WO2022087154A1 - Mhc class ii peptide multimers and uses thereof - Google Patents
Mhc class ii peptide multimers and uses thereof Download PDFInfo
- Publication number
- WO2022087154A1 WO2022087154A1 PCT/US2021/055882 US2021055882W WO2022087154A1 WO 2022087154 A1 WO2022087154 A1 WO 2022087154A1 US 2021055882 W US2021055882 W US 2021055882W WO 2022087154 A1 WO2022087154 A1 WO 2022087154A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mhcii
- peptide
- multimer
- mhc
- cells
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/70503—Immunoglobulin superfamily
- C07K14/70539—MHC-molecules, e.g. HLA-molecules
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/20—Fusion polypeptide containing a tag with affinity for a non-protein ligand
- C07K2319/22—Fusion polypeptide containing a tag with affinity for a non-protein ligand containing a Strep-tag
Definitions
- MHC multimers have been used for detection of antigen-responsive T cells since Altman (Altman et al. (1996) SCIENCE 274:94-96) showed that tetramerization of peptide- loaded MHC class I (pMHCI) molecules provided sufficient stability to T cell receptor (TCR)-pMHC interactions, allowing detection of fluorescently -labeled MHC multimer- binding T cells using flow cytometry.
- pMHCI-based technologies were initially restricted by the tedious production of molecules in which each peptide required an individual folding and purification procedure (Bakker et al. (2005) CURR. OPIN. IMMUNOL. 17:428-433).
- MHCI multimers, and libraries thereof have been prepared using biotinylated peptide-MHCI monomers that then associate with the biotin-binding site on streptavidin to form tetramers (see e.g., Leisner et al. (2008) PLoS ONE 3(2):el678).
- MHC Class I libraries approaches have been described in which oligonucleotide barcode labels have been conjugated to the streptavidin.
- existing strategies involve complex and/or costly approaches that limit the facile production of large libraries.
- streptavidin precursors must be barcoded individually by overlap extension PCR prior to tetramerization of biotinylated peptide-HLA monomers (Zhang et al. (2016) NATURE BIOTECH. 2018; doi: 10.1038. nbt.4282).
- streptavidin-conjugated dextran which is a costly reagent, is used to create a dextramer to which both the biotinylated peptide-HLA monomers and the biotinylated barcode oligonucleotide are complexed (Bentzen et al. 2016) NATURE BIOTECH.
- soluble MHC class II molecules also have been used to prepare pMHCII tetramers, which have been used in the study of the antigenic specificity of CD4+ T helper cells (as reviewed in, for example, Nepom et al. (2002) ARTHRIT. RHEUMAT. 46:5-12; Vollers and Stem (2008) IMMUNOL. 123:305-313; Cecconi et al. (2008) CYTOMETRY 73A:1010-1018).
- soluble biotinylated MHCII a/ dimers are recombinantly expressed and then tetramerized by binding to streptavidin or avidin through their biotin-binding sites. Fluorescent labeling of the streptavidin or avidin then allows for isolation of T cells that bind the pMHCII multimers by flow cytometry.
- antigenic peptide loading of the MHCII molecules in one approach, a peptide is attached to the MHCII a/p dimers covalently.
- the present disclosure provides methods for producing barcoded, peptide loaded MHCII multimers (e.g, tetramers), including libraries thereof, as well as methods of using such multimers.
- the approach of the present disclosure involves attaching MHCII-binding peptides to a multimerization domain, to thereby create a multimer composition, which is then loaded with soluble MHCII molecules to create MHCII multimers.
- the peptide-multimerization domain composition can be made recombinantly, using a nucleic acid construct co-encoding the peptide and multimerization domain, which typically are linked by inclusion of a spacer linker between the two.
- the peptide can be attached to the multimerization domain through chemical conjugation, again typically using a spacer linker in between.
- the MHCII molecules are provided as soluble alpha/beta chains, either “empty” dimers (i. e. , not loaded with peptide) or, more typically, dimers with a digestible placeholder peptide loaded into the peptide-binding groove.
- the empty MHCII molecules then can be loaded onto the peptide-multimerization domain composition or, for peptide-loaded dimers, they can be loaded by digestion of the placeholder peptide, followed by peptide exchange, thereby producing MHCII multimers.
- the methods provide MHCII multimers that allow for ease of peptide exchange and barcode labeling of the multimers to thereby allow for efficient preparation of large MHCII multimer libraries. Accordingly, the compositions and methods described herein are suitable for routine laboratory research, as well as large scale industrial and clinical applications, in all circumstances where MHCII multimers are useful, e.g., for analysis of CD4+ T cell antigen recognition. Moreover, the MHCII multimers of the disclosure can be labeled with individual identifiers, such as oligonucleotide barcodes, to facilitate identification of library members.
- biotinbinding sites within streptavidin are not being used for multimerization of the monomers, these biotin-binding sites are available for easy labeling using biotinylated oligonucleotide barcodes.
- the disclosure pertains to a method of producing a Major Histocompatibility Complex Class II (MHCII) multimer, the method comprising:
- the multimer composition is a tetramer comprising streptavidin or avidin as the multimerization domain.
- the multimer composition further comprises a biotinylated nucleic acid that binds the multimerization domain, wherein the biotinylated nucleic acid encodes the MHCII-binding peptides.
- the MHCII-binding peptides are produced from the biotinylated nucleic acid by in vitro transcription/translation (IVTT).
- the MHCII-binding peptides are attached to the multimerization domain by covalent linkage using a spacer linker.
- the spacer linker comprises an amino acid sequence selected from the group consisting of SEQ ID NOs: 6-8 and 72-80.
- the MHC Class II alpha chain comprises an amino acid sequence selected from the group of sequences shown in SEQ ID NOs: 42-46.
- the MHC Class II beta chain comprises an amino acid sequence selected from the group of sequences shown in SEQ ID NOs: 47-71.
- the MHC Class II alpha chain and the MHC Class II beta chain comprise the amino acid sequences set forth in SEQ ID NOs: 42 and 47; 42 and 48; 42 and 49; 42 and 50; 42 and 51; 42 and 52; 42 and 53; 42 and 54; 42 and 55; 42 and 56; 42 and 57; 42 and 58; 42 and 59; 42 and 60; 42 and 61; 44 and 62; 44 and 63; 45 and 64; 46 and 65; or 144 and 146, respectively.
- each MHCII molecule is loaded with a digestible placeholder peptide, and wherein the MHCII molecules are contacted with the multimer composition under conditions for cleavage of the placeholder peptide, thereby to produce an MHCII multimer by peptide exchange with the multimer composition.
- the digestible placeholder peptide is thermolabile, labile at acidic pH, enzymatically cleavable or photocleavable.
- the digestible placeholder peptide comprises a placeholder peptide linked to the MHCII molecule by an digestible linker.
- the digestible placeholder peptide is a CLIP peptide comprising the amino acid sequence KPVSKMRMATPLLMQA (SEQ ID NO: 3) or ATPLLMQALPMGA (SEQ ID NO: 134).
- peptide exchange is achieved by digestion of the placeholder peptide (e.g., by protease cleavage or UV -mediated cleavage) and combining the multimer composition and the MHCII molecules under low pH conditions.
- digestion of the placeholder peptide involves cleavage of a linker that connects the placeholder peptide to the soluble MHCII molecules.
- the MHCII molecule is an empty molecule, i.e. no peptide is bound prior to the peptide exchange.
- the method further comprises labeling the multimer composition with an oligonucleotide barcode (e.g., a biotinylated oligonucleotide barcode, which is bound to biotin-binding sites on the multimerization domain).
- step (a) provides multimers comprising a plurality of MHCII - binding peptides, thereby to produce a library of MHCII multimers (i.e., a composition comprising a plurality of MHCII multimers).
- each member of the library utilizes the same MHCII molecule.
- each member of the library utilizes different MHCII-binding peptides.
- the disclosure pertains to a multimer composition
- a multimer composition comprising a plurality of MHCII-binding peptides attached to a multimerization domain, wherein each MHCII-binding peptide within the plurality has the same amino acid sequence.
- each multimer composition comprises multiple copies of the same MHCII-binding peptide.
- the multimerization domain is not covalently linked to a MHCII molecule.
- the multimer composition is a tetramer, e.g., the multimerization domain comprises streptavidin or avidin.
- the multimer composition comprises four copies of the same MHCII-binding peptide linked to streptavidin or avidin.
- the multimer comprises a biotinylated nucleic acid that binds the multimerization domain, wherein the biotinylated nucleic acid encodes the MHCII-binding peptides.
- the MHCII-binding peptides are produced from the biotinylated nucleic acid by IVTT.
- the MHCII-binding peptides are attached to the multimerization domain by covalent linkage using a spacer linker, for example prepared by recombinant expression or chemical conjugation.
- the spacer linker used to link the peptide to the multimerization domain comprises an amino acid sequences selected from the group consisting of SEQ ID NOs: 6-8 and 72-80.
- the multimer composition further comprises MHCII molecules bound to the MHCII-binding peptides, each MHCII molecule comprising an alpha chain and a beta chain, to thereby create an MHCII multimer (i.e., the peptide-multimerization domain composition is loaded with MHCII molecules to thereby create MHCII multimers).
- the MHC Class II alpha chain comprises an amino acid sequence selected from the group of sequences shown in SEQ ID NOs: 42-46.
- the MHC Class II beta chain comprises an amino acid sequence selected from the group of sequences shown in SEQ ID NOs: 47-71.
- the MHC Class II alpha chain and the MHC Class II beta chain comprise the amino acid sequences set forth in SEQ ID NOs: 42 and 47; 42 and 48; 42 and 49; 42 and 50; 42 and 51; 42 and 52; 42 and 53; 42 and 54; 42 and 55; 42 and 56; 42 and 57; 42 and 58; 42 and 59; 42 and 60; 42 and 61; 44 and 62; 44 and 63; 45 and 64; 46 and 65; or 144 and 146, respectively.
- Libraries of MHCII multimers can be made comprising a plurality of the MHCII multimers.
- each member of the library utilizes the same MHCII molecule.
- each member of the library utilizes different MHCII-binding peptides.
- the multimer composition is labeled with an oligonucleotide barcode (e.g, a biotinylated oligonucleotide barcode is bound to biotin-binding sites on the multimerization domain).
- an oligonucleotide barcode e.g, a biotinylated oligonucleotide barcode is bound to biotin-binding sites on the multimerization domain.
- the disclosure pertains to a nucleic acid construct encoding a multimer composition subunit, wherein the nucleic acid construct encodes a polypeptide comprising an MHCII-binding peptide and a multimerization domain, linked by a spacer linker.
- the polypeptide does not comprise a MHCII molecule.
- the multimerization domain comprises streptavidin or avidin.
- the nucleic acid construct further comprises a biotin moiety.
- the spacer linker comprises an amino acid sequence selected from the group consisting of SEQ ID NOs: 6-8 and 72-80.
- the multimer composition (comprising the peptide linked to the multimerization domain) can be expressed by standard methods, e.g., by in vitro transcription/translation (IVTT) or by recombinant expression in a host cell using an expression vector comprising the nucleic acid construct.
- IVTT in vitro transcription/translation
- recombinant expression in a host cell using an expression vector comprising the nucleic acid construct.
- the disclosure pertains to a method of isolating MHCII- multimer bound lymphocytes comprising:
- each compartment comprises a lymphocyte bound to an MHCII multimer of the library
- each member of the library of MHCII multimers is labeled with an oligonucleotide barcode and the method further comprises decoding the oligonucleotide barcode of the isolated MHCII-multimer bound to the lymphocyte. This allows for identification of the peptide sequence recognized by the lymphocyte.
- FIG. 1 is a schematic diagram of recombinant MHC Class II alpha and beta chains loaded with a placeholder CLIP peptide via a cleavable linker.
- FIG. 2A-2B are schematic diagrams of streptavidin-peptide reagents.
- FIG. 2A is a schematic diagram of a nucleic acid construct encoding a streptavidin-peptide monomer.
- FIG. 2B is a schematic diagram of a streptavidin-peptide tetramer resulting from in vitro transcription/translation (IVTT) of the streptavidin-peptide monomer nucleic acid construct.
- IVTT in vitro transcription/translation
- FIG. 3 is a schematic diagram showing cleavage of the placeholder linker of p*MHCII by a protease and exchange with a rescue peptide that is fused to SA tetramer.
- FIG. 4A is a schematic diagram showing preparation of a placeholder peptide-loaded MHCII (p*MHCII) and protease cleavage thereof, preparation of an SA-peptide tetramer comprising a rescue peptide and low pH-mediated peptide exchange, wherein single-template encapsulation is achieved by either drop-based or well-based methods.
- FIG. 4B is a schematic diagram showing an exemplary barcoded SA-peptide loaded MHCII tetramer binding a cognate T cell receptor (TCR) in solution or on the surface of a cell. The figure shows an exemplary TCR molecule bound to the peptide-loaded MHCII tetramer. It is contemplated that the tetramer can also bind two, three, or four TCR molecules (e.g., soluble TCRs or transmembrane TCRs on a T cell surface).
- TCR T cell receptor
- FIG. 5A-5B show results of analysis of p*MHCII recombinant production and Factor Xa protease cleavage.
- FIG. 5A shows the elution profile of recombinant p*MHCII following purification by size exclusion chromatography.
- FIG. 5B shows the results of SDS-PAGE analysis of p*MHCII before and after cleavage with Factor Xa.
- FIG. 6A-6C show preparation and analysis of three SA-peptide constructs.
- FIG. 6A schematically illustrates the three constructs, each using a different linker, referred to as SA GS ' HA , SA Pro ' HA and SA GS ' Pro ' HA .
- FIG. 6B shows the results of SDS-PAGE analysis of the expressed constructs.
- Lanes 1, 4 and 7 represent the supernatant fraction after cell lysis.
- Lanes 2, 5 and 8 represent the unbound fraction following binding to anti-FLAG resin.
- Lanes 3, 6 and 9 represent the elution fraction following incubation with 3xFLAG competitor peptide. Each variant is indicated at the bottom of the gel.
- FIG. 6C shows the size exclusion chromatography elution profile of the three constructs following FLAG affinity purification.
- FIG. 7 shows the results of SDS-PAGE analysis following peptide exchange between pj,MHCII monomers and SA-peptide tetramers, showing the formation of an SDS-resistant complex following the peptide exchange reaction.
- FIG. 8A-8B shows the detection of pMHCII-SA-HA using ELISA.
- FIG. 8A illustrates the two different ELISA formats tested. In the first format, (FIG. 8A, upper panel), L243 was used as a capturing antibody while biotin-HRP binding to newly-exchanged SA-peptide was used for detection. In the second format (FIG. 8A, lower panel), antistreptavidin was used as a capturing antibody while L243-HRP binding to MHCII was used for detection.
- FIG. 8B shows the corresponding ELISA results for each capturing/detection format.
- an “altered peptide ligand” or “APL” refers to an altered or mutated version of a peptide ligand, such as an MHC binding peptide.
- the altered or mutated version of the peptide ligand contains at least one structural modification (e.g., amino acid substitution) as compared to the peptide ligand from which it is derived.
- a panel of APLs can be prepared by systematic or random mutation of a known MHC binding peptide, to thereby create a pool of APLs that can be used as a library of MHC binding peptides for loading onto MHC Multimers as described herein.
- antigenic determinant refers to a site on an antigen to which the variable domain of a T-cell receptor, an MHC molecule or antibody specifically binds.
- Epitopes can be formed both from contiguous amino acids or noncontiguous amino acids juxtaposed by tertiary folding of a protein. Epitopes formed from contiguous amino acids are typically retained on exposure to denaturing solvents, whereas epitopes formed by tertiary folding are typically lost on treatment with denaturing solvents.
- An epitope typically includes at least 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 or 15 amino acids in a unique spatial conformation and typically can include up to about 25 amino acids.
- epitope mapping Methods for determining what epitopes are bound by a given TCR or antibody (i.e., epitope mapping) are well known in the art and include, for example, immunoblotting and immunoprecipitation assays, wherein overlapping or contiguous peptides from the antigen are tested for reactivity with the given TCR or immunoglobulin.
- Methods of determining spatial conformation of epitopes include techniques in the art and those described herein, for example, x-ray crystallography nuclear magnetic resonance, cryogenic electron microscopy (cryo-EM), hydrogen deuterium exchange mass spectrometry (HDX-MS), and site-directed mutagenesis (see, e.g., EPITOPE MAPPING PROTOCOLS IN METHODS IN MOLECULAR BIOLOGY, Vol. 66, G. E. Morris, Ed. (1996)).
- the term “avidity” as used herein, refers to the binding strength of as a function of the cooperative interactivity of multiple binding sites of a multivalent molecule (e.g., a soluble multimeric pMHC-immunoglobulin protein) with a target molecule.
- a multivalent molecule e.g., a soluble multimeric pMHC-immunoglobulin protein
- a number of technologies exist to characterize the avidity of molecular interactions including switchSENSE and surface plasmon resonance (Gjelstrup et al. (2012) J. IMMUNOL. 188:1292- 1306; Vorup-Jensen (2012) ADV. DRUG. DELIV. REV. 64:1759-1781).
- a “barcode”, also referred to as an oligonucleotide barcode, is a typically short nucleotide sequence (e.g, about 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 or 15 nucleotides long or longer) that identifies a molecule to which it is conjugated. Barcodes can be used, for example, to identify molecules in a reaction mixture. Barcodes uniquely identify the molecule to which it is conjugated, for example, by performing reverse transcription using primers that each contain a “unique molecular identifier” barcode. In other embodiments, primers can be utilized that contain “molecular barcodes” unique to each molecule.
- a “DNA barcode” is a DNA sequence used to identify a target molecule during DNA sequencing.
- a library of DNA barcodes is generated randomly, for example, by assembling oligos in pools.
- the library of DNA barcodes is rationally designed in silico and then manufactured.
- Binding affinity generally refers to the strength of the sum total of noncovalent interactions between a single binding site of a molecule (e.g., a TCR, pMHC) and its binding partner. Unless indicated otherwise, as used herein, “binding affinity” refers to intrinsic binding affinity which reflects a 1: 1 interaction between members of a binding pair (e.g., TCR and peptide-MHC).
- the affinity of a molecule X for its partner Y can generally be represented by the dissociation constant (Kd).
- the Kd can be about 200 nM, 150 nM, 100 nM, 60 nM, 50 nM, 40 nM, 30 nM, 20 nM, 10 nM, 8 nM, 6 nM, 4 nM, 2 nM, 1 nM, or stronger, including up to 1 pM.
- Affinity can be measured by common methods known in the art, including those described herein. A variety of methods of measuring binding affinity are known in the art, any of which can be used for purposes of the present disclosure.
- bioorthogonal chemistry refers to any chemical reaction that can occur inside of living systems without interfering with native biochemical processes.
- the term includes chemical reactions that are chemical reactions that occur in vitro at physiological pH in, or in the presence of water. To be considered bioorthogonol, the reactions are selective and avoid side-reactions with other functional groups found in the starting compounds.
- the resulting covalent bond between the reaction partners should be strong and chemically inert to biological reactions and should not affect the biological activity of the desired molecule.
- carrier and “pharmaceutically acceptable carrier” includes any and all solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents, and the like that are physiologically compatible.
- cleavage site refers to a site, a motif or sequence that is cleavable, such as by an enzyme (e.g., a protease) or by particular reaction conditions.
- the cleavage moiety comprises a protein, e.g, enzymatic, cleavage site.
- the cleavage moiety comprises a chemical cleavage site, e.g., through exposure to oxidation/reduction conditions, light/sound, temperature, pH, pressure, etc.
- click chemistry refers to a set of reliable and selective bioorthogonal reactions for the rapid synthesis of new compounds and combinatorial libraries. Properties of click reactions include modularity, wideness in scope, high yielding, stereospecificity and simple product isolation (separation from inert by-products by non-chromatographic methods) to produce compounds that are stable under physiological conditions.
- click chemistry is a generic term for a set of labeling reactions which make use of selective and modular building blocks and enable chemoselective ligations to radiolabel biologically relevant compounds in the absence of catalysts.
- a “click reaction” can be with copper, or it can be a copper-free click reaction.
- cross-linking unit can refer to a molecule that links to another (same or different) molecule.
- the cross-linking unit is a monomer.
- the cross-link is a chemical bond.
- the cross-link is a covalent bond.
- the cross-link is an ionic bond.
- the cross-link alters at least one physical property of the linked molecules, e.g., a polymer’s physical property.
- endoprotease refers to a protease that cleaves a peptide bond of a non-terminal amino acid.
- epitope refers to a portion of an antigen (e.g., antigenic protein) that binds to (interacts with or is recognized by) an immune receptor.
- an antigen e.g., antigenic protein
- a T cell receptor recognizes and binds to an MHC molecule complexed with (loaded with) a peptide epitope.
- exchangeable pMHC polypeptide refers to MHC monomers and MHC multimers, comprising a placeholder peptide in the binding groove of the MHC polypeptide, and are also referred to as “p*MHC” monomers or multimers.
- Exchangeable refers to the property of a p*MHC monomer or p*MHC multimer allowing for the exchange of the placeholder peptide with an antigenic peptide.
- the exchangeable pMHC or p*MHC polypeptide comprises an MHC Class II molecule with an MHC Class Il-binding peptide in the binding groove of the MHC Class II molecule.
- expression construct refers to a vector designed for gene expression, e.g., in a host cell.
- An expression vector promotes the expression (i.e., transcription/translation) of an encoded polypeptide (e.g., fusion polypeptide).
- the vector is a plasmid, although other suitable vectors, including viral and non-viral vectors are also encompassed by the term “expression construct.”
- a “fusion protein” or “fusion polypeptide” as used interchangeably herein refers to a recombinant protein prepared by linking or fusing two polypeptides into a single protein molecule.
- isolated refers to an MHC glycoprotein, which is in other than its native state, for example, not associated with the cell membrane of a cell that normally expresses MHC. This term embraces a full length subunit chain, as well as a functional fragment of the MHC monomer.
- a functional fragment is one comprising an antigen binding site and sequences necessary for recognition by the appropriate T cell receptor. It typically comprises at least about 60-80%, typically 90-95% of the sequence of the full-length chain.
- An “isolated” MHC subunit component may be recombinantly produced or solubilized from the appropriate cell source.
- the “isolated” MHC monomer is an MHC Class II monomer, such as a soluble form of the MHC Class II a/p chains.
- identifier refers to a readable representation of data that provides information, such as an identity, that corresponds with the identifier.
- linker sequence refers to a nucleotide sequence, and corresponding encoded amino acid sequence, within an expression construct that serves to link or separate two polypeptides, such as two polypeptide domains of a fusion protein.
- an intervening linker sequence can serve to provide flexibility and/or additional space between the two polypeptides that flank the linker.
- operatively linked and “operably linked” are used interchangeably to describe configurations between sequences within an expression construct that allow for particular operations to carried out.
- a regulatory sequence when a regulatory sequence is “operatively linked” to a coding sequence within an expression construct, the regulatory sequence operates to regulate the expression of the coding sequence.
- a cleavage sequence site
- cleavage at the cleavage sequence operates to cleave the peptide sequence away from the rest of the polypeptide encoded by the expression construct.
- MHC Major Histocompatibility Complex
- MHC classical class I and class II molecules that regulate the immune response by presenting peptides of fragmented proteins to circulating cytotoxic and helper T lymphocytes, respectively.
- HLA human leukocyte antigen
- Human MHC class I genes encode, for example, HLA-A, HLA-B and HLA- C molecules.
- HLA-A is one of three major types of human MHC class I cell surface receptors. The others are HLA-B and HLA-C.
- the HLA-A protein is a heterodimer, and is composed of a heavy a chain and smaller [3 chain.
- the a chain is encoded by a variant HLA- A gene, and the P chain is an invariant 2 microglobulin ( 2m) polypeptide.
- the 2 microglobulin polypeptide is coded for by a separate region of the human genome.
- HLA- A*02 (A*02) is a human leukocyte antigen serotype within the HLA-A serotype group. The serotype is determined by the antibody recognition of the a2 domain of the HLA-A a-chain.
- A*02 the a chain is encoded by the HLA-A*02 gene and the chain is encoded by the B2M locus.
- Human MHC class II genes encode, for example, HLA-DPA1, HLA-DPB1, HLA-DQA1, HLA-DQB1, HLA-DRA and HLA-DRB1.
- the complete nucleotide sequence and gene map of the human major histocompatibility complex is publicly available (e.g, The MHC sequencing consortium, Nature 401:921-923, 1999).
- MHC molecule and “MHC protein” are used herein to refer to the polymorphic glycoproteins encoded by the MHC class I and MHC class II genes, which are involved in the presentation of peptide epitopes to T cells.
- MHC class I or “MHC I” are used interchangeably to refer to protein molecules comprising an a chain composed of three domains (al, a2 and a3), and a second, invariant P2-microglobulin. The a3 domain is linked to the transmembrane domain, anchoring the MHC class I molecule to the cell membrane.
- MHC Class I molecules such as HLA-A are part of a process that presents short polypeptides to the immune system. These polypeptides are typically 9-11 amino acids in length and originate from proteins being expressed by the cell, which can be endogenous proteins or exogenous proteins (e.g., viral or bacterial proteins, vaccine proteins). MHC class I molecules present antigen to CD8+ cytotoxic T cells.
- MHC class II and “MHC II” are used interchangeably to refer to protein molecules containing an a chain with two domains (al and a2) and a P chain with two domains (pi and P2).
- the peptide-binding groove is formed by the al/pi heterodimer.
- MHC class II molecules present antigen to specific CD4+ T cells. Antigens delivered endogenously to APCs are processed primarily for association with MHC class I. Antigens delivered exogenously to APCs are processed primarily for association with MHC class II.
- MHC proteins also includes MHC variants which contain amino acid substitutions, deletions or insertions and yet which still bind MHC peptide epitopes (MHC Class I or MHC Class II peptide epitopes).
- MHC Class I or MHC Class II peptide epitopes MHC Class I or MHC Class II peptide epitopes.
- the term also includes fragments of all these proteins, for example, the extracellular domain, which retain peptide binding.
- MHC protein also includes MHC proteins of non-human species of vertebrates.
- MHC proteins of non-human species of vertebrates play a role in the examination and healing of diseases of these species of vertebrates, for example, in veterinary medicine and in animal tests in which human diseases are examined on an animal model, for example, EAE (experimental autoimmune encephalomyelitis) in mice (mus musculus), which is an animal model of the human disease multiple sclerosis.
- EAE experimental autoimmune encephalomyelitis
- mice mus musculus
- Non-human species of vertebrates are, for example, and more specifically mice (mus musculus), rats (rattus norvegicus), cows (bos taurus), horses (equus equus) and green monkeys (macaca mulatta).
- MHC proteins of mice are, for example, referred to as H-2 -proteins, wherein the MHC class I proteins are encoded by the gene loci H2K, H2L and H2D and the MHC class II proteins are encoded by the gene loci H2I.
- a “peptide free MHC polypeptide” or “peptide free MHC multimer” as used herein refers to an MHC monomer or MHC multimer which does not contain a peptide in binding groove of the MHC polypeptide. Peptide free MHC monomers and multimers are also referred to as “empty”. In one embodiment, the peptide free MHC polypeptide or multimer is an MHC Class I polypeptide or multimer. In another embodiment, the peptide free MHC polypeptide or multimer is an MHC Class II polypeptide or multimer.
- the term “multimer” refers to a plurality of units. In some embodiments, the multimer comprises one or more different units. In some embodiments, the units in the multimer are the same. In some embodiments, the units in the multimer are different. In some embodiments, the multimer comprises a mixture of units that are the same and different.
- multimer composition refers to a composition comprising a plurality of MHCII-binding peptides attached to a multimerization domain (e.g., streptavidin) thereby creating a multimer (e.g., tetramer) displaying the plurality e.g., four) MHCII- binding peptides.
- a multimerization domain e.g., streptavidin
- Such a multimer composition can be loaded with MHCII molecules as described herein, thereby creating MHCII multimers.
- peptide epitope refers to an MHC ligand that can bind in the peptide binding groove of an MHC molecule.
- the peptide epitope can typically be presented by the MHC molecule.
- a peptide epitope typically has between 8 and 25 amino acids that are linked via peptide bonds.
- the peptide can contain modification such as, but not limited to, the side chains of the amino acid residues, the presence of a label or tag, the presence of a synthetic amino acid, a functional equivalent of an amino acid, or the like. Typical modifications include those as produced by the cellular machinery, such as glycan addition and phosphorylation. However, other types of modification are also within the scope of the disclosure.
- peptide exchange refers to a competition assay wherein a placeholder peptide is removed and replaced by a “exchanged peptide” (or “exchange peptide epitope”) also referred to herein as a “rescue peptide” (or “rescue peptide epitope”) or “competitor peptide” (or “competitor peptide epitope).
- peptide exchange occurs under conditions in which the placeholder peptide is released by cleavage of the peptide or under suitable conditions allowing rescue peptides to compete for binding to the binding pocket of an MHC monomer or multimer.
- peptide exchange can be accomplished by temperature-induced exchange, UV -induced exchange, dipeptide-induced exchange, pH-induced exchange, or other exchange methods known in the art, and disclosed herein.
- the term “peptide library” refers to a plurality of peptides.
- the library comprises one or more peptides with unique sequences.
- each peptide in the library has a different sequence.
- the library comprises a mixture of peptides with the same and different sequences.
- high diversity peptide library refers to a peptide library with a high degree of peptide variety.
- a high diversity peptide library comprises about 10 3 , about 10 4 , about 10 5 , about 10 6 , about 10 7 , about 10 8 , about 10 9 , about IO 10 , about 10 11 , about 10 12 , about 10 13 , about 10 14 , about 10 15 , about 10 16 , about 10 17 , about 10 18 , about 10 19 , about IO 20 , or more different peptides.
- library peptide refers to a single peptide in the library.
- placeholder peptide or “exchangeable peptide” are used interchangeably to refer to a peptide or peptide-like compound that binds with sufficient affinity to an MHC protein (e.g, MHCI or MHCII protein) and which causes or promotes proper folding of the MHC protein from the unfolded state or stabilization of the folded MHC protein.
- the placeholder peptide can subsequently be exchanged with a different peptide of interest (referred to as an exchange peptide or rescue peptide). This exchange can be accomplished by, for example, UV-induced exchange, dipeptide-induced exchange, temperature-induced exchange, pH-induced exchange, or other exchange methods known in the art.
- polypeptide “peptide”, and “protein” are used interchangeably herein to refer to a polymer of amino acid residues.
- the terms apply to amino acid polymers in which one or more amino acid residue is an artificial chemical mimetic of a corresponding naturally occurring amino acid, as well as to naturally occurring amino acid polymers and non- naturally occurring amino acid polymer.
- isolated protein and “isolated polypeptide” are used interchangeably to refer to a protein (e.g, a soluble, multimeric protein) which has been separated or purified from other components (e.g, proteins, cellular material) and/or chemicals.
- a polypeptide is purified when it constitutes at least 60 (e.g, at least 65, 70, 75, 80, 85, 90, 92, 95, 97, or 99) % by weight of the total protein in the sample.
- protein folding refers to spatial organization of a peptide.
- the amino acid sequence influences the spatial organization or folding of the peptide.
- a peptide may be folded in a functional conformation.
- a folded peptide has one or more biological functions.
- a folded peptide acquires a three-dimensional structure.
- N-terminus amino acid residue refers to one or more amino acids at the N-terminus of a polypeptide.
- small ubiquitin-like modifier moiety or “SUMO domain” or “SUMO moiety” are used interchangeably and refer to a specific protease recognition moiety.
- a tag refers to an oligonucleotide component, generally DNA, that provides a means of addressing a target molecule (e.g., an MHC Multimer) to which it is joined.
- a tag comprises a nucleotide sequence that permits identification, recognition, and/or molecular or biochemical manipulation of the molecule to which the tag is attached (e.g, by providing a unique sequence, and/or a site for annealing an oligonucleotide, such as a primer for extension by a DNA polymerase, or an oligonucleotide for capture or for a ligation reaction).
- a tag can be a barcode, an adapter sequence, a primer hybridization site, or a combination thereof.
- T cell refers to a type of white blood cell that can be distinguised from other white blood cells by the presence of a T cell receptor on the cell surface.
- T helper cells a.k.a.
- TH cells or CD4 + T cells and subtypes, including THI, TH2, TH3, TH17, TH9, and TFH cells, cytotoxic T cells (a.k.a Tc cells, CD8 + T cells, cytotoxic T lymphocytes, T-killer cells, killer T cells), memory T cells and subtypes, including central memory T cells (TCM cells), effector memory T cells (TEM and TEMRA cells), and resident memory T cells (TRM cells), regulatory T cells (a.k.a.
- Treg cells or suppressor T cells and subtypes, including CD4 + FOXP3 + Treg cells, CD4 + FOXP3‘ Treg cells, Tri cells, Th3 cells, and T re gl7 cells, natural killer T cells (a.k.a. NKT cells), mucosal associated invariant T cells (MAITs), and gamma delta T cells (y8 T cells), including Vy9/V62 T cells.
- T cell cytotoxicity includes any immune response that is mediated by CD8+ T cell activation.
- T cell receptor and the term “TCR” refer to a surface protein of a T cell that allows the T cell to recognize an antigen and/or an epitope thereof, typically bound to one or more major histocompatibility complex (MHC) molecules.
- MHC major histocompatibility complex
- TCRs are heterodimers comprising two different protein chains.
- the TCR comprises an alpha (a) chain and a beta ((3) chain.
- Each chain comprises two extracellular domains: a variable (V) region and a constant (C) region, the latter of which is membrane-proximal.
- the variable domains of a-chains and of [3-chains consist of three hypervariable regions that are also referred to as the complementarity determining regions (CDRs).
- the CDRs are primarily responsible for contacting antigens and thus define the specificity of the TCR, although CDR1 of the a-chain can interact with the N-terminal part of the antigen, and CDR1 of the [3-chain interacts with the C-terminal part of the antigen.
- Approximately 5% of T cells have TCRs made up of gamma and delta (y/5) chains. All numbering of the amino acid sequences and designation of protein loops and sheets of the TCRs is according to the IMGT numbering scheme (IMGT, the international ImMunoGeneTics information system@imgt.cines.fr; http://imgt.cines.fr; Lefranc et al. (2003) DEV. COMP. IMMUNOL. 27:55 77.; Lefranc et al. (2005) DEV. COMP. IMMUNOL. 29:185-203).
- IMGT the international ImMunoGeneTics information system@imgt.cines.fr
- soluble T-cell receptor and “sTCR” refer to heterodimeric truncated variants of TCRs, which comprise extracellular portions of the TCR a-chain and [3- chain (e.g., linked by a disulfide bond), but which lack the transmembrane and cytosolic domains of the full-length protein.
- the sequence (amino acid or nucleic acid) of the soluble TCR a-chain and [3-chains may be identical to the corresponding sequences in a native TCR or may comprise variant soluble TCR a-chain and [3-chain sequences, as compared to the corresponding native TCR sequences.
- soluble T-cell receptor encompasses soluble TCRs with variant or non-variant soluble TCR a-chain and [3-chain sequences.
- the variations may be in the variable or constant regions of the soluble TCR a- chain and [3-chain sequences and can include, but are not limited to, amino acid deletion, insertion, substitution mutations as well as changes to the nucleic acid sequence, which do not alter the amino acid sequence. Variants retain the binding functionality of their parent molecules.
- a “TCR/pMHC complex” refers to a protein complex formed by binding between T cell receptor (TCR), or soluble portion thereof, and a peptide-loaded MHC molecule.
- a “component of a TCR/pMHC complex” refers to one or more subunits of a TCR (e.g, Va, V[3, Ca, C ), or to one or more subunits of an MHC or pMHC class I or II molecule.
- the term “unbiased” refers to lacking one or more selective criteria.
- This disclosure provides methods and compositions for the high-throughput generation of libraries containing peptide-loaded MHCII multimers containing a plurality of unique peptides in the MHC binding groove and having oligonucleotide barcode labeling to facilitate identification of library members.
- a multimer composition is provided that comprises a plurality of MHCII-binding peptides attached to a multimerization domain. Upon expression, multimerization mediated by the multimerization domain occurs such that a multimer composition is produced that displays a plurality of MHCII-binding peptides.
- MHCII-binding peptides typically include a library of MHCII-binding peptides.
- a soluble MHCII molecule optionally including a cleavable placeholder peptide in the peptide-binding groove is provided.
- the placeholder peptide can be cleaved and peptide exchange is performed such that the MHCII molecules bind to the peptides of the multimer composition.
- a binding site on the multimerization domain e.g., the biotin-binding site of streptavidin or avidin
- unique identifiers e.g., biotinylated oligonucleotide barcodes
- MHCII multimers are useful in a range of therapeutic, diagnostic, and research applications, essentially in any situation in which MHCII multimers are useful.
- MHCII multimers as described herein can be used in a variety of methods, for example, to identify and isolate specific T-cells in a wide array of applications, e.g., for determining the antigenic specificity of CD4+ T cells (e.g., helper T cells).
- the MHCII multimers of the disclosure are prepared using a peptide-multimerization domain composition as the “scaffold” onto which soluble MHCII molecules are loaded to thereby create the MHCII multimers.
- the peptide-multimerization domain molecule is also referred to herein as a “multimer composition” and comprises MHCII-binding peptides attached to a multimerization domain, typically with a spacer linker (e.g., a flexible linker) linking the peptide to the multimerization domain.
- the peptide (and, optionally, linker) is attached to the N-terminus of the multimerization domain.
- the peptide (and, optionally, linker) is attached to the C-terminus of the multimerization domain.
- a non-limiting representative example of a multimer composition is shown schematically in FIG. 2B, in which four copies of an MHCII-binding peptide are attached to four streptavidin (SA)subunits to create an MHCII-binding peptide-SA tetramer. Preparation of various peptide-SA tetramers is also described in detail in Example 3. The components of the multimer composition, and methods of making the composition, are described further below.
- Multimerization domains for use in producing the multimer compositions provided herein include proteins, polypeptide or other multimeric moieties suitable for the attachment of two or more MHCII-binding peptides, which do not interfere with binding of the MHCII- binding peptides to cells MHCII molecules.
- the multimerization domain comprises protein subunits.
- the multimerization domain is a homomultimer of protein subunits.
- the multimerization domain is a heteromultimer of protein subunits.
- the multimer is a dimer, trimer, tetramer, pentamer, hexamer, octamer decamer or dodecamer. In one embodiment, the multimer is a tetramer.
- binding entities are streptavidin (SA) and avidin and derivatives thereof, biotin, polymers, immunoglobulins, antibodies (monoclonal, polyclonal, and recombinant), antibody fragments and derivatives thereof, leucine zipper domain of AP-1 (jun and fos), hexa-his (metal chelate moiety), hexa-hat GST (glutathione S-transferase) glutathione affinity, Calmodulin-binding peptide (CBP), Strep-tag®, Cellulose Binding Domain, Maltose Binding Protein, S-Peptide Tag, Chitin Binding Tag, Immuno-reactive Epitopes, Epitope Tags, E2Tag, HA Epitope Tag, Myc Epitope, FLAG Epitope, AU1 and AU5 Epitopes, Glu-Glu Epitope, KT3 Epitope, IRS Epitope, Btag Epitope, Protein Kinas
- SA streptavi
- Con A Canavaliaensi formis
- WGA wheat germ agglutinin
- tetranectin Protein A or G
- antibody affinity coiled-coil polypeptides e.g. leucine zipper. Combinations of such binding entities are also included.
- the multimerization domain is a tetramer of streptavidin (SA or SAv) or a derivative thereof. In some embodiments, the multimerization domain is tetrameric streptavidin. In some embodiments, the tetramer comprises Strep-tactin®, an engineered form of streptavidin that binds an engineered peptide sequence referred to as Strep-tag®. Strep-tag® and Strep-tactin® are described in U.S. Patent No. 5,506,121 and U.S. Patent No. 6,103,493, respectively, and are commercially available from a number of sources.
- an avitag can be incorporated into the peptide, for example at the C-terminal end, such that the peptide can be biotinylated through the avitag.
- avitag sequences include SEQ ID NO: 85 (avitag with Myc tag), SEQ ID NO: 86 (avitag with Myc tag and 6xHis tag) and SEQ ID NO: 87 (avitag with 6xHis Tag and FLAG tag).
- the multimerization domain comprises full-length streptavidin.
- the multimerization domain comprises a natural streptavidin core polypeptide.
- the multimerization domain comprises a recombinant streptavidin core polypeptide, such as STV25 or STV13 (e.g., as described in Sano et al. (1995) J. BIOL. CHEM. 270:28204-28209).
- STV25 or STV13 e.g., as described in Sano et al. (1995) J. BIOL. CHEM. 270:28204-28209.
- the multimerization domain is a polymer (i.e., a compound composed of repeating subunits), such as dextran, polyethylene glycol (PEG) and the like.
- the polymer is a sugar polymer, e.g, a polysaccharide, such as dextran.
- the dextran is a modified dextran, wherein the dextran backbone has been modified to carry acceptor sites, such as a VogelmerTM (Immunodex).
- MHCII-binding peptides known in the art, identified based on the MHCII allele to which they bind. Any such known MHCII-binding peptides can be utilized in a multimer composition of the disclosure.
- a non-limiting example of such a known MHCII-binding peptide is an analog of a hemagglutinin (HA) peptide from Influenza A virus having the amino acid sequence shown in SEQ ID NO: 5, which HA peptide binds to an MHCII molecule comprising HLA-DRA*01:01 and HLA-DRB1 *01:01 (as described in Examples 1 and 3).
- HA hemagglutinin
- Protein sequences for the desired antigen can be analyzed for potential HLA specific antigens by using SYFPEITHI (Rammensee et al. (1999) IMMUNOGENETICS 50:213-219), and the artificial neural network (ANN) and stabilized matrix method (SMM) algorithms from IEDB (Peters et al. (2005) PLoS BIOL. 3:e91). Peptides are selected based on a predicted binding value of either >21 for SYFPEITHI, ⁇ 6000 for ANN, or ⁇ 600 for SMM. Selected peptides are synthesized. Other suitable methods for analyzing protein sequences for potential HLA specific antigens also are known in the art and are suitable for use in identifying such HLA specific examples, such as NetMHCpan.
- Binding assays can be performed using a fluorescence polarization (FP) assay as previously described (e.g, Buchi et al. (2004) BIOCHEMISTRY 43:14852-14863; Sette et al. (1994) MOL. IMMUNOL. 31:813-822).
- FP fluorescence polarization
- An epitope library can comprise peptides containing natural amino acids, non-natural amino acids, or a combination of natural and non-natural amino acids.
- Non-natural amino acids can be included to facilitate post-translational modifications, including but not limited to glycosylation, methylation, deamidation, oxidation, reduction and the like. Methods for preparing epitope libraries including non-natural amino acids are established in the art.
- the peptides used in the multimer compositions are from an unbiased library of peptides.
- the MHC-binding peptides can be 8mers, 9mers, lOmers, timers, 12mers, 13mers, Miners, Miners, Miners, Miners, 18mers, 19mers, 20mers, 21mers, 22mers, 23mers, 24mers or 25mers.
- MHCII-binding peptides are 13mers-18mers.
- the library comprises all k-mer peptides produced by transcription and translation of any polynucleotide sequence of interest, for example, in silica production of the transcription and translation products of both the forward and reverse strands of a genome or metagenome in all six reading frames.
- a library of the disclosure comprises all k-mer peptides that can be derived from in silica translation of an exome of interest.
- a library of the disclosure comprises all k-mer peptides that can be derived from in silica translation of a transcriptome of interest.
- a library of the disclosure comprises all k-mer peptides that can be derived from a proteome of interest. [0103] In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from in silico translation of an ORFeome of interest.
- an algorithm can be used to select peptides in a peptide library. For example, an algorithm can be used to predict peptides most likely to fold or dock in an MHC binding pocket, and peptides above a certain threshold value can be selected for inclusion in the library.
- a library of the disclosure comprises all peptides that can be derived from in silico transcription and translation or translation of a group of genomes, proteomes, transcriptomes, ORFeomes, or any combination thereof.
- the peptides are derived from in silico transcription and translation or translation of polynucleotide sequences from a group of samples, for example, clinical samples from a patient population, or a group of pathogen genomes.
- the peptides are derived from a differential genome, proteome, transcriptome, ORFeome, or any combination thereof, where two or more genomes, proteomes, transcriptomes, ORFeomes, or a combination thereof are compared to identify sequences that are differential sequences (e.g., that differ between them).
- the peptide sequences are identified by comparing tissues of interest.
- the peptide sequences are identified by comparing cells of interest.
- the peptide sequences are identified by comparing diseased versus healthy cells or tissues.
- the diseased cells or tissues are cancer cells or tissues.
- the diseased cells are derived from an individual with an autoimmune disorder.
- the peptides are derived from homologous sequences of genomes, proteomes, transcriptomes, ORFeomes, or any combination thereof, where two or more genomes, proteomes, transcriptomes, ORFeomes, or a combination thereof are compared to identify sequences that are homologous sequences.
- the peptides are derived from mutations in a sequence of interest, for example, all 9-mer peptides that can be generated from single nucleotide mutations in a polynucleotide sequence encoding an antigen or epitope.
- the peptide an overlapping peptide library, comprising overlapping peptides from a template sequence (e.g., in silico translated genome), wherein overlapping peptides of a set length are offset by a defined number of residues.
- selection of peptides comprises prioritizing peptides based on predicted binding affinity for a certain HLA type.
- selection of peptides for a library of the disclosure prioritizes HLA types or alleles based on prevalence in a population, e.g., a human population.
- the library comprises all k-mer peptides produced by transcription and translation of any polynucleotide sequence of interest, for example, in silico production of the transcription and translation products of both the forward and reverse strands of a genome or metagenome in all six reading frames.
- a library of the disclosure comprises all k-mer peptides that can be derived from in silico transcription and translation of a mammalian genome, for example, a mouse genome, a human genome, a patient genome, an autoimmune patient genome, or a cancer genome.
- a library of the disclosure comprises all k-mer peptides that can be derived from in silico transcription and translation of a microorganism genome, for example, a bacterial genome, a viral genome, a protozoan genome, a protist genome, a yeast genome, an archaeal genome, or a bacteriophage genome.
- a library of the disclosure comprises all k-mer peptides that can be derived from in silico transcription and translation of a pathogen genome, for example, a bacterial pathogen genome, a viral pathogen genome, a fungal pathogen genome, an opportunistic pathogen genome, a conditional pathogen genome, or a eukaryotic parasite genome.
- a library of the disclosure can be derived from a plant genome or a fungal genome.
- a library of the disclosure comprises k-mer peptides derived from in silico transcription and translation of a genome, wherein the genome is modified during in silico transcription and translation, for example, in silico mutated to produce k-mer peptides comprising mutations (e.g. substitutions, insertions, deletions).
- a library of the disclosure comprises all k-mer peptides that can be derived from in silico translation of an exome of interest, for example, a mammalian exome, a human exome, a mouse exome, a patient exome, an autoimmune patient exome, a cancer exome, a viral exome, a protozoan exome, a protist exome, a yeast exome, a pathogen exome, a eukaryotic parasite exome, a plant exome, or a fungal exome.
- an exome of interest for example, a mammalian exome, a human exome, a mouse exome, a patient exome, an autoimmune patient exome, a cancer exome, a viral exome, a protozoan exome, a protist exome, a yeast exome, a pathogen exome, a eukaryotic parasite exome, a plant exome, or a fungal exome.
- a library of the disclosure comprises k-mer peptides derived from in silico translation of a exome, wherein the exome is modified during in silico translation, for example, in silico mutated to produce k-mer peptides comprising mutations (e.g., substitutions, insertions, deletions).
- a library of the disclosure comprises all k-mer peptides that can be derived from in silico translation of a transcriptome of interest, for example, a mammalian transcriptome, a human transcriptome, a mouse transcriptome, a patient transcriptome, an autoimmune patient transcriptome, a cancer trans criptome, a microorganism trans criptome, a bacterial transcriptome, a viral trans criptome, a protozoan transcriptome, a protist transcriptome, a yeast transcriptome, an archaeal transcriptome, a bacteriophage trans criptome, a pathogen transcriptome, a eukaryotic parasite transcriptome, a plant transcriptome, a fungal trans criptome, a transcriptome derived from RNA sequencing, a microbiome transcriptome, or a transcriptome derived from metagenomic RNA-sequencing.
- a mammalian transcriptome for example, a mammalian transcriptome, a human transcriptome, a mouse transcript
- a library of the disclosure comprises k-mer peptides derived from in silico translation of a transcriptome, wherein the transcriptome is modified during in silico translation, for example, in silico mutated to produce k-mer peptides comprising mutations (e.g. substitutions, insertions, deletions).
- a library of the disclosure comprises all k-mer peptides that can be derived from a proteome of interest, for example, a mammalian proteome, a human proteome, a mouse proteome, a patient proteome, an autoimmune patient proteome, a cancer proteome, a microorganism proteome, a bacterial proteome, a viral proteome, a protozoan proteome, a protist proteome, a yeast proteome, an archaeal proteome, a bacteriophage proteome, a pathogen proteome, a eukaryotic parasite proteome, a plant proteome or a fungal proteome.
- a mammalian proteome for example, a mammalian proteome, a human proteome, a mouse proteome, a patient proteome, an autoimmune patient proteome, a cancer proteome, a microorganism proteome, a bacterial proteome, a viral proteome, a protozoan proteome, a pro
- a library of the disclosure comprises k-mer peptides derived from a proteome wherein the k-mer peptides are modified from the proteome sequence, for example, k-mer peptides comprising mutations (e.g., substitutions, insertions, deletions).
- a library of the disclosure comprises all k-mer peptides that can be derived from in silico translation of an ORFeome of interest, for example, a mammalian ORFeome, a human ORFeome, a mouse ORFeome, a patient ORFeome, an autoimmune patient ORFeome, a cancer ORFeome, a microorganism ORFeome, a bacterial ORFeome, a viral ORFeome, a protozoan ORFeome, a protist ORFeome, a yeast ORFeome, an archaeal ORFeome, a bacteriophage ORFeome, a pathogen ORFeome, a eukaryotic parasite ORFeome, a plant ORFeome or a fungal ORFeome, an ORFeome derived from nextgen sequencing, a microbiome ORFeome, or an ORFeome derived from metagenomic sequencing
- a library of the disclosure comprises k-mer peptides derived from in silico translation of an ORFeome, wherein the ORFeome is modified during in silico translation, for example, in silico mutated to produce k-mer peptides comprising mutations (e.g. substitutions, insertions, deletions).
- a library of the disclosure comprises all k-mer peptides that can be derived from in silico transcription and translation or translation of a group of genomes, proteomes, transcriptomes, ORFeomes, or any combination thereof.
- a library of the disclosure comprises all k-mer peptides that can be derived from in silico transcription and translation or translation of polynucleotide sequences from a group of samples, for example, clinical samples from a patient population, or a group of pathogen genomes.
- a library of the disclosure comprises all k-mer peptides that can be derived from in silico transcription and translation of a group of viral genomes, for example, the human virome.
- a library of the disclosure comprises all k-mer peptides that can be derived from in silico transcription and translation of a group of genomes, proteomes, transcriptomes, ORFeomes, or any combination thereof, wherein the source sequences are modified during in silico translation, for example, in silico mutated to produce k-mer peptides comprising mutations (e.g., substitutions, insertions, deletions).
- a library of the disclosure comprises all k-mer peptides that can be derived from a differential genome, proteome, transcriptome, ORFeome, or any combination thereof, where two or more genomes, proteomes, trans criptomes, ORFeomes, or a combination thereof are compared to identify sequences that are differential sequences (e.g., that differ between them), for example, differing in nucleotide sequence, amino acid sequence, nucleotide abundance, or protein abundance.
- differential sequences of a genome, proteome, trans criptome, or ORFeome are generated by comparing tissues of interest.
- differential sequences of a genome, proteome, transcriptome, or ORFeome are generated by comparing sequences from cells of interest (e.g., a healthy cell versus a cancer cell). In some embodiments, differential sequences of a genome, proteome, transcriptome, or ORFeome are generated by comparing sequences of organisms of interest. In some embodiments, differential sequences of a genome, proteome, transcriptome, or ORFeome can be generated by comparing subjects of interest (e.g., diseased versus healthy subjects).
- a library of the disclosure comprises all k-mer peptides that can be derived from homologous sequences of genomes, proteomes, transcriptomes, ORFeomes, or any combination thereof, where two or more genomes, proteomes, transcriptomes, ORFeomes, or a combination thereof are compared to identify sequences that are homologous sequences (e.g, that share a degree of homology), for example, homologous nucleotide sequences, homologous amino acid sequences, homologous nucleotide abundance, or homologous protein abundance.
- homologous sequences of genomes, proteomes, transcriptomes, or ORFeomes are generated by comparing tissues of interest.
- homologous sequences of genomes, proteomes, transcriptomes, or ORFeomes are generated by comparing sequences from cells of interest (e.g, a healthy cell versus a involved in autoimmunity cell (e.g., a cell that induces autoimmunity or a cell that is targeted during autoimmunity).
- homologous sequences of genomes, proteomes, transcriptomes, or ORFeomes are generated by comparing sequences of organisms of interest.
- homologous sequences of genomes, proteomes, transcriptomes, or ORFeomes are generated by comparing subjects of interest (e.g., diseased versus healthy subjects).
- a library of the disclosure comprises all k-mer peptides that can be derived from a polypeptide sequence of interest, for example, all possible 9-mer peptides covering the complete protein sequence of a viral protein.
- a library of the disclosure comprises k-mer peptides that can be generated from a polypeptide sequence of interest, wherein the polypeptide sequence of interest is modified, e.g. in silico mutated to produce k-mer peptides comprising mutations (e.g., substitutions, insertions, deletions).
- a library of the disclosure comprises all k-mer peptides that can be derived from mutations in a sequence of interest, for example, all 9-mer peptides that can be generated from single nucleotide mutations in a polynucleotide sequence encoding an antigen or epitope.
- a library of the disclosure comprises all 9-mer peptides that can be generated from two, three, four, five, six, seven, eight, or nine nucleotide mutations in a polynucleotide sequence encoding an antigen or epitope.
- a library of the disclosure comprises all k-mer peptides that can be derived from alanine substitutions, for example, alanine substitutions at any position in any of the sequences described herein (e.g., a protein, a group of proteins, a proteome, an in silico transcripted and translated genome).
- a library of the disclosure comprises a positional scanning library, wherein selected amino acid residues are sequentially substituted with all other natural amino acids.
- a library of the disclosure comprises a combinatorial positional scanning library, wherein selected amino acid residues are sequentially substituted with all other natural amino acids, two or more positions at a time.
- a library of the disclosure comprises an overlapping peptide library, comprising overlapping peptides from a template sequence (e.g., in silico translated genome), wherein overlapping peptides of a set length are offset by a defined number of residues.
- a library of the disclosure comprises a T cell truncated peptide library, wherein each replicate of the library comprises equimolar mixtures of peptides with truncations at one terminus (e.g., 8- mers, 9-mers, 10-mers and 11-mers that can be derived from C-terminal truncations of a nominal 11-mer).
- a library of the disclosure comprises a customized set of peptides, wherein the customized set of peptides are provided in a list.
- a genome, exome, trans criptome, proteome, or ORFeome of the disclosure is a viral genome, exome, transcriptome, proteome, or ORFeome.
- viruses include Adenovirus, Adeno-associated virus, Aichi virus, Australian bat lyssavirus, BK polyomavirus, Banna virus, Barmah forest virus, Bunyamwera virus, Bunyavirus La Crosse, Bunyavirus snowshoe hare, Cercopithecine herpesvirus, Chandipura virus, Chikungunya virus, Cosavirus A, Cowpox virus, Coxsackievirus, Crimean-Congo hemorrhagic fever virus, Cytomegalovirus (CMV), Dengue virus, Dhori virus, Dugbe virus, Duvenhage virus, Eastern equine encephalitis virus, Ebolavirus, Echovirus, Encephalomyocarditis virus, Epstein-Barr virus (EBV).
- Adenovirus Adeno-
- HTLV-1, HTLV -2, HTLV-3 Human torovirus, Influenza A virus, Influenza B virus, Influenza C virus, Isfahan virus, JC polyomavirus, Japanese encephalitis virus, Junin arenavirus, KI Polyomavirus, Kunjin virus, Lagos bat virus, Lake Victoria Marburgvirus, Langat virus, Lassa virus, Lordsdale virus, Louping ill virus, Lymphocytic choriomeningitis virus, Machupo virus, Mayaro virus, MERS coronavirus, Measles virus, Mengo encephalomyocarditis virus, Merkel cell polyomavirus, Mokola virus, Molluscum contagiosum virus, Monkeypox virus, Mumps virus, Murray valley encephalitis virus, New York virus, Nipah virus, Norovirus, Norwalk virus, O’nyong-nyong virus, Orf virus, Oropouche virus, Pichinde virus, Poliovirus, Punta toro phle
- louis encephalitis virus Tick-home powassan vims, Torque teno vims, Toscana virus, Uukuniemi vims, Vaccinia virus, Varicella-zoster virus, Variola virus, Venezuelan equine encephalitis vims, Vesicular stomatitis vims, Western equine encephalitis vims, WU polyomavirus, West Nile virus, Yaba monkey tumor virus, Yaba-like disease virus, Yellow fever virus, and Zika virus.
- a genome, exome, trans criptome, proteome, or ORFeome of the disclosure is a cancer genome, exome, trans criptome, proteome, or ORFeome.
- a library of the disclosure comprises known cancer neoepitopes.
- a library of the disclosure comprises all k-mer peptides that can be derived from known cancer antigenic proteins.
- a library of the disclosure comprises all k-mer peptides that can be derived from genes involved in epithelial- mesenchymal transition.
- a library of the disclosure comprises all k- mer peptides that can be derived from cancer implicated genes.
- a library of the disclosure comprises all k-mer peptides that can be derived from mutational cancer driver genes. In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from proto-oncogenes, oncogenes, or tumor suppressor genes. In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from proto-oncogenes, oncogenes, or tumor suppressor genes, wherein the k-mers comprise mutations as described herein (e.g, amino acid substitutions, alanine substitutions, positional scanning, combinatorial positional scanning etc.).
