Lambia <signal fly> Species 0.000 description 1
- 206010023825 Laryngeal cancer Diseases 0.000 description 1
- 241000589248 Legionella Species 0.000 description 1
- 208000007764 Legionnaires' Disease Diseases 0.000 description 1
- 241000222722 Leishmania <genus> Species 0.000 description 1
- 241000222727 Leishmania donovani Species 0.000 description 1
- 206010024238 Leptospirosis Diseases 0.000 description 1
- 102100032913 Leukocyte surface antigen CD47 Human genes 0.000 description 1
- 102100020943 Leukocyte-associated immunoglobulin-like receptor 1 Human genes 0.000 description 1
- 208000022010 Lhermitte-Duclos disease Diseases 0.000 description 1
- 206010061523 Lip and/or oral cavity cancer Diseases 0.000 description 1
- 206010062038 Lip neoplasm Diseases 0.000 description 1
- 208000002404 Liver Cell Adenoma Diseases 0.000 description 1
- 208000016604 Lyme disease Diseases 0.000 description 1
- 206010052178 Lymphocytic lymphoma Diseases 0.000 description 1
- 229940123628 Lysine (K)-specific demethylase 1A inhibitor Drugs 0.000 description 1
- 102000001291 MAP Kinase Kinase Kinase Human genes 0.000 description 1
- 108091054455 MAP kinase family Proteins 0.000 description 1
- 102000043136 MAP kinase family Human genes 0.000 description 1
- 108060006687 MAP kinase kinase kinase Proteins 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 208000006644 Malignant Fibrous Histiocytoma Diseases 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 208000000172 Medulloblastoma Diseases 0.000 description 1
- 102000007557 Melanoma-Specific Antigens Human genes 0.000 description 1
- 108010071463 Melanoma-Specific Antigens Proteins 0.000 description 1
- 208000002030 Merkel cell carcinoma Diseases 0.000 description 1
- 206010027480 Metastatic malignant melanoma Diseases 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- QXKHYNVANLEOEG-UHFFFAOYSA-N Methoxsalen Chemical compound C1=CC(=O)OC2=C1C=C1C=COC1=C2OC QXKHYNVANLEOEG-UHFFFAOYSA-N 0.000 description 1
- FQISKWAFAHGMGT-SGJOWKDISA-M Methylprednisolone sodium succinate Chemical compound [Na+].C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2[C@@H](O)C[C@]2(C)[C@@](O)(C(=O)COC(=O)CCC([O-])=O)CC[C@H]21 FQISKWAFAHGMGT-SGJOWKDISA-M 0.000 description 1
- GCKMFJBGXUYNAG-HLXURNFRSA-N Methyltestosterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@](C)(O)[C@@]1(C)CC2 GCKMFJBGXUYNAG-HLXURNFRSA-N 0.000 description 1
- 208000032818 Microsatellite Instability Diseases 0.000 description 1
- 102100028199 Mitogen-activated protein kinase kinase kinase kinase 1 Human genes 0.000 description 1
- 241000235395 Mucor Species 0.000 description 1
- 241000235388 Mucorales Species 0.000 description 1
- 208000008770 Multiple Hamartoma Syndrome Diseases 0.000 description 1
- 241000711386 Mumps virus Species 0.000 description 1
- 101100381978 Mus musculus Braf gene Proteins 0.000 description 1
- 101100407308 Mus musculus Pdcd1lg2 gene Proteins 0.000 description 1
- 101100481410 Mus musculus Tek gene Proteins 0.000 description 1
- 208000014767 Myeloproliferative disease Diseases 0.000 description 1
- HSHXDCVZWHOWCS-UHFFFAOYSA-N N'-hexadecylthiophene-2-carbohydrazide Chemical compound CCCCCCCCCCCCCCCCNNC(=O)c1cccs1 HSHXDCVZWHOWCS-UHFFFAOYSA-N 0.000 description 1
- QJMCKEPOKRERLN-UHFFFAOYSA-N N-3,4-tridhydroxybenzamide Chemical compound ONC(=O)C1=CC=C(O)C(O)=C1 QJMCKEPOKRERLN-UHFFFAOYSA-N 0.000 description 1
- 108700026495 N-Myc Proto-Oncogene Proteins 0.000 description 1
- 102000055056 N-Myc Proto-Oncogene Human genes 0.000 description 1
- 102100029166 NT-3 growth factor receptor Human genes 0.000 description 1
- 101150117329 NTRK3 gene Proteins 0.000 description 1
- 241000224438 Naegleria fowleri Species 0.000 description 1
- 206010028729 Nasal cavity cancer Diseases 0.000 description 1
- 206010029266 Neuroendocrine carcinoma of the skin Diseases 0.000 description 1
- 241001126259 Nippostrongylus brasiliensis Species 0.000 description 1
- QUPUOWXJTJFVCC-UHFFFAOYSA-N OCC1=NC(Br)=CC=C1OCCN1CCCC1 Chemical compound OCC1=NC(Br)=CC=C1OCCN1CCCC1 QUPUOWXJTJFVCC-UHFFFAOYSA-N 0.000 description 1
- 208000035327 Oestrogen receptor positive breast cancer Diseases 0.000 description 1
- 201000010133 Oligodendroglioma Diseases 0.000 description 1
- 208000010191 Osteitis Deformans Diseases 0.000 description 1
- 208000000035 Osteochondroma Diseases 0.000 description 1
- 239000012661 PARP inhibitor Substances 0.000 description 1
- 229940124780 PI3K delta inhibitor Drugs 0.000 description 1
- 208000027067 Paget disease of bone Diseases 0.000 description 1
- 241001631646 Papillomaviridae Species 0.000 description 1
- 241000526686 Paracoccidioides brasiliensis Species 0.000 description 1
- 208000000821 Parathyroid Neoplasms Diseases 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 206010061336 Pelvic neoplasm Diseases 0.000 description 1
- 208000002471 Penile Neoplasms Diseases 0.000 description 1
- 239000004264 Petrolatum Substances 0.000 description 1
- 229940122907 Phosphatase inhibitor Drugs 0.000 description 1
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 1
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 1
- 102000014750 Phosphorylase Kinase Human genes 0.000 description 1
- 108010064071 Phosphorylase Kinase Proteins 0.000 description 1
- 208000007641 Pinealoma Diseases 0.000 description 1
- 208000007913 Pituitary Neoplasms Diseases 0.000 description 1
- 201000005746 Pituitary adenoma Diseases 0.000 description 1
- 206010061538 Pituitary tumour benign Diseases 0.000 description 1
- 206010035148 Plague Diseases 0.000 description 1
- 241000223810 Plasmodium vivax Species 0.000 description 1
- 102100026547 Platelet-derived growth factor receptor beta Human genes 0.000 description 1
- 241000233872 Pneumocystis carinii Species 0.000 description 1
- 229940121906 Poly ADP ribose polymerase inhibitor Drugs 0.000 description 1
- 101710179684 Poly [ADP-ribose] polymerase Proteins 0.000 description 1
- 102100023712 Poly [ADP-ribose] polymerase 1 Human genes 0.000 description 1
- 229920000776 Poly(Adenosine diphosphate-ribose) polymerase Polymers 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- 108700030875 Programmed Cell Death 1 Ligand 2 Proteins 0.000 description 1
- 102100024213 Programmed cell death 1 ligand 2 Human genes 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 241000588769 Proteus <enterobacteria> Species 0.000 description 1
- 102000016971 Proto-Oncogene Proteins c-kit Human genes 0.000 description 1
- 108010014608 Proto-Oncogene Proteins c-kit Proteins 0.000 description 1
- 241000125945 Protoparvovirus Species 0.000 description 1
- 241000589516 Pseudomonas Species 0.000 description 1
- 241000589517 Pseudomonas aeruginosa Species 0.000 description 1
- 229940044606 RIG-I agonist Drugs 0.000 description 1
- 241000711798 Rabies lyssavirus Species 0.000 description 1
- 101100372762 Rattus norvegicus Flt1 gene Proteins 0.000 description 1
- 101710100969 Receptor tyrosine-protein kinase erbB-3 Proteins 0.000 description 1
- 102100029986 Receptor tyrosine-protein kinase erbB-3 Human genes 0.000 description 1
- 102100029981 Receptor tyrosine-protein kinase erbB-4 Human genes 0.000 description 1
- 101710100963 Receptor tyrosine-protein kinase erbB-4 Proteins 0.000 description 1
- 102100020718 Receptor-type tyrosine-protein kinase FLT3 Human genes 0.000 description 1
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 1
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 1
- 208000006265 Renal cell carcinoma Diseases 0.000 description 1
- 241000725643 Respiratory syncytial virus Species 0.000 description 1
- 208000005678 Rhabdomyoma Diseases 0.000 description 1
- 241000606651 Rickettsiales Species 0.000 description 1
- 241000702670 Rotavirus Species 0.000 description 1
- 241000710799 Rubella virus Species 0.000 description 1
- 241000315672 SARS coronavirus Species 0.000 description 1
- 208000004337 Salivary Gland Neoplasms Diseases 0.000 description 1
- 206010061934 Salivary gland cancer Diseases 0.000 description 1
- 241000607142 Salmonella Species 0.000 description 1
- 201000010208 Seminoma Diseases 0.000 description 1
- 102100027103 Serine/threonine-protein kinase B-raf Human genes 0.000 description 1
- 102100023085 Serine/threonine-protein kinase mTOR Human genes 0.000 description 1
- 241000607720 Serratia Species 0.000 description 1
- 208000000097 Sertoli-Leydig cell tumor Diseases 0.000 description 1
- 241000700584 Simplexvirus Species 0.000 description 1
- 208000021712 Soft tissue sarcoma Diseases 0.000 description 1
- 229920002125 Sokalan® Polymers 0.000 description 1
- 241001149962 Sporothrix Species 0.000 description 1
- 241000295644 Staphylococcaceae Species 0.000 description 1
- 241000191967 Staphylococcus aureus Species 0.000 description 1
- 241000282898 Sus scrofa Species 0.000 description 1
- 206010042971 T-cell lymphoma Diseases 0.000 description 1
- 208000027585 T-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- 102100035268 T-cell surface protein tactile Human genes 0.000 description 1
- 108091005729 TAM receptors Proteins 0.000 description 1
- 108010065917 TOR Serine-Threonine Kinases Proteins 0.000 description 1
- 239000005463 Tandutinib Substances 0.000 description 1
- NAVMQTYZDKMPEU-UHFFFAOYSA-N Targretin Chemical compound CC1=CC(C(CCC2(C)C)(C)C)=C2C=C1C(=C)C1=CC=C(C(O)=O)C=C1 NAVMQTYZDKMPEU-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 206010043376 Tetanus Diseases 0.000 description 1
- 101150115851 Tff2 gene Proteins 0.000 description 1
- 206010043515 Throat cancer Diseases 0.000 description 1
- 206010062129 Tongue neoplasm Diseases 0.000 description 1
- 241000223996 Toxoplasma Species 0.000 description 1
- 102100039630 Transmembrane protein PVRIG Human genes 0.000 description 1
- MSLJYSGFUMYUDX-UHFFFAOYSA-N Trimidox Chemical compound ON=C(N)C1=CC(O)=C(O)C(O)=C1 MSLJYSGFUMYUDX-UHFFFAOYSA-N 0.000 description 1
- 241000223105 Trypanosoma brucei Species 0.000 description 1
- 241000223109 Trypanosoma cruzi Species 0.000 description 1
- 101150098329 Tyro3 gene Proteins 0.000 description 1
- 102100039094 Tyrosinase Human genes 0.000 description 1
- 108060008724 Tyrosinase Proteins 0.000 description 1
- 102100021657 Tyrosine-protein phosphatase non-receptor type 6 Human genes 0.000 description 1
- 208000015778 Undifferentiated pleomorphic sarcoma Diseases 0.000 description 1
- 208000023915 Ureteral Neoplasms Diseases 0.000 description 1
- 206010046392 Ureteric cancer Diseases 0.000 description 1
- 206010046431 Urethral cancer Diseases 0.000 description 1
- 229940124674 VEGF-R inhibitor Drugs 0.000 description 1
- 208000009311 VIPoma Diseases 0.000 description 1
- 241000700618 Vaccinia virus Species 0.000 description 1
- 201000003761 Vaginal carcinoma Diseases 0.000 description 1
- 102000016663 Vascular Endothelial Growth Factor Receptor-3 Human genes 0.000 description 1
- 206010048214 Xanthoma Diseases 0.000 description 1
- 206010048215 Xanthomatosis Diseases 0.000 description 1
- PCWZKQSKUXXDDJ-UHFFFAOYSA-N Xanthotoxin Natural products COCc1c2OC(=O)C=Cc2cc3ccoc13 PCWZKQSKUXXDDJ-UHFFFAOYSA-N 0.000 description 1
- 241000607479 Yersinia pestis Species 0.000 description 1
- 102100025093 Zinc fingers and homeoboxes protein 2 Human genes 0.000 description 1
- CTCBPRXHVPZNHB-VQFZJOCSSA-N [[(2r,3s,4r,5r)-5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] phosphono hydrogen phosphate;(2r,3r,4s,5r)-2-(6-aminopurin-9-yl)-5-(hydroxymethyl)oxolane-3,4-diol Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O.C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@H]1O CTCBPRXHVPZNHB-VQFZJOCSSA-N 0.000 description 1
- AIWRTTMUVOZGPW-HSPKUQOVSA-N abarelix Chemical compound C([C@@H](C(=O)N[C@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCNC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N[C@H](C)C(N)=O)N(C)C(=O)[C@H](CO)NC(=O)[C@@H](CC=1C=NC=CC=1)NC(=O)[C@@H](CC=1C=CC(Cl)=CC=1)NC(=O)[C@@H](CC=1C=C2C=CC=CC2=CC=1)NC(C)=O)C1=CC=C(O)C=C1 AIWRTTMUVOZGPW-HSPKUQOVSA-N 0.000 description 1
- 229960002184 abarelix Drugs 0.000 description 1
- 108010023617 abarelix Proteins 0.000 description 1
- 229950001573 abemaciclib Drugs 0.000 description 1
- 239000003070 absorption delaying agent Substances 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- YBCVMFKXIKNREZ-UHFFFAOYSA-N acoh acetic acid Chemical compound CC(O)=O.CC(O)=O YBCVMFKXIKNREZ-UHFFFAOYSA-N 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000011149 active material Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 208000002718 adenomatoid tumor Diseases 0.000 description 1
- 229940121359 adenosine receptor antagonist Drugs 0.000 description 1
- 238000009098 adjuvant therapy Methods 0.000 description 1
- 208000024447 adrenal gland neoplasm Diseases 0.000 description 1
- 229940009456 adriamycin Drugs 0.000 description 1
- 108010081667 aflibercept Proteins 0.000 description 1
- 101150045355 akt1 gene Proteins 0.000 description 1
- 108700025316 aldesleukin Proteins 0.000 description 1
- 229960005310 aldesleukin Drugs 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 229960001445 alitretinoin Drugs 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001447 alkali salts Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 125000003342 alkenyl group Chemical group 0.000 description 1
- 150000003973 alkyl amines Chemical class 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- OFCNXPDARWKPPY-UHFFFAOYSA-N allopurinol Chemical compound OC1=NC=NC2=C1C=NN2 OFCNXPDARWKPPY-UHFFFAOYSA-N 0.000 description 1
- 229960003459 allopurinol Drugs 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 229960003437 aminoglutethimide Drugs 0.000 description 1
- ROBVIMPUHSLWNV-UHFFFAOYSA-N aminoglutethimide Chemical compound C=1C=C(N)C=CC=1C1(CC)CCC(=O)NC1=O ROBVIMPUHSLWNV-UHFFFAOYSA-N 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 229960001220 amsacrine Drugs 0.000 description 1
- XCPGHVQEEXUHNC-UHFFFAOYSA-N amsacrine Chemical compound COC1=CC(NS(C)(=O)=O)=CC=C1NC1=C(C=CC=C2)C2=NC2=CC=CC=C12 XCPGHVQEEXUHNC-UHFFFAOYSA-N 0.000 description 1
- 239000004037 angiogenesis inhibitor Substances 0.000 description 1
- 229940121369 angiogenesis inhibitor Drugs 0.000 description 1
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 230000006023 anti-tumor response Effects 0.000 description 1
- 239000000611 antibody drug conjugate Substances 0.000 description 1
- 229940124691 antibody therapeutics Drugs 0.000 description 1
- 229940049595 antibody-drug conjugate Drugs 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 125000002029 aromatic hydrocarbon group Chemical group 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 239000010425 asbestos Substances 0.000 description 1
- 229960003272 asparaginase Drugs 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-M asparaginate Chemical compound [O-]C(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-M 0.000 description 1
- 229940120638 avastin Drugs 0.000 description 1
- 229960003005 axitinib Drugs 0.000 description 1
- RITAVMQDGBJQJZ-FMIVXFBMSA-N axitinib Chemical compound CNC(=O)C1=CC=CC=C1SC1=CC=C(C(\C=C\C=2N=CC=CC=2)=NN2)C2=C1 RITAVMQDGBJQJZ-FMIVXFBMSA-N 0.000 description 1
- 229960002756 azacitidine Drugs 0.000 description 1
- 125000003725 azepanyl group Chemical group 0.000 description 1
- 208000007456 balantidiasis Diseases 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 229960002707 bendamustine Drugs 0.000 description 1
- YTKUWDBFDASYHO-UHFFFAOYSA-N bendamustine Chemical compound ClCCN(CCCl)C1=CC=C2N(C)C(CCCC(O)=O)=NC2=C1 YTKUWDBFDASYHO-UHFFFAOYSA-N 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 208000001119 benign fibrous histiocytoma Diseases 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- 125000004603 benzisoxazolyl group Chemical group O1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- HSDAJNMJOMSNEV-UHFFFAOYSA-N benzyl chloroformate Chemical compound ClC(=O)OCC1=CC=CC=C1 HSDAJNMJOMSNEV-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 229960002938 bexarotene Drugs 0.000 description 1
- 125000002618 bicyclic heterocycle group Chemical group 0.000 description 1
- LASLVGACQUUOEB-UHFFFAOYSA-N bicyclo[1.1.0]butane Chemical compound C1C2CC21 LASLVGACQUUOEB-UHFFFAOYSA-N 0.000 description 1
- JSMRMEYFZHIPJV-UHFFFAOYSA-N bicyclo[2.1.1]hexane Chemical compound C1C2CC1CC2 JSMRMEYFZHIPJV-UHFFFAOYSA-N 0.000 description 1
- BVCRERJDOOBZOH-UHFFFAOYSA-N bicyclo[2.2.1]heptanyl Chemical group C1C[C+]2CC[C-]1C2 BVCRERJDOOBZOH-UHFFFAOYSA-N 0.000 description 1
- GPRLTFBKWDERLU-UHFFFAOYSA-N bicyclo[2.2.2]octane Chemical compound C1CC2CCC1CC2 GPRLTFBKWDERLU-UHFFFAOYSA-N 0.000 description 1
- JAPMJSVZDUYFKL-UHFFFAOYSA-N bicyclo[3.1.0]hexane Chemical compound C1CCC2CC21 JAPMJSVZDUYFKL-UHFFFAOYSA-N 0.000 description 1
- SHOMMGQAMRXRRK-UHFFFAOYSA-N bicyclo[3.1.1]heptane Chemical compound C1C2CC1CCC2 SHOMMGQAMRXRRK-UHFFFAOYSA-N 0.000 description 1
- AWYMFBJJKFTCFO-UHFFFAOYSA-N bicyclo[3.2.0]heptane Chemical compound C1CCC2CCC21 AWYMFBJJKFTCFO-UHFFFAOYSA-N 0.000 description 1
- LPCWKMYWISGVSK-UHFFFAOYSA-N bicyclo[3.2.1]octane Chemical compound C1C2CCC1CCC2 LPCWKMYWISGVSK-UHFFFAOYSA-N 0.000 description 1
- RPZUBXWEQBPUJR-UHFFFAOYSA-N bicyclo[4.2.0]octane Chemical compound C1CCCC2CCC21 RPZUBXWEQBPUJR-UHFFFAOYSA-N 0.000 description 1
- 239000000227 bioadhesive Substances 0.000 description 1
- 230000008827 biological function Effects 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 239000012472 biological sample Substances 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 206010005084 bladder transitional cell carcinoma Diseases 0.000 description 1
- 201000001528 bladder urothelial carcinoma Diseases 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 208000016738 bone Paget disease Diseases 0.000 description 1
- RMHDLBZYPISZOI-UHFFFAOYSA-N borane;methylsulfanylmethane Chemical compound B.CSC RMHDLBZYPISZOI-UHFFFAOYSA-N 0.000 description 1
- 229960003736 bosutinib Drugs 0.000 description 1
- UBPYILGKFZZVDX-UHFFFAOYSA-N bosutinib Chemical compound C1=C(Cl)C(OC)=CC(NC=2C3=CC(OC)=C(OCCCN4CCN(C)CC4)C=C3N=CC=2C#N)=C1Cl UBPYILGKFZZVDX-UHFFFAOYSA-N 0.000 description 1
- 201000009480 botryoid rhabdomyosarcoma Diseases 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 201000003149 breast fibroadenoma Diseases 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 208000003362 bronchogenic carcinoma Diseases 0.000 description 1
- 201000002143 bronchus adenoma Diseases 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- DLDJFQGPPSQZKI-UHFFFAOYSA-N but-2-yne-1,4-diol Chemical compound OCC#CCO DLDJFQGPPSQZKI-UHFFFAOYSA-N 0.000 description 1
- NFLBOYSTEVDVQC-UHFFFAOYSA-N butyl (4-nitrophenyl) carbonate Chemical compound CCCCOC(=O)OC1=CC=C([N+]([O-])=O)C=C1 NFLBOYSTEVDVQC-UHFFFAOYSA-N 0.000 description 1
- 229960001292 cabozantinib Drugs 0.000 description 1
- ONIQOQHATWINJY-UHFFFAOYSA-N cabozantinib Chemical compound C=12C=C(OC)C(OC)=CC2=NC=CC=1OC(C=C1)=CC=C1NC(=O)C1(C(=O)NC=2C=CC(F)=CC=2)CC1 ONIQOQHATWINJY-UHFFFAOYSA-N 0.000 description 1
- KVUAALJSMIVURS-ZEDZUCNESA-L calcium folinate Chemical compound [Ca+2].C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)N[C@@H](CCC([O-])=O)C([O-])=O)C=C1 KVUAALJSMIVURS-ZEDZUCNESA-L 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- IVFYLRMMHVYGJH-PVPPCFLZSA-N calusterone Chemical compound C1C[C@]2(C)[C@](O)(C)CC[C@H]2[C@@H]2[C@@H](C)CC3=CC(=O)CC[C@]3(C)[C@H]21 IVFYLRMMHVYGJH-PVPPCFLZSA-N 0.000 description 1
- 229950009823 calusterone Drugs 0.000 description 1
- 229940112129 campath Drugs 0.000 description 1
- 229940088954 camptosar Drugs 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- 238000009566 cancer vaccine Methods 0.000 description 1
- 229940022399 cancer vaccine Drugs 0.000 description 1
- 229950005852 capmatinib Drugs 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 231100000504 carcinogenesis Toxicity 0.000 description 1
- BLMPQMFVWMYDKT-NZTKNTHTSA-N carfilzomib Chemical group C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)[C@]1(C)OC1)NC(=O)CN1CCOCC1)CC1=CC=CC=C1 BLMPQMFVWMYDKT-NZTKNTHTSA-N 0.000 description 1
- 229960002438 carfilzomib Drugs 0.000 description 1
- 108010021331 carfilzomib Proteins 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000012876 carrier material Substances 0.000 description 1
- 230000025084 cell cycle arrest Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 208000025997 central nervous system neoplasm Diseases 0.000 description 1
- 230000010129 centrosome duplication Effects 0.000 description 1
- 210000003679 cervix uteri Anatomy 0.000 description 1
- 229910052729 chemical element Inorganic materials 0.000 description 1
- 229960002559 chlorotrianisene Drugs 0.000 description 1
- BFPSDSIWYFKGBC-UHFFFAOYSA-N chlorotrianisene Chemical compound C1=CC(OC)=CC=C1C(Cl)=C(C=1C=CC(OC)=CC=1)C1=CC=C(OC)C=C1 BFPSDSIWYFKGBC-UHFFFAOYSA-N 0.000 description 1
- 201000005217 chondroblastoma Diseases 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 208000024207 chronic leukemia Diseases 0.000 description 1
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 238000011284 combination treatment Methods 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 229940028617 conventional vaccine Drugs 0.000 description 1
- 229960001334 corticosteroids Drugs 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- DYNHJHQFHQTFTP-UHFFFAOYSA-N crenolanib Chemical compound C=1C=C2N(C=3N=C4C(N5CCC(N)CC5)=CC=CC4=CC=3)C=NC2=CC=1OCC1(C)COC1 DYNHJHQFHQTFTP-UHFFFAOYSA-N 0.000 description 1
- 229950009240 crenolanib Drugs 0.000 description 1
- 201000010305 cutaneous fibrous histiocytoma Diseases 0.000 description 1
- 208000035250 cutaneous malignant susceptibility to 1 melanoma Diseases 0.000 description 1
- 208000017763 cutaneous neuroendocrine carcinoma Diseases 0.000 description 1
- 229940043378 cyclin-dependent kinase inhibitor Drugs 0.000 description 1
- 125000001047 cyclobutenyl group Chemical group C1(=CCC1)* 0.000 description 1
- 125000000522 cyclooctenyl group Chemical group C1(=CCCCCCC1)* 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 208000002445 cystadenocarcinoma Diseases 0.000 description 1
- 239000000824 cytostatic agent Substances 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 229960002465 dabrafenib Drugs 0.000 description 1
- BFSMGDJOXZAERB-UHFFFAOYSA-N dabrafenib Chemical compound S1C(C(C)(C)C)=NC(C=2C(=C(NS(=O)(=O)C=3C(=CC=CC=3F)F)C=CC=2)F)=C1C1=CC=NC(N)=N1 BFSMGDJOXZAERB-UHFFFAOYSA-N 0.000 description 1
- 229940018872 dalteparin sodium Drugs 0.000 description 1
- DEZRYPDIMOWBDS-UHFFFAOYSA-N dcm dichloromethane Chemical compound ClCCl.ClCCl DEZRYPDIMOWBDS-UHFFFAOYSA-N 0.000 description 1
- 125000004855 decalinyl group Chemical group C1(CCCC2CCCCC12)* 0.000 description 1
- 229960003603 decitabine Drugs 0.000 description 1
- 210000004443 dendritic cell Anatomy 0.000 description 1
- 229960002923 denileukin diftitox Drugs 0.000 description 1
- 108010017271 denileukin diftitox Proteins 0.000 description 1
- CFCUWKMKBJTWLW-UHFFFAOYSA-N deoliosyl-3C-alpha-L-digitoxosyl-MTM Natural products CC=1C(O)=C2C(O)=C3C(=O)C(OC4OC(C)C(O)C(OC5OC(C)C(O)C(OC6OC(C)C(O)C(C)(O)C6)C5)C4)C(C(OC)C(=O)C(O)C(C)O)CC3=CC2=CC=1OC(OC(C)C1O)CC1OC1CC(O)C(O)C(C)O1 CFCUWKMKBJTWLW-UHFFFAOYSA-N 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 230000002074 deregulated effect Effects 0.000 description 1
- 230000003831 deregulation Effects 0.000 description 1
- 239000007933 dermal patch Substances 0.000 description 1
- 229960000605 dexrazoxane Drugs 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 125000003963 dichloro group Chemical group Cl* 0.000 description 1
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- RGLYKWWBQGJZGM-ISLYRVAYSA-N diethylstilbestrol Chemical compound C=1C=C(O)C=CC=1C(/CC)=C(\CC)C1=CC=C(O)C=C1 RGLYKWWBQGJZGM-ISLYRVAYSA-N 0.000 description 1
- 229960000452 diethylstilbestrol Drugs 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- 206010013023 diphtheria Diseases 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- JZBWUTVDIDNCMW-UHFFFAOYSA-L dipotassium;oxido sulfate Chemical compound [K+].[K+].[O-]OS([O-])(=O)=O JZBWUTVDIDNCMW-UHFFFAOYSA-L 0.000 description 1
- NYDXNILOWQXUOF-UHFFFAOYSA-L disodium;2-[[4-[2-(2-amino-4-oxo-1,7-dihydropyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]amino]pentanedioate Chemical compound [Na+].[Na+].C=1NC=2NC(N)=NC(=O)C=2C=1CCC1=CC=C(C(=O)NC(CCC([O-])=O)C([O-])=O)C=C1 NYDXNILOWQXUOF-UHFFFAOYSA-L 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 239000002612 dispersion medium Substances 0.000 description 1
- CNXMDTWQWLGCPE-UHFFFAOYSA-N ditert-butyl-(2-phenylphenyl)phosphane Chemical compound CC(C)(C)P(C(C)(C)C)C1=CC=CC=C1C1=CC=CC=C1 CNXMDTWQWLGCPE-UHFFFAOYSA-N 0.000 description 1
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 229940112141 dry powder inhaler Drugs 0.000 description 1
- 238000010410 dusting Methods 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 229960002224 eculizumab Drugs 0.000 description 1
- 239000012636 effector Substances 0.000 description 1
- 229940121647 egfr inhibitor Drugs 0.000 description 1
- 229940120655 eloxatin Drugs 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 201000009409 embryonal rhabdomyosarcoma Diseases 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 230000002124 endocrine Effects 0.000 description 1
- 210000000750 endocrine system Anatomy 0.000 description 1
- 230000002357 endometrial effect Effects 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 229940007078 entamoeba histolytica Drugs 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 229940125532 enzyme inhibitor Drugs 0.000 description 1
- XBRDBODLCHKXHI-UHFFFAOYSA-N epolamine Chemical compound OCCN1CCCC1 XBRDBODLCHKXHI-UHFFFAOYSA-N 0.000 description 1
- 229930013356 epothilone Natural products 0.000 description 1
- HESCAJZNRMSMJG-KKQRBIROSA-N epothilone A Chemical class C/C([C@@H]1C[C@@H]2O[C@@H]2CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 HESCAJZNRMSMJG-KKQRBIROSA-N 0.000 description 1
- 229940082789 erbitux Drugs 0.000 description 1
- 201000005619 esophageal carcinoma Diseases 0.000 description 1
- 210000003238 esophagus Anatomy 0.000 description 1
- 201000007281 estrogen-receptor positive breast cancer Diseases 0.000 description 1
- OCLXJTCGWSSVOE-UHFFFAOYSA-N ethanol etoh Chemical compound CCO.CCO OCLXJTCGWSSVOE-UHFFFAOYSA-N 0.000 description 1
- 229960002568 ethinylestradiol Drugs 0.000 description 1
- VCYZVXRKYPKDQB-UHFFFAOYSA-N ethyl 2-fluoroacetate Chemical compound CCOC(=O)CF VCYZVXRKYPKDQB-UHFFFAOYSA-N 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 1
- OJCSPXHYDFONPU-UHFFFAOYSA-N etoac etoac Chemical compound CCOC(C)=O.CCOC(C)=O OJCSPXHYDFONPU-UHFFFAOYSA-N 0.000 description 1
- 229960000752 etoposide phosphate Drugs 0.000 description 1
- LIQODXNTTZAGID-OCBXBXKTSA-N etoposide phosphate Chemical compound COC1=C(OP(O)(O)=O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 LIQODXNTTZAGID-OCBXBXKTSA-N 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 230000005713 exacerbation Effects 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 208000024519 eye neoplasm Diseases 0.000 description 1
- 201000001343 fallopian tube carcinoma Diseases 0.000 description 1
- 229960004207 fentanyl citrate Drugs 0.000 description 1
- IVLVTNPOHDFFCJ-UHFFFAOYSA-N fentanyl citrate Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O.C=1C=CC=CC=1N(C(=O)CC)C(CC1)CCN1CCC1=CC=CC=C1 IVLVTNPOHDFFCJ-UHFFFAOYSA-N 0.000 description 1
- 229940125829 fibroblast growth factor receptor inhibitor Drugs 0.000 description 1
- 229960004177 filgrastim Drugs 0.000 description 1
- 239000010408 film Substances 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 229960005304 fludarabine phosphate Drugs 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 229960001751 fluoxymesterone Drugs 0.000 description 1
- YLRFCQOZQXIBAB-RBZZARIASA-N fluoxymesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1CC[C@](C)(O)[C@@]1(C)C[C@@H]2O YLRFCQOZQXIBAB-RBZZARIASA-N 0.000 description 1
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 1
- 229960002074 flutamide Drugs 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 235000008191 folinic acid Nutrition 0.000 description 1
- 239000011672 folinic acid Substances 0.000 description 1
- WBJINCZRORDGAQ-UHFFFAOYSA-N formic acid ethyl ester Natural products CCOC=O WBJINCZRORDGAQ-UHFFFAOYSA-N 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 244000053095 fungal pathogen Species 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- GAEKPEKOJKCEMS-UHFFFAOYSA-N gamma-valerolactone Chemical compound CC1CCC(=O)O1 GAEKPEKOJKCEMS-UHFFFAOYSA-N 0.000 description 1
- 208000015419 gastrin-producing neuroendocrine tumor Diseases 0.000 description 1
- 201000000052 gastrinoma Diseases 0.000 description 1
- 201000011243 gastrointestinal stromal tumor Diseases 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 229960003297 gemtuzumab ozogamicin Drugs 0.000 description 1
- 102000034356 gene-regulatory proteins Human genes 0.000 description 1
- 108091006104 gene-regulatory proteins Proteins 0.000 description 1
- 201000003115 germ cell cancer Diseases 0.000 description 1
- GYQYAJJFPNQOOW-UHFFFAOYSA-N gilteritinib Chemical compound N1=C(NC2CCOCC2)C(CC)=NC(C(N)=O)=C1NC(C=C1OC)=CC=C1N(CC1)CCC1N1CCN(C)CC1 GYQYAJJFPNQOOW-UHFFFAOYSA-N 0.000 description 1
- 229960002913 goserelin Drugs 0.000 description 1
- 229960003690 goserelin acetate Drugs 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 230000003394 haemopoietic effect Effects 0.000 description 1
- 201000009277 hairy cell leukemia Diseases 0.000 description 1
- 125000004438 haloalkoxy group Chemical group 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 210000003128 head Anatomy 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 201000005787 hematologic cancer Diseases 0.000 description 1
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 description 1
- 208000005252 hepatitis A Diseases 0.000 description 1
- 208000002672 hepatitis B Diseases 0.000 description 1
- 208000006359 hepatoblastoma Diseases 0.000 description 1
- 201000002735 hepatocellular adenoma Diseases 0.000 description 1
- 229940022353 herceptin Drugs 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229960003911 histrelin acetate Drugs 0.000 description 1
- BKEMVGVBBDMHKL-VYFXDUNUSA-N histrelin acetate Chemical compound CC(O)=O.CC(O)=O.CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC(N=C1)=CN1CC1=CC=CC=C1 BKEMVGVBBDMHKL-VYFXDUNUSA-N 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 108091008039 hormone receptors Proteins 0.000 description 1
- MHLPKAGDPWUOOT-UHFFFAOYSA-N housane Chemical compound C1CC2CC21 MHLPKAGDPWUOOT-UHFFFAOYSA-N 0.000 description 1
- 229920002674 hyaluronan Polymers 0.000 description 1
- 229960003160 hyaluronic acid Drugs 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- 229960002899 hydroxyprogesterone Drugs 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 229940121569 ieramilimab Drugs 0.000 description 1
- 125000002962 imidazol-1-yl group Chemical group [*]N1C([H])=NC([H])=C1[H] 0.000 description 1
- 125000001841 imino group Chemical group [H]N=* 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 230000002163 immunogen Effects 0.000 description 1
- 229960003444 immunosuppressant agent Drugs 0.000 description 1
- 239000003018 immunosuppressive agent Substances 0.000 description 1
- 201000004933 in situ carcinoma Diseases 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 206010022498 insulinoma Diseases 0.000 description 1
- 229960003521 interferon alfa-2a Drugs 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 108010093036 interleukin receptors Proteins 0.000 description 1
- 102000002467 interleukin receptors Human genes 0.000 description 1
- 210000002570 interstitial cell Anatomy 0.000 description 1
- 201000002313 intestinal cancer Diseases 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 238000007917 intracranial administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 230000002601 intratumoral effect Effects 0.000 description 1
- 238000007915 intraurethral administration Methods 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 238000007914 intraventricular administration Methods 0.000 description 1
- 201000010985 invasive ductal carcinoma Diseases 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 229960005386 ipilimumab Drugs 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- NIZHERJWXFHGGU-UHFFFAOYSA-N isocyanato(trimethyl)silane Chemical compound C[Si](C)(C)N=C=O NIZHERJWXFHGGU-UHFFFAOYSA-N 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 210000001117 keloid Anatomy 0.000 description 1
- 125000000468 ketone group Chemical group 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 208000022013 kidney Wilms tumor Diseases 0.000 description 1
- 230000002147 killing effect Effects 0.000 description 1
- 150000003951 lactams Chemical class 0.000 description 1
- 150000002596 lactones Chemical class 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229960001320 lapatinib ditosylate Drugs 0.000 description 1
- 206010023841 laryngeal neoplasm Diseases 0.000 description 1
- 229960003784 lenvatinib Drugs 0.000 description 1
- WOSKHXYHFSIKNG-UHFFFAOYSA-N lenvatinib Chemical compound C=12C=C(C(N)=O)C(OC)=CC2=NC=CC=1OC(C=C1Cl)=CC=C1NC(=O)NC1CC1 WOSKHXYHFSIKNG-UHFFFAOYSA-N 0.000 description 1
- YPJRHEKCFKOVRT-UHFFFAOYSA-N lerociclib Chemical compound C1CN(C(C)C)CCN1C(C=N1)=CC=C1NC1=NC=C(C=C2N3C4(CCCCC4)CNC2=O)C3=N1 YPJRHEKCFKOVRT-UHFFFAOYSA-N 0.000 description 1
- 229940121577 lerociclib Drugs 0.000 description 1
- 229950001845 lestaurtinib Drugs 0.000 description 1
- 229960001691 leucovorin Drugs 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- MPVGZUGXCQEXTM-UHFFFAOYSA-N linifanib Chemical compound CC1=CC=C(F)C(NC(=O)NC=2C=CC(=CC=2)C=2C=3C(N)=NNC=3C=CC=2)=C1 MPVGZUGXCQEXTM-UHFFFAOYSA-N 0.000 description 1
- 229950002216 linifanib Drugs 0.000 description 1
- 208000012987 lip and oral cavity carcinoma Diseases 0.000 description 1
- 201000006721 lip cancer Diseases 0.000 description 1
- 206010024627 liposarcoma Diseases 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- 201000007270 liver cancer Diseases 0.000 description 1
- DHMTURDWPRKSOA-RUZDIDTESA-N lonafarnib Chemical compound C1CN(C(=O)N)CCC1CC(=O)N1CCC([C@@H]2C3=C(Br)C=C(Cl)C=C3CCC3=CC(Br)=CN=C32)CC1 DHMTURDWPRKSOA-RUZDIDTESA-N 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 201000005243 lung squamous cell carcinoma Diseases 0.000 description 1
- 201000011649 lymphoblastic lymphoma Diseases 0.000 description 1
- 229940124302 mTOR inhibitor Drugs 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 201000004792 malaria Diseases 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- 201000010893 malignant breast melanoma Diseases 0.000 description 1
- 201000004593 malignant giant cell tumor Diseases 0.000 description 1
- 201000000289 malignant teratoma Diseases 0.000 description 1
- 239000003628 mammalian target of rapamycin inhibitor Substances 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 229960004961 mechlorethamine Drugs 0.000 description 1
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical compound ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- PSGAAPLEWMOORI-PEINSRQWSA-N medroxyprogesterone acetate Chemical compound C([C@@]12C)CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2CC[C@]2(C)[C@@](OC(C)=O)(C(C)=O)CC[C@H]21 PSGAAPLEWMOORI-PEINSRQWSA-N 0.000 description 1
- 229960002985 medroxyprogesterone acetate Drugs 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 210000002418 meninge Anatomy 0.000 description 1
- COTNUBDHGSIOTA-UHFFFAOYSA-N meoh methanol Chemical compound OC.OC COTNUBDHGSIOTA-UHFFFAOYSA-N 0.000 description 1
- 229960000901 mepacrine Drugs 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 208000037819 metastatic cancer Diseases 0.000 description 1
- 208000011575 metastatic malignant neoplasm Diseases 0.000 description 1
- 208000021039 metastatic melanoma Diseases 0.000 description 1
- OKKJLVBELUTLKV-VMNATFBRSA-N methanol-d1 Chemical compound [2H]OC OKKJLVBELUTLKV-VMNATFBRSA-N 0.000 description 1
- 229960004469 methoxsalen Drugs 0.000 description 1
- AYMMPTHLFMQVFH-UHFFFAOYSA-N methyl 6-bromo-5-fluoropyridine-2-carboxylate Chemical compound COC(=O)C1=CC=C(F)C(Br)=N1 AYMMPTHLFMQVFH-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 229960004584 methylprednisolone Drugs 0.000 description 1
- 229960001566 methyltestosterone Drugs 0.000 description 1
- 239000004530 micro-emulsion Substances 0.000 description 1
- 238000012737 microarray-based gene expression Methods 0.000 description 1
- 229950010895 midostaurin Drugs 0.000 description 1
- BMGQWWVMWDBQGC-IIFHNQTCSA-N midostaurin Chemical compound CN([C@H]1[C@H]([C@]2(C)O[C@@H](N3C4=CC=CC=C4C4=C5C(=O)NCC5=C5C6=CC=CC=C6N2C5=C43)C1)OC)C(=O)C1=CC=CC=C1 BMGQWWVMWDBQGC-IIFHNQTCSA-N 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 239000003595 mist Substances 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 208000010492 mucinous cystadenocarcinoma Diseases 0.000 description 1
- 210000004877 mucosa Anatomy 0.000 description 1
- 238000012243 multiplex automated genomic engineering Methods 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 201000000050 myeloid neoplasm Diseases 0.000 description 1
- 208000009091 myxoma Diseases 0.000 description 1
- PEECTLLHENGOKU-UHFFFAOYSA-N n,n-dimethylpyridin-4-amine Chemical compound CN(C)C1=CC=NC=C1.CN(C)C1=CC=NC=C1 PEECTLLHENGOKU-UHFFFAOYSA-N 0.000 description 1
- AZBFJBJXUQUQLF-UHFFFAOYSA-N n-(1,5-dimethylpyrrolidin-3-yl)pyrrolidine-1-carboxamide Chemical compound C1N(C)C(C)CC1NC(=O)N1CCCC1 AZBFJBJXUQUQLF-UHFFFAOYSA-N 0.000 description 1
- BLCLNMBMMGCOAS-UHFFFAOYSA-N n-[1-[[1-[[1-[[1-[[1-[[1-[[1-[2-[(carbamoylamino)carbamoyl]pyrrolidin-1-yl]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-[(2-methylpropan-2-yl)oxy]-1-oxopropan-2-yl]amino]-3-(4-hydroxyphenyl)-1-oxopropan-2-yl]amin Chemical compound C1CCC(C(=O)NNC(N)=O)N1C(=O)C(CCCN=C(N)N)NC(=O)C(CC(C)C)NC(=O)C(COC(C)(C)C)NC(=O)C(NC(=O)C(CO)NC(=O)C(CC=1C2=CC=CC=C2NC=1)NC(=O)C(CC=1NC=NC=1)NC(=O)C1NC(=O)CC1)CC1=CC=C(O)C=C1 BLCLNMBMMGCOAS-UHFFFAOYSA-N 0.000 description 1
- UZWDCWONPYILKI-UHFFFAOYSA-N n-[5-[(4-ethylpiperazin-1-yl)methyl]pyridin-2-yl]-5-fluoro-4-(7-fluoro-2-methyl-3-propan-2-ylbenzimidazol-5-yl)pyrimidin-2-amine Chemical compound C1CN(CC)CCN1CC(C=N1)=CC=C1NC1=NC=C(F)C(C=2C=C3N(C(C)C)C(C)=NC3=C(F)C=2)=N1 UZWDCWONPYILKI-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- WOOWBQQQJXZGIE-UHFFFAOYSA-N n-ethyl-n-propan-2-ylpropan-2-amine Chemical compound CCN(C(C)C)C(C)C.CCN(C(C)C)C(C)C WOOWBQQQJXZGIE-UHFFFAOYSA-N 0.000 description 1
- FWLMVFUGMHIOAA-UHFFFAOYSA-N n-methyl-4-[[4-[[3-[methyl(methylsulfonyl)amino]pyrazin-2-yl]methylamino]-5-(trifluoromethyl)pyrimidin-2-yl]amino]benzamide Chemical compound C1=CC(C(=O)NC)=CC=C1NC1=NC=C(C(F)(F)F)C(NCC=2C(=NC=CN=2)N(C)S(C)(=O)=O)=N1 FWLMVFUGMHIOAA-UHFFFAOYSA-N 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- UBWXUGDQUBIEIZ-QNTYDACNSA-N nandrolone phenpropionate Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@H]4CCC(=O)C=C4CC3)CC[C@@]21C)C(=O)CCC1=CC=CC=C1 UBWXUGDQUBIEIZ-QNTYDACNSA-N 0.000 description 1
- 229960001133 nandrolone phenpropionate Drugs 0.000 description 1
- 229960000513 necitumumab Drugs 0.000 description 1
- 210000003739 neck Anatomy 0.000 description 1
- IXOXBSCIXZEQEQ-UHTZMRCNSA-N nelarabine Chemical compound C1=NC=2C(OC)=NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1O IXOXBSCIXZEQEQ-UHTZMRCNSA-N 0.000 description 1
- 229960000801 nelarabine Drugs 0.000 description 1
- 201000008026 nephroblastoma Diseases 0.000 description 1
- 208000007538 neurilemmoma Diseases 0.000 description 1
- 210000002569 neuron Anatomy 0.000 description 1
- 208000004649 neutrophil actin dysfunction Diseases 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- 229960001346 nilotinib Drugs 0.000 description 1
- HHZIURLSWUIHRB-UHFFFAOYSA-N nilotinib Chemical compound C1=NC(C)=CN1C1=CC(NC(=O)C=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)=CC(C(F)(F)F)=C1 HHZIURLSWUIHRB-UHFFFAOYSA-N 0.000 description 1
- 239000002353 niosome Substances 0.000 description 1
- PCHKPVIQAHNQLW-CQSZACIVSA-N niraparib Chemical compound N1=C2C(C(=O)N)=CC=CC2=CN1C(C=C1)=CC=C1[C@@H]1CCCNC1 PCHKPVIQAHNQLW-CQSZACIVSA-N 0.000 description 1
- 229950011068 niraparib Drugs 0.000 description 1
- PSACHCMMPFMFAJ-UHFFFAOYSA-N nmm n-methylmorpholine Chemical compound CN1CCOCC1.CN1CCOCC1 PSACHCMMPFMFAJ-UHFFFAOYSA-N 0.000 description 1
- UMRZSTCPUPJPOJ-KNVOCYPGSA-N norbornane Chemical compound C1C[C@H]2CC[C@@H]1C2 UMRZSTCPUPJPOJ-KNVOCYPGSA-N 0.000 description 1
- 229960003347 obinutuzumab Drugs 0.000 description 1
- 201000002575 ocular melanoma Diseases 0.000 description 1
- 229960000572 olaparib Drugs 0.000 description 1
- FAQDUNYVKQKNLD-UHFFFAOYSA-N olaparib Chemical compound FC1=CC=C(CC2=C3[CH]C=CC=C3C(=O)N=N2)C=C1C(=O)N(CC1)CCN1C(=O)C1CC1 FAQDUNYVKQKNLD-UHFFFAOYSA-N 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 238000012379 oncolytic virotherapy Methods 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 208000003388 osteoid osteoma Diseases 0.000 description 1
- 208000008798 osteoma Diseases 0.000 description 1
- 229940127084 other anti-cancer agent Drugs 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 210000003101 oviduct Anatomy 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000003551 oxepanyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 125000003544 oxime group Chemical group 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- HWXVIOGONBBTBY-ONEGZZNKSA-N pacritinib Chemical compound C=1C=C(C=2)NC(N=3)=NC=CC=3C(C=3)=CC=CC=3COC\C=C\COCC=2C=1OCCN1CCCC1 HWXVIOGONBBTBY-ONEGZZNKSA-N 0.000 description 1
- 229950011410 pacritinib Drugs 0.000 description 1
- 229960004390 palbociclib Drugs 0.000 description 1
- AHJRHEGDXFFMBM-UHFFFAOYSA-N palbociclib Chemical compound N1=C2N(C3CCCC3)C(=O)C(C(=O)C)=C(C)C2=CN=C1NC(N=C1)=CC=C1N1CCNCC1 AHJRHEGDXFFMBM-UHFFFAOYSA-N 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 229940046231 pamidronate Drugs 0.000 description 1
- WRUUGTRCQOWXEG-UHFFFAOYSA-N pamidronate Chemical compound NCCC(O)(P(O)(O)=O)P(O)(O)=O WRUUGTRCQOWXEG-UHFFFAOYSA-N 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 201000002094 pancreatic adenocarcinoma Diseases 0.000 description 1
- 201000002530 pancreatic endocrine carcinoma Diseases 0.000 description 1
- 208000021255 pancreatic insulinoma Diseases 0.000 description 1
- 244000045947 parasite Species 0.000 description 1
- 210000002990 parathyroid gland Anatomy 0.000 description 1
- 229950007073 parsaclisib Drugs 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 229960000639 pazopanib Drugs 0.000 description 1
- CUIHSIWYWATEQL-UHFFFAOYSA-N pazopanib Chemical compound C1=CC2=C(C)N(C)N=C2C=C1N(C)C(N=1)=CC=NC=1NC1=CC=C(C)C(S(N)(=O)=O)=C1 CUIHSIWYWATEQL-UHFFFAOYSA-N 0.000 description 1
- HQQSBEDKMRHYME-UHFFFAOYSA-N pefloxacin mesylate Chemical compound [H+].CS([O-])(=O)=O.C1=C2N(CC)C=C(C(O)=O)C(=O)C2=CC(F)=C1N1CCN(C)CC1 HQQSBEDKMRHYME-UHFFFAOYSA-N 0.000 description 1
- 229960001744 pegaspargase Drugs 0.000 description 1
- 108010001564 pegaspargase Proteins 0.000 description 1
- 229960001373 pegfilgrastim Drugs 0.000 description 1
- 108010044644 pegfilgrastim Proteins 0.000 description 1
- 229960003349 pemetrexed disodium Drugs 0.000 description 1
- 229940121317 pemigatinib Drugs 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 238000010647 peptide synthesis reaction Methods 0.000 description 1
- 239000002304 perfume Substances 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 229940066842 petrolatum Drugs 0.000 description 1
- JGWRKYUXBBNENE-UHFFFAOYSA-N pexidartinib Chemical compound C1=NC(C(F)(F)F)=CC=C1CNC(N=C1)=CC=C1CC1=CNC2=NC=C(Cl)C=C12 JGWRKYUXBBNENE-UHFFFAOYSA-N 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 125000001792 phenanthrenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3C=CC12)* 0.000 description 1
- 239000002953 phosphate buffered saline Substances 0.000 description 1
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 229950010773 pidilizumab Drugs 0.000 description 1
- 208000024724 pineal body neoplasm Diseases 0.000 description 1
- 201000004123 pineal gland cancer Diseases 0.000 description 1
- 208000021310 pituitary gland adenoma Diseases 0.000 description 1
- 239000004584 polyacrylic acid Substances 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 231100000683 possible toxicity Toxicity 0.000 description 1
- 230000001323 posttranslational effect Effects 0.000 description 1
- IUBQJLUDMLPAGT-UHFFFAOYSA-N potassium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([K])[Si](C)(C)C IUBQJLUDMLPAGT-UHFFFAOYSA-N 0.000 description 1
- WFIZEGIEIOHZCP-UHFFFAOYSA-M potassium formate Chemical compound [K+].[O-]C=O WFIZEGIEIOHZCP-UHFFFAOYSA-M 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 229940126457 povorcitinib Drugs 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 229960005205 prednisolone Drugs 0.000 description 1
- OIGNJSKKLXVSLS-VWUMJDOOSA-N prednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OIGNJSKKLXVSLS-VWUMJDOOSA-N 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 208000016800 primary central nervous system lymphoma Diseases 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 201000005825 prostate adenocarcinoma Diseases 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 239000000296 purinergic P1 receptor antagonist Substances 0.000 description 1
- 125000002206 pyridazin-3-yl group Chemical group [H]C1=C([H])C([H])=C(*)N=N1 0.000 description 1
- QLULGIRFKAWHOJ-UHFFFAOYSA-N pyridin-4-ylboronic acid Chemical compound OB(O)C1=CC=NC=C1 QLULGIRFKAWHOJ-UHFFFAOYSA-N 0.000 description 1
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 description 1
- 125000004527 pyrimidin-4-yl group Chemical group N1=CN=C(C=C1)* 0.000 description 1
- 125000004528 pyrimidin-5-yl group Chemical group N1=CN=CC(=C1)* 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- GPKJTRJOBQGKQK-UHFFFAOYSA-N quinacrine Chemical compound C1=C(OC)C=C2C(NC(C)CCCN(CC)CC)=C(C=CC(Cl)=C3)C3=NC2=C1 GPKJTRJOBQGKQK-UHFFFAOYSA-N 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 229950001626 quizartinib Drugs 0.000 description 1
- CVWXJKQAOSCOAB-UHFFFAOYSA-N quizartinib Chemical compound O1C(C(C)(C)C)=CC(NC(=O)NC=2C=CC(=CC=2)C=2N=C3N(C4=CC=C(OCCN5CCOCC5)C=C4S3)C=2)=N1 CVWXJKQAOSCOAB-UHFFFAOYSA-N 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 229960002633 ramucirumab Drugs 0.000 description 1
- 229960003876 ranibizumab Drugs 0.000 description 1
- 229960000424 rasburicase Drugs 0.000 description 1
- 108010084837 rasburicase Proteins 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 229940044601 receptor agonist Drugs 0.000 description 1
- 239000000018 receptor agonist Substances 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 229960004836 regorafenib Drugs 0.000 description 1
- FNHKPVJBJVTLMP-UHFFFAOYSA-N regorafenib Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=C(F)C(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 FNHKPVJBJVTLMP-UHFFFAOYSA-N 0.000 description 1
- 201000007444 renal pelvis carcinoma Diseases 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 230000004043 responsiveness Effects 0.000 description 1
- 208000029922 reticulum cell sarcoma Diseases 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 229940120975 revlimid Drugs 0.000 description 1
- 229950003687 ribociclib Drugs 0.000 description 1
- 229910052895 riebeckite Inorganic materials 0.000 description 1
- 238000007142 ring opening reaction Methods 0.000 description 1
- HMABYWSNWIZPAG-UHFFFAOYSA-N rucaparib Chemical compound C1=CC(CNC)=CC=C1C(N1)=C2CCNC(=O)C3=C2C1=CC(F)=C3 HMABYWSNWIZPAG-UHFFFAOYSA-N 0.000 description 1
- 229950004707 rucaparib Drugs 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 description 1
- 206010039667 schwannoma Diseases 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 239000008299 semisolid dosage form Substances 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 208000004548 serous cystadenocarcinoma Diseases 0.000 description 1
- 208000037968 sinus cancer Diseases 0.000 description 1
- 201000000849 skin cancer Diseases 0.000 description 1
- 210000003625 skull Anatomy 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- 235000009518 sodium iodide Nutrition 0.000 description 1
- RMBAVIFYHOYIFM-UHFFFAOYSA-M sodium methanethiolate Chemical compound [Na+].[S-]C RMBAVIFYHOYIFM-UHFFFAOYSA-M 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 201000011096 spinal cancer Diseases 0.000 description 1
- 210000000278 spinal cord Anatomy 0.000 description 1
- 208000014618 spinal cord cancer Diseases 0.000 description 1
- 125000003003 spiro group Chemical group 0.000 description 1
- OGNAOIGAPPSUMG-UHFFFAOYSA-N spiro[2.2]pentane Chemical compound C1CC11CC1 OGNAOIGAPPSUMG-UHFFFAOYSA-N 0.000 description 1
- FOEYMRPOKBCNCR-UHFFFAOYSA-N spiro[2.5]octane Chemical compound C1CC11CCCCC1 FOEYMRPOKBCNCR-UHFFFAOYSA-N 0.000 description 1
- DVDJICIUXXAIKJ-UHFFFAOYSA-N spiro[2.6]nonane Chemical compound C1CC11CCCCCC1 DVDJICIUXXAIKJ-UHFFFAOYSA-N 0.000 description 1
- PLDXRPSSERMPSV-UHFFFAOYSA-N spiro[3.6]decane Chemical compound C1CCC21CCCCCC2 PLDXRPSSERMPSV-UHFFFAOYSA-N 0.000 description 1
- PHICBFWUYUCFKS-UHFFFAOYSA-N spiro[4.4]nonane Chemical compound C1CCCC21CCCC2 PHICBFWUYUCFKS-UHFFFAOYSA-N 0.000 description 1
- NECLQTPQJZSWOE-UHFFFAOYSA-N spiro[5.5]undecane Chemical compound C1CCCCC21CCCCC2 NECLQTPQJZSWOE-UHFFFAOYSA-N 0.000 description 1
- CTDQAGUNKPRERK-UHFFFAOYSA-N spirodecane Chemical compound C1CCCC21CCCCC2 CTDQAGUNKPRERK-UHFFFAOYSA-N 0.000 description 1
- 208000017572 squamous cell neoplasm Diseases 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 235000011044 succinic acid Nutrition 0.000 description 1
- 150000003444 succinic acids Chemical class 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000005846 sugar alcohols Polymers 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-N sulfuric acid group Chemical class S(O)(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 1
- 239000002511 suppository base Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 230000009747 swallowing Effects 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 229940121503 tafasitamab Drugs 0.000 description 1
- UXXQOJXBIDBUAC-UHFFFAOYSA-N tandutinib Chemical compound COC1=CC2=C(N3CCN(CC3)C(=O)NC=3C=CC(OC(C)C)=CC=3)N=CN=C2C=C1OCCCN1CCCCC1 UXXQOJXBIDBUAC-UHFFFAOYSA-N 0.000 description 1
- 229950009893 tandutinib Drugs 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 229940126625 tavolimab Drugs 0.000 description 1
- 229940063683 taxotere Drugs 0.000 description 1
- 208000001608 teratocarcinoma Diseases 0.000 description 1
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 1
- GKGFAEREWWZBKY-UHFFFAOYSA-N tert-butyl n-(4-bromobutyl)carbamate Chemical compound CC(C)(C)OC(=O)NCCCCBr GKGFAEREWWZBKY-UHFFFAOYSA-N 0.000 description 1
- OWLBQQTUOQLZST-UHFFFAOYSA-N tert-butyl n-[4-(hydroxymethyl)-1,3-thiazol-2-yl]carbamate Chemical compound CC(C)(C)OC(=O)NC1=NC(CO)=CS1 OWLBQQTUOQLZST-UHFFFAOYSA-N 0.000 description 1
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 1
- MUQNAPSBHXFMHT-UHFFFAOYSA-N tert-butylhydrazine Chemical compound CC(C)(C)NN MUQNAPSBHXFMHT-UHFFFAOYSA-N 0.000 description 1
- 210000001550 testis Anatomy 0.000 description 1
- 229960003604 testosterone Drugs 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 229950006410 tezacitabine Drugs 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- 125000001583 thiepanyl group Chemical group 0.000 description 1
- 125000002053 thietanyl group Chemical group 0.000 description 1
- 201000002510 thyroid cancer Diseases 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- PLHJCIYEEKOWNM-HHHXNRCGSA-N tipifarnib Chemical compound CN1C=NC=C1[C@](N)(C=1C=C2C(C=3C=C(Cl)C=CC=3)=CC(=O)N(C)C2=CC=1)C1=CC=C(Cl)C=C1 PLHJCIYEEKOWNM-HHHXNRCGSA-N 0.000 description 1
- 201000006134 tongue cancer Diseases 0.000 description 1
- 239000012049 topical pharmaceutical composition Substances 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 231100000583 toxicological profile Toxicity 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 231100000765 toxin Toxicity 0.000 description 1
- 108700012359 toxins Proteins 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 229950007217 tremelimumab Drugs 0.000 description 1
- IUCJMVBFZDHPDX-UHFFFAOYSA-N tretamine Chemical compound C1CN1C1=NC(N2CC2)=NC(N2CC2)=N1 IUCJMVBFZDHPDX-UHFFFAOYSA-N 0.000 description 1
- 229950001353 tretamine Drugs 0.000 description 1
- 229960001727 tretinoin Drugs 0.000 description 1
- 229960005294 triamcinolone Drugs 0.000 description 1
- GFNANZIMVAIWHM-OBYCQNJPSA-N triamcinolone Chemical compound O=C1C=C[C@]2(C)[C@@]3(F)[C@@H](O)C[C@](C)([C@@]([C@H](O)C4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 GFNANZIMVAIWHM-OBYCQNJPSA-N 0.000 description 1
- 125000003866 trichloromethyl group Chemical group ClC(Cl)(Cl)* 0.000 description 1
- 229940086542 triethylamine Drugs 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- 229950007127 trilaciclib Drugs 0.000 description 1
- XZZNDPSIHUTMOC-UHFFFAOYSA-N triphenyl phosphate Chemical compound C=1C=CC=CC=1OP(OC=1C=CC=CC=1)(=O)OC1=CC=CC=C1 XZZNDPSIHUTMOC-UHFFFAOYSA-N 0.000 description 1
- 229940086984 trisenox Drugs 0.000 description 1
- 201000007423 tubular adenocarcinoma Diseases 0.000 description 1
- 208000022271 tubular adenoma Diseases 0.000 description 1
- 230000005747 tumor angiogenesis Effects 0.000 description 1
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 241001529453 unidentified herpesvirus Species 0.000 description 1
- 229950005972 urelumab Drugs 0.000 description 1
- 208000023747 urothelial carcinoma Diseases 0.000 description 1
- 210000004291 uterus Anatomy 0.000 description 1
- 229950003520 utomilumab Drugs 0.000 description 1
- 238000002255 vaccination Methods 0.000 description 1
- 210000001215 vagina Anatomy 0.000 description 1
- 229960000653 valrubicin Drugs 0.000 description 1
- ZOCKGBMQLCSHFP-KQRAQHLDSA-N valrubicin Chemical compound O([C@H]1C[C@](CC2=C(O)C=3C(=O)C4=CC=CC(OC)=C4C(=O)C=3C(O)=C21)(O)C(=O)COC(=O)CCCC)[C@H]1C[C@H](NC(=O)C(F)(F)F)[C@H](O)[C@H](C)O1 ZOCKGBMQLCSHFP-KQRAQHLDSA-N 0.000 description 1
- JNAHVYVRKWKWKQ-CYBMUJFWSA-N veliparib Chemical compound N=1C2=CC=CC(C(N)=O)=C2NC=1[C@@]1(C)CCCN1 JNAHVYVRKWKWKQ-CYBMUJFWSA-N 0.000 description 1
- 229950011257 veliparib Drugs 0.000 description 1
- 229960003862 vemurafenib Drugs 0.000 description 1
- GPXBXXGIAQBQNI-UHFFFAOYSA-N vemurafenib Chemical compound CCCS(=O)(=O)NC1=CC=C(F)C(C(=O)C=2C3=CC(=CN=C3NC=2)C=2C=CC(Cl)=CC=2)=C1F GPXBXXGIAQBQNI-UHFFFAOYSA-N 0.000 description 1
- 208000009540 villous adenoma Diseases 0.000 description 1
- 229960004355 vindesine Drugs 0.000 description 1
- UGGWPQSBPIFKDZ-KOTLKJBCSA-N vindesine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(N)=O)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1N=C1[C]2C=CC=C1 UGGWPQSBPIFKDZ-KOTLKJBCSA-N 0.000 description 1
- 210000001835 viscera Anatomy 0.000 description 1
- WAEXFXRVDQXREF-UHFFFAOYSA-N vorinostat Chemical compound ONC(=O)CCCCCCC(=O)NC1=CC=CC=C1 WAEXFXRVDQXREF-UHFFFAOYSA-N 0.000 description 1
- 229960000237 vorinostat Drugs 0.000 description 1
- 210000003905 vulva Anatomy 0.000 description 1
- 208000013013 vulvar carcinoma Diseases 0.000 description 1
- 239000003871 white petrolatum Substances 0.000 description 1
- 229940053867 xeloda Drugs 0.000 description 1
- UGOMMVLRQDMAQQ-UHFFFAOYSA-N xphos Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 UGOMMVLRQDMAQQ-UHFFFAOYSA-N 0.000 description 1
- 229960002760 ziv-aflibercept Drugs 0.000 description 1
- XRASPMIURGNCCH-UHFFFAOYSA-N zoledronic acid Chemical compound OP(=O)(O)C(P(O)(O)=O)(O)CN1C=CN=C1 XRASPMIURGNCCH-UHFFFAOYSA-N 0.000 description 1
- 229960004276 zoledronic acid Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/12—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains three hetero rings
- C07D491/18—Bridged systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Definitions
- CDKs Cyclin-Dependent Kinases
- CDKs are relatively small proteins with molecular weights between about 34-40 kDa. They contain little more than the kinase domain, and are essentially inactive when not in complex with a class of regulatory proteins called cyclins. CDK levels remain relatively constant throughout the cell cycle, and most regulation is post-translational, most prominently by binding to cyclins.
- CDK2 is of particular interest because deregulation of CDK2 activity occurs frequently in a variety of human cancers.
- CDK2 plays a crucial role in promoting G1/S transition and S phase progression.
- CCNE cyclin E
- CDK2 phosphorylates retinoblastoma pocket protein family members (p107, p130, pRb) , leading to de-repression of E2F transcription factors, expression of G1/S transition related genes and transition from G1 to S phase (Henley, S.A. and F.A. Dick, Cell Div, 2012, 7 (1) : 10) .
- CDK2/cyclin A which phosphorylates endogenous substrates that permit DNA synthesis, replication and centrosome duplication. It has been reported that the CDK2 pathway influences tumorigenesis mainly through amplification and/or overexpression of CCNE1 and mutations that inactivate CDK2 endogenous inhibitors (e.g., p27) , respectively (Xu, X., et al., Biochemistry, 1999, 38 (27) : 8713-22) .
- CCNE1 copy-number gain and overexpression have been identified in ovarian, gastric, endometrial, breast and other cancers and been associated with poor outcomes in these tumors (Keyomarsi, K., et al., N Engl J Med, 2002, 347 (20) : 1566-75; Nakayama, N., et al., Cancer, 2010, 116 (11) : 2621-34; Au-Yeung, G., et al., Clin Cancer Res, 2017, 23 (7) : 1862-1874; Rosen, D.G., et al., Cancer, 2006, 106 (9) : 1925-32) .
- Amplification and/or overexpression of CCNE1 also reportedly contribute to trastuzumab resistance in HER2+ breast cancer and resistance to CDK4/6 inhibitors in estrogen receptor-positive breast cancer (Scaltriti, M., et al., Proc Natl Acad Sci USA, 2011, 108 (9) : 3761-6; Herrera-Abreu, M. T., et al., Cancer Res, 2016, 76 (8) : 2301-13) .
- CDK2 Various approaches targeting CDK2 have been shown to induce cell cycle arrest and tumor growth inhibition (Chen, Y N., et al., Proc Natl Acad Sci USA, 1999, 96 (8) : 4325-9; Mendoza, N., et al., Cancer Res, 2003, 63 (5) : 1020-4) . Inhibition of CDK2 also reportedly restores sensitivity to trastuzumab treatment in resistant HER2+ breast tumors in a preclinical model (Scaltriti, supra) .
- CDK2 CDK1
- CDK1 CDK1
- CDK inhibitors having novel activity profiles, in particular those specifically or selectively targeting CDK2.
- Described herein are compounds of Formula (I') or (I) that inhibit (e.g., selectively inhibit) the activity of CDK2, and pharmaceutically acceptable salts, or stereoisomers thereof.
- the present disclosure provides a compound of Formula (I') , a pharmaceutically acceptable salt, or a stereoisomer thereof:
- the present disclosure provides a compound of Formula (I) , a pharmaceutically acceptable salt, or a stereoisomer thereof:
- compositions comprising a compound of Formula (I') or (I) , a pharmaceutically acceptable salt, or a stereoisomer thereof and a pharmaceutically acceptable carrier or excipient.
- the present disclsoure further provides methods of inhibiting CDK2 in a patient, comprising administering to the patient a compound of Formula (I') or (I) , or a pharmaceutically acceptable salt, or a stereoisomer thereof.
- the present disclsoure also provides methods of treating a disease or or condition modulated at least in part by CDK2 in a subject, comprising administering to the subject a therapeutically effective amount of a compound of Formula (I') or (I) , a pharmaceutically acceptable salt, or a stereoisomer thereof.
- the present disclosure further provides a method of treating cancer in a patient in need thereof, comprising administering to the patient an effective amount of (1) a compound of Formula (I') or (I) , a pharmaceutically acceptable salt, or a stereoisomer thereof; or (2) a pharmaceutically acceptable composition comprising a compound of Formula (I') or (I) , a pharmaceutically acceptable salt, or a stereoisomer thereof, and a pharmaceutically acceptable carrier.
- the cancer is treatable by inhibiting (e.g., selectively inhibiting) CDK2, such as a cancer selected from the group consisting of: ovarian cancer, breast cancer (such as hormone receptor positive, HER2/neu negative advanced or metastatic breast cancer, HER2 positive breast cancer and triple negative breast cancer ) , lung cancer, endometrial cancer, neuroblastoma, gastric cancer, colorectal cancer, prostate cancer, glioblastoma, melanoma, mantel cell lymphoma, chronic myeloid leukemia and acute myeloid leukemia.
- CDK2 such as a cancer selected from the group consisting of: ovarian cancer, breast cancer (such as hormone receptor positive, HER2/neu negative advanced or metastatic breast cancer, HER2 positive breast cancer and triple negative breast cancer )
- lung cancer endometrial cancer
- neuroblastoma gastric cancer
- colorectal cancer colorectal cancer
- prostate cancer glioblastoma, melanoma
- the cancer exhibits abnormally up-regulated CCNE1 /Cyclin E activity, through overexpression of Cyclin Eor duplication of the Cyclin E-coding CCNE1 gene. In certain embodiments, the cancer exhibits abnormally up-regulated Cyclin A2 activity.
- the cancer can be treated by inhibiting (e.g., selectively inhibiting) the activity of CDK2.
- the compounds of the invention are administered with any one of a second therapeutic agent as described herein that also treats the same cancer.
- the present disclosure also provides a use of a compound of Formula (I') or (I) , a pharmaceutically acceptable salt, or a stereoisomer thereof or a pharmaceutical composition comprising the same in any of the methods described herein.
- a compound of Formula (I') or (I) or a pharmaceutically acceptable salt or a stereoisomer thereof or a pharmaceutical composition comprising the same for use in any of the methods described herein.
- a compound of Formula (I') or (I) or a pharmaceutically acceptable salt or a stereoisomer thereof or a pharmaceutical composition comprising the same for the manufacture of a medicament for any of the methods described herein.
- the present disclosure provides a compound represented by Formula (I') :
- ring A is 6-10 membered aryl or 5-10 memberaed heteroaryl; wherein said 6-10 membered aryl or 5-10 membered heteroaryl represented by ring A is optionally substituted by one or more R 1 ;
- R 1 is halogen, -CN, C 1-6 alkyl, C 1-6 haloalkyl, C 2-6 alkenyl, C 2-6 alknyl, C 1-6 alkyleneamine, C 1-6 alkylenehydroxyl, -C (O) R 1a , -C (O) OR 1a , -C (O) NR 1a R 1b , -OR 1a , -SR 1a , -NR 1a R 1b , -NR 1a C (O) R 1b , -NR 1a C (O) OR 1b , -NR 1a SO 2 R 1b , -NR 1a SO 2 NR 1b R 1c , -SO 2 R 1a , -SO 2 NR 1a R 1b , or -P (O) R 1a R 1b , 3-6 membered carbocyclyl, 4-8 membered heterocyclyl, 6-10 membered aryl, 5-10 membered heteroaryl
- R 10 in each occurrence, is independently selected from the group consisting of halogen, -CN, C 1-6 alkyl, C 1-6 haloalkyl, C 1-6 alkoxyl, and 4-6 membered heterocyclyl;
- R 1a , R 1b , and R 1c are independently selected from the group consisting of hydrogen, C 1-6 alkyl, C 1-6 haloalkyl, C 2-6 alkenyl, C 2-6 alkynyl, 3-6 membered carbocyclyl, and -C 0-6 alkyl-4-6 membered heterocyclyl;
- R 2 in each occurrence, is independently hydrogen, C 1-6 alkyl, or C 1-6 haloalkyl
- W is absent, C 1-6 alkylene, C 2-6 alkenylene, or C 2-6 alkynylene; wherein said C 1- 6 alkylene, C 2-6 alkenylene, or C 2-6 alkynylene represented by W is optionally substituted by one or more R 3 ;
- L is absent, -O-, -NH-, 3-12 membered carbocyclyl, 3-12 membered heterocyclyl, 6-10 membered aryl, or 5-10 membered heteroaryl; wherein said -NH-, 3-12 membered carbocyclyl, 3-12 membered heterocyclyl, 6-10 membered aryl, or 5-10 membered heteroaryl represented by L is optionally substituted by one or more R 3 ; and
- Z is absent, C 1-6 alkylene, C 2-6 alkenylene, or C 2-6 alkynylene; wherein said C 1- 6 alkylene, C 2-6 alkenylene, or C 2-6 alkynylene represented by Z is optionally substituted by one or more R 3 ; wherein
- R 3a and R 3b are independently selected from the group consisting of hydrogen, C 1-6 alkyl, C 1-6 haloalkyl, C 2-6 alkenyl, C 2-6 alkynyl, 3-6 membered carbocyclyl, and 4-6 membered heterocyclyl;
- R 22 is hydrogen, C 1-6 alkyl, or C 1-6 haloalkyl
- R 4 is hydrogen or C 1-6 alkyl
- each R 5 is independently hydrogen or C 1-6 alkyl
- V O or S.
- the present disclosure provides a compound according to the first embodiment, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein the compound is represented by Formula II':
- the present disclosure provides a compound according to the first or second embodiment, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein the compound is represented by Formula (II'A ) or (II'B) :
- the present disclosure provides a compound according to any one of the first through third embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
- ring A is phenyl or nitrogen containing 5-10 membered heteroaryl; wherein said phenyl or nitrogen containing 5-10 membered heteroaryl represented by ring A is optionally substituted by one to three R 1 ; wherein
- R 1 is halogen, -CN, C 1-4 alkyl, C 1-4 haloalkyl, C 1-4 alkyleneamine, C 1-4 alkylenehydroxyl, -C (O) NR 1a R 1b , -OR 1a , -SR 1a , -SO 2 R 1a , 4-6 membered carbocyclyl, 4-6 membered heterocyclyl, phenyl, or 5-6 membered heteroaryl; wherein said C 1-4 alkyleneamine, C 1-4 alkylenehydroxyl, 4-6 membered carbocyclyl, 4-6 membered heterocyclyl, phenyl, or 5-6 membered heteroaryl represented by R 1 is optionally substituted by one to three R 10 ; wherein
- R 10 in each occurrence, is independently selected from the group consisting of halogen, -CN, C 1-4 alkyl, C 1-4 haloalkyl, C 1-4 alkoxyl, and 5-6 membered heterocyclyl; and
- R 1a and R 1b are independently selected from the group consisting of hydrogen, C 1-4 alkyl, C 1-4 haloalkyl, C 2-4 alkenyl, C 2-4 alkynyl, and -C 0-3 alkyl-4-6 membered heterocyclyl.
- the present disclosure provides a compound of any one of the first through fourth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
- ring A is phenyl, nitrogen containing 5-6 membered heteroaryl, or nitrogen containing 9 membered bicyclic heteroaryl; wherein said phenyl, nitrogen containing 5-6 membered heteroaryl, or nitrogen containing 9 membered bicyclic heteroaryl represented by ring A is optionally substituted by one to two R 1 ; wherein
- R 1 is halogen, -CN, C 1-3 alkyl, C 1-2 haloalkyl, C 1-2 alkyleneamine, C 1-2 alkylenehydroxyl, -C (O) NR 1a R 1b , -OR 1a , -SR 1a , -SO 2 R 1a , 5-6 membered carbocyclyl, 5-6 membered heterocyclyl, phenyl, or 5-6 membered heteroaryl; wherein said C 1-2 alkyleneamine, C 1-2 alkylenehydroxyl, 5-6 membered carbocyclyl, 5-6 membered heterocyclyl, phenyl, or 5-6 membered heteroaryl represented by R 1 is optionally substituted by one to three R 10 ; wherein
- R 10 in each occurrence, is independently selected from the group consisting of F, Cl, -CN, C 1-2 alkyl, C 1-2 haloalkyl, C 1-3 alkoxyl, and 5-6 membered heterocyclyl; and
- R 1a and R 1b are independently selected from the group consisting of hydrogen, C 1-2 alkyl, C 1-2 haloalkyl, and -C 0-2 alkyl-5 membered heterocyclyl.
- the present disclosure provides a compound of any one of first through fifth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein ring A is selected from a group consisting of phenyl, pyridyl, pyrimidinyl, pyrazinyl, thiophenyl, thiazolyl, pyrrolopyridinyl, pyrazolopyrimidinyl, pyrazolopyrazinyl, thienopyridinyl, and imidazo [4, 5-c] pyridinyl; wherein said phenyl, pyridyl, pyrimidinyl, pyrazinyl, thiophenyl, thiazolyl, pyrrolopyridinyl, pyrazolopyrimidinyl, pyrazolopyrazinyl, thienopyridinyl, and imidazo [4, 5-c] pyridinyl is optionally substituted by one to
- R 1 is F, Cl, Br, -CN, -CH 3 , -CH (CH 3 ) 2 , -CF 3 , -OR 1a , -SR 1a , -SO 2 R 1a , -C (O) NR 1a R 1b , -CH 2 NH 2 , -CH 2 OH, cyclopentanyl, cyclohexanyl, cyclohexenyl, piperidinyl, tetrahydropyridinyl, 3, 6-dihydro-2H-thiopyranyl, thiophenyl, pyrazolyl, phenyl, or pyridyl; wherein said -CH 2 NH 2 , -CH 2 OH, cyclopentanyl, cyclohexanyl, cyclohexenyl, piperidinyl, tetrahydropyridinyl, 3, 6-dihydro-2H-thiopyranyl, thiopheny
- R 10 in each occurrence, is independently selected from the group consisting of F, Cl, -CN, -CH 3 , -CF 3 , -CH 2 CF 3 , -OCH 2 CH 3 , -OCH (CH 3 ) 2 , and morpholinyl; and
- R 1a and R 1b are independently selected from the group consisting of hydrogen, -CH 3 , -OCHF 2 , tetrahydrofuranyl, and - (CH 2 ) 2 -pyrrolidinyl.
- the present disclosure provides a compound of any one of the first through sixth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein ring A is selected from the group consisting of
- each of them is optionally substituted by one to two R 1 ;
- the present disclosure provides a compound of any one of the first through the seventh embodiments, a pharmaceutically acceptable salt thereof, or a stereoisomer thereof, wherein ring A is pyridyl; wherein said pyridyl is optionally substituted by one R 1 .
- the definitions of the remaining variables are provided in the first through the seventh embodiments.
- the present disclosure provides a compound of any one of the first through the eighth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
- R 2 in each occurrence, is independently hydrogen, C 1-3 alkyl, or C 1-3 haloalkyl.
- the present disclosure provides a compound of any one of the first through the ninth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
- R 2 in each occurrence, is independently hydrogen, -CH 3 , -CF 3 , or isopropyl.
- the present disclosure provides a compound of any one of the first through the tenth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein X is absent or ⁇ -CH 2 -O- ⁇ ; wherein
- the present disclosure provides a compound of any one of the first through the eleventh embodiments, or a pharmaceutically acceptable salt or a stereoisomer thereof, wherein
- W is absent, C 1-4 alkylene, C 2-4 alkenylene, or C 2-4 alkynylene; wherein said C 1-4 alkylene, C 2-4 alkenylene, or C 2-4 alkynylene represented by W is optionally substituted by one to three R 3 ;
- L is absent, -O-, -NH-, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 6-10 membered aryl, or 5-10 membered heteroaryl; wherein said -NH-, 3-8 membered carbocyclyl, 3-6 membered heterocyclyl, 6-10 membered aryl, and 5-10 membered heteroaryl represented by L is optionally substituted by one to three R 3 ; and
- Z is absent, C 1-4 alkylene, C 2-4 alkenylene, or C 2-4 alkynylene; wherein said C 1-4 alkylene, C 2-4 alkenylene, or C 2-4 alkynylene represented by Z is optionally substituted by one to three R 3 ; wherein
- R 3a is hydrogen or C 1-4 alkyl.
- the present disclosure provides a compound of any one of the first through the twelfth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
- W is absent, C 1-3 alkylene, C 2-3 alkenylene, or C 2-3 alkynylene; wherein said C 1-3 alkylene, C 2-3 alkenylene, or C 2-3 alkynylene represented by W is optionally substituted by one to two R 3 ;
- L is absent, -O-, -NH-, 3-6 membered monocyclic carbocyclyl, 6-8 membered bicyclic carbocyclyl; 4-6 membered monocyclic heterocyclyl, phenyl, and 5-6 membered monocyclic heteroaryl; wherein said 3-6 membered monocyclic carbocyclyl, 6-8 membered bicyclic carbocyclyl; 4-6 membered monocyclic heterocyclyl, phenyl, and 5-6 membered monocyclic heteroaryl represented by L is optionally substituted by one R 3 ; and
- Z is absent, C 1-3 alkylene, C 2-3 alkenylene, or C 2-3 alkynylene; wherein said C 1-3 alkylene, C 2-3 alkenylene, or C 2-3 alkynylene represented by Z is optionally substituted by one to two R 3 ; wherein
- the present disclosure provides a compound of any one of the first through the thirteenth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
- W is C 1-3 alkylene, C 1-3 haloalkylene, C 2-3 alkenylene, or C 2-3 alkynylene;
- L is absent, -NH-or -O-;
- Z is C 1-3 alkylene, C 1-3 haloalkylene, C 2-3 alkenylene, or C 2-3 alkynylene.
- the present disclosure provides a compound of any one of the first through the fourteenth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
- W is C 3-4 alkylene, C 3-4 haloalkylene, C 3-4 alkenylene, or C 3-4 alkynylene;
- the present disclosure provides a compound of any one of the first through the fifteenth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
- W is absent, C 1-2 alkylene, or C 1-2 haloalkylene
- L is 4-6 membered carbocyclyl, 4-6 membered heterocyclyl, or 5-6 membered heteroaryl;
- Z is absent or methylene.
- the present disclosure provides a compound of any one of the first through the sixteenth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
- L is absent, -O-, -NH-, cyclobutyl, cyclohexyl, spiro [3.3] heptanyl, azetidinyl, piperazinyl, piperidinyl, oxazolyl, isoxazolyl, imidazolyl, triazolyl, or thiozolyl; wherein said -NH-, cyclobutyl, cyclohexyl, spiro [3.3] heptanyl, azetidinyl, piperazinyl, piperidinyl, oxazolyl, isoxazolyl, imidazolyl, triazolyl, or thiozolyl is optionally substituted by one R 3 ;
- the present disclosure provides a compound of any one of the first through the thirteenth and seventeenth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein *-W-L-Z-**is selected from the group consisting of
- the present disclosure provides a compound of any one of the first through the thirteenth, seventeenth, and eighteenth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein *-W-L-Z-**is selected from the group consisting of wherein
- the present disclosure provides a compound of any one of the first through the nineteenth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein is represented by Formula A, B, C, or D,
- R 22 is hydrogen, C 1-4 alkyl, or C 1-4 haloalkyl
- R 4 is hydrogen or C 1-4 alkyl; or R 4 and one R 3 attached to Z, together with the atoms to which they are attached, form 4-6 membered heterocyclyl.
- the present disclosure provides a compound of the twentieth embodiment, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein is
- R 4 is hydrogen or -CH 3 ; or R 4 and one R 3 attached to Z together with the atoms to which they are attached, form azetidinyl or pyrrolidinyl.
- the present disclosure provides a compound of the twenty-first embodiment, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein R 4 is hydrogen.
- R 4 is hydrogen.
- the present disclosure provides a compound of any one of the first through the nineteenth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein is represented by Formula E:
- R 5 is hydrogen or isopropyl.
- the present disclosure provides a compound selected from the compounds disclosed in examples and Table 1, a pharmaceutically acceptable salt or a stereoisomer thereof.
- ring A is 6-10 membered aryl or 5-10 membered heteroaryl; wherein said 6-10 membered aryl or 5-10 membered heteroaryl represented by ring A is optionally substituted by one or more R 1 ; wherein
- R 1 is halogen, -CN, C 1-6 alkyl, C 1-6 haloalkyl, C 2-6 alkenyl, C 2-6 alknyl, -C (O) R 1a , -C (O) OR 1a , -C (O) NR 1a R 1b , -OR 1a , -NR 1a R 1b , -NR 1a C (O) R 1b , -NR 1a C (O) OR 1b , -NR 1a SO 2 R 1b , -NR 1a SO 2 NR 1b R 1c , -SO 2 R 1a , -SO 2 NR 1a R 1b , or -P (O) R 1a R 1b ; wherein
- R 1a , R 1b , and R 1c are independently selected from the group consisting of hydrogen, C 1-6 alkyl, C 1-6 haloalkyl, C 2-6 alkenyl, C 2-6 alkynyl, 3-6 membered carbocyclyl, and 4-6 membered heterocyclyl;
- X is absent, -O-, -NR 2 -, -C (O) -, -NR 2 C (O) -, or -C (O) NR 2 -;
- R 2 in each occurrence, is independently hydrogen, C 1-6 alkyl, or C 1-6 haloalkyl
- Y is –O-or NR 22 -;
- R 22 is hydrogen, C 1-6 alkyl, or C 1-6 haloalkyl
- W is absent, C 1-6 alkylene, C 2-6 alkenylene, or C 2-6 alkynylene; wherein said C 1-6 alkylene, C 2-6 alkenylene, or C 2-6 alkynylene represented by W is optionally substituted by one or more R 3 ;
- L is absent, -O-, -NH-, 3-12 membered carbocyclyl, 3-12 membered heterocyclyl, 6-10 membered aryl, or 5-10 membered heteroaryl; wherein said -NH-, 3-12 membered carbocyclyl, 3-12 membered heterocyclyl, 6-10 membered aryl, or 5-10 membered heteroaryl represented by L is optionally substituted by one or more R 3 ; and
- Z is absent, C 1-6 alkylene, C 2-6 alkenylene, or C 2-6 alkynylene; wherein said C 1-6 alkylene, C 2-6 alkenylene, or C 2-6 alkynylene represented by Z is optionally substituted by one or more R 3 ; wherein
- R 3 in each occurrence, is independently halogen, CN, C 1-6 alkyl, -OR 3a , or -NR 3a R 3b ; wherein
- R 3a and R 3b are independently selected from the group consisting of hydrogen, C 1-6 alkyl, C 1-6 haloalkyl, C 2-6 alkenyl, C 2-6 alkynyl, 3-6 membered carbocyclyl, and 4-6 membered heterocyclyl;
- R 4 is hydrogen or C 1-6 alkyl; or one R 3 and R 4 together with the atoms to which they are attached form 4-8 membered heterocyclyl;
- heterocyclyl comprises 1-3 heteroatoms selected from oxygen, nitrogen, and sulfur; and said heteroaryl comprises 1-4 heteroatoms selected from oxygen, nitrogen, and sulfur.
- ring A is phenyl or nitrogen containing 5-10 membered heteroaryl; wherein said phenyl or nitrogen containing 5-10 membered heteroaryl represented by ring A is optionally substituted by one to three R 1 ; wherein
- R 1 is halogen, -CN, C 1-4 alkyl, C 1-4 haloalkyl, C 2-4 alkenyl, C 2-4 alkynyl, -C (O) R 1a , -C (O) OR 1a , -C (O) NR 1a R 1b , -OR 1a , -NR 1a R 1b , -NR 1a C (O) R 1b , -NR 1a C (O) OR 1b , -NR 1a SO 2 R 1b , -NR 1a SO 2 NR 1b R 1c , -SO 2 R 1a , -SO 2 NR 1a R 1b , or -P (O) R 1a R 1b ; wherein
- R 1a , R 1b , and R 1c are independently selected from the group consisting of hydrogen, C 1-4 alkyl, C 1-4 haloalkyl, C 2-4 alkenyl, C 2-4 alkynyl, 4-6 membered cycloalkyl, and 4-6 membered heterocyclyl.
- ring A is phenyl or nitrogen containing 5-6 membered heteroaryl; wherein said phenyl or nitrogen containing 5-6 membered heteroaryl represented by ring A is optionally substituted by one to two R 1 ;
- R 1 is halogen, -CN, C 1-4 alkyl, C 1-4 haloalkyl, -OR 1a , or -NR 1a R 1b ; wherein
- R 1a or R 1b are independently selected from the group consisting of hydrogen, C 1-4 alkyl, C 1-4 haloalkyl, C 2-4 alkenyl, and C 2-4 alkynyl.
- ring A is selected from a group consisting of phenyl, pyridyl, pyrimidinyl, pyrazinyl, thiophenyl, thiazolyl, and imidazo [4, 5-c] pyridinyl; wherein said phenyl, pyridyl, pyrimidinyl, pyrazinyl, thiophenyl, thiazolyl, or imidazo [4, 5-c] pyridinyl is optionally substituted by one to two R 1 ; wherein R 1 is halogen, -CN, C 1-4 alkyl, C 1-4 haloalkyl, or -OC 1-4 alkyl.
- X is absent, -O-, -NR 2 -, -C (O) -, -NHC (O) -, or -C (O) NH-; wherein R 2 in each occurrence, is independently hydrogen, C 1-4 alkyl, or C 1-4 haloalkyl; and
- Y is -O-or NR 22 -; wherein R 22 is hydrogen, C 1-4 alkyl, or C 1-4 haloalkyl.
- W is absent, C 1-4 alkylene, C 2-4 alkenylene, or C 2-4 alkynylene; wherein said C 1-4 alkylene, C 2-4 alkenylene, or C 2-4 alkynylene represented by W is optionally substituted by one to three R 3 ;
- L is absent, -O-, -NH-, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 6-10 membered aryl, or 5-10 membered heteroaryl; wherein said -NH-, 3-8 membered carbocyclyl, 3-6 membered heterocyclyl, 6-10 membered aryl, and 5-10 membered heteroaryl represented by L is optionally substituted by one to three R 3 ; and
- Z is absent, C 1-4 alkylene, C 2-4 alkenylene, or C 2-4 alkynylene; wherein said C 1-4 alkylene, C 2-4 alkenylene, or C 2-4 alkynylene represented by Z is optionally substituted by one to three R 3 ; wherein R 3 , in each occurrence, is independently halogen or C 1-4 alkyl; and
- R 4 is hydrogen, C 1-4 alkyl, or C 1-4 haloalkyl; or one R 3 and R 4 together with the atoms to which they are attached form 5-6 membered heterocyclyl.
- W is absent, C 1-3 alkylene, C 2-3 alkenylene, or C 2-3 alkynylene; wherein said C 1-3 alkylene, C 2-3 alkenylene, or C 2-3 alkynylene represented by W is optionally substituted by one to two R 3 ;
- L is absent, -O-, -NH-, 3-6 membered monocyclic carbocyclyl, 6-8 membered bicyclic carbocyclyl; 4-6 membered monocyclic heterocyclyl, phenyl, and 5-6 membered monocyclic heteroaryl; wherein said 3-6 membered monocyclic carbocyclyl, 6-8 membered bicyclic carbocyclyl; 4-6 membered monocyclic heterocyclyl, phenyl, and 5-6 membered monocyclic heteroaryl represented by L is optionally substituted by one R 3 ; and
- Z is absent, C 1-3 alkylene, C 2-3 alkenylene, or C 2-3 alkynylene; wherein said C 1-3 alkylene, C 2-3 alkenylene, or C 2-3 alkynylene represented by Z is optionally substituted by one R 3 ; wherein
- R 3 in each occurrence, is independently halogen or C 1-3 alkyl
- R 4 is hydrogen, methyl, or -CF 3 ; or one R 3 and R 4 together with the atoms to which they are attached form 6 membered heterocyclyl.
- W is C 1-3 alkylene, C 1-3 haloalkylene, C 2-3 alkenylene, or C 2-3 alkynylene;
- L is absent or -O-
- Z is C 1-3 alkylene, C 1-3 haloalkylene, C 2-3 alkenylene, or C 2-3 alkynylene.
- W is C 3-4 alkylene, C 3-4 haloalkylene, C 3-4 alkenylene, or C 3-4 alkynylene;
- W is absent, C 1-2 alkylene, or C 1-2 haloalkylene
- L is 4-6 membered carbocyclyl, 4-6 membered heterocyclyl, or 5-6 membered heteroaryl;
- Z is absent or methylene.
- L is absent, -O-, -NH-, cyclobutyl, cyclohexyl, spiro [3.3] heptanyl, azetidinyl, piperazinyl, piperidinyl, imidazolyl, triazolyl, or thiozolyl; wherein said -NH-, cyclobutyl, cyclohexyl, spiro [3.3] heptanyl, azetidinyl, piperazinyl, piperidinyl, imidazolyl, triazolyl, or thiozolyl is optionally substituted by one R 3 ;
- R 4 is hydrogen; or one R 3 and R 4 together with the atoms to which they are attached form a piperazinyl ring.
- halogen refers to fluoride, chloride, bromide, or iodide.
- alkyl used alone or as part of a larger moiety, such as “alkoxy” or “haloalkyl” and the like, means saturated aliphatic straight-chain or branched monovalent hydrocarbon radical of formula -C n H (2n+1) .
- an alkyl group typically has 1-6 carbon atoms, i.e. C 1 - 6 alkyl.
- a “C 1-6 alkyl” group means a radical having from 1 to 6 carbon atoms in a linear or branched arrangement.
- Examples include methyl, ethyl, n-propyl, iso-propyl, n-butyl, iso-butyl, tert-butyl, n-pentyl, isopentyl, hexyl, and the like.
- haloalkyl means alkyl, as the case may be, substituted with one or more halogen atoms. In one embodiment, the alkyl can be substituted by one to three halogens. Examples of haloalkyl, include, but are not limited to, trifluoromethyl, trichloromethyl, pentafluoroethyl and the like.
- alkylene as used herein, means a straight or branched chain divalent hydrocarbon group of formula -C n H 2n -. Non-limiting examples include ethylene, and propylene.
- haloalkylene means alkylene, as the case may be, substituted with one or more halogen atoms. In one embodiment, the alkylene can be substituted by one to three halogens.
- alkenyl means an alkyl group in which one or more carbon/carbon single bond is replaced by a double bond.
- alkynyl means an alkyl group in which one or more carbon/carbon single bond is replaced by a triple bond.
- carbocyclyl refers to a 3-14 membered non-aromatic hydrocarbon ring system and may exist as a monocylic ring or a polycylic ring (e.g., a bicyclic ring (including fused, spiro or bridged carbocyclic rings) or a tricyclic ring) .
- carbocyclyl is 3-, 4-, 5-, 6-, 7-, or 8-membered monocyclic or bicyclic or 7-, 8-, 9-, 10-, 11-, or 12-membered bicyclic or tricyclic hydrocarbon ring, any of which may be saturated, partially unsaturated.
- Any substitutable ring atom can be substituted (e.g., by one or more substituents) .
- carbocycles include, but are not limited to, cyclopropyl, cyclobutyl, cyclobutenyl, cyclopentyl, cyclopentenyl, cyclohexyl, cycloheptenyl, cycloheptyl, cycloheptenyl, adamantyl, cyclooctyl, cyclooctenyl, and cyclooctadienyl.
- carbocyclyl is intended to include, bridged, fused, and spirocyclic rings.
- a spirocyclic carbocyclyl In a spirocyclic carbocyclyl, one atom is common to two different rings.
- An example of a spirocyclic carbocyclyl is spiro [3.3] heptanyl.
- a bridged carbocyclyl the rings share at least two common non-adjacent atoms.
- bridged carbocyclyls include bicyclo [2.2.1] heptanyl, bicyclo [2.2.1] hept-2-enyl, and adamantanyl.
- two or more rings may be fused together, such that two rings share one common bond.
- Examples of two-or three-fused ring carbocyclyls include naphthalenyl, tetrahydronaphthalenyl (tetralinyl) , indenyl, indanyl (dihydroindenyl) , anthracenyl, phenanthrenyl, and decalinyl.
- the term “carbocyclyl” as used herein, includes groups in which a carbocyclyl ring is fused to one or more aryl, where the radical or point of attachment is on the carbocyclyl ring. Nonlimiting examples of such fused ring systems include:
- cycloalkyl refers to a cyclic, bicyclic, tricyclic, or polycyclic saturated hydrocarbon groups having 3 to 12 ring carbons. In one embodiment, cycloalkyl may have 3 to 7 ring cabons. Any substitutable ring atom can be substituted (e.g., by one or more substituents) .
- Examples of cycloalkyl groups include, without limitation, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and cyclooctyl. Cycloalkyl may include multiple fused and/or bridged rings.
- Non-limiting examples of fused/bridged cycloalkyl include: bicyclo [1.1.0] butane, bicyclo [2.1.0] pentane, bicyclo [1.1.0] pentane, bicyclo [3.1.0] hexane, bicyclo [2.1.1] hexane, bicyclo [3.2.0] heptane, bicyclo [4.1.0] heptane, bicyclo [2.2.1] heptane, bicyclo [3.1.1] heptane, bicyclo [4.2.0] octane, bicyclo [3.2.1] octane, bicyclo [2.2.2] octane, and the like.
- Cycloalkyl also includes spirocyclic rings (e.g., spirocyclic bicycle wherein two rings are connected through just one atom) .
- spirocyclic cycloalkyls include spiro [2.2] pentane, spiro [2.5] octane, spiro [3.5] nonane, spiro [3.5] nonane, spiro [3.5] nonane, spiro [4.4] nonane, spiro [2.6] nonane, spiro [4.5] decane, spiro [3.6] decane, spiro [5.5] undecane, and the like.
- heterocyclyl refers to a radical of a 3-to 12-membered non-aromatic ring system having ring carbon atoms and 1 to 4 ring heteroatoms, wherein each heteroatom is independently selected from nitrogen, quaternary nitrogen, oxidized nitrogen (e.g., NO) , oxygen, and sulfur, including sulfoxide and sulfone ( “3-12 membered heterocyclyl” ) .
- a heterocyclyl group is a 3-7 membered non-aromatic ring system having ring carbon atoms and 1-4 ring heteroatoms, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur ( “3-7 membered heterocyclyl” ) .
- a heterocyclyl group comprises 1-3 heteroatoms selected from oxygen, nitrogen, and sulfur.
- the point of attachment can be a carbon or nitrogen atom, as valency permits.
- a heterocyclyl group can either be monocyclic ( “monocyclic heterocyclyl” ) or polycyclic (e.g., a bicyclic system ( “bicyclic heterocyclyl” ) or tricyclic system ( “tricyclic heterocyclyl” ) ; polycyclic ring systems include fused, bridged, or spiro ring systems) .
- Exemplary monocyclic heterocyclyl groups include azetidinyl, oxetanyl, thietanyl, tetrahydrofuranyl, pyrrolidinyl, piperidinyl, tetrahydropyranyl, piperazinyl, morpholinyl, azepanyl, oxepanyl, thiepanyl, tetrahydropyridinyl, and the like.
- Heterocyclyl polycyclic ring systems can include heteroatoms in one or more rings in the polycyclic ring system. Substituents may be present on one or more rings in the polycyclic ring system.
- Spiro heterocyclyl refers to 5 to 12 membered polycyclic heterocyclyl with rings connected through one common carbon atom (called as spiro atom) , wherein said rings have one or more heteroatoms selected from the group consisting of nitrogen, quaternary nitrogen, oxidized nitrogen (e.g., NO) , oxygen, and sulfur, including sulfoxide and sulfone, the remaining ring atoms being C, wherein one or more rings may contain one or more double bonds, but none of the rings has a completely conjugated pi-electron system.
- spiro heterocyclyl include, but are not limited to the following groups:
- Fused heterocyclyl refers to a 5 to 12 membered polycyclic heterocyclyl group, wherein each ring in the group shares an adjacent pair of carbon atoms with another ring in the group, wherein one or more rings can contain one or more double bonds, but none of the rings has a completely conjugated ⁇ -electron system, and wherein said rings have one or more heteroatoms selected from the group consisting of nitrogen, quaternary nitrogen, oxidized nitrogen (e.g., NO) , oxygen, and sulfur, including sulfoxide and sulfone, the remaining ring atoms being C.
- fused heterocyclyl include, but are not limited to the following groups:
- a fused heterocyclyl include groups in which a heterocyclyl ring is fused to one or more aryl or heteroaryl, where the radical or point of attachment is on the heterocyclyl ring.
- fused heterocyclyl ring systems include:
- Bridged heterocyclyl refers to a 5 to 12 membered polycyclic heterocyclyl group, wherein any two rings in the group share two disconnected atoms, the rings can have one or more double bonds but have no completely conjugated ⁇ -electron system, and the rings have one or more heteroatoms selected from the group consisting of nitrogen, quaternary nitrogen, oxidized nitrogen (e.g., NO) , oxygen, and sulfur, including sulfoxide and sulfone as ring atoms, the remaining ring atoms being C.
- Representive examples of bridged heterocyclyl include, but are not limited to the following groups:
- the carbocyclyl, the cycloalkyl, or the heterocyclyl may be unsubstituted, or be substituted with one or more substituents as valency allows, wherein the substituents can be independently selected from a number of groups such as oxo, -CN, halogen, alkyl and alkoxyl, opotionally, the alkyl substitution may be further substituted.
- aryl refers to a 6 to 12 membered all-carbon monocyclic ring or a polycyclic fused ring (a “fused” ring system means that each ring in the system shares an adjacent pair of carbon atoms with other ring in the system) group, and has a completely conjugated ⁇ -electron system.
- aryl may be used interchangeably with the terms “aryl ring” “carbocyclic aromatic ring” , “aryl group” and “carbocyclic aromatic group” .
- Representive examples of aryl are phenyl and naphthyl.
- aryl as used herein, includes groups in which an aromatic ring is fused to one or more non-aromatic carbocyclyl ring, where the radical or point of attachment is on the aromatic ring.
- fused ring systems include:
- heteroaryl refers to a monocyclic or multicyclic aromatic hydrocarbon in which at least one of the ring carbon atoms has been replaced with a heteroatom independently selected from oxygen, nitrogen and sulfur.
- the heteroaryl is based on a C 5-10 aryl with one or more of its ring carbon atoms replaced by the heteroatom.
- heteroaryl comprises 1-4 heteroatoms selected from oxygen, nitrogen, and sulfur.
- a heteroaryl group may be attached through a ring carbon atom or, where valency permits, through a ring nitrogen atom.
- the heteroaryl may be unsubstituted, or be substituted with one or more substituents as valency allows with the substituents being independently selected from halogen, OH, alkyl, alkoxyl, and amino (e.g., NH 2 , NHalkyl, N (alkyl) 2 ) , optionally, the alkyl may be further substituted.
- Examples of monocyclic 5-6 membered heteroaryl groups include furanyl (e.g., 2-furanyl, 3-furanyl) , imidazolyl (e.g., N-imidazolyl, 2-imidazolyl, 4-imidazolyl, 5-imidazolyl) , isoxazolyl (e.g., 3-isoxazolyl, 4-isoxazolyl, 5-isoxazolyl) , oxadiazolyl (e.g., 2-oxadiazolyl, 5-oxadiazolyl) , oxazolyl (e.g., 2-oxazolyl, 4-oxazolyl, 5-oxazolyl) , pyrazolyl (e.g., 3-pyrazolyl, 4-pyrazolyl) , pyrrolyl (e.g., 1-pyrrolyl, 2-pyrrolyl, 3-pyrrolyl) , pyridyl (e.g., 2-pyrid
- polycyclic aromatic heteroaryl groups examples include carbazolyl, benzimidazolyl, benzothienyl, benzofuranyl, indolyl, quinolinyl, benzotriazolyl, benzothiazolyl, benzoxazolyl, benzimidazolyl, isoquinolinyl, indolyl, isoindolyl, acridinyl, or benzisoxazolyl.
- a “substituted heteroaryl group” is substituted at any one or more substitutable ring atom, which is a ring carbon or ring nitrogen atom bonded to a hydrogen.
- moieties e.g., alkyl, alkylene, cycloalkyl, aryl, heteroaryl, or heterocyclyl
- substituents any substituents that are suitable to attach to the moiety.
- -NR a1 C ( O) R b1 , -
- Each R a1 and each R b1 are independently selected from –H and C 1-5 alkyl, optionally substituted with hydroxyl or C 1-3 alkoxy;
- R c1 is –H, C 1-5 haloalkyl or C 1-5 alkyl, wherein the C 1-5 alkyl is optionally substituted with hydroxyl or C 1 -C 3 alkoxy.
- pharmaceutically-acceptable salt refers to a pharmaceutical salt that is, within the scope of sound medical judgment, suitable for use in contact with the tissues of humans and lower animals without undue toxicity, irritation, and allergic response, and is commensurate with a reasonable benefit/risk ratio.
- Pharmaceutically-acceptable salts are well known in the art. For example, S. M. Berge et al. describes pharmacologically acceptable salts in J. Pharm. Sci., 1977, 66, 1–19.
- compositions of any one of the formulae described above include acid addition and base salts.
- Suitable pharmaceutically acceptable salts of the compounds disclosed herein can form pharmaceutically acceptable salts with pharmaceutically acceptable acid (s) .
- Suitable pharmaceutically acceptable acid addition salts of the compounds described herein include salts of inorganic acids (such as hydrochloric, hydrobromic, phosphoric, metaphosphoric, nitric, and sulfuric acids) and of organic acids (such as acetic, benzenesulfonic, benzoic, ethanesulfonic, methanesulfonic, and succinic acids) .
- Compounds of the present teachings with acidic groups such as carboxylic acids can form pharmaceutically acceptable salts with pharmaceutically acceptable base (s) .
- Suitable pharmaceutically acceptable basic salts include ammonium salts, alkali metal salts (such as sodium and potassium salts) and alkaline earth metal salts (such as magnesium and calcium salts) .
- the resulting salt may precipitate out and be collected by filtration or may be recovered by evaporation of the solvent.
- the degree of ionisation in the resulting salt may vary from completely ionised to almost non-ionised.
- the compounds of any one of the formulae described above may exhibit one or more kinds of isomerism (e.g. optical, geometric or tautomeric isomerism) .
- isomerism e.g. optical, geometric or tautomeric isomerism
- Stereoisomers are compounds that differ only in their spatial arrangement. Stereoisomers include all diastereomeric and enantiomeric forms of a compound. Enantiomers are stereoisomers that are mirror images of each other. Diastereomers are stereoisomers having two or more chiral centers that are not identifcal and are not mirror images of each other.
- a compound When a compound is designated by its chemical name (e.g., where the configuration is indicated in the chemical name by “R” or “S” ) or its structure (e.g., the configuration is indicated by “wedge” bonds) that indicates a single enantiomer, unless indicated otherwise, the compound is at least 60%, 70%, 80%, 90%, 99%or 99.9%optically pure (also referred to as “enantiomerically pure” ) .
- Optical purity is the weight in the mixture of the named or depicted enantiomer divided by the total weight in the mixture of both enantiomers.
- stereochemistry of a disclosed compound is named or depicted by structure, and the named or depicted structure encompasses more than one stereoisomer (e.g., as in a diastereomeric pair)
- the stereoisomeric purity of the named or depicted stereoisomers at least 60%, 70%, 80%, 90%, 99%or 99.9%by weight.
- the stereoisomeric purity in this case is determined by dividing the total weight in the mixture of the stereoisomers encompassed by the name or structure by the total weight in the mixture of all of the stereoisomers.
- a disclosed compound having a chiral center is depicted by a structure without showing a configuration at that chiral center, the structure is meant to encompass the compound with the S configuration at that chiral center, the compound with the R configuration at that chiral center, or the compound with a mixture of the R and S configuration at that chiral center.
- a disclosed compound having a chiral center is depicted by its chemical name without indicating a configuration at that chiral center with “S” or “R”
- the name is meant to encompass the compound with the S configuration at that chiral center, the compound with the R configuration at that chiral center or the compound with a mixture of the R and S configuration at that chiral center.
- Racemic mixture means 50%of one enantiomer and 50%of the corresponding enantiomer.
- a compound with one chiral center is named or depicted without indicating the stereochemistry of the chiral center, it is understood that the name or structure encompasses both possible enantiomeric forms (e.g., both enantiomerically-pure, enantiomerically-enriched or racemic) of the compound.
- geometric isomer means isomers that differ in the orientation of substituent atoms in relationship to a carbon-carbon double bond, to a carbocyclic ring, or to a bridged bicyclic system.
- Substituent atoms (other than hydrogen) on each side of a carbon-carbon double bond may be in an E or Z configuration according to the Cahn-Ingold-Prelog priority rules. In the “E” configuration, the substituents having the highest priorities are on opposite sides in relationship to the carbon-carbon double bond. In the “Z” configuration, the substituents having the highest priorities are oriented on the same side in relationship to the carbon-carbon double bond.
- Substituents around a carbon-carbon double bond can also be referred to as “cis” or “trans, ” where “cis” represents substituents on the same side of the double bond and “trans” represents substituents on opposite sides of the double bond.
- the arrangement of substituents around a carbocyclic ring can also be designated as “cis” or “trans. ”
- the term “cis” represents substituents on the same side of the plane of the ring, and the term “trans” represents substituents on opposite sides of the plane of the ring.
- Mixtures of compounds wherein the substituents are disposed on both the same and opposite sides of plane of the ring are designated “cis/trans. ”
- tautomeric isomerism ( “tautomerism” ) can occur. This can take the form of proton tautomerism in compounds of any one of the formulae described above containing, for example, an imino, keto, or oxime group, or so-called valence tautomerism in compounds which contain an aromatic moiety. It follows that a single compound may exhibit more than one type of isomerism.
- geometric isomer When a geometric isomer is depicted by name or structure, it is to be understood that the named or depicted isomer exists to a greater degree than another isomer, that is that the geometric isomeric purity of the named or depicted geometric isomer is greater than 50%, such as at least 60%, 70%, 80%, 90%, 99%, or 99.9%pure by weight. Geometric isomeric purity is determined by dividing the weight of the named or depicted geometric isomer in the mixture by the total weight of all of the geomeric isomers in the mixture.
- Cis/trans isomers may be separated by conventional techniques well known to those skilled in the art, for example, chromatography and fractional crystallisation.
- racemate or a racemic precursor
- HPLC high pressure liquid chromatography
- the racemate (or a racemic precursor) may be reacted with a suitable optically active compound, for example, an alcohol, or, in the case where the compound of any one of the formulae described above contains an acidic or basic moiety, a base or acid such as 1-phenylethylamine or tartaric acid.
- the resulting diastereomeric mixture may be separated by chromatography and/or fractional crystallization and one or both of the diastereoisomers converted to the corresponding pure enantiomer (s) by means well known to a skilled person.
- Chiral compounds of any one of the formulae described above (and chiral precursors thereof) may be obtained in enantiomerically-enriched form using chromatography, typically HPLC, on an asymmetric resin with a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from 0 to 50%by volume of isopropanol, typically from 2%to 20%, and from 0 to 5%by volume of an alkylamine, typically 0.1%diethylamine.
- a compound of the present disclosure is administered in an amount effective to treat a condition as described herein.
- the compounds of the present disclosure can be administered as compound per se, or alternatively, as a pharmaceutically acceptable salt.
- the compound per se or pharmaceutically acceptable salt thereof will simply be referred to as the compounds of the present disclosure.
- the compounds of the present disclosure are administered by any suitable route in the form of a pharmaceutical composition adapted to such a route, and in a dose effective for the treatment intended.
- the compounds of the present disclosure may be administered orally, rectally, vaginally, parenterally, or topically.
- the compounds of the present disclosure may be administered orally.
- Oral administration may involve swallowing, so that the compound enters the gastrointestinal tract, or buccal or sublingual administration may be employed by which the compound enters the bloodstream directly from the mouth.
- the compounds of the present disclosure may also be administered directly into the bloodstream, into muscle, or into an internal organ.
- Suitable means for parenteral administration include intravenous, intraarterial, intraperitoneal, intrathecal, intraventricular, intraurethral, intrasternal, intracranial, intramuscular and subcutaneous.
- Suitable devices for parenteral administration include needle (including microneedle) injectors, needle-free injectors and infusion techniques.
- the compounds of the present disclosure may also be administered topically to the skin or mucosa, that is, dermally or transdermally.
- the compounds of the present disclosure can also be administered intranasally or by inhalation.
- the compounds of the present disclosure may be administered rectally or vaginally.
- the compounds of the present disclosure may also be administered directly to the eye or ear.
- the dosage regimen for the compounds of the present disclosure and/or compositions containing said compounds is based on a variety of factors, including the type, age, weight, sex and medical condition of the patient; the severity of the condition; the route of administration; and the activity of the particular compound employed. Thus the dosage regimen may vary widely.
- the total daily dose of a compound of the present disclosure is typically from about 0.001 to about 100 mg/kg (i.e., mg compound of the present disclosure per kg body weight) for the treatment of the indicated conditions discussed herein.
- compositions may be provided in the form of tablets containing 0.1-500 milligrams of the active ingredient for the symptomatic adjustment of the dosage to the patient.
- a medicament typically contains from about 0.01 mg to about 500 mg of the active ingredient.
- doses may range from about 0.01 to about 10 mg/kg/minute during a constant rate infusion.
- Suitable subjects according to the present disclosure include mammalian subjects, including non-human mammal such as primates, rodents (mice, rats, hamsters, rabbits etc) .
- humans are suitable subjects. Human subjects may be of either gender and at any stage of development.
- the present disclosure comprises pharmaceutical compositions.
- Such pharmaceutical compositions comprise a compound of the present disclosure presented, a pharmaceutically acceptable salt, or a stereoisomer thereof with a pharmaceutically acceptable carrier or excipient.
- Other pharmacologically active substances can also be present.
- pharmaceutically acceptable carrier or excipient includes any and all solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents, and the like that are physiologically compatible.
- pharmaceutically acceptable carriers include one or more of water, saline, phosphate buffered saline, dextrose, glycerol, ethanol and the like, as well as combinations thereof, and may include isotonic agents, for example, sugars, sodium chloride, or polyalcohols such as mannitol, or sorbitol in the composition.
- Pharmaceutically acceptable substances such as wetting agents or minor amounts of auxiliary substances such as wetting or emulsifying agents, preservatives or buffers, which enhance the shelf life or effectiveness of the antibody or antibody portion.
- compositions of present disclosure may be in a variety of forms. These include, for example, liquid, semi-solid and solid dosage forms, such as liquid solutions (e.g., injectable and infusible solutions) , dispersions or suspensions, tablets, pills, powders, liposomes and suppositories.
- liquid solutions e.g., injectable and infusible solutions
- dispersions or suspensions tablets, pills, powders, liposomes and suppositories.
- the form depends on the intended mode of administration and therapeutic application.
- compositions are in the form of injectable or infusible solutions, such as compositions similar to those used for passive immunization of humans with antibodies in general.
- One mode of administration is parenteral (e.g. intravenous, subcutaneous, intraperitoneal, intramuscular) .
- the antibody is administered by intravenous infusion or injection.
- the antibody is administered by intramuscular or subcutaneous injection.
- Oral administration of a solid dose form may be, for example, presented in discrete units, such as hard or soft capsules, pills, cachets, lozenges, or tablets, each containing a predetermined amount of at least one compound of the present disclosure.
- the oral administration may be in a powder or granule form.
- the oral dose form is sub-lingual, such as, for example, a lozenge.
- the compounds of any one of the formulae described above are ordinarily combined with one or more adjuvants.
- Such capsules or tablets may contain a controlled release formulation.
- the dosage forms also may comprise buffering agents or may be prepared with enteric coatings.
- oral administration may be in a liquid dose form.
- Liquid dosage forms for oral administration include, for example, pharmaceutically acceptable emulsions, solutions, suspensions, syrups, and elixirs containing inert diluents commonly used in the art (e.g., water) .
- Such compositions also may comprise adjuvants, such as wetting, emulsifying, suspending, flavoring (e.g., sweetening) , and/or perfuming agents.
- the present disclosure comprises a parenteral dose form.
- Parenter administration includes, for example, subcutaneous injections, intravenous injections, intraperitoneally, intramuscular injections, intrasternal injections, and infusion.
- injectable preparations i.e., sterile injectable aqueous or oleaginous suspensions
- suitable dispersing, wetting agents, and/or suspending agents may be formulated according to the known art using suitable dispersing, wetting agents, and/or suspending agents.
- the present disclosure comprises a topical dose form.
- Topical administration includes, for example, transdermal administration, such as via transdermal patches or iontophoresis devices, intraocular administration, or intranasal or inhalation administration.
- Compositions for topical administration also include, for example, topical gels, sprays, ointments, and creams.
- a topical formulation may include a compound which enhances absorption or penetration of the active ingredient through the skin or other affected areas.
- Typical formulations for this purpose include gels, hydrogels, lotions, solutions, creams, ointments, dusting powders, dressings, foams, films, skin patches, wafers, implants, sponges, fibres, bandages and microemulsions. Liposomes may also be used.
- Typical carriers include alcohol, water, mineral oil, liquid petrolatum, white petrolatum, glycerin, polyethylene glycol and propylene glycol.
- Penetration enhancers may be incorporated -see, for example, Finnin and Morgan, J. Pharm. Sci., 88: 955-958, 1999.
- Formulations suitable for topical administration to the eye include, for example, eye drops wherein the compound of present disclosure is dissolved or suspended in a suitable carrier.
- a typical formulation suitable for ocular or aural administration may be in the form of drops of a micronized suspension or solution in isotonic, pH-adjusted, sterile saline.
- Other formulations suitable for ocular and aural administration include ointments, biodegradable (i.e., absorbable gel sponges, collagen) and non-biodegradable (i.e., silicone) implants, wafers, lenses and particulate or vesicular systems, such as niosomes or liposomes.
- a polymer such as crossed linked polyacrylic acid, polyvinyl alcohol, hyaluronic acid, a cellulosic polymer, for example, hydroxypropylmethylcellulose, hydroxyethylcellulose, or methylcellulose, or a heteropolysaccharide polymer, for example, gelan gum, may be incorporated together with a preservative, such as benzalkonium chloride.
- a preservative such as benzalkonium chloride.
- Such formulations may also be delivered by iontophoresis.
- the compounds of the present disclosure are conveniently delivered in the form of a solution or suspension from a pump spray container that is squeezed or pumped by the patient or as an aerosol spray presentation from a pressurized container or a nebulizer, with the use of a suitable propellant.
- Formulations suitable for intranasal administration are typically administered in the form of a dry powder (either alone, as a mixture, for example, in a dry blend with lactose, or as a mixed component particle, for example, mixed with phospholipids, such as phosphatidylcholine) from a dry powder inhaler or as an aerosol spray from a pressurized container, pump, spray, atomizer (preferably an atomizer using electrohydrodynamics to produce a fine mist) , or nebulizer, with or without the use of a suitable propellant, such as 1, 1, 1, 2-tetrafluoroethane or 1, 1, 1, 2, 3, 3, 3-heptafluoropropane.
- the powder may comprise a bioadhesive agent, for example, chitosan or cyclodextrin.
- the present disclosure comprises a rectal dose form.
- rectal dose form may be in the form of, for example, a suppository. Cocoa butter is a traditional suppository base, but various alternatives may be used as appropriate.
- compositions of the present disclosure may be prepared by any of the well-known techniques of pharmacy, such as effective formulation and administration procedures.
- Compounds of the present disclosure can inhibit CDK2 and therefore are useful for treating diseases wherein the underlying pathology is, wholly or partially, mediated by CDK2.
- diseases include cancer and other diseases with proliferation disorder.
- the present disclosure provides treatment of an individual or a patient in vivo using a compound of Formula (I') or (I) , or a pharmaceutically acceptable salt, or a stereoisomer thereof such that growth of cancerous tumors is inhibited.
- a compound of Formula (I') or (I) or of any of the formulae as described herein, or a compound as recited in any of the claims and described herein, or a pharmaceutically acceptable salt or a stereoisomer thereof can be used to inhibit the growth of cancerous tumors with aberrations that activate the CDK2 kinase activity.
- CCNE1 diseases
- diseases e.g., cancers
- CCNE1 cyclin E1
- the patient has been previously determined to have an amplification of the cyclin E1 (CCNE1) gene and/or an expression level of CCNE1 in a biological sample obtained from the human subject that is higher than a control expression level of CCNE1.
- a compound of Formula (I') or (I) or of any of the formulae as described herein, or a compound as recited in any of the claims and described herein, or a pharmaceutically acceptable salt or a stereoisomer thereof can be used in conjunction with other agents or standard cancer treatments, as described below.
- the present disclosure provides a method for inhibiting growth of tumor cells in vitro. The method includes contacting the tumor cells in vitro with a compound of Formula (I') or (I) or of any of the formulae as described herein, or of a compound as recited in any of the claims and described herein, or of a pharmaceutically acceptable salt or a stereoisomer thereof.
- the present disclosure provides a method for inhibiting growth of tumor cells with CCNE1 amplification and overexpression in an individual or a patient.
- the method includes administering to the individual or patient in need thereof a therapeutically effective amount of a compound of Formula (I') or (I) or of any of the formulae as described herein, or of a compound as recited in any of the claims and described herein, or a pharmaceutically acceptable salt or a stereoisomer thereof.
- compounds of the present disclosure selectively inhibit CDK2 over CDK1, with a ratio of IC 50 values for the latter (CDK1) against the former (CDK2) of at least about 2, 5, 10, 15, 20, 40, 50, 60, 80, 100 or more.
- provided herein is a method of inhibiting CDK2, comprising contacting the CDK2 with a compound of Formula (I') or (I) or any of the formulae as described herein, a compound as recited in any of the claims and described herein, or a pharmaceutically acceptable salt, or a stereoisomer thereof.
- a method of inhibiting CDK2 in a patient comprising administering to the patient a compound of Formula (I') or (I) or any of the formulae as described herein, a compound as recited in any of the claims and described herein, or a pharmaceutically acceptable salt or a stereoisomer thereof.
- a method for treating cancer includes administering to a patient (in need thereof) , a therapeutically effective amount of a compound of Formula (I') or (I) or any of the formulae as described herein, a compound as recited in any of the claims and described herein, or a pharmaceutically acceptable salt or a stereoisomer thereof.
- the cancer is characterized by amplification or overexpression of CCNE1.
- the cancer is characterized by inactivation of a CDK2 inhibitor, such as p21Cip1 or p27Kip1.
- the cancer is ovarian cancer or breast cancer, characterized by amplification or overexpression of CCNE1.
- the patient has been diagnosed with a cancer characterized by amplification or overexpression of CCNE1, and/or loss of function of p21Cip1 or p27Kip1.
- the method further comprises determining the status of expression of CCNE1, p21Cip1 and/or p27Kip1.
- the method further comprises selecting patients characterized by amplification or overexpression of CCNE1, and/or loss of function of p21Cip1 or p27Kip1 for treatment.
- provided herein is a method of treating a disease or disorder associated with CDK2 in a patient, comprising administering to the patient a therapeutically effective amount of a compound of Formula (I') or (I) or any of the formulae as described herein, a compound as recited in any of the claims and described herein, or a pharmaceutically acceptable salt or a stereoisomer thereof.
- the disease or disorder associated with CDK2 is associated with an amplification of the cyclin E1 (CCNE1) gene and/or overexpression of CCNE1.
- CCNE1 cyclin E1
- the disease or disorder associated with CDK2 is N-myc amplified neuroblastoma cells (See Molenaar et al., Proc Natl Acad Sci USA, 106 (31) : 12968-12973) , K-Ras mutant lung cancers (see Hu, S., et al., Mol Cancer Ther, 2015, 14 (11) : 2576-85, and cancers with FBW7 mutation and CCNE1 overexpression (see Takada, et al., Cancer Res, 2017, 77 (18) : 4881-4893) .
- the disease or disorder associated with CDK2 is breast, lung, colorectal, gastric, or bone cancer, leukemia or lymphoma.
- the disease or disorder associated with CDK2 is lung squamous cell carcinoma, lung adenocarcinoma, pancreatic adenocarcinoma, breast invasive carcinoma, uterine carcinosarcoma, ovarian serous cystadenocarcinoma, stomach adenocarcinoma, esophageal carcinoma, bladder urothelial carcinoma, mesothelioma, or sarcoma.
- the disease or disorder associated with CDK2 is lung adenocarcinoma, breast invasive carcinoma, uterine carcinosarcoma, ovarian serous cystadenocarcinoma, or stomach adenocarcinoma.
- the disease or disorder associated with CDK2 is an adenocarcinoma, carcinoma, or cystadenocarcinoma.
- the disease or disorder associated with CDK2 is uterine cancer, ovarian cancer, stomach cancer, esophageal cancer, lung cancer, bladder cancer, pancreatic cancer, or breast cancer.
- the disease or disorder associated with CDK2 is a cancer.
- the cancer is characterized by amplification or overexpression of CCNE1.
- the cancer is ovarian cancer or breast cancer, characterized by amplification or overexpression of CCNE1.
- the breast cancer is chemotherapy or radiotherapy resistant breast cancer, endocrine resistant breast cancer, trastuzumab resistant breast cancer, or breast cancer demonstrating primary or acquired resistance to CDK4/6 inhibition.
- the breast cancer is advanced or metastatic breast cancer.
- cancers that are treatable using the compounds of the present disclosure include, but are not limited to, bone cancer, pancreatic cancer, skin cancer, cancer of the head or neck, cutaneous or intraocular malignant melanoma, uterine cancer, ovarian cancer, rectal cancer, cancer of the anal region, stomach cancer, testicular cancer, uterine cancer, carcinoma of the fallopian tubes, carcinoma of the endometrium, endometrial cancer, carcinoma of the cervix, carcinoma of the vagina, carcinoma of the vulva, Hodgkin's Disease, non-Hodgkin's lymphoma, cancer of the esophagus, cancer of the small intestine, cancer of the endocrine system, cancer of the thyroid gland, cancer of the parathyroid gland, cancer of the adrenal gland, sarcoma of soft tissue, cancer of the urethra, cancer of the penis, chronic or acute leukemias including acute myeloid leukemia
- cancers treatable with compounds of the present disclosure include melanoma (e.g., metastatic malignant melanoma, BRAF and HSP90 inhibition-resistant melanoma) , renal cancer (e.g., clear cell carcinoma) , prostate cancer (e.g., hormone refractory prostate adenocarcinoma) , breast cancer, colon cancer, lung cancer (e.g., non-small cell lung cancer and small cell lung cancer) , squamous cell head and neck cancer, urothelial cancer (e.g., bladder) and cancers with high microsatellite instability (MSI high ) . Additionally, the disclosure includes refractory or recurrent malignancies whose growth may be inhibited using the compounds of the disclosure.
- melanoma e.g., metastatic malignant melanoma, BRAF and HSP90 inhibition-resistant melanoma
- renal cancer e.g., clear cell carcinoma
- prostate cancer e.g., hormone re
- cancers that are treatable using the compounds of the present disclosure include, but are not limited to, solid tumors (e.g., prostate cancer, colon cancer, esophageal cancer, endometrial cancer, ovarian cancer, uterine cancer, renal cancer, hepatic cancer, pancreatic cancer, gastric cancer, breast cancer, lung cancer, cancers of the head and neck, thyroid cancer, glioblastoma, sarcoma, bladder cancer, etc.
- solid tumors e.g., prostate cancer, colon cancer, esophageal cancer, endometrial cancer, ovarian cancer, uterine cancer, renal cancer, hepatic cancer, pancreatic cancer, gastric cancer, breast cancer, lung cancer, cancers of the head and neck, thyroid cancer, glioblastoma, sarcoma, bladder cancer, etc.
- lymphoma e.g., lymphoma, leukemia such as acute lymphoblastic leukemia (ALL) , acute myelogenous leukemia (AML) , chronic lymphocytic leukemia (CLL) , chronic myelogenous leukemia (CML) , DLBCL, mantle cell lymphoma, Non-Hodgkin lymphoma (including relapsed or refractory NHL and recurrent follicular) , Hodgkin lymphoma or multiple myeloma) and combinations of said cancers.
- ALL acute lymphoblastic leukemia
- AML acute myelogenous leukemia
- CLL chronic lymphocytic leukemia
- CML chronic myelogenous leukemia
- DLBCL mantle cell lymphoma
- Non-Hodgkin lymphoma including relapsed or refractory NHL and recurrent follicular
- cancers that are treatable using the compounds of the present disclosure include, but are not limited to, cholangiocarcinoma, bile duct cancer, triple negative breast cancer, rhabdomyosarcoma, small cell lung cancer, leiomyosarcoma, hepatocellular carcinoma, Ewing’s sarcoma, brain cancer, brain tumor, astrocytoma, neuroblastoma, neurofibroma, basal cell carcinoma, chondrosarcoma, epithelioid sarcoma, eye cancer, Fallopian tube cancer, gastrointestinal cancer, gastrointestinal stromal tumors, hairy cell leukemia, intestinal cancer, islet cell cancer, oral cancer, mouth cancer, throat cancer, laryngeal cancer, lip cancer, mesothelioma, neck cancer, nasal cavity cancer, ocular cancer, ocular melanoma, pelvic cancer, rectal cancer, renal cell carcinoma, salivary gland cancer, sinus cancer, spinal cancer, tongue cancer, tubular carcinoma, ure
- diseases and indications that are treatable using the compounds of the present disclosure include, but are not limited to hematological cancers, sarcomas, lung cancers, gastrointestinal cancers, genitourinary tract cancers, liver cancers, bone cancers, nervous system cancers, gynecological cancers, and skin cancers.
- Exemplary hematological cancers include lymphomas and leukemias such as acute lymphoblastic leukemia (ALL) , acute myelogenous leukemia (AML) , acute promyelocytic leukemia (APL) , chronic lymphocytic leukemia (CLL) , chronic myelogenous leukemia (CML) , diffuse large B-cell lymphoma (DLBCL) , mantle cell lymphoma, Non-Hodgkin lymphoma (including relapsed or refractory NHL and recurrent follicular) , Hodgkin lymphoma, myeloproliferative diseases (e.g., primary myelofibrosis (PMF) , polycythemia vera (PV) , and essential thrombocytosis (ET) ) , myelodysplasia syndrome (MDS) , T-cell acute lymphoblastic lymphoma (T-ALL) and multiple myelo
- Exemplary sarcomas include chondrosarcoma, Ewing’s sarcoma, osteosarcoma, rhabdomyosarcoma, angiosarcoma, fibrosarcoma, liposarcoma, myxoma, rhabdomyoma, rhabdosarcoma, fibroma, lipoma, harmatoma, and teratoma.
- Exemplary lung cancers include non-small cell lung cancer (NSCLC) , small cell lung cancer (SCLC) , bronchogenic carcinoma, squamous cell, undifferentiated small cell, undifferentiated large cell, adenocarcinoma, alveolar (bronchiolar) carcinoma, bronchial adenoma, chondromatous hamartoma, and mesothelioma.
- NSCLC non-small cell lung cancer
- SCLC small cell lung cancer
- bronchogenic carcinoma squamous cell
- undifferentiated small cell undifferentiated large cell
- adenocarcinoma undifferentiated small cell
- adenocarcinoma alveolar (bronchiolar) carcinoma
- bronchial adenoma chondromatous hamartoma
- mesothelioma mesothelioma.
- Exemplary gastrointestinal cancers include cancers of the esophagus (squamous cell carcinoma, adenocarcinoma, leiomyosarcoma, lymphoma) , stomach (carcinoma, lymphoma, leiomyosarcoma) , pancreas (ductal adenocarcinoma, insulinoma, glucagonoma, gastrinoma, carcinoid tumors, vipoma) , small bowel (adenocarcinoma, lymphoma, carcinoid tumors, Kaposi's sarcoma, leiomyoma, hemangioma, lipoma, neurofibroma, fibroma) , large bowel (adenocarcinoma, tubular adenoma, villous adenoma, hamartoma, leiomyoma) , and colorectal cancer.
- esophagus squamous cell carcinoma, a
- Exemplary genitourinary tract cancers include cancers of the kidney (adenocarcinoma, Wilm's tumor [nephroblastoma] ) , bladder and urethra (squamous cell carcinoma, transitional cell carcinoma, adenocarcinoma) , prostate (adenocarcinoma, sarcoma) , and testis (seminoma, teratoma, embryonal carcinoma, teratocarcinoma, choriocarcinoma, sarcoma, interstitial cell carcinoma, fibroma, fibroadenoma, adenomatoid tumors, lipoma) .
- liver cancers include hepatoma (hepatocellular carcinoma) , cholangiocarcinoma, hepatoblastoma, angiosarcoma, hepatocellular adenoma, and hemangioma.
- Exemplary bone cancers include, for example, osteogenic sarcoma (osteosarcoma) , fibrosarcoma, malignant fibrous histiocytoma, chondrosarcoma, Ewing's sarcoma, malignant lymphoma (reticulum cell sarcoma) , multiple myeloma, malignant giant cell tumor chordoma, osteochronfroma (osteocartilaginous exostoses) , benign chondroma, chondroblastoma, chondromyxofibroma, osteoid osteoma, and giant cell tumors.
- osteogenic sarcoma osteosarcoma
- fibrosarcoma malignant fibrous histiocytoma
- chondrosarcoma chondrosarcoma
- Ewing's sarcoma malignant lymphoma
- multiple myeloma malignant giant cell tumor chordoma
- Exemplary nervous system cancers include cancers of the skull (osteoma, hemangioma, granuloma, xanthoma, osteitis deformans) , meninges (meningioma, meningiosarcoma, gliomatosis) , brain (astrocytoma, medulloblastoma, glioma, ependymoma, germinoma (pinealoma) , glioblastoma, glioblastoma multiform, oligodendroglioma, schwannoma, retinoblastoma, congenital tumors) , and spinal cord (neurofibroma, meningioma, glioma, sarcoma) , as well as neuroblastoma and Lhermitte-Duclos disease.
- skull osteoma, hemangioma, granuloma,
- Exemplary gynecological cancers include cancers of the uterus (endometrial carcinoma) , cervix (cervical carcinoma, pre -tumor cervical dysplasia) , ovaries (ovarian carcinoma (serous cystadenocarcinoma, mucinous cystadenocarcinoma, unclassified carcinoma) , granulosa-thecal cell tumors, Sertoli-Leydig cell tumors, dysgerminoma, malignant teratoma) , vulva (squamous cell carcinoma, intraepithelial carcinoma, adenocarcinoma, fibrosarcoma, melanoma) , vagina (clear cell carcinoma, squamous cell carcinoma, botryoid sarcoma (embryonal rhabdomyosarcoma) , and fallopian tubes (carcinoma) .
- endometrial carcinoma endometrial carcinoma
- cervix cervical carcinoma,
- Exemplary skin cancers include melanoma, basal cell carcinoma, Merkel cell carcinoma, squamous cell carcinoma, Kaposi's sarcoma, moles dysplastic nevi, lipoma, angioma, dermatofibroma, and keloids.
- diseases and indications that are treatable using the compounds of the present disclosure include, but are not limited to, sickle cell disease (e.g., sickle cell anemia) , triple-negative breast cancer (TNBC) , myelodysplastic syndromes, testicular cancer, bile duct cancer, esophageal cancer, and urothelial carcinoma.
- compounds of Formula (I') or (I) may possess satisfactory pharmacological profile and promising biopharmaceutical properties, such as toxicological profile, metabolism and pharmacokinetic properties, solubility, and permeability. It will be understood that determination of appropriate biopharmaceutical properties is within the knowledge of a person skilled in the art, e.g., determination of cytotoxicity in cells or inhibition of certain targets or channels to determine potential toxicity.
- mice preferably mice, rats, other rodents, rabbits, dogs, cats, swine, cattle, sheep, horses, or primates, and most preferably humans.
- treatment refers to reversing, alleviating, or inhibiting the progress of a disease described herein.
- treatment may be administered after one or more signs or symptoms of the disease have developed or have been observed (i.e., therapeutic treatment) .
- treatment may be administered in the absence of signs or symptoms of the disease.
- treatment may be administered to a susceptible subject prior to the onset of symptoms (i.e., prophylactic treatment) (e.g., in light of a history of symptoms and/or in light of exposure to a pathogen) .
- Treatment may also be continued after symptoms have resolved, for example, to delay or prevent recurrence.
- condition ” “disease, ” and “disorder” are used interchangeably.
- administer refers to methods introducing a compound disclosed herein, or a composition thereof, in or on a patient. These methods include, but are not limited to, intraarticular (in the joints) , intravenous, intramuscular, intratumoral, intradermal, intraperitoneal, subcutaneous, orally, topically, intrathecally, inhalationally, transdermally, rectally, and the like. Administration techniques that can be employed with the agents and methods described herein are found in e.g., Goodman and Gilman, The Pharmacological Basis of Therapeutics, current ed.; Pergamon; and Remington’s, Pharmaceutical Sciences (current edition) , Mack Publishing Co., Easton, Pa.
- an effective amount of a compound taught herein varies depending upon various factors, such as the given drug or compound, the pharmaceutical formulation, the route of administration, the type of disease or disorder, the identity of the subject or host being treated, and the like, but can nevertheless be routinely determined by one skilled in the art.
- An effective amount of a compound of the present teachings may be readily determined by one of ordinary skill by routine methods known in the art.
- terapéuticaally effective amount means an amount when administered to the subject which results in beneficial or desired results, including clinical results, e.g., inhibits, suppresses or reduces the symptoms of the condition being treated in the subject as compared to a control.
- a therapeutically effective amount can be an amount effective for detectable killing or inhibition of the growth or spread of cancer cells; the size or number of tumors; or other measure of the level, stage, progression or severity of the cancer.
- the exact amount required will vary from subject to subject, depending on the species, age, and general condition of the subject, the severity of the disease, the particular anticancer agent, its mode of administration, combination treatment with other therapies, and the like.
- Cancer cell growth and survival can be impacted by dysfunction in multiple signaling pathways.
- Targeting more than one signaling pathway (or more than one biological molecule involved in a given signaling pathway) may reduce the likelihood of drug-resistance arising in a cell population, and/or reduce the toxicity of treatment.
- One or more additional pharmaceutical agents such as, for example, chemotherapeutics, anti-estrogen agents, anti-inflammatory agents, steroids, immunosuppressants, immune-oncology agents, metabolic enzyme inhibitors, chemokine receptor inhibitors, and phosphatase inhibitors, as well as targeted therapies such as Bcr-Abl, Flt-3, EGFR, HER2, JAK, c-MET, VEGFR, PDGFR, c-Kit, IGF-1R, RAF, FAK, and CDK4/6 kinase inhibitors such as, for example, those described in WO 2006/056399 can be used in combination with the compounds of the present disclosure for treatment of CDK2-associated diseases, disorders or conditions.
- Other agents such as therapeutic antibodies can be used in combination with the compounds of the present disclosure for treatment of CDK2- associated diseases, disorders or conditions.
- the one or more additional pharmaceutical agents can be administered to a patient simultaneously or sequentially.
- the CDK2 inhibitor is administered or used in combination with an anti-estrogen agent or a CDK4/6 inhibitor or a mTOR inhibitor or a BCL2 inhibitor or a chemotherapy.
- the compounds as disclosed herein can be used in combination with one or more other enzyme/protein/receptor inhibitors therapies for the treatment of diseases, such as cancer and other diseases or disorders described herein.
- diseases and indications treatable with combination therapies include those as described herein.
- cancers include solid tumors and non-solid tumors, such as liquid tumors, blood cancers.
- infections include viral infections, bacterial infections, fungus infections or parasite infections.
- the compounds of the present disclosure can be combined with one or more inhibitors of the following kinases for the treatment of cancer: Akt1, Akt2, Akt3, BCL2, CDK4/6, TGF-DR, PKA, PKG, PKC, CaM-kinase, phosphorylase kinase, MEKK, ERK, MAPK, mTOR, EGFR, HER2, HER3, HER4, INS-R, IDH2, IGF-1R, IR-R, PDGF ⁇ R, PDGF ⁇ R, PI3K (alpha, beta, gamma, delta, and multiple or selective) , CSF1R, KIT, FLK-II, KDR/FLK-1, FLK-4, flt-1, FGFR1, FGFR2, FGFR3, FGFR4, c-Met, PARP, Ron, Sea, TRKA, TRKB, TRKC, TAM kinases (Axl, Mer, Tyro3) , FLT3, VEGFR/
- the compounds of the present disclosure can be combined with one or more of the following inhibitors for the treatment of cancer or infections.
- inhibitors that can be combined with the compounds of the present disclosure for treatment of cancer and infections include an FGFR inhibitor (FGFR1, FGFR2, FGFR3 or FGFR4, e.g., pemigatinib (INCB54828) , INCB62079) , an EGFR inhibitor (also known as ErB-1 or HER-1; e.g., erlotinib, gefitinib, vandetanib, orsimertinib, cetuximab, necitumumab, or panitumumab) , a VEGFR inhibitor or pathway blocker (e.g., bevacizumab, pazopanib, sunitinib, sorafenib, axitinib, regorafenib, ponatinib, cabozantinib,
- FGFR inhibitor
- the compound or salt described herein is administered with a PI3K6 inhibitor. In some embodiments, the compound or salt described herein is administered with a JAK inhibitor. In some embodiments, the compound or salt described herein is administered with a JAK1 or JAK2 inhibitor (e.g., baricitinib or ruxolitinib) . In some embodiments, the compound or salt described herein is administered with a JAK1 inhibitor. In some embodiments, the compound or salt described herein is administered with a JAK1 inhibitor, which is selective over JAK2.
- Example antibodies for use in combination therapy include, but are not limited to, trastuzumab (e.g., anti-HER2) , ranibizumab (e.g., anti-VEGF-A) , bevacizumab (AVASTINTM , e.g., anti-VEGF) , panitumumab (e.g., anti-EGFR) , cetuximab (e.g., anti-EGFR) , rituxan (e.g., anti-CD20) , and antibodies directed to c-MET.
- trastuzumab e.g., anti-HER2
- ranibizumab e.g., anti-VEGF-A
- bevacizumab AVASTINTM , e.g., anti-VEGF
- panitumumab e.g., anti-EGFR
- cetuximab e.g., anti-EGFR
- rituxan e.g., anti
- cytostatic agent cisplatin, doxorubicin, taxotere, taxol, etoposide, irinotecan, camptosar, topotecan, paclitaxel, docetaxel, epothilones, tamoxifen, 5-fluorouracil, methotrexate, temozolomide, cyclophosphamide, SCH 66336, R115777, L778, 123, BMS 214662, IRESSATM (gefitinib) , TARCEVATM (erlotinib) , antibodies to EGFR, intron, ara-C, adriamycin, cytoxan, gemcitabine, uracil mustard, chlormethine, ifosfamide, melphalan, chlorambucil, pipobroman, triethylenemelamine
- the compounds of the present disclosure can further be used in combination with other methods of treating cancers, for example by chemotherapy, irradiation therapy, tumor-targeted therapy, adjuvant therapy, immunotherapy or surgery.
- immunotherapy include cytokine treatment (e.g., interferons, GM-CSF, G-CSF, IL-2) , CRS-207 immunotherapy, cancer vaccine, monoclonal antibody, bispecific or multi-specific antibody, antibody drug conjugate, adoptive T cell transfer, Toll receptor agonists, RIG-I agonists, oncolytic virotherapy and immunomodulating small molecules, including thalidomide or JAK1/2 inhibitor, PI3Kd inhibitor and the like.
- the compounds can be administered in combination with one or more anti-cancer drugs, such as a chemotherapeutic agent.
- chemotherapeutics include any of: abarelix, aldesleukin, alemtuzumab, alitretinoin, allopurinol, altretamine, anastrozole, arsenic trioxide, asparaginase, azacitidine, bevacizumab, bexarotene, baricitinib, bleomycin, bortezomib, busulfan intravenous, busulfan oral, calusterone, capecitabine, carboplatin, carmustine, cetuximab, chlorambucil, cisplatin, cladribine, clofarabine, cyclophosphamide, cytarabine, dacarbazine, dactinomycin, dalteparin sodium, dasatinib, daunorubicin, decitabine,
- chemotherapeutics include proteasome inhibitors (e.g., bortezomib) , thalidomide, revlimid, and DNA-damaging agents such as melphalan, doxorubicin, cyclophosphamide, vincristine, etoposide, carmustine, and the like.
- proteasome inhibitors e.g., bortezomib
- thalidomide thalidomide
- revlimid thalidomide
- DNA-damaging agents such as melphalan, doxorubicin, cyclophosphamide, vincristine, etoposide, carmustine, and the like.
- Example steroids include corticosteroids such as dexamethasone or prednisone.
- Example Bcr-Abl inhibitors include imatinib mesylate (GLEEVAC TM ) , nilotinib, dasatinib, bosutinib, and ponatinib, and pharmaceutically acceptable salts.
- Other example suitable Bcr-Abl inhibitors include the compounds, and pharmaceutically acceptable salts thereof, of the genera and species disclosed in U.S. Pat. No. 5,521,184, WO 04/005281, and U.S. Ser. No. 60/578,491.
- Example suitable Flt-3 inhibitors include midostaurin, lestaurtinib, linifanib, sunitinib, sunitinib, maleate, sorafenib, quizartinib, crenolanib, pacritinib, tandutinib, PLX3397 and ASP2215, and their pharmaceutically acceptable salts.
- Other example suitable Flt-3 inhibitors include compounds, and their pharmaceutically acceptable salts, as disclosed in WO 03/037347, WO 03/099771, and WO 04/046120.
- Example suitable RAF inhibitors include dabrafenib, sorafenib, and vemurafenib, and their pharmaceutically acceptable salts.
- Other example suitable RAF inhibitors include compounds, and their pharmaceutically acceptable salts, as disclosed in WO 00/09495 and WO 05/028444.
- Example suitable FAK inhibitors include VS-4718, VS-5095, VS-6062, VS-6063, BI853520, and GSK2256098, and their pharmaceutically acceptable salts.
- Other example suitable FAK inhibitors include compounds, and their pharmaceutically acceptable salts, as disclosed in WO 04/080980, WO 04/056786, WO 03/024967, WO 01/064655, WO 00/053595, and WO 01/014402.
- Example suitable CDK4/6 inhibitors include palbociclib, ribociclib, trilaciclib, lerociclib, and abemaciclib, and their pharmaceutically acceptable salts.
- Other example suitable CDK4/6 inhibitors include compounds, and their pharmaceutically acceptable salts, as disclosed in WO 09/085185, WO 12/129344, WO 11/101409, WO 03/062236, WO 10/075074, and WO 12/061156.
- the compounds of the disclosure can be used in combination with one or more other kinase inhibitors including imatinib, particularly for treating patients resistant to imatinib or other kinase inhibitors.
- the compounds of the disclosure can be used in combination with a chemotherapeutic in the treatment of cancer, and may improve the treatment response as compared to the response to the chemotherapeutic agent alone, without exacerbation of its toxic effects.
- the compounds of the disclosure can be used in combination with a chemotherapeutic provided herein.
- additional pharmaceutical agents used in the treatment of multiple myeloma can include, without limitation, melphalan, melphalan plus prednisone [MP] , doxorubicin, dexamethasone, and Velcade (bortezomib) .
- the agent is an alkylating agent, a proteasome inhibitor, a corticosteroid, or an immunomodulatory agent.
- an alkylating agent include cyclophosphamide (CY) , melphalan (MEL) , and bendamustine.
- the proteasome inhibitor is carfilzomib.
- the corticosteroid is dexamethasone (DEX) .
- the immunomodulatory agent is lenalidomide (LEN) or pomalidomide (POM) . Additive or synergistic effects are desirable outcomes of combining a CDK2 inhibitor of the present disclosure with an additional agent.
- the agents can be combined with the present compound in a single or continuous dosage form, or the agents can be administered simultaneously or sequentially as separate dosage forms.
- the compounds of the present disclosure can be used in combination with one or more other inhibitors or one or more therapies for the treatment of infections.
- infections include viral infections, bacterial infections, fungus infections or parasite infections.
- a corticosteroid such as dexamethasone is administered to a patient in combination with the compounds of the disclosure where the dexamethasone is administered intermittently as opposed to continuously.
- the compounds of Formula (I') or (I) or any of the formulae as described herein, a compound as recited in any of the claims and described herein, or salts thereof can be combined with another immunogenic agent, such as cancerous cells, purified tumor antigens (including recombinant proteins, peptides, and carbohydrate molecules) , cells, and cells transfected with genes encoding immune stimulating cytokines.
- tumor vaccines include peptides of melanoma antigens, such as peptides of gp100, MAGE antigens, Trp-2, MARTI and/or tyrosinase, or tumor cells transfected to express the cytokine GM-CSF.
- tumor vaccines include the proteins from viruses implicated in human cancers such as Human Papilloma Viruses (HPV) , Hepatitis Viruses (HBV and HCV) and Kaposi's Herpes Sarcoma Virus (KHSV) .
- HPV Human Papilloma Viruses
- HBV and HCV Hepatitis Viruses
- KHSV Kaposi's Herpes Sarcoma Virus
- the compounds of the present disclosure can be used in combination with tumor specific antigen such as heat shock proteins isolated from tumor tissue itself.
- the compounds of Formula (I') or (I) or any of the formulae as described herein, a compound as recited in any of the claims and described herein, or salts thereof can be combined with dendritic cells immunization to activate potent anti-tumor responses.
- the compounds of the present disclosure can be used in combination with bispecific macrocyclic peptides that target Fe alpha or Fe gamma receptor-expressing effectors cells to tumor cells.
- the compounds of the present disclosure can also be combined with macrocyclic peptides that activate host immune responsiveness.
- combinations of the compounds of the disclosure with other therapeutic agents can be administered to a patient prior to, during, and/or after a bone marrow transplant or stem cell transplant.
- the compounds of the present disclosure can be used in combination with bone marrow transplant for the treatment of a variety of tumors of hematopoietic origin.
- the compounds of Formula (I') or (I) or any of the formulae as described herein, a compound as recited in any of the claims and described herein, or salts thereof can be used in combination with vaccines, to stimulate the immune response to pathogens, toxins, and self-antigens.
- pathogens for which this therapeutic approach may be particularly useful include pathogens for which there is currently no effective vaccine, or pathogens for which conventional vaccines are less than completely effective. These include, but are not limited to, HIV, Hepatitis (A, B, & C) , Influenza, Herpes, Giardia, Malaria, Leishmania, Staphylococcus aureus, Pseudomonas Aeruginosa.
- Viruses causing infections treatable by methods of the present disclosure include, but are not limit to human papillomavirus, influenza, hepatitis A, B, C or D viruses, adenovirus, poxvirus, herpes simplex viruses, human cytomegalovirus, severe acute respiratory syndrome virus, Ebola virus, measles virus, herpes virus (e.g., VZV, HSV-1, HAV-6, HSV-II, and CMV, Epstein Barr virus) , flaviviruses, echovirus, rhinovirus, coxsackie virus, cornovirus, respiratory syncytial virus, mumps virus, rotavirus, measles virus, rubella virus, parvovirus, vaccinia virus, HTLV virus, dengue virus, papillomavirus, molluscum virus, poliovirus, rabies virus, JC virus and arboviral encephalitis virus.
- human papillomavirus influenza, hepatitis A
- Pathogenic bacteria causing infections treatable by methods of the disclosure include, but are not limited to, chlamydia, rickettsial bacteria, mycobacteria, staphylococci, streptococci, pneumococci, meningococci and conococci, klebsiella, proteus, serratia, pseudomonas, legionella, diphtheria, salmonella, bacilli, cholera, tetanus, botulism, anthrax, plague, leptospirosis, and Lyme's disease bacteria.
- Pathogenic fungi causing infections treatable by methods of the disclosure include, but are not limited to, Candida (albicans, krusei, glabrata, tropicalis, etc. ) , Cryptococcus neoformans, Aspergillus (fumigatus, niger, etc. ) , Genus Mucorales (mucor, absidia, rhizophus) , Sporothrix schenkii, Blastomyces dermatitidis, Paracoccidioides brasiliensis, Coccidioides immitis and Histoplasma capsulatum.
- Candida albicans, krusei, glabrata, tropicalis, etc.
- Cryptococcus neoformans Aspergillus (fumigatus, niger, etc. )
- Genus Mucorales micor, absidia, rhizophus
- Sporothrix schenkii Blastomyces dermatitidis
- Pathogenic parasites causing infections treatable by methods of the disclosure include, but are not limited to, Entamoeba histolytica, Balantidium coli, Naegleriafowleri, Acanthamoeba sp., Giardia lambia, Cryptosporidium sp., Pneumocystis carinii, Plasmodium vivax, Babesia microti, Trypanosoma brucei, Trypanosoma cruzi, Leishmania donovani, Toxoplasma gondi, and Nippostrongylus brasiliensis.
- more than one pharmaceutical agent When more than one pharmaceutical agent is administered to a patient, they can be administered simultaneously, separately, sequentially, or in combination (e.g., for more than two agents) .
- immune checkpoint inhibitors include inhibitors against immune checkpoint molecules such as CBL-B, CD20, CD28, CD40, CD122, CD96, CD73, CD47, GITR, CSF1R, JAK, PI3K delta, PI3K gamma, TAM, arginase, HPK1, CD137 (also known as 4-1BB) , ICOS, A2AR, B7-H3, B7-H4, BTLA, CTLA-4, LAG3, TIM3, TIGIT, CD112R, VISTA, PD-1, PD-L1 and PD-L2.
- immune checkpoint inhibitors include inhibitors against immune checkpoint molecules such as CBL-B, CD20, CD28, CD40, CD122, CD96, CD73, CD47, GITR, CSF1R, JAK, PI3K delta, PI3K gamma, TAM, arginase, HPK1, CD137 (also known as 4-1BB) , ICOS, A2AR, B7
- the immune checkpoint molecule is a stimulatory checkpoint molecule selected from CD27, CD28, CD40, ICOS, OX40, GITR and CD137.
- the immune checkpoint molecule is an inhibitory checkpoint molecule selected from A2AR, B7-H3, B7-H4, BTLA, CTLA-4, IDO, KIR, LAG3, PD-1, TIM3, TIGIT, and VISTA.
- the compounds provided herein can be used in combination with one or more agents selected from KIR inhibitors, TIGIT inhibitors, LAIR1 inhibitors, CD160 inhibitors, 2B4 inhibitors and TGFR beta inhibitors.
- the compounds provided herein can be used in combination with one or more agonists of immune checkpoint molecules, e.g., OX40, CD27, GITR, and CD137 (also known as 4-1B) .
- immune checkpoint molecules e.g., OX40, CD27, GITR, and CD137 (also known as 4-1B) .
- the inhibitor of an immune checkpoint molecule is anti-PD1 antibody, anti-PD-L1 antibody, or anti-CTLA-4 antibody.
- the inhibitor of an immune checkpoint molecule is an inhibitor of PD-1, e.g., an anti-PD-1 monoclonal antibody.
- the anti-PD-1 monoclonal antibody is nivolumab, pembrolizumab (also known as MK-3475) , pidilizumab, SHR-1210, PDR001, MGA012, PDR001, AB122, or AMP-224.
- the anti-PD-1 monoclonal antibody is nivolumab or pembrolizumab.
- the anti-PD1 antibody is pembrolizumab.
- the anti-PD-1 monoclonal antibody is MGA012.
- the anti-PD1 antibody is SHR-1210.
- Other anti-cancer agent (s) include antibody therapeutics such as 4-1BB (e.g., urelumab, utomilumab) .
- the inhibitor of an immune checkpoint molecule is an inhibitor of PD-L1, e.g., an anti-PD-L1 monoclonal antibody.
- the anti-PD-L1 monoclonal antibody is BMS-935559, MEDI4736, MPDL3280A (also known as RG7446) , or MSB0010718C.
- the anti-PD-L1 monoclonal antibody is MPDL3280A or MEDI4736.
- the inhibitor of an immune checkpoint molecule is an inhibitor of PD-1 and PD-L1, e.g., an anti-PD-1/PD-L1 bispecific antibody.
- the anti-PD-1/PD-L1 is MCLA-136.
- the inhibitor is MCLA-145.
- the inhibitor of an immune checkpoint molecule is an inhibitor of CTLA-4, e.g., an anti-CTLA-4 antibody.
- the anti-CTLA-4 antibody is ipilimumab, tremelimumab, AGEN1884, or CP-675, 206.
- the inhibitor of an immune checkpoint molecule is an inhibitor of LAG3, e.g., an anti-LAG3 antibody.
- the anti-LAG3 antibody is BMS-986016, LAG525, or INCAGN2385.
- the inhibitor of an immune checkpoint molecule is an inhibitor of TIM3, e.g., an anti-TIM3 antibody.
- the anti-TIM3 antibody is INCAGN2390, MBG453, or TSR-022.
- the inhibitor of an immune checkpoint molecule is an inhibitor of GITR, e.g., an anti-GITR antibody.
- the anti-GITR antibody is TRX518, MK-4166, INCAGN1876, MK-1248, AMG228, BMS-986156, GWN323, or MEDI1873.
- the inhibitor of an immune checkpoint molecule is an agonist of OX40, e.g., OX40 agonist antibody or OX40L fusion protein.
- OX40 e.g., OX40 agonist antibody or OX40L fusion protein.
- the anti-OX40 antibody is MEDI0562, MOXR-0916, PF-04518600, GSK3174998, or BMS-986178.
- the OX40L fusion protein is MEDI6383.
- the inhibitor of an immune checkpoint molecule is an inhibitor of CD20, e.g., an anti-CD20 antibody.
- the anti-CD20 antibody is obinutuzumab or rituximab.
- the compounds of the present disclosure can be used in combination with bispecific antibodies.
- one of the domains of the bispecific antibody targets PD-1, PD-L1, CTLA-4, GITR, OX40, TIM3, LAG3, CD137, ICOS, CD3 or TGFb receptor.
- the compounds of the disclosure can be used in combination with one or more metabolic enzyme inhibitors.
- the metabolic enzyme inhibitor is an inhibitor of IDO1, TDO, or arginase.
- IDO1 inhibitors include epacadostat, NLG919, BMS-986205, PF-06840003, IOM2983, RG-70099 and LY338196.
- the additional compounds, inhibitors, agents, etc. can be combined with the present compound in a single or continuous dosage form, or they can be administered simultaneously or sequentially as separate dosage forms.
- kits for conveniently and effectively carrying out the methods or uses in accordance with the present invention.
- the pharmaceutical pack or kit comprises one or more containers filled with one or more of the ingredients of the pharmaceutical compositions of the invention.
- kits are especially suited for the delivery of solid oral forms such as tablets or capsules.
- a kit preferably includes a number of unit dosages, and may also include a card having the dosages oriented in the order of their intended use.
- a memory aid can be provided, for example in the form of numbers, letters, or other markings or with a calendar insert, designating the days in the treatment schedule in which the dosages can be administered.
- Optionally associated with such container (s) can be a notice in the form prescribed by a governmental agency regulating the manufacture, use or sale of pharmaceutical products, which notice reflects approval by the agency of manufacture, use or sale for human administration.
- the compounds of any one of the formulae described above may be prepared by the general and specific methods described below, using the common general knowledge of one skilled in the art of synthetic organic chemistry. Such common general knowledge can be found in standard reference books such as Comprehensive Organic Chemistry, Ed. Barton and Ollis, Elsevier; Comprehensive Organic Transformations: A Guide to Functional Group Preparations, Larock, John Wiley and Sons; and Compendium of Organic Synthetic Methods, Vol. I-XII (published by Wiley-Interscience) .
- the starting materials used herein are commercially available or may be prepared by routine methods known in the art.
- certain compounds contain primary amines or carboxylic acid functionalities which may interfere with reactions at other sites of the molecule if left unprotected. Accordingly, such functionalities may be protected by an appropriate protecting group which may be removed in a subsequent step.
- Suitable protecting groups for amine and carboxylic acid protection include those protecting groups commonly used in peptide synthesis (such as N-t-butoxycarbonyl (Boc) , benzyloxycarbonyl (Cbz) , and 9-fluorenylmethylenoxycarbonyl (Fmoc) for amines, and lower alkyl or benzyl esters for carboxylic acids) which are generally not chemically reactive under the reaction conditions described and can typically be removed without chemically altering other functionality in the any one of the formulae described above compounds.
- Solvent A 0.1%formic acid (FA) in water
- Step 1 methyl 3, 3-dimethoxycyclopentane-1-carboxylate
- Step 2 3- (3, 3-dimethoxycyclopentyl) -3-oxopropanenitrile
- Step 3 1- (tert-butyl) -3- (3, 3-dimethoxycyclopentyl) -1H-pyrazol-5-amine
- Step 4 benzyl (1- (tert-butyl) -3- (3, 3-dimethoxycyclopentyl) -1H-pyrazol-5-yl) carbamate
- Step 5 benzyl (1- (tert-butyl) -3- (3-oxocyclopentyl) -1H-pyrazol-5-yl) carbamate
- Step 6 (rac, cis) -benzyl (1- (tert-butyl) -3- (3-hydroxycyclopentyl) -1H-pyrazol-5-yl) carbamate
- Step 7 (rac, cis) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol
- Step 1 (rac, cis) -benzyl (1- (tert-butyl) -3- (3- (1, 3-dioxoisoindolin-2-yl) cyclopentyl) -1H-pyrazol-5- yl) carbamate:
- Step 2 (rac, cis) -2- (3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl) isoindoline-1, 3-dione:
- Step 1 (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol
- Step 2 1- (tert-butyl) -3- ( (1S, 3R) -3- ( (tert-butyldimethylsilyl) oxy) cyclopentyl) -1H-pyrazol- 5-amine
- Step 1 tert-butyl N- [4- [ (6-bromo-2-pyridyl) oxy] butyl] carbamate
- Step 2 (rac, cis) -tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- (3-hydroxycyclopentyl) -1H-pyrazol-5- yl) amino) pyridin-2-yl) oxy) butyl) carbamate
- Step 3 (rac, cis) -tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- (3- ( ( (4- nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2- yl) oxy) butyl) carbamate
- Step 4 [ (rac, cis) -3- [3- [ [6- (4-aminobutoxy) -2-pyridyl] amino] -1H-pyrazol-5-yl] cyclopentyl] (4- nitrophenyl) carbonate
- Step 5 (rac, cis) -2 1 H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) - cyclopentanacyclododecaphan-11-one
- Step 1 (rac, cis) -tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- (3- (1, 3-dioxoisoindolin-2-yl) cyclopentyl) -1H- pyrazol-5-yl) amino) pyridin-2-yl) oxy) butyl) carbamate
- Step 2 (rac, cis) -tert-butyl (4- ( (6- ( (3- (3-aminocyclopentyl) -1- (tert-butyl) -1H-pyrazol-5- yl) amino) pyridin-2-yl) oxy) butyl) carbamate
- Step 3 (rac, cis) -tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- (3- ( ( (4- nitrophenoxy) carbonyl) amino) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2- yl) oxy) butyl) carbamate
- Step 4 (rac, cis) - (4-nitrophenyl) N- [3- [3- [ [6- (4-aminobutoxy) -2-pyridyl] amino] -1H-pyrazol-5- yl] cyclopentyl] carbamate
- Step 5 (rac, cis) -2 1 H-5-oxa-3, 10, 12-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) - cyclopentanacyclododecaphan-11-one
- Step 1 (rac, cis) -tert-butyl (5- ( (6- ( (1- (tert-butyl) -3- (3- (1, 3-dioxoisoindolin-2-yl) cyclopentyl) -1H- pyrazol-5-yl) amino) pyridin-2-yl) oxy) pentyl) carbamate
- Step 2 (rac, cis) -tert-butyl (5- ( (6- ( (3- (3-aminocyclopentyl) -1- (tert-butyl) -1H-pyrazol-5- yl) amino) pyridin-2-yl) oxy) pentyl) carbamate
- Step 3 (rac, cis) -4-nitrophenyl (3- (5- ( (6- ( (5- ( (tert-butoxycarbonyl) amino) pentyl) oxy) pyridin-2- yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl) carbamate
- Step 4 (rac, cis) -4-nitrophenyl (3- (3- ( (6- ( (5-aminopentyl) oxy) pyridin-2-yl) amino) -1H-pyrazol-5- yl) cyclopentyl) carbamate
- Step 5 (rac, cis) -2 1 H-5-oxa-3, 11, 13-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) - cyclopentanacyclotridecaphan-12-one
- Step 1 2-bromo-6- (4- ( (tert-butyldimethylsilyl) oxy) butoxy) pyridine
- Step 2 (rac, cis) -2- ( (1R, 3S) -3- (1- (tert-butyl) -5- ( (6- (4- ( (tert- butyldimethylsilyl) oxy) butoxy) pyridin-2-yl) amino) -1H-pyrazol-3-yl) cyclopentyl) isoindoline-1, 3- dione
- Step 3 (rac, cis) -N- (3- ( (3-aminocyclopentyl) -1- (tert-butyl) -1H-pyrazol-5-yl) -6- (4- ( (tert- butyldimethylsilyl) oxy) butoxy) pyridin-2-amine
- Step 4 (rac, cis) -tert-butyl (3- (1- (tert-butyl) -5- ( (6- (4- ( (tert- butyldimethylsilyl) oxy) butoxy) pyridin-2-yl) amino) -1H-pyrazol-3-yl) cyclopentyl) carbamate
- Step 5 (rac, cis) -tert-butyl (3- (1- (tert-butyl) -5- ( (6- (4-hydroxybutoxy) pyridin-2-yl) amino) -1H- pyrazol-3-yl) cyclopentyl) carbamate
- Step 6 (rac, cis) -tert-butyl (3- (1- (tert-butyl) -5- ( (6- (4- ( ( (4- nitrophenoxy) carbonyl) oxy) butoxy) pyridin-2-yl) amino) -1H-pyrazol-3-yl) cyclopentyl) carbamate
- Step 7 (rac, cis) -4- ( (6- ( (3- (3-aminocyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) oxy) butyl (4-nitrophenyl) carbonate
- Step 8 (rac, cis) -2 1 H-5, 10-dioxa-3, 12-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) - cyclopentanacyclododecaphan-11-one
- Step 1 (rac, cis) -4- ( (6- ( (3- ( (3-aminocyclopentyl) -1- (tert-butyl) -1H-pyrazol-5-yl) amino) pyridin- 2-yl) oxy) butan-1-ol
- Step 2 (rac, cis) -tert-butyl (3- (1- (tert-butyl) -5- ( (6- (4-hydroxybutoxy) pyridin-2-yl) amino) -1H- pyrazol-3-yl) cyclopentyl) carbamate
- Step 3 (rac, cis) -4- ( (6- ( (3- (3- ( (tert-butoxycarbonyl) amino) cyclopentyl) -1- (tert-butyl) -1H- pyrazol-5-yl) amino) pyridin-2-yl) oxy) butyl 4-methylbenzenesulfonate
- Step 4 (rac, cis) -4- ( (6- ( (3- (3-aminocyclopentyl) -1- (tert-butyl) -1H-pyrazol-5-yl) amino) pyridin-2- yl) oxy) butyl 4-methylbenzenesulfonate
- Step 5 (rac, cis) -tert-butyl-21H-5-oxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) - cyclopentanacyclodecaphane
- Step 6 (rac, cis) -21- (tert-butyl) -21H-5-oxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) - cyclopentanacyclodecaphane-10-carboxamide
- Step 7 (rac, cis) -21H-5-oxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) - cyclopentanacyclodecaphane-10-carboxamide
- Step 2 tert-butyl (4- ( (6-chloro-4-phenylpyridin-2-yl) oxy) butyl) carbamate
- Step 3 tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( (tert-butyldiphenylsilyl) oxy) cyclopentyl) - 1H-pyrazol-5-yl) amino) -4-phenylpyridin-2-yl) oxy) butyl) carbamate
- Step 4 tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5- yl) amino) -4-phenylpyridin-2-yl) oxy) butyl) carbamate
- Step 5 (1R, 3S) -3- (5- ( (6- (4- ( (tert-butoxycarbonyl) amino) butoxy) -4-phenylpyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
- tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -4-phenylpyridin-2-yl) oxy) butyl) carbamate (45.0 mg, 80 ⁇ mol) in DCM (1.0 mL) were sequentially added DMAP (5.0 mg, 40 ⁇ mol) , di (imidazol-1-yl) methanone (65.0 mg, 400 ⁇ mol) and N-ethyl-N-isopropyl-propan-2-amine (70 ⁇ L, 52.0 mg, 400 ⁇ mol) at 25 °C.
- Step 6 (1R, 3S) -3- (5- ( (6- (4-aminobutoxy) -4-phenylpyridin-2-yl) amino) -1- (tert-butyl) -1H- pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
- Step 7 (1 1 S, 1 3 R, Z) -2 1 - (tert-butyl) -4 4 -phenyl-2 1 H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) - pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
- Step 8 (1 1 S, 1 3 R, Z) -4 4 -phenyl-2 1 H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) - cyclopentanacyclododecaphan-11-one
- Step 2 tert-butyl (4- ( (6-bromo-3-iodopyridin-2-yl) oxy) butyl) carbamate
- Step 3 tert-butyl (4- ( (6-bromo-3-phenylpyridin-2-yl) oxy) butyl) carbamate
- Step 4 tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5- yl) amino) -3-phenylpyridin-2-yl) oxy) butyl) carbamate
- reaction mixture was warmed up to 80 °C and stirred at that temperature for 16 h under N 2 before it was cooled to 25 °C, the mixture was diluted with water (5 mL) and extracted with EtOAc (5 mL ⁇ 3) . The combined organic layer was washed with brine (5 mL) , dried over Na 2 SO 4 , filtered and concentrated under reduced pressure.
- Step 5 (1R, 3S) -3- (5- ( (6- (4- ( (tert-butoxycarbonyl) amino) butoxy) -5-phenylpyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
- Step 6 (1R, 3S) -3- (3- ( (6- (4-aminobutoxy) -5-phenylpyridin-2-yl) amino) -1H-pyrazol-5- yl) cyclopentyl 1H-imidazole-1-carboxylate
- Step 7 (1 1 S, 1 3 R, Z) -4 5 -phenyl-2 1 H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) - cyclopentanacyclododecaphan-11-one
- Step 1 tert-butyl (4- ( ( (6-bromopyridin-2-yl) oxy) methyl) thiazol-2-yl) carbamate
- Step 2 tert-butyl (4- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5- yl) amino) pyridin-2-yl) oxy) methyl) thiazol-2-yl) carbamate
- the reaction was warmed to 90 °C and stirred at this temperature for 6 h.
- the mixture was cooled to 25 °C and was diluted with water (150 mL) , the aqueous layer was extracted with EtOAc (50 mL ⁇ 3) .
- the combined organic layer was washed with brine (50 mL) , dried over Na 2 SO 4 , filtered and concentrated.
- Step 3 (1R, 3S) -3- (5- ( (6- ( (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) methoxy) pyridin-2- yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
- Step 4 (1R, 3S) -3- (5- ( (6- ( (2-aminothiazol-4-yl) methoxy) pyridin-2-yl) amino) -1H-pyrazol-3- yl) cyclopentyl 1H-imidazole-1-carboxylate
- Step 5 (1 1 S, 1 3 R, 2 4 Z, 7 2 Z) -2 1 H-5, 10-dioxa-3, 8-diaza-7 (4, 2) -thiazola-4 (2, 6) -pyridina-2 (5, 3) - pyrazola-1 (1, 3) -cyclopentanacyclodecaphan-9-one
- Step 1 tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5- yl) amino) pyridin-2-yl) oxy) but-2-yn-1-yl) carbamate
- Step 2 tert-butyl ( (Z) -4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5- yl) amino) pyridin-2-yl) oxy) but-2-en-1-yl) carbamate
- Step 3 (1R, 3S) -3- (5- ( (6- ( ( (Z) -4- ( (tert-butoxycarbonyl) amino) but-2-en-1-yl) oxy) pyridin-2- yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
- Step 4 (1R, 3S) -3- (5- ( (6- ( ( (Z) -4-aminobut-2-en-1-yl) oxy) pyridin-2-yl) amino) -1- (tert-butyl) -1H- pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
- Step 5 (1 1 S, 1 3 R, 2 4 Z, 7Z) -2 1 - (tert-butyl) -2 1 H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) - pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-7-en-11-one
- Step 6 (1 1 S, 1 3 R, 2 4 Z, 7Z) -2 1 H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) - cyclopentanacyclododecaphan-7-en-11-one
- Step 1 4- ( (6-bromopyridin-2-yl) oxy) but-2-yn-1-ol
- Step 2 4- ( (6-bromopyridin-2-yl) oxy) but-2-yn-1-amine
- Step 3 tert-butyl (4- ( (6-bromopyridin-2-yl) oxy) but-2-yn-1-yl) carbamate
- Step 4 tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5- yl) amino) pyridin-2-yl) oxy) but-2-yn-1-yl) carbamate
- Step 5 (1R, 3S) -3- (5- ( (6- ( (4- ( (tert-butoxycarbonyl) amino) but-2-yn-1-yl) oxy) pyridin-2-yl) amino) - 1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
- Step 6 (1R, 3S) -3- (3- ( (6- ( (4-aminobut-2-yn-1-yl) oxy) pyridin-2-yl) amino) -1H-pyrazol-5- yl) cyclopentyl 1H-imidazole-1-carboxylate
- Step 7 (7S, 10R) ⁇ 11, 18 ⁇ dioxa ⁇ 2, 4, 5, 13, 23 ⁇ pentaazatetracyclo [17.3.1.1 3, 6 .1 7, 10 ] pentacosa ⁇ 1 (23) , 3, 6 (25) , 19, 21 ⁇ pentaen ⁇ 15 ⁇ yn ⁇ 12 ⁇ one
- Step 4 (3- ( ( (6-bromopyridin-2-yl) oxy) methyl) cyclobutyl) methanol
- Step 5 (3- ( ( (6-bromopyridin-2-yl) oxy) methyl) cyclobutyl) methyl methanesulfonate
- Step 6 2- ( (3- (azidomethyl) cyclobutyl) methoxy) -6-bromopyridine
- Step 7 (3- ( ( (6-bromopyridin-2-yl) oxy) methyl) cyclobutyl) methanamine
- Step 8 tert-butyl ( (3- ( ( (6-bromopyridin-2-yl) oxy) methyl) cyclobutyl) methyl) carbamate
- Step 9 tert-butyl ( (3- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5- yl) amino) pyridin-2-yl) oxy) methyl) cyclobutyl) methyl) carbamate
- Step 10 tert-butyl ( (3- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4- nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2- yl) oxy) methyl) cyclobutyl) methyl) carbamate
- Step 11 (1R, 3S) -3- (5- ( (6- ( (3- (aminomethyl) cyclobutyl) methoxy) pyridin-2-yl) amino) -1- (tert- butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate
- Step 12 (1 1 S, 1 3 R, 7 1 R, 7 3 S, Z) -2 1 - (tert-butyl) -2 1 H-5, 11-dioxa-3, 9-diaza-4 (2, 6) -pyridina-2 (3, 5) - pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacycloundecaphan-10-one and (1 1 S, 1 3 R, 7 1 S, 7 3 R, Z) -2 1 - (tert-butyl) -2 1 H-5, 11-dioxa-3, 9-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola- 1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacycloundecaphan-10-one
- Step 13 (1 1 S, 1 3 R, 7 1 R, 7 3 S, Z) -2 1 H-5, 11-dioxa-3, 9-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) - cyclopentana-7 (1, 3) -cyclobutanacycloundecaphan-10-one
- Step 14 (1 1 S, 1 3 R, 7 1 S, 7 3 R, Z) -2 1 H-5, 11-dioxa-3, 9-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) - cyclopentana-7 (1, 3) -cyclobutanacycloundecaphan-10-one
- Step 3 tert-butyl (2, 4-dimethoxybenzyl) (4-hydroxypentyl) carbamate
- Step 4 tert-butyl (4- ( (6-bromopyridin-2-yl) oxy) pentyl) (2, 4-dimethoxybenzyl) carbamate
- Step 5 tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5- yl) amino) pyridin-2-yl) oxy) pentyl) (2, 4-dimethoxybenzyl) carbamate
- Step 6 (1R, 3S) -3- (5- ( (6- ( (5- ( (tert-butoxycarbonyl) (2, 4-dimethoxybenzyl) amino) pentan-2- yl) oxy) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1- carboxylate
- Step 7 (1R, 3S) -3- (3- ( (6- ( (5-aminopentan-2-yl) oxy) pyridin-2-yl) amino) -1- (tert-butyl) -1H- pyrazol-5-yl) cyclopentyl 1H-imidazole-1-carboxylate
- Step 8 (1 1 S, 1 3 R, 6S, Z) -21- (tert-butyl) -6-methyl-2 1 H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) - pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one & (1 1 S, 1 3 R, 6R, Z) -21- (tert-butyl) -6-methyl- 2 1 H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan- 11-one
- Step 9 (1 1 S, 1 3 R, 6S, Z) -6-methyl-2 1 H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola- 1 (1, 3) -cyclopentanacyclododecaphan-11-one
- Step 10 (1 1 S, 1 3 R, 6R, Z) -6-methyl-2 1 H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola- 1 (1, 3) -cyclopentanacyclododecaphan-11-one
- Step 1 tert-butyl (4- ( (6-chloro-4-cyanopyridin-2-yl) oxy) butyl) carbamate
- Step 2 tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5- yl) amino) -4-cyanopyridin-2-yl) oxy) butyl) carbamate
- Step 3 (1R, 3S) -3- (5- ( (6- (4- ( (tert-butoxycarbonyl) amino) butoxy) -4-cyanopyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
- Step 4 (1R, 3S) -3- (5- ( (6- (4-aminobutoxy) -4-cyanopyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol- 3-yl) cyclopentyl 1H-imidazole-1-carboxylate
- Step 5 (1 1 S, 1 3 R, Z) -2 1 - (tert-butyl) -11-oxo-2 1 H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) - pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-4 4 -carbonitrile
- Step 6 (1 1 S, 1 3 R, Z) -11-oxo-2 1 H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) - cyclopentanacyclododecaphane-4 4 -carbonitrile and (1 1 S, 1 3 R, Z) -11-oxo-2 1 H-5, 12-dioxa-3, 10- diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-4 4 -carboxamide
- Step 1 tert-butyl (4- ( (6-chloro-3-cyanopyridin-2-yl) oxy) butyl) carbamate
- Step 2 tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5- yl) amino) -3-cyanopyridin-2-yl) oxy) butyl) carbamate
- the reaction was heated to 95 °C and stirred at 95 °Cunder nitrogen for 16 h.
- the mixture was diluted with water (50 mL) and extracted with EtOAc (30 mL ⁇ 3) .
- the combined organic layer was washed with brine (50 mL) , dried over Na 2 SO 4 , filtered and concentrated.
- Step 3 (1R, 3S) -3- (5- ( (6- (4- ( (tert-butoxycarbonyl) amino) butoxy) -5-cyanopyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
- Step 4 (1R, 3S) -3- (5- ( (6- (4-aminobutoxy) -5-cyanopyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol- 3-yl) cyclopentyl 1H-imidazole-1-carboxylate
- Step 5 (1 1 S, 1 3 R, Z) -2 1 - (tert-butyl) -11-oxo-2 1 H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) - pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-4 5 -carbonitrile
- Step 6 (1 1 S, 1 3 R, Z) -11-oxo-2 1 H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) - cyclopentanacyclododecaphane-4 5 -carbonitrile
- Step 1 tert-butyl N- [4- (6-bromopyrrolo [2, 3-b] pyridin-1-yl) butyl] carbamate
- Step 2 tert-butyl N- [4- [6- [ [1-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3- yl] amino] pyrrolo [2, 3-b] pyridin-1-yl] butyl] carbamate
- Step 3 [ (1R, 3S) -3- [5- [ [1- [4- (tert-butoxycarbonylamino) butyl] pyrrolo [2, 3-b] pyridin-6-yl] amino] - 2-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate
- Step 4 [ (1R, 3S) -3- [3- [ [1- (4-aminobutyl) pyrrolo [2, 3-b] pyridin-6-yl] amino] -1H-pyrazol-5- yl] cyclopentyl] imidazole-1-carboxylate
- Step 5 (1 1 S, 1 3 R, Z) -2 1 H, 4 1 H-11-oxa-3, 9-diaza-4 (6, 1) -pyrrolo [2, 3-b] pyridina-2 (5, 3) -pyrazola- 1 (1, 3) -cyclopentanacycloundecaphan-10-one
- Step 1 tert-butyl (3- ( (6-chloropyrazin-2-yl) methoxy) propyl) carbamate
- Step 2 tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5- yl) amino) pyrazin-2-yl) methoxy) propyl) carbamate
- Step 3 (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) pyrazin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
- Step 4 (1R, 3S) -3- (5- ( (6- ( (3-aminopropoxy) methyl) pyrazin-2-yl) amino) -1- (tert-butyl) -1H- pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
- Step 5 (1 1 S, 1 3 R, Z) -2 1 - (tert-butyl) -2 1 H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyrazina-2 (3, 5) -pyrazola- 1 (1, 3) -cyclopentanacyclododecaphan-11-one
- Step 6 (1 1 S, 1 3 R, Z) -2 1 H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyrazina-2 (5, 3) -pyrazola-1 (1, 3) - cyclopentanacyclododecaphan-11-one
- Step 1 tert-butyl (3- ( (2-bromothiazol-4-yl) methoxy) propyl) carbamate
- Step 2 tert-butyl (3- ( (2- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5- yl) amino) thiazol-4-yl) methoxy) propyl) carbamate
- Step 3 (1R, 3S) -3- (5- ( (4- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) thiazol-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
- Step 4 (1R, 3S) -3- (5- ( (4- ( (3-aminopropoxy) methyl) thiazol-2-yl) amino) -1- (tert-butyl) -1H-pyrazol- 3-yl) cyclopentyl 1H-imidazole-1-carboxylate
- Step 5 (1 1 S, 1 3 R, 2 4 Z, 4 2 Z) -2 1 - (tert-butyl) -2 1 H-6, 12-dioxa-3, 10-diaza-4 (2, 4) -thiazola-2 (3, 5) - pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
- Step 6 (1 1 S, 1 3 R, 2 4 Z, 4 2 Z) -2 1 H-6, 12-dioxa-3, 10-diaza-4 (2, 4) -thiazola-2 (5, 3) -pyrazola-1 (1, 3) - cyclopentanacyclododecaphan-11-one
- Step 3 tert-butyl (3- ( (5-fluoro-6-oxo-1, 6-dihydropyrimidin-2-yl) methoxy) propyl) carbamate
- Step 4 tert-butyl (3- ( (4-chloro-5-fluoropyrimidin-2-yl) methoxy) propyl) carbamate
- Step 5 tert-butyl (3- ( (4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5- yl) amino) -5-fluoropyrimidin-2-yl) methoxy) propyl) carbamate
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present disclosure provides a compound represented by structural formula (I') or a pharmaceutically acceptable salt, or a stereoisomer thereof and their use in, e.g. treating a disease or disorder associated with CDK2. This disclosure also features compositions containing the same as well as methods of using and making the same.
Description
CROSS-REFERENCE TO RELATED APPLICATIONS
This application claims the benefit of priority to International Patent Application No. PCT/CN2021/084358, filed on March 31, 2021. The entire contents of the aforementioned application are incorporated herein by reference.
Cyclin-Dependent Kinases (CDKs) are a family of protein kinases first discovered for their roles in regulating cell cycle. They have since been identified to play roles in regulating a number of other biological functions such as transcription, mRNA processing, and the differentiation of nerve cells.
CDKs are relatively small proteins with molecular weights between about 34-40 kDa. They contain little more than the kinase domain, and are essentially inactive when not in complex with a class of regulatory proteins called cyclins. CDK levels remain relatively constant throughout the cell cycle, and most regulation is post-translational, most prominently by binding to cyclins.
CDK2 is of particular interest because deregulation of CDK2 activity occurs frequently in a variety of human cancers. CDK2 plays a crucial role in promoting G1/S transition and S phase progression. In complex with cyclin E (CCNE) , CDK2 phosphorylates retinoblastoma pocket protein family members (p107, p130, pRb) , leading to de-repression of E2F transcription factors, expression of G1/S transition related genes and transition from G1 to S phase (Henley, S.A. and F.A. Dick, Cell Div, 2012, 7 (1) : 10) . This in turn enables activation of CDK2/cyclin A, which phosphorylates endogenous substrates that permit DNA synthesis, replication and centrosome duplication (Ekholm, S.V. and S.I. Reed, Curr Opin Cell Biol, 2000, 12 (6) : 676-84) . It has been reported that the CDK2 pathway influences tumorigenesis mainly through amplification and/or overexpression of CCNE1 and mutations that inactivate CDK2 endogenous inhibitors (e.g., p27) , respectively (Xu, X., et al., Biochemistry, 1999, 38 (27) : 8713-22) .
CCNE1 copy-number gain and overexpression have been identified in ovarian, gastric, endometrial, breast and other cancers and been associated with poor outcomes in these tumors (Keyomarsi, K., et al., N Engl J Med, 2002, 347 (20) : 1566-75; Nakayama, N., et al., Cancer, 2010, 116 (11) : 2621-34; Au-Yeung, G., et al., Clin Cancer Res, 2017, 23 (7) : 1862-1874; Rosen, D.G., et al., Cancer, 2006, 106 (9) : 1925-32) . Amplification and/or overexpression of CCNE1 also reportedly contribute to trastuzumab resistance in HER2+ breast cancer and resistance to CDK4/6 inhibitors in estrogen receptor-positive breast cancer (Scaltriti, M., et al., Proc Natl Acad Sci USA, 2011, 108 (9) : 3761-6; Herrera-Abreu, M. T., et al., Cancer Res, 2016, 76 (8) : 2301-13) . Various approaches targeting CDK2 have been shown to induce cell cycle arrest and tumor growth inhibition (Chen, Y N., et al., Proc Natl Acad Sci USA, 1999, 96 (8) : 4325-9; Mendoza, N., et al., Cancer Res, 2003, 63 (5) : 1020-4) . Inhibition of CDK2 also reportedly restores sensitivity to trastuzumab treatment in resistant HER2+ breast tumors in a preclinical model (Scaltriti, supra) .
These data provide a rationale for considering CDK2 as a potential target for new drug development in cancer associated with deregulated CDK2 activity. In the last decade there has been increasing interest in the development of CDK selective inhibitors. Despite significant efforts, there are no approved agents targeting CDK2 to date (Cicenas, J., et al., Cancers (Basel) , 2014, 6 (4) : 2224-42) .
Identifying selective CDK2 inhibitors is very difficult, partly due to the extreme similarity between the active sites of CDK2 and other CDKs, especially CDK1, which is the only essential CDK in the cell cycle. Inhibition of CDK1 could lead to many unintended side effects.
Therefore, it remains a need to discover CDK inhibitors having novel activity profiles, in particular those specifically or selectively targeting CDK2.
SUMMARY
Described herein are compounds of Formula (I') or (I) that inhibit (e.g., selectively inhibit) the activity of CDK2, and pharmaceutically acceptable salts, or stereoisomers thereof.
In one aspect, the present disclosure provides a compound of Formula (I') , a pharmaceutically acceptable salt, or a stereoisomer thereof:

wherein ring A, linkers 1 and 2, X, and R
4 are as defined herein.
In another aspect, the present disclosure provides a compound of Formula (I) , a pharmaceutically acceptable salt, or a stereoisomer thereof:

wherein ring A, linker, X, Y, and R
4 are as defined herein.
Also provided are pharmaceutical compositions comprising a compound of Formula (I') or (I) , a pharmaceutically acceptable salt, or a stereoisomer thereof and a pharmaceutically acceptable carrier or excipient.
The present disclsoure further provides methods of inhibiting CDK2 in a patient, comprising administering to the patient a compound of Formula (I') or (I) , or a pharmaceutically acceptable salt, or a stereoisomer thereof.
The present disclsoure also provides methods of treating a disease or or condition modulated at least in part by CDK2 in a subject, comprising administering to the subject a therapeutically effective amount of a compound of Formula (I') or (I) , a pharmaceutically acceptable salt, or a stereoisomer thereof.
The present disclosure further provides a method of treating cancer in a patient in need thereof, comprising administering to the patient an effective amount of (1) a compound of Formula (I') or (I) , a pharmaceutically acceptable salt, or a stereoisomer thereof; or (2) a pharmaceutically acceptable composition comprising a compound of Formula (I') or (I) , a pharmaceutically acceptable salt, or a stereoisomer thereof, and a pharmaceutically acceptable carrier. In certain embodiments, the cancer is treatable by inhibiting (e.g., selectively inhibiting) CDK2, such as a cancer selected from the group consisting of: ovarian cancer, breast cancer (such as hormone receptor positive, HER2/neu negative advanced or metastatic breast cancer, HER2 positive breast cancer and triple negative breast cancer ) , lung cancer, endometrial cancer, neuroblastoma, gastric cancer, colorectal cancer, prostate cancer, glioblastoma, melanoma, mantel cell lymphoma, chronic myeloid leukemia and acute myeloid leukemia. In certain embodiments, the cancer exhibits abnormally up-regulated CCNE1 /Cyclin E activity, through overexpression of Cyclin Eor duplication of the Cyclin E-coding CCNE1 gene. In certain embodiments, the cancer exhibits abnormally up-regulated Cyclin A2 activity.
In certain embodiments of the methods of the invention, the cancer can be treated by inhibiting (e.g., selectively inhibiting) the activity of CDK2.
In certain embodiments of the methods of the invention, the compounds of the invention are administered with any one of a second therapeutic agent as described herein that also treats the same cancer.
The present disclosure also provides a use of a compound of Formula (I') or (I) , a pharmaceutically acceptable salt, or a stereoisomer thereof or a pharmaceutical composition comprising the same in any of the methods described herein. In one embodiment, provided is a compound of Formula (I') or (I) or a pharmaceutically acceptable salt or a stereoisomer thereof or a pharmaceutical composition comprising the same for use in any of the methods described herein. In another embodiment, provided is use of a compound of Formula (I') or (I) or a pharmaceutically acceptable salt or a stereoisomer thereof or a pharmaceutical composition comprising the same for the manufacture of a medicament for any of the methods described herein.
1. Compounds
In a first embodiment, the present disclosure provides a compound represented by Formula (I') :

a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein:
ring A is 6-10 membered aryl or 5-10 memberaed heteroaryl; wherein said 6-10 membered aryl or 5-10 membered heteroaryl represented by ring A is optionally substituted by one or more R
1;
wherein
R
1 is halogen, -CN, C
1-6alkyl, C
1-6haloalkyl, C
2-6alkenyl, C
2-6alknyl, C
1-6alkyleneamine, C
1-6alkylenehydroxyl, -C (O) R
1a, -C (O) OR
1a, -C (O) NR
1aR
1b, -OR
1a, -SR
1a, -NR
1aR
1b, -NR
1aC (O) R
1b, -NR
1aC (O) OR
1b, -NR
1aSO
2R
1b, -NR
1aSO
2NR
1bR
1c, -SO
2R
1a, -SO
2NR
1aR
1b, or -P (O) R
1aR
1b, 3-6 membered carbocyclyl, 4-8 membered heterocyclyl, 6-10 membered aryl, 5-10 membered heteroaryl; wherein said C
1-6alkyleneamine, C
1-6alkylenehydroxyl, 3-6 membered carbocyclyl, 4-8 membered heterocyclyl, 6-10 membered aryl, or 5-10 membered heteroaryl represented by R
1 is optionally substituted by one or more R
10, wherein
R
10, in each occurrence, is independently selected from the group consisting of halogen, -CN, C
1-6alkyl, C
1-6haloalkyl, C
1-6alkoxyl, and 4-6 membered heterocyclyl;
R
1a, R
1b, and R
1c are independently selected from the group consisting of hydrogen, C
1-6alkyl, C
1-6haloalkyl, C
2-6alkenyl, C
2-6alkynyl, 3-6 membered carbocyclyl, and -C
0-6alkyl-4-6 membered heterocyclyl;
X is absent, ^- (CH
2)
0-1-O-^^, -NR
2-, ^- (CH
2)
0-1-C (O) -^^, ^- (CH
2)
0-1-NR
2C (O) -^^, ^- (CH
2)
0-1-C (O) NR
2-^^, ^-SO
2-^^, or ^-C (R
2) =N-O-^^; wherein
^-represents the point which attaches to ring A; -^^ represents the point which attaches to
 and
and
R
2, in each occurrence, is independently hydrogen, C
1-6alkyl, or C
1-6haloalkyl;
*-W-L-Z-**;
wherein *-represents the point which attaches to X; -**represents the point which attaches to

W is absent, C
1-6alkylene, C
2-6alkenylene, or C
2-6alkynylene; wherein said C
1-
6alkylene, C
2-6alkenylene, or C
2-6alkynylene represented by W is optionally substituted by one or more R
3;
L is absent, -O-, -NH-, 3-12 membered carbocyclyl, 3-12 membered heterocyclyl, 6-10 membered aryl, or 5-10 membered heteroaryl; wherein said -NH-, 3-12 membered carbocyclyl, 3-12 membered heterocyclyl, 6-10 membered aryl, or 5-10 membered heteroaryl represented by L is optionally substituted by one or more R
3; and
Z is absent, C
1-6alkylene, C
2-6alkenylene, or C
2-6alkynylene; wherein said C
1-
6alkylene, C
2-6alkenylene, or C
2-6alkynylene represented by Z is optionally substituted by one or more R
3; wherein
R
3, in each occurrence, is independently halogen, CN, C
1-6alkyl, C
1-6haloalkyl, -OR
3a, -C (=O) R
3a, or -NR
3aR
3b; wherein
R
3a and R
3b are independently selected from the group consisting of hydrogen, C
1-6alkyl, C
1-6haloalkyl, C
2-6alkenyl, C
2-6alkynyl, 3-6 membered carbocyclyl, and 4-6 membered heterocyclyl;
wherein W, L, and Z are not absent simultaneously;

wherein @-represents the point which attaches to the cyclopentyl shown in Formula (I’) ; -@@represents the point which attaches to

when U is connected with
 Y is –O-or -NR
22-; and U is –O-or -NR
4-;
Y is –O-or -NR
22-; and U is –O-or -NR
4-;
wherein
R
22 is hydrogen, C
1-6alkyl, or C
1-6haloalkyl; and
R
4 is hydrogen or C
1-6alkyl; or
R
4 and one R
3 attached to Z together with the atoms to which they are attached form 4-8 membered heterocyclyl;
when Y is connected with
 Y is N; and U is –N (R
5)
2-; each R
5 is independently hydrogen or C
1-6alkyl; and
Y is N; and U is –N (R
5)
2-; each R
5 is independently hydrogen or C
1-6alkyl; and
V is O or S.
In a second embodiment, the present disclosure provides a compound according to the first embodiment, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein the compound is represented by Formula II':

The definitions of the variables are provided in the first embodiment.
In a third embodiment, the present disclosure provides a compound according to the first or second embodiment, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein the compound is represented by Formula (II'A ) or (II'B) :

The definitions of the remaining variables are provided in the first or second embodiment.
In a fourth embodiment, the present disclosure provides a compound according to any one of the first through third embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
ring A is phenyl or nitrogen containing 5-10 membered heteroaryl; wherein said phenyl or nitrogen containing 5-10 membered heteroaryl represented by ring A is optionally substituted by one to three R
1; wherein
R
1 is halogen, -CN, C
1-4alkyl, C
1-4haloalkyl, C
1-4alkyleneamine, C
1-4alkylenehydroxyl, -C (O) NR
1aR
1b, -OR
1a, -SR
1a, -SO
2R
1a, 4-6 membered carbocyclyl, 4-6 membered heterocyclyl, phenyl, or 5-6 membered heteroaryl; wherein said C
1-4alkyleneamine, C
1-4alkylenehydroxyl, 4-6 membered carbocyclyl, 4-6 membered heterocyclyl, phenyl, or 5-6 membered heteroaryl represented by R
1 is optionally substituted by one to three R
10; wherein
R
10, in each occurrence, is independently selected from the group consisting of halogen, -CN, C
1-4alkyl, C
1-4haloalkyl, C
1-4alkoxyl, and 5-6 membered heterocyclyl; and
R
1a and R
1b are independently selected from the group consisting of hydrogen, C
1-4alkyl, C
1-4haloalkyl, C
2-4alkenyl, C
2-4alkynyl, and -C
0-3alkyl-4-6 membered heterocyclyl.
The definitions of the remaining variables are provided in the first through third embodiments.
In a fifth embodiment, the present disclosure provides a compound of any one of the first through fourth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
ring A is phenyl, nitrogen containing 5-6 membered heteroaryl, or nitrogen containing 9 membered bicyclic heteroaryl; wherein said phenyl, nitrogen containing 5-6 membered heteroaryl, or nitrogen containing 9 membered bicyclic heteroaryl represented by ring A is optionally substituted by one to two R
1; wherein
R
1 is halogen, -CN, C
1-3alkyl, C
1-2haloalkyl, C
1-2alkyleneamine, C
1-2alkylenehydroxyl, -C (O) NR
1aR
1b, -OR
1a, -SR
1a, -SO
2R
1a, 5-6 membered carbocyclyl, 5-6 membered heterocyclyl, phenyl, or 5-6 membered heteroaryl; wherein said C
1-2alkyleneamine, C
1-2alkylenehydroxyl, 5-6 membered carbocyclyl, 5-6 membered heterocyclyl, phenyl, or 5-6 membered heteroaryl represented by R
1 is optionally substituted by one to three R
10; wherein
R
10, in each occurrence, is independently selected from the group consisting of F, Cl, -CN, C
1-2alkyl, C
1-2haloalkyl, C
1-3alkoxyl, and 5-6 membered heterocyclyl; and
R
1a and R
1b are independently selected from the group consisting of hydrogen, C
1-2alkyl, C
1-2haloalkyl, and -C
0-2alkyl-5 membered heterocyclyl.
The definitions of the remaining variables are provided in the first through fourth embodiments.
In a sixth embodiment, the present disclosure provides a compound of any one of first through fifth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein ring A is selected from a group consisting of phenyl, pyridyl, pyrimidinyl, pyrazinyl, thiophenyl, thiazolyl, pyrrolopyridinyl, pyrazolopyrimidinyl, pyrazolopyrazinyl, thienopyridinyl, and imidazo [4, 5-c] pyridinyl; wherein said phenyl, pyridyl, pyrimidinyl, pyrazinyl, thiophenyl, thiazolyl, pyrrolopyridinyl, pyrazolopyrimidinyl, pyrazolopyrazinyl, thienopyridinyl, and imidazo [4, 5-c] pyridinyl is optionally substituted by one to two R
1; wherein
R
1 is F, Cl, Br, -CN, -CH
3, -CH (CH
3)
2, -CF
3, -OR
1a, -SR
1a, -SO
2R
1a, -C (O) NR
1aR
1b, -CH
2NH
2, -CH
2OH, cyclopentanyl, cyclohexanyl, cyclohexenyl, piperidinyl, tetrahydropyridinyl, 3, 6-dihydro-2H-thiopyranyl, thiophenyl, pyrazolyl, phenyl, or pyridyl; wherein said -CH
2NH
2, -CH
2OH, cyclopentanyl, cyclohexanyl, cyclohexenyl, piperidinyl, tetrahydropyridinyl, 3, 6-dihydro-2H-thiopyranyl, thiophenyl, pyrazolyl, phenyl, or pyridyl is optionally substituted by one to three R
10; wherein
R
10, in each occurrence, is independently selected from the group consisting of F, Cl, -CN, -CH
3, -CF
3, -CH
2CF
3, -OCH
2CH
3, -OCH (CH
3)
2, and morpholinyl; and
R
1a and R
1b are independently selected from the group consisting of hydrogen, -CH
3, -OCHF
2, tetrahydrofuranyl, and - (CH
2)
2-pyrrolidinyl.
The definitions of the remaining variables are provided in the first through fifth embodiments.
In a seventh embodiment, the present disclosure provides a compound of any one of the first through sixth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein ring A is selected from the group consisting of

wherein
each of them is optionally substituted by one to two R
1; and
The definitions of the remaining variables are provided in the first through the sixth embodiments.
In an eighth embodiment, the present disclosure provides a compound of any one of the first through the seventh embodiments, a pharmaceutically acceptable salt thereof, or a stereoisomer thereof, wherein ring A is pyridyl; wherein said pyridyl is optionally substituted by one R
1. The definitions of the remaining variables are provided in the first through the seventh embodiments.
In a ninth embodiment, the present disclosure provides a compound of any one of the first through the eighth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
X is absent, -O-, ^-CH
2-O-^^, ^-CH
2-C (O) -^^, ^-NR
2C (O) -^^, ^-CH
2-NR
2C (O) -^^, ^-C (O) NR
2-^^, ^-CH
2-C (O) NR
2-^^, ^-SO
2-^^, or ^-C (R
2) =N-O-^^; wherein
^-represents the point which attaches to ring A; -^^ represents the point which attaches to

R
2, in each occurrence, is independently hydrogen, C
1-3alkyl, or C
1-3haloalkyl.
The definitions of the remaining variables are provided in the first through the eighth embodiments.
In a tenth embodiment, the present disclosure provides a compound of any one of the first through the ninth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
X is absent, -O-, ^-CH
2-O-^^, ^-CH
2-C (O) -^^, ^-NHC (O) -^^, ^-CH
2-NHC (O) -^^, ^-C (O) NH-^^, ^-CH
2-C (O) NH-^^, ^-SO
2-^^, or ^-C (R
2) =N-O-^^; wherein
^-represents the point which attaches to ring A; -^^ represents the point which attaches to
 and
and
R
2, in each occurrence, is independently hydrogen, -CH
3, -CF
3, or isopropyl.
The definitions of the remaining variables are provided in the first through the ninth embodiments.
In an eleventh embodiment, the present disclosure provides a compound of any one of the first through the tenth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein X is absent or ^-CH
2-O-^^; wherein
^-represents the point which attaches to ring A; -^^ represents the point which attaches to

The definitions of the remaining variables are provided in the first through the tenth embodiments.
In a twelfth embodiment, the present disclosure provides a compound of any one of the first through the eleventh embodiments, or a pharmaceutically acceptable salt or a stereoisomer thereof, wherein
W is absent, C
1-4alkylene, C
2-4alkenylene, or C
2-4alkynylene; wherein said C
1-4alkylene, C
2-4alkenylene, or C
2-4alkynylene represented by W is optionally substituted by one to three R
3;
L is absent, -O-, -NH-, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 6-10 membered aryl, or 5-10 membered heteroaryl; wherein said -NH-, 3-8 membered carbocyclyl, 3-6 membered heterocyclyl, 6-10 membered aryl, and 5-10 membered heteroaryl represented by L is optionally substituted by one to three R
3; and
Z is absent, C
1-4alkylene, C
2-4alkenylene, or C
2-4alkynylene; wherein said C
1-4alkylene, C
2-4alkenylene, or C
2-4alkynylene represented by Z is optionally substituted by one to three R
3; wherein
R
3, in each occurrence, is independently halogen, C
1-4alkyl, C
1-4haloalkyl, or-C (=O) R
3a; wherein
R
3a is hydrogen or C
1-4alkyl.
The definitions of the remaining variables are provided in the first through the eleventh embodiments.
In a thirteenth embodiment, the present disclosure provides a compound of any one of the first through the twelfth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
W is absent, C
1-3alkylene, C
2-3alkenylene, or C
2-3alkynylene; wherein said C
1-3alkylene, C
2-3alkenylene, or C
2-3alkynylene represented by W is optionally substituted by one to two R
3;
L is absent, -O-, -NH-, 3-6 membered monocyclic carbocyclyl, 6-8 membered bicyclic carbocyclyl; 4-6 membered monocyclic heterocyclyl, phenyl, and 5-6 membered monocyclic heteroaryl; wherein said 3-6 membered monocyclic carbocyclyl, 6-8 membered bicyclic carbocyclyl; 4-6 membered monocyclic heterocyclyl, phenyl, and 5-6 membered monocyclic heteroaryl represented by L is optionally substituted by one R
3; and
Z is absent, C
1-3alkylene, C
2-3alkenylene, or C
2-3alkynylene; wherein said C
1-3alkylene, C
2-3alkenylene, or C
2-3alkynylene represented by Z is optionally substituted by one to two R
3; wherein
R
3, in each occurrence, is independently halogen, C
1-3alkyl, C
1-2haloalkyl, or -C (=O) CH
3.
The definitions of the remaining variables are provided in the first through the twelfth embodiments.
In a fourteenth embodiment, the present disclosure provides a compound of any one of the first through the thirteenth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
W is C
1-3alkylene, C
1-3haloalkylene, C
2-3alkenylene, or C
2-3alkynylene;
L is absent, -NH-or -O-; and
Z is C
1-3alkylene, C
1-3haloalkylene, C
2-3alkenylene, or C
2-3alkynylene.
The definitions of the remaining variables are provided in the first through the thirteenth embodiments.
In a fifteenth embodiment, the present disclosure provides a compound of any one of the first through the fourteenth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
W is C
3-4alkylene, C
3-4haloalkylene, C
3-4alkenylene, or C
3-4alkynylene;
L is absent; and
Z is absent.
The definitions of the remaining variables are provided in the first through the fourteenth embodiments.
In a sixteenth embodiment, the present disclosure provides a compound of any one of the first through the fifteenth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
W is absent, C
1-2alkylene, or C
1-2haloalkylene;
L is 4-6 membered carbocyclyl, 4-6 membered heterocyclyl, or 5-6 membered heteroaryl; and
Z is absent or methylene.
The definitions of the remaining variables are provided in the first through the fifteenth embodiments.
In a seventeenth embodiment, the present disclosure provides a compound of any one of the first through the sixteenth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
W is absent, -CH
2-, -CH
2CH
2-, -CH
2CH
2CH
2-, -CH=CH-, -CH
2CH=CH-, or -CH
2C≡C-; wherein said -CH
2-, -CH
2CH
2-, -CH
2CH
2CH
2-, -CH=CH-, -CH
2CH=CH-, or -CH
2C≡C-represented by W is optionally substituted by one to two R
3
L is absent, -O-, -NH-, cyclobutyl, cyclohexyl, spiro [3.3] heptanyl, azetidinyl, piperazinyl, piperidinyl, oxazolyl, isoxazolyl, imidazolyl, triazolyl, or thiozolyl; wherein said -NH-, cyclobutyl, cyclohexyl, spiro [3.3] heptanyl, azetidinyl, piperazinyl, piperidinyl, oxazolyl, isoxazolyl, imidazolyl, triazolyl, or thiozolyl is optionally substituted by one R
3;
Z is absent, -CH
2-, -CH
2CH
2-, -CH
2CH
2CH
2-, -CH=CH-, -CH=CHCH
2-, or -C≡CCH-;
wherein
R
3 is F, -CH
3, -CF
3, -CH
2CH
3, -CH (CH
3)
2, -CH
2CF
3, or –C (=O) CH
3.
The definitions of the remaining variables are provided in the first through the sixteenth embodiments.
In an eighteenth embodiment, the present disclosure provides a compound of any one of the first through the thirteenth and seventeenth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein *-W-L-Z-**is selected from the group consisting of

wherein each of them is optionally substituted by one to two R
3; wherein
*-represents the point which attaches to X; -**represents the point which attaches to

The definitions of the remaining variables are provided in the first through the thirteenth and seventeenth embodiments.
In a nineteenth embodiment, the present disclosure provides a compound of any one of the first through the thirteenth, seventeenth, and eighteenth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein *-W-L-Z-**is selected from the group consisting of
 wherein
wherein
*-represents the point which attaches to X; -**represents the point which attaches to

The definitions of the remaining variables are provided in the first through the thirteenth, seventeenth, and eighteenth embodiments.
In a twentieth embodiment, the present disclosure provides a compound of any one of the first through the nineteenth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
 is represented by Formula A, B, C, or D,
is represented by Formula A, B, C, or D,

wherein
@-represents the point which attaches to the cyclopentyl shown in Formula (I’) ; -@@represents the point which attaches to

R
22 is hydrogen, C
1-4alkyl, or C
1-4haloalkyl; and
R
4 is hydrogen or C
1-4alkyl; or R
4 and one R
3 attached to Z, together with the atoms to which they are attached, form 4-6 membered heterocyclyl.
The definitions of the remaining variables are provided in the first through the nineteenth embodiments.
In a twenty-first embodiment, the present disclosure provides a compound of the twentieth embodiment, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
 is
is

wherein
@-represents the point which attaches to the cyclopentyl shown in Formula (I’ ) ;
-@@represents the point which attaches to
 and
and
R
4 is hydrogen or -CH
3; or R
4 and one R
3 attached to Z together with the atoms to which they are attached, form azetidinyl or pyrrolidinyl.
The definitions of the remaining variables are provided in the twentieth embodiment.
In a twenty-second embodiment, the present disclosure provides a compound of the twenty-first embodiment, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein R
4 is hydrogen. The definitions of the remaining variables are provided in the twenty-first embodiments.
In a twenty-third embodiment, the present disclosure provides a compound of any one of the first through the nineteenth embodiments, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
 is represented by Formula E:
is represented by Formula E:

wherein
@-represents the point which attaches to the cyclopentyl shown in Formula (I’) ;
-@@represents the point which attaches to
 and
and
R
5 is hydrogen or isopropyl.
In one embodiment, the present disclosure provides a compound selected from the compounds disclosed in examples and Table 1, a pharmaceutically acceptable salt or a stereoisomer thereof.
Table 1













The present disclosure also provides other embodiments described in the text below.
(1) . A compound of formula I:

a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein:
ring A is 6-10 membered aryl or 5-10 membered heteroaryl; wherein said 6-10 membered aryl or 5-10 membered heteroaryl represented by ring A is optionally substituted by one or more R
1; wherein
R
1 is halogen, -CN, C
1-6alkyl, C
1-6haloalkyl, C
2-6alkenyl, C
2-6alknyl, -C (O) R
1a, -C (O) OR
1a, -C (O) NR
1aR
1b, -OR
1a, -NR
1aR
1b, -NR
1aC (O) R
1b, -NR
1aC (O) OR
1b, -NR
1aSO
2R
1b, -NR
1aSO
2NR
1bR
1c, -SO
2R
1a, -SO
2NR
1aR
1b, or -P (O) R
1aR
1b; wherein
R
1a, R
1b, and R
1c are independently selected from the group consisting of hydrogen, C
1-6alkyl, C
1-6haloalkyl, C
2-6alkenyl, C
2-6alkynyl, 3-6 membered carbocyclyl, and 4-6 membered heterocyclyl;
X is absent, -O-, -NR
2-, -C (O) -, -NR
2C (O) -, or -C (O) NR
2-; wherein
R
2, in each occurrence, is independently hydrogen, C
1-6alkyl, or C
1-6haloalkyl;
Y is –O-or NR
22-; wherein
R
22 is hydrogen, C
1-6alkyl, or C
1-6haloalkyl;
*-W-L-Z-**;
wherein *-represents the point which attaches to the variable X; -**represents the point which attaches to the moiety -NR
4-;
W is absent, C
1-6alkylene, C
2-6alkenylene, or C
2-6alkynylene; wherein said C
1-6alkylene, C
2-6alkenylene, or C
2-6alkynylene represented by W is optionally substituted by one or more R
3;
L is absent, -O-, -NH-, 3-12 membered carbocyclyl, 3-12 membered heterocyclyl, 6-10 membered aryl, or 5-10 membered heteroaryl; wherein said -NH-, 3-12 membered carbocyclyl, 3-12 membered heterocyclyl, 6-10 membered aryl, or 5-10 membered heteroaryl represented by L is optionally substituted by one or more R
3; and
Z is absent, C
1-6alkylene, C
2-6alkenylene, or C
2-6alkynylene; wherein said C
1-6alkylene, C
2-6alkenylene, or C
2-6alkynylene represented by Z is optionally substituted by one or more R
3; wherein
R
3, in each occurrence, is independently halogen, CN, C
1-6alkyl, -OR
3a, or -NR
3aR
3b; wherein
R
3a and R
3b are independently selected from the group consisting of hydrogen, C
1-6alkyl, C
1-6haloalkyl, C
2-6alkenyl, C
2-6alkynyl, 3-6 membered carbocyclyl, and 4-6 membered heterocyclyl;
R
4 is hydrogen or C
1-6alkyl; or one R
3 and R
4 together with the atoms to which they are attached form 4-8 membered heterocyclyl;
wherein W, L, and Z are not absent simultaneously; and
wherein said heterocyclyl comprises 1-3 heteroatoms selected from oxygen, nitrogen, and sulfur; and said heteroaryl comprises 1-4 heteroatoms selected from oxygen, nitrogen, and sulfur.
(2) . The compound of embodiment (1) above, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein the compound is represented by Formula II:

(3) . The compound of embodiment (1) or (2) above, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
ring A is phenyl or nitrogen containing 5-10 membered heteroaryl; wherein said phenyl or nitrogen containing 5-10 membered heteroaryl represented by ring A is optionally substituted by one to three R
1; wherein
R
1 is halogen, -CN, C
1-4alkyl, C
1-4haloalkyl, C
2-4alkenyl, C
2-4alkynyl, -C (O) R
1a, -C (O) OR
1a, -C (O) NR
1aR
1b, -OR
1a, -NR
1aR
1b, -NR
1aC (O) R
1b, -NR
1aC (O) OR
1b, -NR
1aSO
2R
1b, -NR
1aSO
2NR
1bR
1c, -SO
2R
1a, -SO
2NR
1aR
1b, or -P (O) R
1aR
1b; wherein
R
1a, R
1b, and R
1c are independently selected from the group consisting of hydrogen, C
1-4alkyl, C
1-4haloalkyl, C
2-4alkenyl, C
2-4alkynyl, 4-6 membered cycloalkyl, and 4-6 membered heterocyclyl.
(4) . The compound of any one of claims 1-3, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
ring A is phenyl or nitrogen containing 5-6 membered heteroaryl; wherein said phenyl or nitrogen containing 5-6 membered heteroaryl represented by ring A is optionally substituted by one to two R
1;
R
1 is halogen, -CN, C
1-4alkyl, C
1-4haloalkyl, -OR
1a, or -NR
1aR
1b; wherein
R
1a or R
1b are independently selected from the group consisting of hydrogen, C
1-4alkyl, C
1-4haloalkyl, C
2-4alkenyl, and C
2-4alkynyl.
(5) . The compound of any one of embodiments (1) to (4) above, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein ring A is selected from a group consisting of phenyl, pyridyl, pyrimidinyl, pyrazinyl, thiophenyl, thiazolyl, and imidazo [4, 5-c] pyridinyl; wherein said phenyl, pyridyl, pyrimidinyl, pyrazinyl, thiophenyl, thiazolyl, or imidazo [4, 5-c] pyridinyl is optionally substituted by one to two R
1; wherein R
1 is halogen, -CN, C
1-4alkyl, C
1-4haloalkyl, or -OC
1-4alkyl.
(6) . The compound of any one of embodiments (1) , (2) , or (5) above, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein ring A is selected from a group consisting of


wherein
 represents the point which attaches to the moiety –NH-;
represents the point which attaches to the moiety –NH-;
 represents the point which attaches to variable X.
represents the point which attaches to variable X.
(7) . The compound of any one of embodiments (1) - (5) above, a pharmaceutically acceptable salt thereof, or a stereoisomer thereof, wherein ring A is
 optionally substituted by one to two R
1, wherein R
1 is F or -OCH
3; wherein
optionally substituted by one to two R
1, wherein R
1 is F or -OCH
3; wherein
 represents the point which attaches to the moiety –NH-;
represents the point which attaches to the moiety –NH-;
 represents the point which attaches to variable X.
represents the point which attaches to variable X.
(8) . The compound of any one of embodiments (1) - (7) above, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
X is absent, -O-, -NR
2-, -C (O) -, -NHC (O) -, or -C (O) NH-; wherein R
2 in each occurrence, is independently hydrogen, C
1-4alkyl, or C
1-4haloalkyl; and
Y is -O-or NR
22-; wherein R
22 is hydrogen, C
1-4alkyl, or C
1-4haloalkyl.
(9) . The compound of any one of embodiments (1) - (8) above, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein X is -O-or -NH-and Y is -O-or -NH-.
(10) . The compound of any one of embodiments (1) - (9) above, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein X is -O-and Y is O.
(11) . The compound of any one of embodiments (1) - (10) above, or a pharmaceutically acceptable salt or a stereoisomer thereof, wherein
W is absent, C
1-4alkylene, C
2-4alkenylene, or C
2-4alkynylene; wherein said C
1-4alkylene, C
2-4alkenylene, or C
2-4alkynylene represented by W is optionally substituted by one to three R
3;
L is absent, -O-, -NH-, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 6-10 membered aryl, or 5-10 membered heteroaryl; wherein said -NH-, 3-8 membered carbocyclyl, 3-6 membered heterocyclyl, 6-10 membered aryl, and 5-10 membered heteroaryl represented by L is optionally substituted by one to three R
3; and
Z is absent, C
1-4alkylene, C
2-4alkenylene, or C
2-4alkynylene; wherein said C
1-4alkylene, C
2-4alkenylene, or C
2-4alkynylene represented by Z is optionally substituted by one to three R
3; wherein R
3, in each occurrence, is independently halogen or C
1-4alkyl; and
R
4 is hydrogen, C
1-4alkyl, or C
1-4haloalkyl; or one R
3 and R
4 together with the atoms to which they are attached form 5-6 membered heterocyclyl.
(12) . The compound of any one of embodiments (1) - (11) above, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
W is absent, C
1-3alkylene, C
2-3alkenylene, or C
2-3alkynylene; wherein said C
1-3alkylene, C
2-3alkenylene, or C
2-3alkynylene represented by W is optionally substituted by one to two R
3;
L is absent, -O-, -NH-, 3-6 membered monocyclic carbocyclyl, 6-8 membered bicyclic carbocyclyl; 4-6 membered monocyclic heterocyclyl, phenyl, and 5-6 membered monocyclic heteroaryl; wherein said 3-6 membered monocyclic carbocyclyl, 6-8 membered bicyclic carbocyclyl; 4-6 membered monocyclic heterocyclyl, phenyl, and 5-6 membered monocyclic heteroaryl represented by L is optionally substituted by one R
3; and
Z is absent, C
1-3alkylene, C
2-3alkenylene, or C
2-3alkynylene; wherein said C
1-3alkylene, C
2-3alkenylene, or C
2-3alkynylene represented by Z is optionally substituted by one R
3; wherein
R
3, in each occurrence, is independently halogen or C
1-3alkyl; and
R
4 is hydrogen, methyl, or -CF
3; or one R
3 and R
4 together with the atoms to which they are attached form 6 membered heterocyclyl.
(13) . The compound of any one of embodiments (1) - (12) above, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
W is C
1-3alkylene, C
1-3haloalkylene, C
2-3alkenylene, or C
2-3alkynylene;
L is absent or -O-; and
Z is C
1-3alkylene, C
1-3haloalkylene, C
2-3alkenylene, or C
2-3alkynylene.
(14) . The compound of any one of embodiments (1) - (12) above, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
W is C
3-4alkylene, C
3-4haloalkylene, C
3-4alkenylene, or C
3-4alkynylene;
L is absent; and
Z is absent.
(15) . The compound of any one of embodiments (1) - (12) above, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
W is absent, C
1-2alkylene, or C
1-2haloalkylene;
L is 4-6 membered carbocyclyl, 4-6 membered heterocyclyl, or 5-6 membered heteroaryl; and
Z is absent or methylene.
(16) . The compound of any one of embodiments (1) - (12) above, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
W is absent, -CH
2-, -CH
2CH
2-, -CH
2CH
2CH
2-, -CH=CH-, -CH
2CH=CH-, or -CH
2C≡C-;
L is absent, -O-, -NH-, cyclobutyl, cyclohexyl, spiro [3.3] heptanyl, azetidinyl, piperazinyl, piperidinyl, imidazolyl, triazolyl, or thiozolyl; wherein said -NH-, cyclobutyl, cyclohexyl, spiro [3.3] heptanyl, azetidinyl, piperazinyl, piperidinyl, imidazolyl, triazolyl, or thiozolyl is optionally substituted by one R
3;
Z is absent, -CH
2-, -CH
2CH
2-, -CH
2CH
2CH
2-, -CH=CH-, -CH=CHCH
2-, or -C≡CCH-;
R
4 is hydrogen; or one R
3 and R
4 together with the atoms to which they are attached form a piperazinyl ring.
(17) . The compound of any one of embodiments (1) - (16) above, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein R
4 is hydrogen.
(18) . The compound of any one of embodiments (1) - (12) , (16) , and (17) above, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
 is selected from the group consisting of
is selected from the group consisting of

wherein *-represents the point which attaches to variable X; -**represents the point which attaches to the moiety -NR
4-.
(19) . The compound of embodiment (18) above, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein
 is selected from the group consisting of
is selected from the group consisting of

wherein *-represents the point which attaches to variable X; -**represents the point which attaches to the moiety -NR
4-.
2. Definitions
The term “halogen, ” as used herein, refers to fluoride, chloride, bromide, or iodide.
The term “alkyl” used alone or as part of a larger moiety, such as “alkoxy” or “haloalkyl” and the like, means saturated aliphatic straight-chain or branched monovalent hydrocarbon radical of formula -C
nH
(2n+1) . Unless otherwise specified, an alkyl group typically has 1-6 carbon atoms, i.e. C
1-
6alkyl. As used herein, a “C
1-6alkyl” group means a radical having from 1 to 6 carbon atoms in a linear or branched arrangement. Examples include methyl, ethyl, n-propyl, iso-propyl, n-butyl, iso-butyl, tert-butyl, n-pentyl, isopentyl, hexyl, and the like.
The terms “haloalkyl” means alkyl, as the case may be, substituted with one or more halogen atoms. In one embodiment, the alkyl can be substituted by one to three halogens. Examples of haloalkyl, include, but are not limited to, trifluoromethyl, trichloromethyl, pentafluoroethyl and the like.
The term “alkylene” as used herein, means a straight or branched chain divalent hydrocarbon group of formula -C
nH
2n-. Non-limiting examples include ethylene, and propylene.
The terms “haloalkylene” means alkylene, as the case may be, substituted with one or more halogen atoms. In one embodiment, the alkylene can be substituted by one to three halogens.
The term “alkenyl” means an alkyl group in which one or more carbon/carbon single bond is replaced by a double bond.
The term “alkynyl” means an alkyl group in which one or more carbon/carbon single bond is replaced by a triple bond.
The term “carbocyclyl” refers to a 3-14 membered non-aromatic hydrocarbon ring system and may exist as a monocylic ring or a polycylic ring (e.g., a bicyclic ring (including fused, spiro or bridged carbocyclic rings) or a tricyclic ring) . In one embodiment, carbocyclyl is 3-, 4-, 5-, 6-, 7-, or 8-membered monocyclic or bicyclic or 7-, 8-, 9-, 10-, 11-, or 12-membered bicyclic or tricyclic hydrocarbon ring, any of which may be saturated, partially unsaturated. Any substitutable ring atom can be substituted (e.g., by one or more substituents) . Examples of such carbocycles include, but are not limited to, cyclopropyl, cyclobutyl, cyclobutenyl, cyclopentyl, cyclopentenyl, cyclohexyl, cycloheptenyl, cycloheptyl, cycloheptenyl, adamantyl, cyclooctyl, cyclooctenyl, and cyclooctadienyl. In one embodiment, carbocyclyl is intended to include, bridged, fused, and spirocyclic rings. In a spirocyclic carbocyclyl, one atom is common to two different rings. An example of a spirocyclic carbocyclyl is spiro [3.3] heptanyl. In a bridged carbocyclyl, the rings share at least two common non-adjacent atoms. Examples of bridged carbocyclyls include bicyclo [2.2.1] heptanyl, bicyclo [2.2.1] hept-2-enyl, and adamantanyl. In a fused-ring carbocyclyl system, two or more rings may be fused together, such that two rings share one common bond. Examples of two-or three-fused ring carbocyclyls include naphthalenyl, tetrahydronaphthalenyl (tetralinyl) , indenyl, indanyl (dihydroindenyl) , anthracenyl, phenanthrenyl, and decalinyl. The term “carbocyclyl” as used herein, includes groups in which a carbocyclyl ring is fused to one or more aryl, where the radical or point of attachment is on the carbocyclyl ring. Nonlimiting examples of such fused ring systems include:

The term “cycloalkyl” refers to a cyclic, bicyclic, tricyclic, or polycyclic saturated hydrocarbon groups having 3 to 12 ring carbons. In one embodiment, cycloalkyl may have 3 to 7 ring cabons. Any substitutable ring atom can be substituted (e.g., by one or more substituents) . Examples of cycloalkyl groups include, without limitation, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and cyclooctyl. Cycloalkyl may include multiple fused and/or bridged rings. Non-limiting examples of fused/bridged cycloalkyl include: bicyclo [1.1.0] butane, bicyclo [2.1.0] pentane, bicyclo [1.1.0] pentane, bicyclo [3.1.0] hexane, bicyclo [2.1.1] hexane, bicyclo [3.2.0] heptane, bicyclo [4.1.0] heptane, bicyclo [2.2.1] heptane, bicyclo [3.1.1] heptane, bicyclo [4.2.0] octane, bicyclo [3.2.1] octane, bicyclo [2.2.2] octane, and the like. Cycloalkyl also includes spirocyclic rings (e.g., spirocyclic bicycle wherein two rings are connected through just one atom) . Non-limiting examples of spirocyclic cycloalkyls include spiro [2.2] pentane, spiro [2.5] octane, spiro [3.5] nonane, spiro [3.5] nonane, spiro [3.5] nonane, spiro [4.4] nonane, spiro [2.6] nonane, spiro [4.5] decane, spiro [3.6] decane, spiro [5.5] undecane, and the like.
The term “heterocyclyl” or “heterocyclic” refers to a radical of a 3-to 12-membered non-aromatic ring system having ring carbon atoms and 1 to 4 ring heteroatoms, wherein each heteroatom is independently selected from nitrogen, quaternary nitrogen, oxidized nitrogen (e.g., NO) , oxygen, and sulfur, including sulfoxide and sulfone ( “3-12 membered heterocyclyl” ) . In some embodiments, a heterocyclyl group is a 3-7 membered non-aromatic ring system having ring carbon atoms and 1-4 ring heteroatoms, wherein each heteroatom is independently selected from nitrogen, oxygen, and sulfur ( “3-7 membered heterocyclyl” ) . In some embodiments, a heterocyclyl group comprises 1-3 heteroatoms selected from oxygen, nitrogen, and sulfur. In heterocyclyl groups that contain one or more nitrogen atoms, the point of attachment can be a carbon or nitrogen atom, as valency permits. A heterocyclyl group can either be monocyclic ( “monocyclic heterocyclyl” ) or polycyclic (e.g., a bicyclic system ( “bicyclic heterocyclyl” ) or tricyclic system ( “tricyclic heterocyclyl” ) ; polycyclic ring systems include fused, bridged, or spiro ring systems) . Exemplary monocyclic heterocyclyl groups include azetidinyl, oxetanyl, thietanyl, tetrahydrofuranyl, pyrrolidinyl, piperidinyl, tetrahydropyranyl, piperazinyl, morpholinyl, azepanyl, oxepanyl, thiepanyl, tetrahydropyridinyl, and the like. Heterocyclyl polycyclic ring systems can include heteroatoms in one or more rings in the polycyclic ring system. Substituents may be present on one or more rings in the polycyclic ring system.
Spiro heterocyclyl refers to 5 to 12 membered polycyclic heterocyclyl with rings connected through one common carbon atom (called as spiro atom) , wherein said rings have one or more heteroatoms selected from the group consisting of nitrogen, quaternary nitrogen, oxidized nitrogen (e.g., NO) , oxygen, and sulfur, including sulfoxide and sulfone, the remaining ring atoms being C, wherein one or more rings may contain one or more double bonds, but none of the rings has a completely conjugated pi-electron system. Representive examples of spiro heterocyclyl include, but are not limited to the following groups:

Fused heterocyclyl refers to a 5 to 12 membered polycyclic heterocyclyl group, wherein each ring in the group shares an adjacent pair of carbon atoms with another ring in the group, wherein one or more rings can contain one or more double bonds, but none of the rings has a completely conjugated π-electron system, and wherein said rings have one or more heteroatoms selected from the group consisting of nitrogen, quaternary nitrogen, oxidized nitrogen (e.g., NO) , oxygen, and sulfur, including sulfoxide and sulfone, the remaining ring atoms being C. Representive examples of fused heterocyclyl include, but are not limited to the following groups:

In some embodiments, a fused heterocyclyl include groups in which a heterocyclyl ring is fused to one or more aryl or heteroaryl, where the radical or point of attachment is on the heterocyclyl ring. Nonlimiting examples of such fused heterocyclyl ring systems include:

Bridged heterocyclyl refers to a 5 to 12 membered polycyclic heterocyclyl group, wherein any two rings in the group share two disconnected atoms, the rings can have one or more double bonds but have no completely conjugated π-electron system, and the rings have one or more heteroatoms selected from the group consisting of nitrogen, quaternary nitrogen, oxidized nitrogen (e.g., NO) , oxygen, and sulfur, including sulfoxide and sulfone as ring atoms, the remaining ring atoms being C. Representive examples of bridged heterocyclyl include, but are not limited to the following groups:

Generally, the carbocyclyl, the cycloalkyl, or the heterocyclyl may be unsubstituted, or be substituted with one or more substituents as valency allows, wherein the substituents can be independently selected from a number of groups such as oxo, -CN, halogen, alkyl and alkoxyl, opotionally, the alkyl substitution may be further substituted.
The term “aryl” refers to a 6 to 12 membered all-carbon monocyclic ring or a polycyclic fused ring (a “fused” ring system means that each ring in the system shares an adjacent pair of carbon atoms with other ring in the system) group, and has a completely conjugated π-electron system. The term “aryl” may be used interchangeably with the terms “aryl ring” “carbocyclic aromatic ring” , “aryl group” and “carbocyclic aromatic group” . Representive examples of aryl are phenyl and naphthyl. In some embodiments, two adjacent substituents on an aryl ring, taken together with the intervening ring atoms, form an optionally substituted fused 5-to 6-membered aromatic or 4-to 8-membered non-aromatic carbocyclyl ring. Thus, the term "aryl" , as used herein, includes groups in which an aromatic ring is fused to one or more non-aromatic carbocyclyl ring, where the radical or point of attachment is on the aromatic ring. Nonlimiting examples of such fused ring systems include:

The term “heteroaryl, ” as used herein, refers to a monocyclic or multicyclic aromatic hydrocarbon in which at least one of the ring carbon atoms has been replaced with a heteroatom independently selected from oxygen, nitrogen and sulfur. Preferably, the heteroaryl is based on a C
5-10 aryl with one or more of its ring carbon atoms replaced by the heteroatom. In some embodiments, heteroaryl comprises 1-4 heteroatoms selected from oxygen, nitrogen, and sulfur. A heteroaryl group may be attached through a ring carbon atom or, where valency permits, through a ring nitrogen atom. Generally, the heteroaryl may be unsubstituted, or be substituted with one or more substituents as valency allows with the substituents being independently selected from halogen, OH, alkyl, alkoxyl, and amino (e.g., NH
2, NHalkyl, N (alkyl)
2) , optionally, the alkyl may be further substituted.
Examples of monocyclic 5-6 membered heteroaryl groups include furanyl (e.g., 2-furanyl, 3-furanyl) , imidazolyl (e.g., N-imidazolyl, 2-imidazolyl, 4-imidazolyl, 5-imidazolyl) , isoxazolyl (e.g., 3-isoxazolyl, 4-isoxazolyl, 5-isoxazolyl) , oxadiazolyl (e.g., 2-oxadiazolyl, 5-oxadiazolyl) , oxazolyl (e.g., 2-oxazolyl, 4-oxazolyl, 5-oxazolyl) , pyrazolyl (e.g., 3-pyrazolyl, 4-pyrazolyl) , pyrrolyl (e.g., 1-pyrrolyl, 2-pyrrolyl, 3-pyrrolyl) , pyridyl (e.g., 2-pyridyl, 3-pyridyl, 4-pyridyl) , pyrimidinyl (e.g., 2-pyrimidinyl, 4-pyrimidinyl, 5-pyrimidinyl) , pyridazinyl (e.g., 3-pyridazinyl) , thiazolyl (e.g., 2-thiazolyl, 4-thiazolyl, 5-thiazolyl) , triazolyl (e.g., 2-triazolyl, 5-triazolyl) , tetrazolyl (e.g., tetrazolyl) , thienyl (e.g., 2-thienyl, 3-thienyl) , pyrimidinyl, pyridinyl and pyridazinyl. Examples of polycyclic aromatic heteroaryl groups include carbazolyl, benzimidazolyl, benzothienyl, benzofuranyl, indolyl, quinolinyl, benzotriazolyl, benzothiazolyl, benzoxazolyl, benzimidazolyl, isoquinolinyl, indolyl, isoindolyl, acridinyl, or benzisoxazolyl. A “substituted heteroaryl group” is substituted at any one or more substitutable ring atom, which is a ring carbon or ring nitrogen atom bonded to a hydrogen.
As used herein, many moieties (e.g., alkyl, alkylene, cycloalkyl, aryl, heteroaryl, or heterocyclyl ) are referred to as being either “substituted” or “optionally substituted” . When a moiety is modified by one of these terms, unless otherwise noted, it denotes that any portion of the moiety that is known to one skilled in the art as being available for substitution can be substituted, which includes one or more substituents. Where if more than one substituent is present, then each substituent may be independently selected. Such means for substitution are well-known in the art and/or taught by the instant disclosure. The optional substituents can be any substituents that are suitable to attach to the moiety.
Where suitable substituents are not specifically enumerated, exemplary substituents include, but are not limited to: C
1-5alkyl, C
1-5hydroxyalkyl, C
1-5haloalkyl, C
1-5alkoxy, C
1-5 haloalkoxy, halogen, hydroxyl, cyano, amino, -CN, -NO
2, -OR
c1, -NR
a1R
b1, -S (O)
iR
a1, -NR
a1S (O)
iR
b1, -S (O)
iNR
a1R
b1, -C (=O) OR
a1, -OC (=O) OR
a1, -C (=S) OR
a1, -O (C=S) R
a1, -C (=O) NR
a1R
b1, |-NR
a1C (=O) R
b1, -C (=S) NR
a1R
b1, -C (=O) R
a1, -C (=S) R
a1, NR
a1C (=S) R
b1, -O (C=O) NR
a1R
b1, -NR
a1 (C=S) OR
b1, -O (C=S) NR
a1R
b1, -NR
a1 (C=O) NR
a1R
b1, -NR
a1 (C=S) NR
a1R
b1, phenyl, or 5-6 membered heteroaryl. Each R
a1 and each R
b1 are independently selected from –H and C
1-5alkyl, optionally substituted with hydroxyl or C
1-3alkoxy; R
c1 is –H, C
1-5haloalkyl or C
1-5alkyl, wherein the C
1-5alkyl is optionally substituted with hydroxyl or C
1-C
3alkoxy.
The symbol
 as used herein, refers to the point where the moiety attaches.
as used herein, refers to the point where the moiety attaches.
Pharmaceutically Acceptable Salts
The term “pharmaceutically-acceptable salt” refers to a pharmaceutical salt that is, within the scope of sound medical judgment, suitable for use in contact with the tissues of humans and lower animals without undue toxicity, irritation, and allergic response, and is commensurate with a reasonable benefit/risk ratio. Pharmaceutically-acceptable salts are well known in the art. For example, S. M. Berge et al. describes pharmacologically acceptable salts in J. Pharm. Sci., 1977, 66, 1–19.
Pharmaceutically acceptable salts of the compounds of any one of the formulae described above include acid addition and base salts.
Included in the present teachings are pharmaceutically acceptable salts of the compounds disclosed herein. Compounds having basic groups can form pharmaceutically acceptable salts with pharmaceutically acceptable acid (s) . Suitable pharmaceutically acceptable acid addition salts of the compounds described herein include salts of inorganic acids (such as hydrochloric, hydrobromic, phosphoric, metaphosphoric, nitric, and sulfuric acids) and of organic acids (such as acetic, benzenesulfonic, benzoic, ethanesulfonic, methanesulfonic, and succinic acids) . Compounds of the present teachings with acidic groups such as carboxylic acids can form pharmaceutically acceptable salts with pharmaceutically acceptable base (s) . Suitable pharmaceutically acceptable basic salts include ammonium salts, alkali metal salts (such as sodium and potassium salts) and alkaline earth metal salts (such as magnesium and calcium salts) .
Pharmaceutically acceptable salts of compounds of any one of the formulae described above may be prepared by one or more of three methods:
(i) by reacting the compound of any one of the formulae described above with the desired acid or base;
(ii) by removing an acid-or base-labile protecting group from a suitable precursor of the compound of any one of the formulae described above or by ring-opening a suitable cyclic precursor, for example, a lactone or lactam, using the desired acid or base; or
(iii) by converting one salt of the compound of any one of the formulae described above to another by reaction with an appropriate acid or base or by means of a suitable ion exchange column.
All three reactions are typically carried out in solution. The resulting salt may precipitate out and be collected by filtration or may be recovered by evaporation of the solvent. The degree of ionisation in the resulting salt may vary from completely ionised to almost non-ionised.
The compounds of any one of the formulae described above, and pharmaceutically acceptable salts thereof, may exist in unsolvated and solvated forms.
Stereoisomers and Other Variations
The compounds of any one of the formulae described above may exhibit one or more kinds of isomerism (e.g. optical, geometric or tautomeric isomerism) . Such variation is implicit to the compounds of any one of the formulae described above defined as they are by reference to their structural features and therefore within the scope of the present disclosure.
Compounds having one or more chiral centers can exist in various stereoisomeric forms, i.e., each chiral center can have an R or S configuration, or can be a mixture of both. Stereoisomers are compounds that differ only in their spatial arrangement. Stereoisomers include all diastereomeric and enantiomeric forms of a compound. Enantiomers are stereoisomers that are mirror images of each other. Diastereomers are stereoisomers having two or more chiral centers that are not identifcal and are not mirror images of each other.
When a compound is designated by its chemical name (e.g., where the configuration is indicated in the chemical name by “R” or “S” ) or its structure (e.g., the configuration is indicated by “wedge” bonds) that indicates a single enantiomer, unless indicated otherwise, the compound is at least 60%, 70%, 80%, 90%, 99%or 99.9%optically pure (also referred to as “enantiomerically pure” ) . Optical purity is the weight in the mixture of the named or depicted enantiomer divided by the total weight in the mixture of both enantiomers.
When the stereochemistry of a disclosed compound is named or depicted by structure, and the named or depicted structure encompasses more than one stereoisomer (e.g., as in a diastereomeric pair) , it is to be understood that one of the encompassed stereoisomers or any mixture of the encompassed stereoisomers is included. It is to be further understood that the stereoisomeric purity of the named or depicted stereoisomers at least 60%, 70%, 80%, 90%, 99%or 99.9%by weight. The stereoisomeric purity in this case is determined by dividing the total weight in the mixture of the stereoisomers encompassed by the name or structure by the total weight in the mixture of all of the stereoisomers.
When two stereoisomers are depicted by their chemical names or structures, and the chemical names or structures are connected by an “and” , a mixture of the two stereoisomers is intended.
When two stereoisomers are depicted by their chemical names or structures, and the names or structures are connected by an “or” , one or the other of the two stereoisomers is intended, but not both.
When a disclosed compound having a chiral center is depicted by a structure without showing a configuration at that chiral center, the structure is meant to encompass the compound with the S configuration at that chiral center, the compound with the R configuration at that chiral center, or the compound with a mixture of the R and S configuration at that chiral center. When a disclosed compound having a chiral center is depicted by its chemical name without indicating a configuration at that chiral center with “S” or “R” , the name is meant to encompass the compound with the S configuration at that chiral center, the compound with the R configuration at that chiral center or the compound with a mixture of the R and S configuration at that chiral center.
Racemic mixture means 50%of one enantiomer and 50%of the corresponding enantiomer. When a compound with one chiral center is named or depicted without indicating the stereochemistry of the chiral center, it is understood that the name or structure encompasses both possible enantiomeric forms (e.g., both enantiomerically-pure, enantiomerically-enriched or racemic) of the compound. When a compound with two or more chiral centers is named or depicted without indicating the stereochemistry of the chiral centers, it is understood that the name or structure encompasses all possible diasteriomeric forms (e.g., diastereomerically pure, diastereomerically enriched and equimolar mixtures of one or more diastereomers (e.g., racemic mixtures) of the compound.
The term “geometric isomer” means isomers that differ in the orientation of substituent atoms in relationship to a carbon-carbon double bond, to a carbocyclic ring, or to a bridged bicyclic system. Substituent atoms (other than hydrogen) on each side of a carbon-carbon double bond may be in an E or Z configuration according to the Cahn-Ingold-Prelog priority rules. In the “E” configuration, the substituents having the highest priorities are on opposite sides in relationship to the carbon-carbon double bond. In the “Z” configuration, the substituents having the highest priorities are oriented on the same side in relationship to the carbon-carbon double bond.
Substituents around a carbon-carbon double bond can also be referred to as “cis” or “trans, ” where “cis” represents substituents on the same side of the double bond and “trans” represents substituents on opposite sides of the double bond. The arrangement of substituents around a carbocyclic ring can also be designated as “cis” or “trans. ” The term “cis” represents substituents on the same side of the plane of the ring, and the term “trans” represents substituents on opposite sides of the plane of the ring. Mixtures of compounds wherein the substituents are disposed on both the same and opposite sides of plane of the ring are designated “cis/trans. ”
Where structural isomers are interconvertible via a low energy barrier, tautomeric isomerism ( “tautomerism” ) can occur. This can take the form of proton tautomerism in compounds of any one of the formulae described above containing, for example, an imino, keto, or oxime group, or so-called valence tautomerism in compounds which contain an aromatic moiety. It follows that a single compound may exhibit more than one type of isomerism.
In certain instances tautomeric forms of the disclosed compounds exist, such as the tautomeric structures shown below:
When a geometric isomer is depicted by name or structure, it is to be understood that the named or depicted isomer exists to a greater degree than another isomer, that is that the geometric isomeric purity of the named or depicted geometric isomer is greater than 50%, such as at least 60%, 70%, 80%, 90%, 99%, or 99.9%pure by weight. Geometric isomeric purity is determined by dividing the weight of the named or depicted geometric isomer in the mixture by the total weight of all of the geomeric isomers in the mixture.
Cis/trans isomers may be separated by conventional techniques well known to those skilled in the art, for example, chromatography and fractional crystallisation.
Conventional techniques for the preparation/isolation of individual enantiomers/diastereomers include chiral synthesis from a suitable optically pure precursor or resolution of the racemate (or the racemate of a salt or derivative) using, for example, chiral high pressure liquid chromatography (HPLC) . Alternatively, the racemate (or a racemic precursor) may be reacted with a suitable optically active compound, for example, an alcohol, or, in the case where the compound of any one of the formulae described above contains an acidic or basic moiety, a base or acid such as 1-phenylethylamine or tartaric acid. The resulting diastereomeric mixture may be separated by chromatography and/or fractional crystallization and one or both of the diastereoisomers converted to the corresponding pure enantiomer (s) by means well known to a skilled person. Chiral compounds of any one of the formulae described above (and chiral precursors thereof) may be obtained in enantiomerically-enriched form using chromatography, typically HPLC, on an asymmetric resin with a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from 0 to 50%by volume of isopropanol, typically from 2%to 20%, and from 0 to 5%by volume of an alkylamine, typically 0.1%diethylamine. Concentration of the eluate affords the enriched mixture. Chiral chromatography using sub-and supercritical fluids may be employed. Methods for chiral chromatography useful in some embodiments of the present disclosure are known in the art (see, for example, Smith, Roger M., Loughborough University, Loughborough, UK; Chromatographic Science Series (1998) , 75 (Supercritical Fluid Chromatography with Packed Columns) , pp. 223-249 and references cited therein) . Columns can be obtained from Chiral Technologies, Inc, West Chester, Pa., USA, a subsidiary of
 Chemical Industries, Ltd., Tokyo, Japan.
Chemical Industries, Ltd., Tokyo, Japan.
It must be emphasized that the compounds of any one of the formulae described above have been drawn herein in a single tautomeric form, all possible tautomeric forms are included within the scope of the present disclosure.
3. Administration and Dosing
Typically, a compound of the present disclosure is administered in an amount effective to treat a condition as described herein. The compounds of the present disclosure can be administered as compound per se, or alternatively, as a pharmaceutically acceptable salt. For administration and dosing purposes, the compound per se or pharmaceutically acceptable salt thereof will simply be referred to as the compounds of the present disclosure.
The compounds of the present disclosure are administered by any suitable route in the form of a pharmaceutical composition adapted to such a route, and in a dose effective for the treatment intended. The compounds of the present disclosure may be administered orally, rectally, vaginally, parenterally, or topically.
The compounds of the present disclosure may be administered orally. Oral administration may involve swallowing, so that the compound enters the gastrointestinal tract, or buccal or sublingual administration may be employed by which the compound enters the bloodstream directly from the mouth.
In another embodiment, the compounds of the present disclosure may also be administered directly into the bloodstream, into muscle, or into an internal organ. Suitable means for parenteral administration include intravenous, intraarterial, intraperitoneal, intrathecal, intraventricular, intraurethral, intrasternal, intracranial, intramuscular and subcutaneous. Suitable devices for parenteral administration include needle (including microneedle) injectors, needle-free injectors and infusion techniques.
In another embodiment, the compounds of the present disclosure may also be administered topically to the skin or mucosa, that is, dermally or transdermally. In another embodiment, the compounds of the present disclosure can also be administered intranasally or by inhalation. In another embodiment, the compounds of the present disclosure may be administered rectally or vaginally. In another embodiment, the compounds of the present disclosure may also be administered directly to the eye or ear.
The dosage regimen for the compounds of the present disclosure and/or compositions containing said compounds is based on a variety of factors, including the type, age, weight, sex and medical condition of the patient; the severity of the condition; the route of administration; and the activity of the particular compound employed. Thus the dosage regimen may vary widely. In one embodiment, the total daily dose of a compound of the present disclosure is typically from about 0.001 to about 100 mg/kg (i.e., mg compound of the present disclosure per kg body weight) for the treatment of the indicated conditions discussed herein.
For oral administration, the compositions may be provided in the form of tablets containing 0.1-500 milligrams of the active ingredient for the symptomatic adjustment of the dosage to the patient. A medicament typically contains from about 0.01 mg to about 500 mg of the active ingredient. Intravenously, doses may range from about 0.01 to about 10 mg/kg/minute during a constant rate infusion.
Suitable subjects according to the present disclosure include mammalian subjects, including non-human mammal such as primates, rodents (mice, rats, hamsters, rabbits etc) . In one embodiment, humans are suitable subjects. Human subjects may be of either gender and at any stage of development.
4. Pharmaceutical Compositions
In another embodiment, the present disclosure comprises pharmaceutical compositions. Such pharmaceutical compositions comprise a compound of the present disclosure presented, a pharmaceutically acceptable salt, or a stereoisomer thereof with a pharmaceutically acceptable carrier or excipient. Other pharmacologically active substances can also be present.
As used herein, “pharmaceutically acceptable carrier or excipient” includes any and all solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents, and the like that are physiologically compatible. Examples of pharmaceutically acceptable carriers include one or more of water, saline, phosphate buffered saline, dextrose, glycerol, ethanol and the like, as well as combinations thereof, and may include isotonic agents, for example, sugars, sodium chloride, or polyalcohols such as mannitol, or sorbitol in the composition. Pharmaceutically acceptable substances such as wetting agents or minor amounts of auxiliary substances such as wetting or emulsifying agents, preservatives or buffers, which enhance the shelf life or effectiveness of the antibody or antibody portion.
The compositions of present disclosure may be in a variety of forms. These include, for example, liquid, semi-solid and solid dosage forms, such as liquid solutions (e.g., injectable and infusible solutions) , dispersions or suspensions, tablets, pills, powders, liposomes and suppositories. The form depends on the intended mode of administration and therapeutic application.
Typical compositions are in the form of injectable or infusible solutions, such as compositions similar to those used for passive immunization of humans with antibodies in general. One mode of administration is parenteral (e.g. intravenous, subcutaneous, intraperitoneal, intramuscular) . In another embodiment, the antibody is administered by intravenous infusion or injection. In yet another embodiment, the antibody is administered by intramuscular or subcutaneous injection.
Oral administration of a solid dose form may be, for example, presented in discrete units, such as hard or soft capsules, pills, cachets, lozenges, or tablets, each containing a predetermined amount of at least one compound of the present disclosure. In another embodiment, the oral administration may be in a powder or granule form. In another embodiment, the oral dose form is sub-lingual, such as, for example, a lozenge. In such solid dosage forms, the compounds of any one of the formulae described above are ordinarily combined with one or more adjuvants. Such capsules or tablets may contain a controlled release formulation. In the case of capsules, tablets, and pills, the dosage forms also may comprise buffering agents or may be prepared with enteric coatings.
In another embodiment, oral administration may be in a liquid dose form. Liquid dosage forms for oral administration include, for example, pharmaceutically acceptable emulsions, solutions, suspensions, syrups, and elixirs containing inert diluents commonly used in the art (e.g., water) . Such compositions also may comprise adjuvants, such as wetting, emulsifying, suspending, flavoring (e.g., sweetening) , and/or perfuming agents.
In another embodiment, the present disclosure comprises a parenteral dose form.
“Parenteral administration” includes, for example, subcutaneous injections, intravenous injections, intraperitoneally, intramuscular injections, intrasternal injections, and infusion. Injectable preparations (i.e., sterile injectable aqueous or oleaginous suspensions) may be formulated according to the known art using suitable dispersing, wetting agents, and/or suspending agents.
In another embodiment, the present disclosure comprises a topical dose form.
“Topical administration” includes, for example, transdermal administration, such as via transdermal patches or iontophoresis devices, intraocular administration, or intranasal or inhalation administration. Compositions for topical administration also include, for example, topical gels, sprays, ointments, and creams. A topical formulation may include a compound which enhances absorption or penetration of the active ingredient through the skin or other affected areas. When the compounds of present disclosure are administered by a transdermal device, administration will be accomplished using a patch either of the reservoir and porous membrane type or of a solid matrix variety. Typical formulations for this purpose include gels, hydrogels, lotions, solutions, creams, ointments, dusting powders, dressings, foams, films, skin patches, wafers, implants, sponges, fibres, bandages and microemulsions. Liposomes may also be used. Typical carriers include alcohol, water, mineral oil, liquid petrolatum, white petrolatum, glycerin, polyethylene glycol and propylene glycol. Penetration enhancers may be incorporated -see, for example, Finnin and Morgan, J. Pharm. Sci., 88: 955-958, 1999.
Formulations suitable for topical administration to the eye include, for example, eye drops wherein the compound of present disclosure is dissolved or suspended in a suitable carrier. A typical formulation suitable for ocular or aural administration may be in the form of drops of a micronized suspension or solution in isotonic, pH-adjusted, sterile saline. Other formulations suitable for ocular and aural administration include ointments, biodegradable (i.e., absorbable gel sponges, collagen) and non-biodegradable (i.e., silicone) implants, wafers, lenses and particulate or vesicular systems, such as niosomes or liposomes. A polymer such as crossed linked polyacrylic acid, polyvinyl alcohol, hyaluronic acid, a cellulosic polymer, for example, hydroxypropylmethylcellulose, hydroxyethylcellulose, or methylcellulose, or a heteropolysaccharide polymer, for example, gelan gum, may be incorporated together with a preservative, such as benzalkonium chloride. Such formulations may also be delivered by iontophoresis.
For intranasal administration or administration by inhalation, the compounds of the present disclosure are conveniently delivered in the form of a solution or suspension from a pump spray container that is squeezed or pumped by the patient or as an aerosol spray presentation from a pressurized container or a nebulizer, with the use of a suitable propellant. Formulations suitable for intranasal administration are typically administered in the form of a dry powder (either alone, as a mixture, for example, in a dry blend with lactose, or as a mixed component particle, for example, mixed with phospholipids, such as phosphatidylcholine) from a dry powder inhaler or as an aerosol spray from a pressurized container, pump, spray, atomizer (preferably an atomizer using electrohydrodynamics to produce a fine mist) , or nebulizer, with or without the use of a suitable propellant, such as 1, 1, 1, 2-tetrafluoroethane or 1, 1, 1, 2, 3, 3, 3-heptafluoropropane. For intranasal use, the powder may comprise a bioadhesive agent, for example, chitosan or cyclodextrin.
In another embodiment, the present disclosure comprises a rectal dose form. Such rectal dose form may be in the form of, for example, a suppository. Cocoa butter is a traditional suppository base, but various alternatives may be used as appropriate.
Other carrier materials and modes of administration known in the pharmaceutical art may also be used. Pharmaceutical compositions of the present disclosure may be prepared by any of the well-known techniques of pharmacy, such as effective formulation and administration procedures.
The above considerations in regard to effective formulations and administration procedures are well known in the art and are described in standard textbooks. Formulation of drugs is discussed in, for example, Hoover, John E., Remington’s Pharmaceutical Sciences, Mack Publishing Co., Easton, Pa., 1975; Liberman et al., Eds., Pharmaceutical Dosage Forms, Marcel Decker, New York, N.Y., 1980; and Kibbe et al., Eds., Handbook of Pharmaceutical Excipients (3
rd Ed. ) , American Pharmaceutical Association, Washington, 1999.
5. Method of Treatment
Compounds of the present disclosure can inhibit CDK2 and therefore are useful for treating diseases wherein the underlying pathology is, wholly or partially, mediated by CDK2. Such diseases include cancer and other diseases with proliferation disorder.
In some embodiments, the present disclosure provides treatment of an individual or a patient in vivo using a compound of Formula (I') or (I) , or a pharmaceutically acceptable salt, or a stereoisomer thereof such that growth of cancerous tumors is inhibited. A compound of Formula (I') or (I) or of any of the formulae as described herein, or a compound as recited in any of the claims and described herein, or a pharmaceutically acceptable salt or a stereoisomer thereof, can be used to inhibit the growth of cancerous tumors with aberrations that activate the CDK2 kinase activity. These include, but not limited to, diseases (e.g., cancers) that are characterized by amplification or overexpression of CCNE1 such as ovarian cancer, uterine carcinosarcoma and breast cancer and p27 inactivation such as breast cancer and melanomas. Accordingly, in some embodiments of the methods, the patient has been previously determined to have an amplification of the cyclin E1 (CCNE1) gene and/or an expression level of CCNE1 in a biological sample obtained from the human subject that is higher than a control expression level of CCNE1. Alternatively, a compound of Formula (I') or (I) or of any of the formulae as described herein, or a compound as recited in any of the claims and described herein, or a pharmaceutically acceptable salt or a stereoisomer thereof, can be used in conjunction with other agents or standard cancer treatments, as described below. In one embodiment, the present disclosure provides a method for inhibiting growth of tumor cells in vitro. The method includes contacting the tumor cells in vitro with a compound of Formula (I') or (I) or of any of the formulae as described herein, or of a compound as recited in any of the claims and described herein, or of a pharmaceutically acceptable salt or a stereoisomer thereof. In another embodiment, the present disclosure provides a method for inhibiting growth of tumor cells with CCNE1 amplification and overexpression in an individual or a patient. The method includes administering to the individual or patient in need thereof a therapeutically effective amount of a compound of Formula (I') or (I) or of any of the formulae as described herein, or of a compound as recited in any of the claims and described herein, or a pharmaceutically acceptable salt or a stereoisomer thereof.
In certain embodiments, compounds of the present disclosure selectively inhibit CDK2 over CDK1, with a ratio of IC
50 values for the latter (CDK1) against the former (CDK2) of at least about 2, 5, 10, 15, 20, 40, 50, 60, 80, 100 or more.
In some embodiments, provided herein is a method of inhibiting CDK2, comprising contacting the CDK2 with a compound of Formula (I') or (I) or any of the formulae as described herein, a compound as recited in any of the claims and described herein, or a pharmaceutically acceptable salt, or a stereoisomer thereof. In some embodiments, provided herein is a method of inhibiting CDK2 in a patient, comprising administering to the patient a compound of Formula (I') or (I) or any of the formulae as described herein, a compound as recited in any of the claims and described herein, or a pharmaceutically acceptable salt or a stereoisomer thereof.
In some embodiments, provided herein is a method for treating cancer. The method includes administering to a patient (in need thereof) , a therapeutically effective amount of a compound of Formula (I') or (I) or any of the formulae as described herein, a compound as recited in any of the claims and described herein, or a pharmaceutically acceptable salt or a stereoisomer thereof. In another embodiment, the cancer is characterized by amplification or overexpression of CCNE1. In some embodiments, the cancer is characterized by inactivation of a CDK2 inhibitor, such as p21Cip1 or p27Kip1. In some embodiments, the cancer is ovarian cancer or breast cancer, characterized by amplification or overexpression of CCNE1.
In certain embodiments, the patient has been diagnosed with a cancer characterized by amplification or overexpression of CCNE1, and/or loss of function of p21Cip1 or p27Kip1.
In certain embodiments, the method further comprises determining the status of expression of CCNE1, p21Cip1 and/or p27Kip1.
In certain embodiments, the method further comprises selecting patients characterized by amplification or overexpression of CCNE1, and/or loss of function of p21Cip1 or p27Kip1 for treatment.
In some embodiments, provided herein is a method of treating a disease or disorder associated with CDK2 in a patient, comprising administering to the patient a therapeutically effective amount of a compound of Formula (I') or (I) or any of the formulae as described herein, a compound as recited in any of the claims and described herein, or a pharmaceutically acceptable salt or a stereoisomer thereof.
In some embodiments, the disease or disorder associated with CDK2 is associated with an amplification of the cyclin E1 (CCNE1) gene and/or overexpression of CCNE1.
In some embodiments, the disease or disorder associated with CDK2 is N-myc amplified neuroblastoma cells (See Molenaar et al., Proc Natl Acad Sci USA, 106 (31) : 12968-12973) , K-Ras mutant lung cancers (see Hu, S., et al., Mol Cancer Ther, 2015, 14 (11) : 2576-85, and cancers with FBW7 mutation and CCNE1 overexpression (see Takada, et al., Cancer Res, 2017, 77 (18) : 4881-4893) .
In some embodiments, the disease or disorder associated with CDK2 is breast, lung, colorectal, gastric, or bone cancer, leukemia or lymphoma.
In some embodiments, the disease or disorder associated with CDK2 is lung squamous cell carcinoma, lung adenocarcinoma, pancreatic adenocarcinoma, breast invasive carcinoma, uterine carcinosarcoma, ovarian serous cystadenocarcinoma, stomach adenocarcinoma, esophageal carcinoma, bladder urothelial carcinoma, mesothelioma, or sarcoma.
In some embodiments, the disease or disorder associated with CDK2 is lung adenocarcinoma, breast invasive carcinoma, uterine carcinosarcoma, ovarian serous cystadenocarcinoma, or stomach adenocarcinoma.
In some embodiments, the disease or disorder associated with CDK2 is an adenocarcinoma, carcinoma, or cystadenocarcinoma.
In some embodiments, the disease or disorder associated with CDK2 is uterine cancer, ovarian cancer, stomach cancer, esophageal cancer, lung cancer, bladder cancer, pancreatic cancer, or breast cancer.
In some embodiments, the disease or disorder associated with CDK2 is a cancer.
In some embodiments, the cancer is characterized by amplification or overexpression of CCNE1.
In some embodiments, the cancer is ovarian cancer or breast cancer, characterized by amplification or overexpression of CCNE1.
In some embodiments, the breast cancer is chemotherapy or radiotherapy resistant breast cancer, endocrine resistant breast cancer, trastuzumab resistant breast cancer, or breast cancer demonstrating primary or acquired resistance to CDK4/6 inhibition.
In some embodiments, the breast cancer is advanced or metastatic breast cancer. Examples of cancers that are treatable using the compounds of the present disclosure include, but are not limited to, bone cancer, pancreatic cancer, skin cancer, cancer of the head or neck, cutaneous or intraocular malignant melanoma, uterine cancer, ovarian cancer, rectal cancer, cancer of the anal region, stomach cancer, testicular cancer, uterine cancer, carcinoma of the fallopian tubes, carcinoma of the endometrium, endometrial cancer, carcinoma of the cervix, carcinoma of the vagina, carcinoma of the vulva, Hodgkin's Disease, non-Hodgkin's lymphoma, cancer of the esophagus, cancer of the small intestine, cancer of the endocrine system, cancer of the thyroid gland, cancer of the parathyroid gland, cancer of the adrenal gland, sarcoma of soft tissue, cancer of the urethra, cancer of the penis, chronic or acute leukemias including acute myeloid leukemia, chronic myeloid leukemia, acute lymphoblastic leukemia, chronic lymphocytic leukemia, solid tumors of childhood, lymphocytic lymphoma, cancer of the bladder, cancer of the kidney or urethra, carcinoma of the renal pelvis, neoplasm of the central nervous system (CNS) , primary CNS lymphoma, tumor angiogenesis, spinal axis tumor, brain stem glioma, pituitary adenoma, Kaposi's sarcoma, epidermoid cancer, squamous cell cancer, T-cell lymphoma, environmentally induced cancers including those induced by asbestos, and combinations of said cancers. The compounds of the present disclosure are also useful for the treatment of metastatic cancers.
In some embodiments, cancers treatable with compounds of the present disclosure include melanoma (e.g., metastatic malignant melanoma, BRAF and HSP90 inhibition-resistant melanoma) , renal cancer (e.g., clear cell carcinoma) , prostate cancer (e.g., hormone refractory prostate adenocarcinoma) , breast cancer, colon cancer, lung cancer (e.g., non-small cell lung cancer and small cell lung cancer) , squamous cell head and neck cancer, urothelial cancer (e.g., bladder) and cancers with high microsatellite instability (MSI
high) . Additionally, the disclosure includes refractory or recurrent malignancies whose growth may be inhibited using the compounds of the disclosure.
In some embodiments, cancers that are treatable using the compounds of the present disclosure include, but are not limited to, solid tumors (e.g., prostate cancer, colon cancer, esophageal cancer, endometrial cancer, ovarian cancer, uterine cancer, renal cancer, hepatic cancer, pancreatic cancer, gastric cancer, breast cancer, lung cancer, cancers of the head and neck, thyroid cancer, glioblastoma, sarcoma, bladder cancer, etc. ) , hematological cancers (e.g., lymphoma, leukemia such as acute lymphoblastic leukemia (ALL) , acute myelogenous leukemia (AML) , chronic lymphocytic leukemia (CLL) , chronic myelogenous leukemia (CML) , DLBCL, mantle cell lymphoma, Non-Hodgkin lymphoma (including relapsed or refractory NHL and recurrent follicular) , Hodgkin lymphoma or multiple myeloma) and combinations of said cancers.
In some embodiments, cancers that are treatable using the compounds of the present disclosure include, but are not limited to, cholangiocarcinoma, bile duct cancer, triple negative breast cancer, rhabdomyosarcoma, small cell lung cancer, leiomyosarcoma, hepatocellular carcinoma, Ewing’s sarcoma, brain cancer, brain tumor, astrocytoma, neuroblastoma, neurofibroma, basal cell carcinoma, chondrosarcoma, epithelioid sarcoma, eye cancer, Fallopian tube cancer, gastrointestinal cancer, gastrointestinal stromal tumors, hairy cell leukemia, intestinal cancer, islet cell cancer, oral cancer, mouth cancer, throat cancer, laryngeal cancer, lip cancer, mesothelioma, neck cancer, nasal cavity cancer, ocular cancer, ocular melanoma, pelvic cancer, rectal cancer, renal cell carcinoma, salivary gland cancer, sinus cancer, spinal cancer, tongue cancer, tubular carcinoma, urethral cancer, and ureteral cancer. In some embodiments, the compounds of the present disclosure can be used to treat sickle cell disease and sickle cell anemia.
In some embodiments, diseases and indications that are treatable using the compounds of the present disclosure include, but are not limited to hematological cancers, sarcomas, lung cancers, gastrointestinal cancers, genitourinary tract cancers, liver cancers, bone cancers, nervous system cancers, gynecological cancers, and skin cancers.
Exemplary hematological cancers include lymphomas and leukemias such as acute lymphoblastic leukemia (ALL) , acute myelogenous leukemia (AML) , acute promyelocytic leukemia (APL) , chronic lymphocytic leukemia (CLL) , chronic myelogenous leukemia (CML) , diffuse large B-cell lymphoma (DLBCL) , mantle cell lymphoma, Non-Hodgkin lymphoma (including relapsed or refractory NHL and recurrent follicular) , Hodgkin lymphoma, myeloproliferative diseases (e.g., primary myelofibrosis (PMF) , polycythemia vera (PV) , and essential thrombocytosis (ET) ) , myelodysplasia syndrome (MDS) , T-cell acute lymphoblastic lymphoma (T-ALL) and multiple myeloma (MM) .
Exemplary sarcomas include chondrosarcoma, Ewing’s sarcoma, osteosarcoma, rhabdomyosarcoma, angiosarcoma, fibrosarcoma, liposarcoma, myxoma, rhabdomyoma, rhabdosarcoma, fibroma, lipoma, harmatoma, and teratoma.
Exemplary lung cancers include non-small cell lung cancer (NSCLC) , small cell lung cancer (SCLC) , bronchogenic carcinoma, squamous cell, undifferentiated small cell, undifferentiated large cell, adenocarcinoma, alveolar (bronchiolar) carcinoma, bronchial adenoma, chondromatous hamartoma, and mesothelioma.
Exemplary gastrointestinal cancers include cancers of the esophagus (squamous cell carcinoma, adenocarcinoma, leiomyosarcoma, lymphoma) , stomach (carcinoma, lymphoma, leiomyosarcoma) , pancreas (ductal adenocarcinoma, insulinoma, glucagonoma, gastrinoma, carcinoid tumors, vipoma) , small bowel (adenocarcinoma, lymphoma, carcinoid tumors, Kaposi's sarcoma, leiomyoma, hemangioma, lipoma, neurofibroma, fibroma) , large bowel (adenocarcinoma, tubular adenoma, villous adenoma, hamartoma, leiomyoma) , and colorectal cancer.
Exemplary genitourinary tract cancers include cancers of the kidney (adenocarcinoma, Wilm's tumor [nephroblastoma] ) , bladder and urethra (squamous cell carcinoma, transitional cell carcinoma, adenocarcinoma) , prostate (adenocarcinoma, sarcoma) , and testis (seminoma, teratoma, embryonal carcinoma, teratocarcinoma, choriocarcinoma, sarcoma, interstitial cell carcinoma, fibroma, fibroadenoma, adenomatoid tumors, lipoma) .
Exemplary liver cancers include hepatoma (hepatocellular carcinoma) , cholangiocarcinoma, hepatoblastoma, angiosarcoma, hepatocellular adenoma, and hemangioma.
Exemplary bone cancers include, for example, osteogenic sarcoma (osteosarcoma) , fibrosarcoma, malignant fibrous histiocytoma, chondrosarcoma, Ewing's sarcoma, malignant lymphoma (reticulum cell sarcoma) , multiple myeloma, malignant giant cell tumor chordoma, osteochronfroma (osteocartilaginous exostoses) , benign chondroma, chondroblastoma, chondromyxofibroma, osteoid osteoma, and giant cell tumors.
Exemplary nervous system cancers include cancers of the skull (osteoma, hemangioma, granuloma, xanthoma, osteitis deformans) , meninges (meningioma, meningiosarcoma, gliomatosis) , brain (astrocytoma, medulloblastoma, glioma, ependymoma, germinoma (pinealoma) , glioblastoma, glioblastoma multiform, oligodendroglioma, schwannoma, retinoblastoma, congenital tumors) , and spinal cord (neurofibroma, meningioma, glioma, sarcoma) , as well as neuroblastoma and Lhermitte-Duclos disease.
Exemplary gynecological cancers include cancers of the uterus (endometrial carcinoma) , cervix (cervical carcinoma, pre -tumor cervical dysplasia) , ovaries (ovarian carcinoma (serous cystadenocarcinoma, mucinous cystadenocarcinoma, unclassified carcinoma) , granulosa-thecal cell tumors, Sertoli-Leydig cell tumors, dysgerminoma, malignant teratoma) , vulva (squamous cell carcinoma, intraepithelial carcinoma, adenocarcinoma, fibrosarcoma, melanoma) , vagina (clear cell carcinoma, squamous cell carcinoma, botryoid sarcoma (embryonal rhabdomyosarcoma) , and fallopian tubes (carcinoma) .
Exemplary skin cancers include melanoma, basal cell carcinoma, Merkel cell carcinoma, squamous cell carcinoma, Kaposi's sarcoma, moles dysplastic nevi, lipoma, angioma, dermatofibroma, and keloids. In some embodiments, diseases and indications that are treatable using the compounds of the present disclosure include, but are not limited to, sickle cell disease (e.g., sickle cell anemia) , triple-negative breast cancer (TNBC) , myelodysplastic syndromes, testicular cancer, bile duct cancer, esophageal cancer, and urothelial carcinoma.
It is believed that compounds of Formula (I') or (I) , or any of the embodiments thereof, may possess satisfactory pharmacological profile and promising biopharmaceutical properties, such as toxicological profile, metabolism and pharmacokinetic properties, solubility, and permeability. It will be understood that determination of appropriate biopharmaceutical properties is within the knowledge of a person skilled in the art, e.g., determination of cytotoxicity in cells or inhibition of certain targets or channels to determine potential toxicity.
The terms “individual” or “patient, ” used interchangeably, refer to any animal, including mammals, preferably mice, rats, other rodents, rabbits, dogs, cats, swine, cattle, sheep, horses, or primates, and most preferably humans.
The terms “treatment, ” “treat, ” and “treating” refer to reversing, alleviating, or inhibiting the progress of a disease described herein. In some embodiments, treatment may be administered after one or more signs or symptoms of the disease have developed or have been observed (i.e., therapeutic treatment) . In other embodiments, treatment may be administered in the absence of signs or symptoms of the disease. For example, treatment may be administered to a susceptible subject prior to the onset of symptoms (i.e., prophylactic treatment) (e.g., in light of a history of symptoms and/or in light of exposure to a pathogen) . Treatment may also be continued after symptoms have resolved, for example, to delay or prevent recurrence.
The terms “condition, ” “disease, ” and “disorder” are used interchangeably.
The term “administer, ” “administering, ” or “administration” refers to methods introducing a compound disclosed herein, or a composition thereof, in or on a patient. These methods include, but are not limited to, intraarticular (in the joints) , intravenous, intramuscular, intratumoral, intradermal, intraperitoneal, subcutaneous, orally, topically, intrathecally, inhalationally, transdermally, rectally, and the like. Administration techniques that can be employed with the agents and methods described herein are found in e.g., Goodman and Gilman, The Pharmacological Basis of Therapeutics, current ed.; Pergamon; and Remington’s, Pharmaceutical Sciences (current edition) , Mack Publishing Co., Easton, Pa.
Generally, an effective amount of a compound taught herein varies depending upon various factors, such as the given drug or compound, the pharmaceutical formulation, the route of administration, the type of disease or disorder, the identity of the subject or host being treated, and the like, but can nevertheless be routinely determined by one skilled in the art. An effective amount of a compound of the present teachings may be readily determined by one of ordinary skill by routine methods known in the art.
The term “therapeutically effective amount” means an amount when administered to the subject which results in beneficial or desired results, including clinical results, e.g., inhibits, suppresses or reduces the symptoms of the condition being treated in the subject as compared to a control. For example, a therapeutically effective amount can be an amount effective for detectable killing or inhibition of the growth or spread of cancer cells; the size or number of tumors; or other measure of the level, stage, progression or severity of the cancer. The exact amount required will vary from subject to subject, depending on the species, age, and general condition of the subject, the severity of the disease, the particular anticancer agent, its mode of administration, combination treatment with other therapies, and the like.
6. Combination Therapies
I. Cancer Therapies
Cancer cell growth and survival can be impacted by dysfunction in multiple signaling pathways. Thus, it is useful to combine different enzyme/protein/receptor inhibitors, exhibiting different preferences in the targets which they modulate the activities of, to treat such conditions. Targeting more than one signaling pathway (or more than one biological molecule involved in a given signaling pathway) may reduce the likelihood of drug-resistance arising in a cell population, and/or reduce the toxicity of treatment.
One or more additional pharmaceutical agents such as, for example, chemotherapeutics, anti-estrogen agents, anti-inflammatory agents, steroids, immunosuppressants, immune-oncology agents, metabolic enzyme inhibitors, chemokine receptor inhibitors, and phosphatase inhibitors, as well as targeted therapies such as Bcr-Abl, Flt-3, EGFR, HER2, JAK, c-MET, VEGFR, PDGFR, c-Kit, IGF-1R, RAF, FAK, and CDK4/6 kinase inhibitors such as, for example, those described in WO 2006/056399 can be used in combination with the compounds of the present disclosure for treatment of CDK2-associated diseases, disorders or conditions. Other agents such as therapeutic antibodies can be used in combination with the compounds of the present disclosure for treatment of CDK2- associated diseases, disorders or conditions. The one or more additional pharmaceutical agents can be administered to a patient simultaneously or sequentially.
In some embodiments, the CDK2 inhibitor is administered or used in combination with an anti-estrogen agent or a CDK4/6 inhibitor or a mTOR inhibitor or a BCL2 inhibitor or a chemotherapy.
The compounds as disclosed herein can be used in combination with one or more other enzyme/protein/receptor inhibitors therapies for the treatment of diseases, such as cancer and other diseases or disorders described herein. Examples of diseases and indications treatable with combination therapies include those as described herein. Examples of cancers include solid tumors and non-solid tumors, such as liquid tumors, blood cancers. Examples of infections include viral infections, bacterial infections, fungus infections or parasite infections. For example, the compounds of the present disclosure can be combined with one or more inhibitors of the following kinases for the treatment of cancer: Akt1, Akt2, Akt3, BCL2, CDK4/6, TGF-DR, PKA, PKG, PKC, CaM-kinase, phosphorylase kinase, MEKK, ERK, MAPK, mTOR, EGFR, HER2, HER3, HER4, INS-R, IDH2, IGF-1R, IR-R, PDGFαR, PDGF βR, PI3K (alpha, beta, gamma, delta, and multiple or selective) , CSF1R, KIT, FLK-II, KDR/FLK-1, FLK-4, flt-1, FGFR1, FGFR2, FGFR3, FGFR4, c-Met, PARP, Ron, Sea, TRKA, TRKB, TRKC, TAM kinases (Axl, Mer, Tyro3) , FLT3, VEGFR/Flt2, Flt4, EphA1, EphA2, EphA3, EphB2, EphB4, Tie2, Src, Fyn, Lck, Fgr, Btk, Fak, SYK, FRK, JAK, ABL, ALK and B-Raf. In some embodiments, the compounds of the present disclosure can be combined with one or more of the following inhibitors for the treatment of cancer or infections. Non-limiting examples of inhibitors that can be combined with the compounds of the present disclosure for treatment of cancer and infections include an FGFR inhibitor (FGFR1, FGFR2, FGFR3 or FGFR4, e.g., pemigatinib (INCB54828) , INCB62079) , an EGFR inhibitor (also known as ErB-1 or HER-1; e.g., erlotinib, gefitinib, vandetanib, orsimertinib, cetuximab, necitumumab, or panitumumab) , a VEGFR inhibitor or pathway blocker (e.g., bevacizumab, pazopanib, sunitinib, sorafenib, axitinib, regorafenib, ponatinib, cabozantinib, vandetanib, ramucirumab, lenvatinib, ziv-aflibercept) , a PARP inhibitor (e.g., olaparib, rucaparib, veliparib or niraparib) , a JAK inhibitor (JAK1 and/or JAK2, e.g., ruxolitinib or baricitinib; JAK1, e.g., itacitinib (INCB39110) , INCB052793, or INCB054707) , an IDO inhibitor (e.g., epacadostat, NLG919, or BMS-986205, MK7162) , an LSD1 inhibitor (e.g., GSK2979552, INCB59872 and INCB60003) , a TDO inhibitor, a PI3K-delta inhibitor (e.g., parsaclisib (INCB50465) or INCB50797) , a PI3K-gamma inhibitor such as PI3K-gamma selective inhibitor, a Pim inhibitor (e.g., INCB53914) , a CSF1R inhibitor, a TAM receptor tyrosine kinases (Tyro-3, Axl, and Mer; e.g., INCB081776) , an adenosine receptor antagonist (e.g., A2a/A2b receptor antagonist) , an HPK1 inhibitor, a chemokine receptor inhibitor (e.g., CCR2 or CCR5 inhibitor) , a SHP1/2 phosphatase inhibitor, a histone deacetylase inhibitor (HDAC) such as an HDAC8 inhibitor, an angiogenesis inhibitor, an interleukin receptor inhibitor, bromo and extra terminal family members inhibitors (for example, bromodomain inhibitors or BET inhibitors such as INCB54329 and INCB57643) , c-MET inhibitors (e.g., capmatinib) , an anti-CD19 antibody (e.g., tafasitamab) , an ALK2 inhibitor (e.g., INCB00928) ; or combinations thereof.
In some embodiments, the compound or salt described herein is administered with a PI3K6 inhibitor. In some embodiments, the compound or salt described herein is administered with a JAK inhibitor. In some embodiments, the compound or salt described herein is administered with a JAK1 or JAK2 inhibitor (e.g., baricitinib or ruxolitinib) . In some embodiments, the compound or salt described herein is administered with a JAK1 inhibitor. In some embodiments, the compound or salt described herein is administered with a JAK1 inhibitor, which is selective over JAK2. Example antibodies for use in combination therapy include, but are not limited to, trastuzumab (e.g., anti-HER2) , ranibizumab (e.g., anti-VEGF-A) , bevacizumab (AVASTINTM , e.g., anti-VEGF) , panitumumab (e.g., anti-EGFR) , cetuximab (e.g., anti-EGFR) , rituxan (e.g., anti-CD20) , and antibodies directed to c-MET. One or more of the following agents may be used in combination with the compounds of the present disclosure and are presented as a non-limiting list: a cytostatic agent, cisplatin, doxorubicin, taxotere, taxol, etoposide, irinotecan, camptosar, topotecan, paclitaxel, docetaxel, epothilones, tamoxifen, 5-fluorouracil, methotrexate, temozolomide, cyclophosphamide, SCH 66336, R115777, L778, 123, BMS 214662, IRESSATM (gefitinib) , TARCEVATM (erlotinib) , antibodies to EGFR, intron, ara-C, adriamycin, cytoxan, gemcitabine, uracil mustard, chlormethine, ifosfamide, melphalan, chlorambucil, pipobroman, triethylenemelamine, triethylenethiophosphoramine, busulfan, carmustine, lomustine, streptozocin, dacarbazine, floxuridine, cytarabine, 6-mercaptopurine, 6-thioguanine, fludarabine phosphate, oxaliplatin, leucovirin, ELOXATIN
TM (oxaliplatin) , pentostatine, vinblastine, vincristine, vindesine, bleomycin, dactinomycin, daunorubicin, doxorubicin, epirubicin, idarubicin, mithramycin, deoxycoformycin, mitomycin-C, L-asparaginase, teniposide 17. alpha. -ethinylestradiol, diethylstilbestrol, testosterone, Prednisone, Fluoxymesterone, Dromostanolone propionate, testolactone, megestrolacetate, methylprednisolone, methyltestosterone, prednisolone, triamcinolone, chlorotrianisene, hydroxyprogesterone, aminoglutethimide, estramustine, medroxyprogesteroneacetate, leuprolide, flutamide, toremifene, goserelin, carboplatin, hydroxyurea, amsacrine, procarbazine, mitotane, mitoxantrone, levamisole, navelbene, anastrazole, letrazole, capecitabine, reloxafine, droloxafine, hexamethylmelamine, avastin, HERCEPTIN
TM (trastuzumab) , BEXXAR
TM (tositumomab) , VELCADE
TM (bortezomib) , ZEVALIN
TM (ibritumomab tiuxetan) , TRISENOX
TM (arsenic trioxide) , XELODA
TM (capecitabine) , vinorelbine, porfimer, ERBITUX
TM (cetuximab) , thiotepa, altretamine, melphalan, trastuzumab, lerozole, fulvestrant, exemestane, ifosfomide, rituximab, C225 (cetuximab) , Campath (alemtuzumab) , clofarabine, cladribine, aphidicolon, rituxan, sunitinib, dasatinib, tezacitabine, Sml1, fludarabine, pentostatin, triapine, didox, trimidox, amidox, 3-AP, and MDL-101, 731.
The compounds of the present disclosure can further be used in combination with other methods of treating cancers, for example by chemotherapy, irradiation therapy, tumor-targeted therapy, adjuvant therapy, immunotherapy or surgery. Examples of immunotherapy include cytokine treatment (e.g., interferons, GM-CSF, G-CSF, IL-2) , CRS-207 immunotherapy, cancer vaccine, monoclonal antibody, bispecific or multi-specific antibody, antibody drug conjugate, adoptive T cell transfer, Toll receptor agonists, RIG-I agonists, oncolytic virotherapy and immunomodulating small molecules, including thalidomide or JAK1/2 inhibitor, PI3Kd inhibitor and the like. The compounds can be administered in combination with one or more anti-cancer drugs, such as a chemotherapeutic agent. Examples of chemotherapeutics include any of: abarelix, aldesleukin, alemtuzumab, alitretinoin, allopurinol, altretamine, anastrozole, arsenic trioxide, asparaginase, azacitidine, bevacizumab, bexarotene, baricitinib, bleomycin, bortezomib, busulfan intravenous, busulfan oral, calusterone, capecitabine, carboplatin, carmustine, cetuximab, chlorambucil, cisplatin, cladribine, clofarabine, cyclophosphamide, cytarabine, dacarbazine, dactinomycin, dalteparin sodium, dasatinib, daunorubicin, decitabine, denileukin, denileukin diftitox, dexrazoxane, docetaxel, doxorubicin, dromostanolone propionate, eculizumab, epirubicin, erlotinib, estramustine, etoposide phosphate, etoposide, exemestane, fentanyl citrate, filgrastim, floxuridine, fludarabine, fluorouracil, fulvestrant, gefitinib, gemcitabine, gemtuzumab ozogamicin, goserelin acetate, histrelin acetate, ibritumomab tiuxetan, idarubicin, ifosfamide, imatinib mesylate, interferon alfa 2a, irinotecan, lapatinib ditosylate, lenalidomide, letrozole, leucovorin, leuprolide acetate, levamisole, lomustine, meclorethamine, megestrol acetate, melphalan, mercaptopurine, methotrexate, methoxsalen, mitomycin C, mitotane, mitoxantrone, nandrolone phenpropionate, nelarabine, nofetumomab, oxaliplatin, paclitaxel, pamidronate, panitumumab, pegaspargase, pegfilgrastim, pemetrexed disodium, pentostatin, pipobroman, plicamycin, procarbazine, quinacrine, rasburicase, rituximab, ruxolitinib, sorafenib, streptozocin, sunitinib, sunitinib maleate, tamoxifen, temozolomide, teniposide, testolactone, thalidomide, thioguanine, thiotepa, topotecan, toremifene, tositumomab, trastuzumab, tretinoin, uracil mustard, valrubicin, vinblastine, vincristine, vinorelbine, vorinostat, and zoledronate.
Additional examples of chemotherapeutics include proteasome inhibitors (e.g., bortezomib) , thalidomide, revlimid, and DNA-damaging agents such as melphalan, doxorubicin, cyclophosphamide, vincristine, etoposide, carmustine, and the like.
Example steroids include corticosteroids such as dexamethasone or prednisone. Example Bcr-Abl inhibitors include imatinib mesylate (GLEEVAC
TM) , nilotinib, dasatinib, bosutinib, and ponatinib, and pharmaceutically acceptable salts. Other example suitable Bcr-Abl inhibitors include the compounds, and pharmaceutically acceptable salts thereof, of the genera and species disclosed in U.S. Pat. No. 5,521,184, WO 04/005281, and U.S. Ser. No. 60/578,491.
Example suitable Flt-3 inhibitors include midostaurin, lestaurtinib, linifanib, sunitinib, sunitinib, maleate, sorafenib, quizartinib, crenolanib, pacritinib, tandutinib, PLX3397 and ASP2215, and their pharmaceutically acceptable salts. Other example suitable Flt-3 inhibitors include compounds, and their pharmaceutically acceptable salts, as disclosed in WO 03/037347, WO 03/099771, and WO 04/046120.
Example suitable RAF inhibitors include dabrafenib, sorafenib, and vemurafenib, and their pharmaceutically acceptable salts. Other example suitable RAF inhibitors include compounds, and their pharmaceutically acceptable salts, as disclosed in WO 00/09495 and WO 05/028444.
Example suitable FAK inhibitors include VS-4718, VS-5095, VS-6062, VS-6063, BI853520, and GSK2256098, and their pharmaceutically acceptable salts. Other example suitable FAK inhibitors include compounds, and their pharmaceutically acceptable salts, as disclosed in WO 04/080980, WO 04/056786, WO 03/024967, WO 01/064655, WO 00/053595, and WO 01/014402.
Example suitable CDK4/6 inhibitors include palbociclib, ribociclib, trilaciclib, lerociclib, and abemaciclib, and their pharmaceutically acceptable salts. Other example suitable CDK4/6 inhibitors include compounds, and their pharmaceutically acceptable salts, as disclosed in WO 09/085185, WO 12/129344, WO 11/101409, WO 03/062236, WO 10/075074, and WO 12/061156.
In some embodiments, the compounds of the disclosure can be used in combination with one or more other kinase inhibitors including imatinib, particularly for treating patients resistant to imatinib or other kinase inhibitors.
In some embodiments, the compounds of the disclosure can be used in combination with a chemotherapeutic in the treatment of cancer, and may improve the treatment response as compared to the response to the chemotherapeutic agent alone, without exacerbation of its toxic effects. In some embodiments, the compounds of the disclosure can be used in combination with a chemotherapeutic provided herein. For example, additional pharmaceutical agents used in the treatment of multiple myeloma, can include, without limitation, melphalan, melphalan plus prednisone [MP] , doxorubicin, dexamethasone, and Velcade (bortezomib) . Further additional agents used in the treatment of multiple myeloma include Bcr-Abl, Flt-3, RAF and FAK kinase inhibitors. In some embodiments, the agent is an alkylating agent, a proteasome inhibitor, a corticosteroid, or an immunomodulatory agent. Examples of an alkylating agent include cyclophosphamide (CY) , melphalan (MEL) , and bendamustine. In some embodiments, the proteasome inhibitor is carfilzomib. In some embodiments, the corticosteroid is dexamethasone (DEX) . In some embodiments, the immunomodulatory agent is lenalidomide (LEN) or pomalidomide (POM) . Additive or synergistic effects are desirable outcomes of combining a CDK2 inhibitor of the present disclosure with an additional agent.
The agents can be combined with the present compound in a single or continuous dosage form, or the agents can be administered simultaneously or sequentially as separate dosage forms.
The compounds of the present disclosure can be used in combination with one or more other inhibitors or one or more therapies for the treatment of infections. Examples of infections include viral infections, bacterial infections, fungus infections or parasite infections.
In some embodiments, a corticosteroid such as dexamethasone is administered to a patient in combination with the compounds of the disclosure where the dexamethasone is administered intermittently as opposed to continuously.
The compounds of Formula (I') or (I) or any of the formulae as described herein, a compound as recited in any of the claims and described herein, or salts thereof can be combined with another immunogenic agent, such as cancerous cells, purified tumor antigens (including recombinant proteins, peptides, and carbohydrate molecules) , cells, and cells transfected with genes encoding immune stimulating cytokines. Non-limiting examples of tumor vaccines that can be used include peptides of melanoma antigens, such as peptides of gp100, MAGE antigens, Trp-2, MARTI and/or tyrosinase, or tumor cells transfected to express the cytokine GM-CSF.
The compounds of Formula (I') or (I) or any of the formulae as described herein, a compound as recited in any of the claims and described herein, or salts thereof can be used in combination with a vaccination protocol for the treatment of cancer. In some embodiments, the tumor cells are transduced to express GM-CSF. In some embodiments, tumor vaccines include the proteins from viruses implicated in human cancers such as Human Papilloma Viruses (HPV) , Hepatitis Viruses (HBV and HCV) and Kaposi's Herpes Sarcoma Virus (KHSV) . In some embodiments, the compounds of the present disclosure can be used in combination with tumor specific antigen such as heat shock proteins isolated from tumor tissue itself. In some embodiments, the compounds of Formula (I') or (I) or any of the formulae as described herein, a compound as recited in any of the claims and described herein, or salts thereof can be combined with dendritic cells immunization to activate potent anti-tumor responses.
The compounds of the present disclosure can be used in combination with bispecific macrocyclic peptides that target Fe alpha or Fe gamma receptor-expressing effectors cells to tumor cells. The compounds of the present disclosure can also be combined with macrocyclic peptides that activate host immune responsiveness.
In some further embodiments, combinations of the compounds of the disclosure with other therapeutic agents can be administered to a patient prior to, during, and/or after a bone marrow transplant or stem cell transplant.
The compounds of the present disclosure can be used in combination with bone marrow transplant for the treatment of a variety of tumors of hematopoietic origin.
The compounds of Formula (I') or (I) or any of the formulae as described herein, a compound as recited in any of the claims and described herein, or salts thereof can be used in combination with vaccines, to stimulate the immune response to pathogens, toxins, and self-antigens. Examples of pathogens for which this therapeutic approach may be particularly useful, include pathogens for which there is currently no effective vaccine, or pathogens for which conventional vaccines are less than completely effective. These include, but are not limited to, HIV, Hepatitis (A, B, & C) , Influenza, Herpes, Giardia, Malaria, Leishmania, Staphylococcus aureus, Pseudomonas Aeruginosa.
Viruses causing infections treatable by methods of the present disclosure include, but are not limit to human papillomavirus, influenza, hepatitis A, B, C or D viruses, adenovirus, poxvirus, herpes simplex viruses, human cytomegalovirus, severe acute respiratory syndrome virus, Ebola virus, measles virus, herpes virus (e.g., VZV, HSV-1, HAV-6, HSV-II, and CMV, Epstein Barr virus) , flaviviruses, echovirus, rhinovirus, coxsackie virus, cornovirus, respiratory syncytial virus, mumps virus, rotavirus, measles virus, rubella virus, parvovirus, vaccinia virus, HTLV virus, dengue virus, papillomavirus, molluscum virus, poliovirus, rabies virus, JC virus and arboviral encephalitis virus.
Pathogenic bacteria causing infections treatable by methods of the disclosure include, but are not limited to, chlamydia, rickettsial bacteria, mycobacteria, staphylococci, streptococci, pneumococci, meningococci and conococci, klebsiella, proteus, serratia, pseudomonas, legionella, diphtheria, salmonella, bacilli, cholera, tetanus, botulism, anthrax, plague, leptospirosis, and Lyme's disease bacteria.
Pathogenic fungi causing infections treatable by methods of the disclosure include, but are not limited to, Candida (albicans, krusei, glabrata, tropicalis, etc. ) , Cryptococcus neoformans, Aspergillus (fumigatus, niger, etc. ) , Genus Mucorales (mucor, absidia, rhizophus) , Sporothrix schenkii, Blastomyces dermatitidis, Paracoccidioides brasiliensis, Coccidioides immitis and Histoplasma capsulatum.
Pathogenic parasites causing infections treatable by methods of the disclosure include, but are not limited to, Entamoeba histolytica, Balantidium coli, Naegleriafowleri, Acanthamoeba sp., Giardia lambia, Cryptosporidium sp., Pneumocystis carinii, Plasmodium vivax, Babesia microti, Trypanosoma brucei, Trypanosoma cruzi, Leishmania donovani, Toxoplasma gondi, and Nippostrongylus brasiliensis.
When more than one pharmaceutical agent is administered to a patient, they can be administered simultaneously, separately, sequentially, or in combination (e.g., for more than two agents) .
Methods for the safe and effective administration of most of these chemotherapeutic agents are known to those skilled in the art. In addition, their administration is described in the standard literature. For example, the administration of many of the chemotherapeutic agents is described in the "Physicians'Desk Reference" (PDR, e.g., 1996 edition, Medical Economics Company, Montvale, NJ) , the disclosure of which is incorporated herein by reference as if set forth in its entirety.
II. Immune-checkpoint therapies
Compounds of the present disclosure can be used in combination with one or more immune checkpoint inhibitors for the treatment of diseases, such as cancer or infections. Exemplary immune checkpoint inhibitors include inhibitors against immune checkpoint molecules such as CBL-B, CD20, CD28, CD40, CD122, CD96, CD73, CD47, GITR, CSF1R, JAK, PI3K delta, PI3K gamma, TAM, arginase, HPK1, CD137 (also known as 4-1BB) , ICOS, A2AR, B7-H3, B7-H4, BTLA, CTLA-4, LAG3, TIM3, TIGIT, CD112R, VISTA, PD-1, PD-L1 and PD-L2. In some embodiments, the immune checkpoint molecule is a stimulatory checkpoint molecule selected from CD27, CD28, CD40, ICOS, OX40, GITR and CD137. In some embodiments, the immune checkpoint molecule is an inhibitory checkpoint molecule selected from A2AR, B7-H3, B7-H4, BTLA, CTLA-4, IDO, KIR, LAG3, PD-1, TIM3, TIGIT, and VISTA. In some embodiments, the compounds provided herein can be used in combination with one or more agents selected from KIR inhibitors, TIGIT inhibitors, LAIR1 inhibitors, CD160 inhibitors, 2B4 inhibitors and TGFR beta inhibitors.
In some embodiments, the compounds provided herein can be used in combination with one or more agonists of immune checkpoint molecules, e.g., OX40, CD27, GITR, and CD137 (also known as 4-1B) .
In some embodiments, the inhibitor of an immune checkpoint molecule is anti-PD1 antibody, anti-PD-L1 antibody, or anti-CTLA-4 antibody.
In some embodiments, the inhibitor of an immune checkpoint molecule is an inhibitor of PD-1, e.g., an anti-PD-1 monoclonal antibody. In some embodiments, the anti-PD-1 monoclonal antibody is nivolumab, pembrolizumab (also known as MK-3475) , pidilizumab, SHR-1210, PDR001, MGA012, PDR001, AB122, or AMP-224. In some embodiments, the anti-PD-1 monoclonal antibody is nivolumab or pembrolizumab. In some embodiments, the anti-PD1 antibody is pembrolizumab. In some embodiments, the anti-PD-1 monoclonal antibody is MGA012. In some embodiments, the anti-PD1 antibody is SHR-1210. Other anti-cancer agent (s) include antibody therapeutics such as 4-1BB (e.g., urelumab, utomilumab) .
In some embodiments, the inhibitor of an immune checkpoint molecule is an inhibitor of PD-L1, e.g., an anti-PD-L1 monoclonal antibody. In some embodiments, the anti-PD-L1 monoclonal antibody is BMS-935559, MEDI4736, MPDL3280A (also known as RG7446) , or MSB0010718C. In some embodiments, the anti-PD-L1 monoclonal antibody is MPDL3280A or MEDI4736. In some embodiments, the inhibitor of an immune checkpoint molecule is an inhibitor of PD-1 and PD-L1, e.g., an anti-PD-1/PD-L1 bispecific antibody. In some embodiments, the anti-PD-1/PD-L1 is MCLA-136. In some embodiments, the inhibitor is MCLA-145.
In some embodiments, the inhibitor of an immune checkpoint molecule is an inhibitor of CTLA-4, e.g., an anti-CTLA-4 antibody. In some embodiments, the anti-CTLA-4 antibody is ipilimumab, tremelimumab, AGEN1884, or CP-675, 206.
In some embodiments, the inhibitor of an immune checkpoint molecule is an inhibitor of LAG3, e.g., an anti-LAG3 antibody. In some embodiments, the anti-LAG3 antibody is BMS-986016, LAG525, or INCAGN2385.
In some embodiments, the inhibitor of an immune checkpoint molecule is an inhibitor of TIM3, e.g., an anti-TIM3 antibody. In some embodiments, the anti-TIM3 antibody is INCAGN2390, MBG453, or TSR-022.
In some embodiments, the inhibitor of an immune checkpoint molecule is an inhibitor of GITR, e.g., an anti-GITR antibody. In some embodiments, the anti-GITR antibody is TRX518, MK-4166, INCAGN1876, MK-1248, AMG228, BMS-986156, GWN323, or MEDI1873.
In some embodiments, the inhibitor of an immune checkpoint molecule is an agonist of OX40, e.g., OX40 agonist antibody or OX40L fusion protein. In some embodiments, the anti-OX40 antibody is MEDI0562, MOXR-0916, PF-04518600, GSK3174998, or BMS-986178. In some embodiments, the OX40L fusion protein is MEDI6383.
In some embodiments, the inhibitor of an immune checkpoint molecule is an inhibitor of CD20, e.g., an anti-CD20 antibody. In some embodiments, the anti-CD20 antibody is obinutuzumab or rituximab. The compounds of the present disclosure can be used in combination with bispecific antibodies.
In some embodiments, one of the domains of the bispecific antibody targets PD-1, PD-L1, CTLA-4, GITR, OX40, TIM3, LAG3, CD137, ICOS, CD3 or TGFb receptor.
In some embodiments, the compounds of the disclosure can be used in combination with one or more metabolic enzyme inhibitors. In some embodiments, the metabolic enzyme inhibitor is an inhibitor of IDO1, TDO, or arginase. Examples of IDO1 inhibitors include epacadostat, NLG919, BMS-986205, PF-06840003, IOM2983, RG-70099 and LY338196.
As provided throughout, the additional compounds, inhibitors, agents, etc. can be combined with the present compound in a single or continuous dosage form, or they can be administered simultaneously or sequentially as separate dosage forms.
7. Treatment Kits
One aspect of the present invention relates to a kit for conveniently and effectively carrying out the methods or uses in accordance with the present invention. In general, the pharmaceutical pack or kit comprises one or more containers filled with one or more of the ingredients of the pharmaceutical compositions of the invention. Such kits are especially suited for the delivery of solid oral forms such as tablets or capsules. Such a kit preferably includes a number of unit dosages, and may also include a card having the dosages oriented in the order of their intended use. If desired, a memory aid can be provided, for example in the form of numbers, letters, or other markings or with a calendar insert, designating the days in the treatment schedule in which the dosages can be administered. Optionally associated with such container (s) can be a notice in the form prescribed by a governmental agency regulating the manufacture, use or sale of pharmaceutical products, which notice reflects approval by the agency of manufacture, use or sale for human administration.
The following representative examples contain important additional information, exemplification and guidance which can be adapted to the practice of this invention in its various embodiments and the equivalents thereof. These examples are intended to help illustrate the invention, and are not intended to, nor should they be construed to, limit its scope. Indeed, various modifications of the invention, and many further embodiments thereof, in addition to those shown and described herein, will become apparent to those skilled in the art upon review of this document, including the examples which follow and the references to the scientific and patent literature cited herein.
The contents of the cited references are incorporated herein by reference to help illustrate the state of the art.
In addition, for purposes of this invention, the chemical elements are identified in accordance with the Periodic Table of the Elements, CAS version, Handbook of Chemistry and Physics, 75
th Ed., inside cover. Additionally, general principles of organic chemistry, as well as specific functional moieties and reactivity, are described in “Organic Chemistry, ” Thomas Sorrell, University Science Books, Sausalito: 1999, and “Organic Chemistry, ” Morrison & Boyd (3d Ed) , the entire contents of both of which are incorporated herein by reference.
8. Preparation
The compounds of any one of the formulae described above, may be prepared by the general and specific methods described below, using the common general knowledge of one skilled in the art of synthetic organic chemistry. Such common general knowledge can be found in standard reference books such as Comprehensive Organic Chemistry, Ed. Barton and Ollis, Elsevier; Comprehensive Organic Transformations: A Guide to Functional Group Preparations, Larock, John Wiley and Sons; and Compendium of Organic Synthetic Methods, Vol. I-XII (published by Wiley-Interscience) . The starting materials used herein are commercially available or may be prepared by routine methods known in the art.
In the preparation of the compounds of any one of the formulae described above, it is noted that some of the preparation methods described herein may require protection of remote functionality (e.g., primary amine, secondary amine, carboxyl in any one of the formulae described above precursors) . The need for such protection will vary depending on the nature of the remote functionality and the conditions of the preparation methods. The need for such protection is readily determined by one skilled in the art. The use of such protection/deprotection methods is also within the skill in the art. For a general description of protecting groups and their use, see Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991.
For example, certain compounds contain primary amines or carboxylic acid functionalities which may interfere with reactions at other sites of the molecule if left unprotected. Accordingly, such functionalities may be protected by an appropriate protecting group which may be removed in a subsequent step. Suitable protecting groups for amine and carboxylic acid protection include those protecting groups commonly used in peptide synthesis (such as N-t-butoxycarbonyl (Boc) , benzyloxycarbonyl (Cbz) , and 9-fluorenylmethylenoxycarbonyl (Fmoc) for amines, and lower alkyl or benzyl esters for carboxylic acids) which are generally not chemically reactive under the reaction conditions described and can typically be removed without chemically altering other functionality in the any one of the formulae described above compounds.
The Schemes described below are intended to provide a general description of the methodology employed in the preparation of the compounds of the present disclosure. Some of the compounds of the present present disclosure may contain single or multiple chiral centers with the stereochemical designation (R) or (S) . It will be apparent to one skilled in the art that all of the synthetic transformations can be conducted in a similar manner whether the materials are enantioenriched or racemic. Moreover, the resolution to the desired optically active material may take place at any desired point in the sequence using well known methods such as described herein and in the chemistry literature.
EXAMPLES
Abbreviations
ATP Adenosine triphosphate
ACN Acetonitrile
AcOH Acetic acid
BSA Bovine serum albumin
CDI 1, 1'-Carbonyldiimidazole
DCE 1, 2-Dichloromethane
DCM Dichloromethane
DIPEA N, N-Diisopropylethylamine
DMAP 4-Dimethylaminopyridine
DMF Dimethylformamide
DMSO Dimethyl sulfoxide
DTT Dithiothreitol
EDCI N- (3-dimethylaminopropyl) -N'-ethylcarbodiimide hydrochloride
EtOH Ethanol
EDTA Ethylenediaminetetraacetic acid
EtOAc Ethyl acetate
FA Formic acid
FBS Fetal bovine serum
FCC Flash column chromatrography
HATU N- [ (Dimethylamino) -lH-l, 2, 3-triazolo- [4, 5-b] pyridin-l-ylmethylene] -N
methylmethanaminium hexafluorophosphate N-oxide
HOBT 1-Hydroxybenzotriazole
HEPES 4- (2-hydroxyethyl) -1-piperazineethanesulfonic acid
HPLC High performance liquid chromatography
Prep-HPLC Preparative High-performance liquid chromatography
LC-MS Liquid chromatography -mass spectrometry
MeOH Methanol
NBS N-bromosuccinimide
NMM N-Methylmorpholine
Pd
2 (dba)
3 Tris (dibenzylideneacetone) dipalladium
Pd (dppf) Cl
2 Dichloro [l, l'-bis (diphenylphosphino) ferrocene] palladium
Pd (PPh
3)
4 Tetrakis (triphenylphosphine) palladium
PE Petroleum ether
rac Racemic
SFC Supercritical fluid chromatography
TEA Triethyl amine
THF Tetrahydrofuran
TFA Trifluoroacetic acid
XantPhos 4, 5-Bis (diphenylphosphino) -9, 9-dimethylxanthene
wt. % Weight percentage
General Equipment Description
1H NMR spectra were recorded on a Bruker Ascend 400 spectrometer. Chemical shifts are expressed in parts per million (ppm, δ units) . Coupling constants are in units of hertz (Hz) . Splitting patterns describe apparent multiplicities and are designated as s (singlet) , d (doublet) , t (triplet) , q (quartet) , quint (quintet) , m (multiplet) , br (broad) .
The analytical low-resolution mass spectra (MS) were recorded on Waters ACQUITY UPLC with SQ Detectors using a Waters CORTECS C18+, 2.7 μm 4.6×30 mm using a gradient elution method.
Solvent A: 0.1%formic acid (FA) in water
Solvent B: 0.1%FA in acetonitrile
5%ACN to 95%ACN in 1.0 min, hold 1.0 min,
Total 2.5 min; Flow rate: 1.8 mL/min; Column Temp 40 degree.
Benzyl (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) carbamate, Benzyl (1- (tert-butyl) -3- ( (1R, 3S) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) carbamate and (trans, rac) -3- (5- ( ( (benzyloxy) carbonyl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl methanesulfonate were purchased from PharmaBlock.
Intermediate 1

Step 1: methyl 3, 3-dimethoxycyclopentane-1-carboxylate
To a solution of 3-oxocyclopentanecarboxylic acid (15 g, 117.1 mmol) in MeOH (30 mL) was added trimethoxymethane (74.5 g, 702.4 mmol, 77.0 mL) and p-Toluenesulfonic acid monohydrate (445.4 mg, 2.3 mmol, 359.2 μL) at 0 ℃. The mixture was stirred at 20 ℃ for 16 hours. The mixture was quenched with NaHCO3 aq. (2x50 mL) , extracted with EtOAc (40 mL) , dried over Na
2SO
4, filtered, concentrated under reduced pressure to give methyl 3, 3-dimethoxycyclopentanecarboxylate (19 g, crude) as a yellow oil.
Step 2: 3- (3, 3-dimethoxycyclopentyl) -3-oxopropanenitrile
To a solution of n-BuLi (2.4 M, 84.1 mL) in THF (30 mL) was added acetonitrile (8.3 g, 201.9 mmol, 10.5 mL) at -78 ℃. The mixture was stirred at -78 ℃ under N
2 for 1 hour. Then methyl 3, 3-dimethoxycyclopentanecarboxylate (19 g, 100.9 mmol) was added and stirred for 1.5 hours. The mixture was quenched with water, adjusted pH to 7 by HCl (1 M) , extracted with EtOAc (50 mL) , dried over Na
2SO
4, filtered, concentrated under reduced pressure to give 3- (3, 3-dimethoxycyclopentyl) -3-oxo-propanenitrile (18.9 g, crude) as a red oil.
Step 3: 1- (tert-butyl) -3- (3, 3-dimethoxycyclopentyl) -1H-pyrazol-5-amine
To a solution of tert-butylhydrazine·hydrochloride (14.33 g, 114.99 mmol) in EtOH (25 mL) was added NaOH (4.6 g, 114.9 mmol) at 0 ℃ and stirred for 1 hours. Then 3- (3, 3-dimethoxycyclopentyl) -3-oxo-propanenitrile (18.9 g, 95.8 mmol) was added and stirred at 75 ℃ for 16 hours. LCMS showed the desired mass peak was found. The mixture was filtered, the filtrate was concentrated under reduced pressure. The residue was purified by FCC (120 g silica gel, 0~20%~50%EtOAc in PE) to give 2-tert-butyl-5- (3, 3-dimethoxycyclopentyl) pyrazol-3-amine (8.8 g, 32.8 mmol, 34%yield) as a red oil and 2-tert-butyl-5- [ (3Z) -3- (tert-butylhydrazono) cyclopentyl] pyrazol-3-amine (2.0 g, 6.9 mmol, 7%yield) as a red oil. LC-MS: m/z 222 [M+H]
+.
Step 4: benzyl (1- (tert-butyl) -3- (3, 3-dimethoxycyclopentyl) -1H-pyrazol-5-yl) carbamate
To a solution of 2-tert-butyl-5- (3, 3-dimethoxycyclopentyl) pyrazol-3-amine (8.8 g, 32.8 mmol) in CH
3CN (30 mL) was added benzyl carbonochloridate (11.2 g, 65.7 mmol, 9.3 mL) at 0 ℃. The mixture was stirred at 20 ℃ for 2 hours. Then Sodium bicarbonate (8.8 g, 105.1 mmol) was added and stirred at 20 ℃ for 16 hours. LCMS showed the desired mass peak was found. The mixture was filtered, concentrated under reduced pressure. The residue was extracted with EtOAc (40 mL) , washed with water (2x50 mL) . The organic phase was concentrated under reduced pressure to give benzyl N- [2-tert-butyl-5- (3, 3-dimethoxycyclopentyl) pyrazol-3-yl] carbamate (17.9 g, crude) as a red oil. The crude product was used to the next step directly. LC-MS: m/z 402 [M+H]
+.
Step 5: benzyl (1- (tert-butyl) -3- (3-oxocyclopentyl) -1H-pyrazol-5-yl) carbamate
A solution of benzyl N- [2-tert-butyl-5- (3, 3-dimethoxycyclopentyl) pyrazol-3-yl] carbamate (17.9 g, 44.8 mmol) and p-Toluenesulfonic acid monohydrate (1.1 g, 5.8 mmol) in acetone (30 mL) and H
2O (30 mL) was stirred at 60 ℃ for 16 hours. LCMS showed the reaction was completed. The mixture was extracted with EtOAc (45 mL) , washed with water (2x55 mL) . The organic phase was concentrated under reduced pressure. The residue was purified by FCC (120 g silica gel, 0~35%EtOAc in PE) to give benzyl N- [2-tert-butyl-5- (3-oxocyclopentyl) pyrazol-3-yl] carbamate (8.0 g, 22.7 mmol, 50%yield) as a yellow solid. LC-MS: m/z 356 [M+H]
+.
Step 6: (rac, cis) -benzyl (1- (tert-butyl) -3- (3-hydroxycyclopentyl) -1H-pyrazol-5-yl) carbamate
To a solution of benzyl N- [2-tert-butyl-5- (3-oxocyclopentyl) pyrazol-3-yl] carbamate (8.0 g, 22.7 mmol) in THF (25 mL) was added Lithium triethylborohydride (1.0 M, 45.4 mL) at -65 ℃ dropwise. The mixture was stirred at -65 ℃ under N
2 for 1.5 hours. LCMS showed the desired mass peak was found. The mixture was quenched with NaHCO
3 aq. at -40 ℃, extracted with EtOAc (35 mL) . The organic phase was concentrated under reduced pressure. The residue was purified by FCC (120 g silica gel, 0~50%DCM in EtOAc) to give (rac, cis) -benzyl (1- (tert-butyl) -3- (3-hydroxycyclopentyl) -1H-pyrazol-5-yl) carbamate (5.6 g, 15.7 mmol, 69%yield) as a white solid. And (rac, trans) -benzyl (1- (tert-butyl) -3- (3-hydroxycyclopentyl) -1H-pyrazol-5-yl) carbamate (2.0 g, 5.6 mmol, 24%yield) as a light yellow solid. LC-MS: m/z 358 [M+H]
+.
Step 7: (rac, cis) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol
To a solution of (rac, cis) -benzyl N- [2-tert-butyl-5- [3-hydroxycyclopentyl] pyrazol-3-yl] carbamate (2.8 g, 6.4 mmol) in THF (18 mL) was added Pd/C (300 mg) at 20 ℃. The mixture was stirred at 20 ℃ for 4 hours under H
2. LCMS showed the reaction was completed. The mixture was filtered, concentrated to give (rac, cis) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (1.7 g, 5.7 mmol, 88%yield) as a yellow oil. LC-MS: m/z 224 [M+H]
+.
Intermediate 2

Step 1: (rac, cis) -benzyl (1- (tert-butyl) -3- (3- (1, 3-dioxoisoindolin-2-yl) cyclopentyl) -1H-pyrazol-5-
yl) carbamate:
To a stirred solution of (rac, trans) -benzyl N- [1-tert-butyl-3- [3-hydroxycyclopentyl] pyrazol-5-yl] carbamate (400 mg, 1.12 mmol) in THF (10 mL) were sequentially added isoindoline-1, 3-dione (247 mg, 1.68 mmol) , triphenyl phosphate (730 mg, 2.24 mmol) and diethyl azodicarboxylate (292 mg, 275 μL, 1.68 mmol) at 0 ℃. The resulting mixture was warmed to 25 ℃ and stirred at that temperature for 3 h. The mixture was quenched with saturated aq. NaHCO
3 (0.1 mL) and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/petroleum ether with EtOAc from 0 to 75%in 20 min to give (rac, cis) -benzyl N- [1-tert-butyl-3- [-3- (1, 3-dioxoisoindolin-2-yl) cyclopentyl] pyrazol-5-yl] carbamate (230 mg, 42%yield) as a colorless oil. LC-MS: m/z 487.8 [M+H]
+.
Step 2: (rac, cis) -2- (3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl) isoindoline-1, 3-dione:
To a stirred solution of (rac, cis) -benzyl N- [1-tert-butyl-3- [-3- (1, 3-dioxoisoindolin-2-yl) cyclopentyl] pyrazol-5-yl] carbamate (230 mg, 0.472 mmol) in Methanol (5.0 mL) was added Pd/C (50.3 mg, 5%wt., 0.0236 mmol) . The reaction mixture was stirred under hydrogen atmosphere (balloon) at 25℃ for 2 h. The mixture was filtered through a pad of Celite. The filtrate was concentrated under reduced pressure to afford (rac, cis) -2- [3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentyl] isoindoline-1, 3-dione (150 mg, 90%yield) as a brown oil which was used directly in the next step without further purification. LC-MS: m/z 353.8 [M+H]
+.
Intermediate 3 and 4

Step 1: (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol
To a suspension of benzyl (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) carbamate (20.0 g, 55.9 mmol) in THF (300 mL) was added Pd/C (10.0 g, 50 wt. %) at 25℃ and stirred at that temperature for 16 h under H
2. The reaction mixture was filtered and the filtrate was concentrated under reduced pressure to afford (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (12.0 g, 96%yield) as a white solid. LC-MS: m/z [M+H] + 224.2.
Step 2: 1- (tert-butyl) -3- ( (1S, 3R) -3- ( (tert-butyldimethylsilyl) oxy) cyclopentyl) -1H-pyrazol-
5-amine
To a solution of (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (4.00 g, 17.9 mmol) and Imidazole (2.44 g, 35.8 mmol) in CH
2Cl
2 (100 mL) was added tert-butyl-chloro-dimethyl-silane (6.67 mL, 5.40 g, 35.8 mmol) at 25 ℃. The mixture was stirred at that temperature for 16 h. The mixture was washed with water (100 mL × 2) . The organic layer was dried over Na
2SO
4 and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 25 %in 25 min) to afford 1- (tert-butyl) -3- ( (1S, 3R) -3- ( (tert-butyldimethylsilyl) oxy) cyclopentyl) -1H-pyrazol-5-amine (6.00 g, 99%yield) as a yellow oil. LC-MS: m/z [M+H] + 338.0.
Intermediate 5

A solution of benzyl (1- (tert-butyl) -3- ( (1R, 3S) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) car bamate (5.00 g, 14.0 mmol) in THF (20.0 mL) and EtOAc (20.0 mL) was stirred at 25 ℃ for 2 h under H
2 atmosphere (balloon) before it was filtered through a pad of Celite. The filtrate was concentrated under reduced pressure to give (1S, 3R) -3- (5-amin o-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (4.70 g, crude) as a white solid. LC-MS: m/z [M+H]
+224.2.
Synthetic Examples
Synthetic Example 1

Step 1: tert-butyl N- [4- [ (6-bromo-2-pyridyl) oxy] butyl] carbamate
To a solution of tert-butyl N- (4-hydroxybutyl) carbamate (240 mg, 1.3 mmol) in THF (5 mL) was added sodium hydride (55 mg, 1.4 mmol, 60%purity) at 0 ℃ under nitrogen. The reaction was stirred at 0 ℃for 30 min and then 2-bromo-6-fluoro-pyridine (200 mg, 1.1 mmol) was added. The reaction was stirred at 0 ℃ for 2 hours. The mixture was quenched with saturated NH
4Cl solution (50 mL) and extracted with EtOAc (3x30 mL) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrated. The residue was purified by flash chromatography on silica gel eluting with 0-40%EtOAc in PE to afford tert-butyl N- [4- [ (6-bromo-2-pyridyl) oxy] butyl] carbamate (295 mg, 75%yield) as colorless oil. LC-MS: m/z 344.7 [M+H]
+.
Step 2: (rac, cis) -tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- (3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) oxy) butyl) carbamate
To a mixture of tert-butyl N- [4- [ (6-bromo-2-pyridyl) oxy] butyl] carbamate (150 mg, 434 μmol) , (rac, cis) -3- (5-amino-2-tert-butyl-pyrazol-3-yl) cyclopentanol (110 mg, 492 μmol) and Cs
2CO
3 (290 mg, 890 μmol) in dioxane (5 mL) was added XantPhos (25 mg, 43 μmol) and Pd
2 (dba)
3 (20 mg, 22 μmol) under nitrogen. The reaction was stirred at 100 ℃ under nitrogen for 12 hours. The mixture was diluted with water (50 mL) and extracted with EtOAc (30 mL x 3) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrated. The residue was purified by flash chromatography on silica gel eluting with 0-40%EtOAc in PE to afford (rac, cis) -tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- (3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) oxy) butyl) carbamate (100 mg, 47%yield) as a light brown solid. LC-MS: m/z 487.9 [M+H]
+.
Step 3: (rac, cis) -tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- (3- ( ( (4-
nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-
yl) oxy) butyl) carbamate
A mixture of (rac, cis) -tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- (3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) oxy) butyl) carbamate (100 mg, 205 μmol) , 4-Nitrophenyl chloroformate (125 mg, 620 μmol) , Et
3N (145 μL, 1.04 mmol) and DMAP (5 mg, 41 μmol) in DCE (5 mL) was stirred at 70 ℃ for 12 hours. The mixture was diluted with water (50 mL) and extracted with DCM (3x30 mL) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrated. The residue was purified by flash chromatography on silica gel eluting with 0-30%EtOAc in PE to afford (rac, cis) -tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- (3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) oxy) butyl) carbamate (85 mg, 63%yield) as a light brown solid. LC-MS: m/z 653.8 [M+H]
+.
Step 4: [ (rac, cis) -3- [3- [ [6- (4-aminobutoxy) -2-pyridyl] amino] -1H-pyrazol-5-yl] cyclopentyl] (4-
nitrophenyl) carbonate
A mixture of (rac, cis) -tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- (3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) oxy) butyl) carbamate (85 mg, 130 μmol) in TFA (5 mL) was stirred at 100 ℃ for 5 hours. The solvent was evaporated to afford [ (rac, cis) -3- [3- [ [6- (4-aminobutoxy) -2-pyridyl] amino] -1H-pyrazol-5-yl] cyclopentyl] (4-nitrophenyl) carbonate; 2, 2, 2-trifluoroacetic acid (80 mg, 100%yield) as colorless oil, which was used in the next step directly. LC-MS: m/z 496.8 [M+H]
+.
Step 5: (rac, cis) -2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one
To a mixture of [ (rac, cis) -3- [3- [ [6- (4-aminobutoxy) -2-pyridyl] amino] -1H-pyrazol-5-yl] cyclopentyl] (4-nitrophenyl) carbonate; 2, 2, 2-trifluoroacetic acid (80 mg, 131 μmol) in MeCN (10 mL) was added Et
3N (100 μL, 717 μmol) at 15 ℃. The reaction was stirred at 15 ℃ for 1 hour. The solvent was evaporated and the residue was purified by prep-HPLC (C18, 35-70%MeCN in 0.05%FA/water, 40 mL/min) to afford (rac, cis) -2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) - cyclopentanacyclododecaphan-11-one (11.8 mg, 25%yield) as a white solid. LC-MS: m/z 357.9 [M+H]
+.
1H-NMR (400 MHz, DMSO-d6) δ 11.82 (s, 1H) , 9.34 -9.10 (m, 1H) , 7.36 (t, J = 8.0 Hz, 1H) , 6.88 (t, J = 6.0 Hz, 1H) , 6.66 -6.44 (m, 1H) , 6.31 (d, J = 8.0 Hz, 1H) , 5.99 (d, J = 8.0 Hz, 1H) , 5.12 -4.94 (m, 1H) , 4.49 –3.94 (m, 2H) , 3.24 –2.70 (m, 3H) , 2.08 –1.43 (m, 10H) .
The compounds in below table were prepared in accordance with the synthetic protocols set forth in Example 1 using the appropriate starting materials.
Synthetic Example 2

LC-MS: m/z 371.8 [M+H]
+.
1H-NMR (400 MHz, CDCl
3) δ 7.39 (t, J = 8.0 Hz, 1H) , 6.91 (s, 1H) , 6.75 (s, 1H) , 6.65 (s, 1H) , 6.14 (d, J = 8.0 Hz, 2H) , 5.26 -5.06 (m, 1H) , 4.77 -4.60 (m, 1H) , 4.49 –4.22 (m, 2H) , 4.16 -4.06 (m, 1H) , 3.62 -3.46 (m, 1H) , 3.38 -3.24 (m, 1H) , 3.20 -2.51 (m, 5H) , 2.21 -2.07 (m, 2H) , 1.68 -1.37 (m, 4H) .
Synthetic Example 3

LC-MS: m/z 385.9 [M+H]
+.
1H-NMR (400 MHz, CDCl
3) δ 7.40 (t, J = 8.0 Hz, 1H) , 6.96 -6.63 (m, 1H) , 6.17 (d, J = 8.0 Hz, 1H) , 6.14 (d, J = 7.6 Hz, 1H) , 5.31 -5.07 (m, 1H) , 4.62 –4.17 (m, 2H) , 3.58 -3.37 (m, 1H) , 3.26 -2.90 (m, 2H) , 2.73 –2.52 (m, 1H) , 2.16 –1.86 (m, 8H) , 1.58 -1.37 (m, 6H) .
Synthetic Example 4

LC-MS: m/z 343.8 [M+H]
+.
1H-NMR (400 MHz, DMSO-d6) δ 9.35 -9.12 (m, 1H) , 8.14 (s, 1H) , 7.35 (t, J = 8.0 Hz, 1H) , 7.10 (d, J = 8.0 Hz, 1H) , 6.43 (s, 1H) , 6.32 (d, J = 7.8 Hz, 1H) , 5.98 (d, J = 8.0 Hz, 1H) , 4.90 -4.78 (m, 1H) , 3.82 -3.71 (m, 1H) , 3.50 -3.42 (m, 1H) , 3.22 -3.07 (m, 2H) , 2.99 -2.86 (m, 1H) , 2.41 -2.30 (m, 1H) , 2.18 -2.06 (m, 1H) , 2.05 -1.95 (m, 1H) , 1.90 -1.59 (m, 5H) .
Synthetic Example 5

LC-MS: m/z 395.8 [M+H]
+.
1H NMR (400 MHz, DMSO-d
6) δ 11.91 (s, 1H) , 9.08 (d, J = 58.2 Hz, 1H) , 7.84 –6.71 (m, 1H) , 6.62 –6.14 (m, 2H) , 5.97 (t, J = 8.0 Hz, 1H) , 4.96 (d, J = 7.1 Hz, 1H) , 4.70 (s, 1H) , 3.62 (dt, J = 7.5, 3.5 Hz, 1H) , 3.02 (s, 1H) , 2.82 –2.58 (m, 1H) , 2.34 –2.18 (m, 1H) , 2.15 –1.91 (m, 10H) , 1.77 (d, J = 8.9 Hz, 2H) .
Synthetic Example 6

LC-MS: m/z 401.8 [M+H]
+.
1H-NMR (400 MHz, DMSO-d6) δ 11.92 (s, 1H) , 7.20 (d, J = 8.0 Hz, 1H) , 6.93 (d, J = 9.2 Hz, 1H) , 6.83 (brs, 1H) , 6.60 -6.43 (m, 1H) , 6.04 (d, J = 8.4 Hz, 1H) , 5.08 -4.83 (m, 1H) , 4.42 –3.89 (m, 2H) , 3.79 (s, 3H) , 3.18 -2.68 (m, 2H) , 2.64 -2.55 (m, 1H) , 2.14 –1.26 (m, 12H) .
Synthetic Example 7

LC-MS: m/z 373.8 [M+H]
+.
1H-NMR (400 MHz, MeOD) δ 7.42 (t, J = 8.0 Hz, 1H) , 6.67 (s, 1H) , 6.31 (d, J = 8.0 Hz, 1H) , 6.11 (d, J = 8.0 Hz, 1H) , 5.15 -5.02 (m, 1H) , 4.55 -4.40 (m, 1H) , 4.25 -4.14 (m, 1H) , 3.92 -3.73 (m, 2H) , 3.70 -3.45 (m, 3H) , 3.23 -2.99 (m, 2H) , 2.66 -2.53 (m, 1H) , 2.16 -1.97 (m, 2H) , 1.97 -1.73 (m, 3H) .
Synthetic Example 8

LC-MS: m/z 370.9 [M+H]
+.
Synthetic Example 9

LC-MS: m/z 375.8 [M+H]
+.
1H-NMR (400 MHz, DMSO-d6) δ 11.97 (s, 1H) , 9.01 -8.59 (m, 1H) , 7.41 (t, J = 9.6 Hz, 1H) , 6.92 -6.79 (m, 1H) , 6.67 -6.44 (m, 1H) , 6.01 (d, J = 8.4 Hz, 1H) , 5.12 -4.92 (m, 1H) , 4.54 -3.89 (m, 2H) , 3.22 -3.05 (m, 1H) , 3.04 -2.71 (m, 2H) , 2.61 -2.53 (m, 1H) , 2.07 -1.93 (m, 1H) , 1.91 -1.63 (m, 6H) , 1.60 -1.43 (m, 2H) .
Synthetic Example 10

LC-MS: m/z 393.8 [M+H]
+.
1H-NMR (400 MHz, DMSO-d6) δ 11.97 (s, 1H) , 9.14 -8.62 (m, 1H) , 7.74 (t, J = 9.6 Hz, 1H) , 7.35 -6.74 (m, 1H) , 6.62 -6.30 (m, 1H) , 5.13 -4.92 (m, 1H) , 4.66 -3.94 (m, 2H) , 3.22 -3.02 (m, 1H) , 3.03 -2.70 (m, 2H) , 2.60 -2.54 (m, 1H) , 2.05 -1.41 (m, 9H) .
Synthetic Example 11

Step 1: (rac, cis) -tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- (3- (1, 3-dioxoisoindolin-2-yl) cyclopentyl) -1H-
pyrazol-5-yl) amino) pyridin-2-yl) oxy) butyl) carbamate
To a stirred solution of (rac, cis) -2- (3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl) isoindoline-1, 3-dione (100 mg, 0.284 mmol) in 1, 4-dioxane (3.0 mL) were sequentially added tert-butyl N- [4- [ (6-bromo-2-pyridyl) oxy] butyl] carbamate (108 mg, 0.312 mmol) , 5-diphenylphosphanyl-9, 9-dimethyl-xanthen-4-yl-diphenyl-phosphane (16.4 mg, 0.0284 mmol) , dicesium carbonate (185 mg, 0.567 mmol) and 1, 5-diphenylpenta-1, 4-dien-3-one palladium (13.0 mg, 0.142 mmol) at 25 ℃. The reaction mixture was warmed to 100 ℃ and stirred at that temperature for 8 h. The reaction mixture was cooled to 25 ℃ and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/petroleum ether with EtOAc from 0 to 50%in 20 min to give (rac, cis) -tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- (3- (1, 3-dioxoisoindolin-2-yl) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) oxy) butyl) carbamate (100 mg, 57%yield) as a colorless oil. LC-MS: m/z 617.8 [M+H]
+ .
Step 2: (rac, cis) -tert-butyl (4- ( (6- ( (3- (3-aminocyclopentyl) -1- (tert-butyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) oxy) butyl) carbamate
To a solution of (rac, cis) -tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- (3- (1, 3-dioxoisoindolin-2-yl) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) oxy) butyl) carbamate (100 mg, 0.162 mmol) in Ethanol (4.0 mL) were sequentially added hydrazine hydrate (29.6 μL, 80%wt., 0.486 mmol) at 25 ℃. The reaction mixture was warmed to 80 ℃ and stirred at that temperature for 3 h. The reaction mixture was cooled to 25 ℃ and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with MeOH/CH
2Cl
2 with MeOH from 0 to 20%in 20 min to give (rac, cis) -tert-butyl (4- ( (6- ( (3- (3-aminocyclopentyl) -1- (tert-butyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) oxy) butyl) carbamate (60 mg, 76%yield) as a colorless oil. LC-MS: m/z 487.9 [M+H]
+ .
Step 3: (rac, cis) -tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- (3- ( ( (4-
nitrophenoxy) carbonyl) amino) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-
yl) oxy) butyl) carbamate
To a stirred solution of (rac, cis) -tert-butyl (4- ( (6- ( (3- (3-aminocyclopentyl) -1- (tert-butyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) oxy) butyl) carbamate (50 mg, 0.10 mmol) in dichloromethane (10 mL) were sequentially added (4-nitrophenyl) carbonochloridate (22.8 mg, 0.113 mmol) , N, N-diethylethanamine (15.6 mg, 21.5 μL, 0.154 mmol) and N, N-dimethylpyridin-4-amine (1.3 mg, 0.010 mmol) at 25 ℃. The reaction mixture was stirred at that temperature for 8 h before it was quenched with saturated aq. NaHCO
3 (0.1 mL) . The mixture was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/petroleum ether with EtOAc from 0 to 75 %in 20 min to give (rac, cis) -tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- (3- ( ( (4-nitrophenoxy) carbonyl) amino) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) oxy) butyl) carbamate (50 mg, 75%yield) as a colorless oil. LC-MS: m/z 652.7 [M+H]
+ .
Step 4: (rac, cis) - (4-nitrophenyl) N- [3- [3- [ [6- (4-aminobutoxy) -2-pyridyl] amino] -1H-pyrazol-5-
yl] cyclopentyl] carbamate
To a Biotage pressure tube were sequentially added (rac, cis) -tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- (3- ( ( (4-nitrophenoxy) carbonyl) amino) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) oxy) butyl) carbamate (45 mg, 0.069 mmol) and trifluoroacetic acid (10 mL) at 25 ℃. The mixture was sealed, warmed to 80 ℃ under microwave and stirred at that temperature for 30 min. The mixture was cooled to 25 ℃and concentrated under reduced pressure to give (rac, cis) - (4-nitrophenyl) N- [3- [3- [ [6- (4-aminobutoxy) -2-pyridyl] amino] -1H-pyrazol-5-yl] cyclopentyl] carbamate (34 mg, 100%yield) as a colorless oil which was used directly in the next step without further purification. LC-MS: m/z 496.8 [M+H]
+.
Step 5: (rac, cis) -2
1H-5-oxa-3, 10, 12-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one
To a stirred solution of (rac, cis) - (4-nitrophenyl) N- [3- [3- [ [6- (4-aminobutoxy) -2-pyridyl] amino] -1H-pyrazol-5-yl] cyclopentyl] carbamate (35 mg, 0.071 mmol) in acetonitrile (10 mL) was added N, N-diethylethanamine (14.3 mg, 19.8 μL, 0.141 mmol) at 25 ℃. The resulting mixture was stirred at that temperature for 2 h before it was concentrated under reduced pressure. The residue was purified by Pre-HPLC eluting with CH
3CN in water with CH
3CN from 5%to 95%in 9 min to give (rac, cis) -2
1H-5-oxa-3, 10, 12-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (5.0 mg, 20%yield) as a white solid.
1H NMR (400 MHz, DMSO-d
6) : δ 9.16 (s, 1H) , 8.13 (s, 1H) , 7.34 (t, J = 7.9 Hz, 1H) , 6.56 (s, 1H) , 6.30 (d, J = 7.9 Hz, 1H) , 5.98 (d, J = 7.7 Hz, 1H) , 5.90 –5.78 (m, 2H) , 4.38 (d, J = 9.6 Hz, 1H) , 4.14 –4.03 (m, 1H) , 4.01 –3.88 (m, 1H) , 3.13 (dd, J = 11.2, 4.4 Hz, 1H) , 2.82 (dd, J = 13.8, 5.6 Hz, 1H) , 2.37 –2.23 (m, 1H) , 1.97 –1.87 (m, 1H) , 1.86 –1.64 (m, 5H) , 1.60 –1.47 (m, 3H) . LC-MS: m/z 357.8 [M+H]
+ .
Example 12
(rac, cis) -2
1H-5-oxa-3, 11, 13-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclotridecaphan-12-one

Step 1: (rac, cis) -tert-butyl (5- ( (6- ( (1- (tert-butyl) -3- (3- (1, 3-dioxoisoindolin-2-yl) cyclopentyl) -1H-
pyrazol-5-yl) amino) pyridin-2-yl) oxy) pentyl) carbamate
To a Biotage pressure tube were sequentially added tert-butyl N- [5- [ (6-bromo-2-pyridyl) oxy] pentyl] carbamate (224 mg, 0.624 mmol) , (rac, cis) -2- [3- (5-amino-2-tert-butyl-pyrazol-3-yl) cyclopentyl] isoindoline-1, 3-dione (200 mg, 0.567 mmol) , Cs
2CO
3 (370 mg, 1.13 mmol) , Pd
2 (dba)
3 (26.0 mg, 0.0284 mmol) , Xantphos (32.8 mg, 0.0568 mmol) and 1, 4-dioxane (6.0 mL) at 25 ℃. The mixture was bubbled with N
2 for 10 mins before it was sealed and warmed to 100 ℃ and stirred at that temperature for 6 h. The reaction mixture was cooled to 25 ℃ and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/petroleum ether with EtOAc from 0 to 100%in 20 min to give (rac, cis) -tert-butyl N- [5- [ [6- [ [1-tert-butyl-5- [3- (1, 3-dioxoisoindolin-2-yl) cyclopentyl] pyrazol-3-yl] amino] -2-pyridyl] oxy] pentyl] carbamate (310 mg, 87%yield) as a brown oil. LC-MS: m/z 631.8 [M+H]
+ .
Step 2: (rac, cis) -tert-butyl (5- ( (6- ( (3- (3-aminocyclopentyl) -1- (tert-butyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) oxy) pentyl) carbamate
To a stirred solution of (rac, cis) -tert-butyl N- [5- [ [6- [ [1-tert-butyl-5- [3- (1, 3-dioxoisoindolin-2-yl) cyclopentyl] pyrazol-3-yl] amino] -2-pyridyl] oxy] pentyl] carbamate (300 mg, 0.476 mmol) in Ethanol (5.0 mL) was added hydrazine hydrate (89.3 mg, 86.7 μL, 1.43 mmol) at 25 ℃. The reaction mixture was warmed to 80 ℃ and stirred at that temperature for 3 h. The reaction mixture was cooled to 25 ℃, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with MeOH/CH
2Cl
2 with MeOH from 0 to 20%in 20 min to give (rac, cis) -tert-butyl N- [5- [ [6- [ [5- [3-aminocyclopentyl] -1-tert-butyl-pyrazol-3-yl] amino] -2-pyridyl] oxy] pentyl] carbamate (220 mg, 92%yield) as a colorless oil. LC-MS: m/z 501.9 [M+H]
+ .
Step 3: (rac, cis) -4-nitrophenyl (3- (5- ( (6- ( (5- ( (tert-butoxycarbonyl) amino) pentyl) oxy) pyridin-2-
yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl) carbamate
To a stirred solution of (rac, cis) -tert-butyl N- [5- [ [6- [ [5- [3-aminocyclopentyl] -1-tert-butyl-pyrazol-3-yl] amino] -2-pyridyl] oxy] pentyl] carbamate (108 mg, 0.216 mmol) in dichloromethane (10 mL) were sequentially added (4-nitrophenyl) carbonochloridate (65.2 mg, 0.324 mmol) , Et
3N (65.5 mg, 90.2 μL, 0.647 mmol) and DMAP (65.5 mg, 0.647 mmol) at 25 ℃. The reaction mixture was stirred at that temperature for 8 h before it was quenched with saturated aq. NaHCO
3 (0.1 mL) . The mixture was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/petroleum ether with EtOAc from 0 to 75 %in 20 min to give (rac, cis) - (4-nitrophenyl) N- [3- [5- [ [6- [5- (tert-butoxycarbonylamino) pentoxy] -2-pyridyl] amino] -2-tert-butyl-pyrazol-3-yl] cyclopentyl] carbamate (110 mg, 77%yield) as a color oil. LC-MS: m/z 666.8 [M+H]
+ .
Step 4: (rac, cis) -4-nitrophenyl (3- (3- ( (6- ( (5-aminopentyl) oxy) pyridin-2-yl) amino) -1H-pyrazol-5-
yl) cyclopentyl) carbamate
To a Biotage pressure tube were sequentially added (rac, cis) - (4-nitrophenyl) N- [3- [5- [ [6- [5- (tert-butoxycarbonylamino) pentoxy] -2-pyridyl] amino] -2-tert-butyl-pyrazol-3-yl] cyclopentyl] carbamate (110 mg, 0.165 mmol) and TFA (10 mL) at 25 ℃. The mixture was sealed, warmed to 80 ℃ under microwave and stirred at that temperature for 30 min. The mixture was cooled to 25 ℃and concentrated under reduced pressure to give (rac, cis) - (4-nitrophenyl) N- [3- [3- [ [6- (5-aminopentoxy) -2-pyridyl] amino] -1H-pyrazol-5-yl] cyclopentyl] carbamate (84.0 mg, 0.165 mmol, 99%yield) as a colorless oil which was used directly in the next step without further purification. LC-MS: m/z 510.8 [M+H]
+.
Step 5: (rac, cis) -2
1H-5-oxa-3, 11, 13-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclotridecaphan-12-one
To a stirred solution of (rac, cis) - (4-nitrophenyl) N- [3- [3- [ [6- (5-aminopentoxy) -2-pyridyl] amino] -1H-pyrazol-5-yl] cyclopentyl] carbamate (84 mg, 0.165 mmol) in acetonitrile (10 mL) was added Et
3N (16.7 mg, 23.0 μL, 165 mmol) at 25 ℃. The resulting mixture was stirred at that temperature for 2 h before it was concentrated under reduced pressure. The residue was purified by Pre-HPLC eluting with CH
3CN in water with CH
3CN from 5%to 95%in 9 min to give (rac, cis) -2
1H-5-oxa-3, 11, 13-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclotridecaphan-12-one (18.5 mg, 30%yield) as a white solid. LC-MS: m/z 371.9 [M+H]
+.
Example 13
(rac, cis) -2
1H-5, 10-dioxa-3, 12-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one

Step 1: 2-bromo-6- (4- ( (tert-butyldimethylsilyl) oxy) butoxy) pyridine
To a stirred solution of 4- [tert-butyl (dimethyl) silyl] oxybutan-1-ol (1.05 g, 5.14 mmol) in THF (20.0 mL) were sequentially added sodium hydride (256 mg, 6.68 mmol, 60%wt. ) and 2-bromo-6-fluoro-pyridine (994 mg, 5.65 mmol) at 0 ℃. The reaction mixture was stirred at that temperature for 10 min before it was warmed to 50 ℃ and stirred at that temperature for 4 h. The reaction mixture was cooled to 25 ℃, quenched with saturated aq. NH
4Cl (30 mL) and extracted with EtOAc (50 mL × 3) . The combined organic phases were washed with brine, dried over anhydrous Na
2SO
4, and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/petroleum ether (with EtOAc from 0 to 50%) in 20 min to give 4- [ (6-bromo-2-pyridyl) oxy] butoxy-tert-butyl-dimethyl-silane (1.51 g, 82%yield) as a yellow oil. LC-MS: m/z 359 . 8 [M+H]
+ .
Step 2: (rac, cis) -2- ( (1R, 3S) -3- (1- (tert-butyl) -5- ( (6- (4- ( (tert-
butyldimethylsilyl) oxy) butoxy) pyridin-2-yl) amino) -1H-pyrazol-3-yl) cyclopentyl) isoindoline-1, 3-
dione
To a Biotage pressure tube were sequentially added 4- [ (6-bromo-2-pyridyl) oxy] butoxy-tert-butyl-dimethyl-silane (225 mg, 0.624 mmol) , (rac, cis) -2- [3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentyl] isoindoline-1, 3-dione (200 mg, 0.567 mmol) , Cs
2CO
3 (370 mg, 1.13 mmol) , Pd
2 (dba)
3 (26.0 mg, 0.0284 mmol) , Xantphos (32.8 mg, 0.0568 mmol) and 1, 4-dioxane (6.0 mL) at 25 ℃. The mixture was bubbled with N
2 for 10 min before it was sealed and warmed to 100 ℃ and stirred at that temperature for 6 h. The reaction mixture was cooled to 25 ℃ and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/petroleum ether with EtOAc from 0 to 100%in 20 min to give (rac, cis) -2- [3- [1-tert-butyl-5- [ [6- [4- [tert-butyl (dimethyl) silyl] oxybutoxy] -2-pyridyl] amino] pyrazol-3-yl] cyclopentyl] isoindoline-1, 3-dione (285 mg, 79%yield) as a brown oil. LC-MS: m/z 632.8 [M+H]
+ .
Step 3: (rac, cis) -N- (3- ( (3-aminocyclopentyl) -1- (tert-butyl) -1H-pyrazol-5-yl) -6- (4- ( (tert-
butyldimethylsilyl) oxy) butoxy) pyridin-2-amine
To a stirred solution of (rac, cis) -2- [3- [1-tert-butyl-5- [ [6- [4- [tert-butyl (dimethyl) silyl] oxybutoxy] -2-pyridyl] amino] pyrazol-3-yl] cyclopentyl] isoindoline-1, 3-dione (285 mg, 0.451 mmol) in Ethanol (10 mL) was added hydrazine hydrate (84.7 mg, 82.2 μL, 1.35 mmol) at 25 ℃. The mixture was warmed to 80 ℃ and stirred at that temperature for 3 h before it was filtered through a pad of Celite. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with MeOH/CH
2Cl
2 containing 1%Et
3N (with MeOH from 1%to 10%) in 20 min to give (rac, cis) -N- [5- [3-aminocyclopentyl] -2-tert-butyl-pyrazol-3-yl] -6- [4- [tert-butyl (dimethyl) silyl] oxybutoxy] pyridin-2-amine (200 mg, 88%yield) as a colorless oil. LC-MS: m/z 502.9 [M+H]
+ .
Step 4: (rac, cis) -tert-butyl (3- (1- (tert-butyl) -5- ( (6- (4- ( (tert-
butyldimethylsilyl) oxy) butoxy) pyridin-2-yl) amino) -1H-pyrazol-3-yl) cyclopentyl) carbamate
To a stirred solution of (rac, cis) -N- [5- [3-aminocyclopentyl] -2-tert-butyl-pyrazol-3-yl] -6- [4- [tert-butyl (dimethyl) silyl] oxybutoxy] pyridin-2-amine (200 mg, 0.399 mmol) in CH
2Cl
2 (5.0 mL) were sequentially added Et
3N (121mg, 167 μL, 1.20 mmol) , (Boc)
2O (130mg, 137 μL, 598 mmol) at 25 ℃. The mixture was stirred at that temperature for 3 h before it was quenched with saturated aq. NaHCO
3 (20 mL) and extracted with EtOAc (20 mL × 3) . The combined organic phases were washed with brine, dried over anhydrous Na
2SO
4, and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/petroleum ether (with EtOAc from 0 to 100%) in 20 min to give (rac, cis) -tert-butyl N- [3- [1-tert-butyl-5- [ [6- [4- [tert-butyl (dimethyl) silyl] oxybutoxy] -2-pyridyl] amino] pyrazol-3-yl] cyclopentyl] carbamate (220 mg, 92%yield) as a colorless oil. LC-MS: m/z 601.9 [M+H]
+.
Step 5: (rac, cis) -tert-butyl (3- (1- (tert-butyl) -5- ( (6- (4-hydroxybutoxy) pyridin-2-yl) amino) -1H-
pyrazol-3-yl) cyclopentyl) carbamate
To a stirred solution of (rac, cis) -tert-butyl N- [3- [1-tert-butyl-5- [ [6- [4- [tert-butyl (dimethyl) silyl] oxybutoxy] -2-pyridyl] amino] pyrazol-3-yl] cyclopentyl] carbamate (220 mg, 0.366 mmol) in THF (1.0 mL) was added TBAF (731 μL, 1.0 M, 0.731 mmol) at 25 ℃. The mixture was stirred at that temperature for 2 h before it was quenched with saturated aq. NaHCO
3 (0.1 mL) and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/petroleum ether (with EtOAc from 0 to 100%) in 20 min to give (rac, cis) -tert-butyl N- [3- [1-tert-butyl-5- [ [6- (4-hydroxybutoxy) -2-pyridyl] amino] pyrazol-3-yl] cyclopentyl] carbamate (168 mg, 0.345 mmol, 94%yield) as a colorless oil. LC-MS: m/z 488.9 [M+H]
+.
Step 6: (rac, cis) -tert-butyl (3- (1- (tert-butyl) -5- ( (6- (4- ( ( (4-
nitrophenoxy) carbonyl) oxy) butoxy) pyridin-2-yl) amino) -1H-pyrazol-3-yl) cyclopentyl) carbamate
To a stirred solution of (rac, cis) -tert-butyl N- [3- [1-tert-butyl-5- [ [6- (4-hydroxybutoxy) -2-pyridyl] amino] pyrazol-3-yl] cyclopentyl] carbamate (100 mg, 0.205 mmol) were sequentially added (4-nitrophenyl) carbonochloridate (124 mg, 0.615 mmol) , Et
3N (125 mg, 171 μL, 1.23 mmol) at 25 ℃. The reaction mixture was stirred at that temperature for 8 h before it was quenched with saturated aq. NaHCO
3 (0.1 mL) . The mixture was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/petroleum ether with EtOAc from 0 to 75 %in 20 min to give (rac, cis) -4- [ [6- [ [5- [3- (tert-butoxycarbonylamino) cyclopentyl] -2-tert-butyl-pyrazol-3-yl] amino] -2-pyridyl] oxy] butyl (4-nitrophenyl) carbonate (80.0 mg, 60%yield) as a color oil. LC-MS: m/z 653.8 [M+H]
+.
Step 7: (rac, cis) -4- ( (6- ( (3- (3-aminocyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) oxy) butyl
(4-nitrophenyl) carbonate
To a Biotage pressure tube were sequentially added (rac, cis) -4- [ [6- [ [5- [3- (tert-butoxycarbonylamino) cyclopentyl] -2-tert-butyl-pyrazol-3-yl] amino] -2-pyridyl] oxy] butyl (4-nitrophenyl) carbonate (50.0 mg, 0.766 mmol) and TFA (5.0 mL) at 25 ℃. The mixture was sealed, warmed to 80 ℃ under microwave and stirred at that temperature for 30 min. The mixture was cooled to 25 ℃ and concentrated under reduced pressure to give (rac, cis) -4- [ [6- [ [5- [3-aminocyclopentyl] -1H-pyrazol-3-yl] amino] -2-pyridyl] oxy] butyl (4-nitrophenyl) carbonate (37.0 mg, 97%yield) as a colorless oil which was used directly in the next step without further purification. LC-MS: m/z 497.8 [M+H]
+.
Step 8: (rac, cis) -2
1H-5, 10-dioxa-3, 12-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one
To a stirred solution of (rac, cis) -4- [ [6- [ [3- [3-aminocyclopentyl] -1H-pyrazol-5-yl] amino] -2-pyridyl] oxy] butyl (4-nitrophenyl) carbonate (37.0 mg, 0.0745 mmol) in acetonitrile (3.0 mL) was added Et
3N (75.4 mg, 104 μL, 0.745 mmol) at 25 ℃. The resulting mixture was stirred at that temperature for 2 h before it was concentrated under reduced pressure. The residue was purified by Pre-HPLC eluting with CH
3CN in water with CH
3CN from 5%to 95%in 9 min to give (rac, cis) -2
1H-5, 10-dioxa-3, 12-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (3.5 mg, 13%yield) as a white solid. LC-MS: m/z 358.9 [M+H]
+.
Example 202
(rac, cis) -2
1H-5-oxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclodecaphane-10-carboxamide


Step 1: (rac, cis) -4- ( (6- ( (3- ( (3-aminocyclopentyl) -1- (tert-butyl) -1H-pyrazol-5-yl) amino) pyridin-
2-yl) oxy) butan-1-ol
To a stirred solution of (rac, cis) -N- [5- [3-aminocyclopentyl] -2-tert-butyl-pyrazol-3-yl] -6- [4- [tert-butyl (dimethyl) silyl] oxybutoxy] pyridin-2-amine (100 mg, 0.199 mmol) in THF (2.0 mL) was added TBAF (598 μL, 1.0 M in THF, 0.598 mmol) at 25 ℃. The mixture was stirred at that temperature for 1 h before it was quenched with saturated aq. NaHCO
3 (0.1 mL) and concentrated under vacuum. The residue was purified by silica gel chromatography eluting with MeOH/CH
2Cl
2 containing 1%Et
3N (with MeOH from 0 to 10%) in 20 min to give to (rac, cis) -4- [ [6- [ [5- [3-aminocyclopentyl] -2-tert-butyl-pyrazol-3-yl] amino] -2-pyridyl] oxy] butan-1-ol (65.0 mg, 84%yield) as a colorless oil. LC-MS: m/z 388.9 [M+H]
+.
Step 2: (rac, cis) -tert-butyl (3- (1- (tert-butyl) -5- ( (6- (4-hydroxybutoxy) pyridin-2-yl) amino) -1H-
pyrazol-3-yl) cyclopentyl) carbamate
To a stirred solution of (rac, cis) -4- [ [6- [ [5- [3-aminocyclopentyl] -2-tert-butyl-pyrazol-3-yl] amino] -2-pyridyl] oxy] butan-1-ol (55.0 mg, 0.142 mmol) in CH
2Cl
2 (3.0 mL) were sequentially added Et
3N (79 μL, 57.5 mg, 0.568 mmol) , (Boc)
2O (65 μL, 62.0 mg, 0.284 mmol) at 0 ℃. The mixture was stirred at that temperature for 1 h before it was quenched with saturated aq. NaHCO
3 (20 mL) and extracted with EtOAc (20 mL × 3) . The combined organic phases were washed with brine, dried over anhydrous Na
2SO
4, and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/petroleum ether (with EtOAc from 0 to 100%) in 20 min to give (rac, cis) -tert-butyl N- [3- [1-tert-butyl-5- [ [6- (4-hydroxybutoxy) -2-pyridyl] amino] pyrazol-3-yl] cyclopentyl] carbamate (61.0 mg, 88 yield) as a colorless oil. LC-MS: m/z 488.9 [M+H]
+.
Step 3: (rac, cis) -4- ( (6- ( (3- (3- ( (tert-butoxycarbonyl) amino) cyclopentyl) -1- (tert-butyl) -1H-
pyrazol-5-yl) amino) pyridin-2-yl) oxy) butyl 4-methylbenzenesulfonate
To a stirred solution of (rac, cis) -tert-butyl N- [3- [1-tert-butyl-5- [ [6- (4-hydroxybutoxy) -2-pyridyl] amino] pyrazol-3-yl] cyclopentyl] carbamate (61.0 mg, 0.125 mmol) in CH
2Cl
2 (5.0 mL) were sequentially added Et
3N (52 μL, 38.0 mg, 0.375 mmol) , TsCl (35.8 mg, 0.188 mmol) and DMAP (1.5 mg, 0.013 mmol) at 25 ℃. The reaction mixture was stirred at that temperature for 3 h before it was quenched with saturated aq. NaHCO
3 (0.1 mL) and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/petroleum ether (with EtOAc from 0 to 100%) in 20 min to (rac, cis) -4- [ [6- [ [5- [3- (tert-butoxycarbonylamino) cyclopentyl] -2-tert-butyl-pyrazol-3-yl] amino] -2-pyridyl] oxy] butyl 4-methylbenzenesulfonate (66.0 mg, 82 %yield) as a white solid. LC-MS: m/z 642.8 [M+H]
+.
Step 4: (rac, cis) -4- ( (6- ( (3- (3-aminocyclopentyl) -1- (tert-butyl) -1H-pyrazol-5-yl) amino) pyridin-2-
yl) oxy) butyl 4-methylbenzenesulfonate
To a stirred solution of (rac, cis) -4- [ [6- [ [5- [3- (tert-butoxycarbonylamino) cyclopentyl] -2-tert-butyl-pyrazol-3-yl] amino] -2-pyridyl] oxy] butyl 4-methylbenzenesulfonate (66.0 mg, 0.103 mmol) in CH
2Cl
2 (2.0 mL) was added TFA (396 μL, 586 mg, 5.14 mmol) at 25 ℃. The mixture was stirred at that temperature for 3 h before it was concentrated under reduced pressure to give (rac, cis) -4- [ [6- [ [5- [3-aminocyclopentyl] -2-tert-butyl-pyrazol-3-yl] amino] -2-pyridyl] oxy] butyl 4-methylbenzenesulfonate (55.0 mg, 99 %yield) as a colorless oil which was used in the next step without further purification. LC-MS: m/z 542.8 [M+H]
+.
Step 5: (rac, cis) -tert-butyl-21H-5-oxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -
cyclopentanacyclodecaphane
To a stirred solution of (rac, cis) -4- [ [6- [ [5- [3-aminocyclopentyl] -2-tert-butyl-pyrazol-3-yl] amino] -2-pyridyl] oxy] butyl 4-methylbenzenesulfonate (50 mg, 92.30 umol) in MeCN (10 mL) were sequentially added K
2CO
3 (54.9 mg, 0.554 mmol) and NaI (4.2 mg, 0.028 mmol) at 25 ℃. The reaction mixture was warmed to 60 ℃ and stirred at that temperature for 8 h. The reaction mixture was cooled to 25 ℃, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with MeOH/CH
2Cl
2 containing 1%Et
3N (with MeOH from 0 to 10%) in 20 min to give (rac, cis) -tert-butyl-2
1H-5-oxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclodecaphane (30.0 mg, 88%yield) as a colorless oil. LC-MS: m/z 370.9 [M+H]
+.
Step 6: (rac, cis) -21- (tert-butyl) -21H-5-oxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -
cyclopentanacyclodecaphane-10-carboxamide
To a stirred solution of (rac, cis) -tert-butyl-2
1H-5-oxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclodecaphane (30.0 mg, 0.0812 mmol) in iPrOH (3.0 mL) was added isocyanato (trimethyl) silane (55 μL, 46.8 mg, 0.406 mmol) at 25 ℃. The mixture was stirred at that temperature for 12 h before it was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/petroleum ether (with EtOAc from 0 to 100%) in 20 min to give (rac, cis) -21- (tert-butyl) -2
1H-5-oxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclodecaphane-10-carboxamid (31.0 mg, 93%yield) as a white solid. LC-MS: m/z 413.9 [M+H]
+.
Step 7: (rac, cis) -21H-5-oxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclodecaphane-10-carboxamide
To a Biotage pressure tube were sequentially added (rac, cis) -2
1- (tert-butyl) -21H-5-oxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclodecaphane-10-carboxamid (31.0 mg, 0.0752 mmol) and TFA (3.0 mL) at 25 ℃. The mixture was sealed and warmed to 80 ℃ by microwave radiation and stirred at that temperature for 30 min. The reaction mixture was cooled to 25 ℃ and concentrated under reduced pressure. The residue was purified by Pre-HPLC eluting with CH
3CN in water with CH
3CN from 5%to 95%in 10 min to give (rac, cis) -2
1H-5-oxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclodecaphane-10-carboxamide (5.0 mg, 19%yield) as a white solid. LC-MS: m/z 357.9 [M+H]
+.
The following compounds were prepared using the similar procedure disclosed in synthetic example 202.

