WO2020112195A1 - Compositions, technologies and methods of using plerixafor to enhance gene editing - Google Patents
Compositions, technologies and methods of using plerixafor to enhance gene editing Download PDFInfo
- Publication number
- WO2020112195A1 WO2020112195A1 PCT/US2019/049018 US2019049018W WO2020112195A1 WO 2020112195 A1 WO2020112195 A1 WO 2020112195A1 US 2019049018 W US2019049018 W US 2019049018W WO 2020112195 A1 WO2020112195 A1 WO 2020112195A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- gene
- cells
- gene editing
- triplex
- plerixafor
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/16—Hydrolases (3) acting on ester bonds (3.1)
- C12N9/22—Ribonucleases [RNase]; Deoxyribonucleases [DNase]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/15—Nucleic acids forming more than 2 strands, e.g. TFOs
- C12N2310/152—Nucleic acids forming more than 2 strands, e.g. TFOs on a single-stranded target, e.g. fold-back TFOs
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/31—Chemical structure of the backbone
- C12N2310/318—Chemical structure of the backbone where the PO2 is completely replaced, e.g. MMI or formacetal
- C12N2310/3181—Peptide nucleic acid, PNA
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2320/00—Applications; Uses
- C12N2320/30—Special therapeutic applications
- C12N2320/31—Combination therapy
Definitions
- Substitution at the gamma position creates chirality and provides helical pre-organization to the PNA oligomer, and may yield substantially increased binding affinity to the target DNA (Rapireddy, et al., Biochemistry, 50(19):3913-8 (2011), He et al.,“The Structure of a g-modified peptide nucleic acid duplex”, Mol. BioSyst. 6:1619-1629 (2010); and Sahu et al., “Synthesis and Characterization of Conformationally Preorganized, (R)- Diethylene Glycol-Containing g-Peptide Nucleic Acids with Superior Hybridization Properties and Water Solubility”, J. Org. Chem, 76:5614- 5627) (2011)).
- Other advantageous properties can be conferred depending on the chemical nature of the specific substitution at the gamma position (the “R” group in the illustration of the Chiral gRNA, above).
- PNAs may require even fewer purines to a form a triple helix.
- a triple helix may be formed with a target sequence containing fewer than 8 purines. Therefore, PNAs may be designed to target a site on duplex nucleic acid containing between 6-30 polypurine:polypyrimidines, preferably, 6-25 polypurine:polypyrimidines, more preferably 6-20
- Specificity and binding affinity of the pseudocomplemetary oligonucleotides may vary from oligomer to oligomer, depending on factors such as length, the number of G:C and A:T base pairs, and the formulation.
- recombination for example, a substitution, a deletion, or an insertion of one or more nucleotides.
- Successful recombination of the donor sequence results in a change of the sequence of the target region.
- This strategy exploits the ability of a triplex to provoke DNA repair, potentially increasing the probability of recombination with the homologous donor DNA. It is understood in the art that in most cases, a greater number of homologous positions within the donor fragment will increase the probability that the donor fragment will be recombined into the target sequence, target region, or target site.
- Non-tethered or unlinked fragments may range in length from 20 nucleotides to several thousand.
- the donor oligonucleotide molecules, whether linked or unlinked, can exist in single stranded (ss) or double stranded form (ds) (e.g., ssDNA, dsDNA).
- the donor fragment to be recombined can be linked or un-linked to the triplex-forming molecules.
- the linked donor fragment may range in length from 4 nucleotides to 100 nucleotides, preferably from 4 to 80 nucleotides in length.
- the unlinked donor fragments may have a much broader range, from 20 nucleotides to several thousand nucleotides in length.
- the oligonucleotide donor is between 25 and 80 nucleobases.
- the non-tethered donor nucleotide is about 50 to 60 nucleotides in length.
- the outer surface of the particle may be treated using a mannose amine, thereby mannosylating the outer surface of the particle. This treatment may cause the particle to bind to the target cell or tissue at a mannose receptor on the antigen presenting cell surface.
- the nanoparticles may further include epithelial cell targeting molecules, such as, antibodies or bioactive fragments thereof that recognize and bind to epitopes displayed on the surface of epithelial cells, or ligands which bind to an epithelial cell surface receptor.
- epithelial cell targeting molecules such as, antibodies or bioactive fragments thereof that recognize and bind to epitopes displayed on the surface of epithelial cells, or ligands which bind to an epithelial cell surface receptor.
- suitable receptors include, but are not limited to, IgE Fc receptors, EpCAM, selected carbohydrate specificites, dipeptidyl peptidase, and E-cadherin.
- the plerixafor is contacted with the target cell prior to the gene editing technology and/or donor oligonucleotide.
- the plerixafor can be contacted with the target cell, for example, 1, 2, 3, 4, 5, 6, 8, 10, 12, 18, or 24 hours, or 1, 2, 3, 4, 5, 6, or 7 days, or any combination thereof prior to the gene editing technology and/or donor oligonucleotide.
- somatic cells may be harvested from a host.
- somatic cells may be harvested from a host.
- PNA and donor DNA were mixed at a 2:1 molar ratio and added dropwise to the PLGA solution under vortex.
- DNA was added dropwise at a molar ratio of 2 nmoles/mg of polymer.
- the resulting mixture was sonicated three times for 10 seconds using an amplitude of 38%.
- the water-in-oil emulsion was subsequently added dropwise to a surfactant solution containing polyvinyl alcohol (5% w/v). Following the second emulsion, the sonication step was repeated as described.
- the resulting nanoparticle solution was added to 25 ml of a 0.3% PVA solution and allowed to stir for 3 hours at room temperature.
- the Townes mouse model was developed by Ryan TM, Ciavatta DJ, Townes TM.,“Knockout-transgenic mouse model of sickle cell disease.” Science. 1997 Oct 31 ;278(5339): 873-6. PMID: 9346487.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Zoology (AREA)
- Organic Chemistry (AREA)
- Biomedical Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Wood Science & Technology (AREA)
- Medicinal Chemistry (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Biochemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Microbiology (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Physics & Mathematics (AREA)
- Biophysics (AREA)
- Plant Pathology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Compositions, technologies and methods for improved gene editing are disclosed. In a preferred method, gene editing involves use of plerixafor as an agent to enhance targeted editing by a gene editing technology, such as a PNA, in combination with a donor oligonucleotide. Genomic modification occurs at a higher frequency when cells are contacted with plerixafor, gene editing technology and donor oligonucleotide, as compared to the absence of the plerixafor. The methods are suitable for in vivo, in vitro or ex vivo approaches to gene editing. Cells modified according to the disclosed methods can be administered to a subject in need thereof in an effective amount to treat a symptom of a disease or disorder. Also disclosed are nanoparticle compositions for intracellular delivery of the gene editing technologies.
Description
COMPOSITIONS, TECHNOLOGIES AND METHODS OF USING PLERIXAFOR TO ENHANCE GENE EDITING
CROSS-REFERENCE TO RELATED APPLICATION
This application claims the benefit of and priority to U.S.S.N.
62/773,977, filed November 30, 2018, which is specifically incorporated by reference herein in its entirety.
STATEMENT REGARDING FEDERALLY SPONSORED RESEARCH
This invention was made with Government Support under HL125892 and All 12443 awarded by the National Institutes of Health. The
Government has certain rights in the invention.
REFERENCE TO THE SEQUENCE LISTING
The Sequence Listing submitted as a text file named
“YU_7505_PCT” created on August 28, 2019, and having a size of 3,002 bytes is hereby incorporated by reference pursuant to 37 C.F.R. § 1.52(e)(5).
FIELD OF THE INVENTION
The invention is generally related to the field of gene editing technology, and more particularly, to methods of improving gene editing by use of plerixafor, or a compound related thereto.
BACKGROUND OF THE INVENTION
Gene editing provides an attractive strategy for treatment of inherited genetic disorders such as sickle cell anemia and b-thalassemia. Genes can be selectively edited by several methods, including targeted nucleases such as zinc finger nucleases (ZFNs) (Haendel, et al., Gene Ther., 11:28-37 (2011)) and CRISPRs (Yin, et al., Nat. Biotechnol., 32:551-553 (2014)), short fragment homologous recombination (SFHR) (Goncz, et al.,
Oligonucleotides, 16:213-224 (2006)), or triplex-forming oligonucleotides (TFOs) (Vasquez, et al., Science, 290:530-533 (2000)). While there has been widespread focus on targeted nucleases such as CRISPR/Cas9 technology because of its ease of use and facile reagent design (Doudna, et al., Science, 346:1258096 (2014)), the CRISPR approach introduces an active nuclease into cells, which can lead to off-target cleavage in the genome (Cradick, et
al., Nucleic Acids Res., 41:9584-9592 (2013)), a problem that so far has not been eliminated.
Alternatives have been developed such as triplex-forming peptide nucleic acid (PNA) oligomers which recruit the cell’s endogenous DNA repair systems to initiate site-specific modification of the genome when single-stranded“donor DNAs” are co-delivered as templates (Rogers, et al., Proc. Natl. Acad. Sci. USA, 99:16695-16700 (2002)).
Historically however, the efficiency of gene modification is generally low. Accordingly, there remains a need for compositions, technologies and methods to increase the efficiency of gene editing.
It is therefore an object of the invention to provide compositions, technologies and methods to increase the frequency of gene modification.
SUMMARY OF THE INVENTION
Plerixafor increases the efficiency of gene editing technologies in a cell or a subject. In vivo, plerixafor (MOZOBIL®) has been shown to release stem cells from the bone marrow and mobilize them into the peripheral circulation by selectively blocking the interaction between CXCR4 (on the surface of stem cells) with SDF-1 (on the surface of bone marrow stromal cells). However, the experiments described in the Examples show that plerixafor has a direct effect of enhancing the efficiency of targeted gene editing, even when the cells are treated in vitro or ex vivo or otherwise outside the bone marrow niche. Without being bound by theory, plerixafor appears to stimulate gene editing by PNAs/donor DNAs by a mechanism independent of its use as a stem cell mobilizer in the body of a subject.
Thus, compositions and technologies for enhanced, targeted gene editing and methods of use thereof are provided. An exemplary method of modifying the genome of a cell can include in vivo, in vitro, or ex vivo contacting the cell with an effective amount of (i) plerixafor or a compound related thereto such as an analog, derivative, or pharmaceutically acceptable salt thereof (collectively referred to herein as“plerixafor” unless otherwise specified), and (ii) a gene editing technology that can induce genomic modification of the cell (e.g. , triplex-forming molecules,
pseudocomplementary oligonucleotides, CRISPR systems, zinc finger nucleases (ZFN), transcription activator-like effector nucleases (TALEN),
small fragment homologous replacement (e.g., polynucleotide small DNA fragments (SDFs)), single-stranded oligodeoxynucleotide-mediated gene modification (e.g., ssODN/SSOs) and intron encoded meganucleases.
Genomic modification can occur at a higher frequency in a population of cells contacted with both (i) and (ii), than in an equivalent population contacted with (ii) in the absence of (i).
The method can include contacting the cells with a donor oligonucleotide including, for example, a sequence that corrects or induces a mutation(s) in the cell’s genome by insertion or recombination of the donor induced or enhanced by the gene editing technology. The donor
oligonucleotide (e.g., DNA) may be single stranded or double stranded. Preferably, the donor oligonucleotide is single stranded DNA. The plerixafor, gene editing technology, and/or donor oligonucleotide can be contacted with the cell in any order. A preferred gene editing technology is a triplex forming molecule, such as a peptide nucleic acid (PNA).
In some embodiments, the cell’s genome has a mutation underlying a disease or disorder, for example, genetic disorders such as hemophilia, muscular dystrophy, globinopathies, cystic fibrosis, xeroderma
pigmentosum, lysosomal storage diseases, immune deficiency syndromes such as X-linked severe combined immunodeficiency and ADA deficiency, tyrosinemia, Fanconi anemia, the red cell disorder spherocytosis, alpha- 1- anti-trypsin deficiency, Wilson’s disease, Leber’s hereditary optic neuropathy, and chronic granulomatous disorder. The globinopathy can be sickle cell anemia or beta-thalassemia. The lysosomal storage disease can be Gaucher's disease, Fabry disease, or Hurler syndrome. In some
embodiments, the method induces a mutation that reduces HIV infection, for example, by reducing an activity of a cell surface receptor that facilitates entry of HIV into the cell.
The contacting of the compositions or technologies with the cell can occur in vivo, in vitro or ex vivo. The ex vzvo-treated cells may be hematopoietic stem cells or hematopoietic progenitor cells, which then can be administered to a subject in need thereof in an effective amount to treat one or more symptoms of a disease or disorder.
Any of the compositions including the plerixafor, gene editing technology, and/or donor oligonucleotide can be packaged together or separately in nanoparticles. The nanoparticles may be formed from polyhydroxy acids, such as poly(lactic-co-glycolic acid) (PLGA), alone or in a blend with poly (beta- amino) esters (PBAEs). The nanoparticles may be prepared by double emulsion or nanoprecipitation. The gene editing technology, the donor oligonucleotide or a combination thereof may be complexed with a polycation prior to preparation of the nanoparticles.
Functional molecules such as targeting moieties, cell-penetrating peptides, or a combination thereof can be associated with, linked, conjugated, or otherwise directly or indirectly attached to the plerixafor, the gene editing technology, the donor oligonucleotide, nanoparticles, or a combination thereof.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1A is a schematic representation of the binding site position of tcPNA2 targeting the beta globin gene in the vicinity of the sickle cell disease (SCD) mutation, and includes the sequences of SCD-tcPNA2 ((SEQ ID NO:4) without gamma side chain substitutions) and SCD-tcPNA2A ((SEQ ID NO:4) with mini-PEG gamma side chain substitutions (underlined residues)). Figure IB is a schematic representation of the experimental procedure for performing and assessing gene editing in bone marrow cells, as described in Example 1. Figure 1C is a bar graph showing the percentage of gene editing following treatment of bone marrow cells with or without tcPNA2/donor DNA-containing nanoparticles. Figure ID is a bar graph showing the percentage of gene editing in bone marrow cells from Townes mice treated with tcPNA2A/donor DNA-containing nanoparticles in combination with stem cell factor (SCF), erythropoietin (EPO), or Plerixafor.
DETAIFED DESCRIPTION OF THE INVENTION
I. Definitions
As used herein, the term“modulate” means to regulate, alter, adapt, or adjust to a certain measure or proportion. Modulation encompasses inhibition, reduction or repression, activation, promotion or enhancement, and competition.
As used herein, the term“subject” means any individual who is the target of administration. The subject can be a vertebrate, for example, a mammal. Thus, the subject can be a human. The term does not denote a particular age or sex.
As used herein, the terms“effective amount” or“therapeutically effective amount” means that the amount of the composition or technology used is of sufficient quantity to ameliorate one or more causes or symptoms of a disease or disorder. Such amelioration only requires a reduction or alteration, not necessarily elimination. The precise dosage will vary according to a variety of factors such as subject-dependent variables (e.g. , age, immune system health, etc.), the disease or disorder being treated, as well as the route of administration and the pharmacokinetics of the agent being administered.
As used herein, the term“treat” refers to the medical management of a patient with the intent to cure, ameliorate, stabilize, or prevent a disease, pathological condition, or disorder. This term includes active treatment, that is, treatment directed specifically toward the improvement of a disease, pathological condition, or disorder, and also includes causal treatment, that is, treatment directed toward removal of the cause of the associated disease, pathological condition, or disorder. In addition, this term includes palliative treatment, that is, treatment designed for the relief of symptoms rather than the curing of the disease, pathological condition, or disorder; preventative treatment, that is, treatment directed to minimizing or partially or completely inhibiting the development of the associated disease, pathological condition, or disorder; and supportive treatment, that is, treatment employed to supplement another specific therapy directed toward the improvement of the associated disease, pathological condition, or disorder.
As used herein,“targeting moiety” is a substance which can direct a composition or compound such as a nanoparticle or gene editing technology to a receptor site on a selected cell or tissue type or serve to couple or attach another molecule to the composition or compound. As used herein,“direct” refers to causing a molecule to preferentially attach to a selected cell or tissue type. This can be used to direct cellular materials, molecules, or drugs, as discussed below.
As used herein, the term“inhibit” or“reduce” means to decrease an activity, response, condition, disease, or other biological parameter. This can include, but is not limited to, the complete ablation of the activity, response, condition, or disease. This may also include, for example, a 10% reduction in the activity, response, condition, or disease as compared to the native or control level. Thus, the reduction can be a 10, 20, 30, 40, 50, 60, 70, 80, 90, 100%, or any amount of reduction in between as compared to native or control levels.
As used herein, the term“small molecule” generally refers to an organic molecule that is less than about 2000 g/mol in molecular weight, less than about 1500 g/mol, less than about 1000 g/mol, less than about 800 g/mol, or less than about 500 g/mol. Small molecules are non-polymeric and/or non-oligomeric.
Recitation of ranges of values herein are merely intended to serve as a shorthand method of referring individually to each separate value falling within the range, unless otherwise indicated herein, and each separate value is incorporated into the specification as if it were individually recited herein. II. Plerixafor and Compounds Related Thereto
As illustrated in the Examples below, it has been discovered that plerixafor and compounds related thereto (jointly referred to herein as “plerixafor” unless otherwise specified), can be used to increase the efficiency of gene editing technologies. Accordingly, compositions, technologies and methods of increasing the efficiency of a gene editing technology, such as a triplex-forming PNA and donor DNA (optionally in a nanoparticle composition), a CRISPR Cas9 system and donor DNA, or others have been developed.
The plerixafor can be a compound of Formula (I):
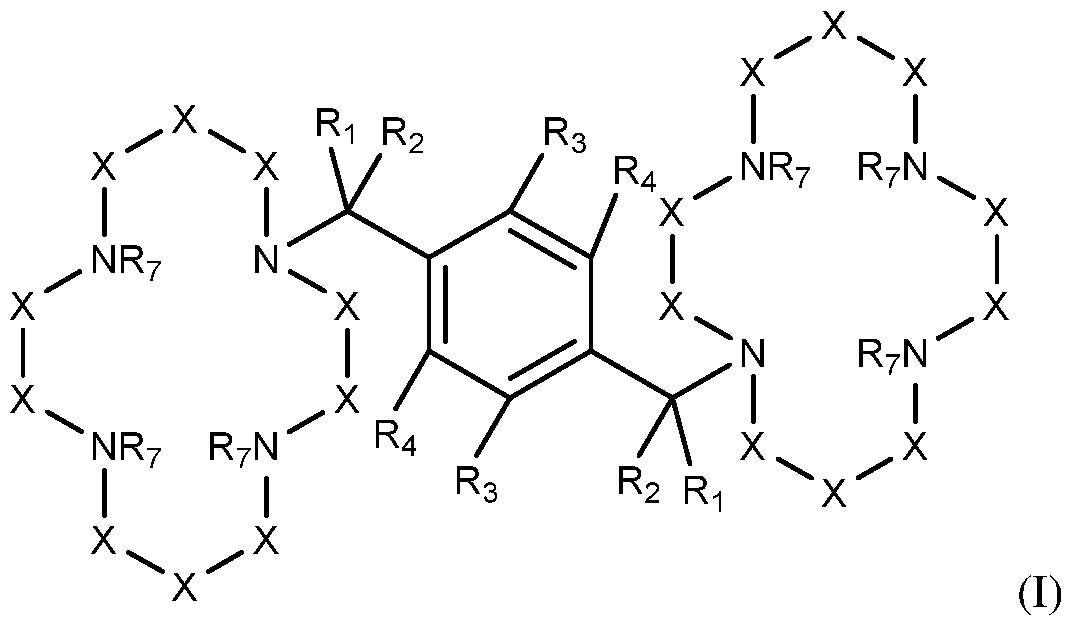
or a pharmaceutically acceptable salt thereof, wherein,
each Ri or R2 is independently hydrogen, deuterium or fluoro;
each R3 or R4 is independently hydrogen, deuterium, or halo;
or R3 and R4 taken together form a cyclic aromatic or a saturated or unsaturated non-aromatic ring of 4 to 7 atoms, wherein the aromatic or non aromatic ring can optionally be heterocyclic;
each X is independently -C( RsRe). C=0 or C=S, wherein each R5 and each R6 is independently hydrogen, deuterium, OH, fluoro, alkyl, or alkoxy; and
each R7 is independently, hydrogen, deuterium, or alkyl. In some embodiments, each Ri or R2 is independently hydrogen. In some embodiments, each R3 or R4 is independently hydrogen. In some embodiments, each X is independently C( RsRe) (e.g., CH2). In some embodiments, each R7 is independently hydrogen.
In some embodiments, the compound of Formula (I) is plerixafor or a pharmaceutically acceptable salt thereof. Plerixafor is a hematopoietic stem cell mobilizer with the chemical name 1, l'-[l,4-phenylenebis (methylene)] - bis- 1,4,8,11- tetraazacyclotetradecane. It has the molecular formula
C28H54N8. The molecular weight of plerixafor is 502.79 g/mol. The structural formula is provided below:

Plerixafor is provided commercially as a white to off-white crystalline solid. It is hygroscopic. Plerixafor has a typical melting point of 131.5 °C. The partition coefficient of plerixafor between 1-octanol and pH 7 aqueous buffer is < 0.1. Plerixafor is a strong base; all eight nitrogen atoms accept protons readily. The two macrocyclic rings form chelate complexes with bivalent metal ions, especially zinc, copper and nickel, as well as cobalt and rhodium. See also U.S. Patent No. 5,021,409, Murrer et al.
