WO2021046426A1 - Cyclic dinucleotide sting agonists tethered to a pd-1 or ctla-4 antibodies - Google Patents
Cyclic dinucleotide sting agonists tethered to a pd-1 or ctla-4 antibodies Download PDFInfo
- Publication number
- WO2021046426A1 WO2021046426A1 PCT/US2020/049513 US2020049513W WO2021046426A1 WO 2021046426 A1 WO2021046426 A1 WO 2021046426A1 US 2020049513 W US2020049513 W US 2020049513W WO 2021046426 A1 WO2021046426 A1 WO 2021046426A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- antibody
- drug conjugate
- alkyl
- aryl
- heteroaryl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC*N(*)C(*(C)*)=O Chemical compound CC*N(*)C(*(C)*)=O 0.000 description 23
- XLHDGJPPMZUJIM-UHFFFAOYSA-N CC(C)=NNC(C)=O Chemical compound CC(C)=NNC(C)=O XLHDGJPPMZUJIM-UHFFFAOYSA-N 0.000 description 1
- XHGVIKOVBUUEFD-UHFFFAOYSA-N CC(C)C(CC(C(C1)SC)=O)C1=O Chemical compound CC(C)C(CC(C(C1)SC)=O)C1=O XHGVIKOVBUUEFD-UHFFFAOYSA-N 0.000 description 1
- TYVCNNQZVKVJAG-CAMSNTMWSA-N CC(C)C(OCc(cc1)cc(C)c1O[C@H]([C@@H]([C@H](C1O)O)O)O[C@@H]1C(O)=O)=O Chemical compound CC(C)C(OCc(cc1)cc(C)c1O[C@H]([C@@H]([C@H](C1O)O)O)O[C@@H]1C(O)=O)=O TYVCNNQZVKVJAG-CAMSNTMWSA-N 0.000 description 1
- VHJYHFFABVQRGJ-UHFFFAOYSA-N CC(CNC(CCCCNC(N)=O)=O)=O Chemical compound CC(CNC(CCCCNC(N)=O)=O)=O VHJYHFFABVQRGJ-UHFFFAOYSA-N 0.000 description 1
- SURZCVYFPAXNGN-UHFFFAOYSA-N CCOC(NC)=O Chemical compound CCOC(NC)=O SURZCVYFPAXNGN-UHFFFAOYSA-N 0.000 description 1
- ZWOJWTRMUGXACV-SSDOTTSWSA-N NC(NCCCCC(N[C@@H](CO)C=O)=O)=N Chemical compound NC(NCCCCC(N[C@@H](CO)C=O)=O)=N ZWOJWTRMUGXACV-SSDOTTSWSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/6807—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug or compound being a sugar, nucleoside, nucleotide, nucleic acid, e.g. RNA antisense
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6849—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a receptor, a cell surface antigen or a cell surface determinant
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6889—Conjugates wherein the antibody being the modifying agent and wherein the linker, binder or spacer confers particular properties to the conjugates, e.g. peptidic enzyme-labile linkers or acid-labile linkers, providing for an acid-labile immuno conjugate wherein the drug may be released from its antibody conjugated part in an acidic, e.g. tumoural or environment
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/04—Linear peptides containing only normal peptide links
- C07K7/06—Linear peptides containing only normal peptide links having 5 to 11 amino acids
Definitions
- ADCs Antibody Drug Conjugates
- mAbs monoclonal antibodies
- ADCs By combining the unique targeting capabilities of mAbs with the cancer-killing ability of cytotoxic drugs, ADCs allow sensitive discrimination between healthy and diseased tissue. Accordingly, ADCs represent an important class of biopharmaceutical drugs designed to act as a targeted therapy for the treatment of subjects with various disease states (Ducry, L. Bioconjugate Chemistry (2010) 21:5-13).
- ADCs are comprised of a drug like small molecule, covalently linked to an antibody.
- the antibody represents a targeting mechanism tuned to a specific site of action.
- the ADC Upon reaching the site, the ADC is designed to release a small molecule, the drug, allowing it to perform its designed function in a targeted manner, as opposed to diffusing systemically through the entire body of the subject. This targeted approach allows for treatment with drugs that would otherwise require doses so high as to be toxic when administered systemically.
- a key feature of the innate immune system is the recognition and elimination of foreign substances. Identification of these pathogenic invaders occurs through host recognition of evolutionarily conserved microbial structures known as pathogen-associated molecular patterns (PAMPs) (Jensen, S. and Thomsen, A.R. J Virol (2012) 86:2900-2910). Host recognition may occur by multiple pathways, such as activation of pattern recognition receptors (PRRs), which ultimately lead to downstream signaling events and culminate in the mounting of an immune response.
- PAMPs pathogen-associated molecular patterns
- the retinoic acid-inducible gene-I (RIG-I) protein is a RNA helicase that also functions as a sensor of microbial -derived RNA.
- RIG-I serves as a biomarker for the prediction of prognosis for certain types of cancer, such as hepatocellular carcinoma (Hou, J. et al, Cancer Cell (2014) 25:49-63).
- Recent publications have highlighted the importance of RIG-I and STING as mediators of innate and adaptive immunity, and RIG-I and STING agonists have been recognized as immuno-oncology agents in cancer therapy (Li, X. Y.
- RIG-I is involved in the regulation of basic cellular processes such as hematopoietic proliferation and differentiation, maintenance of leukemic sternness, and tumorigenesis of hepatocellular carcinoma, indicating that RIG-I performs an essential function as a tumor suppressor.
- STING pathway of cytosolic DNA sensing has been shown to play an important mechanistic role in innate immune sensing, driving type I IFN production in cancer and in the context of immune-oncology applications, including therapeutics and diagnostics.
- PD-1 Programmed death-1
- T cells T cells
- PD-L1 or PD-L2 A major functional role of the PD-1 signaling pathway is the inhibition of self-reactive T cells, which serve to protect against autoimmune diseases.
- Elimination of the PD-1 pathway can therefore result in the breakdown of immune tolerance that can ultimately lead to the development of pathogenic autoimmunity.
- tumor cells can at times co-opt the PD-1 pathway to escape from immune-surveillance mechanisms. Therefore, blockade of the PD-1 pathway has become an attractive target in cancer therapy.
- Cyclic dinucleotide compounds compositions comprising cyclic dinucleotide compounds, and related methods of use are described herein.
- the disclosure features an antibody-drug conjugate of Formula (I): or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
- Ab is an anti-PD-1 antibody or an anti-CTLA-4 antibody; each of B 1 and B 2 is independently a purinyl nucleobase or pyrimidinyl nucleobase; each of X 1 and X 2 is independently O or S; each of Y 1 and Y 2 is independently O, S, or N(R 5 ); each of Z 1 is indpendently O or S; each of Z 2 and Z 3 is independently absent, -C 1 -C 20 - alkylene (e.g., -C 1 -C 6 - alkylene), C 1 -C 20 heteroalkylene (e.g., -C 1 -C 6 - heteroalkylene), -OC(O)OC 1 -C 20 - alkylene (e.g., -C 1 -C 6 - alkylene), -cycloalkylene-, -heterocyclyl-, -aryl-, or -heteroaryl-, wherein each -cyclo
- Z 4 is self-immolative group-C 1 -C 20 -alkylene-Q 1 (e.g., -self-immolative group-C 1 -C 6 - alkylene-Q 1 ), heterocyclyl-C 1 -C 20 -alkylene-Q (e.g., heterocyclyl-C 1 -C 6 -alkylene-Q),- OH, -N(R 5 ) 2 , SR 5 , -CHO, -C(O)N(R 5 ) 2 , -OC(O)N(R 5 ) 2 , -N(R 5 )C(O)OR 5 , aryl , heteroaryl, -S(O)R 5 ,
- Z 5 is a self-immolative group or absent
- T is a spacer group or absent
- L 1 is absent, -C 1 -C 6 - alkylene (e.g., -C 1 -C 3 - alkylene) or -C 1 -C 6 - heteroalkylene;
- L 2 is absent, -C 1 -C 6 - alkylene (e.g., -C 1 -C 3 - alkylene) or -C 1 -C 6 - heteroalkylene, wherein each alkylene and heteroalkyl is optionally substituted with one or more R 6 ;
- L 3 is absent, -C 1 -C 20 - alkylene, -O-, -N(R 5 )-, -S-, -S(O)-, -S(O) 2 -, -S(O)N(R 5 )-,
- L 4 is -C 1 -C 20 - alkylene (e.g., -C 1 -C 6 - alkylene), -C 1 -C 20 - heteroalkylene (e.g., -C 1 -C 6 - heteroalkylene), -C 1 -C 20 - alkenylene (e.g., -C 2 -C 6 - alkenylene), -C 1 -C 20 - alkynylene (e.g., - -C 2 -C 6 - alkynylene), or an oligopeptide, wherein the oligopeptide is optionally substituted by one or more R 16 ;
- L 5 is a linker connecting Z 4 and Z 5 , or is absent;
- Q 1 is C(O), C(S), or CH 2 ; each of R 1 and R 2 is independently hydrogen, halo, -CN, -C 1 -C 20 alkyl (e.g., C 1 -C 6 alkyl), or - OR 7 ;
- R 3 is hydrogen, -C 1 -C 20 - alkyl (e.g., -C 1 -C 6 alkyl), -C 1 -C 20 heteroalkyl (e.g., -C 1 -C 6 heteroalkyl), -OC(O)OC 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, heteroalkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R 8 ; each R 4 is independently hydrogen, -C 1 -C 20 alkyl, -O-C 1 -C 20 alkyl, -C 1 -C 20 heteroalkyl, halo, -CN, -NO 2 or -OH;
- R 5 is hydrogen or -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl);
- R 6 is halo, -CN, -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -OR 7 , oxo, cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R 9 ;
- R 7 is hydrogen, -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R 9 ; each R 8 is independently -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C 1 -C 20 heteroalkyl,
- -C(O)-C 1 -C 20 alkyl -OC(O)-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C(O)O-C 1 -C 20 alkyl (e.g ⁇ ,
- -C 1 -C 6 alkyl -OC(O)O-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C(O)N(R 5 )-C 1 -C 20 alkyl (e.g ⁇ ,
- -C 1 -C 6 alkyl -N(R 5 )C(O)-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -OC(O)N(R 5 )-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -O-aryl, -O-heteroaryl, -C(O)-aryl, -C(O)-heteroaryl, - OC(O)-aryl,
- each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R 9 ; each R 9 is independently -C 1 -C 20 alkyl, -O-C 1 -C 20 alkyl, -C 1 -C 20 heteroalkyl, halo,
- each R 16 is independently, -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C 1 -C 20 heteroalkyl (e.g., -C 1 -C 6 heteroalkyl), -OC(O)OC 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), C(O)N(R 4 ) 2 cycloalkyl, heterocyclyl, aryl, or heteroaryl.
- -C 1 -C 20 alkyl e.g., -C 1 -C 6 alkyl
- -C 1 -C 20 heteroalkyl e.g., -C 1 -C 6 heteroalkyl
- -OC(O)OC 1 -C 20 alkyl e.g., -C 1 -C 6 alkyl
- C(O)N(R 4 ) 2 cycloalkyl, heterocyclyl, ary
- the disclosure features an antibody-drug conjugate of Formula (IV): or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
- Ab is an anti-PD-1 antibody or an anti-CTLA-4 antibody; each of B 1 and B 2 is independently a purinyl nucleobase or pyrimidinyl nucleobase; each of X 1 and X 2 is independently O or S; each of Y 1 and Y 2 is independently O, S, or N(R 5 ); each of Z 1 is independently O or S;
- Z 2 is -O-, -N(R 5 )-, -S-, -C(O)-, -C(O)N(R 5 )-, -OC(O)N(R 5 )-, -N(R 5 )C(O)O-, -aryl-, -heteroaryl-, -S(O)-, -S(O) 2 -, -S(O)N(R 5 )-, -S(O) 2 N(R 5 )- or -N(R 5 )S(O)-;
- Z 3 is absent, hydrogen, heterocyclyl, heterocyclyl-C 1 -C 20 -alkylene-Q 1 , -OH, -N(R 5 ) 2 , SR 5 , - CHO, -C(O)N(R 5 ) 2 , -OC(O)N(R 5 ) 2 , -N(R 5 )C(O)OR 5 , aryl , heteroaryl, -S(O)R 5 , - S(O) 2 R 5 , -S(O)N(R 5 ) 2 , -S(O) 2 N(R 5 ) 2 , -N(R 5 )S(O)R 5 , -OSi(C 1 -C 4 alkyl) 3 , or -C(O)C 2 - C 6 alkenyl (e.g., -C 2 -C 4 alkenyl);
- Z 4 is a self-immolative group or absent
- T is a absent or spacer group; each L 1 and L 2 is absent, -C 1 -C 6 - alkylene (e.g., -C 1 -C 3 - alkylene) or -C 1 -C 6 - heteroalkyl ene, wherein each alkylene and heteroalkyl is optionally substituted with one or more R 6 ;
- L 3 is oligiopeptide-C(O)-, oligiopeptide-aryl-C 1 -C 6 -alkylene-, oligiopeptide-aryl-C 1 -C 6 - alklyene-C(O)-, oligiopeptide-aryl-C 1 -C 6 -heteroalkylene, oligiopeptide-C 1 -C 6 - alkylene-C(O)-, oligiopeptide-C 1 -C 6 -heteroalkylene-C(O)-, -C 1 -C 40 - alkylene (e.g., - C 1 -C 20 - alkylene), -C 1 -C 40 - heteroalkyl ene (e.g., -C 1 -C 20 - heteroalkyl), -C 1 -C 40 - alkenylene (e.g., -C 2 -C 20 - alkenylene), or -C 1 -
- L 4 is absent or a linker connecting Z 3 and Z 4 ;
- Q 1 is C(O), C(S), or CH 2 ; each of R 1 and R 2 is independently hydrogen, halo, -CN, -C 1 -C 20 alkyl (e.g., C 1 -C 6 alkyl), or - OR 7 ; each R 3 and R 4 is independently hydrogen, -C 1 -C 20 - alkyl (e.g., -C 1 -C 6 alkyl), -C 1 -C 20 heteroalkyl (e.g., -C 1 -C 6 heteroalkyl), -OC(O)OC 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, heteroalkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R 8 ;
- R 5 is hydrogen or -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl);
- R 6 is halo, -CN, -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -OR 7 , oxo, cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R 9 ;
- R 7 is hydrogen, -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R 9 ; each R 8 is independently -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C 1 -C 20 heteroalkyl,
- -C(O)-C 1 -C 20 alkyl -OC(O)-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C(O)O-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -OC(O)O-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C(O)N(R 5 )-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -N(R 5 )C(O)-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -OC(O)N(R 5 )- C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -O-aryl, -O-hetero
- each R 16 is independently, -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C 1 -C 20 heteroalkyl (e.g., -C 1 -C 6 heteroalkyl), -OC(O)OC 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), C(O)N(R 4 ) 2 cycloalkyl, heterocyclyl, aryl, or heteroaryl.
- -C 1 -C 20 alkyl e.g., -C 1 -C 6 alkyl
- -C 1 -C 20 heteroalkyl e.g., -C 1 -C 6 heteroalkyl
- -OC(O)OC 1 -C 20 alkyl e.g., -C 1 -C 6 alkyl
- C(O)N(R 4 ) 2 cycloalkyl, heterocyclyl, ary
- the disclosure features an antibody-drug conjugate of Formula (Vlla) or Formula (Vllb): or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
- Ab is an anti-PD-1 antibody or an anti-CTLA-4 antibody; each of B 1 and B 2 is independently a purinyl nucleobase or pyrimidinyl nucleobase; each of X 1 and X 2 is independently O or S; each of Y 1 and Y 2 is independently O, S, or N(R 5 ); each of Z 1 is independently O or S;
- Z 2 is -O-, -N(R 5 )-, -S-, -C(O)-, -C(O)N(R 5 )-, -OC(O)N(R 5 )-, -N(R 5 )C(O)O-, -aryl-, -heteroaryl-, -S(O)-, -S(O) 2 -, -S(O)N(R 5 )-, -S(O) 2 N(R 5 )- or -N(R 5 )S(O)-;
- Z 3 is absent, hydrogen, heterocyclyl, heterocyclyl-C 1 -C 20 -alkylene-Q 1 , -OH, -N(R 5 ) 2 , SR 5 , - CHO, -C(O)N(R 5 ) 2 , -OC(O)N(R 5 ) 2 , -N(R 5 )C(O)OR 5 , aryl , heteroaryl, -S(O)R 5 , - S(O) 2 R 5 , -S(O)N(R 5 ) 2 , -S(O) 2 N(R 5 ) 2 , -N(R 5 )S(O)R 5 , -OSi(C 1 -C 4 alkyl) 3 , or -C(O)C 2 - C 6 alkenyl (e.g., -C 2 -C 4 alkenyl);
- Z 4 is a self-immolative group or absent
- T is a absent or spacer group; each L 1 and L 2 is absent, -C 1 -C 6 - alkylene (e.g., -C 1 -C 3 - alkylene) or -C 1 -C 6 - heteroalkyl ene, wherein each alkylene and heteroalkyl is optionally substituted with one or more R 6 ; L 3 is oligiopeptide-C(O)-, oligiopeptide-aryl-C 1 -C 6 -alkylene-, oligiopeptide-aryl-C 1 -C 6 - alklyene-C(O)-, oligiopeptide-aryl-C 1 -C 6 -heteroalkylene, oligiopeptide-C 1 -C 6 - alkylene-C(O)-, oligiopeptide-C 1 -C 6 -heteroalkylene-C(O)-, oligiopeptide-C 1 -C 6 -heteroal
- L 4 is absent or a linker connecting Z 3 and Z 4 ;
- Q 1 is C(O), C(S), or CH 2 ; each of R 1 and R 2 is independently hydrogen, halo, -CN, -C 1 -C 20 alkyl (e.g., C 1 -C 6 alkyl), or - OR 7 ; each R 3 and R 4 is independently hydrogen, -C 1 -C 20 - alkyl (e.g., -C 1 -C 6 alkyl), -C 1 -C 20 heteroalkyl (e.g., -C 1 -C 6 heteroalkyl), -OC(O)OC 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, heteroalkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R 8 ;
- R 5 is hydrogen or -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl);
- R 6 is halo, -CN, -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -OR 7 , oxo, cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R 9 ;
- R 7 is hydrogen, -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R 9 ; each R 8 is independently -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C 1 -C 20 heteroalkyl,
- -C(O)-C 1 -C 20 alkyl -OC(O)-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C(O)O-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -OC(O)O-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C(O)N(R 5 )-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -N(R 5 )C(O)-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -OC(O)N(R 5 )- C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -O-aryl, -O-hetero
- each R 16 is independently, -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C 1 -C 20 heteroalkyl (e.g., -C 1 -C 6 heteroalkyl), -OC(O)OC 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), C(O)N(R 4 ) 2 cycloalkyl, heterocyclyl, aryl, or heteroaryl.
- -C 1 -C 20 alkyl e.g., -C 1 -C 6 alkyl
- -C 1 -C 20 heteroalkyl e.g., -C 1 -C 6 heteroalkyl
- -OC(O)OC 1 -C 20 alkyl e.g., -C 1 -C 6 alkyl
- C(O)N(R 4 ) 2 cycloalkyl, heterocyclyl, ary
- the disclosure features an antibody-drug conjugate of Formula (Xa) or Formula (Xb): or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
- Ab is an anti-PD-1 antibody or an anti-CTLA-4 antibody; each of B 1 and B 2 is independently a purinyl nucleobase or pyrimidinyl nucleobase; each of X 1 and X 2 is independently O or S; each of Y 1 and Y 2 is independently O, S, or N(R 5 ); each of Z 1 is independently O or S; Z 2 is -O-, -N(R 5 )-, -S-, -C(O)-, -C(O)N(R 5 )-, -OC(O)N(R 5 )-, -N(R 5 )C(O)O-, -aryl-, -heteroaryl-, -S(O)-, -S(O) 2 -, -S(O)N(R 5 )-, -S(O) 2 N(R 5 )- or -N(R 5 )S(O)-;
- Z 3 is absent, hydrogen, heterocyclyl, heterocyclyl-C 1 -C 20 -alkylene-Q 1 , -OH, -N(R 5 ) 2 , SR 5 , - CHO, -C(O)N(R 5 ) 2 , -OC(O)N(R 5 ) 2 , -N(R 5 )C(O)OR 5 , aryl , heteroaryl, -S(O)R 5 , - S(O) 2 R 5 , -S(O)N(R 5 ) 2 , -S(O) 2 N(R 5 ) 2 , -N(R 5 )S(O)R 5 , -OSi(C 1 -C 4 alkyl) 3 , or -C(O)C 2 - C 6 alkenyl (e.g., -C 2 -C 4 alkenyl);
- Z 4 is a self-immolative group or absent
- T is a absent or spacer group; each L 1 and L 2 is absent, -C 1 -C 6 - alkylene (e.g., -C 1 -C 3 - alkylene) or -C 1 -C 6 - heteroalkyl ene, wherein each alkylene and heteroalkyl is optionally substituted with one or more R 6 ;
- L 3 is oligiopeptide-C(O)-, oligiopeptide-aryl-C 1 -C 6 -alkylene-, oligiopeptide-aryl-C 1 -C 6 - alklyene-C(O)-, oligiopeptide-aryl-C 1 -C 6 -heteroalkylene, oligiopeptide-C 1 -C 6 - alkylene-C(O)-, oligiopeptide-C 1 -C 6 -heteroalkylene-C(O)-, -C 1 -C 40 - alkylene (e.g., - C 1 -C 20 - alkylene), -C 1 -C 40 - heteroalkyl ene (e.g., -C 1 -C 20 - heteroalkyl), -C 1 -C 40 - alkenylene (e.g., -C 2 -C 20 - alkenylene), or -C 1 -
- L 4 is absent or a linker connecting Z 3 and Z 4 ;
- Q 1 is C(O), C(S), or CH 2 ; each of R 1 and R 2 is independently hydrogen, halo, -CN, -C 1 -C 20 alkyl (e.g., C 1 -C 6 alkyl), or - OR 7 ; each R 3 and R 4 is independently hydrogen, -C 1 -C 20 - alkyl (e.g., -C 1 -C 6 alkyl), -C 1 -C 20 heteroalkyl (e.g., -C 1 -C 6 heteroalkyl), -OC(O)OC 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, heteroalkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R 8 ;
- R 5 is hydrogen or -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl);
- R 6 is halo, -CN, -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -OR 7 , oxo, cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R 9 ;
- R 7 is hydrogen, -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R 9 ; each R 8 is independently -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C 1 -C 20 heteroalkyl,
- -C(O)-C 1 -C 20 alkyl -OC(O)-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C(O)O-C 1 -C 20 alkyl (e.g, -C 1 -C 6 alkyl), -OC(O)O-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C(O)N(R 5 )-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -N(R 5 )C(O)-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -OC(O)N(R 5 )- C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -O-aryl, -O-heteroary
- each R 16 is independently, -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C 1 -C 20 heteroalkyl (e.g., -C 1 -C 6 heteroalkyl), -OC(O)OC 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), C(O)N(R 4 ) 2 cycloalkyl, heterocyclyl, aryl, or heteroaryl.
- -C 1 -C 20 alkyl e.g., -C 1 -C 6 alkyl
- -C 1 -C 20 heteroalkyl e.g., -C 1 -C 6 heteroalkyl
- -OC(O)OC 1 -C 20 alkyl e.g., -C 1 -C 6 alkyl
- C(O)N(R 4 ) 2 cycloalkyl, heterocyclyl, ary
- certain diseases e.g., cancer
- agent is used herein to denote a chemical compound (such as an organic or inorganic compound, a mixture of chemical compounds), a biological macromolecule (such as a nucleic acid, an antibody, including parts thereof as well as humanized, chimeric and human antibodies and monoclonal antibodies, a protein or portion thereof, e.g., a peptide, a lipid, a carbohydrate), or an extract made from biological materials such as bacteria, plants, fungi, or animal (particularly mammalian) cells or tissues.
- Agents include, for example, agents whose structure is known, and those whose structure is not known. The ability of such agents to inhibit AR or promote AR degradation may render them suitable as “therapeutic agents” in the methods and compositions of this disclosure.
- a “patient,” “subject,” or “individual” are used interchangeably and refer to either a human or a non-human animal. These terms include mammals, such as humans, primates, livestock animals (including bovines, porcines, etc.), companion animals (e.g., canines, felines, etc.) and rodents (e.g., mice and rats).
- Treating” a condition or patient refers to taking steps to obtain beneficial or desired results, including clinical results.
- treatment is an approach for obtaining beneficial or desired results, including clinical results.
- Beneficial or desired clinical results can include, but are not limited to, alleviation or amelioration of one or more symptoms or conditions, diminishment of extent of disease, stabilized (i.e. not worsening) state of disease, preventing spread of disease, delay or slowing of disease progression, amelioration or palliation of the disease state, and remission (whether partial or total), whether detectable or undetectable.
- Treatment can also mean prolonging survival as compared to expected survival if not receiving treatment.
- preventing is art-recognized, and when used in relation to a condition, such as a local recurrence (e.g., pain), a disease such as cancer, a syndrome complex such as heart failure or any other medical condition, is well understood in the art, and includes administration of a composition which reduces the frequency of, or delays the onset of, symptoms of a medical condition in a subject relative to a subject which does not receive the composition.
- a condition such as a local recurrence (e.g., pain)
- a disease such as cancer
- a syndrome complex such as heart failure or any other medical condition
- prevention of cancer includes, for example, reducing the number of detectable cancerous growths in a population of patients receiving a prophylactic treatment relative to an untreated control population, and/or delaying the appearance of detectable cancerous growths in a treated population versus an untreated control population, e.g., by a statistically and/or clinically significant amount.
- administering or “administration of’ a substance, a compound or an agent to a subject can be carried out using one of a variety of methods known to those skilled in the art.
- a compound or an agent can be administered, intravenously, arterially, intradermally, intramuscularly, intraperitoneally, subcutaneously, ocularly, sublingually, orally (by ingestion), intranasally (by inhalation), intraspinally, intracerebrally, and transdermally (by absorption, e.g., through a skin duct).
- a compound or agent can also appropriately be introduced by rechargeable or biodegradable polymeric devices or other devices, e.g., patches and pumps, or formulations, which provide for the extended, slow or controlled release of the compound or agent.
- Administering can also be performed, for example, once, a plurality of times, and/or over one or more extended periods.
- a compound or an agent is administered orally, e.g., to a subject by ingestion.
- the orally administered compound or agent is in an extended release or slow release formulation, or administered using a device for such slow or extended release.
- the phrase “conjoint administration” refers to any form of administration of two or more different therapeutic agents such that the second agent is administered while the previously administered therapeutic agent is still effective in the body (e.g., the two agents are simultaneously effective in the patient, which may include synergistic effects of the two agents).
- the different therapeutic compounds can be administered either in the same formulation or in separate formulations, either concomitantly or sequentially.
- an individual who receives such treatment can benefit from a combined effect of different therapeutic agents.
- a “therapeutically effective amount” or a “therapeutically effective dose” of a drug or agent is an amount of a drug or an agent that, when administered to a subject will have the intended therapeutic effect.
- the full therapeutic effect does not necessarily occur by administration of one dose, and may occur only after administration of a series of doses.
- a therapeutically effective amount may be administered in one or more administrations.
- the precise effective amount needed for a subject will depend upon, for example, the subject’s size, health and age, and the nature and extent of the condition being treated, such as cancer or MDS. The skilled worker can readily determine the effective amount for a given situation by routine experimentation.
- the terms “optional” or “optionally” mean that the subsequently described event or circumstance may occur or may not occur, and that the description includes instances where the event or circumstance occurs as well as instances in which it does not.
- “optionally substituted alkyl” refers to the alkyl may be substituted as well as where the alkyl is not substituted.
- substituents and substitution patterns on the compounds of the present disclosure can be selected by one of ordinary skilled person in the art to result chemically stable compounds which can be readily synthesized by techniques known in the art, as well as those methods set forth below, from readily available starting materials. If a substituent is itself substituted with more than one group, it is understood that these multiple groups may be on the same carbon or on different carbons, so long as a stable structure results.
- the term “optionally substituted” refers to the replacement of one to six hydrogen radicals in a given structure with the radical of a specified substituent including, but not limited to: hydroxyl, hydroxyalkyl, alkoxy, halogen, alkyl, nitro, silyl, acyl, acyloxy, aryl, cycloalkyl, heterocyclyl, amino, aminoalkyl, cyano, haloalkyl, haloalkoxy, -OCO-CH 2 -O- alkyl, -OP(O)(O-alkyl) 2 or -CH 2 -OP(O)(O-alkyl) 2 .
- “optionally substituted” refers to the replacement of one to four hydrogen radicals in a given structure with the substituents mentioned above. More preferably, one to three hydrogen radicals are replaced by the substituents as mentioned above. It is understood that the substituent can be further substituted.
- alkyl refers to saturated aliphatic groups, including but not limited to C 1 -C 10 straight-chain alkyl groups or C 1 -C 10 branched-chain alkyl groups.
- the “alkyl” group refers to C 1 -C 6 straight-chain alkyl groups or C 1 -C 6 branched- chain alkyl groups.
- the “alkyl” group refers to C 1 -C 4 straight-chain alkyl groups or C 1 -C 4 branched-chain alkyl groups.
- alkyl examples include, but are not limited to, methyl, ethyl, 1 -propyl, 2-propyl, n-butyl, sec-butyl, tert-butyl, 1 -pentyl, 2-pentyl, 3 -pentyl, neo-pentyl, 1 -hexyl, 2-hexyl, 3 -hexyl, 1-heptyl, 2-heptyl, 3-heptyl, 4-heptyl, 1 -octyl, 2-octyl, 3-octyl or 4-octyl and the like.
- the “alkyl” group may be optionally substituted.
- acyl is art-recognized and refers to a group represented by the general formula hydrocarbylC(O)-, preferably alkylC(O)-.
- acylamino is art-recognized and refers to an amino group substituted with an acyl group and may be represented, for example, by the formula hydrocarbylC(O)NH-.
- acyloxy is art-recognized and refers to a group represented by the general formula hydrocarbylC(O)O-, preferably alkylC(O)O-.
- alkoxy refers to an alkyl group having an oxygen attached thereto.
- Representative alkoxy groups include methoxy, ethoxy, propoxy, tert-butoxy and the like.
- alkoxyalkyl refers to an alkyl group substituted with an alkoxy group and may be represented by the general formula alkyl -O-alkyl.
- alkyl refers to saturated aliphatic groups, including straight-chain alkyl groups, branched-chain alkyl groups, cycloalkyl (alicyclic) groups, alkyl -substituted cycloalkyl groups, and cycloalkyl-substituted alkyl groups.
- a straight chain or branched chain alkyl has 30 or fewer carbon atoms in its backbone (e.g., C 1 - 30 for straight chains, C 3-30 for branched chains), and more preferably 20 or fewer.
- alkyl as used throughout the specification, examples, and claims is intended to include both unsubstituted and substituted alkyl groups, the latter of which refers to alkyl moieties having substituents replacing a hydrogen on one or more carbons of the hydrocarbon backbone, including haloalkyl groups such as trifluoromethyl and 2,2,2- trifluoroethyl, etc.
- C x-y or “C x -C y ”, when used in conjunction with a chemical moiety, such as, acyl, acyloxy, alkyl, alkenyl, alkynyl, or alkoxy is meant to include groups that contain from x to y carbons in the chain.
- Coalkyl indicates a hydrogen where the group is in a terminal position, a bond if internal.
- a C 1-6 alkyl group for example, contains from one to six carbon atoms in the chain.
- alkylamino refers to an amino group substituted with at least one alkyl group.
- alkylthio refers to a thiol group substituted with an alkyl group and may be represented by the general formula alkylS-.
- amide refers to a group wherein R 9 and R 10 each independently represent a hydrogen or hydrocarbyl group, or R 9 and R 10 taken together with the N atom to which they are attached complete a heterocycle having from 4 to 8 atoms in the ring structure.
- amine and “amino” are art-recognized and refer to both unsubstituted and substituted amines and salts thereof, e.g., a moiety that can be represented by wherein R 9 , R 10 , and R 10 ’ each independently represent a hydrogen or a hydrocarbyl group, or R 9 and R 10 taken together with the N atom to which they are attached complete a heterocycle having from 4 to 8 atoms in the ring structure.
- aminoalkyl refers to an alkyl group substituted with an amino group.
- aralkyl refers to an alkyl group substituted with an aryl group.
- aryl as used herein include substituted or unsubstituted single-ring aromatic groups in which each atom of the ring is carbon.
- the ring is a 5- to 7-membered ring, more preferably a 6-membered ring.
- aryl also includes polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is aromatic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls.
- Aryl groups include benzene, naphthalene, phenanthrene, phenol, aniline, and the like.
- the aromatic ring may be substituted at one or more ring positions with one or more substituents, such as halogen, azide, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, alkoxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, sulfonamido, ketone, aldehyde, ester, heterocyclyl, aromatic or heteroaromatic moieties, fluoroalkyl (such as trifluromethyl), cyano, or the like.
- substituents such as halogen, azide, alkyl, aralkyl, alkenyl, alky
- carboxylate is art-recognized and refers to a group wherein R 9 and R 10 independently represent hydrogen or a hydrocarbyl group.
- Carbocyclylalkyl refers to an alkyl group substituted with a carbocycle group.
- Carbocycle includes 5-7 membered monocyclic and 8-12 membered bicyclic rings. Each ring of a bicyclic carbocycle may be selected from saturated, unsaturated and aromatic rings. Carbocycle includes bicyclic molecules in which one, two or three or more atoms are shared between the two rings.
- fused carbocycle refers to a bicyclic carbocycle in which each of the rings shares two adjacent atoms with the other ring. Each ring of a fused carbocycle may be selected from saturated, unsaturated and aromatic rings.
- an aromatic ring e.g., phenyl
- a saturated or unsaturated ring e.g., cyclohexane, cyclopentane, or cyclohexene.
- Exemplary “carbocycles” include cyclopentane, cyclohexane, bicyclo[2.2.1]heptane, 1,5-cyclooctadiene, 1,2,3,4-tetrahydronaphthalene, bicyclo[4.2.0]oct- 3-ene, naphthalene and adamantane.
- Exemplary fused carbocycles include decalin, naphthalene, 1,2,3,4-tetrahydronaphthalene, bicyclo[4.2.0]octane, 4,5,6,7-tetrahydro-lH- indene and bicyclo[4.1.0]hept-3-ene.
- “Carbocycles” may be substituted at any one or more positions capable of bearing a hydrogen atom.
- Carbocyclylalkyl refers to an alkyl group substituted with a carbocycle group.
- carbonate is art-recognized and refers to a group -OCO 2 -.
- esters refers to a group -C(O)OR 9 wherein R 9 represents a hydrocarbyl group.
- ether refers to a hydrocarbyl group linked through an oxygen to another hydrocarbyl group. Accordingly, an ether substituent of a hydrocarbyl group may be hydrocarbyl-O-. Ethers may be either symmetrical or unsymmetrical. Examples of ethers include, but are not limited to, heterocycle-O-heterocycle and aryl-O-heterocycle. Ethers include “alkoxyalkyl” groups, which may be represented by the general formula alkyl- O-alkyl.
- halo and “halogen” as used herein means halogen and includes chloro, fluoro, bromo, and iodo.
- heteroalkyl and “heteroaralkyl”, as used herein, refers to an alkyl group substituted with a hetaryl group.
- heteroaryl and “hetaryl” include substituted or unsubstituted aromatic single ring structures, preferably 5- to 7-membered rings, more preferably 5- to 6-membered rings, whose ring structures include at least one heteroatom, preferably one to four heteroatoms, more preferably one or two heteroatoms.
- heteroaryl and “hetaryl” also include polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is heteroaromatic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls.
- Heteroaryl groups include, for example, pyrrole, furan, thiophene, imidazole, oxazole, thiazole, pyrazole, pyridine, pyrazine, pyridazine, pyrimidine, and the like.
- the heteroaromatic ring may be substituted at one or more ring positions with one or more substituents, such as halogen, azide, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, alkoxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, sulfonamido, ketone, aldehyde, ester, heterocyclyl, aromatic or heteroaromatic moieties, fluoroalkyl (such as trifluromethyl), cyano, or the like.
- substituents such as halogen, azide, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, alkoxyl, amino, nitro, sulfhydry
- heteroatom as used herein means an atom of any element other than carbon or hydrogen. Preferred heteroatoms are nitrogen, oxygen, and sulfur.
- heterocyclylalkyl refers to an alkyl group substituted with a heterocycle group.
- heterocyclyl refers to substituted or unsubstituted non-aromatic ring structures, preferably 3- to 10-membered rings, more preferably 3- to 7-membered rings, whose ring structures include at least one heteroatom, preferably one to four heteroatoms, more preferably one or two heteroatoms.
- heterocyclyl and “heterocyclic” also include polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is heterocyclic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls.
- Heterocyclyl groups include, for example, piperidine, piperazine, pyrrolidine, morpholine, lactones, lactams, and the like.
- Hydrocarbyl groups include, but are not limited to aryl, heteroaryl, carbocycle, heterocycle, alkyl, alkenyl, alkynyl, and combinations thereof.
- hydroxyalkyl refers to an alkyl group substituted with a hydroxy group.
- lower when used in conjunction with a chemical moiety, such as, acyl, acyloxy, alkyl, alkenyl, alkynyl, or alkoxy is meant to include groups where there are ten or fewer atoms in the substituent, preferably six or fewer.
- acyl, acyloxy, alkyl, alkenyl, alkynyl, or alkoxy substituents defined herein are respectively lower acyl, lower acyloxy, lower alkyl, lower alkenyl, lower alkynyl, or lower alkoxy, whether they appear alone or in combination with other substituents, such as in the recitations hydroxyalkyl and aralkyl (in which case, for example, the atoms within the aryl group are not counted when counting the carbon atoms in the alkyl substituent).
- polycyclyl refers to two or more rings (e.g., cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls) in which two or more atoms are common to two adjoining rings, e.g., the rings are “fused rings”.
- Each of the rings of the polycycle can be substituted or unsubstituted.
- each ring of the polycycle contains from 3 to 10 atoms in the ring, preferably from 5 to 7.
- sulfate is art-recognized and refers to the group -OSO 3 H, or a pharmaceutically acceptable salt thereof.
- sulfonamide is art-recognized and refers to the group represented by the general formulae wherein R 9 and R 10 independently represents hydrogen or hydrocarbyl.
- sulfoxide is art-recognized and refers to the group-S(O)-.
- sulfonate is art-recognized and refers to the group SO 3 H, or a pharmaceutically acceptable salt thereof.
- substituted refers to moieties having substituents replacing a hydrogen on one or more carbons of the backbone. It will be understood that “substitution” or “substituted with” includes the implicit proviso that such substitution is in accordance with permitted valence of the substituted atom and the substituent, and that the substitution results in a stable compound, e.g., which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, etc. As used herein, the term “substituted” is contemplated to include all permissible substituents of organic compounds.
- the permissible substituents include acyclic and cyclic, branched and unbranched, carbocyclic and heterocyclic, aromatic and non-aromatic substituents of organic compounds.
- the permissible substituents can be one or more and the same or different for appropriate organic compounds.
- the heteroatoms such as nitrogen may have hydrogen substituents and/or any permissible substituents of organic compounds described herein which satisfy the valences of the heteroatoms.
- Substituents can include any substituents described herein, for example, a halogen, a hydroxyl, a carbonyl (such as a carboxyl, an alkoxycarbonyl, a formyl, or an acyl), a thiocarbonyl (such as a thioester, a thioacetate, or a thioformate), an alkoxyl, a phosphoryl, a phosphate, a phosphonate, a phosphinate, an amino, an amido, an amidine, an imine, a cyano, a nitro, an azido, a sulfhydryl, an alkylthio, a sulfate, a sulfonate, a sulfamoyl, a sulfonamido, a sulfonyl, a heterocyclyl, an aralkyl, or an aromatic or heteroaromatic mo
- thioalkyl refers to an alkyl group substituted with a thiol group.
- thioester refers to a group -C(O)SR 9 or -SC(O)R 9 wherein R 9 represents a hydrocarbyl.
- thioether is equivalent to an ether, wherein the oxygen is replaced with a sulfur.
- urea is art-recognized and may be represented by the general formula wherein R 9 and R 10 independently represent hydrogen or a hydrocarbyl.
- modulate includes the inhibition or suppression of a function or activity (such as cell proliferation) as well as the enhancement of a function or activity.
- compositions, excipients, adjuvants, polymers and other materials and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- “Pharmaceutically acceptable salt” or “salt” is used herein to refer to an acid addition salt or a basic addition salt which is suitable for or compatible with the treatment of patients.
- pharmaceutically acceptable acid addition salt means any non-toxic organic or inorganic salt of any base compounds represented by Formula I.
- Illustrative inorganic acids which form suitable salts include hydrochloric, hydrobromic, sulfuric and phosphoric acids, as well as metal salts such as sodium monohydrogen orthophosphate and potassium hydrogen sulfate.
- Illustrative organic acids that form suitable salts include mono-, di-, and tricarboxylic acids such as glycolic, lactic, pyruvic, malonic, succinic, glutaric, fumaric, malic, tartaric, citric, ascorbic, maleic, benzoic, phenylacetic, cinnamic and salicylic acids, as well as sulfonic acids such as p-toluene sulfonic and methanesulfonic acids. Either the mono or di-acid salts can be formed, and such salts may exist in either a hydrated, solvated or substantially anhydrous form.
- mono-, di-, and tricarboxylic acids such as glycolic, lactic, pyruvic, malonic, succinic, glutaric, fumaric, malic, tartaric, citric, ascorbic, maleic, benzoic, phenylacetic, cinnamic and salicylic acids, as well as sul
- the acid addition salts of compounds of Formula I are more soluble in water and various hydrophilic organic solvents, and generally demonstrate higher melting points in comparison to their free base forms.
- the selection of the appropriate salt will be known to one skilled in the art.
- Other non-pharmaceutically acceptable salts e.g., oxalates, may be used, for example, in the isolation of compounds of Formula I for laboratory use, or for subsequent conversion to a pharmaceutically acceptable acid addition salt.
- pharmaceutically acceptable basic addition salt means any non-toxic organic or inorganic base addition salt of any acid compounds represented by Formula I or any of their intermediates.
- Illustrative inorganic bases which form suitable salts include lithium, sodium, potassium, calcium, magnesium, or barium hydroxide.
- Illustrative organic bases which form suitable salts include aliphatic, alicyclic, or aromatic organic amines such as methylamine, trimethylamine and picoline or ammonia. The selection of the appropriate salt will be known to a person skilled in the art.
- stereogenic center in their structure.
- This stereogenic center may be present in a R or a S configuration, said R and S notation is used in correspondence with the rules described in Pure Appl. Chem. (1976), 45, 11-30.
- the disclosure contemplates all stereoisomeric forms such as enantiomeric and diastereoisomeric forms of the compounds, salts, prodrugs or mixtures thereof (including all possible mixtures of stereoisomers). See, e.g., WO 01/062726.
- Prodrug or “pharmaceutically acceptable prodrug” refers to a compound that is metabolized, for example hydrolyzed or oxidized, in the host after administration to form the compound of the present disclosure (e.g., compounds of formula I).
- Typical examples of prodrugs include compounds that have biologically labile or cleavable (protecting) groups on a functional moiety of the active compound.
- Prodrugs include compounds that can be oxidized, reduced, aminated, deaminated, hydroxylated, dehydroxyl ated, hydrolyzed, dehydrolyzed, alkylated, dealkylated, acylated, deacylated, phosphorylated, or dephosphorylated to produce the active compound.
- prodrugs using ester or phosphoramidate as biologically labile or cleavable (protecting) groups are disclosed in U.S. Patents 6,875,751, 7,585,851, and 7,964,580, the disclosures of which are incorporated herein by reference.
- the prodrugs of this disclosure are metabolized to produce a compound of Formula I.
- the present disclosure includes within its scope, prodrugs of the compounds described herein. Conventional procedures for the selection and preparation of suitable prodrugs are described, for example, in “Design of Prodrugs” Ed. H. Bundgaard, Elsevier, 1985.
- pharmaceutically acceptable carrier means a pharmaceutically acceptable material, composition or vehicle, such as a liquid or solid filter, diluent, excipient, solvent or encapsulating material useful for formulating a drug for medicinal or therapeutic use.
- Log of solubility is used in the art to quantify the aqueous solubility of a compound.
- the aqueous solubility of a compound significantly affects its absorption and distribution characteristics. A low solubility often goes along with a poor absorption.
- LogS value is a unit stripped logarithm (base 10) of the solubility measured in mol/liter.
- the terms “induce” or “induction of’ refer to the increase or enhancement of a function, e.g., the increase or enhancement of the expression of a pattern recognition receptor (e.g, STING).
- “induction of PRR expression” refers to induction of transcription of PRR RNA, e.g., STING RNA (e.g., mRNA, e.g., an increase or enhancement of), or the translation of a PRR protein, e.g., the STING protein (e.g., an increase or enhancement of).
- induction of PRR expression refers to the increase or enhancement of the concentration of a PRR RNA, e.g., or STING RNA (e.g., mRNA) or the STING protein, e.g., in a cell.
- induction of PRR expression refers to the increase of the number of copies of PRR RNA, e.g., STING RNA (e.g., mRNA) or PRR protein, e.g., the STING protein, e.g., in a cell.
- to induce expression of a PRR may refer to the initiation of PRR RNA (e.g., STING RNA (e.g., mRNA)) or transcription or PRR protein (e.g., STING protein) translation.
- PRR protein e.g., STING protein
- to induce expression of a PRR may refer to an increase in the rate of PRR RNA (e.g., STING RNA (e.g., mRNA)) transcription or an increase in the rate of PRR protein (e.g., STING protein) expression.
- activation refers to the stimulation or triggering of a function, e.g., of a downstream pathway, e.g., a downstream signaling pathway.
- activation of a pattern recognition receptor refers to the stimulation of a specific protein or pathway, e.g., through interaction with a downstream signaling partner (e.g., IFN-b promoter stimulator 1 (IPS-1), IRF3, IRF7, NF-KB, interferons (e.g., IFN-a or IFN-b) and/or cytokines).
- IPS-1 IFN-b promoter stimulator 1
- IRF3, IRF7 e.g., IFN-a or IFN-b
- cytokines interferons
- a PRR may be activated without resulting in an induction of PRR expression (e.g., expression of STING).
- activation may include induction of expression of a PRR (e.g., STING).
- activation of a PRR may trigger the induction of expression of a PRR (e.g., STING) by about 0.1%, about 0.5%, about 1%, about 5%, about 10%, about 15%, about 20%, about 25%, about 30%, about 40%, about 50%, about 60%, about 70%, about 80%, about 90%, about 95%, or more compared to a reference standard (e.g., basal expression levels of a PRR (e.g., STING)).
- a reference standard e.g., basal expression levels of a PRR (e.g., STING)
- Cmd refers to the word “compound” or “Compound”, and all of the terms are used interchangeably.
- nucleobase is a nitrogen-containing biological compound found linked to a sugar within a nucleoside — the basic building blocks of deoxyribonucleic acid (DNA) and ribonucleic acid (RNA).
- the primary, or naturally occurring, nucleobases are cytosine (DNA and RNA), guanine (DNA and RNA), adenine (DNA and RNA), thymine (DNA) and uracil (RNA), abbreviated as C, G, A, T, and U, respectively. Because A, G, C, and T appear in the DNA, these molecules are called DNA-bases; A, G, C, and U are called RNA- bases.
- Adenine and guanine belong to the double-ringed class of molecules called purines (abbreviated as R). Cytosine, thymine, and uracil are all pyrimidines. Other nucleobases that do not function as normal parts of the genetic code are termed non-naturally occurring.
- ADC refers to an antibody drug conjugate.
- Link means a chemical moiety comprising a covalent bond or a chain of atoms that covalently attaches two groups, e.g., an antibody to a drug moiety.
- the term “monoclonal antibody” refers to a homogeneous antibody population involved in the highly specific recognition and binding of a single antigenic determinant or epitope. This is in contrast to polyclonal antibodies that typically include different antibodies directed against a variety of different antigenic determinants.
- the term “monoclonal antibody” includes 33 antibody fragments (such as Fab, Fab', F(ab') 2 , Fd, Fv), single chain (scFv) mutants, fusion proteins including an antibody portion, and any other modified immunoglobulin molecule including an antigen recognition site as well as both intact and full- length monoclonal antibodies, but are not limited thereto. Additionally, “monoclonal antibody” refers to such antibodies made in any number of methods including but not limited to hybridoma, phage selection, recombinant expression, and transgenic animals.
- humanized antibody refers to forms of non-human (e.g., murine) antibodies that are specific immunoglobulin chains, chimeric immunoglobulins, or fragments thereof that contain minimal non-human (e.g., murine) sequences.
- humanized antibodies are human immunoglobulins in which residues from complementary determining region (CDR) are replaced by residues from CDR of a non-human species (e.g., mouse, rat, rabbit, and hamster) having the desired specificity, affinity, and capability (ref: Jones et al., 1986, Nature, 321:522-525; Riechmann et al., 1988, Nature, 332:323-327; Verhoeyen et al., 1988, Science, 239:1534-1536).
- Fv framework region (FR) residues of a human immunoglobulin are replaced with the corresponding residues in an antibody from a non- human species having the desired specificity, affinity, and/or binding capability.
- the humanized 34 antibody may be further modified by the substitution of additional residues either in the Fv framework region and/or within the replaced non-human residues to refine and optimize antibody specificity, affinity, and/or binding capability.
- the humanized antibody includes substantially all of at least one, and typically two or three, variable domains containing all or substantially all of the CDRs that correspond to the non-human immunoglobulin whereas all or substantially all of the framework regions (FRs) have those of a human immunoglobulin consensus sequence.
- the humanized antibody may also include at least a portion of an immunoglobulin constant region or domain (Fc), typically that of a human immunoglobulin. Examples of methods used to generate humanized antibodies are described in U.S. Patent No. 5,225,539.
- human antibody refers to an antibody produced by a human or an antibody having an amino acid sequence corresponding to an antibody produced by a human using any technique known in the art. This definition of the human antibody includes intact or fulllength antibodies, fragments thereof, and/or antibodies including at least one human heavy and/or light chain polypeptide such as, for example, an antibody including murine light chain and human heavy chain polypeptides.
- chimeric antibody refers to an antibody wherein an amino acid sequence of an immunoglobulin molecule is derived from two or more species.
- variable regions of both light and heavy chains correspond to variable regions of antibodies derived from one species of mammals (e.g., mouse, rat, rabbit, etc) with the desired specificity, affinity, and capability, while constant regions are homologous to the sequences in antibodies derived from another species (usually human) to avoid eliciting an immune response in that species.
- mammals e.g., mouse, rat, rabbit, etc
- constant regions are homologous to the sequences in antibodies derived from another species (usually human) to avoid eliciting an immune response in that species.
- self-eliminating linker or “self-immolative linker” refers to a temporary extender, spacer, or placeholder unit attaching two or more molecules together by chemical bonds that are cleaved under defined conditions to release the two molecules.
- a self-eliminating or self-immolative linker may be linear or branched, and may link two or more of the same molecules together, or may link two or more different molecules together.
- a self- immolative moiety may be defined as a bifunctional chemical group which is capable of covalently linking together two spaced chemical moieties into a normally stable molecule, releasing one of said spaced chemical moieties from the molecule by means of enzymatic cleavage; and following said enzymatic cleavage, spontaneously cleaving from the remainder of the bifunctional chemical group to release the other of said spaced chemical moieties.
- the self-immolative refers to a heterocyclic self-immolative moiety.
- Exemplary self-immolative linkers include His- Ala, /;-ami nobenzyl oxycarbonyl (PABC), 2,4- bis(hydroxymethyl)aniline, -NH-(CH 2 ) 4 -C(O)- and -NH-(CH 2 ) 3 -C(O)-.
- PABC amino nobenzyl oxycarbonyl
- cleaveable group is refers to a moiety that is unstable in vivo.
- the “cleaveable group” allows for activation of the marker or therapeutic agent by cleaving the marker or agent from the rest of the conjugate.
- the linker is preferably cleaved in vivo by the biological environment. The cleavage may come from any process without limitation, e.g., enzymatic, reductive, pH, etc.
- the cleaveable group is selected so that activation occurs at the desired site of action, which can be a site in or near the target cells (e.g., carcinoma cells) or tissues such as at the site of therapeutic action or marker activity.
- Such cleavage may be enzymatic and exemplary enzymatically cleaveable groups include natural amino acids or peptide sequences that end with a natural amino acid, and are attached at their carboxyl terminus to the linker. While the degree of cleavage rate enhancement is not critical to the invention, preferred examples of cleaveable linkers are those in which at least about 10% of the cleaveable groups are cleaved in the blood stream within 24 hours of administration, most preferably at least about 35%.
- spacer group refers any chemical group designed to facilitate the attachment of the drug conjugates to an antibody, e.g., in order to overcome steric hinderance.
- PPRs Patter Recognition Receptors
- the present disclosure relates to methods of activating and/or inducing the expression of PRRs (e.g., STING) in a subject, in particular for the treatment of a microbial infection or a proliferative disease (e.g., cancer).
- the method comprises administration of an antibody-drug conjugate of the disclosure or pharmaceutically acceptable salt thereof. It is to be noted that induction of any PRR with these compounds can stimulate interferon and/or NF-KB production which can induce the expression of a variety of PRRs which are inducible genes by feedback mechanism.
- PRR expression e.g., STING expression
- a subject e.g., a subject with a proliferative disease (e.g., cancer).
- Pattern recognition receptors are a broad class of proteins which recognize pathogen-associated molecular patterns (PAMPs) conserved within pathogenic invaders. PAMPs are typically products of biosynthetic pathways that are essential to the survival and/or infectivity of the pathogen, e.g., lipopolysaccharides, glycoproteins, and nucleic acids. Recognition of PAMPs by their cognate PRRs activates signaling pathways that result in the production of immune defense factors such as pro-inflammatory and anti-inflammatory cytokines, type I interferons (IFN-a, IFN-b), and/or interferon stimulated genes (ISGs). It is well known that induction of innate immune signaling also results in the activation of T cell responses as well as the induction of adaptive immunity.
- IFN-a, IFN-b type I interferons
- ISGs interferon stimulated genes
- the stimulator of interferon genes is a cytosolic microbial-derived DNA sensor that has been shown to be particularly sensitive to double-stranded DNA and cyclic dinucleotides (e.g., cyclic di-GMP) (Burdette, D. L. and Vance, R. E. (2013) Nat Immunol 14: 19-26).
- cyclic di-GMP cyclic dinucleotides
- Two molecules of STING form a homodimer mediated by an a-helix present in the C-terminal dimerization domain, and molecular binding studies have revealed that each STING dimer binds one molecule of microbial nucleic acids, e.g., DNA or a cyclic dinucleotide.
- STING has been shown to play a major role in the innate immune response against tumor antigens, driving dendritic cell activation and subsequent T cell priming in several cancers (Woo, S.R. et al. Trends in Immunol (2015) 36:250-256).
- RIG- 1 is the founding member of a family of PRRs termed RIG-I-like receptors (RLRs) that primarily detect RNA derived from foreign sources. It is a critical sensor of microbial infection (e.g., viral infection) in most cells and is constitutively expressed at low levels in the cytosol. After ligand binding, the expression of RIG-I is rapidly enhanced, leading to increased RIG-I concentrations in the cell (Jensen, S. and Thomsen, A.R. J Virol (2012) 86:2900-2910; Yoneyama M. et al. Nat Immunol (2004) 5:730- 737).
- RIG-I-like receptors RLRs
- RIG-I is an ATP-dependent helicase containing a central DExD/H box ATPase domain and tandem N-terminal caspase-recruiting domains (CARDs) that mediate downstream signaling.
- the C-terminus of RIG-I comprises an ssRNA/dsRNA-binding domain that when unbound acts to silence CARD function at the N-terminus.
- IFN-b promoter stimulator 1 IPS-1
- MAVS mitochondrial antiviral signaling molecule
- CARD IF mitochondrial antiviral signaling molecule
- NOD2 nucleotide-binding and oligomerization domain
- NLR family Caruso, R. et al, Immunity (2014) 41:898-908
- NOD2 is composed of an N-terminal CARD, a centrally-located nucleotide-binding oligomerization domain, and a C-terminal leucine rich repeat domain that is responsible for binding microbial PAMPs, such as bacterial peptidoglycan fragments and microbial nucleic acids.
- NOD2 Ligand binding activates NOD2 and is believed to drive interaction with the CARD-containing kinase RIPK2, which in turn activates a number of downstream proteins including NF-KB, MAPK, IRF7, and IRF3, the latter of which results in the induction of type 1 interferons.
- NOD2 is expressed in a diverse set of cell types, including macrophages, dendritic cells, paneth cells, epithelial cells (e.g., lung epithelial cells, intestinal epithelia), and osteoblasts.
- ADCs Antibody drug conjugates
- ADCs are an important class of biopharmaceutical drugs, which are designed to act as a targeted therapy for the treatment of subjects with various disease states (Ducry, L. Bioconjugate Chemistry (2010) 21:5-13).
- ADCs are comprised of a small molecule covalently linked to an antibody.
- the covalent linkage comprises a point of attachment to the small molecule, a biocompatible linker unit, and a self-immolative group attached to the antibody.
- the disclosure features an antibody-drug conjugate of Formula (I): or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
- Ab is an anti-PD-1 antibody or an anti-CTLA-4 antibody; each of B 1 and B 2 is independently a purinyl nucleobase or pyrimidinyl nucleobase; each of X 1 and X 2 is independently O or S; each of Y 1 and Y 2 is independently O, S, or N(R 5 ); each of Z 1 is indpendently O or S; each of Z 2 and Z 3 is independently absent, -C 1 -C 20 - alkylene (e.g., -C 1 -C 6 - alkylene), C 1 -C 20 heteroalkylene (e.g., -C 1 -C 6 - heteroalkylene), -OC(O)OC 1 -C 20 - alkylene (e.g., -C 1 -C 6 - alkylene), -cycloalkylene-, -heterocyclyl-, -aryl-, or -heteroaryl-, wherein each -cyclo
- Z 4 is self-immolative group-C 1 -C 20 -alkylene-Q 1 (e.g., -self-immolative group-C 1 -C 6 - alkylene-Q 1 ), heterocyclyl-C 1 -C 20 -alkylene-Q (e.g., heterocyclyl-C 1 -C 6 -alkylene-Q),- OH, -N(R 5 ) 2 , SR 5 , -CHO, -C(O)N(R 5 ) 2 , -OC(O)N(R 5 ) 2 , -N(R 5 )C(O)OR 5 , aryl , heteroaryl, -S(O)R 5 ,
- Z 5 is a self-immolative group or absent
- T is a spacer group or absent
- L 1 is absent, -C 1 -C 6 - alkylene (e.g., -C 1 -C 3 - alkylene) or -C 1 -C 6 - heteroalkylene;
- L 2 is absent, -C 1 -C 6 - alkylene (e.g., -C 1 -C 3 - alkylene) or -C 1 -C 6 - heteroalkylene, wherein each alkylene and heteroalkyl is optionally substituted with one or more R 6 ;
- L 3 is absent, -C 1 -C 20 - alkylene, -O-, -N(R 5 )-, -S-, -S(O)-, -S(O) 2 -, -S(O)N(R 5 )-,
- L 4 is -C 1 -C 20 - alkylene (e.g., -C 1 -C 6 - alkylene), -C 1 -C 20 - heteroalkylene (e.g., -C 1 -C 6 - heteroalkylene), -C 1 -C 20 - alkenylene (e.g., -C 2 -C 6 - alkenylene), -C 1 -C 20 - alkynylene (e.g., -C 2 -C 6 - alkynylene), or an oligopeptide, wherein the oligopeptide is optionally substituted by one or more R 16 ;
- L 5 is a linker connecting Z 4 and Z 5 , or is absent;
- Q 1 is C(O), C(S), or CH 2 ; each of R 1 and R 2 is independently hydrogen, halo, -CN, -C 1 -C 20 alkyl (e.g., C 1 -C 6 alkyl), or - OR 7 ;
- R 3 is hydrogen, -C 1 -C 20 - alkyl (e.g., -C 1 -C 6 alkyl), -C 1 -C 20 heteroalkyl (e.g., -C 1 -C 6 heteroalkyl), -OC(O)OC 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, heteroalkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R 8 ; each R 4 is independently hydrogen, -C 1 -C 20 alkyl, -O-C 1 -C 20 alkyl, -C 1 -C 20 heteroalkyl, halo, -CN, -NO 2 or -OH;
- R 5 is hydrogen or -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl);
- R 6 is halo, -CN, -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -OR 7 , oxo, cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R 9 ;
- R 7 is hydrogen, -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R 9 ; each R 8 is independently -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C 1 -C 20 heteroalkyl,
- -C(O)-C 1 -C 20 alkyl -OC(O)-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C(O)O-C 1 -C 20 alkyl (e.g ⁇ ,
- -C 1 -C 6 alkyl -OC(O)O-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C(O)N(R 5 )-C 1 -C 20 alkyl (e.g ⁇ ,
- -C 1 -C 6 alkyl -N(R 5 )C(O)-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -OC(O)N(R 5 )-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -O-aryl, -O-heteroaryl, -C(O)-aryl, -C(O)-heteroaryl, - OC(O)-aryl,
- each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R 9 ; each R 9 is independently -C 1 -C 20 alkyl, -O-C 1 -C 20 alkyl, -C 1 -C 20 heteroalkyl, halo,
- each R 16 is independently, -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C 1 -C 20 heteroalkyl (e.g., -C 1 -C 6 heteroalkyl), -OC(O)OC 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), C(O)N(R 4 ) 2 cycloalkyl, heterocyclyl, aryl, or heteroaryl.
- -C 1 -C 20 alkyl e.g., -C 1 -C 6 alkyl
- -C 1 -C 20 heteroalkyl e.g., -C 1 -C 6 heteroalkyl
- -OC(O)OC 1 -C 20 alkyl e.g., -C 1 -C 6 alkyl
- C(O)N(R 4 ) 2 cycloalkyl, heterocyclyl, ary
- the antibody-drug conjugate is represented by formula (Il-a),
- each of B 1 or B 2 is selected from a naturally occurring nucleobase or a modified nucleobase. In some embodiments, each of B 1 or B 2 is selected from adenosinyl, guanosinyl, cytosinyl, thyminyl, uracilyl, 5’-methylcytosinyl, 5’-fluorouracilyl, 5’- propynyluracilyl, and 7-deazaadenosinyl. In some embodiments, each ofB 1 or B 2 is selected from:
- one of B 1 or B 2 is selected from a naturally occurring nucleobase and the other of B 1 or B 2 is a modified nucleobase.
- one of B 1 or B 2 is adenosinyl, guanosinyl, thyminyl, cytosinyl, or uracilyl, and the other of B 1 or B 2 is 5’- methylcytosinyl, 5’-fluorouracilyl, 5’-propynyluracilyl, or 7-deazaadenosinyl.
- B 1 or B 2 is a purinyl nucleobase. In some embodiments, each of B 1 or B 2 is independently a purinyl nucleobase. In some embodiments, B 1 is a purinyl nucleobase. In some embodiments, B 2 is a pyrimidinyl nucleobase. In some embodiments, B 1 is a purinyl nucleobase and B 2 is a pyrimidinyl nucleobase.
- B 1 is adenosinyl or guanosinyl.
- B 2 is cytosinyl, thyminyl, or uracilyl.
- B 1 is adenosinyl or guanosinyl and B 2 is cytosinyl, thyminyl, or uracilyl.
- each of B 1 and B 2 is independently uracilyl.
- each of B 1 and B 2 is independently adenosinyl.
- each of R 1 and R 2 is independently hydrogen, halo, or OR 7 . In some embodiments, each of R 1 and R 2 is independently halo (e.g., fluoro). In some embodiments, each of R 1 and R 2 is not hydrogen or OR 7 .
- X 1 is O. In some embodiments, X 2 is O. In some embodiments, each of X 1 and X 2 is independently O.
- Y 1 is O or S. In some embodiments, Y 2 is O or S. In some embodiments, each of Y 1 and Y 2 is independently O or S. In some embodiments, one of Y 1 or Y 2 is O and the other of Y 1 or Y 2 is S. In some embodiments, each of Y 1 or Y 2 is independently S. In some embodiments, each of Y 1 or Y 2 is independently O.
- R 3 is hydrogen, aryl, or heteroaryl, wherein aryl and heteroaryl is optionally substituted with 1-5 R 8 .
- R 3 is aryl or heteroaryl, each of which is optionally substituted with 1-5 R 8 .
- R 3 is phenyl substituted with 1 R 8 .
- R 4 is hydrogen.
- each of Y 1 and Y 2 is O and R 3 is hydrogen. In some embodiments, each of Y 1 and Y 2 is independently S and R 3 is substituted with 1 R 8 . In some embodiments, Y 1 is S and R 3 is substituted with 1 R 8 .
- each R 8 is independently C 1 -C 20 alkyl (e.g., C 1 -C 6 alkyl), C 1 -C 20 heteroalkyl, C(O)-C 1 -C 20 alkyl, OC(O)-C 1 -C 20 alkyl, OC(O)O-C 1 -C 20 alkyl, OC(O)N(R 5 )-C 1 - C 20 alkyl, O-aryl, C(O)-aryl, OC(O)-aryl, or C(O)N(R 5 )-aryl, wherein each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R 9 .
- each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R 9 .
- R 8 is OC(O)-aryl optionally substituted by 1-5 R 9 (e.g., 1 R 9 ).
- R 9 is O-C 1 -C 12 alkyl (e.g., O-CH 2 (CH 2 ) 8 CH 3 ). In some embodiments, R 9 is O-C 1 -C 10 alkyl (e.g., O-CH 2 (CH 2 ) 8 CH 3 ). In some embodiments, R 9 is O- C 1 -C 6 alkyl (e.g., O-CH 2 (CH 2 ) 8 CH 3 ). In some embodiments, R 9 is O-C 1 -C 6 alkyl (e.g., O- CH 2 (CH 2 ) 4 CH 3 ).
- R 16 is independently, -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C 1 -C 20 heteroalkyl (e.g., -C 1 -C 6 heteroalkyl), -OC(O)OC 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), C(O)N(R 4 ) 2 cycloalkyl, heterocyclyl, aryl, or heteroaryl.
- -C 1 -C 20 alkyl e.g., -C 1 -C 6 alkyl
- -C 1 -C 20 heteroalkyl e.g., -C 1 -C 6 heteroalkyl
- -OC(O)OC 1 -C 20 alkyl e.g., -C 1 -C 6 alkyl
- C(O)N(R 4 ) 2 cycloalkyl cycloalkyl
- each of L 1 and L 2 is independently C 1 -C 6 alkylene (e.g., C 1 alkylene) or absent.
- L 3 is absent, -N(H)-, -O(O)C-, or -O-. In some embodiments, L 3 is -C(O)-.
- L 4 is an -C 1 -C 20 - alkylene (e.g., -C 1 -C 18 - alkylene), or oligiopeptide.
- L 4 is an oligopeptide.
- the oligopeptide is a dipeptide.
- the oligopeptide comprises 3 amino acid residues, 4 amino acid residues, 5 amino acid residues, 6 amino acid residues, 7 amino acid residues, 8 amino acid residues, 9 amino acid residues, or 10 amino acid residues.
- the oligopeptide comprises 8 amino acid residues.
- the oligopeptide comprises an amino acid selected from Tyr, Ser, Thr, Arg, Leu, lie, Gly, Val, and Ala. In some embodiments, the oligopeptide comprises an amino acid selected from Phe, Tyr, Arg, Gly, Ser, and Leu. In some embodiments, L 4 is In other embodiments, L 4 is
- L 4 is selected from the group consisting of and In some embodiments, L 4 is
- L 4 -C 1 -C 20 - alkylene e.g., -C 1 -C 18 - alkylene.
- L 4 is -C 1 -C 20 - heteroalkylene (e.g., -C 1 -C 18 - heteroalkylene).
- L 4 is substituted with one or more instances of R 10 .
- R 10 is C(O)NH 2 .
- L 5 is -C 1 -C 40 - alkylene (e.g., -C 1 -C 20 - alkylene), -C 1 -C 40 - heteroalkylene (e.g., -C 1 -C 20 - heteroalkyl), -C 1 -C 40 - alkenylene (e.g., -C 2 -C 20 - alkenylene), or -C 1 -C 40 - alkynylene (e.g., -C 2 -C 20 - alkynylene).
- L5 is an oligopeptide comprising of 1-40 amino acid residues.
- L 5 further further comprises one sub-unit.
- L 5 further comprises one sub-unit.
- L 5 further comprises one sub-unit.
- L 5 further comprises one sub-unit.
- L 5 further comprises one sub-unit.
- each Z 1 is oxygen
- Z 2 is -aryl-. In some embodiments, Z 2 is -OC(O)OC 1 -C 20 - alkylene (e.g., -C 1 -C 6 - alkylene).
- Z 3 is -aryl-. In some embodiments, Z 3 is -OC(O)OC 1 -C 20 - alkylene (e.g., -C 1 -C 6 - alkylene). In some embodiments, Z 3 is aryl substituted with -OC 1 -C 20 - heteroalkylene (e.g., -C 1 -C 12 -heteroalkylene). In some embodiments, Z 3 is aryl substituted with
- Z 4 is Heterocyclyl-C 1 -C 20 - alkylene-Q. In some embodiments,
- Z 4 is or In some embodiments, Q is C(O).
- Z 4 is -0-, -N(R 5 )- or -heteroaryl-.
- Z 5 is In some embodiments, Z 5 is In some embodiments, Z 5 is In some embodiments, Z 5 is In some embodiments, Z 5 is In some embodiments, Z 5 is In some embodiments, Z 5 is In some embodiments, Z 5 is In some embodiments, Z 5 is in some embodiments, Z 5 is
- Z 5 is represented by formula (III-a), (III-b), or (III-c): wherein:
- U is O, S or NR 13 ;
- Q 2 is CR 13 or N;
- each V 1 , V 2 and V 3 are independently CR 17 or N provided that for formula (Ill-b) and (III-c) at least one of Q 2 , V 1 and V 2 is N;
- R 11 is the side chain of an amino acid and is optionally protected with a protecting group
- Each R 10 , R 14 , and R 15 are independently selected from hydrogen, halo, OH, -N(R 12 ) 2 , - N(R 12 ) 3 + , C 1 -C 8 heteroalkyl, carboxylate, sulfate, sulfamate, sulfonate, -SO 2 R 5 , - S(O)R 12 ,
- R 12 and R 13 are independently selected from hydrogen, C 1 -C 8 alkyl, C 2 -C 8 alkenyl, C 2 -C 8 alkynyl, aryl, heterocycle, wherein each alkyl, alkenyl, alkynyl, aryl, and heterocycle are independently substituted with one or more substituents selected from halo, OH, - N(R 12 ) 2 , -N(R 12 ) 3 +, heteroalkyl, carboxylate, sulfate, sulfamate, sulfonate, 4- dialkylaminopyridinium, alkyl, -SO 2 R 12 , -S(O)R 12 , -SR 12 , -SO 2 N(R 12 ) 2 , C(O)R 12 , CO 2 R 12 , C(O)N(R 12 ) 2 , -CN, -N 3 , -NO 2 , C 3 -C 12 carbocycle, ary
- Q 2 is N; V 1 is CH, and V 2 is CH. In some embodiments, Q 2 is CH; V 1 is CH and V 2 is N. In some embodiments, Q 2 is N; V 1 is CH, and V 2 is N. In some embodiments, Q 2 is N; and V 1 is N and V 2 is N. In some embodiments, Q 2 is N; V 1 is N and V 2 is CH. In some embodiments, Q 2 is N; and V 1 , V 2 and V 3 are each CH. In some embodiments, Q 2 is CH; and V 1 , V 2 and V 3 are each CH. In some embodiments, Q 2 is CH; and V 1 , V 2 and V 3 are each CH.
- T is absent, -C 1 -C 20 - alkylene (e.g., -C 1 -C 6 - alkylene), -C 1 -C 20 - heteroalkylene (e.g., -C 1 -C 6 - heteroalkylene), -C 1 -C 20 - alkenylene (e.g., -C 2 -C 6 - alkenylene), - C 1 -C 20 - alkynylene (e.g., -C 2 -C 6 - alkynyl), -aryl- or -heteroaryl-.
- -C 1 -C 20 - alkylene e.g., -C 1 -C 6 - alkylene
- -C 1 -C 20 - heteroalkylene e.g., -C 1 -C 6 - heteroalkylene
- -C 1 -C 20 - alkenylene e.g., -C
- anti -PD- 1 antibody or anti-CTLA-4 antibody is a monoclonal antibody, a domain antibody (dAb), a single chain antibody (scAb), a Fab fragment, a F(ab’) 2 fragment, a single chain variable fragment (scFv), a scFv-Fc fragment, a single domain heavy chain antibody, a single domain light chain antibody, a variant antibody, a multimeric antibody, or a bispecific antibody.
- the antibody is an anti -PD- 1 antibody.
- the anti -PD- 1 antibody is pembrolizumab, nivolumab, cemiplimab, Anti -mouse PD-1 antibody Clone J43, Anti-mouse PD-1 antibody Clone RMP1-14, mouse anti -PD- 1 antibody Clone EH 12, ANB011, MDX-1 106, AMP-514, AMP-224, or Pidilizumab.
- the anti-PD-1 antibody is pembrolizumab or nivolumab.
- the antibody is an anti-CTLA-4 antibody.
- the anti-CTLA-4 antibody is ipilimumab, clone 9H10, tremelimumab, or clone BNI3.
- the antibody-drug conjugate is selected from Table 1:
- the disclosure features an antibody-drug conjugate of Formula (IV): or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
- Ab is an anti-PD-1 antibody or an anti-CTLA-4 antibody; each of B 1 and B 2 is independently a purinyl nucleobase or pyrimidinyl nucleobase; each of X 1 and X 2 is independently O or S; each of Y 1 and Y 2 is independently O, S, or N(R 5 ); each of Z 1 is independently O or S;
- Z 2 is -O-, -N(R 5 )-, -S-, -C(O)-, -C(O)N(R 5 )-, -OC(O)N(R 5 )-, -N(R 5 )C(O)O-, -aryl-, -heteroaryl-, -S(O)-, -S(O) 2 -, -S(O)N(R 5 )-, -S(O) 2 N(R 5 )- or -N(R 5 )S(O)-;
- Z 3 is absent, hydrogen, heterocyclyl, heterocyclyl-C 1 -C 20 -alkylene-Q 1 , -OH, -N(R 5 ) 2 , SR 5 , - CHO, -C(O)N(R 5 ) 2 , -OC(O)N(R 5 ) 2 , -N(R 5 )C(O)OR 5 , aryl , heteroaryl, -S(O)R 5 , - S(O) 2 R 5 , -S(O)N(R 5 ) 2 , -S(O) 2 N(R 5 ) 2 , -N(R 5 )S(O)R 5 , -OSi(C 1 -C 4 alkyl) 3 , or -C(O)C 2 - C 6 alkenyl (e.g., -C 2 -C 4 alkenyl);
- Z 4 is a self-immolative group or absent
- T is a absent or spacer group; each L 1 and L 2 is absent, -C 1 -C 6 - alkylene (e.g., -C 1 -C 3 - alkylene) or -C 1 -C 6 - heteroalkyl ene, wherein each alkylene and heteroalkyl is optionally substituted with one or more R 6 ; L 3 is oligiopeptide-C(O)-, oligiopeptide-aryl-C 1 -C 6 -alkylene-, oligiopeptide-aryl-C 1 -C 6 - alklyene-C(O)-, oligiopeptide-aryl-C 1 -C 6 -heteroalkylene, oligiopeptide-C 1 -C 6 - alkylene-C(O)-, oligiopeptide-C 1 -C 6 -heteroalkylene-C(O)-, oligiopeptide-C 1 -C 6 -heteroal
- L 4 is absent or a linker connecting Z 3 and Z 4 ;
- Q 1 is C(O), C(S), or CH 2 ; each of R 1 and R 2 is independently hydrogen, halo, -CN, -C 1 -C 20 alkyl (e.g., C 1 -C 6 alkyl), or - OR 7 ; each R 3 and R 4 is independently hydrogen, -C 1 -C 20 - alkyl (e.g., -C 1 -C 6 alkyl), -C 1 -C 20 heteroalkyl (e.g., -C 1 -C 6 heteroalkyl), -OC(O)OC 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, heteroalkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R 8 ;
- R 5 is hydrogen or -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl);
- R 6 is halo, -CN, -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -OR 7 , oxo, cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R 9 ;
- R 7 is hydrogen, -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R 9 ; each R 8 is independently -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C 1 -C 20 heteroalkyl,
- -C(O)-C 1 -C 20 alkyl -OC(O)-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C(O)O-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -OC(O)O-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C(O)N(R 5 )-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -N(R 5 )C(O)-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -OC(O)N(R 5 )- C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -O-aryl, -O-hetero
- each R 16 is independently, -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C 1 -C 20 heteroalkyl (e.g., -C 1 -C 6 heteroalkyl), -OC(O)OC 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), C(O)N(R 4 ) 2 cycloalkyl, heterocyclyl, aryl, or heteroaryl.
- -C 1 -C 20 alkyl e.g., -C 1 -C 6 alkyl
- -C 1 -C 20 heteroalkyl e.g., -C 1 -C 6 heteroalkyl
- -OC(O)OC 1 -C 20 alkyl e.g., -C 1 -C 6 alkyl
- C(O)N(R 4 ) 2 cycloalkyl, heterocyclyl, ary
- the antibody-drug conjugate is an antibody-drug conjugate of formula (V-e), (V-f), (V-g), (V-h), (V-i) or (V-j):
- W 1 is N(R 5 ), -O- CH; and W 2 is N or CH.
- each of B 1 or B 2 is selected from a naturally occurring nucleobase or a modified nucleobase. In some embodiments, each of B 1 or B 2 is selected from adenosinyl, guanosinyl, cytosinyl, thyminyl, uracilyl, 5’ -methyl cytosinyl, 5’- fluorouracilyl, 5’-propynyluracilyl, and 7-deazaadenosinyl. In some embodiments, each of B 1 or B 2 is selected from: wherein indicates the linkage of the nucleobase to the ribose ring.
- one of B 1 or B 2 is selected from a naturally occurring nucleobase and the other of B 1 or B 2 is a modified nucleobase.
- one of B 1 or B 2 is adenosinyl, guanosinyl, thyminyl, cytosinyl, or uracilyl, and the other of B 1 or B 2 is 5’- methylcytosinyl, 5’-fluorouracilyl, 5’-propynyluracilyl, or 7-deazaadenosinyl.
- B 1 is adenosinyl or guanosinyl.
- B 2 is cytosinyl, thyminyl, or uracilyl.
- B 1 is adenosinyl or guanosinyl and B 2 is cytosinyl, thyminyl, or uracilyl.
- each of B 1 and B 2 is independently uracilyl.
- each of B 1 and B 2 is independently adenosinyl.
- each of R 1 and R 2 is independently hydrogen, halo, or OR 7 . In some embodiments, each of R 1 and R 2 is independently halo (e.g., fluoro). In some embodiments, each of R 1 and R 2 is not hydrogen or OR 7 .
- X 1 is O. In some embodiments, X 2 is O. In some embodiments, each of X 1 and X 2 is independently O.
- Y 1 is O or S. In some embodiments, Y 2 is O or S. In some embodiments, each of Y 1 and Y 2 is independently O or S. In some embodiments, one of Y 1 or Y 2 is O and the other of Y 1 or Y 2 is S. In some embodiments, each of Y 1 or Y 2 is independently S. In some embodiments, each of Y 1 or Y 2 is independently O.
- L 1 is C 1 -C 6 alkyl (e.g., CH 2 ). In some embodiments, L 2 is C 1 - C 6 alkyl (e.g., CH 2 ). In some embodiments, each of L 1 and L 2 is independently C 1 -C 6 alkyl (e.g., CH 2 ).
- R 3 is hydrogen, aryl, or heteroaryl, wherein aryl and heteroaryl is optionally substituted with 1-5 R 8 . In some embodiments, R 3 is aryl or heteroaryl, each of which is optionally substituted with 1-5 R 8 . In some embodiments, R 3 is phenyl substituted with 1 R 8 .
- R 4 is independently hydrogen, aryl, or heteroaryl, wherein aryl and heteroaryl is optionally substituted with 1-5 R 8 . In some embodiments, R 4 is aryl or heteroaryl, each of which is optionally substituted with 1-5 R 8 . In some embodiments, R 4 is phenyl substituted with 1 R 8 .
- each of R 3 and R 4 is independently hydrogen, aryl, or heteroaryl, wherein aryl and heteroaryl is optionally substituted with 1-5 R 8 .
- R 3 is aryl or heteroaryl, each of which is optionally substituted with 1-5 R 8 , and R 4 is hydrogen.
- R 3 is phenyl substituted with 1 R 8 and R 4 is hydrogen.
- each of R 3 and R 4 is independently phenyl substituted with 1 R 8 .
- each of Y 1 and Y 2 is O and each of R 3 and R 4 is independently hydrogen. In some embodiments, Y 2 is O and R 4 is hydrogen. In some embodiments, each of Y 1 and Y 2 is independently S and each of R 3 and R 4 is independently substituted with 1 R 8 . In some embodiments, Y 1 is S and R 3 is substituted with 1 R 8 .
- each R 8 is independently C 1 -C 20 alkyl (e.g., C 1 -C 6 alkyl), C 1 -C 20 heteroalkyl, C(O)-C 1 -C 20 alkyl, OC(O)-C 1 -C 20 alkyl, OC(O)O-C 1 -C 20 alkyl, OC(O)N(R 5 )-C 1 - C 20 alkyl, O-aryl, C(O)-aryl, OC(O)-aryl, or C(O)N(R 5 )-aryl, wherein each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R 9 .
- each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R 9 .
- R 8 is OC(O)-aryl optionally substituted by 1-5 R 9 (e.g., 1 R 9 ).
- R 9 is O-C 1 -C 12 alkyl (e.g., O-CH 2 (CH 2 ) 8 CH 3 ).
- R 9 is O-C 1 -C 10 alkyl (e.g., O-CH 2 (CH 2 ) 8 CH 3 ).
- R 9 is O-C 1 -C 8 alkyl (e.g., O-CH 2 (CH 2 ) 6 CH 3 ).
- R 9 is O-C 1 -C 6 alkyl (e.g., O-CH 2 (CH 2 )4CH 3 ).
- each of L 1 and L 2 is independently C 1 -C 6 alkylene (e.g., C 1 alkylene).
- L 3 is -C 1 -C 20 - alkylene (e.g., -C 1 -C 18 - alkylene). In some embodiments, L 3 is -C 1 -C 20 - heteroalkylene (e.g., -C 1 -C 18 - heteroalkylene).
- L 3 is an oligiopeptide-C(O)-, oligiopeptide-aryl-C 1 -C 6 -alkylene-, oligiopeptide- aryl-C 1 -C 6 -heteroalkylene, oligiopeptide-aryl-C 1 -C 6 -alklyene-C(O)-, oligiopeptide-C 1 -C 6 - alkylene-C(O)-, oligiopeptide-C 1 -C 6 -heteroalkylene-C(O)-.
- the aryl is phenyl.
- the oligiopeptide is a dipeptide.
- the oligopeptide comprises 3 amino acid residues, 4 amino acid residues, 5 amino acid residues, 6 amino acid residues, 7 amino acid residues, 8 amino acid residues, 9 amino acid residues, or 10 amino acid residues. In some embodiments, the oligopeptide comprises 8 amino acid residues. In some embodiments, the oligopeptide comprises an amino acid selected from Tyr, Ser, Thr, Arg, Leu, lie, Gly, Val, and Ala. In some embodiments, he oligopeptide comprises an amino acid selected from Phe, Tyr, Arg, Gly, Ser, and Leu. In some embodiments, L 3 is or In other embodiments, L 3 is In yet other embodiments, L 3 is
- L 3 is
- the oligiopeptide is substituted by one or more instances of R 16 .
- R 16 is C(O)NH 2 .
- L 4 is -C 1 -C 40 - alkylene (e.g., -C 1 -C 20 - alkylene), -C 1 -C 40 - heteroalkylene (e.g., -C 1 -C 20 - heteroalkyl), -C 1 -C 40 - alkenylene (e.g., -C 2 -C 20 - alkenylene), or -C 1 -C 40 - alkynylene (e.g., -C 2 -C 20 - alkynylene).
- L 4 is an oligopeptide comprising of 1 -40 amino acid residues.
- L 4 further comprises one sub-units.
- L 4 further comprises one sub- unit.
- L 4 further comprises one sub-unit.
- L 4 further comprises one sub-unit.
- L 4 further comprises one sub-unit.
- L 4 further comprises one sub-unit.
- L 4 further comprises one sub-unit.
- L 4 further comprises one sub-unit
- each Z 1 is oxygen.
- Z 3 is -0-, -N(R 5 )- or -heteroaryl-.
- Z 3 is In some embodiments, Z 3 is In some embodiments, Q 1 is C(O).
- Z 4 is In some embodiments, Z 4 is In some embodiments, Z 4 is In some embodiments, Z 4 is In some embodiments, Z 4 is . In some embodiments, Z 4 is In some embodiments, Z 4 is wherein:
- U is O, S or NR 13 ;
- Q is CR 13 orN; each V 1 , V 2 and V 3 are independently CR 17 or N provided that for formula (Ill-b) and (III-c) at least one of Q, V 1 and V 2 is N;
- R 11 is the side chain of an amino acid and is optionally protected with a protecting group;
- Each R 10 , R 14 , and R 15 are independently selected from hydrogen, halo, OH, -N(R 12 ) 2 , - N(R 12 ) 3 + , C 1 -C 8 heteroalkyl, carboxylate, sulfate, sulfamate, sulfonate, -SO 2 R 5 , - S(O)R 12 ,
- R 12 and R 13 are independently selected from hydrogen, C 1 -C 8 alkyl, C 2 -C 8 alkenyl, C 2 -C 8 alkynyl, aryl, heterocycle, wherein each alkyl, alkenyl, alkynyl, aryl, and heterocycle are independently substituted with one or more substituents selected from halo, OH, - N(R 12 ) 2 , -N(R 12 ) 3 +, heteroalkyl, carboxylate, sulfate, sulfamate, sulfonate, 4- dialkylaminopyridinium, alkyl, -SO 2 R 12 , -S(O)R 12 , -SR 12 , -SO 2 N(R 12 ) 2 , C(O)R 12 , CO 2 R 12 , C(O)N(R 12 ) 2 , -CN, -N 3 ,
- Q is N; V 1 is CH, and V 2 is CH. In some embodiments, Q is CH; V 1 is CH and V 2 is N. In some embodiments, Q is N; V 1 is CH, and V 2 is N. In some embodiments, Q is N; and V 1 is N and V 2 is N. In some embodiments, Q is N; V 1 is N and V 2 is CH. In some embodiments, Q is N; and V 1 , V 2 and V 3 are each CH. In some embodiments, Q is CH; and V 1 , V 2 and V 3 are each CH. In some embodiments, Z 4 is absent.
- T is -C 1 -C 20 - alkylene (e.g., -C 1 -C 6 - alkylene), -C 1 -C 20 - heteroalkylene (e.g., -C 1 -C 6 - heteroalkylene), -C 1 -C 20 - alkenylene (e.g., -C 2 -C 6 - alkenylene), - C 1 -C 20 - alkynylene (e.g., -C 2 -C 6 - alkynyl), -aryl- or -heteroaryl-.
- T is absent.
- anti -PD- 1 antibody or anti-CTLA-4 antibody is a monoclonal antibody, a domain antibody (dAb), a single chain antibody (scAb), a Fab fragment, a F(ab’) 2 fragment, a single chain variable fragment (scFv), a scFv-Fc fragment, a single domain heavy chain antibody, a single domain light chain antibody, a variant antibody, a multimeric antibody, or a bispecific antibody.
- the antibody is an anti -PD- 1 antibody.
- the anti -PD- 1 antibody is pembrolizumab, nivolumab, cemiplimab, Anti -mouse PD-1 antibody Clone J43, Anti-mouse PD-1 antibody Clone RMP1-14, mouse anti-PD-1 antibody Clone EH12, ANB011, MDX-1 106, AMP-514, AMP-224, or Pidilizumab.
- the anti-PD-1 antibody is pembrolizumab or nivolumab.
- the antibody is an anti-CTLA-4 antibody.
- the anti-CTLA-4 antibody is ipilimumab, clone 9H10, tremelimumab, or clone BNI3.
- the antibody-drug conjugate is selected from Table 2:
- Ab is an anti -PD- 1 antibody or an anti- CTLA-4 antibody.
- the disclosure features an antibody-drug conjugate of Formula (Vlla) or Formula (Vllb):
- Ab is an anti-PD-1 antibody or an anti-CTLA-4 antibody; each of B 1 and B 2 is independently a purinyl nucleobase or pyrimidinyl nucleobase; each of X 1 and X 2 is independently O or S; each of Y 1 and Y 2 is independently O, S, or N(R 5 ); each of Z 1 is independently O or S;
- Z 2 is -O-, -N(R 5 )-, -S-, -C(O)-, -C(O)N(R 5 )-, -OC(O)N(R 5 )-, -N(R 5 )C(O)O-, -aryl-, -heteroaryl-, -S(O)-, -S(O) 2 -, -S(O)N(R 5 )-, -S(O) 2 N(R 5 )- or -N(R 5 )S(O)-;
- Z 3 is absent, hydrogen, heterocyclyl, heterocyclyl-C 1 -C 20 -alkylene-Q 1 , -OH, -N(R 5 ) 2 , SR 5 , - CHO, -C(O)N(R 5 ) 2 , -OC(O)N(R 5 ) 2 , -N(R 5 )C(O)OR 5 , aryl, heteroaryl, -S(O)R 5 , - S(O) 2 R 5 , -S(O)N(R 5 ) 2 , -S(O) 2 N(R 5 ) 2 , -N(R 5 )S(O)R 5 , -OSi(C 1 -C 4 alkyl) 3 , or -C(O)C 2 - C 6 alkenyl (e.g., -C 2 -C 4 alkenyl);
- Z 4 is a self-immolative group or absent
- T is a absent or spacer group; each L 1 and L 2 is absent, -C 1 -C 6 - alkylene (e.g., -C 1 -C 3 - alkylene) or -C 1 -C 6 - heteroalkyl ene, wherein each alkylene and heteroalkyl is optionally substituted with one or more R 6 ;
- L 3 is oligiopeptide-C(O)-, oligiopeptide-aryl-C 1 -C 6 -alkylene-, oligiopeptide-aryl-C 1 -C 6 - alklyene-C(O)-, oligiopeptide-aryl-C 1 -C 6 -heteroalkylene, oligiopeptide-C 1 -C 6 - alkylene-C(O)-, oligiopeptide-C 1 -C 6 -heteroalkylene-C(O)-, -C 1 -C 40 - alkylene (e.g., - C 1 -C 20 - alkylene), -C 1 -C 40 - heteroalkyl ene (e.g., -C 1 -C 20 - heteroalkyl), -C 1 -C 40 - alkenylene (e.g., -C 2 -C 20 - alkenylene), or -C 1 -
- L 4 is absent or a linker connecting Z 3 and Z 4 ;
- Q 1 is C(O), C(S), or CH 2 ;
- each of R 1 and R 2 is independently hydrogen, halo, -CN, -C 1 -C 20 alkyl (e.g., C 1 -C 6 alkyl), or - OR 7 ;
- each R 3 and R 4 is independently hydrogen, -C 1 -C 20 - alkyl (e.g., -C 1 -C 6 alkyl), -C 1 -C 20 heteroalkyl (e.g., -C 1 -C 6 heteroalkyl), -OC(O)OC 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, heteroalkyl, cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each
- R 5 is hydrogen or -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl);
- R 6 is halo, -CN, -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -OR 7 , oxo, cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R 9 ;
- R 7 is hydrogen, -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R 9 ; each R 8 is independently -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C 1 -C 20 heteroalkyl,
- -C(O)-C 1 -C 20 alkyl -OC(O)-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C(O)O-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -OC(O)O-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C(O)N(R 5 )-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -N(R 5 )C(O)-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -OC(O)N(R 5 )- C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -O-aryl, -O-hetero
- each R 16 is independently, -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C 1 -C 20 heteroalkyl (e.g., -C 1 -C 6 heteroalkyl), -OC(O)OC 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), C(O)N(R 4 ) 2 cycloalkyl, heterocyclyl, aryl, or heteroaryl.
- -C 1 -C 20 alkyl e.g., -C 1 -C 6 alkyl
- -C 1 -C 20 heteroalkyl e.g., -C 1 -C 6 heteroalkyl
- -OC(O)OC 1 -C 20 alkyl e.g., -C 1 -C 6 alkyl
- C(O)N(R 4 ) 2 cycloalkyl, heterocyclyl, ary
- the antibody-drug conjugate is an antibody-drug conjugate of formula (VllI-a), (VllI-b), (VIII-c), (VllI-d), (VllI-e), (VllI-f), (VIII-g), or (Vlll-h):
- each of B 1 or B 2 is selected from a naturally occurring nucleobase or a modified nucleobase. In some embodiments, each of B 1 or B 2 is selected from adenosinyl, guanosinyl, cytosinyl, thyminyl, uracilyl, 5’-methylcytosinyl, 5’-fluorouracilyl, 5’- propynyluracilyl, and 7-deazaadenosinyl. In some embodiments, each of B 1 or B 2 is selected from: wherein ndicates the linkage of the nucleobase to the ribose ring.
- one of B 1 or B 2 is selected from a naturally occurring nucleobase and the other of B 1 or B 2 is a modified nucleobase.
- one of B 1 or B 2 is adenosinyl, guanosinyl, thyminyl, cytosinyl, or uracilyl, and the other of B 1 or B 2 is 5’- methylcytosinyl, 5’-fluorouracilyl, 5’-propynyluracilyl, or 7-deazaadenosinyl.
- B 1 is adenosinyl or guanosinyl.
- B 2 is cytosinyl, thyminyl, or uracilyl.
- B 1 is adenosinyl or guanosinyl and B 2 is cytosinyl, thyminyl, or uracilyl.
- each of B 1 and B 2 is independently uracilyl.
- each of B 1 and B 2 is independently adenosinyl.
- each of R 1 and R 2 is independently hydrogen, halo, or OR 7 . In some embodiments, each of R 1 and R 2 is independently halo (e.g., fluoro). In some embodiments, each of R 1 and R 2 is not hydrogen or OR 7 .
- X 1 is O. In some embodiments, X 2 is O. In some embodiments, each of X 1 and X 2 is independently O.
- Y 1 is O or S. In some embodiments, Y 2 is O or S. In some embodiments, each of Y 1 and Y 2 is independently O or S. In some embodiments, one of Y 1 or Y 2 is O and the other of Y 1 or Y 2 is S. In some embodiments, each of Y 1 or Y 2 is independently S. In some embodiments, each of Y 1 or Y 2 is independently O.
- L 1 is C 1 -C 6 alkyl (e.g., CH 2 ). In some embodiments, L 2 is C 1 - C 6 alkyl (e.g., CH 2 ). In some embodiments, each of L 1 and L 2 is independently C 1 -C 6 alkyl (e.g., CH 2 ).
- R 3 is hydrogen, aryl, or heteroaryl, wherein aryl and heteroaryl is optionally substituted with 1-5 R 8 . In some embodiments, R 3 is aryl or heteroaryl, each of which is optionally substituted with 1-5 R 8 . In some embodiments, R 3 is phenyl substituted with 1 R 8 .
- R 4 is independently hydrogen, aryl, or heteroaryl, wherein aryl and heteroaryl is optionally substituted with 1-5 R 8 . In some embodiments, R 4 is aryl or heteroaryl, each of which is optionally substituted with 1-5 R 8 . In some embodiments, R 4 is phenyl substituted with 1 R 8 .
- each of R 3 and R 4 is independently hydrogen, aryl, or heteroaryl, wherein aryl and heteroaryl is optionally substituted with 1-5 R 8 .
- R 3 is aryl or heteroaryl, each of which is optionally substituted with 1-5 R 8
- R 4 is hydrogen.
- R 3 is phenyl substituted with 1 R 8 and R 4 is hydrogen.
- each of R 3 and R 4 is independently phenyl substituted with 1 R 8 .
- each of Y 1 and Y 2 is O and each of R 3 and R 4 is independently hydrogen.
- Y 2 is O and R 4 is hydrogen.
- each of Y 1 and Y 2 is independently S and each of R 3 and R 4 is independently substituted with 1 R 8 .
- Y 1 is S and R 3 is substituted with 1 R 8 .
- each R 8 is independently C 1 -C 20 alkyl (e.g., C 1 -C 6 alkyl), C 1 -C 20 heteroalkyl, C(O)-C 1 -C 20 alkyl, OC(O)-C 1 -C 20 alkyl, OC(O)O-C 1 -C 20 alkyl, OC(O)N(R 5 )-C 1 - C 20 alkyl, O-aryl, C(O)-aryl, OC(O)-aryl, or C(O)N(R 5 )-aryl, wherein each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R 9 .
- each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R 9 .
- R 8 is OC(O)-aryl optionally substituted by 1-5 R 9 (e.g., 1 R 9 ).
- R 9 is O-C 1 -C 12 alkyl (e.g., O-CH 2 (CH 2 ) 8 CH 3 ).
- R 9 is O-C 1 -C 10 alkyl (e.g., O-CH 2 (CH 2 ) 8 CH 3 ).
- R 9 is O-C 1 -C 8 alkyl (e.g., O-CH 2 (CH 2 ) 6 CH 3 ).
- R 9 is O-C 1 -C 6 alkyl (e.g., O-CH 2 (CH 2 )4CH 3 ).
- each of L 1 and L 2 is independently C 1 -C 6 alkylene (e.g., C 1 alkylene).
- L 3 is -C 1 -C 20 - alkylene (e.g., -C 1 -C 18 - alkylene). In some embodiments, L 3 is -C 1 -C 20 - heteroalkylene (e.g., -C 1 -C 18 - heteroalkylene).
- L 3 is an oligiopeptide-C(O)-, oligiopeptide-aryl-C 1 -C 6 -alkylene-, oligiopeptide- aryl-C 1 -C 6 -heteroalkylene, oligiopeptide-aryl-C 1 -C 6 -alklyene-C(O)-, oligiopeptide-C 1 -C 6 - alkylene-C(O)-, oligiopeptide-C 1 -C 6 -heteroalkylene-C(O)-.
- the aryl is phenyl.
- the oligiopeptide is a dipeptide.
- the oligopeptide comprises 3 amino acid residues, 4 amino acid residues, 5 amino acid residues, 6 amino acid residues, 7 amino acid residues, 8 amino acid residues, 9 amino acid residues, or 10 amino acid residues. In some embodiments, the oligopeptide comprises 8 amino acid residues. In some embodiments, the oligopeptide comprises an amino acid selected from Tyr, Ser, Thr, Arg, Leu, lie, Gly, Val, and Ala. In some embodiments, he oligopeptide comprises an amino acid selected from Phe, Tyr, Arg, Gly, Ser, and Leu.
- L 3 is or In other embodiments, L 3 is In yet other embodiments, L 3 is selected from the group consisting of and In some embodiments, L 3 is In some embodiments, the oligiopeptide is substituted by one or more instances of R 16 . In some embodiments, R 16 is C(0)NH 2 .
- L 4 is -C 1 -C 40 - alkylene (e.g., -C 1 -C 20 - alkyl ene), -C 1 -C 40 - heteroalkylene (e.g., -C 1 -C 20 - heteroalkyl), -C 1 -C 40 - alkenylene (e.g., -C2-C20- alkenylene), or -C 1 -C 40 - alkynylene (e.g., -C 2 -C 20 - alkynylene).
- L 4 is an oligopeptide comprising of 1-40 amino acid residues.
- L 4 further comprises one sub-units.
- L further comprises one sub unit.
- L 4 further comprises one sub-unit.
- L 4 further comprises one sub-unit.
- L 4 is absent.
- each Z 1 is oxygen
- Z 3 is -0-, -N(R 5 )- or -heteroaryl-. In some embodiments, Z 3 is In some embodiments, Z 3 is . In some embodiments, Q 1 is C(0).
- Z 4 is In some embodiments, Z 4 is In some embodiments, Z 4 is In some embodiments, Z 4 is In some embodiments, Z 4 is In some embodiments, Z 4 is In some embodiments, Z 4 is In some embodiments, Z 4 is In some embodiments, Z 4 is In some embodiments, Z 4 is represented by formula (IX-a), (IX-b), or (IX-c): wherein:
- U is O, S or NR 13 ;
- Q is CR 13 orN; each V 1 , V 2 and V 3 are independently CR 17 or N provided that for formula (Ill-b) and (III-c) at least one of Q, V 1 and V 2 is N;
- R 11 is the side chain of an amino acid and is optionally protected with a protecting group
- Each R 10 , R 14 , and R 15 are independently selected from hydrogen, halo, OH, -N(R 12 ) 2 , - N(R 12 ) 3 + , C 1 -C 8 heteroalkyl, carboxylate, sulfate, sulfamate, sulfonate, -SO 2 R 5 , - S(O)R 12 ,
- R 12 and R 13 are independently selected from hydrogen, C 1 -Cx alkyl, C 2 -C 8 alkenyl, C 2 -C 8 alkynyl, aryl, heterocycle, wherein each alky
- Q is N; V 1 is CH, and V 2 is CH. In some embodiments, Q is CH; V 1 is CH and V 2 is N. In some embodiments, Q is N; V 1 is CH, and V 2 is N. In some embodiments, Q is N; and V 1 is N and V 2 is N. In some embodiments, Q is N; V 1 is N and V 2 is CH. In some embodiments, Q is N; and V 1 , V 2 and V 3 are each CH. In some embodiments, Q is CH; and V 1 , V 2 and V 3 are each CH. In some embodiments, Q is CH; and V 1 , V 2 and V 3 are each CH.
- Z 4 is absent.
- T is -C 1 -C 20 - alkylene (e.g., -C 1 -C 6 - alkylene), -C 1 -C 20 - heteroalkylene (e.g., -C 1 -C 6 - heteroalkylene), -C 1 -C 20 - alkenylene (e.g., -C 2 -C 6 - alkenylene), - C 1 -C 20 - alkynylene (e.g., -C 2 -C 6 - alkynyl), -aryl- or -heteroaryl-.
- T is absent.
- anti -PD- 1 antibody or anti-CTLA-4 antibody is a monoclonal antibody, a domain antibody (dAb), a single chain antibody (scAb), a Fab fragment, a F(ab’) 2 fragment, a single chain variable fragment (scFv), a scFv-Fc fragment, a single domain heavy chain antibody, a single domain light chain antibody, a variant antibody, a multimeric antibody, or a bispecific antibody.
- the antibody is an anti -PD- 1 antibody.
- the anti -PD- 1 antibody is pembrolizumab, nivolumab, cemiplimab, Anti -mouse PD-1 antibody Clone J43, Anti-mouse PD-1 antibody Clone RMP1-14, mouse anti-PD-1 antibody Clone EH12, ANB011, MDX-1 106, AMP-514, AMP-224, or Pidilizumab.
- the anti-PD-1 antibody is pembrolizumab or nivolumab.
- the antibody is an anti-CTLA-4 antibody.
- the anti-CTLA-4 antibody is ipilimumab, clone 9H10, tremelimumab, or clone BNI3.
- the antibody-drug conjugate is selected from Table 2: Table 3: Exemplary Antibody-Drug Conjugates of the Present Invention
- the disclosure features an antibody-drug conjugate of Formula (Xa) or Formula (Xb): or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
- Ab is an anti-PD-1 antibody or an anti-CTLA-4 antibody; each of B 1 and B 2 is independently a purinyl nucleobase or pyrimidinyl nucleobase; each of X 1 and X 2 is independently O or S; each of Y 1 and Y 2 is independently O, S, or N(R 5 ); each of Z 1 is independently O or S;
- Z 2 is -O-, -N(R 5 )-, -S-, -C(O)-, -C(O)N(R 5 )-, -OC(O)N(R 5 )-, -N(R 5 )C(O)O-, -aryl-, -heteroaryl-, -S(O)-, -S(O) 2 -, -S(O)N(R 5 )-, -S(O) 2 N(R 5 )- or -N(R 5 )S(O)-;
- Z 3 is absent, hydrogen, heterocyclyl, heterocyclyl-C 1 -C 20 -alkylene-Q 1 , -OH, -N(R 5 ) 2 , SR 5 , - CHO, -C(O)N(R 5 ) 2 , -OC(O)N(R 5 ) 2 , -N(R 5 )C(O)OR 5 , aryl , heteroaryl, -S(O)R 5 , - S(O) 2 R 5 , -S(O)N(R 5 ) 2 , -S(O) 2 N(R 5 ) 2 , -N(R 5 )S(O)R 5 , -OSi(C 1 -C 4 alkyl) 3 , or -C(O)C 2 - C 6 alkenyl (e.g., -C 2 -C 4 alkenyl);
- Z 4 is a self-immolative group or absent
- T is a absent or spacer group; each L 1 and L 2 is absent, -C 1 -C 6 - alkylene (e.g., -C 1 -C 3 - alkylene) or -C 1 -C 6 - heteroalkyl ene, wherein each alkylene and heteroalkyl is optionally substituted with one or more R 6 ;
- L 3 is oligiopeptide-C(O)-, oligiopeptide-aryl-C 1 -C 6 -alkylene-, oligiopeptide-aryl-C 1 -C 6 - alklyene-C(O)-, oligiopeptide-aryl-C 1 -C 6 -heteroalkylene, oligiopeptide-C 1 -C 6 - alkylene-C(O)-, oligiopeptide-C 1 -C 6 -heteroalkylene-C(O)-, -C 1 -C 40 - alkylene (e.g., - C 1 -C 20 - alkylene), -C 1 -C 40 - heteroalkyl ene (e.g., -C 1 -C 20 - heteroalkyl), -C 1 -C 40 - alkenylene (e.g., -C 2 -C 20 - alkenylene), or -C 1 -
- L 4 is absent or a linker connecting Z 3 and Z 4 ;
- Q 1 is C(O), C(S), or CH 2 ; each of R 1 and R 2 is independently hydrogen, halo, -CN, -C 1 -C 20 alkyl (e.g., C 1 -C 6 alkyl), or - OR 7 ; each R 3 and R 4 is independently hydrogen, -C 1 -C 20 - alkyl (e.g., -C 1 -C 6 alkyl), -C 1 -C 20 heteroalkyl (e.g., -C 1 -C 6 heteroalkyl), -OC(O)OC 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, heteroalkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R 8 ; R 5 is hydrogen or -C 1 -C 20 al
- R 6 is halo, -CN, -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -OR 7 , oxo, cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R 9 ;
- R 7 is hydrogen, -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R 9 ; each R 8 is independently -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C 1 -C 20 heteroalkyl,
- -C(O)-C 1 -C 20 alkyl -OC(O)-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C(O)O-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -OC(O)O-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C(O)N(R 5 )-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -N(R 5 )C(O)-C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -OC(O)N(R 5 )- C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -O-aryl, -O-hetero
- each R 16 is independently, -C 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), -C 1 -C 20 heteroalkyl (e.g., -C 1 -C 6 heteroalkyl), -OC(O)OC 1 -C 20 alkyl (e.g., -C 1 -C 6 alkyl), C(O)N(R 4 ) 2 cycloalkyl, heterocyclyl, aryl, or heteroaryl.
- -C 1 -C 20 alkyl e.g., -C 1 -C 6 alkyl
- -C 1 -C 20 heteroalkyl e.g., -C 1 -C 6 heteroalkyl
- -OC(O)OC 1 -C 20 alkyl e.g., -C 1 -C 6 alkyl
- C(O)N(R 4 ) 2 cycloalkyl, heterocyclyl, ary
- the antibody-drug conjugate is an antibody-drug conjugate of formula (Xl-a), (CI-b), (XI-c), (CI-d), (XI-e), (XI-f), (XI-g), or (Xl-h):
- the antibody-drug conjugate is an antibody-drug conjugate of formula (CI-i), (Xl-j), (CI-k), (XI-1), (Xl-m), (CI-n), (XI-o), (XI-p), (Xl-q), (CI-r), (XI-s), or (XI-t):
- W 1 is N(R 5 ), -O- CH; and W 2 is N or CH.
- each of B 1 or B 2 is selected from a naturally occurring nucleobase or a modified nucleobase. In some embodiments, each of B 1 or B 2 is selected from adenosinyl, guanosinyl, cytosinyl, thyminyl, uracilyl, 5’ -methyl cytosinyl, 5’-fluorouracilyl, 5’- propynyluracilyl, and 7-deazaadenosinyl. In some embodiments, each ofB 1 or B 2 is selected from: wherein indicates the linkage of the nucleobase to the ribose ring.
- one of B 1 or B 2 is selected from a naturally occurring nucleobase and the other of B 1 or B 2 is a modified nucleobase.
- one of B 1 or B 2 is adenosinyl, guanosinyl, thyminyl, cytosinyl, or uracilyl, and the other of B 1 or B 2 is 5’- methyl cytosinyl, 5’-fluorouracilyl, 5’-propynyluracilyl, or 7-deazaadenosinyl.
- B 1 is adenosinyl or guanosinyl.
- B 2 is cytosinyl, thyminyl, or uracilyl.
- B 1 is adenosinyl or guanosinyl and B 2 is cytosinyl, thyminyl, or uracilyl.
- each of B 1 and B 2 is independently uracilyl.
- each of B 1 and B 2 is independently adenosinyl.
- each of R 1 and R 2 is independently hydrogen, halo, or OR 7 . In some embodiments, each of R 1 and R 2 is independently halo (e.g., fluoro). In some embodiments, each of R 1 and R 2 is not hydrogen or OR 7 .
- X 1 is O. In some embodiments, X 2 is O. In some embodiments, each of X 1 and X 2 is independently O.
- Y 1 is O or S. In some embodiments, Y 2 is O or S. In some embodiments, each of Y 1 and Y 2 is independently O or S. In some embodiments, one of Y 1 or Y 2 is O and the other of Y 1 or Y 2 is S. In some embodiments, each of Y 1 or Y 2 is independently S. In some embodiments, each of Y 1 or Y 2 is independently O.
- L 1 is C 1 -C 6 alkyl (e.g., CH 2 ). In some embodiments, L 2 is C 1 - C 6 alkyl (e.g., CH 2 ). In some embodiments, each of L 1 and L 2 is independently C 1 -C 6 alkyl (e.g., CH 2 ).
- R 3 is hydrogen, aryl, or heteroaryl, wherein aryl and heteroaryl is optionally substituted with 1-5 R 8 . In some embodiments, R 3 is aryl or heteroaryl, each of which is optionally substituted with 1-5 R 8 . In some embodiments, R 3 is phenyl substituted with 1 R 8 .
- R 4 is independently hydrogen, aryl, or heteroaryl, wherein aryl and heteroaryl is optionally substituted with 1-5 R 8 . In some embodiments, R 4 is aryl or heteroaryl, each of which is optionally substituted with 1-5 R 8 . In some embodiments, R 4 is phenyl substituted with 1 R 8 .
- each of R 3 and R 4 is independently hydrogen, aryl, or heteroaryl, wherein aryl and heteroaryl is optionally substituted with 1-5 R 8 .
- R 3 is aryl or heteroaryl, each of which is optionally substituted with 1-5 R 8 , and R 4 is hydrogen.
- R 3 is phenyl substituted with 1 R 8 and R 4 is hydrogen.
- each of R 3 and R 4 is independently phenyl substituted with 1 R 8 .
- each of Y 1 and Y 2 is O and each of R 3 and R 4 is independently hydrogen. In some embodiments, Y 2 is O and R 4 is hydrogen. In some embodiments, each of Y 1 and Y 2 is independently S and each of R 3 and R 4 is independently substituted with 1 R 8 . In some embodiments, Y 1 is S and R 3 is substituted with 1 R 8 .
- each R 8 is independently C 1 -C 20 alkyl (e.g., C 1 -C 6 alkyl), C 1 -C 20 heteroalkyl, C(O)-C 1 -C 20 alkyl, OC(O)-C 1 -C 20 alkyl, OC(O)O-C 1 -C 20 alkyl, OC(O)N(R 5 )-C 1 - C 20 alkyl, O-aryl, C(O)-aryl, OC(O)-aryl, or C(O)N(R 5 )-aryl, wherein each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R 9 .
- each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R 9 .
- R 8 is OC(O)-aryl optionally substituted by 1-5 R 9 (e.g., 1 R 9 ).
- R 9 is O-C 1 -C 12 alkyl (e.g., O-CH 2 (CH 2 ) 8 CH 3 ).
- R 9 is O-C 1 -C 10 alkyl (e.g., O-CH 2 (CH 2 ) 8 CH 3 ).
- R 9 is O-C 1 -C 8 alkyl (e.g., O-CH 2 (CH 2 ) 6 CH 3 ).
- R 9 is O-C 1 -C 6 alkyl (e.g., O-CH 2 (CH 2 )4CH 3 ).
- each of L 1 and L 2 is independently C 1 -C 6 alkylene (e.g., C 1 alkylene).
- L 3 is -C 1 -C 20 - alkylene (e.g., -C 1 -C 18 - alkylene). In some embodiments, L 3 is -C 1 -C 20 - heteroalkylene (e.g., -C 1 -C 18 - heteroalkylene).
- L 3 is an oligiopeptide-C(O)-, oligiopeptide-aryl-C 1 -C 6 -alkylene-, oligiopeptide- aryl-C 1 -C 6 -heteroalkylene, oligiopeptide-aryl-C 1 -C 6 -alklyene-C(O)-, oligiopeptide-C 1 -C 6 - alkylene-C(O)-, oligiopeptide-C 1 -C 6 -heteroalkylene-C(O)-.
- the aryl is phenyl.
- the oligiopeptide is a dipeptide.
- the oligopeptide comprises 3 amino acid residues, 4 amino acid residues, 5 amino acid residues, 6 amino acid residues, 7 amino acid residues, 8 amino acid residues, 9 amino acid residues, or 10 amino acid residues. In some embodiments, the oligopeptide comprises 8 amino acid residues. In some embodiments, the oligopeptide comprises an amino acid selected from Tyr, Ser, Thr, Arg, Leu, lie, Gly, Val, and Ala. In some embodiments, he oligopeptide comprises an amino acid selected from Phe, Tyr, Arg, Gly, Ser, and Leu. In some embodiments, L 3 is or In other embodiments, L 3 is In yet other embodiments, L 3 is
- L 3 is
- the oligiopeptide is substituted by one or more instances of R 16 .
- R 16 is C(O)NH 2 .
- L 4 is -C 1 -C 40 - alkylene (e.g., -C 1 -C 20 - alkylene), -C 1 -C 40 - heteroalkylene (e.g., -C 1 -C 20 - heteroalkyl), -C 1 -C 40 - alkenylene (e.g., -C 2 -C 20 - alkenylene), or -C 1 -C 40 - alkynylene (e.g., -C 2 -C 20 - alkynylene).
- L 4 is an oligopeptide comprising of 1 -40 amino acid residues.
- L 4 further comprises one sub-units.
- L 4 further comprises one sub- unit.
- L 4 further comprises one sub-unit.
- L 4 further comprises one sub-unit.
- L 4 further comprises one sub-unit.
- L 4 further comprises one sub-unit.
- L 4 further comprises one sub-unit.
- L 4 further comprises one sub-unit
- each Z 1 is oxygen.
- Z 3 is -0-, -N(R 5 )- or -heteroaryl-.
- Z 3 is In some embodiments, Z 3 is In some embodiments, Q 1 is C(O).
- Z 4 is In some embodiments, Z 4 is In some embodiments, Z 4 is In some embodiments, Z 4 is In some embodiments, Z 4 is In some embodiments, Z 4 is In some embodiments, Z 4 is In some embodiments, Z 4 is In some embodiments, Z 4 is represented by formula (Xll-a), (Xll-b), or (XII- c):
- U is O, S or NR 13 ;
- Q is CR 13 orN; each V 1 , V 2 and V 3 are independently CR 17 or N provided that for formula (Ill-b) and (III-c) at least one of Q, V 1 and V 2 is N;
- R 11 is the side chain of an amino acid and is optionally protected with a protecting group;
- Each R 10 , R 14 , and R 15 are independently selected from hydrogen, halo, OH, -N(R 12 ) 2 , - N(R 12 ) 3 + , C 1 -C 8 heteroalkyl, carboxylate, sulfate, sulfamate, sulfonate, -SO 2 R 5 , - S(O)R 12 ,
- R 12 and R 13 are independently selected from hydrogen, C 1 -C 8 alkyl, C 2 -C 8 alkenyl, C 2 -C 8 alkynyl, aryl, heterocycle, wherein each alkyl, alkenyl, alkynyl, aryl, and heterocycle are independently substituted with one or more substituents selected from halo, OH, - N(R 12 ) 2 , -N(R 12 ) 3 +, heteroalkyl, carboxylate, sulfate, sulfamate, sulfonate, 4- dialkylaminopyridinium, alkyl, -SO 2 R 12 , -S(O)R 12 , -SR 12 , -SO 2 N(R 12 ) 2 , C(O)R 12 , CO 2 R 12 , C(O)N(R 12 ) 2 , -CN, -N 3 ,
- Q is N; V 1 is CH, and V 2 is CH. In some embodiments, Q is CH; V 1 is CH and V 2 is N. In some embodiments, Q is N; V 1 is CH, and V 2 is N. In some embodiments, Q is N; and V 1 is N and V 2 is N. In some embodiments, Q is N; V 1 is N and V 2 is CH. In some embodiments, Q is N; and V 1 , V 2 and V 3 are each CH. In some embodiments, Q is CH; and V 1 , V 2 and V 3 are each CH. In some embodiments, Q is CH; and V 1 , V 2 and V 3 are each CH.
- Z 4 is absent.
- T is -C 1 -C 20 - alkylene (e.g., -C 1 -C 6 - alkylene), -C 1 -C 20 - heteroalkylene (e.g., -C 1 -C 6 - heteroalkylene), -C 1 -C 20 - alkenylene (e.g., -C 2 -C 6 - alkenylene), - C 1 -C 20 - alkynylene (e.g., -C 2 -C 6 - alkynyl), -aryl- or -heteroaryl-.
- T is absent.
- anti -PD- 1 antibody or anti-CTLA-4 antibody is a monoclonal antibody, a domain antibody (dAb), a single chain antibody (scAb), a Fab fragment, a F(ab’) 2 fragment, a single chain variable fragment (scFv), a scFv-Fc fragment, a single domain heavy chain antibody, a single domain light chain antibody, a variant antibody, a multimeric antibody, or a bispecific antibody.
- the antibody is an anti -PD- 1 antibody.
- the anti -PD- 1 antibody is pembrolizumab, nivolumab, cemiplimab, Anti -mouse PD-1 antibody Clone J43, Anti-mouse PD-1 antibody Clone RMP1-14, mouse anti-PD-1 antibody Clone EH12, ANB011, MDX-1 106, AMP-514, AMP-224, or Pidilizumab.
- the anti-PD-1 antibody is pembrolizumab or nivolumab.
- the antibody is an anti-CTLA-4 antibody.
- the anti-CTLA-4 antibody is ipilimumab, clone 9H10, tremelimumab, or clone BNI3.
- a antibody-drug conjugate described herein is in the form of a pharmaceutically acceptable salt.
- Exemplary salts are described herein, such as ammonium salts.
- the antibody-drug conjugate is a mono-salt
- the antibody-drug conjugate is a di-salt.
- the antibody- drug conjugate described herein e.g., a antibody-drug conjugate in Table 1 or Table 2 is not a salt (e.g., is a free acid or free base).
- the antibody-drug conjugate provided herein may contain one or more asymmetric centers and thus occur as racemates and racemic mixtures, single enantiomers, individual diastereomers, and diastereomeric mixtures. All such isomeric forms of these antibody-drug conjugate are expressly included within the scope. Unless otherwise indicated when a antibody-drug conjugate is named or depicted by a structure without specifying the stereochemistry and has one or more chiral centers, it is understood to represent all possible stereoisomers of the antibody-drug conjugate.
- the antibody-drug conjugate provided herewith may also contain linkages (e.g., carbon-carbon bonds, phosphorus-oxygen bonds, or phosphorus-sulfur bonds) or substituents that can restrict bond rotation, e.g., restriction resulting from the presence of a ring or double bond.
- the method described herein comprises administration of an antibody-drug conjugate of the disclosure or a pharmaceutically acceptable salt thereof.
- the method described herein comprises administration of an antibody-drug conjugate of the disclosure or a pharmaceutically acceptable salt thereof.
- the antibody-drug conjugate of the disclosure comprises an isomer (e.g., an Rp- isomer or Sp isomer) or a mixture of isomers (e.g., Rp-isomers or Sp isomers).
- an isomer e.g., an Rp- isomer or Sp isomer
- a mixture of isomers e.g., Rp-isomers or Sp isomers
- PD-1 Programmed cell death-1
- PD-L1 or PD-L2 are members of the CD28 superfamily that delivers negative signals upon interaction with its two ligands, PD-L1 or PD-L2.
- PD-1 and its ligands are broadly expressed and exert a wider range of immunoregulatory roles in T cells activation and tolerance compared with other CD28 members.
- PD-1 and its ligands are involved in attenuating infectious immunity and tumor immunity and facilitating chronic infection and tumor progression.
- the biological significance of PD-1 and its ligand suggests the therapeutic potential of manipulation of PD-1 pathway against various human diseases (Hyun-Tak Jin, et al., Curr Top Microbiol Immunol. (2011); 350:17-37).
- T-cell activation and dysfunction relies on direct and modulated receptors. Based on their functional outcome, co-signaling molecules can be divided as co-stimulators and co- inhibitors, which positively and negatively control the priming, growth, differentiation and functional maturation of a T-cell response (Li Shi, et al., Journal of Hematology & Oncology 2013, 6:74).
- PD-1 Programmed death-1
- PD-L1 or PD-L2 The binding of PD-1 to its ligands, PD-L1 or PD-L2, is vital for the physiological regulation of the immune system.
- a major functional role of the PD-1 signaling pathway is the inhibition of self-reactive T cells, which serve to protect against autoimmune diseases. Elimination of the PD-1 pathway can therefore result in the breakdown of immune tolerance that can ultimately lead to the development of pathogenic autoimmunity.
- tumor cells can at times co- opt the PD-1 pathway to escape from immunosurveillance mechanisms. Therefore, blockade of the PD-1 pathway has become an attractive target in cancer therapy.
- T-cell activation is a complex process that requires >1 stimulatory signal.
- TCR binding to MHC provides specificity to T-cell activation, but further costimulatory signals are required.
- Binding of B7-1 (CD80) or B7-2 (CD86) molecules on the APC with CD28 molecules on the T cell leads to signaling within the T cell.
- Sufficient levels of CD28:B7-l/2 binding lead to proliferation of T cells, increased T-cell survival, and differentiation through the production of growth cytokines such as interleukin-2 (IL-2), increased energy metabolism, and upregulation of cell survival genes.
- IL-2 interleukin-2
- CTLA-4 is a CD28 homolog with much higher binding affinity for B7 and is thought to regulate T-cell proliferation early in an immune response, primarily in lymph nodes. Inhibition of these CTLA-4, resulting in increased activation of the immune system and has led to new immunotherapies for melanoma, non-small cell lung cancer, and other cancers.
- the anti-CTLA-4 antibody blocks the binding of CTLA-4 to CD80 (B7-1) and/or CD86 (B7-2) expressed on antigen presenting cells.
- Exemplary antibodies against CTLA-4 include: Bristol Meyers Squibb’s anti-CTLA-4 antibody ipilimumab (also known as Yervoy, MDX-010, BMS-734016 and MDX-101); anti-CTLA4 Antibody, clone 9H10 from Millipore; Pfizer’s tremelimumab (CP-675,206, ticilimumab); and anti-CTLA-4 antibody clone BNI3 from Abeam.
- the anti-CTLA-4 antibody is an anti-CTLA-4 antibody disclosed in any of the following patent publications (herein incorporated by reference): WO 2001014424; WO 2004035607; US2005/0201994; EP 1212422 B 1; W02003086459; W02012120125; W02000037504; W02009100140; W0200609649; W02005092380; W02007123737; W02006029219; W020100979597; W0200612168; and WO1997020574. Additional CTLA-4 antibodies are described in U.S. Patent Nos. 5,811,097, 5,855,887, 6,051,227, and 6,984,720; in PCT Publication Nos.
- the anti-CTLA-4 antibody is an, for example, those disclosed in: WO 98/42752; U.S. Patent Nos. 6,682,736 and 6,207, 156; Hurwitz et al, Proc. Natl. Acad. Sci. USA, 95(17): 10067-10071 (1998); Camacho et al, J. Clin. Oncol., 22(145): Abstract No. 2505 (2004) (antibody CP- 675206); Mokyr et al, Cancer Res., 58:5301-5304 (1998) (incorporated herein by reference).
- T cell infiltrate includes tumor antigen-specific T cells that have been activated spontaneously in response to the growing tumor, perhaps through immune surveillance mechanisms. This attempted host immune response, even if it does not eliminate the tumor completely, is thought to delay tumor progression and thus yield improved clinical outcome.
- innate immune mechanisms can lead to adaptive T cell response against tumor antigens even in the absence of exogenous infection.
- human cancer gene expression profiling studies reveal an association between a type I IFN signature, T cell infiltration, and clinical outcome.
- innate immune sensing pathways that trigger type I IFN production might represent crucial intermediate mechanistic step.
- gene expression profiling of melanoma two major subsets of tumor microenvironment has been found that represent either the presence or absence of a transcriptional profile indicative of T cell infiltrate.
- CD8+ T cells, macrophages, as well as of some B cells and plasma cells in these lesions in melanoma metastases is similar to the phenotype described in early-stage colon cancer and other tumors in which activated T cells have been associated with favorable prognosis.
- CD8+ T cells were required for the up- regulation of all immune factors within the tumor micro-environment. Studies indicate that IFN production is necessary for optimal T cell priming against tumor antigens. There are many PRRs that trigger IFN-b production by host DC 8 in response to a growing tumor in vivo including STING.
- STING is an adapter protein that is activated by cyclic dinucleotides generated by cyclic GMP-AMP synthase (cGAS), which in turn is directly activated by cytosolic DNA.
- cGAS cyclic GMP-AMP synthase
- STING is translocated from the endoplasmic reticulum to various perinuclear components; for example, palmitoylation of STING at the Golgi has been shown to be essential for STING activation (Mukai, K. et al (2016) Nat Commun doi:10.1038/ncommsl l932).
- STING forms aggregates, activates TBK1, which in turn phosphorylates interferon regulatory factor 3 (IRF3) that directly contributes to type I IFN gene transcription.
- IRF3 interferon regulatory factor 3
- This pathway has been implicated in the sensing of DNA viruses, and also in selected autoimmune models.
- activating mutations of STING have recently been identified in human patients with a vasculitis/pulmonary inflammation syndrome that is characterized by increased type I IFN production.
- Mechanistic studies using mouse transplantable tumor models revealed that STING-knockout mice, and IRF3 -knockout mice showed defective spontaneous T cell priming against tumor antigens in vivo, and rejection of immunogenic tumors was ablated.
- tumor-derived DNA was found within the cytosol of a major population of tumor-infiltrating DC 8 , and this was associated with STING pathway activation and IFN-b production. Therefore, the host STING pathway appears to be an important innate immune sensing pathway that detects the presence of a tumor and to drive DC activation and subsequent T cell priming against tumor-associated antigens in vivo. A functional role for the STING pathway in vivo has also been reported in other mouse-tumor systems. An inducible glioma model was shown to result in induction of a type I IFN gene signature as part of the host response. This induction was substantially reduced in STING-knockout mice, and tumors grew more aggressively, leading to shorter mouse survival.
- PRRs such as cGAS, RIG-I and/STING stimulates the production of type I interferons (e.g., IFN-a or IFN-b), thus triggering a series of downstream signaling events that may lead to apoptosis in susceptible cells.
- type I interferons e.g., IFN-a or IFN-b
- PRRs such as cGAS, RIG-I and/STING
- RIG-I expression may be useful as a biomarker for prediction of prognosis and response to immunotherapy.
- induction of RIG-I expression has been shown to induce immunogenic cell death of pancreatic cancer cells, prostate cancer cells, breast cancer cells, skin cancer cells, and lung cancer cells (Duewell, P. et al, Cell Death Differ (2014) 21:1825-1837; Besch, R. et al, J Clin Invest (2009) 119:2399-2411; Kaneda, Y. Oncoimmunology (2013) 2:e23566; Li, X.Y. et al ,Mol Cell Oncol (2014) l:e968016), highlighting a new approach in immune-mediated cancer treatment.
- STING is recognized as the key adapter protein in the cGAS-STING-IFN cascade, although it is also reported to be a sensor for DNA.
- a role for STING in the stimulation of innate immunity in response to cancer has also been identified.
- Recent studies have revealed the presence of tumor-derived DNA in the cytosol of certain antigen-presenting cells, such as tumor-infiltrating dendritic cells, likely generated through tumor cell stress or cell death.
- This tumor-derived DNA is known to activate cGAS which causes the production of cyclic nucleotides that have been shown to activate STING, resulting in production of associated type 1 interferons (Woo, S.R. et al, Immunity (2014) 41:830-842).