- Non-limiting examples of cancers include Acute Lymphoblastic Leukemia (ALL), Acute Myeloid Leukemia (AML), Adrenocortical Carcinoma, AIDS-Related Cancers, AIDS- Related Lymphoma, Anal Cancer, Appendix Cancer, Astrocytoma, Atypical Teratoid/Rhabdoid Tumor, Basal Cell Carcinoma, Bile Duct Cancer, Bladder Cancer, Bone Cancer, Brain Tumor, Breast Cancer, Bronchial Tumors, Burkitt Lymphoma, Carcinoid Tumor, Carcinoma of Unknown Primary, Cardiac Tumor, Central Nervous System cancer, Cervical Cancer, Cholangiocarcinoma, Chordoma, Chronic Lymphocytic Leukemia (CLL), Chronic Myelogenous Leukemia (CML), Chronic Myeloproliferative Neoplasms, Colorectal Cancer, Craniopharyngioma, Cutaneous T-Cell Lymphoma, Du
- a genome, exome, trans criptome, proteome, or ORFeome of the disclosure is an inflammatory or autoimmunogenic genome, exome, transcriptome, proteome, or ORFeome.
- a library of the disclosure comprises known inflammatory or autoimmunogenic neoepitopes or self-epitopes.
- a library of the disclosure comprises all k-mer peptides that can be derived from known inflammatory or autoimmunogenic antigenic proteins.
- a library of the disclosure comprises all k-mer peptides that can be derived from inflammatory or autoimmune-implicated genes.
- a library of the disclosure comprises all k-mer peptides that can be derived from mutation of inflammatory or autoimmune-related driver genes.
- Non-limiting examples of inflammatory or autoimmune diseases or conditions include Acute Disseminated Encephalomyelitis (ADEM); Acute necrotizing hemorrhagic leukoencephalitis; Addison’s disease; Adjuvant-induced arthritis; Agammaglobulinemia; Alopecia areata; Amyloidosis; Ankylosing spondylitis; Anti-GBM/Anti-TBM nephritis; Antiphospholipid syndrome (APS); Autoimmune angioedema; Autoimmune aplastic anemia; Autoimmune dysautonomia; Autoimmune gastric atrophy; Autoimmune hemolytic anemia; Autoimmune hepatitis; Autoimmune hyperlipidemia; Autoimmune immunodeficiency; Autoimmune inner ear disease (AIED); Autoimmune myocarditis; Autoimmune oophoritis; Autoimmune pancreatitis; Autoimmune retinopathy; Autoimmune
- a spacer linker is positioned between the MHCII binding peptide and the multimerization domain.
- the term “spacer linker” denotes a linear amino acid chain of natural and/or synthetic origin.
- the linker has the function to ensure that polypeptides conjugated to each other can perform their biological activity by allowing the polypeptides to fold correctly and to be presented properly.
- the spacer linker may contain repetitive amino acid sequences or sequences of naturally occurring polypeptides.
- the peptide linker has a length of from 2 to 50 amino acids.
- the peptide linker is between 3 and 30 amino acids, between 5 to 25 amino acids, between 5 to 20 amino acids, or between 10 and 20 amino acids.
- the spacer linker is a flexible linker, e.g., composed of a glycine- serine-rich sequence, such as the linker shown in SEQ ID NO: 6.
- the spacer linker is a rigid linker, e.g., composed of a proline-rich sequence, such as the linker shown in SEQ ID NO: 7.
- the spacer linker is a flexible-rigid linker, comprising both a flexible region (e.g., a glycine-serine-rich sequence) and a rigid region (e.g., a proline-rich sequence), such as the linker shown in SEQ ID NO: 8.
- the peptide linker is rich in glycine, glutamine, and/or serine residues. These residues are arranged e.g. in small repetitive units of up to five amino acids. This small repetitive unit may be repeated for one to five times. At the amino- and/or carboxy -terminal ends of the multimeric unit up to six additional arbitrary, naturally occurring amino acids may be added. Other synthetic peptidic linkers are composed of a single amino acid, which is repeated between 10 to 20 times and may comprise at the amino- and/or carboxy -terminal end up to six additional arbitrary, naturally occurring amino acids. All peptidic linkers can be encoded by a nucleic acid molecule and therefore can be recombinantly expressed. As the linkers are themselves peptides, the polypeptide connected by the linker are connected to the linker via a peptide bond that is formed between two amino acids.
- Suitable peptide linkers are well known in the art, and are disclosed in, e.g., US2010/0210511 US2010/0179094, and US2012/0094909, which are herein incorporated by reference in its entirety.
- Other linkers are provided, for example, in U.S. Pat. Nos. 5,525,491; Alfthan et al. (1995) PROTEIN ENG. 8:725-731; Shan etal. (1999) J. IMMUNOL. 162:6589- 6595; Newton et al. (1996) BIOCHEMISTRY 35:545-553; Megeed et al. (2006) BIOMACROMOLECULES 7:999-1004; and Perisic et al.
- the spacer linker is synthetic.
- synthetic with respect to a spacer linker includes peptides (or polypeptides) which comprise an amino acid sequence (which may or may not be naturally occurring) that is linked in a linear sequence of amino acids to a sequence (which may or may not be naturally occurring) to which it is not naturally linked in nature.
- the spacer linker may comprise non-naturally occurring polypeptides which are modified forms of naturally occurring polypeptides (e.g., comprising a mutation such as an addition, substitution or deletion) or which comprise a first amino acid sequence (which may or may not be naturally occurring).
- a spacer linker will be relatively non-immunogenic and not inhibit any non- covalent association among monomer subunits of a binding protein.
- the linker is a Gly-Ser polypeptide linker, i.e., a peptide that consists of glycine and serine residues.
- Another exemplary Gly-Ser polypeptide linker comprises the sequence SSSSGSSSSGSAA (SEQ ID NO: 73).
- Another linker comprises only glycine, e.g., Gs linkers (GGGGG; SEQ ID NO: 74).
- Gs linkers GGGGG; SEQ ID NO: 74.
- n l.
- n 2.
- n 3, i.e., Ser(Gly4Ser)3.
- n 4, i.e., Ser(Gly4Ser)4.
- n 5.
- n 6.
- n 7.
- n 8.
- exemplary linkers include GS linkers (i.e., (GS)n), GGSG linkers (i.e., (GGSG)n) (SEQ ID NO: 76), GSAT linkers (SEQ ID NO: 77), SEG linkers, GGS linkers (i.e., (GGSGGS)n) (SEQ ID NO: 78), wherein n is a positive integer (e.g., 1, 2, 3, 4, or 5), (Gly 4 Ser)4 (GGGGSGGGGSGGGGSGGGGS; SEQ ID NO: 79) and (GS) 2 AG 2 SGSG 3 S linkers (GSGSAGGSGSGGGS; SEQ ID NO: 80).
- linker Database is a database of inter-domain linkers in multi-functional enzymes which serve as potential linkers in novel multimeric fusion proteins (see, e.g., George et al. (2002) PROTEIN ENGINEERING 15:871-9).
- the spacer linker comprises an amino acid sequence selected from the group consisting of SEQ ID NOs: 6-8 and 72-80.
- a multimer composition of the disclosure is prepared by standard recombinant DNA techniques using a nucleic acid construct that encodes the MHCII-binding peptide operatively linked to the multimerization domain (MD), typically with sequences encoding a spacer linker positioned between the sequences encoding the peptide and the MD.
- a non-limiting representative nucleic acid construct encoding a multimer composition is shown schematically in FIG. 2A.
- the peptide is operatively linked to the N-terminus of the MD.
- the peptide is operatively linked to the C- terminus of the MD.
- Such regulatory elements include a transcriptional promoter, an optional operator sequence to control transcription, a sequence encoding suitable mRNA ribosomal binding site, and sequences that control the termination of transcription and translation.
- a transcriptional promoter an optional operator sequence to control transcription
- a sequence encoding suitable mRNA ribosomal binding site and sequences that control the termination of transcription and translation.
- the ability to replicate in a host, usually conferred by an origin of replication, and a selection gene to facilitate recognition of transformants is additionally incorporated.
- the nucleic acid construct is designed to be suitable for in vitro transcription/translation (IVTT).
- the nucleic acid is designed to be suitable for recombinant expression in a host cell.
- Appropriate cloning and expression vectors for use with bacterial, fungal, yeast, and mammalian cellular hosts can be found in CLONING VECTORS: A LABORATORY MANUAL (Elsevier, New York (1985)), the relevant disclosure of which is hereby incorporated by reference.
- the multimer composition is synthesized utilizing an in vitro transcription/translation (IVTT) system that can both transcribe, for example, a DNA construct into RNA, and then translate the RNA into a protein.
- IVTT can allow for protein production in a cell-free environment directly from a DNA or RNA template.
- An IVTT method used herein can be performed using, for example, a PCR product, a linear DNA plasmid, a circular DNA plasmid, or an mRNA template with a ribosome-binding site (RBS) sequence.
- transcription components can be added to the template including, for example, ribonucleotide triphosphates, and RNA polymerase.
- translation components can be added, which can be found in, for example, rabbit reticulocyte lysate, or wheat germ extract.
- the transcription and translation can occur during a single step, in which purified translation components found in, for example, rabbit reticulocyte lysate or wheat germ extract are added at the same time as adding the transcription components to the nucleic acid template.
- the nucleic acid sequence is incorporated into a vector, such as a plasmid vector, a viral vector or a non-viral vector.
- the vector is selected to be suitable for use in the intended host cell (i.e., the vector incudes all necessary transcriptional regulatory elements to allow for expression of the encoded multimer composition in the host cell).
- Suitable vectors, including transcriptional regulatory elements for use in various host cells, including mammalian host cells, are well established in the art.
- nucleic acid sequence encoding a protein described herein may be modified slightly in sequence and yet still encode its respective gene product.
- Nucleic acids encoding any of the various proteins or polypeptides described herein may be synthesized chemically or prepared through standard recombinant DNA techniques. Codon usage may be selected so as to improve expression in a cell. Such codon usage will depend on the cell type selected.
- the vector is designed for expression in a prokaryotic host cell (e.g, E. coll). In one embodiment, the vector is designed for expression in a eukaryotic host cell (e.g., yeast). In one embodiment, the vector is designed for expression in a mammalian host cell. In one embodiment, the mammalian host cells are human host cells. In one embodiment, the human host cells are human embryonic kidney (HEK) cells. In one embodiment, the HEK cells are 293 cells or are a 293 -derived HEK strain. Such HEK cells are commercially available in the art, a non-limiting example of which is the Expi293FTM cell line (Fisher ThermoScientific).
- HEK human embryonic kidney
- the mammalian host cell is a CHO cell line.
- the mammalian cell line such as a HEK or CHO cell line is stably transfected with the expression construct with a virus, for example a lentivirus or a retrovirus.
- the signal sequence used in the expression construct is derived from a mammalian protein.
- the transcriptional regulatory sequences used in the vector are selected for their effectiveness in mammalian host cell expression.
- Other expression systems include stable Drosophila cell transfectants and baculovirus infected insect-cells suitable for expression of proteins.
- the signal sequence is substituted by a prokaryotic signal sequence selected, for example, from the group of the alkaline phosphatase, penicillinase, 1 pp, or heat-stable enterotoxin II leaders.
- the native signal sequence may be substituted by, e.g., a yeast invertase leader, a factor leader (including Saccharomyces and Kluyveromyces alpha-factor leaders), or acid phosphatase leader, the C. albicans glucoamylase leader, or the signal sequence described in U.S. Pat. No. 5,631,144.
- yeast invertase leader e.g., a yeast invertase leader, a factor leader (including Saccharomyces and Kluyveromyces alpha-factor leaders), or acid phosphatase leader, the C. albicans glucoamylase leader, or the signal sequence described in U.S. Pat. No. 5,631,144.
- mammalian signal sequences as well as viral secretory leaders for example, the herpes simplex gD signal, are available.
- the DNA for such precursor regions may be ligated in reading frame to DNA encoding the protein.
- Both expression and cloning vectors contain a nucleic acid sequence that enables the vector to replicate in one or more selected host cells.
- this sequence is one that enables the vector to replicate independently of the host chromosomal DNA, and includes origins of replication or autonomously replicating sequences.
- origins of replication or autonomously replicating sequences are well known for a variety of bacteria, yeast, and viruses.
- the origin of replication from the plasmid pBR322 is suitable for most Gram-negative bacteria, the 2 micron plasmid origin is suitable for yeast, and various viral origins (SV40, polyoma, adenovirus, VSV or BPV) are useful for cloning vectors in mammalian cells.
- the origin of replication component is not needed for mammalian expression vectors (the SV40 origin may typically be used only because it contains the early promoter).
- Expression and cloning vectors may contain a selection gene, also termed a selectable marker.
- Typical selection genes encode proteins that (a) confer resistance to antibiotics or other toxins, e.g., ampicillin, neomycin, methotrexate, or tetracycline, (b) complement auxotrophic deficiencies, or (c) supply critical nutrients not available from complex media, e.g., the gene encoding D-alanine racemase for Bacilli.
- Expression and cloning vectors usually contain a promoter that is recognized by the host organism and is operably linked to the nucleic acid encoding the MHC multimer described herein.
- Promoters suitable for use with prokaryotic hosts include the phoA promoter, beta-lactamase and lactose promoter systems, alkaline phosphatase, a tryptophan (trp) promoter system, and hybrid promoters such as the tan promoter.
- trp tryptophan
- Other known bacterial promoters are suitable.
- Promoters for use in bacterial systems also will contain a Shine-Dai gamo (S.D.) sequence operably linked to the DNA encoding the protein described herein. Promoter sequences are known for eukaryotes.
- Virtually all eukaryotic genes have an AT-rich region located approximately 25 to 30 bases upstream from the site where transcription is initiated. Another sequence found 70 to 80 bases upstream from the start of transcription of many genes is a CNCAAT region where N may be any nucleotide. At the 3’ end of most eukaryotic genes is an AATAAA sequence that may be the signal for addition of the poly A tail to the 3’ end of the coding sequence. All of these sequences are suitably inserted into eukaryotic expression vectors.
- suitable promoting sequences for use with yeast hosts include the promoters for 3-phosphogly cerate kinase or other glycolytic enzymes, such as enolase, glyceraldehyde-3-phosphate dehydrogenase, hexokinase, pyruvate decarboxylase, phosphofructokinase, glucose-6-phosphate isomerase, 3-phosphogly cerate mutase, pyruvate kinase, triosephosphate isomerase, phosphoglucose isomerase, and glucokinase.
- 3-phosphogly cerate kinase or other glycolytic enzymes such as enolase, glyceraldehyde-3-phosphate dehydrogenase, hexokinase, pyruvate decarboxylase, phosphofructokinase, glucose-6-phosphate isomerase, 3-phosphogly cerate mutase, pyruvate
- Transcription from vectors in mammalian host cells can be controlled, for example, by promoters obtained from the genomes of viruses such as polyoma virus, fowlpox virus, adenovirus (such as Adenovirus 2), bovine papilloma virus, avian sarcoma virus, cytomegalovirus, a retrovirus, hepatitis-B virus and most preferably Simian Virus 40 (SV40), from heterologous mammalian promoters, e.g., the actin promoter or an immunoglobulin promoter, from heat-shock promoters, provided such promoters are compatible with the host cell systems.
- viruses such as polyoma virus, fowlpox virus, adenovirus (such as Adenovirus 2), bovine papilloma virus, avian sarcoma virus, cytomegalovirus, a retrovirus, hepatitis-B virus and most preferably Simian Virus 40 (SV
- Enhancer sequences are now known from mammalian genes (globin, elastase, albumin, a-fetoprotein, and insulin). Typically, however, one will use an enhancer from a eukaryotic cell virus.
- Examples include the SV40 enhancer on the late side of the replication origin (bp 100-270), the cytomegalovirus early promoter enhancer, the polyoma enhancer on the late side of the replication origin, and adenovirus enhancers. See also Yaniv (1982) NATURE 297:17-18 on enhancing elements for activation of eukaryotic promoters.
- the enhancer may be spliced into the vector at a position 5’ or 3’ to the peptide-encoding sequence, but is preferably located at a site 5’ from the promoter.
- Expression vectors used in eukaryotic host cells will also contain sequences necessary for the termination of transcription and for stabilizing the mRNA. Such sequences are commonly available from the 5’ and, occasionally 3’, untranslated regions of eukaryotic or viral DNAs or cDNAs. These regions contain nucleotide segments transcribed as polyadenylated fragments in the untranslated portion of mRNA encoding the protein described herein.
- One useful transcription termination component is the bovine growth hormone polyadenylation region. See WO 94/11026 and the expression vector disclosed therein.
- the expression construct comprises a signal sequence operatively linked upstream from the sequences encoding the multimer composition to thereby facilitate secretion of the multimer composition from a host cell.
- the signal sequence is from an Ig supergroup member.
- the signal sequence is an immunoglobulin chain signal sequence.
- the signal sequence is an Ig Kappa chain V-III region CLL signal peptide, e.g., having the sequence MEAPAQLLFLLLLWLPDTTG (SEQ ID NO: 88).
- Suitable signal sequences include a human CD4 signal peptide, e.g., having the sequence MNRGVPFRHLLLVLQLALLPAAT (SEQ ID NO: 89), a mouse Ig kappa chain V-III region signal peptide, e.g., having the sequence METDTLLLWVLLLWVPGSTG (SEQ ID NO: 90), a mouse H-2Kb signal peptide, e.g., having the sequence MVPCTLLLLLAAALAPTQTRA (SEQ ID NO: 91), a human serum albumin signal peptide, e.g., having the sequence MKWVTFISLLFLFSSAYS (SEQ ID NO: 92), a human IL-2 signal peptide, e.g., having the sequence MYRMQLLSCIALSLALVTNS (SEQ ID NO: 93), a human HLA-A*02:01 signal peptide, e g., having the sequence MAVMAPRTLLLLLSGALALTQTWA (
- the expression construct is introduced into the host cell using a method appropriate to the host cell, as will be apparent to one of skill in the art.
- a variety of methods for introducing nucleic acids into host cells are known in the art, including, but not limited to, electroporation; transfection employing calcium chloride, rubidium chloride, calcium phosphate, DEAE-dextran, or other substances; microprojectile bombardment; lipofection; and infection (where the vector is an infectious agent).
- Suitable host cells include prokaryotes, yeast, mammalian cells, or bacterial cells.
- Suitable bacteria include gram negative or gram positive organisms, for example, E. coli or Bacillus spp. Yeast, preferably from the Saccharomyces species, such as 5. cerevisiae, may also be used for production of polypeptides.
- Various mammalian or insect cell culture systems can also be employed to express recombinant proteins. Baculovirus systems for production of heterologous proteins in insect cells are reviewed by Luckow et al. ((1988) BIO/TECHNOLOGY 6:47).
- suitable mammalian host cell lines include endothelial cells, COS-7 monkey kidney cells, CV-1, L cells, C127, 3T3, Chinese hamster ovary (CHO), human embryonic kidney cells, HeLa, 293, 293T, and BHK cell lines.
- Purified polypeptides are prepared by culturing suitable host/vector systems to express the recombinant proteins. For many applications, the small size of many of the polypeptides described herein would make expression in E. coli as the preferred method for expression. The protein is then purified from culture media or cell extracts.
- the host cells used to produce the proteins of this invention may be cultured in a variety of media.
- Commercially available media such as Ham’s F10 (Sigma), Minimal Essential Medium ((MEM), (Sigma), RPMI-1640 (Sigma), and Dulbecco’s Modified Eagle’s Medium ((DMEM), Sigma)) are suitable for culturing the host cells.
- MEM Minimal Essential Medium
- RPMI-1640 Sigma
- DMEM Dulbecco’s Modified Eagle’s Medium
- 4,767,704, 4,657,866, 4,927,762, 4,560,655, 5,122,469, 6,048,728, 5,672,502, or U.S. Pat. No. RE 30,985 may be used as culture media for the host cells. Any of these media may be supplemented as necessary with hormones and/or other growth factors (such as insulin, transferrin, or epidermal growth factor), salts (such as sodium chloride, calcium, magnesium, and phosphate), buffers (such as HEPES), nucleotides (such as adenosine and thymidine), antibiotics (such as Gentamycin drug), trace elements (defined as inorganic compounds usually present at final concentrations in the micromolar range), and glucose or an equivalent energy source. Any other necessary supplements may also be included at appropriate concentrations that would be known to those skilled in the art.
- the culture conditions, such as temperature, pH, and the like, are those previously used with the host cell selected for expression, and will be apparent to the ordinarily skilled artisan.
- Proteins described herein can also be produced using cell-free translation systems.
- the nucleic acids encoding the polypeptide must be modified to allow in vitro transcription to produce mRNA and to allow cell-free translation of the mRNA in the particular cell-free system being utilized (eukaryotic such as a mammalian or yeast cell-free translation system or prokaryotic such as a bacterial cell-free translation system).
- Proteins described herein can also be produced by chemical synthesis (e.g, by the methods described in SOLID PHASE PEPTIDE SYNTHESIS, 2nd Edition, The Pierce Chemical Co., Rockford, Ill. (1984)). Modifications to the protein can also be produced by chemical synthesis.
- the proteins of the present invention can be purified by isolation/purification methods for proteins generally known in the field of protein chemistry.
- Non-limiting examples include extraction, recrystallization, salting out (e.g., with ammonium sulfate or sodium sulfate), centrifugation, dialysis, ultrafiltration, adsorption chromatography, ion exchange chromatography, hydrophobic chromatography, normal phase chromatography, reversed- phase chromatography, get filtration, gel permeation chromatography, affinity chromatography, electrophoresis, countercurrent distribution or any combinations of these.
- polypeptides may be exchanged into different buffers and/or concentrated by any of a variety of methods known to the art, including, but not limited to, filtration and dialysis.
- the purified polypeptide is preferably at least 85% pure, or preferably at least 95% pure, and most preferably at least 98% pure. Regardless of the exact numerical value of the purity, the polypeptide is sufficiently pure for its intended use.
- the expression construct includes at least one tag sequence, most typically as at the C-terminal end of the coding region, although inclusion of a tag at the N-terminal end (alternative to or in addition to the C-terminal end) is also encompassed. Suitable tag sequences are known in the art and described further herein.
- a protein tag sequence that may be useful e.g., for labeling the multimer composition and/or for purifying the composition. Examples of protein tags include, but are not limited to, a histidine tag (6x His), a FLAG tag, a myc tag, an HA tag, a GST tag, and combinations thereof.
- Non-limiting examples of suitable tags include FLAG tags (e.g., having the amino acid sequence shown in SEQ ID NO: 96), 6xHis tags (e.g., having the amino acid sequence shown in SEQ ID NO: 97), V5 tags (e.g., having the amino acid sequence shown in SEQ ID NO: 98), Strep-Tags (e.g., having the amino acid sequence shown in SEQ ID NO: 99), Protein C tags (e.g., having the amino acid sequence shown in SEQ ID NO: 100) and/or myc tags (e.g., having the amino acid sequence shown in SEQ ID NO: 101).
- FLAG tags e.g., having the amino acid sequence shown in SEQ ID NO: 96
- 6xHis tags e.g., having the amino acid sequence shown in SEQ ID NO: 97
- V5 tags e.g., having the amino acid sequence shown in SEQ ID NO: 98
- Strep-Tags e.g., having the amino acid sequence shown
- Additional tags suitable for use in the methods and compositions provided herein include affinity tags, including but not limited to enzymes, protein domains, or small polypeptides which bind with high specificity to a range of substrates, such as carbohydrates, small biomolecules, metal chelates, antibodies, etc. to allow rapid and efficient purification of proteins.
- Solubility tags enhance proper folding and solubility of a protein and are frequently used in tandem with affinity tags. Sequences encoding such a tag(s) can be incorporated into an expression construct of the disclosure, such as at the C-terminus or N-terminus of the peptide- multimer-encoding regions to thereby incorporate a detectable tag into the expressed polypeptide.
- a multimer composition of the disclosure is produced by covalent conjugation of the MHCII-binding peptide to the multimerization domain.
- a multimer composition is produced by covalent conjugation of the peptide to the N-terminus of the multimerization domain.
- a multimer composition is produced by covalent conjugation of the peptide to the C-terminus of the multimerization domain.
- the peptide and multimerization domain (MD) components can be prepared either recombinantly or chemically.
- peptides can be generated according to synthesis methods known in the art, or synthetically produced by a commercial vendor or using a peptide synthesizer according to manufacturer’s instructions.
- peptides can be made by in silico production methods.
- peptides can be synthesized via chemical methods, for example, tea bag synthesis, digital photolithography, pin synthesis, and SPOT synthesis.
- an array of peptides can be generated via SPOT synthesis, where amino acid chains are built on a cellulose membrane by repeated cycles of adding amino acids, and cleaving side-chain protection groups.
- peptides and/or MDs can be expressed using recombinant DNA technology, for example, introducing an expression construct into bacterial cells, insect cells, or mammalian cells, and purifying the recombinant protein from cell extracts, e.g., as described above.
- the chemical conjugation is mediated by cysteine bioconjugation of the MHCII-binding peptide (e.g., including a spacer linker) to the multimerization domain.
- the cysteine bioconjugation is mediated by cysteine alkylation.
- the cysteine bioconjugation is mediated by cysteine oxidation.
- the cysteine bioconjugation is mediated by a desulfurization reaction.
- cysteine bioconjugation is mediated by iodoacetamide.
- the cysteine bioconjugation is mediated by maleimide.
- the multimer compositions are produced by chemical modification of amino acids other than cysteine, including but not limited to lysine, tyrosine, arginine, glutamate, aspartate, serine, threonine, methionine, histidine and tryptophan sidechains, as well as N-terminal amines or C-terminal carboxyls, as previously described (Basle et al. (2010) M CHEM BIOL. 17(3):213-27; Hu e/ a/. (2016) CHEM SOC REV. 45(6): 1691-719; Lin et al. (2017) SCIENCE 355(6325): 597-602).
- amino acids other than cysteine including but not limited to lysine, tyrosine, arginine, glutamate, aspartate, serine, threonine, methionine, histidine and tryptophan sidechains, as well as N-terminal amines or C-terminal carboxyls, as previously described
- the multimer compositions are produced by native chemical ligation (NCL), wherein each peptide (or peptide-spacer linker) comprises a C-terminal thioester, and each subunit of the multimerization domain comprises an N-terminal cysteine residue, or functional equivalent thereof, wherein the reaction between the cysteine sidechain and the thioester irreversibly forms a native peptide bond, thus ligating the peptides to the multimerization domain.
- NCL native chemical ligation
- [3- and/or y-thio amino acids are incorporated into the peptides (or peptide-spacer linker).
- [3- and/or y-thio amino acids replace the cysteine-like residue at an N-terminal position of each subunit of the multimerization domain, e.g., to provide a reactive thiol for trans-thioesterifi cation.
- Desulfurization protocols can then produce the desired native side-chain.
- NCL is performed at an alanine residue. In other embodiments, NCL is performed at phenylalanine (Crich & Banerjee, 2007), valine (Chen et al.
- the multimer compositions are produced by bioorthogonal conjugation between the conjugation moiety at the C-terminus of each peptide (or peptide- spacer linker) and the conjugation moiety at the N-terminus of each subunit of the multimerization domain.
- the bioorthongonal conjugation is mediated by “click chemistry.” (see, e.g., Kolb, Finn and Sharpless (2001) ANGEWANDTE CHEMIE INTERNATIONAL EDITION 40: 2004-2021).
- Conjugation moieties suitable for click chemistry, reaction conditions, and associated methods are available in the art (e.g., Kolb et al.
- a click chemistry moiety may comprise or consist of a terminal alkyne, azide, strained alkyne, diene, dieneophile, alkoxyamine, carbonyl, phosphine, hydrazide, thiol, or alkene moiety.
- the azide is a copperchelating azide.
- the copper-chelating azide is a picolyl azide, such as Gly-Gly-Gly-(PEG)4-Picolyl-Azide.
- Reagents for use in click chemistry reactions are commercially available, such as from Click Chemistry Tools (Scottsdale, AZ) or GenScript (Piscataway, NJ).
- the click chemistry moieties of the components have to be reactive with each other, for example, in that the reactive group of one of the click chemistry moiety of each peptide (or peptide-spacer linker) reacts with the reactive group of the second click chemistry moiety on a subunit of the multimerization domain to form a covalent bond.
- each peptide (or peptide-spacer linker) conjugation moiety can be covalently conjugated under click chemistry reaction conditions to the conjugation moiety of each subunit of the multimerization domain.
- a sortase-mediated conjugation is used to install a first click chemistry moiety at the C-terminus of each peptide (or peptide-spacer linker), and a second click chemistry moiety reaction to each subunit of the multimerization domain.
- two or more peptides (or peptide- spacer linkers) containing the first click chemistry moiety are conjugated to the second click chemistry moiety at the C-terminus of each subunit of the multimerization domain under click chemistry conditions.
- Methods of attaching click chemistry moieties utilizing sortase are described, for example, in W02013/00355, the entire contents of which is hereby incorporated by reference.
- an intein-mediated conjugation is used to install a first click chemistry moiety at the C-terminus of each peptide (or peptide-spacer linker), and a second click chemistry moiety reaction to each subunit of the multimerization domain.
- the methods of click chemistry mediated covalent conjugation of the peptide (or peptide-spacer linker) to the multimerization domain provided herein comprise native chemical ligation of C-terminal thioesters with -amino thiols (Xiao J, Tolbert TJ (2009) ORG LETT. 11(18):4144-7).
- the click chemistry used to produce the multimer composition comprises 1,3-dipolar cycloaddition (e.g., the Cu(I)-catalyzed stepwise variant, often referred to simply as the “click reaction”; see, e.g., Tomoe et al.
- Copper and ruthenium are the commonly used catalysts in the reaction.
- the use of copper as a catalyst results in the formation of 1,4-regioisomer whereas ruthenium results in formation of the 1,5-regioisomer.
- the peptides are ligated to an alkynated peptide by expressed protein ligation (EPL) and then conjugated to an azide- labeled multimerization domain by Cu(I)-catalyzed terminal azide-alkyne cycloaddition (CuAAC).
- EPL expressed protein ligation
- CuAAC Cu(I)-catalyzed terminal azide-alkyne cycloaddition
- the click chemistry conjugation comprises a cycloaddition reaction, such as the Diels-Alder reaction.
- the MHCII-binding peptide and multimerization domain are conjugated by azide-alkyne 1,3-dipolar cycloaddition (“click chemistry).
- the cycloaddition is promoted by the presence of Cu(I)-catalyzed cycloaddition (CuAAC).
- the click chemistry conjugation comprises nucleophilic addition to small strained rings like epoxides and aziridines.
- the cycloaddition is promoted by strained cyclooctyne systems, for example, as described in Agard NJ, Prescher JA, Bertozzi CR (2004) J AM CHEM SOC. 126(46): 15046-7.
- the click chemistry conjugation comprises nucleophilic addition to activated carbonyl groups.
- the conjugation of the peptide (or peptide-spacer linker) and multimerization domain occurs by a bioorthogonal reaction.
- the MHC and multimerization domain are conjugated by inverse-electron demand Diels-Alder reactions between strained dienophiles and tetrazine dienes, for example, as described in Blackman ML, Royzen M, Fox JM (2008) J AM CHEM SOC. 130(41): 13518-9; and Devaraj NK, Weissleder R, Hilderbrand SA (2008) BlOCONJUG CHEM. 19(12):2297-9).
- the dienophile is a trans-cyclooctene.
- the dienophile is a norbomene.
- conjugation between the peptide (or peptide-spacer linker) and the multimerization domain is mediated by a cysteine transpeptidase.
- the cysteine transpeptidase is a sortase, or enzymatically active fragment thereof.
- sortase enzymes have been described and are commercially available (e.g., Antos et al. (2016) CURR. OPIN. STRUCT. BIOL. 38:111-118).
- Sortases recognize and cleave an amino acid motif, referred to as a “sortag”, to produce a peptide bond between the acyl donor and acceptor site on two polypeptides, resulting in the ligation of different polypeptides which contain N- or C- terminal sortags.
- sortases join a C-terminal LPETGG recognition motif (SEQ ID NO: 133) to an N-terminal GGG (oligoglycine) motif.
- the recognition motif is added to the C-terminus of each peptide (or peptide-spacer linker), and an oligo-glycine motif is added to the N- terminus of each of the subunits of the mutimerization domain.
- the recognition motif is added to the C-terminus of each of the subunits of the multimerization domain and the oligo-glycine motif is added to the N-terminus of each peptide (or peptide-spacer linker).
- sortase to the mixture of peptide (or peptide-spacer linker) and multimerization domains, the polypeptides are covalently linked through a native peptide bond.
- the peptide (or peptide-spacer linker) and/or multimerization domain are expressed in frame with the sortags.
- additional tags may be included, for example, a 6x-His tag (Sinisi et al. (2012) BlOCONJUG. CHEM 23: 1119-1126), a nucleophilic fluorochrome (Nair et al. (2013) IMMUN. INFLAMM. DIS. 1:3-13), and/or a FLAG tag (Greineder et al. (2016) BlOCONJUG. CHEM. 29:56-66).
- the sortag contains a modified amino acid suitable for chemical conjugation between the MHC monomers and the mutimerization domain. In some embodiments, the sortag contains a C-terminal azidolysine residue to enable oriented clickclick chemistry conjugation as described herein.
- the peptide and/or multimerization domains comprise a linker between the polypeptide and the sortag.
- each peptide and each subunit of the multimerization domain comprises a sortag with a linker.
- Suitable linkers have been described herein and in the art, for example, in Greineder et al. (2016) BlOCONJUG. CHEM. 29:56-66.
- the linker is a flexible, rigid, or semi-rigid linker, e.g., a linker having an amino acid sequence shown in any one of SEQ ID NOs: 6-8 and 72- 80.
- the sortag contains a fluorophore-modified lysine residue to facilitate measurement of reaction progression and efficiency.
- the sortase is Ca2+ dependent. In some embodiments, the sortase is Ca2+ independent.
- the sortag comprises the amino acid sequence LPXTG (SEQ ID NO: 102), wherein X is any amino acid, and the sortase cleaves between the threonine and glycine backbone within the motif.
- the sortase recognizes a sortag comprising an amino acid sequence selected from IPKTG (SEQ ID NO: 103), MPXTG (SEQ ID NO: 104), wherein X is any amino acid, LAETG (SEQ ID NO: 105), LPXAG (SEQ ID NO: 106), wherein X is any amino acid, LPESG (SEQ ID NO: 107), LPELG (SEQ ID NO: 108) or LPEVG (SEQ ID NO: 109).
- the sortase is a SrtAstaph mutant.
- the SrtAstaph mutant is F40, and the recognition motif is XPKTG (SEQ ID NO: 110), wherein X is any amino acid (Piotukh et al. (2011) J. AM. CHEM. SOC. 133:17536-17539).
- the SrtAstaph mutant is F40 and the recognition motif is APKTG (SEQ ID NO: 111), DPKTG (SEQ ID NO: 112) or SPKTG (SEQ ID NO: 113).
- the SrtAstaph mutant is SrtAstaph pentamutant and the recognition motif is LPXTG (SEQ ID NO: 114), wherein X is any amino acid, LPEXG, (SEQ ID NO: 115), wherein X is any amino acid, or LAETG (SEQ ID NO: 116).
- the mutant is SrtAstaph pentamutant and the recognition motif is LPEAG (SEQ ID NO: 117), LPECG (SEQ ID NO: 118) or LPESG (SEQ ID NO: 119).
- the SrtAstaph mutant is 2A-9 and the recognition motif is LAETG (SEQ ID NO: 116).
- the sortase is a soluble fragment of the wild-type sortase. In some embodiments, the sortase is a soluble fragment of a modified sortase A (Mao H, Hart SA, Schink A, Pollok BA (2004) J AM CHEM SOC. 126(9):2670-l A).
- the sortase is a variant or homolog of 5. aureus sortase A (Antos JM, Truttmann MC, Ploegh HL (2016) CURR OPIN STRUCT BIOL. 38: 111-8; Dorr BM, Ham HO, An C, Chaikof EL, Liu DR (2014) PROC NATL ACAD SCI USA 111(37): 13343-8; Glasgow JE, Salit ML, Cochran JR (2016) J AM CHEM SOC. 138(24):7496-9). [0197] Methods of conjugation of sortags into proteins have also been described.
- the aminoglycine peptide fragment generated by the sortase reaction is removed by dialysis or centrifugation, e.g., while the reaction is proceeding (Freiburger L, Stanford M, Hennig J, Li J, Zou P, Sattler M (2015) J BlOMOL NMR 63(1): 1- 8).
- affinity immobilization strategies or flow-based platforms are used for the selective removal of reaction components (Policarpo RL, Kang H, Liao X, Rabideau AE, Simon MD, Pentelute BL (2014) ANGEW CHEM INT ED ENGL 53(35): 9203-8).
- the equilibrium of the reaction can be controlled by ligation product or by-product deactivation.
- the reaction is controlled by ligation of a WTWTW (SEQ ID NO: 122) motif added to the donor and acceptor as described in Yamamura Y, Hirakawa H, Yamaguchi S, Nagamune T (2011) CHEM COMMUN (CAMB) 47(16): 4742 -4).
- by-products are deactivated by chemical modification of the acyl donor glycine as described, for example, in Liu F, Luo EY, Flora DB, Mezo AR (2014) J ORG CHEM 79(2):487-92; and Williamson DJ, Webb ME, Turnbull WB (2014) NAT PROTOC 9(2):253-62).
- Inteins are naturally occurring, self-splicing protein subdomains that are capable of excising out their own protein subdomain from a larger protein structure while simultaneously joining the two formerly flanking peptide regions (“exteins”) together to form a mature host protein. Intein-based methods of protein modification and ligation have been developed.
- An intein is an internal protein sequence capable of catalyzing a protein splicing reaction that excises the intein sequence from a precursor protein and joins the flanking sequences (N- and C-exteins) with a peptide bond.
- split intein refers to any intein in which one or more peptide bond breaks exists between the N-terminal intein segment and the C-terminal intein segment such that the N-terminal and C-terminal intein segments become separate molecules that cannon-covalently reassociate, or reconstitute, into an intein that is functional for splicing or cleaving reactions.
- Any catalytically active intein, or fragment thereof, may be used to derive a split intein for usein the systems and methods disclosed herein.
- the split intein may be derived from a eukaryotic intein.
- the split intein may be derived from a bacterial intein. In another aspect, the split intein may be derived from an archaeal intein. Preferably, the split intein so-derived will possess only the amino acid sequences essential for catalyzing splicing reactions.
- N-terminal intein segment refers to any intein sequence that comprises an N-terminal amino acid sequence that is functional for splicing and/or cleaving reactions when combined with a corresponding C-terminal intein segment.
- An N-terminal intein segment thus also comprises a sequence that is spliced out when splicing occurs.
- An N-terminal intein segment can comprise a sequence that is a modification of the N-terminal portion of a naturally occurring (native) intein sequence.
- an N-terminal intein segment can comprise additional amino acid residues and/or mutated residues so long as the inclusion of such additional and/or mutated residues does not render the intein non-functional for splicing or cleaving.
- the inclusion of the additional and/or mutated residues improves or enhances the splicing activity and/or controllability of the intein.
- Non-intein residues can also be genetically fused to intein segments to provide additional functionality, such as the ability to be affinity purified or to be covalently immobilized.
- the “C-terminal intein segment” refers to any intein sequence that comprises a C-terminal amino acid sequence that is functional for splicing or cleaving reactions when combined with a corresponding N-terminal intein segment.
- the C-terminal intein segment comprises a sequence that is spliced out when splicing occurs.
- the C-terminal intein segment is cleaved from a peptide sequence fused to its C-terminus.
- a C-terminal intein segment can comprise a sequence that is a modification of the C-terminal portion of a naturally occurring (native) intein sequence.
- a C terminal intein segment can comprise additional amino acid residues and/or mutated residues so long as the inclusion of such additional and/or mutated residues does not render the C- terminal intein segment non-functional for splicing or cleaving.
- Expressed protein ligation refers to a native chemical ligation between a recombinant protein with a C-terminal thioester and a second agent with an N-terminal cysteine.
- the C-terminal thioester can readily be introduced onto any recombinant protein (i.e. , the targeting ligand) through the use of auto-processing, also known as protein-splicing, mediated by an intein (intervening protein).
- Inteins are proteins that can excise themselves from a larger precursor polypeptide chain, utilizing a process that results in the formation of a native peptide bond between the flanking extein (external protein) fragments.
- thiols e.g., 2- mercaptoethanesulfonic acid, MESNA
- MESNA 2- mercaptoethanesulfonic acid
- EPL operates in a site-specific manner, and the reaction is known to be very efficient if both functional groups are in high concentrations, (reviewed in Elias et al. Small 6:2460-2468).
- MHCII-binding peptides are ligated to an alkynated peptide by expressed protein ligation (EPL) and then conjugated to an azide-labeled multimerization domain by Cu(I)-catalyzed terminal azidealkyne cycloaddition (CuAAC).
- EPL expressed protein ligation
- CuAAC Cu(I)-catalyzed terminal azidealkyne cycloaddition
- the peptides are conjugated to the multimerization domain by an intein peptide tag.
- the peptide (or peptides-spacer linker) comprises a C-terminal thioester
- the multimerization domain comprises an N-extein fused to a modified intein lacking the ability to perform trans- esterification and /ram-esterification occurs by the addition of exogenous thiol.
- inteins have now been described including, but not limited to MxeGyrA (Frutos et al. (2010); Southworth et al. (1999); SspDnaE (Shah et al. (2012); Wu et al. (1998); NpuDnaE (Shah et al. (2012); Vila-Perello et al. (2013); AvaDnaE (David etal. (2015); Shah et al. (2012); Cfa (consensus DnaE split intein) (Stevens et al. (2016)); gp41-l and gp41-8 (Carvajal -Vallejos etal.
- NrdJ-1 Carvajal -Vallejos et al. (2012)
- IMPDH-1 Carvajal -Vallejos et al. (2012)
- AceL-TerL Thiel et al. (2014). The properties and use of these inteins are summarized in Table 1.
- the intein is the 198-residue gyrase A intein from Mycobacterium xenopi (Mxe GyrA) (Southworth MW, Amaya K, Evans TC, Xu MQ, Perler FB (1999) BIOTECHNIQUES 27(l):110-4, 116, 118-20).
- the intein is from cyanobacterium Synechocystis sp. strain PCC6803 (Ssp).
- the intein is a split intein pair.
- the split intein pair is an orthogonal split intein pair (Carvajal -Vallejos P, Pallisse R, Mootz HD, Schmidt SR (2012) J BIOL CHEM 287(34):28686-96; Shah NH, Vila-Perello M, Muir TW (2011) ANGEW CHEM INT ED ENGL 50(29):6511-5).
- the split intein pair is an artificially split intein pair that are as short as six or eleven residues (Appleby JH, Zhou K, Volkmann G, Liu XQ (2009) J BIOL CHEM 284(10):6194-9; Ludwig C, Pfeiff M, Linne U, Mootz HD (2006) ANGEW CHEM INT ED ENGL 45(31):5218-21).
- the intein is a DnaE intein.
- the DnaE intein is from Nostoc punctiforme (Npu).
- the intein is the gp41-l intein.
- the intein is the gp41-8 intein.
- the intein is the IMPDH-1 intein.
- the intein is the NrdJ Intein.
- the split intein pair is AceL-TerL (Thiel IV, Volkmann G, Pietrokovski S, Mootz HD (2014) ANGEW CHEM INT ED ENGL 53(5): 1306-10).
- the intein comprises consensus split intein sequence (Cfa) (Stevens AJ, Brown ZZ, Shah NH, Sekar G, Cowbum D, Muir TW (2016) JOURNAL OF THE AMERICAN CHEMICAL SOCIETY 138(7):2162-2165).
- the conjugation of the peptide (or peptide-spacer linker) and multimerization domain is mediated enzymatically.
- the enzyme is formylglycine generating enzyme (FGE) that recognizes the CXPXR amino acid sequence motif and converts the cysteine residue to formylglycine, thus introducing an aldehyde functional group (Wu P, Shui W, Carlson BL, Hu N, Rabuka D, Lee J, Bertozzi CR (2009) PROC NATL ACAD SCI USA 106(9):3000-5), which is subjected to bio-orthogonal transformations such as oximation and Hydrazino-Pictet-Spengler reactions (Agarwal P, Kudirka R, Albers AE, Barfield RM, de Hart GW, Drake PM, Jones LC, Rabuka D (2013) BIOCONJUG CHEM 24(6): 846-51; Dirksen A, Dawson PE (2008) BlO
- FGE formylglycine
- Site-specific bioconjugation strategies offer many possibilities for directed protein modifications.
- formylglycine-generating enzymes allow to posttranslationally introduce the amino acid Ca-formylglycine (FGly) into recombinant proteins, starting from cysteine or serine residues within distinct consensus motifs.
- the aldehyde-bearing FGly-residue displays orthogonal reactivity to all other natural amino acids and can, therefore, be used for site-specific labeling reactions on protein scaffolds.
- Formylglycine generating enzyme recognizes a pentapeptide consensus sequence, CxPxR, and it specifically oxidizes the cysteine in this sequence to an unusual aldehyde-bearing formylglyine.
- the FGE recognition sequence, or aldehyde tag can be inserted into heterologous recombinant proteins produced in either prokaryotic or eukaryotic expression systems.
- cysteine to formylglycine is accomplished by co- overexpression of FGE, either transiently or as a stable cell line, and the resulting aldehyde can be selectively reacted with a-nucleophiles to generate a site-selectively modified bioconjugate (Rabuka et al. (2012) NAT PROTOC 7(6): 1052-1067).
- the enzyme is lipoic acid ligase, an enzyme that modifies a lysine side-chain within the 13-residue target sequence (Uttamapinant C, White KA, Baruah H, Thompson S, Fernandez-Suarez M, Puthenveetil S, Ting AY (2010) PROC NATL ACAD SCI USA 107(24): 10914-9) to introduce bio-orthogonal groups, including azides, aryl aldehydes and hydrazines, p-iodophenyl derivatives, norbomenes, and trans-cyclooctenes (reviewed in Debelouchina et al. (2017) Q. REV BIOPHYS. 50 e7. doi:10.1017/S0033583517000021).
- the enzyme is biotin ligase, famesyltransferase, transglutaminase or N-myristoyltransferase (reviewed in Rashidian M, Dozier JK, Distefano MD (2013) BIOCONJUG CHEM 24(8): 1277-94).
- the multimer composition (comprising peptide-MD) is “loaded” with MHCII molecules, e.g., soluble MHCII dimers.
- the soluble MHCII molecules can be “empty”, i.e., not loaded with a peptide in the peptide binding groove (e.g., certain MHCII molecules may be sufficiently stable as empty soluble molecules to enable loading onto the peptide-MD compositions).
- the MHCII molecules are prepared as soluble molecules with a cleavable placeholder peptide in the antigen-binding groove (referred to herein as p*MHC) to enhance the stability of the MHCII molecules.
- FIG. 1 A representative schematic diagram of a soluble MHCII molecule loaded with a cleavable placeholder peptide is shown in FIG. 1 and preparation thereof is described in detail in Example 2. The components of the MHCII molecules are described in further detail below.
- MHC class II molecules are heterodimers composed of an a chain and a chain, both of which are encoded by the MHC.
- the alpha chain is comprised of al and a2 domains.
- the beta chain is comprised of P 1 and 2 domains.
- the al and pi domains of the chains interact noncovalently to form a membrane-distal peptide-binding domain, whereas the a2 and P2 domains form membrane-proximal immunoglobulin-like domains.
- the antigen binding groove, where a peptide epitope binds is made up of two a-helices and a P-sheet. Since the antigen binding groove of MHC class II molecules is open at both ends, the groove can accommodate longer peptide epitopes than MHC class I molecules.
- Peptide epitopes presented by MHC class II molecules typically are about 15-24 amino acid residues in length.
- the MHCII molecule can suitably be a vertebrate MHCII molecule such as a human, a mouse, a rat, a porcine, a bovine or an avian MHCII molecule.
- the multimeric MHCII multimers described herein, the MHC molecule is a human MHC class II protein: HLA-DR, HLA-DQ, HLA-DX, HLA-DO, HLA- DZ, and HLA-DP.
- the amino acid sequences of the MHCII a and P chains from a variety of vertebrate species, including humans, are known in the art and publicly available.
- the human MHCII molecule is of an allotype selected from the group consisting of DRBl*0101 (see, e.g., Cameron et al. (2002) J. IMMUNOL. METHODS 268:51-69; Cunliffe et al. (2002) EUR. J. IMMUNOL. 32:3366-3375; Washington et al. (2003) J.
- IMMUNOL. 171:3163-3169 DRBl*1501 (see, e.g., Day et al. (2003) J. CLIN. INVEST 112:831-842), DRB5*0101 (see, e.g., Day et al., ibid), DRBl*0301 (see, e.g., Bronke et al. (2005) HUM. IMMUNOL. 66:950-961), DRBl*0401 (see, e.g., Meyer etal. (2000) PNAS 97:11433-11438; Novak et al. (1999) J. CLIN. INVEST 104:R63-R67; Kotzin et al.
- DRBl*0402 see, e.g, Veldman et al. (2007) CLIN. IMMUNOL. 122:330- 337), DRBl*0404 (see, e.g, Gebe etal. (2001) J. IMMUNOL. 167:3250-3256), DRBl*1101 (see, e.g., Cunliffe, ibid; Moro et al. (2005) BMC IMMUNOL. 6:24), DRB1*13O2 (see, e.g., Laughlin et al. (2007) INFECT. IMMUNOL.
- DRBl*0701 see, e.g., Cartoon, ibid
- DQAl*0102 see, e.g, Kwok et al. (2000) J. IMMUNOL. 164:4244-4249
- DQBl*0602 see, e.g., Kwok, ibid
- DQAl*0501 see, e.g., Quarsten et al. (2001) J. IMMUNOL. 167:4861- 4868
- DQBl*0201 see, e.g., Quarsten, ibid
- DPAl*0103 see, e.g., Zhang et al. (2005) EUR. J. IMMUNOL. 35:1066-1075; Yang et al. (2005) J. CLIN. IMMUNOL. 25:428-436
- DPBl*0401 see, e.g., Zhang, ibid; Yang, ibid).
- the MHCII molecule is human, and comprise, for example, an MHCII alpha and beta chains selected from the group consisting of HLA-DRA*01 :01, HLA- DRBl*01:01, HLA-DRBl*01:02, HLA-DRBl*03:01, HLA-DRBl*04:01, HLA- DRBl*04:03, HLA-DRBl*04:04, HLA-DRBl*07:01, HLA-DRBl*08:01, HLA- DRBl*08:02, HLA-DRB 1*09:01, HLA-DRBl*10:01, HLA-DRB1*11:O1, HLA- DRB1*11:O4, HLA-DRB1*13:O1, HLA-DRB 1*13:02, HLA-DRB1*14:O1, HLA- DRB1*15:O1, HLA-DRB 1*15:02, HLA-DRA*01 :01, H
- the full-length amino acid sequence (including signal sequence and transmembrane domain) of a representative HLA-DP beta chain is shown in SEQ ID NO: 145.
- the amino acid sequence of a soluble form of this MHCII chain (lacking signal sequence and transmembrane domain) is shown in SEQ ID NO: 146.
- heterodimerization pairs can be appended to the C-teriminal sequence of the alpha and/or beta chains of the MHCII molecule.
- heterodimerization pair sequences include Fos and Jun (e.g., having the amino acid sequences shown in SEQ ID NOs: 123 and 124, respectively), acidic and basic leucine zippers (e.g., having the amino acid sequences shown in SEQ ID NOs: 125 and 126, respectively), knob and hole sequences (e.g, having the amino acid sequences shown in SEQ ID NOs: 127 and 128, respectively) for knobs-into-holes technology or spytab and spycatcher sequences (e.g, having the amino acid sequences shown in SEQ ID NOs: 129 and 130, respectively).
- Fos and Jun e.g., having the amino acid sequences shown in SEQ ID NOs: 123 and 124, respectively
- acidic and basic leucine zippers e.g., having the amino acid sequences shown in SEQ ID NOs
- an MHCII-binding placeholder peptide is included in the expression construct for one of the MHCII chains, preferably the beta chain, such that the placeholder peptide and a digestible (cleavable) linker are encoded in the construct upstream of (N-terminally) and in operative linkage with the coding sequences for the MHCII chain.
- the expression construct can encode (from N- to C-terminus): a placeholder peptide, a digestible linker, the MHCII chain (e.g., beta chain) and a C-terminal tag.
- an N-terminal tag is also appended upstream of the placeholder peptide, which allows for removal of non-exchanged peptide species following peptide exchange.
- an MHCII multimer described herein comprises the al and a2 domains of an MHCII alpha chain and the pi and [32 domains of an MHCII beta chain.
- an MHCII multimer described herein comprises only the al and pi domains of an MHCII heavy chain.
- an MHCII multimer comprises an alpha-chain and a beta-chain combined with a peptide.
- Other embodiments include an MHCII molecule comprised only of alpha-chain and beta-chain (so-called “empty” MHC II without loaded peptide), a truncated alpha-chain (e.g.
- al domain combined with full-length betachain, either empty or loaded with a peptide
- a truncated beta-chain e.g. the pi domain
- a full-length alpha-chain either empty or loaded with a peptide
- a truncated alpha-chain combined with a truncated beta-chain (e.g. al and pi domain), either empty or loaded with a peptide.
- the MHCII alpha and beta chains comprise a HLA-DR alpha chain paired with a HLA-DR beta chain.
- the HLA-DR alpha chain is HLA-DRA*01:01 and the HLA-DR beta chain is for example HLA-DRBl*01:01, HLA- DRBl*01:02, HLA-DRB 1*03:01, HLA-DRBl*04:01, HLA-DRBl*04:03, HLA- DRBl*04:04, HLA-DRB 1*07:01, HLA-DRB 1*08: 01, HLA-DRB 1*08: 02, HLA- DRBl*09:01, HLA-DRBl*10:01, HLA-DRBl*ll:01, HLA-DRB1 *11:04, HLA- DRBl*13:01, HLA-DRB 1*13:02, HLA-DRB1*14:O1, HLA-DR
- the MHCII alpha and beta chains comprise a HLA-DQ alpha chain paired with a HLA-DQ beta chain.
- the HLA-DQ alpha chain is HLA-DQA1 *01:01
- the HLA-DQ beta chain is for example HLA-DQBl*05:01, HLA- DQBl*06:02, HLA-DQB 1*03:02, HLA-DQBl*02:01, HLA-DQBl*03:01, HLA- DQBl*03:03, HLA-DQB 1*04:02, HLA-DQB 1*05: 03, HLA-DQB1 *06:03, or HLA- DQBl*06:04.
- the HLA-DQ alpha chain is HLA-DQAl*01:02
- the HLA-DQ beta chain is for example HLA-DQB1 *05:01, HLA-DQB 1*06: 02, HLA- DQBl*03:02, HLA-DQB1 *02:01, HLA-DQBl*03:01, HLA-DQBl*03:03, HLA- DQBl*04:02, HLA-DQB 1*05:03, HLA-DQB1 *06:03, or HLA-DQB 1*06: 04.
- the HLA-DQ alpha chain is HLA-DQAl*03:01
- the HLA-DQ beta chain is for example HLA-DQB1 *05:01, HLA-DQB 1*06: 02, HLA-DQB 1*03: 02, HLA- DQBl*02:01, HLA-DQB1 *03:01, HLA-DQBl*03:03, HLA-DQB1 *04:02, HLA- DQBl*05:03, HLA-DQB 1*06: 03, or HLA-DQB 1*06: 04.
- the HLA- DQ alpha chain is HLA-DQAl*05:01
- the HLA-DQ beta chain is for example HLA- DQBl*05:01, HLA-DQB 1*06:02, HLA-DQB 1*03: 02, HLA-DQBl*02:01, HLA- DQBl*03:01, HLA-DQB 1*03:03, HLA-DQB1 *04:02, HLA-DQBl*05:03, HLA- DQBl*06:03 ,or HLA-DQB 1*06:04.
- the MHCII alpha and beta chains comprise a HLA-DP alpha chain paired with a HLA-DP beta chain.
- the HLA-DP alpha chain is HLA-DPAl*01:03
- the HLA-DP beta chain is HLA-DPB1 *04:01.
- the MHCII alpha and beta chains comprise the amino acid sequences set forth in SEQ ID NOs: 42 and 47; 42 and 48; 42 and 49; 42 and 50; 42 and 51; 42 and 52; 42 and 53; 42 and 54; 42 and 55; 42 and 56; 42 and 57; 42 and 58; 42 and 59; 42 and 60; 42 and 61; 44 and 62; 44 and 63; 45 and 64; 46 and 65; or 144 and 146, respectively.
- the MHCII molecule comprises a soluble MHCII polypeptide. In some embodiments the MHCII molecule comprises a soluble MHCII lacking transmembrane and intracellular domains.
- the alpha-chain and beta-chain may be expressed in separate cells as individual polypeptides or in the same cell as a fusion protein.
- the peptide of the MHC Il-peptide complex may be produced separately and added following purification of whole MHC complexes or added during in vitro refolding or expressed together with alphachain and/or beta-chain connected to either chain through a linker.
- the genetic material can encode all or only a fragment of MHC class II alpha- and beta-chains.
- the genetic material may be fused with genes encoding other proteins, including proteins useful in purification of the expressed polypeptide chains (c.g.
- purification tags proteins useful in increasing/decreasing solubility of the polypeptide(s), proteins useful in detection of polypeptide(s), proteins involved in coupling of MHC complex to multimerization domains and/or coupling of labels to MHC complex and/or MHC multimer.
- MHC II complexes are not easily refolded after denaturation in vitro. Only some MHC II alleles can be expressed in E. coli and refolded in vitro. Therefore, preferred expression systems for production of MHC II molecules are eukaryotic systems where refolding after expression of protein is not necessary.
- Preferred expression systems include mammalian expression systems, such as CHO cells, HEK cells or other mammalian cell lines suitable for expression of human proteins.
- Other expression systems include stable Drosophila cell transfectants, baculovirus infected insect-cells or other mammalian cell lines suitable for expression of proteins.
- the MHC II complexes are “empty,” i.e., without intentional loading of peptide into the class II antigen-binding groove.
- Methods for production of empty MHC II complexes have been described, such as expression in insect cells (see e.g., Novak et al. (1999) J. CLIN. INVEST. 104:63-67; Nepom etal. (2002) ARTHRIT. RHEUMAT. 46:5-12; and Moro et al. (2005) BMC IMMUNOLOGY 6:24).