Also included are pharmaceutically acceptable salts of plerixafor or a structurally related compounds. The term“plerixafor and compounds related thereto” includes plerixafor, analogs of plerixafor, and derivatives of plerixafor in any form, such as a base (zwitter ion), pharmaceutically acceptable salts of plerixafor, e.g., pharmaceutically acceptable acid addition salts, hydrates or solvates of the base or salt, as well as anhydrates, and also amorphous, or crystalline forms.
Without being bound by theory, it is believed that plerixafor acts by inhibiting or antagonizing the CXCR4 chemokine receptor and blocking binding of its cognate ligand, stromal cell-derived factor- la (SDF-la), to mobilize stem cells. SDF-la and CXCR4 play a role in the trafficking and homing of human hematopoietic stem cells (HSCs) to the marrow compartment. Once in the marrow, stem cell CXCR4 can act to help anchor these cells to the marrow matrix, either directly via SDF-la or through the induction of other adhesion molecules. Disruption of the SDF- 1(CXCL12)/CXCR4 retention axis by plerixafor in the bone marrow can release a whole host of progenitor cells without the necessity of priming. Treatment with plerixafor has resulted in leukocytosis and elevations in circulating hematopoietic progenitor cells in mice, dogs and humans. CD34+
cells mobilized by plerixafor were capable of engraftment with long-term repopulating capacity up to one year in canine transplantation models.
In some embodiments, the compound is a compound other than plerixafor. Exemplary compounds include, for example,
tetrahydroquinolines, N-substituted indoles, l,4-phenylenebis(methylene) derivatives, diketopiperazine mimetics, and peptides, for example
AMD3465, AMD070, T140, FC131, FC122. Additional exemplary compounds are known in the art; see, for example, (e.g., Ko, et ah, Nano Research., 11(4):2159-2172 (2018); Hatse, et ah, Biochem,
Pharmacol., 70(5):752-61 (2005); and Debnath, et al. Theranostics, 3(1):41- 75 (2013).
Dosages for plerixafor are known in the art, and have been disclosed in clinical trials for mobilization of stem cells (e.g., DiPersio, et ah, J. Clin. Oncol., 27(28):4767-73, (2009); DiPersio, et ah, Blood., 113(23):5720-6, (2009)).
For in vivo application, the dosage can be selected by the practitioner based on known, preferred human dosages. Exemplary dosages for plerixafor and compounds related thereto may be about 0.01 mg/kg to about 10 mg/kg. Plerixafor or a compound related thereto, administered in an amount from about 0.01 mg/kg to about 1.0 mg/kg, or any amount in between. For example, plerixafor is administered in a dosage of 0.16 to 0.24 mg/kg for cancer therapy.
For in vitro and ex vivo applications, the concentration of plerixafor or a compound related thereto is typically between about 10 nM and about 10 mM, but other concentrations may be used.
In the in vitro DNA repair assays discussed below, the concentration of plerixafor was between 1 and 900 mM. Ex vivo studies were performed in the presence of plerixafor at a concentration of about 100 mM.
Thus, a preferred in vitro/ex vivo concentration based on current results is 100 mM.
III. Gene Editing Technology
A described herein, gene editing technologies are preferably used in combination with plerixafor. Exemplary gene editing technologies include,
but are not limited to, triplex-forming compositions, pseudocomplementary oligonucleotides, CRISPR/Cas, zinc finger nucleases, and TALENs, each of which are discussed in more detail below. Some gene editing technologies are used in combination with a donor oligonucleotide. In some
embodiments, the gene editing technology is the donor oligonucleotide, which can be used alone to modify genes. Strategies include, but are not limited to, small fragment homologous replacement (e.g., polynucleotide small DNA fragments (SDFs)), single-stranded oligodeoxynucleotide- mediated gene modification (e.g., ssODN/SSOs) and other described in Sargent, Oligonucleotides, 21(2): 55-75 (2011)), and elsewhere. Other suitable gene editing technologies include, but are not limited to, intron encoded meganucleases that are engineered to change their target specificity. See, e.g., Arnould, et al., Protein Eng. Des. Sel., 24(l-2):27-31 (2011)).
In some embodiments, the gene editing technology modifies a target sequence within a genome by reducing or preventing expression of the target sequence. The gene editing technology can induce single-stranded or double- stranded breaks in the target sequence. The gene editing technology can induce formation of a triplex within the target sequence.
Gene editing technologies include CRISPR systems, zinc finger nucleases (ZFN), transcription activator-like effector nucleases (TALEN), small fragment homologous replacement (e.g., polynucleotide small DNA fragments (SDFs)), single-stranded oligodeoxynucleotide-mediated gene modification (e.g., ssODN/SSOs), and intron encoded meganucleases. The gene editing technology can be a triplex forming composition. The gene editing technology can be a pseudocomplementary oligonucleotide or pseudocomplementary PNA oligomer.
A. Triplex-Forming Molecules
1. Compositions
Compositions containing“triplex- forming molecules,” that bind to duplex DNA in a sequence- specific manner to form a triple- stranded structure include, but are not limited to, triplex-forming oligonucleotides (TFOs), peptide nucleic acids (PNA), and“tail clamp” PNA (tcPNA) are provided. The triplex-forming molecules can be used to induce site-specific homologous recombination in mammalian cells when combined with donor
DNA molecules. The donor DNA molecules can contain mutated nucleic acids relative to the target DNA sequence. This is useful to activate, inactivate, or otherwise alter the function of a polypeptide or protein encoded by the targeted duplex DNA. Triplex-forming molecules include triplex forming oligonucleotides and peptide nucleic acids (PNAs). Triplex-forming molecules are described in U.S. Patents 5,962,426, 6,303,376, 7,078,389, 7,279,463, 8,658,608, U.S. Published Application Nos. 2003/0148352, 2010/0172882, 2011/0268810, 2011/0262406, 2011/0293585, and published PCT application numbers WO 1995/001364, WO 1996/040898, WO 1996/039195, WO 2003/052071, WO 2008/086529, WO 2010/123983, WO 2011/053989, WO 2011/133802, WO 2011/13380, Rogers, et al„ Proc Natl Acad Sci USA, 99:16695-16700 (2002), Majumdar, et al., Nature Genetics, 20:212-214 (1998), Chin, et al., Proc Natl Acad Sci USA, 105:13514-13519 (2008), and Schleifman, et al., Chem Biol., 18:1189-1198 (2011). As discussed in more detail below, triplex- forming molecules are typically single- stranded oligonucleotides that bind to polypyrimidine :polypurine target motif in a double stranded nucleic acid molecule to form a triple- stranded nucleic acid molecule. The single-stranded
oligonucleotide/oligomer typically includes a sequence substantially complementary to the polypurine strand of the polypyrimidine:polypurine target motif via Hoogsteen or reverse Hoogsteen binding.
a. Triplex-forming Oligonucleotides (TFOs)
Triplex-forming oligonucleotides (TFOs) are defined as
oligonucleotides which bind as third strands to duplex DNA in a sequence specific manner. The oligonucleotides are synthetic or isolated nucleic acid molecules which selectively bind to or hybridize with a predetermined target sequence, target region, or target site within or adjacent to a human gene so as to form a triple-stranded structure.
Preferably, the oligonucleotide is a single-stranded nucleic acid molecule between 7 and 40 nucleotides in length, most preferably 10 to 20 nucleotides in length for in vitro mutagenesis and 20 to 30 nucleotides in length for in vivo mutagenesis. The nucleobase (sometimes referred to herein simply as“base”) composition may be homopurine or
homopyrimidine. Alternatively, the nucleobase composition may be polypurine or polypyrimidine. However, other compositions are also useful.
The oligonucleotides are preferably generated using known DNA synthesis procedures. In one embodiment, oligonucleotides are generated synthetically. Oligonucleotides can also be chemically modified using standard methods that are well known in the art.
The nucleobase sequence of the oligonucleotides/oligomer is selected based on the sequence of the target sequence, the physical constraints imposed by the need to achieve binding of the oligonucleotide/oligomer within the major groove of the target region, and the need to have a low dissociation constant (Kd) for the oligo/target sequence complex. The oligonucleotides/oligomers have a nucleobase composition which is conducive to triple-helix formation and is generated based on one of the known structural motifs for third strand binding (e.g. Hoogsteen binding). The most stable complexes are formed on polypurine:polypyrimidine elements, which are relatively abundant in mammalian genomes. Triplex formation by TFOs can occur with the third strand oriented either parallel or anti-parallel to the purine strand of the nucleic acid duplex. In the anti parallel, purine motif, the triplets are G.G:C and A.A:T, whereas in the parallel pyrimidine motif, the canonical triplets are C+.G:C and T.A:T. The triplex structures can be stabilized by one, two or three Hoogsteen hydrogen bonds (depending on the nucleobase) between the bases in the TFO strand and the purine strand in the duplex. A review of base compositions and binding properties for third strand binding oligonucleotides and/or peptide nucleic acids is provided in, for example, U.S. Patent No. 5,422,251, Bentin et al., Nucl. Acids Res. , 34(20): 5790-5799 (2006), and Hansen et al., Nucl. Acids Res., 37(13): 4498-4507 (2009).
Preferably, the oligonucleotide/oligomer binds to or hybridizes to the target sequence under conditions of high stringency and specificity. Most preferably, the oligonucleotides/oligomers bind in a sequence-specific manner within the major groove of duplex DNA. Reaction conditions for in vitro triple helix formation of an oligonucleotide/oligomer to a double stranded nucleic acid sequence vary from oligo to oligo, depending on factors such as polymer length, the number of G:C and A:T base pairs, and
the composition of the buffer utilized in the hybridization reaction. An oligonucleotide substantially complementary, based on the third strand binding code, to the target region of the double- stranded nucleic acid molecule is preferred.
As used herein, a triplex forming molecule is said to be substantially complementary to a target region when the oligonucleotide has a nucleobase composition which allows for the formation of a triple-helix with the target region. As such, an oligonucleotide/oligomer can be substantially complementary to a target region even when there are non-complementary bases present in the oligonucleotide/oligomer. As stated above, there are a variety of structural motifs available which can be used to determine the nucleobase sequence of a substantially complementary
oligonucleotide/oligomer
b. Peptide nucleic acids (PNA)
In another embodiment, the triplex- forming molecules are peptide nucleic acids (PNAs). Peptide nucleic acids can be considered polymeric molecules in which the sugar phosphate backbone of an oligonucleotide has been replaced in its entirety by repeating substituted or unsubstituted N-(2- aminoethyl)-glycine residues that are linked by amide bonds. In some embodiments, the various nucleobases are linked to the backbone by methylene carbonyl linkages. PNAs maintain spacing of the nucleobases in a manner that is similar to that of an oligonucleotide (DNA or RNA), but because the sugar phosphate backbone has been replaced, classic
(unsubstituted) PNAs are achiral and neutrally charged molecules. Peptide nucleic acids are composed of peptide nucleic acid residues (sometimes referred to as‘residues’). The nucleobases can be any of the standard bases (uracil, thymine, cytosine, adenine and guanine) or any of the modified heterocyclic nucleobases described below.
PNAs can bind to DNA via Watson-Crick hydrogen bonds, but may have binding affinities significantly higher than those of a corresponding nucleotide composed of DNA or RNA. The neutral backbone of PNAs decreases electrostatic repulsion between the PNA and target DNA phosphates. Under in vitro or in vivo conditions that promote opening of the
duplex DNA, PNAs can mediate strand invasion of duplex DNA resulting in displacement of one DNA strand to form a D-loop.
Highly stable triplex PNA:DNA:PNA structures can be formed from a homopurine DNA strand and two PNA strands. The two PNA strands may be two separate PNA molecules (see Bentin et al., Nucl. Acids Res., 34(20): 5790-5799 (2006) and Hansen et al., Nucl. Acids Res., 37(13): 4498-4507 (2009)), or two PNA molecules linked together by a linker of sufficient flexibility to form a single bis-PNA molecule (See: US Pat. No: 6,441,130). In both cases, the PNA molecule(s) may form a triplex“clamp” with one of the strands of the target duplex while displacing the other strand of the duplex target. In this structure, one strand forms Watson-Crick base pairs with the DNA strand in the anti-parallel orientation (the Watson-Crick binding portion), whereas the other strand forms Hoogsteen base pairs to the DNA strand in the parallel orientation (the Hoogsteen binding portion). A homopurine strand allows formation of a stable PNA/DNA/PNA triplex. PNA clamps can form at shorter homopurine sequences than those required by triplex-forming oligonucleotides (TFOs) and also do so with greater stability.
Suitable molecules for use in linkers of bis-PNA molecules include, but are not limited to, 8-amino-3,6-dioxaoctanoic acid, referred to as an O- linker, and 6-aminohexanoic acid. Poly(ethylene) glycol monomers can also be used in bis-PNA linkers. A bis-PNA linker can contain multiple linker residues in any combination of two or more of the foregoing. In some embodiments, the PNA oligomers are linked by three 8-amino-2, 6, 10- trioxaoctanoic acid, three 8-amino-3,6-dioxaoctanoic acid, or three 6- aminohexanoic acid molecules.
PNAs can also include other positively charged moieties to increase the solubility of the PNA and increase the affinity of the PNA for duplex DNA. Commonly used positively charged moieties include the amino acids lysine and arginine (e.g., as additional substituents attached to the C- or N- terminus of the PNA oligomer (or a segment thereof) or as a side-chain modification of the backbone (see Huang et al., Arch. Pharm. Res. 35(3): 517-522 (2012) and Jain et al., JOC, 79(20): 9567-9577 (2014)), although other positively charged moieties may also be useful (See for Example: US
6,326,479). In some embodiments, the PNA oligomer can have one or more‘miniPEG’ side chain modifications of the backbone (see, for example, US Pat. No. 9,193,758 and Sahu et al„ JOC, 76: 5614-5627 (2011)).
Peptide nucleic acids are unnatural synthetic polyamides, prepared using known methodologies, generally as adapted from peptide synthesis processes.
c. Tail clamp peptide nucleic acids (tcPNA)
Although polypurine:polypyrimidine stretches do exist in mammalian genomes, it is desirable to target triplex formation in the absence of this requirement. In some embodiments such as PNA, triplex-forming molecules include a“tail” added to the end of the Watson-Crick binding portion.
Adding additional nucleobases, known as a“tail” or“tail clamp”, to the Watson-Crick binding portion that bind to the target strand outside the triple helix further reduces the requirement for a polypurine:polypyrimidine stretch. This reduced requirement for a polypurine :polypyrimidine stretch can increases the number of potential target sites while also increasing the specificity for the potential target site. The tail is most typically added to the end of the Watson-Crick binding sequence furthest from the linker. This molecule therefore may mediate a mode of binding to DNA that
encompasses both triplex and duplex formation (Kaihatsu, et a ,
Biochemistry, 42(47): 13996-4003 (2003); Bentin, et ak, Biochemistry,
42(47): 13987-95 (2003)). For example, if the triplex-forming molecules are tail clamp PNA (tcPNA), the PNA/DNA/PNA triple helix portion and the PNA/DNA duplex portion both produce displacement of the pyrimidine-rich strand, creating an altered helical structure that strongly provokes the nucleotide excision repair pathway and activating the site for recombination with a donor DNA molecule (Rogers, et ak, Proc. Natl. Acad. Sci. U.S.A., 99(26): 16695-700 (2002)).
Tails added to clamp PNAs (sometimes referred to as bis-PNAs) form tail-clamp PNAs (referred to as tcPNAs) that have been described, for example, by Kaihatsu, et ak, Biochemistry, 42(47): 13996-4003 (2003); Bentin, et ak, Biochemistry, 42(47): 13987-95 (2003). tcPNAs are known to bind to DNA more efficiently due to low dissociation constants. The addition of the tail may also increase binding specificity and binding
stringency of the triplex-forming molecules to the target duplex. It has also been found that the addition of a tail to clamp PNA may improve the frequency of recombination of the donor oligonucleotide at the target site compared to PNA without the tail.
In some embodiments a PNA tail clamp system includes one or more the following, preferable in the specified orientation/order:
a positively charged region including one or more positively charged amino acids such as lysine;
a region including a number of PNA subunits with Hoogsteen homology with a target sequence;
a linker;
a region including a number of PNA subunits having Watson Crick homology binding with the target sequence;
a region including a number of PNA subunits having Watson Crick homology binding with a tail target sequence;
a positively charged region including one or more positively charged amino acids subunits, such as lysine.
In some embodiments, one or more PNA monomers of the tail target sequence is modified as disclosed herein.
d. PNA Modifications
PNAs can also include other positively charged moieties to increase the solubility of the PNA and increase the affinity of the PNA for duplex DNA. Commonly used positively charged moieties include the amino acids lysine and arginine, although other positively charged moieties may also be useful. Lysine and arginine residues can be added to a bis-PNA linker or can be added to the carboxy and/or the N-terminus of a PNA strand. Common modifications to PNA are discussed in Sugiyama and Kittaka, Molecules, 18:287-310 (2013)) and Sahu, et ak, J. Org. Chem., 76, 5614-5627 (2011), each of which are specifically incorporated by reference in their entireties, and include, but are not limited to, incorporation of charged amino acid residues, such as lysine at the termini or in the interior part of the oligomer; inclusion of polar groups in the backbone, carboxymethylene bridge, and in the nucleobases; chiral PNAs bearing substituents on the original N-(2- aminoethyl)glycine backbone; replacement of the original aminoethylglycyl
backbone skeleton with a negatively-charged scaffold; conjugation of high molecular weight polyethylene glycol (PEG) to one of the termini; fusion of PNA to DNA to generate a chimeric oligomer, redesign of the backbone architecture, conjugation of PNA to DNA or RNA. These modifications improve solubility but often result in reduced binding affinity and/or sequence specificity.
Triplex-forming peptide nucleic acid (PNA) oligomers having a g (also referred to as“gamma”) modification (also referred to as
“substitution”) in one or more PNA residues (also referred to as“subunits”) of the PNA oligomer are also provided. In some embodiments, the some or all of the PNA residues are modified at the gamma position in the polyamide backbone (yPNAs) as illustrated below (wherein“B” is a nucleobase and “R” is a substitution at the gamma position).
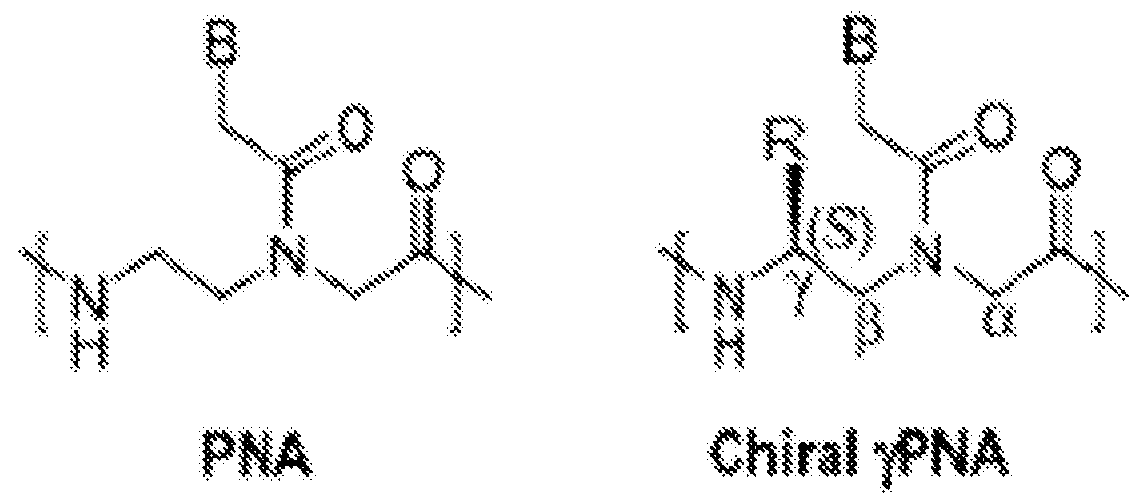
Substitution at the gamma position creates chirality and provides helical pre-organization to the PNA oligomer, and may yield substantially increased binding affinity to the target DNA (Rapireddy, et al., Biochemistry, 50(19):3913-8 (2011), He et al.,“The Structure of a g-modified peptide nucleic acid duplex”, Mol. BioSyst. 6:1619-1629 (2010); and Sahu et al., “Synthesis and Characterization of Conformationally Preorganized, (R)- Diethylene Glycol-Containing g-Peptide Nucleic Acids with Superior Hybridization Properties and Water Solubility”, J. Org. Chem, 76:5614- 5627) (2011)). Other advantageous properties can be conferred depending on the chemical nature of the specific substitution at the gamma position (the “R” group in the illustration of the Chiral gRNA, above).