Example 14
(1
1S, 1
3R, Z) -4
4-phenyl-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one

Step 1: 2, 6-dichloro-4-phenyl-pyridine
To a stirred solution of 4-bromo-2, 6-dichloro-pyridine (5.00 g, 22.04 mmol) in 1, 4-dioxane (75.0 mL) and H
2O (15.0 mL) were sequentially added phenylboronic acid (2.96 g, 24.24 mmol) , Pd (PPh
3)
4 (2.55 g, 2.20 mmol) and K
2CO
3 (4.57 g, 33.06 mmol) at 25 ℃. The reaction mixture was warmed to 80 ℃ and stirred at that temperature for 2 h before it was cooled to 25 ℃ and concentrated under reduced pressure. The reaction mixture was filtered through a pad of Celite with EtOAc (100 mL) , and the combined organic phases were concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with DCM/PE (with DCM from 0 to 25%in 25 min) to give 2, 6-dichloro-4-phenyl-pyridine (2.37 g, 48 %yield) as an off-white solid. LC-MS: m/z [M+H]
+224.1.
Step 2: tert-butyl (4- ( (6-chloro-4-phenylpyridin-2-yl) oxy) butyl) carbamate
To a stirred solution of tert-butyl N- (4-hydroxybutyl) carbamate (802 mg, 4.24 mmol) in THF (15.0 mL) was added NaH (535 mg, 13.39 mmol, 60 wt. %in mineral oil) at 0 ℃. The reaction mixture was stirred at that temperature for 30 min before 2, 6-dichloro-4-phenyl-pyridine (1.00 g, 4.46 mmol) in THF (5.0 mL) was added at that temperature. The reaction mixture was warmed to 60 ℃and stirred at that temperature for 2 h before it was concentrated under reduced pressure, purified by silica gel chromatography eluting with EtOAc /PE (with EtOAc from 0 to 25%in 25 min) to give tert-butyl (4- ( (6-chloro-4-phenylpyridin-2-yl) oxy) butyl) carbamate (700 mg, 42 %yield) as a colourless oil. LC-MS: m/z [M+H]
+ 377.2.
Step 3: tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( (tert-butyldiphenylsilyl) oxy) cyclopentyl) -
1H-pyrazol-5-yl) amino) -4-phenylpyridin-2-yl) oxy) butyl) carbamate
To a stirred solution of tert-butyl (4- ( (6-chloro-4-phenylpyridin-2-yl) oxy) butyl) carbamate (100 mg, 265 μmol) were sequentially added 2-tert-butyl-5- [ (1S, 3R) -3- [tert-butyl (diphenyl) silyl] oxycyclopentyl] pyrazol-3-amine (98.0 mg, 212 μmol) , XantPhos Pd G
3 (18.5 mg, 53.1 μmol) and Cs
2CO
3 (173 mg, 531 μmol) in 1, 4-dioxane (4.0 mL) at 25 ℃. The reaction mixture was warmed to 100 ℃ and stirred at that temperature for 2 h before it was cooled to 25 ℃and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with DCM /PE (with DCM from 0 to 15%in 15 min) to give tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( (tert-butyldiphenylsilyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) -4-phenylpyridin-2-yl) oxy) butyl) carbamate (107 mg, 50 %yield) as a light yellow oil. LC-MS: m/z [M+H]
+ 801.7.
Step 4: tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) -4-phenylpyridin-2-yl) oxy) butyl) carbamate
To a stirred solution of tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( (tert-butyldiphenylsilyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) -4-phenylpyridin-2- yl) oxy) butyl) carbamate (107 mg, 133.40 μmol) in THF (3.0 mL) was added TBAF (115 μL, 105 mg, 400 μmol, 1M in THF) at 25 ℃. The resulting mixture was stirred at that temperature for 16 h before it was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 50%in 25 min) to give tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -4-phenylpyridin-2-yl) oxy) butyl) carbamate (50 mg, 66 %yield) as a pale yellow solid. LC-MS: m/z [M+H]
+ 564.4.
Step 5: (1R, 3S) -3- (5- ( (6- (4- ( (tert-butoxycarbonyl) amino) butoxy) -4-phenylpyridin-2-yl) amino) -1-
(tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a stirred solution of tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -4-phenylpyridin-2-yl) oxy) butyl) carbamate (45.0 mg, 80 μmol) in DCM (1.0 mL) were sequentially added DMAP (5.0 mg, 40 μmol) , di (imidazol-1-yl) methanone (65.0 mg, 400 μmol) and N-ethyl-N-isopropyl-propan-2-amine (70 μL, 52.0 mg, 400 μmol) at 25 ℃. The reaction mixture was stirred at that temperature for 2 h before it was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/DCM (with EtOAc from 0 to 30%in 20 min) to give (1R, 3S) -3- (5- ( (6- (4- ( (tert-butoxycarbonyl) amino) butoxy) -4-phenylpyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (34 mg, 65 %yield) as a pale yellow solid. LC-MS: m/z [M+H]
+ 657.8.
Step 6: (1R, 3S) -3- (5- ( (6- (4-aminobutoxy) -4-phenylpyridin-2-yl) amino) -1- (tert-butyl) -1H-
pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a stirred solution of (1R, 3S) -3- (5- ( (6- (4- ( (tert-butoxycarbonyl) amino) butoxy) -4-phenylpyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (34.0 mg, 52 μmol) in DCM (1.0 mL) was added TFA (39.8 μL, 58.9 mg, 517 μmol ) at 25 ℃. The reaction mixture was stirred at that temperature for 1 h before it was concentrated under reduced pressure to afford (1R, 3S) -3- (5- ( (6- (4-aminobutoxy) -4-phenylpyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (28 mg, 97%yield) as a white solid. LC-MS: m/z [M+H]
+ 558.3.
Step 7: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4-phenyl-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a stirred solution of (1R, 3S) -3- (5- ( (6- (4-aminobutoxy) -4-phenylpyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (28.0 mg, 50.0 μmol) in CH
3CN (20.0 mL) was added Et
3N (7 μL, 5.0 mg, 50 μmol) at 25 ℃. The reaction mixture was warmed to 70 ℃and stirred at that temperature for 16 h before it was cooled to 25 ℃ and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 40%in 20 min) to give (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4-phenyl-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (13 mg, 53%yield) as a light yellow solid. LC-MS: m/z [M+H]
+ 489.9.
Step 8: (1
1S, 1
3R, Z) -4
4-phenyl-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one
A stirred solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4-phenyl-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (13 mg, 27 μmol) in TFA (2.0 mL) was warmed to 70 ℃ and stirred at that temperature for 16 h before it was cooled to 25 ℃ and concentrated under reduced pressure. The residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 5%to 60%in 50 min) to (1
1S, 1
3R, Z) -4
4-phenyl-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (4.3 mg, 10 μmol, 37 %yield) as a light pink solid. LC-MS: m/z [M+H]
+ 434.2.
Example 15
(1
1S, 1
3R, Z) -4
5-phenyl-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one

Step 1: 6-bromo-2-fluoro-3-iodopyridine
To a stirred solution of 2-bromo-6-fluoro-pyridine (10.0 g, 56.8 mmol, 5.86 mL) in THF (80.0 mL) was added LDA (31.3 mL, 6.70 g, 62.5 mmol, 2.0 M) slowly at -78 ℃ under N
2 atmosphere and the resulting mixture was stirred for 1 hours at that time before a solution of I
2 (14.4 g, 56.8 mmol) in THF (80.0 mL) was added slowly at -78 ℃. The reaction mixture was warmed to -60 ℃ within 1.5 h before it was quenched with a saturated aqueous of Na
2S
2O
3 (100 mL) at -60 ℃. The reaction mixture was warmed to 25 ℃ and extracted with EtOAc (200 mL × 2) . The combined organic layers were washed with brine (200 mL) and dried over Na
2SO
4, filtered, concentrated under reduced pressure. The residue was purified by flash chromatography eluting with EtOAc/PE (with EtOAc from 0 to 30%in 15 min) to afford 6-bromo-2-fluoro-3-iodopyridine (11.3 g, 66%yield) as a white solid. LC-MS: m/z [M+H]
+ 302.3.
Step 2: tert-butyl (4- ( (6-bromo-3-iodopyridin-2-yl) oxy) butyl) carbamate
To a solution of tert-butyl N- (4-hydroxybutyl) carbamate (627 mg, 3.31 mmol) in THF (20.0 mL) was added NaH (265 mg, 60 wt. %in mineral oil, 6.63 mmol) at 0 ℃. The reaction mixture was stirred at that temperature for 30 min before 6-bromo-2-fluoro-3-iodo-pyridine (1.0 g, 3.31 mmol) was added at 0 ℃. The reaction mixture was warmed to 25 ℃ and stirred at that temperature for 16 h before it was poured into saturated aqueous of NH
4Cl (20 mL) and extracted with EtOAc (20 mL × 3) . The combined organic phases were dried over Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 15%in 20 min) to afford tert-butyl (4- ( (6-bromo-3-iodopyridin-2-yl) oxy) butyl) carbamate (900 mg, 58 %yield) as a white solid. LC-MS: m/z [M+H]
+ 414.5, 416.5.
Step 3: tert-butyl (4- ( (6-bromo-3-phenylpyridin-2-yl) oxy) butyl) carbamate
To a stirred solution of tert-butyl (4- ( (6-bromo-3-iodopyridin-2-yl) oxy) butyl) carbamate (450 mg, 955 μmol) and phenylboronic acid (175 mg, 1.43 mmol) in CH
3CN (6.0 mL) and H
2O (1.0 mL) were sequentially added K
3PO
4 (405 mg, 1.91 mmol) , PPh
3 (50.1 mg, 191 μmol) and Pd (OAc)
2 (21.4 mg, 95.5 μmol) at 25 ℃. The mixture was warmed to 55 ℃ and stirred at that temperature for 16 h under N
2 before it was cooled to 25 ℃. The reaction mixture was concentrated under reduced pressure and purified by flash silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 10%in 15 min) to give tert-butyl (4- ( (6-bromo-3-phenylpyridin-2-yl) oxy) butyl) carbamate (230 mg, 57%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 421.9.
Step 4: tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) -3-phenylpyridin-2-yl) oxy) butyl) carbamate
To a stirred solution of tert-butyl (4- ( (6-bromo-3-phenylpyridin-2-yl) oxy) butyl) carbamate (396 mg, 940 μmol) , (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (140 mg, 627 μmol) and Cs
2CO
3 (408 mg, 1.25 mmol) in 1, 4 -dioxane (10.0 mL) were sequentially added Pd
2 (dba)
3 (57.4 mg, 62.7 μmol) and XantPhos (36.3 mg, 62.7 μmol) at 25 ℃. The reaction mixture was warmed up to 80 ℃ and stirred at that temperature for 16 h under N
2 before it was cooled to 25 ℃, the mixture was diluted with water (5 mL) and extracted with EtOAc (5 mL × 3) . The combined organic layer was washed with brine (5 mL) , dried over Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 40%in 25 min) to afford tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -3-phenylpyridin-2-yl) oxy) butyl) carbamate (230 mg, 65%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 564.3.
Step 5: (1R, 3S) -3- (5- ( (6- (4- ( (tert-butoxycarbonyl) amino) butoxy) -5-phenylpyridin-2-yl) amino) -1-
(tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a stirred solution of tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -3-phenylpyridin-2-yl) oxy) butyl) carbamate (200 mg, 355 μmol) in CH
2Cl
2 (3.0 mL) were sequentially added CDI (173 mg, 1.06 mmol) , DMAP (4.33 mg, 35.5 μmol) and DIPEA (247 μL, 179 mg, 1.77 mmol) at 25 ℃. The mixture was warmed to 35 ℃ and stirred at that temperature for 2 h before it was diluted with water (5 mL) and extracted with EtOAc (5 mL × 3) . The combined organic layer was washed with brine (10 mL) , dried over Na
2SO
4, filtered and concentrated to give (1R, 3S) -3- (5- ( (6- (4- ( (tert-butoxycarbonyl) amino) butoxy) -5-phenylpyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (200 mg, 86%yield) as yellow oil which was directly used to the next step without any further purification. LC-MS: m/z [M+H]
+ 658.3.
Step 6: (1R, 3S) -3- (3- ( (6- (4-aminobutoxy) -5-phenylpyridin-2-yl) amino) -1H-pyrazol-5-
yl) cyclopentyl 1H-imidazole-1-carboxylate
A stirred solution of (1R, 3S) -3- (5- ( (6- (4- ( (tert-butoxycarbonyl) amino) butoxy) -5-phenylpyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (200 mg, 304 μmol) in TFA (2.0 mL) was warmed to 70 ℃ and stirred at that temperature for 2 h before it was cooled to 25 ℃. The mixture was concentrated under reduced pressure to give (1R, 3S) -3- (3- ( (6- (4-aminobutoxy) -5-phenylpyridin-2-yl) amino) -1H-pyrazol-5-yl) cyclopentyl 1H-imidazole-1-carboxylate (150 mg) as yellow oil which was directly used to the next step without any further purification. LC-MS: m/z [M+H]
+ 502.
Step 7: (1
1S, 1
3R, Z) -4
5-phenyl-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one
To a stirred solution of (1R, 3S) -3- (3- ( (6- (4-aminobutoxy) -5-phenylpyridin-2-yl) amino) -1H-pyrazol-5-yl) cyclopentyl 1H-imidazole-1-carboxylate (150 mg, 299 μmol) in CH
3CN (1.0 mL) was added Et
3N (0.1 mL) at 25 ℃. The mixture was warmed to 80 ℃ and stirred at that temperature for 5 h before it was cooled to 25 ℃. The mixture was concentrated under reduced pressure and purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 0%to 40%in 40 min) to afford (1
1S, 1
3R, Z) -4
5-phenyl-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (40.1 mg, 31%yield) as a white solid. LC-MS: m/z [M+H]
+434.2.
The following compounds were prepared using the similar procedure disclosed in synthetic example 15.

Example 19
(1
1S, 1
3R, 2
4Z, 7
2Z) -2
1H-5, 10-dioxa-3, 8-diaza-7 (4, 2) -thiazola-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-
1 (1, 3) -cyclopentanacyclodecaphan-9-one

Step 1: tert-butyl (4- ( ( (6-bromopyridin-2-yl) oxy) methyl) thiazol-2-yl) carbamate
To a solution of tert-butyl (4- (hydroxymethyl) thiazol-2-yl) carbamate (1.0 g, 4.34 mmol) in THF (20 mL) was added NaH (869 mg, 21.7 mmol, 60%purity) at 0 ℃, the mixture was stirred at 0 ℃ for 30 min, 2-bromo-6-fluoro-pyridine (1.15 g, 6.51 mmol) was added, the mixture was warmed to 25 ℃ and stirred at this temperature for 16 h.
The mixture was quenched with saturated NH
4Cl solution (100 mL) and extracted with EtOAc (50 mL × 2) . The combined organic layer was washed with brine (100 mL) , dried over Na
2SO
4, filtered and concentrated. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 40%in 25 min) to afford tert-butyl (4- ( ( (6-bromopyridin-2-yl) oxy) methyl) thiazol-2-yl) carbamate (1.46 g, 87%yield) as a colorless oil. LC-MS: (ESI) m/z 386.0 [M+H]
+.
Step 2: tert-butyl (4- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) oxy) methyl) thiazol-2-yl) carbamate
To a mixture of tert-butyl (4- ( ( (6-bromopyridin-2-yl) oxy) methyl) thiazol-2-yl) carbamate (300 mg, 0.78 mmol) and (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (173 mg, 0.78 mmol) in 1, 4-dioxane (10 mL) was added Pd
2dba
3 (142 mg, 0.155 mmol) , Xantphos (180 mg, 0.31 mmol) and Cs
2CO
3 (632 mg, 1.94 mmol) at 25 ℃ under nitrogen. The reaction was warmed to 90 ℃ and stirred at this temperature for 6 h. The mixture was cooled to 25 ℃ and was diluted with water (150 mL) , the aqueous layer was extracted with EtOAc (50 mL × 3) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrated. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 60%in 25 min) to afford tert-butyl (4- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) oxy) methyl) thiazol-2-yl) carbamate (310 mg, 75%yield) as a yellow oil. LC-MS: m/z 529.2 [M+H]
+.
Step 3: (1R, 3S) -3- (5- ( (6- ( (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) methoxy) pyridin-2-
yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of tert-butyl (4- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) oxy) methyl) thiazol-2-yl) carbamate (50.0 mg, 94.6 μmol) in DCM (3.0 mL) was added Et
3N (47.8 mg, 473 μmol) and CDI (46.1 mg, 284 μmol) at 25 ℃, the mixture was warmed to 35 ℃ and stirred at this temperature for 1 h. The mixture was cooled to 25 ℃ and diluted with water (50 mL) , the aqueous layer was extracted with DCM (30 mL × 3) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrated to afford (1R, 3S) -3- (5- ( (6- ( (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) methoxy) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (50 mg, crude) as yellow oil. LC-MS: m/z 623.3 [M+H]
+.
Step 4: (1R, 3S) -3- (5- ( (6- ( (2-aminothiazol-4-yl) methoxy) pyridin-2-yl) amino) -1H-pyrazol-3-
yl) cyclopentyl 1H-imidazole-1-carboxylate
The mixture of (1R, 3S) -3- (5- ( (6- ( (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) methoxy) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (50.0 mg, 94.7 μmol) in TFA (2.0 mL) was warmed to 70 ℃ and stirred at this temperature for 4 h. The mixture was cooled to 25 ℃ and was concentrated under reduced pressure to afford (1R, 3S) -3- (5- ( (6- ( (2-aminothiazol-4-yl) methoxy) pyridin-2-yl) amino) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (40 mg, crude) as a yellow oil, which was used in the next step directly. LC-MS: m/z 467.1 [M+H]
+.
Step 5: (1
1S, 1
3R, 2
4Z, 7
2Z) -2
1H-5, 10-dioxa-3, 8-diaza-7 (4, 2) -thiazola-4 (2, 6) -pyridina-2 (5, 3) -
pyrazola-1 (1, 3) -cyclopentanacyclodecaphan-9-one
To a solution of (1R, 3S) -3- (5- ( (6- ( (2-aminothiazol-4-yl) methoxy) pyridin-2-yl) amino) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (30.0 mg, 64.3 μmol) in ACN (3.0 mL) was added Et
3N (65.1 mg, 643 μmol) and NaH (25.7 mg, 643 μmol, 60%purity) at 25 ℃, after addition, the mixture was stirred at 25 ℃ for 1 h. The mixture was concentrated under reduced pressure and the residue was purified by prep-HPLC (with CH
3CN from 22%to 32%in 12 min) to afford (1
1S, 1
3R, 2
4Z, 7
2Z) -2
1H-5, 10-dioxa-3, 8-diaza-7 (4, 2) -thiazola-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclodecaphan-9-one (0.5 mg, 2%yield) as a white solid. LC-MS: m/z 399.1 [M+H]
+.
Example 20
(1
1S, 1
3R, 2
4Z, 7Z) -2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-7-en-11-one

Step 1: tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) oxy) but-2-yn-1-yl) carbamate
To a solution of tert-butyl (4- ( (6-bromopyridin-2-yl) oxy) but-2-yn-1-yl) carbamate (160 mg, 469 μmol) and (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (105 mg, 469 μmol) in dioxane (5.0 mL) was added Pd
2 (dba)
3 (42.9 mg, 46.9 μmol) , XantPhos (54.3 mg, 93.8 μmol) and Cs
2CO
3 (458 mg, 1.41 mmol) under the atmosphere of N
2. Then the reaction mixture was heated to 100 ℃and stirred at 100 ℃ under the atmosphere of N
2 for 3 hours. The mixture was concentrated under reduced pressure, and the residue was purified by flash column chromatography eluting with EtOAc in PE (with EtOAc from 0~60%) in 20 min to afford the product tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) oxy) but-2-yn-1-yl) carbamate (134 mg, 59%yield) as a yellow gum. LC-MS: (ESI) m/z [M+H]
+ 484.2.
Step 2: tert-butyl ( (Z) -4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) oxy) but-2-en-1-yl) carbamate
To a solution of tert-butyl N- [4- [ [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -2-pyridyl] oxy] but-2-ynyl] carbamate (110 mg, 227 μmol) in EtOAc (5.0 mL) was added Pd/CaCO
3 (40 . 0 mg, 18.8 μmol, 5%purity) . Then the reaction mixture was degassed with H
2 (balloon) for 3 times. Then the reaction mixture was stirred at 20 ℃ under the atmosphere of H
2 for 15 h. The mixture was filtered and concentrated under reduced pressure, the residue was purified by flash column chromatography eluting with EtOAc in PE (with EtOAc from 0~50%) in 20 min to afford the product tert-butyl N- [ (Z) -4- [ [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -2-pyridyl] oxy] but-2-enyl] carbamate (61.0 mg, 55%yield) as a yellow solid. LC-MS: (ESI) m/z [M+H]
+ 486.2.
Step 3: (1R, 3S) -3- (5- ( (6- ( ( (Z) -4- ( (tert-butoxycarbonyl) amino) but-2-en-1-yl) oxy) pyridin-2-
yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of tert-butyl N- [ (Z) -4- [ [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -2-pyridyl] oxy] but-2-enyl] carbamate (61.0 mg, 126 μmol) in DCM (5.0 mL) was added Et
3N (63.6 mg, 628 μmol, 87.5 μL) at 20 ℃, then di (imidazol-1-yl) methanone (40.7 mg, 251 μmol) was added to the reaction mixture. The mixture was stirred at 35 ℃ for 5 h. The mixture was quenched with ice water (80 mL) , then the mixture was extracted with EtOAc (100 mL × 3) . The combined organic layer was dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure, and the residue was purified by flash column chromatography eluting with EtAOc in PE (with EtOAc from 0~60%) in 15 min to afford the product [ (1R, 3S) -3- [5- [ [6- [ (Z) -4- (tert-butoxycarbonylamino) but-2-enoxy] -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (60.0 mg, 82%yield) as a yellow gum. LC-MS: (ESI) m/z [M+H]
+ 580.2.
Step 4: (1R, 3S) -3- (5- ( (6- ( ( (Z) -4-aminobut-2-en-1-yl) oxy) pyridin-2-yl) amino) -1- (tert-butyl) -1H-
pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of [ (1R, 3S) -3- [5- [ [6- [ (Z) -4- (tert-butoxycarbonylamino) but-2-enoxy] -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (60.0 mg, 103 μmol) in DCM (5.0 mL) was added TFA (740mg, 6.49 mmol, 0.5 mL) . Then the reaction mixture was stirred at 20 ℃ for 3 h. The mixture was concentrated under reduced pressure to afford the product [ (1R, 3S) -3- [5- [ [6- [ (Z) -4-aminobut-2-enoxy] -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (60.0 mg, crude, TFA) as a yellow oil. LC-MS: (ESI) m/z [M+H]
+ 480.2.
Step 5: (1
1S, 1
3R, 2
4Z, 7Z) -2
1- (tert-butyl) -2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-7-en-11-one
To a solution of [ (1R, 3S) -3- [5- [ [6- [ (Z) -4-aminobut-2-enoxy] -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (60.0 mg, 101 μmol, TFA) in ACN (12 mL) was added Et
3N (10.2 mg, 101 μmol, 14.1 μL) . Then the reaction mixture was heated to 80 ℃ and stirred at 80 ℃ for 12 h. The mixture was concentrated under reduced pressure, and the residue was purified by flash column eluting with EtAOc in PE (with EtOAc from 0~50%) in 15 min to afford the product (1
1S, 1
3R, 2
4Z, 7Z) -2
1- (tert-butyl) -2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-7-en-11-one (12.0 mg, 29%yield) as a yellow solid. LC-MS: (ESI) m/z [M+H]
+ 412.1.
Step 6: (1
1S, 1
3R, 2
4Z, 7Z) -2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-7-en-11-one
The solution of (1
1S, 1
3R, 2
4Z, 7Z) -2
1- (tert-butyl) -2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-7-en-11-one (12.0 mg, 29.2 μmol) in TFA (5.0 mL) was heated to 70 ℃ and stirred at 70 ℃ for 12 h. The mixture was concentrated under reduced pressure, and the residue was sent to purified by prep-HPLC (with CH
3CN from 25%to 55%in 8 min) to afford the product (1
1S, 1
3R, 2
4Z, 7Z) -2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-7-en-11-one (3.00 mg, 29%yield) as a white solid. LC-MS: (ESI) m/z [M+H]
+ 356.1.
Example 21
(7S, 10R) ‐11, 18‐dioxa‐2, 4, 5, 13, 23‐pentaazatetracyclo [17.3.1.1
3, 6.1
7, 10] pentacosa‐
1 (23) , 3, 6 (25) , 19, 21‐pentaen‐15‐yn‐12‐one


Step 1: 4- ( (6-bromopyridin-2-yl) oxy) but-2-yn-1-ol
To a solution of but-2-yne-1, 4-diol (500 mg, 5.81 mmol) in DMF (10 mL) was added Sodium hydride (278 mg, 11.6 mmol) at 0 ℃. The mixture was stirred at 25 ℃ for 0.5 h. Then the mixture was added 2-bromo-6-fluoro-pyridine (1.02 g, 5.81 mmol, 598 μL) at 25 ℃ and stirred at 25 ℃ for 1 h. The mixture was added H
2O (30 mL) and extracted with EtOAc (30 mL × 3) . The combined organic layer was washed with brine (20 mL) and dried over anhydrous Na
2SO
4. The mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography eluting with EtAOc in PE (with EtOAc from 0~40%) in 15 min to afford 4- ( (6-bromopyridin-2-yl) oxy) but-2-yn-1-ol (1.26 g, 90%yield) as a colorless oil. LC-MS: m/z 242.0 [M+H]
+.
Step 2: 4- ( (6-bromopyridin-2-yl) oxy) but-2-yn-1-amine
To a mixture of 4- ( (6-bromopyridin-2-yl) oxy) but-2-yn-1-ol (1.26 g, 5.22 mmol) and Et
3N (1.06 g, 10.4 mmol, 1.45 mL) in DCM (15 mL) was added MsCl (717 mg, 6.26 mmol, 485 μL) dropwise at 0 ℃. The mixture was stirred at 25 ℃ for 0.5 h. Then the mixture was quenched with H
2O (5.0 mL) and extracted with DCM (30 mL × 3) . The combined organic layer was washed with brine (20 mL) and dried over anhydrous Na
2SO
4. The mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was added NH
3-MeOH (70 mmol, 10 mL, 7 mol/L in MeOH) at 25 ℃and stirred at 25 ℃ for 6 h. The mixture was concentrated under reduced pressure to afford 4- ( (6-bromopyridin-2-yl) oxy) but-2-yn-1-amine (800 mg, crude) as a yellow oil, which was used directly into the next step without further treatment. LC-MS: m/z 241.1 [M+H]
+.
Step 3: tert-butyl (4- ( (6-bromopyridin-2-yl) oxy) but-2-yn-1-yl) carbamate
To a mixture of 4- ( (6-bromopyridin-2-yl) oxy) but-2-yn-1-amine (800 mg, 3.32 mmol) and (Boc)
2O (1.09 g, 4.98 mmol, 1.14 mL) in THF (10 mL) was added Et
3N (1.01 g, 9.96 mmol, 1.39 mL) at 25 ℃. The mixture was stirred at 25 ℃ for 16 h. The mixture was added H
2O (10 mL) and extracted with EtOAc (30 mL × 3) . The combined organic layers were washed with brine (20 mL) and dried over anhydrous Na
2SO
4. The mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography eluting with EtAOc in PE (with EtOAc from 0~25%) in 15 min to afford the desired product tert-butyl (4- ( (6-bromopyridin-2-yl) oxy) but-2-yn-1-yl) carbamate (326 mg, 29%yield) as a yellow solid. LC-MS: m/z 363.0 [M+Na]
+.
Step 4: tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) oxy) but-2-yn-1-yl) carbamate
To a mixture of tert-butyl (4- ( (6-bromopyridin-2-yl) oxy) but-2-yn-1-yl) carbamate (150 mg, 439 μmol) and (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (98.1 mg, 439 μmol) in dioxane (5.0 mL) was added Pd
2 (dba)
3 (80.5 mg, 87.9 μmol) , Xantphos (101 mg, 175 μmol) and Cs
2CO
3 (429 mg, 1.32 mmol) at 25 ℃. The mixture was heated to 95 ℃ and stirred at 95 ℃ for 6 h. The mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography eluting with EtAOc in PE (with EtOAc from 0~80%) in 25 min to afford tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) oxy) but-2-yn-1-yl) carbamate (171 mg, 80%yield) as a yellow oil. LC-MS: m/z 484.2 [M+H]
+.
Step 5: (1R, 3S) -3- (5- ( (6- ( (4- ( (tert-butoxycarbonyl) amino) but-2-yn-1-yl) oxy) pyridin-2-yl) amino) -
1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a mixture of tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) oxy) but-2-yn-1-yl) carbamate (151 mg, 312 μmol) and 1, 1'-Carbonyldiimidazole (253 mg, 1.56 mmol) in DCM (5.0 mL) was added Et
3N (157 mg, 1.56 mmol, 217.60 μL) at 25 ℃. The mixture was stirred at 35 ℃ for 16 h. The mixture was concentrated under reduced pressure to afford (1R, 3S) -3- (5- ( (6- ( (4- ( (tert-butoxycarbonyl) amino) but-2-yn-1-yl) oxy) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (175 mg, crude) as a brown oil. LC-MS: m/z 578.3 [M+H]
+.
Step 6: (1R, 3S) -3- (3- ( (6- ( (4-aminobut-2-yn-1-yl) oxy) pyridin-2-yl) amino) -1H-pyrazol-5-
yl) cyclopentyl 1H-imidazole-1-carboxylate
A solution of (1R, 3S) -3- (5- ( (6- ( (4- ( (tert-butoxycarbonyl) amino) but-2-yn-1-yl) oxy) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (175 mg, 302 μmol) in TFA (2.96 g, 25.9 mmol, 2.0 mL) was heated to 70 ℃ and stirred at 70 ℃ for 1 h. The mixture was concentrated under reduced pressure to afford (1R, 3S) -3- (3- ( (6- ( (4-aminobut-2-yn-1-yl) oxy) pyridin-2-yl) amino) -1H-pyrazol-5-yl) cyclopentyl 1H-imidazole-1-carboxylate (125 mg, crude) as a brown oil. LC-MS: m/z 422.1 [M+H]
+.
Step 7: (7S, 10R) ‐11, 18‐dioxa‐2, 4, 5, 13, 23‐pentaazatetracyclo [17.3.1.1
3, 6.1
7, 10] pentacosa‐
1 (23) , 3, 6 (25) , 19, 21‐pentaen‐15‐yn‐12‐one
To a solution of (1R, 3S) -3- (3- ( (6- ( (4-aminobut-2-yn-1-yl) oxy) pyridin-2-yl) amino) -1H-pyrazol-5-yl) cyclopentyl 1H-imidazole-1-carboxylate (65.0 mg, 154.23 μmol) in Acetonitrile (5.0 mL) was added Et
3N (726.00 mg, 7.17 mmol, 1.0 mL) at 25 ℃. The mixture was heated to 50 ℃ and stirred at 50 ℃ for 16 h. The mixture was purified by Prep-TLC (DCM/MeOH=15: 1) to afford RGT002-940-102-01 (1.10 mg, 2%yield) as a white solid. LC-MS: m/z 354.2 [M+H]
+.
Example 22 and 23
(1
1S, 1
3R, 7
1R, 7
3S, Z) -2
1H-5, 11-dioxa-3, 9-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentana-7 (1, 3) -cyclobutanacycloundecaphan-10-one and (1
1S, 1
3R, 7
1S, 7
3R, Z) -2
1H-5, 11-
dioxa-3, 9-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -
cyclobutanacycloundecaphan-10-one

Step 1: 3-methylenecyclobutane-1-carboxylic acid
The mixture of 3-methylenecyclobutanecarbonitrile (1.00 g, 10.7 mmol) and potassium hydroxide (2.41 g, 42.9 mmol) in EtOH (5.0 mL) and H
2O (5.0 mL) was heated to 90 ℃ and stirred at 90 ℃ for 17 h. The mixture was concentrated under reduced pressure. To the residue was added H
2O (10 mL) , and the pH value of the aqueous layer was adjusted to 1 by adding conc. HCl. The aqueous layer was extracted with EtOAc (10 mL × 3) . The combined organic layer was dried over Na
2SO
4, filtered and concentrated to give 3-methylenecyclobutanecarboxylic acid (800 mg, 66%yield) as a colorless oil.
1H NMR (400 MHz, CDCl
3) δ 10.76 (brs, 1H) , 4.82 (q, J = 2.4 Hz, 2H) , 3.17-3.15 (m, 1H) , 3.08-3.00 (m, 2H) , 2.98-2.90 (m, 2H) .
Step 2: (3-methylenecyclobutyl) methanol
To a stirred solution of 3-methylenecyclobutanecarboxylic acid (1.23 g, 10.9 mmol) in THF (10 mL) at 0 ℃ was added a solution of lithium aluminium hydride in THF (13.1 mL, 13.1 mmol) slowly. After addition, the mixture was stirred at 25 ℃ for 2 h. The mixture was cooled down to 0 ℃ and was quenched with ice-water (50 mL) . THF was removed under reduced pressure and the aqueous layer was extracted with EtOAc (15 mL × 3) , the combined organic layer was dried over Na
2SO
4, filtered and concentrated to give (3-methylenecyclobutyl) methanol (800 mg, 74%yield) as a yellow oil.
1H NMR (400 MHz, CDCl
3) δ 4.77 (q, J = 2.4 Hz, 2H) , 3.66 (d, J = 6.8 Hz, 2H) , 2.77-2.74 (m, 2H) , 2.51-2.47 (m, 1H) , 2.43-2.38 (m, 2H) .
Step 3: cyclobutane-1, 3-diyldimethanol
To a stirring solution of (3-methylenecyclobutyl) methanol (6.20 g, 63.1 mmol) in THF (50 mL) at 0 ℃ was added BH
3-THF (1.0 M, 94.7 mL) slowly. After addition, the mixture was stirred at 25 ℃ for 17 h. The mixture was cooled down to 0 ℃. The aqueous potassium hydroxide (3.0 M, 42.1 mL) solution was added to the above mixture slowly. After addition, the mixture was stirred at 25 ℃for 1 h. 30%hydrogen peroxide (13.0 mL, 126 mmol) was added to the mixture. The mixture was stirred for further 2 h. To the mixture was added H
2O (100 mL) , the aqueous layer was extracted with EtOAc (80 mL × 4) . The combined organic layer was dried over Na
2SO
4, filtered and concentrated. The residue was purified by silica gel chromatography eluting with EtOAc/petroleum ether (with EtOAc from 0 to 50%) in 25 mins to give (trans+cis) -cyclobutane-1, 3-diyldimethanol (3.20 g, 44%yield) as a colorless oil.
1H NMR (400 MHz, CDCl
3) δ 3.66 (d, J = 6.8 Hz, 2H) , 3.56 (d, J = 6.0 Hz, 2H) , 2.51-2.38 (m, 2H) , 2.16-2.09 (m, 1H) , 1.87 (t, J = 7.2 Hz, 2H) , 1.60-1.54 (m, 1H) .
Step 4: (3- ( ( (6-bromopyridin-2-yl) oxy) methyl) cyclobutyl) methanol
To a stirring solution of cyclobutane-1, 3-diyldimethanol (99.01 mg, 852.34 μmol) in THF (5.0 mL) at 0 ℃ was added KHMDs in THF (1.0 M, 511.40 μL) portionwise. After addition, the mixture was stirred at 25 ℃ for 30 min. To the mixture was added 2-bromo-6-fluoro-pyridine (75.0 mg, 426 μmol) . The mixture was stirred at 25 ℃ for 3 h. The mixture was quenched with aqueous NH
4Cl solution (10 mL) . The aqueous layer was extracted with EtOAc (15 mL × 3) . The combined organic layer was dried over Na
2SO
4, filtered and concentrated. The residue was purified by silica gel chromatography eluting with EtOAc in petroleum ether (with EtOAc from 0 to 25 %) in 20 min to give (3- ( ( (6-bromopyridin-2-yl) oxy) methyl) cyclobutyl) methanol (40.0 mg, 34%yield) as a colorless oil. LC-MS: m/z 272.0 [M+H]
+.
Step 5: (3- ( ( (6-bromopyridin-2-yl) oxy) methyl) cyclobutyl) methyl methanesulfonate
To a stirring solution of (3- ( ( (6-bromopyridin-2-yl) oxy) methyl) cyclobutyl) methanol (130 mg, 477 μmol) and Et
3N (133 μL, 955 μmol, ) in DCM (15 mL) at 0 ℃ was added methanesulfonyl chloride (71.1 mg, 621 μmol) . After addition, the mixture was stirred at 25 ℃ for 2 h. The mixture was quenched with saturated aqueous NaHCO
3 (25 mL) and the aqueous layer was extracted with DCM (10 mL × 2) . The combined organic layer was dried, filtered. The residue was concentrated to give (3- ( ( (6-bromopyridin-2-yl) oxy) methyl) cyclobutyl) methyl methanesulfonate (160 mg, 96%yield) as a yellow oil, which was used directly into the next step without further purification. LC-MS: m/z 350.0 [M+H]
+
Step 6: 2- ( (3- (azidomethyl) cyclobutyl) methoxy) -6-bromopyridine
The mixture of (3- ( ( (6-bromopyridin-2-yl) oxy) methyl) cyclobutyl) methyl methanesulfonate (150 mg, 428 μmol) and sodium azide (55.6 mg, 856 μmol) in DMF (8.0 mL) was heated to 100 ℃ and stirred at 100 ℃ for 17 h. After cooled down to room temperature, the mixture was poured into ice-water (30 mL) . The aqueous layer was extracted with EtOAc (10 mL × 2) , the combined organic layer was dried over Na
2SO
4, filtered and concentrated. The residue was purified by silica gel chromatography eluting with EtOAc in petroleum ether (with EtOAc from 0 to 20%) in 20 min to give 2- ( (3- (azidomethyl) cyclobutyl) methoxy) -6-bromopyridine (70.0 mg, 55%yield) as a colorless oil. LC-MS: m/z 297.0 [M+H]
+.
Step 7: (3- ( ( (6-bromopyridin-2-yl) oxy) methyl) cyclobutyl) methanamine
The mixture of 2- ( (3- (azidomethyl) cyclobutyl) methoxy) -6-bromopyridine (70.0 mg, 235 μmol) and triphenylphosphane (185 mg, 706 μmol) in THF (2.0 mL) and H
2O (2.0 mL) was stirred at 25 ℃ for 2 h. The solution was used directly in the next step without further purification. LC-MS: m/z 271.0 [M+H]
+.
Step 8: tert-butyl ( (3- ( ( (6-bromopyridin-2-yl) oxy) methyl) cyclobutyl) methyl) carbamate
To a stirred mixture of (3- ( ( (6-bromopyridin-2-yl) oxy) methyl) cyclobutyl) methanamine (100 mg, 368 μmol) and sodium carbonate (390 mg, 3.69 mmol) in THF (5.0 mL) and H
2O (5.0 mL) was added Boc
2O (241 mg, 1.11 mmol) . The mixture was stirred at 25 ℃ for 17 h. The aqueous layer was extracted with EtOAc (5.0 mL × 2) . The combined organic layer was dried over Na
2SO
4, filtered and concentrated. The residue was purified by silica gel chromatography eluting with EtOAc in petroleum ether (with EtOAc from 0 to 20%) in 20 minutes to give tert-butyl ( (3- ( ( (6-bromopyridin-2-yl) oxy) methyl) cyclobutyl) methyl) carbamate (35.0 mg, 26%yield) as a colorless oil. LC-MS: m/z 393.0 [M+Na]
+.
Step 9: tert-butyl ( (3- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) oxy) methyl) cyclobutyl) methyl) carbamate
The mixture of tert-butyl ( (3- ( ( (6-bromopyridin-2-yl) oxy) methyl) cyclobutyl) methyl) carbamate (300 mg, 808 μmol) , (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (180 mg, 808 μmol) , Cs
2CO
3 (789 mg, 2.42 mmol) , Pd
2 (dba)
3 (73.9 mg, 80.8 μmol) and XantPhos (93.4 mg, 161 μmol) in 1, 4-dioxane (10 mL) was heated to 110 ℃ and stirred at 100 ℃ for 6 h under N
2. The mixture was concentrated under reduced pressure and the residue was purified by silica gel chromatography eluting with EtOAc in petroleum ether (with EtOAc from 0 to 50%) in 25 minutes to give tert-butyl ( (3- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) oxy) methyl) cyclobutyl) methyl) carbamate (280 mg, 67%yield) as a yellow solid. LC-MS: m/z 514.3 [M+H]
+.
Step 10: tert-butyl ( (3- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-
nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-
yl) oxy) methyl) cyclobutyl) methyl) carbamate
The mixture of tert-butyl ( (3- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) oxy) methyl) cyclobutyl) methyl) carbamate (280 mg, 545 μmol) , (4-nitrophenyl) carbonochloridate (329 mg, 1.64 mmol) , Et
3N (275 mg, 2.73 mmol) and DMAP (13.3 mg, 109 μmol) in DCE (9.8 mL) was heated to 85 ℃ and stirred at 85 ℃ for 17 h. The mixture was concentrated under reduced pressure and the residue was purified by Prep-TLC (EtOAc/PE = 1/1) to give tert-butyl ( (3- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) oxy) methyl) cyclobutyl) methyl) carbamate (150 mg, 41%yield) as a yellow oil. LC-MS: m/z 679.3 [M+H]
+.
Step 11: (1R, 3S) -3- (5- ( (6- ( (3- (aminomethyl) cyclobutyl) methoxy) pyridin-2-yl) amino) -1- (tert-
butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate
The mixture of tert-butyl ( (3- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) oxy) methyl) cyclobutyl) methyl) carbamate (150 mg, 221 μmol) in DCM (3.0 mL) and trifluoroacetic acid (3.0 mL) was stirred at 25 ℃ for 1 h. The mixture was concentrated in vacuo to give (1R, 3S) -3- (5- ( (6- ( (3- (aminomethyl) cyclobutyl) methoxy) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate (150 mg, 70%yield) as a brown oil, which was used directly into the next step without further purification. LC-MS: m/z 579.2 [M+H]
+.
Step 12: (1
1S, 1
3R, 7
1R, 7
3S, Z) -2
1- (tert-butyl) -2
1H-5, 11-dioxa-3, 9-diaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacycloundecaphan-10-one and
(1
1S, 1
3R, 7
1S, 7
3R, Z) -2
1- (tert-butyl) -2
1H-5, 11-dioxa-3, 9-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-
1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacycloundecaphan-10-one
The mixture of (1R, 3S) -3- (5- ( (6- ( (3- (aminomethyl) cyclobutyl) methoxy) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate (150 mg, 259 μmol) and Et
3N (524 mg, 5.18 mmol) in acetonitrile (70 mL) was stirred at 25 ℃ for 17 h. The mixture was concentrated under reduced pressure and the residue was purified by Prep-TLC (EtOAc/PE= 1/1) to give (1
1S, 1
3R, 7
1S, 7
3R, Z) -2
1- (tert-butyl) -2
1H-5, 11-dioxa-3, 9-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacycloundecaphan-10-one (relative configuration arbitrarily assigned, 46.0 mg, Rf = 0.35, 40%yield) as a yellow oil and (1
1S, 1
3R, 7
1R, 7
3S, Z) -2
1- (tert-butyl) -2
1H-5, 11-dioxa-3, 9-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacycloundecaphan-10-one (relative configuration arbitrarily assigned, 29.0 mg, Rf = 0.3, 25%yield) as a yellow oil. LC-MS: m/z 440.2 [M+H]
+.
Step 13: (1
1S, 1
3R, 7
1R, 7
3S, Z) -2
1H-5, 11-dioxa-3, 9-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentana-7 (1, 3) -cyclobutanacycloundecaphan-10-one
The mixture of (1
1S, 1
3R, 7
1R, 7
3S, Z) -2
1- (tert-butyl) -2
1H-5, 11-dioxa-3, 9-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacycloundecaphan-10-one (29.0 mg, 65.9 μmol) in trifluoroacetic acid (5.0 mL) was heated to 80 ℃ and stirred at 80 ℃ for 6 h. The mixture was concentrated under reduced pressure and the residue was purified Prep-HPLC (with CH
3CN from 50%to 80%in 20 min) to give (1
1S, 1
3R, 7
1R, 7
3S, Z) -2
1H-5, 11-dioxa-3, 9-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacycloundecaphan-10-one (8.6 mg, 36%yield) as a white solid. LC-MS: m/z 384.2 [M+H]
+.
Step 14: (1
1S, 1
3R, 7
1S, 7
3R, Z) -2
1H-5, 11-dioxa-3, 9-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentana-7 (1, 3) -cyclobutanacycloundecaphan-10-one
The mixture of (1
1S, 1
3R, 7
1S, 7
3R, Z) -2
1- (tert-butyl) -2
1H-5, 11-dioxa-3, 9-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacycloundecaphan-10-one (46 mg, 104.65 μmol) in trifluoroacetic acid (5 mL) was heated 80 ℃ for 6 h. The mixture was concentrated under reduced pressure and the residue was purified Prep-HPLC eluting with CH
3CN in water with CH
3CN from 50%to 80%in 20 min to give (1
1S, 1
3R, 7
1S, 7
3R, Z) -2
1H-5, 11-dioxa-3, 9-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacycloundecaphan-10-one (20.4 mg, 51%yield) as a white solid. LC-MS: m/z 384.2 [M+H]
+.
Example 24 and 25
(1
1S, 1
3R, 6S, Z) -6-methyl-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one and (1
1S, 1
3R, 6R, Z) -6-methyl-2
1H-5, 12-dioxa-3, 10-diaza-
4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one

Step 1: N- (2, 4-dimethoxybenzyl) -4-hydroxypentanamide
The mixture of 5-methyltetrahydrofuran-2-one (1.00 g, 9.99 mmol, 950 μL) and (2, 4-dimethoxyphenyl) methanamine (1.84 g, 10.9 mmol) was heated to 85 ℃ and stirred at 85 ℃ for 16 h. The reaction was diluted with water (20 mL) and then extracted with EA (50 mL × 3) . The organic phase was dried over Na
2SO
4, filtered and concentrated. The residue was purified by flash chromatography eluting with MeOH in DCM (with MeOH from 0 to 5%) in 15 min to give N- [ (2, 4-dimethoxyphenyl) methyl] -4-hydroxy-pentanamide (1.90 g, 71%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 268.2.
Step 2: 5- ( (2, 4-dimethoxybenzyl) amino) pentan-2-ol
The mixture of N- [ (2, 4-dimethoxyphenyl) methyl] -4-hydroxy-pentanamide (1.70 g, 6.36 mmol) and borane; methylsulfanylmethane (966 mg, 12.7 mmol, 1.21 mL) in THF (20 mL) was heated to 70 ℃ and stirred at 70 ℃ for 3 h. The reaction was diluted with water (50 mL) and then extracted with EtOAc (50 mL × 3) . The organic solution was dried over Na
2SO
4, filtered and concentrated. The residue was purified by flash chromatography eluting with MeOH in DCM (with MeOH from 0 to 10%) in 25 min to give 5- [ (2, 4-dimethoxyphenyl) methylamino] pentan-2-ol (400 mg, 24%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 254.2.
Step 3: tert-butyl (2, 4-dimethoxybenzyl) (4-hydroxypentyl) carbamate
To a stirring solution of 5- [ (2, 4-dimethoxyphenyl) methylamino] pentan-2-ol (230 mg, 907 μmol) and Na
2CO
3 (288 mg, 2.72 mmol, 114 μL) in THF (2.5 mL) and water (5.0 mL) at 25 ℃ was added tert-butoxycarbonyl tert-butyl carbonate (396 mg, 1.82 mmol, 41 μL) . After addition, the mixture was stirred at 25 ℃ for 16 h. The reaction was diluted with water (50 mL) and then extracted with EtOAc (50 mL × 3) . The organic solution was dried over Na
2SO
4, filtered and concentrated. The residue was purified by flash chromatography eluting with EtOAc in PE (with EtOAc from 0 to 10%) in 15 min to give tert-butyl N- [ (2, 4-dimethoxyphenyl) methyl] -N- (4-hydroxypentyl) carbamate (300 mg, 93%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 376.3.
Step 4: tert-butyl (4- ( (6-bromopyridin-2-yl) oxy) pentyl) (2, 4-dimethoxybenzyl) carbamate
To a stirring solution of tert-butyl N- [ (2, 4-dimethoxyphenyl) methyl] -N- (4-hydroxypentyl) carbamate (300 mg, 848 μmol) and 2-bromo-6-fluoro-pyridine (179 mg, 1.02 mmol, 105 μL) in DMF (8.0 mL) at 0 ℃ was added KHMDS (253 mg, 1.27 mmol, 288 μL) . After addition, the mixture was stirred at 25 ℃ for 2 h. The reaction was diluted with water (50 mL) and then extracted with EtOAc (50 mL ×3) . The organic solution was dried over Na
2SO
4, filtered and concentrated. The residue was purified by flash chromatography eluting with EtOAc in PE (with EtOAc from 0 to 30%) in 20 min to give tert-butyl N- [4- [ (6-bromo-2-pyridyl) oxy] pentyl] -N- [ (2, 4-dimethoxyphenyl) methyl] carbamate (300 mg, 69%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 531.1.
Step 5: tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) oxy) pentyl) (2, 4-dimethoxybenzyl) carbamate
The mixture of tert-butyl N- [4- [ (6-bromo-2-pyridyl) oxy] pentyl] -N- [ (2, 4-dimethoxyphenyl) methyl] carbamate (350 mg, 687 μmol) , (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (138 mg, 618 μmol) , dicesium; carbonate (671 mg, 2.06 mmol) , (1E, 4E) -1, 5-diphenylpenta-1, 4-dien-3-one; palladium (62.9 mg, 68.7 μmol) and (5-diphenylphosphanyl-9, 9-dimethyl-xanthen-4-yl) -diphenyl-phosphane (79.5 mg, 137 μmol) in dioxane (8.0 mL) was heated to 100 ℃ and stirred at 100 ℃ for 3 h. The reaction was diluted with water (50 mL) and then extracted with EtOAc (50 mL × 3) . The organic solution was dried over Na
2SO
4, filtered and concentrated. The residue was purified by flash chromatography eluting with EtOAc in PE (with EtOAc from 0 to 50%) in 20 min to give tert-butyl N- [4- [ [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -2-pyridyl] oxy] pentyl] -N- [ (2, 4-dimethoxyphenyl) methyl] carbamate (250 mg, 55%yield) as a yellow oil. LC-MS: m/z [M+H]
+652.3.
Step 6: (1R, 3S) -3- (5- ( (6- ( (5- ( (tert-butoxycarbonyl) (2, 4-dimethoxybenzyl) amino) pentan-2-
yl) oxy) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-
carboxylate
To a stirring solution of tert-butyl N- [4- [ [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -2-pyridyl] oxy] pentyl] -N- [ (2, 4-dimethoxyphenyl) methyl] carbamate (300 mg, 460 μmol) and Et
3N (232 mg, 2.30 mmol, 320 μL) in DCM (8.0 mL) at 25 ℃ was added CDI (198 mg, 1.38 mmol) . After addition, the mixture was stirred at 35 ℃ for 2 h. The reaction was diluted with water (30 mL) and then extracted with DCM (40 mL × 3) . The organic solution was dried over Na
2SO
4, filtered and concentrated. The residue was purified by flash chromatography eluting with EtOAc in PE (with EtOAc from 0 to 50%) in 20 min to give [ (1R, 3S) -3- [5- [ [6- [4- [tert-butoxycarbonyl- [ (2, 4-dimethoxyphenyl) methyl] amino] -1-methyl-butoxy] -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (250 mg, 72%yield) as a yellow solid. LC-MS: m/z [M+H]
+ 746.3.
Step 7: (1R, 3S) -3- (3- ( (6- ( (5-aminopentan-2-yl) oxy) pyridin-2-yl) amino) -1- (tert-butyl) -1H-
pyrazol-5-yl) cyclopentyl 1H-imidazole-1-carboxylate
The mixture of [ (1R, 3S) -3- [5- [ [6- [4- [tert-butoxycarbonyl- [ (2, 4-dimethoxyphenyl) methyl] amino] -1-methyl-butoxy] -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (200 mg, 268.13 μmol) in TFA (5.0 mL) was stirred at 25 ℃ for 16 h. The reaction was concentrated to give [ (1R, 3S) -3- [5- [ [6- (4-amino-1-methyl-butoxy) -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (100 mg, 75%yield) as a yellow oil. LC-MS: m/z [M+H]
+496.3.
Step 8: (1
1S, 1
3R, 6S, Z) -21- (tert-butyl) -6-methyl-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one & (1
1S, 1
3R, 6R, Z) -21- (tert-butyl) -6-methyl-
2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-
11-one
To a stirring solution of [ (1R, 3S) -3- [5- [ [6- (4-amino-1-methyl-butoxy) -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (75.0 mg, 151 μmol) in MeCN (2.0 mL) at 25 ℃was added Et
3N (15.3 mg, 151 μmol, 21.0 μL) . After addition, the mixture was heated to 60 ℃ and stirred at 60 ℃ for 17 h. The reaction was concentrated. The residue was purified by prep-TLC (petroleum ether/EtOAc = 1: 1) to give (1
1S, 1
3R, 6S, Z) -21- (tert-butyl) -6-methyl-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (absolute configuration is arbitrarily assigned, 25.0 mg, Rf = 0.5, 38%yield) as a white solid. LC-MS: m/z [M+H]
+ 428.3. And (1
1S, 1
3R, 6R, Z) -2
1- (tert-butyl) -6-methyl-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (absolute configuration is arbitrarily assigned, 25.0 mg, Rf = 0.3, 38%yield) as a white solid. LC-MS: m/z [M+H]
+ 428.3.
Step 9: (1
1S, 1
3R, 6S, Z) -6-methyl-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphan-11-one
The mixture of (1
1S, 1
3R, 6S, Z) -21- (tert-butyl) -6-methyl-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (21.5 mg, 50.4 μmol) in TFA (3.0 mL) was heated to 60 ℃ and stirred at 60 ℃ for 17 h. The reaction was concentrated. The residue was purified by prep-HPLC (with CH
3CN from 25%to 55%in 8 min) to give (1
1S, 1
3R, 6S, Z) -6-methyl-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (4.00 mg, 21%yield) as a white solid. LC-MS: m/z [M+H]
+ 372.1.
Step 10: (1
1S, 1
3R, 6R, Z) -6-methyl-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphan-11-one
The mixture of (1
1S, 1
3R, 6S, Z) -2
1- (tert-butyl) -6-methyl-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (21.5 mg, 50.4 μmol) in TFA (3 mL) was heated to 60 ℃ and stirred at 60 ℃ for 17 h. The reaction was concentrated. The residue was purified by prep-HPLC (with CH
3CN from 25%to 55%in 8 min) to give (1
1S, 1
3R, 6R, Z) -6-methyl-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (4.20 mg, 21%yield) as a white solid. LC-MS: m/z [M+H]
+ 372.1.
Example 26 and 27
(1
1S, 1
3R, Z) -11-oxo-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-4
4-carbonitrile and (1
1S, 1
3R, Z) -11-oxo-2
1H-5, 12-dioxa-3, 10-
diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-4
4-carboxamide

Step 1: tert-butyl (4- ( (6-chloro-4-cyanopyridin-2-yl) oxy) butyl) carbamate
To a solution of tert-butyl N- (4-hydroxybutyl) carbamate (1.31 g, 6.94 mmol) in THF (10 mL) was added NaH (1.16 g, 28.9 mmol, 60%purity) at 0 ℃, then the reaction mixture was stirred at 0 ℃for 30 minutes. Then 2, 6-dichloropyridine-4-carbonitrile (1.00 g, 5.78 mmol) was added to the reaction mixture. The mixture was stirred at 20 ℃ for 3 h. The mixture was not quenched (the reaction mixture cannot contact with water and MeOH) and the mixture was filtered and concentrated under reduced pressure, and the residue was purified by flash column chromatography eluting with EtOAc in PE (with EtOAc from 0-80%) in 25 min to afford the product tert-butyl (4- ( (6-chloro-4-cyanopyridin-2-yl) oxy) butyl) carbamate (1.40 g, 74%yield) as a colorless oil. LC-MS: (ESI) m/z [M+Na]
+ 348.0.
Step 2: tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) -4-cyanopyridin-2-yl) oxy) butyl) carbamate
To a solution of tert-butyl N- [4- [ (6-chloro-4-cyano-2-pyridyl) oxy] butyl] carbamate (230 mg, 705 μmol) and (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (150 mg, 671 μmol) in dioxane (5.0 mL) was added Pd
2 (dba)
3 (61.5 mg, 67.2 μmol) , XantPhos (77.7 mg, 134 μmol) and Cs
2CO
3 (656 mg, 2.02 mmol) under the atmosphere of N
2. Then the reaction mixture was heated to 100 ℃ and stirred at 100 ℃ under the atmosphere of N
2 for 5 h. The mixture was concentrated under reduced pressure, and the residue was purified by flash column chromatography eluting with EtOAc in PE (with EtOAc from 0-60%) in 15 min to afford tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -4-cyanopyridin-2-yl) oxy) butyl) carbamate (253 mg, 73%yield) as a yellow solid. LC-MS: (ESI) m/z [M+H]
+ 513.2.
Step 3: (1R, 3S) -3- (5- ( (6- (4- ( (tert-butoxycarbonyl) amino) butoxy) -4-cyanopyridin-2-yl) amino) -1-
(tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -4-cyanopyridin-2-yl) oxy) butyl) carbamate (230 mg, 449 μmol) in DCM (5.0 mL) was added Et
3N (227 mg, 2.24 mmol, 312 μL) at 20 ℃. Then di (imidazol-1-yl) methanone (145 mg, 897 μmol) was added to the reaction mixture. The mixture was stirred at 35 ℃ for 12 h. The mixture was quenched with ice water (80 mL) , then the mixture was extracted with EtOAc (100 mL × 3) . The combined organic layer was dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure, and the residue was purified by flash column chromatography eluting with EtOAc in PE (with EtOAc from 0-60%) in 15 min to afford the product [ (1R, 3S) -3- [5- [ [6- [4- (tert-butoxycarbonylamino) butoxy] -4-cyano-2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (260 mg, 96%yield) as a yellow gum. LC-MS: (ESI) m/z [M+H]
+ 607.2.
Step 4: (1R, 3S) -3- (5- ( (6- (4-aminobutoxy) -4-cyanopyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-
3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of [ (1R, 3S) -3- [5- [ [6- [4- (tert-butoxycarbonylamino) butoxy] -4-cyano-2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (190 mg, 313 μmol) in DCM (5.0 mL) was added TFA (178 mg, 1.57 mmol, 121 μL) at 20 ℃. Then the reaction mixture was stirred at 20 ℃ for 3 h. The mixture was concentrated under reduced pressure to afford the product (1R, 3S) -3- (5- ( (6- (4-aminobutoxy) -4-cyanopyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (200 mg, crude) as a yellow gum. LC-MS: (ESI) m/z [M+H]
+ 507.2.
Step 5: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -11-oxo-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-4
4-carbonitrile
To a solution of (1R, 3S) -3- (5- ( (6- (4-aminobutoxy) -4-cyanopyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (200 mg, 322 μmol) in CH
3CN (8.0 mL) was added Et
3N (1.09 g, 10.8 mmol, 1.50 mL) . Then the reaction mixture was heated to 80 ℃ and stirred at 80 ℃ for 15 h. The mixture was concentrated under reduced pressure, and the residue was purified by flash column chromatography eluting with EtOAc in PE (with EtOAc from 0-50%) in 15 min to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -11-oxo-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-4
4-carbonitrile (78.0 mg, 55%yield) as a yellow solid. LC-MS: (ESI) m/z [M+H]
+ 439.2
Step 6: (1
1S, 1
3R, Z) -11-oxo-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-4
4-carbonitrile and (1
1S, 1
3R, Z) -11-oxo-2
1H-5, 12-dioxa-3, 10-
diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-4
4-carboxamide
The solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -11-oxo-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-4
4-carbonitrile (78.0 mg, 178 μmol) in TFA (5.0 mL) was heated to 70 ℃ and stirred at 70 ℃ for 32 h. The mixture was concentrated under reduced pressure, and the residue was sent to purified by Prep-HPLC (with CH
3CN from 30%to 60%in 10 min) to afford (1
1S, 1
3R, Z) -11-oxo-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-4
4-carbonitrile (23.0 mg, 34%yield) as a pale yellow solid. LC-MS: (ESI) m/z [M+H]
+ 383.1. And to afford (1
1S, 1
3R, Z) -11-oxo-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-4
4-carboxamide (18.0 mg, 25%yield) as a white solid. LC-MS: (ESI) m/z [M+H]
+ 401.2.
Example 28
(1
1S, 1
3R, Z) -1
1-oxo-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-4
5-carbonitrile

Step 1: tert-butyl (4- ( (6-chloro-3-cyanopyridin-2-yl) oxy) butyl) carbamate
To a solution of tert-butyl (4-hydroxybutyl) carbamate (457 mg, 2.64 mmol) in THF (10 mL) was added sodium hydride (211 mg, 5.28 mmol, 60%purity) at 0 ℃ under nitrogen. The reaction was stirred at 0 ℃ for 30 minutes and then 2, 6-dichloronicotinonitrile (500 mg, 2.64 mmol) was added. The reaction was stirred at 25 ℃ for 2 h. The mixture was quenched with saturated NH
4Cl solution (50 mL) and extracted with EtOAc (30 mL × 3) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrated. The residue was purified by flash chromatography on silica gel eluting with EtOAc in PE (with EtOAc from 0 to 40%) in 15 min to afford tert-butyl (4- ( (6-chloro-3-cyanopyridin-2-yl) oxy) butyl) carbamate (285 mg, 33%yield) as colorless oil. LC-MS: m/z 348.1 [M+H]
+.
Step 2: tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) -3-cyanopyridin-2-yl) oxy) butyl) carbamate
To a mixture of (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (150 mg, 671 μmol) , tert-butyl (4- ( (6-chloro-3-cyanopyridin-2-yl) oxy) butyl) carbamate (243 mg, 746 μmol) and Cs
2CO
3 (730 mg, 2.24 mmol) in dioxane (15 mL) was added XantPhos (173 mg, 298 μmol) and Pd
2 (dba)
3 (137 mg, 149 μmol) under nitrogen. The reaction was heated to 95 ℃ and stirred at 95 ℃under nitrogen for 16 h. The mixture was diluted with water (50 mL) and extracted with EtOAc (30 mL × 3) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrated. The residue was purified by flash chromatography on silica gel eluting with EtOAc in PE (with EtOAc from 0-50%) in 20 min to afford tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -3-cyanopyridin-2-yl) oxy) butyl) carbamate (136 mg, 36%yield) as a light yellow solid. LC-MS: m/z 513.3 [M+H]
+.
Step 3: (1R, 3S) -3- (5- ( (6- (4- ( (tert-butoxycarbonyl) amino) butoxy) -5-cyanopyridin-2-yl) amino) -1-
(tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
A mixture of tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -3-cyanopyridin-2-yl) oxy) butyl) carbamate (130 mg, 254 μmol) , CDI (822 mg, 5.07 mmol) , DIPEA (164 mg, 1.27 mmol) and DSC (649 mg, 2.54 mmol) in DCE (10 mL) was heated to 70 ℃and stirred at 70 ℃ for 2 h. The mixture was diluted with water (50 mL) and extracted with DCM (30 mL × 3) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrated. The residue was purified by flash chromatography on silica gel eluting with EtOAc in PE (with EtOAc from 0-100%) in 15 min to afford (1R, 3S) -3- (5- ( (6- (4- ( (tert-butoxycarbonyl) amino) butoxy) -5-cyanopyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (89 mg, 58%yield) as a yellow solid. LC-MS: m/z 607.3 [M+H]
+.
Step 4: (1R, 3S) -3- (5- ( (6- (4-aminobutoxy) -5-cyanopyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-
3-yl) cyclopentyl 1H-imidazole-1-carboxylate
A solution of (1R, 3S) -3- (5- ( (6- (4- ( (tert-butoxycarbonyl) amino) butoxy) -5-cyanopyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (89 mg, 146 μmol) in TFA (3.0 mL) and DCM (10 mL) was stirred at 25 ℃ for 1 h. The solvent was evaporated to afford (1R, 3S) -3- (5- ( (6- (4-aminobutoxy) -5-cyanopyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (74 mg, 100%yield) as light brown oil, which was used in the next step directly. LC-MS: m/z 507.2 [M+H]
+.
Step 5: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -11-oxo-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-4
5-carbonitrile
To a solution of [ (1R, 3S) -3- [5- [ [6- (4-aminobutoxy) -5-cyano-2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (140 mg, 276 μmol) in CH
3CN (6.0 mL) was added N, N-Diisopropylethylamine (107 mg, 829 μmol, 144 μL) . The mixture was heated to 80 ℃ and stirred at 80 ℃ for 12 h. LCMS showed the starting material was consumed and desired product was formed. The mixture was concentrated under reduced pressure. The residue was diluted with H
2O (30 mL) and extracted with EtOAc (30 mL × 2) . The combined organic layer was washed by brine (50 mL) and dried over Na
2SO
4, filtered and concentrated . The residue was purified by flash chromatography eluting with MeOH in DCM (with MeOH from 0 to 5%) in 15 min to provide the (1
1S, 1
3R, Z) -2
1- (tert-butyl) -11-oxo-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-45-carbonitrile (20 mg, 16%yield) as a white solid. LC-MS: m/z 439.3 [M+H]
+.
Step 6: (1
1S, 1
3R, Z) -11-oxo-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-4
5-carbonitrile
The mixture of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -11-oxo-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-4
5-carbonitrile (15.0 mg, 34.2 μmol) in FA (3.0 mL) was heated to 100 ℃ and stirred at 100 ℃ for 12 h in a Schlenk tube. LCMS showed the starting material was consumed and desired product was formed. The residue was purified by prep-HPLC (with CH
3CN from 10%to 40%in 8 min) to provide (1
1S, 1
3R, Z) -11-oxo-2
1H-5, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-4
5-carbonitrile (1.00 mg, 7%yield) as a colorless solid. LC-MS: m/z 383.1 [M+H]
+.
Example 29
(1
1S, 1
3R, Z) -2
1H, 4
1H-11-oxa-3, 9-diaza-4 (6, 1) -pyrrolo [2, 3-b] pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacycloundecaphan-10-one

Step 1: tert-butyl N- [4- (6-bromopyrrolo [2, 3-b] pyridin-1-yl) butyl] carbamate
To a suspension of 6-bromo-1H-pyrrolo [2, 3-b] pyridine (300 mg, 1.52 mmol) in DMF (12.0 mL) was added slowly NaH (91.3 mg, 60 wt. %in mineral oil, 2.28 mmol) at 0 ℃ and stirred for 30 min under N
2. Then tert-butyl N- (4-bromobutyl) carbamate (575 mg, 2.28 mmol) was added at 0 ℃. The reaction was stirred at 25 ℃ for 24 h. Then it was quenched with ice-cold water (20 mL) and extracted with EtOAc (3 × 20 mL) . The organic phase was washed with brine (50 mL) and dried over anhydrous Na
2SO
4, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by flash column chromatography eluting with EtOAc/PE (with EtOAc from 0 to 25%in 25 min) to afford tert-butyl N- [4- (6-bromopyrrolo [2, 3-b] pyridin-1-yl) butyl] carbamate (410 mg, 73%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 367.9.
Step 2: tert-butyl N- [4- [6- [ [1-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-
yl] amino] pyrrolo [2, 3-b] pyridin-1-yl] butyl] carbamate
To a suspension of tert-butyl N- [4- (6-bromopyrrolo [2, 3-b] pyridin-1-yl) butyl] carbamate (250 mg, 0.678 mmol) in toluene (10.0 mL) was sequentially added (1R, 3S) -3- (5-amino-2-tert-butyl-pyrazol-3-yl) cyclopentanol (151 mg, 0.678 mmol) , Pd
2 (dba)
3 (62.2 mg, 0.067 mmol) , XantPhos (78 mg, 0.135 mmol) and Cs
2CO
3 (663 mg, 2.04 mmol) . The reaction was warmed to 80 ℃ and stirred at that temperature for 16 h under N
2. Then it was concentrated under reduced pressure and purified by flash column chromatography eluting with EtOAc/PE (with EtOAc from 0 to 50%in 25 min) to afford tert-butyl N- [4- [6- [ [1-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] pyrrolo [2, 3-b] pyridin-1-yl] butyl] carbamate (110 mg, 31%yield) as a brown oil. LC-MS: m/z [M+H]
+ 511.0.
Step 3: [ (1R, 3S) -3- [5- [ [1- [4- (tert-butoxycarbonylamino) butyl] pyrrolo [2, 3-b] pyridin-6-yl] amino] -
2-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate
To a suspension of tert-butyl N- [4- [6- [ [1-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] pyrrolo [2, 3-b] pyridin-1-yl] butyl] carbamate (100 mg, 0.195 mmol) in CH
2Cl
2 (5.0 mL) was added CDI (84.6 mg, 0.587 mmol) and Et
3N (136 μL, 99.0 mg, 0.979 mmol) at 35 ℃ and stirred for 16 h. Then the reaction mixture was quenched with ice-cold water (10 mL) and extracted with CH
2Cl
2 (3 × 10 mL) . The organic phase was dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The crude product [ (1R, 3S) -3- [5- [ [1- [4- (tert-butoxycarbonylamino) butyl] pyrrolo [2, 3-b] pyridin-6-yl] amino] -2-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate was used in the next step without further purification. LC-MS: m/z [M+H]
+605.0.
Step 4: [ (1R, 3S) -3- [3- [ [1- (4-aminobutyl) pyrrolo [2, 3-b] pyridin-6-yl] amino] -1H-pyrazol-5-
yl] cyclopentyl] imidazole-1-carboxylate
A mixture of [ (1R, 3S) -3- [5- [ [1- [4- (tert-butoxycarbonylamino) butyl] pyrrolo [2, 3-b] pyridin-6-yl] amino] -2-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate in TFA (5.0 mL) was stirred for 16 h at 70 ℃. Then it was concentrated to give crude [ (1R, 3S) -3- [3- [ [1- (4-aminobutyl) pyrrolo [2, 3-b] pyridin-6-yl] amino] -1H-pyrazol-5-yl] cyclopentyl] imidazole-1-carboxylate as a brown oil, which was used in the next step without further purification. LC-MS: m/z [M+H]
+448.9.
Step 5: (1
1S, 1
3R, Z) -2
1H, 4
1H-11-oxa-3, 9-diaza-4 (6, 1) -pyrrolo [2, 3-b] pyridina-2 (5, 3) -pyrazola-
1 (1, 3) -cyclopentanacycloundecaphan-10-one
To a suspension of [ (1R, 3S) -3- [3- [ [1- (4-aminobutyl) pyrrolo [2, 3-b] pyridin-6-yl] amino] -1H-pyrazol-5-yl] cyclopentyl] imidazole-1-carboxylate in CH
3CN (5.0 mL) was added Et
3N (0.27 mL, 196 mg, 1.94 mmol) at room temperature. The mixture was stirred at 80 ℃ for 5 h in a sealed tube. Then it was concentrated and purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 30%to 55%in 10 min (0.1%HCO
2H condition) ) to afford (1
1S, 1
3R, Z) -2
1H, 4
1H-11-oxa-3, 9-diaza-4 (6, 1) -pyrrolo [2, 3-b] pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacycloundecaphan-10-one (13.2 mg, 17%yield) as an off-white solid. LC-MS: m/z [M+H]
+ 380.8.
Example 30
(1
1S, 1
3R, Z) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyrazina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one

Step 1: tert-butyl (3- ( (6-chloropyrazin-2-yl) methoxy) propyl) carbamate
To a solution of NaH (110 mg, 2.77 mmol, 60%purity) in DMF (15 mL) was added (6-chloropyrazin-2-yl) methanol (200 mg, 1.38 mmol) in DMF (1.0 mL) . The mixture was stirred for 10 min at -20 ℃under N
2. Then tert-butyl N- (3-bromopropyl) carbamate (362 mg, 1.52 mmol) in DMF (1.0 mL) was added to this mixture, then it was stirred at -20℃ for 20 min. LCMS showed the starting material was consumed and desired product was detected. The reaction was quenched with addition of saturation water (50 mL) , extracted with EtOAc (50 mL × 3) . The combined organic layers were washed with brine (50 mL × 3) , dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by flash column chromatography eluting with EtOAc in PE from 0 to 50%to give tert-butyl (3- ( (6-chloropyrazin-2-yl) methoxy) propyl) carbamate (160 mg, 38%yield) as yellow oil. LC-MS: m/z [M-Boc+H]
+ 202.1.
Step 2: tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyrazin-2-yl) methoxy) propyl) carbamate
To a suspension of tert-butyl (3- ( (6-chloropyrazin-2-yl) methoxy) propyl) carbamate (140 mg, 464 μmol) in dioxane (20 mL) was added (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (103 mg, 464μmol) , Pd
2 (dba)
3 (84.9 mg, 92.7 μmol) , XantPhos (107 mg, 185 μmol) and K
2CO
3 (192 mg, 1.39 mmol) at 25 ℃. Then the reaction mixture was stirred at 100 ℃ for 3 h under N
2. The resulting mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography eluting with MeOH/DCM (with MeOH from 0 to 5%in 20 min) to give tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyrazin-2-yl) methoxy) propyl) carbamate (87.0 mg, 38%yield) as a yellow solid. LC-MS: m/z [M+H]
+ 489.3.
Step 3: (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) pyrazin-2-yl) amino) -1-
(tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyrazin-2-yl) methoxy) propyl) carbamate (67.0 mg, 137 μmol) in DCM (3.0 mL) was added Et
3N (138 mg, 1.37 mmol, 191 μL) and di (imidazol-1-yl) methanone (66.7 mg, 411 μmol) . The mixture was stirred at 40 ℃ for 2 h under N
2. The resulting mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography eluting with MeOH/DCM (MeOH from 0 to 5%in 15 min) to give (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) pyrazin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (70.0 mg, 88%yield) as yellow solid. LC-MS: m/z [M+H]
+ 583.3.
Step 4: (1R, 3S) -3- (5- ( (6- ( (3-aminopropoxy) methyl) pyrazin-2-yl) amino) -1- (tert-butyl) -1H-
pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) pyrazin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (60.0 mg, 102 μmol) in DCM (3.0 mL) was added TFA (3.0 mL) . The mixture was stirred at 25 ℃ for 0.5 h under N
2. LCMS showed the starting material was consumed and desired product was detected. The crude product was used immediately in the next step without purification. LC-MS: m/z [M+H]
+ 483.2.
Step 5: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyrazina-2 (3, 5) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a solution of (1R, 3S) -3- (5- ( (6- ( (3-aminopropoxy) methyl) pyrazin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (60.0 mg, 124 μmol) in CH
3CN (30 mL) was added DIPEA (5.0 mL) . The mixture was heated to 90 ℃ and stirred at 90 ℃ for 12 h under N
2. The resulting mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography eluting with MeOH/DCM (with MeOH from 0 to 5%in 15 min) to give (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyrazina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (30.0 mg, 58%yield) as a yellow oil. LC-MS: m/z [M+H]
+415.2.
Step 6: (1
1S, 1
3R, Z) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyrazina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one
A suspension of give (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyrazina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (30.0 mg, 72.3 μmol) in TFA (5.0 mL) . The mixture was heated to 80 ℃ and stirred at 80 ℃ for 20 h under N
2. LCMS showed the starting material was consumed and desired product was detected. The crude product was purified by prep-HPLC (with CH
3CN from 14%to 44%in 9 min) to give (1
1S, 1
3R, Z) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyrazina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (4.90 mg, 19%yield) as a yellow solid. LC-MS: m/z [M+H]
+ 359.1.
Example 31
(1
1S, 1
3R, 2
4Z, 4
2Z) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 4) -thiazola-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one

Step 1: tert-butyl (3- ( (2-bromothiazol-4-yl) methoxy) propyl) carbamate
To a solution of (2-bromothiazol-4-yl) methanol (350 mg, 1.80 mmol) and tert-butyl (3-bromopropyl) carbamate (859 mg, 3.61 mmol) in DMF (10 mL) was added NaH (216 mg, 9.02 mmol) . The mixture was stirred at 25 ℃ for 2 h. The mixture was diluted with H
2O (20 ml) , and extrated with EtOAc (20 mL × 2) . Organic layers were combined and dired over Na
2SO
4, filtered and concentrated to get a residue, which was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 50%in 12 min) to afford the product tert-butyl (3- ( (2-bromothiazol-4-yl) methoxy) propyl) carbamate (600 mg, 95%yield) as a white gum. LC-MS: m/z 372.9 [M+Na]
+.
Step 2: tert-butyl (3- ( (2- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) thiazol-4-yl) methoxy) propyl) carbamate
To a solution of tert-butyl N- [3- [ (2-bromothiazol-4-yl) methoxy] propyl] carbamate (200 mg, 569 μmol) and (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (150 mg, 672 μmol) in toluene (2.0 mL) was added XantPhos (132 mg, 228 μmol) , Pd
2 (dba)
3 (104 mg, 114 μmol) and K
3PO
4 (363 mg, 1.71 mmol) . The mixture was heated to 100 ℃ and stirred at 100 ℃ for 15 h. The mixture was cooled to room temperature and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 100%in 15 min) to afford tert-butyl (3- ( (2- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) thiazol-4-yl) methoxy) propyl) carbamate (90.0 mg, 32%yield) as a brown liquid. LC-MS: m/z 494.3 [M+H]
+.
Step 3: (1R, 3S) -3- (5- ( (4- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) thiazol-2-yl) amino) -1-
(tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of tert-butyl N- [3- [ [2- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] thiazol-4-yl] methoxy] propyl] carbamate (90.0 mg, 182 μmol) and di (1H-imidazol-1-yl) methanone (74.0 mg, 456 μmol) in DCM (5.0 mL) was added DIPEA (71.0 mg, 547 μmol) . The mixture was stirred at 35 ℃ for 2 h. The mixture was cooled to room temperature and concentrated under reduced pressure to give (1R, 3S) -3- (5- ( (4- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) thiazol-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (100 mg, crude) . LC-MS: m/z 588.3 [M+H]
+.
Step 4: (1R, 3S) -3- (5- ( (4- ( (3-aminopropoxy) methyl) thiazol-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-
3-yl) cyclopentyl 1H-imidazole-1-carboxylate
A mixture of [ (1R, 3S) -3- [5- [ [4- [3- (tert-butoxycarbonylamino) propoxymethyl] thiazol-2-yl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (100 mg, 170 μmol) in DCM : TFA = 2: 1 (6.0 mL) was stirred at 25 ℃ for 0.5 h. The mixture was concentrated under reduced pressure to give (1R, 3S) -3- (5- ( (4- ( (3-aminopropoxy) methyl) thiazol-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (80.0 mg, crude) LC-MS: m/z 488.1 [M+H]
+.
Step 5: (1
1S, 1
3R, 2
4Z, 4
2Z) -2
1- (tert-butyl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 4) -thiazola-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a solution of [ (1R, 3S) -3- [5- [ [4- (3-aminopropoxymethyl) thiazol-2-yl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (80.0 mg, 164 μmol) in CH
3CN (15 mL) was added Et
3N (332 mg, 3.28 mmol) . The mixture was heated to 80 ℃ and stirred at 80 ℃ for 16 h. The mixture was cooled to room temperature and concentrated under reduced pressure to give (1
1S, 1
3R, 2
4Z, 4
2Z) -2
1- (tert-butyl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 4) -thiazola-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (15.0 mg, 20%yield) . LC-MS: m/z 420.2 [M+H]
+.
Step 6: (1
1S, 1
3R, 2
4Z, 4
2Z) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 4) -thiazola-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one
A mixture of (1
1S, 1
3R, 2
4Z, 4
2Z) -2
1- (tert-butyl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 4) -thiazola-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (15.0 mg, 35.8 μmol) in TFA (2.0 mL) was heated to 80 ℃ and stirred at 80 ℃ for 2 h. The mixture was cooled to room temperature and concentrated under reduced pressure. The residue was purified by Pre-HPLC (with CH
3CN CH
3CN from 30%to 70%in 15 min) to give (1
1S, 1
3R, 2
4Z, 4
2Z) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 4) -thiazola-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (1.00 mg, 8%yield) as a white solid. LC-MS: (ESI) [M+H]
+364.1.
Example 32
(1
1S, 1
3R, Z) -4
5-fluoro-2
1H-6, 12-dioxa-3, 10-diaza-4 (4, 2) -pyrimidina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one

Step 1: ethyl 2-fluoro-3-oxopropanoate
To a suspension of NaH (5.65 g, 141 mmol, 60%purity) in THF (30 mL) was added dropwise of the mixture solution of ethyl 2-fluoroacetate (5.00 g, 47.1 mmol, 4.57 mL) and ethyl formate (5.24 g, 70.7 mmol, 5.69 mL) in THF (5.0 mL) at 0 ℃. Then the reaction mixture was warmed to 25 ℃ and stirred at 25 ℃ for 13 h. The mixture was concentrated under reduced pressure to afford the crude product ethyl 2-fluoro-3-oxo-propanoate (6.30 g, crude) as a yellow solid, which was used for next step directly. LC-MS: (ESI) m/z No Ms.
Step 2: 2- (chloromethyl) -5-fluoropyrimidin-4 (3H) -one
The mixture suspension of ethyl 2-fluoro-3-oxo-propanoate (6.30 g, crude) , 2-chloroacetamidine (7.88 g, 61.1 mmol, HCl) and NaOEt (4.80 g, 70.5 mmol) in Ethanol (40 mL) was stirred at 85 ℃ for 3 h. The mixture was concentrated under reduced pressure, and the residue was dissolved in EtOAc (100 mL) , then the mixture was filtered, and the filter cake was washed with EtOAc (200 mL) . The filtrate was concentrated under reduced pressure, and the residue was purified by silica gel chromatography eluting with EtOAc/DCM with EtOAc from 0 to 100%in 20 min to afford the product 2- (chloromethyl) -5-fluoropyrimidin-4 (3H) -one (700 mg, 9%yield) as a pale-yellow gum. LC-MS: (ESI) m/z [M+H]
+ 163.0.
Step 3: tert-butyl (3- ( (5-fluoro-6-oxo-1, 6-dihydropyrimidin-2-yl) methoxy) propyl) carbamate
To a solution of 2- (chloromethyl) -5-fluoro-1H-pyrimidin-6-one (1.00 g, 6.15 mmol) and tert-butyl N-(3-hydroxypropyl) carbamate (1.13 g, 6.46 mmol) in THF (15 mL) was added NaH (984 mg, 24.6 mmol, 60%purity) at 0 ℃. Then the reaction was allowed warmed to 20 ℃ and stirred at 20 ℃ for 3 h. The mixture was quenched with 5.0 mL water, adjusted the pH to 6 with 1 N HCl, then the mixture was concentrated under reduced pressure, and the residue was purified by silica gel chromatography eluting with EtOAc/PE with EtOAc from 0 to 100%in 20 min to afford the product tert-butyl (3- ( (5-fluoro-6-oxo-1, 6-dihydropyrimidin-2-yl) methoxy) propyl) carbamate (690 mg, 37%yield) as a pale yellow solid. LC-MS: (ESI) m/z [M+H]
+ 302.1.
Step 4: tert-butyl (3- ( (4-chloro-5-fluoropyrimidin-2-yl) methoxy) propyl) carbamate
To a solution of tert-butyl (3- ( (5-fluoro-6-oxo-1, 6-dihydropyrimidin-2-yl) methoxy) propyl) carbamate (470 mg, 1.56 mmol) in toluene (13 mL) was added DIPEA (1.74 g, 13.5 mmol, 2.35 mL) , followed by POCl
3 (1.32 g, 8.58 mmol) at 25 ℃. Then the reaction mixture was stirred at 25 ℃ for 6 h. The mixture was diluted with EtOAc (100 mL) , then the mixture was washed with brine (80 mL) , the organic layer was dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure, the residue was purified by silica gel chromatography eluting with EtOAc/PE with EtOAc from 0 to 50%in 15 min to afford the product tert-butyl (3- ( (4-chloro-5-fluoropyrimidin-2-yl) methoxy) propyl) carbamate (166 mg, 33%yield) as a pale yellow gum. LC-MS: (ESI) m/z [M+Na]
+ 342.1.
Step 5: tert-butyl (3- ( (4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) -5-fluoropyrimidin-2-yl) methoxy) propyl) carbamate
To a solution of tert-butyl (3- ( (4-chloro-5-fluoropyrimidin-2-yl) methoxy) propyl) carbamate (160 mg, 500 μmol) and (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (111 mg, 500 μmol) in dioxane (5.0 mL) was added Pd
2 (dba)
3 (68.7 mg, 75.1 μmol) , XanPhos (86.9 mg, 150 μmol) and K
2CO
3 (207 mg, 1.50 mmol) under the atmosphere of N
2. Then the reaction mixture was stirred at 100 ℃ under the atmosphere of N
2 for 5 h. The mixture was concentrated under reduced pressure, and the residue was purified by silica gel chromatography eluting with EtOAc/PE with EtOAc from 0 to 100%in 20 min to afford the product tert-butyl (3- ( (4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -5-fluoropyrimidin-2-yl) methoxy) propyl) carbamate (230 mg, 91%yield) as a yellow solid. LC-MS: (ESI) m/z [M+H]
+ 507.3.
Step 6: (1R, 3S) -3- (5- ( (2- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) -5-fluoropyrimidin-4-
yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of tert-butyl (3- ( (4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -5-fluoropyrimidin-2-yl) methoxy) propyl) carbamate (200 mg, 395 μmol) in DCM (10 mL) was added DIPEA (371 mg, 2.87 mmol, 0.500 mL) at 20 ℃, then di (imidazol-1-yl) methanone (192 mg, 1.18 mmol) was added to the reaction mixture, the mixture was heated to 60 ℃ and stirred at 60 ℃ for 5 h. The mixture was concentrated under reduced pressure to afford the product (1R, 3S) -3- (5- ( (2- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) -5-fluoropyrimidin-4-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (250 mg, crude) as a yellow gum. LC-MS: (ESI) m/z [M+H]
+ 601.3.
Step 7: (1R, 3S) -3- (5- ( (2- ( (3-aminopropoxy) methyl) -5-fluoropyrimidin-4-yl) amino) -1- (tert-butyl) -
1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of (1R, 3S) -3- (5- ( (2- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) -5-fluoropyrimidin-4-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (250 mg, 416 μmol) in DCM (5.0 mL) was added TFA (4.44 g, 38.94 mmol, 3.00 mL) at 20 ℃. Then the reaction mixture was stirred at 20 ℃ for 3 h. The mixture was concentrated under reduced pressure to afford the product (1R, 3S) -3- (5- ( (2- ( (3-aminopropoxy) methyl) -5-fluoropyrimidin-4-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (210 mg, crude) as a yellow gum. LC-MS: (ESI) m/z [M+H]
+ 501.2.
Step 8: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5-fluoro-2
1H-6, 12-dioxa-3, 10-diaza-4 (4, 2) -pyrimidina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
The solution of (1R, 3S) -3- (5- ( (2- ( (3-aminopropoxy) methyl) -5-fluoropyrimidin-4-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (210 mg, 420 μmol) in CH
3CN (30 mL) was added DIPEA (3.71 g, 28.7 mmol, 5.00 mL) at 20 ℃. Then the reaction mixture was heated to 80 ℃ and stirred at 80 ℃ for 15 h. The mixture was concentrated under reduced pressure, and the residue was purified by silica gel chromatography eluting with EtOAc/PE with EtOAc from 0 to 100%in 20 min to afford the product (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5-fluoro-2
1H-6, 12-dioxa-3, 10-diaza-4 (4, 2) -pyrimidina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (150 mg, 83%yield) as a yellow gum. LC-MS: (ESI) m/z [M+H]
+ 433.2.
Step 9: (1
1S, 1
3R, Z) -4
5-fluoro-2
1H-6, 12-dioxa-3, 10-diaza-4 (4, 2) -pyrimidina-2 (5, 3) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphan-11-one
The solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5-fluoro-2
1H-6, 12-dioxa-3, 10-diaza-4 (4, 2) -pyrimidina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (150 mg, 347 μmol) in TFA (5.0 mL) was stirred at 80 ℃ for 5 h. The mixture was concentrated under reduced pressure, and the residue was purified by Prep-HPLC (eluting with CH
3CN in water with CH
3CN from 20%to 50%in 9 min) to afford the product (1
1S, 1
3R, Z) -4
5-fluoro-2
1H-6, 12-dioxa-3, 10-diaza-4 (4, 2) -pyrimidina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (56.0 mg, 43%yield) as a white solid. LC-MS: (ESI) m/z [M+H]
+ 377.1.
Example 33
(1
1S, 1
3R, Z) -2
1H-6, 12-dioxa-3, 10-diaza-4 (4, 2) -pyrimidina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one

Step 1: tert-butyl (3- ( (6-oxo-1, 6-dihydropyrimidin-2-yl) methoxy) propyl) carbamate
To a solution of tert-butyl N- (3-hydroxypropyl) carbamate (299 mg, 1.71 mmol) in THF (10 mL) was added NaH (263 mg, 6.57 mmol, 60%purity) at 0 ℃. Then the reaction mixture was stirred at 0 ℃ for 10 min. Then 2- (chloromethyl) -1H-pyrimidin-6-one (190 mg, 1.31 mmol) was added to the reaction mixture. The reaction mixture was stirred at 20 ℃ for 3 h. The reaction mixture was quenched with MeOH (5 mL) , then the mixture was concentrated under reduced pressure, and the residue was purified by flash column chromatography eluting with PE/EtOAc (with EtOAc from 0~60%in 15 min) to afford tert-butyl (3- ( (6-oxo-1, 6-dihydropyrimidin-2-yl) methoxy) propyl) carbamate (295 mg, 79%yield) as a pale yellow oil. LC-MS: (ESI) m/z [M+H]
+284.2.
Step 2: tert-butyl (3- ( (4-chloropyrimidin-2-yl) methoxy) propyl) carbamate
To a solution of tert-butyl (3- ( (6-oxo-1, 6-dihydropyrimidin-2-yl) methoxy) propyl) carbamate (200 mg, 706 μmol) in toluene (8.0 mL) was added DIPEA (456 mg, 3.53 mmol, 614 μL) and POCl
3 (216 mg, 1.41 mmol) at 25 ℃. Then the reaction mixture was stirred at 25 ℃ for 72 h. The mixture was diluted with EtOAc (100 mL) , then the mixture was washed with brine (80 mL) , the organic layer was dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure, the residue was purified by flash column chromatography eluting with PE/EtOAc (with EtOAc from 0~50%in 15 min) to afford tert-butyl (3- ( (4-chloropyrimidin-2-yl) methoxy) propyl) carbamate (80.0 mg, 38%yield) as a pale yellow gum. LC-MS: (ESI) m/z [M+H]
+ 302.1.
Step 3: tert-butyl (3- ( (4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyrimidin-2-yl) methoxy) propyl) carbamate
To a solution of tert-butyl (3- ( (4-chloropyrimidin-2-yl) methoxy) propyl) carbamate (80.0 mg, 265 μmol) and (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (59.2 mg, 265 μmol) in dioxane (8.0 mL) was added Pd
2 (dba)
3 (24.3 mg, 26.5 μmol) , XantPhos (30.7 mg, 53.0 μmol) and Cs
2CO
3 (216 mg, 663 μmol) under the atmosphere of N
2. Then the reaction mixture was heated to 100 ℃ and stirred at 100 ℃ under the atmosphere of N
2 for 5 h. The mixture was concentrated under reduced pressure, and the residue was purified by flash column chromatography eluting with PE/EtAOc (with EtOAc from 0~60%in 15 min) to afford tert-butyl (3- ( (4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyrimidin-2-yl) methoxy) propyl) carbamate (60.0 mg, 46%yield) as a yellow solid. LC-MS: (ESI) m/z [M+H]
+ 489.2.
Step 4: tert-butyl (3- ( (4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-
nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyrimidin-2-
yl) methoxy) propyl) carbamate
To a solution of tert-butyl N- [3- [ [4- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] pyrimidin-2-yl] methoxy] propyl] carbamate (60.0 mg, 123 μmol) in DCE (37 mL) was added DMAP (3.00 mg, 24.56 μmol) and DIPEA (79.3 mg, 614 μmol, 107 μL) , then (4-nitrophenyl) carbonochloridate (74.3 mg, 368 μmol) was added to the reaction mixture. The reaction mixture was heated to 80 ℃ and stirred at 80 ℃ for 15 h. The mixture was not disposed for the weekend. The mixture was diluted with DCM (100 mL) , the mixture was washed with brine (150 mL) , dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure, and the residue was purified by flash column chromatography eluting with PE /EtOAc (with EtOAc from 0~60%in 10 min) to afford [ (1R, 3S) -3- [5- [ [2- [3- (tert-butoxycarbonylamino) propoxymethyl] pyrimidin-4-yl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] (4-nitrophenyl) carbonate (36.0 mg, 45%yield) as a yellow solid. LC-MS: (ESI) m/z [M+H]
+ 654.3.
Step 5: (1R, 3S) -3- (5- ( (2- ( (3-aminopropoxy) methyl) pyrimidin-4-yl) amino) -1- (tert-butyl) -1H-
pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate
To a solution of [ (1R, 3S) -3- [5- [ [2- [3- (tert-butoxycarbonylamino) propoxymethyl] pyrimidin-4-yl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] (4-nitrophenyl) carbonate (36.0 mg, 55.1 μmol) in DCM (5.0 mL) was added TFA (37.6 mg, 330 μmol, 25.5 μL) at 20 ℃. Then the reaction mixture was stirred at 20 ℃ for 3 h. The mixture was concentrated under reduced pressure to afford [ (1R, 3S) -3- [5- [ [2- (3-aminopropoxymethyl) pyrimidin-4-yl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] (4-nitrophenyl) carbonate (40.0 mg, crude, TFA) as a yellow gum. LC-MS: (ESI) m/z [M+H]
+ 554.2.
Step 6: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (4, 2) -pyrimidina-2 (3, 5) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a solution of [ (1R, 3S) -3- [5- [ [2- (3-aminopropoxymethyl) pyrimidin-4-yl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] (4-nitrophenyl) carbonate (40.0 mg, 59.9 μmol, TF) in CH
3CN (15 mL) was added dropwise the DIPEA (77.4 mg, 599 μmol, 104 μL) slowly. Then the reaction mixture was stirred at 20 ℃ for 15 h. The mixture was concentrated under reduced pressure, and the residue was purified by flash column chromatography eluting with PE/EtOAc (with EtOAc from 0~50%in 10 min) to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (4, 2) -pyrimidina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (18.0 mg, 72%yield) as a yellow solid, LC-MS: (ESI) m/z [M+H]
+ 415.2.
Step 7: (1
1S, 1
3R, Z) -2
1H-6, 12-dioxa-3, 10-diaza-4 (4, 2) -pyrimidina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one
The solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (4, 2) -pyrimidina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (18.0 mg, 43.43 μmol) in TFA (5.0 mL) was heated to 70 ℃ and stirred at 70 ℃ for 12 h. The mixture was concentrated under reduced pressure, and the residue was sent to purified by Prep-HPLC (with CH
3CN from 10%to 20%in 8 min) to afford (1
1S, 1
3R, Z) -2
1H-6, 12-dioxa-3, 10-diaza-4 (4, 2) -pyrimidina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (7.00 mg, 45%yield) as a white solid. LC-MS: (ESI) m/z [M+H]
+ 359.1.
Example 34, 35 and 36
(1
1S, 1
3R, Z) -4
4- (methylsulfonyl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphan-11-one, (1
1S, 1
3R, Z) -4
4-chloro-2
1H-6, 12-dioxa-3, 10-diaza-
4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one and (1
1S, 1
3R, Z) -4
4-
(methylthio) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one

Step 1: methyl 4, 6-dichloropicolinate
To a stirring solution of 4, 6-dichloropicolinic acid (3.00 g, 15.6 mmol) in Methanol (30 mL) was added conc. H
2SO
4 (1.80 g, 18.6 mmol) at 25 ℃. The reaction mixture was heated to 70 ℃ and stirred at 70 ℃ for 2 h. The reaction mixture was concentrated under reduce pressure, diluted with EtOAc (100 mL) and poured into saturated NaHCO
3 solution (80 mL) . The aqueous layer was extracted with EtOAc (2 × 150 mL) . The combined organic layer was washed with H
2O (100 mL) and brine (100 mL) . The organic layers were dried over Na
2SO
4 and concentrated under reduced pressure to afford methyl 4, 6-dichloropicolinate (2.80 g, 87%yield) as a yellow solid. LC-MS: m/z 206.7 [M+H]
+.
Step 2: (4, 6-dichloropyridin-2-yl) methanol
To a solution of methyl 4, 6-dichloropyridine-2-carboxylate (3.00 g, 14.6 mmol) in Methanol (12 mL) was added NaBH
4 (2.20 g, 58.2 mmol) at 0 ℃. The mixture was heated to 70 ℃ and stirred at 70 ℃ for 16 h. The mixture was cooled to 25 ℃, concentrated under reduce pressure, portioned into H
2O (50 mL) and extracted with DCM (50 mL × 3) . The organic layer was dried over Na
2SO
4 and concentrated to afford (4, 6-dichloropyridin-2-yl) methanol (2.40 g, 93%yield) as a white solid. LC-MS: m/z 178.7 [M+H]
+.
Step 3: tert-butyl (3- ( (4, 6-dichloropyridin-2-yl) methoxy) propyl) carbamate
To a solution of NaH (202 mg, 8.4 mmol) in DMF (25 mL) was added (4, 6-dichloropyridin-2-yl) methanol (1 g, 5.62 mmol) at 0 ℃. The mixture was stirred at this temperature under N
2 for 0.5 hour. After adding tert-butyl (3-bromopropyl) carbamate (1.6 g, 6.74 mmol) at 0 ℃, the mixture was stirred at 25 ℃ under N
2 for 2 h. The mixture was diluted with H
2O (20 ml) and extracted with EtOAc (20 mL × 2) . Organic layer was washed with LiCl solution (20 mL) , then concentrated to get a residue, which was purified by flash chromatography eluting with EtOAc/PE (with EtOAc from 0~50%in 10 min) to afford tert-butyl (3- ( (4, 6-dichloropyridin-2-yl) methoxy) propyl) carbamate (370 mg, 20%yield) as a yellow liquid. LC-MS: m/z 335.7 [M+H]
+.
Step 4: tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) -4-chloropyridin-2-yl) methoxy) propyl) carbamate
To a solution of tert-butyl N- [3- [ (4, 6-dichloro-2-pyridyl) methoxy] propyl] carbamate (450 mg, 1.34 mmol) and (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (250 mg, 1.12 mmol) in 1, 4-dioxane (20 mL) was added Pd
2 (dba)
3 (205 mg, 223 μmol) , XantPhos (259 mg, 447 μmol) and Cs
2CO
3 (1.10 g, 3.36 mmol) . The mixture was heated to 100 ℃ and stirred at 100 ℃under N
2 for 8 h. The mixture was filtered and concentrated under reduced pressure. The residue was purified by flash chromatograthy eluting with EtOAc/PE (with EtOAc from 0~50%in 15 min) to get tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -4-chloropyridin-2-yl) methoxy) propyl) carbamate (420 mg, 72%yield) as a yellow liquid. LC-MS: m/z 522.7 [M+H]
+.
Step 5: (1S, 3R) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) -4-chloropyridin-2-
yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of tert-butyl N- [3- [ [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -4-chloro-2-pyridyl] methoxy] propyl] carbamate (100 mg, 191 μmol) in DCM (10 mL) was added Et
3N (58.0 mg, 574 μmol) . The mixture was stirred at 50 ℃ for 3 h. The mixture was diluted with NaCl solution (10 mL) and extracted with EtOAc (10 mL × 2) . Organic layers were dried with Na
2SO
4 and concentrated under reduce pressure. The residue was purified by flash chromatography eluting with EtOAc/PE (with EtOAc from 0~70%in 15 min) to afford [ (1S, 3R) -3- [5- [ [6- [3- (tert-butoxycarbonylamino) propoxymethyl] -4-chloro-2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (110 mg, 93%yield) as a yellow liquid. LC-MS: m/z 616.7 [M+H]
+.
Step 6: (1S, 3R) -3- (5- ( (6- ( (3-aminopropoxy) methyl) -4-chloropyridin-2-yl) amino) -1- (tert-butyl) -
1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of [ (1S, 3R) -3- [5- [ [6- [3- (tert-butoxycarbonylamino) propoxymethyl] -4-chloro-2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (110 mg, 178 μmol) in DCM (4.0 mL) was added TFA (2.0 mL) . The mixture was stirred at 25 ℃ for 1 h. The mixture was concentrated to get (1R, 3S) -3- (5- ( (6- ( (3-aminopropoxy) methyl) -4-chloropyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (90.0 mg, 98%yield) as a yellow liquid. LC-MS: m/z [M+H]
+ 516.7.
Step 7: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4-chloro-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a solution of [ (1S, 3R) -3- [5- [ [6- (3-aminopropoxymethyl) -4-chloro-2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (80.0 mg, 155 μmol) in CH
3CN (615 μL) was added Et
3N (313 mg, 3.10 mmol) . The mixture was stirred at 80 ℃ under N
2 for 16 hours. The mixture was concentrated to get a residue. The residue was purified by flash chromatography column eluting with EtOAc/PE (with EtOAc from 0~70%in 15 min) to get (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4-chloro-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (60.0 mg, 86%yield) as a yellow liquid. LC-MS: m/z [M+H]
+448.7.
Step 8: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- (methylthio) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-
2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4-chloro-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3-cyclopentanacyclododecaphan-11-one (50.0 mg, 111 μmol) in Ethanol/H
2O =1: 1 (2.0 mL) was added sodium; methane thiolate (31.0 mg, 446 μmol) . The mixture was heated to 140 ℃ and stirred at 140 ℃ for 16 h. The mixture was diluted with H
2O (5.0 mL) and extracted with EtOAc (5 mL × 2) . Organic layers were combined and dried over Na
2SO
4, filtered and concentrated to get a residue, which was purified by flash chromatography column eluting with EtOAc/PE (with EtOAc from 0~70%in 15 min) to get (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- (methylthio) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (29.0 mg, 56%yield) as a yellow solid. LC-MS: (ESI) [M+H]
+ = 460.2.
Step 9: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- (methylsulfonyl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-
2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- (methylthio) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (20.0 mg, 43.5 μmol) in acetone (2.0 mL) was added Potassium Peroxomonosulfate 4.5% (actice oxygen) (80.0 mg, 130 μmol) at 0 ℃. The mixture was stirred at 25 ℃ for 5 h. The mixture was filtered and concentrated under reduced pressure to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- (methylsulfonyl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (18.0 mg, 84%yield) as a yellow liquid. LC-MS: (ESI) [M+H]
+ = 492.1.
Step 10: (1
1S, 1
3R, Z) -4
4- (methylsulfonyl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
A mixture of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- (methylsulfonyl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (15.0 mg, 30.5 μmol) in TFA (2.0 mL) was heated to 80 ℃ and stirred at 80 ℃ for 1 h. The mixture was concentrated under reduced pressure. The residue was purified by Prep-HPLC (with CH
3CN from 25%to 75%in 8 min) to give (1
1S, 1
3R, Z) -4
4- (methylsulfonyl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (8.00 mg, 60%yield) as a white solid. LC-MS: (ESI) [M+H]
+ = 436.1.
Step 11: (1
1S, 1
3R, Z) -4
4-chloro-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one
A mixture of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4-chloro-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (59.0 mg, 131 μmol) in TFA (5.0 mL) was heated to 80 ℃ and stirred at 80 ℃ for 2 h. The mixture was concentrated under reduced pressure. The residue was purified by Prep-HPLC (with CH
3CN from 40%to 60%in 8 min) to give (1
1S, 1
3R, Z) -4
4-chloro-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) cyclopentanacyclododecaphan-11-one (15.0 mg, 29%yield) as a white solid. LC-MS: (ESI) [M+H]
+=392.1.
Step 12: (1
1S, 1
3R, Z) -4
4- (methylthio) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphan-11-one
A mixture of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- (methylthio) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (10.0 mg, 21.7 μmol) in TFA (1.0 mL) was heated to 80 ℃ and stirred at 80 ℃ for 1 h. The mixture was concentrated under reduced pressure. The residue was purified by Prep-HPLC (with CH
3CN from 40%to 60%in 8 min) to give (1
1S, 1
3R, Z) -4
4- (methylthio) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) - cyclopentanacyclododecaphan-11-one (6.00 mg, 68%yield) as a white solid. LC-MS: (ESI) [M+H]
+= 404.1.
Example 37 and 38
(1
1S, 1
3R, Z) -44- (1-methyl-1, 2, 3, 6-tetrahydropyridin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -
pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one and (1
1S, 1
3R, Z) -4
4- (1-
methylpiperidin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one

Step 1: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- (1-methyl-1, 2, 3, 6-tetrahydropyridin-4-yl) -2
1H-6, 12-dioxa-
3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -44-chloro-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (30.0 mg, 67.0 μmol) in dioxane (2.0 mL) was added 1-methyl-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -1, 2, 3, 6-tetrahydropyridine (30.0 mg, 134 μmol) , Pd (OAc)
2 (3.00 mg, 13.4 μmol) , X-Phos (12.0 mg, 26.8 μmol) , K
2CO
3 (27.0 mg, 200 μmol) and H
2O (0.10 mL) . The suspension was degassed with N
2 for 5 times. The reaction mixture was heated to 110 ℃ and stirred at 110 ℃ for 10 h. The mixture was concentrated to dryness. The mixture was diluted with water (50 mL) and extracted with DCM (50 mL × 3) . The combined organic layer was washed with brine (50 mL × 2) , dried over Na
2SO
4, filtered and concentrated to dryness. The residue was purified by flash column chromatography eluting with DCM/MeOH (with MeOH from 0~9%in 10 min) to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- (1-methyl-1, 2, 3, 6-tetrahydropyridin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (22.0 mg, 64%yield) as a light yellow solid. LC-MS: (ESI) m/z [M+H]
+ 509.3.
Step 2: (1
1S, 1
3R, Z) -44- (1-methyl-1, 2, 3, 6-tetrahydropyridin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-
4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
The mixture of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- (1-methyl-1, 2, 3, 6-tetrahydropyridin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (20.0 mg, 39.3 μmol) in TFA (1.5 mL) was heated to 75 ℃ and stirred at 75 ℃ for 2 h. The mixture was concentrated to afford crude product, which was purified by prep-HPLC (with CH
3CN from 30%to 100%in 8 min) to afford the product (1
1S, 1
3R, Z) -4
4- (1-methyl-1, 2, 3, 6-tetrahydropyridin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (5.80 mg, 32%yield) as a white solid. LC-MS: (ESI) m/z [M+H]
+ 453.2.
Step 3: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- (1-methylpiperidin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -
pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- (1-methyl-1, 2, 3, 6-tetrahydropyridin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (25.0 mg, 49.2 μmol) in Methanol (2.0 mL) was added Pd/C (3.00 mg, 19.7 μmol) . The mixture was stirred at 25 ℃ for 3 h. The mixture was filtered and concentrated under reduced pressure to give (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- (1-methylpiperidin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (20.0 mg, 80%yield) as a yellow gum. LC-MS: m/z 511.3 [M+H]
+.
Step 4: (1R, 3S) -3- (2-cyclohexyl-1H-pyrrolo [2, 3-b] pyridin-5-yl) cyclopentyl isopropylcarbamate
A mixture of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- (1-methylpiperidin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (25.0 mg, 48.9 μmol) in TFA (2.0 mL) was heated to 80 ℃ and stirred at 80 ℃ for 3 h. The mixture was concentrated under reduced pressure. The residue was purified by Prep-HPLC (with CH
3CN from 40%to 60%in 12 min) to give (1
1S, 1
3R, Z) -4
4- (1-methylpiperidin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (6.00 mg, 30%yield) as a white solid. LC-MS: (ESI) [M+H]
+=455.2.
The following compounds were prepared using the similar procedure disclosed in synthetic example 38.


Example 44
(1
1S, 1
3R, Z) -4
4- (pyridin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one

Step 1: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- (pyridin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-
2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4-chloro-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (40.0 mg, 89.2 μmol) in dioxane (5.0 mL) and H
2O (1.0 mL) was added K
2CO
3 (37.0 mg, 267 μmol) , Pd (dppf) Cl
2 (11.5 mg, 17.8 μmol) and 4-pyridylboronic acid (21.9 mg, 178 μmol) . The reaction mixture was irradiated in a microwave reactor at 120 ℃ for 0.5 h. The residue was purified by flash column chromatography eluting with EtOAc/PE (with EtOAc from 0 to 50%in 15 min) to give (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- (pyridin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (40.0 mg, 91%yield) as yellow solid. LC-MS: m/z [M+H]
+ 491.2.
Step 2: (1
1S, 1
3R, Z) -4
4- (pyridin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphan-11-one
A suspension of (1
1S, 1
3R, Z) -21- (tert-butyl) -4
4- (pyridin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (30.0 mg, 61.1 μmol) in TFA (5.0 mL) was heated to 80 ℃ and stirred at 80 ℃ for 1 h under N
2. LCMS showed the starting material was consumed and desired product was detected. The crude product was purified by prep-HPLC (with CH
3CN from 8%to 38%in 9 min) to give (1
1S, 1
3R, Z) -4
4- (pyridin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (13.9 mg, 52%yield) as yellow solid. LC-MS: m/z [M+H]
+ 435.2.
The following compounds were prepared using the similar procedure disclosed in synthetic example 44.

