WO2012027065A2 - Combination therapy for treatment of disease - Google Patents
Combination therapy for treatment of disease Download PDFInfo
- Publication number
- WO2012027065A2 WO2012027065A2 PCT/US2011/045917 US2011045917W WO2012027065A2 WO 2012027065 A2 WO2012027065 A2 WO 2012027065A2 US 2011045917 W US2011045917 W US 2011045917W WO 2012027065 A2 WO2012027065 A2 WO 2012027065A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- seq
- inhibitor
- polypeptide
- actriib
- actriia
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *c1ccc[o]1 Chemical compound *c1ccc[o]1 0.000 description 2
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/177—Receptors; Cell surface antigens; Cell surface determinants
- A61K38/179—Receptors; Cell surface antigens; Cell surface determinants for growth factors; for growth regulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
Definitions
- compositions comprising inhibitors of the Type II Activin Receptor ("ActRII") and kits comprising such compositions. Also provided herein are methods of treatment of disease comprising administration of one or more of the
- compositions described herein and administration of a second active agent are provided.
- Activins were originally discovered as gonadal peptides involved in the regulation of follicle stimulating hormone synthesis, and are now believed to be involved in the regulation of a number of biological activities. Activins bind and signal through a combination of activin type II recptors (ActRII), including activin type IIA (ActRIIA) and activin type IIB (ActRIIB) receptors, both of which are transmembrane serine/threonine kinases (Harrison et al, J. Biol. Chem. 279, 28036-28044 (2004)). Inhibition of activin and/or activin receptors has shown promise in the treatment of disease.
- ActRII activin type II recptors
- ActRIIA activin type IIA
- ActRIIB activin type IIB
- Inhibitors of ActRII receptors are known in the art, and include soluble polypeptides that comprise the activin-binding domains of ActRII receptors (See, e.g., International Patent Application Publication Nos. WO/2002/088171, WO/2002/032925, WO/2005/037989; WO/2006/055689, WO 2006/012627 and WO 2010/019261; U.S. Patent Application Publication Nos. 2003/0133939, US 2005/0238646 and US 2010/0068215).
- Such polypeptides possess the ability to sequester ActRII ligands, thereby preventing signaling through the ActRII receptors, and may comprise all or a portion of the extracellular domain of an ActRII receptor (e.g., all or a portion of the extracellular domain of ActRIIA or all or a portion of the extracellular domain of ActRIIB).
- Multiple myeloma (MM; also known as myeloma, plasma cell myeloma, or Kahler's disease) is a type of cancer of plasma cells, which are antibody-producing immune system cells. Symptoms of multiple myeloma include bone pain, infection, renal failure, anemia, and bone lesions. Types of multiple myeloma include relapsed multiple myeloma, refractory multiple myeloma, and newly diagnosed multiple myeloma.
- MDS Myelodysplasia syndrome
- AML acute myelogenous leukemia
- Anemia the most common disorder of the blood, is a decrease in number of red blood cells (RBCs) or less than the normal quantity of hemoglobin in the blood. Symptoms of anemia include feelings of weakness and/or fatigue, general malaise and sometimes poor concentration. In very severe anemia, the body may compensate for the lack of oxygen- carrying capability of the blood by increasing cardiac output, resulting in symptoms such as palpitations, angina, intermittent claudication of the legs, and symptoms of heart failure.
- RBCs red blood cells
- compositions comprising inhibitors of ActRII receptors (e.g., the ActRIIA and ActRIIB inhibitors provided in Section 4.2, below).
- the compositions comprising inhibitors of ActRII receptors are administered in combination with a second active agent (e.g., the second active agents provided in Section 4.3, below).
- compositions comprising inhibitors of ActRII receptors that can be administered in combination with a second active agent, wherein the ActRII receptor inhibitor is a polypeptide comprising all or a portion of an ActRII extracellular domain (e.g., the extracellular domain of ActRIIA or ActRIIB) and wherein the second active agent is an IMiD® compound (such as lenalidomide (Revlimid®), i.e., 3-(4- amino-l-oxo-l,3-dihydro-isoindol-2-yl)-piperidine-2,6-dione), cyclophosphamide, bisphosphonate, thalidomide, dexamethasone, melphalan, prednisone, bortezomib, azacitidine, doxorubicin, vincristine, proteasome inhibitor and dacogen.
- the ActRII receptor inhibitor is a polypeptide comprising all or a portion of an Act
- composition provided herein comprises (i) an
- ActRIIA inhibitor wherein the ActRIIA inhibitor comprises or consists of a polypeptide selected from the group consisting of: a. a polypeptide at least 90% identical to SEQ ID NO:2; b. a polypeptide at least 95% identical to SEQ ID NO:2; c. a polypeptide at least 98% identical to SEQ ID NO:2; d. SEQ ID NO:2; e. a polypeptide at least 90% identical to SEQ ID NO:3; f. a polypeptide at least 95% identical to SEQ ID NO:3; g. a polypeptide at least 98% identical to SEQ ID NO:3; h. SEQ ID NO:3; i.
- the ActRIIA inhibitor is a polypeptide comprising or consisting of the amino acid sequence of SEQ ID NO:7.
- a composition provided herein comprises an ActRIIB inhibitor, wherein the ActRIIB inhibitor comprises or consists of a polypeptide selected from the group consisting of: a. a polypeptide at least 90%> identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; b. a polypeptide at least 95% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; c. a polypeptide at least 98% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; d.
- the ActRIIB inhibitor comprises or consists of a polypeptide selected from the group consisting of: a. a polypeptide at least 90%> identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; b. a polypeptide at least 95% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29,
- the ActRIIB inhibitor is a polypeptide comprising or consisting of SEQ ID NO:23. In another specific embodiment, the ActRIIB inhibitor is a polypeptide comprising or consisting of SEQ ID NO:25.
- a composition provided herein comprises an ActRIIA inhibitor and an ActRIIB inhibitor, wherein the ActRIIA inhibitor comprises or consists of a polypeptide selected from the group consisting of: a. a polypeptide at least 90%> identical to SEQ ID NO:2; b. a polypeptide at least 95% identical to SEQ ID NO:2; c. a polypeptide at least 98% identical to SEQ ID NO:2; d. SEQ ID NO:2; e. a polypeptide at least 90% identical to SEQ ID NO:3; f. a polypeptide at least 95% identical to SEQ ID NO:3; g.
- the ActRIIB inhibitor comprises or consists of a polypeptide selected from the group consisting of: a. a polypeptide at least 90% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; b. a polypeptide at least 95% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; c.
- polypeptide at least 98% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; d. SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; e. a polypeptide 90% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; f a polypeptide 95% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; g. a polypeptide 98% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; and h.
- the ActRIIA inhibitor is a polypeptide comprising or consisting of SEQ ID NO: 7 and the ActRIIB inhibitor is a polypeptide comprising or consisting of SEQ ID NO:23.
- the ActRIIA inhibitor is a polypeptide comprising or consisting of SEQ ID NO: 7 and the ActRIIB inhibitor is a polypeptide comprising or consisting of SEQ ID NO:25.
- the ActRII receptor inhibitor when administered in combination with a second active agent, can be administered parenterally, and the second active agent, e.g., 3-(4-amino-l-oxo-l,3- dihydro- isoindol-2-yl) -piperidine -2,6 -dione (i.e., lenalidomide (Revlimid®)), can be administered orally.
- the second active agent e.g., 3-(4-amino-l-oxo-l,3- dihydro- isoindol-2-yl) -piperidine -2,6 -dione (i.e., lenalidomide (Revlimid®)
- the second active agent e.g., 3-(4-amino-l-oxo-l,3- dihydro- isoindol-2-yl) -piperidine -2,6 -dione (i.e., len
- the second active agent e.g., 3-(4-amino-l-oxo-l,3- dihydro- isoindol-2-yl)-piperidine-2,6-dione (i.e., lenalidomide (Revlimid®)
- the second active agent e.g., 3-(4-amino-l-oxo-l,3-dihydro- isoindol-2-yl) -piperidine -2,6 -dione (i.e., lenalidomide (Revlimid®)
- the ActRIIA inhibitor can comprise or consist of a polypeptide comprising an amino acid sequence selected from the group consisting of: a. a polypeptide at least 90% identical to SEQ ID NO:2; b. a polypeptide at least 95% identical to SEQ ID NO:2; c. a polypeptide at least 98% identical to SEQ ID NO:2; d. SEQ ID NO:2; e. a polypeptide at least 90% identical to SEQ ID NO:3; f. a polypeptide at least 95% identical to SEQ ID NO:3; g.
- the second active agent can be one or more of the following: an IMiD® compound (such as lenalidomide (Revlimid®), i.e., 3-(4-amino-l-oxo-l,3-dihydro-isoindol-2-yl)-piperidine-2,6- dione); pomalidomide (4-(amino)-2-(2,6-dioxo(3-piperidyl))-isoindoline-l,3-dione); and 3- (5-amino-2-methyl-4-oxo-4H-quinazolin-3-yl)-piperidine-2,6-dione), cyclophosphamide, bisphosphonate, thalidomide, dexamethasone, melphalan, prednisone, bortezomib, azacitidine, doxorubicin, vincristine, proteasome inhibitor, and dacogen.
- a method for treating and/or preventing disease comprising administering an ActRIIB inhibitor and administering a second active agent.
- the ActRIIB inhibitor can comprise or consist of a polypeptide comprising an amino acid sequence selected from the group consisting of: a. a polypeptide at least 90% identical to SEQ ID NO:17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; b. a polypeptide at least 95% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; c.
- the second active agent can be one or more of the following: an IMiD® compound (such as lenalidomide (Revlimid®), i.e., 3-(4-amino-l-oxo-l,3-dihydro-isoindol-2-yl)-piperidine-2,6- dione); pomalidomide (4-(amino)-2-(2,6-dioxo(3-piperidyl))-isoindoline-l,3-dione); and 3- (5-amino-2-methyl-4-oxo-4H-quinazolin-3-yl)-piperidine-2,6-dione), cyclophosphamide, bisphosphonate, thalidomide, dexamethasone, melphalan, prednisone, bortezomib, azacit
- the ActRIIB inhibitor is a polypeptide comprising or consisting of the amino acid sequence of SEQ ID NO:23 and the second active agent is lenalidomide (Revlimid®). In a specific embodiment, the ActRIIB inhibitor is a polypeptide comprising or consisting of the amino acid sequence of SEQ ID NO:25 and the second active agent is lenalidomide (Revlimid®).
- the ActRIIA inhibitor can comprise or consist of a polypeptide comprising an amino acid sequence selected from the group consisting of: a. a polypeptide at least 90%> identical to SEQ ID NO:2; b. a polypeptide at least 95% identical to SEQ ID NO:2; c. a polypeptide at least 98% identical to SEQ ID NO:2; d. SEQ ID NO:2; e. a polypeptide at least 90% identical to SEQ ID NO:3; f. a polypeptide at least 95% identical to SEQ ID NO:3; g.
- the ActRIIB inhibitor can comprise or consist of a polypeptide comprising an amino acid sequence selected from the group consisting of: a. a polypeptide at least 90% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; b. a polypeptide at least 95% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; c. a polypeptide at least 98% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; d. SEQ ID NO: 17, 18,
- the ActRIIA inhibitor is a polypeptide comprising or consisting of SEQ ID NO: 7 and the ActRIIB inhibitor is a polypeptide comprising or consisting of the amino acid sequence of SEQ ID NO:23.
- the ActRIIA inhibitor is a polypeptide comprising or consisting of SEQ ID NO: 7 and the ActRIIB inhibitor is a polypeptide comprising or consisting of the amino acid sequence of SEQ ID NO:25.
- a method for treating and/or preventing disease comprising administering an ActRIIA inhibitor and an ActRIIB inhibitor, and a second active agent.
- the ActRIIA inhibitor can comprise or consist of a polypeptide comprising an amino acid sequence selected from the group consisting of: a. a polypeptide at least 90% identical to SEQ ID NO:2; b. a polypeptide at least 95% identical to SEQ ID NO:2; c. a polypeptide at least 98% identical to SEQ ID NO:2; d. SEQ ID NO:2; e. a polypeptide at least 90% identical to SEQ ID NO:3; f.
- the ActRIIB inhibitor can comprise or consist of a polypeptide comprising an amino acid sequence selected from the group consisting of: a. a polypeptide at least 90%> identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; b.
- the second active agent can be one or more of the following: an IMiD® compound (such as lenalidomide (Revlimid®), i.e., 3-(4-amino-l-oxo-l,3-dihydro-isoindol-2-yl)-piperidine-2,6-dione);
- an IMiD® compound such as lenalidomide (Revlimid®), i.e., 3-(4-amino-l-oxo-l,3-dihydro-isoindol-2-yl)-piperidine-2,6-dione
- pomalidomide (4-(amino)-2-(2,6-dioxo(3-piperidyl))-isoindoline-l,3-dione); and 3-(5-amino- 2-methyl-4-oxo-4H-quinazolin-3-yl)-piperidine-2,6-dione)), cyclophosphamide,
- the ActRIIA inhibitor is a polypeptide comprising or consisting of SEQ ID NO:7
- the ActRIIB inhibitor is a polypeptide comprising or consisting of the amino acid sequence of SEQ ID NO:23
- the second active agent is lenalidomide (Revlimid®).
- the ActRIIA inhibitor is a polypeptide comprising or consisting of SEQ ID NO:7
- the ActRIIB inhibitor is a polypeptide comprising or consisting of the amino acid sequence of SEQ ID NO:25
- the second active agent is lenalidomide
- the methods of treating disease provided herein comprise parenteral administration of an ActRIIA inhibitor comprising or consisting of a polypeptide that comprises SEQ ID NO:7 and oral administration (e.g., in the form of a capsule or tablet) of lenalidomide (Revlimid®) at an amount of from about 5 to about 25 mg per day.
- the methods of treating disease provided herein comprise parenteral administration of an ActRIIB inhibitor comprising a polypeptide that comprises or consists of SEQ ID NO:23 and oral administration (e.g., in the form of a capsule or tablet) of lenalidomide (Revlimid®) at an amount of from about 5 to about 25 mg per day.
- the methods of treating disease provided herein comprise parenteral administration of an ActRIIB inhibitor comprising a polypeptide that comprises or consists of SEQ ID NO:25 and oral administration (e.g., in the form of a capsule or tablet) of lenalidomide (Revlimid®) at an amount of from about 5 to about 25 mg per day.
- an ActRIIB inhibitor comprising a polypeptide that comprises or consists of SEQ ID NO:25
- oral administration e.g., in the form of a capsule or tablet
- lenalidomide Revlimid®
- provided herein is a method of treating multiple myeloma, comprising administering an ActRII inhibitor and administering a second active agent.
- the ActRII inhibitor is an ActRIIA inhibitor, wherein said ActRIIA inhibitor is a polypeptide that comprises or consists of SEQ ID NO:7; and the second active agent is Revlimid®.
- the ActRII inhibitor is an ActRIIB inhibitor, wherein said ActRIIB inhibitor is a polypeptide that comprises or consists of SEQ ID NO:25; and the second active agent is Revlimid®.
- the ActRII inhibitor is an ActRIIA inhibitor, wherein said ActRIIA inhibitor is a polypeptide that comprises or consists of SEQ ID NO:7; and the second active agent is pomalidomide.
- the ActRII inhibitor is an ActRIIB inhibitor, wherein said ActRIIB inhibitor is a polypeptide that comprises or consists of SEQ ID NO:25; and the second active agent is pomalidomide.
- a method of treating myelodysplasia syndrome comprising administering an ActRII inhibitor and administering a second active agent.
- the ActRII inhibitor is an ActRIIA inhibitor, wherein said ActRIIA inhibitor is a polypeptide that comprises or consists of SEQ ID NO:7; and the second active agent is Revlimid®.
- the ActRII inhibitor is an ActRIIB inhibitor, wherein said ActRIIB inhibitor is a polypeptide that comprises or consists of SEQ ID NO:25; and the second active agent is Revlimid®.
- the ActRII inhibitor is an ActRIIA inhibitor, wherein said ActRIIA inhibitor is a polypeptide that comprises or consists of SEQ ID NO:7; and the second active agent is pomalidomide.
- the ActRII inhibitor is an ActRIIB inhibitor, wherein said ActRIIB inhibitor is a polypeptide that comprises or consists of SEQ ID NO:25; and the second active agent is pomalidomide.
- a method of treating anemia comprising administering an ActRII inhibitor and administering a second active agent.
- the ActRII inhibitor is an ActRIIA inhibitor, wherein said ActRIIA inhibitor is a polypeptide that comprises or consists of SEQ ID NO:7; and the second active agent is Revlimid® or pomalidomide.
- the ActRII inhibitor is an ActRIIB inhibitor, wherein said ActRIIB inhibitor is a polypeptide that comprises or consists of SEQ ID NO:25; and the second active agent is Revlimid® or pomalidomide.
- the ActRII inhibitor(s) and the second active agent(s) are administered at the same time. In certain embodiments, the ActRII inhibitor and the second active agent are administered in the same formulation. In other embodiments, the ActRII inhibitor and the second active agent are administered sequentially.
- the present invention provides a method for improving the safety of a treatment with 3-(4-amino-l-oxo-l,3-dihydro-isoindol-2-yl)-piperidine-2,6- dione (i.e., lenalidomide (Revlimid®)) in a patient, wherein the method comprises administering an ActRII inhibitor(s) to the patient being treated with 3-(4-amino-l-oxo-l,3- dihydro-isoindol-2-yl)-piperidine-2,6-dione (i.e., lenalidomide (Revlimid®)).
- an ActRII inhibitor(s) to the patient being treated with 3-(4-amino-l-oxo-l,3- dihydro-isoindol-2-yl)-piperidine-2,6-dione (i.e., lenalidomide (Revlimid®)).
- compositions comprising ActRII receptor inhibitors (e.g., ActRIIA or ActRIIB inhibitors) that can be administered in combination with each other and/or in combination with second active agents, as well as methods for the treatment of disease comprising administration of one or more ActRII receptor inhibitors or one or more ActRII receptor inhibitors and a second active agent, such as thalidomide, dexamethasone, melphalan, prednisone, bortezomib, cyclophosphamide, bisphosphonate, dacogen, azacitidine, doxorubicin, vincristine, proteasome inhibitor, and/or an IMiD® compound (e.g., lenalidomide (also known as Revlimid® or 3-(4-amino-l-oxo-l,3-dihydro-isoindol-2-yl)- piperidine-2,6-dione); pomalidomide (4-
- Exemplary diseases that can be treated using the methods provided herein include cancer, multiple myeloma (e.g., relapsed multiple myeloma, refractory multiple myeloma, and newly diagnosed multiple myeloma), myelodysplastic syndrome (MDS), and anemia.
- the ActRII receptor inhibitor(s) can be administered separately from the second active agent(s), or the ActRII receptor inhibitor(s) can be administered at the same time as, or in the same pharmaceutical formulation as, the second active agent(s).
- Inhibitors of ActRII receptors are described in Section 4.2.
- Second active agents are described in Section 4.3.
- Methods of treatment of disease, as well as dosage and administration regimens, are described in Section 4.4.
- Patient populations are described in Section 4.5.
- Pharmaceutical compositions are described in Section 4.7. 4.2 INHIBITORS OF ACTRII RECEPTORS
- Inhibitors of ActRII receptors encompassed herein include ActRIIA inhibitors and ActRIIB inhibitors (see below).
- an ActRII receptor inhibitor is specific to ActRIIA.
- an ActRII receptor inhibitor is specific to ActRIIB.
- an ActRII receptor inhibitor preferentially inhibits ActRIIA.
- an ActRII receptor inhibitor preferenctially inhibits ActRIIB.
- an ActRII receptor inhibitor inhibits both ActRIIA and ActRIIB.
- inhibitors of ActRII receptors can be polypeptides comprising activin-binding domains of ActRII.
- activin-binding domain comprising polypeptides sequester activin and thereby prevent activin signaling.
- These activin-binding domain comprising polypeptides may comprise all or a portion of the extracellular domain of an ActRII receptor (i.e., all or a portion of the extracellular domain of ActRIIA or all or a portion of the extracellular domain of ActRIIB).
- the extracellular domain of an ActRII receptor is soluble.
- the activin-binding domain comprising polypeptides are linked to an Fc portion of an antibody (i.e., a conjugate comprising an activin-binding domain comprising polypeptide of an ActRII receptor and an Fc portion of an antibody is generated).
- the antibody portion confers increased stability on the conjugate.
- the activin-binding domain is linked to an Fc portion of an antibody via a linker, e.g., a peptide linker.
- the inhibitors of ActRII receptors used in the compositions and methods described herein comprise molecules that inhibit ActRIIA and/or ActRIIB, directly or indirectly, either extracellularly or intracellularly.
- the inhibitors of ActRIIA and/or ActRIIB used in the compositions and methods described herein inhibit ActRIIA and/or ActRIIB via interactions with the receptor(s) itself.
- the inhibitors of ActRIIA and/or ActRIIB used in the compositions and methods described herein inhibit ActRIIA and/or ActRIIB via interactions with an ActRIIA and/or ActRIIB ligand, e.g., Activin.
- ActRIIA refers to a family of activin receptor type Ila
- ActRIIA proteins from any species and variants derived from such ActRIIA proteins by mutagenesis or other modification.
- Reference to ActRIIA herein is understood to be a reference to any one of the currently identified forms.
- Members of the ActRIIA family are generally transmembrane proteins, composed of a ligand-binding extracellular domain with a cysteine-rich region, a transmembrane domain, and a cytoplasmic domain with predicted serine/threonine kinase activity.
- ActRIIA inhibitors to be used in the compositions and methods described herein include, without limitation, activin-binding soluble ActRIIA polypeptides; antibodies that bind to activin (particularly the activin A or B subunits, also referred to as BA or B B ) and disrupt ActRIIA binding; antibodies that bind to ActRIIA and disrupt activin binding; non- antibody proteins selected for activin or ActRIIA binding (see e.g., WO/2002/088171 , WO/2006/055689, WO/2002/032925, WO/2005/037989, US 2003/0133939, and US
- two or more different proteins (or other moieties) with activin or ActRIIA binding activity may be linked together to create a bifunctional or multifunctional binding molecule that inhibits ActRIIA and thus can be used in the compositions and methods described herein include.
- activin- ActRIIA signaling axis antagonists that inhibit ActRIIA include nucleic acid aptamers, small molecules and other agents are used in the compositions and methods described herein include.
- ActRIIA polypeptide includes polypeptides comprising any naturally occurring polypeptide of an ActRIIA family member as well as any variants thereof
- ActRIIA polypeptides include polypeptides derived from the sequence of any known ActRIIA having a sequence at least about 80% identical to the sequence of an ActRIIA polypeptide, and optionally at least 85%, 90%, 95%, 97%, 98%, 99% or greater identity.
- an ActRIIA polypeptide may bind to and inhibit the function of an ActRIIA protein and/or activin.
- ActRIIA polypeptides examples include human ActRIIA precursor polypeptide (SEQ ID NO: 1) and soluble human ActRIIA polypeptides (e.g., SEQ ID NOs: 2, 3, 7 and 12).
- SEQ ID NO: 1 the signal peptide of the human ActRIIA precursor polypeptide located at amino acid positions 1 to 20; the extracellular domain is located at amino acid positions 21 to 135 and the N-linked glycosylation sites of the human ActRIIA precursor polypeptide (SEQ ID NO: 1) are located at amino acid positions 43 and 56 of SEQ ID NO: 1.
- the nucleic acid sequence encoding the human ActRIIB precursor polypeptide of SEQ ID NO: l is disclosed as SEQ ID NO:4 (nucleotides 164-1705 of
- Genbank entry NM 001616 The nucleic acid sequence encoding the soluble human ActRIIA polypeptide of SEQ ID NO:2 is disclosed as SEQ ID NO:5. See Table 1 for a description of the sequences.
- the ActRIIA polypeptides used in the compositions and methods described herein are soluble ActRIIA polypeptides.
- An extracellular domain of an ActRIIA protein can bind to activin and is generally soluble, and thus can be termed a soluble, activin-binding ActRIIA polypeptide.
- soluble ActRIIA polypeptide generally refers to polypeptides comprising an extracellular domain of an ActRIIA protein, including any naturally occurring extracellular domain of an ActRIIA protein as well as any variants thereof (including mutants, fragments and peptidomimetic forms).
- Soluble ActRIIA polypeptides can bind to activin; however, the wild type ActRIIA protein does not exhibit significant selectivity in binding to activin versus myostatin (growth differentiation factor (GDF) 8) or bone morphogenic protein 11 (BMP1 1; also known as GDF11).
- GDF growth differentiation factor
- BMP1 1 bone morphogenic protein 11
- Native or altered ActRIIA proteins may be given added specificity for activin by coupling them with a second, activin-selective binding agent.
- Examples of soluble, activin- binding ActRIIA polypeptides include the soluble polypeptides illustrated in SEQ ID NOs: 2, 3, 7, 12 and 13.
- soluble, activin-binding ActRIIA polypeptides comprise a signal sequence in addition to the extracellular domain of an ActRIIA protein, for example, the honey bee mellitin leader sequence (SEQ ID NO: 8), the tissue plasminogen activator (TP A) leader (SEQ ID NO: 9) or the native ActRIIA leader (SEQ ID NO: 10).
- the ActRIIA- hFc polypeptide illustrated in SEQ ID NO: 13 uses a TP A leader.
- the inhibitors of ActRIIA used in the compositions and methods described herein comprise a conjugate/fusion protein comprising an activin-binding domain of ActRIIA linked to an Fc portion of an antibody.
- the activin-binding domain is linked to an Fc portion of an antibody via a linker, e.g. , a peptide linker.
- the Fc domain has one or more mutations at residues such as Asp-265, lysine 322, and Asn-434.
- the mutant Fc domain having one or more of these mutations (e.g., an Asp-265 mutation) has a reduced ability to bind to the Fey receptor relative to a wild-type Fc domain.
- the mutant Fc domain having one or more of these mutations has an increased ability to bind to the MHC class I- related Fc-receptor (FcR ) relative to a wild-type Fc domain.
- FcR MHC class I- related Fc-receptor
- Exemplary fusion proteins comprising a soluble extracellular domain of ActRIIA fused to an Fc domain are set forth in SEQ ID NOs:6, 7, 12, and 13.
- the ActRIIA inhibitors used in the compositions and methods described herein comprise the extracellular domain of ActRIIA, or a portion thereof, linked to an Fc portion of an antibody, wherein said ActRIIA inhibitor comprises an amino acid sequence that is at least 75% identical to an amino acid sequence selected from SEQ ID NOs:6, 7, 12, and 13.
- the ActRIIA inhibitors used in the compositions and methods described herein comprise the extracellular domain of ActRIIA, or a portion thereof, linked to an Fc portion of an antibody, wherein said ActRIIA inhibitor comprises an amino acid sequence that is at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical to an amino acid sequence selected from SEQ ID NOs:6, 7, 12, and 13.
- the inhibitors of ActRIIA used in the compositions and methods described herein comprise a truncated form of an extracellular domain of ActRIIA.
- the truncation can be at the carboxy terminus and/or the amino terminus of the ActRIIA polypeptide.
- the truncation can be 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 amino acids long relative to the mature ActRIIB polypeptide extracellular domain.
- the truncation can be 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 N-terminal amino acids of the mature ActRIIA polypeptide extracellular domain.
- the truncation can be 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 C-terminal amino acids of the mature ActRIIA polypeptide extracellular domain.
- truncated forms of ActRIIA include polypeptides with amino acids 20-119; 20-128; 20-129; 20-130; 20-131; 20-132; 20-133; 20-134; 20-131; 21- 131; 22-131; 23-131; 24-131; and 25-131, wherein the amino acid positions refer to the amino acid positions in SEQ ID NO: 1.
- the inhibitors of ActRIIA used in the compositions and methods described herein comprise an extracellular domain of ActRIIA with one or more amino acid substitutions. In certain embodiments, the inhibitors of ActRIIA used in the compositions and methods described herein comprise a truncated form of an ActRIIA extracellular domain that also carries an amino acid substitution.
- the ActRIIA inhibitor to be used in the compositions and methods described herein is a fusion protein between the extracellular domain of the human ActRIIA receptor and the Fc portion of IgGl .
- the ActRIIA inhibitor to be used in the compositions and methods described herein is a fusion protein between a truncated extracellular domain of the human ActRIIA receptor and the Fc portion of IgGl .
- the ActRIIA inhibitor to be used in the compositions and methods described herein is a fusion protein between a truncated extracellular domain of the human ActRIIA receptor and the Fc portion of IgGl, wherein the truncated extracellular domain of the human ActRIIA receptor possesses one or more amino acid substitutions.
- Functionally active fragments of ActRIIA polypeptides can be obtained, for example, by screening polypeptides recombinantly produced from the corresponding fragment of the nucleic acid encoding an ActRIIA polypeptide.
- fragments can be chemically synthesized using techniques known in the art such as conventional Merrifield solid phase f-Moc or t-Boc chemistry. The fragments can be produced (recombinantly or by chemical synthesis) and tested to identify those peptidyl fragments that can function as antagonists (inhibitors) of ActRIIA protein or signaling mediated by activin.
- a functional variant of ActRIIA polypeptides can be obtained, for example, by screening libraries of modified polypeptides recombinantly produced from the corresponding mutagenized nucleic acids encoding an ActRIIA polypeptide. The variants can be produced and tested to identify those that can function as antagonists (inhibitors) of ActRIIA protein or signaling mediated by activin.
- a functional variant of the ActRIIA polypeptides comprises an amino acid sequence that is at least 75% identical to an amino acid sequence selected from SEQ ID NOs: 2 or 3.
- the functional variant has an amino acid sequence at least 80%, 85%, 90%, 95%, 97%, 98%, 99% or 100% identical to an amino acid sequence selected from SEQ ID NOs: 2 or 3.
- Functional variants may be generated, for example, by modifying the structure of an ActRIIA polypeptide for such purposes as enhancing therapeutic efficacy, or stability (e.g., ex vivo shelf life and resistance to proteolytic degradation in vivo). Such modified ActRIIA polypeptides when selected to retain activin binding, are considered functional equivalents of the naturally-occurring ActRIIA polypeptides. Modified ActRIIA
- polypeptides can also be produced, for instance, by amino acid substitution, deletion, or addition. For instance, it is reasonable to expect that an isolated replacement of a leucine with an isoleucine or valine, an aspartate with a glutamate, a threonine with a serine, or a similar replacement of an amino acid with a structurally related amino acid (e.g., conservative mutations) will not have a major effect on the biological activity of the resulting molecule. Conservative replacements are those that take place within a family of amino acids that are related in their side chains.
- Whether a change in the amino acid sequence of an ActRIIA polypeptide results in a functional homolog can be readily determined by assessing the ability of the variant ActRIIA polypeptide to produce a response in cells in a fashion similar to the wild-type ActRIIA polypeptide.
- telomere sequence Asparagine-linked glycosylation recognition sites generally comprise a tripeptide sequence, asparagine-X-threonine (or asparagines-X-serine) (where "X" is any amino acid) which is specifically recognized by appropriate cellular glycosylation enzymes.
- the alteration may also be made by the addition of, or substitution by, one or more serine or threonine residues to the sequence of the wild-type ActRIIA polypeptide (for O- linked glycosylation sites).
- a variety of amino acid substitutions or deletions at one or both of the first or third amino acid positions of a glycosylation recognition site (and/or amino acid deletion at the second position) results in non-glycosylation at the modified tripeptide sequence.
- Another means of increasing the number of carbohydrate moieties on an ActRIIA polypeptide is by chemical or enzymatic coupling of glycosides to the ActRIIA polypeptide.
- the sugar(s) may be attached to (a) arginine and histidine; (b) free carboxyl groups; (c) free sulfhydryl groups such as those of cysteine; (d) free hydroxyl groups such as those of serine, threonine, or hydroxyproline; (e) aromatic residues such as those of phenylalanine, tyrosine, or tryptophan; or (f) the amide group of glutamine.
- arginine and histidine free carboxyl groups
- free sulfhydryl groups such as those of cysteine
- free hydroxyl groups such as those of serine, threonine, or hydroxyproline
- aromatic residues such as those of phenylalanine, tyrosine, or tryptophan
- the amide group of glutamine are described in WO 87/05330 published Sep. 11, 1987, and in Aplin and Wriston (1981) CRC Crit. Rev. Biochem., pp. 259
- trifluoromethanesulfonic acid or an equivalent compound. This treatment results in the cleavage of most or all sugars except the linking sugar (N-acetylglucosamine or N- acetylgalactosamine), while leaving the amino acid sequence intact.
- ActRIIA polypeptide may be adjusted, as appropriate, depending on the type of expression system used, as mammalian, yeast, insect and plant cells may all introduce differing glycosylation patterns that can be affected by the amino acid sequence of the peptide.
- ActRIIA proteins for use in humans will be expressed in a mammalian cell line that provides proper glycosylation, such as HEK293 or CHO cell lines, although other expression systems, such as other mammalian expression cell lines, yeast cell lines with engineered glycosylation enzymes and insect cells, are expected to be useful as well.
- mutants particularly sets of combinatorial mutants of an ActRIIA polypeptide, as well as truncation mutants; pools of combinatorial mutants are especially useful for identifying functional variant sequences.
- the purpose of screening such combinatorial libraries may be to generate, for example, ActRIIA polypeptide variants which can act as either agonists or antagonist, or alternatively, which possess novel activities all together.
- a variety of screening assays are provided below, and such assays may be used to evaluate variants.
- an ActRIIA polypeptide variant may be screened for ability to bind to an ActRIIA ligand, to prevent binding of an ActRIIA ligand to an ActRIIA polypeptide or to interfere with signaling caused by an ActRIIA ligand.
- Combinatorially-derived variants can be generated which have a selective or generally increased potency relative to a naturally occurring ActRIIA polypeptide.
- mutagenesis can give rise to variants which have intracellular half-lives dramatically different than the corresponding a wild-type ActRIIA polypeptide.
- the altered protein can be rendered either more stable or less stable to proteolytic degradation or other cellular processes which result in destruction of, or otherwise inactivation of a native ActRIIA polypeptide.
- Such variants, and the genes which encode them can be utilized to alter ActRIIA polypeptide levels by modulating the half- life of the ActRIIA polypeptides.
- a short half-life can give rise to more transient biological effects and can allow tighter control of recombinant ActRIIA polypeptide levels within the patient.
- mutations may be made in the linker (if any) and/or the Fc portion to alter the half- life of the protein.
- a combinatorial library may be produced by way of a degenerate library of genes encoding a library of polypeptides which each include at least a portion of potential ActRIIA polypeptide sequences.
- a mixture of synthetic oligonucleotides can be enzymatically ligated into gene sequences such that the degenerate set of potential ActRIIA polypeptide nucleotide sequences are expressible as individual polypeptides, or alternatively, as a set of larger fusion proteins (e.g., for phage display).
- the library of potential homologs can be generated from a degenerate oligonucleotide sequence.
- Chemical synthesis of a degenerate gene sequence can be carried out in an automatic DNA synthesizer, and the synthetic genes then be ligated into an appropriate vector for expression. The synthesis of degenerate
- oligonucleotides is well known in the art (see for example, Narang, S A (1983) Tetrahedron 39:3; Itakura et al, (1981) Recombinant DNA, Proc. 3rd Cleveland Sympos.
- ActRIIA polypeptide variants can be generated and isolated from a library by screening using, for example, alanine scanning mutagenesis and the like (Ruf et al, (1994) Biochemistry 33: 1565-1572; Wang et al, (1994) J. Biol. Chem.
- the most widely used techniques for screening large gene libraries typically comprises cloning the gene library into replicable expression vectors, transforming appropriate cells with the resulting library of vectors, and expressing the combinatorial genes under conditions in which detection of a desired activity facilitates relatively easy isolation of the vector encoding the gene whose product was detected.
- Preferred assays include activin binding assays and activin-mediated cell signaling assays.
- ActRIIA polypeptides may further comprise post- translational modifications in addition to any that are naturally present in the ActRIIA polypeptides. Such modifications include, but are not limited to, acetylation, carboxylation, glycosylation, phosphorylation, lipidation, and acylation. As a result, the modified ActRIIA polypeptides may contain non-amino acid elements, such as polyethylene glycols, lipids, poly- or mono-saccharide, and phosphates. Effects of such non-amino acid elements on the functionality of a ActRIIA polypeptide may be tested by any method known to the skilled artisan.
- an ActRIIA polypeptide When an ActRIIA polypeptide is produced in cells by cleaving a nascent form of the ActRIIA polypeptide, post-translational processing may also be important for correct folding and/or function of the protein.
- Different cells such as CHO, HeLa, MDCK, 293, W138, NIH-3T3 or HEK293 have specific cellular machinery and characteristic mechanisms for such post-translational activities and may be chosen to ensure the correct modification and processing of the ActRIIA polypeptides.
- fusion proteins having at least a portion of the ActRIIA polypeptides and one or more fusion domains.
- fusion domains include, but are not limited to, polyhistidine, Glu-Glu, glutathione S transferase (GST), thioredoxin, protein A, protein G, an immunoglobulin heavy chain constant region (Fc), maltose binding protein (MBP), or human serum albumin.
- a fusion domain may be selected so as to confer a desired property. For example, some fusion domains are particularly useful for isolation of the fusion proteins by affinity chromatography.
- matrices for affinity chromatography such as glutathione-, amylase-, and nickel- or cobalt-conjugated resins are used.
- Many of such matrices are available in "kit” form, such as the Pharmacia GST purification system and the QIAexpressTM system (Qiagen) useful with (HIS 6 ) fusion partners.
- a fusion domain may be selected so as to facilitate detection of the ActRIIA polypeptides. Examples of such detection domains include the various fluorescent proteins (e.g., GFP) as well as "epitope tags," which are usually short peptide sequences for which a specific antibody is available.
- an ActRIIA polypeptide is fused with a domain that stabilizes the ActRIIA polypeptide in vivo (a "stabilizer" domain).
- stabilizing is meant anything that increases serum half life, regardless of whether this is because of decreased destruction, decreased clearance by the kidney, or other
- Fusions with the Fc portion of an immunoglobulin are known to confer desirable pharmacokinetic properties on a wide range of proteins.
- fusions to human serum albumin can confer desirable properties.
- Other types of fusion domains that may be selected include multimerizing (e.g., dimerizing, tetramerizing) domains and functional domains (that confer an additional biological function, such as further stimulation of bone growth or muscle growth, as desired).
- multimerizing e.g., dimerizing, tetramerizing domains
- functional domains that confer an additional biological function, such as further stimulation of bone growth or muscle growth, as desired.
- an ActRIIA polypeptide may be placed C-terminal to a heterologous domain, or, alternatively, a heterologous domain may be placed C-terminal to an ActRIIA polypeptide.
- the ActRIIA polypeptide domain and the heterologous domain need not be adjacent in a fusion protein, and additional domains or amino acid sequences may be included C- or N-terminal to either domain or between the domains.
- the ActRIIA polypeptides described herein contain one or more modifications that are capable of stabilizing the ActRIIA polypeptides.
- modifications enhance the in vitro half life of the ActRIIA polypeptides, enhance circulatory half life of the ActRIIA polypeptides or reduce proteolytic degradation of the ActRIIA polypeptides.
- Such stabilizing modifications include, but are not limited to, fusion proteins (including, for example, fusion proteins comprising an ActRIIA polypeptide and a stabilizer domain), modifications of a glycosylation site (including, for example, addition of a glycosylation site to an ActRIIA polypeptide), and modifications of carbohydrate moiety (including, for example, removal of carbohydrate moieties from an ActRIIA polypeptide).
- fusion proteins including, for example, fusion proteins comprising an ActRIIA polypeptide and a stabilizer domain
- modifications of a glycosylation site including, for example, addition of a glycosylation site to an ActRIIA polypeptide
- modifications of carbohydrate moiety including, for example, removal of carbohydrate moieties from an ActRIIA polypeptide.
- an ActRIIA polypeptide is fused to a stabilizer domain such as an IgG molecule (e.g., an Fc domain).
- stabilizer domain not only refers to a fusion domain (e.g., Fc) as in the case of fusion proteins, but also includes nonproteinaceous modifications such as a carbohydrate moiety, or nonproteinaceous polymer, such as polyethylene glycol.
- isolated and/or purified forms of the ActRIIA are isolated and/or purified forms of the ActRIIA.
- ActRIIA polypeptides which are isolated from, or otherwise substantially free of, other proteins can be used with the methods and compositions described herein. ActRIIA polypeptides will generally be produced by expression from recombinant nucleic acids.
- nucleic acids encoding any of the ActRIIA polypeptides (e.g., soluble ActRIIA polypeptides), including fragments, functional variants and fusion proteins disclosed herein.
- SEQ ID NO: 4 encodes the naturally occurring human ActRIIA precursor polypeptide
- SEQ ID NO: 5 encodes the processed extracellular domain of ActRIIA.
- the subject nucleic acids may be single-stranded or double stranded.
- Such nucleic acids may be DNA or RNA molecules. These nucleic acids may be used, for example, in methods for making ActRIIA polypeptides or as direct therapeutic agents (e.g., in a gene therapy approach).
- the subject nucleic acids encoding ActRIIA polypeptides are further understood to include nucleic acids that are variants of SEQ ID NO: 4 or 5.
- Variant nucleotide sequences include sequences that differ by one or more nucleotide substitutions, additions or deletions, such as allelic variants.
- nucleic acid sequences that are at least 80%, 85%, 90%, 95%, 97%, 98%, 99% or 100% identical to SEQ ID NO: 4 or 5.
- nucleic acid sequences complementary to SEQ ID NO: 4 or 5, and variants of SEQ ID NO: 4 or 5 are also encompassed herein.
- the nucleic acid sequences provided herein can be isolated, recombinant, and/or fused with a heterologous nucleotide sequence, or in a DNA library.
- nucleic acids provided herein also include nucleotide sequences that hybridize under highly stringent conditions to the nucleotide sequence designated in SEQ ID NO: 4 or 5, complement sequence of SEQ ID NO: 4 or 5, or fragments thereof.
- appropriate stringency conditions which promote DNA hybridization can be varied.
- appropriate stringency conditions which promote DNA hybridization can be varied. For example, one can perform the hybridization at 6.0 times sodium chloride/sodium citrate (SSC) at about 45 degrees Celsius, followed by a wash of 2.0 times SSC at 50 degrees Celsius.
- SSC sodium chloride/sodium citrate
- the salt concentration in the wash step can be selected from a low stringency of about 2.0 times SSC at 50 degrees Celsius to a high stringency of about 0.2 times SSC at 50 degrees Celsius.
- the temperature in the wash step can be increased from low stringency conditions at room temperature, about 22 degrees Celsius, to high stringency conditions at about 65 degrees Celsius. Both temperature and salt may be varied, or temperature or salt concentration may be held constant while the other variable is changed.
- nucleic acids which hybridize under low stringency conditions of 6 times SSC at room temperature followed by a wash at 2 times SSC at room temperature can be used with the methods and compositions described herein.
- Isolated nucleic acids which differ from the nucleic acids as set forth in SEQ ID NOs: 4 or 5 due to degeneracy in the genetic code are also encompassed herein.
- a number of amino acids are designated by more than one triplet. Codons that specify the same amino acid, or synonyms (for example, CAU and CAC are synonyms for histidine) may result in "silent" mutations which do not affect the amino acid sequence of the protein.
- CAU and CAC are synonyms for histidine
- nucleotides up to about 3-5% of the nucleotides
- nucleic acids encoding a particular protein may exist among individuals of a given species due to natural allelic variation. Any and all such nucleotide variations and resulting amino acid polymorphisms are encompassed herein.
- the recombinant nucleic acids may be operably linked to one or more regulatory nucleotide sequences in an expression construct. Regulatory nucleotide sequences will generally be appropriate to the host cell used for expression.
- said one or more regulatory nucleotide sequences may include, but are not limited to, promoter sequences, leader or signal sequences, ribosomal binding sites, transcriptional start and termination sequences, translational start and termination sequences, and enhancer or activator sequences.
- the promoters may be either naturally occurring promoters, or hybrid promoters that combine elements of more than one promoter.
- An expression construct may be present in a cell on an episome, such as a plasmid, or the expression construct may be inserted in a chromosome.
- the expression vector contains a selectable marker gene to allow the selection of transformed host cells. Selectable marker genes are well known in the art and will vary with the host cell used.
- the subject nucleic acid is provided in an expression vector comprising a nucleotide sequence encoding an ActRIIA polypeptide and operably linked to at least one regulatory sequence.
- Regulatory sequences are art-recognized and are selected to direct expression of the ActRIIA polypeptide. Accordingly, the term regulatory sequence includes promoters, enhancers, and other expression control elements. Exemplary regulatory sequences are described in Goeddel; Gene Expression Technology: Methods in Enzymology, Academic Press, San Diego, Calif. (1990).
- any of a wide variety of expression control sequences that control the expression of a DNA sequence when operatively linked to it may be used in these vectors to express DNA sequences encoding an ActRIIA polypeptide.
- useful expression control sequences include, for example, the early and late promoters of SV40, tet promoter, adenovirus or cytomegalovirus immediate early promoter, RSV promoters, the lac system, the trp system, the TAC or TRC system, T7 promoter whose expression is directed by T7 RNA polymerase, the major operator and promoter regions of phage lambda, the control regions for fd coat protein, the promoter for 3-phosphoglycerate kinase or other glycolytic enzymes, the promoters of acid phosphatase, e.g., Pho5, the promoters of the yeast .alpha.
- the design of the expression vector may depend on such factors as the choice of the host cell to be transformed and/or the type of protein desired to be expressed. Moreover, the vector's copy number, the ability to control that copy number and the expression of any other protein encoded by the vector, such as antibiotic markers, should also be considered.
- a recombinant nucleic acid provided herein can be produced by ligating the cloned gene, or a portion thereof, into a vector suitable for expression in either prokaryotic cells, eukaryotic cells (yeast, avian, insect or mammalian), or both.
- Expression vehicles for production of a recombinant ActRIIA polypeptide include plasmids and other vectors.
- suitable vectors include plasmids of the types: pBR322-derived plasmids, pEMBL- derived plasmids, pEX-derived plasmids, pBTac-derived plasmids and pUC-derived plasmids for expression in prokaryotic cells, such as E. coli.
- Some mammalian expression vectors contain both prokaryotic sequences to facilitate the propagation of the vector in bacteria, and one or more eukaryotic transcription units that are expressed in eukaryotic cells.
- the pcDNAI/amp, pcDNAI/neo, pRc/CMV, pSV2gpt, pSV2neo, pSV2-dhfr, pTk2, pRSVneo, pMSG, pSVT7, pko-neo and pHyg derived vectors are examples of mammalian expression vectors suitable for transfection of eukaryotic cells.
- vectors are modified with sequences from bacterial plasmids, such as pBR322, to facilitate replication and drug resistance selection in both prokaryotic and eukaryotic cells.
- bacterial plasmids such as pBR322
- derivatives of viruses such as the bovine papilloma virus (BPV-1), or Epstein-Barr virus (pHEBo, pREP-derived and p205) can be used for transient expression of proteins in eukaryotic cells.
- BBV-1 bovine papilloma virus
- pHEBo Epstein-Barr virus
- pREP-derived and p205 Epstein-Barr virus
- examples of other viral (including retroviral) expression systems can be found below in the description of gene therapy delivery systems.
- the various methods employed in the preparation of the plasmids and in transformation of host organisms are well known in the art.
- baculovirus expression systems include pVL-derived vectors (such as pVL1392, pVL1393 and pVL941), pAcUW-derived vectors (such as pAcUWl), and pBlueBac-derived vectors (such as the .beta. -gal containing pBlueBac III).
- a vector will be designed for production of the subject ActRIIA polypeptides in CHO cells, such as a Pcmv-Script vector (Stratagene, La Jolla, Calif), pcDNA4 vectors (Invitrogen, Carlsbad, Calif.) and pCI-neo vectors (Promega, Madison, Wis.).
- a vector will be designed for production of the subject ActRIIA polypeptides in CHO cells, such as a Pcmv-Script vector (Stratagene, La Jolla, Calif), pcDNA4 vectors (Invitrogen, Carlsbad, Calif.) and pCI-neo vectors (Promega, Madison, Wis.).
- the subject gene constructs can be used to cause expression of the subject ActRIIA polypeptides in cells propagated in culture, e.g., to produce proteins, including fusion proteins or variant proteins, for purification.
- This disclosure also pertains to a host cell transfected with a recombinant gene including a coding sequence (e.g., SEQ ID NO: 4 or 5) for one or more of the subject ActRIIA polypeptides.
- the host cell may be any prokaryotic or eukaryotic cell.
- an ActRIIA polypeptide provided herein may be expressed in bacterial cells such as E. coli, insect cells (e.g., using a baculovirus expression system), yeast, or mammalian cells. Other suitable host cells are known to those skilled in the art.
- a host cell transfected with an expression vector encoding an ActRIIA polypeptide can be cultured under appropriate conditions to allow expression of the ActRIIA polypeptide to occur.
- the ActRIIA polypeptide may be secreted and isolated from a mixture of cells and medium containing the ActRIIA polypeptide.
- the ActRIIA polypeptide may be retained cytoplasmically or in a membrane fraction and the cells harvested, lysed and the protein isolated.
- a cell culture includes host cells, media and other byproducts. Suitable media for cell culture are well known in the art.
- the subject ActRIIA polypeptides can be isolated from cell culture medium, host cells, or both, using techniques known in the art for purifying proteins, including ion-exchange chromatography, gel filtration chromatography, ultrafiltration, electrophoresis, immunoaffinity purification with antibodies specific for particular epitopes of the ActRIIA polypeptides and affinity purification with an agent that binds to a domain fused to the ActRIIA polypeptide (e.g., a protein A column may be used to purify an ActRIIA-Fc fusion).
- the ActRIIA ion-exchange chromatography, gel filtration chromatography, ultrafiltration, electrophoresis, immunoaffinity purification with antibodies specific for particular epitopes of the ActRIIA polypeptides and affinity purification with an agent that binds to a domain fused to the ActRIIA polypeptide (e.g., a protein A column may be used to purify an ActRIIA-Fc fusion).
- polypeptide is a fusion protein containing a domain which facilitates its purification.
- purification is achieved by a series of column chromatography steps, including, for example, three or more of the following, in any order: protein A
- ActRIIA-hFc protein was purified to a purity of >98% as determined by size exclusion chromatography and >95% as determined by SDS PAGE. This level of purity was sufficient to achieve desirable effects on bone in mice and an acceptable safety profile in mice, rats and non-human primates.
- a fusion gene coding for a purification leader sequence such as a poly-(His)/enterokinase cleavage site sequence at the N-terminus of the desired portion of the recombinant ActRIIA polypeptide, can allow purification of the expressed fusion protein by affinity chromatography using a Ni 2+ metal resin.
- the purification leader sequence can then be subsequently removed by treatment with enterokinase to provide the purified ActRIIA polypeptide (e.g., see Hochuli et al, (1987) J. Chromatography 411 : 177; and Janknecht et al, PNAS USA 88:8972).
- fusion genes are well known. Essentially, the joining of various DNA fragments coding for different polypeptide sequences is performed in accordance with conventional techniques, employing blunt-ended or stagger-ended termini for ligation, restriction enzyme digestion to provide for appropriate termini, filling-in of cohesive ends as appropriate, alkaline phosphatase treatment to avoid undesirable joining, and enzymatic ligation.
- the fusion gene can be synthesized by conventional techniques including automated DNA synthesizers.
- ActRIIA-Fc fusion protein can be expressed in stably transfected CHO-DUKX Bl 1 cells from a pAID4 vector (SV40 ori/enhancer, CMV promoter), using, for example, a tissue plasminogen leader sequence of SEQ ID NO:9.
- the Fc portion can be a human IgGl Fc sequence.
- the protein contained has, on average, between about 1.5 and 2.5 moles of sialic acid per molecule of ActRIIA-Fc fusion protein.
- the long serum half-life of an ActRIIA-Fc fusion can be 25-32 days in human patients.
- the CHO cell expressed material can have a higher affinity for activin B ligand than that reported for an ActRIIA-hFc fusion protein expressed in human 293 cells (del Re et al., J Biol Chem. 2004 Dec 17;279(51):53126-35).
- the use of the TPA leader sequence provided greater production than other leader sequences and, unlike ActRIIA-Fc expressed with a native leader, may provide a highly pure N-terminal sequence. Use of the native leader sequence may result in two major species of ActRIIA-Fc, each having a different N-terminal sequence.
- ActRIIB refers to a family of activin receptor type IIB (ActRIIB) proteins from any species and variants derived from such ActRIIB proteins by mutagenesis or other modification.
- Reference to ActRIIB herein is understood to be a reference to any one of the currently identified forms of the receptor.
- Members of the ActRIIB family are generally transmembrane proteins, composed of a ligand-binding extracellular domain with a cysteine -rich region, a transmembrane domain, and a cytoplasmic domain with predicted serine/threonine kinase activity.
- ActRIIB inhibitors to be used in the compositions and methods described herein include, without limitation, activin-binding soluble ActRIIB polypeptides; antibodies that bind to activin (particularly the activin A or B subunits, also referred to as BA or BB) and disrupt ActRIIB binding; antibodies that bind to ActRIIB and disrupt activin binding; non- antibody proteins selected for activin or ActRIIB binding; and randomized peptides selected for activin or ActRIIB binding, which can be conjugated to an Fc domain.
- two or more different proteins (or other moieties) with activin or ActRIIB binding activity may be linked together to create a bifunctional or multifunctional binding molecule that inhibits ActRIIB and thus can be used in the compositions and methods described herein include.
- activin- ActRIIB signaling axis antagonists that inhibit ActRIIB include nucleic acid aptamers, small molecules and other agents are used in the compositions and methods described herein include.
- ActRIIB polypeptide refers to polypeptides comprising any naturally occurring polypeptide of an ActRIIB family member as well as any variants thereof (including mutants, fragments, fusions, and peptidomimetic forms) that retain a useful activity.
- ActRIIB polypeptides include polypeptides derived from the sequence of any known ActRIIB receptor having a sequence at least about 80% identical to the sequence of an ActRIIB polypeptide, and optionally at least 85%, 90%, 95%, 96%, 97%, 98%, 99% or greater identity.
- an ActRIIB polypeptide may bind to and inhibit the function of an ActRIIB protein and/or activin.
- an example of an ActRIIB polypeptide includes the human ActRIIB precursor polypeptide (SEQ ID NO: 16 or SEQ ID NO:28).
- the signal peptide of the ActRIIB precursor polypeptide is located at amino acids 1 to 18; the extracellular domain is located at amino acids 19 to 134 and the potential N-linked glycosylation sites are located at amino acid positions 42 and 65.
- SEQ ID NO: 19 The nucleic acid sequence encoding the human ActRIIB precursor polypeptide of SEQ ID NO: 16 is disclosed as SEQ ID NO: 19 (SEQ ID NO: 19 provides an alanine at the codon corresponding to amino acid position 64, but could be readily modified by one of skill in the art using methods known in the art to provide an arginine at the codon corresponding to amino acid position 64 instead). See Table 1 for a description of the sequences.
- the numbering of amino acids for all of the ActRIIB -related polypeptides described herein is based on the amino acid numbering for SEQ ID NO: 16 and SEQ ID NO:28 (which only differ in the amino acid expressed at position 64), unless specifically designated otherwise.
- SEQ ID NO: 16 and SEQ ID NO:28 which only differ in the amino acid expressed at position 64
- position 79 refers to the 79 th amino acid in SEQ ID NO: 16 or SEQ ID NO:28, from which the ActRIIB polypeptide is derived.
- an ActRIIB polypeptide is described as having an alanine or an arginine at amino acid position 64, then it is to be understood that position 64 refers to the 64 th amino acid in SEQ ID NO: 16 or SEQ ID NO:28, from which the ActRIIB polypeptide is derived.
- the inhibitors of ActRIIB used in the compositions and methods described herein comprise polypeptides comprising an activin-binding domain of ActRIIB.
- the activin-binding domains of ActRIIB comprise the extracellular domain of ActRIIB, or a portion thereof.
- the extracellular domain or portion thereof of ActRIIB is soluble.
- the ActRIIB polypeptides used in the compositions and methods described herein are soluble ActRIIB polypeptides.
- the term "soluble ActRIIB polypeptide” generally refers to polypeptides comprising an extracellular domain of an ActRIIB protein, including any naturally occurring extracellular domain of an ActRIIB protein as well as any variants thereof (including mutants, fragments and peptidomimetic forms). Soluble ActRIIB polypeptides can bind to activin; however, the wild type ActRIIB protein does not exhibit significant selectivity in binding to activin versus GDF8/11. In certain embodiments, altered forms of ActRIIB with different binding properties can be used in the methods provided herein.
- exemplary soluble ActRIIB polypeptides include the extracellular domain of a human ActRIIB polypeptide (e.g., SEQ ID NOs: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, and 43).
- A64 has been demonstrated to possess a relatively low affinity for activin and GDF-11.
- an Fc fusion protein with an arginine at position 64 of the ActRIIB precursor amino acid sequence (herein referred to as "R64") has an affinity for activin and GDF-11 in the low nanomolar to high picomolar range (see, e.g., U.S. Patent Application Publication No. 20100068215, the disclosure of which is herein incorporated in its entirety).
- An ActRIIB precursor amino acid sequence with an arginine at position 64 is presented in SEQ ID NO:28.
- the ActRIIB polypeptides used in accordance with the compositions and methods described herein may comprise either (i) an alanine at the position corresponding to amino acid 64 of the ActRIIB precursor amino acid sequence, i.e., SEQ ID NO: 16; or (ii) an arginine at position 64 of the ActRIIB precursor amino acid sequence, i.e., SEQ ID NO: 28.
- the ActRIIB polypeptides used in accordance with the compositions and methods described herein may comprise an amino acid that is not alanine or arginine at the position
- An ActRIIB-Fc fusion protein containing amino acids 20-119 of SEQ ID NO: 28 i.e., SEQ ID NO:32
- ActRIIB(20-l 19)-Fc has reduced binding to GDF-11 and activin relative to an ActRIIB-Fc fusion protein containing amino acids 20-134 of SEQ ID NO: 28 (i.e., SEQ ID NO:31), "ActRIIB(20-134)-Fc", which includes the proline knot region and the complete juxtamembrane domain.
- ActRIIB-Fc fusion protein containing amino acids 20-129 of SEQ ID NO: 28, "ActRIIB(20- 129)-Fc" retains similar but somewhat reduced activity relative to the non-truncated extracellular domain of ActRIIB, even though the proline knot region is disrupted.
- ActRIIB polypeptides comprising extracellular domains that stop at amino acid 134, 133, 132, 131, 130 and 129 of SEQ ID NO: 28 (or SEQ ID NO: 16) are all expected to be active, but constructs stopping at amino acid 134 or 133 may be most active.
- the ActRIIB polypeptides used in accordance with the methods and compositions described herein may end as early as amino acid 109 (i.e., the final cysteine) of SEQ ID NO:28 (or SEQ ID NO: 16), however, forms ending at or between amino acid positions 109 and 119 of SEQ ID NO:28 (or SEQ ID NO: 16) are expected to have reduced ligand binding ability.
- Amino acid 29 of SEQ ID NO: 16 and SEQ ID NO:28 represents the initial cysteine in the ActRIIB precursor sequence. It is expected that an ActRIIB polypeptide beginning at amino acid 29 of the N-terminus of SEQ ID NO: 16 or SEQ ID NO:28, or before these amino acid positions, will retain ligand binding activity. An alanine to asparagine mutation at position 24 of SEQ ID NO: 16 or SEQ ID NO:28 introduces an N-linked glycosylation sequence without substantially affecting ligand binding.
- the active portions (i.e., ActRIIB polypeptides) of the ActRIIB precursor protein (i.e., SEQ ID NO: 16 or SEQ ID NO:28) to be used in accordance with the methods and compositions described herein will generally comprise amino acids 29-109 of SEQ ID NO: 16 or SEQ ID NO:28, and such ActRIIB polypeptides may, for example, begin at a residue corresponding to any one of amino acids 19-29 of SEQ ID NO: 16 or SEQ ID NO:28 and end at a position corresponding to any one of amino acids 109-134 of SEQ ID NO: 16 or SEQ ID NO:28.
- ActRIIB polypeptides encompassed herein include those that begin at an amino acid position from 19-29, 20-29 or 21-29 of SEQ ID NO: 16 or SEQ ID NO:28 and end at an amino acid position from 119-134, 119-133 or 129- 134, 129-133 of SEQ ID NO:16 or SEQ ID NO:28.
- ActRIIB polypeptides encompassed herein include those that begin at an amino acid position from 20- 24 (or 21-24, or 22-25) of SEQ ID NO: 16 or SEQ ID NO:28 and end at an amino acid position from 109-134 (or 109-133), 119-134 (or 119-133) or 129-134 (or 129-133) of SEQ ID NO: 16 or SEQ ID NO:28.
- Variant ActRIIB polypeptides falling within these ranges are also contemplated, particularly those having at least 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%), 96%), 97%), 98%), or 99%> sequence identity or sequence homology to the corresponding portion of SEQ ID NO: 16 or SEQ ID NO:28.
- the inhibitors of ActRIIB used in the compositions and methods described herein comprise a truncated form of an extracellular domain of ActRIIB.
- the truncation can be at the carboxy terminus and/or the amino terminus of the ActRIIB polypeptide.
- the truncation can be 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 amino acids long relative to the mature ActRIIB polypeptide extracellular domain.
- the truncation can be 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 N-terminal amino acids of the mature ActRIIB polypeptide extracellular domain.
- the truncation can be 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 C-terminal amino acids of the mature ActRIIB polypeptide extracellular domain.
- truncated forms of ActRIIB include polypeptides with amino acids 20-119; 20-128; 20-129; 20-130; 20-131; 20-132; 20-133; 20-134; 20-131; 21- 131; 22-131; 23-131; 24-131; and 25-131, wherein the amino acid positions refer to the amino acid positions in SEQ ID NO: 16 or SEQ ID NO:28.
- Additional exemplary truncated forms of ActRIIB include (i) polypeptides beginning at amino acids at any of amino acids 21-29 of SEQ ID NO: 16 or SEQ ID NO:28 (optionally beginning at 22-25 of SEQ ID NO: 16 or SEQ ID NO:28) and ending at any of amino acids 109-134 of SEQ ID NO: 16 or SEQ ID NO:28; (ii) polypeptides beginning at any of amino acids 20-29 of SEQ ID NO: 16 or SEQ ID NO:28 (optionally beginning at 22-25 of SEQ ID NO: 16 or SEQ ID NO:28) and ending at any of amino acids 109-133 of SEQ ID NO: 16 or SEQ ID NO:28; (iii) polypeptides beginning at any of amino acids 20-24 of SEQ ID NO: 16 or SEQ ID NO:28 (optionally beginning at 22-25 of SEQ ID NO: 16 or SEQ ID NO:28) and ending at any of amino acids 109-133 of SEQ ID NO: 16 or SEQ ID NO:28
- an ActRIIB polypeptides comprises, consists essentially of, or consists of, an amino acid sequence beginning at amino acid position 25 of SEQ ID NO: 16 or SEQ ID NO:28 and ending at amino acid position 131 of SEQ ID NO: 16 or SEQ ID NO:28.
- an ActRIIB polypeptide consists of, or consists essentially of, the amino acid sequence of SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43.
- any of the ActRIIB polypeptides disclosed herein may be produced as a homodimer. Any of the ActRIIB polypeptides disclosed herein may be formulated as a fusion protein having a heterologous portion that comprises a constant region from an IgG heavy chain, such as an Fc domain. Any of the ActRIIB polypeptides disclosed herein may comprise an acidic amino acid at the position corresponding to position 79 of SEQ ID NO: 16 or SEQ ID NO:28, optionally in combination with one or more additional amino acid substitutions, deletions or insertions relative to SEQ ID NO: 16 or SEQ ID NO:28.
- the inhibitors of ActRIIB used in the compositions and methods described herein comprise an extracellular domain of ActRIIB with one or more amino acid substitutions/mutations.
- an amino acid substitution/mutation can be, for example, an exchange from the leucine at amino acid position 79 of SEQ ID NO: 16 or SEQ ID NO:28 to an acidic amino acid, such as aspartic acid or glutamic acid.
- position L79 of SEQ ID NO: 16 or SEQ ID NO:28 may be altered in ActRIIB extracellular domain polypeptides to confer altered activin-myostatin (GDF-11) binding properties.
- L79A and L79P mutations reduce GDF-11 binding to a greater extent than activin binding.
- L79E and L79D mutations retain GDF-11 binding, while demonstrating greatly reduced activin binding.
- the inhibitors of ActRIIB used in the compositions and methods described herein comprise a truncated form of an ActRIIB extracellular domain that also carries an amino acid substitution, e.g., an exchange from the leucine at amino acid position 79 of SEQ ID NO: 16 or SEQ ID NO:28 to an acidic amino acid, such as aspartic acid or glutamic acid.
- the truncated form of an extracellular domain of ActRIIB polypeptide that also carries an amino acid substitution used in the compositions and methods described herein is SEQ ID NO:23.
- Forms of ActRIIB that are truncated and/or carry one or more amino acid substitutions can be linked to an Fc domain of an antibody as discussed above.
- Functionally active fragments of ActRIIB polypeptides can be obtained, for example, by screening polypeptides recombinantly produced from the corresponding fragment of the nucleic acid encoding an ActRIIB polypeptide.
- fragments can be chemically synthesized using techniques known in the art such as conventional Merrifield solid phase f-Moc or t-Boc chemistry. The fragments can be produced (recombinantly or by chemical synthesis) and tested to identify those peptidyl fragments that can function as antagonists (inhibitors) of ActRIIB protein or signaling mediated by activin.
- a functional variant of ActRIIB polypeptides can be obtained, for example, by screening libraries of modified polypeptides recombinantly produced from the corresponding mutagenized nucleic acids encoding an ActRIIB polypeptide. The variants can be produced and tested to identify those that can function as antagonists (inhibitors) of ActRIIB protein or signaling mediated by activin.
- a functional variant of the ActRIIB polypeptides comprises an amino acid sequence that is at least 75% identical to an amino acid sequence selected from SEQ ID NO:17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43.
- the functional variant has an amino acid sequence at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical to an amino acid sequence selected from SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43.
- Functional variants may be generated, for example, by modifying the structure of an ActRIIB polypeptide for such purposes as enhancing therapeutic efficacy, or stability (e.g., ex vivo shelf life and resistance to proteolytic degradation in vivo).
- modified ActRIIB polypeptides when selected to retain activin binding, are considered functional equivalents of the naturally-occurring ActRIIB polypeptides.
- Modified ActRIIB polypeptides can also be produced, for instance, by amino acid substitution, deletion, or addition.
- mutants particularly sets of combinatorial mutants of an ActRIIB polypeptide, as well as truncation mutants; pools of combinatorial mutants are especially useful for identifying functional variant sequences.
- the purpose of screening such combinatorial libraries may be to generate, for example, ActRIIB polypeptide variants which can act as either agonists or antagonist, or alternatively, which possess novel activities all together.
- the ligand binding pocket of ActRIIB is defined by residues Y31, N33, N35, L38 through T41, E47, E50, Q53 through K55, L57, H58, Y60, S62, K74, W78 through N83, Y85, R87, A92, and E94 through F101 of SEQ ID NO: 16 or SEQ ID NO:28.
- R40A, K55A, F82A and mutations at position L79 is a K in Xenopus, indicating that basic amino acids at this position will be tolerated.
- an ActRIIB polypeptide for use in the methods and compositions described herein is one that comprises amino acids 29-109 of SEQ ID NO: 16 or SEQ ID NO:28, but optionally beginning at an amino acid position ranging from 20-24 or 22-25 of SEQ ID NO: 16 or SEQ ID NO:28 and ending at an amino acid position ranging from 129- 134 of SEQ ID NO: 16 or SEQ ID NO:28, and comprising no more than 1, 2, 5, or 15 conservative amino acid changes in the ligand binding pocket, and zero, one or more non- conservative alterations at amino acid positions 40, 53, 55, 74, 79 and/or 82 of SEQ ID NO: 16 or SEQ ID NO:28 in the ligand binding pocket.
- Such an ActRIIB polypeptide may retain greater than 80%, 90%, 95%> or 99%> sequence identity or sequence homology to the sequence of amino acids 29-109 of SEQ ID NO: 16 or SEQ ID NO:28.
- Sites outside the binding pocket, at which variability may be particularly well tolerated, include the amino and carboxy termini of the extracellular domain of ActRIIB, and positions 42-46 and 65-73.
- An asparagine to alanine alteration at position 65 of SEQ ID NO: 16 or SEQ ID NO:28 (N65A) actually improves ligand binding in the A64 background, and is thus expected to have no detrimental effect on ligand binding in the R64 background.
- the positively-charged amino acid residue Asp (D80) of the ligand-binding domain of ActRIIB can be mutated to a different amino acid residue such that the variant ActRIIB polypeptide preferentially binds to GDF8, but not activin.
- the D80 residue is changed to an amino acid residue selected from the group consisting of: an uncharged amino acid residue, a negative amino acid residue, and a hydrophobic amino acid residue.
- the hydrophobic residue L79 can be altered to the acidic amino acids aspartic acid or glutamic acid to greatly reduce activin binding while retaining GDF11 binding.
- most of the described mutations, variants or modifications may be made at the nucleic acid level or, in some cases, by post translational modification or chemical synthesis. Such techniques are well known in the art.
- the inhibitors of ActRIIB used in the compositions and methods described herein comprise a conjugate/fusion protein comprising an extracellular domain (e.g., an activin-binding domain) of an ActRIIB receptor linked to an Fc portion of an antibody.
- conjugate/fusion proteins may comprise any of the ActRIIB polypeptides disclosed herein (e.g., any of SEQ ID NOs: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43), any ActRIIB polypeptides known in the art, or any ActRIIB polypeptides generated using methods known in the art and/or provided herein.
- the extracellular domain is linked to an Fc portion of an antibody via a linker, e.g. , a peptide linker.
- linkers include short polypeptide sequences such as 2-10, 2-5, 2-4, 2-3 amino acid residues (e.g., glycine residues), such as, for example, a Gly-Gly-Gly linker.
- the linker comprises the amino acid sequence Gly-Gly-Gly (GGG).
- the linker comprises the amino acid sequence Thr-Gly-Gly-Gly (TGGG).
- the Fc domain has one or more mutations at residues such as Asp-265, lysine 322, and Asn-434.
- the mutant Fc domain having one or more of these mutations has a reduced ability to bind to the Fey receptor relative to a wild-type Fc domain.
- the mutant Fc domain having one or more of these mutations has an increased ability to bind to the MHC class I- related Fc-receptor (FcRN) relative to a wild- type Fc domain.
- Exemplary fusion proteins comprising a soluble extracellular domain of ActRIIB fused to an Fc domain are set forth in SEQ ID NOs:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, and 47.
- the ActRIIB inhibitors used in the compositions and methods described herein comprise the extracellular domain of ActRIIB, or a portion thereof, linked to an Fc portion of an antibody, wherein said ActRIIB inhibitor comprises an amino acid sequence that is at least 75% identical to an amino acid sequence selected from
- the ActRIIB inhibitors used in the compositions and methods described herein comprise the extracellular domain of ActRIIB, or a portion thereof, linked to an Fc portion of an antibody, wherein said ActRIIB inhibitor comprises an amino acid sequence that is at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical to an amino acid sequence selected from SEQ ID NOs:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, and 47.
- the ActRIIB inhibitor to be used in the compositions and methods described herein is a fusion protein between the extracellular domain of the human ActRIIB receptor and the Fc portion of IgGl .
- the ActRIIB inhibitor to be used in the compositions and methods described herein is a fusion protein between a truncated extracellular domain of the human ActRIIB receptor and the Fc portion of IgGl .
- the ActRIIB inhibitor to be used in the compositions and methods described herein is a fusion protein between a truncated extracellular domain of the human ActRIIB receptor and the Fc portion of IgGl, wherein the truncated extracellular domain of the human ActRIIB receptor possesses an amino acid substitution at the amino acid position corresponding to amino acid 79 of SEQ ID NO: 16 or SEQ ID NO:28.
- the amino acid substitution at the amino acid position corresponding to amino acid 79 of SEQ ID NO: 16 or SEQ ID NO:28 is substitution of Leucine for Aspartic Acid (i.e., an L79D mutation).
- the ActRIIB inhibitor to be used in the compositions and methods described herein is SEQ ID NO:24 or 25, which represents a fusion protein between the extracellular domain of the human ActRIIB receptor and the Fc portion of IgGl, wherein said ActRIIB extracellular domain comprises amino acids 25-131 of SEQ ID NO:28 with an L79D mutation.
- the nucleic acid sequence encoding the ActRIIB-Fc fusion protein of SEQ ID NO:24 is presented in SEQ ID NO:45.
- the ActRIIB inhibitor to be used in the compositions and methods described herein is SEQ ID NO:34 or 35, which represents a fusion protein between the extracellular domain of the human ActRIIB receptor and the Fc portion of IgGl, wherein said ActRIIB extracellular domain comprises amino acids 25-131 of SEQ ID NO: 16 with an L79D mutation.
- Asparagine-linked glycosylation recognition sites generally comprise a tripeptide sequence, asparagine-X-threonine (or asparagine-X-serine) (where "X" is any amino acid) which is specifically recognized by appropriate cellular glycosylation enzymes.
- the alteration may also be made by the addition of, or substitution by, one or more serine or threonine residues to the sequence of the wild-type ActRIIB polypeptide (for O-linked glycosylation sites).
- a variety of amino acid substitutions or deletions at one or both of the first or third amino acid positions of a glycosylation recognition site (and/or amino acid deletion at the second position) results in non-glycosylation at the modified tripeptide sequence.
- Another means of increasing the number of carbohydrate moieties on an ActRIIB polypeptide is by chemical or enzymatic coupling of glycosides to the ActRIIB polypeptide.
- the sugar(s) may be attached to (a) arginine and histidine; (b) free carboxyl groups; (c) free sulfhydryl groups such as those of cysteine; (d) free hydroxyl groups such as those of serine, threonine, or hydroxyproline; (e) aromatic residues such as those of phenylalanine, tyrosine, or tryptophan; or (f) the amide group of glutamine.
- Removal of one or more carbohydrate moieties present on an ActRIIB polypeptide may be accomplished chemically and/or enzymatically.
- Chemical deglycosylation may involve, for example, exposure of the ActRIIB polypeptide to the compound trifluoromethanesulfonic acid, or an equivalent compound. This treatment results in the cleavage of most or all sugars except the linking sugar (N-acetylglucosamine or N-acetylgalactosamine), while leaving the amino acid sequence intact.
- Chemical deglycosylation is further described by Hakimuddin et al. (1987) Arch. Biochem. Biophys. 259:52 and by Edge et al. (1981) Anal. Biochem. 118: 131.
- Enzymatic cleavage of carbohydrate moieties on ActRIIB polypeptides can be achieved by the use of a variety of endo- and exo-glycosidases as described by Thotakura et al. (1987) Meth. Enzymol. 138:350.
- the sequence of an ActRIIB polypeptide may be adjusted, as appropriate, depending on the type of expression system used, as mammalian, yeast, insect and plant cells may all introduce differing glycosylation patterns that can be affected by the amino acid sequence of the peptide.
- ActRIIB proteins for use in humans will be expressed in a mammalian cell line that provides proper glycosylation, such as HEK293 or CHO cell lines, although other expression systems, such as other mammalian expression cell lines, yeast cell lines with engineered glycosylation enzymes and insect cells, are expected to be useful as well.
- mutated ActRIIB polypeptides comprising the addition of a further N-linked glycosylation site (N-X-S/T) that increases the serum half-life of an ActRIIB-Fc fusion protein, relative to the ActRIIB(R64)-Fc form.
- N-X-S/T N-linked glycosylation site
- introduction of an asparagine at position 24 of SEQ ID NO: 16 or SEQ ID NO:28 (A24N) results in the creation of an NXT sequence that confers a longer half- life.
- NX(T/S) sequences can be found at 42-44 (NQS) and 65-67 (NSS), although the latter may not be efficiently glycosylated with the R at position 64 (i.e., in R64 polypeptides).
- N- X-S/T sequences may be generally introduced at positions outside the ligand binding pocket of ActRIIB, which is detailed above. Particularly suitable sites for the introduction of non- endogenous N-X-S/T sequences include amino acids 20-29, 20-24, 22-25, 109-134, 120-134 or 129-134 of SEQ ID NO: 16 or SEQ ID NO:28.
- N-X-S/T sequences may also be introduced into the linker between the ActRIIB sequence and the Fc or other fusion component.
- Such a site may be introduced with minimal effort by introducing an N in the correct position with respect to a pre-existing S or T, or by introducing an S or T at a position corresponding to a pre-existing N.
- desirable alterations that would create an N-linked glycosylation site are: A24N, R64N, S67N (possibly combined with an N65A alteration), E106N, Rl 12N, G120N, E123N, P129N, A132N, Rl 12S and Rl 12T (with all amino acid positions corresponding to the positions they can be found in SEQ ID NO: 16 or SEQ ID NO:28).
- Any S that is predicted to be glycosylated may be altered to a T without creating an immunogenic site, because of the protection afforded by the glycosylation.
- any T that is predicted to be glycosylated may be altered to an S.
- the alterations S67T and S44T are encompassed herein.
- an S26T alteration may be used.
- an ActRIIB polypeptide may include one or more additional, non-endogenous N-linked glycosylation consensus sequences.
- an ActRIIB polypeptide variant may be screened for ability to bind to an ActRIIB ligand, to prevent binding of an ActRIIB ligand to an ActRIIB polypeptide or to interfere with signaling caused by an ActRIIB ligand.
- the activity of an ActRIIB polypeptide or its variants may also be tested in a cell-based or in vivo assay.
- Combinatorially-derived variants can be generated which have a selective or generally increased potency relative to a naturally occurring ActRIIB polypeptide.
- mutagenesis can give rise to variants which have intracellular half-lives dramatically different than the corresponding wild-type ActRIIB polypeptide.
- the altered protein can be rendered either more stable or less stable to proteolytic degradation or other cellular processes which result in destruction of, or otherwise inactivation of a native ActRIIB polypeptide.
- Such variants, and the genes which encode them can be utilized to alter ActRIIB polypeptide levels by modulating the half-life of the ActRIIB polypeptides.
- a short half-life can give rise to more transient biological effects and can allow tighter control of recombinant ActRIIB polypeptide levels within the patient.
- mutations may be made in the linker (if any) and/or the Fc portion to alter the half- life of the protein.
- a combinatorial library may be produced by way of a degenerate library of genes encoding a library of polypeptides which each include at least a portion of potential ActRIIB polypeptide sequences.
- a mixture of synthetic oligonucleotides can be enzymatically ligated into gene sequences such that the degenerate set of potential ActRIIB polypeptide nucleotide sequences are expressible as individual polypeptides, or alternatively, as a set of larger fusion proteins (e.g., for phage display).
- the library of potential homologs can be generated from a degenerate oligonucleotide sequence.
- Chemical synthesis of a degenerate gene sequence can be carried out in an automatic DNA synthesizer, and the synthetic genes then be ligated into an appropriate vector for expression.
- the synthesis of degenerate oligonucleotides is well known in the art (see for example, Narang, S A (1983) Tetrahedron 39:3; Itakura et al, (1981) Recombinant DNA, Proc. 3rd Cleveland Sympos.
- ActRIIB polypeptide variants can be generated and isolated from a library by screening using, for example, alanine scanning mutagenesis and the like (Ruf et al, (1994) Biochemistry 33: 1565-1572; Wang et al, (1994) J. Biol. Chem.
- the most widely used techniques for screening large gene libraries typically comprises cloning the gene library into replicable expression vectors, transforming appropriate cells with the resulting library of vectors, and expressing the combinatorial genes under conditions in which detection of a desired activity facilitates relatively easy isolation of the vector encoding the gene whose product was detected.
- Preferred assays include activin binding assays and activin-mediated cell signaling assays.
- ActRIIB polypeptides may further comprise post- translational modifications in addition to any that are naturally present in the ActRIIB polypeptides. Such modifications include, but are not limited to, acetylation, carboxylation, glycosylation, phosphorylation, lipidation, and acylation. As a result, the modified ActRIIB polypeptides may contain non-amino acid elements, such as polyethylene glycols, lipids, poly- or mono-saccharide, and phosphates. Effects of such non-amino acid elements on the functionality of a ActRIIB polypeptide may be tested by any method known to the skilled artisan.
- an ActRIIB polypeptide When an ActRIIB polypeptide is produced in cells by cleaving a nascent form of the ActRIIB polypeptide, post-translational processing may also be important for correct folding and/or function of the protein.
- Different cells such as CHO, HeLa, MDCK, 293, W138, NIH-3T3 or HEK293 have specific cellular machinery and characteristic mechanisms for such post-translational activities and may be chosen to ensure the correct modification and processing of the ActRIIB polypeptides.
- fusion proteins having at least a portion of the ActRIIB polypeptides and one or more fusion domains.
- fusion domains include, but are not limited to, polyhistidine, Glu-Glu, glutathione S transferase (GST), thioredoxin, protein A, protein G, an immunoglobulin heavy chain constant region (Fc), maltose binding protein (MBP), or human serum albumin.
- a fusion domain may be selected so as to confer a desired property. For example, some fusion domains are particularly useful for isolation of the fusion proteins by affinity chromatography.
- matrices for affinity chromatography such as glutathione-, amylase-, and nickel- or cobalt-conjugated resins are used.
- Many of such matrices are available in "kit” form, such as the Pharmacia GST purification system and the QIAexpressTM system (Qiagen) useful with (HIS 6 ) fusion partners.
- a fusion domain may be selected so as to facilitate detection of the ActRIIB polypeptides. Examples of such detection domains include the various fluorescent proteins (e.g., GFP) as well as "epitope tags," which are usually short peptide sequences for which a specific antibody is available.
- fusion domains have a protease cleavage site, such as for Factor Xa or Thrombin, which allows the relevant protease to partially digest the fusion proteins and thereby liberate the recombinant proteins therefrom. The liberated proteins can then be isolated from the fusion domain by subsequent chromatographic separation.
- an ActRIIB polypeptide is fused with a domain that stabilizes the ActRIIB polypeptide in vivo (a "stabilizer” domain).
- stabilizing is meant anything that increases serum half life, regardless of whether this is because of decreased destruction, decreased clearance by the kidney, or other
- Fusions with the Fc portion of an immunoglobulin are known to confer desirable pharmacokinetic properties on a wide range of proteins.
- fusions to human serum albumin can confer desirable properties.
- Other types of fusion domains that may be selected include multimerizing (e.g., dimerizing, tetramerizing) domains and functional domains (that confer an additional biological function, such as further stimulation of bone growth or muscle growth, as desired).
- an ActRIIB polypeptide may be placed C-terminal to a heterologous domain, or, alternatively, a heterologous domain may be placed C-terminal to an ActRIIB polypeptide.
- the ActRIIB polypeptide domain and the heterologous domain need not be adjacent in a fusion protein, and additional domains or amino acid sequences may be included C- or N-terminal to either domain or between the domains.
- the ActRIIB polypeptides contain one or more modifications that are capable of stabilizing the ActRIIB polypeptides.
- modifications enhance the in vitro half life of the ActRIIB polypeptides, enhance circulatory half life of the ActRIIB polypeptides or reduce proteolytic degradation of the ActRIIB polypeptides.
- Such stabilizing modifications include, but are not limited to, fusion proteins (including, for example, fusion proteins comprising an ActRIIB polypeptide and a stabilizer domain), modifications of a glycosylation site (including, for example, addition of a glycosylation site to an ActRIIB polypeptide), and modifications of carbohydrate moiety (including, for example, removal of carbohydrate moieties from an ActRIIB polypeptide).
- fusion proteins including, for example, fusion proteins comprising an ActRIIB polypeptide and a stabilizer domain
- modifications of a glycosylation site including, for example, addition of a glycosylation site to an ActRIIB polypeptide
- modifications of carbohydrate moiety including, for example, removal of carbohydrate moieties from an ActRIIB polypeptide.
- an ActRIIB polypeptide is fused to a stabilizer domain such as an IgG molecule (e.g., an Fc domain).
- stabilizer domain not only refers to a fusion domain (e.g., Fc) as in the case of fusion proteins, but also includes nonproteinaceous modifications such as a carbohydrate moiety, or nonproteinaceous polymer, such as polyethylene glycol.
- ActRIIB polypeptides which are isolated from, or otherwise substantially free of, other proteins can be used with the methods and compositions described herein. ActRIIB polypeptides will generally be produced by expression from recombinant nucleic acids.
- nucleic acids encoding any of the ActRIIB polypeptides (e.g., soluble ActRIIB polypeptides), including fragments, functional variants and fusion proteins disclosed herein.
- SEQ ID NO: 19 encodes the naturally occurring human ActRIIB precursor polypeptide.
- the subject nucleic acids may be single-stranded or double stranded.
- Such nucleic acids may be DNA or RNA molecules. These nucleic acids may be used, for example, in methods for making ActRIIB polypeptides or as direct therapeutic agents (e.g., in a gene therapy approach).
- nucleic acids encoding ActRIIB polypeptides are further understood to include nucleic acids that are variants of SEQ ID NO: 19 as well as variants of those nucleic acid sequences that encode soluble ActRIIB polypeptides (e.g., nucleic acids that encode SEQ ID NOs: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, and 43).
- Variant nucleotide sequences include sequences that differ by one or more nucleotide substitutions, additions or deletions, such as allelic variants.
- the isolated or recombinant nucleic acid sequences that can be used are at least 80%, 85%, 90%, 95%, 97%, 98%, 99% or 100% identical to SEQ ID NO: 19 or those nucleic acid sequences that encode soluble ActRIIB polypeptides (e.g., nucleic acids that encode SEQ ID NOs: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, and 43).
- nucleic acid sequences are at least 80%, 85%, 90%, 95%, 97%, 98%, 99% or 100% identical to SEQ ID NO: 19 or those nucleic acid sequences that encode soluble ActRIIB polypeptides (e.g., nucleic acids that encode SEQ ID NOs: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, and 43).
- nucleic acid sequences that encode soluble ActRIIB polypeptides can be used with the methods and compositions described herein.
- nucleic acid sequences can be isolated, recombinant, and/or fused with a heterologous nucleotide sequence, or in a DNA library.
- nucleic acids that can be used also include nucleotide sequences that hybridize under highly stringent conditions to the nucleotide sequence designated in SEQ ID NO: 19 or those nucleic acid sequences that encode soluble ActRIIB polypeptides (e.g., nucleic acids that encode SEQ ID NOs: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, and 43), complement sequence of SEQ ID NO: 19 or those nucleic acid sequences that encode soluble ActRIIB polypeptides (e.g., nucleic acids that encode SEQ ID NOs: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, and 43), or fragments thereof.
- nucleic acids that encode SEQ ID NOs: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, and 43 or fragments thereof.
- appropriate stringency conditions which promote DNA hybridization can be varied.
- appropriate stringency conditions which promote DNA hybridization can be varied. For example, one can perform the hybridization at 6.0 times sodium chloride/sodium citrate (SSC) at about 45 degree Celsius, followed by a wash of 2.0 times SSC at 50 degree Celsius.
- the salt concentration in the wash step can be selected from a low stringency of about 2.0 times SSC at 50 degree Celsius to a high stringency of about 0.2 times SSC at 50 degree Celsius.
- the temperature in the wash step can be increased from low stringency conditions at room temperature, about 22 degree Celsius, to high stringency conditions at about 65 degree Celsius.
- Both temperature and salt may be varied, or temperature or salt concentration may be held constant while the other variable is changed.
- nucleic acids which hybridize under low stringency conditions of 6 times SSC at room temperature followed by a wash at 2 times SSC at room temperature can be used with the methods and compositions described herein.
- Isolated nucleic acids which differ from the nucleic acids as set forth in SEQ ID NO: 19 or those nucleic acid sequences that encode soluble ActRIIB polypeptides (e.g., nucleic acids that encode SEQ ID NOs: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, and 43) due to degeneracy in the genetic code can also be used.
- a number of amino acids are designated by more than one triplet. Codons that specify the same amino acid, or synonyms (for example, CAU and CAC are synonyms for histidine) may result in "silent" mutations which do not affect the amino acid sequence of the protein.
- DNA sequence polymorphisms that do lead to changes in the amino acid sequences of the subject proteins will exist among mammalian cells.
- these variations in one or more nucleotides (up to about 3-5% of the nucleotides) of the nucleic acids encoding a particular protein may exist among individuals of a given species due to natural allelic variation. Any and all such nucleotide variations and resulting amino acid polymorphisms can be used with the methods and compositions described herein.
- the recombinant nucleic acids may be operably linked to one or more regulatory nucleotide sequences in an expression construct. Regulatory nucleotide sequences will generally be appropriate to the host cell used for expression.
- said one or more regulatory nucleotide sequences may include, but are not limited to, promoter sequences, leader or signal sequences, ribosomal binding sites, transcriptional start and termination sequences, translational start and termination sequences, and enhancer or activator sequences.
- promoters as known in the art can be used with the methods and compositions described herein.
- the promoters may be either naturally occurring promoters, or hybrid promoters that combine elements of more than one promoter.
- An expression construct may be present in a cell on an episome, such as a plasmid, or the expression construct may be inserted in a chromosome.
- the expression vector contains a selectable marker gene to allow the selection of transformed host cells. Selectable marker genes are well known in the art and will vary with the host cell used.
- the subject nucleic acid is provided in an expression vector comprising a nucleotide sequence encoding an ActRIIB polypeptide and operably linked to at least one regulatory sequence.
- Regulatory sequences are art-recognized and are selected to direct expression of the ActRIIB polypeptide. Accordingly, the term regulatory sequence includes promoters, enhancers, and other expression control elements. Exemplary regulatory sequences are described in Goeddel; Gene Expression Technology: Methods in Enzymology, Academic Press, San Diego, Calif. (1990).
- any of a wide variety of expression control sequences that control the expression of a DNA sequence when operatively linked to it may be used in these vectors to express DNA sequences encoding an ActRIIB polypeptide.
- useful expression control sequences include, for example, the early and late promoters of SV40, tet promoter, adenovirus or cytomegalovirus immediate early promoter, RSV promoters, the lac system, the trp system, the TAC or TRC system, T7 promoter whose expression is directed by T7 RNA polymerase, the major operator and promoter regions of phage lambda, the control regions for fd coat protein, the promoter for 3-phosphoglycerate kinase or other glycolytic enzymes, the promoters of acid phosphatase, e.g., Pho5, the promoters of the yeast .alpha.
- the design of the expression vector may depend on such factors as the choice of the host cell to be transformed and/or the type of protein desired to be expressed. Moreover, the vector's copy number, the ability to control that copy number and the expression of any other protein encoded by the vector, such as antibiotic markers, should also be considered.
- a recombinant nucleic acid can be produced by ligating the cloned gene, or a portion thereof, into a vector suitable for expression in either prokaryotic cells, eukaryotic cells (yeast, avian, insect or mammalian), or both.
- Expression vehicles for production of a recombinant ActRIIB polypeptide include plasmids and other vectors.
- suitable vectors include plasmids of the types: pBR322-derived plasmids, pEMBL-derived plasmids, pEX-derived plasmids, pBTac-derived plasmids and pUC-derived plasmids for expression in prokaryotic cells, such as E. coli.
- Some mammalian expression vectors contain both prokaryotic sequences to facilitate the propagation of the vector in bacteria, and one or more eukaryotic transcription units that are expressed in eukaryotic cells.
- the pcDNAI/amp, pcDNAI/neo, pRc/CMV, pSV2gpt, pSV2neo, pSV2-dhfr, pTk2, pRSVneo, pMSG, pSVT7, pko-neo and pHyg derived vectors are examples of mammalian expression vectors suitable for transfection of eukaryotic cells.
- vectors are modified with sequences from bacterial plasmids, such as pBR322, to facilitate replication and drug resistance selection in both prokaryotic and eukaryotic cells.
- bacterial plasmids such as pBR322
- derivatives of viruses such as the bovine papilloma virus (BPV-1), or Epstein-Barr virus (pHEBo, pREP-derived and p205) can be used for transient expression of proteins in eukaryotic cells.
- BBV-1 bovine papilloma virus
- pHEBo Epstein-Barr virus
- pREP-derived and p205 Epstein-Barr virus
- examples of other viral (including retroviral) expression systems can be found below in the description of gene therapy delivery systems.
- the various methods employed in the preparation of the plasmids and in transformation of host organisms are well known in the art.
- baculovirus expression systems include pVL-derived vectors (such as pVL1392, pVL1393 and pVL941), pAcUW-derived vectors (such as pAcUWl), and pBlueBac-derived vectors (such as the .beta. -gal containing pBlueBac III).
- a vector will be designed for production of the subject ActRIIB polypeptides in CHO cells, such as a Pcmv-Script vector (Stratagene, La Jolla, Calif), pcDNA4 vectors (Invitrogen, Carlsbad, Calif.) and pCI-neo vectors (Promega, Madison, Wis.).
- a vector will be designed for production of the subject ActRIIB polypeptides in CHO cells, such as a Pcmv-Script vector (Stratagene, La Jolla, Calif), pcDNA4 vectors (Invitrogen, Carlsbad, Calif.) and pCI-neo vectors (Promega, Madison, Wis.).
- the subject gene constructs can be used to cause expression of the subject ActRIIB polypeptides in cells propagated in culture, e.g., to produce proteins, including fusion proteins or variant proteins, for purification.
- This disclosure also pertains to a host cell transfected with a recombinant gene including a coding sequence (e.g., SEQ ID NO: 19 or those nucleic acid sequences that encode soluble ActRIIB polypeptides (e.g., nucleic acids that encode SEQ ID NOs: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, and 43)) for one or more of the subject ActRIIB polypeptides.
- the host cell may be any prokaryotic or eukaryotic cell.
- an ActRIIB polypeptide may be expressed in bacterial cells such as E. coli, insect cells (e.g., using a baculovirus expression system), yeast, or mammalian cells. Other suitable host cells are known to those skilled in the art.
- a host cell transfected with an expression vector encoding an ActRIIB polypeptide can be cultured under appropriate conditions to allow expression of the ActRIIB polypeptide to occur.
- the ActRIIB polypeptide may be secreted and isolated from a mixture of cells and medium containing the ActRIIB polypeptide.
- the ActRIIB polypeptide may be retained cytoplasmically or in a membrane fraction and the cells harvested, lysed and the protein isolated.
- a cell culture includes host cells, media and other byproducts. Suitable media for cell culture are well known in the art.
- the subject ActRIIB polypeptides can be isolated from cell culture medium, host cells, or both, using techniques known in the art for purifying proteins, including ion-exchange chromatography, gel filtration chromatography, ultrafiltration, electrophoresis, immunoaffinity purification with antibodies specific for particular epitopes of the ActRIIB polypeptides and affinity purification with an agent that binds to a domain fused to the ActRIIB polypeptide (e.g., a protein A column may be used to purify an ActRIIB-Fc fusion).
- the ActRIIB e.g., a protein A column may be used to purify an ActRIIB-Fc fusion.
- polypeptide is a fusion protein containing a domain which facilitates its purification.
- purification is achieved by a series of column chromatography steps, including, for example, three or more of the following, in any order: protein A
- ActRIIB -hFc protein was purified to a purity of >98% as determined by size exclusion chromatography and >95% as determined by SDS PAGE. This level of purity was sufficient to achieve desirable effects on bone in mice and an acceptable safety profile in mice, rats and non-human primates.
- a fusion gene coding for a purification leader sequence such as a poly-(His)/enterokinase cleavage site sequence at the N-terminus of the desired portion of the recombinant ActRIIB polypeptide, can allow purification of the expressed fusion protein by affinity chromatography using a Ni 2+ metal resin.
- the purification leader sequence can then be subsequently removed by treatment with enterokinase to provide the purified ActRIIB polypeptide (e.g., see Hochuli et al, (1987) J. Chromatography 411 : 177; and Janknecht et al, PNAS USA 88:8972).
- fusion genes are well known. Essentially, the joining of various DNA fragments coding for different polypeptide sequences is performed in accordance with conventional techniques, employing blunt-ended or stagger-ended termini for ligation, restriction enzyme digestion to provide for appropriate termini, filling-in of cohesive ends as appropriate, alkaline phosphatase treatment to avoid undesirable joining, and enzymatic ligation.
- the fusion gene can be synthesized by conventional techniques including automated DNA synthesizers.
- PCR amplification of gene fragments can be carried out using anchor primers which give rise to complementary overhangs between two consecutive gene fragments which can subsequently be annealed to generate a chimeric gene sequence (see, for example, Current Protocols in Molecular Biology, eds. Ausubel et al, John Wiley & Sons: 1992).
- ActRIIB -Fc fusion protein can be expressed in stably transfected CHO-DUKX Bl
- the Fc portion can comprise a human IgGl Fc sequence, as shown in SEQ ID NO:7.
- the protein contained upon expression, has, on average, between about 1.5 and 2.5 moles of sialic acid per molecule of ActRIIB-Fc fusion protein.
- the long serum half-life of an ActRIIB-Fc fusion can be 25-32 days in human patients.
- the CHO cell expressed material can have a higher affinity for activin B ligand than that reported for an ActRIIB-hFc fusion protein expressed in human 293 cells (del Re et al., J Biol Chem. 2004 Dec 17;279(51):53126-35).
- the use of the TPA leader sequence provided greater production than other leader sequences and, unlike ActRIIB-Fc expressed with a native leader, may provide a highly pure N-terminal sequence. Use of the native leader sequence may result in two major species of ActRIIB-Fc, each having a different N-terminal sequence.
- compositions and methods described herein are nucleic acid compounds.
- nucleic acid compounds that inhibit ActRII receptors include antisense nucleic acids, siRNA or RNAi constructs and catalytic nucleic acid constructs.
- a nucleic acid compound may be single- or double-stranded.
- a double-stranded compound may also include regions of overhang or non-complementarity, where one or the other of the strands is single-stranded.
- a single-stranded compound may include regions of self-complementarity, meaning that the compound may form a so-called "hairpin” or "stem- loop” structure, with a region of double helical structure.
- the nucleic acid compounds that inhibit ActRII receptors may comprise a nucleotide sequence that is complementary to a region consisting of no more than 1000, no more than 500, no more than 250, no more than 100 or no more than 50, 35, 30, 25, 22, 20 or 18 nucleotides of the full-length ActRII receptor nucleic acid sequence or activin nucleic acid sequence (e.g., the nucleic acid sequence of an activin A or activin B subunit, also referred to as BA or B B ).
- complementarity will be at least 8 nucleotides, and optionally at least 10 or at least 15 nucleotides, and optionally between 15 and 25 nucleotides.
- a region of complementarity may fall within an intron, a coding sequence or a noncoding sequence of the target transcript, such as the coding sequence portion.
- a nucleic acid compound that inhibits an ActRII receptor will have a length of about 8 to about 500 nucleotides or base pairs in length, and optionally the length will be about 14 to about 50 nucleotides.
- a nucleic acid compound that inhibits an ActRII receptor may be a DNA (particularly for use as an antisense), an RNA, or an RNA:DNA hybrid.
- Any one strand may include a mixture of DNA and RNA, as well as modified forms that cannot readily be classified as either DNA or RNA.
- a double stranded nucleic acid compound may be DNA:DNA, DNA:RNA, or RNA:RNA, and any one strand may also include a mixture of DNA and RNA, as well as modified forms that cannot readily be classified as either DNA or RNA.
- the nucleic acid compounds that inhibit an ActRII receptor may include any of a variety of modifications, including one or modifications to the backbone (the sugar-phosphate portion in a natural nucleic acid, including internucleotide linkages) or the base portion (the purine or pyrimidine portion of a natural nucleic acid).
- an antisense nucleic acid compound will have a length of about 15 to about 30 nucleotides and will often contain one or more modifications to improve certain characteristics, such as stability in the serum, stability in in a cell, or stability in in a place where the compound is likely to be delivered, such as, e.g., the stomach in the case of orally delivered compounds and the lung for inhaled compounds.
- the strand complementary to the target transcript will generally be RNA or modifications thereof.
- the other strand may be RNA, DNA, or any other variation.
- the duplex portion of double stranded or single stranded "hairpin" RNAi construct may, in certain embodiments, have a length of 18 to 40 nucleotides in length and optionally about 21 to 23 nucleotides in length, so long as it serves as a Dicer substrate.
- Catalytic or enzymatic nucleic acids may be ribozymes or DNA enzymes and may also contain modified forms.
- nucleic acid compounds that inhibit ActRII receptors may inhibit expression of their target by about 50%, 60%>, 70%>, 75%, 80%>, 85%), 90%), 95%o, 99%o, or more under physiological conditions and at a concentration where a nonsense or sense control has little or no effect.
- Concentrations for testing the effect of nucleic acid compounds include 1, 5, 10 micromolar, or more.
- the inhibitors of ActRII receptors used in the compositions and methods described herein are antibodies.
- Such antibodies include antibodies that bind to activin (particularly the activin A or B subunits, also referred to as BA or B B ) and disrupt ActRII receptor binding; and antibodies that bind to ActRII receptor polypeptides (e.g., a soluble ActRIIA or soluble ActRIIB polypeptide) and disrupt activin binding.
- anti-protein/anti-peptide antisera or monoclonal antibodies can be made by standard protocols (see, for example, Antibodies: A Laboratory Manual ed. by Harlow and Lane (Cold Spring Harbor Press: 1988)).
- a mammal such as a mouse, a hamster or rabbit can be immunized with an immunogenic form of the ActRII receptor polypeptide, an antigenic fragment which is capable of eliciting an antibody response, or a fusion protein.
- Techniques for conferring immunogenicity on a protein or peptide include conjugation to carriers or other techniques well known in the art.
- An immunogenic portion of an ActRII receptor or activin polypeptide can be administered in the presence of adjuvant. The progress of immunization can be monitored by detection of antibody titers in plasma or serum.
- Standard ELISA or other immunoassays can be used with the immunogen as antigen to assess the levels of antibodies.
- antisera can be obtained and, if desired, polyclonal antibodies can be isolated from the serum.
- antibody-producing cells lymphocytes
- myeloma cells can be harvested from an immunized animal and fused by standard somatic cell fusion procedures with immortalizing cells such as myeloma cells to yield hybridoma cells.
- Hybridoma cells can be screened immunochemically for production of antibodies specifically reactive with an ActRII receptor polypeptide and monoclonal antibodies isolated from a culture comprising such hybridoma cells.
- antibody as used herein is intended to include fragments thereof which are also specifically reactive with a subject polypeptide. Antibodies can be fragmented using conventional techniques and the fragments screened for utility in the same manner as described above for whole antibodies. For example, F(ab) 2 fragments can be generated by treating antibody with pepsin. The resulting F(ab) 2 fragment can be treated to reduce disulfide bridges to produce Fab fragments.
- An antibody is further intended to include bispecific, single-chain, chimeric, humanized and fully human molecules having affinity for an ActRII receptor or activin polypeptide conferred by at least one CDR region of the antibody.
- An antibody may further comprise a label attached thereto and able to be detected (e.g., the label can be a radioisotope, fluorescent compound, enzyme or enzyme co-factor).
- the antibody is a recombinant antibody, which term encompasses any antibody generated in part by techniques of molecular biology, including CDR-grafted or chimeric antibodies, human or other antibodies assembled from library- selected antibody domains, single chain antibodies and single domain antibodies (e.g., human V H proteins or camelid V HH proteins).
- an antibody can be a monoclonal antibody, and in certain embodiments.
- a method for generating a monoclonal antibody that binds specifically to an ActRII receptor polypeptide or activin polypeptide may comprise administering to a mouse an amount of an immunogenic composition comprising the antigen polypeptide effective to stimulate a detectable immune response, obtaining antibody-producing cells (e.g., cells from the spleen) from the mouse and fusing the antibody-producing cells with myeloma cells to obtain antibody-producing hybridomas, and testing the antibody-producing hybridomas to identify a hybridoma that produces a monocolonal antibody that binds specifically to the antigen.
- antibody-producing cells e.g., cells from the spleen
- a hybridoma can be propagated in a cell culture, optionally in culture conditions where the hybridoma-derived cells produce the monoclonal antibody that binds specifically to the antigen.
- the monoclonal antibody may be purified from the cell culture.
- the adjective "specifically reactive with” as used in reference to an antibody is intended to mean, as is generally understood in the art, that the antibody is sufficiently selective between the antigen of interest (e.g., an ActRII receptor polypeptide) and other antigens that are not of interest that the antibody is useful for, at minimum, detecting the presence of the antigen of interest in a particular type of biological sample. In certain methods employing the antibody, such as therapeutic applications, a higher degree of specificity in binding may be desirable. Monoclonal antibodies generally have a greater tendency (as compared to polyclonal antibodies) to discriminate effectively between the desired antigens and cross-reacting polypeptides.
- the antigen of interest e.g., an ActRII receptor polypeptide
- Monoclonal antibodies generally have a greater tendency (as compared to polyclonal antibodies) to discriminate effectively between the desired antigens and cross-reacting polypeptides.
- One characteristic that influences the specificity of an antibody:antigen interaction is the affinity of the antibody for the antigen. Although the desired specificity may be reached with a range of different affinities, generally preferred antibodies will have an affinity (a dissociation constant) of about 10 ⁇ 6 , 10 ⁇ 7 , 10 ⁇ 8 , 10 " 9 or less. Given the extraordinarily tight binding between activin and an ActRII receptor, it is expected that a neutralizing anti-activin or anti- ActRII receptor antibody would generally have a dissociation constant of 10 ⁇ 10 or less.
- the techniques used to screen antibodies in order to identify a desirable antibody may influence the properties of the antibody obtained. For example, if an antibody is to be used for binding an antigen in solution, it may be desirable to test solution binding.
- a variety of different techniques are available for testing interaction between antibodies and antigens to identify particularly desirable antibodies. Such techniques include ELISAs, surface plasmon resonance binding assays (e.g., the Biacore.TM. binding assay, Biacore AB, Uppsala, Sweden), sandwich assays (e.g., the paramagnetic bead system of IGEN International, Inc., Gaithersburg, Md.), Western blots, immunoprecipitation assays, and immunohistochemistry.
- ActRII receptor inhibitors to be used in the compositions and methods described herein include alternative forms of activin, particularly those with alterations in the type I receptor binding domain can bind to type II receptors and fail to form an active ternary complex.
- nucleic acids such as antisense molecules, siRNAs or ribozymes that inhibit activin A, B, C or E, or, particularly, ActRII receptor expression, can be used in the compositions and methods described herein.
- the ActRII receptor inhibitors to be used in the compositions and methods described herein exhibit selectivity for inhibiting activin-mediated signaling versus other members of the TGF-beta family, particularly with respect to GDF8 and GDF11.
- the inhibitors of ActRII receptors used in the compositions and methods described herein are non-antibody proteins with ActRII receptor antagonist activity, including inhibin (i.e., inhibin alpha subunit), follistatin (e.g., follistatin-288 and follistatin-315), Cerberus, follistatin related protein ("FSRP"), endoglin, activin C, alpha(2)- macroglobulin, and an Ml 08 A (methionine to alanine change at position 108) mutant activin A.
- inhibin i.e., inhibin alpha subunit
- follistatin e.g., follistatin-288 and follistatin-315
- Cerberus Cerberus
- follistatin related protein follistatin related protein
- endoglin activin C
- alpha(2)- macroglobulin alpha(2)- macroglobulin
- Ml 08 A methionine to alanine change at position 10
- the ActRII receptor inhibitor to be used in the compositions and methods described herein is a follistatin polypeptide that antagonizes activin bioactivity and/or binds to activin.
- follistatin polypeptide includes polypeptides comprising any naturally occurring polypeptide of follistatin as well as any variants thereof (including mutants, fragments, fusions, and peptidomimetic forms) that retain a useful activity, and further includes any functional monomer or multimer of follistatin. Variants of follistatin polypeptides that retain activin binding properties can be identified based on previous studies involving follistatin and activin interactions.
- WO2008/030367 discloses specific follistatin domains ("FSDs") that are shown to be important for activin binding.
- Follistatin polypeptides include polypeptides derived from the sequence of any known follistatin having a sequence at least about 80% identical to the sequence of a follistatin polypeptide, and optionally at least 85%, 90%, 95%, 96%, 97%, 98%, 99% or greater identity.
- follistatin polypeptides include the mature follistatin polypeptide or shorter isoforms or other variants of the human follistatin precursor polypeptide as described, for example, in
- the ActRII receptor inhibitor to be used in the compositions and methods described herein is a follistatin-like related gene (FLRG) that antagonizes activin bioactivity and/or binds to activin.
- FLRG polypeptide includes polypeptides comprising any naturally occurring polypeptide of FLRG as well as any variants thereof (including mutants, fragments, fusions, and peptidomimetic forms) that retain a useful activity. Variants of FLRG polypeptides that retain activin binding properties can be identified using routine methods to assay FLRG and activin interactions. See, for example, U.S. Pat. No. 6,537,966, which is included by reference herein in its entirety.
- FLRG polypeptides include polypeptides derived from the sequence of any known FLRG having a sequence at least about 80%> identical to the sequence of an FLRG polypeptide, and optionally at least 85%, 90%, 95%, 96%, 97%, 98%, 99% or greater identity.
- follistatin polypeptides and FLRG polypeptides include fusion proteins having at least a portion of the follistatin polypeptides or FLRG polypeptides and one or more fusion domains, such as, for example, domains that facilitate isolation, detection, stabilization or multimerization of the polypeptide. Suitable fusion domains are discussed in detail above with reference to the ActRII A and ActRIIB polypeptides.
- an ActRII receptor inhibitor is a fusion protein comprising an activin binding portion of a follistaton polypeptide fused to an Fc domain.
- an ActRII receptor inhibitor is a fusion protein comprising an activin binding portion of an FLRG polypeptide fused to an Fc domain.
- second active agent can be used as second active agent with the methods and compositions of the present invention. See subsection (a) below.
- Other second active agents that can be used with the methods and formulations of the present invention include thalidomide,
- low-dose dexamethasone (see Rajkumar et al. 2010, The Lancet 11 :29-37) is combined with an Activin- ActRII inhibitor.
- a third active agent can be used with the methods and formulations.
- lenalidomide also known as Revlimid® or 3-(4-amino-l-oxo-l,3-dihydro-isoindol-2-yl)-piperidine-2,6-dione
- dexamethasone melphalan
- prednisone bortezomib
- cyclophosphamide bisphosphonate
- bisphosphonate bisphosphonate
- azacytidine is combined with an Activin-ActRII inhibitor.
- a combination of Revlimid®, dexamethasone, bisphosonate, and an ActRII inhibitor is used with the methods of the invention (e.g., to treat multiple myeloma).
- the invention also provides a pharmaceutical composition comprising Revlimid®, dexamethasone, bisphosonate, and an ActRII inhibitor.
- a combination of Revlimid®, melphalan, prednisone, and an ActRII inhibitor is used with the methods of the invention (e.g., to treat multiple myeloma).
- the invention also provides a pharmaceutical composition comprising
- Revlimid® dexamethasone, bisphosonate, and an ActRII inhibitor.
- lenalidomide is combined with doxorubicin (Doxil®), vincristine and/or dexamethasone (Decadron®), and an ActRII inhibitor.
- Second active agents can either be commercially purchased or prepared according to the methods described in the patents or patent publications disclosed herein. Further, optically pure compounds can be asymmetrically synthesized or resolved using known resolving agents or chiral columns as well as other standard synthetic organic chemistry techniques.
- the term "pharmaceutically acceptable salt” encompasses non-toxic acid and base addition salts of the compound to which the term refers.
- Acceptable non-toxic acid addition salts include those derived from organic and inorganic acids or bases know in the art, which include, for example, hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid, methanesulphonic acid, acetic acid, tartaric acid, lactic acid, succinic acid, citric acid, malic acid, maleic acid, sorbic acid, aconitic acid, salicylic acid, phthalic acid, embolic acid, enanthic acid, and the like.
- bases that can be used to prepare pharmaceutically acceptable base addition salts of such acidic compounds are those that form non-toxic base addition salts, i.e., salts containing pharmacologically acceptable cations such as, but not limited to, alkali metal or alkaline earth metal salts and the calcium, magnesium, sodium or potassium salts in particular.
- Suitable organic bases include, but are not limited to,
- ⁇ , ⁇ -dibenzylethylenediamine chloroprocaine, choline, diethanolamine, ethylenediamine, meglumaine (N-methylglucamine), lysine, and procaine.
- prodrug means a derivative of a compound that can hydrolyze, oxidize, or otherwise react under biological conditions (in vitro or in vivo) to provide the compound.
- prodrugs include, but are not limited to, derivatives of immunomodulatory compounds that comprise
- biohydrolyzable moieties such as biohydrolyzable amides, biohydrolyzable esters, biohydrolyzable carbamates, biohydrolyzable carbonates, biohydrolyzable ureides, and biohydrolyzable phosphate analogues.
- Other examples of prodrugs include derivatives of immunomodulatory compounds that comprise -NO, -N0 2 , -ONO, or -ON0 2 moieties.
- Prodrugs can typically be prepared using well-known methods, such as those described in 1 Burger 's Medicinal Chemistry and Drug Discovery, 172-178, 949-982 (Manfred E. Wolff ed., 5th ed. 1995), and Design of Prodrugs (H. Bundgaard ed., Elselvier, New York 1985).
- biohydrolyzable amide As used herein and unless otherwise indicated, the terms “biohydrolyzable amide,” “biohydrolyzable ester,” “biohydrolyzable carbamate,” “biohydrolyzable carbonate,”
- biohydrolyzable ureide means an amide, ester, carbamate, carbonate, ureide, or phosphate, respectively, of a compound that either: 1) does not interfere with the biological activity of the compound but can confer upon that compound
- biohydrolyzable esters include, but are not limited to, lower alkyl esters, lower acyloxyalkyl esters (such as acetoxylmethyl, acetoxyethyl, aminocarbonyloxymethyl, pivaloyloxymethyl, and pivaloyloxyethyl esters), lactonyl esters (such as phthalidyl and thiophthalidyl esters), lower alkoxy acyloxyalkyl esters (such as methoxycarbonyl-oxymethyl, ethoxycarbonyloxyethyl and isopropoxycarbonyloxyethyl esters), alkoxyalkyl esters, choline esters, and acylamino alkyl esters (such as acetamidomethyl esters).
- lower alkyl esters such as acetoxylmethyl, acetoxyethyl, aminocarbonyloxymethyl, pivaloyloxymethyl, and pivaloyloxyethyl
- biohydrolyzable amides include, but are not limited to, lower alkyl amides, a-amino acid amides, alkoxyacyl amides, and alkylaminoalkylcarbonyl amides. Examples of
- biohydrolyzable carbamates include, but are not limited to, lower alkylamines, substituted ethylenediamines, amino acids, hydroxy alkylamines, heterocyclic and heteroaromatic amines, and polyether amines.
- Various immunomodulatory compounds contain one or more chiral centers, and can exist as racemic mixtures of enantiomers or mixtures of diastereomers. Stereomerically pure forms of such compounds, as well as the use of mixtures of those forms, can be used with the methods and compositions of the present invention. For example, mixtures comprising equal or unequal amounts of the enantiomers of a particular immunomodulatory compounds may be used in methods and compositions of the invention. These isomers may be asymmetrically synthesized or resolved using standard techniques such as chiral columns or chiral resolving agents. See, e.g., Jacques, J., et al., Enantiomers, Racemates and
- stereomerically pure means a composition that comprises one stereoisomer of a compound and is substantially free of other stereoisomers of that compound.
- a stereomerically pure composition of a compound having one chiral center will be substantially free of the opposite enantiomer of the compound.
- a stereomerically pure composition of a compound having two chiral centers will be substantially free of other diastereomers of the compound.
- a typical stereomerically pure compound comprises greater than about 80% by weight of one stereoisomer of the compound and less than about 20% by weight of other stereoisomers of the compound, more preferably greater than about 90% by weight of one stereoisomer of the compound and less than about 10%> by weight of the other stereoisomers of the compound, even more preferably greater than about 95% by weight of one stereoisomer of the compound and less than about 5% by weight of the other stereoisomers of the compound, and most preferably greater than about 97% by weight of one stereoisomer of the compound and less than about 3% by weight of the other stereoisomers of the compound.
- stereomerically enriched means a composition that comprises greater than about 60% by weight of one stereoisomer of a compound, preferably greater than about 70% by weight, more preferably greater than about 80% by weight of one stereoisomer of a compound.
- stereoisomer preferably greater than about 70% by weight, more preferably greater than about 80% by weight of one stereoisomer of a compound.
- enantiomerically pure means a stereomerically pure composition of a compound having one chiral center.
- stereomerically enriched means a stereomerically enriched composition of a compound having one chiral center.
- IMiD® compounds include, but are not limited to, the substituted 2-(2,6-dioxopiperidin-3-yl) phthalimides and substituted 2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindoles described in United States Patent Nos. 6,281,230 and 6,316,471, both to G.W. Muller, et al.
- the IMiD® compounds that can be used with the methods and compositions of the present invention is Lenalidomide (Revlimid®; 3-(4-amino- l-oxo-l,3-dihydro-isoindol-2-yl)-piperidine-2,6-dione).
- the IMiD® compound is pomalidomide (4-(amino)-2-(2,6-dioxo(3-piperidyl))-isoindoline-l,3-dione).
- the IMiD® compound is 3-(5-amino-2-methyl-4-oxo-4H-quinazolin-3- yl)-piperidine-2,6-dione.
- Compounds used with the present invention include immunomodulatory compounds that are racemic, stereomerically enriched or stereomerically pure, and pharmaceutically acceptable salts, solvates, hydrates, stereoisomers, clathrates, and prodrugs thereof.
- Preferred compounds used with the methods and compositions of the invention are small organic molecules having a molecular weight less than about 1 ,000 g/mol, and are not proteins, peptides, oligonucleotides, oligosaccharides or other macromolecules.
- immunomodulatory compounds and “IMiDs®” compounds (Celgene Corporation) encompasses small organic molecules that markedly inhibit TNF-a, LPS induced monocyte IL1B and IL12, and partially inhibit IL6 production. Specific immunomodulatory compounds are discussed below.
- TNF-a is an inflammatory cytokine produced by macrophages and monocytes during acute inflammation. TNF-a is responsible for a diverse range of signaling events within cells. TNF-a may play a pathological role in cancer. Without being limited by theory, one of the biological effects exerted by the immunomodulatory compounds is the reduction of synthesis of TNF-a.
- immunomodulatory compounds enhance the degradation of TNF-a mRNA.
- immunomodulatory compounds used in the invention may also be potent co-stimulators of T cells and increase cell proliferation dramatically in a dose dependent manner. Immunomodulatory compounds may also have a greater co-stimulatory effect on the CD8+ T cell subset than on the CD4+ T cell subset. In addition, the compounds preferably have anti-inflammatory properties, and efficiently co- stimulate T cells.
- immunomodulatory compounds include, but are not limited to, cyano and carboxy derivatives of substituted styrenes such as those disclosed in U.S. patent no. 5,929, 1 17; l-oxo-2-(2,6-dioxo-3-fiuoropiperidin-3yl) isoindolines and 1 ,3-dioxo-
- 4-methyl derivatives of thalidomide and EM- 12 including, but not limited to, those disclosed in U.S. patent no. 5,635,517; and a class of non-polypeptide cyclic amides disclosed in U.S. patent nos. 5,698,579 and 5,877,200; analogs and derivatives of thalidomide, including hydrolysis products, metabolites, derivatives and precursors of thalidomide, such as those described in U.S. patent nos.
- immunomodulatory compounds include, but are not limited to:
- R 1 is hydrogen or methyl.
- the enantiomerically pure forms e.g. optically pure (R) or (S) enantiomers
- R optically pure
- S S enantiomers
- R 1 is H, (Ci-C 8 )alkyl, (C 3 -C 7 )cycloalkyl, (C 2 -C 8 )alkenyl, (C 2 -C 8 )alkynyl, benzyl, aryl, (C 0 -C 4 )alkyl-(Ci-C 6 )heterocycloalkyl, (C 0 -C 4 )alkyl-(C 2 -C 5 )heteroaryl, C(0)R 3 , C(S)R 3 , C(0)OR 4 , (Ci-C 8 )alkyl-N(R 6 ) 2 , (C C 8 )alkyl-OR 5 , (C C 8 )alkyl-C(0)OR 5 , C(0)NHR 3 , C(S)NHR 3 , C(0)NR 3 R 3' , C(S)NR 3 R 3' or (Ci-C 8 )alkyl-0(CO)R 5
- R 2 is H, F, benzyl, (Ci-C 8 )alkyl, (C 2 -C 8 )alkenyl, or (C 2 -C 8 )alkynyl;
- R 3 and R 3' are independently (Ci-C 8 )alkyl, (C 3 -C 7 )cycloalkyl, (C 2 -C 8 )alkenyl, (C 2 - C 8 )alkynyl, benzyl, aryl, (C 0 -C 4 )alkyl-(Ci-C 6 )heterocycloalkyl, (C 0 -C 4 )alkyl-(C 2 - C 5 )heteroaryl, (C 0 -C 8 )alkyl-N(R 6 ) 2 , (Ci-C 8 )alkyl-OR 5 , (Ci-C 8 )alkyl-C(0)OR 5 , (Ci-C 8 )alkyl- 0(CO)R 5 , or C(0)OR 5 ;
- R 4 is (Ci-C 8 )alkyl, (C 2 -C 8 )alkenyl, (C 2 -C 8 )alkynyl, (Ci-C 4 )alkyl-OR 5 , benzyl, aryl, (C 0 -C 4 )alkyl-(Ci-C 6 )heterocycloalkyl, or (C 0 -C 4 )alkyl-(C 2 -C 5 )heteroaryl;
- R 5 is (Ci-C 8 )alkyl, (C 2 -C 8 )alkenyl, (C 2 -C 8 )alkynyl, benzyl, aryl, or (C 2 -C 5 )heteroaryl; each occurrence of R 6 is independently H, (Ci-C 8 )alkyl, (C 2 -C 8 )alkenyl, (C 2 - C 8 )alkynyl, benzyl, aryl, (C 2 -Cs)heteroaryl, or (Co-C 8 )alkyl-C(0)0-R 5 or the R 6 groups can join to form a heterocycloalkyl group;
- n 0 or 1 ;
- R 1 is (C 3 -C 7 )cycloalkyl, (C 2 -C 8 )alkenyl, (C 2 -Cg)alkynyl, benzyl, aryl, (Co-C 4 )alkyl-(Ci-C6)heterocycloalkyl, (Co- C 4 )alkyl-(C 2 -C 5 )heteroaryl, C(0)R 3 , C(0)OR 4 , (Ci-C 8 )alkyl-N(R 6 ) 2 , (C C 8 )alkyl-OR 5 , (C C 8 )alkyl-C(0)OR 5 , C(S)NHR 3 , or (Ci-C 8 )alkyl-0(CO)R 5 ;
- R 2 is H or (Ci-C 8 )alkyl
- R 3 is (Ci-C 8 )alkyl, (C 3 -C 7 )cycloalkyl, (C 2 -C 8 )alkenyl, (C 2 -C 8 )alkynyl, benzyl, aryl, (C 0 -C 4 )alkyl-(Ci -C 6 )heterocycloalkyl, (C 0 -C 4 )alkyl-(C 2 -C 5 )heteroaryl, (C 5 -C 8 )alkyl-N(R 6 ) 2 ; (C 0 -C 8 )alkyl-NH-C(O)O-R 5 ; (Ci-C 8 )alkyl-OR 5 , (Ci-C 8 )alkyl-C(0)OR 5 , (Ci-C 8 )alkyl- 0(CO)R 5 , or C(0)OR 5 ; and the other variables have the same definitions.
- R 2 is H or (Ci-C 4 )alkyl.
- R 1 is (Ci-C 8 )alkyl or benzyl.
- R 1 is H, (Ci-C 8 )alkyl, benzyl,
- R 1 is
- Q is O or S, and each occurrence of R is independently H, (Ci-Cg)alkyl, benzyl, CH 2 OCH 3 , or CH 2 CH 2 OCH 3 .
- R 1 is C(0)R 3 .
- R 3 is (C0-C4)alkyl-(C2- C5)heteroaryl, (Cl-C8)alkyl, aryl, or (C 0 -C 4 )alkyl-OR 5 .
- heteroaryl is pyridyl, furyl, or thienyl.
- R 1 is C(0)OR 4 .
- the H of C(0)NHC(0) can be replaced with (Ci-C 4 )alkyl, aryl, or benzyl.
- R is H or CH 2 OCOR'
- each of R 1 , R 2 , R 3 , or R 4 independently of the others, is halo, alkyl of 1 to 4 carbon atoms, or alkoxy of 1 to 4 carbon atoms or (ii) one of R 1 , R 2 , R 3 , or R 4 is nitro or -NHR 5 and the remaining of R 1 , R 2 , R 3 , or R 4 are hydrogen; R 5 is hydrogen or alkyl of 1 to 8 carbons
- R 6 hydrogen, alkyl of 1 to 8 carbon atoms, benzo, chloro, or fluoro;
- R' is R 7 -CHR 10 -N(R 8 R 9 );
- R 7 is m-phenylene or p-phenylene or -(C n H 2n )- in which n has a value of 0 to 4;
- each of R 8 and R 9 taken independently of the other is hydrogen or alkyl of 1 to 8 carbon atoms, or R 8 and R 9 taken together are tetramethylene, pentamethylene,
- R 10 is hydrogen, alkyl of to 8 carbon atoms, or phenyl
- immunomodulatory compounds are 4-(amino)-2-(2,6-dioxo(3-piperidyl))- isoindoline- 1 ,3-dione and 3-(4-amino- 1 -oxo- 1 ,3-dihydro-isoindol-2-yl)-piperidine-2,6-dione.
- the compounds can be obtained via standard, synthetic methods (see e.g. , United States Patent No. 5,635,517, incorporated herein by reference). The compounds are available from Celgene Corporation, Summit, NJ.
- 4-(Amino)-2-(2,6-dioxo(3-piperidyl))-isoindoline-l ,3- dione has the following chemical structure:
- Another compound provided herein is 3-(5-amino-2-methyl-4-oxo-4H-quinazolin- 3-yl)-piperidine-2,6-dione, which has the following structure:
- 3-(5-Amino-2-methyl-4-oxo-4H-quinazolin-3-yl)-piperidine-2,6-dione can be prepared according to the methods described in the Examples provided herein or as described in U.S. Pat. No. 7,635,700, the disclosure of which is incorporated herein by reference in its entirety.
- the compound can be also synthesized according to other methods apparent to those of skill in the art based upon the teaching herein.
- 3-(5-amino-2- methyl-4-oxo-4H-quinazolin-3-yl)-piperidine-2,6-dione is in a crystalline form described in U.S. Provisional Pat. App. No. 61/451,806, filed March 11, 2011, which is incorporated herein by reference in its entirety.
- the hydrochloride salt of the compound is used in the methods provided herein.
- the immunomodulatory compound can be, for example, a compound of formula IV, V, VI, VII, VIII, IX, X, XI, XII, XIII, XIV, XV, XVI, XVII, XVIII, XIX, XX, XXI, XXIII, XIV, XXV, XXVI, XXVII, XXVIII, XXIX, XXX, and XXXI or a pharmaceutically acceptable salt, solvate, or stereoisomer thereof:
- each of R 1 , R 2 , R 3 , and R 4 independently of the others, is halo, alkyl of 1 to 4 carbon atoms, or alkoxy of 1 to 4 carbon atoms, or (ii) one of R 1 , R 2 , R 3 , and R 4 is -NHR 5 and the remaining of R 1 , R 2 , R 3 , and R 4 are hydrogen;
- R 5 is hydrogen or alkyl of 1 to 8 carbon atoms
- R 6 is hydrogen, alkyl of 1 to 8 carbon atoms, benzyl, or halo
- R 1 is H, (Ci-Cs )alkyl, (C 3 -C 7 )cycloalkyl, (C 2 -C 8 )alkenyl, (C 2 -C 8 )alkynyl, benzyl, aryl, (C 0 -C 4 )alkyl-(Ci-C 6 )heterocycloalkyl, (C 0 -C 4 )alkyl-(C 2 -C 5 )heteroaryl, C(0)R 3 , C(S)R 3 , C(0)OR 4 , (Ci-C 8 )alkyl-N(R 6 ) 2 , (Ci-C 8 )alkyl-OR 5 , (C C 8 )alkyl-C(0)OR 5 ,
- R 2 is H, F, benzyl, (Ci-C 8 )alkyl, (C 2 -C 8 )alkenyl, or (C 2 -C 8 )alkynyl;
- R 3 and R 3' are independently (Ci-C 8 )alkyl, (C 3 -C 7 )cycloalkyl, (C 2 -C 8 )alkenyl, (C 2 -C 8 )alkynyl, benzyl, aryl, (C 0 -C 4 )alkyl-(Ci-C 6 )heterocycloalkyl, (C 0 -C 4 )alkyl-(C 2 - C 5 )heteroaryl, (C 0 -C 8 )alkyl-N(R 6 ) 2 , (Ci-C 8 )alkyl-OR 5 , (Ci-C 8 )alkyl-C(0)OR 5 , (Ci-C 8 )alkyl- 0(CO)R 5 , or C(0)OR 5 ;
- R 4 is (Ci-C 8 )alkyl, (C 2 -C 8 )alkenyl, (C 2 -C 8 )alkynyl, (Ci-C 4 )alkyl-OR 5 , benzyl, aryl, (C 0 -C 4 )alkyl-(Ci-C 6 )heterocycloalkyl, or (C 0 -C 4 )alkyl-(C 2 -C 5 )heteroaryl;
- R 5 is (Ci-C 8 )alkyl, (C 2 -C 8 )alkenyl, (C 2 -C 8 )alkynyl, benzyl, aryl, or (C 2 -
- R 6 is independently H, (Ci-Cg)alkyl, (C 2 -Cg)alkenyl, (C 2 -Cg)alkynyl, benzyl, aryl, (C 2 -C 5 )heteroaryl, or (C 0 -C 8 )alkyl-C(O)O-R 5 or the R 6 groups can join to form a heterocycloalkyl group;
- n 0 or 1 ;
- R is H or CH 2 OCOR'
- each of R 1 , R 2 , R 3 , or R 4 independently of the others, is halo, alkyl of 1 to 4 carbon atoms, or alkoxy of 1 to 4 carbon atoms or (ii) one of R 1 , R 2 , R 3 , or R 4 is nitro or -NHR 5 and the remaining of R 1 , R 2 , R 3 , or R 4 are hydrogen;
- R 5 is hydrogen or alkyl of 1 to 8 carbons
- R 6 hydrogen, alkyl of 1 to 8 carbon atoms, benzo, chloro, or fluoro;
- R' is R 7 -CHR 10 -N(R 8 R 9 );
- R 7 is m-phenylene or p-phenylene or -(C n H 2n )- in which n has a value of 0 to each of R 8 and R 9 taken independently of the other is hydrogen or alkyl of 1 to
- R 8 and R 9 taken together are tetramethylene, pentamethylene,
- R 10 is hydrogen, alkyl of to 8 carbon atoms, or phenyl
- each of R 1 , R 2 , R 3 , or R 4 independently of the others, is halo, alkyl of 1 to 4 carbon atoms, or alkoxy of 1 to 4 carbon atoms or (ii) one of R 1 , R 2 , R 3 , and R 4 is -NHR 5 and the remaining of R 1 , R 2 , R 3 , and R 4 are hydrogen;
- R 5 is hydrogen or alkyl of 1 to 8 carbon atoms
- R 6 is hydrogen, alkyl of 1 to 8 carbon atoms, benzo, chloro, or fluoro;
- R 7 is m-phenylene or p-phenylene or -(C n H 2n )- in which n has a value of 0 to
- each of R 8 and R 9 taken independently of the other is hydrogen or alkyl of 1 to 8 carbon atoms, or R 8 and R 9 taken together are tetramethylene, pentamethylene,
- hexamethylene or -CH 2 CH 2 X 1 CH 2 CH 2 - in which X 1 is -0-, -S-, or -NH-;
- R 10 is hydrogen, alkyl of to 8 carbon atoms, or phenyl
- each of R 1 , R 2 , R 3 , and R 4 independently of the others, is halo, alkyl of 1 to 4 carbon atoms, or alkoxy of 1 to 4 carbon atoms or (ii) one of R 1 , R 2 , R 3 , and R 4 is nitro or protected amino and the remaining R 2 , R 3 , and R 4 are hydrogen; and
- R 6 is hydrogen, alkyl of 1 to 8 carbon atoms, benzo, chloro, or fluoro;
- each of R 1 , R 2 , R 3 , and R 4 independently of the others, is halo, alkyl of 1 to 4 carbon atoms, or alkoxy of 1 to 4 carbon atoms or (ii) one of R 1 , R 2 , R 3 , and R 4 is -NHR 5 and the remaining of R 1 , R 2 , R 3 , and R 4 are hydrogen;
- R 5 is hydrogen, alkyl of 1 to 8 carbon atoms, or CO-R 7 -CH(R 10 )NR 8 R 9 in which each of R 7 , R 8 , R 9 , and R 10 is as herein defined;
- R 6 is alkyl of 1 to 8 carbon atoms, benzo, chloro, or fluoro;
- R 6 is hydrogen, alkyl of 1 to 8 carbon atoms, benzyl, chloro, or fluoro;
- R 7 is m-phenylene, p-phenylene or -(C n H 2n )- in which n has a value of 0 to 4; each of R 8 and R 9 taken independently of the other is hydrogen or alkyl of 1 to 8 carbon atoms, or R 8 and R 9 taken together are tetramethylene, pentamethylene, hexamethylene, or -CH 2 CH 2 X 1 CH 2 CH 2 - in which X 1 is -0-, -S- or -NH-; and
- R is hydrogen, alkyl of 1 to 8 carbon atoms, or phenyl
- R 1 , R 2 , R 3 , and R 4 independently of the others, is hydrogen, halo, alkyl of 1 to 4 carbon atoms, alkoxy of 1 to 4 carbon atoms, or amino;
- each of R 1 , R 2 , R 3 , and R 4 is halo, alkyl of 1 to 4 carbon atoms, or alkoxy of 1 to 4 carbon atoms;
- Y is oxygen or H 2 ,
- a first of R 1 and R 2 is halo, alkyl, alkoxy, alkylamino, dialkylamino, cyano, or carbamoyl
- the second of R 1 and R 2 independently of the first, is hydrogen, halo, alkyl, alkoxy, alkylamino, dialkylamino, cyano, or carbamoyl
- R 3 is hydrogen, alkyl, or benzyl
- a first of R 1 and R 2 is halo, alkyl of from 1 to 4 carbon atoms, alkoxy of from 1 to 4 carbon atoms, dialkylamino in which each alkyl is of from 1 to 4 carbon atoms, cyano, or carbamoyl;
- the second of R 1 and R 2 independently of the first, is hydrogen, halo, alkyl of from 1 to 4 carbon atoms, alkoxy of from 1 to 4 carbon atoms, alkylamino in which alkyl is of from 1 to 4 carbon atoms, dialkylamino in which each alkyl is of from 1 to 4 carbon atoms, cyano, or carbamoyl; and
- R 3 is hydrogen, alkyl of from 1 to 4 carbon atoms, or benzyl
- n when n is not zero and R 1 is not the same as R 2 , C* is a center of chirality; one of X 1 and X 2 is amino, nitro, alkyl of one to six carbons, or NH-Z, and the other of X 1 or X 2 is hydrogen;
- each of R 1 and R 2 independent of the other, is hydroxy or NH-Z;
- R 3 is hydrogen, alkyl of one to six carbons, halo, or haloalkyl;
- Z is hydrogen, aryl, alkyl of one to six carbons, formyl, or acyl of one to six carbons;
- n has a value of 0, 1 , or 2;
- C* is a center of chirality; one of X 1 and X 2 is amino, nitro, alkyl of one to six carbons, or NH-Z, and the other of X 1 or X 2 is hydrogen;
- each of R 1 and R 2 independent of the other, is hydroxy or NH-Z;
- R 3 is alkyl of one to six carbons, halo, or hydrogen;
- Z is hydrogen, aryl or an alkyl or acyl of one to six carbons; and n has a value of 0, 1 , or 2;
- C* is a center of chirality; one of X 1 and X 2 is amino, nitro, alkyl of one to six carbons, or NH-Z, and the other of X 1 or X 2 is hydrogen;
- each of R 1 and R 2 independent of the other, is hydroxy or NH-Z;
- R 3 is alkyl of one to six carbons, halo, or hydrogen;
- Z is hydrogen, aryl, or an alkyl or acyl of one to six carbons; and n has a value of 0, 1 , or 2;
- one of X 1 and X 2 is nitro, or NH-Z, and the other of X 1 or X 2 is hydrogen; each of R 1 and R 2 , independent of the other, is hydroxy or NH-Z; R 3 is alkyl of one to six carbons, halo, or hydrogen;
- Z is hydrogen, phenyl, an acyl of one to six carbons, or an alkyl of one to six n has a value of 0, 1 , or 2;
- C is a center of chirality
- one of X 1 and X 2 is alkyl of one to six carbons
- each of R 1 and R 2 is hydroxy or NH-Z;
- R 3 is alkyl of one to six carbons, halo, or hydrogen
- Z is hydrogen, phenyl, an acyl of one to six carbons, or an alkyl of one to six n has a value of 0, 1 , or 2;
- C is a center of chirality
- the carbons are centers of chirality
- X is -C(O)- or -CH 2 -;
- R 1 is alkyl of 1 to 8 carbon atoms or -NHR 3 ;
- R 2 is hydrogen, alkyl of 1 to 8 carbon atoms, or halogen; and R 3 is hydrogen, alkyl of 1 to 8 carbon atoms, unsubstituted or substituted with alkoxy of 1 to 8 carbon atoms, halo, amino, or alkylamino of 1 to 4 carbon atoms, cycloalkyl of 3 to 18 carbon atoms, phenyl, unsubstituted or substituted with alkyl of 1 to 8 carbon atoms, alkoxy of 1 to 8 carbon atoms, halo, amino, or alkylamino of 1 to 4 carbon atoms, benzyl, unsubstituted or substituted with alkyl of 1 to 8 carbon atoms, alkoxy of 1 to 8 carbon atoms, halo, amino, or alkylamino of 1 to 4 carbon atoms, or -COR 4 , wherein
- R 4 is hydrogen, alkyl of 1 to 8 carbon atoms, unsubstituted or substituted with alkoxy of 1 to 8 carbon atoms, halo, amino, or alkylamino of 1 to 4 carbon atoms, cycloalkyl of 3 to 18 carbon atoms, phenyl, unsubstituted or substituted with alkyl of 1 to 8 carbon atoms, alkoxy of 1 to 8 carbon atoms, halo, amino, or alkylamino of 1 to 4 carbon atoms, or benzyl, unsubstituted or substituted with alkyl of 1 to 8 carbon atoms, alkoxy of 1 to 8 carbon atoms, halo, amino, or alkylamino of 1 to 4 carbon atoms.
- the immunomodulatory compound can be, for example, l-oxo-2-(2,6-dioxopiperidin-3-yl)-4- aminoisoindoline or 1 ,3-dioxo-2-(2,6-dioxopiperidin-3-yl)-4-aminoisoindoline.
- R 1 is -Y-R 2 ;
- Y is aryl, heteroaryl or heterocycle, optionally substituted with one or more halogen
- R 2 is: -(CH 2 ) n -heterocycle or -0-(CH 2 ) n -heterocycle, wherein the heterocycle is optionally substituted with one or more (Ci-C 6 )alkyl, oxo, amino, hydroxyl, deuterium, or -COO-(Ci-C 6 )alkyl;
- n 0, 1, 2 or 3;
- R 3 is unsubstituted 9 to 10 membered bicyclic ring selected from the group consisting of benzothiazole, quinoline, isoquinoline, naphthalene, 2,3-dihydro-lH-indene, imidazo[l,2-a]pyridine, benzofuran, 2,3-dihydrobenzofuran, benzothiophene, benzo[d]oxazole isoindoline and chroman;
- R 4 , R 5 and R 6 are each independently hydrogen, halogen, nitro, carbamoyl, amino, -S0 2 R 7 , -CONR 8 R 9 , -(Ci-C 6 )alkyl or -(Ci-Ce)alkoxy, said alkyl or alkoxy may be optionally substituted with one or more halogen, amino, hydroxyl, or NR 8 R 9 ;
- R 7 is (Ci-C 6 )alkyl or amino optionally substituted with (Ci-Ce)alkyl;
- R 8 and R 9 are each independently hydrogen, 6 to 10 membered aryl, -COO-(Ci-Ce)alkyl, -(Co-C 6 )alkyl-CHO, -(C 0 -C 6 )alkyl-NR 8' R 9' , -(C 0 -C 6 )alkyl-(5 to 10 membered heterocycle), -(Ci-C 6 )alkyl-OH, -(Ci-C 6 )alkyl-0-(Ci-C 6 )alkyl, (Ci-C 6 )alkyl, or (C3-C 6 )cycloalkyl; or
- R 8 and R 9 together may form a 5 to 6 membered ring
- R 8' and R 9' are each independently hydrogen or (Ci-C 6 )alkyl
- X is N or C
- R 1 and R 2 are each independently hydrogen, (Ci-C 6 )alkyl, (Ci-C 6 )alkyl-(C
- R 1 is : hydrogen; halo; -(CH 2 ) n OH; (Ci-C 6 )alkyl, optionally substituted with one
- R a is:
- aryl or heteroaryl is optionally substituted with one or more of: halo; -SCF 3 ; (Ci-C 6 )alkyl, itself optionally substituted with one or more halo; or (Ci-C 6 )alkoxy, itself optionally substituted with one or more halo;
- R b and R c are each independently:
- R 2 is: hydrogen; -(CH 2 ) n OH; phenyl; -0-(Ci-C 6 )alkyl; or (Ci-C 6 )alkyl, optionally substituted with one or more halo;
- R 3 is: hydrogen; or (Ci-C 6 )alkyl, optionally substituted with one or more halo;
- R 4 is: hydrogen; halo; -(CH 2 ) n OH; (Ci-C 6 )alkyl, optionally substituted with one or more halo; or (Ci-C 6 )alkoxy, optionally substituted with one or more halo;
- R 5 is: hydrogen; -(CH 2 ) n OH; phenyl; -0-(Ci-C 6 )alkyl; or (Ci-C 6 )alkyl, optionally substituted with one or more halo;
- R 6 is: hydrogen; or (Ci-C 6 )alkyl, optionally substituted with one or more halo;
- n 0, 1, or 2;
- R d is:
- (Ci-C 6 )alkyl optionally substituted with one or more -C(0)-(Ci-Cg)alkyl, wherein the alkyl is optionally substituted with one or more halo;
- R e and R f are each independently:
- (Ci-C 6 )alkyl optionally substituted with one or more halo; or (Ci-C 6 )alkoxy, optionally substituted with one or more halo; or -C(0)-(CH 2 ) n -0-(Ci-C 6 )alkyl.
- R 7 is: hydrogen; -(CH 2 ) n OH; phenyl; -0-(Ci-C 6 )alkyl; or (Ci-C 6 )alkyl, optionally substituted with one or more halo;
- R 8 is: hydrogen; or (Ci-C 6 )alkyl, optionally substituted with one or more halo;
- n 0, 1, or 2;
- R g is:
- heteroaryl wherein the aryl or heteroaryl is optionally substituted with one or more of: halo; -SCF 3 ; (Ci-C 6 )alkyl, itself optionally substituted with one or more halo; or (Ci-C 6 )alkoxy, itself optionally substituted with one or more halo;
- R h is:
- (Ci-C 6 )alkyl itself optionally substituted with one or more halo; or (Ci-C 6 )alkoxy, itself optionally substituted with one or more halo; or
- R 9 is: hydrogen; -(CH 2 ) n OH; phenyl; -0-(Ci-C 6 )alkyl; or (Ci-C 6 )alkyl, optionally substituted with one or more halo; R is: hydrogen; or (Ci-C 6 )alkyl, optionally substituted with one or more halo; and n is 0, 1, or 2;
- R 1 is hydrogen, alkyl, alkenyl, alkynyl, aryl, aralkyl, cycloalkyl, cycloalkylalkyl,
- heterocyclyl heterocyclylalkyl, heteroaryl, heteroarylalkyl, arylaminocarbonyl,
- alkylcarbonyl alkylammocarbonyl, dialkylaminocarbonyl, alkoxycarbonyl,
- R 1 is optionally substituted with one or more groups selected from alkoxy, halo, alkyl, carboxy, alkylammocarbonyl, alkoxycarbonyl, nitro, amine, nitrile, haloalkyl, hydroxy, and alkylsulfonyl; and
- R 5 is aryl or heteroaryl, optionally substituted with one, two or three groups seleted from alkyl, halo, alkoxy, carboxy, alkylammocarbonyl, alkoxycarbonyl, nitro, amine, nitrile, haloalkyl, hydroxy, and alkylsulfonyl; and ni is 0-5.
- the methods comprise (i) administering to a patient in need of treatment an inhibitor of ActRII (see Section 4.2) and administering to the patient in need of treatment a second active agent (see Section 4.3); (ii) administering to a patient in need of treatment an inhibitor of ActRIIA (see Section 4.2) and an inhibitor of ActRIIB (see Section 4.2); and/or (iii) administering to a patient in need of treatment an inhibitor of ActRIIA (see Section 4.2), an inhibitor of ActRIIB (see Section 4.2), and a second active agent (see Section 4.3).
- the methods for the treatment and/or prevention of disease provided herein are sufficient to ameliorate one or more symptoms of the disease to be treated. In certain embodiments, the methods for the treatment and/or prevention of disease provided herein are sufficient to prevent one or more symptoms of the disease to be treated from worsening.
- the methods for the treatment and/or prevention of disease provided herein comprise administering to a patient in need of treatment a therapeutically effective amount of an inhibitor of ActRII (see Section 4.2) and administering to the patient in need of treatment a therapeutically effective amount of a second active agent (see Section 4.3).
- the methods for the treatment and/or prevention of disease provided herein comprise administering to a patient in need of treatment a therapeutically effective amount of an inhibitor of ActRIIA (see Section 4.2) and
- the methods for the treatment and/or prevention of disease provided herein comprise administering to a patient in need of treatment a therapeutically effective amount of an inhibitor of ActRIIA (see Section 4.2), administering to the patient in need of treatment a therapeutically effective amount of an inhibitor of ActRIIB (see Section 4.2), and administering to the patient in need of treatment a
- Also encompassed herein is a method of increasing the dosage of a second active agent that can be safely and effectively administered to a patient, by combining the administration regimen of the second active agent with an administration regimen of an ActRII inhibitor.
- the present invention provides a method for increasing the dose of Revlimid® that can be administered to a patient with a disease by at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 150%, 200%, 250%, 300%, 350%, 400%, 450%, 500%, 600%, 700%, 800%, 900%, or by at least 1000% without increasing the severity of any side effects or without causing any side effects.
- Also encompassed herein is a method of increasing the dosage of a second active agent that can be safely and effectively administered to a patient, by combining the administration regimen of the second active agent with an administration regimen of an ActRII inhibitor.
- the present invention provides a method for increasing the dose of Revlimid® that can be administered to a patient with a disease by at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 150%, 200%, 250%, 300%, 350%, 400%, 450%, 500%, 600%, 700%, 800%, 900%, or by at least 1000% without increasing the severity of any side effects or without causing any side effects.
- Also encompassed herein is a method of decreasing the dosage of a second active administered to a patient, so as to achieve efficacy of treatment of to reach a given endpoint, by combining the administration regimen of the second active agent with an administration regimen of an ActRII inhibitor.
- the present invention provides a method for decreasing the dose of Revlimid® administered to a patient with a disease by at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 150%, 200%, 250%, 300%, 350%, 400%, 450%, 500%, 600%, 700%, 800%, 900%, or by at least 1000%.
- a method for improving the safety of a treatment with lenalidomide comprising administering an ActRII inhibitor to the patient being treated with lenalidomide (Revlimid®).
- safety is improved if the severity of any side effects of treatment with lenalidomide is reduced by at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 150%, 200%, 250%, 300%, 350%, 400%, 450%, 500%, 600%, 700%, 800%, 900%, or by at least 1000%.
- a method of treating, preventing and/or managing a disease which comprises administering a second active agent and an ActRII inhibitor in combination with conventional therapy including, but not limited to, surgery, immunotherapy, biological therapy, radiation therapy, or other non-drug based therapy presently used to treat, prevent or manage the disease.
- a method of treatment described herein comprises administering to a patient an Activin-ActRII inhibitor and one or more second active agents, wherein the one or more second active agents include, without limitation, Revlimid ®
- the Activin-ActRII is administered with one second active agent. In another embodiment, the Activin-ActRII is administered with two second active agents. In another embodiment, the Activin-ActRII is administered with three second active agents. In another embodiment, the Activin-ActRII is administered with more than three second active agents.
- a method of treatment described herein comprises administering to a patient an Activin-ActRII inhibitor and Revlimid ® (lenalidomide). In another specific embodiment, a method of treatment described herein comprises
- a method of treatment described herein comprises administering to a patient an Activin-ActRII inhibitor, Revlimid ® (lenalidomide) and melphalan.
- a method of treatment described herein comprises administering to a patient an Activin-ActRII inhibitor, Revlimid ® (lenalidomide), and prednisone.
- a method of treatment described herein comprises administering to a patient an Activin-ActRII inhibitor, Revlimid ® (lenalidomide), melphalan and prednisone.
- a method of treatment described herein comprises administering to a patient an Activin-ActRII inhibitor and dexamethasone (e.g., a low dose regimen of dexamethasone or a high dose regimen of dexamethasone).
- a method of treatment described herein comprises administering to a patient an Activin-ActRII inhibitor, Revlimid ® (lenalidomide), and dexamethasone (e.g., a low dose regimen of dexamethasone or a high dose regimen of dexamethasone).
- a method of treatment described herein comprises administering to a patient an Activin-ActRII inhibitor and Velcade ® (bortezomib). In another specific embodiment, a method of treatment described herein comprises administering to a patient an Activin-ActRII inhibitor, Revlimid ® (lenalidomide), and Velcade ® (bortezomib). [00195] In a specific embodiment, a method of treatment described herein comprises administering to a patient an Activin-ActRII inhibitor and Dacogen ® (decitabine). In another specific embodiment, a method of treatment described herein comprises administering to a patient an Activin-ActRII inhibitor, Revlimid ® (lenalidomide), and Dacogen ® (decitabine).
- a method of treatment described herein comprises administering to a patient an Activin-ActRII inhibitor and bisphosphonate.
- a method of treatment described herein comprises administering to a patient an Activin-ActRII inhibitor, Revlimid ® (lenalidomide), and bisphosphonate.
- treatment of disease by administering a combination of ActRII inhibitors; or by administration of a combination of one or more ActRII inhibitors and a second active agent may provide additive or synergistic effects relative to the administration of an ActRII inhibitor alone or a second active agent alone.
- Such synergistic effects can be demonstrated, e.g., using the assays set forth in Section 4.6.
- any dose of the second active agent(s) deemed suitable for administration or known in the art to treat the disease to be treated can be used. Dosage and administration regimes are described below.
- methods of treating cancer comprising (i) administering to a patient in need of treatment an inhibitor of ActRII (see Section 4.2) and administering to the patient in need of treatment a second active agent (see Section 4.3); (ii) administering to a patient in need of treatment an inhibitor of ActRIIA (see Section 4.2) and an inhibitor of ActRIIB (see Section 4.2); and/or (iii) administering to a patient in need of treatment an inhibitor of ActRIIA (see Section 4.2), an inhibitor of ActRIIB (see Section 4.2), and a second active agent (see Section 4.3).
- the second active agent is lenalidomide (Revlimid®).
- the ActRII inhibitor is an ActRIIA inhibitor.
- the ActRII inhibitor is an ActRIIA inhibitor and the second active agent is lenalidomide (Revlimid®).
- the ActRII inhibitor is an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and the second active agent is lenalidomide (Revlimid®).
- the ActRIl inhibitor is an ActRIIA inhibitor and the second active agent is pomalidomide.
- the ActRIl inhibitor is an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and the second active agent is pomalidomide.
- the ActRIl inhibitor is an ActRIIB inhibitor.
- the ActRIl inhibitor is an ActRIIB inhibitor and the second active agent is lenalidomide (Revlimid®).
- the ActRIl inhibitor is an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:23 and the second active agent is lenalidomide (Revlimid®).
- the ActRIl inhibitor is an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and the second active agent is
- the ActRIl inhibitor is an ActRIIB inhibitor and the second active agent is pomalidomide.
- the ActRIl inhibitor is an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:23 and the second active agent is
- the ActRIl inhibitor is an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and the second active agent is pomalidomide.
- a method of treating cancer comprising administering to a patient in need of treatment an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
- an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
- a method of treating cancer comprising administering to a patient in need of treatment an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
- an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
- kits for treating cancer comprising administering to a patient in need of treatment an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and administering to the patient in need of treatment an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25.
- kits for treating cancer comprising administering to a patient in need of treatment an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:7, administering to the patient in need of treatment an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25, and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
- a method of treating, preventing and/or managing cancer which comprises administering a second active agent and an ActRII inhibitor in combination with conventional therapy including, but not limited to, surgery, immunotherapy, biological therapy, radiation therapy, or other non-drug based therapy presently used to treat, prevent or manage cancer.
- Cancers that can be treated in accordance with the methods described herein include, without limitation, leukemia (e.g., chronic lymphocytic leukemia, chronic myeloid leukemia, acute lymphocytic leukemia, and acute myeloid leukemia), lymphoma (e.g., Hodgkin's lymphoma and non-Hodgkin's lymphoma), myeloma (e.g., multiple myeloma), bone and connective tissue sarcomas, brain cancer, breast cancer, ovarian cancer, kidney cancer, pancreatic cancer, esophageal cancer, stomach cancer, lung cancer (e.g, small cell lung cancer (SCLC), non-small cell lung cancer (NSCLC), throat cancer, and mesothelioma), and prostate cancer.
- leukemia e.g., chronic lymphocytic leukemia, chronic myeloid leukemia, acute lymphocytic leukemia, and acute myeloid leukemia
- multiple myeloma e.g., relapsed multiple myeloma, refractory multiple myeloma, and newly diagnosed multiple myeloma
- methods of treating multiple myeloma comprising (i) administering to a patient in need of treatment an inhibitor of ActRII (see Section 4.2) and administering to the patient in need of treatment a second active agent (see Section 4.3); (ii) administering to a patient in need of treatment an inhibitor of ActRIIA (see Section 4.2) and an inhibitor of ActRIIB (see Section 4.2); and/or (iii) administering to a patient in need of treatment an inhibitor of ActRIIA (see Section 4.2), an inhibitor of ActRIIB (see Section 4.2), and a second active agent (see Section 4.3).
- the second active agent is lenalidomide (Revlimid®). In another embodiment, the second active agent is pomalidomide. In another embodiment, the second active agent is 3-(5-amino-2-methyl-4-oxo-4H-quinazolin-3-yl)-piperidine-2,6-dione.
- the ActRIl inhibitor is an ActRIIA inhibitor.
- the ActRIl inhibitor is an ActRIIA inhibitor and the second active agent is lenalidomide (Revlimid®).
- the ActRIl inhibitor is an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and the second active agent is lenalidomide (Revlimid®).
- the ActRIl inhibitor is an ActRIIA inhibitor and the second active agent is pomalidomide.
- the ActRIl inhibitor is an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and the second active agent is pomalidomide.
- the ActRIl inhibitor is an ActRIIB inhibitor.
- the ActRIl inhibitor is an ActRIIB inhibitor and the second active agent is lenalidomide (Revlimid®).
- the ActRIl inhibitor is an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:23 and the second active agent is lenalidomide (Revlimid®).
- the ActRIl inhibitor is an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and the second active agent is
- the ActRIl inhibitor is an ActRIIB inhibitor and the second active agent is pomalidomide.
- the ActRIl inhibitor is an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:23 and the second active agent is
- the ActRIl inhibitor is an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and the second active agent is pomalidomide.
- a method of treating multiple myeloma comprising administering to a patient in need of treatment an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
- an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
- a method of treating multiple myeloma comprising administering to a patient in need of treatment an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
- an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
- kits for treating multiple myeloma comprising administering to a patient in need of treatment an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and administering to the patient in need of treatment an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25.
- an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:7,
- ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25, and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
- administering results in at least 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 98%, 99%, or 100% reduction of malignant plasma cells present in the patient relative to the beginning of treatment or relative to an untreated patient.
- a second active agent e.g., lenalidomide
- the methods of treating multiple myeloma provided herein comprise administering to a patient in need of treatment a therapeutically effective amount of an inhibitor of ActRII (see Section 4.2) and administering to the patient in need of treatment a therapeutically effective amount of a second active agent (see Section 4.3).
- a therapeutically effective amount of an ActRII inhibitor increases osteoblast differentiation or osteoblastogenesis in the patient by at least 5%, 10%>, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 98%, 99%, or 100% relative to the beginning of treatment or relative to an untreated patient.
- a therapeutically effective amount of an ActRII inhibitor increases the red blood cell level and / or hemoglobin levels in the patient by at least 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 98%, 99%, or 100% relative to the beginning of treatment or relative to an untreated patient.
- a therapeutically effective amount of an ActRII inhibitor decreases osteolytic lesions in the patient by at least 5%, 10%>, 15%, 20%>, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 98%, 99%, or 100% relative to the beginning of treatment or relative to an untreated patient.
- a therapeutically effective amount of a second active agent results in at least 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 98%, 99%, or 100% reduction of malignant plasma cells present in the patient relative to the beginning of treatment or relative to an untreated patient.
- a method of treating, preventing and/or managing multiple myeloma which comprises administering a second active agent and an ActRII inhibitor in combination with conventional therapy including, but not limited to, surgery, immunotherapy, biological therapy, radiation therapy, or other non- drug based therapy presently used to treat, prevent or manage multiple myeloma.
- multiple myeloma e.g., relapsed multiple myeloma, refractory multiple myeloma, and newly diagnosed multiple myeloma
- conventional therapy including, but not limited to, surgery, immunotherapy, biological therapy, radiation therapy, or other non- drug based therapy presently used to treat, prevent or manage multiple myeloma.
- a second active agent can be administered in an amount of from about 0.1 to about 150 mg, and preferably from about 1 to about 25 mg, more preferably from about 2 to about 10 mg orally and daily, or about 5, 10, 15, 20, or about 25 mg in combination with an ActRII inhibitor, prior to, during, or after the use of conventional therapy.
- MDS myelodysplastic syndrome
- a patient in need of treatment an inhibitor of ActRII (see Section 4.2) and administering to the patient in need of treatment a second active agent (see Section 4.3); (ii) administering to a patient in need of treatment an inhibitor of ActRIIA (see Section 4.2) and an inhibitor of ActRIIB (see Section 4.3).
- the second active agent is lenalidomide (Revlimid®).
- the ActRIl inhibitor is an ActRIIA inhibitor.
- the ActRIl inhibitor is an ActRIIA inhibitor and the second active agent is lenalidomide (Revlimid®).
- the ActRIl inhibitor is an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and the second active agent is lenalidomide (Revlimid®).
- the ActRIl inhibitor is an ActRIIA inhibitor and the second active agent is pomalidomide.
- the ActRIl inhibitor is an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and the second active agent is pomalidomide.
- the ActRIl inhibitor is an ActRIIB inhibitor.
- the ActRIl inhibitor is an ActRIIB inhibitor and the second active agent is lenalidomide (Revlimid®).
- the ActRIl inhibitor is an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:23 and the second active agent is lenalidomide (Revlimid®).
- the ActRIl inhibitor is an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and the second active agent is
- the ActRIl inhibitor is an ActRIIB inhibitor and the second active agent is pomalidomide.
- the ActRIl inhibitor is an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:23 and the second active agent is
- the ActRIl inhibitor is an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and the second active agent is pomalidomide.
- a method of treating MDS comprising administering to a patient in need of treatment an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
- an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
- a method of treating MDS comprising administering to a patient in need of treatment an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
- methods of treating MDS comprising administering to a patient in need of treatment an ActRllA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and administering to the patient in need of treatment an ActRIlB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25.
- kits for treating MDS comprising administering to a patient in need of treatment an ActRllA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:7, administering to the patient in need of treatment an ActRIlB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25, and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
- a method of treating, preventing and/or managing MDS which comprises administering a second active agent and an ActRII inhibitor in combination with conventional therapy including, but not limited to, surgery, immunotherapy, biological therapy, radiation therapy, or other non-drug based therapy presently used to treat, prevent or manage MDS.
- kits for treating anemia comprising (i) administering to a patient in need of treatment an inhibitor of ActRII (see Section 4.2) and administering to the patient in need of treatment a second active agent (see Section 4.3); (ii) administering to a patient in need of treatment an inhibitor of ActRllA (see Section 4.2) and an inhibitor of ActRIlB (see Section 4.2); and/or (iii) administering to a patient in need of treatment an inhibitor of ActRllA (see Section 4.2), an inhibitor of ActRIlB (see Section 4.2), and a second active agent (see Section 4.3).
- the second active agent is lenalidomide (Revlimid®).
- the second active agent is pomalidomide.
- the ActRII inhibitor is an ActRllA inhibitor.
- the ActRII inhibitor is an ActRllA inhibitor and the second active agent is lenalidomide (Revlimid®).
- the ActRII inhibitor is an ActRllA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and the second active agent is lenalidomide (Revlimid®).
- the ActRIl inhibitor is an ActRIIA inhibitor and the second active agent is pomalidomide.
- the ActRIl inhibitor is an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and the second active agent is pomalidomide.
- the ActRIl inhibitor is an ActRllB inhibitor.
- the ActRIl inhibitor is an ActRllB inhibitor and the second active agent is lenalidomide (Revlimid®).
- the ActRIl inhibitor is an ActRllB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:23 and the second active agent is lenalidomide (Revlimid®).
- the ActRIl inhibitor is an ActRllB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and the second active agent is
- the ActRIl inhibitor is an ActRllB inhibitor and the second active agent is pomalidomide.
- the ActRIl inhibitor is an ActRllB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:23 and the second active agent is
- the ActRIl inhibitor is an ActRllB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and the second active agent is pomalidomide.
- a method of treating anemia comprising administering to a patient in need of treatment an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
- a method of treating anemia comprising administering to a patient in need of treatment an ActRllB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
- kits for treating anemia comprising administering to a patient in need of treatment an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and administering to the patient in need of treatment an ActRllB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25.
- kits for treating anemia comprising administering to a patient in need of treatment an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:7, administering to the patient in need of treatment an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25, and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
- a method of treating, preventing and/or managing anemia which comprises administering a second active agent and an ActRII inhibitor in combination with conventional therapy presently used to treat, prevent or manage anemia.
- the anemia treated in accordance with the methods described herein is caused by, or is a symptom of, cancer (e.g., multiple myeloma).
- the anemia treated in accordance with the methods described herein is caused by, or is a symptom of, MDS.
- the anemia treated in accordance with the methods described herein is not caused by, or is not a symptom of, cancer (e.g., multiple myeloma) or MDS.
- the dose of an ActRII inhibitor described herein or a second active agent described herein that will be effective in the treatment or prevention of disease can be determined by standard clinical techniques. In vitro or in vivo assays may optionally be employed to help identify optimal dosage ranges. The precise dose to be employed will also depend, e.g., on the route of administration, the type of disease, and the seriousness of the disease, and should be decided according to the judgment of the practitioner and each patient's or subject's circumstances.
- the dosage of an ActRII inhibitor described herein or a second active agent described herein is determined by extrapolating from the "no observed adverse effective level" (NOAEL), as determined in animal studies. This extrapolated dosage is useful in determining the maximum recommended starting dose for human clinical trials.
- NOAELs can be extrapolated to determine human equivalent dosages (HED).
- HED is extrapolated from a non-human animal dosage based on the doses that are normalized to body surface area (i.e., mg/m 2 ).
- the NOAELs are determined in mice, hamsters, rats, ferrets, guinea pigs, rabbits, dogs, primates, primates (monkeys, marmosets, squirrel monkeys, baboons), micropigs or minipigs.
- NOAELs are determined in mice, hamsters, rats, ferrets, guinea pigs, rabbits, dogs, primates, primates (monkeys, marmosets, squirrel monkeys, baboons), micropigs or minipigs.
- an ActRII inhibitor described herein or a second active agent described herein, or composition thereof is administered at a dose that is lower than the human equivalent dosage (HED) of the NOAEL over a period of 1 week, 2 weeks, 3 weeks, 1 month, 2 months, three months, four months, six months, nine months, 1 year, 2 years, 3 years, 4 years or more.
- HED human equivalent dosage
- a dosage regime for a human subject can be extrapolated from animal model studies using the dose at which 10% of the animals die (LDio).
- LDio dose at which 10% of the animals die
- a standard measure of toxicity of a drug in preclinical testing is the percentage of animals that die because of treatment. It is well within the skill of the art to correlate the LDio in an animal study with the maximal-tolerated dose (MTD) in humans, adjusted for body surface a basis to extrapolate a starting human dose.
- MTD maximal-tolerated dose
- the interrelationship of dosages for one animal model can be converted for use in another animal, including humans, using conversion factors (based on milligrams per meter squared of body surface) as described, e.g., in Freireich et al. , Cancer Chemother. Rep., 1966, 50:219-244.
- Body surface area may be approximately determined from height and weight of the patient. See, e.g., Scientific Tables, Geigy Pharmaceuticals, Ardley, N. Y., 1970, 537.
- the adjustment for body surface area includes host factors such as, for example, surface area, weight, metabolism, tissue distribution, absorption rate, and excretion rate.
- the route of administration, excipient usage, and the specific disease to be treated are also factors to consider.
- the standard conservative starting dose is about 1/10 the murine LDio, although it may be even lower if other species ⁇ i.e., dogs) were more sensitive to the ActRII inhibitor or second active.
- the standard conservative starting dose is about 1/100, 1/95, 1/90, 1/85, 1/80, 1/75, 1/70, 1/65, 1/60, 1/55, 1/50, 1/45, 1/40, 1/35, 1/30, 1/25, 1/20, 1/15, 2/10, 3/10, 4/10, or 5/10 of the murine LDi 0 .
- a starting dose amount of an ActRII inhibitor described herein or a second active agent described herein in a human is lower than the dose extrapolated from animal model studies. In another embodiment, a starting dose amount of an ActRII inhibitor described herein or a second active agent described herein in a human is higher than the dose extrapolated from animal model studies. It is well within the skill of the art to start doses of the active composition at relatively low levels, and increase or decrease the dosage as necessary to achieve the desired effect with minimal toxicity.
- the ActRII inhibitor is dosed at intervals and amounts sufficient to achieve serum concentrations of 0.2 microgram/kg or greater, and serum levels of 1 microgram/kg or 2 microgram/kg or greater are desirable for achieving significant effects on bone density and strength.
- Dosing regimens may be designed to reach serum concentrations of between 0.2 and 15 microgram/kg, and optionally between 1 and 5 microgram/kg.
- serum levels of 0.2 microgram kg may be achieved with a single dose of 0.1 mg/kg or greater and serum levels of 1 microgram/kg may be achieved with a single dose of 0.3 mg/kg or greater.
- the observed serum half-life of the molecule is between about 20 and 30 days, substantially longer than most Fc fusion proteins, and thus a sustained effective serum level may be achieved, for example, by dosing with 0.2-0.4 mg/kg on a weekly or biweekly basis, or higher doses may be used with longer intervals between dosings.
- doses of 1-3 mg/kg might be used on a monthly or bimonthly basis, and the effect on bone may be sufficiently durable that dosing is necessary only once every 3, 4, 5, 6, 9, 12 or more months.
- Serum levels of the ActRII inhibitor can be measured by any means known to the skilled artisan.
- antibodies against the ActRII inhibitor can be used to determine the serum levels of the ActRII inhibitor using, e.g., an ELISA.
- the dose of the ActRII inhibitor ranges from 0.01 to 3.0 mg/kg intravenously or from 0.03 to 0.1 mg/kg subcutaneously. In certain embodiments, the dose of ActRII inhibitor is about 0.01 mg/kg, about 0.1 mg/kg, about 0.2 mg/kg, about 0.3 mg/kg, about 0.4 mg/kg, about 0.5 mg/kg, about 1.0 mg/kg, about 1.5 mg/kg, about 2.0 mg/kg, about 2.5 mg/kg, about 3.0 mg/kg, about 3.5 mg/kg, about 4.0 mg/kg, about 4.5 mg/kg, or about 5.0 mg/kg.
- the dose of of ActRII inhibitor is about 10.0 mg/kg, about 15.0 mg/kg, about 20.0 mg/kg, about 25.0 mg/kg, or about 30.0 mg/kg. In certain embodiments, the dose of ActRII inhibitor is between 0.01 mg/kg and 0.1 mg/kg, between 0.1 mg/kg and 0.3 mg/kg, between 0.3 mg/kg and 0.5 mg/kg, between 0.5 mg/kg and 1.0 mg/kg, between 1.0 mg/kg and 2.0 mg/kg, between 1.0 mg/kg and 3.0 mg/kg, between 2.0 mg/kg and 3.0 mg/kg, between 2.0 mg/kg and 4.0 mg/kg, between 3.0 mg/kg and 5.0 mg/kg, between 5.0 mg/kg and 10.0 mg/kg, between 10.0 mg/kg and 15.0 mg/kg, between 10.0 mg/kg and 20.0 mg/kg, between 15.0 mg/kg and 20.0 mg/kg, or between 20.0 mg/kg and 30.0 mg/kg.
- the dose of ActRII inhibitor is between 0.01 mg/kg
- Typical dosage forms comprise a second active agent, or a pharmaceutically acceptable salt, solvate, stereoisomer, or prodrug thereof, in an amount of from about 0.001 to about 150 mg.
- dosage forms comprise a second active agent, or a pharmaceutically acceptable salt, solvate, stereoisomer, or prodrug thereof, in an amount of from about 0.001 to about 150 mg.
- dosage forms comprise a second active agent, or a pharmaceutically acceptable salt, solvate, stereoisomer, or prodrug thereof, in an amount of from about 0.001 to about 150 mg.
- dosage forms comprise a second active agent, or a
- pharmaceutically acceptable salt, solvate, stereoisomer, or prodrug thereof in an amount of about 0.001, 0.01, 0.1, 1, 2, 5, 7.5, 10, 12.5, 15, 17.5, 20, 25, 50, 100, 150 or 200 mg.
- unit dosage formulations of a second active agent that comprise between about 1 mg and about 2000 mg, about 1 mg and 200 mg, about 35 mg and about 1400 mg, about 125 mg and about 1000 mg, about 250 mg and about 1000 mg, or about 500 mg and about 1000 mg of a second active agent.
- unit dosage formulations of a second active agent that comprise 1 mg, 2.5 mg, 5 mg, 10 mg, 15 mg, 20 mg, 30 mg, 35 mg, 50 mg, 70 mg, 100 mg, 125 mg, 140 mg, 175 mg, 200 mg, 250 mg, 280 mg, 350 mg, 500 mg, 560 mg, 700 mg, 750 mg, 1000 mg or 1400 mg of a second active agent.
- suitable dosage ranges of a second active agent for oral administration are about 0.001 milligram to about 500 milligrams of a second active agent, per kilogram body weight per day.
- the oral dose is about 0.01 milligram to about 100 milligrams per kilogram body weight per day, about 0.1 milligram to about 75 milligrams per kilogram body weight per day or about 0.5 milligram to 5 milligrams per kilogram body weight per day.
- the dosage amounts described herein refer to total amounts administered; that is, if more than one second active agent is administered, then, in some embodiments, the dosages correspond to the total amount administered.
- oral compositions contain about 10% to about 95% of a second active agent by weight.
- suitable dosage ranges for intravenous (i.v.) administration of a second active agent are about 0.01 milligram to about 100 milligrams per kilogram body weight per day, about 0.1 milligram to about 35 milligrams per kilogram body weight per day, and about 1 milligram to about 10 milligrams per kilogram body weight per day.
- suitable dosage ranges for intranasal administration are about 0.01 pg/kg body weight per day to about 1 mg/kg body weight per day.
- Suppositories generally contain about 0.01 milligram to about 50 milligrams of a second active agent per kilogram body weight per day and comprise active ingredient in the range of about 0.5% to about 10% by weight.
- suitable dosages for intradermal, intramuscular, intraperitoneal, subcutaneous, epidural, sublingual, intracerebral, intravaginal, transdermal administration or administration by inhalation of a second active agent are in the range of about 0.001 milligram to about 500 milligrams per kilogram of body weight per day.
- a second active agent can be administered orally and in single or divided daily doses in an amount of from about 0.10 to about 150 mg/day.
- Exemplary doses of Revlimid ® include, but are not limited to, 0.001, 0.01, 0.1, 1, 2, 5, 10, 25 or 50 mg. Exemplary dose ranges of Revlimid ®
- Revlimid ® (lenalidomide) include, but are not limited to, 0.001 to 0.01 mg, 0.01 to 0.1 mg, 0.1 to 1 mg, 1 to 2 mg, 2 to 5 mg, 5 to 10 mg, 5 to 25 mg, or 5 to 50 mg.
- Revlimid ® (lenalidomide) may be administered at a dose of 5 to 50 mg per day, or alternatively from about 10 to about 50 mg every other day.
- 3 (4 amino 1 oxo 1,3 dihydro isoindol 2 yl piperidine 2,6 dione (Revlimid®) may be administered in an amount of from about 5 to 25 mg per day, or alternatively from about 10 to about 50 mg every other day.
- 3 (4 amino 1 oxo 1,3 dihydro isoindol 2 yl) piperidine 2,6 dione may be administered initially in an amount of 5 mg/day and the dose can be escalated every week to 10, 20, 25, 30 and 50 mg/day.
- Revlimid® may be orally administered in an amount of about 25 mg per day for 21 days followed by seven days rest in a 28 day cycle.
- Exemplary dose ranges of pomalidomide include, but are not limited to, 0.1 to 1 mg, 1 to 2 mg, 2 to 4, 2.5 to 5 mg, or 5 to 10 mg per day.
- pomalidomide may be orally
- pomalidomide may be orally administered in an amount of about 0.5 mg to 4 mg per day on days 1 through 28 in a 28 day cycle.
- Exemplary dose ranges of 3-(5-amino-2-methyl-4-oxo-4H-quinazolin-3-yl)- piperidine-2,6-dione include, but are not limited to, 0.001 to 0.01 mg, 0.01 to 0.1 mg, 0.1 to 1 mg, 1 to 2 mg, 2 to 5 mg, 5 to 10 mg, 5 to 25 mg, or 5 to 50 mg per day.
- Exemplary doses of melphalan include, but are not limited to, 0.01 mg/kg, 0.05 mg/kg, 0.1 mg/kg, 0.15 mg/kg, 0.16 mg/kg, 0.17 mg/kg, 0.18 mg/kg, 0.19 mg/kg, 0.2 mg/kg, 0.25 mg/kg, 0.3 mg/kg, 0.35 mg/kg, 0.4 mg/kg, 0.45 mg/kg, or 0.5 mg/kg.
- Exemplary dose ranges of melphalan include, but are not limited to, 0.01 to 0.05 mg/kg, 0.05 to 0.1 mg/kg, 0.1 to 0.15 mg/kg, 0.1 to 0.2 mg/kg, 0.1 to 0.25 mg/kg, 0.2 to 0.3 mg/kg, 0.2 to 0.35 mg/kg, 0.3 to 0.4 mg/kg, 0.3 to 0.45 mg/kg, or 0.4 to 0.5 mg/kg.
- melphalan may be administered at a dose of 0.1 to 0.25 mg/kg per day.
- Exemplary doses of prednisone include, but are not limited to, 0.1 mg/kg, 0.5 mg/kg, 1 mg/kg, 1.5 mg/kg, 2 mg/kg, 2.5 mg/kg, 3 mg/kg, 3.5 mg/kg, 4 mg/kg, 4.5 mg/kg, or 5 mg/kg.
- Exemplary dose ranges of prednisone include, but are not limited to, 0.1 to 1 mg/kg, 0.1 to 2 mg/kg, 1 to 2 mg/kg, 1 to 3 mg/kg, 2 to 3 mg/kg, 2 to 4 mg/kg, 2 to 5, or 3 to 5 mg/kg.
- prednisone may be administered at a dose of 1 to 3 mg/kg per day.
- Exemplary doses of dexamethasone include, but are not limited to, 5 mg, 10 mg, 15 mg, 20 mg, 25 mg, 30 mg, 35 mg, 40 mg, 45 mg, 50 mg, 55 mg, 60 mg, 65 mg, or 70 mg.
- Exemplary dose ranges of dexamethasone include, but are not limited to, 5 to 20 mg, 10 to 30 mg, 20 to 40 mg, 30 to 50 mg, 40 to 60 mg, or 50 to 70 mg.
- dexamethasone may be administered at a dose of 40 mg on days 1 to 4, 9-12, and 17-20 of a 28-day treatment cycle (i.e., high-dose dexamethasone treatment).
- dexamethasone may be administered at a dose of 40 mg on days 1, 8, 15, and 22 of a 28-day treatment cycle (i.e., low-dose dexamethasone treatment).
- Exemplary doses of Velcade ® include, but are not limited to, 0.1 mg/m 2 (of body surface area), 0.5 mg/m 2 , 0.6 mg/m 2 , 0.7 mg/m 2 , 0.8 mg/m 2 , 0.9 mg/m 2 , 1 mg/m 2 , 1.1 mg/m 2 , 1.2 mg/m 2 , 1.3 mg/m 2 , 1.4 mg/m 2 , 1.5 mg/m 2 , 2 mg/m2 , 2.5 mg/m 2 , or 3 mg/m 2 .
- Exemplary dose ranges of Velcade ® (bortezomib) include, but are not limited to, 0.1 mg/m 2 (of body surface area), 0.5 mg/m 2 , 0.6 mg/m 2 , 0.7 mg/m 2 , 0.8 mg/m 2 , 0.9 mg/m 2 , 1 mg/m 2 , 1.1 mg/m 2 , 1.2 mg/m 2 , 1.3 mg/m 2 , 1.4 mg/m
- Velcade ® (bortezomib) may be administered at a dose of 0.7 to 1.3 mg/m 2 daily, every other day, every third day, or weekly.
- Exemplary doses of Dacogen include, but are not limited to, 5 mg/m 2 (of body surface area), 10 mg/m 2 , 15 mg/m 2 , 20 mg/m 2 , or 25 mg/m 2.
- Dacogen ® includes, but are not limited to, 5 to 10 mg/m 2 , 5 to 15 mg/m 2 , 10 to 15 mg/m 2 , 10 to 20 mg/m 2 , 15 to 20 mg/m 2 , or 15 to 25 mg/m 2.
- Dacogen ® (decitabine) may be administered at a dose of 10 to 20 mg/m 2 daily for five days.
- Exemplary doses of bisphosphonate include, but are not limited to 1 mg, 5 mg, 10 mg, 15 mg, 20 mg, 25 mg, 30 mg, 40 mg, 50 mg, 60 mg, 70 mg, 80 mg, 90 mg, 100 mg, 110 mg, 120 mg, 130 mg, 140 mg, 150 mg, or 200 mg. Exemplary dose ranges of
- bisphosphonate include, but are not limited to, 1 to 10 mg, 1 to 25 mg, 1 to 50 mg, 10 to 25 mg, 10 to 50 mg, 25 to 50 mg, 25 to 75 mg, 50 to 75 mg, 50 to 100 mg, 75 to 100 mg, 75 to 150 mg, 100 to 150 mg, 100 to 200 mg, or 150 to 200 mg.
- an ActRII inhibitor and a second active agent are administered simultanteously, e.g., as part of the same formulation.
- an ActRII inhibitor and a second active agent are administered less than 5 minutes apart, less than 30 minutes apart, 1 hour apart, at about 1 hour apart, at about 1 to about 2 hours apart, at about 2 hours to about 3 hours apart, at about 3 hours to about 4 hours apart, at about 4 hours to about 5 hours apart, at about 5 hours to about 6 hours apart, at about 6 hours to about 7 hours apart, at about 7 hours to about 8 hours apart, at about 8 hours to about 9 hours apart, at about 9 hours to about 10 hours apart, at about 10 hours to about 11 hours apart, at about 11 hours to about 12 hours apart, at about 12 hours to 18 hours apart, 18 hours to 24 hours apart, 24 hours to 36 hours apart, 36 hours to 48 hours apart, 48 hours to 52 hours apart, 52 hours to 60 hours apart, 60 hours to 72 hours apart, 72 hours
- an ActRII inhibitor and a second active agent (i) an ActRII inhibitor and a second active agent; (ii) an ActRIIA inhibitor and an ActRIIB inhibitor; or (iii) an ActRIIA inhibitor, an ActRIIB inhibitor, and a second active agent are administered within the same patent visit.
- the interval of time between the administration of (i) an ActRII inhibitor and a second active agent; (ii) an ActRIIA inhibitor and an ActRIIB inhibitor; or (iii) an ActRIIA inhibitor, an ActRIIB inhibitor, and a second active agent and the administration of a second active agent may be about 1-5 minutes, 1-30 minutes, 30 minutes to 60 minutes, 1 hour, 1-2 hours, 2-6 hours, 2-12 hours, 12-24 hours, 1-2 days, 2 days, 3 days, 4 days, 5 days, 6 days, 7 days, 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 7 weeks, 8 weeks, 9 weeks, 10 weeks, 15 weeks, 20 weeks, 26 weeks, 52 weeks, 11- 15 weeks, 15-20 weeks, 20-30 weeks, 30-40 weeks, 40-50 weeks, 1 month, 2 months, 3 months, 4 months 5 months, 6 months, 7 months, 8 months, 9 months, 10 months, 11 months, 12 months, 1 year, 2 years, or any period of time in between.
- an ActRII inhibitor and a second active agent are administered less than 1 day, 1 week, 2 weeks, 3 weeks, 4 weeks, one month, 2 months, 3 months, 6 months, 1 year, 2 years, or 5 years apart.
- a dose(s) of an ActRII inhibitor described herein or composition thereof or a second active agent described herein or composition thereof is administered for 2 days, 3 days, 5 days, 7 days, 14 days, or 21 days.
- a dose an ActRII inhibitor described herein or composition thereof or a second active agent described herein or composition thereof is administered for 1 month, 1.5 months, 2 months, 2.5 months, 3 months, 4 months, 5 months, 6 months or more.
- an ActRII inhibitor described herein or composition thereof and the administration of a second active agent described herein or composition thereof can be performed concurrently, or on alternative schedules.
- an ActRII inhibitor described herein or composition thereof may be administered on a specific day or at a specific time
- a second active agent described herein or composition thereof may be administered on a different specific day or at a different specific time.
- the administration schedules of an ActRII inhibitor described herein or composition thereof and a second active agent described herein or composition thereof overlap.
- the administration schedules of an ActRII inhibitor described herein or composition thereof and a second active agent described herein or composition thereof do not overlap.
- the methods of treatment and prevention of disease provided herein involve administering (i) an ActRII inhibitor and a second active agent; (ii) an ActRIIA inhibitor and an ActRIIB inhibitor; or (iii) an ActRIIA inhibitor, an ActRIIB inhibitor, and a second active agent daily, three times a week, twice a week, or once a week; and administering a second active agent daily, three times a week, twice a week, once every 2 weeks, once every 3 weeks, once every 4 weeks, once every month, once every 2 months (e.g., approximately 8 weeks), once every 3 months (e.g., approximately 12 weeks), or once every 4 months (e.g., approximately 16 weeks).
- an ActRII inhibitor and a second active agent are cyclically administered to a subject.
- Cycling therapy involves the administration of an ActRII inhibitor or composition thereof (or an ActRIIA inhibitor and an ActRIIB inhibitor) for a period of time, followed by the administration of a second active agent for a period of time, and repeating this sequential administration.
- cycling therapy may also include a period of rest where the ActRII inhibitor or composition thereof (or an ActRIIA inhibitor and an ActRIIB inhibitor) or the second active agent is not administered for a period of time (e.g., 2 days, 3 days, 4 days, 5 days, 6 days, 7 days, 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 10 weeks, 20 weeks, 1 month, 2 months, 3 months, 4 months, 5 months, 6 months, 7 months, 8 months, 9 months, 10 months, 11 months, 12 months, 2 years, or 3 years).
- the number of cycles administered is from 1 to 12 cycles, from 2 to 10 cycles, or from 2 to 8 cycles.
- the methods of treatment and prevention of disease provided herein comprise administering an ActRII inhibitor or composition thereof (or an ActRIIA inhibitor and an ActRIIB inhibitor) as a single agent for a period of time prior to administering the ActRII inhibitor or composition thereof (or an ActRIIA inhibitor and an ActRllB inhibitor) in combination with a second active agent.
- the methods of treatment and prevention of disease provided herein comprise administering a second active agent alone for a period of time prior to administering the second active agent in combination with an ActRII inhibitor or composition thereof (or an ActRIlA inhibitor and an ActRllB inhibitor).
- an ActRII inhibitor or an ActRIlA inhibitor and an ActRllB inhibitor
- a second active agent can be administered to a subject in the same
- an ActRII inhibitor or composition thereof or an ActRIlA inhibitor and an ActRllB inhibitor
- a second active agent can be administered concurrently to a subject in separate pharmaceutical compositions.
- An ActRII inhibitor or composition thereof or an ActRIlA inhibitor and an ActRllB inhibitor
- a second active agent can be administered sequentially to a subject in separate pharmaceutical compositions.
- An ActRII inhibitor or composition thereof or an ActRIlA inhibitor and an ActRllB inhibitor
- a second active agent may also be administered to a subject by the same or different routes of administration.
- a second active agent can be administered in an amount of from about 0.1 to about 150 mg, and preferably from about 1 to about 25 mg, more preferably from about 2 to about 10 mg orally and daily, or about 5, 10, 15, 20, or about 25 mg in combination with an ActRII inhibitor (or an ActRIlA inhibitor and an ActRllB inhibitor), prior to, during, or after the use of conventional therapy.
- a second active agent is administered daily in a single or divided doses in a four to six week cycle with a rest period of about a week or two weeks. The invention further allows the frequency, number, and length of dosing cycles to be increased.
- another specific embodiment encompasses the administration of a second active agent (in combination with an ActRII inhibitor) for more cycles than are typical when it is administered alone.
- a second active agent is administered (in combination with an ActRII inhibitor) for a greater number of cycles that would typically cause dose-limiting toxicity in a patient to whom a second active ingredient is not also being administered.
- a second active agent is administered (in combination with an ActRII inhibitor) daily and continuously for three or four weeks at a dose of from about 0.1 to about 150 mg/d followed by a break of one or two weeks.
- Revlimid® is administered (in combination with an ActRII inhibitor) in an amount of about 5, 10, or 25mg/day, preferably in an amount of about 25 mg/day for three to four weeks, followed by one week or two weeks of rest in a four or six week cycle.
- a second active agent is administered orally, with
- one cycle comprises the administration of from about 10 to about 25 mg/day of Revlimid® and from about 50 to about 200 mg/m 2 /day of an ActRII inhibitor daily for three to four weeks and then one or two weeks of rest.
- one or more ActRII inhibitors described herein may be administered to a na ' ive subject, i.e., a subject that does not have a disease.
- a second active agent e.g., lenalidomide (Revlimid®); see Section 4.3
- a composition described herein that comprises one or more ActRII inhibitors described herein or one or more ActRII inhibitors described herein and a second active agent may be administered to a na ' ive subject, i.e., a subject that does not have a disease.
- one or more ActRII inhibitors described herein are administered to a patient who has been diagnosed with cancer, e.g., the patient has been diagnosed with leukemia (e.g., chronic lymphocytic leukemia, chronic myeloid leukemia, acute lymphocytic leukemia, and acute myeloid leukemia), lymphoma (e.g., Hodgkin's lymphoma and non-Hodgkin's lymphoma), myeloma (e.g., multiple myeloma), bone and connective tissue sarcomas, brain cancer, breast cancer, ovarian cancer,
- leukemia e.g., chronic lymphocytic leukemia, chronic myeloid leukemia, acute lymphocytic leukemia, and acute myeloid leukemia
- lymphoma e.g., Hodgkin's lymphoma and non-Hodgkin's lymphoma
- myeloma e.g.
- one or more ActRII inhibitors described herein (see Section 4.2) or one or more ActRII inhibitors described herein and a second active agent (e.g., lenalidomide (Revlimid®); see Section 4.3) or a composition described herein that comprises one or more ActRII inhibitors described herein or one or more ActRII inhibitors described herein and a second active agent are administered to a patient who has been diagnosed with multiple myeloma ⁇ e.g., relapsed multiple myeloma, refractory multiple myeloma, and newly diagnosed multiple myeloma).
- a second active agent e.g., lenalidomide (Revlimid®); see Section 4.3
- a composition described herein that comprises one or more ActRII inhibitors described herein or one or more ActRII inhibitors described herein and a second active agent are administered to a patient who has been diagnosed with multiple myeloma ⁇ e.g., relapse
- one or more ActRII inhibitors described herein (see Section 4.2) or one or more ActRII inhibitors described herein and a second active agent (e.g., lenalidomide (Revlimid®); see Section 4.3) or a composition described herein that comprises one or more ActRII inhibitors described herein or one or more ActRII inhibitors described herein and a second active agent are administered to a patient who has been diagnosed with myelodysplastic syndrome (MDS).
- MDS myelodysplastic syndrome
- one or more ActRII inhibitors described herein (see Section 4.2) or one or more ActRII inhibitors described herein and a second active agent (e.g., lenalidomide (Revlimid®); see Section 4.3) or a composition described herein that comprises one or more ActRII inhibitors described herein or one or more ActRII inhibitors described herein and a second active agent are administered to a patient who has been diagnosed with anemia.
- a second active agent e.g., lenalidomide (Revlimid®); see Section 4.3
- a composition described herein that comprises one or more ActRII inhibitors described herein or one or more ActRII inhibitors described herein and a second active agent are administered to a patient who has been diagnosed with anemia.
- one or more ActRII inhibitors described herein (see Section 4.2) or one or more ActRII inhibitors described herein and a second active agent (e.g., lenalidomide (Revlimid®); see Section 4.3) or a composition described herein that comprises one or more ActRII inhibitors described herein or one or more ActRII inhibitors described herein and a second active agent are administered to a patient with a disease (e.g., multiple myeloma, MDS, or anemia) before symptoms of the disease manifest or before symptoms of the disease become severe (e.g., before the patient requires hospitalization).