One class of g substitution is miniPEG, but other residues and side chains can be considered, and even mixed substitutions can be used to tune the properties of the oligomers. “MiniPEG” and“MP” refer to a short
polyethylene glycol moiety, such as a diethylene glycol. MiniPEG- containing yPNAs are conformationally preorganized PNAs that exhibit superior hybridization properties and water solubility as compared to the original PNA design and other chiral yPNAs. Sahu et al., describes yPNAs prepared from E-amino acids that adopt a right-handed helix, and yPNAs prepared from D-amino acids that adopt a left-handed helix. Only the right- handed helical yPNAs hybridize to DNA or RNA with high affinity and sequence selectivity. In the most preferred embodiments, some or all of the PNA residues are miniPEG-containing yPNAs (Sahu, et al., J. Org. Chem., 76, 5614-5627 (2011). In some embodiments, tcPNAs are prepared wherein every other PNA residue on the Watson-Crick binding side of the linker is a miniPEG-containing gRNA. Accordingly, for these embodiments, the tail clamp side of the PNA has alternating classic PNA and miniPEG-containing gRNA residues.
In some embodiments PNA-mediated gene editing are achieved via additional or alternative g substitutions or other PNA chemical modifications including but limited to those introduced above and below. Examples of g substitution with other side chains include that of alanine, serine, threonine, cysteine, valine, leucine, isoleucine, methionine, proline, phenylalanine, tyrosine, aspartic acid, glutamic acid, asparagine, glutamine, histidine, lysine, arginine, and the derivatives thereof. The“derivatives thereof’ herein are defined as those chemical moieties that are covalently attached to these amino acid side chains, for instance, to that of serine, cysteine, threonine, tyrosine, aspartic acid, glutamic acid, asparagine, glutamine, lysine, and arginine.
In addition to yPNAs showing consistently improved gene editing potency the level of off-target effects in the genome remains extremely low. This is in keeping with the lack of any intrinsic nuclease activity in the PNAs (in contrast to ZFNs or CRISPR/Cas9 or TALENS), and may reflect the mechanism of triplex-induced gene editing, which is believed to involve the formation of an altered structure at the target-binding site that engages endogenous high fidelity DNA repair pathways. As discussed above, the
SCF/c-Kit pathway also stimulates these same pathways, providing for enhanced gene editing without increasing off-target risk or cellular toxicity.
Additionally, any of the triplex forming sequences can be modified to include guanidine-G-clamp (“G-clamp”) PNA residues(s) to enhance PNA binding, wherein the G-clamp is linked to the backbone as any other nucleobase would be. yPNAs with substitution of cytosine by G-clamp (9- (2-guanidinoethoxy) phenoxazine), a cytosine analog that can form five Pi- bonds with guanine, and can also provide extra base stacking due to the expanded phenoxazine ring system and substantially increased binding affinity. In vitro studies indicate that a single G-clamp substitution for C can substantially enhance the binding of a PNA-DNA duplex by 23°C (Kuhn, et ak, Artificial DNA, PNA & XNA, 1(1):45-53(2010)). As a result, yPNAs containing G-clamp substitutions can have further increased activity.
The structure of a G-clamp monomer-to-G base pair (G-clamp indicated by the“X”) is illustrated below in comparison to C-G base pair.

Some studies have shown improvements using D-amino acids in peptide synthesis.
In particular embodiments, the gene editing technology includes at least one peptide nucleic acid (PNA) oligomer. The at least one PNA oligomer can be a modified PNA oligomer including at least one modification at a gamma position of a backbone carbon. The modified PNA oligomer can include at least one miniPEG modification at a gamma position of a backbone carbon. The gene editing technology can include at least one
donor oligonucleotide. The gene editing composition can modify a target sequence within a fetal genome.
The PNA can include a Hoogsteen binding peptide nucleic acid (PNA) segment and a Watson-Crick binding PNA segment collectively totaling no more than 50 nucleobases in length, wherein the two segments bind or hybridize to a target region of a genomic DNA comprising a polypurine stretch to induce strand invasion, displacement, and formation of a triple- stranded composition among the two PNA segments and the polypurine stretch of the genomic DNA, wherein the Hoogsteen binding segment binds to the target region by Hoogsteen binding for a length of least five nucleobases, and wherein the Watson-Crick binding segment binds to the target region by Watson-Crick binding for a length of least five nucleobases.
The PNA segments can include a gamma modification of a backbone carbon. The gamma modification can be a gamma miniPEG modification. The Hoogsteen binding segment can include one or more chemically modified cytosines selected from the group consisting of pseudocytosine, pseudoisocytosine, and 5-methylcytosine. The Watson-Crick binding segment can include a sequence of up to fifteen nucleobases that binds to the target duplex by Watson-Crick binding outside of the triplex. The two segments can be linked by a linker. In some embodiments, all of the peptide nucleic acid residues in the Hoogsteen-binding segment only, in the Watson- Crick-binding segment only, or across the entire PNA oligomer include a gamma modification of a backbone carbon. In some embodiments, one or more of the peptide nucleic acid residues in the Hoogsteen-binding segment only or in the Watson-Crick-binding segment only of the PNA oligomer include a gamma modification of a backbone carbon. In some embodiments, alternating peptide nucleic acid residues in the Hoogsteen-binding portion only, in the Watson-Crick-binding portion only, or across the entire PNA oligomer include a gamma modification of a backbone carbon.
In some embodiments, least one gamma modification of the backbone carbon is a gamma miniPEG modification. In some embodiments, at least one gamma modification is a side chain of an amino acid selected from the group consisting of alanine, serine, threonine, cysteine, valine,
leucine, isoleucine, methionine, phenylalanine, tyrosine, aspartic acid, glutamic acid, asparagine, glutamine, histidine, lysine, arginine, and the derivatives thereof. In some embodiments, all gamma modifications are gamma miniPEG modifications. Optionally, at least one PNA segment comprises a G-clamp (9-(2-guanidinoethoxy) phenoxazine).
2. Triplex-forming Target Sequence Considerations
The triplex-forming molecules bind to a predetermined target region referred to herein as the“target sequence,”“target region,” or“target site.” The target sequence for the triplex-forming molecules can be within or adjacent to a human gene encoding, for example the beta globin, cystic fibrosis transmembrane conductance regulator (CFTR) or other gene discussed in more detail below, or an enzyme necessary for the metabolism of lipids, glycoproteins, or mucopolysaccharides, or another gene in need of correction. The target sequence can be within the coding DNA sequence of the gene or within an intron. The target sequence can also be within DNA sequences which regulate expression of the target gene, including promoter or enhancer sequences or sites that regulate RNA splicing.
The nucleotide sequences of the triplex- forming molecules can be selected based on the sequence of the target sequence, the physical constraints, and the preference for a low dissociation constant (Kd) for the triplex- forming molecules/target sequence. As used herein, triplex- forming molecules can be substantially complementary to a target region when the triplex-forming molecules has a nucleobase composition which allows for the formation of a triple-helix with the target region. A triplex-forming molecule can be substantially complementary to a target region even when there are non-complementary nucleobases present in the triplex- forming molecules.
There are a variety of structural motifs available which can be used to determine the nucleotide sequence of a substantially complementary oligonucleotide. Preferably, the triplex-forming molecules bind to or hybridize to the target sequence under conditions of high stringency and specificity. Reaction conditions for in vitro triple helix formation of an triplex- forming molecules probe or primer to a nucleic acid sequence vary from triplex-forming molecules to triplex-forming molecules, depending on
factors such as the length triplex-forming molecules, the number of G:C and A:T base pairs, and the composition of the buffer utilized in the
hybridization reaction.
a. Target sequence considerations for TFOs
Preferably, the TFO is a single- stranded nucleic acid molecule between 7 and 40 nucleotides in length, most preferably 10 to 20 nucleotides in length for in vitro mutagenesis and 20 to 30 nucleotides in length for in vivo mutagenesis. The base composition may be homopurine or
homopyrimidine. Alternatively, the base composition may be polypurine or polypyrimidine. However, other compositions are also useful. Most preferably, the oligonucleotides bind in a sequence- specific manner within the major groove of duplex DNA. An oligonucleotide substantially complementary, based on the third strand binding code, to the target region of the double-stranded nucleic acid molecule may be preferred. The oligonucleotides may have a base composition which is conducive to triple helix formation and may be generated based on one of the known structural motifs for third strand binding. The most stable complexes may be formed on polypurine:polypyrimidine elements, which are relatively abundant in mammalian genomes. Triplex formation by TFOs can occur with the third strand oriented either parallel or anti-parallel to the purine strand of the duplex. In the anti-parallel, purine motif, the triplets are G.G:C and A.A:T, whereas in the parallel pyrimidine motif, the canonical triplets are C+.G:C and T.A:T. The triplex structures are stabilized by two Hoogsteen hydrogen bonds between the bases in the TFO strand and the purine strand in the duplex. A review of base compositions for third strand binding
oligonucleotides is provided in US Patent No. 5,422,251.
TFOs are preferably generated using known DNA and/or PNA synthesis procedures. In one embodiment, oligonucleotides are generated synthetically. Oligonucleotides can also be chemically modified using standard methods that are well known in the art.
b. Target sequence considerations for PNAs
Some triplex-forming molecules, such as PNA (e.g., PNA clamps and tail clamp PNAs (tcPNAs)) invade the target duplex, with displacement of the polypyrimidine strand, and induce triplex formation with the polypurine
strand of the target duplex by both Watson-Crick and Hoogsteen binding. Preferably, both the Watson-Crick and Hoogsteen binding portions of the triplex-forming molecules are substantially complementary to the target sequence. Although, as with triplex-forming oligonucleotides, a homopurine strand is needed to allow formation of a stable PNA/DNA/PNA triplex, PNA clamps may form at shorter homopurine sequences than those required by triplex-forming oligonucleotides and also do so with greater stability.
Preferably, PNAs may be between 6 and 50 nucleobase-containing residues in length. The Watson-Crick portion maybe 9 or more nucleobase- containing residues in length, optionally including a tail sequence. More preferably, the Watson-Crick binding portion may be between about 9 and 30 nucleobase-containing residues in length, optionally including a tail sequence of between 0 and about 15 nucleobase-containing residues. More preferably, the Watson-Crick binding portion may be between about 10 and 25 nucleobase-containing residues in length, optionally including a tail sequence of between 0 and about 10 nucleobase-containing residues in length. More preferably, the Watson-Crick binding portion may be between 15 and 25 nucleobase-containing residues in length, optionally including a tail sequence of between 5 and 10 nucleobase-containing residues in length. The Hoogsteen binding portion may be 6 or more nucleobase residues in length. Most preferably, the Hoogsteen binding portion may be between about 6 and 15 nucleobase-containing residues in length, inclusive.
The triplex-forming molecules are designed to target the polypurine strand of a polypurine:polypyrimidine stretch in the target duplex nucleotide. Therefore, the base composition of the triplex-forming molecules may be homopyrimidine. Alternatively, the base composition may be
polypyrimidine. The addition of a“tail” reduces the requirement for polypurine:polypyrimidine ran. Adding additional nucleobase-containing residues, known as a“tail,” to the Watson-Crick binding portion of the triplex-forming molecules may allow the Watson-Crick binding portion to bind/hybridize to the target strand outside the site of polypurine sequence for triplex formation. These additional bases further reduce the requirement for the polypurine:polypyrimidine stretch in the target duplex and therefore increase the number of potential target sites. Triplex-forming molecules
(TFMs) including, e.g., triplex-forming oligonucleotides (TFOs) and helix- invading peptide nucleic acids (bis-PNAs and tcPNAs), also generally utilize a polypurine :polypyrimidine sequence to a form a triple helix. Traditional nucleic acid TFOs may need a stretch of at least 15 and preferably 30 or more nucleobase-containing residues. Peptide nucleic acids need fewer purines to a form a triple helix, although at least 10 or preferably more may be needed. Peptide nucleic acids including a tail, also referred to tail clamp PNAs, or tcPNAs, may require even fewer purines to a form a triple helix. A triple helix may be formed with a target sequence containing fewer than 8 purines. Therefore, PNAs may be designed to target a site on duplex nucleic acid containing between 6-30 polypurine:polypyrimidines, preferably, 6-25 polypurine:polypyrimidines, more preferably 6-20
polypurine :polypyrimidines .
The addition of a“mixed-sequence” tail to the Watson-Crick-binding strand of the triplex-forming molecules such as PNAs may also increase the length of the triplex-forming molecule and, correspondingly, the length of the binding site. This may increase the target specificity and size of the lesion created at the target site and may disrupt the helix in the duplex nucleic acid, while maintaining a low requirement for a stretch of polypurine:polypyrimidines. Increasing the length of the target sequence may improve specificity for the target, for example, a target of 17 base pairs will statistically be unique in the human genome. Relative to a smaller lesion, it is likely that a larger triplex lesion with greater disruption of the underlying DNA duplex will be detected and processed more quickly and efficiently by the endogenous DNA repair machinery that facilitates recombination of the donor oligonucleotide.
The triple-forming molecules are preferably generated using known synthesis procedures. In one embodiment, triplex-forming molecules are generated synthetically. Triplex-forming molecules can also be chemically modified using standard methods that are well known in the art.
B. Pseudocomplementary Oligonucleotides/PNAs
The gene editing technology may include pseudocomplementary oligonucleotides such as those disclosed in U.S. Patent No. 8,309,356.
“Double duplex-forming molecules,” are oligonucleotides that bind to duplex
DNA in a sequence- specific manner to form a four-stranded structure. Double duplex-forming molecules, such as a pair of pseudocomplementary oligonucleotides/PNAs, can induce recombination with a donor
oligonucleotide at a chromosomal site in mammalian cells.
Pseudocomplementary oligonucleotides/PNAs are complementary oligonucleotides/PNAs that contain one or more modifications such that they do not recognize or hybridize to each other, for example due to steric hindrance, but each can recognize and hybridize to its complementary nucleic acid strands at the target site. As used herein the term‘pseudocomplementary oligonucleotide(s)’ include pseudocomplementary peptide nucleic acids (pcPNAs). A pseudocomplementary oligonucleotide is said to be
substantially complementary to a target region when the oligonucleotide has a base composition which allows for the formation of a double duplex with the target region. As such, an oligonucleotide can be substantially
complementary to a target region even when there are non-complementary bases present in the pseudocomplementary oligonucleotide.
This strategy can be more efficient and may provide increased flexibility over other methods of induced recombination such as triple-helix oligonucleotides and bis-peptide nucleic acids which can prefer a polypurine sequence in the target double-stranded DNA. The design ensures that the pseudocomplementary oligonucleotides do not pair with each other but instead bind the cognate nucleic acids at the target site, inducing the formation of a double duplex.
The predetermined region that the double duplex-forming molecules bind to can be referred to as a“double duplex target sequence,”“double duplex target region,” or“double duplex target site.” The double duplex target sequence (DDTS) for the double duplex-forming molecules can be, for example, within or adjacent to a human gene in need of induced gene correction. The DDTS can be within the coding DNA sequence of the gene or within introns. The DDTS can also be within DNA sequences which regulate expression of the target gene, including promoter or enhancer sequences.
The nucleotide/nucleobase sequence of the pseudocomplementary oligonucleotides is selected based on the sequence of the DDTS. Therapeutic administration of pseudocomplementary oligonucleotides may involve two
single stranded oligonucleotides unlinked, or linked by a linker. One pseudocomplementary oligonucleotide strand is complementary to the DDTS, while the other is complementary to the displaced DNA strand. The use of pseudocomplementary oligonucleotides, particularly pcPNAs are not subject to limitation on sequence choice and/or target length and specificity as are triplex-forming oligonucleotides, helix-invading peptide nucleic acids (bis- PNAs and tcPNAs) and side-by-side minor groove binders.
Pseudocomplementary oligonucleotides do not require third-strand
Hoogsteen-binding, and therefore are not restricted to homopurine targets. Pseudocomplementary oligonucleotides can be designed for mixed, general sequence recognition of a desired target site. Preferably, the target site contains an A:T base pair content of about 40% or greater. Preferably pseudocomplementary oligonucleotides are between about 8 and 50 nucleobase-containing residues in length, more preferably 8 to 30, even more preferably between about 8 and 20 nucleobase-containing residues in length.
The pseudocomplementary oligonucleotides may be designed to bind to the target site (DDTS) at a distance of between about 1 to 800 bases from the target site of the donor oligonucleotide. More preferably, the
pseudocomplementary oligonucleotides may bind at a distance of between about 25 and 75 bases from the donor oligonucleotide. Most preferably, the pseudocomplementary oligonucleotides may bind at a distance of about 50 bases from the donor oligonucleotide. Preferred pcPNA sequences for targeted repair of a mutation in the b-globin intron IVS2 (G to A) are described in U.S. Patent 8,309,356.
Preferably, the pseudocomplementary oligonucleotides may bind/hybridize to the target nucleic acid molecule under conditions of high stringency and specificity. Most preferably, the oligonucleotides may bind in a sequence-specific manner and induce the formation of double duplex.
Specificity and binding affinity of the pseudocomplemetary oligonucleotides may vary from oligomer to oligomer, depending on factors such as length, the number of G:C and A:T base pairs, and the formulation.
C. CRISPR/Cas
In some embodiments, the gene editing technology is the
CRISPR/Cas system. CRISPR (Clustered Regularly Interspaced Short
Palindromic Repeats) is an acronym for DNA loci that contain multiple, short, direct repetitions of base sequences. The prokaryotic CRISPR/Cas system has been adapted for use as gene editing (silencing, enhancing or changing specific genes) for use in eukaryotes (see, for example, Cong, Science, 15 : 339(6121):819— 823 (2013) and Jinek, et a , Science,
337(6096):816-21 (2012)). By transfecting a cell with the required elements including a cas gene and specifically designed CRISPRs, the organism's genome can be cut and modified at any desired location. Methods of preparing compositions for use in genome editing using the CRISPR/Cas systems are described in detail in WO 2013/176772 and WO 2014/018423.
In general,“CRISPR system” refers collectively to transcripts and other elements involved in the expression of or directing the activity of CRISPR-associated (“Cas”) genes, including sequences encoding a Cas gene, a tracr (trans-activating CRISPR) sequence (e.g., tracrRNA or an active partial tracrRNA), a tracr-mate sequence (encompassing a“direct repeat” and a tracrRNA-processed partial direct repeat in the context of an endogenous CRISPR system), a guide sequence (also referred to as a “spacer” in the context of an endogenous CRISPR system), or other sequences and transcripts from a CRISPR locus. One or more tracr mate sequences operably linked to a guide sequence (e.g., direct repeat-spacer- direct repeat) can also be referred to as pre-crRNA (pre-CRISPR RNA) before processing or crRNA after processing by a nuclease.
In some embodiments, a tracrRNA and crRNA are linked and form a chimeric crRNA-tracrRNA hybrid where a mature crRNA is fused to a partial tracrRNA via a synthetic stem loop to mimic the natural
crRNAdracrRNA duplex as described in Cong, Science, 15:339(6121):819- 823 (2013) and Jinek, et ak, Science, 337(6096):816-21 (2012)). A single fused crRNA-tracrRNA construct can also be referred to as a guide RNA or gRNA (or single-guide RNA (sgRNA)). Within a sgRNA, the crRNA portion can be identified as the“target sequence” and the tracrRNA is often referred to as the“scaffold.”
There are many resources available for helping practitioners determine suitable target sites once a desired DNA target sequence is identified. For example, numerous public resources, including a
bioinformatically generated list of about 190,000 potential sgRNAs, targeting more than 40% of human exons, are available to aid practitioners in selecting target sites and designing the associate sgRNA to affect a nick or double strand break at the site. See also, crispr.u-psud.fr/, a tool designed to help scientists find CRISPR targeting sites in a wide range of species and generate the appropriate crRNA sequences.
In some embodiments, one or more vectors driving expression of one or more elements of a CRISPR system are introduced into a target cell such that expression of the elements of the CRISPR system direct formation of a CRISPR complex at one or more target sites. While the specifics can be varied in different engineered CRISPR systems, the overall methodology is similar. A practitioner interested in using CRISPR technology to target a DNA sequence can insert a short DNA fragment containing the target sequence into a guide RNA expression plasmid. The sgRNA expression plasmid contains the target sequence (about 20 nucleotides), a form of the tracrRNA sequence (the scaffold) as well as a suitable promoter and necessary elements for proper processing in eukaryotic cells. Such vectors are commercially available (see, for example, Addgene). Many of the systems rely on custom, complementary oligomers that are annealed to form a double stranded DNA and then cloned into the sgRNA expression plasmid. Co-expression of the sgRNA and the appropriate Cas enzyme from the same or separate plasmids in transfected cells results in a single or double strand break (depending of the activity of the Cas enzyme) at the desired target site.