- Stimulation of STING and resulting downstream signaling pathways also likely contributes to effector T cell recruitment into the inflamed tumor microenvironment (Woo, S. R. Trends in Immunol (2015) 36:250- 256).
- STING activation in the tumor microenvironment can induce adaptive immune response leading to anti-tumor activity.
- the described herein can still have anti-tumor activity through activation of antigen-presenting cells and dendritic cells, (APC 8 and DC 8 ) and induction of adaptive immune response.
- ADC 8 In the field of cancer chemotherapeutics, ADC 8 combine the specificity of antibodies with the potent anti-tumor effects of cytotoxic drugs. In recent literature, ADC 8 have emerged as powerful methods for the targeted treatment of cancer. Two ADC products, brentuximab vedotin (Adcetris ® ) and trastuzumab emtansine (Kadcyla ® ), have received FDA approval and there are more than 40 ADC candidates in clinical trials for the treatment of various cancers (Yejin, K. J Pharm. Investigation. (2016) 46:341-349).
- the methods of inducing expression of a PRR comprise administering a therapeutically effective amount of an antibody- drug conjugate of the disclosure or a pharmaceutically acceptable salt thereof to a subject suffering from cancer.
- the methods of inducing expression of STING disclosed herein comprise administering a therapeutically effective amount of an antibody-drug conjugate of the disclosure or a pharmaceutically acceptable salt thereof to a subject suffering from cancer.
- the methods of inducing expression of RIG-I disclosed herein comprise administering a therapeutically effective amount of an antibody-drug conjugate of the disclosure or a pharmaceutically acceptable salt thereof to a subject suffering from cancer.
- the methods of inducing expression of NOD2 disclosed herein comprise administering a therapeutically effective amount of an antibody-drug conjugate of the disclosure or a pharmaceutically acceptable salt thereof to a subject suffering from cancer.
- the cancer is selected from a cancer of the breast, bone, brain, cervix, colon, gastrointestinal tract, eye, gall bladder, lymph nodes, blood, lung, liver, skin, mouth, prostate, ovary, penis, pancreas, uterus, testicles, stomach, thymus, thyroid, or other part of the body.
- the cancer comprises a solid tumor (e.g., a carcinoma, a sarcoma, or a lymphoma).
- the cancer is a hepatocellular carcinoma or other cancer of the liver.
- the cancer is a leukemia or other cancer of the blood.
- the cancer comprises breast cancer, renal cell carcinoma, colon cancer, melanoma, ovarian cancer, head and neck squamous cell carcinoma, pancreatic cancer, prostate cancer, lung cancer, brain cancer, thyroid cancer, renal cancer, testis cancer, stomach cancer, urothelial cancer, skin cancer, cervical cancer, endometrial cancer, liver cancer, lung cancer, lymphoma or gastrointestinal stromal cancer and solid tumors.
- the cancer cells e.g., tumor cells
- the methods of inducing expression of a PRR e.g., STING, RIG-I, MDA5, LGP2
- a PRR e.g., STING, RIG-I, MDA5, LGP2
- an increase in PRR expression e.g., STING expression
- expression of a PRR is induced by a factor of about 1.1, about 1.2, about 1.3, about 1.4, about 1.5, about 1.6, about 1.7, about 1.8, about 1.9, about 2, about 2.5, about 3, about 4, about 5, about 7.5, about 10, about 15, about 20, about 25, about 30, about 40, about 50, about 75, about 100, about 150, about 200, about 250, about 500, about 1000, about 1500, about 2500, about 5000, about 10,000, or more.
- induction of expression of a PRR occurs within about 5 minutes of administration of an antibody-drug conjugate of the disclosure or a pharmaceutically acceptable salt thereof.
- induction of expression of a PRR occurs within about 10 minutes, about 15 minutes, about 20 minutes, about 25 minutes, about 30 minutes, about 45 minutes, about 1 hour, about 1.5 hours, about 2 hours, about 3 hours, about 4 hours, about 5 hours, about 6 hours, about 7 hours, about 8 hours, about 10 hours, about 12 hours or more following administration of an antibody-drug conjugate of the disclosure or a pharmaceutically acceptable salt thereof.
- STING activation of STING by compounds may lead to induction of expression of other PRRs such as RIG-I, MDA5, NOD2 etc. which may further amplify IFN production in the tumor microenvironment and prime T-cells for enhanced anti-tumor activity.
- PRRs such as RIG-I, MDA5, NOD2 etc.
- compositions and methods of the present disclosure may be utilized to treat an individual in need thereof.
- the individual is a mammal such as a human, or a non-human mammal.
- the composition or the antibody-drug conjugate is preferably administered as a pharmaceutical composition comprising, for example, an antibody-drug conjugate of the disclosure and a pharmaceutically acceptable carrier.
- Pharmaceutically acceptable carriers are well known in the art and include, for example, aqueous solutions such as water or physiologically buffered saline or other solvents or vehicles such as glycols, glycerol, oils such as olive oil, or injectable organic esters.
- the aqueous solution is pyrogen-free, or substantially pyrogen-free.
- the excipients can be chosen, for example, to effect delayed release of an agent or to selectively target one or more cells, tissues or organs.
- the pharmaceutical composition can be in dosage unit form such as tablet, capsule (including sprinkle capsule and gelatin capsule), granule, lyophile for reconstitution, powder, solution, syrup, suppository, injection or the like.
- the composition can also be present in a transdermal delivery system, e.g., a skin patch.
- the composition can also be present in a solution suitable for topical administration, such as a lotion, cream, or ointment.
- a pharmaceutically acceptable carrier can contain physiologically acceptable agents that act, for example, to stabilize, increase solubility or to increase the absorption of an antibody-drug conjugate of the invention.
- physiologically acceptable agents include, for example, carbohydrates, such as glucose, sucrose or dextrans, antioxidants, such as ascorbic acid or glutathione, chelating agents, low molecular weight proteins or other stabilizers or excipients.
- the choice of a pharmaceutically acceptable carrier, including a physiologically acceptable agent depends, for example, on the route of administration of the composition.
- the preparation or pharmaceutical composition can be a selfemulsifying drug delivery system or a selfmicroemulsifying drug delivery system.
- the pharmaceutical composition also can be a liposome or other polymer matrix, which can have incorporated therein, for example, an antibody-drug conjugate of the invention.
- Liposomes for example, which comprise phospholipids or other lipids, are nontoxic, physiologically acceptable and metabolizable carriers that are relatively simple to make and administer.
- phrases "pharmaceutically acceptable” is employed herein to refer to those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- pharmaceutically acceptable carrier means a pharmaceutically acceptable material, composition or vehicle, such as a liquid or solid filler, diluent, excipient, solvent or encapsulating material. Each carrier must be “acceptable” in the sense of being compatible with the other ingredients of the formulation and not injurious to the patient.
- materials which can serve as pharmaceutically acceptable carriers include: (1) sugars, such as lactose, glucose and sucrose; (2) starches, such as com starch and potato starch; (3) cellulose, and its derivatives, such as sodium carboxym ethyl cellulose, ethyl cellulose and cellulose acetate; (4) powdered tragacanth; (5) malt; (6) gelatin; (7) talc; (8) excipients, such as cocoa butter and suppository waxes; (9) oils, such as peanut oil, cottonseed oil, safflower oil, sesame oil, olive oil, com oil and soybean oil; (10) glycols, such as propylene glycol; (11) polyols, such as glycerin, sorbitol, mannitol and polyethylene glycol; (12) esters, such as ethyl oleate and ethyl laurate; (13) agar; (14) buffering agents, such as magnesium hydrox
- a pharmaceutical composition can be administered to a subject by any of a number of routes of administration including, for example, orally (for example, drenches as in aqueous or non-aqueous solutions or suspensions, tablets, capsules (including sprinkle capsules and gelatin capsules), boluses, powders, granules, pastes for application to the tongue); absorption through the oral mucosa (e.g., sublingually); subcutaneously; transdermally (for example as a patch applied to the skin); and topically (for example, as a cream, ointment or spray applied to the skin).
- the antibody-drug conjugate may also be formulated for inhalation.
- an antibody-drug conjugate may be simply dissolved or suspended in sterile water. Details of appropriate routes of administration and compositions suitable for same can be found in, for example, U.S. Pat. Nos. 6,110,973, 5,763,493, 5,731,000, 5,541,231, 5,427,798, 5,358,970 and 4,172,896, as well as in patents cited therein.
- the formulations may conveniently be presented in unit dosage form and may be prepared by any methods well known in the art of pharmacy.
- the amount of active ingredient which can be combined with a carrier material to produce a single dosage form will vary depending upon the host being treated, the particular mode of administration.
- the amount of active ingredient that can be combined with a carrier material to produce a single dosage form will generally be that amount of the antibody-drug conjugate which produces a therapeutic effect. Generally, out of one hundred percent, this amount will range from about 1 percent to about ninety-nine percent of active ingredient, preferably from about 5 percent to about 70 percent, most preferably from about 10 percent to about 30 percent.
- Methods of preparing these formulations or compositions include the step of bringing into association an active compound, such as an antibody-drug conjugate of the invention, with the carrier and, optionally, one or more accessory ingredients.
- the formulations are prepared by uniformly and intimately bringing into association an antibody-drug conjugate of the present disclosure with liquid carriers, or finely divided solid carriers, or both, and then, if necessary, shaping the product.
- Formulations of the disclosure suitable for oral administration may be in the form of capsules (including sprinkle capsules and gelatin capsules), cachets, pills, tablets, lozenges (using a flavored basis, usually sucrose and acacia or tragacanth), lyophile, powders, granules, or as a solution or a suspension in an aqueous or non-aqueous liquid, or as an oil-in-water or water-in-oil liquid emulsion, or as an elixir or syrup, or as pastilles (using an inert base, such as gelatin and glycerin, or sucrose and acacia) and/or as mouth washes and the like, each containing a predetermined amount of an antibody-drug conjugate of the present disclosure as an active ingredient.
- Compositions or compounds may also be administered as a bolus, electuary or paste.
- the active ingredient is mixed with one or more pharmaceutically acceptable carriers, such as sodium citrate or dicalcium phosphate, and/or any of the following: (1) fillers or extenders, such as starches, lactose, sucrose, glucose, mannitol, and/or silicic acid; (2) binders, such as, for example, carboxymethylcellulose, alginates, gelatin, polyvinyl pyrrolidone, sucrose and/or acacia; (3) humectants, such as glycerol; (4) disintegrating agents, such as agar-agar, calcium carbonate, potato or tapioca starch, alginic acid, certain silicates, and sodium carbonate; (5) solution retarding agents, such as paraffin; (6) absorption accelerators, such as quaternary ammonium compounds; (7) wetting agents,
- pharmaceutically acceptable carriers such as sodium citrate or dicalcium phosphate, and/or any of the following: (1) fillers or extenders, such as starches, lactose
- the pharmaceutical compositions may also comprise buffering agents.
- Solid compositions of a similar type may also be employed as fillers in soft and hard- filled gelatin capsules using such excipients as lactose or milk sugars, as well as high molecular weight polyethylene glycols and the like.
- a tablet may be made by compression or molding, optionally with one or more accessory ingredients.
- Compressed tablets may be prepared using binder (for example, gelatin or hydroxypropylmethyl cellulose), lubricant, inert diluent, preservative, disintegrant (for example, sodium starch glycolate or cross-linked sodium carboxymethyl cellulose), surface- active or dispersing agent.
- Molded tablets may be made by molding in a suitable machine a mixture of the powdered antibody-drug conjugate moistened with an inert liquid diluent.
- the tablets, and other solid dosage forms of the pharmaceutical compositions may optionally be scored or prepared with coatings and shells, such as enteric coatings and other coatings well known in the pharmaceutical-formulating art. They may also be formulated so as to provide slow or controlled release of the active ingredient therein using, for example, hydroxypropylmethyl cellulose in varying proportions to provide the desired release profile, other polymer matrices, liposomes and/or microspheres.
- compositions may be sterilized by, for example, filtration through a bacteria-retaining filter, or by incorporating sterilizing agents in the form of sterile solid compositions that can be dissolved in sterile water, or some other sterile injectable medium immediately before use.
- These compositions may also optionally contain opacifying agents and may be of a composition that they release the active ingredient(s) only, or preferentially, in a certain portion of the gastrointestinal tract, optionally, in a delayed manner.
- embedding compositions that can be used include polymeric substances and waxes.
- the active ingredient can also be in micro-encapsulated form, if appropriate, with one or more of the above-described excipients.
- Liquid dosage forms useful for oral administration include pharmaceutically acceptable emulsions, lyophiles for reconstitution, microemulsions, solutions, suspensions, syrups and elixirs.
- the liquid dosage forms may contain inert diluents commonly used in the art, such as, for example, water or other solvents, cyclodextrins and derivatives thereof, solubilizing agents and emulsifiers, such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3- butylene glycol, oils (in particular, cottonseed, groundnut, corn, germ, olive, castor and sesame oils), glycerol, tetrahydrofuryl alcohol, polyethylene glycols and fatty acid esters of sorbitan, and mixtures thereof.
- inert diluents commonly used in the art, such
- the oral compositions can also include adjuvants such as wetting agents, emulsifying and suspending agents, sweetening, flavoring, coloring, perfuming and preservative agents.
- adjuvants such as wetting agents, emulsifying and suspending agents, sweetening, flavoring, coloring, perfuming and preservative agents.
- Suspensions in addition to the active compounds, may contain suspending agents as, for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar and tragacanth, and mixtures thereof.
- Dosage forms for the topical or transdermal administration include powders, sprays, ointments, pastes, creams, lotions, gels, solutions, patches and inhalants.
- the active ingredient may be mixed under sterile conditions with a pharmaceutically acceptable carrier, and with any preservatives, buffers, or propellants that may be required.
- the ointments, pastes, creams and gels may contain, in addition to an active compound, excipients, such as animal and vegetable fats, oils, waxes, paraffins, starch, tragacanth, cellulose derivatives, polyethylene glycols, silicones, bentonites, silicic acid, talc and zinc oxide, or mixtures thereof.
- excipients such as animal and vegetable fats, oils, waxes, paraffins, starch, tragacanth, cellulose derivatives, polyethylene glycols, silicones, bentonites, silicic acid, talc and zinc oxide, or mixtures thereof.
- Powders and sprays can contain, in addition to an active compound, excipients such as lactose, talc, silicic acid, aluminum hydroxide, calcium silicates and polyamide powder, or mixtures of these substances.
- Sprays can additionally contain customary propellants, such as chlorofluorohydrocarbons and volatile unsubstituted hydrocarbons, such as butane and propane.
- parenteral administration and “administered parenterally” as used herein means modes of administration other than enteral and topical administration, usually by injection, and includes, without limitation, intravenous, intramuscular, intraarterial, intrathecal, intracapsular, intraorbital, intracardiac, intradermal, intraperitoneal, transtracheal, subcutaneous, subcuticular, intraarticular, subcapsular, subarachnoid, intraspinal and intrastemal injection and infusion.
- compositions suitable for parenteral administration comprise one or more active compounds in combination with one or more pharmaceutically acceptable sterile isotonic aqueous or nonaqueous solutions, dispersions, suspensions or emulsions, or sterile powders which may be reconstituted into sterile injectable solutions or dispersions just prior to use, which may contain antioxidants, buffers, bacteriostats, solutes which render the formulation isotonic with the blood of the intended recipient or suspending or thickening agents.
- aqueous and nonaqueous carriers examples include water, ethanol, polyols (such as glycerol, propylene glycol, polyethylene glycol, and the like), and suitable mixtures thereof, vegetable oils, such as olive oil, and injectable organic esters, such as ethyl oleate.
- polyols such as glycerol, propylene glycol, polyethylene glycol, and the like
- vegetable oils such as olive oil
- injectable organic esters such as ethyl oleate.
- Proper fluidity can be maintained, for example, by the use of coating materials, such as lecithin, by the maintenance of the required particle size in the case of dispersions, and by the use of surfactants.
- compositions may also contain adjuvants such as preservatives, wetting agents, emulsifying agents and dispersing agents. Prevention of the action of microorganisms may be ensured by the inclusion of various antibacterial and antifungal agents, for example, paraben, chlorobutanol, phenol sorbic acid, and the like. It may also be desirable to include isotonic agents, such as sugars, sodium chloride, and the like into the compositions. In addition, prolonged absorption of the injectable pharmaceutical form may be brought about by the inclusion of agents that delay absorption such as aluminum monostearate and gelatin.
- the absorption of the drug in order to prolong the effect of a drug, it is desirable to slow the absorption of the drug from subcutaneous or intramuscular injection. This may be accomplished by the use of a liquid suspension of crystalline or amorphous material having poor water solubility. The rate of absorption of the drug then depends upon its rate of dissolution, which, in turn, may depend upon crystal size and crystalline form. Alternatively, delayed absorption of a parenterally administered drug form is accomplished by dissolving or suspending the drug in an oil vehicle.
- Injectable depot forms are made by forming microencapsulated matrices of the subject compounds in biodegradable polymers such as polylactide-polyglycolide. Depending on the ratio of drug to polymer, and the nature of the particular polymer employed, the rate of drug release can be controlled. Examples of other biodegradable polymers include poly(orthoesters) and poly(anhydrides). Depot injectable formulations are also prepared by entrapping the drug in liposomes or microemulsions that are compatible with body tissue.
- active compounds can be given per se or as a pharmaceutical composition containing, for example, 0.1 to 99.5% (more preferably, 0.5 to 90%) of active ingredient in combination with a pharmaceutically acceptable carrier.
- Methods of introduction may also be provided by rechargeable or biodegradable devices.
- Various slow release polymeric devices have been developed and tested in vivo in recent years for the controlled delivery of drugs, including proteinaceous biopharmaceuticals.
- a variety of biocompatible polymers including hydrogels, including both biodegradable and non-degradable polymers, can be used to form an implant for the sustained release of an antibody-drug conjugate at a particular target site.
- Actual dosage levels of the active ingredients in the pharmaceutical compositions may be varied so as to obtain an amount of the active ingredient that is effective to achieve the desired therapeutic response for a particular patient, composition, and mode of administration, without being toxic to the patient.
- the selected dosage level will depend upon a variety of factors including the activity of the particular antibody-drug conjugate or combination of antibody-drug conjugates employed, or the ester, salt or amide thereof, the route of administration, the time of administration, the rate of excretion of the particular compound(s) being employed, the duration of the treatment, other drugs, compounds and/or materials used in combination with the particular compound(s) employed, the age, sex, weight, condition, general health and prior medical history of the patient being treated, and like factors well known in the medical arts.
- a physician or veterinarian having ordinary skill in the art can readily determine and prescribe the therapeutically effective amount of the pharmaceutical composition required.
- the physician or veterinarian could start doses of the pharmaceutical composition or antibody-drug conjugate at levels lower than that required in order to achieve the desired therapeutic effect and gradually increase the dosage until the desired effect is achieved.
- therapeutically effective amount is meant the concentration of the antibody-drug conjugate that is sufficient to elicit the desired therapeutic effect. It is generally understood that the effective amount of the antibody-drug conjugate will vary according to the weight, sex, age, and medical history of the subject.
- the effective amount may include, but are not limited to, the severity of the patient's condition, the disorder being treated, the stability of the compound, and, if desired, another type of therapeutic agent being administered with the antibody-drug conjugate of the invention.
- a larger total dose can be delivered by multiple administrations of the agent. Methods to determine efficacy and dosage are known to those skilled in the art (Isselbacher et al. (1996) Harrison’s Principles of Internal Medicine 13 ed., 1814-1882, herein incorporated by reference).
- a suitable daily dose of an active antibody-drug conjugate used in the compositions and methods of the disclosure will be that amount of the antibody-drug conjugate that is the lowest dose effective to produce a therapeutic effect. Such an effective dose will generally depend upon the factors described above.
- the effective daily dose of the antibody-drug conjugate may be administered as one, two, three, four, five, six or more sub-doses administered separately at appropriate intervals throughout the day, optionally, in unit dosage forms.
- the antibody-drug conjugate may be administered two or three times daily. In preferred embodiments, the antibody-drug conjugate will be administered once daily.
- the patient receiving this treatment is any animal in need, including primates, in particular humans; and other mammals such as equines, cattle, swine, sheep, cats, and dogs; poultry; and pets in general.
- compounds of the disclosure may be used alone or conjointly administered with another type of therapeutic agent.
- the present disclosure includes the use of pharmaceutically acceptable salts of compounds of the disclosure in the compositions and methods of the present invention.
- contemplated salts of the disclosure include, but are not limited to, alkyl, dialkyl, trialkyl or tetra-alkyl ammonium salts.
- contemplated salts of the disclosure include, but are not limited to, L-arginine, benenthamine, benzathine, betaine, calcium hydroxide, choline, deanol, diethanolamine, diethylamine, 2-(diethylamino)ethanol, ethanolamine, ethylenediamine, N-methylglucamine, hydrabamine, lH-imidazole, lithium, L- lysine, magnesium, 4-(2-hydroxyethyl)morpholine, piperazine, potassium, 1-(2- hydroxyethyl)pyrrolidine, sodium, triethanolamine, tromethamine, and zinc salts.
- contemplated salts of the disclosure include, but are not limited to, Na, Ca, K, Mg, Zn or other metal salts.
- contemplated salts of the disclosure include, but are not limited to, l-hydroxy-2-naphthoic acid, 2,2-dichloroacetic acid, 2- hydroxyethanesulfonic acid, 2-oxoglutaric acid, 4-acetamidobenzoic acid, 4-aminosalicylic acid, acetic acid, adipic acid, 1-ascorbic acid, 1-aspartic acid, benzenesulfonic acid, benzoic acid, (+)-camphoric acid, (+)-camphor-10-sulfonic acid, capric acid (decanoic acid), caproic acid (hexanoic acid), caprylic acid (octanoic acid), carbonic acid, cinnamic acid, citric acid, cyclamic acid, dodecylsulfuric acid, e
- the pharmaceutically acceptable acid addition salts can also exist as various solvates, such as with water, methanol, ethanol, dimethylformamide, and the like. Mixtures of such solvates can also be prepared.
- the source of such solvate can be from the solvent of crystallization, inherent in the solvent of preparation or crystallization, or adventitious to such solvent.
- wetting agents such as sodium lauryl sulfate and magnesium stearate, as well as coloring agents, release agents, coating agents, sweetening, flavoring and perfuming agents, preservatives and antioxidants can also be present in the compositions.
- antioxidants examples include: (1) water-soluble antioxidants, such as ascorbic acid, cysteine hydrochloride, sodium bisulfate, sodium metabi sulfite, sodium sulfite and the like; (2) oil-soluble antioxidants, such as ascorbyl palmitate, butylated hydroxyanisole (BHA), butylated hydroxytoluene (BHT), lecithin, propyl gallate, alpha-tocopherol, and the like; and (3) metal-chelating agents, such as citric acid, ethylenediamine tetraacetic acid (EDTA), sorbitol, tartaric acid, phosphoric acid, and the like.
- water-soluble antioxidants such as ascorbic acid, cysteine hydrochloride, sodium bisulfate, sodium metabi sulfite, sodium sulfite and the like
- oil-soluble antioxidants such as ascorbyl palmitate, butylated hydroxyanisole (BHA), butylated hydroxytoluene (
- the present disclosure features pharmaceutical composition comprising an antibody-drug conjugate of the disclosure and a pharmaceutically acceptable excipient.
- the composition is administered directly to the tumor (e.g., intratumoral administration).
- the composition is administered orally. In other embodiments, the composition is administered parenterally (e.g., intravenous, subcutaneous, intraperitoneal, or intramuscular administration). In some embodiments, the composition is administered intraperitoneally.
- the methods of the present disclosure described herein entail administration of an antibody-drug conjugate of the disclosure or a pharmaceutically acceptable salt thereof to a subject to activate the PRR for IFNs, ISGs and cytokines production or additionally induce the expression of PRRs (e.g., RIG-I, STING).
- PRRs e.g., RIG-I, STING
- the subject is suffering from or is diagnosed with a condition, e.g., a proliferative disease, e.g., cancer.
- a patient and/or subject can be selected for treatment using an antibody-drug conjugate of the disclosure or a pharmaceutically acceptable salt thereof by first evaluating the patient and/or subject to determine whether the subject is infected with a proliferative disease, e.g., cancer.
- a subject can be evaluated as infected with a proliferative disease (e.g., cancer) using methods known in the art.
- the subject can also be monitored, for example, subsequent to administration of an antibody-drug conjugate of the disclosure herein.
- the subject is a mammal. In some embodiments, the subject is a human. In some embodiments, the subject is an adult. In some embodiments, the subject has a proliferative disease, e.g., cancer. In some embodiments, the subject has a cancer of the of the breast, bone, brain, cervix, colon, gastrointestinal tract, eye, gall bladder, lymph nodes, blood, lung, liver, skin, mouth, prostate, ovary, penis, pancreas, uterus, testicles, stomach, thymus, thyroid, or other part of the body.
- a proliferative disease e.g., cancer.
- the subject has a cancer of the of the breast, bone, brain, cervix, colon, gastrointestinal tract, eye, gall bladder, lymph nodes, blood, lung, liver, skin, mouth, prostate, ovary, penis, pancreas, uterus, testicles, stomach, thymus, thyroid, or other part of the body.
- the subject has a cancer comprising a solid tumor (e.g., a carcinoma, a sarcoma, or a lymphoma).
- the subject has a hepatocellular carcinoma or other cancer of the liver.
- the subject has a leukemia or other cancer of the blood.
- the subject has a breast cancer, renal cell carcinoma, colon cancer, melanoma, ovarian cancer, head and neck squamous cell carcinoma, pancreatic cancer, prostate cancer, lung cancer, brain cancer, or gastrointestinal stromal cancer.
- the subject has cancer cells (e.g., tumor cells) comprising specific cancer-associated antigens that induce a T-cell response.
- the subject is treatment naive. In some embodiments, the subject has been previously treated for a proliferative disease (e.g., a cancer). In some embodiments, the subject has relapsed.
- a proliferative disease e.g., a cancer
- an antibody-drug conjugate of the disclosure described herein may be used in combination with other known therapies.
- Administered “in combination”, as used herein, means that two (or more) different treatments are delivered to the subject during the course of the subject's affliction with the disorder, e.g., the two or more treatments are delivered after the subject has been diagnosed with the disorder and before the disorder has been cured or eliminated or treatment has ceased for other reasons.
- the delivery of one treatment is still occurring when the delivery of the second begins, so that there is overlap in terms of administration. This is sometimes referred to herein as “simultaneous” or “concurrent delivery”.
- the delivery of one treatment ends before the delivery of the other treatment begins.
- the treatment is more effective because of combined administration.
- the second treatment is more effective, e.g., an equivalent effect is seen with less of the second treatment, or the second treatment reduces symptoms to a greater extent, than would be seen if the second treatment were administered in the absence of the first treatment, or the analogous situation is seen with the first treatment.
- delivery is such that the reduction in a symptom, or other parameter related to the disorder is greater than what would be observed with one treatment delivered in the absence of the other.
- the effect of the two treatments can be partially additive, wholly additive, or greater than additive.
- the delivery can be such that an effect of the first treatment delivered is still detectable when the second is delivered.
- An antibody-drug conjugate described herein and the at least one additional therapeutic agent can be administered simultaneously, in the same or in separate compositions, or sequentially.
- the compound described herein can be administered first, and the additional agent can be administered second, or the order of administration can be reversed.
- the combination of an antibody-drug conjugate or a pharmaceutically acceptable salt thereof and the additional agent has a synergistic or additive effect.
- the term “additive” refers to an outcome wherein when two agents are used in combination, the combination of the agents acts in a manner equal to but not greater than the sum of the individual activity of each agent.
- the terms “synergy” or “synergistic” refer to an outcome wherein when two agents are used in combination, the combination of the agents acts so as to require a lower concentration of each individual agent than the concentration required to be efficacious in the absence of the other agent.
- a synergistic effect results in a reduced in a reduced minimum inhibitory concentration of one or both agents, such that the effect is greater than the sum of the effects.
- a synergistic effect is greater than an additive effect.
- the agents in the composition herein may exhibit a synergistic effect, wherein the activity at a particular concentration is greater than at least about 1.25, 1.5, 1.75, 2, 2.5, 3, 4, 5, 10, 12, 15, 20, 25, 50, or 100 times the activity of either agent alone.
- any of the methods described herein may further comprise the administration of a therapeutically effective amount of an additional agent.
- additional pharmaceutical agents include, but are not limited to, anti-proliferative agents, anti-cancer agents, anti-diabetic agents, anti-inflammatory agents, immunosuppressant agents, and a pain-relieving agent.
- Pharmaceutical agents include small organic molecules such as drug compounds ( e.g ., compounds approved by the U.S.
- the additional agent is an anti-cancer agent, e.g., an alkylating agent (e.g., cyclophosphamide).
- the additional agent is an immunooncology agent, for example, an agent that activate the immune system, e.g., making it able to recognize cancer cells and destroy them.
- the agent is a cell based agent such as CAR-t therapy.
- a and B were treated with 5-(ethylthio)-1H-tetrazole in acetonitrile and the Iyer- Beaucage reagent. Following this, the product was detritlyated to yield C. C was then treated with sodium iodide in acetone to yield deprotected product D. D was treated with 1 -mesityl ene- 2-sulfonyl-3-nitro- 1,2, 4-triazole and pyridine to yield cyclized product E.
- Cyclic phosphoromonothio diphosphate (100 mg, 0.145 mmol) was dissolved in HPLC-water (200 mL). A solution of alkylating agent (110 mg, 0.174 mmol) in a mixture of THF : Acetone (1 : 1, 3.0 mL) was added to the reaction mixture. The solution was stirred at room temperature for three days. LC/MS analysis showed completion of the reaction. Solvents were removed under reduced pressure to dryness. Compound was re-dissolved in THF:acetone (1 : 1, 5.0 mL). Solvents were evaporated under reduced pressure to dryness.
- Cyclic phosphoromonothio diphosphate (100 mg, 0.145 mmol) was dissolved in HPLC-water (500 mL). A solution of alkylating agent (150 mg, 0.192 mmol) in a mixture of THF : Acetone (1 : 1, 3.5 mL) was added to the reaction mixture. The solution was stirred at room temperature for two days. LCMS analysis showed completion of the reaction. Solvents were removed under reduced pressure to dryness. Compound was re-dissolved in THF:acetone (1 : 1, 5.0 mL) and precipitated by adding to diethyl ether (20 mL). The precipitate was collected by centrifugation to get product as off white solid.
- Step 1 5'O-DMT-2'F-3'Phosphramidite-dA (15.0 g, 17.12 mmol) was co-evaporated with anhydrous acetonitrile (2 x 100 mL), and dried under high vacuum for 1 h. Argon was flushed over the residue in the flask. Acetonitrile (150 mL, anhydrous) was added to residue under argon. Allyl alcohol (Aldrich, 99%) (2.32 mL, 34.24 mmol) was added to the solution followed by ETT (2.22 g, 17.12 mmol) in acetonitrile (20 mL). The reaction mixture was stirred at room temperature under argon for 2.5 hours.
- the reaction mixture was partitioned between DCM (150 mL) and water (100 mL). The organic layer was separated and water layer was extracted with DCM (50 mL). The combined organic layers were dried over NaiSCL and filtered to remove the Na 2 SO 4 salt.
- Step 2 Detritylation: The solution of crude DMT-N-bz-3'-O-Allyl-2'-FdA obtained above (200 mL) in DCM was cooled in an ice- water bath.
- Step 1 Coupling Reaction for Synthesis of Phosphorothioate Dimer: A mixture of C (1.09 g, 2.0 mmol) and E (1.5 g, 2.0 mmol) was co-evaporated with anhydrous acetonitrile (2 x 40 mL) and dried under high vacuum for 1 h. Argon was flushed over the round bottom flask and anhydrous acetonitrile (40 mL) was added to reaction mixture. ETT (260 mg, 2.0 mmol) in acetonitrile (2.0 mL) was added to the mixture of C and E, under argon. The mixture was stirred at room temperature under argon for 2h.
- Step 3 Detritylation: The dried DCM solution (50 mL) was cooled to approximately 0°C in a round bottom flask.
- Step 1 To a suspension of benzoic acid derivative (10 g, 0.054 mol) in a 250 mL single neck flask in toluene, thionyl chloride (7.8 mL) was added slowly and stirred at r.t. for 15 minutes followed by heating in an oil bath at 80-85°C to obtain a clear solution that was maintained for ⁇ 3h. The reaction mixture was cooled to RT and excess thionyl chloride was removed in vacuo. The toluene was concentrated using a rotavap at 40-45°C. It was then co- evaporated twice with ethyl acetate (25 mL). The residue was taken up in ethyl acetate (15ml).
- Step 2 To a suspension of 4- hydroxyl benzyl alcohol coupled derivative (9.0 g, 0.026 mol) in a mixture of anhydrous acetonitrile (80 mL) and anhydrous dichloromethane (30 mL) in a 250 mL single neck flask, was added C 8 l (18.2 g, 0.078 mol) in one portion. To this, BF3.Et 2 0 (8.7 mL) was added slowly and stirred in the dark (covered with aluminum foil) under argon overnight at room temperature. The reaction was found to be complete by TLC Hex: EtOAc (7:3).
- Step 1 Deprotection of cyclic phosphoromonothio diphosphate: Fully protected cyclic phosphoro monothio diphosphate (70 mg) was dissolved in a mixture of cone. NH 4 OH (2.0 mL) and DCM (5.0 mL) stirred at room temperature overnight. LC-MS analysis showed completion of the reaction. Reaction mixture was transferred to separatory funnel and the DCM layer was removed. The aqueous layer was evaporated under reduced pressure to remove ammonia and was then washed with ethyl acetate (3 x 5 mL) to remove benzamide byproduct completely. The product was isolated from the aqueous layer by lyophilization to yield 60 mg of as white solid.
- Step 2 Cyclic phosphoromonothio diphosphate (50 mg, 0.072 mmol) was dissolved in water (500 uL). A solution of I (53 mg, 0.108 mmol) in a mixture of THF: Acetone (1:1, 3.5 mL) was added to the reaction mixture. The solution was stirred at room temperature for two days. Solvents were removed under reduced pressur. The crude product was re-dissolved in THF: acetone (1 :1, 5.0 mL) and precipitated by adding to diethyl ether (10 mL) to remove unreacted iodo-compound. The precipitate was collected by centrifugation to yield product as an off white solid.
- THP1 dual cells grown in complete media were treated with various concentrations of a compound of the present disclosure or DMSO control.
- Dual cells carry both secreted embryonic alkaline phosphatase (SEAP) reporter gene under the control of an IFN-b minimal promoter fused to five copies of the NF-kB consensus transcriptional response element to measure NF-kB activity and Lucia reporter gene under the control of an ISG54 minimal promoter to measure IRF activity.
- SEAP embryonic alkaline phosphatase
- IRF activity was assessed using QUANTI-luc to measure levels of Lucia and NF-kB activity was determined by measure SEAP levels at 620-655 nm.
- % induction was calculated from fold change in luminescence/absorbance compared to DMSO treated sample.
- EC 50 values were generated by curve fit in Xlfit.
- Cells grown in complete media were treated with various concentrations of a compound of the disclosure or DMSO control.
- Dual cells carry both secreted embryonic alkaline phosphatase (SEAP) reporter gene under the control of an IFN- ⁇ minimal promoter fused to five copies of the NF-kB consensus transcriptional response element to measure NF-kB activity and Lucia reporter gene under the control of an ISG54 minimal promoter to measure IRF activity.
- SEAP secreted embryonic alkaline phosphatase
- IRF activity was assessed using QUANTI-luc to measure levels of Lucia and NF-kB activity was determined by measure SEAP levels at 620-655 nm.
- % induction was calculated from fold change in luminescence/absorbance compared to DMSO treated sample. EC 50 values are generated by curve fit in Xlfit.
- Table 3 EC 50 values for exemplary compounds of the disclosure.
- A represents an EC50 of less than 50 nM;
- B an EC 8 o of between 50 nM and 500 nM;
- C an EC 8 o of between 500 nM and 1 mM;
- D an EC 8 o of between 1 mM and 2 pM;
- E an EC 8 o of greater than 2 pM.
- Example 4 Study to Determine the Efficacy of Compound 4* in a CT26 Murine Colon Carcinoma Model Using Female BALB/c Mice
- mice Female BALB/c mice (BALB/c AnNcrl, Charles River) were eight weeks old on Day 1 of the study and had a body weight range of 15.1 to 19.7 g. The animals were fed ad libitum water (reverse osmosis, 1 ppm Cl) and a NIH 31 Modified and Irradiated Lab Diet consisting of 18.0% crude protein, 5.0% crude fat, and 5.0% crude fiber.
- ad libitum water reverse osmosis, 1 ppm Cl
- NIH 31 Modified and Irradiated Lab Diet consisting of 18.0% crude protein, 5.0% crude fat, and 5.0% crude fiber.
- CT26 murine colon carcinoma cells were grown in RPMI-1640 medium containing 10% fetal bovine serum, 2 mM glutamine, 100 units/mL penicillin G sodium, 100 mg/mL streptomycin sulfate, and 25 mg/mL gentamicin. The cells were cultured in tissue culture flasks in a humidified incubator at 37 °C, in an atmosphere of 5% CO 2 and 95% air.
- Compound 4 was dissolved by adding the appropriate volume of sterile saline (vehicle) into each tube, vortexing, incubating at 37 °C for 2-5 minutes, followed by sonication, if needed. Preparations of Compound 4 resulted in the appropriate 0.2 and 0.6 mg/mL dosing solutions which provided 1 and 3 mg/kg doses in a dosing volume of 5 mL/kg, adjusted to the body weight of the animal. A fresh vial was prepared on each day of dosing.
- the study endpoint was a tumor volume of 2000 mm 3 or Day 30, whichever came first. The study ended on Day 29.
- the study protocol specified a tumor growth delay assay based on the median time-to-endpoint (TTE) in a treated group versus the control group. Tumors were measured using calipers twice per week, and each animal was euthanized for tumor progression (TP) when its tumor reached the 2000 mm 3 volume endpoint.
- TTE for each mouse was calculated with the following equation: where b is the intercept and m is the slope of the line obtained by linear regression of a log- transformed tumor growth data set. The data set is comprised of the first observation that exceeded the study endpoint volume and the three consecutive observations that immediately preceded the attainment of the endpoint volume.
- TTE value equal to the last day of the study (Day 29).
- a linear interpolation was performed to approximate TTE.
- MTV (n) was defined as the median tumor volume of the number of animals, n, that survived to the last day and whose tumors had not reached the volume endpoint. Any animal determined to have died from treatment-related (TR) causes was to be assigned a TTE value equal to the day of death. Any animal that died from non- treatment-related (NTR) causes was to be excluded from the analysis. Treatment outcome was evaluated from tumor growth delay (TGD), which was defined as the increase in the median TTE for a treatment group compared to the control group:
- TGD T - C expressed in days, or as a percentage of the median TTE of the control group: wherein T is the median TTE for a treatment group and C is the median TTE for the control group.
- TGI Tumor Growth Inhibition
- the data set for TGI analysis includes all animals in a group, except those euthanized for sample collection (ES) and those that die due to treatment-related (TR) or non-treatment- related (NTR) causes.
- Treatment efficacy was also determined from the number of regression responses. Treatment may cause partial regression (PR) or complete regression (CR) of the tumor in an animal.
- PR partial regression
- CR complete regression
- the tumor volume is 50% or less of its Day 1 volume for three consecutive measurements during the course of the study, and equal to or greater than 13.5 mm for one or more of these three measurements.
- a CR response the tumor volume is less than 13.5 mm for three consecutive measurements during the course of the study. Animals were scored only once during the study for a PR or CR event and only as CR if both PR and CR criteria were satisfied. Any animal with a CR response on the last day of the study is additionally classified as a tumor-free-survivor (TFS).
- TFS tumor-free-survivor
- a death was to be classified as TR if it was attributable to treatment side effects as evidenced by clinical signs and/or necropsy, or may also be classified as TR if due to unknown causes during the dosing period or within 14 days of the last dose.
- a death was classified as NTR if there was evidence that the death was related to the tumor model, rather than treatment-related. NTR deaths are further categorized as NTRa (due to accident or human error), NTRm (due to necropsy-confirmed tumor dissemination by invasion or metastasis), and NTRu (due to unknown causes).
- Table 1 displays the study design as of Day 1 of the study. Vehicle is saline.
- the protected compound E' was taken in THF (1 mL). LiOH (5 eq) in water (0.5 mL) was added and stirred at room temperature for 3 hours. The solvent was then removed in vacuo and the residue was neutralized with 1 N HC 1 . The crude organic product was extracted with EtOAc (2 x 5 mL). Combined organic layers were dried (Na 2 S04), concentrated in vacuo and acid F ' (2 mg, 36%) formed was used as such in the next step.
- Synthesis of peptide, linked to a STING agonist could be achieved through the conversion of Ftnoc dipeptide intermediate F to the corresponding iodo derivative C*. Alkylation of STING agonist with G could yield the peptide linked STING agonist. Subsequent removal ofFmoc group would yield H.
- the protected compound E' could be taken up in THF (1 mL). LiOH (5eq) in water (0.5mL) could then be added and stirred at room temperature for 3 hours. The solvent could then be removed in vacuo and the residue could be neutralized with 1 N HCl. The crude organic product could be extracted with EtOAc (2 x 5 mL). Combined organic layers could be dried (Na 2 SO4), concentrated in vacuo to yield acid F'
- Example 9 Exemplary synthesis of b-glucuronide drug-linker unit:
- Aryl b-glucuronide was acylated with acid chloride 1, and then converted to p- nitrophenyl (PNP) carbonate 2 (US 2017/0189542; incorporated by reference).
- PNP p- nitrophenyl
- Example 11 Exemplary synthesis of heterocyclic self-immolative groups
- Compound 2A (1.5 g) was dissolved in 45 mL methanol. To the solution was added 8 mL of HCI in dioxane (4.0M). After stirring at room temperature r.t. for 2 hours, the solution was concentrated down at a rotary evaporator under reduced pressure. The crude product was dried under vacuum for additional 18 hours at r.t. and used for next reaction without further purification (US 2010/0273843; incorporated by reference).
- Compound 3A (1.2 g), 1.1 grams of Boc-valine, 1.4 mL of DIE A, and 3.3 g of HBTU were dissolved in 36 mL DMF. After stirring at r.t.
- Example 12 Exemplary synthesis of malemide self-immolative groups
- H-Lys(boc)-OH (246 mg, 1 mmol) and maleic anhydride (98 mg, 1 mmol) were dissolved in 1 mL acetic acid and the solution was stirred at room temperature for 3 hours.
- the reaction mixture was concentrated to an oil on the rotovap, and the product was precipitated by adding ⁇ 10 mL dichloromethane.
- the precipitate was collected by vacuum filtration, washed with dichloromethane, and dried overnight in the vacuum oven. 270mg of product was recovered as a white powder (85% yield) (WO 2013/173337; incorporated by reference).
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Molecular Biology (AREA)
- Organic Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Animal Behavior & Ethology (AREA)
- Biochemistry (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Immunology (AREA)
- Cell Biology (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biotechnology (AREA)
- Peptides Or Proteins (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
Disclosed are antibody-drug conjugates and compositions thereof for the activation or induction of expression of a pattern recognition receptor (e.g., STING, RIG-I, MDA5), and methods of use thereof.
Description
CYCLIC DINUCLEOTIDE STING AGONISTS TETHERED TO A PD-1 OR CTLA-4 ANTIBODY
RELATED APPLICATIONS
This application claims the benefit of priority to U.S. Provisional Patent Application No.: 62/896,983, filed September 6, 2019, the contents of which are hereby incorporated by reference.
BACKGROUND
Antibody Drug Conjugates (ADCs) are monoclonal antibodies (mAbs) attached to biologically active drugs by chemical linkers with labile bonds (Lu, J. Int. J. Mol. Sci. (2016) 17:561-583; Perez, H. Drug Discov. Today (2013) 00:1-13; Jain, N. Pharm. Res. (2015) 323526-3540). By combining the unique targeting capabilities of mAbs with the cancer-killing ability of cytotoxic drugs, ADCs allow sensitive discrimination between healthy and diseased tissue. Accordingly, ADCs represent an important class of biopharmaceutical drugs designed to act as a targeted therapy for the treatment of subjects with various disease states (Ducry, L. Bioconjugate Chemistry (2010) 21:5-13).
ADCs are comprised of a drug like small molecule, covalently linked to an antibody. The antibody represents a targeting mechanism tuned to a specific site of action. Upon reaching the site, the ADC is designed to release a small molecule, the drug, allowing it to perform its designed function in a targeted manner, as opposed to diffusing systemically through the entire body of the subject. This targeted approach allows for treatment with drugs that would otherwise require doses so high as to be toxic when administered systemically.
A key feature of the innate immune system is the recognition and elimination of foreign substances. Identification of these pathogenic invaders occurs through host recognition of evolutionarily conserved microbial structures known as pathogen-associated molecular patterns (PAMPs) (Jensen, S. and Thomsen, A.R. J Virol (2012) 86:2900-2910). Host recognition may occur by multiple pathways, such as activation of pattern recognition receptors (PRRs), which ultimately lead to downstream signaling events and culminate in the mounting of an immune response.
To date, several PRRs have been identified that serve as sensors of pathogenic infection. For example, the retinoic acid-inducible gene-I (RIG-I) protein is a RNA helicase that also functions as a sensor of microbial -derived RNA. RIG-I serves as a biomarker for the prediction of prognosis for certain types of cancer, such as hepatocellular carcinoma (Hou, J. et al, Cancer
Cell (2014) 25:49-63). Recent publications have highlighted the importance of RIG-I and STING as mediators of innate and adaptive immunity, and RIG-I and STING agonists have been recognized as immuno-oncology agents in cancer therapy (Li, X. Y. et al, Mol Cell Oncol (2014) 1 :e968016; Woo, S. R. Trends in Immunol (2015) 36:250-256). In particular, RIG-I is involved in the regulation of basic cellular processes such as hematopoietic proliferation and differentiation, maintenance of leukemic sternness, and tumorigenesis of hepatocellular carcinoma, indicating that RIG-I performs an essential function as a tumor suppressor. Importantly, the STING pathway of cytosolic DNA sensing has been shown to play an important mechanistic role in innate immune sensing, driving type I IFN production in cancer and in the context of immune-oncology applications, including therapeutics and diagnostics.
An important part of the immune system is its ability to tell between normal cells in the body and those it sees as “foreign.” This lets the immune system attack the foreign cells while leaving the normal cells alone. To do this, the immune uses “checkpoints” - molecules on certain immune cells that need to be activated (or inactivated) to start an immune response. For example, Programmed death-1 (PD-1) is a co-receptor that is expressed predominantly by T cells. The binding of PD-1 to its ligands, PD-L1 or PD-L2, is vital for the physiological regulation of the immune system. A major functional role of the PD-1 signaling pathway is the inhibition of self-reactive T cells, which serve to protect against autoimmune diseases. Elimination of the PD-1 pathway can therefore result in the breakdown of immune tolerance that can ultimately lead to the development of pathogenic autoimmunity. Conversely, tumor cells can at times co-opt the PD-1 pathway to escape from immune-surveillance mechanisms. Therefore, blockade of the PD-1 pathway has become an attractive target in cancer therapy.
In view of the forgoing, there exists a need for therapies that combine both the advantages associated with both STING agonists and immune checkpoint inhibitors.
SUMMARY OF THE INVENTION
Cyclic dinucleotide compounds, compositions comprising cyclic dinucleotide compounds, and related methods of use are described herein. In one aspect, the disclosure features an antibody-drug conjugate of Formula (I):
 or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
Ab is an anti-PD-1 antibody or an anti-CTLA-4 antibody; each of B1 and B2 is independently a purinyl nucleobase or pyrimidinyl nucleobase; each of X1 and X2 is independently O or S; each of Y1 and Y2 is independently O, S, or N(R5); each of Z1 is indpendently O or S; each of Z2 and Z3 is independently absent, -C1-C20- alkylene (e.g., -C1-C6- alkylene), C1-C20 heteroalkylene (e.g., -C1-C6- heteroalkylene), -OC(O)OC1-C20- alkylene (e.g., -C1-C6- alkylene), -cycloalkylene-, -heterocyclyl-, -aryl-, or -heteroaryl-, wherein each -cycloakyl-, -heterocyclyl-, -aryl- or -heteroaryl- is optionally substituted with one or more R4;
Z4 is self-immolative group-C1-C20-alkylene-Q1 (e.g., -self-immolative group-C1-C6- alkylene-Q1), heterocyclyl-C1-C20-alkylene-Q (e.g., heterocyclyl-C1-C6-alkylene-Q),- OH, -N(R5)2, SR5, -CHO, -C(O)N(R5)2, -OC(O)N(R5)2, -N(R5)C(O)OR5, aryl, heteroaryl, -S(O)R5,
-S(O)2R5, -S(O)N(R5)2, -S(O)2N(R5)2, -N(R5)S(O)R5, -OSi(C1-C4 alkyl)3, or -C(O)C2- C6 alkenyl (e.g., -C2-C4 alkenyl);
Z5 is a self-immolative group or absent;
T is a spacer group or absent;
L1 is absent, -C1-C6- alkylene (e.g., -C1-C3- alkylene) or -C1-C6- heteroalkylene;
L2 is absent, -C1-C6- alkylene (e.g., -C1-C3- alkylene) or -C1-C6- heteroalkylene, wherein each alkylene and heteroalkyl is optionally substituted with one or more R6;
L3 is absent, -C1-C20- alkylene, -O-, -N(R5)-, -S-, -S(O)-, -S(O)2-, -S(O)N(R5)-,
-S(O)2N(R5)-, -N(R5)S(O)-, -N(R5)S(O)2-, -C(O)-, -C(O)O-, OC(O) -, -C(O)N(R5)-, -N(R5)C(O)-;
L4 is -C1-C20- alkylene (e.g., -C1-C6- alkylene), -C1-C20- heteroalkylene (e.g., -C1-C6- heteroalkylene), -C1-C20- alkenylene (e.g., -C2-C6- alkenylene), -C1-C20- alkynylene (e.g., - -C2-C6- alkynylene), or an oligopeptide, wherein the oligopeptide is optionally substituted by one or more R16;
L5 is a linker connecting Z4 and Z5, or is absent;
Q1 is C(O), C(S), or CH2; each of R1 and R2 is independently hydrogen, halo, -CN, -C1-C20 alkyl (e.g., C1-C6 alkyl), or - OR7;
R3 is hydrogen, -C1-C20- alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl (e.g., -C1-C6 heteroalkyl), -OC(O)OC1-C20 alkyl (e.g., -C1-C6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, heteroalkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R8; each R4 is independently hydrogen, -C1-C20 alkyl, -O-C1-C20 alkyl, -C1-C20 heteroalkyl, halo, -CN, -NO2 or -OH;
R5 is hydrogen or -C1-C20 alkyl (e.g., -C1-C6 alkyl);
R6 is halo, -CN, -C1-C20 alkyl (e.g., -C1-C6 alkyl), -OR7, oxo, cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R9;
R7 is hydrogen, -C1-C20 alkyl (e.g., -C1-C6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R9; each R8 is independently -C1-C20 alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl,
-C(O)-C1-C20 alkyl, -OC(O)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -C(O)O-C1-C20 alkyl (e.g·,
-C1-C6 alkyl), -OC(O)O-C1-C20 alkyl (e.g., -C1-C6 alkyl), -C(O)N(R5)-C1-C20 alkyl (e.g·,
-C1-C6 alkyl), -N(R5)C(O)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -OC(O)N(R5)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -O-aryl, -O-heteroaryl, -C(O)-aryl, -C(O)-heteroaryl, - OC(O)-aryl,
-C(O)O-aryl, -OC(O)-heteroaryl, -C(O)O-heteroaryl, -C(O)O-aryl,
-C(O)O-heteroaryl, -C(O)N(R5)-aryl, -C(O)N(R5)-heteroaryl, -N(R5)C(O)-aryl,
-N(R5)2C(O)-aryl, or -N(R5)C(O)-heteroaryl, -S(O)2N(R5)-aryl, wherein each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R9; each R9 is independently -C1-C20 alkyl, -O-C1-C20 alkyl, -C1-C20 heteroalkyl, halo,
-CN, -OH, oxo, aryl, heteroaryl, -O-aryl, or -O-heteroaryl; and each R16 is independently, -C1-C20 alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl (e.g., -C1-C6 heteroalkyl), -OC(O)OC1-C20 alkyl (e.g., -C1-C6 alkyl), C(O)N(R4)2 cycloalkyl, heterocyclyl, aryl, or heteroaryl.
In another aspect, the disclosure features an antibody-drug conjugate of Formula (IV):
 or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
Ab is an anti-PD-1 antibody or an anti-CTLA-4 antibody; each of B1 and B2 is independently a purinyl nucleobase or pyrimidinyl nucleobase; each of X1 and X2 is independently O or S; each of Y1 and Y2 is independently O, S, or N(R5); each of Z1 is independently O or S;
Z2 is -O-, -N(R5)-, -S-, -C(O)-, -C(O)N(R5)-, -OC(O)N(R5)-, -N(R5)C(O)O-, -aryl-, -heteroaryl-, -S(O)-, -S(O)2-, -S(O)N(R5)-, -S(O)2N(R5)- or -N(R5)S(O)-;
Z3 is absent, hydrogen, heterocyclyl, heterocyclyl-C1-C20-alkylene-Q1, -OH, -N(R5)2, SR5, - CHO, -C(O)N(R5)2, -OC(O)N(R5)2, -N(R5)C(O)OR5, aryl, heteroaryl, -S(O)R5, - S(O)2R5, -S(O)N(R5)2, -S(O)2N(R5)2, -N(R5)S(O)R5, -OSi(C1-C4 alkyl)3, or -C(O)C2- C6 alkenyl (e.g., -C2-C4 alkenyl);
Z4 is a self-immolative group or absent;
T is a absent or spacer group;
each L1 and L2 is absent, -C1-C6- alkylene (e.g., -C1-C3- alkylene) or -C1-C6- heteroalkyl ene, wherein each alkylene and heteroalkyl is optionally substituted with one or more R6;
L3 is oligiopeptide-C(O)-, oligiopeptide-aryl-C1-C6-alkylene-, oligiopeptide-aryl-C1-C6- alklyene-C(O)-, oligiopeptide-aryl-C1-C6-heteroalkylene, oligiopeptide-C1-C6- alkylene-C(O)-, oligiopeptide-C1-C6-heteroalkylene-C(O)-, -C1-C40- alkylene (e.g., - C1-C20- alkylene), -C1-C40- heteroalkyl ene (e.g., -C1-C20- heteroalkyl), -C1-C40- alkenylene (e.g., -C2-C20- alkenylene), or -C1-C40- alkynylene (e.g., -C2-C20- alkynylene), wherein the oligopeptide is optionally substituted by one or more R14;
L4 is absent or a linker connecting Z3 and Z4;
Q1 is C(O), C(S), or CH2; each of R1 and R2 is independently hydrogen, halo, -CN, -C1-C20 alkyl (e.g., C1-C6 alkyl), or - OR7; each R3 and R4 is independently hydrogen, -C1-C20- alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl (e.g., -C1-C6 heteroalkyl), -OC(O)OC1-C20 alkyl (e.g., -C1-C6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, heteroalkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R8;
R5 is hydrogen or -C1-C20 alkyl (e.g., -C1-C6 alkyl);
R6 is halo, -CN, -C1-C20 alkyl (e.g., -C1-C6 alkyl), -OR7, oxo, cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R9;
R7 is hydrogen, -C1-C20 alkyl (e.g., -C1-C6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R9; each R8 is independently -C1-C20 alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl,
-C(O)-C1-C20 alkyl, -OC(O)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -C(O)O-C1-C20 alkyl (e.g., -C1-C6 alkyl), -OC(O)O-C1-C20 alkyl (e.g., -C1-C6 alkyl), -C(O)N(R5)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -N(R5)C(O)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -OC(O)N(R5)- C1-C20 alkyl (e.g., -C1-C6 alkyl), -O-aryl, -O-heteroaryl, -C(O)-aryl, -C(O)-heteroaryl, -OC(O)-aryl, -C(O)O-aryl, -OC(O)-heteroaryl, -C(O)O-heteroaryl, -C(O)O-aryl, - C(O)O-heteroaryl, -C(O)N(R5)-aryl, -C(O)N(R5)-heteroaryl, -N(R5)C(O)-aryl, - N(R5)2C(O)-aryl, or -N(R5)C(O)-heteroaryl, -S(O)2N(R5)-aryl, wherein each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R9;
each R9 is independently -C1-C20 alkyl, -O-C1-C20 alkyl, -C1-C20 heteroalkyl, halo,
-CN, -OH, oxo, aryl, heteroaryl, -O-aryl, or -O-heteroaryl; and each R16 is independently, -C1-C20 alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl (e.g., -C1-C6 heteroalkyl), -OC(O)OC1-C20 alkyl (e.g., -C1-C6 alkyl), C(O)N(R4)2 cycloalkyl, heterocyclyl, aryl, or heteroaryl.
In yet another aspect, the disclosure features an antibody-drug conjugate of Formula (Vlla) or Formula (Vllb):
 or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
Ab is an anti-PD-1 antibody or an anti-CTLA-4 antibody; each of B1 and B2 is independently a purinyl nucleobase or pyrimidinyl nucleobase; each of X1 and X2 is independently O or S; each of Y1 and Y2 is independently O, S, or N(R5); each of Z1 is independently O or S;
Z2 is -O-, -N(R5)-, -S-, -C(O)-, -C(O)N(R5)-, -OC(O)N(R5)-, -N(R5)C(O)O-, -aryl-, -heteroaryl-, -S(O)-, -S(O)2-, -S(O)N(R5)-, -S(O)2N(R5)- or -N(R5)S(O)-;
Z3 is absent, hydrogen, heterocyclyl, heterocyclyl-C1-C20-alkylene-Q1, -OH, -N(R5)2, SR5, - CHO, -C(O)N(R5)2, -OC(O)N(R5)2, -N(R5)C(O)OR5, aryl, heteroaryl, -S(O)R5, - S(O)2R5, -S(O)N(R5)2, -S(O)2N(R5)2, -N(R5)S(O)R5, -OSi(C1-C4 alkyl)3, or -C(O)C2- C6 alkenyl (e.g., -C2-C4 alkenyl);
Z4 is a self-immolative group or absent;
T is a absent or spacer group; each L1 and L2 is absent, -C1-C6- alkylene (e.g., -C1-C3- alkylene) or -C1-C6- heteroalkyl ene, wherein each alkylene and heteroalkyl is optionally substituted with one or more R6;
L3 is oligiopeptide-C(O)-, oligiopeptide-aryl-C1-C6-alkylene-, oligiopeptide-aryl-C1-C6- alklyene-C(O)-, oligiopeptide-aryl-C1-C6-heteroalkylene, oligiopeptide-C1-C6- alkylene-C(O)-, oligiopeptide-C1-C6-heteroalkylene-C(O)-, -C1-C40- alkylene (e.g., - C1-C20- alkylene), -C1-C40- heteroalkyl ene (e.g., -C1-C20- heteroalkyl), -C1-C40- alkenylene (e.g., -C2-C20- alkenylene), or -C1-C40- alkynylene (e.g., -C2-C20- alkynylene), wherein the oligopeptide is optionally substituted by one or more R14;
L4 is absent or a linker connecting Z3 and Z4;
Q1 is C(O), C(S), or CH2; each of R1 and R2 is independently hydrogen, halo, -CN, -C1-C20 alkyl (e.g., C1-C6 alkyl), or - OR7; each R3 and R4 is independently hydrogen, -C1-C20- alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl (e.g., -C1-C6 heteroalkyl), -OC(O)OC1-C20 alkyl (e.g., -C1-C6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, heteroalkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R8;
R5 is hydrogen or -C1-C20 alkyl (e.g., -C1-C6 alkyl);
R6 is halo, -CN, -C1-C20 alkyl (e.g., -C1-C6 alkyl), -OR7, oxo, cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R9;
R7 is hydrogen, -C1-C20 alkyl (e.g., -C1-C6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R9; each R8 is independently -C1-C20 alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl,
-C(O)-C1-C20 alkyl, -OC(O)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -C(O)O-C1-C20 alkyl (e.g., -C1-C6 alkyl), -OC(O)O-C1-C20 alkyl (e.g., -C1-C6 alkyl), -C(O)N(R5)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -N(R5)C(O)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -OC(O)N(R5)- C1-C20 alkyl (e.g., -C1-C6 alkyl), -O-aryl, -O-heteroaryl, -C(O)-aryl, -C(O)-heteroaryl, -OC(O)-aryl, -C(O)O-aryl, -OC(O)-heteroaryl, -C(O)O-heteroaryl, -C(O)O-aryl, - C(O)O-heteroaryl, -C(O)N(R5)-aryl, -C(O)N(R5)-heteroaryl, -N(R5)C(O)-aryl, - N(R5)2C(O)-aryl, or -N(R5)C(O)-heteroaryl, -S(O)2N(R5)-aryl, wherein each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R9; each R9 is independently -C1-C20 alkyl, -O-C1-C20 alkyl, -C1-C20 heteroalkyl, halo,
-CN, -OH, oxo, aryl, heteroaryl, -O-aryl, or -O-heteroaryl; and
each R16 is independently, -C1-C20 alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl (e.g., -C1-C6 heteroalkyl), -OC(O)OC1-C20 alkyl (e.g., -C1-C6 alkyl), C(O)N(R4)2 cycloalkyl, heterocyclyl, aryl, or heteroaryl.
In yet another aspect, the disclosure features an antibody-drug conjugate of Formula (Xa) or Formula (Xb):
 or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
Ab is an anti-PD-1 antibody or an anti-CTLA-4 antibody; each of B1 and B2 is independently a purinyl nucleobase or pyrimidinyl nucleobase; each of X1 and X2 is independently O or S; each of Y1 and Y2 is independently O, S, or N(R5); each of Z1 is independently O or S;
Z2 is -O-, -N(R5)-, -S-, -C(O)-, -C(O)N(R5)-, -OC(O)N(R5)-, -N(R5)C(O)O-, -aryl-, -heteroaryl-, -S(O)-, -S(O)2-, -S(O)N(R5)-, -S(O)2N(R5)- or -N(R5)S(O)-;
Z3 is absent, hydrogen, heterocyclyl, heterocyclyl-C1-C20-alkylene-Q1, -OH, -N(R5)2, SR5, - CHO, -C(O)N(R5)2, -OC(O)N(R5)2, -N(R5)C(O)OR5, aryl, heteroaryl, -S(O)R5, - S(O)2R5, -S(O)N(R5)2, -S(O)2N(R5)2, -N(R5)S(O)R5, -OSi(C1-C4 alkyl)3, or -C(O)C2- C6 alkenyl (e.g., -C2-C4 alkenyl);
Z4 is a self-immolative group or absent;
T is a absent or spacer group; each L1 and L2 is absent, -C1-C6- alkylene (e.g., -C1-C3- alkylene) or -C1-C6- heteroalkyl ene, wherein each alkylene and heteroalkyl is optionally substituted with one or more R6;
L3 is oligiopeptide-C(O)-, oligiopeptide-aryl-C1-C6-alkylene-, oligiopeptide-aryl-C1-C6- alklyene-C(O)-, oligiopeptide-aryl-C1-C6-heteroalkylene, oligiopeptide-C1-C6- alkylene-C(O)-, oligiopeptide-C1-C6-heteroalkylene-C(O)-, -C1-C40- alkylene (e.g., - C1-C20- alkylene), -C1-C40- heteroalkyl ene (e.g., -C1-C20- heteroalkyl), -C1-C40- alkenylene (e.g., -C2-C20- alkenylene), or -C1-C40- alkynylene (e.g., -C2-C20- alkynylene), wherein the oligopeptide is optionally substituted by one or more R14;
L4 is absent or a linker connecting Z3 and Z4;
Q1 is C(O), C(S), or CH2; each of R1 and R2 is independently hydrogen, halo, -CN, -C1-C20 alkyl (e.g., C1-C6 alkyl), or - OR7; each R3 and R4 is independently hydrogen, -C1-C20- alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl (e.g., -C1-C6 heteroalkyl), -OC(O)OC1-C20 alkyl (e.g., -C1-C6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, heteroalkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R8;
R5 is hydrogen or -C1-C20 alkyl (e.g., -C1-C6 alkyl);
R6 is halo, -CN, -C1-C20 alkyl (e.g., -C1-C6 alkyl), -OR7, oxo, cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R9;
R7 is hydrogen, -C1-C20 alkyl (e.g., -C1-C6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R9; each R8 is independently -C1-C20 alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl,
-C(O)-C1-C20 alkyl, -OC(O)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -C(O)O-C1-C20 alkyl
(e.g, -C1-C6 alkyl), -OC(O)O-C1-C20 alkyl (e.g., -C1-C6 alkyl), -C(O)N(R5)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -N(R5)C(O)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -OC(O)N(R5)- C1-C20 alkyl (e.g., -C1-C6 alkyl), -O-aryl, -O-heteroaryl, -C(O)-aryl, -C(O)-heteroaryl, -OC(O)-aryl, -C(O)O-aryl, -OC(O)-heteroaryl, -C(O)O-heteroaryl, -C(O)O-aryl, - C(O)O-heteroaryl, -C(O)N(R5)-aryl, -C(O)N(R5)-heteroaryl, -N(R5)C(O)-aryl, - N(R5)2C(O)-aryl, or -N(R5)C(O)-heteroaryl, -S(O)2N(R5)-aryl, wherein each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R9; each R9 is independently -C1-C20 alkyl, -O-C1-C20 alkyl, -C1-C20 heteroalkyl, halo,
-CN, -OH, oxo, aryl, heteroaryl, -O-aryl, or -O-heteroaryl; and each R16 is independently, -C1-C20 alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl (e.g., -C1-C6 heteroalkyl), -OC(O)OC1-C20 alkyl (e.g., -C1-C6 alkyl), C(O)N(R4)2 cycloalkyl, heterocyclyl, aryl, or heteroaryl.
Also disclosed herein are methods of using the antibody-drug conjugates of the disclosure in the treatment of certain diseases (e.g., cancer).
BRIEF DESCRIPTION OF THE DRAWINGS Figure 1 depicts a mechanism of cleavage of an antibody-STING agonist conjugate.
DETATEED DESCRIPTION OF THE INVENTION
Definitions
Unless otherwise defined herein, scientific and technical terms used in this application shall have the meanings that are commonly understood by those of ordinary skill in the art. Generally, nomenclature used in connection with, and techniques of, chemistry, cell and tissue culture, molecular biology, cell and cancer biology, neurobiology, neurochemistry, virology, immunology, microbiology, pharmacology, genetics and protein and nucleic acid chemistry, described herein, are those well known and commonly used in the art.
The methods and techniques of the present disclosure are generally performed, unless otherwise indicated, according to conventional methods well known in the art and as described in various general and more specific references that are cited and discussed throughout this specification. See, e.g. “Principles of Neural Science”, McGraw-Hill Medical, New York, N. Y. (2000); Motulsky, “Intuitive Biostatistics”, Oxford University Press, Inc. (1995); Lodish et ah, “Molecular C6ll Biology, 4th ed ”, W. H. Freeman & Co., New York (2000); Griffiths et ah, “Introduction to Genetic Analysis, 7th ed ”, W. H. Freeman & Co., N.Y. (1999); and Gilbert et ak, “Developmental Biology, 6th ed.”, Sinauer Associates, Inc., Sunderland, MA (2000).
Chemistry terms used herein, unless otherwise defined herein, are used according to conventional usage in the art, as exemplified by “The McGraw-Hill Dictionary of Chemical Terms”, Parker S., Ed., McGraw-Hill, San Francisco, C.A. (1985).
All of the above, and any other publications, patents and published patent applications referred to in this application are specifically incorporated by reference herein. In case of conflict, the present specification, including its specific definitions, will control.
The term “agent” is used herein to denote a chemical compound (such as an organic or inorganic compound, a mixture of chemical compounds), a biological macromolecule (such as a nucleic acid, an antibody, including parts thereof as well as humanized, chimeric and human antibodies and monoclonal antibodies, a protein or portion thereof, e.g., a peptide, a lipid, a carbohydrate), or an extract made from biological materials such as bacteria, plants, fungi, or animal (particularly mammalian) cells or tissues. Agents include, for example, agents whose structure is known, and those whose structure is not known. The ability of such agents to inhibit AR or promote AR degradation may render them suitable as “therapeutic agents” in the methods and compositions of this disclosure.
A “patient,” “subject,” or “individual” are used interchangeably and refer to either a human or a non-human animal. These terms include mammals, such as humans, primates, livestock animals (including bovines, porcines, etc.), companion animals (e.g., canines, felines, etc.) and rodents (e.g., mice and rats).
“Treating” a condition or patient refers to taking steps to obtain beneficial or desired results, including clinical results. As used herein, and as well understood in the art, “treatment” is an approach for obtaining beneficial or desired results, including clinical results. Beneficial or desired clinical results can include, but are not limited to, alleviation or amelioration of one or more symptoms or conditions, diminishment of extent of disease, stabilized (i.e. not worsening) state of disease, preventing spread of disease, delay or slowing of disease progression, amelioration or palliation of the disease state, and remission (whether partial or total), whether detectable or undetectable. “Treatment” can also mean prolonging survival as compared to expected survival if not receiving treatment.
The term “preventing” is art-recognized, and when used in relation to a condition, such as a local recurrence (e.g., pain), a disease such as cancer, a syndrome complex such as heart failure or any other medical condition, is well understood in the art, and includes administration of a composition which reduces the frequency of, or delays the onset of, symptoms of a medical condition in a subject relative to a subject which does not receive the composition. Thus, prevention of cancer includes, for example, reducing the number of detectable cancerous
growths in a population of patients receiving a prophylactic treatment relative to an untreated control population, and/or delaying the appearance of detectable cancerous growths in a treated population versus an untreated control population, e.g., by a statistically and/or clinically significant amount.
“Administering” or “administration of’ a substance, a compound or an agent to a subject can be carried out using one of a variety of methods known to those skilled in the art. For example, a compound or an agent can be administered, intravenously, arterially, intradermally, intramuscularly, intraperitoneally, subcutaneously, ocularly, sublingually, orally (by ingestion), intranasally (by inhalation), intraspinally, intracerebrally, and transdermally (by absorption, e.g., through a skin duct). A compound or agent can also appropriately be introduced by rechargeable or biodegradable polymeric devices or other devices, e.g., patches and pumps, or formulations, which provide for the extended, slow or controlled release of the compound or agent. Administering can also be performed, for example, once, a plurality of times, and/or over one or more extended periods.
Appropriate methods of administering a substance, a compound or an agent to a subject will also depend, for example, on the age and/or the physical condition of the subject and the chemical and biological properties of the compound or agent (e.g., solubility, digestibility, bioavailability, stability and toxicity). In some embodiments, a compound or an agent is administered orally, e.g., to a subject by ingestion. In some embodiments, the orally administered compound or agent is in an extended release or slow release formulation, or administered using a device for such slow or extended release.
As used herein, the phrase “conjoint administration” refers to any form of administration of two or more different therapeutic agents such that the second agent is administered while the previously administered therapeutic agent is still effective in the body (e.g., the two agents are simultaneously effective in the patient, which may include synergistic effects of the two agents). For example, the different therapeutic compounds can be administered either in the same formulation or in separate formulations, either concomitantly or sequentially. Thus, an individual who receives such treatment can benefit from a combined effect of different therapeutic agents.
A “therapeutically effective amount” or a “therapeutically effective dose” of a drug or agent is an amount of a drug or an agent that, when administered to a subject will have the intended therapeutic effect. The full therapeutic effect does not necessarily occur by administration of one dose, and may occur only after administration of a series of doses. Thus, a therapeutically effective amount may be administered in one or more administrations. The
precise effective amount needed for a subject will depend upon, for example, the subject’s size, health and age, and the nature and extent of the condition being treated, such as cancer or MDS. The skilled worker can readily determine the effective amount for a given situation by routine experimentation.
As used herein, the terms “optional” or “optionally” mean that the subsequently described event or circumstance may occur or may not occur, and that the description includes instances where the event or circumstance occurs as well as instances in which it does not. For example, “optionally substituted alkyl” refers to the alkyl may be substituted as well as where the alkyl is not substituted.
It is understood that substituents and substitution patterns on the compounds of the present disclosure can be selected by one of ordinary skilled person in the art to result chemically stable compounds which can be readily synthesized by techniques known in the art, as well as those methods set forth below, from readily available starting materials. If a substituent is itself substituted with more than one group, it is understood that these multiple groups may be on the same carbon or on different carbons, so long as a stable structure results.
As used herein, the term “optionally substituted” refers to the replacement of one to six hydrogen radicals in a given structure with the radical of a specified substituent including, but not limited to: hydroxyl, hydroxyalkyl, alkoxy, halogen, alkyl, nitro, silyl, acyl, acyloxy, aryl, cycloalkyl, heterocyclyl, amino, aminoalkyl, cyano, haloalkyl, haloalkoxy, -OCO-CH2-O- alkyl, -OP(O)(O-alkyl)2 or -CH2 -OP(O)(O-alkyl)2. Preferably, “optionally substituted” refers to the replacement of one to four hydrogen radicals in a given structure with the substituents mentioned above. More preferably, one to three hydrogen radicals are replaced by the substituents as mentioned above. It is understood that the substituent can be further substituted.
As used herein, the term “alkyl” refers to saturated aliphatic groups, including but not limited to C1-C10 straight-chain alkyl groups or C1-C10 branched-chain alkyl groups. Preferably, the “alkyl” group refers to C1-C6 straight-chain alkyl groups or C1-C6 branched- chain alkyl groups. Most preferably, the “alkyl” group refers to C1-C4 straight-chain alkyl groups or C1-C4 branched-chain alkyl groups. Examples of “alkyl” include, but are not limited to, methyl, ethyl, 1 -propyl, 2-propyl, n-butyl, sec-butyl, tert-butyl, 1 -pentyl, 2-pentyl, 3 -pentyl, neo-pentyl, 1 -hexyl, 2-hexyl, 3 -hexyl, 1-heptyl, 2-heptyl, 3-heptyl, 4-heptyl, 1 -octyl, 2-octyl, 3-octyl or 4-octyl and the like. The “alkyl” group may be optionally substituted.
The term “acyl” is art-recognized and refers to a group represented by the general formula hydrocarbylC(O)-, preferably alkylC(O)-.
The term “acylamino” is art-recognized and refers to an amino group substituted with an acyl group and may be represented, for example, by the formula hydrocarbylC(O)NH-.
The term “acyloxy” is art-recognized and refers to a group represented by the general formula hydrocarbylC(O)O-, preferably alkylC(O)O-.
The term “alkoxy” refers to an alkyl group having an oxygen attached thereto. Representative alkoxy groups include methoxy, ethoxy, propoxy, tert-butoxy and the like.
The term “alkoxyalkyl” refers to an alkyl group substituted with an alkoxy group and may be represented by the general formula alkyl -O-alkyl.
The term “alkyl” refers to saturated aliphatic groups, including straight-chain alkyl groups, branched-chain alkyl groups, cycloalkyl (alicyclic) groups, alkyl -substituted cycloalkyl groups, and cycloalkyl-substituted alkyl groups. In preferred embodiments, a straight chain or branched chain alkyl has 30 or fewer carbon atoms in its backbone (e.g., C1- 30 for straight chains, C3-30 for branched chains), and more preferably 20 or fewer.
Moreover, the term “alkyl” as used throughout the specification, examples, and claims is intended to include both unsubstituted and substituted alkyl groups, the latter of which refers to alkyl moieties having substituents replacing a hydrogen on one or more carbons of the hydrocarbon backbone, including haloalkyl groups such as trifluoromethyl and 2,2,2- trifluoroethyl, etc.
The term “Cx-y” or “Cx-Cy”, when used in conjunction with a chemical moiety, such as, acyl, acyloxy, alkyl, alkenyl, alkynyl, or alkoxy is meant to include groups that contain from x to y carbons in the chain. Coalkyl indicates a hydrogen where the group is in a terminal position, a bond if internal. A C1-6alkyl group, for example, contains from one to six carbon atoms in the chain.
The term “alkylamino”, as used herein, refers to an amino group substituted with at least one alkyl group.
The term “alkylthio”, as used herein, refers to a thiol group substituted with an alkyl group and may be represented by the general formula alkylS-.
The term “amide”, as used herein, refers to a group
 wherein R9 and R10 each independently represent a hydrogen or hydrocarbyl group, or R9 and R10 taken together with the N atom to which they are attached complete a heterocycle having from 4 to 8 atoms in the ring structure.
The terms “amine” and “amino” are art-recognized and refer to both unsubstituted and substituted amines and salts thereof, e.g., a moiety that can be represented by
wherein R9 and R10 each independently represent a hydrogen or hydrocarbyl group, or R9 and R10 taken together with the N atom to which they are attached complete a heterocycle having from 4 to 8 atoms in the ring structure.
The terms “amine” and “amino” are art-recognized and refer to both unsubstituted and substituted amines and salts thereof, e.g., a moiety that can be represented by
 wherein R9, R10, and R10’ each independently represent a hydrogen or a hydrocarbyl group, or R9 and R10 taken together with the N atom to which they are attached complete a heterocycle having from 4 to 8 atoms in the ring structure.
wherein R9, R10, and R10’ each independently represent a hydrogen or a hydrocarbyl group, or R9 and R10 taken together with the N atom to which they are attached complete a heterocycle having from 4 to 8 atoms in the ring structure.
The term “aminoalkyl”, as used herein, refers to an alkyl group substituted with an amino group.
The term “aralkyl”, as used herein, refers to an alkyl group substituted with an aryl group.
The term “aryl” as used herein include substituted or unsubstituted single-ring aromatic groups in which each atom of the ring is carbon. Preferably the ring is a 5- to 7-membered ring, more preferably a 6-membered ring. The term “aryl” also includes polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is aromatic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls. Aryl groups include benzene, naphthalene, phenanthrene, phenol, aniline, and the like. The aromatic ring may be substituted at one or more ring positions with one or more substituents, such as halogen, azide, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, alkoxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, sulfonamido, ketone, aldehyde, ester, heterocyclyl, aromatic or heteroaromatic moieties, fluoroalkyl (such as trifluromethyl), cyano, or the like.
The term “carbamate” is art-recognized and refers to a group
 wherein R9 and R10 independently represent hydrogen or a hydrocarbyl group.
wherein R9 and R10 independently represent hydrogen or a hydrocarbyl group.
The term “carbocyclylalkyl”, as used herein, refers to an alkyl group substituted with a carbocycle group.
The term “carbocycle” includes 5-7 membered monocyclic and 8-12 membered bicyclic rings. Each ring of a bicyclic carbocycle may be selected from saturated, unsaturated and aromatic rings. Carbocycle includes bicyclic molecules in which one, two or three or more
atoms are shared between the two rings. The term “fused carbocycle” refers to a bicyclic carbocycle in which each of the rings shares two adjacent atoms with the other ring. Each ring of a fused carbocycle may be selected from saturated, unsaturated and aromatic rings. In an exemplary embodiment, an aromatic ring, e.g., phenyl, may be fused to a saturated or unsaturated ring, e.g., cyclohexane, cyclopentane, or cyclohexene. Any combination of saturated, unsaturated and aromatic bicyclic rings, as valence permits, is included in the definition of carbocyclic. Exemplary “carbocycles” include cyclopentane, cyclohexane, bicyclo[2.2.1]heptane, 1,5-cyclooctadiene, 1,2,3,4-tetrahydronaphthalene, bicyclo[4.2.0]oct- 3-ene, naphthalene and adamantane. Exemplary fused carbocycles include decalin, naphthalene, 1,2,3,4-tetrahydronaphthalene, bicyclo[4.2.0]octane, 4,5,6,7-tetrahydro-lH- indene and bicyclo[4.1.0]hept-3-ene. “Carbocycles” may be substituted at any one or more positions capable of bearing a hydrogen atom.
The term “carbocyclylalkyl”, as used herein, refers to an alkyl group substituted with a carbocycle group.
The term “carbonate” is art-recognized and refers to a group -OCO2-.
The term “carboxy”, as used herein, refers to a group represented by the formula -CO2H.
The term “ester”, as used herein, refers to a group -C(O)OR9 wherein R9 represents a hydrocarbyl group.
The term “ether”, as used herein, refers to a hydrocarbyl group linked through an oxygen to another hydrocarbyl group. Accordingly, an ether substituent of a hydrocarbyl group may be hydrocarbyl-O-. Ethers may be either symmetrical or unsymmetrical. Examples of ethers include, but are not limited to, heterocycle-O-heterocycle and aryl-O-heterocycle. Ethers include “alkoxyalkyl” groups, which may be represented by the general formula alkyl- O-alkyl.
The terms “halo” and “halogen” as used herein means halogen and includes chloro, fluoro, bromo, and iodo.
The terms “hetaralkyl” and “heteroaralkyl”, as used herein, refers to an alkyl group substituted with a hetaryl group.
The terms “heteroaryl” and “hetaryl” include substituted or unsubstituted aromatic single ring structures, preferably 5- to 7-membered rings, more preferably 5- to 6-membered rings, whose ring structures include at least one heteroatom, preferably one to four heteroatoms, more preferably one or two heteroatoms. The terms “heteroaryl” and “hetaryl” also include polycyclic ring systems having two or more cyclic rings in which two or more carbons are
common to two adjoining rings wherein at least one of the rings is heteroaromatic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls. Heteroaryl groups include, for example, pyrrole, furan, thiophene, imidazole, oxazole, thiazole, pyrazole, pyridine, pyrazine, pyridazine, pyrimidine, and the like. The heteroaromatic ring may be substituted at one or more ring positions with one or more substituents, such as halogen, azide, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, alkoxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, sulfonamido, ketone, aldehyde, ester, heterocyclyl, aromatic or heteroaromatic moieties, fluoroalkyl (such as trifluromethyl), cyano, or the like.
The term “heteroatom” as used herein means an atom of any element other than carbon or hydrogen. Preferred heteroatoms are nitrogen, oxygen, and sulfur.
The term “heterocyclylalkyl”, as used herein, refers to an alkyl group substituted with a heterocycle group.
The terms “heterocyclyl”, “heterocycle”, and “heterocyclic” refer to substituted or unsubstituted non-aromatic ring structures, preferably 3- to 10-membered rings, more preferably 3- to 7-membered rings, whose ring structures include at least one heteroatom, preferably one to four heteroatoms, more preferably one or two heteroatoms. The terms “heterocyclyl” and “heterocyclic” also include polycyclic ring systems having two or more cyclic rings in which two or more carbons are common to two adjoining rings wherein at least one of the rings is heterocyclic, e.g., the other cyclic rings can be cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls. Heterocyclyl groups include, for example, piperidine, piperazine, pyrrolidine, morpholine, lactones, lactams, and the like.
The term “hydrocarbyl”, as used herein, refers to a group that is bonded through a carbon atom that does not have a =0 or =S substituent, and typically has at least one carbon- hydrogen bond and a primarily carbon backbone, but may optionally include heteroatoms. Thus, groups like methyl, ethoxyethyl, 2-pyridyl, and even trifluoromethyl are considered to be hydrocarbyl for the purposes of this application, but substituents such as acetyl (which has a =0 substituent on the linking carbon) and ethoxy (which is linked through oxygen, not carbon) are not. Hydrocarbyl groups include, but are not limited to aryl, heteroaryl, carbocycle, heterocycle, alkyl, alkenyl, alkynyl, and combinations thereof.
The term “hydroxyalkyl”, as used herein, refers to an alkyl group substituted with a hydroxy group.
The term “lower” when used in conjunction with a chemical moiety, such as, acyl, acyloxy, alkyl, alkenyl, alkynyl, or alkoxy is meant to include groups where there are ten or
fewer atoms in the substituent, preferably six or fewer. A “lower alkyl”, for example, refers to an alkyl group that contains ten or fewer carbon atoms, preferably six or fewer. In certain embodiments, acyl, acyloxy, alkyl, alkenyl, alkynyl, or alkoxy substituents defined herein are respectively lower acyl, lower acyloxy, lower alkyl, lower alkenyl, lower alkynyl, or lower alkoxy, whether they appear alone or in combination with other substituents, such as in the recitations hydroxyalkyl and aralkyl (in which case, for example, the atoms within the aryl group are not counted when counting the carbon atoms in the alkyl substituent).
The terms “polycyclyl”, “polycycle”, and “polycyclic” refer to two or more rings (e.g., cycloalkyls, cycloalkenyls, cycloalkynyls, aryls, heteroaryls, and/or heterocyclyls) in which two or more atoms are common to two adjoining rings, e.g., the rings are “fused rings”. Each of the rings of the polycycle can be substituted or unsubstituted. In certain embodiments, each ring of the polycycle contains from 3 to 10 atoms in the ring, preferably from 5 to 7.
The term “sulfate” is art-recognized and refers to the group -OSO3H, or a pharmaceutically acceptable salt thereof.
The term “sulfonamide” is art-recognized and refers to the group represented by the general formulae
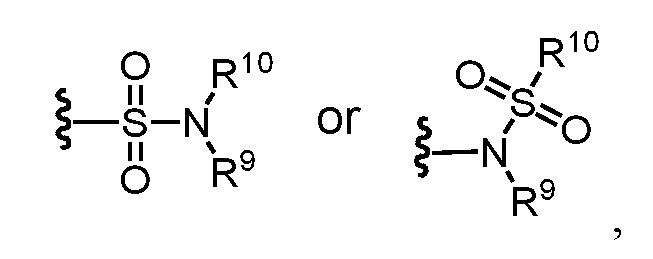 wherein R9 and R10 independently represents hydrogen or hydrocarbyl.
wherein R9 and R10 independently represents hydrogen or hydrocarbyl.
The term “sulfoxide” is art-recognized and refers to the group-S(O)-.
The term “sulfonate” is art-recognized and refers to the group SO3H, or a pharmaceutically acceptable salt thereof.
The term “sulfone” is art-recognized and refers to the group -S(O)2-.
The term “substituted” refers to moieties having substituents replacing a hydrogen on one or more carbons of the backbone. It will be understood that “substitution” or “substituted with” includes the implicit proviso that such substitution is in accordance with permitted valence of the substituted atom and the substituent, and that the substitution results in a stable compound, e.g., which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, etc. As used herein, the term “substituted” is contemplated to include all permissible substituents of organic compounds. In a broad aspect, the permissible substituents include acyclic and cyclic, branched and unbranched, carbocyclic and heterocyclic, aromatic and non-aromatic substituents of organic compounds. The permissible substituents can be one or more and the same or different for appropriate organic
compounds. For purposes of this invention, the heteroatoms such as nitrogen may have hydrogen substituents and/or any permissible substituents of organic compounds described herein which satisfy the valences of the heteroatoms. Substituents can include any substituents described herein, for example, a halogen, a hydroxyl, a carbonyl (such as a carboxyl, an alkoxycarbonyl, a formyl, or an acyl), a thiocarbonyl (such as a thioester, a thioacetate, or a thioformate), an alkoxyl, a phosphoryl, a phosphate, a phosphonate, a phosphinate, an amino, an amido, an amidine, an imine, a cyano, a nitro, an azido, a sulfhydryl, an alkylthio, a sulfate, a sulfonate, a sulfamoyl, a sulfonamido, a sulfonyl, a heterocyclyl, an aralkyl, or an aromatic or heteroaromatic moiety. It will be understood by those skilled in the art that the moieties substituted on the hydrocarbon chain can themselves be substituted, if appropriate.
The term “thioalkyl”, as used herein, refers to an alkyl group substituted with a thiol group.
The term “thioester”, as used herein, refers to a group -C(O)SR9 or -SC(O)R9 wherein R9 represents a hydrocarbyl.
The term “thioether”, as used herein, is equivalent to an ether, wherein the oxygen is replaced with a sulfur.
The term “urea” is art-recognized and may be represented by the general formula
 wherein R9 and R10 independently represent hydrogen or a hydrocarbyl.
wherein R9 and R10 independently represent hydrogen or a hydrocarbyl.
The term “modulate” as used herein includes the inhibition or suppression of a function or activity (such as cell proliferation) as well as the enhancement of a function or activity.
The phrase “pharmaceutically acceptable” is art-recognized. In certain embodiments, the term includes compositions, excipients, adjuvants, polymers and other materials and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
“Pharmaceutically acceptable salt” or “salt” is used herein to refer to an acid addition salt or a basic addition salt which is suitable for or compatible with the treatment of patients.
The term “pharmaceutically acceptable acid addition salt” as used herein means any non-toxic organic or inorganic salt of any base compounds represented by Formula I. Illustrative inorganic acids which form suitable salts include hydrochloric, hydrobromic, sulfuric and phosphoric acids, as well as metal salts such as sodium monohydrogen
orthophosphate and potassium hydrogen sulfate. Illustrative organic acids that form suitable salts include mono-, di-, and tricarboxylic acids such as glycolic, lactic, pyruvic, malonic, succinic, glutaric, fumaric, malic, tartaric, citric, ascorbic, maleic, benzoic, phenylacetic, cinnamic and salicylic acids, as well as sulfonic acids such as p-toluene sulfonic and methanesulfonic acids. Either the mono or di-acid salts can be formed, and such salts may exist in either a hydrated, solvated or substantially anhydrous form. In general, the acid addition salts of compounds of Formula I are more soluble in water and various hydrophilic organic solvents, and generally demonstrate higher melting points in comparison to their free base forms. The selection of the appropriate salt will be known to one skilled in the art. Other non-pharmaceutically acceptable salts, e.g., oxalates, may be used, for example, in the isolation of compounds of Formula I for laboratory use, or for subsequent conversion to a pharmaceutically acceptable acid addition salt.
The term “pharmaceutically acceptable basic addition salt” as used herein means any non-toxic organic or inorganic base addition salt of any acid compounds represented by Formula I or any of their intermediates. Illustrative inorganic bases which form suitable salts include lithium, sodium, potassium, calcium, magnesium, or barium hydroxide. Illustrative organic bases which form suitable salts include aliphatic, alicyclic, or aromatic organic amines such as methylamine, trimethylamine and picoline or ammonia. The selection of the appropriate salt will be known to a person skilled in the art.
Many of the compounds useful in the methods and compositions of this disclosure have at least one stereogenic center in their structure. This stereogenic center may be present in a R or a S configuration, said R and S notation is used in correspondence with the rules described in Pure Appl. Chem. (1976), 45, 11-30. The disclosure contemplates all stereoisomeric forms such as enantiomeric and diastereoisomeric forms of the compounds, salts, prodrugs or mixtures thereof (including all possible mixtures of stereoisomers). See, e.g., WO 01/062726.
Furthermore, certain compounds which contain alkenyl groups may exist as Z (zusammen) or E (entgegen) isomers. In each instance, the disclosure includes both mixture and separate individual isomers.
Some of the compounds may also exist in tautomeric forms. Such forms, although not explicitly indicated in the formulae described herein, are intended to be included within the scope of the present disclosure.
“Prodrug” or “pharmaceutically acceptable prodrug” refers to a compound that is metabolized, for example hydrolyzed or oxidized, in the host after administration to form the compound of the present disclosure (e.g., compounds of formula I). Typical examples of
prodrugs include compounds that have biologically labile or cleavable (protecting) groups on a functional moiety of the active compound. Prodrugs include compounds that can be oxidized, reduced, aminated, deaminated, hydroxylated, dehydroxyl ated, hydrolyzed, dehydrolyzed, alkylated, dealkylated, acylated, deacylated, phosphorylated, or dephosphorylated to produce the active compound. Examples of prodrugs using ester or phosphoramidate as biologically labile or cleavable (protecting) groups are disclosed in U.S. Patents 6,875,751, 7,585,851, and 7,964,580, the disclosures of which are incorporated herein by reference. The prodrugs of this disclosure are metabolized to produce a compound of Formula I. The present disclosure includes within its scope, prodrugs of the compounds described herein. Conventional procedures for the selection and preparation of suitable prodrugs are described, for example, in “Design of Prodrugs” Ed. H. Bundgaard, Elsevier, 1985.
The phrase “pharmaceutically acceptable carrier” as used herein means a pharmaceutically acceptable material, composition or vehicle, such as a liquid or solid filter, diluent, excipient, solvent or encapsulating material useful for formulating a drug for medicinal or therapeutic use.
The term “Log of solubility”, “LogS” or “logS” as used herein is used in the art to quantify the aqueous solubility of a compound. The aqueous solubility of a compound significantly affects its absorption and distribution characteristics. A low solubility often goes along with a poor absorption. LogS value is a unit stripped logarithm (base 10) of the solubility measured in mol/liter.
As used herein, the terms “induce” or “induction of’ refer to the increase or enhancement of a function, e.g., the increase or enhancement of the expression of a pattern recognition receptor (e.g, STING). In some embodiments, “induction of PRR expression” refers to induction of transcription of PRR RNA, e.g., STING RNA (e.g., mRNA, e.g., an increase or enhancement of), or the translation of a PRR protein, e.g., the STING protein (e.g., an increase or enhancement of). In some embodiments, induction of PRR expression (e.g., STING expression) refers to the increase or enhancement of the concentration of a PRR RNA, e.g., or STING RNA (e.g., mRNA) or the STING protein, e.g., in a cell. In some embodiments, induction of PRR expression (e.g., STING expression) refers to the increase of the number of copies of PRR RNA, e.g., STING RNA (e.g., mRNA) or PRR protein, e.g., the STING protein, e.g., in a cell. In some embodiments, to induce expression of a PRR (e.g., STING) may refer to the initiation of PRR RNA (e.g., STING RNA (e.g., mRNA)) or transcription or PRR protein (e.g., STING protein) translation. In some embodiments, to induce expression of a PRR (e.g.,
STING) may refer to an increase in the rate of PRR RNA (e.g., STING RNA (e.g., mRNA)) transcription or an increase in the rate of PRR protein (e.g., STING protein) expression.
As used herein, the terms “activate” or “activation” refer to the stimulation or triggering of a function, e.g., of a downstream pathway, e.g., a downstream signaling pathway. In some embodiments, activation of a pattern recognition receptor (PRR) (e.g., STING) refers to the stimulation of a specific protein or pathway, e.g., through interaction with a downstream signaling partner (e.g., IFN-b promoter stimulator 1 (IPS-1), IRF3, IRF7, NF-KB, interferons (e.g., IFN-a or IFN-b) and/or cytokines). In some embodiments, activation is distinct from the induction of expression of a PRR. In some embodiments, a PRR may be activated without resulting in an induction of PRR expression (e.g., expression of STING). In some embodiments, activation may include induction of expression of a PRR (e.g., STING). In some embodiments, activation of a PRR may trigger the induction of expression of a PRR (e.g., STING) by about 0.1%, about 0.5%, about 1%, about 5%, about 10%, about 15%, about 20%, about 25%, about 30%, about 40%, about 50%, about 60%, about 70%, about 80%, about 90%, about 95%, or more compared to a reference standard (e.g., basal expression levels of a PRR (e.g., STING)).
As used herein, the term “Cmd” refers to the word “compound” or “Compound”, and all of the terms are used interchangeably.
The term “nucleobase” as used herein, is a nitrogen-containing biological compound found linked to a sugar within a nucleoside — the basic building blocks of deoxyribonucleic acid (DNA) and ribonucleic acid (RNA). The primary, or naturally occurring, nucleobases are cytosine (DNA and RNA), guanine (DNA and RNA), adenine (DNA and RNA), thymine (DNA) and uracil (RNA), abbreviated as C, G, A, T, and U, respectively. Because A, G, C, and T appear in the DNA, these molecules are called DNA-bases; A, G, C, and U are called RNA- bases. Adenine and guanine belong to the double-ringed class of molecules called purines (abbreviated as R). Cytosine, thymine, and uracil are all pyrimidines. Other nucleobases that do not function as normal parts of the genetic code are termed non-naturally occurring.
The term “ADC” refers to an antibody drug conjugate.
As used herein, the term “Linker”, “Linker Unit”, or “link” means a chemical moiety comprising a covalent bond or a chain of atoms that covalently attaches two groups, e.g., an antibody to a drug moiety.
The term “monoclonal antibody” refers to a homogeneous antibody population involved in the highly specific recognition and binding of a single antigenic determinant or epitope. This is in contrast to polyclonal antibodies that typically include different antibodies
directed against a variety of different antigenic determinants. The term “monoclonal antibody” includes 33 antibody fragments (such as Fab, Fab', F(ab')2 , Fd, Fv), single chain (scFv) mutants, fusion proteins including an antibody portion, and any other modified immunoglobulin molecule including an antigen recognition site as well as both intact and full- length monoclonal antibodies, but are not limited thereto. Additionally, “monoclonal antibody” refers to such antibodies made in any number of methods including but not limited to hybridoma, phage selection, recombinant expression, and transgenic animals.
The term “humanized antibody” refers to forms of non-human (e.g., murine) antibodies that are specific immunoglobulin chains, chimeric immunoglobulins, or fragments thereof that contain minimal non-human (e.g., murine) sequences. In general, humanized antibodies are human immunoglobulins in which residues from complementary determining region (CDR) are replaced by residues from CDR of a non-human species (e.g., mouse, rat, rabbit, and hamster) having the desired specificity, affinity, and capability (ref: Jones et al., 1986, Nature, 321:522-525; Riechmann et al., 1988, Nature, 332:323-327; Verhoeyen et al., 1988, Science, 239:1534-1536). In some instances, Fv framework region (FR) residues of a human immunoglobulin are replaced with the corresponding residues in an antibody from a non- human species having the desired specificity, affinity, and/or binding capability. The humanized 34 antibody may be further modified by the substitution of additional residues either in the Fv framework region and/or within the replaced non-human residues to refine and optimize antibody specificity, affinity, and/or binding capability. In general, the humanized antibody includes substantially all of at least one, and typically two or three, variable domains containing all or substantially all of the CDRs that correspond to the non-human immunoglobulin whereas all or substantially all of the framework regions (FRs) have those of a human immunoglobulin consensus sequence. The humanized antibody may also include at least a portion of an immunoglobulin constant region or domain (Fc), typically that of a human immunoglobulin. Examples of methods used to generate humanized antibodies are described in U.S. Patent No. 5,225,539.
The term “human antibody” as used herein refers to an antibody produced by a human or an antibody having an amino acid sequence corresponding to an antibody produced by a human using any technique known in the art. This definition of the human antibody includes intact or fulllength antibodies, fragments thereof, and/or antibodies including at least one human heavy and/or light chain polypeptide such as, for example, an antibody including murine light chain and human heavy chain polypeptides.
The term “chimeric antibody” refers to an antibody wherein an amino acid sequence of an immunoglobulin molecule is derived from two or more species. In general, variable regions of both light and heavy chains correspond to variable regions of antibodies derived from one species of mammals (e.g., mouse, rat, rabbit, etc) with the desired specificity, affinity, and capability, while constant regions are homologous to the sequences in antibodies derived from another species (usually human) to avoid eliciting an immune response in that species.
The term “self-eliminating linker” or “self-immolative linker” refers to a temporary extender, spacer, or placeholder unit attaching two or more molecules together by chemical bonds that are cleaved under defined conditions to release the two molecules. In general, a self-eliminating or self-immolative linker may be linear or branched, and may link two or more of the same molecules together, or may link two or more different molecules together. A self- immolative moiety may be defined as a bifunctional chemical group which is capable of covalently linking together two spaced chemical moieties into a normally stable molecule, releasing one of said spaced chemical moieties from the molecule by means of enzymatic cleavage; and following said enzymatic cleavage, spontaneously cleaving from the remainder of the bifunctional chemical group to release the other of said spaced chemical moieties. In some embodiments, the self-immolative refers to a heterocyclic self-immolative moiety. Exemplary self-immolative linkers include His- Ala, /;-ami nobenzyl oxycarbonyl (PABC), 2,4- bis(hydroxymethyl)aniline, -NH-(CH2)4-C(O)- and -NH-(CH2)3-C(O)-.
The term “cleaveable group” is refers to a moiety that is unstable in vivo. Preferably, the “cleaveable group” allows for activation of the marker or therapeutic agent by cleaving the marker or agent from the rest of the conjugate. Operatively defined, the linker is preferably cleaved in vivo by the biological environment. The cleavage may come from any process without limitation, e.g., enzymatic, reductive, pH, etc. Preferably, the cleaveable group is selected so that activation occurs at the desired site of action, which can be a site in or near the target cells (e.g., carcinoma cells) or tissues such as at the site of therapeutic action or marker activity. Such cleavage may be enzymatic and exemplary enzymatically cleaveable groups include natural amino acids or peptide sequences that end with a natural amino acid, and are attached at their carboxyl terminus to the linker. While the degree of cleavage rate enhancement is not critical to the invention, preferred examples of cleaveable linkers are those in which at least about 10% of the cleaveable groups are cleaved in the blood stream within 24 hours of administration, most preferably at least about 35%.
The term “spacer group” refers any chemical group designed to facilitate the attachment of the drug conjugates to an antibody, e.g., in order to overcome steric hinderance.
Patter Recognition Receptors (PPRs)
The present disclosure relates to methods of activating and/or inducing the expression of PRRs (e.g., STING) in a subject, in particular for the treatment of a microbial infection or a proliferative disease (e.g., cancer). In some embodiments, the method comprises administration of an antibody-drug conjugate of the disclosure or pharmaceutically acceptable salt thereof. It is to be noted that induction of any PRR with these compounds can stimulate interferon and/or NF-KB production which can induce the expression of a variety of PRRs which are inducible genes by feedback mechanism.
The disclosure presented herein features methods for the activation and induction of PRR expression (e.g., STING expression) in a subject, e.g., a subject with a proliferative disease (e.g., cancer).
Pattern recognition receptors (PRRs) are a broad class of proteins which recognize pathogen-associated molecular patterns (PAMPs) conserved within pathogenic invaders. PAMPs are typically products of biosynthetic pathways that are essential to the survival and/or infectivity of the pathogen, e.g., lipopolysaccharides, glycoproteins, and nucleic acids. Recognition of PAMPs by their cognate PRRs activates signaling pathways that result in the production of immune defense factors such as pro-inflammatory and anti-inflammatory cytokines, type I interferons (IFN-a, IFN-b), and/or interferon stimulated genes (ISGs). It is well known that induction of innate immune signaling also results in the activation of T cell responses as well as the induction of adaptive immunity.
The stimulator of interferon genes (STING) is a cytosolic microbial-derived DNA sensor that has been shown to be particularly sensitive to double-stranded DNA and cyclic dinucleotides (e.g., cyclic di-GMP) (Burdette, D. L. and Vance, R. E. (2013) Nat Immunol 14: 19-26). Two molecules of STING form a homodimer mediated by an a-helix present in the C-terminal dimerization domain, and molecular binding studies have revealed that each STING dimer binds one molecule of microbial nucleic acids, e.g., DNA or a cyclic dinucleotide. STING has been shown to play a major role in the innate immune response against tumor antigens, driving dendritic cell activation and subsequent T cell priming in several cancers (Woo, S.R. et al. Trends in Immunol (2015) 36:250-256).
Another class of PRRs includes RIG- 1, which is the founding member of a family of PRRs termed RIG-I-like receptors (RLRs) that primarily detect RNA derived from foreign sources. It is a critical sensor of microbial infection (e.g., viral infection) in most cells and is constitutively expressed at low levels in the cytosol. After ligand binding, the expression of RIG-I is rapidly enhanced, leading to increased RIG-I concentrations in the cell (Jensen, S. and
Thomsen, A.R. J Virol (2012) 86:2900-2910; Yoneyama M. et al. Nat Immunol (2004) 5:730- 737). RIG-I is an ATP-dependent helicase containing a central DExD/H box ATPase domain and tandem N-terminal caspase-recruiting domains (CARDs) that mediate downstream signaling. The C-terminus of RIG-I comprises an ssRNA/dsRNA-binding domain that when unbound acts to silence CARD function at the N-terminus. Without wishing to be bound by theory, it is believed that upon recognition of target RNA structures, two N-terminal CARDs are exposed, allowing for interaction with the CARD of a downstream binding partner, IFN-b promoter stimulator 1 (IPS-1), also known as mitochondrial antiviral signaling molecule (MAVS) and CARD IF. This interaction in turn triggers further downstream signaling, such as induction of IRF3, IRF7, NF-KB, IFNS, and cytokine production that results in the initiation of the host immune response.
Another class of PRRs encompasses the nucleotide-binding and oligomerization domain (NOD)-like receptors, or NLR family (Caruso, R. et al, Immunity (2014) 41:898-908), which includes the microbial sensor NOD2. NOD2 is composed of an N-terminal CARD, a centrally-located nucleotide-binding oligomerization domain, and a C-terminal leucine rich repeat domain that is responsible for binding microbial PAMPs, such as bacterial peptidoglycan fragments and microbial nucleic acids. Ligand binding activates NOD2 and is believed to drive interaction with the CARD-containing kinase RIPK2, which in turn activates a number of downstream proteins including NF-KB, MAPK, IRF7, and IRF3, the latter of which results in the induction of type 1 interferons. NOD2 is expressed in a diverse set of cell types, including macrophages, dendritic cells, paneth cells, epithelial cells (e.g., lung epithelial cells, intestinal epithelia), and osteoblasts.
Antibody Drug Conjugates
Antibody drug conjugates (ADCs) are an important class of biopharmaceutical drugs, which are designed to act as a targeted therapy for the treatment of subjects with various disease states (Ducry, L. Bioconjugate Chemistry (2010) 21:5-13). ADCs are comprised of a small molecule covalently linked to an antibody. The covalent linkage comprises a point of attachment to the small molecule, a biocompatible linker unit, and a self-immolative group attached to the antibody. In some aspects, there is an optional biocompatible spacer group between the self-immolative group and the antibody.
Antibody-Drug Conjugates of Formula I
In one aspect, the disclosure features an antibody-drug conjugate of Formula (I):
 or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
Ab is an anti-PD-1 antibody or an anti-CTLA-4 antibody; each of B1 and B2 is independently a purinyl nucleobase or pyrimidinyl nucleobase; each of X1 and X2 is independently O or S; each of Y1 and Y2 is independently O, S, or N(R5); each of Z1 is indpendently O or S; each of Z2 and Z3 is independently absent, -C1-C20- alkylene (e.g., -C1-C6- alkylene), C1-C20 heteroalkylene (e.g., -C1-C6- heteroalkylene), -OC(O)OC1-C20- alkylene (e.g., -C1-C6- alkylene), -cycloalkylene-, -heterocyclyl-, -aryl-, or -heteroaryl-, wherein each -cycloakyl-, -heterocyclyl-, -aryl- or -heteroaryl- is optionally substituted with one or more R4;
Z4 is self-immolative group-C1-C20-alkylene-Q1 (e.g., -self-immolative group-C1-C6- alkylene-Q1), heterocyclyl-C1-C20-alkylene-Q (e.g., heterocyclyl-C1-C6-alkylene-Q),- OH, -N(R5)2, SR5, -CHO, -C(O)N(R5)2, -OC(O)N(R5)2, -N(R5)C(O)OR5, aryl, heteroaryl, -S(O)R5,
-S(O)2R5, -S(O)N(R5)2, -S(O)2N(R5)2, -N(R5)S(O)R5, -OSi(C1-C4 alkyl)3, or -C(O)C2- C6 alkenyl (e.g., -C2-C4 alkenyl);
Z5 is a self-immolative group or absent;
T is a spacer group or absent;
L1 is absent, -C1-C6- alkylene (e.g., -C1-C3- alkylene) or -C1-C6- heteroalkylene;
L2 is absent, -C1-C6- alkylene (e.g., -C1-C3- alkylene) or -C1-C6- heteroalkylene, wherein each alkylene and heteroalkyl is optionally substituted with one or more R6;
L3 is absent, -C1-C20- alkylene, -O-, -N(R5)-, -S-, -S(O)-, -S(O)2-, -S(O)N(R5)-,
-S(O)2N(R5)-, -N(R5)S(O)-, -N(R5)S(O)2-, -C(O)-, -C(O)O-, OC(O) -, -C(O)N(R5)-, -N(R5)C(O)-;
L4 is -C1-C20- alkylene (e.g., -C1-C6- alkylene), -C1-C20- heteroalkylene (e.g., -C1-C6- heteroalkylene), -C1-C20- alkenylene (e.g., -C 2-C6- alkenylene), -C1-C20- alkynylene (e.g., -C 2-C6- alkynylene), or an oligopeptide, wherein the oligopeptide is optionally substituted by one or more R16;
L5 is a linker connecting Z4 and Z5, or is absent;
Q1 is C(O), C(S), or CH2; each of R1 and R2 is independently hydrogen, halo, -CN, -C1-C20 alkyl (e.g., C1-C6 alkyl), or - OR7;
R3 is hydrogen, -C1-C20- alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl (e.g., -C1-C6 heteroalkyl), -OC(O)OC1-C20 alkyl (e.g., -C1-C6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, heteroalkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R8; each R4 is independently hydrogen, -C1-C20 alkyl, -O-C1-C20 alkyl, -C1-C20 heteroalkyl, halo, -CN, -NO2 or -OH;
R5 is hydrogen or -C1-C20 alkyl (e.g., -C1-C6 alkyl);
R6 is halo, -CN, -C1-C20 alkyl (e.g., -C1-C6 alkyl), -OR7, oxo, cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R9;
R7 is hydrogen, -C1-C20 alkyl (e.g., -C1-C6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R9; each R8 is independently -C1-C20 alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl,
-C(O)-C1-C20 alkyl, -OC(O)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -C(O)O-C1-C20 alkyl (e.g·,
-C1-C6 alkyl), -OC(O)O-C1-C20 alkyl (e.g., -C1-C6 alkyl), -C(O)N(R5)-C1-C20 alkyl (e.g·,
-C1-C6 alkyl), -N(R5)C(O)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -OC(O)N(R5)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -O-aryl, -O-heteroaryl, -C(O)-aryl, -C(O)-heteroaryl, - OC(O)-aryl,
-C(O)O-aryl, -OC(O)-heteroaryl, -C(O)O-heteroaryl, -C(O)O-aryl,
-C(O)O-heteroaryl, -C(O)N(R5)-aryl, -C(O)N(R5)-heteroaryl, -N(R5)C(O)-aryl,
-N(R5)2C(O)-aryl, or -N(R5)C(O)-heteroaryl, -S(O)2N(R5)-aryl, wherein each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R9; each R9 is independently -C1-C20 alkyl, -O-C1-C20 alkyl, -C1-C20 heteroalkyl, halo,
-CN, -OH, oxo, aryl, heteroaryl, -O-aryl, or -O-heteroaryl; and each R16 is independently, -C1-C20 alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl (e.g., -C1-C6 heteroalkyl), -OC(O)OC1-C20 alkyl (e.g., -C1-C6 alkyl), C(O)N(R4)2 cycloalkyl, heterocyclyl, aryl, or heteroaryl.
In some embodiments, the antibody-drug conjugate is represented by formula (Il-a),
(Il-b), (II-c), or (II-d):
 or a pharmaceutically acceptable salt or stereoisomer thereof.
or a pharmaceutically acceptable salt or stereoisomer thereof.
In some embodiments, each of B1 or B2 is selected from a naturally occurring nucleobase or a modified nucleobase. In some embodiments, each of B1 or B2 is selected from adenosinyl, guanosinyl, cytosinyl, thyminyl, uracilyl, 5’-methylcytosinyl, 5’-fluorouracilyl, 5’- propynyluracilyl, and 7-deazaadenosinyl. In some embodiments, each ofB1 or B2 is selected from:

In some embodiments, one of B1 or B2 is selected from a naturally occurring nucleobase and the other of B1 or B2 is a modified nucleobase. In some embodiments, one of B1 or B2 is adenosinyl, guanosinyl, thyminyl, cytosinyl, or uracilyl, and the other of B1 or B2 is 5’- methylcytosinyl, 5’-fluorouracilyl, 5’-propynyluracilyl, or 7-deazaadenosinyl.
In some embodiments, at least one of B1 or B2 is a purinyl nucleobase. In some embodiments, each of B1 or B2 is independently a purinyl nucleobase. In some embodiments, B1 is a purinyl nucleobase. In some embodiments, B2 is a pyrimidinyl nucleobase. In some embodiments, B1 is a purinyl nucleobase and B2 is a pyrimidinyl nucleobase.
In some embodiments, B1 is adenosinyl or guanosinyl. In some embodiments, B2 is cytosinyl, thyminyl, or uracilyl. In some embodiments, B1 is adenosinyl or guanosinyl and B2 is cytosinyl, thyminyl, or uracilyl. In some embodiments, each of B1 and B2 is independently uracilyl. In some embodiments, each of B1 and B2 is independently adenosinyl.
In some embodiments, each of R1 and R2 is independently hydrogen, halo, or OR7. In some embodiments, each of R1 and R2 is independently halo (e.g., fluoro). In some embodiments, each of R1 and R2 is not hydrogen or OR7.
In some embodiments, X1 is O. In some embodiments, X2 is O. In some embodiments, each of X1 and X2 is independently O.
In some embodiments, Y1 is O or S. In some embodiments, Y2 is O or S. In some embodiments, each of Y1 and Y2 is independently O or S. In some embodiments, one of Y1 or Y2 is O and the other of Y1 or Y2 is S. In some embodiments, each of Y1 or Y2 is independently S. In some embodiments, each of Y1 or Y2 is independently O.
In some embodiments, R3 is hydrogen, aryl, or heteroaryl, wherein aryl and heteroaryl is optionally substituted with 1-5 R8. In some embodiments, R3 is aryl or heteroaryl, each of which is optionally substituted with 1-5 R8. In some embodiments, R3 is phenyl substituted with 1 R8.
In some embodiments, R4 is hydrogen.
In some embodiments, each of Y1 and Y2 is O and R3 is hydrogen. In some embodiments, each of Y1 and Y2 is independently S and R3 is substituted with 1 R8. In some embodiments, Y1 is S and R3 is substituted with 1 R8.
In some embodiments, each R8 is independently C1-C20 alkyl (e.g., C1-C6 alkyl), C1-C20 heteroalkyl, C(O)-C1-C20 alkyl, OC(O)-C1-C20 alkyl, OC(O)O-C1-C20 alkyl, OC(O)N(R5)-C1- C20 alkyl, O-aryl, C(O)-aryl, OC(O)-aryl, or C(O)N(R5)-aryl, wherein each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R9.
In some embodiments, R8 is OC(O)-aryl optionally substituted by 1-5 R9 (e.g., 1 R9).
In some embodiments, R9 is O-C1-C12 alkyl (e.g., O-CH2(CH2)8CH3). In some embodiments, R9 is O-C1-C10 alkyl (e.g., O-CH2(CH2)8CH3). In some embodiments, R9 is O- C1-C6 alkyl (e.g., O-CH2(CH2)8CH3). In some embodiments, R9 is O-C1-C6 alkyl (e.g., O- CH2(CH2)4CH3).
In some embodiments, R16 is independently, -C1-C20 alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl (e.g., -C1-C6 heteroalkyl), -OC(O)OC1-C20 alkyl (e.g., -C1-C6 alkyl), C(O)N(R4)2 cycloalkyl, heterocyclyl, aryl, or heteroaryl.
In some embodiments, each of L1 and L2 is independently C1-C6 alkylene (e.g., C1 alkylene) or absent.
In some embodiments, L3 is absent, -N(H)-, -O(O)C-, or -O-. In some embodiments, L3 is -C(O)-.
In some embodiments, L4 is an -C1-C20- alkylene (e.g., -C1-C18- alkylene), or oligiopeptide. In some embodiments, L4 is an oligopeptide. In some embodiments, the oligopeptide is a dipeptide. In other embodiments, the oligopeptide comprises 3 amino acid residues, 4 amino acid residues, 5 amino acid residues, 6 amino acid residues, 7 amino acid residues, 8 amino acid residues, 9 amino acid residues, or 10 amino acid residues. In some embodiments, the oligopeptide comprises 8 amino acid residues. In some embodiments, the oligopeptide comprises an amino acid selected from Tyr, Ser, Thr, Arg, Leu, lie, Gly, Val, and Ala. In some embodiments, the oligopeptide comprises an amino acid selected from Phe, Tyr, Arg, Gly, Ser, and Leu.
In some embodiments, L4 is
 In other embodiments, L4 is
In other embodiments, L4 is

