WO2017088746A1 - New epidermal growth factor receptor inhibitor and application thereof - Google Patents
New epidermal growth factor receptor inhibitor and application thereof Download PDFInfo
- Publication number
- WO2017088746A1 WO2017088746A1 PCT/CN2016/106862 CN2016106862W WO2017088746A1 WO 2017088746 A1 WO2017088746 A1 WO 2017088746A1 CN 2016106862 W CN2016106862 W CN 2016106862W WO 2017088746 A1 WO2017088746 A1 WO 2017088746A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- group
- amino
- alkylamino
- cycloalkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- AKGMBEQNHFJEDM-UHFFFAOYSA-N BrCCC[n](cc1)c2c1cccc2 Chemical compound BrCCC[n](cc1)c2c1cccc2 AKGMBEQNHFJEDM-UHFFFAOYSA-N 0.000 description 1
- 0 CC(CCCC*CC1=**)=C([C@]2NC(N)=NC(*)=C2*)C1=**=C Chemical compound CC(CCCC*CC1=**)=C([C@]2NC(N)=NC(*)=C2*)C1=**=C 0.000 description 1
- RJUQFOXGKUDISB-UHFFFAOYSA-N CC(C[n]1c2ccccc22)N(C)Cc1c2-c1ccnc(Nc(cc(c(N(C)CCN(C)C)c2)NC(C=C)=O)c2OC)n1 Chemical compound CC(C[n]1c2ccccc22)N(C)Cc1c2-c1ccnc(Nc(cc(c(N(C)CCN(C)C)c2)NC(C=C)=O)c2OC)n1 RJUQFOXGKUDISB-UHFFFAOYSA-N 0.000 description 1
- AXUCJUWACLKWAG-UHFFFAOYSA-N CN(C)CCN(C)c(c(NC(C=C)=O)c1)cc(OC)c1Nc1nc(-c2c(C(C3)C3C3)[n]3c3ccccc23)ccn1 Chemical compound CN(C)CCN(C)c(c(NC(C=C)=O)c1)cc(OC)c1Nc1nc(-c2c(C(C3)C3C3)[n]3c3ccccc23)ccn1 AXUCJUWACLKWAG-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5383—1,4-Oxazines, e.g. morpholine ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/54—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one sulfur as the ring hetero atoms, e.g. sulthiame
- A61K31/542—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one sulfur as the ring hetero atoms, e.g. sulthiame ortho- or peri-condensed with heterocyclic ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
Definitions
- the invention belongs to the field of medical chemistry, and in particular relates to a novel class of epidermal growth factor receptor inhibitors, a pharmaceutical composition containing the same, and the use of the inhibitor or pharmaceutical composition as a therapeutic drug for cancer.
- the Epidermal Growth Factor Receptor is an expression product of the proto-oncogene C-erbB-1 and is a member of the EGFR family.
- the EGFR family includes four members, EGFR (HER-1), ERBB2 (HER-2), ERBB3 (HER-3), and ERBB4 (HER-4).
- Overexpression or mutation of EGFR has been shown to generally trigger tumors.
- EGFR mutations lead to sustained activation of EGFR, enhanced autocrine loops, disruption of receptor down-regulation, activation of abnormal signaling pathways, and play an important role in tumor progression.
- EGFR-tyrosine kinase inhibitor for EGFR has become the gold standard in the field of non-small cell lung cancer treatment.
- clinical use has found that most patients will have different degrees of drug resistance 6-12 months after treatment with EGFR-TKI inhibitors such as gefitinib and erlotinib, resulting in a significant reduction in drug efficacy and tumor progression.
- EGFR-TKI inhibitors such as gefitinib and erlotinib
- methionine has a larger space occupying than threonine, it forms steric hindrance and changes the affinity of ATP in EGFR kinase domain, which leads to EGFR-TKI small molecule drug can not effectively block EGFR activation signal, leading to drug resistance.
- the production At the same time, the first generation of EGFR inhibitors lacked the selectivity of wild-type EGFR and mutant EGFR, and generally had side effects such as rash and diarrhea, which affected patient compliance.
- Another object of the present invention is to provide a compound of the formula II or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, which is prepared by using the compound of the formula II as a key intermediate.
- Compound I mild reaction conditions, high yield and purity,
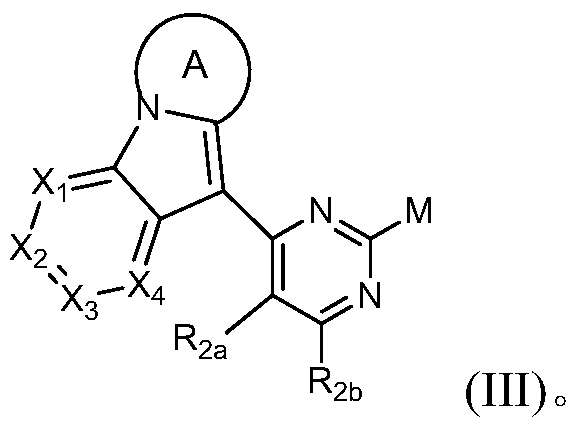
- a third object of the present invention is to provide a compound of the formula III or a salt thereof, which is a key intermediate of the compound of the formula III, which has a small number of reaction steps, mild conditions, and a relatively good yield and purity. high,
- a fourth object of the present invention is to provide a process for the preparation of a compound of the formula I, II or III of the present invention, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof.
- a fifth object of the present invention is to provide a pharmaceutical composition comprising a compound of the formula I of the present invention, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, and a pharmaceutically acceptable carrier, and a pharmaceutical composition comprising the same
- a pharmaceutical composition of a compound of formula I of the invention, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, and another tumor suppressor is provided.
- a sixth object of the present invention is to provide a compound of the formula I of the present invention or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, and a pharmaceutical composition of the present invention for treating and/or preventing cancer Methods and their use in the preparation of a medicament for the treatment and/or prevention of cancer.
- the present invention provides the following technical solutions:
- the present invention provides a compound of Formula I, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof,
- Ring A is a C 4 -C 8 nitrogen-containing heterocyclic group optionally substituted by one or more halogen, oxo, alkyl, cycloalkyl, haloalkyl, alkoxy, haloalkoxy, nitro, cyano , hydroxy, amino, monoalkylamino, dialkylamino, aminoalkyl, monoalkylaminoalkyl, dialkylaminoalkyl, hydroxyalkyl, alkoxyalkyl, alkyl acyl, amino acyl, a monoalkylaminoacyl group, a dialkylaminoacyl group, an aryl group, a heteroaryl group, and a heterocycloalkyl group.
- two adjacent substituents may be substituted together with the atoms to which they are attached.
- an unsubstituted cycloalkyl, heterocycloalkyl, aryl, heteroaryl group, or two substituents attached to the same atom may form a substituted or unsubstituted cycloalkyl group with the atoms to which they are attached.
- X 1 , X 2 , X 3 and X 4 are each independently selected from C(R 1 ) and N;
- R 1 , R 2a , R 2b , R 3a , R 3b and R 3c are each independently selected from the group consisting of hydrogen, halogen, alkyl, cycloalkyl, haloalkyl, alkoxy, haloalkoxy, nitro, cyano, Hydroxy, amino, monoalkylamino, dialkylamino, aminoalkyl, monoalkylaminoalkyl, dialkylaminoalkyl, hydroxyalkyl, alkoxyalkyl, alkyl acyl, amino acyl, single An alkylamino acyl group and a dialkylamino acyl group;
- R 4 , R 5 and R 6 are each independently selected from the group consisting of hydrogen, halogen, cyano, alkyl, alkoxy and cycloalkyl, said alkyl, alkoxy and cycloalkyl optionally being one or more Halogen, alkyl, haloalkyl, alkoxy, haloalkoxy, amino, monoalkylamino, dialkylamino, hydroxy, aminoalkyl, monoalkylaminoalkyl, dialkylaminoalkyl or hydroxy Alkyl substitution
- R 7 is selected from the group consisting of hydrogen, alkyl, cycloalkyl and haloalkyl
- R 8 is selected from the group consisting of hydrogen, alkyl and cycloalkyl, optionally substituted by one or more halogen, alkyl, haloalkyl, alkoxy, amino, monoalkylamino, dioxane Substituted by a base or a hydroxy group; or
- the present invention provides a compound of Formula II, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof,
- Rings A, X 1 , X 2 , X 3 , X 4 , R 2a , R 2b , R 3a , R 3b , R 3c , R 7 and R 8 have the definitions in Formula I.
- the present invention provides a compound of Formula III or a pharmaceutically acceptable salt thereof,
- Rings A, X 1 , X 2 , X 3 , X 4 , R 2a , R 2b have the definitions in Formula I, and M is a halogen.
- the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein Ring A is C 4 -C 8 a nitrogen-containing heteromonocycloalkyl group optionally substituted by one or more of halogen, oxo, alkyl, cycloalkyl, haloalkyl, alkoxy, haloalkoxy, nitro, cyano, hydroxy, amino, singly Alkylamino, dialkylamino, aminoalkyl, monoalkylaminoalkyl, dialkylaminoalkyl, hydroxyalkyl, alkoxyalkyl, alkylacyl, aminoacyl, monoalkylaminoacyl, a dialkylamino acyl group, an aryl group, a heteroaryl group and a heterocycloalkyl group.
- Ring A is C 4 -C 8 a nitrogen-containing heteromonocycloalkyl group optionally substitute
- two adjacent substituents may form a substituted or unsubstituted naphthenic ring together with the atoms to which they are attached.
- a heterocyclic alkyl group, an aryl group, a heteroaryl group, or two substituents bonded to the same atom may form a substituted or unsubstituted cycloalkyl group or a heterocycloalkyl group with the atoms to which they are attached.
- the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein Ring A is a nitrogen atom only C 4 -C 8 nitrogen-containing heteromonocycloalkyl optionally substituted by one or more halogen, oxo, alkyl, cycloalkyl, haloalkyl, alkoxy, haloalkoxy, nitro, cyano , hydroxy, amino, monoalkylamino, dialkylamino, aminoalkyl, monoalkylaminoalkyl, dialkylaminoalkyl, hydroxyalkyl, alkoxyalkyl, alkyl acyl, amino acyl, a monoalkylaminoacyl group, a dialkylaminoacyl group, an aryl group, a heteroaryl group, and a heterocycloalkyl group.
- Ring A is a nitrogen atom only C 4 -C 8 nitrogen-containing hetero
- two adjacent substituents may be substituted together with the atoms to which they are attached.
- an unsubstituted cycloalkyl, heterocycloalkyl, aryl, heteroaryl group, or two substituents attached to the same atom may form a substituted or unsubstituted cycloalkyl group with the atoms to which they are attached. Cycloalkyl.
- the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein Ring A is n is selected from 1, 2, 3, 4, 5 and 6, m is selected from 1, 2, 3 and 4, and R a is selected from the group consisting of hydrogen, C 1-6 alkyl, C 3-8 cycloalkyl, halogen C 1-6 alkyl, C 1-6 alkoxy, halogenated C 1-6 alkoxy, nitro, cyano, hydroxy, amino, mono C 1-6 alkylamino, di C 1-6 alkyl Amino group, amino C 1-6 alkyl group, mono C 1-6 alkylamino C 1-6 alkyl group, di C 1-6 alkylamino C 1-6 alkyl group, hydroxy C 1-6 alkyl group, C 1 -6 alkoxy C 1-6 alkyl, C 1-6 alkyl acyl, amino acyl, mono C 1-6 alkylamino
- the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein Ring A is n is selected from 1, 2, 3 and 4, m is selected from 1, 2, 3 and 4, and R a is selected from the group consisting of hydrogen, C 1-3 alkyl, C 3-6 cycloalkyl, halogenated C 1-3 alkane , C 1-3 alkoxy, halo C 1-3 alkoxy, nitro, cyano, hydroxy, amino, mono C 1-3 alkylamino, di C 1-3 alkylamino, amino C 1-3 alkyl, mono C 1-3 alkylamino C 1-3 alkyl, di C 1-3 alkylamino C 1-3 alkyl, hydroxy C 1-3 alkyl, C 1-3 alkoxy a group C 1-3 alkyl, C 1-3 alkyl acyl, amino acyl, mono C 1-3 alkylamino acyl, di C
- the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein Ring A is two nitrogens A C 4 -C 8 nitrogen-containing heteromonocycloalkyl group of an atom optionally substituted by one or more halogen, oxo, alkyl, cycloalkyl, haloalkyl, alkoxy, haloalkoxy, nitro, cyanide Base, hydroxy, amino, monoalkylamino, dialkylamino, aminoalkyl, monoalkylaminoalkyl, dialkylaminoalkyl, hydroxyalkyl, alkoxyalkyl, alkyl acyl, amino acyl a monoalkylaminoacyl group, a dialkylaminoacyl group, an aryl group, a heteroaryl group, and a heterocycloalkyl group.
- Ring A is two nitrogens
- two adjacent substituents may be bonded together with the atoms to which they are attached.
- a substituted or unsubstituted cycloalkyl group, a heterocycloalkyl group, an aryl group, a heteroaryl group, or two substituents bonded to the same atom may form a substituted or unsubstituted cycloalkyl group with an atom to which they are attached, Heterocycloalkyl.
- the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein Ring A is m is selected from 1, 2, 3 and 4, p is selected from 0, 1 and 2, q is selected from 0, 1 and 2, and R a is selected from the group consisting of hydrogen, C 1-6 alkyl, C 3-8 cycloalkyl, halo-C 1-6 alkyl, C 1-6 alkoxy, halo C 1-6 alkoxy, nitro, cyano, hydroxy, amino, mono-C 1-6 alkylamino, di C 1- 6 alkylamino group, amino C 1-6 alkyl group, mono C 1-6 alkylamino C 1-6 alkyl group, di C 1-6 alkylamino C 1-6 alkyl group, hydroxy C 1-6 alkyl group , C 1-6 alkoxy C 1-6 alkyl, C 1-6 alkyl acyl, amino acyl, mono C 1-6
- the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein Ring A is m is selected from 1, 2, 3 and 4, p is selected from 0, 1 and 2, q is selected from 0, 1 and 2, and R a is selected from the group consisting of hydrogen, C 1-3 alkyl, C 3-6 cycloalkyl, Halogen C 1-3 alkyl, C 1-3 alkoxy, halo C 1-3 alkoxy, nitro, cyano, hydroxy, amino, mono C 1-3 alkylamino, di C 1- 3 alkylamino group, amino C 1-3 alkyl group, mono C 1-3 alkylamino C 1-3 alkyl group, di C 1-3 alkylamino C 1-3 alkyl group, hydroxy C 1-3 alkyl group , C 1-3 alkoxy C 1-3 alkyl, C 1-3 alkyl acyl, amino acyl, mono C 1-3 alkyla
- the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein Ring A is a nitrogen atom And a C 4 -C 8 nitrogen-containing heteromonocycloalkyl group having one oxygen atom, optionally substituted by one or more halogen, oxo, alkyl, cycloalkyl, haloalkyl, alkoxy, haloalkoxy, nitrate Base, cyano, hydroxy, amino, monoalkylamino, dialkylamino, aminoalkyl, monoalkylaminoalkyl, dialkylaminoalkyl, hydroxyalkyl, alkoxyalkyl, alkyl acyl , aminoacyl, monoalkylaminoacyl, dialkylaminoacyl, aryl, heteroaryl and heterocycloalkyl substituted, when two or more substituents are present, two adjacent substituents may be attached there
- the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein Ring A is m is selected from 1, 2, 3 and 4, p is selected from 0, 1 and 2, q is selected from 0, 1 and 2, and R a is selected from the group consisting of hydrogen, C 1-6 alkyl, C 3-8 cycloalkyl, Halogen C 1-6 alkyl, C 1-6 alkoxy, halo C 1-6 alkoxy, nitro, cyano, hydroxy, amino, mono C 1-6 alkylamino, di C 1- 6 alkylamino group, amino C 1-6 alkyl group, mono C 1-6 alkylamino C 1-6 alkyl group, di C 1-6 alkylamino C 1-6 alkyl group, hydroxy C 1-6 alkyl group , C 1-6 alkoxy C 1-6 alkyl, C 1-6 alkyl acyl, amino acyl, mono C 1-6 alkyla
- the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein Ring A is m is selected from 1, 2, 3 and 4, p is selected from 0, 1 and 2, q is selected from 0, 1 and 2, and R a is selected from the group consisting of hydrogen, C 1-3 alkyl, C 3-6 cycloalkyl, Halogen C 1-3 alkyl, C 1-3 alkoxy, halo C 1-3 alkoxy, nitro, cyano, hydroxy, amino, mono C 1-3 alkylamino, di C 1- 3 alkylamino group, amino C 1-3 alkyl group, mono C 1-3 alkylamino C 1-3 alkyl group, di C 1-3 alkylamino C 1-3 alkyl group, hydroxy C 1-3 alkyl group , C 1-3 alkoxy C 1-3 alkyl, C 1-3 alkyl acyl, amino acyl, mono C 1-3 alkyla
- the present invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein Ring A is a nitrogen atom and a C 4 -C 8 nitrogen-containing heteromonocycloalkyl group of a sulfur atom optionally substituted by one or more halogen, oxo, alkyl, cycloalkyl, haloalkyl, alkoxy, haloalkoxy, nitro , cyano, hydroxy, amino, monoalkylamino, dialkylamino, aminoalkyl, monoalkylaminoalkyl, dialkylaminoalkyl, hydroxyalkyl, alkoxyalkyl, alkyl acyl, Aminoacyl, monoalkylaminoacyl, dialkylaminoacyl, aryl, heteroaryl and heterocycloalkyl substituted, when two or more substituents are present, the two adjacent substituents may be
- the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein X 1 , X 2 , X 3 And X 4 are both C(R 1 ).
- the invention provides a compound of formula Ia, IIa, and IIIa, or a pharmaceutically acceptable salt, isomer, solvate, crystal, or prodrug thereof,
- Rings A, R 1 , R 2a , R 2b , R 3a , R 3b , R 3c , R 4 , R 5 , R 6 , R 7 , R 8 and M have the definitions of Formulas I, II and III above , n is selected from integers of 1, 2, 3 and 4.
- the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein X 1 , X 2 , X At least one of 3 and X 4 is N.
- the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein X 1 , X 2 , X Two of 3 and X 4 are N.
- the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein X 1 , X 2 , X Three of 3 and X 4 are N.
- the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein X 1 , X 2 , X 3 and X 4 are both N.
- the present invention of general formula I, II or III compound or a pharmaceutically acceptable salt, isomer, solvate, crystallization or prodrug thereof, wherein R 1, R 2a and R 2b Each is independently selected from the group consisting of hydrogen, halogen, C 1-6 alkyl, C 3-8 cycloalkyl, halo C 1-6 alkyl, C 1-6 alkoxy, halo C 1-6 alkoxy , nitro, cyano, hydroxy, amino, mono C 1-6 alkylamino, di C 1-6 alkylamino, amino C 1-6 alkyl, mono C 1-6 alkylamino C 1-6 alkane Base, di-C 1-6 alkylamino C 1-6 alkyl, hydroxy C 1-6 alkyl, C 1-6 alkoxy C 1-6 alkyl, C 1-6 alkyl acyl, amino acyl, Mono C 1-6 alkylamino acyl and di C 1-6 alkylamino acyl.
- the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein each R 1 , R 2a And R 2b are each independently independently selected from the group consisting of hydrogen, halogen, C 1-3 alkyl, C 3-6 cycloalkyl, halo C 1-3 alkyl, C 1-3 alkoxy, halo C 1 -3 alkoxy, nitro, cyano, hydroxy, amino, mono C 1-3 alkyl amino, di C 1-3 alkyl amino, amino C 1-3 alkyl, mono C 1-3 alkyl amino C 1-6 alkyl, di C 1-3 alkylamino C 1-3 alkyl, hydroxy C 1-3 alkyl, C 1-3 alkoxy C 1-3 alkyl, C 1-3 alkyl An acyl group, an aminoacyl group, a mono C 1-3 alkylamino acyl group and a di C 1-3 alkylamino
- the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein R 1 , R 2a And R 2b are each independently selected from the group consisting of hydrogen, fluorine, chlorine, bromine, iodine, methyl, ethyl, propyl, isopropyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, trifluoromethyl , trifluoroethyl, difluoromethyl, difluoroethyl, methoxy, ethoxy, propoxy, trifluoromethyloxy, nitro, cyano, hydroxy, amino, methylamino, ethylamino , propylamino, isopropylamino, dimethylamino, diethylamino, dipropylamino, diisopropylamino, N-methyl
- the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein R 1 , R 2a And R 2b are each independently selected from the group consisting of hydrogen, fluorine, chlorine, bromine, methyl, ethyl, propyl, isopropyl, trifluoromethyl, trifluoroethyl, methoxy, ethoxy, hydroxy, amino , methylamino, ethylamino, propylamino, dimethylamino, diethylamino, aminomethyl, aminoethyl, aminopropyl, methylaminomethyl, methylaminoethyl, ethylaminomethyl, ethylaminoethyl, Dimethylaminomethyl, diethylaminomethyl, hydroxymethyl and hydroxyethyl.
- R 1 , R 2a And R 2b are each independently selected from the group consisting of
- the invention provides a compound of Formula I or II, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein R 3a , R 3b and R 3c Each is independently selected from the group consisting of hydrogen, halogen, C 1-6 alkyl, C 3-8 cycloalkyl, halo C 1-6 alkyl, C 1-6 alkoxy, halo C 1-6 alkoxy , nitro, cyano, hydroxy, amino, mono C 1-6 alkylamino, di C 1-6 alkylamino, amino C 1-6 alkyl, mono C 1-6 alkylamino C 1-6 alkane Base, di-C 1-6 alkylamino C 1-6 alkyl, hydroxy C 1-6 alkyl, C 1-6 alkoxy C 1-6 alkyl, C 1-6 alkyl acyl, amino acyl, Mono C 1-6 alkylamino acyl and di C 1-6 alkylamino acyl.
- the present invention provides a compound of Formula I or II, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein each R 3a , R 3b and R 3c are each independently selected from the group consisting of hydrogen, halogen, C 1-3 alkyl, C 3-6 cycloalkyl, halo C 1-3 alkyl, C 1-3 alkoxy, halo C 1-3 alkane Oxy, nitro, cyano, hydroxy, amino, mono C 1-3 alkylamino, di C 1-3 alkylamino, amino C 1-3 alkyl, mono C 1-3 alkylamino C 1- 6 alkyl, di C 1-3 alkyl amino C 1-3 alkyl, hydroxy C 1-3 alkyl, C 1-3 alkoxy C 1-3 alkyl, C 1-3 alkyl group, an amino group An acyl group, a mono C 1-3 alkylamino acyl group and a di C 1-3 alkyla
- the invention provides a compound of Formula I or II, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein R 3a , R 3b and R 3c are each independently selected from the group consisting of hydrogen, fluorine, chlorine, bromine, iodine, methyl, ethyl, propyl, isopropyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, trifluoromethyl, tri Fluoroethyl, difluoromethyl, difluoroethyl, methoxy, ethoxy, propoxy, trifluoromethyloxy, nitro, cyano, hydroxy, amino, methylamino, ethylamino, propyl Amino, isopropylamino, dimethylamino, diethylamino, dipropylamino, diisopropylamino, N-methyl-N
- the invention provides a compound of Formula I or II, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein R 3a , R 3b and R 3c are each independently selected from the group consisting of hydrogen, fluorine, chlorine, bromine, methyl, ethyl, propyl, isopropyl, trifluoromethyl, trifluoroethyl, methoxy, ethoxy, hydroxy, amino, A Amino, ethylamino, propylamino, dimethylamino, diethylamino, aminomethyl, aminoethyl, aminopropyl, methylaminomethyl, methylaminoethyl, ethylaminomethyl, ethylaminoethyl, dimethyl Aminomethyl, diethylaminomethyl, hydroxymethyl and hydroxyethyl.
- R 3a , R 3b and R 3c are each independently selected from the group consisting of hydrogen,
- the invention provides a compound of Formula I or II, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein R 7 is selected from the group consisting of hydrogen, C 1-6 Alkyl, C 3-6 cycloalkyl and halogenated C 1-6 alkyl.
- the present invention provides Formula I or II compound as shown or a pharmaceutically acceptable salt, isomer, solvate, crystallization or prodrug thereof, wherein R 7 is selected from hydrogen, C 1 a -3 alkyl group, a C 3-6 cycloalkyl group, and a halogenated C 1-3 alkyl group.
- the present invention provides Formula I or II compound as shown or a pharmaceutically acceptable salt, isomer, solvate, crystallization or prodrug thereof, wherein R 7 is selected from hydrogen, methyl , ethyl, propyl, isopropyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and trifluoromethyl.
- the present invention provides Formula I or II compound as shown or a pharmaceutically acceptable salt, isomer, solvate, crystallization or prodrug thereof, wherein R 7 is methyl.
- the present invention provides a formula I or II compound represented by or a pharmaceutically acceptable salt, isomer, solvate, crystallization or prodrug thereof, wherein R 8 is selected from hydrogen, C 1-6 An alkyl group and a C 3-6 cycloalkyl group, said C 1-6 alkyl group and a C 3-6 cycloalkyl group optionally being one or more halogen, C 1-6 alkyl, halogenated C 1-6 Alkyl, C 1-6 alkoxy, amino, mono C 1-6 alkylamino, di C 1-6 alkylamino or hydroxy substituted.
- R 8 is selected from hydrogen, C 1-6 An alkyl group and a C 3-6 cycloalkyl group, said C 1-6 alkyl group and a C 3-6 cycloalkyl group optionally being one or more halogen, C 1-6 alkyl, halogenated C 1-6 Alkyl, C 1-6 alkoxy, amino, mono C 1-6 alkylamino, di C
- the present invention provides a formula I or II or a compound represented by the pharmaceutically acceptable salts, isomers, solvates, crystalline or prodrug thereof, wherein R 8 is selected from hydrogen, C 1 a -3 alkyl group and a C 3-6 cycloalkyl group, said C 1-3 alkyl group and a C 3-6 cycloalkyl group optionally being one or more halogen, C 1-3 alkyl group, halogenated C 1 a -3 alkyl group, a C 1-3 alkoxy group, an amino group, a mono C 1-3 alkylamino group, a di C 1-3 alkylamino group or a hydroxy group.
- the present invention provides a formula I or II compound represented by or a pharmaceutically acceptable salt, isomer, solvate, crystallization or prodrug thereof, wherein R 8 is selected from hydrogen, C 1 -3 alkyl and C 3-6 cycloalkyl, said C 1-3 alkyl and C 3-6 cycloalkyl optionally being one or more halogen, methyl, ethyl, propyl, isopropyl Base, trifluoromethyl, difluoromethyl, trifluoroethyl, methoxy, ethoxy, propoxy, isopropyloxy, amino, methylamino, ethylamino, propylamino, dimethylamino, Diethylamine, dipropylamino, methylethylamino, methylpropylamino, ethylpropylamino or hydroxy substituted.
- R 8 is selected from hydrogen, C 1 -3 alkyl and C 3-6 cycloalkyl, said
- the present invention provides a formula I or II compound represented by or a pharmaceutically acceptable salt, isomer, solvate, crystallization or prodrug thereof, wherein R 8 is selected from hydrogen, methyl , ethyl and propyl, said methyl, ethyl and propyl optionally being one or more amino, methylamino, ethylamino, propylamino, dimethylamino, diethylamino, dipropylamino, A Substituted by ethylamino, methylpropylamino or ethylpropylamino.
- the present invention provides a formula I or II compound represented by or a pharmaceutically acceptable salt, isomer, solvate, crystallization or prodrug thereof, wherein R 8 is substituted with dimethylamino Ethyl.
- the present invention provides a compound of general formula I or II, or a pharmaceutically acceptable salt thereof shown, isomer, solvate, crystallization or prodrug thereof, wherein R 7, R 8 and connected N constitutes a four to eight membered azacycloalkyl group, and the four to eight membered azacycloalkyl group is optionally substituted by one or more halogen, C 1-6 alkyl, halo C 1-6 alkyl, C 1- 6 alkoxy, amino, mono C 1-6 alkylamino, di C 1-6 alkylamino, hydroxy, amino C 1-6 alkyl, mono C 1-6 alkylamino C 1-6 alkyl, Di-C 1-6 alkylamino C 1-6 alkyl or hydroxyalkyl substituted.
- the present invention provides a compound of Formula I or II, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein R 7 , R 8 and their linkages N constitutes a four to eight membered azacycloalkyl group, and the four to eight membered azacycloalkyl group is optionally substituted by one or more halogen, C 1-3 alkyl, halo C 1-3 alkyl, C 1 -3 alkoxy, amino, mono C 1-3 alkylamino, di C 1-3 alkylamino, hydroxy, amino C 1-3 alkyl, mono C 1-3 alkylamino C 1-3 alkyl , a di-C 1-3 alkylamino C 1-3 alkyl group or a hydroxyalkyl group.
- the present invention provides a compound of Formula I or II, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein R 7 , R 8 and their linkages N-constituting five to six yuan azacycloalkyl, said azacycloalkyl five to six yuan optionally substituted with one or more halo, C 1-3 alkyl, halo C 1-3 alkyl, C 1-3 alkoxy, amino, mono C 1-3 alkylamino, di C 1-3 alkylamino, hydroxy, amino C 1-3 alkyl, mono C 1-3 alkylamino C 1-3 Alkyl, di-C 1-3 alkylamino C 1-3 alkyl or hydroxyalkyl substituted.
- the invention provides a compound of Formula I, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein R 4 , R 5 and R 6 are each independently selected From hydrogen, halogen, cyano, C 1-6 alkyl, C 1-6 alkoxy, C 3-8 cycloalkyl, said C 1-6 alkyl, C 1-6 alkoxy and ring
- the alkyl group is optionally selected from one or more of halogen, C 1-6 alkyl, halo C 1-6 alkyl, C 1-6 alkoxy, halo C 1-6 alkoxy, amino, mono C 1 -6 alkylamino, di C 1-6 alkylamino, hydroxy, amino C 1-6 alkyl, mono C 1-6 alkylamino C 1-6 alkyl, di C 1-6 alkylamino C 1 -6 alkyl and hydroxy C 1-6 alkyl substituted.
- the invention provides a compound of Formula I, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein R 4 , R 5 and R 6 are each independently Is selected from the group consisting of hydrogen, halogen, cyano, C 1-3 alkyl, C 1-3 alkoxy, C 3-6 cycloalkyl, said C 1-3 alkyl, C 1-3 alkoxy and C 3-6 cycloalkyl optionally substituted with one or more halo, C 1-3 alkyl, halo C 1-3 alkyl, C 1-3 alkoxy, a C 1-3 alkoxy group, halo , amino, mono C 1-3 alkylamino, di C 1-3 alkylamino, hydroxy, amino C 1-3 alkyl, mono C 1-3 alkylamino C 1-3 alkyl, di C 1- 3 alkylamino C 1-3 alkyl or hydroxy C 1-3 alkyl substituted.
- R 4 , R 5 and R 6 are each
- the invention provides a compound of Formula I, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein R 4 , R 5 and R 6 are hydrogen.
- the invention provides a compound of Formula III, or a pharmaceutically acceptable salt thereof, wherein M is selected from the group consisting of fluorine, chlorine, bromine, and iodine.
- the invention provides a compound of Formula III, or a pharmaceutically acceptable salt thereof, wherein M is selected from the group consisting of chlorine, bromine, and iodine.
- the present invention provides a compound of Formula I, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein
- n is selected from 1, 2, 3 and 4;
- n is selected from 1, 2, 3 and 4;
- R a is selected from the group consisting of hydrogen, C 1-3 alkyl, C 3-6 cycloalkyl, halogenated C 1-3 alkyl, C 1-3 alkoxy, halo C 1-3 alkoxy, nitro , cyano, hydroxy, amino, mono C 1-3 alkylamino, di C 1-3 alkylamino, amino C 1-3 alkyl, mono C 1-3 alkylamino C 1-3 alkyl, two C 1-3 alkylamino C 1-3 alkyl, hydroxy C 1-3 alkyl, C 1-3 alkoxy C 1-3 alkyl, C 1-3 alkyl acyl, amino acyl, mono C 1 -3 alkylaminoacyl, di C 1-3 alkylamino acyl, C 6-10 aryl, C 5-10 heteroaryl and C 3-6 heterocycloalkyl, when m is selected from 2, 3 and 4 When two R a groups of two adjacent carbon atoms may form a C 3-6 cycloalkyl
- X 1 , X 2 , X 3 and X 4 are each independently selected from C(R 1 ) and N;
- R 1 , R 2a , R 2b , R 3a , R 3b and R 3c are each independently selected from the group consisting of hydrogen, halogen, C 1-3 alkyl, halo C 1-3 alkyl, C 1-3 alkoxy, Halogen C 1-3 alkoxy, nitro, cyano, hydroxy, amino, mono C 1-3 alkylamino, di C 1-3 alkylamino, amino C 1-3 alkyl, mono C 1- 3 alkylamino C 1-6 alkyl, di C 1-3 alkylamino C 1-3 alkyl, hydroxy C 1-3 alkyl, C 1-3 alkoxy C 1-3 alkyl, C 1 a -3 alkyl acyl group, an amino acyl group, a mono C 1-3 alkylamino acyl group and a di C 1-3 alkylamino acyl group;
- R 4 , R 5 and R 6 are all hydrogen
- R 7 is a methyl group
- R 8 is an ethyl group substituted by dimethylamino.
- the present invention provides a compound of Formula I, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein
- n is selected from 1, 2, 3 and 4;
- p is selected from 0, 1, and 2;
- R a is selected from hydrogen, C 1-3 alkyl, C 3-6 cycloalkyl, halogenated C 1-3 alkyl, C 1-3 alkoxy, halogen C 1-3 alkoxy, nitro, cyano, hydroxy, amino, mono C 1-3 alkylamino, di C 1-3 alkylamino, amino C 1-3 alkyl, mono C 1-3 alkyl C 1-3 alkyl amino, di C 1-3 alkyl amino C 1-3 alkyl, hydroxy C 1-3 alkyl, C 1-3 alkoxy C 1-3 alkyl, C 1-3 alkoxy Alkanoyl group, aminoacyl group, mono C 1-3 alkylamino acyl group, di C 1-3 alkylamino acyl group, C 6-10 aryl group, C 5-10 heteroaryl group and C 3-6 heterocycloalkyl group, When m is selected from 2, 3 and 4, two R a on two adjacent atoms may together with the atom to
- X 1 , X 2 , X 3 and X 4 are each independently selected from C(R 1 ) and N;
- R 1 , R 2a , R 2b , R 3a , R 3b and R 3c are each independently selected from the group consisting of hydrogen, halogen, C 1-3 alkyl, halo C 1-3 alkyl, C 1-3 alkoxy, Halogen C 1-3 alkoxy, nitro, cyano, hydroxy, amino, mono C 1-3 alkylamino, di C 1-3 alkylamino, amino C 1-3 alkyl, mono C 1- 3 alkylamino C 1-6 alkyl, di C 1-3 alkylamino C 1-3 alkyl, hydroxy C 1-3 alkyl, C 1-3 alkoxy C 1-3 alkyl, C 1 a -3 alkyl acyl group, an amino acyl group, a mono C 1-3 alkylamino acyl group and a di C 1-3 alkylamino acyl group;
- R 4 , R 5 and R 6 are all hydrogen
- R 7 is a methyl group
- R 8 is an ethyl group substituted by dimethylamino.
- the present invention provides a compound of Formula I, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein
- n is selected from 1, 2, 3 and 4;
- p is selected from 0, 1, and 2;
- R a is selected from hydrogen, C 1-3 alkyl, C 3-6 cycloalkyl, halogenated C 1-3 alkyl, C 1-3 alkoxy, halogen C 1-3 alkoxy, nitro, cyano, hydroxy, amino, mono C 1-3 alkylamino, di C 1-3 alkylamino, amino C 1-3 alkyl, mono C 1-3 alkyl Amino C 1-3 alkyl, di C 1-3 alkylamino C 1-3 alkyl, hydroxy C 1-3 alkyl, C 1-3 alkoxy C 1-3 alkyl, C 1-3 alkane an acyl group, an amino group, a mono C 1-3 alkyl amino group, a di C 1-3 alkyl amino group, C 6-10 aryl, C 5-10 heteroaryl, and C 3-6 heterocycloalkyl, When m is selected from 2, 3 and 4, two R a on two adjacent atoms may form a C 3-6 cycloalkyl, When m is selected from 2,
- X 1 , X 2 , X 3 and X 4 are each independently selected from C(R 1 ) and N;
- R 1 , R 2a , R 2b , R 3a , R 3b and R 3c are each independently selected from the group consisting of hydrogen, halogen, C 1-3 alkyl, halo C 1-3 alkyl, C 1-3 alkoxy, Halogen C 1-3 alkoxy, nitro, cyano, hydroxy, amino, mono C 1-3 alkylamino, di C 1-3 alkylamino, amino C 1-3 alkyl, mono C 1- 3 alkylamino C 1-6 alkyl, di C 1-3 alkylamino C 1-3 alkyl, hydroxy C 1-3 alkyl, C 1-3 alkoxy C 1-3 alkyl, C 1 a -3 alkyl acyl group, an amino acyl group, a mono C 1-3 alkylamino acyl group and a di C 1-3 alkylamino acyl group;
- R 4 , R 5 and R 6 are all hydrogen
- R 7 is a methyl group
- R 8 is an ethyl group substituted by dimethylamino.
- the invention provides the following compounds, or pharmaceutically acceptable salts, isomers, solvates, crystals or prodrugs thereof:
- the invention provides the following compounds, or pharmaceutically acceptable salts, isomers, solvates, crystals or prodrugs thereof:
- the invention provides a process for the preparation of a compound of formula I, II and III of the invention, comprising the steps of:
- the invention provides a process for the preparation of a compound of formula I of the invention, which process comprises the use of a compound of formula II of the invention.
- the invention provides a process for the preparation of a compound of formula I of the invention, which process comprises the use of a compound of formula III of the invention.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula I of the invention, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, and a pharmaceutically acceptable carrier.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound, isomer, solvate, crystal or prodrug of Formula I of the invention, further comprising one or more compounds selected from the group consisting of: Gefitinib, erlotinib, lapatinib, afatinib, vandetanib, carnitinib, apatinib, dacomitinib, pelitinib , WZ4002, AG-490, AZD8931, AZD9291 and so on.
- the compound, isomer, solvate, crystal or prodrug of the formula I of the present invention may be admixed with a pharmaceutically acceptable carrier to prepare a pharmaceutical preparation suitable for oral or parenteral administration.
- Methods of administration include, but are not limited to, intradermal, intramuscular, intraperitoneal, intravenous, subcutaneous, intranasal, and oral routes.
- the formulation may be administered by any route, for example by infusion or bolus injection, by a route of absorption through the epithelium or skin mucosa (e.g., oral mucosa or rectum, etc.). Administration can be systemic or topical.
- orally administered preparations include solid or liquid dosage forms, specifically, tablets, pills, granules, powders, capsules, syrups, emulsions, suspensions and the like.
- the formulation can be prepared by methods known in the art and comprises a carrier conventionally used in the field of pharmaceutical formulations.
- the present invention provides a method of treating and/or preventing a tumor of a compound, isomer, solvate, crystal or prodrug of the present invention or a pharmaceutical composition of the present invention, and in the preparation of a treatment and/or prevention Use in a tumor drug, comprising administering to a tumor-prone population or a tumor patient a compound, isomer, solvate, crystal or prodrug of the formula I of the invention or a compound, isomer comprising the formula I of the invention, A pharmaceutical composition of a solvate, crystal or prodrug to effectively reduce the incidence of tumors and prolong the life of a tumor patient.
- the invention provides a method of treating a tumor having resistance to a compound, a isomer, a solvate, a crystal or a prodrug of the invention, or a pharmaceutical composition of the invention, comprising A pharmaceutically acceptable tumor patient is administered a therapeutically effective amount of a compound, isomer, solvate, crystal or prodrug of the formula I according to the invention or a compound, isomer, solvate, crystal or pre-compound comprising the formula I of the invention Pharmaceutical composition of the drug.
- the invention provides a compound, isomer, solvate, crystal or prodrug of the invention, or a pharmaceutical composition of the invention, in the manufacture of a medicament for treating a tumor having drug resistance application.
- the drug-resistant tumor may be a tumor resistant to a plurality of drugs, preferably a tumor resistant to an EGFR inhibitor, for example, to a first, second, and third generation EGFR inhibitor, such as gefitinib. , erlotinib and lapatinib have drug-resistant tumors.
- Such tumors include, but are not limited to, solid tumors, preferably lung cancer, head and neck tumors, colorectal cancer, bladder cancer, pancreatic cancer, breast cancer, prostate cancer, gastric cancer, oral cancer, liver cancer, ovarian cancer. More preferably, the tumor is non-small cell lung cancer.
- the invention provides a method of treating a tumor having resistance to a compound, isomer, solvate, crystal or prodrug of Formula I of the invention, wherein the tumor carries an EGFR mutated gene.
- the EGFR mutated gene carried by the tumor is a T790M mutation in exon 20.
- the EGFR mutated gene carried by the tumor is a L858R mutation and/or a deletion/insertion mutation in exon 21.
- the EGFR mutated gene carried by the tumor is a T790M and L858R double mutation.
- the invention provides a compound, isomer, solvate, crystal or a compound of the formula I of the invention for use in treating a tumor A prodrug or a pharmaceutical composition of the invention wherein the therapeutic effect of the tumor is manifested in a prominent therapeutic effect, a high degree of selectivity and/or less side effects.
- the present invention provides a method of treating a tumor of a compound, isomer, solvate, crystal or prodrug of the present invention or a pharmaceutical composition of the present invention, the method comprising administering it A therapeutically effective amount of a compound, isomer, solvate, crystal or prodrug of the present invention or a pharmaceutical composition of the present invention, which produces a therapeutic effect in the treatment of a tumor, which is highly effective. Selectivity and / or fewer side effects.
- “Isomers” of the present invention include cis-trans isomers of the cis or trans configuration, as well as enantiomers and diastereomers derived from chiral carbons.
- the "pharmaceutically acceptable salt” of the present invention means a pharmaceutically acceptable salt formed by the compound of the present invention and an acid, and the acid may be selected from the group consisting of phosphoric acid, sulfuric acid, hydrochloric acid, hydrobromic acid, citric acid, Maleic acid, malonic acid, mandelic acid, succinic acid, fumaric acid, acetic acid, lactic acid, nitric acid, sulfonic acid, p-toluenesulfonic acid, malic acid, methanesulfonic acid and the like.
- Crystal as used in the present invention refers to various solid forms formed by the compounds of the present invention, including crystalline forms and amorphous forms.
- the "prodrug” of the present invention refers to a compound which is converted into a compound of the present invention by a reaction with an enzyme, gastric acid or the like under physiological conditions of a living body, that is, a compound which is converted into the invention by oxidation, reduction, hydrolysis or the like of the enzyme and/or A compound which is converted into a compound of the invention by a hydrolysis reaction such as gastric acid or the like.
- a "pharmaceutical composition” is meant to comprise any one of the compounds described herein, including isomers, prodrugs, solvates, pharmaceutically acceptable salts or chemically protected forms thereof, and one or more A mixture of pharmaceutically acceptable carriers.
- the "application in the preparation of a medicament for treating and/or preventing a tumor” of the present invention means that the growth, development and/or metastasis of a tumor can be inhibited, and a therapeutically effective dose of the present invention is mainly administered to a human or animal in need thereof.
- a compound inhibits, slows or reverses the growth, progression or spread of a tumor in a subject.
- alkyl refers to a straight or branched saturated hydrocarbon group, preferably a hydrocarbon group of 6 or less carbon atoms.
- alkyl groups include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, tert-butyl, n-pentyl, isopentyl, neopentyl, n-hexyl, isohexyl, 2,2-Methylbutyl and 2,3-dimethylbutyl.
- C 1-6 alkyl refers to a straight or branched saturated hydrocarbon group containing from 1 to 6 carbon atoms.
- C 1-3 alkyl means a straight or branched saturated hydrocarbon group having 1 to 3 carbon atoms.
- the "cycloalkyl group” of the present invention means a cyclic saturated, partially saturated hydrocarbon group, preferably a cycloalkyl group of 12 or less carbon atoms, more preferably a cycloalkyl group of 8 or less carbon atoms, still more preferably 6 carbon atoms.
- the following cycloalkyl group examples include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl.
- the "C 3-8 cycloalkyl group” of the present invention means a cyclic saturated hydrocarbon group having 3 to 8 carbon atoms.
- alkoxy group of the present invention means an -O-alkyl group.
- the sulfur atom is replaced by an oxo group
- the sulfur atom is replaced by two oxo groups
- halogen of the present invention means fluorine, chlorine, bromine or iodine.
- haloalkoxy group of the present invention means an alkoxy group substituted with at least one halogen, preferably a C 1-6 alkoxy group substituted with at least one halogen, and further preferably a C 1-3 alkane substituted with at least one halogen.
- Oxyl, a suitable halogenated C 1-3 alkoxy group is chloromethoxy, fluoromethoxy, dichloromethoxy, difluoromethoxy, trichloromethoxy, trifluoromethoxy; Chloroethoxy, difluoroethoxy, trichloroethoxy, trifluoroethoxy.
- the "monoalkylamino group” of the present invention means -NH-alkyl group, preferably -NH-C 1-6 alkyl group, and more preferably -NH-C 1-3 alkyl group.
- dialkylamino group means -N-(alkyl)(alkyl), preferably -N-(C 1-6 alkyl)(C 1-6 alkyl), further preferably -N -(C 1-3 alkyl) (C 1-3 alkyl).
- aminoalkyl group of the present invention means -alkyl-NH 2 .
- the "monoalkylaminoalkyl group" of the present invention means an -alkyl-NH-alkyl group.
- dialkylaminoalkyl group of the present invention means -alkyl-N-(alkyl)(alkyl).
- hydroxyalkyl group of the present invention means -alkyl-OH.
- alkyl acyl group of the present invention means a -C(O)-alkyl group.
- the "monoalkylaminoacyl" of the present invention means -C(O)-NH-alkyl.
- dialkylaminoacyl group of the present invention means -C(O)-N-(alkyl)(alkyl).
- heterocycloalkyl group of the present invention means a substituted or unsubstituted saturated, partially saturated cyclic alkyl group containing at least one hetero atom selected from N, O, and S.
- the "aryl group” of the present invention means an aromatic system which may contain a monocyclic or polycondensed ring such as a bicyclic or tricyclic aromatic ring, wherein at least a part of the fused ring forms a conjugated aromatic system containing 5 to 50 One carbon atom, preferably from about 6 to about 14 carbon atoms.
- Suitable aryl groups include, but are not limited to, phenyl, naphthyl, biphenyl, anthracenyl, tetrahydronaphthyl, anthracenyl, indanyl, biphenylene, and anthracenyl.
- the "C 6 -C 10 aryl group” of the present invention means an aromatic system having 6 to 10 carbon atoms.
- heteroaryl of the present invention means an aromatic monocyclic or polycondensed ring such as an aromatic group in which at least one carbon atom of a bicyclic or tricyclic ring is replaced by a hetero atom, and the hetero atom is O, S, N.
- Suitable heteroaryl groups include, but are not limited to, imidazolyl, benzimidazolyl, imidazopyridyl, quinazolinone, pyrrolyl, imidazolidinyl, furyl, thienyl, pyrazolyl, oxazolyl, thiazole Base, isoxazolyl, isothiazolyl, oxadiazolyl, triazolyl and the like.
- the "C 5 -C 10 heteroaryl group” of the present invention means a heteroaryl group having 5 to 10 atoms.
- the "C 4 -C 8 nitrogen-containing heterocyclic group” of the present invention means a substituted or unsubstituted saturated, partially saturated and fully unsaturated total ring atom number of 4, 5, 6, 7 or containing at least one nitrogen atom.
- the heterocyclic group of 8, for example, a C 4 nitrogen-containing heterocyclic group means a substituted or unsubstituted saturated, partially saturated and fully unsaturated total nitrogen-containing heterocyclic group having 4 ring atoms.
- the "C 4 -C 8 nitrogen-containing heteromonocycloalkyl group” of the present invention means a substituted or unsubstituted saturated, partially saturated and fully unsaturated total ring atom number of 4, 5, 6, containing at least one nitrogen atom.
- Suitable heterocycloalkyl groups of 7 or 8 include, but are not limited to, substituted or unsubstituted azetidinyl, azacyclopentyl, azacyclohexyl, azepanyl, azacyclooctyl , azacyclopentyl, azacyclohexyl, aziridine, azacyclooctyl, azathiolanyl, azathioheptyl, azathioheptyl, a nitrogen sulfur a heterocyclic octyl group, a diazacyclopentyl group, a diazacyclohexyl group, a diazepanyl group or a diazacyclooctyl group; and a substituted or unsubstituted nitrogen-containing heteroaryl group, for example, substituted or unsubstituted Pyridyl, imidazolyl, pyrazolyl, pyrrolyl, pyrida
- the obtained product of the step b was sequentially added 1-(3-iodopropyl)-1H-indole (5.44 g, 19.83 mmol), potassium phosphate (8.4 g, 39.67 mmol), tetratriphenylphosphine.
- Palladium (2.3 g, 1.98 mmol) and 1,4-dioxane (80 ml) were argon-protected and refluxed overnight. After the reaction was completed, the mixture was evaporated.
- step d 9-(2-chloropyrimidin-4-yl)-2,3-dihydropyrrolo[1,2-a]indole
- step 2 In a 100 ml reaction flask, aluminum trichloride (2.18 g, 16.35 mmol), ethylene glycol dimethyl ether (50 ml), 2,4-dichloropyrimidine (2.44 g, 16.35 mmol) and step 2 were successively added. 3-Dihydropyrrolo[1,2-a]indole (2.57 g, 16.35 mmol), refluxed for 2 h. After completion of the reaction, the reaction solution was cooled to room temperature, filtered, and the filter cake was washed with water and dried to give the title compound.
- step e In a 50 ml reaction flask, the resulting product of step e was sequentially added N-(4-fluoro-2-methoxy-5-nitrophenyl)-4-(2,3-dihydropyrrolo[1,2-a ⁇ -9-yl)-pyrimidine-2-amine (2.5 g, 6 mmol), N,N,N'-trimethylethylenediamine (0.61 g, 6 mmol), diisopropylethylamine (2.3 g , 18 mmol), and 10 ml of dioxane, refluxing at 110 ° C for 3 h. After completion of the reaction, concentration and purification by column chromatography
- step N was added N-(4-((2-(dimethylamino)ethyl))(methyl)amino)-2-methoxy-5-nitrophenyl) 4-(2,3-dihydropyrrolo[1,2-a]indol-9-yl)-pyrimidin-2-amine (2.5 g, 5 mmol), 10% Pd-C (20 mg) and 30 mL methanol Hydrogen was reduced for 1 h at 1 standard atmosphere. After the reaction was completed, it was filtered and concentrated to give the title compound.
- step g was added N-(4-((2-(dimethylamino)ethyl)(methyl)amino)-2-methoxy-5-aminophenyl)-4 -(2,3-dihydropyrrolo[1,2-a]indol-9-yl)-pyrimidin-2-amine (2 g, 4.2 mmol), diisopropylethylamine (0.53 g, 4.2 mmol)
- 20 ml of anhydrous dichloromethane was added, and 5 ml of a solution of allylic acid chloride (0.37 g, 4.2 mmol) in methylene chloride was added slowly, and the mixture was reacted for 10 min.
- step b In a 250 mL three-necked flask, the obtained product of step b was added 1-(4-iodobutyl)-1H-indole (5.93 g, 19.83 mmol), potassium phosphate (8.4 g, 39.67 mmol), tetratriphenyl. Palladium phosphate (2.3 g, 1.98 mmol), 1,4-dioxane (80 mL), argon-protected, refluxed overnight. After the reaction was completed, the mixture was evaporated.
- step c In a 100 mL reaction flask, aluminum trichloride (2.18 g, 16.35 mmol), ethylene glycol dimethyl ether (50 mL), 2,4-dichloropyrimidine (2.44 g, 16.35 mmol), and the obtained product of step c were successively added. , 7,8,9-tetrahydropyrido[1,2-a]indole (2.8 g, 16.35 mmol), refluxed for 2 h. After completion of the reaction, the reaction solution was cooled to room temperature, filtered, and the filter cake was washed with water and dried to give the title compound.
- the product obtained in the step d is 10-(2-chloropyrimidin-4-yl)-6,7,8,9-tetrahydropyrido[1,2-a]indole, 4-fluoro-2-methoxy- 5-Nitroaniline, N,N,N'-trimethylethylenediamine and allyl chloride were used as starting materials, and the title compound was obtained according to the following procedure:
- Step b N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((5-methyl-4-(6,7,8) Synthesis of 9-tetrahydropyrido[1,2-a]indol-10-ylpyrimidin-2-yl)amino)phenyl)propanamide
- the product obtained in the step a is 10-(2-chloro-5-methylpyrimidin-4-yl)-6,7,8,9-tetrahydropyrido[1,2-a]indole, 4-fluoro-2. -Methoxy-5-nitroaniline, N,N,N'-trimethylethylenediamine and allyl chloride as starting materials, the title compound was obtained according to the procedure of Example 1, Steps e, f, g and h .
- Step c N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(3,4-dihydro-1H-[ Synthesis of 1,4]oxazino[4,3-a]indole-10-ylpyrimidin-2-yl)amino)phenyl)propanamide
- Step b Synthesis of N-((1-propenyl-1H-indol-2-yl)methenyl)-4-methylbenzenesulfonylhydrazide:
- Step c Synthesis of 1,1,2,8b-tetrahydrocyclopropyl[3,4]pyrrolo[1,2-a]indole:
- N-((1-propenyl-1H-indol-2-yl)methenyl)-4-methylbenzenesulfonyl hydrazide (2.2 g, 6.23 mmol) obtained in step b was added, and carbonic acid was added.
- Potassium (1.29 g, 9.35 mmol) and 60 ml of dioxane were refluxed in an oil bath at 100 ° C for 12 h. Filtration, concentration, addition of 50 ml of water, extraction with 3X 40 ml of ethanol, washing with 30 ml of water, concentration and column chromatography to give 1.0 g of the title product. Used directly in the next step.
- Step d N-(2-((2-(Dimethylamino)ethyl)(methyl)amino-4-methoxy-5-((4(1,1a,2,8b-tetrahydrocyclo)) Synthesis of propyl[3,4]pyrrolo[1,2-a]indole-8-yl)pyrimidin-2-yl)amino)phenyl)acrylamide
- EGFR WT kinase purchased from Carna Corporation
- EGFR T790M/L858R kinase was purchased from Invitrogen.
- ATP Adenosine triphosphate
- Peptide FAM-P22 purchased from GL Biochem
- Ethylenediaminetetraacetic acid was purchased from Sigma.
- Caliper EZ reader microfluidic chip instrument purchased from Caliper Life Sciences, Inc.
- 1 ⁇ kinase base buffer (for EGFR WT ): 50 mM HEPES, pH 7.5, 0.0015% Brij-35, 10 mM MgCl 2 , 10 mM MnCl 2 , 2 mM DTT;
- 1 ⁇ kinase base buffer for EGFR T790M/L858R : 50 mM HEPES, pH 7.5, 0.0015% Brij-35, 10 mM MgCl 2 , 2 mM DTT;
- Stop buffer 100 mM HEPES, pH 7.5, 0.0015% Brij-35, 0.2% Coating Reagent #3, 50 mM EDTA.
- the compounds of the present invention were separately dissolved in 10 mM with 100% DMSO, diluted to 50 ⁇ M with complete medium, and then diluted to 5 ⁇ M with complete medium containing 0.1% DMSO, followed by 3-fold dilution for a total of 10 concentrations (for EGFR WT );
- the compound of the present invention was separately dissolved to 10 mM with 100% DMSO, diluted to 50 ⁇ M with complete medium, and then diluted to 1 ⁇ M with complete medium containing 0.1% DMSO, and then diluted 3 times in total for 10 concentrations (for EGFR T790M/L858R );
- the 96-well plate used above was labeled as the source plate.
- 10 ⁇ l of the solution was transferred from the source plate into a new 96-well plate as an intermediate plate, and 90 ⁇ l of 1 ⁇ kinase buffer was added to each well of the intermediate plate, and vortexed and mixed for 10 minutes.
- a non-kinase-free compound control group (containing DMSO, 1 ⁇ basal buffer and 2.5 ⁇ peptide solution, and a kinase-free compound control group (including DMSO, 2.5 ⁇ kinase solution, and 2.5 ⁇ peptide solution) was also provided.
- Inhibition rate % (max-com)/(max-min) ⁇ 100 Calculate the inhibition rate, where “max” represents a kinase-free compound control group, “com” represents a test compound group, and “min” represents a kinase-free compound. Control group.
- the compound of the present invention has a good inhibitory activity against a mutant EGFR kinase such as EGFR L858R/T790M kinase, and has an IC 50 value of less than 1 nM, and has a small effect on wild-type EGFR kinase and has good selectivity.
- a mutant EGFR kinase such as EGFR L858R/T790M kinase
- the experimental cell line NCI-H1975 (EGFR double mutant cells with L858R and T790M mutations) and A431 (EGFR wild type cells) were purchased from ATCC.
- RPMI1640 medium purchased from Invitrogen
- DMEM medium purchased from Invitrogen
- Fetal bovine serum purchased from Invitrogen;
- NCI-H1975 cells were cultured in RPMI1640 medium containing 10% inactivated fetal bovine serum (GIBCO), containing penicillin 100 IU/mL and streptomycin 100 ⁇ g/mL;
- GEBCO inactivated fetal bovine serum
- A431 cells were cultured in DMEM medium containing 10% inactivated fetal bovine serum (GIBCO) containing penicillin 100 IU/mL and streptomycin 100 ⁇ g/mL.
- GEBCO inactivated fetal bovine serum
- NCI-H1975 cells and A431 cells After digesting NCI-H1975 cells and A431 cells in logarithmic growth phase, they were blown into single cell suspensions, seeded in 96-well culture plates, 100 ⁇ L per well medium, and each cell line was composed of 3 96-well plates. NCI-H1975 cells were seeded with 3 ⁇ 10 3 cells per well, and A431 cells were seeded with 4 ⁇ 10 3 cells per well. The inoculated NCI-H1975 cells and A431 cells were cultured in a 5% CO 2 incubator for 16-24 hours. After the cells were attached, the test compound was added at the following concentration (the highest test of the compound on NCI-H1975 cells).
- the concentration was 4 ⁇ M, 3 fold dilution, a total of 9 concentrations; the highest test concentration on A431 cells was 10 ⁇ M, 3 fold dilution, a total of 9 concentrations), and cultured for an additional 72 hours in an incubator.
- a blank control group only medium, no cell and DMSO solution
- a DMSO control group cells and 0.5% DMSO solution were added to the medium.
- Add 100 ⁇ L of CTG solution shake it for 2 min in the dark, and incubate for 10 min.
- the multi-mode microplate reader reads the plate, records the luminescence reading results, and calculates the inhibition rate according to the following formula:
- Inhibitor (%) (1-(RLU com - RLU blank ) / (RLU DMSO - RLU blank )) ⁇ 100%,
- RLU com represents the absorbance of the test compound group
- RLU blank represents the absorbance of the blank control group
- RLU DMSO represents the absorbance of the DMSO control group
- the XEFFit curve fitting software was used to plot the pharmacodynamic inhibition rate curve and calculate the IC 50 value. The results are shown in Table 2.
- the compounds of the present invention have a better inhibitory effect on double mutant cells (NCI-H1975), and have less inhibition on EGFR wild type cells (A431), and the selectivity is similar to or significantly superior to AZD9291.
- One of the major side effects of currently marketed EGFR inhibitors is rash, diarrhea, etc., which are associated with inhibition of wild-type EGFR and poor selectivity. Therefore, the compound of the present invention is expected to be a drug having a specific therapeutic effect against a tumor resistant to EGFR mutation and having a small side effect.
- the compound of the present invention was dissolved in 25 mM sodium citrate-sodium citrate buffer (pH 4.5) to prepare a clear solution having a concentration of 1.25 mg/mL, and was set as a test compound group; 25 mM citric acid was used.
- - sodium citrate buffer (pH 4.5) was set as a vehicle control group;
- mice Female BALB/C mice, 5 in each group, weighing 18-22 g, were provided by Shanghai Xipuer-Beikai Experimental Animal Co., Ltd. The test mice were given an environmental adaptation period of 2 to 4 days before the experiment, and fasted for 8-12 hours before administration.
- intragastric administration administration of the compound of the invention 25mg / kg;
- AUC [(A 15min +A 0 ) ⁇ 7.5]+[(A 30min + A 15min ) ⁇ 7.5]+[(A 60min +A 30min ) ⁇ 15]+[(A 120min +A 60min ) ⁇ 30]
- AUC value and increase % [(AUC compound- AUC solvent ) according to the formula AUC/ AUC solvent ] ⁇ 100% calculated the AUC growth rate, where “A 0 ” represents the blood glucose level at 0 min after gavage, “A 15 min ” represents the blood glucose level after 15 min, and “A 30 min ” represents the blood glucose after 30 min.
- EGFR inhibitors may cause side effects such as hyperglycemia and impaired insulin signaling.
- AZD9291 and Clociletinib (CO1686) of Clovis Oncology have a certain degree of side effects on blood glucose (Management of hyperglycemia from epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) targeting T790M-mediated resistance, Jeryl Villadolid , et al. Translational Lung Cancer Research (2015), 4(5), 576-583).
- EGFR epidermal growth factor receptor
- TKIs tyrosine kinase inhibitors
- the compounds of the invention are formulated into either oral and injectable formulations.
- the oral drug formulation was dissolved in 25 mM sodium citrate-sodium citrate buffer (pH 4.5) to prepare a 1.25 mg/mL clear solution; the tail vein injection drug formulation was citrate-sodium citrate buffer solution.
- a mixed solution of (pH 4.5) and physiological saline volume ratio of 1:1 was prepared into a 0.2 mg/mL solution.
- mice Female BALB/C mice, 5 in each group, weighing 18-22 g, were provided by Shanghai Xipuer-Beikai Experimental Animal Co., Ltd. The test mice were given an environmental adaptation period of 2 to 4 days before the experiment, and were fasted for 8-12 hours before administration, and given water for 2 hours after administration, and fed after 4 hours.
- Acetonitrile (chromatographically pure): produced by Spectrum;
- intragastric administration administration of the compound of the invention 25mg / kg;
- mice in step 2.1 the intravenous vein (Intravenous administration, I.V.) administered the compound of the invention 2mg / kg;
- the compounds of the invention have good pharmacokinetic data and should have good clinical application prospects.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description
本发明属于医药化学领域,具体涉及一类新的表皮生长因子受体抑制剂、含有该抑制剂的药物组合物,以及所述抑制剂或药物组合物作为癌症治疗药物的用途。The invention belongs to the field of medical chemistry, and in particular relates to a novel class of epidermal growth factor receptor inhibitors, a pharmaceutical composition containing the same, and the use of the inhibitor or pharmaceutical composition as a therapeutic drug for cancer.
表皮生长因子受体(Epidermal Growth Factor Receptor,EGFR)是原癌基因C-erbB-1的表达产物,为EGFR家族成员之一。EGFR家族包括EGFR(HER-1)、ERBB2(HER-2)、ERBB3(HER-3)和ERBB4(HER-4)四个成员。已表明EGFR过表达或突变一般会引发肿瘤。EGFR突变导致EGFR的持续活化,自分泌环的作用增强,受体下调机制破坏,异常信号传导通路激活,在肿瘤的演进中起着重要作用。The Epidermal Growth Factor Receptor (EGFR) is an expression product of the proto-oncogene C-erbB-1 and is a member of the EGFR family. The EGFR family includes four members, EGFR (HER-1), ERBB2 (HER-2), ERBB3 (HER-3), and ERBB4 (HER-4). Overexpression or mutation of EGFR has been shown to generally trigger tumors. EGFR mutations lead to sustained activation of EGFR, enhanced autocrine loops, disruption of receptor down-regulation, activation of abnormal signaling pathways, and play an important role in tumor progression.
针对EGFR采用EGFR-酪氨酸激酶抑制剂(EGFR-TKI)治疗已经成为非小细胞肺癌治疗领域里的金标准。不过,临床使用发现,多数患者会在吉非替尼、厄洛替尼等EGFR-TKI抑制剂治疗后6-12个月发生不同程度的耐药现象,进而导致药物的疗效显著降低,肿瘤进展。研究显示EGFR-TKI耐药性的产生与EGFR基因的二次突变有关,其中最常见的突变是EGFR基因20号外显子第790位点的突变,即T790M基因突变。由于甲硫氨酸比苏氨酸空间占位大,因此形成空间位阻,改变了EGFR激酶区ATP的亲和性,导致EGFR-TKI小分子药物不能有效阻断EGFR活化信号,导致耐药性的产生。同时,第一代EGFR抑制剂缺乏野生型EGFR与突变型EGFR的选择性,普遍存在皮疹、腹泻等副作用,影响患者依从性。The use of EGFR-tyrosine kinase inhibitor (EGFR-TKI) for EGFR has become the gold standard in the field of non-small cell lung cancer treatment. However, clinical use has found that most patients will have different degrees of drug resistance 6-12 months after treatment with EGFR-TKI inhibitors such as gefitinib and erlotinib, resulting in a significant reduction in drug efficacy and tumor progression. . Studies have shown that the production of EGFR-TKI resistance is associated with a second mutation in the EGFR gene, the most common of which is the mutation at position 790 of exon 20 of the EGFR gene, the T790M gene mutation. Because methionine has a larger space occupying than threonine, it forms steric hindrance and changes the affinity of ATP in EGFR kinase domain, which leads to EGFR-TKI small molecule drug can not effectively block EGFR activation signal, leading to drug resistance. The production. At the same time, the first generation of EGFR inhibitors lacked the selectivity of wild-type EGFR and mutant EGFR, and generally had side effects such as rash and diarrhea, which affected patient compliance.
因此,开发对因EGFR二次突变产生耐药的肿瘤患者有效的药物,特别是开发能进一步提高对野生型EGFR与突变型EGFR选择性,增强疗效,降低副作用的药物,将具有良好的应用前景。Therefore, the development of drugs that are effective against tumor patients who are resistant to secondary mutations in EGFR, especially the development of drugs that can further improve the selectivity, enhance efficacy, and reduce side effects of wild-type EGFR and mutant EGFR, will have good application prospects. .
发明内容Summary of the invention
本发明的目的是提供通式I的表皮生长因子抑制剂或其药学可接受的盐、异构体、溶剂合物、结晶或前药,该类化合物表现出良好的EGFR抑制活性,尤其是针对突变的EGFR, It is an object of the present invention to provide an epidermal growth factor inhibitor of the formula I, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, which exhibits good EGFR inhibitory activity, especially Mutant EGFR,

