WO2011056542A1 - Cancer therapy with combinations of fts with hdac inhibitors - Google Patents
Cancer therapy with combinations of fts with hdac inhibitors Download PDFInfo
- Publication number
- WO2011056542A1 WO2011056542A1 PCT/US2010/054042 US2010054042W WO2011056542A1 WO 2011056542 A1 WO2011056542 A1 WO 2011056542A1 US 2010054042 W US2010054042 W US 2010054042W WO 2011056542 A1 WO2011056542 A1 WO 2011056542A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cancer
- fts
- ras
- hdac inhibitor
- cells
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/164—Amides, e.g. hydroxamic acids of a carboxylic acid with an aminoalcohol, e.g. ceramides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/165—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/165—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide
- A61K31/166—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide having the carbon of a carboxamide group directly attached to the aromatic ring, e.g. procainamide, procarbazine, metoclopramide, labetalol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/165—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide
- A61K31/167—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide having the nitrogen of a carboxamide group directly attached to the aromatic ring, e.g. lidocaine, paracetamol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/17—Amides, e.g. hydroxamic acids having the group >N—C(O)—N< or >N—C(S)—N<, e.g. urea, thiourea, carmustine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/18—Sulfonamides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/192—Carboxylic acids, e.g. valproic acid having aromatic groups, e.g. sulindac, 2-aryl-propionic acids, ethacrynic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/21—Esters, e.g. nitroglycerine, selenocyanates
- A61K31/215—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids
- A61K31/22—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids of acyclic acids, e.g. pravastatin
- A61K31/222—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids of acyclic acids, e.g. pravastatin with compounds having aromatic groups, e.g. dipivefrine, ibopamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
- A61K31/4045—Indole-alkylamines; Amides thereof, e.g. serotonin, melatonin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4184—1,3-Diazoles condensed with carbocyclic rings, e.g. benzimidazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4406—Non condensed pyridines; Hydrogenated derivatives thereof only substituted in position 3, e.g. zimeldine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/60—Salicylic acid; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/60—Salicylic acid; Derivatives thereof
- A61K31/609—Amides, e.g. salicylamide
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- Ras proteins are central to the control of growth in both normal cells and cancerous or malignant cells alike. They are known to be mutated in approximately 30% of all human cancers . In order to exert their activity, normal Ras proteins must undergo at least three changes, the first of which is a chemical modification that entails attachment of a prenyl group. When that prenyl group is the triterpenoid farnesyl group, the attachment step is known as farnesylation . This reaction is catalyzed by an enzyme known as farnesyl transferase. Normal farnesylated Ras then undergoes additional enzyme-catalyzed cleavage of several amino acid residues, a step that is promoted by its interaction with growth factor receptors. Then, the Ras proteins must become activated, which entails docking or anchoring to galectin which is a protein located on the inner surface of the cell membrane.
- cell division is controlled in part by the amount of activated farnesylated Ras attached to the cell membrane, which is in balance with inactive farnesylated Ras in the cytosol.
- that balance is abnormal and is shifted toward greater amounts of membrane-bound Ras, especially mutated Ras, which remains anchored to the cell membrane. This imbalance results in uncontrolled cell division that is characteristic of this disease .
- Ras proteins Activation of Ras proteins and the ensuing downstream events have been extensively researched, particularly in the context of cancer.
- inhibitors of Ras proteins have been developed with the goal of suppressing Ras activity.
- farnesyl transferase inhibitors growth factor receptor inhibitors which hinder the maturation of farnesylated Ras
- Ras antagonists that target the anchoring of farnesylated Ras to galectin have all been the subject of human clinical trials.
- the Ras antagonist S-trans trans- farnesylthiosalicylic acid (FTS) has proven effective in clinical trials to date in connection with several cancers.
- FTS trans- farnesylthiosalicylic acid
- a first aspect of the present invention is directed to a composition that includes S-trans , trans- farnesylthiosalicylic acid (also referred to herein as FTS or Salirasib) or an FTS analog, which together are defined by the formula described herein, an inhibitor of a histone deacetylase enzyme (referred to herein as an HDAC inhibitor) , and a pharmaceutically acceptable carrier.
- FTS and the HDAC inhibitor are present in the composition in effective amounts.
- a second aspect of the present invention is directed to a method of treating a cancer patient that includes co ⁇ administration of FTS or an FTS analog, and the HDAC inhibitor.
- the treatment is carried out by administration of these anti-cancer agents in a single composition .
- Fig. 1 is a bar graph that shows growth inhibition of three different cancer cell lines, namely human A549 cells (non-small cell lung cancer cells), DLD1 cells (human colon adenocarcinoma cells) and ARO cells (thyroid carcinoma cells), in the presence of 0.1% Me 2 S0 4 (control) or the indicated concentrations of FTS, and the HDAC inhibitor valproic acid (VPA) or a combination of the two inhibitors, wherein the number of viable cells in the treated cultures is expressed as a percentage of the total viable number of cells counted in the control .
- human A549 cells non-small cell lung cancer cells
- DLD1 cells human colon adenocarcinoma cells
- ARO cells thyroid carcinoma cells
- Figs. 2A and B are graphs that show synergistic growth inhibitory effect of VPA plus FTS on A549 and DLDl cells which had been incubated with FTS and VPA for 16 days, and wherein cells were counted at days 7, 12 and 16 , and wherein the number of cells that are in S-Phase of mitosis in the treated cultures was determined by FACS analysis and is expressed as a percentage of the control number of S-phase cells .
- Fig. 2C is a bar graph showing, via FACS analysis, that VPA plus FTS achieved a synergistic growth inhibition of A549, DLDl and ARO cells in the S-phase (after 72 hrs) .
- Figs. 3A and B show that combined treatment of FTS and VPA inhibits signaling in A549 and DLDl cells following treatment with the indicated concentrations of FTS and VPA for 72h, wherein Fig. 3A shows photos of electrophoretic gels showing total levels of Cyclin Dl, Ras, Survivin and ⁇ -Tubulin after lysing the cells and then immunoblotting, and wherein Fig. 3B is a bar graph showing real-time PCR analysis of survivin transcripts in control A549 cells and in FTS, VPA and VPA plus FTS A549-treated cells, wherein data are expressed as normalized survivin transcripts in drug-treated cells relative to the normalized control levels in percentage.
- Fig. 4 is a bar graph that shows that the combination of VPA and FTS synergistically inhibits aurora kinase A transcription and interferes with mitosis in A549 cells, via real-time PCR analysis of aurora A (Aurk-A) transcripts in control, FTS, VPA and VPA plus FTS A549 treated cells (72h) , wherein data are expressed as percentage of normalized aurora A transcripts in drug-treated cells relative to the normalized control levels .
- Fig. 4 is a bar graph that shows that the combination of VPA and FTS synergistically inhibits aurora kinase A transcription and interferes with mitosis in A549 cells, via real-time PCR analysis of aurora A (Aurk-A) transcripts in control, FTS, VPA and VPA plus FTS A549 treated cells (72h) , wherein data are expressed as percentage of normalized aurora A transcripts in drug-treated cells relative to the normalized control levels .
- FIG. 5 is a bar graph that shows real-time PCR analysis of NuSAP transcripts in control FTS, VPA and VPA-plus- FTS A549-treated cells (72h) , wherein the level of NuSAP transcripts was normalized to the expression of the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene, and wherein data are expressed as normalized AurK-A transcripts in drug-treated cells relative to the normalized control levels in percentage .
- GPDH glyceraldehyde-3-phosphate dehydrogenase
- Figs. 6A and B are graphs showing the synergistic growth inhibitory effect of SAHA plus FTS on A549 and SW-480 cells respectively, at the indicated concentrations, for 18 days, wherein all cells were counted at days 7, 14 and 18.
- Ras proteins e.g., H-, N- and K- ras, act as on-off switches that regulate signal-transduction pathways controlling cell growth, differentiation, and survival.
- They are anchored to the inner leaflet of the plasma membrane, where activation of cell-surface receptors, such as receptor tyrosine kinase, induces the exchange of guanosine diphosphate (GDP) for guanosine triphosphate (GTP) on Ras and the conversion of inactive Ras-GDP to active Ras-GTP.
- GDP guanosine diphosphate
- GTP guanosine triphosphate
- the active Ras protein promotes oncogenesis through activation of multiple Ras effectors that contribute to deregulated cell growth, differentiation, and increased survival, migration and invasion.
- Ras effectors that contribute to deregulated cell growth, differentiation, and increased survival, migration and invasion.
- FTS is known as a Ras inhibitor that acts in a rather specific manner on the active, GTP-bound forms of H-, N-, and K- Ras proteins.
- Ras inhibitor that acts in a rather specific manner on the active, GTP-bound forms of H-, N-, and K- Ras proteins.
- FTS competes with Ras-GTP for binding to specific saturable binding sites in the plasma membrane, resulting in mislocalization of active Ras and facilitating Ras degradation.
- Ras antaqonists useful in the present invention include FTS and its structural analoqs, are described below.