In yet other embodiments, L4 is selected from the group consisting of
 and
and
 In some embodiments, L4 is
In some embodiments, L4 is

In some embodiments, is L4 -C1-C20- alkylene (e.g., -C1-C18- alkylene). In some embodiments, L4 is -C1-C20- heteroalkylene (e.g., -C1-C18- heteroalkylene).
In some embodiments, L4 is substituted with one or more instances of R10. In some embodiments, R10 is C(O)NH2.
In some embodiments, L5 is -C1-C40- alkylene (e.g., -C1-C20- alkylene), -C1-C40- heteroalkylene (e.g., -C1-C20- heteroalkyl), -C1-C40- alkenylene (e.g., -C2-C20- alkenylene), or -C1-C40- alkynylene (e.g., -C2-C20- alkynylene). In some embodiments, L5 is an oligopeptide comprising of 1-40 amino acid residues. In some embodiments, L5 further further comprises one
 sub-unit. In some embodiments, L5 further comprises one
sub-unit. In some embodiments, L5 further comprises one
 sub-unit. In some embodiments, L5 further comprises one
sub-unit. In some embodiments, L5 further comprises one
 sub-unit. In some embodiments, L5 further comprises one
sub-unit. In some embodiments, L5 further comprises one
 sub-unit.
sub-unit.
In some embodiments, each Z1 is oxygen.
In some embodiments, Z2 is -aryl-. In some embodiments, Z2 is -OC(O)OC1-C20- alkylene (e.g., -C1-C6- alkylene).
In some embodiments, Z3 is -aryl-. In some embodiments, Z3 is -OC(O)OC1-C20- alkylene (e.g., -C1-C6- alkylene). In some embodiments, Z3 is aryl substituted with -OC1-C20- heteroalkylene (e.g., -C1-C12-heteroalkylene). In some embodiments, Z3 is aryl substituted with
-OC1-C20-NH- (e g., -C1-C12-NH-).
In some embodiments, Z4 is Heterocyclyl-C1-C20- alkylene-Q. In some embodiments,
Z4 is
 or
or
 In some embodiments, Q is C(O).
In some embodiments, Q is C(O).
In some embodiments, Z4 is -0-, -N(R5)- or -heteroaryl-.
In some embodiments, Z5 is In some embodiments, Z5 is
 In
In
 some embodiments, Z5 is In some embodiments, Z5 is
some embodiments, Z5 is In some embodiments, Z5 is
 In some
In some
 embodiments, Z5 is In some embodiments, Z5 is
embodiments, Z5 is In some embodiments, Z5 is
In some embodiments, Z5 is represented by formula (III-a), (III-b), or (III-c):
 wherein:
wherein:
U is O, S or NR13;
Q2 is CR13 or N; each V1, V2 and V3 are independently CR17 or N provided that for formula (Ill-b) and (III-c) at least one of Q2, V1 and V2 is N;
R11 is the side chain of an amino acid and is optionally protected with a protecting group;
Each R10, R14, and R15 are independently selected from hydrogen, halo, OH, -N(R12)2, - N(R12)3 +, C1-C8 heteroalkyl, carboxylate, sulfate, sulfamate, sulfonate, -SO2R5, - S(O)R12,
-SR12, -SO2N(R12)2, -C(O)R5, -CO2R12, -C(O)N(R12)2, -CN, -N3, -NO2, C1-C8 heteroalkyl, polyethyl eneoxy, phosphonate, phosphate, C1-C8 alkyl, C2-C8, C2-C8 alkenyl, C2-C8 alkynl, C2-C8, aryl, and heterocycle; or when taken together, R14 and R15 form a carbonyl (=0), or spiro-carbocyclic ring comprising of 3 to 7 carbon atoms; and
R12 and R13 are independently selected from hydrogen, C1-C8 alkyl, C2-C8 alkenyl, C2-C8 alkynyl, aryl, heterocycle, wherein each alkyl, alkenyl, alkynyl, aryl, and heterocycle are independently substituted with one or more substituents selected from halo, OH, - N(R12)2, -N(R12)3+, heteroalkyl, carboxylate, sulfate, sulfamate, sulfonate, 4- dialkylaminopyridinium, alkyl, -SO2R12, -S(O)R12, -SR12, -SO2N(R12)2, C(O)R12, CO2R12, C(O)N(R12)2, -CN, -N3, -NO2, C3-C12 carbocycle, aryl, heterocycle, polyethyleneoxy, phosphonate, and phosphate.
In some embodiments, Q2 is N; V1 is CH, and V2 is CH. In some embodiments, Q2 is CH; V1 is CH and V2 is N. In some embodiments, Q2 is N; V1 is CH, and V2 is N. In some embodiments, Q2 is N; and V1 is N and V2 is N. In some embodiments, Q2 is N; V1 is N and V2 is CH. In some embodiments, Q2 is N; and V1, V2 and V3 are each CH. In some embodiments, Q2 is CH; and V1, V2 and V3 are each CH.
In some embodiments, T is absent, -C1-C20- alkylene (e.g., -C1-C6- alkylene), -C1-C20- heteroalkylene (e.g., -C1-C6- heteroalkylene), -C1-C20- alkenylene (e.g., -C 2-C6- alkenylene), - C1-C20- alkynylene (e.g., -C 2-C6- alkynyl), -aryl- or -heteroaryl-.
In some embodiments, anti -PD- 1 antibody or anti-CTLA-4 antibody is a monoclonal antibody, a domain antibody (dAb), a single chain antibody (scAb), a Fab fragment, a F(ab’)2 fragment, a single chain variable fragment (scFv), a scFv-Fc fragment, a single domain heavy chain antibody, a single domain light chain antibody, a variant antibody, a multimeric antibody, or a bispecific antibody.
In some embodiments, the antibody is an anti -PD- 1 antibody. In some embodiments, the anti -PD- 1 antibody is pembrolizumab, nivolumab, cemiplimab, Anti -mouse PD-1 antibody
Clone J43, Anti-mouse PD-1 antibody Clone RMP1-14, mouse anti -PD- 1 antibody Clone EH 12, ANB011, MDX-1 106, AMP-514, AMP-224, or Pidilizumab. In some embodiments, the anti-PD-1 antibody is pembrolizumab or nivolumab.
In some embodiments, the antibody is an anti-CTLA-4 antibody. In some embodiments, the anti-CTLA-4 antibody is ipilimumab, clone 9H10, tremelimumab, or clone BNI3.
In some embodiments, the antibody-drug conjugate is selected from Table 1:
Table 1 : Exemplary Antibody-Drug Conjugates of the Present Invention


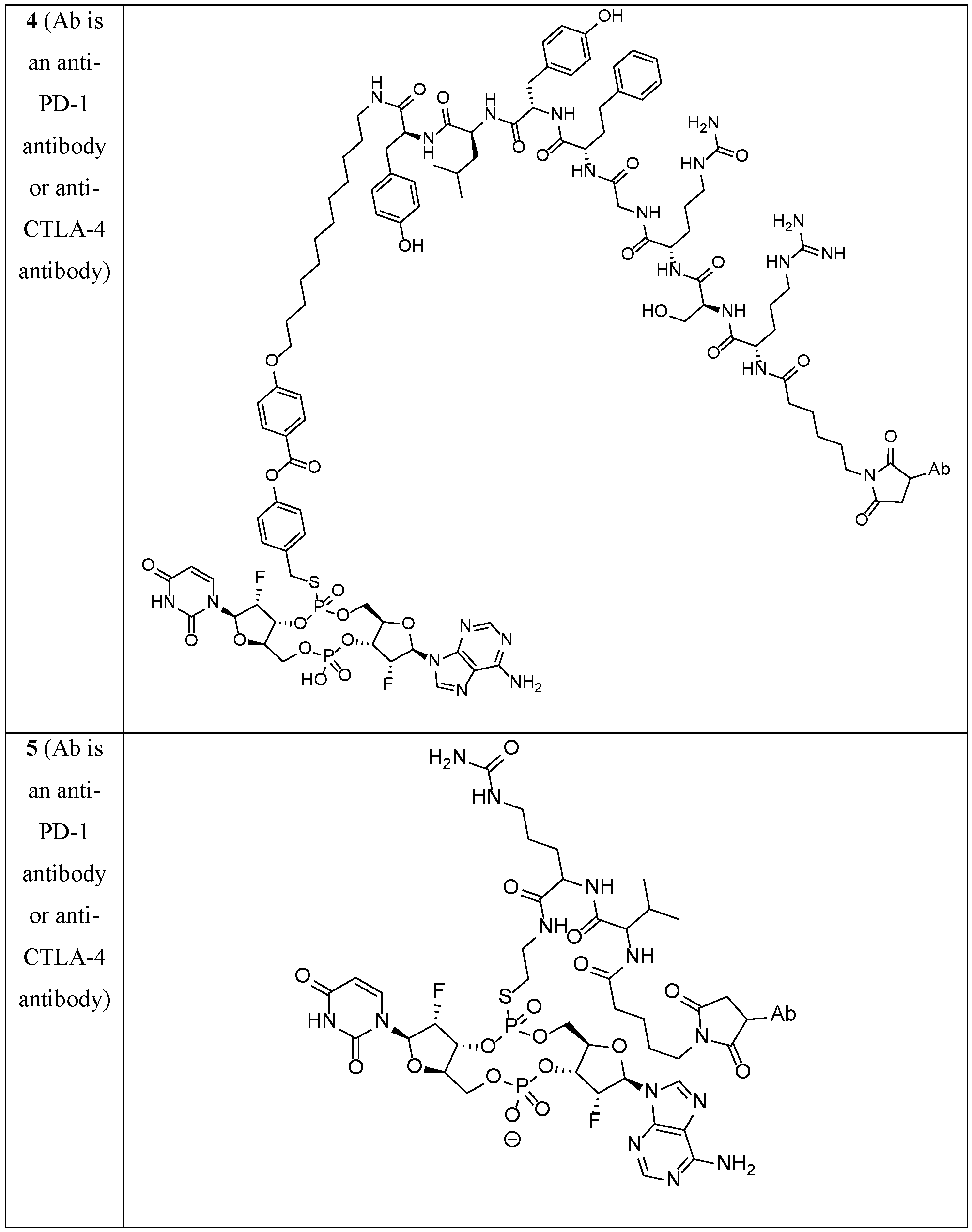

In another aspect, the disclosure features an antibody-drug conjugate of Formula (IV):
 or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
Ab is an anti-PD-1 antibody or an anti-CTLA-4 antibody; each of B1 and B2 is independently a purinyl nucleobase or pyrimidinyl nucleobase; each of X1 and X2 is independently O or S; each of Y1 and Y2 is independently O, S, or N(R5); each of Z1 is independently O or S;
Z2 is -O-, -N(R5)-, -S-, -C(O)-, -C(O)N(R5)-, -OC(O)N(R5)-, -N(R5)C(O)O-, -aryl-, -heteroaryl-, -S(O)-, -S(O)2-, -S(O)N(R5)-, -S(O)2N(R5)- or -N(R5)S(O)-;
Z3 is absent, hydrogen, heterocyclyl, heterocyclyl-C1-C20-alkylene-Q1, -OH, -N(R5)2, SR5, - CHO, -C(O)N(R5)2, -OC(O)N(R5)2, -N(R5)C(O)OR5, aryl, heteroaryl, -S(O)R5, - S(O)2R5, -S(O)N(R5)2, -S(O)2N(R5)2, -N(R5)S(O)R5, -OSi(C1-C4 alkyl)3, or -C(O)C2- C6 alkenyl (e.g., -C2-C4 alkenyl);
Z4 is a self-immolative group or absent;
T is a absent or spacer group; each L1 and L2 is absent, -C1-C6- alkylene (e.g., -C1-C3- alkylene) or -C1-C6- heteroalkyl ene, wherein each alkylene and heteroalkyl is optionally substituted with one or more R6; L3 is oligiopeptide-C(O)-, oligiopeptide-aryl-C1-C6-alkylene-, oligiopeptide-aryl-C1-C6- alklyene-C(O)-, oligiopeptide-aryl-C1-C6-heteroalkylene, oligiopeptide-C1-C6- alkylene-C(O)-, oligiopeptide-C1-C6-heteroalkylene-C(O)-, -C1-C40- alkylene (e.g., - C1-C20- alkylene), -C1-C40- heteroalkyl ene (e.g., -C1-C20- heteroalkyl), -C1-C40-
alkenylene (e.g., -C2-C20- alkenylene), or -C1-C40- alkynylene (e.g., -C2-C20- alkynylene), wherein the oligopeptide is optionally substituted by one or more R14;
L4 is absent or a linker connecting Z3 and Z4;
Q1 is C(O), C(S), or CH2; each of R1 and R2 is independently hydrogen, halo, -CN, -C1-C20 alkyl (e.g., C1-C6 alkyl), or - OR7; each R3 and R4 is independently hydrogen, -C1-C20- alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl (e.g., -C1-C6 heteroalkyl), -OC(O)OC1-C20 alkyl (e.g., -C1-C6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, heteroalkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R8;
R5 is hydrogen or -C1-C20 alkyl (e.g., -C1-C6 alkyl);
R6 is halo, -CN, -C1-C20 alkyl (e.g., -C1-C6 alkyl), -OR7, oxo, cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R9;
R7 is hydrogen, -C1-C20 alkyl (e.g., -C1-C6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R9; each R8 is independently -C1-C20 alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl,
-C(O)-C1-C20 alkyl, -OC(O)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -C(O)O-C1-C20 alkyl (e.g., -C1-C6 alkyl), -OC(O)O-C1-C20 alkyl (e.g., -C1-C6 alkyl), -C(O)N(R5)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -N(R5)C(O)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -OC(O)N(R5)- C1-C20 alkyl (e.g., -C1-C6 alkyl), -O-aryl, -O-heteroaryl, -C(O)-aryl, -C(O)-heteroaryl, -OC(O)-aryl, -C(O)O-aryl, -OC(O)-heteroaryl, -C(O)O-heteroaryl, -C(O)O-aryl, - C(O)O-heteroaryl, -C(O)N(R5)-aryl, -C(O)N(R5)-heteroaryl, -N(R5)C(O)-aryl, - N(R5)2C(O)-aryl, or -N(R5)C(O)-heteroaryl, -S(O)2N(R5)-aryl, wherein each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R9; each R9 is independently -C1-C20 alkyl, -O-C1-C20 alkyl, -C1-C20 heteroalkyl, halo,
-CN, -OH, oxo, aryl, heteroaryl, -O-aryl, or -O-heteroaryl; and each R16 is independently, -C1-C20 alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl (e.g., -C1-C6 heteroalkyl), -OC(O)OC1-C20 alkyl (e.g., -C1-C6 alkyl), C(O)N(R4)2 cycloalkyl, heterocyclyl, aryl, or heteroaryl.
In some embodiments, wherein the antibody-drug conjugate is an antibody-drug conjugate of formula (V-e), (V-f), (V-g), (V-h), (V-i) or (V-j):

W1 is N(R5), -O- CH; and W2 is N or CH.
In some embodiments, each of B1 or B2 is selected from a naturally occurring nucleobase or a modified nucleobase. In some embodiments, each of B1 or B2 is selected
from adenosinyl, guanosinyl, cytosinyl, thyminyl, uracilyl, 5’ -methyl cytosinyl, 5’- fluorouracilyl, 5’-propynyluracilyl, and 7-deazaadenosinyl. In some embodiments, each of B1 or B2 is selected from:
 wherein indicates the linkage of the nucleobase to the ribose ring.
wherein indicates the linkage of the nucleobase to the ribose ring.