- a disease e.g., multiple myeloma, MDS, or anemia
- one or more ActRII inhibitors described herein (see Section 4.2) or one or more ActRII inhibitors described herein and a second active agent (e.g., lenalidomide (Revlimid®); see Section 4.3) or a composition described herein that comprises one or more ActRII inhibitors described herein or one or more ActRII inhibitors described herein and a second active agent are administered to a patient with a disease after symptoms of the disease manifest or after symptoms of the disease become severe (e.g., after the patient requires hospitalization).
- a second active agent e.g., lenalidomide (Revlimid®); see Section 4.3
- a composition described herein that comprises one or more ActRII inhibitors described herein or one or more ActRII inhibitors described herein and a second active agent are administered to a patient with a disease after symptoms of the disease manifest or after symptoms of the disease become severe (e.g., after the patient requires hospitalization).
- a subject to be administered one or more ActRIl inhibitors described herein see Section 4.2) or one or more ActRIl inhibitors described herein and a second active agent (e.g., lenalidomide (Revlimid®); see Section 4.3) or a composition described herein that comprises one or more ActRIl inhibitors described herein or one or more ActRIl inhibitors described herein and a second active agent is a human toddler.
- a second active agent e.g., lenalidomide (Revlimid®); see Section 4.3
- a composition described herein that comprises one or more ActRIl inhibitors described herein or one or more ActRIl inhibitors described herein and a second active agent is a human toddler.
- a subject to be administered one or more ActRIl inhibitors described herein see Section 4.2) or one or more ActRIl inhibitors described herein and a second active agent (e.g., lenalidomide (Revlimid®); see Section 4.3) or a composition described herein that comprises one or more ActRIl inhibitors described herein or one or more ActRIl inhibitors described herein and a second active agent is a human child.
- a second active agent e.g., lenalidomide (Revlimid®); see Section 4.3
- a composition described herein that comprises one or more ActRIl inhibitors described herein or one or more ActRIl inhibitors described herein and a second active agent is a human child.
- the treatment regimens described herein can be tested in suitable in vitro and in vivo assays prior to use in humans.
- a number assays are known in the art for assessing the efficacy of treatment of disease, e.g., cancer, multiple myeloma (e.g., relapsed multiple myeloma, refractory multiple myeloma, and newly diagnosed multiple myeloma), and MDS.
- diseases e.g., cancer
- multiple myeloma e.g., relapsed multiple myeloma, refractory multiple myeloma, and newly diagnosed multiple myeloma
- MDS multi-limiting examples of such assays are provided below.
- In vitro assays used for testing the efficacy of one or more multiple myeloma treatments described herein can utilize one or more multiple myeloma cell lines.
- Such multiple myeloma cell lines are known in the art and include, without limitation, MM. IS, MM.1R, RPMI 8226 (RPMI), RPMI-Dox40 (Dox40), NCI-H929, KMS-11, OPM-2, and U266.
- the efficacy of a multiple myeloma treatment regimen described herein can be tested using assays in which the treatment regimen is added to multiple myeloma cells grown in vitro and one or more of cell growth, cell apoptosis, and cell migration are assessed as compared to multiple myeloma cells that have not been exposed to the treatment regimen.
- cell growth, cell apoptosis, and cell migration assays are described in Podar et al, 2007, Blood 109(4): 1669-1677.
- the efficacy of a multiple myeloma treatment regimen described herein can be tested using in vivo animal model systems.
- animal model systems include, but are not limited to, rats, mice, chicken, cows, monkeys, pigs, dogs, rabbits, etc.
- animal models for multiple myeloma include, but are not limited to, xenograft mouse models (see, e.g., Podar et al, 2007, Blood 109(4): 1669-1677); the NOD/SCID human chimeric mouse model (see, e.g., Huang et al, 2004, American Journal of Pathology 164(2):747-756); and aging mice of the mouse strain C57BL/KaLwRij strain (see, e.g., Radl et al., 1988, American Journal of Pathology 132(3):593-597).
- the efficacy of a treatment regimen in a human multiple myeloma patient can be assessed using gross measurements including, without limitation, computed tomography (CT), magnetic resonance imaging (MRI), X-ray imaging, PET scan, and bone scans.
- CT computed tomography
- MRI magnetic resonance imaging
- X-ray imaging PET scan
- bone scans bone scans.
- the toxicity of the therapeutic regimens described herein can be determined using standard pharmaceutical procedures in cell cultures or experimental animals, e.g., for determining the LD50 (the dose lethal to 50% of the population) and the ED50 (the dose therapeutically effective in 50% of the population).
- the dose ratio between toxic and therapeutic effects is the therapeutic index and it can be expressed as the ratio LD50/ED50.
- MDS Myelodysplastic Syndrome
- myelodysplastic syndrome cell lines are known in the art and include, without limitation, MUTZ-1, MDS-L, PC-MDS, MDS92, M-TAT, and TER-3.
- the efficacy of a myelodysplastic syndrome treatment regimen described herein can be tested using in vivo animal model systems.
- animal model systems include, but are not limited to, rats, mice, chicken, cows, monkeys, pigs, dogs, rabbits, etc.
- animal models for myelodysplastic syndrome include, but are not limited to, those described in Beachy et al, 2010, Hematol. Oncol. Clin. North Am. 24(2):361-375.
- the efficacy of a treatment regimen in a human myelodysplastic syndrome patient can be assessed using standard laboratory procedures used for diagnosis of myelodysplastic syndrome including, but not limited to complete blood count, bone marrow aspiration/examination, cytogenetics (chromosomal studies) performed on bone marrow aspirates, complete blood count, and flow cytometry using cells obtained from bone marrow aspirates.
- the efficacy of treatment can be assessed by comparing the results of the assay used to the results of the same assay in the patient prior to treatment.
- mice are ovariectomized, which causes the mice to lose substantial bone mineral content and bone mineral density, with the trabecular bone losing roughly 50% of bone mineral density. Bone density could be increased in the ovariectomized mice by administration of factors such as parathyroid hormone.
- fracture healing assays that are known in the art can be used. These assays include fracture technique, histological analysis, and biomechanical analysis, which are described in, for example, U.S. Pat. No. 6,521,750, which is incorporated by reference in its entirety for its disclosure of experimental protocols for causing as well as measuring the extent of fractures, and the repair process.
- the efficacy of the methods of treatment described herein can be tested for biological activity using animal models for cancer.
- animal model systems include, but are not limited to, rats, mice, chicken, cows, monkeys, pigs, dogs, rabbits, etc.
- the efficacy of the methods of treatment described herein are tested in a mouse model system.
- Such model systems are widely used and well-known to the skilled artisan such as the SCID mouse model or transgenic mice.
- the efficacy of the methods of treatment described herein can be determined by administering an ActRII inhibitor described herein and a second active agent (e.g., lenalidomide (Revlimid®)) or a composition described herein that comprises an ActRII inhibitor described herein and a second active agent (e.g., lenalidomide (Revlimid®)) to an animal model and verifying that such administration is effective in reducing the severity of cancer in said animal model.
- a second active agent e.g., lenalidomide (Revlimid®)
- a composition described herein that comprises an ActRII inhibitor described herein and a second active agent (e.g., lenalidomide (Revlimid®)
- animal models for cancer in general include, include, but are not limited to, spontaneously occurring tumors of companion animals (see, e.g., Vail & MacEwen, 2000, Cancer Invest 18(8):781-92).
- animal models for lung cancer include, but
- Panc02 murine pancreatic adenocarcinoma see, e.g., Wang et al., 2001, Int. J. Pancreatol.
- mice generated in subcutaneous pancreatic tumors
- SCID severe combined immunodeficiency
- IgHmu-HOXl 1 transgenic mouse see, e.g., Hough et al, 1998, Proc. Natl. Acad. Sci. USA 95(23): 13853-8).
- the ActRII inhibitor(s) (see Section 4.2) and the second active agent(s) (see Section 4.3) are formulated separately. In other embodiments, the ActRII inhibitor(s) and the second active agent(s) are formulated together.
- a therapeutic method provided herein includes administering the second active agent and / or the ActRII inhibitor systemically, or locally as an implant or device.
- the therapeutic composition for use in this invention is in a pyrogen-free, physiologically acceptable form.
- a pharmaceutical composition comprising an ActRII inhibitor comprising or consisting of a polypeptide comprising the amino acid sequence of SEQ ID NO:23 and Revlimid®. In certain embodiments, provided herein is a pharmaceutical composition comprising or consisting of a polypeptide comprising the ActRII inhibitor comprising an amino acid sequence of SEQ ID NO:23 and Revlimid® and a pharmaceutically acceptable carrier. In certain embodiments, provided herein is a pharmaceutical composition comprising an ActRII inhibitor comprising or consisting of a polypeptide comprising the amino acid sequence of SEQ ID NO:25 and Revlimid®. In certain embodiments, provided herein is a pharmaceutical composition comprising or consisting of a polypeptide comprising the ActRII inhibitor comprising an amino acid sequence of SEQ ID NO:25 and Revlimid® and a pharmaceutically acceptable carrier.
- the ActRII inhibitor is administered parenterally.
- compositions suitable for parenteral administration may comprise one or more ActRII polypeptides in combination with one or more pharmaceutically acceptable sterile isotonic aqueous or nonaqueous solutions, dispersions, suspensions or emulsions, or sterile powders which may be reconstituted into sterile injectable solutions or dispersions just prior to use, which may contain antioxidants, buffers, bacteriostats, solutes which render the formulation isotonic with the blood of the intended recipient or suspending or thickening agents.
- compositions of the present invention may be encapsulated or injected in a form for delivery to a target tissue site (e.g., bone).
- a target tissue site e.g., bone
- compositions of the present invention may include a matrix capable of delivering one or more therapeutic compounds
- matrices may be formed of materials presently in use for other implanted medical applications.
- the choice of matrix material is based on biocompatibility, biodegradability, mechanical properties, cosmetic appearance and interface properties. The particular application of the subject compositions will define the appropriate formulation.
- Potential matrices for the compositions may be biodegradable and chemically defined calcium sulfate, tricalciumphosphate, hydroxyapatite, polylactic acid and polyanhydrides. Other potential materials are biodegradable and biologically well defined, such as bone or dermal collagen. Further matrices are comprised of pure proteins or extracellular matrix components. Other potential matrices are non-biodegradable and chemically defined, such as sintered
- administration of the ActRII inhibitor can be orally, e.g., in the form of capsules, cachets, pills, tablets, lozenges (using a flavored basis, usually sucrose and acacia or tragacanth), powders, granules, or as a solution or a suspension in an aqueous or non-aqueous liquid, or as an oil-in- water or water-in-oil liquid emulsion, or as an elixir or syrup, or as pastilles (using an inert base, such as gelatin and glycerin, or sucrose and acacia) and/or as mouth washes and the like, each containing a predetermined amount of an agent as an active ingredient.
- An agent may also be administered as a bolus, electuary or paste.
- one or more therapeutic compounds may be mixed with one or more pharmaceutically acceptable carriers, such as sodium citrate or dicalcium phosphate, and/or any of the following: (1) fillers or extenders, such as starches, lactose, sucrose, glucose, mannitol, and/or silicic acid; (2) binders, such as, for example,
- compositions may also comprise buffering agents.
- Solid compositions of a similar type may also be employed as fillers in soft and hard- filled gelatin capsules using such excipients as lactose or milk sugars, as well as high molecular weight polyethylene glycols and the like.
- Liquid dosage forms for oral administration include pharmaceutically acceptable emulsions, microemulsions, solutions, suspensions, syrups, and elixirs.
- the liquid dosage forms may contain inert diluents commonly used in the art, such as water or other solvents, solubilizing agents and emulsifiers, such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3-butylene glycol, oils (in particular, cottonseed, groundnut, corn, germ, olive, castor, and sesame oils), glycerol, tetrahydrofuryl alcohol, polyethylene glycols and fatty acid esters of sorbitan, and mixtures thereof.
- the oral compositions can also include adjuvants such as wetting agents, emulsifying
- compositions for use with the methods the invention may also contain adjuvants, such as preservatives, wetting agents, emulsifying agents and dispersing agents. Prevention of the action of microorganisms may be ensured by the inclusion of various antibacterial and antifungal agents, for example, paraben, chlorobutanol, phenol sorbic acid, and the like. It may also be desirable to include isotonic agents, such as sugars, sodium chloride, and the like into the compositions. In addition, prolonged absorption of the injectable pharmaceutical form may be brought about by the inclusion of agents which delay absorption, such as aluminum monostearate and gelatin.
- adjuvants such as preservatives, wetting agents, emulsifying agents and dispersing agents. Prevention of the action of microorganisms may be ensured by the inclusion of various antibacterial and antifungal agents, for example, paraben, chlorobutanol, phenol sorbic acid, and the like. It may also be desirable to include isotonic agents, such
- RNA virus such as a retrovirus
- retroviral vector is a derivative of a murine or avian retrovirus.
- retroviral vectors in which a single foreign gene can be inserted include, but are not limited to: Moloney murine leukemia virus (MoMuLV), Harvey murine sarcoma virus (HaMuSV), murine mammary tumor virus (MuMTV), and Rous Sarcoma Virus (RSV).
- MoMuLV Moloney murine leukemia virus
- HaMuSV Harvey murine sarcoma virus
- MuMTV murine mammary tumor virus
- RSV Rous Sarcoma Virus
- Retroviral vectors can transfer or incorporate a gene for a selectable marker so that transduced cells can be identified and generated.
- Retroviral vectors can be made target-specific by attaching, for example, a sugar, a glycolipid, or a protein. Preferred targeting is accomplished by using an antibody.
- specific polynucleotide sequences can be inserted into the retroviral genome or attached to a viral envelope to allow target specific delivery of the retroviral vector containing the ActRIIa polynucleotide.
- the vector is targeted to bone or cartilage.
- colloidal dispersion systems include macromolecule complexes, nanocapsules, microspheres, beads, and lipid-based systems including oil-in-water emulsions, micelles, mixed micelles, and liposomes.
- a colloidal system is a liposome. Liposomes are artificial membrane vesicles which are useful as delivery vehicles in vitro and in vivo. RNA, DNA and intact virions can be encapsulated within the aqueous interior and be delivered to cells in a biologically active form (see e.g., Fraley, et al, Trends Biochem. Sci., 6:77, 1981).
- compositions of the liposome is usually a combination of phospholipids, usually in combination with steroids, especially cholesterol. Other phospholipids or other lipids may also be used.
- the physical characteristics of liposomes depend on pH, ionic strength, and the presence of divalent cations.
- lipids useful in liposome production include phosphatidyl compounds, such as phosphatidylglycerol, phosphatidylcholine, phosphatidylserine, phosphatidylethanolamine, sphingolipids, cerebrosides, and gangliosides.
- Illustrative phospholipids include egg phosphatidylcholine, dipalmitoylphosphatidylcholine, and distearoylphosphatidylcholine.
- the targeting of liposomes is also possible based on, for example, organ-specificity, cell-specificity, and organelle-specificity and is known in the art.
- the ActRII inhibitor is substantially pure in a
- compositions and dosage forms to be used with the methods of the invention comprise an a second active agent, or a pharmaceutically acceptable salt, solvate, hydrate, stereoisomer, clathrate, or prodrug thereof.
- Pharmaceutical compositions and dosage forms can further comprise one or more excipients.
- Single unit dosage forms are suitable for oral, mucosal (e.g., nasal, sublingual, vaginal, buccal, or rectal), parenteral (e.g., subcutaneous, intravenous, bolus injection, intramuscular, or intraarterial), topical (e.g., eye drops or other ophthalmic preparations), transdermal or transcutaneous administration to a patient.
- mucosal e.g., nasal, sublingual, vaginal, buccal, or rectal
- parenteral e.g., subcutaneous, intravenous, bolus injection, intramuscular, or intraarterial
- topical e.g., eye drops or other ophthalmic preparations
- transdermal or transcutaneous administration e.g., transcutaneous administration to a patient.
- dosage forms include, but are not limited to: tablets; caplets; capsules, such as soft elastic gelatin capsules; cachets; troches; lozenges; dispersions; suppositories; powders; aerosols (e.g., nasal sprays or inhalers); gels; liquid dosage forms suitable for oral or mucosal administration to a patient, including suspensions (e.g., aqueous or non-aqueous liquid suspensions, oil-in-water emulsions, or a water-in-oil liquid emulsions), solutions, and elixirs; liquid dosage forms suitable for parenteral administration to a patient; eye drops or other ophthalmic preparations suitable for topical administration; and sterile solids (e.g., crystalline or amorphous solids) that can be reconstituted to provide liquid dosage forms suitable for parenteral administration to a patient.
- suspensions e.g., aqueous or non-aqueous liquid suspensions, oil-in-water e
- composition, shape, and type of dosage forms will typically vary depending on their use.
- a dosage form used in the acute treatment of a disease may contain larger amounts of one or more of the active ingredients it comprises than a dosage form used in the chronic treatment of the same disease.
- a parenteral dosage form may contain smaller amounts of one or more of the active ingredients it comprises than an oral dosage form used to treat the same disease.
- Typical pharmaceutical compositions and dosage forms comprise one or more excipients.
- Suitable excipients are well known to those skilled in the art of pharmacy, and non-limiting examples of suitable excipients are provided herein. Whether a particular excipient is suitable for incorporation into a pharmaceutical composition or dosage form depends on a variety of factors well known in the art including, but not limited to, the way in which the dosage form will be administered to a patient.
- oral dosage forms such as tablets may contain excipients not suited for use in parenteral dosage forms. The suitability of a particular excipient may also depend on the specific active ingredients in the dosage form.
- the decomposition of some active ingredients may be accelerated by some excipients such as lactose, or when exposed to water.
- Active ingredients that comprise primary or secondary amines are particularly susceptible to such accelerated decomposition. Consequently, pharmaceutical compositions and dosage forms can contain little, if any, lactose other mono- or di-saccharides.
- lactose-free means that the amount of lactose present, if any, is insufficient to substantially increase the degradation rate of an active ingredient.
- Lactose-free compositions comprise excipients that are well known in the art and are listed, for example, in the U.S. Pharmacopeia (USP) 25 NF20 (2002).
- lactose-free compositions comprise active ingredients, a binder/filler, and a lubricant in
- Anhydrous pharmaceutical compositions and dosage forms comprising a second active agent can also be used, since water can facilitate the degradation of some compounds.
- water e.g., 5%
- water is widely accepted in the pharmaceutical arts as a means of simulating long-term storage in order to determine characteristics such as shelf- life or the stability of formulations over time. See, e.g., Jens T. Carstensen, Drug Stability: Principles & Practice, 2d. Ed., Marcel Dekker, NY, NY, 1995, pp. 379-80.
- water and heat accelerate the decomposition of some compounds.
- the effect of water on a formulation can be of great significance since moisture and/or humidity are commonly encountered during manufacture, handling, packaging, storage, shipment, and use of formulations.
- anhydrous pharmaceutical composition should be prepared and stored such that its anhydrous nature is maintained. Accordingly, anhydrous compositions are preferably packaged using materials known to prevent exposure to water such that they can be included in suitable formulary kits. Examples of suitable packaging include, but are not limited to, hermetically sealed foils, plastics, unit dose containers (e.g., vials), blister packs, and strip packs. [00326] Like the amounts and types of excipients, the amounts and specific types of active ingredients in a dosage form may differ depending on factors such as, but not limited to, the route by which it is to be administered to patients.
- typical dosage forms comprise an immunomodulatory compound or a pharmaceutically acceptable salt, solvate, hydrate, stereoisomer, clathrate, or prodrug thereof in an amount of from about 0.10 to about 150 mg.
- Typical dosage forms comprise an immunomodulatory compound or a pharmaceutically acceptable salt, solvate, hydrate, stereoisomer, clathrate, or prodrug thereof in an amount of about 0.1, 1, 2, 5, 7.5, 10, 12.5, 15, 17.5, 20, 25, 50, 100, 150 or 200 mg.
- a preferred dosage form comprises 4-(amino)-2-(2,6-dioxo(3-piperidyl))- isoindoline-l,3-dione (ActimidTM) in an amount of about 1, 2, 5, 10, 25 or 50mg.
- ActimidTM 4-(amino)-2-(2,6-dioxo(3-piperidyl))- isoindoline-l,3-dione
- ActimidTM 4-(amino)-2-(2,6-dioxo(3-piperidyl))- isoindoline-l,3-dione
- ActimidTM isoindoline-l,3-dione
- a preferred dosage form comprises 3 (4 amino 1 oxo 1,3 dihydro isoindol 2 yl)-piperidine 2,6 dione (RevimidTM) in an amount of about 5, 10, 25 or 50mg.
- Typical dosage forms comprise the second active ingredient in an
- immunomodulatory compound and any optional additional active agents concurrently administered to the patient.
- compositions that are suitable for oral administration can be presented as discrete dosage forms, such as, but are not limited to, tablets (e.g., chewable tablets), caplets, capsules, and liquids (e.g., flavored syrups).
- dosage forms contain predetermined amounts of active ingredients, and may be prepared by methods of pharmacy well known to those skilled in the art. See generally, Remington's Pharmaceutical Sciences, 18th ed., Mack Publishing, Easton PA (1990).
- Typical oral dosage forms are prepared by combining the active ingredients in an intimate admixture with at least one excipient according to conventional pharmaceutical compounding techniques.
- Excipients can take a wide variety of forms depending on the form of preparation desired for administration.
- excipients suitable for use in oral liquid or aerosol dosage forms include, but are not limited to, water, glycols, oils, alcohols, flavoring agents, preservatives, and coloring agents.
- excipients suitable for use in solid oral dosage forms include, but are not limited to, starches, sugars, micro-crystalline cellulose, diluents, granulating agents, lubricants, binders, and disintegrating agents.
- tablets and capsules represent the most advantageous oral dosage unit forms, in which case solid excipients are employed. If desired, tablets can be coated by standard aqueous or nonaqueous techniques. Such dosage forms can be prepared by any of the methods of pharmacy. In general, pharmaceutical compositions and dosage forms are prepared by uniformly and intimately admixing the active ingredients with liquid carriers, finely divided solid carriers, or both, and then shaping the product into the desired presentation if necessary.
- a tablet can be prepared by compression or molding.
- Compressed tablets can be prepared by compressing in a suitable machine the active ingredients in a free- flowing form such as powder or granules, optionally mixed with an excipient.
- Molded tablets can be made by molding in a suitable machine a mixture of the powdered compound moistened with an inert liquid diluent.
- excipients that can be used in oral dosage forms include, but are not limited to, binders, fillers, disintegrants, and lubricants.
- Binders suitable for use in pharmaceutical compositions and dosage forms include, but are not limited to, corn starch, potato starch, or other starches, gelatin, natural and synthetic gums such as acacia, sodium alginate, alginic acid, other alginates, powdered tragacanth, guar gum, cellulose and its derivatives (e.g., ethyl cellulose, cellulose acetate, carboxymethyl cellulose calcium, sodium carboxymethyl cellulose), polyvinyl pyrrolidone, methyl cellulose, pre-gelatinized starch, hydroxypropyl methyl cellulose, (e.g., Nos. 2208, 2906, 2910), microcrystalline cellulose, and mixtures thereof.
- Suitable forms of microcrystalline cellulose include, but are not limited to, the materials sold as AVICEL-PH-101, AVICEL-PH-103 AVICEL RC-581, AVICEL-PH-105 (available from FMC Corporation, American Viscose Division, Avicel Sales, Marcus Hook, PA), and mixtures thereof.
- a specific binder is a mixture of microcrystalline cellulose and sodium carboxymethyl cellulose sold as AVICEL RC-581.
- Suitable anhydrous or low moisture excipients or additives include AVICEL-PH-103TM and Starch 1500 LM.
- fillers suitable for use in the pharmaceutical compositions and dosage forms disclosed herein include, but are not limited to, talc, calcium carbonate (e.g., granules or powder), microcrystalline cellulose, powdered cellulose, dextrates, kaolin, mannitol, silicic acid, sorbitol, starch, pre-gelatinized starch, and mixtures thereof.
- the binder or filler in pharmaceutical compositions is typically present in from about 50 to about 99 weight percent of the pharmaceutical composition or dosage form.
- Disintegrants are used in the compositions to provide tablets that disintegrate when exposed to an aqueous environment. Tablets that contain too much disintegrant may disintegrate in storage, while those that contain too little may not disintegrate at a desired rate or under the desired conditions. Thus, a sufficient amount of disintegrant that is neither too much nor too little to detrimentally alter the release of the active ingredients should be used to form solid oral dosage forms.
- the amount of disintegrant used varies based upon the type of formulation, and is readily discernible to those of ordinary skill in the art.
- Typical pharmaceutical compositions comprise from about 0.5 to about 15 weight percent of disintegrant, preferably from about 1 to about 5 weight percent of disintegrant.
- Disintegrants that can be used in pharmaceutical compositions and dosage forms include, but are not limited to, agar-agar, alginic acid, calcium carbonate, microcrystalline cellulose, croscarmellose sodium, crospovidone, polacrilin potassium, sodium starch glycolate, potato or tapioca starch, other starches, pre-gelatinized starch, other starches, clays, other algins, other celluloses, gums, and mixtures thereof.
- Lubricants that can be used in pharmaceutical compositions and dosage forms include, but are not limited to, calcium stearate, magnesium stearate, mineral oil, light mineral oil, glycerin, sorbitol, mannitol, polyethylene glycol, other glycols, stearic acid, sodium lauryl sulfate, talc, hydrogenated vegetable oil (e.g., peanut oil, cottonseed oil, sunflower oil, sesame oil, olive oil, corn oil, and soybean oil), zinc stearate, ethyl oleate, ethyl laureate, agar, and mixtures thereof.
- a second active agent can be administered by controlled release means or by delivery devices that are well known to those of ordinary skill in the art. Examples include, but are not limited to, those described in U.S. Patent Nos.: 3,845,770; 3,916,899; 3,536,809; 3,598,123; and 4,008,719, 5,674,533, 5,059,595, 5,591,767, 5,120,548, 5,073,543, 5,639,476, 5,354,556, and 5,733,566, each of which is incorporated herein by reference.
- Such dosage forms can be used to provide slow or controlled-release of one or more active ingredients using, for example, hydropropylmethyl cellulose, other polymer matrices, gels, permeable membranes, osmotic systems, multilayer coatings, microparticles, liposomes, microspheres, or a combination thereof to provide the desired release profile in varying proportions.
- active ingredients for example, hydropropylmethyl cellulose, other polymer matrices, gels, permeable membranes, osmotic systems, multilayer coatings, microparticles, liposomes, microspheres, or a combination thereof to provide the desired release profile in varying proportions.
- Suitable controlled-release formulations known to those of ordinary skill in the art, including those described herein, can be readily selected for use with the second active agent.
- Single unit dosage forms suitable for oral administration such as, but not limited to, tablets, capsules, gelcaps, and caplets that are adapted for controlled-release can be used with the methods and compositions of the present invention.
- controlled-release pharmaceutical products have a common goal of improving drug therapy over that achieved by their non-controlled counterparts.
- the use of an optimally designed controlled-release preparation in medical treatment is characterized by a minimum of drug substance being employed to cure or control the condition in a minimum amount of time.
- Advantages of controlled-release formulations include extended activity of the drug, reduced dosage frequency, and increased patient compliance.
- controlled-release formulations can be used to affect the time of onset of action or other characteristics, such as blood levels of the drug, and can thus affect the occurrence of side (e.g., adverse) effects.
- Controlled-release formulations are designed to initially release an amount of drug (active ingredient) that promptly produces the desired therapeutic effect, and gradually and continually release of other amounts of drug to maintain this level of therapeutic or prophylactic effect over an extended period of time.
- drug active ingredient
- Controlled-release of an active ingredient can be stimulated by various conditions including, but not limited to, H, temperature, enzymes, water, or other physiological conditions or compounds.
- Parenteral dosage forms can be administered to patients by various routes including, but not limited to, subcutaneous, intravenous (including bolus injection), intramuscular, and intraarterial. Because their administration typically bypasses patients' natural defenses against contaminants, parenteral dosage forms are preferably sterile or capable of being sterilized prior to administration to a patient. Examples of parenteral dosage forms include, but are not limited to, solutions ready for injection, dry products ready to be dissolved or suspended in a pharmaceutically acceptable vehicle for injection, suspensions ready for injection, and emulsions.
- Suitable vehicles that can be used to provide parenteral dosage forms are well known to those skilled in the art. Examples include, but are not limited to: Water for Injection USP; aqueous vehicles such as, but not limited to, Sodium Chloride Injection, Ringer's Injection, Dextrose Injection, Dextrose and Sodium Chloride Injection, and Lactated Ringer's Injection; water-miscible vehicles such as, but not limited to, ethyl alcohol, polyethylene glycol, and polypropylene glycol; and non-aqueous vehicles such as, but not limited to, corn oil, cottonseed oil, peanut oil, sesame oil, ethyl oleate, isopropyl myristate, and benzyl benzoate.
- water for Injection USP Water for Injection USP
- aqueous vehicles such as, but not limited to, Sodium Chloride Injection, Ringer's Injection, Dextrose Injection, Dextrose and Sodium Chloride Injection,
- Compounds that increase the solubility of a second active agent can also be incorporated into the parenteral dosage forms.
- cyclodextrin and its derivatives can be used to increase the solubility of an immunomodulatory compound and its derivatives. See, e.g., U.S. Patent No. 5,134,127, which is incorporated herein by reference.
- Topical and mucosal dosage forms include, but are not limited to, sprays, aerosols, solutions, emulsions, suspensions, eye drops or other ophthalmic preparations, or other forms known to one of skill in the art. See, e.g., Remington's Pharmaceutical Sciences, 16th and 18th eds., Mack Publishing, Easton PA (1980 & 1990); and Introduction to Pharmaceutical Dosage Forms, 4th ed., Lea & Febiger, Philadelphia (1985). Dosage forms suitable for treating mucosal tissues within the oral cavity can be formulated as mouthwashes or as oral gels.
- Moisturizers or humectants can also be added to pharmaceutical compositions and dosage forms if desired. Examples of such additional ingredients are well known in the art. See, e.g., Remington's Pharmaceutical Sciences, 16th and 18th eds., Mack Publishing, Easton PA (1980 & 1990).
- stearates can serve as a lipid vehicle for the formulation, as an emulsifying agent or surfactant, and as a delivery-enhancing or penetration-enhancing agent.
- Different salts, hydrates or solvates of the active ingredients can be used to further adjust the properties of the resulting composition.
- an ActRII inhibitor and a second active agent are not administered to a patient at the same time or by the same route of administration.
- kits which, when used by the medical practitioner, can simplify the administration of appropriate amounts of active ingredients to a patient.
- a typical kit provided herein comprises a dosage form of one or more ActRII inhibitors (i.e., and inhibitor of ActRIIA or ActRIIB; see Section 4.2) or a dosage form of one or more ActRII inhibitors (i.e., and inhibitor of ActRIIA or ActRIIB; see Section 4.2) and a dosage form of a a second active agent (see Section 4.3) or a pharmaceutically acceptable salt salt, solvate, hydrate, stereoisomer, prodrug, or clathrate thereof.
- a kit provided herein comprises a dosage form of an ActRIIA inhibitor comprising or consisting of a polypeptide that comprises the amino acid sequence of SEQ ID NO:7 and a dosage form of Revlimid®.
- a kit provided herein comprises a dosage form of an ActRIIB inhibitor comprising or consisting of a polypeptide that comprises the amino acid sequence of SEQ ID NO:23 and a dosage form of Revlimid®.
- kits provided herein comprises a dosage form of an ActRIIB inhibitor comprising or consisting of a polypeptide that comprises the amino acid sequence of SEQ ID NO:25 and a dosage form of Revlimid®.
- a kit provided herein comprises a dosage form of an ActRIIA inhibitor comprising and a dosage form of an ActRIIB inhibitor.
- a kit may additionally comprise a dosage form of a second active agent.
- a kit provided herein comprises a dosage form of an ActRIIA inhibitor comprising or consisting of a polypeptide that comprises the amino acid sequence of SEQ ID NO: 7 and a dosage form of an ActRIIB inhibitor comprising or consisting of a polypeptide that comprises the amino acid sequence of SEQ ID NO:25.
- a kit provided herein comprises a dosage form of an ActRIIA inhibitor comprising or consisting of a polypeptide that comprises the amino acid sequence of SEQ ID NO:7, a dosage form of an ActRIIB inhibitor comprising or consisting of a polypeptide that comprises the amino acid sequence of SEQ ID NO:25, and a dosage form of Revlimid®.
- the selected form employs the TPA leader and has the following unprocessed amino acid sequence is set forth in SEQ ID NO: 13. This polypeptide is encoded by SEQ ID NO: 14.
- ActRIIb-Fc has a sequence SEQ ID NO:20.
- the growth inhibitory effects of ActRIIA inhibitors and lenalidomide against multiple myeloma cell lines and CD 138+ cells from multiple myeloma patients' bone marrow aspirates can be assessed by culturing the cell lines and cells with ActRIIA inhibitors and lenalidomide alone and in co-culture with BMSC, OC and OB; followed by measuring growth inhibitory effects via MTT assay, [3H]-thymidine uptake, and cytokine secretion with ELISA.
- the molecular mechanisms of action of a combination of ActRIIA inhibitors and lenalidomide can be assessed by detecting signaling pathways and transcription factor expression in the different components of the tumor niche using Western blotting and quantitative PCR analysis.
- amino acids 20-134 of SEQ ID NO: 16 amino acids 20-134 of SEQ ID NO: 16
- amino acids 20-134 of SEQ ID NO:28 amino acids 20-134 of SEQ ID NO:28
- amino acids 20-134 of SEQ ID NO:28 amino acids 20-134 of SEQ ID NO:28 with L79D mutation fused to an Fc domain with a GGG linker
- amino acids 20-134 of SEQ ID NO: 16 amino acids 20-134 of SEQ ID NO: 16 with L79D mutation fused to an Fc domain
- amino acids 20-134 of SEQ ID NO:28 with L79D mutation fused to an Fc domain and with TPA leader sequence
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Cell Biology (AREA)
- Zoology (AREA)
- Gastroenterology & Hepatology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Provided herein are compositions comprising inhibitors of the Type II Activin Receptor ("ActRII") and kits comprising such compositions. Also provided herein are methods of treatment of disease comprising administration of one or more of the compositions described herein and administration of a second active agent.
Description
COMBINATION THERAPY FOR
TREATMENT OF DISEASE
[0001] This application claims priority to U.S. Provisional Application No. 61/377,718, filed August 27, 2010, which is incorporated herein by reference in its entirety.
1. INTRODUCTION
[0002] Provided herein are compositions comprising inhibitors of the Type II Activin Receptor ("ActRII") and kits comprising such compositions. Also provided herein are methods of treatment of disease comprising administration of one or more of the
compositions described herein and administration of a second active agent.
2. BACKGROUND
[0003] Activins were originally discovered as gonadal peptides involved in the regulation of follicle stimulating hormone synthesis, and are now believed to be involved in the regulation of a number of biological activities. Activins bind and signal through a combination of activin type II recptors (ActRII), including activin type IIA (ActRIIA) and activin type IIB (ActRIIB) receptors, both of which are transmembrane serine/threonine kinases (Harrison et al, J. Biol. Chem. 279, 28036-28044 (2004)). Inhibition of activin and/or activin receptors has shown promise in the treatment of disease.
[0004] Inhibitors of ActRII receptors are known in the art, and include soluble polypeptides that comprise the activin-binding domains of ActRII receptors (See, e.g., International Patent Application Publication Nos. WO/2002/088171, WO/2002/032925, WO/2005/037989; WO/2006/055689, WO 2006/012627 and WO 2010/019261; U.S. Patent Application Publication Nos. 2003/0133939, US 2005/0238646 and US 2010/0068215). Such polypeptides possess the ability to sequester ActRII ligands, thereby preventing signaling through the ActRII receptors, and may comprise all or a portion of the extracellular domain of an ActRII receptor (e.g., all or a portion of the extracellular domain of ActRIIA or all or a portion of the extracellular domain of ActRIIB).
[0005] Multiple myeloma (MM; also known as myeloma, plasma cell myeloma, or Kahler's disease) is a type of cancer of plasma cells, which are antibody-producing immune system cells. Symptoms of multiple myeloma include bone pain, infection, renal failure,
anemia, and bone lesions. Types of multiple myeloma include relapsed multiple myeloma, refractory multiple myeloma, and newly diagnosed multiple myeloma.
[0006] Myelodysplasia syndrome (MDS) is a diverse collection of hematological (blood- related) medical conditions that involve ineffective production (or dysplasia) of the myeloid class of blood cells. Patients with MDS often develop severe anemia and require frequent blood transfusions. In most cases, the disease worsens and the patient develops cytopenias (low blood counts) due to progressive bone marrow failure. About one third of patients with MDS develop acute myelogenous leukemia (AML) within months or years.
[0007] Anemia, the most common disorder of the blood, is a decrease in number of red blood cells (RBCs) or less than the normal quantity of hemoglobin in the blood. Symptoms of anemia include feelings of weakness and/or fatigue, general malaise and sometimes poor concentration. In very severe anemia, the body may compensate for the lack of oxygen- carrying capability of the blood by increasing cardiac output, resulting in symptoms such as palpitations, angina, intermittent claudication of the legs, and symptoms of heart failure.
3. SUMMARY
[0008] In one aspect, provided herein are compositions comprising inhibitors of ActRII receptors (e.g., the ActRIIA and ActRIIB inhibitors provided in Section 4.2, below). In certain embodiments, the compositions comprising inhibitors of ActRII receptors are administered in combination with a second active agent (e.g., the second active agents provided in Section 4.3, below).
[0009] In one embodiment, provided herein are compositions comprising inhibitors of ActRII receptors that can be administered in combination with a second active agent, wherein the ActRII receptor inhibitor is a polypeptide comprising all or a portion of an ActRII extracellular domain (e.g., the extracellular domain of ActRIIA or ActRIIB) and wherein the second active agent is an IMiD® compound (such as lenalidomide (Revlimid®), i.e., 3-(4- amino-l-oxo-l,3-dihydro-isoindol-2-yl)-piperidine-2,6-dione), cyclophosphamide, bisphosphonate, thalidomide, dexamethasone, melphalan, prednisone, bortezomib, azacitidine, doxorubicin, vincristine, proteasome inhibitor and dacogen.
[0010] In a specific embodiment, a composition provided herein comprises (i) an
ActRIIA inhibitor, wherein the ActRIIA inhibitor comprises or consists of a polypeptide selected from the group consisting of: a. a polypeptide at least 90% identical to SEQ ID
NO:2; b. a polypeptide at least 95% identical to SEQ ID NO:2; c. a polypeptide at least 98% identical to SEQ ID NO:2; d. SEQ ID NO:2; e. a polypeptide at least 90% identical to SEQ ID NO:3; f. a polypeptide at least 95% identical to SEQ ID NO:3; g. a polypeptide at least 98% identical to SEQ ID NO:3; h. SEQ ID NO:3; i. a polypeptide at least 90% identical to SEQ ID NO:6; j. a polypeptide at least 95% identical to SEQ ID NO:6; k. a polypeptide at least 98% identical to SEQ ID NO:6; 1. SEQ ID NO:6; m. a polypeptide at least 90% identical to SEQ ID NO:7; n. a polypeptide at least 95% identical to SEQ ID NO:7; o. a polypeptide at least 98% identical to SEQ ID NO:7; p. SEQ ID NO:7; q. a polypeptide at least 90% identical to SEQ ID NO: 12; r. a polypeptide at least 95% identical to SEQ ID NO: 12; s. a polypeptide at least 98% identical to SEQ ID NO: 12; and t. SEQ ID NO: 12. In a specific embodiment, the ActRIIA inhibitor is a polypeptide comprising or consisting of the amino acid sequence of SEQ ID NO:7.
[0011] In another specific embodiment, a composition provided herein comprises an ActRIIB inhibitor, wherein the ActRIIB inhibitor comprises or consists of a polypeptide selected from the group consisting of: a. a polypeptide at least 90%> identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; b. a polypeptide at least 95% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; c. a polypeptide at least 98% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; d. SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; e. a polypeptide 90% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; f. a polypeptide 95% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; g. a polypeptide 98% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; and h. SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47. In a specific embodiment, the ActRIIB inhibitor is a polypeptide comprising or consisting of SEQ ID NO:23. In another specific embodiment, the ActRIIB inhibitor is a polypeptide comprising or consisting of SEQ ID NO:25.
[0012] In another specific embodiment, a composition provided herein comprises an ActRIIA inhibitor and an ActRIIB inhibitor, wherein the ActRIIA inhibitor comprises or consists of a polypeptide selected from the group consisting of: a. a polypeptide at least 90%> identical to SEQ ID NO:2; b. a polypeptide at least 95% identical to SEQ ID NO:2; c. a polypeptide at least 98% identical to SEQ ID NO:2; d. SEQ ID NO:2; e. a polypeptide at least 90% identical to SEQ ID NO:3; f. a polypeptide at least 95% identical to SEQ ID NO:3;
g. a polypeptide at least 98% identical to SEQ ID NO:3; h. SEQ ID NO:3; i. a polypeptide at least 90% identical to SEQ ID NO:6; j. a polypeptide at least 95% identical to SEQ ID NO:6; k. a polypeptide at least 98% identical to SEQ ID NO:6; 1. SEQ ID NO:6; m. a polypeptide at least 90% identical to SEQ ID NO:7; n. a polypeptide at least 95% identical to SEQ ID NO:7; o. a polypeptide at least 98% identical to SEQ ID NO:7; p. SEQ ID NO:7; q. a polypeptide at least 90% identical to SEQ ID NO: 12; r. a polypeptide at least 95% identical to SEQ ID NO: 12; s. a polypeptide at least 98% identical to SEQ ID NO: 12; and t. SEQ ID NO: 12; and wherein the ActRIIB inhibitor comprises or consists of a polypeptide selected from the group consisting of: a. a polypeptide at least 90% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; b. a polypeptide at least 95% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; c. a polypeptide at least 98% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; d. SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; e. a polypeptide 90% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; f a polypeptide 95% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; g. a polypeptide 98% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; and h. SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47. In a specific embodiment, the ActRIIA inhibitor is a polypeptide comprising or consisting of SEQ ID NO: 7 and the ActRIIB inhibitor is a polypeptide comprising or consisting of SEQ ID NO:23. In another specific embodiment, the ActRIIA inhibitor is a polypeptide comprising or consisting of SEQ ID NO: 7 and the ActRIIB inhibitor is a polypeptide comprising or consisting of SEQ ID NO:25.
[0013] In certain embodiments, when administered in combination with a second active agent, the ActRII receptor inhibitor, or composition comprising the ActRII receptor inhibitor, can be administered parenterally, and the second active agent, e.g., 3-(4-amino-l-oxo-l,3- dihydro- isoindol-2-yl) -piperidine -2,6 -dione (i.e., lenalidomide (Revlimid®)), can be administered orally. More specifically, the second active agent, e.g., 3-(4-amino-l-oxo-l,3- dihydro- isoindol-2-yl)-piperidine-2,6-dione (i.e., lenalidomide (Revlimid®)), can be administered in the form of a capsule or tablet. In certain embodiments, the second active agent, e.g., 3-(4-amino-l-oxo-l,3-dihydro- isoindol-2-yl) -piperidine -2,6 -dione (i.e., lenalidomide (Revlimid®)), can be administered in an amount of from about 5 to about 25 mg per day.
[0014] In another aspect, provided herein are methods for treating and/or preventing disease, wherein the methods comprise administering one or more ActRII receptor inhibitors, or administering one or more ActRII receptor inhibitors in combination with the
administration of one or more second active agents.
[0015] In a specific embodiment, provided herein is a method for treating and/or preventing disease comprising administering an ActRIIA inhibitor and administering a second active agent. The ActRIIA inhibitor can comprise or consist of a polypeptide comprising an amino acid sequence selected from the group consisting of: a. a polypeptide at least 90% identical to SEQ ID NO:2; b. a polypeptide at least 95% identical to SEQ ID NO:2; c. a polypeptide at least 98% identical to SEQ ID NO:2; d. SEQ ID NO:2; e. a polypeptide at least 90% identical to SEQ ID NO:3; f. a polypeptide at least 95% identical to SEQ ID NO:3; g. a polypeptide at least 98% identical to SEQ ID NO:3; h. SEQ ID NO:3; i. a polypeptide at least 90% identical to SEQ ID NO:6; j. a polypeptide at least 95% identical to SEQ ID NO:6; k. a polypeptide at least 98% identical to SEQ ID NO:6; 1. SEQ ID NO:6; m. a polypeptide at least 90% identical to SEQ ID NO:7; n. a polypeptide at least 95% identical to SEQ ID NO:7; o. a polypeptide at least 98% identical to SEQ ID NO:7; p. SEQ ID NO:7; q. a polypeptide at least 90% identical to SEQ ID NO: 12; r. a polypeptide at least 95% identical to SEQ ID NO: 12; s. a polypeptide at least 98% identical to SEQ ID NO: 12; and t. SEQ ID NO: 12. The second active agent can be one or more of the following: an IMiD® compound (such as lenalidomide (Revlimid®), i.e., 3-(4-amino-l-oxo-l,3-dihydro-isoindol-2-yl)-piperidine-2,6- dione); pomalidomide (4-(amino)-2-(2,6-dioxo(3-piperidyl))-isoindoline-l,3-dione); and 3- (5-amino-2-methyl-4-oxo-4H-quinazolin-3-yl)-piperidine-2,6-dione), cyclophosphamide, bisphosphonate, thalidomide, dexamethasone, melphalan, prednisone, bortezomib, azacitidine, doxorubicin, vincristine, proteasome inhibitor, and dacogen. In a specific embodiment, the ActRIIA inhibitor is a polypeptide comprising or consisting of the amino acid sequence of SEQ ID NO: 7 and the second active agent is lenalidomide (Revlimid®).
[0016] In another specific embodiment, provided herein is a method for treating and/or preventing disease comprising administering an ActRIIB inhibitor and administering a second active agent. The ActRIIB inhibitor can comprise or consist of a polypeptide comprising an amino acid sequence selected from the group consisting of: a. a polypeptide at least 90% identical to SEQ ID NO:17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; b. a polypeptide at least 95% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36,
37, 42, or 43; c. a polypeptide at least 98% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; d. SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; e. a polypeptide 90% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; f. a polypeptide 95% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; g. a polypeptide 98% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; and h. SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47. The second active agent can be one or more of the following: an IMiD® compound (such as lenalidomide (Revlimid®), i.e., 3-(4-amino-l-oxo-l,3-dihydro-isoindol-2-yl)-piperidine-2,6- dione); pomalidomide (4-(amino)-2-(2,6-dioxo(3-piperidyl))-isoindoline-l,3-dione); and 3- (5-amino-2-methyl-4-oxo-4H-quinazolin-3-yl)-piperidine-2,6-dione), cyclophosphamide, bisphosphonate, thalidomide, dexamethasone, melphalan, prednisone, bortezomib, azacitidine, doxorubicin, vincristine, proteasome inhibitor, and dacogen. In a specific embodiment, the ActRIIB inhibitor is a polypeptide comprising or consisting of the amino acid sequence of SEQ ID NO:23 and the second active agent is lenalidomide (Revlimid®). In a specific embodiment, the ActRIIB inhibitor is a polypeptide comprising or consisting of the amino acid sequence of SEQ ID NO:25 and the second active agent is lenalidomide (Revlimid®).
[0017] In another specific embodiment, provided herein is a method for treating and/or preventing disease comprising administering an ActRIIA inhibitor and an ActRIIB inhibitor. The ActRIIA inhibitor can comprise or consist of a polypeptide comprising an amino acid sequence selected from the group consisting of: a. a polypeptide at least 90%> identical to SEQ ID NO:2; b. a polypeptide at least 95% identical to SEQ ID NO:2; c. a polypeptide at least 98% identical to SEQ ID NO:2; d. SEQ ID NO:2; e. a polypeptide at least 90% identical to SEQ ID NO:3; f. a polypeptide at least 95% identical to SEQ ID NO:3; g. a polypeptide at least 98% identical to SEQ ID NO:3; h. SEQ ID NO:3; i. a polypeptide at least 90% identical to SEQ ID NO:6; j. a polypeptide at least 95% identical to SEQ ID NO:6; k. a polypeptide at least 98% identical to SEQ ID NO:6; 1. SEQ ID NO:6; m. a polypeptide at least 90% identical to SEQ ID NO:7; n. a polypeptide at least 95% identical to SEQ ID NO:7; o. a polypeptide at least 98% identical to SEQ ID NO:7; p. SEQ ID NO:7; q. a polypeptide at least 90% identical to SEQ ID NO: 12; r. a polypeptide at least 95% identical to SEQ ID NO: 12; s. a polypeptide at least 98% identical to SEQ ID NO: 12; and t. SEQ ID NO: 12. The ActRIIB inhibitor can comprise or consist of a polypeptide comprising an amino acid sequence selected from the
group consisting of: a. a polypeptide at least 90% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; b. a polypeptide at least 95% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; c. a polypeptide at least 98% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; d. SEQ ID NO: 17, 18,
23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; e. a polypeptide 90% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; f. a polypeptide 95% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; g. a polypeptide 98% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; and h. SEQ ID NO:20, 21,
24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47. In a specific embodiment, the ActRIIA inhibitor is a polypeptide comprising or consisting of SEQ ID NO: 7 and the ActRIIB inhibitor is a polypeptide comprising or consisting of the amino acid sequence of SEQ ID NO:23. In another specific embodiment, the ActRIIA inhibitor is a polypeptide comprising or consisting of SEQ ID NO: 7 and the ActRIIB inhibitor is a polypeptide comprising or consisting of the amino acid sequence of SEQ ID NO:25.
[0018] In another specific embodiment, provided herein is a method for treating and/or preventing disease comprising administering an ActRIIA inhibitor and an ActRIIB inhibitor, and a second active agent. The ActRIIA inhibitor can comprise or consist of a polypeptide comprising an amino acid sequence selected from the group consisting of: a. a polypeptide at least 90% identical to SEQ ID NO:2; b. a polypeptide at least 95% identical to SEQ ID NO:2; c. a polypeptide at least 98% identical to SEQ ID NO:2; d. SEQ ID NO:2; e. a polypeptide at least 90% identical to SEQ ID NO:3; f. a polypeptide at least 95% identical to SEQ ID NO:3; g. a polypeptide at least 98% identical to SEQ ID NO:3; h. SEQ ID NO:3; i. a polypeptide at least 90% identical to SEQ ID NO:6; j. a polypeptide at least 95% identical to SEQ ID NO:6; k. a polypeptide at least 98% identical to SEQ ID NO:6; 1. SEQ ID NO:6; m. a polypeptide at least 90% identical to SEQ ID NO:7; n. a polypeptide at least 95% identical to SEQ ID NO:7; o. a polypeptide at least 98% identical to SEQ ID NO:7; p. SEQ ID NO:7; q. a polypeptide at least 90% identical to SEQ ID NO: 12; r. a polypeptide at least 95% identical to SEQ ID NO: 12; s. a polypeptide at least 98% identical to SEQ ID NO: 12; and t. SEQ ID NO: 12. The ActRIIB inhibitor can comprise or consist of a polypeptide comprising an amino acid sequence selected from the group consisting of: a. a polypeptide at least 90%> identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; b. a polypeptide at least 95% identical to SEQ ID NO:17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; c. a
polypeptide at least 98% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; d. SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; e. a polypeptide 90% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; f. a polypeptide 95% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; g. a polypeptide 98% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; and h. SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47. The second active agent can be one or more of the following: an IMiD® compound (such as lenalidomide (Revlimid®), i.e., 3-(4-amino-l-oxo-l,3-dihydro-isoindol-2-yl)-piperidine-2,6-dione);
pomalidomide (4-(amino)-2-(2,6-dioxo(3-piperidyl))-isoindoline-l,3-dione); and 3-(5-amino- 2-methyl-4-oxo-4H-quinazolin-3-yl)-piperidine-2,6-dione)), cyclophosphamide,
bisphosphonate, thalidomide, dexamethasone, melphalan, prednisone, bortezomib, azacitidine, doxorubicin, vincristine, proteasome inhibitor, and dacogen. In a specific embodiment, the ActRIIA inhibitor is a polypeptide comprising or consisting of SEQ ID NO:7, the ActRIIB inhibitor is a polypeptide comprising or consisting of the amino acid sequence of SEQ ID NO:23, and the second active agent is lenalidomide (Revlimid®). In another specific embodiment, the ActRIIA inhibitor is a polypeptide comprising or consisting of SEQ ID NO:7, the ActRIIB inhibitor is a polypeptide comprising or consisting of the amino acid sequence of SEQ ID NO:25, and the second active agent is lenalidomide
(Revlimid®).
[0019] In a specific embodiment, the methods of treating disease provided herein comprise parenteral administration of an ActRIIA inhibitor comprising or consisting of a polypeptide that comprises SEQ ID NO:7 and oral administration (e.g., in the form of a capsule or tablet) of lenalidomide (Revlimid®) at an amount of from about 5 to about 25 mg per day. In another specific embodiment, the methods of treating disease provided herein comprise parenteral administration of an ActRIIB inhibitor comprising a polypeptide that comprises or consists of SEQ ID NO:23 and oral administration (e.g., in the form of a capsule or tablet) of lenalidomide (Revlimid®) at an amount of from about 5 to about 25 mg per day. In another specific embodiment, the methods of treating disease provided herein comprise parenteral administration of an ActRIIB inhibitor comprising a polypeptide that comprises or consists of SEQ ID NO:25 and oral administration (e.g., in the form of a capsule or tablet) of lenalidomide (Revlimid®) at an amount of from about 5 to about 25 mg per day.
[0020] In one embodiment, provided herein is a method of treating multiple myeloma, comprising administering an ActRII inhibitor and administering a second active agent. In a specific embodiment, the ActRII inhibitor is an ActRIIA inhibitor, wherein said ActRIIA inhibitor is a polypeptide that comprises or consists of SEQ ID NO:7; and the second active agent is Revlimid®. In another specific embodiment, the ActRII inhibitor is an ActRIIB inhibitor, wherein said ActRIIB inhibitor is a polypeptide that comprises or consists of SEQ ID NO:25; and the second active agent is Revlimid®. In a specific embodiment, the ActRII inhibitor is an ActRIIA inhibitor, wherein said ActRIIA inhibitor is a polypeptide that comprises or consists of SEQ ID NO:7; and the second active agent is pomalidomide. In another specific embodiment, the ActRII inhibitor is an ActRIIB inhibitor, wherein said ActRIIB inhibitor is a polypeptide that comprises or consists of SEQ ID NO:25; and the second active agent is pomalidomide.
[0021] In one embodiment, provided herein is a method of treating myelodysplasia syndrome, comprising administering an ActRII inhibitor and administering a second active agent. In a specific embodiment, the ActRII inhibitor is an ActRIIA inhibitor, wherein said ActRIIA inhibitor is a polypeptide that comprises or consists of SEQ ID NO:7; and the second active agent is Revlimid®. In another specific embodiment, the ActRII inhibitor is an ActRIIB inhibitor, wherein said ActRIIB inhibitor is a polypeptide that comprises or consists of SEQ ID NO:25; and the second active agent is Revlimid®. In another specific
embodiment, the ActRII inhibitor is an ActRIIA inhibitor, wherein said ActRIIA inhibitor is a polypeptide that comprises or consists of SEQ ID NO:7; and the second active agent is pomalidomide. In another specific embodiment, the ActRII inhibitor is an ActRIIB inhibitor, wherein said ActRIIB inhibitor is a polypeptide that comprises or consists of SEQ ID NO:25; and the second active agent is pomalidomide.
[0022] In one embodiment, provided herein is a method of treating anemia, comprising administering an ActRII inhibitor and administering a second active agent. In a specific embodiment, the ActRII inhibitor is an ActRIIA inhibitor, wherein said ActRIIA inhibitor is a polypeptide that comprises or consists of SEQ ID NO:7; and the second active agent is Revlimid® or pomalidomide. In another specific embodiment, the ActRII inhibitor is an ActRIIB inhibitor, wherein said ActRIIB inhibitor is a polypeptide that comprises or consists of SEQ ID NO:25; and the second active agent is Revlimid® or pomalidomide.
[0023] In certain embodiments, the ActRII inhibitor(s) and the second active agent(s) are administered at the same time. In certain embodiments, the ActRII inhibitor and the second active agent are administered in the same formulation. In other embodiments, the ActRII inhibitor and the second active agent are administered sequentially.
[0024] In certain embodiments, the present invention provides a method for improving the safety of a treatment with 3-(4-amino-l-oxo-l,3-dihydro-isoindol-2-yl)-piperidine-2,6- dione (i.e., lenalidomide (Revlimid®)) in a patient, wherein the method comprises administering an ActRII inhibitor(s) to the patient being treated with 3-(4-amino-l-oxo-l,3- dihydro-isoindol-2-yl)-piperidine-2,6-dione (i.e., lenalidomide (Revlimid®)).
4. DETAILED DESCRIPTION
4.1 OVERVIEW
[0025] Provided herein are compositions comprising ActRII receptor inhibitors (e.g., ActRIIA or ActRIIB inhibitors) that can be administered in combination with each other and/or in combination with second active agents, as well as methods for the treatment of disease comprising administration of one or more ActRII receptor inhibitors or one or more ActRII receptor inhibitors and a second active agent, such as thalidomide, dexamethasone, melphalan, prednisone, bortezomib, cyclophosphamide, bisphosphonate, dacogen, azacitidine, doxorubicin, vincristine, proteasome inhibitor, and/or an IMiD® compound (e.g., lenalidomide (also known as Revlimid® or 3-(4-amino-l-oxo-l,3-dihydro-isoindol-2-yl)- piperidine-2,6-dione); pomalidomide (4-(amino)-2-(2,6-dioxo(3-piperidyl))-isoindoline-l ,3- dione); and 3-(5-amino-2-methyl-4-oxo-4H-quinazolin-3-yl)-piperidine-2,6-dione)).
Exemplary diseases that can be treated using the methods provided herein include cancer, multiple myeloma (e.g., relapsed multiple myeloma, refractory multiple myeloma, and newly diagnosed multiple myeloma), myelodysplastic syndrome (MDS), and anemia. The ActRII receptor inhibitor(s) can be administered separately from the second active agent(s), or the ActRII receptor inhibitor(s) can be administered at the same time as, or in the same pharmaceutical formulation as, the second active agent(s). Inhibitors of ActRII receptors are described in Section 4.2. Second active agents are described in Section 4.3. Methods of treatment of disease, as well as dosage and administration regimens, are described in Section 4.4. Patient populations are described in Section 4.5. Pharmaceutical compositions are described in Section 4.7.
4.2 INHIBITORS OF ACTRII RECEPTORS
[0026] Inhibitors of ActRII receptors (also referred to herein as "activin-ActRII inhibitors") encompassed herein include ActRIIA inhibitors and ActRIIB inhibitors (see below). In certain embodiments, an ActRII receptor inhibitor is specific to ActRIIA. In other embodiments, an ActRII receptor inhibitor is specific to ActRIIB. In certain embodiments, an ActRII receptor inhibitor preferentially inhibits ActRIIA. In other embodiments, an ActRII receptor inhibitor preferenctially inhibits ActRIIB. In certain embodiments, an ActRII receptor inhibitor inhibits both ActRIIA and ActRIIB.
[0027] In certain embodiments, inhibitors of ActRII receptors can be polypeptides comprising activin-binding domains of ActRII. Without being bound by theory, such activin- binding domain comprising polypeptides sequester activin and thereby prevent activin signaling. These activin-binding domain comprising polypeptides may comprise all or a portion of the extracellular domain of an ActRII receptor (i.e., all or a portion of the extracellular domain of ActRIIA or all or a portion of the extracellular domain of ActRIIB). In specific embodiments, the extracellular domain of an ActRII receptor is soluble.
[0028] In certain embodiments, the activin-binding domain comprising polypeptides are linked to an Fc portion of an antibody (i.e., a conjugate comprising an activin-binding domain comprising polypeptide of an ActRII receptor and an Fc portion of an antibody is generated). Without being bound by theory, the antibody portion confers increased stability on the conjugate. In certain embodiments, the activin-binding domain is linked to an Fc portion of an antibody via a linker, e.g., a peptide linker.
[0029] The inhibitors of ActRII receptors used in the compositions and methods described herein comprise molecules that inhibit ActRIIA and/or ActRIIB, directly or indirectly, either extracellularly or intracellularly. In some embodiments, the inhibitors of ActRIIA and/or ActRIIB used in the compositions and methods described herein inhibit ActRIIA and/or ActRIIB via interactions with the receptor(s) itself. In other embodiments, the inhibitors of ActRIIA and/or ActRIIB used in the compositions and methods described herein inhibit ActRIIA and/or ActRIIB via interactions with an ActRIIA and/or ActRIIB ligand, e.g., Activin.
(a) INHIBITORS OF ACTRIIA
[0030] As used herein, the term "ActRIIA" refers to a family of activin receptor type Ila
(ActRIIA) proteins from any species and variants derived from such ActRIIA proteins by
mutagenesis or other modification. Reference to ActRIIA herein is understood to be a reference to any one of the currently identified forms. Members of the ActRIIA family are generally transmembrane proteins, composed of a ligand-binding extracellular domain with a cysteine-rich region, a transmembrane domain, and a cytoplasmic domain with predicted serine/threonine kinase activity.
[0031] ActRIIA inhibitors to be used in the compositions and methods described herein include, without limitation, activin-binding soluble ActRIIA polypeptides; antibodies that bind to activin (particularly the activin A or B subunits, also referred to as BA or BB) and disrupt ActRIIA binding; antibodies that bind to ActRIIA and disrupt activin binding; non- antibody proteins selected for activin or ActRIIA binding (see e.g., WO/2002/088171 , WO/2006/055689, WO/2002/032925, WO/2005/037989, US 2003/0133939, and US
2005/0238646, each of which is incorporated herein by reference in its entirety, for examples of such proteins and methods for design and selection of same); and randomized peptides selected for activin or ActRIIA binding, which can be conjugated to an Fc domain.
[0032] In certain embodiments, two or more different proteins (or other moieties) with activin or ActRIIA binding activity, especially activin binders that block the type I (e.g., a soluble type I activin receptor) and type II (e.g., a soluble type II activin receptor) binding sites, respectively, may be linked together to create a bifunctional or multifunctional binding molecule that inhibits ActRIIA and thus can be used in the compositions and methods described herein include. In certain embodiments, Activin- ActRIIA signaling axis antagonists that inhibit ActRIIA include nucleic acid aptamers, small molecules and other agents are used in the compositions and methods described herein include.
(i) ActRIIA Inhibitors Comprising ActRIIA Polypeptides
[0033] The term "ActRIIA polypeptide" includes polypeptides comprising any naturally occurring polypeptide of an ActRIIA family member as well as any variants thereof
(including mutants, fragments, fusions, and peptidomimetic forms) that retain a useful activity. For example, ActRIIA polypeptides include polypeptides derived from the sequence of any known ActRIIA having a sequence at least about 80% identical to the sequence of an ActRIIA polypeptide, and optionally at least 85%, 90%, 95%, 97%, 98%, 99% or greater identity. For example, an ActRIIA polypeptide may bind to and inhibit the function of an ActRIIA protein and/or activin. Examples of ActRIIA polypeptides include human ActRIIA precursor polypeptide (SEQ ID NO: 1) and soluble human ActRIIA polypeptides (e.g., SEQ
ID NOs: 2, 3, 7 and 12). With respect to the ActRIIA precursor polypeptide whose amino acid sequence is depicted at SEQ ID NO: l, the signal peptide of the human ActRIIA precursor polypeptide located at amino acid positions 1 to 20; the extracellular domain is located at amino acid positions 21 to 135 and the N-linked glycosylation sites of the human ActRIIA precursor polypeptide (SEQ ID NO: 1) are located at amino acid positions 43 and 56 of SEQ ID NO: 1. The nucleic acid sequence encoding the human ActRIIB precursor polypeptide of SEQ ID NO: l is disclosed as SEQ ID NO:4 (nucleotides 164-1705 of
Genbank entry NM 001616). The nucleic acid sequence encoding the soluble human ActRIIA polypeptide of SEQ ID NO:2 is disclosed as SEQ ID NO:5. See Table 1 for a description of the sequences.
[0034] In specific embodiments, the ActRIIA polypeptides used in the compositions and methods described herein are soluble ActRIIA polypeptides. An extracellular domain of an ActRIIA protein can bind to activin and is generally soluble, and thus can be termed a soluble, activin-binding ActRIIA polypeptide. Thus, as used herein, the term "soluble ActRIIA polypeptide" generally refers to polypeptides comprising an extracellular domain of an ActRIIA protein, including any naturally occurring extracellular domain of an ActRIIA protein as well as any variants thereof (including mutants, fragments and peptidomimetic forms). Soluble ActRIIA polypeptides can bind to activin; however, the wild type ActRIIA protein does not exhibit significant selectivity in binding to activin versus myostatin (growth differentiation factor (GDF) 8) or bone morphogenic protein 11 (BMP1 1; also known as GDF11). Native or altered ActRIIA proteins may be given added specificity for activin by coupling them with a second, activin-selective binding agent. Examples of soluble, activin- binding ActRIIA polypeptides include the soluble polypeptides illustrated in SEQ ID NOs: 2, 3, 7, 12 and 13. Other examples of soluble, activin-binding ActRIIA polypeptides comprise a signal sequence in addition to the extracellular domain of an ActRIIA protein, for example, the honey bee mellitin leader sequence (SEQ ID NO: 8), the tissue plasminogen activator (TP A) leader (SEQ ID NO: 9) or the native ActRIIA leader (SEQ ID NO: 10). The ActRIIA- hFc polypeptide illustrated in SEQ ID NO: 13 uses a TP A leader.
[0035] In certain embodiments, the inhibitors of ActRIIA used in the compositions and methods described herein comprise a conjugate/fusion protein comprising an activin-binding domain of ActRIIA linked to an Fc portion of an antibody. In certain embodiments, the activin-binding domain is linked to an Fc portion of an antibody via a linker, e.g. , a peptide
linker. Optionally, the Fc domain has one or more mutations at residues such as Asp-265, lysine 322, and Asn-434. In certain cases, the mutant Fc domain having one or more of these mutations (e.g., an Asp-265 mutation) has a reduced ability to bind to the Fey receptor relative to a wild-type Fc domain. In other cases, the mutant Fc domain having one or more of these mutations (e.g., an Asn-434 mutation) has an increased ability to bind to the MHC class I- related Fc-receptor (FcR ) relative to a wild-type Fc domain. Exemplary fusion proteins comprising a soluble extracellular domain of ActRIIA fused to an Fc domain are set forth in SEQ ID NOs:6, 7, 12, and 13.
[0036] In a specific embodiment, the ActRIIA inhibitors used in the compositions and methods described herein comprise the extracellular domain of ActRIIA, or a portion thereof, linked to an Fc portion of an antibody, wherein said ActRIIA inhibitor comprises an amino acid sequence that is at least 75% identical to an amino acid sequence selected from SEQ ID NOs:6, 7, 12, and 13. In another specific embodiment, the ActRIIA inhibitors used in the compositions and methods described herein comprise the extracellular domain of ActRIIA, or a portion thereof, linked to an Fc portion of an antibody, wherein said ActRIIA inhibitor comprises an amino acid sequence that is at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical to an amino acid sequence selected from SEQ ID NOs:6, 7, 12, and 13.
[0037] In certain embodiments, the inhibitors of ActRIIA used in the compositions and methods described herein comprise a truncated form of an extracellular domain of ActRIIA. The truncation can be at the carboxy terminus and/or the amino terminus of the ActRIIA polypeptide. In certain embodiments, the truncation can be 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 amino acids long relative to the mature ActRIIB polypeptide extracellular domain. In certain embodiments, the truncation can be 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 N-terminal amino acids of the mature ActRIIA polypeptide extracellular domain. In certain
embodiments, the truncation can be 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 C-terminal amino acids of the mature ActRIIA polypeptide extracellular domain. For example, truncated forms of ActRIIA include polypeptides with amino acids 20-119; 20-128; 20-129; 20-130; 20-131; 20-132; 20-133; 20-134; 20-131; 21- 131; 22-131; 23-131; 24-131; and 25-131, wherein the amino acid positions refer to the amino acid positions in SEQ ID NO: 1.
[0038] In certain embodiments, the inhibitors of ActRIIA used in the compositions and methods described herein comprise an extracellular domain of ActRIIA with one or more amino acid substitutions. In certain embodiments, the inhibitors of ActRIIA used in the compositions and methods described herein comprise a truncated form of an ActRIIA extracellular domain that also carries an amino acid substitution.
[0039] In a specific embodiment, the ActRIIA inhibitor to be used in the compositions and methods described herein is a fusion protein between the extracellular domain of the human ActRIIA receptor and the Fc portion of IgGl . In another specific embodiment, the ActRIIA inhibitor to be used in the compositions and methods described herein is a fusion protein between a truncated extracellular domain of the human ActRIIA receptor and the Fc portion of IgGl . In another specific embodiment, the ActRIIA inhibitor to be used in the compositions and methods described herein is a fusion protein between a truncated extracellular domain of the human ActRIIA receptor and the Fc portion of IgGl, wherein the truncated extracellular domain of the human ActRIIA receptor possesses one or more amino acid substitutions.
[0040] Functionally active fragments of ActRIIA polypeptides can be obtained, for example, by screening polypeptides recombinantly produced from the corresponding fragment of the nucleic acid encoding an ActRIIA polypeptide. In addition, fragments can be chemically synthesized using techniques known in the art such as conventional Merrifield solid phase f-Moc or t-Boc chemistry. The fragments can be produced (recombinantly or by chemical synthesis) and tested to identify those peptidyl fragments that can function as antagonists (inhibitors) of ActRIIA protein or signaling mediated by activin.
[0041] In addition, functionally active variants of ActRIIA polypeptides can be obtained, for example, by screening libraries of modified polypeptides recombinantly produced from the corresponding mutagenized nucleic acids encoding an ActRIIA polypeptide. The variants can be produced and tested to identify those that can function as antagonists (inhibitors) of ActRIIA protein or signaling mediated by activin. In certain embodiments, a functional variant of the ActRIIA polypeptides comprises an amino acid sequence that is at least 75% identical to an amino acid sequence selected from SEQ ID NOs: 2 or 3. In certain cases, the functional variant has an amino acid sequence at least 80%, 85%, 90%, 95%, 97%, 98%, 99% or 100% identical to an amino acid sequence selected from SEQ ID NOs: 2 or 3.
[0042] Functional variants may be generated, for example, by modifying the structure of an ActRIIA polypeptide for such purposes as enhancing therapeutic efficacy, or stability (e.g., ex vivo shelf life and resistance to proteolytic degradation in vivo). Such modified ActRIIA polypeptides when selected to retain activin binding, are considered functional equivalents of the naturally-occurring ActRIIA polypeptides. Modified ActRIIA
polypeptides can also be produced, for instance, by amino acid substitution, deletion, or addition. For instance, it is reasonable to expect that an isolated replacement of a leucine with an isoleucine or valine, an aspartate with a glutamate, a threonine with a serine, or a similar replacement of an amino acid with a structurally related amino acid (e.g., conservative mutations) will not have a major effect on the biological activity of the resulting molecule. Conservative replacements are those that take place within a family of amino acids that are related in their side chains. Whether a change in the amino acid sequence of an ActRIIA polypeptide results in a functional homolog can be readily determined by assessing the ability of the variant ActRIIA polypeptide to produce a response in cells in a fashion similar to the wild-type ActRIIA polypeptide.
[0043] In certain embodiments, provided herein are specific mutations of the ActRIIA polypeptides that can alter the glycosylation of the polypeptide. Such mutations may be selected so as to introduce or eliminate one or more glycosylation sites, such as O-linked or N-linked glycosylation sites. Asparagine-linked glycosylation recognition sites generally comprise a tripeptide sequence, asparagine-X-threonine (or asparagines-X-serine) (where "X" is any amino acid) which is specifically recognized by appropriate cellular glycosylation enzymes. The alteration may also be made by the addition of, or substitution by, one or more serine or threonine residues to the sequence of the wild-type ActRIIA polypeptide (for O- linked glycosylation sites). A variety of amino acid substitutions or deletions at one or both of the first or third amino acid positions of a glycosylation recognition site (and/or amino acid deletion at the second position) results in non-glycosylation at the modified tripeptide sequence. Another means of increasing the number of carbohydrate moieties on an ActRIIA polypeptide is by chemical or enzymatic coupling of glycosides to the ActRIIA polypeptide. Depending on the coupling mode used, the sugar(s) may be attached to (a) arginine and histidine; (b) free carboxyl groups; (c) free sulfhydryl groups such as those of cysteine; (d) free hydroxyl groups such as those of serine, threonine, or hydroxyproline; (e) aromatic residues such as those of phenylalanine, tyrosine, or tryptophan; or (f) the amide group of
glutamine. These methods are described in WO 87/05330 published Sep. 11, 1987, and in Aplin and Wriston (1981) CRC Crit. Rev. Biochem., pp. 259-306, incorporated by reference herein. Removal of one or more carbohydrate moieties present on an ActRIIA polypeptide may be accomplished chemically and/or enzymatically. Chemical deglycosylation may involve, for example, exposure of the ActRIIA polypeptide to the compound
trifluoromethanesulfonic acid, or an equivalent compound. This treatment results in the cleavage of most or all sugars except the linking sugar (N-acetylglucosamine or N- acetylgalactosamine), while leaving the amino acid sequence intact. Chemical
deglycosylation is further described by Hakimuddin et al. (1987) Arch. Biochem. Biophys. 259:52 and by Edge et al. (1981) Anal. Biochem. 118: 131. Enzymatic cleavage of carbohydrate moieties on ActRIIA polypeptides can be achieved by the use of a variety of endo- and exo-glycosidases as described by Thotakura et al. (1987) Meth. Enzymol. 138:350. The sequence of an ActRIIA polypeptide may be adjusted, as appropriate, depending on the type of expression system used, as mammalian, yeast, insect and plant cells may all introduce differing glycosylation patterns that can be affected by the amino acid sequence of the peptide. In general, ActRIIA proteins for use in humans will be expressed in a mammalian cell line that provides proper glycosylation, such as HEK293 or CHO cell lines, although other expression systems, such as other mammalian expression cell lines, yeast cell lines with engineered glycosylation enzymes and insect cells, are expected to be useful as well.
[0044] Further provided herein are methods of generating mutants, particularly sets of combinatorial mutants of an ActRIIA polypeptide, as well as truncation mutants; pools of combinatorial mutants are especially useful for identifying functional variant sequences. The purpose of screening such combinatorial libraries may be to generate, for example, ActRIIA polypeptide variants which can act as either agonists or antagonist, or alternatively, which possess novel activities all together. A variety of screening assays are provided below, and such assays may be used to evaluate variants. For example, an ActRIIA polypeptide variant may be screened for ability to bind to an ActRIIA ligand, to prevent binding of an ActRIIA ligand to an ActRIIA polypeptide or to interfere with signaling caused by an ActRIIA ligand.
[0045] Combinatorially-derived variants can be generated which have a selective or generally increased potency relative to a naturally occurring ActRIIA polypeptide. Likewise, mutagenesis can give rise to variants which have intracellular half-lives dramatically different than the corresponding a wild-type ActRIIA polypeptide. For example, the altered protein
can be rendered either more stable or less stable to proteolytic degradation or other cellular processes which result in destruction of, or otherwise inactivation of a native ActRIIA polypeptide. Such variants, and the genes which encode them, can be utilized to alter ActRIIA polypeptide levels by modulating the half- life of the ActRIIA polypeptides. For instance, a short half-life can give rise to more transient biological effects and can allow tighter control of recombinant ActRIIA polypeptide levels within the patient. In an Fc fusion protein, mutations may be made in the linker (if any) and/or the Fc portion to alter the half- life of the protein.
[0046] A combinatorial library may be produced by way of a degenerate library of genes encoding a library of polypeptides which each include at least a portion of potential ActRIIA polypeptide sequences. For instance, a mixture of synthetic oligonucleotides can be enzymatically ligated into gene sequences such that the degenerate set of potential ActRIIA polypeptide nucleotide sequences are expressible as individual polypeptides, or alternatively, as a set of larger fusion proteins (e.g., for phage display).
[0047] There are many ways by which the library of potential homologs can be generated from a degenerate oligonucleotide sequence. Chemical synthesis of a degenerate gene sequence can be carried out in an automatic DNA synthesizer, and the synthetic genes then be ligated into an appropriate vector for expression. The synthesis of degenerate
oligonucleotides is well known in the art (see for example, Narang, S A (1983) Tetrahedron 39:3; Itakura et al, (1981) Recombinant DNA, Proc. 3rd Cleveland Sympos.
Macromolecules, ed. AG Walton, Amsterdam: Elsevier pp 273-289; Itakura et al, (1984) Annu. Rev. Biochem. 53:323; Itakura et al, (1984) Science 198: 1056; Ike et al, (1983) Nucleic Acid Res. 11 :477). Such techniques have been employed in the directed evolution of other proteins (see, for example, Scott et al, (1990) Science 249:386-390; Roberts et al, (1992) PNAS USA 89:2429-2433; Devlin et al, (1990) Science 249: 404-406; Cwirla et al, (1990) PNAS USA 87: 6378-6382; as well as U.S. Pat. Nos. 5,223,409, 5,198,346, and 5,096,815).
[0048] Alternatively, other forms of mutagenesis can be utilized to generate a
combinatorial library. For example, ActRIIA polypeptide variants can be generated and isolated from a library by screening using, for example, alanine scanning mutagenesis and the like (Ruf et al, (1994) Biochemistry 33: 1565-1572; Wang et al, (1994) J. Biol. Chem.
269:3095-3099; Balint et al, (1993) Gene 137: 109-118; Grodberg et al, (1993) Eur. J.
Biochem. 218:597-601; Nagashima et al, (1993) J. Biol. Chem. 268:2888-2892; Lowman et al, (1991) Biochemistry 30: 10832-10838; and Cunningham et al, (1989) Science 244:1081- 1085), by linker scanning mutagenesis (Gustin et al, (1993) Virology 193:653-660; Brown et al, (1992) Mol. Cell Biol. 12:2644-2652; McKnight et al, (1982) Science 232:316); by saturation mutagenesis (Meyers et al, (1986) Science 232:613); by PCR mutagenesis (Leung et al., (1989) Method Cell Mol Biol 1 : 11-19); or by random mutagenesis, including chemical mutagenesis, etc. (Miller et al, (1992) A Short Course in Bacterial Genetics, CSHL Press, Cold Spring Harbor, N.Y.; and Greener et al, (1994) Strategies in Mol Biol 7:32-34). Linker scanning mutagenesis, particularly in a combinatorial setting, is an attractive method for identifying truncated (bioactive) forms of ActRIIA polypeptides.
[0049] A wide range of techniques are known in the art for screening gene products of combinatorial libraries made by point mutations and truncations, and, for that matter, for screening cDNA libraries for gene products having a certain property. Such techniques will be generally adaptable for rapid screening of the gene libraries generated by the
combinatorial mutagenesis of ActRIIA polypeptides. The most widely used techniques for screening large gene libraries typically comprises cloning the gene library into replicable expression vectors, transforming appropriate cells with the resulting library of vectors, and expressing the combinatorial genes under conditions in which detection of a desired activity facilitates relatively easy isolation of the vector encoding the gene whose product was detected. Preferred assays include activin binding assays and activin-mediated cell signaling assays.
[0050] In certain embodiments, ActRIIA polypeptides may further comprise post- translational modifications in addition to any that are naturally present in the ActRIIA polypeptides. Such modifications include, but are not limited to, acetylation, carboxylation, glycosylation, phosphorylation, lipidation, and acylation. As a result, the modified ActRIIA polypeptides may contain non-amino acid elements, such as polyethylene glycols, lipids, poly- or mono-saccharide, and phosphates. Effects of such non-amino acid elements on the functionality of a ActRIIA polypeptide may be tested by any method known to the skilled artisan. When an ActRIIA polypeptide is produced in cells by cleaving a nascent form of the ActRIIA polypeptide, post-translational processing may also be important for correct folding and/or function of the protein. Different cells (such as CHO, HeLa, MDCK, 293, W138, NIH-3T3 or HEK293) have specific cellular machinery and characteristic mechanisms for
such post-translational activities and may be chosen to ensure the correct modification and processing of the ActRIIA polypeptides.
[0051] In certain aspects, functional variants or modified forms of the ActRIIA polypeptides include fusion proteins having at least a portion of the ActRIIA polypeptides and one or more fusion domains. Well known examples of such fusion domains include, but are not limited to, polyhistidine, Glu-Glu, glutathione S transferase (GST), thioredoxin, protein A, protein G, an immunoglobulin heavy chain constant region (Fc), maltose binding protein (MBP), or human serum albumin. A fusion domain may be selected so as to confer a desired property. For example, some fusion domains are particularly useful for isolation of the fusion proteins by affinity chromatography. For the purpose of affinity purification, relevant matrices for affinity chromatography, such as glutathione-, amylase-, and nickel- or cobalt-conjugated resins are used. Many of such matrices are available in "kit" form, such as the Pharmacia GST purification system and the QIAexpress™ system (Qiagen) useful with (HIS6) fusion partners. As another example, a fusion domain may be selected so as to facilitate detection of the ActRIIA polypeptides. Examples of such detection domains include the various fluorescent proteins (e.g., GFP) as well as "epitope tags," which are usually short peptide sequences for which a specific antibody is available. Well known epitope tags for which specific monoclonal antibodies are readily available include FLAG, influenza virus haemagglutinin (HA), and c-myc tags. In some cases, the fusion domains have a protease cleavage site, such as for Factor Xa or Thrombin, which allows the relevant protease to partially digest the fusion proteins and thereby liberate the recombinant proteins therefrom. The liberated proteins can then be isolated from the fusion domain by subsequent chromatographic separation. In certain preferred embodiments, an ActRIIA polypeptide is fused with a domain that stabilizes the ActRIIA polypeptide in vivo (a "stabilizer" domain). By "stabilizing" is meant anything that increases serum half life, regardless of whether this is because of decreased destruction, decreased clearance by the kidney, or other
pharmacokinetic effect. Fusions with the Fc portion of an immunoglobulin are known to confer desirable pharmacokinetic properties on a wide range of proteins. Likewise, fusions to human serum albumin can confer desirable properties. Other types of fusion domains that may be selected include multimerizing (e.g., dimerizing, tetramerizing) domains and functional domains (that confer an additional biological function, such as further stimulation of bone growth or muscle growth, as desired).
[0052] It is understood that different elements of the fusion proteins may be arranged in any manner that is consistent with the desired functionality. For example, an ActRIIA polypeptide may be placed C-terminal to a heterologous domain, or, alternatively, a heterologous domain may be placed C-terminal to an ActRIIA polypeptide. The ActRIIA polypeptide domain and the heterologous domain need not be adjacent in a fusion protein, and additional domains or amino acid sequences may be included C- or N-terminal to either domain or between the domains.
[0053] In certain embodiments, the ActRIIA polypeptides described herein contain one or more modifications that are capable of stabilizing the ActRIIA polypeptides. For example, such modifications enhance the in vitro half life of the ActRIIA polypeptides, enhance circulatory half life of the ActRIIA polypeptides or reduce proteolytic degradation of the ActRIIA polypeptides. Such stabilizing modifications include, but are not limited to, fusion proteins (including, for example, fusion proteins comprising an ActRIIA polypeptide and a stabilizer domain), modifications of a glycosylation site (including, for example, addition of a glycosylation site to an ActRIIA polypeptide), and modifications of carbohydrate moiety (including, for example, removal of carbohydrate moieties from an ActRIIA polypeptide). In the case of fusion proteins, an ActRIIA polypeptide is fused to a stabilizer domain such as an IgG molecule (e.g., an Fc domain). As used herein, the term "stabilizer domain" not only refers to a fusion domain (e.g., Fc) as in the case of fusion proteins, but also includes nonproteinaceous modifications such as a carbohydrate moiety, or nonproteinaceous polymer, such as polyethylene glycol.
[0054] In certain embodiments, isolated and/or purified forms of the ActRIIA
polypeptides, which are isolated from, or otherwise substantially free of, other proteins can be used with the methods and compositions described herein. ActRIIA polypeptides will generally be produced by expression from recombinant nucleic acids.
[0055] In certain aspects, provided herein are isolated and/or recombinant nucleic acids encoding any of the ActRIIA polypeptides (e.g., soluble ActRIIA polypeptides), including fragments, functional variants and fusion proteins disclosed herein. For example, SEQ ID NO: 4 encodes the naturally occurring human ActRIIA precursor polypeptide, while SEQ ID NO: 5 encodes the processed extracellular domain of ActRIIA. The subject nucleic acids may be single-stranded or double stranded. Such nucleic acids may be DNA or RNA
molecules. These nucleic acids may be used, for example, in methods for making ActRIIA polypeptides or as direct therapeutic agents (e.g., in a gene therapy approach).
[0056] In certain aspects, the subject nucleic acids encoding ActRIIA polypeptides are further understood to include nucleic acids that are variants of SEQ ID NO: 4 or 5. Variant nucleotide sequences include sequences that differ by one or more nucleotide substitutions, additions or deletions, such as allelic variants.
[0057] In certain embodiments, provided herein are isolated or recombinant nucleic acid sequences that are at least 80%, 85%, 90%, 95%, 97%, 98%, 99% or 100% identical to SEQ ID NO: 4 or 5. One of ordinary skill in the art will appreciate that nucleic acid sequences complementary to SEQ ID NO: 4 or 5, and variants of SEQ ID NO: 4 or 5 are also encompassed herein. In further embodiments, the nucleic acid sequences provided herein can be isolated, recombinant, and/or fused with a heterologous nucleotide sequence, or in a DNA library.
[0058] In other embodiments, nucleic acids provided herein also include nucleotide sequences that hybridize under highly stringent conditions to the nucleotide sequence designated in SEQ ID NO: 4 or 5, complement sequence of SEQ ID NO: 4 or 5, or fragments thereof. As discussed above, one of ordinary skill in the art will readily understand that appropriate stringency conditions which promote DNA hybridization can be varied. One of ordinary skill in the art will understand that appropriate stringency conditions which promote DNA hybridization can be varied. For example, one can perform the hybridization at 6.0 times sodium chloride/sodium citrate (SSC) at about 45 degrees Celsius, followed by a wash of 2.0 times SSC at 50 degrees Celsius. For example, the salt concentration in the wash step can be selected from a low stringency of about 2.0 times SSC at 50 degrees Celsius to a high stringency of about 0.2 times SSC at 50 degrees Celsius. In addition, the temperature in the wash step can be increased from low stringency conditions at room temperature, about 22 degrees Celsius, to high stringency conditions at about 65 degrees Celsius. Both temperature and salt may be varied, or temperature or salt concentration may be held constant while the other variable is changed. In one embodiment, nucleic acids which hybridize under low stringency conditions of 6 times SSC at room temperature followed by a wash at 2 times SSC at room temperature can be used with the methods and compositions described herein.
[0059] Isolated nucleic acids which differ from the nucleic acids as set forth in SEQ ID NOs: 4 or 5 due to degeneracy in the genetic code are also encompassed herein. For
example, a number of amino acids are designated by more than one triplet. Codons that specify the same amino acid, or synonyms (for example, CAU and CAC are synonyms for histidine) may result in "silent" mutations which do not affect the amino acid sequence of the protein. However, it is expected that DNA sequence polymorphisms that do lead to changes in the amino acid sequences of the subject proteins will exist among mammalian cells. One skilled in the art will appreciate that these variations in one or more nucleotides (up to about 3-5% of the nucleotides) of the nucleic acids encoding a particular protein may exist among individuals of a given species due to natural allelic variation. Any and all such nucleotide variations and resulting amino acid polymorphisms are encompassed herein.
[0060] In certain embodiments, the recombinant nucleic acids may be operably linked to one or more regulatory nucleotide sequences in an expression construct. Regulatory nucleotide sequences will generally be appropriate to the host cell used for expression.
Numerous types of appropriate expression vectors and suitable regulatory sequences are known in the art for a variety of host cells. Typically, said one or more regulatory nucleotide sequences may include, but are not limited to, promoter sequences, leader or signal sequences, ribosomal binding sites, transcriptional start and termination sequences, translational start and termination sequences, and enhancer or activator sequences.
Constitutive or inducible promoters as known in the art are contemplated herein. The promoters may be either naturally occurring promoters, or hybrid promoters that combine elements of more than one promoter. An expression construct may be present in a cell on an episome, such as a plasmid, or the expression construct may be inserted in a chromosome. In a preferred embodiment, the expression vector contains a selectable marker gene to allow the selection of transformed host cells. Selectable marker genes are well known in the art and will vary with the host cell used.
[0061] In certain aspects, the subject nucleic acid is provided in an expression vector comprising a nucleotide sequence encoding an ActRIIA polypeptide and operably linked to at least one regulatory sequence. Regulatory sequences are art-recognized and are selected to direct expression of the ActRIIA polypeptide. Accordingly, the term regulatory sequence includes promoters, enhancers, and other expression control elements. Exemplary regulatory sequences are described in Goeddel; Gene Expression Technology: Methods in Enzymology, Academic Press, San Diego, Calif. (1990). For instance, any of a wide variety of expression control sequences that control the expression of a DNA sequence when operatively linked to
it may be used in these vectors to express DNA sequences encoding an ActRIIA polypeptide. Such useful expression control sequences, include, for example, the early and late promoters of SV40, tet promoter, adenovirus or cytomegalovirus immediate early promoter, RSV promoters, the lac system, the trp system, the TAC or TRC system, T7 promoter whose expression is directed by T7 RNA polymerase, the major operator and promoter regions of phage lambda, the control regions for fd coat protein, the promoter for 3-phosphoglycerate kinase or other glycolytic enzymes, the promoters of acid phosphatase, e.g., Pho5, the promoters of the yeast .alpha. -mating factors, the polyhedron promoter of the baculovirus system and other sequences known to control the expression of genes of prokaryotic or eukaryotic cells or their viruses, and various combinations thereof. It should be understood that the design of the expression vector may depend on such factors as the choice of the host cell to be transformed and/or the type of protein desired to be expressed. Moreover, the vector's copy number, the ability to control that copy number and the expression of any other protein encoded by the vector, such as antibiotic markers, should also be considered.
[0062] A recombinant nucleic acid provided herein can be produced by ligating the cloned gene, or a portion thereof, into a vector suitable for expression in either prokaryotic cells, eukaryotic cells (yeast, avian, insect or mammalian), or both. Expression vehicles for production of a recombinant ActRIIA polypeptide include plasmids and other vectors. For instance, suitable vectors include plasmids of the types: pBR322-derived plasmids, pEMBL- derived plasmids, pEX-derived plasmids, pBTac-derived plasmids and pUC-derived plasmids for expression in prokaryotic cells, such as E. coli.
[0063] Some mammalian expression vectors contain both prokaryotic sequences to facilitate the propagation of the vector in bacteria, and one or more eukaryotic transcription units that are expressed in eukaryotic cells. The pcDNAI/amp, pcDNAI/neo, pRc/CMV, pSV2gpt, pSV2neo, pSV2-dhfr, pTk2, pRSVneo, pMSG, pSVT7, pko-neo and pHyg derived vectors are examples of mammalian expression vectors suitable for transfection of eukaryotic cells. Some of these vectors are modified with sequences from bacterial plasmids, such as pBR322, to facilitate replication and drug resistance selection in both prokaryotic and eukaryotic cells. Alternatively, derivatives of viruses such as the bovine papilloma virus (BPV-1), or Epstein-Barr virus (pHEBo, pREP-derived and p205) can be used for transient expression of proteins in eukaryotic cells. Examples of other viral (including retroviral) expression systems can be found below in the description of gene therapy delivery systems.
The various methods employed in the preparation of the plasmids and in transformation of host organisms are well known in the art. For other suitable expression systems for both prokaryotic and eukaryotic cells, as well as general recombinant procedures, see Molecular Cloning A Laboratory Manual, 3rd Ed., ed. by Sambrook, Fritsch and Maniatis (Cold Spring Harbor Laboratory Press, 2001). In some instances, it may be desirable to express the recombinant polypeptides by the use of a baculovirus expression system. Examples of such baculovirus expression systems include pVL-derived vectors (such as pVL1392, pVL1393 and pVL941), pAcUW-derived vectors (such as pAcUWl), and pBlueBac-derived vectors (such as the .beta. -gal containing pBlueBac III).
[0064] In a preferred embodiment, a vector will be designed for production of the subject ActRIIA polypeptides in CHO cells, such as a Pcmv-Script vector (Stratagene, La Jolla, Calif), pcDNA4 vectors (Invitrogen, Carlsbad, Calif.) and pCI-neo vectors (Promega, Madison, Wis.). As will be apparent, the subject gene constructs can be used to cause expression of the subject ActRIIA polypeptides in cells propagated in culture, e.g., to produce proteins, including fusion proteins or variant proteins, for purification.
[0065] This disclosure also pertains to a host cell transfected with a recombinant gene including a coding sequence (e.g., SEQ ID NO: 4 or 5) for one or more of the subject ActRIIA polypeptides. The host cell may be any prokaryotic or eukaryotic cell. For example, an ActRIIA polypeptide provided herein may be expressed in bacterial cells such as E. coli, insect cells (e.g., using a baculovirus expression system), yeast, or mammalian cells. Other suitable host cells are known to those skilled in the art.
[0066] Accordingly, provided herein are methods of producing the ActRIIA
polypeptides. For example, a host cell transfected with an expression vector encoding an ActRIIA polypeptide can be cultured under appropriate conditions to allow expression of the ActRIIA polypeptide to occur. The ActRIIA polypeptide may be secreted and isolated from a mixture of cells and medium containing the ActRIIA polypeptide. Alternatively, the ActRIIA polypeptide may be retained cytoplasmically or in a membrane fraction and the cells harvested, lysed and the protein isolated. A cell culture includes host cells, media and other byproducts. Suitable media for cell culture are well known in the art. The subject ActRIIA polypeptides can be isolated from cell culture medium, host cells, or both, using techniques known in the art for purifying proteins, including ion-exchange chromatography, gel filtration chromatography, ultrafiltration, electrophoresis, immunoaffinity purification with antibodies
specific for particular epitopes of the ActRIIA polypeptides and affinity purification with an agent that binds to a domain fused to the ActRIIA polypeptide (e.g., a protein A column may be used to purify an ActRIIA-Fc fusion). In a preferred embodiment, the ActRIIA
polypeptide is a fusion protein containing a domain which facilitates its purification. In a preferred embodiment, purification is achieved by a series of column chromatography steps, including, for example, three or more of the following, in any order: protein A
chromatography, Q sepharose chromatography, phenylsepharose chromatography, size exclusion chromatography, and cation exchange chromatography. The purification could be completed with viral filtration and buffer exchange. As demonstrated herein, ActRIIA-hFc protein was purified to a purity of >98% as determined by size exclusion chromatography and >95% as determined by SDS PAGE. This level of purity was sufficient to achieve desirable effects on bone in mice and an acceptable safety profile in mice, rats and non-human primates.
[0067] In another embodiment, a fusion gene coding for a purification leader sequence, such as a poly-(His)/enterokinase cleavage site sequence at the N-terminus of the desired portion of the recombinant ActRIIA polypeptide, can allow purification of the expressed fusion protein by affinity chromatography using a Ni2+ metal resin. The purification leader sequence can then be subsequently removed by treatment with enterokinase to provide the purified ActRIIA polypeptide (e.g., see Hochuli et al, (1987) J. Chromatography 411 : 177; and Janknecht et al, PNAS USA 88:8972).
[0068] Techniques for making fusion genes are well known. Essentially, the joining of various DNA fragments coding for different polypeptide sequences is performed in accordance with conventional techniques, employing blunt-ended or stagger-ended termini for ligation, restriction enzyme digestion to provide for appropriate termini, filling-in of cohesive ends as appropriate, alkaline phosphatase treatment to avoid undesirable joining, and enzymatic ligation. In another embodiment, the fusion gene can be synthesized by conventional techniques including automated DNA synthesizers. Alternatively, PCR amplification of gene fragments can be carried out using anchor primers which give rise to complementary overhangs between two consecutive gene fragments which can subsequently be annealed to generate a chimeric gene sequence (see, for example, Current Protocols in Molecular Biology, eds. Ausubel et al, John Wiley & Sons: 1992).
[0069] ActRIIA-Fc fusion protein can be expressed in stably transfected CHO-DUKX Bl 1 cells from a pAID4 vector (SV40 ori/enhancer, CMV promoter), using, for example, a tissue plasminogen leader sequence of SEQ ID NO:9. The Fc portion can be a human IgGl Fc sequence. In certain embodiments, upon expression, the protein contained has, on average, between about 1.5 and 2.5 moles of sialic acid per molecule of ActRIIA-Fc fusion protein.
[0070] In certain embodiments, the long serum half-life of an ActRIIA-Fc fusion can be 25-32 days in human patients. Additionally, the CHO cell expressed material can have a higher affinity for activin B ligand than that reported for an ActRIIA-hFc fusion protein expressed in human 293 cells (del Re et al., J Biol Chem. 2004 Dec 17;279(51):53126-35). Additionally, without being bound by theory, the use of the TPA leader sequence provided greater production than other leader sequences and, unlike ActRIIA-Fc expressed with a native leader, may provide a highly pure N-terminal sequence. Use of the native leader sequence may result in two major species of ActRIIA-Fc, each having a different N-terminal sequence.
(b) INHIBITORS OF ACTRIIB
[0071] As used herein, the term "ActRIIB" refers to a family of activin receptor type IIB (ActRIIB) proteins from any species and variants derived from such ActRIIB proteins by mutagenesis or other modification. Reference to ActRIIB herein is understood to be a reference to any one of the currently identified forms of the receptor. Members of the ActRIIB family are generally transmembrane proteins, composed of a ligand-binding extracellular domain with a cysteine -rich region, a transmembrane domain, and a cytoplasmic domain with predicted serine/threonine kinase activity.
[0072] ActRIIB inhibitors to be used in the compositions and methods described herein include, without limitation, activin-binding soluble ActRIIB polypeptides; antibodies that bind to activin (particularly the activin A or B subunits, also referred to as BA or BB) and disrupt ActRIIB binding; antibodies that bind to ActRIIB and disrupt activin binding; non- antibody proteins selected for activin or ActRIIB binding; and randomized peptides selected for activin or ActRIIB binding, which can be conjugated to an Fc domain.
[0073] In certain embodiments, two or more different proteins (or other moieties) with activin or ActRIIB binding activity, especially activin binders that block the type I (e.g., a soluble type I activin receptor) and type II (e.g., a soluble type II activin receptor) binding
sites, respectively, may be linked together to create a bifunctional or multifunctional binding molecule that inhibits ActRIIB and thus can be used in the compositions and methods described herein include. In certain embodiments, Activin- ActRIIB signaling axis antagonists that inhibit ActRIIB include nucleic acid aptamers, small molecules and other agents are used in the compositions and methods described herein include.
(i) ActRIIB Inhibitors Comprising ActRIIB Polypeptides
[0074] As used herein, the term "ActRIIB polypeptide" refers to polypeptides comprising any naturally occurring polypeptide of an ActRIIB family member as well as any variants thereof (including mutants, fragments, fusions, and peptidomimetic forms) that retain a useful activity. For example, ActRIIB polypeptides include polypeptides derived from the sequence of any known ActRIIB receptor having a sequence at least about 80% identical to the sequence of an ActRIIB polypeptide, and optionally at least 85%, 90%, 95%, 96%, 97%, 98%, 99% or greater identity. For example, an ActRIIB polypeptide may bind to and inhibit the function of an ActRIIB protein and/or activin. An example of an ActRIIB polypeptide includes the human ActRIIB precursor polypeptide (SEQ ID NO: 16 or SEQ ID NO:28). With respect to the ActRIIB precursor polypeptide whose amino acid sequence is depicted as SEQ ID NO: 16 or SEQ ID NO:28 (i.e., the human ActRIIB precursor polypeptide), the signal peptide of the ActRIIB precursor polypeptide is located at amino acids 1 to 18; the extracellular domain is located at amino acids 19 to 134 and the potential N-linked glycosylation sites are located at amino acid positions 42 and 65. The nucleic acid sequence encoding the human ActRIIB precursor polypeptide of SEQ ID NO: 16 is disclosed as SEQ ID NO: 19 (SEQ ID NO: 19 provides an alanine at the codon corresponding to amino acid position 64, but could be readily modified by one of skill in the art using methods known in the art to provide an arginine at the codon corresponding to amino acid position 64 instead). See Table 1 for a description of the sequences.
[0075] The numbering of amino acids for all of the ActRIIB -related polypeptides described herein is based on the amino acid numbering for SEQ ID NO: 16 and SEQ ID NO:28 (which only differ in the amino acid expressed at position 64), unless specifically designated otherwise. For example, if an ActRIIB polypeptide is described as having a substitution/mutation at amino acid position 79, then it is to be understood that position 79 refers to the 79th amino acid in SEQ ID NO: 16 or SEQ ID NO:28, from which the ActRIIB polypeptide is derived. Likewise, if an ActRIIB polypeptide is described as having an
alanine or an arginine at amino acid position 64, then it is to be understood that position 64 refers to the 64th amino acid in SEQ ID NO: 16 or SEQ ID NO:28, from which the ActRIIB polypeptide is derived.
[0076] In certain embodiments, the inhibitors of ActRIIB used in the compositions and methods described herein comprise polypeptides comprising an activin-binding domain of ActRIIB. In some embodiments, the activin-binding domains of ActRIIB comprise the extracellular domain of ActRIIB, or a portion thereof. In specific embodiments, the extracellular domain or portion thereof of ActRIIB is soluble. Illustrative modified forms of ActRIIB polypeptides are disclosed in U.S. Patent Application Publication Nos.
20090005308 and 20100068215, the disclosures of which are incorporated herein by reference in their entireties.
[0077] In specific embodiments, the ActRIIB polypeptides used in the compositions and methods described herein are soluble ActRIIB polypeptides. The term "soluble ActRIIB polypeptide" generally refers to polypeptides comprising an extracellular domain of an ActRIIB protein, including any naturally occurring extracellular domain of an ActRIIB protein as well as any variants thereof (including mutants, fragments and peptidomimetic forms). Soluble ActRIIB polypeptides can bind to activin; however, the wild type ActRIIB protein does not exhibit significant selectivity in binding to activin versus GDF8/11. In certain embodiments, altered forms of ActRIIB with different binding properties can be used in the methods provided herein. Such altered forms are disclosed, e.g. , in international patent application publication Nos. WO 2006/012627 and WO 2010/019261, the disclosures of which are incorporated herein by reference in their entireties. Native or altered ActRIIB proteins may be given added specificity for activin by coupling them with a second, activin- selective binding agent. Exemplary soluble ActRIIB polypeptides include the extracellular domain of a human ActRIIB polypeptide (e.g., SEQ ID NOs: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, and 43).
[0078] An Fc fusion protein having the ActRIIB extracellular sequence disclosed by
Hilden et al. (Blood, 1994, 83(8):2163-70), which has an alanine at the position
corresponding to amino acid 64 of the ActRIIB precursor amino acid sequence, i.e., SEQ ID
NO: 16 (herein referred to as "A64"), has been demonstrated to possess a relatively low affinity for activin and GDF-11. By contrast, an Fc fusion protein with an arginine at position 64 of the ActRIIB precursor amino acid sequence (herein referred to as "R64") has
an affinity for activin and GDF-11 in the low nanomolar to high picomolar range (see, e.g., U.S. Patent Application Publication No. 20100068215, the disclosure of which is herein incorporated in its entirety). An ActRIIB precursor amino acid sequence with an arginine at position 64 is presented in SEQ ID NO:28. As such, in certain embodiments, the ActRIIB polypeptides used in accordance with the compositions and methods described herein may comprise either (i) an alanine at the position corresponding to amino acid 64 of the ActRIIB precursor amino acid sequence, i.e., SEQ ID NO: 16; or (ii) an arginine at position 64 of the ActRIIB precursor amino acid sequence, i.e., SEQ ID NO: 28. In other embodiments, the ActRIIB polypeptides used in accordance with the compositions and methods described herein may comprise an amino acid that is not alanine or arginine at the position
corresponding to amino acid 64 of the ActRIIB precursor amino acid sequence, i.e., SEQ ID NO: 16 or SEQ ID NO:28.
[0079] It has been shown that a deletion of the proline knot at the C-terminus of the extracellular domain of ActRIIB reduces the affinity of the receptor for activin (see, e.g., Attisano et al., Cell, 1992, 68(1):97-108). An ActRIIB-Fc fusion protein containing amino acids 20-119 of SEQ ID NO: 28 (i.e., SEQ ID NO:32), "ActRIIB(20-l 19)-Fc" has reduced binding to GDF-11 and activin relative to an ActRIIB-Fc fusion protein containing amino acids 20-134 of SEQ ID NO: 28 (i.e., SEQ ID NO:31), "ActRIIB(20-134)-Fc", which includes the proline knot region and the complete juxtamembrane domain. However, an ActRIIB-Fc fusion protein containing amino acids 20-129 of SEQ ID NO: 28, "ActRIIB(20- 129)-Fc" retains similar but somewhat reduced activity relative to the non-truncated extracellular domain of ActRIIB, even though the proline knot region is disrupted. Thus, ActRIIB polypeptides comprising extracellular domains that stop at amino acid 134, 133, 132, 131, 130 and 129 of SEQ ID NO: 28 (or SEQ ID NO: 16) are all expected to be active, but constructs stopping at amino acid 134 or 133 may be most active. Similarly, mutations at any of residues 129-134 are not expected to alter ligand binding affinity by large margins, as indicated by the fact that mutations of P129 and P130 of SEQ ID NO: 28 do not substantially decrease ligand binding. Therefore, the ActRIIB polypeptides used in accordance with the methods and compositions described herein may end as early as amino acid 109 (i.e., the final cysteine) of SEQ ID NO:28 (or SEQ ID NO: 16), however, forms ending at or between amino acid positions 109 and 119 of SEQ ID NO:28 (or SEQ ID NO: 16) are expected to have reduced ligand binding ability.
[0080] Amino acid 29 of SEQ ID NO: 16 and SEQ ID NO:28 represents the initial cysteine in the ActRIIB precursor sequence. It is expected that an ActRIIB polypeptide beginning at amino acid 29 of the N-terminus of SEQ ID NO: 16 or SEQ ID NO:28, or before these amino acid positions, will retain ligand binding activity. An alanine to asparagine mutation at position 24 of SEQ ID NO: 16 or SEQ ID NO:28 introduces an N-linked glycosylation sequence without substantially affecting ligand binding. This confirms that mutations in the region between the signal cleavage peptide and the cysteine cross-linked region, corresponding to amino acids 20-29 of SEQ ID NO: 16 or SEQ ID NO:28, are well tolerated. In particular, ActRIIB polypeptides beginning at amino acid position 20, 21, 22, 23 and 24 of SEQ ID NO: 16 or SEQ ID NO:28 will retain activity, and ActRIIB polypeptides beginning at amino acid positions 25, 26, 27, 28 and 29 of SEQ ID NO: 16 or SEQ ID NO:28 are also expected to retain activity. An ActRIIB polypeptide beginning at amino acid position 22, 23, 24 or 25 of SEQ ID NO: 16 or SEQ ID NO:28 will have the most activity.
[0081] Taken together, the active portions (i.e., ActRIIB polypeptides) of the ActRIIB precursor protein (i.e., SEQ ID NO: 16 or SEQ ID NO:28) to be used in accordance with the methods and compositions described herein will generally comprise amino acids 29-109 of SEQ ID NO: 16 or SEQ ID NO:28, and such ActRIIB polypeptides may, for example, begin at a residue corresponding to any one of amino acids 19-29 of SEQ ID NO: 16 or SEQ ID NO:28 and end at a position corresponding to any one of amino acids 109-134 of SEQ ID NO: 16 or SEQ ID NO:28. Specific examples of ActRIIB polypeptides encompassed herein include those that begin at an amino acid position from 19-29, 20-29 or 21-29 of SEQ ID NO: 16 or SEQ ID NO:28 and end at an amino acid position from 119-134, 119-133 or 129- 134, 129-133 of SEQ ID NO:16 or SEQ ID NO:28. Other specific examples of ActRIIB polypeptides encompassed herein include those that begin at an amino acid position from 20- 24 (or 21-24, or 22-25) of SEQ ID NO: 16 or SEQ ID NO:28 and end at an amino acid position from 109-134 (or 109-133), 119-134 (or 119-133) or 129-134 (or 129-133) of SEQ ID NO: 16 or SEQ ID NO:28. Variant ActRIIB polypeptides falling within these ranges are also contemplated, particularly those having at least 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%), 96%), 97%), 98%), or 99%> sequence identity or sequence homology to the corresponding portion of SEQ ID NO: 16 or SEQ ID NO:28.
[0082] In certain embodiments, the inhibitors of ActRIIB used in the compositions and methods described herein comprise a truncated form of an extracellular domain of ActRIIB.
The truncation can be at the carboxy terminus and/or the amino terminus of the ActRIIB polypeptide. In certain embodiments, the truncation can be 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 amino acids long relative to the mature ActRIIB polypeptide extracellular domain. In certain embodiments, the truncation can be 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 N-terminal amino acids of the mature ActRIIB polypeptide extracellular domain. In certain
embodiments, the truncation can be 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, or 25 C-terminal amino acids of the mature ActRIIB polypeptide extracellular domain. For example, truncated forms of ActRIIB include polypeptides with amino acids 20-119; 20-128; 20-129; 20-130; 20-131; 20-132; 20-133; 20-134; 20-131; 21- 131; 22-131; 23-131; 24-131; and 25-131, wherein the amino acid positions refer to the amino acid positions in SEQ ID NO: 16 or SEQ ID NO:28.
[0083] Additional exemplary truncated forms of ActRIIB include (i) polypeptides beginning at amino acids at any of amino acids 21-29 of SEQ ID NO: 16 or SEQ ID NO:28 (optionally beginning at 22-25 of SEQ ID NO: 16 or SEQ ID NO:28) and ending at any of amino acids 109-134 of SEQ ID NO: 16 or SEQ ID NO:28; (ii) polypeptides beginning at any of amino acids 20-29 of SEQ ID NO: 16 or SEQ ID NO:28 (optionally beginning at 22-25 of SEQ ID NO: 16 or SEQ ID NO:28) and ending at any of amino acids 109-133 of SEQ ID NO: 16 or SEQ ID NO:28; (iii) polypeptides beginning at any of amino acids 20-24 of SEQ ID NO: 16 or SEQ ID NO:28 (optionally beginning at 22-25 of SEQ ID NO: 16 or SEQ ID NO:28) and ending at any of amino acids 109-133 of SEQ ID NO: 16 or SEQ ID NO:28; (iv) polypeptides beginning at any of amino acids 21-24 of SEQ ID NO: 16 or SEQ ID NO:28 and ending at any of amino acids 109-134 of SEQ ID NO: 16 or SEQ ID NO:28; (v) polypeptides beginning at any of amino acids 20-24 of SEQ ID NO: 16 or SEQ ID NO:28 and ending at any of amino acids 118-133 of SEQ ID NO: 16 or SEQ ID NO:28; (vi) polypeptides beginning at any of amino acids 21-24 of SEQ ID NO: 16 or SEQ ID NO:28 and ending at any of amino acids 118-134 of SEQ ID NO: 16 or SEQ ID NO:28; (vii) polypeptides beginning at any of amino acids 20-24 of SEQ ID NO: 16 or SEQ ID NO:28 and ending at any of amino acids 128-133 of SEQ ID NO: 16 or SEQ ID NO:28; (viii) polypeptides beginning at any of amino acids 20-24 of SEQ ID NO: 16 or SEQ ID NO:28 and ending at any of amino acids 128-133 of SEQ ID NO: 16 or SEQ ID NO:28; (ix) polypeptides beginning at any of amino acids 21-29 of SEQ ID NO: 16 or SEQ ID NO:28 and ending at
any of amino acids 118-134 of SEQ ID NO: 16 or SEQ ID NO:28; (x) polypeptides beginning at any of amino acids 20-29 of SEQ ID NO: 16 or SEQ ID NO:28 and ending at any of amino acids 118-133 of SEQ ID NO: 16 or SEQ ID NO:28; (xi) polypeptides beginning at any of amino acids 21-29 of SEQ ID NO: 16 or SEQ ID NO:28 and ending at any of amino acids 128-134 of SEQ ID NO: 16 or SEQ ID NO:28; and (xii) polypeptides beginning at any of amino acids 20-29 of SEQ ID NO: 16 or SEQ ID NO:28 and ending at any of amino acids 128-133 of SEQ ID NO: 16 or SEQ ID NO:28. In a specific embodiment, an ActRIIB polypeptides comprises, consists essentially of, or consists of, an amino acid sequence beginning at amino acid position 25 of SEQ ID NO: 16 or SEQ ID NO:28 and ending at amino acid position 131 of SEQ ID NO: 16 or SEQ ID NO:28. In another specific embodiment, an ActRIIB polypeptide consists of, or consists essentially of, the amino acid sequence of SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43.
[0084] Any of the ActRIIB polypeptides disclosed herein may be produced as a homodimer. Any of the ActRIIB polypeptides disclosed herein may be formulated as a fusion protein having a heterologous portion that comprises a constant region from an IgG heavy chain, such as an Fc domain. Any of the ActRIIB polypeptides disclosed herein may comprise an acidic amino acid at the position corresponding to position 79 of SEQ ID NO: 16 or SEQ ID NO:28, optionally in combination with one or more additional amino acid substitutions, deletions or insertions relative to SEQ ID NO: 16 or SEQ ID NO:28.
[0085] In specific embodiments, the inhibitors of ActRIIB used in the compositions and methods described herein comprise an extracellular domain of ActRIIB with one or more amino acid substitutions/mutations. Such an amino acid substitution/mutation can be, for example, an exchange from the leucine at amino acid position 79 of SEQ ID NO: 16 or SEQ ID NO:28 to an acidic amino acid, such as aspartic acid or glutamic acid. For example, position L79 of SEQ ID NO: 16 or SEQ ID NO:28 may be altered in ActRIIB extracellular domain polypeptides to confer altered activin-myostatin (GDF-11) binding properties. L79A and L79P mutations reduce GDF-11 binding to a greater extent than activin binding. L79E and L79D mutations retain GDF-11 binding, while demonstrating greatly reduced activin binding.
[0086] In certain embodiments, the inhibitors of ActRIIB used in the compositions and methods described herein comprise a truncated form of an ActRIIB extracellular domain that also carries an amino acid substitution, e.g., an exchange from the leucine at amino acid
position 79 of SEQ ID NO: 16 or SEQ ID NO:28 to an acidic amino acid, such as aspartic acid or glutamic acid. In a specific embodiment, the truncated form of an extracellular domain of ActRIIB polypeptide that also carries an amino acid substitution used in the compositions and methods described herein is SEQ ID NO:23. Forms of ActRIIB that are truncated and/or carry one or more amino acid substitutions can be linked to an Fc domain of an antibody as discussed above.
[0087] Functionally active fragments of ActRIIB polypeptides can be obtained, for example, by screening polypeptides recombinantly produced from the corresponding fragment of the nucleic acid encoding an ActRIIB polypeptide. In addition, fragments can be chemically synthesized using techniques known in the art such as conventional Merrifield solid phase f-Moc or t-Boc chemistry. The fragments can be produced (recombinantly or by chemical synthesis) and tested to identify those peptidyl fragments that can function as antagonists (inhibitors) of ActRIIB protein or signaling mediated by activin.
[0088] In addition, functionally active variants of ActRIIB polypeptides can be obtained, for example, by screening libraries of modified polypeptides recombinantly produced from the corresponding mutagenized nucleic acids encoding an ActRIIB polypeptide. The variants can be produced and tested to identify those that can function as antagonists (inhibitors) of ActRIIB protein or signaling mediated by activin. In certain embodiments, a functional variant of the ActRIIB polypeptides comprises an amino acid sequence that is at least 75% identical to an amino acid sequence selected from SEQ ID NO:17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43. In certain embodiments, the functional variant has an amino acid sequence at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical to an amino acid sequence selected from SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43.
[0089] Functional variants may be generated, for example, by modifying the structure of an ActRIIB polypeptide for such purposes as enhancing therapeutic efficacy, or stability (e.g., ex vivo shelf life and resistance to proteolytic degradation in vivo). Such modified ActRIIB polypeptides when selected to retain activin binding, are considered functional equivalents of the naturally-occurring ActRIIB polypeptides. Modified ActRIIB polypeptides can also be produced, for instance, by amino acid substitution, deletion, or addition. For instance, it is reasonable to expect that an isolated replacement of a leucine with an isoleucine or valine, an aspartate with a glutamate, a threonine with a serine, or a similar replacement of an amino acid with a structurally related amino acid (e.g., conservative mutations) will not have a
major effect on the biological activity of the resulting molecule. Conservative replacements are those that take place within a family of amino acids that are related in their side chains. Whether a change in the amino acid sequence of an ActRIIB polypeptide results in a functional homolog can be readily determined by assessing the ability of the variant ActRIIB polypeptide to produce a response in cells in a fashion similar to the wild-type ActRIIB polypeptide.
[0090] Provided herein are methods of generating mutants, particularly sets of combinatorial mutants of an ActRIIB polypeptide, as well as truncation mutants; pools of combinatorial mutants are especially useful for identifying functional variant sequences. The purpose of screening such combinatorial libraries may be to generate, for example, ActRIIB polypeptide variants which can act as either agonists or antagonist, or alternatively, which possess novel activities all together.
[0091] It has been demonstrated that the ligand binding pocket of ActRIIB is defined by residues Y31, N33, N35, L38 through T41, E47, E50, Q53 through K55, L57, H58, Y60, S62, K74, W78 through N83, Y85, R87, A92, and E94 through F101 of SEQ ID NO: 16 or SEQ ID NO:28. At these positions, it is expected that conservative mutations will be tolerated, although a K74A mutation is well-tolerated, as are R40A, K55A, F82A and mutations at position L79. R40 is a K in Xenopus, indicating that basic amino acids at this position will be tolerated. Q53 is R in bovine ActRIIB and K in Xenopus ActRIIB, and therefore amino acids including R, K, Q, N and H will be tolerated at this position. Thus, a general formula for an ActRIIB polypeptide for use in the methods and compositions described herein is one that comprises amino acids 29-109 of SEQ ID NO: 16 or SEQ ID NO:28, but optionally beginning at an amino acid position ranging from 20-24 or 22-25 of SEQ ID NO: 16 or SEQ ID NO:28 and ending at an amino acid position ranging from 129- 134 of SEQ ID NO: 16 or SEQ ID NO:28, and comprising no more than 1, 2, 5, or 15 conservative amino acid changes in the ligand binding pocket, and zero, one or more non- conservative alterations at amino acid positions 40, 53, 55, 74, 79 and/or 82 of SEQ ID NO: 16 or SEQ ID NO:28 in the ligand binding pocket. Such an ActRIIB polypeptide may retain greater than 80%, 90%, 95%> or 99%> sequence identity or sequence homology to the sequence of amino acids 29-109 of SEQ ID NO: 16 or SEQ ID NO:28. Sites outside the binding pocket, at which variability may be particularly well tolerated, include the amino and carboxy termini of the extracellular domain of ActRIIB, and positions 42-46 and 65-73. An
asparagine to alanine alteration at position 65 of SEQ ID NO: 16 or SEQ ID NO:28 (N65A) actually improves ligand binding in the A64 background, and is thus expected to have no detrimental effect on ligand binding in the R64 background. This change probably eliminates glycosylation at N65 in the A64 background, thus demonstrating that a significant change in this region is likely to be tolerated. While an R64A change is poorly tolerated, R64K is well- tolerated, and thus another basic residue, such as H may be tolerated at position 64.
[0092] As a specific example of an ActRIIB polypeptide with a mutation in the ligand binding domain, the positively-charged amino acid residue Asp (D80) of the ligand-binding domain of ActRIIB can be mutated to a different amino acid residue such that the variant ActRIIB polypeptide preferentially binds to GDF8, but not activin. In a specific
embodiment, the D80 residue is changed to an amino acid residue selected from the group consisting of: an uncharged amino acid residue, a negative amino acid residue, and a hydrophobic amino acid residue. As a further specific example, the hydrophobic residue L79 can be altered to the acidic amino acids aspartic acid or glutamic acid to greatly reduce activin binding while retaining GDF11 binding. As will be recognized by one of skill in the art, most of the described mutations, variants or modifications may be made at the nucleic acid level or, in some cases, by post translational modification or chemical synthesis. Such techniques are well known in the art.
[0093] In specific embodiments, the inhibitors of ActRIIB used in the compositions and methods described herein comprise a conjugate/fusion protein comprising an extracellular domain (e.g., an activin-binding domain) of an ActRIIB receptor linked to an Fc portion of an antibody. Such conjugate/fusion proteins may comprise any of the ActRIIB polypeptides disclosed herein (e.g., any of SEQ ID NOs: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43), any ActRIIB polypeptides known in the art, or any ActRIIB polypeptides generated using methods known in the art and/or provided herein.
[0094] In certain embodiments, the extracellular domain is linked to an Fc portion of an antibody via a linker, e.g. , a peptide linker. Exemplary linkers include short polypeptide sequences such as 2-10, 2-5, 2-4, 2-3 amino acid residues (e.g., glycine residues), such as, for example, a Gly-Gly-Gly linker. In a specific embodiment, the linker comprises the amino acid sequence Gly-Gly-Gly (GGG). In another specific embodiment, the linker comprises the amino acid sequence Thr-Gly-Gly-Gly (TGGG). Optionally, the Fc domain has one or more mutations at residues such as Asp-265, lysine 322, and Asn-434. In certain cases, the mutant
Fc domain having one or more of these mutations (e.g., an Asp-265 mutation) has a reduced ability to bind to the Fey receptor relative to a wild-type Fc domain. In other cases, the mutant Fc domain having one or more of these mutations (e.g., an Asn-434 mutation) has an increased ability to bind to the MHC class I- related Fc-receptor (FcRN) relative to a wild- type Fc domain. Exemplary fusion proteins comprising a soluble extracellular domain of ActRIIB fused to an Fc domain are set forth in SEQ ID NOs:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, and 47.
[0095] In a specific embodiment, the ActRIIB inhibitors used in the compositions and methods described herein comprise the extracellular domain of ActRIIB, or a portion thereof, linked to an Fc portion of an antibody, wherein said ActRIIB inhibitor comprises an amino acid sequence that is at least 75% identical to an amino acid sequence selected from
SEQ ID NOs:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, and 47. In another specific embodiment, the ActRIIB inhibitors used in the compositions and methods described herein comprise the extracellular domain of ActRIIB, or a portion thereof, linked to an Fc portion of an antibody, wherein said ActRIIB inhibitor comprises an amino acid sequence that is at least 80%, 85%, 90%, 95%, 96%, 97%, 98%, or 99% identical to an amino acid sequence selected from SEQ ID NOs:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, and 47.
[0096] In a specific embodiment, the ActRIIB inhibitor to be used in the compositions and methods described herein is a fusion protein between the extracellular domain of the human ActRIIB receptor and the Fc portion of IgGl . In another specific embodiment, the ActRIIB inhibitor to be used in the compositions and methods described herein is a fusion protein between a truncated extracellular domain of the human ActRIIB receptor and the Fc portion of IgGl . In another specific embodiment, the ActRIIB inhibitor to be used in the compositions and methods described herein is a fusion protein between a truncated extracellular domain of the human ActRIIB receptor and the Fc portion of IgGl, wherein the truncated extracellular domain of the human ActRIIB receptor possesses an amino acid substitution at the amino acid position corresponding to amino acid 79 of SEQ ID NO: 16 or SEQ ID NO:28. In one embodiment, the amino acid substitution at the amino acid position corresponding to amino acid 79 of SEQ ID NO: 16 or SEQ ID NO:28 is substitution of Leucine for Aspartic Acid (i.e., an L79D mutation).
[0097] In a specific embodiment, the ActRIIB inhibitor to be used in the compositions and methods described herein is SEQ ID NO:24 or 25, which represents a fusion protein
between the extracellular domain of the human ActRIIB receptor and the Fc portion of IgGl, wherein said ActRIIB extracellular domain comprises amino acids 25-131 of SEQ ID NO:28 with an L79D mutation. The nucleic acid sequence encoding the ActRIIB-Fc fusion protein of SEQ ID NO:24 is presented in SEQ ID NO:45.
[0098] In another specific embodiment, the ActRIIB inhibitor to be used in the compositions and methods described herein is SEQ ID NO:34 or 35, which represents a fusion protein between the extracellular domain of the human ActRIIB receptor and the Fc portion of IgGl, wherein said ActRIIB extracellular domain comprises amino acids 25-131 of SEQ ID NO: 16 with an L79D mutation.
[0099] Asparagine-linked glycosylation recognition sites generally comprise a tripeptide sequence, asparagine-X-threonine (or asparagine-X-serine) (where "X" is any amino acid) which is specifically recognized by appropriate cellular glycosylation enzymes. The alteration may also be made by the addition of, or substitution by, one or more serine or threonine residues to the sequence of the wild-type ActRIIB polypeptide (for O-linked glycosylation sites). A variety of amino acid substitutions or deletions at one or both of the first or third amino acid positions of a glycosylation recognition site (and/or amino acid deletion at the second position) results in non-glycosylation at the modified tripeptide sequence. Another means of increasing the number of carbohydrate moieties on an ActRIIB polypeptide is by chemical or enzymatic coupling of glycosides to the ActRIIB polypeptide. Depending on the coupling mode used, the sugar(s) may be attached to (a) arginine and histidine; (b) free carboxyl groups; (c) free sulfhydryl groups such as those of cysteine; (d) free hydroxyl groups such as those of serine, threonine, or hydroxyproline; (e) aromatic residues such as those of phenylalanine, tyrosine, or tryptophan; or (f) the amide group of glutamine. These methods are described in International Patent Application No. WO
87/05330 published Sep. 11, 1987, and in Aplin and Wriston (1981) CRC Crit. Rev.
Biochem., pp. 259-306, incorporated by reference herein. Removal of one or more carbohydrate moieties present on an ActRIIB polypeptide may be accomplished chemically and/or enzymatically. Chemical deglycosylation may involve, for example, exposure of the ActRIIB polypeptide to the compound trifluoromethanesulfonic acid, or an equivalent compound. This treatment results in the cleavage of most or all sugars except the linking sugar (N-acetylglucosamine or N-acetylgalactosamine), while leaving the amino acid sequence intact. Chemical deglycosylation is further described by Hakimuddin et al. (1987)
Arch. Biochem. Biophys. 259:52 and by Edge et al. (1981) Anal. Biochem. 118: 131.
Enzymatic cleavage of carbohydrate moieties on ActRIIB polypeptides can be achieved by the use of a variety of endo- and exo-glycosidases as described by Thotakura et al. (1987) Meth. Enzymol. 138:350. The sequence of an ActRIIB polypeptide may be adjusted, as appropriate, depending on the type of expression system used, as mammalian, yeast, insect and plant cells may all introduce differing glycosylation patterns that can be affected by the amino acid sequence of the peptide. In general, ActRIIB proteins for use in humans will be expressed in a mammalian cell line that provides proper glycosylation, such as HEK293 or CHO cell lines, although other expression systems, such as other mammalian expression cell lines, yeast cell lines with engineered glycosylation enzymes and insect cells, are expected to be useful as well.
[00100] In specific embodiments, encompassed herein are mutated ActRIIB polypeptides comprising the addition of a further N-linked glycosylation site (N-X-S/T) that increases the serum half-life of an ActRIIB-Fc fusion protein, relative to the ActRIIB(R64)-Fc form. In a specific embodiment, introduction of an asparagine at position 24 of SEQ ID NO: 16 or SEQ ID NO:28 (A24N) results in the creation of an NXT sequence that confers a longer half- life. Other NX(T/S) sequences can be found at 42-44 (NQS) and 65-67 (NSS), although the latter may not be efficiently glycosylated with the R at position 64 (i.e., in R64 polypeptides). N- X-S/T sequences may be generally introduced at positions outside the ligand binding pocket of ActRIIB, which is detailed above. Particularly suitable sites for the introduction of non- endogenous N-X-S/T sequences include amino acids 20-29, 20-24, 22-25, 109-134, 120-134 or 129-134 of SEQ ID NO: 16 or SEQ ID NO:28. N-X-S/T sequences may also be introduced into the linker between the ActRIIB sequence and the Fc or other fusion component. Such a site may be introduced with minimal effort by introducing an N in the correct position with respect to a pre-existing S or T, or by introducing an S or T at a position corresponding to a pre-existing N. Thus, desirable alterations that would create an N-linked glycosylation site are: A24N, R64N, S67N (possibly combined with an N65A alteration), E106N, Rl 12N, G120N, E123N, P129N, A132N, Rl 12S and Rl 12T (with all amino acid positions corresponding to the positions they can be found in SEQ ID NO: 16 or SEQ ID NO:28). Any S that is predicted to be glycosylated may be altered to a T without creating an immunogenic site, because of the protection afforded by the glycosylation. Likewise, any T that is predicted to be glycosylated may be altered to an S. Thus the alterations S67T and S44T are
encompassed herein. Likewise, in an A24N variant, an S26T alteration may be used.
Accordingly, an ActRIIB polypeptide may include one or more additional, non-endogenous N-linked glycosylation consensus sequences.
[00101] A variety of screening assays are provided herein, and such assays may be used to evaluate ActRIIB polypeptide variants. For example, an ActRIIB polypeptide variant may be screened for ability to bind to an ActRIIB ligand, to prevent binding of an ActRIIB ligand to an ActRIIB polypeptide or to interfere with signaling caused by an ActRIIB ligand. The activity of an ActRIIB polypeptide or its variants may also be tested in a cell-based or in vivo assay.
[00102] Combinatorially-derived variants can be generated which have a selective or generally increased potency relative to a naturally occurring ActRIIB polypeptide. Likewise, mutagenesis can give rise to variants which have intracellular half-lives dramatically different than the corresponding wild-type ActRIIB polypeptide. For example, the altered protein can be rendered either more stable or less stable to proteolytic degradation or other cellular processes which result in destruction of, or otherwise inactivation of a native ActRIIB polypeptide. Such variants, and the genes which encode them, can be utilized to alter ActRIIB polypeptide levels by modulating the half-life of the ActRIIB polypeptides. For instance, a short half-life can give rise to more transient biological effects and can allow tighter control of recombinant ActRIIB polypeptide levels within the patient. In an Fc fusion protein, mutations may be made in the linker (if any) and/or the Fc portion to alter the half- life of the protein.
[00103] A combinatorial library may be produced by way of a degenerate library of genes encoding a library of polypeptides which each include at least a portion of potential ActRIIB polypeptide sequences. For instance, a mixture of synthetic oligonucleotides can be enzymatically ligated into gene sequences such that the degenerate set of potential ActRIIB polypeptide nucleotide sequences are expressible as individual polypeptides, or alternatively, as a set of larger fusion proteins (e.g., for phage display).
[00104] There are many ways by which the library of potential homologs can be generated from a degenerate oligonucleotide sequence. Chemical synthesis of a degenerate gene sequence can be carried out in an automatic DNA synthesizer, and the synthetic genes then be ligated into an appropriate vector for expression. The synthesis of degenerate
oligonucleotides is well known in the art (see for example, Narang, S A (1983) Tetrahedron 39:3; Itakura et al, (1981) Recombinant DNA, Proc. 3rd Cleveland Sympos.
Macromolecules, ed. AG Walton, Amsterdam: Elsevier pp 273-289; Itakura et al, (1984) Annu. Rev. Biochem. 53:323; Itakura et al, (1984) Science 198: 1056; Ike et al, (1983) Nucleic Acid Res. 11 :477). Such techniques have been employed in the directed evolution of other proteins (see, for example, Scott et al, (1990) Science 249:386-390; Roberts et al, (1992) PNAS USA 89:2429-2433; Devlin et al, (1990) Science 249: 404-406; Cwirla et al, (1990) PNAS USA 87: 6378-6382; as well as U.S. Pat. Nos. 5,223,409, 5,198,346, and 5,096,815).
[00105] Alternatively, other forms of mutagenesis can be utilized to generate a
combinatorial library. For example, ActRIIB polypeptide variants can be generated and isolated from a library by screening using, for example, alanine scanning mutagenesis and the like (Ruf et al, (1994) Biochemistry 33: 1565-1572; Wang et al, (1994) J. Biol. Chem.
269:3095-3099; Balint et al, (1993) Gene 137: 109-118; Grodberg et al, (1993) Eur. J.
Biochem. 218:597-601; Nagashima et al, (1993) J. Biol. Chem. 268:2888-2892; Lowman et al, (1991) Biochemistry 30: 10832-10838; and Cunningham et al, (1989) Science 244:1081- 1085), by linker scanning mutagenesis (Gustin et al, (1993) Virology 193:653-660; Brown et al, (1992) Mol. Cell Biol. 12:2644-2652; McKnight et al, (1982) Science 232:316); by saturation mutagenesis (Meyers et al, (1986) Science 232:613); by PCR mutagenesis (Leung et al., (1989) Method Cell Mol Biol 1 : 11-19); or by random mutagenesis, including chemical mutagenesis, etc. (Miller et al, (1992) A Short Course in Bacterial Genetics, CSHL Press, Cold Spring Harbor, N.Y.; and Greener et al, (1994) Strategies in Mol Biol 7:32-34). Linker scanning mutagenesis, particularly in a combinatorial setting, is an attractive method for identifying truncated (bioactive) forms of ActRIIB polypeptides.
[00106] A wide range of techniques are known in the art for screening gene products of combinatorial libraries made by point mutations and truncations, and, for that matter, for screening cDNA libraries for gene products having a certain property. Such techniques will be generally adaptable for rapid screening of the gene libraries generated by the
combinatorial mutagenesis of ActRIIB polypeptides. The most widely used techniques for screening large gene libraries typically comprises cloning the gene library into replicable expression vectors, transforming appropriate cells with the resulting library of vectors, and expressing the combinatorial genes under conditions in which detection of a desired activity
facilitates relatively easy isolation of the vector encoding the gene whose product was detected. Preferred assays include activin binding assays and activin-mediated cell signaling assays.
[00107] In certain embodiments, ActRIIB polypeptides may further comprise post- translational modifications in addition to any that are naturally present in the ActRIIB polypeptides. Such modifications include, but are not limited to, acetylation, carboxylation, glycosylation, phosphorylation, lipidation, and acylation. As a result, the modified ActRIIB polypeptides may contain non-amino acid elements, such as polyethylene glycols, lipids, poly- or mono-saccharide, and phosphates. Effects of such non-amino acid elements on the functionality of a ActRIIB polypeptide may be tested by any method known to the skilled artisan. When an ActRIIB polypeptide is produced in cells by cleaving a nascent form of the ActRIIB polypeptide, post-translational processing may also be important for correct folding and/or function of the protein. Different cells (such as CHO, HeLa, MDCK, 293, W138, NIH-3T3 or HEK293) have specific cellular machinery and characteristic mechanisms for such post-translational activities and may be chosen to ensure the correct modification and processing of the ActRIIB polypeptides.
[00108] In certain aspects, functional variants or modified forms of the ActRIIB polypeptides include fusion proteins having at least a portion of the ActRIIB polypeptides and one or more fusion domains. Well known examples of such fusion domains include, but are not limited to, polyhistidine, Glu-Glu, glutathione S transferase (GST), thioredoxin, protein A, protein G, an immunoglobulin heavy chain constant region (Fc), maltose binding protein (MBP), or human serum albumin. A fusion domain may be selected so as to confer a desired property. For example, some fusion domains are particularly useful for isolation of the fusion proteins by affinity chromatography. For the purpose of affinity purification, relevant matrices for affinity chromatography, such as glutathione-, amylase-, and nickel- or cobalt-conjugated resins are used. Many of such matrices are available in "kit" form, such as the Pharmacia GST purification system and the QIAexpress™ system (Qiagen) useful with (HIS6) fusion partners. As another example, a fusion domain may be selected so as to facilitate detection of the ActRIIB polypeptides. Examples of such detection domains include the various fluorescent proteins (e.g., GFP) as well as "epitope tags," which are usually short peptide sequences for which a specific antibody is available. Well known epitope tags for which specific monoclonal antibodies are readily available include FLAG,
influenza virus haemagglutinin (HA), and c-myc tags. In some cases, the fusion domains have a protease cleavage site, such as for Factor Xa or Thrombin, which allows the relevant protease to partially digest the fusion proteins and thereby liberate the recombinant proteins therefrom. The liberated proteins can then be isolated from the fusion domain by subsequent chromatographic separation. In certain preferred embodiments, an ActRIIB polypeptide is fused with a domain that stabilizes the ActRIIB polypeptide in vivo (a "stabilizer" domain). By "stabilizing" is meant anything that increases serum half life, regardless of whether this is because of decreased destruction, decreased clearance by the kidney, or other
pharmacokinetic effect. Fusions with the Fc portion of an immunoglobulin are known to confer desirable pharmacokinetic properties on a wide range of proteins. Likewise, fusions to human serum albumin can confer desirable properties. Other types of fusion domains that may be selected include multimerizing (e.g., dimerizing, tetramerizing) domains and functional domains (that confer an additional biological function, such as further stimulation of bone growth or muscle growth, as desired).
[00109] It is understood that different elements of the fusion proteins may be arranged in any manner that is consistent with the desired functionality. For example, an ActRIIB polypeptide may be placed C-terminal to a heterologous domain, or, alternatively, a heterologous domain may be placed C-terminal to an ActRIIB polypeptide. The ActRIIB polypeptide domain and the heterologous domain need not be adjacent in a fusion protein, and additional domains or amino acid sequences may be included C- or N-terminal to either domain or between the domains.
[00110] In certain embodiments, the ActRIIB polypeptides contain one or more modifications that are capable of stabilizing the ActRIIB polypeptides. For example, such modifications enhance the in vitro half life of the ActRIIB polypeptides, enhance circulatory half life of the ActRIIB polypeptides or reduce proteolytic degradation of the ActRIIB polypeptides. Such stabilizing modifications include, but are not limited to, fusion proteins (including, for example, fusion proteins comprising an ActRIIB polypeptide and a stabilizer domain), modifications of a glycosylation site (including, for example, addition of a glycosylation site to an ActRIIB polypeptide), and modifications of carbohydrate moiety (including, for example, removal of carbohydrate moieties from an ActRIIB polypeptide). In the case of fusion proteins, an ActRIIB polypeptide is fused to a stabilizer domain such as an IgG molecule (e.g., an Fc domain). As used herein, the term "stabilizer domain" not only
refers to a fusion domain (e.g., Fc) as in the case of fusion proteins, but also includes nonproteinaceous modifications such as a carbohydrate moiety, or nonproteinaceous polymer, such as polyethylene glycol.
[00111] In certain embodiments, isolated and/or purified forms of the ActRIIB
polypeptides, which are isolated from, or otherwise substantially free of, other proteins can be used with the methods and compositions described herein. ActRIIB polypeptides will generally be produced by expression from recombinant nucleic acids.
[00112] In certain aspects, provided herein are isolated and/or recombinant nucleic acids encoding any of the ActRIIB polypeptides (e.g., soluble ActRIIB polypeptides), including fragments, functional variants and fusion proteins disclosed herein. For example, SEQ ID NO: 19 encodes the naturally occurring human ActRIIB precursor polypeptide. The subject nucleic acids may be single-stranded or double stranded. Such nucleic acids may be DNA or RNA molecules. These nucleic acids may be used, for example, in methods for making ActRIIB polypeptides or as direct therapeutic agents (e.g., in a gene therapy approach).
[00113] In certain aspects, the nucleic acids encoding ActRIIB polypeptides are further understood to include nucleic acids that are variants of SEQ ID NO: 19 as well as variants of those nucleic acid sequences that encode soluble ActRIIB polypeptides (e.g., nucleic acids that encode SEQ ID NOs: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, and 43). Variant nucleotide sequences include sequences that differ by one or more nucleotide substitutions, additions or deletions, such as allelic variants.
[00114] In certain embodiments, the isolated or recombinant nucleic acid sequences that can be used are at least 80%, 85%, 90%, 95%, 97%, 98%, 99% or 100% identical to SEQ ID NO: 19 or those nucleic acid sequences that encode soluble ActRIIB polypeptides (e.g., nucleic acids that encode SEQ ID NOs: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, and 43). One of ordinary skill in the art will appreciate that nucleic acid sequences
complementary to SEQ ID NO: 19 or those nucleic acid sequences that encode soluble ActRIIB polypeptides (e.g., nucleic acids that encode SEQ ID NOs: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, and 43), and variants of SEQ ID NO: 19 or those nucleic acid sequences that encode soluble ActRIIB polypeptides (e.g., nucleic acids that encode SEQ ID NOs: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, and 43) can be used with the methods and compositions described herein. In further embodiments, the nucleic acid sequences can
be isolated, recombinant, and/or fused with a heterologous nucleotide sequence, or in a DNA library.
[00115] In other embodiments, nucleic acids that can be used also include nucleotide sequences that hybridize under highly stringent conditions to the nucleotide sequence designated in SEQ ID NO: 19 or those nucleic acid sequences that encode soluble ActRIIB polypeptides (e.g., nucleic acids that encode SEQ ID NOs: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, and 43), complement sequence of SEQ ID NO: 19 or those nucleic acid sequences that encode soluble ActRIIB polypeptides (e.g., nucleic acids that encode SEQ ID NOs: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, and 43), or fragments thereof. As discussed above, one of ordinary skill in the art will readily understand that appropriate stringency conditions which promote DNA hybridization can be varied. One of ordinary skill in the art will understand that appropriate stringency conditions which promote DNA hybridization can be varied. For example, one can perform the hybridization at 6.0 times sodium chloride/sodium citrate (SSC) at about 45 degree Celsius, followed by a wash of 2.0 times SSC at 50 degree Celsius. For example, the salt concentration in the wash step can be selected from a low stringency of about 2.0 times SSC at 50 degree Celsius to a high stringency of about 0.2 times SSC at 50 degree Celsius. In addition, the temperature in the wash step can be increased from low stringency conditions at room temperature, about 22 degree Celsius, to high stringency conditions at about 65 degree Celsius. Both temperature and salt may be varied, or temperature or salt concentration may be held constant while the other variable is changed. In one embodiment, nucleic acids which hybridize under low stringency conditions of 6 times SSC at room temperature followed by a wash at 2 times SSC at room temperature can be used with the methods and compositions described herein.
[00116] Isolated nucleic acids which differ from the nucleic acids as set forth in SEQ ID NO: 19 or those nucleic acid sequences that encode soluble ActRIIB polypeptides (e.g., nucleic acids that encode SEQ ID NOs: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, and 43) due to degeneracy in the genetic code can also be used. For example, a number of amino acids are designated by more than one triplet. Codons that specify the same amino acid, or synonyms (for example, CAU and CAC are synonyms for histidine) may result in "silent" mutations which do not affect the amino acid sequence of the protein. However, it is expected that DNA sequence polymorphisms that do lead to changes in the amino acid sequences of the subject proteins will exist among mammalian cells. One skilled in the art
will appreciate that these variations in one or more nucleotides (up to about 3-5% of the nucleotides) of the nucleic acids encoding a particular protein may exist among individuals of a given species due to natural allelic variation. Any and all such nucleotide variations and resulting amino acid polymorphisms can be used with the methods and compositions described herein.
[00117] In certain embodiments, the recombinant nucleic acids may be operably linked to one or more regulatory nucleotide sequences in an expression construct. Regulatory nucleotide sequences will generally be appropriate to the host cell used for expression.
Numerous types of appropriate expression vectors and suitable regulatory sequences are known in the art for a variety of host cells. Typically, said one or more regulatory nucleotide sequences may include, but are not limited to, promoter sequences, leader or signal sequences, ribosomal binding sites, transcriptional start and termination sequences, translational start and termination sequences, and enhancer or activator sequences.
Constitutive or inducible promoters as known in the art can be used with the methods and compositions described herein. The promoters may be either naturally occurring promoters, or hybrid promoters that combine elements of more than one promoter. An expression construct may be present in a cell on an episome, such as a plasmid, or the expression construct may be inserted in a chromosome. In a preferred embodiment, the expression vector contains a selectable marker gene to allow the selection of transformed host cells. Selectable marker genes are well known in the art and will vary with the host cell used.
[00118] In certain aspects, the subject nucleic acid is provided in an expression vector comprising a nucleotide sequence encoding an ActRIIB polypeptide and operably linked to at least one regulatory sequence. Regulatory sequences are art-recognized and are selected to direct expression of the ActRIIB polypeptide. Accordingly, the term regulatory sequence includes promoters, enhancers, and other expression control elements. Exemplary regulatory sequences are described in Goeddel; Gene Expression Technology: Methods in Enzymology, Academic Press, San Diego, Calif. (1990). For instance, any of a wide variety of expression control sequences that control the expression of a DNA sequence when operatively linked to it may be used in these vectors to express DNA sequences encoding an ActRIIB polypeptide. Such useful expression control sequences, include, for example, the early and late promoters of SV40, tet promoter, adenovirus or cytomegalovirus immediate early promoter, RSV promoters, the lac system, the trp system, the TAC or TRC system, T7 promoter whose
expression is directed by T7 RNA polymerase, the major operator and promoter regions of phage lambda, the control regions for fd coat protein, the promoter for 3-phosphoglycerate kinase or other glycolytic enzymes, the promoters of acid phosphatase, e.g., Pho5, the promoters of the yeast .alpha. -mating factors, the polyhedron promoter of the baculovirus system and other sequences known to control the expression of genes of prokaryotic or eukaryotic cells or their viruses, and various combinations thereof. It should be understood that the design of the expression vector may depend on such factors as the choice of the host cell to be transformed and/or the type of protein desired to be expressed. Moreover, the vector's copy number, the ability to control that copy number and the expression of any other protein encoded by the vector, such as antibiotic markers, should also be considered.
[00119] A recombinant nucleic acid can be produced by ligating the cloned gene, or a portion thereof, into a vector suitable for expression in either prokaryotic cells, eukaryotic cells (yeast, avian, insect or mammalian), or both. Expression vehicles for production of a recombinant ActRIIB polypeptide include plasmids and other vectors. For instance, suitable vectors include plasmids of the types: pBR322-derived plasmids, pEMBL-derived plasmids, pEX-derived plasmids, pBTac-derived plasmids and pUC-derived plasmids for expression in prokaryotic cells, such as E. coli.
[00120] Some mammalian expression vectors contain both prokaryotic sequences to facilitate the propagation of the vector in bacteria, and one or more eukaryotic transcription units that are expressed in eukaryotic cells. The pcDNAI/amp, pcDNAI/neo, pRc/CMV, pSV2gpt, pSV2neo, pSV2-dhfr, pTk2, pRSVneo, pMSG, pSVT7, pko-neo and pHyg derived vectors are examples of mammalian expression vectors suitable for transfection of eukaryotic cells. Some of these vectors are modified with sequences from bacterial plasmids, such as pBR322, to facilitate replication and drug resistance selection in both prokaryotic and eukaryotic cells. Alternatively, derivatives of viruses such as the bovine papilloma virus (BPV-1), or Epstein-Barr virus (pHEBo, pREP-derived and p205) can be used for transient expression of proteins in eukaryotic cells. Examples of other viral (including retroviral) expression systems can be found below in the description of gene therapy delivery systems. The various methods employed in the preparation of the plasmids and in transformation of host organisms are well known in the art. For other suitable expression systems for both prokaryotic and eukaryotic cells, as well as general recombinant procedures, see Molecular Cloning A Laboratory Manual, 3rd Ed., ed. by Sambrook, Fritsch and Maniatis (Cold Spring
Harbor Laboratory Press, 2001). In some instances, it may be desirable to express the recombinant polypeptides by the use of a baculovirus expression system. Examples of such baculovirus expression systems include pVL-derived vectors (such as pVL1392, pVL1393 and pVL941), pAcUW-derived vectors (such as pAcUWl), and pBlueBac-derived vectors (such as the .beta. -gal containing pBlueBac III).
[00121] In a preferred embodiment, a vector will be designed for production of the subject ActRIIB polypeptides in CHO cells, such as a Pcmv-Script vector (Stratagene, La Jolla, Calif), pcDNA4 vectors (Invitrogen, Carlsbad, Calif.) and pCI-neo vectors (Promega, Madison, Wis.). As will be apparent, the subject gene constructs can be used to cause expression of the subject ActRIIB polypeptides in cells propagated in culture, e.g., to produce proteins, including fusion proteins or variant proteins, for purification.
[00122] This disclosure also pertains to a host cell transfected with a recombinant gene including a coding sequence (e.g., SEQ ID NO: 19 or those nucleic acid sequences that encode soluble ActRIIB polypeptides (e.g., nucleic acids that encode SEQ ID NOs: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, and 43)) for one or more of the subject ActRIIB polypeptides. The host cell may be any prokaryotic or eukaryotic cell. For example, an ActRIIB polypeptide may be expressed in bacterial cells such as E. coli, insect cells (e.g., using a baculovirus expression system), yeast, or mammalian cells. Other suitable host cells are known to those skilled in the art.
[00123] Accordingly, provided herein are methods of producing the ActRIIB polypeptides.
For example, a host cell transfected with an expression vector encoding an ActRIIB polypeptide can be cultured under appropriate conditions to allow expression of the ActRIIB polypeptide to occur. The ActRIIB polypeptide may be secreted and isolated from a mixture of cells and medium containing the ActRIIB polypeptide. Alternatively, the ActRIIB polypeptide may be retained cytoplasmically or in a membrane fraction and the cells harvested, lysed and the protein isolated. A cell culture includes host cells, media and other byproducts. Suitable media for cell culture are well known in the art. The subject ActRIIB polypeptides can be isolated from cell culture medium, host cells, or both, using techniques known in the art for purifying proteins, including ion-exchange chromatography, gel filtration chromatography, ultrafiltration, electrophoresis, immunoaffinity purification with antibodies specific for particular epitopes of the ActRIIB polypeptides and affinity purification with an agent that binds to a domain fused to the ActRIIB polypeptide (e.g., a protein A column may
be used to purify an ActRIIB-Fc fusion). In a preferred embodiment, the ActRIIB
polypeptide is a fusion protein containing a domain which facilitates its purification. In a preferred embodiment, purification is achieved by a series of column chromatography steps, including, for example, three or more of the following, in any order: protein A
chromatography, Q sepharose chromatography, phenylsepharose chromatography, size exclusion chromatography, and cation exchange chromatography. The purification could be completed with viral filtration and buffer exchange. As demonstrated herein, ActRIIB -hFc protein was purified to a purity of >98% as determined by size exclusion chromatography and >95% as determined by SDS PAGE. This level of purity was sufficient to achieve desirable effects on bone in mice and an acceptable safety profile in mice, rats and non-human primates.
[00124] In another embodiment, a fusion gene coding for a purification leader sequence, such as a poly-(His)/enterokinase cleavage site sequence at the N-terminus of the desired portion of the recombinant ActRIIB polypeptide, can allow purification of the expressed fusion protein by affinity chromatography using a Ni2+ metal resin. The purification leader sequence can then be subsequently removed by treatment with enterokinase to provide the purified ActRIIB polypeptide (e.g., see Hochuli et al, (1987) J. Chromatography 411 : 177; and Janknecht et al, PNAS USA 88:8972).
[00125] Techniques for making fusion genes are well known. Essentially, the joining of various DNA fragments coding for different polypeptide sequences is performed in accordance with conventional techniques, employing blunt-ended or stagger-ended termini for ligation, restriction enzyme digestion to provide for appropriate termini, filling-in of cohesive ends as appropriate, alkaline phosphatase treatment to avoid undesirable joining, and enzymatic ligation. In another embodiment, the fusion gene can be synthesized by conventional techniques including automated DNA synthesizers. Alternatively, PCR amplification of gene fragments can be carried out using anchor primers which give rise to complementary overhangs between two consecutive gene fragments which can subsequently be annealed to generate a chimeric gene sequence (see, for example, Current Protocols in Molecular Biology, eds. Ausubel et al, John Wiley & Sons: 1992).
[00126] ActRIIB -Fc fusion protein can be expressed in stably transfected CHO-DUKX Bl
1 cells from a pAID4 vector (SV40 ori/enhancer, CMV promoter), using a tissue
plasminogen leader sequence of SEQ ID NO:8. The Fc portion can comprise a human IgGl
Fc sequence, as shown in SEQ ID NO:7. In certain embodiments, upon expression, the protein contained has, on average, between about 1.5 and 2.5 moles of sialic acid per molecule of ActRIIB-Fc fusion protein.
[00127] In certain embodiments, the long serum half-life of an ActRIIB-Fc fusion can be 25-32 days in human patients. Additionally, the CHO cell expressed material can have a higher affinity for activin B ligand than that reported for an ActRIIB-hFc fusion protein expressed in human 293 cells (del Re et al., J Biol Chem. 2004 Dec 17;279(51):53126-35). Additionally, without being bound by theory, the use of the TPA leader sequence provided greater production than other leader sequences and, unlike ActRIIB-Fc expressed with a native leader, may provide a highly pure N-terminal sequence. Use of the native leader sequence may result in two major species of ActRIIB-Fc, each having a different N-terminal sequence.
(c) Other ActRII Receptor Inhibitors
[00128] In certain embodiments, the inhibitors of ActRII receptors used in the
compositions and methods described herein are nucleic acid compounds.
[00129] Examples of categories of nucleic acid compounds that inhibit ActRII receptors include antisense nucleic acids, siRNA or RNAi constructs and catalytic nucleic acid constructs. A nucleic acid compound may be single- or double-stranded. A double-stranded compound may also include regions of overhang or non-complementarity, where one or the other of the strands is single-stranded. A single-stranded compound may include regions of self-complementarity, meaning that the compound may form a so-called "hairpin" or "stem- loop" structure, with a region of double helical structure.
[00130] In certain embodiments, the nucleic acid compounds that inhibit ActRII receptors may comprise a nucleotide sequence that is complementary to a region consisting of no more than 1000, no more than 500, no more than 250, no more than 100 or no more than 50, 35, 30, 25, 22, 20 or 18 nucleotides of the full-length ActRII receptor nucleic acid sequence or activin nucleic acid sequence (e.g., the nucleic acid sequence of an activin A or activin B subunit, also referred to as BA or BB). In specific embodiments, the region of
complementarity will be at least 8 nucleotides, and optionally at least 10 or at least 15 nucleotides, and optionally between 15 and 25 nucleotides. A region of complementarity may fall within an intron, a coding sequence or a noncoding sequence of the target transcript,
such as the coding sequence portion. Generally, a nucleic acid compound that inhibits an ActRII receptor will have a length of about 8 to about 500 nucleotides or base pairs in length, and optionally the length will be about 14 to about 50 nucleotides. A nucleic acid compound that inhibits an ActRII receptor may be a DNA (particularly for use as an antisense), an RNA, or an RNA:DNA hybrid. Any one strand may include a mixture of DNA and RNA, as well as modified forms that cannot readily be classified as either DNA or RNA. Likewise, a double stranded nucleic acid compound may be DNA:DNA, DNA:RNA, or RNA:RNA, and any one strand may also include a mixture of DNA and RNA, as well as modified forms that cannot readily be classified as either DNA or RNA.
[00131] The nucleic acid compounds that inhibit an ActRII receptor may include any of a variety of modifications, including one or modifications to the backbone (the sugar-phosphate portion in a natural nucleic acid, including internucleotide linkages) or the base portion (the purine or pyrimidine portion of a natural nucleic acid). In certain embodiments, an antisense nucleic acid compound will have a length of about 15 to about 30 nucleotides and will often contain one or more modifications to improve certain characteristics, such as stability in the serum, stability in in a cell, or stability in in a place where the compound is likely to be delivered, such as, e.g., the stomach in the case of orally delivered compounds and the lung for inhaled compounds. In the case of an RNAi construct, the strand complementary to the target transcript will generally be RNA or modifications thereof. The other strand may be RNA, DNA, or any other variation. The duplex portion of double stranded or single stranded "hairpin" RNAi construct may, in certain embodiments, have a length of 18 to 40 nucleotides in length and optionally about 21 to 23 nucleotides in length, so long as it serves as a Dicer substrate. Catalytic or enzymatic nucleic acids may be ribozymes or DNA enzymes and may also contain modified forms. In certain embodiments, nucleic acid compounds that inhibit ActRII receptors may inhibit expression of their target by about 50%, 60%>, 70%>, 75%, 80%>, 85%), 90%), 95%o, 99%o, or more under physiological conditions and at a concentration where a nonsense or sense control has little or no effect. Concentrations for testing the effect of nucleic acid compounds include 1, 5, 10 micromolar, or more.
[00132] In other embodiments, the inhibitors of ActRII receptors used in the compositions and methods described herein are antibodies. Such antibodies include antibodies that bind to activin (particularly the activin A or B subunits, also referred to as BA or BB) and disrupt ActRII receptor binding; and antibodies that bind to ActRII receptor polypeptides (e.g., a
soluble ActRIIA or soluble ActRIIB polypeptide) and disrupt activin binding.
[00133] By using immunogens derived from an ActRII receptor polypeptide or an activin polypeptide, anti-protein/anti-peptide antisera or monoclonal antibodies can be made by standard protocols (see, for example, Antibodies: A Laboratory Manual ed. by Harlow and Lane (Cold Spring Harbor Press: 1988)). A mammal, such as a mouse, a hamster or rabbit can be immunized with an immunogenic form of the ActRII receptor polypeptide, an antigenic fragment which is capable of eliciting an antibody response, or a fusion protein. Techniques for conferring immunogenicity on a protein or peptide include conjugation to carriers or other techniques well known in the art. An immunogenic portion of an ActRII receptor or activin polypeptide can be administered in the presence of adjuvant. The progress of immunization can be monitored by detection of antibody titers in plasma or serum.
Standard ELISA or other immunoassays can be used with the immunogen as antigen to assess the levels of antibodies.
[00134] Following immunization of an animal with an antigenic preparation of an ActRII receptor polypeptide, antisera can be obtained and, if desired, polyclonal antibodies can be isolated from the serum. To produce monoclonal antibodies, antibody-producing cells (lymphocytes) can be harvested from an immunized animal and fused by standard somatic cell fusion procedures with immortalizing cells such as myeloma cells to yield hybridoma cells. Such techniques are well known in the art, and include, for example, the hybridoma technique (originally developed by Kohler and Milstein, (1975) Nature, 256: 495-497), the human B cell hybridoma technique (Kozbar et al., (1983) Immunology Today, 4: 72), and the EBV-hybridoma technique to produce human monoclonal antibodies (Cole et al., (1985) Monoclonal Antibodies and Cancer Therapy, Alan R. Liss, Inc. pp. 77-96). Hybridoma cells can be screened immunochemically for production of antibodies specifically reactive with an ActRII receptor polypeptide and monoclonal antibodies isolated from a culture comprising such hybridoma cells.
[00135] The term "antibody" as used herein is intended to include fragments thereof which are also specifically reactive with a subject polypeptide. Antibodies can be fragmented using conventional techniques and the fragments screened for utility in the same manner as described above for whole antibodies. For example, F(ab)2 fragments can be generated by treating antibody with pepsin. The resulting F(ab)2 fragment can be treated to reduce disulfide bridges to produce Fab fragments. An antibody is further intended to include
bispecific, single-chain, chimeric, humanized and fully human molecules having affinity for an ActRII receptor or activin polypeptide conferred by at least one CDR region of the antibody. An antibody may further comprise a label attached thereto and able to be detected (e.g., the label can be a radioisotope, fluorescent compound, enzyme or enzyme co-factor).
[00136] In certain embodiments, the antibody is a recombinant antibody, which term encompasses any antibody generated in part by techniques of molecular biology, including CDR-grafted or chimeric antibodies, human or other antibodies assembled from library- selected antibody domains, single chain antibodies and single domain antibodies (e.g., human VH proteins or camelid VHH proteins). In certain embodiments, an antibody can be a monoclonal antibody, and in certain embodiments. For example, a method for generating a monoclonal antibody that binds specifically to an ActRII receptor polypeptide or activin polypeptide may comprise administering to a mouse an amount of an immunogenic composition comprising the antigen polypeptide effective to stimulate a detectable immune response, obtaining antibody-producing cells (e.g., cells from the spleen) from the mouse and fusing the antibody-producing cells with myeloma cells to obtain antibody-producing hybridomas, and testing the antibody-producing hybridomas to identify a hybridoma that produces a monocolonal antibody that binds specifically to the antigen. Once obtained, a hybridoma can be propagated in a cell culture, optionally in culture conditions where the hybridoma-derived cells produce the monoclonal antibody that binds specifically to the antigen. The monoclonal antibody may be purified from the cell culture.
[00137] The adjective "specifically reactive with" as used in reference to an antibody is intended to mean, as is generally understood in the art, that the antibody is sufficiently selective between the antigen of interest (e.g., an ActRII receptor polypeptide) and other antigens that are not of interest that the antibody is useful for, at minimum, detecting the presence of the antigen of interest in a particular type of biological sample. In certain methods employing the antibody, such as therapeutic applications, a higher degree of specificity in binding may be desirable. Monoclonal antibodies generally have a greater tendency (as compared to polyclonal antibodies) to discriminate effectively between the desired antigens and cross-reacting polypeptides. One characteristic that influences the specificity of an antibody:antigen interaction is the affinity of the antibody for the antigen. Although the desired specificity may be reached with a range of different affinities, generally preferred antibodies will have an affinity (a dissociation constant) of about 10~6, 10~7, 10~8, 10"
9 or less. Given the extraordinarily tight binding between activin and an ActRII receptor, it is expected that a neutralizing anti-activin or anti- ActRII receptor antibody would generally have a dissociation constant of 10~10 or less.
[00138] In addition, the techniques used to screen antibodies in order to identify a desirable antibody may influence the properties of the antibody obtained. For example, if an antibody is to be used for binding an antigen in solution, it may be desirable to test solution binding. A variety of different techniques are available for testing interaction between antibodies and antigens to identify particularly desirable antibodies. Such techniques include ELISAs, surface plasmon resonance binding assays (e.g., the Biacore.TM. binding assay, Biacore AB, Uppsala, Sweden), sandwich assays (e.g., the paramagnetic bead system of IGEN International, Inc., Gaithersburg, Md.), Western blots, immunoprecipitation assays, and immunohistochemistry.
[00139] In certain embodiments, ActRII receptor inhibitors to be used in the compositions and methods described herein include alternative forms of activin, particularly those with alterations in the type I receptor binding domain can bind to type II receptors and fail to form an active ternary complex. In certain embodiments, nucleic acids, such as antisense molecules, siRNAs or ribozymes that inhibit activin A, B, C or E, or, particularly, ActRII receptor expression, can be used in the compositions and methods described herein. In certain embodiments, the ActRII receptor inhibitors to be used in the compositions and methods described herein exhibit selectivity for inhibiting activin-mediated signaling versus other members of the TGF-beta family, particularly with respect to GDF8 and GDF11.
[00140] In other embodiments, the inhibitors of ActRII receptors used in the compositions and methods described herein are non-antibody proteins with ActRII receptor antagonist activity, including inhibin (i.e., inhibin alpha subunit), follistatin (e.g., follistatin-288 and follistatin-315), Cerberus, follistatin related protein ("FSRP"), endoglin, activin C, alpha(2)- macroglobulin, and an Ml 08 A (methionine to alanine change at position 108) mutant activin A.
[00141] In a specific embodiment, the ActRII receptor inhibitor to be used in the compositions and methods described herein is a follistatin polypeptide that antagonizes activin bioactivity and/or binds to activin. The term "follistatin polypeptide" includes polypeptides comprising any naturally occurring polypeptide of follistatin as well as any variants thereof (including mutants, fragments, fusions, and peptidomimetic forms) that retain
a useful activity, and further includes any functional monomer or multimer of follistatin. Variants of follistatin polypeptides that retain activin binding properties can be identified based on previous studies involving follistatin and activin interactions. For example, WO2008/030367, which is included by reference herein in its entirety, discloses specific follistatin domains ("FSDs") that are shown to be important for activin binding. Follistatin polypeptides include polypeptides derived from the sequence of any known follistatin having a sequence at least about 80% identical to the sequence of a follistatin polypeptide, and optionally at least 85%, 90%, 95%, 96%, 97%, 98%, 99% or greater identity. Examples of follistatin polypeptides include the mature follistatin polypeptide or shorter isoforms or other variants of the human follistatin precursor polypeptide as described, for example, in
WO2005/025601, which is included by reference herein in its entirety.
[00142] In a specific embodiment, the ActRII receptor inhibitor to be used in the compositions and methods described herein is a follistatin-like related gene (FLRG) that antagonizes activin bioactivity and/or binds to activin. The term "FLRG polypeptide" includes polypeptides comprising any naturally occurring polypeptide of FLRG as well as any variants thereof (including mutants, fragments, fusions, and peptidomimetic forms) that retain a useful activity. Variants of FLRG polypeptides that retain activin binding properties can be identified using routine methods to assay FLRG and activin interactions. See, for example, U.S. Pat. No. 6,537,966, which is included by reference herein in its entirety. FLRG polypeptides include polypeptides derived from the sequence of any known FLRG having a sequence at least about 80%> identical to the sequence of an FLRG polypeptide, and optionally at least 85%, 90%, 95%, 96%, 97%, 98%, 99% or greater identity.
[00143] In certain embodiments, functional variants or modified forms of the follistatin polypeptides and FLRG polypeptides include fusion proteins having at least a portion of the follistatin polypeptides or FLRG polypeptides and one or more fusion domains, such as, for example, domains that facilitate isolation, detection, stabilization or multimerization of the polypeptide. Suitable fusion domains are discussed in detail above with reference to the ActRII A and ActRIIB polypeptides. In one embodiment, an ActRII receptor inhibitor is a fusion protein comprising an activin binding portion of a follistaton polypeptide fused to an Fc domain. In another embodiment, an ActRII receptor inhibitor is a fusion protein comprising an activin binding portion of an FLRG polypeptide fused to an Fc domain.
4.3 SECOND ACTIVE AGENTS
[00144] Immunomodulatory compounds known as IMiDs® compounds (Celgene
Corporation) can be used as second active agent with the methods and compositions of the present invention. See subsection (a) below. Other second active agents that can be used with the methods and formulations of the present invention include thalidomide,
dexamethasone, melphalan, prednisone, bortezomib, cyclophosphamide, bisphosphonate, azacitidine, doxorubicin, vincristine, proteasome inhibitor, and dacogen.
[00145] In a specific embodiment, low-dose dexamethasone (see Rajkumar et al. 2010, The Lancet 11 :29-37) is combined with an Activin- ActRII inhibitor.
[00146] In certain embodiments, a third active agent can be used with the methods and formulations. In a specific embodiment, one or more of the following: lenalidomide (also known as Revlimid® or 3-(4-amino-l-oxo-l,3-dihydro-isoindol-2-yl)-piperidine-2,6-dione), dexamethasone, melphalan, prednisone, bortezomib, cyclophosphamide, bisphosphonate, and azacytidine is combined with an Activin-ActRII inhibitor.
[00147] In a specific embodiment, a combination of Revlimid®, dexamethasone, bisphosonate, and an ActRII inhibitor is used with the methods of the invention (e.g., to treat multiple myeloma). The invention also provides a pharmaceutical composition comprising Revlimid®, dexamethasone, bisphosonate, and an ActRII inhibitor.
[00148] In a specific embodiment, a combination of Revlimid®, melphalan, prednisone, and an ActRII inhibitor is used with the methods of the invention (e.g., to treat multiple myeloma). The invention also provides a pharmaceutical composition comprising
Revlimid®, dexamethasone, bisphosonate, and an ActRII inhibitor.
[00149] In certain embodiments, lenalidomide is combined with doxorubicin (Doxil®), vincristine and/or dexamethasone (Decadron®), and an ActRII inhibitor.
[00150] Second active agents can either be commercially purchased or prepared according to the methods described in the patents or patent publications disclosed herein. Further, optically pure compounds can be asymmetrically synthesized or resolved using known resolving agents or chiral columns as well as other standard synthetic organic chemistry techniques.
[00151] As used herein and unless otherwise indicated, the term "pharmaceutically acceptable salt" encompasses non-toxic acid and base addition salts of the compound to
which the term refers. Acceptable non-toxic acid addition salts include those derived from organic and inorganic acids or bases know in the art, which include, for example, hydrochloric acid, hydrobromic acid, phosphoric acid, sulfuric acid, methanesulphonic acid, acetic acid, tartaric acid, lactic acid, succinic acid, citric acid, malic acid, maleic acid, sorbic acid, aconitic acid, salicylic acid, phthalic acid, embolic acid, enanthic acid, and the like.
[00152] Compounds that are acidic in nature are capable of forming salts with various pharmaceutically acceptable bases. The bases that can be used to prepare pharmaceutically acceptable base addition salts of such acidic compounds are those that form non-toxic base addition salts, i.e., salts containing pharmacologically acceptable cations such as, but not limited to, alkali metal or alkaline earth metal salts and the calcium, magnesium, sodium or potassium salts in particular. Suitable organic bases include, but are not limited to,
Ν,Ν-dibenzylethylenediamine, chloroprocaine, choline, diethanolamine, ethylenediamine, meglumaine (N-methylglucamine), lysine, and procaine.
[00153] As used herein and unless otherwise indicated, the term "prodrug" means a derivative of a compound that can hydrolyze, oxidize, or otherwise react under biological conditions (in vitro or in vivo) to provide the compound. Examples of prodrugs include, but are not limited to, derivatives of immunomodulatory compounds that comprise
biohydrolyzable moieties such as biohydrolyzable amides, biohydrolyzable esters, biohydrolyzable carbamates, biohydrolyzable carbonates, biohydrolyzable ureides, and biohydrolyzable phosphate analogues. Other examples of prodrugs include derivatives of immunomodulatory compounds that comprise -NO, -N02, -ONO, or -ON02 moieties.
Prodrugs can typically be prepared using well-known methods, such as those described in 1 Burger 's Medicinal Chemistry and Drug Discovery, 172-178, 949-982 (Manfred E. Wolff ed., 5th ed. 1995), and Design of Prodrugs (H. Bundgaard ed., Elselvier, New York 1985).
[00154] As used herein and unless otherwise indicated, the terms "biohydrolyzable amide," "biohydrolyzable ester," "biohydrolyzable carbamate," "biohydrolyzable carbonate,"
"biohydrolyzable ureide," "biohydrolyzable phosphate" mean an amide, ester, carbamate, carbonate, ureide, or phosphate, respectively, of a compound that either: 1) does not interfere with the biological activity of the compound but can confer upon that compound
advantageous properties in vivo, such as uptake, duration of action, or onset of action; or 2) is biologically inactive but is converted in vivo to the biologically active compound. Examples of biohydrolyzable esters include, but are not limited to, lower alkyl esters, lower
acyloxyalkyl esters (such as acetoxylmethyl, acetoxyethyl, aminocarbonyloxymethyl, pivaloyloxymethyl, and pivaloyloxyethyl esters), lactonyl esters (such as phthalidyl and thiophthalidyl esters), lower alkoxy acyloxyalkyl esters (such as methoxycarbonyl-oxymethyl, ethoxycarbonyloxyethyl and isopropoxycarbonyloxyethyl esters), alkoxyalkyl esters, choline esters, and acylamino alkyl esters (such as acetamidomethyl esters). Examples of
biohydrolyzable amides include, but are not limited to, lower alkyl amides, a-amino acid amides, alkoxyacyl amides, and alkylaminoalkylcarbonyl amides. Examples of
biohydrolyzable carbamates include, but are not limited to, lower alkylamines, substituted ethylenediamines, amino acids, hydroxy alkylamines, heterocyclic and heteroaromatic amines, and polyether amines.
[00155] Various immunomodulatory compounds contain one or more chiral centers, and can exist as racemic mixtures of enantiomers or mixtures of diastereomers. Stereomerically pure forms of such compounds, as well as the use of mixtures of those forms, can be used with the methods and compositions of the present invention. For example, mixtures comprising equal or unequal amounts of the enantiomers of a particular immunomodulatory compounds may be used in methods and compositions of the invention. These isomers may be asymmetrically synthesized or resolved using standard techniques such as chiral columns or chiral resolving agents. See, e.g., Jacques, J., et al., Enantiomers, Racemates and
Resolutions (Wiley-lnterscience, New York, 1981); Wilen, S. H., et al, Tetrahedron 33:2725 (1977); Eliel, E. L., Stereochemistry of Carbon Compounds (McGraw-Hill, NY, 1962); and Wilen, S. H., Tables of Resolving Agents and Optical Resolutions p. 268 (E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN, 1972).
[00156] As used herein and unless otherwise indicated, the term "stereomerically pure" means a composition that comprises one stereoisomer of a compound and is substantially free of other stereoisomers of that compound. For example, a stereomerically pure composition of a compound having one chiral center will be substantially free of the opposite enantiomer of the compound. A stereomerically pure composition of a compound having two chiral centers will be substantially free of other diastereomers of the compound. A typical stereomerically pure compound comprises greater than about 80% by weight of one stereoisomer of the compound and less than about 20% by weight of other stereoisomers of the compound, more preferably greater than about 90% by weight of one stereoisomer of the compound and less than about 10%> by weight of the other stereoisomers of the compound,
even more preferably greater than about 95% by weight of one stereoisomer of the compound and less than about 5% by weight of the other stereoisomers of the compound, and most preferably greater than about 97% by weight of one stereoisomer of the compound and less than about 3% by weight of the other stereoisomers of the compound. As used herein and unless otherwise indicated, the term "stereomerically enriched" means a composition that comprises greater than about 60% by weight of one stereoisomer of a compound, preferably greater than about 70% by weight, more preferably greater than about 80% by weight of one stereoisomer of a compound. As used herein and unless otherwise indicated, the term
"enantiomerically pure" means a stereomerically pure composition of a compound having one chiral center. Similarly, the term "stereomerically enriched" means a stereomerically enriched composition of a compound having one chiral center.
(a) Immunomodulatory Compounds (IMiD® Compounds)
[00157] A number of studies have been conducted with the aim of providing compounds that can safely and effectively be used to treat diseases associated with abnormal production of TNF a. See, e.g., Marriott, J.B., et al, Expert Opin. Biol. Ther. 1(4): 1-8 (2001); G.W. Muller, et al, Journal of Medicinal Chemistry 39(17): 3238-3240 (1996); and G.W. Muller, et al, Bioorganic & Medicinal Chemistry Letters 8: 2669-2674 (1998). Some studies have focused on a group of compounds selected for their capacity to potently inhibit TNF-a production by LPS stimulated PBMC. L.G. Corral, et al, Ann. Rheum. Dis. 58:(Suppl I) 1107-1113 (1999). These compounds, which are referred to as IMiD® compounds (Celgene Corporation) or Immunomodulatory Drugs, show not only potent inhibition of TNF-a but also marked inhibition of LPS induced monocyte IL1B and IL12 production. LPS induced IL6 is also inhibited by immunomodulatory compounds, albeit partially. These compounds are potent stimulators of LPS induced IL10. Id. Particular examples of IMiD® compounds include, but are not limited to, the substituted 2-(2,6-dioxopiperidin-3-yl) phthalimides and substituted 2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindoles described in United States Patent Nos. 6,281,230 and 6,316,471, both to G.W. Muller, et al.
[00158] In a specific embodiment, the IMiD® compounds that can be used with the methods and compositions of the present invention is Lenalidomide (Revlimid®; 3-(4-amino- l-oxo-l,3-dihydro-isoindol-2-yl)-piperidine-2,6-dione). In another embodiment, the IMiD® compound is pomalidomide (4-(amino)-2-(2,6-dioxo(3-piperidyl))-isoindoline-l,3-dione). In
another embodiment, the IMiD® compound is 3-(5-amino-2-methyl-4-oxo-4H-quinazolin-3- yl)-piperidine-2,6-dione.
[00159] Compounds used with the present invention include immunomodulatory compounds that are racemic, stereomerically enriched or stereomerically pure, and pharmaceutically acceptable salts, solvates, hydrates, stereoisomers, clathrates, and prodrugs thereof. Preferred compounds used with the methods and compositions of the invention are small organic molecules having a molecular weight less than about 1 ,000 g/mol, and are not proteins, peptides, oligonucleotides, oligosaccharides or other macromolecules. As used herein and unless otherwise indicated, the terms "immunomodulatory compounds" and "IMiDs®" compounds (Celgene Corporation) encompasses small organic molecules that markedly inhibit TNF-a, LPS induced monocyte IL1B and IL12, and partially inhibit IL6 production. Specific immunomodulatory compounds are discussed below. TNF-a is an inflammatory cytokine produced by macrophages and monocytes during acute inflammation. TNF-a is responsible for a diverse range of signaling events within cells. TNF-a may play a pathological role in cancer. Without being limited by theory, one of the biological effects exerted by the immunomodulatory compounds is the reduction of synthesis of TNF-a.
Without being bound by theory, immunomodulatory compounds enhance the degradation of TNF-a mRNA.
[00160] Further, without being limited by theory, immunomodulatory compounds used in the invention may also be potent co-stimulators of T cells and increase cell proliferation dramatically in a dose dependent manner. Immunomodulatory compounds may also have a greater co-stimulatory effect on the CD8+ T cell subset than on the CD4+ T cell subset. In addition, the compounds preferably have anti-inflammatory properties, and efficiently co- stimulate T cells.
[00161] Specific examples of immunomodulatory compounds, include, but are not limited to, cyano and carboxy derivatives of substituted styrenes such as those disclosed in U.S. patent no. 5,929, 1 17; l-oxo-2-(2,6-dioxo-3-fiuoropiperidin-3yl) isoindolines and 1 ,3-dioxo-
2-(2,6-dioxo-3-fluoropiperidine-3-yl) isoindolines such as those described in U.S. patent no.
5,874,448; the tetra substituted 2-(2,6-dioxopiperdin-3-yl)-l-oxoisoindolines described in
U.S. patent no. 5,798,368; 1-oxo and l ,3-dioxo-2-(2,6-dioxopiperidin-3-yl) isoindolines (e.g.,
4-methyl derivatives of thalidomide and EM- 12), including, but not limited to, those disclosed in U.S. patent no. 5,635,517; and a class of non-polypeptide cyclic amides
disclosed in U.S. patent nos. 5,698,579 and 5,877,200; analogs and derivatives of thalidomide, including hydrolysis products, metabolites, derivatives and precursors of thalidomide, such as those described in U.S. patent nos. 5,593,990, 5,629,327, and 6,071,948 to D'Amato; aminothalidomide, as well as analogs, hydrolysis products, metabolites, derivatives and precursors of aminothalidomide, and substituted 2-(2,6-dioxopiperidin-3-yl) phthalimides and substituted 2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindoles such as those described in U.S. patent nos. 6,281,230 and 6,316,471; isoindole-imide compounds such as those described in U.S. patent application no. 09/972,487 filed on October 5, 2001, U.S. patent application no. 10/032,286 filed on December 21, 2001, and International Application No. PCT/USO 1/50401 (International Publication No. WO 02/059106). The entireties of each of the patents and patent applications identified herein are incorporated herein by reference.
[00162] Other specific immunomodulatory compounds include, but are not limited to, 1- oxo-and 1,3 dioxo-2-(2,6-dioxopiperidin-3-yl) isoindolines substituted with amino in the benzo ring as described in U.S. Patent no. 5,635,517 which is incorporated herein by reference. These compounds have the structure I:

in which one of X and Y is C=0, the other of X and Y is C=0 or CH2 , and R2 is hydrogen or lower alkyl, in particular methyl. Specific immunomodulatory compounds include, but are not limited to:
l-oxo-2-(2,6-dioxopiperidin-3-yl)-4-aminoisoindoline;
l-oxo-2-(2,6-dioxopiperidin-3-yl)-5-aminoisoindoline;
l-oxo-2-(2,6-dioxopiperidin-3-yl)-6-aminoisoindoline;
l-oxo-2-(2,6-dioxopiperidin-3-yl)-7-aminoisoindoline;
1 ,3-dioxo-2-(2,6-dioxopiperidin-3-yl)-4-aminoisoindoline; and
l,3-dioxo-2-(2,6-dioxopiperidin-3-yl)-5-aminoisoindoline.
[00163] Other specific immunomodulatory compounds belong to a class of substituted 2- (2,6-dioxopiperidin-3-yl) phthalimides and substituted 2-(2,6-dioxopiperidin-3-yl)-l- oxoisoindoles, such as those described in U.S. patent nos. 6,281,230; 6,316,471; 6,335,349; and 6,476,052, and International Patent Application No. PCT/US97/13375 (International
Publication No. WO 98/03502), each of which is incorporated herein by reference.
Compounds representative of this class are of the formulas:
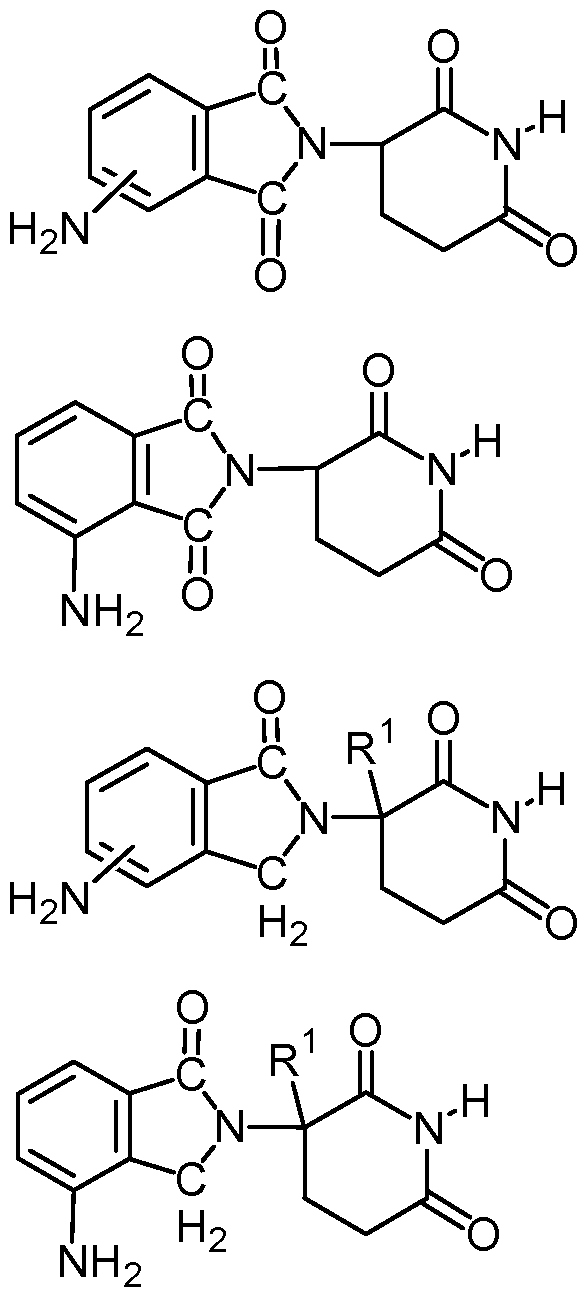
wherein R1 is hydrogen or methyl. In a separate embodiment, the enantiomerically pure forms (e.g. optically pure (R) or (S) enantiomers) of these compounds are used with the methods and formulations of the present invention.
[00164] Still other specific immunomodulatory compounds belong to a class of isoindole- imides disclosed in U.S. patent application nos. 10/032,286 and 09/972,487, and International Application No. PCT/USO 1/50401 (International Publication No. WO 02/059106), each of which are incorporated herein by reference. Representative compounds are of formula II:
R1 is H, (Ci-C8 )alkyl, (C3-C7)cycloalkyl, (C2-C8)alkenyl, (C2-C8)alkynyl, benzyl, aryl, (C0-C4)alkyl-(Ci-C6)heterocycloalkyl, (C0-C4)alkyl-(C2-C5)heteroaryl, C(0)R3 , C(S)R3, C(0)OR4, (Ci-C8)alkyl-N(R6)2, (C C8)alkyl-OR5, (C C8)alkyl-C(0)OR5, C(0)NHR3, C(S)NHR3, C(0)NR3R3', C(S)NR3R3' or (Ci-C8)alkyl-0(CO)R5;
R2 is H, F, benzyl, (Ci-C8)alkyl, (C2-C8)alkenyl, or (C2-C8)alkynyl;
R3 and R3' are independently (Ci-C8)alkyl, (C3-C7)cycloalkyl, (C2-C8)alkenyl, (C2- C8)alkynyl, benzyl, aryl, (C0-C4)alkyl-(Ci-C6)heterocycloalkyl, (C0-C4)alkyl-(C2- C5)heteroaryl, (C0-C8)alkyl-N(R6)2, (Ci-C8)alkyl-OR5, (Ci-C8)alkyl-C(0)OR5, (Ci-C8)alkyl- 0(CO)R5, or C(0)OR5;
R4 is (Ci-C8)alkyl, (C2-C8)alkenyl, (C2-C8)alkynyl, (Ci-C4)alkyl-OR5, benzyl, aryl, (C0-C4)alkyl-(Ci-C6)heterocycloalkyl, or (C0-C4)alkyl-(C2-C5)heteroaryl;
R5 is (Ci-C8)alkyl, (C2-C8)alkenyl, (C2-C8)alkynyl, benzyl, aryl, or (C2-C5)heteroaryl; each occurrence of R6 is independently H, (Ci-C8)alkyl, (C2-C8)alkenyl, (C2- C8)alkynyl, benzyl, aryl, (C2-Cs)heteroaryl, or (Co-C8)alkyl-C(0)0-R5 or the R6 groups can join to form a heterocycloalkyl group;
n is 0 or 1 ; and
* represents a chiral-carbon center.
[00165] In specific compounds of formula II, when n is 0 then R1 is (C3-C7)cycloalkyl, (C2-C8)alkenyl, (C2-Cg)alkynyl, benzyl, aryl, (Co-C4)alkyl-(Ci-C6)heterocycloalkyl, (Co- C4)alkyl-(C2-C5)heteroaryl, C(0)R3, C(0)OR4, (Ci-C8)alkyl-N(R6)2, (C C8)alkyl-OR5, (C C8)alkyl-C(0)OR5, C(S)NHR3, or (Ci-C8)alkyl-0(CO)R5;
R2 is H or (Ci-C8)alkyl; and
R3 is (Ci-C8)alkyl, (C3-C7)cycloalkyl, (C2-C8)alkenyl, (C2-C8)alkynyl, benzyl, aryl, (C0-C4)alkyl-(Ci -C6)heterocycloalkyl, (C0-C4)alkyl-(C2-C5)heteroaryl, (C5-C8)alkyl-N(R6)2 ; (C0-C8)alkyl-NH-C(O)O-R5; (Ci-C8)alkyl-OR5, (Ci-C8)alkyl-C(0)OR5, (Ci-C8)alkyl- 0(CO)R5, or C(0)OR5; and the other variables have the same definitions.
[00166] In other specific compounds of formula II, R2 is H or (Ci-C4)alkyl.
[00167] In other specific compounds of formula II, R1 is (Ci-C8)alkyl or benzyl.
[00168] In other specific compounds of formula II, R1 is H, (Ci-C8)alkyl, benzyl,
CH2OCH3, CH2CH2OCH3, or