Example 46
(1
1S, 1
3R, Z) -4
5- (1-methyl-1, 2, 3, 6-tetrahydropyridin-4-yl) -21H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -
pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one

Step 1: methyl 3-bromo-6-chloropicolinate
To a stirred solution of 3-bromo-6-chloropicolinic acid (100 mg, 423 umol) in MeOH (2.0 mL) was added H
2SO
4 (0.1 mL, 42 mg, 423 umol) at 25 ℃. The reaction mixture was warmed to 90 ℃ and stirred at that temperature for 16 h before it was treated with ice-cold water (20 mL) and extracted with EtOAc (50 mL × 3) . The combined organic phases were washed with brine (20 mL) , dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography eluting with PE/EtOAc with EtOAc from 0 to 20%in 15 min to afford methyl 3-bromo-6-chloropicolinate (80.0 mg, 76%yield) as a white solid. LC-MS: m/z [M+H]
+ 249.9.
Step 2: (3-bromo-6-chloropyridin-2-yl) methanol
To a suspension of methyl 3-bromo-6-chloropicolinate (100 mg, 399 umol) in MeOH (2.0 mL) was slowly added NaBH
4 (75.9 mg, 2.00 mmol) at 0 ℃. The mixture was warmed to 25 ℃ and stirred at that temperature for 16 h before it was treated with ice-cold water (20 mL) and extracted with EtOAc (50 mL ×3) . The organic phases was washed with brine (20 mL) and dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography eluting with PE/EtOAc with EtOAc from 0 to 30%in 25 min to afford (3-bromo-6-chloropyridin-2-yl) methanol (70.0 mg, 79%yield) as a colorless oil. LC-MS: m/z [M+H]
+ 222.0.
Step 3: tert-butyl (3- ( (3-bromo-6-chloropyridin-2-yl) methoxy) propyl) carbamate
To a stirred solution of NaH (2.59 g, 64.7 mmol, 60%purity) in THF (80.0 mL) was added (3-bromo-6-chloropyridin-2-yl) methanol (4.80 g, 21.6 mmol) . The mixture was stirred at 0 ℃ for 20 min under N
2. Then tert-butyl (3-bromopropyl) carbamate (5.14 g, 21.6 mmol) in THF (80.0 mL) was added to the mixture. The resulting mixture was stirred at 25 ℃ for 20 h under N
2 before it was quenched with water (100 mL) and then extracted with EtOAc (150 mL × 2) . The combined organic phases were dried over Na
2SO
4 and then filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE with EtOAc from 0 to 10%in 20 min to give tert-butyl (3- ( (3-bromo-6-chloropyridin-2-yl) methoxy) propyl) carbamate (3.00 g, 37%yield) as a yellow oil. LC-MS: m/z [M+Na]
+ 400.5.
Step 4: tert-butyl (3- ( (6-chloro-1'-methyl-1', 2', 3', 6'-tetrahydro- [3, 4'-bipyridin] -2-
yl) methoxy) propyl) carbamate
To a stirred solution of tert-butyl (3- ( (3-bromo-6-chloropyridin-2-yl) methoxy) propyl) carbamate (1.00 g, 2.63 mmol) in 1, 4-dioxane (10.0 mL) were sequentially added 1-methyl-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -1, 2, 3, 6-tetrahydropyridine (588 mg, 2.63 mmol) , Pd (dppf) Cl
2 (193 mg, 263 μmol) and Na
2CO
3 (698 mg, 6.58 mmol) and H
2O (2.0 mL) at 25 ℃. The mixture was warmed to 95 ℃ and stirred at that temperature for 3 h under N
2 before it was cooled to 25 ℃, diluted with water (25 mL) and extracted with EtOAc (50 mL × 2) . The combined organic phases were dried over Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE with EtOAc from 0 to 65%in 25 min to give tert-butyl (3- ( (6-chloro-1'-methyl-1', 2', 3', 6'-tetrahydro- [3, 4'-bipyridin] -2-yl) methoxy) propyl) carbamate (500 mg, 48%yield) as a brown solid. LC-MS: m/z [M+H]
+ 396.1.
Step 5: tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) -1', 2', 3', 6'-tetrahydro- [3, 4'-bipyridin] -2-yl) methoxy) propyl) carbamate
To a stirred solution of tert-butyl (3- ( (6-chloro-1'-methyl-1', 2', 3', 6'-tetrahydro- [3, 4'-bipyridin] -2-yl) methoxy) propyl) carbamate (355 mg, 896 μmol) in 1, 4-dioxane (10.0 mL) were sequentially added (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (100 mg, 448 μmol) , Pd
2 (dba)
3 (41.0 mg, 44.8 μmol) , XantPhos (25.9 mg, 44.8 μmol) and Cs
2CO
3 (292 mg, 896 μmol) at 25 ℃. The reaction mixture was warmed to 100℃ and stirred at that temperature for 3 h under N
2 atomosphere before it was cooled and concentrated under reduced pressure. The residue was purified by flash column chromatography eluting with petroleum ether/EtOAc with EtOAc from 0 to 70%in 25 min to afford tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H- pyrazol-5-yl) amino) -1', 2', 3', 6'-tetrahydro- [3, 4'-bipyridin] -2-yl) methoxy) propyl) carbamate (60.0 mg, 22%yield) as a yellow solid. LC-MS: m/z [1/2M+H]
+ 292.0.
Step 6: (1R, 3S) -3- (5- ( (2- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) -1'-methyl-1', 2', 3', 6'-
tetrahydro- [3, 4'-bipyridin] -6-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-
imidazole-1-carboxylate
To a suspension tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -1', 2', 3', 6'-tetrahydro- [3, 4'-bipyridin] -2-yl) methoxy) propyl) carbamate (60.0 mg, 103 μmol) in CH
2Cl
2 (2.0 mL) was added CDI (44.5 mg, 309 μmol) and Et
3N (72 μL, 52.1 mg, 515 μmol) at 25 ℃ and stirred for 2 h before it was quenched with ice-cold water (20 mL) and extracted with CH
2Cl
2 (20 mL × 3) . The organic phases were dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue (1R, 3S) -3- (5- ( (2- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) -1'-methyl-1', 2', 3', 6'-tetrahydro- [3, 4'-bipyridin] -6-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate was used in the next step without further purification. LC-MS: m/z [M+H]
+ 677.6.
Step 7: (1R, 3S) -3- (5- ( (2- ( (3-aminopropoxy) methyl) -1'-methyl-1', 2', 3', 6'-tetrahydro- [3, 4'-
bipyridin] -6-yl) amino) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
A mixture of (1R, 3S) -3- (5- ( (2- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) -1'-methyl-1', 2', 3', 6'-tetrahydro- [3, 4'-bipyridin] -6-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (crude) in TFA (4.0 mL) was stirred for 16 h at 70 ℃. Then it was concentrated to give (1R, 3S) -3- (5- ( (2- ( (3-aminopropoxy) methyl) -1'-methyl-1', 2', 3', 6'-tetrahydro- [3, 4'-bipyridin] -6-yl) amino) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate as a brown oil which was used in the next step without further purification. LC-MS: m/z [M+H]
+ 520.7.
Step 8: (1
1S, 1
3R, Z) -4
5- (1-methyl-1, 2, 3, 6-tetrahydropyridin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-
4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a suspension of (1R, 3S) -3- (5- ( (2- ( (3-aminopropoxy) methyl) -1'-methyl-1', 2', 3', 6'-tetrahydro- [3, 4'-bipyridin] -6-yl) amino) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (crude) in CH
3CN (5.0 mL) was added Et
3N (269 μL, 196 mg, 1.94 mmol) at 25 ℃. The mixture was warmed to 80 ℃and stirred at that temperature for 5 h. Then it was concentrated and purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 5%to 70%in 30 min) to afford (1
1S, 1
3R, Z) -45- (1-methyl-1, 2, 3, 6-tetrahydropyridin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (1.30 mg, 4%yield) as an light yellow solid. LC-MS: m/z [M+H]
+ 453.3.
Example 47
(1
1S, 1
3R, Z) -4
5-bromo-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one

Step 1: 3, 6-dibromo-2- (bromomethyl) pyridine
A mixture of 3, 6-dibromo-2-methylpyridine (10.0 g, 39.9 mmol) , AIBN (6.54 g, 39.9 mmol) and NBS (14.2 g, 79.7 mmol) in CCl
4 (100.0 mL) was stirred at 90 ℃ for 16 h before it was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 10%in 20 min) to give 3, 6-dibromo-2- (bromomethyl) pyridine (9.00 g, 69%yield) . LC-MS: m/z [M+H]
+ 329.4.
Step 2: tert-butyl (3- ( (3, 6-dibromopyridin-2-yl) methoxy) propyl) carbamate
To a stirred solution of NaH (0.67 g, 16.7 mmol, 60%purity) in THF (30.0 mL) was added tert-butyl (3-hydroxypropyl) carbamate (2.93 g, 16.7 mmol) at 0 ℃. The reaction mixture was stirred at that temperature for 30 min before a solution of 3, 6-dibromo-2- (bromomethyl) pyridine (5.00 g, 15.2 mmol) in THF (50.0 mL) was added. The resulting mixture was stirred at 0 ℃ for 1 h before it was diluted with H
2O (50 mL) and warmed to 25 ℃. The mixture was extracted with EtOAc (150 mL × 2) . The combined organic phases were washed with brine (100 mL) , dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 50%in 20 min) to afford tert-butyl (3- ( (3, 6-dibromopyridin-2-yl) methoxy) propyl) carbamate (2.10 g, 33%yield) as a light-yellow oil. LC-MS: m/z [M+H]
+ 424.9.
Step 3: tert-butyl (3- ( (3-bromo-6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-
5-yl) amino) pyridin-2-yl) methoxy) propyl) carbamate
To a mixture of tert-butyl (3- ( (3, 6-dibromopyridin-2-yl) methoxy) propyl) carbamate (360 mg, 849 μmol) , (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (190 mg, 849 μmol) and Cs
2CO
3 (553 mg, 1.70 mmol) in 1, 4-dioxane (8.0 mL) were sequentially added XantPhos (49.2 mg, 84.9 μmol) and Pd
2 (dba)
3 (77.8 mg, 84.9 μmol) at 25 ℃. The reaction mixture was stirred at 80 ℃under N
2 atmosphere for 3 h before it was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 80%in 20 min) to afford tert-butyl (3- ( (3-bromo-6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methoxy) propyl) carbamate (280 mg, 58%yield) as a light-yellow oil. LC-MS: m/z [M+H]
+ 565.6.
Step 4: (1R, 3S) -3- (5- ( (5-bromo-6- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) pyridin-2-
yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a stirred solution of tert-butyl (3- ( (3-bromo-6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methoxy) propyl) carbamate (280 mg, 494 μmol) in CH
2Cl
2 (12.0 mL) were sequentially added Et
3N (207 μL, 150 mg, 1.48 mmol) and CDI (213 mg, 1.48 mmol) at 25 ℃. The reaction mixture was warmed to 35 ℃ and stirred at that temperature for 2 h before it was cooled, diluted with H
2O (30 mL) and extracted with CH
2Cl
2 (50 mL × 2) . The combined organic phase was washed with brine (20 mL) and dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 80%in 20 min) to afford (1R, 3S) -3- (5- ( (5-bromo-6- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (280 mg, 85%yield) as a light-yellow solid. LC-MS: m/z [M+H]
+ 659.6.
Step 5: (1R, 3S) -3- (5- ( (6- ( (3-aminopropoxy) methyl) -5-bromopyridin-2-yl) amino) -1- (tert-butyl) -
1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a stirred solution of (1R, 3S) -3- (5- ( (5-bromo-6- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (280 mg, 424 μmol) in CH
2Cl
2 (20.0 mL) was added TFA (97.96 μL, 145 mg, 1.27 mmol) at 25 ℃. The reaction mixture was stirred at that temperature for 2 h before it was concentrated under reduced pressure to afford (1R, 3S) -3- (5- ( (6- ( (3-aminopropoxy) methyl) -5-bromopyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (crude) as a light-yellow oil. LC-MS: m/z [M+H]
+ 559.5.
Step 6: (1
1S, 1
3R, Z) -4
5-bromo-2
1- (tert-butyl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a stirred solution (1R, 3S) -3- (5- ( (6- ( (3-aminopropoxy) methyl) -5-bromopyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (crude) in CH
3CN (10.0 mL) was added Et
3N (1 mL) at 25 ℃ . The reaction mixture was warmed to 70 ℃ and stirred at that temperature for 16 h. The reaction mixture was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 80%in 20 min) to afford (1
1S, 1
3R, Z) -4
5-bromo-2
1- (tert-butyl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (150 mg, 62%yield for 2 steps) as a light-yellow oil. LC-MS: m/z [M+H]
+ 492.0.
Step 7: (1
1S, 1
3R, Z) -4
5-bromo-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one
A mixture of (1
1S, 1
3R, Z) -4
5-bromo-2
1- (tert-butyl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (25.0 mg, 50.9 mmol) in TFA (2.0 mL) was stirred for 16 h at 70 ℃. Then it was concentrated and purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 5%to 45%in 40 min) to give (1
1S, 1
3R, Z) -4
5-bromo-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (4.6 mg, 21%yield) . LC-MS: m/z [M+H]
+ 435.9.
Example 48
(1
1S, 1
3R, Z) -4
5- ( ( (2, 2, 2-trifluoroethyl) amino) methyl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-
2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one

Step 1: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -11-oxo-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-4
5-carbaldehyde
To a stirred solution of (1
1S, 1
3R, Z) -4
5-bromo-2
1- (tert-butyl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (100.0 mg, 0.20 mmol) and Pd (OAc)
2 (13.7 mg, 0.06 mmol) , JohnPhos (18.2 mg, 0.06 mmol) , HCOOK (199 mg, 2.03 mmol ) and t-BuNC (2.81 mg, 0.41 mmol) were sequentially added DMF (5.0 mL) under N
2 atmosphere at 25 ℃. The mixture was warmed to 60 ℃ and stirred at that temperature for 12 h before it was cooled to 25 ℃. The mixture was poured into H
2O (5 mL) and extracted with EtOAc (5 mL × 3) . The combined organic layers were washed with brine (5 mL) , dried over Na
2SO
4, filtered and concentrated. The mixture was concentrated and purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 40 %in 30 min) to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -11-oxo-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-4
5-carbaldehyde (50.0 mg, 56%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 442.24.
Step 2: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5- ( ( (2, 2, 2-trifluoroethyl) amino) methyl) -2
1H-6, 12-dioxa-3, 10-
diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a stirred solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -11-oxo-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-4
5-carbaldehyde (50.0 mg, 0.11 mmol) , 2, 2, 2-trifluoroethanamine (56.1 mg, 56.6 mmol) , Sodium cyanoborohydride (28.0 mg, 0.453 mmol) in DME (2.0 mL) was added acetic acid (2 drops) at 25 ℃ and the reaction mixture was stirred at that temperature for 12 h under N
2 atmosphere. The mixture was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 60 %in 25 min) to give (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5- ( ( (2, 2, 2-trifluoroethyl) amino) methyl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one afford (30.0 mg, 51%yield) as a brown oil. LC-MS: m/z [M+H]
+ 525.3.
Step 3: (1
1S, 1
3R, Z) -4
5- ( ( (2, 2, 2-trifluoroethyl) amino) methyl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -
pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
A stirred solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5- ( ( (2, 2, 2-trifluoroethyl) amino) methyl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one in TFA (2.0 mL) was stirred at 70 ℃ for 12 h before it was concentrated under reduced pressure. The residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 0%to 40%in 40 min) to give (1
1S, 1
3R, Z) -4
5- ( ( (2, 2, 2-trifluoroethyl) amino) methyl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (2.60 mg, 9.7%yield) as an off-white solid. LC-MS: m/z [M+H]
+ 469.2.
The following compounds were prepared using the similar procedure disclosed in synthetic example 48.


Example 50
(1
1S, 1
3R, Z) -4
5- (1-methylpiperidin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one

Step 1: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5- (1-methyl-1, 2, 3, 6-tetrahydropyridin-4-yl) -2
1H-6, 12-dioxa-
3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a suspension of (1
1S, 1
3R, Z) -4
5-bromo-2
1- (tert-butyl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (35.0 mg, 0.071 mmol) in 1, 4-dioxane (2.0 mL) and water (0.2 mL) was added 1-methyl-4- (4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl) -1,2, 3, 6-tetrahydropyridine (31.7 mg, 0.142 mmol) , Na
2CO
3 (22.6 mg, 0.213 mmol) and Pd (dppf) Cl
2 (10.4 mg, 0.014 mmol) at room temperature. The reaction was stirred at 80 ℃ for 2 h under N
2, before it was filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography eluting with MeOH/CH
2Cl
2 (with MeOH from 0 to 10%in 30 min) to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5- (1-methyl-1, 2, 3, 6-tetrahydropyridin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza- 4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (25.0 mg, 70%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 509.3.
Step 2: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5- (1-methylpiperidin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -
pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a suspension of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5- (1-methyl-1, 2, 3, 6-tetrahydropyridin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (25.0 mg, 0.049 mmol) in MeOH (2.0 mL) was added slowly Pd/C (11.0 mg, 0.098 mmol) at room temperature and stirred for 16 h under H
2. After completion of the reaction as judged by LCMS, reaction mixture was filtered and concentrated under reduced pressure. The crude product (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5- (1-methylpiperidin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (25.0 mg, crude) was used in the next step without further purification. LC-MS: m/z [M+H]
+ 511.0.
Step 3: (1
1S, 1
3R, Z) -4
5- (1-methylpiperidin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
A mixture of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5- (1-methylpiperidin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (25.0 mg, crude) in HCO
2H (2.0 mL) was stirred for 2 h at 80 ℃, until the reaction was complete as indicated by LCMS, the reaction mixture was concentrated under reduced pressure, purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 2%to 15%in 30 min) to give (1
1S, 1
3R, Z) -4
5- (1-methylpiperidin-4-yl) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (1.5 mg) as a white solid. LC-MS: m/z [M+H]
+ 455.3.
The following compounds were prepared using the similar procedure disclosed in synthetic example 50.


Example 54
(1
1S, 1
3R, Z) -4
5- (2- (pyrrolidin-1-yl) ethoxy) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one


Step 1: methyl 3-hydroxypicolinate
To a stirred solution of 3-hydroxypyridine-2-carboxylic acid (7.0 g, 50.3 mmol) in MeOH (400 mL) was added H
2SO
4 (4.0 mL) at 25 ℃. The mixture was warmed to 60 ℃ and stirred at that temperature for 16 h before it was cooled to 25 ℃. The mixture was diluted with water (100 mL) , neutralized with saturated aqueous of NaHCO
3, extracted with EtOAc (50 mL × 3) . The combined organic phases were washed with brine (10 mL) , dried over Na
2SO
4, filtered, and concentrated to give methyl 3-hydroxypyridine-2-carboxylate (5.6 g) as a yellow solid, which was directly used into the next step without any further purification. LC-MS: m/z [M+H]
+ 154.1.
Step 2: methyl 6-bromo-3-hydroxypicolinate
To a stirred solution of methyl 3-hydroxypyridine-2-carboxylate (5.60 g, 36.6 mmol) in H
2O (400 mL) was added bromine (8.20 g, 51.31 mmol) at 25 ℃ and the mixture was stirred at that temperature for 3 h. The mixture was extracted with CH
2Cl
2 (100 mL × 2) . The combined organic phases were washed with water (20 mL) , brine (20 mL) and dried over Na
2SO
4, filtered, and concentrated under reduced pressure to give methyl 6-bromo-3-hydroxy-pyridine-2-carboxylate (6.50 g) as a yellow oil which was directly used into the next step without any further purification. LC-MS: m/z [M+H]
+231.8.
Step 3: methyl 6-bromo-3- (2- (pyrrolidin-1-yl) ethoxy) picolinate
To a stirred mixture of methyl 6-bromo-3-hydroxy-pyridine-2-carboxylate (500 mg, 2.15 mmol) in THF (10.0 mL) were sequentially added 2-pyrrolidin-1-ylethanol (377 μL, 372 mg, 3.23 mmol) and PPh
3 (848 mg, 3.23 mmol) , DIAD (479 mg, 2.37 mmol) at 25 ℃. The mixture was stirred at that temperature for 2 h before it was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with CH
2Cl
2/MeOH (with MeOH from 0 to 5%in 25 min) to give methyl 6-bromo-3- (2-pyrrolidin-1-ylethoxy) pyridine-2-carboxylate (500 mg, 70%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 328.7.
Step 4: (6-bromo-3- (2- (pyrrolidin-1-yl) ethoxy) pyridin-2-yl) methanol
To a stirred solution of methyl 6-bromo-3- (2-pyrrolidin-1-ylethoxy) pyridine-2-carboxylate (500 mg, 152 μmol) in MeOH (10.0 mL) was added NaBH
4 (17.2 mg, 456 μmol) at 25 ℃ and the resulting mixture was stirred at that temperature for 12 h. The mixture was diluted with water (50 mL) , extracted with CH
2Cl
2 (50 mL × 3) . The combined organic layers were washed with brine (50 mL) , dried over Na
2SO
4 filtered, and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with CH
2Cl
2/MeOH (with MeOH from 0 to 10%in 25 min) to give (6-bromo-3- (2-pyrrolidin-1-ylethoxy) -2-pyridyl] methanol (400 mg, 87%yield) as a colorless oil. LC-MS: m/z [M+H]
+ 300.8.
Step 5: tert-butyl (3- ( (6-bromo-3- (2- (pyrrolidin-1-yl) ethoxy) pyridin-2-yl) methoxy) propyl)
carbamate
To a stirred solution of (6-bromo-3- (2-pyrrolidin-1-ylethoxy) -2-pyridyl] methanol (400 mg, 1.33 mmol) in THF (10.0 mL) was added NaH (80.0 mg, 60 wt. %in mineral oil, 2.0 mmol) at 0 ℃ under N
2 atmosphere. The reaction mixture was stirred for 1 h at that temperature before tert-butyl N- (3-bromopropyl) carbamate (474 mg, 2.0 mmol) was added. The resulting mixture was stirred at 0 ℃ for 2 h before it was quenched with H
2O (30 mL) , extracted with EtOAc (30 mL × 3) . The combined organic phases were washed with brine (10 mL) , dried over Na
2SO
4, filtered, and concentrated under reduced pressure to give tert-butyl (3- ( (6-bromo-3- (2- (pyrrolidin-1-yl) ethoxy) pyridin-2-yl) methoxy) propyl) carbamate (600 mg) as a yellow oil which was directly used into the next step without any further purification. LC-MS: m/z [M+H]
+ 457.9.
Step 6: tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) -3- (2- (pyrrolidin-1-yl) ethoxy) pyridin-2-yl) methoxy) propyl) carbamate
To a stirred solution of (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (100 mg, 447. μmol) in 1, 4-dioxane (5.0 mL) were sequentially added tert-butyl (3- ( (6-bromo-3- (2- (pyrrolidin-1-yl) ethoxy) pyridin-2-yl) methoxy) propyl) carbamate (307 mg, 672 μmol) , Pd
2 (dba)
3 (82.0 mg, 89.6 μmol) , XantPhos (104 mg, 179 μmol) and Cs
2CO
3 (438 mg, 1.34 mmol) at 25 ℃. The reaction mixture was warmed up to 80 ℃ and stirred at that temperature for 16 h under N
2 atmosphere before it was cooled to 25 ℃ and concentrated under reduced pressure. The residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 0%to 60%in 50 min) to give tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -3- (2- (pyrrolidin-1-yl) ethoxy) pyridin-2-yl) methoxy) propyl) carbamate (110 mg, 41%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 601.9.
Step 7: (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) -5- (2- (pyrrolidin-1-
yl) ethoxy) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-
carboxylate
To a stirred solution of tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -3- (2- (pyrrolidin-1-yl) ethoxy) pyridin-2-yl) methoxy) propyl) carbamate (90.0 mg, 150 μmol) in CH
2Cl
2 (5.0 mL) were sequentially added CDI (72.9 mg, 449 μmol) , Et
3N (62.3 μL, 45.5 mg, 449 μmol) and DMAP (18.3 mg, 150 μmol) at 25 ℃ and the mixture was stirred at that temperature for 1 h. The mixture was concentrated under reduced pressure to give (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) -5- (2- (pyrrolidin-1-yl) ethoxy) pyridin-2-yl) amino) - 1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (90 mg) as a yellow oil which was directly used into the next step without any further purification. LC-MS: m/z [M+H]
+ 694.9.
Step 8: (1
1S, 1
3R, Z) -4
5- (2- (pyrrolidin-1-yl) ethoxy) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-
2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
A stirred solution of (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) -5- (2- (pyrrolidin-1-yl) ethoxy) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (90.0 mg, 129 μmol) in TFA (3.0 mL) was warmed up to 80 ℃ and stirred at that temperature for 2 h before it was cooled to 25 ℃. The mixture was concentrated under reduced pressure. The residue was dissolved in CH
3CN (3.0 mL) , and then Et
3N (89.7 μL, 65.5 mg, 648 μmol) was added into this mixture at 25 ℃. The mixture was warmed up to 80 ℃ and stirred at that temperature for 4 h before it was cooled to 25 ℃ and concentrated under reduced pressure. The residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 0%to 45%in 45 min) to give (1
1S, 1
3R, Z) -4
5- (2- (pyrrolidin-1-yl) ethoxy) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (5.0 mg, 8.0%yield) as a colorless oil. LC-MS: m/z [M+H]
+ 470.9.
Example 55
(1
1S, 1
3R, Z) -4
5-methoxy-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one


Step 1: 4, 6-dibromo-2-methylpyridin-3-ol
To a suspension of 2-methylpyridin-3-ol (3.0 g, 27.5 mmol) in CH
3CN (30.0 mL) was added NBS (9.79 g, 54.9 mmol) at 25 ℃. The reaction mixture was warmed to 90 ℃ and stirred at that temperature for 2 h. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The crude product was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 15 %in 25 min) to afford 4, 6-dibromo-2-methylpyridin-3-ol (5.0 g, 68%yield) as a yellow solid. C-MS: m/z [M+H] + 265.8
Step 2: 6-bromo-2-methylpyridin-3-ol
To a stirred suspension of 4, 6-dibromo-2-methylpyridin-3-ol (4.50 g, 16.9 mmol) in THF (30.0 mL) was stirred at -78 ℃ for 10 min under N
2. Then n-BuLi (13.5 mL, 2.5 M, 33.8 mmol) was added slowly to the solution and stirred another 2h at that temperature. The reaction mixture was quenched with ice-cold water (20 mL) and extracted with EtOAc (20 mL× 3) . The organic phase was washed with brine (20 mL) and dried over anhydrous Na
2SO
4, filtered and the filtrate was concentrated under reduced pressure. The crude product was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 12 %in 25 min) to afford 6-bromo-2-methylpyridin-3-ol (2.60 g, 83%yield) as a white solid. LC-MS: m/z [M+H] + 188.0
Step 3: 6-bromo-3-methoxy-2-methylpyridine
To a suspension of 6-bromo-2-methylpyridin-3-ol (2.60 g, 13.9 mmol) in acetone (40.0 mL) was added CH
3I (1.29 mL, 2.94 g, 20.9 mmol) and K
2CO
3 (5.76 g, 41.7 mmol) at 25 ℃. The reaction mixture was warmed to 50 ℃ and stirred at that temperature for 2 h. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 8 %in 25 min) to afford 6-bromo-3-methoxy-2-methylpyridine (1.60 g, 57%yield) as a white solid. LC-MS: m/z [M+H] + 202.0
Step 4: 6-bromo-2- (bromomethyl) -3-methoxypyridine
To a suspension of 6-bromo-3-methoxy-2-methylpyridine (1.60 g, 7.96 mmol) in CCl
4 (20.0 mL) was added NBS (2.12 g, 11.9 mmol) and AIBN (261 mg, 1.59 mmol) at 25 ℃. The reaction mixture was warmed to 70 ℃ and stirred at that temperature for 2 h. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 10 %in 25 min) to afford 6-bromo-2-(bromomethyl) -3-methoxypyridine (2.0 g, 50%yield) as a white solid. LC-MS: m/z [M+H] + 279.7
Step 5: tert-butyl (3- ( (6-bromo-3-methoxypyridin-2-yl) methoxy) propyl) carbamate
To a stirred suspension of tert-butyl (3-hydroxypropyl) carbamate (3.70 g, 21.4 mmol) in THF (20.0 mL) was added NaH (860 mg, 60 wt. %in mineral oil, 21.4 mmol) at 0 ℃. The mixture was stirred for 30 min at that temperature before 6-bromo-2- (bromomethyl) -3-methoxypyridine (2.0 g, 7.14 mmol) was added. The mixture was stirred at 25 ℃ for 2 h. Then it was quenched with ice-water (20 mL) and extracted with EtOAc (20 mL × 3) . The organic phase was washed with brine (20 mL) and dried over anhydrous Na
2SO
4, filtered and the filtrate was concentrated under reduced pressure. The crude product was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 10 %in 25 min) to afford tert-butyl (3- ( (6-bromo-3-methoxypyridin-2-yl) methoxy) propyl) carbamate (1.90 g, 71%yield) as a white solid. LC-MS: m/z [M+H] + 374.9
Step 6: tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) -3-methoxypyridin-2-yl) methoxy) propyl) carbamate
To a stirred suspension of tert-butyl (3- ( (6-bromo-3-methoxypyridin-2-yl) methoxy) propyl) carbamate (600 mg, 1.60 mmol) in 1, 4-dioxane (10.0 mL) were sequentially added (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (358 mg, 1.60 mmol) , Cs
2CO
3 (1.56 g, 4.80 mmol) , XantPhos (185 mg, 320 μmol) and Pd
2 (dba)
3 (146 mg, 160 μmol) at 25 ℃. The reaction mixture was warmed to 100 ℃ and stirred at that temperature for 12 h. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 30 %in 25 min) to afford tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -3-methoxypyridin-2-yl) methoxy) propyl) carbamate (400 mg, 48%yield) as a yellow oil. LC-MS: m/z [M+H] + 518.0
Step 7: (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) -5-methoxypyridin-2-
yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a stirred suspension of tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -3-methoxypyridin-2-yl) methoxy) propyl) carbamate (400 mg, 770 μmol) in CH
2Cl
2 (10.0 mL) was added CDI (374 mg, 2.31 mmol) and DIPEA (663 μL, 500 mg, 3.85 mmol) at 25 ℃. The reaction mixture was warmed to 35 ℃ and stirred at that temperature for 12 h. The reaction mixture was quenched with ice-cold water (10 mL) and extracted with EtOAc (10 mL × 3) . The combined organic phases were dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure to afford (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) -5-methoxypyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (crude) as a yellow oil, which was used in the next step without further purification. LC-MS: m/z [M+H]
+ 612.0
Step 8: (1R, 3S) -3- (5- ( (6- ( (3-aminopropoxy) methyl) -5-methoxypyridin-2-yl) amino) -1- (tert-butyl) -
1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a stirred suspension of (1R, 3S) -3- (5- ( (6- ( (3-aminopropoxy) methyl) -5-methoxypyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate in CH
2Cl
2 (10.0 mL) was added slowly TFA (396 μL, 586 mg, 5.14 mmol) at 25 ℃. The mixture was stirred at that temperature for 1 h before it was concentrated under reduced pressure to afford (1R, 3S) -3- (5- ( (6- ( (3-aminopropoxy) methyl) -5-methoxypyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate as a yellow oil, which was used in the next step without further purification. LC-MS: m/z [M+H]
+ 512.1
Step 9: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5-methoxy-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a suspension of (1R, 3S) -3- (5- ( (6- ( (3-aminopropoxy) methyl) -5-methoxypyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate in CH
3CN (10.0 mL) was added Et
3N (1.23 mL, 902 mg, 8.91 mmol) at 25 ℃. The reaction mixture was warmed to 80 ℃ and stirred at that temperature for 12 h. The reaction mixture was concentrated under reduced pressure to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5-methoxy-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one as a yellow oil. LC-MS: m/z [M+H] + 444.0
Step 10: (1
1S, 1
3R, Z) -4
5-methoxy-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphan-11-one
A solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5-methoxy-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one in HCO
2H (5.0 mL) was warmed to 100 ℃ and stirred at that temperature for 5 h before it was cooled, concentrated under reduced pressure. The residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 5%to 25%in 20 min) to afford (1
1S, 1
3R, Z) -4
5-methoxy-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (25.4 mg, 4.1%yield) as a white solid. LC-MS: m/z [M+H] + 387.9.
Example 56
(1
1S, 1
3R, Z) -4
5-fluoro-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one

Step 1: 6-bromo-2- (bromomethyl) -3-fluoropyridine
A stirred solution of 6-bromo-3-fluoro-2-methylpyridine (1.0 g, 5.26 mmol) , BPO (63.7 mg, 263 μmol) and NBS (1.03 g, 5.79 mmol) in CCl
4 (50.0 mL) was warmed to 90 ℃ and stirred for 4 h at that time before it was cooled to 25 ℃. The mixture was concentrated to afford 6-bromo-2-(bromomethyl) -3-fluoropyridine as a yellow solid which was directly used into the next step without further purification. (1.40 g, 99%yield) .
1H NMR (400 MHz, CDCl
3) δ 7.44 (dd, J = 8.4, 3.6 Hz, 1H) , 7.32 (d, J = 8.4 Hz, 1H) , 4.53 (s, 2H) .
Step 2: tert-butyl (3- ( (6-bromo-3-fluoropyridin-2-yl) methoxy) propyl) carbamate
To a stirred solution of NaH (53.6 mg, 60 wt. %in mineral oil, 2.23 mmol) in THF (10.0 mL) solution was added tert-butyl (3-hydroxypropyl) carbamate (360 mg, 2.23 mmol) at 0 ℃. The mixture was stirred at 0 ℃ for 30 min before a solution of 6-bromo-2- (bromomethyl) -3-fluoropyridine (500 mg, 1.86 mmol) in THF (10.0 mL) was added dropwise. The mixture was stirred at that temperature for 1 h before it was diluted with H
2O (50 mL) and warmed to 25 ℃. The mixture was extracted with EtOAc (15 mL × 3) . The combined organic phases were washed with brine (20 mL) , dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by flash silica gel column chromatography eluting with EtOAc/PE (with EtOAc from 0 to 30 %in 25 min) to afford tert-butyl (3- ( (6-bromo-3-fluoropyridin-2-yl) methoxy) propyl) carbamate (350 mg, 52 %yield) as a light-yellow oil. LC-MS: m/z [M+H]
+ 363.0.
Step 3: tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) -3-fluoropyridin-2-yl) methoxy) propyl) carbamate
To a stirred mixture of tert-butyl (3- ( (3, 6-dibromopyridin-2-yl) methoxy) propyl) carbamate (350 mg, 849 μmol) , (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (189 mg, 849 μmol) and Cs
2CO
3 (553 mg, 1.70 mmol) in 1, 4-dioxane (10.0 mL) were sequentially added XantPhos (49.2 mg, 84.9 μmol) and Pd
2 (dba)
3 (77.8 mg, 84.9 μmol) at 25 ℃. The mixture was warmed to 80 ℃ and stirred at that temperature for 3 h under N
2 atmosphere before it was cooled to 25 ℃ and concentrated under reduced pressure. The residue was purified by silica gel column chromatography eluting with EtOAc/PE (with EtOAc from 0 to 50 %in 30 min) to afford tert-butyl (3- ( (3-bromo-6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methoxy) propyl) carbamate (280 mg, 58%yield) as a light-yellow oil. LC-MS: m/z [M+H]
+ 506.3.
Step 4: (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) -5-fluoropyridin-2-
yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a stirred solution of tert-butyl (3- ( (3-bromo-6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methoxy) propyl) carbamate (260 mg, 395 μmol) in CH
2Cl
2 (12.0 mL) were sequentially added Et
3N (214 μL, 156 mg, 1.54 mmol) and CDI (167 mg, 1.03 mmol) at 25 ℃ . The mixture was warmed to 40 ℃ and stirred at that temperature for 2 h. The mixture was concentrated under reduced pressure to afford (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) -5-fluoropyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (350 mg) as a light-yellow solid which was used to the next step without further purification. LC-MS: m/z [M+H]
+ 600.3.
Step 5: (1R, 3S) -3- (5- ( (6- ( (3-aminopropoxy) methyl) -5-fluoropyridin-2-yl) amino) -1- (tert-butyl) -
1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a stirred solution of (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) -5-fluoropyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (340 mg, 567 μmol ) in CH
2Cl
2 (10.0 mL) was added TFA (2.0 mL) at 25 ℃ and the mixture was stirred at that temperature for 2 h. The mixture was concentrated to afford (1R, 3S) -3- (5- ( (6- ( (3-aminopropoxy) methyl) -5-fluoropyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (300 mg) as a light-yellow oil which was used to the next step without further purification. LC-MS: m/z [M+H]
+ 500.1.
Step 6: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5-fluoro-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a stirred solution (1R, 3S) -3- (5- ( (6- ( (3-aminopropoxy) methyl) -5-fluoropyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (290 mg, 580 μmol) in CH
3CN (10.0 mL) was added Et
3N (1.0 mL) at 25 ℃ . The mixture was warmed to 70 ℃ and stirred at that temperature for 16 h before it was cooled to 25 ℃ and concentrated under reduced pressure. The residue was purified by silica gel column chromatography eluting with EtOAc/PE (with EtOAc from 0 to 40 %in 30 min) to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5-fluoro-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (150 mg, 60%yield ) as a light-yellow oil. LC-MS: m/z [M+H]
+ 432.2.
Step 7: (1
1S, 1
3R, Z) -4
5-fluoro-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one
To a stirred solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5-fluoro-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (100 mg, 232 μmol) in TFA (2.0 mL) at 25 ℃. The mixture was warmed to 70 ℃ and stirred at that temperature for 12 h before it was cooled to 25 ℃ and concentrated under reduced pressure. The residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 0%to 40%in 45 min) to give (1
1S, 1
3R, Z) -4
5-fluoro-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (7.40 mg, 8.5%yield) as a white solid. LC-MS: m/z [M+H]
+376.0.
Example 57
(1
1S, 1
3R, Z) -4
3-fluoro-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one

Step 1: (6-bromo-5-fluoropyridin-2-yl) methanol
To a solution of methyl 6-bromo-5-fluoropicolinate (1.00 g, 4.27 mmol) in Methanol (20 mL) was added Sodium borohydride (485 mg, 12.82 mmol) at 0 ℃. The reaction was stirred at 25 ℃ for 4 h under nitrogen atmosphere. The mixture was quenched with water (50 mL) and extracted with EtOAc (50 mL × 3) . The combined organic layer was washed with brine (20 mL) , dried over Na
2SO
4. The mixture was filtered and concentrated to dryness. The residue was purified by flash chromatography eluting with EtOAc/PE (with EtOAc from 0 to 35%in 15 min) to afford (6-bromo-5-fluoropyridin-2-yl) methanol (795 mg, 90%yield) as a white solid. LC-MS: m/z 206.0 [M+H]
+
Step 2: tert-butyl (3- ( (6-bromo-5-fluoropyridin-2-yl) methoxy) propyl) carbamate
A solution of (6-bromo-5-fluoropyridin-2-yl) methanol (745 mg, 3.62 mmol) in DMF (2.0 mL) was added to a stirring suspension of NaH (289 mg, 7.23 mmol, 60%purity) and 18-Crown-6 (191mg, 723 μmol) in DMF (4.0 mL) at 0 ℃. The mixture was stirred at 0 ℃ for 50 min. Then a solution of tert-butyl (3-bromopropyl) carbamate (1.29 g, 5.42 mmol) in DMF (4.0 mL) was added to the above reaction solution . The reaction mixtrue was stirred at 25℃ for 3 h. The mixture was quenched with saturated NH
4Cl solution (50 mL) and extracted with EtOAc (50 mL × 2) . The combined organic layer was washed with saturated LiCl solution (50 mL × 2) , dried over Na
2SO
4. The mixture was filtered and concentrated to dryness. The residue was purified by flash chromatography eluting with EtOAc/PE (with EtOAc from 0 to 20%in 20 min) to afford tert-butyl (3- ( (6-bromo-5-fluoropyridin-2-yl) methoxy) propyl) carbamate (300 mg, 23%yield) as a yellow solid. LC-MS: m/z 385.0 [M+Na]
+
Step 3: tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) -5-fluoropyridin-2-yl) methoxy) propyl) carbamate
To a suspension of tert-butyl (3- ( (6-bromo-5-fluoropyridin-2-yl) methoxy) propyl) carbamate (200 mg, 551 μmol) and (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (135 mg, 606 μmol) in dioxane (6.0 mL) was added Pd
2 (dba)
3 (100 mg, 110 μmol) , Cs
2CO
3 (538 mg, 1.65 mmol) and XantPhos (127 mg, 220 μmol) . The suspension was degassed with N
2 for 5 times. The mixture was heated to 90 ℃ and stirred at 90 ℃ for 6 h. The mixture was diluted with water (50 mL) and extracted with EtOAc (50 mL ×2) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4. The mixture was filtered and concentrated to dryness. The residue was purified by flash chromatography eluting with EtOAc/PE (with EtOAc from 0 to 72%in 15 min) to afford tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -5-fluoropyridin-2-yl) methoxy) propyl) carbamate (277 mg, 99%yield) as a yellow oil. LC-MS: m/z 506.2 [M+H]
+
Step 4: (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) -3-fluoropyridin-2-
yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -5-fluoropyridin-2-yl) methoxy) propyl) carbamate (257 mg, 508 μmol) in DCM (8.0 mL) was added 1, 1'-Carbonyldiimidazole (494 mg, 3.05 mmol) at 25 ℃. The reaction was stirred at 45 ℃for 3 h in a sealed tube. The residue was purified by flash chromatography eluting with EtOAc/PE (with EtOAc from 0 to 62%in 15 min) to afford (1R, 3S) -3- (5- ( (6- ( (3- ( (tert- butoxycarbonyl) amino) propoxy) methyl) -3-fluoropyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (300 mg, 98%yield) as a yellow oil. LC-MS: m/z 600.3 [M+H]
+
Step 5: (1R, 3S) -3- (5- ( (6- ( (3-aminopropoxy) methyl) -3-fluoropyridin-2-yl) amino) -1- (tert-butyl) -
1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) -3-fluoropyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (250 mg, 417 μmol) in DCM (4.0 mL) was added TFA (2.0 mL) at 0 ℃. The reaction was stirred under nitrogen atmosphere at 25 ℃ for 2 h. The mixture was concentrated to provide (1R, 3S) -3- (5- ( (6- ( (3-aminopropoxy) methyl) -3-fluoropyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (500 mg, crude) as a yellow oil. LC-MS: m/z 500.1 [M+H]
+
Step 6: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
3-fluoro-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a solution of (1R, 3S) -3- (5- ( (6- ( (3-aminopropoxy) methyl) -3-fluoropyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (500 mg, 1.00 mmol) in MeCN (20 mL) was added Et
3N (1.29 g, 10.0 mmol, 1.74 mL) at 0 ℃. The reaction was heated to 80 ℃ and stirred at 80 ℃ for 18 h under nitrogen atmosphere. The mixture was concentrated to dryness. The residue was purified by flash chromatography eluting with EtOAc/PE (with EtOAc from 0 to 70%in 15 min) to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
3-fluoro-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (146 mg, 34%yield) as a yellow solid. LC-MS: m/z 432.1 [M+H]
+
Step 7: (1
1S, 1
3R, Z) -4
3-fluoro-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one
A solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
3-fluoro-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (126 mg, 292 μmol) in FA (2.0 mL) was heated to 80 ℃ and stirred at 80 ℃ for 2 h. The mixture was concentrated to dryness. The residue was purified by Prep-HPLC (with CH
3CN from 50%to 70%in 8 min) to afford (1
1S, 1
3R, Z) -4
3-fluoro-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (2.30 mg, 3%yield) as a yellow solid. LC-MS: m/z 376.1 [M+H]
+.
Example 58
(1
1S, 1
3R, Z) -5, 5-dimethyl-2
1H-12-oxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one

Step 1: 2- (6-bromopyridin-2-yl) propan-2-ol
To a stirred solution of methyl 6-bromopyridine-2-carboxylate (5.00 g, 23.1 mmol) in THF (80.0 mL) was added MeMgBr (30.9 mL, 92.6 mmol, 3 M in diethyl ether) at 0 ℃. The reaction mixture was warmed to 25 ℃ and stirred at that temperature for 16 h. The reaction mixture was cooled to 0 ℃and quenched with aqueous 10%HCl (100 mL) and extracted with EtOAc (100 mL × 3) . The combined organic phases were washed with brine (50 mL) , dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 20%in 25 min) to afford 2- (6-bromo-2-pyridyl) propan-2-ol (3.30 g, 66%yield) . LC-MS: m/z [M+H]
+ 216.0.
Step 2: tert-butyl (3- ( (2- (6-bromopyridin-2-yl) propan-2-yl) oxy) propyl) carbamate
To a stirred solution of 2- (6-bromo-2-pyridyl) propan-2-ol (2.70 g, 12.5 mmol) in THF (50.0 mL ) was added NaH (1.00 g, 60 wt. %in mineral oil, 25.0 mmol) at 0 ℃. The reaction mixture was stirred for 30 min before tert-butyl N- (3-bromopropyl) carbamate (8.93 g, 37.5 mmol) and KI (6.22 g, 37.5 mmol) were added at that temperature. The reaction mixture was warmed to 25 ℃ and stirred at that temperature for 12 h. The reaction mixture was quenched with ice-cold water (100 mL) and extracted with EtOAc (100 mL × 3) . The combined organic phases were washed with brine (100 mL) , dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 30 %in 20 min) to afford tert-butyl N- [3- [1- (6-bromo-2-pyridyl) -1-methyl-ethoxy] propyl] carbamate (2.00 g, 43%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 372.9.
Step 3: tert-butyl (3- ( (2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) propan-2-yl) oxy) propyl) carbamate
To a stirred solution of tert-butylN- [4- [ [6-bromo-3- (1-methylpyrazol-4-yl) -2-pyridyl] oxy] butyl] carbamate (351 mg, 940 μmol) in 1, 4-dioxane (20.0 mL) were sequentially added (1R, 3S) -3- (5-amino-2-tert-butyl-pyrazol-3-yl) cyclopentanol (140 mg, 627 μmol) , Pd
2 (dba)
3 (57.4 mg, 62.7 μmol) , XantPhos (36.3 mg, 62.7 μmol) and Cs
2CO
3 (409 mg, 1.25 mmol) at 25 ℃. The reaction was warmed to 100 ℃ and stirred at that temperature for 16 h under N
2 atmosphere before it was cooled to 25 ℃ and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 50 %in 25 min) to afford tert-butyl (3- ( (2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) propan-2-yl) oxy) propyl) carbamate (230 mg, 57%yield) as a yellow oil. LC-MS: m/z [M+H]
+516.4.
Step 4: (1R, 3S) -3- (5- ( (6- (2- (3- ( (tert-butoxycarbonyl) amino) propoxy) propan-2-yl) pyridin-2-
yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a stirred solution of tert-butyl (3- ( (2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) propan-2-yl) oxy) propyl) carbamate (230 mg, 446 μmol) in CH
2Cl
2 (5.0 mL) were sequentially added CDI (217 mg, 1.34 mmol) , DMAP (16.4 mg, 134 μmol) and Et
3N (311 μL, 226 mg, 2.23 mmol) at 25 ℃. The reaction mixture was stirred at that temperature for 2 h before it was quenched with ice-cold water (20 mL) and extracted with CH
2Cl
2 (20 mL × 3) . The combined organic phases were dried over anhydrous Na
2SO
4, filtered. The filtrate was concentrated under reduced pressure to give product (1R, 3S) -3- (5- ( (6- (2- (3- ( (tert-butoxycarbonyl) amino) propoxy) propan-2-yl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate which was directly used in the next step without further purification. LC-MS: m/z [M+H]
+ 610.4.
Step 5: (1R, 3S) -3- (3- ( (6- (2- (3-aminopropoxy) propan-2-yl) pyridin-2-yl) amino) -1H-pyrazol-5-
yl) cyclopentyl 1H-imidazole-1-carboxylate
A solution of (1R, 3S) -3- (5- ( (6- (2- (3- ( (tert-butoxycarbonyl) amino) propoxy) propan-2-yl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (crude) in TFA (4.0 mL) was warmed to 70 ℃ and stirred at that temperature for 16 h. The reaction mixture was concentrated to give (1R, 3S) -3- (3- ( (6- (2- (3-aminopropoxy) propan-2-yl) pyridin-2-yl) amino) -1H-pyrazol-5-yl) cyclopentyl 1H-imidazole-1-carboxylate as a brown oil, which was directly used in the next step without further purification. LC-MS: m/z [M+H]
+ 453.7.
Step 6: (1
1S, 1
3R, Z) -5, 5-dimethyl-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a stirred solution of (1R, 3S) -3- (3- ( (6- (2- (3-aminopropoxy) propan-2-yl) pyridin-2-yl) amino) -1H-pyrazol-5-yl) cyclopentyl 1H-imidazole-1-carboxylate (crude) in CH
3CN (5.0 mL) was added Et
3N (271 μL, 196 mg, 1.94 mmol) at 25 ℃. The mixture was warmed to 80 ℃ and stirred at that temperature for 5 h before it was cooled to 25 ℃ and concentrated under reduced pressure. The residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 0%to 50%in 40 min) to afford (1
1S, 1
3R, Z) -5, 5-dimethyl-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (9.60 mg, 8.1%yield) as an off-white solid. LC-MS: m/z [M+H]
+ 385.8.
Example 59
(1
1S, 1
3R, Z) -2
1H-6, 13-dioxa-3, 11-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclotridecaphan-12-one

Step 1: 2-bromo-6- (bromomethyl) pyridine
To a solution of 2-bromo-6-methyl-pyridine (5.00 g, 29.1 mmol) in CCl
4 (50 mL) was added NBS (5.43 g, 30.5 mmol) and 2- [ (E) - (1-cyano-1-methyl-ethyl) azo] -2-methyl-propanenitrile (238 mg, 1.45 mmol) . The mixture was heated to 80 ℃ and stirred at 80 ℃ for 16 h. The mixture was concentrated under reduce pressure. The reisdue was purified with flash column chromatography eluting with DCM/PE (with DCM from 0 to 20%in 25 min) to give 2-bromo-6- (bromomethyl) pyridine (4.10 g, 56%yield) as a white solid. LC-MS: m/z 251.1 [M+H]
+
Step 2: tert-butyl (4- ( (6-bromopyridin-2-yl) methoxy) butyl) carbamate
To a solution of tert-butyl N- (4-hydroxybutyl) carbamate (226 mg, 1.20 mmol) in DMF (5.0 mL) was added NaH (37.3 mg, 1.55 mmol) . The mixture was stirred at 0 ℃ for 1 h under N
2. Then 2-bromo-6- (bromomethyl) pyridine (300 mg, 1.20 mmol) was added. The mixture was warmed to 25 ℃ for 1 h. The mixture was quenched by NH
4Cl (2 mol/L in water, 3.0 mL) , extracted with ethyl acetate (5.0 mL × 2) . The combined organic layer was concentrated under reduced pressure, the residue was purified by flash column chromatography eluting with EtOAc/PE (with EtOAc from 0 to 30%in 20 min) to give tert-butyl N- [4- [ (6-bromo-2-pyridyl) methoxy] butyl] carbamate (278 mg, 64%yield) as an off-white solid. LC-MS: m/z 361.1 [M+H]
+.
Step 3: tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) methoxy) butyl) carbamate
The solution of tert-butyl N- [4- [ (6-bromo-2-pyridyl) methoxy] butyl] carbamate (50.0 mg, 139 μmol) , (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (31.0 mg, 139 μmol) , (1E, 4E) -1, 5-diphenylpenta-1, 4-dien-3-one; palladium (12.7 mg, 13.9 μmol) , Cs
2CO
3 (136 mg, 417 μmol) and XantPhos (16.1 mg, 27.8 μmol) in 1, 4-Dioxane (1.5 mL) was heated to 120 ℃ and stirred at 120 ℃for 18 h under N
2. The mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography eluting with EtOAc/PE (with EtOAc from 0 to 80%in 20 min) to give tert-butyl N- [4- [ [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -2-pyridyl] methoxy] butyl] carbamate (31.0 mg, 44%yield) as a yellow oil. LC-MS: m/z 502.3 [M+H]
+.
Step 4: (1R, 3S) -3- (5- ( (6- ( (4- ( (tert-butoxycarbonyl) amino) butoxy) methyl) pyridin-2-yl) amino) -1-
(tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
The stirring solution of tert-butyl N- [4- [ [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -2-pyridyl] methoxy] butyl] carbamate (234 mg, 466 μmol) and di (imidazol-1-yl) methanone (227 mg, 1.40 mmol) in DCM (645 μL) was added DIPEA (301 mg, 2.33 mmol) at 25 ℃ for 2 h under N
2. The mixture was concentrated under reduced pressure to give [ (1R, 3S) -3- [5- [ [6- [4- (tert-butoxycarbonylamino) butoxymethyl] -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (270 mg, 82%yield) as a yellow oil. LC-MS: m/z 596.3 [M+H]
+.
Step 5: (1R, 3S) -3- (5- ( (6- ( (4-aminobutoxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-
3-yl) cyclopentyl 1H-imidazole-1-carboxylate
The solution of [ (1R, 3S) -3- [5- [ [6- [4- (tert-butoxycarbonylamino) butoxymethyl] -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (230 mg, 386 μmol) in 2, 2, 2-trifluoroacetic acid (440 mg, 3.86 mmol) was stirred at 25 ℃ for 5 min. The mixture was concentrated under reduced pressure to give [ (1R, 3S) -3- [5- [ [6- (4-aminobutoxymethyl) -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (190 mg, 99%yield) as a yellow oil. LC-MS: m/z 496.3 [M+H]
+.
Step 6: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-6, 13-dioxa-3, 11-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-
1 (1, 3) -cyclopentanacyclotridecaphan-12-one
The solution [ (1R, 3S) -3- [5- [ [6- (4-aminobutoxymethyl) -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (190 mg, 319 μmol) in CH
3CN (3.0 mL) was added DIPEA (206 mg, 1.59 mmol) The mixture was heated to ℃ and stirred at 80 ℃ for 18 h. The mixture was concentrated under reduced pressure, the residue was purified by prep-TLC (petroleum ether/EtOAc = 2: 1) to give (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-6, 13-dioxa-3, 11-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclotridecaphan-12-one (48.0 mg, 35%yield) as a yellow solid. LC-MS: m/z 428.2 [M+H]
+.
Step 7: (1
1S, 1
3R, Z) -2
1H-6, 13-dioxa-3, 11-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclotridecaphan-12-one
The solution (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-6, 13-dioxa-3, 11-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclotridecaphan-12-one (45.0 mg, 105 μmol) in trifluoroacetic acid (120 mg, 1.05 mmol) was heated to 80 ℃ and stirred at 80 ℃ for 4 h. The mixture was concentrated under reduced pressure. The residue was purified by prep-HPLC (with CH
3CN from 20%to 30%in 8 min) to afford (1
1S, 1
3R, Z) -2
1H-6, 13-dioxa-3, 11-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclotridecaphan-12-one (6.60 mg, 16%yield) as a white solid. LC-MS: m/z 372.2 [M+H]
+.
The following compounds were prepared using the similar procedure disclosed in synthetic example 59.

Example 60
(1
1S, 1
3R, Z) -2
1H-6, 11-dioxa-3, 9-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacycloundecaphan-10-one

Step 1: (6-bromopyridin-2-yl) methyl methane sulfonate
To a solution of (6-bromopyridin-2-yl) methanol (1.00 g, 5.32 mmol) and Et
3N (700 mg, 6.91 mmol, 963 μL) in DCM (20 mL) was drop-wised added methane sulfonyl chloride (640 mg, 5.58 mmol, 433 μL) at 0 ℃. The reaction was stirred at 0 ℃ for 2 h. The mixture was quenched with saturated NaHCO
3 solution (50 mL) and extracted with DCM (50 mL × 2) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4. The mixture was filtered and concentrated to afford (6-bromopyridin-2-yl) methyl methane sulfonate (1.60 g, crude) as a yellow oil. LC-MS: m/z 267.1 [M+H]
+
Step 2: tert-butyl (2- ( (6-bromopyridin-2-yl) methoxy) ethyl) carbamate
A solution of tert-butyl (2-hydroxyethyl) carbamate (200 mg, 1.24 mmol) in THF (1.0 mL) was added to a stirring suspension of NaH (99.2 mg, 2.48 mmol, 60%purity) in THF (5.0 mL) at 0℃. The mixture was stirred at 0 ℃ for 30 mi. After that, a solution of (6-bromopyridin-2-yl) methyl methane sulfonate (660 mg, 2.48 mmol) in THF (2 mL) was added to the above reaction solution. The mixture was stirred at 25 ℃ for 16 h. The mixture was quenched with saturated NH
4Cl solution (50 mL) and extracted with EtOAc (50 mL × 2) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4. The mixture was filtered and concentrated to dryness. The residue was purified by flash chromatography eluting with EtOAc/PE (with EtOAc from 0 to 18%in 15 min) to afford tert-butyl (2- ( (6-bromopyridin-2-yl) methoxy) ethyl) carbamate (345 mg, 64%yield) as a yellow oil. LC-MS: m/z 331.0 [M+H]
+
Step 3: tert-butyl (2- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) methoxy) ethyl) carbamate
To a suspension of tert-butyl (2- ( (6-bromopyridin-2-yl) methoxy) ethyl) carbamate (240 mg, 725 μmol) and (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (178 mg, 797 μmol) in dioxane (8.0 mL) was added Pd
2 (dba)
3 (133 mg, 290 μmol) , Cesium carbonate (708 mg, 2.17 mmol) and XantPhos (168 mg, 290 μmol) . The suspension was degassed with N
2 for 5 times. The mixture was heated to 90 ℃ and stirred at 90 ℃ for 6 h. The mixture was diluted with water (50 mL) and extracted with EtOAc (50 mL × 2) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4. The mixture was filtered and concentrated to dryness. The residue was purified by flash chromatography eluting with MeOH/DCM (with MeOH from 0 to 5%in 15 min) to afford tert-butyl (2- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methoxy) ethyl) carbamate (300 mg, 87%yield) as a yellow solid. LC-MS: m/z 474.2 [M+H]
+.
Step 4: tert-butyl (2- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy)
carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methoxy) ethyl) carbamate
To a solution of tert-butyl (2- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methoxy) ethyl) carbamate (210 mg, 443 μmol) and 4-nitrophenyl carbonochloridate (358 mg, 1.77 mmol) in DCE (8.0 mL) was added DMAP (10.8 mg, 88.7 μmol) and Et
3N (224 mg, 2.22 mmol, 309 μL) at 25 ℃. The reaction was stirred under nitrogen atmosphere at 80 ℃ for 20 hours. The mixture was washed by brine (50 mL) and extracted with DCM (50 mL ×2) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4. The mixture was filtered and concentrated to dryness. The residue was purified by flash chromatography on silica gel eluting with EtOAc/PE (with EtOAc from 0 to 50%in 15 min) to afford tert-butyl (2- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methoxy) ethyl) carbamate (114 mg, 41%yield) as a yellow oil. LC-MS: m/z 639.3 [M+H]
+
Step 5: (1R, 3S) -3- (5- ( (6- ( (2-aminoethoxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-
3-yl) cyclopentyl (4-nitrophenyl) carbonate
To a solution of tert-butyl (2- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methoxy) ethyl) carbamate (114 mg, 178 μmol) in DCM (1.0 mL) was added TFA (0.50 mL) at 0 ℃. The reaction was stirred at 25 ℃ for 2 h. The mixture was concentrated to afford (1R, 3S) -3- (5- ( (6- ( (2-aminoethoxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate (140 mg, crude) as a yellow oil. LC-MS: m/z 539.2 [M+H]
+
Step 6: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-6, 11-dioxa-3, 9-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-
1 (1, 3) -cyclopentanacycloundecaphan-10-one
To a solution of (1R, 3S) -3- (5- ( (6- ( (2-aminoethoxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate (140 mg, 260 μmol) in MeCN (11 mL) was added Et
3N (526 mg, 5.20 mmol, 724 μL) at 0 ℃. The reaction was stirred under nitrogen atmosphere at 25 ℃ for 16 h. The mixture was concentrated to dryness. The residue was purified by flash chromatography eluting with EtOAc/PE (with EtOAc from 0 to 80%in 10 min) to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-6, 11-dioxa-3, 9-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacycloundecaphan-10-one (46.0 mg, 44%yield) as a yellow oil. LC-MS: m/z 400.2 [M+H]
+
Step 7: (1
1S, 1
3R, Z) -2
1H-6, 11-dioxa-3, 9-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacycloundecaphan-10-one
A solution of (1
1S, 1
3R, Z) -21- (tert-butyl) -2
1H-6, 11-dioxa-3, 9-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacycloundecaphan-10-one (46.0 mg, 115 μmol) in TFA (2.0 mL) was heated to 80 ℃ and stirred at 80 ℃ for 18 h. The mixture was concentrated to afford the crude solid, which was purified by prep-HPLC (with CH
3CN from 22%to 32%in 8 min) to afford (11S, 13R, Z) -2
1H-6, 11-dioxa-3, 9-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacycloundecaphan-10-one (15.6 mg, 39%yield) as a white solid. LC-MS: m/z 344.2 [M+H]
+.
Example 61
(1
1S, 1
3R, 2
4Z, 8Z) -2
1H-6, 13-dioxa-3, 11-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclotridecaphan-8-en-12-one


Step 1: 4- ( (6-bromopyridin-2-yl) methoxy) but-2-yn-1-ol
To a solution of but-2-yne-1, 4-diol (1.37 g, 15.9 mmol) in DMF (10 mL) was added NaH (1.59 g, 39.9 mmol, 60%purity) at 0 ℃. The reaction mixture was stirred at 0 ℃ for 10 min. Then the solution of 2-bromo-6- (bromomethyl) pyridine (2.00 g, 7.97 mmol) in DMF (5.0 mL) was added dropwise to the reaction mixture. Then the reaction mixture was warmed to 20 ℃ and stirred at 20 ℃for 2 h. The mixture was quenched with ice water (80 mL) , the mixture was extracted with EtOAc (100 mL) . The organic layer was dried over anhydrous Na
2SO
4, filtered, and concentrated under reduced pressure, and the residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 50%in 20 min) to afford 4- ( (6-bromopyridin-2-yl) methoxy) but-2-yn-1-ol (1.50 g, 73%yield) as a pale yellow oil. LC-MS: (ESI) m/z [M+H]
+ 256.0.
Step 2: 4- ( (6-bromopyridin-2-yl) methoxy) but-2-yn-1-amine
To a solution of 4- ( (6-bromopyridin-2-yl) methoxy) but-2-yn-1-ol (500 mg, 1.95 mmol) in DCM (10 mL) was added DIPEA (757 mg, 5.86 mmol, 1.02 mL) , then methanesulfonyl chloride (447 mg, 3.90 mmol, 303 μL) was added to the reaction mixture at 15 ℃. The reaction mixture was stirred at 15 ℃for 2 h. Then the reaction was diluted with DCM (50 mL) , washed with brine (50 mL) , the organic layer was dried over anhydrous Na
2SO
4, filtered, and concentrated under reduced pressure to afford the crude intermediate. The intermediate was dissolved in NH
3 (7 M in MeOH, 10 mL) , then reaction mixture was stirred at 15 ℃ for 12 h. The mixture was concentrated under reduced pressure and the residue was redissolved in DCM (100 mL) , washed with brine (100 mL) , dried over anhydrous Na
2SO
4, filtered, and concentrated under reduced pressure to afford the crude product 4- ( (6-bromopyridin-2-yl) methoxy) but-2-yn-1-amine (500 mg, crude) as a yellow oil. LC-MS: (ESI) m/z [M+H]
+ 255.0.
Step 3: tert-butyl (4- ( (6-bromopyridin-2-yl) methoxy) but-2-yn-1-yl) carbamate
To a solution of 4- ( (6-bromopyridin-2-yl) methoxy) but-2-yn-1-amine (500 mg, 1.96 mmol) in THF (10 mL) was added TEA (992 mg, 9.80 mmol, 1.37 mL) and tert-butoxycarbonyl tert-butyl carbonate (1.28 g, 5.88 mmol, 1.35 mL) . Then the reaction mixture was stirred at 25 ℃ for 5 h. The mixture was concentrated under reduced pressure, the residue was purified by silica gel chromatography eluting with EtOAc/PE with EtOAc from 0 to 30%in 15 min to afford the product tert-butyl (4- ( (6-bromopyridin-2-yl) methoxy) but-2-yn-1-yl) carbamate (320 mg, 46%yield) as a yellow solid. LC-MS: (ESI) m/z [M+H]
+ 355.1.
Step 4: tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) methoxy) but-2-yn-1-yl) carbamate
To a solution of tert-butyl (4- ( (6-bromopyridin-2-yl) methoxy) but-2-yn-1-yl) carbamate (320 mg, 901 μmol) and (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (201 mg, 901 μmol) in dioxane (8.0 mL) was added Pd
2 (dba)
3 (123 mg, 135 μmol) , XantPhos (156 mg, 270 μmol) and Cs
2CO
3 (881 mg, 2.70 mmol) under the atmosphere of N
2. Then the reaction mixture was stirred at 100 ℃ under the atmosphere of N
2 for 3 h. The mixture was concentrated under reduced pressure, and the residue was purified by silica gel chromatography eluting with EtOAc/PE with EtOAc from 0 to 60%in 15 min to afford the product tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methoxy) but-2-yn-1-yl) carbamate (320 mg, 71%yield) as a yellow gum. LC-MS: (ESI) m/z [M+H]
+ 498.3.
Step 5: tert-butyl ( (Z) -4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) methoxy) but-2-en-1-yl) carbamate
To a solution of tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methoxy) but-2-yn-1-yl) carbamate (380 mg, 763.62 μmol) in EtOAc (10 mL) was added Pd/CaCO
3 (100 mg, 46.9 μmol, 5%purity) , then the reaction mixture was degassed with H
2 (ballon) for 3 times. Then the reaction mixture was stirred at 20 ℃ under the atmosphere of H
2 for 5 h. The mixture was filtered and concentrated under reduced pressure, the residue was purified by silica gel chromatography eluting with EtOAc/PE with EtOAc from 0 to 50%in 15 min to afford the product tert-butyl ( (Z) -4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methoxy) but-2-en-1-yl) carbamate (380 mg, 70%yield) as a yellow solid. LC-MS: (ESI) m/z [M+H] + 500.3.
Step 6: (1R, 3S) -3- (5- ( (6- ( ( ( (Z) -4- ( (tert-butoxycarbonyl) amino) but-2-en-1-yl) oxy) methyl) pyridin-
2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of tert-butyl ( (Z) -4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methoxy) but-2-en-1-yl) carbamate (200 mg, 400 μmol) in dioxane (10 mL) was added DIPEA (742 mg, 5.74 mmol, 1.00 mL) at 20 ℃, then di (imidazol-1-yl) methanone (195 mg, 1.20 mmol) was added to the reaction mixture, the mixture was stirred at 35 ℃ for 5 h. The mixture was concentrated under reduced pressure to afford the product (1R, 3S) -3- (5- ( (6- ( ( ( (Z) -4- ( (tert-butoxycarbonyl) amino) but-2-en-1-yl) oxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (230 mg, crude) as a yellow gum. LC-MS: (ESI) m/z [M+H]
+ 594.3.
Step 7: (1R, 3S) -3- (5- ( (6- ( ( ( (Z) -4-aminobut-2-en-1-yl) oxy) methyl) pyridin-2-yl) amino) -1- (tert-
butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of (1R, 3S) -3- (5- ( (6- ( ( ( (Z) -4- ( (tert-butoxycarbonyl) amino) but-2-en-1-yl) oxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (230 mg, 387 μmol) in DCM (5.0 mL) was added TFA (2.96 g, 25.96 mmol, 2.00 mL) at 20 ℃. Then the reaction mixture was stirred at 20 ℃ for 13 h. The mixture was concentrated under reduced pressure to afford the mixture of product (1R, 3S) -3- (5- ( (6- ( ( ( (Z) -4-aminobut-2-en-1-yl) oxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (190 mg, crude) as a yellow gum. LC-MS: (ESI) m/z [M+H]
+ 494.3.
Step 8: (1
1S, 1
3R, 2
4Z, 8Z) -2
1- (tert-butyl) -2
1H-6, 13-dioxa-3, 11-diaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclotridecaphan-8-en-12-one
The solution of (1R, 3S) -3- (5- ( (6- ( ( ( (Z) -4-aminobut-2-en-1-yl) oxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (190 mg, 192 μmol) in CH
3CN (30 mL) was added DIPEA (1.86 g, 14.4 mmol, 2.50 mL) at 20 ℃. Then the reaction mixture was heated to 80 ℃ and stirred at 80 ℃ for 5 h. The mixture was concentrated under reduced pressure, and the residue was purified by silica gel chromatography eluting with EtOAc/PE with EtOAc from 0 to 70%in 20 min to afford the product (1
1S, 1
3R, 2
4Z, 8Z) -2
1- (tert-butyl) -2
1H-6, 13-dioxa-3, 11-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclotridecaphan-8-en-12-one (25.0 mg, 31%yield) as a yellow gum. LC-MS: (ESI) m/z [M+H]
+ 426.2.
Step 9: (1
1S, 1
3R, 2
4Z, 8Z) -2
1H-6, 13-dioxa-3, 11-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclotridecaphan-8-en-12-one
The solution of (1
1S, 1
3R, 2
4Z, 8Z) -2
1- (tert-butyl) -2
1H-6, 13-dioxa-3, 11-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclotridecaphan-8-en-12-one (25.0 mg, 58.8 μmol) in TFA (5.0 mL) was stirred at 80 ℃ for 5 h. The mixture was concentrated under reduced pressure, and the residue was sent to purified by Prep-HPLC eluting with CH
3CN in water with CH
3CN from 17%to 27%in 9 min to afford the product 1 (1
1S, 1
3R, 2
4Z, 8Z) -2
1H-6, 13-dioxa-3, 11-diaza-4 (2, 6) -pyridina-2 (5, 3) - pyrazola-1 (1, 3) -cyclopentanacyclotridecaphan-8-en-12-one (2.50 mg, 12%yield) as a white solid. LC-MS: (ESI) m/z [M+H]
+ 370.1.
Example 62
(1
1S, 1
3R, 2
4Z, 8E) -2
1H-6, 13-dioxa-3, 11-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclotridecaphan-8-en-12-one

Step 1: tert-butyl (E) - (4- ( (6-bromopyridin-2-yl) methoxy) but-2-en-1-yl) carbamate
To a solution of tert-butyl (E) - (4-hydroxybut-2-en-1-yl) carbamate (507 mg, 2.71 mmol) in DMF (10 mL) was added NaH (141 mg, 3.52 mmol) at 0 ℃, after addition, the mixture was stirred at 0 ℃ for 1 h. To the mixture was added 2-bromo-6- (bromomethyl) pyridine (680 mg, 2.71 mmol) , the mixture was warmed to 25 ℃ and stirred at this temperature for 17 h. The mixture was quenched with aqueous NH
4Cl (50 mL) , and the aqueous layer was extracted with EtOAc (35 mL) . The combined organic layer was dried over Na
2SO
4, concentrated under reduced pressure. the residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 10 to 30%in 20 min) to give tert-butyl (E) - (4- ( (6-bromopyridin-2-yl) methoxy) but-2-en-1-yl) carbamate (380 mg, 39%yield) as a colorless oil. LC-MS: m/z 357.1 [M+H
++.
Step 2: tert-butyl ( (E) -4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) methoxy) but-2-en-1-yl) carbamate
The mixture of tert-butyl (E) - (4- ( (6-bromopyridin-2-yl) methoxy) but-2-en-1-yl) carbamate (150 mg, 0.42 mmol) , (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (93.8 mg, 0.42 mmol) , XantPhos (48.6 mg, 84.0 μmol) , Pd
2 (dba)
3 (38.5 mg, 42.0 μmol) and Cs
2CO
3 (274 mg, 0.84 mmol) in 1, 4-dioxane (10 mL) was heated to and stirred at 100 ℃ for 6 h under N
2. The mixture was cooled to 25 ℃ and was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with MeOH/DCM (with MeOH from 0 to 7%in 20 min) to give tert-butyl ( (E) -4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methoxy) but-2-en-1-yl) carbamate (180 mg, 86%yield) as a brown oil. LC-MS: m/z 500.4 [M+H]
+.
Step 3: (1R, 3S) -3- (5- ( (6- ( ( ( (E) -4- ( (tert-butoxycarbonyl) amino) but-2-en-1-yl) oxy) methyl) pyridin-
2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a stirred solution of tert-butyl ( (E) -4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methoxy) but-2-en-1-yl) carbamate (140 mg, 0.28 mmol) and DIEA (2.0 mL, 11.5 mmol) in 1, 4-dioxane (5 mL) was added CDI (136 mg, 0.84 mmol) at 25 ℃. After addition, the mixture was stirred at 100 ℃ for 17 h. The mixture was cooled to 25 ℃ and was concentrated under reduced pressure to give (1R, 3S) -3- (5- ( (6- ( ( ( (E) -4- ( (tert-butoxycarbonyl) amino) but-2-en-1-yl) oxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (160 mg, 96%yield) as a brown oil, which was used directly into the next step without further purification. LC-MS: m/z [M+H]
+ 594.3
Step 4: (1R, 3S) -3- (5- ( (6- ( ( ( (E) -4-aminobut-2-en-1-yl) oxy) methyl) pyridin-2-yl) amino) -1- (tert-
butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a stirred solution of (1R, 3S) -3- (5- ( (6- ( ( ( (E) -4- ( (tert-butoxycarbonyl) amino) but-2-en-1-yl) oxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (160 mg, 0.27 mmol) in DCM (4 mL) at 0 ℃ was added TFA (2.0 mL, 26.0 mmol) . After addtion, the mixture was warmed to 25 ℃ and stirred at this temperature for 1 h. The mixture was concentrated to give (1R, 3S) -3- (5- ( (6- ( ( ( (E) -4-aminobut-2-en-1-yl) oxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (150 mg, 85%yield) as a brown oil, which was used directly into the next step without further purification. LC-MS: m/z [M+H]
+494.2.
Step 5: (1
1S, 1
3R, 2
4Z, 8E) -2
1- (tert-butyl) -2
1H-6, 13-dioxa-3, 11-diaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclotridecaphan-8-en-12-one
The mixture of (1R, 3S) -3- (5- ( (6- ( ( ( (E) -4-aminobut-2-en-1-yl) oxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (150 mg, 0.30 mmol) and DIEA (393 mg, 3.03 mmol) in 1, 4-dioxane (5 mL) was heated 100 ℃ for 17 h. The mixture was cooled to 25 ℃and was concentrated under reduced pressure. The residue was purified by prep-TLC (EtOAc/PE = 1: 1) to give (1
1S, 1
3R, 2
4Z, 8E) -2
1- (tert-butyl) -2
1H-6, 13-dioxa-3, 11-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclotridecaphan-8-en-12-one (25.0 mg, 19%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 426.1
Step 6: (1
1S, 1
3R, 2
4Z, 8E) -2
1H-6, 13-dioxa-3, 11-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclotridecaphan-8-en-12-one
The mixture of (1
1S, 1
3R, 2
4Z, 8E) -2
1- (tert-butyl) -2
1H-6, 13-dioxa-3, 11-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclotridecaphan-8-en-12-one (25 mg, 58.8 μmol) and HCOOH (5.0 mL, 132 mmol) was heated to 90 ℃ and stirred at 90 ℃ for 2 h. The mixture was cooled to 25 ℃ and was concentrated under reduced pressure, the residue was purified by Pre-HPLC eluting with CH
3CN in water with CH
3CN from 5%to 35%in 8 min to give (1
1S, 1
3R, 2
4Z, 8E) -2
1H-6, 13-dioxa-3, 11-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclotridecaphan-8-en-12-one (2.40 mg, 11%yield) as a white solid. LC-MS: m/z [M+H]
+ 370.2.
Example 63 and 64
(1
1S, 1
3R, 7R, Z) -7-methyl-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one and (1
1S, 1
3R, 7S, Z) -7-methyl-2
1H-6, 12-dioxa-3, 10-diaza-
4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one


Step 1: tert-butyl (3- ( (6-bromopyridin-2-yl) methoxy) butyl) carbamate
To a solution of tert-butyl N- (3-hydroxybutyl) carbamate (317 mg, 1.67 mmol) in THF (30 mL) was added NaH (167 mg, 4.18 mmol, 60%purity) at 0 ℃ under nitrogen. The reaction was stirred at 0 ℃for 30 min and then 2-bromo-6- (bromomethyl) pyridine (350 mg, 1.39 mmol) was added. The reaction was warmed to 25 ℃ and stirred at 25 ℃ for 17 h. The mixture was quenched with saturated NH
4Cl solution (50 mL) and extracted with EtOAc (25 mL × 3) . The combined organic layer was dried over Na
2SO
4, filtered and concentrated. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 15%in 25 min) to afford tert-butyl (3- ( (6-bromopyridin-2-yl) methoxy) butyl) carbamate (305 mg, 61%yield) as a colorless oil. LC-MS: m/z 359.1 [M+H]
+.
Step 2: tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) methoxy) butyl) carbamate
To a mixture of tert-butyl (3- ( (6-bromopyridin-2-yl) methoxy) butyl) carbamate (386 mg, 1.07 mmol) , (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (200 mg, 896 μmol) and Cs
2CO
3 (584 mg, 1.79 mmol) in 1, 4-dioxane (10 mL) was added Pd
2 (dba)
3 (82.0 mg, 89.6 μmol) and Xantphos (104 mg, 179 μmol) under nitrogen. The reaction was heated to 100 ℃ and stirred at 100 ℃under nitrogen for 6 h. The mixture was concentrated under reduced pressure and the residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 80%) in 25 min to afford tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methoxy) butyl) carbamate (306 mg, 68%yield) as a brown oil. LC-MS: m/z [M+H]
+ 502.3.
Step 3: (1R, 3S) -3- (5- ( (6- ( ( (4- ( (tert-butoxycarbonyl) amino) butan-2-yl) oxy) methyl) pyridin-2-
yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methoxy) butyl) carbamate (270 mg, 538 μmol) in DCM (15 mL) was added CDI (262 mg, 1.61 mmol) and DIPEA (2.69 mmol, 469 μL) at 25 ℃. Then the reaction mixture was heated to 35 ℃ and stirred at 35 ℃ for 1 h. The mixture was concentrated under reduced pressure to afford (1R, 3S) -3- (5- ( (6- ( ( (4- ( (tert-butoxycarbonyl) amino) butan-2-yl) oxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (310 mg, crude) as a brown oil, which was used directly in the next step without further purification. LC-MS: m/z [M+H]
+ 596.3.
Step 4: (1R, 3S) -3- (5- ( (6- ( ( (4-aminobutan-2-yl) oxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-
pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a stirring solution of (1R, 3S) -3- (5- ( (6- ( ( (4- ( (tert-butoxycarbonyl) amino) butan-2-yl) oxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (300 mg, 504 μmol) in DCM (10 mL) at 25 ℃ was added TFA (10 mL) . After addition, the mixture was stirred at this temperature for 10 min. The mixture was concentrated under reduced pressure to afford (1R, 3S) -3- (5- ( (6- ( ( (4-aminobutan-2-yl) oxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (240 mg, crude) as a brown oil, which was used directly in the next step without further purification. LC-MS: m/z [M+H]
+ 496.3.
Step 5: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -7-methyl-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a solution of (1R, 3S) -3- (5- ( (6- ( ( (4-aminobutan-2-yl) oxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (240 mg, 484 μmol) in CH
3CN (10 mL) was added DIPEA (2.97 g, 22.97 mmol) . The mixture was warmed to 80 ℃ and stirred at this temperature for 6 h. The mixture was cooled down to 25 ℃ and concentrated under reduced pressure, and the residue was purified by flash chromatography on silica gel eluting with EtOAc/petroleum ether (with EtOAc from 0 to 80%) in 25 min to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -7-methyl-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (90.0 mg, 43%yield) as a colorless oil. LC-MS: m/z [M+H]
+ 428.3.
Step 6: (1
1S, 1
3R, 7R, Z) -7-methyl-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphan-11-one and (1
1S, 1
3R, 7S, Z) -7-methyl-2
1H-6, 12-dioxa-3, 10-
diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
The mixture of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -7-methyl-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (80.0 mg, 187 μmol) in TFA (3.0 mL) was heated to 90 ℃ and stirred at 90 ℃ for 6 h. The mixture was cooled to 25 ℃ and was concentrated under reduced pressure. The residue was purified by Prep-HPLC (with CH
3CN from 13%to 23%in 8 min) to afford (1
1S, 1
3R, 7R, Z) -7-methyl-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (retention time = 5.7 min, 7.5 mg, absolute configuration arbitrarily assigned, 11%yield) as a white solid. LC-MS: m/z 372.2 [M+H]
+. And (1
1S, 1
3R, 7S, Z) -7-methyl-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (retention time = 6.5 min, 11.1 mg, absolute configuration arbitrarily assigned, 16%yield) as a white solid. LC-MS: m/z 372.2 [M+H]
+.
Example 65 and 66
(11S, 13R, 5S, Z) -43-fluoro-5-methyl-21H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphan-11-one and (11S, 13R, 5R, Z) -43-fluoro-5-methyl-21H-6, 12-
dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one

Step 1: 2-chloro-6- (1-ethoxyvinyl) -3-fluoropyridine
To a solution of 6-bromo-2-chloro-3-fluoropyridine (800 mg, 3.80 mmol) in dioxane (50 mL) was added PdCl
2 (Ph
3P)
2 (534 mg, 760 μmol) , CuI (145 mg, 761 μmol, 25.8 μL) tributyl (1-ethoxyvinyl) stannane (2.06 g, 5.70 mmol, 1.93 mL) at 25 ℃ under N
2. The mixture was heated to 80℃ and stirred at 80 ℃ under N
2 for 2 h. The reaction was cooled to room temperature and filtered. The filter cake was washed with acetonitrile and the organic layers were combined. The resulting solution was concentrated under reduced pressure to purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 10%in 15 min) to afford 2-chloro-6- (1-ethoxyvinyl) -3-fluoropyridine (740 mg, 96%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 202.1.
Step 2: 1- (6-chloro-5-fluoropyridin-2-yl) ethan-1-one
To a solution of 2-chloro-6- (1-ethoxyvinyl) -3-fluoropyridine (730 mg, 3.62 mmol) in dioxane (20 mL) was added HCl (2.5 M, 10 mL) at 25 ℃. The reaction mixture was stirred at 25 ℃ for 1 h. The mixture was diluted with water (20 mL) and extracted with EtOAc (5.0 mL × 3) . The combined organic layer was washed with brine (20 mL) , dried over anhydrous Na
2SO
4, filtered and concentrated. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 10%in 15 min) to afford 1- (6-chloro-5-fluoropyridin-2-yl) ethan-1-one (477 mg, 76%yield) as a yellow oil. LC-MS: m/z 174.0 [M+H]
+.
Step 3: 1- (6-chloro-5-fluoropyridin-2-yl) ethan-1-ol
To a solution of 1- (6-chloro-5-fluoropyridin-2-yl) ethan-1-one (477 mg, 2.75 mmol) in MeOH (50 mL) was added sodium borohydride (197 mg, 5.50 mmol) under N
2 at 0 ℃. The mixture was warmed to 25℃ and stirred at 25 ℃ under N
2 for 1 h. The mixture was quenched with saturated NaCl solution (100 mL) and then extracted with ethyl acetate (30 mL × 3) . The combined organic layer was dried over anhydrous Na
2SO
4, filtered and concentrated. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 40%in 20 min) to afford 1- (6-chloro-5-fluoropyridin-2-yl) ethan-1-ol (380 mg, 79%yield) as a colorless oil. LC-MS: m/z [M+H]
+ 176.1.
Step 4: tert-butyl (3- (1- (6-chloro-5-fluoropyridin-2-yl) ethoxy) propyl) carbamate
To a solution of 1- (6-chloro-5-fluoropyridin-2-yl) ethan-1-ol (380 mg, 2.16 mmol) in DMF (4.0 mL) was added NaH (173 mg, 4.33 mmol) at 0 ℃ under nitrogen. The reaction was stirred at 0 ℃ for 30 min and then tert-butyl (3-bromopropyl) carbamate (1.03 g, 4.33 mmol) was added. The reaction was stirred at 25 ℃ for 1 h. The mixture was quenched with saturated NH
4Cl solution (20 mL) and extracted with EtOAc (10 mL × 3) . The combined organic layer was washed with brine (30 mL) , dried over anhydrous Na
2SO
4, filtered and concentrated. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 30%in 15 min) to afford tert-butyl (3- (1- (6-chloro-5-fluoropyridin-2-yl) ethoxy) propyl) carbamate (450 mg, 62 %yield) as a colorless oil. LC-MS: m/z 355.1 [M+Na]
+.
Step 5: tert-butyl (3- (1- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) -5-fluoropyridin-2-yl) ethoxy) propyl) carbamate
To a solution of (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (130 mg, 582 μmol) in dioxane (10 mL) was added tert-butyl (3- (1- (6-chloro-5-fluoropyridin-2-yl) ethoxy) propyl) carbamate (232 mg, 699 μmol) , Cs
2CO
3 (379 mg, 1.16 mmol) Pd
2 (dba)
3 (107 mg, 116 μmol) and XantPhos (67.4 mg, 116 μmol) under N
2 at 25 ℃. The reaction was heated to 100 ℃and stirred at 100 ℃ under N
2 for 12 h. The mixture was cooled to room temperature. The mixture was diluted with water (20 mL) and extracted with EtOAc (5.0 mL × 3) . The combined organic layer was dried over anhydrous Na
2SO
4, filtered and concentrated. The residue was purified by silica gel chromatography eluting with MeOH/DCM (with MeOH from 0 to 7%in 15 min) to afford tert-butyl (3- (1- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -5-fluoropyridin-2-yl) ethoxy) propyl) carbamate (253 mg, 84%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 520.3.
Step 6: tert-butyl (3- (1- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-
nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) -5-fluoropyridin-2-
yl) ethoxy) propyl) carbamate
To a solution of tert-butyl (3- (1- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -5-fluoropyridin-2-yl) ethoxy) propyl) carbamate (230 mg, 443 μmol) in DCE (20 mL) was added 4-nitrophenyl carbonochloridate (268 mg, 1.33 mmol) , DMAP (54.1 mg, 443 μmol) and DIPEA (172 mg, 1.33 mmol, 232 μL) at 25 ℃. The reaction was heated to 85 ℃ and stirred at 85 ℃under N
2 for 16 h. The mixture was cooled to room temperature. The mixture was diluted with water (50 mL) and extracted with DCM (30 mL × 3) . The combined organic layer dried over anhydrous Na
2SO
4, filtered and concentrated. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 50%in 20 min) to afford tert-butyl (3- (1- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) -5-fluoropyridin-2-yl) ethoxy) propyl) carbamate (170 mg, 56%yield) as a brown oil. LC-MS: m/z 685.3 [M+H]
+.
Step 7: (1R, 3S) -3- (5- ( (6- (1- (3-aminopropoxy) ethyl) -3-fluoropyridin-2-yl) amino) -1- (tert-butyl) -
1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate
To a solution of tert-butyl (3- (1- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) -5-fluoropyridin-2-yl) ethoxy) propyl) carbamate (170 mg, 248μmol) in DCM/TFA=1: 1 (10 mL) was stirred at 25 ℃ for 1 h. The mixture was concentrated under reduced pressure to afford (1R, 3S) -3- (5- ( (6- (1- (3-aminopropoxy) ethyl) -3-fluoropyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate (200 mg, crude) as a yellow oil, which was used directly in the next step. LC-MS: m/z [M+H]
+ 585.2.
Step 8: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
3-fluoro-5-methyl-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-
2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a solution of (1R, 3S) -3- (5- ( (6- (1- (3-aminopropoxy) ethyl) -3-fluoropyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate (200 mg, 205 μmol) in CH
3CN (100 mL) was added DIPEA (2.23 g, 17.2 mmol, 3.00 mL) at 25 ℃. The reaction was heated to 85 ℃ and stirred at 85 ℃ for 12 h. The mixture was cooled to room temperature and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 40%in 15 min) to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
3-fluoro-5-methyl-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (80.0 mg, 87%yield) as a yellow solid. LC-MS: m/z [M+H]
+ 446.2.
Step 9: (1
1S, 1
3R, 5S, Z) -4
3-fluoro-5-methyl-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one & (1
1S, 1
3R, 5R, Z) -4
3-fluoro-5-methyl-2
1H-
6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-
one
The solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
3-fluoro-5-methyl-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (80.0 mg, 180 μmol) in FA (5.0 mL) was heated to 80 ℃ and stirred at 80 ℃ for 2 h. The mixture was cooled to room temperature and concentrated under reduced pressure. The residue was purified by Prep-HPLC (with CH
3CN from 25%to 55%in 8 min) to afford the product (1
1S, 1
3R, 5S, Z) -4
3-fluoro-5-methyl-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (retention time = 4.6 min, 10.6 mg, absolute configuration arbitrarily assigned, 15%yield) as white solid. LC-MS: m/z [M+H] + 390.1. And (1
1S, 1
3R, 5R, Z) -4
3-fluoro-5-methyl-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (retention time = 6.9 min, 9.40 mg, absolute configuration arbitrarily assigned, 13%yield) as white solid. LC-MS: m/z [M+H]
+ 390.1.
Example 67
(1
1S, 1
3R, Z) -8, 8-dimethyl-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one

Step 1: tert-butyl (3- ( (6-bromopyridin-2-yl) methoxy) -2, 2-dimethylpropyl) carbamate
To a solution of tert-butyl (3-hydroxy-2, 2-dimethylpropyl) carbamate (200 mg, 984 μmol) in THF (10 mL) was added NaH (157 mg, 3.94 mmol, 60%purity) at 0 ℃ under nitrogen. The reaction was stirred at 0 ℃ for 30 min and then 2-bromo-6- (bromomethyl) pyridine (296 mg, 1.18 mmol) was added. The reaction was warmed to 25 ℃ stirred at 25 ℃ for 1 h. The mixture was quenched with saturated NH
4Cl solution (30 mL) and extracted with EtOAc (10 mL × 3) . The combined organic layer was dried over Na
2SO
4, filtered and concentrated. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 20%in 15 min) to afford tert-butyl (3- ( (6-bromopyridin-2-yl) methoxy) -2, 2-dimethylpropyl) carbamate (182 mg, 49%yield) as a brown oil. LC-MS: m/z 374.1 [M+H]
+.
Step 2: tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) methoxy) -2, 2-dimethylpropyl) carbamate
To a mixture of tert-butyl (3- ( (6-bromopyridin-2-yl) methoxy) -2, 2-dimethylpropyl) carbamate (167 mg, 448 μmol) , (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (100 mg, 448 μmol) and Cs
2CO
3 (292 mg, 896 μmol) in 1, 4-dioxane (10 mL) was added Pd
2 (dba)
3 (82.0 mg, 90.0 μmol) and Xantphos (51.8 mg, 89.6 μmol) under nitrogen. The reaction was heated to 100 ℃ stirred at 100 ℃ under nitrogen for 12 h. The mixture was diluted with ice water (30 mL) and extracted with EtOAc (10 mL × 3) . The combined organic layer was dried over Na
2SO
4, filtered and concentrated. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 70%) in 15 min to afford tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methoxy) -2, 2-dimethylpropyl) carbamate (190 mg, 82%yield) as a brown oil. LC-MS: m/z [M+H]
+ 516.4.
Step 3: (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) -2, 2-dimethylpropoxy) methyl) pyridin-
2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methoxy) -2, 2-dimethylpropyl) carbamate (190 mg, 368 μmol) in DCM (10 mL) was added CDI (179 mg, 1.11 mmol) and DIPEA (1.84 mmol, 321 μL) at 25 ℃. Then the reaction mixture was heated to 35 ℃ and stirred at 35 ℃ for 1 h. The mixture was concentrated under reduced pressure to afford (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) -2, 2-dimethylpropoxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (198 mg, crude) as a yellow solid, which was used directly in the next step. LC-MS: m/z [M+H]
+ 610.3.
Step 4: (1R, 3S) -3- (5- ( (6- ( (3-amino-2, 2-dimethylpropoxy) methyl) pyridin-2-yl) amino) -1- (tert-
butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
A solution of (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) -2, 2-dimethylpropoxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (198 mg, crude) in TFA (5.0 mL) was stirred at 25 ℃ for 30 min. The mixture was concentrated under reduced pressure to afford (1R, 3S) -3- (5- ( (6- ( (3-amino-2, 2-dimethylpropoxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (170 mg, crude) as a brown oil, which was used directly in the next step. LC-MS: m/z [M+H]
+ 510.3.
Step 5: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -8, 8-dimethyl-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a solution of (1R, 3S) -3- (5- ( (6- ( (3-amino-2, 2-dimethylpropoxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (170 mg, 267 μmol) in CH
3CN (30 mL) was added DIPEA (103 mg, 801 μmol) . The mixture was stirred at 25 ℃ for 12 h. The mixture was concentrated under reduced pressure, and the residue was purified by flash chromatography on silica gel eluting with EtOAc/petroleum ether (with EtOAc from 0 to 40%) in 15 min to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -8, 8-dimethyl-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (60.0 mg, 51%yield) as a yellow solid. LC-MS: m/z [M+H]
+ 442.2.
Step 6: (1
1S, 1
3R, Z) -8, 8-dimethyl-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphan-11-one
A solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -8, 8-dimethyl-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (50.0 mg, 113 μmol) in TFA (6.0 mL) was heated to 80 ℃ and stirred at 80 ℃ for 12 h. The mixture was concentrated under reduced pressure. The residue was purified by Prep-HPLC (with CH
3CN from 10%to 40%in 8 min) to afford (1
1S, 1
3R, Z) -8, 8-dimethyl-2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (4.00 mg, 9%yield) as a white solid. LC-MS: m/z 386.2 [M+H]
+.
Example 68 and 69
(1
1S, 1
3R, 7
1R, 7
3S, Z) -2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-9-one and (1
1S, 1
3R, 7
1S, 7
3R, Z) -2
1H-6, 10-dioxa-
3, 8-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-
9-one

Step 1: tert-butyl N- [3- [ (6-bromo-2-pyridyl) methoxy] cyclobutyl] carbamate
To a suspension of tert-butyl N- (3-hydroxycyclobutyl) carbamate (400 mg, 2.14 mmol) in THF (10.0 mL) was added slowly NaH (76.0 mg, 60 wt. %in mineral oil, 3.20 mmol) at 0 ℃. The suspension was stirred at that temperature for 15 min under N
2 before 2-bromo-6- (bromomethyl) pyridine (562 mg, 2.24 mmol) was added and the reaction was stirred at 20 ℃ for 16 h. The reaction mixture was quenched with ice-cold water (20 mL) and extracted with EtOAc (3 × 20 mL) . The organic phase was concentrated under reduced pressure. The crude product was purified by flash chromatography eluting with EtOAc/PE (with EtOAc from 0 to 25%in 25 min) to afford tert-butyl N- [3- [ (6-bromo-2-pyridyl) methoxy] cyclobutyl] carbamate (600 mg, 78%yield) as a yellow solid. LC-MS: m/z 356.8 [M+H]
+.
Step 2: tert-butyl N- [3- [ [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -2-
pyridyl] methoxy] cyclobutyl] carbamate
To a suspension of tert-butyl N- [3- [ (6-bromo-2-pyridyl) methoxy] cyclobutyl] carbamate (600 mg, 1.68 mmol) in 1, 4-dioxane (10.0 mL) was sequentially added (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (375 mg, 1.68 mmol) , Cs
2CO
3 (1.64 g, 5.04 mmol) , XantPhos (194 mg, 0.335 mmol) and Pd
2 (dba)
3 (153 mg, 0.167 mmol) at room temperature and the reaction was stirred at 100 ℃ for 16 h under N
2. After completion of the reaction as judged by LCMS, the reaction mixture was filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography eluting with EtOAc/PE (with EtOAc from 0 to 40%in 30 min) to afford tert-butyl N- [3- [ [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -2-pyridyl] methoxy] cyclobutyl] carbamate (450 mg, 53%yield) as a yellow solid. LC-MS: m/z 500.0 [M+H]
+.
Step 3: [ (1R, 3S) -3- [5- [ [6- [ [3- (tert-butoxycarbonylamino) cyclobutoxy] methyl] -2-pyridyl] amino] -
1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate
To a suspension of tert-butyl N- [3- [ [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -2-pyridyl] methoxy] cyclobutyl] carbamate (450 mg, 0.90 mmol) in CH
2Cl
2 (5.0 mL) was added CDI (388 mg, 2.70 mmol) and DIPEA (781 μL, 580 mg, 4.50 mmol) at 35 ℃and the reaction was stirred for 5 h. After completion of the reaction as judged by LCMS, the reaction mixture was quenched with ice-cold water (10 mL) and extracted with EtOAc (3 × 10 mL) . The organic phase was washed with brine (20 mL) and dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure to afford [ (1R, 3S) -3- [5- [ [6- [ [3- (tert-butoxycarbonylamino) cyclobutoxy] methyl] -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (crude) as a yellow oil. The crude product was used in the next step without further purification. LC-MS: m/z 594.1 [M+H]
+.
Step 4: [ (1R, 3S) -3- [5- [ [6- [ (3-aminocyclobutoxy) methyl] -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-
yl] cyclopentyl] imidazole-1-carboxylate
To a suspension of [ (1R, 3S) -3- [5- [ [6- [ [3- (tert-butoxycarbonylamino) cyclobutoxy] methyl] -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (crude) in CH
2Cl
2 (3.0 mL) was added slowly TFA (1.0 mL) at 25 ℃ and the reaction was stirred for 1 h. After completion of the reaction as judged by LCMS, the reaction mixture was concentrated under reduced pressure to afford [ (1R, 3S) -3- [5- [ [6- [ (3-aminocyclobutoxy) methyl] -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (crude) as a yellow oil. The crude product was used in the next step without further purification. LC-MS: m/z 494.1 [M+H]
+.
Step 5: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-
1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-9-one
To a suspension of [ (1R, 3S) -3- [5- [ [6- [ (3-aminocyclobutoxy) methyl] -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (crude) in CH
3CN (5.0 mL) was added Et
3N (1.24 mL, 902 mg, 8.91 mmol) and the reaction was stirred at 80 ℃ for 16 h. After completion of the reaction as judged by LCMS, the reaction mixture was concentrated under reduced pressure. The crude product was purified by flash column chromatography eluting with EtOAc/PE (with EtOAc from 0 to 80%in 30 min) to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-9-one (100 mg) as a yellow solid. m/z 426.1 [M+H]
+.
Step 6: (1
1S, 1
3R, 7
1R, 7
3S, Z) -2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-9-one and (1
1S, 1
3R, 7
1S, 7
3R, Z) -2
1H-6, 10-dioxa-
3, 8-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-
9-one
A mixture of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-9-one (90.0 mg, 211 μmol) in HCO
2H (3.0 mL) was stirred for 5 h at 100 ℃. The reaction mixture was concentrated under reduced pressure. The residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 2%to 15%in 10 min (0.1%HCO
2H) and further purified by SFC eluting with CO
2 in EtOH (AD-H column, CO
2/EtOH = 60/40, EtOH with 0.2%NH
4OH, 40 g/min, 40 ℃) to afford the desired product (1
1S, 1
3R, 7
1R, 7
3S, Z) -2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-9-one (relative configuraton arbitrarily assigned, retention time = 5.4 min, 8.4 mg, 10%yield) , LC-MS: m/z 370.0 [M+H]
+, and (1
1S, 1
3R, 7
1S, 7
3R, Z) -2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-9-one (relative configuraton arbitrarily assigned, retention time = 8.8 min, 2.0 mg, 3%yield) as a white solid. LC-MS: m/z 370.0 [M+H]
+.
Example 70
(1
1S, 1
3R, 7
1R, 7
3S, Z) -9-oxo-2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentana-7 (1, 3) -cyclobutanacyclodecaphane-4
4-carbonitrile


Step 1: 2, 4-dibromo-6- (bromomethyl) pyridine
To a stirred solution of 2-bromoisonicotinonitrile (5.00 g, 19.9 mmol) in CCl
4 (50.0 mL) were sequentially added AIBN (654 mg, 3.99 mmol) and NBS (4.26 g, 23.9 mmol) at 25 ℃. The reaction mixture was warmed to 80 ℃ and stirred at that temperature for 16 h under N
2 atmosphere. The mixture was cooled and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 50%in 30 min) to afford 2, 4-dibromo-6- (bromomethyl) pyridine (5.00 g, crude) as a yellow solid. LC-MS: m/z [M+H]
+ 327.7.
Step 2: tert-butyl ( (1r, 3r) -3- ( (4, 6-dibromopyridin-2-yl) methoxy) cyclobutyl) carbamate
To a stirred solution of 2, 4-dibromo-6- (bromomethyl) pyridine (5.00 g, 15.2 mmol) in THF (100 mL) was added NaH (1.21 g, 60%wt. in mineral oil, 30.3 mmol) at 0 ℃. The reaction mixture was stirred at 0 ℃ for 30 min before tert-butyl ( (1r, 3r) -3-hydroxycyclobutyl) carbamate (2.84 g, 15.2 mmol) was added at that temperature. The reaction mixture was warmed to 25 ℃ and stirred at that temperature for 16 h before it was quenched with water (200 mL) and extracted with EtOAc (200 mL × 2) . The combined organic phases were dried over Na
2SO
4 and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 30%in 25 min) to afford tert-butyl ( (1r, 3r) -3- ( (4, 6-dibromopyridin-2-yl) methoxy) cyclobutyl) carbamate (2.50 g, 36%yield ) as a yellow oil. LC-MS: m/z [M+Na]
+ 458.8.
Step 3: tert-butyl ( (1S, 3r) -3- ( (4-bromo-6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-
pyrazol-5-yl) amino) pyridin-2-yl) methoxy) cyclobutyl) carbamate
To a stirred solution of tert-butyl N- [3- [ (4, 6-dibromo-2-pyridyl) methoxy] cyclobutyl] carbamate (2.50 g, 5.73 mmol) in 1, 4-Dioxane (60.0 mL) were sequentially added (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (1.28 g, 5.73 mmol) , Pd
2 (dba)
3 (525 mg, 0.573 mmol) , XantPhos (663 mg, 1.15 mmol) and Cs
2CO
3 (3.74 g, 11.5 mmol) at 25 ℃. The reaction mixture was warmed to 80 ℃ and stirred at that temperature for 3 h before it was cooled to 25 ℃ and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with MeOH/DCM (with MeOH from 0 to 10%in 25 min) and concentrated under reduced pressure. The residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 30%to 70%in 60 min) to give tert-butyl ( (1S, 3r) -3- ( (4-bromo-6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methoxy) cyclobutyl) carbamate (1.80 g, 52%yield) as a yellow solid. LC-MS: m/z [M+H]
+ 578.1.
Step 4: (1R, 3S) -3- (5- ( (4-bromo-6- ( ( (1r, 3S) -3- ( (tert-
butoxycarbonyl) amino) cyclobutoxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-
yl) cyclopentyl 1H-imidazole-1-carboxylate
To a stirred suspension of tert-butyl N- [3- [ [4-bromo-6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -2-pyridyl] methoxy] cyclobutyl] carbamate (1.71 g, 2.96 mmol) in CH
2Cl
2 (50.0 mL) were sequentially added CDI (1.28 g, 8.87 mmol) , Et
3N (822 μL, 599 mg, 5.91 mmol) and DMAP (289 mg, 2.36 mmol) at 25 ℃. The reaction mixture was warmed to 40 ℃ and stirred at that temperature for 16 h before it was cooled to 25 ℃. The reaction mixture was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 80 %in 30 min) to afford (1R, 3S) -3- (5- ( (4-bromo-6- ( ( (1r, 3S) -3- ( (tert-butoxycarbonyl) amino) cyclobutoxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (1.40 g, 66%yield) as a colorless oil. LC-MS: m/z [M+H]
+ 672.2.
Step 5: (1R, 3S) -3- (5- ( (6- ( ( (1r, 3S) -3-aminocyclobutoxy) methyl) -4-bromopyridin-2-yl) amino) -1-
(tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a stirred solution of (1R, 3S) -3- (5- ( (4-bromo-6- ( ( (1r, 3S) -3- ( (tert-butoxycarbonyl) amino) cyclobutoxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (1.50 g, 2.23 mmol) in CH
2Cl
2 (20.0 mL) were added TFA (20.0 mL) at 25 ℃. The resulting mixture was stirred at that temperature for 2 h. The mixture was concentrated under reduced pressure to give (1R, 3S) -3- (5- ( (6- ( ( (1r, 3S) -3-aminocyclobutoxy) methyl) -4-bromopyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (1.50 g, crude) as a colorless oil. LC-MS: m/z [M+H]
+ 572.0.
Step 6: (1
1S, 1
3R, 7
1R, 7
3S, Z) -4
4-bromo-2
1- (tert-butyl) -2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-
2 (3, 5) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-9-one
To a stirred solution of (1R, 3S) -3- (5- ( (6- ( ( (1r, 3S) -3-aminocyclobutoxy) methyl) -4-bromopyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (1.50 g, 2.62 mmol) in CH
3CN (20.0 mL) was added Et
3N (1.09 mL, 795 mg, 7.86 mmol) at 25 ℃. The resulting mixture was stirred at that temperature for 16 h before it was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0%to 80%in 30 min) to give (1
1S, 1
3R, 7
1R, 7
3S, Z) -4
4-bromo-2
1- (tert-butyl) -2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-9-one (450 mg, 31%yield) as a brown solid. LC-MS: m/z [M+H]
+ 504.0.
Step 7: (1
1S, 1
3R, 7
1R, 7
3S, Z) -2
1- (tert-butyl) -9-oxo-2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphane-4
4-carbonitrile
To a stirred solution of (1
1S, 1
3R, 7
1R, 7
3S, Z) -4
4-bromo-2
1- (tert-butyl) -2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-9-one (180 mg, 357 μmol) in 1, 4-Dioxane (10.0 mL) were sequentially added Potassium hexacyanoferrate (II) trihydrate (226 mg, 535 μmol) , Xphos Pd G3 (18.1 mg, 21.4 μmol) , KOAc (75.5 mg, 785 μmol) and H
2O (2.0 mL) at 25 ℃. The reaction mixture was warmed to 100 ℃ and stirred at that temperature for 16 h. The mixture was cooled to 25 ℃ and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 50%in 30 min) to afford (1
1S, 1
3R, 71
R, 7
3S, Z) -2
1- (tert-butyl) -9-oxo-2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphane-4
4-carbonitrile (120 mg, 63%yield) as a brown solid. LC-MS: m/z [M+H]
+ 451.1.
Step 8: (1
1S, 1
3R, 7
1R, 7
3S, Z) -9-oxo-2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-
1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphane-4
4-carbonitrile
A solution of (1
1S, 1
3R, 71
R, 7
3S, Z) -2
1- (tert-butyl) -9-oxo-2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphane-4
4-carbonitrile (120 mg, 266 μmol) in TFA (10.0 mL) was warmed to 50 ℃ and stirred at that temperature for 3 h before it was cooled to 25 ℃. The reaction mixture was concentrated under reduced pressure. The residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 30%to 70%in 40 min) to give (1
1S, 1
3R, 7
1R, 7
3S, Z) -9-oxo-2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphane-4
4-carbonitrile (50.0 mg, 47%yield) as a yellow solid. LC-MS: m/z [M+H]
+ 395.1.
Example 182
(1
1S, 1
3R, 7
1R, 7
3S, Z) -4
4- (aminomethyl) -2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-
1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-9-one

To a stirred solution of (1
1S, 1
3R, 7
1R, 7
3S, Z) -9-oxo-2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphane-4
4-carbonitrile (30.0 mg, 76.1 μmol) and HOAc (21.8 μL , 22.8 mg, 380 μmol, ) in MeOH (20.0 mL) was added Raney Ni (22.3 mg, 380 μmol) at 25℃. The reaction mixture was warmed to 50 ℃ and stirred at that temperature for 16 h before it was cooled to 25 ℃. The reaction mixture was filtered through a pad of Celite before the filtrate was concentrated under reduced pressure. The residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 30%to 70%in 40 min) to give (1
1S, 1
3R, 7
1R, 7
3S, Z) -4
4- (aminomethyl) -2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-9-one (7.90 mg, 26%yield) as a white solid. LC-MS: m/z [M+H] + 399.0.
Example 71
(1
1S, 1
3R, 7
1R, 7
3S, Z) -4
4- (hydroxymethyl) -2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (5, 3) -
pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-9-one

Step 1: (1
1S, 1
3R, 7
1R, 7
3S, Z) -2
1- (tert-butyl) -9-oxo-2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphane-4
4-carbaldehyde
To a stirred solution of (1
1S, 1
3R, 7
1R, 7
3S, Z) -4
4-bromo-2
1- (tert-butyl) -2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-9-one (180 mg, 357 μmol) in THF (20.0 mL) was added n-BuLi (1.78 mL, 356 μmol, 2M in THF) at -78℃. The reaction mixture was stirred at that temperature for 0.5 h under N
2 atmosphere before DMF (27.6 μL, 26.1 mg, 357 μmol) was added at that temperature. The reaction mixture was stirred at -78℃ for 0.5 h under N
2 atmosphere before it was warmed to 25 ℃. The reaction mixture was quenched with H
2O (30 mL) and extracted with EtOAc (100 mL × 3) . The combined organic phases were washed with brine (50 mL) , dried over Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 100 %in 40 min) to give (1
1S, 1
3R, 7
1R, 7
3S, Z) -2
1- (tert-butyl) -9-oxo-2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphane-4
4-carbaldehyde (110 mg, 68%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 454.1.
Step 2: (1
1S, 1
3R, 7
1R, 7
3S, Z) -2
1- (tert-butyl) -4
4- (hydroxymethyl) -2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -
pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-9-one
To a stirred solution of (1
1S, 1
3R, 7
1R, 7
3S, Z) -2
1- (tert-butyl) -9-oxo-2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphane-4
4-carbaldehyde (60.0 mg, 132 μmol) in MeOH (15.0 mL) was added NaBH
4 (15.0 mg, 397 μmol) at 0 ℃. The reaction mixture was warmed to 25 ℃ and stirred at that temperature for 1 h. The reaction mixture was quenched with H
2O (15 mL) and extracted with EtOAc (15 mL × 3) . The combined organic phases were washed with brine (15 mL) , dried over Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with MeOH/CH
2Cl
2 (with MeOH from 0 to 10 %in 20 min) to give (1
1S, 1
3R, 7
1R, 7
3S, Z) -2
1- (tert-butyl) -4
4- (hydroxymethyl) -2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-9-one (36.0 mg, 51%yield) as a colorless oil. LC-MS: m/z [M+H]
+456.2.
Step 3: (1
1S, 1
3R, 7
1R, 7
3S, Z) -4
4- (hydroxymethyl) -2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (5, 3) -
pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-9-one
A stirred solution of (1
1S, 1
3R, 7
1R, 7
3S, Z) -2
1- (tert-butyl) -4
4- (hydroxymethyl) -2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-9-one (28.0 mg, 61.5 μmol) in TFA (5.0 mL) at 25 ℃. The resulting mixture was warmed to 80 ℃ and stirred at that temperature for 3 h. The mixture was cooled to 25 ℃. The residue was concentrated under reduced pressure and purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 30%to 70%in 40 min) to give (1
1S, 1
3R, 7
1R, 7
3S, Z) -4
4- (hydroxymethyl) -2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-9-one (5.4 mg, 22%yield) as a white solid. LC-MS: m/z [M+H]
+ 400.1.
Example 72
(1
1S, 1
3R, 7
1R, 7
3S, Z) -4
3-fluoro-2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-9-one


Step 1: 2-bromo-6- (bromomethyl) -3-fluoropyridine
To a solution of (6-bromo-5-fluoropyridin-2-yl) methanol (600 mg, 2.91 mmol) in THF (20 mL) was added carbon tetrabromide (1.93 g, 5.82 mmol) and triphenylphosphane (1.53 g, 5.82 mmol) . The mixture was stirred at 25 ℃ for 20 h. The mixture was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/petroleum ether (with EtOAc from 0 to 30%) in 20 min to give 2-bromo-6- (bromomethyl) -3-fluoropyridine (700 mg, 89%yield) as a white solid. LC-MS: m/z [M+H]
+ 269.9.
Step 2: tert-butyl (3- ( (6-bromo-5-fluoropyridin-2-yl) methoxy) cyclobutyl) carbamate
To a solution of tert-butyl (3-hydroxycyclobutyl) carbamate (406 mg, 2.17 mmol) and NaH (260 mg, 10.8 mmol) in THF (5.0 mL) was added 2-bromo-6- (bromomethyl) -3-fluoro-pyridine (700 mg, 2.60 mmol) . The mixture was stirred at 25 ℃ for 16 h. The mixture was dulited with H
2O (10 mL) and extracted with EtOAc (10 ml × 2) . The combined organic layer was dried over N
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/petroleum ether (with EtOAc from 0 to 50%) in 15 min to give tert-butyl (3- ( (6-bromo-5-fluoropyridin-2-yl) methoxy) cyclobutyl) carbamate (700 mg, 86%yield) as a yellow liquid. LC-MS: m/z [M+Na]
+ 397.0.
Step 3: tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) -5-fluoropyridin-2-yl) methoxy) cyclobutyl) carbamate
To a solution of tert-butyl N- [3- [ (6-bromo-5-fluoro-2-pyridyl) methoxy] cyclobutyl] carbamate (200 mg, 533 μmol) and (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (108 mg, 484 μmol) in 1, 4-dioxane (2.0 mL) was added Cs
2CO
3 (474 mg, 1.45 mmol) , Pd
2 (dba)
3 (88.7 mg, 96.9 μmol) and (5-diphenylphosphanyl-9, 9-dimethyl-xanthen-4-yl) -diphenyl-phosphane (112 mg, 194 μmol) . The mixture was heated to 100 ℃ and stirred at 100 ℃ under N
2 for 5 h. The mixture was cooled to room temperature and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/petroleum ether (with EtOAc from 0 to 70%) in 15 min to give tert-butyl N- [3- [ [6- [ [2-tert-butyl-5- [ (1R, 3S) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -5-fluoro-2-pyridyl] methoxy] cyclobutyl] carbamate (180 mg, 72%yield) as a yellow liquid. LC-MS: m/z [M+H]
+ 518.3.
Step 4: (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) cyclobutoxy) methyl) -3-fluoropyridin-2-
yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of tert-butyl N- [3- [ [6- [ [2-tert-butyl-5- [ (1R, 3S) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -5-fluoro-2-pyridyl] methoxy] cyclobutyl] carbamate (180 mg, 348 μmol) and di (1H-imidazol-1-yl) methanone (169 mg, 1.04 mmol) in DCM (10 mL) was added DIPEA (225 mg, 1.74 mmol) . The mixture was warmed to 40 ℃ and stirred at 40 ℃ for 5 h. The mixture was cooled to room temperature and concentrated under reduced pressure to get (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) cyclobutoxy) methyl) -3-fluoropyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (200 mg, 94%yield) as a yellow gum. LC-MS: m/z [M+H]
+ 612.3.
Step 5: (1R, 3S) -3- (5- ( (6- ( (3-aminocyclobutoxy) methyl) -3-fluoropyridin-2-yl) amino) -1- (tert-
butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
A mixture of [ (1R, 3S) -3- [5- [ [6- [ [3- (tert-butoxycarbonylamino) cyclobutoxy] methyl] -3-fluoro-2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (200 mg, 327 μmol) in DCM: TFA = 2: 1 (15 mL) was stirred at 25 ℃ for 1 h. The mixture was concentrated under reduced pressure to get [ (1R, 3S) -3- [5- [ [6- [ (3-aminocyclobutoxy) methyl] -3-fluoro-2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (150 mg, 90%yield) as a yellow gum. LC-MS: m/z [M+H]
+ 512.3.
Step 6: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
3-fluoro-2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-9-one
To a solution of [ (1R, 3S) -3- [5- [ [6- [ (3-aminocyclobutoxy) methyl] -3-fluoro-2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (150 mg, 293 μmol) in CH
3CN (20 mL) was added DIPEA (148 mg, 1.15 mmol) . The mixture was heated to 80 ℃ and stirred at 80 ℃ for 16 h. The mixture was cooled to room temperature and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/petroleum ether (with EtOAc from 0 to 50%) in 15 min to give (1
1S, 1
3R, 7
1R, 7
3S, Z) -2
1- (tert-butyl) -4
3-fluoro-2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-9-one (90.0 mg, 69%yield) as a white solid. LC-MS: m/z [M+H]
+ 444.2.
Step 7: (1
1S, 1
3R, 7
1R, 7
3S, Z) -4
3-fluoro-2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-
1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-9-one
A mixture of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
3-fluoro-2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-9-one (150 mg, 338 μmol) in FA (8.0 mL) was heated to 80 ℃ and stirred at 80 ℃ for 2 h. The mixture was cooled to room temperature and concentrated under reduced pressure. The residue was washed with CH
3CN (3.0 mL) and filtered to get (1
1S, 1
3R, 7
1R, 7
3S, Z) -4
3-fluoro-2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentana-7 (1, 3) -cyclobutanacyclodecaphan-9-one (68.0 mg, 52%yield) as a white solid. LC-MS: m/z [M+H]
+ 388.1.
Example 73
(1
1S, 1
3R, 7
1S, 7
3S, Z) -2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1, 7 (1, 3) -
dicyclopentanacyclodecaphan-9-one

Step 1: tert-butyl ( (1S, 3S) -3- ( (6-bromopyridin-2-yl) methoxy) cyclopentyl) carbamate
To a stirred suspension of tert-butyl ( (1S, 3S) -3-hydroxycyclopentyl) carbamate (401 mg, 1.99 mmol) in THF (5.0 mL) was added slowly NaH (159 mg, 60 wt. %in mineral oil, 3.99 mmol) at 0 ℃ under N
2. The mixture was stirred at 25 ℃ for 30 min before 2-bromo-6- (bromomethyl) pyridine (500 mg, 1.99 mmol) in THF (5.0 mL) was added to it. The reaction mixture was stirred at 25 ℃ under N
2 for 16 h. The mixture was quenched with ice-cold water (20 mL) and extracted with EtOAc (3 × 20 mL) . The organic phase was washed with brine (50 mL) and dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The crude product was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 30%in 15 min) to afford tert-butyl ( (1S, 3S) -3- ( (6-bromopyridin-2-yl) methoxy) cyclopentyl) carbamate (700 mg, 94%yield) as a yellow solid. LC-MS: m/z [M-55]
+ 314.8.
Step 2: tert-butyl ( (1S, 3S) -3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) methoxy) cyclopentyl) carbamate
To a stirred solution of tert-butyl ( (1S, 3S) -3- ( (6-bromopyridin-2-yl) methoxy) cyclopentyl) carbamate (400 mg, 1.08 mmol) and (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (241 mg, 1.08 mmol) in dioxane (8.0 mL) were sequentially added Pd
2 (dba)
3 (98.7 mg, 108 μmol) , XantPhos (125 mg, 215 μmol) and Cs
2CO
3 (1.05 g, 3.23 mmol) . The reaction mixture was warmed to 100 ℃ and stirred at that temperature for 12 h. The reaction mixture was cooled to 25 ℃ and filtered under reduced pressure. The filtrated was concentrated under reduced pressure and the residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 30 to 80 %in 20 min) to give tert-butyl ( (1S, 3S) -3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methoxy) cyclopentyl) carbamate (350 mg, 63%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 514.3
Step 3: (1R, 3S) -3- (5- ( (6- ( ( ( (1S, 3S) -3- ( (tert-
butoxycarbonyl) amino) cyclopentyl) oxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-
yl) cyclopentyl 1H-imidazole-1-carboxylate
To a suspension of tert-butyl ( (1S, 3S) -3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methoxy) cyclopentyl) carbamate (350 mg, 681 μmol) and DIPEA (475 μL, 352 mg, 2.73 mmol) in CH
2Cl
2 (10.0 mL) was added CDI (442 mg, 2.73 mmol) at 25 ℃. The mixture was heated to 40 ℃ and stirred at that temperature for 6 h. The reaction mixture was quenched with ice-cold water (20 mL) and extracted with CH
2Cl
2 (3 × 20 mL) . The organic phase was dried over anhydrous Na
2SO
4 and concentrated under reduced pressure. The crude product was used next step without further purification. LC-MS: m/z [M+H]
+ 608.4.
Step 4: (1R, 3S) -3- (5- ( (6- ( ( ( (1S, 3S) -3-aminocyclopentyl) oxy) methyl) pyridin-2-yl) amino) -1- (tert-
butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a stirred solution of crude (1R, 3S) -3- (5- ( (6- ( ( ( (1S, 3S) -3- ( (tert-butoxycarbonyl) amino) cyclopentyl) oxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate in CH
2Cl
2 (3.0 mL) was added TFA (1 mL, 1.48 g, 12.98 mmol) at 25 ℃. The mixture was stirred at that temperature for 1 h. The mixture was concentrated under reduced pressure. The crude product was used next step without further purification. LC-MS: m/z [M+H]
+ 508.3.
Step 5: (1
1S, 1
3R, 7
1S, 7
3S, Z) -2
1- (tert-butyl) -2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1, 7 (1, 3) -dicyclopentanacyclodecaphan-9-one
To a stirred solution of crude (1R, 3S) -3- (5- ( (6- ( ( ( (1S, 3S) -3-aminocyclopentyl) oxy) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate in CH
3CN (40.0 mL) was added DIPEA (1.17 mL, 865 mg, 6.70 mmol) at 25 ℃. The mixture was heated to 80 ℃ and stirred at that temperature for 16 h. The mixture was concentrated under reduced pressure and the residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 25 to 80 %in 20 min) to afford (1
1S, 1
3R, 7
1S, 7
3S, Z) -2
1- (tert-butyl) -2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1, 7 (1, 3) -dicyclopentanacyclodecaphan-9-one (100 mg, 33%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 440.3.
Step 6: (1
1S, 1
3R, 7
1S, 7
3S, Z) -2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1, 7 (1, 3) -
dicyclopentanacyclodecaphan-9-one
A solution of (1
1S, 1
3R, 7
1S, 7
3S, Z) -2
1- (tert-butyl) -2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1, 7 (1, 3) -dicyclopentanacyclodecaphan-9-one (100 mg, 227 μmol) in HCO
2H (2.0 mL, 2.44 g, 53.01 mmol) was heated to 90 ℃ and stirred at that temperature for 6 h. The mixture was concentrated under reduced pressure and the residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 10%to 30%in 30 min) to afford (1
1S, 1
3R, 7
1S, 7
3S, Z) -2
1H-6, 10-dioxa-3, 8-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1, 7 (1, 3) -dicyclopentanacyclodecaphan-9-one (60.7 mg, 69%yield) as a white solid. LC-MS: m/z [M+H]
+ 383.9.
The following compounds were prepared using the similar procedure disclosed in synthetic example 73.