In some embodiments, one of B1 or B2 is selected from a naturally occurring nucleobase and the other of B1 or B2 is a modified nucleobase. In some embodiments, one of B1 or B2 is adenosinyl, guanosinyl, thyminyl, cytosinyl, or uracilyl, and the other of B1 or B2 is 5’- methylcytosinyl, 5’-fluorouracilyl, 5’-propynyluracilyl, or 7-deazaadenosinyl.
In some embodiments, B1 is adenosinyl or guanosinyl. In some embodiments, B2 is cytosinyl, thyminyl, or uracilyl. In some embodiments, B1 is adenosinyl or guanosinyl and B2 is cytosinyl, thyminyl, or uracilyl. In some embodiments, each of B1 and B2 is independently uracilyl. In some embodiments, each of B1 and B2 is independently adenosinyl.
In some embodiments, each of R1 and R2 is independently hydrogen, halo, or OR7. In some embodiments, each of R1 and R2 is independently halo (e.g., fluoro). In some embodiments, each of R1 and R2 is not hydrogen or OR7.
In some embodiments, X1 is O. In some embodiments, X2 is O. In some embodiments, each of X1 and X2 is independently O.
In some embodiments, Y1 is O or S. In some embodiments, Y2 is O or S. In some embodiments, each of Y1 and Y2 is independently O or S. In some embodiments, one of Y1 or Y2 is O and the other of Y1 or Y2 is S. In some embodiments, each of Y1 or Y2 is independently S. In some embodiments, each of Y1 or Y2 is independently O.
In some embodiments, L1 is C1-C6 alkyl (e.g., CH2). In some embodiments, L2 is C1- C6 alkyl (e.g., CH2). In some embodiments, each of L1 and L2 is independently C1-C6 alkyl (e.g., CH2).
In some embodiments, R3 is hydrogen, aryl, or heteroaryl, wherein aryl and heteroaryl is optionally substituted with 1-5 R8. In some embodiments, R3 is aryl or heteroaryl, each of which is optionally substituted with 1-5 R8. In some embodiments, R3 is phenyl substituted with 1 R8.
In some embodiments, R4 is independently hydrogen, aryl, or heteroaryl, wherein aryl and heteroaryl is optionally substituted with 1-5 R8. In some embodiments, R4 is aryl or heteroaryl, each of which is optionally substituted with 1-5 R8. In some embodiments, R4 is phenyl substituted with 1 R8.
In some embodiments, each of R3 and R4 is independently hydrogen, aryl, or heteroaryl, wherein aryl and heteroaryl is optionally substituted with 1-5 R8. In some embodiments, R3 is aryl or heteroaryl, each of which is optionally substituted with 1-5 R8, and R4 is hydrogen. In some embodiments, R3 is phenyl substituted with 1 R8 and R4 is hydrogen. In some embodiments, each of R3 and R4 is independently phenyl substituted with 1 R8.
In some embodiments, each of Y1 and Y2 is O and each of R3 and R4 is independently hydrogen. In some embodiments, Y2 is O and R4 is hydrogen. In some embodiments, each of Y1 and Y2 is independently S and each of R3 and R4 is independently substituted with 1 R8. In some embodiments, Y1 is S and R3 is substituted with 1 R8.
In some embodiments, each R8 is independently C1-C20 alkyl (e.g., C1-C6 alkyl), C1-C20 heteroalkyl, C(O)-C1-C20 alkyl, OC(O)-C1-C20 alkyl, OC(O)O-C1-C20 alkyl, OC(O)N(R5)-C1- C20 alkyl, O-aryl, C(O)-aryl, OC(O)-aryl, or C(O)N(R5)-aryl, wherein each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R9.
In some embodiments, R8 is OC(O)-aryl optionally substituted by 1-5 R9 (e.g., 1 R9). In some embodiments, R9 is O-C1-C12 alkyl (e.g., O-CH2(CH2)8CH3). In some embodiments, R9 is O-C1-C10 alkyl (e.g., O-CH2(CH2)8CH3). In some embodiments, R9 is O-C1-C8 alkyl (e.g., O-CH2(CH2)6CH3). In some embodiments, R9 is O-C1-C6 alkyl (e.g., O-CH2(CH2)4CH3).
In some embodiments, each of L1 and L2 is independently C1-C6 alkylene (e.g., C1 alkylene).
In some embodiments, L3 is -C1-C20- alkylene (e.g., -C1-C18- alkylene). In some embodiments, L3 is -C1-C20- heteroalkylene (e.g., -C1-C18- heteroalkylene). In some embodiments, L3 is an oligiopeptide-C(O)-, oligiopeptide-aryl-C1-C6-alkylene-, oligiopeptide- aryl-C1-C6-heteroalkylene, oligiopeptide-aryl-C1-C6-alklyene-C(O)-, oligiopeptide-C1-C6- alkylene-C(O)-, oligiopeptide-C1-C6-heteroalkylene-C(O)-. In some embodiments, the aryl is phenyl. In some embodiments, the oligiopeptide is a dipeptide. In some embodiments, the oligopeptide comprises 3 amino acid residues, 4 amino acid residues, 5 amino acid residues, 6
amino acid residues, 7 amino acid residues, 8 amino acid residues, 9 amino acid residues, or 10 amino acid residues. In some embodiments, the oligopeptide comprises 8 amino acid residues. In some embodiments, the oligopeptide comprises an amino acid selected from Tyr, Ser, Thr, Arg, Leu, lie, Gly, Val, and Ala. In some embodiments, he oligopeptide comprises an amino acid selected from Phe, Tyr, Arg, Gly, Ser, and Leu. In some embodiments, L3 is
 or In other embodiments, L3 is
or In other embodiments, L3 is
 In yet other embodiments, L3 is
In yet other embodiments, L3 is
selected from the group consisting of
 and
and
 In some embodiments, L3 is
In some embodiments, L3 is

In some embodiments, the oligiopeptide is substituted by one or more instances of R16. In some embodiments, R16 is C(O)NH2.
In some embodiments, L4 is -C1-C40- alkylene (e.g., -C1-C20- alkylene), -C1-C40- heteroalkylene (e.g., -C1-C20- heteroalkyl), -C1-C40- alkenylene (e.g., -C2-C20- alkenylene), or -C1-C40- alkynylene (e.g., -C2-C20- alkynylene). In some embodiments, L4 is an oligopeptide comprising of 1 -40 amino acid residues. In some embodiments, L4 further comprises one sub-units. In some embodiments, L4 further comprises one
 sub-
sub-
 unit. In some embodiments, L4 further comprises one
unit. In some embodiments, L4 further comprises one
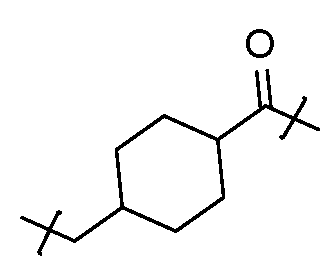 sub-unit. In some embodiments, L4 further comprises one sub-unit. In some embodiments,
sub-unit. In some embodiments, L4 further comprises one sub-unit. In some embodiments,

L4 is absent.
In some embodiments, each Z1 is oxygen.
In some embodiments, Z3 is -0-, -N(R5)- or -heteroaryl-. In some embodiments, Z3 is
 In some embodiments, Z3 is In
In some embodiments, Z3 is In
 some embodiments, Q1 is C(O).
some embodiments, Q1 is C(O).
In some embodiments, Z4 is In some embodiments, Z4 is In

 some embodiments, Z4 is
some embodiments, Z4 is
 In some embodiments, Z4 is In some
In some embodiments, Z4 is In some
 embodiments, Z4 is
embodiments, Z4 is
 . In some embodiments, Z4 is
. In some embodiments, Z4 is
 In some embodiments, Z4 is
In some embodiments, Z4 is

 wherein:
wherein:
U is O, S or NR13;
Q is CR13 orN; each V1, V2 and V3 are independently CR17 or N provided that for formula (Ill-b) and (III-c) at least one of Q, V1 and V2 is N;
R11 is the side chain of an amino acid and is optionally protected with a protecting group; Each R10, R14, and R15 are independently selected from hydrogen, halo, OH, -N(R12)2, - N(R12)3 +, C1-C8 heteroalkyl, carboxylate, sulfate, sulfamate, sulfonate, -SO2R5, - S(O)R12,
-SR12, -SO2N(R12)2, -C(O)R5, -CO2R12, -C(O)N(R12)2, -CN, -N3, -NO2, C1-C8 heteroalkyl, polyethyl eneoxy, phosphonate, phosphate, C1-C8 alkyl, C2-C8, C2-C8 alkenyl, C2-C8 alkynl, C2-C8, aryl, and heterocycle; or when taken together, R14 and R15 form a carbonyl (=0), or spiro-carbocyclic ring comprising of 3 to 7 carbon atoms; and
R12 and R13 are independently selected from hydrogen, C1-C8 alkyl, C2-C8 alkenyl, C2-C8 alkynyl, aryl, heterocycle, wherein each alkyl, alkenyl, alkynyl, aryl, and heterocycle are independently substituted with one or more substituents selected from halo, OH, - N(R12)2, -N(R12)3+, heteroalkyl, carboxylate, sulfate, sulfamate, sulfonate, 4- dialkylaminopyridinium, alkyl, -SO2R12, -S(O)R12, -SR12, -SO2N(R12)2, C(O)R12, CO2R12, C(O)N(R12)2, -CN, -N3,
-NO2, C3-C12 carbocycle, aryl, heterocycle, polyethyleneoxy, phosphonate, and phosphate.
In some embodiments, Q is N; V1 is CH, and V2 is CH. In some embodiments, Q is CH; V1 is CH and V2 is N. In some embodiments, Q is N; V1 is CH, and V2 is N. In some embodiments, Q is N; and V1 is N and V2 is N. In some embodiments, Q is N; V1 is N and V2 is CH. In some embodiments, Q is N; and V1, V2 and V3 are each CH. In some embodiments, Q is CH; and V1, V2 and V3 are each CH.
In some embodiments, Z4 is absent.
In some embodiments, T is -C1-C20- alkylene (e.g., -C1-C6- alkylene), -C1-C20- heteroalkylene (e.g., -C1-C6- heteroalkylene), -C1-C20- alkenylene (e.g., -C 2-C6- alkenylene), - C1-C20- alkynylene (e.g., -C 2-C6- alkynyl), -aryl- or -heteroaryl-. In some embodiments, T is absent.
In some embodiments, anti -PD- 1 antibody or anti-CTLA-4 antibody is a monoclonal antibody, a domain antibody (dAb), a single chain antibody (scAb), a Fab fragment, a F(ab’)2 fragment, a single chain variable fragment (scFv), a scFv-Fc fragment, a single domain heavy chain antibody, a single domain light chain antibody, a variant antibody, a multimeric antibody, or a bispecific antibody.
In some embodiments, the antibody is an anti -PD- 1 antibody. In some embodiments, the anti -PD- 1 antibody is pembrolizumab, nivolumab, cemiplimab, Anti -mouse PD-1 antibody Clone J43, Anti-mouse PD-1 antibody Clone RMP1-14, mouse anti-PD-1 antibody Clone EH12, ANB011, MDX-1 106, AMP-514, AMP-224, or Pidilizumab. In some embodiments, the anti-PD-1 antibody is pembrolizumab or nivolumab.
In some embodiments, the antibody is an anti-CTLA-4 antibody. In some embodiments, the anti-CTLA-4 antibody is ipilimumab, clone 9H10, tremelimumab, or clone BNI3.
In some embodiments, the antibody-drug conjugate is selected from Table 2:
Table 2: Exemplary Antibody-Drug Conjugates of the Present Invention




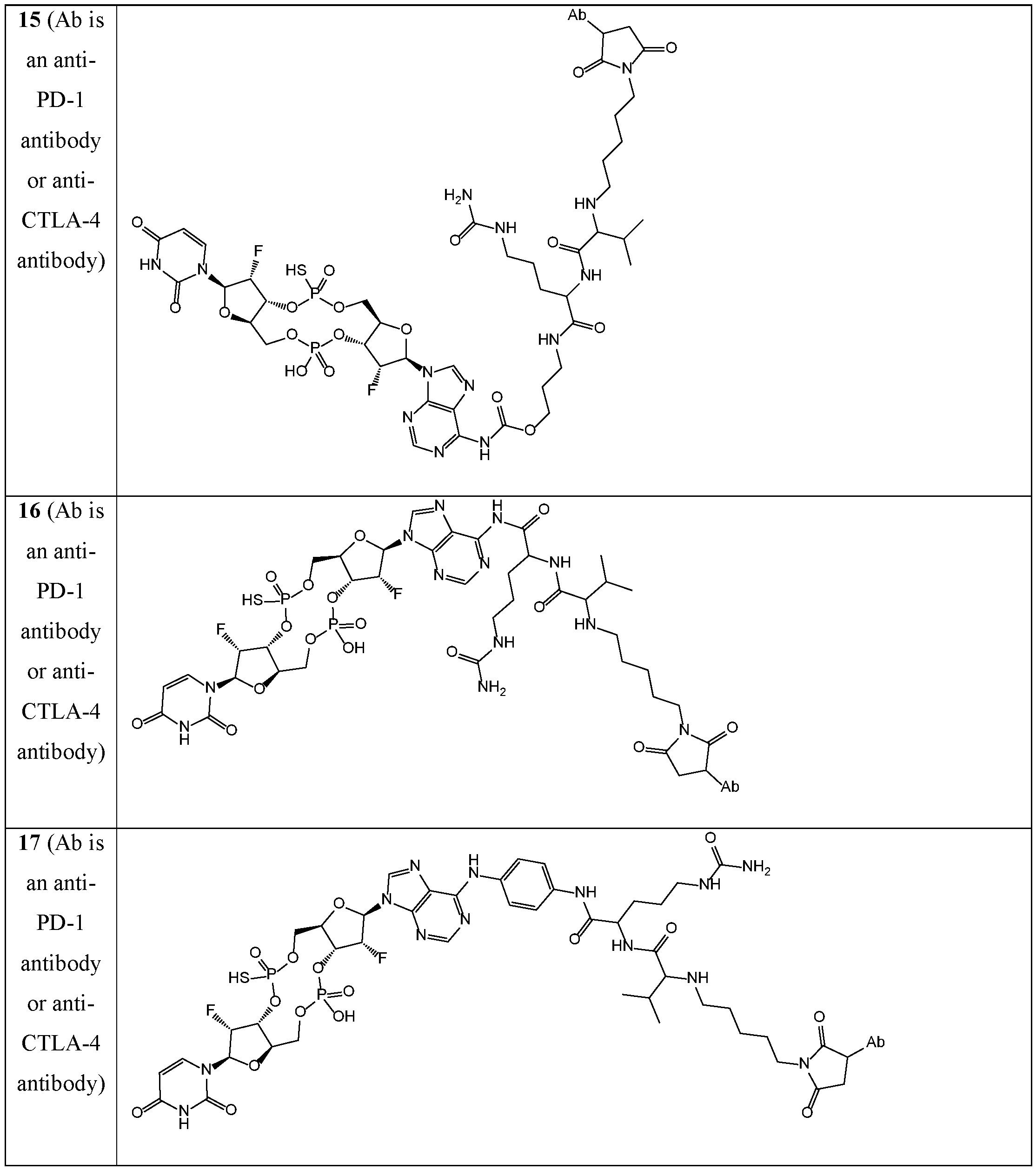

Antibody-Drug Conjugates of Formula VII
In another aspect, the disclosure features an antibody-drug conjugate of Formula (Vlla) or Formula (Vllb):

Ab is an anti-PD-1 antibody or an anti-CTLA-4 antibody; each of B1 and B2 is independently a purinyl nucleobase or pyrimidinyl nucleobase; each of X1 and X2 is independently O or S; each of Y1 and Y2 is independently O, S, or N(R5); each of Z1 is independently O or S;
Z2 is -O-, -N(R5)-, -S-, -C(O)-, -C(O)N(R5)-, -OC(O)N(R5)-, -N(R5)C(O)O-, -aryl-, -heteroaryl-, -S(O)-, -S(O)2-, -S(O)N(R5)-, -S(O)2N(R5)- or -N(R5)S(O)-;
Z3 is absent, hydrogen, heterocyclyl, heterocyclyl-C1-C20-alkylene-Q1, -OH, -N(R5)2, SR5, - CHO, -C(O)N(R5)2, -OC(O)N(R5)2, -N(R5)C(O)OR5, aryl, heteroaryl, -S(O)R5, - S(O)2R5, -S(O)N(R5)2, -S(O)2N(R5)2, -N(R5)S(O)R5, -OSi(C1-C4 alkyl)3, or -C(O)C2- C6 alkenyl (e.g., -C2-C4 alkenyl);
Z4 is a self-immolative group or absent;
T is a absent or spacer group; each L1 and L2 is absent, -C1-C6- alkylene (e.g., -C1-C3- alkylene) or -C1-C6- heteroalkyl ene, wherein each alkylene and heteroalkyl is optionally substituted with one or more R6;
L3 is oligiopeptide-C(O)-, oligiopeptide-aryl-C1-C6-alkylene-, oligiopeptide-aryl-C1-C6- alklyene-C(O)-, oligiopeptide-aryl-C1-C6-heteroalkylene, oligiopeptide-C1-C6- alkylene-C(O)-, oligiopeptide-C1-C6-heteroalkylene-C(O)-, -C1-C40- alkylene (e.g., - C1-C20- alkylene), -C1-C40- heteroalkyl ene (e.g., -C1-C20- heteroalkyl), -C1-C40- alkenylene (e.g., -C2-C20- alkenylene), or -C1-C40- alkynylene (e.g., -C2-C20- alkynylene), wherein the oligopeptide is optionally substituted by one or more R14;
L4 is absent or a linker connecting Z3 and Z4;
Q1 is C(O), C(S), or CH2; each of R1 and R2 is independently hydrogen, halo, -CN, -C1-C20 alkyl (e.g., C1-C6 alkyl), or - OR7; each R3 and R4 is independently hydrogen, -C1-C20- alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl (e.g., -C1-C6 heteroalkyl), -OC(O)OC1-C20 alkyl (e.g., -C1-C6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, heteroalkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R8;
R5 is hydrogen or -C1-C20 alkyl (e.g., -C1-C6 alkyl);
R6 is halo, -CN, -C1-C20 alkyl (e.g., -C1-C6 alkyl), -OR7, oxo, cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R9;
R7 is hydrogen, -C1-C20 alkyl (e.g., -C1-C6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R9; each R8 is independently -C1-C20 alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl,
-C(O)-C1-C20 alkyl, -OC(O)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -C(O)O-C1-C20 alkyl (e.g., -C1-C6 alkyl), -OC(O)O-C1-C20 alkyl (e.g., -C1-C6 alkyl), -C(O)N(R5)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -N(R5)C(O)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -OC(O)N(R5)- C1-C20 alkyl (e.g., -C1-C6 alkyl), -O-aryl, -O-heteroaryl, -C(O)-aryl, -C(O)-heteroaryl, -OC(O)-aryl, -C(O)O-aryl, -OC(O)-heteroaryl, -C(O)O-heteroaryl, -C(O)O-aryl, - C(O)O-heteroaryl, -C(O)N(R5)-aryl, -C(O)N(R5)-heteroaryl, -N(R5)C(O)-aryl, - N(R5)2C(O)-aryl, or -N(R5)C(O)-heteroaryl, -S(O)2N(R5)-aryl, wherein each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R9; each R9 is independently -C1-C20 alkyl, -O-C1-C20 alkyl, -C1-C20 heteroalkyl, halo,
-CN, -OH, oxo, aryl, heteroaryl, -O-aryl, or -O-heteroaryl; and each R16 is independently, -C1-C20 alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl (e.g., -C1-C6 heteroalkyl), -OC(O)OC1-C20 alkyl (e.g., -C1-C6 alkyl), C(O)N(R4)2 cycloalkyl, heterocyclyl, aryl, or heteroaryl.
In some embodiments, wherein the antibody-drug conjugate is an antibody-drug conjugate of formula (VllI-a), (VllI-b), (VIII-c), (VllI-d), (VllI-e), (VllI-f), (VIII-g), or (Vlll-h):


In some embodiments, each of B1 or B2 is selected from a naturally occurring nucleobase or a modified nucleobase. In some embodiments, each of B1 or B2 is selected from adenosinyl, guanosinyl, cytosinyl, thyminyl, uracilyl, 5’-methylcytosinyl, 5’-fluorouracilyl, 5’- propynyluracilyl, and 7-deazaadenosinyl. In some embodiments, each of B1 or B2 is selected from:
 wherein ndicates the linkage of the nucleobase to the ribose ring.
wherein ndicates the linkage of the nucleobase to the ribose ring.
 In some embodiments, one of B1 or B2 is selected from a naturally occurring nucleobase and the other of B1 or B2 is a modified nucleobase. In some embodiments, one of B1 or B2 is adenosinyl, guanosinyl, thyminyl, cytosinyl, or uracilyl, and the other of B1 or B2 is 5’- methylcytosinyl, 5’-fluorouracilyl, 5’-propynyluracilyl, or 7-deazaadenosinyl.
In some embodiments, one of B1 or B2 is selected from a naturally occurring nucleobase and the other of B1 or B2 is a modified nucleobase. In some embodiments, one of B1 or B2 is adenosinyl, guanosinyl, thyminyl, cytosinyl, or uracilyl, and the other of B1 or B2 is 5’- methylcytosinyl, 5’-fluorouracilyl, 5’-propynyluracilyl, or 7-deazaadenosinyl.
In some embodiments, B1 is adenosinyl or guanosinyl. In some embodiments, B2 is cytosinyl, thyminyl, or uracilyl. In some embodiments, B1 is adenosinyl or guanosinyl and B2 is cytosinyl, thyminyl, or uracilyl. In some embodiments, each of B1 and B2 is independently uracilyl. In some embodiments, each of B1 and B2 is independently adenosinyl.
In some embodiments, each of R1 and R2 is independently hydrogen, halo, or OR7. In some embodiments, each of R1 and R2 is independently halo (e.g., fluoro). In some embodiments, each of R1 and R2 is not hydrogen or OR7.
In some embodiments, X1 is O. In some embodiments, X2 is O. In some embodiments, each of X1 and X2 is independently O.
In some embodiments, Y1 is O or S. In some embodiments, Y2 is O or S. In some embodiments, each of Y1 and Y2 is independently O or S. In some embodiments, one of Y1 or Y2 is O and the other of Y1 or Y2 is S. In some embodiments, each of Y1 or Y2 is independently S. In some embodiments, each of Y1 or Y2 is independently O.
In some embodiments, L1 is C1-C6 alkyl (e.g., CH2). In some embodiments, L2 is C1- C6 alkyl (e.g., CH2). In some embodiments, each of L1 and L2 is independently C1-C6 alkyl (e.g., CH2).
In some embodiments, R3 is hydrogen, aryl, or heteroaryl, wherein aryl and heteroaryl is optionally substituted with 1-5 R8. In some embodiments, R3 is aryl or heteroaryl, each of which is optionally substituted with 1-5 R8. In some embodiments, R3 is phenyl substituted with 1 R8.
In some embodiments, R4 is independently hydrogen, aryl, or heteroaryl, wherein aryl and heteroaryl is optionally substituted with 1-5 R8. In some embodiments, R4 is aryl or heteroaryl, each of which is optionally substituted with 1-5 R8. In some embodiments, R4 is phenyl substituted with 1 R8.
In some embodiments, each of R3 and R4 is independently hydrogen, aryl, or heteroaryl, wherein aryl and heteroaryl is optionally substituted with 1-5 R8. In some embodiments, R3 is aryl or heteroaryl, each of which is optionally substituted with 1-5 R8, and R4 is hydrogen. In some embodiments, R3 is phenyl substituted with 1 R8 and R4 is hydrogen. In some embodiments, each of R3 and R4 is independently phenyl substituted with 1 R8.
In some embodiments, each of Y1 and Y2 is O and each of R3 and R4 is independently hydrogen. In some embodiments, Y2 is O and R4 is hydrogen. In some embodiments, each of Y1 and Y2 is independently S and each of R3 and R4 is independently substituted with 1 R8. In some embodiments, Y1 is S and R3 is substituted with 1 R8.
In some embodiments, each R8 is independently C1-C20 alkyl (e.g., C1-C6 alkyl), C1-C20 heteroalkyl, C(O)-C1-C20 alkyl, OC(O)-C1-C20 alkyl, OC(O)O-C1-C20 alkyl, OC(O)N(R5)-C1- C20 alkyl, O-aryl, C(O)-aryl, OC(O)-aryl, or C(O)N(R5)-aryl, wherein each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R9.
In some embodiments, R8 is OC(O)-aryl optionally substituted by 1-5 R9 (e.g., 1 R9). In some embodiments, R9 is O-C1-C12 alkyl (e.g., O-CH2(CH2)8CH3). In some embodiments, R9 is O-C1-C10 alkyl (e.g., O-CH2(CH2)8CH3). In some embodiments, R9 is O-C1-C8 alkyl (e.g., O-CH2(CH2)6CH3). In some embodiments, R9 is O-C1-C6 alkyl (e.g., O-CH2(CH2)4CH3).
In some embodiments, each of L1 and L2 is independently C1-C6 alkylene (e.g., C1 alkylene).
In some embodiments, L3 is -C1-C20- alkylene (e.g., -C1-C18- alkylene). In some embodiments, L3 is -C1-C20- heteroalkylene (e.g., -C1-C18- heteroalkylene). In some embodiments, L3 is an oligiopeptide-C(O)-, oligiopeptide-aryl-C1-C6-alkylene-, oligiopeptide- aryl-C1-C6-heteroalkylene, oligiopeptide-aryl-C1-C6-alklyene-C(O)-, oligiopeptide-C1-C6- alkylene-C(O)-, oligiopeptide-C1-C6-heteroalkylene-C(O)-. In some embodiments, the aryl is phenyl. In some embodiments, the oligiopeptide is a dipeptide. In some embodiments, the oligopeptide comprises 3 amino acid residues, 4 amino acid residues, 5 amino acid residues, 6 amino acid residues, 7 amino acid residues, 8 amino acid residues, 9 amino acid residues, or 10 amino acid residues. In some embodiments, the oligopeptide comprises 8 amino acid residues. In some embodiments, the oligopeptide comprises an amino acid selected from Tyr, Ser, Thr, Arg, Leu, lie, Gly, Val, and Ala. In some embodiments, he oligopeptide comprises an amino acid selected from Phe, Tyr, Arg, Gly, Ser, and Leu. In some embodiments, L3 is

 or
or
 In other embodiments, L3 is
In other embodiments, L3 is
 In yet other embodiments, L3 is selected from the group consisting of
In yet other embodiments, L3 is selected from the group consisting of
 and
and
 In some embodiments, L3 is
In some embodiments, L3 is
 In some embodiments, the oligiopeptide is substituted by one or more instances of R16. In some embodiments, R16 is C(0)NH2.
In some embodiments, the oligiopeptide is substituted by one or more instances of R16. In some embodiments, R16 is C(0)NH2.
In some embodiments, L4is -C1-C40- alkylene (e.g., -C1-C20- alkyl ene), -C1-C40- heteroalkylene (e.g., -C1-C20- heteroalkyl), -C1-C40- alkenylene (e.g., -C2-C20- alkenylene), or -C1-C40- alkynylene (e.g., -C2-C20- alkynylene). In some embodiments, L4 is an oligopeptide
comprising of 1-40 amino acid residues. In some embodiments, L4 further comprises one
 sub-units. In some embodiments, L further comprises one
sub-units. In some embodiments, L further comprises one
 sub unit. In some embodiments, L4 further comprises one
sub unit. In some embodiments, L4 further comprises one
 sub-unit. In some embodiments, L4 further comprises one
sub-unit. In some embodiments, L4 further comprises one
 sub-unit. In some embodiments, L4 is absent.
sub-unit. In some embodiments, L4 is absent.
In some embodiments, each Z1 is oxygen.
In some embodiments, Z3 is -0-, -N(R5)- or -heteroaryl-. In some embodiments, Z3 is
 In some embodiments, Z3 is . In
In some embodiments, Z3 is . In
 some embodiments, Q1 is C(0).
some embodiments, Q1 is C(0).
In some embodiments, Z4 is
 In some embodiments, Z4 is
In some embodiments, Z4 is
 In some embodiments, Z4 is In some embodiments, Z4 is In some
In some embodiments, Z4 is In some embodiments, Z4 is In some

 embodiments, Z4 is
embodiments, Z4 is
 In some embodiments, Z4 is In some embodiments, Z4 is represented by formula (IX-a), (IX-b), or (IX-c):
In some embodiments, Z4 is In some embodiments, Z4 is represented by formula (IX-a), (IX-b), or (IX-c):

 wherein:
wherein:
U is O, S or NR13;
Q is CR13 orN; each V1, V2 and V3 are independently CR17 or N provided that for formula (Ill-b) and (III-c) at least one of Q, V1 and V2 is N;
R11 is the side chain of an amino acid and is optionally protected with a protecting group;
Each R10, R14, and R15 are independently selected from hydrogen, halo, OH, -N(R12)2, - N(R12)3 +, C1-C8 heteroalkyl, carboxylate, sulfate, sulfamate, sulfonate, -SO2R5, - S(O)R12,
-SR12, -SO2N(R12)2, -C(O)R5, -CO2R12, -C(O)N(R12)2, -CN, -N3, -NO2, C1-C8 heteroalkyl, polyethyl eneoxy, phosphonate, phosphate, C1-C8 alkyl, C2-C8, C2-C8 alkenyl, C2-C8 alkynl, C2-C8, aryl, and heterocycle; or when taken together, R14 and R15 form a carbonyl (=O), or spiro-carbocyclic ring comprising of 3 to 7 carbon atoms; and
R12 and R13 are independently selected from hydrogen, C1-Cx alkyl, C2-C8 alkenyl, C2-C8 alkynyl, aryl, heterocycle, wherein each alkyl, alkenyl, alkynyl, aryl, and heterocycle are independently substituted with one or more substituents selected from halo, OH, - N(Ri2)2, -N(R12)3+, heteroalkyl, carboxylate, sulfate, sulfamate, sulfonate, 4- dialkylaminopyridinium, alkyl, -SO2R12, -S(O)R12, -SR12, -SO2N(R12)2, C(O)R12, CO2R12, C(O)N(R12)2, -CN, -N3,
-NO2, C3-C12 carbocycle, aryl, heterocycle, polyethyleneoxy, phosphonate, and phosphate.
In some embodiments, Q is N; V1 is CH, and V2 is CH. In some embodiments, Q is CH; V1 is CH and V2 is N. In some embodiments, Q is N; V1 is CH, and V2 is N. In some embodiments, Q is N; and V1 is N and V2 is N. In some embodiments, Q is N; V1 is N and V2 is CH. In some embodiments, Q is N; and V1, V2 and V3 are each CH. In some embodiments, Q is CH; and V1, V2 and V3 are each CH.
In some embodiments, Z4 is absent.
In some embodiments, T is -C1-C20- alkylene (e.g., -C1-C6- alkylene), -C1-C20- heteroalkylene (e.g., -C1-C6- heteroalkylene), -C1-C20- alkenylene (e.g., -C 2-C6- alkenylene), - C1-C20- alkynylene (e.g., -C 2-C6- alkynyl), -aryl- or -heteroaryl-. In some embodiments, T is absent.
In some embodiments, anti -PD- 1 antibody or anti-CTLA-4 antibody is a monoclonal antibody, a domain antibody (dAb), a single chain antibody (scAb), a Fab fragment, a F(ab’)2 fragment, a single chain variable fragment (scFv), a scFv-Fc fragment, a single domain heavy chain antibody, a single domain light chain antibody, a variant antibody, a multimeric antibody, or a bispecific antibody.
In some embodiments, the antibody is an anti -PD- 1 antibody. In some embodiments, the anti -PD- 1 antibody is pembrolizumab, nivolumab, cemiplimab, Anti -mouse PD-1 antibody Clone J43, Anti-mouse PD-1 antibody Clone RMP1-14, mouse anti-PD-1 antibody Clone EH12, ANB011, MDX-1 106, AMP-514, AMP-224, or Pidilizumab. In some embodiments, the anti-PD-1 antibody is pembrolizumab or nivolumab.
In some embodiments, the antibody is an anti-CTLA-4 antibody. In some embodiments, the anti-CTLA-4 antibody is ipilimumab, clone 9H10, tremelimumab, or clone BNI3.
In some embodiments, the antibody-drug conjugate is selected from Table 2:
Table 3: Exemplary Antibody-Drug Conjugates of the Present Invention






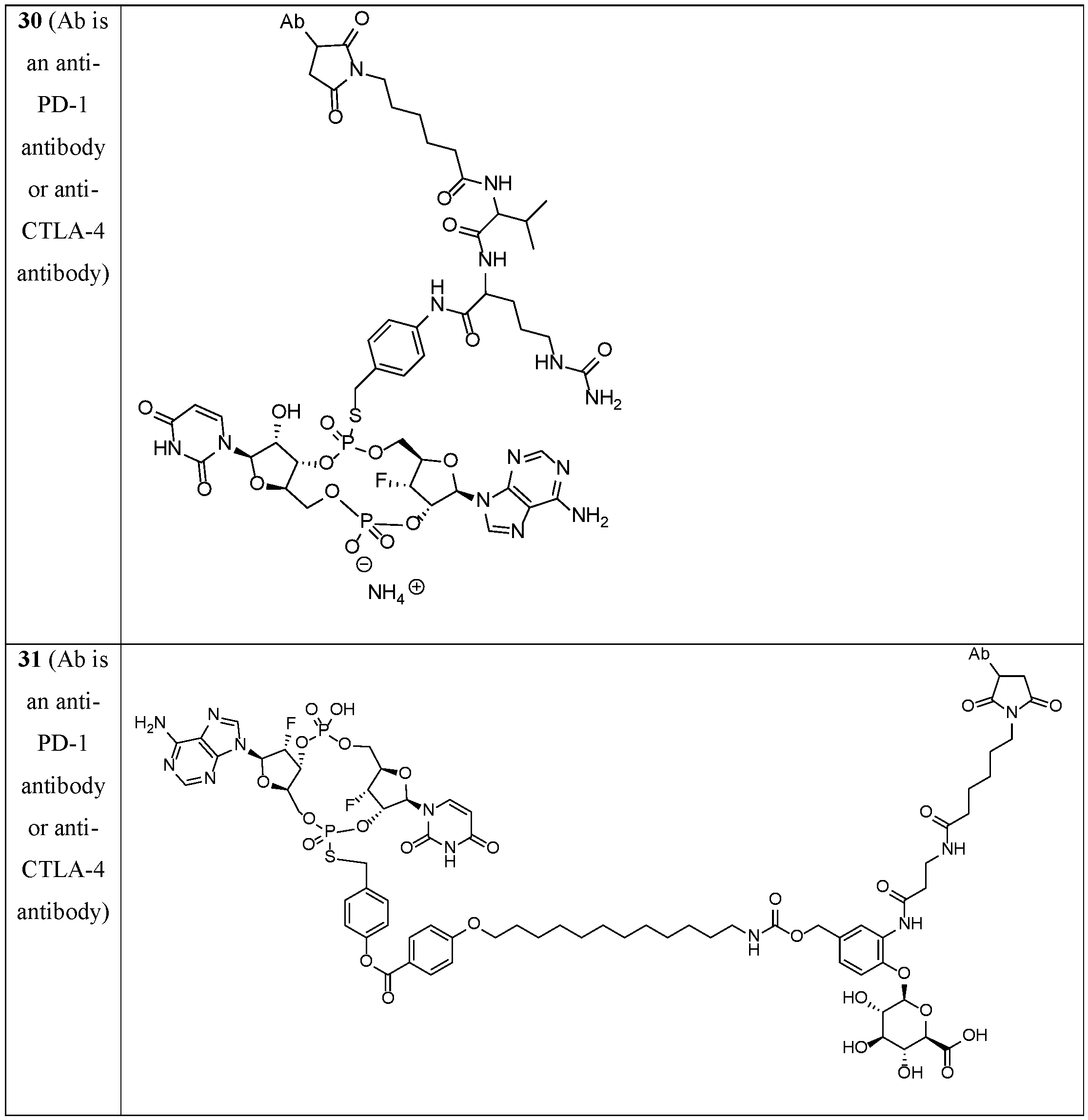

Antibody-Drug Conjugates of Formula X
In another aspect, the disclosure features an antibody-drug conjugate of Formula (Xa) or Formula (Xb):

 or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
Ab is an anti-PD-1 antibody or an anti-CTLA-4 antibody; each of B1 and B2 is independently a purinyl nucleobase or pyrimidinyl nucleobase; each of X1 and X2 is independently O or S; each of Y1 and Y2 is independently O, S, or N(R5); each of Z1 is independently O or S;
Z2 is -O-, -N(R5)-, -S-, -C(O)-, -C(O)N(R5)-, -OC(O)N(R5)-, -N(R5)C(O)O-, -aryl-, -heteroaryl-, -S(O)-, -S(O)2-, -S(O)N(R5)-, -S(O)2N(R5)- or -N(R5)S(O)-;
Z3 is absent, hydrogen, heterocyclyl, heterocyclyl-C1-C20-alkylene-Q1, -OH, -N(R5)2, SR5, - CHO, -C(O)N(R5)2, -OC(O)N(R5)2, -N(R5)C(O)OR5, aryl, heteroaryl, -S(O)R5, - S(O)2R5, -S(O)N(R5)2, -S(O)2N(R5)2, -N(R5)S(O)R5, -OSi(C1-C4 alkyl)3, or -C(O)C2- C6 alkenyl (e.g., -C2-C4 alkenyl);
Z4 is a self-immolative group or absent;
T is a absent or spacer group; each L1 and L2 is absent, -C1-C6- alkylene (e.g., -C1-C3- alkylene) or -C1-C6- heteroalkyl ene, wherein each alkylene and heteroalkyl is optionally substituted with one or more R6;
L3 is oligiopeptide-C(O)-, oligiopeptide-aryl-C1-C6-alkylene-, oligiopeptide-aryl-C1-C6- alklyene-C(O)-, oligiopeptide-aryl-C1-C6-heteroalkylene, oligiopeptide-C1-C6- alkylene-C(O)-, oligiopeptide-C1-C6-heteroalkylene-C(O)-, -C1-C40- alkylene (e.g., - C1-C20- alkylene), -C1-C40- heteroalkyl ene (e.g., -C1-C20- heteroalkyl), -C1-C40- alkenylene (e.g., -C2-C20- alkenylene), or -C1-C40- alkynylene (e.g., -C2-C20- alkynylene), wherein the oligopeptide is optionally substituted by one or more R14;
L4 is absent or a linker connecting Z3 and Z4;
Q1 is C(O), C(S), or CH2; each of R1 and R2 is independently hydrogen, halo, -CN, -C1-C20 alkyl (e.g., C1-C6 alkyl), or - OR7; each R3 and R4 is independently hydrogen, -C1-C20- alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl (e.g., -C1-C6 heteroalkyl), -OC(O)OC1-C20 alkyl (e.g., -C1-C6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, heteroalkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R8;
R5 is hydrogen or -C1-C20 alkyl (e.g., -C1-C6 alkyl);
R6 is halo, -CN, -C1-C20 alkyl (e.g., -C1-C6 alkyl), -OR7, oxo, cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R9;
R7 is hydrogen, -C1-C20 alkyl (e.g., -C1-C6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R9; each R8 is independently -C1-C20 alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl,
-C(O)-C1-C20 alkyl, -OC(O)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -C(O)O-C1-C20 alkyl (e.g., -C1-C6 alkyl), -OC(O)O-C1-C20 alkyl (e.g., -C1-C6 alkyl), -C(O)N(R5)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -N(R5)C(O)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -OC(O)N(R5)- C1-C20 alkyl (e.g., -C1-C6 alkyl), -O-aryl, -O-heteroaryl, -C(O)-aryl, -C(O)-heteroaryl, -OC(O)-aryl, -C(O)O-aryl, -OC(O)-heteroaryl, -C(O)O-heteroaryl, -C(O)O-aryl, - C(O)O-heteroaryl, -C(O)N(R5)-aryl, -C(O)N(R5)-heteroaryl, -N(R5)C(O)-aryl, - N(R5)2C(O)-aryl, or -N(R5)C(O)-heteroaryl, -S(O)2N(R5)-aryl, wherein each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R9; each R9 is independently -C1-C20 alkyl, -O-C1-C20 alkyl, -C1-C20 heteroalkyl, halo,
-CN, -OH, oxo, aryl, heteroaryl, -O-aryl, or -O-heteroaryl; and each R16 is independently, -C1-C20 alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl (e.g., -C1-C6 heteroalkyl), -OC(O)OC1-C20 alkyl (e.g., -C1-C6 alkyl), C(O)N(R4)2 cycloalkyl, heterocyclyl, aryl, or heteroaryl.
In some embodiments, wherein the antibody-drug conjugate is an antibody-drug conjugate of formula (Xl-a), (CI-b), (XI-c), (CI-d), (XI-e), (XI-f), (XI-g), or (Xl-h):


In some embodiments, wherein the antibody-drug conjugate is an antibody-drug conjugate of formula (CI-i), (Xl-j), (CI-k), (XI-1), (Xl-m), (CI-n), (XI-o), (XI-p), (Xl-q), (CI-r), (XI-s), or (XI-t):
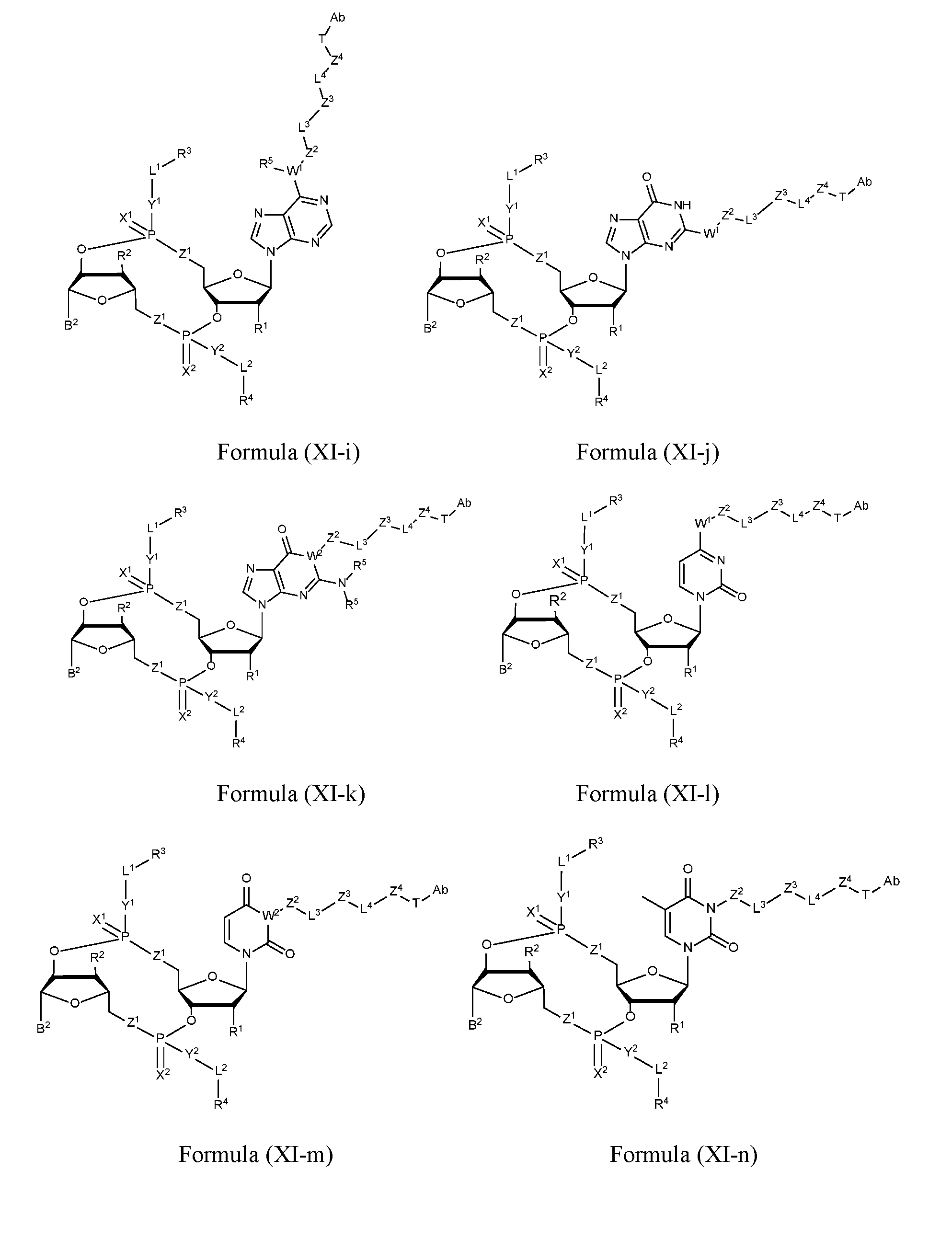

W1 is N(R5), -O- CH; and W2 is N or CH.
In some embodiments, each of B1 or B2 is selected from a naturally occurring nucleobase or a modified nucleobase. In some embodiments, each of B1 or B2 is selected from
adenosinyl, guanosinyl, cytosinyl, thyminyl, uracilyl, 5’ -methyl cytosinyl, 5’-fluorouracilyl, 5’- propynyluracilyl, and 7-deazaadenosinyl. In some embodiments, each ofB1 or B2 is selected from:
 wherein indicates the linkage of the nucleobase to the ribose ring.
wherein indicates the linkage of the nucleobase to the ribose ring.

In some embodiments, one of B1 or B2 is selected from a naturally occurring nucleobase and the other of B1 or B2 is a modified nucleobase. In some embodiments, one of B1 or B2 is adenosinyl, guanosinyl, thyminyl, cytosinyl, or uracilyl, and the other of B1 or B2 is 5’- methyl cytosinyl, 5’-fluorouracilyl, 5’-propynyluracilyl, or 7-deazaadenosinyl.
In some embodiments, B1 is adenosinyl or guanosinyl. In some embodiments, B2 is cytosinyl, thyminyl, or uracilyl. In some embodiments, B1 is adenosinyl or guanosinyl and B2 is cytosinyl, thyminyl, or uracilyl. In some embodiments, each of B1 and B2 is independently uracilyl. In some embodiments, each of B1 and B2 is independently adenosinyl.
In some embodiments, each of R1 and R2 is independently hydrogen, halo, or OR7. In some embodiments, each of R1 and R2 is independently halo (e.g., fluoro). In some embodiments, each of R1 and R2 is not hydrogen or OR7.
In some embodiments, X1 is O. In some embodiments, X2 is O. In some embodiments, each of X1 and X2 is independently O.
In some embodiments, Y1 is O or S. In some embodiments, Y2 is O or S. In some embodiments, each of Y1 and Y2 is independently O or S. In some embodiments, one of Y1 or Y2 is O and the other of Y1 or Y2 is S. In some embodiments, each of Y1 or Y2 is independently S. In some embodiments, each of Y1 or Y2 is independently O.
In some embodiments, L1 is C1-C6 alkyl (e.g., CH2). In some embodiments, L2 is C1- C6 alkyl (e.g., CH2). In some embodiments, each of L1 and L2 is independently C1-C6 alkyl (e.g., CH2).
In some embodiments, R3 is hydrogen, aryl, or heteroaryl, wherein aryl and heteroaryl is optionally substituted with 1-5 R8. In some embodiments, R3 is aryl or heteroaryl, each of which is optionally substituted with 1-5 R8. In some embodiments, R3 is phenyl substituted with 1 R8.
In some embodiments, R4 is independently hydrogen, aryl, or heteroaryl, wherein aryl and heteroaryl is optionally substituted with 1-5 R8. In some embodiments, R4 is aryl or heteroaryl, each of which is optionally substituted with 1-5 R8. In some embodiments, R4 is phenyl substituted with 1 R8.
In some embodiments, each of R3 and R4 is independently hydrogen, aryl, or heteroaryl, wherein aryl and heteroaryl is optionally substituted with 1-5 R8. In some embodiments, R3 is aryl or heteroaryl, each of which is optionally substituted with 1-5 R8, and R4 is hydrogen. In some embodiments, R3 is phenyl substituted with 1 R8 and R4 is hydrogen. In some embodiments, each of R3 and R4 is independently phenyl substituted with 1 R8.
In some embodiments, each of Y1 and Y2 is O and each of R3 and R4 is independently hydrogen. In some embodiments, Y2 is O and R4 is hydrogen. In some embodiments, each of Y1 and Y2 is independently S and each of R3 and R4 is independently substituted with 1 R8. In some embodiments, Y1 is S and R3 is substituted with 1 R8.
In some embodiments, each R8 is independently C1-C20 alkyl (e.g., C1-C6 alkyl), C1-C20 heteroalkyl, C(O)-C1-C20 alkyl, OC(O)-C1-C20 alkyl, OC(O)O-C1-C20 alkyl, OC(O)N(R5)-C1- C20 alkyl, O-aryl, C(O)-aryl, OC(O)-aryl, or C(O)N(R5)-aryl, wherein each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R9.
In some embodiments, R8 is OC(O)-aryl optionally substituted by 1-5 R9 (e.g., 1 R9). In some embodiments, R9 is O-C1-C12 alkyl (e.g., O-CH2(CH2)8CH3). In some embodiments, R9 is O-C1-C10 alkyl (e.g., O-CH2(CH2)8CH3). In some embodiments, R9 is O-C1-C8 alkyl (e.g., O-CH2(CH2)6CH3). In some embodiments, R9 is O-C1-C6 alkyl (e.g., O-CH2(CH2)4CH3).
In some embodiments, each of L1 and L2 is independently C1-C6 alkylene (e.g., C1 alkylene).
In some embodiments, L3 is -C1-C20- alkylene (e.g., -C1-C18- alkylene). In some embodiments, L3 is -C1-C20- heteroalkylene (e.g., -C1-C18- heteroalkylene). In some embodiments, L3 is an oligiopeptide-C(O)-, oligiopeptide-aryl-C1-C6-alkylene-, oligiopeptide- aryl-C1-C6-heteroalkylene, oligiopeptide-aryl-C1-C6-alklyene-C(O)-, oligiopeptide-C1-C6- alkylene-C(O)-, oligiopeptide-C1-C6-heteroalkylene-C(O)-. In some embodiments, the aryl is phenyl. In some embodiments, the oligiopeptide is a dipeptide. In some embodiments, the oligopeptide comprises 3 amino acid residues, 4 amino acid residues, 5 amino acid residues, 6
amino acid residues, 7 amino acid residues, 8 amino acid residues, 9 amino acid residues, or 10 amino acid residues. In some embodiments, the oligopeptide comprises 8 amino acid residues. In some embodiments, the oligopeptide comprises an amino acid selected from Tyr, Ser, Thr, Arg, Leu, lie, Gly, Val, and Ala. In some embodiments, he oligopeptide comprises an amino acid selected from Phe, Tyr, Arg, Gly, Ser, and Leu. In some embodiments, L3 is

 or
or
 In other embodiments, L3 is In yet other embodiments, L3 is
In other embodiments, L3 is In yet other embodiments, L3 is

selected from the group consisting of
 and
and
 . In some embodiments, L3 is
. In some embodiments, L3 is

In some embodiments, the oligiopeptide is substituted by one or more instances of R16. In some embodiments, R16 is C(O)NH2.
In some embodiments, L4 is -C1-C40- alkylene (e.g., -C1-C20- alkylene), -C1-C40- heteroalkylene (e.g., -C1-C20- heteroalkyl), -C1-C40- alkenylene (e.g., -C2-C20- alkenylene), or -C1-C40- alkynylene (e.g., -C2-C20- alkynylene). In some embodiments, L4 is an oligopeptide comprising of 1 -40 amino acid residues. In some embodiments, L4 further comprises one
 sub-units. In some embodiments, L4 further comprises one
sub-units. In some embodiments, L4 further comprises one
 sub- unit. In some embodiments, L4 further comprises one
sub- unit. In some embodiments, L4 further comprises one
 sub-unit. In some embodiments, L4 further comprises one
sub-unit. In some embodiments, L4 further comprises one
 sub-unit. In some embodiments,
sub-unit. In some embodiments,
L4 is absent.
In some embodiments, each Z1 is oxygen.
In some embodiments, Z3 is -0-, -N(R5)- or -heteroaryl-. In some embodiments, Z3 is
 In some embodiments, Z3 is In
In some embodiments, Z3 is In
 some embodiments, Q1 is C(O).
some embodiments, Q1 is C(O).
In some embodiments, Z4 is
 In some embodiments, Z4 is
In some embodiments, Z4 is
 In some embodiments, Z4 is In some embodiments, Z4 is In some
In some embodiments, Z4 is In some embodiments, Z4 is In some
 embodiments, Z4 is In some embodiments, Z4 is
embodiments, Z4 is In some embodiments, Z4 is

 In some embodiments, Z4 is represented by formula (Xll-a), (Xll-b), or (XII- c):
In some embodiments, Z4 is represented by formula (Xll-a), (Xll-b), or (XII- c):


U is O, S or NR13;
Q is CR13 orN; each V1, V2 and V3 are independently CR17 or N provided that for formula (Ill-b) and (III-c) at least one of Q, V1 and V2 is N;
R11 is the side chain of an amino acid and is optionally protected with a protecting group; Each R10, R14, and R15 are independently selected from hydrogen, halo, OH, -N(R12)2, - N(R12)3 +, C1-C8 heteroalkyl, carboxylate, sulfate, sulfamate, sulfonate, -SO2R5, - S(O)R12,
-SR12, -SO2N(R12)2, -C(O)R5, -CO2R12, -C(O)N(R12)2, -CN, -N3, -NO2, C1-C8 heteroalkyl, polyethyl eneoxy, phosphonate, phosphate, C1-C8 alkyl, C2-C8, C2-C8 alkenyl, C2-C8 alkynl, C2-C8, aryl, and heterocycle; or when taken together, R14 and R15 form a carbonyl (=O), or spiro-carbocyclic ring comprising of 3 to 7 carbon atoms; and
R12 and R13 are independently selected from hydrogen, C1-C8 alkyl, C2-C8 alkenyl, C2-C8 alkynyl, aryl, heterocycle, wherein each alkyl, alkenyl, alkynyl, aryl, and heterocycle are independently substituted with one or more substituents selected from halo, OH, - N(R12)2, -N(R12)3+, heteroalkyl, carboxylate, sulfate, sulfamate, sulfonate, 4- dialkylaminopyridinium, alkyl, -SO2R12, -S(O)R12, -SR12, -SO2N(R12)2, C(O)R12, CO2R12, C(O)N(R12)2, -CN, -N3,
-NO2, C3-C12 carbocycle, aryl, heterocycle, polyethyleneoxy, phosphonate, and phosphate.
In some embodiments, Q is N; V1 is CH, and V2 is CH. In some embodiments, Q is CH; V1 is CH and V2 is N. In some embodiments, Q is N; V1 is CH, and V2 is N. In some embodiments, Q is N; and V1 is N and V2 is N. In some embodiments, Q is N; V1 is N and V2
is CH. In some embodiments, Q is N; and V1, V2 and V3 are each CH. In some embodiments, Q is CH; and V1, V2 and V3 are each CH.
In some embodiments, Z4 is absent.
In some embodiments, T is -C1-C20- alkylene (e.g., -C1-C6- alkylene), -C1-C20- heteroalkylene (e.g., -C1-C6- heteroalkylene), -C1-C20- alkenylene (e.g., -C2-C6- alkenylene), - C1-C20- alkynylene (e.g., -C2-C6- alkynyl), -aryl- or -heteroaryl-. In some embodiments, T is absent.
In some embodiments, anti -PD- 1 antibody or anti-CTLA-4 antibody is a monoclonal antibody, a domain antibody (dAb), a single chain antibody (scAb), a Fab fragment, a F(ab’)2 fragment, a single chain variable fragment (scFv), a scFv-Fc fragment, a single domain heavy chain antibody, a single domain light chain antibody, a variant antibody, a multimeric antibody, or a bispecific antibody.
In some embodiments, the antibody is an anti -PD- 1 antibody. In some embodiments, the anti -PD- 1 antibody is pembrolizumab, nivolumab, cemiplimab, Anti -mouse PD-1 antibody Clone J43, Anti-mouse PD-1 antibody Clone RMP1-14, mouse anti-PD-1 antibody Clone EH12, ANB011, MDX-1 106, AMP-514, AMP-224, or Pidilizumab. In some embodiments, the anti-PD-1 antibody is pembrolizumab or nivolumab.
In some embodiments, the antibody is an anti-CTLA-4 antibody. In some embodiments, the anti-CTLA-4 antibody is ipilimumab, clone 9H10, tremelimumab, or clone BNI3.
Table 3: Exemplary Antibody-Drug Conjugates of the Present Invention