本发明的另一个目的是提供通式II的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,以所述通式II的化合物为关键中间体,制备通式I的化合物,反应条件温和,收率和纯度较高,Another object of the present invention is to provide a compound of the formula II or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, which is prepared by using the compound of the formula II as a key intermediate. Compound I, mild reaction conditions, high yield and purity,

本发明的第三个目的是提供通式III的化合物或其盐,以所述通式III的化合物为关键中间体,制备通式I的化合物,反应步骤少,条件温和,收率和纯度较高,A third object of the present invention is to provide a compound of the formula III or a salt thereof, which is a key intermediate of the compound of the formula III, which has a small number of reaction steps, mild conditions, and a relatively good yield and purity. high,
本发明的第四个目的是提供制备本发明的通式I、II或III的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药的方法。A fourth object of the present invention is to provide a process for the preparation of a compound of the formula I, II or III of the present invention, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof.
本发明的第五个目的是提供包含本发明的通式I的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药和药学可接受的载体的药物组合物以及包含本发明的通式I的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药和另一种肿瘤抑制剂的药物组合物。A fifth object of the present invention is to provide a pharmaceutical composition comprising a compound of the formula I of the present invention, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, and a pharmaceutically acceptable carrier, and a pharmaceutical composition comprising the same A pharmaceutical composition of a compound of formula I of the invention, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, and another tumor suppressor.
本发明的第六个目的是提供本发明的通式I的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药以及本发明的药物组合物治疗和/或预防癌症的方法及其在制备用于治疗和/或预防癌症的药物中的应用。A sixth object of the present invention is to provide a compound of the formula I of the present invention or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, and a pharmaceutical composition of the present invention for treating and/or preventing cancer Methods and their use in the preparation of a medicament for the treatment and/or prevention of cancer.
针对上述发明目的,本发明提供以下技术方案:In view of the above object, the present invention provides the following technical solutions:
第一方面,本发明提供通式I所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药, In a first aspect, the present invention provides a compound of Formula I, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof,
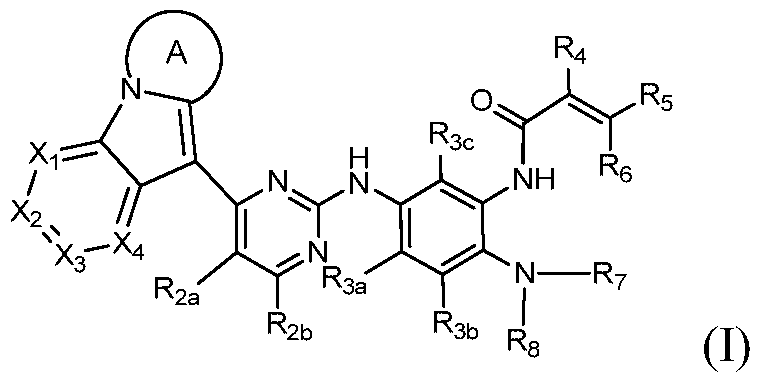
其中,among them,
环A为C4-C8含氮杂环基,其任选被一个或多个卤素、氧代、烷基、环烷基、卤代烷基、烷氧基、卤代烷氧基、硝基、氰基、羟基、氨基、单烷基氨基、二烷基氨基、氨基烷基、单烷基氨基烷基、二烷基氨基烷基、羟基烷基、烷氧基烷基、烷基酰基、氨基酰基、单烷基氨基酰基、二烷基氨基酰基、芳基、杂芳基和杂环烷基取代,当取代基有两个以上时,相邻的两个取代基可以与它们连接的原子一起构成取代或未取代的环烷基、杂环烷基、芳基、杂芳基,或连接在同一个原子上的两个取代基可以与它们共同连接的原子形成取代或未取代的环烷基、杂环烷基;Ring A is a C 4 -C 8 nitrogen-containing heterocyclic group optionally substituted by one or more halogen, oxo, alkyl, cycloalkyl, haloalkyl, alkoxy, haloalkoxy, nitro, cyano , hydroxy, amino, monoalkylamino, dialkylamino, aminoalkyl, monoalkylaminoalkyl, dialkylaminoalkyl, hydroxyalkyl, alkoxyalkyl, alkyl acyl, amino acyl, a monoalkylaminoacyl group, a dialkylaminoacyl group, an aryl group, a heteroaryl group, and a heterocycloalkyl group. When two or more substituents are present, two adjacent substituents may be substituted together with the atoms to which they are attached. Or an unsubstituted cycloalkyl, heterocycloalkyl, aryl, heteroaryl group, or two substituents attached to the same atom may form a substituted or unsubstituted cycloalkyl group with the atoms to which they are attached. Cycloalkyl;
X1、X2、X3和X4各自独立选自C(R1)和N;X 1 , X 2 , X 3 and X 4 are each independently selected from C(R 1 ) and N;
R1、R2a、R2b、R3a、R3b和R3c各自独立地选自氢、卤素、烷基、环烷基、卤代烷基、烷氧基、卤代烷氧基、硝基、氰基、羟基、氨基、单烷基氨基、二烷基氨基、氨基烷基、单烷基氨基烷基、二烷基氨基烷基、羟基烷基、烷氧基烷基、烷基酰基、氨基酰基、单烷基氨基酰基和二烷基氨基酰基;R 1 , R 2a , R 2b , R 3a , R 3b and R 3c are each independently selected from the group consisting of hydrogen, halogen, alkyl, cycloalkyl, haloalkyl, alkoxy, haloalkoxy, nitro, cyano, Hydroxy, amino, monoalkylamino, dialkylamino, aminoalkyl, monoalkylaminoalkyl, dialkylaminoalkyl, hydroxyalkyl, alkoxyalkyl, alkyl acyl, amino acyl, single An alkylamino acyl group and a dialkylamino acyl group;
R4、R5和R6各自独立地选自氢、卤素、氰基、烷基、烷氧基和环烷基,所述的烷基、烷氧基和环烷基任选被一个或多个卤素、烷基、卤代烷基、烷氧基、卤代烷氧基、氨基、单烷基氨基、二烷基氨基、羟基、氨基烷基、单烷基氨基烷基、二烷基氨基烷基或羟基烷基取代;R 4 , R 5 and R 6 are each independently selected from the group consisting of hydrogen, halogen, cyano, alkyl, alkoxy and cycloalkyl, said alkyl, alkoxy and cycloalkyl optionally being one or more Halogen, alkyl, haloalkyl, alkoxy, haloalkoxy, amino, monoalkylamino, dialkylamino, hydroxy, aminoalkyl, monoalkylaminoalkyl, dialkylaminoalkyl or hydroxy Alkyl substitution
R7选自氢、烷基、环烷基和卤代烷基;和R 7 is selected from the group consisting of hydrogen, alkyl, cycloalkyl and haloalkyl;
R8选自氢、烷基和环烷基,所述的烷基和环烷基任选被一个或多个卤素、烷基、卤代烷基、烷氧基、氨基、单烷基氨基、二烷基氨基或羟基取代;或R 8 is selected from the group consisting of hydrogen, alkyl and cycloalkyl, optionally substituted by one or more halogen, alkyl, haloalkyl, alkoxy, amino, monoalkylamino, dioxane Substituted by a base or a hydroxy group; or
R7、R8及其连接的N原子一起构成含氮杂环烷基,所述的含氮杂环烷基任选被一个或多个卤素、烷基、卤代烷基、烷氧基、氨基、单烷基氨基、二烷基氨基、羟基、氨基烷基、单烷基氨基烷基、二烷基氨基烷基或羟基烷基取代。R 7 , R 8 and the N atom to which they are attached together form a nitrogen-containing heterocycloalkyl group, which is optionally substituted by one or more halogen, alkyl, haloalkyl, alkoxy, amino, Monoalkylamino, dialkylamino, hydroxy, aminoalkyl, monoalkylaminoalkyl, dialkylaminoalkyl or hydroxyalkyl substituted.
第二方面,本发明提供通式II的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,In a second aspect, the present invention provides a compound of Formula II, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof,
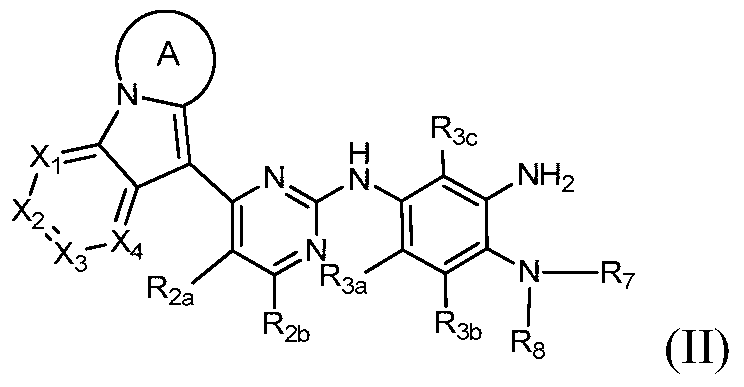
其中环A、X1、X2、X3、X4、R2a、R2b、R3a、R3b、R3c、R7和R8具有通式I中的定义。 Wherein Rings A, X 1 , X 2 , X 3 , X 4 , R 2a , R 2b , R 3a , R 3b , R 3c , R 7 and R 8 have the definitions in Formula I.
第三方面,本发明提供通式III的化合物或其药学可接受的盐,In a third aspect, the present invention provides a compound of Formula III or a pharmaceutically acceptable salt thereof,
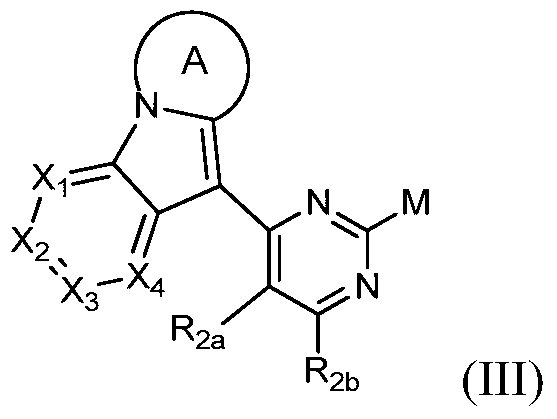
其中环A、X1、X2、X3、X4、R2a、R2b具有通式I中的定义,M为卤素。Wherein Rings A, X 1 , X 2 , X 3 , X 4 , R 2a , R 2b have the definitions in Formula I, and M is a halogen.
在一些实施方案中,本发明提供通式I、II或III所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中环A为C4-C8含氮杂单环烷基,其任选被一个或多个卤素、氧代、烷基、环烷基、卤代烷基、烷氧基、卤代烷氧基、硝基、氰基、羟基、氨基、单烷基氨基、二烷基氨基、氨基烷基、单烷基氨基烷基、二烷基氨基烷基、羟基烷基、烷氧基烷基、烷基酰基、氨基酰基、单烷基氨基酰基、二烷基氨基酰基、芳基、杂芳基和杂环烷基取代,当取代基有两个以上时,相邻的两个取代基可以与它们连接的原子一起构成取代或未取代的环烷基、杂环烷基、芳基、杂芳基,或连接在同一个原子上的两个取代基可以与它们共同连接的原子形成取代或未取代的环烷基、杂环烷基。In some embodiments, the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein Ring A is C 4 -C 8 a nitrogen-containing heteromonocycloalkyl group optionally substituted by one or more of halogen, oxo, alkyl, cycloalkyl, haloalkyl, alkoxy, haloalkoxy, nitro, cyano, hydroxy, amino, singly Alkylamino, dialkylamino, aminoalkyl, monoalkylaminoalkyl, dialkylaminoalkyl, hydroxyalkyl, alkoxyalkyl, alkylacyl, aminoacyl, monoalkylaminoacyl, a dialkylamino acyl group, an aryl group, a heteroaryl group and a heterocycloalkyl group. When two or more substituents are present, two adjacent substituents may form a substituted or unsubstituted naphthenic ring together with the atoms to which they are attached. A heterocyclic alkyl group, an aryl group, a heteroaryl group, or two substituents bonded to the same atom may form a substituted or unsubstituted cycloalkyl group or a heterocycloalkyl group with the atoms to which they are attached.
在一些实施方案中,本发明提供通式I、II或III所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中环A为只含有一个氮原子的C4-C8含氮杂单环烷基,其任选被一个或多个卤素、氧代、烷基、环烷基、卤代烷基、烷氧基、卤代烷氧基、硝基、氰基、羟基、氨基、单烷基氨基、二烷基氨基、氨基烷基、单烷基氨基烷基、二烷基氨基烷基、羟基烷基、烷氧基烷基、烷基酰基、氨基酰基、单烷基氨基酰基、二烷基氨基酰基、芳基、杂芳基和杂环烷基取代,当取代基有两个以上时,相邻的两个取代基可以与它们连接的原子一起构成取代或未取代的环烷基、杂环烷基、芳基、杂芳基,或连接在同一个原子上的两个取代基可以与它们共同连接的原子形成取代或未取代的环烷基、杂环烷基。In some embodiments, the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein Ring A is a nitrogen atom only C 4 -C 8 nitrogen-containing heteromonocycloalkyl optionally substituted by one or more halogen, oxo, alkyl, cycloalkyl, haloalkyl, alkoxy, haloalkoxy, nitro, cyano , hydroxy, amino, monoalkylamino, dialkylamino, aminoalkyl, monoalkylaminoalkyl, dialkylaminoalkyl, hydroxyalkyl, alkoxyalkyl, alkyl acyl, amino acyl, a monoalkylaminoacyl group, a dialkylaminoacyl group, an aryl group, a heteroaryl group, and a heterocycloalkyl group. When two or more substituents are present, two adjacent substituents may be substituted together with the atoms to which they are attached. Or an unsubstituted cycloalkyl, heterocycloalkyl, aryl, heteroaryl group, or two substituents attached to the same atom may form a substituted or unsubstituted cycloalkyl group with the atoms to which they are attached. Cycloalkyl.
在一些优选的实施方案中,本发明提供通式I、II或III所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中环A为![]()
![]()
在一些优选的实施方案中,本发明提供通式I、II或III所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中环A为![]()
![]()
在另一些实施方案中,本发明提供通式I、II或III所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中环A为含有两个氮原子的C4-C8含氮杂单环烷基,其任选被一个或多个卤素、氧代、烷基、环烷基、卤代烷基、烷氧基、卤代烷氧基、硝基、氰基、羟基、氨基、单烷基氨基、二烷基氨基、氨基烷基、单烷基氨基烷基、二烷基氨基烷基、羟基烷基、烷氧基烷基、烷基酰基、氨基酰基、单烷基氨基酰基、二烷基氨基酰基、芳基、杂芳基和杂环烷基取代,当取代基有两个以上时,相邻的两个取代基可以与它们连接的原子一起构成取代或未取代的环烷基、杂环烷基、芳基、杂芳基,或连接在同一个原子上的两个取代基可以与它们共同连接的原子形成取代或未取代的环烷基、杂环烷基。In other embodiments, the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein Ring A is two nitrogens A C 4 -C 8 nitrogen-containing heteromonocycloalkyl group of an atom optionally substituted by one or more halogen, oxo, alkyl, cycloalkyl, haloalkyl, alkoxy, haloalkoxy, nitro, cyanide Base, hydroxy, amino, monoalkylamino, dialkylamino, aminoalkyl, monoalkylaminoalkyl, dialkylaminoalkyl, hydroxyalkyl, alkoxyalkyl, alkyl acyl, amino acyl a monoalkylaminoacyl group, a dialkylaminoacyl group, an aryl group, a heteroaryl group, and a heterocycloalkyl group. When two or more substituents are present, two adjacent substituents may be bonded together with the atoms to which they are attached. a substituted or unsubstituted cycloalkyl group, a heterocycloalkyl group, an aryl group, a heteroaryl group, or two substituents bonded to the same atom may form a substituted or unsubstituted cycloalkyl group with an atom to which they are attached, Heterocycloalkyl.
在一些优选的实施方案中,本发明提供通式I、II或III所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中环A为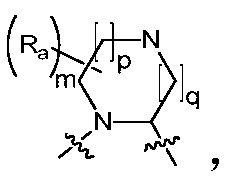
在一些优选的实施方案中,本发明提供通式I、II或III所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中环A为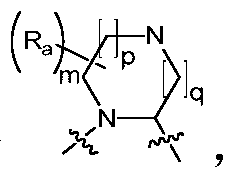
在另一些实施方案中,本发明提供通式I、II或III所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中环A为含有一个氮原子和一个氧原子的C4-C8含氮杂单环烷基,其任选被一个或多个卤素、氧代、烷基、环烷基、卤代烷基、烷氧基、卤代烷氧基、硝基、氰基、羟基、氨基、单烷基氨基、二烷基氨基、氨基烷基、单烷基氨基烷基、二烷基氨基烷基、羟基烷基、烷氧基烷基、烷基酰基、氨基酰基、单烷基氨基酰基、二烷基氨基酰基、芳基、杂芳基和杂环烷基取代,当取代基有两个以上时,相邻的两个取代基可以与它们连接的原子一起构成取代或未取代的环烷基、杂环烷基、芳基、杂芳基,或连接在同一个原子上的两个取代基可以与它们共同连接的原子形成取代或未取代的环烷基、杂环烷基。In other embodiments, the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein Ring A is a nitrogen atom And a C 4 -C 8 nitrogen-containing heteromonocycloalkyl group having one oxygen atom, optionally substituted by one or more halogen, oxo, alkyl, cycloalkyl, haloalkyl, alkoxy, haloalkoxy, nitrate Base, cyano, hydroxy, amino, monoalkylamino, dialkylamino, aminoalkyl, monoalkylaminoalkyl, dialkylaminoalkyl, hydroxyalkyl, alkoxyalkyl, alkyl acyl , aminoacyl, monoalkylaminoacyl, dialkylaminoacyl, aryl, heteroaryl and heterocycloalkyl substituted, when two or more substituents are present, two adjacent substituents may be attached thereto The atoms together form a substituted or unsubstituted cycloalkyl, heterocycloalkyl, aryl, heteroaryl group, or two substituents attached to the same atom may form a substituted or unsubstituted ring with the atoms to which they are attached. Alkyl, heterocycloalkyl.
在一些优选的实施方案中,本发明提供通式I、II或III所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中环A为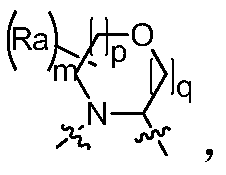
在一些优选的实施方案中,本发明提供通式I、II或III所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中环A为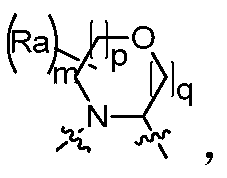
在一些实施方案中,本发明提供通式I、II或III所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中X1、X2、X3和X4均为C(R1)。In some embodiments, the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein X 1 , X 2 , X 3 And X 4 are both C(R 1 ).
在一些具体的实施方案中,本发明提供以下通式Ia、IIa和IIIa所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,In some specific embodiments, the invention provides a compound of formula Ia, IIa, and IIIa, or a pharmaceutically acceptable salt, isomer, solvate, crystal, or prodrug thereof,
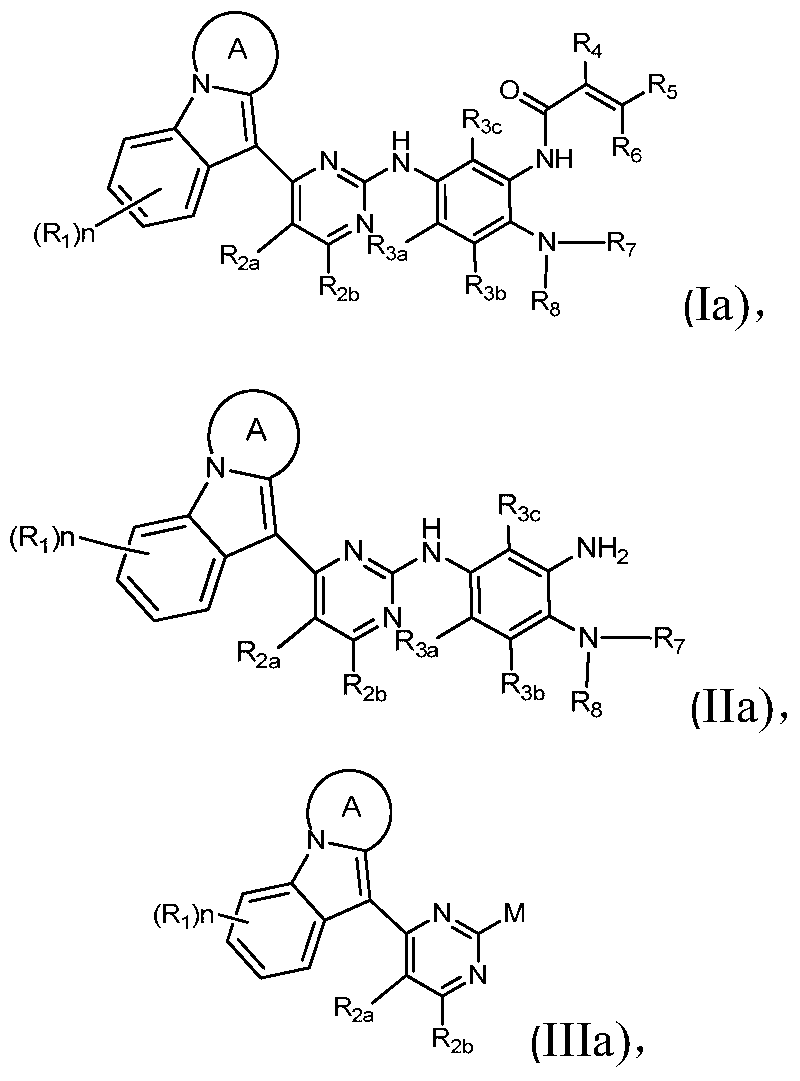
其中环A、R1、R2a、R2b、R3a、R3b、R3c、R4、R5、R6、R7、R8和M具有以上通式I、II和III中的定义,n选自1、2、3和4的整数。Wherein Rings A, R 1 , R 2a , R 2b , R 3a , R 3b , R 3c , R 4 , R 5 , R 6 , R 7 , R 8 and M have the definitions of Formulas I, II and III above , n is selected from integers of 1, 2, 3 and 4.
在另一些实施方案中,本发明提供通式I、II或III所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中X1、X2、X3和X4中至少有一个为N。In other embodiments, the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein X 1 , X 2 , X At least one of 3 and X 4 is N.
在另一些实施方案中,本发明提供通式I、II或III所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中X1、X2、X3和X4中有两个为N。In other embodiments, the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein X 1 , X 2 , X Two of 3 and X 4 are N.
在另一些实施方案中,本发明提供通式I、II或III所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中X1、X2、X3和X4中有三个为N。In other embodiments, the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein X 1 , X 2 , X Three of 3 and X 4 are N.
在另一些实施方案中,本发明提供通式I、II或III所示的化合物或其药学可接受的盐、 异构体、溶剂合物、结晶或前药,其中X1、X2、X3和X4均为N。In other embodiments, the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein X 1 , X 2 , X 3 and X 4 are both N.
在一些实施方案中,本发明提供通式I、II或III所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中R1、R2a和R2b各自独立地选自氢、卤素、C1-6烷基、C3-8环烷基、卤代C1-6烷基、C1-6烷氧基、卤代C1-6烷氧基、硝基、氰基、羟基、氨基、单C1-6烷基氨基、二C1-6烷基氨基、氨基C1-6烷基、单C1-6烷基氨基C1-6烷基、二C1-6烷基氨基C1-6烷基、羟基C1-6烷基、C1-6烷氧基C1-6烷基、C1-6烷基酰基、氨基酰基、单C1-6烷基氨基酰基和二C1-6烷基氨基酰基。In some embodiments, the present invention of general formula I, II or III compound or a pharmaceutically acceptable salt, isomer, solvate, crystallization or prodrug thereof, wherein R 1, R 2a and R 2b Each is independently selected from the group consisting of hydrogen, halogen, C 1-6 alkyl, C 3-8 cycloalkyl, halo C 1-6 alkyl, C 1-6 alkoxy, halo C 1-6 alkoxy , nitro, cyano, hydroxy, amino, mono C 1-6 alkylamino, di C 1-6 alkylamino, amino C 1-6 alkyl, mono C 1-6 alkylamino C 1-6 alkane Base, di-C 1-6 alkylamino C 1-6 alkyl, hydroxy C 1-6 alkyl, C 1-6 alkoxy C 1-6 alkyl, C 1-6 alkyl acyl, amino acyl, Mono C 1-6 alkylamino acyl and di C 1-6 alkylamino acyl.
在一些优选的实施方案中,本发明提供通式I、II或III所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中各R1、R2a和R2b各自独立地独立地选自氢、卤素、C1-3烷基、C3-6环烷基、卤代C1-3烷基、C1-3烷氧基、卤代C1-3烷氧基、硝基、氰基、羟基、氨基、单C1-3烷基氨基、二C1-3烷基氨基、氨基C1-3烷基、单C1-3烷基氨基C1-6烷基、二C1-3烷基氨基C1-3烷基、羟基C1-3烷基、C1-3烷氧基C1-3烷基、C1-3烷基酰基、氨基酰基、单C1-3烷基氨基酰基和二C1-3烷基氨基酰基。In some preferred embodiments, the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein each R 1 , R 2a And R 2b are each independently independently selected from the group consisting of hydrogen, halogen, C 1-3 alkyl, C 3-6 cycloalkyl, halo C 1-3 alkyl, C 1-3 alkoxy, halo C 1 -3 alkoxy, nitro, cyano, hydroxy, amino, mono C 1-3 alkyl amino, di C 1-3 alkyl amino, amino C 1-3 alkyl, mono C 1-3 alkyl amino C 1-6 alkyl, di C 1-3 alkylamino C 1-3 alkyl, hydroxy C 1-3 alkyl, C 1-3 alkoxy C 1-3 alkyl, C 1-3 alkyl An acyl group, an aminoacyl group, a mono C 1-3 alkylamino acyl group and a di C 1-3 alkylamino acyl group.
在另一些优选的实施方案中,本发明提供通式I、II或III所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中R1、R2a和R2b各自独立地选自氢、氟、氯、溴、碘、甲基、乙基、丙基、异丙基、环丙基、环丁基、环戊基、环己基、三氟甲基、三氟乙基、二氟甲基、二氟乙基、甲氧基、乙氧基、丙氧基、三氟甲基氧基、硝基、氰基、羟基、氨基、甲氨基、乙氨基、丙氨基、异丙基氨基、二甲氨基、二乙氨基、二丙氨基、二异丙氨基、N-甲基-N-乙基氨基、N-甲基-N-丙基氨基、N-甲基-N-异丙基氨基、N-乙基-N-丙基氨基、N-乙基-N-异丙基氨基、N-丙基-N-异丙基氨基、氨基甲基、氨基乙基、氨基丙基、甲氨基甲基、甲氨基乙基、乙氨基甲基、乙氨基乙基、二甲氨基甲基、二甲氨基乙基、二乙氨基甲基、二乙氨基乙基、羟甲基、羟乙基、甲氧基甲基、乙氧基甲基、丙氧基甲基、甲氧基乙基和乙氧基乙基。In still other preferred embodiments, the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein R 1 , R 2a And R 2b are each independently selected from the group consisting of hydrogen, fluorine, chlorine, bromine, iodine, methyl, ethyl, propyl, isopropyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, trifluoromethyl , trifluoroethyl, difluoromethyl, difluoroethyl, methoxy, ethoxy, propoxy, trifluoromethyloxy, nitro, cyano, hydroxy, amino, methylamino, ethylamino , propylamino, isopropylamino, dimethylamino, diethylamino, dipropylamino, diisopropylamino, N-methyl-N-ethylamino, N-methyl-N-propylamino, N- Methyl-N-isopropylamino, N-ethyl-N-propylamino, N-ethyl-N-isopropylamino, N-propyl-N-isopropylamino, aminomethyl, amino Ethyl, aminopropyl, methylaminomethyl, methylaminoethyl, ethylaminomethyl, ethylaminoethyl, dimethylaminomethyl, dimethylaminoethyl, diethylaminomethyl, diethylaminoethyl , hydroxymethyl, hydroxyethyl, methoxy , Ethoxymethyl, propoxymethyl, methoxyethyl and ethoxyethyl.
在另一些优选的实施方案中,本发明提供通式I、II或III所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中R1、R2a和R2b各自独立地选自氢、氟、氯、溴、甲基、乙基、丙基、异丙基、三氟甲基、三氟乙基、甲氧基、乙氧基、羟基、氨基、甲氨基、乙氨基、丙氨基、二甲氨基、二乙氨基、氨基甲基、氨基乙基、氨基丙基、甲氨基甲基、甲氨基乙基、乙氨基甲基、乙氨基乙基、二甲氨基甲基、二乙氨基甲基、羟甲基和羟乙基。In still other preferred embodiments, the invention provides a compound of Formula I, II or III, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein R 1 , R 2a And R 2b are each independently selected from the group consisting of hydrogen, fluorine, chlorine, bromine, methyl, ethyl, propyl, isopropyl, trifluoromethyl, trifluoroethyl, methoxy, ethoxy, hydroxy, amino , methylamino, ethylamino, propylamino, dimethylamino, diethylamino, aminomethyl, aminoethyl, aminopropyl, methylaminomethyl, methylaminoethyl, ethylaminomethyl, ethylaminoethyl, Dimethylaminomethyl, diethylaminomethyl, hydroxymethyl and hydroxyethyl.
在一些优选的实施方案中,本发明提供通式I或II所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中R3a、R3b和R3c各自独立地选自氢、卤素、C1-6烷基、C3-8环烷基、卤代C1-6烷基、C1-6烷氧基、卤代C1-6烷氧基、硝基、氰基、羟基、氨基、单C1-6烷基氨基、二C1-6烷基氨基、氨基C1-6烷基、单C1-6烷基氨基C1-6烷基、二C1-6烷基氨基C1-6烷基、羟基C1-6烷基、C1-6烷氧基C1-6烷基、C1-6烷基酰基、氨基酰基、单C1-6烷基氨基酰基和二C1-6烷基氨基酰基。 In some preferred embodiments, the invention provides a compound of Formula I or II, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein R 3a , R 3b and R 3c Each is independently selected from the group consisting of hydrogen, halogen, C 1-6 alkyl, C 3-8 cycloalkyl, halo C 1-6 alkyl, C 1-6 alkoxy, halo C 1-6 alkoxy , nitro, cyano, hydroxy, amino, mono C 1-6 alkylamino, di C 1-6 alkylamino, amino C 1-6 alkyl, mono C 1-6 alkylamino C 1-6 alkane Base, di-C 1-6 alkylamino C 1-6 alkyl, hydroxy C 1-6 alkyl, C 1-6 alkoxy C 1-6 alkyl, C 1-6 alkyl acyl, amino acyl, Mono C 1-6 alkylamino acyl and di C 1-6 alkylamino acyl.
在另一些优选的实施方案中,本发明提供通式I或II所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中各R3a、R3b和R3c各自独立地选自氢、卤素、C1-3烷基、C3-6环烷基、卤代C1-3烷基、C1-3烷氧基、卤代C1-3烷氧基、硝基、氰基、羟基、氨基、单C1-3烷基氨基、二C1-3烷基氨基、氨基C1-3烷基、单C1-3烷基氨基C1-6烷基、二C1-3烷基氨基C1-3烷基、羟基C1-3烷基、C1-3烷氧基C1-3烷基、C1-3烷基酰基、氨基酰基、单C1-3烷基氨基酰基和二C1-3烷基氨基酰基。In still other preferred embodiments, the present invention provides a compound of Formula I or II, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein each R 3a , R 3b and R 3c are each independently selected from the group consisting of hydrogen, halogen, C 1-3 alkyl, C 3-6 cycloalkyl, halo C 1-3 alkyl, C 1-3 alkoxy, halo C 1-3 alkane Oxy, nitro, cyano, hydroxy, amino, mono C 1-3 alkylamino, di C 1-3 alkylamino, amino C 1-3 alkyl, mono C 1-3 alkylamino C 1- 6 alkyl, di C 1-3 alkyl amino C 1-3 alkyl, hydroxy C 1-3 alkyl, C 1-3 alkoxy C 1-3 alkyl, C 1-3 alkyl group, an amino group An acyl group, a mono C 1-3 alkylamino acyl group and a di C 1-3 alkylamino acyl group.