In some embodiments, a vector includes a regulatory element operably linked to an enzyme-coding sequence encoding a CRISPR enzyme, such as a Cas protein. Non-limiting examples of Cas proteins include Casl, CaslB, Cas2, Cas3, Cas4, Cas5, Cas6, Cas7, Cas8, Cas9 (also known as Csnl and Csxl2), CaslO, Csyl, Csy2, Csy3, Csel, Cse2, Cscl, Csc2, Csa5, Csn2, Csm2, Csm3, Csm4, Csm5, Csm6, Cmrl, Cmr3, Cmr4, Cmr5, Cmr6, Csbl, Csb2, Csb3, Csxl7, Csxl4, CsxlO, Csxl6, CsaX, Csx3, Csxl, Csxl5, Csfl, Csf2, Csf3, Csf4, Cpfl, homologues thereof, or modified versions thereof. In some embodiments, the unmodified CRISPR enzyme has DNA cleavage activity, such as Cas9. In some embodiments, the CRISPR enzyme directs cleavage of one or both strands at the location of a target sequence, such as
within the target sequence and/or within the complement of the target sequence. In some embodiments, the CRISPR enzyme directs cleavage of one or both strands within about 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 50,
100, 200, 500, or more base pairs from the first or last nucleotide of a target sequence.
The CRISPR/Cas system may contain an enzyme that is mutated with respect to a corresponding wild-type enzyme such that the mutated CRISPR enzyme lacks the ability to cleave one or both strands of a target
polynucleotide containing a target sequence. By independently mutating one of the two Cas9 nuclease domains, the Cas9 nickase was developed. For example, an aspartate-to-alanine substitution (D10A) in the RuvC I catalytic domain of Cas9 from S. pyogenes converts Cas9 from a nuclease that cleaves both strands to a nickase (cleaves a single strand). Other residues can be mutated to achieve the above effects (i.e. inactivate one or the other nuclease portions). As non-limiting examples, residues D10, G12, G17, E762, H840, N854, N863, H982, H983, A984, D986, and/or A987 can be substituted. Specific mutations that render Cas9 a nickase include, without limitation, H840A, N854A, and N863A. Mutations other than alanine substitutions are also suitable. Two or more catalytic domains of Cas9 (RuvC I, RuvC II, and RuvC III) can be mutated to produce a mutated Cas9 substantially lacking all DNA cleavage activity. A D10A mutation may be combined with one or more of H840A, N854A, or N863A mutations to produce a Cas9 enzyme substantially lacking all DNA cleavage activity (e.g., when activity of the mutated enzyme is less than about 25%, 10%, 5%>, 1%>, 0.1 %>, 0.01%, or lower with respect to its non-mutated form).
Preferably, variants of Cas9, such as for example, a Cas9 nickase are employed in the gene editing technologies containing a CRISPR Cas system. Nickases can lower the probability of off-target editing, for example, when used with two adjacent gRNAs. A Cas9 nickase having a D10A mutation cleaves only the target strand. Conversely, a Cas9 nickase having an H840A mutation in the HNH domain creates a non-target strand-cleaving nickase. Instead of cutting both strands bluntly with WT Cas9 and one gRNA, one can create a staggered cut using a Cas9 nickase and two gRNAs. This provides even greater control over precise gene integration and insertion.
Because both nicking Cas9 enzymes must effectively nick their target DNA, paired nickases have significantly lower off-target effects compared to the double-strand-cleaving Cas9 system, and are generally more effective tools. In a preferred embodiment, the gene editing technology is a Crispr/Cas9 nickase (e.g., D10A, H840A, N854A, and N863A nickase). In a more preferred embodiment, the gene editing technology is a Crispr/Cas9 D10A nickase.
D. Zinc Finger Nucleases
In some embodiments, the element that induces a single or a double strand break in the target cell’s genome is a nucleic acid construct or constructs encoding a zinc finger nucleases (ZFNs). ZFNs are typically fusion proteins that include a DNA-binding domain derived from a zinc- finger protein linked to a cleavage domain.
The most common cleavage domain is the Type IIS enzyme Fokl. Fokl catalyzes double-stranded cleavage of DNA, at 9 nucleotides from its recognition site on one strand and 13 nucleotides from its recognition site on the other. See, for example, U.S. Pat. Nos. 5,356,802; 5,436, 150 and 5,487,994; as well as Li et al. Proc., Natl. Acad. Sci. USA 89 (1992):4275- 4279; Li et al. Proc. Natl. Acad. Sci. USA, 90:2764-2768 (1993); Kim et al. Proc. Natl. Acad. Sci. USA. 91:883-887 (1994a); Kim et al. J. Biol. Chem. 269:31 ,978-31,982 (1994b). One or more of these enzymes (or
enzymatically functional fragments thereof) can be used as a source of cleavage domains.
The DNA-binding domain, which can, in principle, be designed to target any genomic location of interest, can be a tandem array of Cys2His2 zinc fingers, each of which generally recognizes three to four nucleotides in the target DNA sequence. The Cys2His2 domain has a general structure: Phe (sometimes Tyr)-Cys-(2 to 4 amino acids)-Cys-(3 amino acids)- Phe(sometimes Tyr)-(5 amino acids)-Leu-(2 amino acids)-His-(3 amino acids)-His. By linking together multiple fingers (the number varies: three to six fingers have been used per monomer in published studies), ZFN pairs can be designed to bind to genomic sequences 18-36 nucleotides long.
Engineering methods include, but are not limited to, rational design and various types of empirical selection methods. Rational design includes,
for example, using databases including triplet (or quadruplet) nucleotide sequences and individual zinc finger amino acid sequences, in which each triplet or quadruplet nucleotide sequence is associated with one or more amino acid sequences of zinc fingers which bind the particular triplet or quadruplet sequence. See, for example, U.S. Pat. Nos. 6, 140,081; 6,453,242; 6,534,261; 6,610,512; 6,746,838; 6,866,997; 7,067,617; U.S. Published Application Nos. 2002/0165356; 2004/0197892; 2007/0154989;
2007/0213269; and International Patent Application Publication Nos. WO 98/53059 and WO 2003/016496.
E. Transcription Activator-Like Effector Nucleases
In some embodiments, the element that induces a single or a double strand break in the target cell’s genome is a nucleic acid construct or constructs encoding a transcription activator- like effector nuclease
(TALEN). TALENs have an overall architecture similar to that of ZFNs, with the main difference that the DNA-binding domain comes from TAL effector proteins, transcription factors from plant pathogenic bacteria. The DNA-binding domain of a TALEN is a tandem array of amino acid repeats, each about 34 residues long. The repeats are very similar to each other; typically they differ principally at two positions (amino acids 12 and 13, called the repeat variable diresidue, or RVD). Each RVD specifies preferential binding to one of the four possible nucleotides, meaning that each TALEN repeat binds to a single base pair, though the NN RVD is known to bind adenines in addition to guanine. TAL effector DNA binding is mechanistically less well understood than that of zinc-finger proteins, but their seemingly simpler code could prove very beneficial for engineered- nuclease design. TALENs also cleave as dimers, have relatively long target sequences (the shortest reported so far binds 13 nucleotides per monomer) and appear to have less stringent requirements than ZFNs for the length of the spacer between binding sites. Monomeric and dimeric TALENs can include more than 10, more than 14, more than 20, or more than 24 repeats.
Methods of engineering TAL to bind to specific nucleic acids are described in Cermak, et al, Nucl. Acids Res. 1-11 (2011). U.S. Published Application No. 2011/0145940, which discloses TAL effectors and methods of using them to modify DNA. Miller et al. Nature Biotechnol 29: 143
(2011) reported making TALENs for site-specific nuclease architecture by linking TAL truncation variants to the catalytic domain of Fokl nuclease.
The resulting TALENs were shown to induce gene modification in immortalized human cells. General design principles for TALEN binding domains can be found in, for example, WO 2011/072246.
IV. Donor Oligonucleotides
In some embodiments, the gene editing technology includes or is administered in combination with a donor oligonucleotide. Generally, in the case of gene therapy, the donor oligonucleotide includes a sequence that can correct a mutation(s) in the host genome, though in some embodiments, the donor introduces a mutation that can, for example, reduce expression of an oncogene or a receptor that facilitates HIV infection. In addition to containing a sequence designed to introduce the desired correction or mutation, the donor oligonucleotide may also contain synonymous (silent) mutations (e.g., 7 to 10). The additional silent mutations can facilitate detection of the corrected target sequence using allele-specific PCR of genomic DNA isolated from treated cells.
The donor oligonucleotide can exist in single stranded (ss) or double stranded (ds) form (e.g., ssDNA, dsDNA). The donor oligonucleotide can be of any length. For example, the size of the donor oligonucleotide may be between 1 to 800 nucleotides. In one embodiment, the donor oligonucleotide is between 25 and 200 nucleotides. In some embodiments, the donor oligonucleotide is between 100 and 150 nucleotides. In a further
embodiment, the donor nucleotide is about 40 to 80 nucleotides in length.
The donor oligonucleotide may be about 60 nucleotides in length. ssDNAs of length 25-200 are active. Most studies have been with ssDNAs of length 60- 70. Longer ssDNAs of length 70-150 may also be effective. In some embodiments, the preferred length is about 60 nucleotides.
Successful recombination of the donor sequence results in a change of the sequence of the target region. Donor oligonucleotides are also referred to as donor fragments, donor nucleic acids, donor DNA, or donor DNA fragments. It is understood in the art that a greater number of homologous positions within the donor fragment will increase the probability that the donor fragment will be recombined into the target sequence, target region, or
target site. Target sequences can be within the coding DNA sequence of the gene or within introns. Target sequences can also be within DNA sequences which regulate expression of the target gene, including promoter or enhancer sequences or sequences that regulate RNA splicing.
The donor sequence can contain one or more nucleic acid sequence alterations compared to the sequence of the region targeted for
recombination, for example, a point mutation, a substitution, a deletion, or an insertion of one or more nucleotides. Deletions and insertions can result in frameshift mutations or deletions. Point mutations can cause missense or nonsense mutations. These mutations may disrupt, reduce, stop, increase, improve, or otherwise alter the expression of the target gene.
The donor oligonucleotide may correspond to the wild type sequence of a gene (or a portion thereof), for example, a mutated gene involved with a disease or disorder (e.g., hemophilia, muscular dystrophy, globinopathies, cystic fibrosis, xeroderma pigmentosum, lysosomal storage diseases, immune deficiency syndromes such as X-linked severe combined immunodeficiency and ADA deficiency, tyrosinemia, Fanconi anemia, the red cell disorder spherocytosis, alpha- 1-anti-trypsin deficiency, Wilson’s disease, Leber’ s hereditary optic neuropathy, and chronic granulomatous disorder).
One or more (e.g., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 or more) different donor oligonucleotide sequences may be used in accordance with the disclosed methods. This may be useful, for example, to create a heterozygous target gene where the two alleles contain different modifications.
Donor oligonucleotides are preferably DNA oligonucleotides, composed of the principal naturally-occurring nucleotides (uracil, thymine, cytosine, adenine and guanine) as the heterocyclic bases, deoxyribose as the sugar moiety, and phosphate ester linkages. Donor oligonucleotides may include modifications to nucleobases, sugar moieties, or backbone/linkages, depending on the desired structure of the replacement sequence at the site of recombination or to provide some resistance to degradation by nucleases. For example, the terminal two or three inter-nucleoside linkages at each end of a ssDNA oligonucleotide (both 5’ and 3’ ends) may be replaced with phosphorothioate linkages in lieu of the usual phosphodiester linkages,
thereby providing increased resistance to exonucleases. Modifications to the donor oligonucleotide should not prevent the donor oligonucleotide from successfully recombining at the recombination target sequence.
Donor oligonucleotides can be either single stranded or double stranded, and can target one or both strands of the genomic sequence at a target locus. The donors are typically presented as single stranded DNA sequences targeting one strand of the target genomic locus. However, even where not expressly provided, the reverse complement of each donor, and double stranded DNA sequences, based on the provided sequences may also be used. In some embodiments, the donor oligonucleotide is a functional fragment of the disclosed sequence, or the reverse complement, or double stranded DNA thereof.
In some embodiments, the donor oligonucleotide includes 1, 2, 3, 4,
5, 6, or more optional phosphorothioate internucleoside linkages. In some embodiments, the donor includes phosphorothioate intemucleoside linkages between first 2, 3, 4 or 5 nucleotides, and/or the last 2, 3, 4, or 5 nucleotides in the donor oligonucleotide.
A. Preferred Donor Oligonucleotide Design for
Triplex and Double-Duplex based Technologies
The triplex-forming molecules including peptide nucleic acids may be administered in combination with, or tethered to, a donor oligonucleotide via a mixed sequence linker or used in conjunction with a non-tethered donor oligonucleotide that is substantially homologous to the target sequence. Triplex-forming molecules can induce recombination of a donor
oligonucleotide sequence up to several hundred base pairs away. It is preferred that the donor oligonucleotide sequence is between 1 to 800 bases from the target binding site of the triplex-forming molecules. More preferably the donor oligonucleotide sequence is between 25 to 75 bases from the target binding site of the triplex-forming molecules. Most preferably, the donor oligonucleotide sequence is about 50 nucleotides from the target binding site of the triplex-forming molecules.
The donor sequence can contain one or more nucleic acid sequence alterations compared to the sequence of the region targeted for
recombination, for example, a substitution, a deletion, or an insertion of one
or more nucleotides. Successful recombination of the donor sequence results in a change of the sequence of the target region. This strategy exploits the ability of a triplex to provoke DNA repair, potentially increasing the probability of recombination with the homologous donor DNA. It is understood in the art that in most cases, a greater number of homologous positions within the donor fragment will increase the probability that the donor fragment will be recombined into the target sequence, target region, or target site. Tethering of a donor oligonucleotide to a triplex-forming molecule facilitates target site recognition via triple helix formation while at the same time positioning the tethered donor fragment for possible recombination and information transfer. Triplex-forming molecules may also effectively induce homologous recombination of non-tethered donor oligonucleotides.
Non-tethered or unlinked fragments may range in length from 20 nucleotides to several thousand. The donor oligonucleotide molecules, whether linked or unlinked, can exist in single stranded (ss) or double stranded form (ds) (e.g., ssDNA, dsDNA). The donor fragment to be recombined can be linked or un-linked to the triplex-forming molecules. The linked donor fragment may range in length from 4 nucleotides to 100 nucleotides, preferably from 4 to 80 nucleotides in length. However, the unlinked donor fragments may have a much broader range, from 20 nucleotides to several thousand nucleotides in length. In one embodiment the oligonucleotide donor is between 25 and 80 nucleobases. In a further embodiment, the non-tethered donor nucleotide is about 50 to 60 nucleotides in length.
Compositions including triplex-forming molecules such as tcPNA may include one or more (e.g., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 or more) donor oligonucleotides. More than one donor oligonucleotides may be administered with triplex-forming molecules in a single transfection, or sequential transfections.
B. Preferred Donor Oligonucleotides Design for
Nuclease-based Technologies
The nuclease activity of some of the genome editing systems described herein cleave target DNA to produce single or double strand
breaks in the target DNA. Double strand breaks can be repaired by the cell in one of two ways: non-homologous end joining, and homology-directed repair. In non-homologous end joining (NHEJ), the double-strand breaks are repaired by direct ligation of the break ends to one another. As such, no new nucleic acid material is inserted into the site, although some nucleic acid material may be lost, resulting in a deletion. In homology-directed repair, a donor polynucleotide with homology to the cleaved target DNA sequence is used as a template for repair of the cleaved target DNA sequence, resulting in the transfer of genetic information from a donor polynucleotide to the target DNA. As such, new nucleic acid material can be inserted/copied into the site. The modifications of the target DNA due to NHEJ and/or homology-directed repair can be used to induce gene correction, gene replacement, gene tagging, transgene insertion, nucleotide deletion, gene disruption, gene mutation, etc.
A polynucleotide including a donor sequence to be inserted at the cleavage site is provided to the cell to be edited. The donor polynucleotide typically contains sufficient homology to a genomic sequence at the cleavage site, e.g., 70%, 80%, 85%, 90%, 95%, or 100% homology with the nucleotide sequences flanking the cleavage site, e.g., within about 50 bases or less of the cleavage site, e.g., within about 30 bases, within about 15 bases, within about 10 bases, within about 5 bases, or immediately flanking the cleavage site, to support homology-directed repair between it and the genomic sequence to which it bears homology.
The donor sequence may or may not be identical to the genomic sequence that it replaces. The donor sequence may correspond to the wild type sequence (or a portion thereof) of the target sequence (e.g., a gene). The donor sequence may contain at least one or more single base changes, insertions, deletions, inversions or rearrangements with respect to the genomic sequence, so long as sufficient homology is present to support homology-directed repair. In some embodiments, the donor sequence includes a non-homologous sequence flanked by two regions of homology, such that homology-directed repair between the target DNA region and the two flanking sequences results in insertion of the non-homologous sequence at the target region.
When the genome editing composition or technology includes a donor polynucleotide sequence that includes at least a segment with homology to the target DNA sequence, the methods can be used to add, i.e., insert or replace, nucleic acid material to a target DNA sequence (e.g., to “knock in” a nucleic acid that encodes for a protein, an siRNA, an miRNA, etc.), to add a tag (e.g., 6xHis, a fluorescent protein (e.g., a green fluorescent protein; a yellow fluorescent protein, etc.), hemagglutinin (HA), FLAG, etc.), to add a regulatory sequence to a gene (e.g., promoter, polyadenylation signal, internal ribosome entry sequence (IRES), 2A peptide, start codon, stop codon, splice signal, localization signal, etc.), or to modify a nucleic acid sequence (e.g., introduce a mutation).
C. Oligonucleotide Variations
Any of the disclosed gene editing technologies, components thereof, donor oligonucleotides, or other nucleic acids can include one or more modifications or substitutions to the nucleobases or linkages. Although modifications are particularly preferred for use with triplex-forming technologies and typically discussed below with reference thereto, any of the modifications can be utilized in the construction of any of the disclosed gene editing technologies, donor oligonucleotides, other nucleotides, etc.
Modifications should not prevent, and preferably enhance the activity, persistence, or function of the gene editing technology. For example, modifications to oligonucleotides for use as triplex-forming should not prevent, and preferably enhance duplex invasion, strand displacement, and/or stabilize triplex formation as described above by increasing specificity or binding affinity of the triplex-forming molecules to the target site. Modified bases and base analogues, modified sugars and sugar analogues and/or various suitable linkages known in the art are also suitable for use in the molecules disclosed herein.
1. Heterocyclic Bases
The principal naturally-occurring nucleotides include uracil, thymine, cytosine, adenine and guanine as the heterocyclic bases. Gene editing molecules can include chemical modifications to their nucleotide or nucleobase constituents. For example, target sequences with adjacent cytosines can be problematic. Triplex stability may be compromised by runs
of cytosines, thought to be due to repulsion between the positive charge resulting from the N3 protonation or perhaps because of competition for protons by the adjacent cytosines. Chemical modification of nucleotides including triplex-forming molecules such as PNAs may be useful to increase binding affinity of triplex- forming molecules and/or triplex stability under physiologic conditions.
Chemical modifications of heterocyclic bases or heterocyclic base analogs may be effective to increase the binding affinity of a nucleotide or its stability in a triplex. Chemically-modified heterocyclic bases include, but are not limited to, inosine, 5-(l-propynyl) uracil (pU), 5-(l-propynyl) cytosine (pC), 5-methylcytosine, 8-oxo-adenine, pseudocytosine, pseudoisocytosine,
5 and 2-amino-5-(2,-deoxy- -D-ribofuranosyl)pyridine (2-aminopyridine), and various pyrrolo- and pyrazolopyrimidine derivatives. Substitution of 5- methylcytosine or pseudoisocytosine for cytosine in triplex-forming molecules such as PNAs helps to stabilize triplex formation at neutral and/or physiological pH, especially in triplex-forming molecules with isolated cytosines. This is because the positive charge may partially reduce the negative charge repulsion between the triplex- forming molecules and the target duplex, and allows for Hoogsteen binding.
2. Backbone
The nucleotide subunits of the oligonucleotides may contain certain modifications. For example, the phosphate backbone of the oligonucleotide may be replaced in its entirety by repeating N-(2-aminoethyl)-glycine units and/or phosphodiester bonds may be replaced by peptide bonds or phosphorothioate linkages, either partial or complete. For example, in PNAs, the phosphate backbone of the oligonucleotide is replaced in its entirety by repeating N-(2-aminoethyl)-glycine units and phosphodiester bonds are typically replaced by peptide bonds. The various heterocyclic bases are linked to the backbone by methylene carbonyl bonds, which allow them to form PNA-DNA or PNA-RNA duplexes via Watson-Crick base pairing with high affinity and sequence-specificity. PNAs maintain spacing of heterocyclic bases that is similar to conventional DNA oligonucleotides. Peptide nucleic acids are composed of peptide nucleic acid monomers.
Other backbone modifications include peptide and amino acid variations and modifications. The backbone constituents of donor oligonucleotides may be peptide linkages, or alternatively, they may be non peptide linkages. Examples include acetyl caps, amino spacers such as 8- amino-3,6-dioxaoctanoic acid (referred to herein as O-linkers), amino acids such as lysine are particularly useful if positive charges are desired in the oligonucleotide (e.g., PNA) and the like. Methods for the chemical assembly of PNAs are well known. See, for example, U.S. Patent No. 5,539,082, 5,527,675, 5,623,049, 5,714,331, 5,736,336, 5,773,571 and 5,786,571.