[00169] In another embodiment of the compounds of formula II, R1 is
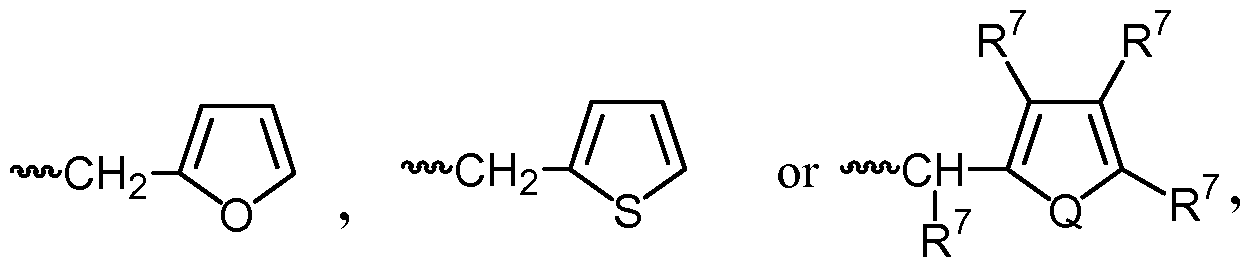
wherein Q is O or S, and each occurrence of R is independently H, (Ci-Cg)alkyl, benzyl, CH2OCH3, or CH2CH2OCH3.
[00170] In other specific compounds of formula II, R1 is C(0)R3.
[00171] In other specific compounds of formula II, R3 is (C0-C4)alkyl-(C2- C5)heteroaryl, (Cl-C8)alkyl, aryl, or (C0-C4)alkyl-OR5.
[00172] In other specific compounds of formula II, heteroaryl is pyridyl, furyl, or thienyl.
[00173] In other specific compounds of formula II, R1 is C(0)OR4.
[00174] In other specific compounds of formula II, the H of C(0)NHC(0) can be replaced with (Ci-C4)alkyl, aryl, or benzyl.
[00175] Still other specific immunomodulatory compounds belong to a class of isoindole- imides disclosed in U.S. patent application no. 09/781,179, International Publication No. WO 98/54170, and United States Patent No. 6,395,754, each of which are incorporated herein by reference. Representative compounds are of formula III:

and pharmaceutically acceptable salts, hydrates, solvates, clathrates, enantiomers, diastereomers, racemates, and mixtures of stereoisomers thereof, wherein:
one of X and Y is C=0 and the other is CH2 or C=0;
R is H or CH2OCOR';
(i) each of R1, R2, R3, or R4, independently of the others, is halo, alkyl of 1 to 4 carbon atoms, or alkoxy of 1 to 4 carbon atoms or (ii) one of R1, R2, R3, or R4 is nitro or -NHR5 and the remaining of R1, R2, R3, or R4 are hydrogen;
R5 is hydrogen or alkyl of 1 to 8 carbons
R6 hydrogen, alkyl of 1 to 8 carbon atoms, benzo, chloro, or fluoro;
R' is R7-CHR10-N(R8R9);
R7 is m-phenylene or p-phenylene or -(Cn H2n)- in which n has a value of 0 to 4;
each of R8 and R9 taken independently of the other is hydrogen or alkyl of 1 to 8 carbon atoms, or R8 and R9 taken together are tetramethylene, pentamethylene,
hexamethylene, or -CH2CH2[X]XiCH2CH2- in which [X]Xi is -0-, -S-, or -NH-;
R10 is hydrogen, alkyl of to 8 carbon atoms, or phenyl; and
* represents a chiral-carbon center.
[00176] Other immunomodulatory compounds are 4-(amino)-2-(2,6-dioxo(3-piperidyl))- isoindoline- 1 ,3-dione and 3-(4-amino- 1 -oxo- 1 ,3-dihydro-isoindol-2-yl)-piperidine-2,6-dione. The compounds can be obtained via standard, synthetic methods (see e.g. , United States Patent No. 5,635,517, incorporated herein by reference). The compounds are available from Celgene Corporation, Summit, NJ. 4-(Amino)-2-(2,6-dioxo(3-piperidyl))-isoindoline-l ,3- dione (pomalidomide or ACTIMID™) has the following chemical structure:
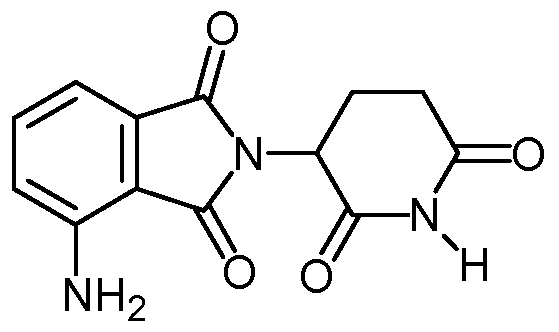
The compound 3-(4-amino-l-oxo-l ,3-dihydro-isoindol-2-yl)-piperidine-2,6-dione
(REVIMID™) has the following chemical structure:

[00177] Another compound provided herein is 3-(5-amino-2-methyl-4-oxo-4H-quinazolin- 3-yl)-piperidine-2,6-dione, which has the following structure:
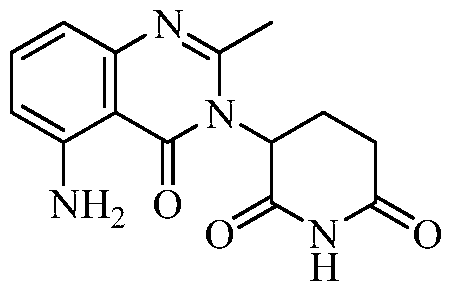
or an enantiomer or a mixture of enantiomers thereof; or a pharmaceutically acceptable
salt, solvate, hydrate, co-crystal, clathrate, or polymorph thereof.
[00178] 3-(5-Amino-2-methyl-4-oxo-4H-quinazolin-3-yl)-piperidine-2,6-dione can be prepared according to the methods described in the Examples provided herein or as described in U.S. Pat. No. 7,635,700, the disclosure of which is incorporated herein by reference in its entirety. The compound can be also synthesized according to other methods apparent to those of skill in the art based upon the teaching herein. In certain embodiments, 3-(5-amino-2- methyl-4-oxo-4H-quinazolin-3-yl)-piperidine-2,6-dione is in a crystalline form described in U.S. Provisional Pat. App. No. 61/451,806, filed March 11, 2011, which is incorporated herein by reference in its entirety. In some embodiments, the hydrochloride salt of the compound is used in the methods provided herein.
[00179] In some embodiments, the immunomodulatory compound can be, for example, a compound of formula IV, V, VI, VII, VIII, IX, X, XI, XII, XIII, XIV, XV, XVI, XVII, XVIII, XIX, XX, XXI, XXII, XXIII, XXIV, XXV, XXVI, XXVII, XXVIII, XXIX, XXX, and XXXI or a pharmaceutically acceptable salt, solvate, or stereoisomer thereof:
(IV)
wherein:
one of X and Y is C=0, the other of X and Y is C=0 or CH2;
hydrogen or lower alkyl;
(V)
wherein:
one of X and Y is C=0 and the other of X and Y is C=0 or CH2;
(i) each of R1, R2, R3, and R4, independently of the others, is halo, alkyl of 1 to 4 carbon atoms, or alkoxy of 1 to 4 carbon atoms, or (ii) one of R1, R2, R3, and R4 is -NHR5 and the remaining of R1, R2, R3, and R4 are hydrogen;
R5 is hydrogen or alkyl of 1 to 8 carbon atoms;
R6 is hydrogen, alkyl of 1 to 8 carbon atoms, benzyl, or halo;
provided that R6 is other than hydrogen if X and Y are C=0 and (i) each of R1, R2, R3, and R4 is fluoro or (ii) one of R1, R2, R3, or R4 is amino;

