WO2015091532A1 - Pyrrolopyrimidine derivatives as pi3k inhibitors - Google Patents
Pyrrolopyrimidine derivatives as pi3k inhibitors Download PDFInfo
- Publication number
- WO2015091532A1 WO2015091532A1 PCT/EP2014/078038 EP2014078038W WO2015091532A1 WO 2015091532 A1 WO2015091532 A1 WO 2015091532A1 EP 2014078038 W EP2014078038 W EP 2014078038W WO 2015091532 A1 WO2015091532 A1 WO 2015091532A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- branched
- linear
- alkyl
- phenyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC(c1nc2c(*)c(*)c(*)nc2[n]1-c1c(*)c(*)c(*)c(O*)c1[Re])Nc1c(c(*c(c(*)c2*)c(cc(*)[n]3*)c3c2[Rh])c[n]2)c2ncn1 Chemical compound CC(c1nc2c(*)c(*)c(*)nc2[n]1-c1c(*)c(*)c(*)c(O*)c1[Re])Nc1c(c(*c(c(*)c2*)c(cc(*)[n]3*)c3c2[Rh])c[n]2)c2ncn1 0.000 description 8
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
Definitions
- Phosphoinositide 3-Kinases are among the enzymes involved in early signalling events to a plethora of different types of stimuli.
- PI3Ks phosphorylate the 3-hydroxyl group of the inositol ring of phosphatidylinositol (Ptdlns), Ptdlns-4-phosphate (Ptdlns4P), and Ptdlns-4,5-bisphosphate (Ptdlns(4,5)P2).
- the resulting 3-phosphoinositides mediate correct localization and subsequent activation of a number of downstream effector proteins that bind to the lipids via specific lipid binding sequences such as the pleckstrin homology (PH) domain (Vanhaesebroeck B, 2010, Nat Rev Mol Cell Biol 5:11381-6).
- PH pleckstrin homology
- PI3K class I The PI3K family is divided into 3 different classes (PI3K class I, class II, and class III), depending on substrate preference and structural features.
- the best characterized is the PI3K class I with the preferential substrate Ptdlns- (4,5)P2. It englobes 4 different isoforms which originally were further subdivided into class IA (p1 10a, p1 10b, p1 10d), binding to a p85 type of regulatory subunit, and class IB (p1 10g) which is regulated by p101 and p87 subunits.
- p1 10a PI3Ka or PI3Ka
- p1 10b PI3Kb or ⁇ 3 ⁇
- p1 10g PI3Kg or ⁇ 3 ⁇
- p1 10d PI3Kd or PI3K5
- PI3Kd and PI3Kg are involved in activation of immune cells by a large variety of different stimuli.
- Pharmacological inhibition or genetic deficiency in active p1 10d has been shown to inhibit T cell proliferation and cytokine production in response to different stimuli such as anti-CD3, anti-CD3/CD28, superantigen or antigen in vitro (Ji H, Blood 2007; Okkenhaug K, Science 2002; Garcon F, 2009; Soond DR, Blood 2010; Herman SEM, Blood June 3, 2010; William O, Chemistry & Biology 17, 2010) and to suppress concanavalin A and anti-CD3 induced cytokine production as well as antigen- dependent tissue retention in vivo (Soond DR, Blood 2010; Jarmin SJ, JCI 2008).
- B cell function is critically dependent on functional PI3Kd activity as demonstrated by suppressed B cell proliferation and cytokine release in vitro in response to anti-lgM (Bilancio A, Blood 107, 2006), toll like receptor agonists such as LPS and oligodeoxynucleotides (Dil N, Mol Immunol 46, 2009) or impaired ability to stimulate antigen-specific T cells (Al-Alwan M, Jl 2007) in the absence of functional p1 10d or pharmacological inhibition.
- PI3Kg deficient mice display partially suppressed antibody production upon immunization (Garcon F, 2009; Durand CA, Jl
- mast cell degranulation was reduced in cells from mice with inactivated PI3Kd or by pharmacological inhibition of PI3Kd (AN K, Nature 431 :1007-101 1 , 2004; AN K, Journal of Immunology 180:2538-2544, 2008) and basophil activation via the FcE receptor is suppressed by pharmacological inhibition of PI3Kd (Lannutti BJ, Blood Oct.
- PI3Kd inhibition inhibits migration of mouse neutrophils to fMLP in an under-agarose migration assay by inhibiting cell polarization and directional movement (Sadhu C, Jl 170, 2003) and mouse PI3Kd deficient or inhibitor treated neutrophils show slightly (25%) reduced in vitro chemotaxis to LTB4, whereas in vivo accumulation in the lung in response to LPS was reduced by more than 80%, indicating an important role of PI3Kd in endothelial cells for mediating PMN transendothelial migration (Puri KD, Blood 103, 2004).
- TNF induced neutrophil infiltration to an air pouch in mice and elastase release is partially inhibited by a PI3Kd selective inhibitor (Sadhu C, Biochem Biophys Res Comm 308, 2003).
- PI3Kd selective inhibitor Sadhu C, Biochem Biophys Res Comm 308, 2003.
- TNF mediated priming of oxidative burst by human neutrophils depends on PI3Kd activity (Condliffe AM, Blood 106, 2005).
- PI3Kg seems to affect primarily chemotaxis of different immune cells induced by various mediators and chemokines (Martin AL, Jl 180, 2008; Thomas MS, J Leukoc Biol 84, 2008; Jarmin SJ, JCI 2008; Matthew T, Immunology 126, 2008), as well as degranulation and oxidative burst of innate immune cells induced by GPCR mediated stimuli such as fMLP, IL-8 or C5a (Condliffe AM, Blood 106, 2005; Yum HK, Jl 167, 2001 ; Pinho V, Jl 179, 2007
- PI3Kd or dual PI3Kd/PI3Kg pharmacological inhibition represents a promising approach for treating a variety of diseases such as respiratory diseases (asthma, chronic obstructive pulmonary disease (COPD), cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis), allergic diseases (allergic rhinitis), inflammatory or autoimmune diseases (rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, myastenia gravias, acute disseminated encephalomyelitis, idiopathic thrombocytopenic purpura, Sjoegren's syndrome, autoimmune hemolytic anemia, type I diabetes, psoriasis, acrodermatitis, angiodermatitis, atopic dermatitis, contact dermatitis, ec
- COPD chronic
- PI3Kd inhibition Puri KD, Blood 2004;103:3448
- inflammation in response to LPS or tobacco smoke exposure is suppressed by a dual PI3Kd/g inhibitor
- PI3Kd seems to be involved in the reduction of responsiveness to corticosteroid treatment associated with oxidative stress and chronic obstructive pulmonary disease (COPD).
- COPD chronic obstructive pulmonary disease
- PI3Kd selective inhibitor To Y, AJRCCM 182:897- 904, 2010.
- in vitro induction of corticosteroid resistance by oxidative stress is prevented by PI3Kd inhibition (To Y, AJRCCM 2010).
- lung macrophages display increased expression of PI3Kd and phosphorylation of its downstream effector Akt and non-selective PI3K or PI3Kd- selective inhibition restored the impaired inhibitory efficacy of dexamethasone in PBMC from COPD patients (To Y, AJRCCM 182:897-904, 2010; Marwick JA, JACI 125:1 146-53, 2010).
- PI3Kd inhibition was effective in a model of contact hypersensitivity (Soond DR, Blood Jan 2010).
- Soond DR Blood Jan 2010
- PI3Kd deficiency or pharmacological inhibition of PI3Kd attenuated T cell activation and function and reduced T cell numbers in the CNS, suggesting a therapeutic benefit of PI3Kd inhibitor in multiple sclerosis and other Th17-mediated autoimmune diseases (Haylock-Jacobs S, J. Autoimmun 2010).
- genetic deficiency or pharmacological inhibition of PI3Kd diminished joint erosion in a mouse model of inflammatory arthritis (Randis TM, Eur J Immunol 38, 2008).
- PI3Kd overexpression seems to contribute to excessive vascular contraction and PI3Kd inhibition normalized vascular contractive responses in a mouse model of type I diabetes, suggesting a therapeutic potential of PI3Kd blockade to treat vascular dysfunction in diabetic patients (Pinho JF, Br. J. Pharmacol 161 , 2010).
- PI3Kd or dual PI3Kd/g dual inhibition is effective in the treatment of cancers including but not restricted to leukemias, such as chronic lymphocytic leukemia, B-cell acute lymphoblastic leukemia, T-cell acute lymphoblastic leukaemia, non-hodgkins lymphoma, B-cell lymphoma, acute myeloid leukaemia, myelo-dysplastic syndrome or myelo-proliferative diseases.
- leukemias such as chronic lymphocytic leukemia, B-cell acute lymphoblastic leukemia, T-cell acute lymphoblastic leukaemia, non-hodgkins lymphoma, B-cell lymphoma, acute myeloid leukaemia, myelo-dysplastic syndrome or myelo-proliferative diseases.
- the selective PI3Kd inhibitor CAL-101 demonstrated anti-proliferative properties on different tumor cells in vitro and efficacy in cancer patients with a dysregulated PI3Kd activity, such as chronic lymphocytic leukemia (Hermann SE, Blood 1 16:2078-88, 2010; Lannutti BJ, Blood Oct. 2010).
- a dysregulated PI3Kd activity such as chronic lymphocytic leukemia (Hermann SE, Blood 1 16:2078-88, 2010; Lannutti BJ, Blood Oct. 2010).
- Conditions in which targeting of the PI3K pathway or modulation of the PI3 Kinases, particularly PI3Kd or PI3Kd/g, are contemplated to be therapeutically useful for the treatment or prevention of diseases includes: respiratory diseases (asthma, chronic obstructive pulmonary disease (COPD), cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis), allergic diseases (allergic rhinitis), inflammatory or autoimmune-mediated diseases (rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, myastenia gravias, acute disseminated encephalomyelitis, idiopathic thromocytopenic purpura, Sjoegren's syndrome, autoimmune hemolytic anemia, type I diabetes, psoriasis, acrodermatitis, angiodermatitis,
- novel pyrrolopyrimidine derivatives for use in the treatment of conditions in which targeting of the PI3K pathway or inhibition of PI3 Kinases can be therapeutically useful.
- the compounds described in the present invention are potent PI3K inhibitors, particularly PI3Kd or dual PK3Kd/g inhibitors.
- pulmonary diseases asthma, chronic obstructive pulmonary disease (COPD), cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis), allergic diseases (allergic rhinitis), inflammatory or autoimmune diseases (rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, myastenia gravias, acute disseminated encephalomyelitis, idiopathic thromocytopenic purpura, Sjoegren's syndrome, autoimmune hemolytic anemia, type I diabetes, psoriasis, acrodermatitis, angiodermatitis, atopic dermatitis, contact dermatitis, eczema, acne, chronic urticaria, scleroderma, cutaneous vascu
- the compounds described in the present invention are particularly useful for the treatment or prevention of pathological conditions or diseases such as neoplastic diseases (e.g. leukemia, lymphomas, solid tumors); transplant rejection, bone marrow transplant applications (e.g., graft- versus-host disease); autoimmune diseases (e.g.
- rheumatoid arthritis multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, cutaneous vasculitis, cutaneous lupus erythematosus, dermatomyositis and blistering diseases including but not limited to pemphigus vulgaris, bullous pemphigoid and epidermolysis bullosa; respiratory inflammation diseases (e.g.
- asthma chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis
- skin inflammatory diseases e.g., atopic dermatitis, contact dermatitis, eczema or psoriasis
- premalignant and malignant skin conditions e.g. basal cell carcinoma (BCC), squamous cell carcinoma (SCC) or actinic keratosis (AK)
- BCC basal cell carcinoma
- SCC squamous cell carcinoma
- AK actinic keratosis
- neurological disorders and pain such as pain associated with rheumatoid arthritis or osteoarthritis, back pain, general inflammatory pain, inflammatory neuropathic pain, trigeminal neuralgia or central pain
- the compounds described in the present invention are particularly useful for the treatment or prevention of pathological conditions or diseases selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, cutaneous vasculitis, cutaneous lupus erythematosus, dermatomyositis, blistering diseases including but not limited to pemphigus vulgaris, bullous pemphigoid and epidermolysis bullosa, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic ker
- the invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising the compounds of the invention in association with a pharmaceutically acceptable diluent or carrier.
- the invention also provides a combination product comprising (i) the compounds of the invention as described herein; and (ii) one or more additional active substances which are known to be useful in the treatment of respiratory diseases; allergic diseases; inflammatory or autoimmune-mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (MPDs); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors; more in particular wherein the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid artritis (RA), multiple sclerosis (MS), amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, cutaneous vasculitis, cutaneous lupus erythematosus,
- A represents a group selected from i) a group of formula (A-1 ), ii) a group of formula (A-2), iii) a group of formula (A-3),
- Ri represents a phenyl group or a 5- to 6- membered heteroaryl group containing at least one heteroatom selected from O, S and N,
- phenyl and heteroaryl groups are unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched Ci-C 4 alkyl group, a C1-C4 haloalkyi group, a C1-C4 hydroxyalkyi group, a C3-C1 0 cycloalkyl group, a -(CH 2 )i- 3 CN group, a -(CH 2 )o-30R 7 group, a -(CH 2 ) 0 -3NR 7 R8 group, a -C(0)-(CH 2 )i -3 -CN group, a -C(0)-(CH 2 )o-3-R 7 group, a -C(O)- (CH 2 )o-3-NR 7 R8 group, a -S(CH 2 ) 0 - 3 R 7 group, a -S(O)(CH 2
- R2, R3, R 4 , R5, and R 6 each independently represent a hydrogen atom, a C1-C4 alkoxy group, a C1-C4 haloalkyi group, a C1-C4 hydroxyalkyi group, a C3-C1 0 cycloalkyl group, a -(CH 2 ) 0 - 3 CN group, a -C(0)-(CH 2 ) 1-3 -CN group, a -C(O)- (CH 2 )o-3-R' group, a -C(0)-(CH 2 ) 0 - 3 -NR 9 Rio, a -(CH 2 ) 0 - 3 NR 9 Rio group, or a linear or branched C1-C4 alkyl group, which alkyl group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkoxy group, a cyano group or a C3-C10 cycloalky
- L represents a direct bound or a linker selected from -0-, -S-, a -N R'- group, a C(O)- N R'- group, a C(0)-0-R"- group or a -(CH 2 )i-4 group; wherein R' represents hydrogen or a linear or branched C1-C4 alkyl group, and R" represents a linear or branched C C 4 alkyl group;
- B represents a phenyl group or a 5- to 14- membered heteroaryl group containing at least one heteroatom selected from O, S and N,
- phenyl and heteroaryl groups are unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyl group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group, a -(CH 2 )o-3-0(Ci-C 4 alkyl) group, a -(CH 2 )o-3-0(Ci-C 4 haloalkyl) group, a -(CH 2 )o-3-0-(CH2)i-3-0(Ci-C 4 alkyl) group, a -(CH 2 ) 0 - 3 -O- (CH 2 ) 1-3 -0(Ci-C 4 haloalkyl) group, a -(CH 2 ) 0 - 3 -O-(CH 2 ) 1-3 -0(Ci-C
- Ci-C 6 alkyl embraces linear or branched radicals having 1 to 6 carbon atoms, preferably 1 to 4 carbon atoms. Examples include methyl, ethyl, n- propyl, i-propyl, n-butyl, sec-butyl, t-butyl, n-pentyl, 1 -methylbutyl, 2-methylbutyl, isopentyl, 1 -ethylpropyl, 1 ,1 -dimethylpropyl, 1 ,2-dimethylpropyl, n-hexyl, 1 -ethylbutyl, 2- ethylbutyl, 1 ,1 -dimethylbutyl, 1 ,2-dimethylbutyl, 1 ,3-dimethylbutyl, 2,2-dimethylbutyl, 2,3-dimethylbutyl, 2-methylpentyl, 3-methylpentyl and iso-hexyl radical
- alkyl radical may be optionally substituted it is meant to include linear or branched alkyl radical as defined above, which may be unsubstituted or substituted in any position by one or more substituents, for example by 1 , 2 or 3 substituents. When two or more substituents are present, each substituent may be the same or different.
- C1-C4 haloalkyl group is an alkyl group, for example a C1-C4 or C1-C2 alkyl group, which is bonded to one or more, preferably 1 , 2 or 3 halogen atoms.
- said haloakyl group is chosen from -CCI 3 , -CHF 2 and -CF 3 .
- C1-C4 hydroxyalkyl embraces linear or branched alkyl radicals having 1 to 4 carbon atoms, any one of which may be substituted by one or more, preferably 1 or 2, more preferably 1 hydroxyl radicals. Examples of such radicals include hydroxymethyl, hydroxyethyl, hydroxypropyl, and hydroxybutyl.
- C1-C4 alkoxy (or alkyloxy) embraces linear or branched oxy- containing radicals each having alkyl portions of 1 to 4 carbon atoms.
- C3-C1 0 cycloalkyl embraces saturated monocyclic or polycyclic carbocyclic radicals having from 3 to 10 carbon atoms, preferably from 3 to 7 carbon atoms.
- An optionally substituted C3-C1 0 cycloalkyl radical is typically unsubstituted or substituted by 1 , 2 or 3 substituents which may be the same or different.
- substituents may be the same or different.
- the substituents on a C3-C1 0 cycloalkyl group are themselves unsubstituted.
- Polycyclic cycloalkyl radicals contains two or more fused cycloalkyl groups, preferably two cycloalkyi groups.
- polycyclic cycloalkyi radicals are selected from decahydronaphthyl (decalyl), bicyclo[2.2.2]octyl, adamantly, camphyl or bornyl groups.
- Examples of monocyclic cycloalkyi groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl and cyclodecyl.
- the term 5- to 14- membered heteroaryl radical embraces typically a 5- to 14- membered ring system, preferably a 5- to 10- membered ring system, more preferably a 5- to 9- membered ring system, even more preferably a 5- to 6- membered ring system, comprising at least one heteroaromatic ring and containing at least one heteroatom selected from O, S and N.
- a 5- to 14- membered heteroaryl radical may be a single ring or two fused rings wherein at least one ring contains a heteroatom.
- a said optionally substituted 5- to 14- membered heteroaryl radical is typically unsubstituted or substituted by 1 , 2 or 3 substituents which may be the same or different.
- substituents may be the same or different.
- the substituents on a 5- to 14- membered heteroaryl radical are typically themselves unsubstituted.
- Examples include pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, furyl, benzofuranyl, oxadiazolyl, oxazolyl, isoxazolyl, benzoxazolyl, imidazolyl, benzimidazolyl, thiazolyl, thiadiazolyl, thienyl, pyrrolyl, benzothiazolyl, indolyl, indazolyl, purinyl, quinolyl, isoquinolyl, phthalazinyl, naphthyridinyl, quinoxalinyl, quinazolinyl, quinolizinyl, cinnolinyl, triazolyl, indolizinyl, indolinyl, isoindolinyl, isoindolyl, imidazolidinyl, pteridinyl, thianthrenyl, pyrazolyl, 2
- atoms, radicals, moieties, chains and cycles present in the general structures of the invention are "optionally substituted".
- substituents can be either unsubstituted or substituted in any position by one or more, for example 1 , 2, 3 or 4, substituents, whereby the hydrogen atoms bound to the unsubstituted atoms, radicals, moieties, chains and cycles are replaced by chemically acceptable atoms, radicals, moieties, chains and cycles.
- substituents When two or more substituents are present, each substituent may be the same or different. The substituents are typically themselves unsubstituted.
- halogen atom embraces chlorine, fluorine, bromine and iodine atoms.
- a halogen atom is typically a fluorine, chlorine or bromine atom, most preferably chlorine or fluorine.
- the term halo when used as a prefix has the same meaning.
- Compounds containing one or more chiral centre may be used in enantiomerically or diastereoisomerically pure form, in the form of racemic mixtures and in the form of mixtures enriched in one or more stereoisomer.
- the scope of the invention as described and claimed encompasses the racemic forms of the compounds as well as the individual enantiomers, diastereomers, and stereoisomer-enriched mixtures.
- enantiomers include chiral synthesis from a suitable optically pure precursor or resolution of the racemate using, for example, chiral high pressure liquid chromatography (HPLC).
- HPLC high pressure liquid chromatography
- the racemate (or a racemic precursor) may be reacted with a suitable optically active compound, for example, an alcohol, or, in the case where the compound contains an acidic or basic moiety, an acid or base such as tartaric acid or 1 -phenylethylamine.
- a suitable optically active compound for example, an alcohol, or, in the case where the compound contains an acidic or basic moiety, an acid or base such as tartaric acid or 1 -phenylethylamine.
- the resulting diastereomeric mixture may be separated by chromatography and/or fractional crystallization and one or both of the diastereoisomers converted to the corresponding pure enantiomer(s) by means well known to one skilled in the art.
- Chiral compounds of the invention may be obtained in enantiomerically-enriched form using chromatography, typically HPLC, on an asymmetric resin with a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from 0 to 50% isopropanol, typically from 2 to 20%, and from 0 to 5% of an alkylamine, typically 0.1 % diethylamine. Concentration of the eluate affords the enriched mixture.
- Stereoisomer conglomerates may be separated by conventional techniques known to those skilled in the art. See, e.g. "Stereochemistry of Organic Compounds" by Ernest L. Eliel (Wiley, New York, 1994).
- Atropisomers are stereoisomers resulting from hindered rotation about single bonds where the steric strain barrier to rotation is high enough to allow for the isolation of the conformers.
- Oki (Oki, M; Topics in Stereochemistry 1983, 1 ) defined atropisomers as conformers that interconvert with a half-life of more than 1000 seconds at a given temperature.
- the scope of the invention as described and claimed encompasses the racemic forms of the compounds as well as the individual atropisomers (an atropisomer "substantially free" of his corresponding enantiomer) and stereoisomer-enriched mixtures, i.e. mixtures of atropisomers. Separation of atropisomers is possibly by chiral resolution methods such as selective crystallization.
- Atroposelective synthesis may be carried out by use of chiral auxiliaries like a Corey-Bakshi-Shibata (CBS) catalyst (asymmetric catalyst derived from proline) in the total synthesis of knipholone or by approaches based on thermodynamic equilibration when an isomerization reaction favors one atropisomer over the other.
- CBS Corey-Bakshi-Shibata
- the term pharmaceutically acceptable salt refers to a salt prepared from a base or acid which is acceptable for administration to a patient, such as a mammal.
- Such salts can be derived from pharmaceutically-acceptable inorganic or organic bases and from pharmaceutically-acceptable inorganic or organic acids.
- Pharmaceutically acceptable acids include both inorganic acids, for example hydrochloric, sulphuric, phosphoric, diphosphoric, hydrobromic, hydroiodic and nitric acid; and organic acids, for example citric, fumaric, gluconic, glutamic, lactic, maleic, malic, mandelic, mucic, ascorbic, oxalic, pantothenic, succinic, tartaric, benzoic, acetic, methanesulphonic, ethanesulphonic, benzenesulphonic, p-toluenesulphonic acid, xinafoic (1 -hydroxy-2-naphthoic acid), napadisilic (1 ,5-naphthalenedisulfonic acid) and the like. Particularly preferred are salts derived from fumaric, hydrobromic, hydrochloric, acetic, sulfuric, methanesulfonic, xinafoic, and tartaric acids.
- Salts derived from pharmaceutically-acceptable inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic, manganous, potassium, sodium, zinc and the like. Particularly preferred are ammonium, calcium, magnesium, potassium and sodium salts.
- Salts derived from pharmaceutically-acceptable organic bases include salts of primary, secondary and tertiary amines, including alkyl amines, arylalkyl amines, heterocyclyl amines, cyclic amines, naturally-occurring amines and the like, such as arginine, betaine, caffeine, choline, ⁇ , ⁇ '-dibenzylethylenediamine, diethylamine, 2- diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N- ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine and the
- X " may be an anion of various mineral acids such as, for example, chloride, bromide, iodide, sulphate, nitrate, phosphate, or an anion of an organic acid such as, for example, acetate, maleate, fumarate, citrate, oxalate, succinate, tartrate, malate, mandelate, trifluoroacetate, methanesulphonate and p-toluenesulphonate.
- mineral acids such as, for example, chloride, bromide, iodide, sulphate, nitrate, phosphate
- organic acid such as, for example, acetate, maleate, fumarate, citrate, oxalate, succinate, tartrate, malate, mandelate, trifluoroacetate, methanesulphonate and p-toluenesulphonate.
- X " is preferably an anion selected from chloride, bromide, iodide, sulphate, nitrate, acetate, maleate, oxalate, succinate or trifluoroacetate. More preferably X " is chloride, bromide, trifluoroacetate or methanesulphonate.
- an N-oxide is formed from the tertiary basic amines or imines present in the molecule, using a convenient oxidising agent.
- the present invention also embraces tautomeric forms of the compounds of formula (I), or pharmaceutically acceptable salts, solvates, N-oxides, stereoisomers or deuterated derivatives thereof.
- the compounds of the invention may exist in both unsolvated and solvated forms.
- solvate is used herein to describe a molecular complex comprising a compound of the invention and an amount of one or more pharmaceutically acceptable solvent molecules.
- hydrate is employed when said solvent is water.
- solvate forms include, but are not limited to, compounds of the invention in association with water, acetone, dichloromethane, 2-propanol, ethanol, methanol, dimethylsulfoxide (DMSO), ethyl acetate, acetic acid, ethanolamine, or mixtures thereof. It is specifically contemplated that in the present invention one solvent molecule can be associated with one molecule of the compounds of the present invention, such as a hydrate.
- solvates of the present invention are contemplated as solvates of compounds of the present invention that retain the biological effectiveness of the non- solvate form of the compounds.
- the invention also includes isotopically-labeled compounds of the invention, wherein one or more atoms is replaced by an atom having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number usually found in nature.
- isotopes suitable for inclusion in the compounds of the invention include isotopes of hydrogen, such as 2 H and 3 H, carbon, such as 11 C, 13 C and 14 C, chlorine, such as 36 CI, fluorine, such as 18 F, iodine, such as 123 l and 125 l, nitrogen, such as 13 N and 15 N, oxygen, such as 15 0, 17 0 and 18 0, phosphorus, such as 32 P, and sulfur, such as 35 S.
- isotopically-labeled compounds of the invention for example, those incorporating a radioactive isotope, are useful in drug and/or substrate tissue distribution studies.
- the radioactive isotopes tritium, 3 H, and carbon- 14, 14 C are particularly useful for this purpose in view of their ease of incorporation and ready means of detection. Substitution with heavier isotopes such as deuterium, 2 H, may afford certain therapeutic advantages resulting from greater metabolic stability, for example, increased in vivo half-life or reduced dosage requirements, and hence may be preferred in some circumstances. Substitution with positron emitting isotopes, such as 11 C, 18 F, 15 0 and 13 N, can be useful in Positron Emission Topography (PET) studies for examining substrate receptor occupancy.
- PET Positron Emission Topography
- Isotopically-labeled compounds of the invention can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described herein, using an appropriate isotopically-labeled reagent in place of the non-labeled reagent otherwise employed.
- Preferred isotopically-labeled compounds include deuterated derivatives of the compounds of the invention.
- deuterated derivative embraces compounds of the invention where in a particular position at least one hydrogen atom is replaced by deuterium.
- Deuterium (D or 2 H) is a stable isotope of hydrogen which is present at a natural abundance of 0.015 molar %.
- Hydrogen deuterium exchange (deuterium incorporation) is a chemical reaction in which a covalently bonded hydrogen atom is replaced by a deuterium atom. Said exchange (incorporation) reaction can be total or partial.
- a deuterated derivative of a compound of the invention has an isotopic enrichment factor (ratio between the isotopic abundance and the natural abundance of that isotope, i.e. the percentage of incorporation of deuterium at a given position in a molecule in the place of hydrogen) for each deuterium present at a site designated as a potential site of deuteration on the compound of at least 3500 (52.5% deuterium incorporation).
- isotopic enrichment factor ratio between the isotopic abundance and the natural abundance of that isotope, i.e. the percentage of incorporation of deuterium at a given position in a molecule in the place of hydrogen
- the isotopic enrichment factor is at least 5000 (75% deuterium). In a more preferred embodiment, the isotopic enrichment factor is at least 6333.3 (95% deuterium incorporation). In a most preferred embodiment, the isotopic enrichment factor is at least 6633.3 (99.5% deuterium incorporation). It is understood that the isotopic enrichment factor of each deuterium present at a site designated as a site of deuteration is independent from the other deuteration sites.
- the isotopic enrichment factor can be determined using conventional analytical methods known to an ordinary skilled in the art, including mass spectrometry (MS) and nuclear magnetic resonance (NMR).
- Prodrugs of the compounds described herein are also within the scope of the invention.
- certain derivatives of the compounds of the present invention which derivatives may have little or no pharmacological activity themselves, when administered into or onto the body may be converted into compounds of the present invention having the desired activity, for example, by hydrolytic cleavage.
- Such derivatives are referred to as 'prodrugs'. Further information on the use of prodrugs may be found in Pro-drugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T. Higuchi and W.
- Prodrugs in accordance with the invention can, for example, be produced by replacing appropriate functionalities present in the compounds of the present invention with certain moieties known to those skilled in the art as 'pro-moieties' as described, for example, in Design of Prodrugs by H. Bundgaard (Elsevier, 1985).
- inventive compounds and salts may exist in different crystalline or polymorphic forms, or in an amorphous form, all of which are intended to be within the scope of the present invention.
- the term PI3Kd inhibitor generally refers to a compound that inhibits the activity of the PI3Kd isoform more effectively than other isoforms of the PI3K family.
- the term PI3Kd/g inhibitor generally refers to a compound that inhibits the activity of both the PI3Kd isoform and the PI3Kg isoform more effectively than other isoforms of the PI3K family.
- the relative efficacies of compounds as inhibitors of an enzyme activity (or other biological activity) can be established by determining the concentrations at which each compound inhibits the activity to a predefined extent and then comparing the results.
- the preferred determination is the concentration that inhibits 50% of the activity in a biochemical assay, i.e., the 50% inhibitory concentration or "IC 50 .”
- IC 50 determinations can be accomplished using conventional techniques known in the art. In general, an IC 50 can be determined by measuring the activity of a given enzyme in the presence of a range of concentrations of the inhibitor under study. The experimentally obtained values of enzyme activity then are plotted against the inhibitor concentrations used. The concentration of the inhibitor that shows 50% enzyme activity (as compared to the activity in the absence of any inhibitor) is taken as the IC 50 value.
- a PI3Kd inhibitor alternatively can be understood to refer to a compound that exhibits a 50% inhibitory concentration (IC 50 ) with respect to PI3Kd that is at least of less than about 100 ⁇ , preferably of less than about 50 ⁇ , more preferably of less than about 20 ⁇ , even more preferably of less than about 10 ⁇ PI3K HTRF assay (as described in Gray et al. Anal Biochem, 2003; 313: 234-45).
- IC 50 50% inhibitory concentration
- Ri represents a phenyl group wherein the phenyl group is unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyl group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group, a C3-C10 cycloalkyl group, a -(CH 2 )i-3CN group, a -(CH 2 )o-30R 7 group, a -(CH 2 )o-3NR 7 R8 group, a -C(0)-(CH 2 )i-3-CN group, a -C(0)-(CH 2 )o-3-R 7 group, a -C(O)-(CH 2 ) 0 - 3 -NR 7 R 8 group, a - S(CH 2 )o -3 R 7 group
- Ri represents a phenyl group wherein the phenyl group is unsubstituted or substituted by one or more halogen atoms.
- A represents a group selected from: -1a), group of formula (A-2a),
- R a , R b , R c , Rd and R e independently represents a hydrogen atom or a substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyl group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group, a C3-C1 0 cycloalkyl group, a -(CH 2 )i- 3 CN group, a -(CH 2 ) 0 -3OR 7 group, a -(CH 2 )o-3N R 7 R8 group, a -C(0)-(CH 2 )i- 3 -CN group, a - C(0)-(CH 2 )o-3-R 7 group, a -C(O)-(CH 2 ) 0
- R 2 , R3, R 4 , R 5 , and R 6 each independently represent a hydrogen atom, a C1-C4 alkoxy group, or a linear or branched C1-C4 alkyi group, which alkyi group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkoxy group, a cyano group or a C3-C1 0 cycloalkyl group. More preferably R 2 , R3, R 4 , R5, and R 6 each independently represent a hydrogen atom, a methoxy, or methyl group.
- L is preferably a direct bond.
- B represents a phenyl group, or a 5-to 9-membered heteroaryl group containing at least one heteroatom selected from 0,S and N , wherein said heteroaryl group is preferably pyridyl group, a pyrazole group or an indole group, and wherein the phenyl, pyridyl, pyrazole or indole groups are unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyi group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group, a -(CH 2 )o-3-0(Ci-C 4 alkyi) group, a -(CH 2 )o- 3-0(Ci-C 4 haloalkyl) group, a -(CH 2 )o-
- NRnR 12 group a C3-C1 0 cycloalkyl group, a -(CH 2 ) 0 - 3 NRn Ri 2 group, a -(CH 2 )o-3-C(0)- (CH 2 ) 0 -3-NRiiRi 2 group, a -(CH 2 ) 0 - 3 -C(0)0-(CH 2 ) o-3Ri i group, a -(CH 2 ) o- 3 NRii-S(0) 2 Ri 2 group, a -(CH 2 ) 0 - 3 - S(O) 2 (CH 2 ) 0 - 3 -Rn group, a -(CH 2 ) 0 - 3 -SH group, a -(CH 2 ) 0 -3-S-(CH 2 )o- 3 - R11 group or a -(CH 2 ) 0- 3-(5- to 7- membered heterocyclyl group containing at least one heterocycl
- B represents a phenyl, pyridyl, pyrazole or indole groups, unsubstituted or substituted by one or more substituents selected from a hydroxyl group, a linear or branched C1-C4 alkyl group, a C1-C4 hydroxyalkyl group, a -(CH 2 )o-3- 0(Ci-C 4 alkyl) group, a -(CH 2 )o-3NRn Ri 2 group, a -(CH 2 )o-3NR i S(0) 2 Ri 2 group or a - (CH 2 )o-3-(5- to 7- membered heterocyclyl group containing at least one heteroatom selected from O, S and N); which heterocyclyl group is unsubstituted or substituted by one or more substituents selected from a halogen atom, a linear or branched C1-C4 alkyl group, a -(CH 2 )o- 5 NR
- A represents a group selected from i) a group of formula (A-1 a), ii) a group of formula (A-2a),
- R2, R3, R 4 , R5, and R 6 each independently represent a hydrogen atom, a C1-C4 alkoxy group, or a linear or branched C1-C4 alkyi group, which alkyi group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkoxy group, a cyano group or a C3-C1 0 cycloalkyl group; each R a , R b , R c , R d and R e independently represents a hydrogen atom or a substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyi group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group, a C3-C1 0 cycloalkyl group, a -(CH 2 )i- 3 CN group, a
- A is bonded to the CH(CH 3 )-N H-pyrrolopyrimidine group
- B represents a phenyl group, a pyrazole group or an indole group, wherein the phenyl, pyridyl, pyrazole or indole groups are unsubstituted or substituted by one or more substituents selected from a hydroxyl group, a linear or branched C1-C4 alkyl group, a C1-C4 hydroxyalkyl group, a -(CH 2 ) 0 -3-O(Ci-C 4 alkyl) group, a -(CH 2 )o- 3 NRn R 12 group, a - (CH 2 )o- 3 NR i S(0) 2 Ri 2 group or a -(CH 2 ) 0- 3-(5- to 7- membered heterocyclyl group containing at least one heteroatom selected from O, S and N); which heterocyclyl group is un
- L, Ri, R 2 , R3, R 4 , R 5 , and R 6 are as defined for general formula (I) and each of R f , R g , R h , R and R j independently represent a hydrogen atom, halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyl group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group, a -(CH 2 )o-3-0(Ci-C 4 alkyl) group, a -(CH 2 )o- 3-0(Ci-C 4 haloalkyl) group, a -(CH 2 )o-3-0-(CH2)i-3-0(Ci-C 4 alkyl) group, a -(CH 2 ) 0 - 3 -O- (CH 2 )i-3-0(Ci-C 4 haloalkyl) group, a
- each R a , R b , R c , R d and R e independently represents a hydrogen atom or a substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyl group, a C1-C4 haloalkyi group, a C1-C4 hydroxyalkyl group, a C 3 - C10 cycloalkyl group, a -(CH 2 )i-3CN group, a -(CH 2 )o-30R 7 group, a -(CH 2 )o-3NR 7 R8 group, a -C(0)-(CH 2 )i -3 -CN group, a -C(0)-(CH 2 )o-3-R 7 group, a -C(O)-(CH 2 ) 0 - 3 -NR 7 R 8 group, a -S(CH 2 ) 0 - 3 R 7 group,
- each of R f , R g , R h , R, and R j independently represent a hydrogen atom, halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyl group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group, a -(CH 2 )o-3-0(Ci-C 4 alkyl) group, a -(CH 2 )o-3- 0(Ci-C 4 haloalkyl) group, a -(CH 2 )o-3-0-(CH2)i-3-0(Ci-C 4 alkyl) group, a -(CH 2 ) 0 - 3 -O- (CH 2 )i-3-0(Ci-C 4 haloalkyl) group, a -(CH 2 ) 0 - 3 -0-(CH 2 ) o-3 i group,
- N ii i2 group a C3-C10 cycloalkyl group, a -(CH 2 ) o-3NRn Ri 2 group, a -(CH 2 ) 0 - 3 -C(O)- (CH 2 ) 0 -3-NRii Ri 2 group, a -(CH 2 ) 0 - 3 -C(O)O-(CH 2 ) 0 - 3 Rn group, a -(CH 2 ) o- 3 NRn-S(0) 2 Ri 2 group, a -(CH 2 ) 0 - 3 - S(O) 2 (CH 2 ) 0 - 3 -Rn group, a -(CH 2 ) 0 - 3 -SH group, a -(CH 2 ) 0 -3-S-(CH 2 )o- 3 - R11 group or a -(CH 2 ) 0- 3-(5- to 7- membered heterocyclyl group containing
- A represents a group selected from i ),
- Ri represents a phenyl group or a 5- to 6- membered heteroaryl group containing at least one heteroatom selected from O, S and N,
- phenyl and heteroaryl groups are unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched Ci-C 4 alkyl group, a Ci-C 4 haloalkyl group, a C C 4 hydroxyalkyl group, a C 3 -Ci 0 cycloalkyl group, a -(CH 2 )i- 3 CN group, a -(CH 2 )o-30R 7 group, a -(CH 2 )o-3NR 7 R8 group, a -C(0)-(CH 2 )i -3 -CN group, a -C(0)-(CH 2 )o-3-R 7 group, a -C(O)- (CH 2 )o-3-NR 7 R8 group, a -S(CH 2 ) 0 - 3 R 7 group, a -S(O)(CH 2 ) 0
- R2, R3, R 4 , R5, and R 6 each independently represent a hydrogen atom, a C1-C4 alkoxy group, a C1-C4 haloalkyi group, a C1-C4 hydroxyalkyl group, a C3-C10 cycloalkyl group, a -(CH 2 )o- 3 CN group, a -C(0)-(CH 2 )i-3-CN group, a -C(O)- (CH 2 )o-3-R' group, a -C(0)-(CH 2 )o- 3 -N R 9 Rio, a -(CH 2 ) 0 - 3 N R 9 Rio group, or a linear or branched C1-C4 alkyl group, which alkyl group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkyl group;
- L represents a direct bound or a linker selected from -0-, -S-, a -N R'- group, a C(O)- N R'- group, a C(0)-0-R"- group or a -(CH 2 ) 1 -4 group; wherein R' represents hydrogen or a linear or branched C1-C4 alkyl group, and R" represents a linear or branched C C 4 alkyl group;
- B represents a phenyl group or a 5- to 14- membered heteroaryl group containing at least one heteroatom selected from O, S and N,
- phenyl and heteroaryl groups are unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyl group, a C1-C4 haloalkyi group, a C1-C4 hydroxyalkyl group, a -(CH 2 ) 0 -3-O(Ci-C 4 alkyl) group, a -(CH 2 ) 0 -3-O(Ci-C 4 haloalkyi) group, a -(CH 2 )o-3-0-(CH 2 ) 1-3 -0(Ci-C4 alkyl) group, a -(CH 2 ) 0 - 3 -O- (CH 2 ) 1-3 -0(Ci-C 4 haloalkyi) group, a -(CH 2 ) 0 - 3 -O-(CH 2 )
- B represents a phenyl group, or a 5-to 9- membered heteroaryl group containing at least one heteroatom selected from 0,S and N, wherein said heteroaryl group is preferably a pyrazole group or an indole group, and wherein the phenyl, pyrazole or indole groups are unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched CrC 4 alkyl group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyi group, a -(CH 2 )o-3-0(Ci-C 4 alkyl) group, a -(CH 2 )o-3-0(Ci-C 4 haloalkyl) group, a -(CH 2 )o- 3-0-(CH 2 )i-3-0(Ci-C 4 alkyl)
- B preferably represents a phenyl group, a pyrazole group or an indole group, substituted by one or more substituents selected from a hydroxyl group, a linear or branched Ci-C 4 alkyl group, a Ci-C 4 hydroxyalkyi group, a -(CH 2 ) 0 - 3 NRn-S(O) 2 Ri 2 group wherein Rn and Ri 2 each independently represent a hydrogen atom, a linear or branched C1-C4 alkyl group, a -(CH 2 ) 0 - 3 NR 13 R 14 group and wherein R 3 and Ri 4 each represent a hydrogen atom.
- A represents a group selected from i) a group of formula (A-1a), ii) a group of formula (A-2a),
- R2, R3, R 4 , R5, and R 6 each independently represent a hydrogen atom, a C1-C4 alkoxy group, or a linear or branched C1-C4 alkyi group, which alkyi group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkoxy group, a cyano group or a C3-C1 0 cycloalkyl group; each R a , R b , R c , R d and R e independently represents a hydrogen atom or a substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyi group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group, a C3-C1 0 cycloalkyl group, a -(CH 2 )i- 3 CN group, a
- preferred compounds of the invention can be represented by the various subformulas:
- N ii i2 group a C3-C10 cycloalkyl group, a -(CH 2 ) o-3N n i 2 group, a -(CH 2 ) 0 - 3 -C(O)- (CH 2 ) 0 -3-NRiiRi 2 group, a -(CH 2 ) 0 -3-C(0)0-(CH 2 )o- 3 Rii group, a -(CH 2 ) o- 3 NRii-S(0) 2 Ri 2 group, a -(CH 2 ) 0 - 3 - S(O) 2 (CH 2 ) 0 - 3 -Rn group, a -(CH 2 ) 0 - 3 -SH group, or a -(CH 2 ) 0 - 3 -S- (CH 2 )o-3-Rii group; wherein Rn and R12 each independently represent a hydrogen atom, a hydroxyl group, a linear or branched
- each R a , R b , R c , R d and R e independently represents a hydrogen atom or a substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyl group, a C1-C4 haloalkyi group, a C1-C4 hydroxyalkyl group, a C 3 - C10 cycloalkyl group, a -(CH 2 )i-3CN group, a -(CH 2 )o-30R 7 group, a -(CH 2 )o-3NR 7 R8 group, a -C(0)-(CH 2 )i -3 -CN group, a -C(0)-(CH 2 )o-3-R 7 group, a -C(O)-(CH 2 ) 0 - 3 -NR 7 R 8 group, a -S(CH 2 ) 0 - 3 R 7 group,
- R f , R g , R h , R, and R j independently represent a hydrogen atom, a halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyl group, a C1-C4 haloalkyi group, a C1-C4 hydroxyalkyl group, a -(CH 2 ) 0 -3-O(Ci-C 4 alkyl) group, a -(CH 2 )o- 3-0(Ci-C 4 haloalkyi) group, a -(CH 2 )o-3-0-(CH 2 ) 1-3 -0(Ci-C4 alkyl) group, a -(CH 2 ) 0 - 3 -O- (CH 2 )i-3-0(Ci-C 4 haloalkyi) group, a -(CH 2 ) 0 - 3 -0-(CH 2 ) o
- NRnR 12 group a C3-C1 0 cycloalkyl group, a -(CH 2 ) 0 - 3 NRn Ri 2 group, a -(CH 2 )o-3-C(0)- (CH 2 ) 0 -3-NRiiRi 2 group, a -(CH 2 ) 0 -3-C(0)0-(CH 2 )o- 3 Rii group, a -(CH 2 ) o- 3 NRii-S(0) 2 Ri 2 group, a -(CH 2 ) 0 - 3 - S(O) 2 (CH 2 ) 0 - 3 -Rn group, a -(CH 2 ) 0 - 3 -SH group, or a -(CH 2 ) 0 - 3 -S- (CH 2 )o-3-Rii group; wherein Rn and R12 each independently represent a hydrogen atom, a hydroxyl group, a linear or branched C1-C1
- Particular individual compounds of the invention include:
- particular individual compounds of the invention include:
- the invention is also directed to the compounds of the invention as described herein, for use in the treatment of a pathological condition or disease susceptible to amelioration by inhibiton of Phosphoinositide 3-Kinases (PI3Ks), in particular wherein the pathological condition or disease is selected from respiratory diseases; allergic diseases; inflammatory or autoimmune-mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (MPDs); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors; more in particular wherein the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid artritis (RA), multiple sclerosis (MS), amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type
- the invention is also directed to use of the compounds of the invention as described herein, in the manufacture of a medicament for treatment of a pathological condition or disease susceptible to amelioration by inhibiton of Phosphoinositide 3-Kinases (PI3Ks), in particular wherein the pathological condition or disease is selected from respiratory diseases; allergic diseases; inflammatory or autoimmune-mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (MPDs); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors; more in particular wherein the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid artritis (RA), multiple sclerosis (MS), amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune
- the invention also provides a method of treatment of a pathological condition or disease susceptible to amelioration by inhibiton of Phosphoinositide 3-Kinases (PI3Ks), in particular wherein the pathological condition or disease is selected from respiratory diseases; allergic diseases; inflammatory or autoimmune-mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (MPDs); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors; more in particular wherein the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid artritis (RA), multiple sclerosis (MS), amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, cutaneous vasculitis, cutaneous
- the invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising the compounds of the invention in association with a pharmaceutically acceptable diluent or carrier.
- the term pharmaceutical composition refers to a mixture of one or more of the compounds described herein, or physiologically/pharmaceutically acceptable salts, solvates, N-oxides, stereoisomers, deuterated derivatives thereof or prodrugs thereof, with other chemical components, such as physiologically/pharmaceutically acceptable carriers and excipients.
- the purpose of a pharmaceutical composition is to facilitate administration of a compound to an organism.
- a physiologically/pharmaceutically acceptable diluent or carrier refers to a carrier or diluent that does not cause significant irritation to an organism and does not abrogate the biological activity and properties of the administered compound.
- the invention further provides pharmaceutical compositions comprising the compounds of the invention in association with a pharmaceutically acceptable diluent or carrier together with one or more other therapeutic agents for use in the treatment of a pathological condition or disease susceptible to amelioration by inhibiton of Phosphoinositide 3-Kinases (PI3Ks), such as the ones previously described.
- PI3Ks Phosphoinositide 3-Kinases
- the invention is also directed to pharmaceutical compositions of the invention for use in the treatment of a pathological condition or disease susceptible to amelioration by inhibiton of Phosphoinositide 3-Kinases (PI3Ks), in particular wherein the pathological condition or disease is selected from respiratory diseases; allergic diseases; inflammatory or autoimmune-mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (MPDs such as polycythemia vera, essential thrombocythemia or mielofibrosis); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors; more in particular wherein the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus
- the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic keratosis.
- the invention also encompasses the use of a pharmaceutical composition of the invention for the manufacture of a medicament for treating these diseases.
- the invention also provides a method of treatment of a pathological condition or disease susceptible to amelioration by inhibiton of Phosphoinositide 3-Kinases (PI3Ks), in particular wherein the pathological condition or disease is selected from respiratory diseases; allergic diseases; inflammatory or autoimmune-mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (MPDs such as polycythemia vera, essential thrombocythemia or mielofibrosis); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors; more in particular wherein the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis,
- compositions which comprise, as an active ingredient, at least a compound of formula (I) or a pharmaceutically acceptable salt thereof in association with a pharmaceutically acceptable excipient such as a carrier or diluent.
- the active ingredient may comprise 0.001 % to 99% by weight, preferably 0.01 % to 90% by weight, of the composition depending upon the nature of the formulation and whether further dilution is to be made prior to application.
- the compositions are made up in a form suitable for oral, inhalation, topical, nasal, rectal, percutaneous or injectable administration.
- compositions suitable for the delivery of compounds of the invention and methods for their preparation will be readily apparent to those skilled in the art. Such compositions and methods for their preparation can be found, for example, in Remington: The Science and Practice of Pharmacy, 21 st Edition, Lippincott Williams & Wilkins, Philadelphia, Pa., 2001 .
- compositions of this invention are well-known per se and the actual excipients used depend inter alia on the intended method of administering the compositions.
- excipients include calcium carbonate, calcium phosphate, various sugars and types of starch, cellulose derivatives, gelatin, vegetable oils and polyethylene glycols.
- compositions for oral administration may take the form of tablets, retard tablets, sublingual tablets, capsules, inhalation aerosols, inhalation solutions, dry powder inhalation, or liquid preparations, such as mixtures, solutions, elixirs, syrups or suspensions, all containing the compound of the invention; such preparations may be made by methods well-known in the art.
- the active ingredient may also be presented as a bolus, electuary or paste.
- composition is in the form of a tablet
- any pharmaceutical carrier routinely used for preparing solid formulations may be used.
- examples of such carriers include magnesium stearate, talc, gelatine, acacia, stearic acid, starch, lactose and sucrose.
- a tablet may be made by compression or moulding, optionally with one or more accessory ingredients.
- Compressed tablets may be prepared by compressing in a suitable machine the active ingredient in a free-flowing form such as a powder or granules, optionally mixed with a binder, lubricant, inert diluent, lubricating, surface active or dispersing agent.
- Moulded tablets may be made by moulding in a suitable machine a mixture of the powdered compound moistened with an inert liquid diluent.
- the tablets may optionally be coated or scored and may be formulated so as to provide slow or controlled release of the active ingredient therein.
- the drug may make up from 1 wt% to 80 wt% of the dosage form, more typically from 5 wt% to 60 wt% of the dosage form.
- tablets generally contain a disintegrant.
- disintegrants include sodium starch glycolate, sodium carboxymethyl cellulose, calcium carboxymethyl cellulose, croscarmellose sodium, crospovidone, polyvinylpyrrolidone, methyl cellulose, microcrystalline cellulose, lower alkyl- substituted hydroxypropyl cellulose, starch, pregelatinized starch and sodium alginate.
- the disintegrant will comprise from 1 wt% to 25 wt%, preferably from 5 wt% to 20 wt% of the dosage form.
- Binders are generally used to impart cohesive qualities to a tablet formulation. Suitable binders include microcrystalline cellulose, gelatin, sugars, polyethylene glycol, natural and synthetic gums, polyvinylpyrrolidone, pregelatinized starch, hydroxypropyl cellulose and hydroxypropyl methylcellulose. Tablets may also contain diluents, such as lactose (monohydrate, spray-dried monohydrate, anhydrous and the like), mannitol, xylitol, dextrose, sucrose, sorbitol, microcrystalline cellulose, starch and dibasic calcium phosphate dihydrate.
- lactose monohydrate, spray-dried monohydrate, anhydrous and the like
- mannitol xylitol
- dextrose sucrose
- sorbitol microcrystalline cellulose
- starch dibasic calcium phosphate dihydrate
- Tablets may also optionally include surface active agents, such as sodium lauryl sulfate and polysorbate 80, and glidants such as silicon dioxide and talc.
- surface active agents such as sodium lauryl sulfate and polysorbate 80
- glidants such as silicon dioxide and talc.
- surface active agents are typically in amounts of from 0.2 wt% to 5 wt% of the tablet, and glidants typically from 0.2 wt% to 1 wt% of the tablet.
- Tablets also generally contain lubricants such as magnesium stearate, calcium stearate, zinc stearate, sodium stearyl fumarate, and mixtures of magnesium stearate with sodium lauryl sulphate.
- Lubricants generally are present in amounts from 0.25 wt% to 10 wt%, preferably from 0.5 wt% to 3 wt% of the tablet.
- Other conventional ingredients include anti-oxidants, colorants, flavoring agents, preservatives and taste- masking agents.
- Exemplary tablets contain up to about 80 wt% drug, from about 10 wt% to about 90 wt% binder, from about 0 wt% to about 85 wt% diluent, from about 2 wt% to about 10 wt% disintegrant, and from about 0.25 wt% to about 10 wt% lubricant.
- Tablet blends may be compressed directly or by roller to form tablets. Tablet blends or portions of blends may alternatively be wet-, dry-, or melt-granulated, melt congealed, or extruded before tabletting.
- the final formulation may include one or more layers and may be coated or uncoated; or encapsulated.
- composition is in the form of a capsule
- any routine encapsulation is suitable, for example using the aforementioned carriers in a hard gelatine capsule.
- composition is in the form of a soft gelatine capsule
- any pharmaceutical carrier routinely used for preparing dispersions or suspensions may be considered, for example aqueous gums, celluloses, silicates or oils, and are incorporated in a soft gelatine capsule.
- Solid formulations for oral administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- Suitable modified release formulations are described in U.S. Patent No. 6,106,864. Details of other suitable release technologies such as high energy dispersions and osmotic and coated particles can be found in Verma et al, Pharmaceutical Technology On-line, 25(2), 1 -14 (2001 ). The use of chewing gum to achieve controlled release is described in WO 00/35298. The disclosures of these references are incorporated herein by reference in their entireties.
- Liquid formulations include suspensions, solutions, syrups and elixirs. Such formulations may be used as fillers in soft or hard capsules and typically include a carrier, for example, water, ethanol, polyethylene glycol, propylene glycol, methylcellulose, or a suitable oil, and one or more emulsifying agents and/or suspending agents.
- the solutions may be aqueous solutions of a soluble salt or other derivative of the active compound in association with, for example, sucrose to form a syrup.
- the suspensions may comprise an insoluble active compound of the invention or a pharmaceutically acceptable salt thereof in association with water, together with a suspending agent or flavouring agent.
- Liquid formulations may also be prepared by the reconstitution of a solid, for example, from a sachet. ii) Oral mucosal administration
- the compounds of the invention can also be administered via the oral mucosal.
- delivery of drugs is classified into three categories: (a) sublingual delivery, which is systemic delivery of drugs through the mucosal membranes lining the floor of the mouth, (b) buccal delivery, which is drug administration through the mucosal membranes lining the cheeks (buccal mucosa), and (c) local delivery, which is drug delivery into the oral cavity.
- Pharmaceutical products to be administered via the oral mucosal can be designed using mucoadhesive, quick dissolve tablets and solid lozenge formulations, which are formulated with one or more mucoadhesive (bioadhesive) polymers (such as hydroxy propyl cellulose, polyvinyl pyrrolidone, sodium carboxymethyl cellulose, hydroxy propyl methyl cellulose, hydroxy ethyl cellulose, polyvinyl alcohol, polyisobutylene or polyisoprene); and oral mucosal permeation enhancers (such as butanol, butyric acid, propranolol, sodium lauryl sulphate and others) iii) Inhaled administration
- mucoadhesive polymers such as hydroxy propyl cellulose, polyvinyl pyrrolidone, sodium carboxymethyl cellulose, hydroxy propyl methyl cellulose, hydroxy ethyl cellulose, polyvinyl alcohol, polyisobutylene or poly
- the compounds of the invention can also be administered by inhalation, typically in the form of a dry powder (either alone, as a mixture, for example, in a dry blend with lactose, or as a mixed component particle, for example, mixed with phospholipids, such as phosphatidylcholine) from a dry powder inhaler or as an aerosol spray from a pressurized container, pump, spray, atomizer (preferably an atomizer using electrohydrodynamics to produce a fine mist), or nebulizer, with or without the use of a suitable propellant, such as 1 ,1 ,1 ,2-tetrafluoroethane or 1 ,1 ,1 ,2,3,3,3- heptafluoropropane.
- the powder may include a bioadhesive agent, for example, chitosan or cyclodextrin.
- Dry powder compositions for topical delivery to the lung by inhalation may, for example, be presented in capsules and cartridges of for example gelatine or blisters of for example laminated aluminium foil, for use in an inhaler or insufflator.
- Formulations generally contain a powder mix for inhalation of the compound of the invention and a suitable powder base (carrier substance) such as lactose or starch. Use of lactose is preferred.
- a suitable powder base such as lactose or starch.
- lactose is preferred.
- Each capsule or cartridge may generally contain between 0.001 -50 mg, more preferably 0.01 -5 mg of active ingredient or the equivalent amount of a pharmaceutically acceptable salt thereof.
- the active ingredient (s) may be presented without excipients.
- Packaging of the formulation may be suitable for unit dose or multi-dose delivery.
- the formulation can be pre-metered or metered in use. Dry powder inhalers are thus classified into three groups: (a) single dose, (b) multiple unit dose and (c) multi dose devices.
- inhalers of the first type single doses have been weighed by the manufacturer into small containers, which are mostly hard gelatine capsules.
- a capsule has to be taken from a separate box or container and inserted into a receptacle area of the inhaler.
- the capsule has to be opened or perforated with pins or cutting blades in order to allow part of the inspiratory air stream to pass through the capsule for powder entrainment or to discharge the powder from the capsule through these perforations by means of centrifugal force during inhalation.
- the emptied capsule has to be removed from the inhaler again.
- disassembling of the inhaler is necessary for inserting and removing the capsule, which is an operation that can be difficult and burdensome for some patients.
- Some capsule inhalers have a magazine from which individual capsules can be transferred to a receiving chamber, in which perforation and emptying takes place, as described in WO 92/03175.
- Other capsule inhalers have revolving magazines with capsule chambers that can be brought in line with the air conduit for dose discharge (e. g. WO91/02558 and GB 2242134). They comprise the type of multiple unit dose inhalers together with blister inhalers, which have a limited number of unit doses in supply on a disk or on a strip.
- Blister inhalers provide better moisture protection of the medicament than capsule inhalers. Access to the powder is obtained by perforating the cover as well as the blister foil, or by peeling off the cover foil.
- a blister strip is used instead of a disk, the number of doses can be increased, but it is inconvenient for the patient to replace an empty strip. Therefore, such devices are often disposable with the incorporated dose system, including the technique used to transport the strip and open the blister pockets.
- Multi-dose inhalers do not contain pre-measured quantities of the powder formulation. They consist of a relatively large container and a dose measuring principle that has to be operated by the patient. The container bears multiple doses that are isolated individually from the bulk of powder by volumetric displacement.
- rotatable membranes Ex. EP0069715
- disks Ex. GB 2041763; EP 0424790; DE 4239402 and EP 0674533