- R represents farnesyl, or qeranyl- qeranyl
- R 2 is COOR 7 , CONR 7 R 8 , or COOCHR 9 OR 10
- R 7 and R 8 are each independently hydroqen, alkyl, or alkenyl, includinq linear and branched alkyl or alkenyl, which in some embodiments includes C1-C4 alkyl or alkenyl
- R 9 represents H or alkyl
- R 10 represents alkyl, includinq linear and branched alkyl and which in some embodiments represents C1-C4 alkyl
- R 3 , R 4 , R 5 and R 6 are each independently hydroqen, alkyl, alkenyl, alkoxy (includinq linear and branched alkyl, alkenyl or alkoxy and which in some embodiments are Cl- C4 alkyl, alkenyl or alkoxy) , halo, trifluoromethyl, trifluorome
- any of R 7 , R 8 , R 9 and/or R 10 represents alkyl, it is preferably a methyl or ethyl qroup .
- the Ras antaqonists may be present in the form of a pharmaceutically acceptable salt, or any other form in which it is therapeutically effective.
- the Ras antaqonist is S- trans , trans-farnesylthiosalicylic acid or FTS (wherein R 1 is farnesyl, R 2 is COOR 7 , and R 7 is hydroqen) .
- the FTS analog is halogenated, e.g., 5-chloro-FTS (wherein R 1 is farnesyl, R 2 is COOR 7 , R 4 is chloro, and R 7 is hydrogen) , and 5-fluoro-FTS (wherein R 1 is farnesyl, R 2 is COOR 7 , R 4 is fluoro, and R 7 is hydrogen) .
- the FTS analog is FTS-methyl ester (wherein R 1 represents farnesyl, R 2 represents COOR 7 , and R 7 represents methyl) , FTS-amide (wherein R 1 represents farnesyl, R 2 represents CONR 7 R 8 , and R 7 and R 8 both represent hydrogen) ; FTS-methylamide (wherein R 1 represents farnesyl, R 2 represents CONR 7 R 8 , R 7 represents hydrogen and R 8 represents methyl); and FTS-dimethylamide (wherein R 1 represents farnesyl, R 2 represents CONR 7 R 8 , and R 7 and R 8 each represents methyl) .
- the Ras antagonist is an alkoxyalkyl S-prenylthiosalicylate or an FTS-alkoxyalkyl ester (wherein R 2 represents COOCHR 9 OR 10 ) .
- Representative examples include methoxymethyl S-farnesylthiosalicylate (wherein R 1 is farnesyl, R 9 is H, and R 10 is methyl); methoxymethyl S- geranylgeranylthiosalicylate (wherein R 1 is geranylgeranyl, R 9 is H, and R 10 is methyl) ; methoxymethyl 5-fluoro-S- farnesylthiosalicylate (wherein R 1 is farnesyl, R 5 is fluoro, R 9 is H, and R 10 is methyl) ; and ethoxymethyl S- farnesylthiosalicyate (wherein R 1 is farnesyl, R 9 is methyl and R 10 is ethyl) .
- each of R 3 , R 4 , R 5 and R 6 represents hydrogen.
- Histones are small proteins that complex with DNA.
- Two of each of the histones known as H2A, H2B, H3 and H4 are tightly complexed with DNA, typically in amounts of about 150 base pairs, to form a nucleosome. This structure is further connected by linker DNA to form a solenoid.
- Histone acetyltransferases (HATs) and histone deacetylases (HDACs) represent two enzyme families that control the level of histone tail acetylation by the addition and removal respectively, of an acetyl group from the lysine residues of core nucleosomal histories.
- Histones extending from the nucleosomal core are thus enzymatically modified, affecting chromatin structure and gene expression.
- HATs via acetylation of histones, allow transcription and gene expression since acetylated histones can recruit transcription factors and other co-activator proteins.
- HDACs are usually associated with DNA hyper-methylation and gene silencing.
- HDACs play an important role in cell proliferation and differentiation. Acetylated histones are relatively less effective in facilitating DNA transcription, whereas deacetylated histones are relatively more effective in facilitating this process. Acetylation and deacetylation are in balance in normal cells. However, in malignant cells, there is an excess of HDAC activity compared to HAT activity, resulting in an imbalance of deacetylated histones to acetylated histones. This imbalance results in excessive DNA transcription and uncontrolled cell proliferation. The involvement of HDACs in the control of cell proliferation and differentiation suggests that aberrant HDAC activity may play a role in cancer .
- the HDAC enzyme family includes at least 18 enzymes, grouped in four (4) classes (Classes I, Ila, lib, III and IV) .
- Class I HDACs include HDACs 1, 2, 3, and 8.
- Class I HDACs can be found in the nucleus and are believed to be involved with transcriptional control repressors and co-factors.
- Class Ila HDACs include HDACS 4, 5, 7 and 9, and
- Class lib HDACs include HDACs 6 and 10. These enzymes can be found in both the cytoplasm as well as the nucleus.
- Certain class I and class II HDACs are overexpressed in tumors relative to normal tissues. See, Johnstone, Nature Reviews Drug Disovery 1:287-99 (2002) .
- Class III HDACs are believed to be NAD-dependent proteins and include members of the sirtuin family of proteins .
- Non-limiting examples of sirtuin proteins include SIRT1-7.
- Class IV HDACs include HDAC 11.
- HDAC inhibitor refers to a compound that has the ability to inhibit histone deacetylase activity. This therapeutic class is able to block angiogenesis and cell cycling, and promote apoptosis and differentiation. HDAC inhibitors exhibit targeted anticancer activity and as disclosed herein, act synergistically with FTS and its analogs in the treatment of cancer.
- Selective and non-selective HDAC inhibitors alike may be useful in the present invention.
- the term "selective HDAC inhibitor” refers to an HDAC inhibitor that does not significantly interact with all three HDAC classes.
- a "Class I selective HDAC” refers to an HDAC inhibitor that interacts with one or more of HDACs 1, 2, 3 or 8, but does not significantly interact with the Class II HDACs (i.e., HDACs 4-7, 9 and 10) .
- the HDAC inhibitor is selective HDAC inhibitor, such as a Class I selective HDAC inhibitor.
- a Class I selective HDAC inhibitor Representative examples of such inhibitors that may be useful in the present invention include benzamides such as MGCD-0103 (N- (2-amino-phenyl) -4- [ (4-pyridin- 3-yl-pyrimidin-2-ylamino) -methyl] -benzamide) , also known as Mocetinostat , and related compounds as disclosed in U.S.
- Patent 6, 897, 220 benzamides such as MS-275 ( (N- ( 2-aminophenyl ) -4- (N- (pyridin-3-ylmethoxycarbonyl) aminomethyl) benzamide), also known as entinostat or "SNDX-275”), and related compounds as disclosed in e.g., U.S. Patent 6,174,905), spiruchostatin A, SK7041 and SK7068 (Class I HDAC inhibitors; Park, et al . , Clin. Cancer Res. 10:5271 (2004) ) and 6-amino nicotinamides.
- the HDAC is a non-selective HDAC inhibitor.
- HDAC inhibitors include hydroxamic acids including trichostatin analogs such as trichostatin A (TSA) and trichostatin C (TSC) (Koghe, et al., Biochem. Pharmacol. 55:1359-64 (1998), salicylihydroxamic acid (SBHA) (U.S.
- Patent 5,608,108 azelaic bishydroxamic acid (ABHA) , azelaic-1- hydroxamate-9-analide (AAHA) , 6- (3-chlorophenylureido) carpoic hydroxamic acid (3C1-UCHA) , and ' -hydroxy-N-phenyl- octanediamide (suberoylanilide hydroxamic acid, also known as "SAHA” or vorinostat, and related hydroxamic acid compounds as disclosed in U.S.
- TPX trapoxin
- CBHA m- carboxycinnamic acid bishydroxamide
- CBHA Richon, et al . , PNAS 95:3003-7 (1998)
- oxamflatin A-161906, GCK1026
- the non-selective HDAC inhibitor is a small-molecular weight carboxylate such as valproic acid (2-n-propylpentanoic acid, VPA) or a derivative thereof.
- VPA has been reported to inhibit HDACs 1-3 (Class I) and HDACs -8 (Class II) .
- Valproic acid is represented by the
- VPA derivatives suitable for use in the present invention are represented by the formula
- R 1 and R 2 each independently represents a linear or branched, saturated or unsaturated aliphatic 2 -25r preferably C3-25 hydrocarbon chain which optionally comprises one or several heteroatoms and which may be substituted, R 3 is hydroxyl, alkoxy or an optionally alkylated amino group.
- the hydrocarbon chains R 1 and R 2 may contain one or several heteroatoms (e.g., 0, N, S) replacing carbon atoms in the hydrocarbon chain.
- R 1 and R 2 may be substituted, e.g., with hydroxyl, amino, carboxylic and alkoxy groups as well as aryl and heterocyclic groups .
- R 1 and R 2 independently contain 2 to 10, more preferably 3 to 10 or 5 to 10 carbon atoms. It is also preferred that R 1 and R 2 independently are saturated or contain one double bond or one triple bond.
- VPA derivatives that may be particularly suitable include S-4-yn VPA and 2-EHXA (2- Ethyl-hexanoic acid) .
- the HDAC inhibitor may be present in the form of a pharmaceutically acceptable salt, or any other form in which it is therapeutically effective.
- HDAC inhibitors are now in use in clinical cancer trials. Aside from various HDAC inhibitors mentioned above, they include Panobinostat , Belinostat, ITF-2357, PC-24781, phenylbutyrate, SB-939 and JHJ-26481585.