Example 77
(1
1S, 1
3R, Z) -4
4- ( ( (R) -tetrahydrofuran-3-yl) oxy) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one


Step 1: (R) -2-chloro-6-methyl-4- ( (tetrahydrofuran-3-yl) oxy) pyridine
To a solution of (3R) -tetrahydrofuran-3-ol (2.00 g, 22.7 mmol) in DMF (15 mL) was added NaH (2.47 g, 61.7 mmol) at 0 ℃. Then the reaction mixture was stirred at 0 ℃ for 10 min. Then the solution of 2, 4-dichloro-6-methyl-pyridine (2.00 g, 12.3 mmol) in DMF (1.0 mL) was added dropwise to the reaction mixture. Then the reaction mixture was allowed warmed to 20 ℃ and stirred at 20 ℃for 2 h. The mixture was quenched with ice water (80 mL) . The mixture was extracted with EtOAc (100 mL) . The organic layer was dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure, and the residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 50%) in 15 min to afford 2-chloro-6-methyl-4- [ (3R) -tetrahydrofuran-3-yl] oxy-pyridine (1.80 g, 68%yield) as a pale yellow solid. LC-MS: (ESI) m/z [M+H]
+ 214.1.
Step 2: (R) -2- (bromomethyl) -6-chloro-4- ( (tetrahydrofuran-3-yl) oxy) pyridine
To a solution of 2-chloro-6-methyl-4- [ (3R) -tetrahydrofuran-3-yl] oxy-pyridine (500 mg, 2.34 mmol) in CCl
4 (15 mL) was added NBS (625 mg, 3.51 mmol, 298 μL) and AIBN (76.9 mg, 468 μmol) at 20 ℃. Then the reaction mixture was heated to 80 ℃ and stirred at 80 ℃ under the atmosphere of N
2 for 3 h. The mixture was used for next step without further purification. LC-MS: (ESI) m/z [M+H]
+292.0. 244.1.
Step 3: tert-butyl (R) - (3- ( (6-chloro-4- ( (tetrahydrofuran-3-yl) oxy) pyridin-2-
yl) methoxy) propyl) carbamate
To the solution of tert-butyl N- (3-hydroxypropyl) carbamate (620 mg, 3.54 mmol) in THF (8.0 mL) was added NaH (236 mg, 5.90 mmol) at 0 ℃. The reaction mixture was stirred at 0 ℃ for 30 min. Then the mixture was added to the suspension of 2- (bromomethyl) -6-chloro-4- [ (3R) -tetrahydrofuran-3-yl] oxy-pyridine (690 mg, 2.36 mmol) in CCl
4 (13 mL) at 20 ℃. The reaction mixture was stirred at 20 ℃ for 12 h. The mixture was diluted with DCM (150 mL) . The mixture was washed with brine (100 mL) , dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 50%) in 15 min to afford tert-butyl (R) - (3- ( (6-chloro-4- ( (tetrahydrofuran-3-yl) oxy) pyridin-2-yl) methoxy) propyl) carbamate (210 mg, 23%yield) as a yellow gum. LC-MS: (ESI) m/z [M+H]
+387.1.
Step 4: tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) -4- ( ( (R) -tetrahydrofuran-3-yl) oxy) pyridin-2-yl) methoxy) propyl) carbamate
To a solution of tert-butyl (R) - (3- ( (6-chloro-4- ( (tetrahydrofuran-3-yl) oxy) pyridin-2-yl) methoxy) propyl) carbamate (200 mg, 517 μmol) and (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (121 mg, 543 μmol) in dioxane (5.0 mL) was added Pd
2 (dba)
3 (71.0 mg, 77.6 μmol) , XantPhos (89.7 mg, 155 μmol) and Cs
2CO
3 (505 mg, 1.55 mmol) under the atmosphere of N
2. Then the reaction mixture was heated to 100 ℃ and stirred at 100 ℃ under the atmosphere of N
2 for 5 h. The mixture was concentrated under reduced pressure, and the residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 80%) in 15 min to afford tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -4- ( ( (R) -tetrahydrofuran-3-yl) oxy) pyridin-2-yl) methoxy) propyl) carbamate (240 mg, 81%yield) as a yellow solid. LC-MS: (ESI) m/z [M+H]
+ 574.3.
Step 5: (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) -4- ( ( (R) -
tetrahydrofuran-3-yl) oxy) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-
imidazole-1-carboxylate
To a solution of tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -4- ( ( (R) -tetrahydrofuran-3-yl) oxy) pyridin-2-yl) methoxy) propyl) carbamate (180 mg, 313 μmol) in dioxane (8.0 mL) was added DIPEA (324 mg, 437 μL) at 20 ℃, then di (imidazol-1-yl) methanone (153 mg, 941 μmol) was added to the reaction mixture. The mixture was heated to 80 ℃ and stirred at 80 ℃ for 8 h. The mixture was concentrated under reduced pressure to afford (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) -4- ( ( (R) -tetrahydrofuran-3-yl) oxy) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (210 mg, crude) as a yellow gum. LC-MS: (ESI) m/z [M+H]
+ 668.3.
Step 6: (1R, 3S) -3- (5- ( (6- ( (3-aminopropoxy) methyl) -4- ( ( (R) -tetrahydrofuran-3-yl) oxy) pyridin-2-
yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propoxy) methyl) -4- ( ( (R) -tetrahydrofuran-3-yl) oxy) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (210 mg, 314 μmol) in DCM (3.0 mL) was added TFA (2.22 g, 19.5 mmol, 1.50 mL) at 20 ℃. Then the reaction mixture was stirred at 20 ℃ for 3 h. The mixture was concentrated under reduced pressure to afford (1R, 3S) -3- (5- ( (6- ( (3-aminopropoxy) methyl) -4- ( ( (R) -tetrahydrofuran-3-yl) oxy) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H- imidazole-1-carboxylate (220 mg, crude, TF) as a yellow gum, which was used in next step without further purification. LC-MS: (ESI) m/z [M+H]
+ 568.3.
Step 7: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- ( ( (R) -tetrahydrofuran-3-yl) oxy) -2
1H-6, 12-dioxa-3, 10-diaza-
4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
The solution of (1R, 3S) -3- (5- ( (6- ( (3-aminopropoxy) methyl) -4- ( ( (R) -tetrahydrofuran-3-yl) oxy) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (220 mg, 387 μmol) in CH
3CN (20 mL) was added DIPEA (2.23 g, 17.2 mmol, 3.00 mL) . Then the reaction mixture was heated to 80 ℃ and stirred at 80 ℃ for 13 h. The mixture was concentrated under reduced pressure, and the residue was diluted with EtOAc (100 mL) , the mixture was washed brine (80 mL) . The organic layer was dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure, the residue was purified silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 100%) in 20 min to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- ( ( (R) -tetrahydrofuran-3-yl) oxy) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (50.0 mg, 26%yield) as a yellow gum. LC-MS: (ESI) m/z [M+H]
+ 500.2.
Step 8: (1
1S, 1
3R, Z) -4
4- ( ( (R) -tetrahydrofuran-3-yl) oxy) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -
pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
The solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- ( ( (R) -tetrahydrofuran-3-yl) oxy) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (35.0 mg, 70.1 μmol) in FA (1.0 mL) and dioxane (1.0 mL) was stirred at 90 ℃ for 5 h. The mixture was concentrated under reduced pressure, and the residue was sent to purified by Prep-HPLC (with CH
3CN from 30%to 60%in 9 min) to afford the product (1
1S, 1
3R, Z) -4
4- ( ( (R) -tetrahydrofuran-3-yl) oxy) -2
1H-6, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (6.00 mg, 19%yield) as a white solid. LC-MS: (ESI) m/z [M+H]
+ 444.3.
Example 78
(1
1R, 1
3S, Z) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one

Step 1: tert-butyl (3- ( ( (6-bromopyridin-2-yl) methyl) amino) propyl) carbamate
To a stirred solution of tert-butyl (3-aminopropyl) carbamate (2.00 g, 2.00 mL , 11.5 mmol) in CH
2Cl
2 (20.0 mL) were sequentially added 6-bromopyridine-2-carbaldehyde (2.14 g, 11.5 mmol) and HOAc (0.5 mL) at 25 ℃. The resulting mixture was stirred at that temperature for 2 h before NaBH (OAc)
3 (2.92 g, 13.8 mmol) was added at 25 ℃. The reaction mixture was stirred at 25 ℃ for 16 h before it was quenched with water (100 mL) and extracted with EtOAc (100 mL × 2) . The combined organic phases were dried over Na
2SO
4 and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 50%in 25 min) to afford tert-butyl (3- ( ( (6-bromopyridin-2-yl) methyl) amino) propyl) carbamate (2.00 g, 54%yield) as a colorless oil. LC-MS: m/z [M+H]
+ 344.0.
Step 2: benzyl ( (6-bromopyridin-2-yl) methyl) (3- ( (tert-butoxycarbonyl) amino) propyl) carbamate
To a stirred solution of tert-butyl (3- ( ( (6-bromopyridin-2-yl) methyl) amino) propyl) carbamate (1.00 g, 2.90 mmol) in THF (10.0 mL) and H
2O (2.0 mL) were sequentially added Na
2CO
3 (308 mg, 2.90 mmol) and CbzCl (496 mg, 2.90 mmol) at 25℃. The resulting mixture was stirred at that temperature for 16 h before it was quenched with water (100 mL) and extracted with EtOAc (100 mL × 2) . The combined organic phases were dried over Na
2SO
4 and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 30%in 25 min) to afford benzyl ( (6-bromopyridin-2-yl) methyl) (3- ( (tert-butoxycarbonyl) amino) propyl) carbamate (0.65 g, 47%yield) as a colorless oil. LC-MS: m/z [M+H]
+477.6.
Step 3: benzyl (3- ( (tert-butoxycarbonyl) amino) propyl) ( (6- ( (1- (tert-butyl) -3- ( (1R, 3S) -3-
hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methyl) carbamate
To a stirred solution of benzyl ( (6-bromopyridin-2-yl) methyl) (3- ( (tert-butoxycarbonyl) amino) propyl) carbamate (600 mg, 1.25 mmol) in 1, 4-Dioxane (10.0 mL) were sequentially added XantPhos (72.3 mg, 0.125 mmol) , Pd
2 (dba)
3 (115 mg, 0.125 mmol) , Cs
2CO
3 (815 mg, 2.50 mmol) and (1S, 3R) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (279 mg, 1.25 mmol) at 25 ℃. The reaction mixture was warmed to 100 ℃ and stirred at that temperature for 16 h before it was cooled to 25 ℃ and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with MeOH/DCM (with MeOH from 0 to 10%in 25 min) to give benzyl (3- ( (tert-butoxycarbonyl) amino) propyl) ( (6- ( (1- (tert-butyl) -3- ( (1R, 3S) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methyl) carbamate (550 mg, 71%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 620.7
Step 4: (1S, 3R) -3- (5- ( (6- ( ( ( (benzyloxy) carbonyl) (3- ( (tert-
butoxycarbonyl) amino) propyl) amino) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-
yl) cyclopentyl 1H-imidazole-1-carboxylate
To a stirred suspension of benzyl (3- ( (tert-butoxycarbonyl) amino) propyl) ( (6- ( (1- (tert-butyl) -3- ( (1R, 3S) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methyl) carbamate (500 mg, 0.805 mmol) in CH
2Cl
2 (10.0 mL) were sequentially added CDI (580 mg, 4.03 mmol) and Et
3N (560 μL, 408 mg, 4.03 mmol) at 25 ℃. The reaction mixture was warmed to 40 ℃ and stirred at that temperature for 16 h before it was cooled to 25 ℃. The reaction mixture was concentrated under reduced pressure to afford (1S, 3R) -3- (5- ( (6- ( ( ( (benzyloxy) carbonyl) (3- ( (tert-butoxycarbonyl) amino) propyl) amino) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (550 mg, 96%yield) as a yellow oil which was directly used for the next step without further purification. LC-MS: m/z [M+H]
+ 715.3.
Step 5: (1S, 3R) -3- (5- ( (6- ( ( (3-aminopropyl) ( (benzyloxy) carbonyl) amino) methyl) pyridin-2-
yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a stirred solution of (1S, 3R) -3- (5- ( (6- ( ( ( (benzyloxy) carbonyl) (3- ( (tert-butoxycarbonyl) amino) propyl) amino) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (550 mg, 0.770 mmol) in CH
2Cl
2 (10.0 mL) was added TFA (10.0 mL) at 25 ℃. The resulting mixture was stirred at that temperature for 12 h. The mixture was concentrated under reduced pressure to give (1S, 3R) -3- (5- ( (6- ( ( (3-aminopropyl) ( (benzyloxy) carbonyl) amino) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (450 mg, crude) as a colorless oil. LC-MS: m/z [M+H]
+615.3.
Step 6: (1
1R, 1
3S, Z) -2
1- (tert-butyl) -11-oxo-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-6-carboxylate
To a stirred solution of (1S, 3R) -3- (5- ( (6- ( ( (3-aminopropyl) ( (benzyloxy) carbonyl) amino) methyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (400 mg, 0.651 mmol) in CH
3CN (5.0 mL) was added Et
3N (0.45 mL, 330 mg, 3.25 mmol) at 25 ℃. The reaction mixture was warmed to 70 ℃ and stirred at that temperature for 2 h. The mixture was cooled to 25 ℃ and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0%to 80%in 30 min) to give (1
1R, 1
3S, Z) -21- (tert-butyl) -11-oxo-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-6-carboxylate (200 mg, 59%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 546.7.
Step 7: (1
1R, 1
3S, Z) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one
A stirred solution of (1
1R, 1
3S, Z) -21- (tert-butyl) -11-oxo-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-6-carboxylate (50.0 mg, 91.0 μmol) in TFA (5.0 mL) at 25 ℃ was warmed to 70 ℃ and stirred at that temperature for 2 h before it was cooled to 25 ℃. The reaction mixture was concentrated under reduced pressure. The residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 30%to 70%in 40 min) to give (1
1R, 1
3S, Z) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (2.0 mg, 6%yield) as a white solid. LC-MS: m/z [M+H]
+357.2.
Example 79
(1
1R, 1
3S, Z) -6- (2, 2, 2-trifluoroethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphan-11-one

Step 1: (1
1R, 1
3S, Z) -2
1- (tert-butyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a stirred solution of benzyl (1
1R, 1
3S, Z) -2
1- (tert-butyl) -11-oxo-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-6-carboxylate (200 mg, 265 μmol) in HOAc (3.0 mL) was added HBr (gas) in AcOH (5.0 mL) at 25 ℃. The resulting mixture was stirred at that temperature for 4 h before it was quenched with water (100 mL) and extracted with EtOAc (100 mL × 2) . The combined organic phases were dried over Na
2SO
4 and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 50%in 25 min) to afford (1
1R, 1
3S, Z) -2
1- (tert-butyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (100 mg, 63%yield) as a colorless oil. LC-MS: m/z [M+H]
+ 413.2.
Step 2: (1
1R, 1
3S, Z) -2
1- (tert-butyl) -6- (2, 2, 2-trifluoroethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -
pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a stirred solution of (1
1R, 1
3S, Z) -2
1- (tert-butyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (100 mg, 242 μmol) in CH
3CN (5.0 mL) were sequentially added 2, 2, 2-trifluoroethyl trifluoromethanesulfonate (84.0 mg, 363 μmol) and K
2CO
3 (100 mg, 727 μmol) at 25℃. The reaction mixture was warmed to 80 ℃ and stirred at that temperature for 16 h before it was cooled to 25 ℃ and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 30%in 25 min) to afford (1
1R, 1
3S, Z) -2
1- (tert-butyl) -6- (2, 2, 2-trifluoroethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (30.0 mg, 23%yield) as a colorless oil. LC-MS: m/z [M+H]
+ 495.3.
Step 3: (1
1R, 1
3S, Z) -6- (2, 2, 2-trifluoroethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
A stirred solution of (1
1R, 1
3S, Z) -21- (tert-butyl) -6- (2, 2, 2-trifluoroethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (30.0 mg, 60.0 μmol) in TFA (8.0 mL) was warmed to 50 ℃ and stirred at that temperature for 16 h. The mixture was cooled to 25 ℃ before it was concentrated under reduced pressure. The residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 30%to 70%in 40 min) to give (1
1R, 1
3S, Z) -6- (2, 2, 2-trifluoroethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (7.2 mg, 27%yield) as a white solid. LC-MS: m/z [M+H]
+439.2.
Example 80
(1
1R, 1
3S, Z) -6-isopropyl-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one

Step 1: (1
1R, 1
3S, Z) -2
1- (tert-butyl) -6-isopropyl-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a solution of (1
1R, 1
3S, Z) -21- (tert-butyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (60 mg, 0.145 mmol) in acetone (0.2 mL) and MeOH (8 mL) was added AcOH (3 drops) . The reaction solution was stirred at 25 ℃ for 2 hours and NaBH
3CN (27 mg, 0.435 mmol) was added. The resulting solution was stirred at 25 ℃ for 16 hours. Then it was concentrated in vacuo and the residue was purified by flash column chromatography eluting with MeOH/CH
2Cl
2 from 0 to 12%in 15 min to afford (1
1R, 1
3S, Z) -2
1- (tert-butyl) -6-isopropyl-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (45 mg, 68%yield) as a white solid. LC-MS: m/z 455.3 [M+H]
+.
Step 2: (1
1R, 1
3S, Z) -6-isopropyl-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one
A solution of (1
1R, 1
3S, Z) -2
1- (tert-butyl) -6-isopropyl-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (45 mg, 0.099 mmol) in FA (6 mL) was stirred at 80℃ for 12 hours. The reaction solution was concentrated in vacuo and the residue was purified by prep-HPLC (C18, 10-30%MeCN in 0.1%FA/water) to afford (1
1R, 1
3S, Z) -6-isopropyl-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (16.3 mg, 41%yield) as a white solid. LC-MS: m/z 399.2 [M+H]
+.
The following compounds were prepared using the similar procedure disclosed in synthetic example 80.

Example 83
(1
1R, 1
3S, Z) -6-acetyl-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one

Step 1: (1
1R, 1
3S, Z) -6-acetyl-2
1- (tert-butyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a suspension of (1
1R, 1
3S, Z) -2
1- (tert-butyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (60 mg, 145 μmol) and Ac
2O (22 mg, 218 μmol, 20 μL) in DCM (1 mL) was added slowly TEA (74 mg, 727 μmol, 101 μL) at 0 ℃ and then it was warmed up to room temperature and stirred for 1 hour under N
2. After completion of the reaction as judged by LCMS, reaction mixture was quenched with ice-cold water (20 mL) and extracted with EtOAc (3 × 20 mL) . The combined organic phase was washed with brine (50 mL) and dried over anhydrous Na
2SO
4, filtered and concentrated in vacuo. The crude product was purified by flash chromatography (SiO
2, DCM/MeOH: 12: 1) to afford (1
1R, 1
3S, Z) -6-acetyl-2
1- (tert-butyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (35 mg, 77 μmol, 53%yield) as a colorless liquid. LC-MS: m/z 455.2 [M+H]
+ .
Step 2: (1
1R, 1
3S, Z) -6-acetyl-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one
A mixture of (1
1R, 1
3S, Z) -6-acetyl-2
1- (tert-butyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (35 mg, 80 μmol) in TFA (3 mL) was stirred for 16 hours at 70 ℃ under N
2, until the reaction was complete as indicated by LCMS, the reaction mixture was concentrated in vacuo, purified by column chromatography followed by Prep-HPLC purification 2-35%MeCN in H
2O (0.1%FA) to give the desired product (1
1R, 1
3S, Z) -6-acetyl-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (5.5 mg, 14 μmol, 17%yield) as a white solid. LC-MS: m/z 399.2 [M+H]
+.
Example 84
(1
1S, 1
3R, Z) -6-methyl-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one

Step 1: tert-butyl (3- ( ( (6-bromopyridin-2-yl) methyl) (methyl) amino) propyl) carbamate
To a solution of 6-bromopicolinaldehyde (1.0 g, 5.38 mmol) in THF (10 mL) was added tert-butyl (3- (methylamino) propyl) carbamate (1.1 g, 5.64 mmol) at 25 ℃ under nitrogen. The reaction mixture was stirred at 25 ℃ for 1 h and then a solution of NaBH (OAc)
3 (1.2 g, 5.64 mmol) in DMSO (10 mL) was added. The reaction mixture was stirred at 25 ℃ for 2.5 h. The mixture was quenched with H
2O (20 mL) and extracted with EtOAc (30 mL × 3) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrated. The residue was purified by flash chromatography on silica gel eluting with 0-30%EtOAc in PE to afford tert-butyl (3- ( ( (6-bromopyridin-2-yl) methyl) (methyl) amino) propyl) carbamate (1.7 g, 88%yield) as colorless oil. LC-MS: m/z 358.1 [M+H]
+.
Step 2: tert-butyl (3- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) methyl) (methyl) amino) propyl) carbamate
To a mixture of tert-butyl (3- ( ( (6-bromopyridin-2-yl) methyl) (methyl) amino) propyl) carbamate (300 mg, 837 μmol) , (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (187 mg, 837 μmol) and Cs
2CO
3 (818 mg, 2.5 mmol) in dioxane (5 mL) was added XantPhos (97 mg, 167 μmol) and Pd
2 (dba)
3 (153 mg, 167 μmol) under nitrogen. The reaction was stirred at 90 ℃ under nitrogen for 16 h. The mixture was diluted with water (50 mL) and extracted with EtOAc (30 mL × 3) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrated. The residue was purified by flash chromatography on silica gel eluting with 0-9%MeOH in DCM to afford tert-butyl (3- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methyl) (methyl) amino) propyl) carbamate (370 mg, 88%yield) as yellow oil. LC-MS: m/z 501.3 [M+H]
+.
Step 3: tert-butyl (3- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-
nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-
yl) methyl) (methyl) amino) propyl) carbamate
A mixture of tert-butyl (3- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methyl) (methyl) amino) propyl) carbamate (100 mg, 200 μmol) , 4-nitrophenyl carbonochloridate (121 mg, 600 μmol) , Et
3N (101 mg, 1.0 mmol) and DMAP (5 mg, 40 μmol) in DCE (5 mL) was stirred at 70 ℃ for 16 h. The mixture was diluted with water (50 mL) and extracted with DCM (30 mL × 3) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrated. The residue was purified by flash chromatography on silica gel eluting with 0-90%EtOAc in PE to afford tert-butyl (3- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methyl) (methyl) amino) propyl) carbamate (106 mg, 80%yield) as yellow oil. LC-MS: m/z 666.2 [M+H]
+.
Step 4: (1R, 3S) -3- (3- ( (6- ( ( (3-aminopropyl) (methyl) amino) methyl) pyridin-2-yl) amino) -1H-
pyrazol-5-yl) cyclopentyl (4-nitrophenyl) carbonate
A solution of tert-butyl (3- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methyl) (methyl) amino) propyl) carbamate (106 mg, 159 μmol) in TFA (5 mL) was stirred at 70 ℃for 2 h. The solvent was removed under reduced pressure to afford (1R, 3S) -3- (3- ( (6- ( ( (3-aminopropyl) (methyl) amino) methyl) pyridin-2-yl) amino) -1H-pyrazol-5-yl) cyclopentyl (4-nitrophenyl) carbonate (Crude 80 mg) as yellow oil, which was used in the next step directly. LC-MS: m/z 510.2 [M+H]
+.
Step 5: (1
1S, 1
3R, Z) -6-methyl-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one
To a mixture of (1R, 3S) -3- (3- ( (6- ( ( (3-aminopropyl) (methyl) amino) methyl) pyridin-2-yl) amino) -1H-pyrazol-5-yl) cyclopentyl (4-nitrophenyl) carbonate (80 mg, 157 μmol) in CH
3CN (10 mL) was added Et
3N (364 mg, 3.60 mmol) at 25 ℃. The reaction mixture was stirred at 25 ℃ for 1 h. The solvent was evaporated and the residue was purified by prep-HPLC (C18, 13%to 23%CH
3CN in 0.1%TFA/water, 20 mL/min) to afford (1
1S, 1
3R, Z) -6-methyl-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (3.0 mg, 5%yield) as colorless gum. LC-MS: m/z 371.1 [M+H]
+.
The following compounds were prepared using the similar procedure disclosed in synthetic example 84.

Example 85
(1
1S, 1
3R, Z) -6-methyl-44- (trifluoromethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one

Step 1: 2-bromo-6- (bromomethyl) -4- (trifluoromethyl) pyridine
To a suspension of 2-bromo-6-methyl-4- (trifluoromethyl) pyridine (1 g, 4.2 mmol) and NBS (822 mg, 4.62 mmol) in CCl
4 (30 mL) was added AIBN (689 mg, 4.2 mmol) . The reaction mixture was stirred at 80 ℃ for 4 hours. After completion of the reaction as judged by LCMS. The reaction solution was concentrated in vacuo. The residue was purified by flash column chromatography eluting with ethyl acetate/petroleum ether from 0 to 10%in 15 min to afford 2-bromo-6- (bromomethyl) -4- (trifluoromethyl) pyridine (360 mg, 42%yield) as a light red oil.
Step 2: tert-butyl (3- ( ( (6-bromo-4- (trifluoromethyl) pyridin-2-
yl) methyl) (methyl) amino) propyl) carbamate
To a suspension of 2-bromo-6- (bromomethyl) -4- (trifluoromethyl) pyridine (360 mg, 1.13 mmol) and tert-butyl (3- (methylamino) propyl) carbamate (212 mg, 1.13 mmol) in CH
3CN (20 mL) was added K
2CO
3 (312 mg, 2.26 mmol) . The reaction mixture was stirred at 80 ℃ for 1 hour. After completion of the reaction as judged by LCMS. The reaction solution was concentrated in vacuo. The residue was purified by flash column chromatography eluting with ethyl acetate/petroleum ether from 0 to 10%in 15 min to afford tert-butyl (3- ( ( (6-bromo-4- (trifluoromethyl) pyridin-2-yl) methyl) (methyl) amino) propyl) carbamate (350 mg, 73%yield) as a light yellow solid. LC-MS: m/z 425.7 [M+H]
+.
Step 3: tert-butyl (3- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) -4- (trifluoromethyl) pyridin-2-yl) methyl) (methyl) amino) propyl) carbamate
To a suspension of tert-butyl (3- ( ( (6-bromo-4- (trifluoromethyl) pyridin-2-yl) methyl) (methyl) amino) propyl) carbamate (350 mg, 0.82 mmol) , (1R, 3S) -3- (3-amino-1- (tert-butyl) -1H-pyrazol-5-yl) cyclopentan-1-ol (183 mg, 0.82 mmol) in toluene (20 mL) was added Pd
2 (dba)
3 (75 mg, 0.082 mmol) , XantPhos (95 mg, 0.164 mmol) and Cs
2CO
3 (535 mg, 1.64 mmol) . The reaction mixture was stirred at 80 ℃ for 16 hours under N
2. Then it was concentrated in vacuo and the residue was purified by flash column chromatography eluting with ethyl acetate/petroleum ether from 0 to 80%in 25 min to afford tert-butyl (3- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -4- (trifluoromethyl) pyridin-2-yl) methyl) (methyl) amino) propyl) carbamate (200 mg, 43%yield) as a red oil. LC-MS: m/z 568.9 [M+H]
+.
Step 4: (1R, 3S) -3- (5- ( (6- ( ( (3- ( (tert-butoxycarbonyl) amino) propyl) (methyl) amino) methyl) -4-
(trifluoromethyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-
carboxylate
To a suspension of tert-butyl (3- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -4- (trifluoromethyl) pyridin-2-yl) methyl) (methyl) amino) propyl) carbamate (200 mg, 0.35 mmol) and CDI (57 mg, 0.35 mmol) in DCM (15 mL) was added TEA (139 mg, 1.38 mmol) and the reaction mixture was stirred at 35 ℃ for 2 hours. The reaction solution was concentrated in vacuo to afford (1R, 3S) -3- (5- ( (6- ( ( (3- ( (tert-butoxycarbonyl) amino) propyl) (methyl) amino) methyl) -4- (trifluoromethyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (400 mg, crude) as a light red solid. LC-MS: m/z 662.8 [M+H]
+.
Step 5: (1R, 3S) -3- (5- ( (6- ( ( (3-aminopropyl) (methyl) amino) methyl) -4- (trifluoromethyl) pyridin-2-
yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
A mixture of (1R, 3S) -3- (5- ( (6- ( ( (3- ( (tert-butoxycarbonyl) amino) propyl) (methyl) amino) methyl) -4- (trifluoromethyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (400 mg, crude) in TFA/DCM (3 mL/9 mL) was stirred at 25℃ for 1 hour. The reaction solution was concentrated in vacuo to give (1R, 3S) -3- (5- ( (6- ( ( (3-aminopropyl) (methyl) amino) methyl) -4- (trifluoromethyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (500 mg, crude) as a red oil, which was used in the next step without further purification. LC-MS: m/z 562.9 [M+H]
+.
Step 6: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -6-methyl-4
4- (trifluoromethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -
pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a solution of (1R, 3S) -3- (5- ( (6- ( ( (3-aminopropyl) (methyl) amino) methyl) -4- (trifluoromethyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (500 mg, crude) in CH
3CN (30 mL) was added TEA (270 mg, 2.67 mmol) . The reaction mixture was stirred at 50 ℃ for 20 hours. Then it was concentrated in vacuo and the residue was purified by flash column chromatography eluting with MeOH/CH
2Cl
2 from 0 to 5%in 10 min to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -6-methyl-4
4- (trifluoromethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (250 mg, purity: 70%) as a white solid. LC-MS: m/z 494.8 [M+H]
+.
Step 7: (1
1S, 1
3R, Z) -6-methyl-44- (trifluoromethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-
2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
A mixture of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -6-methyl-4
4- (trifluoromethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (250 mg, purity: 70%) in FA (10 mL) was stirred at 80℃ for 6 hours. The reaction solution was concentrated in vacuo to give the residue, which was purified by prep-HPLC (10-40%MeCN in 0.1%FA/water) to afford (1
1S, 1
3R, Z) -6-methyl-4
4- (trifluoromethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (13 mg, 8%yield) as a white solid. LC-MS: m/z 438.8 [M+H]
+.
Example 86 and 87
(1
1S, 1
3R, 5S, Z) -5- (trifluoromethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphan-11-one and (1
1S, 1
3R, 5R, Z) -5- (trifluoromethyl) -2
1H-12-oxa-
3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one


Step 1: tert-butyl N- [3- [ (Z) - [1- (6-bromo-2-pyridyl) -2, 2, 2-trifluoro-
ethylidene] amino] propyl] carbamate
To a suspension of 1- (6-bromo-2-pyridyl) -2, 2, 2-trifluoro-ethanone (500 mg, 1.97 mmol) in toluene (10.0 mL) was slowly added tert-butyl N- (3-aminopropyl) carbamate (342 mg, 1.97 mmol) and Ti(O-i-Pr)
4 (615 mg, 2.17 mmol) at room temperature. The reaction was stirred for 3 h at 80 ℃, before it was quenched with ice-cold water (20 mL) and extracted with CH
2Cl
2 (3 × 20 mL) . The organic phase was concentrated under reduced pressure to give the crude product tert-butyl N- [3- [ (Z) - [1- (6-bromo-2-pyridyl) -2, 2, 2-trifluoro-ethylidene] amino] propyl] carbamate, which was used in the next step without further purification. LC-MS: m/z [M-100]
+ 309.8.
Step 2: tert-butyl N- [3- [ [1- (6-bromo-2-pyridyl) -2, 2, 2-trifluoro-ethyl] amino] propyl] carbamate
To a suspension of tert-butyl N- [3- [ (Z) - [1- (6-bromo-2-pyridyl) -2, 2, 2-trifluoro-ethylidene] amino] propyl] carbamate in MeOH (15.0 mL) was added slowly NaBH
4 (148 mg, 3.90 mmol) and NiCl
2
.6H
2O (695 mg, 2.93 mmol) at 0 ℃ and stirred for 2 h at 25 ℃. After completion of the reaction as judged by LCMS, reaction mixture was quenched with ice-cold water (20 mL) and extracted with EtOAc (3 × 20 mL) . The organic phase was concentrated under reduced pressure. The crude product was purified by flash chromatography eluting with EtOAc/PE (with EtOAc from 0 to 30%in 30 min) to afford tert-butyl N- [3- [ [1- (6-bromo-2-pyridyl) -2, 2, 2-trifluoro-ethyl] amino] propyl] carbamate (400 mg) as a yellow oil. LC-MS: m/z [M+H]
+ 411.9.
Step 3: tert-butyl N- [3- [ [1- [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-
yl] amino] -2-pyridyl] -2, 2, 2-trifluoro-ethyl] amino] propyl] carbamate
To a suspension of tert-butyl N- [3- [ [1- (6-bromo-2-pyridyl) -2, 2, 2-trifluoro-ethyl] amino] propyl] carbamate (200 mg, 0.485 mmol) in 1, 4-dioxane (10.0 mL) was sequentially added (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (108 mg, 0.485 mmol) , Cs
2CO
3 (474 mg, 1.46 mmol) , XantPhos (56.0 mg, 0.097 mmol) and Pd
2 (dba)
3 (44.4 mg, 0.048 mmol) at room temperature. The reaction was stirred at 100 ℃ for 16 h under N
2, before it was filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography eluting with EtOAc/PE (with EtOAc from 0 to 60%in 30 min) to afford tert-butyl N- [3- [ [1- [6- [ [2-tert-butyl- 5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -2-pyridyl] -2, 2, 2-trifluoro-ethyl] amino] propyl] carbamate (200 mg, 74%yield) as a yellow solid. LC-MS: m/z [M+H]
+ 555.0.
Step 4: [ (1R, 3S) -3- [5- [ [6- [1- [3- (tert-butoxycarbonylamino) propylamino] -2, 2, 2-trifluoro-ethyl] -2-
pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate
To a suspension of tert-butyl N- [3- [ [1- [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -2-pyridyl] -2, 2, 2-trifluoro-ethyl] amino] propyl] carbamate (200 mg, 0.360 mmol) in CH
2Cl
2 (15.0 mL) was added CDI (175 mg, 1.08 mmol) and DIPEA (313 μL, 232 mg, 1.80 mmol) at 35 ℃and stirred for 2 h. After completion of the reaction as judged by LCMS, reaction mixture was quenched with ice-cold water (20 mL) and extracted with EtOAc (3 × 20 mL) . The organic phase was washed with brine (50 mL) and dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure to afford [ (1R, 3S) -3- [5- [ [6- [1- [3- (tert-butoxycarbonylamino) propylamino] -2, 2, 2-trifluoro-ethyl] -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate as a yellow oil. The crude product was used in the next step without further purification. LC-MS: m/z [M+H]
+ 648.9.
Step 5: [ (1R, 3S) -3- [5- [ [6- [1- (3-aminopropylamino) -2, 2, 2-trifluoro-ethyl] -2-pyridyl] amino] -1-tert-
butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate
To a suspension of [ (1R, 3S) -3- [5- [ [6- [1- [3- (tert-butoxycarbonylamino) propylamino] -2, 2, 2-trifluoro-ethyl] -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (crude) in CH
2Cl
2 (3.0 mL) was added slowly TFA (1.0 mL) at 25 ℃. The reaction was stirred for 1 h, before it was concentrated under reduced pressure to afford [ (1R, 3S) -3- [5- [ [6- [1- (3-aminopropylamino) -2, 2, 2-trifluoro-ethyl] -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate as a yellow oil. The crude product was used in the next step without further purification. LC-MS: m/z [M+H]
+ 548.9.
Step 6: (1
1S, 1
3R, 5S, Z) -2
1- (tert-butyl) -5- (trifluoromethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -
pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one and (1
1S, 1
3R, 5R, Z) -2
1-
(tert-butyl) -5- (trifluoromethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one
To a suspension of [ (1R, 3S) -3- [5- [ [6- [1- (3-aminopropylamino) -2, 2, 2-trifluoro-ethyl] -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (crude) in CH
3CN (3.0 mL) was added Et
3N (379 μL, 276 mg, 2.73 mmol) and the reaction was stirred at 80 ℃ for 16 h. After completion of the reaction as judged by LCMS, reaction mixture was concentrated under reduced pressure. The crude product was purified by flash column chromatography eluting with MeOH/CH
2Cl
2 (with MeOH from 0 to 10%in 20 min) to afford the mixture of (1
1S, 1
3R, 5S, Z) -2
1- (tert-butyl) -5- (trifluoromethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one and (1
1S, 1
3R, 5R, Z) -2
1- (tert-butyl) -5- (trifluoromethyl) -2
1H-12- oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (130 mg) as a yellow solid. LC-MS: m/z [M+H]
+ 480.9.
Step 7: (1
1S, 1
3R, 5S, Z) -5- (trifluoromethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one and (1
1S, 1
3R, 5R, Z) -5- (trifluoromethyl) -
2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-
11-one
A mixture of (1
1S, 1
3R, 5S, Z) -2
1- (tert-butyl) -5- (trifluoromethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one and (1
1S, 1
3R, 5R, Z) -2
1- (tert-butyl) -5- (trifluoromethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (130 mg) in TFA (3.0 mL) was stirred for 24 h at 100 ℃, until the reaction was complete as indicated by LCMS, the reaction mixture was concentrated under reduced pressure, purified by prep-HPLC eluting with CH
3CN in water (with CH
3CN from 15%to 40%in 30 min) to give (1
1S, 1
3R, 5S, Z) -5- (trifluoromethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (24.6 mg, retention time = 8.5 min, absolute configuration arbitrarily assigned) , LC-MS: m/z [M+H]
+ 424.9, and (1
1S, 1
3R, 5R, Z) -5- (trifluoromethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (22.6 mg, retention time = 10.9 min, absolute configuration arbitrarily assigned) as a white solid. LC-MS: m/z [M+H]
+ 424.9.
Example 88
(1
1S, 1
3R, Z) -2
1H-12-oxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one

Step 1: 5- (6-bromopyridin-2-yl) pentanenitrile
To a round-bottom flask containing zinc (2.98 g, 45.6 mmol) , 1, 2-dibromoethane (327 μL, 714 mg, 3.80 mmol) was added, and the resulting mixture was warmed to 60 ℃ and then allowed to cool for 1 min. This heating/cooling process was repeated three more times, and then the flask was allowed to cool for an additional 3 min. TMSCl (119 μL, 102 mg, 937 μmol) in THF (20.0 mL) was added. The resulting mixture was warmed to 60 ℃, and a solution of 5-iodopentanenitrile (3.18 g, 15.2 mmol) in THF (2.5 mL) was added. The mixture was stirred at 60 ℃ for 1 h. The resulting solution of alkylzinc iodide was transferred to a second flask charged with 2, 6-dibromopyridine (1.20 g, 5.07 mmol) and Pd (PPh
3)
2Cl
2 (178 mg, 253 μmol) . The resulting mixture was stirred at 60 ℃ under N
2 for 18 h, cooled to 25 ℃, and the reaction quenched with saturated aqueous NH
4Cl solution (5 mL) . The resulting mixture was stirred at 25 ℃ for 20 min and diluted with EtOAc (100 mL) . The organic phase was washed with saturated aqueous NH
4Cl (50 mL) , brine (50 mL) , and dried over Na
2SO
4. The organic phase was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 40 %in 20 min) to afford 5- (6-bromopyridin-2-yl) pentanenitrile (390 mg, 32%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 239.1.
Step 2: 5- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-
yl) pentanenitrile
To a mixture of 5- (6-bromopyridin-2-yl) pentanenitrile (390 mg, 1.63 mmol) and (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (219 mg, 978 μmol) in 1, 4-dioxane (5.0 mL) were sequentially added Pd
2 (dba)
3 (149 mg, 160 μmol) , XantPhos (189 mg, 326 μmol) and Cs
2CO
3 (1.06 g, 3.26 mmol) . The mixture was heated to 100 ℃ and stirred at that temperature for 3 h. The reaction mixture was filtered and the combined organics were concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 15 to 70 %in 15 min) to afford 5- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) pentanenitrile (290 mg, 46%yield) as yellow solid. LC-MS: m/z [M+H]
+382.3.
Step 3: tert-butyl (5- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) pentyl) carbamate
A mixture of 5- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) pentanenitrile (290 mg, 760 μmol) and NiCl (9.85 mg, 76.0 μmol) in MeOH (5.0 mL) was cooled down to 0 ℃. To the stirring solution were sequentially added NaBH
4 (173 mg, 4.56 mmol) and Boc
2O (349 μL, 332 mg, 1.52 mmol) . The mixture was stirred for 1 h at 0 ℃. The reaction mixture was quenched with cold water (20 mL) and extracted with EA (30 mL × 3) . The combined organics were dried over Na
2SO
4 and concentrated under reduced pressure. The residue was by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 25 %in 15 min) to afford tert-butyl (5- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) pentyl) carbamate (150 mg, 40%yield) as yellow solid. LC-MS: m/z [M+H]
+ 486.4.
Step 4: (1R, 3S) -3- (5- ( (6- (5- ( (tert-butoxycarbonyl) amino) pentyl) pyridin-2-yl) amino) -1- (tert-
butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a mixture of tert-butyl (5- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) pentyl) carbamate (160 mg, 329 μmol) in CH
2Cl
2 (5.0 mL) were sequentially added CDI (107 mg, 659 μmol) and Et
3N (459 μL, 333 mg, 3.29 mmol) . The mixture was heated to 40 ℃ and stirred at that temperature for 5 h. The mixture was washed with H
2O (10 mL × 2) and dried over Na
2SO
4, the combined organics were concentrated under reduced pressure to give crude (1R, 3S) -3- (5- ( (6- (5- ( (tert-butoxycarbonyl) amino) pentyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate as yellow oil. LC-MS: m/z [M+H]
+ 580.4.
Step 5: (1R, 3S) -3- (5- ( (6- (5-aminopentyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-
yl) cyclopentyl 1H-imidazole-1-carboxylate
To a suspension of (1R, 3S) -3- (5- ( (6- (5- ( (tert-butoxycarbonyl) amino) pentyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate in CH
2Cl
2 (3.0 mL) was added slowly TFA (1.0 mL, 1.49 g, 13.1 mmol) . The mixture was stirred for 30 min at 25 ℃. The reaction mixture was concentrated under reduced pressure to afford crude (1R, 3S) -3- (5- ( (6- (5-aminopentyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate as a yellow oil. LC-MS: m/z [M+H]
+.
Step 6: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-12-oxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one
To a solution of (1R, 3S) -3- (5- ( (6- (5-aminopentyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate in CH
3CN (10.0 mL) was added Et
3N (872 μL, 633 mg, 6.26 mmol) . The mixture was heated to 80 ℃ and stirred at that temperature for 5 h. The reaction mixture was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 15 to 75 %in 20 min) to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-12-oxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (90.0 mg, 69%yield) as pale yellow solid. LC-MS: m/z [M+H]
+ 412.3.
Step 7: (1
1S, 1
3R, Z) -2
1H-12-oxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one
A mixture of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-12-oxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (90.0 mg, 219 μmol) in TFA (2.0 mL) was heated to 100 ℃ and stirred at that temperature for 5 h. The reaction mixture was concentrated under reduced pressure. The residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 10%to 30%in 30 min) to afford (1
1S, 1
3R, Z) -2
1H-12-oxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (31.0 mg, 39%yield) as white solid. LC-MS: m/z [M+H]
+ 356.2.
Example 89
(11S, 13R, Z) -21H-7, 13-dioxa-3, 11-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclotridecaphan-12-one

Step 1: tert-butyl (3- (2- (6-bromopyridin-2-yl) ethoxy) propyl) carbamate
To a solution of 2- (6-bromopyridin-2-yl) ethan-1-ol (350 mg, 1.73 mmol) in MeOH (18 mL) was sequentially added KOH (485 mg, 8.66 mmol) and tert-butyl (3-bromopropyl) carbamate (1.24 g, 5.20 mmol) at 25 ℃. The reaction mixture was heated to 35 ℃ and stirred at 35 ℃ for 16 h. The reaction mixture was cooled to room temperature, filtered, and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 25%in 20 min) to afford the product tert-butyl (3- (2- (6-bromopyridin-2-yl) ethoxy) propyl) carbamate (500 mg, 80%yield) as a colorless oil. LC-MS: (ESI) m/z [M+H]
+ 359.0.
Step 2: tert-butyl (3- (2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) ethoxy) propyl) carbamate
To a solution of tert-butyl (3- (2- (6-bromopyridin-2-yl) ethoxy) propyl) carbamate (300 mg, 835 μmol) in 1, 4-dioxane (8.0 mL) was sequentially added (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol- 3-yl) cyclopentan-1-ol (242 mg, 1.09 mmol) , Pd
2 (dba)
3 (153 mg, 167 μmol) , XantPhos (193 mg, 334 μmol) and Cs
2CO
3 (816 mg, 2.51 mmol) at 25 ℃. The suspension was degassed with N
2 for 5 times. The reaction mixture was heated to 80 ℃ and stirred at 80 ℃ under the atmosphere of N
2 for 16 h. The mixture was cooled to room temperature, concentrated under reduced pressure, diluted with water (30 mL) and extracted with DCM (30 mL × 3) . The combined organic phases were washed with brine (50 mL) , dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 67%in 20 min) to afford the product tert-butyl (3- (2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) ethoxy) propyl) carbamate (400 mg, 95%yield) as a light yellow solid. LC-MS: (ESI) m/z [M+H]
+ 502.0.
Step 3: (1R, 3S) -3- (5- ( (6- (2- (3- ( (tert-butoxycarbonyl) amino) propoxy) ethyl) pyridin-2-yl) amino) -1-
(tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of tert-butyl (3- (2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) ethoxy) propyl) carbamate (350 mg, 697 μmol) in DCM (6.0 mL) was sequentially added Et
3N (353 mg, 3.49 mmol) and di (1H-imidazol-1-yl) methanone (452 mg, 2.79 mmol) at 25 ℃. The reaction mixture was heated to 50 ℃ and stirred at 50 ℃ for 3 h. The mixture was cooled to room temperature and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 76%in 20 min) to afford the product (1R, 3S) -3- (5- ( (6- (2- (3- ( (tert-butoxycarbonyl) amino) propoxy) ethyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (100 mg, 24%yield) as a yellow oil. LC-MS: (ESI) m/z [M+H]
+ 596.3.
Step 4: (1R, 3S) -3- (5- ( (6- (2- (3-aminopropoxy) ethyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-
pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
A solution of [ (1R, 3S) -3- [5- [ [6- [2- [3- (tert-butoxycarbonylamino) propoxy] ethyl] -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (100 mg, 167 μmol) and TFA (95.0 mg, 839 μmol) in DCM (2.0 mL) was stirred at 25 ℃ for 2 h. The mixture was concentrated under reduced pressure to afford the product (1R, 3S) -3- (5- ( (6- (2- (3-aminopropoxy) ethyl) pyridin-2-yl) amino) -1- (tert-butyl) -1i-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (60.0 mg, 73%yield) as a yellow oil, which was used in the next step without further purification. LC-MS: (ESI) m/z [M+H]
+ 496.3.
Step 5: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-7, 13-dioxa-3, 11-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-
1 (1, 3) -cyclopentanacyclotridecaphan-12-one
The solution of [ (1R, 3S) -3- [5- [ [6- [2- (3-aminopropoxy) ethyl] -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (300 mg, 605 μmol) and DIPEA (782 mg, 6.05 mmol) in CH
3CN (40 mL) was heated to 90 ℃ and stirred at 90 ℃ for 16 h. The mixture was concentrated under reduced pressure, diluted with water (30 mL) and extracted with DCM (30 mL × 3) . The combined organic phase was washed with brine (30 mL × 2) , dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 75%in 20 min) to give the product (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-7, 13-dioxa-3, 11-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclotridecaphan-12-one (130 mg, 50%yield) as a yellow solid. LC-MS: (ESI) m/z [M+H]
+ 428.2.
Step 6: (1
1S, 1
3R, Z) -2
1H-7, 13-dioxa-3, 11-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclotridecaphan-12-one
The solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-7, 13-dioxa-3, 11-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclotridecaphan-12-one (130 mg, 304 μmol) in TFA (10 mL) was heated to 80 ℃ and stirred at 80 ℃ for 2 h. The mixture was concentrated under reduced pressure. The residue was purified by prep-HPLC (with CH
3CN from 30%to 100%in 8 min) to give the product (1
1S, 1
3R, Z) -2
1H-7, 13-dioxa-3, 11-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclotridecaphan-12-one (34.0 mg, 30%yield) as a white solid. LC-MS: m/z [M+H]
+372.2.
Example 90
(1
1S, 1
3R, Z) -2
1H-7, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one

Step 1: 2- (6-bromopyridin-2-yl) ethan-1-ol
To a solution of (diisopropylamino) lithium (14.24 mL, 28.48 mmol, 2M in THF) in THF (15 mL) was added dropwise a solution of 2-bromo-6-methyl-pyridine (3.5 g, 20.35 mmol) in THF (15 mL) and the resulting mixture was stirred at -70 ℃ for 0.5 hour. Then a solution of N, N-dimethylformamide (1.49 g, 20.35 mmol, 1.58 mL) in THF (5 mL) was added dropwise. After a stirring at -70 ℃ for 0.5 hour, MeOH (30 mL) , AcOH (1.22 g, 20.35 mmol) and NaBH
4 (769.75 mg, 20.35 mmol) were added sequentially at -70 ℃ and the resulting mixture was stirred at 20 ℃ for 11 hours. Then it was poured into saturated aqueous NaHCO
3 (100 mL) and extracted with EtOAc (3 × 100 mL) . The combined organic layer was washed with brine (100 mL) , dried over Na
2SO
4, filtered and concentrated. The residue was purified by column chromatography to give 2- (6-bromopyridin-2-yl) ethan-1-ol (3.2 g, 15.84 mmol, 77.84%yield) as a yellow oil. LC-MS: m/z 202.0 [M+H]
+.
Step 2: 2- (2- (6-bromopyridin-2-yl) ethoxy) acetonitrile
To a solution of 2- (6-bromopyridin-2-yl) ethan-1-ol (2 g, 9.90 mmol) in THF was added dropwise lithium tert-butoxide, 99.9% (metals basis) (9.00 mL, 18 mmol, 2.2 M in THF) and 2-bromoacetonitrile (2.37 g, 19.80 mmol, 1.38 mL) at 20 ℃ simultaneously and the resulting mixture was stirred at 20 ℃ for 12 hours. The mixture was poured into H
2O (50 mL) and extracted with EtOAc (3 × 50 mL) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrated. The residue was purified by column chromatography to give 2- (2- (6-bromo-2-pyridyl) ethoxy) acetonitrile (1 g, 4.15 mmol, 41.90%yield) as a yellow oil. LC-MS: m/z 241.0 [M+H]
+.
Step 3: 2- (2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-
2-yl) ethoxy) acetonitrile
To a stirred solution of 2- (2- (6-bromo-2-pyridyl) ethoxy) acetonitrile (500 mg, 2.07 mmol) and (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (250 mg, 1.12 mmol) in dioxane (2 mL) was added Cs
2CO
3 (729.51 mg, 2.24 mmol) , Pd
2 (dba)
3 (205.03 mg, 223.90 μmol) and XantPhos (194.33 mg, 335.85 μmol) at 25 ℃. The mixture was stirred at 100 ℃ for 12 hours under N
2. The reaction mixture was concentrated under reduced pressure to give the residue. The residue was purified by silica gel chromatography eluting with EtOAc/petroleum ether with EtOAc from 0 to 60%in 20 min to give 2- (2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) ethoxy) acetonitrile (220 mg, 573.68 μmol, 51.24%yield) as a yellow oil. LC-MS: m/z 384.2 [M+H]
+.
Step 4: tert-butyl (2- (2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) ethoxy) ethyl) carbamate
To a stirred solution of 2- (2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) ethoxy) acetonitrile (200 mg, 521.53 μmol) in methanol (5 mL) was added Boc
2O (341.47 mg, 1.56 mmol) and Pd/C (100 mg, 82.34 μmol, 10%purity) at 20 ℃. The mixture was stirred at 20 ℃ for 12 hours under H
2. The reaction solution was filtered, concentrated under reduced pressure to give the residue. The residue was purified by silica gel chromatography eluting with EtOAc/petroleum ether with EtOAc from 0 to 50%in 20 min to give tert-butyl (2- (2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) ethoxy) ethyl) carbamate (60 mg, 123.04 μmol, 23.59%yield) as a yellow oil. LC-MS: m/z 488.0 [M+H]
+.
Step 5: (1R, 3S) -3- (5- ( (6- (2- (2- ( (tert-butoxycarbonyl) amino) ethoxy) ethyl) pyridin-2-yl) amino) -1-
(tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a stirred solution of tert-butyl (2- (2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) ethoxy) ethyl) carbamate (55 mg, 112.79 μmol) in DCM (1.96 mL) was added Et
3N (34.24 mg, 338.37 μmol, 47.16 μL) , CDI (54.87 mg, 338.37 μmol) and DMAP (6.89 mg, 56.39 μmol) at 25 ℃. The mixture was stirred at 25 ℃ for 2 hours. The reaction solution was concentrated under reduced pressure to give the crude product (1R, 3S) -3- (5- ( (6- (2- (2- ( (tert-butoxycarbonyl) amino) ethoxy) ethyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (65 mg, 111.74 μmol, 99.07%yield) as a yellow oil, which was used into next step without further purification. LC-MS: m/z 582.3 [M+H]
+.
Step 6: (1R, 3S) -3- (5- ( (6- (2- (2-aminoethoxy) ethyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-
3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a stirred solution of (1R, 3S) -3- (5- ( (6- (2- (2- ( (tert-butoxycarbonyl) amino) ethoxy) ethyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (65 mg, 111.74 μmol) in DCM (3 mL) was added TFA (1.48 g, 12.98 mmol, 1 mL) at 25 ℃. The mixture was stirred at 25 ℃ for 12 hours. The reaction solution was concentrated under reduced pressure to give the crude product (1R, 3S) -3- (5- ( (6- (2- (2-aminoethoxy) ethyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (50 mg, 103.82 μmol, 92.91%yield) as a yellow oil, which was used into next step without further purification. LC-MS: m/z 482.3 [M+H]
+.
Step 7: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-7, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a stirred solution of (1R, 3S) -3- (5- ( (6- (2- (2-aminoethoxy) ethyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (50 mg, 103.82 μmol) in MeCN (2 mL) was added TEA (363.00 mg, 3.59 mmol, 0.5 mL) at 25 ℃. The mixture was stirred at 80 ℃ for 5 hours. The reaction solution was cooled, concentrated under reduced pressure to give the residue. The residue was purified by prep-TLC to afford the product (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-7, 12-dioxa- 3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (15 mg, 36.27 μmol, 34.94%yield) as a yellow oil. LC-MS: m/z 562.2 [M+H]
+.
Step 8: (1
1S, 1
3R, Z) -2
1H-7, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one
A solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-7, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (15 mg, 36.27 μmol) in TFA (1 mL) was stirred at 70 ℃for 16 hours. The reaction solution was concentrated under reduced pressure to give the residue. The residue was purified by prep-HPLC (FA condition) to afford the product (1
1S, 1
3R, Z) -2
1H-7, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentana cyclododecaphan-11-one (0.9 mg, 2.52 μmol, 6.94%yield) as a yellow oil. LC-MS: m/z 358.1 [M+H]
+.
The following compounds were prepared using the similar procedure disclosed in synthetic example 90.

Example 91
(1
1S, 1
3R, Z) -5, 5-difluoro-2
1H-7, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one


Step 1: ethyl 2- (6-bromopyridin-2-yl) -2, 2-difluoroacetate
To a suspension of 2, 6-dibromopyridine (1 g, 4.2 mmol) and ethyl 2-bromo-2, 2-difluoroacetate (1 g, 5 mmol) in DMSO (10 mL) was added Cu (538 mg, 8.4 mmol) . The reaction mixture was stirred at 100 ℃ for 4 h in a sealed tube. The reaction mixture was filtered and the filtrate was concentrated in vacuo. The residue was purified by flash column chromatography eluting with EtOAc/PE (with EtOAc from 0 to 8%in 10 min) to afford ethyl 2- (6-bromopyridin-2-yl) -2, 2-difluoroacetate (520 mg, 44%yield) as a colorless liquid. LC-MS: m/z 280.0 [M+H]
+.
Step 2: 2- (6-bromopyridin-2-yl) -2, 2-difluoroethan-1-ol
To a suspension of ethyl 2- (6-bromopyridin-2-yl) -2, 2-difluoroacetate (480 mg, 1.71 mmol) in MeOH (10.0 mL) was added NaBH
4 (130 mg, 3.42 mmol) . The reaction mixture was stirred at 25 ℃ for 1 h. The reaction solution was concentrated in vacuo. The residue was purified by flash column chromatography eluting with EtOAc/PE (with EtOAc from 0 to 20%in 15 min) to afford 2- (6-bromopyridin-2-yl) -2, 2-difluoroethan-1-ol (310 mg, 76%yield) as a colorless liquid. LC-MS: m/z 238.0 [M+H]
+.
Step 3: 2- (2- (6-bromopyridin-2-yl) -2, 2-difluoroethoxy) acetonitrile
To a suspension of 2- (6-bromopyridin-2-yl) -2, 2-difluoroethan-1-ol (250 mg, 1.05 mmol) in THF (10.0 mL) was added NaH (84 mg, 60 wt. %in mineral oil, 2.1 mmol) . The reaction mixture was stirred at 25 ℃ for 0.5 h before 2-bromoacetonitrile (150 mg, 1.26 mmol) was added to the reaction mixture. The solution was stirred at 25 ℃ for another 2 h before it was poured into ice water (50 mL) and extracted with CH
2Cl
2 (20 mL*3) . The organic layer was collected and concentrated in vacuo. The residue was purified by flash column chromatography eluting with EtOAc/PE (with EtOAc from 0 to 20%in 15 min) to afford 2- (2- (6-bromopyridin-2-yl) -2, 2-difluoroethoxy) acetonitrile (160 mg, 55%yield) as a light yellow liquid. LC-MS: m/z 277.0 [M+H]
+.
Step 4: 2- (2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-
2-yl) -2, 2-difluoroethoxy) acetonitrile
To a suspension of 2- (2- (6-bromopyridin-2-yl) -2, 2-difluoroethoxy) acetonitrile (110 mg, 0.4 mmol) and (1R, 3S) -3- (3-amino-1- (tert-butyl) -1H-pyrazol-5-yl) cyclopentan-1-ol (89 mg, 0.4 mmol) in toluene (8.0 mL) was added Pd
2 (dba)
3 (37 mg, 0.04 mmol) , XantPhos (46 mg, 0.08 mmol) and Cs
2CO
3 (261 mg, 0.8 mmol) . The reaction mixture was stirred at 80 ℃ for 16 h under N
2 atmosphere. Then it was concentrated in vacuo and the residue was purified by flash column chromatography eluting with EtOAc/PE (with EtOAc from 0 to 50%in 25 min) to afford 2- (2- (6- ( (1- (tert-butyl) -3- ( (1R, 3S) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) -2, 2-difluoroethoxy) acetonitrile (140 mg, 84%yield) as a light yellow solid. LC-MS: m/z 420.2 [M+H]
+.
Step 5: tert-butyl (2- (2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) -2, 2-difluoroethoxy) ethyl) carbamate
To a suspension of 2- (2- (6- ( (1- (tert-butyl) -3- ( (1R, 3S) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) -2, 2-difluoroethoxy) acetonitrile (140 mg, 0.33 mmol) and Boc
2O (144 mg, 0.66 mmol) in MeOH (10 mL) was added Pd/C (70 mg, 10%wt) . The reaction mixture was stirred at 20 ℃ for 1 h under H
2 atmosphere, before it was filtrated and the filtrate was concentrated in vacuo. The residue was purified by flash column chromatography eluting with EtOAc/PE (with EtOAc from 0 to 70%in 25 min) to afford tert-butyl (2- (2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) -2, 2-difluoroethoxy) ethyl) carbamate (70 mg, 40%yield) as a yellow solid. LC-MS: m/z 524.3 [M+H]
+.
Step 6: (1R, 3S) -3- (5- ( (6- (2- (2- ( (tert-butoxycarbonyl) amino) ethoxy) -1, 1-difluoroethyl) pyridin-2-
yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a suspension of tert-butyl (6- (2- ( (1- (tert-butyl) -3- ( (1R, 3S) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) carbamoyl) pyrrolidin-1-yl) hexyl) carbamate (70 mg, 0.13 mmol) and CDI (105 mg, 0.65 mmol) in DCM (15 mL) was added DIEA (84 mg, 0.65 mmol) . The reaction mixture was stirred at 35 ℃ for 3 h. The reaction solution was concentrated in vacuo to afford (1R, 3S) -3- (5- ( (6- (2- (2- ( (tert-butoxycarbonyl) amino) ethoxy) -1, 1-difluoroethyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (110 mg, crude) as a light red oil. LC-MS: m/z 618.3 [M+H]
+.
Step 7: (1R, 3S) -3- (5- ( (6- (2- (2-aminoethoxy) -1, 1-difluoroethyl) pyridin-2-yl) amino) -1- (tert-butyl) -
1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
A mixture of (1R, 3S) -3- (5- ( (6- (2- (2- ( (tert-butoxycarbonyl) amino) ethoxy) -1, 1-difluoroethyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate in TFA/DCM (3 mL/9 mL) was stirred at 20 ℃ for 1 h. The reaction solution was concentrated in vacuo to afford (1R, 3S) -3- (5- ( (6- (2- (2-aminoethoxy) -1, 1-difluoroethyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (90 mg, crude) as a red oil, which was used in the next step without further purification. LC-MS: m/z 518.2 [M+H]
+.
Step 8: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -5, 5-difluoro-2
1H-7, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one
To a suspension of (1R, 3S) -3- (5- ( (6- (2- (2-aminoethoxy) -1, 1-difluoroethyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate in CH
3CN (20.0 mL) was added TEA (0.12 mL, 86 mg, 0.85 mmol) . The reaction mixture was stirred at 60 ℃ for 24 h. Then it was concentrated in vacuo and the residue was purified by flash column chromatography eluting with EtOAc/PE (with EtOAc from 0 to 80%in 25 min) to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -5, 5-difluoro-2
1H-7, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (25 mg) as a light yellow solid. LC-MS: m/z 450.2 [M+H]
+.
Step 9: (1
1S, 1
3R, Z) -5, 5-difluoro-2
1H-7, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphan-11-one
A mixture of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -5, 5-difluoro-2
1H-7, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (25 mg, 0.056 mmol) in HCO
2H (5.0 mL) was stirred at 80 ℃ for 1 h. The reaction solution was concentrated in vacuo, the residue was purified by prep-HPLC eluting with CH
3CN in water (with CH
3CN from 15%to 50%in 10 min (0.1%FA condition) ) to afford (1
1S, 1
3R, Z) -5, 5-difluoro-2
1H-7, 12-dioxa-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one (2.3 mg, 10%yield) as a white solid. LC-MS: m/z 394.1 [M+H]
+.
Example 187
(1
1S, 1
3R, Z) -2
1H-7, 12-dioxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione

Step 1: tert-butyl 6-bromopicolinate
A mixture of 6-bromopyridine-2-carboxylic acid (500 mg, 2.48 mmol) , 1, 1-ditert-butoxy-N, N-dimethyl-methanamine (2.01 g, 9.90 mmol) in dioxane (20 mL) was stirred for 12 hours at 100 ℃in a RBF under N
2, until the reaction was complete as indicated by LCMS, the reaction mixture was concentrated in vacuo. The residue was purified by flash chromatography on silica gel eluting with EtOAc/petroleum ether with EtOAc from 0 to 10%in 15 min to give the desired product tert-butyl 6-bromopyridine-2-carboxylate (350 mg, 1.36 mmol, 54.78%yield) as a white solid. LC-MS: m/z 280.0 [M+Na]
+.
Step 2: tert-butyl 6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) picolinate
A mixture of tert-butyl 6-bromopyridine-2-carboxylate (350 mg, 1.36 mmol) , (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (302.81 mg, 1.36 mmol) , Pd
2 (dba)
3 (124.07 mg, 135.60 μmol) , XantPhos (157.03 mg, 271.20 μmol) , Cs
2CO
3 (881.40 mg, 2.71 mmol) in dioxane (30 mL) was stirred for 2 hours at 80 ℃ in a RBF under N
2, until the reaction was complete as indicated by LCMS, the reaction mixture was filtered through a pad of Celite with EtOAc, and the combined organics were concentrated in vacuo. The residue was purified by flash chromatography on silica gel eluting with EtOAc/petroleum ether with EtOAc from 0 to 30%in 25 min to give the desired product tert-butyl 6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) picolinate (500 mg, 1.25 mmol, 92.06%yield) as a pale yellow oil. LC-MS: m/z 400.8 [M+H]
+.
Step 3: tert-butyl 6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -
1H-pyrazol-5-yl) amino) picolinate
A mixture of tert-butyl 6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) picolinate (200 mg, 499.36 μmol) , 4-nitrophenyl carbonochloridate (301.96 mg, 1.50 mmol) , DMAP (122.01 mg, 998.72 μmol) and NMM (252.55 mg, 2.50 mmol, 274.51 μL) in MeCN (10 mL) was stirred for 1 hour at 25 ℃ in a RBF under N
2, until the reaction was complete as indicated by LCMS, the reaction mixture was concentrated in vacuo. The residue was purified by flash chromatography on silica gel eluting with EtOAc/petroleum ether with EtOAc from 0 to 30%in 25 min to give the desired product tert-butyl 6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) picolinate (220 mg, 388.96 μmol, 77.89%yield) as a pale yellow solid. LC-MS: m/z 566.2 [M+H]
+.
Step 4: tert-butyl 6- ( (3- ( (1S, 3R) -3- ( ( (2- ( ( (tert-
butoxycarbonyl) amino) oxy) ethyl) carbamoyl) oxy) cyclopentyl) -1- (tert-butyl) -1H-pyrazol-5-
yl) amino) picolinate
A mixture of tert-butyl 6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) picolinate (220 mg, 388.96 μmol) , tert-butyl N- (2-aminoethoxy) carbamate (137.08 mg, 0.78mmol) , TEA (118.08 mg, 1.17 mmol, 162.64 μL) in MeCN (10 mL) was stirred for 1 hour at 25 ℃ in a RBF under N
2, until the reaction was complete as indicated by LCMS, the reaction mixture concentrated in vacuo. The residue was purified by flash chromatography on silica gel eluting with EtOAc/petroleum ether with EtOAc from 0 to 50%in 25 min to give the desired product tert-butyl 6- ( (3- ( (1S, 3R) -3- ( ( (2- ( ( (tert-butoxycarbonyl) amino) oxy) ethyl) carbamoyl) oxy) cyclopentyl) -1- (tert-butyl) -1H-pyrazol-5-yl) amino) picolinate (140 mg, 232.28 μmol, 59.72%yield) as a pale yellow oil. LC-MS: m/z 603.4 [M+H]
+.
Step 5: 6- ( (3- ( (1S, 3R) -3- ( ( (2- (aminooxy) ethyl) carbamoyl) oxy) cyclopentyl) -1- (tert-butyl) -1H-
pyrazol-5-yl) amino) picolinic acid
A mixture of tert-butyl 6- ( (3- ( (1S, 3R) -3- ( ( (2- ( ( (tert-butoxycarbonyl) amino) oxy) ethyl) carbamoyl) oxy) cyclopentyl) -1- (tert-butyl) -1H-pyrazol-5-yl) amino) picolinate (140 mg, 232.28 μmol) , TFA (1.32 g, 11.61 mmol, 894.75 μL) in DCM (10 mL) was stirred for 12 hours at 25 ℃ in a RBF under N
2, until the reaction was complete as indicated by LCMS, the reaction mixture was concentrated in vacuo to give the desired crude product 6- ( (3- ( (1S, 3R) -3- ( ( (2- (aminooxy) ethyl) carbamoyl) oxy) cyclopentyl) -1- (tert-butyl) -1H-pyrazol-5-yl) amino) picolinic acid (120 mg, 161.25 μmol, 69.42%yield, 60%purity) as a pale yellow oil as TFA salt. LC-MS: m/z 447.3 [M+H]
+.
Step 6: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-7, 12-dioxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
A mixture of 6- ( (3- ( (1S, 3R) -3- ( ( (2- (aminooxy) ethyl) carbamoyl) oxy) cyclopentyl) -1- (tert-butyl) -1H-pyrazol-5-yl) amino) picolinic acid (140 mg, 313.55 μmol) , TCFH (175.59 mg, 627.10 μmol) , NMI (128.56 mg, 1.57 mmol) in MeCN (50 mL) was stirred for 2 hours at 25 ℃ in a RBF under N
2, until the reaction was complete as indicated by LCMS, the reaction mixture was concentrated in vacuo.
The residue was purified by flash chromatography on silica gel eluting with EtOAc/petroleum ether with EtOAc from 0 to 100%in 25 min to give the desired product (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-7, 12-dioxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (40 mg, 93.35 μmol, 29.77%yield) as a pale yellow solid. LC-MS: m/z 429.2 [M+H]
+.
Step 7: (1
1S, 1
3R, Z) -2
1H-7, 12-dioxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione
A mixture of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-7, 12-dioxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (40 mg, 93.35 μmol) in TFA (10 mL) was stirred for 4 hours at 70 ℃ in a RBF under N
2, until the reaction was complete as indicated by LCMS, the reaction mixture was concentrated in vacuo. The residue was purified by prep-HPLC to give the desired product (1
1S, 1
3R, Z) -2
1H-7, 12-dioxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (11.7 mg, 31.42 μmol, 33.66%yield) as a white solid. LC-MS: m/z 373.0 [M+H]
+.
Example 188
(1
1S, 1
3R, Z) -2
1H-12-oxa-3, 6, 7, 10-tetraaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione

Step 1: 6-bromopicolinohydrazide
To a stirred suspension of methyl 6-bromopyridine-2-carboxylate (5.0 g, 23.14 mmol) in MeOH (50 mL) was sequentially added hydrazine hydrate (1.16 g, 23.14 mmol) at 25 ℃. The resulting mixture was stirred at that temperature overnight. The reaction mixture was quenched with water (30 mL) and then extracted with EA (3 × 20 mL) . The combined organic layers were dried over Na
2SO
4 and filtered. The filtrate was concentrated and purified by silica gel chromatography eluting with EtOAc/PE with MeOH from 0 to 60 %in 20 min to give 6-bromopicolinohydrazide (2.0 g, 9.26 mmol, 66.67%yield) as a colorless oil. LC-MS: m/z 215.9 [M+H]
+.
Step 2: tert-butyl (2- (2- (6-bromopicolinoyl) hydrazineyl) ethyl) carbamate
To a stirred suspension of tert-butyl (2-oxoethyl) carbamate (957.89 mg, 6.02 mmol) in DCM (20 mL) was added 6-bromopicolinohydrazide (1.3 g, 6.02 mmol) and AcOH (0.5 mL) at 0 ℃. The resulting mixture was warmed to 25℃ and stirred at that temperature for 2 h. Then NaBH (OAc)
3 (1.53 g, 7.22 mmol) was added. The reaction mixture was kept at room temperature overnight. it was quenched with water (30 mL) and then extracted with EA (20 mL × 3) . The combined organic layers were dried over Na
2SO
4 and filtered. The filtrate was concentrated under vacuum. The residue was purified by silica gel chromatography eluting with EtOAc/petroleum ether with EtOAc from 0 to 50 %in 40 min to give (2- (2- (6-bromopicolinoyl) hydrazineyl) ethyl) carbamate (2 g, 5.57 mmol, 92.52%yield) as a colorless oil. LC-MS: m/z 258.9 [M+H]
+
Step 3: benzyl 2- (6-bromopicolinoyl) -1- (2- ( (tert-butoxycarbonyl) amino) ethyl) hydrazine-1-
carboxylate
To a stirred solution of tert-butyl (2- (2- (6-bromopicolinoyl) hydrazineyl) ethyl) carbamate (2 g, 5.57 mmol) in THF (10 mL) and H
2O (2 mL) were sequentially added Na
2CO
3 (1.18 g, 11.14 mmol) and CbzCl (949.80 mg, 5.57 mmol) at 0 ℃. The resulting mixture was warmed to room temperature and stirred at that temperature overnight. The mixture was cooled and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/petroleum ether with EtOAc from 0 to 50 %in 30 min to give benzyl 2- (6-bromopicolinoyl) -1- (2- ( (tert-butoxycarbonyl) amino) ethyl) hydrazine-1-carboxylate (1.2 g, 2.43 mmol, 43.69%yield) as a colorless oil. LC-MS: m/z 515.0 [M+Na]
+
Step 4: benzyl 1- (2- ( (tert-butoxycarbonyl) amino) ethyl) -2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-
hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) picolinoyl) hydrazine-1-carboxylate
To a stirred solution of (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (100 mg, 447.80 μmol) and benzyl 2- (6-bromopicolinoyl) -1- (2- ( (tert-butoxycarbonyl) amino) ethyl) hydrazine-1-carboxylate (397.66 mg, 806.04 μmol) in 1, 4-dioxane (10 mL) was added XantPhos Pd G3 (127.49 mg, 134.34 μmol) and K
2CO
3 (124.49 mg, 895.60 μmol) at 25 ℃. The mixture was stirred at 100 ℃for 12 hours under N
2. The reaction solution was concentrated under reduced pressure to give the residue. The residue was purified by silica gel chromatography eluting with EtOAc/petroleum ether with EtOAc from 0 to 60%in 20 min to give benzyl 1- (2- ( (tert-butoxycarbonyl) amino) ethyl) -2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) picolinoyl) hydrazine-1-carboxylate (100 mg, 157.29 μmol, 35.13%yield) as a yellow oil. LC-MS: m/z 636.3 [M+H]
+.
Step 5: (1R, 3S) -3- (5- ( (6- (2- ( (benzyloxy) carbonyl) -2- (2- ( (tert-
butoxycarbonyl) amino) ethyl) hydrazine -1-carbonyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-
pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a stirred solution of benzyl 1- (2- ( (tert-butoxycarbonyl) amino) ethyl) -2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) picolinoyl) hydrazine-1-carboxylate (100 mg, 157.29 μmol) in DCM (3.0 mL) was added DIEA (31.83 mg, 314.59 μmol, ) , CDI (51.01 mg, 314.59 μmol) and DMAP (3.84 mg, 31.46 μmol) at 25 ℃. The reaction mixture was stirred at 25 ℃ for 2 hours. The mixture was poured into H
2O (5 mL) and extracted with DCM (3 × 5 mL) . The combined organic layer was washed with brine (5 mL) , dried over Na
2SO
4, filtered and concentrated to give (1R, 3S) -3- (5- ( (6- (2- ( (benzyloxy) carbonyl) -2- (2- ( (tert-butoxycarbonyl) amino) ethyl) hydrazine-1-carbonyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (110 mg, 150.72 μmol, 95.82%yield) as a yellow oil which was used directly for the next step without further purification. LC-MS: m/z 729.8 [M+H]
+.
Step 6: (1R, 3S) -3- (5- ( (6- (2- (2-aminoethyl) -2- ( (benzyloxy) carbonyl) hydrazine-1-
carbonyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-
carboxylate
To a stirred solution of (1R, 3S) -3- (5- ( (6- (2- ( (benzyloxy) carbonyl) -2- (2- ( (tert-butoxycarbonyl) amino) ethyl) hydrazine-1-carbonyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (110 mg, 150.72 μmol) in DCM (2.0 mL) was added TFA (407.00 mg, 3.57 mmol) at 25 ℃. The mixture was stirred at 25 ℃ for 2 hours. The mixture was concentrated to give (1R, 3S) -3- (5- ( (6- (2- (2-aminoethyl) -2- ( (benzyloxy) carbonyl) hydrazine-1-carbonyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (90 mg, 142.92 μmol, 94.83%yield) as a yellow oil which was used directly for the next step without further purification. LC-MS: m/z [M+H]
+ 631.3.
Step 7: benzyl (1
1S, 1
3R, Z) -2
1- (tert-butyl) -5, 11-dioxo-2
1H-12-oxa-3, 6, 7, 10-tetraaza-4 (2, 6) -
pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-7-carboxylate
To a stirred solution of (1R, 3S) -3- (5- ( (6- (2- (2-aminoethyl) -2- ( (benzyloxy) carbonyl) hydrazine-1-carbonyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (90 mg, 142.92 μmol) in ACN (2 mL) was added TEA (145.20 mg, 1.43 mmol, 0.2 mL) at 25 ℃. The mixture was stirred at 80 ℃ for 3 hours. The mixture was concentrated and purified by prep-TLC to give benzyl (1
1S, 1
3R, Z) -2
1- (tert-butyl) -5, 11-dioxo-2
1H-12-oxa-3, 6, 7, 10-tetraaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-7-carboxylate (30 mg, 53.42 μmol, 37.37%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 562.2.
Step 8: benzyl (1
1S, 1
3R, Z) -5, 11-dioxo-2
1H-12-oxa-3, 6, 7, 10-tetraaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-7-carboxylate
A solution of benzyl (1
1S, 1
3R, Z) -2
1- (tert-butyl) -5, 11-dioxo-2
1H-12-oxa-3, 6, 7, 10-tetraaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-7-carboxylate (30 mg, 53.42 μmol) in HCOOH (1 mL) was stirred at 70 ℃ for 24 hours. The mixture was concentrated and purified by prep-HPLC to give benzyl (1
1S, 1
3R, Z) -5, 11-dioxo-2
1H-12-oxa-3, 6, 7, 10-tetraaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-7-carboxylate (10 mg, 19.78 μmol, 37.03%yield) as a white solid. LC-MS: m/z [M+H]
+ 506.2.
Step 9: (1
1S, 1
3R, Z) -2
1H-12-oxa-3, 6, 7, 10-tetraaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione
To a solution of benzyl (1
1S, 1
3R, Z) -5, 11-dioxo-2
1H-12-oxa-3, 6, 7, 10-tetraaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-7-carboxylate (10 mg, 19.78 μmol) in MeOH (2 mL) was added Pd/C (10 mg, 8.23 μmol, 10%purity) and the resulting mixture was stirred at 25 ℃ for 0.5 hour under H
2 (15 psi) . The reaction mixture was filtered and concentrated to give the residue. The residue was purified by prep-HPLC (FA condition) to afford the product (1
1S, 1
3R, Z) -2
1H-12-oxa-3, 6, 7, 10-tetraaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (1.4 mg, 3.70 μmol, 18.69%yield, 98.06%purity) as a white solid. LC-MS: m/z [M+H]
+372.2.
Example 189
(1
1S, 1
3R, Z) -2
1H-7, 12-dioxa-3, 5, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-6, 11-dione

Step 1: 2- ( (tert-butoxycarbonyl) amino) ethyl (6-bromopyridin-2-yl) carbamate
To a solution of 6-bromopyridin-2-amine (600 mg, 3.47 mmol) in THF (20 mL) was added Triphosgene (514 mg, 1.73 mmol) and Et
3N (1.05 g, 10.4 mmol, 1.45 mL) at 0 ℃ under nitrogen. The reaction was stirred at 0 ℃ for 5 minutes and then tert-butyl (2-hydroxyethyl) carbamate (559 mg, 3.47 mmol, 536 μL) was added. The reaction was stirred at 25 ℃ for 20 minutes. The reaction was diluted with water (40 mL) and then extracted with EtOAc (10 mL × 3) . The combined organic layer was washed with brine (30 mL) , dried over Na
2SO
4, filtered and concentrated. The residue was purified by silica gel chromatography eluting with EtOAc/petroleum ether with EtOAc from 0 to 20%in 10 minutes to afford 2- ( (tert-butoxycarbonyl) amino) ethyl (6-bromopyridin-2-yl) carbamate (820 mg, 65%yield) as a white solid. LC-MS: m/z 361.0 [M+H]
+
Step 2: 2- ( (tert-butoxycarbonyl) amino) ethyl (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -
1H-pyrazol-5-yl) amino) pyridin-2-yl) carbamate
To a mixture of 2- ( (tert-butoxycarbonyl) amino) ethyl (6-bromopyridin-2-yl) carbamate (242 mg, 671 μmol) , (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (150 mg, 671 μmol) and C
S2CO
3 (656 mg, 2.02 mmol) in 1, 4-dioxane (2 mL) was added Pd
2 (dba)
3 (123 mg, 134 μmol) and XantPhos (38.8 mg, 67.2 μmol) under nitrogen. The reaction was stirred at 100 ℃ under nitrogen for 8 hours. The mixture was diluted with water (20 mL) and extracted with EtOAc (5 mL × 3) . The combined organic layer was washed with brine (20 mL) , dried over Na
2SO
4, filtered and concentrated. The residue was purified by flash chromatography on silica gel eluting with MeOH/DCM with MeOH from 0 to 8%in 10 minutes to afford 2- ( (tert-butoxycarbonyl) amino) ethyl (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) carbamate (263 mg, 77%yield) as a yellow solid. LC-MS: m/z 503.2 [M+H]
+.
Step 3: 2- ( (tert-butoxycarbonyl) amino) ethyl (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-
nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) carbamate
A mixture of 2- ( (tert-butoxycarbonyl) amino) ethyl (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) carbamate (263 mg, 523 μmol) , 4-nitrophenyl carbonochloridate (316 mg, 1.57 mmol) , DIPEA (202 mg, 1.57 mmol, 273 μL) and DMAP (63.9 mg, 523 μmol) in DCE (10 mL) was stirred at 85 ℃ for 8 hours. The mixture was quenched with water (10 mL) and then extracted with DCM (5 mL × 3) . The combined organic phase was dried over sodium sulfate and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel eluting with MeOH/DCM with MeOH from 0 to 6%in 15 minutes to afford 2- ( (tert-butoxycarbonyl) amino) ethyl (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) carbamate (280 mg, 80%yield) as a yellow solid. LC-MS: m/z 668.2 [M+H]
+ .
Step 4: 2-aminoethyl (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-
nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) carbamate
A mixture of 2- ( (tert-butoxycarbonyl) amino) ethyl (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) carbamate (280 mg, 419 μmol) in TFA/DCM=1/1 (6 mL) was stirred at 25 ℃ for 30 minutes. The mixture was concentrated under reduced pressure to afford 2-aminoethyl (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4- nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) carbamate (360 mg, crude) as a yellow oil, which was used directly in the next step. LC-MS: m/z 568.3 [M+H]
+.
Step 5: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-7, 12-dioxa-3, 5, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphane-6, 11-dione
To a solution of 2-aminoethyl (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) carbamate (360 mg, 634 μmol) in ACN (50 mL) was added DIPEA (320 mg, 2.48 mmol, 432 μL) . Then the reaction mixture was stirred at 25 ℃ for 12 hours. The mixture was concentrated under reduced pressure, and the residue was purified by flash chromatography on silica gel eluting with EtOAc/petroleum ether with MeOH/DCM with MeOH from 0 to 10%in 20 minutes to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-7, 12-dioxa-3, 5, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-6, 11-dione (80 mg, 29%yield) as a yellow solid. LC-MS: m/z 429.2 [M+H]
+.
Step 6: (1
1S, 1
3R, Z) -2
1H-7, 12-dioxa-3, 5, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-6, 11-dione
The solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-7, 12-dioxa-3, 5, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-6, 11-dione (80 mg, 187 μmol) in TFA (6 mL) was stirred at 80 ℃ for 2 hours. The mixture was concentrated under reduced pressure, and the residue was sent to purified by Prep-HPLC (Chromatographic columns: Xbridge-C18 150 × 19 mm, 5 μm; Mobile Phase: ACN/H
2O (0.1%FA) ; Gradient: 10%-20%) to afford (1
1S, 1
3R, Z) -2
1H-7, 12-dioxa-3, 5, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-6, 11-dione (13.2 mg, 19%yield) as a white solid. LC-MS: m/z 373.1 [M+H]
+.
Example 190
(1
1S, 1
3R, Z) -2
1H-13-oxa-3, 5, 11-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclotridecaphane-6, 12-dione

Step 1: tert-butyl (5- ( (6-bromopyridin-2-yl) amino) -5-oxopentyl) carbamate
A solution of 5- (tert-butoxycarbonylamino) pentanoic acid (753 mg, 3.47 mmol) and 1-methylimidazole (1.66 g, 20.23 mmol, 1.61 mL) in DCM (10 mL) was stirred at 0 ℃. Then MsCl (370 mg, 3.23 mmol, 250.51 μL) was added and the resulting mixture was stirred at 0 ℃ for 30 min. Then 6-bromopyridin-2-amine (500 mg, 2.89 mmol) was added and the mixture was stirred at room temperature overnight. The resulting solution was diluted with water (20 mL) , then extracted with DCM (3 × 20 mL) . The organic layers were combined, washed with brine (20 mL) , dried and concentrated in vacuo. The residue was applied on a silica gel column and eluted with PE/EA (4/1) to give tert-butyl (5- ( (6-bromopyridin-2-yl) amino) -5-oxopentyl) carbamate (1.0 g, 2.69 mmol, 92.95%yield) as a colorless oil. LC-MS: m/z 371.8. [M+H]
+.
Step 2: tert-butyl (5- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) amino) -5-oxopentyl) carbamate
To a solution of (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (130 mg, 582.14 μmol) and tert-butyl (5- ( (6-bromopyridin-2-yl) amino) -5-oxopentyl) carbamate (325.06 mg, 873.21 μmol) in toluene (10 mL) was added Cs
2CO
3 (378.39 mg, 1.16 mmol) , Pd
2 (dba)
3 (106.62 mg, 116.43 μmol) and XantPhos (101.05 mg, 174.64 μmol) at 20 ℃ and stirred at 80 ℃ for 12 hours under N
2. The reaction solution was concentrated under reduced pressure to get the residue. The residue was purified by prep-HPLC (FA condition) to afford tert-butyl (5- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) amino) -5-oxopentyl) carbamate (100 mg, 194.30 μmol, 33.38%yield) as a white solid. LC-MS: m/z 515.3. [M+H]
+.
Step 3: tert-butyl (5- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-
nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) amino) -5-
oxopentyl) carbamate
To a solution of tert-butyl (5- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) amino) -5-oxopentyl) carbamate (70 mg, 136.01 μmol) in MeCN (2 mL) was added 4-methylmorpholine (68.79 mg, 680.06 μmol, 74.77 μL) , DMAP (33.23 mg, 272.03 μmol) and (4-nitrophenyl) carbonochloridate (54.83 mg, 272.03 μmol) at 20 ℃ and the reaction mixture was stirred at 20 ℃ for 12 hours. The reaction solution was concentrated under reduced pressure to give tert-butyl (5- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridine -2-yl) amino) -5-oxopentyl) carbamate (85 mg, 125.04 μmol, 91.94%yield) as a yellow oil, which was used in the next step without further purification. LC-MS: m/z 680.3 [M+H]
+.
Step 4: (1R, 3S) -3- (5- ( (6- (5-aminopentanamido) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-
yl) cyclopentyl (4-nitrophenyl) carbonate
To a solution of tert-butyl (5- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridine-2-yl) amino) -5-oxopentyl) carbamate (85 mg, 125.04 μmol) in DCM (3 mL) was added TFA (722.56 mg, 6.34 mmol, 488.22 μL) and the reaction mixture was stirred at 20 ℃ for 2 hours. The reaction solution was concentrated under reduced pressure to give (1R, 3S) -3- (5- ( (6- (5-aminopentanamido) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate (70 mg, 120.76 μmol, 96.58%yield) as a yellow oil, which was used in the next step without further purification. LC-MS: m/z 680.3 [M+H]
+.
Step 5: (1
1S, 1
3R, Z) -21- (tert-butyl) -2
1H-13-oxa-3, 5, 11-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-
1 (1, 3) -cyclopentanacyclotridecaphane-6, 12-dione
To a solution of (1R, 3S) -3- (5- ( (6- (5-aminopentanamido) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate (70 mg, 120.76 μmol) in MeCN (2 mL) was added TEA (217.80 mg, 2.15 mmol, 0.3 mL) and the reaction solution was stirred at 80 ℃ for 5 hours. The reaction solution was concentrated under reduced pressure and purified by prep-TLC to afford the product (11S, 13R, Z) -21- (tert-butyl) -21H-13-oxa-3, 5, 11-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclotridecaphane-6, 12-dione (30 mg, 68.10 μmol, 56.39%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 441.2.
Step 6: (1
1S, 1
3R, Z) -2
1H-13-oxa-3, 5, 11-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclotridecaphane-6, 12-dione
A solution of (1
1S, 1
3R, Z) -21- (tert-butyl) -2
1H-13-oxa-3, 5, 11-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclotridecaphane-6, 12-dione (30 mg, 68.10 μmol) in TFA (1 mL) was stirred at 70 ℃ for 12 hours. The reaction solution was cooled, concentrated under reduced pressure and the residue was diluted with DCM (5 mL) , washed with saturated aqueous solution of NaHCO
3 (5 mL) . The precipitate was collected by filtration and the filter cake was washed with MeCN (2 mL) and dried in vacuo to afford (11S, 13R, Z) -21H-13-oxa-3, 5, 11-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclotridecaphane-6, 12-dione (8 mg, 30.6%yield) as yellow solid. LC-MS: m/z [M+H]
+ 385.2.
The following compounds were prepared using the similar procedure disclosed in synthetic example 190.

Example 192
(1
1S, 1
3R, Z) -2
1H-12-oxa-5-thia-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one 5, 5-dioxide


Step 1: 2- (4-sulfanylbutyl) isoindoline-1, 3-dione
A mixture of 2- (4-bromobutyl) isoindoline-1, 3-dione (1.8 g, 6.38 mmol) , TBAB (617 mg, 1.91 mmol) and NaSH (562 mg, 7.02 mmol, 70%purity) in DMF (20 mL) was stirred for 16 hours at 20 ℃ under N
2, until the reaction was complete as indicated by LCMS. The reaction mixture was quenched with ice-cold water (200 mL) and extracted with EtOAc (3 × 300 mL) . The combined organic phase was washed with brine (500 mL) and dried over anhydrous Na
2SO
4, filtered and concentrated in vacuo. The crude product was purified by flash chromatography (SiO
2, hexane/ethyl acetate 10: 1) to afford 2- (4-sulfanylbutyl) isoindoline-1, 3-dione (0.95 g, 4.04 mmol, 63%yield) as a colorless liquid. LC-MS: m/z 236.1 [M+H]
+ .
Step 2: 2- (4- ( (6-bromopyridin-2-yl) thio) butyl) isoindoline-1, 3-dione
A mixture of 2-bromo-6-fluoro-pyridine (647 mg, 3.68 mmol, 379 μL) , 2- (4-sulfanylbutyl) isoindoline-1, 3-dione (865 mg, 3.68 mmol) , K
2CO
3 (1.01 g, 7.35 mmol) in DMF (30 mL) was stirred for 12 hours at 25 ℃. After completion of the reaction as judged by LCMS, reaction mixture was quenched with ice-cold water (100 mL) and extracted with EtOAc (3 × 50 mL) . The organic phase was washed with brine (50 mL) and dried over anhydrous Na
2SO
4, filtered and concentrated in vacuo. The crude product was purified by flash chromatography (SiO
2, hexane/ethyl acetate 3: 1) to afford 2- [4- [ (6-bromo-2-pyridyl) sulfanyl] butyl] isoindoline-1, 3-dione (300 mg, 767 μmol, 21%yield) as a white solid. LC-MS: m/z 391.0 [M+H]
+.
Step 3: 2- (4- ( (6-bromopyridin-2-yl) sulfonyl) butyl) isoindoline-1, 3-dione
A mixture of 2- [4- [ (6-bromo-2-pyridyl) sulfanyl] butyl] isoindoline-1, 3-dione (270 mg, 690 μmol) , m-CPBA (475 mg, 2.76 mmol) in DCM (20 mL) was stirred for 12 hours at 25 ℃ in a RBF under N
2, until the reaction was complete as indicated by LCMS, the reaction mixture was diluted with EtOAc (60 mL) and washed with brine (20 mL) . The organic phase was concentrated in vacuo. The crude product was purified by flash chromatography (SiO
2, hexane/ethyl acetate 1: 1) to afford 2- (4- ( (6-bromopyridin-2-yl) sulfonyl) butyl) isoindoline-1, 3-dione (250 mg, 0.59 mmol, 85%yield) as a white solid. LC-MS: m/z 423.0 [M+H]
+.
Step 4: 2- (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) sulfonyl) butyl) isoindoline-1, 3-dione
A mixture of 2- [4- [ (6-bromo-2-pyridyl) sulfonyl] butyl] isoindoline-1, 3-dione (210 mg, 496 μmol) , (1R) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (111 mg, 496 μmol) , Pd
2 (dba)
3 (45 mg, 50 μmol) and XantPhos (57 mg, 99 μmol) , Cs
2CO
3 (322 mg, 992 μmol) in dioxane (20 mL) was stirred for 4 hours at 100 ℃ in a RBF under N
2, until the reaction was complete as indicated by LCMS, the reaction mixture was filtered through a pad of Celite with EtOAc (50 mL) , and the combined organics were concentrated in vacuo, purified by silica gel chromatography (SiO
2, hexane/ethyl acetate 1: 1) to give the desired product 2- (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) sulfonyl) butyl) isoindoline-1, 3-dione (150 mg, 265 μmol, 53%yield) as a pale yellow solid. LC-MS: m/z 566.2 [M+H]
+.
Step 5: (1R, 3S) -3- (5- ( (6- ( (4-aminobutyl) sulfonyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-
3-yl) cyclopentan-1-ol
To a suspension of 2- (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) sulfonyl) butyl) isoindoline-1, 3-dione (150 mg, 264 μmol) in EtOH (100 mL) was added NH
2NH
2. H
2O (330 mg, 5.28 mmol, 320 μL, 80%purity) at room temperature and the resulting mixture was stirred for 3 hours at 80 ℃ under N
2. After completion of the reaction as judged by LCMS, reaction mixture was concentrated in vacuo. The crude product was purified by flash chromatography (SiO
2, DCM/MeOH: 10: 1) to afford (1R, 3S) -3- (5- ( (6- ( (4-aminobutyl) sulfonyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (100 mg, 230 μmol, 87%yield) as a colorless oil. LC-MS: m/z 436.2 [M+H]
+.
Step 6: tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) sulfonyl) butyl) carbamate
A mixture of (1R, 3S) -3- (5- ( (6- ( (4-aminobutyl) sulfonyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (100 mg, 230 μmol) , Boc
2O (100 mg, 460 μmol, 105 μL) and DIEA (148 mg, 1.15 mmol, 190 μL) in DCM (2 mL) was stirred for 1 hour at room temperature under N
2, until the reaction was complete as indicated by LCMS, the reaction mixture was concentrated in vacuo, purified by silica gel chromatography (DCM/MeOH= 20: 1) to give the desired product tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) sulfonyl) butyl) carbamate (110 mg, 205 μmol, 89%yield) as a colorless oil. LC-MS: m/z 536.3 [M+H]
+.
Step 7: tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-
nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-
yl) sulfonyl) butyl) carbamate
To a suspension of tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) sulfonyl) butyl) carbamate (100 mg, 186 μmol) , (4-nitrophenyl) carbonochloridate (113 mg, 560 μmol) and 4-methylmorpholine (189 mg, 1.87 mmol, 205 μL) in MeCN (5 mL) was added DMAP (46 mg, 373 μmol) at room temperature and the resulting mixture was stirred for 2 hours under N
2. After completion of the reaction as judged by LCMS, reaction mixture was concentrated in vacuo. The crude product was purified by flash chromatography (SiO
2, hexane/ethyl acetate 1: 1) to afford tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) sulfonyl) butyl) carbamate (60 mg, 86 μmol, 46%yield) as a colorless oil. LC-MS: m/z 701.2 [M+H]
+.
Step 8: (1R, 3S) -3- (5- ( (6- ( (4-aminobutyl) sulfonyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-
3-yl) cyclopentyl (4-nitrophenyl) carbonate
A mixture of tert-butyl (4- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) sulfonyl) butyl) carbamate (60 mg, 86 μmol) in DCM (4.7 mL) and TFA (488 mg, 4.28 mmol, 330 μL) was stirred for 20 minutes at room temperature under N
2, until the reaction was complete as indicated by LCMS, the reaction mixture was concentrated in vacuo to give the desired product (1R, 3S) -3- (5- ( (6- ( (4-aminobutyl) sulfonyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate (50 mg, 83 μmol, 97%yield) as a colorless oil which was used to the next step without further purification. LC-MS: m/z 601.2 [M+H]
+.
Step 9: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-12-oxa-5-thia-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphan-11-one 5, 5-dioxide
A mixture of (1R, 3S) -3- (5- ( (6- ( (4-aminobutyl) sulfonyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate (50 mg, 83.24 μmol) and TEA (336.92 mg, 3.33 mmol, 464.07 μL) in MeCN (4.6 mL) was stirred for 6 hours at room temperature under N
2, until the reaction was complete as indicated by LCMS, the reaction mixture was concentrated in vacuo, purified by silica gel chromatography (DCM/MeOH = 10: 1) to give the desired product (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-12-oxa-5-thia-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one 5, 5-dioxide (25 mg, 54 μmol, 65%yield) as a white solid. LC-MS: m/z 462.2 [M+H]
+.
Step 10: (1
1S, 1
3R, Z) -2
1H-12-oxa-5-thia-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one 5, 5-dioxide
A mixture of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-12-oxa-5-thia-3, 10-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one 5, 5-dioxide (25 mg, 54.16 μmol) in TFA (4 mL) was stirred for 16 hours at 70 ℃ under N
2, until the reaction was complete as indicated by LCMS, the reaction mixture was concentrated in vacuo, purified by column chromatography followed by prep. HPLC purification 2 -40%MeCN in H
2O (0.1%FA) to give the desired product (1
1S, 1
3R, Z) -2
1H-12-oxa-5-thia-3, 10-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one 5, 5-dioxide (10.2 mg, 25 μmol, 46%yield) as an off-white solid. LC-MS: m/z 406.1 [M+H]
+.
Example 193
(1
1S, 1
3R, Z) -2
1H-12-oxa-5-thia-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one 5, 5-dioxide

Step 1: tert-butyl (3- ( (6-bromopyridine) -2-sulfonamido) propyl) carbamate
To a suspension of 6-bromopyridine-2-sulfonyl chloride (500 mg, 1.95 mmol) in DCM (15 mL) was added tert-butyl N- (3-aminopropyl) carbamate (339.64 mg, 1.95 mmol) and TEA (394.50 mg, 3.90 mmol) . The reaction mixture was stirred at 25 ℃ for 16 hours. Then it was concentrated in vacuo and purified by flash column chromatography eluting with ethyl acetate/petroleum ether from 0 to 30%in 25 min to afford tert-butyl N- [3- [ (6-bromo-2-pyridyl) sulfonylamino] propyl] carbamate (690 mg, 1.75 mmol, 89.78%yield) . LC-MS: m/z 294.0 [M+H]
+.
Step 2: tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridine) -2-sulfonamido) propyl) carbamate
To a suspension of tert-butyl N- [3- [ (6-bromo-2-pyridyl) sulfonylamino] propyl] carbamate (330 mg, 836.96 μmol) in toluene (10 mL) was added (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (150 mg, 671.70 μmol) , XantPhos Pd G3 (150 mg, 430.43 μmol) and K
2CO
3 (210 mg, 1.52 mmol) . The reaction was stirred at 80 ℃ for 16 hours under nitrogen. Then it was concentrated in vacuo and purified by flash column chromatography eluting with ethyl acetate/petroleum ether from 0 to 60%in 25 min to afford tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridine) -2-sulfonamido) propyl) carbamate (300 mg, 558.99 μmol, 83.22%yield) as a brown oil.
LC-MS: m/z 536.7 [M+H]
+.
Step 3: (1R, 3S) -3- (5- ( (6- (N- (3- ( (tert-butoxycarbonyl) amino) propyl) sulfamoyl) pyridin-2-
yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a suspension of tert-butyl (3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridine) -2-sulfonamido) propyl) carbamate (137 mg, 255.27 μmol) in DCM (4 mL) was added CDI (82.78 mg, 510.54 μmol) , DMAP (31.19 mg, 255.27 μmol) and TEA (129.15 mg, 1.28 mmol, 177.90 μL) and it was stirred for 16 hours at 35 ℃. Then the reaction mixture was quenched with ice-cold water (10 mL) and extracted with DCM (3 × 10 mL) . The organic phase was dried over anhydrous Na
2SO
4, filtered and concentrated in vacuo. The crude product (1R, 3S) -3- (5- ( (6- (N- (3- ( (tert-butoxycarbonyl) amino) propyl) sulfamoyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate was used in the next step without further purification. LC-MS: m/z 630.6 [M+H]
+.
Step 4: (1R, 3S) -3- (5- ( (6- (N- (3-aminopropyl) sulfamoyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-
pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
A mixture of (1R, 3S) -3- (5- ( (6- (N- (3- ( (tert-butoxycarbonyl) amino) propyl) sulfamoyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (crude) in DCM (2 ml) and TFA (2 mL) was stirred at 25 ℃ for 1 hour. Then it was concentrated to afford crude (1R, 3S) -3- (5- ( (6- (N- (3-aminopropyl) sulfamoyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate as a brown oil, which was used in the next step without further purification. LC-MS: m/z 530.6 [M+H]
+.
Step 5: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-12-oxa-5-thia-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one 5, 5-dioxide
To a suspension of (1R, 3S) -3- (5- ( (6- (N- (3-aminopropyl) sulfamoyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (crude) in CH
3CN (5 mL) was added TEA (196 mg, 1.94 mmol) at room temperature. The reaction was stirred at 80 ℃ for 16 hours. Then it was cooled, concentrated in vacuo and purified by flash column chromatography eluting with methyl alcohol/dichloromethane ether from 0 to 4.6%in 25 min to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-12-oxa-5-thia-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one 5, 5-dioxide (13 mg, 28.10 μmol, 14.91%yield) as a colorless oil.. LC-MS: m/z 462.7 [M+H]
+.
Step 6: (1
1S, 1
3R, Z) -2
1H-12-oxa-5-thia-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-11-one 5, 5-dioxide
A solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-12-oxa-5-thia-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one 5, 5-dioxide (13 mg, 28.10 μmol) in FA (2 mL) was stirred at 80 ℃ for 5 hours. Then it was concentrated and purified by Prep-HPLC (FA; CH
3CN: water: 30%~55%) to afford (1
1S, 1
3R, Z) -2
1H-12-oxa-5-thia-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-11-one 5, 5-dioxide (5 mg, 12.30 μmol, 43.77%yield) as an off-white solid. LC-MS: m/z 406.7 [M+H]
+.
Example 194
(1
4Z, 4
4Z, 5
1S, 5
3R) -4
1H-6-oxa-3, 8-diaza-1 (3, 5) -isoxazola-2 (2, 6) -pyridina-4 (3, 5) -pyrazola-5 (1, 3) -
cyclopentanacyclononaphan-7-one

Step 1: tert-butyl ( (3- (6-bromopyridin-2-yl) isoxazol-5-yl) methyl) carbamate
To a suspension of (2E) -6-bromopyridine-2-carbaldehyde oxime (400 mg, 2.0 mmol) in DMF (10 mL) was added slowly three portions 1-chloropyrrolidine-2, 5-dione (266 mg, 2.0 mmol, 161 μL) at 0 ℃ and stirred for 1 h at 50 ℃ under N
2. The reaction was cooled to 0 ℃ and a solution of tert-butyl N-prop-2-ynylcarbamate (309 mg, 2.0 mmol) in DCM (10 mL) and N, N-diethylethanamine (201 mg, 2.0 mmol, 277 μL) was added dropwise. The reaction mixture was stirred at ambient temperature for 3 h, quenched with saturated sodium bicarbonate (50 mL) and extracted with DCM (3 x 50 mL) . The combined organic layer was washed with brine (50 mL X 3) , dried over anhydrous sodiusulfate and evaporated under reduced pressure. LCMS showed the starting material was consumed and desired product was detected. The resulting mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography eluting with EtOAc in PE from 0 to 10%to give tert-butyl ( (3- (6-bromopyridin-2-yl) isoxazol-5-yl) methyl) carbamate (323 mg, 0.9 mmol, 46%yield) as white solid. LC-MS: m/z [M+H]
+ 354.0.
Step 2: tert-butyl ( (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) isoxazol-5-yl) methyl) carbamate
A mixture of tert-butyl N- [ [3- (6-bromo-2-pyridyl) isoxazol-5-yl] methyl] carbamate (262 mg, 738 μmol) , (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (150 mg, 671 μmol) , Tris (Dibenzylideneacetone) Dipalladium (O) (123 mg, 134 μmol) , (5-diphenylphosphanyl-9, 9-dimethyl-xanthen-4-yl) -diphenyl-phosphane (155 mg, 268 μmol) and Cesium carbonate (657 mg, 2.02 mmol) in dioxane (15 mL) was stirred for 3 h at 100 ℃ in under N
2 until the reaction was complete as indicated by LCMS. The resulting mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography eluting with EtOAc in PE from 0 to 50%to give tert-butyl ( (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) isoxazol-5-yl) methyl) carbamate (320 mg, 644 μmol, 96%yield) as yellow oil. LC-MS: m/z [M+H]
+ 497.2.
Step 3: tert-butyl ( (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-
nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) isoxazol-5-
yl) methyl) carbamate
To a suspension of tert-butyl N- [ [3- [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -2-pyridyl] isoxazol-5-yl] methyl] carbamate (300 mg, 604 μmol) in DCE (10 mL) was added slowly (4-nitrophenyl) carbonochloridate (365 mg, 1.81 mmol) and DMAP (15 mg, 120 μmol) at 25 ℃ and stirred for 6 hours at 85 ℃ under N
2. LCMS showed the starting material was consumed and the desired product was detetced. The resulting mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography eluting with EtOAc in PE from 0 to 50%, then it was purified by flash column chromatography eluting with MeOH in DCM from 0 to 1%to give tert-butyl ( (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) isoxazol-5-yl) methyl) carbamate (250 mg, 377 μmol, 62%yield) as a yellow solid. LC-MS: m/z [M+H]
+ 662.2.
Step 4: (1R, 3S) -3- (5- ( (6- (5- (aminomethyl) isoxazol-3-yl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-
pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate
To a stirred solution of tert-butyl ( (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) isoxazol-5-yl) methyl) carbamate (240 mg, 362 μmol) in DCM (5 mL) solution at 25 ℃ was added TFA (5 mL) . The reaction mixture was stirred at 25 ℃ for 1 hour. LCMS showed the desired product was detected. The resulting mixture was concentrated under reduced pressure. The crude product was used immediately in the next step without further purification. LC-MS: m/z [M+H]
+ 562.2.
Step 5: (1
4Z, 4
4Z, 5
1S, 5
3R) -4
1- (tert-butyl) -4
1H-6-oxa-3, 8-diaza-1 (3, 5) -isoxazola-2 (2, 6) -pyridina-
4 (3, 5) -pyrazola-5 (1, 3) -cyclopentanacyclononaphan-7-one
To a stirred solution of (1R, 3S) -3- (5- ( (6- (5- (aminomethyl) isoxazol-3-yl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate (200 mg, 356 μmol) in CH
3CN (30 mL) solution at 25 ℃ was added TEA (5 mL) The reaction mixture was stirred at 50 ℃ for 12 h under N
2. LCMS showed the starting material was consumed and desired product was detected. The resulting mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography eluting with EtOAc in PE from 0 to 50%to give (1
4Z, 4
4Z, 5
1S, 5
3R) -41- (tert-butyl) -4
1H-6-oxa-3, 8-diaza-1 (3, 5) -isoxazola-2 (2, 6) -pyridina-4 (3, 5) -pyrazola-5 (1, 3) -cyclopentanacyclononaphan-7-one (60 mg, 142 μmol, 40%yield) as yellow solid and crude pruduct (150 mg crude) . LC-MS: m/z [M+H]
+ 423.2.
Step 6: (1
4Z, 4
4Z, 5
1S, 5
3R) -4
1H-6-oxa-3, 8-diaza-1 (3, 5) -isoxazola-2 (2, 6) -pyridina-4 (3, 5) -pyrazola-
5 (1, 3) -cyclopentanacyclononaphan-7-one
A suspension of (1
4Z, 4
4Z, 5
1S, 5
3R) -4
1- (tert-butyl) -4
1H-6-oxa-3, 8-diaza-1 (3, 5) -isoxazola-2 (2, 6) -pyridina-4 (3, 5) -pyrazola-5 (1, 3) -cyclopentanacyclononaphan-7-one (60 mg, 142 μmol) in TFA (4 mL) at 0 ℃ and stirred for 3 hours at 70 ℃ under N
2. LCMS showed the desired product was detected. The product was further purified by prep-HPLC (C18, 20-50%MeCN in 0.1%FA/water, 20 mL/min) to give (1
4Z, 4
4Z, 5
1S, 5
3R) -4
1H-6-oxa-3, 8-diaza-1 (3, 5) -isoxazola-2 (2, 6) -pyridina-4 (3, 5) -pyrazola-5 (1, 3) -cyclopentanacyclononaphan-7-one (20 mg, 54.8 μmol, 39 %yield) as yellow solid. LC-MS: m/z [M+H]
+ 367.1.
Example 92
(1
2Z, 4
4Z, 5
1S, 5
3R) -4
1H-6-oxa-3, 8-diaza-1 (2, 4) -oxazola-2 (2, 6) -pyridina-4 (3, 5) -pyrazola-5 (1, 3) -
cyclopentanacyclodecaphan-7-one