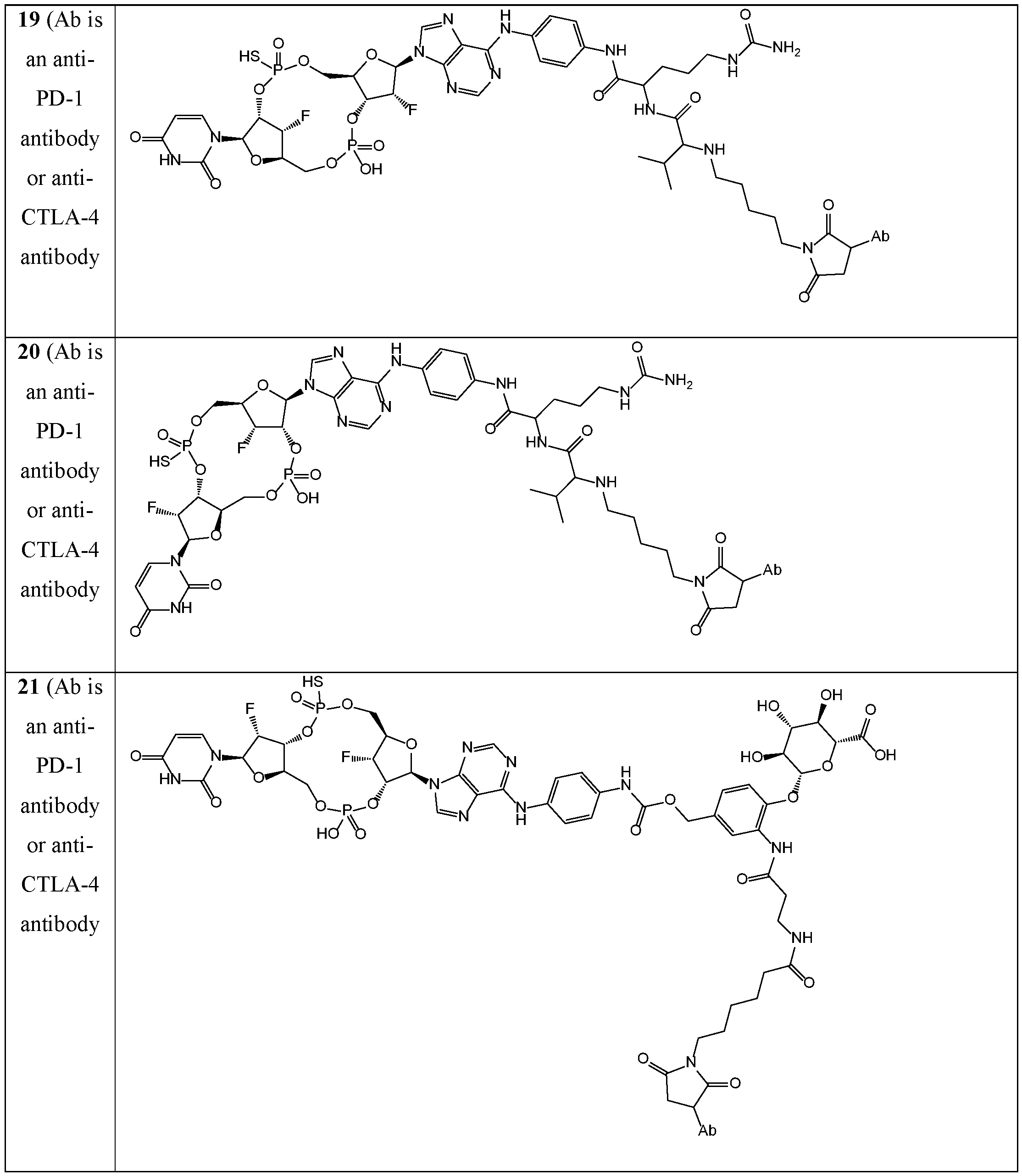


Salts and Isomers
In an embodiment, a antibody-drug conjugate described herein is in the form of a pharmaceutically acceptable salt. Exemplary salts are described herein, such as ammonium salts. In some embodiments, the antibody-drug conjugate is a mono-salt In some embodiments, the antibody-drug conjugate is a di-salt. In some embodiments, the antibody- drug conjugate described herein (e.g., a antibody-drug conjugate in Table 1 or Table 2) is not a salt (e.g., is a free acid or free base).
The antibody-drug conjugate provided herein may contain one or more asymmetric centers and thus occur as racemates and racemic mixtures, single enantiomers, individual diastereomers, and diastereomeric mixtures. All such isomeric forms of these antibody-drug conjugate are expressly included within the scope. Unless otherwise indicated when a antibody-drug conjugate is named or depicted by a structure without specifying the stereochemistry and has one or more chiral centers, it is understood to represent all possible stereoisomers of the antibody-drug conjugate. The antibody-drug conjugate provided herewith may also contain linkages (e.g., carbon-carbon bonds, phosphorus-oxygen bonds, or phosphorus-sulfur bonds) or substituents that can restrict bond rotation, e.g., restriction resulting from the presence of a ring or double bond.
In some embodiments, the method described herein comprises administration of an antibody-drug conjugate of the disclosure or a pharmaceutically acceptable salt thereof. In some embodiments, the method described herein comprises administration of an antibody-drug conjugate of the disclosure or a pharmaceutically acceptable salt thereof. In some embodiments, the antibody-drug conjugate of the disclosure comprises an isomer (e.g., an Rp- isomer or Sp isomer) or a mixture of isomers (e.g., Rp-isomers or Sp isomers).
PD-1 Antibodies
Programmed cell death-1 (PD-1) is a member of the CD28 superfamily that delivers negative signals upon interaction with its two ligands, PD-L1 or PD-L2. PD-1 and its ligands are broadly expressed and exert a wider range of immunoregulatory roles in T cells activation and tolerance compared with other CD28 members. PD-1 and its ligands are involved in attenuating infectious immunity and tumor immunity and facilitating chronic infection and tumor progression. The biological significance of PD-1 and its ligand suggests the therapeutic potential of manipulation of PD-1 pathway against various human diseases (Hyun-Tak Jin, et al., Curr Top Microbiol Immunol. (2011); 350:17-37).
T-cell activation and dysfunction relies on direct and modulated receptors. Based on their functional outcome, co-signaling molecules can be divided as co-stimulators and co- inhibitors, which positively and negatively control the priming, growth, differentiation and functional maturation of a T-cell response (Li Shi, et al., Journal of Hematology & Oncology 2013, 6:74).
Programmed death-1 (PD-1) is a co-receptor that is expressed predominantly by T cells. The binding of PD-1 to its ligands, PD-L1 or PD-L2, is vital for the physiological regulation of the immune system. A major functional role of the PD-1 signaling pathway is the inhibition of self-reactive T cells, which serve to protect against autoimmune diseases. Elimination of the PD-1 pathway can therefore result in the breakdown of immune tolerance that can ultimately lead to the development of pathogenic autoimmunity. Conversely, tumor cells can at times co- opt the PD-1 pathway to escape from immunosurveillance mechanisms. Therefore, blockade of the PD-1 pathway has become an attractive target in cancer therapy. Current approaches include six agents that are either PD-1 and PD-L1 targeted neutralizing antibodies or fusion proteins. More than forty clinical trials are underway to better define the role of PD-1 blockade in variety of tumor types (Ariel Pedoeem et al., Clinical Immunology (2014), 153(1), 145-152).
International applications W02002086083, W02004004771, W02004056875, W02006121168, W02008156712, W02010077634, W02011066389, W02014055897 and
W02014100079 report PD-1, PD-L1 inhibitory antibodies and/or methods of identifying such antibodies; the contents of each of these publications are hereby incorporated by reference in their entirety. Further, US patents such as US8735553 and US8168757 report PD-1 or PD-L1 inhibitory antibodies and/or fusion proteins; the contents of each of these patents are hereby incorporated by reference in their entirety.
CTLA-4 Inhibitors
T-cell activation is a complex process that requires >1 stimulatory signal. TCR binding to MHC provides specificity to T-cell activation, but further costimulatory signals are required. Binding of B7-1 (CD80) or B7-2 (CD86) molecules on the APC with CD28 molecules on the T cell leads to signaling within the T cell. Sufficient levels of CD28:B7-l/2 binding lead to proliferation of T cells, increased T-cell survival, and differentiation through the production of growth cytokines such as interleukin-2 (IL-2), increased energy metabolism, and upregulation of cell survival genes.
CTLA-4 is a CD28 homolog with much higher binding affinity for B7 and is thought to regulate T-cell proliferation early in an immune response, primarily in lymph nodes. Inhibition of these CTLA-4, resulting in increased activation of the immune system and has led to new immunotherapies for melanoma, non-small cell lung cancer, and other cancers.
In one embodiment, the anti-CTLA-4 antibody blocks the binding of CTLA-4 to CD80 (B7-1) and/or CD86 (B7-2) expressed on antigen presenting cells. Exemplary antibodies against CTLA-4 include: Bristol Meyers Squibb’s anti-CTLA-4 antibody ipilimumab (also known as Yervoy, MDX-010, BMS-734016 and MDX-101); anti-CTLA4 Antibody, clone 9H10 from Millipore; Pfizer’s tremelimumab (CP-675,206, ticilimumab); and anti-CTLA-4 antibody clone BNI3 from Abeam.
In some embodiments, the anti-CTLA-4 antibody is an anti-CTLA-4 antibody disclosed in any of the following patent publications (herein incorporated by reference): WO 2001014424; WO 2004035607; US2005/0201994; EP 1212422 B 1; W02003086459; W02012120125; W02000037504; W02009100140; W0200609649; W02005092380; W02007123737; W02006029219; W020100979597; W0200612168; and WO1997020574. Additional CTLA-4 antibodies are described in U.S. Patent Nos. 5,811,097, 5,855,887, 6,051,227, and 6,984,720; in PCT Publication Nos. WO 01/14424 and WO 00/37504; and in U.S. Publication Nos. 2002/0039581 and 2002/086014; and/or U.S. Patent Nos. 5,977,318, 6,682,736, 7, 109,003, and 7,132,281, incorporated herein by reference). In some embodiments, the anti-CTLA-4 antibody is an, for example, those disclosed in: WO 98/42752;
U.S. Patent Nos. 6,682,736 and 6,207, 156; Hurwitz et al, Proc. Natl. Acad. Sci. USA, 95(17): 10067-10071 (1998); Camacho et al, J. Clin. Oncol., 22(145): Abstract No. 2505 (2004) (antibody CP- 675206); Mokyr et al, Cancer Res., 58:5301-5304 (1998) (incorporated herein by reference).
Methods of Use
It has been reported that many patients with advanced solid tumors show a spontaneous T cell-inflamed tumor microenvironment, which is predictive of prognosis and clinical response to immunotherapies. Recent findings suggest the STING pathway of cytosolic DNA sensing is an important innate immune sensing mechanism driving type I IFN production in the tumor context. Knowledge of this pathway is guiding the further development of novel immunotherapeutic strategies.
It has been reported that in early-stage colorectal cancer, the presence of activated CD8+ T cells within the tumor microenvironment significant positive prognostic outcome. Patients with other solid tumor histology also appear to have a spontaneous T cell infiltrate that may have similar positive prognostic value. These include breast cancer, renal cell carcinoma, melanoma, ovarian cancer, and gastrointestinal tumors. It is believed that T cell infiltrate includes tumor antigen-specific T cells that have been activated spontaneously in response to the growing tumor, perhaps through immune surveillance mechanisms. This attempted host immune response, even if it does not eliminate the tumor completely, is thought to delay tumor progression and thus yield improved clinical outcome. Furthermore, the innate immune mechanisms can lead to adaptive T cell response against tumor antigens even in the absence of exogenous infection. In this regard, human cancer gene expression profiling studies reveal an association between a type I IFN signature, T cell infiltration, and clinical outcome. Thus, innate immune sensing pathways that trigger type I IFN production might represent crucial intermediate mechanistic step. In gene expression profiling of melanoma, two major subsets of tumor microenvironment has been found that represent either the presence or absence of a transcriptional profile indicative of T cell infiltrate. In fact, CD8+ T cells, macrophages, as well as of some B cells and plasma cells in these lesions in melanoma metastases is similar to the phenotype described in early-stage colon cancer and other tumors in which activated T cells have been associated with favorable prognosis. CD8+ T cells were required for the up- regulation of all immune factors within the tumor micro-environment. Studies indicate that IFN production is necessary for optimal T cell priming against tumor antigens. There are many PRRs that trigger IFN-b production by host DC8 in response to a growing tumor in vivo
including STING. STING is an adapter protein that is activated by cyclic dinucleotides generated by cyclic GMP-AMP synthase (cGAS), which in turn is directly activated by cytosolic DNA. In the presence of these cyclic dinucleotides and/or DNA, STING is translocated from the endoplasmic reticulum to various perinuclear components; for example, palmitoylation of STING at the Golgi has been shown to be essential for STING activation (Mukai, K. et al (2016) Nat Commun doi:10.1038/ncommsl l932).
Activated STING forms aggregates, activates TBK1, which in turn phosphorylates interferon regulatory factor 3 (IRF3) that directly contributes to type I IFN gene transcription. This pathway has been implicated in the sensing of DNA viruses, and also in selected autoimmune models. Moreover, activating mutations of STING have recently been identified in human patients with a vasculitis/pulmonary inflammation syndrome that is characterized by increased type I IFN production. Mechanistic studies using mouse transplantable tumor models revealed that STING-knockout mice, and IRF3 -knockout mice showed defective spontaneous T cell priming against tumor antigens in vivo, and rejection of immunogenic tumors was ablated. Similarly, tumor-derived DNA was found within the cytosol of a major population of tumor-infiltrating DC8, and this was associated with STING pathway activation and IFN-b production. Therefore, the host STING pathway appears to be an important innate immune sensing pathway that detects the presence of a tumor and to drive DC activation and subsequent T cell priming against tumor-associated antigens in vivo. A functional role for the STING pathway in vivo has also been reported in other mouse-tumor systems. An inducible glioma model was shown to result in induction of a type I IFN gene signature as part of the host response. This induction was substantially reduced in STING-knockout mice, and tumors grew more aggressively, leading to shorter mouse survival. Exogenous delivery of cyclic dinucleotides as STING agonists exerted a therapeutic effect in vivo. A crucial role for host type I IFNs and the host STING pathway was also confirmed in the B16.0VA and EL4.0VA models in response to cryo-ablation. Interestingly, the mechanisms involved paralleled what was observed in the Bml2 mouse model of lupus because host STING was also required for maximal production of anti-DNA antibodies. Thus, the antitumor immune response triggered in part by tumor DNA has overlap with the mechanisms involved in autoimmunity driven by extracellular DNA. A role for STING also has been explored in an inducible colon cancer model. It seems likely that the ability of a cancer in an individual patient to support STING pathway activation is linked to the spontaneous generation of a T cell-inflamed tumor microenvironment. Because this phenotype is associated with improved prognosis of early-
stage cancer patients, and also with clinical response to immunotherapies in the metastatic setting, failed STING activation may therefore represent an early functional block, and thus itself may have prognostic/predictive value as a biomarker. Second, strategies that activate or mimic the output of the host STING pathway should have immunotherapeutic potential in the clinic. In as much as non-T cell-inflamed tumors appear to lack evidence of a type I IFN transcriptional signature, strategies to promote robust innate signaling via APC8 in the tumor microenvironment might facilitate improved cross-priming of tumor antigen-specific CD8+ T cells, and also augment chemokine production for subsequent oncolytic activity.
Recognition of nucleic acid ligands by a PRRs such as cGAS, RIG-I and/STING stimulates the production of type I interferons (e.g., IFN-a or IFN-b), thus triggering a series of downstream signaling events that may lead to apoptosis in susceptible cells. In recent years, a connection between the induction of PRR expression and a number of cancers has been discovered. For example, RIG-I expression has been shown to be significantly downregulated in hepatocellular carcinoma, and patients exhibiting low RIG-I expression in tumors had shorter survival and poorer responses to IFN-a therapy (Hou, J. etal, Cancer Cell (2014) 25:49- 63). As such, it has been suggested that the level of RIG-I expression may be useful as a biomarker for prediction of prognosis and response to immunotherapy. In other cases, induction of RIG-I expression has been shown to induce immunogenic cell death of pancreatic cancer cells, prostate cancer cells, breast cancer cells, skin cancer cells, and lung cancer cells (Duewell, P. et al, Cell Death Differ (2014) 21:1825-1837; Besch, R. et al, J Clin Invest (2009) 119:2399-2411; Kaneda, Y. Oncoimmunology (2013) 2:e23566; Li, X.Y. et al ,Mol Cell Oncol (2014) l:e968016), highlighting a new approach in immune-mediated cancer treatment.
STING is recognized as the key adapter protein in the cGAS-STING-IFN cascade, although it is also reported to be a sensor for DNA. A role for STING in the stimulation of innate immunity in response to cancer has also been identified. Recent studies have revealed the presence of tumor-derived DNA in the cytosol of certain antigen-presenting cells, such as tumor-infiltrating dendritic cells, likely generated through tumor cell stress or cell death. This tumor-derived DNA is known to activate cGAS which causes the production of cyclic nucleotides that have been shown to activate STING, resulting in production of associated type 1 interferons (Woo, S.R. et al, Immunity (2014) 41:830-842). Stimulation of STING and resulting downstream signaling pathways also likely contributes to effector T cell recruitment into the inflamed tumor microenvironment (Woo, S. R. Trends in Immunol (2015) 36:250- 256). STING activation in the tumor microenvironment can induce adaptive immune response leading to anti-tumor activity. Hence, in those tumors that are STING-deficient, the described
herein can still have anti-tumor activity through activation of antigen-presenting cells and dendritic cells, (APC8 and DC8) and induction of adaptive immune response.
Cancer and Antibody Drug Conjugates
In the field of cancer chemotherapeutics, ADC8 combine the specificity of antibodies with the potent anti-tumor effects of cytotoxic drugs. In recent literature, ADC8 have emerged as powerful methods for the targeted treatment of cancer. Two ADC products, brentuximab vedotin (Adcetris®) and trastuzumab emtansine (Kadcyla®), have received FDA approval and there are more than 40 ADC candidates in clinical trials for the treatment of various cancers (Yejin, K. J Pharm. Investigation. (2016) 46:341-349).
The combination of the PRR modulators described herein and the target specificity of ADC8 represents an attractive and powerful new approach to the treatment of diseases such as cancer, when compared to existing methods of treatment.
In some embodiments, the methods of inducing expression of a PRR (e.g., a PRR described herein) comprise administering a therapeutically effective amount of an antibody- drug conjugate of the disclosure or a pharmaceutically acceptable salt thereof to a subject suffering from cancer.
In some embodiments, the methods of inducing expression of STING disclosed herein comprise administering a therapeutically effective amount of an antibody-drug conjugate of the disclosure or a pharmaceutically acceptable salt thereof to a subject suffering from cancer.
In some embodiments, the methods of inducing expression of RIG-I disclosed herein comprise administering a therapeutically effective amount of an antibody-drug conjugate of the disclosure or a pharmaceutically acceptable salt thereof to a subject suffering from cancer.
In some embodiments, the methods of inducing expression of NOD2 disclosed herein comprise administering a therapeutically effective amount of an antibody-drug conjugate of the disclosure or a pharmaceutically acceptable salt thereof to a subject suffering from cancer.
In some embodiments, the cancer is selected from a cancer of the breast, bone, brain, cervix, colon, gastrointestinal tract, eye, gall bladder, lymph nodes, blood, lung, liver, skin, mouth, prostate, ovary, penis, pancreas, uterus, testicles, stomach, thymus, thyroid, or other part of the body. In some embodiments, the cancer comprises a solid tumor (e.g., a carcinoma, a sarcoma, or a lymphoma). In some embodiments, the cancer is a hepatocellular carcinoma or other cancer of the liver. In some embodiments, the cancer is a leukemia or other cancer of the blood. In some embodiments, the cancer comprises breast cancer, renal cell carcinoma, colon cancer, melanoma, ovarian cancer, head and neck squamous cell carcinoma, pancreatic cancer,
prostate cancer, lung cancer, brain cancer, thyroid cancer, renal cancer, testis cancer, stomach cancer, urothelial cancer, skin cancer, cervical cancer, endometrial cancer, liver cancer, lung cancer, lymphoma or gastrointestinal stromal cancer and solid tumors. In some embodiments, the cancer cells (e.g., tumor cells) comprise specific cancer-associated antigens that induce a T-cell-mediated anti -turn or response.
In some embodiments, the methods of inducing expression of a PRR (e.g., STING, RIG-I, MDA5, LGP2) in a subject suffering from a cancer disclosed herein result in an increase in PRR expression (e.g., STING expression). In some embodiments, expression of a PRR (e.g., STING) is induced by a factor of about 1.1, about 1.2, about 1.3, about 1.4, about 1.5, about 1.6, about 1.7, about 1.8, about 1.9, about 2, about 2.5, about 3, about 4, about 5, about 7.5, about 10, about 15, about 20, about 25, about 30, about 40, about 50, about 75, about 100, about 150, about 200, about 250, about 500, about 1000, about 1500, about 2500, about 5000, about 10,000, or more.
In some embodiments, induction of expression of a PRR (e.g., STING) occurs within about 5 minutes of administration of an antibody-drug conjugate of the disclosure or a pharmaceutically acceptable salt thereof.
In some embodiments, induction of expression of a PRR (e.g., STING) occurs within about 10 minutes, about 15 minutes, about 20 minutes, about 25 minutes, about 30 minutes, about 45 minutes, about 1 hour, about 1.5 hours, about 2 hours, about 3 hours, about 4 hours, about 5 hours, about 6 hours, about 7 hours, about 8 hours, about 10 hours, about 12 hours or more following administration of an antibody-drug conjugate of the disclosure or a pharmaceutically acceptable salt thereof.
It is recognized that activation of STING by compounds may lead to induction of expression of other PRRs such as RIG-I, MDA5, NOD2 etc. which may further amplify IFN production in the tumor microenvironment and prime T-cells for enhanced anti-tumor activity.
Pharmaceutical Compositions
The compositions and methods of the present disclosure may be utilized to treat an individual in need thereof. In certain embodiments, the individual is a mammal such as a human, or a non-human mammal. When administered to an animal, such as a human, the composition or the antibody-drug conjugate is preferably administered as a pharmaceutical composition comprising, for example, an antibody-drug conjugate of the disclosure and a pharmaceutically acceptable carrier. Pharmaceutically acceptable carriers are well known in the art and include, for example, aqueous solutions such as water or physiologically buffered
saline or other solvents or vehicles such as glycols, glycerol, oils such as olive oil, or injectable organic esters. In preferred embodiments, when such pharmaceutical compositions are for human administration, particularly for invasive routes of administration (i.e., routes, such as injection or implantation, that circumvent transport or diffusion through an epithelial barrier), the aqueous solution is pyrogen-free, or substantially pyrogen-free. The excipients can be chosen, for example, to effect delayed release of an agent or to selectively target one or more cells, tissues or organs. The pharmaceutical composition can be in dosage unit form such as tablet, capsule (including sprinkle capsule and gelatin capsule), granule, lyophile for reconstitution, powder, solution, syrup, suppository, injection or the like. The composition can also be present in a transdermal delivery system, e.g., a skin patch. The composition can also be present in a solution suitable for topical administration, such as a lotion, cream, or ointment.
A pharmaceutically acceptable carrier can contain physiologically acceptable agents that act, for example, to stabilize, increase solubility or to increase the absorption of an antibody-drug conjugate of the invention. Such physiologically acceptable agents include, for example, carbohydrates, such as glucose, sucrose or dextrans, antioxidants, such as ascorbic acid or glutathione, chelating agents, low molecular weight proteins or other stabilizers or excipients. The choice of a pharmaceutically acceptable carrier, including a physiologically acceptable agent, depends, for example, on the route of administration of the composition. The preparation or pharmaceutical composition can be a selfemulsifying drug delivery system or a selfmicroemulsifying drug delivery system. The pharmaceutical composition (preparation) also can be a liposome or other polymer matrix, which can have incorporated therein, for example, an antibody-drug conjugate of the invention. Liposomes, for example, which comprise phospholipids or other lipids, are nontoxic, physiologically acceptable and metabolizable carriers that are relatively simple to make and administer.
The phrase "pharmaceutically acceptable" is employed herein to refer to those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
The phrase "pharmaceutically acceptable carrier" as used herein means a pharmaceutically acceptable material, composition or vehicle, such as a liquid or solid filler, diluent, excipient, solvent or encapsulating material. Each carrier must be "acceptable" in the sense of being compatible with the other ingredients of the formulation and not injurious to the patient. Some examples of materials which can serve as pharmaceutically acceptable carriers
include: (1) sugars, such as lactose, glucose and sucrose; (2) starches, such as com starch and potato starch; (3) cellulose, and its derivatives, such as sodium carboxym ethyl cellulose, ethyl cellulose and cellulose acetate; (4) powdered tragacanth; (5) malt; (6) gelatin; (7) talc; (8) excipients, such as cocoa butter and suppository waxes; (9) oils, such as peanut oil, cottonseed oil, safflower oil, sesame oil, olive oil, com oil and soybean oil; (10) glycols, such as propylene glycol; (11) polyols, such as glycerin, sorbitol, mannitol and polyethylene glycol; (12) esters, such as ethyl oleate and ethyl laurate; (13) agar; (14) buffering agents, such as magnesium hydroxide and aluminum hydroxide; (15) alginic acid; (16) pyrogen-free water; (17) isotonic saline; (18) Ringer's solution; (19) ethyl alcohol; (20) phosphate buffer solutions; and (21) other non-toxic compatible substances employed in pharmaceutical formulations.
A pharmaceutical composition (preparation) can be administered to a subject by any of a number of routes of administration including, for example, orally (for example, drenches as in aqueous or non-aqueous solutions or suspensions, tablets, capsules (including sprinkle capsules and gelatin capsules), boluses, powders, granules, pastes for application to the tongue); absorption through the oral mucosa (e.g., sublingually); subcutaneously; transdermally (for example as a patch applied to the skin); and topically (for example, as a cream, ointment or spray applied to the skin). The antibody-drug conjugate may also be formulated for inhalation. In certain embodiments, an antibody-drug conjugate may be simply dissolved or suspended in sterile water. Details of appropriate routes of administration and compositions suitable for same can be found in, for example, U.S. Pat. Nos. 6,110,973, 5,763,493, 5,731,000, 5,541,231, 5,427,798, 5,358,970 and 4,172,896, as well as in patents cited therein.
The formulations may conveniently be presented in unit dosage form and may be prepared by any methods well known in the art of pharmacy. The amount of active ingredient which can be combined with a carrier material to produce a single dosage form will vary depending upon the host being treated, the particular mode of administration. The amount of active ingredient that can be combined with a carrier material to produce a single dosage form will generally be that amount of the antibody-drug conjugate which produces a therapeutic effect. Generally, out of one hundred percent, this amount will range from about 1 percent to about ninety-nine percent of active ingredient, preferably from about 5 percent to about 70 percent, most preferably from about 10 percent to about 30 percent.
Methods of preparing these formulations or compositions include the step of bringing into association an active compound, such as an antibody-drug conjugate of the invention, with the carrier and, optionally, one or more accessory ingredients. In general, the formulations are
prepared by uniformly and intimately bringing into association an antibody-drug conjugate of the present disclosure with liquid carriers, or finely divided solid carriers, or both, and then, if necessary, shaping the product.
Formulations of the disclosure suitable for oral administration may be in the form of capsules (including sprinkle capsules and gelatin capsules), cachets, pills, tablets, lozenges (using a flavored basis, usually sucrose and acacia or tragacanth), lyophile, powders, granules, or as a solution or a suspension in an aqueous or non-aqueous liquid, or as an oil-in-water or water-in-oil liquid emulsion, or as an elixir or syrup, or as pastilles (using an inert base, such as gelatin and glycerin, or sucrose and acacia) and/or as mouth washes and the like, each containing a predetermined amount of an antibody-drug conjugate of the present disclosure as an active ingredient. Compositions or compounds may also be administered as a bolus, electuary or paste.
To prepare solid dosage forms for oral administration (capsules (including sprinkle capsules and gelatin capsules), tablets, pills, dragees, powders, granules and the like), the active ingredient is mixed with one or more pharmaceutically acceptable carriers, such as sodium citrate or dicalcium phosphate, and/or any of the following: (1) fillers or extenders, such as starches, lactose, sucrose, glucose, mannitol, and/or silicic acid; (2) binders, such as, for example, carboxymethylcellulose, alginates, gelatin, polyvinyl pyrrolidone, sucrose and/or acacia; (3) humectants, such as glycerol; (4) disintegrating agents, such as agar-agar, calcium carbonate, potato or tapioca starch, alginic acid, certain silicates, and sodium carbonate; (5) solution retarding agents, such as paraffin; (6) absorption accelerators, such as quaternary ammonium compounds; (7) wetting agents, such as, for example, cetyl alcohol and glycerol monostearate; (8) absorbents, such as kaolin and bentonite clay; (9) lubricants, such a talc, calcium stearate, magnesium stearate, solid polyethylene glycols, sodium lauryl sulfate, and mixtures thereof; (10) complexing agents, such as, modified and unmodified cyclodextrins; and (11) coloring agents. In the case of capsules (including sprinkle capsules and gelatin capsules), tablets and pills, the pharmaceutical compositions may also comprise buffering agents. Solid compositions of a similar type may also be employed as fillers in soft and hard- filled gelatin capsules using such excipients as lactose or milk sugars, as well as high molecular weight polyethylene glycols and the like.
A tablet may be made by compression or molding, optionally with one or more accessory ingredients. Compressed tablets may be prepared using binder (for example, gelatin or hydroxypropylmethyl cellulose), lubricant, inert diluent, preservative, disintegrant (for example, sodium starch glycolate or cross-linked sodium carboxymethyl cellulose), surface-
active or dispersing agent. Molded tablets may be made by molding in a suitable machine a mixture of the powdered antibody-drug conjugate moistened with an inert liquid diluent.
The tablets, and other solid dosage forms of the pharmaceutical compositions, such as dragees, capsules (including sprinkle capsules and gelatin capsules), pills and granules, may optionally be scored or prepared with coatings and shells, such as enteric coatings and other coatings well known in the pharmaceutical-formulating art. They may also be formulated so as to provide slow or controlled release of the active ingredient therein using, for example, hydroxypropylmethyl cellulose in varying proportions to provide the desired release profile, other polymer matrices, liposomes and/or microspheres. They may be sterilized by, for example, filtration through a bacteria-retaining filter, or by incorporating sterilizing agents in the form of sterile solid compositions that can be dissolved in sterile water, or some other sterile injectable medium immediately before use. These compositions may also optionally contain opacifying agents and may be of a composition that they release the active ingredient(s) only, or preferentially, in a certain portion of the gastrointestinal tract, optionally, in a delayed manner. Examples of embedding compositions that can be used include polymeric substances and waxes. The active ingredient can also be in micro-encapsulated form, if appropriate, with one or more of the above-described excipients.
Liquid dosage forms useful for oral administration include pharmaceutically acceptable emulsions, lyophiles for reconstitution, microemulsions, solutions, suspensions, syrups and elixirs. In addition to the active ingredient, the liquid dosage forms may contain inert diluents commonly used in the art, such as, for example, water or other solvents, cyclodextrins and derivatives thereof, solubilizing agents and emulsifiers, such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3- butylene glycol, oils (in particular, cottonseed, groundnut, corn, germ, olive, castor and sesame oils), glycerol, tetrahydrofuryl alcohol, polyethylene glycols and fatty acid esters of sorbitan, and mixtures thereof.
Besides inert diluents, the oral compositions can also include adjuvants such as wetting agents, emulsifying and suspending agents, sweetening, flavoring, coloring, perfuming and preservative agents.
Suspensions, in addition to the active compounds, may contain suspending agents as, for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar and tragacanth, and mixtures thereof.
Dosage forms for the topical or transdermal administration include powders, sprays, ointments, pastes, creams, lotions, gels, solutions, patches and inhalants. The active ingredient may be mixed under sterile conditions with a pharmaceutically acceptable carrier, and with any preservatives, buffers, or propellants that may be required.
The ointments, pastes, creams and gels may contain, in addition to an active compound, excipients, such as animal and vegetable fats, oils, waxes, paraffins, starch, tragacanth, cellulose derivatives, polyethylene glycols, silicones, bentonites, silicic acid, talc and zinc oxide, or mixtures thereof.
Powders and sprays can contain, in addition to an active compound, excipients such as lactose, talc, silicic acid, aluminum hydroxide, calcium silicates and polyamide powder, or mixtures of these substances. Sprays can additionally contain customary propellants, such as chlorofluorohydrocarbons and volatile unsubstituted hydrocarbons, such as butane and propane.
The phrases "parenteral administration" and "administered parenterally" as used herein means modes of administration other than enteral and topical administration, usually by injection, and includes, without limitation, intravenous, intramuscular, intraarterial, intrathecal, intracapsular, intraorbital, intracardiac, intradermal, intraperitoneal, transtracheal, subcutaneous, subcuticular, intraarticular, subcapsular, subarachnoid, intraspinal and intrastemal injection and infusion. Pharmaceutical compositions suitable for parenteral administration comprise one or more active compounds in combination with one or more pharmaceutically acceptable sterile isotonic aqueous or nonaqueous solutions, dispersions, suspensions or emulsions, or sterile powders which may be reconstituted into sterile injectable solutions or dispersions just prior to use, which may contain antioxidants, buffers, bacteriostats, solutes which render the formulation isotonic with the blood of the intended recipient or suspending or thickening agents.
Examples of suitable aqueous and nonaqueous carriers that may be employed in the pharmaceutical compositions of the disclosure include water, ethanol, polyols (such as glycerol, propylene glycol, polyethylene glycol, and the like), and suitable mixtures thereof, vegetable oils, such as olive oil, and injectable organic esters, such as ethyl oleate. Proper fluidity can be maintained, for example, by the use of coating materials, such as lecithin, by the maintenance of the required particle size in the case of dispersions, and by the use of surfactants.
These compositions may also contain adjuvants such as preservatives, wetting agents, emulsifying agents and dispersing agents. Prevention of the action of microorganisms may be
ensured by the inclusion of various antibacterial and antifungal agents, for example, paraben, chlorobutanol, phenol sorbic acid, and the like. It may also be desirable to include isotonic agents, such as sugars, sodium chloride, and the like into the compositions. In addition, prolonged absorption of the injectable pharmaceutical form may be brought about by the inclusion of agents that delay absorption such as aluminum monostearate and gelatin.
In some cases, in order to prolong the effect of a drug, it is desirable to slow the absorption of the drug from subcutaneous or intramuscular injection. This may be accomplished by the use of a liquid suspension of crystalline or amorphous material having poor water solubility. The rate of absorption of the drug then depends upon its rate of dissolution, which, in turn, may depend upon crystal size and crystalline form. Alternatively, delayed absorption of a parenterally administered drug form is accomplished by dissolving or suspending the drug in an oil vehicle.
Injectable depot forms are made by forming microencapsulated matrices of the subject compounds in biodegradable polymers such as polylactide-polyglycolide. Depending on the ratio of drug to polymer, and the nature of the particular polymer employed, the rate of drug release can be controlled. Examples of other biodegradable polymers include poly(orthoesters) and poly(anhydrides). Depot injectable formulations are also prepared by entrapping the drug in liposomes or microemulsions that are compatible with body tissue.
For use in the methods of this invention, active compounds can be given per se or as a pharmaceutical composition containing, for example, 0.1 to 99.5% (more preferably, 0.5 to 90%) of active ingredient in combination with a pharmaceutically acceptable carrier.
Methods of introduction may also be provided by rechargeable or biodegradable devices. Various slow release polymeric devices have been developed and tested in vivo in recent years for the controlled delivery of drugs, including proteinaceous biopharmaceuticals. A variety of biocompatible polymers (including hydrogels), including both biodegradable and non-degradable polymers, can be used to form an implant for the sustained release of an antibody-drug conjugate at a particular target site.
Actual dosage levels of the active ingredients in the pharmaceutical compositions may be varied so as to obtain an amount of the active ingredient that is effective to achieve the desired therapeutic response for a particular patient, composition, and mode of administration, without being toxic to the patient.
The selected dosage level will depend upon a variety of factors including the activity of the particular antibody-drug conjugate or combination of antibody-drug conjugates employed, or the ester, salt or amide thereof, the route of administration, the time of
administration, the rate of excretion of the particular compound(s) being employed, the duration of the treatment, other drugs, compounds and/or materials used in combination with the particular compound(s) employed, the age, sex, weight, condition, general health and prior medical history of the patient being treated, and like factors well known in the medical arts.
A physician or veterinarian having ordinary skill in the art can readily determine and prescribe the therapeutically effective amount of the pharmaceutical composition required. For example, the physician or veterinarian could start doses of the pharmaceutical composition or antibody-drug conjugate at levels lower than that required in order to achieve the desired therapeutic effect and gradually increase the dosage until the desired effect is achieved. By “therapeutically effective amount” is meant the concentration of the antibody-drug conjugate that is sufficient to elicit the desired therapeutic effect. It is generally understood that the effective amount of the antibody-drug conjugate will vary according to the weight, sex, age, and medical history of the subject. Other factors which influence the effective amount may include, but are not limited to, the severity of the patient's condition, the disorder being treated, the stability of the compound, and, if desired, another type of therapeutic agent being administered with the antibody-drug conjugate of the invention. A larger total dose can be delivered by multiple administrations of the agent. Methods to determine efficacy and dosage are known to those skilled in the art (Isselbacher et al. (1996) Harrison’s Principles of Internal Medicine 13 ed., 1814-1882, herein incorporated by reference).
In general, a suitable daily dose of an active antibody-drug conjugate used in the compositions and methods of the disclosure will be that amount of the antibody-drug conjugate that is the lowest dose effective to produce a therapeutic effect. Such an effective dose will generally depend upon the factors described above.
If desired, the effective daily dose of the antibody-drug conjugate may be administered as one, two, three, four, five, six or more sub-doses administered separately at appropriate intervals throughout the day, optionally, in unit dosage forms. In certain embodiments of the present invention, the antibody-drug conjugate may be administered two or three times daily. In preferred embodiments, the antibody-drug conjugate will be administered once daily.
The patient receiving this treatment is any animal in need, including primates, in particular humans; and other mammals such as equines, cattle, swine, sheep, cats, and dogs; poultry; and pets in general.
In certain embodiments, compounds of the disclosure may be used alone or conjointly administered with another type of therapeutic agent.
The present disclosure includes the use of pharmaceutically acceptable salts of compounds of the disclosure in the compositions and methods of the present invention. In certain embodiments, contemplated salts of the disclosure include, but are not limited to, alkyl, dialkyl, trialkyl or tetra-alkyl ammonium salts. In certain embodiments, contemplated salts of the disclosure include, but are not limited to, L-arginine, benenthamine, benzathine, betaine, calcium hydroxide, choline, deanol, diethanolamine, diethylamine, 2-(diethylamino)ethanol, ethanolamine, ethylenediamine, N-methylglucamine, hydrabamine, lH-imidazole, lithium, L- lysine, magnesium, 4-(2-hydroxyethyl)morpholine, piperazine, potassium, 1-(2- hydroxyethyl)pyrrolidine, sodium, triethanolamine, tromethamine, and zinc salts. In certain embodiments, contemplated salts of the disclosure include, but are not limited to, Na, Ca, K, Mg, Zn or other metal salts. In certain embodiments, contemplated salts of the disclosure include, but are not limited to, l-hydroxy-2-naphthoic acid, 2,2-dichloroacetic acid, 2- hydroxyethanesulfonic acid, 2-oxoglutaric acid, 4-acetamidobenzoic acid, 4-aminosalicylic acid, acetic acid, adipic acid, 1-ascorbic acid, 1-aspartic acid, benzenesulfonic acid, benzoic acid, (+)-camphoric acid, (+)-camphor-10-sulfonic acid, capric acid (decanoic acid), caproic acid (hexanoic acid), caprylic acid (octanoic acid), carbonic acid, cinnamic acid, citric acid, cyclamic acid, dodecylsulfuric acid, ethane- 1,2-disulfonic acid, ethanesulfonic acid, formic acid, fumaric acid, galactaric acid, gentisic acid, d-glucoheptonic acid, d-gluconic acid, d-glucuronic acid, glutamic acid, glutaric acid, glycerophosphoric acid, glycolic acid, hippuric acid, hydrobromic acid, hydrochloric acid, isobutyric acid, lactic acid, lactobionic acid, lauric acid, maleic acid, 1-malic acid, malonic acid, mandelic acid, methanesulfonic acid , naphthalene- 1, 5-disulfonic acid, naphthalene-2-sulfonic acid, nicotinic acid, nitric acid, oleic acid, oxalic acid, palmitic acid, pamoic acid, phosphoric acid, proprionic acid, 1-pyroglutamic acid, salicylic acid, sebacic acid, stearic acid, succinic acid, sulfuric acid, 1-tartaric acid, thiocyanic acid, p-toluenesulfonic acid, trifluoroacetic acid, and undecylenic acid acid salts.
The pharmaceutically acceptable acid addition salts can also exist as various solvates, such as with water, methanol, ethanol, dimethylformamide, and the like. Mixtures of such solvates can also be prepared. The source of such solvate can be from the solvent of crystallization, inherent in the solvent of preparation or crystallization, or adventitious to such solvent.
Wetting agents, emulsifiers and lubricants, such as sodium lauryl sulfate and magnesium stearate, as well as coloring agents, release agents, coating agents, sweetening, flavoring and perfuming agents, preservatives and antioxidants can also be present in the compositions.
Examples of pharmaceutically acceptable antioxidants include: (1) water-soluble antioxidants, such as ascorbic acid, cysteine hydrochloride, sodium bisulfate, sodium metabi sulfite, sodium sulfite and the like; (2) oil-soluble antioxidants, such as ascorbyl palmitate, butylated hydroxyanisole (BHA), butylated hydroxytoluene (BHT), lecithin, propyl gallate, alpha-tocopherol, and the like; and (3) metal-chelating agents, such as citric acid, ethylenediamine tetraacetic acid (EDTA), sorbitol, tartaric acid, phosphoric acid, and the like.
In one aspect, the present disclosure features pharmaceutical composition comprising an antibody-drug conjugate of the disclosure and a pharmaceutically acceptable excipient.
In some embodiments, the composition is administered directly to the tumor (e.g., intratumoral administration).
In some embodiments, the composition is administered orally. In other embodiments, the composition is administered parenterally (e.g., intravenous, subcutaneous, intraperitoneal, or intramuscular administration). In some embodiments, the composition is administered intraperitoneally.
Patient Selection and Monitoring
The methods of the present disclosure described herein entail administration of an antibody-drug conjugate of the disclosure or a pharmaceutically acceptable salt thereof to a subject to activate the PRR for IFNs, ISGs and cytokines production or additionally induce the expression of PRRs (e.g., RIG-I, STING).
In some embodiments, the subject is suffering from or is diagnosed with a condition, e.g., a proliferative disease, e.g., cancer. Accordingly, a patient and/or subject can be selected for treatment using an antibody-drug conjugate of the disclosure or a pharmaceutically acceptable salt thereof by first evaluating the patient and/or subject to determine whether the subject is infected with a proliferative disease, e.g., cancer. A subject can be evaluated as infected with a proliferative disease (e.g., cancer) using methods known in the art. The subject can also be monitored, for example, subsequent to administration of an antibody-drug conjugate of the disclosure herein.
In some embodiments, the subject is a mammal. In some embodiments, the subject is a human. In some embodiments, the subject is an adult. In some embodiments, the subject has a proliferative disease, e.g., cancer. In some embodiments, the subject has a cancer of the of the breast, bone, brain, cervix, colon, gastrointestinal tract, eye, gall bladder, lymph nodes, blood, lung, liver, skin, mouth, prostate, ovary, penis, pancreas, uterus, testicles, stomach, thymus, thyroid, or other part of the body. In some embodiments, the subject has a cancer
comprising a solid tumor (e.g., a carcinoma, a sarcoma, or a lymphoma). In some embodiments, the subject has a hepatocellular carcinoma or other cancer of the liver. In some embodiments, the subject has a leukemia or other cancer of the blood. In some embodiments, the subject has a breast cancer, renal cell carcinoma, colon cancer, melanoma, ovarian cancer, head and neck squamous cell carcinoma, pancreatic cancer, prostate cancer, lung cancer, brain cancer, or gastrointestinal stromal cancer. In some embodiments, the subject has cancer cells (e.g., tumor cells) comprising specific cancer-associated antigens that induce a T-cell response.
In some embodiments, the subject is treatment naive. In some embodiments, the subject has been previously treated for a proliferative disease (e.g., a cancer). In some embodiments, the subject has relapsed.
Combination Therapies
An antibody-drug conjugate of the disclosure described herein may be used in combination with other known therapies. Administered “in combination”, as used herein, means that two (or more) different treatments are delivered to the subject during the course of the subject's affliction with the disorder, e.g., the two or more treatments are delivered after the subject has been diagnosed with the disorder and before the disorder has been cured or eliminated or treatment has ceased for other reasons. In some embodiments, the delivery of one treatment is still occurring when the delivery of the second begins, so that there is overlap in terms of administration. This is sometimes referred to herein as “simultaneous” or “concurrent delivery”. In other embodiments, the delivery of one treatment ends before the delivery of the other treatment begins. In some embodiments, of either case, the treatment is more effective because of combined administration. For example, the second treatment is more effective, e.g., an equivalent effect is seen with less of the second treatment, or the second treatment reduces symptoms to a greater extent, than would be seen if the second treatment were administered in the absence of the first treatment, or the analogous situation is seen with the first treatment. In some embodiments, delivery is such that the reduction in a symptom, or other parameter related to the disorder is greater than what would be observed with one treatment delivered in the absence of the other. The effect of the two treatments can be partially additive, wholly additive, or greater than additive. The delivery can be such that an effect of the first treatment delivered is still detectable when the second is delivered.
An antibody-drug conjugate described herein and the at least one additional therapeutic agent can be administered simultaneously, in the same or in separate compositions, or sequentially. For sequential administration, the compound described herein can be
administered first, and the additional agent can be administered second, or the order of administration can be reversed.
In some embodiments, the combination of an antibody-drug conjugate or a pharmaceutically acceptable salt thereof and the additional agent has a synergistic or additive effect.
In some embodiments, the term “additive” refers to an outcome wherein when two agents are used in combination, the combination of the agents acts in a manner equal to but not greater than the sum of the individual activity of each agent. In some embodiments, the terms “synergy” or “synergistic” refer to an outcome wherein when two agents are used in combination, the combination of the agents acts so as to require a lower concentration of each individual agent than the concentration required to be efficacious in the absence of the other agent. In some embodiments, a synergistic effect results in a reduced in a reduced minimum inhibitory concentration of one or both agents, such that the effect is greater than the sum of the effects. A synergistic effect is greater than an additive effect. In some embodiments, the agents in the composition herein may exhibit a synergistic effect, wherein the activity at a particular concentration is greater than at least about 1.25, 1.5, 1.75, 2, 2.5, 3, 4, 5, 10, 12, 15, 20, 25, 50, or 100 times the activity of either agent alone.
For example, any of the methods described herein may further comprise the administration of a therapeutically effective amount of an additional agent. Exemplary additional pharmaceutical agents include, but are not limited to, anti-proliferative agents, anti-cancer agents, anti-diabetic agents, anti-inflammatory agents, immunosuppressant agents, and a pain-relieving agent. Pharmaceutical agents include small organic molecules such as drug compounds ( e.g ., compounds approved by the U.S. Food and Drug Administration as provided in the Code of Federal Regulations (CFR)), peptides, proteins, carbohydrates, monosaccharides, oligosaccharides, polysaccharides, nucleoproteins, mucoproteins, lipoproteins, synthetic polypeptides or proteins, small molecules linked to proteins, glycoproteins, steroids, nucleic acids, DNAs, RNAs, nucleotides, nucleosides, oligonucleotides, antisense oligonucleotides, lipids, hormones, vitamins, and cells. In some embodiments, the additional agent is an anti-cancer agent, e.g., an alkylating agent (e.g., cyclophosphamide).
In an embodiment, the additional agent is an immunooncology agent, for example, an agent that activate the immune system, e.g., making it able to recognize cancer cells and destroy them. In another embodiment, the agent is a cell based agent such as CAR-t therapy.
EXAMPLES
The embodiments now being generally described, it will be more readily understood by reference to the following examples which are included merely for purposes of illustration of certain aspects and embodiments of the present invention, and are not intended to limit the invention.
Example 1 : Preparation of Exemplary Compounds

A and B were treated with 5-(ethylthio)-1H-tetrazole in acetonitrile and the Iyer- Beaucage reagent. Following this, the product was detritlyated to yield C. C was then treated with sodium iodide in acetone to yield deprotected product D. D was treated with 1 -mesityl ene- 2-sulfonyl-3-nitro- 1,2, 4-triazole and pyridine to yield cyclized product E.

Synthesis of compound 8
Cyclic phosphoromonothio diphosphate (100 mg, 0.145 mmol) was dissolved in HPLC-water (200 mL). A solution of alkylating agent (110 mg, 0.174 mmol) in a mixture of THF : Acetone (1 : 1, 3.0 mL) was added to the reaction mixture. The solution was stirred at room temperature for three days. LC/MS analysis showed completion of the reaction. Solvents were removed under reduced pressure to dryness. Compound was re-dissolved in THF:acetone (1 : 1, 5.0 mL). Solvents were evaporated under reduced pressure to dryness. Product was re- dissolved in 20% water in acetonitrile (15 mL) and hexane (20 mL) was added and vortexed, evaporated under reduced pressure and dried under high vacuum for several hours to yield 80 mg of product as off-white solid. MS, m/z 1151.2 (M-H)+
Synthesis of compound 2
Cyclic phosphoromonothio diphosphate (100 mg, 0.145 mmol) was dissolved in HPLC-water (500 mL). A solution of alkylating agent (150 mg, 0.192 mmol) in a mixture of THF : Acetone (1 : 1, 3.5 mL) was added to the reaction mixture. The solution was stirred at room temperature for two days. LCMS analysis showed completion of the reaction. Solvents were removed under reduced pressure to dryness. Compound was re-dissolved in THF:acetone (1 : 1, 5.0 mL) and precipitated by adding to diethyl ether (20 mL). The precipitate was collected by centrifugation to get product as off white solid. Product was re-dissolved in IPA:DCM (1 : 1, 20 mL) and mixed with water (20 mL) and vortexed to obtain an emulsion. Saturated sodium chloride (100 mL) was added to achieve separation of the two phases. The organic layer was
collected and the aqueous layer was re-extracted with IPA:DCM (1:1, 20 mL). The combined organic layers were dried over Na2SO4 , filtered, and concentrated under reduced pressure to get 100 mg of an off-white solid. The solidwas dissolved in IPA:DCM (1:2) mixture (15 mL). Reaction mixture was cooled to ice-water bath. 5% PTSA solution in IPA:DCM (1:1, 2.0 mL) was added to reaction mixture. Reaction mixture was quenched by adding water (5.0 mL) and stirred for 5 min. Water layer was separated, organic layer was washed with aq. NaHCO3 (5%, 5.0 mL). The organic layer was separated and dried over Na2SO4 and concentrated under reduced pressure to give product as off-white solid. Product was purified on C18 sep-pack column (2.0 gr) product was eluted at 1:1 mixture of acetonitrile:water. Pure product fractions were collected and lyophilized to yield 25 mg of pure product as white solid. MS, m/z 1036.4 (M-H)+
Example 2: Preparation of Exemplary Compounds
 Synthesis of Allyl (( 2R, 3R, 4R, 5R)-5-( 6-henzamido-9H-purin-9-yl)-4-fluoro-2- (hydroxymethyl)tetrahydrofuran-3-yl) (2-cyanoethyl) phosphate (C)
Synthesis of Allyl (( 2R, 3R, 4R, 5R)-5-( 6-henzamido-9H-purin-9-yl)-4-fluoro-2- (hydroxymethyl)tetrahydrofuran-3-yl) (2-cyanoethyl) phosphate (C)

Step 1: 5'O-DMT-2'F-3'Phosphramidite-dA (15.0 g, 17.12 mmol) was co-evaporated with anhydrous acetonitrile (2 x 100 mL), and dried under high vacuum for 1 h. Argon was flushed over the residue in the flask. Acetonitrile (150 mL, anhydrous) was added to residue under argon. Allyl alcohol (Aldrich, 99%) (2.32 mL, 34.24 mmol) was added to the solution followed by ETT (2.22 g, 17.12 mmol) in acetonitrile (20 mL). The reaction mixture was stirred at room temperature under argon for 2.5 hours. TLC analysis (98:2 DCM: MeOH, multiple runs) showed completion of the reaction. It was then cooled in an ice water bath to 0-5C. Tert- butyl hydroperoxide (TBHP, 5-6 M solution in nonane, 2.0 equivalents) was added to the reaction mixture dr op wise at 0-5°C (ice water bath). The mixture was allowed to warm to room temperature and stirred for an additional 30 minutes at room temperature. Excess TBHP was quenched by cooling the solution followed by the addition of saturated thiosulfate solution (10 mL). Reaction mixture was warmed up to room temperature and solvents were evaporated under reduced pressure to remove acetonitrile. The reaction mixture was partitioned between DCM (150 mL) and water (100 mL). The organic layer was separated and water layer was extracted with DCM (50 mL). The combined organic layers were dried over NaiSCL and filtered to remove the Na2SO4 salt.
Step 2 Detritylation: The solution of crude DMT-N-bz-3'-O-Allyl-2'-FdA obtained above (200 mL) in DCM was cooled in an ice- water bath. Para-Toluene Sulfonic Acid (PTSA) (10.0 g) was dissolved in MeOH (60 mL) and diluted with DCM (140 mL) to make 5% PTSA solution in DCM : MeOH (7:3, 200 mL) and added to DMT-N-bz-3'-0-Allyl-2'-FdA. This was stirred at 0-5C for about 30 minutes and checked for reaction completion by TLC (95:5 DCM: MeOH, Rf = 0.2). When DMT deprotection was completed, water (100 mL) was added
and stirred for 15 min whilst the reaction was allowed to warm to room temperature. The mixture was transferred to a separatory funnel and the layers were separated. The aqueous layer was extracted with DCM (25 mL) and the combined organic layers were washed with aq. NaHCO3 (5%, 2 x 100 mL). The organic layer was then washed with saturated brine (100 mL) and dried over Na2SO4. After filtering the salts, the solution was concentrated in vacuo to yield the crude product which was dried under high vacuum to yield a foamy solid. The crude product was dissolved in DCM (30 mL) and added to t-butyl methyl ether (180 mL) to yield a white precipitate, which was collected by filtration. After the first isolation, the product was triturated with t-butyl methyl ether (150 mL) and filtered to obtain a white powder, which was dried under high vacuum for overnight to get 9.3 g (99% yield) of pure product C as white solid.
Synthesis of allyl ((2R,3R,4R,5R)-5-(6-benzamido-9H-purin-9-yl)-2-((((2- cyanoethoxy)(((2R, 3S, 4R, 5R)-2-(2, 4-dioxo-3, 4-dihydropyrimidin-l(2H)-yl)-4-fluoro-5- (hydroxymethyl)tetrahydrofuran-3-yl)oxy)phosphorothioyl)oxy)methyl)-4- fluorotetrahydrofuran-3-yl) (2-cyanoethyl) phosphate (F)

Step 1 Coupling Reaction for Synthesis of Phosphorothioate Dimer: A mixture of C (1.09 g, 2.0 mmol) and E (1.5 g, 2.0 mmol) was co-evaporated with anhydrous acetonitrile (2 x 40 mL) and dried under high vacuum for 1 h. Argon was flushed over the round bottom flask and anhydrous acetonitrile (40 mL) was added to reaction mixture. ETT (260 mg, 2.0 mmol) in acetonitrile (2.0 mL) was added to the mixture of C and E, under argon. The mixture was stirred at room temperature under argon for 2h. TLC analysis (95:5 DCM: MeOH, Rf = 0.5) indicated reaction completion. Deoxygenated water was added to the reaction mixture (72 mL, 2 equivalents to E).
Step 2 Sulfurization: In a silanized flask, Iyer-Beaucage reagent (3H-BD) (800 mg, 4.0 mmol) was dissolved in acetonitrile (10.0 mL). The reaction mixture of C and E from above was added to a solution of sulfurizing reagent (3H-BD) under argon and stirred at room temperature for 45 minutes to complete the sulfurization reaction. Methanol (10 mL) was added to reaction mixture and it was stirred for 30 min followed by concentration under reduced pressure until dryness. The dried residue was dissolved in DCM (50 mL) and washed with water (50 mL). The DCM layer was collected and dried over Na2SO4 and filtered.
Step 3 Detritylation: The dried DCM solution (50 mL) was cooled to approximately 0°C in a round bottom flask. PTSA (2.5 g) was dissolved in methanol (15 mL) and diluted with DCM (35 mL) to make 5% PTSA solution in DCM:MeOH (7:3, 50 mL) which was added to DCM reaction mixture solution and stirred for 15-20 min in an ice water bath. Reaction progress was monitored by TLC (95:5 DCM : MeOH, Rf = 0.15). Water (50 mL) was added and mixed for another 15 minutes. The mixtures were transferred to separator funnel, the water layer was separated and the organic layer was collected. The water layer was extracted with DCM (25.0 mL). The combined organic layers were washed with 5% NaHCO3 solution (2 x 50 mL) to ensure the pH of the aqueous layer was > 7.0. The organic layers were then washed with saturated brine and dried over Na2SO4, filtered, and concentrated under reduced pressure to give crude product, which was dried under high vacuum. Crude product was purified by combiflash silica gel column chromatography using 0-5% MeOH in DCM to give 550 mg of the desired product F as off white solid.
Synthesis of (2S, 3S, 4S, 5S)-5-( 6-henzamido-9H-purin-9-yl)-2-( (((2- cyanoethoxy)(((2S, 3R, 4S, 5S)-2-(2, 4-dioxo-3, 4-dihydropyrimidin-l(2H)-yl)-4-fluoro-5- (hydroxymethyl)tetrahydrofuran-3-yl)oxy)phosphorothioyl)oxy)methyl)-4- fluorotetrahydrofuran-3-yl (2-cyanoethyl) hydrogen phosphate (G)
 To a solution of 3'-Allyl protected dimer (500 mg, 0.565 mmol) in acetone (10 mL) was added sodium iodide (810 mg, 5.41 mmol) and the resulting solution was stirred at 60°C for 1 h. TLC analysis (80:20 DCM : MeOH, Rf = 0.15) showed completion of the reaction. The reaction mixture was cooled to room temperature. DCM (10 mL) was added to the suspension to precipitate of the product. Product was collected by centrifugation, which was triturate with DCM (25 mL) then centrifuged a second time to obtain the product. The product was dried under high vacuum to yield an off white solid. This solid was triturated with 20% MeOH in DCMTButyl methyl ether (1:1, 25 mL) and collected by centrifugation, which was dried under high vacuum to get 500 mg of product as off white solid.
To a solution of 3'-Allyl protected dimer (500 mg, 0.565 mmol) in acetone (10 mL) was added sodium iodide (810 mg, 5.41 mmol) and the resulting solution was stirred at 60°C for 1 h. TLC analysis (80:20 DCM : MeOH, Rf = 0.15) showed completion of the reaction. The reaction mixture was cooled to room temperature. DCM (10 mL) was added to the suspension to precipitate of the product. Product was collected by centrifugation, which was triturate with DCM (25 mL) then centrifuged a second time to obtain the product. The product was dried under high vacuum to yield an off white solid. This solid was triturated with 20% MeOH in DCMTButyl methyl ether (1:1, 25 mL) and collected by centrifugation, which was dried under high vacuum to get 500 mg of product as off white solid.
Synthesis of (H)

Dinucleotide G (500 mg, 0.565 mmol) was co-evaporated with anhydrous pyridine (2 x 20 mL), dried under high vacuum, flushed with argon (3 times) and dissolved in anhydrous pyridine (20 mL). l-Mesitylene-2-sulfonyl-3-nitro-1,2,4-triazole (MSNT) (0.838 g, 2.82 mmol) was added to the solution of G at room temperature. The resulting mixture was stirred at room temperature for 1.5 h. Reaction progress was monitored by TLC analysis (90: 10 DCM: MeOH) showed completion of the cyclization after 1.5 hours. Toluene (20 mL) was added to the reaction mixture. Solvents were evaporated under reduced pressure to give crude product. The resulting mixture was dissolved in 25% IPA in DCM (50 mL) and washed with water (50 mL). The aqueous layer was extracted with 25% IPA in DCM (50 mL), and the combined organic layers were washed with saturated aq. NaHCO3 (10 mL) and brine (10 mL). Organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure to give crude product. Crude product was dissolved in 10% MeOH in DCM (5 mL) and precipitated by adding to t-butyl methyl ether (10 mL) (to remove colored impurities). The precipitate was collected by centrifugation. Product was triturated with DCM: t-butyl methyl ether (1:1, 15 mL) and product was collected by centrifugation to give light yellow product. Crude product
was purified by combiflash silica gel column chromatography (gradient 0-10% MeOH in DCM) to yield 80 mg of product H as off white solid.
Synthesis of 4-(Iodomethyl)phenyl 4-(decyloxy)benzoate (I)

Step 1: To a suspension of benzoic acid derivative (10 g, 0.054 mol) in a 250 mL single neck flask in toluene, thionyl chloride (7.8 mL) was added slowly and stirred at r.t. for 15 minutes followed by heating in an oil bath at 80-85°C to obtain a clear solution that was maintained for ~ 3h. The reaction mixture was cooled to RT and excess thionyl chloride was removed in vacuo. The toluene was concentrated using a rotavap at 40-45°C. It was then co- evaporated twice with ethyl acetate (25 mL). The residue was taken up in ethyl acetate (15ml). 4-Hydroxybenzyl alcohol (4.5 g, 0.054 mol) was suspended in ethyl acetate (25 mL) and cooled in an ice bath. TEA (5.5 mL) was added with stirring followed by the addition of the ethyl acetate solution of acid chloride. A suspension forms and this was stirred overnight. The insoluble solids were removed by filtration and the filtrate was transferred to a separatory funnel. The filtrate was diluted with ethyl acetate (200 mL), washed with water (50 mL), and the organic layer washed with brine (50 mL). Concentration after drying gave the crude product, which was taken up in 200 mL of 4: 1 Hexane (or Heptanes) :EtO Ac, and stirred for 2 h to precipitate the product. The precipitated product was filtered and the solid dried under high vacuum to yield 9.0 g (67% yield) of the desired product.
Step 2: To a suspension of 4- hydroxyl benzyl alcohol coupled derivative (9.0 g, 0.026 mol) in a mixture of anhydrous acetonitrile (80 mL) and anhydrous dichloromethane (30 mL) in a 250 mL single neck flask, was added C8l (18.2 g, 0.078 mol) in one portion. To this, BF3.Et20 (8.7 mL) was added slowly and stirred in the dark (covered with aluminum foil) under argon overnight at room temperature. The reaction was found to be complete by TLC Hex: EtOAc (7:3). The product was concentrated and the reaction mixture was worked up by adding water (50 mL) followed by extraction with DCM (200 mL) in a separatory funnel. The organic layer was washed with saturated sodium bicarbonate (25 mL), followed by washing with NaHSO3 (5%, 30 mL). The organic layer was dried over anhydrous Na2SO4, filtered, concentrated to a film and later dried in high vacuum for two days to give 9.6 g (85% yield) the desired product I.
Synthesis of Example 4*

Step 1 Deprotection of cyclic phosphoromonothio diphosphate: Fully protected cyclic phosphoro monothio diphosphate (70 mg) was dissolved in a mixture of cone. NH4OH (2.0 mL) and DCM (5.0 mL) stirred at room temperature overnight. LC-MS analysis showed completion of the reaction. Reaction mixture was transferred to separatory funnel and the DCM layer was removed. The aqueous layer was evaporated under reduced pressure to remove ammonia and was then washed with ethyl acetate (3 x 5 mL) to remove benzamide byproduct completely. The product was isolated from the aqueous layer by lyophilization to yield 60 mg of as white solid.
Step 2: Cyclic phosphoromonothio diphosphate (50 mg, 0.072 mmol) was dissolved in water (500 uL). A solution of I (53 mg, 0.108 mmol) in a mixture of THF: Acetone (1:1, 3.5 mL) was added to the reaction mixture. The solution was stirred at room temperature for two days. Solvents were removed under reduced pressur. The crude product was re-dissolved in THF: acetone (1 :1, 5.0 mL) and precipitated by adding to diethyl ether (10 mL) to remove unreacted iodo-compound. The precipitate was collected by centrifugation to yield product as an off white solid. This was re-dissolved in IPA:DCM (1 : 1, 20 mL) and mixed with water (20 mL), which was formed as a single-phase solution. Saturated sodium chloride (5 mL) was added to achieve separation of the two phases. The organic layer was collected (lower layer) and aqueous layer was re-extracted with IPA:DCM (1:1, 2x10 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure to yield the product as off white solid. The product was re-dissolved in 5% acetonitrile in water (2.0 mL) and lyophilized to obtain 76 mg of product 4* as off white solid.
Example 3: Evaluation of Induction of IRF and NF-KB
THP1 dual cells grown in complete media were treated with various concentrations of a compound of the present disclosure or DMSO control. Dual cells carry both secreted embryonic alkaline phosphatase (SEAP) reporter gene under the control of an IFN-b minimal promoter fused to five copies of the NF-kB consensus transcriptional response element to
measure NF-kB activity and Lucia reporter gene under the control of an ISG54 minimal promoter to measure IRF activity. After 20 h incubation, IRF activity was assessed using QUANTI-luc to measure levels of Lucia and NF-kB activity was determined by measure SEAP levels at 620-655 nm. % induction was calculated from fold change in luminescence/absorbance compared to DMSO treated sample. Any negative values were given base value 1 for plotting data in log scale for accurate demonstration of dose response. EC50 values were generated by curve fit in Xlfit. Cells grown in complete media were treated with various concentrations of a compound of the disclosure or DMSO control. Dual cells carry both secreted embryonic alkaline phosphatase (SEAP) reporter gene under the control of an IFN-β minimal promoter fused to five copies of the NF-kB consensus transcriptional response element to measure NF-kB activity and Lucia reporter gene under the control of an ISG54 minimal promoter to measure IRF activity. After 20 h incubation, IRF activity was assessed using QUANTI-luc to measure levels of Lucia and NF-kB activity was determined by measure SEAP levels at 620-655 nm. % induction was calculated from fold change in luminescence/absorbance compared to DMSO treated sample. EC50 values are generated by curve fit in Xlfit.
Table 3: EC50 values for exemplary compounds of the disclosure. “A” represents an EC50 of less than 50 nM; “B” an EC8o of between 50 nM and 500 nM; “C” an EC8o of between 500 nM and 1 mM; “D” an EC8o of between 1 mM and 2 pM; “E” an EC8o of greater than 2 pM.