Backbone modifications of oligonucleotides should not prevent the molecules from binding with high specificity to the DNA target site and mediating information transfer. For example, modifications of triplex forming molecules should not prevent the molecules from binding with high specificity to the target site and creating a triplex with the target duplex nucleic acid by displacing one strand of the target duplex and forming a clamp around the other strand of the target duplex.
3. Modified Nucleic Acids
Modified nucleic acids in addition to peptide nucleic acids are also useful as triplex-forming molecules. Oligonucleotides are composed of a chain of nucleotides which are linked to one another. Canonical nucleotides typically include a heterocyclic base (nucleic acid base), a sugar moiety attached to the heterocyclic base, and a phosphate moiety which esterifies a hydroxyl function of the sugar moiety. The principal naturally-occurring nucleotides include uracil, thymine, cytosine, adenine and guanine as the heterocyclic bases, and ribose or deoxyribose sugar linked by phosphodiester bonds. As used herein“modified nucleotide” or“chemically modified nucleotide” defines a nucleotide that has a chemical modification of one or more of the heterocyclic base, sugar moiety or phosphate moiety
constituents. The charge of the modified nucleotide may be reduced compared to DNA or RNA oligonucleotides of the same nucleobase sequence. For example, the triplex-forming molecules may have low negative charge, no charge, or positive charge such that electrostatic repulsion with the nucleotide duplex at the target site is reduced compared to DNA or RNA oligonucleotides with the corresponding nucleobase sequence.
Examples of modified nucleotides with reduced charge include modified intemucleotide linkages such as phosphate analogs having achiral and uncharged intersubunit linkages (e.g., Sterchak, E. P. et ak, Organic Chem., 52:4202, (1987)), and uncharged morpholino-based polymers having achiral intersubunit linkages (see, e.g., U.S. Patent No. 5,034,506). Some internucleotide linkage analogs include morpholidate, acetal, and polyamide- linked heterocycles. Locked nucleic acids (LNA) are modified RNA nucleotides (see, for example, Braasch, et ak, Chem. Biol., 8(1): 1-7 (2001)). LNAs form hybrids with DNA which are more stable than DNA/DNA hybrids, a property similar to that of peptide nucleic acid (PNA)/DNA hybrids. Therefore, LNA can be used just as PNA molecules would be. LNA binding efficiency can be increased in some embodiments by adding positive charges to it. Commercial nucleic acid synthesizers and standard
phosphoramidite chemistry are used to make LNAs.
Molecules may also include nucleotides with modified heterocyclic bases, sugar moieties or sugar moiety analogs. Modified nucleotides may include modified heterocyclic bases or base analogs as described above with respect to peptide nucleic acids. Sugar moiety modifications include, but are not limited to, 2’-(9-aminoethoxy, 2’-(9-amonioethyl (2’-OAE), T -O- methoxy, 2’-(9-methyl, 2-guanidoethyl (2’-OGE), 2’-(9,4’-C-methylene (LNA), 2’-0-(methoxyethyl) (2’-OME) and 2’-0-(N-(methyl)acetamido) (2’-OMA). 2’-(9-aminoethyl sugar moiety substitutions are especially preferred because they are protonated at neutral pH and thus suppress the charge repulsion between the triplex- forming molecule and the target duplex. V. Nanoparticle Delivery
The compositions and technologies including, but not limited to, plerixafor, gene editing molecules, donor oligonucleotides, etc., can be delivered to the target cells using a nanoparticle delivery vehicle. In some embodiments, some of the compositions or technologies are packaged in nanoparticles and some are not. Lor example, in some embodiments, the gene editing technology and/or donor oligonucleotide is incorporated into nanoparticles while the plerixafor is not. In some embodiments, the gene editing technology and/or donor oligonucleotide, and the plerixafor are packaged in nanoparticles. The different compositions or technologies can be
packaged in the same nanoparticles or different nanoparticles. For example, the compositions or technologies can be mixed and packaged together. In some embodiments, the different compositions or technologies are packaged separately into separate nanoparticles wherein the nanoparticles are similarly or identically composed and/or manufactured. In some embodiments, the different compositions or technologies are packaged separately into separate nanoparticles wherein the nanoparticles are differentially composed and/or manufactured.
Nanoparticles generally refers to particles having a mean diameter that is between 25 and 500 nm, more preferably having a diameter that is between 50 and 300 nm. Cellular internalization of polymeric particles may be highly dependent upon their size, with nanoparticulate polymeric particles being internalized by cells with much higher efficiency than microparticulate polymeric particles. For example, Desai, et al. have demonstrated that about 2.5 times more nanoparticles that are 100 nm in diameter are taken up by cultured Caco-2 cells as compared to microparticles having a diameter on 1 mM (Desai, et ak, Pharm. Res., 14:1568-73 (1997)). Nanoparticles also have a greater ability to diffuse deeper into tissues in vivo.
Nanoparticles can be formed of polymers, lipids, inorganic materials or combinations thereof.
A. Particle Composition
The polymer that forms the core of the nanoparticle may be any biodegradable or non-biodegradable synthetic or natural polymer. In a preferred embodiment, the polymer is FDA approved. In the most preferred embodiment, the polymer is a biodegradable polymer.
Examples of preferred biodegradable polymers include synthetic polymers that degrade by hydrolysis such as poly(hydroxy acids), such as polymers and copolymers of lactic acid and glycolic acid, other degradable polyesters, poly anhydrides, poly(ortho)esters, polyesters, polyurethanes, poly(butic acid), poly(valeric acid), poly(caprolactone),
poly(hydroxyalkanoates), poly(lactide-co-caprolactone), and poly(amine-co- ester) polymers, such as those described in Zhou, et ak, Nature Materials,
11:82-90 (2012) and WO 2013/082529, U.S. Published Application No. 2014/0342003, and PCT/US2015/061375.
In some embodiments, non-biodegradable polymers can be used, especially hydrophobic polymers. Examples of preferred non-biodegradable polymers include ethylene vinyl acetate, poly(meth) acrylic acid, copolymers of maleic anhydride with other unsaturated polymerizable monomers, poly (butadiene maleic anhydride), polyamides, copolymers and mixtures thereof, and dextran, cellulose and derivatives thereof. Other suitable biodegradable and non-biodegradable polymers are known in the art. These materials may be used alone, as physical mixtures (blends), or as co polymers.
The nanoparticle formulation can be selected based on the considerations including the targeted tissue or cells. For example, in embodiments directed to treatment of treating or correcting beta-thalassemia (e.g. when the target cells are, for example, hematopoietic stem cells), a preferred nanoparticle formulation comprises PLGA. In a preferred embodiment, the nanoparticles are formed of polymers fabricated from polylactides (PLA) and copolymers of lactide and glycolide (PLGA). These have established commercial use in humans and have a long safety record (Jiang, et al., Adv. Drug Deliv. Rev. , 57(3):391-410); Aguado and Lambert, Immunobiology , 184(2-3): 113-25 (1992); Bramwell, et al., Adv. Drug Deliv. Rev., 57(9): 1247-65 (2005)).
Other preferred nanoparticle formulations, particularly preferred for treating cystic fibrosis, are described in McNeer, et al., Nature Commun., 6:6952. doi: 10.1038/ncomms7952 (2015), and Fields, et al., Adv Healthc Mater., 4(3):361-6 (2015). doi: 10.1002/adhm.201400355 (2015) Epub 2014. Such nanoparticles are composed of a blend of poly (beta- amino) esters (PBAEs) and poly(lactic-co-glycolic acid) (PLGA). Therefore, in some embodiments, the nanoparticles utilized to deliver the compositions include PBAE or a blend of PBAE and a polyhydroxy acid such as PLGA.
PLGA and PBAE/PLGA blended nanoparticles loaded with gene editing technology can be formulated using a double-emulsion solvent evaporation technique such as that described in McNeer, et al., Nature Commun., 6:6952. doi: 10.1038/ncomms7952 (2015) and Fields, et al., Adv
Healthc Mater., 4(3):361-6 (2015). doi: 10.1002/adhm.201400355 (2015) Epub 2014. Poly(beta amino ester) (PBAE) can synthesized by a Michael addition reaction of 1,4-butanediol diacrylate and 4,4'- trimethylenedipiperidine as described in Akinc, et al., Bioconjug Chem., 14:979-988 (2003). In some embodiments, PBAE blended particles such as PLGA/PBAE blended particles contain between about 1 and 99, or between about 1 and 50, or between about 5 and 25, or between about 5 and 20, or between about 10 and 20, or about 15 percent PBAE (wt%).
B. Polycations
The nucleic acids, peptide nucleic acids, or other nanoparticle cargo can be complexed to polycations to increase the encapsulation efficiency of the cargo into the nanoparticles. The term“polycation” refers to a compound having a positive charge, preferably at least 2 positive charges, at a selected pH, preferably physiological pH. Polycationic moieties have between about 2 to about 15 positive charges, preferably between about 2 to about 12 positive charges, and more preferably between about 2 to about 8 positive charges at selected pH values.
Many polycations are known in the art. Suitable constituents of polycations include basic amino acids and their derivatives such as arginine, asparagine, glutamine, lysine and histidine; cationic dendrimers; and amino polysaccharides. Suitable polycations can be linear, such as linear tetralysine, branched or dendrimeric in structure.
Exemplary polycations include, but are not limited to, synthetic polycations based on acrylamide and 2-acrylamido-2- methylpropanetrimethylamine, poly(N-ethyl-4-vinylpyridine) or similar quarternized polypyridine, diethylaminoethyl polymers and dextran conjugates, polymyxin B sulfate, lipopoly amines, poly(allylamines) such as the strong polycation poly(dimethyldiallylammonium chloride),
polyethyleneimine, polybrene, and polypeptides such as protamine, the histone polypeptides, polylysine, polyarginine and polyornithine.
In some embodiments, the particles themselves are a polycation (e.g., a blend of PLGA and poly(beta amino ester).
C. Functional/Targeting Molecules
Targeting molecules can be associated with, linked, conjugated, or otherwise attached directly or indirectly to the gene editing molecule, or to a nanoparticle or other delivery vehicle thereof. Targeting molecules can be proteins, peptides, nucleic acid molecules, saccharides or polysaccharides that bind to a receptor or other molecule on the surface of a targeted cell. The degree of specificity and the avidity of binding can be modulated through the selection of the targeting molecule.
Examples of moieties include, for example, targeting moieties which provide for the delivery of molecules to specific cells, e.g., antibodies to hematopoietic stem cells, CD34+ cells, T cells or any other preferred cell type, as well as receptor and ligands expressed on the preferred cell type. Preferably, the moieties may target hematopoeitic stem cells. Examples of molecules targeting extracellular matrix (“ECM”) include
glycosaminoglycan (“GAG”) and collagen. In one embodiment, the external surface of polymer particles may be modified to enhance the ability of the particles to interact with selected cells or tissue. In some embodiments, an adaptor element conjugated to a targeting molecule is inserted into the particle. In another embodiment, the outer surface of a polymer micro- or nanoparticle having a carboxy terminus may be linked to targeting molecules that have a free amine terminus.
Other useful ligands attached to polymeric micro- and nanoparticles include pathogen-associated molecular patterns (PAMPs). PAMPs target Toll-like Receptors (TLRs) on the surface of the cells or tissue, or signal the cells or tissue internally, thereby potentially increasing uptake. PAMPs conjugated to the particle surface or co-encapsulated may include:
unmethylated CpG DNA (bacterial), double-stranded RNA (viral), lipopolysacharride (bacterial), peptidoglycan (bacterial), lipoarabinomannin (bacterial), zymosan (yeast), mycoplasmal lipoproteins such as MALP-2 (bacterial), flagellin (bacterial) poly(inosinic-cytidylic) acid (bacterial), lipoteichoic acid (bacterial) or imidazoquinolines (synthetic).
In another embodiment, the outer surface of the particle may be treated using a mannose amine, thereby mannosylating the outer surface of the particle. This treatment may cause the particle to bind to the target cell
or tissue at a mannose receptor on the antigen presenting cell surface.
Alternatively, surface conjugation with an immunoglobulin molecule containing an Fc portion (targeting Fc receptor), heat shock protein moiety (HSP receptor), phosphatidylserine (scavenger receptors), and
lipopolysaccharide (LPS) are additional receptor targets on cells or tissue.
Lectins can be covalently attached to micro- and nanoparticles to render them target specific to the mucin and mucosal cell layer.
The choice of targeting molecule will depend on the method of administration of the nanoparticle composition and the cells or tissues to be targeted. The targeting molecule may generally increase the binding affinity of the particles for cell or tissues or may target the nanoparticle to a particular tissue in an organ or a particular cell type in a tissue. The covalent attachment of any of the natural components of mucin in either pure or partially purified form to the particles would decrease the surface tension of the bead-gut interface and increase the solubility of the bead in the mucin layer. The attachment of polyamino acids containing extra pendant carboxylic acid side groups, e.g., polyaspartic acid and poly glutamic acid, may also provide a useful means of increasing bioadhesiveness. Using polyamino acids in the 15,000 to 50,000 kDa molecular weight range yields chains of 120 to 425 amino acid residues attached to the surface of the particles. The polyamino chains may increase bioadhesion by means of chain entanglement in mucin strands as well as by increased carboxylic charge.
In some embodiments, the nanoparticles may further include epithelial cell targeting molecules, such as, antibodies or bioactive fragments thereof that recognize and bind to epitopes displayed on the surface of epithelial cells, or ligands which bind to an epithelial cell surface receptor. Examples of suitable receptors include, but are not limited to, IgE Fc receptors, EpCAM, selected carbohydrate specificites, dipeptidyl peptidase, and E-cadherin.
The efficiency of nanoparticle delivery systems can also be improved by the attachment of functional ligands to the NP surface. Potential ligands include, but are not limited to, small molecules, cell-penetrating peptides (CPPs), targeting peptides, antibodies or aptamers (Yu, et al., PLoS One., 6:e24077 (2011), Cu, et al., J Control Release, 156:258-264 (2011), Nie, et
al., J Control Release, 138:64-70 (2009), Cruz, et al., J Control Release, 144:118-126 (2010)). In some embodiments, the functional molecule is a CPP such as mTAT (HIV-1 (with histidine modification)
HHHHRKKRRQRRRRHHHHH (SEQ ID NO:l) (Yamano, et al., J Control Release, 152:278-285 (2011)); or bPrPp (Bovine prion)
M VKS KIGS WILVLFV AMWSD V GLCKKRPKP (SEQ ID NO:2)
(Magzoub, et al., Biochem Biophys Res Commun., 348:379-385 (2006)); or MPG (Synthetic chimera: SV40 Lg T. Ant.+HIV gb41 coat)
GALFLGFLGAAGSTMGAWS QPKKKRKV (SEQ ID NOG) (Endoh, et al., Adv Drug Deliv Rev., 61:704-709 (2009)). Attachment of these moieties serves a variety of different functions; such as inducing intracellular uptake, endosome disruption, and delivery to the nucleus.
VI. Methods
The compositions and technologies can be used for in vivo, in vitro, and ex vivo gene editing. The methods typically include contacting a cell with an effective amount of gene editing technology, in combination with plerixafor to modify the cell’s genome. The preferred methods typically include contacting cells, for example, in vivo, in vitro or ex vivo, for example, isolated cells, with an effective amount of plerixafor or a compound related thereto.
In one preferred embodiment, the method includes contacting a population of target cells with an effective amount of gene editing technology and optional donor oligonucleotide, in combination with plerixafor, to modify the genomes of a sufficient number of cells to achieve a therapeutic result.
A therapeutically effective amount can be an in vivo dosage of the compositions or technology or of in vitro or ex vivo cells modified by the composition or technology sufficient to treat, inhibit, or alleviate one or more symptoms of a disease or disorder, or to otherwise provide a desired pharmacologic and/or physiologic effect, for example, reducing, inhibiting, or reversing one or more of the pathophysiological mechanisms underlying a disease or disorder.
In some embodiments, when the gene editing technology is triplex forming molecules, the molecules can be administered in an effective
amount to induce formation of a triple helix at the target site. An effective amount of gene editing technology such as triplex-forming molecules may also be an amount effective to increase the rate of recombination of a donor fragment relative to administration of the donor fragment in the absence of the gene editing technology. The formulation of the plerixafor, gene editing technology, and donor oligonucleotide is made to suit the mode of delivery.
The compositions or technologies can be contacted with a target cell once, twice, or three time daily; one, two, three, four, five, six, seven times a week, one, two, three, four, five, six, seven or eight times a month.
The compositions or technologies may or may not be administered at the same time. In some embodiments, the plerixafor is contacted with the target cell prior to the gene editing technology and/or donor oligonucleotide. The plerixafor can be contacted with the target cell, for example, 1, 2, 3, 4, 5, 6, 8, 10, 12, 18, or 24 hours, or 1, 2, 3, 4, 5, 6, or 7 days, or any combination thereof prior to the gene editing technology and/or donor oligonucleotide.
In some embodiments, the gene editing technology and/or donor oligonucleotide is contacted with the target cell prior to the plerixafor. The gene editing technology can be contacted with the target cell, for example, 1 , 2, 3, 4, 5, 6, 8, 10, 12, 18, or 24 hours, or 1, 2, 3, 4, 5, 6, or 7 days, or any combination thereof prior to the plerixafor
In preferred embodiments, the compositions or technologies are contacted with a target cell in an amount effective to induce gene
modification in at least one target allele to occur at frequency of at least 0.01, 0.02. 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.1, 0.2. 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25% of target cells. In some embodiments, particularly ex vivo applications, gene modification occurs in at least one target allele at a frequency of about 0.1-25%, or 0.5-25%, or 1-25% 2-25%, or 3-25%, or 4- 25% or 5-25% or 6-25%, or 7-25%, or 8-25%, or 9-25%, or 10-25%, 11- 25%, or 12-25%, or 13%-25% or 14%-25% or 15-25%, or 2-20%, or 3-20%, or 4-20% or 5-20% or 6-20%, or 7-20%, or 8-20%, or 9-20%, or 10-20%, 11-20%, or 12-20%, or 13%-20% or 14%-20% or 15-20%, 2-15%, or 3-15%, or 4-15% or 5-15% or 6-15%, or 7-15%, or 8-15%, or 9-15%, or 10-15%, 11-15%, or 12-15%, or 13%-15% or 14%-15%.
In some embodiments, gene modification occurs in at least one target allele at a frequency of about 0.1% to about 10%, or about 0.2% to about 10%, or about 0.3% to about 10%, or about 0.4% to about 10%, or about 0.5% to about 10%, or about 0.6% to about 10%, or about 0.7% to about 10%, or about 0.8% to about 10%, or about 0.9% to about 10%, or about 1.0% to about 10% , or about 1.1% to about 10%, or about 1.1% to about 10%, 1.2% to about 10%, or about 1.3% to about 10%, or about 1.4% to about 10%, or about 1.5% to about 10%, or about 1.6% to about 10%, or about 1.7% to about 10%, or about 1.8% to about 10%, or about 1.9% to about 10%, or about 2.0% to about 10%, or about 2.5% to about 10% , or about 3.0% to about 10%, or about 3.5% to about 10%, or about 4.0% to about 10%, or about 4.5% to about 10%, or about 5.0% to about 10%, or about 1% to about 5%, about 1.5% to about 5%, about 2.0% to about 5%, or about 2.5% to about 5% , or about 3.0% to about 5%, or about 3.5% to about 5%, or about 4.0% to about 5%, or about 4.5% to about 5%.
In some embodiments, particularly in vivo applications, gene modification occurs in at least one target allele at a frequency of about 0.1% to about 15%, or about 0.2% to about 15%, or about 0.3% to about 15%, or about 0.4% to about 15%, or about 0.5% to about 15%, or about 0.6% to about 15%, or about 0.7% to about 15%, or about 0.8% to about 15%, or about 0.9% to about 15%, or about 1.0% to about 15% , or about 1.1% to about 15%, or about 1.1% to about 15%, 1.2% to about 15%, or about 1.3% to about 15%, or about 1.4% to about 15%, or about 1.5% to about 15%, or about 1.6% to about 15%, or about 1.7% to about 15%, or about 1.8% to about 15%, or about 1.9% to about 15%, or about 2.0% to about 15%, or about 2.5% to about 15% , or about 3.0% to about 15%, or about 3.5% to about 15%, or about 4.0% to about 15%, or about 4.5% to about 15%, or about 5.0% to about 15%, or about 1% to about 15%, about 1.5% to about 15%, about 2.0% to about 15%, or about 2.5% to about 15% , or about 3.0% to about 15%, or about 3.5% to about 15%, or about 4.0% to about 15%, or about 4.5% to about 15%.