- rotatable cylinders Ex. EP 0166294; GB 2165159 and WO 92/09322
- rotatable frustums Ex. WO 92/00771
- Other multi dose devices have measuring slides (Ex. US 5201308 and WO 97/00703) or measuring plungers with a local or circumferential recess to displace a certain volume of powder from the container to a delivery chamber or an air conduit (Ex.
- Multi dose inhalers can contain a much higher number of doses, whereas the number of handlings to prime a dose is generally lower. Because the inspiratory air stream in multi-dose devices is often straight across the dose measuring cavity, and because the massive and rigid dose measuring systems of multi dose inhalers can not be agitated by this inspiratory air stream, the powder mass is simply entrained from the cavity and little de-agglomeration is obtained during discharge.
- compositions of the invention can be administered in aerosols which operate via propellant gases or by means of so-called atomisers, via which solutions of pharmacologically-active substances can be sprayed under high pressure so that a mist of inhalable particles results.
- atomisers such as the Respimat® which is described, for example, in PCT Patent Applications Nos. W0 91/14468 and WO 97/12687, reference here is being made to the contents thereof.
- Spray compositions for topical delivery to the lung by inhalation may for example be formulated as aqueous solutions or suspensions or as aerosols delivered from pressurised packs, such as a metered dose inhaler, with the use of a suitable liquefied propellant.
- Aerosol compositions suitable for inhalation can be either a suspension or a solution and generally contain the active ingredient (s) and a suitable propellant such as a fluorocarbon or hydrogen-containing chlorofluorocarbon or mixtures thereof, particularly hydrofluoroalkanes, e. g.
- dichlorodifluoromethane trichlorofluoromethane, dichlorotetra-fluoroethane, especially 1 ,1 , 1 , 2-tetrafluoroethane, 1 ,1 , 1 ,2, 3,3, 3- heptafluoro-n-propane or a mixture thereof.
- Carbon dioxide or other suitable gas may also be used as propellant.
- the aerosol composition may be excipient free or may optionally contain additional formulation excipients well known in the art such as surfactants (eg oleic acid or lecithin) and cosolvens (eg ethanol).
- Pressurised formulations will generally be retained in a canister (eg an aluminium canister) closed with a valve (eg a metering valve) and fitted into an actuator provided with a mouthpiece.
- Medicaments for administration by inhalation desirably have a controlled particle size.
- the optimum particle size for inhalation into the bronchial system is usually 1 -10 ⁇ , preferably 2-5 ⁇ . Particles having a size above 20 ⁇ are generally too large when inhaled to reach the small airways.
- the particles of the active ingredient as produced may be size reduced by conventional means eg by micronisation.
- the desired fraction may be separated out by air classification or sieving.
- the particles will be crystalline.
- an excipient such as lactose or glucose is generally employed.
- the particle size of the excipient will usually be much greater than the inhaled medicament within the present invention.
- lactose it will typically be present as milled lactose, preferably crystalline alpha lactose monohydrate.
- Pressurized aerosol compositions will generally be filled into canisters fitted with a valve, especially a metering valve.
- Canisters may optionally be coated with a plastics material e. g. a fluorocarbon polymer as described in W096/32150.
- Canisters will be fitted into an actuator adapted for buccal delivery.
- Nasal mucosal administration The compounds of the invention may also be administered via the nasal mucosal.
- compositions for nasal mucosa administration are typically applied by a metering, atomizing spray pump and are in the form of a solution or suspension in an inert vehicle such as water optionally in combination with conventional excipients such as buffers, anti-microbials, tonicity modifying agents and viscosity modifying agents.
- an inert vehicle such as water
- excipients such as buffers, anti-microbials, tonicity modifying agents and viscosity modifying agents.
- the compounds of the invention may also be administered directly into the blood stream, into muscle, or into an internal organ.
- Suitable means for parenteral administration include intravenous, intraarterial, intraperitoneal, intrathecal, intraventricular, intraurethral, intrasternal, intracranial, intramuscular and subcutaneous.
- Suitable devices for parenteral administration include needle (including microneedle) injectors, needle-free injectors and infusion techniques.
- Parenteral formulations are typically aqueous solutions which may contain excipients such as salts, carbohydrates and buffering agents (preferably to a pH of from 3 to 9), but, for some applications, they may be more suitably formulated as a sterile nonaqueous solution or as a dried form to be used in conjunction with a suitable vehicle such as sterile, pyrogen-free water.
- excipients such as salts, carbohydrates and buffering agents (preferably to a pH of from 3 to 9)
- a suitable vehicle such as sterile, pyrogen-free water.
- parenteral formulations under sterile conditions may readily be accomplished using standard pharmaceutical techniques well known to those skilled in the art.
- solubility of compounds of the invention used in the preparation of parenteral solutions may be increased by the use of appropriate formulation techniques, such as the incorporation of solubility-enhancing agents.
- Formulations for parenteral administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- compounds of the invention may be formulated as a solid, semi-solid, or thixotropic liquid for administration as an implanted depot providing modified release of the active compound. Examples of such formulations include drug-coated stents and PGLA microspheres.
- the compounds of the invention may also be administered topically to the skin or mucosa, that is, dermally or transdermally.
- Typical formulations for this purpose include gels, hydrogels, lotions, solutions, creams, ointments, dusting powders, dressings, foams, films, skin patches, wafers, implants, sponges, fibers, bandages and microemulsions.
- Liposomes may also be used.
- Typical carriers include alcohol, water, mineral oil, liquid petrolatum, white petrolatum, glycerin, polyethylene glycol and propylene glycol.
- Penetration enhancers may be incorporated; see, for example, J Pharm Sci, 88 (10), 955-958 by Finnin and Morgan (October 1999).
- Other means of topical administration include delivery by electroporation, iontophoresis, phonophoresis, sonophoresis and microneedle or needle-free injection.
- Formulations for topical administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release. vii) Rectal/lntravaginal Administration
- Compounds of the invention may be administered rectally or vaginally, for example, in the form of a suppository, pessary, or enema. Cocoa butter is a traditional suppository base, but various alternatives may be used as appropriate.
- Formulations for rectal/vaginal administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release. viii) Ocular Administration
- Compounds of the invention may also be administered directly to the eye or ear, typically in the form of drops of a micronized suspension or solution in isotonic, pH- adjusted, sterile saline.
- Other formulations suitable for ocular and aural administration include ointments, biodegradable ⁇ e.g. absorbable gel sponges, collagen) and nonbiodegradable (e.g. silicone) implants, wafers, lenses and particulate or vesicular systems, such as niosomes or liposomes.
- a polymer such as crossed-linked polyacrylic acid, polyvinylalcohol, hyaluronic acid, a cellulosic polymer, for example, hydroxypropylmethylcellulose, hydroxyethylcellulose, or methyl cellulose, or a heteropolysaccharide polymer, for example, gelan gum, may be incorporated together with a preservative, such as benzalkonium chloride.
- a preservative such as benzalkonium chloride.
- Such formulations may also be delivered by iontophoresis.
- Formulations for ocular/aural administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted, or programmed release. ix) Other Technologies
- Compounds of the invention may be combined with soluble macromolecular entities, such as cyclodextrin and suitable derivatives thereof or polyethylene glycol-containing polymers, in order to improve their solubility, dissolution rate, taste-masking, bioavailability and/or stability for use in any of the aforementioned modes of administration.
- soluble macromolecular entities such as cyclodextrin and suitable derivatives thereof or polyethylene glycol-containing polymers
- the amount of the active compound administered will be dependent on the subject being treated, the severity of the disorder or condition, the rate of administration, the disposition of the compound and the discretion of the prescribing physician. However, an effective dosage is typically in the range of 0.01 -3000 mg, more preferably 0.5-1000 mg of active ingredient or the equivalent amount of a pharmaceutically acceptable salt thereof per day. Daily dosage may be administered in one or more treatments, preferably from 1 to 4 treatments, per day.
- the pharmaceutical compositions of the invention are made up in a form suitable for oral, inhalation or topical administration, being particularly preferred oral or inhalation administration.
- compositions may conveniently be presented in unit dosage form and may be prepared by any of the methods well known in the art of pharmacy.
- the composition is in unit dosage form, for example a tablet, capsule or metered aerosol dose, so that the patient may administer a single dose.
- each active which is required to achieve a therapeutic effect will, of course, vary with the particular active, the route of administration, the subject under treatment, and the particular disorder or disease being treated.
- the invention also provides a combination product comprising (i) the compounds of the invention as described herein; and (ii) one or more additional active substances which are known to be useful in the treatment of respiratory diseases; allergic diseases; inflammatory or autoimmune-mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (MPDs); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors; more in particular wherein the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid artritis (RA), multiple sclerosis (MS), amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, cutaneous vasculitis, cutaneous lupus erythematosus,
- pyrrolopyrimidine derivatives defined herein may also be combined with other active compounds in the treatment of a pathological condition or disease susceptible to amelioration by inhibition of PI3Ks.
- the combinations of the invention can optionally comprise one or more additional active substances which are known to be useful in the treatment of respiratory diseases; allergic diseases; inflammatory or autoimmune-mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (MPDs); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors.
- respiratory diseases allergic diseases
- inflammatory or autoimmune-mediated diseases function disorders and neurological disorders
- cardiovascular diseases viral infection
- metabolism/endocrine function disorders neurological disorders and pain
- bone marrow and organ transplant rejection myelo-dysplastic syndrome
- MPDs myeloproliferative disorders
- the combinations of the invention can optionally comprise one or more additional active substances which are known to be useful in the treatment of neoplastic diseases (e.g. leukemia, lymphomas, solid tumors); transplant rejection, bone marrow transplant applications (e.g., graft- versus-host disease); autoimmune diseases (e.g.
- rheumatoid arthritis multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, cutaneous vasculitis, cutaneous lupus erythematosus, dermatomyositis and blistering diseases including but not limited to pemphigus vulgaris, bullous pemphigoid and epidermolysis bullosa; respiratory inflammation diseases (e.g.
- asthma chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis
- skin inflammatory diseases e.g., atopic dermatitis, contact dermatitis, eczema or psoriasis
- premalignant and malignant skin conditions e.g. basal cell carcinoma (BCC), squamous cell carcinoma (SCC) or actinic keratosis (AK)
- BCC basal cell carcinoma
- SCC squamous cell carcinoma
- AK actinic keratosis
- neurological disorders and pain such as pain associated with rheumatoid arthritis or osteoarthritis, back pain, general inflammatory pain, inflammatory neuropathic pain, trigeminal neuralgia or central pain).
- the combinations of the invention can optionally comprise one or more additional active substances which are known to be useful in the treatment of neoplastic diseases leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, type I diabetes, cutaneous vasculitis, cutaneous lupus erythematosus, dermatomyositis, blistering diseases including but not limited to pemphigus vulgaris, bullous pemphigoid and epidermolysis bullosa, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell
- the combinations of the invention can optionally comprise one or more additional active substances which are known to be useful in the treatment of neoplastic diseases leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, type I diabetes, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic keratosis
- the combinations of the invention comprise (i) a compound of the invention as defined above; and (ii) another compound selected from the group consisting of an Adenoside A 2 A agonist, an agent for treating cardiovascular disorders, an agent for treating diabetes, and an agent for treating liver disease, an anti-allergic agent, an anticholinergic agent, an anti-inflammatory agent, an anti-infective agent, a p2-adrenergic agonist, a Chemoattractant receptor homologous molecule expressed on TH 2 cells (CRTH2) inhibitor, a chemotherapeutic agent, a corticosteroid, an ⁇ / ⁇ (IkB kinase beta or IKK2) inhibitor, an immunosuppressant, a Janus kinase (JAK) inhibitor, a topically acting p38 Mitogen-Activated Protein Kinase (p38 MAPK) inhibitor, a Phosphosdiesterase (PDE) IV inhibitor, and a Spleen tyrosine kinase (Syk
- the combinations of the invention can optionally comprise one or more additional active substances selected from
- Dyhydrofolate reductase inhibitors such as Methotrexate or CH- 1504;
- DHODH Dihydroorotate dehydrogenase
- Immunomodulators such as Glatiramer acetate (Copaxone), Laquinimod or Imiquimod;
- Inhibitors of DNA synthesis and repair such as Mitoxantrone or Cladribine;
- Immunosuppressants such as Imuran (azathioprine) or Purinethol (6-mercaptopurine or 6-MP);
- Anti-alpha 4 integrin antibodies such as Natalizumab (Tysabri) ;
- Alpha 4 integrin antagonists such as R-1295 , TBC-4746, CDP-323, ELND-002, Firategrast or TMC-2003;
- Corticoids and glucocorticoids such as prednisone or methylprednisolone, fluticasone, mometasone, budesonide, ciclesonide or beta-metasone;
- Fumaric acid esters such as BG-12
- Anti-tumor necrosis factor-alpha Anti-TNF-alpha
- monoclonal antibodies such as Infliximab, Adalimumab or Certolizumab pegol
- Soluble Tumor necrosis factor-alpha Antagonists such as Ethanercept
- Anti-CD20 (lymphocyte protein) monoclonal antibodies such as Rituximab, Ocrelizumab Ofatumumab or TRU-015;
- Anti-CD52 (lymphocyte protein) monoclonal antibodies such as alemtuzumab
- Anti-CD25 (lymphocyte protein) such as daclizumab
- Anti-CD88 lymphocyte protein
- eculizumab or pexilizumab a lymphocyte protein
- IL-6R Anti-lnterleukin 6 Receptor
- Inosine-monophosphate dehydrogenase (IMPDH) inhibitors such as mycophenolate mophetyl, ribavirin, mizoribine or mycophenolic acid;
- Cannabinoid receptor agonists such as Sativex
- Chemokine CCR1 antagonists such as MLN-3897 or PS-031291 ;
- Chemokine CCR2 antagonists such as INCB-8696;
- Necrosis factor-kappaB NF-kappaB or NFKB
- Activation Inhibitors such as Sulfasalazine, Iguratimod or MLN-0415;
- Adenosine A 2A agonists such as ATL-313, ATL-146e, CGS-21680,
- S1 P Sphingosine-1 (S1 P) phosphate receptor agonists such as fingolimod, BAF-312, or ACT128800;
- S1 P Sphingosine-1 (S1 P) liase inhibitors such as LX2931 ;
- Spleen tyrosine kinase (Syk) inhibitors such as R-1 12;
- PKC Protein Kinase Inhibitors
- Beta adrenergic agonists such as formoterol, indacaterol or LAS100977 (abediterol);
- MABA molecules with dual activity: beta-adrenergic agonists and muscarinic receptor antagonists
- Histamine 1 (H1 ) receptor antagonists such as azelastine or ebastine
- Cysteinyl leukotriene (CysLT) receptor antagonists such as montelukast
- Mast cell stabilizers such as nedocromil or chromoglycate
- FLAP 5-lipoxygenase-activating protein
- 5-lipoxygenase (5-LO) inhibitors such as WY-50295T
- 5-LO 5-lipoxygenase
- CRTH2 Chemoattractant receptor homologous molecule expressed on TH 2 cells (CRTH2) inhibitors, such as OC-459, AZD-1981 , ACT- 129968, QAV-680;
- Vitamin D derivatives like calcipotriol (Daivonex) ;
- Anti-inflammatory agents such as non-steroidal anti-inflammatory drugs (NSAIDs) or selective cyclooxygenase-2 (COX-2) inhibitors such as aceclofenac, diclofenac, ibuprofen, naproxen, apricoxib, celecoxib, cimicoxib, deracoxib, etoricoxib, lumiracoxib, parecoxib sodium, rofecoxib, selenocoxib-1 or valdecoxib;
- NSAIDs non-steroidal anti-inflammatory drugs
- COX-2 selective cyclooxygenase-2
- Phosphodiestearase (PDE) III inhibitors Phosphodiestearase (PDE) III inhibitors
- Phosphosdiesterase (PDE) IV inhibitors such as roflumilast or GRC-4039;
- Xanthine derivatives such as theophylline or theobromine
- p38 Mitogen-Activated Protein Kinase (p38 MAPK) Inhibitors such as ARRY-797;
- Mitogen-activated extracellular signal regulated kinase kinase (MEK) inhibitor such as ARRY-142886 or ARRY-438162;
- Janus kinase (JAK) inhibitors such as tofacitinib (previously known as tasocitinib or CP-690,550) from Pfizer and INCB-18424, from Incyte;
- Interferons comprising Interferon beta 1 a such as Avonex from Biogen personal, CinnoVex from CinnaGen and Rebif from EMD Serono, and Interferon beta 1 b such as Betaferon from Schering and Betaseron from Berlex;
- Interferon alpha such as Sumiferon MP
- EGFR Epidermal Growth Factor Receptor
- EGFR inhibitors such as erlotinib, Trastuzumab, Herceptin, Avastin, Platins (cisplatin, carboplatin) or Temazolamide;
- Antineoplastic agents such as Docetaxel, Estramustine, Anthracyc lines, (doxorubicin (Adriamycin), epirubicin (Ellence), and liposomal doxorubicin (Doxil)), Taxanes (docetaxel (Taxotere), paclitaxel (Taxol), and protein-bound paclitaxel (Abraxane)), Cyclophosphamide (Cytoxan), Capecitabine (Xeloda), 5 fluorouracil (5 FU), Gemcitabine (Gemzar) or Vinorelbine (Navelbine);
- suitable corticoids and glucocorticoids that can be combined with the PI3K inhibitors of the present invention are prednisolone, methylprednisolone, dexamethasone, dexamethasone cipecilate, naflocort, deflazacort, halopredone acetate,
- Suitable Syk kinase inhibitors that can be combined with the PI3K inhibitors of the present invention are fosfamatinib (from Rigel), R-348 (from Rigel), R- 343 (from Rigel), R-1 12 (from Rigel), piceatannol, 2-(2-Aminoethylamino)-4-[3- (trifluoromethyl)phenylamino] pyrimidine-5-carboxamide, R-091 (from Rigel), 6-[5- Fluoro-2-(3,4,5-trimethoxyphenylamino)pyrimidin-4-ylamino]-2,2-dimethyl-3,4-dihydro- 2H-pyrido[3,2-b][1 ,4]oxazin-3-one benzenesulfonate (R-406 from Rigel), 1 -(2,4,6- Trihydroxyphenyl)-2-(4-methoxyphenyl)ethan-1 -one, N-[4-[6-(6
- M3 antagonists anticholinergics
- beta adrenergic agonists that can be combined with the PI3K inhibitors of the present invention are are terbutaline sulphate, eformoterol fumarate, formoterol fumarate, bambuterol, ibuterol, isoprenaline hydrochloride, dopexamine, metaprotenerol, tulobuterol, procaterol hydrochloride, sibenadet hydrochloride, mabuterol hydrochloride, albuterol sulphate, salbutamol sulphate, salmefamol, salmeterol xinafoate, carmoterol hydrochloride, (R)-albuterol hydrochloride, Levalbuterol hydrochloride; Levosalbutamol hydrochloride; (-)- Salbutamol hydrochloride, formoterol, (R,R)-Formoterol tartrate; Arformoterol tartrate,
- anti-allergic agents that can be combined with the PI3K inhibitors of the present invention are anti-histamines (e.g. Methapyrilene, Mequitazine, Azelastine hydrochloride, Acrivastine, Emedastine difumarate, Emedastine fumarate, Loratadine, Cyproheptadine hydrochloride, Diphenhydramine hydrochloride, Doxepin hydrochloride, Promethazine hydrochloride, Levocabastine hydrochloride, Desloratadine, Cinnarizine, Setastine hydrochloride, Mizolastine, Ebastine, Cetirizine hydrochloride, Epinastine hydrochloride, Olopatadine hydrochloride, Bepotastine besilate,Triprolidine hydrochloride, Rupatadine fumarate, Fexofenadine hydrochloride, Levocetirizine dihydrochloride, Ketotif
- Phosphosdiesterase IV (PDE IV) inhibitors that can be combined with the PI3K inhibitors of the present invention are benafentrine dimaleate, etazolate, denbufylline, rolipram, cipamfylline, zardaverine, arofylline, filaminast, tipelukast, tofimilast, piclamilast, tolafentrine, mesopram, drotaverine hydrochloride, lirimilast, roflumilast, cilomilast, oglemilast, apremilast, tetomilast, filaminast, (R)-(+)-4- [2-(3-Cyclopentyloxy-4-methoxyphenyl)-2-phenylethyl]pyridine (CDP-840), N-(3,5- Dichloro-4-pyridinyl)-2-[1 -(
- Suitable immunosupressants that can be combined with the PI3K inhibitors of the present invention are picremolimus, tacrolimus, cyclosporine A, leflunomide, teriflunomide, vidofludimus, laquinimod, methotrexate, 5-fluorouracil (5- FU), anti-TNF agents and compounds described in PCT patent applications Nos.
- WO 2008/077639, WO 2009/021696, WO 2009/153043, and WO2010083975 (in particular amino(iso)nicotinic acid derivatives selected from the group consisting of 2-(3'-ethoxy- 3-(trifluoromethoxy)biphenyl-4-ylamino)nicotinic acid, 2-(3,5-difluoro-3'- methoxybiphenyl-4-ylamino)nicotinic acid and 2-(3,5-difluoro-2-methylbiphenyl-4- ylamino)nicotinic acid; and azabiphenylaminobenzoic acid derivatives selected from the group consisting of 5-cyclopropyl-2-(2-(2,6-difluorophenyl)pyrimidin-5-ylamino)benzoic acid, 5-cyclopropyl-2-((2-(2-(trifluoromethyl)phenyl)pyrimidin-5-yl)amino)benz
- Particularly preferred combination products according to the invention comprise a compound of formula (I) and a therapeutically effective amount of one or more additional therapeutic agents selected from the group consisting of mometasone furoate, ciclesonide, budesonide, fluticasone propionate, fluticasone furoate, betamethasone valerate, clobetasol propionate, tiotropium salts, glycopyrronium salts, 3-[2-Hydroxy-2,2-bis(2-thienyl)acetoxy]-1 -(3-phenoxypropyl)-1 - azoniabicyclo[2.2.2]octane salts (in particular aclidinium salts, preferably aclidinium bromide), 1 -(2-Phenylethyl)-3-(9H-xanthen-9-ylcarbonyloxy)-1 - azoniabicyclo[2.2.2]octane salts, formoterol, salmeterol, inda
- WO 2008/077639, WO 2009/021696, WO 2009/153043, and WO 2010/083975 (in particular amino(iso)nicotinic acid derivatives selected from the group consisting of 2- (3'-ethoxy-3-(trifluoromethoxy)biphenyl-4-ylamino)nicotinic acid, 2-(3,5-difluoro-3'- methoxybiphenyl-4-ylamino)nicotinic acid and 2-(3,5-difluoro-2-methylbiphenyl-4- ylamino)nicotinic acid; and azabiphenylaminobenzoic acid derivatives selected from the group consisting of 5-cyclopropyl-2-(2-(2,6-difluorophenyl)pyrimidin-5-ylamino)benzoic acid, 5-cyclopropyl-2-((2-(2-(trifluoromethyl)phenyl)pyrimidin-5-yl)amino)benz
- the compounds of formula (I) and the combinations of the invention may be used in the treatment of respiratory diseases; allergic diseases; inflammatory or autoimmune- mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (MPDs such as polycythemia vera, essential thrombocythemia or mielofibrosis); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors, wherein the use of a PI3K inhibitor is expected to have a beneficial effect, for example leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, cutaneous vasculitis, cutaneous lupus ery
- the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic keratosis.
- the active compounds in the combination product may be administered together in the same pharmaceutical composition or in different compositions intended for separate, simultaneous, concomitant or sequential administration by the same or a different route.
- all active agents would be administered at the same time, or very close in time.
- one or two actives could be administered in the morning and the other (s) later in the day.
- one or two actives could be administered twice daily and the other (s) once daily, either at the same time as one of the twice-a-day dosing occurred, or separately.
- at least two, and more preferably all, of the actives would be administered together at the same time.
- at least two, and more preferably all actives would be administered as an admixture.
- the invention is also directed to a combination product of the compounds of the invention together with one or more other therapeutic agents for use in the treatment of a pathological condition or disease susceptible to amelioration by inhibiton of Phosphoinositide 3-Kinases (PI3Ks), in particular wherein the pathological condition or disease is selected from respiratory diseases; allergic diseases; inflammatory or autoimmune-mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo- dysplastic syndrome; myeloproliferative disorders (MPDs such as polycythemia vera, essential thrombocythemia or mielofibrosis); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors; more in particular wherein the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcer
- the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic keratosis.
- the invention also encompasses the use of a combination of the compounds of the invention together with one or more other therapeutic agents for the manufacture of a formulation or medicament for treating these diseases.
- the invention also provides a method of treatment of a pathological condition or disease susceptible to amelioration by inhibiton of Phosphoinositide 3-Kinases (PI3Ks), in particular wherein the pathological condition or disease is selected from respiratory diseases; allergic diseases; inflammatory or autoimmune-mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (MPDs such as polycythemia vera, essential thrombocythemia or mielofibrosis); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors; more in particular wherein the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis
- the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic keratosis.
- the active compounds in the combinations of the invention may be administered by any suitable route, depending on the nature of the disorder to be treated, e.g. orally (as syrups, tablets, capsules, lozenges, controlled-release preparations, fast-dissolving preparations, etc); topically (as creams, ointments, lotions, nasal sprays or aerosols, etc); by injection (subcutaneous, intradermic, intramuscular, intravenous, etc.) or by inhalation (as a dry powder, a solution, a dispersion, etc).
- suitable route e.g. orally (as syrups, tablets, capsules, lozenges, controlled-release preparations, fast-dissolving preparations, etc); topically (as creams, ointments, lotions, nasal sprays or aerosols, etc); by injection (subcutaneous, intradermic, intramuscular, intravenous, etc.) or by inhalation (as a dry powder, a solution, a dispersion,
- the active compounds in the combination i.e. the pyrrolotriazinone derivatives of the invention, and the other optional active compounds may be administered together in the same pharmaceutical composition or in different compositions intended for separate, simultaneous, concomitant or sequential administration by the same or a different route.
- One execution of the present invention consists of a kit of parts comprising a pyrrolopyrimidine derivative of the invention together with instructions for simultaneous, concurrent, separate or sequential use in combination with another active compound useful in the treatment of respiratory diseases; allergic diseases; inflammatory or autoimmune-mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo- dysplastic syndrome; myeloproliferative disorders (MPDs such as polycythemia vera, essential thrombocythemia or mielofibrosis); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors; more in particular wherein the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia
- the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic keratosis.
- Another execution of the present invention consists of a package comprising a imidazopyridine derivative of the invention and another active compound useful in the treatment of respiratory diseases; allergic diseases; inflammatory or autoimmune- mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (MPDs such as polycythemia vera, essential thrombocythemia or mielofibrosis); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors; more in particular wherein the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, cutaneous vasculitis, cutaneous
- the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic keratosis.
- the invention further provides synthetic processes which are useful for preparing said compounds.
- the compounds of the invention can be prepared using the methods and procedures described in WO 2012/146666 or WO 2012/146667, or using similar methods and procedures. It will be appreciated that where typical or preferred process conditions (i.e., reaction temperatures, times, mole ratios of reactants, solvents, pressures, etc.) are given; other process conditions can also be used unless otherwise stated. Optimum reaction conditions may vary with the particular reactants or solvent used, but such conditions can be determined by one skilled in the art by routine optimization procedures.
- protecting groups may be necessary to prevent certain functional groups from undergoing undesired reactions.
- the choice of a suitable protecting group for a particular functional group, as well as suitable conditions for protection and deprotection, are well known in the art. For example, numerous protecting groups, and their introduction and removal are described in T. W. Greene and G. M. Wuts, Protecting Groups in Organic Synthesis, Third Edition, Wiley, New York, 1999, and references cited therein.
- amino-protecting group refers to a protecting group suitable for preventing undesired reactions at amino nitrogen.
- Representative amino-protecting groups include, but are not limited to, formyl; acyl groups, for example alkanoyl groups such as acetyl; alkoxycarbonyl groups such as tert-butoxycarbonyl (Boc); arylmethoxycarbonyl groups such as benzyloxycarbonyl (Cbz) and 9-fluorenylmethoxycarbonyl (Fmoc); arylmethyl groups such as benzyl (Bn), trityl (Tr), and 1 ,1 -di-(4'-methoxyphenyl)methyl; silyl groups such as trimethylsilyl (TMS) and tert-butyldimethylsilyl (TBS); and the like.
- hydroxy-protecting group refers to a protecting group suitable for preventing undesired reactions at a hydroxy group.
- Representative hydroxy-protecting groups include, but are not limited to, alkyl groups, such as methyl, ethyl, and tert-butyl; acyl groups, for example alkanoyl groups, such as acetyl; arylmethyl groups, such as benzyl (Bn), p-methoxybenzyl (PMB), 9-fluorenylmethyl (Fm), and diphenylmethyl (benzhydryl, DPM); silyl groups, such as trimethylsilyl (TMS) and tert-butyldimethylsilyl (TBS); and the like.
- compounds of general Formula (I) may be prepared by the synthetic route illustrated in Scheme 1 , from compounds of Formula (II)
- compounds of Formula (I) can be obtained in two step synthesis from compounds of Formula (IV), where compounds of Formula (IV) can be obtained from reaction of compounds of Formula (II) with compounds of Formula (V) also in the presence of a suitable base such as potassium carbonate, diisopropylethylamine or sodium hydride in an appropriate solvent such as ie f-butanol, /V,/V-dimethylformamide or tetrahydrofuran at temperatures ranging from room temperature to 160 °C, with or without the use of microwaves irradiation and with or without the use of a catalytic amount of caesium fluoride.
- a suitable base such as potassium carbonate, diisopropylethylamine or sodium hydride
- an appropriate solvent such as ie f-butanol, /V,/V-dimethylformamide or tetrahydrofuran at temperatures ranging from room temperature to 160 °C, with or without the use of microwaves ir
- compounds of Formula (I) can be prepared in the two steps synthesis from compounds of Formula (IV) by reaction with the compounds of Formula (VI) where X represents a boronic acid or boronic ester using standard Suzuki coupling conditions.
- compounds of general Formula (I) where L represents a linker selected from -0-, -S-, -NH- group compounds can be obtained from compounds of Formula (IV) by reacting with compounds of Formula (VI) where X represents an hydrogen using copper or palladium catalysed coupling methods well known for those skilled in the art.
- compounds of Formula (I) can be prepared in the two steps synthesis from compounds of Formula (IV) by treatment with a lithiating agent such as n-BuLi, in a non protic solvent such as hexane or tetrahydrofurane, at a temperature between -78°C and 0°C and subsequently treated with compounds of Formula (VI) where X represents an halogen or another leaving group at a temperature between -78°C and room temperature.
- a lithiating agent such as n-BuLi
- a non protic solvent such as hexane or tetrahydrofurane
- Compounds of Formula (VI) can either be commercial or prepared by standard methods, and can be used in a protected form to prevent certain functional groups from undergoing undesired reactions. In these cases, standard methods for the removal of these protecting groups can be used at the suitable step of the synthesis. Numerous protecting groups, their introduction and their removal are described in T. W. Greene and G. M. Wuts, Protecting Groups in Organic Synthesis, Third Edition, Wiley, New York, 1999, and references cited therein. Finally, compounds of general Formula (II) are known and prepared as described in WO 2005/1 13556, US 2009/0312319 and WO 2012/146667.
- Scheme 1 The invention is also directed to a compound of the invention as described herein for use in the treatment of the human or animal body by therapy.
- Compounds of the invention intended for pharmaceutical use may be administered as crystalline or amorphous products, or mixtures thereof. They may be obtained, for example, as solid plugs, powders, or films by methods such as precipitation, crystallization, freeze drying, spray drying, or evaporative drying. Microwave or radio frequency drying may be used for this purpose.
- Reaction products were purified, when necessary, in a Biotage SP1 ® automated purification system. Purifications in normal phase were made in the solvent system as indicated. Purifications in reverse phase were made using a Ci 8 column and a gradient of water-acetonitrile/MeOH (1 :1 ) (0.1 % v/v ammonium formate both phases) from 0% to 100% acetonitrile/MeOH (1 :1 ) in 40 column volumes. The appropriate fractions were collected and the solvents evaporated under reduced pressure and/or liofilized.
- Preparative HPLC-MS were performed on a Waters instrument equipped with a 2767 injector/collector, a 2525 binary gradient pump, a 2996 PDA detector, a 515 pump as a make-up pump and a ZQ4000 Mass spectrometer detector or on a Agilent 1200 Series coupled to an Agilent 6120 Mass spectrometer detector. Both systems were equipped with a Symmetry Prep Ci 8 (19 x 300 mm, 7 ⁇ ) column or a XBridge Prep Ci 8 (19 x 100 mm, 5 ⁇ ) column.
- the mobile phase was formic acid (0.4 mL), ammonia (0.1 mL), methanol (500 mL) and acetonitrile (500 mL) (B) and formic acid (0.5 mL), ammonia (0.125 mL) and water (1000 mL) (A), the specific gradients used are specified in each particular case.
- the flow rate was 20 mL/min.
- the mobile phase was formic acid (0.4 mL), ammonia (0.1 mL), methanol (500 mL) and acetonitrile (500 mL) (B) and formic acid (0.5 mL), ammonia (0.125 mL) and water (1000 mL) (A) and a gradient between 0 to 95% of B was used.
- UPLC ACQUITY UPLC BEH C-18 (2.1 x50mm, 1 .7 m) 1 H Nuclear Magnetic Resonance Spectra were recorded on a Varian Mercury plus operating at a frequency of 400 MHz for the 1 H spectra. Samples were dissolved in the specified deuterated solvent. Tetramethylsilane was used as reference.
- the reaction mixture was stirred at 130°C for 7h. More caesium fluoride (92 mg, 0.60 mmol) and ethyldiisopropylamine (0.21 mL, 1.21 mmol) were added and the mixture was stirred under previous conditions overnight. Water was added (25 mL) and the mixture extracted with ethyl acetate (20 mL, 3x). The organic extracts were collected together and washed with water and brine. The organic phase was dried over anhydrous sodium sulphate, filtered and concentrated in vacuum. The residue was purified using a SP1 Purification System (diethyl ether- methanol from 0% to 5%) to give 31 mg (25% yield) of the title compound.
- SP1 Purification System diethyl ether- methanol from 0% to 5%
- the reaction mixture was stirred at 130°C for 7h. More caesium fluoride (90mg, 0.60mmol) and ethyldiisopropylamine (0.21 mL, 1.21 mmol) were added and the mixture stirred under previous conditions overnight. Water was added (25 mL) and the aqueous phase was extracted with ethyl acetate (20 mL, 3x). The organic extracts were collected together and washed with water and brine. The organic phase was dried over anhydrous sodium sulphate, filtered and concentrated in vacuum. The residue was purified using a SP1 Purification System (hexane-diethyl ether from 0% to 100%) to give 39 mg (27% yield) of the title compound.
- SP1 Purification System hexane-diethyl ether from 0% to 100%
- the reaction mixture was stirred at room temperature for 2h and then poured into water (25 mL) and extracted with ethyl acetate. The organic phase was washed with water and brine, dried, filtered and concentrated in vacuum. The crude obtained was purified by flash chromatography (hexane: ethyl acetate from 0% to 100%) to afford 70 mg (56% yield) of the expected compound.
- Butyldimethylsilyl)oxy)ethyl)-4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 /-/- pyrazole 200 mg, 0.57 mmol
- 2M sodium carbonate 0.3 ml_, 0.60 mmol
- tetrakis(triphenylphosphine)palladium(0) 30 mg, 0.03 mmol
- phenylisoquinolin-1 (2/-/)-one (69 mg, 0.09 mmol) was dissolved in trifluoroacetic acid (1 ml, 13 mmol) and the mixture was stirred at room temperature for 1 h. The solvent was concentrated in vacuum and a solution of ammonia 7N in methanol (1 ml_, 7 mmol) was added. The reaction mixture was stirred at room temperature 2 h and then was evaporated in vacuum. The crude was re-dissolved in dichloromethane and the resulting organic phase was washed with water, a saturated sodium carbonate solution and brine. The combined organics were dried over anhydrous magnesium sulphate, filtered and evaporated under reduced pressure to give 40 mg (70% yield) of the title compound.
- phenylisoquinolin-1 (2/-/)-one (53 mg, 0.06 mmol) was treated with trifluoroacetic acid ( 1 ml_, 13 mmol) and a solution of ammonia 7N in methanol (1 ml_, 7 mmol) according to the method described in Example 10 to give 32 mg (73% yield) of the title compound.
- PI3K ⁇ , ⁇ , ⁇ and ⁇ Enzymatic Inhibition Assays Compounds were screened for their ability to inhibit PI3Ka (PI3Ka), ⁇ 3 ⁇ (PI3Kb), PI3K5 (PI3Kd) and ⁇ 3 ⁇ (PI3Kg) using a cell-free based PI3K HTRF TM assay (Millipore, ref. #33-017).
- PI-3 Kinase HTRF kit (ref. #33-037) and the different PI3K recombinant isoforms (ref. #14-602, ref. #14-603, ref .#14-604, ref.#15-558 for Alpha, Beta, Delta and Gamma respectively) were purchased at Millipore (expressed in insect cells). ATP was purchased at Sigma Aldrich (ref. #A7699).
- the compounds were pre-incubated with the enzyme for 30 min before starting of the catalytic reaction.
- [PIP2] was used at its Km.
- [ATP] was used at 15 ⁇ for all isoforms for technical reasons (Km values varied between 10 and 20 ⁇ depending on the isoform).
- Time of assay and [Enzyme] were optimized to work in the linear range. Stop and Detection mixtures were used as specified in the Millipore PI-3 Kinase kit.
- Reaction time and enzyme concentration in the assay will depend of each batch.
- the compounds of formula (I) are potent inhibitors of Phosphoinositide 3-kinase delta (PI3kd).
- Preferred compounds of the invention possess an IC 50 value for the inhibition of PI3Kd (determined as defined above) of less than 10 ⁇ (10,000 nM), preferably less than 1 ⁇ (1 ,000 nM), even more preferably of less than 0.2 ⁇ (200 nM), most preferably less than 0.05 ⁇ (50 nM) Formulation Examples
- active compound is the compound of example 4. Modifications, which do not affect, alter, change or modify the essential aspects of the compounds, combinations or pharmaceutical compositions described, are included within the scope of the present invention.
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Immunology (AREA)
- Pulmonology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
New pyrrolopyrimidine derivatives having the chemical structure of formula (I) are disclosed; as well as process for their preparation, pharmaceutical compositions comprising them and their use in therapy as inhibitors of Phosphoinositide 3-Kinases (PI3Ks).
Description
PYRROLOPYRIMIDINE DERIVATIVES AS PI3K INHIBITORS
BACKGROUND OF THE INVENTION
When cells are activated by extracellular stimuli, intracellular signaling cascades involving the regulation of second messengers are initiated that eventually produce a response of the cell to the stimuli. Phosphoinositide 3-Kinases (PI3Ks) are among the enzymes involved in early signalling events to a plethora of different types of stimuli. PI3Ks phosphorylate the 3-hydroxyl group of the inositol ring of phosphatidylinositol (Ptdlns), Ptdlns-4-phosphate (Ptdlns4P), and Ptdlns-4,5-bisphosphate (Ptdlns(4,5)P2). The resulting 3-phosphoinositides mediate correct localization and subsequent activation of a number of downstream effector proteins that bind to the lipids via specific lipid binding sequences such as the pleckstrin homology (PH) domain (Vanhaesebroeck B, 2010, Nat Rev Mol Cell Biol 5:11381-6).
The PI3K family is divided into 3 different classes (PI3K class I, class II, and class III), depending on substrate preference and structural features.
The best characterized is the PI3K class I with the preferential substrate Ptdlns- (4,5)P2. It englobes 4 different isoforms which originally were further subdivided into class IA (p1 10a, p1 10b, p1 10d), binding to a p85 type of regulatory subunit, and class IB (p1 10g) which is regulated by p101 and p87 subunits. Whereas p1 10a (PI3Ka or PI3Ka) and p1 10b (PI3Kb or ΡΙ3Κβ) isoforms are expressed ubiquitously, p1 10g (PI3Kg or ΡΙ3Κγ) and especially p1 10d (PI3Kd or PI3K5) have a more restricted expression pattern and seem to play a major role in leukocytes (Kok K, Trends Biochem Science 34:115-127, 2009).
Both, PI3Kd and PI3Kg are involved in activation of immune cells by a large variety of different stimuli. Pharmacological inhibition or genetic deficiency in active p1 10d has been shown to inhibit T cell proliferation and cytokine production in response to different stimuli such as anti-CD3, anti-CD3/CD28, superantigen or antigen in vitro (Ji H, Blood 2007; Okkenhaug K, Science 2002; Garcon F, 2009; Soond DR, Blood 2010; Herman SEM, Blood June 3, 2010; William O, Chemistry & Biology 17, 2010) and to suppress concanavalin A and anti-CD3 induced cytokine production as well as antigen- dependent tissue retention in vivo (Soond DR, Blood 2010; Jarmin SJ, JCI 2008). In addition, B cell function is critically dependent on functional PI3Kd activity as demonstrated by suppressed B cell proliferation and cytokine release in vitro in
response to anti-lgM (Bilancio A, Blood 107, 2006), toll like receptor agonists such as LPS and oligodeoxynucleotides (Dil N, Mol Immunol 46, 2009) or impaired ability to stimulate antigen-specific T cells (Al-Alwan M, Jl 2007) in the absence of functional p1 10d or pharmacological inhibition. In vivo, PI3Kg deficient mice display partially suppressed antibody production upon immunization (Garcon F, 2009; Durand CA, Jl
2009) . Further studies have demonstrated an important role of PI3Kd in inhibition of T cell apoptosis and in TH 17 differentiation (Haylock-Jacobs S, J. Autoimmun 2010).
In addition, mast cell degranulation was reduced in cells from mice with inactivated PI3Kd or by pharmacological inhibition of PI3Kd (AN K, Nature 431 :1007-101 1 , 2004; AN K, Journal of Immunology 180:2538-2544, 2008) and basophil activation via the FcE receptor is suppressed by pharmacological inhibition of PI3Kd (Lannutti BJ, Blood Oct.
2010) . In terms of neutrophil function, PI3Kd inhibition inhibits migration of mouse neutrophils to fMLP in an under-agarose migration assay by inhibiting cell polarization and directional movement (Sadhu C, Jl 170, 2003) and mouse PI3Kd deficient or inhibitor treated neutrophils show slightly (25%) reduced in vitro chemotaxis to LTB4, whereas in vivo accumulation in the lung in response to LPS was reduced by more than 80%, indicating an important role of PI3Kd in endothelial cells for mediating PMN transendothelial migration (Puri KD, Blood 103, 2004). Furthermore, TNF induced neutrophil infiltration to an air pouch in mice and elastase release is partially inhibited by a PI3Kd selective inhibitor (Sadhu C, Biochem Biophys Res Comm 308, 2003). In addition, TNF mediated priming of oxidative burst by human neutrophils depends on PI3Kd activity (Condliffe AM, Blood 106, 2005).
In contrast to the dominant role of PI3Kd in lymphocyte activation, PI3Kg seems to affect primarily chemotaxis of different immune cells induced by various mediators and chemokines (Martin AL, Jl 180, 2008; Thomas MS, J Leukoc Biol 84, 2008; Jarmin SJ, JCI 2008; Matthew T, Immunology 126, 2008), as well as degranulation and oxidative burst of innate immune cells induced by GPCR mediated stimuli such as fMLP, IL-8 or C5a (Condliffe AM, Blood 106, 2005; Yum HK, Jl 167, 2001 ; Pinho V, Jl 179, 2007
The above mentioned findings suggest that selective PI3Kd or dual PI3Kd/PI3Kg pharmacological inhibition represents a promising approach for treating a variety of diseases such as respiratory diseases (asthma, chronic obstructive pulmonary disease (COPD), cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis), allergic diseases
(allergic rhinitis), inflammatory or autoimmune diseases (rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, myastenia gravias, acute disseminated encephalomyelitis, idiopathic thrombocytopenic purpura, Sjoegren's syndrome, autoimmune hemolytic anemia, type I diabetes, psoriasis, acrodermatitis, angiodermatitis, atopic dermatitis, contact dermatitis, eczema, acne, chronic urticaria, blistering diseases including but not limited to bullous pemphigoid, scleroderma, dermatomyositis, etc.), cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain (such as pain associated with rheumatoid arthritis or osteoarthritis, back pain, general inflammatory pain, inflammatory neuropathic pain, trigeminal neuralgia or central pain) as well as in bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (MPDs); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors (such as pancreatic cancer; bladder cancer; colorectal cancer; breast cancer; prostate cancer; renal cancer; hepatocellular cancer; lung cancer; ovarian cancer; cervical cancer; gastric cancer; esophageal cancer; head and neck cancer; non-small cell lung cancer and small-cell lung cancer; melanoma; neuroendocrine cancers; central nervious system cancers; brain tumors; bone cancer; soft tissue sarcoma; chronic lymphocytic leukemia, B-cell acute lymphoblastic leukemia, T-cell acute lymphoblastic leukaemia, non-hodgkins lymphoma, B-cell lymphoma, acute myeloid leukaemia; cutaneous T cell lymphoma, premalignant and malignant skin conditions including but not limited to basal cell carcinoma (BCC), squamous cell carcinoma (SCC) or actinic keratosis (AK)).
There is substantial experimental evidence supporting this view. In rodent models of allergic lung inflammation, genetic or pharmacolocical inactivation of PI3Kd or dual PI3Kd/g dual inhibition reduces cell influx, mucus production, cytokine production and airway hyperreactivity (Nashed et a. 2007, Eur J Immunol 37:416; Lee et al. 2006, FASEB J 20:455 & Lee KS et al. 2006, J Allergy Clin Immunol 1 18:403; Doukas J, JPET 2009;328:758; Par SJ, ERJ 2010). Moreover, LPS induced lung neutrophil infiltration is blocked by PI3Kd inhibition (Puri KD, Blood 2004;103:3448) and inflammation in response to LPS or tobacco smoke exposure is suppressed by a dual PI3Kd/g inhibitor (Doukas J, JPET 2009;328:758). Moreover, PI3Kd seems to be involved in the reduction of responsiveness to corticosteroid treatment associated with oxidative stress and chronic obstructive pulmonary disease (COPD). This notion is based on the findings that tobacco smoke induced inflammation remains responsive to treatment with budesonide, whereas wild type or PI3Kg deficient mice develop resistance to corticosteroid treatment (Marwick JA, JRCCM 179:542-548, 2009).
Similar results were obtained with a PI3Kd selective inhibitor (To Y, AJRCCM 182:897- 904, 2010). In addition, in vitro induction of corticosteroid resistance by oxidative stress is prevented by PI3Kd inhibition (To Y, AJRCCM 2010). In COPD patients, lung macrophages display increased expression of PI3Kd and phosphorylation of its downstream effector Akt and non-selective PI3K or PI3Kd- selective inhibition restored the impaired inhibitory efficacy of dexamethasone in PBMC from COPD patients (To Y, AJRCCM 182:897-904, 2010; Marwick JA, JACI 125:1 146-53, 2010).
Furthermore, PI3Kd inhibition was effective in a model of contact hypersensitivity (Soond DR, Blood Jan 2010). In a model of experimental autoimmune encephalomyelitis, PI3Kd deficiency or pharmacological inhibition of PI3Kd attenuated T cell activation and function and reduced T cell numbers in the CNS, suggesting a therapeutic benefit of PI3Kd inhibitor in multiple sclerosis and other Th17-mediated autoimmune diseases (Haylock-Jacobs S, J. Autoimmun 2010). In line with that, genetic deficiency or pharmacological inhibition of PI3Kd diminished joint erosion in a mouse model of inflammatory arthritis (Randis TM, Eur J Immunol 38, 2008).
Concerning metabolic diseases, PI3Kd overexpression seems to contribute to excessive vascular contraction and PI3Kd inhibition normalized vascular contractive responses in a mouse model of type I diabetes, suggesting a therapeutic potential of PI3Kd blockade to treat vascular dysfunction in diabetic patients (Pinho JF, Br. J. Pharmacol 161 , 2010).
There is also substantial experimental evidence supporting that genetic of pharmacolocical inactivation of PI3Kd or dual PI3Kd/g dual inhibition is effective in the treatment of cancers including but not restricted to leukemias, such as chronic lymphocytic leukemia, B-cell acute lymphoblastic leukemia, T-cell acute lymphoblastic leukaemia, non-hodgkins lymphoma, B-cell lymphoma, acute myeloid leukaemia, myelo-dysplastic syndrome or myelo-proliferative diseases. In this aspect, the selective PI3Kd inhibitor CAL-101 demonstrated anti-proliferative properties on different tumor cells in vitro and efficacy in cancer patients with a dysregulated PI3Kd activity, such as chronic lymphocytic leukemia (Hermann SE, Blood 1 16:2078-88, 2010; Lannutti BJ, Blood Oct. 2010).
Conditions in which targeting of the PI3K pathway or modulation of the PI3 Kinases, particularly PI3Kd or PI3Kd/g, are contemplated to be therapeutically useful for the treatment or prevention of diseases includes: respiratory diseases (asthma, chronic obstructive pulmonary disease (COPD), cystic fibrosis, idiopathic pulmonary fibrosis,
sarcoidosis), allergic diseases (allergic rhinitis), inflammatory or autoimmune-mediated diseases (rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, myastenia gravias, acute disseminated encephalomyelitis, idiopathic thromocytopenic purpura, Sjoegren's syndrome, autoimmune hemolytic anemia, type I diabetes, psoriasis, acrodermatitis, angiodermatitis, atopic dermatitis, contact dermatitis, eczema, acne, chronic urticaria, scleroderma, dermatomyositis and blistering diseases including but not limited to bullous pemphigoid), cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain (such as pain associated with rheumatoid arthritis or osteoarthritis, back pain, general inflammatory pain, inflammatory neuropathic pain, trigeminal neuralgia or central pain) as well as in bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (MPDs); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors (such as pancreatic cancer; bladder cancer; colorectal cancer; breast cancer; prostate cancer; renal cancer; hepatocellular cancer; lung cancer; ovarian cancer; cervical cancer; gastric cancer; esophageal cancer; head and neck cancer; non-small cell lung cancer and small-cell lung cancer; melanoma; neuroendocrine cancers; central nervious system cancers; brain tumors; bone cancer; soft tissue sarcoma; chronic lymphocytic leukemia, B-cell acute lymphoblastic leukemia, T-cell acute lymphoblastic leukaemia, non-hodgkins lymphoma, B-cell lymphoma, acute myeloid leukaemia; cutaneous T cell lymphoma, premalignant and malignant skin conditions including but not limited to basal cell carcinoma (BCC), squamous cell carcinoma (SCC) or actinic keratosis (AK)). In view of the numerous conditions that are contemplated to benefit by treatment involving modulation of the PI3K pathway or modulation of the PI3 Kinases it is immediately apparent that new compounds that modulate PI3K pathways and use of these compounds should provide substantial therapeutic benefits to a wide variety of patients.
BRIEF SUMMARY OF THE INVENTION
Provided herein are novel pyrrolopyrimidine derivatives for use in the treatment of conditions in which targeting of the PI3K pathway or inhibition of PI3 Kinases can be therapeutically useful.
The compounds described in the present invention are potent PI3K inhibitors, particularly PI3Kd or dual PK3Kd/g inhibitors. This property makes them useful for the treatment or prevention of pathological conditions or diseases such as respiratory diseases (asthma, chronic obstructive pulmonary disease (COPD), cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis), allergic diseases (allergic rhinitis), inflammatory or autoimmune diseases (rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, myastenia gravias, acute disseminated encephalomyelitis, idiopathic thromocytopenic purpura, Sjoegren's syndrome, autoimmune hemolytic anemia, type I diabetes, psoriasis, acrodermatitis, angiodermatitis, atopic dermatitis, contact dermatitis, eczema, acne, chronic urticaria, scleroderma, cutaneous vasculitis, cutaneous lupus erythematosus, dermatomyositis and blistering diseases including but not limited to pemphigus vulgaris, bullous pemphigoid and epidermolysis bullosa), cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain (such as pain associated with rheumatoid arthritis or osteoarthritis, back pain, general inflammatory pain, inflammatory neuropathic pain, trigeminal neuralgia or central pain) as well as in bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (such as polycythemia vera, essential thrombocythemia or mielofibrosis); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors (such as pancreatic cancer; bladder cancer; colorectal cancer; breast cancer; prostate cancer; renal cancer; hepatocellular cancer; lung cancer; ovarian cancer; cervical cancer; gastric cancer; esophageal cancer; head and neck cancer; non-small cell lung cancer and small-cell lung cancer; melanoma; neuroendocrine cancers; central nervious system cancers; brain tumors; bone cancer; soft tissue sarcoma; chronic lymphocytic leukemia, B-cell acute lymphoblastic leukemia, T-cell acute lymphoblastic leukaemia, non-hodgkins lymphoma, B-cell lymphoma, acute myeloid leukaemia; cutaneous T cell lymphoma, premalignant and malignant skin conditions including but not limited to basal cell carcinoma (BCC), squamous cell carcinoma (SCC) or actinic keratosis (AK)).
The compounds described in the present invention are particularly useful for the treatment or prevention of pathological conditions or diseases such as neoplastic diseases (e.g. leukemia, lymphomas, solid tumors); transplant rejection, bone marrow transplant applications (e.g., graft- versus-host disease); autoimmune diseases (e.g. rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, cutaneous vasculitis, cutaneous lupus erythematosus, dermatomyositis and
blistering diseases including but not limited to pemphigus vulgaris, bullous pemphigoid and epidermolysis bullosa; respiratory inflammation diseases (e.g. asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis); skin inflammatory diseases (e.g., atopic dermatitis, contact dermatitis, eczema or psoriasis); premalignant and malignant skin conditions (e.g. basal cell carcinoma (BCC), squamous cell carcinoma (SCC) or actinic keratosis (AK)); neurological disorders and pain (such as pain associated with rheumatoid arthritis or osteoarthritis, back pain, general inflammatory pain, inflammatory neuropathic pain, trigeminal neuralgia or central pain)
The compounds described in the present invention are particularly useful for the treatment or prevention of pathological conditions or diseases selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, cutaneous vasculitis, cutaneous lupus erythematosus, dermatomyositis, blistering diseases including but not limited to pemphigus vulgaris, bullous pemphigoid and epidermolysis bullosa, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic keratosis.
Also provided are processes for the preparation of the compounds of the invention.
The invention also provides a pharmaceutical composition comprising the compounds of the invention in association with a pharmaceutically acceptable diluent or carrier.
The invention also provides a combination product comprising (i) the compounds of the invention as described herein; and (ii) one or more additional active substances which are known to be useful in the treatment of respiratory diseases; allergic diseases; inflammatory or autoimmune-mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (MPDs); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors; more in particular wherein the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid artritis (RA), multiple sclerosis (MS), amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, cutaneous vasculitis, cutaneous lupus
erythematosus, dermatomyositis, blistering diseases including but not limited to pemphigus vulgaris, bullous pemphigoid and epidermolysis bullosa, asthma, chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF), idiopathic pulmonary fibrosis, sarcoidosis, atopic dermatitis, allergic rhinitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma (BCC), squamous cell carcinoma (SCC) and actinic keratosis (AK); even more in particular wherein the pathological condition or disease is leukemia, lymphomas and solid tumors, rheumatoid artritis (RA), multiple sclerosis (MS), amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, asthma, chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF), idiopathic pulmonary fibrosis, sarcoidosis, atopic dermatitis, allergic rhinitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma (BCC), squamous cell carcinoma (SCC) and actinic keratosis (AK).
DETAILED DESCRIPTION OF THE INVENTION
It has now been found that certain pyrrolopyrimidine derivatives are novel and potent PI3K inhibitors and can therefore be used in the treatment or prevention of these diseases.
Thus the present invention is directed to compounds of formula (I), or a pharmaceutically acceptable salt, or N-oxide, or isotopically-labeled derivate thereof:

Formula (I) wherein,
A represents a group selected from i) a group of formula (A-1 ), ii) a group of formula (A-2), iii) a group of formula (A-3),

Formula A-1 Formula A-2 Formula A-3 wherein,
Ri represents a phenyl group or a 5- to 6- membered heteroaryl group containing at least one heteroatom selected from O, S and N,
wherein the phenyl and heteroaryl groups are unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched Ci-C4 alkyl group, a C1-C4 haloalkyi group, a C1-C4 hydroxyalkyi group, a C3-C10 cycloalkyl group, a -(CH2)i-3CN group, a -(CH2)o-30R7 group, a -(CH2)0-3NR7R8 group, a -C(0)-(CH2)i-3-CN group, a -C(0)-(CH2)o-3-R7 group, a -C(O)- (CH2)o-3-NR7R8 group, a -S(CH2)0-3R7 group, a -S(O)(CH2)0-3R7 group, a - S(0)(CH2)o-3NR7R8 group, a -S(O)2(CH2)0-3R7 group, a -S(O)2(CH2)0- 3NR7R8 group or a -(CH2)0-3(phenyl)-OR7 group; wherein R7 and R8 each independently represent a hydrogen atom, a C1-C4 haloalkyi group, a C1-C4 hydroxyalkyi group or a linear or branched C1-C4 alkyl group, which alkyl group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkoxy group, a cyano group or a C3- C10 cycloalkyl group;
and (*) represents where A is bonded to the CH(CH3)-N H-pyrrolopyrimidine group;
R2, R3, R4, R5, and R6 each independently represent a hydrogen atom, a C1-C4 alkoxy group, a C1-C4 haloalkyi group, a C1-C4 hydroxyalkyi group, a C3-C10 cycloalkyl group, a -(CH2)0-3CN group, a -C(0)-(CH2)1-3-CN group, a -C(O)- (CH2)o-3-R' group, a -C(0)-(CH2)0-3-NR9Rio, a -(CH2)0-3NR9Rio group, or a linear
or branched C1-C4 alkyl group, which alkyl group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkoxy group, a cyano group or a C3-C10 cycloalkyl group; wherein R9 and R10 each independently represent a hydrogen atom, a hydroxyl group, a C1-C4 alkoxy group or a linear or branched C1-C4 alkyl group;
L represents a direct bound or a linker selected from -0-, -S-, a -N R'- group, a C(O)- N R'- group, a C(0)-0-R"- group or a -(CH2)i-4 group; wherein R' represents hydrogen or a linear or branched C1-C4 alkyl group, and R" represents a linear or branched C C4 alkyl group;
B represents a phenyl group or a 5- to 14- membered heteroaryl group containing at least one heteroatom selected from O, S and N,
wherein the phenyl and heteroaryl groups are unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyl group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group, a -(CH2)o-3-0(Ci-C4 alkyl) group, a -(CH2)o-3-0(Ci-C4 haloalkyl) group, a -(CH2)o-3-0-(CH2)i-3-0(Ci-C4 alkyl) group, a -(CH2)0-3-O- (CH2)1-3-0(Ci-C4 haloalkyl) group, a -(CH2)0-3-O-(CH2) 0-3Rn group, a -(CH2)0-3- 0-(CH2)1 -3N Rn R12 group, a C3-C10 cycloalkyl group, a -(CH2) 0-3NRn Ri2 group, a -(CH2)o-3-C(0)-(CH2) 0-3-N Ri i Ri2group, a -(CH2)0-3-C(0)0-(CH2) o-3Ri i group, a - (CH2) o-3NRii-S(0)2Ri2 group, a -(CH2)0-3- S(O)2(CH2)0-3-Ri i group, a -(CH2)0-3- SH group, a -(CH2)o-3-S-(CH2)0-3-Rn group or a -(CH2)0-3-(5- to 7- membered heterocyclyl group containing at least one heteroatom selected from O, S and N); which heterocyclyl group is unsubstituted or substituted by one or more substituents selected from a halogen atom, a linear or branched C1-C4 alkyl group, a -(CH2)o-5NR15R16 group or a 5- to 7- membered heterocyclyl group containing one or two nitrogen atoms, which heterocyclyl group is unsubstituted or substituted by one or more substituents selected from a linear or branched C1-C4 alkyl groups or a -(CH2)0-3N(CH3)2 group; wherein Rn and R12 each independently represent a hydrogen atom, a hydroxyl group, a linear or branched C1-C4 alkyl group, a -(CH2)o-3NR13R14 group, a phenyl group or a 5- to 7- membered heterocyclyl group containing one or two nitrogen atoms, which phenyl or heterocyclyl groups are unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a C1-C4 hydroxyalkyl group, a C1-C4 haloalkyl group, a C1-C4 alkoxy group, a C3- C10 cycloalkyl group, a linear or branched C1-C4 alkyl group or a -(CH2)0-
5NR15R16 group; wherein R13 and R^ each independently represent a hydrogen atom, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group or a linear or branched C1-C4 alkyl group; and wherein R 5 and Ri6 each independently represent a hydrogen atom or a or a linear or branched C1-C4 alkyl group.
As used herein the term Ci-C6 alkyl embraces linear or branched radicals having 1 to 6 carbon atoms, preferably 1 to 4 carbon atoms. Examples include methyl, ethyl, n- propyl, i-propyl, n-butyl, sec-butyl, t-butyl, n-pentyl, 1 -methylbutyl, 2-methylbutyl, isopentyl, 1 -ethylpropyl, 1 ,1 -dimethylpropyl, 1 ,2-dimethylpropyl, n-hexyl, 1 -ethylbutyl, 2- ethylbutyl, 1 ,1 -dimethylbutyl, 1 ,2-dimethylbutyl, 1 ,3-dimethylbutyl, 2,2-dimethylbutyl, 2,3-dimethylbutyl, 2-methylpentyl, 3-methylpentyl and iso-hexyl radicals.
When it is mentioned that the alkyl radical may be optionally substituted it is meant to include linear or branched alkyl radical as defined above, which may be unsubstituted or substituted in any position by one or more substituents, for example by 1 , 2 or 3 substituents. When two or more substituents are present, each substituent may be the same or different.
As used herein, the term C1-C4 haloalkyl group is an alkyl group, for example a C1-C4 or C1-C2 alkyl group, which is bonded to one or more, preferably 1 , 2 or 3 halogen atoms. Preferably, said haloakyl group is chosen from -CCI3, -CHF2 and -CF3.
As used herein, the term C1-C4 hydroxyalkyl embraces linear or branched alkyl radicals having 1 to 4 carbon atoms, any one of which may be substituted by one or more, preferably 1 or 2, more preferably 1 hydroxyl radicals. Examples of such radicals include hydroxymethyl, hydroxyethyl, hydroxypropyl, and hydroxybutyl.
As used herein, the term C1-C4 alkoxy (or alkyloxy) embraces linear or branched oxy- containing radicals each having alkyl portions of 1 to 4 carbon atoms.
As used herein, the term C3-C10 cycloalkyl embraces saturated monocyclic or polycyclic carbocyclic radicals having from 3 to 10 carbon atoms, preferably from 3 to 7 carbon atoms. An optionally substituted C3-C10 cycloalkyl radical is typically unsubstituted or substituted by 1 , 2 or 3 substituents which may be the same or different. When a C3- C10 cycloalkyl radical carries 2 or more substituents, the substituents may be the same or different. Typically the substituents on a C3-C10 cycloalkyl group are themselves unsubstituted. Polycyclic cycloalkyl radicals contains two or more fused cycloalkyl
groups, preferably two cycloalkyi groups. Typically, polycyclic cycloalkyi radicals are selected from decahydronaphthyl (decalyl), bicyclo[2.2.2]octyl, adamantly, camphyl or bornyl groups.
Examples of monocyclic cycloalkyi groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl and cyclodecyl.
As used herein, the term 5- to 14- membered heteroaryl radical embraces typically a 5- to 14- membered ring system, preferably a 5- to 10- membered ring system, more preferably a 5- to 9- membered ring system, even more preferably a 5- to 6- membered ring system, comprising at least one heteroaromatic ring and containing at least one heteroatom selected from O, S and N. A 5- to 14- membered heteroaryl radical may be a single ring or two fused rings wherein at least one ring contains a heteroatom.
A said optionally substituted 5- to 14- membered heteroaryl radical is typically unsubstituted or substituted by 1 , 2 or 3 substituents which may be the same or different. When a 5- to 14- membered heteroaryl radical carries 2 or more substituents, the substituents may be the same or different. Unless otherwise specified, the substituents on a 5- to 14- membered heteroaryl radical are typically themselves unsubstituted.
Examples include pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, furyl, benzofuranyl, oxadiazolyl, oxazolyl, isoxazolyl, benzoxazolyl, imidazolyl, benzimidazolyl, thiazolyl, thiadiazolyl, thienyl, pyrrolyl, benzothiazolyl, indolyl, indazolyl, purinyl, quinolyl, isoquinolyl, phthalazinyl, naphthyridinyl, quinoxalinyl, quinazolinyl, quinolizinyl, cinnolinyl, triazolyl, indolizinyl, indolinyl, isoindolinyl, isoindolyl, imidazolidinyl, pteridinyl, thianthrenyl, pyrazolyl, 2H-pyrazolo[3,4-d]pyrimidinyl, 1 H-pyrazolo[3,4-d]pyrimidinyl, thieno[2,3-d] pyrimidinyl and the various pyrrolopyridyl radicals.
As used herein, some of the atoms, radicals, moieties, chains and cycles present in the general structures of the invention are "optionally substituted". This means that these atoms, radicals, moieties, chains and cycles can be either unsubstituted or substituted in any position by one or more, for example 1 , 2, 3 or 4, substituents, whereby the hydrogen atoms bound to the unsubstituted atoms, radicals, moieties, chains and cycles are replaced by chemically acceptable atoms, radicals, moieties, chains and cycles. When two or more substituents are present, each substituent may be the same or different. The substituents are typically themselves unsubstituted.
As used herein, the term halogen atom embraces chlorine, fluorine, bromine and iodine atoms. A halogen atom is typically a fluorine, chlorine or bromine atom, most preferably chlorine or fluorine. The term halo when used as a prefix has the same meaning. Compounds containing one or more chiral centre may be used in enantiomerically or diastereoisomerically pure form, in the form of racemic mixtures and in the form of mixtures enriched in one or more stereoisomer. The scope of the invention as described and claimed encompasses the racemic forms of the compounds as well as the individual enantiomers, diastereomers, and stereoisomer-enriched mixtures.
Conventional techniques for the preparation/isolation of individual enantiomers include chiral synthesis from a suitable optically pure precursor or resolution of the racemate using, for example, chiral high pressure liquid chromatography (HPLC). Alternatively, the racemate (or a racemic precursor) may be reacted with a suitable optically active compound, for example, an alcohol, or, in the case where the compound contains an acidic or basic moiety, an acid or base such as tartaric acid or 1 -phenylethylamine. The resulting diastereomeric mixture may be separated by chromatography and/or fractional crystallization and one or both of the diastereoisomers converted to the corresponding pure enantiomer(s) by means well known to one skilled in the art. Chiral compounds of the invention (and chiral precursors thereof) may be obtained in enantiomerically-enriched form using chromatography, typically HPLC, on an asymmetric resin with a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from 0 to 50% isopropanol, typically from 2 to 20%, and from 0 to 5% of an alkylamine, typically 0.1 % diethylamine. Concentration of the eluate affords the enriched mixture. Stereoisomer conglomerates may be separated by conventional techniques known to those skilled in the art. See, e.g. "Stereochemistry of Organic Compounds" by Ernest L. Eliel (Wiley, New York, 1994).
Atropisomers are stereoisomers resulting from hindered rotation about single bonds where the steric strain barrier to rotation is high enough to allow for the isolation of the conformers. Oki (Oki, M; Topics in Stereochemistry 1983, 1 ) defined atropisomers as conformers that interconvert with a half-life of more than 1000 seconds at a given temperature. The scope of the invention as described and claimed encompasses the racemic forms of the compounds as well as the individual atropisomers (an atropisomer "substantially free" of his corresponding enantiomer) and stereoisomer-enriched mixtures, i.e. mixtures of atropisomers.
Separation of atropisomers is possibly by chiral resolution methods such as selective crystallization. In an atropo-enantioselective or atroposelective synthesis one atropisomer is formed at the expense of the other. Atroposelective synthesis may be carried out by use of chiral auxiliaries like a Corey-Bakshi-Shibata (CBS) catalyst (asymmetric catalyst derived from proline) in the total synthesis of knipholone or by approaches based on thermodynamic equilibration when an isomerization reaction favors one atropisomer over the other.
As used herein, the term pharmaceutically acceptable salt refers to a salt prepared from a base or acid which is acceptable for administration to a patient, such as a mammal. Such salts can be derived from pharmaceutically-acceptable inorganic or organic bases and from pharmaceutically-acceptable inorganic or organic acids.
Pharmaceutically acceptable acids include both inorganic acids, for example hydrochloric, sulphuric, phosphoric, diphosphoric, hydrobromic, hydroiodic and nitric acid; and organic acids, for example citric, fumaric, gluconic, glutamic, lactic, maleic, malic, mandelic, mucic, ascorbic, oxalic, pantothenic, succinic, tartaric, benzoic, acetic, methanesulphonic, ethanesulphonic, benzenesulphonic, p-toluenesulphonic acid, xinafoic (1 -hydroxy-2-naphthoic acid), napadisilic (1 ,5-naphthalenedisulfonic acid) and the like. Particularly preferred are salts derived from fumaric, hydrobromic, hydrochloric, acetic, sulfuric, methanesulfonic, xinafoic, and tartaric acids.
Salts derived from pharmaceutically-acceptable inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic, manganous, potassium, sodium, zinc and the like. Particularly preferred are ammonium, calcium, magnesium, potassium and sodium salts.
Salts derived from pharmaceutically-acceptable organic bases include salts of primary, secondary and tertiary amines, including alkyl amines, arylalkyl amines, heterocyclyl amines, cyclic amines, naturally-occurring amines and the like, such as arginine, betaine, caffeine, choline, Ν,Ν'-dibenzylethylenediamine, diethylamine, 2- diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N- ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine and the like.
Other preferred salts according to the invention are quaternary ammonium compounds wherein an equivalent of an anion (X") is associated with the positive charge of the N atom. X" may be an anion of various mineral acids such as, for example, chloride, bromide, iodide, sulphate, nitrate, phosphate, or an anion of an organic acid such as, for example, acetate, maleate, fumarate, citrate, oxalate, succinate, tartrate, malate, mandelate, trifluoroacetate, methanesulphonate and p-toluenesulphonate. X" is preferably an anion selected from chloride, bromide, iodide, sulphate, nitrate, acetate, maleate, oxalate, succinate or trifluoroacetate. More preferably X" is chloride, bromide, trifluoroacetate or methanesulphonate.
As used herein, an N-oxide is formed from the tertiary basic amines or imines present in the molecule, using a convenient oxidising agent.
The present invention also embraces tautomeric forms of the compounds of formula (I), or pharmaceutically acceptable salts, solvates, N-oxides, stereoisomers or deuterated derivatives thereof.
The compounds of the invention may exist in both unsolvated and solvated forms. The term solvate is used herein to describe a molecular complex comprising a compound of the invention and an amount of one or more pharmaceutically acceptable solvent molecules. The term hydrate is employed when said solvent is water. Examples of solvate forms include, but are not limited to, compounds of the invention in association with water, acetone, dichloromethane, 2-propanol, ethanol, methanol, dimethylsulfoxide (DMSO), ethyl acetate, acetic acid, ethanolamine, or mixtures thereof. It is specifically contemplated that in the present invention one solvent molecule can be associated with one molecule of the compounds of the present invention, such as a hydrate.
Furthermore, it is specifically contemplated that in the present invention, more than one solvent molecule may be associated with one molecule of the compounds of the present invention, such as a dihydrate. Additionally, it is specifically contemplated that in the present invention less than one solvent molecule may be associated with one molecule of the compounds of the present invention, such as a hemihydrate. Furthermore, solvates of the present invention are contemplated as solvates of compounds of the present invention that retain the biological effectiveness of the non- solvate form of the compounds.
The invention also includes isotopically-labeled compounds of the invention, wherein one or more atoms is replaced by an atom having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number usually found in nature. Examples of isotopes suitable for inclusion in the compounds of the invention include isotopes of hydrogen, such as 2H and 3H, carbon, such as 11C, 13C and 14C, chlorine, such as 36CI, fluorine, such as 18F, iodine, such as 123l and 125l, nitrogen, such as 13N and 15N, oxygen, such as 150, 170 and 180, phosphorus, such as 32P, and sulfur, such as 35S. Certain isotopically-labeled compounds of the invention, for example, those incorporating a radioactive isotope, are useful in drug and/or substrate tissue distribution studies. The radioactive isotopes tritium, 3H, and carbon- 14, 14C, are particularly useful for this purpose in view of their ease of incorporation and ready means of detection. Substitution with heavier isotopes such as deuterium, 2H, may afford certain therapeutic advantages resulting from greater metabolic stability, for example, increased in vivo half-life or reduced dosage requirements, and hence may be preferred in some circumstances. Substitution with positron emitting isotopes, such as 11C, 18F, 150 and 13N, can be useful in Positron Emission Topography (PET) studies for examining substrate receptor occupancy.
Isotopically-labeled compounds of the invention can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described herein, using an appropriate isotopically-labeled reagent in place of the non-labeled reagent otherwise employed.
Preferred isotopically-labeled compounds include deuterated derivatives of the compounds of the invention. As used herein, the term deuterated derivative embraces compounds of the invention where in a particular position at least one hydrogen atom is replaced by deuterium. Deuterium (D or 2H) is a stable isotope of hydrogen which is present at a natural abundance of 0.015 molar %. Hydrogen deuterium exchange (deuterium incorporation) is a chemical reaction in which a covalently bonded hydrogen atom is replaced by a deuterium atom. Said exchange (incorporation) reaction can be total or partial.
Typically, a deuterated derivative of a compound of the invention has an isotopic enrichment factor (ratio between the isotopic abundance and the natural abundance of that isotope, i.e. the percentage of incorporation of deuterium at a given position in a molecule in the place of hydrogen) for each deuterium present at a site designated as a
potential site of deuteration on the compound of at least 3500 (52.5% deuterium incorporation).
In a preferred embodiment, the isotopic enrichment factor is at least 5000 (75% deuterium). In a more preferred embodiment, the isotopic enrichment factor is at least 6333.3 (95% deuterium incorporation). In a most preferred embodiment, the isotopic enrichment factor is at least 6633.3 (99.5% deuterium incorporation). It is understood that the isotopic enrichment factor of each deuterium present at a site designated as a site of deuteration is independent from the other deuteration sites.
The isotopic enrichment factor can be determined using conventional analytical methods known to an ordinary skilled in the art, including mass spectrometry (MS) and nuclear magnetic resonance (NMR). Prodrugs of the compounds described herein are also within the scope of the invention. Thus certain derivatives of the compounds of the present invention, which derivatives may have little or no pharmacological activity themselves, when administered into or onto the body may be converted into compounds of the present invention having the desired activity, for example, by hydrolytic cleavage. Such derivatives are referred to as 'prodrugs'. Further information on the use of prodrugs may be found in Pro-drugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T. Higuchi and W. Stella) and Bioreversible Carriers in Drug Design, Pergamon Press, 1987 (ed. E. B. Roche, American Pharmaceutical Association). Prodrugs in accordance with the invention can, for example, be produced by replacing appropriate functionalities present in the compounds of the present invention with certain moieties known to those skilled in the art as 'pro-moieties' as described, for example, in Design of Prodrugs by H. Bundgaard (Elsevier, 1985). In the case of compounds that are solids, it is understood by those skilled in the art that the inventive compounds and salts may exist in different crystalline or polymorphic forms, or in an amorphous form, all of which are intended to be within the scope of the present invention. As used herein, the term PI3Kd inhibitor generally refers to a compound that inhibits the activity of the PI3Kd isoform more effectively than other isoforms of the PI3K family.
As used herein, the term PI3Kd/g inhibitor generally refers to a compound that inhibits the activity of both the PI3Kd isoform and the PI3Kg isoform more effectively than other isoforms of the PI3K family. The relative efficacies of compounds as inhibitors of an enzyme activity (or other biological activity) can be established by determining the concentrations at which each compound inhibits the activity to a predefined extent and then comparing the results. Typically, the preferred determination is the concentration that inhibits 50% of the activity in a biochemical assay, i.e., the 50% inhibitory concentration or "IC50." IC50 determinations can be accomplished using conventional techniques known in the art. In general, an IC50 can be determined by measuring the activity of a given enzyme in the presence of a range of concentrations of the inhibitor under study. The experimentally obtained values of enzyme activity then are plotted against the inhibitor concentrations used. The concentration of the inhibitor that shows 50% enzyme activity (as compared to the activity in the absence of any inhibitor) is taken as the IC50 value.
Accordingly, a PI3Kd inhibitor alternatively can be understood to refer to a compound that exhibits a 50% inhibitory concentration (IC50) with respect to PI3Kd that is at least of less than about 100 μΜ, preferably of less than about 50 μΜ, more preferably of less than about 20 μΜ, even more preferably of less than about 10 μΜ PI3K HTRF assay (as described in Gray et al. Anal Biochem, 2003; 313: 234-45).
In a particular and preferred embodiment of the invention Ri represents a phenyl group wherein the phenyl group is unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyl group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group, a C3-C10 cycloalkyl group, a -(CH2)i-3CN group, a -(CH2)o-30R7 group, a -(CH2)o-3NR7R8 group, a -C(0)-(CH2)i-3-CN group, a -C(0)-(CH2)o-3-R7 group, a -C(O)-(CH2)0-3-NR7R8 group, a - S(CH2)o-3R7 group, a -S(O)(CH2)0-3R7 group, a -S(O)(CH2)0-3NR7R8 group, a - S(0)2(CH2)o-3R7 group, a -S(O)2(CH2)0-3NR7R8 group or a -(CH2)0-3(phenyl)-OR7 group; wherein R7 and R8 each independently represent a hydrogen atom, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group or a linear or branched C1-C4 alkyl group, which alkyl group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkoxy group, a cyano group or a C3-C10 cycloalkyl group;
More preferably, Ri represents a phenyl group wherein the phenyl group is unsubstituted or substituted by one or more halogen atoms.
The above particular and preferred embodiment, give rise to the particular embodiment where A represents a group selected from: -1a), group of formula (A-2a),