在另一些优选的实施方案中,本发明提供通式I或II所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中R3a、R3b和R3c各自独立地选自氢、氟、氯、溴、碘、甲基、乙基、丙基、异丙基、环丙基、环丁基、环戊基、环己基、三氟甲基、三氟乙基、二氟甲基、二氟乙基、甲氧基、乙氧基、丙氧基、三氟甲基氧基、硝基、氰基、羟基、氨基、甲氨基、乙氨基、丙氨基、异丙基氨基、二甲氨基、二乙氨基、二丙氨基、二异丙氨基、N-甲基-N-乙基氨基、N-甲基-N-丙基氨基、N-甲基-N-异丙基氨基、N-乙基-N-丙基氨基、N-乙基-N-异丙基氨基、N-丙基-N-异丙基氨基、氨基甲基、氨基乙基、氨基丙基、甲氨基甲基、甲氨基乙基、乙氨基甲基、乙氨基乙基、二甲氨基甲基、二甲氨基乙基、二乙氨基甲基、二乙氨基乙基、羟甲基、羟乙基、甲氧基甲基、乙氧基甲基、丙氧基甲基、甲氧基乙基和乙氧基乙基。In still other preferred embodiments, the invention provides a compound of Formula I or II, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein R 3a , R 3b and R 3c are each independently selected from the group consisting of hydrogen, fluorine, chlorine, bromine, iodine, methyl, ethyl, propyl, isopropyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, trifluoromethyl, tri Fluoroethyl, difluoromethyl, difluoroethyl, methoxy, ethoxy, propoxy, trifluoromethyloxy, nitro, cyano, hydroxy, amino, methylamino, ethylamino, propyl Amino, isopropylamino, dimethylamino, diethylamino, dipropylamino, diisopropylamino, N-methyl-N-ethylamino, N-methyl-N-propylamino, N-methyl -N-isopropylamino, N-ethyl-N-propylamino, N-ethyl-N-isopropylamino, N-propyl-N-isopropylamino, aminomethyl, aminoethyl , aminopropyl, methylaminomethyl, methylaminoethyl, ethylaminomethyl, ethylaminoethyl, dimethylaminomethyl, dimethylaminoethyl, diethylaminomethyl, diethylaminoethyl, hydroxy Methyl, hydroxyethyl, methoxymethyl, Ethoxymethyl, propoxymethyl, methoxyethyl and ethoxyethyl.
在另一些优选的实施方案中,本发明提供通式I或II所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中R3a、R3b和R3c各自独立地选自氢、氟、氯、溴、甲基、乙基、丙基、异丙基、三氟甲基、三氟乙基、甲氧基、乙氧基、羟基、氨基、甲氨基、乙氨基、丙氨基、二甲氨基、二乙氨基、氨基甲基、氨基乙基、氨基丙基、甲氨基甲基、甲氨基乙基、乙氨基甲基、乙氨基乙基、二甲氨基甲基、二乙氨基甲基、羟甲基和羟乙基。In still other preferred embodiments, the invention provides a compound of Formula I or II, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein R 3a , R 3b and R 3c are each independently selected from the group consisting of hydrogen, fluorine, chlorine, bromine, methyl, ethyl, propyl, isopropyl, trifluoromethyl, trifluoroethyl, methoxy, ethoxy, hydroxy, amino, A Amino, ethylamino, propylamino, dimethylamino, diethylamino, aminomethyl, aminoethyl, aminopropyl, methylaminomethyl, methylaminoethyl, ethylaminomethyl, ethylaminoethyl, dimethyl Aminomethyl, diethylaminomethyl, hydroxymethyl and hydroxyethyl.
在一些实施方案中,本发明提供通式I或II所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中R7选自氢、C1-6烷基、C3-6环烷基和卤代C1-6烷基。In some embodiments, the invention provides a compound of Formula I or II, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein R 7 is selected from the group consisting of hydrogen, C 1-6 Alkyl, C 3-6 cycloalkyl and halogenated C 1-6 alkyl.
在一些优选的实施方案中,本发明提供通式I或II所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中R7选自氢、C1-3烷基、C3-6环烷基和卤代C1-3烷基。In some preferred embodiments, the present invention provides Formula I or II compound as shown or a pharmaceutically acceptable salt, isomer, solvate, crystallization or prodrug thereof, wherein R 7 is selected from hydrogen, C 1 a -3 alkyl group, a C 3-6 cycloalkyl group, and a halogenated C 1-3 alkyl group.
在一些优选的实施方案中,本发明提供通式I或II所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中R7选自氢、甲基、乙基、丙基、异丙基、环丙基、环丁基、环戊基、环己基和三氟甲基。In some preferred embodiments, the present invention provides Formula I or II compound as shown or a pharmaceutically acceptable salt, isomer, solvate, crystallization or prodrug thereof, wherein R 7 is selected from hydrogen, methyl , ethyl, propyl, isopropyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and trifluoromethyl.
在一些优选的实施方案中,本发明提供通式I或II所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中R7为甲基。In some preferred embodiments, the present invention provides Formula I or II compound as shown or a pharmaceutically acceptable salt, isomer, solvate, crystallization or prodrug thereof, wherein R 7 is methyl.
在一些实施方案中,本发明提供通式I或II所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中R8选自氢、C1-6烷基和C3-6环烷基,所述的C1-6烷基和C3-6环烷基任选被一个或多个卤素、C1-6烷基、卤代C1-6烷基、C1-6烷氧基、氨基、单C1-6烷基氨基、二C1-6烷基氨基或羟基取代。 In some embodiments, the present invention provides a formula I or II compound represented by or a pharmaceutically acceptable salt, isomer, solvate, crystallization or prodrug thereof, wherein R 8 is selected from hydrogen, C 1-6 An alkyl group and a C 3-6 cycloalkyl group, said C 1-6 alkyl group and a C 3-6 cycloalkyl group optionally being one or more halogen, C 1-6 alkyl, halogenated C 1-6 Alkyl, C 1-6 alkoxy, amino, mono C 1-6 alkylamino, di C 1-6 alkylamino or hydroxy substituted.
在一些优选的实施方案中,本发明提供通式I或II所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中R8选自氢、C1-3烷基和C3-6环烷基,所述的C1-3烷基和C3-6环烷基任选被一个或多个卤素、C1-3烷基、卤代C1-3烷基、C1-3烷氧基、氨基、单C1-3烷基氨基、二C1-3烷基氨基或羟基取代。In some preferred embodiments, the present invention provides a formula I or II or a compound represented by the pharmaceutically acceptable salts, isomers, solvates, crystalline or prodrug thereof, wherein R 8 is selected from hydrogen, C 1 a -3 alkyl group and a C 3-6 cycloalkyl group, said C 1-3 alkyl group and a C 3-6 cycloalkyl group optionally being one or more halogen, C 1-3 alkyl group, halogenated C 1 a -3 alkyl group, a C 1-3 alkoxy group, an amino group, a mono C 1-3 alkylamino group, a di C 1-3 alkylamino group or a hydroxy group.
在一些优选的实施方案中,本发明提供通式I或II所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中R8选自氢、C1-3烷基和C3-6环烷基,所述的C1-3烷基和C3-6环烷基任选被一个或多个卤素、甲基、乙基、丙基、异丙基、三氟甲基、二氟甲基、三氟乙基、甲氧基、乙氧基、丙氧基、异丙基氧基、氨基、甲氨基、乙氨基、丙氨基、二甲氨基、二乙胺基、二丙氨基、甲基乙基氨基、甲基丙基氨基、乙基丙基氨基或羟基取代。In some preferred embodiments, the present invention provides a formula I or II compound represented by or a pharmaceutically acceptable salt, isomer, solvate, crystallization or prodrug thereof, wherein R 8 is selected from hydrogen, C 1 -3 alkyl and C 3-6 cycloalkyl, said C 1-3 alkyl and C 3-6 cycloalkyl optionally being one or more halogen, methyl, ethyl, propyl, isopropyl Base, trifluoromethyl, difluoromethyl, trifluoroethyl, methoxy, ethoxy, propoxy, isopropyloxy, amino, methylamino, ethylamino, propylamino, dimethylamino, Diethylamine, dipropylamino, methylethylamino, methylpropylamino, ethylpropylamino or hydroxy substituted.
在一些优选的实施方案中,本发明提供通式I或II所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中R8选自氢、甲基、乙基和丙基,所述的甲基、乙基和丙基任选被一个或多个氨基、甲氨基、乙氨基、丙氨基、二甲氨基、二乙胺基、二丙氨基、甲基乙基氨基、甲基丙基氨基或乙基丙基氨基取代。In some preferred embodiments, the present invention provides a formula I or II compound represented by or a pharmaceutically acceptable salt, isomer, solvate, crystallization or prodrug thereof, wherein R 8 is selected from hydrogen, methyl , ethyl and propyl, said methyl, ethyl and propyl optionally being one or more amino, methylamino, ethylamino, propylamino, dimethylamino, diethylamino, dipropylamino, A Substituted by ethylamino, methylpropylamino or ethylpropylamino.
在一些优选的实施方案中,本发明提供通式I或II所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中R8为被二甲氨基取代的乙基。In some preferred embodiments, the present invention provides a formula I or II compound represented by or a pharmaceutically acceptable salt, isomer, solvate, crystallization or prodrug thereof, wherein R 8 is substituted with dimethylamino Ethyl.
在另一些实施方案中,本发明提供通式I或II所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中R7、R8及其连接的N构成四至八元氮杂环烷基,所述的四至八元氮杂环烷基任选被一个或多个卤素、C1-6烷基、卤代C1-6烷基、C1-6烷氧基、氨基、单C1-6烷基氨基、二C1-6烷基氨基、羟基、氨基C1-6烷基、单C1-6烷基氨基C1-6烷基、二C1-6烷基氨基C1-6烷基或羟基烷基取代。In other embodiments, the present invention provides a compound of general formula I or II, or a pharmaceutically acceptable salt thereof shown, isomer, solvate, crystallization or prodrug thereof, wherein R 7, R 8 and connected N constitutes a four to eight membered azacycloalkyl group, and the four to eight membered azacycloalkyl group is optionally substituted by one or more halogen, C 1-6 alkyl, halo C 1-6 alkyl, C 1- 6 alkoxy, amino, mono C 1-6 alkylamino, di C 1-6 alkylamino, hydroxy, amino C 1-6 alkyl, mono C 1-6 alkylamino C 1-6 alkyl, Di-C 1-6 alkylamino C 1-6 alkyl or hydroxyalkyl substituted.
在一些优选的实施方案中,本发明提供通式I或II所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中R7、R8及其连接的N构成四至八元氮杂环烷基,所述的四至八元氮杂环烷基任选被一个或多个卤素、C1-3烷基、卤代C1-3烷基、C1-3烷氧基、氨基、单C1-3烷基氨基、二C1-3烷基氨基、羟基、氨基C1-3烷基、单C1-3烷基氨基C1-3烷基、二C1-3烷基氨基C1-3烷基或羟基烷基取代。In some preferred embodiments, the present invention provides a compound of Formula I or II, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein R 7 , R 8 and their linkages N constitutes a four to eight membered azacycloalkyl group, and the four to eight membered azacycloalkyl group is optionally substituted by one or more halogen, C 1-3 alkyl, halo C 1-3 alkyl, C 1 -3 alkoxy, amino, mono C 1-3 alkylamino, di C 1-3 alkylamino, hydroxy, amino C 1-3 alkyl, mono C 1-3 alkylamino C 1-3 alkyl , a di-C 1-3 alkylamino C 1-3 alkyl group or a hydroxyalkyl group.
在一些优选的实施方案中,本发明提供通式I或II所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中R7、R8及其连接的N构成五至六元氮杂环烷基,所述的五至六元氮杂环烷基任选被一个或多个卤素、C1-3烷基、卤代C1-3烷基、C1-3烷氧基、氨基、单C1-3烷基氨基、二C1-3烷基氨基、羟基、氨基C1-3烷基、单C1-3烷基氨基C1-3烷基、二C1-3烷基氨基C1-3烷基或羟基烷基取代。In some preferred embodiments, the present invention provides a compound of Formula I or II, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein R 7 , R 8 and their linkages N-constituting five to six yuan azacycloalkyl, said azacycloalkyl five to six yuan optionally substituted with one or more halo, C 1-3 alkyl, halo C 1-3 alkyl, C 1-3 alkoxy, amino, mono C 1-3 alkylamino, di C 1-3 alkylamino, hydroxy, amino C 1-3 alkyl, mono C 1-3 alkylamino C 1-3 Alkyl, di-C 1-3 alkylamino C 1-3 alkyl or hydroxyalkyl substituted.
在一些实施方案中,本发明提供通式I所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中R4、R5和R6各自独立地选自氢、卤素、氰基、C1-6烷基、C1-6烷氧基、C3-8环烷基,所述的C1-6烷基、C1-6烷氧基和环烷基任选被一个或多个卤素、C1-6烷基、卤代C1-6烷基、C1-6烷氧基、卤代C1-6烷氧基、氨基、单C1-6烷基氨基、二C1-6烷基氨基、羟 基、氨基C1-6烷基、单C1-6烷基氨基C1-6烷基、二C1-6烷基氨基C1-6烷基和羟基C1-6烷基取代。In some embodiments, the invention provides a compound of Formula I, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein R 4 , R 5 and R 6 are each independently selected From hydrogen, halogen, cyano, C 1-6 alkyl, C 1-6 alkoxy, C 3-8 cycloalkyl, said C 1-6 alkyl, C 1-6 alkoxy and ring The alkyl group is optionally selected from one or more of halogen, C 1-6 alkyl, halo C 1-6 alkyl, C 1-6 alkoxy, halo C 1-6 alkoxy, amino, mono C 1 -6 alkylamino, di C 1-6 alkylamino, hydroxy, amino C 1-6 alkyl, mono C 1-6 alkylamino C 1-6 alkyl, di C 1-6 alkylamino C 1 -6 alkyl and hydroxy C 1-6 alkyl substituted.
在一些优选的实施方案中,本发明提供通式I所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中R4、R5和R6各自独立地选自氢、卤素、氰基、C1-3烷基、C1-3烷氧基、C3-6环烷基,所述的C1-3烷基、C1-3烷氧基和C3-6环烷基任选被一个或多个卤素、C1-3烷基、卤代C1-3烷基、C1-3烷氧基、卤代C1-3烷氧基、氨基、单C1-3烷基氨基、二C1-3烷基氨基、羟基、氨基C1-3烷基、单C1-3烷基氨基C1-3烷基、二C1-3烷基氨基C1-3烷基或羟基C1-3烷基取代。In some preferred embodiments, the invention provides a compound of Formula I, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein R 4 , R 5 and R 6 are each independently Is selected from the group consisting of hydrogen, halogen, cyano, C 1-3 alkyl, C 1-3 alkoxy, C 3-6 cycloalkyl, said C 1-3 alkyl, C 1-3 alkoxy and C 3-6 cycloalkyl optionally substituted with one or more halo, C 1-3 alkyl, halo C 1-3 alkyl, C 1-3 alkoxy, a C 1-3 alkoxy group, halo , amino, mono C 1-3 alkylamino, di C 1-3 alkylamino, hydroxy, amino C 1-3 alkyl, mono C 1-3 alkylamino C 1-3 alkyl, di C 1- 3 alkylamino C 1-3 alkyl or hydroxy C 1-3 alkyl substituted.
在一些优选的实施方案中,本发明提供通式I所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中R4、R5和R6均为氢。In some preferred embodiments, the invention provides a compound of Formula I, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein R 4 , R 5 and R 6 are hydrogen.
在一些实施方案中,本发明提供通式III所示的化合物或其药学可接受的盐,其中M选自氟、氯、溴和碘。In some embodiments, the invention provides a compound of Formula III, or a pharmaceutically acceptable salt thereof, wherein M is selected from the group consisting of fluorine, chlorine, bromine, and iodine.
在一些优选的实施方案中,本发明提供通式III所示的化合物或其药学可接受的盐,其中M选自氯、溴和碘。In some preferred embodiments, the invention provides a compound of Formula III, or a pharmaceutically acceptable salt thereof, wherein M is selected from the group consisting of chlorine, bromine, and iodine.
在一些具体的实施方案中,本发明提供通式I所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中In some specific embodiments, the present invention provides a compound of Formula I, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein
环A为![]()
![]()
n选自1、2、3和4;n is selected from 1, 2, 3 and 4;
m选自1、2、3和4;m is selected from 1, 2, 3 and 4;
Ra选自氢、C1-3烷基、C3-6环烷基、卤代C1-3烷基、C1-3烷氧基、卤代C1-3烷氧基、硝基、氰基、羟基、氨基、单C1-3烷基氨基、二C1-3烷基氨基、氨基C1-3烷基、单C1-3烷基氨基C1-3烷基、二C1-3烷基氨基C1-3烷基、羟基C1-3烷基、C1-3烷氧基C1-3烷基、C1-3烷基酰基、氨基酰基、单C1-3烷基氨基酰基、二C1-3烷基氨基酰基、C6-10芳基、C5-10杂芳基和C3-6杂环烷基,当m选自2、3和4时,相邻两个碳原子上的两个Ra可以与它们所连接的碳原子一起构成C3-6环烷基或C3-6杂环烷基,或同一个碳原子上的两个Ra可以与它们共同连接的碳原子一起构成C3-6环烷基或C3-6杂环烷基;R a is selected from the group consisting of hydrogen, C 1-3 alkyl, C 3-6 cycloalkyl, halogenated C 1-3 alkyl, C 1-3 alkoxy, halo C 1-3 alkoxy, nitro , cyano, hydroxy, amino, mono C 1-3 alkylamino, di C 1-3 alkylamino, amino C 1-3 alkyl, mono C 1-3 alkylamino C 1-3 alkyl, two C 1-3 alkylamino C 1-3 alkyl, hydroxy C 1-3 alkyl, C 1-3 alkoxy C 1-3 alkyl, C 1-3 alkyl acyl, amino acyl, mono C 1 -3 alkylaminoacyl, di C 1-3 alkylamino acyl, C 6-10 aryl, C 5-10 heteroaryl and C 3-6 heterocycloalkyl, when m is selected from 2, 3 and 4 When two R a groups of two adjacent carbon atoms may form a C 3-6 cycloalkyl group or a C 3-6 heterocycloalkyl group together with the carbon atom to which they are attached, or two on the same carbon atom. R a may together with the carbon atom to which they are attached together constitute a C 3-6 cycloalkyl group or a C 3-6 heterocycloalkyl group;
X1、X2、X3和X4各自独立选自C(R1)和N;X 1 , X 2 , X 3 and X 4 are each independently selected from C(R 1 ) and N;
R1、R2a、R2b、R3a、R3b和R3c各自独立地选自氢、卤素、C1-3烷基、卤代C1-3烷基、C1-3烷氧基、卤代C1-3烷氧基、硝基、氰基、羟基、氨基、单C1-3烷基氨基、二C1-3烷基氨基、氨基C1-3烷基、单C1-3烷基氨基C1-6烷基、二C1-3烷基氨基C1-3烷基、羟基C1-3烷基、C1-3烷氧基C1-3烷基、C1-3烷基酰基、氨基酰基、单C1-3烷基氨基酰基和二C1-3烷基氨基酰基;R 1 , R 2a , R 2b , R 3a , R 3b and R 3c are each independently selected from the group consisting of hydrogen, halogen, C 1-3 alkyl, halo C 1-3 alkyl, C 1-3 alkoxy, Halogen C 1-3 alkoxy, nitro, cyano, hydroxy, amino, mono C 1-3 alkylamino, di C 1-3 alkylamino, amino C 1-3 alkyl, mono C 1- 3 alkylamino C 1-6 alkyl, di C 1-3 alkylamino C 1-3 alkyl, hydroxy C 1-3 alkyl, C 1-3 alkoxy C 1-3 alkyl, C 1 a -3 alkyl acyl group, an amino acyl group, a mono C 1-3 alkylamino acyl group and a di C 1-3 alkylamino acyl group;
R4、R5和R6均为氢;R 4 , R 5 and R 6 are all hydrogen;
R7为甲基; R 7 is a methyl group;
R8为被二甲氨基取代的乙基。R 8 is an ethyl group substituted by dimethylamino.
在另一些具体的实施方案中,本发明提供通式I所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中In other specific embodiments, the present invention provides a compound of Formula I, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein
环A为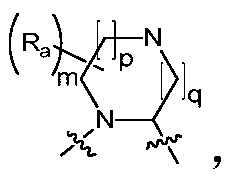
m选自1、2、3和4;m is selected from 1, 2, 3 and 4;
p选自0、1和2;p is selected from 0, 1, and 2;
q选自0、1和2;Ra选自氢、C1-3烷基、C3-6环烷基、卤代C1-3烷基、C1-3烷氧基、卤代C1-3烷氧基、硝基、氰基、羟基、氨基、单C1-3烷基氨基、二C1-3烷基氨基、氨基C1-3烷基、单C1-3烷基氨基C1-3烷基、二C1-3烷基氨基C1-3烷基、羟基C1-3烷基、C1-3烷氧基C1-3烷基、C1-3烷基酰基、氨基酰基、单C1-3烷基氨基酰基、二C1-3烷基氨基酰基、C6-10芳基、C5-10杂芳基和C3-6杂环烷基,当m选自2、3和4时,相邻两个原子上的两个Ra可以与它们所连接的原子一起构成C3-6环烷基或C3-6杂环烷基,或同一个原子上的两个Ra可以与它们共同连接的原子一起构成C3-6环烷基或C3-6杂环烷基;q is selected from 0, 1, and 2; R a is selected from hydrogen, C 1-3 alkyl, C 3-6 cycloalkyl, halogenated C 1-3 alkyl, C 1-3 alkoxy, halogen C 1-3 alkoxy, nitro, cyano, hydroxy, amino, mono C 1-3 alkylamino, di C 1-3 alkylamino, amino C 1-3 alkyl, mono C 1-3 alkyl C 1-3 alkyl amino, di C 1-3 alkyl amino C 1-3 alkyl, hydroxy C 1-3 alkyl, C 1-3 alkoxy C 1-3 alkyl, C 1-3 alkoxy Alkanoyl group, aminoacyl group, mono C 1-3 alkylamino acyl group, di C 1-3 alkylamino acyl group, C 6-10 aryl group, C 5-10 heteroaryl group and C 3-6 heterocycloalkyl group, When m is selected from 2, 3 and 4, two R a on two adjacent atoms may together with the atom to which they are attached form a C 3-6 cycloalkyl group or a C 3-6 heterocycloalkyl group, or the same Two R a on one atom may together with the atoms to which they are attached form a C 3-6 cycloalkyl group or a C 3-6 heterocycloalkyl group;
X1、X2、X3和X4各自独立选自C(R1)和N;X 1 , X 2 , X 3 and X 4 are each independently selected from C(R 1 ) and N;
R1、R2a、R2b、R3a、R3b和R3c各自独立地选自氢、卤素、C1-3烷基、卤代C1-3烷基、C1-3烷氧基、卤代C1-3烷氧基、硝基、氰基、羟基、氨基、单C1-3烷基氨基、二C1-3烷基氨基、氨基C1-3烷基、单C1-3烷基氨基C1-6烷基、二C1-3烷基氨基C1-3烷基、羟基C1-3烷基、C1-3烷氧基C1-3烷基、C1-3烷基酰基、氨基酰基、单C1-3烷基氨基酰基和二C1-3烷基氨基酰基;R 1 , R 2a , R 2b , R 3a , R 3b and R 3c are each independently selected from the group consisting of hydrogen, halogen, C 1-3 alkyl, halo C 1-3 alkyl, C 1-3 alkoxy, Halogen C 1-3 alkoxy, nitro, cyano, hydroxy, amino, mono C 1-3 alkylamino, di C 1-3 alkylamino, amino C 1-3 alkyl, mono C 1- 3 alkylamino C 1-6 alkyl, di C 1-3 alkylamino C 1-3 alkyl, hydroxy C 1-3 alkyl, C 1-3 alkoxy C 1-3 alkyl, C 1 a -3 alkyl acyl group, an amino acyl group, a mono C 1-3 alkylamino acyl group and a di C 1-3 alkylamino acyl group;
R4、R5和R6均为氢;R 4 , R 5 and R 6 are all hydrogen;
R7为甲基;R 7 is a methyl group;
R8为被二甲氨基取代的乙基。R 8 is an ethyl group substituted by dimethylamino.
在另一些具体的实施方案中,本发明提供通式I所示的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药,其中In other specific embodiments, the present invention provides a compound of Formula I, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, wherein
环A为![]()
![]()
m选自1、2、3和4;m is selected from 1, 2, 3 and 4;
p选自0、1和2;p is selected from 0, 1, and 2;
q选自0、1和2;Ra选自氢、C1-3烷基、C3-6环烷基、卤代C1-3烷基、C1-3烷氧基、卤代C1-3烷氧基、硝基、氰基、羟基、氨基、单C1-3烷基氨基、二C1-3烷基氨基、氨基C1-3烷基、单C1-3烷基氨基C1-3烷基、二C1-3烷基氨基C1-3烷基、羟基C1-3烷基、C1-3烷氧基C1-3烷基、C1-3烷基酰基、氨基酰基、单C1-3烷基氨基酰基、二C1-3烷基氨基酰基、C6-10芳基、C5-10杂芳基和C3-6杂环烷基,当m选自2、3和4时,相邻两个原子上的两个Ra可以与它们 所连接的原子一起构成C3-6环烷基或C3-6杂环烷基,或同一个原子上的两个Ra可以与它们共同连接的原子一起构成C3-6环烷基或C3-6杂环烷基;q is selected from 0, 1, and 2; R a is selected from hydrogen, C 1-3 alkyl, C 3-6 cycloalkyl, halogenated C 1-3 alkyl, C 1-3 alkoxy, halogen C 1-3 alkoxy, nitro, cyano, hydroxy, amino, mono C 1-3 alkylamino, di C 1-3 alkylamino, amino C 1-3 alkyl, mono C 1-3 alkyl Amino C 1-3 alkyl, di C 1-3 alkylamino C 1-3 alkyl, hydroxy C 1-3 alkyl, C 1-3 alkoxy C 1-3 alkyl, C 1-3 alkane an acyl group, an amino group, a mono C 1-3 alkyl amino group, a di C 1-3 alkyl amino group, C 6-10 aryl, C 5-10 heteroaryl, and C 3-6 heterocycloalkyl, When m is selected from 2, 3 and 4, two R a on two adjacent atoms may form a C 3-6 cycloalkyl group or a C 3-6 heterocycloalkyl group together with the atom to which they are attached, or the same Two R a on one atom may together with the atoms to which they are attached form a C 3-6 cycloalkyl group or a C 3-6 heterocycloalkyl group;
X1、X2、X3和X4各自独立选自C(R1)和N;X 1 , X 2 , X 3 and X 4 are each independently selected from C(R 1 ) and N;
R1、R2a、R2b、R3a、R3b和R3c各自独立地选自氢、卤素、C1-3烷基、卤代C1-3烷基、C1-3烷氧基、卤代C1-3烷氧基、硝基、氰基、羟基、氨基、单C1-3烷基氨基、二C1-3烷基氨基、氨基C1-3烷基、单C1-3烷基氨基C1-6烷基、二C1-3烷基氨基C1-3烷基、羟基C1-3烷基、C1-3烷氧基C1-3烷基、C1-3烷基酰基、氨基酰基、单C1-3烷基氨基酰基和二C1-3烷基氨基酰基;R 1 , R 2a , R 2b , R 3a , R 3b and R 3c are each independently selected from the group consisting of hydrogen, halogen, C 1-3 alkyl, halo C 1-3 alkyl, C 1-3 alkoxy, Halogen C 1-3 alkoxy, nitro, cyano, hydroxy, amino, mono C 1-3 alkylamino, di C 1-3 alkylamino, amino C 1-3 alkyl, mono C 1- 3 alkylamino C 1-6 alkyl, di C 1-3 alkylamino C 1-3 alkyl, hydroxy C 1-3 alkyl, C 1-3 alkoxy C 1-3 alkyl, C 1 a -3 alkyl acyl group, an amino acyl group, a mono C 1-3 alkylamino acyl group and a di C 1-3 alkylamino acyl group;
R4、R5和R6均为氢;R 4 , R 5 and R 6 are all hydrogen;
R7为甲基;R 7 is a methyl group;
R8为被二甲氨基取代的乙基。R 8 is an ethyl group substituted by dimethylamino.
在一些具体的实施方案中,本发明提供了以下化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药:In some specific embodiments, the invention provides the following compounds, or pharmaceutically acceptable salts, isomers, solvates, crystals or prodrugs thereof:
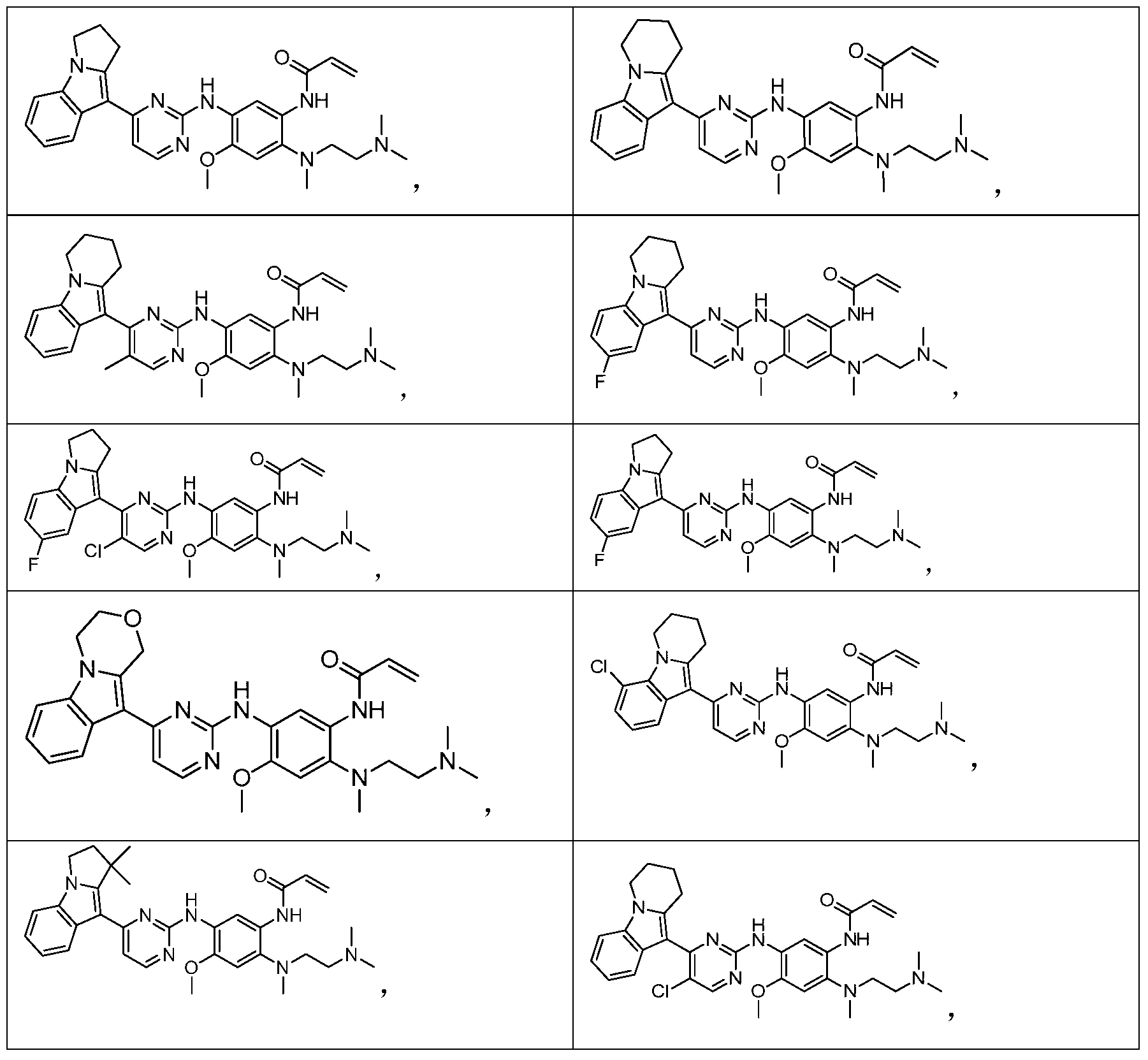
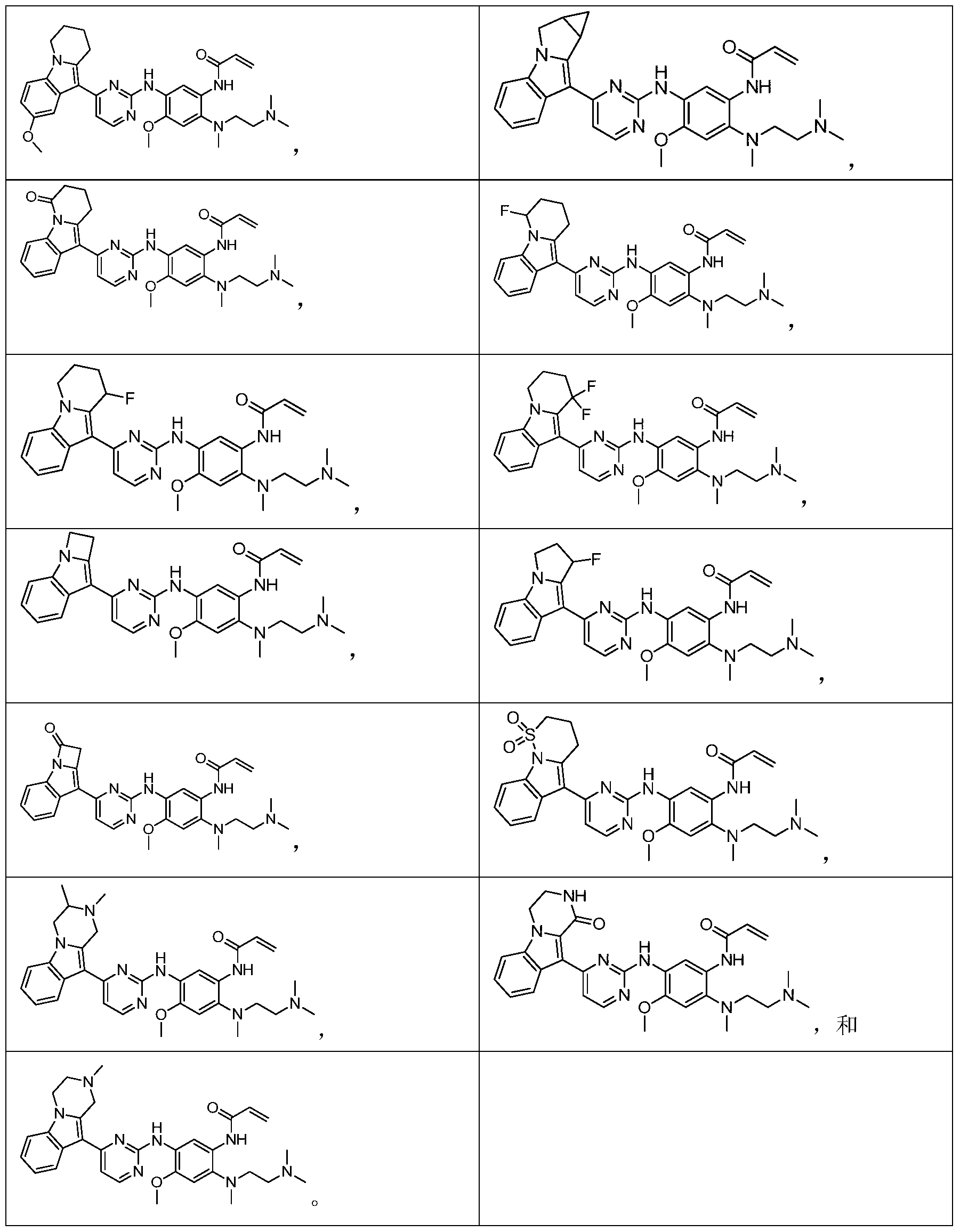
在另一些具体的实施方案中,本发明提供了以下化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药:In other specific embodiments, the invention provides the following compounds, or pharmaceutically acceptable salts, isomers, solvates, crystals or prodrugs thereof:
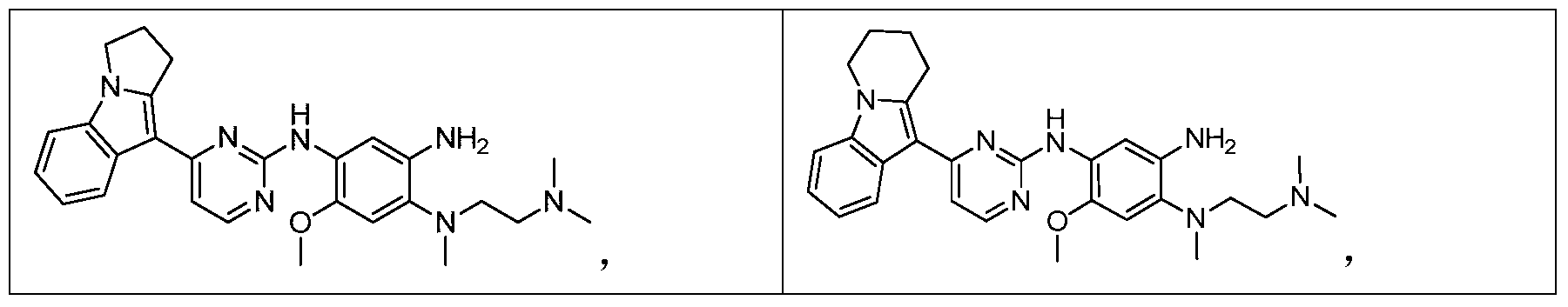