Example 4: Study to Determine the Efficacy of Compound 4* in a CT26 Murine Colon Carcinoma Model Using Female BALB/c Mice
Mice
Female BALB/c mice (BALB/c AnNcrl, Charles River) were eight weeks old on Day 1 of the study and had a body weight range of 15.1 to 19.7 g. The animals were fed ad libitum
water (reverse osmosis, 1 ppm Cl) and a NIH 31 Modified and Irradiated Lab Diet consisting of 18.0% crude protein, 5.0% crude fat, and 5.0% crude fiber.
Tumor Cell Culture
CT26 murine colon carcinoma cells were grown in RPMI-1640 medium containing 10% fetal bovine serum, 2 mM glutamine, 100 units/mL penicillin G sodium, 100 mg/mL streptomycin sulfate, and 25 mg/mL gentamicin. The cells were cultured in tissue culture flasks in a humidified incubator at 37 °C, in an atmosphere of 5% CO2 and 95% air.
In Vivo Implantation and Tumor Growth
On the day of implantation, cultured CT26 cells were harvested during log phase growth and resuspended in phosphate buffered saline, pH 7.4 (PBS) at a concentration of 3 x 106 cells/mL. Each mouse was injected subcutaneously in the right flank with 3 x 105 tumor cells (0.1 mL cell suspension) and tumors were monitored as their volumes approached the target range of 80 to 120 mm3. Eleven days after tumor cell implantation, on Day 1 of the study, animals were sorted into three groups (n=8/group) with individual tumor volumes of 63 to 126 mm3, and a group mean tumor volume of 105 mm3. Tumors were measured with a caliper twice weekly for the duration of the study. Tumor size was calculated using the formula:
 wherein w is width and 1 is length, in mm, of a tumor. Tumor weight may be estimated with the assumption that 1 mg is equivalent to 1 mm3 of tumor volume.
wherein w is width and 1 is length, in mm, of a tumor. Tumor weight may be estimated with the assumption that 1 mg is equivalent to 1 mm3 of tumor volume.
Test Articles
Compound 4 was dissolved by adding the appropriate volume of sterile saline (vehicle) into each tube, vortexing, incubating at 37 °C for 2-5 minutes, followed by sonication, if needed. Preparations of Compound 4 resulted in the appropriate 0.2 and 0.6 mg/mL dosing solutions which provided 1 and 3 mg/kg doses in a dosing volume of 5 mL/kg, adjusted to the body weight of the animal. A fresh vial was prepared on each day of dosing.
Treatment
On Day 1 of the study, three groups of BALB/c mice (n = 8) began dosing according to the protocol in Figure 20. Compound 4 and vehicle were administered intravenously (i.v.). Group 1 received vehicle on Days 1, 5, 9, and 14. Groups 2 and 3 received CMD 4 at 1 and 3 mg/kg, respectively, on Days 1, 5, 9, and 14.
Tumor Growth Delay Endpoint
The study endpoint was a tumor volume of 2000 mm3 or Day 30, whichever came first. The study ended on Day 29. The study protocol specified a tumor growth delay assay based on the median time-to-endpoint (TTE) in a treated group versus the control group. Tumors were measured using calipers twice per week, and each animal was euthanized for tumor progression (TP) when its tumor reached the 2000 mm3 volume endpoint. The TTE for each mouse was calculated with the following equation:
 where b is the intercept and m is the slope of the line obtained by linear regression of a log- transformed tumor growth data set. The data set is comprised of the first observation that exceeded the study endpoint volume and the three consecutive observations that immediately preceded the attainment of the endpoint volume. Any animal that did not reach endpoint was euthanized at the end of the study and assigned a TTE value equal to the last day of the study (Day 29). In instances in which the log-transformed calculated TTE preceded the day prior to reaching endpoint or exceeded the day of reaching tumor volume endpoint, a linear interpolation was performed to approximate TTE.
where b is the intercept and m is the slope of the line obtained by linear regression of a log- transformed tumor growth data set. The data set is comprised of the first observation that exceeded the study endpoint volume and the three consecutive observations that immediately preceded the attainment of the endpoint volume. Any animal that did not reach endpoint was euthanized at the end of the study and assigned a TTE value equal to the last day of the study (Day 29). In instances in which the log-transformed calculated TTE preceded the day prior to reaching endpoint or exceeded the day of reaching tumor volume endpoint, a linear interpolation was performed to approximate TTE.
On Day 29, MTV (n) was defined as the median tumor volume of the number of animals, n, that survived to the last day and whose tumors had not reached the volume endpoint. Any animal determined to have died from treatment-related (TR) causes was to be assigned a TTE value equal to the day of death. Any animal that died from non- treatment-related (NTR) causes was to be excluded from the analysis. Treatment outcome was evaluated from tumor growth delay (TGD), which was defined as the increase in the median TTE for a treatment group compared to the control group:
TGD = T - C expressed in days, or as a percentage of the median TTE of the control group:
 wherein T is the median TTE for a treatment group and C is the median TTE for the control group.
Tumor Growth Inhibition (TGI) Analysis
wherein T is the median TTE for a treatment group and C is the median TTE for the control group.
Tumor Growth Inhibition (TGI) Analysis
The study endpoint was defined as a mean tumor volume of 2000 mm3 in the control group (sum of both flank tumors) or 30 days, whichever came first. The study reached TGI endpoint on Day 18. Treatment efficacy was determined using data from the final day that all control animals remained on study (Day 18). The MTV (n), the median tumor volume for the number of animals, n, on the final day, was determined for each group. Percent tumor growth inhibition (%TGI) was defined as the difference between the MTV of the designated control group (Group 1) and the MTV of the drug-treated group, expressed as a percentage of the MTV of the control group: = [ 1 —(MTV drug-treated/MTV control)] X 100

The data set for TGI analysis includes all animals in a group, except those euthanized for sample collection (ES) and those that die due to treatment-related (TR) or non-treatment- related (NTR) causes.
Criteria for Regression Responses
Treatment efficacy was also determined from the number of regression responses. Treatment may cause partial regression (PR) or complete regression (CR) of the tumor in an animal. In a PR response, the tumor volume is 50% or less of its Day 1 volume for three consecutive measurements during the course of the study, and equal to or greater than 13.5 mm for one or more of these three measurements. In a CR response, the tumor volume is less than 13.5 mm for three consecutive measurements during the course of the study. Animals were scored only once during the study for a PR or CR event and only as CR if both PR and CR criteria were satisfied. Any animal with a CR response on the last day of the study is additionally classified as a tumor-free-survivor (TFS).
Toxicity
Animals were weighed daily for the first five days of the study and twice weekly thereafter. The mice were observed frequently for health and overt signs of any adverse treatment related (TR) side effects, and noteworthy clinical observations were recorded. Individual body weight loss was monitored per protocol, and any animal with weight loss exceeding 30% for one measurement, or exceeding 25% for three measurements, was to be euthanized for health as a TR death. If group mean body weight recovered, dosing may resume
in that group, but at a lower dose or less frequent dosing schedule. Acceptable toxicity was defined as a group mean BW loss of less than 20% during the study and not more than one TR death among ten treated animals, or 10%. Any dosing regimen resulting in greater toxicity is considered above the maximum tolerated dose (MTD). A death was to be classified as TR if it was attributable to treatment side effects as evidenced by clinical signs and/or necropsy, or may also be classified as TR if due to unknown causes during the dosing period or within 14 days of the last dose. A death was classified as NTR if there was evidence that the death was related to the tumor model, rather than treatment-related. NTR deaths are further categorized as NTRa (due to accident or human error), NTRm (due to necropsy-confirmed tumor dissemination by invasion or metastasis), and NTRu (due to unknown causes).
Study Design
Table 2: Protocol Design for the CT26 Study

Table 1 displays the study design as of Day 1 of the study. Vehicle is saline.
Example 5: Synthesis of Peptide Linked Phosphorothioate Compounds Synthesis of C*

Scheme 1 : Synthesis of C*
Step 1
To a solution of the acid A* (0.0397 g, 0.02 mmol) in DMF (1 mL) was added triethylamine (leq) followed by HATU (1 eq). The mixture was stirred at RT for 15min, after
which time the mixture was treated with the SMNH (1 eq). The reaction mixture was stirred at RT for 2.5 hours. The mixture was diluted with EtOAc (10 mL) and washed with IN HCI (5 mL), sat. NaCl (5 mL), dried over Na2SO4, and concentrated. The resulting solid B* was used in the next step. Yield [35 mg, 54%].
Step 2
To compound B* (0.033 g. 0.012 mmol) was added 1 mL of trill uoroaeetie acid: trlethylsilane and H2O in the ratio of TFA:TES: H2O, 95:0.25:0.25. The reaction was stirred for 1 hour. Solvent was evaporated and the mixture was diluted with EtOAc (10 mL) and washed with sat NaHCO3 (2 mL), sat NaCl (5 mL), dried (NazSQO, and concentrated. The resulting crude solid was taken in DSMO and subjected to HPLC purification which provided two fractions which were concentrated to provide two isomers of compound C*. Isomer 1 [6.5 rug, 23% Yield], isomer 2 [6.2 mg, 22% Yield].
Synthesis of G

Scheme 2: Synthesis of G'
Step 1
To a solution of amine A' (250 mg, 0.5 mmol) in C2Cl2 (3 mL) was added acid B' (1 eq), EEDQ (1.2 eq). The mixture was stirred at room temperature overnight Saturated aqueouC NaHCO3 (3 ml..) was added to the mixture and the organic product extracted with C2Cl2 (3 mL). The combined organic layers were dried Na2SO4 .filtered through ceitie and concentrated in vacuo. Purification of the residue on silica gel eluted with MeOH:CHCl2 · [0 to 100%] provided product C' (200mg, 48% Yield),
Step 2
To a solution of amide C' (100mg, 0.13 mmol) in DMF (1 mL) was added DIPEA (1.2 eq). The solution was stirred for 15min. Then bis(4-nitrophenyl)carbonate (1.5 eq) was added and the mixture was stirred at room temperature for 18h. Saturated aqueous NaHCO3 (2 mL) was added to the mixture and the organic product extracted with C2Cl2 (3 mL). The combined organic layers were dried ( Na2SO4), filtered through Celite and concentrated in vacuo to provide product D' (39mg, 32% Yield).
Step 3
A solution of the compound D' (0.023mg, 0.025mmol) in DMF (0.5 mL) was treated with SMNH (1.1 eq), and DIPEA (1.5 eq) and the mixture was stirred at room temperature for 2.5h. The solvent was removed under reduced pressure and to the residue was added a saturated aqueous solution of NaHCO3 (1 mL). The organic product was extracted with EtOAc (2 x 3 mL). The combined organic layers were dried (Na2SO4), concentrated in vacuo and the residue was purified on silica gel eluted with MeOH: C2Cl2: [0 to 100%] to provide two fractions of compound 5 having 10mg each. [Combined yield, 20 mg, 43%].
Step 4
The protected compound E' was taken in THF (1 mL). LiOH (5 eq) in water (0.5 mL) was added and stirred at room temperature for 3 hours. The solvent was then removed in vacuo and the residue was neutralized with 1 N HC1. The crude organic product was extracted with EtOAc (2 x 5 mL). Combined organic layers were dried (Na2S04), concentrated in vacuo and acid F' (2 mg, 36%) formed was used as such in the next step.
Step 5
A solution of F' (2 mg, 0.0013 mmol) and compound H' (N-hydroxysuccinimide (NHS) ester; 1.1 eq) was taken in N-methyl morpholine (0.5 mL). Diisopropyl ethylamine (1 eq) was added the reaction was stirred at 60°C for 4h. The resulting solution was concentrated to afford a quantitative yield of product G\
Example 6: Exemplary Synthesis of Peptide Linked Phosphorothioate Compounds
Fmoc-protected valine as succinimide ester A was coupled with citrulline B in dimethoxyethane and sodium bicarbonate led to dipeptide C. This dipeptide was condensed with 4-aminobenzyl alcohol in the presence of EEDQ in DCM:MeOH (2:1) at r. t. for 36 h resulted in intermediate D.

Scheme 1 : Synthesis of D
Synthesis of peptide, directly linked to phosphorothioate, was achieved through the conversion of Fmoc dipeptide intermediate D to the corresponding iodo derivative E. Alkylation of phosphorothioate with E yielded peptide linked phosphorothioate. Subsequent removal of Fmoc group from 6 led to the isolation of peptide linked phosphorothioate with terminal free amino group F.

Scheme 2: Preparation of Peptide linked Phosphorothioate
Subsequent removal of Fmoc group from D using diethyl amine in DMF at room temperature afforded the dipeptide G, with a free amino group. Maleimidocaproic acid as its succimidie ester was coupled with G in DMF at r. t. to yield maleimido derivative H. The terminal benzyl alcohol in H was converted to the benzyl iodide derivative I with phosphonium iodide, which was used to alkylate phosphorothioates in a mixture of DCM-DMF to give the S-alkylated prodrugs J.

Scheme 3: Preparation of Maleimide-Peptide linked Phosphorothioate
Key intermediate L, linking 2-aminoethanol with Fmoc-Val-CitOH K has been prepared. Further subsequent reactions can be carried out to synthesis compounds M, N, O and Q following the protocols, described above with benzyl alcohol derivative.

Scheme 4: Exemplary synthesis of compounds N, O, and Q. Example 7: Exemplary Synthesis of Compounds of the Disclosure Coupling of Fmoc protected valine as suceinimide ester A was coupled with citnsllioe B in dimethoxyethane and sodium bicarbonate led to dipeptide C. This dipeptide was condensed with 4-aminobenzyl alcohol in the presence ofEEDQ in DCM;MeOH (2:1) at r. t.
for 36 h resulted in intermediate D. Intermediate D could then be coupled (e.g., via a GDI or phosgene mediated coupling) with STING agonist to yield E.

Scheme 1 : Exemplary Synthesis of E
Synthesis of peptide, linked to a STING agonist could be achieved through the conversion of Ftnoc dipeptide intermediate F to the corresponding iodo derivative C*. Alkylation of STING agonist with G could yield the peptide linked STING agonist. Subsequent removal ofFmoc group would yield H.

Scheme 2: Preparation of Peptide linked Phosphoroiliioate
Subsequent removal of Frnoc group from I using dieihyl amine in DMF at room temperature afforded dipeptide X with free amino group. Maleimidocaproic acid as its soceimldie ester was coupled with J in DMF at r t. to yield maleioiklo derivative K. The terminal benzyl alcohol in K was converted to the benzyl iodide derivative L using phosphonium iodide. This could be used to alkylate a STING agonist give the N-alkylated M.

Scheme 3: Preparation of Maleimide-Peptide linked Phosphorothioate (Scheme 3)
Key intermediate O, linking 2-aminoethanol with Fmoc-Val-CitOH N has been prepared. Further subsequent reactions can be carried out to synthesis the compounds illustrated below following protocols similar to those described above with benzyl alcohol derivative.

Scheme 4: Prophetic Synthesis of peptide linked STING agonists Q and S.
Example 8; Prophetic Synthesis of Peptide-Linked Phosphorothioate Compounds Prophetic Synthesis of C*

Scheme 1: Prophetic Synthesis of C*
Intermediate A* could be coupled (e.g., via a CDI or phosgene mediated coupling) with STING agonist (SMNH) and then deprotected (e.g., via TEA. TES in H2O) to yield C*.
Synthesis of G'

Scheme 2: Synthesis of G'
Step 1
To a solution of amine A" (250 mg, 0,5 mmol) in C2Cl2 (3 mL) was added acid B' (I eq), EEDQ (1.2 eq). The mixture was stirred at room temperature overnight. Saturated aqueous NaHCO3 (3 mL) was added to the mixture and the organic product extracted with C2Cl2 (3 mL). The combined organic layers were dried (NarSOi). tillered through eellte and
concentrated in vacuo. Purification of the residue on silica gel eluted with MeOH:CH2Cl2 [0 to 100%] provided product C' (200mg, 48% Yield).
Step 2
To a solution of amide C' (100 mg, 0.13 mmol) in DMF (1 mL) was added DIPEA (1.2 eq). The solution was stirred for 15min. Then bis(4-nitrophenyl)carbonate (1.5 eq) was added and the mixture was stirred at room temperature for 18 h. Saturated aqueous NaHCO3 (2 mL) was added to the mixture and the organic product extracted with C2Cl2 (3 mL). The combined organic layers were dried (Na2SO4), filtered through C6lite and concentrated in vacuo to provide product D' (39mg, 32% Yield).
Step 3
Intermediate D' could then be coupled (e.g., via a CDI or phosgene mediated coupling) with a STING agonist to yield E'
Step 4
The protected compound E' could be taken up in THF (1 mL). LiOH (5eq) in water (0.5mL) could then be added and stirred at room temperature for 3 hours. The solvent could then be removed in vacuo and the residue could be neutralized with 1 N HCl. The crude organic product could be extracted with EtOAc (2 x 5 mL). Combined organic layers could be dried (Na2SO4), concentrated in vacuo to yield acid F'
Step 5
A solution of F' (2 mg, 0.0013 mmol) and compound H' (N-hydroxysuccinimide (NHS) ester; 1.1 eq) could be taken up in N-methyl morpholine (0.5 mL). Diisopropyl ethylamine (1 eq) could then be added to the reaction mixture and it could be stirred at 60°C for 4 h. The resulting solution could then be concentrated to afford product G'.
Example 9: Exemplary synthesis of b-glucuronide drug-linker unit:

Aryl b-glucuronide was acylated with acid chloride 1, and then converted to p- nitrophenyl (PNP) carbonate 2 (US 2017/0189542; incorporated by reference).
Example 10: Exemplary synthesis of methylene carbamate drug-linker unit utilizing the Curtis Rearrangement Reaction

Exemplary synthesis utilizing the Curitus Rearrangement reaction for methylene carbamate linkers wherein A represents a connection to the drug unit and and B represents a connection to a selfimmolative unit (US 2016/0303254; incorporated by reference).
Example 11 : Exemplary synthesis of heterocyclic self-immolative groups
NMR was obtained on a Varian-300 spectrometer, with 1H 300 MH2 in deuterated DMSO unless otherwise specified. All chemical shifts are referenced to tetramethylsilane. Mass spectra were determined on a PE SCIEX, API 2000 LC-MS spectrometer. Solutions in organic solvents were dried with anhydrous Na2SO4 . Solvents were evaporated under reduced pressure on a Buchi rotary evaporator. TLC was carried out on glass-backed silica gel plates (Merck 60 F254) with visualization of components by UV light (254 nm). Flash colmnn chromatography was performed on silica gel (Merck 230-400 mesh) (US 2010/0273843; incorporated by reference).

8.6 g of Boc-Citrulline was dissolved in 250 mL of DMF. The solution was added 7.2 mL of DIEA and 6.7 g of CDI. After stirred at r.t. for 30 min, the solution was added 5 g of ethyl-2-amino-4-(trifluoromethyl)-5-thiazolecarboxylate (Matrix Scientific, Columbia S.C. USA). The reaction was quenched after additional 2 hours at r.t. by the addition of 25 mL of water. The mixture was diluted with 250 mL of EtOAc. The organic layer was washed with IN HC1, brine and worked up as described in General Procedure. Pure title compound 1A was
obtained by purification on a fresh silica gel column eluted with 5% MeOH in DCM (5.6 g, yield 54%) (US 2010/0273843; incorporated by reference).

3 g of 1A was dissolved in 90 mL ofTHF. The solution was added 12 mL of lithium aluminum hydride (1.0M solution in THF) at 0° C. After stirring at 0°C for 2 hours, the reaction was quenched by addition of 10 mL of water, diluted with 250 mL of EtOAc and filtered through a celite pad. The organic layer was washed with brine and worked up as described in General Procedure. Pure title compound 2A was obtained by purification on a fresh silica gel column eluted with 5% MeOH in DCM (2.2 grams yield 80%) (US 2010/0273843; incorporated by reference).

Compound 2A (1.5 g) was dissolved in 45 mL methanol. To the solution was added 8 mL of HCI in dioxane (4.0M). After stirring at room temperature r.t. for 2 hours, the solution was concentrated down at a rotary evaporator under reduced pressure. The crude product was dried under vacuum for additional 18 hours at r.t. and used for next reaction without further purification (US 2010/0273843; incorporated by reference).
 Compound 3A (1.2 g), 1.1 grams of Boc-valine, 1.4 mL of DIE A, and 3.3 g of HBTU were dissolved in 36 mL DMF. After stirring at r.t. for 18 hours, the reaction was quenched by addition of 5 mL of water and diluted with 400 mL of EtOAc. The organic layer was washed with brine and worked up as described in General Procedure. Pure title compound 4A was obtained by purification on a fresh silica gel column eluted with 10% MeOH in DCM (1.4 g, yield 75%) (US 2010/0273843; incorporated by reference).
Compound 3A (1.2 g), 1.1 grams of Boc-valine, 1.4 mL of DIE A, and 3.3 g of HBTU were dissolved in 36 mL DMF. After stirring at r.t. for 18 hours, the reaction was quenched by addition of 5 mL of water and diluted with 400 mL of EtOAc. The organic layer was washed with brine and worked up as described in General Procedure. Pure title compound 4A was obtained by purification on a fresh silica gel column eluted with 10% MeOH in DCM (1.4 g, yield 75%) (US 2010/0273843; incorporated by reference).

4A (50 mg) was dissolved in 0.5 mL of THF and DCM (1 : 1). To the solution was added 30 mg of 4-nitrophenyl isocyanate. After stirring at RT for 48 hours, the mixture was directly charged to a silica gel column eluted with 5% MeOH in DCM to give the pure title compound 5A (US 2010/0273843; incorporated by reference).
Example 12: Exemplary synthesis of malemide self-immolative groups

In a 50 mL round bottom flask H-Lys(boc)-OH (246 mg, 1 mmol) and maleic anhydride (98 mg, 1 mmol) were dissolved in 1 mL acetic acid and the solution was stirred at room temperature for 3 hours. The reaction mixture was concentrated to an oil on the rotovap, and the product was precipitated by adding ~10 mL dichloromethane. The precipitate was collected by vacuum filtration, washed with dichloromethane, and dried overnight in the vacuum oven. 270mg of product was recovered as a white powder (85% yield) (WO 2013/173337; incorporated by reference).
 Maleyl-Lys(boc)-OH (100 mg, 0.29 mmol) was suspended in toluene (3 mL) and triethylamine (224 uL) over molecular sieves in a 50 mL round bottom flask equipped with a condenser. D1VIA (-150 uL) was added to aid solubility. The solution was heated to 125°C and refluxed tor 4 hours after which the reaction was shown to be complete by LCMS. The reaction mixture was concentrated to dryness on the rotovap, redissolved in DMSO and purified by preparative HPLC. 56 mg of product was isolated as a white powder (60% yield) (WO 2013/173337; incorporated by reference).
Maleyl-Lys(boc)-OH (100 mg, 0.29 mmol) was suspended in toluene (3 mL) and triethylamine (224 uL) over molecular sieves in a 50 mL round bottom flask equipped with a condenser. D1VIA (-150 uL) was added to aid solubility. The solution was heated to 125°C and refluxed tor 4 hours after which the reaction was shown to be complete by LCMS. The reaction mixture was concentrated to dryness on the rotovap, redissolved in DMSO and purified by preparative HPLC. 56 mg of product was isolated as a white powder (60% yield) (WO 2013/173337; incorporated by reference).
INCORPORATION BY REFERENCE
All publications and patents mentioned herein are hereby incorporated by reference in their entirety as if each individual publication or patent was specifically and individually indicated to be incorporated by reference. In case of conflict, the present application, including any definitions herein, will control.
EQUIVALENTS
While specific embodiments of the subject disclosure have been discussed, the above specification is illustrative and not restrictive. Many variations of the disclosure will become apparent to those skilled in the art upon review of this specification and the claims below. The full scope of the disclosure should be determined by reference to the claims, along with their full scope of equivalents, and the specification, along with such variations.
Claims
1. An antibody-drug conjugate of Formula (I):
 or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
Ab is an anti-PD-1 antibody or an anti-CTLA-4 antibody; each of B1 and B2 is independently a purinyl nucleobase or pyrimidinyl nucleobase; each of X1 and X2 is independently O or S; each of Y1 and Y2 is independently O, S, or N(R5); each of Z1 is independently O or S; each of Z2 and Z3 is independently absent, -C1-C20- alkylene (e.g., -C1-C6- alkylene), C1-C20 heteroalkylene (e.g., -C1-C6- heteroalkylene), -OC(O)OC1-C20- alkylene (e.g., -C1-C6- alkylene), -cycloalkylene-, -heterocyclyl-, -aryl-, or -heteroaryl-, wherein each -cycloakyl-, -heterocyclyl-, -aryl- or -heteroaryl- is optionally substituted with one or more R4;
Z4 is self-immolative group-C1-C20-alkylene-Q1 (e.g., -self-immolative group-C1-C6- alkylene-Q1), heterocyclyl-C1-C20-alkylene-Q (e.g., heterocyclyl-C1-C6-alkylene-Q),- OH, -N(R5)2, SR5, -CHO, -C(O)N(R5)2, -OC(O)N(R5)2, -N(R5)C(O)OR5, aryl, heteroaryl, -S(O)R5,
-S(O)2R5, -S(O)N(R5)2, -S(O)2N(R5)2, -N(R5)S(O)R5, -OSi(C1-C4 alkyl)3, or -C(O)C2- C6 alkenyl (e.g., -C2-C4 alkenyl);
Z5 is a self-immolative group or absent;
T is a spacer group or absent;
L1 is absent, -C1-C6- alkylene (e.g., -C1-C3- alkylene) or -C1-C6- heteroalkylene;
L2 is absent, -C1-C6- alkylene (e.g., -C1-C3- alkylene) or -C1-C6- heteroalkylene, wherein each alkylene and heteroalkyl is optionally substituted with one or more R6;
L3 is absent, -C1-C20- alkylene, -O-, -N(R5)-, -S-, -S(O)-, -S(O)2-, -S(O)N(R5)-,
-S(O)2N(R5)-, -N(R5)S(O)-, -N(R5)S(O)2-, -C(O)-, -C(O)O-, -OC(O)-, -C(O)N(R5)-, -N(R5)C(O)-;
L4 is -C1-C20- alkylene (e.g., -C1-C6- alkylene), -C1-C20- heteroalkylene (e.g., -C1-C6- heteroalkylene), -C1-C20- alkenylene (e.g., -C 2-C6- alkenylene), -C1-C20- alkynylene (e.g., -C 2-C6- alkynylene), or an oligopeptide, wherein the oligopeptide is optionally substituted by one or more R16;
L5 is a linker connecting Z4 and Z5, or is absent;
Q1 is C(O), C(S), or CH2; each of R1 and R2 is independently hydrogen, halo, -CN, -C1-C20 alkyl (e.g., C1-C6 alkyl), or - OR7;
R3 is hydrogen, -C1-C20- alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl (e.g., -C1-C6 heteroalkyl), -OC(O)OC1-C20 alkyl (e.g., -C1-C6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, heteroalkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R8; each R4 is independently hydrogen, -C1-C20 alkyl, -O-C1-C20 alkyl, -C1-C20 heteroalkyl, halo, -CN, -NO2 or -OH;
R5 is hydrogen or -C1-C20 alkyl (e.g., -C1-C6 alkyl);
R6 is halo, -CN, -C1-C20 alkyl (e.g., -C1-C6 alkyl), -OR7, oxo, cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R9;
R7 is hydrogen, -C1-C20 alkyl (e.g., -C1-C6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R9; each R8 is independently -C1-C20 alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl,
-C(O)-C1-C20 alkyl, -OC(O)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -C(O)O-C1-C20 alkyl
(e g·,
-C1-C6 alkyl), -OC(O)O-C1-C20 alkyl (e.g., -C1-C6 alkyl), -C(O)N(R5)-C1-C20 alkyl
(e g·,
-C1-C6 alkyl), -N(R5)C(O)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -OC(O)N(R5)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -O-aryl, -O-heteroaryl, -C(O)-aryl, -C(O)-heteroaryl, -
OC(O)-aryl,
-C(O)O-aryl, -OC(O)-heteroaryl, -C(O)O-heteroaryl, -C(O)O-aryl, -C(O)O-heteroaryl, -C(O)N(R5)-aryl, -C(O)N(R5)-heteroaryl, -N(R5)C(O)-aryl, -N(R5)2C(O)-aryl, or -N(R5)C(O)-heteroaryl, -S(O)2N(R5)-aryl, wherein each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R9; each R9 is independently -C1-C20 alkyl, -O-C1-C20 alkyl, -C1-C20 heteroalkyl, halo,
-CN, -OH, oxo, aryl, heteroaryl, -O-aryl, or -O-heteroaryl; and each R16 is independently, -C1-C20 alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl (e.g., -C1-C6 heteroalkyl), -OC(O)OC1-C20 alkyl (e.g., -C1-C6 alkyl), C(O)N(R4)2 cycloalkyl, heterocyclyl, aryl, or heteroaryl.
2. The antibody-drug conjugate of claim 1, wherein the antibody-drug conjugate is represented by formula (Il-a), (Il-b), (II-c), or (Il-d):
 or a pharmaceutically acceptable salt or stereoisomer thereof.
or a pharmaceutically acceptable salt or stereoisomer thereof.
3. The antibody-drug conjugate of claim 1 or 2, wherein Z1 is O.
4. The antibody-drug conjugate of any one of claims 1-3, wherein B1 is a purinyl nucleobase and B2 is a pyrimidinyl nucleobase.
5. The antibody-drug conjugate of any one of claims 1-4, wherein B1 is adenosinyl or guanosinyl and B2 is cytosinyl, thyminyl, or uracilyl.
6. The antibody-drug conjugate of any one of claims 1-5, wherein B1 is adenosinyl, and B2 is uracilyl.
7. The antibody-drug conjugate of any one of claims 1-6, wherein each of R1 and R2 is independently hydrogen, halo, or OR7.
8. The antibody-drug conjugate of any one of claims 1-7, wherein each of R1 and R2 is independently halo (e.g., fluoro).
9. The antibody-drug conjugate of any one of claims 1-7, wherein each of R1 and R2 is independently halo, -CN, -C1-C20 alkyl, or (e.g., C1-C6 alkyl).
10. The antibody-drug conjugate of any one of claims 1-9, wherein each of X1 and X2 is independently O.
11. The antibody-drug conjugate of any one of claims 1-10, wherein each of Y1 and Y2 is independently O or S.
12. The antibody-drug conjugate of any one of claims 1-11, wherein one of Y1 or Y2 is O and the other is S.
13. The antibody-drug conjugate of any one of claims 1-11, wherein each of Y1 or Y2 is independently S.
14. The antibody-drug conjugate of any one of claims 1-11, wherein each of Y1 or Y2 is independently O.
15. The antibody-drug conjugate of any one of claims 1-14, wherein each of L1 and L2 is independently C1-C6 alkylene (e.g., C1 alkylene).
16. The antibody-drug conjugate of any one of claims 1-14, wherein L1 is absent.
17. The antibody-drug conjugate of any one of claims 1-16, wherein R3 is independently hydrogen, aryl, or heteroaryl, wherein aryl and heteroaryl is optionally substituted with 1-5 R8.
18. The antibody-drug conjugate of any one of claims 1-17, wherein R3 is aryl or heteroaryl, each of which is optionally substituted with 1-5 R8.
19. The antibody-drug conjugate of any one of claims 1-18, wherein R3 is phenyl substituted with one R8.
20. The antibody-drug conjugate of any one of claims 1-11 and 15-19, wherein each of Y1 and Y2 is independently S and R3 is independently substituted with one R8.
21. The antibody-drug conjugate of claim 20, wherein Y1 is S and R3 is substituted with one R8.
22. The antibody-drug conjugate of any one of claims 1-11 and 14-19, wherein each of Y1 and Y2 is O and R3 is hydrogen.
23. The antibody-drug conjugate of any one of claims 17-22, wherein R8 is -OC(O)-aryl, and the aryl is optionally substituted by 1-5 R9 (e.g., 1 R9).
24. The antibody-drug conjugate of claim 23, wherein R9 is -O-C1-C12 alkyl (e.g., O- CH2(CH2)8CH3).
25. The antibody-drug conjugate of any one of claims 1-24, wherein Z2 is -aryl-.
26. The antibody-drug conjugate of any one of claims 1-24, wherein Z2 is -OC(O)OC1- C20- alkylene (e.g., -C1-C6- alkylene).
27. The antibody-drug conjugate of any one of claims 1-24, wherein Z2 is absent.
28. The antibody-drug conjugate of any one of claims 1-27, wherein R4 is hydrogen.
29. The antibody-drug conjugate of any one of claims 1-28, wherein L3 is -O(O)C-.
30. The antibody-drug conjugate of any one of claims 1-28, wherein L3 is absent
31. The antibody-drug conjugate of any one of claims 1-30, wherein Z3 is -aryl-.
32. The antibody-drug conjugate of any one of claims 1-31, wherein Z3 is aryl substituted with -OC1-C20-heteroalkylene (e.g., -C i-C 12- heteroalkyl ene).
33. The antibody-drug conjugate of any one of claims 1-32, wherein Z3 is aryl substituted with -OC1-C20-NH- (e.g., -C1-C12-NH-).
34. The antibody-drug conjugate of any one of claims 1-30, wherein Z3 is -OC(O)OC1- C20-alkylene (e.g., -C1-C6- alkylene).
35. The antibody-drug conjugate of any one of claims 1-30, wherein Z3 is -CThCThlN^H)-
36. The antibody-drug conjugate of any one of claims 1-30, wherein Z3 is absent.
37. The antibody-drug conjugate of any one of claims 1-36, wherein L4 is a -C1-C20- alkylene (e.g., -C1-C18- alkylene).
38. The antibody-drug conjugate of any one of claims 1-36, wherein L4 is an oligiopeptide.
39. The antibody-drug conjugate of claim 38, wherein the oligopeptide is a dipeptide.
40. The antibody-drug conjugate of claim 38 or 39, wherein the oligopeptide comprises 3 amino acid residues, 4 amino acid residues, 5 amino acid residues, 6 amino acid residues, 7 amino acid residues, 8 amino acid residues, 9 amino acid residues, or 10 amino acid residues.
41. The antibody-drug conjugate of claim 40, wherein the oligopeptide comprises 8 amino acid residues.
42. The antibody-drug conjugate of any one of claims 38-41, wherein the oligopeptide comprises an amino acid selected from Tyr, Ser, Thr, Arg, Leu, He, Gly, Val, and Ala.
43. The antibody-drug conjugate of any one of claims 38-42, wherein the oligopeptide comprises an amino acid selected from Phe, Tyr, Arg, Gly, Ser, and Leu.
44. The antibody-drug conjugate of any one of claims 38-43, wherein the oligopeptide is substituted with one R16.
45. The antibody-drug conjugate of claim 44, wherein R16 is C(0)NH2.
46. The antibody-drug conjugate of claim 44 or 45, wherein L4 is

47. The antibody-drug conjugate of claim 38, wherein L4 is
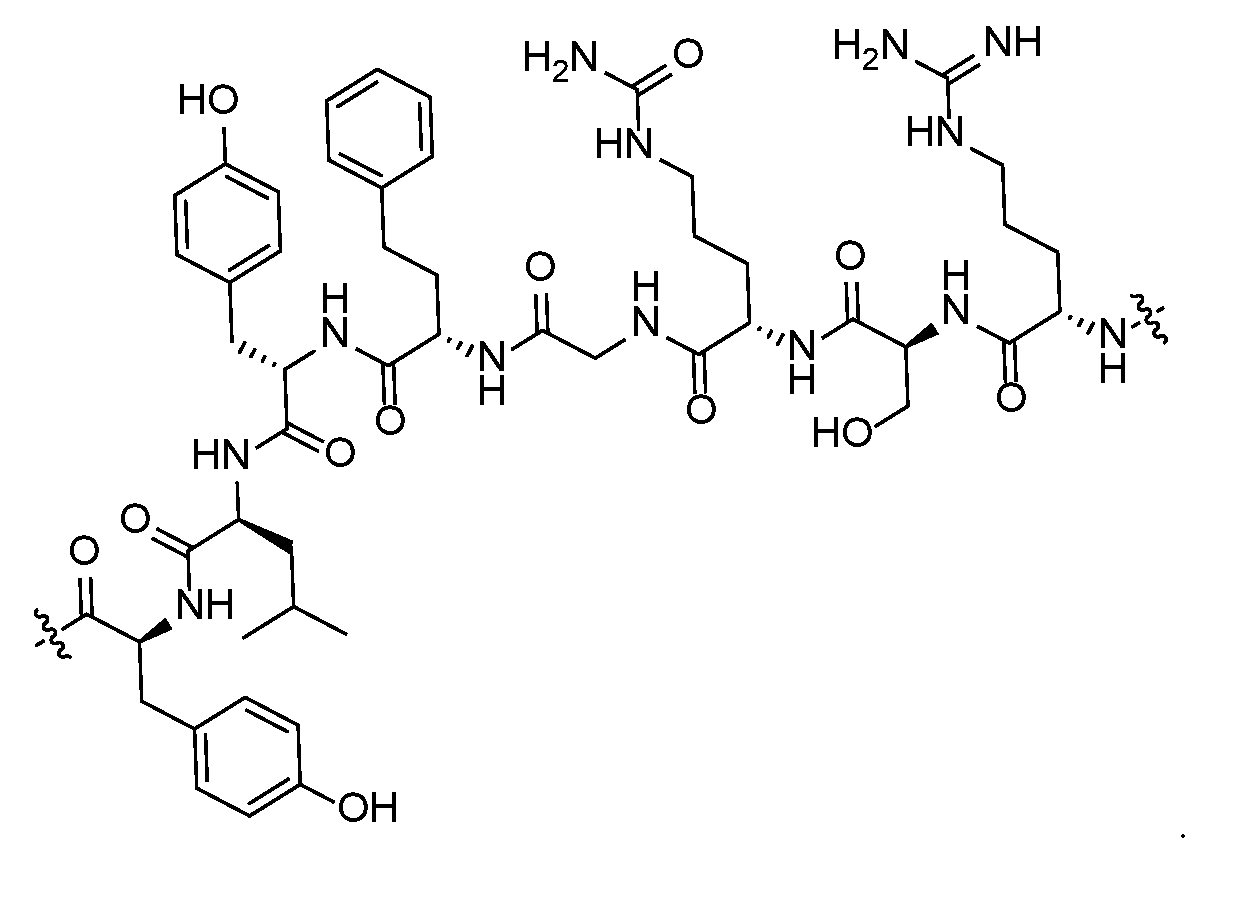
48. The antibody-drug conjugate of claim 38, wherein L4 is selected from the group consisting of and


49. The antibody-drug conjugate of claim 38, wherein L4 is

50. The antibody-drug conjugate of any one of claims 1-49, wherein Z4 is heterocyclyl- C1-C20- alkylene-Q.
51. The antibody-drug conjugate of any one of claims 1-49, wherein Z4 is

52. The antibody-drug conjugate of claims 51, wherein Q1 is C(O).
53. The antibody-drug conjugate of any one of claims 1-49, wherein Z4 is -OR5, -N(R5)2,
-Ns, -OSi(C1-C4 alkyl)3, or -C(0)C2-C6 alkenyl (e.g., -C2-C4 alkenyl).
54. The antibody-drug conjugate of any one of claims 1-54, wherein L5 is -C1-C40- alkylene (e.g., -C1-C20- alkylene), -C1-C40- heteroalkylene (e.g., -C1-C20- heteroalkyl), -C1-
C40- alkenylene (e.g., -C2-C20- alkenylene), or -C1-C40- alkynylene (e.g., -C2-C20- alkynylene).
55. The antibody-drug conjugate of any one of claims 1-54, wherein L5 is an oligopeptide comprising of 1-40 amino acid residues.
56. The antibody-drug conjugate of any one of claims 1-55, wherein L5 further comprises one
 sub-unit.
sub-unit.
57. The antibody-drug conjugate of any one of claims 1-56, wherein L5 further comprises one
 sub-unit.
sub-unit.
58. The antibody-drug conjugate of any one of claims 1-57, wherein L5 further comprises one
 sub-unit.
sub-unit.
59. The antibody-drug conjugate of any one of claims 1-58, wherein L5 further comprises one
 sub-unit.
sub-unit.
60. The antibody-drug conjugate of any one of claims 1-53, wherein L is absent.
61. The antibody-drug conjugate of any one of claims 1-60, wherein Z5 is absent.
62. The antibody-drug conjugate of any one of claims 1-60, wherein Z5 is


63. The antibody-drug conjugate of any one of claims 1-60, wherein Z5 is

64. The antibody-drug conjugate of any one of claims 1-60, wherein Z5 is

65. The antibody-drug conjugate of any one of claims 1-60, wherein Z5 is

66. The antibody-drug conjugate of any one of claims 1-60, wherein Z5 is

67. The antibody-drug conjugate of any one of claims 1-60, wherein Z5 is

68. The antibody-drug conjugate of any one of claims 1-60, wherein Z5 is selected from the group consisting of
 and
and

69. The antibody-drug conjugate of any one of claims 1-60, wherein Z5 is represented by formula (Ill-a), (Ill-b), or (III-c):


U is O, S or NR12;
Q2 is CR13 or N; each V1, V2 and V3 are independently CR13 or N provided that for formula (Ill-b) and (III-c) at least one of Q2, V1 and V2 is N;
R11 is the side chain of an amino acid and is optionally protected with a protecting group; each R10, R14, and R15 are independently selected from hydrogen, halo, -OH, -N(R12)2, - N(R12)3 +, -C1-C8 heteroalkyl, carboxylate, sulfate, sulfamate, sulfonate, -SO2R5, - S(O)R12, -SR12, -SO2N(R12)2, -C(O)R5, -CO2R12, -C(O)N(R12)2, -CN, -N3, -NO2, -C1- C8 heteroalkyl, polyethyleneoxy, phosphonate, phosphate, -C1-C8 alkyl, -C2-C8 heteroalkyl, -C2-C8 alkenyl, -C2-C8 alkynl, aryl, and heterocycle; or when taken together, R14 and R15 form a carbonyl (=0), or spiro-carbocyclic ring comprising of 3 to 7 carbon atoms;
R12 and R13 are independently selected from hydrogen, C1-C8 alkyl, C2-C8 alkenyl, C2-C8 alkynyl, aryl, heterocycle, wherein each alkyl, alkenyl, alkynyl, aryl, and heterocycle are independently substituted with one or more substituents selected from halo, OH, -N(R12)2, -N(R12)3+, heteroalkyl, carboxylate, sulfate, sulfamate, sulfonate, 4- dialkylaminopyridinium, alkyl, -SO2R12, -S(O)R12, -SR12, -SO2N(R12)2, C(O)R12,
CO2R12, C(O)N(R12)2, -CN, -N3, -NO2, C3-C12 carbocycle, aryl, heterocycle, polyethyleneoxy, phosphonate, and phosphate; and m is 1-6.
70. The antibody-drug conjugate of claim 69, wherein Q2 is N; V1 is CH, and V2 is CH.
71. The antibody-drug conjugate of claim 69, wherein Q2 is CH; V1 is CH and V2 is N.
72. The antibody-drug conjugate of claim 69, wherein Q2 is N; V1 is CH, and V2 is N.
73. The antibody-drug conjugate of claim 69, wherein Q2 is N; and V1 is N and V2 is N.
74. The antibody-drug conjugate of claim 69, wherein Q2 is N; V1 is N and V2 is CH.
75. The antibody-drug conjugate of claim 69, wherein Q2 is N; and V1, V2 and V3 are each CH.
76. The antibody-drug conjugate of claim 69, wherein Q2 is CH; and V1, V2 and V3 are each CH.
77. The antibody-drug conjugate of any one of claims 1-76, wherein T is -C1-C20- alkylene (e.g., -C1-C6- alkylene), -C1-C20- heteroalkylene (e.g., -C1-C6- heteroalkylene), -C1- C20- alkenylene (e.g., -C 2-C6- alkenylene), -C1-C20- alkynylene (e.g., -C 2-C6- alkynyl), -aryl- or -heteroaryl-.
78. The antibody-drug conjugate of claim 1, wherein the antibody-drug conjugate is selected from the group consisting of:





; or a pharmaceutically acceptable salt thereof; wherein Ab is an anti -PD 1 antibody, or an anti-CTLA-4 antibody.
79. The antibody-drug conjugate of any one of claims 1-78, wherein the anti -PD -1 antibody or anti-CTLA-4 antibody is a monoclonal antibody, a domain antibody (dAb), a single chain antibody (scAb), a Fab fragment, a F(ab’)2 fragment, a single chain variable
fragment (scFv), a scFv-Fc fragment, a single domain heavy chain antibody, a single domain light chain antibody, a variant antibody, a multimeric antibody, or a bispecific antibody.
80. The antibody-drug conjugate of any one of claims 1-79, wherein the antibody is an anti -PD -1 antibody.
81. The antibody-drug conjugate of any one of claims 1-80, wherein the anti-PD-1 antibody is pembrolizumab, nivolumab, cemiplimab, Anti-mouse PD-1 antibody Clone J43, Anti-mouse PD-1 antibody Clone RMP1-14, mouse anti-PD-1 antibody Clone EH12, ANB011, MDX-1 106, AMP-514, AMP-224, or Pidilizumab.
82. The antibody-drug conjugate of any one of claims 1-81, wherein the anti-PD-1 antibody is pembrolizumab or nivolumab.
83. The antibody-drug conjugate of any one of claims 1-79, wherein the antibody is an anti-CTLA-4 antibody.
84. The antibody-drug conjugate of any one of claims 1-79 and 83, wherein the anti- CTLA-4 antibody is ipilimumab, clone 9H10, tremelimumab, or clone BNI3.
85. The antibody-drug conjugate of any one of claims 1-79, 83 and 84, wherein the anti- CTLA-4 antibody is ipilimumab.
86. An antibody-drug conjugate of Formula (IV):
 or a pharmaceutically acceptable salt or stereoisomer thereof,
wherein:
or a pharmaceutically acceptable salt or stereoisomer thereof,
wherein:
Ab is an anti-PD-1 antibody or an anti-CTLA-4 antibody; each of B1 and B2 is independently a purinyl nucleobase or pyrimidinyl nucleobase; each of X1 and X2 is independently O or S; each of Y1 and Y2 is independently O, S, or N(R5); each of Z1 is independently O or S;
Z2 is -O-, -N(R5)-, -S-, -C(O)-, -C(O)N(R5)-, -OC(O)N(R5)-, -N(R5)C(O)O-, -aryl-, -heteroaryl-, -S(O)-, -S(O)2-, -S(O)N(R5)-, -S(O)2N(R5)- or -N(R5)S(O)-;
Z3 is absent, hydrogen, heterocyclyl, heterocyclyl-C1-C20-alkylene-Q1, -OH, -N(R5)2, SR5, - CHO, -C(O)N(R5)2, -OC(O)N(R5)2, -N(R5)C(O)OR5, aryl, heteroaryl, -S(O)R5, - S(O)2R5, -S(O)N(R5)2, -S(O)2N(R5)2, -N(R5)S(O)R5, -OSi(C1-C4 alkyl)3, or -C(O)C2- C6 alkenyl (e.g., -C2-C4 alkenyl);
Z4 is a self-immolative group or absent;
T is a absent or spacer group; each L1 and L2 is absent, -C1-C6- alkylene (e.g., -C1-C3- alkylene) or -C1-C6- heteroalkyl ene, wherein each alkylene and heteroalkyl is optionally substituted with one or more R6;
L3 is oligiopeptide-C(O)-, oligiopeptide-aryl-C1-C6-alkylene-, oligiopeptide-aryl-C1-C6- alklyene-C(O)-, oligiopeptide-aryl-C1-C6-heteroalkylene, oligiopeptide-C1-C6- alkylene-C(O)-, oligiopeptide-C1-C6-heteroalkylene-C(O)-, -C1-C40- alkylene (e.g., - C1-C20- alkylene), -C1-C40- heteroalkyl ene (e.g., -C1-C20- heteroalkyl), -C1-C40- alkenylene (e.g., -C2-C20- alkenylene), or -C1-C40- alkynylene (e.g., -C2-C20- alkynylene), wherein the oligopeptide is optionally substituted by one or more R14;
L4 is absent or a linker connecting Z3 and Z4;
Q1 is C(O), C(S), or CH2; each of R1 and R2 is independently hydrogen, halo, -CN, -C1-C20 alkyl (e.g., C1-C6 alkyl), or - OR7; each R3 and R4 is independently hydrogen, -C1-C20- alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl (e.g., -C1-C6 heteroalkyl), -OC(O)OC1-C20 alkyl (e.g., -C1-C6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, heteroalkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R8;
R5 is hydrogen or -C1-C20 alkyl (e.g., -C1-C6 alkyl);
R6 is halo, -CN, -C1-C20 alkyl (e.g., -C1-C6 alkyl), -OR7, oxo, cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R9;
R7 is hydrogen, -C1-C20 alkyl (e.g., -C1-C6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R9; each R8 is independently -C1-C20 alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl,
-C(O)-C1-C20 alkyl, -OC(O)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -C(O)O-C1-C20 alkyl (e.g., -C1-C6 alkyl), -OC(O)O-C1-C20 alkyl (e.g., -C1-C6 alkyl), -C(O)N(R5)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -N(R5)C(O)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -OC(O)N(R5)- C1-C20 alkyl (e.g., -C1-C6 alkyl), -O-aryl, -O-heteroaryl, -C(O)-aryl, -C(O)-heteroaryl, -OC(O)-aryl, -C(O)O-aryl, -OC(O)-heteroaryl, -C(O)O-heteroaryl, -C(O)O-aryl, - C(O)O-heteroaryl, -C(O)N(R5)-aryl, -C(O)N(R5)-heteroaryl, -N(R5)C(O)-aryl, - N(R5)2C(O)-aryl, or -N(R5)C(O)-heteroaryl, -S(O)2N(R5)-aryl, wherein each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R9; each R9 is independently -C1-C20 alkyl, -O-C1-C20 alkyl, -C1-C20 heteroalkyl, halo,
-CN, -OH, oxo, aryl, heteroaryl, -O-aryl, or -O-heteroaryl; and each R16 is independently, -C1-C20 alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl (e.g., -C1-C6 heteroalkyl), -OC(O)OC1-C20 alkyl (e.g., -C1-C6 alkyl), C(O)N(R4)2 cycloalkyl, heterocyclyl, aryl, or heteroaryl.
87. The antibody-drug conjugate of claim 86, wherein the antibody-drug conjugate is represented by formula (V-a), (V-b), (V-c), or (V-d):

88. The antibody-drug conjugate of claim 86 or 87, wherein the antibody-drug conjugate is an antibody-drug conjugate of formula (V-e), (V-f), (V-g), (V-h), (V-i) or (V-j):
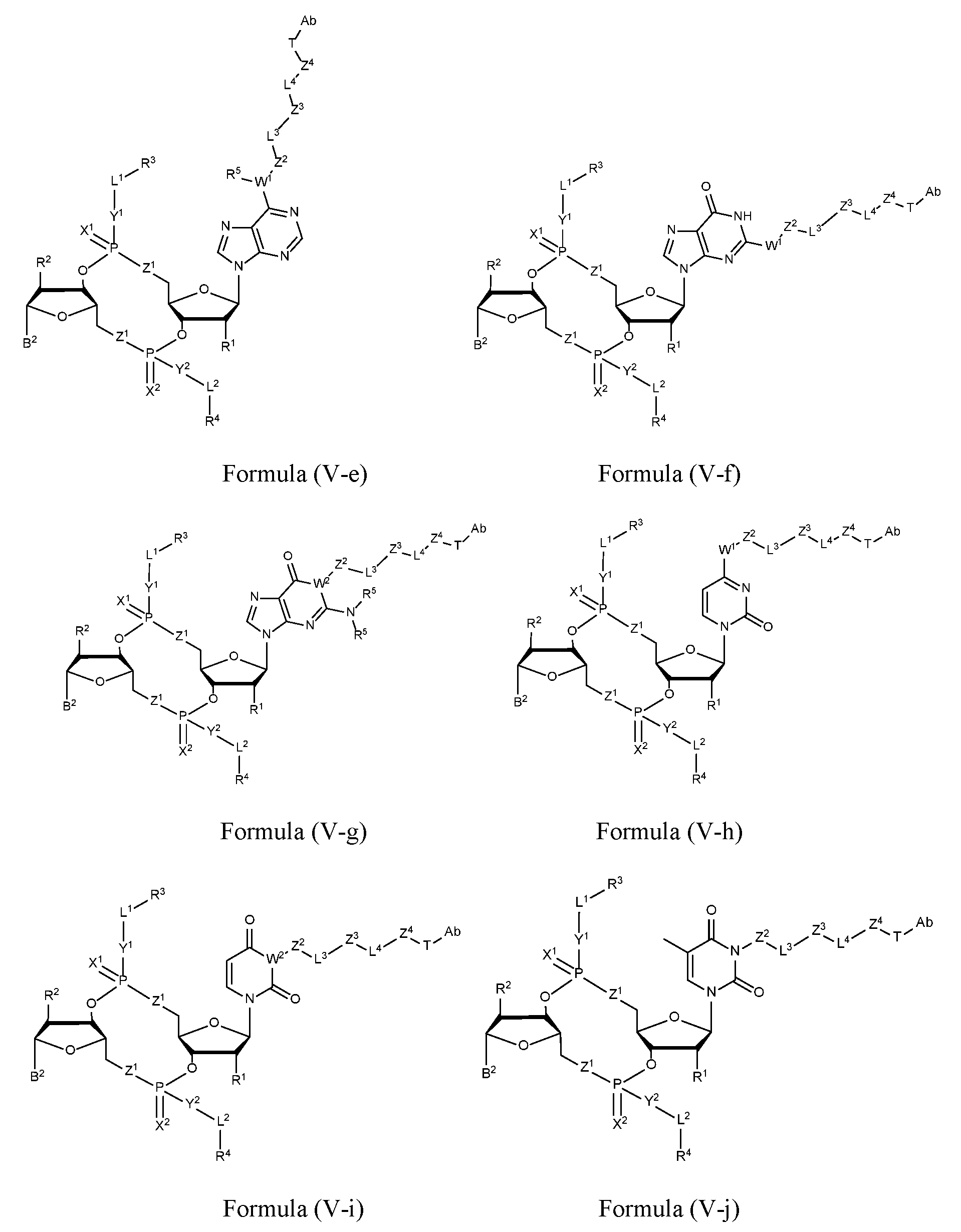 or a pharmaceutically acceptable salt or stereoisomer thereof, wherein: W1 is N(R5), -O- CH; and W2 is N or CH.
or a pharmaceutically acceptable salt or stereoisomer thereof, wherein: W1 is N(R5), -O- CH; and W2 is N or CH.
89. The antibody-drug conjugate of any one of claims 86-88, wherein each Z1 is O.
90. The antibody-drug conjugate of any one of claims 86-89, wherein B1 is a purinyl nucleobase, and B2 is a pyrimidinyl nucleobase.
91. The antibody-drug conjugate of any one of claims 86-90, wherein B1 is adenosinyl or guanosinyl, and B2 is cytosinyl, thyminyl, or uracilyl.
92. The antibody-drug conjugate of any one of claims 86-91, wherein B1 is adenosinyl, and B2 is uracilyl.
93. The antibody-drug conjugate of any one of claims 86-92, wherein each of R1 and R2 is independently hydrogen, halo, or OR7.
94. The antibody-drug conjugate of any one of claims 86-93, wherein each of R1 and R2 is independently halo (e.g., fluoro).
95. The antibody-drug conjugate of any one of claims 86-94, wherein each of R1 and R2 is independently halo, -CN, -C1-C20 alkyl, or (e.g., C1-C6 alkyl).
96. The antibody-drug conjugate of any one of claims 86-95, wherein each of X1 and X2 is independently O.
97. The antibody-drug conjugate of any one of claims 86-96, wherein each of Y1 and Y2 is independently O or S.
98. The antibody-drug conjugate of any one of claims 86-97, wherein one of Y1 or Y2 is O and the other of Y1 or Y2 is S.
99. The antibody-drug conjugate of any one of claims 86-97, wherein each of Y1 or Y2 is independently S.
100. The antibody-drug conjugate of any one of claims 86-97, wherein each of Y1 or Y2 is independently O.
101. The antibody-drug conjugate of any one of claims 86-100, wherein each of L1 and L2 is independently C1-C6 alkylene (e.g., C1 alkylene).
102. The antibody-drug conjugate of any one of claims 86-100, wherein L1 is absent.
103. The antibody-drug conjugate of any one of claims 86-102, wherein each of R3 and R4 is independently hydrogen, aryl, or heteroaryl, wherein aryl and heteroaryl is optionally substituted with 1-5 R8
104. The antibody-drug conjugate of any one of claims 86-103, wherein R3 is aryl or heteroaryl, each of which is optionally substituted with 1-5 R8, and R4 is hydrogen.
105. The antibody-drug conjugate of any one of claims 86-104, wherein R3 is phenyl substituted with 1 R8 and R4 is hydrogen.
106. The antibody-drug conjugate of any one of claims 86-103, wherein each of R3 and R4 is independently phenyl substituted with 1 R8.
107. The antibody-drug conjugate of any one of claims 86-97 and 101-107, wherein each of Y1 and Y2 is O and each of R3 and R4 is independently hydrogen.
108. The antibody-drug conjugate of any one of claims 86-97, 99, and 101-106, wherein each of Y1 and Y2 is independently S and each of R3 and R4 is independently substituted with 1 R8.
109. The antibody-drug conjugate of any one of claims 86-108, wherein R8 is -OC(O)-aryl, and the aryl is optionally substituted by 1-5 R9 (e.g., 1 R9).
110. The antibody-drug conjugate of any one of claims 86-109, wherein R9 is -O-C1-C12 alkyl (e.g, O-CH2(CH2)8CH3).
111. The antibody-drug conjugate of any one of claims 86-110, wherein L3 -C1-C20- alkylene (e.g., -C1-C18- alkylene).
112. The antibody-drug conjugate of any one of claims 86-111, wherein L3 -C1-C20- heteroalkylene (e.g., -C1-C18- heteroalkylene).
113. The antibody-drug conjugate of any one of claims 86-112, wherein L3 is an oligopeptide-C(O)-, oligiopeptide-aryl-C1-C6-alkylene-, oligiopeptide-aryl-C1-C6- heteroalkylene, oligiopeptide-aryl-C1-C6-alklyene-C(O)-, oligiopeptide-C1-C6-alkylene-C(O)- , oligiopeptide-C1-C6-heteroalkylene-C(O)-.
114. The antibody-drug conjugate of claim 113, wherein the aryl is phenyl.
115. The antibody-drug conjugate of claim 113 or 114, wherein the oligopeptide is a dipeptide.
116. The antibody-drug conjugate of any one of claims 113-115, wherein the oligopeptide comprises 3 amino acid residues, 4 amino acid residues, 5 amino acid residues, 6 amino acid residues, 7 amino acid residues, 8 amino acid residues, 9 amino acid residues, or 10 amino acid residues.
117. The antibody-drug conjugate of any one of claims 113-116, wherein the oligopeptide comprises 8 amino acid residues.
118. The antibody-drug conjugate of any one of claims 113-117, wherein the oligopeptide comprises an amino acid selected from Tyr, Ser, Thr, Arg, Leu, He, Gly, Val, and Ala.
119. The antibody-drug conjugate of any one of claims 113-118, wherein the oligopeptide comprises an amino acid selected from Phe, Tyr, Arg, Gly, Ser, and Leu.
120. The antibody-drug conjugate of any one of claims 113-119, wherein the oligopeptide is substituted by one or more R16.
121. The antibody-drug conjugate of claim 120, wherein R16 is C(O)NH2.
122. The antibody-drug conjugate of any one of claims 86-110, wherein L3 is
 or
or


123. The antibody-drug conjugate of any one of claims 86-110, wherein L3 is

124. The antibody-drug conjugate of any one of claims 86-110, wherein L3 is selected from the group consisting of and


125. The antibody-drug conjugate of any one of claims 86-110, wherein L3 is

126. The antibody-drug conjugate of any one of claims 86-125, wherein Z3 is -O-, -N(R5)- or -heteroaryl-.
127. The antibody-drug conjugate of any one of claims 86-125, wherein Z3 is heterocyclyl- C1-C20-alkylene-Q.
128. The antibody-drug conjugate of any one of claims 86-125, wherein Z3 is

129. The antibody-drug conjugate of claim 128, wherein Q1 is C(O).
130. The antibody-drug conjugate of any one of claims 86-129, wherein L4 is -C1-C40- alkylene (e.g., -C1-C20- alkylene), -C1-C40- heteroalkylene (e.g., -C1-C20- heteroalkyl), -C1- C40- alkenylene (e.g., -C2-C20- alkenylene), or -C1-C40- alkynylene (e.g., -C2-C20- alkynylene).
131. The antibody-drug conjugate of any one of claims 86-129, wherein L4 is an oligopeptide comprising of 1-40 amino acid residues.
132. The antibody-drug conjugate of any one of claims 86-131, wherein L4 further comprises one
 sub-unit.
sub-unit.
133. The antibody-drug conjugate of any one of claims 86-132, wherein L4 further comprises one sub-unit.