Formula A-1 a Formula A-2a group of formula (A-3a),

Formula A-3a where Ri, R2, R3, R4, R5, and R6 are as defined above and each Ra, Rb, Rc, Rd and Re independently represents a hydrogen atom or a substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyl group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group, a C3-C10 cycloalkyl group, a -(CH2)i-3CN group, a -(CH2)0-3OR7 group, a -(CH2)o-3N R7R8 group, a -C(0)-(CH2)i-3-CN group, a - C(0)-(CH2)o-3-R7 group, a -C(O)-(CH2)0-3-NR7R8 group, a -S(CH2)0-3R7 group, a - S(0)(CH2)o-3R7 group, a -S(O)(CH2)0-3NR7R8 group, a -S(O)2(CH2)0-3R7 group, a - S(0)2(CH2)o-3N R7R8 group or a -(CH2)0-3(phenyl)-OR7 group; wherein R7 and R8 each
independently represent a hydrogen atom, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group or a linear or branched C1-C4 alkyi group, which alkyi group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkoxy group, a cyano group or a C3-C10 cycloalkyl group;
and (*) represents where A is bonded to the CH(CH3)-NH-pyrrolopyrimidine group;
In another particular and preferred embodiment R2, R3, R4, R5, and R6 each independently represent a hydrogen atom, a C1-C4 alkoxy group, or a linear or branched C1-C4 alkyi group, which alkyi group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkoxy group, a cyano group or a C3-C10 cycloalkyl group. More preferably R2, R3, R4, R5, and R6 each independently represent a hydrogen atom, a methoxy, or methyl group.
In a particular embodiment, L is preferably a direct bond.
In yet another particular and preferred embodiment B represents a phenyl group, or a 5-to 9-membered heteroaryl group containing at least one heteroatom selected from 0,S and N , wherein said heteroaryl group is preferably pyridyl group, a pyrazole group or an indole group, and wherein the phenyl, pyridyl, pyrazole or indole groups are unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyi group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group, a -(CH2)o-3-0(Ci-C4 alkyi) group, a -(CH2)o- 3-0(Ci-C4 haloalkyl) group, a -(CH2)o-3-0-(CH2)i-3-0(Ci-C4 alkyi) group, a -(CH2)0-3-O- (CH2)i-3-0(Ci-C4 haloalkyl) group, a -(CH2)0-3-0-(CH2) o-3 i i group, a -(ΟΗ2)Μ-0-(ΟΗ2)Ι. 3NRnR12 group, a C3-C10 cycloalkyl group, a -(CH2) 0-3NRn Ri2 group, a -(CH2)o-3-C(0)- (CH2) 0-3-NRiiRi2group, a -(CH2)0-3-C(0)0-(CH2) o-3Ri i group, a -(CH2) o-3NRii-S(0)2Ri2 group, a -(CH2)0-3- S(O)2(CH2)0-3-Rn group, a -(CH2)0-3-SH group, a -(CH2)0-3-S-(CH2)o-3- R11 group or a -(CH2)0-3-(5- to 7- membered heterocyclyl group containing at least one heteroatom selected from O, S and N); which heterocyclyl group is unsubstituted or substituted by one or more substituents selected from a halogen atom, a linear or branched C1-C4 alkyi group, a -(CH2)o-5NR15R16 group or a 5- to 7- membered heterocyclyl group containing one or two nitrogen atoms, which heterocyclyl group is unsubstituted or substituted by one or more substituents selected from a linear or branched C1-C4 alkyi groups or a -(CH2)0-3N(CH3)2 group; wherein Rn and R12 each independently represent a hydrogen atom, a hydroxyl group, a linear or branched C C4 alkyi group, a -(CH2)o-3NR13R14 group, a phenyl group or a 5- to 7- membered heterocyclyl group containing one or two nitrogen atoms, which phenyl or heterocyclyl
groups are unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a C1-C4 hydroxyalkyl group, a C1-C4 haloalkyi group, a C1-C4 alkoxy group, a C3-C10 cycloalkyl group, a linear or branched C1-C4 alkyl group or a -(CH2)o-5NRi5 i6 group; wherein R13 and Ri4 each independently represent a hydrogen atom, a C1-C4 haloalkyi group, a C1-C4 hydroxyalkyl group or a linear or branched C1-C4 alkyl group; and wherein R 5 and Ri6 each independently represent a hydrogen atom or a linear or branched C1-C4 alkyl group.
More preferably B represents a phenyl, pyridyl, pyrazole or indole groups, unsubstituted or substituted by one or more substituents selected from a hydroxyl group, a linear or branched C1-C4 alkyl group, a C1-C4 hydroxyalkyl group, a -(CH2)o-3- 0(Ci-C4 alkyl) group, a -(CH2)o-3NRn Ri2 group, a -(CH2)o-3NRi S(0)2Ri2 group or a - (CH2)o-3-(5- to 7- membered heterocyclyl group containing at least one heteroatom selected from O, S and N); which heterocyclyl group is unsubstituted or substituted by one or more substituents selected from a halogen atom, a linear or branched C1-C4 alkyl group, a -(CH2)o-5NR15R16 group or a 5- to 7- membered heterocyclyl group containing one or two nitrogen atoms, which heterocyclyl group is unsubstituted or substituted by one or more linear or branched C1-C4 alkyl groups; wherein Rn and R12 each independently represent a hydrogen atom a linear or branched C1-C4 alkyl group, a -(CH2)o-3NR13R14 group or a 5- to 7- membered heterocyclyl group containing one or two nitrogen atoms, which heterocyclyl group is unsubstituted or substituted by one or more substituents selected from a C1-C4 haloalkyi group, a C1-C4 alkoxy group, a linear or branched C1-C4 alkyl group or a -(CH2)o-5NR15R16 group; wherein R13 and R14 each independently represent a hydrogen atom; and wherein R 5 and Ri6 each independently represent a hydrogen atom or a linear or branched C1-C4 alkyl group.
In the more preferred embodiment of the invention A represents a group selected from i) a group of formula (A-1 a), ii) a group of formula (A-2a),

group of formula (A-3a),

Formula A-3a
R2, R3, R4, R5, and R6 each independently represent a hydrogen atom, a C1-C4 alkoxy group, or a linear or branched C1-C4 alkyi group, which alkyi group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkoxy group, a cyano group or a C3-C10 cycloalkyl group; each Ra, Rb, Rc, Rd and Re independently represents a hydrogen atom or a substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyi group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group, a C3-C10 cycloalkyl group, a -(CH2)i-3CN group, a -(CH2)o- 3OR7 group, a -(CH2)o-3NR7R8 group, a -C(0)-(CH2)i-3-CN group, a -C(O)-(CH2)0- 3-R7 group, a -C(0)-(CH2)o-3-N R7R8 group, a -S(CH2)0-3R7 group, a -S(O)(CH2)0-
3R7 group, a -S(0)(CH2)o-3NR7R8 group, a -S(0)2(CH2)o-3R7 group, a - S(0)2(CH2)o-3NR7R8 group or a -(CH2)o-3(phenyl)-OR7 group; wherein R7 and R8 each independently represent a hydrogen atom, a C1-C4 haloalkyi group, a d- C4 hydroxyalkyl group or a linear or branched C1-C4 alkyl group, which alkyl group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkoxy group, a cyano group or a C3-C10 cycloalkyl group;
and (*) represents where A is bonded to the CH(CH3)-N H-pyrrolopyrimidine group; B represents a phenyl group, a pyrazole group or an indole group, wherein the phenyl, pyridyl, pyrazole or indole groups are unsubstituted or substituted by one or more substituents selected from a hydroxyl group, a linear or branched C1-C4 alkyl group, a C1-C4 hydroxyalkyl group, a -(CH2)0-3-O(Ci-C4 alkyl) group, a -(CH2)o-3NRn R12 group, a - (CH2)o-3NRi S(0)2Ri2 group or a -(CH2)0-3-(5- to 7- membered heterocyclyl group containing at least one heteroatom selected from O, S and N); which heterocyclyl group is unsubstituted or substituted by one or more substituents selected from a halogen atom, a linear or branched C1-C4 alkyl group, a -(CH2)o-5NR15R16 group or a 5- to 7- membered heterocyclyl group containing one or two nitrogen atoms, which heterocyclyl group is unsubstituted or substituted by one or more substituents selected from a linear or branched C1-C4 alkyl groups or a -(CH2)0-3N(CH3)2 group; wherein Rn and R12 each independently represent a hydrogen atom a linear or branched C1-C4 alkyl group, a - (CH2)o-3NR13R14 group or a 5- to 7- membered heterocyclyl group containing one or two nitrogen atoms, which heterocyclyl group is unsubstituted or substituted by one or more substituents selected from a C1-C4 haloalkyi group, a C1-C4 alkoxy group, a linear or branched C1-C4 alkyl group or a -(CH2)o-5NR15R16 group; wherein R13 and R14 each independently represent a hydrogen atom; and wherein R 5 and Ri6 each independently represent a hydrogen atom or a linear or branched C1-C4 alkyl group.
The more preferred compounds of the invention can be represented by the various subformulas:
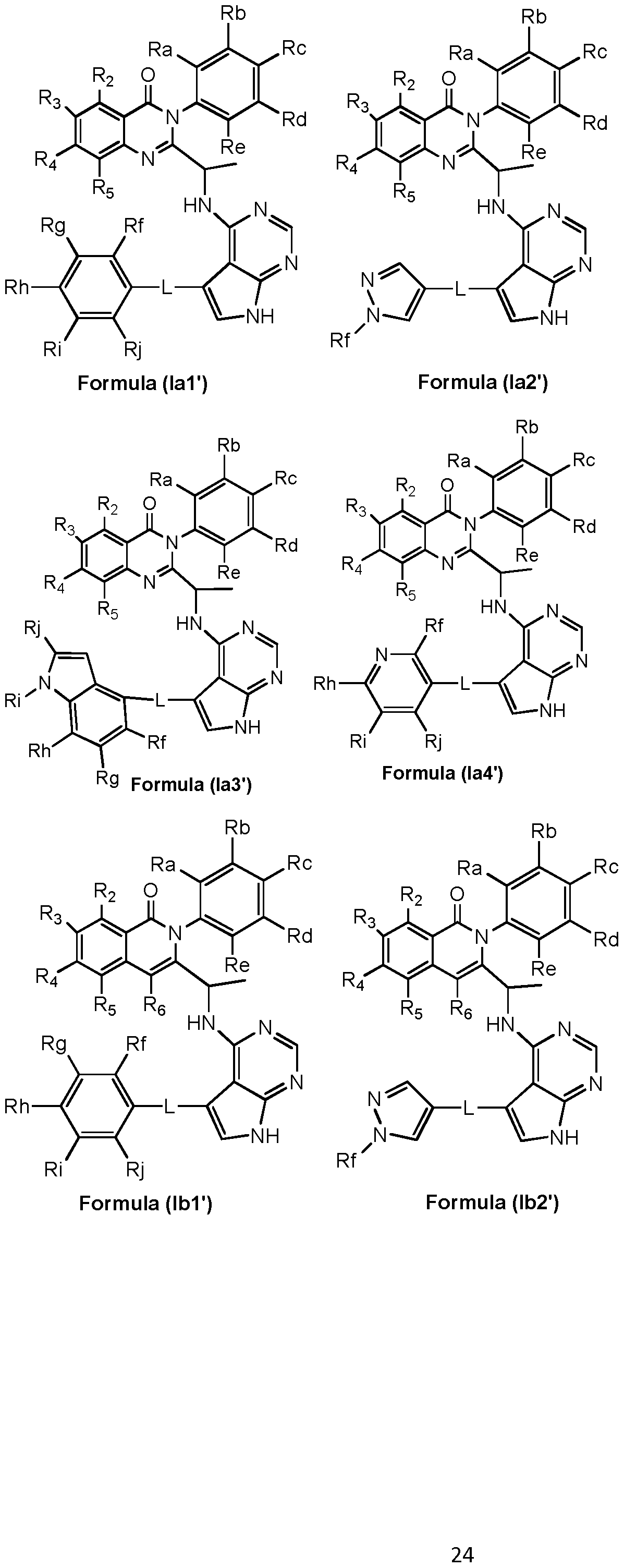

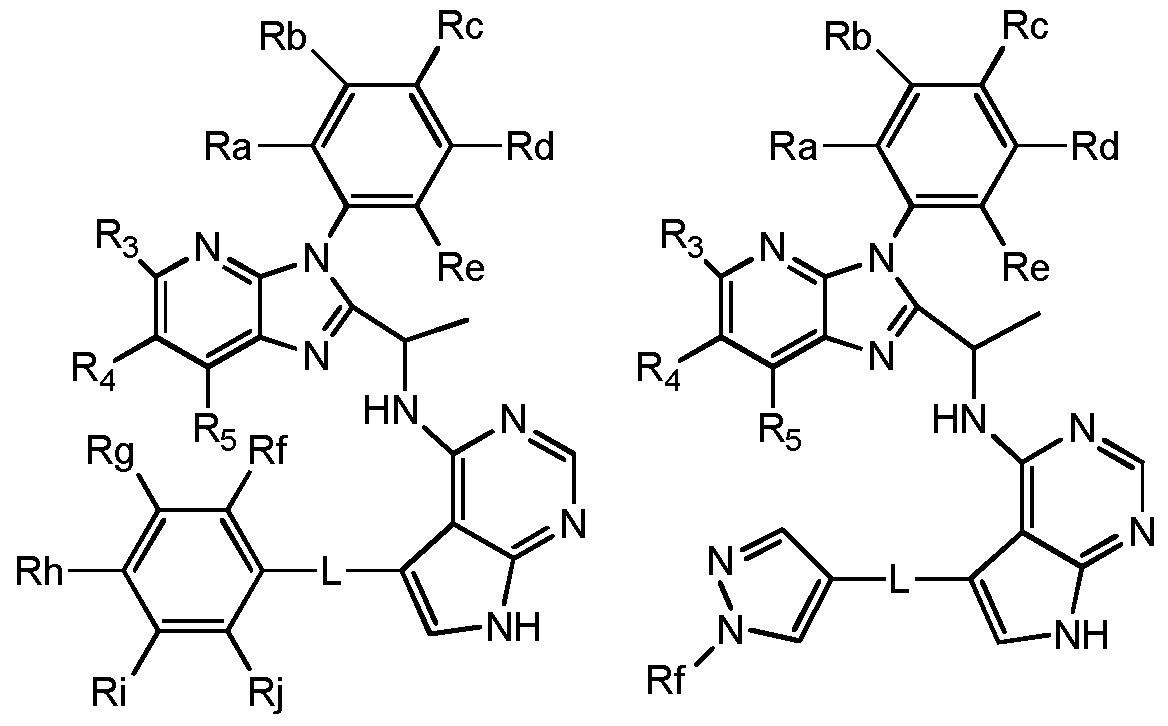
Formu la (Id ') Formu la (Ic2')
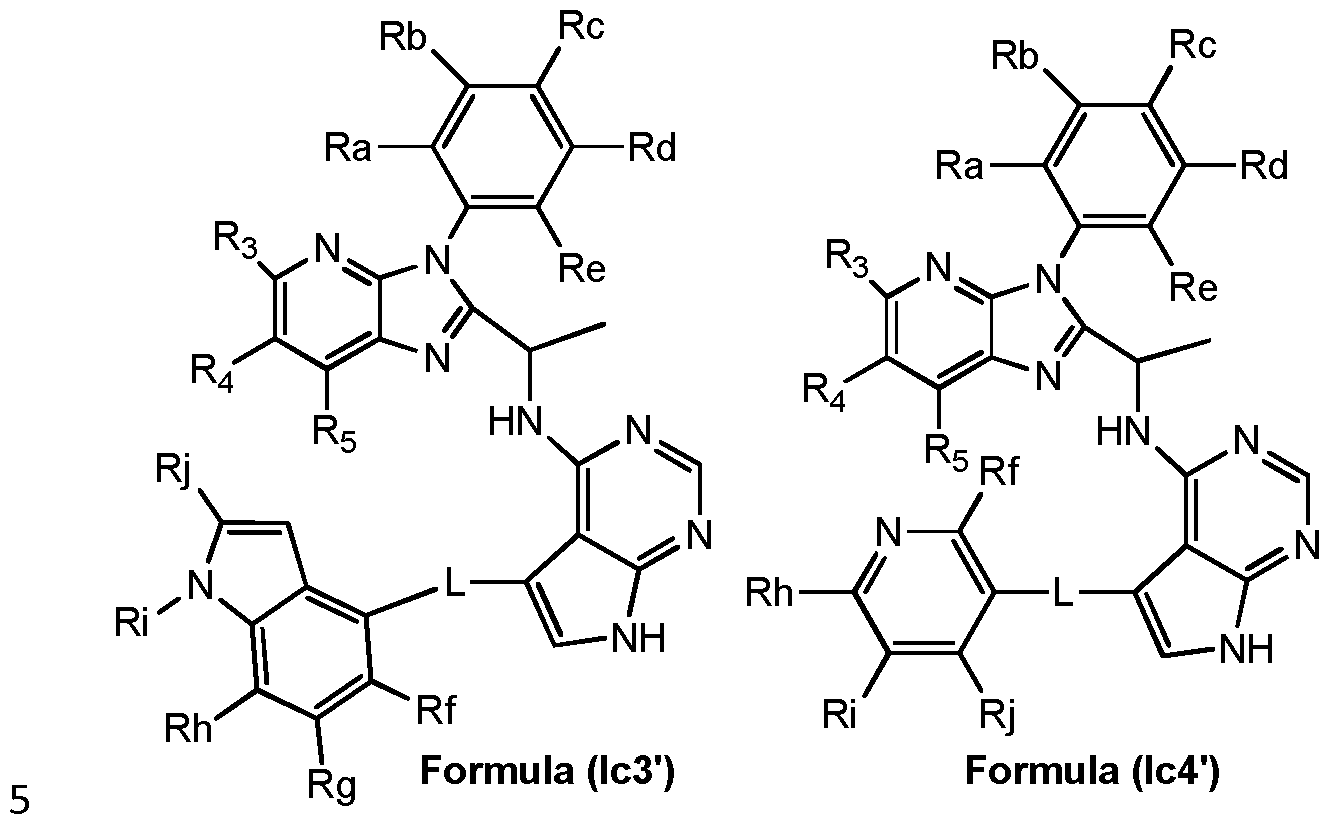

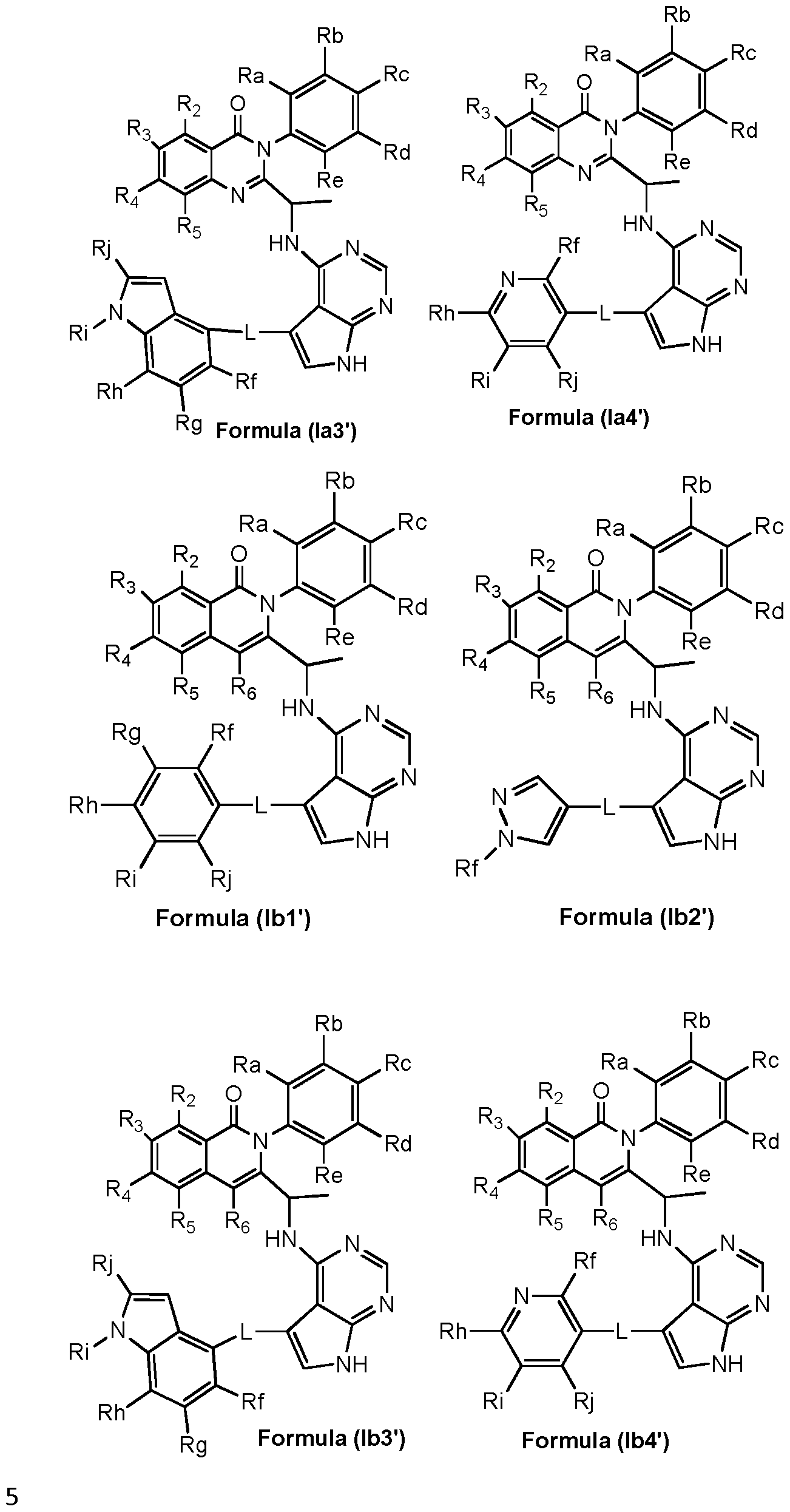

Formu la (Id ') Formu la (Ic2')
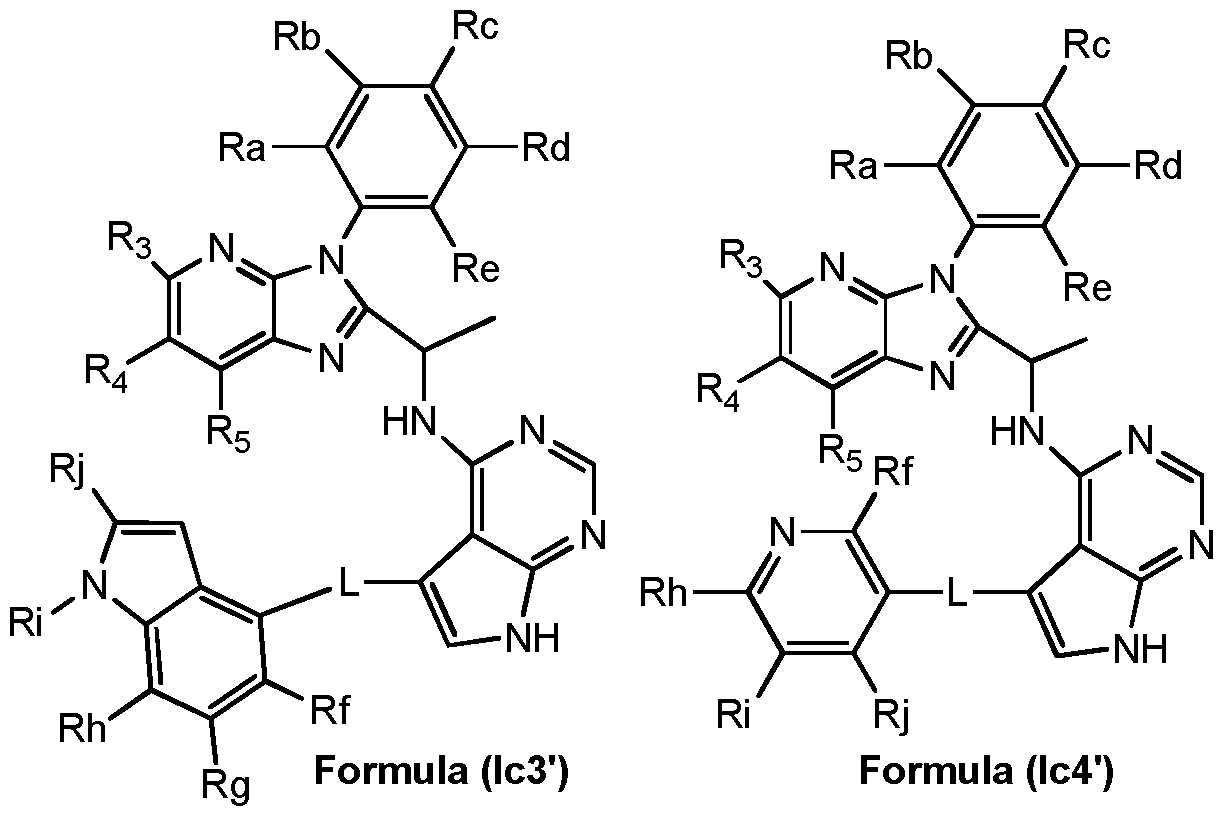
each Ra, Rb, Rc, Rd and Re independently represents a hydrogen atom or a substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyl group, a C1-C4 haloalkyi group, a C1-C4 hydroxyalkyl group, a C3- C10 cycloalkyl group, a -(CH2)i-3CN group, a -(CH2)o-30R7 group, a -(CH2)o-3NR7R8 group, a -C(0)-(CH2)i-3-CN group, a -C(0)-(CH2)o-3-R7 group, a -C(O)-(CH2)0-3-NR7R8 group, a -S(CH2)0-3R7 group, a -S(O)(CH2)0-3R7 group, a -S(O)(CH2)0-3NR7R8 group, a - S(0)2(CH2)o-3R7 group, a -S(O)2(CH2)0-3NR7R8 group or a -(CH2)0-3(phenyl)-OR7 group; wherein R7 and R8 each independently represent a hydrogen atom, a C1-C4 haloalkyi group, a C1-C4 hydroxyalkyl group or a linear or branched C1-C4 alkyl group, which alkyl group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkoxy group, a cyano group or a C3-C10 cycloalkyl group;
and each of Rf, Rg, Rh, R, and Rj independently represent a hydrogen atom, halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyl group, a C1-C4
haloalkyl group, a C1-C4 hydroxyalkyl group, a -(CH2)o-3-0(Ci-C4 alkyl) group, a -(CH2)o-3- 0(Ci-C4 haloalkyl) group, a -(CH2)o-3-0-(CH2)i-3-0(Ci-C4 alkyl) group, a -(CH2)0-3-O- (CH2)i-3-0(Ci-C4 haloalkyl) group, a -(CH2)0-3-0-(CH2) o-3 i i group, a -(ΟΗ2)Μ-0-(ΟΗ2)Ι. 3N ii i2 group, a C3-C10 cycloalkyl group, a -(CH2) o-3NRn Ri2 group, a -(CH2)0-3-C(O)- (CH2) 0-3-NRii Ri2group, a -(CH2)0-3-C(O)O-(CH2) 0-3Rn group, a -(CH2) o-3NRn-S(0)2Ri2 group, a -(CH2)0-3- S(O)2(CH2)0-3-Rn group, a -(CH2)0-3-SH group, a -(CH2)0-3-S-(CH2)o-3- R11 group or a -(CH2)0-3-(5- to 7- membered heterocyclyl group containing at least one heteroatom selected from O, S and N); which heterocyclyl group is unsubstituted or substituted by one or more substituents selected from a halogen atom, a linear or branched C1-C4 alkyl group, a -(CH2)o-5NR15R16 group or a 5- to 7- membered heterocyclyl group containing one or two nitrogen atoms, which heterocyclyl group is unsubstituted or substituted by one or more substituents selected from a linear or branched C1-C4 alkyl groups or a -(CH2)0-3N(CH3)2 group; wherein Rn and R12 each independently represent a hydrogen atom, a hydroxyl group, a linear or branched C1-C4 alkyl group, a -(CH2)o-3NR13R14 group, a phenyl group or a 5- to 7- membered heterocyclyl group containing one or two nitrogen atoms, which phenyl or heterocyclyl groups are unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a C1-C4 hydroxyalkyl group, a C1-C4 haloalkyl group, a C1-C4 alkoxy group, a C3-C10 cycloalkyl group, a linear or branched C1-C4 alkyl group or a -(CH2)o-5NR15R16 group; wherein R13 and Ri4 each independently represent a hydrogen atom, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group or a linear or branched C1-C4 alkyl group; and wherein R 5 and Ri6 each independently represent a hydrogen atom or a or a linear or branched C1-C4 alkyl group.
In a particular embodiment, in the compound of formula (I) A represents a group selected from i ),
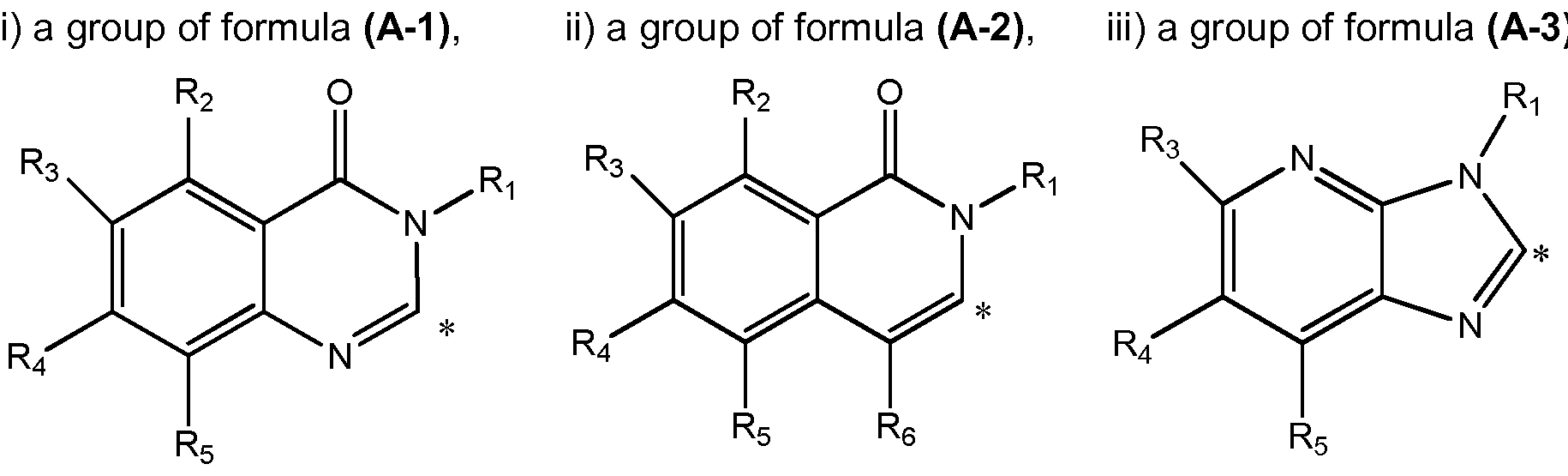
Formula A-1 Formula A-2 Formula A-3 wherein,
Ri represents a phenyl group or a 5- to 6- membered heteroaryl group containing at least one heteroatom selected from O, S and N,
wherein the phenyl and heteroaryl groups are unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched Ci-C4 alkyl group, a Ci-C4 haloalkyl group, a C C4 hydroxyalkyl group, a C3-Ci0 cycloalkyl group, a -(CH2)i-3CN group, a -(CH2)o-30R7 group, a -(CH2)o-3NR7R8 group, a -C(0)-(CH2)i-3-CN group, a -C(0)-(CH2)o-3-R7 group, a -C(O)- (CH2)o-3-NR7R8 group, a -S(CH2)0-3R7 group, a -S(O)(CH2)0-3R7 group, a - S(0)(CH2)o-3NR7R8 group, a -S(O)2(CH2)0-3R7 group, a -S(O)2(CH2)0- 3NR7R8 group or a -(CH2)0-3(phenyl)-OR7 group; wherein R7 and R8 each independently represent a hydrogen atom, a C C4 haloalkyl group, a Ci-C4 hydroxyalkyl group or a linear or branched C C4 alkyl group, which alkyl group is unsubstituted or substituted by one or more substituents selected from a C C4 alkoxy group, a cyano group or a C3- Cio cycloalkyl group;
and (*) represents where A is bonded to the CH(CH3)-NH-pyrrolopyrimidine group;
R2, R3, R4, R5, and R6 each independently represent a hydrogen atom, a C1-C4 alkoxy group, a C1-C4 haloalkyi group, a C1-C4 hydroxyalkyl group, a C3-C10 cycloalkyl group, a -(CH2)o-3CN group, a -C(0)-(CH2)i-3-CN group, a -C(O)- (CH2)o-3-R' group, a -C(0)-(CH2)o-3-N R9Rio, a -(CH2)0-3N R9Rio group, or a linear or branched C1-C4 alkyl group, which alkyl group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkoxy group, a cyano group or a C3-C10 cycloalkyl group; wherein R9 and R10 each independently represent a hydrogen atom, a hydroxyl group, a C1-C4 alkoxy group or a linear or branched C1-C4 alkyl group;
L represents a direct bound or a linker selected from -0-, -S-, a -N R'- group, a C(O)- N R'- group, a C(0)-0-R"- group or a -(CH2)1 -4 group; wherein R' represents hydrogen or a linear or branched C1-C4 alkyl group, and R" represents a linear or branched C C4 alkyl group;
B represents a phenyl group or a 5- to 14- membered heteroaryl group containing at least one heteroatom selected from O, S and N,
wherein the phenyl and heteroaryl groups are unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyl group, a C1-C4 haloalkyi group, a C1-C4 hydroxyalkyl group, a -(CH2)0-3-O(Ci-C4 alkyl) group, a -(CH2)0-3-O(Ci-C4 haloalkyi) group, a -(CH2)o-3-0-(CH2)1-3-0(Ci-C4 alkyl) group, a -(CH2)0-3-O- (CH2)1-3-0(Ci-C4 haloalkyi) group, a -(CH2)0-3-O-(CH2) 0-3Rn group, a -(CH2)0-3- 0-(CH2)1 -3N Rn R12 group, a C3-C10 cycloalkyl group, a -(CH2) 0-3NRn Ri2 group, a -(CH2)o-3-C(0)-(CH2) 0-3-N Ri i Ri2group, a -(CH2)0-3-C(0)0-(CH2) o-3Ri i group, a - (CH2) o-3NRii-S(0)2Ri2 group, a -(CH2)0-3- S(O)2(CH2)0-3-Ri i group, a -(CH2)0-3- SH group, or a -(CH2)o-3-S-(CH2)0-3-Rn group; wherein Rn and R12 each independently represent a hydrogen atom, a hydroxyl group, a linear or branched C1-C4 alkyl group, a -(CH2)o-3NR13R14 group or a phenyl group, which phenyl group is unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a C1-C4 hydroxyalkyl group, a C1-C4 haloalkyi group, a C1-C4 alkoxy group or a C3-C10 cycloalkyl group; wherein R 3 and Ri4 each independently represent a hydrogen atom, a C1-C4 haloalkyi group, a C1-C4 hydroxyalkyl group or a linear or branched C1-C4 alkyl group.
In yet another particular embodiment B represents a phenyl group, or a 5-to 9- membered heteroaryl group containing at least one heteroatom selected from 0,S and N, wherein said heteroaryl group is preferably a pyrazole group or an indole group, and wherein the phenyl, pyrazole or indole groups are unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched CrC4 alkyl group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyi group, a -(CH2)o-3-0(Ci-C4 alkyl) group, a -(CH2)o-3-0(Ci-C4 haloalkyl) group, a -(CH2)o- 3-0-(CH2)i-3-0(Ci-C4 alkyl) group, a -(CH2)o-3-0-(CH2)1-3-0(Ci-C4 haloalkyl) group, a - (CH2)o-3-0-(CH2) o-3Ri i group, a -(CH2)0-3-O-(CH2)1-3NR11 R12 group, a C3-Ci0 cycloalkyl group, a -(CH2) 0-3NRnRi2 group, a -(CH2)0-3-C(O)-(CH2) 0-3-NRn egroup, a -(CH2)0-3- C(0)0-(CH2) o-3Rii group, a -(CH2) o-3NRn-S(0)2Ri2 group, a -(CH2)0-3- S(O)2(CH2)0-3- Rii group, a -(CH2)0-3-SH group, or a -(CH2)o-3-S-(CH2)0-3-Rn group; wherein Rn and Ri2 each independently represent a hydrogen atom, a hydroxyl group, a linear or branched C1-C4 alkyl group, a -(CH2)o-3NR13R14 group or a phenyl group, which phenyl group is unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a C1-C4 hydroxyalkyi group, a C1-C4 haloalkyl group, a C1-C4 alkoxy group or a C3-Ci0 cycloalkyl group; wherein R13 and Ri4 each independently represent a hydrogen atom, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyi group or a linear or branched Ci-C4 alkyl group.
In yet another particular embodiment, B preferably represents a phenyl group, a pyrazole group or an indole group, substituted by one or more substituents selected from a hydroxyl group, a linear or branched Ci-C4 alkyl group, a Ci-C4 hydroxyalkyi group, a -(CH2) 0-3NRn-S(O)2Ri2 group wherein Rn and Ri2 each independently represent a hydrogen atom, a linear or branched C1-C4 alkyl group, a -(CH2)0-3NR13R14 group and wherein R 3 and Ri4 each represent a hydrogen atom.
In another particular embodiment of the invention A represents a group selected from i) a group of formula (A-1a), ii) a group of formula (A-2a),
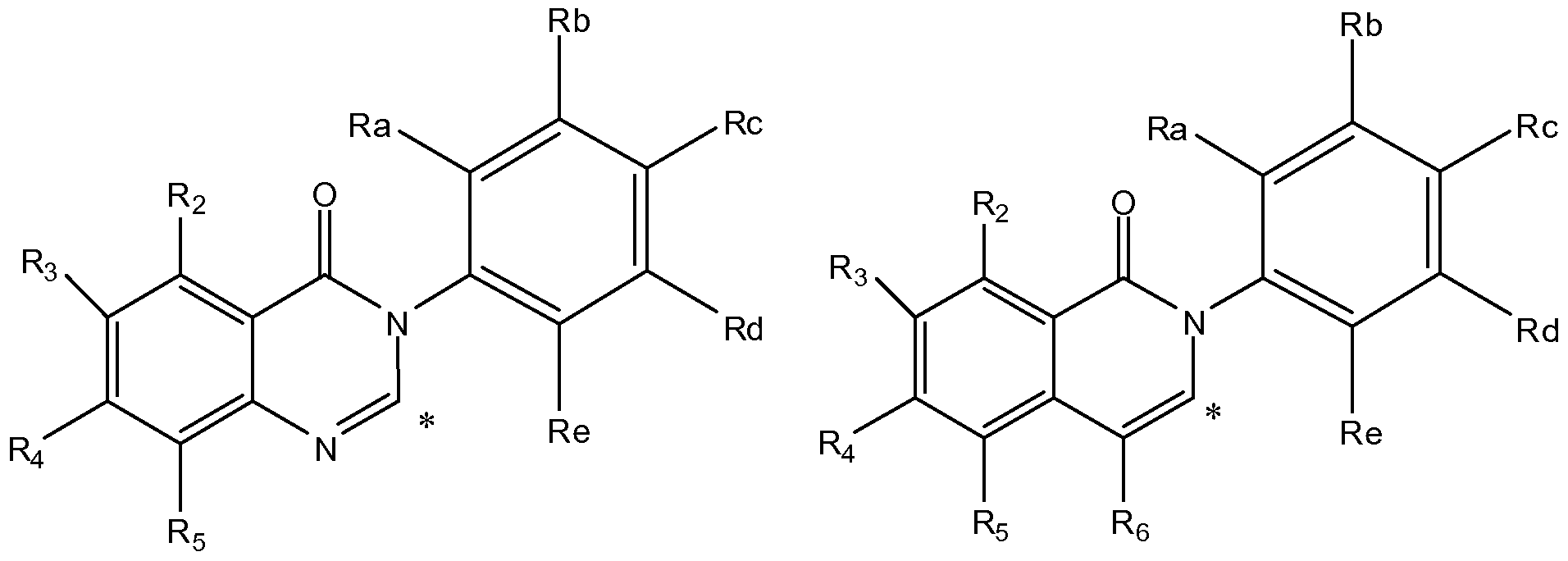
group of formula (A-3a),
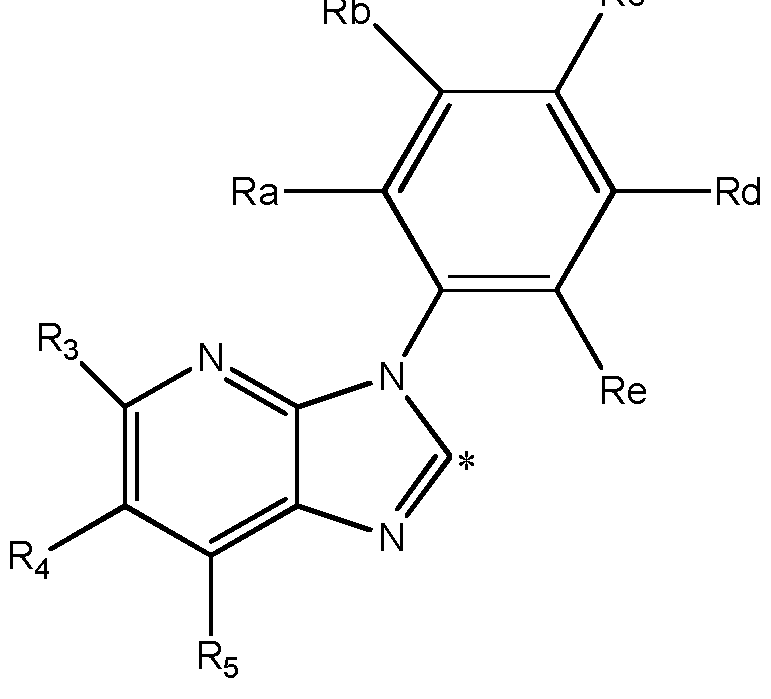
Formula A-3a
R2, R3, R4, R5, and R6 each independently represent a hydrogen atom, a C1-C4 alkoxy group, or a linear or branched C1-C4 alkyi group, which alkyi group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkoxy group, a cyano group or a C3-C10 cycloalkyl group; each Ra, Rb, Rc, Rd and Re independently represents a hydrogen atom or a substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyi group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group, a C3-C10 cycloalkyl group, a -(CH2)i-3CN group, a -(CH2)o- 3OR7 group, a -(CH2)o-3NR7R8 group, a -C(0)-(CH2)i-3-CN group, a -C(O)-(CH2)0- 3-R7 group, a -C(0)-(CH2)o-3-N R7R8 group, a -S(CH2)0-3R7 group, a -S(O)(CH2)0-
3R7 group, a -S(0)(CH2)o-3NR7R8 group, a -S(0)2(CH2)o-3R7 group, a - S(0)2(CH2)o-3NR7R8 group or a -(CH2)o-3(phenyl)-OR7 group; wherein R7 and R8 each independently represent a hydrogen atom, a C1-C4 haloalkyl group, a d- C4 hydroxyalkyl group or a linear or branched C1-C4 alkyl group, which alkyl group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkoxy group, a cyano group or a C3-C10 cycloalkyl group; and (*) represents where A is bonded to the CH(CH3)-N H-pyrrolopyrimidine group; B represents a phenyl group, a pyrazole group or an indole group, where the phenyl, pyrazole or indole groups are unsubstituted or substituted by one or more substituents selected from a hydroxyl group, a linear or branched C1-C4 alkyl group, a C1-C4 hydroxyalkyl group, a -(CH2) 0-3NRn-S(O)2Ri2 group wherein Rn and R12 each independently represent a hydrogen atom a linear or branched C1-C4 alkyl group, a - (CH2)o-3NR13R14 group, R13 and R14 being a hydrogen atom.
In another particular embodiment of the invention, preferred compounds of the invention can be represented by the various subformulas:
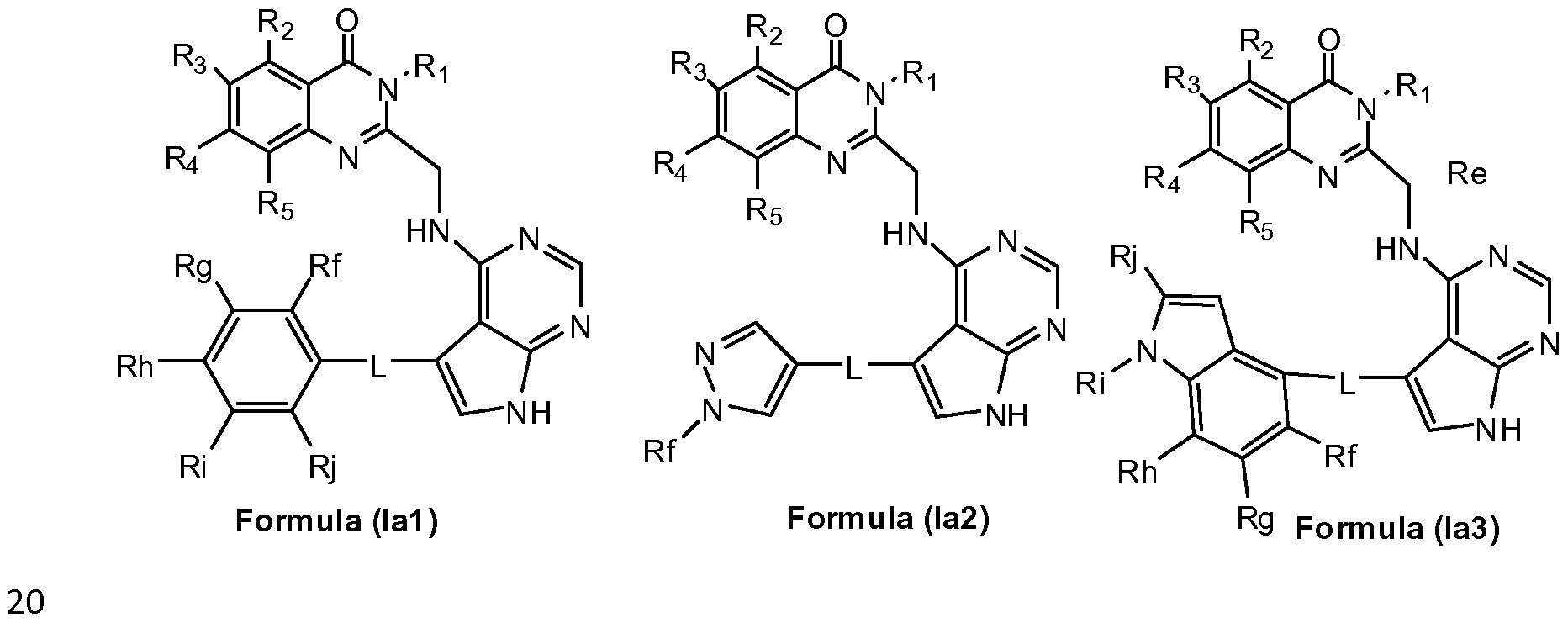
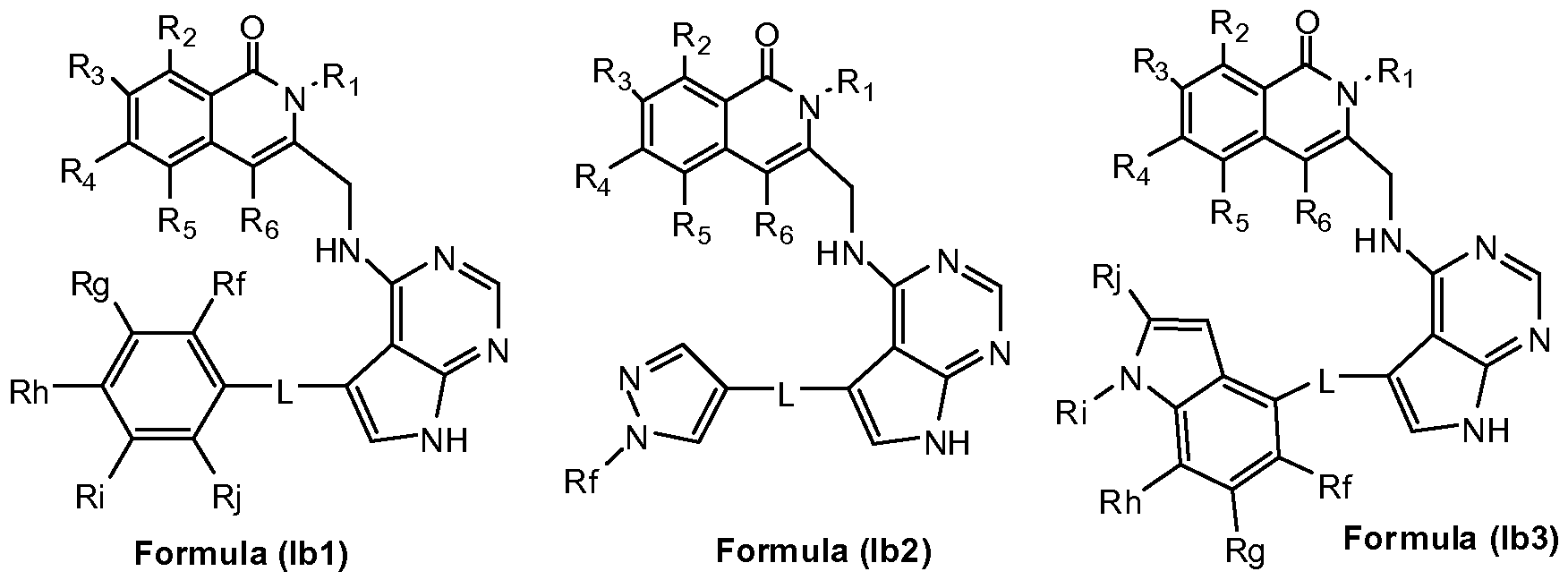