第四方面,本发明提供本发明的通式I、II和III的化合物的制备方法,包括如下步骤:In a fourth aspect, the invention provides a process for the preparation of a compound of formula I, II and III of the invention, comprising the steps of:
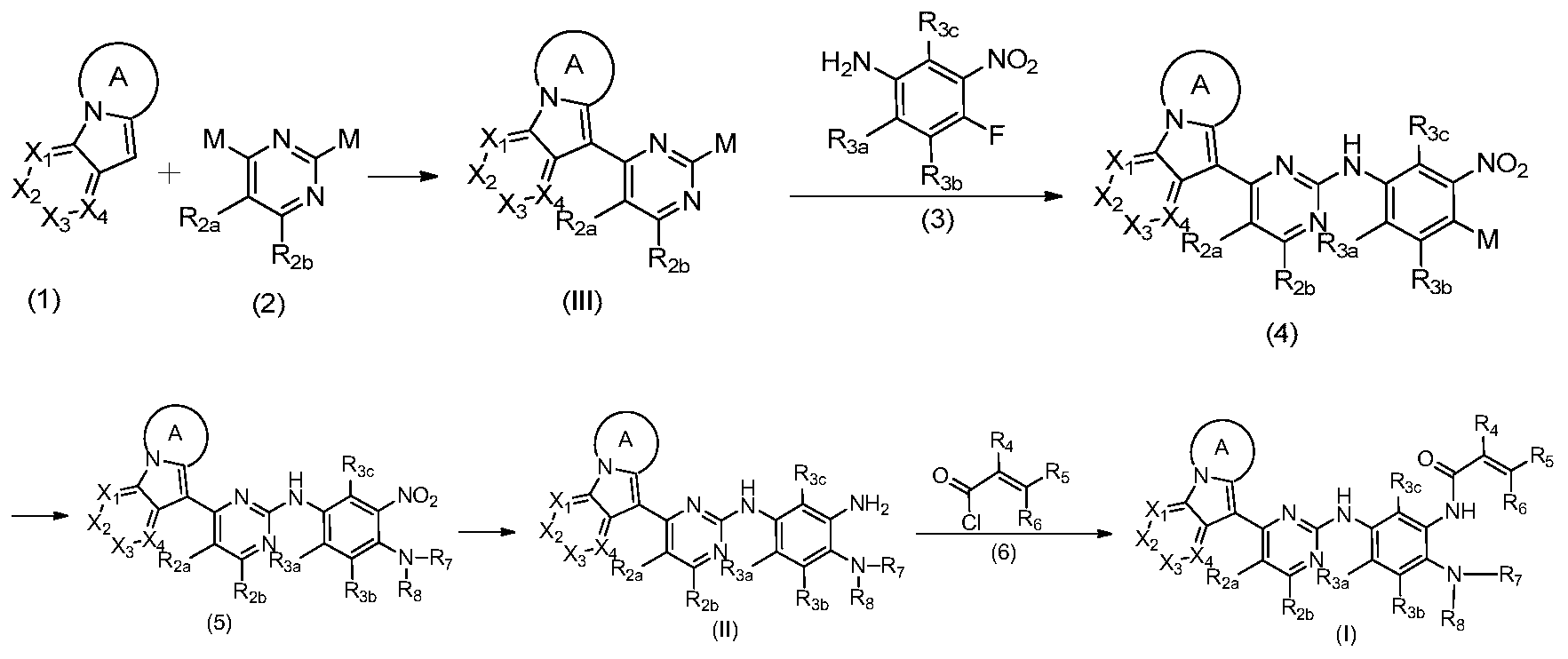
A)式1的原料和式2的原料发生亲核反应制得式III的中间体;A) the starting material of formula 1 and the starting material of formula 2 are nucleophilic to produce an intermediate of formula III;
B)式III的中间体与式3的原料反应制得式4的中间体;B) an intermediate of formula III is reacted with a starting material of formula 3 to produce an intermediate of formula 4;
C)式4的中间体经过亲核反应制得式5的中间体;C) an intermediate of formula 4 is subjected to a nucleophilic reaction to produce an intermediate of formula 5;
D)式5的中间体经过还原反应制得式II的中间体;D) an intermediate of formula 5 is subjected to a reduction reaction to produce an intermediate of formula II;
E)式II的中间体与式6的原料反应制得式I的化合物。E) An intermediate of formula II is reacted with a starting material of formula 6 to produce a compound of formula I.
上述环A、X1、X2、X3、X4、R2a、R2b、R3a、R3b、R3c、R4、R5、R6、R7、R8和M具有通式I、II、III中的定义。The above rings A, X 1 , X 2 , X 3 , X 4 , R 2a , R 2b , R 3a , R 3b , R 3c , R 4 , R 5 , R 6 , R 7 , R 8 and M have the formula Definitions in I, II, III.
在一些实施方案中,本发明提供制备本发明的通式I的化合物的制备方法,所述方法包括使用本发明的通式II的化合物。In some embodiments, the invention provides a process for the preparation of a compound of formula I of the invention, which process comprises the use of a compound of formula II of the invention.
在一些实施方案中,本发明提供制备本发明的通式I的化合物的制备方法,所述方法包括使用本发明的通式III的化合物。 In some embodiments, the invention provides a process for the preparation of a compound of formula I of the invention, which process comprises the use of a compound of formula III of the invention.
第五方面,本发明提供药物组合物,其包含本发明通式I的化合物或其药学可接受的盐、异构体、溶剂合物、结晶或前药和药学可接受的载体。In a fifth aspect, the invention provides a pharmaceutical composition comprising a compound of formula I of the invention, or a pharmaceutically acceptable salt, isomer, solvate, crystal or prodrug thereof, and a pharmaceutically acceptable carrier.
在一些实施方案中,本发明提供药物组合物,其包含本发明通式I的化合物、异构体、溶剂合物、结晶或前药,还包含选自下列组成的一种或多种化合物:吉非替尼、厄洛替尼、拉帕替尼、阿法替尼、凡德他尼、卡奈替尼、阿帕替尼、达卡替尼(dacomitinib)、培利替尼(pelitinib)、WZ4002、AG-490、AZD8931、AZD9291等。In some embodiments, the invention provides a pharmaceutical composition comprising a compound, isomer, solvate, crystal or prodrug of Formula I of the invention, further comprising one or more compounds selected from the group consisting of: Gefitinib, erlotinib, lapatinib, afatinib, vandetanib, carnitinib, apatinib, dacomitinib, pelitinib , WZ4002, AG-490, AZD8931, AZD9291 and so on.
可以将本发明通式I的化合物、异构体、溶剂合物、结晶或前药与药学上可接受的载体混合制备成药物制剂,以适合于经口或胃肠外给药。给药方法包括,但不限于皮内、肌内、腹膜内、静脉内、皮下、鼻内和经口途径。所述制剂可以通过任何途径施用,例如通过输注或推注,通过经上皮或皮肤粘膜(例如口腔粘膜或直肠等)吸收的途径施用。给药可以是全身的或局部的。经口施用制剂的实例包括固体或液体剂型,具体而言,包括片剂、丸剂、粒剂、粉剂、胶囊剂、糖浆、乳剂、混悬剂等。所述制剂可通过本领域已知的方法制备,且包含药物制剂领域常规使用的载体。The compound, isomer, solvate, crystal or prodrug of the formula I of the present invention may be admixed with a pharmaceutically acceptable carrier to prepare a pharmaceutical preparation suitable for oral or parenteral administration. Methods of administration include, but are not limited to, intradermal, intramuscular, intraperitoneal, intravenous, subcutaneous, intranasal, and oral routes. The formulation may be administered by any route, for example by infusion or bolus injection, by a route of absorption through the epithelium or skin mucosa (e.g., oral mucosa or rectum, etc.). Administration can be systemic or topical. Examples of the orally administered preparations include solid or liquid dosage forms, specifically, tablets, pills, granules, powders, capsules, syrups, emulsions, suspensions and the like. The formulation can be prepared by methods known in the art and comprises a carrier conventionally used in the field of pharmaceutical formulations.
第六方面,本发明提供本发明通式I的化合物、异构体、溶剂合物、结晶或前药或本发明的药物组合物治疗和/或预防肿瘤的方法和在制备治疗和/或预防肿瘤药物中的应用,包括向肿瘤易发人群或肿瘤患者施用本发明通式I的化合物、异构体、溶剂合物、结晶或前药或者包含本发明通式I的化合物、异构体、溶剂合物、结晶或前药的药物组合物,以有效降低肿瘤发生率、延长肿瘤患者生命。In a sixth aspect, the present invention provides a method of treating and/or preventing a tumor of a compound, isomer, solvate, crystal or prodrug of the present invention or a pharmaceutical composition of the present invention, and in the preparation of a treatment and/or prevention Use in a tumor drug, comprising administering to a tumor-prone population or a tumor patient a compound, isomer, solvate, crystal or prodrug of the formula I of the invention or a compound, isomer comprising the formula I of the invention, A pharmaceutical composition of a solvate, crystal or prodrug to effectively reduce the incidence of tumors and prolong the life of a tumor patient.
在一些实施方案中,本发明提供本发明的通式I的化合物、异构体、溶剂合物、结晶或前药或本发明的药物组合物治疗具有抗药性的肿瘤的方法,包括向具有抗药性的肿瘤患者施用治疗有效量的本发明通式I的化合物、异构体、溶剂合物、结晶或前药或者包含本发明通式I的化合物、异构体、溶剂合物、结晶或前药的药物组合物。In some embodiments, the invention provides a method of treating a tumor having resistance to a compound, a isomer, a solvate, a crystal or a prodrug of the invention, or a pharmaceutical composition of the invention, comprising A pharmaceutically acceptable tumor patient is administered a therapeutically effective amount of a compound, isomer, solvate, crystal or prodrug of the formula I according to the invention or a compound, isomer, solvate, crystal or pre-compound comprising the formula I of the invention Pharmaceutical composition of the drug.
在另一些实施方案中,本发明提供本发明的通式I的化合物、异构体、溶剂合物、结晶或前药或本发明的药物组合物在制备治疗具有抗药性的肿瘤的药物中的应用。所述具有抗药性的肿瘤可以是对多种药物具有抗药性的肿瘤,优选对EGFR抑制剂抗药的肿瘤,例如对第一、第二、第三代EGFR抑制剂,例如对吉非替尼、厄洛替尼和拉帕替尼具有抗药性的肿瘤。所述肿瘤包括但不限于实体瘤,优选为肺癌、头颈部肿瘤、结直肠癌、膀胱癌、胰腺癌、乳腺癌、前列腺癌、胃癌、口腔癌、肝癌、卵巢癌。更优选地,所述肿瘤为非小细胞肺癌。在一些实施方案中,本发明提供本发明的通式I的化合物、异构体、溶剂合物、结晶或前药治疗具有抗药性的肿瘤的方法,其中所述肿瘤携带EGFR突变基因。在一个实施方案中,所述肿瘤携带的EGFR突变基因是第20号外显子存在T790M突变。在另一个实施方案中,所述肿瘤携带的EGFR突变基因是第21号外显子存在L858R突变和/或缺失/插入突变。在另一个实施方案中,所述肿瘤携带的EGFR突变基因是T790M和L858R双重突变。在另一些实施方案中,本发明提供用于治疗肿瘤的本发明的通式I的化合物、异构体、溶剂合物、结晶或 前药或本发明的药物组合物,其中治疗肿瘤作用表现在突出的疗效,高度的选择性和/或较少的副作用。在再一些实施方案中,本发明提供本发明的通式I的化合物、异构体、溶剂合物、结晶或前药或本发明的药物组合物治疗肿瘤的方法,所述方法包括给予需要其的患者治疗有效量的本发明的通式I的化合物、异构体、溶剂合物、结晶或前药或本发明的药物组合物,所产生的治疗肿瘤方面的作用表现在突出的疗效,高度的选择性和/或较少的副作用。In other embodiments, the invention provides a compound, isomer, solvate, crystal or prodrug of the invention, or a pharmaceutical composition of the invention, in the manufacture of a medicament for treating a tumor having drug resistance application. The drug-resistant tumor may be a tumor resistant to a plurality of drugs, preferably a tumor resistant to an EGFR inhibitor, for example, to a first, second, and third generation EGFR inhibitor, such as gefitinib. , erlotinib and lapatinib have drug-resistant tumors. Such tumors include, but are not limited to, solid tumors, preferably lung cancer, head and neck tumors, colorectal cancer, bladder cancer, pancreatic cancer, breast cancer, prostate cancer, gastric cancer, oral cancer, liver cancer, ovarian cancer. More preferably, the tumor is non-small cell lung cancer. In some embodiments, the invention provides a method of treating a tumor having resistance to a compound, isomer, solvate, crystal or prodrug of Formula I of the invention, wherein the tumor carries an EGFR mutated gene. In one embodiment, the EGFR mutated gene carried by the tumor is a T790M mutation in exon 20. In another embodiment, the EGFR mutated gene carried by the tumor is a L858R mutation and/or a deletion/insertion mutation in exon 21. In another embodiment, the EGFR mutated gene carried by the tumor is a T790M and L858R double mutation. In other embodiments, the invention provides a compound, isomer, solvate, crystal or a compound of the formula I of the invention for use in treating a tumor A prodrug or a pharmaceutical composition of the invention wherein the therapeutic effect of the tumor is manifested in a prominent therapeutic effect, a high degree of selectivity and/or less side effects. In still other embodiments, the present invention provides a method of treating a tumor of a compound, isomer, solvate, crystal or prodrug of the present invention or a pharmaceutical composition of the present invention, the method comprising administering it A therapeutically effective amount of a compound, isomer, solvate, crystal or prodrug of the present invention or a pharmaceutical composition of the present invention, which produces a therapeutic effect in the treatment of a tumor, which is highly effective. Selectivity and / or fewer side effects.
术语定义Definition of Terms
除非另外定义,本文使用的所有技术和科学术语具有与本发明所属领域的普通技术人员通常理解的相同的含义。Unless otherwise defined, all technical and scientific terms used herein have the same meaning meaning meaning
本发明的“异构体”包括顺式或反式构型的顺反异构体,也包括手性碳产生的对映异构体和非对映异构体。"Isomers" of the present invention include cis-trans isomers of the cis or trans configuration, as well as enantiomers and diastereomers derived from chiral carbons.
本发明的“药学可接受的盐”是指本发明所述的化合物与酸形成的药学上可接受的盐,所述的酸可选自:磷酸、硫酸、盐酸、氢溴酸、柠檬酸、马来酸、丙二酸、扁桃酸、琥珀酸、富马酸、醋酸、乳酸、硝酸、磺酸、对甲苯磺酸、苹果酸、甲烷磺酸等。The "pharmaceutically acceptable salt" of the present invention means a pharmaceutically acceptable salt formed by the compound of the present invention and an acid, and the acid may be selected from the group consisting of phosphoric acid, sulfuric acid, hydrochloric acid, hydrobromic acid, citric acid, Maleic acid, malonic acid, mandelic acid, succinic acid, fumaric acid, acetic acid, lactic acid, nitric acid, sulfonic acid, p-toluenesulfonic acid, malic acid, methanesulfonic acid and the like.
本发明的“溶剂合物”是指通过与溶剂分子配位形成固态或液态的配合物的本发明化合物的形式。水合物是溶剂合物的特殊形式,其中与水发生配位。在本发明范围内,溶剂合物优选是水合物。The "solvate" of the present invention means a form of the compound of the present invention which forms a solid or liquid complex by coordination with a solvent molecule. Hydrates are a special form of solvates in which coordination occurs with water. Within the scope of the invention, the solvate is preferably a hydrate.
本发明的“结晶”是指本发明所述的化合物形成的各种固体形态,包括晶型、无定形。"Crystalline" as used in the present invention refers to various solid forms formed by the compounds of the present invention, including crystalline forms and amorphous forms.
本发明的“前药”是指在生物体的生理条件下,由于与酶、胃酸等反应而转化成本发明的化合物,即通过酶的氧化、还原、水解等转化成本发明的化合物和/或通过胃酸等的水解反应等转化成本发明的化合物的化合物。The "prodrug" of the present invention refers to a compound which is converted into a compound of the present invention by a reaction with an enzyme, gastric acid or the like under physiological conditions of a living body, that is, a compound which is converted into the invention by oxidation, reduction, hydrolysis or the like of the enzyme and/or A compound which is converted into a compound of the invention by a hydrolysis reaction such as gastric acid or the like.
本发明的“药物组合物”是指包含任何一种本文所述的化合物,包括异构体、前药、溶剂合物、药学上可接受的盐或其化学的保护形式,和一种或多种药学上可接受载体的混合物。A "pharmaceutical composition" according to the invention is meant to comprise any one of the compounds described herein, including isomers, prodrugs, solvates, pharmaceutically acceptable salts or chemically protected forms thereof, and one or more A mixture of pharmaceutically acceptable carriers.
本发明的“在制备用于治疗和/或预防肿瘤的药物中的应用”是指可以抑制肿瘤的生长、发展和/或转移,主要向所需要的人或动物给予治疗有效剂量的本发明的化合物以抑制、减慢或逆转受治疗者肿瘤的生长、发展或扩散。The "application in the preparation of a medicament for treating and/or preventing a tumor" of the present invention means that the growth, development and/or metastasis of a tumor can be inhibited, and a therapeutically effective dose of the present invention is mainly administered to a human or animal in need thereof. A compound inhibits, slows or reverses the growth, progression or spread of a tumor in a subject.
术语“烷基”是指直链或支链的饱和烃基,优选6个碳原子以下的烃基。烷基的实施例包括甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、正戊基、异戊基、新戊基、正己基、异己基、2,2-甲基丁基和2,3-二甲基丁基。术语“C1-6烷基”是指含有1-6个碳原子的直链或支链的饱和烃基。术语“C1-3烷基”是指含有1-3个碳原子的直链或支链的饱和烃基。The term "alkyl" refers to a straight or branched saturated hydrocarbon group, preferably a hydrocarbon group of 6 or less carbon atoms. Examples of alkyl groups include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, tert-butyl, n-pentyl, isopentyl, neopentyl, n-hexyl, isohexyl, 2,2-Methylbutyl and 2,3-dimethylbutyl. The term "C 1-6 alkyl" refers to a straight or branched saturated hydrocarbon group containing from 1 to 6 carbon atoms. The term "C 1-3 alkyl" means a straight or branched saturated hydrocarbon group having 1 to 3 carbon atoms.
本发明的“环烷基”是指环状的饱和的、部分饱和的烃基,优选12个碳原子以下的环烷基,进一步优选8个碳原子以下的环烷基,更进一步优选6个碳原子以下的环烷基。环烷基的实施例包括环丙基、环丁基、环戊基、环己基、环庚基。本发明的“C3-8环烷基”是指含有3-8个碳原子的环状的饱和烃基。 The "cycloalkyl group" of the present invention means a cyclic saturated, partially saturated hydrocarbon group, preferably a cycloalkyl group of 12 or less carbon atoms, more preferably a cycloalkyl group of 8 or less carbon atoms, still more preferably 6 carbon atoms. The following cycloalkyl group. Examples of cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl. The "C 3-8 cycloalkyl group" of the present invention means a cyclic saturated hydrocarbon group having 3 to 8 carbon atoms.
本发明的“烷氧基”是指-O-烷基。The "alkoxy group" of the present invention means an -O-alkyl group.
本发明的“氧代”是指O=,例如碳原子被氧代基团取代形成![]()
![]()
![]()
![]()
![]()
![]()
本发明的“卤素”是指氟、氯、溴、碘。The "halogen" of the present invention means fluorine, chlorine, bromine or iodine.
本发明的“卤代烷基”是指至少被一个卤素原子取代的烷基。The "haloalkyl group" of the present invention means an alkyl group substituted with at least one halogen atom.
本发明的“卤代烷氧基”是指至少被一个卤素取代的烷氧基,优选为至少被一个卤素取代的C1-6烷氧基,进一步优选为至少被一个卤素取代的C1-3烷氧基,合适的卤代C1-3烷氧基为氯甲氧基、氟甲氧基、二氯甲氧基、二氟甲氧基、三氯甲氧基、三氟甲氧基;二氯乙氧基、二氟乙氧基、三氯乙氧基、三氟乙氧基。The "haloalkoxy group" of the present invention means an alkoxy group substituted with at least one halogen, preferably a C 1-6 alkoxy group substituted with at least one halogen, and further preferably a C 1-3 alkane substituted with at least one halogen. Oxyl, a suitable halogenated C 1-3 alkoxy group is chloromethoxy, fluoromethoxy, dichloromethoxy, difluoromethoxy, trichloromethoxy, trifluoromethoxy; Chloroethoxy, difluoroethoxy, trichloroethoxy, trifluoroethoxy.
本发明的“单烷基氨基”是指-NH-烷基,优选为-NH-C1-6烷基,进一步优选为-NH-C1-3烷基。The "monoalkylamino group" of the present invention means -NH-alkyl group, preferably -NH-C 1-6 alkyl group, and more preferably -NH-C 1-3 alkyl group.
本发明的“二烷基氨基”是指-N-(烷基)(烷基),优选为-N-(C1-6烷基)(C1-6烷基),进一步优选为-N-(C1-3烷基)(C1-3烷基)。The "dialkylamino group" of the present invention means -N-(alkyl)(alkyl), preferably -N-(C 1-6 alkyl)(C 1-6 alkyl), further preferably -N -(C 1-3 alkyl) (C 1-3 alkyl).
本发明的“氨基烷基”是指-烷基-NH2。The "aminoalkyl group" of the present invention means -alkyl-NH 2 .
本发明的“单烷基氨基烷基”是指-烷基-NH-烷基。The "monoalkylaminoalkyl group" of the present invention means an -alkyl-NH-alkyl group.
本发明的“二烷基氨基烷基”是指-烷基-N-(烷基)(烷基)。The "dialkylaminoalkyl group" of the present invention means -alkyl-N-(alkyl)(alkyl).
本发明的“羟基烷基”是指-烷基-OH。The "hydroxyalkyl group" of the present invention means -alkyl-OH.
本发明的“烷基酰基”是指-C(O)-烷基。The "alkyl acyl group" of the present invention means a -C(O)-alkyl group.
本发明的“氨基酰基”是指-C(O)-NH2。The "aminoacyl group" of the present invention means -C(O)-NH 2 .
本发明的“单烷基氨基酰基”是指-C(O)-NH-烷基。The "monoalkylaminoacyl" of the present invention means -C(O)-NH-alkyl.
本发明的“二烷基氨基酰基”是指-C(O)-N-(烷基)(烷基)。The "dialkylaminoacyl group" of the present invention means -C(O)-N-(alkyl)(alkyl).
本发明的“杂环烷基”是指取代或未取代的至少含有一个杂原子的饱和的、部分饱和的环状烷基,所述的杂原子选自N、O、S。The "heterocycloalkyl group" of the present invention means a substituted or unsubstituted saturated, partially saturated cyclic alkyl group containing at least one hetero atom selected from N, O, and S.
本发明的“芳基”是指可以包含单环或多稠环例如二环或三环的芳香环的芳香系,其中至少稠合的环的一部分形成共轭的芳香系,其含有5至50个碳原子,优选约6至约14个碳原子。合适的芳基包括但不限于苯基、萘基、联苯基、蒽基、四氢萘基、芴基、茚满基、亚联苯基和苊基。本发明的“C6-C10芳基”是指含有6-10个碳原子的芳香系。The "aryl group" of the present invention means an aromatic system which may contain a monocyclic or polycondensed ring such as a bicyclic or tricyclic aromatic ring, wherein at least a part of the fused ring forms a conjugated aromatic system containing 5 to 50 One carbon atom, preferably from about 6 to about 14 carbon atoms. Suitable aryl groups include, but are not limited to, phenyl, naphthyl, biphenyl, anthracenyl, tetrahydronaphthyl, anthracenyl, indanyl, biphenylene, and anthracenyl. The "C 6 -C 10 aryl group" of the present invention means an aromatic system having 6 to 10 carbon atoms.
本发明的“杂芳基”是指芳族单环或多稠环如二环或三环的至少有一个碳原子被杂原子替代的芳香性基团,所述的杂原子为O、S、N。合适的杂芳基包括但不限于咪唑基、苯并咪唑基、咪唑并吡啶基、喹唑啉酮基、吡咯基、咪唑酮基、呋喃基、噻吩基、吡唑基、噁唑基、噻唑基、异噁唑基、异噻唑基、噁二唑基、三唑基等。本发明的“C5-C10杂芳基”是指含有5-10个原子的杂芳基。The "heteroaryl" of the present invention means an aromatic monocyclic or polycondensed ring such as an aromatic group in which at least one carbon atom of a bicyclic or tricyclic ring is replaced by a hetero atom, and the hetero atom is O, S, N. Suitable heteroaryl groups include, but are not limited to, imidazolyl, benzimidazolyl, imidazopyridyl, quinazolinone, pyrrolyl, imidazolidinyl, furyl, thienyl, pyrazolyl, oxazolyl, thiazole Base, isoxazolyl, isothiazolyl, oxadiazolyl, triazolyl and the like. The "C 5 -C 10 heteroaryl group" of the present invention means a heteroaryl group having 5 to 10 atoms.
本发明的“C4-C8含氮杂环基”是指含有至少一个氮原子的取代或未取代的饱和、部分饱和
和完全不饱和的总环原子数为4、5、6、7或8的杂环基团,例如C4含氮杂环基是指取代或未取代的饱和、部分饱和和完全不饱和的总环原子数为4的含氮杂环基团。本发明的“C4-C8含氮杂单环烷基”是指含有至少一个氮原子的取代或未取代的饱和、部分饱和和完全不饱和的总环原子数为4、5、6、7或8的单杂环烷基,合适的例子包括但不限于取代或未取代的氮杂环丁基、氮杂环戊基、氮杂环己基、氮杂环庚基、氮杂环辛基、氮氧杂环戊基、氮氧杂环己基、氮氧杂环庚基、氮氧杂环辛基、氮硫杂环戊基、氮硫杂环己基、氮硫杂环庚基、氮硫杂环辛基、二氮杂环戊基、二氮杂环己基、二氮杂环庚基或二氮杂环辛基;以及取代或未取代的含氮杂芳基,例如取代或未取代的吡啶基、咪唑基、吡唑基、吡咯基、哒嗪基、嘧啶基、吡嗪基,其中所述取代基选自烷基、氧代、环烷基、羟基、羟烷基、烷氧基、氨基、单烷基氨基、双烷基氨基、酰氨基、烷基酰氨基、芳基酰氨基、杂芳基酰氨基、卤素、卤代烷基、卤代烷氧基。本文中,所述C4-C8含氮杂环基与吲哚环构成氮杂环并吲哚结构,例如,合适的氮杂环并吲哚结构包括但不限于![]()
![]()
![]()
![]()
![]()
![]()
下面代表性的实施例是为了更好地说明本发明,而非用于限制本发明的保护范围。The following representative examples are intended to better illustrate the invention and are not intended to limit the scope of the invention.
实施例1 N-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(2,3-二氢-1H-吡咯并[1,2-a]-吲哚-9-基)嘧啶-2-基)氨基)苯基)烯丙酰胺的合成Example 1 N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(2,3-dihydro-1H-) Synthesis of pyrrolo[1,2-a]-indol-9-yl)pyrimidin-2-yl)amino)phenyl)allylamide
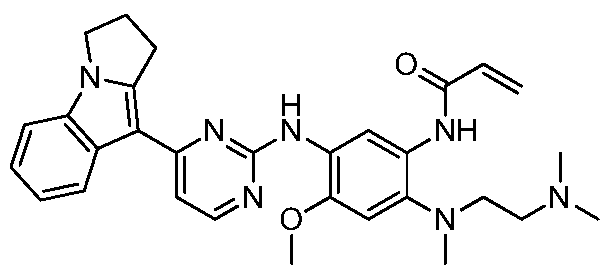
步骤a 1-(3-溴丙基)-1H-吲哚的合成Step a Synthesis of 1-(3-bromopropyl)-1H-indole