- the Ras antagonist and the HDAC inhibitor are co ⁇ administered, which as used herein, encompasses treatment regimens in which these anti-cancer agents are administered to the cancer patient at the same or different times (i.e., substantially simultaneously or sequentially) , and by the same or different route of administration, such that both agents and/or their metabolites are present in the patient at the same time in order to achieve the benefits of their combined therapeutic effect.
- Co-administration thus includes simultaneous administration in separate compositions, administration at different times in separate compositions, and/or administration in a composition that contains both agents.
- the Ras antagonist and the HDAC inhibitor are administered in a single composition.
- the term "therapeutically effective amounts”, as used herein, refers to a sufficient amount of each of the Ras antagonist and the HDAC inhibitor that will ameliorate at least one symptom of the cancer and its associated manifestations, diminish extent or severity of the disease, delay or retard disease progression, achieve partial or complete remission, prolong survival and combinations thereof.
- combinations of the Ras antagonist and the HDAC inhibitor achieve synergy, i.e., a greater than additive effect, at least with respect to inhibition of the growth of the cancer cells in vitro. Applicants believe that these results reflect tumor/cancer cell growth inhibitory activity in vivo, and ultimately result in more effective cancer therapy and a commensurate improvement in one or more of these clinical manifestations of the disease.
- Appropriate "effective" amounts for any cancer patient can be determined using techniques, such as a dose escalation study. Specific dose levels for any particular patient will depend on several factors such as the potency of the Ras antagonist and the HDAC inhibitor, the age, weight, and general health of the patient, and the severity of the cancer.
- the average daily dose of the Ras antagonists of the present invention generally ranges from about 200 mg to about 2000 mg (e.g., 200 mg, 400 mg, 600 mg, 800 mg, 1000 mg, 1200 mg, 1400 mg, 1600 mg, 1800 mg and 2000 mg) , in some embodiments from about 400 to about 1600 mg, and in some other embodiments from about 600 to about 1200 mg, and in yet other embodiments, from about 800 mg to about 1200 mg .
- the average daily dose of the HDAC inhibitor will vary, depending on the specific agent.
- the average daily dose for valproic acid (and its derivatives) for example, generally ranges from about 500 mg to about 3500 mg, in some embodiments from about 750 to 3000 mg, and in some other embodiments from about 750 mg to about 1500 mg .
- the average daily dose for vorinostat (and its derivatives), for example, generally ranges from about 100 mg to about 600 mg, in some other embodiments from about 200 to about 500 mg, and in some other embodiments from about 300 mg to about 400 mg .
- the average weekly dose for SNDX-275 (and its derivatives) for example, generally ranges from about 2.5 mg to about 10 mg and in some embodiments from about 5 mg to about 10 mg, once every two weeks .
- administration, " and the like, as used herein, refer to the methods that may be used to enable delivery of compounds or compositions to the desired site of biological action.
- Medically acceptable administration techniques suitable for use in the present invention are known in the art. See, e.g., Goodman and Gilman, The Pharmacological Basis of Therapeutics, current ed . ; Pergamon; and Remington's, Pharmaceutical Sciences (current edition), Mack Publishing Co., Easton, Pa.
- at least one or both active agents are administered orally.
- at least one or both active agents are administered parenterally (which for purposes of the present invention, includes intravenous, subcutaneous, intraperitoneal, intramuscular, intravascular and infusion) .
- Other administration routes such as topical and rectal administration may also be suitable.
- pharmaceutically acceptable refers to a material, such as a carrier and other non-active excipients, which does not abrogate the biological activity or properties of the active agent (s), and is relatively nontoxic.
- composition refers to the Ras antagonist and/or the HDAC inhibitor, optionally combined (e.g., mixed) with a pharmaceutically acceptable carrier. These ingredients are non-toxic, physiologically inert and do not adversely interact with the active agent (s) present in the composition. Carriers facilitate formulation and/or administration of the active agents. Pharmaceutical compositions of the present invention may further contain one or more excipients.
- compositions for the Ras antagonist and/or the HDAC inhibitor can be prepared by bringing the agent (s) into association with (e.g., mixing with) the carrier, the selection of which is based on the mode of administration.
- Carriers are generally solid or liquid. In some cases, compositions may contain solid and liquid carriers.
- Compositions suitable for oral administration that contain the active are preferably in solid dosage forms such as tablets (e.g., including film-coated, sugar-coated, controlled or sustained release) , capsules, e.g., hard gelatin capsules (including controlled or sustained release) and soft gelatin capsules, powders and granules.
- compositions may be contained in other carriers that enable administration to a patient in other oral forms, e.g., a liquid or gel. Regardless of the form, the composition is divided into individual or combined doses containing predetermined quantities of the active ingredient or ingredient s .
- Oral dosage forms may be prepared by mixing the active pharmaceutical ingredient or ingredients with one or more appropriate carriers (optionally with one or more other pharmaceutically acceptable excipients), and then formulating the composition into the desired dosage form e.g., compressing the composition into a tablet or filling the composition into a capsule or a pouch.
- Typical carriers and excipients include bulking agents or diluents, binders, buffers or pH adjusting agents, disintegrants (including crosslinked and super disintegrants such as croscarmellose) , glidants, and/or lubricants, including lactose, starch, mannitol, microcrystalline cellulose, ethylcellulose , sodium carboxymethylcellulose , hydroxypropylmethylcellulose, dibasic calcium phosphate, acacia, gelatin, stearic acid, magnesium stearate, corn oil, vegetable oils, and polyethylene glycols.
- Coating agents such as sugar, shellac, and synthetic polymers may be employed, as well as colorants and preservatives. See, Remington' s Pharmaceutical Sciences, The Science and Practice of Pharmacy, 20th Edition (2000) .
- Liquid form compositions include, for example, solutions, suspensions, emulsions, syrups, elixirs and pressurized compositions.
- the active agent (s) for example, can be dissolved or suspended in a pharmaceutically acceptable liquid carrier such as water, an organic solvent (and mixtures thereof), and/or pharmaceutically acceptable oils or fats.
- liquid carriers for oral administration include water (particularly containing additives as above, e.g., cellulose derivatives, preferably in suspension in sodium carboxymethyl cellulose solution) , alcohols (including monohydric alcohols (including monohydric alcohols and polyhydric alcohols, e.g., glycerin and non-toxic glycols) and their derivatives, and oils (e.g., fractionated coconut oil and arachis oil) .
- the liquid composition can contain other suitable pharmaceutical additives such as solubilizers, emulsifiers, buffers, preservatives, sweeteners, flavoring agents, suspending agents, thickening agents, colorants, viscosity regulators, stabilizers or osmoregulators .
- Carriers suitable for preparation of compositions for parenteral administration include Sterile Water for Injection, Bacteriostatic Water for Injection, Sodium Chloride Injection (0.45%, 0.9%), Dextrose Injection (2.5%, 5%, 10%), Lactated Ringer's Injection, and the like. Dispersions can also be prepared in glycerol, liquid polyethylene glycols and mixtures thereof, and in oils.
- Compositions may also contain tonicity agents (e.g., sodium chloride and mannitol) , antioxidants (e.g., sodium bisulfite, sodium metabisulfite and ascorbic acid) and preservatives (e.g., benzyl alcohol, methyl paraben, propyl paraben and combinations of methyl and propyl parabens) .
- tonicity agents e.g., sodium chloride and mannitol
- antioxidants e.g., sodium bisulfite, sodium metabisulfite and ascorbic acid
- preservatives e.g., benzyl alcohol, methyl paraben, propyl paraben and combinations of methyl and propyl parabens
- the pharmaceutical composition containing the Ras antagonist and the HDAC inhibitor, or first and second compositions containing the Ras antagonist and the HDAC inhibitor respectively, may be packaged and sold in the form of a kit.
- the composition might be in the form of one or more oral dosage forms such as tablets or capsules (e.g., hard or soft gelatin capsules) containing one or both of the active agents.
- the kit may also contain written instructions for carrying out the inventive methods as described herein.
- the Ras antagonist is administered by dosing orally on a daily basis (in single or divided doses) for three weeks, followed by a one-week "off period", and repeating until remission is achieved.
- the HDAC inhibitor may be present in the same composition.
- VPA for example, is administered daily in single or divided dosages (e.g., 2 or 3 times daily) .
- Vorinostat may be dosed once daily, e.g., at an initial dose of about 400 mg, which then is reduced to a daily dose of 300 mg and then may be continued at 300 mg every 5 consecutive days.
- SNDX may be administered every two weeks in an amount of about 5 or 10 mg .
- Cancer generally refers to a disease caused by the uncontrolled, abnormal growth of cells that can spread to adjoining tissues or other parts of the body.
- Cancer cells can form a solid tumor, in which the cancer cells are massed together, or they can exist as dispersed cells, as in leukemia. Normal cells divide (reproduce) until maturation is attained and then only as necessary for replacement of damaged or dead cells. Cancer cells are often referred to as "malignant", because they divide endlessly, eventually crowding out nearby cells and spreading to other parts of the body. Malignant cancer cells eventually metastasize and spread to other parts of the body via the bloodstream or lymphatic system, where they can multiply and form new tumors. Malignant tumors are divided into carcinomas (which arise from epithelial precursor cells), sarcomas (which arise largely from mesenchymal tissues) and lymphomas (which arise from precursors of red and white blood cells) .