- soluble domains of the alpha and beta subunits of MHC II complexes are co-expressed and secreted from insect cells infected or stably transformed with baculovirus. Since the insect cells lack the mammalian loading machinery for MHC II complexes, the complexes are essentially empty after assembly. The overexpressed complexes can then be purified from the cell culture medium.
- the alpha-chain and the beta-chain may be each fused with a dimerization domain such as a leucine zipper, IgG constant region, or an enzymatic biotinylation site e.g., Bir) and dimerized after expression and purification.
- a dimerization domain such as a leucine zipper, IgG constant region, or an enzymatic biotinylation site e.g., Bir
- MHCII monomers are prepared in which a placeholder peptide is covalently linked to the MHCII molecule.
- a placeholder peptide is covalently linked to the MHCII molecule.
- one approach is the covalent synthesis of single-chain MHC class II chain-peptide complexes, directed by engineering peptide-specific complementary DNA (cDNA) sequences proximal to the beta-chain cDNA (as described in Crawford et al. (1999) IMMUNITY 8:675-682).
- cDNA peptide-specific complementary DNA
- the resulting polypeptide refolds with the peptide sequence extended from the amino terminus of the class II molecule.
- a tethering linker sequence in the peptide allows enough flexibility for the peptide to occupy the peptide binding groove in the mature class II molecule.
- a cleavable linker can be used to allow for cleavage of the covalent linkage between the peptide and the MHCII molecule e.g., as described in Day et al. (2003) J. CLIN. INVEST. 112:831-842), thereby allowing for peptide exchange and loading of the MHCII molecule with other peptides (e.g, a library of different peptides).
- the MHCII complexes can be purified directly as whole MHCII or MHCII-placeholder peptide monomers from MHCII expressing cells.
- the MHCII monomers may be expressed on the surface of cells, and are then isolated by disruption of the cell membrane using, e.g., detergent followed by purification of the MHCII.
- MHC monomers are expressed into the periplasm and expressing cells are lysed and released MHCII monomers purified.
- MHC monomers may be purified from the supernatant of cells secreting expressed proteins into culture supernatant.
- Methods for purifying MHCII monomers are well known in the art, for example, via the use of affinity tags together with affinity chromatography, beads coated with ant-tag and/or other techniques involving immobilization of MHCII protein to affinity matrix; size exclusion chromatography using, e.g, gel filtration, ion exchange or other methods able to separate MHC molecules from cells and/or cell lysates.
- recombinant expression of MHCII polypeptides allow a number of modifications of the MHC monomers.
- recombinant techniques provide methods for carboxy terminal truncation which deletes the hydrophobic transmembrane domain.
- the carboxy termini can also be arbitrarily chosen to facilitate the conjugation of ligands or labels, for example, by introducing cysteine and/or lysine residues into the molecule.
- the synthetic gene will typically include restriction sites to aid insertion into expression vectors and manipulation of the gene sequence.
- the genes encoding the appropriate monomers are then inserted into expression vectors, expressed in an appropriate host, such as E. coli, yeast, insect, or other suitable cells, and the recombinant proteins are obtained.
- the MHCII monomers are biotinylated on either their alpha or beta chain. In some embodiments, the MHCII monomers are biotinylated before loading of the peptide either by refolding or peptide exchange.
- Biotinylation of the MHC monomers can be achieved as known in the art, e.g. by attaching biotin to a specific attachment site which is the recognition site of a biotinylating enzyme.
- the biotinylating enzyme is BirA.
- biotinylation is carried out on the desired protein chain in vivo as a post translational modification during protein expression.
- the MHCII molecules are prepared with a placeholder peptide to facilitate proper folding of the MHCII monomers to produce placeholder-peptide loaded MHCII (p*MHCII) prior to multimerization.
- the placeholder peptide is peptide that binds HLA-DR, HLA-DQ, HLA-DX, HLA-DO, HLA-DZ or HLA-DP.
- the placeholder peptide is a synthetic peptide.
- the placeholder peptide is tethered to the MHCII molecules using a linker, e.g, a linker containing a cleavage site that can be used to remove the placeholder peptide from the MHCII molecules.
- a linker e.g, a linker containing a cleavage site that can be used to remove the placeholder peptide from the MHCII molecules.
- the placeholder peptide, or linker thereof is digestible, thereby allowing release of the peptide from the antigen-binding groove of the MHCII molecule, for example as part of peptide exchange with the multimer compositions of the disclosure.
- the placeholder peptide, or linker thereof is thermolabile, labile at acidic pH, enzymatically cleavable (e.g., using a protease) or photocl eav able (e.g., using UV light) as means for digestion of the placeholder peptide, or linker thereof.
- the placeholder peptide, or linker thereof is cleavable by a protease that recognizes a protease cleavage site within the placeholder peptide, or linker thereof.
- protease cleavage sites include a Factor Xa cleavage site (e.g., having the amino acid sequence shown in SEQ ID NO: 131) and an enterokinase cleavage site (e.g, having the amino acid sequence shown in SEQ ID NO: 132).
- the affinity of the placeholder peptide for the binding groove of MHCII is lower than the rescue peptide(s). In some embodiments, the affinity of the placeholder peptide for the MHCII binding groove is about 10-fold lower than the rescue peptide(s).
- the placeholder peptide is thermolabile. In some embodiments, the placeholder peptide is thermolabile at a temperature between about 30- 37°C. In some embodiments, the placeholder peptide is labile at a temperature at or above 30°C, at or above 32°C, at or above 34°C, at or above 35°C, at or above 36°C, or at about 37°C.
- Thermal labile placeholder peptides and methods of identifying and producing thermal labile placeholder peptides have been described (e.g, WO 93/10220; WO 2005/047902; US 2008/0206789; Luimstra et al. (2019) CURR. PROTOC. IMMUNOL. 126(l):e85; Luimstra et al. (2016) J. EXP. MED. 215(5): 1493-1504).
- the placeholder peptide is labile at an acidic pH. In some embodiments, the placeholder peptide is labile between about pH 2.5 and 6.5. In some embodiments, the placeholder peptide is labile at a pH of about 2.5-6.0, 3.0-6.0, 3.0-6.5, 3.5- 6.0 3.5-6.5, 4.0-6.0, 4.0-6.5, 4.5-6.0, 4.5-6.5, 5.0-6.0, 5.0-6.5, 5.0, 5.5., 6.0 or 6.5. In some embodiments, the placeholder peptide is labile at a basic pH. In some embodiments, the placeholder peptide is labile between about pH 9-11.
- the placeholder peptide is labile at or above pH 9, at or above pH 9.5, at or about pH 10, at or about pH 10.5, or at or about pH 11.
- Methods of generating and using pH sensitive placeholder peptides are publicly available, for example, as described in WO 93/10220; US 2008/0206789; and Cameron etal. (2002) J. IMMUNOL. METH. 268:51-59.
- the placeholder peptide comprises a cleavable moiety.
- cleavable moieties include, for example, moieties that are cleaved by photoirradiation, enzymes, nucleophilic or electrophilic agents, reducing and oxidizing reagents (e.g, reviewed in Leriche et al. (2012) BlORG. MED. CHEM. 20(2):571- 582).
- the placeholder peptide is fused to a degradation tag and peptide exchange is promoted by proteolysis in the presence of a corresponding protease (the digests the degradation tag) along with the presence of the rescue peptide(s).
- the cleavable placeholder peptide is a photocleavable peptide, e.g., cleaved upon exposure to UV light.
- the placeholder peptide can comprise one or more photocleavable photocleavable non-natural amino acids.
- MHCII-binding photocleavable peptides e.g, that incorporate the UV-sensitive amino acid analog 3-amino- 3-(2-nitrophenyl)-propionate have been described (see e.g., Negroni and Stem (2016) PLos ONE 13(7):e0199704).
- the MHCII placeholder peptide is a CLIP peptide, such as having the amino acid sequence KPVSKMRMATPLLMQA (SEQ ID NO: 3) or ATPLLMQALPMGA (SEQ ID NO: 134).
- the CLIP peptide, or linker thereof is cleavable.
- the CLIP peptide, or linker thereof is cleavable by Factor Xa.
- the MHCII monomers are synthesized with the cleavable CLIP peptide covalently attached, such as by synthesis of single-chain MHC class II chain- peptide complexes, directed by engineering peptide-specific complementary DNA (cDNA) sequences proximal to the beta-chain cDNA (see e.g., Day et al. (2003) J. CLIN. INVEST.
- cDNA peptide-specific complementary DNA
- MHCII binding peptides have been described in the art that can be used as placeholder peptides, based on appropriate pairing of an MHCII molecule and its known MHCII binding peptide.
- Non-limiting examples of known MHCII molecule/MHCII binding peptide pairs include: DRAl*0101/DRB 1*0401 and the immunodominant peptide of hemagglutinin, HA307-319 (see Novak et al. (1999) J. CLIN. INVEST.
- HLA- DR*1101 and tetanus-toxoid (TT)-derived p2 peptide having the amino acid sequence QIYKANSKFIGITEL (SEQ ID NO: 120) (see Cecconi et al. (2008) CYTOMETRY 73 A: 1010-1018).
- the multimer composition (as described in section I above) and the soluble MHCII molecule (as described in section II above) are combined such that peptide exchange occurs, wherein the placeholder peptide bound to the MHCII molecule is released and the MHCII molecule is loaded onto the peptides of the multimer composition.
- the peptides of the multimer composition serve as “rescue peptides” that replace the placeholder peptides to create the MHCII multimers.
- Peptide exchange is described in detail in Example 4 and non-limiting representative schematic diagrams of this approach are shown in FIG. 3, FIG. 4A and FIG. 4B.
- One aspect of peptide exchange involves digestion of the placeholder peptide such that it is removed from the antigen-binding groove of MHCII.
- the digestible placholder peptide is thermolabile and digestion is achieved by adjusting the temperature such that the peptide is removed from the antigen-binding groove.
- the digestible placholder peptide is labile at acidic pH and digestion is achieved by adjusting the pH such that the peptide is removed from the antigen-binding groove.
- the digestible placholder peptide is enzymatically cleavable and digestion is achieved by enzymatic cleavage (e.g., protease cleavage) such that the peptide is removed from the antigen-binding groove.
- the digestible placholder peptide is photocleavable and digestion is achieved by photocleavage (e.g., UV light cleavage) such that the peptide is removed from the antigen-binding groove.
- Another aspect of peptide exchange involves loading the MHCII molecules onto the peptides of the multimer composition, which can be achieved, for example, by combining the MHCII molecules and the multimer composition under acidic pH conditions.
- the multimer compositions and placeholder peptide-loaded soluble MHCII molecules of the disclosure can be used to generate a library of or microarray of MHCII multimers loaded with a diversity of unique peptide epitopes by in situ or in vitro peptide exchange reactions as described herein.
- the peptide exchange reactions are performed in multiwell formats and under native conditions.
- Peptide binding, and thus peptide exchange can be determined by a number of techniques, such as ELISA or Differential scanning fluorimetry (DSF), which monitors the stability of the MHCII structure, or by biophysical techniques that monitor peptide binding, such as fluorescence polarization.
- a fluorescently labeled placeholder peptide is used in exchange reactions in the presence of unlabeled exchange peptides. Aliquots of fluorescently labeled p*MHCII multimers are either left untreated or exposed to peptide exchange conditions for different time periods. The amount of remaining p*MHCII-containing the placeholder peptide is monitored by fluorescence analysis to monitor the reduction in p*MHCII complexes.
- the placeholder peptide has a lower affinity for the MHCII peptide binding groove than the exchanged (rescue) peptide epitope, and peptide exchange comprises contacting the p*MHCII molecule with an excess of (rescue) peptide epitope in a competition assay.
- the placeholder peptide has a KD that is about 10- fold lower than the exchanged peptide epitope.
- Peptides that bind to the peptide binding groove of the MHCII molecule can be a naturally occurring peptide but can also be synthetically created using the knowledge of the binding specificity of the binding pocket of the particular MHCII molecule or the supertype family it belongs to. Suitable ligands can be generated using the available 3D structures of MHCII complexes and the knowledge on the binding pocket specificity of the respective MHCII molecules.
- peptide exchange is induced by elevating the temperature of the mixture to between about 30°-37°C. In some embodiments, the mixture is elevated to 31°, 32°, 33°, 34°, 35°, 36° or 37°
- peptide exchange is induced by reducing the pH of the mixture to between about pH 2.5-6. In some embodiments, peptide exchange is induced by increasing the pH of the mixture to about pH 9-11.
- the MHCII placeholder peptide is a CLIP peptide, such as having the amino acid sequence KPVSKMRMATPLLMQA (SEQ ID NO: 3) or ATPLLMQALPMGA (SEQ ID NO: 134).
- the placeholder peptide further comprises a fluorescent label.
- the fluorescent label is attached to a cysteine residue in the placeholder peptide.
- MHCII peptide exchange is performed in multiwell format for high- throughput screening of peptide ligands as described herein.
- Peptide exchange can be monitored by a number of techniques such as ELISA or fluorescence polarization, for example, as generally described in Rodenko et al. ((2006) NAT. PROTOCOL. 1 : 1120-1132).
- the disclosure pertains to methods of producing a library of MHC II multimers comprising a diversity of loaded peptide epitopes.
- steps in the preparation of peptide-exchanged, barcoded MHC libraries have been described in the art. These steps use standard methods known in the art for preparing barcoded libraries, including use of single-cell sequencing, use of porous hydrogels, use of single template PCR to generate peptide-encoding amplicons (barcodes) and use of in-drop in vitro transcription/translation (IVTT).
- Libraries of MHCII multimers can be prepared using single template encapsulation methods known in the art.
- the single template encapsulation method is drop-based (e.g., using a hydrogel).
- the single template encapsulation method is well-based (e.g., using a 96-well plate).
- MHCII multimers can be conjugated with a fluorescent label, allowing for identification of T cells that bind the MHCII multimer, for example, via flow cytometry or microscopy. T cells can also be selected based on a fluorescence label through, e.g., fluorescence or magnetic activated cell sorting.
- one or more detectable labels are conjugated to a linker.
- a “detectable label” is any molecule or functional group that allows for the detection of a biological or chemical characteristic or change in a system, such as the presence of a target substance in the sample.
- detectable labels examples include fluorophores, chromophores, electro chemiluminescent labels, bioluminescent labels, polymers, polymer particles, bead or other solid surfaces, gold or other metal particles or heavy atoms, spin labels, radioisotopes, enzyme substrates, haptens, antigens, Quantum Dots, aminohexyl, pyrene, nucleic acids or nucleic acid analogs, or proteins ,such as receptors, peptide ligands or substrates, enzymes, and antibodies(including antibody fragments).
- polymer particles labels which may be used include micro particles, beads, or latex particles of polystyrene, PMMA or silica, which can be embedded with fluorescent dyes, or polymer micelles or capsules which contain dyes, enzymes or substrates.
- metal particles which may be used include gold particles and coated gold particles, which can be converted by silver stains.
- haptens that may be conjugated in some embodiments are fluorophores, myc, nitrotyrosine, biotin, avidin, streptavidin, 2,4-dinitrophenyl, digoxigenin, bromodeoxy uridine, sulfonate, acetylaminoflurene, mercury trintrophonol, and estradiol.
- Examples of enzymes which may be used comprise horse radish peroxidase (HRP), alkaline phosphatase (AP), beta-galactosidase (GAL), glucose-6-phosphate dehydrogenase, beta-N-acetylglucosaminidase, Pglucuronidase, invertase, Xanthine Oxidase, firefly luciferase and glucose oxidase (GO).
- HRP horse radish peroxidase
- AP alkaline phosphatase
- GAL beta-galactosidase
- glucose-6-phosphate dehydrogenase beta-N-acetylglucosaminidase
- Pglucuronidase invertase
- Xanthine Oxidase firefly luciferase
- glucose oxidase GO
- HRP horse radish peroxidase
- DAB diaminobenzi dine
- AEC 3-amino-9-ethylcarbazole
- BDHC Benzidine dihydrochloride
- Hanker- Yates reagent Hanker- Yates reagent
- IB Indophane blue
- TMB tetramethylbenzidine
- CN 4-chloro-l-naphtol
- CN alpha-naphtol pyronin
- Examples of commonly used substrates for Alkaline Phosphatase include Naphthol-AS-Bl-phosphateZfast red TR (NABPZFR),Naphthol-AS-MX-phosphateZfast red TR (NAMPZFR),Naphthol-AS-Bl- phosphateZfast red TR (NABPZFR),Naphthol-AS-MX-phosphateZfast red TR (NAMPZFR),Naphthol-AS-Bl-phosphateZnew fuschin (NABPZNF), bromochloroindolylphosphate/nitroblue tetrazolium (BCIP/NBT), b-Bromo-chloro-S-indolyl- beta-delta-galactopyranoside (BCIG).
- NABPZFR Naphthol-AS-Bl-phosphateZfast red TR
- NAMPZFR Naphthol-AS-MX-phosphateZfast red TR
- luminescent labels which may be used include luminol, isoluminol, acridinium esters, 1 ,2-dioxetanes and pyridopyridazines.
- electrochemiluminescent labels include ruthenium derivatives.
- radioactive labels which may be used include radioactive isotopes of iodide, cobalt, selenium, hydrogen, carbon, sulfur, and phosphorous.
- Some “detectable labels” also include “colour labels,” in which the biological change or event in the system may be assayed by the presence of a colour, or a change in colour.
- colours labels are chromophores, fluorophores, chemiluminescent compounds, electrochemiluminescent labels, bioluminescent labels, and enzymes that catalyze a colour change in a substrate.
- Fluorophores as described herein are molecules that emit detectable electromagnetic radiation upon excitation with electro-magnetic radiation at one or more wavelengths.
- a large variety of fluorophores are known in the art and are developed by chemists for use as detectable molecular labels and can be conjugated to the MHCII multimers provided herein.
- FLUORESCEINTM or its derivatives, such as FLUORESCEIN®-5-isothiocyanate (FITC), 5-(and6)-carboxyFLUORESCEIN®, 5- or 6- carboxyFLUORESCEIN®,6-(FLUORESCEIN®)-5-(and 6)-carboxamido hexanoic acid, FLUORESCEIN® isothiocyanate, rhodamine or its derivatives such as tetramethyl rhodamine and tetramethylrhodamine-5-(and -6) isothiocyanate (TRITC).
- FLUORESCEINTM FLUORESCEINTM, or its derivatives, such as FLUORESCEIN®-5-isothiocyanate (FITC), 5-(and6)-carboxyFLUORESCEIN®, 5- or 6- carboxyFLUORESCEIN®,6-(FLUORESCEIN®)-5-(and 6)-carboxamido
- fluorophores include: coumarin dyes such as (diethyl-amino)coumarin or7-amino-4-methylcoumarin-3- acetic acid, succinimidyl ester (AMCA); sulforhodamine 101 sulfonyl chloride (TexasRed® or TexasRed® sulfonyl chloride; 5-(and-6)-carboxyrhodamine 101, succinimidyl ester, also known as 5-(and-6)-carboxy-X-rhodamine, succinimidyl ester (CXR); lissamine or lissamine derivatives such as lissamine rhodamine B sulfonyl Chloride (LisR); 5-(and-6)- carboxyFLUORESCEIN®, succinimidyl ester(CFI); FLUORESCEIN®5-isothiocyanate (FITC);7-diethylaminocoumarin-3-carboxy
- fluorescent proteins such as green fluorescent protein and its analogs or derivatives, fluorescent amino acids such as tyrosine and tryptophan and their analogs, fluorescent nucleosides, and other fluorescent molecules such as Cy2,Cy3, Cy 3.5, CY5.TM., CY5.TM.5, Cy 7, IR dyes, Dyomics dyes, phycoerythrine, Oregon green 488, pacific blue, rhodamine green, and Alexa dyes.
- fluorescent labels include conjugates of R-phycoerythrin orallophycoerythrin, inorganic fluorescent labels such as particles based on semiconductor material like coated CdSe nanocrystallites.
- the detectable label can be detected by numerous methods, including, for example, reflectance, transmittance, light scatter, optical rotation, and fluorescence or combinations hereof in the case of optical labels or by film, scintillation counting, or phosphorimaging in the case of radioactive labels. See, e.g., Larsson (1988) IMMUNOCYTOCHEMISTRY: THEORY AND PRACTICE (CRC Press, Boca Raton, Fla.); METHODS IN MOLECULAR BIOLOGY, vol. 80 (1998), John D. Pound (ed.) (Humana Press, Totowa, N.J.). In some embodiments, more than one detectable labels employed.
- an MHCII multimer of the disclosure comprises an identifier tag or label, such as an oligonucleotide barcode, that facilitates identification of the MHCII multimer.
- the identifier tag e.g., oligonucleotide barcode
- the identifier tag is attached to the multimerization domain of the MHCII multimer, such as through a binding moiety on the identifier tag, e.g., oligonucleotide barcode, that binds to a binding site on the multimerization domain.
- the MHCII multimer when the multimerization domain is streptavidin or avidin, since the MHCII-binding peptides are conjugated to the multimerization domain at a site other than the biotin-binding site, the MHCII multimer can be labeled with an identifier tag, e.g., oligonucleotide barcode, using a biotinylated form of the identifier tag, e.g., a biotinylated oligonucleotide barcode. Labeling of the MHCII multimer is easily achieved by incubation of the multimer composition with the biotinylated identifier tag, e.g., biotinylated oligonucleotide barcode.
- an identifier tag e.g., oligonucleotide barcode
- an oligonucleotide barcode is a unique oligonucleotide sequence ranging for 10 to more than 50 nucleotides.
- the barcode has shared amplification sequences in the 3’ and 5’ ends, and a unique sequence in the middle. This sequence can be revealed by sequencing and can serve as a specific barcode for a given molecule.
- the nucleic acid component of the barcode (typically DNA) has a special structure.
- the at least one nucleic acid molecule is composed of at least a 5’ first primer region, a central region (barcode region), and a 3’ second primer region. In this way the central region (the barcode region) can be amplified by a primer set.
- the length of the nucleic acid molecule may also vary.
- the at least one nucleic acid molecule has a length in the range 20-100 nucleotides, such as 30-100, such as 30-80, such as 30-50 nucleotides.
- the nucleic acid identifier is from 40 nucleotides to 120 nucleotides in length.
- the coupling of the oligonucleotide barcode to the multimer composition may also vary.
- the at least one oligonucleotide barcode is linked to said multimer composition via a biotin binding domain interacting with streptavidin or avidin within the multimer composition.
- Other coupling moieties may also be used, depending on the availability of an appropriate binding site with the multimer composition (e.g., within the multimerization domain) and an appropriate corresponding binding domain that can be attached to the oligonucleotide barcodes molecules to facilitate attachment.
- the at least oligonucleotide barcode molecule comprises or consists of DNA, RNA, and/or artificial nucleotides such as PLA or LNA.
- DNA Preferably DNA, but other nucleotides may be included to e.g. increase stability.
- barcode technology is well known in the art, see for example Shiroguchi et al. (2012) PROC. NATL. ACAD. Set. USA 109(4): 1347-52; and Smith et al. (2010) NUCLEIC ACIDS RESEARCH 38(13)1 l:el42. Further methods and compositions for using barcode technology include those described in U.S. 2016/0060621. Use of barcode technology specifically to label MHC multimers also has been described, see for example Bentzen et al. (2016) NATURE BIOTECH. 34:10: 1037-1045; Bentzen and Hadrup (2017) CANCER IMMUNOL. IMMUNOTHERAP. 66:657-666.
- Standard methods for preparing barcode oligonucleotides, including conjugating them with a suitable binding moiety (e.g, biotinylation) that can bind the MHC multimer, are known in the art and can be applied to preparing barcode oligonucleotides for labeling the MHC multimers.
- a suitable binding moiety e.g, biotinylation
- Methods for generating customizable DNA barcode libraries are publicly available. Programs include Generator and nxCode, consisting of 96-587 barcodes, respectively, as well as The DNA Barcodes Package and TagD software (reporting generating libraries consisting of 100,000 barcodes).
- the unique molecular identifier barcode is encoded by a contiguous sequence of nucleotides tagged to one end of a target nucleic acid.
- the unique molecular identifier (UMI) barcode is encoded by a non-contiguous sequence. Noncontiguous UMIs can have a portion of the barcode at a first end of the target nucleic acid and a portion of the barcode at a second end of the target nucleic acid.
- the UMI is a non-contiguous barcode containing a variable length barcode sequence at a first end and a second identifier sequence at a second end of the target nucleic acid.
- the UMI is a non-contiguous barcode having a variable length barcode sequence at a first end and a second identifier sequence at a second end of the target nucleic acid, wherein the second identifier sequence is determined by a position of a transposase fragmentation event, e.g., a transposase fragmentation site and transposon end insertion event.
- a transposase fragmentation event e.g., a transposase fragmentation site and transposon end insertion event.
- the barcode is a “variable length barcode.”
- a variable length barcode is an oligonucleotide that differs from other variable length barcode oligonucleotides in a population, by length, which can be identified by the number of contiguous nucleotides in the barcode.
- additional barcode complexity for the variable length barcode can be provided by the use of variable nucleotide sequence, as described in the paragraphs above, in addition to the variable length.
- a variable length barcode can have a length of from 0 to no more than 5 nucleotides.
- variable length barcode can be denoted by the term “[0- 5].”
- a population of target nucleic acids that are attached to such a variable length barcode is expected to include at least one target nucleic acid attached to a variable length barcode that has at least 1 nucleotide (e.g., attached to a variable length barcode having only 1, only 2, only 3, only 4, or only 5 nucleotides).
- a population of target nucleic acids that are attached to such a variable length barcode can include at least one target nucleic acid that contains no variable length barcode (i. e.
- variable length barcode having a length of 0 a variable length barcode having a length of 0
- at least one target nucleic acid that contains a variable length barcode having only 1 nucleotide and/or at least one target nucleic acid that contains a variable length barcode having only 2 nucleotides, and/or at least one target nucleic acid that contains a variable length barcode having only 3 nucleotides, and/or at least one target nucleic acid that contains a variable length barcode having only 4 nucleotides, and/or and at least one target nucleic acid that contains a variable length barcode having only 5 nucleotides.
- the [0-5] variable length barcode can uniquely identify (differentiate), by itself, 5 different target nucleic acid molecules of the same sequence. Further, in such an embodiment, the [0-5] variable length barcode can uniquely identify (differentiate) 5 different target nucleic molecules of a first sequence, 5 different target nucleic acid molecules of a second sequence, etc. for each different target nucleic acid sequence. Furthermore, barcode labelled MHCII-multimers can be used in combination with single-cell sorting and TCR sequencing, where the specificity of the TCR can be determined by the co-attached barcode.
- TCR specificity for potentially 1000+different antigen responsive T-cells in parallel from the same sample, and match the TCR sequence to the antigen specificity.
- the future potential of this technology relates to the ability to predict antigen responsiveness based on the TCR sequence.
- the barcode is co-attached to the multimer and serves as a specific label for a particular peptide-MHCII complex.
- a specific label for a particular peptide-MHCII complex at least, for example, 10 or 100 or 1000 or 10,000 or more different peptide-MHCII multimers can be mixed, allow specific interaction with T-cells from blood or other biological specimens, wash-out unbound MHCII-multimers and determine the sequence of the DNA-barcodes.
- the sequence of barcodes present above background level will provide a fingerprint for identification of the antigen responsive cells present in the given cell-population.
- the number of sequence-reads for each specific barcode will correlate with the frequency of specific T-cells, and the frequency can be estimated by comparing the frequency of reads to the input-frequency of T-cells.
- the DNA-barcode serves as a specific label for the antigen specific T-cells and can be used to determine the specificity of a T-cell after e.g. single-cell sorting, functional analyses or phenotypical assessments. In this way antigen specificity can be linked to both the T-cell receptor sequence (that can be revealed by single-cell sequencing methods) and functional and phenotypical characteristics of the antigen specific cells.
- Barcode labeled MHCII multimer libraries can be used for the quantitative assessment of MHCII multimer binding to a given T-cell clone or TCR transduced/transfected cells.
- this strategy can be used to determine the avidity of a given TCR relative to a library of related peptide-MHCII multimers.
- the relative contribution of the different DNA-barcode sequences in the final readout is determined based on the quantitative contribution of the TCR binding for each of the different peptide-MHCII multimers in the library.
- Via titration based analyses it is possible to determine the quantitative binding properties of a TCR in relation to a large library of peptide-MHCII multimers, all merged into a single sample.
- the MHCII multimer library may specifically hold related peptide sequences or alanine-substitution peptide libraries.
- unique identifiers can be used for each sample of a plurality of samples.
- identifiers can be shared between two or more samples.
- identifiers can comprise some sequences that are shared between all samples, and other sequences that are unique to one sample.
- an identifier can comprise a sequence shared between all samples, and a sequence unique to one sample.
- a sequence shared between samples can be used for identifier amplification (e.g., PCR amplification with suitable primers).
- a sequence unique to one sample or shared between a subset of samples can be used for detection or quantification via qPCR (e.g., sequences for hydrolysis probes, such as TaqMan probes).
- a sequence unique to one sample or shared between a subset of samples can be used for detection or quantification via sequencing.
- an identifier can comprise a unique, in sti/co-generated sequence; each identifier sequence can be assigned to a sample of a plurality of samples and the identifier-sample assignment can be stored in a database.
- an identifier can comprise a nucleotide sequence that codes for all or part of a peptide or protein.
- an identifier can comprise a nucleotide sequence that codes for an open reading frame. In some embodiments, an identifier can comprise a nucleotide sequence that includes a promoter sequence. In some embodiments, an identifier can comprise a nucleotide sequence that includes a binding site for a DNA-binding protein, e.g. a transcription factor or polymerase enzyme. In some embodiments, an identifier can comprise one or more sequences targeted by a nuclease, e.g. a restriction enzyme. In some embodiments, an identifier can comprise all sequence elements necessary for in vitro transcription and translation of a sequence. In some embodiments, an identifier does not comprise all sequence elements necessary for in vitro transcription and translation of a sequence.
- an identifier can comprise a biotinylated nucleotide sequence.
- an identifier can be biotinylated by PCR amplification with a biotinylated primer(s).
- an identifier can be biotinylated by enzymatic incorporation of a biotinylated label, e.g. a biotin dUTP label, by use of KI enow DNA polymerase enzyme, nick translation or mixed primer labeling RNA polymerases, including T7, T3, and SP6 RNA polymerases.
- an identifier can be biotinylated by photobiotinylation, e.g. photoactivatable biotin can be added to the sample, and the sample irradiated with UV light.
- an identifier can be generated from a template polynucleotide, e.g. via PCR amplification of a template DNA.
- a template polynucleotide can comprise a nucleotide sequence that codes for an open reading frame.
- a template polynucleotide can comprise a nucleotide sequence that includes a promoter sequence.
- a template polynucleotide can comprise a nucleotide sequence that includes a binding site for a DNA-binding protein, e.g. a transcription factor or polymerase enzyme.
- a template polynucleotide can comprise one or more sequences targeted by a nuclease, e.g. a restriction enzyme. In some embodiments, a template polynucleotide can comprise all sequence elements necessary for in vitro transcription and translation of a sequence. In some embodiments, a template polynucleotide does not comprise all sequence elements necessary for in vitro transcription and translation of a sequence.
- MHCII multimers with attached identifiers can be incubated with a plurality of T cells, followed by sorting of T cells into single-cell compartments.
- T cells are lysed, and nucleic acids from lysed T cells comprising identifiers are produced. Nucleic acids are pooled and sequenced. Identifiers allow matching of peptide identifiers to T cell sequences from the same compartment.
- TCR-antigen specificity profiles are determined by identifying a TCR sequence (e.g., variable region, hypervariable region, or CDR) from a compartment, and quantifying peptide identifier reads from the same compartment.
- TCRs can be identified that exhibit binding affinity for peptides of the peptide library, and multiple peptides can be identified that exhibit binding affinity for specific TCRs.
- Epitope mutations in an antigen of an identified TCR-antigen pair can be identified that result in increased TCR binding affinity.
- Peptides and TCR sequences can be identified that are associated with control of disease associated protein, and can be used to design vaccines and cell therapies.
- TCR sequences are identified. Multiple TCRs are identified that exhibit binding affinity for some peptides of the peptide library, and multiple peptides are identified that exhibit binding affinity for some TCRs. Subjects are followed longitudinally and results of assays are compared to identify peptides and TCR sequences that are associated with successful response to immunotherapy.
- kits for use in the methods described herein Any of the compositions of the disclosure can be formulated into a kit, including the components and instructs for use of the components for the desired use.
- the disclosure provides a kit comprising at least one expression construct encoding a peptide-multimerization domain (a multimer composition of the disclosure).
- the kit can further comprise at least one expression construct encoding soluble MHCII with a placeholder peptide.
- the kit can further comprise instructions for expressing the multimer composition(s) and the soluble MHCII molecule(s) and for preparing MHCII multimers by peptide exchange.
- the kit comprises a library of expression constructs (e.g., a library of multimer compositions encoding a plurality of different MHCII- binding peptides).
- the disclosure provides a kit comprising one or more peptide- multimerization domain compositions (multimer compositions of the disclosure).
- the kit can further comprise one or more soluble MHCII molecules loaded with a placeholder peptide.
- the kit can further comprise instructions for preparing MHCII multimers from the multimer composition(s) and the soluble MHCII molecule(s) by peptide exchange.
- the kit comprises a library of multimer compositions encoding a plurality of different MHCII-binding peptides.
- the multimerization domain of the multimer composition is a tetramer.
- the tetramer is streptavidin or avidin.
- the tetramer further comprises a biotinylated oligonucleotide barcode bound to the biotinbinding site of streptavidin or avidin.
- Another aspect of the invention relates to methods for detecting antigen responsive T cells, for example in a sample.
- the methods comprise providing a plurality of MHC multimers of the disclosure; contacting the MHC multimers with said sample; and detecting binding of the MHC multimers to antigen responsive T cells within the sample, thereby detecting T cells responsive to an antigenic peptide present in the plurality of MHC multimers.
- binding is detected by amplifying the barcode region of the oligonucleotide barcode linked to the MHC multimer.
- the antigen responsive T cell is a CD4+ T cell, whose TCRs recognize peptide-bound MHC Class II molecules.
- This MHC multimer technology allows for detection of multiple (potentially >1000) different antigen-specific T cells in a single sample.
- the technology can be used, for example, for T-cell epitope mapping, immune-recognition discovery, diagnostics tests and measuring immune reactivity after vaccination or immune-related therapies.
- the MHC multimers allow for identification and selection of antigen-specific T cells to be administered for therapy, such as for adoptive T cell transfer therapy.
- MHC multimers can be used for detection of individual T-cells in fluid samples using flow cytometry or flow cytometry-like analysis.
- Liquid cell samples can be analyzed using a flow cytometer, able to detect and count individual cells passing in a stream through a laser beam.
- a flow cytometer able to detect and count individual cells passing in a stream through a laser beam.
- cells are stained with fluorescently labeled MHC multimer by incubating cells with MHC multimer and then forcing the cells with a large volume of liquid through a nozzle creating a stream of spaced cells. Each cell passes through a laser beam and any fluorochrome bound to the cell is excited and thereby fluoresces.
- Sensitive photomultipliers detect emitted fluorescence, providing information about the amount of MHC multimer bound to the cell.
- MHC multimers can be used to identify individual T-cells and/or specific T-cell populations in liquid samples.
- Cell samples capable of being analyzed by MHC multimers in flow cytometry analysis include, but is not limited to, blood samples or fractions thereof, T-cell lines (hybridomas, transfected cells) and homogenized tissues like spleen, lymph nodes, tumors, brain or any other tissue comprising T-cells.
- T-cell lines hybridas, transfected cells
- homogenized tissues like spleen, lymph nodes, tumors, brain or any other tissue comprising T-cells.
- gating reagent When analyzing blood samples whole blood can be used with or without lysis of red blood cells prior to analysis on flow cytometer. Lysing reagent can be added before or after staining with MHC multimers. When analyzing blood samples without lysis of red blood cells one or more gating reagents may be included to distinguish lymphocytes from red blood cells. Preferred gating reagent are marker molecules specific for surface proteins on red blood cells, enabling subtraction of this cell population from the remaining cells of the sample. As an example, a fluorochrome labelled CD45 specific marker molecule e.g. an antibody can be used to set the trigger discriminator to allow the flow cytometer to distinguish between red blood corpuscles and stained white blood cells.
- a fluorochrome labelled CD45 specific marker molecule e.g. an antibody can be used to set the trigger discriminator to allow the flow cytometer to distinguish between red blood corpuscles and stained white blood cells.
- lymphocytes can be purified before flow cytometry analysis e.g. using standard procedures like a FICOLL®-Hypaque gradient.
- Another possibility is to isolate T-cells from the blood sample, for example, by adding the sample to antibodies or other T-cell specific markers immobilized on solid support. Marker specific T-cells are then attached to the solid support and following washing specific T-cells can be eluted. This purified T-cell population can then be used for flow cytometry analysis together with MHCII multimers.
- T-cells may also be purified from other lymphocytes or blood cells by rosetting.
- Human T-cells form spontaneous rosettes with sheep erythrocytes, also called E-rossette formation.
- E-rossette formation can be carried out by incubating lymphocytes with sheep red erythrocytes followed by purification over a density gradient e.g. a FICOLL® Hypaque gradient.
- unwanted cells like B-cells, NK cells or other cell populations can be removed prior to the analysis.
- a preferred method for removal of unwanted cells is to incubate the sample with marker molecules specific or one or more surface proteins on the unwanted cells immobilized unto solid support.
- An example includes use of beads coated with antibodies or other marker molecule specific for surface receptors on the unwanted cells e.g. markers directed against CD19, CD56, CD14, CD15 or others. Briefly beads coated with the specific surface marker(s) are added to the cell sample. Cells different from the wanted T-cells with appropriate surface receptors will bind the beads. Beads are removed by e.g. centrifugation or magnetic withdrawal (when using magnetic beads) and remaining cell are enriched for T-cells.
- Another example is affinity chromatography using columns with material coated with antibodies or other markers specific for the unwanted cells.
- specific antibodies or markers can be added to the blood sample together with complement, thereby killing cells recognized by the antibodies or markers.
- Gating reagents here means labeled antibodies or other labelled marker molecules identifying subsets of cells by binding to unique surface proteins or intracellular components or intracellular secreted components.
- Preferred gating reagents when using MHC multimers are antibodies and marker molecules directed against CD2, CD3, CD4, and CD8 identifying major subsets of T-cells.
- gating reagents are antibodies and markers against CDlla, CD14, CD15, CD19, CD25, CD30, CD37, CD49a, CD49e,CD56, CD27, CD28, CD45, CD45RA, CD45RO, CD45RB, CCR7, CCR5, CD62L, CD75, CD94, CD99, CD107b, CD109, CD152, CD153, CD154, CD160, CD161, CD178, CDwl97, CDw217, Cd229, CD245, CD247, Foxp3, or other antibodies or marker molecules recognizing specific proteins unique for different lymphocytes, lymphocyte populations or other cell populations. Also included are antibodies and markers directed against interleukins e.g. IL-2, IL-4, IL-6, IL-10, IL-12, IL-21; Interferons e.g., INFy, TNFa, TNF[3. or other cytokine or chemokines.
- interleukins e.g. IL-2, IL-4
- Gating reagents can be added before, after or simultaneous with addition of MHC multimer to the sample. Following labelling with MHC multimers and before analysis on a flow cytometer stained cells can be treated with a fixation reagent (e.g., formaldehyde, ethanol or methanol) to cross-link bound MHC multimer to the cell surface. Stained cells can also be analyzed directly without fixation.
- a fixation reagent e.g., formaldehyde, ethanol or methanol
- the flow cytometer can in one embodiment be equipped to separate and collect particular types of cells. This is called cell sorting.
- MHC multimers in combination with sorting on a flow cytometer can be used to isolate antigen specific T-cell populations. Gating reagents as described above can be included further specifying the T-cell population to be isolated.
- Magentic-activated cell sorting can also be used to isolate specific T cell populations, wherein T cells are stained with the MHC multimers, followed by capture with magnetic beads coated with an anti-MHC multimer antibody (e.g, anti-Flag or other tag, anti-streptavidin, anti-MHC, etc.). Isolated and collected specific T-cell populations can then be further manipulated as described elsewhere herein, e.g. expanded in vitro.
- the concentration of MHC-peptide specific T-cells in a sample can be obtained by staining blood cells or other cell samples with MHC multimers and relevant gating reagents followed by addition of an exact amount of counting beads of known concentration.
- the counting beads are microparticles with scatter properties that put them in the context of the cells of interest when registered by a flow cytometer. They can be either labelled with antibodies, fluorochromes or other marker molecules or they may be unlabelled.
- the beads are polystyrene beads with molecules embedded in the polymer that are fluorescent in most channels of the flow-cytometer. Inhere the terms “counting bead” and “microparticle” are used interchangeably.
- Beads or microparticles suitable for use include those which are used for gel chromatography, for example, gel filtration media such as SEPHADEX®.
- Suitable microbeads of this sort include, but is not limited to, SEPHADEX® G-10 having a bead size of 40-120 pm (SigmaAldrich catalogue number 27, 103-9), SEPHADEX®. G-15 having a bead size of 40-120 pm (Sigma Aldrich catalogue number 27, 104-7), SEPHADEX®. G-25 having a bead size of 20-50 pm (Sigma Aldrich catalogue number 27, 106-3), SEPHADEX®.
- G-25 having a bead size of 20-80 pm (Sigma Aldrich catalogue number 27, 107-1), SEPHADEX®.
- G-25 having a bead size of 50-150 gm (Sigma Aldrich catalogue number 27, 109-8), SEPHADEX.®.
- plastic microbeads are usually solid, they may also be hollow inside and could be vesicles and other microcarriers. They do not have to be perfect spheres in order to function in the methods described here.
- Plastic materials such as polystyrene, polyacrylamide and other latex materials may be employed for fabricating the beads, but other plastic materials such as polyvinylchloride, polypropylene and the like may also be used.
- the counting beads are used as reference population to measure the exact volume of analyzed sample.
- the sample(s) are analyzed on a flow cytometer and the amount of MHC- specific T-cell is determined using e.g., a predefined gating strategy and then correlating this number to the number of counted counting beads in the same sample.
- Detection of specific T-cells in a sample combined with simultaneous detection of activation status of T-cells can also be measured using marker molecules specific for up- or down-regulated surface exposed receptors together with MHC multimers.
- the marker molecule and MHC multimer can be labelled with the same label or different labelling molecules and added to the sample simultaneously or sequentially or separately. 1. Detection of Individual T-Cells in Fluid Samples Using Microscopy
- Microscopy comprises any type of microscopy including optical, electron and scanning probe microscopy, Bright field microscopy, Dark field microscopy, Phase contrast microscopy, Differential interference contrast microscopy, Fluorescence microscopy, Confocal laser scanning microscopy, X-ray microscopy, Transmission electron microscopy, Scanning electron microscopy, atomic force microscope, Scanning tunneling microscope and photonic force microscope. This can be done as follows: A suspension of T-cells are added to MHC multimers, the sample washed and then the amount of MHC multimer bound to each cell is measured. Bound MHC multimers may be labelled directly or measured through addition of labelled marker molecules.
- the sample is then spread out on a slide or similar in a thin layer able to distinguish individual cells and labelled cells identified using a microscope.
- a microscope is used for the analysis.
- MHC multimers can be labeled with a flourochrome or bound MHC multimer detected with a fluorescent antibody. Cells with bound fluorescent MHC multimers can then be visualized using e.g. an immunofluorescence microscope or a confocal fluorescence microscope.
- IHC is a method where MHC multimers can be used to directly detect specific T-cells e.g. in sections of solid tissue.
- sections of fixed or frozen tissue sample are incubated with MHC multimer allowing MHC multimer to bind specific T-cells in the tissue.
- the MHC multimer may be labelled with a fluorochrome, chromophore, or any other labelling molecule that can be detected.
- the labeling of the MHC multimer may be directly or through a second marker molecule.
- the MHC multimer can be labelled with a tag that can be recognized by e.g. a secondary antibody, optionally labelled with HRP or another label.
- the bound MHC multimer is then detected by its fluorescence or absorbance (for fluorophore or chromophore), or by addition of an enzyme-labelled antibody directed against this tag, or another component of the MHC multimer (e.g. one of the protein chains, a label on the one or more multimerization domain).
- the enzyme can e.g. be Horseradish Peroxidase (HRP) or Alkaline Phosphatase (AP), both of which convert a colorless substrate into a colored reaction product in situ. This colored deposit identifies the binding site of the MHC multimer and can be visualized under e.g. alight microscope.
- the MHC multimer can also be directly labelled with e.g. HRP or AP, and used in IHC without an additional antibody.
- the detection of T-cells in solid tissue includes use of tissue embedded in paraffin, from which tissue sections are made and fixed in formalin before staining.
- Antibodies are standard reagents used for staining of formalin-fixed tissue sections; these antibodies often recognize linear epitopes.
- most MHC multimers are expected to recognize a conformational epitope on the TCR. In this case, the native structure of TCR needs to be at least partly preserved in the fixed tissue.
- staining performed tissue sections from frozen tissue blocks. In this type of staining fixation is done after MHC multimer staining.
- MHC multimers can be used to identify specific T-cells in sections of solid tissue. Instead of visualization of bound MHC multimer by an enzymatic reaction, MHC multimers are labelled with a fluorochrome or bound MHC multimer are detected by a fluorescent antibody. Cells with bound fluorescent MHC multimers can be visualized in an immunofluorescence microscope or in a confocal fluorescence microscope. This method can also be used for detection of T-cells in fluid samples using the principles described for detection of T-cells in fluid sample described elsewhere herein.
- MHC multimers may also be used for detection of T-cells in solid tissue in vivo.
- labeled MHC multimers are injected into the body of the individual to be investigated.
- the MHC multimers may be labeled with e.g. a paramagnetic isotope.
- MRI magnetic resonance imaging
- ESRjscanner MHC multimer binding T-cells can then be measured and localized.
- any conventional method for diagnostic imaging visualization can be utilized.
- gamma and positron emitting radioisotopes are used for camera and paramagnetic isotopes for MRI.
- Such support may be any which is suited for immobilization, separation etc.
- Non-limiting examples include particles, beads, biodegradable particles, sheets, gels, filters, membranes (e. g. nylon membranes), fibres, capillaries, needles, microtitre strips, tubes, plates or wells, combs, pipette tips, microarrays, chips, slides, or indeed any solid surface material.
- the solid or semi-solid support may be labelled, if this is desired.
- the support may also have scattering properties or sizes, which enable discrimination among supports of the same nature, e.g. particles of different sizes or scattering properties, color or intensities.
- MHC multimers can be used for detection of immobilized T-cells.
- ELISA Enzyme-Linked ImmunosorbentAssay
- ELISA is a binding assay originally used for detection of antibody-antigen interaction. Detection is based on an enzymatic reaction, and commonly used enzymes are e.g. HRP and AP.
- MHC multimers can be used in ELISA-based assays for analysis of purified TCR’s and T-cells immobilized in wells of a microtiter plate.
- the bound MHC multimers can be labelled either by direct chemical coupling of e.g. HRP or AP to the MHC multimer (e.g.
- the one or more multimerization domain or the MHC proteins e.g. by an HRP- or AP-coupled antibody or other marker molecule that binds to the MHC multimer.
- Detection of the enzyme-label is then by addition of a substrate (e.g. colorless) that is turned into a detectable product (e.g. colored) by the HRP or AP enzyme.
- the solid support may be made of e.g. glass, silica, latex, plastic or any polymeric material.
- the support may also be made from a biodegradable material. Generally speaking, the nature of the support is not critical and a variety of materials may be used.
- the surface of support may be hydrophobic or hydrophilic. Non-magnetic polymer beads may also be applicable. Such are available from a wide range of manufactures, e.g. Dynal Particles AS, Qiagen, Amersham Biosciences, Serotec, Seradyne, Merck, Nippon Paint, Chemagen, Promega, Prolabo, Polysciences, Agowa, and Bangs Laboratories.
- Magnetic beads or particles Another example of a suitable support is magnetic beads or particles.
- the term “magnetic” as used everywhere herein is intended to mean that the support is capable of having a magnetic moment imparted to it when placed in a magnetic field, and thus is displaceable under the action of that magnetic field.
- a support comprising magnetic beads or particles may readily be removed by magnetic aggregation, which provides a quick, simple and efficient way of separating out the beads or particles from a solution.
- Magnetic beads and particles may suitably be paramagnetic or superparamagnetic.
- Superparamagnetic beads and particles are e.g. described in EP 0 106 873. Magnetic beads and particles are available from several manufacturers, e.g. Dynal Biotech ASA (Oslo, Norway, previously Dynal AS, e.g. DYNABEADS.RTM.).
- a microarray of MHC multimers can be formed, by immobilization of different MHC multimers on solid support, to form a spatial array where the position specifies the identity of the MHC-peptide complex or specific empty MHC immobilized at this position.
- the microarray e.g., blood cells
- the cells carrying TCRs specific for MHC multimers in the microarray will become immobilized.
- the label will thus be located at specific regions of the microarray, which will allow identification of the MHC multimers that bind the cells, and thus, allows the identification of e.g. T-cells with recognition specificity for the immobilized MHC multimers.
- the cells can be labelled after they have been bound to the MHC multimers.
- the label can be specific for the type of cell that is expected to bind the MHC multimer, or the label can stain cells in general (e.g. a label that binds DNA).
- cytokine capture antibodies can be co-spotted together with MHC on the solid support and the cytokine secretion from bound antigen specific T-cells analyzed. This is possible because T-cells are stimulated to secrete cytokines when recognizing and binding specific MHC-peptide complexes.
- T-cells in a sample may also be detected indirectly using MHC multimers.
- indirect detection the number or activity of T-cells are measured, by detection of events that are the result of TCR-MHC-peptide complex interaction. Interaction between MHC multimer and T- cell may stimulate the T-cell resulting in activation of T-cells, in cell division and proliferation of T-cell populations or alternatively result in inactivation of T-cells. All these mechanisms can be measured using detection methods able to detect these events.
- Example measurement of activation include measurement of secretion of specific soluble factor e.g. cytokine that can be measured using flowcytometry as described in the section with flow cytometry, measurement of expression of activation markers e.g. measurement of expression of CD27 and CD28 and/or other receptors by e.g. flow cytometry and/or ELISA-like methods and measurement of T-cell effector function e.g. CD8 T-cell cytotoxicity that can be measured in cytotoxicity assays like chromium release assay’s know by persons skilled in the art.
- Example measurement of proliferation include but is not limited to measurement of mRNA, measurement of incorporation of thymidine or incorporation of other molecules like bromo-2’-deoxyuridine (BrdU).
- Example measurements of inactivation of T-cells include but is not limited to measurement of effect of blockade of specific TCR and measurement of apoptosis.
- T cells When contacted with a diverse population of T cells, such as is contained in a sample of the peripheral blood lymphocytes (PBLs) of a subject, those tetramers containing pMHCs that are recognized by a T cell in the sample will bind to the matched T cell. Contents of the reaction is analyzed using fluorescence flow cytometry, to determine, quantify and/or isolate those T-cells having an MHC tetramer bound thereto.
- PBLs peripheral blood lymphocytes
- the MHC multimers of the disclosure can be used in a variety of different screening assays.
- a library of fluorescently-labeled peptides derived from one or more antigens is applied to MHC multimers comprising a placeholder peptide under conditions to induce release of the placeholder peptide and binding of the antigen- derived peptides.
- Peptide exchange is monitored by fluorescence polarization assay.
- the use of placeholder peptides permits the generation of empty, peptide-receptive MHC multimers under physiological conditions.
- This screening approach can be used to identify peptide ligands that bind to an MHC molecule.
- Peptide exchange reactions can be performed in multiwell formats and under native conditions.
- Binding can be determined by a number of techniques, such as ELISA, which monitors the stability of the MHC structure, or by biophysical techniques that monitor peptide binding, such as fluorescence polarization. This screening approach can also be used to scan peptide sets (such as those derived from pathogen genomes, tumor-associated antigens or autoimmune antigens) for MHC ligands.
- the MHC multimers, and libraries thereof, disclosed herein can be used in a number of screening methods that allow for the convenient detection and quantification of antigenspecific binding to immune cell receptors.
- Such MHC multimer libraries can allow, for example, detection of T cells specific for a given antigen, multiplex detection of T cell specificities in a given sample, matching of TCR sequence with specificity (e.g, via single cell sequencing), comparative TCR affinity determination, determination of a consensus specificity sequence of a given TCR, or mapping of antigen responsiveness of T cells against sequences of interest.
- the resulting MHC multimer libraries may be used in T cell screens to determine antigen-reactive T cells as described, for example, in Simon et al. (2014) CANCER IMMUNOL RES 2(12): 1230-1244.
- the TCR-expressing cell is a T cell, e.g., a CD4+ T cell when using a MHCII multimer library.
- a cell can be transfected or transduced to express a TCR.
- a non-lymphocyte cell can be transfected or transduced to express TCR.
- the MHC multimers of the disclosure can be used to identify antigen-specific T cells of interest, for example by screening a plurality of T cells with a library of MHC multimers.
- the library comprises MHC multimers loaded with a diversity of more than 10, more than 100, more than 500, 1000, more than 2,000, more than 5,000, more than 10,000, more than 10 6 , more than 10 7 , more than 10 8 , more than 10 9 , or more than 10 10 unique peptides.
- the identification approach can comprise compartmentalizing a cell of the plurality of cells bound to a MHC multimer of the library in a single compartment, wherein the MHC multimer comprises a unique identifier; and determining the unique identifier for each MHC multimer bound to the compartmentalized cell.
- a compartment can be a separate space, e.g., a well, a plate, a divided boundary, a phase shift, a vessel, a vesicle, a cell, etc.
- compositions and methods disclosed herein can be used to identify a plurality of peptides that bind to a TCR. In some embodiments, the compositions and methods disclosed herein can be used to identify a plurality of TCRs that bind an MHC. In some embodiments, the compositions and methods disclosed herein can be used to identify a plurality of TCRs that bind a plurality of MHCs (for example, a plurality of TCRs that bind to MHC multimers derived from a pathogen library, cancer library, or autoimmune library).
- compositions and methods disclosed herein are used for identifying TCR-antigen specificity.
- identity of a TCR on a selected T cell is determined by sequencing (e.g., sequencing a variable, hypervariable region or complementarity determining region (CDR) of a TCR).
- identity of the peptide of the MHC bound which binds to a TCR is determined by sequencing (e.g, using an identifier as disclosed herein).
- MHC multimers of the disclosure can be used for the detection of antigen-specific T cells by flow cytometry or for can be used for T-cell purification.
- the compositions and methods of the disclosure allow for the production of very large collections of peptide-loaded MHC multimers that are well suited for rapid identification of helper T cell (i.e. , CD4+ T cell) antigens when using MHCII multimers.
- MHC multimers that are attached to solid surfaces can be used to probe T cell function.
- the peptide-MHC antigenic complexes fixed to the solid surface can function to stimulate T cell activity through the TCR, thereby allowing for study of downstream T cell functions subsequent to TCR stimulation.
- compositions and methods disclosed herein are used to determine how mutations in an identified MHC-binding peptide affect TCR binding. In some embodiments, the compositions and methods disclosed herein are used to identify mutations in an identified MHC-binding peptide that result in enhanced or reduced TCR binding affinity. In some embodiments, the compositions and methods disclosed herein are used to identify mutations in an identified MHC-binding peptide that retain TCR binding affinity. In some embodiments, the compositions and methods disclosed herein are used to identify mutations in an identified MHC-binding peptide that result in loss of TCR binding affinity.
- compositions and methods disclosed herein are used to determine how mutations in a TCR identified using the methods described herein alter the binding of a peptide epitope. In some embodiments, the compositions and methods disclosed herein are used to identify mutations in a TCR that result in decreased or increased binding affinity for a peptide epitope. In some embodiments, the compositions and methods disclosed herein can be used to identify mutations in a TCR that retain binding of a peptide epitope. In some embodiments, the compositions and methods disclosed herein can be used to identify mutations in a TCR that result in loss of binding of a peptide epitope.
- the methods disclosed herein are performed on T cells from a plurality of subjects.
- analysis of data from multiple subjects allows identification of MHC-binding peptide epitopes recognized by multiple subjects.
- analysis of data from multiple subjects allows identification of MHC-binding peptide epitopes recognized by multiple TCR clonotypes.
- analysis of data from multiple subjects allows identification of MHC-binding peptide epitopes recognized by multiple patients, e.g., multiple cancer patients, multiple patients with an autoimmune condition, or multiple patients with protective immunity against a pathogen.
- analysis of data from multiple subjects allows identification of MHC- binding peptide epitopes recognized in subjects comprising different HLA types or alleles. In some embodiments, analysis of data from multiple subjects allows identification of distinct hypervariable or complementarity determining region sequences of TCRs that exhibit convergent antigen binding.
- the methods disclosed herein are performed using a plurality of libraries.
- analysis of data from multiple libraries allows identification of shared reactive MHC-binding peptide epitopes between libraries, e.g., antigens exhibiting TCR affinity that are present in multiple strains of a pathogen, multiple cancer types, multiple cancer patients, multiple autoimmune diseases, or multiple autoimmune conditions.
- analysis of data from multiple libraries allows identification of distinct reactive MHC-binding peptide epitopes among libraries, e.g., antigens present in a subset of pathogen strains, cancers, conditions, or patients.
- T cells identified using an MHC multimer library of the disclosure are subjected to gene expression analysis (e.g., RNA-seq, qPCR).
- gene expression analysis is conducted on cells identified as possessing a receptor exhibiting specificity for a peptide in a library of the disclosure.
- cells determined to express TCRs that bind to an MHC multimer derived from a pathogen library, cancer library, or autoimmune library are subjected to gene expression analysis.
- Gene expression analysis can be global or targeted.
- Genes analyzed for expression include, but are not limited to, genes with known functions, genes coding for immune effector molecules (e.g., perforin, granzyme, cytokines, chemokines), immune checkpoint molecules, pro- inflammatory molecules, anti-inflammatory molecules, lineage markers, integrins, selectins, lymphocyte memory markers, death receptors, caspases, cell cycle checkpoint molecules, enzymes, phosphatases, kinases, lipases, and metabolic genes.
- immune effector molecules e.g., perforin, granzyme, cytokines, chemokines
- immune checkpoint molecules e.g., pro- inflammatory molecules, anti-inflammatory molecules, lineage markers, integrins, selectins, lymphocyte memory markers, death receptors, caspases, cell cycle checkpoint molecules, enzymes, phosphatases, kinases, lipases, and metabolic genes.