在100ml反应瓶中,依次加入NaH(60%含量,1.23g,30.73mmol)和DMF(10ml),室温搅拌5min后冷却至0-4℃,缓慢加入10mL溶解有吲哚(3g,25.61mmol)的DMF溶液,加毕,升至室温反应20min制得吲哚活化溶液。Add NaH (60% content, 1.23 g, 30.73 mmol) and DMF (10 ml) in a 100 ml reaction flask, stir at room temperature for 5 min, then cool to 0-4 ° C, slowly add 10 mL of dissolved hydrazine (3 g, 25.61 mmol) The DMF solution was added and added to room temperature for 20 min to prepare a hydrazine activation solution.
另取250ml反应瓶,加入1,3-二溴丙烷(15.51g,76.82mmol)和DMF(50ml)。0-4℃下缓慢滴加上述制得的吲哚活化溶液,滴毕,室温下反应0.5h。反应结束后,加入水(100ml)淬灭反 应,乙酸乙酯萃取,合并乙酸乙酯层,无水硫酸钠干燥,过滤,浓缩,柱层析纯化得标题化合物。Another 250 ml reaction flask was taken, and 1,3-dibromopropane (15.51 g, 76.82 mmol) and DMF (50 ml) were added. The hydrazine activating solution prepared above was slowly added dropwise at 0-4 ° C, and the reaction was carried out for 0.5 h at room temperature. After the reaction was over, water (100 ml) was added to quench the reaction. The title compound was obtained after EtOAc.
ESI-Ms m/z:239.1[M+H]。ESI-Ms m/z: 239.1 [M+H].
步骤b 1-(3-碘丙基)-1H-吲哚的合成Step b Synthesis of 1-(3-iodopropyl)-1H-indole
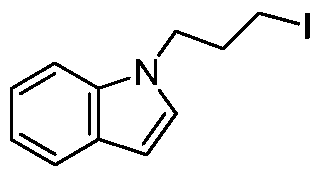
在250ml反应瓶中,依次加入步骤a所得物1-(3-溴丙基)-1H-吲哚(4.72g,19.83mmol)、碘化钠(13.39g,89.93mmol)和乙腈(100ml),回流过夜。反应结束后,加水,乙酸乙酯萃取,合并乙酸乙酯层,无水硫酸钠干燥,过滤,浓缩干燥得标题物,直接用于下一步。In a 250 ml reaction flask, 1-(3-bromopropyl)-1H-indole (4.72 g, 19.83 mmol), sodium iodide (13.39 g, 89.93 mmol) and acetonitrile (100 ml) were obtained. Reflux overnight. After completion of the reaction, water was added, and ethyl acetate was evaporated.
ESI-Ms m/z:286.1[M+H]。ESI-Ms m/z: 286.1 [M+H].
步骤c 2,3-二氢吡咯并[1,2-a]吲哚的合成Step c Synthesis of 2,3-dihydropyrrolo[1,2-a]indole
在250ml三颈瓶中,依次加入步骤b所得物1-(3-碘丙基)-1H-吲哚(5.44g,19.83mmol)、磷酸钾(8.4g,39.67mmol)、四三苯基磷钯(2.3g,1.98mmol)和1,4-二氧六环(80ml),氩气保护,回流过夜反应。反应结束后,加水淬灭反应,乙酸乙酯萃取,合并乙酸乙酯层,无水硫酸钠干燥,过滤,浓缩,柱层析纯化得标题化合物。In a 250 ml three-necked flask, the obtained product of the step b was sequentially added 1-(3-iodopropyl)-1H-indole (5.44 g, 19.83 mmol), potassium phosphate (8.4 g, 39.67 mmol), tetratriphenylphosphine. Palladium (2.3 g, 1.98 mmol) and 1,4-dioxane (80 ml) were argon-protected and refluxed overnight. After the reaction was completed, the mixture was evaporated.
ESI-Ms m/z:158.1[M+H]。ESI-Ms m/z: 158.1 [M+H].
步骤d 9-(2-氯嘧啶-4-基)-2,3-二氢吡咯并[1,2-a]吲哚的合成Synthesis of step d 9-(2-chloropyrimidin-4-yl)-2,3-dihydropyrrolo[1,2-a]indole
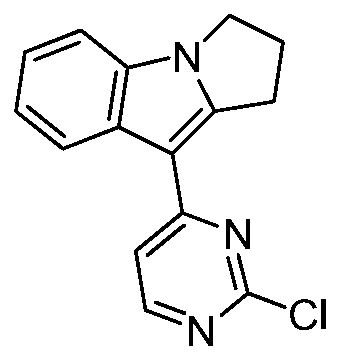
在100ml反应瓶中,依次加入三氯化铝(2.18g,16.35mmol)、乙二醇二甲醚(50ml)、2,4-二氯嘧啶(2.44g,16.35mmol)和步骤c所得物2,3-二氢吡咯并[1,2-a]吲哚(2.57g,16.35mmol),回流反应2h。反应结束后,反应液冷却至室温,过滤,滤饼水洗,干燥后得标题物。In a 100 ml reaction flask, aluminum trichloride (2.18 g, 16.35 mmol), ethylene glycol dimethyl ether (50 ml), 2,4-dichloropyrimidine (2.44 g, 16.35 mmol) and step 2 were successively added. 3-Dihydropyrrolo[1,2-a]indole (2.57 g, 16.35 mmol), refluxed for 2 h. After completion of the reaction, the reaction solution was cooled to room temperature, filtered, and the filter cake was washed with water and dried to give the title compound.
ESI-Ms m/z:270.4[M+H]。ESI-Ms m/z: 270.4 [M+H].
步骤e N-(4-氟-2-甲氧基-5-硝基苯基)-4-(2,3-二氢吡咯并[1,2-a]吲哚-9-基)-嘧啶-2-胺的合成Step e N-(4-Fluoro-2-methoxy-5-nitrophenyl)-4-(2,3-dihydropyrrolo[1,2-a]indol-9-yl)-pyrimidine Synthesis of 2-amine

在250ml反应瓶中,加入步骤d所得9-(2-氯嘧啶-4-基)-2,3-二氢吡咯并[1,2-a]吲哚(2.69g,10mmol)、4-氟-2-甲氧基-5-硝基苯胺(1.86g,10mmol)和对甲苯磺酸(1.71g,10mmol),加入50mL仲丁醇溶解,110℃反应2h,反应结束后,冷却至室温,过滤,仲丁醇洗涤,干燥得标题化合物。In a 250 ml reaction flask, 9-(2-chloropyrimidin-4-yl)-2,3-dihydropyrrolo[1,2-a]indole (2.69 g, 10 mmol), 4-fluoro, obtained in step d 2-methoxy-5-nitroaniline (1.86 g, 10 mmol) and p-toluenesulfonic acid (1.71 g, 10 mmol) were dissolved in 50 mL of sec-butanol and reacted at 110 ° C for 2 h. After the reaction was completed, cooled to room temperature. Filtration, washing with sec-butanol and drying to give the title compound.
ESI-Ms m/z:420.4[M+H]。ESI-Ms m/z: 420.4 [M+H].
步骤f N-(4-((2-(二甲基氨基)乙基)(甲基)氨基)-2-甲氧基-5-硝基苯基)-4-(2,3-二氢吡咯并[1,2-a]吲哚-9-基)-嘧啶-2-胺的合成Step f N-(4-((2-(Dimethylamino)ethyl)(methyl)amino)-2-methoxy-5-nitrophenyl)-4-(2,3-dihydro Synthesis of pyrrolo[1,2-a]fluoren-9-yl)-pyrimidin-2-amine
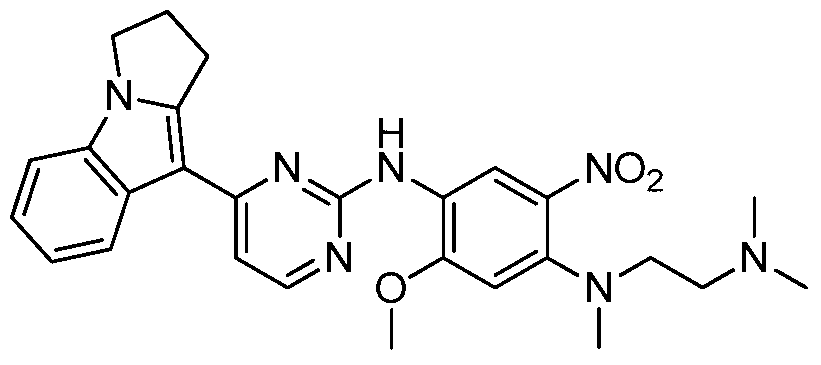
在50ml反应瓶中,依次加入步骤e所得物N-(4-氟-2-甲氧基-5-硝基苯基)-4-(2,3-二氢吡咯并[1,2-a]吲哚-9-基)-嘧啶-2-胺(2.5g,6mmol)、N,N,N’-三甲基乙二胺(0.61g,6mmol)、二异丙基乙胺(2.3g,18mmol)和10ml二氧六环,110℃回流反应3h,反应结束后,浓缩,柱层析纯化得标题化合物。In a 50 ml reaction flask, the resulting product of step e was sequentially added N-(4-fluoro-2-methoxy-5-nitrophenyl)-4-(2,3-dihydropyrrolo[1,2-a吲哚-9-yl)-pyrimidine-2-amine (2.5 g, 6 mmol), N,N,N'-trimethylethylenediamine (0.61 g, 6 mmol), diisopropylethylamine (2.3 g , 18 mmol), and 10 ml of dioxane, refluxing at 110 ° C for 3 h. After completion of the reaction, concentration and purification by column chromatography
ESI-Ms m/z:502.4[M+H]。ESI-Ms m/z: 502.4 [M+H].
步骤g N-(4-((2-(二甲基氨基)乙基)(甲基)氨基)-2-甲氧基-5-氨基苯基)-4-(2,3-二氢吡咯并[1,2-a]吲哚-9-基)-嘧啶-2-胺的合成Step g N-(4-((2-(Dimethylamino)ethyl)(methyl)amino)-2-methoxy-5-aminophenyl)-4-(2,3-dihydropyrrole Synthesis of [1,2-a]fluoren-9-yl)-pyrimidine-2-amine
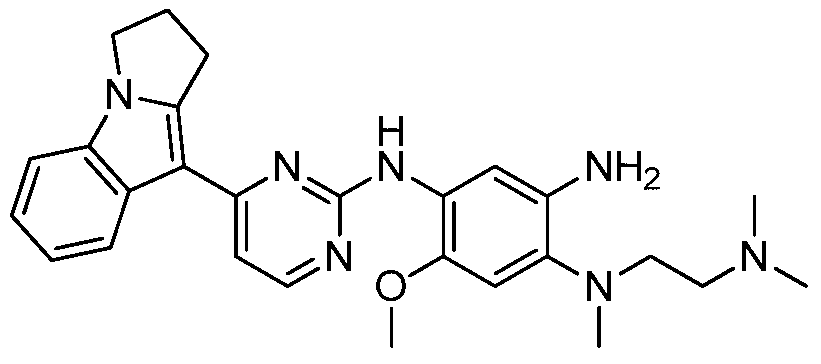
在50ml反应瓶中,依次加入步骤f所得物N-(4-((2-(二甲基氨基)乙基)(甲基)氨基)-2-甲氧基-5-硝基苯基)-4-(2,3-二氢吡咯并[1,2-a]吲哚-9-基)-嘧啶-2-胺(2.5g,5mmol)、10%Pd-C(20mg)和30ml甲醇,在1个标准大气压下,氢气还原1h,反应结束后,过滤,浓缩得标题化合物,直接用于下一步。In a 50 ml reaction flask, the resulting step N was added N-(4-((2-(dimethylamino)ethyl))(methyl)amino)-2-methoxy-5-nitrophenyl) 4-(2,3-dihydropyrrolo[1,2-a]indol-9-yl)-pyrimidin-2-amine (2.5 g, 5 mmol), 10% Pd-C (20 mg) and 30 mL methanol Hydrogen was reduced for 1 h at 1 standard atmosphere. After the reaction was completed, it was filtered and concentrated to give the title compound.
ESI-Ms m/z:472.6[M+H]。ESI-Ms m/z: 472.6 [M+H].
步骤h N-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((5-甲基-4-(2,3-二氢-1H-吡咯并[1,2-a]-吲哚-9-基)嘧啶-2-基)氨基)苯基)烯丙酰胺的合成Step h N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((5-methyl-4-(2,3-di) Synthesis of Hydrogen-1H-pyrrolo[1,2-a]-indol-9-yl)pyrimidin-2-yl)amino)phenyl)propanamide