Formula (Id ) Formula (Ic2) ' Formula (Ic3) wherein L, Ri, R2, R3, R4, R5, and R6 are as defined for general formula (I) and each of Rf, Rg, Rh, R and Rj independently represent a hydrogen atom, a halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyl group, a C1-C4 haloalkyi group, a C1-C4 hydroxyalkyi group, a -(CH2)o-3-0(Ci-C4 alkyl) group, a -(CH2)o- 3-0(Ci-C4 haloalkyi) group, a -(CH2)o-3-0-(CH2)i-3-0(Ci-C4 alkyl) group, a -(CH2)0-3-O- (CH2)i-3-0(Ci-C4 haloalkyi) group, a -(CH2)0-3-0-(CH2) o-3 i i group, a -(ΟΗ2)Μ-0-(ΟΗ2)Ι. 3N ii i2 group, a C3-C10 cycloalkyl group, a -(CH2) o-3N n i2 group, a -(CH2)0-3-C(O)- (CH2) 0-3-NRiiRi2group, a -(CH2)0-3-C(0)0-(CH2)o-3Rii group, a -(CH2) o-3NRii-S(0)2Ri2 group, a -(CH2)0-3- S(O)2(CH2)0-3-Rn group, a -(CH2)0-3-SH group, or a -(CH2)0-3-S- (CH2)o-3-Rii group; wherein Rn and R12 each independently represent a hydrogen atom, a hydroxyl group, a linear or branched C1-C4 alkyl group, a -(CH2)o-3NR13R14 group or a phenyl group, which phenyl group is unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a C1-C4 hydroxyalkyi group, a C1-C4 haloalkyi group, a C1-C4 alkoxy group or a C3-C10
cycloalkyl group; wherein R 3 and R^ each independently represent a hydrogen atom, a C1-C4 haloalkyi group, a C1-C4 hydroxyalkyi group or a linear or branched C1-C4 alkyl group.
In another particular embodiment of the invention, more preferred compounds of the invention can be represented with the following general formulas:


each Ra, Rb, Rc, Rd and Re independently represents a hydrogen atom or a substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyl group, a C1-C4 haloalkyi group, a C1-C4 hydroxyalkyl group, a C3- C10 cycloalkyl group, a -(CH2)i-3CN group, a -(CH2)o-30R7 group, a -(CH2)o-3NR7R8 group, a -C(0)-(CH2)i-3-CN group, a -C(0)-(CH2)o-3-R7 group, a -C(O)-(CH2)0-3-NR7R8 group, a -S(CH2)0-3R7 group, a -S(O)(CH2)0-3R7 group, a -S(O)(CH2)0-3NR7R8 group, a - S(0)2(CH2)o-3R7 group, a -S(O)2(CH2)0-3NR7R8 group or a -(CH2)0-3(phenyl)-OR7 group; wherein R7 and R8 each independently represent a hydrogen atom, a C1-C4 haloalkyi group, a C1-C4 hydroxyalkyl group or a linear or branched C1-C4 alkyl group, which alkyl group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkoxy group, a cyano group or a C3-C10 cycloalkyl group;
and each of Rf, Rg, Rh, R, and Rj independently represent a hydrogen atom, a halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyl group, a C1-C4 haloalkyi group, a C1-C4 hydroxyalkyl group, a -(CH2)0-3-O(Ci-C4 alkyl) group, a -(CH2)o- 3-0(Ci-C4 haloalkyi) group, a -(CH2)o-3-0-(CH2)1-3-0(Ci-C4 alkyl) group, a -(CH2)0-3-O- (CH2)i-3-0(Ci-C4 haloalkyi) group, a -(CH2)0-3-0-(CH2) o-3Ri i group, a -(ΟΗ2)Μ-0-(ΟΗ2)Ι. 3NRnR12 group, a C3-C10 cycloalkyl group, a -(CH2) 0-3NRn Ri2 group, a -(CH2)o-3-C(0)- (CH2) 0-3-NRiiRi2group, a -(CH2)0-3-C(0)0-(CH2)o-3Rii group, a -(CH2) o-3NRii-S(0)2Ri2 group, a -(CH2)0-3- S(O)2(CH2)0-3-Rn group, a -(CH2)0-3-SH group, or a -(CH2)0-3-S- (CH2)o-3-Rii group; wherein Rn and R12 each independently represent a hydrogen atom, a hydroxyl group, a linear or branched C1-C4 alkyl group, a -(CH2)o-3NR13R14 group or a phenyl group, which phenyl group is unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a C1-C4 hydroxyalkyl group, a C1-C4 haloalkyi group, a C1-C4 alkoxy group or a C3-C10
cycloalkyl group; wherein R 3 and R^ each independently represent a hydrogen atom, a C1-C4 haloalkyi group, a C1-C4 hydroxyalkyi group or a linear or branched C1-C4 alkyl group.
Particular individual compounds of the invention include:
• (S)-N-(1 -(3-(3,5-difluorophenyl)-5-methoxy-3H-imidazo[4,5-b]pyridin-2-yl)ethyl)- 5-(1 -methyl-1 H-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine;
• (S)-N-(3-(4-(1 -(3-(3,5-difluorophenyl)-5-methoxy-3H-imidazo[4,5-b]pyridin-2- yl)ethylamino)-7H-pyrrolo[2,3-d]pyrimidin-5-yl)phenyl)methanesulfonamide;
• (S)-N-(3-(4-(1 -(8-methyl-1 -oxo-2-phenyl-1 ,2-dihydroisoquinolin-3- yl)ethylamino)-7H-pyrrolo[2,3-d]pyrimidin-5-yl)phenyl)methanesulfonamide;
• (S)-N-(3-(4-(1 -(5-methyl-4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)ethylamino)- 7H-pyrrolo[2,3-d]pyrimidin-5-yl)phenyl)methanesulfonamide;
• (S)-2-(1 -(5-(1 -(2-hydroxyethyl)-1 H-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-4- ylamino)ethyl)-5-methyl-3-phenylquinazolin-4(3H)-one;
• (S)-3-(1 -(5-(1 -(2-hydroxyethyl)-1 H-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-4- ylamino)ethyl)-8-methyl-2-phenylisoquinolin-1 (2H)-one;
• (S)-N-(3-hydroxy-5-(4-(1 -(5-methyl-4-oxo-3-phenyl-3,4-dihydroquinazolin-2- yl)ethylamino)-7H-pyrrolo[2,3-d]pyrimidin-5-yl)phenyl)methanesulfonamide;
• (S)-N-[4-(4-{[1 -(8-methyl-1 -oxo-2-phenyl-1 ,2-dihydroisoquinolin-3- yl)ethyl]amino}-7H-pyrrolo[2,3-d]pyrimidin-5-yl)-1 H-indol-6-yl]sulfamide;
• (S)-3-(1 -((5-(2-((4-(Dimethylamino)piperidin-1 -yl)methyl)pyridin-4-yl)-7H- pyrrolo[2,3-d]pyrimidin-4-yl)amino)ethyl)-8-methyl-2-phenylisoquinolin-1 (2H)- one;
• (S)-5-Methyl-2-(1 -((5-(5-(4-(1 -methylpiperidin-4-yl)piperazin-1 -yl)pyridin-3-yl)- 7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)ethyl)-3-phenylquinazolin-4(3H)-one;
• (S)-2-(1 -((5-(6-Methoxy-5-(((1 -methylpiperidin-4-yl)amino)methyl)pyridin-3-yl)- 7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)ethyl)-5-methyl-3-phenylquinazolin-4(3H)- one;
• (S)-8-Methyl-3-(1 -((5-(5-(4-(1 -methylpiperidin-4-yl)piperazin-1 -yl)pyridin-3-yl)- 7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)ethyl)-2-phenylisoquinolin-1 (2H)-one;
• (S)-3-(1 -((5-(2-(4-(3-(dimethylamino)propyl)piperazin-1 -yl)pyridin-4-yl)-7H- pyrrolo[2,3-d]pyrimidin-4-yl)amino)ethyl)-8-methyl-2-phenylisoquinolin-1 (2H)- one;
• (S)-3-(1 -((5-(6-Methoxy-5-(((1 -methylpiperidin-4-yl)amino)methyl)pyridin-3-yl)- 7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)ethyl)-8-methyl-2-phenylisoquinolin 1 (2H)-one;
• (S)-2-(1 -((5-(2-(4-(3-(Dimethylamino)propyl)piperazin-1 -yl)pyridin-4-yl)-7H- pyrrolo[2,3-(^pyrimidin-4-yl)amino)ethyl)-5-methyl-3-phenylquinazolin one;
or a pharmaceutically acceptable salt, or solvate, or N-oxide, or stereoisomer or isotopically-labeled derivate thereof.
In one embodiment, particular individual compounds of the invention include:
• (S)-N-(1 -(3-(3,5-difluorophenyl)-5-methoxy-3H-imidazo[4,5-b]pyridin-2-yl)ethyl)- 5-(1 -methyl-1 H-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine;
• (S)-N-(3-(4-(1 -(3-(3,5-difluorophenyl)-5-methoxy-3H-imidazo[4,5-b]pyridin-2- yl)ethylamino)-7H-pyrrolo[2,3-d]pyrimidin-5-yl)phenyl)methanesulfonamide;
• (S)-N-(3-(4-(1 -(8-methyl-1 -oxo-2-phenyl-1 ,2-dihydroisoquinolin-3- yl)ethylamino)-7H-pyrrolo[2,3-d]pyrimidin-5-yl)phenyl)methanesulfonamide;
• (S)-N-(3-(4-(1 -(5-methyl-4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)ethylamino)- 7H-pyrrolo[2,3-d]pyrimidin-5-yl)phenyl)methanesulfonamide;
• (S)-2-(1 -(5-(1 -(2-hydroxyethyl)-1 H-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-4- ylamino)ethyl)-5-methyl-3-phenylquinazolin-4(3H)-one;
• (S)-3-(1 -(5-(1 -(2-hydroxyethyl)-1 H-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-4- ylamino)ethyl)-8-methyl-2-phenylisoquinolin-1 (2H)-one;
• (S)-N-(3-hydroxy-5-(4-(1 -(5-methyl-4-oxo-3-phenyl-3,4-dihydroquinazolin-2- yl)ethylamino)-7H-pyrrolo[2,3-d]pyrimidin-5-yl)phenyl)methanesulfonamide;
• (S)-N-[4-(4-{[1 -(8-methyl-1 -oxo-2-phenyl-1 ,2-dihydroisoquinolin-3- yl)ethyl]amino}-7H-pyrrolo[2,3-d]pyrimidin-5-yl)-1 H-indol-6-yl]sulfamide;
or a pharmaceutically acceptable salt, or solvate, or N-oxide, or stereoisomer or isotopically-labeled derivate thereof.
Therapeutic use
The invention is also directed to the compounds of the invention as described herein, for use in the treatment of a pathological condition or disease susceptible to amelioration by inhibiton of Phosphoinositide 3-Kinases (PI3Ks), in particular wherein the pathological condition or disease is selected from respiratory diseases; allergic diseases; inflammatory or autoimmune-mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine
function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (MPDs); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors; more in particular wherein the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid artritis (RA), multiple sclerosis (MS), amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, cutaneous vasculitis, cutaneous lupus erythematosus, dermatomyositis, blistering diseases including but not limited to pemphigus vulgaris, bullous pemphigoid and epidermolysis bullosa, asthma, chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF), idiopathic pulmonary fibrosis, sarcoidosis, atopic dermatitis, allergic rhinitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma (BCC), squamous cell carcinoma (SCC) and actinic keratosis (AK); even more in particular wherein the pathological condition or disease is leukemia, lymphomas and solid tumors, rheumatoid artritis (RA), multiple sclerosis (MS), amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, asthma, chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF), idiopathic pulmonary fibrosis, sarcoidosis, atopic dermatitis, allergic rhinitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma (BCC), squamous cell carcinoma (SCC) and actinic keratosis (AK).
The invention is also directed to use of the compounds of the invention as described herein, in the manufacture of a medicament for treatment of a pathological condition or disease susceptible to amelioration by inhibiton of Phosphoinositide 3-Kinases (PI3Ks), in particular wherein the pathological condition or disease is selected from respiratory diseases; allergic diseases; inflammatory or autoimmune-mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (MPDs); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors; more in particular wherein the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid artritis (RA), multiple sclerosis (MS), amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, cutaneous vasculitis, cutaneous lupus erythematosus, dermatomyositis, blistering diseases including but not limited to pemphigus vulgaris, bullous pemphigoid and epidermolysis bullosa, asthma, chronic obstructive pulmonary disease (COPD), cystic
fibrosis (CF), idiopathic pulmonary fibrosis, sarcoidosis, atopic dermatitis, allergic rhinitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma (BCC), squamous cell carcinoma (SCC) and actinic keratosis (AK); even more in particular wherein the pathological condition or disease is leukemia, lymphomas and solid tumors, rheumatoid artritis (RA), multiple sclerosis (MS), amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, asthma, chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF), idiopathic pulmonary fibrosis, sarcoidosis, atopic dermatitis, allergic rhinitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma (BCC), squamous cell carcinoma (SCC) and actinic keratosis (AK).
The invention also provides a method of treatment of a pathological condition or disease susceptible to amelioration by inhibiton of Phosphoinositide 3-Kinases (PI3Ks), in particular wherein the pathological condition or disease is selected from respiratory diseases; allergic diseases; inflammatory or autoimmune-mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (MPDs); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors; more in particular wherein the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid artritis (RA), multiple sclerosis (MS), amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, cutaneous vasculitis, cutaneous lupus erythematosus, dermatomyositis, blistering diseases including but not limited to pemphigus vulgaris, bullous pemphigoid and epidermolysis bullosa, asthma, chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF), idiopathic pulmonary fibrosis, sarcoidosis, atopic dermatitis, allergic rhinitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma (BCC), squamous cell carcinoma (SCC) and actinic keratosis (AK); even more in particular wherein the pathological condition or disease is leukemia, lymphomas and solid tumors, rheumatoid artritis (RA), multiple sclerosis (MS), amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, asthma, chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF), idiopathic pulmonary fibrosis, sarcoidosis, atopic dermatitis, allergic rhinitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma (BCC), squamous cell carcinoma (SCC) and actinic keratosis (AK).
Composition
The invention also provides a pharmaceutical composition comprising the compounds of the invention in association with a pharmaceutically acceptable diluent or carrier.
As used herein, the term pharmaceutical composition refers to a mixture of one or more of the compounds described herein, or physiologically/pharmaceutically acceptable salts, solvates, N-oxides, stereoisomers, deuterated derivatives thereof or prodrugs thereof, with other chemical components, such as physiologically/pharmaceutically acceptable carriers and excipients. The purpose of a pharmaceutical composition is to facilitate administration of a compound to an organism.
As used herein, a physiologically/pharmaceutically acceptable diluent or carrier refers to a carrier or diluent that does not cause significant irritation to an organism and does not abrogate the biological activity and properties of the administered compound.
The invention further provides pharmaceutical compositions comprising the compounds of the invention in association with a pharmaceutically acceptable diluent or carrier together with one or more other therapeutic agents for use in the treatment of a pathological condition or disease susceptible to amelioration by inhibiton of Phosphoinositide 3-Kinases (PI3Ks), such as the ones previously described.
The invention is also directed to pharmaceutical compositions of the invention for use in the treatment of a pathological condition or disease susceptible to amelioration by inhibiton of Phosphoinositide 3-Kinases (PI3Ks), in particular wherein the pathological condition or disease is selected from respiratory diseases; allergic diseases; inflammatory or autoimmune-mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (MPDs such as polycythemia vera, essential thrombocythemia or mielofibrosis); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors; more in particular wherein the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, cutaneous vasculitis, cutaneous lupus
erythematosus, dermatomyositis, blistering diseases including but not limited to pemphigus vulgaris, bullous pemphigoid and epidermolysis bullosa, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic keratosis.
In particular the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic keratosis. The invention also encompasses the use of a pharmaceutical composition of the invention for the manufacture of a medicament for treating these diseases.
The invention also provides a method of treatment of a pathological condition or disease susceptible to amelioration by inhibiton of Phosphoinositide 3-Kinases (PI3Ks), in particular wherein the pathological condition or disease is selected from respiratory diseases; allergic diseases; inflammatory or autoimmune-mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (MPDs such as polycythemia vera, essential thrombocythemia or mielofibrosis); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors; more in particular wherein the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, cutaneous vasculitis, cutaneous lupus erythematosus, dermatomyositis, blistering diseases including but not limited to pemphigus vulgaris, bullous pemphigoid and epidermolysis bullosa, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic keratosis; more in particular the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic
anemia, type I diabetes, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic keratosis; comprising administering a therapeutically effective amount of a pharmaceutical composition of the invention.
The present invention also provides pharmaceutical compositions which comprise, as an active ingredient, at least a compound of formula (I) or a pharmaceutically acceptable salt thereof in association with a pharmaceutically acceptable excipient such as a carrier or diluent. The active ingredient may comprise 0.001 % to 99% by weight, preferably 0.01 % to 90% by weight, of the composition depending upon the nature of the formulation and whether further dilution is to be made prior to application. Preferably the compositions are made up in a form suitable for oral, inhalation, topical, nasal, rectal, percutaneous or injectable administration.
Pharmaceutical compositions suitable for the delivery of compounds of the invention and methods for their preparation will be readily apparent to those skilled in the art. Such compositions and methods for their preparation can be found, for example, in Remington: The Science and Practice of Pharmacy, 21 st Edition, Lippincott Williams & Wilkins, Philadelphia, Pa., 2001 .
The pharmaceutically acceptable excipients which are admixed with the active compound or salts of such compound, to form the compositions of this invention are well-known per se and the actual excipients used depend inter alia on the intended method of administering the compositions. Examples, without limitation, of excipients include calcium carbonate, calcium phosphate, various sugars and types of starch, cellulose derivatives, gelatin, vegetable oils and polyethylene glycols.
Additional suitable carriers for formulations of the compounds of the present invention can be found in Remington: The Science and Practice of Pharmacy, 21 st Edition, Lippincott Williams & Wilkins, Philadelphia, Pa., 2001. i) Oral Administration
The compounds of the invention may be administered orally (peroral administration; per os (latin)). Oral administration involve swallowing, so that the compound is absorbed from the gut and delivered to the liver via the portal circulation (hepatic first pass metabolism) and finally enters the gastrointestinal (Gl) tract.
Compositions for oral administration may take the form of tablets, retard tablets, sublingual tablets, capsules, inhalation aerosols, inhalation solutions, dry powder inhalation, or liquid preparations, such as mixtures, solutions, elixirs, syrups or suspensions, all containing the compound of the invention; such preparations may be made by methods well-known in the art. The active ingredient may also be presented as a bolus, electuary or paste.
Where the composition is in the form of a tablet, any pharmaceutical carrier routinely used for preparing solid formulations may be used. Examples of such carriers include magnesium stearate, talc, gelatine, acacia, stearic acid, starch, lactose and sucrose.
A tablet may be made by compression or moulding, optionally with one or more accessory ingredients. Compressed tablets may be prepared by compressing in a suitable machine the active ingredient in a free-flowing form such as a powder or granules, optionally mixed with a binder, lubricant, inert diluent, lubricating, surface active or dispersing agent.
Moulded tablets may be made by moulding in a suitable machine a mixture of the powdered compound moistened with an inert liquid diluent. The tablets may optionally be coated or scored and may be formulated so as to provide slow or controlled release of the active ingredient therein.
For tablet dosage forms, depending on dose, the drug may make up from 1 wt% to 80 wt% of the dosage form, more typically from 5 wt% to 60 wt% of the dosage form. In addition to the drug, tablets generally contain a disintegrant. Examples of disintegrants include sodium starch glycolate, sodium carboxymethyl cellulose, calcium carboxymethyl cellulose, croscarmellose sodium, crospovidone, polyvinylpyrrolidone, methyl cellulose, microcrystalline cellulose, lower alkyl- substituted hydroxypropyl cellulose, starch, pregelatinized starch and sodium alginate. Generally, the disintegrant will comprise from 1 wt% to 25 wt%, preferably from 5 wt% to 20 wt% of the dosage form.
Binders are generally used to impart cohesive qualities to a tablet formulation. Suitable binders include microcrystalline cellulose, gelatin, sugars, polyethylene glycol, natural and synthetic gums, polyvinylpyrrolidone, pregelatinized starch, hydroxypropyl cellulose and hydroxypropyl methylcellulose. Tablets may also contain diluents, such
as lactose (monohydrate, spray-dried monohydrate, anhydrous and the like), mannitol, xylitol, dextrose, sucrose, sorbitol, microcrystalline cellulose, starch and dibasic calcium phosphate dihydrate. Tablets may also optionally include surface active agents, such as sodium lauryl sulfate and polysorbate 80, and glidants such as silicon dioxide and talc. When present, surface active agents are typically in amounts of from 0.2 wt% to 5 wt% of the tablet, and glidants typically from 0.2 wt% to 1 wt% of the tablet.
Tablets also generally contain lubricants such as magnesium stearate, calcium stearate, zinc stearate, sodium stearyl fumarate, and mixtures of magnesium stearate with sodium lauryl sulphate. Lubricants generally are present in amounts from 0.25 wt% to 10 wt%, preferably from 0.5 wt% to 3 wt% of the tablet. Other conventional ingredients include anti-oxidants, colorants, flavoring agents, preservatives and taste- masking agents. Exemplary tablets contain up to about 80 wt% drug, from about 10 wt% to about 90 wt% binder, from about 0 wt% to about 85 wt% diluent, from about 2 wt% to about 10 wt% disintegrant, and from about 0.25 wt% to about 10 wt% lubricant. Tablet blends may be compressed directly or by roller to form tablets. Tablet blends or portions of blends may alternatively be wet-, dry-, or melt-granulated, melt congealed, or extruded before tabletting. The final formulation may include one or more layers and may be coated or uncoated; or encapsulated.
The formulation of tablets is discussed in detail in "Pharmaceutical Dosage Forms: Tablets, Vol. 1 ", by H. Lieberman and L. Lachman, Marcel Dekker, N.Y., 1980.
Where the composition is in the form of a capsule, any routine encapsulation is suitable, for example using the aforementioned carriers in a hard gelatine capsule. Where the composition is in the form of a soft gelatine capsule any pharmaceutical carrier routinely used for preparing dispersions or suspensions may be considered, for example aqueous gums, celluloses, silicates or oils, and are incorporated in a soft gelatine capsule.
Solid formulations for oral administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
Suitable modified release formulations are described in U.S. Patent No. 6,106,864. Details of other suitable release technologies such as high energy dispersions and osmotic and coated particles can be found in Verma et al, Pharmaceutical Technology On-line, 25(2), 1 -14 (2001 ). The use of chewing gum to achieve controlled release is described in WO 00/35298. The disclosures of these references are incorporated herein by reference in their entireties.
Liquid formulations include suspensions, solutions, syrups and elixirs. Such formulations may be used as fillers in soft or hard capsules and typically include a carrier, for example, water, ethanol, polyethylene glycol, propylene glycol, methylcellulose, or a suitable oil, and one or more emulsifying agents and/or suspending agents. The solutions may be aqueous solutions of a soluble salt or other derivative of the active compound in association with, for example, sucrose to form a syrup. The suspensions may comprise an insoluble active compound of the invention or a pharmaceutically acceptable salt thereof in association with water, together with a suspending agent or flavouring agent. Liquid formulations may also be prepared by the reconstitution of a solid, for example, from a sachet. ii) Oral mucosal administration
The compounds of the invention can also be administered via the oral mucosal. Within the oral mucosal cavity, delivery of drugs is classified into three categories: (a) sublingual delivery, which is systemic delivery of drugs through the mucosal membranes lining the floor of the mouth, (b) buccal delivery, which is drug administration through the mucosal membranes lining the cheeks (buccal mucosa), and (c) local delivery, which is drug delivery into the oral cavity.
Pharmaceutical products to be administered via the oral mucosal can be designed using mucoadhesive, quick dissolve tablets and solid lozenge formulations, which are formulated with one or more mucoadhesive (bioadhesive) polymers (such as hydroxy propyl cellulose, polyvinyl pyrrolidone, sodium carboxymethyl cellulose, hydroxy propyl methyl cellulose, hydroxy ethyl cellulose, polyvinyl alcohol, polyisobutylene or polyisoprene); and oral mucosal permeation enhancers (such as butanol, butyric acid, propranolol, sodium lauryl sulphate and others) iii) Inhaled administration
The compounds of the invention can also be administered by inhalation, typically in the form of a dry powder (either alone, as a mixture, for example, in a dry blend with lactose, or as a mixed component particle, for example, mixed with phospholipids, such
as phosphatidylcholine) from a dry powder inhaler or as an aerosol spray from a pressurized container, pump, spray, atomizer (preferably an atomizer using electrohydrodynamics to produce a fine mist), or nebulizer, with or without the use of a suitable propellant, such as 1 ,1 ,1 ,2-tetrafluoroethane or 1 ,1 ,1 ,2,3,3,3- heptafluoropropane. For intranasal use, the powder may include a bioadhesive agent, for example, chitosan or cyclodextrin.
Dry powder compositions for topical delivery to the lung by inhalation may, for example, be presented in capsules and cartridges of for example gelatine or blisters of for example laminated aluminium foil, for use in an inhaler or insufflator. Formulations generally contain a powder mix for inhalation of the compound of the invention and a suitable powder base (carrier substance) such as lactose or starch. Use of lactose is preferred. Each capsule or cartridge may generally contain between 0.001 -50 mg, more preferably 0.01 -5 mg of active ingredient or the equivalent amount of a pharmaceutically acceptable salt thereof. Alternatively, the active ingredient (s) may be presented without excipients.
Packaging of the formulation may be suitable for unit dose or multi-dose delivery. In the case of multi- dose delivery, the formulation can be pre-metered or metered in use. Dry powder inhalers are thus classified into three groups: (a) single dose, (b) multiple unit dose and (c) multi dose devices.
For inhalers of the first type, single doses have been weighed by the manufacturer into small containers, which are mostly hard gelatine capsules. A capsule has to be taken from a separate box or container and inserted into a receptacle area of the inhaler. Next, the capsule has to be opened or perforated with pins or cutting blades in order to allow part of the inspiratory air stream to pass through the capsule for powder entrainment or to discharge the powder from the capsule through these perforations by means of centrifugal force during inhalation. After inhalation, the emptied capsule has to be removed from the inhaler again. Mostly, disassembling of the inhaler is necessary for inserting and removing the capsule, which is an operation that can be difficult and burdensome for some patients.
Other drawbacks related to the use of hard gelatine capsules for inhalation powders are (a) poor protection against moisture uptake from the ambient air, (b) problems with opening or perforation after the capsules have been exposed previously to extreme relative humidity, which causes fragmentation or indenture, and (c) possible inhalation
of capsule fragments. Moreover, for a number of capsule inhalers, incomplete expulsion has been reported (e. g. Nielsen et al, 1997).
Some capsule inhalers have a magazine from which individual capsules can be transferred to a receiving chamber, in which perforation and emptying takes place, as described in WO 92/03175. Other capsule inhalers have revolving magazines with capsule chambers that can be brought in line with the air conduit for dose discharge (e. g. WO91/02558 and GB 2242134). They comprise the type of multiple unit dose inhalers together with blister inhalers, which have a limited number of unit doses in supply on a disk or on a strip.
Blister inhalers provide better moisture protection of the medicament than capsule inhalers. Access to the powder is obtained by perforating the cover as well as the blister foil, or by peeling off the cover foil. When a blister strip is used instead of a disk, the number of doses can be increased, but it is inconvenient for the patient to replace an empty strip. Therefore, such devices are often disposable with the incorporated dose system, including the technique used to transport the strip and open the blister pockets. Multi-dose inhalers do not contain pre-measured quantities of the powder formulation. They consist of a relatively large container and a dose measuring principle that has to be operated by the patient. The container bears multiple doses that are isolated individually from the bulk of powder by volumetric displacement. Various dose measuring principles exist, including rotatable membranes (Ex. EP0069715) or disks (Ex. GB 2041763; EP 0424790; DE 4239402 and EP 0674533), rotatable cylinders (Ex. EP 0166294; GB 2165159 and WO 92/09322) and rotatable frustums (Ex. WO 92/00771 ), all having cavities which have to be filled with powder from the container. Other multi dose devices have measuring slides (Ex. US 5201308 and WO 97/00703) or measuring plungers with a local or circumferential recess to displace a certain volume of powder from the container to a delivery chamber or an air conduit (Ex. EP 0505321 , WO 92/04068 and WO 92/04928), or measuring slides such as the Genuair® (formerly known as Novolizer SD2FL), which is described the following patent applications Nos: WO97/000703, WO03/000325 and WO2006/008027. Reproducible dose measuring is one of the major concerns for multi dose inhaler devices.
The powder formulation has to exhibit good and stable flow properties, because filling of the dose measuring cups or cavities is mostly under the influence of the force of gravity. For reloaded single dose and multiple unit dose inhalers, the dose measuring accuracy and reproducibility can be guaranteed by the manufacturer. Multi dose inhalers on the other hand, can contain a much higher number of doses, whereas the number of handlings to prime a dose is generally lower. Because the inspiratory air stream in multi-dose devices is often straight across the dose measuring cavity, and because the massive and rigid dose measuring systems of multi dose inhalers can not be agitated by this inspiratory air stream, the powder mass is simply entrained from the cavity and little de-agglomeration is obtained during discharge.
Consequently, separate disintegration means are necessary. However in practice, they are not always part of the inhaler design. Because of the high number of doses in multi- dose devices, powder adhesion onto the inner walls of the air conduits and the de- agglomeration means must be minimized and/or regular cleaning of these parts must be possible, without affecting the residual doses in the device. Some multi dose inhalers have disposable drug containers that can be replaced after the prescribed number of doses has been taken (Ex. WO 97/000703). For such semi-permanent multi dose inhalers with disposable drug containers, the requirements to prevent drug accumulation are even more strict.
Apart from applications through dry powder inhalers the compositions of the invention can be administered in aerosols which operate via propellant gases or by means of so- called atomisers, via which solutions of pharmacologically-active substances can be sprayed under high pressure so that a mist of inhalable particles results. The advantage of these atomisers is that the use of propellant gases can be completely dispensed with. Such atomiser is the Respimat® which is described, for example, in PCT Patent Applications Nos. W0 91/14468 and WO 97/12687, reference here is being made to the contents thereof. Spray compositions for topical delivery to the lung by inhalation may for example be formulated as aqueous solutions or suspensions or as aerosols delivered from pressurised packs, such as a metered dose inhaler, with the use of a suitable liquefied
propellant. Aerosol compositions suitable for inhalation can be either a suspension or a solution and generally contain the active ingredient (s) and a suitable propellant such as a fluorocarbon or hydrogen-containing chlorofluorocarbon or mixtures thereof, particularly hydrofluoroalkanes, e. g. dichlorodifluoromethane, trichlorofluoromethane, dichlorotetra-fluoroethane, especially 1 ,1 , 1 , 2-tetrafluoroethane, 1 ,1 , 1 ,2, 3,3, 3- heptafluoro-n-propane or a mixture thereof. Carbon dioxide or other suitable gas may also be used as propellant.
The aerosol composition may be excipient free or may optionally contain additional formulation excipients well known in the art such as surfactants (eg oleic acid or lecithin) and cosolvens (eg ethanol). Pressurised formulations will generally be retained in a canister (eg an aluminium canister) closed with a valve (eg a metering valve) and fitted into an actuator provided with a mouthpiece. Medicaments for administration by inhalation desirably have a controlled particle size. The optimum particle size for inhalation into the bronchial system is usually 1 -10 μηη, preferably 2-5 μηη. Particles having a size above 20 μηη are generally too large when inhaled to reach the small airways. To achieve these particle sizes the particles of the active ingredient as produced may be size reduced by conventional means eg by micronisation. The desired fraction may be separated out by air classification or sieving. Preferably, the particles will be crystalline.
Achieving high dose reproducibility with micronised powders is difficult because of their poor flowability and extreme agglomeration tendency. To improve the efficiency of dry powder compositions, the particles should be large while in the inhaler, but small when discharged into the respiratory tract. Thus, an excipient such as lactose or glucose is generally employed. The particle size of the excipient will usually be much greater than the inhaled medicament within the present invention. When the excipient is lactose it will typically be present as milled lactose, preferably crystalline alpha lactose monohydrate.
Pressurized aerosol compositions will generally be filled into canisters fitted with a valve, especially a metering valve. Canisters may optionally be coated with a plastics material e. g. a fluorocarbon polymer as described in W096/32150. Canisters will be fitted into an actuator adapted for buccal delivery. iv) Nasal mucosal administration
The compounds of the invention may also be administered via the nasal mucosal. Typical compositions for nasal mucosa administration are typically applied by a metering, atomizing spray pump and are in the form of a solution or suspension in an inert vehicle such as water optionally in combination with conventional excipients such as buffers, anti-microbials, tonicity modifying agents and viscosity modifying agents. v) Parenteral Administration
The compounds of the invention may also be administered directly into the blood stream, into muscle, or into an internal organ. Suitable means for parenteral administration include intravenous, intraarterial, intraperitoneal, intrathecal, intraventricular, intraurethral, intrasternal, intracranial, intramuscular and subcutaneous. Suitable devices for parenteral administration include needle (including microneedle) injectors, needle-free injectors and infusion techniques. Parenteral formulations are typically aqueous solutions which may contain excipients such as salts, carbohydrates and buffering agents (preferably to a pH of from 3 to 9), but, for some applications, they may be more suitably formulated as a sterile nonaqueous solution or as a dried form to be used in conjunction with a suitable vehicle such as sterile, pyrogen-free water.
The preparation of parenteral formulations under sterile conditions, for example, by lyophilization, may readily be accomplished using standard pharmaceutical techniques well known to those skilled in the art. The solubility of compounds of the invention used in the preparation of parenteral solutions may be increased by the use of appropriate formulation techniques, such as the incorporation of solubility-enhancing agents.
Formulations for parenteral administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release. Thus compounds of the invention may be formulated as a solid, semi-solid, or thixotropic liquid for administration as an implanted depot providing modified release of the active compound. Examples of such formulations include drug-coated stents and PGLA microspheres. vi) Topical Administration
The compounds of the invention may also be administered topically to the skin or mucosa, that is, dermally or transdermally. Typical formulations for this purpose include gels, hydrogels, lotions, solutions, creams, ointments, dusting powders, dressings,
foams, films, skin patches, wafers, implants, sponges, fibers, bandages and microemulsions. Liposomes may also be used. Typical carriers include alcohol, water, mineral oil, liquid petrolatum, white petrolatum, glycerin, polyethylene glycol and propylene glycol. Penetration enhancers may be incorporated; see, for example, J Pharm Sci, 88 (10), 955-958 by Finnin and Morgan (October 1999). Other means of topical administration include delivery by electroporation, iontophoresis, phonophoresis, sonophoresis and microneedle or needle-free injection.
Formulations for topical administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release. vii) Rectal/lntravaginal Administration
Compounds of the invention may be administered rectally or vaginally, for example, in the form of a suppository, pessary, or enema. Cocoa butter is a traditional suppository base, but various alternatives may be used as appropriate. Formulations for rectal/vaginal administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release. viii) Ocular Administration
Compounds of the invention may also be administered directly to the eye or ear, typically in the form of drops of a micronized suspension or solution in isotonic, pH- adjusted, sterile saline. Other formulations suitable for ocular and aural administration include ointments, biodegradable {e.g. absorbable gel sponges, collagen) and nonbiodegradable (e.g. silicone) implants, wafers, lenses and particulate or vesicular systems, such as niosomes or liposomes. A polymer such as crossed-linked polyacrylic acid, polyvinylalcohol, hyaluronic acid, a cellulosic polymer, for example, hydroxypropylmethylcellulose, hydroxyethylcellulose, or methyl cellulose, or a heteropolysaccharide polymer, for example, gelan gum, may be incorporated together with a preservative, such as benzalkonium chloride. Such formulations may also be delivered by iontophoresis.
Formulations for ocular/aural administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted, or programmed release.
ix) Other Technologies
Compounds of the invention may be combined with soluble macromolecular entities, such as cyclodextrin and suitable derivatives thereof or polyethylene glycol-containing polymers, in order to improve their solubility, dissolution rate, taste-masking, bioavailability and/or stability for use in any of the aforementioned modes of administration.
The amount of the active compound administered will be dependent on the subject being treated, the severity of the disorder or condition, the rate of administration, the disposition of the compound and the discretion of the prescribing physician. However, an effective dosage is typically in the range of 0.01 -3000 mg, more preferably 0.5-1000 mg of active ingredient or the equivalent amount of a pharmaceutically acceptable salt thereof per day. Daily dosage may be administered in one or more treatments, preferably from 1 to 4 treatments, per day. Preferably, the the pharmaceutical compositions of the invention are made up in a form suitable for oral, inhalation or topical administration, being particularly preferred oral or inhalation administration.
The pharmaceutical formulations may conveniently be presented in unit dosage form and may be prepared by any of the methods well known in the art of pharmacy.
Preferably the composition is in unit dosage form, for example a tablet, capsule or metered aerosol dose, so that the patient may administer a single dose.
The amount of each active which is required to achieve a therapeutic effect will, of course, vary with the particular active, the route of administration, the subject under treatment, and the particular disorder or disease being treated.
Combination Product
The invention also provides a combination product comprising (i) the compounds of the invention as described herein; and (ii) one or more additional active substances which are known to be useful in the treatment of respiratory diseases; allergic diseases; inflammatory or autoimmune-mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (MPDs); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors; more in particular wherein the pathological condition or disease is selected from leukemia, lymphomas
and solid tumors, rheumatoid artritis (RA), multiple sclerosis (MS), amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, cutaneous vasculitis, cutaneous lupus erythematosus, dermatomyositis, blistering diseases including but not limited to pemphigus vulgaris, bullous pemphigoid and epidermolysis bullosa, asthma, chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF), idiopathic pulmonary fibrosis, sarcoidosis, atopic dermatitis, allergic rhinitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma (BCC), squamous cell carcinoma (SCC) and actinic keratosis (AK); even more in particular wherein the pathological condition or disease is leukemia, lymphomas and solid tumors, rheumatoid artritis (RA), multiple sclerosis (MS), amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, asthma, chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF), idiopathic pulmonary fibrosis, sarcoidosis, atopic dermatitis, allergic rhinitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma (BCC), squamous cell carcinoma (SCC) and actinic keratosis (AK).
The pyrrolopyrimidine derivatives defined herein may also be combined with other active compounds in the treatment of a pathological condition or disease susceptible to amelioration by inhibition of PI3Ks.
The combinations of the invention can optionally comprise one or more additional active substances which are known to be useful in the treatment of respiratory diseases; allergic diseases; inflammatory or autoimmune-mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (MPDs); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors.
Particularly, the combinations of the invention can optionally comprise one or more additional active substances which are known to be useful in the treatment of neoplastic diseases (e.g. leukemia, lymphomas, solid tumors); transplant rejection, bone marrow transplant applications (e.g., graft- versus-host disease); autoimmune diseases (e.g. rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, cutaneous vasculitis, cutaneous lupus
erythematosus, dermatomyositis and blistering diseases including but not limited to pemphigus vulgaris, bullous pemphigoid and epidermolysis bullosa; respiratory inflammation diseases (e.g. asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis); skin inflammatory diseases (e.g., atopic dermatitis, contact dermatitis, eczema or psoriasis); premalignant and malignant skin conditions (e.g. basal cell carcinoma (BCC), squamous cell carcinoma (SCC) or actinic keratosis (AK)); neurological disorders and pain (such as pain associated with rheumatoid arthritis or osteoarthritis, back pain, general inflammatory pain, inflammatory neuropathic pain, trigeminal neuralgia or central pain).
Preferably, the combinations of the invention can optionally comprise one or more additional active substances which are known to be useful in the treatment of neoplastic diseases leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, type I diabetes, cutaneous vasculitis, cutaneous lupus erythematosus, dermatomyositis, blistering diseases including but not limited to pemphigus vulgaris, bullous pemphigoid and epidermolysis bullosa, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic keratosis.
In particular, the combinations of the invention can optionally comprise one or more additional active substances which are known to be useful in the treatment of neoplastic diseases leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, type I diabetes, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic keratosis
The combinations of the invention comprise (i) a compound of the invention as defined above; and (ii) another compound selected from the group consisting of an Adenoside A2A agonist, an agent for treating cardiovascular disorders, an agent for treating diabetes, and an agent for treating liver disease, an anti-allergic agent, an anticholinergic agent, an anti-inflammatory agent, an anti-infective agent, a p2-adrenergic
agonist, a Chemoattractant receptor homologous molecule expressed on TH2 cells (CRTH2) inhibitor, a chemotherapeutic agent, a corticosteroid, an ΙΚΚβ/ΙΚΒΚΒ (IkB kinase beta or IKK2) inhibitor, an immunosuppressant, a Janus kinase (JAK) inhibitor, a topically acting p38 Mitogen-Activated Protein Kinase (p38 MAPK) inhibitor, a Phosphosdiesterase (PDE) IV inhibitor, and a Spleen tyrosine kinase (Syk) inhibitor, for simultaneous, separate or sequential use in the treatment of the human or animal body.
In a particular embodiment, the combinations of the invention can optionally comprise one or more additional active substances selected from
a) Dyhydrofolate reductase inhibitors, such as Methotrexate or CH- 1504;
b) Dihydroorotate dehydrogenase (DHODH) inhibitors such as leflunomide, teriflunomide, or the compounds described in the International Patent Application Nos. WO2008/077639 and WO2009/021696;
c) Immunomodulators such as Glatiramer acetate (Copaxone), Laquinimod or Imiquimod;
d) Inhibitors of DNA synthesis and repair, such as Mitoxantrone or Cladribine;
e) Immunosuppressants, such as Imuran (azathioprine) or Purinethol (6-mercaptopurine or 6-MP);
f) Anti-alpha 4 integrin antibodies, such as Natalizumab (Tysabri) ; g) Alpha 4 integrin antagonists such as R-1295 , TBC-4746, CDP-323, ELND-002, Firategrast or TMC-2003;
h) Corticoids and glucocorticoids such as prednisone or methylprednisolone, fluticasone, mometasone, budesonide, ciclesonide or beta-metasone;
i) Fumaric acid esters, such as BG-12;
j) Anti-tumor necrosis factor-alpha (Anti-TNF-alpha) monoclonal antibodies such as Infliximab, Adalimumab or Certolizumab pegol; k) Soluble Tumor necrosis factor-alpha (TNF-alpha) Antagonists such as Ethanercept;
I) Anti-CD20 (lymphocyte protein) monoclonal antibodies such as Rituximab, Ocrelizumab Ofatumumab or TRU-015;
m) Anti-CD52 (lymphocyte protein) monoclonal antibodies such as alemtuzumab;
n) Anti-CD25 (lymphocyte protein) such as daclizumab;
o) Anti-CD88 (lymphocyte protein), such as eculizumab or pexilizumab;
p) Anti-lnterleukin 6 Receptor (IL-6R), such as tocilizumab;
q) Anti-lnterleukin 12 Receptor (IL-12R) / Interleukin 23 Receptor (IL-
23R), such as ustekinumab;
r) Calcineurin inhibitors such as cyclosporine A or tacrolimus;
s) Inosine-monophosphate dehydrogenase (IMPDH) inhibitors, such as mycophenolate mophetyl, ribavirin, mizoribine or mycophenolic acid;
t) Cannabinoid receptor agonists such as Sativex;
u) Chemokine CCR1 antagonists such as MLN-3897 or PS-031291 ; v) Chemokine CCR2 antagonists such as INCB-8696;
w) Necrosis factor-kappaB (NF-kappaB or NFKB) Activation Inhibitors such as Sulfasalazine, Iguratimod or MLN-0415;
x) Adenosine A2A agonists, such as ATL-313, ATL-146e, CGS-21680,
Regadenoson or UK-432,097;
y) Sphingosine-1 (S1 P) phosphate receptor agonists such as fingolimod, BAF-312, or ACT128800;
z) Sphingosine-1 (S1 P) liase inhibitors such as LX2931 ;
aa) Spleen tyrosine kinase (Syk) inhibitors, such as R-1 12;
bb) Protein Kinase Inhibitors (PKC) inhibitors, such as NVP-AEB071 ; cc) Anti-cholinergic agents such as tiotropium or aclidinium;
dd) Beta adrenergic agonists such as formoterol, indacaterol or LAS100977 (abediterol);
ee) MABA (molecules with dual activity: beta-adrenergic agonists and muscarinic receptor antagonists)
ff) Histamine 1 (H1 ) receptor antagonists, such as azelastine or ebastine;
gg) Cysteinyl leukotriene (CysLT) receptor antagonists, such as montelukast;
hh) Mast cell stabilizers, such as nedocromil or chromoglycate;
ii) 5-lipoxygenase-activating protein (FLAP) inhibitors, such as MK886 or BAY X 1005;
jj) 5-lipoxygenase (5-LO) inhibitors, such as WY-50295T;
Chemoattractant receptor homologous molecule expressed on TH2 cells (CRTH2) inhibitors, such as OC-459, AZD-1981 , ACT- 129968, QAV-680;
Vitamin D derivatives like calcipotriol (Daivonex) ;
Anti-inflammatory agents, such as non-steroidal anti-inflammatory drugs (NSAIDs) or selective cyclooxygenase-2 (COX-2) inhibitors such as aceclofenac, diclofenac, ibuprofen, naproxen, apricoxib, celecoxib, cimicoxib, deracoxib, etoricoxib, lumiracoxib, parecoxib sodium, rofecoxib, selenocoxib-1 or valdecoxib;
Anti-allergic agents;
Anti-viral agents;
Phosphodiestearase (PDE) III inhibitors;
Phosphosdiesterase (PDE) IV inhibitors such as roflumilast or GRC-4039;
Dual Phosphodiestearase (PDE) lll/IV inhibitors;
Xanthine derivatives, such as theophylline or theobromine;
p38 Mitogen-Activated Protein Kinase (p38 MAPK) Inhibitors such as ARRY-797;
Mitogen-activated extracellular signal regulated kinase kinase (MEK) inhibitor, such as ARRY-142886 or ARRY-438162;
Janus kinase (JAK) inhibitors, such as tofacitinib (previously known as tasocitinib or CP-690,550) from Pfizer and INCB-18424, from Incyte;
Interferons comprising Interferon beta 1 a such as Avonex from Biogen Idee, CinnoVex from CinnaGen and Rebif from EMD Serono, and Interferon beta 1 b such as Betaferon from Schering and Betaseron from Berlex;
Interferon alpha such as Sumiferon MP;
Epidermal Growth Factor Receptor (EGFR) inhibitors such as erlotinib, Trastuzumab, Herceptin, Avastin, Platins (cisplatin, carboplatin) or Temazolamide;
Antineoplastic agents such as Docetaxel, Estramustine, Anthracyc lines, (doxorubicin (Adriamycin), epirubicin (Ellence), and liposomal doxorubicin (Doxil)), Taxanes (docetaxel (Taxotere), paclitaxel (Taxol), and protein-bound paclitaxel (Abraxane)), Cyclophosphamide (Cytoxan), Capecitabine (Xeloda), 5 fluorouracil (5 FU), Gemcitabine (Gemzar) or Vinorelbine (Navelbine);
Specific examples of suitable corticoids and glucocorticoids that can be combined with the PI3K inhibitors of the present invention are prednisolone, methylprednisolone, dexamethasone, dexamethasone cipecilate, naflocort, deflazacort, halopredone acetate, budesonide, beclomethasone dipropionate, hydrocortisone, triamcinolone acetonide, fluocinolone acetonide, fluocinonide, clocortolone pivalate, methylprednisolone aceponate, dexamethasone palmitoate, tipredane, hydrocortisone aceponate, prednicarbate, alclometasone dipropionate, halometasone, methylprednisolone suleptanate, mometasone furoate, rimexolone, prednisolone farnesylate, ciclesonide, butixocort propionate, RPR-106541 , deprodone propionate, fluticasone propionate, fluticasone furoate, halobetasol propionate, loteprednol etabonate, betamethasone butyrate propionate, flunisolide, prednisone, dexamethasone sodium phosphate, triamcinolone, betamethasone 17-valerate, betamethasone, betamethasone dipropionate, hydrocortisone acetate, hydrocortisone sodium succinate, prednisolone sodium phosphate and hydrocortisone probutate.
Specific examples of suitable Syk kinase inhibitors that can be combined with the PI3K inhibitors of the present invention are fosfamatinib (from Rigel), R-348 (from Rigel), R- 343 (from Rigel), R-1 12 (from Rigel), piceatannol, 2-(2-Aminoethylamino)-4-[3- (trifluoromethyl)phenylamino] pyrimidine-5-carboxamide, R-091 (from Rigel), 6-[5- Fluoro-2-(3,4,5-trimethoxyphenylamino)pyrimidin-4-ylamino]-2,2-dimethyl-3,4-dihydro- 2H-pyrido[3,2-b][1 ,4]oxazin-3-one benzenesulfonate (R-406 from Rigel), 1 -(2,4,6- Trihydroxyphenyl)-2-(4-methoxyphenyl)ethan-1 -one, N-[4-[6-(Cyclobutylamino)-9H- purin-2-ylamino]phenyl]-N-methylacetamide (QAB-205 from Novartis), 2-[7-(3,4- Dimethoxyphenyl)imidazo[1 ,2-c]pyrimidin-5-ylamino]pyridine-3-carboxamide
dihydrochloride (BAY-61 -3606 from Bayer) and AVE-0950 (from Sanofi-Aventis).
Specific examples of suitable M3 antagonists (anticholinergics) that can be combined with the PI3K inhibitors of the present invention are tiotropium salts, oxitropium salts, flutropium salts, ipratropium salts, glycopyrronium salts, trospium salts, zamifenacin, revatropate, espatropate, darotropium bromide, CI-923, NPC-14695, BEA-2108, 3-[2- Hydroxy-2,2-bis(2-thienyl)acetoxy]-1 -(3-phenoxypropyl)-1 -azoniabicyclo[2.2.2]octane salts (in particular aclidinium salts, more preferably aclidinium bromide), 1 -(2- Phenylethyl)-3-(9H-xanthen-9-ylcarbonyloxy)-1 -azoniabicyclo[2.2.2]octane salts, 2-oxo- 1 ,2,3,4-tetrahydroquinazoline-3-carboxylic acid endo-8-methyl-8-azabicyclo[3.2.1 ]oct-3- yl ester salts (DAU-5884), 3-(4-Benzylpiperazin-1 -yl)-1 -cyclobutyl-1 -hydroxy-1 - phenylpropan-2-one (NPC-14695), N-[1 -(6-Aminopyridin-2-ylmethyl)piperidin-4-yl]-
2(R)-[3,3-difluoro-1 (R)-cyclopentyl]-2-hydroxy-2-phenylacetamide (J-104135), 2(R)- Cyclopentyl-2-hydroxy-N-[1 -[4(S)-methylhexyl]piperidin-4-yl]-2-phenylacetamide (J- 106366), 2(R)-Cyclopentyl-2-hydroxy-N-[1 -(4-methyl-3-pentenyl)-4-piperidinyl]-2- phenylacetamide (J-104129), 1 -[4-(2-Aminoethyl)piperidin-1 -yl]-2(R)-[3,3- difluorocyclopent-1 (R)-yl]-2-hydroxy-2-phenylethan-1 -one (Banyu-280634), N-[N-[2-[N- [1 -(Cyclohexylmethyl)piperidin-3(R)-ylmethyl]carbamoyl]ethyl]carbamoylmethyl]-3,3,3- triphenylpropionamide (Banyu CPTP), 2(R)-Cyclopentyl-2-hydroxy-2-phenylacetic acid 4-(3-azabicyclo[3.1 .0]hex-3-yl)-2-butynyl ester (Ranbaxy 364057), 3(R)-[4,4-Bis(4- fluorophenyl)-2-oxoimidazolidin-1 -yl]-1 -methyl-1 -[2-oxo-2-(3-thienyl)ethyl]pyrrolidinium iodide, N-[1 -(3-Hydroxybenzyl)-1 -methylpiperidinium-3(S)-yl]-N-[N-[4-
(isopropoxycarbonyl)phenyl]carbamoyl]-L-tyrosinamide trifluoroacetate, UCB-101333, Merck's OrM3, 7-endo-(2-hydroxy-2,2-diphenylacetoxy)-9,9-dimethyl-3-oxa-9- azoniatricyclo[3.3.1 .0(2,4)]nonane salts, 3(R)-[4,4-Bis(4-fluorophenyl)-2- oxoimidazolidin-1 -yl]-1 -methyl-1 -(2-phenylethyl)pyrrolidinium iodide, trans-4-[2- [Hydroxy-2,2-(dithien-2-yl)acetoxy]-1 -methyl-1 -(2-phenoxyethyl)piperidinium bromide from Novartis (412682), 7-(2,2-diphenylpropionyloxy)-7,9,9-trimethyl-3-oxa-9- azoniatricyclo[3.3.1.0*2,4*]nonane salts, 7-hydroxy-7,9,9-trimethyl-3-oxa-9- azoniatricyclo[3.3.1.0*2,4*]nonane 9-methyl-9H-fluorene-9-carboxylic acid ester salts, all of them optionally in the form of their racemates, their enantiomers, their diastereomers and mixtures thereof, and optionally in the form of their pharmacologically-compatible acid addition salts. Among the salts chlorides, bromides, iodides and methanesulphonates are preferred.
Specific examples of suitable beta adrenergic agonists (p2-agonists) that can be combined with the PI3K inhibitors of the present invention are are terbutaline sulphate, eformoterol fumarate, formoterol fumarate, bambuterol, ibuterol, isoprenaline hydrochloride, dopexamine, metaprotenerol, tulobuterol, procaterol hydrochloride, sibenadet hydrochloride, mabuterol hydrochloride, albuterol sulphate, salbutamol sulphate, salmefamol, salmeterol xinafoate, carmoterol hydrochloride, (R)-albuterol hydrochloride, Levalbuterol hydrochloride; Levosalbutamol hydrochloride; (-)- Salbutamol hydrochloride, formoterol, (R,R)-Formoterol tartrate; Arformoterol tartrate, sulfonterol, Bedoradrine sulphate, Indacaterol, Trantinterol hydrochloride, Milveterol hydrochloride, Olodaterol, fenoterol hydrobromide, rimoterol hydrobromide, riproterol hydrochloride, Vilanterol broxaterol, pirbuterol hydrochloride, bitolterol mesylate, clenbuterol hydrochloride, AZD-3199, GSK-159802; GSK-597901 , GSK-678007, GSK- 961081 ; 4-[2-[3-(1 H-Benzimidazol-1 -yl)-1 , 1 -dimethylpropylamino]-1 -hydroxyethyl]-2-(4- methoxybenzylamino)phenol, 1 -[2H-5-hydroxy-3-oxo-4H-1 ,4-benzoxazin-8-yl]-2-[3-(4-
N,N-dimethylaminophenyl)-2-methyl-2-propylamino]ethanol, 1 -[2H-5-hydroxy-3-oxo- 4H-1 ,4-benzoxazin-8-yl]-2-[3-(4-domethoxyphenyl)-2-methyl-2-propylamino]ethanol, 1 - [2H-5-hydroxy-3-oxo-4H-1 ,4-benzoxazin-8-yl]-2-[3-(4-n-butyloxyhenyl)-2-methyl-2- propylamino]ethanol, KUL-1248, HOKU-81 , SM-1 10444, RP-58802B, LAS100977 (abediterol) and compounds described in PCT patent applications Nos. WO 2007/124898, WO 2006/122788A1 , WO 2008/046598, WO 2008095720, WO 2009/068177 and WO 2010/072354.
Specific examples of suitable anti-allergic agents that can be combined with the PI3K inhibitors of the present invention are anti-histamines (e.g. Methapyrilene, Mequitazine, Azelastine hydrochloride, Acrivastine, Emedastine difumarate, Emedastine fumarate, Loratadine, Cyproheptadine hydrochloride, Diphenhydramine hydrochloride, Doxepin hydrochloride, Promethazine hydrochloride, Levocabastine hydrochloride, Desloratadine, Cinnarizine, Setastine hydrochloride, Mizolastine, Ebastine, Cetirizine hydrochloride, Epinastine hydrochloride, Olopatadine hydrochloride, Bepotastine besilate,Triprolidine hydrochloride, Rupatadine fumarate, Fexofenadine hydrochloride, Levocetirizine dihydrochloride, Ketotifen, Azatadine maleate, Dimethindene maleate, Clemastine fumarate, Alcaftadine, Bilastine, Vapitadine hydrochloride, AZD-1744, GSK-1004723D, GSK-835726 or SUN-1334H.
Specific examples of suitable Phosphosdiesterase IV (PDE IV) inhibitors that can be combined with the PI3K inhibitors of the present invention are benafentrine dimaleate, etazolate, denbufylline, rolipram, cipamfylline, zardaverine, arofylline, filaminast, tipelukast, tofimilast, piclamilast, tolafentrine, mesopram, drotaverine hydrochloride, lirimilast, roflumilast, cilomilast, oglemilast, apremilast, tetomilast, filaminast, (R)-(+)-4- [2-(3-Cyclopentyloxy-4-methoxyphenyl)-2-phenylethyl]pyridine (CDP-840), N-(3,5- Dichloro-4-pyridinyl)-2-[1 -(4-fluorobenzyl)-5-hydroxy-1 H-indol-3-yl]-2-oxoacetamide (GSK-842470), 9-(2-Fluorobenzyl)-N6-methyl-2-(trifluoromethyl)adenine (NCS-613), N- (3,5-Dichloro-4-pyridinyl)-8-methoxyquinoline-5-carboxamide (D-4418), 3-[3- (Cyclopentyloxy)-4-methoxybenzyl]-6-(ethylamino)-8-isopropyl-3H-purine hydrochloride (V-1 1294A), 6-[3-(N,N-Dimethylcarbamoyl)phenylsulfonyl]-4-(3-methoxyphenylamino)- 8-methylquinoline-3-carboxamide hydrochloride (GSK-256066), 4-[6,7-Diethoxy-2,3- bis(hydroxymethyl)naphthalen-1 -yl]-1 -(2-methoxyethyl)pyridin-2(1 H)-one (T-440), (-)- trans-2-[3'-[3-(N-Cyclopropylcarbamoyl)-4-oxo-1 ,4-dihydro-1 ,8-naphthyridin-1 -yl]-3- fluorobiphenyl-4-yl]cyclopropanecarboxylic acid, MK-0873, CDC-801 , UK-500001 , BLX-914, 2-carbomethoxy-4-cyano-4-(3-cyclopropylmethoxy-4- difluroromethoxyphenyl)cyclohexan1 -one, cis [4-cyano-4-(3-cyclopropylmethoxy-4-
difluoromethoxyphenyl)cyclohexan-1 -ol, 5(S)-[3-(Cyclopentyloxy)-4-methoxyphenyl]- 3(S)-(3-methylbenzyl)piperidin-2-one (IPL-455903), ONO-6126 (Eur Respir J 2003, 22(Suppl. 45): Abst 2557) and the compounds claimed in the PCT patent applications number WO 03/097613, WO 2004/058729, WO 2005/049581 , WO 2005/123693, WO 2005/123692, and WO 2010/069504.
Specific examples of suitable immunosupressants that can be combined with the PI3K inhibitors of the present invention are picremolimus, tacrolimus, cyclosporine A, leflunomide, teriflunomide, vidofludimus, laquinimod, methotrexate, 5-fluorouracil (5- FU), anti-TNF agents and compounds described in PCT patent applications Nos. WO 2008/077639, WO 2009/021696, WO 2009/153043, and WO2010083975 (in particular amino(iso)nicotinic acid derivatives selected from the group consisting of 2-(3'-ethoxy- 3-(trifluoromethoxy)biphenyl-4-ylamino)nicotinic acid, 2-(3,5-difluoro-3'- methoxybiphenyl-4-ylamino)nicotinic acid and 2-(3,5-difluoro-2-methylbiphenyl-4- ylamino)nicotinic acid; and azabiphenylaminobenzoic acid derivatives selected from the group consisting of 5-cyclopropyl-2-(2-(2,6-difluorophenyl)pyrimidin-5-ylamino)benzoic acid, 5-cyclopropyl-2-((2-(2-(trifluoromethyl)phenyl)pyrimidin-5-yl)amino)benzoic acid and 5-methyl-2-((6-(2,3-difluorophenyl)pyridin-3-yl)amino)benzoic acid) Specific examples of suitable anti-infectives that can be combined with the PI3K inhibitors of the present invention are aclarubicin, actinomycin D, amrubicin, annamycin, adhamycin, bleomycin, daunorubicin, doxorubicin, elsamitrucin, epirubicin, galarubicin, idarubicin, mitomycin C, mupiricin, nemorubicin, neocarzinostatin, peplomycin, pirarubicin, rebeccamycin, retapamulin, stimalamer, streptozocin, valrubicin, zinostatin, amphotericin B, bifonazole, caspofungin, clotrimazole, echinocandin B, econazole, fluconazole, flucytosine, itraconazole, ketoconazole, miconazole, posaconazole, ravuconazole, terbinafine, tioconazole, voriconazole and combinations thereof. Particularly preferred combination products according to the invention comprise a compound of formula (I) and a therapeutically effective amount of one or more additional therapeutic agents selected from the group consisting of mometasone furoate, ciclesonide, budesonide, fluticasone propionate, fluticasone furoate, betamethasone valerate, clobetasol propionate, tiotropium salts, glycopyrronium salts, 3-[2-Hydroxy-2,2-bis(2-thienyl)acetoxy]-1 -(3-phenoxypropyl)-1 - azoniabicyclo[2.2.2]octane salts (in particular aclidinium salts, preferably aclidinium bromide), 1 -(2-Phenylethyl)-3-(9H-xanthen-9-ylcarbonyloxy)-1 -
azoniabicyclo[2.2.2]octane salts, formoterol, salmeterol, indacaterol, carmoterol, LAS 100977 (abediterol), compounds described in PCT patent applications Nos. WO 2008/077639, WO 2009/021696, WO 2009/153043, and WO 2010/083975 (in particular amino(iso)nicotinic acid derivatives selected from the group consisting of 2- (3'-ethoxy-3-(trifluoromethoxy)biphenyl-4-ylamino)nicotinic acid, 2-(3,5-difluoro-3'- methoxybiphenyl-4-ylamino)nicotinic acid and 2-(3,5-difluoro-2-methylbiphenyl-4- ylamino)nicotinic acid; and azabiphenylaminobenzoic acid derivatives selected from the group consisting of 5-cyclopropyl-2-(2-(2,6-difluorophenyl)pyrimidin-5-ylamino)benzoic acid, 5-cyclopropyl-2-((2-(2-(trifluoromethyl)phenyl)pyrimidin-5-yl)amino)benzoic acid and 5-methyl-2-((6-(2,3-difluorophenyl)pyridin-3-yl)amino)benzoic acid), methapyrilene, cetirizine, loratadine, ebastine, desloratadine, fexofenadine, azelastine, levocabastine, olopatadine, Montelukast, picremolimus, tacrolimus, mupiricin, retapamulin, clotrimazole, ketoconazole and terbinafine. The compounds of formula (I) and the combinations of the invention may be used in the treatment of respiratory diseases; allergic diseases; inflammatory or autoimmune- mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (MPDs such as polycythemia vera, essential thrombocythemia or mielofibrosis); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors, wherein the use of a PI3K inhibitor is expected to have a beneficial effect, for example leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, cutaneous vasculitis, cutaneous lupus erythematosus, dermatomyositis, blistering diseases including but not limited to pemphigus vulgaris, bullous pemphigoid and epidermolysis bullosa, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic keratosis.
In particular the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis,
atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic keratosis.
The active compounds in the combination product may be administered together in the same pharmaceutical composition or in different compositions intended for separate, simultaneous, concomitant or sequential administration by the same or a different route.
It is contemplated that all active agents would be administered at the same time, or very close in time. Alternatively, one or two actives could be administered in the morning and the other (s) later in the day. Or in another scenario, one or two actives could be administered twice daily and the other (s) once daily, either at the same time as one of the twice-a-day dosing occurred, or separately. Preferably at least two, and more preferably all, of the actives would be administered together at the same time. Preferably, at least two, and more preferably all actives would be administered as an admixture.
The invention is also directed to a combination product of the compounds of the invention together with one or more other therapeutic agents for use in the treatment of a pathological condition or disease susceptible to amelioration by inhibiton of Phosphoinositide 3-Kinases (PI3Ks), in particular wherein the pathological condition or disease is selected from respiratory diseases; allergic diseases; inflammatory or autoimmune-mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo- dysplastic syndrome; myeloproliferative disorders (MPDs such as polycythemia vera, essential thrombocythemia or mielofibrosis); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors; more in particular wherein the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, cutaneous vasculitis, cutaneous lupus erythematosus, dermatomyositis, blistering diseases including but not limited to pemphigus vulgaris, bullous pemphigoid and epidermolysis bullosa, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic keratosis.
In particular the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic keratosis.
The invention also encompasses the use of a combination of the compounds of the invention together with one or more other therapeutic agents for the manufacture of a formulation or medicament for treating these diseases. The invention also provides a method of treatment of a pathological condition or disease susceptible to amelioration by inhibiton of Phosphoinositide 3-Kinases (PI3Ks), in particular wherein the pathological condition or disease is selected from respiratory diseases; allergic diseases; inflammatory or autoimmune-mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (MPDs such as polycythemia vera, essential thrombocythemia or mielofibrosis); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors; more in particular wherein the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, cutaneous vasculitis, cutaneous lupus erythematosus, dermatomyositis, blistering diseases including but not limited to pemphigus vulgaris, bullous pemphigoid and epidermolysis bullosa, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic keratosis.
In particular the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, asthma, chronic obstructive pulmonary
disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic keratosis. The active compounds in the combinations of the invention may be administered by any suitable route, depending on the nature of the disorder to be treated, e.g. orally (as syrups, tablets, capsules, lozenges, controlled-release preparations, fast-dissolving preparations, etc); topically (as creams, ointments, lotions, nasal sprays or aerosols, etc); by injection (subcutaneous, intradermic, intramuscular, intravenous, etc.) or by inhalation (as a dry powder, a solution, a dispersion, etc).
The active compounds in the combination, i.e. the pyrrolotriazinone derivatives of the invention, and the other optional active compounds may be administered together in the same pharmaceutical composition or in different compositions intended for separate, simultaneous, concomitant or sequential administration by the same or a different route.
One execution of the present invention consists of a kit of parts comprising a pyrrolopyrimidine derivative of the invention together with instructions for simultaneous, concurrent, separate or sequential use in combination with another active compound useful in the treatment of respiratory diseases; allergic diseases; inflammatory or autoimmune-mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo- dysplastic syndrome; myeloproliferative disorders (MPDs such as polycythemia vera, essential thrombocythemia or mielofibrosis); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors; more in particular wherein the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, cutaneous vasculitis, cutaneous lupus erythematosus, dermatomyositis, blistering diseases including but not limited to pemphigus vulgaris, bullous pemphigoid and epidermolysis bullosa, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic keratosis.
In particular the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic keratosis.
Another execution of the present invention consists of a package comprising a imidazopyridine derivative of the invention and another active compound useful in the treatment of respiratory diseases; allergic diseases; inflammatory or autoimmune- mediated diseases; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (MPDs such as polycythemia vera, essential thrombocythemia or mielofibrosis); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors; more in particular wherein the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, cutaneous vasculitis, cutaneous lupus erythematosus, dermatomyositis, blistering diseases including but not limited to pemphigus vulgaris, bullous pemphigoid and epidermolysis bullosa, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic keratosis.
In particular the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic keratosis.
EXPERIMENTAL SECTION
The invention further provides synthetic processes which are useful for preparing said compounds.
The compounds of the invention can be prepared using the methods and procedures described in WO 2012/146666 or WO 2012/146667, or using similar methods and procedures. It will be appreciated that where typical or preferred process conditions (i.e., reaction temperatures, times, mole ratios of reactants, solvents, pressures, etc.) are given; other process conditions can also be used unless otherwise stated. Optimum reaction conditions may vary with the particular reactants or solvent used, but such conditions can be determined by one skilled in the art by routine optimization procedures.
Additionally, as will be apparent to those skilled in the art, conventional protecting groups may be necessary to prevent certain functional groups from undergoing undesired reactions. The choice of a suitable protecting group for a particular functional group, as well as suitable conditions for protection and deprotection, are well known in the art. For example, numerous protecting groups, and their introduction and removal are described in T. W. Greene and G. M. Wuts, Protecting Groups in Organic Synthesis, Third Edition, Wiley, New York, 1999, and references cited therein.
The term amino-protecting group refers to a protecting group suitable for preventing undesired reactions at amino nitrogen. Representative amino-protecting groups include, but are not limited to, formyl; acyl groups, for example alkanoyl groups such as acetyl; alkoxycarbonyl groups such as tert-butoxycarbonyl (Boc); arylmethoxycarbonyl groups such as benzyloxycarbonyl (Cbz) and 9-fluorenylmethoxycarbonyl (Fmoc); arylmethyl groups such as benzyl (Bn), trityl (Tr), and 1 ,1 -di-(4'-methoxyphenyl)methyl; silyl groups such as trimethylsilyl (TMS) and tert-butyldimethylsilyl (TBS); and the like.
The term hydroxy-protecting group refers to a protecting group suitable for preventing undesired reactions at a hydroxy group. Representative hydroxy-protecting groups include, but are not limited to, alkyl groups, such as methyl, ethyl, and tert-butyl; acyl groups, for example alkanoyl groups, such as acetyl; arylmethyl groups, such as benzyl (Bn), p-methoxybenzyl (PMB), 9-fluorenylmethyl (Fm), and diphenylmethyl (benzhydryl, DPM); silyl groups, such as trimethylsilyl (TMS) and tert-butyldimethylsilyl (TBS); and the like.
According to the embodiment of the present invention, compounds of general Formula (I) may be prepared by the synthetic route illustrated in Scheme 1 , from compounds of Formula (II)
by treatment with compounds of Formula (III) in the presence of a suitable base such as potassium carbonate, diisopropylethylamine or sodium hydride in an appropriate solvent such as ie f-butanol, /V,/V-dimethylformamide or tetrahydrofuran at temperatures ranging from room temperature to 160 °C, with or without the use of microwaves irradiation and with or without the use of a catalytic amount of caesium fluoride.
Alternatively, compounds of Formula (I) can be obtained in two step synthesis from compounds of Formula (IV), where compounds of Formula (IV) can be obtained from reaction of compounds of Formula (II) with compounds of Formula (V) also in the presence of a suitable base such as potassium carbonate, diisopropylethylamine or sodium hydride in an appropriate solvent such as ie f-butanol, /V,/V-dimethylformamide or tetrahydrofuran at temperatures ranging from room temperature to 160 °C, with or without the use of microwaves irradiation and with or without the use of a catalytic amount of caesium fluoride. Compounds of Formula (III) can be prepared as shown in Scheme 1 from compounds of Formula (V) following the standard methods described as it follows for compounds of Formula (I), with or without the use of protecting groups.
In the particular case of compounds of general Formula (I) where L represents a direct bond or a -(CH2)i-4 group, compounds of Formula (I) can be prepared in the two steps synthesis from compounds of Formula (IV) by reaction with the compounds of Formula (VI) where X represents a boronic acid or boronic ester using standard Suzuki coupling conditions. In the particular case of compounds of general Formula (I) where L represents a linker selected from -0-, -S-, -NH- group, compounds can be obtained from compounds of Formula (IV) by reacting with compounds of Formula (VI) where X represents an hydrogen using copper or palladium catalysed coupling methods well known for those skilled in the art.
In the particular case of compounds of general Formula (I) where the group L represents a -C(0)-NR'- group or a -C(0)-0-R"- group; compounds of Formula (I) can
be prepared in the two steps synthesis from compounds of Formula (IV) by treatment with a lithiating agent such as n-BuLi, in a non protic solvent such as hexane or tetrahydrofurane, at a temperature between -78°C and 0°C and subsequently treated with compounds of Formula (VI) where X represents an halogen or another leaving group at a temperature between -78°C and room temperature.
Compounds of Formula (VI) can either be commercial or prepared by standard methods, and can be used in a protected form to prevent certain functional groups from undergoing undesired reactions. In these cases, standard methods for the removal of these protecting groups can be used at the suitable step of the synthesis. Numerous protecting groups, their introduction and their removal are described in T. W. Greene and G. M. Wuts, Protecting Groups in Organic Synthesis, Third Edition, Wiley, New York, 1999, and references cited therein. Finally, compounds of general Formula (II) are known and prepared as described in WO 2005/1 13556, US 2009/0312319 and WO 2012/146667.