- gene expression analysis can be conducted concurrently with MHC multimer library screening. In some embodiments, gene expression analysis can be conducted after analysis of MHC multimer library screening results. In some embodiments, gene expression analysis can be conducted before analysis of MHC multimer library screening results. In some embodiments, gene expression analysis allows for immunotyping of cells identified as of interest from MHC-T cell receptor pairings produced using the methods described herein.
- a library comprising a plurality of pMHC multimers as described herein is contacted with a T cell sample, and one or more T cell functions are determined including, but not limited to, T cell proliferation, T cell cytotoxicity, suppression of T cell proliferation, suppression by a T cell, and cytokine production of a T cell.
- MHC multimers that can induce the functional property can then be made into a peptide library subset.
- a library subset can comprise MHC multimers that induce proliferation of a T cell upon binding to TCR, cytotoxicity upon binding to TCR, T cell suppression upon binding to TCR, suppression by a T cell upon binding to TCR, cytokine production upon binding to TCR, or any combination thereof.
- Proliferation can be determined by, for example, a dye-dilution assay (e.g., CFSE dilution assay), or quantification of DNA replication (e.g., BrdU incorporation assay).
- Cytotoxicity can be determined by, for example, assays that are based on release of an intracellular enzyme by dead cells (e.g., lactate dehydrogenase), dye exclusion assays (e.g, propidium iodide), or expression of cytolytic markers (e.g., granzyme, CD 107a) by flow cytometry or qPCR.
- Cytokine production can be determined by, for example, ELISA, multiplex immunoassay, intracellular cytokine staining, ELISPOT, Western Blot, or qPCR.
- T cell suppression can be determined by, for example, co-incubating a T cell clone with effector cells and target antigen, and measuring proliferation, cytotoxicity, cytokine production, expression of activation markers, etc.
- compositions and methods disclosed herein are used to identify antigen-specific T cell effector clones associated with protective immunity, non- protective immunity, or autoimmunity. In some embodiments, compositions and methods disclosed herein are used to identify antigen-specific T cell effector clones that exhibit anergy, exhaustion, tolerogenic properties, autoimmune properties, inflammatory properties, or anti-inflammatory properties (e.g., Tregs).
- compositions and methods disclosed herein are used to identify antigen-specific T cell effector clones that exhibit certain effector or memory properties (e.g., naive, terminal effector, effector memory, central memory, resident memory, Tnl, TH2, TH17, TH9, Tel, Tc2, Tcl7, production of certain cytokines).
- effector or memory properties e.g., naive, terminal effector, effector memory, central memory, resident memory, Tnl, TH2, TH17, TH9, Tel, Tc2, Tcl7, production of certain cytokines.
- a TCR identified using compositions and methods disclosed herein are used as part of a therapeutic intervention.
- a TCR sequence, TCR variable region sequence, or CDR sequence can be transfected or transduced into T cells to generate modified T cells of the same antigenic specificity.
- the modified T cells can be expanded, polarized to a desired effector phenotype (e.g., Tnl, Tel, Treg), and infused into a subject.
- a desired effector phenotype e.g., Tnl, Tel, Treg
- multiple TCRs identified using compositions and methods disclosed herein are used in an oligoclonal therapy.
- a peptide, ligand, agonist, antagonist, antigen, or epitope identified using methods disclosed herein is used as part of a therapeutic intervention.
- a peptide, antigen, or epitope is used to expand a population of cells ex vivo, e.g. using antigen presenting cells, artificial antigen presenting cells, immobilized peptide, or soluble peptide.
- expanded cells are infused into a patient.
- peripheral blood lymphocytes are expanded.
- tumor-infiltrating lymphocytes (TILs) are expanded.
- Tnl cells are expanded.
- cytotoxic T lymphocytes are expanded.
- T regulatory cells are expanded.
- compositions and methods disclosed herein are used to identify MHC-binding antigenic peptides for use in development of a vaccine, e.g. a subunit vaccine, a vaccine eliciting coverage against a range of protective antigens, or a universal vaccine.
- a vaccine e.g. a subunit vaccine, a vaccine eliciting coverage against a range of protective antigens, or a universal vaccine.
- compositions and methods disclosed herein can be used for diagnosis of a medical condition.
- compositions and methods disclosed herein are used to guide clinical decision making, e.g. treatment selection, identification of prognostic factors, monitoring of treatment response or disease progression, or implementation of preventative measures.
- compositions and methods disclosed herein can be used in the selection and/or design of treatments for medical conditions, in particular in the selection of antigen-specific T cells (e.g., CD4+ helper T cells), or TCRs derived therefrom, for use in adoptive transfer T cell therapy.
- antigen-specific T cells e.g., CD4+ helper T cells
- the MHC multimers can be used to identify T cells within a patient sample the react to an antigen(s) of interest, such as a cancer antigen(s) or pathogen antigen(s) to thereby select those cells for expansion in vitro followed by reintroduction into the patient.
- TCRs identified from such antigen-specific T cells can be sequences and recombinantly introduced into T cells to increase the population of cells expressing TCRs that bind to an antigen(s) of therapeutic interest in a patient.
- the first step in this “tetramerization by exchange” is to produce highly pure and homogenous recombinant MHCII loaded with a cleavable placeholder peptide, illustrated schematically in FIG. 1.
- a representative sequence for the MHCII alpha chain is shown in SEQ ID NO: 1.
- This MHCII alpha chain sequence of SEQ ID NO: 1 (HLA-DRA*01:01) includes a Myc tag and a Sorttag.
- a representative starting sequence for the MHCII beta chain is shown in SEQ ID NO: 2, which is the full-length HLA-DRB1 *01:01 sequence, including the signal peptide.
- a placeholder peptide is linked to the N-terminus of the MHCII beta chain via a protease cleavable linker.
- a representative sequence for a suitable placeholder peptide (the CLIP peptide) is shown in SEQ ID NO: 3.
- a representative sequence for the resultant peptide-MHCII beta chain construct is shown in SEQ ID NO: 4.
- the sequence of SEQ ID NO: 4 is the HLA-DRB1 *01:01 beta chain with the CLIP peptide and cleavable linker at the N-terminus and an AviTag and His6 tag at the C-terminus.
- the second piece of the exchange reaction is a variant in which an MHCII restricted peptide is tethered via a linker to streptavidin, referred to as SA-peptide.
- FIG. 2A schematically illustrates the nucleic acid construct encoding the SA-peptide monomer.
- FIG. 2B schematically illustrates the SA-peptide tetramer that results from in vitro transcription/translation (IVTT) of the SA-peptide monomer construct, followed by selfassembly of the tetramer.
- IVTT in vitro transcription/translation
- a representative MHCII restricted peptide sequence for use in the SA-peptide construct is shown in SEQ ID NO: 5, which is an analog of a hemagglutinin (HA) peptide from Influenza A virus.
- Linker #1, #2 and #3 Three representative linker sequences for tethering the peptide to SA, referred to as Linker #1, #2 and #3, are shown in SEQ ID NOs: 6, 7 and 8, respectively.
- pCDNA3.4 was used to express p*MHCII in ExpiCHO cells (Thermo Fisher Scientific).
- p*MHCII was then digested by Factor Xa (NEB) at a ratio of 5: 1 (w/w) overnight at 4°C in the presence of 1 mM CaC12. Then the protease was irreversibly inactivated by the addition of 1,5-Dansyl-Glu-Gly-Arg Chloromethyl Ketone inhibitor according to the manufacturer’s recommendations (Sigma- Aldrich). Undigested and digested samples were analyzed by SDS-PAGE, the results of which are shown in FIG. 5B. Digested samples migrated faster than non-digested samples indicating the removal of the freshly cleaved peptide under SDS-PAGE denaturative conditions.
- a nucleic acid construct was prepared that encodes an MHC Class II- restricted peptide linked to streptavidin, followed by E. coli expression and purification of the construct (as a proof of principle stand-in for in vitro transcription/translation) to thereby express streptavidin-peptide (SA-peptide) monomers, which then self-assemble to form SA- peptide tetramers.
- SA-peptide streptavidin-peptide
- Streptavidin variants were genetically fused to three C-terminal elements in the following order: linker, Influenza hemagglutinin (HA) analog peptide (rescue peptide) and a FLAG tag.
- linker Influenza hemagglutinin (HA) analog peptide (rescue peptide)
- FLAG tag a FLAG tag.
- Variants with three different types of linkers were expressed in E. coli cells: a flexible linker composed of four repeats of G4S blocks (SEQ ID NO: 6), a Proline-rich rigid linker with the GSAPKPAPKPAPAPKPAPKPAP sequence (SEQ ID NO: 7) and a flexible- rigid linker, in which the flexible region is preceding the rigid sequence (SEQ ID NO: 8).
- the amino acid sequences of the SA-peptide constructs using each of these linkers are shown in SEQ ID NOs: 9, 10 and 11, respectively.
- the three variants were named SA GS ' HA , SA Pro ' and SA GS ' Pro_HA , respectively, and are shown schematically in FIG. 6A.
- Bound proteins were eluted using 500 pg/ml of 3xFLAG competitor peptide (Sigma- Aldrich) and were subjected to SDS-PAGE analysis, the results of which are shown in FIG. 6B.
- protease-cleaved MHCII monomers loaded with the CLIP placeholder peptide (p( MHCII) and SA GS ' HA were mixed at a molar ratio of 1:8.
- the reaction was performed over night at 37°C in the presence of exchange buffer that is composed of: 100 mM sodium citrate pH 5.5, 50 mM sodium Chloride, 0.5 M EDTA, 0.1% octyl glucoside (v/v), 0.05% sodium azide (v/v), lx of SIGMAFAST protease inhibitor cocktail (Sigma-Aldrich) and 0.045% iodoacetamide (v/v).
- exchange buffer that is composed of: 100 mM sodium citrate pH 5.5, 50 mM sodium Chloride, 0.5 M EDTA, 0.1% octyl glucoside (v/v), 0.05% sodium azide (v/v), lx of SIGMAFAST protease inhibitor cocktail (Sigma-
- SA GS ' HA (i) and SA GS ' HA (4) represent one subunit of SA-peptide fusion or four subunits, respectively. Arrow indicates newly formed complex that is absent from other lanes. An asterisk represent a p*MHCII SDS species that migrates slower than its expected molecular weight.
- Factor Xa ox represent an oxidized species of the protease consisting of two disulfide-linked chains that fall apart under treatment with reducing buffer.
- ELISA assays were performed in two different formats, illustrated schematically in FIG. 8A.
- the first format was performed for all three variants (FIG. 8A, upper panel) and the second format was carried out only for SA GS-I IA (FIG. 8A, lower panel).
- 150 pl of exchange reactions were prepared in triplicates in a 96-well assay plate, where each well consisted of: lx exchange buffer, 100 ng of pj,MHCII and 5-fold serial dilutions of either free streptavidin, SA GS-I IA .
- the concentrations range of the first three was 10 4 -l 0' 3 nM and 4.5xl0 3 -0.45xl0' 2 for the latter.
- the assay plate was sealed and incubated over-night at 37°C.
- ELISA plates were coated with 100 ng/well of either L243 (conformational sensitive for MHCII) or anti-streptavidin antibodies (Abeam) representing the two ELISA formats, respectively.
- the ELISA plates were sealed and left for over-night incubation at room temperature (L243 plate) or 4C (anti-streptavidin plate).
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Immunology (AREA)
- Organic Chemistry (AREA)
- Biochemistry (AREA)
- Gastroenterology & Hepatology (AREA)
- Zoology (AREA)
- Toxicology (AREA)
- Biophysics (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Medicinal Chemistry (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Cell Biology (AREA)
- Peptides Or Proteins (AREA)
Abstract
MHC Class II peptide multimers are provided in which MHCII-binding peptides are attached to a multimerization domain to thereby create a multimer composition. This multimer composition can then be loaded with MHC Class II molecules to thereby create MHCII multimers. The multimers can further comprise oligonucleotide barcodes. Libraries of the MHCII multimers are also provided. Methods of making and using the MHCII multimers and libraries are also provided.
Description
MHC CLASS II PEPTIDE MULTIMERS AND USES THEREOF
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] This application claims the benefit of and priority to U.S. Provisional Patent Application No. 63/093,934, filed on October 20, 2020, the disclosure of which is hereby incorporated by reference in its entirety for all purposes.
SEQUENCE LISTING
[0002] The instant application contains a Sequence Listing which has been submitted electronically in ASCII format and is hereby incorporated by reference in its entirety. Said ASCII copy, created on October 19, 2021, is named REPT-l llWO_SL.txt and is 186,613 bytes in size.
BACKGROUND
[0003] Identification of peptides recognized by individual T cells is important for the understanding and treatment of immune-related diseases, as well as vaccine development for prevention of diseases. Techniques for the detection of antigen-responsive T cells exploit the interaction between a given TCR and its peptide-MHC (pMHC) recognition motif. The ability to prepare soluble MHC molecules allowed for the preparation of soluble peptide- MHC complexes, which then can be made into multimeric complexes. T cell detection using multimerized pMHC molecules has become the preferred method for detecting antigenspecific T cells in a wide variety of research and clinical situations.
[0004] MHC multimers have been used for detection of antigen-responsive T cells since Altman (Altman et al. (1996) SCIENCE 274:94-96) showed that tetramerization of peptide- loaded MHC class I (pMHCI) molecules provided sufficient stability to T cell receptor (TCR)-pMHC interactions, allowing detection of fluorescently -labeled MHC multimer- binding T cells using flow cytometry. However, since MHC Class I molecules are largely unstable when they are not part of a complex with peptide, pMHCI-based technologies were initially restricted by the tedious production of molecules in which each peptide required an individual folding and purification procedure (Bakker et al. (2005) CURR. OPIN. IMMUNOL. 17:428-433).
[0005] More recently, a variety of MHCI molecules with covalently linked peptides have been reported (e.g., reviewed by Goldberg et al. (2011) J. CELL. MOL. MED. 15:1822-1832). Several types of pMHCI microarrays systems also have been developed, but most work has
focused on optimizing the supporting surface and modifying the conditions applied during binding and/or washing. The use of these systems is also limited due to poor detection limits and low reproducibility compared to existing cytometry -based analyses. For example, a general limitation to such array-based strategies is the propensity of a given T cell to pursue all potential pMHCI interactions displayed on a given array. As a consequence, the frequency of antigen-responsive T cells in the cell preparations typically needs to be >0.1% to allow a robust readout.
[0006] MHCI multimers, and libraries thereof, have been prepared using biotinylated peptide-MHCI monomers that then associate with the biotin-binding site on streptavidin to form tetramers (see e.g., Leisner et al. (2008) PLoS ONE 3(2):el678). For the creation of MHC Class I libraries, approaches have been described in which oligonucleotide barcode labels have been conjugated to the streptavidin. However, existing strategies involve complex and/or costly approaches that limit the facile production of large libraries. For example, in one approach, individual streptavidin precursors must be barcoded individually by overlap extension PCR prior to tetramerization of biotinylated peptide-HLA monomers (Zhang et al. (2018) NATURE BIOTECH. 2018; doi: 10.1038. nbt.4282). In another approach, streptavidin-conjugated dextran, which is a costly reagent, is used to create a dextramer to which both the biotinylated peptide-HLA monomers and the biotinylated barcode oligonucleotide are complexed (Bentzen et al. 2016) NATURE BIOTECH. 34:10: 1037-1045) via the streptavidin conjugated to the dextran backbone. Empty peptide-receptive MHC Class I molecules have also been formed into multimers, which then can be loaded with peptides, but this required manipulation of the native MHCI sequence to stabilize the empty molecules with a disulfide bond to link the a, and co helices close to the F pocket (Saini et al. (2019) Sci. IMMUNOL. 4:eaau9039). Chaperone-mediated peptide exchange, using the molecule chaperone TAPE- PR. has also been used to prepare stable, empty MHCI molecules for multimerization and peptide loading (Overall et al. (2020) NAT. COM UN. 1 1 :1909).
[0007] Similar to the approach with pMHCI tetramers, soluble MHC class II molecules also have been used to prepare pMHCII tetramers, which have been used in the study of the antigenic specificity of CD4+ T helper cells (as reviewed in, for example, Nepom et al. (2002) ARTHRIT. RHEUMAT. 46:5-12; Vollers and Stem (2008) IMMUNOL. 123:305-313; Cecconi et al. (2008) CYTOMETRY 73A:1010-1018). Typically to prepare pMHCII multimers, soluble biotinylated MHCII a/ dimers are recombinantly expressed and then tetramerized by binding to streptavidin or avidin through their biotin-binding sites.
Fluorescent labeling of the streptavidin or avidin then allows for isolation of T cells that bind the pMHCII multimers by flow cytometry. With regard to antigenic peptide loading of the MHCII molecules, in one approach, a peptide is attached to the MHCII a/p dimers covalently. Some groups have generated pMHCII loaded with a covalent but cleavable “stuffer” peptide that can be exchanged with a peptide of interest under acidic conditions (Day et al. (2003) J. CLIN. INVEST. 112(6):831-842).
[0008] In an alternative approach, “empty” MHCII a/ dimers are prepared and then loaded with soluble MHCII-binding peptides (see e.g., Novak et al. (1999) J. CLIN. INVEST. 104:63- 67; Nepom et al. (2002) ARTHRIT. RHEUMAT. 46:5-12; Macaubus et al. (2006) J. IMMUNOL. 176:5069-5077). While this approach allows for greater diversity of peptide loading onto the MHCII a/p dimers, the ability to recombinantly express stable “empty” MHCII a/p dimers is limited, thus again hampering the preparation of large scale pMHCII multimer libraries. For example, production of “empty” MHCII a/p dimers by refolding from A’. coli inclusion bodies or by insect cell or mammalian cell expression has been reported, but with yields that are too low to support high throughput methods (reviewed in Vollers and Stem (2008) IMMUNOLOGY 123: 305-313).
[0009] Accordingly, there remains a need for efficient and cost effective methods of generating peptide-MHCII multimers and libraries thereof, including barcoded libraries, which may be utilized in a variety of methods, for example, screening of T cell specificity for analyses of T cell recognition, for example, at genome-wide levels rather than analyses restricted to a selection of model antigens.
SUMMARY
[0010] The present disclosure provides methods for producing barcoded, peptide loaded MHCII multimers (e.g, tetramers), including libraries thereof, as well as methods of using such multimers. Unlike prior approaches that create MHC multimers by attaching MHC molecules to a multimerization domain, the approach of the present disclosure involves attaching MHCII-binding peptides to a multimerization domain, to thereby create a multimer composition, which is then loaded with soluble MHCII molecules to create MHCII multimers. The peptide-multimerization domain composition can be made recombinantly, using a nucleic acid construct co-encoding the peptide and multimerization domain, which typically are linked by inclusion of a spacer linker between the two. Alternatively, the peptide can be attached to the multimerization domain through chemical conjugation, again
typically using a spacer linker in between. The MHCII molecules are provided as soluble alpha/beta chains, either “empty” dimers (i. e. , not loaded with peptide) or, more typically, dimers with a digestible placeholder peptide loaded into the peptide-binding groove. The empty MHCII molecules then can be loaded onto the peptide-multimerization domain composition or, for peptide-loaded dimers, they can be loaded by digestion of the placeholder peptide, followed by peptide exchange, thereby producing MHCII multimers.
[0011] The methods provide MHCII multimers that allow for ease of peptide exchange and barcode labeling of the multimers to thereby allow for efficient preparation of large MHCII multimer libraries. Accordingly, the compositions and methods described herein are suitable for routine laboratory research, as well as large scale industrial and clinical applications, in all circumstances where MHCII multimers are useful, e.g., for analysis of CD4+ T cell antigen recognition. Moreover, the MHCII multimers of the disclosure can be labeled with individual identifiers, such as oligonucleotide barcodes, to facilitate identification of library members. For example when the multimerization domain is streptavidin, since the biotinbinding sites within streptavidin are not being used for multimerization of the monomers, these biotin-binding sites are available for easy labeling using biotinylated oligonucleotide barcodes.
[0012] Accordingly, in one aspect, the disclosure pertains to a method of producing a Major Histocompatibility Complex Class II (MHCII) multimer, the method comprising:
(a) providing a multimer composition comprising a plurality of MHCII-binding peptides attached to a multimerization domain, wherein each MHCII-binding peptide within the plurality has the same amino acid sequence;
(b) providing a plurality of soluble MHCII molecules comprising an alpha chain and a beta chain (wherein, in certain embodiments, each MHCII molecule is loaded with a digestible placeholder peptide); and
(c) loading the MHCII molecules onto the multimer composition (e.g., by (i) digestion of the placeholder peptide, where applicable, and (ii) peptide exchange with the multimer composition), to thereby produce an MHCII multimer.
[0013] In one embodiment, the multimer composition is a tetramer comprising streptavidin or avidin as the multimerization domain. In one embodiment, the multimer composition further comprises a biotinylated nucleic acid that binds the multimerization domain, wherein the biotinylated nucleic acid encodes the MHCII-binding peptides. In one embodiment, the MHCII-binding peptides are produced from the biotinylated nucleic acid by in vitro
transcription/translation (IVTT). In one embodiment, the MHCII-binding peptides are attached to the multimerization domain by covalent linkage using a spacer linker. In various embodiments, the spacer linker comprises an amino acid sequence selected from the group consisting of SEQ ID NOs: 6-8 and 72-80.
[0014] In one embodiment, the MHC Class II alpha chain comprises an amino acid sequence selected from the group of sequences shown in SEQ ID NOs: 42-46. In one embodiment, the MHC Class II beta chain comprises an amino acid sequence selected from the group of sequences shown in SEQ ID NOs: 47-71. In one embodiment, the MHC Class II alpha chain and the MHC Class II beta chain comprise the amino acid sequences set forth in SEQ ID NOs: 42 and 47; 42 and 48; 42 and 49; 42 and 50; 42 and 51; 42 and 52; 42 and 53; 42 and 54; 42 and 55; 42 and 56; 42 and 57; 42 and 58; 42 and 59; 42 and 60; 42 and 61; 44 and 62; 44 and 63; 45 and 64; 46 and 65; or 144 and 146, respectively.
[0015] In various embodiments, each MHCII molecule is loaded with a digestible placeholder peptide, and wherein the MHCII molecules are contacted with the multimer composition under conditions for cleavage of the placeholder peptide, thereby to produce an MHCII multimer by peptide exchange with the multimer composition. In one embodiment, the digestible placeholder peptide is thermolabile, labile at acidic pH, enzymatically cleavable or photocleavable. In one embodiment, the digestible placeholder peptide comprises a placeholder peptide linked to the MHCII molecule by an digestible linker. In one embodiment, the digestible placeholder peptide is a CLIP peptide comprising the amino acid sequence KPVSKMRMATPLLMQA (SEQ ID NO: 3) or ATPLLMQALPMGA (SEQ ID NO: 134).
[0016] In one embodiment, peptide exchange is achieved by digestion of the placeholder peptide (e.g., by protease cleavage or UV -mediated cleavage) and combining the multimer composition and the MHCII molecules under low pH conditions. In one embodiment, digestion of the placeholder peptide involves cleavage of a linker that connects the placeholder peptide to the soluble MHCII molecules.
[0017] In one embodiment, the MHCII molecule is an empty molecule, i.e. no peptide is bound prior to the peptide exchange.
[0018] In one embodiment, the method further comprises labeling the multimer composition with an oligonucleotide barcode (e.g., a biotinylated oligonucleotide barcode, which is bound to biotin-binding sites on the multimerization domain).
[0019] In one embodiment, step (a) provides multimers comprising a plurality of MHCII - binding peptides, thereby to produce a library of MHCII multimers (i.e., a composition comprising a plurality of MHCII multimers). In one embodiment, each member of the library utilizes the same MHCII molecule. In one embodiment, each member of the library utilizes different MHCII-binding peptides.
[0020] In another aspect, the disclosure pertains to a multimer composition comprising a plurality of MHCII-binding peptides attached to a multimerization domain, wherein each MHCII-binding peptide within the plurality has the same amino acid sequence. Thus, each multimer composition comprises multiple copies of the same MHCII-binding peptide. In one embodiment, the multimerization domain is not covalently linked to a MHCII molecule. In one embodiment, the multimer composition is a tetramer, e.g., the multimerization domain comprises streptavidin or avidin. In one embodiment, the multimer composition comprises four copies of the same MHCII-binding peptide linked to streptavidin or avidin. In one embodiment, the multimer comprises a biotinylated nucleic acid that binds the multimerization domain, wherein the biotinylated nucleic acid encodes the MHCII-binding peptides. In one embodiment, the MHCII-binding peptides are produced from the biotinylated nucleic acid by IVTT.
[0021] In one embodiment, the MHCII-binding peptides are attached to the multimerization domain by covalent linkage using a spacer linker, for example prepared by recombinant expression or chemical conjugation. In one embodiment, the spacer linker used to link the peptide to the multimerization domain comprises an amino acid sequences selected from the group consisting of SEQ ID NOs: 6-8 and 72-80.
[0022] In one embodiment, the multimer composition further comprises MHCII molecules bound to the MHCII-binding peptides, each MHCII molecule comprising an alpha chain and a beta chain, to thereby create an MHCII multimer (i.e., the peptide-multimerization domain composition is loaded with MHCII molecules to thereby create MHCII multimers). In one embodiment, the MHC Class II alpha chain comprises an amino acid sequence selected from the group of sequences shown in SEQ ID NOs: 42-46. In one embodiment, the MHC Class II beta chain comprises an amino acid sequence selected from the group of sequences shown in SEQ ID NOs: 47-71. In one embodiment, the MHC Class II alpha chain and the MHC Class II beta chain comprise the amino acid sequences set forth in SEQ ID NOs: 42 and 47; 42 and 48; 42 and 49; 42 and 50; 42 and 51; 42 and 52; 42 and 53; 42 and 54; 42 and 55; 42 and 56; 42 and 57; 42 and 58; 42 and 59; 42 and 60; 42 and 61; 44 and 62; 44 and 63; 45 and 64; 46
and 65; or 144 and 146, respectively. Libraries of MHCII multimers can be made comprising a plurality of the MHCII multimers. In one embodiment, each member of the library utilizes the same MHCII molecule. In one embodiment, each member of the library utilizes different MHCII-binding peptides.
[0023] In one embodiment, the multimer composition is labeled with an oligonucleotide barcode (e.g, a biotinylated oligonucleotide barcode is bound to biotin-binding sites on the multimerization domain).
[0024] In yet another aspect, the disclosure pertains to a nucleic acid construct encoding a multimer composition subunit, wherein the nucleic acid construct encodes a polypeptide comprising an MHCII-binding peptide and a multimerization domain, linked by a spacer linker. In one embodiment, the polypeptide does not comprise a MHCII molecule. In one embodiment, the multimerization domain comprises streptavidin or avidin. In one embodiment, the nucleic acid construct further comprises a biotin moiety. In one embodiment, the spacer linker comprises an amino acid sequence selected from the group consisting of SEQ ID NOs: 6-8 and 72-80. The multimer composition (comprising the peptide linked to the multimerization domain) can be expressed by standard methods, e.g., by in vitro transcription/translation (IVTT) or by recombinant expression in a host cell using an expression vector comprising the nucleic acid construct.
[0025] In yet another aspect, the disclosure pertains to a method of isolating MHCII- multimer bound lymphocytes comprising:
(a) contacting a plurality of lymphocytes with a library of MHCII multimers of the disclosure;
(b) generating a plurality of compartments, wherein each compartment comprises a lymphocyte bound to an MHCII multimer of the library; and
(c) isolating an MHCII-multimer bound lymphocyte from the compartment. In one embodiment, the lymphocyte is a T cell (e.g., a CD4+ T cell). In one embodiment, each member of the library of MHCII multimers is labeled with an oligonucleotide barcode and the method further comprises decoding the oligonucleotide barcode of the isolated MHCII-multimer bound to the lymphocyte. This allows for identification of the peptide sequence recognized by the lymphocyte.
[0026] For a fuller understanding of the nature and advantages of the present disclosure, reference should be had to the ensuing detailed description taken in conjunction with the
accompanying figures. The present disclosure is capable of modification in various respects without departing from the present disclosure. Accordingly, the figures and description of these embodiments are not restrictive.
BRIEF DESCRIPTION OF THE FIGURES
[0027] The novel features of the invention are set forth with particularity in the appended claims. A better understanding of the features and advantages of the present invention will be obtained by reference to the following detailed description that sets forth illustrative embodiments, in which the principles of the invention are utilized, and the accompanying drawings of which:
[0028] FIG. 1 is a schematic diagram of recombinant MHC Class II alpha and beta chains loaded with a placeholder CLIP peptide via a cleavable linker.
[0029] FIG. 2A-2B are schematic diagrams of streptavidin-peptide reagents. FIG. 2A is a schematic diagram of a nucleic acid construct encoding a streptavidin-peptide monomer. FIG. 2B is a schematic diagram of a streptavidin-peptide tetramer resulting from in vitro transcription/translation (IVTT) of the streptavidin-peptide monomer nucleic acid construct.
[0030] FIG. 3 is a schematic diagram showing cleavage of the placeholder linker of p*MHCII by a protease and exchange with a rescue peptide that is fused to SA tetramer.
[0031] FIG. 4A is a schematic diagram showing preparation of a placeholder peptide-loaded MHCII (p*MHCII) and protease cleavage thereof, preparation of an SA-peptide tetramer comprising a rescue peptide and low pH-mediated peptide exchange, wherein single-template encapsulation is achieved by either drop-based or well-based methods. FIG. 4B is a schematic diagram showing an exemplary barcoded SA-peptide loaded MHCII tetramer binding a cognate T cell receptor (TCR) in solution or on the surface of a cell. The figure shows an exemplary TCR molecule bound to the peptide-loaded MHCII tetramer. It is contemplated that the tetramer can also bind two, three, or four TCR molecules (e.g., soluble TCRs or transmembrane TCRs on a T cell surface).
[0032] FIG. 5A-5B show results of analysis of p*MHCII recombinant production and Factor Xa protease cleavage. FIG. 5A shows the elution profile of recombinant p*MHCII following purification by size exclusion chromatography. FIG. 5B shows the results of SDS-PAGE analysis of p*MHCII before and after cleavage with Factor Xa.
[0033] FIG. 6A-6C show preparation and analysis of three SA-peptide constructs. FIG. 6A schematically illustrates the three constructs, each using a different linker, referred to as SAGS'HA, SAPro'HA and SAGS'Pro'HA. FIG. 6B shows the results of SDS-PAGE analysis of the expressed constructs. Lanes 1, 4 and 7 represent the supernatant fraction after cell lysis. Lanes 2, 5 and 8 represent the unbound fraction following binding to anti-FLAG resin. Lanes 3, 6 and 9 represent the elution fraction following incubation with 3xFLAG competitor peptide. Each variant is indicated at the bottom of the gel. FIG. 6C shows the size exclusion chromatography elution profile of the three constructs following FLAG affinity purification.
[0034] FIG. 7 shows the results of SDS-PAGE analysis following peptide exchange between pj,MHCII monomers and SA-peptide tetramers, showing the formation of an SDS-resistant complex following the peptide exchange reaction.
[0035] FIG. 8A-8B shows the detection of pMHCII-SA-HA using ELISA. FIG. 8A illustrates the two different ELISA formats tested. In the first format, (FIG. 8A, upper panel), L243 was used as a capturing antibody while biotin-HRP binding to newly-exchanged SA-peptide was used for detection. In the second format (FIG. 8A, lower panel), antistreptavidin was used as a capturing antibody while L243-HRP binding to MHCII was used for detection. FIG. 8B shows the corresponding ELISA results for each capturing/detection format.
DETAILED DESCRIPTION
Definitions
[0036] All technical and scientific terms used herein, unless otherwise defined below, are intended to have the same meaning as commonly understood by one of ordinary skill in the art. Mention of techniques employed herein are intended to refer to the techniques as commonly understood in the art, including variations on those techniques or substitutions of equivalent techniques that would be apparent to one of skill in the art. While the following terms are believed to be well understood by one of ordinary skill in the art, the following definitions are set forth to facilitate explanation of the presently disclosed subject matter.
[0037] As used herein, “about” will be understood by persons of ordinary skill and will vary to some extent depending on the context in which it is used. If there are uses of the term which are not clear to persons of ordinary skill given the context in which it is used, “about” will mean up to plus or minus 10% of the particular value.
[0038] As used herein, an “altered peptide ligand” or “APL” refers to an altered or mutated version of a peptide ligand, such as an MHC binding peptide. The altered or mutated version of the peptide ligand contains at least one structural modification (e.g., amino acid substitution) as compared to the peptide ligand from which it is derived. For example, a panel of APLs can be prepared by systematic or random mutation of a known MHC binding peptide, to thereby create a pool of APLs that can be used as a library of MHC binding peptides for loading onto MHC Multimers as described herein.
[0039] As used herein, the term “and/or” when used in the context of a list of entities, refers to the entities being present singly or in any possible combination or subcombination.
[0040] The term “antigenic determinant” or “epitope” refers to a site on an antigen to which the variable domain of a T-cell receptor, an MHC molecule or antibody specifically binds. Epitopes can be formed both from contiguous amino acids or noncontiguous amino acids juxtaposed by tertiary folding of a protein. Epitopes formed from contiguous amino acids are typically retained on exposure to denaturing solvents, whereas epitopes formed by tertiary folding are typically lost on treatment with denaturing solvents. An epitope typically includes at least 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 or 15 amino acids in a unique spatial conformation and typically can include up to about 25 amino acids. Methods for determining what epitopes are bound by a given TCR or antibody (i.e., epitope mapping) are well known in the art and include, for example, immunoblotting and immunoprecipitation assays, wherein overlapping or contiguous peptides from the antigen are tested for reactivity with the given TCR or immunoglobulin. Methods of determining spatial conformation of epitopes include techniques in the art and those described herein, for example, x-ray crystallography nuclear magnetic resonance, cryogenic electron microscopy (cryo-EM), hydrogen deuterium exchange mass spectrometry (HDX-MS), and site-directed mutagenesis (see, e.g., EPITOPE MAPPING PROTOCOLS IN METHODS IN MOLECULAR BIOLOGY, Vol. 66, G. E. Morris, Ed. (1996)).
[0041] The term “avidity” as used herein, refers to the binding strength of as a function of the cooperative interactivity of multiple binding sites of a multivalent molecule (e.g., a soluble multimeric pMHC-immunoglobulin protein) with a target molecule. A number of technologies exist to characterize the avidity of molecular interactions including switchSENSE and surface plasmon resonance (Gjelstrup et al. (2012) J. IMMUNOL. 188:1292- 1306; Vorup-Jensen (2012) ADV. DRUG. DELIV. REV. 64:1759-1781).
[0042] As used herein a “barcode”, also referred to as an oligonucleotide barcode, is a typically short nucleotide sequence (e.g, about 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 or 15 nucleotides long or longer) that identifies a molecule to which it is conjugated. Barcodes can be used, for example, to identify molecules in a reaction mixture. Barcodes uniquely identify the molecule to which it is conjugated, for example, by performing reverse transcription using primers that each contain a “unique molecular identifier” barcode. In other embodiments, primers can be utilized that contain “molecular barcodes” unique to each molecule. The process of labeling a molecule with a barcode is referred to herein as “barcoding.” A “DNA barcode” is a DNA sequence used to identify a target molecule during DNA sequencing. In some embodiments, a library of DNA barcodes is generated randomly, for example, by assembling oligos in pools. In other embodiments, the library of DNA barcodes is rationally designed in silico and then manufactured.
[0043] “Binding affinity” generally refers to the strength of the sum total of noncovalent interactions between a single binding site of a molecule (e.g., a TCR, pMHC) and its binding partner. Unless indicated otherwise, as used herein, “binding affinity” refers to intrinsic binding affinity which reflects a 1: 1 interaction between members of a binding pair (e.g., TCR and peptide-MHC). The affinity of a molecule X for its partner Y can generally be represented by the dissociation constant (Kd). For example, the Kd can be about 200 nM, 150 nM, 100 nM, 60 nM, 50 nM, 40 nM, 30 nM, 20 nM, 10 nM, 8 nM, 6 nM, 4 nM, 2 nM, 1 nM, or stronger, including up to 1 pM. Affinity can be measured by common methods known in the art, including those described herein. A variety of methods of measuring binding affinity are known in the art, any of which can be used for purposes of the present disclosure.
[0044] The term “bioorthogonal chemistry” refers to any chemical reaction that can occur inside of living systems without interfering with native biochemical processes. The term includes chemical reactions that are chemical reactions that occur in vitro at physiological pH in, or in the presence of water. To be considered bioorthogonol, the reactions are selective and avoid side-reactions with other functional groups found in the starting compounds. In addition, the resulting covalent bond between the reaction partners should be strong and chemically inert to biological reactions and should not affect the biological activity of the desired molecule.
[0045] As used herein, the terms “carrier” and “pharmaceutically acceptable carrier” includes any and all solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents, and the like that are physiologically compatible.
[0046] As used herein, the term “cleavage site” or “cleavable moiety” refers to a site, a motif or sequence that is cleavable, such as by an enzyme (e.g., a protease) or by particular reaction conditions. In some embodiments, the cleavage moiety comprises a protein, e.g, enzymatic, cleavage site. In some embodiments, the cleavage moiety comprises a chemical cleavage site, e.g., through exposure to oxidation/reduction conditions, light/sound, temperature, pH, pressure, etc.
[0047] The term “click chemistry” refers to a set of reliable and selective bioorthogonal reactions for the rapid synthesis of new compounds and combinatorial libraries. Properties of click reactions include modularity, wideness in scope, high yielding, stereospecificity and simple product isolation (separation from inert by-products by non-chromatographic methods) to produce compounds that are stable under physiological conditions. In radiochemistry and radiopharmacy, click chemistry is a generic term for a set of labeling reactions which make use of selective and modular building blocks and enable chemoselective ligations to radiolabel biologically relevant compounds in the absence of catalysts. A “click reaction” can be with copper, or it can be a copper-free click reaction.
[0048] As used herein, the term “cross-linking unit” can refer to a molecule that links to another (same or different) molecule. In some embodiments, the cross-linking unit is a monomer. In some embodiments, the cross-link is a chemical bond. In some embodiments, the cross-link is a covalent bond. In some embodiments, the cross-link is an ionic bond. In some embodiments, the cross-link alters at least one physical property of the linked molecules, e.g., a polymer’s physical property.
[0049] As used herein, the term “endoprotease” refers to a protease that cleaves a peptide bond of a non-terminal amino acid.
[0050] As used herein, the term “epitope” (as in “peptide epitope”) refers to a portion of an antigen (e.g., antigenic protein) that binds to (interacts with or is recognized by) an immune receptor. Thus, a T cell receptor recognizes and binds to an MHC molecule complexed with (loaded with) a peptide epitope.
[0051] The terms “exchangeable pMHC polypeptide”, “exchangeable pMHC multimers”, and “placeholder-peptide loaded MHC polypeptide”, which are used interchangeably herein,
refer to MHC monomers and MHC multimers, comprising a placeholder peptide in the binding groove of the MHC polypeptide, and are also referred to as “p*MHC” monomers or multimers. “Exchangeable” refers to the property of a p*MHC monomer or p*MHC multimer allowing for the exchange of the placeholder peptide with an antigenic peptide. In one embodiment, the exchangeable pMHC or p*MHC polypeptide comprises an MHC Class II molecule with an MHC Class Il-binding peptide in the binding groove of the MHC Class II molecule.
[0052] As used herein, the term “expression construct” refers to a vector designed for gene expression, e.g., in a host cell. An expression vector promotes the expression (i.e., transcription/translation) of an encoded polypeptide (e.g., fusion polypeptide). Typically, the vector is a plasmid, although other suitable vectors, including viral and non-viral vectors are also encompassed by the term “expression construct.”
[0053] A “fusion protein” or “fusion polypeptide” as used interchangeably herein refers to a recombinant protein prepared by linking or fusing two polypeptides into a single protein molecule.
[0054] The term “isolated” as applied to MHC monomers herein refers to an MHC glycoprotein, which is in other than its native state, for example, not associated with the cell membrane of a cell that normally expresses MHC. This term embraces a full length subunit chain, as well as a functional fragment of the MHC monomer. A functional fragment is one comprising an antigen binding site and sequences necessary for recognition by the appropriate T cell receptor. It typically comprises at least about 60-80%, typically 90-95% of the sequence of the full-length chain. An “isolated” MHC subunit component may be recombinantly produced or solubilized from the appropriate cell source. In one embodiment, the “isolated” MHC monomer is an MHC Class II monomer, such as a soluble form of the MHC Class II a/p chains.
[0055] As used herein, the term “identifier” refers to a readable representation of data that provides information, such as an identity, that corresponds with the identifier.
[0056] As used herein, the terms “linked,” “conjugated,” “fused,” or “fusion,” are used interchangeably when referring to the joining together of two more elements or components or domains, by whatever means including recombinant or chemical means.
[0057] As used herein, the term “linker sequence” refers to a nucleotide sequence, and corresponding encoded amino acid sequence, within an expression construct that serves to
link or separate two polypeptides, such as two polypeptide domains of a fusion protein. For example, an intervening linker sequence can serve to provide flexibility and/or additional space between the two polypeptides that flank the linker.
[0058] As used herein, the terms “operatively linked” and “operably linked” are used interchangeably to describe configurations between sequences within an expression construct that allow for particular operations to carried out. For example, when a regulatory sequence is “operatively linked” to a coding sequence within an expression construct, the regulatory sequence operates to regulate the expression of the coding sequence. Similarly, when a cleavage sequence (site) is “operatively linked” to a peptide sequence within an expression construct, cleavage at the cleavage sequence operates to cleave the peptide sequence away from the rest of the polypeptide encoded by the expression construct.
[0059] The term “Major Histocompatibility Complex” or “MHC” refers to genomic locus containing a group of genes that encode the polymorphic cell-membrane-bound glycoproteins known as MHC classical class I and class II molecules that regulate the immune response by presenting peptides of fragmented proteins to circulating cytotoxic and helper T lymphocytes, respectively. In humans this group of genes is also called the “human leukocyte antigen” or “HLA” system. Human MHC class I genes encode, for example, HLA-A, HLA-B and HLA- C molecules. HLA-A is one of three major types of human MHC class I cell surface receptors. The others are HLA-B and HLA-C. The HLA-A protein is a heterodimer, and is composed of a heavy a chain and smaller [3 chain. The a chain is encoded by a variant HLA- A gene, and the P chain is an invariant 2 microglobulin ( 2m) polypeptide. The 2 microglobulin polypeptide is coded for by a separate region of the human genome. HLA- A*02 (A*02) is a human leukocyte antigen serotype within the HLA-A serotype group. The serotype is determined by the antibody recognition of the a2 domain of the HLA-A a-chain. For A*02, the a chain is encoded by the HLA-A*02 gene and the chain is encoded by the B2M locus. Human MHC class II genes encode, for example, HLA-DPA1, HLA-DPB1, HLA-DQA1, HLA-DQB1, HLA-DRA and HLA-DRB1. The complete nucleotide sequence and gene map of the human major histocompatibility complex is publicly available (e.g, The MHC sequencing consortium, Nature 401:921-923, 1999).
[0060] As used herein, the terms “MHC molecule” and “MHC protein” are used herein to refer to the polymorphic glycoproteins encoded by the MHC class I and MHC class II genes, which are involved in the presentation of peptide epitopes to T cells. The terms “MHC class I” or “MHC I” are used interchangeably to refer to protein molecules comprising an a chain
composed of three domains (al, a2 and a3), and a second, invariant P2-microglobulin. The a3 domain is linked to the transmembrane domain, anchoring the MHC class I molecule to the cell membrane. Antigen-derived peptide epitopes, which are located in the peptide- binding groove, in the central region of the al/a2 heterodimer. MHC Class I molecules such as HLA-A are part of a process that presents short polypeptides to the immune system. These polypeptides are typically 9-11 amino acids in length and originate from proteins being expressed by the cell, which can be endogenous proteins or exogenous proteins (e.g., viral or bacterial proteins, vaccine proteins). MHC class I molecules present antigen to CD8+ cytotoxic T cells. The terms “MHC class II” and “MHC II” are used interchangeably to refer to protein molecules containing an a chain with two domains (al and a2) and a P chain with two domains (pi and P2). The peptide-binding groove is formed by the al/pi heterodimer. MHC class II molecules present antigen to specific CD4+ T cells. Antigens delivered endogenously to APCs are processed primarily for association with MHC class I. Antigens delivered exogenously to APCs are processed primarily for association with MHC class II.
[0061] As used herein, MHC proteins (MHC Class I or Class II proteins) also includes MHC variants which contain amino acid substitutions, deletions or insertions and yet which still bind MHC peptide epitopes (MHC Class I or MHC Class II peptide epitopes). The term also includes fragments of all these proteins, for example, the extracellular domain, which retain peptide binding.
[0062] The term “MHC protein” also includes MHC proteins of non-human species of vertebrates. MHC proteins of non-human species of vertebrates play a role in the examination and healing of diseases of these species of vertebrates, for example, in veterinary medicine and in animal tests in which human diseases are examined on an animal model, for example, EAE (experimental autoimmune encephalomyelitis) in mice (mus musculus), which is an animal model of the human disease multiple sclerosis. Non-human species of vertebrates are, for example, and more specifically mice (mus musculus), rats (rattus norvegicus), cows (bos taurus), horses (equus equus) and green monkeys (macaca mulatta). MHC proteins of mice are, for example, referred to as H-2 -proteins, wherein the MHC class I proteins are encoded by the gene loci H2K, H2L and H2D and the MHC class II proteins are encoded by the gene loci H2I.
[0063] A “peptide free MHC polypeptide” or “peptide free MHC multimer” as used herein refers to an MHC monomer or MHC multimer which does not contain a peptide in binding groove of the MHC polypeptide. Peptide free MHC monomers and multimers are also
referred to as “empty”. In one embodiment, the peptide free MHC polypeptide or multimer is an MHC Class I polypeptide or multimer. In another embodiment, the peptide free MHC polypeptide or multimer is an MHC Class II polypeptide or multimer.
[0064] As used herein, the term “multimer” refers to a plurality of units. In some embodiments, the multimer comprises one or more different units. In some embodiments, the units in the multimer are the same. In some embodiments, the units in the multimer are different. In some embodiments, the multimer comprises a mixture of units that are the same and different.
[0065] As used herein, the term “multimer composition” refers to a composition comprising a plurality of MHCII-binding peptides attached to a multimerization domain (e.g., streptavidin) thereby creating a multimer (e.g., tetramer) displaying the plurality e.g., four) MHCII- binding peptides. Such a multimer composition can be loaded with MHCII molecules as described herein, thereby creating MHCII multimers.
[0066] The terms “peptide epitope”, “MHC peptide epitope”, “MHC peptide antigen” and “MHC ligand” are used interchangeably herein and refer to an MHC ligand that can bind in the peptide binding groove of an MHC molecule. The peptide epitope can typically be presented by the MHC molecule. A peptide epitope typically has between 8 and 25 amino acids that are linked via peptide bonds. The peptide can contain modification such as, but not limited to, the side chains of the amino acid residues, the presence of a label or tag, the presence of a synthetic amino acid, a functional equivalent of an amino acid, or the like. Typical modifications include those as produced by the cellular machinery, such as glycan addition and phosphorylation. However, other types of modification are also within the scope of the disclosure.
[0067] As used herein, the terms “peptide exchange” refers to a competition assay wherein a placeholder peptide is removed and replaced by a “exchanged peptide” (or “exchange peptide epitope”) also referred to herein as a “rescue peptide” (or “rescue peptide epitope”) or “competitor peptide” (or “competitor peptide epitope). Typically, peptide exchange occurs under conditions in which the placeholder peptide is released by cleavage of the peptide or under suitable conditions allowing rescue peptides to compete for binding to the binding pocket of an MHC monomer or multimer. For example, peptide exchange can be accomplished by temperature-induced exchange, UV -induced exchange, dipeptide-induced
exchange, pH-induced exchange, or other exchange methods known in the art, and disclosed herein.
[0068] As used herein, the term “peptide library” refers to a plurality of peptides. In some embodiments, the library comprises one or more peptides with unique sequences. In some embodiments, each peptide in the library has a different sequence. In some embodiments, the library comprises a mixture of peptides with the same and different sequences.
[0069] As used herein, the term “high diversity peptide library” refers to a peptide library with a high degree of peptide variety. For example, a high diversity peptide library comprises about 103, about 104, about 105, about 106, about 107, about 108, about 109, about IO10, about 1011, about 1012, about 1013, about 1014, about 1015, about 1016, about 1017, about 1018, about 1019, about IO20, or more different peptides.
[0070] As used herein, the term “library peptide” refers to a single peptide in the library.
[0071] As used herein, the terms “placeholder peptide” or “exchangeable peptide” are used interchangeably to refer to a peptide or peptide-like compound that binds with sufficient affinity to an MHC protein (e.g, MHCI or MHCII protein) and which causes or promotes proper folding of the MHC protein from the unfolded state or stabilization of the folded MHC protein. The placeholder peptide can subsequently be exchanged with a different peptide of interest (referred to as an exchange peptide or rescue peptide). This exchange can be accomplished by, for example, UV-induced exchange, dipeptide-induced exchange, temperature-induced exchange, pH-induced exchange, or other exchange methods known in the art.
[0072] The terms “polypeptide,” “peptide”, and “protein” are used interchangeably herein to refer to a polymer of amino acid residues. The terms apply to amino acid polymers in which one or more amino acid residue is an artificial chemical mimetic of a corresponding naturally occurring amino acid, as well as to naturally occurring amino acid polymers and non- naturally occurring amino acid polymer. The terms “isolated protein” and “isolated polypeptide” are used interchangeably to refer to a protein (e.g, a soluble, multimeric protein) which has been separated or purified from other components (e.g, proteins, cellular material) and/or chemicals. Typically, a polypeptide is purified when it constitutes at least 60 (e.g, at least 65, 70, 75, 80, 85, 90, 92, 95, 97, or 99) % by weight of the total protein in the sample.
[0073] As used herein, the term “protein folding” refers to spatial organization of a peptide. In some embodiments, the amino acid sequence influences the spatial organization or folding of the peptide. In some embodiments, a peptide may be folded in a functional conformation. In some embodiments, a folded peptide has one or more biological functions. In some embodiments, a folded peptide acquires a three-dimensional structure.
[0074] As used herein, the term “N-terminus amino acid residue” refers to one or more amino acids at the N-terminus of a polypeptide.
[0075] As used herein, the terms “small ubiquitin-like modifier moiety” or “SUMO domain” or “SUMO moiety” are used interchangeably and refer to a specific protease recognition moiety.
[0076] As used herein, the term “tag” refers to an oligonucleotide component, generally DNA, that provides a means of addressing a target molecule (e.g., an MHC Multimer) to which it is joined. For example, in some embodiments, a tag comprises a nucleotide sequence that permits identification, recognition, and/or molecular or biochemical manipulation of the molecule to which the tag is attached (e.g, by providing a unique sequence, and/or a site for annealing an oligonucleotide, such as a primer for extension by a DNA polymerase, or an oligonucleotide for capture or for a ligation reaction). The process of joining the tag to the target molecule is sometimes referred to herein as “tagging” and a target molecule that undergoes tagging or that contains a tag is referred to as “tagged” (e.g., a “tagged MHC Multimer”).” A tag can be a barcode, an adapter sequence, a primer hybridization site, or a combination thereof.
[0077] The term “T cell” refers to a type of white blood cell that can be distinguised from other white blood cells by the presence of a T cell receptor on the cell surface. There are several subsets of T cells, including, but not limited to, T helper cells (a.k.a. TH cells or CD4+ T cells) and subtypes, including THI, TH2, TH3, TH17, TH9, and TFH cells, cytotoxic T cells (a.k.a Tc cells, CD8+ T cells, cytotoxic T lymphocytes, T-killer cells, killer T cells), memory T cells and subtypes, including central memory T cells (TCM cells), effector memory T cells (TEM and TEMRA cells), and resident memory T cells (TRM cells), regulatory T cells (a.k.a. Treg cells or suppressor T cells) and subtypes, including CD4+ FOXP3+ Treg cells, CD4+FOXP3‘ Treg cells, Tri cells, Th3 cells, and Tregl7 cells, natural killer T cells (a.k.a. NKT cells), mucosal associated invariant T cells (MAITs), and gamma delta T cells (y8 T cells),
including Vy9/V62 T cells. The term “T cell cytotoxicity” includes any immune response that is mediated by CD8+ T cell activation.
[0078] As used herein, the phrase “T cell receptor” and the term “TCR” refer to a surface protein of a T cell that allows the T cell to recognize an antigen and/or an epitope thereof, typically bound to one or more major histocompatibility complex (MHC) molecules. A TCR functions to recognize an antigenic determinant and to initiate an immune response.
Typically, TCRs are heterodimers comprising two different protein chains. In the vast majority of T cells, the TCR comprises an alpha (a) chain and a beta ((3) chain. Each chain comprises two extracellular domains: a variable (V) region and a constant (C) region, the latter of which is membrane-proximal. The variable domains of a-chains and of [3-chains consist of three hypervariable regions that are also referred to as the complementarity determining regions (CDRs). The CDRs, in particular CDR3, are primarily responsible for contacting antigens and thus define the specificity of the TCR, although CDR1 of the a-chain can interact with the N-terminal part of the antigen, and CDR1 of the [3-chain interacts with the C-terminal part of the antigen. Approximately 5% of T cells have TCRs made up of gamma and delta (y/5) chains. All numbering of the amino acid sequences and designation of protein loops and sheets of the TCRs is according to the IMGT numbering scheme (IMGT, the international ImMunoGeneTics information system@imgt.cines.fr; http://imgt.cines.fr; Lefranc et al. (2003) DEV. COMP. IMMUNOL. 27:55 77.; Lefranc et al. (2005) DEV. COMP. IMMUNOL. 29:185-203).
[0079] As used herein, the terms “soluble T-cell receptor” and “sTCR” refer to heterodimeric truncated variants of TCRs, which comprise extracellular portions of the TCR a-chain and [3- chain (e.g., linked by a disulfide bond), but which lack the transmembrane and cytosolic domains of the full-length protein. The sequence (amino acid or nucleic acid) of the soluble TCR a-chain and [3-chains may be identical to the corresponding sequences in a native TCR or may comprise variant soluble TCR a-chain and [3-chain sequences, as compared to the corresponding native TCR sequences. The term “soluble T-cell receptor” as used herein encompasses soluble TCRs with variant or non-variant soluble TCR a-chain and [3-chain sequences. The variations may be in the variable or constant regions of the soluble TCR a- chain and [3-chain sequences and can include, but are not limited to, amino acid deletion, insertion, substitution mutations as well as changes to the nucleic acid sequence, which do not alter the amino acid sequence. Variants retain the binding functionality of their parent molecules.
[0080] As used herein, a “TCR/pMHC complex” refers to a protein complex formed by binding between T cell receptor (TCR), or soluble portion thereof, and a peptide-loaded MHC molecule. Accordingly, a “component of a TCR/pMHC complex” refers to one or more subunits of a TCR (e.g, Va, V[3, Ca, C ), or to one or more subunits of an MHC or pMHC class I or II molecule.
[0081] As used herein, the term “unbiased” refers to lacking one or more selective criteria.
OVERVIEW
[0082] This disclosure provides methods and compositions for the high-throughput generation of libraries containing peptide-loaded MHCII multimers containing a plurality of unique peptides in the MHC binding groove and having oligonucleotide barcode labeling to facilitate identification of library members. In the methods herein, a multimer composition is provided that comprises a plurality of MHCII-binding peptides attached to a multimerization domain. Upon expression, multimerization mediated by the multimerization domain occurs such that a multimer composition is produced that displays a plurality of MHCII-binding peptides. Typically, all peptides displayed by a particular individual multimerization domain have the same amino acid sequence, whereas a collection of such multimerization domains displays an assortment of different MHCII-binding peptides (i. e. , a library of MHCII-binding peptides). To create an MHCII multimer, a soluble MHCII molecule optionally including a cleavable placeholder peptide in the peptide-binding groove is provided. The placeholder peptide can be cleaved and peptide exchange is performed such that the MHCII molecules bind to the peptides of the multimer composition. Moreover, a binding site on the multimerization domain (e.g., the biotin-binding site of streptavidin or avidin) can be used for labeling the MHCII multimers with unique identifiers (e.g., biotinylated oligonucleotide barcodes).
[0083] The libraries of MHCII multimers provided herein are useful in a range of therapeutic, diagnostic, and research applications, essentially in any situation in which MHCII multimers are useful. For example, MHCII multimers as described herein can be used in a variety of methods, for example, to identify and isolate specific T-cells in a wide array of applications, e.g., for determining the antigenic specificity of CD4+ T cells (e.g., helper T cells).
[0084] While prior approaches for making MHCII multimers involve attachment of MHCII molecules to a multimerization domain, the present disclosure provides for attachment of MHCII-binding peptides to a multimerization domain, to create a multimer composition that
then can be loaded with MHCII molecules to thereby create MHCII multimers. This approach is further described in detail in Example 1 and non-limiting representative schematic diagrams of this approach are shown in FIG. 3, FIG. 4A and FIG. 4B. Various components and aspects of the disclosure are described in further detail in the subsections below.
L _ Multimer Compositions
[0085] The MHCII multimers of the disclosure are prepared using a peptide-multimerization domain composition as the “scaffold” onto which soluble MHCII molecules are loaded to thereby create the MHCII multimers. The peptide-multimerization domain molecule is also referred to herein as a “multimer composition” and comprises MHCII-binding peptides attached to a multimerization domain, typically with a spacer linker (e.g., a flexible linker) linking the peptide to the multimerization domain. In one embodiment, the peptide (and, optionally, linker) is attached to the N-terminus of the multimerization domain. In one embodiment, the peptide (and, optionally, linker) is attached to the C-terminus of the multimerization domain. A non-limiting representative example of a multimer composition is shown schematically in FIG. 2B, in which four copies of an MHCII-binding peptide are attached to four streptavidin (SA)subunits to create an MHCII-binding peptide-SA tetramer. Preparation of various peptide-SA tetramers is also described in detail in Example 3. The components of the multimer composition, and methods of making the composition, are described further below.