- Cancers characterized by the presence of elevated wild-type Ras or the presence of mutated Ras proteins are amenable to treatment in accordance with the present invention.
- These cancers include human lymphomas, sarcomas and carcinomas, e.g., fibrosarcoma, myxosarcoma, liposarcoma, chondrosarcoma, osteogenic sarcoma, chordoma, angiosarcoma, endotheliosarcoma, lymphangiosarcoma, synovioma, mesothelioma, lymphangioendotheliosarcoma, Ewing's tumor, leiomyosarcoma, rhabdomyosarcoma, colorectal (e.g., colon) carcinoma, gastrointestinal (e.g., stomach) cancer, pancreatic cancer, thyroid cancer (e.g., follicular, papillary and anaplastic thyroid carcinomas), breast cancer, ovarian cancer, prostate cancer,
- the invention is used to treat a patient afflicted with renal carcinoma, skin cancer, pancreatic cancer, colorectal (e.g., colon) cancer, NSCLC, ovarian cancer, hepatic (liver) cancer, thyroid cancer, seminoma, skin cancer, endometrial cancer, melanoma, leukemia, lymphoma, prostate cancer, bladder and urinary cancers, breast cancer, and brain metastases of these primary tumors, primary brain cancers (such as gliomas and neuroblastomas) and head and neck cancers. See, e.g., Bos, Cancer Res. 49:4682-89 (1989).
- the combination of Ras antagonist and HDAC inhibitor may be used alone or in conjunction with other treatment agents such as biological anti-cancer agents (e.g., antibodies), chemotherapy and radiation.
- the combination may be used as a front-line treatment strategy e.g., as a first course of treatment in a newly diagnosed cancer patient, and whether or not the cancer has metastasized.
- the combination may also be used as a second-line treatment strategy e.g., in the treatment of a cancer patient who has been previously treated using at least one other agent but has not responded to the previous agent (s) or has developed a resistance thereto, which may have resulted in termination of the therapy even before it could achieve an appreciable therapeutic efficacy.
- DLD1 and SW-480 which are human colon adenocarcinoma cells that also express oncogenic K-ras, were cultured in DMEM 10% FCS, 2mM L-Glutamine, 100 U/ml penicillin and 100 yg/ml streptomycin .
- ARO, thyroid carcinoma cells wherein wild-type K-Ras is chronically active, and wherein B-Raf is constitutively active, were cultured in RPMI 1640 medium with 10% FCS, 2mM L- Glutamine, 100 U/ml penicillin and 100 yg/ml streptomycin.
- FTS was prepared as previously described in Rotbalt, Meth. Enzymol. 439:467-89 (2008) .
- VPA was stored at 4°C as a powder and before each experiment it was weighed and diluted with cell medium to the desired concentration.
- SAHA was reconstituted in DMSO to a concentration of 5 mM, divided into 10 ⁇ stocks and stored at 4°C. For each experiment, one stock was diluted to the desired concentration using cell medium.
- A549, DLD1, SW-480 and ARO Cells were plated at a density of 8xl0 3 cells per well in 24-well plates. After 24 hours cells were treated with VPA (Sigma), SAHA (Alexis) or FTS dissolved in 0.1% Me 2 S0 4 (Concordia Pharmaceuticals, Ft. Lauderdale, FL) or a combination of FTS with VPA, or a combination of FTS with SAHA, for the indicated periods of time. Control cells were treated with 0.1% Me 2 S0 4 . In the case of incubation times longer than 3 days the cells were detached, counted and re-plated at a density of 3xl0 5 cells per 10-cm plate. Below are the concentrations of VPA, SAHA and FTS that were used in the different cell lines:
- A549, DLD1 and ARO cells (4xl0 5 cells per 10-cm plate) were plated for 24h and then incubated with VPA and FTS for additional 72h in 10% FCS-containing medium. The cells were then collected and resuspended in PBS containing propidium iodide (5C ⁇ g/mL; Sigma), 0.1% sodium citrate and 0.1% Triton x- 100 (BDH, Poole, United Kingdom) and incubated overnight in the dark at 4°C.
- PBS propidium iodide
- BDH Triton x- 100
- A549, DLD1 and ARO cells were plated at density of 4xl0 5 cells per 10-cm plate and grown for 24h. The cells were then treated 0.1% Me 2 S0 4 (control), FTS, VPA, SAHA, a combined treatment of VPA plus FTS, or a combined treatment of FTS plus SAHA, for 72h.
- the cells were lysed in 300 ⁇ 1 homogenization buffer (50 mmol/L Tris-HCl (pH 7.6), 20mM MgCl 2 , 200mM NaCl, 0.5% NP40, ImM DTT, and protease inhibitors) centrifuged for 10 min at 14,000 rpm at 4°C and the supernatant was collected. Equal amounts of proteins (50-100yg per lane) were subjected to SDS-PAGE, followed by immunoblotting with rabbit anti-cyclin Dl (1:1,000), mouse anti-pan-Ras antibody Ab (Calbiochem) , rabbit anti-Survivin Ab (Santa Cruz, CA) and rabbit anti- -tubulin Ab (Sigma) .
- 300 ⁇ 1 homogenization buffer 50 mmol/L Tris-HCl (pH 7.6), 20mM MgCl 2 , 200mM NaCl, 0.5% NP40, ImM DTT, and protease inhibitors
- A549 cells were plated on glass cover slips placed in 10-cm plates at a density of 4xl0 5 cells/plate for 24h before adding 0.1% Me 2 S0 4 , 75 ⁇ FTS or VPA 0.8mM or VPA+FTS for an additional 24 or 72 hours. Afterwards, the cells were fixed with formaldehyde at room temperature for 30 min and then treated with 0.2% Triton X-100.
- phosphate-buffered saline PBS
- slides were immersed in 1% bovine serum albumin (BSA) with 200yg/mL naive goat IgG (Jackson ImmunoResearch) for 30 min and then incubated with rabbit anti-Aurora A (1:50, Cell Signaling, Danvers, MA), anti- Aurora B (1:50, Bethyl Labs Montgomery, TX) or mouse anti- phosphor-H3 (1:50, Upstate, Charlottesville, VA) overnight at 4°C.
- BSA bovine serum albumin
- rabbit anti-Aurora A 1:50, Cell Signaling, Danvers, MA
- anti- Aurora B 1:50, Bethyl Labs Montgomery, TX
- mouse anti- phosphor-H3 1:50, Upstate, Charlottesville, VA
- the cells were incubated with goat anti-mouse Cy3-conjugated Ab or with donkey anti-rabbit Cy2-conjugated Ab (1:200, Jackson ImmunoResearch) for 1 h in the dark. Then the cells were washed twice in PBS, and further incubated with anti-a-tubulin FITC Ab (1:50, sigma, F2168) for lh at room temperature. Finally, the cells were counterstained with Hoechst 33258 (Fluka AG, CH9470), and examined by fluorescent microscopy at 60x magnification with an LSM 510 META microscope.
- Hoechst 33258 Fluka AG, CH9470
- RNA samples were treated for 72 h with either 75 ⁇ FTS, 0.8mM VPA, 0.1% vehicle (control) or 75 ⁇ FTS plus 0.8mM VPA.
- Total RNA was isolated from cultured cells using protocols and reagents contained in the RNeasy Plus Mini Kit (Qiagene) . The concentration of the RNA samples was determined by measuring the absorbance at 260 nm (A260) in a spectrophotometer. Purified RNA was stored at -70°C in RNase- free water. The purified RNA was used for real-time PCR.
- Extracts of total RNA (1 yg) were reverse-transcribed in a total volume of 20yL using the VersoTM RT-PCR Kits (ABgene) .
- lpL cDNA samples were then used for real-time PCR (QPCR SYBR Green Mix Plus ROX Vial, ABgene) .
- the primers used targeted Survivin, AurK-A and NuSap genes, and the housekeeping gene GAPDH. Primer sequences used for these experiments are set forth in the following table.
- VPA and FTS synergistically in inhibit growth of cells with active Ras pathways.
- VPA and FTS act synergistically to inhibit growth of NSCLC cells with active Ras pathways.
- VPA and FTS synergistically down-regulate survivin in A549 and DLD1 cells
- CPC chromosomal passenger complex
- Typical results of these experiments indicated that FTS but not VPA caused a significant reduction in the levels of Ras and cyclin Dl in A549 and DLD1 cells. These results are in line with the known anti-Ras activity of FTS . Erlich, et al . , Biochem. Pharmacol. 72:427-36 (2006); Blum, et al . , Int. J. Cancer 119:527-38 (2006). VPA did not itself affect the levels of Ras and Cyclin Dl (Fig. 3A) . However, the effect of the combined treatment on Ras, Cyclin Dl, and survivin was clearly stronger than the effect of either drug alone (Fig. 3A) .
- the positive control of Ras on survivin expression can be relieved by FTS (or by dominant-negative Ras) as manifested by a decrease in the level of survivin mRNA and protein in FTS- treated cells.
- FTS or by dominant-negative Ras
- the present results show a similar strong reduction in survivin protein level by FTS in A549 and DLD1 (Fig. 3A) . This result was unexpected at least from the standpoint that in addition to the cell growth cycle, the two drugs affected unrelated cellular pathways .