134. The antibody-drug conjugate of any one of claims 86-133, wherein L4 further comprises one sub-unit.

135. The antibody-drug conjugate of any one of claims 86-134, wherein L4 further comprises one sub-unit.

136. The antibody-drug conjugate of any one of claims 86-129, wherein L4 is absent.
137. The antibody-drug conjugate of any one of claims 86-136, wherein Z4 is

138. The antibody-drug conjugate of any one of claims 86-136, wherein Z4 is

139. The antibody-drug conjugate of any one of claims 86-136, wherein Z4 is
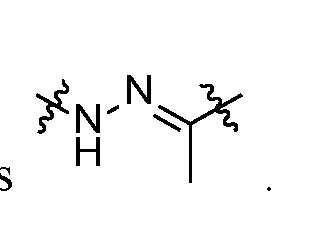
140. The antibody-drug conjugate of any one of claims 86-136, wherein Z4 is

141. The antibody-drug conjugate of any one of claims 86-136, wherein Z4 is

142. The antibody-drug conjugate of any one of claims 86-136, wherein Z4 is

143. The antibody-drug conjugate of any one of claims 86-136, wherein Z4 is selected from the group consisting of
 and
and

144. The antibody-drug conjugate of any one of claims 86-136, wherein Z4 is represented by formula (Vl-a), (VI -b), or (VI -c):
 wherein:
wherein:
U is O, S or NR12;
Q is CR13 orN; each V1, V2 and V3 are independently CR13 or N provided that for formula (Ill-b) and (III-c) at least one of Q, V1 and V2 is N;
R11 is the side chain of an amino acid and is optionally protected with a protecting group; each R10, R14, and R15 are independently selected from hydrogen, halo, -OH, -N(R12)2, - N(R12)3 +, -C1-C8 heteroalkyl, carboxylate, sulfate, sulfamate, sulfonate, -SO2R5, - S(O)R12, -SR12, -SO2N(R12)2, -C(O)R5, -CO2R12, -C(O)N(R12)2, -CN, -N3, -NO2, -C1- C8 heteroalkyl, polyethyleneoxy, phosphonate, phosphate, -C1-C8 alkyl, -C2-C8 heteroalkyl, -C2-C8 alkenyl, -C2-C8 alkynl, aryl, and heterocycle; or when taken together, R14 and R15 form a carbonyl (=0), or spiro-carbocyclic ring comprising of 3 to 7 carbon atoms;
R12 and R13 are independently selected from hydrogen, C1-C8 alkyl, C2-C8 alkenyl, C2-C8 alkynyl, aryl, heterocycle, wherein each alkyl, alkenyl, alkynyl, aryl, and heterocycle are independently substituted with one or more substituents selected from halo, OH, - N(Ri2)2, -N(R12)3+, heteroalkyl, carboxylate, sulfate, sulfamate, sulfonate, 4- dialkylaminopyridinium, alkyl, -SO2R12, -S(O)R12, -SR12, -SO2N(R12)2, C(O)R12, CO2R12, C(O)N(R12)2, -CN, -N3, -NO2, C3-C12 carbocycle, aryl, heterocycle, polyethyleneoxy, phosphonate, and phosphate; and m is 1-6.
145. The antibody-drug conjugate of claim 144, wherein Q is N; V1 is CH, and V2 is CH.
146. The antibody-drug conjugate of claim 144, wherein Q is CH; V1 is CH and V2 is N.
147. The antibody-drug conjugate of claim 144, wherein Q is N; V1 is CH, and V2 is N.
148. The antibody-drug conjugate of claim 144, wherein Q is N; and V1 is N and V2 is N.
149. The antibody-drug conjugate of claim 144, wherein Q is N; V1 is N and V2 is CH.
150. The antibody-drug conjugate of claim 144, wherein Q is N; and V1, V2 and V3 are each CH.
151. The antibody-drug conjugate of claim 144, wherein Q is CH; and V1, V2 and V3 are each CH.
152. The antibody-drug conjugate of any one of claims 86-136, wherein Z4 is absent.
153. The antibody-drug conjugate of any one of claims 86-152, wherein T is -C1-C20- alkylene (e.g., -C1-C6- alkylene), -C1-C20- heteroalkylene (e.g., -C1-C6- heteroalkylene), -C1- C20- alkenylene (e.g., -C 2-C6- alkenylene), -C1-C20- alkynylene (e.g., -C 2-C6- alkynyl), -aryl- or -heteroaryl-.
154. The antibody-drug conjugate of any one of claims 86-152, wherein T is absent.
155. The antibody-drug conjugate of claim 86, wherein the antibody-drug conjugate is selected from:





156. The antibody-drug conjugate of any one of claims 86-155, wherein the anti-PD-1 antibody or anti-CTLA-4 antibody is a monoclonal antibody, a domain antibody (dAb), a single chain antibody (scAb), a Fab fragment, a F(ab’)2 fragment, a single chain variable fragment (scFv), a scFv-Fc fragment, a single domain heavy chain antibody, a single domain light chain antibody, a variant antibody, a multimeric antibody, or a bispecific antibody.
157. The antibody-drug conjugate of any one of claims 86-156, wherein the antibody is an anti -PD -1 antibody.
158. The antibody-drug conjugate of any one of claims 86-157, wherein the anti-PD-1 antibody is pembrolizumab, nivolumab, cemiplimab, Anti-mouse PD-1 antibody Clone J43, Anti-mouse PD-1 antibody Clone RMP1-14, mouse anti-PD-1 antibody Clone EH12, ANB011, MDX-1 106, AMP-514, AMP-224, or Pidilizumab.
159. The antibody-drug conjugate of any one of claims 86-158, wherein the anti-PD-1 antibody is pembrolizumab or nivolumab.
160. The antibody-drug conjugate of any one of claims 86-156, wherein the antibody is an anti-CTLA-4 antibody.
161. The antibody-drug conjugate of any one of claims 86-156 and 160, wherein the anti- CTLA-4 antibody is ipilimumab, clone 9H10, tremelimumab, or clone BNI3.
162. The antibody-drug conjugate of any one of claims 86-156, 160, and 161, wherein the anti-CTLA-4 antibody is ipilimumab.
163. An antibody-drug conjugate of Formula (Vll-a) or Formula (Vll-b):
 or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
Ab is an anti-PD-1 antibody or an anti-CTLA-4 antibody; each of B1 and B2 is independently a purinyl nucleobase or pyrimidinyl nucleobase;
each of X1 and X2 is independently O or S; each of Y1 and Y2 is independently O, S, or N(R5); each of Z1 is independently O or S; each of Z2 and Z3 is independently absent, -C1-C20- alkylene (e.g., -C1-C6- alkylene), C1-C20 heteroalkylene (e.g., -C1-C6- heteroalkylene), -OC(O)OC1-C20- alkylene (e.g., -C1-C6- alkylene), -cycloalkylene-, -heterocyclyl-, -aryl-, or -heteroaryl-, wherein each -cycloakyl-, -heterocyclyl-, -aryl- or -heteroaryl- is optionally substituted with one or more R4;
Z4 is self-immolative group-C1-C20-alkylene-Q1 (e.g., -self-immolative group-C1-C6- alkylene-Q1), heterocyclyl-C1-C20-alkylene-Q (e.g., heterocyclyl-C1-C6-alkylene-Q),- OH, -N(R5)2, SR5, -CHO, -C(O)N(R5)2, -OC(O)N(R5)2, -N(R5)C(O)OR5, aryl, heteroaryl, -S(O)R5,
-S(O)2R5, -S(O)N(R5)2, -S(O)2N(R5)2, -N(R5)S(O)R5, -OSi(C1-C4 alkyl)3, or -C(O)C2- C6 alkenyl (e.g., -C2-C4 alkenyl);
Z5 is a self-immolative group or absent;
T is a spacer group or absent;
L1 is absent, -C1-C6- alkylene (e.g., -C1-C3- alkylene) or -C1-C6- heteroalkylene;
L2 is absent, -C1-C6- alkylene (e.g., -C1-C3- alkylene) or -C1-C6- heteroalkylene, wherein each alkylene and heteroalkyl is optionally substituted with one or more R6;
L3 is absent, -C1-C20- alkylene, -O-, -N(R5)-, -S-, -S(O)-, -S(O)2-, -S(O)N(R5)-,
-S(O)2N(R5)-, -N(R5)S(O)-, -N(R5)S(O)2-, -C(O)-, -C(O)O-, -OC(O)-, -C(O)N(R5)-, -N(R5)C(O)-;
L4 is -C1-C20- alkylene (e.g., -C1-C6- alkylene), -C1-C20- heteroalkylene (e.g., -C1-C6- heteroalkylene), -C1-C20- alkenylene (e.g., -C 2-C6- alkenylene), -C1-C20- alkynylene (e.g., -C2-C6- alkynylene), or an oligopeptide, wherein the oligopeptide is optionally substituted by one or more R16;
L5 is a linker connecting Z4 and Z5, or is absent;
Q1 is C(O), C(S), or CH2; each of R1 and R2 is independently hydrogen, halo, -CN, -C1-C20 alkyl (e.g., C1-C6 alkyl), or - OR7;
R3 is hydrogen, -C1-C20- alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl (e.g., -C1-C6 heteroalkyl), -OC(O)OC1-C20 alkyl (e.g., -C1-C6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, heteroalkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R8;
each R4 is independently hydrogen, -C1-C20 alkyl, -O-C1-C20 alkyl, -C1-C20 heteroalkyl, halo, -CN, -NO2 or -OH;
R5 is hydrogen or -C1-C20 alkyl (e.g., -C1-C6 alkyl);
R6 is halo, -CN, -C1-C20 alkyl (e.g., -C1-C6 alkyl), -OR7, oxo, cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R9;
R7 is hydrogen, -C1-C20 alkyl (e.g., -C1-C6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R9; each R8 is independently -C1-C20 alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl,
-C(O)-C1-C20 alkyl, -OC(O)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -C(O)O-C1-C20 alkyl (e.g·,
-C1-C6 alkyl), -OC(O)O-C1-C20 alkyl (e.g., -C1-C6 alkyl), -C(O)N(R5)-C1-C20 alkyl (e.g·,
-C1-C6 alkyl), -N(R5)C(O)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -OC(O)N(R5)-C1-C2O alkyl (e.g., -C1-C6 alkyl), -O-aryl, -O-heteroaryl, -C(O)-aryl, -C(O)-heteroaryl, - OC(O)-aryl,
-C(O)O-aryl, -OC(O)-heteroaryl, -C(O)O-heteroaryl, -C(O)O-aryl,
-C(O)O-heteroaryl, -C(O)N(R5)-aryl, -C(O)N(R5)-heteroaryl, -N(R5)C(O)-aryl, -N(R5)2C(O)-aryl, or -N(R5)C(O)-heteroaryl, -S(O)2N(R5)-aryl, wherein each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R9; each R9 is independently -C1-C20 alkyl, -O-C1-C20 alkyl, -C1-C20 heteroalkyl, halo,
-CN, -OH, oxo, aryl, heteroaryl, -O-aryl, or -O-heteroaryl; and each R16 is independently, -C1-C20 alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl (e.g., -C1-C6 heteroalkyl), -OC(O)OC1-C20 alkyl (e.g., -C1-C6 alkyl), C(O)N(R4)2 cycloalkyl, heterocyclyl, aryl, or heteroaryl.
164. The antibody-drug conjugate of claim 163, wherein the antibody-drug conjugate is represented by formula (VllI-a), (VllI-b), (VIII-c), (VllI-d), (VllI-e), (VllI-f), (VIII-g), or (Vlll-h):


165. The antibody-drug conjugate of claim 163 or 164, wherein Z1 is O.
166. The antibody-drug conjugate of any one of claims 163-165, wherein B1 is a purinyl nucleobase and B2 is a pyrimidinyl nucleobase.
167. The antibody-drug conjugate of any one of claims 163-166, wherein B1 is adenosinyl or guanosinyl and B2 is cytosinyl, thyminyl, or uracilyl.
168. The antibody-drug conjugate of any one of claims 163-167, wherein B1 is adenosinyl, and B2 is uracilyl.
169. The antibody-drug conjugate of any one of claims 163-168, wherein each of R1 and R2 is independently hydrogen, halo, or OR7.
170. The antibody-drug conjugate of any one of claims 163-169, wherein each of R1 and R2 is independently halo (e.g., fluoro).
171. The antibody-drug conjugate of any one of claims 163-170, wherein each of R1 and R2 is independently halo, -CN, -C1-C20 alkyl, or (e.g., C1-C6 alkyl).
172. The antibody-drug conjugate of any one of claims 163-171, wherein each of X1 and X2 is independently O.
173. The antibody-drug conjugate of any one of claims 163-172, wherein each of Y1 and Y2 is independently O or S.
174. The antibody-drug conjugate of any one of claims 163-173, wherein one of Y1 or Y2 is O and the other is S.
175. The antibody-drug conjugate of any one of claims 163-173, wherein each of Y1 or Y2 is independently S.
176. The antibody-drug conjugate of any one of claims 163-173, wherein each of Y1 or Y2 is independently O.
177. The antibody-drug conjugate of any one of claims 163-176, wherein each of L1 and L2 is independently C1-C6 alkylene (e.g., C1 alkylene).
178. The antibody-drug conjugate of any one of claims 163-176, wherein L1 is absent.
179. The antibody-drug conjugate of any one of claims 163-178, wherein R3 is independently hydrogen, aryl, or heteroaryl, wherein aryl and heteroaryl is optionally substituted with 1-5 R8.
180. The antibody-drug conjugate of any one of claims 163-179, wherein R3 is aryl or heteroaryl, each of which is optionally substituted with 1-5 R8.
181. The antibody-drug conjugate of any one of claims 163-180, wherein R3 is phenyl substituted with one R8.
182. The antibody-drug conjugate of any one of claims 163-173 and 177-181, wherein each of Y1 and Y2 is independently S and R3 is independently substituted with one R8.
183. The antibody-drug conjugate of claim 182, wherein Y1 is S and R3 is substituted with one R8.
184. The antibody-drug conjugate of any one of claims 163-173 and 176-181, wherein each of Y1 and Y2 is O and R3 is hydrogen.
185. The antibody-drug conjugate of any one of claims 179-184, wherein R8 is -OC(O)- aryl, and the aryl is optionally substituted by 1-5 R9 (e.g., 1 R9).
186. The antibody-drug conjugate of claim 185, wherein R9 is -O-C1-C12 alkyl (e.g., O- CH2(CH2)8CH3).
187. The antibody-drug conjugate of any one of claims 163-186, wherein Z2 is -aryl-.
188. The antibody-drug conjugate of any one of claims 163-186, wherein Z2 is -
OC(O)OC1-C20- alkylene (e.g., -C1-C6- alkylene).
189. The antibody-drug conjugate of any one of claims 163-186, wherein Z2 is absent.
190. The antibody-drug conjugate of any one of claims 163-189, wherein R4 is hydrogen.
191. The antibody-drug conjugate of any one of claims 163-190, wherein L3 is -O(O)C-.
192. The antibody-drug conjugate of any one of claims 163-190, wherein L3 is absent
193. The antibody-drug conjugate of any one of claims 163-192, wherein Z3 is -aryl-.
194. The antibody-drug conjugate of any one of claims 163-193, wherein Z3 is aryl substituted with -OC1-C20-heteroalkylene (e.g., -C1-C12-heteroalkylene).
195. The antibody-drug conjugate of any one of claims 163-193, wherein Z3 is aryl substituted with -OC1-C20-NH- (e.g., -C1-C12-NH-).
196. The antibody-drug conjugate of any one of claims 163-193, wherein Z3 is - OC(O)OC1-C20-alkylene (e.g., -C1-C6- alkylene).
197. The antibody-drug conjugate of any one of claims 163-193, wherein Z3 is -CH2CH2N(H)-.
198. The antibody-drug conjugate of any one of claims 163-193, wherein Z3 is absent.
199. The antibody-drug conjugate of any one of claims 163-198, wherein L4 is a -C1-C20- alkylene (e.g., -C1-C18- alkylene).
200. The antibody-drug conjugate of any one of claims 163-198, wherein L4 is an oligiopeptide.
201. The antibody-drug conjugate of claim 200, wherein the oligopeptide is a dipeptide.
202. The antibody-drug conjugate of claim 200 or 201, wherein the oligopeptide comprises 3 amino acid residues, 4 amino acid residues, 5 amino acid residues, 6 amino acid residues, 7 amino acid residues, 8 amino acid residues, 9 amino acid residues, or 10 amino acid residues.
203. The antibody-drug conjugate of claim 202, wherein the oligopeptide comprises 8 amino acid residues.
204. The antibody-drug conjugate of any one of claims 200-203, wherein the oligopeptide comprises an amino acid selected from Tyr, Ser, Thr, Arg, Leu, He, Gly, Val, and Ala.
205. The antibody-drug conjugate of any one of claims 200-204, wherein the oligopeptide comprises an amino acid selected from Phe, Tyr, Arg, Gly, Ser, and Leu.
206. The antibody-drug conjugate of any one of claims 200-205, wherein the oligopeptide is substituted with one R16.
207. The antibody-drug conjugate of claim 206, wherein R16 is C(O)NH2.
208. The antibody-drug conjugate of claim 200 or 201, wherein L4 is

209. The antibody-drug conjugate of claim 200, wherein L4 is

210. The antibody-drug conjugate of claim 200, wherein L4 is selected from the group consisting of and
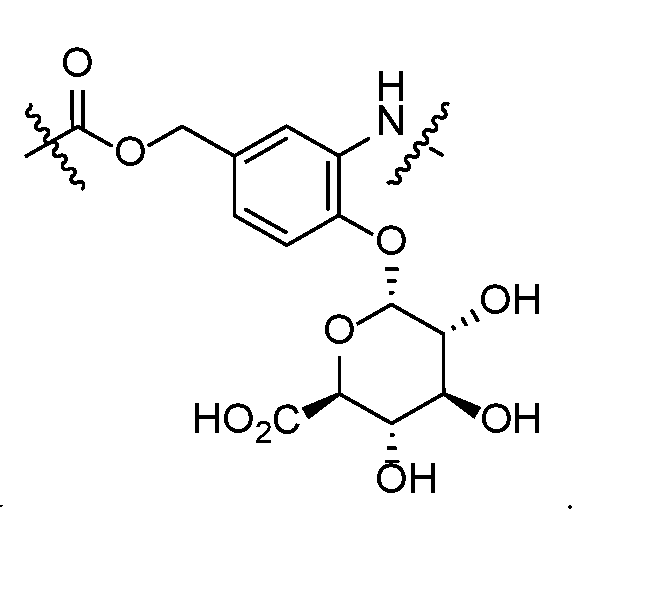

211 The antibody-drug conjugate of claim 200, wherein L4 is

212. The antibody-drug conjugate of any one of claims 163-210, wherein Z4 is heterocyclyl-C1-C20- alkylene-Q.
213. The antibody-drug conjugate of any one of claims 163-210, wherein Z4 is
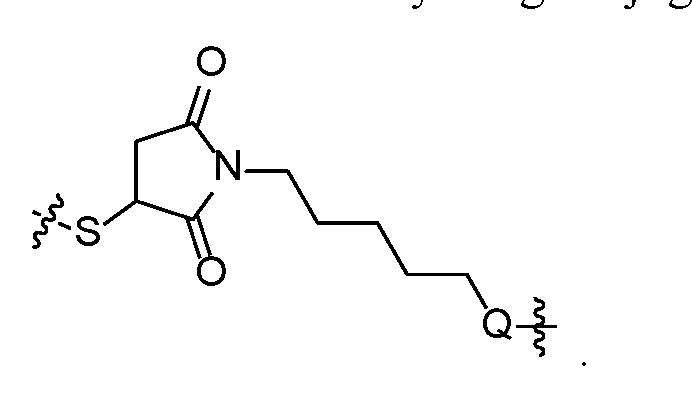
214. The antibody-drug conjugate of claims 213, wherein Q1 is C(O).
215. The antibody-drug conjugate of any one of claims 163-211, wherein Z4 is -OR5, - N(R5)2, -N3, -OSi(C1-C4 alkyl)3, or -C(O)C2-C6 alkenyl (e.g., -C2-C4 alkenyl).
216. The antibody-drug conjugate of any one of claims 163-215, wherein L5 is -C1-C40- alkylene (e.g., -C1-C20- alkylene), -C1-C40- heteroalkylene (e.g., -C1-C20- heteroalkyl), -C1- C40- alkenylene (e.g., -C2-C20- alkenylene), or -C1-C40- alkynylene (e.g., -C2-C20- alkynylene).
217. The antibody-drug conjugate of any one of claims 163-215, wherein L5 is an oligopeptide comprising of 1-40 amino acid residues.
218. The antibody-drug conjugate of any one of claims 163-217, wherein L5 further comprises one sub-unit.

219. The antibody-drug conjugate of any one of claims 163-218, wherein L5 further comprises one
 sub-unit.
sub-unit.
220. The antibody-drug conjugate of any one of claims 163-219, wherein L5 further comprises one
 sub-unit.
sub-unit.
221. The antibody-drug conjugate of any one of claims 163-220, wherein L5 further comprises one
 sub-unit.
sub-unit.
222. The antibody-drug conjugate of any one of claims 163-215, wherein L5 is absent.
223. The antibody-drug conjugate of any one of claims 163-222, wherein Z5 is absent.
224. The antibody-drug conjugate of any one of claims 163-222, wherein Z5 is

225. The antibody-drug conjugate of any one of claims 163-222, wherein Z5 is

226. The antibody-drug conjugate of any one of claims 163-222, wherein Z5 is

227. The antibody-drug conjugate of any one of claims 163-222, wherein Z5 is

228. The antibody-drug conjugate of any one of claims 163-222, wherein Z5 is

229. The antibody-drug conjugate of any one of claims 163-222, wherein Z5 is
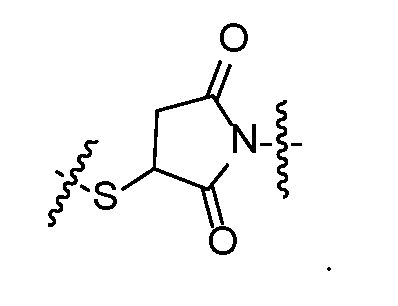
230. The antibody-drug conjugate of any one of claims 163-222, wherein Z5 is selected from the group consisting of
 and
and

231. The antibody-drug conjugate of any one of claims 163-222, wherein Z5 is represented by formula (IX-a), (IX-b), or (IX-c):


U is O, S or NR12;
Q2 is CR13 or N; each V1, V2 and V3 are independently CR13 or N provided that for formula (Ill-b) and (III-c) at least one of Q2, V1 and V2 is N;
R11 is the side chain of an amino acid and is optionally protected with a protecting group; each R10, R14, and R15 are independently selected from hydrogen, halo, -OH, -N(R12)2, - N(R12)3 +, -C1-C8 heteroalkyl, carboxylate, sulfate, sulfamate, sulfonate, -SO2R5, - S(O)R12, -SR12, -SO2N(R12)2, -C(O)R5, -CO2R12, -C(O)N(R12)2, -CN, -N3, -NO2, -C1- C8 heteroalkyl, polyethyleneoxy, phosphonate, phosphate, -C1-C8 alkyl, -C2-C8 heteroalkyl, -C2-C8 alkenyl, -C2-C8 alkynl, aryl, and heterocycle; or when taken together, R14 and R15 form a carbonyl (=0), or spiro-carbocyclic ring comprising of 3 to 7 carbon atoms;
R12 and R13 are independently selected from hydrogen, C1-C8 alkyl, C2-C8 alkenyl, C2-C8 alkynyl, aryl, heterocycle, wherein each alkyl, alkenyl, alkynyl, aryl, and heterocycle are independently substituted with one or more substituents selected from halo, OH, -N(R12)2, -N(R12)3+, heteroalkyl, carboxylate, sulfate, sulfamate, sulfonate, 4- dialkylaminopyridinium, alkyl, -SO2R12, -S(O)R12, -SR12, -SO2N(R12)2, C(O)R12, CO2R12, C(O)N(R12)2, -CN, -N3, -NO2, C3-C12 carbocycle, aryl, heterocycle, polyethyleneoxy, phosphonate, and phosphate; and m is 1-6.
232. The antibody-drug conjugate of claim 231, wherein Q2 is N; V1 is CH, and V2 is CH.
233. The antibody-drug conjugate of claim 231, wherein Q2 is CH; V1 is CH and V2 is N.
234. The antibody-drug conjugate of claim 231, wherein Q2 is N; V1 is CH, and V2 is N.
235. The antibody-drug conjugate of claim 231, wherein Q2 is N; and V1 is N and V2 is N.
236. The antibody-drug conjugate of claim 231, wherein Q2 is N; V1 is N and V2 is CH.
237. The antibody-drug conjugate of claim 231, wherein Q2 is N; and V1, V2 and V3 are each CH.
238. The antibody-drug conjugate of claim 231, wherein Q2 is CH; and V1, V2 and V3 are each CH.
239. The antibody-drug conjugate of any one of claims 163-238, wherein T is -C1-C20- alkylene (e.g., -C1-C6- alkylene), -C1-C20- heteroalkylene (e.g., -C1-C6- heteroalkylene), -C1- C20- alkenylene (e.g., -C 2-C6- alkenylene), -C1-C20- alkynylene (e.g., -C 2-C6- alkynyl), -aryl- or -heteroaryl-.
240. The antibody-drug conjugate of claim 163, wherein the antibody-drug conjugate is selected from the group consisting of:







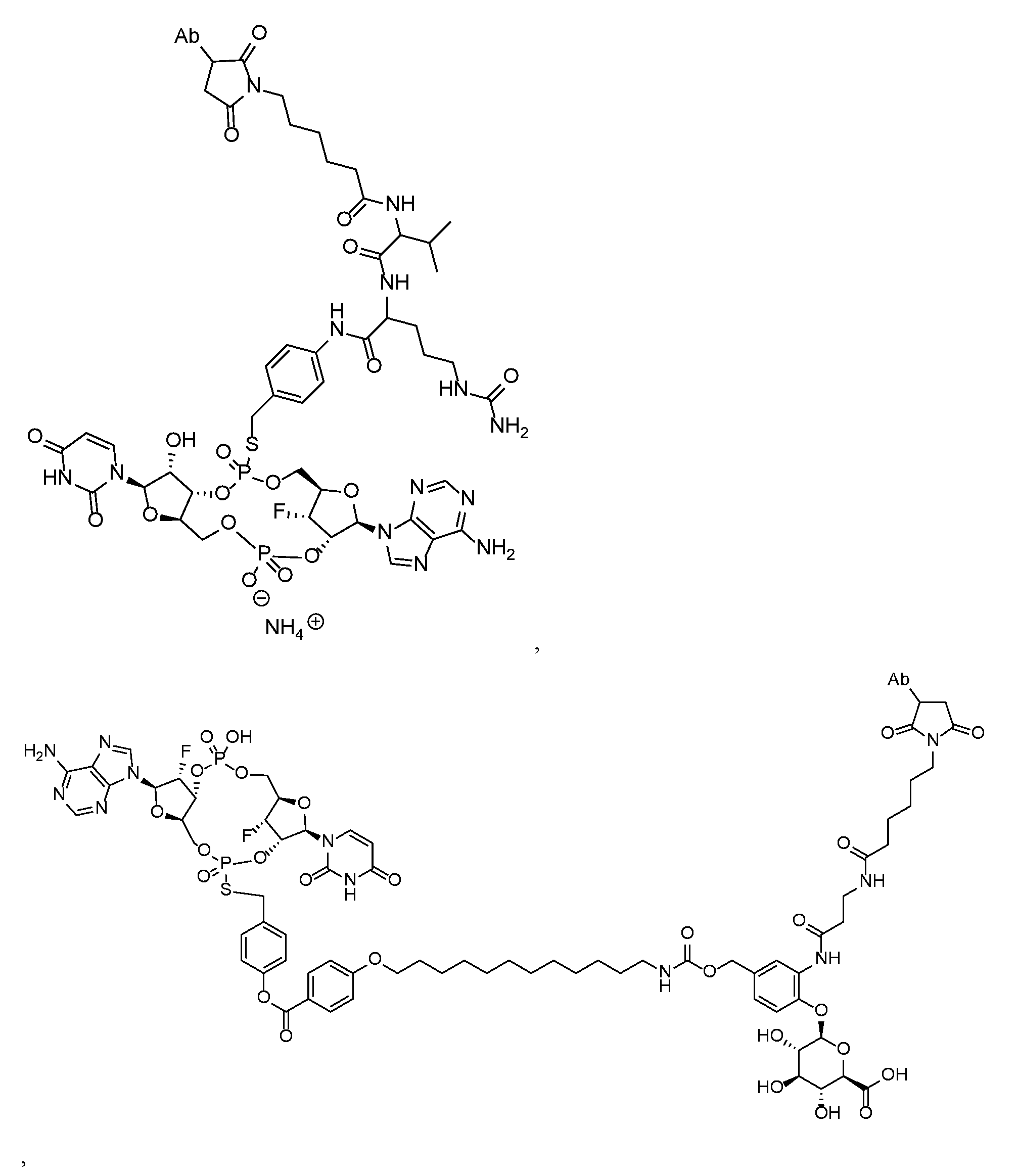

; or a pharmaceutically acceptable salt thereof; wherein Ab is an anti -PD 1 antibody, or an anti-CTLA-4 antibody.
241. The antibody-drug conjugate of any one of claims 163-240, wherein the anti -PD-1 antibody or anti-CTLA-4 antibody is a monoclonal antibody, a domain antibody (dAb), a single chain antibody (scAb), a Fab fragment, a F(ab’)2 fragment, a single chain variable fragment (scFv), a scFv-Fc fragment, a single domain heavy chain antibody, a single domain light chain antibody, a variant antibody, a multimeric antibody, or a bispecific antibody.
242. The antibody-drug conjugate of any one of claims 163-241, wherein the antibody is an anti -PD- 1 antibody.
243. The antibody-drug conjugate of any one of claims 163-242, wherein the anti -PD- 1 antibody is pembrolizumab, nivolumab, cemiplimab, Anti-mouse PD-1 antibody Clone J43, Anti-mouse PD-1 antibody Clone RMP1-14, mouse anti -PD-1 antibody Clone EH 12, ANB011, MDX-1 106, AMP-514, AMP-224, or Pidilizumab.
244. The antibody-drug conjugate of any one of claims 163-243, wherein the anti -PD- 1 antibody is pembrolizumab or nivolumab.
245. The antibody-drug conjugate of any one of claims 163-241, wherein the antibody is an anti-CTLA-4 antibody.
246. The antibody-drug conjugate of any one of claims 163-241 and 245, wherein the anti- CTLA-4 antibody is ipilimumab, clone 9H10, tremelimumab, or clone BNI3.
247. The antibody-drug conjugate of any one of claims 163-241, 247, and 248, wherein the anti-CTLA-4 antibody is ipilimumab.
248. An antibody-drug conjugate of Formula (X-a) or Formula (X-b):
 or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
or a pharmaceutically acceptable salt or stereoisomer thereof, wherein:
Ab is an anti-PD-1 antibody or an anti-CTLA-4 antibody; each of B1 and B2 is independently a purinyl nucleobase or pyrimidinyl nucleobase; each of X1 and X2 is independently O or S; each of Y1 and Y2 is independently O, S, or N(R5); each of Z1 is independently O or S;
Z2 is -O-, -N(R5)-, -S-, -C(O)-, -C(O)N(R5)-, -OC(O)N(R5)-, -N(R5)C(O)O-, -aryl-, -heteroaryl-, -S(O)-, -S(O)2-, -S(O)N(R5)-, -S(O)2N(R5)- or -N(R5)S(O)-;
Z3 is absent, hydrogen, heterocyclyl, heterocyclyl-C1-C20-alkylene-Q1, -OH, -N(R5)2, SR5, - CHO, -C(O)N(R5)2, -OC(O)N(R5)2, -N(R5)C(O)OR5, aryl, heteroaryl, -S(O)R5, - S(O)2R5, -S(O)N(R5)2, -S(O)2N(R5)2, -N(R5)S(O)R5, -OSi(C1-C4 alkyl)3, or -C(O)C2- C6 alkenyl (e.g., -C2-C4 alkenyl);
Z4 is a self-immolative group or absent;
T is a absent or spacer group; each L1 and L2 is absent, -C1-C6- alkylene (e.g., -C1-C3- alkylene) or -C1-C6- heteroalkyl ene, wherein each alkylene and heteroalkyl is optionally substituted with one or more R6;
L3 is oligiopeptide-C(O)-, oligiopeptide-aryl-C1-C6-alkylene-, oligiopeptide-aryl-C1-C6- alklyene-C(O)-, oligiopeptide-aryl-C1-C6-heteroalkylene, oligiopeptide-C1-C6- alkylene-C(O)-, oligiopeptide-C1-C6-heteroalkylene-C(O)-, -C1-C40- alkylene (e.g., - C1-C20- alkylene), -C1-C40- heteroalkyl ene (e.g., -C1-C20- heteroalkyl), -C1-C40- alkenylene (e.g., -C2-C20- alkenylene), or -C1-C40- alkynylene (e.g., -C2-C20- alkynylene), wherein the oligopeptide is optionally substituted by one or more R14;
L4 is absent or a linker connecting Z3 and Z4;
Q1 is C(O), C(S), or CH2; each of R1 and R2 is independently hydrogen, halo, -CN, -C1-C20 alkyl (e.g., C1-C6 alkyl), or - OR7; each R3 and R4 is independently hydrogen, -C1-C20- alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl (e.g., -C1-C6 heteroalkyl), -OC(O)OC1-C20 alkyl (e.g., -C1-C6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, heteroalkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R8;
R5 is hydrogen or -C1-C20 alkyl (e.g., -C1-C6 alkyl);
R6 is halo, -CN, -C1-C20 alkyl (e.g., -C1-C6 alkyl), -OR7, oxo, cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R9;
R7 is hydrogen, -C1-C20 alkyl (e.g., -C1-C6 alkyl), cycloalkyl, heterocyclyl, aryl, or heteroaryl, wherein each alkyl, cycloalkyl, heterocyclyl, aryl, and heteroaryl is optionally substituted with one or more R9; each R8 is independently -C1-C20 alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl,
-C(O)-C1-C20 alkyl, -OC(O)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -C(O)O-C1-C20 alkyl
(e.g, -C1-C6 alkyl), -OC(O)O-C1-C20 alkyl (e.g., -C1-C6 alkyl), -C(O)N(R5)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -N(R5)C(O)-C1-C20 alkyl (e.g., -C1-C6 alkyl), -OC(O)N(R5)- C1-C20 alkyl (e.g., -C1-C6 alkyl), -O-aryl, -O-heteroaryl, -C(O)-aryl, -C(O)-heteroaryl, -OC(O)-aryl, -C(O)O-aryl, -OC(O)-heteroaryl, -C(O)O-heteroaryl, -C(O)O-aryl, - C(O)O-heteroaryl, -C(O)N(R5)-aryl, -C(O)N(R5)-heteroaryl, -N(R5)C(O)-aryl, - N(R5)2C(O)-aryl, or -N(R5)C(O)-heteroaryl, -S(O)2N(R5)-aryl, wherein each alkyl, heteroalkyl, aryl, and heteroaryl is optionally substituted by one or more R9; each R9 is independently -C1-C20 alkyl, -O-C1-C20 alkyl, -C1-C20 heteroalkyl, halo,
-CN, -OH, oxo, aryl, heteroaryl, -O-aryl, or -O-heteroaryl; and each R16 is independently, -C1-C20 alkyl (e.g., -C1-C6 alkyl), -C1-C20 heteroalkyl (e.g., -C1-C6 heteroalkyl), -OC(O)OC1-C20 alkyl (e.g., -C1-C6 alkyl), C(O)N(R4)2 cycloalkyl, heterocyclyl, aryl, or heteroaryl.
249. The antibody-drug conjugate of claim 248, wherein the antibody-drug conjugate is represented by formula (Xl-a), (CI-b), (XI-c), (CI-d), (XI-e), (XI-f), (XI-g), or (Xl-h):


250. The antibody-drug conjugate of claim 248 or 249, wherein the antibody-drug conjugate is an antibody-drug conjugate of formula (CI-i), (XI-j), (XI-k), (XI-1), (Xl-m), (XI- n), (XI-o), (XI-p), (XI-q), (XI-r), (XI-s), or (Xl-t):



W1 is N(R5), -O- CH; and W2 is N or CH.
251. The antibody-drug conjugate of any one of claims 248-250, wherein each Z1 is O.
252. The antibody-drug conjugate of any one of claims 248-251, wherein B1 is a purinyl nucleobase, and B2 is a pyrimidinyl nucleobase.
253. The antibody-drug conjugate of any one of claims 248-252, wherein B1 is adenosinyl or guanosinyl, and B2 is cytosinyl, thyminyl, or uracilyl.
254. The antibody-drug conjugate of any one of claims 248-253, wherein B1 is adenosinyl, and B2 is uracilyl.
255. The antibody-drug conjugate of any one of claims 248-254, wherein each of R1 and R2 is independently hydrogen, halo, or OR7.
256. The antibody-drug conjugate of any one of claims 248-254, wherein each of R1 and R2 is independently halo (e.g., fluoro).
257. The antibody-drug conjugate of any one of claims 248-254, wherein each of R1 and R2 is independently halo, -CN, -C1-C20 alkyl, or (e.g., C1-C6 alkyl).
258. The antibody-drug conjugate of any one of claims 248-257, wherein each of X1 and X2 is independently O.
259. The antibody-drug conjugate of any one of claims 248-258, wherein each of Y1 and Y2 is independently O or S.
260. The antibody-drug conjugate of any one of claims 248-259, wherein one of Y1 or Y2 is O and the other of Y1 or Y2 is S.
261. The antibody-drug conjugate of any one of claims 248-259, wherein each of Y1 or Y2 is independently S.
262. The antibody-drug conjugate of any one of claims 248-259, wherein each of Y1 or Y2 is independently O.
263. The antibody-drug conjugate of any one of claims 248-262, wherein each of L1 and L2 is independently C1-C6 alkylene (e.g., C1 alkylene).
264. The antibody-drug conjugate of any one of claims 248-262, wherein L1 is absent.
265. The antibody-drug conjugate of any one of claims 248-264, wherein each of R3 and R4 is independently hydrogen, aryl, or heteroaryl, wherein aryl and heteroaryl is optionally substituted with 1-5 R8
266. The antibody-drug conjugate of any one of claims 248-265, wherein R3 is aryl or heteroaryl, each of which is optionally substituted with 1-5 R8, and R4 is hydrogen.
267. The antibody-drug conjugate of any one of claims 248-266, wherein R3 is phenyl substituted with 1 R8 and R4 is hydrogen.
268. The antibody-drug conjugate of any one of claims 248-267, wherein each of R3 and R4 is independently phenyl substituted with 1 R8.
269. The antibody-drug conjugate of any one of claims 248-259 and 261-268, wherein each of Y1 and Y2 is O and each of R3 and R4 is independently hydrogen.
270. The antibody-drug conjugate of any one of claims 248-259, 2 and 265-271, wherein each of Y1 and Y2 is independently S and each of R3 and R4 is independently substituted with 1 R8.
271. The antibody-drug conjugate of any one of claims 248-270, wherein R8 is -OC(O)- aryl, and the aryl is optionally substituted by 1-5 R9 (e.g., 1 R9).
272. The antibody-drug conjugate of any one of claims 248-271, wherein R9 is -O-C1-C12 alkyl (e.g, O-CH2(CH2)8CH3).
273. The antibody-drug conjugate of any one of claims 248-272, wherein L3 -C1-C20- alkylene (e.g., -C1-C18- alkylene).
274. The antibody-drug conjugate of any one of claims 248-272, wherein L3 -C1-C20- heteroalkylene (e.g., -C1-C18- heteroalkylene).
275. The antibody-drug conjugate of any one of claims 248-272, wherein L3 is an oligopeptide-C(O)-, oligiopeptide-aryl-C1-C6-alkylene-, oligiopeptide-aryl-C1-C6- heteroalkylene, oligiopeptide-aryl-C1-C6-alklyene-C(O)-, oligiopeptide-C1-C6-alkylene-C(O)- , oligiopeptide-C1-C6-heteroalkylene-C(O)-.
276. The antibody-drug conjugate of claim 275, wherein the aryl is phenyl.
277. The antibody-drug conjugate of claim 275 or 276, wherein the oligopeptide is a dipeptide.
278. The antibody-drug conjugate of any one of claims 275-277, wherein the oligopeptide comprises 3 amino acid residues, 4 amino acid residues, 5 amino acid residues, 6 amino acid
residues, 7 amino acid residues, 8 amino acid residues, 9 amino acid residues, or 10 amino acid residues.
279. The antibody-drug conjugate of any one of claims 275-278, wherein the oligopeptide comprises 8 amino acid residues.
280. The antibody-drug conjugate of any one of claims 275-279, wherein the oligopeptide comprises an amino acid selected from Tyr, Ser, Thr, Arg, Leu, He, Gly, Val, and Ala.
281. The antibody-drug conjugate of any one of claims 275-280, wherein the oligopeptide comprises an amino acid selected from Phe, Tyr, Arg, Gly, Ser, and Leu.
282. The antibody-drug conjugate of any one of claims 275-281, wherein the oligopeptide is substituted by one or more R16.
283. The antibody-drug conjugate of claim 282, wherein R16 is C(O)NH2.
284. The antibody-drug conjugate of any one of claims 248-283, wherein L3 is
 or
or


285. The antibody-drug conjugate of any one of claims 248-283, wherein L is

286. The antibody-drug conjugate of any one of claims 248-283, wherein L is selected from the group consisting of
 and
and

287. The antibody-drug conjugate of any one of claims 248-283, wherein L3 is

288. The antibody-drug conjugate of any one of claims 248-287, wherein Z3 is -O-, - N(R5)- or -heteroaryl-.
289. The antibody-drug conjugate of any one of claims 248-287, wherein Z3 is heterocyclyl-C1-C20-alkylene-Q.
290. The antibody-drug conjugate of any one of claims 248-287, wherein Z3 is

291. The antibody-drug conjugate of claim 290, wherein Q1 is C(O).
292. The antibody-drug conjugate of any one of claims 248-291, wherein L4 is -C1-C40- alkylene (e.g., -C1-C20- alkylene), -C1-C40- heteroalkylene (e.g., -C1-C20- heteroalkyl), -C1- C40- alkenylene (e.g., -C2-C20- alkenylene), or -C1-C40- alkynylene (e.g., -C2-C20- alkynylene).
293. The antibody-drug conjugate of any one of claims 248-291, wherein L4 is an oligopeptide comprising of 1-40 amino acid residues.
294. The antibody-drug conjugate of any one of claims 248-293, wherein L4 further comprises one
 sub-unit.
sub-unit.
295. The antibody-drug conjugate of any one of claims 248-293, wherein L4 further comprises one
 sub-unit.
sub-unit.
296. The antibody-drug conjugate of any one of claims 248-295, wherein L4 further comprises one
 sub-unit.
sub-unit.
297. The antibody-drug conjugate of any one of claims 248-296, wherein L4 further comprises one sub-unit.

298. The antibody-drug conjugate of any one of claims 248-291, wherein L4 is absent.
299. The antibody-drug conjugate of any one of claims 248-298, wherein Z4 is

300. The antibody-drug conjugate of any one of claims 248-298, wherein Z4 is

301. The antibody-drug conjugate of any one of claims 248-298, wherein Z4 is

302. The antibody-drug conjugate of any one of claims 248-298, wherein Z4 is

303. The antibody-drug conjugate of any one of claims 248-298, wherein Z4 is
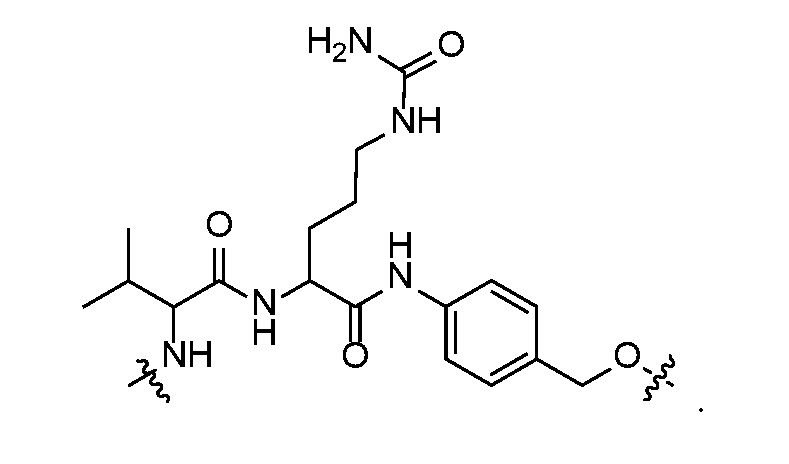
304. The antibody-drug conjugate of any one of claims 248-298, wherein Z4 is

305. The antibody-drug conjugate of any one of claims 248-298, wherein Z4 is selected from the group consisting of and
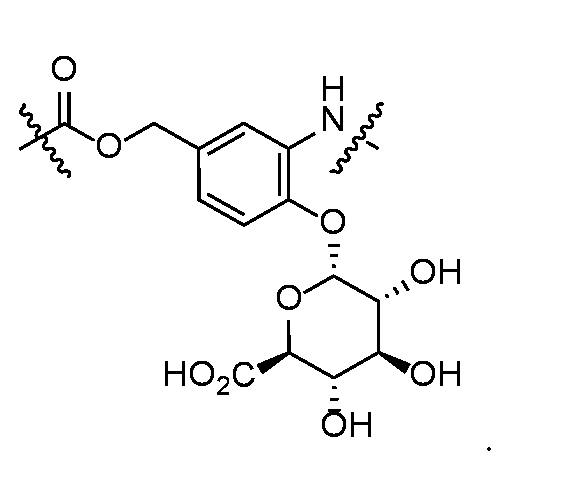

306. The antibody-drug conjugate of any one of claims 248-298, wherein Z4 is represented by formula (Xll-a), (Xll-b), or (XII-c):
 wherein:
wherein:
U is O, S or NR12;
Q is CR13 orN; each V1, V2 and V3 are independently CR13 or N provided that for formula (Ill-b) and (III-c) at least one of Q, V1 and V2 is N;
R11 is the side chain of an amino acid and is optionally protected with a protecting group; each R10, R14, and R15 are independently selected from hydrogen, halo, -OH, -N(R12)2, - N(R12)3 +, -C1-C8 heteroalkyl, carboxylate, sulfate, sulfamate, sulfonate, -SO2R5, - S(O)R12, -SR12, -SO2N(R12)2, -C(O)R5, -CO2R12, -C(O)N(R12)2, -CN, -N3, -NO2, -C1- C8 heteroalkyl, polyethyleneoxy, phosphonate, phosphate, -C1-C8 alkyl, -C2-C8 heteroalkyl, -C2-C8 alkenyl, -C2-C8 alkynl, aryl, and heterocycle; or when taken
together, R14 and R15 form a carbonyl (=0), or spiro-carbocyclic ring comprising of 3 to 7 carbon atoms;
R12 and R13 are independently selected from hydrogen, C1-C8 alkyl, C2-C8 alkenyl, C2-C8 alkynyl, aryl, heterocycle, wherein each alkyl, alkenyl, alkynyl, aryl, and heterocycle are independently substituted with one or more substituents selected from halo, OH, - N(Ri2)2, -N(R12)3+, heteroalkyl, carboxylate, sulfate, sulfamate, sulfonate, 4- dialkylaminopyridinium, alkyl, -SO2R12, -S(O)R12, -SR12, -SO2N(R12)2, C(O)R12, CO2R12, C(O)N(R12)2, -CN, -N3, -NO2, C3-C12 carbocycle, aryl, heterocycle, polyethyleneoxy, phosphonate, and phosphate; and m is 1-6.
307. The antibody-drug conjugate of claim 306, wherein Q is N; V1 is CH, and V2 is CH.
308. The antibody-drug conjugate of claim 306, wherein Q is CH; V1 is CH and V2 is N.
309. The antibody-drug conjugate of claim 306, wherein Q is N; V1 is CH, and V2 is N.
310. The antibody-drug conjugate of claim 306, wherein Q is N; and V1 is N and V2 is N.
311. The antibody-drug conjugate of claim 306, wherein Q is N; V1 is N and V2 is CH.
312. The antibody-drug conjugate of claim 306, wherein Q is N; and V1, V2 and V3 are each CH.
313. The antibody-drug conjugate of claim 306, wherein Q is CH; and V1, V2 and V3 are each CH.
314. The antibody-drug conjugate of any one of claims 248-298, wherein Z4 is absent.
315. The antibody-drug conjugate of any one of claims 248-314, wherein T is -C1-C20- alkylene (e.g., -C1-C6- alkylene), -C1-C20- heteroalkylene (e.g., -C1-C6- heteroalkylene), -C1- C20- alkenylene (e.g., -C 2-C6- alkenylene), -C1-C20- alkynylene (e.g., -C 2-C6- alkynyl), -aryl- or -heteroaryl-.
316. The antibody-drug conjugate of any one of claims 248-314, wherein T is absent.
317. The antibody-drug conjugate of claim 248, wherein the antibody-drug conjugate is selected from:





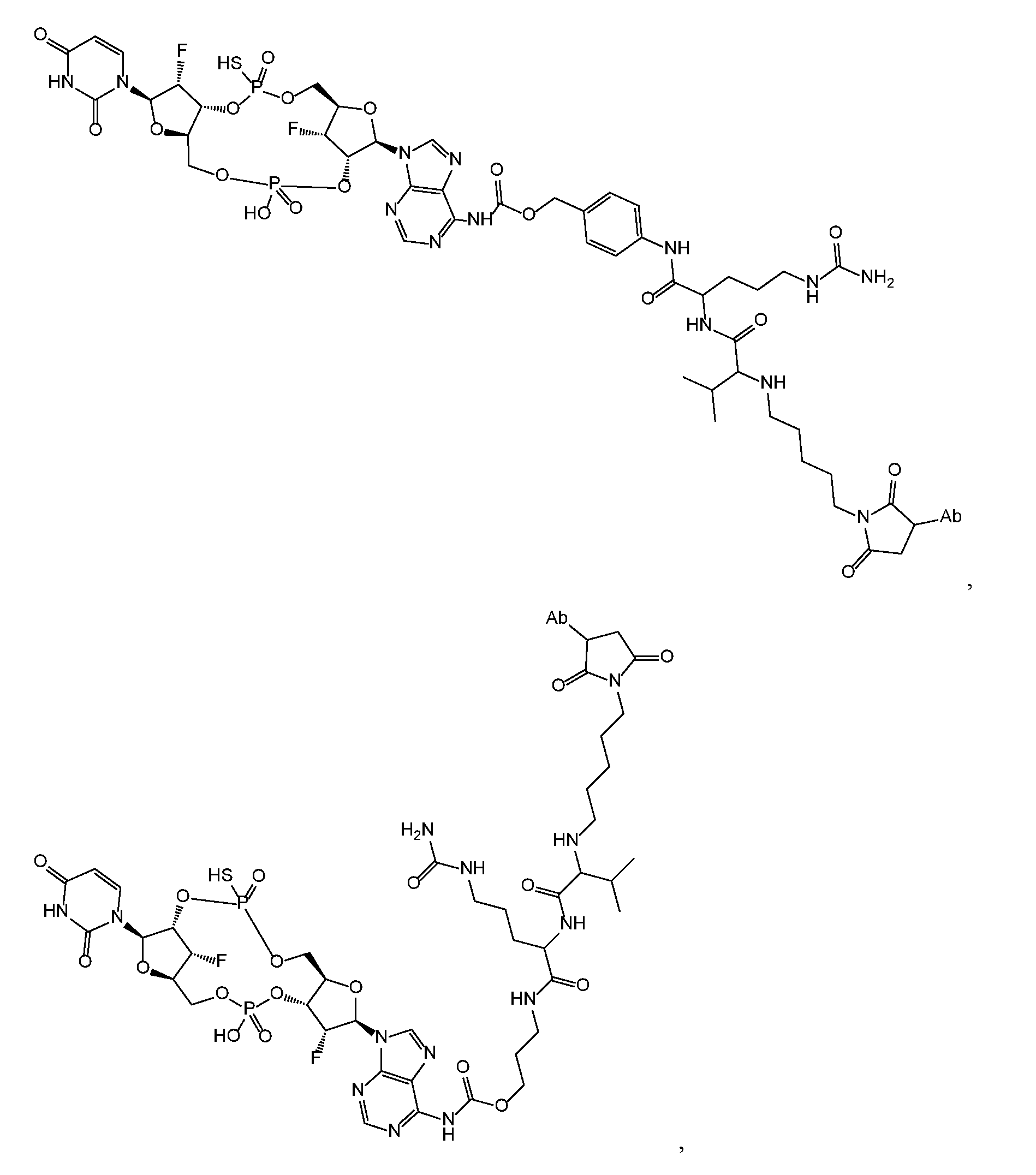




318. The antibody-drug conjugate of any one of claims 248-317, wherein the anti-PD-1 antibody or anti-CTLA-4 antibody is a monoclonal antibody, a domain antibody (dAb), a single chain antibody (scAb), a Fab fragment, a F(ab')2 fragment, a single chain variable fragment (scFv), a scFv-Fc fragment, a single domain heavy chain antibody, a single domain light chain antibody, a variant antibody, a multimeric antibody, or a bispecific antibody.
319. The antibody-drug conjugate of any one of claims 248-318, wherein the antibody is an anti-PD-1 antibody.
320. The antibody-drug conjugate of any one of claims 248-319, wherein the anti-PD-1 antibody is pembrolizumab, nivolumab, cemiplimab, Anti-mouse PD-1 antibody Clone J43, Anti-mouse PD-1 antibody Clone RMP1-14, mouse anti-PD-1 antibody Clone EH12, ANB011, MDX-1 106, AMP-514, AMP-224, or Pidilizumab.
321. The antibody-drug conjugate of any one of claims 248-320, wherein the anti-PD-1 antibody is pembrolizumab or nivolumab.
322. The antibody-drug conjugate of any one of claims 248-314, wherein the antibody is an anti-CTLA-4 antibody.
323. The antibody-drug conjugate of any one of claims 248-314 and 322, wherein the anti- CTLA-4 antibody is ipilimumab, clone 9H10, tremelimumab, or clone BNI3.
324. The antibody-drug conjugate of any one of claims 248-314, 322 and 323, wherein the anti-CTLA-4 antibody is ipilimumab.
325. A pharmaceutical composition comprising the antibody-drug conjugate of any one of claims 1-324; and a pharmaceutically acceptable excipient.
326. A method of treating cancer in a subject, comprising administering to the subject a therapeutically effective amount of the antibody-drug conjugate of any one of claims 1-324.
327. The method of claim 326, wherein the cancer is a cancer of the breast, bone, brain, cervix, colon, gastrointestinal tract, eye, gall bladder, lymph nodes, blood, lung, liver, skin, mouth, prostate, ovary, penis, pancreas, uterus, testicles, stomach, thymus, thyroid, or other part of the body.
328. The method of of claim 326 or 327, wherein the cancer is liver cancer.
329. The method of claim 326 or 327, wherein the cancer is B-cell lymphoma.
330. The method of any one of claims 326-329, wherein the method comprises intratumoral administration of the antibody-drug conjugate.
331. The method of any one of claims 326-329, wherein the method comprises oral administration of the antibody-drug conjugate.
332. The method of any one of claims 326-331, wherein the method comprises parenteral administration (e.g., intravenous, subcutaneous, intraperitoneal, or intramuscular administration) of the antibody-drug conjugate.
333. The method of claim 332, wherein the parenteral administration comprises intraperitoneal administration.
334. The method of any one of claims 326-333, further comprising administration of an additional agent (e.g., an anti cancer agent).
335. The method of claim 334, wherein the additional agent comprises methotrexate, 5- fluorouracil, doxorubicin, vincristine, bleomycin, vinblastine, dacarbazine, toposide, cisplatin, epirubicin, or sorafenib tosylate.
336. The method of any one of claims 326-335, wherein the cancer is relapsed or refractory.
337. A method of inducing the expression of a pattern recognition receptor (PRR) for immune-modulation in a subject, comprising administering to the subject the antibody-drug conjugate of any one of claims 1-324.
338. A method of inducing the expression of a pattern recognition receptors for immunomodulation and inducing a therapeutic response in a subject having cancer, comprising administering to the subject a therapeutically effective amount of the antibody- drug conjugate of any one of claims 1-324.
339. A method of inducing an immune response in a subject, comprising administering to the subject a therapeutically effective amount of the antibody-drug conjugate of any one of claims 1-324.
340. The method of claim 339, wherein the immune response comprises antitumoral immunity.
341. The method of claim 339 or 340, wherein the immune response comprises induction of a PRR (e.g, STING, RIG-I, MDA5).
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201962896983P | 2019-09-06 | 2019-09-06 | |
| US62/896,983 | 2019-09-06 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2021046426A1 true WO2021046426A1 (en) | 2021-03-11 |
| WO2021046426A9 WO2021046426A9 (en) | 2022-02-17 |
Family
ID=74852734
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2020/049513 Ceased WO2021046426A1 (en) | 2019-09-06 | 2020-09-04 | Cyclic dinucleotide sting agonists tethered to a pd-1 or ctla-4 antibodies |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2021046426A1 (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11584774B2 (en) | 2017-09-11 | 2023-02-21 | F-star Therapeutics, Inc. | Compounds, compositions, and methods for the treatment of disease |
| US11707531B2 (en) | 2017-09-11 | 2023-07-25 | F-star Therapeutics, Inc. | Compounds, compositions, and methods for the treatment of disease |
| WO2024100449A1 (en) * | 2022-11-08 | 2024-05-16 | Legochem Biosciences, Inc. | Sting agonists |
| CN118754922A (en) * | 2024-09-09 | 2024-10-11 | 中国人民解放军军事科学院军事医学研究院 | An allyl-cyanoethyl nucleoside dimer and its preparation method and application in oligonucleotide synthesis |
| US12178876B2 (en) | 2023-04-18 | 2024-12-31 | Astrazeneca, Ab | Conjugates comprising cleavable linkers |
| US12398124B2 (en) | 2017-03-29 | 2025-08-26 | Ligachem Biosciences Inc. | Pyrrolobenzodiazepine dimer prodrug and ligand-linker conjugate compound of the same |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20150374816A1 (en) * | 2013-02-18 | 2015-12-31 | Spring Bank Pharmaceuticals, Inc. | Design of short oligonucleotides as vaccine adjuvants and therapeutic agents |
| WO2018100558A2 (en) * | 2016-12-01 | 2018-06-07 | Takeda Pharmaceutical Company Limited | Cyclic dinucleotide |
| WO2018200812A1 (en) * | 2017-04-28 | 2018-11-01 | Novartis Ag | Antibody conjugates comprising sting agonist |
| WO2019051489A1 (en) * | 2017-09-11 | 2019-03-14 | Sperovie Biosciences, Inc. | Compounds, compositions, and methods for the treatment of disease |
-
2020
- 2020-09-04 WO PCT/US2020/049513 patent/WO2021046426A1/en not_active Ceased
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20150374816A1 (en) * | 2013-02-18 | 2015-12-31 | Spring Bank Pharmaceuticals, Inc. | Design of short oligonucleotides as vaccine adjuvants and therapeutic agents |
| WO2018100558A2 (en) * | 2016-12-01 | 2018-06-07 | Takeda Pharmaceutical Company Limited | Cyclic dinucleotide |
| WO2018200812A1 (en) * | 2017-04-28 | 2018-11-01 | Novartis Ag | Antibody conjugates comprising sting agonist |
| WO2019051489A1 (en) * | 2017-09-11 | 2019-03-14 | Sperovie Biosciences, Inc. | Compounds, compositions, and methods for the treatment of disease |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12398124B2 (en) | 2017-03-29 | 2025-08-26 | Ligachem Biosciences Inc. | Pyrrolobenzodiazepine dimer prodrug and ligand-linker conjugate compound of the same |
| US11584774B2 (en) | 2017-09-11 | 2023-02-21 | F-star Therapeutics, Inc. | Compounds, compositions, and methods for the treatment of disease |
| US11707531B2 (en) | 2017-09-11 | 2023-07-25 | F-star Therapeutics, Inc. | Compounds, compositions, and methods for the treatment of disease |
| US12187761B2 (en) | 2017-09-11 | 2025-01-07 | Invox Pharma Limited | Compounds, compositions, and methods for the treatment of disease |
| US12390668B2 (en) | 2017-09-11 | 2025-08-19 | Invox Pharma Limited | Compounds, compositions, and methods for the treatment of disease |
| WO2024100449A1 (en) * | 2022-11-08 | 2024-05-16 | Legochem Biosciences, Inc. | Sting agonists |
| US12178876B2 (en) | 2023-04-18 | 2024-12-31 | Astrazeneca, Ab | Conjugates comprising cleavable linkers |
| US12337038B2 (en) | 2023-04-18 | 2025-06-24 | Astrazeneca Ab | Conjugates comprising cleavable beta-glucuronide-containing linkers |
| CN118754922A (en) * | 2024-09-09 | 2024-10-11 | 中国人民解放军军事科学院军事医学研究院 | An allyl-cyanoethyl nucleoside dimer and its preparation method and application in oligonucleotide synthesis |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2021046426A9 (en) | 2022-02-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2021046426A1 (en) | Cyclic dinucleotide sting agonists tethered to a pd-1 or ctla-4 antibodies | |
| US10392419B2 (en) | Modified cyclic dinucleotide compounds | |
| US20230000993A1 (en) | Silicon based drug conjugates and methods of using same | |
| CA3038860C (en) | Cyclic dinucleotide compounds | |
| JP2023178515A (en) | 1,2,4-oxadiazole and thiadiazole compounds as immunomodulators | |
| TWI759301B (en) | Pegylated carfilzomib compounds | |
| CA2829736C (en) | N-carboxyalkylauristatins and use thereof | |
| KR101413955B1 (en) | Aziridinyl-epothilone compounds | |
| CA3198788A1 (en) | Camptothecine antibody-drug conjugates and methods of use thereof | |
| CN107405336A (en) | 1,3,4 oxadiazoles and thiadiazole compound as immunomodulator | |
| AU2020214507A1 (en) | Bi-ligand drug conjugate and use thereof | |
| CN107427497A (en) | 1,3,4 oxadiazoles and thiadiazole compound as 3 substitutions of immunomodulator | |
| CN107427476A (en) | 1,2,4 oxadiazoles and thiadiazole compound as 3 substitutions of immunomodulator | |
| CN107427491A (en) | Therapeutic cyclic compound as immunomodulator | |
| CA3049791A1 (en) | Tumor targeting conjugates and methods of use thereof | |
| CA3006249A1 (en) | Antibody-drug conjugates comprising branched linkers and methods related thereto | |
| CA2833690A1 (en) | Novel binder-drug conjugates (adcs) and their use | |
| CA3168368A1 (en) | Antibody-drug conjugate including novel cyclic dinucleotide derivative | |
| CA3118026A1 (en) | Fused heterocyclic benzodiazepine derivatives and uses thereof | |
| US20210009627A1 (en) | Cyclic dinucleotide compounds containing 2-aza-hypoxanthine or 6h-pytazolo[1,5-d][1,2,4]trizain-7-one as string agonists | |
| CA3210473A1 (en) | Branched linkers for antibody-drug conjugates and methods of use thereof | |
| US20210024567A1 (en) | Modified cyclic dinucleotide compounds | |
| CA3202759A1 (en) | Mcl-1 inhibitor antibody-drug conjugates and methods of use | |
| CA3224741A1 (en) | Ligand-drug conjugate and use thereof | |
| CN120457136A (en) | STING agonists |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20861407 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 20861407 Country of ref document: EP Kind code of ref document: A1 |