在150ml单口瓶中,加入步骤g所得物N-(4-((2-(二甲基氨基)乙基)(甲基)氨基)-2-甲氧基-5-氨基苯基)-4-(2,3-二氢吡咯并[1,2-a]吲哚-9-基)-嘧啶-2-胺(2g,4.2mmol)、二异丙基乙胺(0.53g,4.2mmol)和20ml无水二氯甲烷,溶解后缓慢加入5ml溶解有烯丙基酰氯(0.37g,4.2mmol)的二氯甲烷溶液,反应10min,反应结束后,浓缩,柱层析纯化得标题化合物。1H NMR(300MHz,DMSO-d6):δ10.17(s,1H),8.69(s,1H),8.28-8.25(d,2H),8.02(s,1H),7.37-7.35(d,1H),7.15-6.97(m,4H),6.44-6.35(m,1H),6.21-6.15(m,1H),5.74-5.70(d,1H),4.1-4.10(t,2H),3.81(s,3H),3.35(s,1H),3.30-3.25(t,1H),2.90(s,2H),2.73(s,3H),2.60-2.56(m,2H),2.33(s,2H),2.22(s,6H)。In a 150 ml single-mouth bottle, the obtained step g was added N-(4-((2-(dimethylamino)ethyl)(methyl)amino)-2-methoxy-5-aminophenyl)-4 -(2,3-dihydropyrrolo[1,2-a]indol-9-yl)-pyrimidin-2-amine (2 g, 4.2 mmol), diisopropylethylamine (0.53 g, 4.2 mmol) After dissolving, 20 ml of anhydrous dichloromethane was added, and 5 ml of a solution of allylic acid chloride (0.37 g, 4.2 mmol) in methylene chloride was added slowly, and the mixture was reacted for 10 min. 1 H NMR (300MHz, DMSO- d 6): δ10.17 (s, 1H), 8.69 (s, 1H), 8.28-8.25 (d, 2H), 8.02 (s, 1H), 7.37-7.35 (d, 1H), 7.15-6.97 (m, 4H), 6.44-6.35 (m, 1H), 6.21-6.15 (m, 1H), 5.74-5.70 (d, 1H), 4.1-4.10 (t, 2H), 3.81 ( s, 3H), 3.35 (s, 1H), 3.30-3.25 (t, 1H), 2.90 (s, 2H), 2.73 (s, 3H), 2.60-2.56 (m, 2H), 2.33 (s, 2H) , 2.22 (s, 6H).
ESI-Ms m/z:526.4[M+H]。ESI-Ms m/z: 526.4 [M+H].
实施例2 N-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)-嘧啶-2-基)氨基)苯基)烯丙酰胺的合成Example 2 N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(6,7,8,9-tetra) Synthesis of Hydropyrido[1,2-a]indole-10-yl)-pyrimidin-2-yl)amino)phenyl)allylamide

步骤a 1-(4-溴丁基)-1H-吲哚的合成Step a Synthesis of 1-(4-bromobutyl)-1H-indole
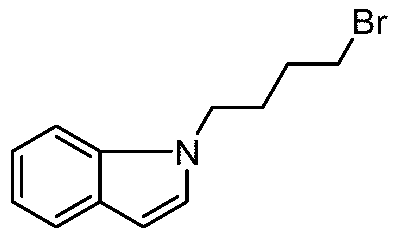
在100mL反应瓶中,依次加入NaH(60%含量,1.23g,30.73mmol)、DMF(10mL),室温搅拌5min后冷却至0-4℃,缓慢加入10mL溶解有吲哚(3g,25.61mmol)的DMF溶液,加毕,升至室温反应20min制得吲哚活化溶液。In a 100 mL reaction flask, NaH (60% content, 1.23 g, 30.73 mmol) and DMF (10 mL) were added sequentially, stirred at room temperature for 5 min, then cooled to 0-4 ° C, and 10 mL of dissolved hydrazine (3 g, 25.61 mmol) was slowly added. The DMF solution was added and added to room temperature for 20 min to prepare a hydrazine activation solution.
另取250mL反应瓶,加入1,4-二溴丁烷(16.59g,76.82mmol)、DMF(50mL)。0-4℃下缓慢滴加上述制得的吲哚活化溶液,滴毕,室温下反应0.5h。反应结束后,加入水(100mL)淬灭,乙酸乙酯萃取,合并乙酸乙酯层,无水硫酸钠干燥,过滤,浓缩,柱层析纯化得标题化合物。Another 250 mL reaction flask was taken, and 1,4-dibromobutane (16.59 g, 76.82 mmol) and DMF (50 mL) were added. The hydrazine activating solution prepared above was slowly added dropwise at 0-4 ° C, and the reaction was carried out for 0.5 h at room temperature. After completion of the reaction, the mixture was evaporated.
ESI-Ms m/z:252.1[M+H]。ESI-Ms m/z: 252.1 [M+H].
步骤b 1-(4-碘丁基)-1H-吲哚的合成Step b Synthesis of 1-(4-iodobutyl)-1H-indole
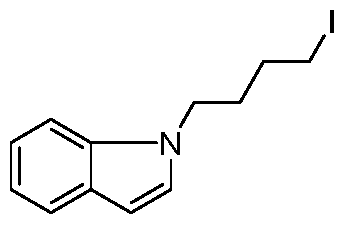
在250mL反应瓶中,依次加入步骤a所得物1-(4-溴丁基)-1H-吲哚(5g,19.83mmol)、碘化钠(13.39g,89.93mmol)、乙腈(100mL),回流过夜。反应结束后,加水,乙酸乙酯萃取,合并乙酸乙酯层,无水硫酸钠干燥,过滤,浓缩干燥得标题物,直接用于下一步。In a 250 mL reaction flask, 1-(4-bromobutyl)-1H-indole (5 g, 19.83 mmol), sodium iodide (13.39 g, 89.93 mmol), acetonitrile (100 mL), and then refluxed. overnight. After completion of the reaction, water was added, and ethyl acetate was evaporated.
ESI-Ms m/z:300.0[M+H]。 ESI-Ms m/z: 300.0 [M+H].
步骤c 6,7,8,9-四氢吡啶并[1,2-a]吲哚的合成Synthesis of step c 6,7,8,9-tetrahydropyrido[1,2-a]indole
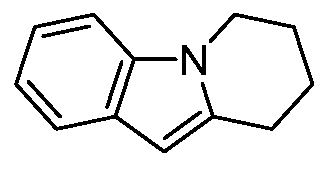
在250mL三颈瓶中,依次加入步骤b所得物1-(4-碘丁基)-1H-吲哚(5.93g,按19.83mmol)、磷酸钾(8.4g,39.67mmol)、四三苯基磷钯(2.3g,1.98mmol)、1,4-二氧六环(80mL),氩气保护,回流过夜反应。反应结束后,加水淬灭反应,乙酸乙酯萃取,合并乙酸乙酯层,无水硫酸钠干燥,过滤,浓缩,柱层析纯化得标题化合物。In a 250 mL three-necked flask, the obtained product of step b was added 1-(4-iodobutyl)-1H-indole (5.93 g, 19.83 mmol), potassium phosphate (8.4 g, 39.67 mmol), tetratriphenyl. Palladium phosphate (2.3 g, 1.98 mmol), 1,4-dioxane (80 mL), argon-protected, refluxed overnight. After the reaction was completed, the mixture was evaporated.
ESI-Ms m/z:172.1[M+H]。ESI-Ms m/z: 172.1 [M+H].
步骤d 10-(2-氯嘧啶-4-基)-6,7,8,9-四氢吡啶并[1,2-a]吲哚的合成Step d Synthesis of 10-(2-chloropyrimidin-4-yl)-6,7,8,9-tetrahydropyrido[1,2-a]indole
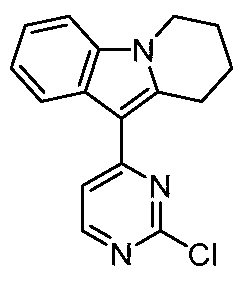
在100mL反应瓶中,依次加入三氯化铝(2.18g,16.35mmol)、乙二醇二甲醚(50mL)、2,4-二氯嘧啶(2.44g,16.35mmol)、步骤c所得物6,7,8,9-四氢吡啶并[1,2-a]吲哚(2.8g,16.35mmol),回流反应2h。反应结束后,反应液冷却至室温,过滤,滤饼水洗,干燥后得标题物。In a 100 mL reaction flask, aluminum trichloride (2.18 g, 16.35 mmol), ethylene glycol dimethyl ether (50 mL), 2,4-dichloropyrimidine (2.44 g, 16.35 mmol), and the obtained product of step c were successively added. , 7,8,9-tetrahydropyrido[1,2-a]indole (2.8 g, 16.35 mmol), refluxed for 2 h. After completion of the reaction, the reaction solution was cooled to room temperature, filtered, and the filter cake was washed with water and dried to give the title compound.
ESI-Ms m/z:284.1[M+H]。ESI-Ms m/z: 284.1 [M+H].
步骤e N-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)烯丙酰胺的合成Step e N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(6,7,8,9-tetrahydro) Synthesis of pyrido[1,2-a]indol-10-ylpyrimidin-2-yl)amino)phenyl)allylamide
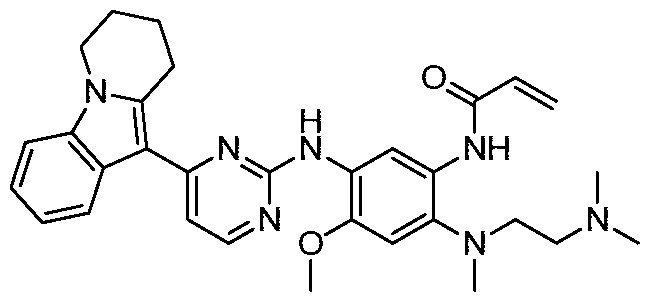
以步骤d所得物10-(2-氯嘧啶-4-基)-6,7,8,9-四氢吡啶并[1,2-a]吲哚、4-氟-2-甲氧基-5-硝基苯胺,N,N,N’-三甲基乙二胺、烯丙基酰氯为原料,按照实施例1步骤e、f、g和h的方法制得标题化合物。The product obtained in the step d is 10-(2-chloropyrimidin-4-yl)-6,7,8,9-tetrahydropyrido[1,2-a]indole, 4-fluoro-2-methoxy- 5-Nitroaniline, N,N,N'-trimethylethylenediamine and allyl chloride were used as starting materials, and the title compound was obtained according
1H NMR(300MHz,DMSO-d6)δ10.20(s,1H),8.65(s,1H),8.34(d,1H),8.11(s,1H),8.06(d,1H),7.43(d,1H),7.19-7.03(m,3H),6.98(s,1H),6.57-6.41(m,1H),6.28-6.15(m,1H),5.82-5.71(m,1H),4.09(t,2H),3.84(s,3H),3.18(t,2H),3.06-2.92(m,2H),2.66(s,3H),2.47-2.40(m,2H),2.27(s,6H),2.08-1.96(m,2H),1.87-1.74(m,2H)。 1 H NMR (300MHz, DMSO- d 6) δ10.20 (s, 1H), 8.65 (s, 1H), 8.34 (d, 1H), 8.11 (s, 1H), 8.06 (d, 1H), 7.43 ( d, 1H), 7.19-7.03 (m, 3H), 6.98 (s, 1H), 6.57-6.41 (m, 1H), 6.28-6.15 (m, 1H), 5.82-5.71 (m, 1H), 4.09 ( t, 2H), 3.84 (s, 3H), 3.18 (t, 2H), 3.06-2.92 (m, 2H), 2.66 (s, 3H), 2.47-2.40 (m, 2H), 2.27 (s, 6H) , 2.08-1.96 (m, 2H), 1.87-1.74 (m, 2H).
ESI-Ms m/z:540.3[M+H]。ESI-Ms m/z: 540.3 [M+H].
实施例3 N-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((5-甲基-4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)烯丙酰胺的合成 Example 3 N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((5-methyl-4-(6,7, Synthesis of 8,9-tetrahydropyrido[1,2-a]indol-10-ylpyrimidin-2-yl)amino)phenyl)allylamide
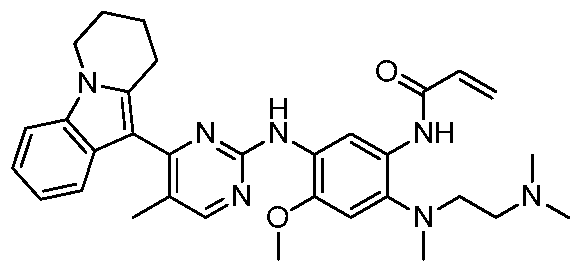
步骤a 10-(2-氯-5-甲基嘧啶-4-基)-6,7,8,9-四氢吡啶并[1,2-a]吲哚的合成Step a Synthesis of 10-(2-chloro-5-methylpyrimidin-4-yl)-6,7,8,9-tetrahydropyrido[1,2-a]indole

以实施例2步骤c所得物6,7,8,9-四氢吡啶并[1,2-a]吲哚和5-甲基-2,4-二氯嘧啶为原料,按照实施例2步骤d的方法制得标题化合物。Using the 6,7,8,9-tetrahydropyrido[1,2-a]indole and 5-methyl-2,4-dichloropyrimidine as the starting materials in the step c of Example 2, according to the procedure of Example 2 The title compound was prepared by the method of d.
步骤b N-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((5-甲基-4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)烯丙酰胺的合成Step b N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((5-methyl-4-(6,7,8) Synthesis of 9-tetrahydropyrido[1,2-a]indol-10-ylpyrimidin-2-yl)amino)phenyl)propanamide
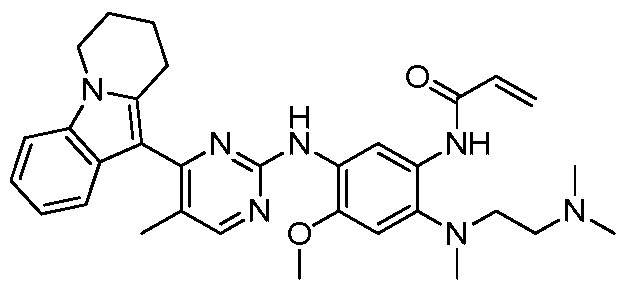
以步骤a所得物10-(2-氯-5-甲基嘧啶-4-基)-6,7,8,9-四氢吡啶并[1,2-a]吲哚、4-氟-2-甲氧基-5-硝基苯胺、N,N,N’-三甲基乙二胺和烯丙基酰氯为原料,按照实施例1步骤e、f、g和h的方法制得标题化合物。The product obtained in the step a is 10-(2-chloro-5-methylpyrimidin-4-yl)-6,7,8,9-tetrahydropyrido[1,2-a]indole, 4-fluoro-2. -Methoxy-5-nitroaniline, N,N,N'-trimethylethylenediamine and allyl chloride as starting materials, the title compound was obtained according to the procedure of Example 1, Steps e, f, g and h .
1H NMR(300MHz,DMSO-d6)δ10.02(s,1H),δ8.84(s,1H),8.33(s,1H),7.77(s,1H),7.42(d,1H),7.26(d,1H),7.10-7.13(m,1H),7.04-7.07(m,1H),6.95(s,1H),6.34-6.39(m,1H),6.19-6.22(m,1H),5.73(d,1H),4.11(t,2H),3.83(s,3H),2.99(s,2H),2.83(t,2H),2.67(s,3H),2.25(t,2H),2.17(s,6H),2.07(s,3H),2.04(m,2H),1.79(m,2H)。 1 H NMR (300MHz, DMSO- d 6) δ10.02 (s, 1H), δ8.84 (s, 1H), 8.33 (s, 1H), 7.77 (s, 1H), 7.42 (d, 1H), 7.26(d,1H), 7.10-7.13 (m,1H), 7.04-7.07 (m,1H), 6.95 (s,1H), 6.34-6.39 (m,1H), 6.19-6.22 (m,1H), 5.73 (d, 1H), 4.11 (t, 2H), 3.83 (s, 3H), 2.99 (s, 2H), 2.83 (t, 2H), 2.67 (s, 3H), 2.25 (t, 2H), 2.17 (s, 6H), 2.07 (s, 3H), 2.04 (m, 2H), 1.79 (m, 2H).
ESI-Ms m/z:554.3[M+H]。ESI-Ms m/z: 554.3 [M+H].
实施例4 N-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(6,7,8,9-四氢吡啶并[1,2-a]-5-氟吲哚-10-基)嘧啶-2-基)氨基)苯基)烯丙酰胺的合成Example 4 N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(6,7,8,9-tetra) Synthesis of Hydropyrido[1,2-a]-5-fluoroindole-10-yl)pyrimidin-2-yl)amino)phenyl)allylamide
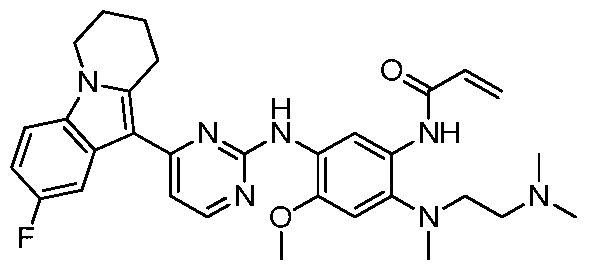
以4-氟-2-甲氧基-5-硝基苯胺、2,4-二氯嘧啶、5-氟吲哚、1,4-二溴丁烷、N,N,N’-三甲基乙二胺、烯丙基酰氯为原料,按照实施例1的方法制得标题化合物。4-fluoro-2-methoxy-5-nitroaniline, 2,4-dichloropyrimidine, 5-fluoroindole, 1,4-dibromobutane, N,N,N'-trimethyl The title compound was obtained according to the method of Example 1 using ethylenediamine and allyl chloride as starting materials.
1H NMR(300MHz,DMSO-d6):δ9.62(s,1H),9.42(s,1H),8.3-8.27(d,2H),7.87(s,1H),7.49(d,1H),7.14(d,1H),7.06(s,2H),6.71-6.63(m,1H),6.22-6.16(m,1H)5.73(d,1H),4.13(s,2H),3.84(s,3H),3.32(s,4H),3.22-3.20(t,2H),2.83 (s,6H),2.64(s,3H),2.02(s,2H),1.85(s,2H)。 1 H NMR (300MHz, DMSO- d 6): δ9.62 (s, 1H), 9.42 (s, 1H), 8.3-8.27 (d, 2H), 7.87 (s, 1H), 7.49 (d, 1H) , 7.14 (d, 1H), 7.06 (s, 2H), 6.71-6.63 (m, 1H), 6.22-6.16 (m, 1H) 5.73 (d, 1H), 4.13 (s, 2H), 3.84 (s, 3H), 3.32 (s, 4H), 3.22-3.20 (t, 2H), 2.83 (s, 6H), 2.64 (s, 3H), 2.02 (s, 2H), 1.85 (s, 2H).
ESI-Ms m/z:558.4[M+H]。ESI-Ms m/z: 558.4 [M+H].
实施例5 N-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-((2,3-二氢-1H-吡咯并[1,2-a]-5-氟吲哚-9-基)-5-氯-嘧啶-2-基)氨基)苯基)烯丙酰胺的合成Example 5 N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-((2,3-dihydro-1H) Synthesis of pyrrolo[1,2-a]-5-fluoroindol-9-yl)-5-chloro-pyrimidin-2-yl)amino)phenyl)allylamide
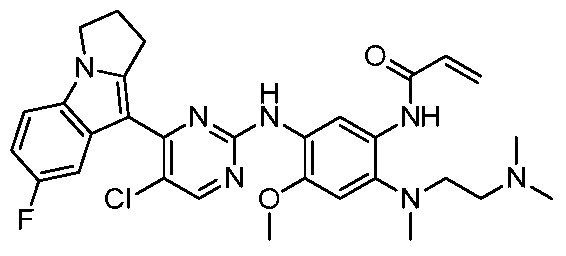
以4-氟-2-甲氧基-5-硝基苯胺、2,4,5-三氯嘧啶、5-氟吲哚、1,3-二溴丙烷、N,N,N’-三甲基乙二胺、烯丙基酰氯为原料,按照实施例1的方法制得标题化合物。4-fluoro-2-methoxy-5-nitroaniline, 2,4,5-trichloropyrimidine, 5-fluoroindole, 1,3-dibromopropane, N,N,N'-trimethyl The title compound was obtained by the method of Example 1 using methylenediamine and allyl chloride as starting materials.
1H NMR(300MHz,DMSO-d6)δ:9.99(s,1H),8.48(s,1H),8.42(s,2H),7.41-7.34(m,2H),6.98-6.91(m,2H),6.44–6.39(m,1H),6.17(m,1H),5.61(d,1H),4.14(t,2H),3.82(s,3H),3.16–3.11(m,2H),2.91-2.87(m,2H),2.68(s,3H),2.57–2.54(m,2H),2.39–2.35(m,2H),2.23(s,6H).1H NMR (300MHz, DMSO-d6) δ: 9.99 (s, 1H), 8.48 (s, 1H), 8.42 (s, 2H), 7.41-7.34 (m, 2H), 6.98-6.91 (m, 2H), 6.44–6.39 (m, 1H), 6.17 (m, 1H), 5.61 (d, 1H), 4.14 (t, 2H), 3.82 (s, 3H), 3.16–3.11 (m, 2H), 2.91-2.87 ( m, 2H), 2.68 (s, 3H), 2.57–2.54 (m, 2H), 2.39–2.35 (m, 2H), 2.23 (s, 6H).
ESI-Ms m/z:578.2[M+H]。ESI-Ms m/z: 578.2 [M+H].
实施例6 N-(2–((2–(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5–((4-(2,3-二氢-1H-吡咯并[1,2-a]-5-氟吲哚-9-基)嘧啶-2-基)氨基)苯基)烯丙酰胺的合成Example 6 N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(2,3-dihydro-1H-) Synthesis of pyrrolo[1,2-a]-5-fluoroindol-9-yl)pyrimidin-2-yl)amino)phenyl)allylamide

以4-氟-2-甲氧基-5-硝基苯胺、2,4-二氯嘧啶、5-氟吲哚、1,3-二溴丙烷、N,N,N’-三甲基乙二胺、烯丙基酰氯为原料,按照实施例1的方法制得标题化合物。4-fluoro-2-methoxy-5-nitroaniline, 2,4-dichloropyrimidine, 5-fluoroindole, 1,3-dibromopropane, N,N,N'-trimethyl The title compound was obtained according to the method of Example 1 using diamine and allyl chloride as starting materials.
1H NMR(300MHz,DMSO-d6)δ10.07(s,1H),8.65(s,1H),8.25(d,1H),8.05–8.02(m,1H),7.36-7.34(m,2H),7.01(s,1H),6.90–6.97(m,2H),6.36-6.34(m,1H),6.17-6.15(m,1H),5.69-5.67(d,1H),4.13(t,2H),3.79(s,3H),2.89–2.88(t,2H),2.73(s,3H),2.62–2.60(t,2H),2.51–2.50(t,2H),2.32-2.30(t,2H),2.21(s,6H).1H NMR (300MHz, DMSO-d6) δ 10.07 (s, 1H), 8.65 (s, 1H), 8.25 (d, 1H), 8.05 - 8.02 (m, 1H), 7.36-7.34 (m, 2H), 7.01(s,1H), 6.90–6.97(m,2H), 6.36-6.34(m,1H), 6.17-6.15(m,1H),5.69-5.67(d,1H), 4.13(t,2H), 3.79(s,3H), 2.89–2.88(t,2H), 2.73(s,3H), 2.62–2.60(t,2H),2.51–2.50(t,2H), 2.32-2.30(t,2H), 2.21(s,6H).
ESI-Ms m/z:544.4[M+H]。ESI-Ms m/z: 544.4 [M+H].
实施例7 N–(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(3,4-二氢-1H-[1,4]噁嗪并[4,3-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)烯丙酰胺的合成Example 7 N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(3,4-dihydro-1H-) Synthesis of [1,4]oxazino[4,3-a]indole-10-yl)pyrimidin-2-yl)amino)phenyl)allylamide
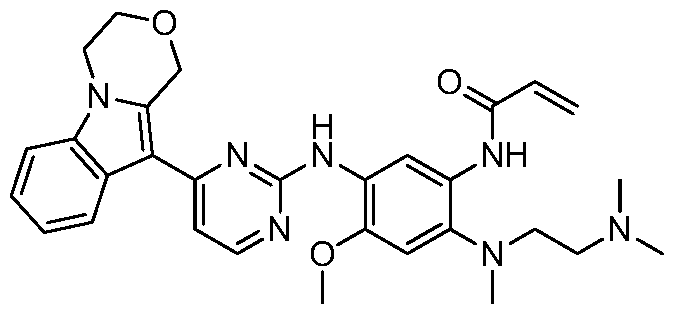
步骤a 2-羟甲基吲哚的合成Step a Synthesis of 2-hydroxymethylhydrazine
在100ml反应瓶中,依次加入四氢铝锂(902.63mg,23.78mmol)、THF(30ml),室温搅拌 5min后冰浴,缓慢加入吲哚-2-羧酸乙酯(3g,15.86mmol),加毕,升至室温反应2h。反应结束后,将体系冷却至0℃,然后将20ml THF以及1.7ml 20%KOH加入其中,搅拌10min后过滤,用20ml THF洗涤滤饼,滤液用饱和NaCl(10ml)洗一次,无水硫酸钠干燥,过滤,浓缩干燥得标题物2.3g,直接用于下一步。In a 100 ml reaction flask, lithium tetrahydroaluminum (902.63 mg, 23.78 mmol) and THF (30 ml) were added in this order, and stirred at room temperature. After 5 min, ice bath was added, and ethyl hydrazine-2-carboxylate (3 g, 15.86 mmol) was slowly added, and the mixture was warmed to room temperature for 2 h. After completion of the reaction, the system was cooled to 0 ° C, then 20 ml of THF and 1.7 ml of 20% KOH were added thereto, stirred for 10 min, filtered, and the filter cake was washed with 20 ml of THF, and the filtrate was washed once with saturated NaCl (10 ml). It was dried, filtered and concentrated to dryness to give the title compound (2.3 g).
ESI-Ms m/z:148.1[M+H]。ESI-Ms m/z: 148.1 [M+H].
步骤b 3,4-二氢-1H-[1,4]噁嗪并[4,3-a]吲哚的合成Step b Synthesis of 3,4-dihydro-1H-[1,4]oxazino[4,3-a]indole
在500ml反应瓶中,依次加入步骤a所得2-羟甲基吲哚(300mg,2.04mmol),KOH(285.91mg,5.10mmol)、CH2Cl2(160ml),冰浴冷却至0℃左右,氮气保护,搅拌10min后。将40mL溶有Diphenyl(vinyl)sulfonium trifluoromethanesulfonate(888.86mg,2.45mmol)的CH2Cl2溶液滴加入上述反应体系中,加毕,升至室温反应。当TLC监测原料消失后,反应结束后,加水淬灭反应,乙酸乙酯萃取,合并乙酸乙酯层,无水硫酸钠干燥,过滤,浓缩,柱层析纯化得标题物300mg。In a 500 ml reaction flask, the 2-hydroxymethyl hydrazine (300 mg, 2.04 mmol) obtained in the step a, KOH (285.91 mg, 5.10 mmol), and CH 2 Cl 2 (160 ml) were sequentially added, and cooled to about 0 ° C in an ice bath. Protected with nitrogen and stirred for 10 min. 40 mL of a solution of Diphenyl(vinyl)sulfonium trifluoromethanesulfonate (888.86 mg, 2.45 mmol) in CH 2 Cl 2 was added dropwise to the above reaction system, and after completion, the mixture was allowed to react to room temperature. After the completion of the reaction, the reaction mixture was quenched with EtOAc (EtOAc)EtOAc.
ESI-Ms m/z:174.1[M+H]。ESI-Ms m/z: 174.1 [M+H].
步骤c N-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(3,4-二氢-1H-[1,4]噁嗪并[4,3-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)烯丙酰胺的合成Step c N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(3,4-dihydro-1H-[ Synthesis of 1,4]oxazino[4,3-a]indole-10-ylpyrimidin-2-yl)amino)phenyl)propanamide
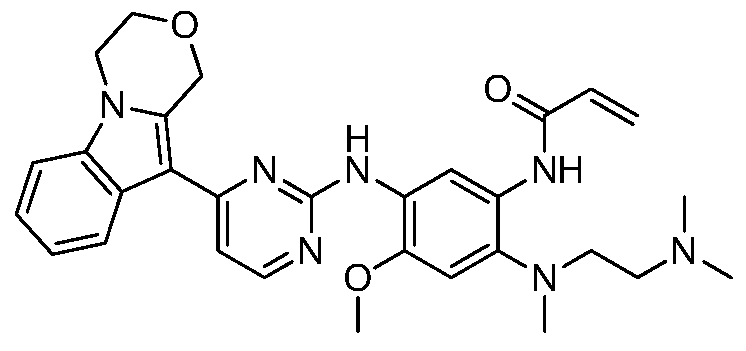
以4-氟-2-甲氧基-5-硝基苯胺、2,4-二氯嘧啶、步骤b所得3,4-二氢-1H-[1,4]噁嗪并[4,3-a]吲哚、N,N,N’-三甲基乙二胺、烯丙基酰氯为原料,按照实施例1的方法制得标题化合物。3-, 4-dihydro-5-nitroaniline, 2,4-dichloropyrimidine, step b, 3,4-dihydro-1H-[1,4]oxazine [4,3- The title compound was obtained according to the procedure of Example 1 using </ RTI> </ RTI> </ RTI> </ RTI> </ RTI> </ RTI> <RTIgt;
1H NMR(300MHz,DMSO-d6)δ10.04(s,1H),8.61(s,1H),8.35(d,1H),8.06(d,1H),8.03(s,1H),7.50-7.49(d,1H),7.24–7.19(m,2H),7.12-7.11(d,1H),7.00(s,1H),6.45(s,1H),6.22–6.19(d,1H),5.73-5.71(d,1H),5.07(s,2H),4.16–4.14(t,2H),4.06–4.04(t,2H),3.81(s,3H),2.96(s,2H),2.70(s,3H),2.30(s,6H),2.35(m,2H).1H NMR (300MHz, DMSO-d6) δ 10.04 (s, 1H), 8.61 (s, 1H), 8.35 (d, 1H), 8.06 (d, 1H), 8.03 (s, 1H), 7.50-7.49 ( d, 1H), 7.24–7.19 (m, 2H), 7.12-7.11 (d, 1H), 7.00 (s, 1H), 6.45 (s, 1H), 6.22–6.19 (d, 1H), 5.73-5.71 ( d,1H),5.07(s,2H),4.16–4.14(t,2H),4.06–4.04(t,2H),3.81(s,3H),2.96(s,2H),2.70(s,3H) , 2.30 (s, 6H), 2.35 (m, 2H).
ESI-Ms m/z:542.2[M+H]。ESI-Ms m/z: 542.2 [M+H].
实施例8 N-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(6,7,8,9-四氢吡啶并[1,2-a]-7-氯吲哚-10-基)嘧啶-2-基)氨基)苯基)烯丙酰胺的合成Example 8 N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(6,7,8,9-tetra) Synthesis of Hydropyrido[1,2-a]-7-chloroindole-10-yl)pyrimidin-2-yl)amino)phenyl)Allylamide
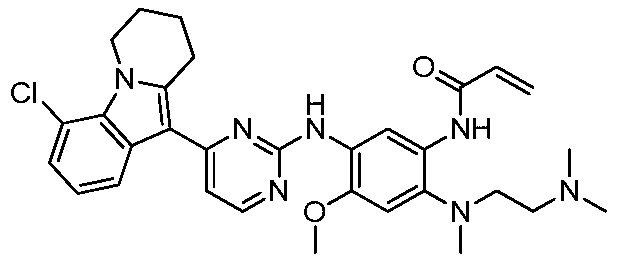
以4-氟-2-甲氧基-5-硝基苯胺、2,4-二氯嘧啶、7-氯吲哚、1,4-二溴丁烷、N,N,N’-三甲基乙二胺、烯丙基酰氯为原料,按照实施例1的方法制得标题化合物。 4-fluoro-2-methoxy-5-nitroaniline, 2,4-dichloropyrimidine, 7-chloroindole, 1,4-dibromobutane, N,N,N'-trimethyl The title compound was obtained according to the method of Example 1 using ethylenediamine and allyl chloride as starting materials.
1H NMR(300MHz,DMSO-d6)δ:10.04(s,1H),8.69(s,1H),8.38–8.36(d,1H),8.26(s,1H),8.00(s,2H),7.14–7.12(m,1H),7.03–6.98(m,2H),6.43–6.35(m,1H),6.17–6.16(d,1H),5.71–5.70(d,1H),4.66–4.62(m,2H),3.34–3.21(m,3H),2.90–2.86(m,3H),2.70(s,4H),2.33–2.31(m,2H),2.20(s,6H),2.20–2.19(m,2H),1.78–1.76(m,2H),1H NMR (300MHz, DMSO-d6) δ: 10.04 (s, 1H), 8.69 (s, 1H), 8.38 - 8.36 (d, 1H), 8.26 (s, 1H), 8.00 (s, 2H), 7.14 - 7.12 (m, 1H), 7.03–6.98 (m, 2H), 6.43–6.35 (m, 1H), 6.17–6.16 (d, 1H), 5.71–5.70 (d, 1H), 4.66–4.62 (m, 2H) ), 3.34–3.21 (m, 3H), 2.90–2.86 (m, 3H), 2.70 (s, 4H), 2.33–2.31 (m, 2H), 2.20 (s, 6H), 2.20–2.19 (m, 2H) ), 1.78–1.76 (m, 2H),
ESI-Ms m/z:575.2[M+H]。ESI-Ms m/z: 575.2 [M+H].
实施例9 N-(5-((4-(1,1-二甲基-2,3-二氢-1H-吡咯并[1,2-a]吲哚-9-基)嘧啶-2-基)氨基)-2-((2-(二甲氨基)乙基)(甲基)氨基)-4-甲氧基苯基)丙烯酰胺的合成Example 9 N-(5-((4-(1,1-Dimethyl-2,3-dihydro-1H-pyrrolo[1,2-a]indol-9-yl)pyrimidin-2-) Synthesis of amide)amino)-2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxyphenyl)acrylamide