A. Multimerization Domains
[0086] Multimerization domains for use in producing the multimer compositions provided herein include proteins, polypeptide or other multimeric moieties suitable for the attachment of two or more MHCII-binding peptides, which do not interfere with binding of the MHCII- binding peptides to cells MHCII molecules. In some embodiments, the multimerization domain comprises protein subunits. In some embodiments, the multimerization domain is a homomultimer of protein subunits. In some embodiments, the multimerization domain is a heteromultimer of protein subunits. In some embodiments, the multimer is a dimer, trimer, tetramer, pentamer, hexamer, octamer decamer or dodecamer. In one embodiment, the multimer is a tetramer.
[0087] Examples of suitable binding entities are streptavidin (SA) and avidin and derivatives thereof, biotin, polymers, immunoglobulins, antibodies (monoclonal, polyclonal, and recombinant), antibody fragments and derivatives thereof, leucine zipper domain of AP-1
(jun and fos), hexa-his (metal chelate moiety), hexa-hat GST (glutathione S-transferase) glutathione affinity, Calmodulin-binding peptide (CBP), Strep-tag®, Cellulose Binding Domain, Maltose Binding Protein, S-Peptide Tag, Chitin Binding Tag, Immuno-reactive Epitopes, Epitope Tags, E2Tag, HA Epitope Tag, Myc Epitope, FLAG Epitope, AU1 and AU5 Epitopes, Glu-Glu Epitope, KT3 Epitope, IRS Epitope, Btag Epitope, Protein Kinase-C Epitope, VSV Epitope, lectins that mediate binding to a diversity of compounds, including carbohydrates, lipids and proteins, e. g., Con A (Canavaliaensi formis) or WGA (wheat germ agglutinin) and tetranectin or Protein A or G (antibody affinity) or coiled-coil polypeptides e.g. leucine zipper. Combinations of such binding entities are also included.
[0088] In some embodiments, the multimerization domain is a tetramer of streptavidin (SA or SAv) or a derivative thereof. In some embodiments, the multimerization domain is tetrameric streptavidin. In some embodiments, the tetramer comprises Strep-tactin®, an engineered form of streptavidin that binds an engineered peptide sequence referred to as Strep-tag®. Strep-tag® and Strep-tactin® are described in U.S. Patent No. 5,506,121 and U.S. Patent No. 6,103,493, respectively, and are commercially available from a number of sources.
[0089] To attach peptides to streptavidin non-covalently via the biotin-binding site of SAv, an avitag can be incorporated into the peptide, for example at the C-terminal end, such that the peptide can be biotinylated through the avitag. Non-limiting examples of avitag sequences include SEQ ID NO: 85 (avitag with Myc tag), SEQ ID NO: 86 (avitag with Myc tag and 6xHis tag) and SEQ ID NO: 87 (avitag with 6xHis Tag and FLAG tag).
[0090] In one embodiment, the multimerization domain comprises full-length streptavidin. In another embodiment, the multimerization domain comprises a natural streptavidin core polypeptide. In another embodiment, the multimerization domain comprises a recombinant streptavidin core polypeptide, such as STV25 or STV13 (e.g., as described in Sano et al. (1995) J. BIOL. CHEM. 270:28204-28209). Accordingly, as used herein, the term “streptavidin” is intended to encompass the full-length protein as well as core portions thereof, including but not limited to the following representative sequences:


[0091] In yet other embodiments, the multimerization domain is a polymer (i.e., a compound composed of repeating subunits), such as dextran, polyethylene glycol (PEG) and the like. In one embodiment, the polymer is a sugar polymer, e.g, a polysaccharide, such as dextran. In one embodiment, the dextran is a modified dextran, wherein the dextran backbone has been modified to carry acceptor sites, such as a Klickmer™ (Immunodex).
B. MHCII-Binding Peptides
[0092] A wide variety of MHCII-binding peptides known in the art, identified based on the MHCII allele to which they bind. Any such known MHCII-binding peptides can be utilized in a multimer composition of the disclosure. A non-limiting example of such a known MHCII-binding peptide is an analog of a hemagglutinin (HA) peptide from Influenza A virus having the amino acid sequence shown in SEQ ID NO: 5, which HA peptide binds to an MHCII molecule comprising HLA-DRA*01:01 and HLA-DRB1 *01:01 (as described in Examples 1 and 3).
[0093] Various processes have been developed for identifying new MHC binding peptides that may be T cell epitopes and many experimental methods start with constructing an overlapping library of peptide fragments from a given protein sequence, by synthesizing a constant length (n-mer) amino acid sequences which are offset from one another along the protein sequence by fixed number of amino acids. The MHC binding properties and potential for activating T cells of each sequence can then be assessed in a number of assays.
[0094] Existing MHC binding peptides that have been identified with the methods outlined above and other methods, such as crystallographic analysis of the conformation of and charge distribution in the MHC binding groove has led to binding motifs being defined for the most common MHC alleles, setting rules for what type of putative MHC binding peptide can actually bind well to MHC molecules of a given allele. These motifs have been translated into predictive computer algorithms for predicting peptide binding to MHC molecules such as the SYFPEITHI algorithm (Rammensee H.-G., et al. (1995) IMMUNOGENETICS 41:178-228).
[0095] Protein sequences for the desired antigen can be analyzed for potential HLA specific antigens by using SYFPEITHI (Rammensee et al. (1999) IMMUNOGENETICS 50:213-219), and
the artificial neural network (ANN) and stabilized matrix method (SMM) algorithms from IEDB (Peters et al. (2005) PLoS BIOL. 3:e91). Peptides are selected based on a predicted binding value of either >21 for SYFPEITHI, <6000 for ANN, or <600 for SMM. Selected peptides are synthesized. Other suitable methods for analyzing protein sequences for potential HLA specific antigens also are known in the art and are suitable for use in identifying such HLA specific examples, such as NetMHCpan.
[0096] Binding assays can be performed using a fluorescence polarization (FP) assay as previously described (e.g, Buchi et al. (2004) BIOCHEMISTRY 43:14852-14863; Sette et al. (1994) MOL. IMMUNOL. 31:813-822). To determine binding capacity of the peptides, percentage inhibition relative to controls can be determined in an FP competition assay with the placeholder peptide.
[0097] An epitope library can comprise peptides containing natural amino acids, non-natural amino acids, or a combination of natural and non-natural amino acids. Non-natural amino acids can be included to facilitate post-translational modifications, including but not limited to glycosylation, methylation, deamidation, oxidation, reduction and the like. Methods for preparing epitope libraries including non-natural amino acids are established in the art.
[0098] In some embodiments, the peptides used in the multimer compositions are from an unbiased library of peptides. In various embodiments, the MHC-binding peptides can be 8mers, 9mers, lOmers, timers, 12mers, 13mers, Miners, Miners, Miners, Miners, 18mers, 19mers, 20mers, 21mers, 22mers, 23mers, 24mers or 25mers. Typically, MHCII-binding peptides are 13mers-18mers.
[0099] In some embodiments, the library comprises all k-mer peptides produced by transcription and translation of any polynucleotide sequence of interest, for example, in silica production of the transcription and translation products of both the forward and reverse strands of a genome or metagenome in all six reading frames.
[0100] In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from in silica translation of an exome of interest.
[0101] In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from in silica translation of a transcriptome of interest.
[0102] In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from a proteome of interest.
[0103] In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from in silico translation of an ORFeome of interest.
[0104] In some embodiments, an algorithm can be used to select peptides in a peptide library. For example, an algorithm can be used to predict peptides most likely to fold or dock in an MHC binding pocket, and peptides above a certain threshold value can be selected for inclusion in the library.
[0105] In some embodiments, a library of the disclosure comprises all peptides that can be derived from in silico transcription and translation or translation of a group of genomes, proteomes, transcriptomes, ORFeomes, or any combination thereof.
[0106] In some embodiments, the peptides are derived from in silico transcription and translation or translation of polynucleotide sequences from a group of samples, for example, clinical samples from a patient population, or a group of pathogen genomes.
[0107] In some embodiments, the peptides are derived from a differential genome, proteome, transcriptome, ORFeome, or any combination thereof, where two or more genomes, proteomes, transcriptomes, ORFeomes, or a combination thereof are compared to identify sequences that are differential sequences (e.g., that differ between them). In some embodiments, the peptide sequences are identified by comparing tissues of interest. In some embodiments, the peptide sequences are identified by comparing cells of interest. In some embodiments, the peptide sequences are identified by comparing diseased versus healthy cells or tissues. In some embodiments, the diseased cells or tissues are cancer cells or tissues. In some embodiments, the diseased cells are derived from an individual with an autoimmune disorder.
[0108] In some embodiments, the peptides are derived from homologous sequences of genomes, proteomes, transcriptomes, ORFeomes, or any combination thereof, where two or more genomes, proteomes, transcriptomes, ORFeomes, or a combination thereof are compared to identify sequences that are homologous sequences.
[0109] In some embodiments, the peptides are derived from mutations in a sequence of interest, for example, all 9-mer peptides that can be generated from single nucleotide mutations in a polynucleotide sequence encoding an antigen or epitope.
[0110] In some embodiments, the peptides an overlapping peptide library, comprising overlapping peptides from a template sequence (e.g., in silico translated genome), wherein overlapping peptides of a set length are offset by a defined number of residues.
[oni] In some embodiments, selection of peptides comprises prioritizing peptides based on predicted binding affinity for a certain HLA type.
[0112] In some embodiments, selection of peptides for a library of the disclosure prioritizes HLA types or alleles based on prevalence in a population, e.g., a human population.
[0113] In some embodiments, the library comprises all k-mer peptides produced by transcription and translation of any polynucleotide sequence of interest, for example, in silico production of the transcription and translation products of both the forward and reverse strands of a genome or metagenome in all six reading frames. In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from in silico transcription and translation of a mammalian genome, for example, a mouse genome, a human genome, a patient genome, an autoimmune patient genome, or a cancer genome. In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from in silico transcription and translation of a microorganism genome, for example, a bacterial genome, a viral genome, a protozoan genome, a protist genome, a yeast genome, an archaeal genome, or a bacteriophage genome. In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from in silico transcription and translation of a pathogen genome, for example, a bacterial pathogen genome, a viral pathogen genome, a fungal pathogen genome, an opportunistic pathogen genome, a conditional pathogen genome, or a eukaryotic parasite genome. In some embodiments, a library of the disclosure can be derived from a plant genome or a fungal genome. In some embodiments, a library of the disclosure comprises k-mer peptides derived from in silico transcription and translation of a genome, wherein the genome is modified during in silico transcription and translation, for example, in silico mutated to produce k-mer peptides comprising mutations (e.g. substitutions, insertions, deletions).
[0114] In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from in silico translation of an exome of interest, for example, a mammalian exome, a human exome, a mouse exome, a patient exome, an autoimmune patient exome, a cancer exome, a viral exome, a protozoan exome, a protist exome, a yeast exome, a pathogen exome, a eukaryotic parasite exome, a plant exome, or a fungal exome. In some embodiments, a library of the disclosure comprises k-mer peptides derived from in silico translation of a exome, wherein the exome is modified during in silico translation, for example, in silico mutated to produce k-mer peptides comprising mutations (e.g., substitutions, insertions, deletions).
[0115] In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from in silico translation of a transcriptome of interest, for example, a mammalian transcriptome, a human transcriptome, a mouse transcriptome, a patient transcriptome, an autoimmune patient transcriptome, a cancer trans criptome, a microorganism trans criptome, a bacterial transcriptome, a viral trans criptome, a protozoan transcriptome, a protist transcriptome, a yeast transcriptome, an archaeal transcriptome, a bacteriophage trans criptome, a pathogen transcriptome, a eukaryotic parasite transcriptome, a plant transcriptome, a fungal trans criptome, a transcriptome derived from RNA sequencing, a microbiome transcriptome, or a transcriptome derived from metagenomic RNA-sequencing. In some embodiments, a library of the disclosure comprises k-mer peptides derived from in silico translation of a transcriptome, wherein the transcriptome is modified during in silico translation, for example, in silico mutated to produce k-mer peptides comprising mutations (e.g. substitutions, insertions, deletions).
[0116] In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from a proteome of interest, for example, a mammalian proteome, a human proteome, a mouse proteome, a patient proteome, an autoimmune patient proteome, a cancer proteome, a microorganism proteome, a bacterial proteome, a viral proteome, a protozoan proteome, a protist proteome, a yeast proteome, an archaeal proteome, a bacteriophage proteome, a pathogen proteome, a eukaryotic parasite proteome, a plant proteome or a fungal proteome. In some embodiments, a library of the disclosure comprises k-mer peptides derived from a proteome wherein the k-mer peptides are modified from the proteome sequence, for example, k-mer peptides comprising mutations (e.g., substitutions, insertions, deletions).
[0117] In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from in silico translation of an ORFeome of interest, for example, a mammalian ORFeome, a human ORFeome, a mouse ORFeome, a patient ORFeome, an autoimmune patient ORFeome, a cancer ORFeome, a microorganism ORFeome, a bacterial ORFeome, a viral ORFeome, a protozoan ORFeome, a protist ORFeome, a yeast ORFeome, an archaeal ORFeome, a bacteriophage ORFeome, a pathogen ORFeome, a eukaryotic parasite ORFeome, a plant ORFeome or a fungal ORFeome, an ORFeome derived from nextgen sequencing, a microbiome ORFeome, or an ORFeome derived from metagenomic sequencing. In some embodiments, a library of the disclosure comprises k-mer peptides derived from in silico translation of an ORFeome, wherein the ORFeome is modified during
in silico translation, for example, in silico mutated to produce k-mer peptides comprising mutations (e.g. substitutions, insertions, deletions).
[0118] In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from in silico transcription and translation or translation of a group of genomes, proteomes, transcriptomes, ORFeomes, or any combination thereof. In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from in silico transcription and translation or translation of polynucleotide sequences from a group of samples, for example, clinical samples from a patient population, or a group of pathogen genomes. In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from in silico transcription and translation of a group of viral genomes, for example, the human virome. In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from in silico transcription and translation of a group of genomes, proteomes, transcriptomes, ORFeomes, or any combination thereof, wherein the source sequences are modified during in silico translation, for example, in silico mutated to produce k-mer peptides comprising mutations (e.g., substitutions, insertions, deletions).
[0119] In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from a differential genome, proteome, transcriptome, ORFeome, or any combination thereof, where two or more genomes, proteomes, trans criptomes, ORFeomes, or a combination thereof are compared to identify sequences that are differential sequences (e.g., that differ between them), for example, differing in nucleotide sequence, amino acid sequence, nucleotide abundance, or protein abundance. In some embodiments, differential sequences of a genome, proteome, trans criptome, or ORFeome are generated by comparing tissues of interest. In some embodiments, differential sequences of a genome, proteome, transcriptome, or ORFeome are generated by comparing sequences from cells of interest (e.g., a healthy cell versus a cancer cell). In some embodiments, differential sequences of a genome, proteome, transcriptome, or ORFeome are generated by comparing sequences of organisms of interest. In some embodiments, differential sequences of a genome, proteome, transcriptome, or ORFeome can be generated by comparing subjects of interest (e.g., diseased versus healthy subjects).
[0120] In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from homologous sequences of genomes, proteomes, transcriptomes, ORFeomes, or any combination thereof, where two or more genomes, proteomes,
transcriptomes, ORFeomes, or a combination thereof are compared to identify sequences that are homologous sequences (e.g, that share a degree of homology), for example, homologous nucleotide sequences, homologous amino acid sequences, homologous nucleotide abundance, or homologous protein abundance. In some embodiments, homologous sequences of genomes, proteomes, transcriptomes, or ORFeomes are generated by comparing tissues of interest. In some embodiments, homologous sequences of genomes, proteomes, transcriptomes, or ORFeomes are generated by comparing sequences from cells of interest (e.g, a healthy cell versus a involved in autoimmunity cell (e.g., a cell that induces autoimmunity or a cell that is targeted during autoimmunity). In some embodiments, homologous sequences of genomes, proteomes, transcriptomes, or ORFeomes are generated by comparing sequences of organisms of interest. In some embodiments, homologous sequences of genomes, proteomes, transcriptomes, or ORFeomes are generated by comparing subjects of interest (e.g., diseased versus healthy subjects).
[0121] In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from a polypeptide sequence of interest, for example, all possible 9-mer peptides covering the complete protein sequence of a viral protein. In some embodiments, a library of the disclosure comprises k-mer peptides that can be generated from a polypeptide sequence of interest, wherein the polypeptide sequence of interest is modified, e.g. in silico mutated to produce k-mer peptides comprising mutations (e.g., substitutions, insertions, deletions).
[0122] In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from mutations in a sequence of interest, for example, all 9-mer peptides that can be generated from single nucleotide mutations in a polynucleotide sequence encoding an antigen or epitope. For example, a library of the disclosure comprises all 9-mer peptides that can be generated from two, three, four, five, six, seven, eight, or nine nucleotide mutations in a polynucleotide sequence encoding an antigen or epitope. In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from alanine substitutions, for example, alanine substitutions at any position in any of the sequences described herein (e.g., a protein, a group of proteins, a proteome, an in silico transcripted and translated genome). In some embodiments, a library of the disclosure comprises a positional scanning library, wherein selected amino acid residues are sequentially substituted with all other natural amino acids. In some embodiments, a library of the disclosure comprises a combinatorial positional scanning library, wherein selected amino acid residues are sequentially substituted with all
other natural amino acids, two or more positions at a time. In some embodiments, a library of the disclosure comprises an overlapping peptide library, comprising overlapping peptides from a template sequence (e.g., in silico translated genome), wherein overlapping peptides of a set length are offset by a defined number of residues. In some embodiments, a library of the disclosure comprises a T cell truncated peptide library, wherein each replicate of the library comprises equimolar mixtures of peptides with truncations at one terminus (e.g., 8- mers, 9-mers, 10-mers and 11-mers that can be derived from C-terminal truncations of a nominal 11-mer). In some embodiments, a library of the disclosure comprises a customized set of peptides, wherein the customized set of peptides are provided in a list.
[0123] In some embodiments, a genome, exome, trans criptome, proteome, or ORFeome of the disclosure is a viral genome, exome, transcriptome, proteome, or ORFeome. Non-limiting examples of viruses include Adenovirus, Adeno-associated virus, Aichi virus, Australian bat lyssavirus, BK polyomavirus, Banna virus, Barmah forest virus, Bunyamwera virus, Bunyavirus La Crosse, Bunyavirus snowshoe hare, Cercopithecine herpesvirus, Chandipura virus, Chikungunya virus, Cosavirus A, Cowpox virus, Coxsackievirus, Crimean-Congo hemorrhagic fever virus, Cytomegalovirus (CMV), Dengue virus, Dhori virus, Dugbe virus, Duvenhage virus, Eastern equine encephalitis virus, Ebolavirus, Echovirus, Encephalomyocarditis virus, Epstein-Barr virus (EBV), European bat lyssavirus, GB virus C/Hepatitis G virus, Hantaan virus, Hendra virus, Hepatitis A virus, Hepatitis B virus, Hepatitis C virus, Hepatitis E virus, Hepatitis delta virus, Horsepox virus, Human adenovirus, Human astrovirus, Human coronavirus, Human cytomegalovirus, Human endogenous retrovirus (HERV), Human enterovirus, Human herpesvirus (e.g., HHV-1, HHV-2, HHV-6A, HHV-6B, HHV-7, HHV-8, Human immunodeficiency virus (e.g., HIV-1, HIV -2), Human papillomavirus (e.g., HPV-1, HPV-2, HPV-16, HPV-18, Human parainfluenza, Human parvovirus Bl 9, Human respiratory syncytial virus (RSV), Human rhinovirus, Human SARS coronavirus, SARS-CoV2, Human spumaretro virus, Human T-lymphotropic virus (HTLV, e.g. HTLV-1, HTLV -2, HTLV-3), Human torovirus, Influenza A virus, Influenza B virus, Influenza C virus, Isfahan virus, JC polyomavirus, Japanese encephalitis virus, Junin arenavirus, KI Polyomavirus, Kunjin virus, Lagos bat virus, Lake Victoria Marburgvirus, Langat virus, Lassa virus, Lordsdale virus, Louping ill virus, Lymphocytic choriomeningitis virus, Machupo virus, Mayaro virus, MERS coronavirus, Measles virus, Mengo encephalomyocarditis virus, Merkel cell polyomavirus, Mokola virus, Molluscum contagiosum virus, Monkeypox virus, Mumps virus, Murray valley encephalitis virus, New
York virus, Nipah virus, Norovirus, Norwalk virus, O’nyong-nyong virus, Orf virus, Oropouche virus, Pichinde virus, Poliovirus, Punta toro phlebovirus, Puumala virus, Rabies virus, Rift valley fever virus, Rosavirus A, Ross river virus, Rotavirus (e.g., rotavirus A, rotavirus B, rotavirus C, rotavirus X), Rubella virus, Sagiyama virus, Salivirus A, Sandfly fever Sicilian virus, Sapporo virus, Semliki forest virus, Seoul virus, Simian foamy virus, Simian virus 5, Sindbis virus, Southampton virus, St. louis encephalitis virus, Tick-home powassan vims, Torque teno vims, Toscana virus, Uukuniemi vims, Vaccinia virus, Varicella-zoster virus, Variola virus, Venezuelan equine encephalitis vims, Vesicular stomatitis vims, Western equine encephalitis vims, WU polyomavirus, West Nile virus, Yaba monkey tumor virus, Yaba-like disease virus, Yellow fever virus, and Zika virus.
[0124] In some embodiments, a genome, exome, trans criptome, proteome, or ORFeome of the disclosure is a cancer genome, exome, trans criptome, proteome, or ORFeome. In some embodiments, a library of the disclosure comprises known cancer neoepitopes. In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from known cancer antigenic proteins. In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from genes involved in epithelial- mesenchymal transition. In some embodiments, a library of the disclosure comprises all k- mer peptides that can be derived from cancer implicated genes. In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from mutational cancer driver genes. In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from proto-oncogenes, oncogenes, or tumor suppressor genes. In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from proto-oncogenes, oncogenes, or tumor suppressor genes, wherein the k-mers comprise mutations as described herein (e.g, amino acid substitutions, alanine substitutions, positional scanning, combinatorial positional scanning etc.).
[0125] Non-limiting examples of cancers include Acute Lymphoblastic Leukemia (ALL), Acute Myeloid Leukemia (AML), Adrenocortical Carcinoma, AIDS-Related Cancers, AIDS- Related Lymphoma, Anal Cancer, Appendix Cancer, Astrocytoma, Atypical Teratoid/Rhabdoid Tumor, Basal Cell Carcinoma, Bile Duct Cancer, Bladder Cancer, Bone Cancer, Brain Tumor, Breast Cancer, Bronchial Tumors, Burkitt Lymphoma, Carcinoid Tumor, Carcinoma of Unknown Primary, Cardiac Tumor, Central Nervous System cancer, Cervical Cancer, Cholangiocarcinoma, Chordoma, Chronic Lymphocytic Leukemia (CLL), Chronic Myelogenous Leukemia (CML), Chronic Myeloproliferative Neoplasms, Colorectal
Cancer, Craniopharyngioma, Cutaneous T-Cell Lymphoma, Ductal Carcinoma In Situ, Embryonal Tumor, Endometrial Cancer, Epithelial Cancer, Ependymoma, Esophageal Cancer, Esthesioneuroblastoma, Ewing Sarcoma, Extracranial Germ Cell Tumor, Extragonadal Germ Cell Tumor, Eye Cancer, Fallopian Tube Cancer, Fibrous Histiocytoma of Bone, Gallbladder Cancer, Gastric Cancer, Gastrointestinal Carcinoid Tumor, Gastrointestinal Stromal Tumors (GIST), Germ Cell Tumors, Gestational Trophoblastic Disease, Hairy Cell Leukemia, Head and Neck Cancer, Hepatocellular Cancer, Histiocytosis, Hodgkin Lymphoma, Hypopharyngeal Cancer, Intraocular Melanoma, Islet Cell Tumors, Kaposi Sarcoma, Kidney (Renal Cell) Cancer, Langerhans Cell Histiocytosis, Laryngeal Cancer, Leukemia, Lip and Oral Cavity Cancer, Liver Cancer, Lung Cancer (Non-Small Cell and Small Cell), Lymphoma, Male Breast Cancer, Malignant Fibrous Histiocytoma of Bone and Osteosarcoma, Melanoma, Merkel Cell Carcinoma, Mesothelioma, Metastatic Cancer, Metastatic Squamous Neck Cancer with Occult Primary, Midline Tract Carcinoma, Mouth Cancer, Multiple Endocrine Neoplasia Syndrome, Multiple Myeloma, Mycosis Fungoides, Myelodysplastic Syndromes, Myelodysplastic/Myeloproliferative Neoplasms, Nasal Cavity Cancer, Nasopharyngeal Cancer, Neuroblastoma, Non-Hodgkin Lymphoma, Non-Small Cell Lung Cancer, Oral Cancer, Lip and Oral Cavity Cancer, Oropharyngeal Cancer, Osteosarcoma, Ovarian Cancer, Pancreatic Cancer, Pancreatic Neuroendocrine Tumors, Papillomatosis, Paraganglioma, Paranasal Sinus Cancer, Parathyroid Cancer, Penile Cancer, Pharyngeal Cancer, Pheochromocytoma, Pituitary Tumor, Plasma Cell Neoplasm, Pleuropulmonary Blastoma, Primary Central Nervous System (CNS) Lymphoma, Primary Peritoneal Cancer, Prostate Cancer, Rectal Cancer, Recurrent Cancer, Retinoblastoma, Rhabdomyosarcoma, Salivary Gland Cancer, Sarcoma, Sezary Syndrome, Skin Cancer, Small Cell Lung Cancer, Small Intestine Cancer, Soft Tissue Sarcoma, Squamous Cell Carcinoma of the Skin, Squamous Neck Cancer with Occult Primary, Stomach Cancer, T- Cell Lymphoma, Testicular Cancer, Throat Cancer, Thymoma and Thymic Carcinoma, Thyroid Cancer, Transitional Cell Cancer, Ureter and Renal Pelvis Cancer, Urethral Cancer, Uterine Cancer, Uterine Sarcoma, Vaginal Cancer, Vascular Tumors, Vulvar Cancer, and Wilms Tumor.
[0126] In some embodiments, a genome, exome, trans criptome, proteome, or ORFeome of the disclosure is an inflammatory or autoimmunogenic genome, exome, transcriptome, proteome, or ORFeome. In some embodiments, a library of the disclosure comprises known inflammatory or autoimmunogenic neoepitopes or self-epitopes. In some embodiments, a
library of the disclosure comprises all k-mer peptides that can be derived from known inflammatory or autoimmunogenic antigenic proteins. In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from inflammatory or autoimmune-implicated genes. In some embodiments, a library of the disclosure comprises all k-mer peptides that can be derived from mutation of inflammatory or autoimmune-related driver genes.
[0127] Non-limiting examples of inflammatory or autoimmune diseases or conditions include Acute Disseminated Encephalomyelitis (ADEM); Acute necrotizing hemorrhagic leukoencephalitis; Addison’s disease; Adjuvant-induced arthritis; Agammaglobulinemia; Alopecia areata; Amyloidosis; Ankylosing spondylitis; Anti-GBM/Anti-TBM nephritis; Antiphospholipid syndrome (APS); Autoimmune angioedema; Autoimmune aplastic anemia; Autoimmune dysautonomia; Autoimmune gastric atrophy; Autoimmune hemolytic anemia; Autoimmune hepatitis; Autoimmune hyperlipidemia; Autoimmune immunodeficiency; Autoimmune inner ear disease (AIED); Autoimmune myocarditis; Autoimmune oophoritis; Autoimmune pancreatitis; Autoimmune retinopathy; Autoimmune thrombocytopenic purpura (ATP); Autoimmune thyroid disease; Autoimmune urticarial; Axonal & neuronal neuropathies; Balo disease; Behcet’s disease; Bullous pemphigoid; Cardiomyopathy;
Castleman disease; Celiac disease; Chagas disease; Chronic inflammatory demyelinating polyneuropathy (CIDP); Chronic recurrent multifocal ostomyelitis (CRMO); Churg-Strauss syndrome; Cicatricial pemphigoid/benign mucosal pemphigoid; Crohn’s disease; Cogans syndrome; Collagen-induced arthritis; Cold agglutinin disease; Congenital heart block; Coxsackie myocarditis; CREST disease; Essential mixed cryoglobulinemia; Demyelinating neuropathies; Dermatitis herpetiformis; Dermatomyositis; Devic’s disease (neuromyelitis optica); Discoid lupus; Dressier’s syndrome; Endometriosis; Eosinophilic esophagitis; Eosinophilic fasciitis; Erythema nodosum Experimental allergic encephalomyelitis; Experimental autoimmune encephalomyelitis; Evans syndrome; Fibromyalgia; Fibrosing alveolitis; Giant cell arteritis (temporal arteritis); Giant cell myocarditis; Glomerulonephritis; Goodpasture’s syndrome; Granulomatosis with Polyangiitis (GPA) (formerly called Wegener’s Granulomatosis); Graves’ disease; Guillain-Barre syndrome; Hashimoto’s encephalitis; Hashimoto’s thyroiditis; Hemolytic anemia; Henoch-Schonlein purpura; Herpes gestationis; Hypogammaglobulinemia; Idiopathic thrombocytopenic purpura (ITP); IgA nephropathy; IgG4-related sclerosing disease; Immunoregulatory lipoproteins; Inclusion body myositis; Interstitial cystitis; Inflammatory bowel disease; Juvenile arthritis; Juvenile
oligoarthritis; Juvenile diabetes (Type 1 diabetes); Juvenile myositis; Kawasaki syndrome; Lambert-Eaton syndrome; Leukocytoclastic vasculitis; Lichen planus; Lichen sclerosus; Ligneous conjunctivitis; Linear IgA disease (LAD); Lupus (SLE); Lyme disease, chronic; Meniere’s disease; Microscopic polyangiitis; Mixed connective tissue disease (MCTD); Mooren’s ulcer; Mucha-Habermann disease; Multiple sclerosis; Myasthenia gravis; Myositis; Narcolepsy; Neuromyelitis optica (Devic’s); Neutropenia; Non-obese diabetes; Ocular cicatricial pemphigoid; Optic neuritis; Palindromic rheumatism; PANDAS (Pediatric Autoimmune Neuropsychiatric Disorders Associated with Streptococcus); Paraneoplastic cerebellar degeneration; Paroxysmal nocturnal hemoglobinuria (PNH); Parry Romberg syndrome; Parsonnage-Tumer syndrome; Pars planitis (peripheral uveitis); Pemphigus; Pemphigus vulgaris; Peripheral neuropathy; Perivenous encephalomyelitis; Pernicious anemia; POEMS syndrome; Polyarteritis nodosa; Type I, II, & III autoimmune polyglandular syndromes; Polymyalgia rheumatic; Polymyositis; Postmyocardial infarction syndrome; Postpericardiotomy syndrome; Progesterone dermatitis; Primary biliary cirrhosis; Primary sclerosing cholangitis; Psoriasis; Plaque Psoriasis; Psoriatic arthritis; Idiopathic pulmonary fibrosis; Pyoderma gangrenosum; Pure red cell aplasia; Raynauds phenomenon; Reactive Arthritis; Reflex sympathetic dystrophy; Reiter’s syndrome; Relapsing polychondritis; Restless legs syndrome; Retroperitoneal fibrosis; Rheumatic fever; Rheumatoid arthritis; Sarcoidosis; Schmidt syndrome; Scleritis; Scleroderma; Sclerosing cholangitis; Sclerosing sialadenitis; Sjogren’s syndrome; Sperm & testicular autoimmunity; Stiff person syndrome; Subacute bacterial endocarditis (SBE); Susac’s syndrome; Sympathetic ophthalmia; Systemic lupus erythematosus (SLE); Systemic sclerosis; Takayasu’s arteritis; Temporal arteritis/Giant cell arteritis; Thrombocytopenic purpura (TTP); Tolosa-Hunt syndrome;Transverse myelitis; Type 1 diabetes; Ulcerative colitis; Undifferentiated connective tissue disease (UCTD); Uveitis; Vasculitis; Vesiculobullous dermatosis; Vitiligo; Wegener’s granulomatosis (now termed Granulomatosis with Poly angiitis (GPA). Non-limiting examples of inflammatory or autoimmune diseases or conditions include infection, such as a chronic infection, latent infection, slow infection, persistent viral infection, bacterial infection, fungal infection, mycoplasma infection or parasitic infection.
C. Spacer Linkers
[0128] In the multimer compositions, typically a spacer linker is positioned between the MHCII binding peptide and the multimerization domain. The term “spacer linker” denotes a linear amino acid chain of natural and/or synthetic origin. The linker has the function to
ensure that polypeptides conjugated to each other can perform their biological activity by allowing the polypeptides to fold correctly and to be presented properly. The spacer linker may contain repetitive amino acid sequences or sequences of naturally occurring polypeptides. In some embodiments, the peptide linker has a length of from 2 to 50 amino acids. In some embodiments, the peptide linker is between 3 and 30 amino acids, between 5 to 25 amino acids, between 5 to 20 amino acids, or between 10 and 20 amino acids.
[0129] In one embodiment, the spacer linker is a flexible linker, e.g., composed of a glycine- serine-rich sequence, such as the linker shown in SEQ ID NO: 6. In another embodiment, the spacer linker is a rigid linker, e.g., composed of a proline-rich sequence, such as the linker shown in SEQ ID NO: 7. In yet another embodiment, the spacer linker is a flexible-rigid linker, comprising both a flexible region (e.g., a glycine-serine-rich sequence) and a rigid region (e.g., a proline-rich sequence), such as the linker shown in SEQ ID NO: 8. These flexible, rigid and flexible-rigid spacer linkers are described further in Example 3 and illustrated schematically in FIG. 6A.
[0130] In various other embodiments, the peptide linker is rich in glycine, glutamine, and/or serine residues. These residues are arranged e.g. in small repetitive units of up to five amino acids. This small repetitive unit may be repeated for one to five times. At the amino- and/or carboxy -terminal ends of the multimeric unit up to six additional arbitrary, naturally occurring amino acids may be added. Other synthetic peptidic linkers are composed of a single amino acid, which is repeated between 10 to 20 times and may comprise at the amino- and/or carboxy -terminal end up to six additional arbitrary, naturally occurring amino acids. All peptidic linkers can be encoded by a nucleic acid molecule and therefore can be recombinantly expressed. As the linkers are themselves peptides, the polypeptide connected by the linker are connected to the linker via a peptide bond that is formed between two amino acids.
[0131] Suitable peptide linkers are well known in the art, and are disclosed in, e.g., US2010/0210511 US2010/0179094, and US2012/0094909, which are herein incorporated by reference in its entirety. Other linkers are provided, for example, in U.S. Pat. Nos. 5,525,491; Alfthan et al. (1995) PROTEIN ENG. 8:725-731; Shan etal. (1999) J. IMMUNOL. 162:6589- 6595; Newton et al. (1996) BIOCHEMISTRY 35:545-553; Megeed et al. (2006) BIOMACROMOLECULES 7:999-1004; and Perisic et al. (1994) STRUCTURE 12:1217-1226; each of which is incorporated by reference in its entirety.
[0132] In some embodiments, the spacer linker is synthetic. As used herein, the term “synthetic” with respect to a spacer linker includes peptides (or polypeptides) which comprise an amino acid sequence (which may or may not be naturally occurring) that is linked in a linear sequence of amino acids to a sequence (which may or may not be naturally occurring) to which it is not naturally linked in nature. For example, the spacer linker may comprise non-naturally occurring polypeptides which are modified forms of naturally occurring polypeptides (e.g., comprising a mutation such as an addition, substitution or deletion) or which comprise a first amino acid sequence (which may or may not be naturally occurring). Preferably, a spacer linker will be relatively non-immunogenic and not inhibit any non- covalent association among monomer subunits of a binding protein.
[0133] In some embodiments, the linker is a Gly-Ser polypeptide linker, i.e., a peptide that consists of glycine and serine residues. One exemplary Gly-Ser polypeptide linker comprises the amino acid sequence (Gly4Ser)n, wherein n=l-6 (SEQ ID NO: 72). In certain embodiments, n=l. In certain embodiments, n=2. In certain embodiments, n=3. In certain embodiments, n=4. In certain embodiments, n=5. In certain embodiments, n=6. Another exemplary Gly-Ser polypeptide linker comprises the sequence SSSSGSSSSGSAA (SEQ ID NO: 73). Another linker comprises only glycine, e.g., Gs linkers (GGGGG; SEQ ID NO: 74). Another exemplary Gly-Ser polypeptide linker comprises the amino acid sequence Ser(Gly4Ser)n, wherein n=l-10 (SEQ ID NO: 75). In certain embodiments, n=l. In certain embodiments, n=2. In certain embodiments, n=3, i.e., Ser(Gly4Ser)3. In certain embodiments, n=4, i.e., Ser(Gly4Ser)4. In certain embodiments, n=5. In certain embodiments, n=6. In certain embodiments, n=7. In certain embodiments, n=8. In certain embodiments, n=9. In certain embodiments, n=10.
[0134] Other exemplary linkers include GS linkers (i.e., (GS)n), GGSG linkers (i.e., (GGSG)n) (SEQ ID NO: 76), GSAT linkers (SEQ ID NO: 77), SEG linkers, GGS linkers (i.e., (GGSGGS)n) (SEQ ID NO: 78), wherein n is a positive integer (e.g., 1, 2, 3, 4, or 5), (Gly4Ser)4 (GGGGSGGGGSGGGGSGGGGS; SEQ ID NO: 79) and (GS)2AG2SGSG3S linkers (GSGSAGGSGSGGGS; SEQ ID NO: 80). Other suitable linkers for use in multimeric fusion proteins can be found using publicly available databases, such as the Linker Database (ibi.vu.nl/programs/linkerdbwww). The Linker Database is a database of inter-domain linkers in multi-functional enzymes which serve as potential linkers in novel multimeric fusion proteins (see, e.g., George et al. (2002) PROTEIN ENGINEERING 15:871-9).
[0135] In one embodiment, the spacer linker comprises an amino acid sequence selected from the group consisting of SEQ ID NOs: 6-8 and 72-80.
D. Recombinant Preparation of Multimer Compositions
[0136] In one embodiment, a multimer composition of the disclosure is prepared by standard recombinant DNA techniques using a nucleic acid construct that encodes the MHCII-binding peptide operatively linked to the multimerization domain (MD), typically with sequences encoding a spacer linker positioned between the sequences encoding the peptide and the MD. A non-limiting representative nucleic acid construct encoding a multimer composition is shown schematically in FIG. 2A. In one embodiment, the peptide is operatively linked to the N-terminus of the MD. In one embodiment, the peptide is operatively linked to the C- terminus of the MD.
[0137] General techniques for nucleic acid manipulation are well established in the art, such as described in, for example, Sambrook et al. (1989) MOLECULAR CLONING: A LABORATORY MANUAL, 2nd Edition, Vols. 1-3, Cold Spring Harbor Laboratory Press, or Ausubel, F. et al. (1987) CURRENT PROTOCOLS IN MOLECULAR BIOLOGY, Green Publishing and Wiley - Interscience, New York and periodic updates, herein incorporated by reference. Generally, the DNA encoding the polypeptide is operably linked to suitable transcriptional or translational regulatory elements derived from mammalian, viral, or insect genes. Such regulatory elements include a transcriptional promoter, an optional operator sequence to control transcription, a sequence encoding suitable mRNA ribosomal binding site, and sequences that control the termination of transcription and translation. The ability to replicate in a host, usually conferred by an origin of replication, and a selection gene to facilitate recognition of transformants is additionally incorporated.
[0138] In one embodiment, the nucleic acid construct is designed to be suitable for in vitro transcription/translation (IVTT). In one embodiment, the nucleic acid is designed to be suitable for recombinant expression in a host cell. Appropriate cloning and expression vectors for use with bacterial, fungal, yeast, and mammalian cellular hosts can be found in CLONING VECTORS: A LABORATORY MANUAL (Elsevier, New York (1985)), the relevant disclosure of which is hereby incorporated by reference.
[0139] In some embodiments, the multimer composition is synthesized utilizing an in vitro transcription/translation (IVTT) system that can both transcribe, for example, a DNA construct into RNA, and then translate the RNA into a protein. IVTT can allow for protein
production in a cell-free environment directly from a DNA or RNA template. An IVTT method used herein can be performed using, for example, a PCR product, a linear DNA plasmid, a circular DNA plasmid, or an mRNA template with a ribosome-binding site (RBS) sequence. After the appropriate template has been isolated, transcription components can be added to the template including, for example, ribonucleotide triphosphates, and RNA polymerase. After transcription has been completed, translation components can be added, which can be found in, for example, rabbit reticulocyte lysate, or wheat germ extract. In some methods, the transcription and translation can occur during a single step, in which purified translation components found in, for example, rabbit reticulocyte lysate or wheat germ extract are added at the same time as adding the transcription components to the nucleic acid template.
[0140] In one embodiment, the nucleic acid sequence is incorporated into a vector, such as a plasmid vector, a viral vector or a non-viral vector. The vector is selected to be suitable for use in the intended host cell (i.e., the vector incudes all necessary transcriptional regulatory elements to allow for expression of the encoded multimer composition in the host cell). Suitable vectors, including transcriptional regulatory elements for use in various host cells, including mammalian host cells, are well established in the art.
[0141] As appreciated by those skilled in the art, because of third base degeneracy, almost every amino acid can be represented by more than one triplet codon in a coding nucleotide sequence. In addition, minor base pair changes may result in a conservative substitution in the amino acid sequence encoded but are not expected to substantially alter the biological activity of the gene product. Therefore, a nucleic acid sequence encoding a protein described herein may be modified slightly in sequence and yet still encode its respective gene product. Nucleic acids encoding any of the various proteins or polypeptides described herein may be synthesized chemically or prepared through standard recombinant DNA techniques. Codon usage may be selected so as to improve expression in a cell. Such codon usage will depend on the cell type selected. Specialized codon usage patterns have been developed for E. coli and other bacteria, as well as mammalian cells, plant cells, yeast cells and insect cells. See for example: Mayfield et al. (2003) PROC. NATL. AC D. SCI. USA 100(2):438-442; Sinclair et al. (2002) PROTEIN EXPR. PURIF. 26(I):96-105; Connell, N.D. (2001) CURR. OPIN. BIOTECHNOL. 12(5):446-449; Makrides et al. (1996) MICROBIOL. REV. 60(3):512-538; and Sharp et al. (1991) YEAST 7(7):657-678.
[0142] In one embodiment, the vector is designed for expression in a prokaryotic host cell (e.g, E. coll). In one embodiment, the vector is designed for expression in a eukaryotic host cell (e.g., yeast). In one embodiment, the vector is designed for expression in a mammalian host cell. In one embodiment, the mammalian host cells are human host cells. In one embodiment, the human host cells are human embryonic kidney (HEK) cells. In one embodiment, the HEK cells are 293 cells or are a 293 -derived HEK strain. Such HEK cells are commercially available in the art, a non-limiting example of which is the Expi293F™ cell line (Fisher ThermoScientific). In yet another embodiment, the mammalian host cell is a CHO cell line. In some embodiments, the mammalian cell line such as a HEK or CHO cell line is stably transfected with the expression construct with a virus, for example a lentivirus or a retrovirus.
[0143] When mammalian host cells are used, typically the signal sequence used in the expression construct is derived from a mammalian protein. Furthermore, the transcriptional regulatory sequences used in the vector are selected for their effectiveness in mammalian host cell expression.
[0144] Other expression systems include stable Drosophila cell transfectants and baculovirus infected insect-cells suitable for expression of proteins.
[0145] For prokaryotic host cells that do not recognize and process a native signal sequence, the signal sequence is substituted by a prokaryotic signal sequence selected, for example, from the group of the alkaline phosphatase, penicillinase, 1 pp, or heat-stable enterotoxin II leaders.
[0146] For yeast secretion the native signal sequence may be substituted by, e.g., a yeast invertase leader, a factor leader (including Saccharomyces and Kluyveromyces alpha-factor leaders), or acid phosphatase leader, the C. albicans glucoamylase leader, or the signal sequence described in U.S. Pat. No. 5,631,144. In mammalian cell expression, mammalian signal sequences as well as viral secretory leaders, for example, the herpes simplex gD signal, are available. The DNA for such precursor regions may be ligated in reading frame to DNA encoding the protein.
[0147] Both expression and cloning vectors contain a nucleic acid sequence that enables the vector to replicate in one or more selected host cells. Generally, in cloning vectors this sequence is one that enables the vector to replicate independently of the host chromosomal DNA, and includes origins of replication or autonomously replicating sequences. Such
sequences are well known for a variety of bacteria, yeast, and viruses. The origin of replication from the plasmid pBR322 is suitable for most Gram-negative bacteria, the 2 micron plasmid origin is suitable for yeast, and various viral origins (SV40, polyoma, adenovirus, VSV or BPV) are useful for cloning vectors in mammalian cells. Generally, the origin of replication component is not needed for mammalian expression vectors (the SV40 origin may typically be used only because it contains the early promoter).
[0148] Expression and cloning vectors may contain a selection gene, also termed a selectable marker. Typical selection genes encode proteins that (a) confer resistance to antibiotics or other toxins, e.g., ampicillin, neomycin, methotrexate, or tetracycline, (b) complement auxotrophic deficiencies, or (c) supply critical nutrients not available from complex media, e.g., the gene encoding D-alanine racemase for Bacilli.
[0149] Expression and cloning vectors usually contain a promoter that is recognized by the host organism and is operably linked to the nucleic acid encoding the MHC multimer described herein. Promoters suitable for use with prokaryotic hosts include the phoA promoter, beta-lactamase and lactose promoter systems, alkaline phosphatase, a tryptophan (trp) promoter system, and hybrid promoters such as the tan promoter. However, other known bacterial promoters are suitable. Promoters for use in bacterial systems also will contain a Shine-Dai gamo (S.D.) sequence operably linked to the DNA encoding the protein described herein. Promoter sequences are known for eukaryotes. Virtually all eukaryotic genes have an AT-rich region located approximately 25 to 30 bases upstream from the site where transcription is initiated. Another sequence found 70 to 80 bases upstream from the start of transcription of many genes is a CNCAAT region where N may be any nucleotide. At the 3’ end of most eukaryotic genes is an AATAAA sequence that may be the signal for addition of the poly A tail to the 3’ end of the coding sequence. All of these sequences are suitably inserted into eukaryotic expression vectors.
[0150] Examples of suitable promoting sequences for use with yeast hosts include the promoters for 3-phosphogly cerate kinase or other glycolytic enzymes, such as enolase, glyceraldehyde-3-phosphate dehydrogenase, hexokinase, pyruvate decarboxylase, phosphofructokinase, glucose-6-phosphate isomerase, 3-phosphogly cerate mutase, pyruvate kinase, triosephosphate isomerase, phosphoglucose isomerase, and glucokinase.
[0151] Transcription from vectors in mammalian host cells can be controlled, for example, by promoters obtained from the genomes of viruses such as polyoma virus, fowlpox virus,
adenovirus (such as Adenovirus 2), bovine papilloma virus, avian sarcoma virus, cytomegalovirus, a retrovirus, hepatitis-B virus and most preferably Simian Virus 40 (SV40), from heterologous mammalian promoters, e.g., the actin promoter or an immunoglobulin promoter, from heat-shock promoters, provided such promoters are compatible with the host cell systems.
[0152] Transcription of a DNA encoding protein described herein by higher eukaryotes is often increased by inserting an enhancer sequence into the vector. Many enhancer sequences are now known from mammalian genes (globin, elastase, albumin, a-fetoprotein, and insulin). Typically, however, one will use an enhancer from a eukaryotic cell virus.
Examples include the SV40 enhancer on the late side of the replication origin (bp 100-270), the cytomegalovirus early promoter enhancer, the polyoma enhancer on the late side of the replication origin, and adenovirus enhancers. See also Yaniv (1982) NATURE 297:17-18 on enhancing elements for activation of eukaryotic promoters. The enhancer may be spliced into the vector at a position 5’ or 3’ to the peptide-encoding sequence, but is preferably located at a site 5’ from the promoter.
[0153] Expression vectors used in eukaryotic host cells (e.g., yeast, fungi, insect, plant, animal, human, or nucleated cells from other multicellular organisms) will also contain sequences necessary for the termination of transcription and for stabilizing the mRNA. Such sequences are commonly available from the 5’ and, occasionally 3’, untranslated regions of eukaryotic or viral DNAs or cDNAs. These regions contain nucleotide segments transcribed as polyadenylated fragments in the untranslated portion of mRNA encoding the protein described herein. One useful transcription termination component is the bovine growth hormone polyadenylation region. See WO 94/11026 and the expression vector disclosed therein.
[0154] In one embodiment, the expression construct comprises a signal sequence operatively linked upstream from the sequences encoding the multimer composition to thereby facilitate secretion of the multimer composition from a host cell. In one embodiment, the signal sequence is from an Ig supergroup member. In one embodiment, the signal sequence is an immunoglobulin chain signal sequence. In one embodiment, the signal sequence is an Ig Kappa chain V-III region CLL signal peptide, e.g., having the sequence MEAPAQLLFLLLLWLPDTTG (SEQ ID NO: 88). Other suitable signal sequences include a human CD4 signal peptide, e.g., having the sequence MNRGVPFRHLLLVLQLALLPAAT (SEQ ID NO: 89), a mouse Ig kappa chain V-III region signal peptide, e.g., having the
sequence METDTLLLWVLLLWVPGSTG (SEQ ID NO: 90), a mouse H-2Kb signal peptide, e.g., having the sequence MVPCTLLLLLAAALAPTQTRA (SEQ ID NO: 91), a human serum albumin signal peptide, e.g., having the sequence MKWVTFISLLFLFSSAYS (SEQ ID NO: 92), a human IL-2 signal peptide, e.g., having the sequence MYRMQLLSCIALSLALVTNS (SEQ ID NO: 93), a human HLA-A*02:01 signal peptide, e g., having the sequence MAVMAPRTLLLLLSGALALTQTWA (SEQ ID NO: 94) and a human b2m signal peptide, e.g., having the sequence MSRSVALAVLALLSLSGLEA (SEQ ID NO: 95).
[0155] The expression construct is introduced into the host cell using a method appropriate to the host cell, as will be apparent to one of skill in the art. A variety of methods for introducing nucleic acids into host cells are known in the art, including, but not limited to, electroporation; transfection employing calcium chloride, rubidium chloride, calcium phosphate, DEAE-dextran, or other substances; microprojectile bombardment; lipofection; and infection (where the vector is an infectious agent).
[0156] Suitable host cells include prokaryotes, yeast, mammalian cells, or bacterial cells. Suitable bacteria include gram negative or gram positive organisms, for example, E. coli or Bacillus spp. Yeast, preferably from the Saccharomyces species, such as 5. cerevisiae, may also be used for production of polypeptides. Various mammalian or insect cell culture systems can also be employed to express recombinant proteins. Baculovirus systems for production of heterologous proteins in insect cells are reviewed by Luckow et al. ((1988) BIO/TECHNOLOGY 6:47). Examples of suitable mammalian host cell lines include endothelial cells, COS-7 monkey kidney cells, CV-1, L cells, C127, 3T3, Chinese hamster ovary (CHO), human embryonic kidney cells, HeLa, 293, 293T, and BHK cell lines. Purified polypeptides are prepared by culturing suitable host/vector systems to express the recombinant proteins. For many applications, the small size of many of the polypeptides described herein would make expression in E. coli as the preferred method for expression. The protein is then purified from culture media or cell extracts.
[0157] The host cells used to produce the proteins of this invention may be cultured in a variety of media. Commercially available media such as Ham’s F10 (Sigma), Minimal Essential Medium ((MEM), (Sigma), RPMI-1640 (Sigma), and Dulbecco’s Modified Eagle’s Medium ((DMEM), Sigma)) are suitable for culturing the host cells. In addition, many of the media described in Ham et al. (1979) METH. ENZYMOL. 58:44, Barites et al. (1980) ANAL. BIOCHEM. 102:255, U.S. Pat. Nos. 4,767,704, 4,657,866, 4,927,762, 4,560,655, 5,122,469,
6,048,728, 5,672,502, or U.S. Pat. No. RE 30,985 may be used as culture media for the host cells. Any of these media may be supplemented as necessary with hormones and/or other growth factors (such as insulin, transferrin, or epidermal growth factor), salts (such as sodium chloride, calcium, magnesium, and phosphate), buffers (such as HEPES), nucleotides (such as adenosine and thymidine), antibiotics (such as Gentamycin drug), trace elements (defined as inorganic compounds usually present at final concentrations in the micromolar range), and glucose or an equivalent energy source. Any other necessary supplements may also be included at appropriate concentrations that would be known to those skilled in the art. The culture conditions, such as temperature, pH, and the like, are those previously used with the host cell selected for expression, and will be apparent to the ordinarily skilled artisan.
[0158] Proteins described herein can also be produced using cell-free translation systems. For such purposes the nucleic acids encoding the polypeptide must be modified to allow in vitro transcription to produce mRNA and to allow cell-free translation of the mRNA in the particular cell-free system being utilized (eukaryotic such as a mammalian or yeast cell-free translation system or prokaryotic such as a bacterial cell-free translation system).
[0159] Proteins described herein can also be produced by chemical synthesis (e.g, by the methods described in SOLID PHASE PEPTIDE SYNTHESIS, 2nd Edition, The Pierce Chemical Co., Rockford, Ill. (1984)). Modifications to the protein can also be produced by chemical synthesis.
[0160] The proteins of the present invention can be purified by isolation/purification methods for proteins generally known in the field of protein chemistry. Non-limiting examples include extraction, recrystallization, salting out (e.g., with ammonium sulfate or sodium sulfate), centrifugation, dialysis, ultrafiltration, adsorption chromatography, ion exchange chromatography, hydrophobic chromatography, normal phase chromatography, reversed- phase chromatography, get filtration, gel permeation chromatography, affinity chromatography, electrophoresis, countercurrent distribution or any combinations of these. After purification, polypeptides may be exchanged into different buffers and/or concentrated by any of a variety of methods known to the art, including, but not limited to, filtration and dialysis.
[0161] The purified polypeptide is preferably at least 85% pure, or preferably at least 95% pure, and most preferably at least 98% pure. Regardless of the exact numerical value of the purity, the polypeptide is sufficiently pure for its intended use.
[0162] In certain embodiments, the expression construct includes at least one tag sequence, most typically as at the C-terminal end of the coding region, although inclusion of a tag at the N-terminal end (alternative to or in addition to the C-terminal end) is also encompassed. Suitable tag sequences are known in the art and described further herein. A protein tag sequence that may be useful e.g., for labeling the multimer composition and/or for purifying the composition. Examples of protein tags include, but are not limited to, a histidine tag (6x His), a FLAG tag, a myc tag, an HA tag, a GST tag, and combinations thereof.
[0163] Non-limiting examples of suitable tags include FLAG tags (e.g., having the amino acid sequence shown in SEQ ID NO: 96), 6xHis tags (e.g., having the amino acid sequence shown in SEQ ID NO: 97), V5 tags (e.g., having the amino acid sequence shown in SEQ ID NO: 98), Strep-Tags (e.g., having the amino acid sequence shown in SEQ ID NO: 99), Protein C tags (e.g., having the amino acid sequence shown in SEQ ID NO: 100) and/or myc tags (e.g., having the amino acid sequence shown in SEQ ID NO: 101).
[0164] Additional tags suitable for use in the methods and compositions provided herein include affinity tags, including but not limited to enzymes, protein domains, or small polypeptides which bind with high specificity to a range of substrates, such as carbohydrates, small biomolecules, metal chelates, antibodies, etc. to allow rapid and efficient purification of proteins. Solubility tags enhance proper folding and solubility of a protein and are frequently used in tandem with affinity tags. Sequences encoding such a tag(s) can be incorporated into an expression construct of the disclosure, such as at the C-terminus or N-terminus of the peptide- multimer-encoding regions to thereby incorporate a detectable tag into the expressed polypeptide.
E. Chemical Conjugation Preparation of Multimer Compositions
[0165] In another embodiment, a multimer composition of the disclosure is produced by covalent conjugation of the MHCII-binding peptide to the multimerization domain. In certain embodiments, a multimer composition is produced by covalent conjugation of the peptide to the N-terminus of the multimerization domain. In certain embodiments, a multimer composition is produced by covalent conjugation of the peptide to the C-terminus of the multimerization domain.
[0166] For chemical conjugation approaches, the peptide and multimerization domain (MD) components can be prepared either recombinantly or chemically. For example, peptides can be generated according to synthesis methods known in the art, or synthetically produced by a
commercial vendor or using a peptide synthesizer according to manufacturer’s instructions. For example, in some embodiments, peptides can be made by in silico production methods. In other embodiments, peptides can be synthesized via chemical methods, for example, tea bag synthesis, digital photolithography, pin synthesis, and SPOT synthesis. For example, an array of peptides can be generated via SPOT synthesis, where amino acid chains are built on a cellulose membrane by repeated cycles of adding amino acids, and cleaving side-chain protection groups.
[0167] In other embodiments, peptides and/or MDs can be expressed using recombinant DNA technology, for example, introducing an expression construct into bacterial cells, insect cells, or mammalian cells, and purifying the recombinant protein from cell extracts, e.g., as described above.
[0168] A number of suitable methods for forming covalent bonds between each MHCII- binding peptide and the multimerization domain are provided herein.
A. Chemical Bioconjugation
[0169] In some embodiments, the chemical conjugation is mediated by cysteine bioconjugation of the MHCII-binding peptide (e.g., including a spacer linker) to the multimerization domain. In some embodiments, the cysteine bioconjugation is mediated by cysteine alkylation. In some embodiments, the cysteine bioconjugation is mediated by cysteine oxidation. In other embodiments, the cysteine bioconjugation is mediated by a desulfurization reaction. In some embodiments, cysteine bioconjugation is mediated by iodoacetamide. In some embodiments, the cysteine bioconjugation is mediated by maleimide. Methods for utilizing cysteine mediated linkage of two moieties which can be used to produce the multimer composition disclosed herein have been described, for example, see Chalker e/ o/. (2009) CHEM ASIAN J. 4(5):630-40; Spicer et al. (2015) NAT COMMUN. 5:4740.
[0170] In some embodiments, the multimer compositions are produced by chemical modification of amino acids other than cysteine, including but not limited to lysine, tyrosine, arginine, glutamate, aspartate, serine, threonine, methionine, histidine and tryptophan sidechains, as well as N-terminal amines or C-terminal carboxyls, as previously described (Basle et al. (2010) M CHEM BIOL. 17(3):213-27; Hu e/ a/. (2016) CHEM SOC REV. 45(6): 1691-719; Lin et al. (2017) SCIENCE 355(6325): 597-602).