- VPA and FTS synergistically inhibit Aurora kinase A transcription blocking mitosis in A549 cells
- VPA and FTS synergistically block Aurora kinase B expression and histone-H3 phosphorylation
- Aurora B is localized to the kinetechores during metaphase and aberrant aurora B expression coincides with reduced levels of phospho histone- H3 (serlO) which is critical for mitosis. Vader, Biochim. Biophys . Acta. 1786:60-12 (2008) .
- Our previous gene profiling analysis in FTS-treated A549 cells (75 ⁇ FTS for 72 h) (Blum, et al . , Cancer Res. 57:3320-8 (2007)) showed a decrease in aurora B expression.
- Other studies showed that HDAC inhibitors reduce the level of aurora B. Zhang, et al . , Cancer Biol. Ther. 7:1388-97 (2008) .
- nucleolar and spindle associated protein is a novel microtubule- associated protein involved in mitotic spindle organization.
- siRNA to NuSap disrupted formation of a normal spindle Raemaekers, et al., J. Cell. Biol. 152:1017-29 (2003).
- FTS active pharmaceutical ingredient 2.0 kg
- sodium valproate active pharmaceutical ingredient 2.3 kg
- microcrystalline cellulose 2.0 kg
- croscarmellose sodium 0.2 kg
- magnesium stearate 0.1 kg
- FTS active pharmaceutical ingredient (1.0 kg), sodium valproate (2.3 kg), microcrystalline cellulose (2.0 kg), croscarmellose sodium (0.2 kg), and magnesium stearate (0.1 kg) are blended to homogeneity and filled into hard gelatin capsules with an encapsulation machine. Assuming a 5% loss on material transfer and encapsulating machine start-up, adjustment, and shut-down, approximately 9,500 capsules are yielded .
- FTS active pharmaceutical ingredient (2.0 kg), vorinostat active pharmaceutical ingredient (2.0 kg), microcrystalline cellulose (2.0 kg), croscarmellose sodium (0.2 kg), and magnesium stearate (0.1 kg) are blended to homogeneity and filled into hard gelatin capsules with an encapsulation machine. Assuming a 5% loss on material transfer and encapsulating machine start-up, adjustment, and shut-down, approximately 19,000 capsules are yielded.
- FTS active pharmaceutical ingredient (2.0 kg) and VPA active pharmaceutical ingredient (2.0 kg) are dissolved in corn oil (4.0 kg) and stirred to form a homogeneous solution.
- the solution is filled into soft gelatin capsules with a form- fill-seal encapsulation machine. Assuming a 5% loss on material transfers and soft-gelatin encapsulating machine start-up, adjustment, and shut-down, approximately 19,000 capsules containing FTS (100 mg) and VPA (100 mg) are yielded.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Hematology (AREA)
- Engineering & Computer Science (AREA)
- Pain & Pain Management (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Oncology (AREA)
- Emergency Medicine (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Medicinal Preparation (AREA)
Abstract
Disclosed are pharmaceutical compositions containing effective amounts of FTS (S-trans, trans-farnesylthiosalicylic acid or Salirasib) or an FTS analog, an inhibitor of histone deacetylase enzyme (HDAC), and a pharmaceutically acceptable carrier. Also disclosed are methods of treating cancer by co- administration of effective amounts of a Ras antagonist comprising FTS, or an analog thereof, and an HDAC inhibitor to a cancer patient.
Description
CANCER THERAPY WITH COMBINATIONS OF FTS WITH HDAC INHIBITORS
CROSS-REFERENCE TO RELATED APPLICATIONS
[ 0001 ] This application claims the benefit of the filing date of United States Provisional Patent Application No. 61/254,872, filed October 26, 2009, the disclosure of which is hereby incorporated herein by reference.
BACKGROUND OF THE INVENTION
[ 0002 ] Ras proteins are central to the control of growth in both normal cells and cancerous or malignant cells alike. They are known to be mutated in approximately 30% of all human cancers . In order to exert their activity, normal Ras proteins must undergo at least three changes, the first of which is a chemical modification that entails attachment of a prenyl group. When that prenyl group is the triterpenoid farnesyl group, the attachment step is known as farnesylation . This reaction is catalyzed by an enzyme known as farnesyl transferase. Normal farnesylated Ras then undergoes additional enzyme-catalyzed cleavage of several amino acid residues, a step that is promoted by its interaction with growth factor receptors. Then, the Ras proteins must become activated, which entails docking or anchoring to galectin which is a protein located on the inner surface of the cell membrane.
[ 0003 ] In normal non-pathological conditions, cell division is controlled in part by the amount of activated farnesylated Ras attached to the cell membrane, which is in balance with inactive farnesylated Ras in the cytosol. In a cancer cell, that balance is abnormal and is shifted toward greater amounts of membrane-bound Ras, especially mutated Ras, which remains anchored to the cell membrane. This imbalance results in uncontrolled cell division that is characteristic of this disease .
[ 0004 ] Activation of Ras proteins and the ensuing downstream events have been extensively researched, particularly in the context of cancer. As a result, inhibitors of Ras proteins
have been developed with the goal of suppressing Ras activity. For example, farnesyl transferase inhibitors, growth factor receptor inhibitors which hinder the maturation of farnesylated Ras, and Ras antagonists that target the anchoring of farnesylated Ras to galectin have all been the subject of human clinical trials. Notably, the Ras antagonist S-trans , trans- farnesylthiosalicylic acid (FTS) has proven effective in clinical trials to date in connection with several cancers.
[0005] Many effective cancer therapies involve a combination of two different anti-cancer agents. Central to the conception and design of therapy is to elect a combination that achieves an additive and preferably a synergistic therapeutic effect. However, predicting beforehand the therapeutic efficacy of any given combination, and especially whether it would be synergistic, remains elusive.
BRIEF SUMMARY OF THE INVENTION
[0006] A first aspect of the present invention is directed to a composition that includes S-trans , trans- farnesylthiosalicylic acid (also referred to herein as FTS or Salirasib) or an FTS analog, which together are defined by the formula described herein, an inhibitor of a histone deacetylase enzyme (referred to herein as an HDAC inhibitor) , and a pharmaceutically acceptable carrier. FTS and the HDAC inhibitor are present in the composition in effective amounts.
[0007] A second aspect of the present invention is directed to a method of treating a cancer patient that includes co¬ administration of FTS or an FTS analog, and the HDAC inhibitor. In some embodiments, the treatment is carried out by administration of these anti-cancer agents in a single composition .
BRIEF DESCRIPTION OF THE DRAWINGS
[0008] Fig. 1 is a bar graph that shows growth inhibition of three different cancer cell lines, namely human A549 cells (non-small cell lung cancer cells), DLD1 cells (human colon adenocarcinoma cells) and ARO cells (thyroid carcinoma cells), in the presence of 0.1% Me2S04 (control) or the indicated
concentrations of FTS, and the HDAC inhibitor valproic acid (VPA) or a combination of the two inhibitors, wherein the number of viable cells in the treated cultures is expressed as a percentage of the total viable number of cells counted in the control .
[ 0009 ] Figs. 2A and B are graphs that show synergistic growth inhibitory effect of VPA plus FTS on A549 and DLDl cells which had been incubated with FTS and VPA for 16 days, and wherein cells were counted at days 7, 12 and 16 , and wherein the number of cells that are in S-Phase of mitosis in the treated cultures was determined by FACS analysis and is expressed as a percentage of the control number of S-phase cells .
[ 0010 ] Fig. 2C is a bar graph showing, via FACS analysis, that VPA plus FTS achieved a synergistic growth inhibition of A549, DLDl and ARO cells in the S-phase (after 72 hrs) .
[ 0011 ] Figs. 3A and B show that combined treatment of FTS and VPA inhibits signaling in A549 and DLDl cells following treatment with the indicated concentrations of FTS and VPA for 72h, wherein Fig. 3A shows photos of electrophoretic gels showing total levels of Cyclin Dl, Ras, Survivin and β-Tubulin after lysing the cells and then immunoblotting, and wherein Fig. 3B is a bar graph showing real-time PCR analysis of survivin transcripts in control A549 cells and in FTS, VPA and VPA plus FTS A549-treated cells, wherein data are expressed as normalized survivin transcripts in drug-treated cells relative to the normalized control levels in percentage.
[ 0012 ] Fig. 4 is a bar graph that shows that the combination of VPA and FTS synergistically inhibits aurora kinase A transcription and interferes with mitosis in A549 cells, via real-time PCR analysis of aurora A (Aurk-A) transcripts in control, FTS, VPA and VPA plus FTS A549 treated cells (72h) , wherein data are expressed as percentage of normalized aurora A transcripts in drug-treated cells relative to the normalized control levels .
[ 0013 ] Fig. 5 is a bar graph that shows real-time PCR analysis of NuSAP transcripts in control FTS, VPA and VPA-plus- FTS A549-treated cells (72h) , wherein the level of NuSAP transcripts was normalized to the expression of the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene, and wherein data are expressed as normalized AurK-A transcripts in drug-treated cells relative to the normalized control levels in percentage .