Formula (V)
B-L-X
Formula (VI)

Formula (IV)
Scheme 1 The invention is also directed to a compound of the invention as described herein for use in the treatment of the human or animal body by therapy. Compounds of the invention intended for pharmaceutical use may be administered as crystalline or amorphous products, or mixtures thereof. They may be obtained, for example, as solid plugs, powders, or films by methods such as precipitation, crystallization, freeze drying, spray drying, or evaporative drying. Microwave or radio frequency drying may be used for this purpose.
Examples
The particular compounds as described in examples 1 -15 (including Preparations 1 -35) are given in order to provide a person skilled in the art with a sufficiently clear and complete explanation of the present invention and about some representative embodiments of compounds encompassed general formula (I), but should not be considered as limiting of the essential aspects of its subject, as set out in the preceding portions of this description. As explained in the general description the compounds were prepared using the methods and procedures described in WO 2012/146666 or WO 2012/146667, or using similar methods and procedures. Reagents, starting materials, and solvents were purchased from commercial suppliers and used as received. Concentration or evaporation refers to evaporation under vacuum using a Buchi rotatory evaporator.
Reaction products were purified, when necessary, in a Biotage SP1® automated purification system. Purifications in normal phase were made in the solvent system as indicated. Purifications in reverse phase were made using a Ci8 column and a gradient of water-acetonitrile/MeOH (1 :1 ) (0.1 % v/v ammonium formate both phases) from 0% to 100% acetonitrile/MeOH (1 :1 ) in 40 column volumes. The appropriate fractions were collected and the solvents evaporated under reduced pressure and/or liofilized.
Preparative HPLC-MS were performed on a Waters instrument equipped with a 2767 injector/collector, a 2525 binary gradient pump, a 2996 PDA detector, a 515 pump as a make-up pump and a ZQ4000 Mass spectrometer detector or on a Agilent 1200 Series coupled to an Agilent 6120 Mass spectrometer detector. Both systems were equipped with a Symmetry Prep Ci8 (19 x 300 mm, 7 μηη) column or a XBridge Prep Ci8 (19 x 100 mm, 5 μηη) column. The mobile phase was formic acid (0.4 mL), ammonia (0.1 mL), methanol (500 mL) and acetonitrile (500 mL) (B) and formic acid (0.5 mL), ammonia (0.125 mL) and water (1000 mL) (A), the specific gradients used are specified in each particular case. The flow rate was 20 mL/min.
Purity and MS identification was performed in a Waters 2795 system coupled to a 2996 Diode array detector and to a Waters ZQ mass spectrometer detector or in a Waters Acquity UPLC system coupled to a SQD mass spectrometer detector. The injection volume was 5 microliter on the HPLC and 0.5 microliter on the UPLC. Chromatograms were processed at 210 nM or 254 nM. Mass spectra of the chromatograms were acquired using positive and negative electrospray ionization. The mobile phase was formic acid (0.4 mL), ammonia (0.1 mL), methanol (500 mL) and acetonitrile (500 mL)
(B) and formic acid (0.5 mL), ammonia (0.125 mL) and water (1000 mL) (A) and a gradient between 0 to 95% of B was used. Columns: HPLC: Waters Symmetry (2.1x50mm, 3.5 m); UPLC: ACQUITY UPLC BEH C-18 (2.1 x50mm, 1 .7 m) 1H Nuclear Magnetic Resonance Spectra were recorded on a Varian Mercury plus operating at a frequency of 400 MHz for the 1H spectra. Samples were dissolved in the specified deuterated solvent. Tetramethylsilane was used as reference.
PREPARATION 1
yV-(3,5-Difluorophenyl)-6-methoxy-3-nitropyridin-2 -amine
3,5-Difluoroaniline (13.35 g, 100.00 mmol) was added to a solution of 2-chloro-6- methoxy-3-nitropyridine (15.00 g, 80.00 mmol) in dry /V,/V-dimethylformamide (60 mL) and was stirred and heated in the microwave at 140°C for 1 h. The reaction was concentrated in vacuum and the residue was partitioned between water and ethyl acetate. The organic phase was dried, filtered and concentrated in vacuum. The residue was purified using a SP1 Purification System (hexane:ethyl acetate from 0% to 5%) to afford 7.20 g (32% yield) of the expected compound.
LRMS (M+1 ): 282
1H NMR (400 MHz, DMSO-d6) δ ppm 3.94 (s, 3 H) 6.48 (d, 1 H) 6.77 - 7.20 (m, 1 H) 7.60 (dd, 2 H) 8.48 (d, 1 H) 10.48 (s, 1 H)
PREPARATION 2
yV2-(3,5-Difluorophenyl)-6-methoxypyridine-2,3-diamine
A mixture of /V-(3,5-difluorophenyl)-6-methoxy-3-nitropyridin-2-amine (7.20 g, 25.60 mmol) and Pd/C 10% (0.27 g, 2.56 mmol) in ethanol (250 mL) was stirred under an atmosphere of hydrogen (ca.1 atm) for 4h, and then filtered through Celite® and concentrated in vacuum to give 6.30 g (98% yield) of the title compound.
LRMS (M+1 ): 252
1H NMR (400 MHz, DMSO-d6) δ ppm 3.75 (s, 3 H) 4.57 (s, 2 H) 6.20 (d, 1 H) 6.51 - 6.68 (m, 1 H) 7.05 (d, 1 H) 7.39 (d, 2 H) 8.22 (s, 1 H)
PREPARATION 3
(S)-ferf-Butyl 1 -((3-amino-6-methoxypyridin-2-yl)(3,5-difluorophenyl)amino)-1 - oxopropan-2-yl carbamate
A mixture of /V2-(3,5-difluorophenyl)-6-methoxypyridine-2,3-diamine (2.00 g, 7.96 mmol), ('S)-2-(ie f-butoxycarbonylamino)propanoic acid (1 .50 g, 7.96 mmol), N-(3- dimethylaminopropyl)-/V'-ethylcarbodiimide hydrochloride (1 .83 g, 9.55 mmol), 1 - hydroxybenzotriazole hydrate (1 .08 g, 7.96 mmol) and /V-methylmorpholine (3.60 mL, 32.74 mmol) in dry Λ/,/V-dimethylformamide (20 mL) was stirred at room temperature overnight. The reaction was poured into a mixture of water/brine (1 :1 ) and extracted with ethyl acetate. The combined organic layers were washed with saturated sodium bicarbonate solution, water and brine and then dried, filtered and concentrated in vacuum to afford 2.86 g (85% yield) of the expected product.
LRMS (M+1 ): 423
1H NMR (400 MHz, DMSO-d6) δ ppm 1.28 (d, 3 H) 1.42 (s, 9 H) 3.88 (s, 3 H) 3.94 - 4.13 (m, 1 H) 6.32 (d, 1 H) 6.62 - 6.77 (m, 1 H) 7.40 (d, 1 H) 7.47 (d, 1 H) 7.51 - 7.67 (m, 2 H) 8.15 (s, 1 H) 9.53 (s, 1 H) PREPARATION 4
(S -l -^-iS.S-DifluorophenylJ-S-methoxy-SH-imida
A solution of (S)-tert-buty\ 1 -((3-amino-6-methoxypyridin-2-yl)(3,5- difluorophenyl)amino)-1 -oxopropan-2-ylcarbamate (2.86 g, 6.77 mmol) in glacial acetic acid (20 mL) was stirred at 130°C overnight. The HPLC-MS of the crude showed a main peak with a mass corresponding to the expected product but acetylated (M+1 : 281 ). This crude was concentrated in vacuum and the residue obtained was hydrolysed by dissolving the crude in methanol (37 mL), adding a 5N hydrochloric acid solution (37 mL) and stirring the mixture at 80°C overnight. The mixture was concentrated in vacuum. The resulting aqueous solution was washed with dichloromethane (2X), neutralised with a 2N sodium hydroxide solution and washed again with dichloromethane (2X). A saturated potassium carbonate solution was added (to reach pH 10-12) and it was extracted with ethyl acetate (3X). The organic phase was dried, filtered and concentrated in vacuum to afford 1 .31 g (60%) of the expected product. LRMS (M+1 ): 305
1H NMR (400 MHz, DMSO-d6) δ ppm 1 .40 (d, 3 H) 2.00 (s, 2 H) 3.75 (s, 3 H) 4.10 (m, 1 H) 6.75 (m, 1 H) 7.50 (m, 1 H) 7.60 (m, 2H) 8.00 (m, 1 H)
PREPARATION 5
1 -Chloro-7-(1 -methyl-1 H-pyrazol-4-yl)-5-((2-(trimethylsilyl)ethoxy)methyl)-5H- pyrrolo[2,3-cf]pyrimidine
4-Chloro-5-iodo-7-((2-(trimethylsilyl)eth^ (500 mg, 1 .22 mmol), 1 -methyl-4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 /-/-pyrazole (305 mg, 1.46 mmol), [1 ,1 '-Bis(diphenylphosphino)ferrocene] dichloropalladium(ll) complex with dichloromethane (99.7 mg, 0.12 mmol), tripotassium phosphate (31 1 mg, 1 .46 mmol) and triethylamine (0.9 mL 6.10 mmol) in a mixture of tetrahydrofurane/water (16 mL/1.6 mL) were stirred at reflux temperature for 3h. The reaction was partitioned between water and ethyl acetate. The organic phase was dried, filtered and concentrated in vacuum. The residue obtained was purified using a SP1 Purification System (dichloromethane:ethyl acetate from 0% to 20%) to afford 380 mg (72% yield) of the expected compound.
LRMS (M+1 ): 364
1H NMR (400 MHz, Chloroform-d) δ ppm 8.65 (s, 1 H), 7.67 (d, J = 0.6 Hz, 1 H), 7.61 (s, 1 H), 7.35 (s, 1 H), 5.67 (s, 2H), 3.98 (s, 3H), 3.65 - 3.51 (m, 2H), 0.99 - 0.88 (m, 2H), - 0.04 (s, 9H).
PREPARATION 6
(S)-W-(1 -(3-(3,5-Difluorophenyl)-5-methox
(1 -methyl-1 H-pyrazol-4-yl)-5-((2-(trimethy^
cf]pyrimidin-1 -amine
To a solution of (S)-1 -(3-(3,5-difluorophenyl)-5-methoxy-3/-/-imidazo[4,5-t)]pyridin-2- yl)ethanamine (60 mg, 0.20 mmol) in ie f-butanol (3.5 mL), 1 -chloro-7-(1 -methyl-1 /-/- pyrazol-4-yl)-5-((2-(trimethylsilyl)ethoxy)methyl)-5/-/-pyrrolo[2,3-c ]pyrimidine (1 10 mg, 0.30 mmol), caesium fluoride (92 mg, 0.60 mmol) and ethyldiisopropylamine (0.21 mL, 1 .21 mmol) were added. The reaction mixture was stirred at 130°C for 7h. More caesium fluoride (92 mg, 0.60 mmol) and ethyldiisopropylamine (0.21 mL, 1.21 mmol) were added and the mixture was stirred under previous conditions overnight. Water was added (25 mL) and the mixture extracted with ethyl acetate (20 mL, 3x). The organic extracts were collected together and washed with water and brine. The organic phase was dried over anhydrous sodium sulphate, filtered and concentrated in vacuum. The residue was purified using a SP1 Purification System (diethyl ether- methanol from 0% to 5%) to give 31 mg (25% yield) of the title compound.
LRMS (M+1 ): 632 PREPARATION 7
W-(3-(4-Chloro-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-<^pyrimi yl)phenyl)methanesulfonamide
To a solution of 4-chloro-5-iodo-7-((2-(trimethylsilyl)ethoxy)methyl)-7/-/-pyrrolo[2,3- c/]pyrimidine (0.5 g, 1 .22 mmol) in tetrahydrofurane/water (160 mL/16 mL), N-(3- (4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)phenyl)methanesulfonamide (0.44 g, 1 .46 mmol), tripotassium phosphate (0.31 g, 1.46 mmol) and triethylamine (0.85 mL, 6.1 mmol) were added. The system was purged and filled in with nitrogen. [1 ,1 - Bis(diphenylphosphino)ferrocene] dichloropalladium(ll) complex with dichloromethane (0.1 g, 0.12 mmol) was added and the inert atmosphere was re-established again. The reaction mixture was stirred at reflux for 3h, then concentrated in vacuum and the aqueous residue was extracted with ethyl acetate. The organic extracts were collected together and washed with brine, dried over anhydrous magnesium sulphate, filtered and concentrated in vacuum. The residue was purified using a SP1 Purification System (dichloromethane-ethyl acetate from 0% to 10%) to give 360 mg (66% yield) of the title compound.
LRMS (M+1 ): 454
PREPARATION 8
(S)-W-(3-(1 -((1 -(3-(3,5-Difluorophenyl)-5-methoxy-3H-imidazo[4,5-fe]pyridin-2- yl)ethyl)amino)-5-((2-(trimethylsilyl)etho
yl)phenyl)methanesulfonamide
To a solution of (S)-1 -(3-(3,5-difluorophenyl)-5-methoxy-3/-/-imidazo[4,5-t)]pyridin-2- yl)ethanamine (60 mg, 0.20 mmol) in ie/f-butanol (3.5 mL), /V-(3-(4-chloro-7-((2- (trimethylsilyl)ethoxy)methyl)-7/-/-pyrrolo[2,3-c/]pyrimidin-5- yl)phenyl)methanesulfonamide (135 mg, 0.30 mmol), caesium fluoride (90 mg, 0.60 mmol) and ethyldiisopropylamine (0.21 mL, 1.21 mmol) were added. The reaction mixture was stirred at 130°C for 7h. More caesium fluoride (90mg, 0.60mmol) and ethyldiisopropylamine (0.21 mL, 1.21 mmol) were added and the mixture stirred under previous conditions overnight. Water was added (25 mL) and the aqueous phase was extracted with ethyl acetate (20 mL, 3x). The organic extracts were collected together and washed with water and brine. The organic phase was dried over anhydrous sodium sulphate, filtered and concentrated in vacuum. The residue was purified using a SP1 Purification System (hexane-diethyl ether from 0% to 100%) to give 39 mg (27% yield) of the title compound.
LRMS (M+1 ): 721
PREPARATION 9
(S)-/V-(3-(4-((1 -(8-Methyl-1 -oxo-2-phenyl-1 ,2-dihydroisoquinolin-3-yl)ethyl)amino)- 7-((2-(trimethylsilyl)ethoxy)methyl)-7H^yrrolo[2,3-<^pyrimidin-5- yl)phenyl)methanesulfonamide To a solution of (S)-3-(1 -aminoethyl)-8-methyl-2-phenylisoquinolin-1 (2/-/)-one (prepared as in US2009/0312319, 80 mg, 0.28 mmol) in ie/f-butanol (5.5 ml_), /V-(3-(4-chloro-7- ((2-(trimethylsilyl)ethoxy)methyl)-7/-/-pyrrolo[2,3-c/]pyrimidin-5- yl)phenyl)methanesulfonamide (1 10 mg, 0.24 mmol), caesium fluoride (145 mg, 0.95 mmol) and ethyldiisopropylamine (410 μΙ, 2.35 mmol) were added. The reaction mixture was stirred at 130° for 44 h. Water was added (20 mL) and the aqueous phase extracted with ethyl acetate (3x15 mL). The combined organic extracts were washed with water and brine, dried over anhydrous magnesium sulphate and the solvent was evaporated under vacuum. The residue was purified using a SP1 Purification System (hexane-diethyl ether from 0% to 100%) to give 37 mg (19% yield) of title compound. LRMS (M+1 ): 695
PREPARATION 10
(S)-W-(3-(4-((1 -(5-Methyl-4-oxo-3^henyl-3,4-dihydroquinazolin-2-yl)ethyl)amino)- 7-((2-(trimethylsilyl)ethoxy)methyl)-7H^yrrolo[2,3-rf]pyrimidin-5- yl)phenyl)methanesulfonamide
To a solution of (S)-2-(1 -aminoethyl)-5-methyl-3-phenylquinazolin-4(3/-/)-one (prepared as in WO2005/1 13556, 90 mg, 0.30 mmol) in ie/f-butanol (6 mL), caesium fluoride (165 mg, 1 .05 mmol) and ethyldiisopropylamine (0.396 mL, 2.77 mmol) were added. The reaction mixture was stirred at 130°C for 52 h. Water was added (20 mL) and the aqueous phase extracted with ethyl acetate (3x15 mL). The combined organic extracts were washed with water and brine, dried over anhydrous magnesium sulphate and the solvent was evaporated under vacuum. The residue was purified using a SP1 Purification System (hexane-diethyl ether from 0% to 100%) to give 57 mg (27% yield) of title compound.
LRMS (M+1 ): 696
PREPARATION 1 1
4-Chloro-5-iodo-7-((2-(trimethylsilyl)ethoxy)methyl)-7H^yrrolo[2,3-<^pyrimidine
Sodium hydride (60 % dispersion in mineral oil, 0.172 g, 4.30 mmol) was suspended in /V,/V-dimethylformamide (5 mL). The mixture was stirred for 10 min and was cooled at
0°C with an ice-bath. 4-Chloro-5-iodo-7/-/-pyrrolo[2,3-c ]pyrimidine (1 g, 3.58 mmol) dissolved in Λ/,/V-dimethylformamide (5 mL) was added drop-wise and the mixture was stirred 30 min. At the same temperature [2-(chloromethoxy)ethyl](trimethyl)silane (0.9 g, 5.4 mmol) dissolved in /V,/V-dimethylformamide (5 mL) was added drop-wise and stirred for 30 min at 0°C. The mixture was poured into water and extracted twice with ethyl acetate. The organics were dried over sodium sulphate and concentrated under reduced pressure. The residue was purified using a SP1 Purification System (hexane- ethyl acetate from 0% to 100%) to give 1.10 g (75% yield) of the title compound as a white solid.
LRMS (M+1 ): 409
PREPARATION 12
(S)-3-(1 -((5-Bromo-7-((2-(trimethylsilyl)etho^
4-yl)amino)ethyl)-8-methyl-2-phenylisoquinolin-1 (2H)-one
To a solution of (S)-3-(1 -aminoethyl)-8-methyl-2-phenylisoquinolin-1 (2/-/)-one (prepared as in US2009/0312319, 1 g, 2.62 mmol) in ie/f-butanol (20 mL), 5-bromo-4- chloro-7-((2-(trimethylsilyl)ethoxy)methyl)-7/-/-pyrrolo[2,3-c/]pyrimidine (prepared as in WO2014/060432, 1.96 g, 5.27 mmol), caesium fluoride (201 mg, 1 .42 mmol) and ethyldiisopropylamine (5.5 mL, 31.58 mmol) were added. The reaction mixture was stirred overnight at 100°C. Further caesium fluoride (210 mg) and ethyldiisopropylamine (1 .25 mL) were added and the reaction mixture stirred overnight at 100°C. The mixture was concentrated under vacuum, a solution of saturated sodium bicarbonate (50 mL) was added and extracted with ethyl acetate (3x25 mL). The combined organic extracts were washed with water and brine. The organic phase was dried over anhydrous magnesium sulphate and the solvent was evaporated under vacuum. The residue was purified using a SP1 Purification System (hexane-ethyl acetate from 0% to 35%) to give 784.8 mg (38% yield) of title compound as a white solid.
LRMS (M+1 ): 604
1H NMR (400 MHz, Chloroform-d) δ ppm 8.13 (s, 1 H), 7.55 - 7.38 (m, 3H), 7.37 - 7.29 (m, 3H), 7.21 (m, 1 H), 7.03 (s, 1 H), 6.62 (s, 1 H), 6.05 (d, J = 7.0 Hz, 1 H), 5.49 (s, 2H), 5.03 (m, 1 H), 3.59 - 3.42 (m, 2H), 2.85 (s, 3H), 1.48 (d, J = 6.8 Hz, 3H), 0.97 - 0.79 (m, 2H), 0.03 - 0.12 (m, 9H).
PREPARATION 13
(S)-3-(1 -((5-(6-Amino-1 H-indol-4-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H- pyrrolo[2,3-<^pyrimidin-4-yl)amino)ethyl)-8-methyl-2^henylisoquinolin-1 (2H)-one
To a solution of (S)-3-(1 -(5-bromo-7-((2-(trimethylsilyl)ethoxy)methyl)-7/-/-pyrrolo[2,3- <^pyrimidin-4-ylamino)ethyl)-8-methyl-2-phenylisoquinolin-1 (2/-/)-one (200 mg, 0.33 mmol) in dimethoxyethane/water (8 mL/2 mL), 4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)-1 H-indol-6-amine (prepared as in WO2014/060432, 215 mg, 0.83 mmol) and sodium carbonate (90 mg, 0.83 mmol) were added. The nitrogen
atmosphere was established and [1 ,1 '-bis(diphenylphosphino)ferrocene]
dichloropalladium(ll) complex with dichloromethane (83 mg, 0.1 mmol) was added. The reaction mixture was stirred at 70°C overnight. The reaction crude was poured into a saturated aqueous solution of ammonium chloride and extracted with ethyl acetate. The organic phase was washed with water and brine, dried, filtered and concentrated in vacuum. The crude obtained was purified by flash chromatography (hexane:ethyl acetate from 0% to 100%) to give 108 mg (50% yield) of the title compound.
LRMS (M+1 )+: 657
1 H-NMR (400MHz, DMSO-d6) δ ppm 0.00 (s, 9H) 0.90 (t, 2H) 1.10 (d, 3H) 2.70 (s, 3H) 3.60 (t, 2H) 4.50 (q, 1 H) 5.60 (s, 2H) 6.10-6.20 (s, 2H), 6.50 (s, 1 H) 6.60 (s, 1 H) 7.10- 7.20 (m, 2H) 7.30-7.60 (m, 12H) 8.20 (s, 1 H) 10.70-10.80 (s, 1 H)
PREPARATION 14
(Sj-/V-(4-(4-((1 -(8-Methyl-1 -oxo-2-phenyl-1 ,2-dihydroisoquinolin-3-yl)ethyl)amino)-
7-((2-(trimethylsilyl)ethoxy)methyl)^
yl)sulfamide
To a solution of (S)-3-(1 -((5-(6-Amino-1 H-indol-4-yl)-7-((2-(trimethylsilyl)ethoxy)methyl)- 7H-pyrrolo[2,3-c/]pyrimidin-4-yl)amino)ethyl)-8-methyl-2-phenylisoquinolin-1 (2/-/)-one (108 mg, 0.16 mmol) in tetrahydrofurane (2 mL), sulfamoyi chloride (50 mg, 0.43 mmol) and pyridine (0.04 mL, 0.49 mmol) were added. The reaction mixture was stirred at room temperature for 2h and then poured into water (25 mL) and extracted with ethyl acetate. The organic phase was washed with water and brine, dried, filtered and concentrated in vacuum. The crude obtained was purified by flash chromatography (hexane: ethyl acetate from 0% to 100%) to afford 70 mg (56% yield) of the expected compound.
LRMS (M+1 ): 736
1H NMR (400 MHz, Chloroform-d) δ ppm 0.9-1 .0 (2H, m), 1 .2 (d, 3H), 2.8 (s, 3H), 3.6 (t, 2H), 4.7-4.8 (q, 2H), 5.0 (d, 1 H), 6.6 (d, 2H), 6.3 (s, 1 H), 6.4 (s, 1 H), 6.5 (s, 1 H), 7.0 (s, 1 1-1)7.2-7.4 (m, 6H), 7.4-7.6 (m 3H), 8.1 (s, 1 H), 8.2 (s, 1 H) PREPARATION 15
(S)-2-(1 -((5-Bromo-7-((2-(trimethylsilyl)etho^
4-yl)amino)ethyl)-5-methyl-3-phenylquinazolin-4(3H)-one
To a solution of (S)-2-(1 -aminoethyl)-5-methyl-3-phenylquinazolin-4(3/-/)-one
hydrochloride (prepared as in WO2005/1 13556, 416 mg, 1 .15 mmol) in ie/f-butanol (7 ml_), 5-bromo-4-chloro-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-c ]pyrimidine (624 mg, 1.72 mmol), caesium fluoride (80 mg, 0.5 mmol) and ethyldiisopropylamine (1 .8 ml_, 10.3 mmol) were added. The reaction mixture was stirred at 100°C overnight and then concentrated in vacuum. Ethyl acetate was added and the organic layer washed with water, a saturated sodium bicarbonate solution and brine and finally dried, filtered and concentrated in vacuum. The crude obtained was purified by column flash chromatography (hexane:ethyl acetate from 0% to 35%) to give 369 mg (53% yield) of the title compound.
LRMS (M+1 ): 607
1H-NMR (400MHz, DMSO-d6) δ ppm 0.00 (s, 9H) 0.90 (t, 2H) 1 .35 (d, 3H) 2.70 (s, 3H) 3.50 (t, 2H) 5.00 (q, 1 H) 5.40 (s, 2H) 7.30 (d, 1 H) 7.50-7.80 (m, 8H) 8.20 (s, 1 H)
PREPARATION 16
1 -(2-((fer Butyldimethylsilyl)oxy)ethyl)-4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan- 2-yl)-1 H-pyrazole
To a solution of 4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 /-/-pyrazole (1 g, 5.15 mmol) in butan-2-one (14 ml_), caesium carbonate (5 g, 15.47mmol) was added. To the resulting suspension, (2-bromoethoxy)(ie f-butyl)dimethylsilane (2.14 ml_, 10.29 mmol) was added and the reaction mixture was heated at 80°C for 3h. The reaction mixture was cooled with and ice-water bath, filtered and the solid washed with butan-2-one and ethyl acetate, providing the product in the filtrate. The combined filtrates were evaporated under vacuum furnishing an oil as a crude. This crude was purified using a SP1 Purification System (hexane-diethyl ether from 0% to 50%) to give 970 mg (50% yield) of the title compound as a pale yellow oil.
LRMS (M+1 ): 353
PREPARATION 17
(S)-2-(1 -((5-(1 -(2-((ferf-Butyldimethylsilyl)oxy)ethyl)-1 H-pyrazol-4-yl)-7-((2-
(trimethylsilyl)ethoxy)methyl)-7H^yrrolo[2,3-<^pyrimidin-4-yl)amino)ethyl)-5- methyl-3-phenylquinazolin-4(3H)-one
(S)-2-(1 -((5-Bromo-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-c/]pyrim yl)amino)ethyl)-5-methyl-3-phenylquinazolin-4(3/-/)-one (150 mg, 0.25
mmol) was dissolved in Λ/,/V-dimethylformamide (5 ml_). 1 -(2-((ie f-
Butyldimethylsilyl)oxy)ethyl)-4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)-1 /-/- pyrazole (200 mg, 0.57 mmol), 2M sodium carbonate (0.3 ml_, 0.60 mmol) and tetrakis(triphenylphosphine)palladium(0) (30 mg, 0.03 mmol) were added under argon atmosphere and the mixture was heated at 100°C for 3 h. The
reaction mixture was poured into water and extracted twice with ethyl acetate. The organics were dried over sodium sulphate, filtered and evaporated under reduced pressure. The residue was purified by reverse phase using a SP1 Purification System to give 1 10mg (57% yield)
of the title compound as an oil.
LRMS (M+1 ): 751
PREPARATION 18
(S -3-(1 -((5-(1 -(2-((ferf-Butyldimethylsilyl)oxy)ethyl)-1 H-pyrazol-4-yl)-7-((2-
(trimethylsilyl)ethoxy)methyl)-7H^yrrolo[2,3-<^pyrimidin-4-yl)amino)ethyl)-8- methyl-2-phenylisoquinolin-1 (2H)-one
(S)-3-(1 -((5-Bromo-7-((2-(trimethylsilyl)ethoxy)methyl)-7/-/-pyrrolo[2,3-c/]pyrimidin-4- yl)amino)ethyl)-8-methyl-2-phenylisoquinolin-1 (2/-/)-one (150 mg, 0.25 mmol) was treated with 1 -(2-((ie/f-butyldimethylsilyl)oxy)ethyl)-4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)-1 /-/-pyrazole, 2M sodium carbonate and tetrakis(triphenylphosphine)palladium(0) according to the method described in Preparation 17. The residue was purified by reverse phase using a SP1 Purification System to give 178 mg (95% yield) of the title compound as a brown oil.
LRMS (M+1 ): 751
PREPARATION 19
(S)-yV-(3-Hydroxy-5-(4-((1 -(5-methyl-4-oxo-3-phenyl-3,4-dihydroquinazolin-2- yl)ethyl)amino)-7-((2-(trimethylsilyl)ethoxy)^
yl)phenyl)methanesulfonamide
(S)-2-(1 -((5-Bromo-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-c/]pyrimid yl)amino)ethyl)-5-methyl-3-phenylquinazolin-4(3/-/)-one (220 mg, 0.36 mmol) was treated with /V-(3-hydroxy-5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2- yl)phenyl)methanesulfonamide (prepared as in WO2014/060432, 178 mg, 0.57 mmol), 2M sodium carbonate (0.450 ml_, 0.9mmol) and tetrakis(triphenylphosphine)palladium(0) (40 mg, 0.1 mmol) according to the method described in Preparation 17. The residue was purified using a SP1 Purification System (hexane-ethyl acetate from 0% to 100%) to give 100 mg (40% yield) of the title compound as an oil.
LRMS (M+1 )+: 713
PREPARATION 20
1 -((4-Bromopyridin-2-yl)methyl)-W,W-dimethylpiperidin-4-amine
4-Bromopicolinaldehyde (300 mg, 1 .61 mmol) was dissolved in dichloroethane (30 ml_). /V,/V-Dimethylpiperidin-4-amine (227 mg, 1.77 mmol) and sodium
triacetoxyborohydride (1.025 g, 4.84 mmol) were added and the mixture was stirred at room temperature overnight. The mixture was partitioned between water and ethyl acetate. The aqueous phase was basified to pH=10 with a diluted sodium hydroxide solution. The aqueous phase was then extracted with dichloromethane. The combined organics were dried over anhydrous magnesium sulphate, filtered and evaporated under reduced pressure to give 390 mg (81 % yield) of the title compound as an orange oil.
LRMS (M+1 ): 298, 300
1H NMR (400 MHz, Chloroform-d) δ ppm 1 .59 (qd, J = 12.2, 3.8 Hz, 3H), 1.73 - 1 .86 (m, 2H), 2.02 - 2.23 (m, 3H), 2.29 (s, 7H), 2.92 (d, J = 1 1.9 Hz, 2H), 3.61 (s, 2H), 7.33 (dd, J = 5.3, 1 .9 Hz, 1 H), 7.63 (d, J = 1 .5 Hz, 1 H), 8.35 (d, J = 5.3 Hz, 1 H). PREPARATION 21
W,W-Dimethyl-1 -((4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridin-2- yl)methyl)piperidin-4-amine
1 -((4-Bromopyridin-2-yl)methyl)-/V,/V-dimethylpiperidin-4-amine (390 mg, 1 .31 mmol), 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi(1 ,3,2-dioxaborolane) (400 mg, 1 .58 mmol), potassium acetate (256 mg, 2.61 mmol) and [1 ,1 '-Bis(diphenylphosphino)ferrocene] dichloropalladium(ll) complex with dichloromethane (107 mg, 0.13 mmol) were
suspended in dioxane (4 mL). The mixture was heated in the microwave at 120°C for 30 minutes. The reaction was diluted with dioxane and filtered through Celite®. The filtrate was concentrated in vacuum and used for the next step without further purification.
LRMS (M+1 ): 346
PREPARATION 22
(S)-3-(1 -((5-(2-((4-(Dimethylamino)piperidin-1 -yl)methyl)pyridin-4-yl)-7-((2- (trimethylsilyl)ethoxy)methyl)-7H^yrrolo[2,3-<^pyrimidin-4-yl)amino)ethyl)-8- methyl-2-phenylisoquinolin-1 (2H)-one
(S)-3-(1 -((5-Bromo-7-((2-(trimethylsilyl)ethoxy)methyl)-7/-/-pyrrolo[2,3-c/]pyrimidin-4- yl)amino)ethyl)-8-methyl-2-phenylisoquinolin-1 (2/-/)-one (80 mg, 0.13 mmol) was dissolved in dioxane (2 mL). /V,/V-Dimethyl-1 -((4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)pyridin-2-yl)methyl)piperidin-4-amine (168 mg, 0.24 mmol), [1 ,1 - Bis(diphenylphosphino)ferrocene] dichloropalladium(ll) complex with dichloromethane (1 1 mg, 0.01 mmol) and 2M caesium carbonate solution (0.19 mL, 0.39 mmol) were added under argon atmosphere and the mixture was heated at 100°C for 1 .5 h. The reaction mixture was evaporated under reduced pressure. The residue was purified by reverse phase using a SP1 Purification System to give 69 mg (71 % yield) of the title compound.
LRMS (M+1 ): 744
1H NMR (400 MHz, Chloroform-d) δ ppm 0.97 (2H, t, J= 8.4 Hz), 1.38 (3H, d, J = 6.8 Hz), 1.76 (2H, m), 1 .94 (2H, t, J= 13.6 Hz), 2.57 (6H, s), 2.83 (4H, m), 3.00 (2H,m), 3.63 (4H, m), 5.03 (1 H, m), 5.1 1 (2H, t, J = 6.8 Hz), 5.64 (2H, m), 6.46 (1 H, s), 7.22- 7.57 (6H, m), 8.25 (1 H, s), 8.63 (1 H, d, J = 4.8Hz).
PREPARATION 23
1 -(5-Bromopyridin-3-yl)-4-(1 -methylpiperidin-4-yl)piperazine
3,5-Dibromopyridine (500 mg, 2.1 1 mmol), 1 -(1 -methylpiperidin-4-yl)piperazine (387 mg, 2.1 1 mmol), bis(dibenzylideneacetone)palladium (38.6 mg, 0.04 mmol), 4,5- Bis(diphenylphosphino)-9,9-dimethylxanthene (73 mg, 0.12 mmol) and sodium tert- butoxide (304 mg, 3.16 mmol) were suspended in toluene (5 mL) under argon atmosphere. The mixture was heated at 100°C for 4h. The solvent was evaporated under reduced pressure. Hydrochloric acid solution 1 M was added and extracted with dichloromethane. The resulting aqueous phase was basified to pH=10 and extracted
with dichloromethane. The combined organics were dried over anhydrous magnesium sulphate, filtered and evaporated under reduced pressure to give 685 mg (96% yield) of the title compound.
LRMS (M+1 ): 339; 341
1H NMR (400 MHz, Chloroform-d) δ ppm 1 .63 (2H, qd, J = 4 Hz,12 Hz), 1 .97 (2H, td, J = 2.4 Hz, 12 Hz), 2.28 (3H, s), 2.71 (4H, m), 2.92 (2H, m), 3.23 (4H, m), 7.28 (1 H, t, J = 2.4 Hz), 8.10 (1 H, d, J = 1.6 Hz), 8.19 (1 H, d, J = 2.4 Hz)
PREPARATION 24
1 -(1 -Methylpiperidin-4-yl)-4-(5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2- yl)pyridin-3-yl)piperazine
1 -(5-Bromopyridin-3-yl)-4-(1 -methylpiperidin-4-yl)piperazine (685 mg, 2.02 mmol), 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi(1 ,3,2-dioxaborolane) (564 mg, 2.22 mmol), potassium acetate (595 mg, 6.06 mmol), 1 ,1 '-Bis(diphenylphosphino)ferrocene (56 mg, 0.1 mmol) and [1 ,1 '-Bis(diphenylphosphino)ferrocene] dichloropalladium(ll) complex with dichloromethane (82 mg, 0.1 mmol) were suspended in dioxane (6 mL). The mixture was heated at 80°C for 16 h. The reaction was filtered and evaporated under reduced pressure. The crude obtained was used for the next step without further purification.
LRMS (M+1 ): 387
PREPARATION 25
(S)-5-Methyl-2-(1 -((5-(5-(4-(1 -methylpiperidin-4-yl)piperazin-1 -yl)pyridin-3-yl)-7-((2- (trimethylsilyl)ethoxy)methyl)-7H^yrrolo[2,3-<^pyrimidin-4-yl)amino)ethyl)-3- phenylquinazolin-4(3H)-one
(S)-2-(1 -((5-Bromo-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-c/]pyrimidin-4- yl)amino)ethyl)-5-methyl-3-phenylquinazolin-4(3/-/)-one (100 mg, 0.16 mmol) was dissolved in dioxane (1 mL). 1 -(1 -Methylpiperidin-4-yl)-4-(5-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)pyridin-3-yl)piperazine (0.33 mmol), [1 ,1 -