B. Native Chemical Ligation
[0171] In some embodiments, the multimer compositions are produced by native chemical ligation (NCL), wherein each peptide (or peptide-spacer linker) comprises a C-terminal thioester, and each subunit of the multimerization domain comprises an N-terminal cysteine residue, or functional equivalent thereof, wherein the reaction between the cysteine sidechain and the thioester irreversibly forms a native peptide bond, thus ligating the peptides to the multimerization domain. Methods for NCL have been described (Hejjaoui et al. (2015) M PROTEIN SCI. 24(7): 1087-99; Mandal et al. (2012) PROC NATL AC D SCI USA 109(37): 14779-84; Torbeev et al. (2013) PROC NATL ACAD SCI USA 110(50): 20051-6).
[0172] In some embodiments, [3- and/or y-thio amino acids are incorporated into the peptides (or peptide-spacer linker). In some embodiments, [3- and/or y-thio amino acids replace the cysteine-like residue at an N-terminal position of each subunit of the multimerization domain, e.g., to provide a reactive thiol for trans-thioesterifi cation. Desulfurization protocols can then produce the desired native side-chain. In some embodiments, NCL is performed at an alanine residue. In other embodiments, NCL is performed at phenylalanine (Crich & Banerjee, 2007), valine (Chen et al. 2008; Haase et al. 2008), leucine (Harpaz et al. 2010; Tan et al. 2010), threonine (Chen et al. 2010b), lysine (El Oualid et al. 2010; Kumar et al. 2009; Yang et al. 2009), proline (Shang et al. 2011), glutamine (Siman et al. 2012), arginine (Malins et al. 2013), tryptophan (Malins etal. 2014), aspartate (Thompson et al. 2013), glutamate (Cergol et al. 2014) and asparagine (Sayers et al. 2015). Ligation/desulfurization approaches that remove purification steps and increase the yield obligated products have been described (Moyal et al. 2013; Thompson et al. 2014).
C. Click Chemistry Mediated Bioorthogonal Conjugation
[0173] In some embodiments, the multimer compositions are produced by bioorthogonal conjugation between the conjugation moiety at the C-terminus of each peptide (or peptide- spacer linker) and the conjugation moiety at the N-terminus of each subunit of the multimerization domain. In some embodiments, the bioorthongonal conjugation is mediated by “click chemistry.” (see, e.g., Kolb, Finn and Sharpless (2001) ANGEWANDTE CHEMIE INTERNATIONAL EDITION 40: 2004-2021). Conjugation moieties suitable for click chemistry, reaction conditions, and associated methods are available in the art (e.g., Kolb et al. )2001) ANGEWANDTE CHEMIE INTERNATIONAL EDITION 40:2004-2021; Evans (2007) AUSTRALIAN JOURNAL OF CHEMISTRY 60: 384-395; Lahann (2009) CLICK CHEMISTRY FOR
BIOTECHNOLOGY AND MATERIALS SCIENCE, John Wiley & Sons Ltd, ISBN 978-0-470- 69970-6). In some embodiments, a click chemistry moiety may comprise or consist of a terminal alkyne, azide, strained alkyne, diene, dieneophile, alkoxyamine, carbonyl, phosphine, hydrazide, thiol, or alkene moiety. In certain embodiments, the azide is a copperchelating azide. In one embodiment, the copper-chelating azide is a picolyl azide, such as Gly-Gly-Gly-(PEG)4-Picolyl-Azide. Reagents for use in click chemistry reactions are commercially available, such as from Click Chemistry Tools (Scottsdale, AZ) or GenScript (Piscataway, NJ).
[0174] For conjugation of each peptide (or peptide-spacer linker) to a subunit of the multimerization domain via click chemistry, the click chemistry moieties of the components have to be reactive with each other, for example, in that the reactive group of one of the click chemistry moiety of each peptide (or peptide-spacer linker) reacts with the reactive group of the second click chemistry moiety on a subunit of the multimerization domain to form a covalent bond.
[0175] In some embodiments, each peptide (or peptide-spacer linker) conjugation moiety can be covalently conjugated under click chemistry reaction conditions to the conjugation moiety of each subunit of the multimerization domain. In some embodiments a sortase-mediated conjugation is used to install a first click chemistry moiety at the C-terminus of each peptide (or peptide-spacer linker), and a second click chemistry moiety reaction to each subunit of the multimerization domain. In the methods provided herein, two or more peptides (or peptide- spacer linkers) containing the first click chemistry moiety are conjugated to the second click chemistry moiety at the C-terminus of each subunit of the multimerization domain under click chemistry conditions. Methods of attaching click chemistry moieties utilizing sortase are described, for example, in W02013/00355, the entire contents of which is hereby incorporated by reference.
[0176] In some embodiments, an intein-mediated conjugation is used to install a first click chemistry moiety at the C-terminus of each peptide (or peptide-spacer linker), and a second click chemistry moiety reaction to each subunit of the multimerization domain.
[0177] In some embodiments, the methods of click chemistry mediated covalent conjugation of the peptide (or peptide-spacer linker) to the multimerization domain provided herein comprise native chemical ligation of C-terminal thioesters with -amino thiols (Xiao J, Tolbert TJ (2009) ORG LETT. 11(18):4144-7).
[0178] In some embodiments, the click chemistry used to produce the multimer composition comprises 1,3-dipolar cycloaddition (e.g., the Cu(I)-catalyzed stepwise variant, often referred to simply as the “click reaction”; see, e.g., Tomoe et al. (2002) JOURNAL OF ORGANIC CHEMISTRY 67: 3057-3064). Copper and ruthenium are the commonly used catalysts in the reaction. The use of copper as a catalyst results in the formation of 1,4-regioisomer whereas ruthenium results in formation of the 1,5-regioisomer.
[0179] In some embodiments, the peptides (or peptide-spacer linkers) are ligated to an alkynated peptide by expressed protein ligation (EPL) and then conjugated to an azide- labeled multimerization domain by Cu(I)-catalyzed terminal azide-alkyne cycloaddition (CuAAC).
[0180] In some embodiments, the click chemistry conjugation comprises a cycloaddition reaction, such as the Diels-Alder reaction. In some embodiments, the MHCII-binding peptide and multimerization domain are conjugated by azide-alkyne 1,3-dipolar cycloaddition (“click chemistry). In some embodiments, the cycloaddition is promoted by the presence of Cu(I)-catalyzed cycloaddition (CuAAC).
[0181] In some embodiments, the click chemistry conjugation comprises nucleophilic addition to small strained rings like epoxides and aziridines. In some embodiments, the cycloaddition is promoted by strained cyclooctyne systems, for example, as described in Agard NJ, Prescher JA, Bertozzi CR (2004) J AM CHEM SOC. 126(46): 15046-7.
[0182] In some embodiments, the click chemistry conjugation comprises nucleophilic addition to activated carbonyl groups.
[0183] In some embodiments, the conjugation of the peptide (or peptide-spacer linker) and multimerization domain occurs by a bioorthogonal reaction. In some embodiments, the MHC and multimerization domain are conjugated by inverse-electron demand Diels-Alder reactions between strained dienophiles and tetrazine dienes, for example, as described in Blackman ML, Royzen M, Fox JM (2008) J AM CHEM SOC. 130(41): 13518-9; and Devaraj NK, Weissleder R, Hilderbrand SA (2008) BlOCONJUG CHEM. 19(12):2297-9). In some embodiments, the dienophile is a trans-cyclooctene. In some embodiments, the dienophile is a norbomene.
D. Sortase Mediated Conjugation
[0184] In some embodiments, conjugation between the peptide (or peptide-spacer linker) and the multimerization domain is mediated by a cysteine transpeptidase. In some embodiments,
the cysteine transpeptidase is a sortase, or enzymatically active fragment thereof. A variety of sortase enzymes have been described and are commercially available (e.g., Antos et al. (2016) CURR. OPIN. STRUCT. BIOL. 38:111-118). Sortases recognize and cleave an amino acid motif, referred to as a “sortag”, to produce a peptide bond between the acyl donor and acceptor site on two polypeptides, resulting in the ligation of different polypeptides which contain N- or C- terminal sortags. In particular, sortases join a C-terminal LPETGG recognition motif (SEQ ID NO: 133) to an N-terminal GGG (oligoglycine) motif.
[0185] Accordingly, in some embodiments, the recognition motif is added to the C-terminus of each peptide (or peptide-spacer linker), and an oligo-glycine motif is added to the N- terminus of each of the subunits of the mutimerization domain. Alternatively, the recognition motif is added to the C-terminus of each of the subunits of the multimerization domain and the oligo-glycine motif is added to the N-terminus of each peptide (or peptide-spacer linker). Upon addition of sortase to the mixture of peptide (or peptide-spacer linker) and multimerization domains, the polypeptides are covalently linked through a native peptide bond.
[0186] In some embodiments, the peptide (or peptide-spacer linker) and/or multimerization domain are expressed in frame with the sortags. In some embodiments, additional tags may be included, for example, a 6x-His tag (Sinisi et al. (2012) BlOCONJUG. CHEM 23: 1119-1126), a nucleophilic fluorochrome (Nair et al. (2013) IMMUN. INFLAMM. DIS. 1:3-13), and/or a FLAG tag (Greineder et al. (2018) BlOCONJUG. CHEM. 29:56-66).
[0187] In some embodiments, the sortag contains a modified amino acid suitable for chemical conjugation between the MHC monomers and the mutimerization domain. In some embodiments, the sortag contains a C-terminal azidolysine residue to enable oriented clickclick chemistry conjugation as described herein.
[0188] In some embodiments, the peptide and/or multimerization domains comprise a linker between the polypeptide and the sortag. In some embodiments, each peptide and each subunit of the multimerization domain comprises a sortag with a linker. Suitable linkers have been described herein and in the art, for example, in Greineder et al. (2018) BlOCONJUG. CHEM. 29:56-66. In some embodiments, the linker is a flexible, rigid, or semi-rigid linker, e.g., a linker having an amino acid sequence shown in any one of SEQ ID NOs: 6-8 and 72- 80.
[0189] In some embodiments, the sortag contains a fluorophore-modified lysine residue to facilitate measurement of reaction progression and efficiency.
[0190] In some embodiments, the sortase is Ca2+ dependent. In some embodiments, the sortase is Ca2+ independent.
[0191] In some embodiments, the sortag comprises the amino acid sequence LPXTG (SEQ ID NO: 102), wherein X is any amino acid, and the sortase cleaves between the threonine and glycine backbone within the motif.
[0192] In some embodiments, the sortase recognizes a sortag comprising an amino acid sequence selected from IPKTG (SEQ ID NO: 103), MPXTG (SEQ ID NO: 104), wherein X is any amino acid, LAETG (SEQ ID NO: 105), LPXAG (SEQ ID NO: 106), wherein X is any amino acid, LPESG (SEQ ID NO: 107), LPELG (SEQ ID NO: 108) or LPEVG (SEQ ID NO: 109).
[0193] In some embodiments, the sortase is a SrtAstaph mutant. In some embodiments, the SrtAstaph mutant is F40, and the recognition motif is XPKTG (SEQ ID NO: 110), wherein X is any amino acid (Piotukh et al. (2011) J. AM. CHEM. SOC. 133:17536-17539). In some embodiments, the SrtAstaph mutant is F40 and the recognition motif is APKTG (SEQ ID NO: 111), DPKTG (SEQ ID NO: 112) or SPKTG (SEQ ID NO: 113).
[0194] In some embodiments, the SrtAstaph mutant is SrtAstaph pentamutant and the recognition motif is LPXTG (SEQ ID NO: 114), wherein X is any amino acid, LPEXG, (SEQ ID NO: 115), wherein X is any amino acid, or LAETG (SEQ ID NO: 116). In some embodiments, the mutant is SrtAstaph pentamutant and the recognition motif is LPEAG (SEQ ID NO: 117), LPECG (SEQ ID NO: 118) or LPESG (SEQ ID NO: 119). In some embodiments, the SrtAstaph mutant is 2A-9 and the recognition motif is LAETG (SEQ ID NO: 116). In some embodiments, the SrtAstaph mutant is 4S-9 and the recognition motif is LPEXG (SEQ ID NO: 121), wherein X = A, C or S.
[0195] In some embodiments, the sortase is a soluble fragment of the wild-type sortase. In some embodiments, the sortase is a soluble fragment of a modified sortase A (Mao H, Hart SA, Schink A, Pollok BA (2004) J AM CHEM SOC. 126(9):2670-l A).
[0196] In some embodiments, the sortase is a variant or homolog of 5. aureus sortase A (Antos JM, Truttmann MC, Ploegh HL (2016) CURR OPIN STRUCT BIOL. 38: 111-8; Dorr BM, Ham HO, An C, Chaikof EL, Liu DR (2014) PROC NATL ACAD SCI USA 111(37): 13343-8; Glasgow JE, Salit ML, Cochran JR (2016) J AM CHEM SOC. 138(24):7496-9).
[0197] Methods of conjugation of sortags into proteins have also been described. (Matsumoto T, Furuta K, Tanaka T, Kondo A (2016) ACS SYNTH BIOL. 5(11): 1284-1289; Williams FP, Milbradt AG, Embrey KJ, Bobby R (2016) PLoS ONE. ll(4):e0154607; and Witte MD, Cragnolini JJ, Dougan SK, Yoder NC, Popp MW, Ploegh HL (2012) PROC NATL AC D SCI USA 109(30): 11993-8; Mao H, Hart SA, Schink A, Pollok BA (2004) J AM CHEM SOC. 126(9):2670-l; Guimaraes CP, Witte MD, Theile CS, Bozkurt G, Kundrat L, Blom AE, Ploegh HL (2013) NAT PROTOC. 8(9): 1787-99; and Theile CS, Witte MD, Blom AE, Kundrat L, Ploegh HL, Guimaraes CP (2013) NAT PROTOC. 8(9): 1800-7.
[0198] In some embodiments, the aminoglycine peptide fragment generated by the sortase reaction, is removed by dialysis or centrifugation, e.g., while the reaction is proceeding (Freiburger L, Sonntag M, Hennig J, Li J, Zou P, Sattler M (2015) J BlOMOL NMR 63(1): 1- 8). In some embodiments, affinity immobilization strategies or flow-based platforms are used for the selective removal of reaction components (Policarpo RL, Kang H, Liao X, Rabideau AE, Simon MD, Pentelute BL (2014) ANGEW CHEM INT ED ENGL 53(35): 9203-8).
[0199] In some embodiments, the equilibrium of the reaction can be controlled by ligation product or by-product deactivation. For example, in some embodiments the reaction is controlled by ligation of a WTWTW (SEQ ID NO: 122) motif added to the donor and acceptor as described in Yamamura Y, Hirakawa H, Yamaguchi S, Nagamune T (2011) CHEM COMMUN (CAMB) 47(16): 4742 -4). In other embodiments, by-products are deactivated by chemical modification of the acyl donor glycine as described, for example, in Liu F, Luo EY, Flora DB, Mezo AR (2014) J ORG CHEM 79(2):487-92; and Williamson DJ, Webb ME, Turnbull WB (2014) NAT PROTOC 9(2):253-62).
E. Intein-Mediated Conjugation
[0200] Inteins are naturally occurring, self-splicing protein subdomains that are capable of excising out their own protein subdomain from a larger protein structure while simultaneously joining the two formerly flanking peptide regions (“exteins”) together to form a mature host protein. Intein-based methods of protein modification and ligation have been developed. An intein is an internal protein sequence capable of catalyzing a protein splicing reaction that excises the intein sequence from a precursor protein and joins the flanking sequences (N- and C-exteins) with a peptide bond.
[0201] As used herein, the term “split intein” refers to any intein in which one or more peptide bond breaks exists between the N-terminal intein segment and the C-terminal intein
segment such that the N-terminal and C-terminal intein segments become separate molecules that cannon-covalently reassociate, or reconstitute, into an intein that is functional for splicing or cleaving reactions. Any catalytically active intein, or fragment thereof, may be used to derive a split intein for usein the systems and methods disclosed herein. For example, in one aspect the split intein may be derived from a eukaryotic intein. In another aspect, the split intein may be derived from a bacterial intein. In another aspect, the split intein may be derived from an archaeal intein. Preferably, the split intein so-derived will possess only the amino acid sequences essential for catalyzing splicing reactions.
[0202] As used herein, the “N-terminal intein segment” refers to any intein sequence that comprises an N-terminal amino acid sequence that is functional for splicing and/or cleaving reactions when combined with a corresponding C-terminal intein segment. An N-terminal intein segment thus also comprises a sequence that is spliced out when splicing occurs. An N-terminal intein segment can comprise a sequence that is a modification of the N-terminal portion of a naturally occurring (native) intein sequence. For example, an N-terminal intein segment can comprise additional amino acid residues and/or mutated residues so long as the inclusion of such additional and/or mutated residues does not render the intein non-functional for splicing or cleaving. Preferably, the inclusion of the additional and/or mutated residues improves or enhances the splicing activity and/or controllability of the intein. Non-intein residues can also be genetically fused to intein segments to provide additional functionality, such as the ability to be affinity purified or to be covalently immobilized.
[0203] As used herein, the “C-terminal intein segment” refers to any intein sequence that comprises a C-terminal amino acid sequence that is functional for splicing or cleaving reactions when combined with a corresponding N-terminal intein segment. In one aspect, the C-terminal intein segment comprises a sequence that is spliced out when splicing occurs. In another aspect, the C-terminal intein segment is cleaved from a peptide sequence fused to its C-terminus. A C-terminal intein segment can comprise a sequence that is a modification of the C-terminal portion of a naturally occurring (native) intein sequence. For example, a C terminal intein segment can comprise additional amino acid residues and/or mutated residues so long as the inclusion of such additional and/or mutated residues does not render the C- terminal intein segment non-functional for splicing or cleaving.
[0204] Expressed protein ligation (EPL) refers to a native chemical ligation between a recombinant protein with a C-terminal thioester and a second agent with an N-terminal cysteine. The C-terminal thioester can readily be introduced onto any recombinant protein
(i.e. , the targeting ligand) through the use of auto-processing, also known as protein-splicing, mediated by an intein (intervening protein). Inteins are proteins that can excise themselves from a larger precursor polypeptide chain, utilizing a process that results in the formation of a native peptide bond between the flanking extein (external protein) fragments. When an autoprocessing protein is cloned downstream of the targeting ligand, thiols (e.g., 2- mercaptoethanesulfonic acid, MESNA) can be used to induce the site-specific cleavage of the auto-processing protein, resulting in the formation of a reactive thioester. The thioester will then react with any agent that has an N-terminal cysteine. EPL operates in a site-specific manner, and the reaction is known to be very efficient if both functional groups are in high concentrations, (reviewed in Elias et al. Small 6:2460-2468).
[0205] Accordingly, in some embodiments, MHCII-binding peptides (or peptides-spacer linkers) are ligated to an alkynated peptide by expressed protein ligation (EPL) and then conjugated to an azide-labeled multimerization domain by Cu(I)-catalyzed terminal azidealkyne cycloaddition (CuAAC).
[0206] In some embodiments, the peptides (or peptides-spacer linkers) are conjugated to the multimerization domain by an intein peptide tag. In some embodiments, the peptide (or peptides-spacer linker) comprises a C-terminal thioester, the multimerization domain comprises an N-extein fused to a modified intein lacking the ability to perform trans- esterification and /ram-esterification occurs by the addition of exogenous thiol.
[0207] A number of inteins have now been described including, but not limited to MxeGyrA (Frutos et al. (2010); Southworth et al. (1999); SspDnaE (Shah et al. (2012); Wu et al. (1998); NpuDnaE (Shah et al. (2012); Vila-Perello et al. (2013); AvaDnaE (David etal. (2015); Shah et al. (2012); Cfa (consensus DnaE split intein) (Stevens et al. (2016)); gp41-l and gp41-8 (Carvajal -Vallejos etal. (2012)); NrdJ-1 (Carvajal -Vallejos et al. (2012)); IMPDH-1 (Carvajal -Vallejos et al. (2012)) and AceL-TerL (Thiel et al. (2014). The properties and use of these inteins are summarized in Table 1.
Table 1


[0208] In some embodiments, the intein is the 198-residue gyrase A intein from Mycobacterium xenopi (Mxe GyrA) (Southworth MW, Amaya K, Evans TC, Xu MQ, Perler FB (1999) BIOTECHNIQUES 27(l):110-4, 116, 118-20). In some embodiments, the intein is from cyanobacterium Synechocystis sp. strain PCC6803 (Ssp).
[0209] In some embodiments, the intein is a split intein pair. In some embodiments, the split intein pair is an orthogonal split intein pair (Carvajal -Vallejos P, Pallisse R, Mootz HD, Schmidt SR (2012) J BIOL CHEM 287(34):28686-96; Shah NH, Vila-Perello M, Muir TW (2011) ANGEW CHEM INT ED ENGL 50(29):6511-5). [0210] In some embodiments, the split intein pair is an artificially split intein pair that are as short as six or eleven residues (Appleby JH, Zhou K, Volkmann G, Liu XQ (2009) J BIOL CHEM 284(10):6194-9; Ludwig C, Pfeiff M, Linne U, Mootz HD (2006) ANGEW CHEM INT ED ENGL 45(31):5218-21).
[0211] In some embodiments, the intein is a DnaE intein. In some embodiments, the DnaE intein is from Nostoc punctiforme (Npu). In some embodiments, the intein is the gp41-l
intein. In some embodiments, the intein is the gp41-8 intein. In some embodiments, the intein is the IMPDH-1 intein. In some embodiments, the intein is the NrdJ Intein.
[0212] In some embodiments, the split intein pair is AceL-TerL (Thiel IV, Volkmann G, Pietrokovski S, Mootz HD (2014) ANGEW CHEM INT ED ENGL 53(5): 1306-10).
[0213] In some embodiments, the intein comprises consensus split intein sequence (Cfa) (Stevens AJ, Brown ZZ, Shah NH, Sekar G, Cowbum D, Muir TW (2016) JOURNAL OF THE AMERICAN CHEMICAL SOCIETY 138(7):2162-2165).
[0214] Suitable intein sequences and protocols for use in protein conjugation have been described in the art, such as in Stevens, et al. (2016) J. AM. CHEM. SOC. 138, 2162-2165; Shah et al. (2012) J. AM. CHEM. SOC. 134, 11338-11341; and Vila-Perello e/ al. (2013) J. AM. CHEM. SOC. 135, 286-292; Batjargal S, Walters CR, Petersson EJ (2015) J AM CHEM SOC 137(5): 1734-7; and Guan D, Ramirez M, Chen Z (2013) BIOTECHNOL BIOENG 110(9):2471- 81, the entire contents of each of which is hereby incorporated by reference.
F. Additional Bioconjugation Methods
[0215] In some embodiments, the conjugation of the peptide (or peptide-spacer linker) and multimerization domain is mediated enzymatically. In some embodiments, the enzyme is formylglycine generating enzyme (FGE) that recognizes the CXPXR amino acid sequence motif and converts the cysteine residue to formylglycine, thus introducing an aldehyde functional group (Wu P, Shui W, Carlson BL, Hu N, Rabuka D, Lee J, Bertozzi CR (2009) PROC NATL ACAD SCI USA 106(9):3000-5), which is subjected to bio-orthogonal transformations such as oximation and Hydrazino-Pictet-Spengler reactions (Agarwal P, Kudirka R, Albers AE, Barfield RM, de Hart GW, Drake PM, Jones LC, Rabuka D (2013) BIOCONJUG CHEM 24(6): 846-51; Dirksen A, Dawson PE (2008) BlOCONJUG CHEM 19(12):2543-8).
[0216] Site-specific bioconjugation strategies offer many possibilities for directed protein modifications. Among the various enzyme-based conjugation protocols, formylglycine- generating enzymes allow to posttranslationally introduce the amino acid Ca-formylglycine (FGly) into recombinant proteins, starting from cysteine or serine residues within distinct consensus motifs. The aldehyde-bearing FGly-residue displays orthogonal reactivity to all other natural amino acids and can, therefore, be used for site-specific labeling reactions on protein scaffolds. (Reviewed in Kruger et al. (2019) BIOL CHEM 400(3):289-297. doi: 10.1515/hsz-2018-0358)
[0217] Formylglycine generating enzyme (FGE) recognizes a pentapeptide consensus sequence, CxPxR, and it specifically oxidizes the cysteine in this sequence to an unusual aldehyde-bearing formylglyine. The FGE recognition sequence, or aldehyde tag, can be inserted into heterologous recombinant proteins produced in either prokaryotic or eukaryotic expression systems. The conversion of cysteine to formylglycine is accomplished by co- overexpression of FGE, either transiently or as a stable cell line, and the resulting aldehyde can be selectively reacted with a-nucleophiles to generate a site-selectively modified bioconjugate (Rabuka et al. (2012) NAT PROTOC 7(6): 1052-1067).
[0218] In some embodiments, the enzyme is lipoic acid ligase, an enzyme that modifies a lysine side-chain within the 13-residue target sequence (Uttamapinant C, White KA, Baruah H, Thompson S, Fernandez-Suarez M, Puthenveetil S, Ting AY (2010) PROC NATL ACAD SCI USA 107(24): 10914-9) to introduce bio-orthogonal groups, including azides, aryl aldehydes and hydrazines, p-iodophenyl derivatives, norbomenes, and trans-cyclooctenes (reviewed in Debelouchina et al. (2017) Q. REV BIOPHYS. 50 e7. doi:10.1017/S0033583517000021).
[0219] In other embodiments, the enzyme is biotin ligase, famesyltransferase, transglutaminase or N-myristoyltransferase (reviewed in Rashidian M, Dozier JK, Distefano MD (2013) BIOCONJUG CHEM 24(8): 1277-94).
II. MHC Class II Molecules
[0220] To create the MHCII multimers of the disclosure, the multimer composition (comprising peptide-MD) is “loaded” with MHCII molecules, e.g., soluble MHCII dimers. The soluble MHCII molecules can be “empty”, i.e., not loaded with a peptide in the peptide binding groove (e.g., certain MHCII molecules may be sufficiently stable as empty soluble molecules to enable loading onto the peptide-MD compositions). More typically, the MHCII molecules are prepared as soluble molecules with a cleavable placeholder peptide in the antigen-binding groove (referred to herein as p*MHC) to enhance the stability of the MHCII molecules. A representative schematic diagram of a soluble MHCII molecule loaded with a cleavable placeholder peptide is shown in FIG. 1 and preparation thereof is described in detail in Example 2. The components of the MHCII molecules are described in further detail below.
A. Soluble MHCII Polypeptides
[0221] MHC class II molecules are heterodimers composed of an a chain and a chain, both of which are encoded by the MHC. The alpha chain is comprised of al and a2 domains.
The beta chain is comprised of P 1 and 2 domains. The al and pi domains of the chains interact noncovalently to form a membrane-distal peptide-binding domain, whereas the a2 and P2 domains form membrane-proximal immunoglobulin-like domains. The antigen binding groove, where a peptide epitope binds, is made up of two a-helices and a P-sheet. Since the antigen binding groove of MHC class II molecules is open at both ends, the groove can accommodate longer peptide epitopes than MHC class I molecules. Peptide epitopes presented by MHC class II molecules typically are about 15-24 amino acid residues in length.
[0222] The MHCII molecule can suitably be a vertebrate MHCII molecule such as a human, a mouse, a rat, a porcine, a bovine or an avian MHCII molecule.
[0223] In some embodiments, the multimeric MHCII multimers described herein, the MHC molecule is a human MHC class II protein: HLA-DR, HLA-DQ, HLA-DX, HLA-DO, HLA- DZ, and HLA-DP. The amino acid sequences of the MHCII a and P chains from a variety of vertebrate species, including humans, are known in the art and publicly available.
[0224] In some embodiments, the human MHCII molecule is of an allotype selected from the group consisting of DRBl*0101 (see, e.g., Cameron et al. (2002) J. IMMUNOL. METHODS 268:51-69; Cunliffe et al. (2002) EUR. J. IMMUNOL. 32:3366-3375; Danke et al. (2003) J.
IMMUNOL. 171:3163-3169), DRBl*1501 (see, e.g., Day et al. (2003) J. CLIN. INVEST 112:831-842), DRB5*0101 (see, e.g., Day et al., ibid), DRBl*0301 (see, e.g., Bronke et al. (2005) HUM. IMMUNOL. 66:950-961), DRBl*0401 (see, e.g., Meyer etal. (2000) PNAS 97:11433-11438; Novak et al. (1999) J. CLIN. INVEST 104:R63-R67; Kotzin et al. (2000) PNAS 97:291-296), DRBl*0402 (see, e.g, Veldman et al. (2007) CLIN. IMMUNOL. 122:330- 337), DRBl*0404 (see, e.g, Gebe etal. (2001) J. IMMUNOL. 167:3250-3256), DRBl*1101 (see, e.g., Cunliffe, ibid; Moro et al. (2005) BMC IMMUNOL. 6:24), DRB1*13O2 (see, e.g., Laughlin et al. (2007) INFECT. IMMUNOL. 75:1852-1860), DRBl*0701 (see, e.g., Danke, ibid), DQAl*0102 (see, e.g, Kwok et al. (2000) J. IMMUNOL. 164:4244-4249), DQBl*0602 (see, e.g., Kwok, ibid), DQAl*0501 (see, e.g., Quarsten et al. (2001) J. IMMUNOL. 167:4861- 4868), DQBl*0201 (see, e.g., Quarsten, ibid), DPAl*0103 (see, e.g., Zhang et al. (2005) EUR. J. IMMUNOL. 35:1066-1075; Yang et al. (2005) J. CLIN. IMMUNOL. 25:428-436), and DPBl*0401 (see, e.g., Zhang, ibid; Yang, ibid).
[0225] In some embodiments, the MHCII molecule is human, and comprise, for example, an MHCII alpha and beta chains selected from the group consisting of HLA-DRA*01 :01, HLA- DRBl*01:01, HLA-DRBl*01:02, HLA-DRBl*03:01, HLA-DRBl*04:01, HLA-
DRBl*04:03, HLA-DRBl*04:04, HLA-DRBl*07:01, HLA-DRBl*08:01, HLA- DRBl*08:02, HLA-DRB 1*09:01, HLA-DRBl*10:01, HLA-DRB1*11:O1, HLA- DRB1*11:O4, HLA-DRB1*13:O1, HLA-DRB 1*13:02, HLA-DRB1*14:O1, HLA- DRB1*15:O1, HLA-DRB 1*15:02, HLA-DRB 1*15:03, HLA-DQAl*01:01, HLA- DQB1*O5:O1, HLA-DQA1 *01:02, HLA-DQBl*06:02 , HLA-DQAl*03:01, HLA- DQBl*03:02, HLA-DQAl*05:01, HLA-DQB1 *02:01, HLA-DQBl*03:01, HLA- DQB1*O3:O3, HLA-DQB 1*04:02, HLA-DQBl*05:03, HLA-DQBl*06:03 and HLA- DQB 1 * 06 : 04, HL A-DP Al * 01 : 03, and HL A-DPB 1 * 04: 01.
[0226] The full-length amino acid sequences (including signal sequence and transmembrane domain) of representative HLA-DR, HLA-DQ, and HLA-DP alpha chains are shown in SEQ ID NOs: 12-16, and SEQ ID NO: 143. The amino acid sequences of soluble forms of these MHCII chains (lacking signal sequence and transmembrane domain) are shown in SEQ ID NOs: 42-46, SEQ ID NO: 144, respectively.
[0227] The full-length amino acid sequence (including signal sequence and transmembrane domain) of a representative HLA-DR alpha chain is shown in SEQ ID NO: 12. The amino acid sequence of the soluble form of this MHCII chain (lacking signal sequence and transmembrane domain) is shown in SEQ ID NO: 42.
[0228] The full-length amino acid sequences (including signal sequence and transmembrane domain) of representative HLA-DQ alpha chains are shown in SEQ ID NOs: 13-16. The amino acid sequences of soluble forms of these MHCII chains (lacking signal sequence and transmembrane domain) are shown in SEQ ID NOs: 43-46, respectively.
[0229] The full-length amino acid sequence (including signal sequence and transmembrane domain) of a representative HLA-DP alpha chain is shown in SEQ ID NOs: 143. The amino acid sequence of a soluble form of this MHCII chain (lacking signal sequence and transmembrane domain) is shown in SEQ ID NOs: 144.
[0230] The full-length amino acid sequences (including signal sequence and transmembrane domain) of representative HLA-DR, HLA-DQ, and HLA-DP beta chains are shown in SEQ ID NOs: 17-41, and SEQ ID NOs: 135, 137, 139, 141, and 145. The amino acid sequences of soluble forms of these MHCII chains (lacking signal sequence and transmembrane domain) are shown in SEQ ID NOs: 47-71, and SEQ ID NOs: 136, 138, 140, 142, and 146, respectively.
[0231] The full-length amino acid sequences (including signal sequence and transmembrane domain) of representative HLA-DR beta chains are shown in SEQ ID NOs: 17-31 and SEQ ID NOs: 135, 137, 139 and 141. The amino acid sequences of soluble forms of these MHCII chains (lacking signal sequence and transmembrane domain) are shown in SEQ ID NOs: 47- 61 and SEQ ID NOs: 136, 138, 140, and 142.
[0232] The full-length amino acid sequences (including signal sequence and transmembrane domain) of representative HLA-DQ beta chains are shown in SEQ ID NOs: 32-41. The amino acid sequences of the soluble forms of these MHCII chains (lacking signal sequence and transmembrane domain) are shown in SEQ ID NOs: 62-71, respectively.
[0233] The full-length amino acid sequence (including signal sequence and transmembrane domain) of a representative HLA-DP beta chain is shown in SEQ ID NO: 145. The amino acid sequence of a soluble form of this MHCII chain (lacking signal sequence and transmembrane domain) is shown in SEQ ID NO: 146.
[0234] In certain embodiments, heterodimerization pairs can be appended to the C-teriminal sequence of the alpha and/or beta chains of the MHCII molecule. Non-limiting examples of such heterodimerization pair sequences include Fos and Jun (e.g., having the amino acid sequences shown in SEQ ID NOs: 123 and 124, respectively), acidic and basic leucine zippers (e.g., having the amino acid sequences shown in SEQ ID NOs: 125 and 126, respectively), knob and hole sequences (e.g, having the amino acid sequences shown in SEQ ID NOs: 127 and 128, respectively) for knobs-into-holes technology or spytab and spycatcher sequences (e.g, having the amino acid sequences shown in SEQ ID NOs: 129 and 130, respectively).
[0235] In certain embodiments, an MHCII-binding placeholder peptide is included in the expression construct for one of the MHCII chains, preferably the beta chain, such that the placeholder peptide and a digestible (cleavable) linker are encoded in the construct upstream of (N-terminally) and in operative linkage with the coding sequences for the MHCII chain. For example, the expression construct can encode (from N- to C-terminus): a placeholder peptide, a digestible linker, the MHCII chain (e.g., beta chain) and a C-terminal tag. In certain embodiments, an N-terminal tag is also appended upstream of the placeholder peptide, which allows for removal of non-exchanged peptide species following peptide exchange.
[0236] In some embodiments, an MHCII multimer described herein comprises the al and a2 domains of an MHCII alpha chain and the pi and [32 domains of an MHCII beta chain. In
some embodiments, an MHCII multimer described herein comprises only the al and pi domains of an MHCII heavy chain. In other embodiments, an MHCII multimer comprises an alpha-chain and a beta-chain combined with a peptide. Other embodiments include an MHCII molecule comprised only of alpha-chain and beta-chain (so-called “empty” MHC II without loaded peptide), a truncated alpha-chain (e.g. the al domain) combined with full-length betachain, either empty or loaded with a peptide, a truncated beta-chain (e.g. the pi domain) combined with a full-length alpha-chain, either empty or loaded with a peptide, or a truncated alpha-chain combined with a truncated beta-chain (e.g. al and pi domain), either empty or loaded with a peptide.
[0237] In some embodiments, the MHCII alpha and beta chains comprise a HLA-DR alpha chain paired with a HLA-DR beta chain. In some embodiments, the HLA-DR alpha chain is HLA-DRA*01:01 and the HLA-DR beta chain is for example HLA-DRBl*01:01, HLA- DRBl*01:02, HLA-DRB 1*03:01, HLA-DRBl*04:01, HLA-DRBl*04:03, HLA- DRBl*04:04, HLA-DRB 1*07:01, HLA-DRB 1*08: 01, HLA-DRB 1*08: 02, HLA- DRBl*09:01, HLA-DRBl*10:01, HLA-DRBl*ll:01, HLA-DRB1 *11:04, HLA- DRBl*13:01, HLA-DRB 1*13:02, HLA-DRB1*14:O1, HLA-DRBl*15:01, HLA- DRB1*15:O2, or HLA-DRB 1*15:03.
[0238] In some embodiments, the MHCII alpha and beta chains comprise a HLA-DQ alpha chain paired with a HLA-DQ beta chain. In some embodiments, the HLA-DQ alpha chain is HLA-DQA1 *01:01, and the HLA-DQ beta chain is for example HLA-DQBl*05:01, HLA- DQBl*06:02, HLA-DQB 1*03:02, HLA-DQBl*02:01, HLA-DQBl*03:01, HLA- DQBl*03:03, HLA-DQB 1*04:02, HLA-DQB 1*05: 03, HLA-DQB1 *06:03, or HLA- DQBl*06:04. In some embodiments, the HLA-DQ alpha chain is HLA-DQAl*01:02, and the HLA-DQ beta chain is for example HLA-DQB1 *05:01, HLA-DQB 1*06: 02, HLA- DQBl*03:02, HLA-DQB1 *02:01, HLA-DQBl*03:01, HLA-DQBl*03:03, HLA- DQBl*04:02, HLA-DQB 1*05:03, HLA-DQB1 *06:03, or HLA-DQB 1*06: 04. In some embodiments, the HLA-DQ alpha chain is HLA-DQAl*03:01, and the HLA-DQ beta chain is for example HLA-DQB1 *05:01, HLA-DQB 1*06: 02, HLA-DQB 1*03: 02, HLA- DQBl*02:01, HLA-DQB1 *03:01, HLA-DQBl*03:03, HLA-DQB1 *04:02, HLA- DQBl*05:03, HLA-DQB 1*06: 03, or HLA-DQB 1*06: 04. In some embodiments, the HLA- DQ alpha chain is HLA-DQAl*05:01, and the HLA-DQ beta chain is for example HLA- DQBl*05:01, HLA-DQB 1*06:02, HLA-DQB 1*03: 02, HLA-DQBl*02:01, HLA- DQBl*03:01, HLA-DQB 1*03:03, HLA-DQB1 *04:02, HLA-DQBl*05:03, HLA-
DQBl*06:03 ,or HLA-DQB 1*06:04. In some embodiments, the HLA-DQ alpha chain DQAl*05:01 pairs with the HLA-DQ beta chain DQBl*02:01. In some embodiments, the HLA-DQ alpha chain DQAl*03:01 pairs with the HLA-DQ beta chain DQBl*03:02. In some embodiments, the HLA-DQ alpha chain DQAl*01:02 pairs with the HLA-DQ beta chain DQBl*06:02. In some embodiments, the HLA-DQ alpha chain DQAl*01:02 pairs with the HLA-DQ beta chain DQBl*05:01.
[0239] In some embodiments, the MHCII alpha and beta chains comprise a HLA-DP alpha chain paired with a HLA-DP beta chain. In some embodiments the HLA-DP alpha chain is HLA-DPAl*01:03, and the HLA-DP beta chain is HLA-DPB1 *04:01.
[0240] In some embodiments, the MHCII alpha and beta chains comprise the amino acid sequences set forth in SEQ ID NOs: 42 and 47; 42 and 48; 42 and 49; 42 and 50; 42 and 51; 42 and 52; 42 and 53; 42 and 54; 42 and 55; 42 and 56; 42 and 57; 42 and 58; 42 and 59; 42 and 60; 42 and 61; 44 and 62; 44 and 63; 45 and 64; 46 and 65; or 144 and 146, respectively.
[0241] In some embodiments, the MHCII molecule comprises a soluble MHCII polypeptide. In some embodiments the MHCII molecule comprises a soluble MHCII lacking transmembrane and intracellular domains.
[0242] The amino acid sequences of numerous MHC Class II proteins, including human MHCII, are known in the art, and the genes have been cloned. Therefore, the alpha and beta chain monomers can be expressed using recombinant methods. Methods for the expression and purification of MHCII molecules have been extensively described (e.g, Crawford et al. (1998) IMMUNITY 8:675-682; Novak etal. (1999) J. CLIN. INVEST. 104:R63-R67; Nepom et al. (2002) ARTHRIT. RHEUM. 46:5-12; Day et al. (2003) J. CLIN. INVEST. 112:831-842;
Vollers and Stem (2008) IMMUNOL. 123:305-313; Cecconi etal. (2008) CYTOMETRY 73A: 1010-1018, the entire contents of each of which is hereby incorporated by reference).
[0243] For MHC II molecules the alpha-chain and beta-chain may be expressed in separate cells as individual polypeptides or in the same cell as a fusion protein. The peptide of the MHC Il-peptide complex may be produced separately and added following purification of whole MHC complexes or added during in vitro refolding or expressed together with alphachain and/or beta-chain connected to either chain through a linker. The genetic material can encode all or only a fragment of MHC class II alpha- and beta-chains. The genetic material may be fused with genes encoding other proteins, including proteins useful in purification of the expressed polypeptide chains (c.g. purification tags), proteins useful in
increasing/decreasing solubility of the polypeptide(s), proteins useful in detection of polypeptide(s), proteins involved in coupling of MHC complex to multimerization domains and/or coupling of labels to MHC complex and/or MHC multimer.
[0244] In contrast to MHC I complexes, MHC II complexes are not easily refolded after denaturation in vitro. Only some MHC II alleles can be expressed in E. coli and refolded in vitro. Therefore, preferred expression systems for production of MHC II molecules are eukaryotic systems where refolding after expression of protein is not necessary. Preferred expression systems include mammalian expression systems, such as CHO cells, HEK cells or other mammalian cell lines suitable for expression of human proteins. Other expression systems include stable Drosophila cell transfectants, baculovirus infected insect-cells or other mammalian cell lines suitable for expression of proteins.
[0245] In some embodiments, the MHC II complexes are “empty,” i.e., without intentional loading of peptide into the class II antigen-binding groove. Methods for production of empty MHC II complexes have been described, such as expression in insect cells (see e.g., Novak et al. (1999) J. CLIN. INVEST. 104:63-67; Nepom etal. (2002) ARTHRIT. RHEUMAT. 46:5-12; and Moro et al. (2005) BMC IMMUNOLOGY 6:24). In this strategy, soluble domains of the alpha and beta subunits of MHC II complexes are co-expressed and secreted from insect cells infected or stably transformed with baculovirus. Since the insect cells lack the mammalian loading machinery for MHC II complexes, the complexes are essentially empty after assembly. The overexpressed complexes can then be purified from the cell culture medium. In some embodiments, the alpha-chain and the beta-chain may be each fused with a dimerization domain such as a leucine zipper, IgG constant region, or an enzymatic biotinylation site e.g., Bir) and dimerized after expression and purification. Such empty MHC II complexes can then be loaded with the desired peptides.
[0246] In some embodiments, MHCII monomers are prepared in which a placeholder peptide is covalently linked to the MHCII molecule. For example, one approach is the covalent synthesis of single-chain MHC class II chain-peptide complexes, directed by engineering peptide-specific complementary DNA (cDNA) sequences proximal to the beta-chain cDNA (as described in Crawford et al. (1999) IMMUNITY 8:675-682). In this strategy, the resulting polypeptide refolds with the peptide sequence extended from the amino terminus of the class II molecule. A tethering linker sequence in the peptide allows enough flexibility for the peptide to occupy the peptide binding groove in the mature class II molecule. A cleavable linker can be used to allow for cleavage of the covalent linkage between the peptide and the
MHCII molecule e.g., as described in Day et al. (2003) J. CLIN. INVEST. 112:831-842), thereby allowing for peptide exchange and loading of the MHCII molecule with other peptides (e.g, a library of different peptides).
[0247] Once expressed, the MHCII complexes can be purified directly as whole MHCII or MHCII-placeholder peptide monomers from MHCII expressing cells. The MHCII monomers may be expressed on the surface of cells, and are then isolated by disruption of the cell membrane using, e.g., detergent followed by purification of the MHCII. In some embodiments, MHC monomers are expressed into the periplasm and expressing cells are lysed and released MHCII monomers purified. Alternatively, MHC monomers may be purified from the supernatant of cells secreting expressed proteins into culture supernatant. Methods for purifying MHCII monomers are well known in the art, for example, via the use of affinity tags together with affinity chromatography, beads coated with ant-tag and/or other techniques involving immobilization of MHCII protein to affinity matrix; size exclusion chromatography using, e.g, gel filtration, ion exchange or other methods able to separate MHC molecules from cells and/or cell lysates.
[0248] In some embodiments, recombinant expression of MHCII polypeptides allow a number of modifications of the MHC monomers. For example, recombinant techniques provide methods for carboxy terminal truncation which deletes the hydrophobic transmembrane domain. The carboxy termini can also be arbitrarily chosen to facilitate the conjugation of ligands or labels, for example, by introducing cysteine and/or lysine residues into the molecule. The synthetic gene will typically include restriction sites to aid insertion into expression vectors and manipulation of the gene sequence. The genes encoding the appropriate monomers are then inserted into expression vectors, expressed in an appropriate host, such as E. coli, yeast, insect, or other suitable cells, and the recombinant proteins are obtained.
[0249] In some embodiments, the MHCII monomers are biotinylated on either their alpha or beta chain. In some embodiments, the MHCII monomers are biotinylated before loading of the peptide either by refolding or peptide exchange. Biotinylation of the MHC monomers can be achieved as known in the art, e.g. by attaching biotin to a specific attachment site which is the recognition site of a biotinylating enzyme. In some embodiments, the biotinylating enzyme is BirA. In some embodiments, biotinylation is carried out on the desired protein chain in vivo as a post translational modification during protein expression.
B. MHC Class II Placeholder Peptides
[0250] In the methods provided herein, the MHCII molecules are prepared with a placeholder peptide to facilitate proper folding of the MHCII monomers to produce placeholder-peptide loaded MHCII (p*MHCII) prior to multimerization. In various embodiments, the placeholder peptide is peptide that binds HLA-DR, HLA-DQ, HLA-DX, HLA-DO, HLA-DZ or HLA-DP. In some embodiments, the placeholder peptide is a synthetic peptide. In some embodiments, the placeholder peptide is tethered to the MHCII molecules using a linker, e.g, a linker containing a cleavage site that can be used to remove the placeholder peptide from the MHCII molecules.
[0251] In various embodiments, the placeholder peptide, or linker thereof, is digestible, thereby allowing release of the peptide from the antigen-binding groove of the MHCII molecule, for example as part of peptide exchange with the multimer compositions of the disclosure. In various embodiments, the placeholder peptide, or linker thereof, is thermolabile, labile at acidic pH, enzymatically cleavable (e.g., using a protease) or photocl eav able (e.g., using UV light) as means for digestion of the placeholder peptide, or linker thereof.
[0252] In one embodiment, the placeholder peptide, or linker thereof, is cleavable by a protease that recognizes a protease cleavage site within the placeholder peptide, or linker thereof. Non-limiting examples of protease cleavage sites include a Factor Xa cleavage site (e.g., having the amino acid sequence shown in SEQ ID NO: 131) and an enterokinase cleavage site (e.g, having the amino acid sequence shown in SEQ ID NO: 132).
[0253] In some embodiments, the affinity of the placeholder peptide for the binding groove of MHCII is lower than the rescue peptide(s). In some embodiments, the affinity of the placeholder peptide for the MHCII binding groove is about 10-fold lower than the rescue peptide(s).
[0254] In some embodiments, the placeholder peptide is thermolabile. In some embodiments, the placeholder peptide is thermolabile at a temperature between about 30- 37°C. In some embodiments, the placeholder peptide is labile at a temperature at or above 30°C, at or above 32°C, at or above 34°C, at or above 35°C, at or above 36°C, or at about 37°C. Thermal labile placeholder peptides and methods of identifying and producing thermal labile placeholder peptides have been described (e.g, WO 93/10220; WO 2005/047902; US
2008/0206789; Luimstra et al. (2019) CURR. PROTOC. IMMUNOL. 126(l):e85; Luimstra et al. (2018) J. EXP. MED. 215(5): 1493-1504).
[0255] In some embodiments the placeholder peptide is labile at an acidic pH. In some embodiments, the placeholder peptide is labile between about pH 2.5 and 6.5. In some embodiments, the placeholder peptide is labile at a pH of about 2.5-6.0, 3.0-6.0, 3.0-6.5, 3.5- 6.0 3.5-6.5, 4.0-6.0, 4.0-6.5, 4.5-6.0, 4.5-6.5, 5.0-6.0, 5.0-6.5, 5.0, 5.5., 6.0 or 6.5. In some embodiments, the placeholder peptide is labile at a basic pH. In some embodiments, the placeholder peptide is labile between about pH 9-11. In some embodiments, the placeholder peptide is labile at or above pH 9, at or above pH 9.5, at or about pH 10, at or about pH 10.5, or at or about pH 11. Methods of generating and using pH sensitive placeholder peptides are publicly available, for example, as described in WO 93/10220; US 2008/0206789; and Cameron etal. (2002) J. IMMUNOL. METH. 268:51-59.
[0256] In some embodiments, the placeholder peptide comprises a cleavable moiety. Various types of cleavable moieties are known in the art and include, for example, moieties that are cleaved by photoirradiation, enzymes, nucleophilic or electrophilic agents, reducing and oxidizing reagents (e.g, reviewed in Leriche et al. (2012) BlORG. MED. CHEM. 20(2):571- 582).
[0257] In one embodiment, the placeholder peptide is fused to a degradation tag and peptide exchange is promoted by proteolysis in the presence of a corresponding protease (the digests the degradation tag) along with the presence of the rescue peptide(s).
[0258] In some embodiments, the cleavable placeholder peptide is a photocleavable peptide, e.g., cleaved upon exposure to UV light. For example, the placeholder peptide can comprise one or more photocleavable photocleavable non-natural amino acids. MHCII-binding photocleavable peptides, e.g, that incorporate the UV-sensitive amino acid analog 3-amino- 3-(2-nitrophenyl)-propionate have been described (see e.g., Negroni and Stem (2018) PLos ONE 13(7):e0199704).
[0259] In one embodiment, the MHCII placeholder peptide is a CLIP peptide, such as having the amino acid sequence KPVSKMRMATPLLMQA (SEQ ID NO: 3) or ATPLLMQALPMGA (SEQ ID NO: 134). In one embodiment, the CLIP peptide, or linker thereof, is cleavable. In one embodiment, the CLIP peptide, or linker thereof, is cleavable by Factor Xa. In one embodiment, the MHCII monomers are synthesized with the cleavable CLIP peptide covalently attached, such as by synthesis of single-chain MHC class II chain-
peptide complexes, directed by engineering peptide-specific complementary DNA (cDNA) sequences proximal to the beta-chain cDNA (see e.g., Day et al. (2003) J. CLIN. INVEST.
112:831-842). Cleavage of the covalent linkage between the CLIP peptide (as the placeholder peptide) and MHCII by Factor Xa (described further in Examples 2 and 4) thus allows for peptide exchange with other MHCII-binding peptides.
[0260] Other MHCII binding peptides have been described in the art that can be used as placeholder peptides, based on appropriate pairing of an MHCII molecule and its known MHCII binding peptide. Non-limiting examples of known MHCII molecule/MHCII binding peptide pairs include: DRAl*0101/DRB 1*0401 and the immunodominant peptide of hemagglutinin, HA307-319 (see Novak et al. (1999) J. CLIN. INVEST. 104:R63-R67) and HLA- DR*1101 and tetanus-toxoid (TT)-derived p2 peptide (TT830-844) having the amino acid sequence QIYKANSKFIGITEL (SEQ ID NO: 120) (see Cecconi et al. (2008) CYTOMETRY 73 A: 1010-1018).
III. Peptide Exchange and MHCII Multimer Library Preparation
[0261] To create an MHCII multimer of the disclosure, the multimer composition (as described in section I above) and the soluble MHCII molecule (as described in section II above) are combined such that peptide exchange occurs, wherein the placeholder peptide bound to the MHCII molecule is released and the MHCII molecule is loaded onto the peptides of the multimer composition. Thus, the peptides of the multimer composition serve as “rescue peptides” that replace the placeholder peptides to create the MHCII multimers. Peptide exchange is described in detail in Example 4 and non-limiting representative schematic diagrams of this approach are shown in FIG. 3, FIG. 4A and FIG. 4B.
[0262] One aspect of peptide exchange involves digestion of the placeholder peptide such that it is removed from the antigen-binding groove of MHCII. In one embodiment, the digestible placholder peptide is thermolabile and digestion is achieved by adjusting the temperature such that the peptide is removed from the antigen-binding groove. In one embodiment, the digestible placholder peptide is labile at acidic pH and digestion is achieved by adjusting the pH such that the peptide is removed from the antigen-binding groove. In one embodiment, the digestible placholder peptide is enzymatically cleavable and digestion is achieved by enzymatic cleavage (e.g., protease cleavage) such that the peptide is removed from the antigen-binding groove. In one embodiment, the digestible placholder peptide is
photocleavable and digestion is achieved by photocleavage (e.g., UV light cleavage) such that the peptide is removed from the antigen-binding groove.
[0263] Another aspect of peptide exchange involves loading the MHCII molecules onto the peptides of the multimer composition, which can be achieved, for example, by combining the MHCII molecules and the multimer composition under acidic pH conditions.
[0264] The multimer compositions and placeholder peptide-loaded soluble MHCII molecules of the disclosure can be used to generate a library of or microarray of MHCII multimers loaded with a diversity of unique peptide epitopes by in situ or in vitro peptide exchange reactions as described herein. In some embodiments, the peptide exchange reactions are performed in multiwell formats and under native conditions. Peptide binding, and thus peptide exchange, can be determined by a number of techniques, such as ELISA or Differential scanning fluorimetry (DSF), which monitors the stability of the MHCII structure, or by biophysical techniques that monitor peptide binding, such as fluorescence polarization.
[0265] In some embodiments, to measure the dissociation efficiency of placeholder peptides or peptide fragments a fluorescently labeled placeholder peptide is used in exchange reactions in the presence of unlabeled exchange peptides. Aliquots of fluorescently labeled p*MHCII multimers are either left untreated or exposed to peptide exchange conditions for different time periods. The amount of remaining p*MHCII-containing the placeholder peptide is monitored by fluorescence analysis to monitor the reduction in p*MHCII complexes.
[0266] In some embodiments, the placeholder peptide has a lower affinity for the MHCII peptide binding groove than the exchanged (rescue) peptide epitope, and peptide exchange comprises contacting the p*MHCII molecule with an excess of (rescue) peptide epitope in a competition assay. In some embodiments, the placeholder peptide has a KD that is about 10- fold lower than the exchanged peptide epitope.
[0267] Peptides that bind to the peptide binding groove of the MHCII molecule can be a naturally occurring peptide but can also be synthetically created using the knowledge of the binding specificity of the binding pocket of the particular MHCII molecule or the supertype family it belongs to. Suitable ligands can be generated using the available 3D structures of MHCII complexes and the knowledge on the binding pocket specificity of the respective MHCII molecules.
[0268] In some embodiments, peptide exchange is induced by elevating the temperature of the mixture to between about 30°-37°C. In some embodiments, the mixture is elevated to 31°, 32°, 33°, 34°, 35°, 36° or 37°
[0269] In some embodiments, peptide exchange is induced by reducing the pH of the mixture to between about pH 2.5-6. In some embodiments, peptide exchange is induced by increasing the pH of the mixture to about pH 9-11.
[0270] In one embodiment, the MHCII placeholder peptide is a CLIP peptide, such as having the amino acid sequence KPVSKMRMATPLLMQA (SEQ ID NO: 3) or ATPLLMQALPMGA (SEQ ID NO: 134).
[0271] In some embodiments, the placeholder peptide further comprises a fluorescent label. In so embodiments, the fluorescent label is attached to a cysteine residue in the placeholder peptide.
[0272] Typically, MHCII peptide exchange is performed in multiwell format for high- throughput screening of peptide ligands as described herein. Peptide exchange can be monitored by a number of techniques such as ELISA or fluorescence polarization, for example, as generally described in Rodenko et al. ((2006) NAT. PROTOCOL. 1 : 1120-1132).
[0273] In another aspect, the disclosure pertains to methods of producing a library of MHC II multimers comprising a diversity of loaded peptide epitopes. Various steps in the preparation of peptide-exchanged, barcoded MHC libraries have been described in the art. These steps use standard methods known in the art for preparing barcoded libraries, including use of single-cell sequencing, use of porous hydrogels, use of single template PCR to generate peptide-encoding amplicons (barcodes) and use of in-drop in vitro transcription/translation (IVTT).
[0274] Libraries of MHCII multimers can be prepared using single template encapsulation methods known in the art. In one embodiment, the single template encapsulation method is drop-based (e.g., using a hydrogel). In one embodiment, the single template encapsulation method is well-based (e.g., using a 96-well plate).
IV. Labeling
[0275] MHCII multimers can be conjugated with a fluorescent label, allowing for identification of T cells that bind the MHCII multimer, for example, via flow cytometry or microscopy. T cells can also be selected based on a fluorescence label through, e.g., fluorescence or magnetic activated cell sorting.
[0276] In some embodiments, one or more detectable labels are conjugated to a linker. According to this invention, a “detectable label” is any molecule or functional group that allows for the detection of a biological or chemical characteristic or change in a system, such as the presence of a target substance in the sample.
[0277] Examples of detectable labels which may be used include fluorophores, chromophores, electro chemiluminescent labels, bioluminescent labels, polymers, polymer particles, bead or other solid surfaces, gold or other metal particles or heavy atoms, spin labels, radioisotopes, enzyme substrates, haptens, antigens, Quantum Dots, aminohexyl, pyrene, nucleic acids or nucleic acid analogs, or proteins ,such as receptors, peptide ligands or substrates, enzymes, and antibodies(including antibody fragments).