以4-氟-2-甲氧基-5-硝基苯胺、2,4-二氯嘧啶、1,3-二溴-3-甲基丁烷、吲哚、N,N,N’-三甲基乙二胺、烯丙基酰氯为原料,按照实施例1的方法制得标题化合物。4-fluoro-2-methoxy-5-nitroaniline, 2,4-dichloropyrimidine, 1,3-dibromo-3-methylbutane, anthracene, N,N,N'-three The title compound was obtained according to the method of Example 1 using methylethylenediamine and allyl chloride as starting materials.
1H NMR(300MHz,DMSO-d6)δ10.00(s,1H),8.58(s,1H),8.36–8.58(m,1H),7.91–7.96(m,2H),7.35–7.40(m,1H),7.09–7.40(m,4H),6.36–6.45(m,1H),6.18–6.23(m,1H),5.71–5.74(m,1H)4.12–4.19(m,2H),3.80(s,3H),2.93(s,2H),2.69(s,3H),2.37–2.44(m,2H),2.28(s,6H),1.56(s,2H),1.37(s,6H).1H NMR (300MHz, DMSO-d6) δ10.00 (s, 1H), 8.58 (s, 1H), 8.36 - 8.58 (m, 1H), 7.91 - 7.96 (m, 2H), 7.35 - 7.40 (m, 1H) ), 7.09 - 7.40 (m, 4H), 6.36 - 6.45 (m, 1H), 6.18 - 6.23 (m, 1H), 5.71 - 5.74 (m, 1H) 4.12 - 4.19 (m, 2H), 3.80 (s, 3H), 2.93 (s, 2H), 2.69 (s, 3H), 2.37 - 2.44 (m, 2H), 2.28 (s, 6H), 1.56 (s, 2H), 1.37 (s, 6H).
ESI-Ms m/z:554.3[M+H]。ESI-Ms m/z: 554.3 [M+H].
实施例10 N-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)-5-氯-嘧啶-2-基)氨基)苯基)烯丙酰胺的合成Example 10 N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(6,7,8,9-tetra) Synthesis of Hydropyrido[1,2-a]indole-10-yl)-5-chloro-pyrimidin-2-yl)amino)phenyl)allylamide
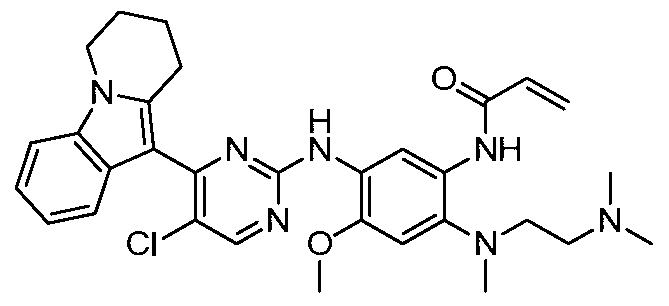
以实施例2步骤c所得物6,7,8,9-四氢吡啶并[1,2-a]吲哚、4-氟-2-甲氧基-5-硝基苯胺、2,4,5-三氯嘧啶、N,N,N’-三甲基乙二胺、烯丙基酰氯为原料,按照实施例1的方法制得标题化合物。The product obtained in the step c of Example 2, 6,7,8,9-tetrahydropyrido[1,2-a]indole, 4-fluoro-2-methoxy-5-nitroaniline, 2,4, The title compound was obtained according to the method of Example 1 using 5-trichloropyrimidine, N,N,N'-trimethylethylenediamine and allyl chloride as starting materials.
1H NMR(300MHz,DMSO-d6)δ:9.63(s,1H),9.47(s,1H),8.3–8.30(d,2H),7.85(s,1H),7.5(d,1H),7.12(d,1H),7.04(s,2H),6.72–6.61(m,1H),6.23–6.15(m,1H),5.7,5(d,1H),4.12(s,2H),3.86(s,3H),3.34(s,4H),3.20–3.17(t,2H),2.86(s,6H),2.66(s,3H),2.01(s,2H),1.86(s,2H).1H NMR (300MHz, DMSO-d6) δ: 9.63 (s, 1H), 9.47 (s, 1H), 8.3 - 8.30 (d, 2H), 7.85 (s, 1H), 7.5 (d, 1H), 7.12 ( d, 1H), 7.04 (s, 2H), 6.72 - 6.61 (m, 1H), 6.23 - 6.15 (m, 1H), 5.7, 5 (d, 1H), 4.12 (s, 2H), 3.86 (s, 3H), 3.34 (s, 4H), 3.20–3.17 (t, 2H), 2.86 (s, 6H), 2.66 (s, 3H), 2.01 (s, 2H), 1.86 (s, 2H).
ESI-Ms m/z:575.2[M+H]。ESI-Ms m/z: 575.2 [M+H].
实施例11 N-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(6,7,8,9-四氢吡啶并[1,2-a]-5-甲氧基-吲哚-10-基)嘧啶-2-基)氨基)苯基)烯丙酰胺的合成Example 11 N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(6,7,8,9-tetra) Synthesis of Hydropyrido[1,2-a]-5-methoxy-indol-1-ylpyrimidin-2-yl)amino)phenyl)allylamide
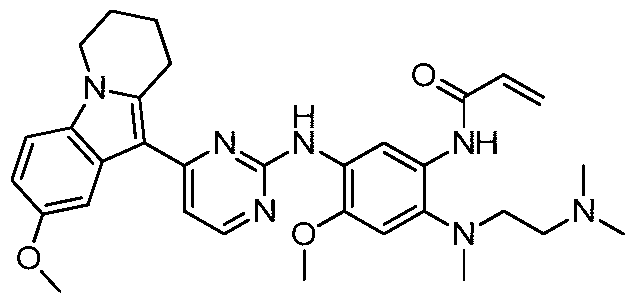
以4-氟-2-甲氧基-5-硝基苯胺、2,4-二氯嘧啶、5-甲氧基吲哚、1,4-二溴丁烷、N,N,N’-三 甲基乙二胺、烯丙基酰氯为原料,按照实施例1的方法制得标题化合物。4-fluoro-2-methoxy-5-nitroaniline, 2,4-dichloropyrimidine, 5-methoxyindole, 1,4-dibromobutane, N,N,N'-three The title compound was obtained according to the method of Example 1 using methylethylenediamine and allyl chloride as starting materials.
1H NMR(300MHz,DMSO-d6)δ:10.02(s,1H),9.53(s,1H),8.40–8.45(d,2H),7.92(s,1H),7.52(d,1H),7.18(d,1H),7.10(s,2H),6.76–6.70(m,1H),6.28–6.20(m,1H)5.72(d,1H),4.12(s,2H),3.85(s,3H),3.72(s,3H),3.32(s,4H),3.21–3.19(t,2H),2.83(s,6H),2.65(s,3H),2.02(s,2H),1.86(s,2H).1H NMR (300MHz, DMSO-d6) δ: 10.02 (s, 1H), 9.53 (s, 1H), 8.40 - 8.45 (d, 2H), 7.92 (s, 1H), 7.52 (d, 1H), 7.18 ( d, 1H), 7.10 (s, 2H), 6.76 - 6.70 (m, 1H), 6.28 - 6.20 (m, 1H) 5.72 (d, 1H), 4.12 (s, 2H), 3.85 (s, 3H), 3.72 (s, 3H), 3.32 (s, 4H), 3.21 - 3.19 (t, 2H), 2.83 (s, 6H), 2.65 (s, 3H), 2.02 (s, 2H), 1.86 (s, 2H) .
ESI-Ms m/z:570.4[M+H]。ESI-Ms m/z: 570.4 [M+H].
实施例12 N-(2-((2-(二甲基氨基)乙基)(甲基)氨基-4-甲氧基-5-((4-(1,1a,2,8b-四氢环丙基[3,4]吡咯并[1,2-a]吲哚-8-基)嘧啶-2-基)氨基)苯基)丙烯酰胺的合成Example 12 N-(2-((2-(Dimethylamino)ethyl)(methyl)amino-4-methoxy-5-((4-(1,1a,2,8b-tetrahydro) Synthesis of cyclopropyl[3,4]pyrrolo[1,2-a]indole-8-yl)pyrimidin-2-yl)amino)phenyl)acrylamide

步骤a 1-烯丙基-1H-吲哚-2-甲醛的合成:Step a 1- Synthesis of 1-allyl-1H-indole-2-carbaldehyde:

在100mL双口瓶中,加入25ml无水DMF,冷却至0-5℃,缓慢加入662mg NaH,搅拌10min。将10mL溶有2g 1H-吲哚-2-甲醛的无水DMF溶液缓慢滴入上述反应液中,气泡产生,溶液颜色变为暗黑色,持续搅拌15min,然后缓慢加入5mL溶有1.82g 3-溴丙烯的无水DMF溶液,室温反应3h,加入50mL水,3X 60ml乙酸乙酯萃取,浓缩,干燥,柱层析得标题物2.0g。In a 100 mL two-necked flask, 25 ml of anhydrous DMF was added, cooled to 0-5 ° C, 662 mg of NaH was slowly added, and stirred for 10 min. 10 mL of an anhydrous DMF solution in which 2 g of 1H-indole-2-carbaldehyde was dissolved was slowly dropped into the above reaction solution, bubbles were generated, the color of the solution became dark black, stirring was continued for 15 min, and then 5 mL was slowly added to dissolve 1.82 g of 3- Anhydrous DMF solution of bromopropene was reacted at room temperature for 3 h, added with 50 mL of water, EtOAc (EtOAc)EtOAc.
ESI-Ms m/z:186.0[M+H]ESI-Ms m/z: 186.0 [M+H]
步骤b:N-((1-丙烯基-1H-吲哚-2-基)甲烯基)-4-甲基苯磺酰肼的合成:Step b: Synthesis of N-((1-propenyl-1H-indol-2-yl)methenyl)-4-methylbenzenesulfonylhydrazide:
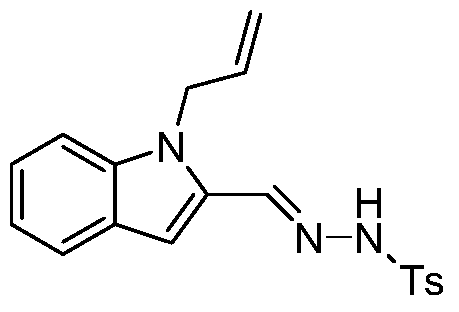
在100mL单口瓶中,依次加入步骤a所得1-烯丙基-1H-吲哚-2-甲醛(1.85g,10mmol)、对甲基苯磺酰肼(1.86g、10mmol)、50mL甲醇,油浴85℃回流反应2h。浓缩得标题粗产物3.6g,直接用于下一步。In a 100 mL single-mouth bottle, 1-allyl-1H-indole-2-carbaldehyde (1.85 g, 10 mmol) obtained in Step a, p-toluenesulfonylhydrazide (1.86 g, 10 mmol), 50 mL of methanol, oil The reaction was refluxed at 85 ° C for 2 h. Concentration gave the title crude product 3.6 g, which was used directly next.
步骤c:1,1a,2,8b-四氢环丙基[3,4]吡咯并[1,2-a]吲哚的合成:Step c: Synthesis of 1,1,2,8b-tetrahydrocyclopropyl[3,4]pyrrolo[1,2-a]indole:

在250mL三口瓶中,加入步骤b所得N-((1-丙烯基-1H-吲哚-2-基)甲烯基)-4-甲基苯磺酰肼(2.2g,6.23mmol)、碳酸钾(1.29g、9.35mmol)、60ml二氧六环,油浴100℃回流反应12h。过滤,浓缩,加入50ml水,3X 40ml乙醇萃取,30ml水洗,浓缩,柱层析得1.0g标题产物, 直接用于下一步。In a 250 mL three-necked flask, N-((1-propenyl-1H-indol-2-yl)methenyl)-4-methylbenzenesulfonyl hydrazide (2.2 g, 6.23 mmol) obtained in step b was added, and carbonic acid was added. Potassium (1.29 g, 9.35 mmol) and 60 ml of dioxane were refluxed in an oil bath at 100 ° C for 12 h. Filtration, concentration, addition of 50 ml of water, extraction with 3X 40 ml of ethanol, washing with 30 ml of water, concentration and column chromatography to give 1.0 g of the title product. Used directly in the next step.
步骤d:N-(2-((2-(二甲基氨基)乙基)(甲基)氨基-4-甲氧基-5-((4(1,1a,2,8b-四氢环丙基[3,4]吡咯并[1,2-a]吲哚-8-基)嘧啶-2-基)氨基)苯基)丙烯酰胺的合成Step d: N-(2-((2-(Dimethylamino)ethyl)(methyl)amino-4-methoxy-5-((4(1,1a,2,8b-tetrahydrocyclo)) Synthesis of propyl[3,4]pyrrolo[1,2-a]indole-8-yl)pyrimidin-2-yl)amino)phenyl)acrylamide
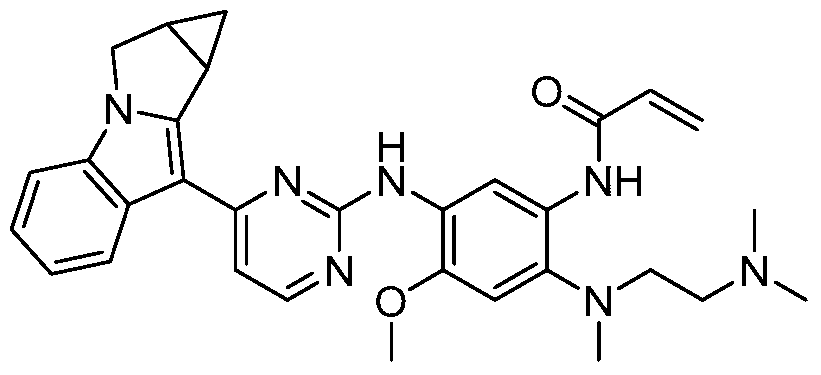
以4-氟-2-甲氧基-5-硝基苯胺、2,4-二氯嘧啶、步骤c所得1,1a,2,8b-四氢环丙基[3,4]吡咯并[1,2-a]吲哚、N,N,N’-三甲基乙二胺和烯丙基酰氯为原料,按照实施例1合成方法制得标题化合物。1,1a,2,8b-tetrahydrocyclopropyl[3,4]pyrrolo[1,4-fluoro-2-methoxy-5-nitroaniline, 2,4-dichloropyrimidine, step c The title compound was obtained according to the procedure of Example 1 using 2-, a, N, N, N'-trimethylethylenediamine and allyl chloride as starting materials.
1H-NMR(300MHz,DMSO-d6)δ:10.03(s,1H),8.70(s,1H),8.32-8.33(d,1H),8.24-8.26(d,1H),8.01(s,1H),7.21-7.28(m,2H),7.10-7.13(m,1H),7.03-7.06(m,2H),6.41-6.47(m,1H),6.20-6.23(d,1H),5.74-5.76(d,1H),4.19-4.28(m,2H),3.86(s,3H),2.97(s,2H),2.95(s,2H),2.74(s,3H),2.29-2.5(m,8H),1.44-1.45(m,1H),0.70-0.71(m,1H)。 1 H-NMR (300MHz, DMSO -d 6) δ: 10.03 (s, 1H), 8.70 (s, 1H), 8.32-8.33 (d, 1H), 8.24-8.26 (d, 1H), 8.01 (s, 1H), 7.21-7.28 (m, 2H), 7.10-7.13 (m, 1H), 7.03-7.06 (m, 2H), 6.41-6.47 (m, 1H), 6.20-6.23 (d, 1H), 5.74 5.76 (d, 1H), 4.19-4.28 (m, 2H), 3.86 (s, 3H), 2.97 (s, 2H), 2.95 (s, 2H), 2.74 (s, 3H), 2.29-2.5 (m, 8H), 1.44-1.45 (m, 1H), 0.70-0.71 (m, 1H).
ESI-Ms m/z:537.9[M+H]ESI-Ms m/z: 537.9 [M+H]
实施例13 N-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(6-氧代-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)-嘧啶-2-基)氨基)苯基)烯丙酰胺的合成Example 13 N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(6-oxo-6,7, Synthesis of 8,9-tetrahydropyrido[1,2-a]indole-10-yl)-pyrimidin-2-yl)amino)phenyl)allylamide
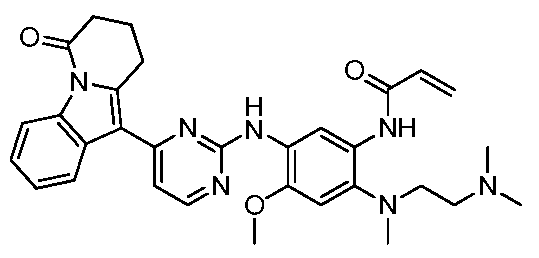
ESI-Ms m/z:554.4[M+H]ESI-Ms m/z: 554.4 [M+H]
实施例14 N-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(6-氟-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)-嘧啶-2-基)氨基)苯基)烯丙酰胺的合成Example 14 N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(6-fluoro-6,7,8) Synthesis of 9-tetrahydropyrido[1,2-a]indol-10-yl)-pyrimidin-2-yl)amino)phenyl)allylamide

ESI-Ms m/z:558.3[M+H]ESI-Ms m/z: 558.3 [M+H]
实施例15 N-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(9-氟-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)-嘧啶-2-基)氨基)苯基)烯丙酰胺的合成Example 15 N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(9-fluoro-6,7,8) Synthesis of 9-tetrahydropyrido[1,2-a]indol-10-yl)-pyrimidin-2-yl)amino)phenyl)allylamide

ESI-Ms m/z:558.5[M+H]ESI-Ms m/z: 558.5 [M+H]
实施例16 N-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(9,9-二氟-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)-嘧啶-2-基)氨基)苯基)烯丙酰胺的合成 Example 16 N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(9,9-difluoro-6, Synthesis of 7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)-pyrimidin-2-yl)amino)phenyl)allylamide
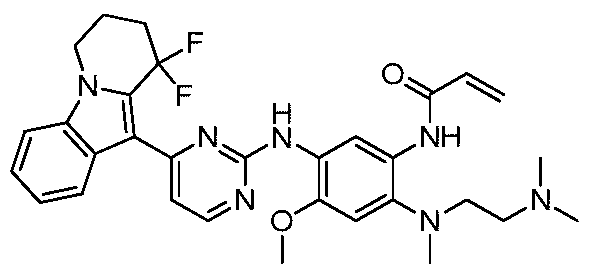
ESI-Ms m/z:575.3[M+H]ESI-Ms m/z: 575.3 [M+H]
实施例17 N-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(1,2-二氢氮杂环丁二烯并[1,2-a]吲哚-8-基)-嘧啶-2-基)氨基)苯基)烯丙酰胺的合成Example 17 N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(1,2-dihydronitrogen) Synthesis of butadieno[1,2-a]indol-8-yl)-pyrimidin-2-yl)amino)phenyl)allylamide
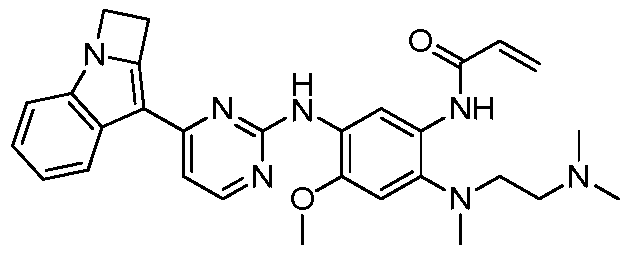
ESI-Ms m/z:511.6[M+H]ESI-Ms m/z: 511.6 [M+H]
实施例18 N-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(1-氟-2,3-二氢-1H-吡咯并[1,2-a]-吲哚-9-基)嘧啶-2-基)氨基)苯基)烯丙酰胺的合成Example 18 N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(1-fluoro-2,3-di) Synthesis of Hydrogen-1H-pyrrolo[1,2-a]-indol-9-yl)pyrimidin-2-yl)amino)phenyl)propanamide
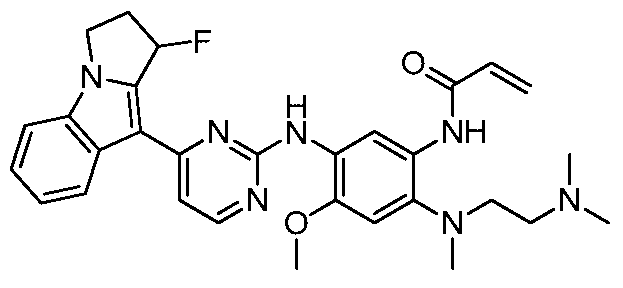
ESI-Ms m/z:543.8[M+H]ESI-Ms m/z: 543.8 [M+H]
实施例19 N-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(2-氧代-1,2-二氢氮杂环丁二烯并[1,2-a]吲哚-8-基)-嘧啶-2-基)氨基)苯基)烯丙酰胺的合成Example 19 N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(2-oxo-1,2-) Synthesis of dihydroazetidino[1,2-a]indol-8-yl)-pyrimidin-2-yl)amino)phenyl)allylamide

ESI-Ms m/z:525.4[M+H]ESI-Ms m/z: 525.4 [M+H]
实施例20 N-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(1,1-二氧代-3,4-二氢-2H-[1,2]噻嗪[2,3-a]吲哚-5-基)嘧啶-2-基)氨基)苯基)烯丙酰胺的合成Example 20 N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(1,1-dioxo-3) Synthesis of 4-dihydro-2H-[1,2]thiazine[2,3-a]indol-5-yl)pyrimidin-2-yl)amino)phenyl)propanamide
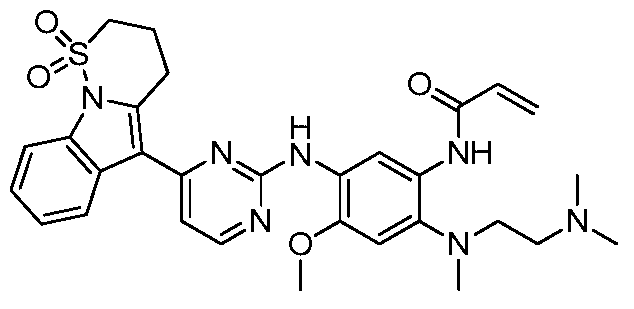
ESI-Ms m/z:590.4[M+H]ESI-Ms m/z: 590.4 [M+H]
实施例21 N-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(2,3-二甲基-1,2,3,4-四氢吡嗪[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)烯丙酰胺的合成 Example 21 N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(2,3-dimethyl-1) Synthesis of 2,3,4-tetrahydropyrazine[1,2-a]indol-10-ylpyrimidin-2-yl)amino)phenyl)allylamide

ESI-Ms m/z:569.5[M+H]ESI-Ms m/z: 569.5 [M+H]
实施例22 N-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(1-氧代-1,2,3,4-四氢吡嗪[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)烯丙酰胺的合成Example 22 N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(1-oxo-1,2, Synthesis of 3,4-tetrahydropyrazine[1,2-a]indol-10-ylpyrimidin-2-yl)amino)phenyl)allylamide

ESI-Ms m/z:555.7[M+H]ESI-Ms m/z: 555.7 [M+H]
实施例23 N-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(2-甲基-1,2,3,4-四氢吡嗪[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)烯丙酰胺的合成Example 23 N-(2-((2-(Dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(2-methyl-1,2, Synthesis of 3,4-tetrahydropyrazine[1,2-a]indol-10-ylpyrimidin-2-yl)amino)phenyl)allylamide
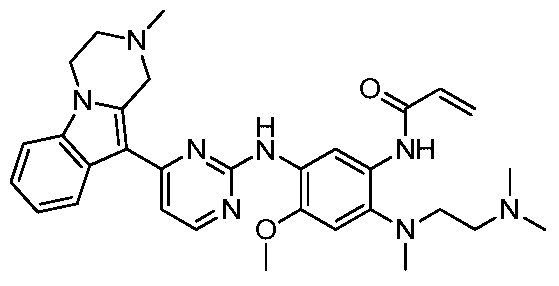
ESI-Ms m/z:554.8[M+H]ESI-Ms m/z: 554.8 [M+H]
实验例1体外激酶活性评价Experimental Example 1 Evaluation of in vitro kinase activity
1实验材料1 experimental material
1.1酶1.1 enzyme
EGFRWT激酶,购于Carna公司;EGFR WT kinase, purchased from Carna Corporation;
EGFRT790M/L858R激酶,购于Invitrogen公司。EGFR T790M/L858R kinase was purchased from Invitrogen.
1.2试剂1.2 reagent
三磷酸腺苷(ATP),购于Sigma公司;Adenosine triphosphate (ATP), purchased from Sigma;
缩氨酸(Peptide FAM-P22),购于GL Biochem公司;Peptide (Peptide FAM-P22), purchased from GL Biochem;
乙二胺四乙酸(EDTA),购于Sigma公司。Ethylenediaminetetraacetic acid (EDTA) was purchased from Sigma.
1.3仪器1.3 instruments
Caliper EZ reader微流控芯片仪器,购于Caliper Life Sciences,Inc.Caliper EZ reader microfluidic chip instrument, purchased from Caliper Life Sciences, Inc.
2实验方法2 experimental methods
2.1准备1×激酶基础缓冲液和终止缓冲液2.1 Preparation of 1× Kinase Base Buffer and Stop Buffer
1×激酶基础缓冲液(对于EGFRWT):50mM HEPES,pH7.5,0.0015%Brij-35,10mM MgCl2,10mM MnCl2,2mM DTT;1×kinase base buffer (for EGFR WT ): 50 mM HEPES, pH 7.5, 0.0015% Brij-35, 10 mM MgCl 2 , 10 mM MnCl 2 , 2 mM DTT;
1×激酶基础缓冲液(对于EGFRT790M/L858R):50mM HEPES,pH7.5,0.0015%Brij-35,10mM MgCl2,2mM DTT; 1×kinase base buffer (for EGFR T790M/L858R ): 50 mM HEPES, pH 7.5, 0.0015% Brij-35, 10 mM MgCl 2 , 2 mM DTT;
终止缓冲液:100mM HEPES,pH7.5,0.0015%Brij-35,0.2%Coating Reagent#3,50mM EDTA。Stop buffer: 100 mM HEPES, pH 7.5, 0.0015% Brij-35, 0.2% Coating Reagent #3, 50 mM EDTA.
2.2准备化合物2.2 Preparation of compounds
用100%DMSO将本发明的化合物分别溶解至10mM,再用完全培养基稀释至50μM,然后用含0.1%DMSO的完全培养基稀释至5μM后,依次3倍稀释,共10个浓度(对于EGFRWT);The compounds of the present invention were separately dissolved in 10 mM with 100% DMSO, diluted to 50 μM with complete medium, and then diluted to 5 μM with complete medium containing 0.1% DMSO, followed by 3-fold dilution for a total of 10 concentrations (for EGFR WT );
用100%DMSO将本发明的化合物分别溶解至10mM,再用完全培养基稀释至50μM,然后用含0.1%DMSO的完全培养基稀释至1μM后,依次3倍稀释,共10个浓度(对于EGFRT790M/L858R);The compound of the present invention was separately dissolved to 10 mM with 100% DMSO, diluted to 50 μM with complete medium, and then diluted to 1 μM with complete medium containing 0.1% DMSO, and then diluted 3 times in total for 10 concentrations (for EGFR T790M/L858R );
在空的孔中加入100μl 100%DMSO用于配制有激酶无化合物对照组和无激酶无化合物对照组;100 μl of 100% DMSO was added to the empty wells for the preparation of a kinase-free compound control group and a non-kinase-free compound control group;
标记以上所用96孔板为来源板。The 96-well plate used above was labeled as the source plate.
2.3准备中间板2.3 Preparing the middle board
从来源板中转移10μl溶液到新的96孔板中,作为中间板,在中间板每孔中加入90μl 1×激酶缓冲液,振荡混匀10min。10 μl of the solution was transferred from the source plate into a new 96-well plate as an intermediate plate, and 90 μl of 1×kinase buffer was added to each well of the intermediate plate, and vortexed and mixed for 10 minutes.
2.4准备实验板2.4 Preparing the experiment board
从96孔中间板中,每孔转移5μl溶液到384孔板中。From the 96-well intermediate plate, transfer 5 μl of solution to each well into a 384-well plate.
2.5激酶反应2.5 kinase reaction
2.5.1.准备2.5×激酶溶液:将EGFRWT激酶和EGFRT790M/L858R激酶原液分别加入1×基础缓冲液中,配制成2.5×激酶溶液;2.5.1. Preparation of 2.5×kinase solution: EGFR WT kinase and EGFR T790M/L858R kinase stock solution were separately added to 1×basal buffer to prepare 2.5×kinase solution;
2.5.2.准备2.5×缩氨酸溶液:将FAM标记的缩氨酸和ATP加到1×基础缓冲液中,配制成2.5×缩氨酸溶液;2.5.2. Preparing a 2.5× peptide solution: adding FAM-labeled peptide and ATP to 1× base buffer to prepare a 2.5× peptide solution;
2.5.3.转移10μl 2.5×激酶溶液到384孔实验板中,室温孵育10min;2.5.3. Transfer 10 μl of 2.5× kinase solution to a 384-well assay plate and incubate for 10 min at room temperature;
2.5.4.转移10μl 2.5×缩氨酸溶液到384孔实验板中,在28℃条件下孵育一段时间,加入25μl终止缓冲液终止反应。2.5.4. Transfer 10 μl of 2.5× peptide solution to a 384-well assay plate, incubate at 28 ° C for a period of time, and add 25 μl of stop buffer to stop the reaction.
同时设置无激酶无化合物对照组(包含DMSO、1×基础缓冲液和2.5×缩氨酸溶液和有激酶无化合物对照组(包括DMSO、2.5×激酶溶液和2.5×缩氨酸溶液)。A non-kinase-free compound control group (containing DMSO, 1×basal buffer and 2.5× peptide solution, and a kinase-free compound control group (including DMSO, 2.5×kinase solution, and 2.5×peptide solution) was also provided.
2.5.5.Caliper仪器读数、拟合曲线,计算抑制率2.5.5. Caliper instrument readings, fitting curves, calculation of inhibition rate
在Caliper仪器上读取数据,并从Caliper程序中获得conversion数据,根据以下公式计算抑制率:Read the data on the Caliper instrument and obtain the conversion data from the Caliper program and calculate the inhibition rate according to the following formula:
抑制率%=(max-com)/(max-min)×100计算抑制率,其中“max”代表有激酶无化合物对照组,“com”代表受试化合物组,“min”代表无激酶无化合物对照组。Inhibition rate %=(max-com)/(max-min)×100 Calculate the inhibition rate, where “max” represents a kinase-free compound control group, “com” represents a test compound group, and “min” represents a kinase-free compound. Control group.
2.5.6.采用Graphpad 5.0数据处理软件计算IC50值。结果见表1。2.5.6. Calculate IC 50 values using Graphpad 5.0 data processing software. The results are shown in Table 1.
表1Table 1