Bis(diphenylphosphino)ferrocene] dichloropalladium(ll) complex with dichloromethane (13 mg, 0.01 mmol) and 2M caesium carbonate solution (0.25 mL, 0.50 mmol) were added under argon atmosphere and the mixture was heated at 100°C for 1 h. The reaction mixture was evaporated under reduced pressure. The residue was purified by reverse phase using a SP1 Purification System to give 36 mg (28% yield) of the title compound.
LRMS (M+1 ): 786
1H NMR (400 MHz, Chloroform-d) δ ppm 0.02 (9H, s), 0.97 (2H, m), 1.38 (3H, d, J = 6.8 Hz), 1 .88-1.73 (4H, m), 2.25 (2H, t, J = 10.4 Hz), 2.4-2.32 (1 H, m), 2.43 (3H, s), 2.66 (4H, t, J = 4.8 Hz), 2.84 (3H, s), 3.1 1 (2H, m), 3.36 (4H, m), 3.62 (2H, m), 5.17 (1 H, q, J = 6.8Hz), 5.64 (2H, s), 6.35 (1 H, d, J = 8 Hz), 7.18 (1 H, s), 7.25 (1 H, d, J = 7.2 Hz), 7.34 (1 H, d, J = 7.6 Hz), 7.38 (1 H, s), 7.40 (1 H, m), 7.46 (1 H, m), 7.53 (1 H, m), 7.70-7.59 (4H, m), 8.35 (1 H, s), 8.41 (1 H, m), 8.44 (1 H, d, J = 2.8 Hz), 8.55 (1 H, s)
PREPARATION 26
W-((5-Bromo-2-methoxypyridin-3-yl)methyl)-1 -methylpiperidin-4-amine
5-Bromo-2-methoxynicotinaldehyde (472 mg, 2.18 mmol) and 1 -methylpiperidin-4- amine (249 mg, 2.18 mmol) were stirred in methanol (10 ml_). Acetic acid (0.12 mL) and sodium cyanoborohydride (137 mg, 2.18 mmol) were then added and the mixture was stirred at room temperature for 22 h. The reaction mixture was evaporated under reduced pressure and the residue was partitioned between dichloromethane and a saturated aqueous sodium hydrogen carbonate solution. The phases were separated. The aqueous layer was extracted with dichloromethane. The combined organics were dried over anhydrous magnesium sulphate, filtered and evaporated under reduced pressure to give 647 mg (94% yield) of the title compound.
LRMS (M+1 ): 314, 316
1H NMR (400 MHz, Chloroform-d) δ ppm 1 .44 (2H, m), 1 .88 (2H, m), 2.26 (3H, s), 2.43 (1 H, m), 2.81 (2H, m), 3.73 (2H, s), 3.93 (3H, s), 7.69 (1 H, d, J = 2.4 Hz), 8.08 (1 H, d, J = 2.4 Hz)
PREPARATION 27
ferf-Butyl ((5-bromo-2-methoxypyridin-3-yl)methyl)(1 -methylpiperidin-4- yl)carbamate /V-((5-Bromo-2-methoxypyridin-3-yl)methyl)-1 -methylpiperidin-4-amine (613 mg, 1.95 mmol) was dissolved in dichloromethane (6 mL). Di-ie f-butyl dicarbonate (510 mg, 2.33 mmol) was added and the mixture was stirred and heated at 50°C for 24h. The mixture was cooled to room temperature and was partitioned between water and dichloromethane. The aqueous phase was extracted with dichloromethane. The combined organics were dried over anhydrous magnesium sulphate, filtered and evaporated under reduced pressure. The residue was purified by reverse phase using
a SP1 Purification System. The product was treated with a saturated aqueous sodium hydrogen carbonate solution to afford 334 mg (41 % yield) of the neutral compound. LRMS (M+1 ): 414; 416
1H NMR (400 MHz, Chloroform-d) δ ppm 1 .58 (9H, s), 1.6-1 .4 (4H, m), 2.0 (2H,m), 2.24 (3H, s), 2.84 (2H, m), 3.92 (3H, s), 4.25 (3H, m), 7.45 (1 H, s), 8.05 (1 H, s)
PREPARATION 28
ferf-Butyl ((2-methoxy-5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridin-3- yl)methyl)(1 -methylpiperidin-4-yl)carbamate ferf-Butyl ((5-bromo-2-methoxypyridin-3-yl)methyl)(1 -methylpiperidin-4-yl)carbamate (288 mg, 0.69 mmol), was treated with 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi(1 ,3,2- dioxaborolane) (195 mg, 0.76 mmol), potassium acetate (206 mg, 2.1 mmol), 1 ,1 '- bis(diphenylphosphino)ferrocene (19 mg, 0.03 mmol) and [1 ,1 - bis(diphenylphosphino)ferrocene] dichloropalladium(ll) complex with dichloromethane (28 mg, 0.03 mmol) according to the method described in Preparation 24. The crude obtained was used for the next step without further purification.
LRMS (M+1 ): 462 PREPARATION 29
(S)-terf-Butyl ((2-methoxy-5-(4-((1 -(5-methyl-4-oxo-3-phenyl-3,4- dihydroquinazolin-2-yl)ethyl)amino)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H- pyrrolo[2,3-<^pyrimidin-5-yl)pyridin-3-yl)methyl)(1 -methylpiperidin-4- yl)carbamate
(S)-2-(1 -((5-Bromo-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-c/]pyrimidin-4- yl)amino)ethyl)-5-methyl-3-phenylquinazolin-4(3/-/)-one (220 mg, 0.36 mmol) was reacted with ferf-butyl ((2-methoxy-5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2- yl)pyridin-3-yl)methyl)(1 -methylpiperidin-4-yl)carbamate (0.76 mmol), [1 ,1 - bis(diphenylphosphino)ferrocene] dichloropalladium(ll) complex with dichloromethane (30 mg, 0.03 mmol) and 2M caesium carbonate solution (0.54 ml_, 1.08 mmol) according to the method described in Preparation 25. The residue was purified by reverse phase using a SP1 Purification System to give 103 mg (33% yield) of the title compound.
LRMS (M+1 ): 861
1H NMR (400 MHz, Chloroform-d) δ ppm 0.97 (2H, t, J = 8.4 Hz), 1 .84 (2H, m), 2.15 (2H, m), 2.84 (3H, s), 3.37 (2H, m), 3.62 (2H, J = 7.6 Hz), 4.13 (3H, s), 4.45 (2H, m),
5.21 (1 H, m), 5.62 (2H, s), 5.49 (1 H, d, J = 7.2 Hz), 7.01 (1 H, s), 7.66-7.2 (10H, m), 8.34 (1 H, s), 8.41 (2H, m)
PREPARATION 30
(S)-8-Methyl-3-(1 -((5-(5-(4-(1 -methylpiperidin-4-yl)piperazin-1 -yl)pyridin-3-yl)-7-((2- (trimethylsilyl)ethoxy)methyl)-7H^yrrolo[2,3-<^pyrimidin-4-yl)amino)ethyl)-2- phenylisoquinolin-1 (2H)-one
(S)-3-(1 -((5-Bromo-7-((2-(trimethylsilyl)etho
yl)amino)ethyl)-8-methyl-2-phenylisoquinolin-1 (2/-/)-one (100 mg, 0.16 mmol) was dissolved in dioxane (1 mL). 1 -(1 -Methylpiperidin-4-yl)-4-(5-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)pyridin-3-yl)piperazine (0.33 mmol), [1 ,1 - bis(diphenylphosphino)ferrocene] dichloropalladium(ll) complex with dichloromethane (13 mg, 0.01 mmol) and 2M caesium carbonate solution (0.25 mL, 0.50 mmol) were added under argon atmosphere and the mixture was heated at 100°C for 4 h. The reaction mixture was evaporated under reduced pressure. The residue was purified by reverse phase using a SP1 Purification System to give 89 mg (69% yield) of the title compound.
LRMS (M+1 ): 785
1H NMR (400 MHz, Chloroform-d) δ ppm 0.03 (9H, s), 0.97 (2H, t, J = 8 Hz), 1 .33 (3H, m), 1 .9-1 .4 (4H, m), 2.46 (3H, s), 2.65 (4H, m), 2.88 (3H, s), 3.15 (2H, m), 3.4-3.22 (4H, m), 3.62 (2H, t, J = 7.6 Hz), 5.04 (2H, d, J = 3.2 Hz), 5.63 (2H, s), 6.43 (1 H, s), 7.16 (1 H, s), 7.56-7.20 (10H, m), 8.27 (2H, m), 8.34 (1 H, J = 2.4 Hz), 8.54 (1 H, s) PREPARATION 31
3- (4-(4-Bromopyridin-2-yl)piperazin-1 -yl)-A/,A/-dimethylpropan-1 -amine
4- Bromo-2-fluoropyridine (0.29 mL, ) was dissolved in dimethyl sulfoxide (10 mL). N,N- dimethyl-3-(piperazin-1 -yl)propan-1 -amine (584 mg, 3.4 mmol) and potassium carbonate (1.96 g, 14.1 mmol) were added and the mixture was heated at 80°C 4.5 h. The reaction was cooled to room temperature and water was added. The mixture was basified to pH=10 and was extracted with ethyl acetate. The combined organics were dried over anhydrous magnesium sulphate, filtered and evaporated under reduced pressure to give 879 mg (95% yield) of the title compound.
LRMS (M+1 ): 327; 329
1H NMR (400 MHz, Chloroform-d) δ ppm 1 .71 (2H, m), 2.23 (6H, s), 2.31 (2H, t, 6.8Hz), 2.40 (2H, m), 2.53 (4H, t, J= 5.6 Hz), 3.54 (4H, t, J = 5.2 Hz), 6.75 (1 H, dd, J = 5.6 Hz.1.6 Hz), 6.78 (1 H, d, J = 1.6 Hz), 7.97 (1 H, d, J = 5.6 Hz) PREPARATION 32
W,W-Dimethyl-3-(4-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridin-2- yl)piperazin-1 -yl)propan-1 -amine
3-(4-(4-Bromopyridin-2-yl)piperazin-1 -yl)-/V,/V-dim (879 mg, 2.68 mmol), 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi(1 ,3,2-dioxaborolane) (749 mg, 2.95 mmol), potassium acetate (789 mg, 8.04 mmol), 1 ,1 '-bis(diphenylphosphino)ferrocene (74 mg, 0.13 mmol) and [1 ,1 '-bis(diphenylphosphino)ferrocene] dichloropalladium(ll) complex with dichloromethane (109 mg, 0.13 mmol) were suspended in dioxane (6 mL). The mixture was heated at 80°C for 21 h. The reaction was filtered and evaporated under reduced pressure. The crude obtained was used for the next step without further purification.
LRMS (M+1 ): 375
PREPARATION 33
(S)-3-(1 -((5-(2-(4-(3-(Dimethylamino)propyl)piperazin-1 -yl)pyridin-4-yl)-7-((2- (trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-<^pyrimidin-4-yl)amino)ethyl)-8- methyl-2-phenylisoquinolin-1 (2H)-one
(S)-3-(1 -((5-Bromo-7-((2-(trimethylsilyl)ethoxy)methyl)-7/-/-pyrrolo[2,3-c/]pyrimidin-4- yl)amino)ethyl)-8-methyl-2-phenylisoquinolin-1 (2/-/)-one (100 mg, 0.16 mmol) was dissolved in dioxane (2 mL). /V,/V-Dimethyl-3-(4-(4-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)pyridin-2-yl)piperazin-1 -yl)propan-1 -amine (0.36 mmol), [1 ,1 - bis(diphenylphosphino)ferrocene] dichloropalladium(ll) complex with dichloromethane (15 mg, 0.01 mmol) and 2M caesium carbonate solution (0.27 mL, 0.54 mmol) were added under argon atmosphere and the mixture was heated at 100°C for 17 h. The reaction mixture was evaporated under reduced pressure. The residue was purified by reverse phase using a SP1 Purification System to give 52 mg (37% yield) of the title compound.
LRMS (M+1 ): 773
1H NMR (400 MHz, Chloroform-d) δ ppm 0.97 (2H, m), 1 .37 (3H, d, J = 6.8Hz), 1.80 (2H, q, J = 7.6 Hz), 2.40 (6H, s), 2.43 (2H, m), 2.54 (6H, m), 2.88 (3H, s), 3.62 (6H, m),
5.07 (1 H, q, J = 6.8 Hz), 5.37 (1 H, d, J = 6.4 Hz), 5.62 (2H, s), 6.44 (1 H, s), 6.75 (1 H, s), 6.79 (1 H, d, J = 5.2 Hz), 7.6-7.22 (9H, m), 8.27 (2H, m), 8.57 (1 H, s)
PREPARATION 34
(Sj-ferf-Butyl ((2-methoxy-5-(4-((1 -(8-methyl-1 -oxo-2-phenyl-1 ,2- dihydroisoquinolin-3-yl)ethyl)amino)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H- pyrrolo[2,3-<^pyrimidin-5-yl)pyridin-3-yl)methyl)(1 -methylpiperidin-4- yl)carbamate (S)-3-(1 -((5-Bromo-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-c/]py
yl)amino)ethyl)-8-methyl-2-phenylisoquinolin-1 (2/-/)-one (1 10 mg, 0.18 mmol) was reacted with ie f-butyl ((2-methoxy-5-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2- yl)pyridin-3-yl)methyl)(1 -methylpiperidin-4-yl)carbamate (0.38 mmol), [1 ,1 - bis(diphenylphosphino)ferrocene] dichloropalladium(ll) complex with dichloromethane (15 mg, 0.01 mmol) and 2M caesium carbonate solution (0.27 ml_, 0.54 mmol) according to the method described in Preparation 30. The residue was purified by reverse phase using a SP1 Purification System to give 23 mg (15% yield) of the title compound.
LRMS (M+1 ): 860
1H NMR (400 MHz, Chloroform-d) δ ppm 0.97 (2H, t, J = 8 Hz), 2.5-1 .77 (m), 2.88 (3H, s), 3.16 (2H, m), 3.62 (2H, t, J = 8.4 Hz), 4.39 (2H, m), 4.95 (1 H, m), 5.06 (1 H, m), 5.63 (2H, s), 6.47 (1 H, s), 7.01 (1 H, s), 7.6-7.2 (8H, m), 8.23 (1 H, s), 8.28 (1 H, s), 8.40 (1 H, s) PREPARATION 35
(S)-2-(1 -((5-(2-(4-(3-(Dimethylamino)propyl)piperazin-1 -yl)pyridin-4-yl)-7-((2-
(trimethylsilyl)ethoxy)methyl)-7H^yrrolo[2,3-<^pyrimidin-4-yl)amino)ethyl)-5- methyl-3-phenylquinazolin-4(3H)-one (S)-2-(1 -((5-Bromo-7-((2-(trimethylsilyl)ethoxy)methyl)-7/-/-pyrrolo[2,3-c/]pyrimidin-4- yl)amino)ethyl)-5-methyl-3-phenylquinazolin-4(3/-/)-one (1 10 mg, 0.18 mmol) was reacted with /V,/V-dimethyl-3-(4-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)pyridin- 2-yl)piperazin-1 -yl)propan-1 -amine (0.36 mmol), [1 ,1 '-bis(diphenylphosphino)ferrocene] dichloropalladium(ll) complex with dichloromethane (15 mg, 0.01 mmol) and 2M caesium carbonate solution (0.27 ml_, 0.54 mmol) according to the method described in Preparation 33. The residue was purified by reverse phase using a SP1 Purification System to give 83 mg (60% yield) of the title compound.
LRMS (M+1 ): 774
1H NMR (400 MHz, Chloroform-d) δ ppm 0.05 (9H, s), 0.97 (2H, t, J = 8 Hz), 1 .41 (3H, d, J = 6.8 Hz), 2.01 (2H, m), 2.49 (2H, t, J = 6.4 Hz), 2.6-2.5 (4H, m), 2.72 (6H, s), 2.85 (3H, s), 2.97 (2H, m), 5.19 (1 H, q, J = 7.2 Hz), 5.64 (2H, s), 6.60 (1 H, d, J = 8 Hz), 6.95 (1 H, s), 6.98 (1 H, m), 7.70-7.24 (8H, m), 8.35 (1 H, s), 8.39 (1 H, m), 8.49 (1 H, s)
EXAMPLE 1
(S)-W-(1 -(3-(3,5-Difluorophenyl)-5-methoxy
(1 -methyl-1 H-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine
(Sj-/V-(1 -(3-(3,5-Difluorophenyl)-5-m^
methyl-1 H-pyrazol-4-yl)-5-((2-(trimethylsilyl)ethoxy)m
1 -amine (31 mg, 0.05mmol) was dissolved in trifluoroacetic acid (1 ml) and stirred at 0- 5°C for 1 h. The reaction mixture was then concentrated in vacuum and a solution of ammonia 7N in methanol (1 ml) was added to the residue obtained. The resulting mixture was stirred at 0-5°C for 1 h and at room temperature for 6h and then concentrated in vacuum. Water was added and the aqueous phase extracted with ethyl acetate. The organic extracts were collected together and washed with brine, dried, filtered and concentrated in vacuum. The crude obtained was purified by flash chromatography and then triturated with hexane to give 7 mg (28% yield) of the title compound.
LRMS (M+1 ): 502
1H NMR (400 MHz, DMSO-d6) δ ppm 1.30 - 1.60 (d, 3 H) 3.80 (s, 3 H) 3.90 (s, 3 H) 5.50 - 5.70 (m, 1 H) 6.10 - 6.30 (d, 1 H) 6.70 - 6.90 (d, 1 H) 7.10 - 7.20 (m, 1 H) 7.40- 7.70 (m, 4 H) 7.90 (s, 1 H) 8.00 (d, 1 H) 8.10 (s, 1 H) 1 1.75 (s, 1 H)
EXAMPLE 2
(S)-W-(3-(4-(1 -(3-(3,5-Difluorophenyl)-5-methoxy-3H-imidazo[4,5-fe]pyridin-2- yl)ethylamino)-7H^yrrolo[2,3-<^pyrimidin-5-yl)phenyl)methanesulfonamide
(S -/V-(3-(1 -((1 -(3-(3,5-Difluorophenyl)-5-methoxy-3H-imidazo[4,5-b]pyridin-2- yl)ethyl)amino)-5-((2-(trimethylsilyl)ethoxy)methyl)-5/-/-pyrrolo[2,3-d]pyrimidin-7- yl)phenyl)methanesulfonamide (39mg, 0.05mmol) was dissolved in trifluoroacetic acid (1 mL) and stirred at 0-5°C for 1 h. The reaction mixture was concentrated in vacuum and a solution of ammonia 7N in methanol (1 mL) was added to the residue obtained. The resulting mixture was stirred at 0-5°C for 6h and then concentrated in vacuum.
Water was added and the aqueous layer was extracted with ethyl acetate. The organic extracts were collected together and washed with brine, dried, filtered and concentrated in vacuum. The crude obtained was purified by flash chromatography to give 19 mg (59% yield) of the title compound.
LRMS (M+1 ): 591
1H NMR (400 MHz, DMSO-d6) δ ppm 1 .31 - 1.58 (d, 3 H) 2.84 - 3.05 (s, 3 H) 3.59 - 3.89 (s, 3 H) 5.48 - 5.68 (m, 1 H) 5.95 (d, 1 H) 6.72 (d, 1 H) 7.09 - 7.33 (m, 4 H) 7.35 - 7.61 (m, 4 H) 7.93 (d, 1 H) 8.01 - 8.26 (m, 1 H) 9.80 - 10.07 (s, 1 H) 1 1 .80 - 12.02 (s, 1 H)
EXAMPLE 3
(S)-/V-(3-(4-(1 -(8-Methyl-1 -oxo-2-phenyl-1 ,2-dihydroisoquinolin-3-yl)ethylamino)- 7H^yrrolo[2,3-rf]pyrimidin-5-yl)phenyl)methanesulfonamide (S)-/V-(3-(4-((1 -(8-Methyl-1 -oxo-2-phenyl-1 ,2-dihydroisoquinolin-3-yl)ethyl)amino)-7-((2- (trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-c/]pyrimidin-5- yl)phenyl)methanesulfonamide (37 mg, 0.05 mmol) was dissolved in trifluoroacetic acid (1 mL) and stirred at room temperature for 30 min. The solvent was evaporated under vacuum. The obtained crude was dissolved in a solution of ammonia 7N in methanol (4 mL) and stirred overnight. The solvent was evaporated under vacuum. Water was added and the aqueous layer was extracted with ethyl acetate. The combined organic extracts were washed with brine. The organic phase was dried over anhydrous magnesium sulphate and the solvent evaporated in vacuum to give 35 mg (99% yield) of the title compound as a white solid .
LRMS (M+1 ): 565
1H NMR (400 MHz, CD3OD) δ ppm 8.03 (s, 1 H), 7.54 - 7.1 1 (m, 12H), 6.72 (s, 1 H), 4.88 (m, 1 H), 2.88 (s, 3H), 2.76 (s, 3H), 1.40 (d, J = 6.8 Hz, 3H).
EXAMPLE 4
(S)-W-(3-(4-(1 -(5-Methyl-4-oxo-3^henyl-3,4-dihydroquinazolin-2-yl)ethylamino)- 7H^yrrolo[2,3- ]pyrimidin-5-yl)phenyl)methanesulfonamide
(S)-/V-(3-(4-((1 -(5-Methyl-4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)ethyl)amino)-7-((2- (trimethylsilyl)ethoxy)methyl)-7/-/-pyrrolo[2,3-c/]pyrimidin-5- yl)phenyl)methanesulfonamide (57 mg, 0.08 mmol) was dissolved in trifluoroacetic acid (1 .5 mL) and was stirred at room temperature for 30 min. The solvent was evaporated
under vacuum. The obtained crude was dissolved in a solution of ammonia 7N in methanol (5 ml) and stirred overnight. The solvent was evaporated under vacuum. Water was added and the aqueous layer was extracted with ethyl acetate. The combined organic extracts were washed with brine, dried over anhydrous magnesium sulphate and the solvent evaporated in vacuum to give 38 mg (82% yield) of the title compound.
LRMS (M+1 ): 566
1H NMR (400 MHz, CD3OD) δ ppm 8.10 (s, 1 H), 7.67 - 7.54 (m, 4H), 7.54 - 7.41 (m, 4H), 7.41 - 7.29 (m, 2H), 7.29 - 7.18 (m, 2H), 7.14 (s, 1 H), 5.02 (q, J = 6.5 Hz, 1 H), 2.95 (s, 3H), 2.74 (s, 3H), 1.37 (d, J = 6.6 Hz, 3H).
EXAMPLE 5
(S)-2-(1 -(5-(1 -(2-Hydroxyethyl)-1 H-pyrazol-4-yl)-7H-pyrrolo[2,3-cf]pyrimidin-4- ylamino)ethyl)-5-methyl-3-phenylquinazolin-4(3H)-one
(S)-2-(1 -((5-(1 -(2-((ferf-Butyldimethylsilyl)oxy)ethyl)-1 H-pyrazol-4-yl)-7-((2- (trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-c/]pyrimidin-4-yl)amino)ethyl)-5-methyl-3- phenylquinazolin-4(3/-/)-one (1 10 mg, 0.15 mmol) was treated with trifluoroacetic acid (2.3 mL, 29.3 mmol) and a solution of ammonia 7N in methanol (4.2 mL, 29.3 mmol) according to the method described in Example 3 to give 15 mg (20% yield) of the title compound as a solid.
LRMS (M+1 ): 507
1H NMR (400 MHz, Chloroform-d) δ ppm 1.21 (t, J = 7.0 Hz, 1 H), 1.39 (d, J = 6.7 Hz, 2H), 2.79 (s, 2H), 3.48 (q, J = 7.0 Hz, 1 H), 4.01 - 4.17 (m, 2H), 4.26 - 4.45 (m, 2H), 5.31 (dd, J = 8.3, 6.7 Hz, 1 H), 6.44 (d, J = 8.4 Hz, 1 H), 6.97 (d, J = 1 .7 Hz, 1 H), 7.22 (d, J = 7.4 Hz, 1 H), 7.30 - 7.35 (m, 1 H), 7.36 - 7.41 (m, 1 H), 7.46 (d, J = 8.1 Hz, 1 H), 7.53 - 7.64 (m, 3H), 7.76 (s, 1 H), 7.85 (s, 1 H), 8.21 (s, 1 H), 9.54 (s, 1 H).
EXAMPLE 6
(S)-3-(1 -(5-(1 -(2-Hydroxyethyl)-1 H-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-4- ylamino)ethyl)-8-methyl-2-phenylisoquinolin-1 (2H)-one.
(S -3-(1 -((5-(1 -(2-((ferf-Butyldimethylsilyl)oxy)ethyl)-1 H-pyrazol-4-yl)-7-((2- (trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-c/]pyrimidin-4-yl)amino)ethyl)-8-methyl-2- phenylisoquinolin-1 (2/-/)-one (50 mg, 0.07 mmol) was treated with trifluoroacetic acid (2 mL, 26 mmol) and a solution of ammonia 7N in methanol (4 mL, 28 mmol)
according to the method described in Example 3, to give 10 mg (29% yield) of the title compound as a solid.
LRMS (M+1 ): 506
1H NMR (400 MHz, Chloroform-d) δ ppm 1 .23 - 1 .28 (m, 1 H), 1 .37 (d, J = 6.8 Hz, 3H), 2.04 (s, 1 H), 2.85 (s, 3H), 3.79 - 4.01 (m, 2H), 4.12 (q, J = 7.1 Hz, 1 H), 4.22 (t, J = 4.8 Hz, 2H), 4.87 - 5.03 (m, 1 H), 5.25 (d, J = 6.9 Hz, 1 H), 6.47 (s, 1 H), 6.96 (d, J = 2.0 Hz, 1 H), 7.20 (d, J = 7.2 Hz, 1 H), 7.27 - 7.32 (m, 2H), 7.32 - 7.38 (m, 1 H), 7.39 - 7.54 (m, 3H), 7.57 (s, 1 H), 7.65 (s, 1 H), 8.16 (s, 1 H), 9.34 (s, 1 H). EXAMPLE 7
(S)-W-(3-Hydroxy-5-(4-(1 -(5-methyl-4-oxo-3-phenyl-3,4-dihydroquinazolin-2- yl)ethylamino)-7H^yrrolo[2,3-d]pyrimidin-5-yl)phenyl)methanesulfonamide.
(S)-/V-(3-Hydroxy-5-(4-((1 -(5-methyl-4-oxo-3-phenyl-3,4-dihydroquinazolin-2- yl)ethyl)amino)-7-((2-(trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-d]pyrimidin-5- yl)phenyl)methanesulfonamide (100 mg, 0.14 mmol) was treated with trifluoroacetic acid (5 mL, 64.9 mmol) and a solution of ammonia 7N in methanol (5 mL, 35 mmol) according to the method described in Example 3 to give 60 mg (72% yield) of the title compound as a solid.
LRMS (M+1 ): 506
1 H NMR (400 MHz, DMSO-d6) δ ppm 1 .29 (d, J = 6.5 Hz, 3H), 2.67 (s, 3H), 2.98 (s, 3H), 4.80 - 4.95 (m, 1 H), 6.66 (s, 1 H), 6.75 (s, 1 H), 6.81 (s, 1 H), 7.21 (d, J = 2.0 Hz, 1 H), 7.27 (t, J = 7.9 Hz, 2H), 7.49 (d, J = 7.0 Hz, 1 H), 7.57 (m, J = 15.0, 7.8 Hz, 5H), 8.1 1 (s, 1 H), 9.79 (d, J = 9.7 Hz, 2H), 1 1.88 (s, 1 H)
EXAMPLE 8
(S)-/V-[4-(4-{[1 -(8-Methyl-1 -oxo-2-phenyl-1 ,2-dihydroisoquinolin-3-yl)ethyl]amino}- 7H-pyrrolo[2,3-d]pyrimidin-5-yl)-1 H-indol-6-yl]sulfamide. (Sj-/V-(4-(4-((1 -(8-Methyl-1 -oxo-2-phenyl-1 ,2-dihydroisoquinolin-3-yl)ethyl)amino)-7-((2- (trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-c/]pyrimidin-5-yl)-1 /-/-indol-6-yl)sulfamide (70 mg, 0.10 mmol) was dissolved in trifluoroacetic acid (4 ml, 52 mmol) and the mixture was stirred at room temperature for 1 h. The solvent was concentrated in vacuum and a solution of ammonia 7N in methanol (8 mL, 56 mmol) was added. The reaction mixture was stirred at room temperature overnight and then concentrated in vacuum. The crude obtained was purified by reverse phase chromatography using a SP1 Purification System to give 32 mg (55% yield) of the title compound.
LRMS (M+1 ): 605
1 H-NMR (DMSO-d6) δ ppm 1 .10 (d, 3H), 2.67 (s, 3H), 4.50 (m, 1 H), 5.50 (d, 1 H), 6.27 (s, 1 H), 6.33 (d, 1 H), 7.00 (s, 2H), 7.03 (d, 1 H), 7.18 (d, 1 H), 7.25-7.55 (m, 9H), 8.17 (s, 1 H), 9.30 (s, 1 H), 1 1 .23 (s, 1 H), 1 1.89 (s, 1 H)
EXAMPLE 9
(S)-3-(1 -((5-(2-((4-(Dimethylamino)piperidin-1 -yl)methyl)pyridin-4-yl)-7H- pyrrolo[2,3-<^pyrimidin-4-yl)amino)ethyl)-8-methyl-2-phenylisoquinolin-1 (2H)-one
((S)-3-(1 -((5-(2-((4-(Dimethylamino)piperidin-1 -yl)methyl)pyridin-4-yl)-7-((2
(trimethylsilyl)ethoxy)methyl)-7H-pyrro^
phenylisoquinolin-1 (2/-/)-one (69 mg, 0.09 mmol) was dissolved in trifluoroacetic acid (1 ml, 13 mmol) and the mixture was stirred at room temperature for 1 h. The solvent was concentrated in vacuum and a solution of ammonia 7N in methanol (1 ml_, 7 mmol) was added. The reaction mixture was stirred at room temperature 2 h and then was evaporated in vacuum. The crude was re-dissolved in dichloromethane and the resulting organic phase was washed with water, a saturated sodium carbonate solution and brine. The combined organics were dried over anhydrous magnesium sulphate, filtered and evaporated under reduced pressure to give 40 mg (70% yield) of the title compound.
LRMS (M+1 ): 614
1H NMR (400 MHz, Chloroform-d) δ ppm 1 .36 (3H, d, J = 6.4 Hz), 1 .58 (2H, qd, J = 3.6 Hz, 1 1 .6 Hz), 1 .77 (m), 2.06 (2H, m), 2.85 (3H, s), 2.93 (2H, t, J = 10 Hz), 3.59 (2H, d, J = 3.6 Hz), 5.06 (1 H, q, J = 7.2 Hz), 5.1 1 (1 H, m), 6.42 (1 H, s), 7.56-7.18 (10H, m), 8.25 (1 H, s), 8.61 (1 H, d, J = 5.2 Hz)
EXAMPLE 10
(S)-5-Methyl-2-(1 -((5-(5-(4-(1 -methylpiperidin-4-yl)piperazin-1 -yl)pyridin-3-yl)-7H- pyrrolo[2,3-<^pyrimidin-4-yl)amino)ethyl)-3-phenylquinazolin-4(3H)-one
(S)-5-Methyl-2-(1 -((5-(5-(4-(1 -methylpiperidin-4-yl)piperazin-1 -yl)pyridin-3-yl)-7-((2- (trimethylsilyl)ethoxy)methyl)-7/-/-pyrrolo[2,3-c/]pyrimidin-4-yl)amino)ethyl)-3- phenylquinazolin-4(3/-/)-one (36 mg, 0.04 mmol) was dissolved in trifluoroacetic acid (2 ml, 26 mmol) and the mixture was stirred at room temperature for 1 h. The solvent was concentrated in vacuum and a solution of ammonia 7N in methanol (1 mL, 7 mmol) was added. The reaction mixture was stirred at room temperature 1 h and then was
evaporated in vacuum. The crude was re-dissolved in dichloromethane and the resulting organic phase was washed with water, a saturated sodium carbonate solution and brine. The combined organics were dried over anhydrous magnesium sulphate, filtered and evaporated under reduced pressure to give 22 mg (73% yield) of the title compound.
LRMS (M+1 ): 656
1H NMR (400 MHz, Chloroform-d) δ ppm 1 .34 (3H, d, J = 6.8 Hz), 1 .58 (2H, m), 1.75 (2H, m), 1.94 (2H, m), 2.26-2.21 (4H, m), 2.62 (4H, m), 2.79 (3H, s), 2.91 (2H, m), 3.33 (4H, m), 5.12 (1 H, q, J = 7.2 Hz), 6.32 (1 H, d, J = 7.6 Hz), 7.1 1 (1 H, s), 7.65-7.18 (10H, m), 8.30 (1 H, s), 8.37 (2H, m), 10.67 (1 H, s)
EXAMPLE 1 1
(S)-2-(1 -((5-(6-Methoxy-5-(((1 -methylpip^
pyrrolo[2,3-<^pyrimidin-4-yl)amino)ethyl)-5-methyl-3^henylquinazolin-4(3H)-one
(S)-tert-Butyl ((2-methoxy-5-(4-((1 -(5-methyl-4-oxo-3-phenyl-3,4-dihydroquinazolin-2- yl)ethyl)amino)-7-((2-(trimethylsilyl)ethoxy)methyl)-7/-/-pyrrolo[2,3-c/]pyrimidin-5- yl)pyridin-3-yl)methyl)(1 -methylpiperidin-4-yl)carbamate (36 mg, 0.04 mmol) was treated with trifluoroacetic acid ( 2 mL, 26 mmol) and a solution of ammonia 7N in methanol (1 mL, 7 mmol) according to the method described in Example 10 to give 44 mg (59% yield) of the title compound.
LRMS (M+1 ): 631
1H NMR (400 MHz, Chloroform-d) δ ppm 1 .34 (3H, d, J = 6.8 Hz), 1.45 (2H,m), 2.04- 1 .75 (4H, m), 2.25 (3H, s), 2.48 (1 H, m), 2.82 (5H, m), 3.82 (2H, dd, J = 30 Hz, 14 Hz), 4.08 (3H, s), 5.16 (1 H, q, J = 7.6 Hz), 6.32 (1 H, d, J = 8Hz), 7.00 (1 H, s), 7,64-7.19 (9H, m), 7.87 (1 H, d, J = 2.4 Hz), 8.26 (1 H,s), 8.32 (1 H, d, J = 2.4 Hz), 10.15 (1 H,s)
EXAMPLE 12
(S)-8-Methyl-3-(1 -((5-(5-(4-(1 -methylpiperidin-4-yl)piperazin-1 -yl)pyridin-3-yl)-7H- pyrrolo[2,3-<^pyrimidin-4-yl)amino)ethyl)-2-phenylisoquinolin-1 (2H)-one
(S)-8-Methyl-3-(1 -((5-(5-(4-(1 -methylpiperidin-4-yl)piperazin-1 -yl)pyridin-3-yl)-7-((2- (trimethylsilyl)ethoxy)methyl)-7/-/-pyrrolo[2,3-c/]pyrimidin-4-yl)amino)ethyl)-2- phenylisoquinolin-1 (2/-/)-one (88 mg, 0.1 1 mmol) was treated with trifluoroacetic acid ( 1 mL, 13 mmol) and a solution of ammonia 7N in methanol ( 1 mL, 7 mmol) according to the method described in Example 10 to give 43 mg (59% yield) of the title
compound.
LRMS (M+1 ): 655
1H NMR (400 MHz, Chloroform-d) δ ppm 1 .31 (3H, d, J = 6.8 Hz), 1 .65 (2H, m), 1.78 (2H, m), 1.98 (2H, m), 2.92-2.21 (4H, m), 2.63 (4H, m), 2.84 (3H, s), 3.24 (4H, m), 5.00 (1 H, q, J = 7.2 Hz), 5.10 (1 H, m), 6.40 (1 H, s), 7.12 (1 H, s), 7.53-7.16 (10H, m), 8.25 (2H, m), 8.31 (1 H, d, J = 2.8 Hz), 1 1.5 (1 H, s)
EXAMPLE 13
(S)-3-(1 -((5-(2-(4-(3-(Dimethylamino)propyl)piperazin-1 -yl)pyridin-4-yl)-7H- pyrrolo[2,3-<^pyrimidin-4-yl)amino)ethyl)-8-methyl-2-phenylisoquinolin-1 (2H)-one
(S)-3-(1 -((5-(2-(4-(3-(Dimethylamino)propyl)piperazin-1 -yl)pyridin-4-yl)-7-((2- (trimethylsilyl)ethoxy)methyl)-7H-pyrrolo^
phenylisoquinolin-1 (2/-/)-one (53 mg, 0.06 mmol) was treated with trifluoroacetic acid ( 1 ml_, 13 mmol) and a solution of ammonia 7N in methanol (1 ml_, 7 mmol) according to the method described in Example 10 to give 32 mg (73% yield) of the title compound.
LRMS (M+1 ): 643
1H NMR (400 MHz, Chloroform-d) δ ppm 1 .33 (3H, d, J = 6.8 Hz), 1 .71 (2H, m), 2.25 (6H, s), 2.38 (4H, m), 2.51 (4H, m), 2.84 (3H, s), 3.59 (4H, m), 5.02 (1 H, q, J = 6.8 Hz), 5.41 (1 H, d, J = 6.4 Hz), 6.41 (1 H, s), 6.75 (2H, m), 7.54-7.17 (9H, m), 8.24 (2H, m), 1 1.3 (1 H, s)
EXAMPLE 14
(S)-3-(1 -((5-(6-Methoxy-5-(((1 -methylpiperi^^
pyrrolo[2,3-<^pyrimidin-4-yl)amino)ethyl)-8-methyl-2-phenylisoquinolin-1 (2H)-one
(S)-ieri-Butyl ((2-methoxy-5-(4-((1 -(8-methyl-1 -oxo-2-phenyl-1 ,2-dihydroisoquinolin-3- yl)ethyl)amino)-7-((2-(trimethylsilyl)ethoxy)methyl)-7/-/-pyrrolo[2,3-c/]pyrimidin-5- yl)pyridin-3-yl)methyl)(1 -methylpiperidin-4-yl)carbamate (23 mg, 0.02 mmol) was treated with trifluoroacetic acid ( 1 mL, 13 mmol) and a solution of ammonia 7N in methanol ( 1 mL,7 mmol) according to the method described in Example 10 to give 9 mg (53% yield) of the title compound.
LRMS (M+1 ): 630
1H NMR (400 MHz, Chloroform-d) δ ppm 1 .26 (3H, d, J = 6.8 Hz), 1.46 (1 H, m), 2.04- 1 .89 (4H, m), 2.28 (3H, s), 2.5 (1 H, m), 2.85 (4H, m), 3.71 (2H, dd, J = 28 Hz, 14 Hz), 3.98 (3H, s), 4.95 (1 H, q, J = 7.2 Hz), 5.03 (1 H, m), 6.42 (1 H, s), 7.02 (1 H, s), 7.54-7.02 (8H, m), 7.75 (1 H, d, J = 2.4 Hz), 8.21 (2H, m)
EXAMPLE 15
(S)-2-(1 -((5-(2-(4-(3-(Dimethylamino)propyl)piperazin-1 -yl)pyridin-4-yl)-7H- pyrrolo[2,3-<^pyrimidin-4-yl)amino)ethyl)-5-methyl-3-phenylquinazolin-4(3H)-one
(S)-2-(1 -((5-(2-(4-(3-(Dimethylamino)propyl)piperazin-1 -yl)pyridin-4-yl)-7-((2- (trimethylsilyl)ethoxy)methyl)-7H-pyrrolo[2,3-(^pyrimidin-4-yl)amin
phenylquinazolin-4(3/-/)-one (83 mg, 0.1 mmol) was treated with trifluoroacetic acid ( 1 ml_, 13 mmol) and a solution of ammonia 7N in methanol (1 ml_,7 mmol) according to the method described in Example 10 to give 44 mg (64% yield) of the title compound. LRMS (M+1 ): 644
1H NMR (400 MHz, Chloroform-d) δ ppm 1 .37 (3H, d, J = 6.8 Hz), 1.68 (2H, m), 2.22 (6H, s), 2.33 (4H, m), 2.49 (4H, m), 2.79 (3H, s), 3.68 (4H, m), 5.14 (1 H, q, J = 6.8 Hz), 6.58 (1 H, d, J = 7.2Hz), 6.91 (2H, m), 7.64-7.18 (9H, m), 8.32 (2H, m)
Pharmacological activity
PI3K α, β, δ and γ Enzymatic Inhibition Assays Compounds were screened for their ability to inhibit PI3Ka (PI3Ka), ΡΙ3Κβ (PI3Kb), PI3K5 (PI3Kd) and ΡΙ3Κγ (PI3Kg) using a cell-free based PI3K HTRF™ assay (Millipore, ref. #33-017).
PI-3 Kinase HTRF kit (ref. #33-037) and the different PI3K recombinant isoforms (ref. #14-602, ref. #14-603, ref .#14-604, ref.#15-558 for Alpha, Beta, Delta and Gamma respectively) were purchased at Millipore (expressed in insect cells). ATP was purchased at Sigma Aldrich (ref. #A7699).
The compounds were pre-incubated with the enzyme for 30 min before starting of the catalytic reaction. [PIP2] was used at its Km. [ATP] was used at 15 μΜ for all isoforms for technical reasons (Km values varied between 10 and 20 μΜ depending on the isoform). Time of assay and [Enzyme] were optimized to work in the linear range. Stop and Detection mixtures were used as specified in the Millipore PI-3 Kinase kit.
• Final Assay conditions

DELTA 8 0.35 15 2 30 1 - 18
GAMMA 8 2.5 15 10 30 1 - 18
Reaction time and enzyme concentration in the assay will depend of each batch.
All experiments were analysed using Activity Base software from IDBS and the four- parameter log equation.
The results are shown in Table 1.