[ 0014 ] Figs. 6A and B are graphs showing the synergistic growth inhibitory effect of SAHA plus FTS on A549 and SW-480 cells respectively, at the indicated concentrations, for 18 days, wherein all cells were counted at days 7, 14 and 18.
DETAILED DESCRIPTION
[ 0015 ] The Ras Antagonists
[ 0016 ] Ras proteins e.g., H-, N- and K- ras, act as on-off switches that regulate signal-transduction pathways controlling cell growth, differentiation, and survival. [Reuther, et al . , Curr. Opin. Cell Biol. 12:157-65 (2000)] . They are anchored to the inner leaflet of the plasma membrane, where activation of cell-surface receptors, such as receptor tyrosine kinase, induces the exchange of guanosine diphosphate (GDP) for guanosine triphosphate (GTP) on Ras and the conversion of inactive Ras-GDP to active Ras-GTP. [Scheffzek, et al. Science 277:333-7 (1997)] . Termination of these signals involves hydrolysis of the Ras-GTP to Ras-GDP. [Scheffzek, et al . , Science 277:333-338 (1997) .] Several mutated forms of Ras are defective in their GTP hydrolysis liability and are therefore constitutively active. [Barbacid, Biochem. 55:779-827 (1987); Box, Eur. J. Cancer 31:1051-1054 (1995) .] These oncogenic Ras proteins, which are found in many cancer types, contribute to malignancy and are therefore considered favored targets for directed therapy. [Bos, Cancer Res. 49:4682-4689 (1989) .] The active Ras protein promotes oncogenesis through activation of multiple Ras effectors that contribute to deregulated cell growth, differentiation, and increased survival, migration and
invasion. [See, e.g., Downward, J., Nat. Rev. Cancer 3:11-22 (2003); Shields, J. M., et al . , Trends Cell Biol. 10:147-541 (2000); and Mitin, . , et al . , Curr. Biol. 15:R563-74 (2005)].]
[ 0017 ] Association with the plasma membrane has been shown to be crucial for Ras activity, in both the wild type and the mutated constitutively active forms alike. [Boguski, et al., Nature, 355:643-654 (1993); Cox, et al . , Curr. Opin. Cell Biol. 4:1008-1016 (1992); Marshall, Curr. Opin. Cell Biol. 3:197-204 (1996) .] At least two structural elements are required for this association; the first is a farnesylcysteine carboxy methyl ester at the carboxy terminal of Ras, and the second element resides at the adjacent upstream sequence and varies among different Ras isoforms. [Hancock, et al., EMBO J.
10:4033-4039 (1991); Hancock, et al . , Cell 57:1167-1177 (1989) .] Normal Ras activity requires specifically the farnesyl isoprenoid moiety [Cox, et al., Curr. Opin. Cell Biol. 4:1008-1016, (1992); Cox, et al . , Mol. Cell. Biol. 12:2606-2615 (1992)] which acts as a specific recognition unit to allow binding of H-Ras with galectin-1 [Elad-Sfadia, et al., J. Biol. Chem. 277:37169-37175 (2002), Rotblat, et al . , J. Biol. Chem. 54:3112-3118 (2004)] and K-Ras with galectin-3 [Elad-Sfadia, et al., J. Biol. Chem. 279:34922-34930 (2004)] promoting strong membrane association and robust signaling.
[ 0018 ] FTS is known as a Ras inhibitor that acts in a rather specific manner on the active, GTP-bound forms of H-, N-, and K- Ras proteins. [Weisz, B., et al . , Oncogene 13:2579-2588 (1999); Gana-Weisz, M., et al . , Clin. Cancer Res. 8:555-65 (2002) ] . More specifically, FTS competes with Ras-GTP for binding to specific saturable binding sites in the plasma membrane, resulting in mislocalization of active Ras and facilitating Ras degradation. [Haklai, et al.,
Biochemistry 37 (5) : 1306-14 (1998)]. This competitive inhibition prevents active Ras from interacting with its prominent downstream effectors and results in reversal of the transformed phenotype in transformed cells that harbor activated Ras. As a
consequence, Ras-dependent cell qrowth and transforminq activities, both in vitro and in vivo, are stronqly inhibited by FTS. [Weisz, B., et al . , supra.; Gana-Weisz, M., et al . , supra . ] .
[ 0019 ] Ras antaqonists useful in the present invention include FTS and its structural analoqs, are described below.
[ 0020 ] The Ras antaqonists are represented by the formula:

[ 0021 ] In some embodiments, the Ras antaqonist is S- trans , trans-farnesylthiosalicylic acid or FTS (wherein R1 is farnesyl, R2 is COOR7, and R7 is hydroqen) .
[ 0022 ] In some embodiments, the FTS analog is halogenated, e.g., 5-chloro-FTS (wherein R1 is farnesyl, R2 is COOR7, R4 is chloro, and R7 is hydrogen) , and 5-fluoro-FTS (wherein R1 is farnesyl, R2 is COOR7, R4 is fluoro, and R7 is hydrogen) .
[ 0023 ] In other embodiments, the FTS analog is FTS-methyl ester (wherein R1 represents farnesyl, R2 represents COOR7, and R7 represents methyl) , FTS-amide (wherein R1 represents farnesyl, R2 represents CONR7R8, and R7 and R8 both represent hydrogen) ; FTS-methylamide (wherein R1 represents farnesyl, R2 represents CONR7R8, R7 represents hydrogen and R8 represents methyl); and FTS-dimethylamide (wherein R1 represents farnesyl, R2 represents CONR7R8, and R7 and R8 each represents methyl) .
[ 0024 ] In yet other embodiments, the Ras antagonist is an alkoxyalkyl S-prenylthiosalicylate or an FTS-alkoxyalkyl ester (wherein R2 represents COOCHR9OR10) . Representative examples include methoxymethyl S-farnesylthiosalicylate (wherein R1 is farnesyl, R9 is H, and R10 is methyl); methoxymethyl S- geranylgeranylthiosalicylate (wherein R1 is geranylgeranyl, R9 is H, and R10 is methyl) ; methoxymethyl 5-fluoro-S- farnesylthiosalicylate (wherein R1 is farnesyl, R5 is fluoro, R9 is H, and R10 is methyl) ; and ethoxymethyl S- farnesylthiosalicyate (wherein R1 is farnesyl, R9 is methyl and R10 is ethyl) . In each of the embodiments described above, unless specifically indicated otherwise, each of R3, R4, R5 and R6 represents hydrogen.
[ 0025 ] HDAC Inhibitors
[ 0026 ] DNA in eukaryotic cells is tightly complexed with proteins to form chromatin. Histones are small proteins that complex with DNA. Two of each of the histones known as H2A, H2B, H3 and H4 are tightly complexed with DNA, typically in amounts of about 150 base pairs, to form a nucleosome. This structure is further connected by linker DNA to form a solenoid. Histone acetyltransferases (HATs) and histone deacetylases (HDACs) represent two enzyme families that control the level of histone tail acetylation by the addition and removal respectively, of an acetyl group from the lysine
residues of core nucleosomal histories. Histones extending from the nucleosomal core are thus enzymatically modified, affecting chromatin structure and gene expression. HATs, via acetylation of histones, allow transcription and gene expression since acetylated histones can recruit transcription factors and other co-activator proteins. HDACs are usually associated with DNA hyper-methylation and gene silencing.
[ 0027 ] HDACs play an important role in cell proliferation and differentiation. Acetylated histones are relatively less effective in facilitating DNA transcription, whereas deacetylated histones are relatively more effective in facilitating this process. Acetylation and deacetylation are in balance in normal cells. However, in malignant cells, there is an excess of HDAC activity compared to HAT activity, resulting in an imbalance of deacetylated histones to acetylated histones. This imbalance results in excessive DNA transcription and uncontrolled cell proliferation. The involvement of HDACs in the control of cell proliferation and differentiation suggests that aberrant HDAC activity may play a role in cancer .
[ 0028 ] The HDAC enzyme family includes at least 18 enzymes, grouped in four (4) classes (Classes I, Ila, lib, III and IV) . Class I HDACs include HDACs 1, 2, 3, and 8. Class I HDACs can be found in the nucleus and are believed to be involved with transcriptional control repressors and co-factors. Class Ila HDACs include HDACS 4, 5, 7 and 9, and Class lib HDACs include HDACs 6 and 10. These enzymes can be found in both the cytoplasm as well as the nucleus. Certain class I and class II HDACs are overexpressed in tumors relative to normal tissues. See, Johnstone, Nature Reviews Drug Disovery 1:287-99 (2002) . Class III HDACs are believed to be NAD-dependent proteins and include members of the sirtuin family of proteins . Non-limiting examples of sirtuin proteins include SIRT1-7. Class IV HDACs include HDAC 11.
[ 0029 ] Broadly, the term "HDAC inhibitor" as used herein refers to a compound that has the ability to inhibit histone
deacetylase activity. This therapeutic class is able to block angiogenesis and cell cycling, and promote apoptosis and differentiation. HDAC inhibitors exhibit targeted anticancer activity and as disclosed herein, act synergistically with FTS and its analogs in the treatment of cancer.
[ 0030 ] Selective and non-selective HDAC inhibitors alike may be useful in the present invention. The term "selective HDAC inhibitor" refers to an HDAC inhibitor that does not significantly interact with all three HDAC classes. As used herein, a "Class I selective HDAC" refers to an HDAC inhibitor that interacts with one or more of HDACs 1, 2, 3 or 8, but does not significantly interact with the Class II HDACs (i.e., HDACs 4-7, 9 and 10) .