Step 1: 6-bromopicolinamide
To a suspension of 6-bromopicolinic acid (5.0 g, 24.8 mmol) in CH
2Cl
2 (25.0 mL) was added NH
4Cl (1.97 g, 37.2 mmol) , HATU (9.40 g, 24.8 mmol) and DIPEA (12.7 mL, 9.59 g, 74.4 mmol) at 25 ℃. The reaction mixture was stirred at that temperature for 2 h before it was quenched with ice-cold water (20 mL) and extracted with CH
2Cl
2 (20 mL × 3) . The combined organic phases were dried over anhydrous Na
2SO
4, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 42 %in 20 min) to afford 6-bromopicolinamide (4.0 g, 80%yield) as a white solid. LC-MS: m/z [M+H] + 201.0
Step 2: 4- (chloromethyl) -2- (6-chloropyridin-2-yl) oxazole
To a suspension of 6-bromopicolinamide (300 mg, 1.50 mmol) in 1, 3-dichloropropan-2-one (950 mg, 7.50 mmol) at 25 ℃. The reaction mixture was warmed to 130 ℃ and stirred at that temperature for 12 h before it was quenched with ice-cold water (20 mL) and extracted with CH
2Cl
2 (20 mL × 3) . The combinedorganic phases were dried over anhydrous Na
2SO
4, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 12 %in 20 min) to afford 4- (chloromethyl) -2- (6-chloropyridin-2-yl) oxazole (40.0 mg, 12%yield) as a white solid. LC-MS: m/z [M+H] + 229.0
Step 3: 2- (2- (6-chloropyridin-2-yl) oxazol-4-yl) acetonitrile
To a suspension of 4- (chloromethyl) -2- (6-chloropyridin-2-yl) oxazole (40.0 mg, 180 μmol) in CH
3CN (10.0 ml) was added TBAF (270 μL, 1 M in THF, 270 μmol) and TMSCN (27.0 mg, 270 μmol) at 25 ℃. The reaction mixture was warmed to 90 ℃ and stirred at that temperature for 30 min. The solution was concentrated under reduced pressure and the residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 10 %in 20 min) to afford 2- (2- (6-chloropyridin-2-yl) oxazol-4-yl) acetonitrile (38.0 mg, 97%yield) as a white solid. LC-MS: m/z [M+Na] + 242.2.
Step 4: 2- (2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-
2-yl) oxazol-4-yl) acetonitrile
To a stirred suspension of 2- (2- (6-chloropyridin-2-yl) oxazol-4-yl) acetonitrile (38.0 mg, 170 μmol) in 1, 4-dioxane (10.0 mL) were sequentially added (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (38.0 mg, 170 μmol) , Cs
2CO
3 (166 mg, 510 μmol) , XantPhos (20.0 mg, 34.0 μmol) and Pd
2 (dba)
3 (16.0 mg, 17.0 μmol) at 25 ℃. The reaction mixture was warmed to 100 ℃ and stirred at that temperature for 12 h. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 25 %in 20 min) to afford 2- (2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) oxazol-4-yl) acetonitrile (50.0 mg, 72%yield) as a yellow oil. LC-MS: m/z [M+H] + 407.2
Step 5: tert-butyl (2- (2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) oxazol-4-yl) ethyl) carbamate
To a suspension of 2- (2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) oxazol-4-yl) acetonitrile (50.0 mg, 120 μmol) in MeOH (10.0 mL) was added NiCl
2 (1.60 mg, 12.0 μmol) , NaBH
4 (28.0 mg, 720 μmol) , (Boc)
2O (0.42 mL, 40.0 mg, 180 μmol) at 0 ℃. The reaction mixture was stirred at that temperature for 2 h before it was filtered and concentrated in vacuo to afford tert-butyl (2- (2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) oxazol-4-yl) ethyl) carbamate (40.0 mg, 65%yield) as a yellow oil. LC-MS: m/z [M+H] + 511.2
Step 6: (1R, 3S) -3- (5- ( (6- (4- (2- ( (tert-butoxycarbonyl) amino) ethyl) oxazol-2-yl) pyridin-2-
yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a suspension of tert-butyl (2- (2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) oxazol-4-yl) ethyl) carbamate (40.0 mg, 78.0 μmol) in CH
2Cl
2 (10.0 mL) were sequentially added CDI (38.0 mg, 240 μmol) and DIPEA (66.0 μL, 50.0 mg, 390 μmol) at 25 ℃. The reaction mixture was warmed to 35 ℃ and stirred at that temperature for 12 h before it was quenched with ice-cold water (20 mL) and extracted with CH
2Cl
2 (20 mL × 3) . The combined organic phases were dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure to afford (1R, 3S) -3- (5- ( (6- (4- (2- ( (tert-butoxycarbonyl) amino) ethyl) oxazol-2-yl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate as a yellow oil.. LC-MS: m/z [M+H] + 605.2
Step 7: (1R, 3S) -3- (5- ( (6- (4- (2-aminoethyl) oxazol-2-yl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-
pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a stirred solution of (1R, 3S) -3- (5- ( (6- (4- (2- ( (tert-butoxycarbonyl) amino) ethyl) oxazol-2-yl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate in CH
2Cl
2 (10.0 ml) was added TFA (396 μL, 586 mg, 5.14 mmol) at 25 ℃. The mixture was stirred at that temperature for 3 h before it was concentrated under reduced pressure to afford (1R, 3S) -3- (5- ( (6- (4- (2-aminoethyl) oxazol-2-yl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate as a yellow oil, which was used in the next step without further purification. LC-MS: m/z [M+H] + 505.2
Step 8: (1
2Z, 4
4Z, 5
1S, 5
3R) -4
1- (tert-butyl) -4
1H-6-oxa-3, 8-diaza-1 (2, 4) -oxazola-2 (2, 6) -pyridina-
4 (5, 3) -pyrazola-5 (1, 3) -cyclopentanacyclodecaphan-7-one
To a stirred suspension of (1R, 3S) -3- (5- ( (6- (4- (2-aminoethyl) oxazol-2-yl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate in CH
3CN (10.0 mL) was added Et
3N (269 μL, 196 mg, 1.94 mmol) at 25 ℃. The reaction mixture was warmed to 80 ℃ and stirred at that temperature for 12 h. The reaction mixture was cooled to 25 ℃, filtered and concentrated under reduced pressure. Then it was concentrated to afford (1
2Z, 4
4Z, 5
1S, 5
3R) -4
1- (tert-butyl) -4
1H-6-oxa-3, 8-diaza-1 (2, 4) -oxazola-2 (2, 6) -pyridina-4 (5, 3) -pyrazola-5 (1, 3) -cyclopentanacyclodecaphan-7-one as a yellow oil, which was used in the next step without further purification. LC-MS: m/z [M+H] + 437.3
Step 9: (1
2Z, 4
4Z, 5
1S, 5
3R) -4
1H-6-oxa-3, 8-diaza-1 (2, 4) -oxazola-2 (2, 6) -pyridina-4 (3, 5) -pyrazola-
5 (1, 3) -cyclopentanacyclodecaphan-7-one
A solution of (1
2Z, 4
4Z, 5
1S, 5
3R) -4
1- (tert-butyl) -4
1H-6-oxa-3, 8-diaza-1 (2, 4) -oxazola-2 (2, 6) -pyridina-4 (5, 3) -pyrazola-5 (1, 3) -cyclopentanacyclodecaphan-7-one in HCO
2H (5.0 mL) was warmed to 100 ℃and stirred at that temperature for 5 h before it was cooled, concentrated under reduced pressure. The residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 5%to 65%in 20 min) to afford (1
2Z, 4
4Z, 5
1S, 5
3R) -4
1H-6-oxa-3, 8-diaza-1 (2, 4) -oxazola-2 (2, 6) -pyridina-4 (3, 5) -pyrazola-5 (1, 3) -cyclopentanacyclodecaphan-7-one (0.5 mg, 1.7%yield) as a white solid. LC-MS: m/z [M+H] + 380.9.
Example 195
(1
1S, 1
3R, Z) -2
1H-12-oxa-3, 7, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-6, 11-dione


Step 1: tert-butyl (2- (2- (6-bromopyridin-2-yl) acetamido) ethyl) carbamate
To a solution of 2- (6-bromopyridin-2-yl) acetic acid (250 mg, 1.16 mmol) in DCM (7 mL) was added tert-butyl (2-aminoethyl) carbamate (241 mg, 1.50 mmol) , EDCI (443 mg, 2.31 mmol) , HOBT (312 mg, 2.31 mmol) and DIPEA (448 mg, 3.47 mmol) in DCM (1 mL) . The reaction mixture was stirred at 25 ℃ for 3 hours. The mixture was diluted with water (50 ml) and extracted with DCM (2 x 50 ml) . The combined organic layers were washed by brine (50 ml) and dried over Na
2SO
4, filtered and concentrated to dryness. The residue was purified by flash column chromatography (12 g silica gel column, Petrol ether/EtOAc with EtOAc from 0~74%) to afford the product tert-butyl (2- (2- (6-bromopyridin-2-yl) acetamido) ethyl) carbamate (400 mg, 1.12 mmol, 96%yield) as a white solid. LC-MS: (ESI) m/z [M+H]
+ 358.0.
Step 2: tert-butyl (2- (2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) acetamido) ethyl) carbamate
To a suspension of tert-butyl (2- (2- (6-bromopyridin-2-yl) acetamido) ethyl) carbamate (210 mg, 586 μmol) and (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (157 mg, 703 μmol) in dioxane (8 mL) was added diacetoxypalladium (26 mg, 117 μmol) , Cesium carbonate (573 mg, 1.76 mmol) and [1- [ (1R) -2-diphenylphosphanyl-1-naphthyl] -2-naphthyl] -diphenyl-phosphane (146 mg, 234 μmol) . The suspension was degassed with N
2 for 5 times. The mixture was stirred at 80 ℃for 16 hour. The mixture was dilutied with water (50 mL) and extracted with EtOAc (50 mL × 2) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrated to dryness. The residue was purified by flash column chromatography (12 g silica gel column, DCM/MeOH with MeOH from 0~10%) to afford tert-butyl (2- (2- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) acetamido) ethyl) carbamate (285 mg, 569 μmol, 97%yield) as a black oil. LC-MS: (ESI) m/z [M+H]
+ 501.3.
Step 3: (1R, 3S) -3- (5- ( (6- (2- ( (2- ( (tert-butoxycarbonyl) amino) ethyl) amino) -2-oxoethyl) pyridin-2-
yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of tert-butyl N- [2- [ [2- [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -2-pyridyl] acetyl] amino] ethyl] carbamate (200 mg, 399 μmol) in DCM (4 mL) was added 1, 1'-Carbonyldiimidazole (324 mg, 2.00 mmol) . The reaction mixture was degassed with N
2 for 5 times. The mixture was stirred at 50 ℃ for 6 hours. The mixture was diluted with water (30 mL) and extracted with DCM (30mL × 3) . The combined organic layer was washed with brine (30 mL × 3) , dried over Na
2SO
4, filtered and concentrated to dryness. The residue was purified by flash column chromatography (12 g silica gel column, Petrol ether/EtOAc with EtOAc from 0~95%) to afford the product (1R, 3S) -3- (5- ( (6- (2- ( (2- ( (tert-butoxycarbonyl) amino) ethyl) amino) -2-oxoethyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (110 mg, 185 μmol, 46%yield) . LC-MS: (ESI) m/z [M+H]
+ 595.2.
Step 4: (1R, 3S) -3- (5- ( (6- (2- ( (2-aminoethyl) amino) -2-oxoethyl) pyridin-2-yl) amino) -1- (tert-butyl) -
1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of (1R, 3S) -3- (5- ( (6- (2- ( (2- ( (tert-butoxycarbonyl) amino) ethyl) amino) -2-oxoethyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (110 mg, 185 μmol) in DCM (2 mL) was added TFA (740 mg, 6.49 mmol) . The mixture was stirred at 25 ℃ for 2 hours. The mixture was concentrated to afford the crude product (1R, 3S) -3- (5- ( (6- (2- ( (2-aminoethyl) amino) -2-oxoethyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (365 mg) as a yellow oil which was used in the next step without purification. LC-MS: (ESI) m/z [M+H]
+ 495.3.
Step 5: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-12-oxa-3, 7, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphane-6, 11-dione
To a solution of [ (1R, 3S) -3- [5- [ [6- [2- (2-aminoethylamino) -2-oxo-ethyl] -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (90 mg, 182 μmol) in MeCN (40 mL) was added Et
3N (368 mg, 3.64 mmol) . The reaction mixture was degassed with N
2 for 5 times. The reaction was stirred at 90 ℃ for 48 hours. The mixture was concentrated to dryness. The mixture was diluted with water (50 mL) and extracted with DCM (50 mL × 3) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrated to dryness. The residue was purified by flash column chromatography (4 g silica gel column, DCM/MeOH with MeOH from 0~6%) to afford the product afford the product (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-12-oxa-3, 7, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-6, 11-dione (75 mg, 175 μmol, 96%yield) as a yellow solid. LC-MS: (ESI) m/z [M+H]
+ 427.2.
Step 6: (1
1S, 1
3R, Z) -2
1H-12-oxa-3, 7, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-6, 11-dione
The solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-12-oxa-3, 7, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-6, 11-dione (70 mg, 164 μmol) in TFA (5 mL) was stirred at 75 ℃ for 5 hours. The mixture was concentrated to afford crude solid which was purified by prep-HPLC (Chromatographic columns: C18 50 x 2.1 mm, Mobile Phase: ACN-H
2O(0.05%TFA) Gradient: 5%-95%) to afford the product (1
1S, 1
3R, Z) -2
1H-12-oxa-3, 7, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-6, 11-dione (10.2 mg, 27.0 μmol, 16%yield, 98%purity) as a white solid. LC-MS: (ESI) m/z [M+H]
+ 371.2.
Example 93
(1
1S, 1
3R, Z) -4
1-methyl-2
1H, 4
1H-11-oxa-3, 6, 9-triaza-4 (5, 3) -pyrrolo [3, 2-b] pyridina-2 (5, 3) -pyrazola-
1 (1, 3) -cyclopentanacycloundecaphane-5, 10-dione

Step 1: 5-chloro-1-methyl-pyrrolo [3, 2-b] pyridine-3-carboxylic acid
To a suspension of methyl 5-chloro-1-methyl-pyrrolo [3, 2-b] pyridine-3-carboxylate (200 mg, 0.89 mmol) in THF (5.0 mL) and H
2O (5.0 mL) was added LiOH
. H
2O (74.7 mg, 1.78 mmol) at 25 ℃and stirred for 16 h. The reaction mixture was concentrated and adjusted to pH = 2~3 with 2 M HCl and extracted with EtOAc (3 × 20 mL) . The organic phase was dried over anhydrous Na
2SO
4, filtered and the filtrate was concentrated under reduced pressure to afford 5-chloro-1-methyl-pyrrolo [3, 2-b] pyridine-3-carboxylic acid (200 mg, crude) as a yellow solid. The crude product was used in the next step without further purification. LC-MS: m/z [M+H]
+ 210.9.
Step 2: tert-butyl N- [2- [ (5-chloro-1-methyl-pyrrolo [3, 2-b] pyridine-3-carbonyl) amino] ethyl]
carbamate
To a suspension of 5-chloro-1-methyl-pyrrolo [3, 2-b] pyridine-3-carboxylic acid (200 mg, crude) in CH
2Cl
2 (20.0 mL) was sequentially added tert-butyl N- (2-aminoethyl) carbamate (152 mg, 0.949 mmol) , HATU (722 mg, 1.9 mmol) and DIPEA (330 μL, 245 mg, 1.90 mmol) at 25 ℃ and stirred for 2 h. After completion of the reaction as judged by LCMS, the reaction mixture was concentrated under reduced pressure. The crude product was purified by flash chromatography eluting with MeOH /CH
2Cl
2 (with MeOH from 0 to 5%in 25 min) to afford tert-butyl N- [2- [ (5-chloro-1-methyl-pyrrolo [3, 2-b] pyridine-3-carbonyl) amino] ethyl] carbamate (280 mg, 83%yield) as a yellow solid. LC-MS: m/z [M+H]
+ 352.9.
Step 3: tert-butyl N- [2- [ [5- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -1-
methyl-pyrrolo [3, 2-b] pyridine-3-carbonyl] amino] ethyl] carbamate
To a suspension of tert-butyl N- [2- [ (5-chloro-1-methyl-pyrrolo [3, 2-b] pyridine-3-carbonyl) amino] ethyl] carbamate (240 mg, 0.680 mmol) in 1, 4-dioxane (20.0 mL) was sequentially added (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (151 mg, 0.680 mmol) , Cs
2CO
3 (665 mg, 2.04 mmol) , XantPhos (78 mg, 0.136 mmol) and Pd
2 (dba)
3 (62 mg, 0.068 mmol) . The reaction was warmed to 100 ℃ stirred at that temperature for 16 h under N
2. After completion of the reaction as judged by LCMS. The reaction was filtered and concentrated under reduced pressure, the crude product was purified by flash column chromatography eluting with MeOH/CH
2Cl
2 (with MeOH from 0 to 5%in 20 min) to afford tert-butyl N- [2- [ [5- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -1-methyl-pyrrolo [3, 2-b] pyridine-3-carbonyl] amino] ethyl] carbamate (200 mg, 54%yield) as a white solid. LC-MS: m/z [M+H]
+ 539.9.
Step 4: [ (1R, 3S) -3- [5- [ [3- [2- (tert-butoxycarbonylamino) ethylcarbamoyl] -1-methyl-pyrrolo [3, 2-
b] pyridin-5-yl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate
To a suspension of tert-butyl N- [2- [ [5- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -1-methyl-pyrrolo [3, 2-b] pyridine-3-carbonyl] amino] ethyl] carbamate (180 mg, 0.333 mmol) in CH
2Cl
2 (10.0 mL) was added Et
3N (231 μL, 168 mg, 1.67 mmol) and CDI (128 mg, 0.888 mmol) at 35 ℃ and stirred for 16 h. After completion of the reaction as judged by LCMS, reaction mixture was quenched with water (20 mL) and extracted with CH
2Cl
2 (3 × 20 mL) . The organic phase was dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The crude product [ (1R, 3S) -3- [5- [ [3- [2- (tert-butoxycarbonylamino) ethylcarbamoyl] -1-methyl-pyrrolo [3, 2-b] pyridin-5-yl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (crude) was used in the next step without further purification. LC-MS: m/z [M+H]
+ 633.9.
Step 5: [ (1R, 3S) -3- [5- [ [3- (2-aminoethylcarbamoyl) -1-methyl-pyrrolo [3, 2-b] pyridin-5-yl] amino] -
1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate
To a suspension of [ (1R, 3S) -3- [5- [ [3- [2- (tert-butoxycarbonylamino) ethylcarbamoyl] -1-methyl-pyrrolo [3, 2-b] pyridin-5-yl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate in CH
2Cl
2 (3.0 mL) was added slowly TFA (1.0 mL) at 25 ℃ and stirred for 1 h. After completion of the reaction as judged by LCMS, reaction mixture was concentrated under reduced pressure to afford [ (1R, 3S) -3- [5- [ [3- (2-aminoethylcarbamoyl) -1-methyl-pyrrolo [3, 2-b] pyridin-5-yl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate as a yellow oil. The crude product was used in the next step without further purification. LC-MS: m/z [M+H]
+ 534.0.
Step 6: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
1-methyl-2
1H, 4
1H-11-oxa-3, 6, 9-triaza-4 (5, 3) -pyrrolo [3, 2-
b] pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacycloundecaphane-5, 10-dione
To a suspension of [ (1R, 3S) -3- [5- [ [3- (2-aminoethylcarbamoyl) -1-methyl-pyrrolo [3, 2-b] pyridin-5-yl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate in CH
3CN (5.0 mL) was added Et
3N (312 μL, 227 mg, 2.25 mmol) at room temperature and the reaction was stirred at 80 ℃for 16 h. After completion of the reaction as judged by LCMS, reaction mixture was concentrated under reduced pressure. The crude product was purified by prep-TLC (CH
2Cl
2/MeOH = 15/1) to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
1-methyl-2
1H, 4
1H-11-oxa-3, 6, 9-triaza-4 (5, 3) -pyrrolo [3, 2-b] pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacycloundecaphane-5, 10-dione (60 mg) as a yellow solid. LC-MS: m/z [M+H]
+ 466.0.
Step 7: (1
1S, 1
3R, Z) -4
1-methyl-2
1H, 4
1H-11-oxa-3, 6, 9-triaza-4 (5, 3) -pyrrolo [3, 2-b] pyridina-2 (5, 3) -
pyrazola-1 (1, 3) -cyclopentanacycloundecaphane-5, 10-dione
A mixture of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
1-methyl-2
1H, 4
1H-11-oxa-3, 6, 9-triaza-4 (5, 3) -pyrrolo [3, 2-b] pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacycloundecaphane-5, 10-dione (50.0 mg, 0.107 mmol) in TFA (3.0 mL) was stirred at 70 ℃ for 6 h. The reaction mixture was concentrated under reduced pressure and purified by flash column chromatography eluting with MeOH/CH
2Cl
2 (with MeOH from 0 to 15%in 25 min) to give (1
1S, 1
3R, Z) -4
1-methyl-2
1H, 4
1H-11-oxa-3, 6, 9-triaza-4 (5, 3) -pyrrolo [3, 2-b] pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacycloundecaphane-5, 10-dione (37.6 mg, 85%yield) as an off-white solid. LC-MS: m/z [M+H]
+ 409.8.
Example 94
(1
1S, 1
3R, Z) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione
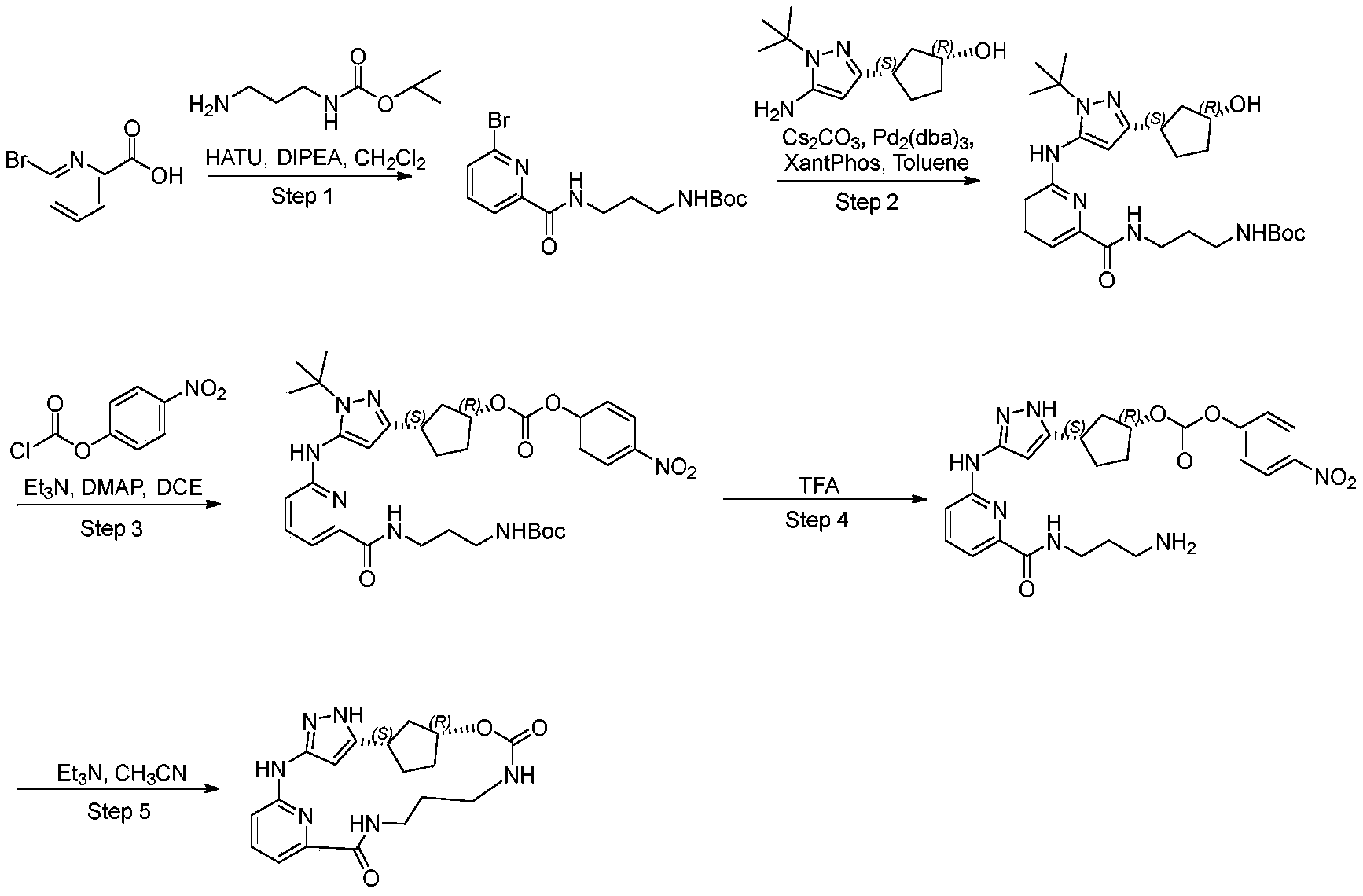
Step 1: tert-butyl (3- (6-bromopicolinamido) propyl) carbamate
To a stirred solution of 6-bromopicolinic acid (2.0 g, 9.90 mmol) and tert-butyl (3-aminopropyl) carbamate (1.72 g, 9.90 mmol) in CH
2Cl
2 (30.0 mL) were sequentially added HATU (7.52 g, 19.8 mmol) and DIPEA (3.44 mL, 2.55 g, 19.8 mmol) at 25 ℃. The reaction mixture was stirred at that temperature for 1 h before it was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 30%in 15 min) to give tert-butyl (3- (6-bromopicolinamido) propyl) carbamate (3.1 g, 88%yield) as a yellow oil. LC-MS: m/z [M+Na]
+ 379.7.
Step 2: tert-butyl (3- (6- ( (1- (tert-butyl) -5- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-3-
yl) amino) picolinamido) propyl) carbamate
To a stirred solution of tert-butyl (3- (6-bromopicolinamido) propyl) carbamate (500 mg, 1.40 mmol) and (1R, 3S) -3- (3-amino-1- (tert-butyl) -1H-pyrazol-5-yl) cyclopentan-1-ol (312 mg, 1.40 mmol) in toluene (20.0 mL) were sequentially added Pd
2 (dba)
3 (128 mg, 140 μmol) , XantPhos (162 mg, 280 μmol) and Cs
2CO
3 (913 mg, 2.80 mmol) at 25 ℃. The reaction mixture was warmed to 80 ℃ and stirred at that temperature for 16 h under N
2 before it was cooled to 25 ℃, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 80 %in 25 min) to give tert-butyl (3- (6- ( (1- (tert-butyl) -5- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-3-yl) amino) picolinamido) propyl) carbamate (500 mg, 71%yield) as a red solid. LC-MS: m/z [M+H]
+ 500.9.
Step 3: tert-butyl (3- (6- ( (1- (tert-butyl) -5- ( (1S, 3R) -3- ( ( (4nitrophenoxy) carbonyl) oxy) cyclopentyl) -
1H-pyrazol-3-yl) amino) picolinamido) propyl) carbamate
To a stirred solution of tert-butyl (3- (6- ( (1- (tert-butyl) -5- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-3-yl) amino) picolinamido) propyl) carbamate (300 mg, 599 μmol) and 4-nitrophenyl carbonochloridate (133 mg, 659 μmol) in DCE (10.0 mL) were sequentially added DMAP (15.0 mg, 120 μmol) and Et
3N (166 μL, 121 mg, 1.20 mmol) at 25 ℃. The reaction mixture was warmed to 70 ℃ and stirred at that temperature for 16 h before it was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 65 %in 20 min) to give tert-butyl (3- (6- ( (1- (tert-butyl) -5- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-3-yl) amino) picolinamido) propyl) carbamate (140 mg, 35%yield) as a light red solid. LC-MS: m/z [M+H]
+ 666.2.
Step 4: (1R, 3S) -3- (3- ( (6- ( (3-aminopropyl) carbamoyl) pyridin-2-yl) amino) -1H-pyrazol-5-
yl) cyclopentyl (4-nitrophenyl) carbonate
A solution of tert-butyl (3- (6- ( (1- (tert-butyl) -5- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-3-yl) amino) picolinamido) propyl) carbamate (120 mg, 180 μmol) in TFA (10.0 mL) was warmed to 100 ℃ and stirred at that temperature for 3 h before it was cooled to 25 ℃ and concentrated under reduced pressure. The crude (1R, 3S) -3- (3- ( (6- ( (3-aminopropyl) carbamoyl) pyridin-2-yl) amino) -1H-pyrazol-5-yl) cyclopentyl (4-nitrophenyl) carbonate (220 mg) was directly used in the next step without further purification. LC-MS: m/z [M+H]
+ 509.8.
Step 5: (1
1S, 1
3R, Z) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione
To a stirred solution of (1R, 3S) -3- (3- ( (6- ( (3-aminopropyl) carbamoyl) pyridin-2-yl) amino) -1H-pyrazol-5-yl) cyclopentyl (4-nitrophenyl) carbonate (220 mg, crude) in CH
3CN (10.0 mL) was added Et
3N (298 μL, 217 mg, 2.15 mmol) at 25 ℃. The reaction mixture was stirred at that temperature for 3 h before it was filtrated and the filter cake was washed with MeOH to give (1
1S, 1
3R, Z) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (27 mg) as an off-white solid. LC-MS: m/z [M+H]
+ 370.8.
The following compounds were prepared using the similar procedure disclosed in synthetic example 94.



Example 112
(1
1S, 1
3R, Z) -4
5-chloro-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione


Step 1: tert-butyl (3- (6-bromo-3-chloropicolinamido) propyl) carbamate
A solution of tert-butyl (3-aminopropyl) carbamate (200 mg, 1.15 mmol) in DCM (1 mL) was added to a solution of 6-bromo-3-chloropicolinic acid (407 mg, 1.72 mmol) in DCM (7 mL) . The reaction mixtrue was stirred at 25 ℃ for 3 hours. The mixture was diluted with water (50 mL) and extracted with DCM (50 mL x 3) . The combined organic layer was washed by brine (50 mL) and dried over Na
2SO
4, filtered and concentrated to dryness. The residue was purified by flash column chromatography (12 g silica gel column, Petrol ether/EtOAc from 0~45%) to afford the product tert-butyl (3- (6-bromo-3-chloropicolinamido) propyl) carbamate (420 mg, 1.07 mmol, 93%yield) as a white solid. LC-MS: (ESI) m/z [M+Na]
+ 416.0.
Step 2: tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) -3-chloropicolinamido) propyl) carbamate
To a suspension of tert-butyl (3- (6-bromo-3-chloropicolinamido) propyl) carbamate (220 mg, 560 μmol) and (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (150 mg, 672 μmol) in dioxane (6 mL) was added Pd
2 (dba)
3 (102 mg, 112 μmol) , Cs
2CO
3 (548 mg, 1.68 mmol) and XantPhos (129 mg, 224 μmol) . The suspension was degassed with N
2 for 5 times and stirred at 90℃for 6 hours. The mixture was dilutied with water (50 mL) and extracted with EtOAc (50 mL x 3) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrated to dryness. The residue was purified by flash column chromatography (12 g silica gel column, Petrol ether/EtOAc from 0~61%) to afford the product tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -3-chloropicolinamido) propyl) carbamate (270 mg, 504 μmol, 90%yield) as a yellow solid. LC-MS: (ESI) m/z [M+H]
+ 535.2.
Step 3: (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propyl) carbamoyl) -5-chloropyridin-2-
yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -3-chloropicolinamido) propyl) carbamate (180 mg, 336 μmol) in THF (6 mL) was added di (1H-imidazol-1-yl) methanone (436 mg, 2.69 mmol) . The reaction mixture was degassed with N
2 for 5 times. The mixture was stirred at 75 ℃ for 5 hours. The mixture was concentrated to dryness. The residue was purified by flash column chromatography (12 g silica gel column, Petrol ether/EtOAc from 0~81%) to afford the product (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propyl) carbamoyl) -5-chloropyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (210 mg, 296 μmol, 88%yield, 89%purity) as a white solid. LC-MS: (ESI) m/z [M+H]
+ 629.2.
Step 4: (1R, 3S) -3- (5- ( (6- ( (3-aminopropyl) carbamoyl) -5-chloropyridin-2-yl) amino) -1- (tert-butyl) -
1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propyl) carbamoyl) -5-chloropyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (210 mg, 334 μmol) in DCM (6.0 mL) was added TFA (2.2 g, 19.4 mmol, 1.49 mL) . The mixture was stirred at 25 ℃ for 2 hours. The mixture was concentrated to afford the product (1R, 3S) -3- (5- ( (6- ( (3-aminopropyl) carbamoyl) -5-chloropyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (454 mg, crude) as a yellow gum. LC-MS: (ESI) m/z [M+H]
+ 529.1.
Step 5: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5-chloro-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
To a solution of (1R, 3S) -3- (5- ( (6- ( (3-aminopropyl) carbamoyl) -5-chloropyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (454 mg, 858 μmol) in MeCN (90.0 mL) was added Et
3N (1.20 mL) at 25 ℃. The reaction mixture was degassed with N
2 for 5 times. The mixture was stirred at 90 ℃ for 6 hours and then concentrated to dryness. The mixture was diluted with water (50 mL) and extracted with DCM (50 mL × 3) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrated to dryness. The residue was purified by flash column chromatography (4 g silica gel column, DCM/MeOH from 0~5%) to afford the product (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5-chloro-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (100 mg, 154 μmol, 18%yield, 71%purity) as a yellow solid. LC-MS: (ESI) m/z [M+H]
+ 461.2.
Step 6: (1
1S, 1
3R, Z) -4
5-chloro-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione
The mixture of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5-chloro-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (90.0 mg, 195 μmol) in TFA (5 mL) was stirred at 75 ℃ for 7 hours. The mixture was concentrated to afford crude solid, which was purified by prep-HPLC (Chromatographic columns: C18 50 x 2.1 mm, Mobile Phase: ACN-H
2O (0.05%TFA) Gradient: 5%-95%) to afford the product (1
1S, 1
3R, Z) -4
5-chloro-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (15.1 mg, 19%yield) as a white solid. LC-MS: (ESI) m/z [M+H]
+ 405.1.
The following compounds were prepared using the similar procedure disclosed in synthetic example 112.
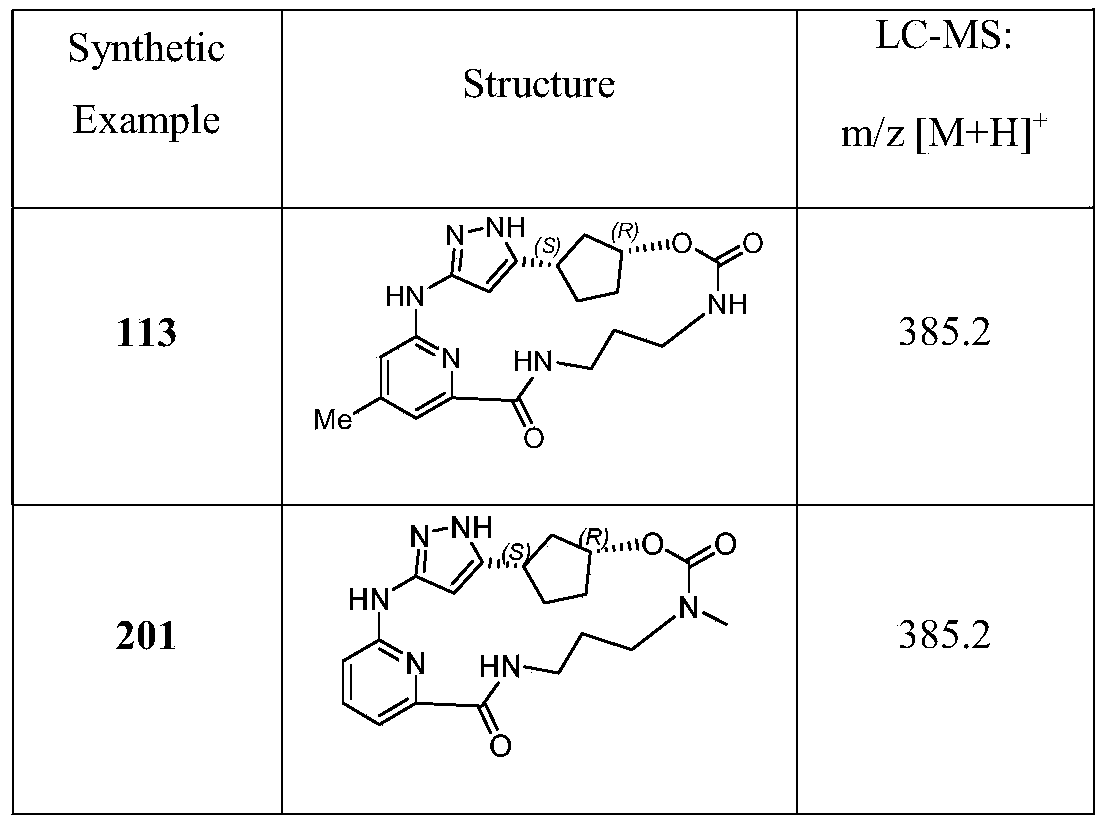
Example 114
(1
1S, 1
3R, Z) -4
4- (2- (trifluoromethyl) pyridin-4-yl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione


Step 1: tert-butyl (3- (6-bromo-4-chloropicolinamido) propyl) carbamate
To a solution of 6-bromo-4-chloro-pyridine-2-carboxylic acid (2.80 g, 11.8 mmol) in DCM (20 mL) was added oxalyl dichloride (2.25 g, 17.8 mmol, 1.59 mL) and one drop of DMF at 25 ℃. The reaction mixture was stirred at 25 ℃ for 2 h. The mixture was concentrated under reduced pressure. The residue was dissolved in DCM (10 mL) . The solution of the intermediate was added dropwise to the solution of tert-butyl N- (3-aminopropyl) carbamate (3.09 g, 17.8 mmol, 3.11 mL) and DIPEA (7.65 g, 59.2 mmol, 10.3 mL) in DCM (20 mL) . Then the reaction mixture was stirred at 25 ℃ for 2 h. The mixture was quenched with MeOH (10 mL) , then the mixture was concentrated under reduced pressure, and the residue was purified by silica gel chromatography eluting with EtOAc/PE with EtOAc from 0 to 40%in 15 min to afford the product tert-butyl N- [3- [ (6-bromo-4-chloro-pyridine-2- carbonyl) amino] propyl] carbamate (3.80 g, 82%yield) as a yellow solid. LC-MS: (ESI) m/z [M+Na]
+413.9.
Step 2: tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) -4-chloropicolinamido) propyl) carbamate
To a solution of tert-butyl N- [3- [ (6-bromo-4-chloro-pyridine-2-carbonyl) amino] propyl] carbamate (3.80 g, 9.68 mmol) and (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (2.16 g, 9.68 mmol) in dioxane (25 mL) was added Pd
2 (dba)
3 (532 mg, 581 μmol) , XantPhos (672 mg, 1.16 mmol) and Cs
2CO
3 (7.88 g, 24.2 mmol) under the atmosphere of N
2 at 20 ℃. Then the reaction mixture was heated to 100 ℃ and stirred at 100 ℃ under the atmosphere of N
2 for 5 h. The mixture was cooled to room temperature and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE with EtOAc from 0 to 60%in 20 min to afford the product tert-butyl N- [3- [ [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -4-chloro-pyridine-2-carbonyl] amino] propyl] carbamate (4.80 g, 93%yield) as a yellow solid. LC-MS: (ESI) m/z [M+H]
+ 535.3.
Step 3: tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-
nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) -4-
chloropicolinamido) propyl) carbamate & 4-nitrophenyl (6- ( (3- ( (tert-
butoxycarbonyl) amino) propyl) carbamoyl) -4-chloropyridin-2-yl) (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-
nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) carbamate
To a solution of tert-butyl N- [3- [ [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -4-chloro-pyridine-2-carbonyl] amino] propyl] carbamate (4.80 g, 8.97 mmol) in DCE (30 mL) was added DMAP (219 mg, 1.79 mmol) and DIPEA (5.80 g, 44.9 mmol, 7.80 mL) , then (4-nitrophenyl) carbonochloridate (5.42 g, 26.9 mmol) was added to the reaction mixture. The reaction mixture was heated to 80 ℃ and stirred at 80 ℃ for 15 h. The mixture was cooled to room temperature and diluted with DCM (150 mL) , then the mixture was washed with brine (150 mL) , dried over anhydrous Na
2SO
4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE with EtOAc from 0 to 60%in 15 min to afford the mixture of product [ (1R, 3S) -3- [5- [ [6- [3- (tert-butoxycarbonylamino) propylcarbamoyl] -4-chloro-2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] (4-nitrophenyl) carbonate and 4-nitrophenyl (6- ( (3- ( (tert-butoxycarbonyl) amino) propyl) carbamoyl) -4-chloropyridin-2-yl) (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) carbamate (5.60 g, 89%yield) as a yellow solid. LC-MS: (ESI) m/z [M+H]
+ 700.3.865.1
Step 4: (1R, 3S) -3- (5- ( (6- ( (3-aminopropyl) carbamoyl) -4-chloropyridin-2-yl) amino) -1- (tert-butyl) -
1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate & 4-nitrophenyl (6- ( (3-
aminopropyl) carbamoyl) -4-chloropyridin-2-yl) (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-
nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) carbamate
To a solution of [ (1R, 3S) -3- [5- [ [6- [3- (tert-butoxycarbonylamino) propylcarbamoyl] -4-chloro-2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] (4-nitrophenyl) carbonate (5.60 g, 8.00 mmol) in DCM (10 mL) was added TFA (5.47 g, 48.0 mmol, 3.71 mL) at 20 ℃. Then the reaction mixture was stirred at 20 ℃ for 3 h. The mixture was concentrated under reduced pressure to afford the mixture of product [ (1R, 3S) -3- [5- [ [6- (3-aminopropylcarbamoyl) -4-chloro-2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] (4-nitrophenyl) carbonate and 4-nitrophenyl (6- ( (3-aminopropyl) carbamoyl) -4-chloropyridin-2-yl) (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) carbamate (6.00 g, crude, TFA) as a yellow gum which was used for next step without further purification. LC-MS: (ESI) m/z [M+H]
+ 600.3, 765.1.
Step 5: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4-chloro-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione & 4-nitrophenyl (1
1S, 1
3R, Z) -2
1- (tert-
butyl) -4
4-chloro-5, 11-dioxo-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-3-carboxylate
To a solution of (1R, 3S) -3- (5- ( (6- ( (3-aminopropyl) carbamoyl) -4-chloropyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate (6.00 g, 8.40 mmol, TFA) and 4-nitrophenyl (6- ( (3-aminopropyl) carbamoyl) -4-chloropyridin-2-yl) (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) carbamate in CH
3CN (80 mL) was added dropwise the DIPEA (10.9 g, 84.0 mmol, 15.0 mL) slowly at 20 ℃. Then the reaction mixture was stirred at 20 ℃ for 72 h. The mixture was concentrated under reduced pressure, and the residue was redissolved in CH
3CN (15 mL) . Then DIPEA (50.0 mL) was added to the reaction mixture. The reaction mixture was stirred at 85 ℃ for 3 d. The mixture was concentrated under reduced pressure, and the residue was purified by silica gel chromatography eluting with EtOAc/DCM with EtOAc from 0 to 50%in 20 min to afford the product (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4-chloro-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (2.00 g, 52%yield) as a yellow solid. LC-MS: (ESI) m/z [M+H]
+ 461.3. And the by-product 4-nitrophenyl (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4-chloro-5, 11-dioxo-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-3-carboxylate (0.500 g, 10%yield) as a yellow solid, LC-MS: (ESI) m/z [M+H] + 626.1.
Step 6: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- (2- (trifluoromethyl) pyridin-4-yl) -2
1H-12-oxa-3, 6, 10-triaza-
4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
To a solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4-chloro-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (30.0 mg, 65.1 μmol) and 4- (4, 4, 5, 5- tetramethyl-1, 3, 2-dioxaborolan-2-yl) -2- (trifluoromethyl) pyridine (35.5 mg, 130 μmol) in dioxane (8.0 mL) was added Pd (OAc)
2 (2.92 mg, 13.0 μmol) , X-Phos (12.4 mg, 26.0 μmol) and Water (0.10 mL) under the atmosphere of N
2. Then the reaction mixture was heated to 100 ℃ and stirred at 100 ℃ for 5 h. The mixture was cooled to room temperature and diluted with DCM (20 mL) , filtered and the filtrate was concentrated under reduced pressure. The residue was purified by Prep-TLC (PE/EtOAc=4: 5) to afford the product (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- (2- (trifluoromethyl) pyridin-4-yl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (36.0 mg, 97%yield) as a yellow solid. LCMS: (ESI) m/z [M+H]
+ 572.2.
Step 7: (1
1S, 1
3R, Z) -4
4- (2- (trifluoromethyl) pyridin-4-yl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -
pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
The solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- (2- (trifluoromethyl) pyridin-4-yl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (36.0 mg, 63.0 μmol) in TFA (8.0 mL) was stirred at 70 ℃ for 4 d. The mixture was concentrated under reduced pressure, and the residue was purified by Prep-HPLC eluting with CH
3CN in water with CH
3CN from 30%to 40%in 8 min to afford the product (1
1S, 1
3R, Z) -4
4- (2- (trifluoromethyl) pyridin-4-yl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (21.0 mg, 65%yield) as a yellow solid. LC-MS: (ESI) m/z [M+H]
+ 516.2.
The following compounds were prepared using the similar procedure disclosed in synthetic example 114.


Example 123
(1
1S, 1
3R, Z) -4
4-morpholino-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione

Step 1: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4-morpholino-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
To a solution of 4-nitrophenyl (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4-chloro-5, 11-dioxo-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-3-carboxylate (150 mg, 239 μmol) and morpholine (41.8 mg, 480 μmol) in dioxane (8 mL) was added Pd2 (dba) 3 (21.9 mg, 23.9 μmol) , XantPhos (27.7 mg, 47.9 μmol) and Cs
2CO
3 (234 mg, 718 μmol) under the atmosphere of N
2. Then the reaction mixture was stirred at 100 ℃ under the atmosphere of N
2 for 5 hours. The mixture was concentrated under reduced pressure, and the residue was purified by flash column chromatography (25 g silica gel column, PE/EtAOc with EtOAc from 0~80%) to afford the product (1
1S, 1
3R, Z) -2
1- (tert-butyl) -44-morpholino-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (27 mg, 22%yield) as a yellow solid. LC-MS: (ESI) m/z [M+H]
+ 512.2.
Step 2: (1
1S, 1
3R, Z) -4
4-morpholino-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
The solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4-morpholino-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (27 mg, 52.77 μmol) in TFA (5 mL) was stirred at 80 ℃ for 6 hours. The mixture was concentrated under reduced pressure, and the residue was sent to purified by Prep-HPLC (Chromatographic columns: -Xbridge-C18 150 × 19 mm, 5 μm; Mobile Phase: ACN/H
2O (0.1%FA) ; Gradient: 20%-50%) to afford the product (1
1S, 1
3R, Z) -4
4-morpholino-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (5 mg, 21%yield) as a white solid. LC-MS: (ESI) m/z [M+H]
+ 456.2.
Example 124
(1
1S, 1
3R, Z) -4
4- (2- (pyrrolidin-1-yl) ethoxy) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione

Step 1: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- (2- (pyrrolidin-1-yl) ethoxy) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -
pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
To a solution of 4-nitrophenyl (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4-chloro-5, 11-dioxo-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-3-carboxylate (100 mg, 159 μmol) in 1-4, dioxane (5 mL) was added 2-pyrrolidin-1-ylethanol (55 mg, 479 μmol) , Pd-118 (10 mg, 15.9 μmol) and 4, 5-Bis (diphenylphosphino) -9, 9-dimethylxanthene (9 mg, 15.9 μmol) . The mixture was stirred at 100 ℃ under N
2 atmosphere for 12 hours. LCMS showed the starting material was consumed and desired product was formed. The mixture was concentrated. The residue was diluted with H
2O (30 mL) and extracted with DCM (30 mL x2) . The combined organic layers were washed by brine (50 mL) and dried over Na
2SO
4, filtered and concentrated. The residue was purified by flash chromatography (5%MeOH in DCM ) to provide the (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- (2- (pyrrolidin-1-yl) ethoxy) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (30 mg, 55.5 μmol, 34%yield) as a white solid. LC-MS: m/z 540.3 [M+H]
+.
Step 2: (1
1S, 1
3R, Z) -4
4- (2- (pyrrolidin-1-yl) ethoxy) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-
2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
The mixture of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- (2- (pyrrolidin-1-yl) ethoxy) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (25 mg, 46.3 μmol) in TFA (5 mL) was stirred at 90 ℃ for 12 hours. LCMS showed the starting material was consumed and desired product was formed. The mixture was concentrated. The residue was purified by prep-HPLC (Chromatographic columns: -Xbridge-C18 250 x 10 mm, Mobile Phase: ACN--H
2O (0.1%FA) ) to provide (1
1S, 1
3R, Z) -4
4- (2- (pyrrolidin-1-yl) ethoxy) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (1 mg, 2.07 μmol, 4%yield) as a colorless solid. LC-MS: m/z 484.3 [M+H]
+.
Example 196
(1
1S, 1
3R, Z) -4
4-hydroxy-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione

Step 1: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4-hydroxy-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
To a solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4-chloro-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (100 mg, 216 μmol) in 1, 4-dioxane (5.0 mL) and H
2O (0.50 mL) was added X-Phos (10.0 mg, 20.0 μmol) , Pd (OAc)
2 (10.0 mg, 44.0 μmol) and Potassium carbonate (60.0 mg, 434 μmol) . The mixture was heated to 100 ℃ and stirred at 100 ℃ under N
2 atmosphere for 10 h. The mixture was concentrated. The residue was diluted with H
2O (30 mL) and extracted with DCM (30 mL × 2) . The combined organic layers were washed by brine (50 mL) and dried over Na
2SO
4, filtered and concentrated. The residue was purified by flash chromatography eluting with MeOH /DCM (with MeOH from 0 to 5%in 10 min) to provide the (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4-hydroxy-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (80.0 mg, 83%yield) as a white solid. LC-MS: m/z 443.3 [M+H]
+.
Step 2: (1
1S, 1
3R, Z) -4
4-hydroxy-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione
The mixture of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4-hydroxy-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (20.0 mg, 45.2 μmol) in TFA (5.0 mL) was heated to 90 ℃ and stirred at 90 ℃ for 12 h. The mixture was concentrated. The residue was purified by prep-HPLC (with CH
3CN from 25%to 55%in 10 min) to provide (1
1S, 1
3R, Z) -4
4-hydroxy-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (6.00 mg, 34%yield) as a white solid. LC-MS: m/z 387.2 [M+H]
+.
Example 197
(1
1S, 1
3R, Z) -4
4- (difluoromethoxy) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione

Step 1: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- (difluoromethoxy) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -
pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
To a mixture of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4-hydroxy-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (25.0 mg, 56.5 μmol) in DMF (5.0 mL) was added ethyl 2-bromo-2, 2-difluoro-acetate (34.4 mg, 169 μmol, 21.7 μL) and Potassium carbonate (23.4 mg, 169 μmol) . The mixture was stirred at 25 ℃ for 12 h. The mixture was concentrated. The residue was diluted with H
2O (30 mL) and extracted with EtOAc (30 mL × 2) . The combined organic layers were washed by brine (50 mL) and dried over Na
2SO
4, filtered and concentrated to provide the (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- (difluoromethoxy) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (20.0 mg, 72%yield) as a white solid. LC-MS: m/z 493.1 [M+H]
+.
Step 2: (1
1S, 1
3R, Z) -4
4- (difluoromethoxy) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
The mixture of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
4- (difluoromethoxy) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (20.0 mg, 40.6 μmol) in TFA (5.0 mL) was heated to 90 ℃ and stirred at 90 ℃ for 12 h. The mixture was concentrated. The residue was purified by prep-HPLC (with CH
3CN from 25%to 55%in 10 min) to provide (1
1S, 1
3R, Z) -4
4- (difluoromethoxy) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (8.00 mg, 45%yield) as a white solid. LC-MS: m/z 437.2 [M+H]
+.
Example 125 and 126
(1
1S, 1
3R, 8R, Z) -8-fluoro-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione and (1
1S, 1
3R, 8S, Z) -8-fluoro-2
1H-12-oxa-3, 6, 10-
triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione

Step 1: 2-fluoromalonamide
A mixture of dimethyl 2-fluoropropanedioate (5.0 g, 33.3 mmol) in NH
3/MeOH (5 mL, 7 mol/L, 35mmol) was stirred for 4 h at 25 ℃, before it was concentrated to give crude 2-fluoropropanediamide as a white solid, which was used in the next step without further purification. LC-MS: m/z [M+H]
+ 121.1.
Step 2: 2-fluoropropane-1, 3-diamine
To a suspension of 2-fluoropropanediamide (3.3 g, 27.5 mmol) in THF (40.0 mL) was added BH
3 in THF (137.5 mL, 1 moL/L, 137mmol) . The reaction was stirred at 70 ℃ for 4h, before it was concentrated in vacuo. The mixture was quenched with KOH aq. (40 mL) and extracted with EtOAc (3 × 50 mL) . The organic phase was washed with brine (50 mL) and dried over anhydrous Na
2SO
4, filtered and concentrated in vacuo to afford 2-fluoropropane-1, 3-diamine (500 mg, crude) as a colorless oil. LC-MS: m/z [M+H]
+ 93.1.
Step 3: N- (3-amino-2-fluoropropyl) -6-bromopicolinamide
To a suspension of 6-bromopyridine-2-carboxylic acid (320 mg, 1.6 mmol) and 2-fluoropropane-1, 3-diamine (434 mg, 4.8 mmol) in DMF (10.0 mL) was added DIPEA (0.83 mL, 613 mg, 4.8 mmol) and HATU (602 mg, 1.6 mmol) at 25 ℃ and stirred for 2 h, before the reaction mixture was diluted with water (20 mL) and extracted with EtOAc (3 × 20 mL) . The organic phase was washed with brine (40 mL) , dried over anhydrous Na
2SO
4, filtered and concentrated in vacuo. The residue was purified by silica gel chromatography eluting with CH
2Cl
2/MeOH (with MeOH from 0 to 20 %in 25 min) to afford N- (3-amino-2-fluoro-propyl) -6-bromo-pyridine-2-carboxamide (320 mg, 73%yield) as a brown oil. LC-MS: m/z [M+H]
+ 275.8.
Step 4: tert-butyl N- [3- [ (6-bromopyridine-2-carbonyl) amino] -2-fluoro-propyl] carbamate
To a suspension of N- (3-amino-2-fluoro-propyl) -6-bromo-pyridine-2-carboxamide (270 mg, 1.0 mmol) and Boc
2O (213 mg, 1.0 mmol) in CH
2Cl
2 (5 mL) was added Et
3N (272 μL, 198 mg, 1.96 mmol) at 25 ℃ and stirred for 2 h. After completion of the reaction as judged by LCMS, reaction mixture was concentrated in vacuo. The crude product was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 35 %in 20 min) to afford tert-butyl N- [3- [ (6-bromopyridine-2-carbonyl) amino] -2-fluoro-propyl] carbamate (220 mg, 59%yield) as a yellow solid. LC-MS: m/z [M-55]
+ 319.9.
Step 5: tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) picolinamido) -2-fluoropropyl) carbamate
To a suspension of tert-butyl N- [3- [ (6-bromopyridine-2-carbonyl) amino] -2-fluoro-propyl] carbamate (220 mg, 0.585 mmol) in 1, 4-dioxane (10.0 mL) was added (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (131 mg, 0.585 mmol) , Pd
2 (dba)
3 (54 mg, 0.058 mmol) , XantPhos (68 mg, 0.117 mmol) and Cs
2CO
3 (381 mg, 1.17 mmol) . The reaction was stirred at 90 ℃ for 2 h under N
2, before it was concentrated in vacuo and purified by silica gel chromatography eluting with CH
2Cl
2/MeOH (with MeOH from 0 to 10 %in 20 min) to afford tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) picolinamido) -2-fluoropropyl) carbamate (230 mg, 75%yield) as a yellow solid. LC-MS: m/z [M+H]
+ 519.2.
Step 6: tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -
1H-pyrazol-5-yl) amino) picolinamido) -2-fluoropropyl) carbamate
To a suspension of tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) picolinamido) -2-fluoropropyl) carbamate (230 mg, 0.443 mmol) in THF (5.0 mL) was added 4-nitrophenyl carbonochloridate (268 mg, 1.33 mmol) , DMAP (108 mg, 0.887 mmol) and NMM (243 μL, 224 mg, 2.21 mmol) at rt. The reaction was stirred for 16 h, before it was concentrated in vacuo and purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 50 %in 20 min) to afford tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) picolinamido) -2-fluoropropyl) carbamate (200 mg, 65%yield) as a yellow solid. LC-MS: m/z [M+H]
+ 684.3.
Step 7: (1R, 3S) -3- (5- ( (6- ( (3-amino-2-fluoropropyl) carbamoyl) pyridin-2-yl) amino) -1- (tert-butyl) -
1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate
A mixture of [ (1R, 3S) -3- [5- [ [6- [ [3- (tert-butoxycarbonylamino) -2-fluoro-propyl] carbamoyl] -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] (4-nitrophenyl) carbonate (200 mg, 0.29 mmol) in TFA (5.0 mL) was stirred for 2 h at 25 ℃, before it was concentrated in vacuo to give crude (1R, 3S) -3- (5- ( (6- ( (3-amino-2-fluoropropyl) carbamoyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate as a brown oil, which was used in the next step without further purification. LC-MS: m/z [M+H]
+ 584.3.
Step 8: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -8-fluoro-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
To a suspension of (1R, 3S) -3- (5- ( (6- ( (3-amino-2-fluoropropyl) carbamoyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) in CH
3CN (3 mL) was added Et
3N (239 μL, 174 mg, 1.72 mmol) at room temperature. The mixture was stirred at room temperature for 2 h, before it was concentrated in vacuo and purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 65 %in 25 min) to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -8-fluoro-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (80 mg) as a yellow solid. LC-MS: m/z [M+H]
+ 445.2.
Step 9: (1
1S, 1
3R, 8R, Z) -8-fluoro-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione and (1
1S, 1
3R, 8S, Z) -8-fluoro-2
1H-12-oxa-3, 6, 10-
triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
A mixture of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -8-fluoro-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (80 mg) in HCO
2H (2.0 mL) was stirred at 100 ℃ for 16 h, before it was concentrated and purified by SFC eluting with CO2 in EtOH (CHIRALPAK OJ-H 250 mm × 20 mm, 5 μm column, CO
2/EtOH = 60/40, EtOH with 0.2%NH
4OH, 40 g/min, 40 ℃) to afford (1
1S, 1
3R, 8R, Z) -8-fluoro-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (7.8 mg, retention time = 2.1 min, absolute configuration arbitrarily assigned) as a white solid, LC-MS: m/z [M+H]
+ 389.2, and (1
1S, 1
3R, 8S, Z) -8-fluoro-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (9.3 mg, retention time = 2.7 min, absolute configuration arbitrarily assigned) as a white solid. LC-MS: m/z [M+H]
+ 389.2.
Example 127
(11S, 13R, Z) -8, 8-difluoro-21H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione

Step 1: 2, 2-difluoromalonamide
Diethyl 2, 2-difluoropropanedioate (1 g, 5.10 mmol) , NH
3 in MeOH (4 M, 38.17 mL) in THF (30 mL) was stirred for 2 hours at 0 ℃ in a RBF under N
2, until the reaction was complete as indicated by LCMS, the reaction mixture was concentrated to give the desired product 2, 2-difluoropropanediamide (600 mg, 4.35 mmol, 85.24%yield) as a white solid. LC-MS: m/z 139.1 [M+H]
+.
Step 2: 2, 2-difluoropropane-1, 3-diamine dihydrochloride
To a suspension of 2, 2-difluoropropanediamide (1 g, 7.24 mmol) in THF (30 mL) was added slowly BH
3 in THF (1 M, 36.21 mL) at 65 ℃ and the resulting mixture was stirred for 30 minutes under N
2. Then the mixture was stirred at 65 ℃ for 4 hours. After completion of the reaction as judged by LCMS, reaction mixture was quenched with MeOH (20 mL) and concentrated in vacuo. The procedure was repeated for 3 times. The crude product was treated with 20 mL HCl (gas, 4N in MeOH) , a white solid appeared. The mixture was filtered to give 2, 2-difluoropropane-1, 3-diamine; dihydrochloride (600 mg, 3.28 mmol, 45.26%yield) as a white solid. LC-MS: m/z 111.1 [M+H]
+.
Step 3: N- (3-amino-2, 2-difluoropropyl) -6-bromopicolinamide
A mixture of 6-bromopyridine-2-carboxylic acid (223 mg, 1.10 mmol) , 2, 2-difluoropropane-1, 3-diamine (364.65 mg, 3.31 mmol) , HATU (629.62 mg, 1.66 mmol) and DIEA (712.04 mg, 5.52 mmol) in DCM (10 mL) was stirred for 1 hour at 25 ℃ in a RBF under N
2, until the reaction was complete as indicated by LCMS. The reaction mixture was concentrated in vacuo. The residue was purified by flash column chromatography eluting with ethyl acetate/petroleum ether from 0 to 100%in 25 min to afford N- (3-amino-2, 2-difluoropropyl) -6-bromopicolinamide (300 mg, 0.91mmol, 83.16%yield, 90%purity) . LC-MS: m/z 294.1 [M+H]
+.
Step 4: tert-butyl (3- (6-bromopicolinamido) -2, 2-difluoropropyl) carbamate
A mixture of N- (3-amino-2, 2-difluoro-propyl) -6-bromo-pyridine-2-carboxamide (300 mg, 1.02 mmol) , Boc
2O (444.75 mg, 2.04 mmol) , TEA (516.11 mg, 5.10 mmol, 710.89 μL) in DCM (10 mL) and MeCN (3 mL) was stirred for 2 hours at 25 ℃ in a RBF under N
2, until the reaction wascomplete as indicated by LCMS, the reaction mixture was concentrated in vacuo. The residue was purified by flash column chromatography eluting with ethyl acetate/petroleum ether from 0 to 100%in 25 min to afford tert-butyl (3- (6-bromopicolinamido) -2, 2-difluoropropyl) carbamate (260 mg, 659.55 μmol, 64.66%yield) as a light-yellow liquid. LC-MS: m/z 416.0 [M+H]
+.
Step 5: tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) picolinamido) -2, 2-difluoropropyl) carbamate
A mixture of tert-butyl N- [3- [ (6-bromopyridine-2-carbonyl) amino] -2, 2-difluoro-propyl] carbamate (240 mg, 608.81 μmol) , (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (135.96 mg, 608.81 μmol) , Pd
2 (dba)
3 (55.71 mg, 60.88 μmol) and XantPhos (70.50 mg, 121.76 μmol) , Cs
2CO
3 (395.73 mg, 1.22 mmol) in dioxane (20 mL) was stirred for 6 hours at 100 ℃ in a RBF under N
2, until the reaction was complete as indicated by LCMS, the reaction mixture was concentrated in vacuo. The crude product was purified by flash column chromatography eluting with ethyl acetate/petroleum ether from 0 to 50%in 25 min to afford to afford tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) picolinamido) -2, 2-difluoropropyl) carbamate (280 mg, 521.79 μmol, 85.71%yield) as a light-yellow oil. LC-MS: m/z 537.3 [M+H]
+.
Step 6: tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -
1H-pyrazol-5-yl) amino) picolinamido) -2, 2-difluoropropyl) carbamate
A mixture of tert-butyl N- [3- [ [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] pyridine-2-carbonyl] amino] -2, 2-difluoro-propyl] carbamate (280 mg, 521.79 μmol) , (4-nitrophenyl) carbonochloridate (315.52 mg, 1.57 mmol) , DMAP (127.49 mg, 1.04 mmol) and NMM (263.90 mg, 2.61 mmol, 286.84 μL) in MeCN (10 mL) was stirred for 2 hours at 25℃ in a RBF under N2, until the reaction was complete as indicated by LCMS, the reaction mixture was concentrated in vacuo. The crude product was purified by flash column chromatography eluting with ethyl acetate/petroleum ether from 0 to 50%in 25 min to afford to afford tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) picolinamido) -2, 2-difluoropropyl) carbamate (240 mg, 342.02 μmol, 65.55%yield) as a colorless oil. LC-MS: m/z 702.2 [M+H]
+.
Step 7: (1R, 3S) -3- (5- ( (6- ( (3-amino-2, 2-difluoropropyl) carbamoyl) pyridin-2-yl) amino) -1- (tert-
butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate
A mixture of tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) picolinamido) -2, 2-difluoropropyl) carbamate (220 mg, 313.52 μmol) , TFA (1.79 g, 15.68 mmol, 1.21 mL) in DCM (8.82 mL) was stirred for 2 hours at 25℃ in a RBF under N
2, until the reaction was complete as indicated by LCMS, the reaction mixture was concentrated in vacuo to give the desired product (1R, 3S) -3- (5- ( (6- ( (3-amino-2, 2-difluoropropyl) carbamoyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate (220 mg, crude) as pale yellow oil. LC-MS: m/z 602.3 [M+H]
+.
Step 8: (11S, 13R, Z) -21- (tert-butyl) -8, 8-difluoro-21H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
A mixture of (1R, 3S) -3- (5- ( (6- ( (3-amino-2, 2-difluoropropyl) carbamoyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate (180 mg, 299.20 μmol) , NMM (151.32 mg, 1.50 mmol, 164.48 μL) , DMAP (73.11 mg, 598.40 μmol) in MeCN (20 mL) was stirred for 1 hour at 25 ℃ in a RBF under N
2, until the reaction was complete as indicated by LCMS, the reaction mixture was concentrated in vacuo. The residue was purified by flash column chromatography eluting with ethyl acetate/petroleum ether from 0 to 100%in 25 min to afford (11S, 13R, Z) -21- (tert-butyl) - 8, 8-difluoro-21H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (74 mg, 160.0 μmol, 53.48%yield) as colorless oil. LC-MS: m/z 462.7 [M+H]
+.
Step 9: (11S, 13R, Z) -8, 8-difluoro-21H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
(11S, 13R, Z) -21- (tert-butyl) -8, 8-difluoro-21H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (74 mg, 160.00 μmol) in TFA (5 mL) was stirred for 12 hours at 70 ℃ in a RBF under N
2, until the reaction was complete as indicated by LCMS, the reaction mixture was concentrated in vacuo. The residue was dissolved in MeOH (5 ml) and was added to H
2O (20 mL) . Then the solid was collected by filtraation to give the desired product (11S, 13R, Z) -8, 8-difluoro-21H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (26 mg, 63.98 μmol, 39.99%yield) as a pale yellow solid. LC-MS: m/z 407.1 [M+H]
+.
The following compounds were prepared using the similar procedure disclosed in synthetic example 127.

Example 129
(1
1S, 1
3R, Z) -11-thioxo-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-5-one

Step 1: tert-butyl (3- (6-bromopicolinamido) propyl) carbamate
To a stirred solution of 6-bromopicolinic acid (2.00 g, 9.90 mmol) and tert-butyl (3-aminopropyl) carbamate (1.72 g, 9.90 mmol) in CH
2Cl
2 (30.0 mL) were added HATU (7.52 g, 19.8 mmol) and DIPEA (2.55 g, 19.8 mmol) . The reaction mixture was stirred at 20 ℃ for 1 h before it was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 30%in 15 min) to afford tert-butyl (3- (6-bromopicolinamido) propyl) carbamate (3.10 g, 88%yield) as a yellow oil. LC-MS: m/z [M+Na]
+379.7.
Step 2: tert-butyl (3- (6- ( (1- (tert-butyl) -5- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-3-
yl) amino) picolinamido) propyl) carbamate
To a stirred solution of tert-butyl (3- (6-bromopicolinamido) propyl) carbamate (500 mg, 1.40 mmol) in toluene (20.0 mL) were sequentially added (1R, 3S) -3- (3-amino-1- (tert-butyl) -1H-pyrazol-5-yl) cyclopentan-1-ol (312 mg, 1.40 mmol) , Pd
2 (dba)
3 (128 mg, 0.140 mmol) , XantPhos (162 mg, 0.280 mmol) and Cs
2CO
3 (913 mg, 2.80 mmol) at 25 ℃. The reaction mixture was warmed to 80 ℃ and stirred at that temperature for 16 h before it was cooled to 25 ℃ and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 70%in 30 min) to afford tert-butyl (3- (6- ( (1- (tert-butyl) -5- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-3-yl) amino) picolinamido) propyl) carbamate (500 mg, 71%yield) as a red solid. LC-MS: m/z [M+H]
+ 500.9.
Step 3: O- ( (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propyl) carbamoyl) pyridin-2-
yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl) 1H-imidazole-1-carbothioate
To a stirred solution of tert-butyl (3- (6- ( (1- (tert-butyl) -5- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-3-yl) amino) picolinamido) propyl) carbamate (300 mg, 0.600 mmol) in CH
2Cl
2 (5.0 mL) were sequentially added di (imidazol-1-yl) methanethione (214 mg, 1.20 mmol) , DMAP (73.2 mg, 0.600 mmol) and Et
3N (251 μL 182 mg, 1.80 mmol) at 25 ℃. The reaction mixture was stirred at that temperature for 2 h before it was quenched with ice-cold water (20 mL) and extracted with CH
2Cl
2 (20 mL × 3) . The combined organic phases were dried over anhydrous Na
2SO
4 and filtered. The filtrate was concentrated under reduced pressure to give O- ( (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propyl) carbamoyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl) 1H-imidazole-1-carbothioate was used in the next step without further purification. LC-MS: m/z [M+H]
+ 610.6.
Step 4: O- ( (1R, 3S) -3- (3- ( (6- ( (3-aminopropyl) carbamoyl) pyridin-2-yl) amino) -1H-pyrazol-5-
yl) cyclopentyl) 1H-imidazole-1-carbothioate
A solution of O- ( (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propyl) carbamoyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl) 1H-imidazole-1-carbothioate (crude) in TFA (2.0 mL) was warmed to 70 ℃ and stirred at that temperature for 16 h. The mixture was concentrated under reduced pressure to give O- ( (1R, 3S) -3- (3- ( (6- ( (3-aminopropyl) carbamoyl) pyridin-2-yl) amino) -1H-pyrazol-5-yl) cyclopentyl) 1H-imidazole-1-carbothioate as a brown oil, which was used in the next step without further purification. LC-MS: m/z [M+H]
+ 454.6.
Step 5: (1
1S, 1
3R, Z) -11-thioxo-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-5-one
To a stirred solution of O- ( (1R, 3S) -3- (3- ( (6- ( (3-aminopropyl) carbamoyl) pyridin-2-yl) amino) -1H-pyrazol-5-yl) cyclopentyl) 1H-imidazole-1-carbothioate (crude) in CH
3CN (5.0 mL) was added Et
3N (270 μL, 196 mg, 1.94 mmol) at 25 ℃. The mixture was warmed to 80 ℃ and stirred at that temperature for 5 h. The mixture was concentrated under reduced pressure. The residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 5%to 50%in 40 min) to afford (1
1S, 1
3R, Z) -11-thioxo-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-5-one (2.00 mg, 4.7%yield) as light yellow solid. LC-MS: m/z [M+H]
+ 387.
Example 130
(rac, cis) -2
1H-3, 6, 10, 12-tetraaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione

Step 1: tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- (1, 3-dioxoisoindolin-2-yl) cyclopentyl) -1H-
pyrazol-5-yl) amino) picolinamido) propyl) carbamate
To a stirred suspension of tert-butyl (3- (6-bromopicolinamido) propyl) carbamate (200 mg, 560 μmol) in 1, 4-dioxane (15.0 mL) were sequentially added 2- ( (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl) isoindoline-1, 3-dione (197 mg, 560 μmol) , Pd
2 (dba)
3 (51.0 mg, 56.0 μmol) , XantPhos (65.0 mg, 112 μmol) and Cs
2CO
3 (547 mg, 1.68 mmol) at 25 ℃. The reaction mixture was warmed to 80 ℃ and stirred at that temperature for 12 h. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 60 %in 25 min) to afford (rac, cis) -tert-butyl (3- (6- ( (1- (tert-butyl) -3- (3- (1, 3-dioxoisoindolin-2-yl) cyclopentyl) -1H-pyrazol-5-yl) amino) picolinamido) propyl) carbamate (300 mg, 85%yield) as a yellow oil. LC-MS: m/z [M+H]
+629.8.
Step 2: (rac, cis) -tert-butyl (3- (6- ( (3- (3-aminocyclopentyl) -1- (tert-butyl) -1H-pyrazol-5-
yl) amino) picolinamido) propyl) carbamate
To a stirred solution of (rac, cis) -tert-butyl (3- (6- ( (1- (tert-butyl) -3- (3- (1, 3-dioxoisoindolin-2-yl) cyclopentyl) -1H-pyrazol-5-yl) amino) picolinamido) propyl) carbamate (300 mg, 480 μmol) in ethanol (15.0 mL) was added NH
2NH
2·H
2O (248 μL, 255 mg, 5.10 mmol, 80%wt) at 25 ℃. The mixture was warmed to 80 ℃ and stirred at that temperature for 3 h before it was filtered through a pad of Celite and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 90 %in 25 min) to afford (rac, cis) -tert-butyl (3- (6- ( (3- (3-aminocyclopentyl) -1- (tert-butyl) -1H-pyrazol-5-yl) amino) picolinamido) propyl) carbamate (150 mg, 63%yield) as a yellow oil. LC-MS: m/z [M+H]
+500.0
Step 3: (rac, cis) -tert-butyl (3- (6- ( (1- (tert-butyl) -3- (3- ( ( (4-
nitrophenoxy) carbonyl) amino) cyclopentyl) -1H-pyrazol-5-
yl) amino) picolinamido) propyl) carbamate
To a suspension of (rac, cis) -tert-butyl (3- (6- ( (3- (3-aminocyclopentyl) -1- (tert-butyl) -1H-pyrazol-5-yl) amino) picolinamido) propyl) carbamate (150 mg, 300 μmol) in DCE (10.0 mL) were sequentially added 4-nitrophenyl carbonochloridate (203 mg, 1.01 mmol) , DMAP (21.0 mg, 170 μmol) and Et
3N (125 μL, 91.0 mg, 900 μmol) at 25 ℃. The reaction mixture was warmed to 70 ℃ and stirred at that temperature for 12 h. The reaction mixture was stirred at that temperature for 2 h before it was quenched with ice-cold water (20 mL) and extracted with CH
2Cl
2 (20 mL × 3) . The combined organic phases were dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 60 %in 25 min) to afford (rac, cis) -tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) amino) cyclopentyl) -1H-pyrazol-5-yl) amino) picolinamido) propyl) carbamate (150 mg, 75%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 664.7
Step 4: (rac, cis) -4-nitrophenyl (3- (5- ( (6- ( (3-aminopropyl) carbamoyl) pyridin-2-yl) amino) -1- (tert-
butyl) -1H-pyrazol-3-yl) cyclopentyl) carbamate
To a stirred solution of (rac, cis) -tert-butyl (3- (6- ( (1- (tert-butyl) -3- (3- ( ( (4-nitrophenoxy) carbonyl) amino) cyclopentyl) -1H-pyrazol-5-yl) amino) picolinamido) propyl) carbamate (150 mg, 230 μmol) in CH
2Cl
2 (10.0 ml) was added TFA (90.0 μL, 131 mg, 1.15 mmol) at 25 ℃. The mixture was stirred at that temperature for 1 h before it was concentrated under reduced pressure to afford (rac, cis) -4-nitrophenyl (3- (5- ( (6- ( (3-aminopropyl) carbamoyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl) carbamate as a brown oil, which was used in the next step without further purification. LC-MS: m/z [M+H]
+ 564.8
Step 5: (rac, cis) -2
1- (tert-butyl) -2
1H-3, 6, 10, 12-tetraaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione
To a suspension of (rac, cis) -4-nitrophenyl (3- (5- ( (6- ( (3-aminopropyl) carbamoyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl) carbamate in CH
3CN (10.0 mL) was added Et
3N (268 μL, 196 mg, 1.94 mmol) at 25 ℃. The mixture was stirred at that temperature for 1 h. Then it was concentrated in vacuo and the residue was purified by silica gel chromatography eluting with MeOH/CH
2Cl
2 (with MeOH from 0 to 2.5 %in 25 min) to afford (rac, cis) -2
1- (tert-butyl) -2
1H-3, 6, 10, 12-tetraaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (80.0 mg, 83%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 425.9
Step 6: (rac, cis) -2
1H-3, 6, 10, 12-tetraaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione
A solution of (rac, cis) -2
1- (tert-butyl) -2
1H-3, 6, 10, 12-tetraaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (70.0 mg, 160 μmol) in TFA (5.0 mL) was warmed to 80 ℃ and stirred at that temperature for 2 h before it was cooled, concentrated under reduced pressure. The residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 5%to 60%in 20 min) to afford (rac, cis) -2
1H-3, 6, 10, 12-tetraaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (5.60 mg, 16%yield) as an off-white solid. LC-MS: m/z [M+H]
+ 370.1.
Example 131
(1
1S, 1
3R, Z) -4
3-fluoro-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione

Step 1: tert-butyl N- [3- [ (6-bromo-5-fluoro-pyridine-2-carbonyl) amino] propyl] carbamate
To a suspension of 6-bromo-5-fluoro-pyridine-2-carboxylic acid (500 mg, 2.27 mmol) in CH
2Cl
2 (20.0 mL) was sequentially added tert-butyl N- (3-aminopropyl) carbamate (396 mg, 2.27 mmol) , HATU (1.73 g, 4.55 mmol) and DIPEA (790 μL, 586 mg, 4.55 mmol) at 25 ℃ and stirred for 2 h. After completion of the reaction as judged by LCMS, reaction mixture was concentrated under reduced pressure. The crude product was purified by flash chromatography eluting with EtOAc/PE (with EtOAc from 0 to 30%in 25 min) to afford tert-butyl N- [3- [ (6-bromo-5-fluoro-pyridine-2-carbonyl) amino] propyl] carbamate (850 mg, 99%yield) as a yellow oil. LC-MS: m/z [M-Boc]
+275.9.
Step 2: tert-butyl N- [3- [ [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -5-
fluoro-pyridine-2-carbonyl] amino] propyl] carbamate
To a suspension of tert-butyl N- [3- [ (6-bromo-5-fluoro-pyridine-2-carbonyl) amino] propyl] carbamate (730 mg, 1.94 mmol) in 1, 4-dioxane (20.0 mL) was sequentially added (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (216 mg, 0.97 mmol) , Cs
2CO
3 (1.90 g, 5.82 mmol) , XantPhos (224 mg, 0.388 mmol) and Pd
2 (dba)
3 (177 mg, 0.194 mmol) at room temperature and the reaction was stirred at 100 ℃ for 16 h under N
2. After completion of the reaction as judged by LCMS, reaction mixture was filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography eluting with EtOAc/PE (with EtOAc from 0 to 50%in 30 min) to afford tert-butyl N- [3- [ [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -5-fluoro-pyridine-2-carbonyl] amino] propyl] carbamate (410 mg, 40%yield) as a yellow solid. LC-MS: m/z [M+H]
+519.0.
Step 3: [ (1R, 3S) -3- [5- [ [6- [3- (tert-butoxycarbonylamino) propylcarbamoyl] -3-fluoro-2-
pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate
To a suspension of tert-butyl N- [3- [ [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -5-fluoro-pyridine-2-carbonyl] amino] propyl] carbamate (380 mg, 0.73 mmol) in CH
2Cl
2 (20.0 mL) was added CDI (356 mg, 2.20 mmol) and DIPEA (636 μL, 472 mg, 3.66 mmol) at 35 ℃and stirred for 2 h. After completion of the reaction as judged by LCMS, the reaction mixture was quenched with ice-cold water (20 mL) and extracted with EtOAc (3 × 20 mL) . The organic phase was washed with brine (50 mL) and dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure to afford [ (1R, 3S) -3- [5- [ [6- [3- (tert-butoxycarbonylamino) propylcarbamoyl] -3-fluoro-2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (crude) as a yellow oil. The crude product was used in the next step without further purification. LC-MS: m/z [M+H]
+ 612.8.
Step 4: [ (1R, 3S) -3- [5- [ [6- (3-aminopropylcarbamoyl) -3-fluoro-2-pyridyl] amino] -1-tert-butyl-
pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate
To a suspension of [ (1R, 3S) -3- [5- [ [6- [3- (tert-butoxycarbonylamino) propylcarbamoyl] -3-fluoro-2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate in CH
2Cl
2 (6.0 mL) was added slowly TFA (2.0 mL) at 25 ℃ and stirred for 1 h. After completion of the reaction as judged by LCMS, reaction mixture was concentrated under reduced pressure to afford [ (1R, 3S) -3- [5- [ [6- (3-aminopropylcarbamoyl) -3-fluoro-2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (crude) as a yellow oil. The crude product was used in the next step without further purification. LC-MS: m/z [M+H]
+ 512.9.
Step 5: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
3-fluoro-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
To a suspension of [ (1R, 3S) -3- [5- [ [6- (3-aminopropylcarbamoyl) -3-fluoro-2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate in CH
3CN (10.0 mL) was added Et
3N (894 μL, 651 mg, 6.44 mmol) and the reaction was stirred at 80 ℃ for 16 h. After completion of the reaction as judged by LCMS, reaction mixture was concentrated under reduced pressure. The crude product was purified by flash column chromatography eluting with MeOH/CH
2Cl
2 (with MeOH from 0 to 10%in 20 min) to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
3-fluoro-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (240 mg) as a yellow solid. LC-MS: m/z [M+H]
+ 444.9.
Step 6: (1
1S, 1
3R, Z) -4
3-fluoro-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione
A mixture of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
3-fluoro-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (240 mg, 0.54 mmol) in HCO
2H (4.0 mL) was stirred for 5 h at 100 ℃, until the reaction was complete as indicated by LCMS, the reaction mixture was concentrated under reduced pressure, purified by prep-HPLC eluting with CH
3CN in water (with CH
3CN from 40%to 95%in 10 min (0.1%HCO
2H condition) ) to give (1
1S, 1
3R, Z) -4
3-fluoro-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (126.9 mg, 60%yield) as a white solid. LC-MS: m/z [M+H]
+ 389.1.
The following compounds were prepared using the similar procedure disclosed in synthetic example 131.


Example 138
(1
1S, 1
3R, Z) -4
5-isopropyl-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione

Step 1: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5- (prop-1-en-2-yl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-
2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
To a mixture of (1
1S, 1
3R, Z) -4
5-bromo-2
1- (tert-butyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (100 mg, 198 μmol) in H
2O (0.15 mL) and 1, 4-dioxane (1.5 mL) were sequentially added 2-isopropenyl-4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolane (133 mg, 791 μmol) , Na
2CO
3 (52.4 mg, 495 μmol) and Pd (dppf) Cl
2 (28.9 mg, 39.6 μmol) . The mixture was heated to 80 ℃ and stirred at that temperature under N
2 for 3 h. The reaction mixture was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 15 to 70%in 15 min) to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5- (prop-1-en-2-yl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (80.0 mg, 86%yield) as a yellow oil. LC-MS: m/z [M+H]
+ 467.3.
Step 2: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5-isopropyl-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
To a solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5- (prop-1-en-2-yl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (80.0 mg, 171 μmol) in MeOH (10.0 mL) was added Pd/C (40.0 mg, 50 wt. %in H
2O) . The mixture was stirred for 2.5 h at 25 ℃ under H
2 atmosphere. The mixture was filtered under reduced pressure and the filtrate was concentrated under reduced pressure to give the crude product as a yellow solid. LC-MS: m/z [M+H]
+469.3.
Step 3: (1
1S, 1
3R, Z) -4
5-isopropyl-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione
A mixture of crude (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5-isopropyl-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione in HCO
2H (1.5 mL) was heated to 100 ℃ and stirred at that temperature for 6 h. The reaction mixture was concentrated under reduced pressure. The residue was purified prep-HPLC eluting with CH
3CN in water (with CH
3CN from 20%to 40%in 30 min) to afford (1
1S, 1
3R, Z) -4
5-isopropyl-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (34.0 mg, 47%yield) as a white solid. LC-MS: m/z [M+H]
+ 413.2.
The following compounds were prepared using the similar procedure disclosed in synthetic example 138.




Example 198
(1
1S, 1
3R, Z) -4
5- ( (dimethylamino) methyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione

Step 1: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -5, 11-dioxo-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-4
5-carbaldehyde
To a solution of (1
1S, 1
3R, Z) -4
5-bromo-2
1- (tert-butyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (100 mg, 197.48 μmol) and t-BuNC (82.02 mg, 987.38 μmol) in DMF (4 mL) was added HCOOK (51.03 mg, 592.43 μmol) , JohnPhos (5.89 mg, 19.75 μmol) and Pd (OAc)
2 (4.42 mg, 19.75 μmol) at 20 ℃, and the mixture was stirred at 60 ℃ for 16 hours. The mixture was poured into H
2O (5 mL) and extracted with EtOAc (3 ×5 mL) . The combined organic layer was washed with brine (5 mL) , dried over Na
2SO
4, filtered and concentrated. The residue was purified by prep-TLC to give (1
1S, 1
3R, Z) -2
1- (tert-butyl) -5, 11-dioxo-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentana cyclododecaphane-4
5-carbaldehyde (60 mg, 131.72 μmol, 66.70%yield) as a yellow oil. LC-MS: m/z 455.3 [M+H]
+.
Step 2: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5- ( (dimethylamino) methyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -
pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
To a solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -5, 11-dioxo-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-4
5-carbaldehyde (60 mg, 0.13 mmol) and N-methylmethanamine (660.04 μL, 1.32 mmol, 2 M in THF) in DCE (3 mL) was added AcOH (15.85 mg, 264.01 μmol) at 20 ℃ and stirred at 20 ℃ for 1 hour. Then NaBH (OAc)
3 (55.98 mg, 264.01 μmol) was added at 20 ℃ and stirred at 20 ℃ for 15 hours. The mixture was poured into saturated aqueous NaHCO
3 (5 mL) and extracted with DCM (3 × 5 mL) . The combined organic layer was washed with brine (5 mL) , dried over Na
2SO
4, filtered and concentrated to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5- ( (dimethylamino) methyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (15 mg, 31.02 μmol, 23.50%yield) as a yellow oil. LC-MS: m/z 484.3 [M+H]
+.
Step 3: (1
1S, 1
3R, Z) -4
5- ( (dimethylamino) methyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
A solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5- ( (dimethylamino) methyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (15 mg, 31.02 μmol) in TFA (1 mL) was stirred at 85 ℃ for 16 hours. The reaction solution was concentrated, the residue was diluted with DCM (5 mL) and washed with saturated aqueous NaHCO
3 (3 × 5 mL) , dried over and concentrated under reduced pressure to give the residue. The residue was purified by Prep-HPLC (FA condition) to afford the product (1
1S, 1
3R, Z) -4
5- ( (dimethylamino) methyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (5.9 mg, 13.56 μmol, 43.71%yield, 98.23%purity) as a pale yellow solid. LC-MS: m/z 428.2 [M+H]
+.
Example 155
(1
1S, 1
3R, Z) -4
5- (2- (pyrrolidin-1-yl) ethoxy) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione

Step 1: tert-butyl (3- (6-bromo-3- (2- (pyrrolidin-1-yl) ethoxy) picolinamido) propyl) carbamate
To a stirred solution of tert-butyl (3-aminopropyl) carbamate (1.43 g, 8.20 mmol) in DCE (10.0 mL) was added AlMe
3 (8.2 mL, 8.20 mmol, 1.0 M in THF) at 0 ℃. The reaction mixture was stirred at that temperature for 30 min before a solution of methyl 6-bromo-3- (2- (pyrrolidin-1-yl) ethoxy) picolinate (900 mg, 2.73 mmol) in DCE (5.0 mL) was added. The resulting mixture was warmed up to 20 ℃ and stirred at that temperature for 12 h before it was quenched with MeOH (3 mL) . The reaction mixture was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 50%in 30 min) to give tert-butyl (3- (6-bromo-3- (2- (pyrrolidin-1-yl) ethoxy) picolinamido) propyl) carbamate as a pale yellow oil (0.95 g, 63%yield) . LC-MS: m/z [M+H]
+ 471.1.
Step 2: tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) -3- (2- (pyrrolidin-1-yl) ethoxy) picolinamido) propyl) carbamate
To a solution of (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (250 mg, 1.12 mmol) and tert-butyl N- [4- [ [6-bromo-3- (2-pyrrolidin-1-ylethoxy) -2-pyridyl] amino] -4-oxo-butyl] carbamate (792 mg, 1.68 mmol) in 1, 4-dioxane (2.0 mL) were sequentially added Cs
2CO
3 (730 mg, 2.24 mmol) , Pd
2 (dba)
3 (205 mg, 224 μmol) and XantPhos (194 mg, 336 μmol) at 20 ℃. The reaction mixture was warmed to 80 ℃ and stirred at that temperature for 12 h under N
2 atmosphere. The reaction mixture was cooled to 20 ℃, concentrated under reduced pressure. The residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 5%to 50%in 50 min) to afford tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -3- (2- (pyrrolidin-1-yl) ethoxy) picolinamido) propyl) carbamate (500 mg, 814.61 μmol, 72.77%yield) as a white solid. LC-MS: m/z [M+H]
+ 614.4.
Step 3: (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propyl) carbamoyl) -5- (2- (pyrrolidin-1-
yl) ethoxy) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-
carboxylate
To a suspension of tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -3- (2- (pyrrolidin-1-yl) ethoxy) picolinamido) propyl) carbamate (300 mg, 488.77 μmol) in CH
2Cl
2 (10.0 mL) were sequentially added CDI (238 mg, 1.47 mmol) , DMAP (59.7 mg, 489 μmol) and DIEA (0.2 mL, 1.47 mmol, 148 mg) at 25 ℃ and the resulting mixture was stirred at that temperature for 12 h. The reaction mixture was quenched with ice-cold water (20 mL) and extracted with EtOAc (20 mL × 3) . The combined organic phases were washed with brine (50 mL) , dried over anhydrous Na
2SO
4, filtered and concentrated to afford (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propyl) carbamoyl) -5- (2- (pyrrolidin-1-yl) ethoxy) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (300 mg) as a yellow oil which was directly used into the next step without further purification. LC-MS: m/z [M+H]
+ 708.3.
Step 4: (1R, 3S) -3- (5- ( (6- ( (3-aminopropyl) carbamoyl) -5- (2- (pyrrolidin-1-yl) ethoxy) pyridin-2-
yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a suspension of (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propyl) carbamoyl) -5- (2- (pyrrolidin-1-yl) ethoxy) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (300 mg, 0.42 mmol) in CH
2Cl
2 (5.0 mL) was added slowly TFA (2.0 mL) at 25 ℃. The reaction mixture was stirred at 25 ℃ for 2 h before it was concentrated to afford (1R, 3S) -3- (5- ( (6- ( (3-aminopropyl) carbamoyl) -5- (2- (pyrrolidin-1-yl) ethoxy) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (300 mg) as a yellow oil which was directly used into the next step without further purification. LC-MS: m/z [M+H]
+ 608.3.
Step 5: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5- (2- (pyrrolidin-1-yl) ethoxy) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -
pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
To a suspension of (1R, 3S) -3- (5- ( (6- ( (3-aminopropyl) carbamoyl) -5- (2- (pyrrolidin-1-yl) ethoxy) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (300 mg, crude) in CH
3CN (5.0 mL) was added Et
3N (1.45 g, 14.4 mmol) at 20 ℃. The reaction was warmed to 80 ℃ and stirred at that temperature for 3 h before it was cooled to 20 ℃ and concentrated under reduced pressure. The residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 5%to 50%in 50 min) to afford (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5- (2- (pyrrolidin-1-yl) ethoxy) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (60.0 mg, 23%yield for 3 steps) as a pale yellow solid. LC-MS: m/z [M+H]
+ 540.3.
Step 6: (1
1S, 1
3R, Z) -4
5- (2- (pyrrolidin-1-yl) ethoxy) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-
2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
A mixture of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5- (2- (pyrrolidin-1-yl) ethoxy) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (60.0 mg, 0.11 mmol) in FA (4.0 mL) was stirred at 70 ℃ for 4 h before it was cooled to 20 ℃ and concentrated under reduced pressure. The residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 5%to 50%in 50 min) to afford (rac, cis) - (1
1S, 1
3R, Z) -2
1H-13-oxa-3, 6, 11-triaza-2 (5, 3) -triazola-4 (1, 3) -benzena-1 (1, 3) -cyclopentanacyclotridecaphane-5, 12-dione (34.7 mg, 65%yield) as a pale yellow solid. LC-MS: m/z [M+H]
+ 484.0.
Example 156 and 157
(1
1S, 1
3R, Z) -4
5- (methoxymethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione and (1
1S, 1
3R, Z) -4
5- (hydroxymethyl) -2
1H-12-oxa-
3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione

Step 1: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5- (methoxymethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-
2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
To a stirred solution of (1
1S, 1
3R, Z) -4
5-bromo-2
1- (tert-butyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (100 mg, 198 μmol) and tributyl (methoxymethyl) stannane (133 mg, 396 μmol) in toluene (1.5 mL) was added Pd (PPh
3)
4 (22.9 mg, 19.8 μmol) at 25 ℃. The reaction mixture was warmed to 100 ℃ and stirred at that temperature for 16 h before it was cooled to 25 ℃. The mixture was poured into H
2O (5 mL) and extracted with EtOAc (5 mL × 3) . The combined organic layers were washed with brine (5 mL) , dried over Na
2SO
4, filtered and concentrated. The residue was purified by Prep-TLC (EtOAc/PE = 2: 1) to give (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5- (methoxymethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (40.0 mg, 43%yield) as a yellow oil. LC-MS: m/z [M+H]
+. 471.3.
Step 2: (1
1S, 1
3R, Z) -4
5- (methoxymethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione and (1
1S, 1
3R, Z) -4
5- (hydroxymethyl) -
2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-
5, 11-dione
A stirred solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5- (methoxymethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (40 mg, 85.00 μmol) in HCO
2H (1.0 mL) was warmed to 70 ℃ and stirred at that temperature for 16 h before it was cooled to 25 ℃. The mixture was concentrated under reduced pressure. The residue was purified by Prep-HPLC HPLC eluting with CH
3CN in water (with CH
3CN from 0%to 45%in 40 min) to afford (1
1S, 1
3R, Z) -4
5- (methoxymethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (1.9 mg, 5.3%yield) as a white solid LC-MS: m/z [M+H]
+ 415.2. And (1
1S, 1
3R, Z) -4
5- (hydroxymethyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (2.4 mg, 6.4%yield) as a white solid. LC-MS: m/z 401.2 [M+H]
+.
Example 158
(1
1S, 1
3R, Z) -4
5- (methylsulfonyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione

Step 1: tert-butyl (3- (6-chloro-3- (methylthio) picolinamido) propyl) carbamate
To a solution of tert-butyl (3- (6-chloro-3-fluoropicolinamido) propyl) carbamate (500 mg, 1.51 mmol) in DMF (5 mL) was added sodium; methanethiolate (105 mg, 1.51 mmol) at 25 ℃. The reaction mixture was stirred at 20 ℃ for 1.5 hours. The mixture was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc in petroleum from 0%to 50% to give tert-butyl (3- (6-chloro-3- (methylthio) picolinamido) propyl) carbamate (527 mg, 1.32 mmol, 87%yield, 90%purity) as a white solid. LC-MS: m/z 382.0 [M+Na]
+.
Step 2: tert-butyl (3- (6-chloro-3- (methylsulfonyl) picolinamido) propyl) carbamate
To a solution of tert-butyl (3- (6-chloro-3- (methylthio) picolinamido) propyl) carbamate (527 mg, 1.5 mmol) in DCM (8 mL) was added m-CPBA (758 mg, 4.3 mmol) at 25 ℃. The reaction mixture was stirred at 25 ℃ for 3 hours. The reaction mixture was poured into saturated NaHSO
3 (50 mL) . The aqueous layer was extracted with DCM (3 x 50 mL) . The combined organic layer was washed with saturated NaHCO
3 (2 x 50 mL) . The combined organic phase was dried over sodium sulfate and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc in petroleum from 0%to 50%to give tert-butyl (3- (6-chloro-3- (methylsulfonyl) picolinamido) propyl) carbamate (490 mg, 1.2 mmol, 81%yield, 95%purity) as a white solid. LC-MS: m/z 414.0 [M+Na]
+.
Step 3: tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) -3- (methylsulfonyl) picolinamido) propyl) carbamate
To a solution of tert-butyl (3- (6-chloro-3- (methylsulfonyl) picolinamido) propyl) carbamate (263 mg, 671 μmol) in dioxane (40 mL) was added (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (150 mg, 671 μmol) , K
2CO
3 (278 mg, 2.02 mmol) , Pd
2 (dba)
3 (61.5 mg, 67.2 μmol) and XantPhos (77.7 mg, 134 μmol) at 25 ℃. The reaction mixture was stirred at 70 ℃ under N
2 for 5 hours. The mixture was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with MeOH in DCM from 0%to 4%to give tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -3-(methylsulfonyl) picolinamido) propyl) carbamate (237 mg, 368 μmol, 54%yield, 90%purity) as a yellow solid. LC-MS: m/z 579.2 [M+H]
+.
Step 4: (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propyl) carbamoyl) -5-
(methylsulfonyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-
carboxylate
To a solution of tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -3- (methylsulfonyl) picolinamido) propyl) carbamate (144 mg, 248 μmol) in DCM (5 mL) was added di (imidazol-1-yl) methanone (121 mg, 746 μmol) and Et
3N (126 mg, 1.24 mmol, 173 μL) at 25 ℃. The reaction mixture was stirred at 25 ℃ for 3 hours. The mixture was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc in petroleum from 0%to 100%to give (1R, 3S) -3- (5- ( (6- ( (3- ( (tert-butoxycarbonyl) amino) propyl) carbamoyl) -5- (methylsulfonyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H- pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (200 mg, 237 μmol, 95%yield, 80%purity) as a white solid. LC-MS: (ESI) m/z [M+H]
+ 673.3.
Step 5: (1R, 3S) -3- (5- ( (6- ( (3-aminopropyl) carbamoyl) -5- (methylsulfonyl) pyridin-2-yl) amino) -1-
(tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
[ (1R, 3S) -3- [5- [ [6- [3- (tert-butoxycarbonylamino) propylcarbamoyl] -5-methylsulfonyl-2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (200 mg, 297 μmol) was dissolved in TFA/DCM=1/1 (8 mL) then stirred at 25 ℃ under N
2 for 1 hours. The mixture was concentrated under reduced pressure to give [ (1R, 3S) -3- [5- [ [6- (3-aminopropylcarbamoyl) -5-methylsulfonyl-2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (200 mg, 279 μmol, 93%yield, 80%purity) as a yellow oil, which was used directly in the next step without further purification. LC-MS: (ESI) m/z [M+H]
+: 573.2.
Step 6: (1
1S, 1
3R, Z) -21- (tert-butyl) -4
5- (methylsulfonyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-
2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
To a solution of [ (1R, 3S) -3- [5- [ [6- (3-aminopropylcarbamoyl) -5-methylsulfonyl-2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (200 mg, 349 μmol) in ACN (80 mL) was added Et
3N (14 mL) at 25 ℃. The reaction mixture was stirred at 80 ℃ for 72 hours. The mixture was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc in petroleum from 0%to 100%to give (1
1S, 1
3R, Z) -2
1- (tert-butyl) -4
5- (methylsulfonyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (150 mg, 297.26 μmol) as a yellow oil. LC-MS: (ESI) m/z [M+H]
+505.2.
Step 7: (1
1S, 1
3R, Z) -4
5- (methylsulfonyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
(1
1S, 1
3R, Z) -21- (tert-butyl) -4
5- (methylsulfonyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (180 mg, 356 μmol) was dissolved in TFA (8 mL) then stirred at 80 ℃ for 48 hours. The mixture was concentrated under reduced pressure. The residue was purified preparative reverse-phase HPLC (Column: Xbridge-C18, 250*21.2 mm 10 um; Mobile phase: MeCN-H
2O (0.1%FA) ; Gradient: 14%to 100%; Flow rate: 25 ml/min) to give (1
1S, 1
3R, Z) -4
5- (methylsulfonyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (36.5 mg, 81.4 μmol, 23%yield) as a white solid. LC-MS: (ESI) m/z [M+H]
+ 449.0.
Example 159
(1
1S, 1
3R, Z) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-7, 11-dione

Step 1: tert-butyl (3- ( ( (6-bromopyridin-2-yl) methyl) amino) -3-oxopropyl) carbamate
To a stirred solution of (6-bromopyridin-2-yl) methanamine (500 mg, 2.69 mmol) in CH
2Cl
2 (15.0 mL) were sequentially added 3- ( (tert-butoxycarbonyl) amino) propanoic acid (510 mg, 2.69 mmol) , HATU (1.50 g, 4.04 mmol) and DIPEA (1.38 mL, 1.04 g, 8.07 mmol) . The mixture was stirred at 25 ℃ for 2 h. The reaction mixture was quenched with water (30 mL) . The organic phase was washed with brine (10 mL) and dried over anhydrous Na
2SO
4, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 50 %in 25 min) to afford tert-butyl (3- ( ( (6-bromopyridin-2-yl) methyl) amino) -3-oxopropyl) carbamate (800 mg, 83%yield) as a white solid. LC-MS: m/z [M+H] + 357.9
Step 2: tert-butyl (3- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) methyl) amino) -3-oxopropyl) carbamate
To a stirred suspension of tert-butyl (3- ( ( (6-bromopyridin-2-yl) methyl) amino) -3-oxopropyl) carbamate (200 mg, 560 μmol) in 1, 4-dioxane (15.0 mL) were sequentially added (1R, 3S) -3- (5-amino-2-tert-butyl-pyrazol-3-yl) cyclopentanol (125 mg, 560 μmol) , Pd
2 (dba)
3 (51.0 mg, 56.0 μmol) , XantPhos (65.0 mg, 112 μmol) and Cs
2CO
3 (550 mg, 1.68 mmol) at 25 ℃. The reaction mixture was warmed to 80 ℃ and stirred at that temperature for 12 h. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 85 %in 25 min) to afford tert-butyl (3- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methyl) amino) -3-oxopropyl) carbamate (240 mg, 85%yield) as a yellow oil. LC-MS: m/z [M+H] + 500.9
Step 3: tert-butyl (3- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-
nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methyl) amino) -3-
oxopropyl) carbamate
To a stirred suspension of tert-butyl (3- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methyl) amino) -3-oxopropyl) carbamate (90.0 mg, 180 μmol) in DCE (15.0 ml) were added 4-nitrophenyl carbonochloridate (110 mg, 540 μmol) , DMAP (13.0 mg, 110 μmol) and Et
3N (74 μL, 55 mg, 540 μmol) . The reaction was warmed to 70 ℃ and stirred for 12 hours. Then the reaction mixture was quenched with ice-cold water (10 mL) and extracted with DCM (3 × 10 mL) . The organic phase was dried over anhydrous Na
2SO
4, filtered and concentrated in vacuo. The crude product tert-butyl (3- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methyl) amino) -3-oxopropyl) carbamate was used in the next step without further purification LC-MS: m/z [M+H] +665.7.
Step 4: (1R, 3S) -3- (3- ( (6- ( (3-aminopropanamido) methyl) pyridin-2-yl) amino) -1H-pyrazol-5-
yl) cyclopentyl (4-nitrophenyl) carbonate
A solution of tert-butyl (3- ( ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methyl) amino) -3-oxopropyl) carbamate in TFA (5.0 mL) was warmed to 100 ℃ and stirred at that temperature for 2 h. The mixture was concentrated under reduced pressure to give (1R, 3S) -3- (3- ( (6- ( (3-aminopropanamido) methyl) pyridin-2-yl) amino) -1H-pyrazol-5-yl) cyclopentyl (4-nitrophenyl) carbonate, which was used in the next step without further purification. LC-MS: m/z [M+H] + 509.8
Step 5: (1
1S, 1
3R, Z) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-7, 11-dione
To a stirred solution of (1R, 3S) -3- (3- ( (6- ( (3-aminopropanamido) methyl) pyridin-2-yl) amino) -1H-pyrazol-5-yl) cyclopentyl (4-nitrophenyl) carbonate in CH
3CN (5.0 mL) was added Et
3N (2.65 mL, 196 mg, 1.94 mmol) . The reaction mixture was stirred at 25 ℃ for 2 h. Then it was concentrated and the residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 3%to 50%in 30 min) to afford (1
1S, 1
3R, Z) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-7, 11-dione (14.2 mg, 22%yield) as an off-white solid. LC-MS: m/z [M+H] + 371.0.
Example 160
(1
1S, 1
3R, Z) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyrazina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione

Step 1: methyl 6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyrazine-2-carboxylate
To a stirred suspension of methyl 6-chloropyrazine-2-carboxylate (500 mg, 2.91 mmol) in 1, 4-dioxane (10.0 mL) were sequentially added (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (648 mg, 2.91 mmol) , Pd
2 (dba)
3 (133 mg, 145 μmol) , XantPhos (168 mg, 290 μmol) and K
3PO
4 (1.85 g, 8.73 mmol) at 25 ℃. The reaction mixture was warmed to 100 ℃ and stirred at that temperature for 12 h. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 80 %in 25 min) to afford methyl 6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyrazine-2-carboxylate (300 mg, 29%yield) as a yellow solid. LC-MS: m/z [M+H] + 360.2
Step 2: 6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyrazine-2-
carboxylic acid
To a suspension of methyl 6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyrazine-2-carboxylate (300 mg, 0.84 mmol) in MeOH/H
2O (5: 1, 12.0 mL) was added LiOH·H
2O (106 mg, 2.52 mmol) at 25 ℃. The reaction mixture was warmed to 60 ℃ and stirred at that temperature for 12 h. The reaction mixture was concentrated and adjusted to pH = 2~3 with 2 M HCl and extracted with EtOAc (20 mL × 3) . The combined organic phases were dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The crude product 6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyrazine-2-carboxylic acid (240 mg, 82%yield) as a colorless oil. was used directly in the next step without further purification. LC-MS: m/z [M+H] + 346.1
Step 3: tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyrazine-2-carboxamido) propyl) carbamate
To a suspension of 6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyrazine-2-carboxylic acid (240 mg, 0.69 mmol) in DMF (10.0 mL) was added tert-butyl (3-aminopropyl) carbamate (180 mg, 1.04 mmol) , HATU (393 mg, 1.03 mmol) and DIPEA (356 μL, 269 mg, 2.07 mmol) at 25 ℃. The reaction mixture was stirred at that temperature for 2 h before it was quenched with ice-cold water (20 mL) and extracted with CH
2Cl
2 (20 mL × 3) . The combined organic phases were dried over anhydrous Na
2SO
4, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 60 %in 20 min) to afford tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyrazine-2-carboxamido) propyl) carbamate (200 mg, 58%yield) as a yellow oil LC-MS: m/z [M+H] + 502.2
Step 4: tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -
1H-pyrazol-5-yl) amino) pyrazine-2-carboxamido) propyl) carbamate
To a stirred suspension of tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyrazine-2-carboxamido) propyl) carbamate (200 mg, 0.40 mmol) in DCE (10.0 mL) was added 4-nitrophenyl carbonochloridate (241 mg, 1.20 mmol) , DMAP (24.0 mg, 200 μmol) and DIPEA (205 μL, 155 mg, 1.20 mmol) at 25 ℃. The mixture was heated to 70 ℃ and stirred at that temperature for 12 h. The reaction mixture was quenched with water (30 mL) . The organic phase was washed with brine (10 mL) and dried over anhydrous Na
2SO
4, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 30 %in 25 min) to give tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyrazine-2-carboxamido) propyl) carbamate (60.0 mg, 23%yield) as a yellow oil. LC-MS: m/z [M+H] + 667.2
Step 5: (1R, 3S) -3- (5- ( (6- ( (3-aminopropyl) carbamoyl) pyrazin-2-yl) amino) -1- (tert-butyl) -1H-
pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate
A solution of tert-butyl (3- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyrazine-2-carboxamido) propyl) carbamate (60.0 mg, 0.90 mmol) in CH
2Cl
2 (10.0 mL) was added slowly TFA (2.0 mL) at 25 ℃. The mixture was stirred at that temperature for 1 h. The reaction mixture was concentrated in vacuo to afford (1R, 3S) -3- (5- ( (6- ( (3-aminopropyl) carbamoyl) pyrazin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate as a yellow oil. The crude product was used in the next step without further purification. LC-MS: m/z [M+H] + 567.2
Step 6: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyrazina-2 (3, 5) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
To a suspension of (1R, 3S) -3- (5- ( (6- ( (3-aminopropyl) carbamoyl) pyrazin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate in CH
3CN (10.0 mL) was added Et
3N (170 μL, 122 mg, 1.21 mmol) at 25 ℃. The reaction was stirred at that temperature for 2 h. The mixture was concentrated in vacuo to afford (1
1S, 1
3R, Z) -21- (tert-butyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyrazina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione as a yellow oil. The crude product was used in the next step without further purification. LC-MS: m/z [M+H] + 428.2
Step 7: (1
1S, 1
3R, Z) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyrazina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione
A solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyrazina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione in HCO
2H (5.0 mL) was warmed to 100 ℃ and stirred at that temperature for 5 h before it was cooled, concentrated under reduced pressure. The residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 5%to 65%in 20 min) to afford (1
1S, 1
3R, Z) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyrazina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (6.2 mg, 18%yield) as a yellow solid. LC-MS: m/z [M+H] + 372.1,
Example 161
(1
1S, 1
3R, Z) -2
1H-12-oxa-3, 6, 10-triaza-4 (4, 2) -pyrimidina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione

Step 1: N- (1- (tert-butyl) -3- ( (1S, 3R) -3- ( (tert-butyldimethylsilyl) oxy) cyclopentyl) -1H-pyrazol-5-
yl) -2-chloropyrimidin-4-amine
To a stirred solution of 2, 4-dichloropyrimidine (700 mg, 4.70 mmol) in 1, 4-dioxane (50.0 mL) were sequentially added 1- (tert-butyl) -3- ( (1S, 3R) -3- ( (tert-butyldimethylsilyl) oxy) cyclopentyl) -1H-pyrazol-5-amine (1.10 g, 3.26 mmol) , Pa
2 (dba)
3
. CHCl
3 (973 mg, 0.940 mmol) , XantPhos (816 mg, 1.41 mmol) and K
3PO
4 (2.00 g, 9.40 mmol) at 25 ℃. The reaction mixture was warmed to 100 ℃ and stirred at that temperature for 16 h under N
2 atmosphere before it was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 50 %in 20 min) to afford N- (1- (tert-butyl) -3- ( (1S, 3R) -3- ( (tert-butyldimethylsilyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) -2-chloropyrimidin-4-amine (350 mg, impure) as a brown oil. LC-MS: m/z [M+H]
+450.2.
Step 2: methyl 4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( (tert-butyldimethylsilyl) oxy) cyclopentyl) -1H-
pyrazol-5-yl) amino) pyrimidine-2-carboxylate
To a stirred solution of N- (1- (tert-butyl) -3- ( (1S, 3R) -3- ( (tert-butyldimethylsilyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) -2-chloropyrimidin-4-amine (250 mg, 555 μmol) in MeOH (15.0 mL) were sequentially added Pd (dppf) Cl
2 (81.3 mg, 111 μmol) and Et
3N (169 mg, 1.67 mmol) at 25 ℃. The reaction mixture was warmed to 100 ℃ and stirred at that temperature for 16 h under CO atmosphere. The reaction mixture was cooled to 25 ℃, concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 50 %in 20 min) to afford methyl 4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( (tert-butyldimethylsilyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyrimidine-2-carboxylate (80.0 mg, 30%yield) as a pale yellow oil. LC-MS: m/z [M+H]
+474.3.
Step 3: tert-butyl (3- (4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( (tert-butyldimethylsilyl) oxy) cyclopentyl) -
1H-pyrazol-5-yl) amino) pyrimidine-2-carboxamido) propyl) carbamate
To a stirred solution of methyl 4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( (tert-butyldimethylsilyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyrimidine-2-carboxylate (95.0 mg, 201 μmol) in CH
2ClCH
2Cl (20.0 mL) were sequentially added tert-butyl (3-aminopropyl) carbamate (35.0 μL, 34.9 mg, 201 μmol) and TMA (28.9 mg, 401 μmol) at 20 ℃ and stirred at that temperature for 16 h. The reaction mixture was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 30 %in 25 min) to afford tert-butyl (3- (4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( (tert-butyldimethylsilyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyrimidine-2-carboxamido) propyl) carbamate (70.0 mg, 57%yield) as a yellow oil. LC-MS: m/z [M+1]
+ 616.4.
Step 4: tert-butyl (3- (4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyrimidine-2-carboxamido) propyl) carbamate
To a stirred solution of tert-butyl (3- (4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( (tert-butyldimethylsilyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyrimidine-2-carboxamido) propyl) carbamate (70.0 mg, 114 μmol) in THF (5.0 mL) was added TBAF (44.6 mg, 170 μmol) at 25 ℃. The reaction mixture was warmed to 40 ℃ and stirred at that temperature for 2 h. The reaction mixture was diluted with water (30 mL) and extracted with EtOAc (20 mL x 3) . The combined organic layers were washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrate under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 60 %in 20 min) to afford tert-butyl (3- (4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyrimidine-2-carboxamido) propyl) carbamate (45.0 mg, 78%yield) as a white solid. LC-MS: m/z [M+H]
+ 502.1.
Step 5: tert-butyl (3- (4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -
1H-pyrazol-5-yl) amino) pyrimidine-2-carboxamido) propyl) carbamate
To a stirred solution of tert-butyl (3- (4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyrimidine-2-carboxamido) propyl) carbamate (40.0 mg, 79.7 μmol) in CH
3CN (4.0 mL) were sequentially added NMM (17.5 μL, 16.1 mg, 159 μmol) , 4-nitrophenyl carbonochloridate (48.2 mg, 239 μmol) and DMAP (9.74 mg, 79.7 μmol) 25 ℃. The reaction mixture was stirred at that temperature for 2 h before it was diluted with water (30 mL) and extracted with DCM (20 mL x 3) . The combined organic layers were washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrate under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 60 %in 20 min) to afford tert-butyl (3- (4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyrimidine-2-carboxamido) propyl) carbamate (20.0 mg, 38%yield) as a pale light yellow solid. LC-MS: m/z [M+H]
+ 667.1.
Step 6: (1R, 3S) -3- (5- ( (2- ( (3-aminopropyl) carbamoyl) pyrimidin-4-yl) amino) -1- (tert-butyl) -1H-
pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate
To a solution of tert-butyl (3- (4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyrimidine-2-carboxamido) propyl) carbamate (20.0 mg, 30.0 μmol) in CH
2Cl
2 (2.0 mL) was added TFA (2.0 mL) at 20 ℃. And the reaction mixture was stirred at that temperature for 1 h before it was concentrated under reduced pressure to give (1R, 3S) -3- (5- ( (2- ( (3-aminopropyl) carbamoyl) pyrimidin-4-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate which was directly used for the next step without further purification. LC-MS: m/z [M+H]
+ 567.3.
Step 7: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (4, 2) -pyrimidina-2 (3, 5) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
To a stirred solution of (1R, 3S) -3- (5- ( (2- ( (3-aminopropyl) carbamoyl) pyrimidin-4-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate (crude) in CH
3CN (2.0 mL) was added TEA (499 μL, 363 mg, 3.59 mmol) at 25 ℃. The reaction mixture was stirred at that temperature for 1 h before it was concentrated under reduced pressure. The ressidue was purified by silica gel chromatography eluting with MeOH/CH
2Cl
2 (with MeOH from 0 to 10%in 25 min) to afford (1
1S, 1
3R, Z) -21- (tert-butyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (4, 2) -pyrimidina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (1.50 mg, 10%yield) as a white solid. LC-MS: m/z [M+H]
+ 428.
Step 8: (1
1S, 1
3R, Z) -2
1H-12-oxa-3, 6, 10-triaza-4 (4, 2) -pyrimidina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione
A solution of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (4, 2) -pyrimidina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (1.50 mg, 3.51 μmol) in TFA (3.0 mL) was warmed to 70 ℃ and stirred at that temperature for 16 h. The reaction mixture was cooled, concentrated under reduced pressure. The residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 5%to 50%in 40 min) to give (1
1S, 1
3R, Z) -2
1H-12-oxa-3, 6, 10-triaza-4 (4, 2) - pyrimidina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (0.900 mg, 69%yield) as a white solid. LC-MS: m/z [M+H]
+ 372.2.
Example 162
(1
1S, 1
3R, 2
4Z, 4
2Z) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 4) -thiazola-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione

Step 1: tert-butyl (3- (2-bromothiazole-4-carboxamido) propyl) carbamate
To a suspension of 2-bromothiazole-4-carboxylic acid (500 mg, 2.42 mmol) in CH
2Cl
2 (15.0 mL) was added tert-butyl (3-aminopropyl) carbamate (630 mg, 3.63 mmol) , HATU (1.38 g, 3.63 mmol) and DIPEA (1.24 mL, 936 mg, 7.26 mmol) at 25 ℃. The reaction mixture was stirred at that temperature for 2 h before it was quenched with ice-cold water (20 mL) and extracted with CH
2Cl
2 (20 mL × 3) . The combined organic phases were dried over anhydrous Na
2SO
4, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 22 %in 20 min) to tert-butyl (3- (2-bromothiazole-4-carboxamido) propyl) carbamate (700 mg, 80%yield) as a white solid. LC-MS: m/z [M-55] + 308.0.
Step 2: tert-butyl (3- (2- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) thiazole-4-carboxamido) propyl) carbamate
To a stirred solution of tert-butyl (3- (2-bromothiazole-4-carboxamido) propyl) carbamate (300 mg, 830 μmol) in 1, 4-dioxane (10.0 mL) were sequentially added (1R, 3S) -3- (5-amino-2-tert-butyl-pyrazol-3-yl) cyclopentanol (185 mg, 830 μmol) , Pd
2 (dba)
3 (75.9 mg, 83.0 μmol) , XantPhos (96.1 mg, 166 μmol) and K
2CO
3 (344 mg, 2.49 mmol) at 25 ℃. The reaction mixture was warmed to 100 ℃ and stirred at that temperature for 12 h. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with MeOH/CH
2Cl
2 (with MeOH from 0 to 4 %in 25 min) to give tert-butyl (3- (2- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) thiazole-4-carboxamido) propyl) carbamate (150 mg, 36%yield) as a yellow oil. LC-MS: m/z [M+H] + 507.3
Step 3: tert-butyl (3- (2- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -
1H-pyrazol-5-yl) amino) thiazole-4-carboxamido) propyl) carbamate
To a stirred suspension of tert-butyl (3- (2- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) thiazole-4-carboxamido) propyl) carbamate (150 mg, 290 μmol) in DCE (15.0 ml) was added 4-nitrophenyl carbonochloridate (175 mg, 870 μmol) , DMAP (35.0 mg, 290 μmol) and DIPEA (149 μL, 112 mg, 870 μmol) at 25 ℃. The mixture was heated to 70 ℃ and stirred at that temperature for 12 h. The reaction mixture was quenched with water (30 mL) . The organic phase was washed with brine (10 mL) and dried over anhydrous Na
2SO
4, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 15 %in 25 min) to afford tert-butyl (3- (2- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) thiazole-4-carboxamido) propyl) carbamate (100 mg, 30%yield) as a yellow oil. LC-MS: m/z [M+H] + 672.2
Step 4: (1R, 3S) -3- (5- ( (4- ( (3-aminopropyl) carbamoyl) thiazol-2-yl) amino) -1- (tert-butyl) -1H-
pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate
To a stirred solution of tert-butyl (3- (2- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) thiazole-4-carboxamido) propyl) carbamate (100 mg, 175 μmol) in CH
2Cl
2 (10.0 ml) was added TFA (396 μL, 586 mg, 5.14 mmol) at 25 ℃. The mixture was stirred at that temperature for 1 h before it was concentrated under reduced pressure to afford (1R, 3S) -3- (5- ( (4- ( (3-aminopropyl) carbamoyl) thiazol-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate as a yellow oil, which was used in the next step without further purification. LC-MS: m/z [M+H] + 572.2
Step 5: (1
1S, 1
3R, 2
4Z, 4
2Z) -2
1- (tert-butyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 4) -thiazola-2 (3, 5) -pyrazola-
1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
To a stirred suspension of (1R, 3S) -3- (5- ( (4- ( (3-aminopropyl) carbamoyl) thiazol-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate in CH
3CN (10.0 mL) was added Et
3N (0.27 mL, 196 mg, 1.94 mmol) at 25 ℃. The mixture was stirred at 25 ℃ for 2 h. The reaction mixture was concentrated in vacuo to afford (1
1S, 1
3R, 2
4Z, 4
2Z) -2
1- (tert-butyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 4) -thiazola-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione, which was used in the next step without further purification. LC-MS: m/z [M+H] + 433.1
Step 6: (1
1S, 1
3R, 2
4Z, 4
2Z) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 4) -thiazola-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione
A solution of (1
1S, 1
3R, 2
4Z, 4
2Z) -2
1- (tert-butyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 4) -thiazola-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione in HCO
2H (5.0 mL) was warmed to 100 ℃ and stirred at that temperature for 5 h before it was cooled, concentrated under reduced pressure. The residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 5%to 75%in 20 min) to afford (1
1S, 1
3R, 2
4Z, 4
2Z) -2
1H-12-oxa-3, 6, 10-triaza-4 (2, 4) -thiazola-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (1.20 mg, 2%yield) as a white solid. LC-MS: m/z [M+H] + 377.1.
Example 163
(1
1S, 1
3R, E) -2
4-fluoro-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione


Step 1: ethyl 1- (2-oxopropyl) -1H-pyrazole-5-carboxylate
To a suspension of ethyl 1H-pyrazole-5-carboxylate (5.5 g, 39.2 mmol) in CH
3CN (100 mL) was added K
2CO
3 (10.9 g, 78.5 mmol) . The reaction was stirred at 25 ℃ for 16 h, before it was concentrated in vacuo. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 10 %in 15 min) to afford ethyl 1- (2-oxopropyl) -1H-pyrazole-5-carboxylate (1.3 g, 18%yield) as a yellow solid. LC-MS: m/z [M+H]
+ 197.2.
Step 2: 6-methylpyrazolo [1, 5-a] pyrazin-4-ol
To a suspension of ethyl 1- (2-oxopropyl) -1H-pyrazole-5-carboxylate (1.0 g, 5.1 mmol) in AcOH (10.0 mL) was added NH
4OAc (3.93 g, 51.0 mmol) . The reaction was stirred at 60 ℃ for 5 h, before it was concentrated in vacuo. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 10 %in 15 min) to afford 6-methylpyrazolo [1, 5-a] pyrazin-4-ol (450 mg, 59%yield) as a yellow solid. LC-MS: m/z [M+H]
+ 150.1.
Step 3: 4-hydroxypyrazolo [1, 5-a] pyrazine-6-carbaldehyde
To a suspension of 6-methylpyrazolo [1, 5-a] pyrazin-4-ol (450 mg, 3.02 mmol) in pyridine (5.0 mL) was added SeO
2 (669 mg, 6.03 mmol) . The reaction was stirred at 100 ℃ for 2 h, before it was concentrated in vacuo. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 5 %in 15 min) to afford 4-hydroxypyrazolo [1, 5-a] pyrazine-6-carbaldehyde (220 mg, 44%yield) as a yellow solid. LC-MS: m/z [M+H]
+ 164.0.
Step 4: 4-hydroxypyrazolo [1, 5-a] pyrazine-6-carboxylic acid
To a suspension of 4-hydroxypyrazolo [1, 5-a] pyrazine-6-carbaldehyde (220 mg, 1.35 mmol) in t-BuOH (3.0 ml) /H
2O (1.0 mL) was added 2-methylbut-2-ene (284 mg, 4.05 mmol) , NaH
2PO
4 (210 mg, 1.35 mmol) and NaClO
2 (366 mg, 4.05 mmol) . The reaction was stirred at 25 ℃ for 2 h, before it was concentrated in vacuo. The residue was purified by silica gel chromatography eluting with MeOH/CH
2Cl
2 (with MeOH from 0 to 10 %in 10 min) to afford 4-hydroxypyrazolo [1, 5-a] pyrazine-6-carboxylic acid (200 mg, 82%yield) as a yellow solid. LC-MS: m/z [M+H]
+ 180.1.
Step 5: methyl 4-hydroxypyrazolo [1, 5-a] pyrazine-6-carboxylate
A mixture of 4-hydroxypyrazolo [1, 5-a] pyrazine-6-carboxylic acid (100 mg, 0.558 mmol) in SOCl
2 (2.0 mL) was stirred for 2 h at 60 ℃, before MeOH (2.0 mL) was added. The mixture was concentrated in vacuo. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 5 %in 15 min) to afford methyl 4-hydroxypyrazolo [1, 5-a] pyrazine-6-carboxylate (100 mg, 92%yield) as a yellow solid. LC-MS: m/z [M+H]
+ 194.2.
Step 6: methyl 4-chloropyrazolo [1, 5-a] pyrazine-6-carboxylate
A mixture of tert-butyl (3- (6- ( (1- (tert-butyl) -4-fluoro-3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) picolinamido) propyl) carbamate (70 mg, 0.102 mmol) in POCl
3 (2.0 mL) was stirred for 2 hours at 80 ℃. The mixture was concentrated in vacuo. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 5 %in 15 min) to afford methyl 4-hydroxypyrazolo [1, 5-a] pyrazine-6-carboxylate (100 mg, 92%yield) as a yellow solid. LC-MS: m/z [M+H]
+ 212.1.
Step 7: methyl 4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyrazolo [1, 5-a] pyrazine-6-carboxylate
To a suspension of methyl 4-hydroxypyrazolo [1, 5-a] pyrazine-6-carboxylate (70 mg, 310 μmol) and (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (69.28 mg, 310 μmol) in 1, 4-dioxane (3.0 mL) was added RuPhos Pd G3 (25.9 mg, 31.0 μmol) , RuPhos (28.9 mg, 62.1 μmol) and Cs
2CO
3 (202 mg, 620 μmol) . The reaction was stirred at 100 ℃ for 16 h under N
2, before it was concentrated in vacuo and purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 35 %in 20 min) to give methyl 4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyrazolo [1, 5-a] pyrazine-6-carboxylate (50 mg, 40%yield) as a yellow solid. LC-MS: m/z [M+H]
+ 399.2.
Step 8: tert-butyl (3- (4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyrazolo [1, 5-a] pyrazine-6-carboxamido) propyl) carbamate
To a suspension of 4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyrazolo [1, 5-a] pyrazine-6-carboxylate (45 mg, 112 μmol) in DCE (2.0 mL) was added dropwise AlMe
3 in THF (0.16 mL, 2.0 M, 0.32 mmol) . The reaction was stirred at 25 ℃ for 5 h under N
2, before it was filtered and concentrated in vacuo. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 45 %in 20 min) to afford tert-butyl (3- (4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyrazolo [1, 5-a] pyrazine-6-carboxamido) propyl) carbamate (35 mg, 57%yield) as a yellow solid. LC-MS: m/z [M+H]
+ 541.1.
Step 9: tert-butyl (3- (4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -
1H-pyrazol-5-yl) amino) pyrazolo [1, 5-a] pyrazine-6-carboxamido) propyl) carbamate
To a suspension of tert-butyl (3- (4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyrazolo [1, 5-a] pyrazine-6-carboxamido) propyl) carbamate (30 mg, 55.4 μmol) in CH
3CN (5.0 mL) was added 4-nitrophenyl carbonochloridate (33.5 mg, 166 μmol) , DMAP (13.5 mg, 111 μmol) and NMM (30.5 μL, 28.1 mg, 277 μmol) . The reaction was stirred at 25 ℃ for 16 h under N
2, before it was concentrated in vacuo and purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 50 %in 20 min) to give tert-butyl (3- (4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyrazolo [1, 5-a] pyrazine-6-carboxamido) propyl) carbamate (20 mg, 51%yield) as a yellow solid. LC-MS: m/z 705.9 [M+H]
+.
Step 10: (1R, 3S) -3- (5- ( (6- ( (3-aminopropyl) carbamoyl) pyrazolo [1, 5-a] pyrazin-4-yl) amino) -1-
(tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate
A mixture of tert-butyl (3- (4- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyrazolo [1, 5-a] pyrazine-6-carboxamido) propyl) carbamate (20 mg, 28.3 μmol) in TFA (1.0 mL) was stirred for 1 h at 25 ℃, before it was concentrated in vacuo to give crude (1R, 3S) -3- (5- ( (6- ( (3-aminopropyl) carbamoyl) pyrazolo [1, 5-a] pyrazin-4-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate as a brown oil, which was used in the next step without further purification. LC-MS: m/z [M+H]
+ 605.8.
Step 11: (1
1S, 1
3R, 2
4Z, 4
4E) -2
1- (tert-butyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (4, 6) -pyrazolo [1, 5-
a] pyrazina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
To a solution of (1R, 3S) -3- (5- ( (6- ( (3-aminopropyl) carbamoyl) pyrazolo [1, 5-a] pyrazin-4-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate in CH
3CN (1.0 mL) was added Et
3N (23μL, 16.7 mg, 165 μmol) at room temperature. The mixture was stirred at room temperature for 2 h, before it was concentrated in vacuo and purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 60 %in 20 min) to afford (1
1S, 1
3R, 2
4Z, 4
4E) -2
1- (tert-butyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (4, 6) -pyrazolo [1, 5-a] pyrazina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (20.0 mg, 26%yield) as a yellow solid. LC-MS: m/z [M+H]
+ 466.9.
Step 12: (1
1S, 1
3R, 2
4Z, 4
4E) -2
1H-12-oxa-3, 6, 10-triaza-4 (4, 6) -pyrazolo [1, 5-a] pyrazina-2 (5, 3) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
A mixture of (1
1S, 1
3R, 2
4Z, 4
4E) -2
1- (tert-butyl) -2
1H-12-oxa-3, 6, 10-triaza-4 (4, 6) -pyrazolo [1, 5-a] pyrazina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (20.0 mg, 21.4 μmol) in HCO
2H (2.0 mL) was stirred at 80 ℃ for 2 h, before it was concentrated and purified by prep-HPLC eluting with CH
3CN in water (with CH
3CN from 15%to 40%in 10 min (0.1%FA condition) ) to afford (1
1S, 1
3R, 2
4Z, 4
4E) -2
1H-12-oxa-3, 6, 10-triaza-4 (4, 6) -pyrazolo [1, 5-a] pyrazina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (1.3 mg, 14%) as a white solid. LC-MS: m/z [M+H]
+ 410.9.
Example 164
(1
1S, 1
3R, Z) -2
1H-11-oxa-3, 6, 9-triaza-4 (5, 3) -thieno [3, 2-b] pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacycloundecaphane-5, 10-dione

Step 1: 5-chlorothieno [3, 2-b] pyridine-3-carbonitrile
To a suspension of 3-bromo-5-chloro-thieno [3, 2-b] pyridine (100 mg, 402 μmol) in DMF (15 mL) at 25 ℃ was added CuCN (113 mg, 1.21 mmol) . Then the reaction mixture was irradiated in a microwave reactor at 160 ℃ for 2 hours. The mixture was quenched with MeOH (5 mL) . After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by flash column chromatography eluting with EtOAc in PE form 0 to 50%, then it was purified by flash column chromatography eluting with methanol in dichloromethane from 0 to 5%to give 5-chlorothieno [3, 2-b] pyridine-3-carbonitrile (78 mg, 401 μmol, 99%yield) as yellow solid. LC-MS: m/z [M+H]
+ 194.9.
Step 2: 5-chlorothieno [3, 2-b] pyridine-3-carboxylic acid
To a suspension of 5-chlorothieno [3, 2-b] pyridine-3-carbonitrile (100 mg, 514 μmol) in AcOH (2 mL) was added H
2O (2 mL) and H
2SO
4 (2 mL) at 25 ℃. Then the reaction mixture was stirred at 110 ℃ for 12 hours in sealed tube. The reaction was quenched with water (30 mL) , extracted with EtOAc (3 × 30 mL) . The combined organic layers were dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure to afford 5-chlorothieno [3, 2-b] pyridine-3-carboxylic acid (90 mg, 421μmol, 82%yield) as yellow solid. LC-MS: m/z [M+H]
+ 213.9.
Step 3: tert-butyl (2- (5-chlorothieno [3, 2-b] pyridine-3-carboxamido) ethyl) carbamate
To a stirred solution of 5-chlorothieno [3, 2-b] pyridine-3-carboxylic acid (90 mg, 421 μmol) in DCM (4 mL) was added tert-butyl N- (2-aminoethyl) carbamate (67.4 mg, 421 μmol) , EDCI (162 mg, 843 μmol) , HOBT (114 mg, 843 μmol) and DIPEA (163 mg, 1.26 mmol, 220 μL) . The reaction mixture was stirred at 25℃ for 12 hours. The reaction was quenched with addition of saturation water (20 mL) , extracted with DCM (20 mL × 3) . The combined organic layers were dried over anhydrous Na
2SO
4. After filtration, the filtrate was concentrated under reduced pressure. The residue was purified by flash column chromatography eluting with EtOAc in PE from 0 to 50%, tnen it was purified by flash column chromatography eluting with methanol in dichloromethane from 0 to 5%to give tert-butyl (2- (5-chlorothieno [3, 2-b] pyridine-3-carboxamido) ethyl) carbamate (130 mg, 365 μmol, 87%yield) as white solid. LC-MS: m/z [M+H]
+ 356.0.
Step 4: tert-butyl (2- (5- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) thieno [3, 2-b] pyridine-3-carboxamido) ethyl) carbamate
To a solution of tert-butyl (2- (5-chlorothieno [3, 2-b] pyridine-3-carboxamido) ethyl) carbamate (110 mg, 309 μmol) in dioxane (20 mL) was added (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (69.0 mg, 309 μmol) , Tris (Dibenzylideneacetone) Dipalladium (O) (56.6 mg, 61.8 μmol) , (5-diphenylphosphanyl-9, 9-dimethyl-xanthen-4-yl) -diphenyl-phosphane (71.5 mg, 123 μmol) and tripotassium; carbonate (128 mg, 927 μmol, 55.9 μL) . The mixture was stirred for 12 hours at 100 ℃ in under N
2. The resulting mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography eluting with EtOAc in PE formed 0 to 50%, then it was purified by flash column chromatography eluting with MeOH in DCM from 0 to 10%to give tert-butyl (2- (5- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) thieno [3, 2-b] pyridine-3-carboxamido) ethyl) carbamate (152 mg, 280 μmol, 91%yield) as yellow solid. LC-MS: m/z [M+H]
+ 543.2.
Step 5: tert-butyl (2- (5- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -
1H-pyrazol-5-yl) amino) thieno [3, 2-b] pyridine-3-carboxamido) ethyl) carbamate
To a suspension of tert-butyl (2- (5- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) thieno [3, 2-b] pyridine-3-carboxamido) ethyl) carbamate (130 mg, 239 μmol) in DCE (15 mL) was added slowly (4-nitrophenyl) carbonochloridate (145 mg, 718 μmol) , DMAP (11.7 mg, 95.8 μmol) and DIPEA (309 mg, 2.40 mmol, 417 μL) at 25 ℃ and stirred for 24 h at 85 ℃ under N
2. The resulting mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography eluting with EtOAc in PE form 0 to 50%, then it was purified by flash column chromatography eluting with methanol in dichloromethane from 0 to 5%to give tert-butyl (2- (5- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5- yl) amino) thieno [3, 2-b] pyridine-3-carboxamido) ethyl) carbamate (125 mg, 176 μmol, 74%yield) as yellow solid. LC-MS: m/z [M+H]
+ 708.2.
Step 6: (1R, 3S) -3- (5- ( (3- ( (2-aminoethyl) carbamoyl) thieno [3, 2-b] pyridin-5-yl) amino) -1- (tert-
butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate
To a stirred solution of tert-butyl (2- (5- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) thieno [3, 2-b] pyridine-3-carboxamido) ethyl) carbamate (100 mg, 141 μmol) in DCM (4 mL) at 25 ℃ was added Et
3N (4 mL) . The reaction mixture was stirred at 25 ℃ for 0.5 hours. The resulting mixture was concentrated under reduced pressure. The crude product was used immediately in the next step without purification. LC-MS: m/z [M+H]
+ 608.1.
Step 7: (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-11-oxa-3, 6, 9-triaza-4 (5, 3) -thieno [3, 2-b] pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacycloundecaphane-5, 10-dione
To a stirred solution of (1R, 3S) -3- (5- ( (3- ( (2-aminoethyl) carbamoyl) thieno [3, 2-b] pyridin-5-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl (4-nitrophenyl) carbonate (85 mg, 139 μmol) in CH
3CN (30 mL) at 25 ℃ was added Et
3N (5 mL) . The reaction mixture was stirred at 25 ℃ for 2 hours. The resulting mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography eluting with EtOAc in PE from 0 to 50%, then it was purified by flash column chromatography eluting with MeOH in DCM from 0 to 5%to give (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-11-oxa-3, 6, 9-triaza-4 (5, 3) -thieno [3, 2-b] pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacycloundecaphane-5, 10-dione (65 mg, 139 μmol, 99%yield) . LC-MS: m/z [M+H]
+ 469.1.
Step 7: (1
1S, 1
3R, Z) -2
1H-11-oxa-3, 6, 9-triaza-4 (5, 3) -thieno [3, 2-b] pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacycloundecaphane-5, 10-dione
A suspension of (1
1S, 1
3R, Z) -2
1- (tert-butyl) -2
1H-11-oxa-3, 6, 9-triaza-4 (5, 3) -thieno [3, 2-b] pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacycloundecaphane-5, 10-dione (65 mg, 138 μmol) at 25 ℃ and stirred for 6 h at 140 ℃ under N
2. The resulting mixture was concentrated under reduced pressure. The product was further purified by prep-HPLC (C18, 30-40%MeCN in 0.1%FA/water, 25 mL/min) to give (1
1S, 1
3R, Z) -2
1H-11-oxa-3, 6, 9-triaza-4 (5, 3) -thieno [3, 2-b] pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacycloundecaphane-5, 10-dione (21 mg, 50.91 μmol, 36.70%yield) . LC-MS: m/z [M+H]
+413.0.
Example 165
(1
1S, 1
3R, Z) -4
4-methoxy-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione

Step 1: 2-bromo-4-methoxy-6-vinylpyridine
To a suspension of 2, 6-dibromo-4-methoxy-pyridine (500 mg, 1.87 mmol) in DME (15.0 mL) was added aq. K
2CO
3 (1.72 mL, 2 M, 3.37 mmol) , 1- (2, 4, 6-trivinyl-1, 3, 5-trioxa-4, 6-dibora-2-boranuidacyclohex-2-yl) pyridin-1-ium (270 mg, 1.12 mmol) and Pd (PPh
3)
4 (43.3 mg, 0.037 mmol) at room temperature. The mixture was warmed to 85 ℃ and stirred at that temperature for 2 hours under N
2, before it was diluted with water (20 mL) and extracted with EtOAc (3 × 20 mL) . The organic phase was dried over anhydrous Na
2SO
4, filtered and the filtrate was concentrated under reduced pressure. The crude product was purified by flash chromatography eluting with EtOAc/PE (with EtOAc from 0 to 15%in 20 min) to afford 2-bromo-4-methoxy-6-vinyl-pyridine (320 mg, 79%yield) as a yellow solid. LC-MS: m/z [M+H]
+ 213.9.
Step 2: 6-bromo-4-methoxypicolinic acid
To a suspension of 2-bromo-4-methoxy-6-vinyl-pyridine (320 mg, 1.49 mmol) in acetone (15.0 mL) was added KMnO
4 (472 mg, 2.99 mmol) at 0 ℃ and stirred for 10 min before it was warmed to 25 ℃. The mixture was stirred for 2 h. After completion of the reaction as judged by LCMS, reaction mixture was filtered, the solid was washed with water and acetone and the filtrate was concentrated. The residue was dissolved in 10%aq. citric acid solution and extracted with EtOAc (3 × 20 mL) . The organic phase was dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure to afford 6-bromo-4-methoxy-pyridine-2-carboxylic acid (300 mg, 86%yield) as a yellow solid. LC-MS: m/z [M+H]
+ 231.8.
Step 3: tert-butyl N- [3- [ (6-bromo-4-methoxy-pyridine-2-carbonyl) amino] propyl] carbamate
To a suspension of 6-bromo-4-methoxy-pyridine-2-carboxylic acid (280 mg, 1.21 mmol) in CH
2Cl
2 (10.0 mL) was sequentially added tert-butyl N- (3-aminopropyl) carbamate (210 mg, 1.21 mmol) , HATU (917mg, 2.41 mmol) and DIPEA (419 μL, 311 mg, 2.41 mmol) at 25 ℃ and stirred for 2 h. After completion of the reaction as judged by LCMS, reaction mixture was concentrated under reduced pressure. The crude product was purified by flash chromatography eluting with EtOAc/PE (with EtOAc from 0 to 30%in 25 min) to afford tert-butyl N- [3- [ (6-bromo-4-methoxy-pyridine-2-carbonyl) amino] propyl] carbamate (300 mg, 64%yield) as a yellow solid. LC-MS: m/z [M-55]
+ 331.8.
Step 4: tert-butyl N- [3- [ [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -4-
methoxy-pyridine-2-carbonyl] amino] propyl] carbamate
To a suspension of tert-butyl N- [3- [ (6-bromo-4-methoxy-pyridine-2-carbonyl) amino] propyl] carbamate (280 mg, 0.721 mmol) in 1, 4-dioxane (15.0 mL) was sequentially added (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (161 mg, 0.721 mmol) , Cs
2CO
3 (705 mg, 2.16 mmol) , XantPhos (83.5 mg, 0.144 mmol) and Pd
2 (dba)
3 (66.0 mg, 0.072 mmol) at room temperature. The reaction was warmed to 100 ℃ and stirred at that temperature for 16 h under N
2, before it was filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography eluting with EtOAc/PE (with EtOAc from 0 to 80%in 30 min) to afford tert-butyl N- [3- [ [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -4-methoxy-pyridine-2-carbonyl] amino] propyl] carbamate (200 mg, 52%yield) as a yellow solid. LC-MS: m/z [M+H]
+ 530.9.
Step 5: [ (1R, 3S) -3- [5- [ [6- [3- (tert-butoxycarbonylamino) propylcarbamoyl] -4-methoxy-2-
pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate
To a suspension of tert-butyl N- [3- [ [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -4-methoxy-pyridine-2-carbonyl] amino] propyl] carbamate (200 mg, 0.376 mmol) in CH
2Cl
2 (10.0 mL) was added CDI (162 mg, 1.13 mmol) and DIPEA (327 μL, 243 mg, 1.88 mmol) at 35 ℃and stirred for 2 h. After completion of the reaction as judged by LCMS, reaction mixture was quenched with ice-cold water (20 mL) and extracted with EtOAc (3 × 20 mL) . The organic phase was washed with brine (50 mL) , dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure to afford [ (1R, 3S) -3- [5- [ [6- [3- (tert-butoxycarbonylamino) propylcarbamoyl] -4-methoxy-2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (crude) as a yellow oil. The crude product was used in the next step without further purification. LC-MS: m/z [M+H]
+624.9.
Step 6: [ (1R, 3S) -3- [5- [ [6- (3-aminopropylcarbamoyl) -4-methoxy-2-pyridyl] amino] -1-tert-butyl-
pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate
To a suspension of [ (1R, 3S) -3- [5- [ [6- [3- (tert-butoxycarbonylamino) propylcarbamoyl] -4-methoxy-2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate in CH
2Cl
2 (3.0 mL) was added slowly TFA (1.0 mL, 12.9 mmol) at 25 ℃ and stirred for 1 h. After completion of the reaction as judged by LCMS, reaction mixture was concentrated under reduced pressure to afford [ (1R, 3S) -3- [5- [ [6- (3-aminopropylcarbamoyl) -4-methoxy-2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (crude) as a yellow oil. The crude product was used in the next step without further purification. LC-MS: m/z [M+H]
+ 525.0.
Step 7: (1
1S, 1
3R, Z) -21- (tert-butyl) -4
4-methoxy-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione
To a suspension of [ (1R, 3S) -3- [5- [ [6- (3-aminopropylcarbamoyl) -4-methoxy-2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (crude) in CH
3CN (3.0 mL) was added Et
3N (397 μL, 289 mg, 2.86 mmol) . The reaction was warmed to 80 ℃ and stirred at that temperature for 16 h, before it was concentrated under reduced pressure. The crude product was purified by flash column chromatography eluting with MeOH/CH
2Cl
2 (with MeOH from 0 to 10%in 20 min) to afford (1
1S, 1
3R, Z) -21- (tert-butyl) -4
4-methoxy-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (85 mg) as a yellow solid. LC-MS: m/z [M+H]
+457.0.
Step 8: (1
1S, 1
3R, Z) -4
4-methoxy-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphane-5, 11-dione
A mixture of (1
1S, 1
3R, Z) -21- (tert-butyl) -4
4-methoxy-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (20.0 mg, 0.043 mmol) in HCO
2H (2.0 mL) was stirred for 5 h at 100 ℃, until the reaction was complete as indicated by LCMS, the reaction mixture was concentrated under reduced pressure, purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 15%to 50%in 10 min (0.1%HCO
2H condition) ) to give the desired product (1
1S, 1
3R, Z) -4
4-methoxy-2
1H-12-oxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphane-5, 11-dione (6.6 mg, 37%yield) as a white solid. LC-MS: m/z [M+H]
+ 401.2.
Example 166
(1
1S, 1
3R, 8
1R, 8
3S, Z) -4
3-fluoro-2
1H-11-oxa-3, 6, 9-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentana-8 (1, 3) -cyclobutanacycloundecaphane-5, 10-dione

Step 1: ( (1r, 3r) -3- ( (tert-butoxycarbonyl) amino) cyclobutyl) methyl 4-methylbenzenesulfonate
To a suspension of tert-butyl ( (1r, 3r) -3- (hydroxymethyl) cyclobutyl) carbamate (1.0 g, 4.97 mmol) in CH
2Cl
2 (15.0 mL) was added TsCl (1.89 g, 9.95 mmol) , Et
3N (1.98 mL, 1.50 g, 14.9 mmol) at 0 ℃. The reaction mixture was warmed to 25 ℃ and stirred at that temperature for 2 h before it was quenched with ice-cold water (20 mL) and extracted with CH
2Cl
2 (20 mL × 3) . The combined organic phases were dried over anhydrous Na
2SO
4, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 12 %in 20 min) to afford ( (1r, 3r) -3- ( (tert-butoxycarbonyl) amino) cyclobutyl) methyl 4-methylbenzenesulfonate (1.50 g, impure) as a yellow solid. LC-MS: m/z [M+Na]
+ 378.0
Step 2: tert-butyl ( (1r, 3r) -3- (azidomethyl) cyclobutyl) carbamate
To a stirred solution of ( (1r, 3r) -3- ( (tert-butoxycarbonyl) amino) cyclobutyl) methyl 4-methylbenzenesulfonate (1.50 g, impure) in DMF (20.0 mL) was added NaN
3 (1.65 g, 25.3 mmol) at 25 ℃. The reaction mixture was warmed to 120 ℃ and stirred at that temperature for 1 h. The solution was quenched with ice-cold water (20 mL) and extracted with CH
2Cl
2 (20 mL × 3) . The combined organic phases were dried over anhydrous Na
2SO
4, filtered and the filtrate was concentrated under reduced pressure to afford tert-butyl ( (1r, 3r) -3- (azidomethyl) cyclobutyl) carbamate (900 mg, crude) as a yellow oil. LC-MS: m/z [M -55] 171.0
Step 3: tert-butyl ( (1r, 3r) -3- (aminomethyl) cyclobutyl) carbamate
To a stirred suspension of tert-butyl ( (1r, 3r) -3- (azidomethyl) cyclobutyl) carbamate in MeOH (15.0 mL) was added Pd/C (900 mg, 10 wt. %in H
2O) at 25 ℃. The reaction mixture was stirred at that temperature for 2 h under hydrogen atmosphere. The mixture was filtered and the filtrate was concentrated under reduced pressure to give tert-butyl ( (1r, 3r) -3- (aminomethyl) cyclobutyl) carbamate, which was used directly in the next step without further purification.
Step 4: tert-butyl ( (1r, 3r) -3- ( (6-bromo-5-fluoropicolinamido) methyl) cyclobutyl) carbamate
To a stirred solution of 6-bromo-5-fluoropicolinic acid (400 mg, 1.83 mmol) in CH
2Cl
2 (10.0 ml) was added tert-butyl ( (1r, 3r) -3- (aminomethyl) cyclobutyl) carbamate (400 mg, crude) , HATU (1.04 g, 2.75 mmol) and DIPEA (940 μL, 708 mg, 5.49 mmol) at 25 ℃. The reaction mixture was stirred at that temperature for 2 h before it was quenched with ice-cold water (20 mL) and extracted with CH
2Cl
2 (20 mL × 3) . The combined organic phases were dried over anhydrous Na
2SO
4, filtered and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 15 %in 20 min) to afford tert-butyl ( (1r, 3r) -3- ( (6-bromo-5-fluoropicolinamido) methyl) cyclobutyl) carbamate (600 mg, 82%yield) as a yellow solid LC-MS: m/z [M + Na]
+ 424.0.
Step 5: tert-butyl ( (1S, 3r) -3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) -5-fluoropicolinamido) methyl) cyclobutyl) carbamate
To a stirred solution of tert-butyl ( (1r, 3r) -3- ( (6-bromo-5-fluoropicolinamido) methyl) cyclobutyl) carbamate (400 mg, 1.00 mmol) in 1, 4-dioxane (10.0 mL) were sequentially added (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (224 mg, 1.00 mmol) , Pd
2 (dba)
3 (100 mg, 100 μmol) , XantPhos (116 mg, 200 μmol) and Cs
2CO
3 (978 mg, 3.00 mmol) at 25 ℃. The reaction mixture was warmed to 100 ℃ and stirred at that temperature for 12 h. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 82 %in 25 min) to afford tert-butyl ( (1S, 3r) -3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -5-fluoropicolinamido) methyl) cyclobutyl) carbamate (430 mg, 79%yield) as a yellow solid. LC-MS: m/z [M+H] + 544.9
Step 6: (1R, 3S) -3- (5- ( (6- ( ( ( (1r, 3S) -3- ( (tert-butoxycarbonyl) amino) cyclobutyl) methyl) carbamoyl) -
3-fluoropyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-
carboxylate
To a stirred suspension of tert-butyl ( (1S, 3r) -3- ( (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) -5-fluoropicolinamido) methyl) cyclobutyl) carbamate (430 mg, 790 μmol) in CH
2Cl
2 (10.0 mL) were sequentially added CDI (384 mg, 2.37 mmol) and DIPEA (675 μL, 509 mg, 3.95 mmol) at 25 ℃. The reaction mixture was stirred at that temperature for 2 h before it was quenched with ice-cold water (20 mL) and extracted with CH
2Cl
2 (20 mL × 3) . The combined organic phases were dried over anhydrous Na
2SO
4, filtered and concentrated under reduced pressure. The crude product (1R, 3S) -3- (5- ( (6- ( ( ( (1r, 3S) -3- ( (tert-butoxycarbonyl) amino) cyclobutyl) methyl) carbamoyl) -3-fluoropyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate was used directly in the next step without further purification. LC-MS: m/z [M+H]
+ 639.3
Step 7: (1R, 3S) -3- (5- ( (6- ( ( ( (1r, 3S) -3-aminocyclobutyl) methyl) carbamoyl) -3-fluoropyridin-2-
yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a stirred solution of (1R, 3S) -3- (5- ( (6- ( ( ( (1r, 3S) -3- ( (tert-butoxycarbonyl) amino) cyclobutyl) methyl) carbamoyl) -3-fluoropyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate in CH
2Cl
2 (10.0 ml) was added TFA (396 μL, 586 mg, 5.14 mmol) at 25 ℃. The mixture was stirred at that temperature for 1 h before it was concentrated under reduced pressure to afford (1R, 3S) -3- (5- ( (6- ( ( ( (1r, 3S) -3-aminocyclobutyl) methyl) carbamoyl) -3-fluoropyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate as a brown oil, which was used in the next step without further purification. LC-MS: m/z [M+H]
+ 539.3
Step 8: (1
1S, 1
3R, 8
1R, 8
3S, Z) -2
1- (tert-butyl) -43-fluoro-2
1H-11-oxa-3, 6, 9-triaza-4 (2, 6) -pyridina-
2 (3, 5) -pyrazola-1 (1, 3) -cyclopentana-8 (1, 3) -cyclobutanacycloundecaphane-5, 10-dione
To a stirred solution of (1R, 3S) -3- (5- ( (6- ( ( ( (1r, 3S) -3-aminocyclobutyl) methyl) carbamoyl) -3-fluoropyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate in CH
3CN (5.0 mL) was added Et
3N (269 μL, 196 mg, 1.94 mmol) at 25 ℃. The reaction mixture was warmed to 80 ℃ and stirred at that temperature for 12 h. The reaction mixture was cooled to 25 ℃, filtered and concentrated under reduced pressure. Then it was concentrated to afford (1
1S, 1
3R, 8
1R, 8
3S, Z) -2
1- (tert-butyl) -43-fluoro-2
1H-11-oxa-3, 6, 9-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentana-8 (1, 3) -cyclobutanacycloundecaphane-5, 10-dione, which was used in the next step without further purification. LC-MS: m/z [M+H] + 471.3
Step 9: (1
1S, 1
3R, 8
1R, 8
3S, Z) -43-fluoro-2
1H-11-oxa-3, 6, 9-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-
1 (1, 3) -cyclopentana-8 (1, 3) -cyclobutanacycloundecaphane-5, 10-dione
A solution of (1
1S, 1
3R, 8
1R, 8
3S, Z) -2
1- (tert-butyl) -43-fluoro-2
1H-11-oxa-3, 6, 9-triaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-1 (1, 3) -cyclopentana-8 (1, 3) -cyclobutanacycloundecaphane-5, 10-dione in HCO
2H (5.0 mL) was warmed to 50 ℃ and stirred at that temperature for 12 h before it was cooled, concentrated under reduced pressure. The residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 5%to 65%in 20 min) to afford (1
1S, 1
3R, 8
1R, 8
3S, Z) -43-fluoro-2
1H-11-oxa-3, 6, 9-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentana-8 (1, 3) -cyclobutanacycloundecaphane-5, 10-dione (36.5 mg, 9%yield) as a white solid. LC-MS: m/z [M+H]
+ 415.2.
Example 167
(1
1S, 1
3R, 2
4Z, 5Z) -5- (trifluoromethyl) -2
1H-7, 11-dioxa-3, 6-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-
9 (3, 1) -azetidina-1 (1, 3) -cyclopentanacycloundecaphan-5-en-10-one

Step 1: tert-butyl 3- ( ( (1, 3-dioxoisoindolin-2-yl) oxy) methyl) azetidine-1-carboxylate
A solution of 2-hydroxyisoindoline-1, 3-dione (1.60 g, 9.81 mmol) , tert-butyl 3-(hydroxymethyl) azetidine-1-carboxylate (1.84 g, 9.81 mmol) and PPh
3 (3.09 g, 11.8 mmol) in THF (20.0 mL) was cooled down to 0 ℃ under N
2 before DIAD (2.3 mL, 2.38 g, 11.7 mmol) was added to the mixture dropwise. The mixture was stirred at 25 ℃ for 16 h. The mixture was diluted with EA (50 mL) and washed with water (20 mL × 3) . The organic layers were dried over Na
2SO
4 and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 10 to 50 %in 15 min) to afford tert-butyl 3- [ (1, 3-dioxoisoindolin-2-yl) oxymethyl] azetidine-1-carboxylate (3.00 g, 92%yield) as a white solid. LC-MS: m/z [M+H]
+277.0.
Step 2: tert-butyl 3- ( (aminooxy) methyl) azetidine-1-carboxylate
To a suspension of tert-butyl 3- [ (1, 3-dioxoisoindolin-2-yl) oxymethyl] azetidine-1-carboxylate (3.00 g, 9.18 mmol) in EtOH (50.0 mL) was added slowly hydrazinium hydroxide (2.2 mL, 2.30 g, 45.9 mmol) . The mixture was heated to 80 ℃ and stirred at that temperature for 3 h. The reaction mixture was filtered under reduced pressure and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with MeOH/CH
2Cl
2 (with MeOH from 0 to 10 %in 15 min) to afford tert-butyl 3- (aminooxymethyl) azetidine-1-carboxylate (1.50 g, 70%yield) as a colorless oil. LC-MS: m/z [M+H]
+147.1.
Step 3: tert-butyl (Z) -3- ( ( ( (1- (6-bromopyridin-2-yl) -2, 2, 2-
trifluoroethylidene) amino) oxy) methyl) azetidine-1-carboxylate
A suspension of 1- (6-bromo-2-pyridyl) -2, 2, 2-trifluoro-ethanone (350 mg, 1.38 mmol) , tert-butyl 3- (aminooxymethyl) azetidine-1-carboxylate (507 mg, 1.38 mmol) and CH
3COOK (406 mg, 4.13 mmol) in EtOH (10.0 mL) was heated to 80 ℃ and stirred at that temperature for 16 h. The reaction mixture was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 0 to 20 %in 15 min) to afford tert-butyl (Z) -3- ( ( ( (1- (6-bromopyridin-2-yl) -2, 2, 2-trifluoroethylidene) amino) oxy) methyl) azetidine-1-carboxylate (520 mg, 86%yield) as a colorless oil. LC-MS: m/z [M+Na]
+ 460.
Step 4: tert-butyl 3- ( ( ( ( (Z) -1- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) -2, 2, 2-trifluoroethylidene) amino) oxy) methyl) azetidine-1-carboxylate
To a mixture of tert-butyl (Z) -3- ( ( ( (1- (6-bromopyridin-2-yl) -2, 2, 2-trifluoroethylidene) amino) oxy) methyl) azetidine-1-carboxylate (520 mg, 1.19 mmol) and (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (159 mg, 710 μmol) in 1, 4-dioxane (5.0 mL) were sequentially added Pd
2 (dba)
3 (109 mg, 120 μmol) , XantPhos (137 mg, 240 μmol) and Cs
2CO
3 (1.16 g, 3.56 mmol) . The mixture was heated to 100 ℃ and stirred at that temperature under N
2 for 6 h. The reaction mixture was filtered and concentrated under reduced pressure. The residue was by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 30 to 60 %in 15 min) to afford tert-butyl 3- ( ( ( ( (Z) -1- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) -2, 2, 2-trifluoroethylidene) amino) oxy) methyl) azetidine-1-carboxylate (650 mg, 94%yield) as yellow solid. LC-MS: m/z [M+H]
+ 581.3.
Step 5: (1R, 3S) -3- (5- ( (6- ( (Z) -1- ( ( (1- (tert-butoxycarbonyl) azetidin-3-yl) methoxy) imino) -2, 2, 2-
trifluoroethyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-
carboxylate
To a mixture of tert-butyl 3- ( ( ( ( (Z) -1- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) -2, 2, 2-trifluoroethylidene) amino) oxy) methyl) azetidine-1-carboxylate (650 mg, 1.12 mmol) in CH
2Cl
2 (10.0 mL) were sequentially added CDI (363 mg, 2.24 mmol) and Et
3N (780 μL, 566 mg, 5.60 mmol) . The mixture was heated to 40 ℃ and stirred at that temperature for 16 h. The reaction mixture was washed with water (10 mL × 2) and dried over Na
2SO
4, concentrated under reduced pressure to give the crude product as pale yellow oil. LC-MS: m/z [M+H]
+ 675.3.
Step 6: (1R, 3S) -3- (5- ( (6- ( (Z) -1- ( (azetidin-3-ylmethoxy) imino) -2, 2, 2-trifluoroethyl) pyridin-2-
yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a suspension of crude (1R, 3S) -3- (5- ( (6- ( (Z) -1- ( ( (1- (tert-butoxycarbonyl) azetidin-3-yl) methoxy) imino) -2, 2, 2-trifluoroethyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate in CH
2Cl
2 (6.0 mL) was added slowly TFA (2.0 mL, 2.96 g, 25.9 mmol) at 25 ℃ and stirred for 30 min. The reaction mixture was concentrated under reduced pressure. The crude product was used for next step without further purification. LC-MS: m/z [M+H]
+575.3.
Step 7: (1
1S, 1
3R, 2
4Z, 5Z) -2
1- (tert-butyl) -5- (trifluoromethyl) -2
1H-7, 11-dioxa-3, 6-diaza-4 (2, 6) -
pyridina-2 (3, 5) -pyrazola-9 (3, 1) -azetidina-1 (1, 3) -cyclopentanacycloundecaphan-5-en-10-one
To a mixture of (1R, 3S) -3- (5- ( (6- ( (Z) -1- ( (azetidin-3-ylmethoxy) imino) -2, 2, 2-trifluoroethyl) pyridin-2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate in CH
3CN (100 mL) was added Et
3N (1.46 mL, 1.06 g, 10.4 mmol) at 25 ℃. The mixture was heated to 80 ℃ and stirred at that temperature for 16 h. The reaction mixture was concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with EtOAc/PE (with EtOAc from 40 to 90 %in 15 min) to afford (1
1S, 1
3R, 2
4Z, 5Z) -2
1- (tert-butyl) -5- (trifluoromethyl) -2
1H-7, 11-dioxa-3, 6-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-9 (3, 1) -azetidina-1 (1, 3) -cyclopentanacycloundecaphan-5-en-10-one (365 mg, 69%yield) as a yellow solid. LC-MS: m/z [M+H]
+ 507.2.
Step 8: (1
1S, 1
3R, 2
4Z, 5Z) -5- (trifluoromethyl) -2
1H-7, 11-dioxa-3, 6-diaza-4 (2, 6) -pyridina-2 (5, 3) -
pyrazola-9 (3, 1) -azetidina-1 (1, 3) -cyclopentanacycloundecaphan-5-en-10-one
A mixture of (1
1S, 1
3R, 2
4Z, 5Z) -2
1- (tert-butyl) -5- (trifluoromethyl) -2
1H-7, 11-dioxa-3, 6-diaza-4 (2, 6) -pyridina-2 (3, 5) -pyrazola-9 (3, 1) -azetidina-1 (1, 3) -cyclopentanacycloundecaphan-5-en-10-one (365 mg, 721 μmol) in HCO
2H (2.0 mL) was heated to 100 ℃ and stirred at that temperature for 16 h. The reaction mixture was concentrated under reduced pressure. The residue was purified by Prep-HPLC eluting with CH
3CN in water (with CH
3CN from 20%to 50%in 30 min) to afford (1
1S, 1
3R, 2
4Z, 5Z) -5- (trifluoromethyl) -2
1H-7, 11-dioxa-3, 6-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-9 (3, 1) -azetidina-1 (1, 3) -cyclopentanacycloundecaphan-5-en-10-one (132 mg, 40%yield) as a white solid. LC-MS: m/z [M+H]
+ 451.2.
The following compounds were prepared using the similar procedure disclosed in synthetic example 167.

Example 169
(11S, 13R, 24Z, 5E) -5-methyl-21H-7, 12-dioxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-5-en-11-one

Step 1: tert-butyl (E) - (2- ( ( (1- (6-bromopyridin-2-yl) ethylidene) amino) oxy) ethyl) carbamate
To a solution of 1- (6-bromo-2-pyridyl) ethanone (1.0 g, 5.0 mmol) and tert-butyl N- (2-chloroethyl) carbamate (1.0 g, 6.0 mmol) in DMSO (25 mL) and H
2O (10 mL) was added hydroxylamine hydrochloride (416 mg, 6.0 mmol) and KOH (2.4 g, 43.0 mmol) at 25℃, The reaction was stirred under nitrogen atmosphere at 80 ℃ for 6 h. The mixture was quenched with saturated NH
4Cl solution (100 mL) and extracted with EtOAc (50 mL × 2) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrated. The residue was purified by flash chromatography on silica gel eluting with 0 to 40%EtOAc in PE to afford tert-butyl N- [2- [ (E) -1- (6-bromo-2-pyridyl) ethylideneamino] oxyethyl] carbamate (1.3 g, 72%yield) as yellow oil. LC-MS: m/z 358.0 [M+H]
+.
Step 2: tert-butyl (2- ( ( ( (E) -1- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) ethylidene) amino) oxy) ethyl) carbamate
To a mixture of tert-butyl N- [2- [ (E) -1- (6-bromo-2-pyridyl) ethylideneamino] oxyethyl] carbamate (600 mg, 1.6 mmol) and (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (486 mg, 2.1 mmol) in 1, 4-dioxane (30 mL) was added Pd
2 (dba)
3 (306 mg, 335 umol) , XantPhos (387 mg, 670 μmol) and Cs
2CO
3 (1.36 g, 4.19 mmol) at 25 ℃ under nitrogen, The reaction was stirred at 90 ℃ for 6 h. The mixture was diluted with water (50 mL) and extracted with EtOAc (30 mL × 3) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrated. The residue was purified by flash chromatography on silica gel eluting with 0 to 40%EtOAc in PE to afford tert-butyl (2- ( ( ( (E) -1- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) ethylidene) amino) oxy) ethyl) carbamate (790 mg, 94%yield) as yellow oil. LC-MS: m/z 501.3 [M+H]
+.
Step 3: tert-butyl (2- ( ( ( (E) -1- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3- ( ( (4-
nitrophenoxy) carbonyl) oxy) cyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-
yl) ethylidene) amino) oxy) ethyl) carbamate
A mixture of tert-butyl N- [2- [ (E) -1- [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -2-pyridyl] ethylideneamino] oxyethyl] carbamate (100 mg, 200 μmol) , 4-Nitrophenyl chloroformate (121 mg, 599 μmol) , Et
3N (101 mg, 999 μmol) and DMAP (12 mg, 100 μmol) in DCE (5 mL) was stirred at 70 ℃ for 12 h. The mixture was diluted with water (50 mL) and extracted with DCM (30 mL × 3) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrated. The residue was purified by flash chromatography on silica gel eluting with 0 to 30%EtOAc in PE to afford [ (1R, 3S) -3- [5- [ [6- [ (E) -N- [2- (tert-butoxycarbonylamino) ethoxy] -C-methyl-carbonimidoyl] -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] (4-nitrophenyl) carbonate (60 mg, 45%yield) as yellow oil. LC-MS: m/z 666.3 [M+H]
+.
Step 4: (1R, 3S) -3- (3- ( (6- ( (E) -1- ( (2-aminoethoxy) imino) ethyl) pyridin-2-yl) amino) -1H-pyrazol-5-
yl) cyclopentyl (4-nitrophenyl) carbonate
A mixture of [ (1R, 3S) -3- [5- [ [6- [ (E) -N- [2- (tert-butoxycarbonylamino) ethoxy] -C-methyl-carbonimidoyl] -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] (4-nitrophenyl) carbonate (60 mg, 90 μmol) in TFA (1 mL) was stirred at 70 ℃ for 4 h. The solvent was removed under reduced pressure to afford (1R, 3S) -3- (3- ( (6- ( (E) -1- ( (2-aminoethoxy) imino) ethyl) pyridin-2-yl) amino) -1H-pyrazol-5-yl) cyclopentyl (4-nitrophenyl) carbonate 2, 2, 2-trifluoroacetic acid (45 mg, 100%yield) as yellow oil, which was used in the next step directly. LC-MS: m/z 510.1 [M+H]
+.
Step 5: (11S, 13R, 24Z, 5E) -5-methyl-21H-7, 12-dioxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -
pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-5-en-11-one
To a mixture of [ (1R, 3S) -3- [3- [ [6- [ (E) -N- (2-aminoethoxy) -C-methyl-carbonimidoyl] -2-pyridyl] amino] -1H-pyrazol-5-yl] cyclopentyl] (4-nitrophenyl) carbonate 2, 2, 2-trifluoroacetic acid (45 mg, 90 μmol) in CH
3CN (10 mL) was added Et
3N (91 mg, 900 μmol) at 25 ℃. The reaction was stirred at 25 ℃ for 1 h. The solvent was removed under reduced pressure. The residue was purified by prep-HPLC (C18, 22%to 32%CH
3CN in 0.1%FA/water, 20 mL/min) to afford (11S, 13R, 24Z, 5E) -5-methyl-21H-7, 12-dioxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-5-en-11-one (4.7 mg, 14%yield) as a white solid. LC-MS: m/z 371.1 [M+H]
+.
Example 170
(1
1S, 1
3R, 2
4Z, 5E) -2
1H-7, 12-dioxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-5-en-11-one

Step 1: (E) -6-bromopicolinaldehyde oxime
To a solution of 6-bromopicolinaldehyde (1.0 g, 5.4 mmol) in ethanol (20 mL) was added hydroxylamine hydrochloride (747 mg, 10.8 mmol) and Potassium Acetate (1.1 g, 10.8 mmol) at 25℃. The reaction was stirred under nitrogen atmosphere at 90 ℃ for 3 hours. The mixture was washed with water (50 mL) and extracted with EtOAc (50 mL ×2) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrated to afford (E) -6-bromopicolinaldehyde oxime (1.0 g, 93%yield) as white solid. LC-MS: m/z 201.0 [M+H]
+
Step 2: tert-butyl (E) - (2- ( ( ( (6-bromopyridin-2-yl) methylene) amino) oxy) ethyl) carbamate
To a solution of (E) -6-bromopicolinaldehyde oxime (500 mg, 2.5 mmol) and tert-butyl (2-bromoethyl) carbamate (669 mg, 3.0 mmol) in DMF (10 mL) was added Potassium carbonate (1.0 g, 7.5 mmol) and Sodium iodide (41 mg, 249 μmol) at 25℃. The reaction was stirred under nitrogen atmosphere at 60 ℃ for 3.5 hours. The mixture was diluted with water (50 mL) and extracted with EtOAc (50 mL ×2) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrated to dryness. The residue was purified by flash chromatography on silica gel eluting with 0 to 45%EtOAc in PE to afford tert-butyl (E) - (2- ( ( ( (6-bromopyridin-2-yl) methylene) amino) oxy) ethyl) carbamate (794 mg, 93%yield) as yellow oil. LC-MS: m/z 288.1 [M-56+H]
+
Step 3: tert-butyl (2- ( ( ( (E) - (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) methylene) amino) oxy) ethyl) carbamate
To a solution of tert-butyl (E) - (2- ( ( ( (6-bromopyridin-2-yl) methylene) amino) oxy) ethyl) carbamate (224 mg, 651 μmol) and (1R, 3S) -3- (5-amino-1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentan-1-ol (160 mg, 716 μmol) in dioxane (8 mL) was added Tris (dibenzylideneacetone) dipalladium (119 mg, 130 μmol) , (5-diphenylphosphanyl-9, 9-dimethyl-xanthen-4-yl) -diphenyl-phosphane (151 mg, 260 μmol) and Cesium carbonate (636 mg, 1.9 mmol) . The suspension was degassed with N2 for 5 times. The mixture was stirred at 90 ℃ for 6 hours. The mixture was diluted with water (50 mL) and extracted with EtOAc (50 mL ×2) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrated to dryness. The residue was purified by flash chromatography on silica gel eluting with EtOAc in PE from 0 to 80%to afford tert-butyl (2- ( ( ( (E) - (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methylene) amino) oxy) ethyl) carbamate (242 mg, 71%yield) as a yellow solid. LC-MS: m/z 487.1 [M+H]
+
Step 4: (1R, 3S) -3- (1- (tert-butyl) -5- ( (6- ( (E) -9, 9-dimethyl-7-oxo-3, 8-dioxa-2, 6-diazadec-1-en-1-
yl) pyridin-2-yl) amino) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of tert-butyl (2- ( ( ( (E) - (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-yl) amino) pyridin-2-yl) methylene) amino) oxy) ethyl) carbamate (210 mg, 432 μmol) in DCM (8 mL) was added carbonyl diimidazole (210 mg, 1.3 mmol) at 25 ℃. The reaction was stirred under nitrogen atmosphere at 40 ℃ for 2 hours. The mixture was diluted with water (50 mL) and extracted with DCM (50 mL ×2) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrated to dryness. The residue was purified by flash chromatography on silica gel eluting with EA in PE from 0 to 60%to afford (1R, 3S) -3- (1- (tert-butyl) -5- ( (6- ( (E) -9, 9-dimethyl-7-oxo-3, 8-dioxa-2, 6-diazadec-1-en-1-yl) pyridin-2-yl) amino) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (231 mg, 80%yield) as a yellow solid. LC-MS: m/z 581.3 [M+H]
+
Step 5: (1R, 3S) -3- (1- (tert-butyl) -5- ( (6- ( (E) -9, 9-dimethyl-7-oxo-3, 8-dioxa-2, 6-diazadec-1-en-1-
yl) pyridin-2-yl) amino) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
A solution of (1R, 3S) -3- (1- (tert-butyl) -5- ( (6- ( (E) -9, 9-dimethyl-7-oxo-3, 8-dioxa-2, 6-diazadec-1-en-1-yl) pyridin-2-yl) amino) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate (231 mg, 398 μmol) in TFA (10 mL) was stirred under nitrogen atmosphere at 75 ℃ for 3 hours. The mixture was concentrated to afford (1R, 3S) -3- (3- ( (6- ( (E) - ( (2- ( (2, 2, 2-trifluoroacetyl) -l4-azaneyl) ethoxy) imino) methyl) pyridin-2-yl) amino) -1H-pyrazol-5-yl) cyclopentyl 1H-imidazole-1-carboxylate (210 mg, crude) as a yellow oil, which was used in the next step without purification. LC-MS: m/z 425.1 [M+H]
+
Step 6: (1
1S, 1
3R, 2
4Z, 5E) -2
1H-7, 12-dioxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -
cyclopentanacyclododecaphan-5-en-11-one
To a solution of (1R, 3S) -3- (3- ( (6- ( (E) - ( (2- ( (2, 2, 2-trifluoroacetyl) -l4-azaneyl) ethoxy) imino) methyl) pyridin-2-yl) amino) -1H-pyrazol-5-yl) cyclopentyl 1H-imidazole-1-carboxylate (210 mg, 495 μmol) in ACN (10 mL) was added Triethylamine (501 mg, 5.0 mmol) at 25℃. The reaction was stirred under nitrogen atmosphere at 90 ℃ for 6 hours. The mixture was concentrated to dryness. The mixture was diluted with water (50 mL) and extracted with DCM (50 mL ×2) . The combined organic layer was washed with brine (50 mL) , dried over Na
2SO
4, filtered and concentrated to dryness. The residue was purified by flash chromatography on silica gel eluting with MeOH in DCM from 0 to 7%to afford the crude solid which was purified by Prep-TLC (6%MeOH in DCM) to give as a yellow solid. The residue was purified by prep-HPLC (C18, 30%to 40%CH
3CN in 0.05%NH
3/water, 20 mL/min) to afford (1
1S, 1
3R, 2
4Z, 5E) -2
1H-7, 12-dioxa-3, 6, 10-triaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-1 (1, 3) -cyclopentanacyclododecaphan-5-en-11-one (9.5 mg, 5%yield ) as a white solid. LC-MS: m/z 357.1 [M+H]
+.
The following compounds were prepared using the similar procedure disclosed in synthetic example 170.


Example 174
(1
1S, 1
3R, 2
4Z, 5E) -5-methyl-2
1H-7, 10-dioxa-3, 6-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-8 (3, 1) -
azetidina-1 (1, 3) -cyclopentanacyclodecaphan-5-en-9-one

Step 1: tert-butyl (E) -3- ( ( (1- (6-bromopyridin-2-yl) ethylidene) amino) oxy) azetidine-1-
carboxylate
To a solution of 1- (6-bromo-2-pyridyl) ethanone (1 g, 5.0 mmol) and tert-butyl 3-iodoazetidine-1-carboxylate (1.4 g, 5.0 mmol) in DMSO (14 mL) and Water (4 mL) were added potassium; hydroxide (2.4 g, 43.0 mmol, 1.2 mL) and hydroxylamine hydrochloride (521 mg, 7.5 mmol, 312 μL) . The mixture was stirred at 90 ℃ for 16 hours. LCMS showed the starting material was consumed and the desired product was detected. The mixture was quenched with water (50 ml) . The mixture was extracted with EtOAc (200 ml *2) . The organic layer was washed with brine (50 ml*2) , dried over Na
2SO
4. The combined organic layer was concentrated under reduced pressure. The residue was purified by flash column chromatography eluting with ethyl acetate in petroleum ether from 0 to 10%in 25 minutes to give tert-butyl 3- [ (E) -1- (6-bromo-2-pyridyl) ethylideneamino] oxyazetidine-1-carboxylate (420 mg, 1.2 mmol, 23%yield, as white solid) . LC-MS: m/z 392.0, 394.0 [M+ Na]
+.
Step 2: tert-butyl 3- ( ( ( (E) -1- (6- ( (1- (tert-butyl) -3- ( (1S, 3R) -3-hydroxycyclopentyl) -1H-pyrazol-5-
yl) amino) pyridin-2-yl) ethylidene) amino) oxy) azetidine-1-carboxylate
To a solution of tert-butyl 3- [ (E) -1- (6-bromo-2-pyridyl) ethylideneamino] oxyazetidine-1-carboxylate (258 mg, 698 μmol) and (1R, 3S) -3- (5-amino-1-tert-butyl-pyrazol-3-yl) cyclopentanol (130 mg, 582 μmol) in Dioxane (3 mL) were added dicesium; carbonate (569 mg, 1.70 mmol) , (1E, 4E) -1, 5-diphenylpenta-1, 4-dien-3-one; palladium (53.3 mg, 58.2 μmol) and XantPhos (67.3 mg, 116 μmol) . The mixture was degassed with N
2 and stirred at 110 ℃ for 6 hours. LCMS showed the starting material was consumed and the desired product was detected. The mixture was filtered through a Celite pad, and the filtrate was concentrated under reduced pressure. The residue was purified by flash column chromatography eluting with ethyl acetate in petroleum ether from 0 to 50%in 25 minutes to give tert-butyl N- [2- [ (E) -1- [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -2-pyridyl] ethylideneamino] oxyethyl] carbamate (248 mg, 495 μmol, 86%yield) as yellow solid. LC-MS: m/z 513.3 [M+H]
+.
Step 3: (1R, 3S) -3- (5- ( (6- ( (E) -1- ( ( (1- (tert-butoxycarbonyl) azetidin-3-yl) oxy) imino) ethyl) pyridin-
2-yl) amino) -1- (tert-butyl) -1H-pyrazol-3-yl) cyclopentyl 1H-imidazole-1-carboxylate
To a solution of tert-butyl 3- [ (E) -1- [6- [ [2-tert-butyl-5- [ (1S, 3R) -3-hydroxycyclopentyl] pyrazol-3-yl] amino] -2-pyridyl] ethylideneamino] oxyazetidine-1-carboxylate (50.1 mg, 97.5 μmol) and N, N-diethylethanamine (49.3 mg, 487 μmol, 67.9 μL) in DCM (4 mL) was added di (imidazol-1-yl) methanone (47.4 mg, 292 μmol) . The resulting mixture was stirred at 35 ℃ for 1 hour. LCMS showed the starting material was consumed and the desired product was detected. The mixture was concentrated under reduced pressure. The mixture was quenched with water (20 mL) . The mixture was extracted with DCM (20 ml *2) . The organic layer was washed with brine (10 mL) , dried over Na
2SO
4. The combined organic layer was concentrated under reduced pressure to give [ (1R, 3S) -3- [5- [ [6- [ (E) -N- (1-tert-butoxycarbonylazetidin-3-yl) oxy-C-methyl-carbonimidoyl] -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (240 mg, crude) as yellow oil. LC-MS: m/z 607.3 [M+H]
+.
Step 4: (1R, 3S) -3- (3- ( (6- ( (E) -1- ( (azetidin-3-yloxy) imino) ethyl) pyridin-2-yl) amino) -1H-pyrazol-
5-yl) cyclopentyl 1H-imidazole-1-carboxylate
A solution of [ (1R, 3S) -3- [5- [ [6- [ (E) -N- (1-tert-butoxycarbonylazetidin-3-yl) oxy-C-methyl-carbonimidoyl] -2-pyridyl] amino] -1-tert-butyl-pyrazol-3-yl] cyclopentyl] imidazole-1-carboxylate (240 mg, 95.6 μmol) in TFA (2 mL) was stirred at 70 ℃ for 1 h. LCMS showed the starting material was consumed and the desired product was detected. The mixture was concentrated under reduced pressure to give [ (1R, 3S) -3- [3- [ [6- [ (E) -N- (azetidin-3-yloxy) -C-methyl-carbonimidoyl] -2-pyridyl] amino] -1H-pyrazol-5-yl] cyclopentyl] imidazole-1-carboxylate (200 mg, crude) as yellow oil. LC-MS: m/z 451.2 [M+H]
+.
Step 5: (1
1S, 1
3R, 2
4Z, 5E) -5-methyl-2
1H-7, 10-dioxa-3, 6-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-
8 (3, 1) -azetidina-1 (1, 3) -cyclopentanacyclodecaphan-5-en-9-one
To a solution of (1R, 3S) -3- (3- ( (6- ( (E) -1- ( (azetidin-3-yloxy) imino) ethyl) pyridin-2-yl) amino) -1H-pyrazol-5-yl) cyclopentyl 1H-imidazole-1-carboxylate (200 mg, 443 μmol) in ACN (20 mL) was added Et
3N (134 mg, 1.3 mmol, 185 μL) . The mixture was stirred at 80 ℃ for 16 hours. LCMS showed the starting material was consumed and the desired product was detected. The mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography eluting with MeOH in DCM from 0 to 6%in 25 minutes to give crude product (20 mg, as yellow oil) , which was further purified by Prep-HPLC eluting with acetonitrile in water (35~45%) containing 0.1%NH
3 to give (1
1S, 1
3R, 2
4Z, 5E) -5-methyl-2
1H-7, 10-dioxa-3, 6-diaza-4 (2, 6) -pyridina-2 (5, 3) -pyrazola-8 (3, 1) -azetidina-1 (1, 3) -cyclopentanacyclodecaphan-5-en-9-one (6.8 mg, 5%yield) as white solid. LC-MS: m/z 383.1 [M+H]
+.
Biological Example 1. Assay for Inhibition of CDK2/Cyclin E1
The homogeneous time-resolved fluorescence (HTRF) assay was performed to detect CDK2/Cyclin E1 catalyzed phosphorylation of peptide substrate in assay buffer containing 50 mM HEPES, PH=7.5, 10 mM MgCl2, 1mM EGTA, 2 mM DTT, 0.01%Tween, 0.1%BSA. The enzymatic reaction was carried out in a 10 μL volume containing 2 nM CDK2/Cyclin E1 enzyme (Merk, 14-475) , 80 μM ATP, 20 nM LANCE Ultra ULight
TM-eIF4E-binding protein 1 (Thr37/46) Peptide (PerkinElmer, TRF0128-M) and 1 %DMSO (or the test compound at appropriate dilutions in DMSO) in the assay buffer. All the components were added to the 384-well plate (PerkinElmer, 6008280) , and incubated at Room Temperature for 4 hours. The reaction was terminated by addition of 10 μL detection buffer (PerkinElmer, CR97-100) containing 20 mM EDTA and 4 nM
 Ultra Europium-anti-phospho-eIF4E-bindingprotein 1 (Thr37/46) (PerkinElmer, TRF0216-M) antibody. Then incubate the plate at Room Temperature for 1 hour. Plate was read using Envision Reader (PerkinElmer, EnVision Multilabel Reader) . The IC
50 values of the test compound were determined by fitting the inhibition curves by 4 parameter sigmoidal dose-response model using the GraphPad Prism 8 software.
Ultra Europium-anti-phospho-eIF4E-bindingprotein 1 (Thr37/46) (PerkinElmer, TRF0216-M) antibody. Then incubate the plate at Room Temperature for 1 hour. Plate was read using Envision Reader (PerkinElmer, EnVision Multilabel Reader) . The IC
50 values of the test compound were determined by fitting the inhibition curves by 4 parameter sigmoidal dose-response model using the GraphPad Prism 8 software.
Biological Example 2. Assay for Inhibition of CDK1/Cyclin A2
The homogeneous time-resolved fluorescence (HTRF) assay was performed to detect CDK1/Cyclin A2 catalyzed phosphorylation of peptide substrate in assay buffer containing 50 mM HEPES, PH=7.5, 10 mM MgCl2, 1mM EGTA, 2 mM DTT, 0.01%Tween, 0.1%BSA. The enzymatic reaction was carried out in a 10 μL volume containing 0.4 nM CDK1/Cyclin A2 enzyme (SignalChem, C22-18G) , 10 μM ATP, 20 nM LANCE Ultra ULight
TM-eIF4E-binding protein 1 (Thr37/46) Peptide (PerkinElmer, TRF0128-M) ) and 1 %DMSO (or the test compound at appropriate dilutions in DMSO) in the assay buffer. All the components were added to the 384-well plate (PerkinElmer, 6008280) , and incubated at Room Temperature for 2 hours. The reaction was terminated by addition of 10 μL detection buffer (PerkinElmer, CR97-100) containing 20 mM EDTA and 4 nM
 Ultra Europium-anti-phospho-eIF4E-bindingprotein 1 (Thr37/46) (PerkinElmer, TRF0216-M) antibody. Then incubate the plate at Room Temperature for 1 hour. Plate was read using Envision Reader (PerkinElmer, EnVision Multilabel Reader) . The IC
50 values of the test compound were determined by fitting the inhibition curves by 4 parameter sigmoidal dose-response model using the GraphPad Prism 8 software.
Ultra Europium-anti-phospho-eIF4E-bindingprotein 1 (Thr37/46) (PerkinElmer, TRF0216-M) antibody. Then incubate the plate at Room Temperature for 1 hour. Plate was read using Envision Reader (PerkinElmer, EnVision Multilabel Reader) . The IC
50 values of the test compound were determined by fitting the inhibition curves by 4 parameter sigmoidal dose-response model using the GraphPad Prism 8 software.
The IC
50 values of each exemplified compound against CDK2 and the ratio of CDK1/CDK2 are provided in the the Table 2. The IC
50 values are indicated as "+" , for values less than or equal to 10 nM; "++" , for values less than or equal to 100 nM; "+++" , for values less than or equal to 1 μM; and "++++" , for values greater than 1 μM, respectively. the ratio of CDK1/CDK2 are indicated as “+” , for values less than or equal to 5 folds; “++” , for values less than or equal to 10 folds; “+++” , for values less than or equal to 20 folds; “++++” , for values greater than 20 folds.
Biological Example 3. Anti-proliferation Assay in OVCAR3 Cell
OVCAR3 cells (ATCC, HTB-161) were plated at 5000 cells/well in 96-well plates respectively, and were incubated in RPMI 1640 medium (Gibco, 31800105) with 10%FBS at 37℃, 5%CO
2. After overnight incubation, baseline values were measured of the samples from one plate using CyQUANT reagent (Invitrogen, C35011) following manufacturer’s recommendations. Cells were incubated with the detection reagent for 1 hour at 37℃, and then the fluorescence was measured with excitation at 485 nm and emission at 535 nm using Envision Multilabel Plate Reader (PerkinElmer) . Other plates were dosed with tested compounds in a 3-fold dilution scheme. On day 6 after compound addition, CyQUANT reagent was added and the fluorescence was measured using Envision. The IC
50 values of the test compound’s anti-proliferation activity was determined from the baseline subtracted viability readout curve using GraphPad Prism 5 software.
The cellular data obtained from biological examples 3 are listed in the Table A below. The IC
50 values are indicated as "+" , for values less than or equal to 100 nM; "++" , for values less than or equal to 500 nM; "+++" , for values less than or equal to 1 μM; and "++++" , for values greater than 1 μM, respectively.
Table 2
| Synthetic Example | CDK2 (IC 50) | CDK1/CDK2 (folds) | OVCAR3 (IC 50) |
| 1 | + | +++ | + |
| 2 | + | ++ | + |
| 3 | + | + | ++ |
| 4 | + | ++++ | ++ |
| 5 | + | ++ | + |
| 6 | ++ | + | ++++ |
| 7 | + | +++ | ++ |
| 8 | + | ++ | + |
| 9 | + | +++ | + |
| 10 | + | +++ | + |
| 11 | + | ++ | N/A |
| 12 | ++ | ++ | ++++ |
| 13 | + | ++++ | ++++ |
| 14 | + | + | + |
| 15 | + | +++ | + |
| 16 | + | +++ | + |
| 17 | + | ++ | + |
| 18 | +++ | ++ | ++++ |
| 19 | + | ++ | + |
| 20 | + | +++ | + |
| 21 | ++ | ++ | ++ |
| 22 | + | + | + |
| 23 | + | + | + |
| 24 | + | ++++ | ++ |
| 25 | + | + | + |
| 26 | + | + | + |
| 27 | + | + | + |
| 28 | + | ++ | + |
| 29 | + | + | + |
| 30 | + | +++ | + |
| 31 | + | ++++ | ++ |
| 32 | + | ++++ | + |
| 33 | + | ++++ | + |
| 34 | + | ++++ | ++ |
| 35 | + | +++ | + |
| 36 | + | +++ | ++ |
| 37 | + | ++ | + |
| 38 | + | +++ | ++ |
| 39 | + | ++ | + |
| 40 | + | ++ | ++ |
| 41 | + | ++ | + |
| 42 | + | ++ | + |
| 43 | + | ++ | + |
| 44 | + | ++ | + |
| 45 | + | ++ | + |
| 46 | + | ++++ | + |
| 47 | + | ++++ | + |
| 48 | + | ++++ | + |
| 49 | + | ++++ | + |
| 50 | + | ++++ | + |
| 51 | + | ++++ | + |
| 52 | + | ++++ | ++ |
| 53 | + | ++++ | ++ |
| 54 | + | ++++ | ++ |
| 55 | + | ++++ | ++ |
| 56 | + | ++++ | + |
| 57 | + | ++++ | ++ |
| 58 | + | ++++ | + |
| 59 | + | +++ | + |
| 60 | + | ++++ | ++ |
| 61 | + | +++ | ++ |
| 62 | + | ++ | ++ |
| 63 | + | ++++ | + |
| 64 | + | ++++ | + |
| 65 | + | ++++ | + |
| 66 | + | ++++ | ++ |
| 67 | + | ++++ | + |
| 68 | + | ++++ | + |
| 69 | + | ++++ | + |
| 70 | + | ++ | + |
| 71 | + | ++++ | + |
| 72 | + | ++++ | ++ |
| 73 | + | ++++ | + |
| 74 | + | ++++ | + |
| 75 | ++ | ++++ | ++++ |
| 76 | + | ++++ | + |
| 77 | ++ | +++ | ++++ |
| 78 | + | +++ | + |
| 79 | + | +++ | + |
| 80 | + | +++ | + |
| 81 | + | +++ | + |
| 82 | + | +++ | + |
| 83 | + | ++ | + |
| 84 | + | ++ | + |
| 85 | + | ++ | + |
| 86 | + | ++ | + |
| 87 | + | ++ | + |
| 88 | + | +++ | + |
| 89 | + | +++ | + |
| 90 | + | ++++ | + |
| 91 | + | ++ | + |
| 92 | ++ | ++++ | ++++ |
| 93 | + | ++++ | ++ |
| 94 | + | ++++ | + |
| 95 | + | +++ | + |
| 96 | + | ++++ | + |
| 97 | + | ++++ | + |
| 98 | + | ++++ | + |
| 99 | + | ++++ | + |
| 100 | ++ | +++ | ++++ |
| 101 | ++ | ++++ | ++++ |
| 102 | ++ | +++ | ++++ |
| 103 | + | ++++ | ++ |
| 104 | + | ++++ | + |
| 105 | + | ++++ | + |
| 106 | + | +++ | + |
| 107 | + | ++++ | +++ |
| 108 | + | ++++ | + |
| 109 | + | ++++ | + |
| 110 | ++ | ++++ | ++++ |
| 111 | + | ++++ | ++++ |
| 112 | + | ++++ | ++ |
| 113 | + | ++++ | + |
| 114 | + | +++ | + |
| 115 | + | ++ | ++ |
| 116 | + | ++ | + |
| 117 | + | ++ | + |
| 118 | + | + | + |
| 119 | + | ++ | + |
| 120 | + | + | + |
| 121 | + | +++ | + |
| 122 | + | ++++ | ++ |
| 123 | + | ++ | + |
| 124 | + | ++++ | ++ |
| 125 | + | ++++ | + |
| 126 | + | ++++ | + |
| 127 | + | ++++ | + |
| 128 | + | ++++ | ++ |
| 129 | + | ++++ | ++ |
| 130 | + | ++++ | ++++ |
| 131 | + | ++++ | + |
| 132 | + | ++++ | ++ |
| 133 | + | ++++ | ++ |
| 134 | + | ++++ | + |
| 135 | + | ++++ | + |
| 136 | + | ++++ | + |
| 137 | + | ++++ | ++ |
| 138 | + | ++++ | ++ |
| 139 | + | ++++ | ++ |
| 140 | + | ++++ | ++++ |
| 141 | + | ++++ | ++++ |
| 142 | + | ++++ | ++ |
| 143 | ++ | +++ | ++++ |
| 144 | + | ++++ | +++ |
| 145 | ++ | ++++ | ++++ |
| 146 | ++ | ++++ | ++ |
| 147 | + | ++++ | ++++ |
| 148 | + | ++++ | ++++ |
| 149 | ++ | ++++ | ++++ |
| 150 | ++ | ++++ | ++++ |
| 151 | + | ++++ | ++++ |
| 152 | ++ | ++++ | ++++ |
| 153 | ++ | ++++ | ++++ |
| 154 | + | ++++ | ++++ |
| 155 | + | ++++ | +++ |
| 156 | + | ++++ | + |
| 157 | + | ++++ | +++ |
| 158 | + | ++++ | ++++ |
| 159 | + | ++++ | ++ |
| 160 | + | ++++ | + |
| 161 | + | ++++ | ++++ |
| 162 | + | ++++ | + |
| 163 | + | ++++ | + |
| 164 | + | ++++ | + |
| 165 | + | ++++ | + |
| 166 | + | ++++ | ++ |
| 167 | + | + | + |
| 168 | + | ++++ | ++ |
| 169 | + | ++++ | + |
| 170 | + | ++++ | + |
| 171 | + | +++ | ++ |
| 172 | + | ++++ | ++ |
| 173 | + | ++++ | ++ |
| 174 | + | ++++ | ++ |
| 175 | ++++ | N/A | N/A |
| 176 | ++ | ++ | ++++ |
| 177 | + | + | + |
| 178 | + | ++++ | + |
| 179 | + | ++++ | + |
| 180+181 | + | ++ | + |
| 182 | + | ++++ | + |
| 183 | + | ++++ | ++ |
| 184 | + | ++++ | + |
| 185 | ++++ | N/A | N/A |
| 186 | ++++ | N/A | N/A |
| 187 | + | ++++ | + |
| 188 | + | ++++ | + |
| 189 | + | ++++ | ++ |
| 190 | + | ++++ | ++ |
| 191 | + | +++ | + |
| 192 | + | ++ | + |
| 193 | + | ++ | + |
| 194 | ++ | +++ | N/A |
| 195 | + | +++ | +++ |
| 196 | + | +++ | + |
| 197 | + | ++++ | +++ |
| 198 | + | ++++ | ++ |
| 199 | + | ++++ | +++ |
| 200 | + | ++++ | +++ |
| 201 | + | ++++ | + |
| 202 | + | + | + |
| 203 | ++ | ++ | ++ |
N/A: not available.
Claims (29)
- A compound of Formula (I') :
 a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein:ring A is 6-10 membered aryl or 5-10 memberaed heteroaryl; wherein said 6-10 membered aryl or 5-10 membered heteroaryl represented by ring A is optionally substituted by one or more R 1; whereinR 1 is halogen, -CN, C 1-6alkyl, C 1-6haloalkyl, C 2-6alkenyl, C 2-6alknyl, C 1-6alkyleneamine, C 1-6alkylenehydroxyl, -C (O) R 1a, -C (O) OR 1a, -C (O) NR 1aR 1b, -OR 1a, -SR 1a, -NR 1aR 1b, -NR 1aC (O) R 1b, -NR 1aC (O) OR 1b, -NR 1aSO 2R 1b, -NR 1aSO 2NR 1bR 1c, -SO 2R 1a, -SO 2NR 1aR 1b, or -P (O) R 1aR 1b, 3-6 membered carbocyclyl, 4-8 membered heterocyclyl, 6-10 membered aryl, 5-10 membered heteroaryl; wherein said C 1-6alkyleneamine, C 1-6alkylenehydroxyl, 3-6 membered carbocyclyl, 4-8 membered heterocyclyl, 6-10 membered aryl, or 5-10 membered heteroaryl represented by R 1 is optionally substituted by one or more R 10, whereinR 10, in each occurrence, is independently selected from the group consisting of halogen, -CN, C 1-6alkyl, C 1-6haloalkyl, C 1-6alkoxyl, and 4-6 membered heterocyclyl;R 1a, R 1b, and R 1c are independently selected from the group consisting of hydrogen, C 1-6alkyl, C 1-6haloalkyl, C 2-6alkenyl, C 2-6alkynyl, 3-6 membered carbocyclyl, and -C 0-6alkyl-4-6 membered heterocyclyl;X is absent, ^- (CH 2) 0-1-O-^^, -NR 2-, ^- (CH 2) 0-1-C (O) -^^, ^- (CH 2) 0-1-NR 2C (O) -^^, ^- (CH 2) 0-1-C (O) NR 2-^^, ^-SO 2-^^, or ^-C (R 2) =N-O-^^; wherein^-represents the point which attaches to ring A; -^^ represents the point which attaches toand
a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein:ring A is 6-10 membered aryl or 5-10 memberaed heteroaryl; wherein said 6-10 membered aryl or 5-10 membered heteroaryl represented by ring A is optionally substituted by one or more R 1; whereinR 1 is halogen, -CN, C 1-6alkyl, C 1-6haloalkyl, C 2-6alkenyl, C 2-6alknyl, C 1-6alkyleneamine, C 1-6alkylenehydroxyl, -C (O) R 1a, -C (O) OR 1a, -C (O) NR 1aR 1b, -OR 1a, -SR 1a, -NR 1aR 1b, -NR 1aC (O) R 1b, -NR 1aC (O) OR 1b, -NR 1aSO 2R 1b, -NR 1aSO 2NR 1bR 1c, -SO 2R 1a, -SO 2NR 1aR 1b, or -P (O) R 1aR 1b, 3-6 membered carbocyclyl, 4-8 membered heterocyclyl, 6-10 membered aryl, 5-10 membered heteroaryl; wherein said C 1-6alkyleneamine, C 1-6alkylenehydroxyl, 3-6 membered carbocyclyl, 4-8 membered heterocyclyl, 6-10 membered aryl, or 5-10 membered heteroaryl represented by R 1 is optionally substituted by one or more R 10, whereinR 10, in each occurrence, is independently selected from the group consisting of halogen, -CN, C 1-6alkyl, C 1-6haloalkyl, C 1-6alkoxyl, and 4-6 membered heterocyclyl;R 1a, R 1b, and R 1c are independently selected from the group consisting of hydrogen, C 1-6alkyl, C 1-6haloalkyl, C 2-6alkenyl, C 2-6alkynyl, 3-6 membered carbocyclyl, and -C 0-6alkyl-4-6 membered heterocyclyl;X is absent, ^- (CH 2) 0-1-O-^^, -NR 2-, ^- (CH 2) 0-1-C (O) -^^, ^- (CH 2) 0-1-NR 2C (O) -^^, ^- (CH 2) 0-1-C (O) NR 2-^^, ^-SO 2-^^, or ^-C (R 2) =N-O-^^; wherein^-represents the point which attaches to ring A; -^^ represents the point which attaches toand R 2, in each occurrence, is independently hydrogen, C 1-6alkyl, or C 1-6haloalkyl;is represented by formula
R 2, in each occurrence, is independently hydrogen, C 1-6alkyl, or C 1-6haloalkyl;is represented by formula *-W-L-Z-**;wherein *-represents the point which attaches to X; -**represents the point which attaches to
*-W-L-Z-**;wherein *-represents the point which attaches to X; -**represents the point which attaches to W is absent, C 1-6alkylene, C 2-6alkenylene, or C 2-6alkynylene; wherein said C 1- 6alkylene, C 2-6alkenylene, or C 2-6alkynylene represented by W is optionally substituted by one or more R 3;L is absent, -O-, -NH-, 3-12 membered carbocyclyl, 3-12 membered heterocyclyl, 6-10 membered aryl, or 5-10 membered heteroaryl; wherein said -NH-, 3-12 membered carbocyclyl,3-12 membered heterocyclyl, 6-10 membered aryl, or 5-10 membered heteroaryl represented by L is optionally substituted by one or more R 3; andZ is absent, C 1-6alkylene, C 2-6alkenylene, or C 2-6alkynylene; wherein said C 1- 6alkylene, C 2-6alkenylene, or C 2-6alkynylene represented by Z is optionally substituted by one or more R 3; whereinR 3, in each occurrence, is independently halogen, CN, C 1-6alkyl, C 1-6haloalkyl, -OR 3a, -C (=O) R 3a, or -NR 3aR 3b; whereinR 3a and R 3b are independently selected from the group consisting of hydrogen, C 1-6alkyl, C 1-6haloalkyl, C 2-6alkenyl, C 2-6alkynyl, 3-6 membered carbocyclyl, and 4-6 membered heterocyclyl;wherein W, L, and Z are not absent simultaneously;is represented by
W is absent, C 1-6alkylene, C 2-6alkenylene, or C 2-6alkynylene; wherein said C 1- 6alkylene, C 2-6alkenylene, or C 2-6alkynylene represented by W is optionally substituted by one or more R 3;L is absent, -O-, -NH-, 3-12 membered carbocyclyl, 3-12 membered heterocyclyl, 6-10 membered aryl, or 5-10 membered heteroaryl; wherein said -NH-, 3-12 membered carbocyclyl,3-12 membered heterocyclyl, 6-10 membered aryl, or 5-10 membered heteroaryl represented by L is optionally substituted by one or more R 3; andZ is absent, C 1-6alkylene, C 2-6alkenylene, or C 2-6alkynylene; wherein said C 1- 6alkylene, C 2-6alkenylene, or C 2-6alkynylene represented by Z is optionally substituted by one or more R 3; whereinR 3, in each occurrence, is independently halogen, CN, C 1-6alkyl, C 1-6haloalkyl, -OR 3a, -C (=O) R 3a, or -NR 3aR 3b; whereinR 3a and R 3b are independently selected from the group consisting of hydrogen, C 1-6alkyl, C 1-6haloalkyl, C 2-6alkenyl, C 2-6alkynyl, 3-6 membered carbocyclyl, and 4-6 membered heterocyclyl;wherein W, L, and Z are not absent simultaneously;is represented by
 wherein @-represents the point which attaches to the cyclopentyl shown in Formula (I’) ; -@@represents the point which attaches to
wherein @-represents the point which attaches to the cyclopentyl shown in Formula (I’) ; -@@represents the point which attaches to when U is connected withY is –O-or -NR 22-; and U is –O-or -NR 4-; wherein
when U is connected withY is –O-or -NR 22-; and U is –O-or -NR 4-; wherein R 22 is hydrogen, C 1-6alkyl, or C 1-6haloalkyl; andR 4 is hydrogen or C 1-6alkyl; orR 4 and one R 3 attached to Z together with the atoms to which they are attached form 4-8 membered heterocyclyl;when Y is connected withY is N; and U is –N (R 5) 2-; each R 5 is independently hydrogen or C 1-6alkyl; and
R 22 is hydrogen, C 1-6alkyl, or C 1-6haloalkyl; andR 4 is hydrogen or C 1-6alkyl; orR 4 and one R 3 attached to Z together with the atoms to which they are attached form 4-8 membered heterocyclyl;when Y is connected withY is N; and U is –N (R 5) 2-; each R 5 is independently hydrogen or C 1-6alkyl; and V is O or S.
V is O or S. - The compound of claim 1, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein the compound is represented by Formula II':

- The compound of claim 1 or 2, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein the compound is represented by Formula (II'A) or (II'B) :


- The compound of any one of claims 1-3, a pharmaceutically acceptable salt, or a stereoisomer thereof, whereinring A is phenyl or nitrogen containing 5-10 membered heteroaryl; wherein said phenyl or nitrogen containing 5-10 membered heteroaryl represented by ring A is optionally substituted by one to three R 1; whereinR 1 is halogen, -CN, C 1-4alkyl, C 1-4haloalkyl, C 1-4alkyleneamine, C 1-4alkylenehydroxyl, -C (O) NR 1aR 1b, -OR 1a, -SR 1a, -SO 2R 1a, 4-6 membered carbocyclyl, 4-6 membered heterocyclyl, phenyl, or 5-6 membered heteroaryl; wherein said C 1-4alkyleneamine, C 1-4alkylenehydroxyl, 4-6 membered carbocyclyl, 4-6 membered heterocyclyl, phenyl, or 5-6 membered heteroaryl represented by R 1 is optionally substituted by one to three R 10; whereinR 10, in each occurrence, is independently selected from the group consisting of halogen, -CN, C 1-4alkyl, C 1-4haloalkyl, C 1-4alkoxyl, and 5-6 membered heterocyclyl; andR 1a and R 1b are independently selected from the group consisting of hydrogen, C 1-4alkyl, C 1-4haloalkyl, C 2-4alkenyl, C 2-4alkynyl, and -C 0-3alkyl-4-6 membered heterocyclyl.
- The compound of any one of claims 1-4, a pharmaceutically acceptable salt, or a stereoisomer thereof, whereinring A is phenyl, nitrogen containing 5-6 membered heteroaryl, or nitrogen containing 9 membered bicyclic heteroaryl; wherein said phenyl, nitrogen containing 5-6 membered heteroaryl, or nitrogen containing 9 membered bicyclic heteroaryl represented by ring A is optionally substituted by one to two R 1; whereinR 1 is halogen, -CN, C 1-3alkyl, C 1-2haloalkyl, C 1-2alkyleneamine, C 1-2alkylenehydroxyl, -C (O) NR 1aR 1b, -OR 1a, -SR 1a, -SO 2R 1a, 5-6 membered carbocyclyl, 5-6 membered heterocyclyl, phenyl, or 5-6 membered heteroaryl; wherein said C 1-2alkyleneamine, C 1-2alkylenehydroxyl, 5-6 membered carbocyclyl, 5-6 membered heterocyclyl, phenyl, or 5-6 membered heteroaryl represented by R 1 is optionally substituted by one to three R 10; whereinR 10, in each occurrence, is independently selected from the group consisting of F, Cl, -CN, C 1-2alkyl, C 1-2haloalkyl, C 1-3alkoxyl, and 5-6 membered heterocyclyl; andR 1a and R 1b are independently selected from the group consisting of hydrogen, C 1-2alkyl, C 1-2haloalkyl, and -C 0-2alkyl-5 membered heterocyclyl.
- The compound of any one of claims 1-5, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein ring A is selected from a group consisting of phenyl, pyridyl, pyrimidinyl, pyrazinyl, thiophenyl, thiazolyl, pyrrolopyridinyl, pyrazolopyrimidinyl, pyrazolopyrazinyl, thienopyridinyl, and imidazo [4, 5-c] pyridinyl; wherein said phenyl, pyridyl, pyrimidinyl, pyrazinyl, thiophenyl, thiazolyl, pyrrolopyridinyl, pyrazolopyrimidinyl, pyrazolopyrazinyl, thienopyridinyl, and imidazo [4, 5-c] pyridinyl is optionally substituted by one to two R 1; whereinR 1 is F, Cl, Br, -CN, -CH 3, -CH (CH 3) 2, -CF 3, -OR 1a, -SR 1a, -SO 2R 1a, -C (O) NR 1aR 1b, -CH 2NH 2, -CH 2OH, cyclopentanyl, cyclohexanyl, cyclohexenyl, piperidinyl, tetrahydropyridinyl, 3, 6-dihydro-2H-thiopyranyl, thiophenyl, pyrazolyl, phenyl, or pyridyl; wherein said -CH 2NH 2, -CH 2OH, cyclopentanyl, cyclohexanyl, cyclohexenyl, piperidinyl, tetrahydropyridinyl, 3, 6-dihydro-2H-thiopyranyl, thiophenyl, pyrazolyl, phenyl, or pyridyl is optionally substituted by one to three R 10; whereinR 10, in each occurrence, is independently selected from the group consisting of F, Cl, -CN, -CH 3, -CF 3, -CH 2CF 3, -OCH 2CH 3, -OCH (CH 3) 2, and morpholinyl; andR 1a and R 1b are independently selected from the group consisting of hydrogen, -CH 3, -OCHF 2, tetrahydrofuranyl, and - (CH 2) 2-pyrrolidinyl.
- The compound of any one of claims 1-6, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein ring A is selected from the group consisting of

 whereineach of them is optionally substituted by one to two R 1; andrepresents the point which attaches to the moiety –NH-;
whereineach of them is optionally substituted by one to two R 1; andrepresents the point which attaches to the moiety –NH-; represents the point which attaches to X.
represents the point which attaches to X.
- The compound of any one of claims 1-6, a pharmaceutically acceptable salt thereof, or a stereoisomer thereof, wherein ring A is pyridyl; wherein said pyridyl is optionally substituted by one R 1.
- The compound of any one of claims 1-8, a pharmaceutically acceptable salt, or a stereoisomer thereof, whereinX is absent, -O-, ^-CH 2-O-^^, ^-CH 2-C (O) -^^, ^-NR 2C (O) -^^, ^-CH 2-NR 2C (O) -^^, ^-C (O) NR 2-^^, ^-CH 2-C (O) NR 2-^^, ^-SO 2-^^, or ^-C (R 2) =N-O-^^; wherein^-represents the point which attaches to ring A; -^^ represents the point which attaches to
 R 2, in each occurrence, is independently hydrogen, C 1-3alkyl, or C 1-3haloalkyl.
R 2, in each occurrence, is independently hydrogen, C 1-3alkyl, or C 1-3haloalkyl. - The compound of any one of claims 1-9, a pharmaceutically acceptable salt, or a stereoisomer thereof, whereinX is absent, -O-, ^-CH 2-O-^^, ^-CH 2-C (O) -^^, ^-NHC (O) -^^, ^-CH 2-NHC (O) -^^, ^-C (O) NH-^^, ^-CH 2-C (O) NH-^^, ^-SO 2-^^, or ^-C (R 2) =N-O-^^; wherein^-represents the point which attaches to ring A; -^^ represents the point which attaches toand
 R 2, in each occurrence, is independently hydrogen, -CH 3, -CF 3, or isopropyl.
R 2, in each occurrence, is independently hydrogen, -CH 3, -CF 3, or isopropyl. - The compound of any one of claims 1-10, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein X is absent or ^-CH 2-O-^^; wherein^-represents the point which attaches to ring A; -^^ represents the point which attaches to

- The compound of any one of claims 1-11 or a pharmaceutically acceptable salt or a stereoisomer thereof, whereinW is absent, C 1-4alkylene, C 2-4alkenylene, or C 2-4alkynylene; wherein said C 1-4alkylene, C 2-4alkenylene, or C 2-4alkynylene represented by W is optionally substituted by one to three R 3;L is absent, -O-, -NH-, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 6-10 membered aryl, or 5-10 membered heteroaryl; wherein said -NH-, 3-8 membered carbocyclyl, 3-6 membered heterocyclyl, 6-10 membered aryl, and 5-10 membered heteroaryl represented by L is optionally substituted by one to three R 3; andZ is absent, C 1-4alkylene, C 2-4alkenylene, or C 2-4alkynylene; wherein said C 1-4alkylene, C 2-4alkenylene, or C 2-4alkynylene represented by Z is optionally substituted by one to three R 3; whereinR 3, in each occurrence, is independently halogen, C 1-4alkyl, C 1-4haloalkyl, or -C (=O) R 3a; whereinR 3a is hydrogen or C 1-4alkyl.
- The compound of any one of claims 1-12, a pharmaceutically acceptable salt, or a stereoisomer thereof, whereinW is absent, C 1-3alkylene, C 2-3alkenylene, or C 2-3alkynylene; wherein said C 1-3alkylene, C 2-3alkenylene, or C 2-3alkynylene represented by W is optionally substituted by one to two R 3;L is absent, -O-, -NH-, 3-6 membered monocyclic carbocyclyl, 6-8 membered bicyclic carbocyclyl; 4-6 membered monocyclic heterocyclyl, phenyl, and 5-6 membered monocyclic heteroaryl; wherein said 3-6 membered monocyclic carbocyclyl, 6-8 membered bicyclic carbocyclyl; 4-6 membered monocyclic heterocyclyl, phenyl, and 5-6 membered monocyclic heteroaryl represented by L is optionally substituted by one R 3; andZ is absent, C 1-3alkylene, C 2-3alkenylene, or C 2-3alkynylene; wherein said C 1-3alkylene, C 2-3alkenylene, or C 2-3alkynylene represented by Z is optionally substituted by one to two R 3; whereinR 3, in each occurrence, is independently halogen, C 1-3alkyl, C 1-2haloalkyl, or -C (=O) CH 3.
- The compound of any one of claims 1-13, a pharmaceutically acceptable salt, or a stereoisomer thereof, whereinW is C 1-3alkylene, C 1-3haloalkylene, C 2-3alkenylene, or C 2-3alkynylene;L is absent, -NH-or -O-; andZ is C 1-3alkylene, C 1-3haloalkylene, C 2-3alkenylene, or C 2-3alkynylene.
- The compound of any one of claims 1-14, a pharmaceutically acceptable salt, or a stereoisomer thereof, whereinW is C 3-4alkylene, C 3-4haloalkylene, C 3-4alkenylene, or C 3-4alkynylene;L is absent; andZ is absent.
- The compound of any one of claims 1-15, a pharmaceutically acceptable salt, or a stereoisomer thereof, whereinW is absent, C 1-2alkylene, or C 1-2haloalkylene;L is 4-6 membered carbocyclyl, 4-6 membered heterocyclyl, or 5-6 membered heteroaryl; andZ is absent or methylene.
- The compound of any one of claims 1-16, a pharmaceutically acceptable salt, or a stereoisomer thereof, whereinW is absent, -CH 2-, -CH 2CH 2-, -CH 2CH 2CH 2-, -CH=CH-, -CH 2CH=CH-, or -CH 2C≡C-; wherein said -CH 2-, -CH 2CH 2-, -CH 2CH 2CH 2-, -CH=CH-, -CH 2CH=CH-, or -CH 2C≡C-represented by W is optionally substituted by one to two R 3L is absent, -O-, -NH-, cyclobutyl, cyclohexyl, spiro [3.3] heptanyl, azetidinyl, piperazinyl, piperidinyl, oxazolyl, isoxazolyl, imidazolyl, triazolyl, or thiozolyl; wherein said -NH-, cyclobutyl, cyclohexyl, spiro [3.3] heptanyl, azetidinyl, piperazinyl, piperidinyl, oxazolyl, isoxazolyl, imidazolyl, triazolyl, or thiozolyl is optionally substituted by one R 3;Z is absent, -CH 2-, -CH 2CH 2-, -CH 2CH 2CH 2-, -CH=CH-, -CH=CHCH 2-, or -C≡CCH-; whereinR 3 is F, -CH 3, -CF 3, -CH 2CH 3, -CH (CH 3) 2, -CH 2CF 3, or –C (=O) CH 3.
- The compound of any one of claims 1-13 and 17, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein *-W-L-Z-**is selected from the group consisting of

 wherein each of them is optionally substituted by one to two R 3; wherein*-represents the point which attaches to X; -**represents the point which attaches to
wherein each of them is optionally substituted by one to two R 3; wherein*-represents the point which attaches to X; -**represents the point which attaches to
- The compound of any one of claims 1-13, 17 and 18, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein *-W-L-Z-**is selected from the group consisting ofwherein
 *-represents the point which attaches to X; -**represents the point which attaches to
*-represents the point which attaches to X; -**represents the point which attaches to
- The compound of any one of claims 1-19, a pharmaceutically acceptable salt, or a stereoisomer thereof, whereinis represented by Formula A, B, C, or D,

 wherein@-represents the point which attaches to the cyclopentyl shown in Formula (I’) ; -@@ represents the point which attaches to
wherein@-represents the point which attaches to the cyclopentyl shown in Formula (I’) ; -@@ represents the point which attaches to R 22 is hydrogen, C 1-4alkyl, or C 1-4haloalkyl; andR 4 is hydrogen or C 1-4alkyl; or R 4 and one R 3 attached to Z, together with the atoms to which they are attached, form 4-6 membered heterocyclyl.
R 22 is hydrogen, C 1-4alkyl, or C 1-4haloalkyl; andR 4 is hydrogen or C 1-4alkyl; or R 4 and one R 3 attached to Z, together with the atoms to which they are attached, form 4-6 membered heterocyclyl. - The compound of claim 20, a pharmaceutically acceptable salt, or a stereoisomer thereof, whereinis

 wherein@-represents the point which attaches to the cyclopentyl shown in Formula (I’) ; -@@ represents the point which attaches toand
wherein@-represents the point which attaches to the cyclopentyl shown in Formula (I’) ; -@@ represents the point which attaches toand R 4 is hydrogen or -CH 3; or R 4 and one R 3 attached to Z together with the atoms to which they are attached, form azetidinyl or pyrrolidinyl.
R 4 is hydrogen or -CH 3; or R 4 and one R 3 attached to Z together with the atoms to which they are attached, form azetidinyl or pyrrolidinyl. - The compound of claim 21, a pharmaceutically acceptable salt, or a stereoisomer thereof, wherein R 4 is hydrogen.
- The compound of any one of claims 1-19, a pharmaceutically acceptable salt, or a stereoisomer thereof, whereinis represented by Formula (E) :

 wherein@-represents the point which attaches to the cyclopentyl shown in Formula (I’) ; -@@ represents the point which attaches toand
wherein@-represents the point which attaches to the cyclopentyl shown in Formula (I’) ; -@@ represents the point which attaches toand R 5 is hydrogen or isopropyl.
R 5 is hydrogen or isopropyl. - A compound of Table 1 or a pharmaceutically acceptable salt or a stereoisomer thereof.
- A pharmaceutical composition comprising the compound of any one of claims 1-24, a pharmaceutically acceptable salt, or a stereoisomer thereof, and a pharmaceutically acceptable carrier or excipient.
- A method of treating a disease or condition modulated at least in part by CDK2 in a subject, comprising administering to the subject in need thereof, the compound of any one of claims 1-24, a pharmaceutically acceptable salt, or a stereoisomer thereof, or the pharmaceutical composition of claim 25.
- A method of inhibiting CDK2 in a patient, comprising administering to the patient the compound of any one of claims 1-24, a pharmaceutically acceptable salt, or a stereoisomer thereof, or the pharmaceutical composition of claim 25.
- A method of treating a disease or disorder associated with CDK2 in a patient, comprising administering to the patient a therapeutically effective amount of the compound of any one of claims 1-24, a pharmaceutically acceptable salt, or a stereoisomer thereof, or the pharmaceutical composition of claim 25; wherein the disease or disorder is associated with an amplification of the cyclin E1 (CCNE1) gene and/or overexpression of CCNE1.
- The method of any one of claims 26-28, wherein the disease or disorder is cancer.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2021084358 | 2021-03-31 | ||
| CNPCT/CN2021/084358 | 2021-03-31 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022206888A1 true WO2022206888A1 (en) | 2022-10-06 |
Family
ID=75625271
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2022/084355 WO2022206888A1 (en) | 2021-03-31 | 2022-03-31 | Cdk2 inhibitors and use thereof |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2022206888A1 (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024123801A1 (en) * | 2022-12-06 | 2024-06-13 | Genesis Therapeutics, Inc. | Compounds for treating cancer |
| US12097261B2 (en) | 2021-05-07 | 2024-09-24 | Kymera Therapeutics, Inc. | CDK2 degraders and uses thereof |
| WO2024178335A3 (en) * | 2023-02-23 | 2024-10-31 | Accutar Biotechnology, Inc. | Novel macrocyclic aminopyrazole compounds as cdk2 inhibitors |
Citations (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5521184A (en) | 1992-04-03 | 1996-05-28 | Ciba-Geigy Corporation | Pyrimidine derivatives and processes for the preparation thereof |
| WO2000009495A1 (en) | 1998-08-11 | 2000-02-24 | Novartis Ag | Isoquinoline derivatives with angiogenesis inhibiting activity |
| WO2000053595A1 (en) | 1999-03-06 | 2000-09-14 | Astrazeneca Ab | Pyrimidine compounds |
| WO2001014402A1 (en) | 1999-08-19 | 2001-03-01 | Isis Pharmaceuticals, Inc. | Antisense modulation of focal adhesion kinase expression |
| WO2001064655A1 (en) | 2000-03-01 | 2001-09-07 | Astrazeneca Ab | 2, 4-di(hetero-)arylamino (-oxy)-5-substituted pyrimidines as antineoplastic agents |
| WO2003024967A2 (en) | 2001-09-19 | 2003-03-27 | Aventis Pharma S.A. | Indolizines as kinase protein inhibitors |
| WO2003037347A1 (en) | 2001-10-30 | 2003-05-08 | Novartis Ag | Staurosporine derivatives as inhibitors of flt3 receptor tyrosine kinase activity |
| WO2003062236A1 (en) | 2002-01-22 | 2003-07-31 | Warner-Lambert Company Llc | 2-(PYRIDIN-2-YLAMINO)-PYRIDO[2,3d]PYRIMIDIN-7-ONES |
| WO2003099771A2 (en) | 2002-05-29 | 2003-12-04 | Novartis Ag | Diaryl urea derivatives useful for the treatment of protein kinase dependent diseases |
| WO2004005281A1 (en) | 2002-07-05 | 2004-01-15 | Novartis Ag | Inhibitors of tyrosine kinases |
| WO2004046120A2 (en) | 2002-11-15 | 2004-06-03 | Vertex Pharmaceuticals Incorporated | Diaminotriazoles useful as inhibitors of protein kinases |
| WO2004056786A2 (en) | 2002-12-20 | 2004-07-08 | Pfizer Products Inc. | Pyrimidine derivates for the treatment of abnormal cell growth |
| WO2004080980A1 (en) | 2003-03-14 | 2004-09-23 | Novartis Ag | 2, 4- di (phenylamino) pyrimidines useful in the treatment of neoplastic diseases, inflammatory and immune system disorders |
| WO2005028444A1 (en) | 2003-09-24 | 2005-03-31 | Novartis Ag | 1,4-disubstituted isoquinilone derivatives as raf-kinase inhibitors useful for the treatment of proliferative diseases |
| WO2006056399A2 (en) | 2004-11-24 | 2006-06-01 | Novartis Ag | Combinations of jak inhibitors and at least one of bcr-abl, flt-3, fak or raf kinase inhibitors |
| WO2009085185A1 (en) | 2007-12-19 | 2009-07-09 | Amgen Inc. | Fused pyridine, pyrimidine and triazine compounds as cell cycle inhibitors |
| WO2010075074A1 (en) | 2008-12-22 | 2010-07-01 | Eli Lilly And Company | Protein kinase inhibitors |
| WO2011101409A1 (en) | 2010-02-19 | 2011-08-25 | Novartis Ag | Pyrrolopyrimidine compounds as inhibitors of cdk4/6 |
| WO2012061156A1 (en) | 2010-10-25 | 2012-05-10 | Tavares Francis X | Cdk inhibitors |
| WO2012129344A1 (en) | 2011-03-23 | 2012-09-27 | Amgen Inc. | Fused tricyclic dual inhibitors of cdk 4/6 and flt3 |
| WO2020157652A2 (en) * | 2019-01-31 | 2020-08-06 | Pfizer Inc. | Cdk2 inhibitors |
-
2022
- 2022-03-31 WO PCT/CN2022/084355 patent/WO2022206888A1/en active Application Filing
Patent Citations (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5521184A (en) | 1992-04-03 | 1996-05-28 | Ciba-Geigy Corporation | Pyrimidine derivatives and processes for the preparation thereof |
| WO2000009495A1 (en) | 1998-08-11 | 2000-02-24 | Novartis Ag | Isoquinoline derivatives with angiogenesis inhibiting activity |
| WO2000053595A1 (en) | 1999-03-06 | 2000-09-14 | Astrazeneca Ab | Pyrimidine compounds |
| WO2001014402A1 (en) | 1999-08-19 | 2001-03-01 | Isis Pharmaceuticals, Inc. | Antisense modulation of focal adhesion kinase expression |
| WO2001064655A1 (en) | 2000-03-01 | 2001-09-07 | Astrazeneca Ab | 2, 4-di(hetero-)arylamino (-oxy)-5-substituted pyrimidines as antineoplastic agents |
| WO2003024967A2 (en) | 2001-09-19 | 2003-03-27 | Aventis Pharma S.A. | Indolizines as kinase protein inhibitors |
| WO2003037347A1 (en) | 2001-10-30 | 2003-05-08 | Novartis Ag | Staurosporine derivatives as inhibitors of flt3 receptor tyrosine kinase activity |
| WO2003062236A1 (en) | 2002-01-22 | 2003-07-31 | Warner-Lambert Company Llc | 2-(PYRIDIN-2-YLAMINO)-PYRIDO[2,3d]PYRIMIDIN-7-ONES |
| WO2003099771A2 (en) | 2002-05-29 | 2003-12-04 | Novartis Ag | Diaryl urea derivatives useful for the treatment of protein kinase dependent diseases |
| WO2004005281A1 (en) | 2002-07-05 | 2004-01-15 | Novartis Ag | Inhibitors of tyrosine kinases |
| WO2004046120A2 (en) | 2002-11-15 | 2004-06-03 | Vertex Pharmaceuticals Incorporated | Diaminotriazoles useful as inhibitors of protein kinases |
| WO2004056786A2 (en) | 2002-12-20 | 2004-07-08 | Pfizer Products Inc. | Pyrimidine derivates for the treatment of abnormal cell growth |
| WO2004080980A1 (en) | 2003-03-14 | 2004-09-23 | Novartis Ag | 2, 4- di (phenylamino) pyrimidines useful in the treatment of neoplastic diseases, inflammatory and immune system disorders |
| WO2005028444A1 (en) | 2003-09-24 | 2005-03-31 | Novartis Ag | 1,4-disubstituted isoquinilone derivatives as raf-kinase inhibitors useful for the treatment of proliferative diseases |
| WO2006056399A2 (en) | 2004-11-24 | 2006-06-01 | Novartis Ag | Combinations of jak inhibitors and at least one of bcr-abl, flt-3, fak or raf kinase inhibitors |
| WO2009085185A1 (en) | 2007-12-19 | 2009-07-09 | Amgen Inc. | Fused pyridine, pyrimidine and triazine compounds as cell cycle inhibitors |
| WO2010075074A1 (en) | 2008-12-22 | 2010-07-01 | Eli Lilly And Company | Protein kinase inhibitors |
| WO2011101409A1 (en) | 2010-02-19 | 2011-08-25 | Novartis Ag | Pyrrolopyrimidine compounds as inhibitors of cdk4/6 |
| WO2012061156A1 (en) | 2010-10-25 | 2012-05-10 | Tavares Francis X | Cdk inhibitors |
| WO2012129344A1 (en) | 2011-03-23 | 2012-09-27 | Amgen Inc. | Fused tricyclic dual inhibitors of cdk 4/6 and flt3 |
| WO2020157652A2 (en) * | 2019-01-31 | 2020-08-06 | Pfizer Inc. | Cdk2 inhibitors |
Non-Patent Citations (22)
| Title |
|---|
| "Pharmaceutical Dosage Forms", 1980, MARCEL DECKER |
| AU-YEUNG, G. ET AL., CLIN CANCER RES, vol. 23, no. 7, 2017, pages 1862 - 1874 |
| CHEN, Y N. ET AL., PROC NATL ACAD SCI USA, vol. 96, no. 8, 1999, pages 4325 - 9 |
| CICENAS, J. ET AL., CANCERS (BASEL, vol. 6, no. 4, 2014, pages 2224 - 42 |
| EKHOLM, S. V.S. I. REED, CURR OPIN CELL BIOL, vol. 12, no. 6, 2000, pages 676 - 84 |
| FINNINMORGAN, J. PHARM. SCI., vol. 88, 1999, pages 955 - 958 |
| GOODMANGILMAN: "The Pharmacological Basis of Therapeutics", 1996, MEDICAL ECONOMICS COMPANY |
| HERRERA-ABREU, M. T. ET AL., CANCER RES, vol. 76, no. 8, 2016, pages 2301 - 13 |
| HOOVER, JOHN E.: "Remington's Pharmaceutical Sciences", 1975, MACK PUBLISHING CO. |
| HU, S. ET AL., MOL CANCER THER, vol. 14, no. 11, 2015, pages 2576 - 85 |
| KEYOMARSI, K. ET AL., N ENGL J MED, vol. 347, no. 20, 2002, pages 1566 - 75 |
| MENDOZA, N ET AL., CANCER RES, vol. 63, no. 5, 2003, pages 1020 - 4 |
| MORRISONBOYD: "Comprehensive Organic Transformations: A Guide to Functional Group Preparations", vol. I-XII, 1991, JOHN WILEY AND SONS |
| NAKAYAMA, N. ET AL., CANCER, vol. 116, no. 11, 2010, pages 2621 - 34 |
| ROSEN, D. G. ET AL., CANCER, vol. 106, no. 9, 2006, pages 1925 - 32 |
| S. M. BERGE ET AL., J. PHARM. SCI., vol. 66, 1977, pages 1 - 19 |
| SCALTRITI, M. ET AL., PROC NATL ACAD SCI USA, vol. 108, no. 9, 2011, pages 3761 - 6 |
| SEE MOLENAAR ET AL., PROC NATL ACAD SCI USA, vol. 106, no. 31, pages 12968 - 12973 |
| SMITH, ROGER M.: "Chromatographic Science Series", vol. 75, 1998, LOUGHBOROUGH UNIVERSITY, pages: 223 - 249 |
| TAKADA ET AL., CANCER RES, vol. 77, no. 18, 2017, pages 4881 - 4893 |
| THOMAS SORRELL: "Handbook of Pharmaceutical Excipients", 1999, PHARMACEUTICAL ASSOCIATION |
| XU, X. ET AL., BIOCHEMISTRY, vol. 38, no. 27, 1999, pages 8713 - 22 |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12097261B2 (en) | 2021-05-07 | 2024-09-24 | Kymera Therapeutics, Inc. | CDK2 degraders and uses thereof |
| WO2024123801A1 (en) * | 2022-12-06 | 2024-06-13 | Genesis Therapeutics, Inc. | Compounds for treating cancer |
| WO2024178335A3 (en) * | 2023-02-23 | 2024-10-31 | Accutar Biotechnology, Inc. | Novel macrocyclic aminopyrazole compounds as cdk2 inhibitors |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11384083B2 (en) | Substituted spiro[cyclopropane-1,5′-pyrrolo[2,3-d]pyrimidin]-6′(7′h)-ones as CDK2 inhibitors | |
| US12083124B2 (en) | Bicyclic heterocycles as FGFR inhibitors | |
| US11866432B2 (en) | Dihydropyrido[2,3-d]pyrimidinone compounds as CDK2 inhibitors | |
| US11591329B2 (en) | Bicyclic heterocycles as FGFR inhibitors | |
| US11713317B2 (en) | Tricyclic heterocyclic compounds as sting activators | |
| US12122767B2 (en) | Bicyclic heterocycles as FGFR inhibitors | |
| US12042495B2 (en) | MAP4K1 inhibitors | |
| US20210355121A1 (en) | Fused tricyclic kras inhibitors | |
| US11427597B2 (en) | Heteroaryl amide compounds as sting activators | |
| WO2022206888A1 (en) | Cdk2 inhibitors and use thereof | |
| AU2017281903A1 (en) | Degradation of bromodomain-containing protein 9 (BRD9) by conjugation of BRD9 inhibitors with E3 ligase ligand and methods of use | |
| US11596692B1 (en) | PD-L1/STING conjugates and methods of use | |
| US20230174555A1 (en) | Naphthyridine compounds as inhibitors of kras | |
| US20240343742A1 (en) | Vinyl imidazole compounds as inhibitors of kras | |
| US11939328B2 (en) | Quinoline compounds as inhibitors of KRAS | |
| US20230056631A1 (en) | Tricyclic compounds as inhibitors of kras | |
| WO2023116884A1 (en) | Cdk2 inhibitors and use thereof | |
| US20230114765A1 (en) | Tricyclic compounds as inhibitors of kras | |
| WO2023168686A1 (en) | Substituted cyclopentanes as cdk2 inhibitors | |
| WO2022155941A1 (en) | Cdk2 inhibitors | |
| US20230203010A1 (en) | Bicyclic amine cdk12 inhibitors | |
| US20240400557A1 (en) | Pyrazoloquinoline kras inhibitors | |
| US12084453B2 (en) | Bicyclic amines as CDK12 inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22714985 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 22714985 Country of ref document: EP Kind code of ref document: A1 |