(VI)
wherein: one of X and Y is C=0 and the other is CH2 or C=0;
R1 is H, (Ci-Cs )alkyl, (C3-C7)cycloalkyl, (C2-C8)alkenyl, (C2-C8)alkynyl, benzyl, aryl, (C0-C4)alkyl-(Ci-C6)heterocycloalkyl, (C0-C4)alkyl-(C2-C5)heteroaryl, C(0)R3, C(S)R3, C(0)OR4, (Ci-C8)alkyl-N(R6)2, (Ci-C8)alkyl-OR5, (C C8)alkyl-C(0)OR5,
C(0)NHR3, C(S)NHR3, C(0)NR3R3', C(S)NR3R3' or (Ci-C8)alkyl-0(CO)R5;
R2 is H, F, benzyl, (Ci-C8)alkyl, (C2-C8)alkenyl, or (C2-C8)alkynyl;
R3 and R3' are independently (Ci-C8)alkyl, (C3-C7)cycloalkyl, (C2-C8)alkenyl, (C2-C8)alkynyl, benzyl, aryl, (C0-C4)alkyl-(Ci-C6)heterocycloalkyl, (C0-C4)alkyl-(C2- C5)heteroaryl, (C0-C8)alkyl-N(R6)2, (Ci-C8)alkyl-OR5, (Ci-C8)alkyl-C(0)OR5, (Ci-C8)alkyl- 0(CO)R5, or C(0)OR5;
R4 is (Ci-C8)alkyl, (C2-C8)alkenyl, (C2-C8)alkynyl, (Ci-C4)alkyl-OR5, benzyl, aryl, (C0-C4)alkyl-(Ci-C6)heterocycloalkyl, or (C0-C4)alkyl-(C2-C5)heteroaryl;
R5 is (Ci-C8)alkyl, (C2-C8)alkenyl, (C2-C8)alkynyl, benzyl, aryl, or (C2-
C5)heteroaryl;
R6 is independently H, (Ci-Cg)alkyl, (C2-Cg)alkenyl, (C2-Cg)alkynyl, benzyl, aryl, (C2-C5)heteroaryl, or (C0-C8)alkyl-C(O)O-R5 or the R6 groups can join to form a heterocycloalkyl group;
n is 0 or 1 ; and
* is a chiral-carbon center;

(VII)
wherein:
one of X and Y is C=0 and the other is CH2 or C=0;
R is H or CH2OCOR';
(i) each of R1, R2, R3, or R4, independently of the others, is halo, alkyl of 1 to 4 carbon atoms, or alkoxy of 1 to 4 carbon atoms or (ii) one of R1, R2, R3, or R4 is nitro or -NHR5 and the remaining of R1, R2, R3, or R4 are hydrogen;
R5 is hydrogen or alkyl of 1 to 8 carbons
R6 hydrogen, alkyl of 1 to 8 carbon atoms, benzo, chloro, or fluoro;
R' is R7-CHR10-N(R8R9);
R7 is m-phenylene or p-phenylene or -(CnH2n)- in which n has a value of 0 to each of R8 and R9 taken independently of the other is hydrogen or alkyl of 1 to
8 carbon atoms, or R8 and R9 taken together are tetramethylene, pentamethylene,
hexamethylene, or -CH2CH2X1CH2CH2- in which Xi is -0-, -S-, or -NH-;
R10 is hydrogen, alkyl of to 8 carbon atoms, or phenyl; and
* represents a chiral-carbon center;
(VIII)
wherein:
one of X and Y is C=0 and the other of X and Y is C=0 or CH2;
(i) each of R1, R2, R3, or R4, independently of the others, is halo, alkyl of 1 to 4 carbon atoms, or alkoxy of 1 to 4 carbon atoms or (ii) one of R1, R2, R3, and R4 is -NHR5 and the remaining of R1, R2, R3, and R4 are hydrogen;
R5 is hydrogen or alkyl of 1 to 8 carbon atoms;
R6 is hydrogen, alkyl of 1 to 8 carbon atoms, benzo, chloro, or fluoro;
R7 is m-phenylene or p-phenylene or -(CnH2n)- in which n has a value of 0 to
4;
each of R8 and R9 taken independently of the other is hydrogen or alkyl of 1 to 8 carbon atoms, or R8 and R9 taken together are tetramethylene, pentamethylene,
hexamethylene, or -CH2CH2 X1CH2CH2- in which X1 is -0-, -S-, or -NH-; and
R10 is hydrogen, alkyl of to 8 carbon atoms, or phenyl;
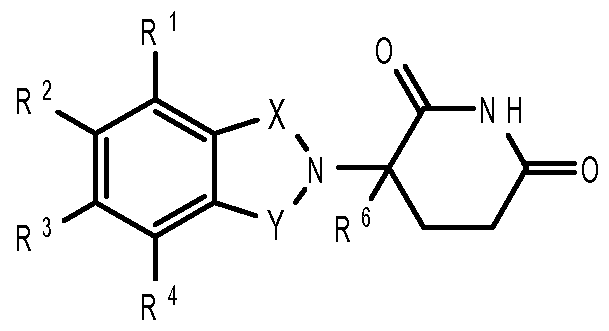
(IX)
wherein:
one of X and Y is C=0 and the other of X and Y is C=0 or CH2;
(i) each of R1, R2, R3, and R4, independently of the others, is halo, alkyl of 1 to 4 carbon atoms, or alkoxy of 1 to 4 carbon atoms or (ii) one of R1, R2, R3, and R4 is nitro or protected amino and the remaining
 R2, R3, and R4 are hydrogen; and
R2, R3, and R4 are hydrogen; and
R6 is hydrogen, alkyl of 1 to 8 carbon atoms, benzo, chloro, or fluoro;

(X)
wherein:
one of X and Y is C=0 and the other of X and Y is C=0 or CH2;
(i) each of R1, R2, R3, and R4, independently of the others, is halo, alkyl of 1 to 4 carbon atoms, or alkoxy of 1 to 4 carbon atoms or (ii) one of R1, R2, R3, and R4 is -NHR5 and the remaining of R1, R2, R3, and R4 are hydrogen;
R5 is hydrogen, alkyl of 1 to 8 carbon atoms, or CO-R7-CH(R10)NR8R9 in which each of R7, R8, R9, and R10 is as herein defined; and
R6 is alkyl of 1 to 8 carbon atoms, benzo, chloro, or fluoro;

(XI)
wherein:
one of X and Y is C=0 and the other of X and Y is C=0 or CH2; R6 is hydrogen, alkyl of 1 to 8 carbon atoms, benzyl, chloro, or fluoro;
R7 is m-phenylene, p-phenylene or -(CnH2n)- in which n has a value of 0 to 4; each of R8 and R9 taken independently of the other is hydrogen or alkyl of 1 to 8 carbon atoms, or R8 and R9 taken together are tetramethylene, pentamethylene,
hexamethylene, or -CH2CH2X1CH2CH2- in which X1 is -0-, -S- or -NH-; and
R is hydrogen, alkyl of 1 to 8 carbon atoms, or phenyl;

(XII)
wherein:
oxygen or H2 and each of R1, R2, R3, and R4, independently of the others, is hydrogen, halo, alkyl of 1 to 4 carbon atoms, alkoxy of 1 to 4 carbon atoms, or amino;
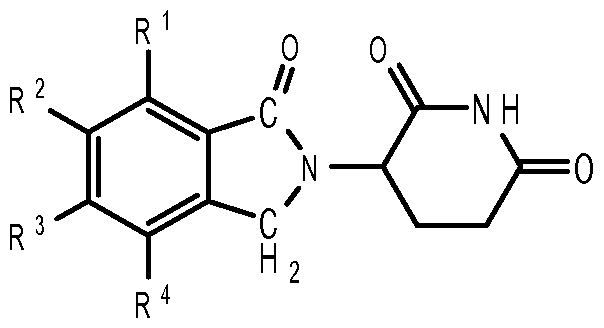
(XIII)
wherein:
each of R1, R2, R3, and R4, independently of the others, is halo, alkyl of 1 to 4 carbon atoms, or alkoxy of 1 to 4 carbon atoms;
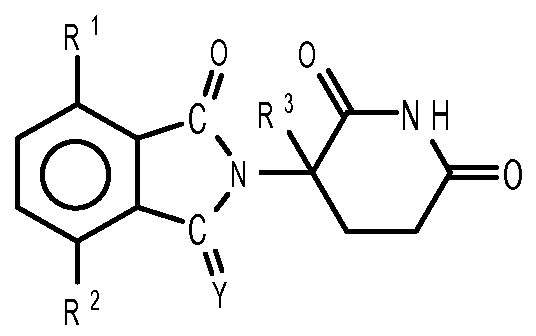
Y is oxygen or H2,
a first of R1 and R2 is halo, alkyl, alkoxy, alkylamino, dialkylamino, cyano, or carbamoyl, the second of R1 and R2, independently of the first, is hydrogen, halo, alkyl, alkoxy, alkylamino, dialkylamino, cyano, or carbamoyl, and
R3 is hydrogen, alkyl, or benzyl;
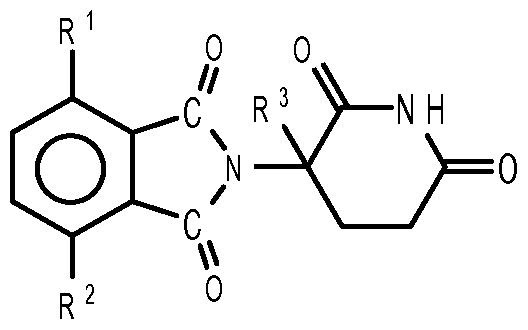
(XV)
wherein:
a first of R1 and R2 is halo, alkyl of from 1 to 4 carbon atoms, alkoxy of from 1 to 4 carbon atoms, dialkylamino in which each alkyl is of from 1 to 4 carbon atoms, cyano, or carbamoyl;
the second of R1 and R2, independently of the first, is hydrogen, halo, alkyl of from 1 to 4 carbon atoms, alkoxy of from 1 to 4 carbon atoms, alkylamino in which alkyl is of from 1 to 4 carbon atoms, dialkylamino in which each alkyl is of from 1 to 4 carbon atoms, cyano, or carbamoyl; and
R3 is hydrogen, alkyl of from 1 to 4 carbon atoms, or benzyl;

(XVI)
wherein:
when n is not zero and R1 is not the same as R2, C* is a center of chirality; one of X1 and X2 is amino, nitro, alkyl of one to six carbons, or NH-Z, and the other of X1 or X2 is hydrogen;
each of R1 and R2 independent of the other, is hydroxy or NH-Z; R3 is hydrogen, alkyl of one to six carbons, halo, or haloalkyl;
Z is hydrogen, aryl, alkyl of one to six carbons, formyl, or acyl of one to six carbons; and
n has a value of 0, 1 , or 2;
provided that if X1 is amino, and n is 1 or 2, then R1 and R2 are not both hydroxy;
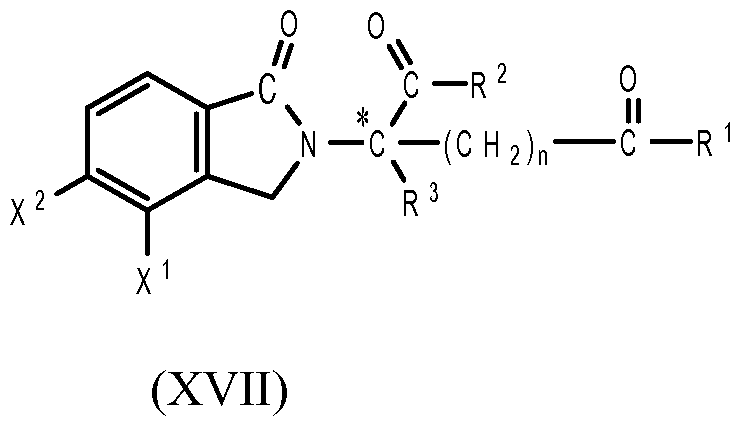
wherein:
when n is not zero and R1 is not R2, C* is a center of chirality; one of X1 and X2 is amino, nitro, alkyl of one to six carbons, or NH-Z, and the other of X1 or X2 is hydrogen;
each of R1 and R2 independent of the other, is hydroxy or NH-Z; R3 is alkyl of one to six carbons, halo, or hydrogen;
Z is hydrogen, aryl or an alkyl or acyl of one to six carbons; and n has a value of 0, 1 , or 2;
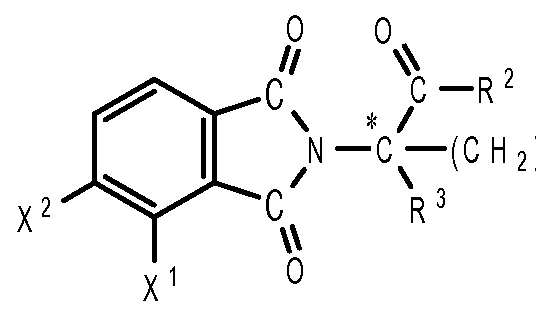
(XVIII)
wherein:
when n is not zero and R1 is not R2, C* is a center of chirality; one of X1 and X2 is amino, nitro, alkyl of one to six carbons, or NH-Z, and the other of X1 or X2 is hydrogen;
each of R1 and R2 independent of the other, is hydroxy or NH-Z; R3 is alkyl of one to six carbons, halo, or hydrogen;
Z is hydrogen, aryl, or an alkyl or acyl of one to six carbons; and n has a value of 0, 1 , or 2;
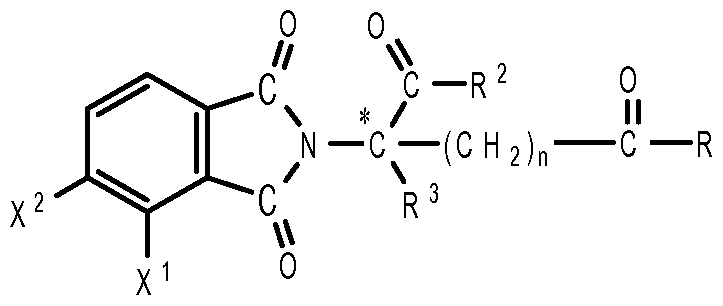
(XIX)
one of X1 and X2 is nitro, or NH-Z, and the other of X1 or X2 is hydrogen; each of R1 and R2, independent of the other, is hydroxy or NH-Z; R3 is alkyl of one to six carbons, halo, or hydrogen;
Z is hydrogen, phenyl, an acyl of one to six carbons, or an alkyl of one to six n has a value of 0, 1 , or 2; and
if -COR2 and -(CH2)„COR1 are different, C is a center of chirality;
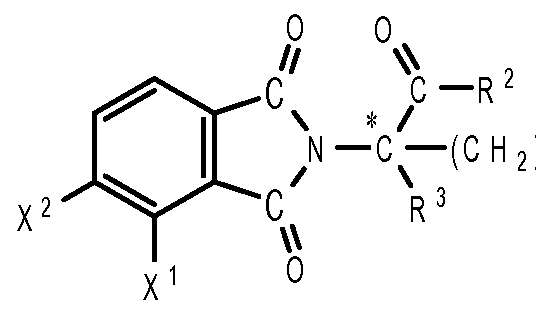
one of X1 and X2 is alkyl of one to six carbons;
each of R1 and R2, independent of the other, is hydroxy or NH-Z;
R3 is alkyl of one to six carbons, halo, or hydrogen;
Z is hydrogen, phenyl, an acyl of one to six carbons, or an alkyl of one to six n has a value of 0, 1 , or 2; and
if -COR2 and -(CH2)„COR1 are different, C is a center of chirality;
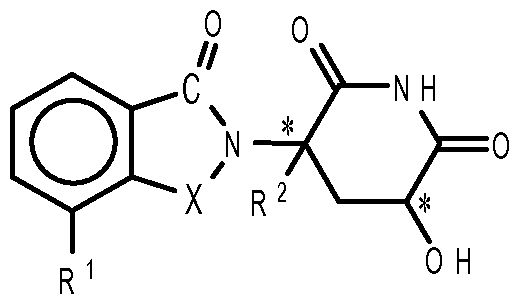
(XXI)
wherein:
the carbons are centers of chirality;
X is -C(O)- or -CH2-;
R1 is alkyl of 1 to 8 carbon atoms or -NHR3;
R2 is hydrogen, alkyl of 1 to 8 carbon atoms, or halogen; and
R3 is hydrogen, alkyl of 1 to 8 carbon atoms, unsubstituted or substituted with alkoxy of 1 to 8 carbon atoms, halo, amino, or alkylamino of 1 to 4 carbon atoms, cycloalkyl of 3 to 18 carbon atoms, phenyl, unsubstituted or substituted with alkyl of 1 to 8 carbon atoms, alkoxy of 1 to 8 carbon atoms, halo, amino, or alkylamino of 1 to 4 carbon atoms, benzyl, unsubstituted or substituted with alkyl of 1 to 8 carbon atoms, alkoxy of 1 to 8 carbon atoms, halo, amino, or alkylamino of 1 to 4 carbon atoms, or -COR4 , wherein
R4 is hydrogen, alkyl of 1 to 8 carbon atoms, unsubstituted or substituted with alkoxy of 1 to 8 carbon atoms, halo, amino, or alkylamino of 1 to 4 carbon atoms, cycloalkyl of 3 to 18 carbon atoms, phenyl, unsubstituted or substituted with alkyl of 1 to 8 carbon atoms, alkoxy of 1 to 8 carbon atoms, halo, amino, or alkylamino of 1 to 4 carbon atoms, or benzyl, unsubstituted or substituted with alkyl of 1 to 8 carbon atoms, alkoxy of 1 to 8 carbon atoms, halo, amino, or alkylamino of 1 to 4 carbon atoms. In an embodiment, the immunomodulatory compound can be, for example, l-oxo-2-(2,6-dioxopiperidin-3-yl)-4- aminoisoindoline or 1 ,3-dioxo-2-(2,6-dioxopiperidin-3-yl)-4-aminoisoindoline.

(XXII)
or a pharmaceutically acceptable salt, solvate or stereoisomer thereof, wherein:
X is C=0 or CH2;
R1 is -Y-R2;
Y is aryl, heteroaryl or heterocycle, optionally substituted with one or more halogen;
R2 is: -(CH2)n-heterocycle or -0-(CH2)n-heterocycle, wherein the heterocycle is optionally substituted with one or more (Ci-C6)alkyl, oxo, amino, hydroxyl, deuterium, or -COO-(Ci-C6)alkyl;
-(CH2)n-heteroaryl or -0-(CH2)n-heteroaryl, wherein the heteroaryl is optionally
substituted with one or more (Ci-C6)alkyl, oxo, amino, hydroxyl, deuterium, or -COO-(Ci-C6)alkyl; and
n is 0, 1, 2 or 3;
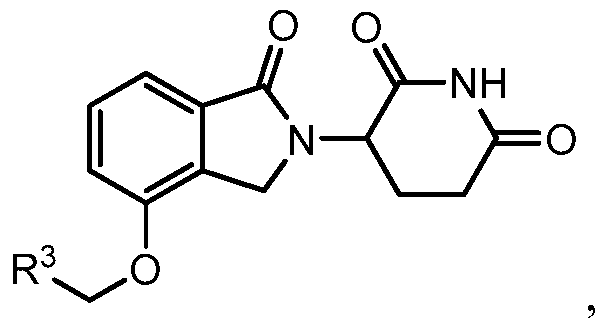
(XXIII)
or a pharmaceutically acceptable salt, solvate or stereoisomer thereof, wherein:
R3 is unsubstituted 9 to 10 membered bicyclic ring selected from the group consisting of benzothiazole, quinoline, isoquinoline, naphthalene, 2,3-dihydro-lH-indene, imidazo[l,2-a]pyridine, benzofuran, 2,3-dihydrobenzofuran, benzothiophene, benzo[d]oxazole isoindoline and chroman;
with the proviso that if the bicyclic ring is benzofuran or benzothiophene, then the ring is not connected to the isoindole ring through the 2-position;

(XXIV)
or a pharmaceutically acceptable salt, solvate or stereoisomer thereof, wherein:
R4, R5 and R6 are each independently hydrogen, halogen, nitro, carbamoyl, amino, -S02R7, -CONR8R9, -(Ci-C6)alkyl or -(Ci-Ce)alkoxy, said alkyl or alkoxy may be optionally substituted with one or more halogen, amino, hydroxyl, or NR8R9;
R7 is (Ci-C6)alkyl or amino optionally substituted with (Ci-Ce)alkyl;
R8 and R9 are each independently hydrogen, 6 to 10 membered aryl, -COO-(Ci-Ce)alkyl, -(Co-C6)alkyl-CHO, -(C0-C6)alkyl-NR8'R9', -(C0-C6)alkyl-(5 to 10 membered heterocycle), -(Ci-C6)alkyl-OH, -(Ci-C6)alkyl-0-(Ci-C6)alkyl, (Ci-C6)alkyl, or (C3-C6)cycloalkyl; or
R8 and R9 together may form a 5 to 6 membered ring; and
R8' and R9' are each independently hydrogen or (Ci-C6)alkyl;
with the proviso that all of R4-R6 cannot be hydrogen; and
with the proviso that if one of R4-R6 is hydrogen and the remaining two of R4-R6 are both chloride, then the two chloride atoms cannot be on 3 and 4 position of the phenyl ring;

(XXV)
or a pharmaceutically acceptable salt, solvate or stereoisomer thereof, wherein:
X is N or C; and
R1 and R2 are each independently hydrogen, (Ci-C6)alkyl, (Ci-C6)alkyl-(C
C6)cycloalkyl, (Ci-C6)alkoxy, -CO(Ci-C6)alkyl, -CO(Ci-C6)cycloalkyl, halogen, - C6)alkyl, phenyl or -S02(Ci-C6)alkyl;
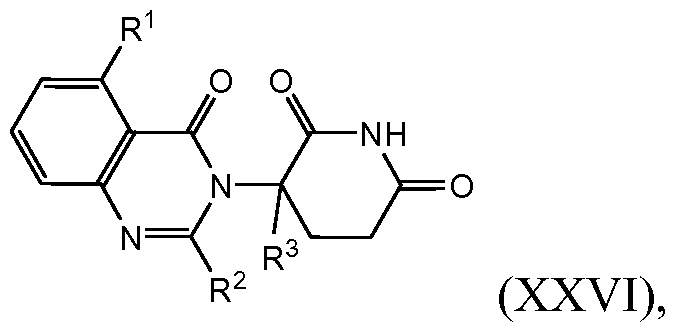
or a pharmaceutically acceptable salt, solvate, or stereoisomer thereof, wherein:
R1 is : hydrogen; halo; -(CH2)nOH; (Ci-C6)alkyl, optionally substituted with one
halo; (Ci- C6)alkoxy, optionally substituted with one or more halo; or
-(CH2)„NHRa, wherein Ra is:
hydrogen;
(Ci-C6)alkyl, optionally substituted with one or more halo;
-(CH2)n-(6 to 10 membered aryl);
-C(0)-(CH2)n-(6 to 10 membered aryl) or -C(0)-(CH2)n-(6 to 10 membered heteroaryl), wherein the aryl or heteroaryl is optionally substituted with one or more of: halo; -SCF3; (Ci-C6)alkyl, itself optionally substituted with one or more halo; or (Ci-C6)alkoxy, itself optionally substituted with one or more halo;
-C(0)-(Ci-Cg)alkyl, wherein the alkyl is optionally substituted with
halo;
-C(0)-(CH2)n-(C3-Cio-cycloalkyl);
-C(0)-(CH2)n-NRbRc, wherein Rb and Rc are each independently:
hydrogen;
(Ci-C6)alkyl, optionally substituted with one or more halo;
(Ci-C6)alkoxy, optionally substituted with one or more halo; or
6 to 10 membered aryl, optionally substituted with one or more of: halo; (Ci-C6)alkyl, itself optionally substituted with one or more halo; or (Ci-C6)alkoxy, itself optionally substituted with one or more halo;
-C(0)-(CH2)n-0-(Ci-C6)alkyl; or
-C(0)-(CH2)„-0-(CH2)„-(6 to 10 membered aryl);
R2 is: hydrogen; -(CH2)nOH; phenyl; -0-(Ci-C6)alkyl; or (Ci-C6)alkyl, optionally substituted with one or more halo;
R3 is: hydrogen; or (Ci-C6)alkyl, optionally substituted with one or more halo; and
is 0, 1, or
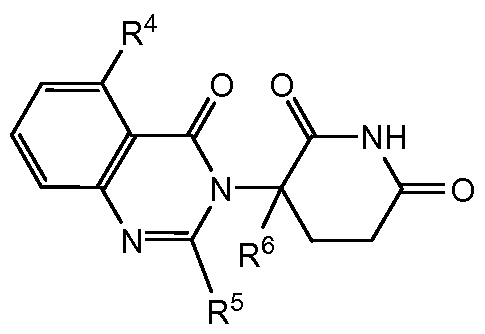
or a pharmaceutically acceptable salt, solvate, or stereoisomer thereof, wherein:
R4 is: hydrogen; halo; -(CH2)nOH; (Ci-C6)alkyl, optionally substituted with one or more halo; or (Ci-C6)alkoxy, optionally substituted with one or more halo;
R5 is: hydrogen; -(CH2)nOH; phenyl; -0-(Ci-C6)alkyl; or (Ci-C6)alkyl, optionally substituted with one or more halo;
R6 is: hydrogen; or (Ci-C6)alkyl, optionally substituted with one or more halo; and
n is 0, 1, or 2;
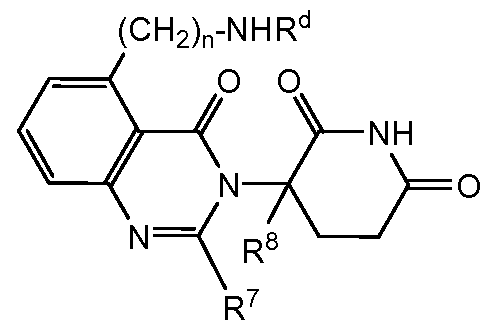
harmaceutically acceptable salt, solvate, or stereoisomer thereof, wherein:
Rd is:
hydrogen;
(Ci-C6)alkyl, optionally substituted with one or more
-C(0)-(Ci-Cg)alkyl, wherein the alkyl is optionally substituted with one or more halo;
-C(0)-(CH2)n-(C3-Cio-cycloalkyl);
-C(0)-(CH2)„-NReRf, wherein Re and Rf are each independently:
hydrogen;
(Ci-C6)alkyl, optionally substituted with one or more halo; or (Ci-C6)alkoxy, optionally substituted with one or more halo; or -C(0)-(CH2)n-0-(Ci-C6)alkyl.
R7 is: hydrogen; -(CH2)nOH; phenyl; -0-(Ci-C6)alkyl; or (Ci-C6)alkyl, optionally substituted with one or more halo;
R8 is: hydrogen; or (Ci-C6)alkyl, optionally substituted with one or more halo; and
n is 0, 1, or 2;
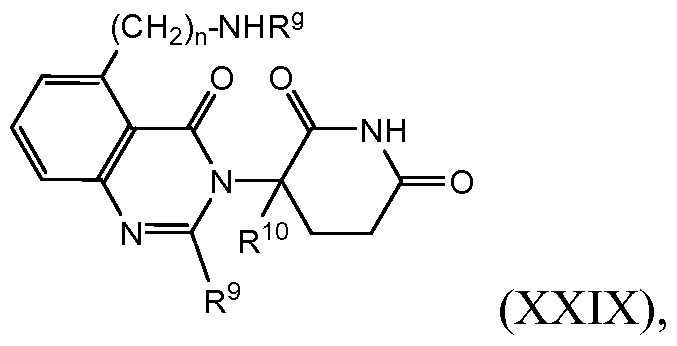
or a pharmaceutically acceptable salt, solvate, or stereoisomer thereof, wherein:
Rg is:
-(CH2)n-(6 to 10 membered aryl);
-C(0)-(CH2)„-(6 to 10 membered aryl) or -C(0)-(CH2)n-(6 to 10 membered
heteroaryl), wherein the aryl or heteroaryl is optionally substituted with one or more of: halo; -SCF3; (Ci-C6)alkyl, itself optionally substituted with one or more halo; or (Ci-C6)alkoxy, itself optionally substituted with one or more halo;
-C(0)-(CH2)n-NHRh, wherein Rh is:
6 to 10 membered aryl, optionally substituted with one or more of: halo;
(Ci-C6)alkyl, itself optionally substituted with one or more halo; or (Ci-C6)alkoxy, itself optionally substituted with one or more halo; or
-C(0)-(CH2)n-0-(CH2)n-(6 to 10 membered aryl);
R9 is: hydrogen; -(CH2)nOH; phenyl; -0-(Ci-C6)alkyl; or (Ci-C6)alkyl, optionally substituted with one or more halo;
R is: hydrogen; or (Ci-C6)alkyl, optionally substituted with one or more halo; and n is 0, 1, or 2;

or a pharmaceutically acceptable salt, solvate, prodrug, clathrate, or stereoisomer thereof, wherein:
Y is C=0 or CH2; and
R1 is hydrogen, alkyl, alkenyl, alkynyl, aryl, aralkyl, cycloalkyl, cycloalkylalkyl,
heterocyclyl, heterocyclylalkyl, heteroaryl, heteroarylalkyl, arylaminocarbonyl,
alkylcarbonyl, alkylammocarbonyl, dialkylaminocarbonyl, alkoxycarbonyl,
cycloalkylcarbonyl, heteroarylcarbonyl or heterocyclylcarbonyl;
and where R1 is optionally substituted with one or more groups selected from alkoxy, halo, alkyl, carboxy, alkylammocarbonyl, alkoxycarbonyl, nitro, amine, nitrile, haloalkyl, hydroxy, and alkylsulfonyl; and