[ 0031 ] In some embodiments of the present invention, the HDAC inhibitor is selective HDAC inhibitor, such as a Class I selective HDAC inhibitor. Representative examples of such inhibitors that may be useful in the present invention include benzamides such as MGCD-0103 (N- (2-amino-phenyl) -4- [ (4-pyridin- 3-yl-pyrimidin-2-ylamino) -methyl] -benzamide) , also known as Mocetinostat , and related compounds as disclosed in U.S. Patent 6, 897, 220), benzamides such as MS-275 ( (N- ( 2-aminophenyl ) -4- (N- (pyridin-3-ylmethoxycarbonyl) aminomethyl) benzamide), also known as entinostat or "SNDX-275"), and related compounds as disclosed in e.g., U.S. Patent 6,174,905), spiruchostatin A, SK7041 and SK7068 (Class I HDAC inhibitors; Park, et al . , Clin. Cancer Res. 10:5271 (2004) ) and 6-amino nicotinamides.
[ 0032 ] In some embodiments, the HDAC is a non-selective HDAC inhibitor. Representative examples of such inhibitors that may be useful in the present invention include hydroxamic acids including trichostatin analogs such as trichostatin A (TSA) and trichostatin C (TSC) (Koghe, et al., Biochem. Pharmacol. 55:1359-64 (1998), salicylihydroxamic acid (SBHA) (U.S. Patent 5,608,108), azelaic bishydroxamic acid (ABHA) , azelaic-1- hydroxamate-9-analide (AAHA) , 6- (3-chlorophenylureido) carpoic hydroxamic acid (3C1-UCHA) , and ' -hydroxy-N-phenyl- octanediamide (suberoylanilide hydroxamic acid, also known as
"SAHA" or vorinostat, and related hydroxamic acid compounds as disclosed in U.S. Patents 5,369,108; 6,087,367; 7,399,787; 7,456,219 and RE38,506), epoxyketones such as trapoxin (TPX, which is a non-competitive HDAC inhibitor) , pyroxamide, m- carboxycinnamic acid bishydroxamide (CBHA; Richon, et al . , PNAS 95:3003-7 (1998)), oxamflatin, A-161906, GCK1026 (scriptaid; Lee, et al . , Int. J. Oncol. 33 (4) : 767-76 (2008)), belinostat (PXD-101, a Class I and II HDAC inhibitor), LAQ-824 (Remiszewski, et al . , J. Med. Chem. 46 (21) : 4609-23 (2003) ) , cyclic hydroxamic acid-containing peptide (CHAP; Furumai, et al., PNAS 98(1) :87-92 (2001), MW2796 and MW2996 (Andrews, et al., Int. J. Parasitol. 30:761-8 (2000)), panobinostat (LBH589) (Tan, et al., J Hematol Oncol. 3:5 (2010)), tacedinaline, CI-994, Acetyldinaline , 4-Acetamido-N- (2-aminophenyl) benzamide (Loprevite, et al. Oncol Res. 15(1) ;39-48 (2005)), BML-210, N- (2-aminophenyl) - ' -phenyl-octanediamide (Savickiene, et al., Eur J Pharmacol. 549 (1-3) : 9-18 (2006) ) M344, D237,
4-Dimethylamino-N- (6-hydroxycarbamoylhexyl) -benzamide (Takai, et al., Gynecol Oncol 101:108-113 (2006)).
[ 0033 ] In some of these embodiments, the non-selective HDAC inhibitor is a small-molecular weight carboxylate such as valproic acid (2-n-propylpentanoic acid, VPA) or a derivative thereof. VPA has been reported to inhibit HDACs 1-3 (Class I) and HDACs -8 (Class II) . Valproic acid is represented by the
formula :
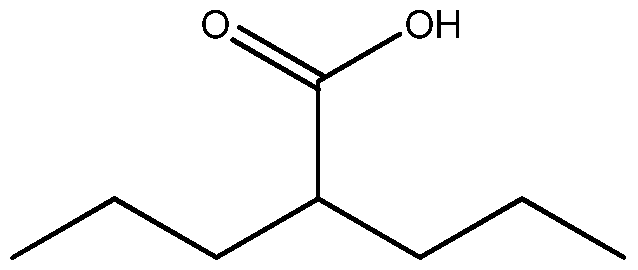
[ 0034 ] VPA derivatives suitable for use in the present invention are represented by the formula

wherein R1 and R2 each independently represents a linear or branched, saturated or unsaturated aliphatic 2-25r preferably
C3-25 hydrocarbon chain which optionally comprises one or several heteroatoms and which may be substituted, R3 is hydroxyl, alkoxy or an optionally alkylated amino group. The hydrocarbon chains R1 and R2 may contain one or several heteroatoms (e.g., 0, N, S) replacing carbon atoms in the hydrocarbon chain. R1 and R2 may be substituted, e.g., with hydroxyl, amino, carboxylic and alkoxy groups as well as aryl and heterocyclic groups . Preferably, R1 and R2 independently contain 2 to 10, more preferably 3 to 10 or 5 to 10 carbon atoms. It is also preferred that R1 and R2 independently are saturated or contain one double bond or one triple bond. VPA derivatives that may be particularly suitable include S-4-yn VPA and 2-EHXA (2- Ethyl-hexanoic acid) .
[0035] The HDAC inhibitor may be present in the form of a pharmaceutically acceptable salt, or any other form in which it is therapeutically effective.
[0036] It is generally believed that the hyperacetylation of the histones caused by HDAC inhibition neutralizes the positive charge of the lysine side chain, causing change of the chromatin structure and the consequential transcriptional activation of a number of genes, such that one ultimate outcome is cell cycle arrest. That is, the cells stop dividing. Numerous HDAC inhibitors are now in use in clinical cancer trials. Aside from various HDAC inhibitors mentioned above, they include Panobinostat , Belinostat, ITF-2357, PC-24781, phenylbutyrate, SB-939 and JHJ-26481585.
[0037] Compositions and Methods
[0038] The Ras antagonist and the HDAC inhibitor are co¬ administered, which as used herein, encompasses treatment regimens in which these anti-cancer agents are administered to the cancer patient at the same or different times (i.e., substantially simultaneously or sequentially) , and by the same or different route of administration, such that both agents and/or their metabolites are present in the patient at the same time in order to achieve the benefits of their combined therapeutic effect. Co-administration thus includes
simultaneous administration in separate compositions, administration at different times in separate compositions, and/or administration in a composition that contains both agents. Thus, in some embodiments, the Ras antagonist and the HDAC inhibitor are administered in a single composition.
[ 0039 ] The term "therapeutically effective amounts", as used herein, refers to a sufficient amount of each of the Ras antagonist and the HDAC inhibitor that will ameliorate at least one symptom of the cancer and its associated manifestations, diminish extent or severity of the disease, delay or retard disease progression, achieve partial or complete remission, prolong survival and combinations thereof. As shown in the working examples, combinations of the Ras antagonist and the HDAC inhibitor achieve synergy, i.e., a greater than additive effect, at least with respect to inhibition of the growth of the cancer cells in vitro. Applicants believe that these results reflect tumor/cancer cell growth inhibitory activity in vivo, and ultimately result in more effective cancer therapy and a commensurate improvement in one or more of these clinical manifestations of the disease. Appropriate "effective" amounts for any cancer patient can be determined using techniques, such as a dose escalation study. Specific dose levels for any particular patient will depend on several factors such as the potency of the Ras antagonist and the HDAC inhibitor, the age, weight, and general health of the patient, and the severity of the cancer.
[ 0040 ] The average daily dose of the Ras antagonists of the present invention generally ranges from about 200 mg to about 2000 mg (e.g., 200 mg, 400 mg, 600 mg, 800 mg, 1000 mg, 1200 mg, 1400 mg, 1600 mg, 1800 mg and 2000 mg) , in some embodiments from about 400 to about 1600 mg, and in some other embodiments from about 600 to about 1200 mg, and in yet other embodiments, from about 800 mg to about 1200 mg . The average daily dose of the HDAC inhibitor will vary, depending on the specific agent. The average daily dose for valproic acid (and its derivatives) for example, generally ranges from about 500 mg to about 3500
mg, in some embodiments from about 750 to 3000 mg, and in some other embodiments from about 750 mg to about 1500 mg . The average daily dose for vorinostat (and its derivatives), for example, generally ranges from about 100 mg to about 600 mg, in some other embodiments from about 200 to about 500 mg, and in some other embodiments from about 300 mg to about 400 mg . The average weekly dose for SNDX-275 (and its derivatives), for example, generally ranges from about 2.5 mg to about 10 mg and in some embodiments from about 5 mg to about 10 mg, once every two weeks .
[ 0041 ] The terms "administer," "administering",
"administration, " and the like, as used herein, refer to the methods that may be used to enable delivery of compounds or compositions to the desired site of biological action. Medically acceptable administration techniques suitable for use in the present invention are known in the art. See, e.g., Goodman and Gilman, The Pharmacological Basis of Therapeutics, current ed . ; Pergamon; and Remington's, Pharmaceutical Sciences (current edition), Mack Publishing Co., Easton, Pa. In some embodiments, at least one or both active agents are administered orally. In other embodiments, at least one or both active agents are administered parenterally (which for purposes of the present invention, includes intravenous, subcutaneous, intraperitoneal, intramuscular, intravascular and infusion) . Other administration routes such as topical and rectal administration may also be suitable.