[0278] Examples of polymer particles labels which may be used include micro particles, beads, or latex particles of polystyrene, PMMA or silica, which can be embedded with fluorescent dyes, or polymer micelles or capsules which contain dyes, enzymes or substrates. Examples of metal particles which may be used include gold particles and coated gold particles, which can be converted by silver stains. Examples of haptens that may be conjugated in some embodiments are fluorophores, myc, nitrotyrosine, biotin, avidin, streptavidin, 2,4-dinitrophenyl, digoxigenin, bromodeoxy uridine, sulfonate, acetylaminoflurene, mercury trintrophonol, and estradiol.
[0279] Examples of enzymes which may be used comprise horse radish peroxidase (HRP), alkaline phosphatase (AP), beta-galactosidase (GAL), glucose-6-phosphate dehydrogenase, beta-N-acetylglucosaminidase, Pglucuronidase, invertase, Xanthine Oxidase, firefly luciferase and glucose oxidase (GO). Examples of commonly used substrates for horse radish peroxidase (HRP) includes.3'-diaminobenzi dine (DAB), diaminobenzidine with nickel enhancement, 3-amino-9-ethylcarbazole (AEC), Benzidine dihydrochloride (BDHC),Hanker- Yates reagent (HYR), Indophane blue (IB), tetramethylbenzidine(TMB), 4-chloro-l-naphtol (CN), alpha-naphtol pyronin (. alpha. -NP),o-dianisidine (OD), 5-bromo-4-chloro-3- indolylphosphate (BCIP), Nitroblue tetrazolium (NBT), 2-(p-iodophenyl)-3-p-nitrophenyl-5- phenyltetrazolium chloride (INT), tetranitro blue tetrazolium (TNBT),. delta. -bromo -chloro- S-indoxyl-beta-D-galactosideZferro-ferricyanide(BCIGZFF). Examples of commonly used substrates for Alkaline Phosphatase include Naphthol-AS-Bl-phosphateZfast red TR (NABPZFR),Naphthol-AS-MX-phosphateZfast red TR (NAMPZFR),Naphthol-AS-Bl- phosphateZfast red TR (NABPZFR),Naphthol-AS-MX-phosphateZfast red TR (NAMPZFR),Naphthol-AS-Bl-phosphateZnew fuschin (NABPZNF),
bromochloroindolylphosphate/nitroblue tetrazolium (BCIP/NBT), b-Bromo-chloro-S-indolyl- beta-delta-galactopyranoside (BCIG).
[0280] Examples of luminescent labels which may be used include luminol, isoluminol, acridinium esters, 1 ,2-dioxetanes and pyridopyridazines. Examples of electrochemiluminescent labels include ruthenium derivatives. Examples of radioactive labels which may be used include radioactive isotopes of iodide, cobalt, selenium, hydrogen, carbon, sulfur, and phosphorous.
[0281] Some “detectable labels” also include “colour labels,” in which the biological change or event in the system may be assayed by the presence of a colour, or a change in colour. Examples of “colour labels” are chromophores, fluorophores, chemiluminescent compounds, electrochemiluminescent labels, bioluminescent labels, and enzymes that catalyze a colour change in a substrate.
[0282] “Fluorophores” as described herein are molecules that emit detectable electromagnetic radiation upon excitation with electro-magnetic radiation at one or more wavelengths. A large variety of fluorophores are known in the art and are developed by chemists for use as detectable molecular labels and can be conjugated to the MHCII multimers provided herein. Examples include FLUORESCEIN™, or its derivatives, such as FLUORESCEIN®-5-isothiocyanate (FITC), 5-(and6)-carboxyFLUORESCEIN®, 5- or 6- carboxyFLUORESCEIN®,6-(FLUORESCEIN®)-5-(and 6)-carboxamido hexanoic acid, FLUORESCEIN® isothiocyanate, rhodamine or its derivatives such as tetramethyl rhodamine and tetramethylrhodamine-5-(and -6) isothiocyanate (TRITC). Other fluorophores include: coumarin dyes such as (diethyl-amino)coumarin or7-amino-4-methylcoumarin-3- acetic acid, succinimidyl ester (AMCA); sulforhodamine 101 sulfonyl chloride (TexasRed® or TexasRed® sulfonyl chloride; 5-(and-6)-carboxyrhodamine 101, succinimidyl ester, also known as 5-(and-6)-carboxy-X-rhodamine, succinimidyl ester (CXR); lissamine or lissamine derivatives such as lissamine rhodamine B sulfonyl Chloride (LisR); 5-(and-6)- carboxyFLUORESCEIN®, succinimidyl ester(CFI); FLUORESCEIN®5-isothiocyanate (FITC);7-diethylaminocoumarin-3-carboxylic acid, succinimidyl ester (DECCA); 5-(and-6)- carboxytetramethyl-rhodamine, succinimidyl ester (CTMR);7-hydroxycoumarin-3-carboxylic acid, succinimidyl ester (HCCA);6->FLUORESCEIN®.-5-(and-6)-carboxamidolhexanoic acid (FCHA);N-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-3-indacenepropionic acid, succinimidyl ester; also known as 5,7-dimethylBODIPY® propionic acid, succinimidyl ester (DMBP); “activated FLUORESCEIN® derivative” (FAP), available from Probes, Inc.; eosin-
5-isothiocyanate (EITC);erythrosin-5-isothiocyanate (ErlTC); and Cascade® Blue acetylazide(CBAA) (the O-acetylazide derivative ofl -hydroxy-3, 6, 8-pyrene-trisulfonic acid). Yet other potential fluorophores useful in this invention include fluorescent proteins such as green fluorescent protein and its analogs or derivatives, fluorescent amino acids such as tyrosine and tryptophan and their analogs, fluorescent nucleosides, and other fluorescent molecules such as Cy2,Cy3, Cy 3.5, CY5.TM., CY5.TM.5, Cy 7, IR dyes, Dyomics dyes, phycoerythrine, Oregon green 488, pacific blue, rhodamine green, and Alexa dyes. Yet other examples of fluorescent labels include conjugates of R-phycoerythrin orallophycoerythrin, inorganic fluorescent labels such as particles based on semiconductor material like coated CdSe nanocrystallites.
[0283] A number of the fluorophores above, as well as others, are available commercially, from companies such as Molecular Probes, Inc. (Eugene, Oreg.), Pierce Chemical Co. (Rockford, Ill.), or Sigma-Aldrich Co. (St.Louis, Mo.).
[0284] The detectable label can be detected by numerous methods, including, for example, reflectance, transmittance, light scatter, optical rotation, and fluorescence or combinations hereof in the case of optical labels or by film, scintillation counting, or phosphorimaging in the case of radioactive labels. See, e.g., Larsson (1988) IMMUNOCYTOCHEMISTRY: THEORY AND PRACTICE (CRC Press, Boca Raton, Fla.); METHODS IN MOLECULAR BIOLOGY, vol. 80 (1998), John D. Pound (ed.) (Humana Press, Totowa, N.J.). In some embodiments, more than one detectable labels employed.
V. Identifiers and Barcoding
[0285] In certain embodiments, an MHCII multimer of the disclosure comprises an identifier tag or label, such as an oligonucleotide barcode, that facilitates identification of the MHCII multimer. Typically, the identifier tag, e.g., oligonucleotide barcode, is attached to the multimerization domain of the MHCII multimer, such as through a binding moiety on the identifier tag, e.g., oligonucleotide barcode, that binds to a binding site on the multimerization domain. For example, when the multimerization domain is streptavidin or avidin, since the MHCII-binding peptides are conjugated to the multimerization domain at a site other than the biotin-binding site, the MHCII multimer can be labeled with an identifier tag, e.g., oligonucleotide barcode, using a biotinylated form of the identifier tag, e.g., a biotinylated oligonucleotide barcode. Labeling of the MHCII multimer is easily achieved by
incubation of the multimer composition with the biotinylated identifier tag, e.g., biotinylated oligonucleotide barcode.
[0286] Typically, an oligonucleotide barcode is a unique oligonucleotide sequence ranging for 10 to more than 50 nucleotides. The barcode has shared amplification sequences in the 3’ and 5’ ends, and a unique sequence in the middle. This sequence can be revealed by sequencing and can serve as a specific barcode for a given molecule.
[0287] In one embodiment, the nucleic acid component of the barcode (typically DNA) has a special structure. Thus, in one embodiment, the at least one nucleic acid molecule is composed of at least a 5’ first primer region, a central region (barcode region), and a 3’ second primer region. In this way the central region (the barcode region) can be amplified by a primer set. The length of the nucleic acid molecule may also vary. Thus, in other embodiments, the at least one nucleic acid molecule has a length in the range 20-100 nucleotides, such as 30-100, such as 30-80, such as 30-50 nucleotides. In one embodiment, the nucleic acid identifier is from 40 nucleotides to 120 nucleotides in length.
[0288] The coupling of the oligonucleotide barcode to the multimer composition may also vary. Thus, in one embodiment, the at least one oligonucleotide barcode is linked to said multimer composition via a biotin binding domain interacting with streptavidin or avidin within the multimer composition. Other coupling moieties may also be used, depending on the availability of an appropriate binding site with the multimer composition (e.g., within the multimerization domain) and an appropriate corresponding binding domain that can be attached to the oligonucleotide barcodes molecules to facilitate attachment.
[0289] In a further embodiment, the at least oligonucleotide barcode molecule comprises or consists of DNA, RNA, and/or artificial nucleotides such as PLA or LNA. Preferably DNA, but other nucleotides may be included to e.g. increase stability.
[0290] The use of barcode technology is well known in the art, see for example Shiroguchi et al. (2012) PROC. NATL. ACAD. Set. USA 109(4): 1347-52; and Smith et al. (2010) NUCLEIC ACIDS RESEARCH 38(13)1 l:el42. Further methods and compositions for using barcode technology include those described in U.S. 2016/0060621. Use of barcode technology specifically to label MHC multimers also has been described, see for example Bentzen et al. (2016) NATURE BIOTECH. 34:10: 1037-1045; Bentzen and Hadrup (2017) CANCER IMMUNOL. IMMUNOTHERAP. 66:657-666. Standard methods for preparing barcode oligonucleotides, including conjugating them with a suitable binding moiety (e.g, biotinylation) that can bind
the MHC multimer, are known in the art and can be applied to preparing barcode oligonucleotides for labeling the MHC multimers.
[0291] Methods for generating customizable DNA barcode libraries are publicly available. Programs include Generator and nxCode, consisting of 96-587 barcodes, respectively, as well as The DNA Barcodes Package and TagD software (reporting generating libraries consisting of 100,000 barcodes).
[0292] Preparation of a variety of large-scale barcode libraries have been described in the art, which approaches can be used to obtain barcode libraries for labeling pMHC multimer libraries. For example, Xu et al. describe a set of 240,000 unique 25-mer oligonucleotides with sequences that have similar amplifications properties while maintaining maximum diversity of their identification motifs (Xu et al. (2008) PNAS 106:2289-2294). Wang et al. describe construction of barcode sets using particle swarm optimization (Wang et al. (2018) IEEE/ ACM TRANS. COMPUT. BIOL. BIOINFORM. 15:999-1002). Lyons describes generation of large-scale libraries of DNA barcodes of up to one million members. (Lyons (2017) SCI. REPORTS 7:13899).
[0293] In some cases, the unique molecular identifier barcode is encoded by a contiguous sequence of nucleotides tagged to one end of a target nucleic acid. In other cases, the unique molecular identifier (UMI) barcode is encoded by a non-contiguous sequence. Noncontiguous UMIs can have a portion of the barcode at a first end of the target nucleic acid and a portion of the barcode at a second end of the target nucleic acid. In some cases, the UMI is a non-contiguous barcode containing a variable length barcode sequence at a first end and a second identifier sequence at a second end of the target nucleic acid. In some cases, the UMI is a non-contiguous barcode having a variable length barcode sequence at a first end and a second identifier sequence at a second end of the target nucleic acid, wherein the second identifier sequence is determined by a position of a transposase fragmentation event, e.g., a transposase fragmentation site and transposon end insertion event.
[0294] In some cases, the barcode is a “variable length barcode.” As used herein, a variable length barcode is an oligonucleotide that differs from other variable length barcode oligonucleotides in a population, by length, which can be identified by the number of contiguous nucleotides in the barcode. In some cases, additional barcode complexity for the variable length barcode can be provided by the use of variable nucleotide sequence, as described in the paragraphs above, in addition to the variable length.
[0295] In an exemplary embodiment, a variable length barcode can have a length of from 0 to no more than 5 nucleotides. Such a variable length barcode can be denoted by the term “[0- 5].” In such an embodiment, it is understood that a population of target nucleic acids that are attached to such a variable length barcode is expected to include at least one target nucleic acid attached to a variable length barcode that has at least 1 nucleotide (e.g., attached to a variable length barcode having only 1, only 2, only 3, only 4, or only 5 nucleotides). In such an embodiment, it is further understood that a population of target nucleic acids that are attached to such a variable length barcode can include at least one target nucleic acid that contains no variable length barcode (i. e. , a variable length barcode having a length of 0), and/or at least one target nucleic acid that contains a variable length barcode having only 1 nucleotide, and/or at least one target nucleic acid that contains a variable length barcode having only 2 nucleotides, and/or at least one target nucleic acid that contains a variable length barcode having only 3 nucleotides, and/or at least one target nucleic acid that contains a variable length barcode having only 4 nucleotides, and/or and at least one target nucleic acid that contains a variable length barcode having only 5 nucleotides. In such an embodiment, the [0-5] variable length barcode can uniquely identify (differentiate), by itself, 5 different target nucleic acid molecules of the same sequence. Further, in such an embodiment, the [0-5] variable length barcode can uniquely identify (differentiate) 5 different target nucleic molecules of a first sequence, 5 different target nucleic acid molecules of a second sequence, etc. for each different target nucleic acid sequence. Furthermore, barcode labelled MHCII-multimers can be used in combination with single-cell sorting and TCR sequencing, where the specificity of the TCR can be determined by the co-attached barcode. This will enable us to identify TCR specificity for potentially 1000+different antigen responsive T-cells in parallel from the same sample, and match the TCR sequence to the antigen specificity. The future potential of this technology relates to the ability to predict antigen responsiveness based on the TCR sequence.
[0296] The complexity of the barcode labeled MHCII multimer libraries will allow for personalized selection of relevant TCRs in a given individual.
[0297] The barcode is co-attached to the multimer and serves as a specific label for a particular peptide-MHCII complex. In this way at least, for example, 10 or 100 or 1000 or 10,000 or more different peptide-MHCII multimers can be mixed, allow specific interaction with T-cells from blood or other biological specimens, wash-out unbound MHCII-multimers and determine the sequence of the DNA-barcodes. When selecting a cell population of
interest, the sequence of barcodes present above background level, will provide a fingerprint for identification of the antigen responsive cells present in the given cell-population. The number of sequence-reads for each specific barcode will correlate with the frequency of specific T-cells, and the frequency can be estimated by comparing the frequency of reads to the input-frequency of T-cells.
[0298] The DNA-barcode serves as a specific label for the antigen specific T-cells and can be used to determine the specificity of a T-cell after e.g. single-cell sorting, functional analyses or phenotypical assessments. In this way antigen specificity can be linked to both the T-cell receptor sequence (that can be revealed by single-cell sequencing methods) and functional and phenotypical characteristics of the antigen specific cells.
[0299] Barcode labeled MHCII multimer libraries can be used for the quantitative assessment of MHCII multimer binding to a given T-cell clone or TCR transduced/transfected cells.
Since sequencing of the barcode label allow several different labels to be determined simultaneously on the same cell population, this strategy can be used to determine the avidity of a given TCR relative to a library of related peptide-MHCII multimers. The relative contribution of the different DNA-barcode sequences in the final readout is determined based on the quantitative contribution of the TCR binding for each of the different peptide-MHCII multimers in the library. Via titration based analyses it is possible to determine the quantitative binding properties of a TCR in relation to a large library of peptide-MHCII multimers, all merged into a single sample. For this particular purpose the MHCII multimer library may specifically hold related peptide sequences or alanine-substitution peptide libraries.
[0300] In some embodiments, unique identifiers can be used for each sample of a plurality of samples. In some embodiments, identifiers can be shared between two or more samples. In some embodiments, identifiers can comprise some sequences that are shared between all samples, and other sequences that are unique to one sample. In some embodiments, an identifier can comprise a sequence shared between all samples, and a sequence unique to one sample. In some embodiments, a sequence shared between samples can be used for identifier amplification (e.g., PCR amplification with suitable primers). In some embodiments, a sequence unique to one sample or shared between a subset of samples can be used for detection or quantification via qPCR (e.g., sequences for hydrolysis probes, such as TaqMan probes). In some embodiments, a sequence unique to one sample or shared between a subset of samples can be used for detection or quantification via sequencing.
[0301] In some embodiments, an identifier can comprise a unique, in sti/co-generated sequence; each identifier sequence can be assigned to a sample of a plurality of samples and the identifier-sample assignment can be stored in a database. In some embodiments, an identifier can comprise a nucleotide sequence that codes for all or part of a peptide or protein. In some embodiments, an identifier can comprise a nucleotide sequence that codes for an open reading frame. In some embodiments, an identifier can comprise a nucleotide sequence that includes a promoter sequence. In some embodiments, an identifier can comprise a nucleotide sequence that includes a binding site for a DNA-binding protein, e.g. a transcription factor or polymerase enzyme. In some embodiments, an identifier can comprise one or more sequences targeted by a nuclease, e.g. a restriction enzyme. In some embodiments, an identifier can comprise all sequence elements necessary for in vitro transcription and translation of a sequence. In some embodiments, an identifier does not comprise all sequence elements necessary for in vitro transcription and translation of a sequence.
[0302] In some embodiments, an identifier can comprise a biotinylated nucleotide sequence. In some embodiments, an identifier can be biotinylated by PCR amplification with a biotinylated primer(s). In some embodiments, an identifier can be biotinylated by enzymatic incorporation of a biotinylated label, e.g. a biotin dUTP label, by use of KI enow DNA polymerase enzyme, nick translation or mixed primer labeling RNA polymerases, including T7, T3, and SP6 RNA polymerases. In some embodiments, an identifier can be biotinylated by photobiotinylation, e.g. photoactivatable biotin can be added to the sample, and the sample irradiated with UV light.
[0303] In some embodiments, an identifier can be generated from a template polynucleotide, e.g. via PCR amplification of a template DNA. In some embodiments, a template polynucleotide can comprise a nucleotide sequence that codes for an open reading frame. In some embodiments, a template polynucleotide can comprise a nucleotide sequence that includes a promoter sequence. In some embodiments, a template polynucleotide can comprise a nucleotide sequence that includes a binding site for a DNA-binding protein, e.g. a transcription factor or polymerase enzyme. In some embodiments, a template polynucleotide can comprise one or more sequences targeted by a nuclease, e.g. a restriction enzyme. In some embodiments, a template polynucleotide can comprise all sequence elements necessary for in vitro transcription and translation of a sequence. In some embodiments, a template
polynucleotide does not comprise all sequence elements necessary for in vitro transcription and translation of a sequence.
[0304] MHCII multimers with attached identifiers (e.g., oligonucleotide barcodes) can be incubated with a plurality of T cells, followed by sorting of T cells into single-cell compartments. T cells are lysed, and nucleic acids from lysed T cells comprising identifiers are produced. Nucleic acids are pooled and sequenced. Identifiers allow matching of peptide identifiers to T cell sequences from the same compartment. TCR-antigen specificity profiles are determined by identifying a TCR sequence (e.g., variable region, hypervariable region, or CDR) from a compartment, and quantifying peptide identifier reads from the same compartment.
[0305] Multiple TCRs can be identified that exhibit binding affinity for peptides of the peptide library, and multiple peptides can be identified that exhibit binding affinity for specific TCRs.
[0306] Epitope mutations in an antigen of an identified TCR-antigen pair can be identified that result in increased TCR binding affinity.
[0307] Peptides and TCR sequences can be identified that are associated with control of disease associated protein, and can be used to design vaccines and cell therapies.
[0308] For assessing response to therapy, for each peptide identifier sequenced, corresponding TCR sequences are identified. Multiple TCRs are identified that exhibit binding affinity for some peptides of the peptide library, and multiple peptides are identified that exhibit binding affinity for some TCRs. Subjects are followed longitudinally and results of assays are compared to identify peptides and TCR sequences that are associated with successful response to immunotherapy.
VI. Kits
[0309] In another aspect, the disclosure comprises kits for use in the methods described herein. Any of the compositions of the disclosure can be formulated into a kit, including the components and instructs for use of the components for the desired use. For example, in one embodiment, the disclosure provides a kit comprising at least one expression construct encoding a peptide-multimerization domain (a multimer composition of the disclosure). The kit can further comprise at least one expression construct encoding soluble MHCII with a placeholder peptide. The kit can further comprise instructions for expressing the multimer composition(s) and the soluble MHCII molecule(s) and for preparing MHCII multimers by
peptide exchange. In another embodiment, the kit comprises a library of expression constructs (e.g., a library of multimer compositions encoding a plurality of different MHCII- binding peptides).
[0310] In another embodiment, the disclosure provides a kit comprising one or more peptide- multimerization domain compositions (multimer compositions of the disclosure). The kit can further comprise one or more soluble MHCII molecules loaded with a placeholder peptide. The kit can further comprise instructions for preparing MHCII multimers from the multimer composition(s) and the soluble MHCII molecule(s) by peptide exchange. In another embodiment, the kit comprises a library of multimer compositions encoding a plurality of different MHCII-binding peptides.
[0311] In one embodiment, the multimerization domain of the multimer composition is a tetramer. In one embodinment, the tetramer is streptavidin or avidin. In one embodiment, the tetramer further comprises a biotinylated oligonucleotide barcode bound to the biotinbinding site of streptavidin or avidin.
VIL Methods of Use
[0312] Another aspect of the invention relates to methods for detecting antigen responsive T cells, for example in a sample. Generally, the methods comprise providing a plurality of MHC multimers of the disclosure; contacting the MHC multimers with said sample; and detecting binding of the MHC multimers to antigen responsive T cells within the sample, thereby detecting T cells responsive to an antigenic peptide present in the plurality of MHC multimers. In one embodiment, binding is detected by amplifying the barcode region of the oligonucleotide barcode linked to the MHC multimer. Typically, for MHCII multimers, the antigen responsive T cell is a CD4+ T cell, whose TCRs recognize peptide-bound MHC Class II molecules.
[0313] This MHC multimer technology allows for detection of multiple (potentially >1000) different antigen-specific T cells in a single sample. The technology can be used, for example, for T-cell epitope mapping, immune-recognition discovery, diagnostics tests and measuring immune reactivity after vaccination or immune-related therapies. For therapeutic use, the MHC multimers allow for identification and selection of antigen-specific T cells to be administered for therapy, such as for adoptive T cell transfer therapy.
A. Assays
[0314] In one embodiment of the present invention MHC multimers can be used for detection of individual T-cells in fluid samples using flow cytometry or flow cytometry-like analysis.
[0315] Liquid cell samples can be analyzed using a flow cytometer, able to detect and count individual cells passing in a stream through a laser beam. For identification of specific T- cells using MHC multimers, cells are stained with fluorescently labeled MHC multimer by incubating cells with MHC multimer and then forcing the cells with a large volume of liquid through a nozzle creating a stream of spaced cells. Each cell passes through a laser beam and any fluorochrome bound to the cell is excited and thereby fluoresces. Sensitive photomultipliers detect emitted fluorescence, providing information about the amount of MHC multimer bound to the cell. By this method MHC multimers can be used to identify individual T-cells and/or specific T-cell populations in liquid samples.
[0316] Cell samples capable of being analyzed by MHC multimers in flow cytometry analysis include, but is not limited to, blood samples or fractions thereof, T-cell lines (hybridomas, transfected cells) and homogenized tissues like spleen, lymph nodes, tumors, brain or any other tissue comprising T-cells.
[0317] When analyzing blood samples whole blood can be used with or without lysis of red blood cells prior to analysis on flow cytometer. Lysing reagent can be added before or after staining with MHC multimers. When analyzing blood samples without lysis of red blood cells one or more gating reagents may be included to distinguish lymphocytes from red blood cells. Preferred gating reagent are marker molecules specific for surface proteins on red blood cells, enabling subtraction of this cell population from the remaining cells of the sample. As an example, a fluorochrome labelled CD45 specific marker molecule e.g. an antibody can be used to set the trigger discriminator to allow the flow cytometer to distinguish between red blood corpuscles and stained white blood cells.
[0318] Alternative to analysis of whole blood, lymphocytes can be purified before flow cytometry analysis e.g. using standard procedures like a FICOLL®-Hypaque gradient. Another possibility is to isolate T-cells from the blood sample, for example, by adding the sample to antibodies or other T-cell specific markers immobilized on solid support. Marker specific T-cells are then attached to the solid support and following washing specific T-cells
can be eluted. This purified T-cell population can then be used for flow cytometry analysis together with MHCII multimers.
[0319] T-cells may also be purified from other lymphocytes or blood cells by rosetting. Human T-cells form spontaneous rosettes with sheep erythrocytes, also called E-rossette formation. E-rossette formation can be carried out by incubating lymphocytes with sheep red erythrocytes followed by purification over a density gradient e.g. a FICOLL® Hypaque gradient.
[0320] Instead of actively isolating T-cells, unwanted cells like B-cells, NK cells or other cell populations can be removed prior to the analysis. A preferred method for removal of unwanted cells is to incubate the sample with marker molecules specific or one or more surface proteins on the unwanted cells immobilized unto solid support. An example includes use of beads coated with antibodies or other marker molecule specific for surface receptors on the unwanted cells e.g. markers directed against CD19, CD56, CD14, CD15 or others. Briefly beads coated with the specific surface marker(s) are added to the cell sample. Cells different from the wanted T-cells with appropriate surface receptors will bind the beads. Beads are removed by e.g. centrifugation or magnetic withdrawal (when using magnetic beads) and remaining cell are enriched for T-cells.
[0321] Another example is affinity chromatography using columns with material coated with antibodies or other markers specific for the unwanted cells.
[0322] Alternatively, specific antibodies or markers can be added to the blood sample together with complement, thereby killing cells recognized by the antibodies or markers.
[0323] Various gating reagents can be included in the analysis. Gating reagents here means labeled antibodies or other labelled marker molecules identifying subsets of cells by binding to unique surface proteins or intracellular components or intracellular secreted components. Preferred gating reagents when using MHC multimers are antibodies and marker molecules directed against CD2, CD3, CD4, and CD8 identifying major subsets of T-cells. Other preferred gating reagents are antibodies and markers against CDlla, CD14, CD15, CD19, CD25, CD30, CD37, CD49a, CD49e,CD56, CD27, CD28, CD45, CD45RA, CD45RO, CD45RB, CCR7, CCR5, CD62L, CD75, CD94, CD99, CD107b, CD109, CD152, CD153, CD154, CD160, CD161, CD178, CDwl97, CDw217, Cd229, CD245, CD247, Foxp3, or other antibodies or marker molecules recognizing specific proteins unique for different lymphocytes, lymphocyte populations or other cell populations. Also included are antibodies
and markers directed against interleukins e.g. IL-2, IL-4, IL-6, IL-10, IL-12, IL-21; Interferons e.g., INFy, TNFa, TNF[3. or other cytokine or chemokines.
[0324] Gating reagents can be added before, after or simultaneous with addition of MHC multimer to the sample. Following labelling with MHC multimers and before analysis on a flow cytometer stained cells can be treated with a fixation reagent (e.g., formaldehyde, ethanol or methanol) to cross-link bound MHC multimer to the cell surface. Stained cells can also be analyzed directly without fixation.
[0325] The flow cytometer can in one embodiment be equipped to separate and collect particular types of cells. This is called cell sorting. MHC multimers in combination with sorting on a flow cytometer can be used to isolate antigen specific T-cell populations. Gating reagents as described above can be included further specifying the T-cell population to be isolated. Magentic-activated cell sorting (MACS) can also be used to isolate specific T cell populations, wherein T cells are stained with the MHC multimers, followed by capture with magnetic beads coated with an anti-MHC multimer antibody (e.g, anti-Flag or other tag, anti-streptavidin, anti-MHC, etc.). Isolated and collected specific T-cell populations can then be further manipulated as described elsewhere herein, e.g. expanded in vitro.
[0326] Direct determination of the concentration of MHC-peptide specific T-cells in a sample can be obtained by staining blood cells or other cell samples with MHC multimers and relevant gating reagents followed by addition of an exact amount of counting beads of known concentration. In general, the counting beads are microparticles with scatter properties that put them in the context of the cells of interest when registered by a flow cytometer. They can be either labelled with antibodies, fluorochromes or other marker molecules or they may be unlabelled. In some embodiments, the beads are polystyrene beads with molecules embedded in the polymer that are fluorescent in most channels of the flow-cytometer. Inhere the terms “counting bead” and “microparticle” are used interchangeably.
[0327] ] Beads or microparticles suitable for use include those which are used for gel chromatography, for example, gel filtration media such as SEPHADEX®. Suitable microbeads of this sort include, but is not limited to, SEPHADEX® G-10 having a bead size of 40-120 pm (SigmaAldrich catalogue number 27, 103-9), SEPHADEX®. G-15 having a bead size of 40-120 pm (Sigma Aldrich catalogue number 27, 104-7), SEPHADEX®. G-25 having a bead size of 20-50 pm (Sigma Aldrich catalogue number 27, 106-3), SEPHADEX®. G-25 having a bead size of 20-80 pm (Sigma Aldrich catalogue number 27, 107-1),
SEPHADEX®. G-25 having a bead size of 50-150 gm (Sigma Aldrich catalogue number 27, 109-8), SEPHADEX.®. G-25 having a bead size of 100-300 gm (Sigma Aldrich catalogue number 27, 110-1), SEPHADEX® G-50 having a bead size of 20-50 gm (Sigma Aldrich catalogue number 27,112-8), SEPHADEX® G-50 having a bead size of 20-80 gm (Sigma Aldrich catalogue number 27, 113-6), SEPHADEX® G-50 having a bead size of 50-150 gm (Sigma Aldrich catalogue number 27, 114-4), SEPHADEX®G-50 having a bead size of 100- 300 gm (SigmaAldrich catalogue number 27, 115-2), SEPHADEX® G-75 having a bead size of 20-50 gm (Sigma Aldrich catalogue number 27, 116-0), SEPHADEX®G-75 having a bead size of 40-120 gm (Sigma Aldrich catalogue number 27, 117-9), SEPHADEX® G-100 having a bead size of 20-50 gm (SigmaAldrich catalogue number 27, 118-7), SEPHADEX® G-100 having a bead size of 40-120 gm (Sigma Aldrich catalogue number 27, 119- 5),SEPHADEX®G-150 having a bead size of 40-120 gm (Sigma Aldrich catalogue number 27, 121-7), and SEPHADEX® G-200 having a bead size of 40-120 gm (Sigma Aldrich catalogue number 27, 123-3).
[0328] Other preferred particles for use in the methods and compositions described here comprise plastic microbeads. While plastic microbeads are usually solid, they may also be hollow inside and could be vesicles and other microcarriers. They do not have to be perfect spheres in order to function in the methods described here. Plastic materials such as polystyrene, polyacrylamide and other latex materials may be employed for fabricating the beads, but other plastic materials such as polyvinylchloride, polypropylene and the like may also be used.
[0329] The counting beads are used as reference population to measure the exact volume of analyzed sample. The sample(s) are analyzed on a flow cytometer and the amount of MHC- specific T-cell is determined using e.g., a predefined gating strategy and then correlating this number to the number of counted counting beads in the same sample.
[0330] [0346] Detection of specific T-cells in a sample combined with simultaneous detection of activation status of T-cells can also be measured using marker molecules specific for up- or down-regulated surface exposed receptors together with MHC multimers. The marker molecule and MHC multimer can be labelled with the same label or different labelling molecules and added to the sample simultaneously or sequentially or separately.
1. Detection of Individual T-Cells in Fluid Samples Using Microscopy
[0331] Another preferred method for detection of individual T-cells in fluid samples is using microscopy. Microscopy comprises any type of microscopy including optical, electron and scanning probe microscopy, Bright field microscopy, Dark field microscopy, Phase contrast microscopy, Differential interference contrast microscopy, Fluorescence microscopy, Confocal laser scanning microscopy, X-ray microscopy, Transmission electron microscopy, Scanning electron microscopy, atomic force microscope, Scanning tunneling microscope and photonic force microscope. This can be done as follows: A suspension of T-cells are added to MHC multimers, the sample washed and then the amount of MHC multimer bound to each cell is measured. Bound MHC multimers may be labelled directly or measured through addition of labelled marker molecules. The sample is then spread out on a slide or similar in a thin layer able to distinguish individual cells and labelled cells identified using a microscope. Depending on the type of label different types of microscopes may be used, e.g. if fluorescent labels are used a fluorescent microscope is used for the analysis. For example, MHC multimers can be labeled with a flourochrome or bound MHC multimer detected with a fluorescent antibody. Cells with bound fluorescent MHC multimers can then be visualized using e.g. an immunofluorescence microscope or a confocal fluorescence microscope.
2. Immunohistochemistry (IHC)
[0332] IHC is a method where MHC multimers can be used to directly detect specific T-cells e.g. in sections of solid tissue. In some embodiments, sections of fixed or frozen tissue sample are incubated with MHC multimer allowing MHC multimer to bind specific T-cells in the tissue. The MHC multimer may be labelled with a fluorochrome, chromophore, or any other labelling molecule that can be detected. The labeling of the MHC multimer may be directly or through a second marker molecule. As an example, the MHC multimer can be labelled with a tag that can be recognized by e.g. a secondary antibody, optionally labelled with HRP or another label. The bound MHC multimer is then detected by its fluorescence or absorbance (for fluorophore or chromophore), or by addition of an enzyme-labelled antibody directed against this tag, or another component of the MHC multimer (e.g. one of the protein chains, a label on the one or more multimerization domain). The enzyme can e.g. be Horseradish Peroxidase (HRP) or Alkaline Phosphatase (AP), both of which convert a colorless substrate into a colored reaction product in situ. This colored deposit identifies the binding site of the MHC multimer and can be visualized under e.g. alight microscope. The
MHC multimer can also be directly labelled with e.g. HRP or AP, and used in IHC without an additional antibody.
[0333] In some embodiments, the detection of T-cells in solid tissue includes use of tissue embedded in paraffin, from which tissue sections are made and fixed in formalin before staining. Antibodies are standard reagents used for staining of formalin-fixed tissue sections; these antibodies often recognize linear epitopes. In contrast, most MHC multimers are expected to recognize a conformational epitope on the TCR. In this case, the native structure of TCR needs to be at least partly preserved in the fixed tissue.
[0334] In other embodiments, staining performed tissue sections from frozen tissue blocks. In this type of staining fixation is done after MHC multimer staining.
3. Immunofluorescence Microscopy
[0335] In some embodiments, MHC multimers can be used to identify specific T-cells in sections of solid tissue. Instead of visualization of bound MHC multimer by an enzymatic reaction, MHC multimers are labelled with a fluorochrome or bound MHC multimer are detected by a fluorescent antibody. Cells with bound fluorescent MHC multimers can be visualized in an immunofluorescence microscope or in a confocal fluorescence microscope. This method can also be used for detection of T-cells in fluid samples using the principles described for detection of T-cells in fluid sample described elsewhere herein.
4. Detection of T-Cells in Solid Tissue In Vivo
[0336] MHC multimers may also be used for detection of T-cells in solid tissue in vivo. For in vivo detection of T-cells labeled MHC multimers are injected into the body of the individual to be investigated. The MHC multimers may be labeled with e.g. a paramagnetic isotope. Using a magnetic resonance imaging (MRI) scanner or electron spin resonance (ESRjscanner MHC multimer binding T-cells can then be measured and localized. In general, any conventional method for diagnostic imaging visualization can be utilized. Usually gamma and positron emitting radioisotopes are used for camera and paramagnetic isotopes for MRI.
5. Detection of T-Cells Immobilized on Solid Support.
[0337] In a number of applications, it may be advantageous immobilize the T-cell onto a solid or semi-solid support. Such support may be any which is suited for immobilization, separation etc. Non-limiting examples include particles, beads, biodegradable particles,
sheets, gels, filters, membranes (e. g. nylon membranes), fibres, capillaries, needles, microtitre strips, tubes, plates or wells, combs, pipette tips, microarrays, chips, slides, or indeed any solid surface material. The solid or semi-solid support may be labelled, if this is desired. The support may also have scattering properties or sizes, which enable discrimination among supports of the same nature, e.g. particles of different sizes or scattering properties, color or intensities.
[0338] An example of a method where MHC multimers can be used for detection of immobilized T-cells is ELISA (Enzyme-Linked ImmunosorbentAssay). ELISA is a binding assay originally used for detection of antibody-antigen interaction. Detection is based on an enzymatic reaction, and commonly used enzymes are e.g. HRP and AP. MHC multimers can be used in ELISA-based assays for analysis of purified TCR’s and T-cells immobilized in wells of a microtiter plate. The bound MHC multimers can be labelled either by direct chemical coupling of e.g. HRP or AP to the MHC multimer (e.g. the one or more multimerization domain or the MHC proteins), or e.g. by an HRP- or AP-coupled antibody or other marker molecule that binds to the MHC multimer. Detection of the enzyme-label is then by addition of a substrate (e.g. colorless) that is turned into a detectable product (e.g. colored) by the HRP or AP enzyme.
[0339] The solid support may be made of e.g. glass, silica, latex, plastic or any polymeric material. The support may also be made from a biodegradable material. Generally speaking, the nature of the support is not critical and a variety of materials may be used. The surface of support may be hydrophobic or hydrophilic. Non-magnetic polymer beads may also be applicable. Such are available from a wide range of manufactures, e.g. Dynal Particles AS, Qiagen, Amersham Biosciences, Serotec, Seradyne, Merck, Nippon Paint, Chemagen, Promega, Prolabo, Polysciences, Agowa, and Bangs Laboratories.
[0340] Another example of a suitable support is magnetic beads or particles. The term “magnetic” as used everywhere herein is intended to mean that the support is capable of having a magnetic moment imparted to it when placed in a magnetic field, and thus is displaceable under the action of that magnetic field. In other words, a support comprising magnetic beads or particles may readily be removed by magnetic aggregation, which provides a quick, simple and efficient way of separating out the beads or particles from a solution. Magnetic beads and particles may suitably be paramagnetic or superparamagnetic. Superparamagnetic beads and particles are e.g. described in EP 0 106 873. Magnetic beads
and particles are available from several manufacturers, e.g. Dynal Biotech ASA (Oslo, Norway, previously Dynal AS, e.g. DYNABEADS.RTM.).
6. Microchip MHC Multimer Technology
[0341] A microarray of MHC multimers can be formed, by immobilization of different MHC multimers on solid support, to form a spatial array where the position specifies the identity of the MHC-peptide complex or specific empty MHC immobilized at this position. When labelled cells are passed over the microarray (e.g., blood cells), the cells carrying TCRs specific for MHC multimers in the microarray will become immobilized. The label will thus be located at specific regions of the microarray, which will allow identification of the MHC multimers that bind the cells, and thus, allows the identification of e.g. T-cells with recognition specificity for the immobilized MHC multimers. Alternatively, the cells can be labelled after they have been bound to the MHC multimers. The label can be specific for the type of cell that is expected to bind the MHC multimer, or the label can stain cells in general (e.g. a label that binds DNA). Alternatively, cytokine capture antibodies can be co-spotted together with MHC on the solid support and the cytokine secretion from bound antigen specific T-cells analyzed. This is possible because T-cells are stimulated to secrete cytokines when recognizing and binding specific MHC-peptide complexes.
7. Indirect Detection of T-Cell Using pMHC Multimers
[0342] T-cells in a sample may also be detected indirectly using MHC multimers. In indirect detection, the number or activity of T-cells are measured, by detection of events that are the result of TCR-MHC-peptide complex interaction. Interaction between MHC multimer and T- cell may stimulate the T-cell resulting in activation of T-cells, in cell division and proliferation of T-cell populations or alternatively result in inactivation of T-cells. All these mechanisms can be measured using detection methods able to detect these events.
[0343] Example measurement of activation include measurement of secretion of specific soluble factor e.g. cytokine that can be measured using flowcytometry as described in the section with flow cytometry, measurement of expression of activation markers e.g. measurement of expression of CD27 and CD28 and/or other receptors by e.g. flow cytometry and/or ELISA-like methods and measurement of T-cell effector function e.g. CD8 T-cell cytotoxicity that can be measured in cytotoxicity assays like chromium release assay’s know by persons skilled in the art.
[0344] Example measurement of proliferation include but is not limited to measurement of mRNA, measurement of incorporation of thymidine or incorporation of other molecules like bromo-2’-deoxyuridine (BrdU).
[0345] Example measurements of inactivation of T-cells include but is not limited to measurement of effect of blockade of specific TCR and measurement of apoptosis.
[0346] When contacted with a diverse population of T cells, such as is contained in a sample of the peripheral blood lymphocytes (PBLs) of a subject, those tetramers containing pMHCs that are recognized by a T cell in the sample will bind to the matched T cell. Contents of the reaction is analyzed using fluorescence flow cytometry, to determine, quantify and/or isolate those T-cells having an MHC tetramer bound thereto.
B. Screening
[0347] The MHC multimers of the disclosure can be used in a variety of different screening assays. For example, in one embodiment, a library of fluorescently-labeled peptides derived from one or more antigens is applied to MHC multimers comprising a placeholder peptide under conditions to induce release of the placeholder peptide and binding of the antigen- derived peptides. Peptide exchange is monitored by fluorescence polarization assay. The use of placeholder peptides permits the generation of empty, peptide-receptive MHC multimers under physiological conditions. This screening approach can be used to identify peptide ligands that bind to an MHC molecule. Peptide exchange reactions can be performed in multiwell formats and under native conditions. Binding can be determined by a number of techniques, such as ELISA, which monitors the stability of the MHC structure, or by biophysical techniques that monitor peptide binding, such as fluorescence polarization. This screening approach can also be used to scan peptide sets (such as those derived from pathogen genomes, tumor-associated antigens or autoimmune antigens) for MHC ligands.
[0348] The MHC multimers, and libraries thereof, disclosed herein can be used in a number of screening methods that allow for the convenient detection and quantification of antigenspecific binding to immune cell receptors. Such MHC multimer libraries can allow, for example, detection of T cells specific for a given antigen, multiplex detection of T cell specificities in a given sample, matching of TCR sequence with specificity (e.g, via single cell sequencing), comparative TCR affinity determination, determination of a consensus specificity sequence of a given TCR, or mapping of antigen responsiveness of T cells against sequences of interest.
[0349] The resulting MHC multimer libraries may be used in T cell screens to determine antigen-reactive T cells as described, for example, in Simon et al. (2014) CANCER IMMUNOL RES 2(12): 1230-1244.
[0350] In some embodiments, the disclosure provides a method for isolating a TCR- expressing cell-MHC pairs comprises contacting a plurality of TCR-expressing cells with a MHC multimer library as described herein; generating a plurality of compartments, wherein a compartment of the plurality comprises a TCR-expressing cell of the plurality of TCR- expressing cells bound to a MHC of the library, thereby isolating the TCR-expressing cell- MHC pair in the compartment. In some embodiments, the TCR-expressing cell is a T cell, e.g., a CD4+ T cell when using a MHCII multimer library. In some embodiments, a cell can be transfected or transduced to express a TCR. In some embodiments, a non-lymphocyte cell can be transfected or transduced to express TCR.
C. Methods of Identifying
[0351] The MHC multimers of the disclosure can be used to identify antigen-specific T cells of interest, for example by screening a plurality of T cells with a library of MHC multimers. In various embodiments, the library comprises MHC multimers loaded with a diversity of more than 10, more than 100, more than 500, 1000, more than 2,000, more than 5,000, more than 10,000, more than 106, more than 107, more than 108, more than 109, or more than 1010 unique peptides. The identification approach can comprise compartmentalizing a cell of the plurality of cells bound to a MHC multimer of the library in a single compartment, wherein the MHC multimer comprises a unique identifier; and determining the unique identifier for each MHC multimer bound to the compartmentalized cell. A compartment can be a separate space, e.g., a well, a plate, a divided boundary, a phase shift, a vessel, a vesicle, a cell, etc.
[0352] In some embodiments, the compositions and methods disclosed herein can be used to identify a plurality of peptides that bind to a TCR. In some embodiments, the compositions and methods disclosed herein can be used to identify a plurality of TCRs that bind an MHC. In some embodiments, the compositions and methods disclosed herein can be used to identify a plurality of TCRs that bind a plurality of MHCs (for example, a plurality of TCRs that bind to MHC multimers derived from a pathogen library, cancer library, or autoimmune library).
[0353] In some embodiments, the compositions and methods disclosed herein are used for identifying TCR-antigen specificity.
[0354] In some embodiments, the identity of a TCR on a selected T cell is determined by sequencing (e.g., sequencing a variable, hypervariable region or complementarity determining region (CDR) of a TCR). In some embodiments, the identity of the peptide of the MHC bound which binds to a TCR is determined by sequencing (e.g, using an identifier as disclosed herein).
[0355] In one embodiment, MHC multimers of the disclosure can be used for the detection of antigen-specific T cells by flow cytometry or for can be used for T-cell purification. The compositions and methods of the disclosure allow for the production of very large collections of peptide-loaded MHC multimers that are well suited for rapid identification of helper T cell (i.e. , CD4+ T cell) antigens when using MHCII multimers.
[0356] In one embodiment, MHC multimers that are attached to solid surfaces can be used to probe T cell function. The peptide-MHC antigenic complexes fixed to the solid surface can function to stimulate T cell activity through the TCR, thereby allowing for study of downstream T cell functions subsequent to TCR stimulation.
[0357] In some embodiments, the compositions and methods disclosed herein are used to determine how mutations in an identified MHC-binding peptide affect TCR binding. In some embodiments, the compositions and methods disclosed herein are used to identify mutations in an identified MHC-binding peptide that result in enhanced or reduced TCR binding affinity. In some embodiments, the compositions and methods disclosed herein are used to identify mutations in an identified MHC-binding peptide that retain TCR binding affinity. In some embodiments, the compositions and methods disclosed herein are used to identify mutations in an identified MHC-binding peptide that result in loss of TCR binding affinity.
[0358] In some embodiments, the compositions and methods disclosed herein are used to determine how mutations in a TCR identified using the methods described herein alter the binding of a peptide epitope. In some embodiments, the compositions and methods disclosed herein are used to identify mutations in a TCR that result in decreased or increased binding affinity for a peptide epitope. In some embodiments, the compositions and methods disclosed herein can be used to identify mutations in a TCR that retain binding of a peptide epitope. In some embodiments, the compositions and methods disclosed herein can be used to identify mutations in a TCR that result in loss of binding of a peptide epitope.
[0359] In some embodiments, the methods disclosed herein are performed on T cells from a plurality of subjects. In some embodiments, analysis of data from multiple subjects allows
identification of MHC-binding peptide epitopes recognized by multiple subjects. In some embodiments, analysis of data from multiple subjects allows identification of MHC-binding peptide epitopes recognized by multiple TCR clonotypes. In some embodiments, analysis of data from multiple subjects allows identification of MHC-binding peptide epitopes recognized by multiple patients, e.g., multiple cancer patients, multiple patients with an autoimmune condition, or multiple patients with protective immunity against a pathogen. In some embodiments, analysis of data from multiple subjects allows identification of MHC- binding peptide epitopes recognized in subjects comprising different HLA types or alleles. In some embodiments, analysis of data from multiple subjects allows identification of distinct hypervariable or complementarity determining region sequences of TCRs that exhibit convergent antigen binding.
[0360] In some embodiments, the methods disclosed herein are performed using a plurality of libraries. In some embodiments, analysis of data from multiple libraries allows identification of shared reactive MHC-binding peptide epitopes between libraries, e.g., antigens exhibiting TCR affinity that are present in multiple strains of a pathogen, multiple cancer types, multiple cancer patients, multiple autoimmune diseases, or multiple autoimmune conditions. In some embodiments, analysis of data from multiple libraries allows identification of distinct reactive MHC-binding peptide epitopes among libraries, e.g., antigens present in a subset of pathogen strains, cancers, conditions, or patients.
[0361] In some embodiments, T cells identified using an MHC multimer library of the disclosure are subjected to gene expression analysis (e.g., RNA-seq, qPCR). In some embodiments, gene expression analysis is conducted on cells identified as possessing a receptor exhibiting specificity for a peptide in a library of the disclosure. For example, cells determined to express TCRs that bind to an MHC multimer derived from a pathogen library, cancer library, or autoimmune library are subjected to gene expression analysis. Gene expression analysis can be global or targeted. Genes analyzed for expression include, but are not limited to, genes with known functions, genes coding for immune effector molecules (e.g., perforin, granzyme, cytokines, chemokines), immune checkpoint molecules, pro- inflammatory molecules, anti-inflammatory molecules, lineage markers, integrins, selectins, lymphocyte memory markers, death receptors, caspases, cell cycle checkpoint molecules, enzymes, phosphatases, kinases, lipases, and metabolic genes.
[0362] In some embodiments, gene expression analysis can be conducted concurrently with MHC multimer library screening. In some embodiments, gene expression analysis can be
conducted after analysis of MHC multimer library screening results. In some embodiments, gene expression analysis can be conducted before analysis of MHC multimer library screening results. In some embodiments, gene expression analysis allows for immunotyping of cells identified as of interest from MHC-T cell receptor pairings produced using the methods described herein.
[0363] The methods and compositions described herein can be used for screening assays. For example, a library comprising a plurality of pMHC multimers as described herein is contacted with a T cell sample, and one or more T cell functions are determined including, but not limited to, T cell proliferation, T cell cytotoxicity, suppression of T cell proliferation, suppression by a T cell, and cytokine production of a T cell.
[0364] In some embodiments, MHC multimers that can induce the functional property can then be made into a peptide library subset. For example, a library subset can comprise MHC multimers that induce proliferation of a T cell upon binding to TCR, cytotoxicity upon binding to TCR, T cell suppression upon binding to TCR, suppression by a T cell upon binding to TCR, cytokine production upon binding to TCR, or any combination thereof. Proliferation can be determined by, for example, a dye-dilution assay (e.g., CFSE dilution assay), or quantification of DNA replication (e.g., BrdU incorporation assay). Cytotoxicity can be determined by, for example, assays that are based on release of an intracellular enzyme by dead cells (e.g., lactate dehydrogenase), dye exclusion assays (e.g, propidium iodide), or expression of cytolytic markers (e.g., granzyme, CD 107a) by flow cytometry or qPCR. Cytokine production can be determined by, for example, ELISA, multiplex immunoassay, intracellular cytokine staining, ELISPOT, Western Blot, or qPCR. T cell suppression can be determined by, for example, co-incubating a T cell clone with effector cells and target antigen, and measuring proliferation, cytotoxicity, cytokine production, expression of activation markers, etc.
[0365] In some embodiments, the compositions and methods disclosed herein are used to identify antigen-specific T cell effector clones associated with protective immunity, non- protective immunity, or autoimmunity. In some embodiments, compositions and methods disclosed herein are used to identify antigen-specific T cell effector clones that exhibit anergy, exhaustion, tolerogenic properties, autoimmune properties, inflammatory properties, or anti-inflammatory properties (e.g., Tregs). In some embodiments, compositions and methods disclosed herein are used to identify antigen-specific T cell effector clones that exhibit certain effector or memory properties (e.g., naive, terminal effector, effector memory,
central memory, resident memory, Tnl, TH2, TH17, TH9, Tel, Tc2, Tcl7, production of certain cytokines).
[0366] In some embodiments, a TCR identified using compositions and methods disclosed herein are used as part of a therapeutic intervention. For example, a TCR sequence, TCR variable region sequence, or CDR sequence can be transfected or transduced into T cells to generate modified T cells of the same antigenic specificity. The modified T cells can be expanded, polarized to a desired effector phenotype (e.g., Tnl, Tel, Treg), and infused into a subject. In some embodiments, multiple TCRs identified using compositions and methods disclosed herein are used in an oligoclonal therapy.
[0367] In some embodiments, a peptide, ligand, agonist, antagonist, antigen, or epitope identified using methods disclosed herein is used as part of a therapeutic intervention. In some embodiments, a peptide, antigen, or epitope is used to expand a population of cells ex vivo, e.g. using antigen presenting cells, artificial antigen presenting cells, immobilized peptide, or soluble peptide. In some embodiments, expanded cells are infused into a patient. In some embodiments, peripheral blood lymphocytes are expanded. In some embodiments, tumor-infiltrating lymphocytes (TILs) are expanded. In some embodiments, Tnl cells are expanded. In some embodiments, cytotoxic T lymphocytes are expanded. In some embodiments, T regulatory cells are expanded.
[0368] In some embodiments, the compositions and methods disclosed herein are used to identify MHC-binding antigenic peptides for use in development of a vaccine, e.g. a subunit vaccine, a vaccine eliciting coverage against a range of protective antigens, or a universal vaccine.
[0369] In some embodiments, the compositions and methods disclosed herein can be used for diagnosis of a medical condition. In some embodiments, the compositions and methods disclosed herein are used to guide clinical decision making, e.g. treatment selection, identification of prognostic factors, monitoring of treatment response or disease progression, or implementation of preventative measures.
[0370] In some embodiments, the compositions and methods disclosed herein can be used in the selection and/or design of treatments for medical conditions, in particular in the selection of antigen-specific T cells (e.g., CD4+ helper T cells), or TCRs derived therefrom, for use in adoptive transfer T cell therapy. For example, the MHC multimers can be used to identify T cells within a patient sample the react to an antigen(s) of interest, such as a cancer antigen(s)
or pathogen antigen(s) to thereby select those cells for expansion in vitro followed by reintroduction into the patient. Moreover, TCRs identified from such antigen-specific T cells can be sequences and recombinantly introduced into T cells to increase the population of cells expressing TCRs that bind to an antigen(s) of therapeutic interest in a patient.
EXAMPLES
[0371] Below are examples of specific embodiments for carrying out the present invention. The examples are offered for illustrative purposes only and are not intended to limit the scope of the present invention.
[0372] The practice of the present invention will employ, unless otherwise indicated, conventional methods of protein chemistry, biochemistry, recombinant DNA techniques and pharmacology, within the skill of the art. Such techniques are explained fully in the literature. See, e.g., T.E. Creighton, PROTEINS: STRUCTURES AND MOLECULAR PROPERTIES (W.H.
Freeman and Company, 1993); A.L. Lehninger, BIOCHEMISTRY (Worth Publishers, Inc., current addition); Sambrook, et al., MOLECULAR CLONING: A LABORATORY MANUAL (2nd Edition, 1989); METHODS IN ENZYMOLOGY (S. Colowick and N. Kaplan eds., Academic Press, Inc.); REMINGTON’S PHARMACEUTICAL SCIENCES, 18th Edition (Easton, Pennsylvania: Mack Publishing Company, 1990); CAREY AND SUNDBERG ADVANCED ORGANIC CHEMISTRY 3rd Ed. (Plenum Press) Vols A and B (1992).
[0373] Unless otherwise stated, all reagents and chemicals were obtained from commercial sources and used without further purification.
Example 1: Loading MHCII exchangeable monomers on streptavidin-peptide tetramers
[0374] In this example, the approach of preparing MHCII-peptide tetramers by loading placeholder peptide-MHCII exchangeable monomers (p*MHCII) on streptavidin-rescue peptide tetramers is described and illustrated schematically.
[0375] The first step in this “tetramerization by exchange” is to produce highly pure and homogenous recombinant MHCII loaded with a cleavable placeholder peptide, illustrated schematically in FIG. 1. A representative sequence for the MHCII alpha chain is shown in SEQ ID NO: 1. This MHCII alpha chain sequence of SEQ ID NO: 1 (HLA-DRA*01:01) includes a Myc tag and a Sorttag. A representative starting sequence for the MHCII beta chain is shown in SEQ ID NO: 2, which is the full-length HLA-DRB1 *01:01 sequence, including the signal peptide. To prepare the recombinant MHCII loaded with a placeholder
peptide, a placeholder peptide is linked to the N-terminus of the MHCII beta chain via a protease cleavable linker. A representative sequence for a suitable placeholder peptide (the CLIP peptide) is shown in SEQ ID NO: 3. A representative sequence for the resultant peptide-MHCII beta chain construct is shown in SEQ ID NO: 4. The sequence of SEQ ID NO: 4 is the HLA-DRB1 *01:01 beta chain with the CLIP peptide and cleavable linker at the N-terminus and an AviTag and His6 tag at the C-terminus.
[0376] The second piece of the exchange reaction is a variant in which an MHCII restricted peptide is tethered via a linker to streptavidin, referred to as SA-peptide. FIG. 2A schematically illustrates the nucleic acid construct encoding the SA-peptide monomer. FIG. 2B schematically illustrates the SA-peptide tetramer that results from in vitro transcription/translation (IVTT) of the SA-peptide monomer construct, followed by selfassembly of the tetramer. A representative MHCII restricted peptide sequence for use in the SA-peptide construct is shown in SEQ ID NO: 5, which is an analog of a hemagglutinin (HA) peptide from Influenza A virus. Three representative linker sequences for tethering the peptide to SA, referred to as Linker #1, #2 and #3, are shown in SEQ ID NOs: 6, 7 and 8, respectively. Three representative SA-peptide sequences, in which the HA peptide is tethered to the C-terminus of SA via Linker #1, #2 or #3, are shown in SEQ ID NOs: 9, 10 and 11, respectively. These SA-peptide sequences also include a FLAG tag at the C-terminus.
[0377] Peptide exchange between the p*MHCII monomers and the SA-rescue peptide tetramers is illustrated schematically in FIG. 3. Cleavage of the protease-sensitive linker in the p*MHCII molecule allows for release of the placeholder peptide during peptide exchange, p], MHCII denotes cleavage of the linker. Incubating p], MHCII and SA-peptide together under exchange compatible conditions, such as an acidic pH, induces removal of the placeholder peptide and loading of four pj,MHCII molecules instead to generate a pMHC-SA tetramer. Preparation of the p*MHCII and SA-peptide components, followed by peptide exchange, are further illustrated schematically in FIG. 4A, wherein single-template encapsulation can be achieved by either drop-based or well-based methods.
Example 2: Preparation and Cleavage of Placeholder Peptide-Loaded MHCII Monomers
[0378] In this example, preparation and cleavage of the placeholder peptide (CLIP peptide)- loaded MHCII monomer, schematically illustrated in FIG. 1, is described.