In some embodiments, gene modification occurs with low off-target effects. In some embodiments, off-target modification is undetectable using routine analysis such as those described in the Examples. In some
embodiments, off-target incidents occur at a frequency of 0-1%, or 0-0.1%, or 0-0.01%, or 0-0.001%, or 0-0.0001%, or 0-0000.1%, or 0-0.000001%. In some embodiments, off-target modification occurs at a frequency that is about 102, 103, 104, or 105 -fold lower than at the target site.
A. Ex vivo Gene Therapy
In some embodiments, ex vivo gene therapy of cells is used for the treatment of a genetic disorder in a subject. For ex vivo gene therapy, cells are isolated from a subject and contacted ex vivo with the compositions or technologies (plerixafor, gene editing technology, and/or donor
oligonucleotide) to produce cells containing altered sequences in or adjacent to genes. In a preferred embodiment, the cells are isolated from the subject to be treated or from a syngeneic host. Target cells may be removed from a subject prior to contacting with a gene editing technology and plerixafor. The cells can be hematopoietic progenitor or stem cells. In a preferred embodiment, the target cells are CD34+ hematopoietic stem cells.
Hematopoietic stem cells (HSCs), such as CD34+ cells are multipotent stem cells that give rise to all the blood cell types including erythrocytes.
Therefore, CD34+ cells can be isolated from a patient with, for example, thalassemia, sickle cell disease, or a lysosomal storage disease, the mutant gene altered or repaired ex-vivo using the compositions or technologies and methods, and the cells reintroduced back into the patient as a treatment or a cure.
Stem cells can be isolated and enriched by one of skill in the art. Methods for such isolation and enrichment of CD34+ and other cells are known in the art and disclosed for example in U.S. Patent Nos. 4,965,204; 4,714,680; 5,061,620; 5,643,741; 5,677,136; 5,716,827; 5,750,397 and 5,759,793. As used herein in the context of compositions enriched in hematopoietic progenitor and stem cells,“enriched” indicates a proportion of a desirable element (e.g. hematopoietic progenitor and stem cells) which is higher than that found in the natural source of the cells. A composition of cells may be enriched over a natural source of the cells by at least one order of magnitude, preferably two or three orders, and more preferably 10, 100, 200 or 1000 orders of magnitude.
In humans, CD34+ cells can be recovered from cord blood, bone marrow or from blood after cytokine mobilization effected by injecting the donor with hematopoietic growth factors such as granulocyte colony stimulating factor (G-CSF), granulocyte-monocyte colony stimulating factor (GM-CSF), stem cell factor (SCF) subcutaneously or intravenously in amounts sufficient to cause movement of hematopoietic stem cells from the bone marrow space into the peripheral circulation. Mobilization can also be achieved by injection with plerixafor or a compound related thereto).
Initially, bone marrow cells may be obtained from any suitable source of bone marrow, e.g. tibiae, femora, spine, and other bone cavities. For isolation of bone marrow, an appropriate solution may be used to flush the bone, which solution will be a balanced salt solution, conveniently supplemented with fetal calf serum or other naturally occurring factors, in conjunction with an acceptable buffer at low concentration, generally from about 5 to 25 mM. Convenient buffers include Hepes, phosphate buffers, lactate buffers, etc.
Cells can be selected by positive and negative selection techniques. Cells can be selected using commercially available antibodies which bind to hematopoietic progenitor or stem cell surface antigens, e.g. CD34, using methods known to those of skill in the art. For example, the antibodies may be conjugated to magnetic beads and immunogenic procedures utilized to recover the desired cell type. Other techniques involve the use of fluorescence activated cell sorting (FACS). The CD34 antigen, which is found on progenitor cells within the hematopoietic system of non-leukemic individuals, is expressed on a population of cells recognized by the monoclonal antibody My- 10 (i.e., express the CD34 antigen) and can be used to isolate stem cell for bone marrow transplantation. My- 10 deposited with the American Type Culture Collection (Rockville, Md.) as HB-8483 is commercially available as anti-HPCA 1. Additionally, negative selection of differentiated and“dedicated” cells from human bone marrow can be utilized, to select against substantially any desired cell marker. For example, progenitor or stem cells, most preferably CD34+ cells, can be characterized as being any of CD3 , CD7 , CD8 , CD10 , CD14 , CD15 , CD19 , CD20 , CD33 , Class II HLA+ and Thy-1+.
Once progenitor or stem cells have been isolated, they may be propagated by growing in any suitable medium. For example, progenitor or stem cells can be grown in conditioned medium from stromal cells, such as those that can be obtained from bone marrow or liver associated with the secretion of factors, or in medium including cell surface factors supporting the proliferation of stem cells. Stromal cells may be freed of hematopoietic cells employing appropriate monoclonal antibodies for removal of the undesired cells.
The isolated cells are contacted ex vivo with a combination of a gene editing technology, plerixafor, and donor oligonucleotide(s) in amounts effective to cause the desired alterations in or adjacent to genes in need of repair or alteration, for example the human beta-globin or a-L-iduronidase gene. These cells are referred to herein as modified cells. Methods for transfection of cells with oligonucleotides are well known in the art
(Koppelhus, et al., Adv. Drug Deliv. Rev., 55(2): 267-280 (2003)). It may be desirable to synchronize the cells in S-phase to further increase the frequency of gene correction. Methods for synchronizing cultured cells, for example, by double thymidine block, are known in the art (Zielke, et al., Methods Cell Biol., 8: 107-121 (1974)).
The modified cells can be maintained or expanded in culture prior to administration to a subject. Culture conditions are generally known in the art depending on the cell type. Conditions for the maintenance of CD34+ in particular have been well studied, and several suitable methods are available. A common approach to ex vivo multi-potential hematopoietic cell expansion is to culture purified progenitor or stem cells in the presence of early-acting cytokines such as interleukin- 3. It has also been shown that inclusion, in a nutritive medium for maintaining hematopoietic progenitor cells ex vivo, of a combination of thrombopoietin (TPO), stem cell factor (SCF), and flt3 ligand (Flt-3L; i.e., the ligand of the flt3 gene product) was useful for expanding primitive (i.e., relatively non-differentiated) human hematopoietic progenitor cells in vitro, and that those cells were capable of engraftment in SCID-hu mice (Luens et al., 1998, Blood 91:1206-1215). In other known methods, cells can be maintained ex vivo in a nutritive medium (e.g., for minutes,
hours, or 3, 6, 9, 13, or more days) including murine prolactin- like protein E (mPLP-E) or murine prolactin- like protein F (mPIP-F; collectively mPLP- E/IF) (U.S. Patent No. 6,261,841). It will be appreciated that other suitable cell culture and expansion methods can be used as well. Cells can also be grown in serum-free medium, as described in U.S. Patent No. 5,945,337.
In another embodiment, the modified hematopoietic stem cells are differentiated ex vivo into CD4+ cells culture using specific combinations of interleukins and growth factors prior to administration to a subject using methods well known in the art. The cells may be expanded ex vivo in large numbers, preferably at least a 5-fold, more preferably at least a 10-fold and even more preferably at least a 20-fold expansion of cells compared to the original population of isolated hematopoietic stem cells.
In another embodiment cells, for ex vivo gene therapy can be dedifferentiated somatic cells. Somatic cells can be reprogrammed to become pluripotent stem-like cells that can be induced to become hematopoietic progenitor cells. The hematopoietic progenitor cells can then be treated with plerixafor and gene editing technology preferably in combination with a donor oligonucleotide to produce recombinant cells having one or more modified genes. Representative somatic cells that can be reprogrammed include, but are not limited to, fibroblasts, adipocytes, and muscles cells. Hematopoietic progenitor cells from induced stem- like cells have been successfully developed in the mouse (Hanna, J. et al. Science, 318:1920- 1923 (2007)).
To produce hematopoietic progenitor cells from induced stem- like cells, somatic cells may be harvested from a host. In a preferred
embodiment, the somatic cells are autologous fibroblasts. The cells are cultured and transduced with vectors encoding Oct4, Sox2, Klf4, and c-Myc transcription factors. The transduced cells may be cultured and screened for embryonic stem cell (ES) morphology and ES cell markers including, but not limited to AP, SSEA1, and Nanog. The transduced ES cells may then be cultured and induced to produce induced stem-like cells. Cells may then be screened for CD41 and c-kit markers (early hematopoietic progenitor markers) as well as markers for myeloid and erythroid differentiation.
The modified hematopoietic stem cells or modified induced hematopoietic progenitor cells are then introduced into a subject. Delivery of the cells may be affected using various methods and includes most preferably intravenous administration by infusion as well as direct depot injection into periosteal, bone marrow and/or subcutaneous sites.
The subject receiving the modified cells may be treated for bone marrow conditioning to enhance engraftment of the cells. The recipient may be treated to enhance engraftment, using a radiation or chemotherapeutic treatment prior to the administration of the cells. Upon administration, the cells will generally require a period of time to engraft. Achieving significant engraftment of hematopoietic stem or progenitor cells may take days, weeks, or months.
A high percentage of engraftment of modified hematopoietic stem cells may not be necessary to achieve significant prophylactic or therapeutic effect. It is believed that the engrafted cells will expand over time following engraftment to increase the percentage of modified cells. For example, in some embodiments, the modified cells have a corrected target gene (e.g., an oc-L-iduronidase gene). For example, in a subject with Hurler syndrome, the modified cells can improve or cure the condition. It is believed that engraftment of only a small number or small percentage of modified hematopoietic stem cells will be required to provide a prophylactic or therapeutic effect.
In preferred embodiments, the cells to be administered to a subject will be autologous, e.g. derived from the subject, or syngeneic.
In some embodiments, the compositions or technologies and methods can be used to edit embryonic genomes in vitro. The methods typically include contacting an embryo in vitro with an effective amount of plerixafor and gene editing technology to induce at least one alteration in the genome of the embryo. Most preferably the embryo is a single cell zygote, however, treatment of male and female gametes prior to and during fertilization, and embryos having 2, 4, 8, or 16 cells and including not only zygotes, but also morulas and blastocytes, are also provided. Typically, the embryo is
contacted with the compositions or technologies on culture days 0-6 during or following in vitro fertilization.
The contacting can be adding the compositions or technologies to liquid media bathing the embryo. For example, the compositions or technologies can be pipetted directly into the embryo culture media, whereupon they are taken up by the embryo.
B. In vivo Gene Therapy
In some embodiments, in vivo gene therapy of cells is used for the treatment of a genetic disorder in a subject. The compositions or
technologies can be administered directly to a subject for in vivo gene therapy.
In general, methods of administering compounds, including plerixafor or a compound related thereto, oligonucleotides and related molecules, are well known in the art. In particular, the routes of
administration already in use for nucleic acid therapeutics, along with formulations in current use, provide preferred routes of administration and formulation for the gene editing technologies described above. Plerixafor is preferably administered by injection or infusion. Preferably the
compositions or technologies are injected or infused into the organism undergoing genetic manipulation, such as an animal requiring gene therapy.
The composition or technologies can be administered by a number of routes including, but not limited to, intravenous, intraperitoneal,
intraamniotic, intramuscular, subcutaneous, or topical (sublingual, rectal, intranasal, pulmonary, rectal mucosa, and vaginal), and oral (sublingual, buccal).
In some embodiments, the compounds are formulated for pulmonary delivery, such as intranasal administration or oral inhalation.
Administration of the formulations may be accomplished by any acceptable method that allows the plerixafor, gene editing technology, and/or donor oligonucleotide to reach their targets. The administration may be localized (i.e., to a particular region, physiological system, tissue, organ, or cell type) or systemic, depending on the condition being treated.
Compositions or technologies and methods for in vivo delivery are also discussed in WO 2017/143042.
The methods can also include administering an effective amount of plerixafor and gene editing technology to an embryo or fetus, or the pregnant mother thereof, in vivo. In some methods, compositions or technologies are delivered in utero by injecting and/or infusing the compositions or technologies into a vein or artery, such as the vitelline vein or the umbilical vein, or into the amniotic sac of an embryo or fetus. See, e.g., Ricciardi, et al., Nat Commun. 2018 Jun 26;9(1):2481. doi: 10.1038/s41467-018-04894-2.
C. Diseases to Be Treated
Gene therapy is apparent when studied in the context of human genetic diseases, for example, cystic fibrosis, hemophilia, muscular dystrophy, globinopathies such as sickle cell anemia and beta-thalassemia, xeroderma pigmentosum, lysosomal storage diseases, immune deficiency syndromes such as X-linked severe combined immunodeficiency and ADA deficiency, tyrosinemia, Fanconi anemia, the red cell disorder spherocytosis, alpha- 1 -anti-trypsin deficiency, Wilson’s disease, Leber’s hereditary optic neuropathy, and chronic granulomatous disorder. The strategies are also useful for treating non-genetic disease such as HIV, in the context of ex vivo- based cell modification and also for in vivo cell modification.
The methods using plerixafor in combination with gene editing technologies and/or donor oligonucleotides may be especially useful to treat genetic deficiencies, disorders and diseases caused by mutations in single genes, for example, to correct genetic deficiencies, disorders and diseases caused by point mutations. If the target gene contains a mutation that is the cause of a genetic disorder, then the disclosed methods can be used for mutagenic repair that may restore the DNA sequence of the target gene to normal. The target sequence can be within the coding DNA sequence of the gene or within an intron. The target sequence can also be within DNA sequences that regulate expression of the target gene, including promoter or enhancer sequences.
In the methods disclosed herein, cells that have been contacted with the plerixafor in combination with the gene editing technology and donor oligonucleotide may be administered to a subject. The subject may have a disease or disorder such as hemophilia, muscular dystrophy, globinopathies, cystic fibrosis, xeroderma pigmentosum, lysosomal storage diseases,
immune deficiency syndromes such as X-linked severe combined immunodeficiency and ADA deficiency, tyrosinemia, Fanconi anemia, the red cell disorder spherocytosis, alpha- 1-anti-trypsin deficiency, Wilson’s disease, Leber’ s hereditary optic neuropathy, or chronic granulomatous disorder. In such embodiments, gene modification may occur in an effective amount to reduce one or more symptoms of the disease or disorder in the subject. Exemplary sequences for triplex-forming molecules and donor oligonucleotides designed to correct mutations in globinopathies, cystic fibrosis, HIV, and lysosomal storage diseases are known in the art and disclosed in, for example, published in international applications WO 2017/143042, WO 2017/143061, and published U.S. Application No.
2017/0283830, each of which is specifically incorporated by reference in its entirety.
D. Combination Therapies
The combination of plerixafor and a gene editing technology and/or donor oligonucleotide can be contacted with cells alone or in further combination with one or more additional active agents. In all cases, the combination of agents can be part of the same admixture or administered as separate composition or technologies.
Examples of preferred additional active agents include other conventional therapies known in the art for treating the desired disease or condition. For example, in the treatment of sickle cell disease, the additional therapy may be hydroxyurea.
In the treatment of cystic fibrosis, the additional therapy may include mucolytics, antibiotics, nutritional agents, etc. Specific drugs are outlined in the Cystic Fibrosis Foundation drug pipeline and include, but are not limited to, CFTR modulators such as KALYDECO® (ivacaftor), ORKAMBI™ (lumacaftor + ivacaftor), ataluren (PTC124), VX-661 + invacaftor, riociguat, QBW251, N91115, and QR-010; agents that improve airway surface liquid such as hypertonic saline, bronchitol, and P-1037; mucus alteration agents such as PULMOZYME® (dornase alfa); anti-inflammatories such as ibuprofen, alpha 1 anti-trypsin, CTX-4430, and JBT-101; anti-infective such as inhaled tobramycin, azithromycin, CAYSTON® (aztreonam for inhalation solution), TOBI inhaled powder, levofloxacin, ARIKACE® (nebulized
liposomal amikacin), AEROVANC® (vancomycin hydrochloride inhalation powder), and gallium; and nutritional supplements such as aquADEKs, pancrelipase enzyme products, liprotamase, and burlulipase.
In the treatment of HIV, the additional therapy maybe an
antiretroviral agents including, but not limited to, a non-nucleoside reverse transcriptase inhibitor (NNRTIs), a nucleoside reverse transcriptase inhibitor (NRTIs), a protease inhibitors (Pis), a fusion inhibitors, a CCR5 antagonists (CCR5s) (also called entry inhibitors), an integrase strand transfer inhibitors (INSTIs), or a combination thereof.
In the treatment of lysosomal storage disease, the additional therapy could include, for example, enzyme replacement therapy, bone marrow transplantation, or a combination thereof.
E. Determining Gene Modification
Sequencing and allele- specific PCR are preferred methods for determining if gene modification has occurred. PCR primers may be designed to distinguish between the original allele, and the new predicted sequence following recombination. Other methods of determining if a recombination event has occurred are known in the art and may be selected based on the type of modification made. Methods include, but are not limited to, analysis of genomic DNA, for example by sequencing, allele-specific PCR, droplet digital PCR, or restriction endonuclease selective PCR (REMS- PCR); analysis of mRNA transcribed from the target gene for example by Northern blot, in situ hybridization, real-time or quantitative reverse transcriptase (RT) PCR; and analysis of the polypeptide encoded by the target gene, for example, by immunostaining, ELISA, or FACS. In some cases, modified cells will be compared to parental controls. Other methods may include testing for changes in the function of the RNA transcribed by, or the polypeptide encoded by the target gene. For example, if the target gene encodes an enzyme, an assay designed to test enzyme function may be used.
EXAMPLES
Example 1: Plerixafor enhances PNA-mediated editing of the beta glohin gene of Townes mouse-derived BM cells
Materials and Methods
PNA and Donor DNA
The sequence of the triplex forming PNA (designated SCD-tcPNA
2A) was H-KKK-TTJJTJT-OOO-TCTCCTTAAACCTGTCTT - KKK-
Nth (SEQ ID NO:4), where, the underlined nucleobases have a gamma mini-PEG side chain substitution, J=pseudoisocytosine, K=lysine, and 0=flexible octanoic acid linker. The relative position of tcPNA2 in the beta globin locus is shown in Fig. 1A. SCD-tcPNA 2 has the same sequence as SCD-tcPNA 2 A, but without the gamma mini-PEG side chain substitutions.
The single-stranded donor DNA oligonucleotide was prepared by standard DNA synthesis. The sequence of the donor DNA was 5’- T(s)T(s)G(s)CCCCACAGGGCAGTAACGGCAGACTTCTCCTCAGGA GTCAGGTGCACCATGGTGTCTGT(s)T(s)T(s)G- 3’ (SEQ ID NO:5) where“(s)” indicates substitution of the usual phosphodiester
intemucleoside linkage with a phosphorothioate linkage, for enhanced resistance to exonucleases.
Nanoparticle synthesis
The polymeric PLGA nanoparticles used to deliver the donor oligonucleotide and PNA were synthesized by a double-emulsion solvent evaporation protocol as described by Bahai, et ak, Nat. Commun., 7:13304 (2016). Briefly, poly(lactic-co-glycolic) acid (PLGA) was dissolved in dichloromethane at a concentration of 40 mg/ml. Prior to encapsulation,
PNA and donor DNA were mixed at a 2:1 molar ratio and added dropwise to the PLGA solution under vortex. For NPs containing donor DNA alone, DNA was added dropwise at a molar ratio of 2 nmoles/mg of polymer. The resulting mixture was sonicated three times for 10 seconds using an amplitude of 38%. The water-in-oil emulsion was subsequently added dropwise to a surfactant solution containing polyvinyl alcohol (5% w/v). Following the second emulsion, the sonication step was repeated as described. The resulting nanoparticle solution was added to 25 ml of a 0.3% PVA solution and allowed to stir for 3 hours at room temperature. After
stirring and particle‘hardening,’ the nanoparticles were washed 3 times via centrifugation (16,100 g, 15 min, 4 °C) before being flash frozen and lyophilized in cryoprotectant (trehalose, mg:mg). Dry nanoparticles were stored at -20 °C until later use.
Mouse Model for Sickle Cells Disease
In sickle cell disease (SCD), the mutation (GAG->GTG) at codon 6 results in glutamic acid being changed to valine. For correction (editing) of this SCD mutation site, studies were performed in the Townes mouse model (Jackson Laboratory).
The Townes mouse model was developed by Ryan TM, Ciavatta DJ, Townes TM.,“Knockout-transgenic mouse model of sickle cell disease.” Science. 1997 Oct 31 ;278(5339): 873-6. PMID: 9346487.
Townes mice exclusively express human sickle hemoglobin (HbS). They were produced by generating transgenic mice expressing human a -, g-, and s-globin that were then bred with knockout mice that had deletions of the murine a- and b-globin genes. Thus, the resulting progeny no longer express mouse a- and b-globin. Instead, they express exclusively human a- and b^IoMh. Hence, the mice express human sickle hemoglobin and possess many of the major hematologic and histopathologic features of individuals with SCD.
Cell culture and treatment
Bone marrow cells were harvested from Townes mice (Jackson Laboratory). Hematopoietic stem cells and progenitor cells were isolated based on their cell surface marker profile (CD5 , CD lib , CD 19 ,
CD45R/B220 , Ly6G/C(Gr-l)’ TER119 ) and enriched via negative selection (EasySep cell separation kit from Stem Cell Technologies).