wherein R5 is aryl or heteroaryl, optionally substituted with one, two or three groups seleted from alkyl, halo, alkoxy, carboxy, alkylammocarbonyl, alkoxycarbonyl, nitro, amine, nitrile, haloalkyl, hydroxy, and alkylsulfonyl; and ni is 0-5.
[00180] It should be noted that if there is a discrepancy between a depicted structure and a name given that structure, the depicted structure is to be accorded more weight. In addition, if the stereochemistry of a structure or a portion of a structure is not indicated with, for example, bold or dashed lines, the structure or portion of the structure is to be interpreted as encompassing all stereoisomers of it.
4.4 METHODS FOR TREATMENT AND PREVENTION OF DISEASE
[00181] Provided herein are methods for the treatment and/or prevention of disease, wherein the methods comprise (i) administering to a patient in need of treatment an inhibitor of ActRII (see Section 4.2) and administering to the patient in need of treatment a second active agent (see Section 4.3); (ii) administering to a patient in need of treatment an inhibitor of ActRIIA (see Section 4.2) and an inhibitor of ActRIIB (see Section 4.2); and/or (iii) administering to a patient in need of treatment an inhibitor of ActRIIA (see Section 4.2), an inhibitor of ActRIIB (see Section 4.2), and a second active agent (see Section 4.3).
[00182] In certain embodiments, the methods for the treatment and/or prevention of disease provided herein are sufficient to ameliorate one or more symptoms of the disease to be treated. In certain embodiments, the methods for the treatment and/or prevention of disease provided herein are sufficient to prevent one or more symptoms of the disease to be treated from worsening.
[00183] In certain embodiments, the methods for the treatment and/or prevention of disease provided herein comprise administering to a patient in need of treatment a therapeutically effective amount of an inhibitor of ActRII (see Section 4.2) and administering to the patient in need of treatment a therapeutically effective amount of a second active agent (see Section 4.3).
[00184] In certain embodiments, the methods for the treatment and/or prevention of disease provided herein comprise administering to a patient in need of treatment a therapeutically effective amount of an inhibitor of ActRIIA (see Section 4.2) and
administering to the patient in need of treatment a therapeutically effective amount of an inhibitor of ActRIIB (see Section 4.2).
[00185] In certain embodiments, the methods for the treatment and/or prevention of disease provided herein comprise administering to a patient in need of treatment a therapeutically effective amount of an inhibitor of ActRIIA (see Section 4.2), administering to the patient in need of treatment a therapeutically effective amount of an inhibitor of ActRIIB (see Section 4.2), and administering to the patient in need of treatment a
therapeutically effective amount of a second active agent (see Section 4.3).
[00186] Also encompassed herein is a method of increasing the dosage of a second active agent that can be safely and effectively administered to a patient, by combining the administration regimen of the second active agent with an administration regimen of an
ActRII inhibitor. In certain embodiments, the present invention provides a method for increasing the dose of Revlimid® that can be administered to a patient with a disease by at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 150%, 200%, 250%, 300%, 350%, 400%, 450%, 500%, 600%, 700%, 800%, 900%, or by at least 1000% without increasing the severity of any side effects or without causing any side effects.
[00187] Also encompassed herein is a method of increasing the dosage of a second active agent that can be safely and effectively administered to a patient, by combining the administration regimen of the second active agent with an administration regimen of an ActRII inhibitor. In certain embodiments, the present invention provides a method for increasing the dose of Revlimid® that can be administered to a patient with a disease by at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 150%, 200%, 250%, 300%, 350%, 400%, 450%, 500%, 600%, 700%, 800%, 900%, or by at least 1000% without increasing the severity of any side effects or without causing any side effects.
[00188] Also encompassed herein is a method of decreasing the dosage of a second active administered to a patient, so as to achieve efficacy of treatment of to reach a given endpoint, by combining the administration regimen of the second active agent with an administration regimen of an ActRII inhibitor. In certain embodiments, the present invention provides a method for decreasing the dose of Revlimid® administered to a patient with a disease by at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 150%, 200%, 250%, 300%, 350%, 400%, 450%, 500%, 600%, 700%, 800%, 900%, or by at least 1000%.
[00189] In certain embodiments, provided herein is a method for improving the safety of a treatment with lenalidomide (Revlimid®), wherein the method comprises administering an ActRII inhibitor to the patient being treated with lenalidomide (Revlimid®). In certain embodiments, safety is improved if the severity of any side effects of treatment with lenalidomide is reduced by at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%, 150%, 200%, 250%, 300%, 350%, 400%, 450%, 500%, 600%, 700%, 800%, 900%, or by at least 1000%.
[00190] In another embodiment, encompassed herein is a method of treating, preventing and/or managing a disease which comprises administering a second active agent and an ActRII inhibitor in combination with conventional therapy including, but not limited to,
surgery, immunotherapy, biological therapy, radiation therapy, or other non-drug based therapy presently used to treat, prevent or manage the disease.
[00191] In certain embodiments, a method of treatment described herein comprises administering to a patient an Activin-ActRII inhibitor and one or more second active agents, wherein the one or more second active agents include, without limitation, Revlimid®
(lenalidomide), pomalidomide, 3-(5-amino-2-methyl-4-oxo-4H-quinazolin-3-yl)-piperidine- 2,6-dione, melphalan, prednisone, dexamethasone, Velcade® (bortezomib), Dacogen® (decitabine), azacitidine, doxorubicin, vincristine, proteasome inhibitor, and bisphosphonate. In one embodiment, the Activin-ActRII is administered with one second active agent. In another embodiment, the Activin-ActRII is administered with two second active agents. In another embodiment, the Activin-ActRII is administered with three second active agents. In another embodiment, the Activin-ActRII is administered with more than three second active agents.
[00192] In a specific embodiment, a method of treatment described herein comprises administering to a patient an Activin-ActRII inhibitor and Revlimid® (lenalidomide). In another specific embodiment, a method of treatment described herein comprises
administering to a patient an Activin-ActRII inhibitor, Revlimid® (lenalidomide) and melphalan. In another specific embodiment, a method of treatment described herein comprises administering to a patient an Activin-ActRII inhibitor, Revlimid® (lenalidomide), and prednisone. In another specific embodiment, a method of treatment described herein comprises administering to a patient an Activin-ActRII inhibitor, Revlimid® (lenalidomide), melphalan and prednisone.
[00193] In a specific embodiment, a method of treatment described herein comprises administering to a patient an Activin-ActRII inhibitor and dexamethasone (e.g., a low dose regimen of dexamethasone or a high dose regimen of dexamethasone). In another specific embodiment, a method of treatment described herein comprises administering to a patient an Activin-ActRII inhibitor, Revlimid® (lenalidomide), and dexamethasone (e.g., a low dose regimen of dexamethasone or a high dose regimen of dexamethasone).
[00194] In a specific embodiment, a method of treatment described herein comprises administering to a patient an Activin-ActRII inhibitor and Velcade® (bortezomib). In another specific embodiment, a method of treatment described herein comprises administering to a patient an Activin-ActRII inhibitor, Revlimid® (lenalidomide), and Velcade® (bortezomib).
[00195] In a specific embodiment, a method of treatment described herein comprises administering to a patient an Activin-ActRII inhibitor and Dacogen® (decitabine). In another specific embodiment, a method of treatment described herein comprises administering to a patient an Activin-ActRII inhibitor, Revlimid® (lenalidomide), and Dacogen® (decitabine).
[00196] In a specific embodiment, a method of treatment described herein comprises administering to a patient an Activin-ActRII inhibitor and bisphosphonate. In another specific embodiment, a method of treatment described herein comprises administering to a patient an Activin-ActRII inhibitor, Revlimid® (lenalidomide), and bisphosphonate.
[00197] Without being limited by theory, it is believed that treatment of disease by administering a combination of ActRII inhibitors; or by administration of a combination of one or more ActRII inhibitors and a second active agent may provide additive or synergistic effects relative to the administration of an ActRII inhibitor alone or a second active agent alone. Such synergistic effects can be demonstrated, e.g., using the assays set forth in Section 4.6.
[00198] In accordance with the methods of treatment described herein which comprise administering to a patient an Activin-ActRII inhibitor and one or more second active agents, any dose of the second active agent(s) deemed suitable for administration or known in the art to treat the disease to be treated can be used. Dosage and administration regimes are described below.
(a) DISEASES
(i) Cancer
[00199] In a specific embodiment, provided herein are methods of treating cancer comprising (i) administering to a patient in need of treatment an inhibitor of ActRII (see Section 4.2) and administering to the patient in need of treatment a second active agent (see Section 4.3); (ii) administering to a patient in need of treatment an inhibitor of ActRIIA (see Section 4.2) and an inhibitor of ActRIIB (see Section 4.2); and/or (iii) administering to a patient in need of treatment an inhibitor of ActRIIA (see Section 4.2), an inhibitor of ActRIIB (see Section 4.2), and a second active agent (see Section 4.3). In a specific embodiment, the second active agent is lenalidomide (Revlimid®).
[00200] In a specific embodiment, the ActRII inhibitor is an ActRIIA inhibitor. In another specific embodiment, the ActRII inhibitor is an ActRIIA inhibitor and the second active agent is lenalidomide (Revlimid®). In another specific embodiment, the ActRII inhibitor is an
ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and the second active agent is lenalidomide (Revlimid®). In another specific embodiment, the ActRIl inhibitor is an ActRIIA inhibitor and the second active agent is pomalidomide. In another specific embodiment, the ActRIl inhibitor is an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and the second active agent is pomalidomide.
[00201] In a specific embodiment, the ActRIl inhibitor is an ActRIIB inhibitor. In another specific embodiment, the ActRIl inhibitor is an ActRIIB inhibitor and the second active agent is lenalidomide (Revlimid®). In another specific embodiment, the ActRIl inhibitor is an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:23 and the second active agent is lenalidomide (Revlimid®). In another specific embodiment, the ActRIl inhibitor is an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and the second active agent is
lenalidomide (Revlimid®). In another specific embodiment, the ActRIl inhibitor is an ActRIIB inhibitor and the second active agent is pomalidomide. In another specific embodiment, the ActRIl inhibitor is an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:23 and the second active agent is
pomalidomide. In another specific embodiment, the ActRIl inhibitor is an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and the second active agent is pomalidomide.
[00202] In a specific embodiment, provided herein is a method of treating cancer comprising administering to a patient in need of treatment an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
[00203] In a specific embodiment, provided herein is a method of treating cancer comprising administering to a patient in need of treatment an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
[00204] In a specific embodiment, provided herein are methods of treating cancer comprising administering to a patient in need of treatment an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and administering to the
patient in need of treatment an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25.
[00205] In a specific embodiment, provided herein are methods of treating cancer comprising administering to a patient in need of treatment an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:7, administering to the patient in need of treatment an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25, and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
[00206] In another embodiment, encompassed herein is a method of treating, preventing and/or managing cancer which comprises administering a second active agent and an ActRII inhibitor in combination with conventional therapy including, but not limited to, surgery, immunotherapy, biological therapy, radiation therapy, or other non-drug based therapy presently used to treat, prevent or manage cancer.
[00207] Cancers that can be treated in accordance with the methods described herein include, without limitation, leukemia (e.g., chronic lymphocytic leukemia, chronic myeloid leukemia, acute lymphocytic leukemia, and acute myeloid leukemia), lymphoma (e.g., Hodgkin's lymphoma and non-Hodgkin's lymphoma), myeloma (e.g., multiple myeloma), bone and connective tissue sarcomas, brain cancer, breast cancer, ovarian cancer, kidney cancer, pancreatic cancer, esophageal cancer, stomach cancer, lung cancer (e.g, small cell lung cancer (SCLC), non-small cell lung cancer (NSCLC), throat cancer, and mesothelioma), and prostate cancer.
(ii) Multiple Myeloma
[00208] In a specific embodiment, provided herein are methods of treating multiple myeloma (e.g., relapsed multiple myeloma, refractory multiple myeloma, and newly diagnosed multiple myeloma) comprising (i) administering to a patient in need of treatment an inhibitor of ActRII (see Section 4.2) and administering to the patient in need of treatment a second active agent (see Section 4.3); (ii) administering to a patient in need of treatment an inhibitor of ActRIIA (see Section 4.2) and an inhibitor of ActRIIB (see Section 4.2); and/or (iii) administering to a patient in need of treatment an inhibitor of ActRIIA (see Section 4.2), an inhibitor of ActRIIB (see Section 4.2), and a second active agent (see Section 4.3). In a specific embodiment, the second active agent is lenalidomide (Revlimid®). In another
embodiment, the second active agent is pomalidomide. In another embodiment, the second active agent is 3-(5-amino-2-methyl-4-oxo-4H-quinazolin-3-yl)-piperidine-2,6-dione.
[00209] In a specific embodiment, the ActRIl inhibitor is an ActRIIA inhibitor. In another specific embodiment, the ActRIl inhibitor is an ActRIIA inhibitor and the second active agent is lenalidomide (Revlimid®). In another specific embodiment, the ActRIl inhibitor is an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and the second active agent is lenalidomide (Revlimid®). In another specific embodiment, the ActRIl inhibitor is an ActRIIA inhibitor and the second active agent is pomalidomide. In another specific embodiment, the ActRIl inhibitor is an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and the second active agent is pomalidomide.
[00210] In a specific embodiment, the ActRIl inhibitor is an ActRIIB inhibitor. In another specific embodiment, the ActRIl inhibitor is an ActRIIB inhibitor and the second active agent is lenalidomide (Revlimid®). In another specific embodiment, the ActRIl inhibitor is an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:23 and the second active agent is lenalidomide (Revlimid®). In another specific embodiment, the ActRIl inhibitor is an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and the second active agent is
lenalidomide (Revlimid®). In another specific embodiment, the ActRIl inhibitor is an ActRIIB inhibitor and the second active agent is pomalidomide. In another specific embodiment, the ActRIl inhibitor is an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:23 and the second active agent is
pomalidomide. In another specific embodiment, the ActRIl inhibitor is an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and the second active agent is pomalidomide.
[00211] In a specific embodiment, provided herein is a method of treating multiple myeloma comprising administering to a patient in need of treatment an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
[00212] In a specific embodiment, provided herein is a method of treating multiple myeloma comprising administering to a patient in need of treatment an ActRIIB inhibitor
comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
[00213] In a specific embodiment, provided herein are methods of treating multiple myeloma comprising administering to a patient in need of treatment an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and administering to the patient in need of treatment an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25.
[00214] In a specific embodiment, provided herein are methods of treating multiple myeloma comprising administering to a patient in need of treatment an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:7,
administering to the patient in need of treatment an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25, and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
[00215] In certain embodiments, administration of one or more ActRII inhibitors or one or more ActRII inhibitors in combination with a second active agent (e.g., lenalidomide) results in at least 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 98%, 99%, or 100% reduction of malignant plasma cells present in the patient relative to the beginning of treatment or relative to an untreated patient.
[00216] In certain embodiments, the methods of treating multiple myeloma provided herein comprise administering to a patient in need of treatment a therapeutically effective amount of an inhibitor of ActRII (see Section 4.2) and administering to the patient in need of treatment a therapeutically effective amount of a second active agent (see Section 4.3).
[00217] In certain embodiments, a therapeutically effective amount of an ActRII inhibitor increases osteoblast differentiation or osteoblastogenesis in the patient by at least 5%, 10%>, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 98%, 99%, or 100% relative to the beginning of treatment or relative to an untreated patient.
[00218] In certain embodiments, a therapeutically effective amount of an ActRII inhibitor increases the red blood cell level and / or hemoglobin levels in the patient by at least 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%,
90%, 95%, 98%, 99%, or 100% relative to the beginning of treatment or relative to an untreated patient.
[00219] In certain embodiments, a therapeutically effective amount of an ActRII inhibitor decreases osteolytic lesions in the patient by at least 5%, 10%>, 15%, 20%>, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 98%, 99%, or 100% relative to the beginning of treatment or relative to an untreated patient.
[00220] In certain embodiments, a therapeutically effective amount of a second active agent results in at least 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 98%, 99%, or 100% reduction of malignant plasma cells present in the patient relative to the beginning of treatment or relative to an untreated patient.
[00221] In another embodiment, encompassed herein is a method of treating, preventing and/or managing multiple myeloma (e.g., relapsed multiple myeloma, refractory multiple myeloma, and newly diagnosed multiple myeloma) which comprises administering a second active agent and an ActRII inhibitor in combination with conventional therapy including, but not limited to, surgery, immunotherapy, biological therapy, radiation therapy, or other non- drug based therapy presently used to treat, prevent or manage multiple myeloma. In one embodiment, a second active agent can be administered in an amount of from about 0.1 to about 150 mg, and preferably from about 1 to about 25 mg, more preferably from about 2 to about 10 mg orally and daily, or about 5, 10, 15, 20, or about 25 mg in combination with an ActRII inhibitor, prior to, during, or after the use of conventional therapy.
(iii) Myelodysplastic Syndrome
[00222] In a specific embodiment, provided herein are methods of treating
myelodysplastic syndrome (MDS) comprising (i) administering to a patient in need of treatment an inhibitor of ActRII (see Section 4.2) and administering to the patient in need of treatment a second active agent (see Section 4.3); (ii) administering to a patient in need of treatment an inhibitor of ActRIIA (see Section 4.2) and an inhibitor of ActRIIB (see Section
4.2) ; and/or (iii) administering to a patient in need of treatment an inhibitor of ActRIIA (see Section 4.2), an inhibitor of ActRIIB (see Section 4.2), and a second active agent (see Section
4.3) . In a specific embodiment, the second active agent is lenalidomide (Revlimid®).
[00223] In a specific embodiment, the ActRIl inhibitor is an ActRIIA inhibitor. In another specific embodiment, the ActRIl inhibitor is an ActRIIA inhibitor and the second active agent is lenalidomide (Revlimid®). In another specific embodiment, the ActRIl inhibitor is an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and the second active agent is lenalidomide (Revlimid®). In another specific embodiment, the ActRIl inhibitor is an ActRIIA inhibitor and the second active agent is pomalidomide. In another specific embodiment, the ActRIl inhibitor is an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and the second active agent is pomalidomide.
[00224] In a specific embodiment, the ActRIl inhibitor is an ActRIIB inhibitor. In another specific embodiment, the ActRIl inhibitor is an ActRIIB inhibitor and the second active agent is lenalidomide (Revlimid®). In another specific embodiment, the ActRIl inhibitor is an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:23 and the second active agent is lenalidomide (Revlimid®). In another specific embodiment, the ActRIl inhibitor is an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and the second active agent is
lenalidomide (Revlimid®). In another specific embodiment, the ActRIl inhibitor is an ActRIIB inhibitor and the second active agent is pomalidomide. In another specific embodiment, the ActRIl inhibitor is an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:23 and the second active agent is
pomalidomide. In another specific embodiment, the ActRIl inhibitor is an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and the second active agent is pomalidomide.
[00225] In a specific embodiment, provided herein is a method of treating MDS comprising administering to a patient in need of treatment an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
[00226] In a specific embodiment, provided herein is a method of treating MDS comprising administering to a patient in need of treatment an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
[00227] In a specific embodiment, provided herein are methods of treating MDS comprising administering to a patient in need of treatment an ActRllA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and administering to the patient in need of treatment an ActRIlB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25.
[00228] In a specific embodiment, provided herein are methods of treating MDS comprising administering to a patient in need of treatment an ActRllA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:7, administering to the patient in need of treatment an ActRIlB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25, and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
[00229] In another embodiment, encompassed herein is a method of treating, preventing and/or managing MDS which comprises administering a second active agent and an ActRII inhibitor in combination with conventional therapy including, but not limited to, surgery, immunotherapy, biological therapy, radiation therapy, or other non-drug based therapy presently used to treat, prevent or manage MDS.
(iv) Anemia
[00230] In a specific embodiment, provided herein are methods of treating anemia comprising (i) administering to a patient in need of treatment an inhibitor of ActRII (see Section 4.2) and administering to the patient in need of treatment a second active agent (see Section 4.3); (ii) administering to a patient in need of treatment an inhibitor of ActRllA (see Section 4.2) and an inhibitor of ActRIlB (see Section 4.2); and/or (iii) administering to a patient in need of treatment an inhibitor of ActRllA (see Section 4.2), an inhibitor of ActRIlB (see Section 4.2), and a second active agent (see Section 4.3). In a specific embodiment, the second active agent is lenalidomide (Revlimid®). In another embodiment, the second active agent is pomalidomide.
[00231] In a specific embodiment, the ActRII inhibitor is an ActRllA inhibitor. In another specific embodiment, the ActRII inhibitor is an ActRllA inhibitor and the second active agent is lenalidomide (Revlimid®). In another specific embodiment, the ActRII inhibitor is an ActRllA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and the second active agent is lenalidomide (Revlimid®). In another specific
embodiment, the ActRIl inhibitor is an ActRIIA inhibitor and the second active agent is pomalidomide. In another specific embodiment, the ActRIl inhibitor is an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and the second active agent is pomalidomide.
[00232] In a specific embodiment, the ActRIl inhibitor is an ActRllB inhibitor. In another specific embodiment, the ActRIl inhibitor is an ActRllB inhibitor and the second active agent is lenalidomide (Revlimid®). In another specific embodiment, the ActRIl inhibitor is an ActRllB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:23 and the second active agent is lenalidomide (Revlimid®). In another specific embodiment, the ActRIl inhibitor is an ActRllB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and the second active agent is
lenalidomide (Revlimid®). In another specific embodiment, the ActRIl inhibitor is an ActRllB inhibitor and the second active agent is pomalidomide. In another specific embodiment, the ActRIl inhibitor is an ActRllB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:23 and the second active agent is
pomalidomide. In another specific embodiment, the ActRIl inhibitor is an ActRllB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and the second active agent is pomalidomide.
[00233] In a specific embodiment, provided herein is a method of treating anemia comprising administering to a patient in need of treatment an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
[00234] In a specific embodiment, provided herein is a method of treating anemia comprising administering to a patient in need of treatment an ActRllB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25 and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
[00235] In a specific embodiment, provided herein are methods of treating anemia comprising administering to a patient in need of treatment an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO: 7 and administering to the patient in need of treatment an ActRllB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25.
[00236] In a specific embodiment, provided herein are methods of treating anemia comprising administering to a patient in need of treatment an ActRIIA inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:7, administering to the patient in need of treatment an ActRIIB inhibitor comprising or consisting of the polypeptide sequence presented in SEQ ID NO:25, and administering to the patient in need of treatment lenalidomide (Revlimid®) or pomalidomide.
[00237] In another embodiment, encompassed herein is a method of treating, preventing and/or managing anemia which comprises administering a second active agent and an ActRII inhibitor in combination with conventional therapy presently used to treat, prevent or manage anemia.
[00238] In one embodiment, the anemia treated in accordance with the methods described herein is caused by, or is a symptom of, cancer (e.g., multiple myeloma). In another embodiment, the anemia treated in accordance with the methods described herein is caused by, or is a symptom of, MDS. In another embodiment, the anemia treated in accordance with the methods described herein is not caused by, or is not a symptom of, cancer (e.g., multiple myeloma) or MDS.
(b) DOSAGES AND ADMINISTRATION
[00239] The dose of an ActRII inhibitor described herein or a second active agent described herein that will be effective in the treatment or prevention of disease can be determined by standard clinical techniques. In vitro or in vivo assays may optionally be employed to help identify optimal dosage ranges. The precise dose to be employed will also depend, e.g., on the route of administration, the type of disease, and the seriousness of the disease, and should be decided according to the judgment of the practitioner and each patient's or subject's circumstances.
[00240] In some embodiments, the dosage of an ActRII inhibitor described herein or a second active agent described herein is determined by extrapolating from the "no observed adverse effective level" (NOAEL), as determined in animal studies. This extrapolated dosage is useful in determining the maximum recommended starting dose for human clinical trials. For instance, the NOAELs can be extrapolated to determine human equivalent dosages (HED). Typically, HED is extrapolated from a non-human animal dosage based on the doses that are normalized to body surface area (i.e., mg/m2). In specific embodiments, the
NOAELs are determined in mice, hamsters, rats, ferrets, guinea pigs, rabbits, dogs, primates, primates (monkeys, marmosets, squirrel monkeys, baboons), micropigs or minipigs. For a discussion on the use of NOAELs and their extrapolation to determine human equivalent doses, See Guidance for Industry Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers, U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER), Pharmacology and Toxicology, July 2005. In one embodiment, an ActRII inhibitor described herein or a second active agent described herein, or composition thereof, is administered at a dose that is lower than the human equivalent dosage (HED) of the NOAEL over a period of 1 week, 2 weeks, 3 weeks, 1 month, 2 months, three months, four months, six months, nine months, 1 year, 2 years, 3 years, 4 years or more.
[00241] In certain embodiments, a dosage regime for a human subject can be extrapolated from animal model studies using the dose at which 10% of the animals die (LDio). In general the starting dose of a Phase I clinical trial is based on preclinical testing. A standard measure of toxicity of a drug in preclinical testing is the percentage of animals that die because of treatment. It is well within the skill of the art to correlate the LDio in an animal study with the maximal-tolerated dose (MTD) in humans, adjusted for body surface a basis to extrapolate a starting human dose. In some embodiments, the interrelationship of dosages for one animal model can be converted for use in another animal, including humans, using conversion factors (based on milligrams per meter squared of body surface) as described, e.g., in Freireich et al. , Cancer Chemother. Rep., 1966, 50:219-244. Body surface area may be approximately determined from height and weight of the patient. See, e.g., Scientific Tables, Geigy Pharmaceuticals, Ardley, N. Y., 1970, 537. In certain embodiments, the adjustment for body surface area includes host factors such as, for example, surface area, weight, metabolism, tissue distribution, absorption rate, and excretion rate. In addition, the route of administration, excipient usage, and the specific disease to be treated are also factors to consider. In one embodiment, the standard conservative starting dose is about 1/10 the murine LDio, although it may be even lower if other species {i.e., dogs) were more sensitive to the ActRII inhibitor or second active. In other embodiments, the standard conservative starting dose is about 1/100, 1/95, 1/90, 1/85, 1/80, 1/75, 1/70, 1/65, 1/60, 1/55, 1/50, 1/45, 1/40, 1/35, 1/30, 1/25, 1/20, 1/15, 2/10, 3/10, 4/10, or 5/10 of the murine LDi0. In other embodiments, a starting dose amount of an ActRII inhibitor described herein or a second
active agent described herein in a human is lower than the dose extrapolated from animal model studies. In another embodiment, a starting dose amount of an ActRII inhibitor described herein or a second active agent described herein in a human is higher than the dose extrapolated from animal model studies. It is well within the skill of the art to start doses of the active composition at relatively low levels, and increase or decrease the dosage as necessary to achieve the desired effect with minimal toxicity.
(i) DOSE OF ACTRII INHIBITOR
[00242] In certain embodiments, the ActRII inhibitor is dosed at intervals and amounts sufficient to achieve serum concentrations of 0.2 microgram/kg or greater, and serum levels of 1 microgram/kg or 2 microgram/kg or greater are desirable for achieving significant effects on bone density and strength. Dosing regimens may be designed to reach serum concentrations of between 0.2 and 15 microgram/kg, and optionally between 1 and 5 microgram/kg. In humans, serum levels of 0.2 microgram kg may be achieved with a single dose of 0.1 mg/kg or greater and serum levels of 1 microgram/kg may be achieved with a single dose of 0.3 mg/kg or greater. The observed serum half-life of the molecule is between about 20 and 30 days, substantially longer than most Fc fusion proteins, and thus a sustained effective serum level may be achieved, for example, by dosing with 0.2-0.4 mg/kg on a weekly or biweekly basis, or higher doses may be used with longer intervals between dosings. For example, doses of 1-3 mg/kg might be used on a monthly or bimonthly basis, and the effect on bone may be sufficiently durable that dosing is necessary only once every 3, 4, 5, 6, 9, 12 or more months. Serum levels of the ActRII inhibitor can be measured by any means known to the skilled artisan. For example, antibodies against the ActRII inhibitor can be used to determine the serum levels of the ActRII inhibitor using, e.g., an ELISA.
[00243] In certain embodiments, the dose of the ActRII inhibitor ranges from 0.01 to 3.0 mg/kg intravenously or from 0.03 to 0.1 mg/kg subcutaneously. In certain embodiments, the dose of ActRII inhibitor is about 0.01 mg/kg, about 0.1 mg/kg, about 0.2 mg/kg, about 0.3 mg/kg, about 0.4 mg/kg, about 0.5 mg/kg, about 1.0 mg/kg, about 1.5 mg/kg, about 2.0 mg/kg, about 2.5 mg/kg, about 3.0 mg/kg, about 3.5 mg/kg, about 4.0 mg/kg, about 4.5 mg/kg, or about 5.0 mg/kg. In certain embodiments, the dose of of ActRII inhibitor is about 10.0 mg/kg, about 15.0 mg/kg, about 20.0 mg/kg, about 25.0 mg/kg, or about 30.0 mg/kg. In certain embodiments, the dose of ActRII inhibitor is between 0.01 mg/kg and 0.1 mg/kg, between 0.1 mg/kg and 0.3 mg/kg, between 0.3 mg/kg and 0.5 mg/kg, between 0.5 mg/kg and
1.0 mg/kg, between 1.0 mg/kg and 2.0 mg/kg, between 1.0 mg/kg and 3.0 mg/kg, between 2.0 mg/kg and 3.0 mg/kg, between 2.0 mg/kg and 4.0 mg/kg, between 3.0 mg/kg and 5.0 mg/kg, between 5.0 mg/kg and 10.0 mg/kg, between 10.0 mg/kg and 15.0 mg/kg, between 10.0 mg/kg and 20.0 mg/kg, between 15.0 mg/kg and 20.0 mg/kg, or between 20.0 mg/kg and 30.0 mg/kg. When used in conjunction with a dose provided herein (e.g., a dose of an ActRII inhibitor or a dose of a second active agent), the word "about" refers to any number within 1, 5 or 10% of the referenced number.
(ii) DOSE OF SECOND ACTIVE AGENT
[00244] Typical dosage forms comprise a second active agent, or a pharmaceutically acceptable salt, solvate, stereoisomer, or prodrug thereof, in an amount of from about 0.001 to about 150 mg. In particular, dosage forms comprise a second active agent, or a
pharmaceutically acceptable salt, solvate, stereoisomer, or prodrug thereof in an amount of about 0.001, 0.01, 0.1, 1, 2, 5, 7.5, 10, 12.5, 15, 17.5, 20, 25, 50, 100, 150 or 200 mg.
[00245] In one embodiment, provided herein are unit dosage formulations of a second active agent that comprise between about 1 mg and about 2000 mg, about 1 mg and 200 mg, about 35 mg and about 1400 mg, about 125 mg and about 1000 mg, about 250 mg and about 1000 mg, or about 500 mg and about 1000 mg of a second active agent.
[00246] In another embodiment, provided herein are unit dosage formulations of a second active agent that comprise 1 mg, 2.5 mg, 5 mg, 10 mg, 15 mg, 20 mg, 30 mg, 35 mg, 50 mg, 70 mg, 100 mg, 125 mg, 140 mg, 175 mg, 200 mg, 250 mg, 280 mg, 350 mg, 500 mg, 560 mg, 700 mg, 750 mg, 1000 mg or 1400 mg of a second active agent.
[00247] In certain embodiments, suitable dosage ranges of a second active agent for oral administration are about 0.001 milligram to about 500 milligrams of a second active agent, per kilogram body weight per day. In specific embodiments, the oral dose is about 0.01 milligram to about 100 milligrams per kilogram body weight per day, about 0.1 milligram to about 75 milligrams per kilogram body weight per day or about 0.5 milligram to 5 milligrams per kilogram body weight per day. The dosage amounts described herein refer to total amounts administered; that is, if more than one second active agent is administered, then, in some embodiments, the dosages correspond to the total amount administered. In a specific embodiment, oral compositions contain about 10% to about 95% of a second active agent by weight.
[00248] In certain embodiments, suitable dosage ranges for intravenous (i.v.) administration of a second active agent are about 0.01 milligram to about 100 milligrams per kilogram body weight per day, about 0.1 milligram to about 35 milligrams per kilogram body weight per day, and about 1 milligram to about 10 milligrams per kilogram body weight per day. In some embodiments, suitable dosage ranges for intranasal administration are about 0.01 pg/kg body weight per day to about 1 mg/kg body weight per day. Suppositories generally contain about 0.01 milligram to about 50 milligrams of a second active agent per kilogram body weight per day and comprise active ingredient in the range of about 0.5% to about 10% by weight.
[00249] In certain embodiments, suitable dosages for intradermal, intramuscular, intraperitoneal, subcutaneous, epidural, sublingual, intracerebral, intravaginal, transdermal administration or administration by inhalation of a second active agent are in the range of about 0.001 milligram to about 500 milligrams per kilogram of body weight per day.
[00250] In one embodiment, a second active agent can be administered orally and in single or divided daily doses in an amount of from about 0.10 to about 150 mg/day.
[00251] Exemplary doses of Revlimid® (lenalidomide) include, but are not limited to, 0.001, 0.01, 0.1, 1, 2, 5, 10, 25 or 50 mg. Exemplary dose ranges of Revlimid®
(lenalidomide) include, but are not limited to, 0.001 to 0.01 mg, 0.01 to 0.1 mg, 0.1 to 1 mg, 1 to 2 mg, 2 to 5 mg, 5 to 10 mg, 5 to 25 mg, or 5 to 50 mg. In certain embodiments, Revlimid® (lenalidomide) may be administered at a dose of 5 to 50 mg per day, or alternatively from about 10 to about 50 mg every other day. In a specific embodiment, 3 (4 amino 1 oxo 1,3 dihydro isoindol 2 yl piperidine 2,6 dione (Revlimid®) may be administered in an amount of from about 5 to 25 mg per day, or alternatively from about 10 to about 50 mg every other day. In another specific embodiment, 3 (4 amino 1 oxo 1,3 dihydro isoindol 2 yl) piperidine 2,6 dione (Revlimid®) may be administered initially in an amount of 5 mg/day and the dose can be escalated every week to 10, 20, 25, 30 and 50 mg/day. In another specific embodiment, Revlimid® may be orally administered in an amount of about 25 mg per day for 21 days followed by seven days rest in a 28 day cycle. Exemplary dose ranges of pomalidomide include, but are not limited to, 0.1 to 1 mg, 1 to 2 mg, 2 to 4, 2.5 to 5 mg, or 5 to 10 mg per day. In another specific embodiment, pomalidomide may be orally
administered in an amount of about 0.5 mg to 4 mg per day for 21 days followed by seven days rest in a 28 day cycle. In another specific embodiment, pomalidomide may be orally
administered in an amount of about 0.5 mg to 4 mg per day on days 1 through 28 in a 28 day cycle. Exemplary dose ranges of 3-(5-amino-2-methyl-4-oxo-4H-quinazolin-3-yl)- piperidine-2,6-dione include, but are not limited to, 0.001 to 0.01 mg, 0.01 to 0.1 mg, 0.1 to 1 mg, 1 to 2 mg, 2 to 5 mg, 5 to 10 mg, 5 to 25 mg, or 5 to 50 mg per day.
[00252] Exemplary doses of melphalan include, but are not limited to, 0.01 mg/kg, 0.05 mg/kg, 0.1 mg/kg, 0.15 mg/kg, 0.16 mg/kg, 0.17 mg/kg, 0.18 mg/kg, 0.19 mg/kg, 0.2 mg/kg, 0.25 mg/kg, 0.3 mg/kg, 0.35 mg/kg, 0.4 mg/kg, 0.45 mg/kg, or 0.5 mg/kg. Exemplary dose ranges of melphalan include, but are not limited to, 0.01 to 0.05 mg/kg, 0.05 to 0.1 mg/kg, 0.1 to 0.15 mg/kg, 0.1 to 0.2 mg/kg, 0.1 to 0.25 mg/kg, 0.2 to 0.3 mg/kg, 0.2 to 0.35 mg/kg, 0.3 to 0.4 mg/kg, 0.3 to 0.45 mg/kg, or 0.4 to 0.5 mg/kg. In certain embodiments, melphalan may be administered at a dose of 0.1 to 0.25 mg/kg per day.
[00253] Exemplary doses of prednisone include, but are not limited to, 0.1 mg/kg, 0.5 mg/kg, 1 mg/kg, 1.5 mg/kg, 2 mg/kg, 2.5 mg/kg, 3 mg/kg, 3.5 mg/kg, 4 mg/kg, 4.5 mg/kg, or 5 mg/kg. Exemplary dose ranges of prednisone include, but are not limited to, 0.1 to 1 mg/kg, 0.1 to 2 mg/kg, 1 to 2 mg/kg, 1 to 3 mg/kg, 2 to 3 mg/kg, 2 to 4 mg/kg, 2 to 5, or 3 to 5 mg/kg. In certain embodiments, prednisone may be administered at a dose of 1 to 3 mg/kg per day.
[00254] Exemplary doses of dexamethasone include, but are not limited to, 5 mg, 10 mg, 15 mg, 20 mg, 25 mg, 30 mg, 35 mg, 40 mg, 45 mg, 50 mg, 55 mg, 60 mg, 65 mg, or 70 mg. Exemplary dose ranges of dexamethasone include, but are not limited to, 5 to 20 mg, 10 to 30 mg, 20 to 40 mg, 30 to 50 mg, 40 to 60 mg, or 50 to 70 mg. In certain embodiments, dexamethasone may be administered at a dose of 40 mg on days 1 to 4, 9-12, and 17-20 of a 28-day treatment cycle (i.e., high-dose dexamethasone treatment). In other embodiments, dexamethasone may be administered at a dose of 40 mg on days 1, 8, 15, and 22 of a 28-day treatment cycle (i.e., low-dose dexamethasone treatment).
[00255] Exemplary doses of Velcade® (bortezomib) include, but are not limited to, 0.1 mg/m 2 (of body surface area), 0.5 mg/m 2 , 0.6 mg/m 2 , 0.7 mg/m 2 , 0.8 mg/m 2 , 0.9 mg/m 2 , 1 mg/m 2 , 1.1 mg/m 2 , 1.2 mg/m 2 , 1.3 mg/m 2 , 1.4 mg/m 2 , 1.5 mg/m 2 , 2 mg/m2 , 2.5 mg/m 2 , or 3 mg/m2. Exemplary dose ranges of Velcade® (bortezomib) include, but are not limited to, 0.1
2 2 2 2 2
to 0.5 mg/m , 0.5 to 1 mg/m , 1 to 1.5 mg/m , 1 to 2 mg/m , 1.5 to 2.5 mg/m , or 2 to 3 mg/m2. In certain embodiments, Velcade® (bortezomib) may be administered at a dose of 0.7 to 1.3 mg/m2 daily, every other day, every third day, or weekly.
[00256] Exemplary doses of Dacogen (decitabine) include, but are not limited to, 5 mg/m 2 (of body surface area), 10 mg/m 2 , 15 mg/m 2 , 20 mg/m 2 , or 25 mg/m 2. Exemplary dose ranges of Dacogen® (decitabine) include, but are not limited to, 5 to 10 mg/m2, 5 to 15 mg/m 2 , 10 to 15 mg/m 2 , 10 to 20 mg/m 2 , 15 to 20 mg/m 2 , or 15 to 25 mg/m 2. In certain embodiments, Dacogen® (decitabine) may be administered at a dose of 10 to 20 mg/m2 daily for five days.
[00257] Exemplary doses of bisphosphonate include, but are not limited to 1 mg, 5 mg, 10 mg, 15 mg, 20 mg, 25 mg, 30 mg, 40 mg, 50 mg, 60 mg, 70 mg, 80 mg, 90 mg, 100 mg, 110 mg, 120 mg, 130 mg, 140 mg, 150 mg, or 200 mg. Exemplary dose ranges of
bisphosphonate include, but are not limited to, 1 to 10 mg, 1 to 25 mg, 1 to 50 mg, 10 to 25 mg, 10 to 50 mg, 25 to 50 mg, 25 to 75 mg, 50 to 75 mg, 50 to 100 mg, 75 to 100 mg, 75 to 150 mg, 100 to 150 mg, 100 to 200 mg, or 150 to 200 mg.
(c) Administration of ActRII Inhibitors and Second Active Agents
[00258] In certain embodiments, (i) an ActRII inhibitor and a second active agent; (ii) an ActRIIA inhibitor and an ActRIIB inhibitor; or (iii) an ActRIIA inhibitor, an ActRIIB inhibitor, and a second active agent are administered simultanteously, e.g., as part of the same formulation.
[00259] In certain embodiments, (i) an ActRII inhibitor and a second active agent; (ii) an ActRIIA inhibitor and an ActRIIB inhibitor; or (iii) an ActRIIA inhibitor, an ActRIIB inhibitor, and a second active agent are administered less than 5 minutes apart, less than 30 minutes apart, 1 hour apart, at about 1 hour apart, at about 1 to about 2 hours apart, at about 2 hours to about 3 hours apart, at about 3 hours to about 4 hours apart, at about 4 hours to about 5 hours apart, at about 5 hours to about 6 hours apart, at about 6 hours to about 7 hours apart, at about 7 hours to about 8 hours apart, at about 8 hours to about 9 hours apart, at about 9 hours to about 10 hours apart, at about 10 hours to about 11 hours apart, at about 11 hours to about 12 hours apart, at about 12 hours to 18 hours apart, 18 hours to 24 hours apart, 24 hours to 36 hours apart, 36 hours to 48 hours apart, 48 hours to 52 hours apart, 52 hours to 60 hours apart, 60 hours to 72 hours apart, 72 hours to 84 hours apart, 84 hours to 96 hours apart, or 96 hours to 120 hours apart. In specific embodiments, (i) an ActRII inhibitor and a second active agent; (ii) an ActRIIA inhibitor and an ActRIIB inhibitor; or (iii) an ActRIIA inhibitor, an ActRIIB inhibitor, and a second active agent are administered within the same patent visit.
[00260] In specific embodiments, the interval of time between the administration of (i) an ActRII inhibitor and a second active agent; (ii) an ActRIIA inhibitor and an ActRIIB inhibitor; or (iii) an ActRIIA inhibitor, an ActRIIB inhibitor, and a second active agent and the administration of a second active agent may be about 1-5 minutes, 1-30 minutes, 30 minutes to 60 minutes, 1 hour, 1-2 hours, 2-6 hours, 2-12 hours, 12-24 hours, 1-2 days, 2 days, 3 days, 4 days, 5 days, 6 days, 7 days, 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 7 weeks, 8 weeks, 9 weeks, 10 weeks, 15 weeks, 20 weeks, 26 weeks, 52 weeks, 11- 15 weeks, 15-20 weeks, 20-30 weeks, 30-40 weeks, 40-50 weeks, 1 month, 2 months, 3 months, 4 months 5 months, 6 months, 7 months, 8 months, 9 months, 10 months, 11 months, 12 months, 1 year, 2 years, or any period of time in between. In certain embodiments, (i) an ActRII inhibitor and a second active agent; (ii) an ActRIIA inhibitor and an ActRIIB inhibitor; or (iii) an ActRIIA inhibitor, an ActRIIB inhibitor, and a second active agent are administered less than 1 day, 1 week, 2 weeks, 3 weeks, 4 weeks, one month, 2 months, 3 months, 6 months, 1 year, 2 years, or 5 years apart.
[00261] In certain embodiments, a dose of an ActRII inhibitor described herein or composition thereof or a second active agent described herein or composition thereof, is administered to a subject every day, every other day, every couple of days, every third day, once a week, twice a week, three times a week, or once every two weeks. In other embodiments, two, three or four doses of an ActRII inhibitor described herein or composition thereof or a second active agent described herein or composition thereof, is administered to a subject every day, every couple of days, every third day, once a week or once every two weeks. In some embodiments, a dose(s) of an ActRII inhibitor described herein or composition thereof or a second active agent described herein or composition thereof, is administered for 2 days, 3 days, 5 days, 7 days, 14 days, or 21 days. In certain embodiments, a dose an ActRII inhibitor described herein or composition thereof or a second active agent described herein or composition thereof, is administered for 1 month, 1.5 months, 2 months, 2.5 months, 3 months, 4 months, 5 months, 6 months or more.
[00262] The administration of an ActRII inhibitor described herein or composition thereof and the administration of a second active agent described herein or composition thereof can be performed concurrently, or on alternative schedules. For example, an ActRII inhibitor described herein or composition thereof may be administered on a specific day or at a specific time, and a second active agent described herein or composition thereof may be
administered on a different specific day or at a different specific time. In certain embodiments, the administration schedules of an ActRII inhibitor described herein or composition thereof and a second active agent described herein or composition thereof overlap. In certain embodiments, the administration schedules of an ActRII inhibitor described herein or composition thereof and a second active agent described herein or composition thereof do not overlap.
[00263] In some embodiments, the methods of treatment and prevention of disease provided herein involve administering (i) an ActRII inhibitor and a second active agent; (ii) an ActRIIA inhibitor and an ActRIIB inhibitor; or (iii) an ActRIIA inhibitor, an ActRIIB inhibitor, and a second active agent daily, three times a week, twice a week, or once a week; and administering a second active agent daily, three times a week, twice a week, once a week, once every 2 weeks, once every 3 weeks, once every 4 weeks, once every month, once every 2 months (e.g., approximately 8 weeks), once every 3 months (e.g., approximately 12 weeks), or once every 4 months (e.g., approximately 16 weeks). In certain embodiments, (i) an ActRII inhibitor and a second active agent; (ii) an ActRIIA inhibitor and an ActRIIB inhibitor; or (iii) an ActRIIA inhibitor, an ActRIIB inhibitor, and a second active agent are cyclically administered to a subject.
[00264] Cycling therapy involves the administration of an ActRII inhibitor or composition thereof (or an ActRIIA inhibitor and an ActRIIB inhibitor) for a period of time, followed by the administration of a second active agent for a period of time, and repeating this sequential administration. In certain embodiments, cycling therapy may also include a period of rest where the ActRII inhibitor or composition thereof (or an ActRIIA inhibitor and an ActRIIB inhibitor) or the second active agent is not administered for a period of time (e.g., 2 days, 3 days, 4 days, 5 days, 6 days, 7 days, 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 10 weeks, 20 weeks, 1 month, 2 months, 3 months, 4 months, 5 months, 6 months, 7 months, 8 months, 9 months, 10 months, 11 months, 12 months, 2 years, or 3 years). In one embodiment, the number of cycles administered is from 1 to 12 cycles, from 2 to 10 cycles, or from 2 to 8 cycles.
[00265] In some embodiments, the methods of treatment and prevention of disease provided herein comprise administering an ActRII inhibitor or composition thereof (or an ActRIIA inhibitor and an ActRIIB inhibitor) as a single agent for a period of time prior to administering the ActRII inhibitor or composition thereof (or an ActRIIA inhibitor and an
ActRllB inhibitor) in combination with a second active agent. In certain embodiments, the methods of treatment and prevention of disease provided herein comprise administering a second active agent alone for a period of time prior to administering the second active agent in combination with an ActRII inhibitor or composition thereof (or an ActRIlA inhibitor and an ActRllB inhibitor).
[00266] In some embodiments, the administration of (i) an ActRII inhibitor and a second active agent; (ii) an ActRIlA inhibitor and an ActRllB inhibitor; or (iii) an ActRIlA inhibitor, an ActRllB inhibitor, and a second active agent in accordance with the methods presented herein have an additive effect relative the administration of the ActRII inhibitor or composition thereof (or an ActRIlA inhibitor and an ActRllB inhibitor) or the second active agent alone. In some embodiments, the administration of (i) an ActRII inhibitor and a second active agent; (ii) an ActRIlA inhibitor and an ActRllB inhibitor; or (iii) an ActRIlA inhibitor, an ActRllB inhibitor, and a second active agent in accordance with the methods presented herein have a synergistic effect relative the administration of the ActRII inhibitor or composition thereof (or an ActRIlA inhibitor and an ActRllB inhibitor) or the second active agent alone.
[00267] The combination of an ActRII inhibitor (or an ActRIlA inhibitor and an ActRllB inhibitor) and a second active agent can be administered to a subject in the same
pharmaceutical composition. Alternatively, an ActRII inhibitor or composition thereof (or an ActRIlA inhibitor and an ActRllB inhibitor) and a second active agent can be administered concurrently to a subject in separate pharmaceutical compositions. An ActRII inhibitor or composition thereof (or an ActRIlA inhibitor and an ActRllB inhibitor) and a second active agent can be administered sequentially to a subject in separate pharmaceutical compositions. An ActRII inhibitor or composition thereof (or an ActRIlA inhibitor and an ActRllB inhibitor) and a second active agent may also be administered to a subject by the same or different routes of administration.
[00268] In one embodiment, a second active agent can be administered in an amount of from about 0.1 to about 150 mg, and preferably from about 1 to about 25 mg, more preferably from about 2 to about 10 mg orally and daily, or about 5, 10, 15, 20, or about 25 mg in combination with an ActRII inhibitor (or an ActRIlA inhibitor and an ActRllB inhibitor), prior to, during, or after the use of conventional therapy.
[00269] In one embodiment, a second active agent is administered daily in a single or divided doses in a four to six week cycle with a rest period of about a week or two weeks. The invention further allows the frequency, number, and length of dosing cycles to be increased. Thus, another specific embodiment encompasses the administration of a second active agent (in combination with an ActRII inhibitor) for more cycles than are typical when it is administered alone. In yet another specific embodiment, a second active agent is administered (in combination with an ActRII inhibitor) for a greater number of cycles that would typically cause dose-limiting toxicity in a patient to whom a second active ingredient is not also being administered.
[00270] In one embodiment, a second active agent is administered (in combination with an ActRII inhibitor) daily and continuously for three or four weeks at a dose of from about 0.1 to about 150 mg/d followed by a break of one or two weeks. In a particular embodiment, Revlimid® is administered (in combination with an ActRII inhibitor) in an amount of about 5, 10, or 25mg/day, preferably in an amount of about 25 mg/day for three to four weeks, followed by one week or two weeks of rest in a four or six week cycle.
[00271] In one embodiment, a second active agent is administered orally, with
administration of the second active agent occurring 30 to 60 minutes prior to or subseqent to administration of an ActRII inhibitor, during a cycle of four to six weeks. In another embodiment, the combination of an ActRII inhibitor and a second active ingredient is administered by intravenous infusion over about 90 minutes every cycle. In a specific embodiment, one cycle comprises the administration of from about 10 to about 25 mg/day of Revlimid® and from about 50 to about 200 mg/m2/day of an ActRII inhibitor daily for three to four weeks and then one or two weeks of rest.
[00272] The above-described administration schedules are provided for illustrative purposes only and should not be considered limiting. A person of ordinary skill in the art will readily understand that all doses are within the scope of the embodiments described herein.
4.5 PATIENT POPULATIONS
[00273] In certain embodiments, one or more ActRII inhibitors described herein (see Section 4.2) or one or more ActRII inhibitors described herein and a second active agent (e.g., lenalidomide (Revlimid®); see Section 4.3) or a composition described herein that comprises one or more ActRII inhibitors described herein or one or more ActRII inhibitors
described herein and a second active agent may be administered to a na'ive subject, i.e., a subject that does not have a disease. In one embodiment, an ActRII inhibitor described herein and a second active agent (e.g., lenalidomide (Revlimid®)) or a composition described herein that comprises an ActRII inhibitor described herein and a second active agent (e.g., lenalidomide (Revlimid®)) may be administered to a na'ive subject that is at risk of acquiring a disease.
[00274] In certain embodiments, one or more ActRII inhibitors described herein (see Section 4.2) or one or more ActRII inhibitors described herein and a second active agent (e.g., lenalidomide (Revlimid®); see Section 4.3) or a composition described herein that comprises one or more ActRII inhibitors described herein or one or more ActRII inhibitors described herein and a second active agent are administered to a patient who has been diagnosed with cancer, e.g., the patient has been diagnosed with leukemia (e.g., chronic lymphocytic leukemia, chronic myeloid leukemia, acute lymphocytic leukemia, and acute myeloid leukemia), lymphoma (e.g., Hodgkin's lymphoma and non-Hodgkin's lymphoma), myeloma (e.g., multiple myeloma), bone and connective tissue sarcomas, brain cancer, breast cancer, ovarian cancer, kidney cancer, pancreatic cancer, esophageal cancer, stomach cancer, lung cancer (e.g, small cell lung cancer (SCLC), non-small cell lung cancer (NSCLC), throat cancer, and mesothelioma), and/or prostate cancer.
[00275] In certain embodiments, one or more ActRII inhibitors described herein (see Section 4.2) or one or more ActRII inhibitors described herein and a second active agent (e.g., lenalidomide (Revlimid®); see Section 4.3) or a composition described herein that comprises one or more ActRII inhibitors described herein or one or more ActRII inhibitors described herein and a second active agent are administered to a patient who has been diagnosed with multiple myeloma {e.g., relapsed multiple myeloma, refractory multiple myeloma, and newly diagnosed multiple myeloma).
[00276] In certain embodiments, one or more ActRII inhibitors described herein (see Section 4.2) or one or more ActRII inhibitors described herein and a second active agent (e.g., lenalidomide (Revlimid®); see Section 4.3) or a composition described herein that comprises one or more ActRII inhibitors described herein or one or more ActRII inhibitors described herein and a second active agent are administered to a patient who has been diagnosed with myelodysplastic syndrome (MDS).
[00277] In certain embodiments, one or more ActRII inhibitors described herein (see Section 4.2) or one or more ActRII inhibitors described herein and a second active agent (e.g., lenalidomide (Revlimid®); see Section 4.3) or a composition described herein that comprises one or more ActRII inhibitors described herein or one or more ActRII inhibitors described herein and a second active agent are administered to a patient who has been diagnosed with anemia.
[00278] In some embodiments, one or more ActRII inhibitors described herein (see Section 4.2) or one or more ActRII inhibitors described herein and a second active agent (e.g., lenalidomide (Revlimid®); see Section 4.3) or a composition described herein that comprises one or more ActRII inhibitors described herein or one or more ActRII inhibitors described herein and a second active agent are administered to a patient with a disease (e.g., multiple myeloma, MDS, or anemia) before symptoms of the disease manifest or before symptoms of the disease become severe (e.g., before the patient requires hospitalization). In some embodiments, one or more ActRII inhibitors described herein (see Section 4.2) or one or more ActRII inhibitors described herein and a second active agent (e.g., lenalidomide (Revlimid®); see Section 4.3) or a composition described herein that comprises one or more ActRII inhibitors described herein or one or more ActRII inhibitors described herein and a second active agent are administered to a patient with a disease after symptoms of the disease manifest or after symptoms of the disease become severe (e.g., after the patient requires hospitalization).
[00279] In some embodiments, a subject to be administered one or more ActRII inhibitors described herein (see Section 4.2) or one or more ActRII inhibitors described herein and a second active agent (e.g., lenalidomide (Revlimid®); see Section 4.3) or a composition described herein that comprises one or more ActRII inhibitors described herein or one or more ActRII inhibitors described herein and a second active agent is an animal. In certain embodiments, the animal is a bird. In certain embodiments, the animal is a canine. In certain embodiments, the animal is a feline. In certain embodiments, the animal is a horse. In certain embodiments, the animal is a cow. In certain embodiments, the animal is a mammal, e.g., a horse, swine, mouse, or primate. In a specific embodiment, the animal is a human.
[00280] In certain embodiments, a subject to be administered one or more ActRII inhibitors described herein (see Section 4.2) or one or more ActRII inhibitors described herein and a second active agent (e.g., lenalidomide (Revlimid®); see Section 4.3) or a
composition described herein that comprises one or more ActRIl inhibitors described herein or one or more ActRIl inhibitors described herein and a second active agent is a human adult. In certain embodiments, a subject to be administered one or more ActRIl inhibitors described herein (see Section 4.2) or one or more ActRIl inhibitors described herein and a second active agent (e.g., lenalidomide (Revlimid®); see Section 4.3) or a composition described herein that comprises one or more ActRIl inhibitors described herein or one or more ActRIl inhibitors described herein and a second active agent is an elderly human subject.
[00281] In certain embodiments, a subject to be administered one or more ActRIl inhibitors described herein (see Section 4.2) or one or more ActRIl inhibitors described herein and a second active agent (e.g., lenalidomide (Revlimid®); see Section 4.3) or a composition described herein that comprises one or more ActRIl inhibitors described herein or one or more ActRIl inhibitors described herein and a second active agent is a human toddler. In certain embodiments, a subject to be administered one or more ActRIl inhibitors described herein (see Section 4.2) or one or more ActRIl inhibitors described herein and a second active agent (e.g., lenalidomide (Revlimid®); see Section 4.3) or a composition described herein that comprises one or more ActRIl inhibitors described herein or one or more ActRIl inhibitors described herein and a second active agent is a human child. In certain embodiments, a subject to be administered one or more ActRIl inhibitors described herein (see Section 4.2) or one or more ActRIl inhibitors described herein and a second active agent (e.g., lenalidomide (Revlimid®); see Section 4.3) or a composition described herein that comprises one or more ActRIl inhibitors described herein or one or more ActRIl inhibitors described herein and a second active agent is a human infant.
4.6 ASSAYS
[00282] The treatment regimens described herein can be tested in suitable in vitro and in vivo assays prior to use in humans. A number assays are known in the art for assessing the efficacy of treatment of disease, e.g., cancer, multiple myeloma (e.g., relapsed multiple myeloma, refractory multiple myeloma, and newly diagnosed multiple myeloma), and MDS. Non-limiting examples of such assays are provided below.
(a) In Vitro Assays for Testing the Efficacy of Multiple Myeloma Treatment
[00283] In vitro assays used for testing the efficacy of one or more multiple myeloma treatments described herein can utilize one or more multiple myeloma cell lines. Such
multiple myeloma cell lines are known in the art and include, without limitation, MM. IS, MM.1R, RPMI 8226 (RPMI), RPMI-Dox40 (Dox40), NCI-H929, KMS-11, OPM-2, and U266.
(b) Three-dimensional culture model
[00284] The efficacy of a multiple myeloma treatment regimen described herein can be tested using the three-dimensional culture model of reconstructed bone marrow described by Kirshner et al, 2008, Blood 112(7):2935-2945. This model reconstructs, in vitro, the human bone marrow environment, allowing for the clonal expansion of multiple myeloma cells within the bone marrow environment. Therapeutic effects can be assessed by determining the effect of a therapy on the multiple myeloma cells in the culture model.
(c) Cell Growth, Cell Apoptosis, and Cell Migration Assays
[00285] The efficacy of a multiple myeloma treatment regimen described herein can be tested using assays in which the treatment regimen is added to multiple myeloma cells grown in vitro and one or more of cell growth, cell apoptosis, and cell migration are assessed as compared to multiple myeloma cells that have not been exposed to the treatment regimen. Non-limiting examples of cell growth, cell apoptosis, and cell migration assays are described in Podar et al, 2007, Blood 109(4): 1669-1677.
(d) In Vivo Assays for Testing the Efficacy of Multiple Myeloma Treatment
[00286] The efficacy of a multiple myeloma treatment regimen described herein can be tested using in vivo animal model systems. Such animal model systems include, but are not limited to, rats, mice, chicken, cows, monkeys, pigs, dogs, rabbits, etc. Examples of animal models for multiple myeloma include, but are not limited to, xenograft mouse models (see, e.g., Podar et al, 2007, Blood 109(4): 1669-1677); the NOD/SCID human chimeric mouse model (see, e.g., Huang et al, 2004, American Journal of Pathology 164(2):747-756); and aging mice of the mouse strain C57BL/KaLwRij strain (see, e.g., Radl et al., 1988, American Journal of Pathology 132(3):593-597).
(e) Assaying Treatment of Multiple Myeloma in Humans
[00287] The efficacy of a multiple myeloma treatment regimen described herein can be tested using human subjects, i.e., human multiple myeloma patients administered a multiple myeloma treatment regimen described herein.
[00288] In some embodiments, the efficacy of a treatment regimen in a human multiple myeloma patient can be assessed using standard laboratory procedures used for diagnosis of multiple myeloma including, but not limited to biopsy, bone marrow aspiration, the serum FLC assay (see, e.g., Sthaneshwar et al, 2009, Clin. Chem. Lab. Med. 47(9): 1101-1107), and complete blood count. In accordance with such methods, the efficacy of treatment can be assessed by comparing the results of the assay used to the results of the same assay in the patient prior to treatment.
[00289] In other embodiments, the efficacy of a treatment regimen in a human multiple myeloma patient can be assessed using gross measurements including, without limitation, computed tomography (CT), magnetic resonance imaging (MRI), X-ray imaging, PET scan, and bone scans. The use of whole body MRI and PET scan for assessing the efficacy of multiple myeloma treatment has been described by Shortt et al., 2009, Am. J. Roentgenol. 192(4):980-986.
(f) Assessing Toxicity
[00290] The toxicity of the therapeutic regimens described herein can be determined using standard pharmaceutical procedures in cell cultures or experimental animals, e.g., for determining the LD50 (the dose lethal to 50% of the population) and the ED50 (the dose therapeutically effective in 50% of the population). The dose ratio between toxic and therapeutic effects is the therapeutic index and it can be expressed as the ratio LD50/ED50.
[00291] The data obtained from the cell culture assays and animal studies can be used in formulating a range of dosage of the therapies for use in humans. The dosage may vary within this range depending upon the dosage form employed and the route of administration utilized.
(g) Myelodysplastic Syndrome (MDS)
[00292] In vitro assays used for testing the efficacy of one or more myelodysplastic syndrome treatments described herein can utilize one or more myelodysplastic syndrome cell lines. Such myelodysplastic syndrome cell lines are known in the art and include, without limitation, MUTZ-1, MDS-L, PC-MDS, MDS92, M-TAT, and TER-3.
[00293] The efficacy of a myelodysplastic syndrome treatment regimen described herein can be tested using assays in which the treatment regimen is added to myelodysplastic
syndrome cells grown in vitro and one or more of cell growth, cell apoptosis, and cell migration are assessed as compared to myelodysplastic syndrome cells that have not been exposed to the treatment regimen.
(h) In Vivo Assays for Testing the Efficacy of Myelodysplastic Syndrome Treatment
[00294] The efficacy of a myelodysplastic syndrome treatment regimen described herein can be tested using in vivo animal model systems. Such animal model systems include, but are not limited to, rats, mice, chicken, cows, monkeys, pigs, dogs, rabbits, etc. Examples of animal models for myelodysplastic syndrome include, but are not limited to, those described in Beachy et al, 2010, Hematol. Oncol. Clin. North Am. 24(2):361-375.
(i) Assaying Treatment of Myelodysplastic Syndrome in Humans
[00295] The efficacy of a myelodysplastic syndrome treatment regimen described herein can be tested using human subjects, i.e., human myelodysplastic syndrome patients administered a myelodysplastic syndrome treatment regimen described herein.
[00296] In some embodiments, the efficacy of a treatment regimen in a human myelodysplastic syndrome patient can be assessed using standard laboratory procedures used for diagnosis of myelodysplastic syndrome including, but not limited to complete blood count, bone marrow aspiration/examination, cytogenetics (chromosomal studies) performed on bone marrow aspirates, complete blood count, and flow cytometry using cells obtained from bone marrow aspirates. In accordance with such methods, the efficacy of treatment can be assessed by comparing the results of the assay used to the results of the same assay in the patient prior to treatment. j) Bone Loss
[00297] Also provided herein are in vivo assays to measure bone or cartilage growth. For example, Namkung-Matthai et al., Bone, 28:80-86 (2001) discloses a rat osteoporotic model in which bone repair during the early period after fracture is studied. Kubo et al, Steroid Biochemistry & Molecular Biology, 68:197-202 (1999) also discloses a rat osteoporotic model in which bone repair during the late period after fracture is studied. Andersson et al., J. Endocrinol. 170:529-537 describe a mouse osteoporosis model in which mice are ovariectomized, which causes the mice to lose substantial bone mineral content and bone mineral density, with the trabecular bone losing roughly 50% of bone mineral density. Bone
density could be increased in the ovariectomized mice by administration of factors such as parathyroid hormone. In certain aspects, fracture healing assays that are known in the art can be used. These assays include fracture technique, histological analysis, and biomechanical analysis, which are described in, for example, U.S. Pat. No. 6,521,750, which is incorporated by reference in its entirety for its disclosure of experimental protocols for causing as well as measuring the extent of fractures, and the repair process.
(k) Anti-Cancer Studies
[00298] The efficacy of the methods of treatment described herein can be tested for biological activity using animal models for cancer. Such animal model systems include, but are not limited to, rats, mice, chicken, cows, monkeys, pigs, dogs, rabbits, etc. In a specific embodiment, the efficacy of the methods of treatment described herein are tested in a mouse model system. Such model systems are widely used and well-known to the skilled artisan such as the SCID mouse model or transgenic mice.
[00299] The efficacy of the methods of treatment described herein can be determined by administering an ActRII inhibitor described herein and a second active agent (e.g., lenalidomide (Revlimid®)) or a composition described herein that comprises an ActRII inhibitor described herein and a second active agent (e.g., lenalidomide (Revlimid®)) to an animal model and verifying that such administration is effective in reducing the severity of cancer in said animal model. Examples of animal models for cancer in general include, include, but are not limited to, spontaneously occurring tumors of companion animals (see, e.g., Vail & MacEwen, 2000, Cancer Invest 18(8):781-92). Examples of animal models for lung cancer include, but are not limited to, lung cancer animal models described by Zhang &
Roth (1994, In-vivo 8(5):755-69) and a transgenic mouse model with disrupted p53 function
(see, e.g. Morris et al, 1998, J La State Med Soc 150(4): 179- 85). An example of an animal model for breast cancer includes, but is not limited to, a transgenic mouse that over expresses cyclin Dl (see, e.g., Hosokawa et al, 2001, Transgenic Res 10(5):471-8). An example of an animal model for colon cancer includes, but is not limited to, a TCR b and p53 double knockout mouse (see, e.g., Kado et al, 2001, Cancer Res. 61(6):2395-8). Examples of animal models for pancreatic cancer include, but are not limited to, a metastatic model of
Panc02 murine pancreatic adenocarcinoma (see, e.g., Wang et al., 2001, Int. J. Pancreatol.
29(1):37- 46) and nu-nu mice generated in subcutaneous pancreatic tumors (see, e.g., Ghaneh et al., 2001, Gene Ther. 8(3): 199-208). Examples of animal models for non-Hodgkin's
lymphoma include, but are not limited to, a severe combined immunodeficiency ("SCID") mouse (see, e.g., Bryant et al, 2000, Lab Invest 80(4):553-73) and an IgHmu-HOXl 1 transgenic mouse (see, e.g., Hough et al, 1998, Proc. Natl. Acad. Sci. USA 95(23): 13853-8). An example of an animal model for esophageal cancer includes, but is not limited to, a mouse transgenic for the human papillomavirus type 16 E7 oncogene (see, e.g., Herber et al, 1996, J. Virol. 70(3): 1873-81). Examples of animal models for colorectal carcinomas include, but are not limited to, Ape mouse models (see, e.g., Fodde & Smits, 2001, Trends Mol Med 7(8):369 73 and Kuraguchi et al, 2000).
4.7 PHARMACEUTICAL COMPOSITIONS
[00300] In certain embodiments, the ActRII inhibitor(s) (see Section 4.2) and the second active agent(s) (see Section 4.3) are formulated separately. In other embodiments, the ActRII inhibitor(s) and the second active agent(s) are formulated together.
[00301] In certain embodiments, a therapeutic method provided herein includes administering the second active agent and / or the ActRII inhibitor systemically, or locally as an implant or device. When administered, the therapeutic composition for use in this invention is in a pyrogen-free, physiologically acceptable form.
[00302] In certain embodiments, provided herein is a pharmaceutical composition comprising more than one ActRII inhibitor (e.g., an inhibitor of ActRIIA (e.g., SEQ ID NO:7) and an inhibitor of ActRIIB (e.g., SEQ ID NO:25)). In certain embodiments, provided herein is a pharmaceutical composition comprising more than one ActRII inhibitor (e.g., an inhibitor of ActRIIA (e.g., SEQ ID NO:7) and an inhibitor of ActRIIB (e.g., SEQ ID NO:25)) and a second active agent (e.g., Revlimid®). In certain embodiments, provided herein is a pharmaceutical composition comprising an ActRII inhibitor and a second active agent. In certain embodiments, provided herein is a pharmaceutical composition comprising an ActRII inhibitor and a second active agent and a pharmaceutically acceptable carrier. In certain embodiments, provided herein is a pharmaceutical composition comprising an ActRII inhibitor comprising or consisting of a polypeptide comprising the amino acid sequence of SEQ ID NO:7 and Revlimid®. In certain embodiments, provided herein is a pharmaceutical composition comprising an ActRII inhibitor comprising or consisting of a polypeptide comprising the amino acid sequence of SEQ ID NO: 7 and Revlimid® and a pharmaceutically acceptable carrier. In certain embodiments, provided herein is a pharmaceutical composition
comprising an ActRII inhibitor comprising or consisting of a polypeptide comprising the amino acid sequence of SEQ ID NO:23 and Revlimid®. In certain embodiments, provided herein is a pharmaceutical composition comprising or consisting of a polypeptide comprising the ActRII inhibitor comprising an amino acid sequence of SEQ ID NO:23 and Revlimid® and a pharmaceutically acceptable carrier. In certain embodiments, provided herein is a pharmaceutical composition comprising an ActRII inhibitor comprising or consisting of a polypeptide comprising the amino acid sequence of SEQ ID NO:25 and Revlimid®. In certain embodiments, provided herein is a pharmaceutical composition comprising or consisting of a polypeptide comprising the ActRII inhibitor comprising an amino acid sequence of SEQ ID NO:25 and Revlimid® and a pharmaceutically acceptable carrier.
(a) ActRII Inhibitor
[00303] In certain embodiments, the ActRII inhibitor is administered parenterally.
Pharmaceutical compositions suitable for parenteral administration may comprise one or more ActRII polypeptides in combination with one or more pharmaceutically acceptable sterile isotonic aqueous or nonaqueous solutions, dispersions, suspensions or emulsions, or sterile powders which may be reconstituted into sterile injectable solutions or dispersions just prior to use, which may contain antioxidants, buffers, bacteriostats, solutes which render the formulation isotonic with the blood of the intended recipient or suspending or thickening agents. Examples of suitable aqueous and nonaqueous carriers which may be employed in the pharmaceutical compositions of the invention include water, ethanol, polyols (such as glycerol, propylene glycol, polyethylene glycol, and the like), and suitable mixtures thereof, vegetable oils, such as olive oil, and injectable organic esters, such as ethyl oleate. Proper fluidity can be maintained, for example, by the use of coating materials, such as lecithin, by the maintenance of the required particle size in the case of dispersions, and by the use of surfactants.
[00304] Further, the composition may be encapsulated or injected in a form for delivery to a target tissue site (e.g., bone). In certain embodiments, compositions of the present invention may include a matrix capable of delivering one or more therapeutic compounds
(e.g., ActRII polypeptides) to a target tissue site (e.g., bone), providing a structure for the developing tissue and optimally capable of being resorbed into the body. For example, the matrix may provide slow release of the ActRII polypeptides. Such matrices may be formed of materials presently in use for other implanted medical applications.
[00305] The choice of matrix material is based on biocompatibility, biodegradability, mechanical properties, cosmetic appearance and interface properties. The particular application of the subject compositions will define the appropriate formulation. Potential matrices for the compositions may be biodegradable and chemically defined calcium sulfate, tricalciumphosphate, hydroxyapatite, polylactic acid and polyanhydrides. Other potential materials are biodegradable and biologically well defined, such as bone or dermal collagen. Further matrices are comprised of pure proteins or extracellular matrix components. Other potential matrices are non-biodegradable and chemically defined, such as sintered
hydroxyapatite, bioglass, aluminates, or other ceramics. Matrices may be comprised of combinations of any of the above mentioned types of material, such as polylactic acid and hydroxyapatite or collagen and tricalciumphosphate. The bioceramics may be altered in composition, such as in calcium-aluminate -phosphate and processing to alter pore size, particle size, particle shape, and biodegradability.
[00306] In certain embodiments, administration of the ActRII inhibitor can be orally, e.g., in the form of capsules, cachets, pills, tablets, lozenges (using a flavored basis, usually sucrose and acacia or tragacanth), powders, granules, or as a solution or a suspension in an aqueous or non-aqueous liquid, or as an oil-in- water or water-in-oil liquid emulsion, or as an elixir or syrup, or as pastilles (using an inert base, such as gelatin and glycerin, or sucrose and acacia) and/or as mouth washes and the like, each containing a predetermined amount of an agent as an active ingredient. An agent may also be administered as a bolus, electuary or paste.
[00307] In solid dosage forms for oral administration (capsules, tablets, pills, dragees, powders, granules, and the like), one or more therapeutic compounds may be mixed with one or more pharmaceutically acceptable carriers, such as sodium citrate or dicalcium phosphate, and/or any of the following: (1) fillers or extenders, such as starches, lactose, sucrose, glucose, mannitol, and/or silicic acid; (2) binders, such as, for example,
carboxymethylcellulose, alginates, gelatin, polyvinyl pyrrolidone, sucrose, and/or acacia; (3) humectants, such as glycerol; (4) disintegrating agents, such as agar-agar, calcium carbonate, potato or tapioca starch, alginic acid, certain silicates, and sodium carbonate; (5) solution retarding agents, such as paraffin; (6) absorption accelerators, such as quaternary ammonium compounds; (7) wetting agents, such as, for example, cetyl alcohol and glycerol
monostearate; (8) absorbents, such as kaolin and bentonite clay; (9) lubricants, such as talc,
calcium stearate, magnesium stearate, solid polyethylene glycols, sodium lauryl sulfate, and mixtures thereof; and (10) coloring agents. In the case of capsules, tablets and pills, the pharmaceutical compositions may also comprise buffering agents. Solid compositions of a similar type may also be employed as fillers in soft and hard- filled gelatin capsules using such excipients as lactose or milk sugars, as well as high molecular weight polyethylene glycols and the like.
[00308] Liquid dosage forms for oral administration include pharmaceutically acceptable emulsions, microemulsions, solutions, suspensions, syrups, and elixirs. In addition to the active ingredient, the liquid dosage forms may contain inert diluents commonly used in the art, such as water or other solvents, solubilizing agents and emulsifiers, such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3-butylene glycol, oils (in particular, cottonseed, groundnut, corn, germ, olive, castor, and sesame oils), glycerol, tetrahydrofuryl alcohol, polyethylene glycols and fatty acid esters of sorbitan, and mixtures thereof. Besides inert diluents, the oral compositions can also include adjuvants such as wetting agents, emulsifying and suspending agents, sweetening, flavoring, coloring, perfuming, and preservative agents.
[00309] Suspensions, in addition to the active compounds, may contain suspending agents such as ethoxylated isostearyl alcohols, polyoxyethylene sorbitol, and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar and tragacanth, and mixtures thereof.
[00310] The compositions for use with the methods the invention may also contain adjuvants, such as preservatives, wetting agents, emulsifying agents and dispersing agents. Prevention of the action of microorganisms may be ensured by the inclusion of various antibacterial and antifungal agents, for example, paraben, chlorobutanol, phenol sorbic acid, and the like. It may also be desirable to include isotonic agents, such as sugars, sodium chloride, and the like into the compositions. In addition, prolonged absorption of the injectable pharmaceutical form may be brought about by the inclusion of agents which delay absorption, such as aluminum monostearate and gelatin.
[00311] It is understood that the dosage regimen will be determined by the attending physician considering various factors which modify the action of the ActRII inhibitor (see Section 4.2). The various factors include, but are not limited to, degree of red blood cell
formation, amount of bone weight desired to be formed, the degree of bone density loss, the site of bone damage, the condition of the damaged bone, the patient's age, sex, and diet, the severity of any disease that may be contributing to bone loss, time of administration, and other clinical factors. Optionally, the dosage may vary with the type of matrix used in the reconstitution and the types of compounds in the composition. Progress can be monitored by periodic assessment of bone growth and/or repair, for example, X-rays (including DEXA), histomorphometric determinations, and tetracycline labeling.
[00312] In certain embodiments,gene therapy for the in vivo production of ActRII polypeptides can be used with the methods and compositions of the present invention. Such therapy would achieve its therapeutic effect by introduction of the ActRII polynucleotide sequences into cells or tissues having the disorders as listed above. Delivery of ActRII polynucleotide sequences can be achieved using a recombinant expression vector such as a chimeric virus or a colloidal dispersion system. Preferred for therapeutic delivery of ActRII polynucleotide sequences is the use of targeted liposomes. The ActRII polypeptides can be ActRIIa and / or ActRIIb polypeptides (see Section 4.2).
[00313] Various viral vectors which can be utilized for gene therapy as taught herein include adenovirus, herpes virus, vaccinia, or, preferably, an RNA virus such as a retrovirus. Preferably, the retroviral vector is a derivative of a murine or avian retrovirus. Examples of retroviral vectors in which a single foreign gene can be inserted include, but are not limited to: Moloney murine leukemia virus (MoMuLV), Harvey murine sarcoma virus (HaMuSV), murine mammary tumor virus (MuMTV), and Rous Sarcoma Virus (RSV). A number of additional retroviral vectors can incorporate multiple genes. All of these vectors can transfer or incorporate a gene for a selectable marker so that transduced cells can be identified and generated. Retroviral vectors can be made target-specific by attaching, for example, a sugar, a glycolipid, or a protein. Preferred targeting is accomplished by using an antibody. Those of skill in the art will recognize that specific polynucleotide sequences can be inserted into the retroviral genome or attached to a viral envelope to allow target specific delivery of the retroviral vector containing the ActRIIa polynucleotide. In a preferred embodiment, the vector is targeted to bone or cartilage.
[00314] Alternatively, tissue culture cells can be directly transfected with plasmids encoding the retroviral structural genes gag, pol and env, by conventional calcium phosphate
transfection. These cells are then transfected with the vector plasmid containing the genes of interest. The resulting cells release the retroviral vector into the culture medium.
[00315] Another targeted delivery system for ActRIIa polynucleotides is a colloidal dispersion system. Colloidal dispersion systems include macromolecule complexes, nanocapsules, microspheres, beads, and lipid-based systems including oil-in-water emulsions, micelles, mixed micelles, and liposomes. A colloidal system is a liposome. Liposomes are artificial membrane vesicles which are useful as delivery vehicles in vitro and in vivo. RNA, DNA and intact virions can be encapsulated within the aqueous interior and be delivered to cells in a biologically active form (see e.g., Fraley, et al, Trends Biochem. Sci., 6:77, 1981). Methods for efficient gene transfer using a liposome vehicle, are known in the art, see e.g., Mannino, et al, Biotechniques, 6:682, 1988. The composition of the liposome is usually a combination of phospholipids, usually in combination with steroids, especially cholesterol. Other phospholipids or other lipids may also be used. The physical characteristics of liposomes depend on pH, ionic strength, and the presence of divalent cations.
[00316] Examples of lipids useful in liposome production include phosphatidyl compounds, such as phosphatidylglycerol, phosphatidylcholine, phosphatidylserine, phosphatidylethanolamine, sphingolipids, cerebrosides, and gangliosides. Illustrative phospholipids include egg phosphatidylcholine, dipalmitoylphosphatidylcholine, and distearoylphosphatidylcholine. The targeting of liposomes is also possible based on, for example, organ-specificity, cell-specificity, and organelle-specificity and is known in the art.
[00317] In certain embodiments, the ActRII inhibitor is substantially pure in a
pharmaceutical composition. Specifically, at most 20%, 10%, 5%, 2.5%, 1%, 0.1 %, or at most 0.05% of the compounds in the pharmaceutical composition are compounds other than the ActRII inhibitor and the pharmaceutical acceptable carrier.
(b) Second Active Agent
[00318] Pharmaceutical compositions and dosage forms to be used with the methods of the invention comprise an a second active agent, or a pharmaceutically acceptable salt, solvate, hydrate, stereoisomer, clathrate, or prodrug thereof. Pharmaceutical compositions and dosage forms can further comprise one or more excipients.
[00319] Single unit dosage forms are suitable for oral, mucosal (e.g., nasal, sublingual, vaginal, buccal, or rectal), parenteral (e.g., subcutaneous, intravenous, bolus injection,
intramuscular, or intraarterial), topical (e.g., eye drops or other ophthalmic preparations), transdermal or transcutaneous administration to a patient. Examples of dosage forms include, but are not limited to: tablets; caplets; capsules, such as soft elastic gelatin capsules; cachets; troches; lozenges; dispersions; suppositories; powders; aerosols (e.g., nasal sprays or inhalers); gels; liquid dosage forms suitable for oral or mucosal administration to a patient, including suspensions (e.g., aqueous or non-aqueous liquid suspensions, oil-in-water emulsions, or a water-in-oil liquid emulsions), solutions, and elixirs; liquid dosage forms suitable for parenteral administration to a patient; eye drops or other ophthalmic preparations suitable for topical administration; and sterile solids (e.g., crystalline or amorphous solids) that can be reconstituted to provide liquid dosage forms suitable for parenteral administration to a patient.
[00320] The composition, shape, and type of dosage forms will typically vary depending on their use. For example, a dosage form used in the acute treatment of a disease may contain larger amounts of one or more of the active ingredients it comprises than a dosage form used in the chronic treatment of the same disease. Similarly, a parenteral dosage form may contain smaller amounts of one or more of the active ingredients it comprises than an oral dosage form used to treat the same disease. These and other ways in which specific dosage forms will vary from one another will be readily apparent to those skilled in the art. See, e.g., Remington's Pharmaceutical Sciences, 18th ed., Mack Publishing, Easton PA (1990).
[00321] Typical pharmaceutical compositions and dosage forms comprise one or more excipients. Suitable excipients are well known to those skilled in the art of pharmacy, and non-limiting examples of suitable excipients are provided herein. Whether a particular excipient is suitable for incorporation into a pharmaceutical composition or dosage form depends on a variety of factors well known in the art including, but not limited to, the way in which the dosage form will be administered to a patient. For example, oral dosage forms such as tablets may contain excipients not suited for use in parenteral dosage forms. The suitability of a particular excipient may also depend on the specific active ingredients in the dosage form. For example, the decomposition of some active ingredients may be accelerated by some excipients such as lactose, or when exposed to water. Active ingredients that comprise primary or secondary amines are particularly susceptible to such accelerated decomposition. Consequently, pharmaceutical compositions and dosage forms can contain
little, if any, lactose other mono- or di-saccharides. As used herein, the term "lactose-free" means that the amount of lactose present, if any, is insufficient to substantially increase the degradation rate of an active ingredient.
[00322] Lactose-free compositions comprise excipients that are well known in the art and are listed, for example, in the U.S. Pharmacopeia (USP) 25 NF20 (2002). In general, lactose- free compositions comprise active ingredients, a binder/filler, and a lubricant in
pharmaceutically compatible and pharmaceutically acceptable amounts. Preferred lactose- free dosage forms comprise active ingredients, microcrystalline cellulose, pre-gelatinized starch, and magnesium stearate.
[00323] Anhydrous pharmaceutical compositions and dosage forms comprising a second active agent can also be used, since water can facilitate the degradation of some compounds. For example, the addition of water (e.g., 5%) is widely accepted in the pharmaceutical arts as a means of simulating long-term storage in order to determine characteristics such as shelf- life or the stability of formulations over time. See, e.g., Jens T. Carstensen, Drug Stability: Principles & Practice, 2d. Ed., Marcel Dekker, NY, NY, 1995, pp. 379-80. In effect, water and heat accelerate the decomposition of some compounds. Thus, the effect of water on a formulation can be of great significance since moisture and/or humidity are commonly encountered during manufacture, handling, packaging, storage, shipment, and use of formulations.
[00324] Anhydrous pharmaceutical compositions and dosage forms can be prepared using anhydrous or low moisture containing ingredients and low moisture or low humidity conditions. Pharmaceutical compositions and dosage forms that comprise lactose and at least one active ingredient that comprises a primary or secondary amine are preferably anhydrous if substantial contact with moisture and/or humidity during manufacturing, packaging, and/or storage is expected.
[00325] An anhydrous pharmaceutical composition should be prepared and stored such that its anhydrous nature is maintained. Accordingly, anhydrous compositions are preferably packaged using materials known to prevent exposure to water such that they can be included in suitable formulary kits. Examples of suitable packaging include, but are not limited to, hermetically sealed foils, plastics, unit dose containers (e.g., vials), blister packs, and strip packs.
[00326] Like the amounts and types of excipients, the amounts and specific types of active ingredients in a dosage form may differ depending on factors such as, but not limited to, the route by which it is to be administered to patients. However, typical dosage forms comprise an immunomodulatory compound or a pharmaceutically acceptable salt, solvate, hydrate, stereoisomer, clathrate, or prodrug thereof in an amount of from about 0.10 to about 150 mg. Typical dosage forms comprise an immunomodulatory compound or a pharmaceutically acceptable salt, solvate, hydrate, stereoisomer, clathrate, or prodrug thereof in an amount of about 0.1, 1, 2, 5, 7.5, 10, 12.5, 15, 17.5, 20, 25, 50, 100, 150 or 200 mg. In a particular embodiment, a preferred dosage form comprises 4-(amino)-2-(2,6-dioxo(3-piperidyl))- isoindoline-l,3-dione (ActimidTM) in an amount of about 1, 2, 5, 10, 25 or 50mg. In a specific embodiment, a preferred dosage form comprises 3 (4 amino 1 oxo 1,3 dihydro isoindol 2 yl)-piperidine 2,6 dione (RevimidTM) in an amount of about 5, 10, 25 or 50mg. Typical dosage forms comprise the second active ingredient in an amount of 1 to about 1000 mg, from about 5 to about 500 mg, from about 10 to about 350 mg, or from about 50 to about 200 mg. Of course, the specific amount of the anti-cancer drug will depend on the specific agent used, the type of cancer being treated or managed, and the amount(s) of an
immunomodulatory compound and any optional additional active agents concurrently administered to the patient.
[00327] Pharmaceutical compositions that are suitable for oral administration can be presented as discrete dosage forms, such as, but are not limited to, tablets (e.g., chewable tablets), caplets, capsules, and liquids (e.g., flavored syrups). Such dosage forms contain predetermined amounts of active ingredients, and may be prepared by methods of pharmacy well known to those skilled in the art. See generally, Remington's Pharmaceutical Sciences, 18th ed., Mack Publishing, Easton PA (1990).
[00328] Typical oral dosage forms are prepared by combining the active ingredients in an intimate admixture with at least one excipient according to conventional pharmaceutical compounding techniques. Excipients can take a wide variety of forms depending on the form of preparation desired for administration. For example, excipients suitable for use in oral liquid or aerosol dosage forms include, but are not limited to, water, glycols, oils, alcohols, flavoring agents, preservatives, and coloring agents. Examples of excipients suitable for use in solid oral dosage forms (e.g., powders, tablets, capsules, and caplets) include, but are not
limited to, starches, sugars, micro-crystalline cellulose, diluents, granulating agents, lubricants, binders, and disintegrating agents.
[00329] Because of their ease of administration, tablets and capsules represent the most advantageous oral dosage unit forms, in which case solid excipients are employed. If desired, tablets can be coated by standard aqueous or nonaqueous techniques. Such dosage forms can be prepared by any of the methods of pharmacy. In general, pharmaceutical compositions and dosage forms are prepared by uniformly and intimately admixing the active ingredients with liquid carriers, finely divided solid carriers, or both, and then shaping the product into the desired presentation if necessary.
[00330] For example, a tablet can be prepared by compression or molding. Compressed tablets can be prepared by compressing in a suitable machine the active ingredients in a free- flowing form such as powder or granules, optionally mixed with an excipient. Molded tablets can be made by molding in a suitable machine a mixture of the powdered compound moistened with an inert liquid diluent.
[00331] Examples of excipients that can be used in oral dosage forms include, but are not limited to, binders, fillers, disintegrants, and lubricants. Binders suitable for use in pharmaceutical compositions and dosage forms include, but are not limited to, corn starch, potato starch, or other starches, gelatin, natural and synthetic gums such as acacia, sodium alginate, alginic acid, other alginates, powdered tragacanth, guar gum, cellulose and its derivatives (e.g., ethyl cellulose, cellulose acetate, carboxymethyl cellulose calcium, sodium carboxymethyl cellulose), polyvinyl pyrrolidone, methyl cellulose, pre-gelatinized starch, hydroxypropyl methyl cellulose, (e.g., Nos. 2208, 2906, 2910), microcrystalline cellulose, and mixtures thereof.
[00332] Suitable forms of microcrystalline cellulose include, but are not limited to, the materials sold as AVICEL-PH-101, AVICEL-PH-103 AVICEL RC-581, AVICEL-PH-105 (available from FMC Corporation, American Viscose Division, Avicel Sales, Marcus Hook, PA), and mixtures thereof. A specific binder is a mixture of microcrystalline cellulose and sodium carboxymethyl cellulose sold as AVICEL RC-581. Suitable anhydrous or low moisture excipients or additives include AVICEL-PH-103™ and Starch 1500 LM.
[00333] Examples of fillers suitable for use in the pharmaceutical compositions and dosage forms disclosed herein include, but are not limited to, talc, calcium carbonate (e.g.,
granules or powder), microcrystalline cellulose, powdered cellulose, dextrates, kaolin, mannitol, silicic acid, sorbitol, starch, pre-gelatinized starch, and mixtures thereof. The binder or filler in pharmaceutical compositions is typically present in from about 50 to about 99 weight percent of the pharmaceutical composition or dosage form.
[00334] Disintegrants are used in the compositions to provide tablets that disintegrate when exposed to an aqueous environment. Tablets that contain too much disintegrant may disintegrate in storage, while those that contain too little may not disintegrate at a desired rate or under the desired conditions. Thus, a sufficient amount of disintegrant that is neither too much nor too little to detrimentally alter the release of the active ingredients should be used to form solid oral dosage forms. The amount of disintegrant used varies based upon the type of formulation, and is readily discernible to those of ordinary skill in the art. Typical pharmaceutical compositions comprise from about 0.5 to about 15 weight percent of disintegrant, preferably from about 1 to about 5 weight percent of disintegrant.
[00335] Disintegrants that can be used in pharmaceutical compositions and dosage forms include, but are not limited to, agar-agar, alginic acid, calcium carbonate, microcrystalline cellulose, croscarmellose sodium, crospovidone, polacrilin potassium, sodium starch glycolate, potato or tapioca starch, other starches, pre-gelatinized starch, other starches, clays, other algins, other celluloses, gums, and mixtures thereof.
[00336] Lubricants that can be used in pharmaceutical compositions and dosage forms include, but are not limited to, calcium stearate, magnesium stearate, mineral oil, light mineral oil, glycerin, sorbitol, mannitol, polyethylene glycol, other glycols, stearic acid, sodium lauryl sulfate, talc, hydrogenated vegetable oil (e.g., peanut oil, cottonseed oil, sunflower oil, sesame oil, olive oil, corn oil, and soybean oil), zinc stearate, ethyl oleate, ethyl laureate, agar, and mixtures thereof. Additional lubricants include, for example, a syloid silica gel (AEROSIL200, manufactured by W.R. Grace Co. of Baltimore, MD), a coagulated aerosol of synthetic silica (marketed by Degussa Co. of Piano, TX), CAB-O-SIL (a pyrogenic silicon dioxide product sold by Cabot Co. of Boston, MA), and mixtures thereof. If used at all, lubricants are typically used in an amount of less than about 1 weight percent of the pharmaceutical compositions or dosage forms into which they are incorporated.
[00337] A preferred solid oral dosage form comprises an immunomodulatory compound, anhydrous lactose, microcrystalline cellulose, polyvinylpyrrolidone, stearic acid, colloidal anhydrous silica, and gelatin.
[00338] A second active agent can be administered by controlled release means or by delivery devices that are well known to those of ordinary skill in the art. Examples include, but are not limited to, those described in U.S. Patent Nos.: 3,845,770; 3,916,899; 3,536,809; 3,598,123; and 4,008,719, 5,674,533, 5,059,595, 5,591,767, 5,120,548, 5,073,543, 5,639,476, 5,354,556, and 5,733,566, each of which is incorporated herein by reference. Such dosage forms can be used to provide slow or controlled-release of one or more active ingredients using, for example, hydropropylmethyl cellulose, other polymer matrices, gels, permeable membranes, osmotic systems, multilayer coatings, microparticles, liposomes, microspheres, or a combination thereof to provide the desired release profile in varying proportions.
Suitable controlled-release formulations known to those of ordinary skill in the art, including those described herein, can be readily selected for use with the second active agent. Single unit dosage forms suitable for oral administration such as, but not limited to, tablets, capsules, gelcaps, and caplets that are adapted for controlled-release can be used with the methods and compositions of the present invention.
[00339] All controlled-release pharmaceutical products have a common goal of improving drug therapy over that achieved by their non-controlled counterparts. Ideally, the use of an optimally designed controlled-release preparation in medical treatment is characterized by a minimum of drug substance being employed to cure or control the condition in a minimum amount of time. Advantages of controlled-release formulations include extended activity of the drug, reduced dosage frequency, and increased patient compliance. In addition, controlled-release formulations can be used to affect the time of onset of action or other characteristics, such as blood levels of the drug, and can thus affect the occurrence of side (e.g., adverse) effects.
[00340] Most controlled-release formulations are designed to initially release an amount of drug (active ingredient) that promptly produces the desired therapeutic effect, and gradually and continually release of other amounts of drug to maintain this level of therapeutic or prophylactic effect over an extended period of time. In order to maintain this constant level of drug in the body, the drug must be released from the dosage form at a rate that will replace the amount of drug being metabolized and excreted from the body. Controlled-release of an
active ingredient can be stimulated by various conditions including, but not limited to, H, temperature, enzymes, water, or other physiological conditions or compounds.
[00341] Parenteral dosage forms can be administered to patients by various routes including, but not limited to, subcutaneous, intravenous (including bolus injection), intramuscular, and intraarterial. Because their administration typically bypasses patients' natural defenses against contaminants, parenteral dosage forms are preferably sterile or capable of being sterilized prior to administration to a patient. Examples of parenteral dosage forms include, but are not limited to, solutions ready for injection, dry products ready to be dissolved or suspended in a pharmaceutically acceptable vehicle for injection, suspensions ready for injection, and emulsions.
[00342] Suitable vehicles that can be used to provide parenteral dosage forms are well known to those skilled in the art. Examples include, but are not limited to: Water for Injection USP; aqueous vehicles such as, but not limited to, Sodium Chloride Injection, Ringer's Injection, Dextrose Injection, Dextrose and Sodium Chloride Injection, and Lactated Ringer's Injection; water-miscible vehicles such as, but not limited to, ethyl alcohol, polyethylene glycol, and polypropylene glycol; and non-aqueous vehicles such as, but not limited to, corn oil, cottonseed oil, peanut oil, sesame oil, ethyl oleate, isopropyl myristate, and benzyl benzoate.
[00343] Compounds that increase the solubility of a second active agent can also be incorporated into the parenteral dosage forms. For example, cyclodextrin and its derivatives can be used to increase the solubility of an immunomodulatory compound and its derivatives. See, e.g., U.S. Patent No. 5,134,127, which is incorporated herein by reference.
[00344] Topical and mucosal dosage forms include, but are not limited to, sprays, aerosols, solutions, emulsions, suspensions, eye drops or other ophthalmic preparations, or other forms known to one of skill in the art. See, e.g., Remington's Pharmaceutical Sciences, 16th and 18th eds., Mack Publishing, Easton PA (1980 & 1990); and Introduction to Pharmaceutical Dosage Forms, 4th ed., Lea & Febiger, Philadelphia (1985). Dosage forms suitable for treating mucosal tissues within the oral cavity can be formulated as mouthwashes or as oral gels.
[00345] Suitable excipients (e.g., carriers and diluents) and other materials that can be used to provide topical and mucosal dosage forms that can be used are well known to those
skilled in the pharmaceutical arts, and depend on the particular tissue to which a given pharmaceutical composition or dosage form will be applied. With that fact in mind, typical excipients include, but are not limited to, water, acetone, ethanol, ethylene glycol, propylene glycol, butane- 1,3-diol, isopropyl myristate, isopropyl palmitate, mineral oil, and mixtures thereof to form solutions, emulsions or gels, which are non-toxic and pharmaceutically acceptable. Moisturizers or humectants can also be added to pharmaceutical compositions and dosage forms if desired. Examples of such additional ingredients are well known in the art. See, e.g., Remington's Pharmaceutical Sciences, 16th and 18th eds., Mack Publishing, Easton PA (1980 & 1990).
[00346] The pH of a pharmaceutical composition or dosage form may also be adjusted to improve delivery of one or more active ingredients. Similarly, the polarity of a solvent carrier, its ionic strength, or tonicity can be adjusted to improve delivery. Compounds such as stearates can also be added to pharmaceutical compositions or dosage forms to
advantageously alter the hydrophilicity or lipophilicity of one or more active ingredients so as to improve delivery. In this regard, stearates can serve as a lipid vehicle for the formulation, as an emulsifying agent or surfactant, and as a delivery-enhancing or penetration-enhancing agent. Different salts, hydrates or solvates of the active ingredients can be used to further adjust the properties of the resulting composition.
4.8 KITS
[00347] In certain embodiments, an ActRII inhibitor and a second active agent are not administered to a patient at the same time or by the same route of administration. Thus, also encompassed herein are kits which, when used by the medical practitioner, can simplify the administration of appropriate amounts of active ingredients to a patient.
[00348] A typical kit provided herein comprises a dosage form of one or more ActRII inhibitors (i.e., and inhibitor of ActRIIA or ActRIIB; see Section 4.2) or a dosage form of one or more ActRII inhibitors (i.e., and inhibitor of ActRIIA or ActRIIB; see Section 4.2) and a dosage form of a a second active agent (see Section 4.3) or a pharmaceutically acceptable salt salt, solvate, hydrate, stereoisomer, prodrug, or clathrate thereof.
[00349] In a specific embodiment, a kit provided herein comprises a dosage form of an ActRIIA inhibitor comprising or consisting of a polypeptide that comprises the amino acid sequence of SEQ ID NO:7 and a dosage form of Revlimid®.
[00350] In another specific embodiment, a kit provided herein comprises a dosage form of an ActRIIB inhibitor comprising or consisting of a polypeptide that comprises the amino acid sequence of SEQ ID NO:23 and a dosage form of Revlimid®.
[00351] In another specific embodiment, a kit provided herein comprises a dosage form of an ActRIIB inhibitor comprising or consisting of a polypeptide that comprises the amino acid sequence of SEQ ID NO:25 and a dosage form of Revlimid®.
[00352] In certain embodiments, a kit provided herein comprises a dosage form of an ActRIIA inhibitor comprising and a dosage form of an ActRIIB inhibitor. Such a kit may additionally comprise a dosage form of a second active agent.
[00353] In a specific embodiment, a kit provided herein comprises a dosage form of an ActRIIA inhibitor comprising or consisting of a polypeptide that comprises the amino acid sequence of SEQ ID NO: 7 and a dosage form of an ActRIIB inhibitor comprising or consisting of a polypeptide that comprises the amino acid sequence of SEQ ID NO:25.
[00354] In another specific embodiment, a kit provided herein comprises a dosage form of an ActRIIA inhibitor comprising or consisting of a polypeptide that comprises the amino acid sequence of SEQ ID NO:7, a dosage form of an ActRIIB inhibitor comprising or consisting of a polypeptide that comprises the amino acid sequence of SEQ ID NO:25, and a dosage form of Revlimid®.
[00355] Kits provided herein can further comprise devices that are used to administer the active ingredients. Examples of such devices include, but are not limited to, syringes, drip bags, patches, and inhalers.
5. EXAMPLES
5.1 EXAMPLE 1
(a) ActRIIa-Fc Fusion Proteins
[00356] A soluble ActRIIa fusion protein that has the extracellular domain of human ActRIIa fused to a human or mouse Fc domain with a minimal linker in between is provided. The constructs are referred to as ActRIIa-hFc and ActRIIa-mFc, respectively. ActRIIa-hFc is provided as SEQ ID NO:7.
[00357] The ActRIIa-hFc and ActRIIa-mFc proteins were expressed in CHO cell lines.
Three different leader sequences were considered:
(i) Honey bee mellitin (HBML): SEQ ID NO: 8
(ii) Tissue Plasminogen Activator (TP A): SEQ ID NO: 9
(iii) Native ActRIIa: SEQ ID NO: 10
[00358] The selected form employs the TPA leader and has the following unprocessed amino acid sequence is set forth in SEQ ID NO: 13. This polypeptide is encoded by SEQ ID NO: 14.
(b) ActRIIb-Fc Fusion Proteins
[00359] Co-crystal structure of an extracellular domain of human ActRIIb fused to a human Fc domain and Activin did not show any role for the final (C-terminal) 15 amino acids (referred to as the "tail" herein) of the extracellular domain in ligand binding. This sequence failed to resolve on the crystal structure, suggesting that these residues are present in a flexible loop that did not pack uniformly in the crystal. Thompson et al. EMBO J. 2003 Apr 1 ;22(7): 1555-66. This sequence is also poorly conserved between ActRIIb and ActRIIa.
Accordingly, these residues were omitted in the basic, or background, ActRIIb-Fc fusion construct. Additionally, position 64 in the background form is occupied by an alanine, which is generally considered the "wild type" form, although a A64R allele occurs naturally. Thus, the background ActRIIb-Fc fusion has the sequence disclosed as SEQ ID NO:21.
[00360] Surprisingly, the C-terminal tail was found to enhance activin and GDF-11 binding, thus a preferred version of ActRIIb-Fc has a sequence SEQ ID NO:20.
[00361] A variety of ActRIIb a variants that may be used according to the methods described herein are described in the International Patent Application published as
WO2006/012627 (see, e.g., pp. 59-60), incorporated herein by reference in its entirety.
5.2 EXAMPLE 2
[00362] The in vitro and in vivo effects of administration of ActRIIA inhibitors in combination with lenalidomide on the multiple myeloma tumor-bone microenvironment can be investigated using experimental models.
[00363] The effects of administration of ActRIIA inhibitors in combination with lenalidomide on osteoblast (OB) formation and activity can be assessed as follows:
mesenchymal stem cells from healthy donor or multiple myeloma patients can be stimulated
with dexamethasone, citric acid and β-glycerol phosphate for 14-21 days. OB number and activity can be analyzed with alkaline-phosphatase and alizarin red staining.
[00364] The effects of ActRIIA inhibitors and lenalidomide on osteoclast (OC) differentiation can be assessed as follows: peripheral blood mononuclear cells from normal donors and bone marrow mononuclear cells from multiple myeloma patients can be stimulated with RANKL and M-CSF. After three weeks, OC number can be evaluated with TRAP staining and OC activity with pit formation assay.
[00365] The growth inhibitory effects of ActRIIA inhibitors and lenalidomide against multiple myeloma cell lines and CD 138+ cells from multiple myeloma patients' bone marrow aspirates can be assessed by culturing the cell lines and cells with ActRIIA inhibitors and lenalidomide alone and in co-culture with BMSC, OC and OB; followed by measuring growth inhibitory effects via MTT assay, [3H]-thymidine uptake, and cytokine secretion with ELISA.
[00366] The molecular mechanisms of action of a combination of ActRIIA inhibitors and lenalidomide can be assessed by detecting signaling pathways and transcription factor expression in the different components of the tumor niche using Western blotting and quantitative PCR analysis.
[00367] The in vivo effects on bone disease and anti-tumor activity induced by ActRIIA inhibitors and lenalidomide alone and in combination can be assessed by analyzing tumor growth by serum immunoglobulin levels and effects on the bone compartment with radiological imaging as well as static and dynamic histomorphometry.
EQUIVALENTS
[00368] Although the invention is described in detail with reference to specific
embodiments thereof, it will be understood that variations which are functionally equivalent are within the scope of this invention. Indeed, various modifications of the invention in addition to those shown and described herein will become apparent to those skilled in the art from the foregoing description and accompanying drawings. Such modifications are intended to fall within the scope of the appended claims. Those skilled in the art will recognize, or be able to ascertain using no more than routine experimentation, many equivalents to the specific embodiments of the invention described herein. Such equivalents are intended to be encompassed by the following claims.
[00369] All publications, patents and patent applications mentioned in this specification are herein incorporated by reference into the specification to the same extent as if each individual publication, patent or patent application was specifically and individually indicated to be incorporated herein by reference in their entireties.
Table 1 : Sequence Information
SEQ ID Description
NO:
1 human ActRIIA precursor polypeptide
2 human ActRIIA soluble (extracellular), processed polypeptide sequence
3 human ActRIIA soluble (extracellular), processed polypeptide sequence with the C-terminal 15 amino acids deleted
4 nucleic acid sequence encoding human ActRIIA precursor protein
5 nucleic acid sequence encoding a human ActRIIA soluble (extracellular)
polypeptide
6 fusion protein comprising a soluble extracellular domain of ActRIIA fused to an Fc domain
7 Extracellular domain of a human ActRIIA fused to a human Fc domain
8 Leader sequence of Honey bee mellitin (HBML)
9 Leader sequence of Tissue Plasminogen Activator (TP A)
10 Native ActRIIA Leader Sequence
11 ActRIIA-hFc and ActRIIA-mFc N-terminal sequence
12 ActRIIA-Fc Protein with deletion of the C-terminal 15 amino acids of the
extracellular domain of ActRIIA
13 Unprocessed ActRIIA-hFc with TPA leader sequence
14 Nucleic acid sequence encoding Unprocessed ActRIIA-hFc with TPA leader sequence
15 human ActRIIB soluble (extracellular), processed polypeptide sequence with the N-terminal 6 amino acids of the EC domain deleted and the C-terminal 4 amino acids of the EC domain deleted (amino acids 25-130 of SEQ ID NO:28) and with an L79D mutation
16 human ActRIIB precursor protein sequence (A64)
17 human ActRIIB soluble (extracellular), processed polypeptide sequence
(amino acids 19-134 of SEQ ID NO: 16)
18 human ActRIIB soluble (extracellular), processed polypeptide sequence with the C-terminal 15 amino acids deleted (amino acids 19-119 of SEQ ID NO: 16)
19 nucleic acid sequence encoding a human ActRIIB (A64) precursor protein
SEQ ID Description
NO:
20 fusion protein comprising a soluble extracellular domain of ActRIIB (A64;
SEQ ID NO: 17) fused to an Fc domain
21 fusion protein comprising a soluble extracellular domain of ActRIIB (A64) with the C-terminal 15 amino acids deleted (SEQ ID NO: 18) fused to an Fc domain
22 human ActRIIB soluble (extracellular), processed polypeptide sequence with the N-terminal 6 amino acids of the EC domain deleted and the C-terminal 5 amino acids of the EC domain deleted (amino acids 25-129 of SEQ ID NO:28) and with an L79D mutation
23 human ActRIIB soluble (extracellular), processed polypeptide sequence with the N-terminal 6 amino acids of the EC domain deleted and the C-terminal 3 amino acids of the EC domain deleted (amino acids 25-131 of SEQ ID NO:28) and with an L79D mutation
24 Unprocessed ActRIIB-Fc fusion protein with the N-terminal 6 amino acids of the EC domain deleted and the C-terminal 3 amino acids of the EC domain deleted (amino acids 25-131 of SEQ ID NO:28) and with an L79D mutation and with TPA leader sequence
25 Processed ActRIIB-Fc fusion protein with the N-terminal 6 amino acids of the
EC domain deleted and the C-terminal 3 amino acids of the EC domain deleted (amino acids 25-131 of SEQ ID NO:28) and with an L79D mutation
26 human ActRIIB soluble (extracellular), processed polypeptide sequence
(amino acids 20-134 of SEQ ID NO: 16)
27 human ActRIIB soluble (extracellular), processed polypeptide sequence with the C-terminal 15 amino acids deleted (amino acids 20-119 of SEQ ID NO: 16)
28 human ActRIIB precursor protein sequence (R64)
29 human ActRIIB soluble (extracellular), processed polypeptide sequence
(amino acids 19-134 of SEQ ID NO:28)
30 human ActRIIB soluble (extracellular), processed polypeptide sequence with the C-terminal 15 amino acids deleted (amino acids 19-119 of SEQ ID NO:28)
31 human ActRIIB soluble (extracellular), processed polypeptide sequence
(amino acids 20-134 of SEQ ID NO:28)
32 human ActRIIB soluble (extracellular), processed polypeptide sequence with the C-terminal 15 amino acids deleted (amino acids 20-119 of SEQ ID NO:28)
33 human ActRIIB soluble (extracellular), processed polypeptide sequence with the N-terminal 6 amino acids of the EC domain deleted and the C-terminal 3 amino acids of the EC domain deleted (amino acids 25-131 of SEQ ID NO: 16) and with an L79D mutation
34 Unprocessed ActRIIB-Fc fusion protein with the N-terminal 6 amino acids of the EC domain deleted and the C-terminal 3 amino acids of the EC domain
SEQ ID Description
NO:
deleted (amino acids 25-131 of SEQ ID NO: 16) and with an L79D mutation and with TPA leader sequence
35 Processed ActRIIB-Fc fusion protein with the N-terminal 6 amino acids of the
EC domain deleted and the C-terminal 3 amino acids of the EC domain deleted (amino acids 25-131 of SEQ ID NO: 16) and with an L79D mutation
36 human ActRIIB soluble (extracellular), processed polypeptide sequence
(amino acids 20-134 of SEQ ID NO:28) with L79D mutation
37 human ActRIIB soluble (extracellular), processed polypeptide sequence
(amino acids 20-134 of SEQ ID NO: 16) with L79D mutation
38 human ActRIIB soluble (extracellular), processed polypeptide sequence
(amino acids 20-134 of SEQ ID NO:28) with L79D mutation fused to an Fc domain with a GGG linker
39 human ActRIIB soluble (extracellular), processed polypeptide sequence
(amino acids 20-134 of SEQ ID NO: 16) with L79D mutation fused to an Fc domain
40 human ActRIIB soluble (extracellular), processed polypeptide sequence
(amino acids 20-134 of SEQ ID NO:28) with L79D mutation fused to an Fc domain and with TPA leader sequence
41 human ActRIIB soluble (extracellular), processed polypeptide sequence
(amino acids 20-134 of SEQ ID NO: 16) with L79D mutation fused to an Fc domain and with TPA leader sequence
42 human ActRIIB soluble (extracellular), processed polypeptide sequence
having a variant C-terminal sequence (disclosed in WO2007/053775)
43 human ActRIIB soluble (extracellular), processed polypeptide sequence
having a variant C-terminal sequence (disclosed in WO2007/053775) having an L79D mutation
44 human ActRIIB soluble (extracellular), processed polypeptide sequence
having a variant C-terminal sequence (disclosed in WO2007/053775) having an L79D mutation fused to an Fc domain with a TGGG linker
45 Nucleic Acid Sequence Encoding SEQ ID NO:24
46 fusion protein comprising a soluble extracellular domain of ActRIIB (R64;
SEQ ID NO:29) fused to an Fc domain
47 fusion protein comprising a soluble extracellular domain of ActRIIB (R64) with the C-terminal 15 amino acids deleted (SEQ ID NO:30) fused to an Fc domain
Claims
1. A method for treating a disease comprising administering to a patient in need of such treatment an ActRIIA inhibitor and a compound of any one of formulas I-XXXI or a pharmaceutically acceptable salt, solvate, or stereoisomer thereof.
2. The method of claim 1 , wherein the ActRIIA inhibitor is a polypeptide comprising an amino acid sequence selected from the group consisting of:
a. 90% identical to SEQ ID NO:2;
b. 95% identical to SEQ ID NO:2;
c. 98% identical to SEQ ID NO:2;
d. SEQ ID NO:2;
e. 90% identical to SEQ ID NO:3;
f. 95% identical to SEQ ID NO:3;
g. 98% identical to SEQ ID NO:3;
h. SEQ ID NO:3;
i. 90% identical to SEQ ID NO:6;
j. 95% identical to SEQ ID NO:6;
k. 98% identical to SEQ ID NO:6;
1. SEQ ID NO:6;
m. 90% identical to SEQ ID NO:7;
n. 95% identical to SEQ ID NO:7;
o. 98% identical to SEQ ID NO:7;
p. SEQ ID NO:7;
q. 90% identical to SEQ ID NO: 12;
r. 95% identical to SEQ ID NO: 12;
s. 98% identical to SEQ ID NO: 12; and
t. SEQ ID NO: 12; and
(ii) the compound is 3-(4-amino-l-oxo-l,3-dihydro-isoindol-2-yl)-piperidine-2,6- dione.
3. The method of claim 2, wherein the ActRIIA inhibitor is a polypeptide comprising the amino acid sequence of SEQ ID NO:7.
4. The method of any one of claims 1, 2, or 3, wherein the ActRIIA inhibitor is administered parenterally.
5. The method of any one of claims 1, 2, or 3, wherein the compound is administered orally.
6. The method of claim 5, wherein the compound is administered in the form of a capsule or tablet.
7. The method of claim 5, wherein the compound is administered in an amount of from about 10 to about 25 mg per day.
8. The method of claim 1 , wherein the ActRIIA inhibitor and the compound are administered at the same time.
9. The method of claim 1 , wherein the ActRIIA inhibitor and the compound are administered sequentially.
10. The method of claim 1, wherein the ActRIIA inhibitor and the compound are administered in the same formulation.
11. The method of claim 1 , wherein the disease is cancer, multiple myeloma, myelodysplasia syndrome, or anemia.
12. The method of claim 1, wherein the disease is multiple myeloma.
13. The method of claim 1, wherein the disease is myelodysplasia syndrome.
14. The method of claim 1, wherein the disease is anemia.
15. The method of claim 1, wherein the method further comprises administration of an ActRIIB inhibitor.
16. The method of claim 15, wherein the ActRIIB inhibitor is a polypeptide comprising an amino acid sequence selected from the group consisting of:
a. 90% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; b.95% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43;
c. 98% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43;
d. SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43;
e. 90% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; f. 95% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; g. 98% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; and
h. SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47.
17. A method for improving the safety of a treatment with 3-(4-amino- 1 -oxo- 1 ,3- dihydro-isoindol-2-yl)-piperidine-2,6-dione) in a patient being treated with 3-(4-amino-l- oxo-l,3-dihydro-isoindol-2-yl)-piperidine-2,6-dione, comprising administering an ActRIlA inhibitor to the patient.
18. The method of claim 17, wherein the ActRIlA inhibitor is a polypeptide comprising an amino acid sequence selected from the group consisting of:
a. 90% identical to SEQ ID NO:2;
b. 95% identical to SEQ ID NO:2;
c. 98% identical to SEQ ID NO:2;
d. SEQ ID NO:2;
e. 90% identical to SEQ ID NO:3;
f. 95% identical to SEQ ID NO:3;
g. 98% identical to SEQ ID NO:3;
h. SEQ ID NO:3;
i. 90% identical to SEQ ID NO:6;
j. 95% identical to SEQ ID NO:6;
k. 98% identical to SEQ ID NO:6;
1. SEQ ID NO:6;
m. 90% identical to SEQ ID NO:7;
n. 95% identical to SEQ ID NO:7;
o. 98% identical to SEQ ID NO:7;
p. SEQ ID NO:7;
q. 90% identical to SEQ ID NO: 12;
r. 95% identical to SEQ ID NO: 12;
s. 98% identical to SEQ ID NO: 12; and
t. SEQ ID NO: 12.
19. The method of claim 18, wherein the ActRIIA inhibitor is a polypeptide comprising the amino acid sequence of SEQ ID NO:7.
20. The method of claim 17, wherein the method further comprises administration of an ActRIIB inhibitor.
21. The method of claim 20, wherein the ActRIIB inhibitor is a polypeptide comprising an amino acid sequence selected from the group consisting of:
a. 90% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; b.95% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43;
c. 98% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43;
d. SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43;
e. 90% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; f. 95% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; g. 98% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; and
h. SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47.
22. A pharmaceutical composition comprising an ActRIIA inhibitor, a compound of any one of formulas I-XXXI, and a pharmaceutically acceptable carrier.
23. The pharmaceutical composition of claim 22, wherein the ActRIIA inhibitor is a polypeptide comprising an amino acid sequence selected from the group consisting of:
a. 90% identical to SEQ ID NO:2;
b. 95% identical to SEQ ID NO:2;
c. 98% identical to SEQ ID NO:2;
d. SEQ ID NO:2;
e. 90% identical to SEQ ID NO:3;
f. 95% identical to SEQ ID NO:3;
g. 98% identical to SEQ ID NO:3;
h. SEQ ID NO:3;
i. 90% identical to SEQ ID NO:6;
j. 95% identical to SEQ ID NO:6;
k. 98% identical to SEQ ID NO:6;
1. SEQ ID NO:6;
m. 90% identical to SEQ ID NO:7;
n. 95% identical to SEQ ID NO:7;
o. 98% identical to SEQ ID NO:7;
p. SEQ ID NO:7;
q. 90% identical to SEQ ID NO: 12;
r. 95% identical to SEQ ID NO: 12;
s. 98% identical to SEQ ID NO: 12; and
t. SEQ ID NO: 12; and
(ii) the compound is 3-(4-amino-l-oxo-l,3-dihydro-isoindol-2-yl)-piperidine-2,6- dione.
24. The pharmaceutical composition of claim 22, further comprising an ActRIIB inhibitor.
25. The pharmaceutical composition of claim 24, wherein the ActRIIB inhibitor is a polypeptide comprising an amino acid sequence selected from the group consisting of: a. 90% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43; b.95% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43;
c. 98% identical to SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43;
d. SEQ ID NO: 17, 18, 23, 26, 27, 29, 30, 31, 32, 33, 36, 37, 42, or 43;
e. 90% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; f. 95% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; g. 98% identical to SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47; and
h. SEQ ID NO:20, 21, 24, 25, 34, 35, 38, 39, 40, 41, 44, 46, or 47.
26. A kit comprising, in one or more containers, the pharmaceutical composition of any one of claims 22 to 25.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US37771810P | 2010-08-27 | 2010-08-27 | |
| US61/377,718 | 2010-08-27 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2012027065A2 true WO2012027065A2 (en) | 2012-03-01 |
| WO2012027065A3 WO2012027065A3 (en) | 2012-05-10 |
Family
ID=45723978
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2011/045917 Ceased WO2012027065A2 (en) | 2010-08-27 | 2011-07-29 | Combination therapy for treatment of disease |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2012027065A2 (en) |
Cited By (39)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014066487A3 (en) * | 2012-10-24 | 2014-06-19 | Celgene Corporation | Methods for treating anemia |
| WO2014152833A1 (en) * | 2013-03-14 | 2014-09-25 | Deuterx, Llc | 3-(substituted-4-oxo-quinazolin-3(4h)-yl)-3-deutero-piperidine-2,6-dione derivatives |
| US9090585B2 (en) | 2011-03-28 | 2015-07-28 | Deuterx, Llc | 2,6-dioxo-3-deutero-piperdin-3-yl-isoindoline compounds |
| WO2015143403A1 (en) * | 2014-03-21 | 2015-09-24 | Acceleron Pharma, Inc. | Methods for increasing red blood cell levels and treating ineffective erythropoiesis by inhibiting activin b and/or gdf11 |
| WO2015192111A1 (en) * | 2014-06-13 | 2015-12-17 | Acceleron Pharma, Inc. | Methods and compositions for treating ulcers |
| US9353356B2 (en) | 2007-09-18 | 2016-05-31 | Acceleron Pharma Inc. | Activin-actriia antagonists for treating a follicle-stimulating horomone-secreting pituitary tumor |
| US9399669B2 (en) | 2007-02-02 | 2016-07-26 | Acceleron Pharma Inc. | Variants derived from ActRIIB |
| US9439945B2 (en) | 2008-08-14 | 2016-09-13 | Acceleron Pharma Inc. | Isolated nucleotide sequences encoding GDF traps |
| WO2016164503A1 (en) * | 2015-04-06 | 2016-10-13 | Acceleron Pharma Inc. | Alk7:actriib heteromultimers and uses thereof |
| US9480742B2 (en) | 2005-11-23 | 2016-11-01 | Acceleron Pharma Inc. | Method of promoting bone growth by an anti-actriia antibody |
| US9493556B2 (en) | 2010-11-08 | 2016-11-15 | Acceleron Pharma Inc. | Actriia binding agents and uses thereof |
| US9505813B2 (en) | 2008-08-14 | 2016-11-29 | Acceleron Pharma Inc. | Use of GDF traps to treat anemia |
| US9526759B2 (en) | 2007-02-01 | 2016-12-27 | Acceleron Pharma Inc. | Activin-actriia antagonists and uses for treating or preventing breast cancer |
| US9540340B2 (en) | 2013-01-14 | 2017-01-10 | Deuterx, Llc | 3-(5-substituted-4-oxoquinazolin-3(4H)-yl)-3-deutero-piperidine-2,6-dione derivatives and compositions comprising and methods of using the same |
| US9572865B2 (en) | 2005-11-23 | 2017-02-21 | Acceleron Pharma Inc. | Activin-actriia antagonists and uses for treating multiple myeloma |
| US9617319B2 (en) | 2009-11-17 | 2017-04-11 | Acceleron Pharma Inc. | ActRIIB proteins and variants and uses therefore relating to utrophin induction for muscular dystrophy therapy |
| US9682952B2 (en) | 2012-09-04 | 2017-06-20 | Celgene Corporation | Isotopologues of 3-(5-amino-2-methyl-4-oxoquinazolin-3(4H)-yl) piperidine-2-6-dione and methods of preparation thereof |
| US9695145B2 (en) | 2013-01-22 | 2017-07-04 | Celgene Corporation | Processes for the preparation of isotopologues of 3-(4-((4- morpholinomethyl)benzyl)oxy)-1-oxoisoindolin-2-yl)piperidine-2,6-dione and pharmaceutically acceptable salts thereof |
| US9732064B2 (en) | 2006-09-26 | 2017-08-15 | Celgene Corporation | 5-substituted quinazolinone derivatives and compositions comprising and methods of using the same |
| WO2017147182A1 (en) * | 2016-02-22 | 2017-08-31 | Acceleron Pharma Inc. | Actrii antagonists for use in increasing immune activity |
| US9751853B2 (en) | 2011-03-11 | 2017-09-05 | Celgene Corporation | Solid forms of 3-(5-amino-2-methyl-4-oxo-4H-quinazolin-3-yl)-piperidine-2,6-dione, and their pharmaceutical compositions and uses |
| WO2017079591A3 (en) * | 2015-11-04 | 2017-09-21 | Acceleron Pharma Inc. | Methods for increasing red blood cell levels and treating ineffective erythropoiesis |
| US9790284B2 (en) | 2009-06-08 | 2017-10-17 | Acceleron Pharma Inc. | Methods for increasing thermogenic adipocytes |
| US9809603B1 (en) | 2015-08-18 | 2017-11-07 | Deuterx, Llc | Deuterium-enriched isoindolinonyl-piperidinonyl conjugates and oxoquinazolin-3(4H)-yl-piperidinonyl conjugates and methods of treating medical disorders using same |
| US9822094B2 (en) | 2010-02-11 | 2017-11-21 | Celgene Corporation | Arylmethoxy isoindoline derivatives and compositions comprising and methods of using the same |
| US10071135B2 (en) | 2005-11-23 | 2018-09-11 | Acceleron Pharma Inc. | Method of identifying an agent that promotes bone growth or increases bone density |
| RU2678117C2 (en) * | 2012-11-02 | 2019-01-23 | Селджин Корпорейшн | Activin-actrii antagonists and uses thereof for treating bone and other disorders |
| CN109789184A (en) * | 2016-07-27 | 2019-05-21 | 阿塞勒隆制药公司 | For treating the method and composition of myelofibrosis |
| US10844039B2 (en) | 2018-11-13 | 2020-11-24 | Biotheryx, Inc. | Substituted isoindolinones |
| US10913782B2 (en) | 2015-04-22 | 2021-02-09 | Biogen Ma Inc. | Hybrid ActRIIB ligand trap proteins for treating muscle wasting diseases |
| US20210052698A1 (en) * | 2018-05-09 | 2021-02-25 | Keros Therapeutics, Inc. | Activin receptor type iia variants and methods of use thereof |
| US11267865B2 (en) | 2016-10-05 | 2022-03-08 | Acceleron Pharma Inc. | Variant ActRIIB proteins and uses thereof |
| US11471510B2 (en) | 2014-12-03 | 2022-10-18 | Celgene Corporation | Activin-ActRII antagonists and uses for treating anemia |
| US11813308B2 (en) | 2014-10-09 | 2023-11-14 | Celgene Corporation | Treatment of cardiovascular disease using ActRII ligand traps |
| US12187783B2 (en) | 2016-10-20 | 2025-01-07 | Alivegen Inc | Methods for treating muscle wasting and bone disease using novel hybrid ActRIIB ligand trap proteins |
| US12269858B2 (en) | 2018-01-12 | 2025-04-08 | Keros Therapeutics, Inc. | Activin receptor type IIB variants and methods of use thereof |
| US12350313B2 (en) | 2016-11-10 | 2025-07-08 | Keros Therapeutics, Inc. | Activin receptor type IIa variants and methods of use thereof |
| US12440539B2 (en) | 2020-03-20 | 2025-10-14 | Keros Therapeutics, Inc. | Methods of using activin receptor type IIB variants |
| US12522646B2 (en) | 2020-03-20 | 2026-01-13 | Keros Therapeutics, Inc. | Activin receptor type II chimeras and methods of use thereof |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7968569B2 (en) * | 2002-05-17 | 2011-06-28 | Celgene Corporation | Methods for treatment of multiple myeloma using 3-(4-amino-1-oxo-1,3-dihydro-isoindol-2-yl)-piperidine-2,6-dione |
| ES2839549T3 (en) * | 2005-11-23 | 2021-07-05 | Acceleron Pharma Inc | Activin-ActRIIa antagonists for use in stimulating bone growth |
| ME02335B (en) * | 2006-12-18 | 2013-04-30 | Acceleron Pharma Inc | ACTIVIN ACTRII ANTAGONISTS AND THEIR USES FOR THE TREATMENT OF ANEMIA |
-
2011
- 2011-07-29 WO PCT/US2011/045917 patent/WO2012027065A2/en not_active Ceased
Cited By (92)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9480742B2 (en) | 2005-11-23 | 2016-11-01 | Acceleron Pharma Inc. | Method of promoting bone growth by an anti-actriia antibody |
| US10239940B2 (en) | 2005-11-23 | 2019-03-26 | Acceleron Pharma Inc. | Method of promoting bone growth by an anti-actriia antibody |
| US10071135B2 (en) | 2005-11-23 | 2018-09-11 | Acceleron Pharma Inc. | Method of identifying an agent that promotes bone growth or increases bone density |
| US9572865B2 (en) | 2005-11-23 | 2017-02-21 | Acceleron Pharma Inc. | Activin-actriia antagonists and uses for treating multiple myeloma |
| US9732064B2 (en) | 2006-09-26 | 2017-08-15 | Celgene Corporation | 5-substituted quinazolinone derivatives and compositions comprising and methods of using the same |
| US9526759B2 (en) | 2007-02-01 | 2016-12-27 | Acceleron Pharma Inc. | Activin-actriia antagonists and uses for treating or preventing breast cancer |
| US10259861B2 (en) | 2007-02-02 | 2019-04-16 | Acceleron Pharma Inc. | Variants derived from ActRIIB and uses therefor |
| US9399669B2 (en) | 2007-02-02 | 2016-07-26 | Acceleron Pharma Inc. | Variants derived from ActRIIB |
| US9353356B2 (en) | 2007-09-18 | 2016-05-31 | Acceleron Pharma Inc. | Activin-actriia antagonists for treating a follicle-stimulating horomone-secreting pituitary tumor |
| US11162085B2 (en) | 2008-08-14 | 2021-11-02 | Acceleron Pharma Inc. | Methods for treating anemia in a subject in need thereof |
| US10377996B2 (en) | 2008-08-14 | 2019-08-13 | Acceleron Pharma Inc. | Methods of identifying ActRIIB variants |
| US9439945B2 (en) | 2008-08-14 | 2016-09-13 | Acceleron Pharma Inc. | Isolated nucleotide sequences encoding GDF traps |
| US11155791B2 (en) | 2008-08-14 | 2021-10-26 | Acceleron Pharma Inc. | Methods for treating anemia in a subject in need thereof |
| US9505813B2 (en) | 2008-08-14 | 2016-11-29 | Acceleron Pharma Inc. | Use of GDF traps to treat anemia |
| US10689427B2 (en) | 2008-08-14 | 2020-06-23 | Acceleron Pharma Inc. | Combined use of GDF traps and erythropoietin receptor activators to increase red blood cell levels |
| US10829532B2 (en) | 2008-08-14 | 2020-11-10 | Acceleron Pharma Inc. | Combined use of gdf traps and erythropoietin receptor activators to increase red blood cell levels |
| US11168311B2 (en) | 2008-08-14 | 2021-11-09 | Acceleron Pharma Inc. | Methods for treating anemia in a subject in need thereof |
| US10889626B2 (en) | 2008-08-14 | 2021-01-12 | Acceleron Pharma Inc. | Combined use of GDF traps and erythropoietin receptor activators to increase red blood cell levels |
| US10829533B2 (en) | 2008-08-14 | 2020-11-10 | Acceleron Pharma Inc. | Combined use of GDF traps and erythropoietin receptor activators to increase red blood cell levels |
| US10968282B2 (en) | 2009-06-08 | 2021-04-06 | Acceleron Pharma Inc. | Methods for screening compounds for increasing thermogenic adipocytes |
| US9790284B2 (en) | 2009-06-08 | 2017-10-17 | Acceleron Pharma Inc. | Methods for increasing thermogenic adipocytes |
| US9617319B2 (en) | 2009-11-17 | 2017-04-11 | Acceleron Pharma Inc. | ActRIIB proteins and variants and uses therefore relating to utrophin induction for muscular dystrophy therapy |
| US10968262B2 (en) | 2009-11-17 | 2021-04-06 | Acceleron Pharma Inc. | Methods of increasing sarcolemmal utrophin |
| US11414399B2 (en) | 2010-02-11 | 2022-08-16 | Celgene Corporation | Arylmethoxy isoindoline derivatives and compositions comprising and methods of using the same |
| US10669257B2 (en) | 2010-02-11 | 2020-06-02 | Celgene Corporation | Arylmethoxy isoindoline derivatives and compositions comprising and methods of using the same |
| US9822094B2 (en) | 2010-02-11 | 2017-11-21 | Celgene Corporation | Arylmethoxy isoindoline derivatives and compositions comprising and methods of using the same |
| US9828361B2 (en) | 2010-02-11 | 2017-11-28 | Celgene Corporation | Arylmethoxy isoindoline derivatives and compositions comprising and methods of using the same |
| US10189814B2 (en) | 2010-02-11 | 2019-01-29 | Celgene Corporation | Arylmethoxy isoindoline derivatives and compositions comprising and methods of using the same |
| US9493556B2 (en) | 2010-11-08 | 2016-11-15 | Acceleron Pharma Inc. | Actriia binding agents and uses thereof |
| US9751853B2 (en) | 2011-03-11 | 2017-09-05 | Celgene Corporation | Solid forms of 3-(5-amino-2-methyl-4-oxo-4H-quinazolin-3-yl)-piperidine-2,6-dione, and their pharmaceutical compositions and uses |
| US9969713B2 (en) | 2011-03-11 | 2018-05-15 | Celgene Corporation | Solid forms of 3-(5-amino-2-methyl-4-oxo-4H-quinazolin-3-yl)-piperidine-2,6-dione, and their pharmaceutical compositions and uses |
| US9090585B2 (en) | 2011-03-28 | 2015-07-28 | Deuterx, Llc | 2,6-dioxo-3-deutero-piperdin-3-yl-isoindoline compounds |
| US9682952B2 (en) | 2012-09-04 | 2017-06-20 | Celgene Corporation | Isotopologues of 3-(5-amino-2-methyl-4-oxoquinazolin-3(4H)-yl) piperidine-2-6-dione and methods of preparation thereof |
| WO2014066487A3 (en) * | 2012-10-24 | 2014-06-19 | Celgene Corporation | Methods for treating anemia |
| RU2678117C2 (en) * | 2012-11-02 | 2019-01-23 | Селджин Корпорейшн | Activin-actrii antagonists and uses thereof for treating bone and other disorders |
| US10195249B2 (en) | 2012-11-02 | 2019-02-05 | Celgene Corporation | Activin-ActRII antagonists and uses for treating bone and other disorders |
| US11040925B2 (en) | 2013-01-14 | 2021-06-22 | Deuterx, Llc | 3-(5-substituted-4-oxoquinazolin-3(4H)-yl)-3-deutero-piperidine-2,6-dione derivatives and compositions comprising and methods of using the same |
| US11820720B2 (en) | 2013-01-14 | 2023-11-21 | Salarius Pharmaceuticals, Inc. | 3-(5-substituted-4-oxoquinazolin-3(4H)-yl)-3-deuteropiperidine-2,6-dione derivatives and compositions comprising and methods of using the same |
| US10202316B2 (en) | 2013-01-14 | 2019-02-12 | Deuterx, Llc | 3-(5-substituted-4-oxoquinazolin-3(4H)-yl)-3-deutero-piperidine-2,6-dione derivatives and compositions comprising and methods of using the same |
| US9540340B2 (en) | 2013-01-14 | 2017-01-10 | Deuterx, Llc | 3-(5-substituted-4-oxoquinazolin-3(4H)-yl)-3-deutero-piperidine-2,6-dione derivatives and compositions comprising and methods of using the same |
| US9969712B2 (en) | 2013-01-22 | 2018-05-15 | Celgene Corporation | Process for the preparation of isotopologues of 3-(4-((4-(morpholinomethyl)benzyl)oxy)-1-oxoisoindolin-2-yl)piperidine-2,6-dione and pharmaceutically acceptable salts thereof |
| US9695145B2 (en) | 2013-01-22 | 2017-07-04 | Celgene Corporation | Processes for the preparation of isotopologues of 3-(4-((4- morpholinomethyl)benzyl)oxy)-1-oxoisoindolin-2-yl)piperidine-2,6-dione and pharmaceutically acceptable salts thereof |
| US9913845B2 (en) | 2013-03-14 | 2018-03-13 | Deuterx, Llc | 3-(substituted-4-oxoquinazolin-3(4H)-yl)-3-deutero-piperidine-2,6-dione derivatives and compositions comprising and methods of using the same |
| US11491156B2 (en) | 2013-03-14 | 2022-11-08 | Deuterx, Llc | 3-(substituted-4-oxoquinazolin-3(4H)-yl)-3-deutero-piperidine-2,6-dione derivatives and compositions comprising and methods of using the same |
| WO2014152833A1 (en) * | 2013-03-14 | 2014-09-25 | Deuterx, Llc | 3-(substituted-4-oxo-quinazolin-3(4h)-yl)-3-deutero-piperidine-2,6-dione derivatives |
| US10555946B2 (en) | 2013-03-14 | 2020-02-11 | Deuterx, Llc | 3-(substituted-4-oxoquinazolin-3(4H)-yl)-3-deutero-piperidine-2,6-dione derivatives and compositions comprising and methods of using the same |
| US9290475B2 (en) | 2013-03-14 | 2016-03-22 | Deuterx, Llc | 3-(substituted-4-oxoquinazolin-3(4H)-yl)-3-deutero-piperidine-2,6-dione derivatives and compositions comprising and methods of using the same |
| CN106659769A (en) * | 2014-03-21 | 2017-05-10 | 阿塞勒隆制药公司 | Methods for increasing red blood cell levels and treating ineffective erythropoiesis by inhibiting activin B and/or GDF11 |
| US20160046690A1 (en) * | 2014-03-21 | 2016-02-18 | Acceleron Pharma, Inc. | Methods for increasing red blood cell levels and treating ineffective erythropoiesis |
| WO2015143403A1 (en) * | 2014-03-21 | 2015-09-24 | Acceleron Pharma, Inc. | Methods for increasing red blood cell levels and treating ineffective erythropoiesis by inhibiting activin b and/or gdf11 |
| US10487144B2 (en) | 2014-06-13 | 2019-11-26 | Acceleron Pharma Inc. | Methods for treating ulcers in a hemoglobinopathy anemia with a soluble actRIIB polypeptide |
| AU2015274277B2 (en) * | 2014-06-13 | 2021-03-18 | Acceleron Pharma, Inc. | Methods and compositions for treating ulcers |
| WO2015192111A1 (en) * | 2014-06-13 | 2015-12-17 | Acceleron Pharma, Inc. | Methods and compositions for treating ulcers |
| CN114699529A (en) * | 2014-06-13 | 2022-07-05 | 阿塞勒隆制药公司 | Methods and compositions for treating ulcers |
| CN107135646B (en) * | 2014-06-13 | 2022-03-15 | 阿塞勒隆制药公司 | Methods and compositions for treating ulcers |
| US11260107B2 (en) | 2014-06-13 | 2022-03-01 | Acceleron Pharma Inc. | Methods and compositions for treating ulcers |
| CN107135646A (en) * | 2014-06-13 | 2017-09-05 | 阿塞勒隆制药公司 | Methods and compositions for treating ulcers |
| US9850298B2 (en) | 2014-06-13 | 2017-12-26 | Acceleron Pharma Inc. | Methods for treating ulcers in thalassemia syndrome with an ActRIIB polypeptide |
| US11813308B2 (en) | 2014-10-09 | 2023-11-14 | Celgene Corporation | Treatment of cardiovascular disease using ActRII ligand traps |
| US11471510B2 (en) | 2014-12-03 | 2022-10-18 | Celgene Corporation | Activin-ActRII antagonists and uses for treating anemia |
| US10227393B2 (en) | 2015-04-06 | 2019-03-12 | Acceleron Pharma Inc. | TGF-beta superfamily type I and type II receptor heteromultimers and uses thereof |
| US10906958B2 (en) | 2015-04-06 | 2021-02-02 | Acceleron Pharma Inc. | TGF-beta superfamily type I and type II receptor heteromultimers and uses thereof |
| CN107709358A (en) * | 2015-04-06 | 2018-02-16 | 阿塞勒隆制药公司 | ALK7: ACTRIIB heteromultimer and its use |
| US11028145B2 (en) | 2015-04-06 | 2021-06-08 | Acceleron Pharma Inc. | ALK7:actriib heteromultimers and uses thereof |
| US11827689B2 (en) | 2015-04-06 | 2023-11-28 | Acceleron Pharma Inc. | ALK7:ActRIIB heteromultimers and uses thereof |
| WO2016164503A1 (en) * | 2015-04-06 | 2016-10-13 | Acceleron Pharma Inc. | Alk7:actriib heteromultimers and uses thereof |
| AU2016246708B2 (en) * | 2015-04-06 | 2020-12-24 | Acceleron Pharma Inc. | ALK7:actRIIB heteromultimers and uses thereof |
| US10227392B2 (en) | 2015-04-06 | 2019-03-12 | Acceleron Pharma Inc. | ALK7:ActRIIB heteromultimers and uses thereof |
| US10913782B2 (en) | 2015-04-22 | 2021-02-09 | Biogen Ma Inc. | Hybrid ActRIIB ligand trap proteins for treating muscle wasting diseases |
| US11292826B2 (en) | 2015-04-22 | 2022-04-05 | Biogen Ma Inc. | Hybrid ActRIIB ligand trap proteins for treating muscle wasting diseases |
| US11932677B2 (en) | 2015-04-22 | 2024-03-19 | Alivegen Inc. | Hybrid ActRIIB ligand trap proteins for treating muscle wasting diseases |
| US9809603B1 (en) | 2015-08-18 | 2017-11-07 | Deuterx, Llc | Deuterium-enriched isoindolinonyl-piperidinonyl conjugates and oxoquinazolin-3(4H)-yl-piperidinonyl conjugates and methods of treating medical disorders using same |
| WO2017079591A3 (en) * | 2015-11-04 | 2017-09-21 | Acceleron Pharma Inc. | Methods for increasing red blood cell levels and treating ineffective erythropoiesis |
| US11123430B2 (en) | 2015-11-04 | 2021-09-21 | Acceleron Pharma Inc. | Methods for increasing red blood cell levels and treating ineffective erythropoiesis |
| JP7058606B2 (en) | 2016-02-22 | 2022-04-22 | アクセルロン ファーマ インコーポレイテッド | ACTRII antagonist for use in increased immune activity |
| WO2017147182A1 (en) * | 2016-02-22 | 2017-08-31 | Acceleron Pharma Inc. | Actrii antagonists for use in increasing immune activity |
| JP2019510001A (en) * | 2016-02-22 | 2019-04-11 | アクセルロン ファーマ, インコーポレイテッド | ACTRII antagonists for use in increased immune activity |
| AU2017302282B2 (en) * | 2016-07-27 | 2024-07-25 | Acceleron Pharma Inc. | Methods and compositions for treating myelofibrosis |
| US12194076B2 (en) | 2016-07-27 | 2025-01-14 | Acceleron Pharma Inc. | Methods and compositions for treating myelofibrosis |
| CN109789184A (en) * | 2016-07-27 | 2019-05-21 | 阿塞勒隆制药公司 | For treating the method and composition of myelofibrosis |
| EP3490582A4 (en) * | 2016-07-27 | 2020-04-01 | Acceleron Pharma Inc. | DISEASE TREATMENT METHODS AND COMPOSITIONS. |
| US12240887B2 (en) | 2016-10-05 | 2025-03-04 | Acceleron Pharma Inc. | Variant ActRIIB proteins and uses thereof |
| US11267865B2 (en) | 2016-10-05 | 2022-03-08 | Acceleron Pharma Inc. | Variant ActRIIB proteins and uses thereof |
| US12187783B2 (en) | 2016-10-20 | 2025-01-07 | Alivegen Inc | Methods for treating muscle wasting and bone disease using novel hybrid ActRIIB ligand trap proteins |
| US12350313B2 (en) | 2016-11-10 | 2025-07-08 | Keros Therapeutics, Inc. | Activin receptor type IIa variants and methods of use thereof |
| US12269858B2 (en) | 2018-01-12 | 2025-04-08 | Keros Therapeutics, Inc. | Activin receptor type IIB variants and methods of use thereof |
| US20210052698A1 (en) * | 2018-05-09 | 2021-02-25 | Keros Therapeutics, Inc. | Activin receptor type iia variants and methods of use thereof |
| US12364737B2 (en) * | 2018-05-09 | 2025-07-22 | Keros Therapeutics, Inc. | Activin receptor type IIA variants and methods of use thereof |
| US10844039B2 (en) | 2018-11-13 | 2020-11-24 | Biotheryx, Inc. | Substituted isoindolinones |
| US11352338B2 (en) | 2018-11-13 | 2022-06-07 | Biotheryx, Inc. | Substituted isoindolinones |
| US12440539B2 (en) | 2020-03-20 | 2025-10-14 | Keros Therapeutics, Inc. | Methods of using activin receptor type IIB variants |
| US12522646B2 (en) | 2020-03-20 | 2026-01-13 | Keros Therapeutics, Inc. | Activin receptor type II chimeras and methods of use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2012027065A3 (en) | 2012-05-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2012027065A2 (en) | Combination therapy for treatment of disease | |
| JP7146850B2 (en) | Biomarkers for use in treating anemia | |
| JP6643432B2 (en) | Activin-ActRIIa antagonists and their use for increasing red blood cell levels | |
| JP5865281B2 (en) | Activin-ActRII antagonist and its use to increase red blood cell levels | |
| TWI472336B (en) | Use GDF traps to increase red blood cell levels | |
| AU2023219885A1 (en) | Methods for increasing red blood cell levels and treating ineffective erythropoiesis by inhibiting activin B and/or GDF11 | |
| JP2020097629A (en) | Methods and compositions for treating ulcers | |
| JP2020183390A (en) | How to treat anemia | |
| EP3415161A1 (en) | The use of an actriib antagonist for the treatment of anemia | |
| BR112016024319B1 (en) | USE OF A COMPOSITION COMPRISING AN ActRII ANTAGONIST FOR THE MANUFACTURING OF A MEDICATION FOR TREATING OR PREVENTING A COMPLICATION OF SICKLE CELL ANEMIA | |
| JP2018501307A (en) | Activin-ActRII antagonist and use for treating anemia | |
| AU2016250354B2 (en) | Activin-ActRII antagonists and uses for increasing red blood cell levels |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11820341 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11820341 Country of ref document: EP Kind code of ref document: A2 |