[0379] pCDNA3.4 was used to express p*MHCII in ExpiCHO cells (Thermo Fisher Scientific). p*MHCII a and P chains having the amino acid sequences shown in SEQ ID NOs: 1 and 4, respectively, were transcribed from a single open reading frame as two chains separated by a T2A ribosomal skipping site and secreted into the expression media as a stable heterodimer. After harvesting and a first step of FPLC purification by Immobilized metal affinity chromatography, relevant fractions were pooled, concentrated and loaded on HiLoad 26/600 Superdex 200 gel filtration column (Sigma-Aldrich). As shown in Fig. 5A, p*MHCII eluted as a clean homogenous peak at -190 ml elution volume. Relevant fractions were pooled, concentrated and the final yield of the protein production was determined as 100 mg per Liter of ExpiCHO media.
[0380] p*MHCII was then digested by Factor Xa (NEB) at a ratio of 5: 1 (w/w) overnight at 4°C in the presence of 1 mM CaC12. Then the protease was irreversibly inactivated by the addition of 1,5-Dansyl-Glu-Gly-Arg Chloromethyl Ketone inhibitor according to the manufacturer’s recommendations (Sigma- Aldrich). Undigested and digested samples were analyzed by SDS-PAGE, the results of which are shown in FIG. 5B. Digested samples migrated faster than non-digested samples indicating the removal of the freshly cleaved peptide under SDS-PAGE denaturative conditions.
Example 3: Preparation of Streptavidin-Rescue Peptide Tetramers
[0381] In this example, a nucleic acid construct was prepared that encodes an MHC Class II- restricted peptide linked to streptavidin, followed by E. coli expression and purification of the construct (as a proof of principle stand-in for in vitro transcription/translation) to thereby express streptavidin-peptide (SA-peptide) monomers, which then self-assemble to form SA- peptide tetramers. The SA-peptide-encoding nucleic acid construct is illustrated schematically in FIG. 2A. The SA-peptide tetramer is illustrated schematically in FIG. 2B.
[0382] Streptavidin variants were genetically fused to three C-terminal elements in the following order: linker, Influenza hemagglutinin (HA) analog peptide (rescue peptide) and a FLAG tag. Variants with three different types of linkers were expressed in E. coli cells: a flexible linker composed of four repeats of G4S blocks (SEQ ID NO: 6), a Proline-rich rigid linker with the GSAPKPAPKPAPAPKPAPKPAP sequence (SEQ ID NO: 7) and a flexible- rigid linker, in which the flexible region is preceding the rigid sequence (SEQ ID NO: 8). The amino acid sequences of the SA-peptide constructs using each of these linkers are shown
in SEQ ID NOs: 9, 10 and 11, respectively. The three variants were named SAGS'HA, SAPro' and SAGS'Pro_HA, respectively, and are shown schematically in FIG. 6A.
[0383] Upon transformation of E. coli BL21(DE3) with an inducible expression plasmid encoding the SA-peptide constructs, and IPTG-induced expression, cell pellets were harvested and the soluble fraction was purified in a batch procedure using Anti-FLAG Tag G1 Affinity Resin following the manufacturer’s protocol (GenScript, Cat. No. L00432).
Bound proteins were eluted using 500 pg/ml of 3xFLAG competitor peptide (Sigma- Aldrich) and were subjected to SDS-PAGE analysis, the results of which are shown in FIG. 6B.
[0384] In the next step, protein samples were loaded on a HiLoad 26/600 Superdex 200 column (Sigma- Aldrich), pre-washed with 20 mM HEPES and 150 NaCl, pH 7.4. The size exclusion chromatography elution profile is shown in FIG. 6C. Fractions from the relevant peaks of each construct were pooled, concentrated and stored at -80°C.
Example 4: Loading of MHCII with Streptavidin-linked peptides
[0385] In this example, peptide exchange between p( MHCII monomers and SA-peptide tetramers is described, which is schematically illustrated in FIG. 3 and FIG. 4A.
[0386] To first test the feasibility of the described design, protease-cleaved MHCII monomers loaded with the CLIP placeholder peptide (p( MHCII) and SAGS'HA were mixed at a molar ratio of 1:8. The reaction was performed over night at 37°C in the presence of exchange buffer that is composed of: 100 mM sodium citrate pH 5.5, 50 mM sodium Chloride, 0.5 M EDTA, 0.1% octyl glucoside (v/v), 0.05% sodium azide (v/v), lx of SIGMAFAST protease inhibitor cocktail (Sigma-Aldrich) and 0.045% iodoacetamide (v/v). A previous study showed that the loading of high affinity peptide on MHCII renders the complex resistant to SDS-induced denaturation (Natarajan et.al. (1999) J. Immunol. 162, 3463-3470). As shown in FIG. 7, SDS-PAGE analysis of samples from the exchange reaction showed the appearance of a newly formed SDS-resistant species representing a stable pMHCII-SAGS'HA complex. Recombinant p( MHCII. recombinant SAGS'HA or the product of incubation of both proteins under exchange conditions were loaded on the gel as either boiled and reduced samples (+ signs at the bottom of the gel) or non-boiled and non reduced samples (- sign at the bottom of the gel). SAGS'HA(i) and SAGS'HA(4) represent one subunit of SA-peptide fusion or four subunits, respectively. Arrow indicates newly formed complex that is absent from other lanes. An asterisk represent a p*MHCII SDS species that migrates slower than its expected molecular weight. Factor Xaox represent an oxidized
species of the protease consisting of two disulfide-linked chains that fall apart under treatment with reducing buffer.
[0387] To further evaluate complex formation quantitatively, ELISA assays were performed in two different formats, illustrated schematically in FIG. 8A. The first format was performed for all three variants (FIG. 8A, upper panel) and the second format was carried out only for SAGS-I IA (FIG. 8A, lower panel). 150 pl of exchange reactions were prepared in triplicates in a 96-well assay plate, where each well consisted of: lx exchange buffer, 100 ng of pj,MHCII and 5-fold serial dilutions of either free streptavidin, SAGS-I IA. SAGS'Pro'HA and SApro-HA The concentrations range of the first three was 104-l 0'3 nM and 4.5xl03-0.45xl0'2 for the latter. The assay plate was sealed and incubated over-night at 37°C. Meanwhile, ELISA plates were coated with 100 ng/well of either L243 (conformational sensitive for MHCII) or anti-streptavidin antibodies (Abeam) representing the two ELISA formats, respectively. The ELISA plates were sealed and left for over-night incubation at room temperature (L243 plate) or 4C (anti-streptavidin plate). In the next morning, the exchange reaction was stopped by neutralizing the acidic pH with the addition of 1:15 (v/v) of 1 M Tris-HCl, pH 10. Using a 96-channel benchtop pipettor, 100 pl from each well of the assay plate were transferred to the corresponding ELISA plates that were pre-washed (3x PBS-T) and pre-blocked with PBS-T supplemented with 2% (v/v) BSA. Following 1 hr incubation at RT, the plates were washed (3x PBS-T) and incubated with either biotin-HRP (for L243 capture) or L243-HRP (for anti-SA capture) for 45 minutes in the dark, washed again (3x PBS-T) and developed using an HRP substrate and stop solution.
[0388] The results of the ELISA analyses are shown if FIG 8B. In the first ELISA format, a positive correlation between the SApeptide concentrations and the levels of HRP signal was observed for all linker variants but not for free SA (FIG. 8B, upper panel). This result indicates that exchange was specific and was mediated by the loading of the HA fusion peptide into the MHCII binding pocket. The exchange of SAGS'HA was slightly more efficient than the other two constructs as it required lower concentrations of ligand to elicit a response that is halfway between the baseline and the maximum. In the second ELISA format a bellshaped curved was observed in which at high concentrations of SAGS-HA a lower signal was detected (FIG. 8B, lower panel). This phenomenon (Hook effect) suggests an excessive binding of SAGS'HAto the anti-SA capture antibody in a way that occludes the L243-HRP detection antibody from binding to its corresponding epitopes. Importantly, this trend was
not observed for free SA, thereby demonstrating that an MHC-restricted HA peptide is required for an exchange to occur.
[0389] Overall, the results indicated successful peptide exchange between pj,MHCII monomers and SA-peptide tetramers, thus demonstrating the ability to load MHCII molecules with tetramers of Streptavidin linked to rescue peptides.
INCORPORATION BY REFERENCE
[001] Each patent, publication, and non-patent literature cited in the application is hereby incorporated by reference in its entirety as if each was incorporated by reference individually.
SEQUENCE LISTING SUMMARY

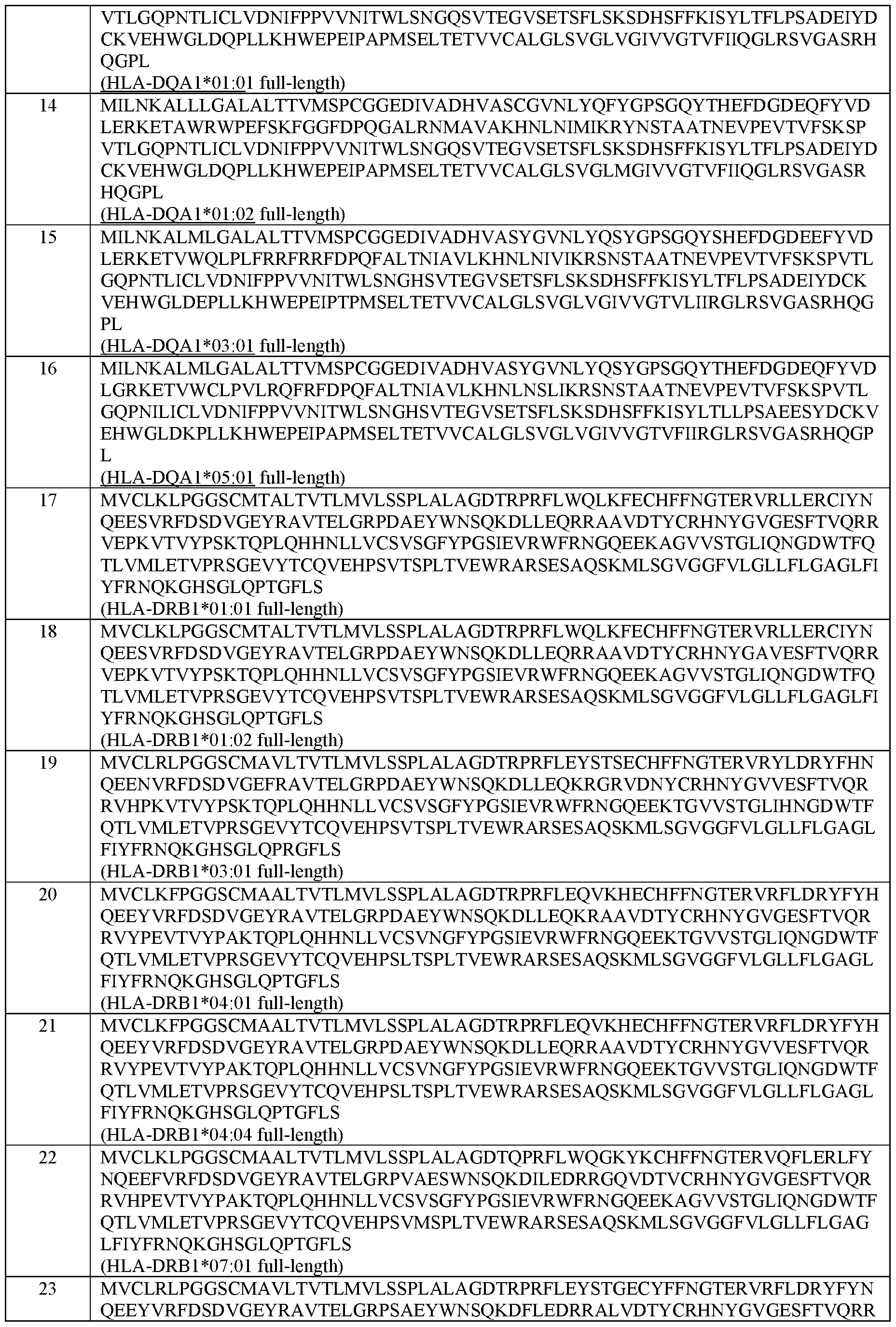
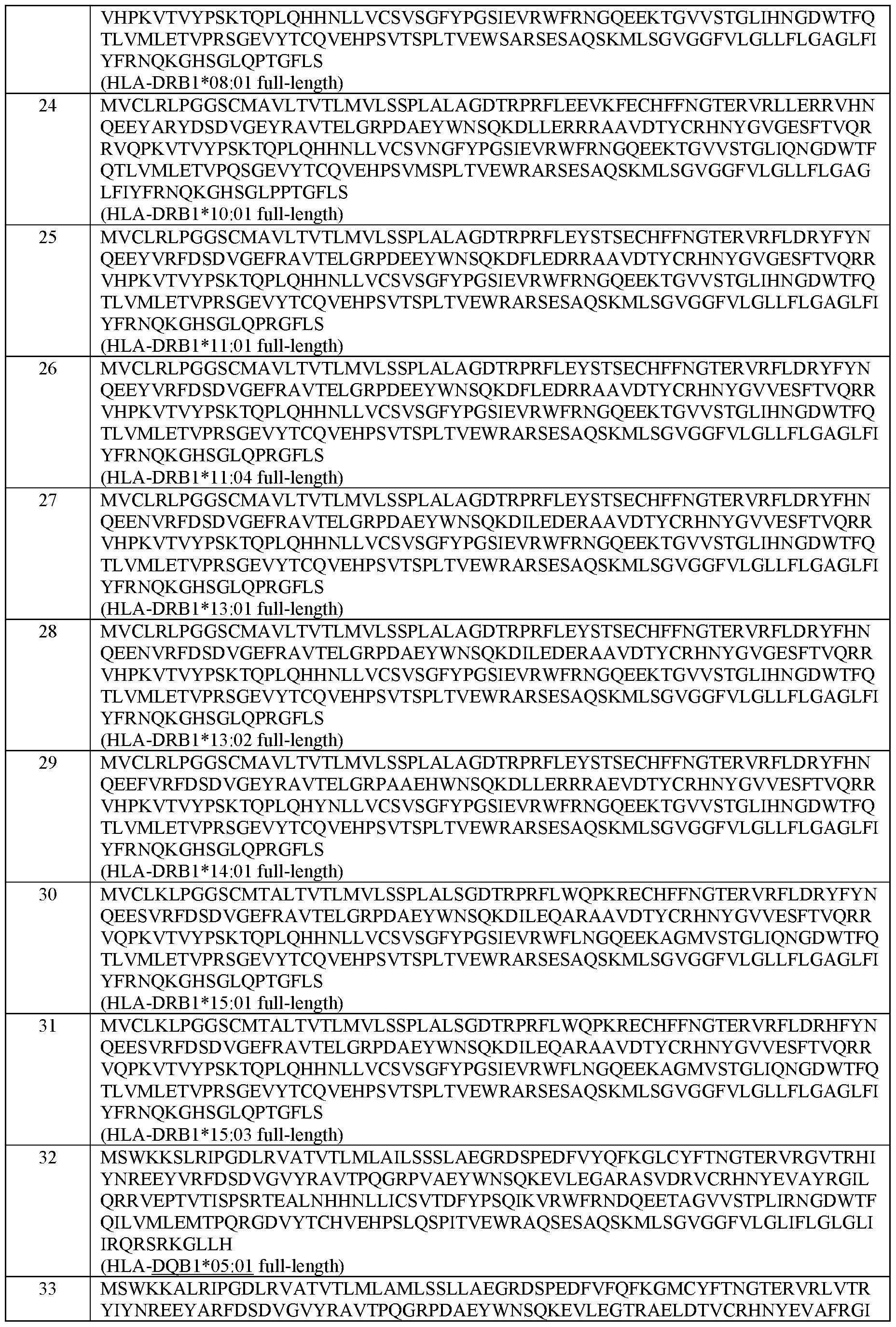
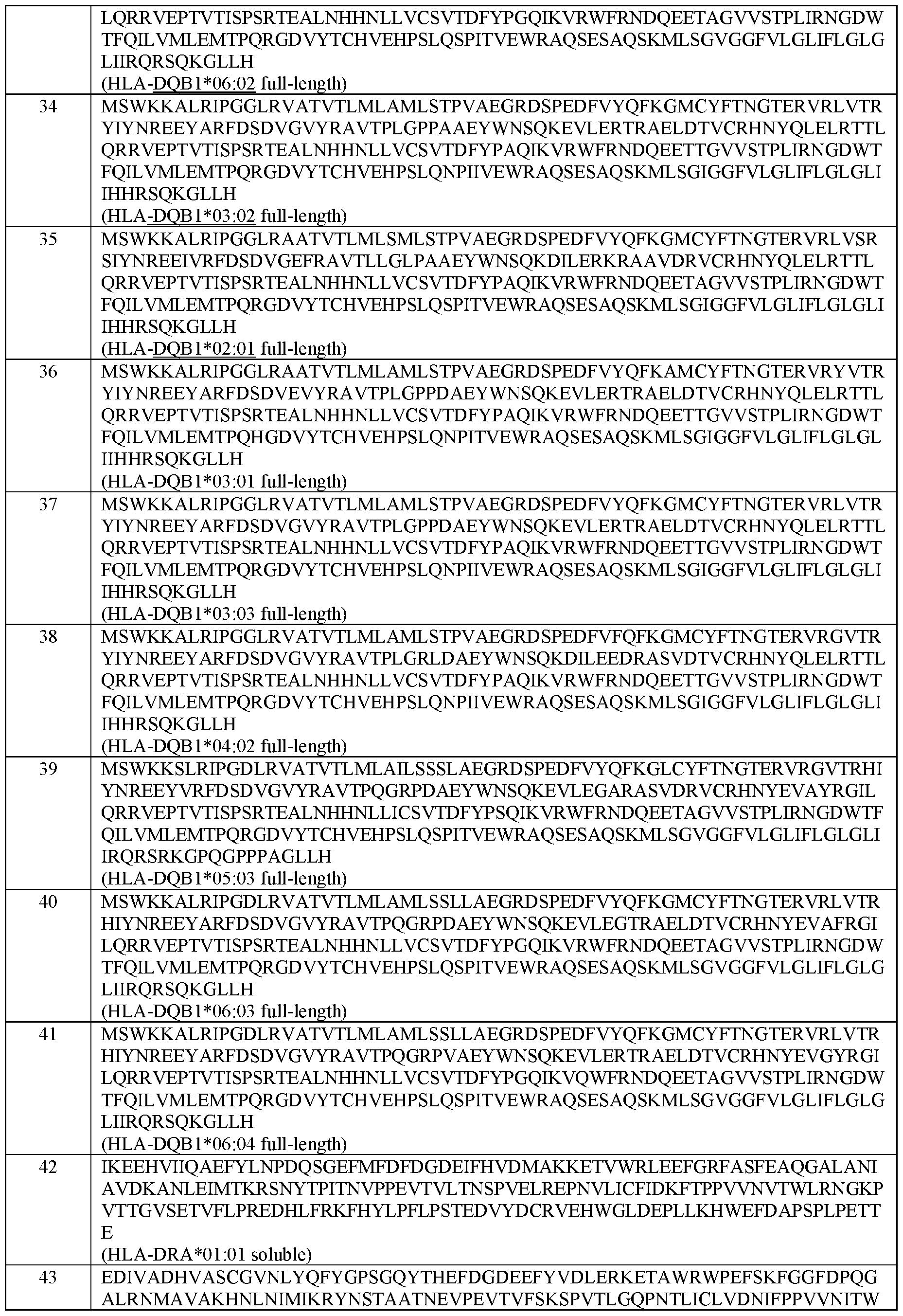

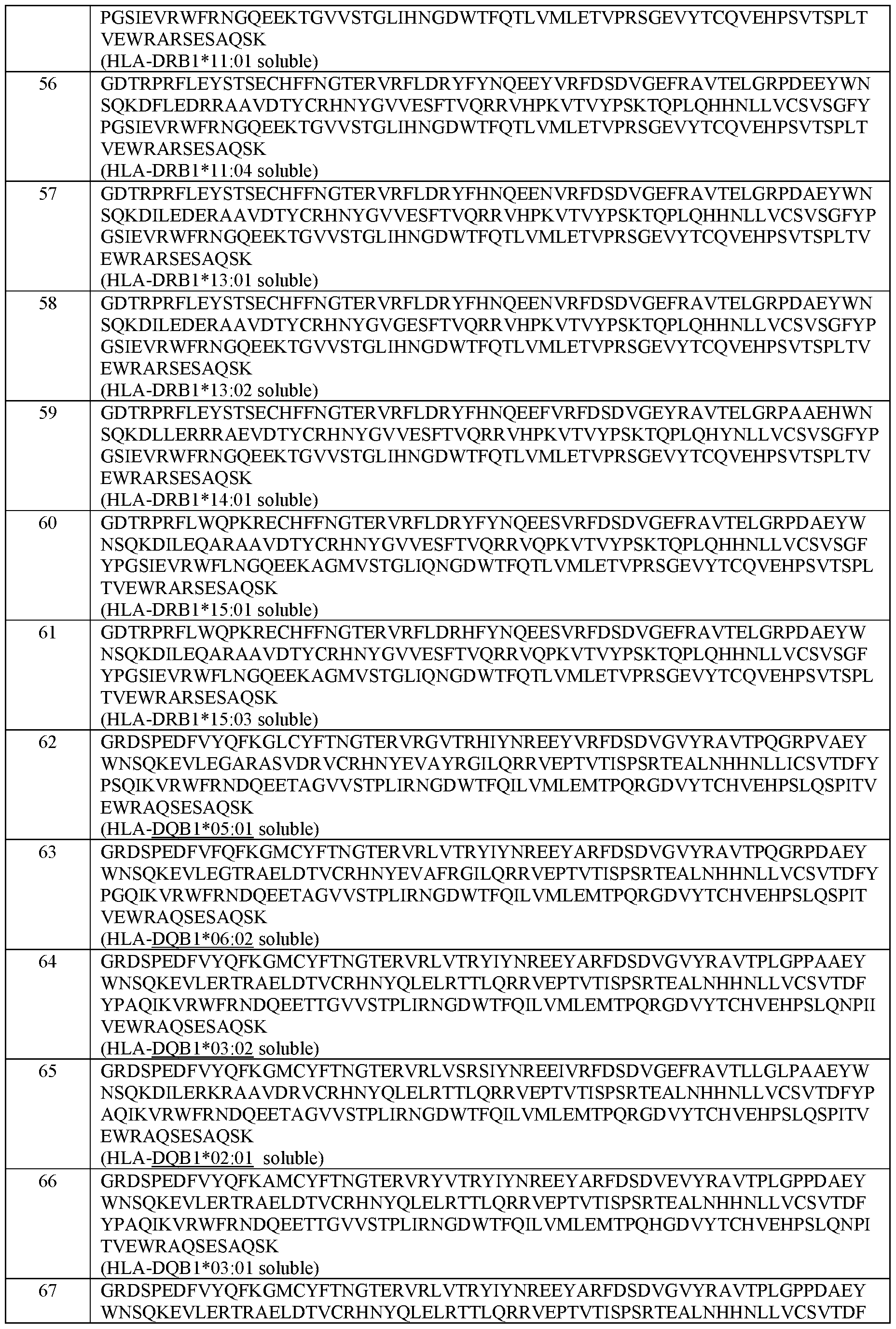




Claims
1. A method of producing a Major Histocompatibility Complex Class II (MHCII) multimer, the method comprising:
(a) providing a multimer composition comprising a plurality of MHCII-binding peptides attached to a multimerization domain, wherein each MHCII-binding peptide within the plurality has the same amino acid sequence;
(b) providing a plurality of soluble MHCII molecules comprising an alpha chain and a beta chain; and
(c) contacting the MHCII molecules with the multimer composition under conditions to produce an MHCII multimer.
2. The method of claim 1 , wherein the multimer composition is a tetramer comprising streptavidin or avidin as the multimerization domain.
3. The method of claim 2, wherein the multimer composition further comprises a biotinylated nucleic acid that binds the multimerization domain, wherein the biotinylated nucleic acid encodes the MHCII-binding peptides.
4. The method of claim 3, wherein the MHCII-binding peptides are produced from the biotinylated nucleic acid by in vitro transcription/translation (IVTT).
5. The method of any one of claims 1-4, wherein the MHCII-binding peptides are attached to the multimerization domain by covalent linkage using a spacer linker.
6. The method of claim 5, wherein the spacer linker comprises an amino acid sequence selected from the group consisting of SEQ ID NOs: 6-8 and 72-80.
7. The method of any one of claims 1-6, wherein each MHCII molecule is loaded with a digestible placeholder peptide, and wherein the MHCII molecules are contacted with the multimer composition under conditions for cleavage of the placeholder peptide, thereby to produce an MHCII multimer by peptide exchange with the multimer composition.
8. The method of claim 7, wherein the digestible placeholder peptide is thermolabile, labile at acidic pH, enzymatically cleavable or photocleavable.
9. The method of claim 8, wherein the digestible placeholder peptide comprises a placeholder peptide linked to the MHCII molecule by an digestible linker.
10. The method of any one of claims 7-9, wherein peptide exchange is achieved by digestion of the placeholder peptide and combining the multimer composition and the MHCII molecules under low pH conditions.
11. The method of any one of claims 1-10, wherein the placeholder peptide is a CLIP peptide comprising the amino acid sequence KPVSKMRMATPLLMQA (SEQ ID NO: 3) or ATPLLMQALPMGA (SEQ ID NO: 134).
12. The method of any one of claims 1-11, wherein the MHC Class II alpha chain comprises an amino acid sequence selected from the group of sequences shown in SEQ ID NOs: 42-46.
13. The method of any one of claims 1-12, wherein the MHC Class II beta chain comprises an amino acid sequence selected from the group of sequences shown in SEQ ID NOs: 47-71.
14. The method of any one of claims 1-13, wherein the MHC Class II alpha chain and the MHC Class II beta chain comprise the amino acid sequences set forth in SEQ ID NOs: 42 and 47; 42 and 48; 42 and 49; 42 and 50; 42 and 51; 42 and 52; 42 and 53; 42 and 54; 42 and 55; 42 and 56; 42 and 57; 42 and 58; 42 and 59; 42 and 60; 42 and 61; 44 and 62; 44 and 63; 45 and 64; 46 and 65; or 144 and 146, respectively.
15. The method of any one of claims 1-14, which further comprises labeling the multimer composition with an oligonucleotide barcode.
16. The method of any one of claims 1-15, wherein step (a) provides multimers comprising a plurality of MHCII-binding peptides, thereby to produce a library of MHCII multimers.
17. A multimer composition comprising a plurality of MHCII-binding peptides attached to a multimerization domain, wherein each MHCII-binding peptide within the plurality has the
same amino acid sequence, wherein the multimerization domain is not covalently linked to a MHCII molecule.
18. The multimer composition of claim 17, which is a tetramer comprising streptavidin or avidin as the multimerization domain.
19. The multimer composition of claim 18, further comprising a biotinylated nucleic acid that binds the multimerization domain, wherein the biotinylated nucleic acid encodes the MHCII-binding peptides.
20. The method of claim 19, wherein the MHCII-binding peptides are produced from the biotinylated nucleic acid by IVTT.
21. The multimer composition of any one of claims 17-20, wherein the MHCII-binding peptides are attached to the multimerization domain by covalent linkage using a spacer linker.
22. The multimer composition of claim 21, wherein the spacer linker comprises an amino acid sequence selected from the group consisting of SEQ ID NOs: 6-8 and 72-80.
23. The multimer composition of any one of claims 17-22, which is labeled with an oligonucleotide barcode.
24. The multimer composition of any one of claims 17-23, which further comprises MHCII molecules bound to the MHCII-binding peptides, each MHCII molecule comprising an alpha chain and a beta chain, to thereby create an MHCII multimer.
25. The multimer composition of claim 24, wherein the MHC Class II alpha chain comprises an amino acid sequence selected from the group of sequences shown in SEQ ID NOs: 42-46.
26. The multimer composition of claim 24 or 25, wherein the MHC Class II beta chain comprises an amino acid sequence selected from the group of sequences shown in SEQ ID NOs: 47-71.
111
27. The multimer composition of any one of claims 24-26, wherein the MHC Class II alpha chain and the MHC Class II beta chain comprise the amino acid sequences set forth in SEQ ID NOs: 42 and 47; 42 and 48; 42 and 49; 42 and 50; 42 and 51; 42 and 52; 42 and 53; 42 and 54; 42 and 55; 42 and 56; 42 and 57; 42 and 58; 42 and 59; 42 and 60; 42 and 61; 44 and 62; 44 and 63; 45 and 64; 46 and 65; or 144 and 146, respectively.
28. A library comprising a plurality of the MHCII multimers of any one of claims 24-27.
29. A nucleic acid construct encoding a multimer composition subunit, wherein the nucleic acid construct encodes a polypeptide comprising an MHCII-binding peptide and a multimerization domain, linked by a spacer linker, wherein the polypeptide does not comprise a MHCII molecule.
30. The nucleic acid construct of claim 29, wherein the multimerization domain comprises streptavidin or avidin.
31. The nucleic acid construct of claim 29 or 30, further comprising a biotin moiety.
32. The nucleic acid construct of any one of claims 29-31, wherein the spacer linker comprises an amino acid sequence selected from the group consisting of SEQ ID NOs: 6-8 and 72-80.
33. A method of isolating MHCII-multimer bound lymphocytes comprising:
(a) contacting a plurality of lymphocytes with the library of MHCII multimers of claim 20;
(b) generating a plurality of compartments, wherein each compartment comprises a lymphocyte bound to a MHCII multimer of the library; and
(c) isolating a MHCII-multimer bound lymphocyte from the compartment.
34. The method of claim 33, wherein the lymphocyte is a T cell.
35. The method of claim 33 or claim 34, wherein each member of the library of MHCII multimers is labeled with an oligonucleotide barcode and the method further comprises
112
decoding the oligonucleotide barcode of the isolated MHCII-multimer bound to the lymphocyte.
113
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202063093934P | 2020-10-20 | 2020-10-20 | |
| US63/093,934 | 2020-10-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022087154A1 true WO2022087154A1 (en) | 2022-04-28 |
Family
ID=78695810
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2021/055882 Ceased WO2022087154A1 (en) | 2020-10-20 | 2021-10-20 | Mhc class ii peptide multimers and uses thereof |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2022087154A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2025125528A1 (en) * | 2023-12-14 | 2025-06-19 | Miltenyi Biotec B.V. & Co. KG | Multiplex mhc peptide binding and t cell detection method / quantifiable multiplex mhc binding assay |
Citations (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| USRE30985E (en) | 1978-01-01 | 1982-06-29 | Serum-free cell culture media | |
| EP0106873A1 (en) | 1982-04-23 | 1984-05-02 | Sintef | METHOD FOR PRODUCING MAGNETIC POLYMER PARTICLES. |
| US4560655A (en) | 1982-12-16 | 1985-12-24 | Immunex Corporation | Serum-free cell culture medium and process for making same |
| US4657866A (en) | 1982-12-21 | 1987-04-14 | Sudhir Kumar | Serum-free, synthetic, completely chemically defined tissue culture media |
| US4767704A (en) | 1983-10-07 | 1988-08-30 | Columbia University In The City Of New York | Protein-free culture medium |
| US4927762A (en) | 1986-04-01 | 1990-05-22 | Cell Enterprises, Inc. | Cell culture medium with antioxidant |
| US5122469A (en) | 1990-10-03 | 1992-06-16 | Genentech, Inc. | Method for culturing Chinese hamster ovary cells to improve production of recombinant proteins |
| WO1993010220A1 (en) | 1991-11-19 | 1993-05-27 | Anergen, Inc. | Soluble mhc molecules and their uses |
| WO1994011026A2 (en) | 1992-11-13 | 1994-05-26 | Idec Pharmaceuticals Corporation | Therapeutic application of chimeric and radiolabeled antibodies to human b lymphocyte restricted differentiation antigen for treatment of b cell lymphoma |
| US5506121A (en) | 1992-11-03 | 1996-04-09 | Institut Fur Bioanalytik Gemeinnutzige Gesellschaft MBH | Fusion peptides with binding activity for streptavidin |
| US5525491A (en) | 1991-02-27 | 1996-06-11 | Creative Biomolecules, Inc. | Serine-rich peptide linkers |
| US5631144A (en) | 1989-04-28 | 1997-05-20 | Transgene S.A. | Application of novel DNA fragments as a coding sequence for a signal peptide for the secretion of mature proteins by recombinant yeast, expression cassettes, transformed yeast and corresponding process for the preparation of proteins |
| US5672502A (en) | 1985-06-28 | 1997-09-30 | Celltech Therapeutics Limited | Animal cell culture |
| US6048728A (en) | 1988-09-23 | 2000-04-11 | Chiron Corporation | Cell culture medium for enhanced cell growth, culture longevity, and product expression |
| US6103493A (en) | 1996-10-10 | 2000-08-15 | Institut Fur Bioanalytic | Streptavidin muteins |
| CA2376192A1 (en) * | 1999-06-07 | 2000-12-14 | Neorx Corporation | Streptavidin expressed gene fusions and methods of use thereof |
| WO2005047902A1 (en) | 2003-11-03 | 2005-05-26 | Beckman Coulter, Inc. | Solution-based methods for detecting mhc-binding peptides |
| US20080206789A1 (en) | 2005-09-19 | 2008-08-28 | Proimmune Limited | Method of Screening Mhc Molecules |
| US20100179094A1 (en) | 2008-11-24 | 2010-07-15 | Bristol-Myers Squibb Company | Bispecific egfr/igfir binding molecules |
| US20100210511A1 (en) | 2009-02-11 | 2010-08-19 | Bristol-Myers Squibb Company | Combination VEGFR2 Therapy with Temozolomide |
| US20120094909A1 (en) | 2010-04-13 | 2012-04-19 | Bristol-Myers Squibb Company | Fibronectin based scaffold domain proteins that bind to pcsk9 |
| WO2013000355A1 (en) | 2011-06-27 | 2013-01-03 | 中国葛洲坝集团股份有限公司 | Lightweight slipform for arc-shaped concrete-bottom pipes or channels |
| US20160060621A1 (en) | 2014-06-24 | 2016-03-03 | Bio-Rad Laboratories, Inc. | Digital pcr barcoding |
-
2021
- 2021-10-20 WO PCT/US2021/055882 patent/WO2022087154A1/en not_active Ceased
Patent Citations (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| USRE30985E (en) | 1978-01-01 | 1982-06-29 | Serum-free cell culture media | |
| EP0106873A1 (en) | 1982-04-23 | 1984-05-02 | Sintef | METHOD FOR PRODUCING MAGNETIC POLYMER PARTICLES. |
| US4560655A (en) | 1982-12-16 | 1985-12-24 | Immunex Corporation | Serum-free cell culture medium and process for making same |
| US4657866A (en) | 1982-12-21 | 1987-04-14 | Sudhir Kumar | Serum-free, synthetic, completely chemically defined tissue culture media |
| US4767704A (en) | 1983-10-07 | 1988-08-30 | Columbia University In The City Of New York | Protein-free culture medium |
| US5672502A (en) | 1985-06-28 | 1997-09-30 | Celltech Therapeutics Limited | Animal cell culture |
| US4927762A (en) | 1986-04-01 | 1990-05-22 | Cell Enterprises, Inc. | Cell culture medium with antioxidant |
| US6048728A (en) | 1988-09-23 | 2000-04-11 | Chiron Corporation | Cell culture medium for enhanced cell growth, culture longevity, and product expression |
| US5631144A (en) | 1989-04-28 | 1997-05-20 | Transgene S.A. | Application of novel DNA fragments as a coding sequence for a signal peptide for the secretion of mature proteins by recombinant yeast, expression cassettes, transformed yeast and corresponding process for the preparation of proteins |
| US5122469A (en) | 1990-10-03 | 1992-06-16 | Genentech, Inc. | Method for culturing Chinese hamster ovary cells to improve production of recombinant proteins |
| US5525491A (en) | 1991-02-27 | 1996-06-11 | Creative Biomolecules, Inc. | Serine-rich peptide linkers |
| WO1993010220A1 (en) | 1991-11-19 | 1993-05-27 | Anergen, Inc. | Soluble mhc molecules and their uses |
| US5506121A (en) | 1992-11-03 | 1996-04-09 | Institut Fur Bioanalytik Gemeinnutzige Gesellschaft MBH | Fusion peptides with binding activity for streptavidin |
| WO1994011026A2 (en) | 1992-11-13 | 1994-05-26 | Idec Pharmaceuticals Corporation | Therapeutic application of chimeric and radiolabeled antibodies to human b lymphocyte restricted differentiation antigen for treatment of b cell lymphoma |
| US6103493A (en) | 1996-10-10 | 2000-08-15 | Institut Fur Bioanalytic | Streptavidin muteins |
| CA2376192A1 (en) * | 1999-06-07 | 2000-12-14 | Neorx Corporation | Streptavidin expressed gene fusions and methods of use thereof |
| WO2005047902A1 (en) | 2003-11-03 | 2005-05-26 | Beckman Coulter, Inc. | Solution-based methods for detecting mhc-binding peptides |
| US20080206789A1 (en) | 2005-09-19 | 2008-08-28 | Proimmune Limited | Method of Screening Mhc Molecules |
| US20100179094A1 (en) | 2008-11-24 | 2010-07-15 | Bristol-Myers Squibb Company | Bispecific egfr/igfir binding molecules |
| US20100210511A1 (en) | 2009-02-11 | 2010-08-19 | Bristol-Myers Squibb Company | Combination VEGFR2 Therapy with Temozolomide |
| US20120094909A1 (en) | 2010-04-13 | 2012-04-19 | Bristol-Myers Squibb Company | Fibronectin based scaffold domain proteins that bind to pcsk9 |
| WO2013000355A1 (en) | 2011-06-27 | 2013-01-03 | 中国葛洲坝集团股份有限公司 | Lightweight slipform for arc-shaped concrete-bottom pipes or channels |
| US20160060621A1 (en) | 2014-06-24 | 2016-03-03 | Bio-Rad Laboratories, Inc. | Digital pcr barcoding |
Non-Patent Citations (138)
| Title |
|---|
| "CAREY AND SUNDBERG ADVANCED ORGANIC CHEMISTRY", 1992, PLENUM PRESS |
| "CLONING VECTORS: A LABORATORY MANUAL", 1985, ELSEVIER |
| "METHODS IN MOLECULAR BIOLOGY", vol. 80, 1998, HUMANA PRESS |
| "SOLID PHASE PEPTIDE SYNTHESIS", 1984, THE PIERCE CHEMICAL CO. |
| "The MHC sequencing consortium", NATURE, vol. 401, 1999, pages 921 - 923 |
| A.L. LEHNINGER: "REMINGTON'S PHARMACEUTICAL SCIENCES", vol. A, B, 1990, MACK PUBLISHING COMPANY |
| AGARD NJPRESCHER JABERTOZZI CR, J AM CHEM SOC., vol. 126, no. 46, 2004, pages 15046 - 7 |
| AGARWAL PKUDIRKA RALBERS AEBARFIELD RMDE HART GWDRAKE PMJONES LCRABUKA D, BIOCONJUG CHEM, vol. 24, no. 8, 2013, pages 1277 - 94 |
| ALFTHAN ET AL., PROTEIN ENG, vol. 8, 1995, pages 725 - 731 |
| ALTMAN ET AL., SCIENCE, vol. 274, 1996, pages 94 - 96 |
| AMALIE KAI BENTZEN ET AL: "Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA barcodes", NATURE BIOTECHNOLOGY, vol. 34, no. 10, 29 August 2016 (2016-08-29), New York, pages 1037 - 1045, XP055372429, ISSN: 1087-0156, DOI: 10.1038/nbt.3662 * |
| ANTOS JMTRUTTMANN MCPLOEGH HL, CURR OPIN STRUCT BIOL, vol. 38, 2016, pages 111 - 8 |
| ANTOS, CURR. OPIN. STRUCT. BIOL., vol. 38, 2016, pages 111 - 118 |
| APPLEBY JHZHOU KVOLKMANN GLIU XQ, J BIOL CHEM, vol. 284, no. 10, 2009, pages 6194 - 9 |
| ARNOLD H BAKKER ET AL: "MHC multimer technology: current status and future prospects", CURRENT OPINION IN IMMUNOLOGY, vol. 17, no. 4, 1 August 2005 (2005-08-01), GB, pages 428 - 433, XP055545280, ISSN: 0952-7915, DOI: 10.1016/j.coi.2005.06.008 * |
| AUSUBEL, F ET AL.: "CURRENT PROTOCOLS IN MOLECULAR BIOLOGY", 1987, GREEN PUBLISHING AND WILEY-INTERSCIENCE |
| BAKKER ET AL., CURR. OPIN. IMMUNOL., vol. 17, 2005, pages 428 - 433 |
| BARITES ET AL., ANAL. BIOCHEM., vol. 102, 1980, pages 255 |
| BASLE, M CHEM BIOL, vol. 17, no. 3, 2010, pages 213 - 27 |
| BATJARGAL SWALTERS CRPETERSSON EJ, J AM CHEM SOC, vol. 137, no. 5, 2015, pages 1734 - 7 |
| BENTZEN ET AL., NATURE BIOTECH, vol. 34, no. 10, 2016, pages 1037 - 1045 |
| BENTZEN ET AL., NATURE BIOTECH., vol. 34, no. 10, 2016, pages 1037 - 1045 |
| BENTZENHADRUP, CANCER IMMUNOL. IMMUNOTHERAP., vol. 66, 2017, pages 657 - 666 |
| BLACKMAN MLROYZEN MFOX JM, J AMCHEM SOC, vol. 130, no. 41, 2008, pages 13518 - 9 |
| BRONKE ET AL., HUM. IMMUNOL., vol. 66, 2005, pages 950 - 961 |
| BUCHI ET AL., BIOCHEMISTRY, vol. 43, 2004, pages 14852 - 14863 |
| CAMERON ET AL., J. IMMUNOL. METH., vol. 268, 2002, pages 51 - 59 |
| CAMERON ET AL., J. IMMUNOL. METHODS, vol. 268, 2002, pages 51 - 69 |
| CAMERON T O ET AL: "Labeling antigen-specific CD4^+ T cells with class II MHC oligomers", JOURNAL OF IMMUNOLOGICAL METHODS, ELSEVIER SCIENCE PUBLISHERS B.V.,AMSTERDAM, NL, vol. 268, no. 1, 1 October 2002 (2002-10-01), pages 51 - 69, XP004379137, ISSN: 0022-1759, DOI: 10.1016/S0022-1759(02)00200-4 * |
| CARVAJAL-VALLEJOS PPALLISSE RMOOTZ HDSCHMIDT SR, J BIOL CHEM, vol. 287, no. 34, 2012, pages 28686 - 96 |
| CECCONI ET AL., CYTOMETRY, vol. 73A, 2008, pages 1010 - 1018 |
| CHALKER ET AL., CHEM ASIAN J, vol. 4, no. 5, 2009, pages 630 - 40 |
| CONNELL, N.D., CURR. OPIN. BIOTECHNOL., vol. 12, no. 5, 2001, pages 446 - 449 |
| CRAWFORD ET AL., IMMUNITY, vol. 8, 1999, pages 675 - 682 |
| CRAWFORD, IMMUNITY, vol. 8, 1998, pages 675 - 682 |
| CUNLIFFE ET AL., EUR. J. IMMUNOL., vol. 32, 2002, pages 3366 - 3375 |
| DANKE ET AL., J. IMMUNOL., vol. 171, 2003, pages 3163 - 3169 |
| DAY ET AL., J. CLIN. INVEST, vol. 112, 2003, pages 831 - 842 |
| DAY ET AL., J. CLIN. INVEST., vol. 112, no. 6, 2003, pages 831 - 842 |
| DEBELOUCHINA ET AL., Q. REVBIOPHYS., vol. 50, 2017, pages e7 |
| DEVARAJ NKWEISSLEDER RHILDERBRAND SA, BIOCONWG CHEM, vol. 19, no. 12, 2008, pages 2297 - 9 |
| DIRKSEN ADAWSON PE, BIOCONJUG CHEM, vol. 19, no. 12, 2008, pages 2543 - 8 |
| DORR BMHAM HOAN CCHAIKOF ELLIU DR, PROC NATL ACAD SCI USA, vol. 111, no. 37, 2014, pages 13343 - 8 |
| ELIAS ET AL., SMALL, vol. 6, pages 2460 - 2468 |
| EVANS, AUSTRALIAN JOURNAL OF CHEMISTRY, vol. 60, 2007, pages 384 - 395 |
| FREIBURGER LSONNTAG MHENNIG JLI JZOU PSATTLER M, J BIOMOL NMR, vol. 63, no. 1, 2015, pages 1 - 8 |
| GEORGE ET AL., PROTEIN ENGINEERING, vol. 15, 2002, pages 871 - 9 |
| GJELSTRUP, J. IMMUNOL., vol. 188, 2012, pages 1292 - 1306 |
| GLASGOW JESALIT MLCOCHRAN JR, J AM CHEM SOC, vol. 138, no. 24, 2016, pages 7496 - 9 |
| GOLDBERG ET AL., J. CELL. MOL. MED., vol. 15, 2011, pages 1822 - 1832 |
| GREINEDER ET AL., BIOCONJUG. CHEM., vol. 29, 2018, pages 56 - 66 |
| GUAN DRAMIREZ MCHEN Z, BIOTECHNOL BIOENG, vol. 110, no. 9, 2013, pages 2471 - 81 |
| GUIMARAES CPWITTE MDTHEILE CSBOZKURT GKUNDRAT LBLOM AEPLOEGH HL, NAT PROTOC, vol. 8, no. 9, 2013, pages 1800 - 99 |
| HAM ET AL., METH. ENZYMOL., vol. 58, 1979, pages 44 |
| HEJJAOUI ET AL., M PROTEIN SCI, vol. 24, no. 7, 2015, pages 1087 - 99 |
| HU ET AL., CHEM SOC REV, vol. 45, no. 6, 2016, pages 1691 - 719 |
| KOLBFINNSHARPLESS, ANGEWANDTE CHEMIE INTERNATIONAL EDITION, vol. 40, 2001, pages 2004 - 2021 |
| KOTZIN ET AL., PNAS, vol. 97, 2000, pages 11433 - 11438 |
| KRUGER ET AL., BIOL CHEM, vol. 400, no. 3, 2019, pages 289 - 297 |
| KWOK ET AL., J. IMMUNOL., vol. 164, 2000, pages 4244 - 4249 |
| LARSSON: "IMMUNOCYTOCHEMISTRY: THEORY AND PRACTICE", 1988, CRC PRESS |
| LAUGHLIN ET AL., INFECT. IMMUNOL., vol. 75, 2007, pages 1852 - 1860 |
| LEFRANC, DEV. COMP. IMMUNOL., vol. 27, 2003, pages 55 - 77 |
| LEFRANC, DEV. COMP. IMMUNOL., vol. 29, 2005, pages 185 - 203 |
| LEISNER ET AL., PLOS ONE, vol. 3, no. 2, 2008, pages el678 |
| LERICHE ET AL., BIORG. MED. CHEM, vol. 20, no. 2, 2012, pages 571 - 582 |
| LIN, SCIENCE, vol. 355, no. 6325, 2017, pages 597 - 602 |
| LIU FLUO EYFLORA DBMEZO AR, J ORG CHEM, vol. 79, no. 2, 2014, pages 487 - 92 |
| LUCKOW ET AL., BIO/TECHNOLOGY, vol. 6, 1988, pages 47 |
| LUDWIG CPFEIFF MLINNE UMOOTZ HD, ANGEW CHEM INT ED ENGL, vol. 45, no. 31, 2006, pages 5218 - 21 |
| LUIMSTRA ET AL., CURR. PROTOC. IMMUNOL., vol. 126, no. 1, 2019, pages e85 |
| LUIMSTRA ET AL., J. EXP. MED, vol. 215, no. 5, 2018, pages 1493 - 1504 |
| MACAUBUS ET AL., J. IMMUNOL., vol. 176, 2006, pages 5069 - 5077 |
| MAKRIDES ET AL., MICROBIOL. REV., vol. 60, no. 3, 1996, pages 512 - 538 |
| MAO HHART SASCHINK APOLLOK BA, J AM CHEM SOC, vol. 126, no. 9, 2004, pages 2670 - 1 |
| MAO HHART SASCHINK APOLLOK BA, JAM CHEM SOC, vol. 126, no. 9, 2004, pages 2670 - 1 |
| MATSUMOTO TFURUTA KTANAKA TKONDO A, ACS SYNTH BIOL, vol. 5, no. 11, 2016, pages 1284 - 1289 |
| MAYFIELD ET AL., PROC. NATL. ACAD. SCI. USA, vol. 100, no. 2, 2003, pages 438 - 442 |
| MEGEED ET AL., BIOMACROMOLECULES, vol. 7, 2006, pages 999 - 1004 |
| MORO ET AL., BMC IMMUNOL., vol. 6, 2005, pages 24 |
| MORO ET AL., BMC IMMUNOLOGY, vol. 6, 2005, pages 24 |
| NAIR ET AL., IMMUN. INFLAMM. DIS., vol. 1, 2013, pages 3 - 13 |
| NATARAJAN, J. IMMUNOL., vol. 162, 1999, pages 3463 - 3470 |
| NEPOM ET AL., ARTHRIT. RHEUM., vol. 46, 2002, pages 5 - 12 |
| NEPOM ET AL., ARTHRIT. RHEUMAT., vol. 46, 2002, pages 5 - 12 |
| NEWTON ET AL., BIOCHEMISTRY, vol. 35, 1996, pages 545 - 553 |
| NOVAK E J ET AL: "MHC class II tetramers identify peptide specific human CD4+ T cells proliferating in response to influenza antigen", THE JOURNAL OF CLINICAL INVESTIGATION, B M J GROUP, GB, vol. 104, 1 January 1999 (1999-01-01), pages R63 - R67, XP002383143, ISSN: 0021-9738, DOI: 10.1172/JCI8476 * |
| NOVAK ET AL., J. CLIN. INVEST, vol. 104, 1999, pages R63 - R67 |
| NOVAK ET AL., J. CLIN. INVEST., vol. 104, 1999, pages R63 - R67 |
| OVERALL ET AL., NAT. COMMUN., vol. 11, 2020, pages 1909 |
| PERISIC ET AL., STRUCTURE, vol. 12, 1994, pages 1217 - 1226 |
| PETERS ET AL., PLOS BIOL, vol. 3, 2005, pages e91 |
| PIOTUKH ET AL., J. AM. CHEM. SOC., vol. 133, 2011, pages 17536 - 17539 |
| POLICARPO RLKANG HLIAO XRABIDEAU AESIMON MDPENTELUTE BL, ANGEW CHEM INT ED ENGL, vol. 53, no. 35, 2014, pages 1306 - 10 |
| QUARSTEN ET AL., J. IMMUNOL., vol. 167, 2001, pages 4861 - 4868 |
| RABUKA ET AL., NAT PROTOC, vol. 7, no. 6, 2012, pages 1052 - 1067 |
| RAMMENSEE H.-G. ET AL., IMMUNOGENETICS, vol. 41, 1995, pages 178 - 228 |
| RAMMENSEE, IMMUNOGENETICS, vol. 50, 1999, pages 213 - 219 |
| RODENKO ET AL., NAT. PROTOCOL., vol. 1, 2006, pages 1120 - 1132 |
| SABRINA S VOLLERS ET AL: "Class II major histocompatibility complex tetramer staining: progress, problems, and prospects", IMMUNOLOGY, WILEY-BLACKWELL PUBLISHING LTD, GB, vol. 123, no. 3, 30 January 2008 (2008-01-30), pages 305 - 313, XP071275710, ISSN: 0019-2805, DOI: 10.1111/J.1365-2567.2007.02801.X * |
| SAINI ET AL., SCI. IMMUNOL., vol. 4, 2019, pages eaau9039 |
| SAMBROOK ET AL.: "MOLECULAR CLONING: A LABORATORY MANUAL", vol. 1-3, 1989, COLD SPRING HARBOR LABORATORY PRESS |
| SANO, J. BIOL. CHEM., vol. 270, 1995, pages 28204 - 28209 |
| SETTE ET AL., MOL. IMMUNOL., vol. 31, 1994, pages 813 - 822 |
| SHAH ET AL., J. AM. CHEM. SOC., vol. 134, 2012, pages 11338 - 11341 |
| SHAH NHVILA-PERELLO MMUIR TW, ANGEW CHEM INT ED ENGL, vol. 50, no. 29, 2011, pages 6511 - 5 |
| SHAN ET AL., J. IMMUNOL., vol. 162, 1999, pages 6589 - 6595 |
| SHARP ET AL., YEAST, vol. 7, no. 7, 1991, pages 657 - 678 |
| SHIROGUCHI ET AL., PROC. NATL. ACAD. SCI. USA, vol. 109, no. 4, 2012, pages 1347 - 52 |
| SIMON ET AL., CANCER IMMUNOL RES, vol. 2, no. 12, 2014, pages 1230 - 1244 |
| SINCLAIR ET AL., PROTEIN EXPR. PURIF., vol. 26, no. I, 2002, pages 96 - 105 |
| SINISI ET AL., BIOCONJUG. CHEM, vol. 23, 2012, pages 1119 - 1126 |
| SMITH ET AL., NUCLEIC ACIDS RESEARCH, vol. 38, no. 13, 2010, pages e142 |
| SOUTHWORTH MWAMAYA KEVANS TCXU MQPERLER FB, BIOTECHNIQUES, vol. 27, no. 1, 1999, pages 110 - 4,116,118-20 |
| SPICER ET AL., NAT COMMUN, vol. 5, 2015, pages 4740 |
| STEVENS AJBROWN ZZSHAH NHSEKAR GCOWBURN DMUIR TW, JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 138, no. 7, 2016, pages 2162 - 2165 |
| STEVENS ET AL., J. AM. CHEM. SOC., vol. 138, 2016, pages 2162 - 2165 |
| T.E. CREIGHTON: "PROTEINS: STRUCTURES AND MOLECULAR PROPERTIES", 1993, W.H. FREEMAN AND COMPANY |
| TORBEEV ET AL., PROC NATL ACAD SCI USA, vol. 110, no. 50, 2013, pages 20051 - 6 |
| TORNOE ET AL., JOURNAL OF ORGANIC CHEMISTRY, vol. 67, 2002, pages 3057 - 3064 |
| UTTAMAPINANT CWHITE KABARUAH HTHOMPSON SFERNANDEZ-SUAREZ MPUTHENVEETIL STING AY, PROC NATL ACAD SCI USA, vol. 107, no. 24, 2010, pages 10914 - 9 |
| VELDMAN ET AL., CLIN. IMMUNOL., vol. 122, 2007, pages 330 - 337 |
| VILA-PERELLO ET AL., J. AM. CHEM. SOC., vol. 135, 2013, pages 286 - 292 |
| VOLLERSSTERN, IMMUNOL, vol. 123, 2008, pages 305 - 313 |
| VOLLERSSTERN, IMMUNOLOGY, vol. 123, 2008, pages 305 - 313 |
| VORUP-JENSEN, ADV. DRUG. DELIV. REV., vol. 64, 2012, pages 1759 - 1781 |
| WANG ET AL., IEEE/ACM TRANS. COMPUT. BIOL. BIOINFORM., vol. 15, 2018, pages 999 - 1002 |
| WILLIAMS FPMILBRADT AGEMBREY KJBOBBY R, PLOS ONE, vol. 11, no. 4, 2016, pages eOI54607 |
| WILLIAMSON DJWEBB METURNBULL WB, NAT PROTOC, vol. 9, no. 2, 2014, pages 253 - 62 |
| WITTE MDCRAGNOLINI JJDOUGAN SKYODER NCPOPP MWPLOEGH HL, PROC NATL ACAD SCI USA, vol. 109, no. 30, 2012, pages 11993 - 84 |
| WU PSHUI WCARLSON BLHU NRABUKA DLEE JBERTOZZI CR, PROC NATL ACAD SCI USA, vol. 106, no. 9, 2009, pages 3000 - 5 |
| XIAO JTOLBERT TJ, ORG LETT, vol. 11, no. 18, 2009, pages 4144 - 7 |
| XU ET AL., PNAS, vol. 106, 2008, pages 2289 - 2294 |
| YAMAMURA YHIRAKAWA HYAMAGUCHI SNAGAMUNE T, CHEM COMMUN (CAMB, vol. 47, no. 16, 2011, pages 4742 - 4 |
| YANG ET AL., J. CLIN. IMMUNOL., vol. 25, 2005, pages 428 - 436 |
| YANIV, NATURE, vol. 297, 1982, pages 17 - 18 |
| ZHANG ET AL., EUR. J. IMMUNOL., vol. 35, 2005, pages 1066 - 1075 |
| ZHANG ET AL., NATURE BIOTECH., 2018, pages 2018 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2025125528A1 (en) * | 2023-12-14 | 2025-06-19 | Miltenyi Biotec B.V. & Co. KG | Multiplex mhc peptide binding and t cell detection method / quantifiable multiplex mhc binding assay |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11668705B2 (en) | General detection and isolation of specific cells by binding of labeled molecules | |
| US20230265157A1 (en) | Mhc multimer expression constructs and uses thereof | |
| US20200347114A1 (en) | MHC Peptide Complexes and Uses Thereof in Infectious Diseases | |
| US10611818B2 (en) | MHC multimers in tuberculosis diagnostics, vaccine and therapeutics | |
| US20230287394A1 (en) | Barcodable exchangeable peptide-mhc multimer libraries | |
| US20220033460A1 (en) | Identification and use of t cell epitopes in designing diagnostic and therapeutic approaches for covid-19 | |
| JP2018500004A (en) | Method for absolute quantification of naturally processed HLA-restricted cancer peptides | |
| US20220088135A1 (en) | Methods of making and using soluble mhc molecules | |
| US20220090297A1 (en) | Peptide libraries and methods of use thereof | |
| WO2022026921A1 (en) | Identification and use of t cell epitopes in designing diagnostic and therapeutic approaches for covid-19 | |
| WO2022087154A1 (en) | Mhc class ii peptide multimers and uses thereof | |
| US20200347103A1 (en) | Mhc multimers in tuberculosis diagnostics, vaccine and therapeutics | |
| HK40095794A (en) | General detection and isolation of specific cells by binding of labeled molecules | |
| US10968269B1 (en) | MHC multimers in borrelia diagnostics and disease |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 21810803 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 21810803 Country of ref document: EP Kind code of ref document: A1 |