Cells were treated with nanoparticles containing the PNA/donor DNA designed to bind to and correct the beta globin gene at the site of the SCD mutation (A:T to T:A). Some samples were additionally treated with stem cell factor (SCF), erythropoietin (EPO), or Plerixafor. Cells were seeded at 500,000 cells per well. Cells were subsequently treated with EPO (1 unit/ml), SCF (3.3 ug/ml), or Plerixafor (100 mM) for 5 minutes. After incubation with SCF, EPO, or Plerixafor, cells were treated with 2 mg/ml NPs.
After 48 hours, the cells were washed prior to genomic DNA isolation (SV Wizard, Promega). Freshly isolated genomic DNA was analyzed by droplet digital PCR (ddPCR) to quantify gene editing frequencies.
Results
As shown in Figure 1C, cells treated with blank nanoparticles yielded no gene editing, while PNA/donor DNA treated cells showed a base editing frequency of approximately 3%. Treatment with SCF or EPO enhanced PNA/donor DNA mediated editing (Fig. ID). Notably, treatment with Plerixafor substantially boosted PNA donor DNA mediated editing, achieving editing frequencies of approximately 13% (Fig. ID).
Unless defined otherwise, all technical and scientific terms used herein have the same meanings as commonly understood by one of skill in the art to which the disclosed invention belongs. Publications cited herein and the materials for which they are cited are specifically incorporated by reference.
Those skilled in the art will recognize, or be able to ascertain using no more than routine experimentation, many equivalents to the specific embodiments of the invention described herein. Such equivalents are intended to be encompassed by the following claims.
Claims
1. A kit comprising
(i) plerixafor in a dosage unit form for administration to an individual or a cell, and
(ii) a gene editing technology that can induce genomic modification of the cell.
2. The kit of claim 1, comprising a donor oligonucleotide that induces a mutation(s) in the cell’s genome by insertion or recombination induced or enhanced by the gene editing technology.
3. The kit of any one of claims 1 or 2, wherein plerixafor is a compound of Formula (I) or a pharmaceutically acceptable salt thereof.
4. The kit of any one of claims 1-3, wherein the kit comprises a pharmaceutically acceptable salt of plerixafor.
5. The kit of any one of claims 1-4, wherein the gene editing technology comprises a triplex- forming molecule or a pseudocomplementary oligonucleotide.
6. The kit of any one of claims 1-5, wherein the gene editing technology comprises a triplex-forming molecule.
7. The kit of claim 6, wherein the triplex-forming molecule comprises a peptide nucleic acid (PNA).
8. The kit of any one of claims 2-7, wherein the donor oligonucleotide comprises DNA.
9. The kit of any one of claims 2-7, wherein the donor oligonucleotide is single stranded or double stranded.
10. The kit of any one of claims 1-4, wherein the gene editing technology comprises, a CRISPR system, a zinc finger nuclease (ZFN), a transcription activator-like effector nuclease (TALEN), small fragment homologous replacement, a single-stranded oligodeoxynucleotide-mediated gene modification, or an intron encoded meganuclease.
11. The kit of any one of claims 1-10, wherein the cell’s genome has a mutation underlying a disease or disorder selected from the group comprising hemophilia, muscular dystrophy, a globinopathy, cystic fibrosis,
xeroderma pigmentosum, a lysosomal storage disease, an immune deficiency syndrome such as X-linked severe combined immunodeficiency or ADA deficiency, tyrosinemia, Fanconi anemia, spherocytosis, alpha- 1-anti-trypsin deficiency, Wilson’ s disease, Leber’ s hereditary optic neuropathy, and chronic granulomatous disorder.
12. The kit of claim 11, wherein the mutation is in a gene encoding coagulation factor VIII, coagulation factor IX, dystrophin, beta-globin, CFTR, XPC, XPD, DNA polymerase eta, Fanconi anemia genes A through L, SPTA1 and other spectrin genes, ANK1 gene, SERPINA1 gene, ATP7B gene, interleukin 2 receptor gamma (IL2RG) gene, ADA gene, FAH gene, and genes linked to chronic granulomatous disease including the CYBA, CYBB, NCF1, NCF2, or NCF4 genes.
13. The kit of claim 12, comprising a donor oligonucleotide wherein the oligonucleotide sequence corresponds to a portion of the wild type sequence of the gene.
14. The kit of any one of claims 1-13, further comprising a nuclease.
15. A composition comprising:
(i) plerixafor, and
(ii) a gene editing technology that can induce genomic modification of a cell.
16. The composition of claim 15, comprising a donor oligonucleotide that induces a mutation(s) in the cell’s genome by insertion or recombination induced or enhanced by the gene editing technology.
17. The composition of claims 15 or 16, wherein plerixafor is a compound of Formula (I) or a pharmaceutically acceptable salt thereof.
18. The composition of any one of claims 15-17, wherein the composition comprises the compound having the formula:
19. The composition of any one of claims 15-18, wherein the gene editing technology comprises a triplex-forming molecule or a
pseudocomplementary oligonucleotide.
20. The composition of claim 19, wherein the gene editing technology comprises a triplex-forming molecule.
21. The composition of claim 20, wherein the triplex-forming molecule comprises a peptide nucleic acid (PNA).
22. The composition of any one of claims 15-18, wherein the gene editing technology comprises a CRISPR system, a zinc finger nuclease (ZFN), a transcription activator-like effector nuclease (TALEN), small fragment homologous replacement, a single- stranded oligodeoxynucleotide- mediated gene modification, or an intron encoded meganuclease.
23. The composition of any one of claims 16-21, wherein the donor oligonucleotide comprises DNA.
24. The composition of any one of claims 16-21, wherein the donor oligonucleotide is single stranded or double stranded.
25. The composition of any one of claims 15-24, wherein the cell’s genome has a mutation underlying a disease or disorder selected from the group comprising hemophilia, muscular dystrophy, a globinopathy, cystic fibrosis, xeroderma pigmentosum, a lysosomal storage disease, an immune deficiency syndrome such as X-linked severe combined immunodeficiency or ADA deficiency, tyrosinemia, Fanconi anemia, spherocytosis, alpha- 1- anti-trypsin deficiency, Wilson’s disease, Leber’s hereditary optic neuropathy, and chronic granulomatous disorder.
26. The composition of claim 25, wherein the mutation is in a gene encoding coagulation factor VIII, coagulation factor IX, dystrophin, beta- globin, CFTR, XPC, XPD, DNA polymerase eta, Fanconi anemia genes A through L, SPTA1 and other spectrin genes, ANK1 gene, SERPINA1 gene, ATP7B gene, interleukin 2 receptor gamma (IL2RG) gene, ADA gene, FAH gene, and genes linked to chronic granulomatous disease including the CYBA, CYBB, NCF1, NCF2, or NCF4 genes.
27. The composition of claim 24, comprising a donor oligonucleotide wherein the oligonucleotide sequence corresponds to a portion of the wild type sequence of the gene.
28. A pharmaceutical composition comprising the composition of any one of claims 15-27 and a pharmaceutically acceptable excipient.
29. The pharmaceutical composition of claim 28, further comprising polymeric nanoparticles.
30. A method of modifying the genome of a cell comprising contacting cells in vivo, in vitro, or ex vivo with an effective amount of plerixafor or a compound related thereto and a gene editing technology that can induce genomic modification of the cells, to increase modification of the cells’ genome compared to contacting the cells with just the gene editing technology.
31. The method of modifying the genome of the cells of claim 30, comprising contacting cells in an individual with the components of the kit of any of claims 1-14.
32. The method of modifying the genome of the cells of claim 30, comprising contacting cells in an individual with the composition of claims 15-29.
33. The method of claim 30 or 31, wherein the gene editing technology and the plerixafor or a compound related thereto are part of the same or different compositions.
34. The method of any one of claims 30-33, wherein (i) plerixafor, (ii) the gene editing technology that can induce genomic modification of the
cells, and optionally donor oligonucleotide are contacted with the cell at the same or different times.
35. The method of any one of claims 30-34 wherein (i) plerixafor, (ii) the gene editing technology that can induce genomic modification of the cells, and optionally donor oligonucleotide are administered separately.
36. The method of any one of claims 30-35, wherein the plerixafor is a compound of Formula (I) or a pharmaceutically acceptable salt thereof as described herein.
37. The method of any one of claims 30-36, wherein the gene editing technology comprises a triplex-forming molecule or a pseudocomplementary oligonucleotide.
38. The method of claim 37, wherein the gene editing technology comprises a triplex-forming molecule.
39. The method of claim 38, wherein the triplex-forming molecule comprises peptide nucleic acids (PNA).
40. The method of any one of claims 30-36, wherein the gene editing technology comprises a CRISPR system, a zinc finger nuclease (ZFN), a transcription activator-like effector nuclease (TALEN), small fragment homologous replacement, a single-stranded oligodeoxynucleotide-mediated gene modification, or an intron encoded meganuclease.
41. The method of any one of claims 30-40, wherein the method comprises contacting the cells in a subject in vivo.
42. The method of any one of claims 30-40, wherein the method comprises contacting the cells in vitro or ex vivo.
43. The method of any one of claims 30-42, wherein the cells’ genome has a mutation underlying a disease or disorder selected from the group consisting of hemophilia, muscular dystrophy, globinopathies, cystic fibrosis, xeroderma pigmentosum, and lysosomal storage diseases, immune deficiency syndromes such as X-linked severe combined immunodeficiency and ADA deficiency, tyrosinemia, Fanconi anemia, the red cell disorder spherocytosis, alpha- 1 -anti-trypsin deficiency, Wilson’s disease, Leber’s hereditary optic neuropathy, and chronic granulomatous disorder.
44. The method of claim 43, wherein the mutation is in a gene encoding coagulation factor VIII, coagulation factor IX, dystrophin, beta-globin, CFTR, XPC, XPD, DNA polymerase eta, Fanconi anemia genes A through L, SPTA1 and other spectrin genes, ANK1 gene, SERPINA1 gene, ATP7B gene, interleukin 2 receptor gamma (IL2RG) gene, ADA gene, FAH gene, and genes linked to chronic granulomatous disease including the CYBA, CYBB, NCF1, NCF2, or NCF4 genes.
45. The method of any one of claims 30-44, comprising a donor oligonucleotide, wherein the oligonucleotide sequence corresponds to a portion of the wild type sequence of the gene.
46. The method of any one of claims 30-45, wherein the cells are hematopoietic stem cells.
47. The method of any one of claims 30-46, further comprising administering the cells to a subject in need thereof.
48. The method of claim 47, wherein the cells are administered to the subject in an effective amount to treat one or more symptoms of a disease or disorder.
49. The method of claim 48, wherein the subject has a disease or disorder selected from the group consisting of hemophilia, muscular dystrophy, globinopathies, cystic fibrosis, xeroderma pigmentosum, and lysosomal storage diseases, immune deficiency syndromes such as X-linked severe combined immunodeficiency and ADA deficiency, tyrosinemia, Fanconi anemia, the red cell disorder spherocytosis, alpha- 1-anti-trypsin deficiency, Wilson’s disease, Leber’s hereditary optic neuropathy, and chronic granulomatous disorder.
50. The method of claim 49, wherein the cells are administered in an effective amount to reduce one or more symptoms of the disease or disorder in the subject.
51. The method of any one of claims 30-50, wherein (i) plerixafor, (ii) the gene editing technology that can induce genomic modification of the cells, and optional donor oligonucleotide, or any combination thereof are encapsulated in nanoparticles together or separately.
52. The method of claim 51, wherein the nanoparticles comprise polyhydroxy acid polymer.
53. The method of claim 52, wherein the nanoparticles comprise poly(lactic-co-glycolic acid) (PLGA).
54. The method of any one of claims 51-53, wherein a targeting moiety, a cell penetrating peptide, or a combination thereof is associated with, linked, conjugated, or otherwise attached directly or indirectly to the nanoparticle.
55. The kit of any one of claims 1-14, wherein the gene editing technology is a triplex forming molecule or a CRISPR system.
56. The kit of claim 55, wherein the CRISPR system comprises
CRISPR/Cas9 D10A nickase.
57. The composition of any one of claims 15-29, wherein the gene editing technology is a triplex forming molecule or a CRISPR system.
58. The composition of claim 57, wherein the CRISPR system comprises CRISPR/Cas9 D10A nickase.
59. The method any one of claims 30-54, wherein the gene editing technology is a triplex forming molecule or a CRISPR system.
60. The method of claim 59, wherein the CRISPR system comprises CRISPR/Cas9 D10A nickase.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862773977P | 2018-11-30 | 2018-11-30 | |
| US62/773,977 | 2018-11-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020112195A1 true WO2020112195A1 (en) | 2020-06-04 |
Family
ID=67982153
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2019/049018 Ceased WO2020112195A1 (en) | 2018-11-30 | 2019-08-30 | Compositions, technologies and methods of using plerixafor to enhance gene editing |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2020112195A1 (en) |
Citations (54)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US150A (en) | 1837-03-25 | Island | ||
| US5436A (en) | 1848-02-08 | Air-heating furnace | ||
| US4714680A (en) | 1984-02-06 | 1987-12-22 | The Johns Hopkins University | Human stem cells |
| US4965204A (en) | 1984-02-06 | 1990-10-23 | The Johns Hopkins University | Human stem cells and monoclonal antibodies |
| US5021409A (en) | 1989-12-21 | 1991-06-04 | Johnson Matthey Plc | Antiviral cyclic polyamines |
| US5034506A (en) | 1985-03-15 | 1991-07-23 | Anti-Gene Development Group | Uncharged morpholino-based polymers having achiral intersubunit linkages |
| US5061620A (en) | 1990-03-30 | 1991-10-29 | Systemix, Inc. | Human hematopoietic stem cell |
| US5356802A (en) | 1992-04-03 | 1994-10-18 | The Johns Hopkins University | Functional domains in flavobacterium okeanokoites (FokI) restriction endonuclease |
| WO1995001364A1 (en) | 1993-06-25 | 1995-01-12 | Yale University | Chemically modified oligonucleotide for site-directed mutagenesis |
| US5422251A (en) | 1986-11-26 | 1995-06-06 | Princeton University | Triple-stranded nucleic acids |
| US5487994A (en) | 1992-04-03 | 1996-01-30 | The Johns Hopkins University | Insertion and deletion mutants of FokI restriction endonuclease |
| US5527675A (en) | 1993-08-20 | 1996-06-18 | Millipore Corporation | Method for degradation and sequencing of polymers which sequentially eliminate terminal residues |
| US5539082A (en) | 1993-04-26 | 1996-07-23 | Nielsen; Peter E. | Peptide nucleic acids |
| WO1996039195A2 (en) | 1995-06-06 | 1996-12-12 | Yale University | Chemically modified oligonucleotide for site-directed mutagenesis |
| WO1996040898A1 (en) | 1995-06-07 | 1996-12-19 | Yale University | Triple-helix forming oligonucleotides for targeted mutagenesis |
| US5623049A (en) | 1993-09-13 | 1997-04-22 | Bayer Aktiengesellschaft | Nucleic acid-binding oligomers possessing N-branching for therapy and diagnostics |
| US5677136A (en) | 1994-11-14 | 1997-10-14 | Systemix, Inc. | Methods of obtaining compositions enriched for hematopoietic stem cells, compositions derived therefrom and methods of use thereof |
| US5714331A (en) | 1991-05-24 | 1998-02-03 | Buchardt, Deceased; Ole | Peptide nucleic acids having enhanced binding affinity, sequence specificity and solubility |
| US5759793A (en) | 1993-09-30 | 1998-06-02 | Systemix, Inc. | Method for mammalian cell separation from a mixture of cell populations |
| US5786571A (en) | 1997-05-09 | 1998-07-28 | Lexmark International, Inc. | Wrapped temperature sensing assembly |
| WO1998053059A1 (en) | 1997-05-23 | 1998-11-26 | Medical Research Council | Nucleic acid binding proteins |
| US5945337A (en) | 1996-10-18 | 1999-08-31 | Quality Biological, Inc. | Method for culturing CD34+ cells in a serum-free medium |
| US6140081A (en) | 1998-10-16 | 2000-10-31 | The Scripps Research Institute | Zinc finger binding domains for GNN |
| US6261841B1 (en) | 1999-06-25 | 2001-07-17 | The Board Of Trustees Of Northwestern University | Compositions, kits, and methods for modulating survival and differentiation of multi-potential hematopoietic progenitor cells |
| US6326479B1 (en) | 1998-01-27 | 2001-12-04 | Boston Probes, Inc. | Synthetic polymers and methods, kits or compositions for modulating the solubility of same |
| US6441130B1 (en) | 1991-05-24 | 2002-08-27 | Isis Pharmaceuticals, Inc. | Linked peptide nucleic acids |
| US6453242B1 (en) | 1999-01-12 | 2002-09-17 | Sangamo Biosciences, Inc. | Selection of sites for targeting by zinc finger proteins and methods of designing zinc finger proteins to bind to preselected sites |
| US20020165356A1 (en) | 2001-02-21 | 2002-11-07 | The Scripps Research Institute | Zinc finger binding domains for nucleotide sequence ANN |
| WO2003016496A2 (en) | 2001-08-20 | 2003-02-27 | The Scripps Research Institute | Zinc finger binding domains for cnn |
| US6534261B1 (en) | 1999-01-12 | 2003-03-18 | Sangamo Biosciences, Inc. | Regulation of endogenous gene expression in cells using zinc finger proteins |
| WO2003052071A2 (en) | 2001-12-14 | 2003-06-26 | Yale University | Intracellular generation of single-stranded dna |
| US6746838B1 (en) | 1997-05-23 | 2004-06-08 | Gendaq Limited | Nucleic acid binding proteins |
| US20040197892A1 (en) | 2001-04-04 | 2004-10-07 | Michael Moore | Composition binding polypeptides |
| US20070154989A1 (en) | 2006-01-03 | 2007-07-05 | The Scripps Research Institute | Zinc finger domains specifically binding agc |
| US20070213269A1 (en) | 2005-11-28 | 2007-09-13 | The Scripps Research Institute | Zinc finger binding domains for tnn |
| US7279463B2 (en) | 1995-06-07 | 2007-10-09 | Yale University | Triple-helix forming oligonucleotides for targeted mutagenesis |
| WO2008086529A2 (en) | 2007-01-11 | 2008-07-17 | Yale University | Compositions and methods for targeted inactivation of hiv cell surface receptors |
| WO2010123983A1 (en) | 2009-04-21 | 2010-10-28 | Yale University | Compostions and methods for targeted gene therapy |
| WO2011013380A1 (en) | 2009-07-31 | 2011-02-03 | Fuji Electric Systems Co., Ltd. | Manufacturing method of semiconductor apparatus and semiconductor apparatus |