It can be seen from Table 1 that the compounds of formula (I) are potent inhibitors of Phosphoinositide 3-kinase delta (PI3kd). Preferred compounds of the invention possess an IC50 value for the inhibition of PI3Kd (determined as defined above) of less than 10 μΜ (10,000 nM), preferably less than 1 μΜ (1 ,000 nM), even more preferably of less than 0.2 μΜ (200 nM), most preferably less than 0.05 μΜ (50 nM) Formulation Examples
Formulation Example 1 (Oral suspension)
Ingredient Amount
Active Compound 3 mg
Citric acid 0.5 g
Sodium chloride 2.0 g
Methyl paraben 0.1 g
Granulated sugar 25 g
Sorbitol (70% solution) 1 1 g
Veegum K 1 .0 g
Flavoring 0.02 g
Dye 0.5 mg
Distilled water q.s. to 100 mL
Formulation Example 2 (Hard gelatine capsule for oral administration)

Formulation Example 3 (Gelatin cartridge for inhalation)

Formulation Example 4 (Formulation for inhalation with a DPI)
Ingredient Amount
Active Compound (micronized) 15 mg
Lactose 3000 mg
Formulation Example 5 (Formulation for a MDI)
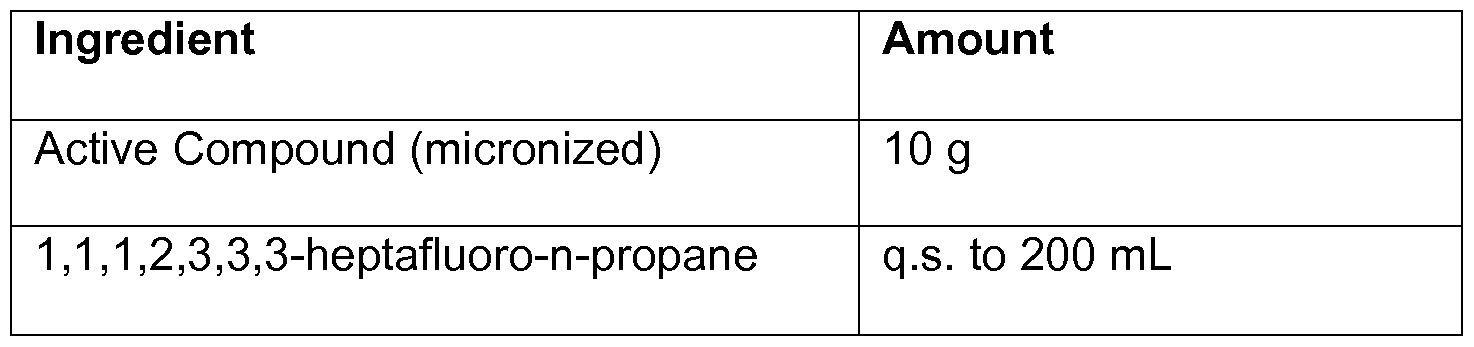
In all the formulation examples, active compound is the compound of example 4. Modifications, which do not affect, alter, change or modify the essential aspects of the compounds, combinations or pharmaceutical compositions described, are included within the scope of the present invention.
Claims
1 . A compound of formula (I), or a pharmaceutically acceptable salt, or solvate, or N- oxide, or stereoisomer or isotopically-labeled derivate thereof:

Formula (I) wherein,
A represents a group selected from i ),

Formula A-1 Formula A-2 Formula A-3 wherein,
Ri represents a phenyl group or a 5- to 6- membered heteroaryl group containing at least one heteroatom selected from O, S and N,
wherein the phenyl and heteroaryl groups are unsubstituted or substituted by one or more substituents selected from a halogen atom, a
hydroxyl group, a cyano group, a linear or branched C1-C4 alkyl group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group, a C3-C10 cycloalkyl group, a -(CH2)i-3CN group, a -(CH2)o-30R7 group, a -(CH2)o-3NR7R8 group, a -C(0)-(CH2)i-3-CN group, a -C(0)-(CH2)o-3-R7 group, a -C(O)- (CH2)o-3-N R7R8 group, a -S(CH2)0-3R7 group, a -S(O)(CH2)0-3R7 group, a -
S(0)(CH2)o-3NR7R8 group, a -S(O)2(CH2)0-3R7 group, a -S(O)2(CH2)0- 3NR7R8 group or a -(CH2)0-3(phenyl)-OR7 group; wherein R7 and R8 each independently represent a hydrogen atom, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group or a linear or branched C1-C4 alkyl group, which alkyl group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkoxy group, a cyano group or a C3- C10 cycloalkyl group;
and (*) represents where A is bonded to the CH(CH3)-NH-pyrrolopyrimidine group;
R2, R3, R4, R5, and R6 each independently represent a hydrogen atom, a C1-C4 alkoxy group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group, a C3-Ci0 cycloalkyl group, a -(CH2)0-3CN group, a -C(0)-(CH2)1-3-CN group, a -C(O)- (CH2)o-3-R' group, a -C(0)-(CH2)o-3-NR9Rio, a -(CH2)0-3NR9Rio group, or a linear or branched C1-C4 alkyl group, which alkyl group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkoxy group, a cyano group or a C3-Cio cycloalkyl group; wherein R9 and R10 each independently represent a hydrogen atom, a hydroxyl group, a C1-C4 alkoxy group or a linear or branched C1-C4 alkyl group;
L represents a direct bound or a linker selected from -0-, -S-, a -NR'- group, a C(O)- NR'- group, a C(0)-0-R"- group or a -(CH2)1-4 group; wherein R' represents hydrogen or a linear or branched C1-C4 alkyl group, and R" represents a linear or branched C C4 alkyl group;
B represents a phenyl group or a 5- to 14- membered heteroaryl group containing at least one heteroatom selected from O, S and N,
wherein the phenyl and heteroaryl groups are unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyl group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group, a -(CH2)0-3-O(Ci-C4 alkyl) group, a -(CH2)0-3-O(Ci-C4 haloalkyl) group, a -(CH2)0-
3-0-(CH2)i-3-0(Ci-C4 alkyl) group, a -(CH2)o-3-0-(CH2)i-3-0(Ci-C4 haloalkyl) group, a - (CH2)o-3-0-(CH2) o-3Ri i group, a -(CH2)o-3-0-(CH2)i-3NR11R12 group, a C3-C10 cycloalkyl group, a -(CH2) 0-3N R11 R12 group, a -(CH2)o-3-C(0)-(CH2) 0-3-NRn egroup, a -(CH2)0-3- C(0)0-(CH2) o-3Ri i group, a -(CH2) o-3NRn-S(0)2Ri2 group, a -(CH2)0-3- S(O)2(CH2)0-3- Rn group, a -(CH2)0-3-SH group, a -(CH2)o-3-S-(CH2)o-3-Ri i group or a -(CH2)0-3-(5- to 7- membered heterocyclyl group containing at least one heteroatom selected from O, S and N); which heterocyclyl group is unsubstituted or substituted by one or more substituents selected from a halogen atom, a linear or branched C C4 alkyl group, a - (CH2)o-5NR15R16 group or a 5- to 7- membered heterocyclyl group containing one or two nitrogen atoms, which heterocyclyl group is unsubstituted or substituted by one or more substituents selected from a linear or branched C C4 alkyl groups or a -(CH2)0- 3N(CH3)2 group; wherein Rn and R12 each independently represent a hydrogen atom, a hydroxyl group, a linear or branched C C4 alkyl group, a -(CH2)0-3NR13R14 group, a phenyl group or a 5- to 7- membered heterocyclyl group containing one or two nitrogen atoms, which phenyl or heterocyclyl groups are unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a Ci-C4 hydroxyalkyl group, a C C4 haloalkyl group, a C C4 alkoxy group, a C3-C10 cycloalkyl group, a linear or branched C C4 alkyl group or a -(CH2)o-5NR15R16 group; wherein R 3 and Ri4 each independently represent a hydrogen atom, a C C4 haloalkyl group, a C C4 hydroxyalkyl group or a linear or branched C C4 alkyl group; and wherein R 5 and Ri6 each independently represent a hydrogen atom or a or a linear or branched C C4 alkyl group.
2. A compound according to claim 1 , wherein:
A represents a group selected from i) a group of formula (A-1 ), ii) a group of formula (A-2), iii) a group of formula (A-3),

Formula A-1 Formula A-2 Formula A-3
wherein,
Ri represents a phenyl group or a 5- to 6- membered heteroaryl group containing at least one heteroatom selected from O, S and N,
wherein the phenyl and heteroaryl groups are unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched Ci-C4 alkyi group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group, a C3-C10 cycloalkyl group, a -(CH2)i-3CN group, a -(CH2)o-30R7 group, a -(CH2)o-3NR7R8 group, a -C(0)-(CH2)i-3-CN group, a -C(0)-(CH2)o-3-R7 group, a -C(O)- (CH2)o-3-NR7R8 group, a -S(CH2)0-3R7 group, a -S(O)(CH2)0-3R7 group, a - S(0)(CH2)o-3NR7R8 group, a -S(O)2(CH2)0-3R7 group, a -S(O)2(CH2)0- 3NR7R8 group or a -(CH2)0-3(phenyl)-OR7 group; wherein R7 and R8 each independently represent a hydrogen atom, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group or a linear or branched C1-C4 alkyi group, which alkyi group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkoxy group, a cyano group or a C3- C10 cycloalkyl group;
and (*) represents where A is bonded to the CH(CH3)-N H-pyrrolopyrimidine group;
R2, R3, R4, R5, and R6 each independently represent a hydrogen atom, a C1-C4 alkoxy group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group, a C3-Ci0 cycloalkyl group, a -(CH2)0-3CN group, a -C(0)-(CH2)1-3-CN group, a -C(O)- (CH2)o-3-R' group, a -C(0)-(CH2)0-3-NR9Rio, a -(CH2)0-3NR9Rio group, or a linear or branched C1-C4 alkyi group, which alkyi group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkoxy group, a cyano group or a C3-Cio cycloalkyl group; wherein R9 and R10 each independently represent a hydrogen atom, a hydroxyl group, a C1-C4 alkoxy group or a linear or branched C1-C4 alkyi group;
L represents a direct bound or a linker selected from -0-, -S-, a -NR'- group, a C(O)- NR'- group, a C(0)-0-R"- group or a -(CH2)1-4 group; wherein R' represents hydrogen or a linear or branched C1-C4 alkyi group, and R" represents a linear or branched C C4 alkyi group;
B represents a phenyl group or a 5- to 14- membered heteroaryl group containing at least one heteroatom selected from O, S and N,
wherein the phenyl and heteroaryl groups are unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched CrC4 alkyl group, a C C4 haloalkyl group, a C C4 hydroxyalkyl group, a -(CH2)o-3-0(Ci-C4 alkyl) group, a -(CH2)o-3-0(Ci-C4 haloalkyl) group, a -(CH2)o- 3-0-(CH2)i-3-0(Ci-C4 alkyl) group, a -(CH2)0-3-O-(CH2)1-3-O(Ci-C4 haloalkyl) group, a - (CH2)o-3-0-(CH2) o-3Ri i group, a -(CH2)0-3-O-(CH2)1-3NR11R12 group, a C3-Ci0 cycloalkyl group, a -(CH2) o-3N n i2 group, a -(CH2)0-3-C(O)-(CH2) 0-3-NRn egroup, a -(CH2)0-3- C(0)0-(CH2) o-3Ri i group, a -(CH2) o-3NRn-S(0)2Ri2 group, a -(CH2)0-3- S(O)2(CH2)0-3- Rii group, a -(CH2)0-3-SH group, or a -(CH2)o-3-S-(CH2)0-3-Rn group; wherein Rn and Ri2 each independently represent a hydrogen atom, a hydroxyl group, a linear or branched C C4 alkyl group, a -(CH2)0-3NR13R14 group or a phenyl group, which phenyl group is unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a C C4 hydroxyalkyl group, a C C4 haloalkyl group, a C C4 alkoxy group or a C3-Ci0 cycloalkyl group; wherein R13 and Ri4 each independently represent a hydrogen atom, a C C4 haloalkyl group, a C C4 hydroxyalkyl group or a linear or branched C C4 alkyl group.
3. A compound according to claim 1 or claim 2, wherein A represents a group selected from -1 a), group of formula (A-2a),

Formula A-1 a Formula A-2a iii) a group of formula (A-3a),

Formula A-3a
where Ri, R2, R3, R4, Rs, and R6 are as defined in claim 1 and each Ra, Rb, Rc, Rd and Re independently represents a hydrogen atom or a substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyi group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group, a C3-C10 cycloalkyl group, a - (CH2)i-3CN group, a -(CH2)o-30R7 group, a -(CH2)o-3NR7R8 group, a -C(0)-(CH2)i-3-CN group, a -C(O)-(CH2)0-3-R7 group, a -C(O)-(CH2)0-3-NR7R8 group, a -S(CH2)0-3R7 group, a -S(0)(CH2)o-3R7 group, a -S(O)(CH2)0-3NR7R8 group, a -S(O)2(CH2)0-3R7 group, a - S(0)2(CH2)o-3NR7R8 group or a -(CH2)0-3(phenyl)-OR7 group; wherein R7 and R8 each independently represent a hydrogen atom, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group or a linear or branched C1-C4 alkyi group, which alkyi group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkoxy group, a cyano group or a C3-Ci0 cycloalkyl group;
and (*) represents where A is bonded to the CH(CH3)-NH-pyrrolopyrimidine group.
4. A compound according to any of claims 1 to 3 wherein R2, R3, R4, R5, and R6 each independently represent a hydrogen atom, a C1-C4 alkoxy group, or a linear or branched C1-C4 alkyi group, which alkyi group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkoxy group, a cyano group or a C3-Ci0 cycloalkyl group.
5. A compound according to any of claims 1 , 3 and 4 wherein B represents a phenyl group, a pyridyl group, a pyrazole group or an indole group,
wherein the phenyl, pyridyl, pyrazole or indole groups are unsubstituted or substituted by one or more substituents selected from a halogen atom, a
hydroxyl group, a cyano group, a linear or branched C1-C4 alkyl group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group, a -(CH2)o-3-0(Ci-C4 alkyl) group, a - (CH2)o-3-0(Ci-C4 haloalkyl) group, a -(CH2)o-3-0-(CH2)i-3-0(Ci-C4 alkyl) group, a -(CH2)o-3-0-(CH2)i-3-0(Ci-C4 haloalkyl) group, a -(CH2)0-3-0-(CH2)o-3 i i group, a -(CH2)o-3-0-(CH2)1-3NR11R12 group, a C3-C10 cycloalkyl group, a -(CH2) 0- 3NRnR12 group, a -(CH2)0-3-C(O)-(CH2) 0-3-NRn Ri2group, a -(CH2)0-3-C(O)O- (CH2) o-3Rii group, a -(CH2) 0-3NRn-S(O)2Ri2 group, a -(CH2)0-3- S(O)2(CH2)0-3- R11 group, a -(CH2)0-3-SH group, a -(CH2)o-3-S-(CH2)o-3-Rii group or a -(CH2)0-3- (5- to 7- membered heterocyclyl group containing at least one heteroatom selected from O, S and N); which heterocyclyl group is unsubstituted or substituted by one or more substituents selected from a halogen atom, a linear or branched C1-C4 alkyl group, a -(CH2)o-5NR15R16 group or a 5- to 7- membered heterocyclyl group containing one or two nitrogen atoms, which heterocyclyl group is unsubstituted or substituted by one or more substituents selected from a linear or branched C1-C4 alkyl groups or a -(CH2)0-3N(CH3)2 group; wherein Rn and R12 each independently represent a hydrogen atom, a hydroxyl group, a linear or branched C1-C4 alkyl group, a -(CH2)o-3N R13R14 group, a phenyl group or a 5- to 7- membered heterocyclyl group containing one or two nitrogen atoms, which phenyl or heterocyclyl groups are unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a C1-C4 hydroxyalkyl group, a C1-C4 haloalkyl group, a C1-C4 alkoxy group, a C3-C10 cycloalkyl group, a linear or branched C1-C4 alkyl group or a -(CH2)o-5NR15R16 group; wherein R13 and Ri4 each independently represent a hydrogen atom, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group or a linear or branched C1-C4 alkyl group; and wherein R 5 and Ri6 each independently represent a hydrogen atom or a linear or branched C1-C4 alkyl group.
6. A compound according to claim 5 where the phenyl, pyridyl, pyrazole or indole groups are unsubstituted or substituted by one or more substituents selected from a hydroxyl group, a linear or branched C1-C4 alkyl group, a C1-C4 hydroxyalkyl group, a - (CH2)o-3-0(Ci-C4 alkyl) group, a
 group or a -(CH2)o-3-(5- to 7- membered heterocyclyl group containing at least one heteroatom selected from O, S and N); which heterocyclyl group is unsubstituted or substituted by one or more substituents selected from a halogen atom, a linear or branched C1-C4 alkyl group, a -(CH2)o-5NR15R16 group or a 5- to 7- membered heterocyclyl group containing one or two nitrogen atoms, which heterocyclyl group is unsubstituted or
substituted by one or more linear or branched C1-C4 alkyl groups; wherein Rn and R12 each independently represent a hydrogen atom a linear or branched C1-C4 alkyl group, a -(CH2)o-3NRi3 i4 group or a 5- to 7- membered heterocyclyl group containing one or two nitrogen atoms, which heterocyclyl group is unsubstituted or substituted by one or more substituents selected from a C1-C4 haloalkyl group, a C1-C4 alkoxy group, a linear or branched C1-C4 alkyl group or a -(CH2)o-5NRi5 i6 group; wherein R13 and R14 each independently represent a hydrogen atom; and wherein R 5 and Ri6 each independently represent a hydrogen atom or a linear or branched C1-C4 alkyl group.
group or a -(CH2)o-3-(5- to 7- membered heterocyclyl group containing at least one heteroatom selected from O, S and N); which heterocyclyl group is unsubstituted or substituted by one or more substituents selected from a halogen atom, a linear or branched C1-C4 alkyl group, a -(CH2)o-5NR15R16 group or a 5- to 7- membered heterocyclyl group containing one or two nitrogen atoms, which heterocyclyl group is unsubstituted or
substituted by one or more linear or branched C1-C4 alkyl groups; wherein Rn and R12 each independently represent a hydrogen atom a linear or branched C1-C4 alkyl group, a -(CH2)o-3NRi3 i4 group or a 5- to 7- membered heterocyclyl group containing one or two nitrogen atoms, which heterocyclyl group is unsubstituted or substituted by one or more substituents selected from a C1-C4 haloalkyl group, a C1-C4 alkoxy group, a linear or branched C1-C4 alkyl group or a -(CH2)o-5NRi5 i6 group; wherein R13 and R14 each independently represent a hydrogen atom; and wherein R 5 and Ri6 each independently represent a hydrogen atom or a linear or branched C1-C4 alkyl group.
7. A compound according to any of claims 2 to 4 wherein B represents a phenyl group, a pyrazole group or an indole group,
wherein the phenyl, pyrazole or indole groups are unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyl group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group, a -(CH2)o-3-0(Ci-C4 alkyl) group, a -(CH2)o-3-0(CrC4 haloalkyl) group, a -(CH2)o-3-0-(CH2)1-3-0(Ci-C4 alkyl) group, a -(CH2)0-3-O- (CH2)i-3-0(Ci-C4 haloalkyl) group, a -(CH2)0-3-O-(CH2) 0-3Rn group, a -(CH2)0-3- 0-(CH2)1 -3N Rn R12 group, a C3-C10 cycloalkyl group, a -(CH2) 0-3NRn Ri2 group, a -(CH2)o-3-C(0)-(CH2) 0-3-N Ri i Ri2group, a -(CH2)0-3-C(0)0-(CH2) o-3Ri i group, a - (CH2) o-3N Ri i-S(0)2Ri2 group, a -(CH2)0-3- S(O)2(CH2)0-3-Ri i group, a -(CH2)0-3-
SH group, or a -(CH2)o-3-S-(CH2)0-3-Rn group; wherein Rn and R12 each independently represent a hydrogen atom, a hydroxyl group, a linear or branched C1-C4 alkyl group, a -(CH2)o-3NR13R14 group or a phenyl group, which phenyl group is unsubstituted or substituted by one or more substituents selected from a halogen atom, a hydroxyl group, a cyano group, a C1-C4 hydroxyalkyl group, a C1-C4 haloalkyl group, a C1-C4 alkoxy group or a C3-C10 cycloalkyl group; wherein R 3 and Ri4 each independently represent a hydrogen atom, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group or a linear or branched C1-C4 alkyl group.
8. A compound according to claim 7 where the phenyl, pyrazole or indole groups are unsubstituted or substituted by one or more substituents selected from a hydroxyl group, a linear or branched C1-C4 alkyl group, a C1-C4 hydroxyalkyl group, a -(CH2) 0- 3N Ri S(0)2Ri2 group wherein Rn and R12 each independently represent a hydrogen atom a linear or branched C1-C4 alkyl group, a -(CH2)0-3N R13R14 group, Ri3 and R14 being a hydrogen atom.
9. A compound according to any of claims 1 and 3 to 6, wherein A represents a group selected from -1 a), ii) a group of formula (A-2a),

Formula A-1 a Formula A -2 a iii) a group of formula (A-3a),

Formula A-3a
R2, R3, R4, Rs, and R6 each independently represent a hydrogen atom, a Ci-C4 alkoxy group, or a linear or branched CrC4 alkyi group, which alkyi group is unsubstituted or substituted by one or more substituents selected from a CrC4 alkoxy group, a cyano group or a C3-Ci0 cycloalkyl group; each Ra, Rb, Rc, Rd and Re independently represents a hydrogen atom or a substituents selected from a halogen atom, a hydroxyl group, a cyano group, a
linear or branched C1-C4 alkyl group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group, a C3-C10 cycloalkyl group, a -(CH2)i-3CN group, a -(CH2)o- 3OR7 group, a -(CH2)o-3NR7R8 group, a -C(0)-(CH2)i-3-CN group, a -C(O)-(CH2)0- 3-R7 group, a -C(0)-(CH2)o-3-N R7R8 group, a -S(CH2)0-3R7 group, a -S(O)(CH2)0- 3R7 group, a -S(0)(CH2)o-3NR7R8 group, a -S(O)2(CH2)0-3R7 group, a -
S(0)2(CH2)o-3NR7R8 group or a -(CH2)0-3(phenyl)-OR7 group; wherein R7 and R8 each independently represent a hydrogen atom, a C1-C4 haloalkyl group, a d- C4 hydroxyalkyl group or a linear or branched C1-C4 alkyl group, which alkyl group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkoxy group, a cyano group or a C3-C10 cycloalkyl group;
and (*) represents where A is bonded to the CH(CH3)-NH-pyrrolopyrimidine group;
B represents a phenyl group, a pyridyl group, a pyrazole group or an indole group, wherein the phenyl, pyridyl, pyrazole or indole groups are unsubstituted or substituted by one or more substituents selected from a hydroxyl group, a linear or branched C1-C4 alkyl group, a C1-C4 hydroxyalkyl group, a -(CH2)0-3-O(Ci-C4 alkyl) group, a -(CH2)0- 3N Rn R12 group, a -(CH2)o-3NRi S(0)2Ri2 group or a -(CH2)0-3-(5- to 7- membered heterocyclyl group containing at least one heteroatom selected from O, S and N); which heterocyclyl group is unsubstituted or substituted by one or more substituents selected from a halogen atom, a linear or branched C1-C4 alkyl group, a -(CH2)o-5NR15R16 group or a 5- to 7- membered heterocyclyl group containing one or two nitrogen atoms, which heterocyclyl group is unsubstituted or substituted by one or more substituents selected from a linear or branched C1-C4 alkyl groups or a -(CH2)0-3N(CH3)2 group; wherein Rn and R12 each independently represent a hydrogen atom a linear or branched C1-C4 alkyl group, a -(CH2)o-3NR13R14 group or a 5- to 7- membered heterocyclyl group containing one or two nitrogen atoms, which heterocyclyl group is unsubstituted or substituted by one or more substituents selected from a C1-C4 haloalkyl group, a C1-C4 alkoxy group, a linear or branched C1-C4 alkyl group or a -(CH2)o-5NR15R16 group; wherein R13 and R14 each independently represent a hydrogen atom; and wherein R 5 and Ri6 each independently represent a hydrogen atom or a linear or branched C1-C4 alkyl group.
10. A compound according to any of the previous claims wherein A represents a group selected from
i) a group of formula (A-1 a), ii) a group of formula (A-2a),

Formula A-1 a Formula A -2 a iii) a group of formula (A-3a),

Formula A-3a
R2, R3, R4, R5, and R6 each independently represent a hydrogen atom, a C1-C4 alkoxy group, or a linear or branched C1-C4 alkyi group, which alkyi group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkoxy group, a cyano group or a C3-C10 cycloalkyl group; each Ra, Rb, Rc, Rd and Re independently represents a hydrogen atom or a substituents selected from a halogen atom, a hydroxyl group, a cyano group, a linear or branched C1-C4 alkyi group, a C1-C4 haloalkyl group, a C1-C4 hydroxyalkyl group, a C3-C10 cycloalkyl group, a -(CH2)i-3CN group, a -(CH2)o- 3OR7 group, a -(CH2)o-3NR7R8 group, a -C(0)-(CH2)i-3-CN group, a -C(O)-(CH2)0-
3-R7 group, a -C(0)-(CH2)o-3-NR7R8 group, a -S(CH2)0-3R7 group, a -S(O)(CH2)0-
3R7 group, a -S(0)(CH2)o-3NR7R8 group, a -S(0)2(CH2)o-3R7 group, a - S(0)2(CH2)o-3NR7R8 group or a -(CH2)o-3(phenyl)-OR7 group; wherein R7 and R8 each independently represent a hydrogen atom, a C1-C4 haloalkyl group, a d- C4 hydroxyalkyl group or a linear or branched C1-C4 alkyl group, which alkyl group is unsubstituted or substituted by one or more substituents selected from a C1-C4 alkoxy group, a cyano group or a C3-C10 cycloalkyl group; and (*) represents where A is bonded to the CH(CH3)-N H-pyrrolopyrimidine group; B represents a phenyl group, a pyrazole group or an indole group, where the phenyl, pyrazole or indole groups are unsubstituted or substituted by one or more substituents selected from a hydroxyl group, a linear or branched C1-C4 alkyl group, a C1-C4 hydroxyalkyl group, a -(CH2) 0-3NRn-S(O)2Ri2 group wherein Rn and R12 each independently represent a hydrogen atom a linear or branched C1-C4 alkyl group, a - (CH2)o-3NR13R14 group, R13 and Ri4 being a hydrogen atom.
1 1 . A compound according to any of claims 1 , 3 to 6 and 9 having one of the following general formulas:



114

Formu la (Id ') Formu la (Ic2')
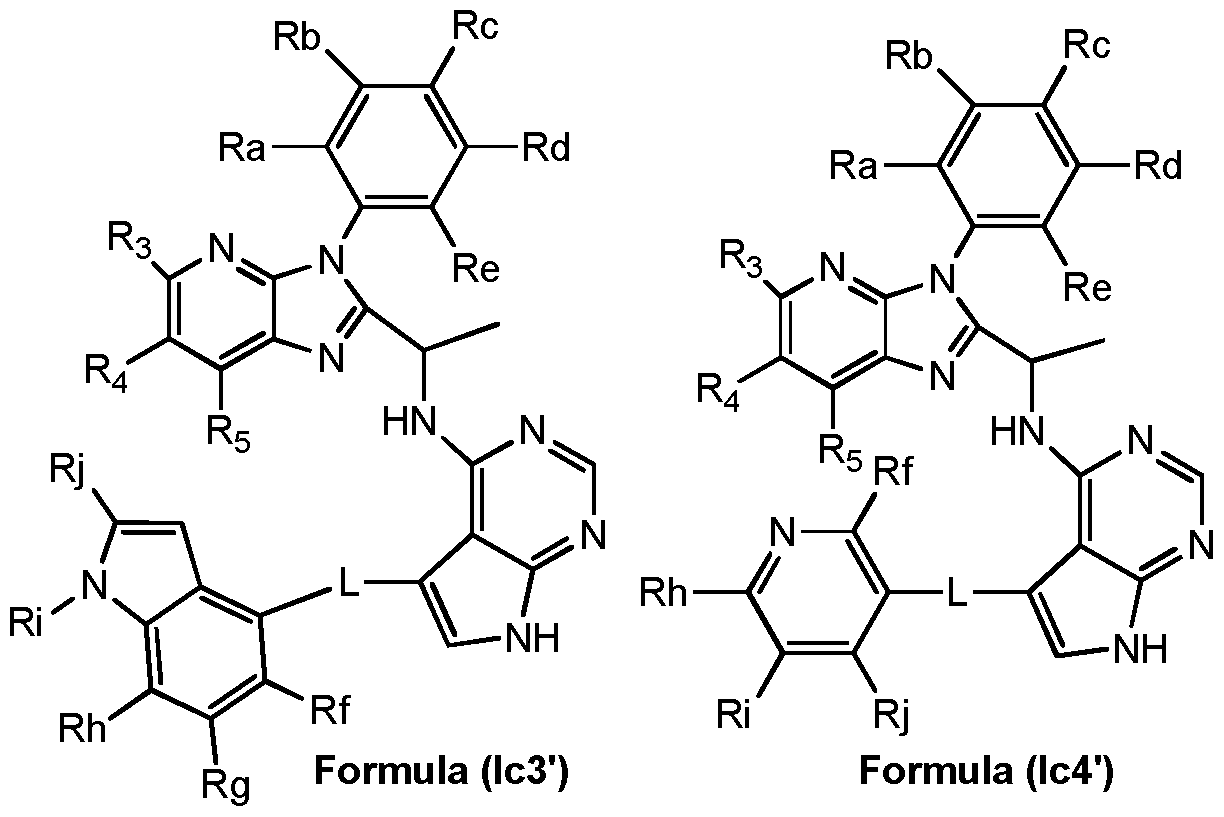
12. A compound according to any of the previous claims having one of the following general formulas:

Formula (\aV) Formula (Ia2') Rg Formula (Ia3')

13. A compound according to any of the previous which is selected from:
• (S)-N-(1 -(3-(3,5-difluorophenyl)-5-methoxy-3H-imidazo[4,5-b]pyridin-2-yl)ethyl)- 5-(1 -methyl-1 H-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine;
• (S)-N-(3-(4-(1 -(3-(3,5-difluorophenyl)-5-methoxy-3H-imidazo[4,5-b]pyridin-2- yl)ethylamino)-7H-pyrrolo[2,3-d]pyrimidin-5-yl)phenyl)methanesulfonamide;
• (S)-N-(3-(4-(1 -(8-methyl-1 -oxo-2-phenyl-1 ,2-dihydroisoquinolin-3- yl)ethylamino)-7H-pyrrolo[2,3-d]pyrimidin-5-yl)phenyl)methanesulfonamide;
• (S)-N-(3-(4-(1 -(5-methyl-4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)ethylamino)- 7H-pyrrolo[2,3-d]pyrimidin-5-yl)phenyl)methanesulfonamide;
• (S)-2-(1 -(5-(1 -(2-hydroxyethyl)-1 H-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-4- ylamino)ethyl)-5-methyl-3-phenylquinazolin-4(3H)-one;
• (S)-3-(1 -(5-(1 -(2-hydroxyethyl)-1 H-pyrazol-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-4- ylamino)ethyl)-8-methyl-2-phenylisoquinolin-1 (2H)-one;
• (S)-N-(3-hydroxy-5-(4-(1 -(5-methyl-4-oxo-3-phenyl-3,4-dihydroquinazolin-2- yl)ethylamino)-7H-pyrrolo[2,3-d]pyrimidin-5-yl)phenyl)methanesulfonamide;
• (S)-N-[4-(4-{[1 -(8-methyl-1 -oxo-2-phenyl-1 ,2-dihydroisoquinolin-3- yl)ethyl]amino}-7H-pyrrolo[2,3-d]pyrimidin-5-yl)-1 H-indol-6-yl]sulfamide;
• (S)-3-(1 -((5-(2-((4-(Dimethylamino)piperidin-1 -yl)methyl)pyridin-4-yl)-7H- pyrrolo[2,3-d]pyrimidin-4-yl)amino)ethyl)-8-methyl-2-phenylisoquinolin-1 (2H)- one;
• (S)-5-Methyl-2-(1 -((5-(5-(4-(1 -methylpiperidin-4-yl)piperazin-1 -yl)pyridin-3-yl)- 7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)ethyl)-3-phenylquinazolin-4(3H)-one;
• (S)-2-(1 -((5-(6-Methoxy-5-(((1 -methylpiperidin-4-yl)amino)methyl)pyridin-3-yl)- 7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)ethyl)-5-methyl-3-phenylquinazolin-4(3H)- one;
• (S)-8-Methyl-3-(1 -((5-(5-(4-(1 -methylpiperidin-4-yl)piperazin-1 -yl)pyridin-3-yl)- 7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)ethyl)-2-phenylisoquinolin-1 (2H)-one;
• (S)-3-(1 -((5-(2-(4-(3-(dimethylamino)propyl)piperazin-1 -yl)pyridin-4-yl)-7H- pyrrolo[2,3-d]pyrimidin-4-yl)amino)ethyl)-8-methyl-2-phenylisoquinolin-1 (2H)- one;
• (S)-3-(1 -((5-(6-Methoxy-5-(((1 -methylpiperidin-4-yl)amino)methyl)pyridin-3-yl)- 7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)ethyl)-8-methyl-2-phenylisoquinolin- 1 (2H)-one;
• (S)-2-(1 -((5-(2-(4-(3-(Dimethylamino)propyl)piperazin-1 -yl)pyridin-4-yl)-7H- pyrrolo[2,3-(^pyrimidin-4-yl)amino)ethyl)-5-methyl-3-phenylquinazolin one; or a pharmaceutically acceptable salt, or solvate, or N-oxide, or stereoisomer or isotopically-labeled derivate thereof.
14. A compound according to any one of claims 1 to 13, for use in the treatment of a pathological condition or disease susceptible to amelioration by inhibition of Phosphoinositide 3-Kinase (PI3K).
15. A compound for use according to claim 14, wherein the pathological condition or disease is selected from respiratory diseases; allergic diseases; inflammatory or autoimmune-mediated; function disorders and neurological disorders; cardiovascular diseases; viral infection; metabolism/endocrine function disorders; neurological disorders and pain; bone marrow and organ transplant rejection; myelo-dysplastic syndrome; myeloproliferative disorders (MPDs); cancer and hematologic malignancies, leukemia, lymphomas and solid tumors.
16. A compound for use according to claims 14 or 15, wherein the pathological condition or disease is selected from leukemia, lymphomas and solid tumors, rheumatoid arthritis, multiple sclerosis, amyotrophic lateral sclerosis, Crohn's disease, ulcerative colitis, systemic lupus erythematosis, autoimmune hemolytic anemia, type I diabetes, cutaneous vasculitis, cutaneous lupus erythematosus, dermatomyositis, blistering diseases including but not limited to pemphigus vulgaris, bullous pemphigoid and epidermolysis bullosa, asthma, chronic obstructive pulmonary disease, cystic fibrosis, idiopathic pulmonary fibrosis, sarcoidosis, allergic rhinitis, atopic dermatitis, contact dermatitis, eczema, psoriasis, basal cell carcinoma, squamous cell carcinoma and actinic keratosis.
17. A pharmaceutical composition comprising a compound as defined in any one of claims 1 to 13 in association with a pharmaceutically acceptable diluent or carrier.
18. Use of a compound as defined in any one of claims 1 to 13, for the manufacture of a medicament for the treatment of a pathological condition or disease as defined in any one of claims 14 to 16.
19. A method for treating a subject afflicted with a pathological condition or disease as defined in any one of claims 14 to 16, which comprises administering to said subject a therapeutically effective amount of a compound as defined in any one of claims 1 to 13, or a pharmaceutical composition as defined in claim 17.
20. A combination product comprising (i) a compound as defined in any one of claims 1 to 13; and (ii) another compound selected from the group consisting of an Adenoside A2A agonist, an agent for treating cardiovascular disorders, an agent for treating diabetes, and an agent for treating liver disease, an anti-allergic agent, an anti- cholinergic agent, an anti-inflammatory agent, an anti-infective agent, a p2-adrenergic agonist, a Chemoattractant receptor homologous molecule expressed on TH2 cells (CRTH2) inhibitor, a chemotherapeutic agent, a corticosteroid, an ΙΚΚβ/ΙΚΒΚΒ (IkB kinase beta or IKK2) inhibitor, an immunosuppressant, a Janus kinase (JAK) inhibitor, a topically acting p38 Mitogen-Activated Protein Kinase (p38 MAPK) inhibitor, a Phosphosdiesterase (PDE) IV inhibitor, and a Spleen tyrosine kinase (Syk) inhibitor, for simultaneous, separate or sequential use in the treatment of the human or animal body.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP13382528 | 2013-12-19 | ||
| EP13382528.1 | 2013-12-19 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015091532A1 true WO2015091532A1 (en) | 2015-06-25 |
Family
ID=49876523
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2014/078038 Ceased WO2015091532A1 (en) | 2013-12-19 | 2014-12-16 | Pyrrolopyrimidine derivatives as pi3k inhibitors |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2015091532A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018094137A1 (en) | 2016-11-18 | 2018-05-24 | Cystic Fibrosis Foundation Therapeutics Inc. | Pyrrolopyrimidines as cftr potentiators |
| US10751339B2 (en) | 2018-01-20 | 2020-08-25 | Sunshine Lake Pharma Co., Ltd. | Substituted aminopyrimidine compounds and methods of use |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011008487A1 (en) * | 2009-06-29 | 2011-01-20 | Incyte Corporation | Pyrimidinones as pi3k inhibitors |
-
2014
- 2014-12-16 WO PCT/EP2014/078038 patent/WO2015091532A1/en not_active Ceased
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011008487A1 (en) * | 2009-06-29 | 2011-01-20 | Incyte Corporation | Pyrimidinones as pi3k inhibitors |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018094137A1 (en) | 2016-11-18 | 2018-05-24 | Cystic Fibrosis Foundation Therapeutics Inc. | Pyrrolopyrimidines as cftr potentiators |
| CN110337294A (en) * | 2016-11-18 | 2019-10-15 | 囊性纤维化基金会治疗公司 | Pyrrolopyrimidines as CFTR synergists |
| JP2020510668A (en) * | 2016-11-18 | 2020-04-09 | システィック ファイブロシス ファンデーション セラピューティクス インコーポレイテッドCystic Fibrosis Foundation Therapeutics Inc. | Pyrrolopyrimidines as CFTR enhancers |
| EP3541390A4 (en) * | 2016-11-18 | 2020-06-24 | Cystic Fibrosis Foundation | PYRROLOPYRIMIDINES FOR USE AS CFTR POTENTIALIZERS |
| RU2757457C2 (en) * | 2016-11-18 | 2021-10-18 | Систик Файбросис Фаундейшн | Pyrrolopyrimidines as cftr potentiators |
| CN110337294B (en) * | 2016-11-18 | 2022-11-01 | 囊性纤维化基金会 | Pyrrolopyrimidines as CFTR potentiators |
| CN115850268A (en) * | 2016-11-18 | 2023-03-28 | 囊性纤维化基金会 | Pyrrolopyrimidines as CFTR potentiators |
| EP4424311A3 (en) * | 2016-11-18 | 2024-12-18 | Cystic Fibrosis Foundation | Pyrrolopyrimidines as cftr potentiators |
| US10751339B2 (en) | 2018-01-20 | 2020-08-25 | Sunshine Lake Pharma Co., Ltd. | Substituted aminopyrimidine compounds and methods of use |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9388189B2 (en) | Pyrrolotriazinone derivatives as PI3K inhibitors | |
| AU2012247491B2 (en) | Pyrrolotriazinone derivatives as PI3k inhibitors | |
| US9034311B2 (en) | Pyridin-2(1 H)-one derivatives as JAK inhibitors | |
| EP2518071A1 (en) | Imidazopyridine derivatives as PI3K inhibitors | |
| EP2714686A1 (en) | Pyridin-2 (1h) -one derivatives useful as medicaments for the treatment of myeloproliferative disorders, transplant rejection, immune-mediated and inflammatory diseases | |
| EP2643321A1 (en) | Imidazo [1,2-b]pyridazine and imidazo [4,5-b]pyridine derivatives as jak inhibitors | |
| WO2014124757A1 (en) | Pyrrolotriazine derivatives as pi3k inhibitors | |
| EP2489663A1 (en) | Compounds as syk kinase inhibitors | |
| WO2014060431A1 (en) | Pyrrolotriazinone derivatives as pi3k inhibitors | |
| WO2015091531A1 (en) | Imidazolopyrimidin-2-yl derivatives as jak inhibitors | |
| WO2015091532A1 (en) | Pyrrolopyrimidine derivatives as pi3k inhibitors | |
| WO2016202800A1 (en) | Pyrrolotriazinone derivatives as pi3k inhibitors | |
| NZ614082B2 (en) | Pyrrolotriazinone derivatives as pi3k inhibitors | |
| NZ618904B2 (en) | Pyridin-2(1h)-one derivatives as jak inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14812271 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 14812271 Country of ref document: EP Kind code of ref document: A1 |