从实验结果可知,本发明的化合物对突变型EGFR激酶,例如EGFRL858R/T790M激酶具有良好的抑制活性,IC50值小于1nM,对野生型EGFR激酶影响小,具有较好的选择性。From the experimental results, the compound of the present invention has a good inhibitory activity against a mutant EGFR kinase such as EGFR L858R/T790M kinase, and has an IC 50 value of less than 1 nM, and has a small effect on wild-type EGFR kinase and has good selectivity.
实验例2体外细胞活性评价Experimental Example 2 Evaluation of in vitro cell viability
2.实验材料2. Experimental materials
1.1细胞和阳性对照药1.1 cells and positive control drugs
实验用细胞株NCI-H1975(EGFR双突变细胞,具有L858R和T790M突变)和A431(EGFR野生型细胞),购自于ATCC。The experimental cell line NCI-H1975 (EGFR double mutant cells with L858R and T790M mutations) and A431 (EGFR wild type cells) were purchased from ATCC.
使用阿斯利康公司生产的已上市高效选择性、不可逆性EGFR突变体抑制剂AZD9291作为阳性对照药,合成方法参见PCT/GB2012/051783(专利号WO 2013/014448)说明书实施例28,并通过氢谱和质谱确认。The commercially available highly potent and selective, irreversible EGFR mutant inhibitor AZD9291 produced by AstraZeneca was used as a positive control. For the synthesis method, see PCT/GB2012/051783 (Patent No. WO 2013/014448), in the specification example 28, and passed hydrogen. Spectral and mass spectrometric confirmation.
1.2试剂1.2 reagent
Cell Titer-Glo luminescent cell viability assay,购自于Promega公司;Cell Titer-Glo luminescent cell viability assay, purchased from Promega;
RPMI1640 medium,购自于Invitrogen公司;RPMI1640 medium, purchased from Invitrogen;
DMEM medium,购自于Invitrogen公司;DMEM medium, purchased from Invitrogen;
胎牛血清,购自于Invitrogen公司;Fetal bovine serum, purchased from Invitrogen;
DMSO,购自于Sigma公司;DMSO, purchased from Sigma;
NCI-H1975细胞培养于含10%灭活的胎牛血清(GIBCO)的RPMI1640培养基中,含青霉素100IU/mL和链霉素100μg/mL;NCI-H1975 cells were cultured in RPMI1640 medium containing 10% inactivated fetal bovine serum (GIBCO), containing penicillin 100 IU/mL and streptomycin 100 μg/mL;
A431细胞培养于含10%灭活的胎牛血清(GIBCO)的DMEM培养基中,含青霉素100IU/mL和链霉素100μg/mL。A431 cells were cultured in DMEM medium containing 10% inactivated fetal bovine serum (GIBCO) containing penicillin 100 IU/mL and streptomycin 100 μg/mL.
2实验方法2 experimental methods
2.1实验过程(CTG assay)2.1 Experimental procedure (CTG assay)
将对数生长期的NCI-H1975细胞和A431细胞消化后,吹打成单细胞悬液,接种于96孔培养板,每孔培养基100μL,每个细胞株各种3块96孔板,其中NCI-H1975细胞每孔接种3X 103个细胞,A431细胞每孔接种4X 103个细胞。将接种后的NCI-H1975细胞和A431细胞在5%CO2培养箱中培养16-24小时,待细胞贴壁后,按以下浓度要求加入受试化合物(化合物在NCI-H1975细胞上的最高测试浓度为4μM,3倍稀释,共9个浓度;在A431细胞上的最高测试浓度为10μM,3倍稀释,共9个浓度),在培养箱中再培养72小时。同时设置空白对照组(只有培养基,不加细胞和DMSO溶液)和DMSO对照组(培养基中加入细胞和0.5%的DMSO溶液)。加入100μL的CTG溶液,避光振荡2min,孵育10min。 After digesting NCI-H1975 cells and A431 cells in logarithmic growth phase, they were blown into single cell suspensions, seeded in 96-well culture plates, 100 μL per well medium, and each cell line was composed of 3 96-well plates. NCI-H1975 cells were seeded with 3 ×10 3 cells per well, and A431 cells were seeded with 4 ×10 3 cells per well. The inoculated NCI-H1975 cells and A431 cells were cultured in a 5% CO 2 incubator for 16-24 hours. After the cells were attached, the test compound was added at the following concentration (the highest test of the compound on NCI-H1975 cells). The concentration was 4 μM, 3 fold dilution, a total of 9 concentrations; the highest test concentration on A431 cells was 10 μM, 3 fold dilution, a total of 9 concentrations), and cultured for an additional 72 hours in an incubator. At the same time, a blank control group (only medium, no cell and DMSO solution) and a DMSO control group (cells and 0.5% DMSO solution were added to the medium). Add 100 μL of CTG solution, shake it for 2 min in the dark, and incubate for 10 min.
2.2读数,计算IC50值2.2 readings, calculate IC 50 values
将培养板放入![]()
![]()
抑制剂(%)=(1-(RLUcom-RLUblank)/(RLUDMSO–RLUblank))×100%,Inhibitor (%) = (1-(RLU com - RLU blank ) / (RLU DMSO - RLU blank )) × 100%,
其中RLUcom表示受试化合物组的吸光值,RLUblank表示空白对照组的吸光值,RLUDMSO表示DMSO对照组的吸光值,Where RLU com represents the absorbance of the test compound group, RLU blank represents the absorbance of the blank control group, and RLU DMSO represents the absorbance of the DMSO control group.
利用XLFit曲线拟合软件绘制药效抑制率曲线并计算IC50值,结果见表2。The XEFFit curve fitting software was used to plot the pharmacodynamic inhibition rate curve and calculate the IC 50 value. The results are shown in Table 2.
表2Table 2
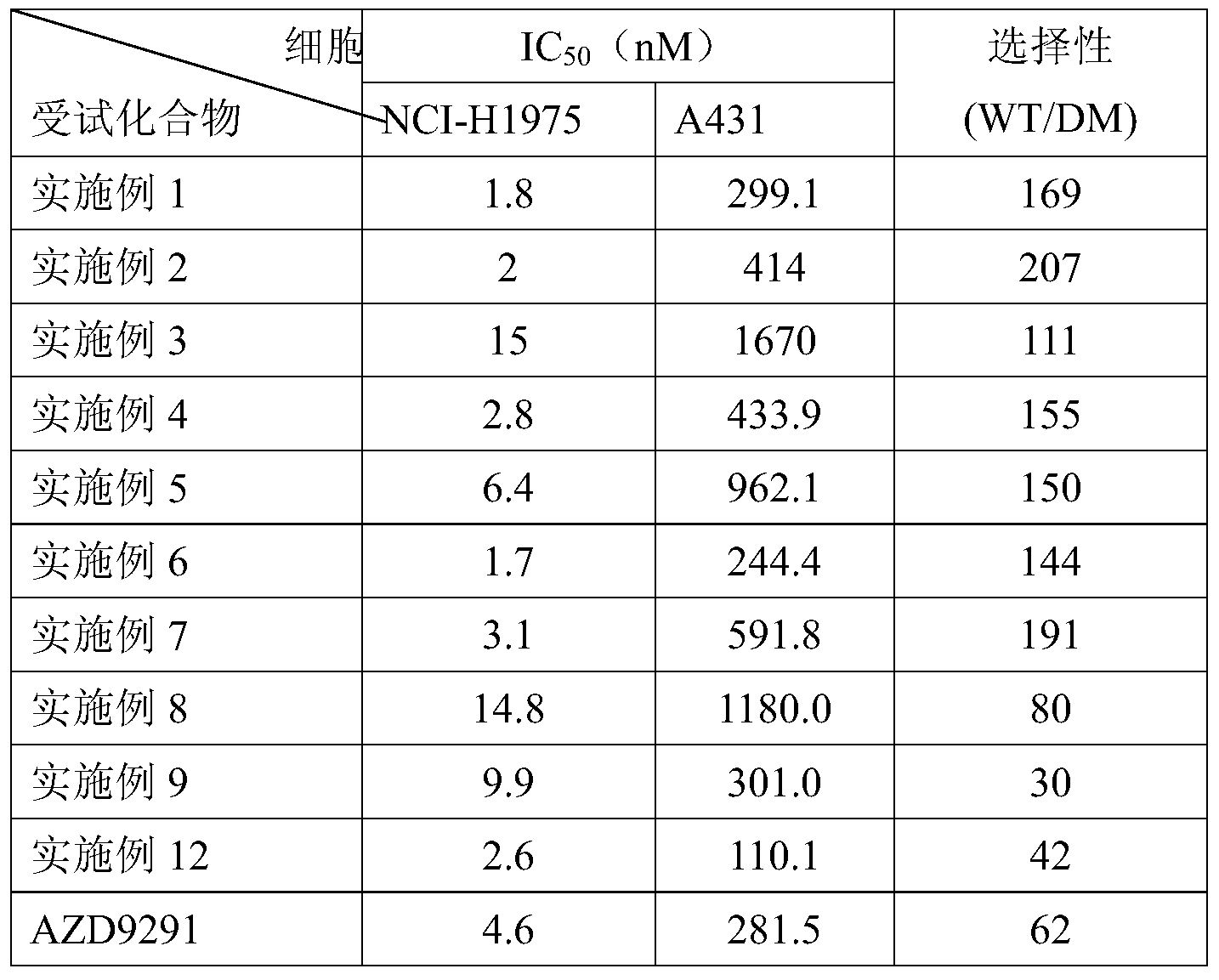
体外细胞实验结果表明,本发明的化合物对双突变型细胞(NCI-H1975)的抑制作用较好,且对EGFR野生型细胞(A431)的抑制小,选择性与AZD9291相似或明显优于AZD9291。已知目前上市的EGFR抑制剂的主要副作用之一为皮疹、腹泻等,这些都与野生型的EGFR被抑制,选择性差有关。因此,本发明的化合物有望成为对抗EGFR突变导致耐药的肿瘤具有特异疗效且副作用较小的药物。The results of in vitro cell experiments showed that the compounds of the present invention have a better inhibitory effect on double mutant cells (NCI-H1975), and have less inhibition on EGFR wild type cells (A431), and the selectivity is similar to or significantly superior to AZD9291. One of the major side effects of currently marketed EGFR inhibitors is rash, diarrhea, etc., which are associated with inhibition of wild-type EGFR and poor selectivity. Therefore, the compound of the present invention is expected to be a drug having a specific therapeutic effect against a tumor resistant to EGFR mutation and having a small side effect.
实验例3血糖影响检测Experimental Example 3 Blood Sugar Impact Test
1实验材料1 experimental material
1.1化合物1.1 compound
将本发明的化合物用25mM的枸橼酸-枸橼酸钠缓冲液(pH4.5)溶解,配制成浓度为1.25mg/mL的澄清溶液,设为受试化合物组;以25mM的枸橼酸-枸橼酸钠缓冲液(pH4.5)设为溶媒对照组;The compound of the present invention was dissolved in 25 mM sodium citrate-sodium citrate buffer (pH 4.5) to prepare a clear solution having a concentration of 1.25 mg/mL, and was set as a test compound group; 25 mM citric acid was used. - sodium citrate buffer (pH 4.5) was set as a vehicle control group;
1.2动物1.2 animals
雌性BALB/C小鼠,每组各5只,体重18-22g,上海西普尔-必凯实验动物有限公司提供。 受试小鼠实验前给予2~4天的环境适应期,给药前禁食8-12h。Female BALB/C mice, 5 in each group, weighing 18-22 g, were provided by Shanghai Xipuer-Beikai Experimental Animal Co., Ltd. The test mice were given an environmental adaptation period of 2 to 4 days before the experiment, and fasted for 8-12 hours before administration.
2实验方法2 experimental methods
2.1小鼠禁食但可自由饮水12小时;2.1 mice fasted but free to drink water for 12 hours;
2.2取步骤2.1中的小鼠5只,灌胃(intragastric administration,I.G.)给予本发明的化合物25mg/kg;2.2 taking 5 mice in step 2.1, intragastric administration (I.G.) administration of the compound of the invention 25mg / kg;
2.3于灌胃后0min,15min,30min,1h,2h眼眶采血,使用罗氏卓越型血糖仪检测动物血糖值A,根据公式AUC=[(A15min+A0)×7.5]+[(A30min+A15min)×7.5]+[(A60min+A30min)×15]+[(A120min+A60min)×30]计算AUC值,并根据公式AUC增长%=[(AUC化合物-AUC溶媒)/AUC溶媒]×100%计算出AUC增长率,其中“A0”代表灌胃后0min的血糖值,“A15min”代表灌胃15min后的血糖值,“A30min”代表灌胃30min后的血糖值,“A60min”代表灌胃60min后的血糖值,“A120min”代表灌胃120min后的血糖值,“AUC化合物”代表受试化合物的AUC值,“AUC溶媒”代表溶媒对照组的AUC值,结果见表3。2.3 Blood was collected from the eyelids at 0 min, 15 min, 30 min, 1 h, 2 h after gavage, and the blood glucose level A of the animals was measured using a Roche superior blood glucose meter according to the formula AUC=[(A 15min +A 0 )×7.5]+[(A 30min + A 15min )×7.5]+[(A 60min +A 30min )×15]+[(A 120min +A 60min )×30] Calculate the AUC value and increase %=[(AUC compound- AUC solvent ) according to the formula AUC/ AUC solvent ] × 100% calculated the AUC growth rate, where “A 0 ” represents the blood glucose level at 0 min after gavage, “A 15 min ” represents the blood glucose level after 15 min, and “A 30 min ” represents the blood glucose after 30 min. Value, "A 60min " represents the blood glucose level after 60 minutes of gavage, "A 120min " represents the blood glucose level after 120 minutes of gavage, "AUC compound " represents the AUC value of the test compound, and "AUC solvent " represents the AUC of the vehicle control group. Value, the results are shown in Table 3.
表3table 3

已知EGFR抑制剂可能会导致在体高血糖以及受损的胰岛素信号传导等副作用。根据实验报道,AZD9291和Clovis Oncology公司的Rociletinib(CO1686)对血糖会有一定程度的升高副作用(Management of hyperglycemia from epidermal growth factor receptor(EGFR)tyrosine kinase inhibitors(TKIs)targeting T790M-mediated resistance,Jeryl Villadolid,et al.Translational Lung Cancer Research(2015),4(5),576-583)。本发明的以上实验结果表明,本发明的化合物 对血糖影响较小,有望克服现有的EGFR抑制剂具有的高血糖副作用,从而有助于降低不良反应和提高患者依从性。It is known that EGFR inhibitors may cause side effects such as hyperglycemia and impaired insulin signaling. According to the experimental report, AZD9291 and Clociletinib (CO1686) of Clovis Oncology have a certain degree of side effects on blood glucose (Management of hyperglycemia from epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) targeting T790M-mediated resistance, Jeryl Villadolid , et al. Translational Lung Cancer Research (2015), 4(5), 576-583). The above experimental results of the present invention indicate that the compound of the present invention It has little effect on blood sugar and is expected to overcome the high blood sugar side effects of existing EGFR inhibitors, thereby helping to reduce adverse reactions and improve patient compliance.
实验例4药物代谢实验Experimental Example 4 Drug Metabolism Experiment
1实验材料1 experimental material
1.1化合物1.1 compound
将本发明的化合物配制成口服和注射两种给药配方。其中,口服药物配方为25mM的枸橼酸-枸橼酸钠缓冲液(pH4.5)溶解,制成1.25mg/mL澄清溶液;尾静脉注射药物配方为枸橼酸-枸橼酸钠缓冲液(pH4.5)与生理盐水体积比为1:1的混合溶液,制成0.2mg/mL溶液。The compounds of the invention are formulated into either oral and injectable formulations. Among them, the oral drug formulation was dissolved in 25 mM sodium citrate-sodium citrate buffer (pH 4.5) to prepare a 1.25 mg/mL clear solution; the tail vein injection drug formulation was citrate-sodium citrate buffer solution. A mixed solution of (pH 4.5) and physiological saline volume ratio of 1:1 was prepared into a 0.2 mg/mL solution.
1.2动物1.2 animals
雌性BALB/C小鼠,每组各5只,体重18-22g,上海西普尔-必凯实验动物有限公司提供。受试小鼠实验前给予2~4天的环境适应期,给药前禁食8-12h,给药2h后给水,4h后给食。Female BALB/C mice, 5 in each group, weighing 18-22 g, were provided by Shanghai Xipuer-Beikai Experimental Animal Co., Ltd. The test mice were given an environmental adaptation period of 2 to 4 days before the experiment, and were fasted for 8-12 hours before administration, and given water for 2 hours after administration, and fed after 4 hours.
1.3试剂1.3 reagent
甲醇(色谱纯):Spectrum公司生产;Methanol (chromatographically pure): produced by Spectrum;
乙腈(色谱纯):Spectrum公司生产;Acetonitrile (chromatographically pure): produced by Spectrum;
其余试剂均为市售分析纯。The remaining reagents were all commercially available analytical grades.
1.4仪器1.4 Instrument
美国AB公司API 4500型三重四级杆液质联用仪,配有电喷雾离子源(ESI),LC-30AD双泵;SIL-30AC自动进样器;CTO-30AC柱温箱;DGU-20A3R脱气机;Analyst QS A01.01色谱工作站;Milli-Q超纯水器(Millipore Inc);Qilinbeier Vortex-5振荡器;HITACHI CF16RⅩⅡ台式高速冷冻离心机。American AB company API 4500 triple quadrupole liquid-mass instrument with electrospray ion source (ESI), LC-30AD double pump; SIL-30AC autosampler; CTO-30AC column thermostat; DGU-20A3R Degasser; Analyst QS A01.01 chromatography workstation; Milli-Q ultrapure water (Millipore Inc); Qilinbeier Vortex-5 oscillator; HITACHI CF16RXII desktop high speed refrigerated centrifuge.
2实验方法2 experimental methods
2.1小鼠禁食但可自由饮水12小时后,采取0时刻空白血浆;2.1 mice fasted but can drink water for 12 hours, take 0 time blank plasma;
2.2取步骤2.1中的小鼠5只,灌胃(intragastric administration,I.G.)给予本发明化合物25mg/kg;2.2 taking 5 mice in step 2.1, intragastric administration (I.G.) administration of the compound of the invention 25mg / kg;
取步骤2.1中的小鼠5只,尾静脉(Intravenousadministration,I.V.)给予本发明的化合物2mg/kg;5 mice in step 2.1, the intravenous vein (Intravenous administration, I.V.) administered the compound of the invention 2mg / kg;
2.3于灌胃后5min,15min,30min,1h,2h,4h,10h,24h,从眼底静脉丛连续取血置于分布有肝素的EP管中,8000rpm/min离心5min后取上层血浆,-20℃冻存,待LC-MS/MS分析;2.3 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 10 h, 24 h after intragastric administration, blood was continuously taken from the fundus venous plexus in an EP tube with heparin distributed, and centrifuged at 8000 rpm/min for 5 min to obtain upper plasma, -20 Cryopreservation at °C until LC-MS/MS analysis;
于尾静脉注射给药后5min,15min,30min,1h,2h,4h,10h,24h,从眼底静脉丛连续取血置于分布有肝素的EP管中,8000rpm/min离心5min后取上层血浆,-20℃冻存,待LC-MS/MS分析;5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 10 h, 24 h after tail vein injection, continuous blood was taken from the fundus venous plexus in an EP tube with heparin distributed, and the supernatant plasma was taken after centrifugation at 8000 rpm/min for 5 min. Cryopreservation at -20 ° C until LC-MS/MS analysis;
2.4根据步骤2.3所得的血药浓度-时间数据,采用WinNonlin软件求算药代动力学参数,见表4。 2.4 According to the plasma concentration-time data obtained in step 2.3, the pharmacokinetic parameters were calculated using WinNonlin software, as shown in Table 4.
表4Table 4
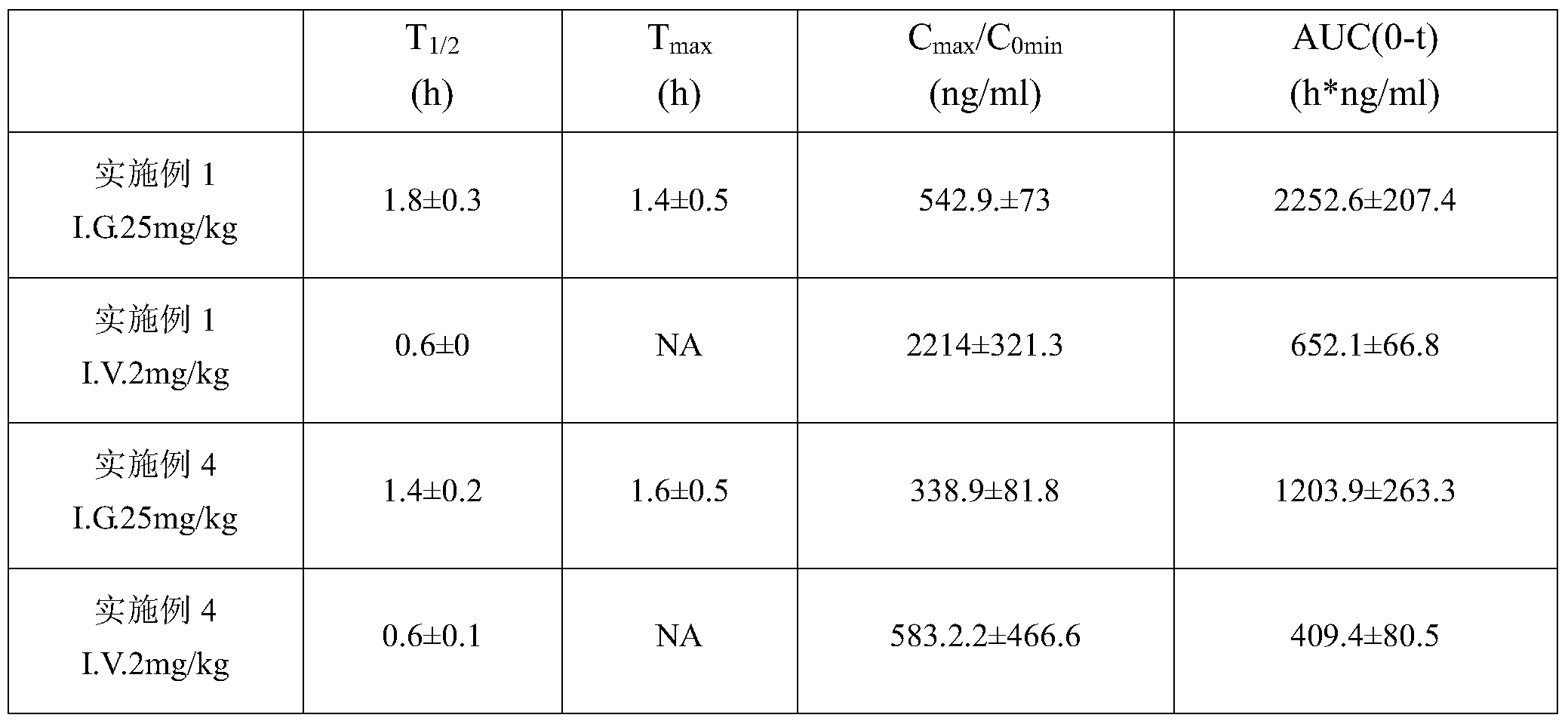
本发明的化合物具有较好的药代动力学数据,应具有较好的临床应用前景。The compounds of the invention have good pharmacokinetic data and should have good clinical application prospects.
尽管以上已经对本发明作了详细描述,但是本领域技术人员理解,在不偏离本发明的精神和范围的前提下可以对本发明进行各种修改和改变。本发明的权利范围并不限于上文所作的详细描述,而应归属于权利要求书。 While the invention has been described hereinabove, it will be understood by those skilled in the art that various modifications and changes of the invention may be made without departing from the spirit and scope of the invention. The scope of the invention is not limited to the details described above, but rather to the claims.
Claims (10)
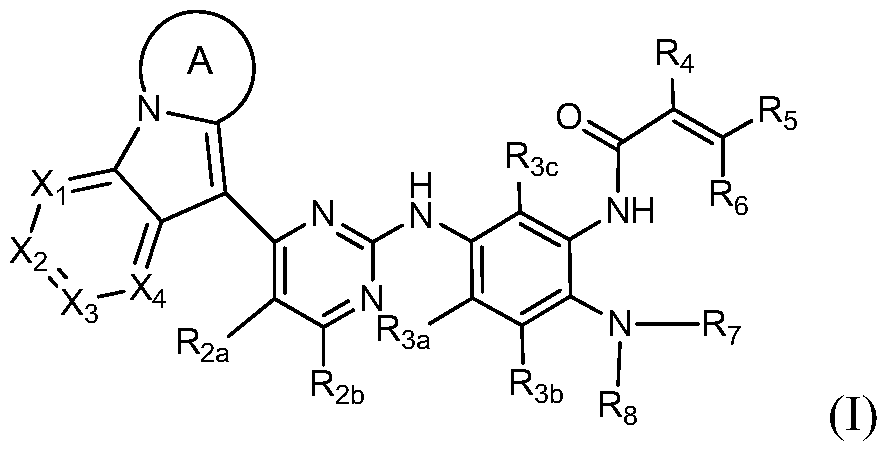

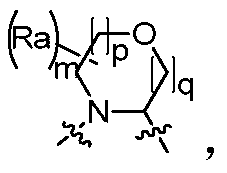
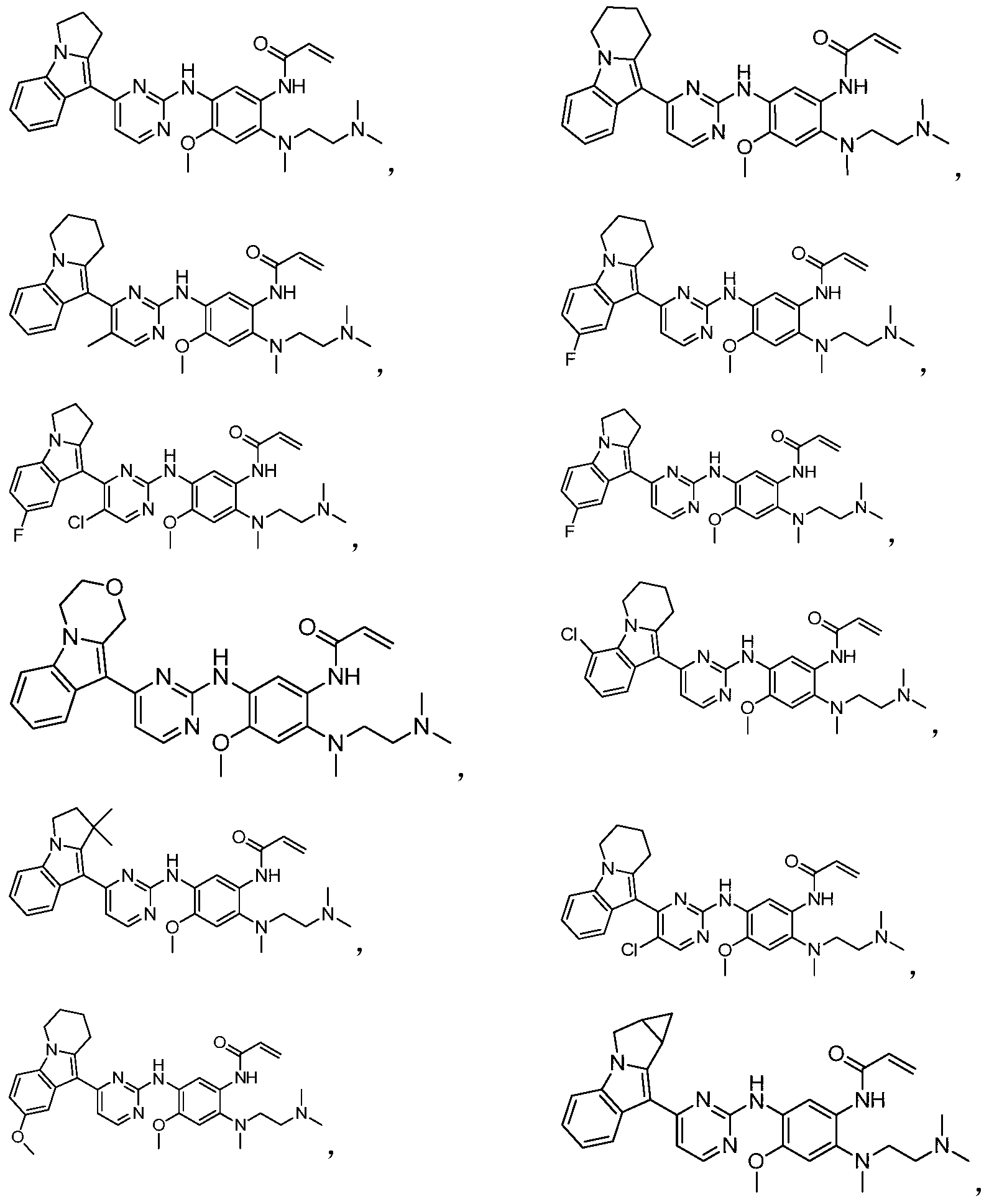
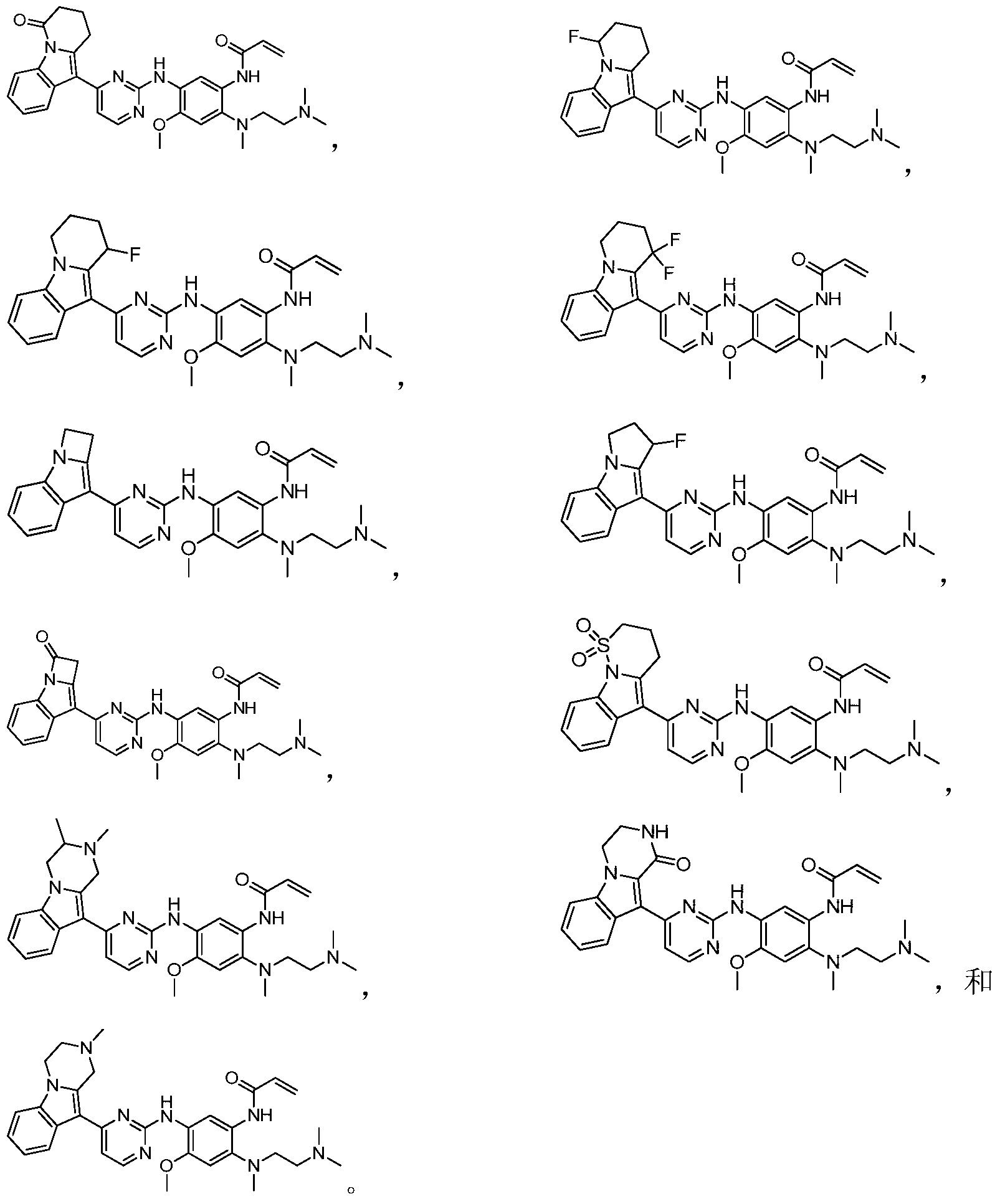
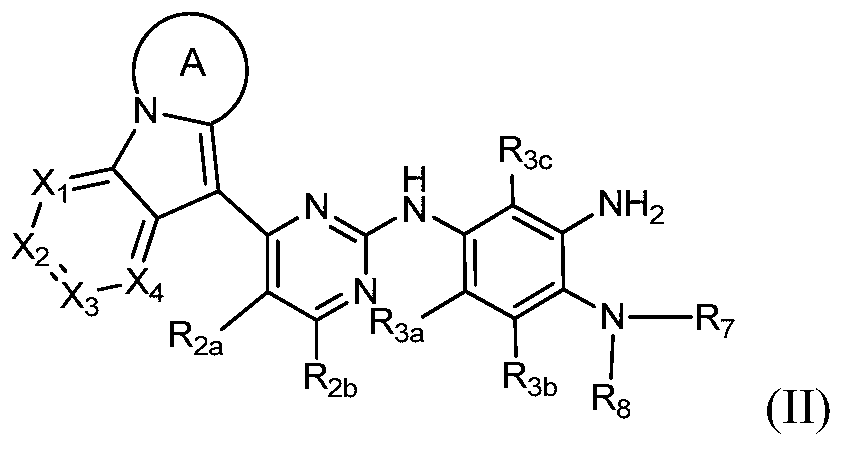

Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201510817624.7 | 2015-11-23 | ||
| CN201510817624 | 2015-11-23 | ||
| CN201610080274.5 | 2016-02-04 | ||
| CN201610080274 | 2016-02-04 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017088746A1 true WO2017088746A1 (en) | 2017-06-01 |
Family
ID=58763786
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2016/106862 Ceased WO2017088746A1 (en) | 2015-11-23 | 2016-11-23 | New epidermal growth factor receptor inhibitor and application thereof |
Country Status (3)
| Country | Link |
|---|---|
| CN (1) | CN106749267B (en) |
| TW (1) | TW201718583A (en) |
| WO (1) | WO2017088746A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3290419A4 (en) * | 2015-04-29 | 2018-04-18 | Guangdong Zhongsheng Pharmaceutical Co., Ltd | Fused-ring or tricyclic aryl pyrimidine compound used as kinase inhibitor |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108602802B (en) | 2016-07-26 | 2020-01-21 | 深圳市塔吉瑞生物医药有限公司 | Aminopyrimidines for Inhibiting Protein Tyrosine Kinase Activity |
| CN107663208B (en) * | 2016-07-27 | 2021-10-15 | 南京圣和药业股份有限公司 | Medicinal salt of novel EGFR kinase inhibitor and preparation method and application thereof |
| CN109705118B (en) * | 2017-10-25 | 2021-12-28 | 南京圣和药业股份有限公司 | Preparation method of tricyclic EGFR kinase inhibitor |
| CN109705117A (en) * | 2017-10-25 | 2019-05-03 | 南京圣和药业股份有限公司 | Tricyclic compounds, preparation method and the usage |
| CN108191861B (en) * | 2018-03-01 | 2020-10-02 | 天津大学 | N-[5-(Pyrimidine-2-amino)-2,4-disubstituted phenyl]-trans-2,4-pentadienamide |
| US20220162205A1 (en) * | 2019-03-12 | 2022-05-26 | Merck Patent Gmbh | Materials for organic electroluminescent devices |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103570731A (en) * | 2012-07-30 | 2014-02-12 | 中国科学院广州生物医药与健康研究院 | Pyrimidotricyclic compounds or pyrimidotetracyclic compounds, pharmaceutical composition and applications thereof |
| CN103702990A (en) * | 2011-07-27 | 2014-04-02 | 阿斯利康(瑞典)有限公司 | 2-(2,4,5-substituted aniline)pyrimidine derivatives as EGFR tuners for the treatment of cancer |
| WO2015003658A1 (en) * | 2013-07-11 | 2015-01-15 | Betta Pharmaceuticals Co., Ltd | Protein tyrosine kinase modulators and methods of use |
| WO2016173438A1 (en) * | 2015-04-29 | 2016-11-03 | 南京明德新药研发股份有限公司 | Fused-ring or tricyclic aryl pyrimidine compound used as kinase inhibitor |
-
2016
- 2016-11-22 CN CN201611025615.5A patent/CN106749267B/en active Active
- 2016-11-22 TW TW105138258A patent/TW201718583A/en unknown
- 2016-11-23 WO PCT/CN2016/106862 patent/WO2017088746A1/en not_active Ceased
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103702990A (en) * | 2011-07-27 | 2014-04-02 | 阿斯利康(瑞典)有限公司 | 2-(2,4,5-substituted aniline)pyrimidine derivatives as EGFR tuners for the treatment of cancer |
| CN103570731A (en) * | 2012-07-30 | 2014-02-12 | 中国科学院广州生物医药与健康研究院 | Pyrimidotricyclic compounds or pyrimidotetracyclic compounds, pharmaceutical composition and applications thereof |
| WO2015003658A1 (en) * | 2013-07-11 | 2015-01-15 | Betta Pharmaceuticals Co., Ltd | Protein tyrosine kinase modulators and methods of use |
| WO2016173438A1 (en) * | 2015-04-29 | 2016-11-03 | 南京明德新药研发股份有限公司 | Fused-ring or tricyclic aryl pyrimidine compound used as kinase inhibitor |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3290419A4 (en) * | 2015-04-29 | 2018-04-18 | Guangdong Zhongsheng Pharmaceutical Co., Ltd | Fused-ring or tricyclic aryl pyrimidine compound used as kinase inhibitor |
| EP3312179A1 (en) * | 2015-04-29 | 2018-04-25 | Guangdong Zhongsheng Pharmaceutical Co., Ltd | Fused-ring or tricyclic aryl pyrimidine compound used as kinase inhibitor |
| US10093656B2 (en) | 2015-04-29 | 2018-10-09 | Nanjing Sanhome Pharmaceutical Co., Ltd | Fused-ring or tricyclic aryl pyrimidine compound used as kinase inhibitor |
| US10494373B2 (en) | 2015-04-29 | 2019-12-03 | Guangdong Zhongsheng Pharmaceutical Co., Ltd | Fused-ring or tricyclic aryl pyrimidine compound used as kinase inhibitor |
Also Published As
| Publication number | Publication date |
|---|---|
| CN106749267B (en) | 2020-08-21 |
| CN106749267A (en) | 2017-05-31 |
| TW201718583A (en) | 2017-06-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN113993590B (en) | New EGFR inhibitors | |
| WO2017088746A1 (en) | New epidermal growth factor receptor inhibitor and application thereof | |
| JP6035423B2 (en) | Novel condensed pyrimidine compound or salt thereof | |
| WO2021088945A1 (en) | Compound as shp2 inhibitor and use thereof | |
| JP7191799B2 (en) | Pyrimidine compounds and pharmaceutical uses thereof | |
| CN111285886B (en) | Substituted pyrazolo [1,5-a ] pyridine compounds, compositions comprising the same and uses thereof | |
| WO2015158310A1 (en) | Tyrosine kinase inhibitor and uses thereof | |
| WO2018205938A1 (en) | Parp inhibitor, pharmaceutical composition, preparation method and use thereof | |
| CN106029659A (en) | glutaminase inhibitors | |
| CN101723936A (en) | Kinase suppressor and pharmaceutical application thereof | |
| WO2022135432A1 (en) | Macrocyclic heterocyclic compounds as egfr inhibitors, and use thereof | |
| WO2019034128A1 (en) | Pyrrolotriazine derivative, preparation method and use thereof | |
| CN106699743A (en) | Pyrimidine derivative and application thereof | |
| CN109970745A (en) | Substituted pyrrolo-triazine class compound and its medical composition and its use | |
| WO2024067744A1 (en) | Heterocyclic substituted quinazoline, preparation method therefor, and use thereof | |
| WO2015058661A1 (en) | Bcr-abl kinase inhibitor and application thereof | |
| TW201805286A (en) | FGFR4 inhibitor and preparation method and application thereof | |
| CN111171049A (en) | Tyrosine kinase inhibitors and uses thereof | |
| WO2015188681A1 (en) | Novel heterocyclic compound and preparation method therefor and use thereof as kinase inhibitor | |
| CN111196814B (en) | Aromatic ring-linked dioxane quinazoline or quinoline compound, composition and application thereof | |
| BR112021005750A2 (en) | fgfr4 inhibitor and its use | |
| CN111763215A (en) | A kind of compound with nitrogen-containing heterocyclic structure and preparation method and use thereof | |
| WO2025011444A1 (en) | Pyrimidinamine nuak inhibitor as well as preparation method therefor and use thereof | |
| WO2024046366A1 (en) | Selective parp1 inhibitor | |
| CN106749193B (en) | Indazole-substituted epidermal growth factor receptor inhibitors and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16867969 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 16867969 Country of ref document: EP Kind code of ref document: A1 |