| WO2011053989A2 (en) | 2009-11-02 | 2011-05-05 | Yale University | Polymeric materials loaded with mutagenic and recombinagenic nucleic acids |
| US20110145940A1 (en) | 2009-12-10 | 2011-06-16 | Voytas Daniel F | Tal effector-mediated dna modification |
| WO2011133802A1 (en) | 2010-04-21 | 2011-10-27 | Helix Therapeutics, Inc. | Compositions and methods for treatment of lysosomal storage disorders |
| US20110262406A1 (en) | 2010-04-21 | 2011-10-27 | Yale University | Compositions and methods for targeted inactivation of hiv cell surface receptors |
| US8309356B2 (en) | 2009-04-01 | 2012-11-13 | Yale University | Pseudocomplementary oligonucleotides for targeted gene therapy |
| WO2013082529A1 (en) | 2011-12-02 | 2013-06-06 | Yale University | Enzymatic synthesis of poly(amine-co-esters) and methods of use thereof for gene delivery |
| WO2013176772A1 (en) | 2012-05-25 | 2013-11-28 | The Regents Of The University Of California | Methods and compositions for rna-directed target dna modification and for rna-directed modulation of transcription |
| WO2014018423A2 (en) | 2012-07-25 | 2014-01-30 | The Broad Institute, Inc. | Inducible dna binding proteins and genome perturbation tools and applications thereof |
| US8658608B2 (en) | 2005-11-23 | 2014-02-25 | Yale University | Modified triple-helix forming oligonucleotides for targeted mutagenesis |
| US20140342003A1 (en) | 2011-12-02 | 2014-11-20 | Yale University | Enzymatic synthesis of poly(amine-co-esters) and methods of use thereof for gene delivery |
| US9193758B2 (en) | 2011-04-08 | 2015-11-24 | Carnegie Mellon University Center For Technology Transfer & Enterprise | Conformationally-preorganized, miniPEG-containing γ-peptide nucleic acids |
| WO2017143042A2 (en) | 2016-02-16 | 2017-08-24 | Yale University | Compositions for enhancing targeted gene editing and methods of use thereof |
| WO2017143061A1 (en) | 2016-02-16 | 2017-08-24 | Yale University | Compositions and methods for treatment of cystic fibrosis |
| WO2018142364A1 (en) * | 2017-02-06 | 2018-08-09 | Novartis Ag | Compositions and methods for the treatment of hemoglobinopathies |
| WO2019199984A1 (en) * | 2018-04-13 | 2019-10-17 | The Regents Of The University Of California | Methods for treating sickle cell disease |
-
2019
- 2019-08-30 WO PCT/US2019/049018 patent/WO2020112195A1/en not_active Ceased
Patent Citations (72)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5436A (en) | 1848-02-08 | Air-heating furnace | ||
| US150A (en) | 1837-03-25 | Island | ||
| US4714680A (en) | 1984-02-06 | 1987-12-22 | The Johns Hopkins University | Human stem cells |
| US4965204A (en) | 1984-02-06 | 1990-10-23 | The Johns Hopkins University | Human stem cells and monoclonal antibodies |
| US4714680B1 (en) | 1984-02-06 | 1995-06-27 | Univ Johns Hopkins | Human stem cells |
| US5034506A (en) | 1985-03-15 | 1991-07-23 | Anti-Gene Development Group | Uncharged morpholino-based polymers having achiral intersubunit linkages |
| US5422251A (en) | 1986-11-26 | 1995-06-06 | Princeton University | Triple-stranded nucleic acids |
| US5021409A (en) | 1989-12-21 | 1991-06-04 | Johnson Matthey Plc | Antiviral cyclic polyamines |
| US5716827A (en) | 1990-03-30 | 1998-02-10 | Systemix, Inc. | Human hematopoietic stem cell |
| US5643741A (en) | 1990-03-30 | 1997-07-01 | Systemix, Inc. | Identification and isolation of human hematopoietic stem cells |
| US5061620A (en) | 1990-03-30 | 1991-10-29 | Systemix, Inc. | Human hematopoietic stem cell |
| US5750397A (en) | 1990-03-30 | 1998-05-12 | Systemix, Inc. | Human hematopoietic stem cell |
| US6441130B1 (en) | 1991-05-24 | 2002-08-27 | Isis Pharmaceuticals, Inc. | Linked peptide nucleic acids |
| US5773571A (en) | 1991-05-24 | 1998-06-30 | Nielsen; Peter E. | Peptide nucleic acids |
| US5714331A (en) | 1991-05-24 | 1998-02-03 | Buchardt, Deceased; Ole | Peptide nucleic acids having enhanced binding affinity, sequence specificity and solubility |
| US5736336A (en) | 1991-05-24 | 1998-04-07 | Buchardt, Deceased; Ole | Peptide nucleic acids having enhanced binding affinity, sequence specificity and solubility |
| US5356802A (en) | 1992-04-03 | 1994-10-18 | The Johns Hopkins University | Functional domains in flavobacterium okeanokoites (FokI) restriction endonuclease |
| US5487994A (en) | 1992-04-03 | 1996-01-30 | The Johns Hopkins University | Insertion and deletion mutants of FokI restriction endonuclease |
| US5539082A (en) | 1993-04-26 | 1996-07-23 | Nielsen; Peter E. | Peptide nucleic acids |
| US7078389B2 (en) | 1993-06-25 | 2006-07-18 | Yale University | Chemically modified oligonucleotide for site-directed mutagenesis |
| US6303376B1 (en) | 1993-06-25 | 2001-10-16 | Yale University | Methods of targeted mutagenesis using triple-helix forming oligonucleotides |
| WO1995001364A1 (en) | 1993-06-25 | 1995-01-12 | Yale University | Chemically modified oligonucleotide for site-directed mutagenesis |
| US5962426A (en) | 1993-06-25 | 1999-10-05 | Yale University | Triple-helix forming oligonucleotides for targeted mutagenesis |
| US5527675A (en) | 1993-08-20 | 1996-06-18 | Millipore Corporation | Method for degradation and sequencing of polymers which sequentially eliminate terminal residues |
| US5623049A (en) | 1993-09-13 | 1997-04-22 | Bayer Aktiengesellschaft | Nucleic acid-binding oligomers possessing N-branching for therapy and diagnostics |
| US5759793A (en) | 1993-09-30 | 1998-06-02 | Systemix, Inc. | Method for mammalian cell separation from a mixture of cell populations |
| US5677136A (en) | 1994-11-14 | 1997-10-14 | Systemix, Inc. | Methods of obtaining compositions enriched for hematopoietic stem cells, compositions derived therefrom and methods of use thereof |
| WO1996039195A2 (en) | 1995-06-06 | 1996-12-12 | Yale University | Chemically modified oligonucleotide for site-directed mutagenesis |
| WO1996040898A1 (en) | 1995-06-07 | 1996-12-19 | Yale University | Triple-helix forming oligonucleotides for targeted mutagenesis |
| US7279463B2 (en) | 1995-06-07 | 2007-10-09 | Yale University | Triple-helix forming oligonucleotides for targeted mutagenesis |
| US5945337A (en) | 1996-10-18 | 1999-08-31 | Quality Biological, Inc. | Method for culturing CD34+ cells in a serum-free medium |
| US5786571A (en) | 1997-05-09 | 1998-07-28 | Lexmark International, Inc. | Wrapped temperature sensing assembly |
| US6746838B1 (en) | 1997-05-23 | 2004-06-08 | Gendaq Limited | Nucleic acid binding proteins |
| WO1998053059A1 (en) | 1997-05-23 | 1998-11-26 | Medical Research Council | Nucleic acid binding proteins |
| US6866997B1 (en) | 1997-05-23 | 2005-03-15 | Gendaq Limited | Nucleic acid binding proteins |
| US6326479B1 (en) | 1998-01-27 | 2001-12-04 | Boston Probes, Inc. | Synthetic polymers and methods, kits or compositions for modulating the solubility of same |
| US6140081A (en) | 1998-10-16 | 2000-10-31 | The Scripps Research Institute | Zinc finger binding domains for GNN |
| US6610512B1 (en) | 1998-10-16 | 2003-08-26 | The Scripps Research Institute | Zinc finger binding domains for GNN |
| US6453242B1 (en) | 1999-01-12 | 2002-09-17 | Sangamo Biosciences, Inc. | Selection of sites for targeting by zinc finger proteins and methods of designing zinc finger proteins to bind to preselected sites |
| US6534261B1 (en) | 1999-01-12 | 2003-03-18 | Sangamo Biosciences, Inc. | Regulation of endogenous gene expression in cells using zinc finger proteins |
| US6261841B1 (en) | 1999-06-25 | 2001-07-17 | The Board Of Trustees Of Northwestern University | Compositions, kits, and methods for modulating survival and differentiation of multi-potential hematopoietic progenitor cells |
| US20020165356A1 (en) | 2001-02-21 | 2002-11-07 | The Scripps Research Institute | Zinc finger binding domains for nucleotide sequence ANN |
| US7067617B2 (en) | 2001-02-21 | 2006-06-27 | The Scripps Research Institute | Zinc finger binding domains for nucleotide sequence ANN |
| US20040197892A1 (en) | 2001-04-04 | 2004-10-07 | Michael Moore | Composition binding polypeptides |
| WO2003016496A2 (en) | 2001-08-20 | 2003-02-27 | The Scripps Research Institute | Zinc finger binding domains for cnn |
| US20030148352A1 (en) | 2001-12-14 | 2003-08-07 | Yale University | Intracellular generation of single-stranded DNA |
| WO2003052071A2 (en) | 2001-12-14 | 2003-06-26 | Yale University | Intracellular generation of single-stranded dna |
| US8658608B2 (en) | 2005-11-23 | 2014-02-25 | Yale University | Modified triple-helix forming oligonucleotides for targeted mutagenesis |
| US20070213269A1 (en) | 2005-11-28 | 2007-09-13 | The Scripps Research Institute | Zinc finger binding domains for tnn |
| US20070154989A1 (en) | 2006-01-03 | 2007-07-05 | The Scripps Research Institute | Zinc finger domains specifically binding agc |
| US20100172882A1 (en) | 2007-01-11 | 2010-07-08 | Glazer Peter M | Compositions and methods for targeted inactivation of hiv cell surface receptors |
| WO2008086529A2 (en) | 2007-01-11 | 2008-07-17 | Yale University | Compositions and methods for targeted inactivation of hiv cell surface receptors |
| US8309356B2 (en) | 2009-04-01 | 2012-11-13 | Yale University | Pseudocomplementary oligonucleotides for targeted gene therapy |
| WO2010123983A1 (en) | 2009-04-21 | 2010-10-28 | Yale University | Compostions and methods for targeted gene therapy |
| WO2011013380A1 (en) | 2009-07-31 | 2011-02-03 | Fuji Electric Systems Co., Ltd. | Manufacturing method of semiconductor apparatus and semiconductor apparatus |
| WO2011053989A2 (en) | 2009-11-02 | 2011-05-05 | Yale University | Polymeric materials loaded with mutagenic and recombinagenic nucleic acids |
| US20110268810A1 (en) | 2009-11-02 | 2011-11-03 | Yale University | Polymeric materials loaded with mutagenic and recombinagenic nucleic acids |
| US20110145940A1 (en) | 2009-12-10 | 2011-06-16 | Voytas Daniel F | Tal effector-mediated dna modification |
| WO2011072246A2 (en) | 2009-12-10 | 2011-06-16 | Regents Of The University Of Minnesota | Tal effector-mediated dna modification |
| WO2011133802A1 (en) | 2010-04-21 | 2011-10-27 | Helix Therapeutics, Inc. | Compositions and methods for treatment of lysosomal storage disorders |
| US20110293585A1 (en) | 2010-04-21 | 2011-12-01 | Helix Therapeutics, Inc. | Compositions and methods for treatment of lysosomal storage disorders |
| US20110262406A1 (en) | 2010-04-21 | 2011-10-27 | Yale University | Compositions and methods for targeted inactivation of hiv cell surface receptors |
| US9193758B2 (en) | 2011-04-08 | 2015-11-24 | Carnegie Mellon University Center For Technology Transfer & Enterprise | Conformationally-preorganized, miniPEG-containing γ-peptide nucleic acids |
| WO2013082529A1 (en) | 2011-12-02 | 2013-06-06 | Yale University | Enzymatic synthesis of poly(amine-co-esters) and methods of use thereof for gene delivery |
| US20140342003A1 (en) | 2011-12-02 | 2014-11-20 | Yale University | Enzymatic synthesis of poly(amine-co-esters) and methods of use thereof for gene delivery |
| WO2013176772A1 (en) | 2012-05-25 | 2013-11-28 | The Regents Of The University Of California | Methods and compositions for rna-directed target dna modification and for rna-directed modulation of transcription |
| WO2014018423A2 (en) | 2012-07-25 | 2014-01-30 | The Broad Institute, Inc. | Inducible dna binding proteins and genome perturbation tools and applications thereof |
| WO2017143042A2 (en) | 2016-02-16 | 2017-08-24 | Yale University | Compositions for enhancing targeted gene editing and methods of use thereof |
| WO2017143061A1 (en) | 2016-02-16 | 2017-08-24 | Yale University | Compositions and methods for treatment of cystic fibrosis |
| US20170283830A1 (en) | 2016-02-16 | 2017-10-05 | Yale University | Compositions for enhancing targeted gene editing and methods of use thereof |
| WO2018142364A1 (en) * | 2017-02-06 | 2018-08-09 | Novartis Ag | Compositions and methods for the treatment of hemoglobinopathies |
| WO2019199984A1 (en) * | 2018-04-13 | 2019-10-17 | The Regents Of The University Of California | Methods for treating sickle cell disease |
Non-Patent Citations (65)
| Title |
|---|
| AGUADOLAMBERT, IMMUNOBIOLOGY, vol. 184, no. 2-3, 1992, pages 113 - 25 |
| AKINC ET AL., BIOCONJUG CHEM., vol. 14, 2003, pages 979 - 988 |
| ARNOULD ET AL., PROTEIN ENG. DES. SEL., vol. 24, no. 1-2, 2011, pages 27 - 31 |
| BAHAL ET AL., NAT. COMMUN., vol. 7, 2016, pages 13304 |
| BENTIN ET AL., NUCL. ACIDS RES., vol. 34, no. 20, 2006, pages 5790 - 5799 |
| BRAASCH ET AL., CHEM. BIOL., vol. 8, no. 1, 2001, pages 1 - 7 |
| BRAMWELL ET AL., ADV. DRUG DELIV. REV., vol. 57, no. 9, 2005, pages 1247 - 410 |
| CERMAK ET AL., NUCL. ACIDS RES., no. 1-11, 2011 |
| CHIN ET AL., PROC NATL ACAD SCI USA, vol. 105, 2008, pages 13514 - 13519 |
| CONG, SCIENCE, vol. 339, no. 6121, 2013, pages 819 - 823 |
| CRADICK ET AL., NUCLEIC ACIDS RES., vol. 41, 2013, pages 9584 - 9592 |
| CRUZ ET AL., J CONTROL RELEASE, vol. 144, 2010, pages 118 - 126 |
| DEBNATH ET AL., THERANOSTICS, vol. 3, no. 1, 2013, pages 41 - 75 |
| DESAI ET AL., PHARM. RES., vol. 14, 1997, pages 1568 - 73 |
| DIPERSIO ET AL., BLOOD., vol. 113, no. 23, 2009, pages 5720 - 6 |
| DIPERSIO ET AL., J. CLIN. ONCOL., vol. 27, no. 28, 2009, pages 4767 - 73 |
| DOUDNA ET AL., SCIENCE, vol. 346, 2014, pages 1258096 |
| ENDOH ET AL., ADV DRUG DELIV REV., vol. 61, 2009, pages 704 - 709 |
| FIELDS ET AL., ADV HEALTHC MATER., vol. 4, no. 3, 2015, pages 361 - 6 |
| GONCZ ET AL., OLIGONUCLEOTIDES, vol. 16, 2006, pages 213 - 224 |
| HAENDEL ET AL., GENE THER., vol. 11, 2011, pages 28 - 37 |
| HANNA, J. ET AL., SCIENCE, vol. 318, 2007, pages 1920 - 1923 |
| HANSEN ET AL., NUCL. ACIDS RES., vol. 37, no. 13, 2009, pages 4498 - 4507 |
| HATSE ET AL., BIOCHEM, PHARMACOL., vol. 70, no. 5, 2005, pages 752 - 61 |
| HE ET AL.: "The Structure of a y-modified peptide nucleic acid duplex", MOL. BIOSYST., vol. 6, 2010, pages 1619 - 1629 |
| HUANG ET AL., ARCH. PHARM. RES., vol. 35, no. 3, 2012, pages 517 - 522 |
| JAIN ET AL., JOC, vol. 79, no. 20, 2014, pages 9567 - 9577 |
| JINEK ET AL., SCIENCE, vol. 337, no. 6096, 2012, pages 816 - 21 |
| KAIHATSU ET AL., BIOCHEMISTRY, vol. 42, no. 47, 2003, pages 13987 - 4003 |
| KIM ET AL., J. BIOL. CHEM., vol. 269, no. 31, 1994, pages 978 - 31,982 |
| KIM ET AL., PROC. NATL. ACAD. SCI. USA., vol. 91, 1994, pages 883 - 887 |
| KO ET AL., NANA RESEARCH., vol. 11, no. 4, 2018, pages 2159 - 2172 |
| KOPPELHUS ET AL., ADV. DRUG DELIV. REV., vol. 55, no. 2, 2003, pages 267 - 280 |
| KUHN ET AL., ARTIFICIAL DNA, PNA & XNA, vol. 1, no. 1, 2010, pages 45 - 53 |
| LI ET AL., PROC. NATL. ACAD. SCI. USA, vol. 90, 1993, pages 2764 - 2768 |
| LI ET AL., PROC., NATL. ACAD. SCI. USA, vol. 89, no. 1992, pages 4275 - 4279 |
| LUENS ET AL., BLOOD, vol. 91, 1998, pages 1206 - 1215 |
| MAGZOUB ET AL., BIOCHEM BIOPHYS RES COMMUN., vol. 348, 2006, pages 379 - 385 |
| MAJUMDAR ET AL., NATURE GENETICS, vol. 20, 1998, pages 212 - 214 |
| MCNEER ET AL., NATURE COMMUN., vol. 6, 2015, pages 6952 |
| MEGAN D HOBAN ET AL: "CRISPR/Cas9-Mediated Correction of the Sickle Mutation in Human CD34+ cells", MOLECULAR THERAPY, vol. 24, no. 9, 1 September 2016 (2016-09-01), pages 1561 - 1569, XP055419337, ISSN: 1525-0016, DOI: 10.1038/mt.2016.148 * |
| MILLER ET AL., NATURE BIOTECHNOL, vol. 29, 2011, pages 143 |
| NIE ET AL., J CONTROL RELEASE, vol. 138, 2009, pages 64 - 70 |
| RAMAN BAHAL ET AL: "In vivo correction of anaemia in β-thalassemic mice by [gamma]PNA-mediated gene editing with nanoparticle delivery", NATURE COMMUNICATIONS, vol. 7, 26 October 2016 (2016-10-26), pages 13304, XP055376685, DOI: 10.1038/ncomms13304 * |
| RAPIREDDY ET AL., BIOCHEMISTRY, vol. 50, no. 19, 2011, pages 3913 - 8 |
| RICCIARDI ET AL., NAT COMMUN., vol. 9, no. 1, 26 June 2018 (2018-06-26), pages 2481 |
| ROGERS ET AL., PROC NATL ACAD SCI USA, vol. 99, 2002, pages 16695 - 16700 |
| ROGERS ET AL., PROC. NATL. ACAD. SCI. U.S.A., vol. 99, no. 26, 2002, pages 16695 - 700 |
| ROGERS ET AL., PROC. NATL. ACAD. SCI. USA, vol. 99, 2002, pages 16695 - 16700 |
| SAHU ET AL., J. ORG. CHEM., vol. 76, 2011, pages 5614 - 5627 |
| SAHU ET AL., JOC, vol. 76, 2011, pages 5614 - 5627 |
| SAHU ET AL.: "Synthesis and Characterization of Conformationally Preorganized, (R)-Diethylene Glycol-Containing y-Peptide Nucleic Acids with Superior Hybridization Properties and Water Solubility", J. ORG. CHEM, vol. 76, 2011, pages 5614 - 5627, XP002731647, doi:10.1021/jo200482d |
| SARGENT, OLIGONUCLEOTIDES, vol. 21, no. 2, 2011, pages 55 - 75 |
| SCHLEIFMAN ET AL., CHEM BIOL., vol. 18, 2011, pages 1189 - 1198 |
| SCIENCE, vol. 278, no. 5339, 31 October 1997 (1997-10-31), pages 873 - 6 |
| SIMON P FRICKER: "A novel CXCR4 antagonist for hematopoietic stem cell mobilization", EXPERT OPINION ON INVESTIGATIONAL DRUGS, vol. 17, no. 11, 15 October 2008 (2008-10-15), UK, pages 1749 - 1760, XP055215855, ISSN: 1354-3784, DOI: 10.1517/13543784.17.11.1749 * |
| STERCHAK, E. P. ET AL., ORGANIC CHEM., vol. 52, 1987, pages 4202 |
| SUGIYAMAKITTAKA, MOLECULES, vol. 18, 2013, pages 287 - 310 |
| THOMAS GAJ ET AL: "ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering", NIH PUBLIC ACCESS AUTHOR MANUSCRIPT, vol. 31, no. 7, 9 May 2013 (2013-05-09), pages 1 - 20, XP055465031, DOI: 10.1016/j.tibtech.2013.04.004 * |
| VASQUEZ ET AL., SCIENCE, vol. 290, 2000, pages 530 - 533 |
| YAMANO ET AL., J CONTROL RELEASE, vol. 152, 2011, pages 278 - 285 |
| YIN ET AL., NAT. BIOTECHNOL., vol. 32, 2014, pages 551 - 553 |
| YU ET AL., PLOS ONE., vol. 6, 2011, pages e24077 |
| ZHOU ET AL., NATURE MATERIALS, vol. 11, 2012, pages 82 - 90 |
| ZIELKE ET AL., METHODS CELL BIOL., vol. 8, 1974, pages 107 - 121 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20230277658A1 (en) | Compositions and methods for enhancing triplex and nuclease-based gene editing | |
| EP3847189B1 (en) | Universal donor cells | |
| US20200299661A1 (en) | Cpf1-related methods and compositions for gene editing | |
| US12427170B2 (en) | Universal donor cells | |
| JP7753197B2 (en) | Universal donor cells | |
| JP2018519801A (en) | Optimized CRISPR / CAS9 system and method for gene editing in stem cells | |
| WO2018117746A1 (en) | Composition for base editing for animal embryo and base editing method | |
| US20230014010A1 (en) | Engineered cells with improved protection from natural killer cell killing | |
| JP2025518552A (en) | Compositions and methods for manipulating cells | |
| US20250197811A1 (en) | Compositions and methods for generating cells with reduced immunogenicity | |
| WO2020112195A1 (en) | Compositions, technologies and methods of using plerixafor to enhance gene editing | |
| Carufe | Applying Gene Editing Technology for Correction of Human Disease | |
| WO2024233505A1 (en) | Compositions and methods for targeting, editing or modifying human genes | |
| HK40058224B (en) | Universal donor cells | |
| HK40058224A (en) | Universal donor cells |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19769637 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 19769637 Country of ref document: EP Kind code of ref document: A1 |