[ 0042 ] The term "pharmaceutically acceptable" as used herein, refers to a material, such as a carrier and other non-active excipients, which does not abrogate the biological activity or properties of the active agent (s), and is relatively nontoxic.
[ 0043 ] The term "pharmaceutical composition, " as used herein, refers to the Ras antagonist and/or the HDAC inhibitor, optionally combined (e.g., mixed) with a pharmaceutically acceptable carrier. These ingredients are non-toxic, physiologically inert and do not adversely interact with the
active agent (s) present in the composition. Carriers facilitate formulation and/or administration of the active agents. Pharmaceutical compositions of the present invention may further contain one or more excipients.
[ 0044 ] Oral compositions for the Ras antagonist and/or the HDAC inhibitor can be prepared by bringing the agent (s) into association with (e.g., mixing with) the carrier, the selection of which is based on the mode of administration. Carriers are generally solid or liquid. In some cases, compositions may contain solid and liquid carriers. Compositions suitable for oral administration that contain the active are preferably in solid dosage forms such as tablets (e.g., including film-coated, sugar-coated, controlled or sustained release) , capsules, e.g., hard gelatin capsules (including controlled or sustained release) and soft gelatin capsules, powders and granules. The compositions, however, may be contained in other carriers that enable administration to a patient in other oral forms, e.g., a liquid or gel. Regardless of the form, the composition is divided into individual or combined doses containing predetermined quantities of the active ingredient or ingredient s .
[ 0045 ] Oral dosage forms may be prepared by mixing the active pharmaceutical ingredient or ingredients with one or more appropriate carriers (optionally with one or more other pharmaceutically acceptable excipients), and then formulating the composition into the desired dosage form e.g., compressing the composition into a tablet or filling the composition into a capsule or a pouch. Typical carriers and excipients include bulking agents or diluents, binders, buffers or pH adjusting agents, disintegrants (including crosslinked and super disintegrants such as croscarmellose) , glidants, and/or lubricants, including lactose, starch, mannitol, microcrystalline cellulose, ethylcellulose , sodium carboxymethylcellulose , hydroxypropylmethylcellulose, dibasic calcium phosphate, acacia, gelatin, stearic acid, magnesium stearate, corn oil, vegetable oils, and polyethylene glycols.
Coating agents such as sugar, shellac, and synthetic polymers may be employed, as well as colorants and preservatives. See, Remington' s Pharmaceutical Sciences, The Science and Practice of Pharmacy, 20th Edition (2000) .
[ 0046 ] Liquid form compositions include, for example, solutions, suspensions, emulsions, syrups, elixirs and pressurized compositions. The active agent (s), for example, can be dissolved or suspended in a pharmaceutically acceptable liquid carrier such as water, an organic solvent (and mixtures thereof), and/or pharmaceutically acceptable oils or fats. Examples of liquid carriers for oral administration include water (particularly containing additives as above, e.g., cellulose derivatives, preferably in suspension in sodium carboxymethyl cellulose solution) , alcohols (including monohydric alcohols (including monohydric alcohols and polyhydric alcohols, e.g., glycerin and non-toxic glycols) and their derivatives, and oils (e.g., fractionated coconut oil and arachis oil) . The liquid composition can contain other suitable pharmaceutical additives such as solubilizers, emulsifiers, buffers, preservatives, sweeteners, flavoring agents, suspending agents, thickening agents, colorants, viscosity regulators, stabilizers or osmoregulators .
[ 0047 ] Carriers suitable for preparation of compositions for parenteral administration include Sterile Water for Injection, Bacteriostatic Water for Injection, Sodium Chloride Injection (0.45%, 0.9%), Dextrose Injection (2.5%, 5%, 10%), Lactated Ringer's Injection, and the like. Dispersions can also be prepared in glycerol, liquid polyethylene glycols and mixtures thereof, and in oils. Compositions may also contain tonicity agents (e.g., sodium chloride and mannitol) , antioxidants (e.g., sodium bisulfite, sodium metabisulfite and ascorbic acid) and preservatives (e.g., benzyl alcohol, methyl paraben, propyl paraben and combinations of methyl and propyl parabens) .
[ 0048 ] The pharmaceutical composition containing the Ras antagonist and the HDAC inhibitor, or first and second
compositions containing the Ras antagonist and the HDAC inhibitor respectively, may be packaged and sold in the form of a kit. For example, the composition might be in the form of one or more oral dosage forms such as tablets or capsules (e.g., hard or soft gelatin capsules) containing one or both of the active agents. The kit may also contain written instructions for carrying out the inventive methods as described herein.
[ 0049 ] In some embodiments, the Ras antagonist is administered by dosing orally on a daily basis (in single or divided doses) for three weeks, followed by a one-week "off period", and repeating until remission is achieved. In these embodiments, the HDAC inhibitor may be present in the same composition. In some other embodiments, VPA for example, is administered daily in single or divided dosages (e.g., 2 or 3 times daily) . Vorinostat may be dosed once daily, e.g., at an initial dose of about 400 mg, which then is reduced to a daily dose of 300 mg and then may be continued at 300 mg every 5 consecutive days. SNDX may be administered every two weeks in an amount of about 5 or 10 mg .
[ 0050 ] Cancer generally refers to a disease caused by the uncontrolled, abnormal growth of cells that can spread to adjoining tissues or other parts of the body. Cancer cells can form a solid tumor, in which the cancer cells are massed together, or they can exist as dispersed cells, as in leukemia. Normal cells divide (reproduce) until maturation is attained and then only as necessary for replacement of damaged or dead cells. Cancer cells are often referred to as "malignant", because they divide endlessly, eventually crowding out nearby cells and spreading to other parts of the body. Malignant cancer cells eventually metastasize and spread to other parts of the body via the bloodstream or lymphatic system, where they can multiply and form new tumors. Malignant tumors are divided into carcinomas (which arise from epithelial precursor cells), sarcomas (which arise largely from mesenchymal tissues) and
lymphomas (which arise from precursors of red and white blood cells) .
[ 0051 ] Cancers characterized by the presence of elevated wild-type Ras or the presence of mutated Ras proteins are amenable to treatment in accordance with the present invention. These cancers include human lymphomas, sarcomas and carcinomas, e.g., fibrosarcoma, myxosarcoma, liposarcoma, chondrosarcoma, osteogenic sarcoma, chordoma, angiosarcoma, endotheliosarcoma, lymphangiosarcoma, synovioma, mesothelioma, lymphangioendotheliosarcoma, Ewing's tumor, leiomyosarcoma, rhabdomyosarcoma, colorectal (e.g., colon) carcinoma, gastrointestinal (e.g., stomach) cancer, pancreatic cancer, thyroid cancer (e.g., follicular, papillary and anaplastic thyroid carcinomas), breast cancer, ovarian cancer, prostate cancer, squamous cell carcinoma, basal cell carcinoma, adenocarcinoma, sweat gland carcinoma, sebaceous gland carcinoma, papillary carcinoma, papillary adenocarcinomas, cystadenocarcinoma, medullary carcinoma, bronchogenic carcinoma, renal cell carcinoma, hepatoma, bile duct carcinoma, choriocarcinoma, seminoma, embryonal carcinoma, Wilms' tumor, cervical cancer, testicular tumor, non-small cell lung carcinoma (NSCLC) , bladder carcinoma, epithelial carcinoma, glioma, astrocytoma, medulloblastoma, craniopharyngioma, ependymoma, pinealoma, hemangioblastoma, acoustic neuroma, oligodendroglioma, meningioma, melanoma, neuroblastoma, retinoblastoma; leukemia, which includes e.g., acute lymphocytic leukemia (ALL) and acute myelocytic leukemia (AML) (myeloblastic, promyelocytic, myelomonocytic, monocytic and erythroleukemia) ; chronic leukemia (chronic myelocytic (granulocytic) leukemia (CML) and chronic lymphocytic leukemia) (CLL) ; and polycythemia vera, lymphoma (which includes, e.g., Hodgkin's disease and non-Hodgkin ' s disease and cutaneous T-cell lymphoma), multiple myeloma, Waldenstrom's macroglobulinemia and heavy chain disease.
[ 0052 ] In some embodiments, the invention is used to treat a patient afflicted with renal carcinoma, skin cancer, pancreatic
cancer, colorectal (e.g., colon) cancer, NSCLC, ovarian cancer, hepatic (liver) cancer, thyroid cancer, seminoma, skin cancer, endometrial cancer, melanoma, leukemia, lymphoma, prostate cancer, bladder and urinary cancers, breast cancer, and brain metastases of these primary tumors, primary brain cancers (such as gliomas and neuroblastomas) and head and neck cancers. See, e.g., Bos, Cancer Res. 49:4682-89 (1989).
[0053] The combination of Ras antagonist and HDAC inhibitor may be used alone or in conjunction with other treatment agents such as biological anti-cancer agents (e.g., antibodies), chemotherapy and radiation. The combination may be used as a front-line treatment strategy e.g., as a first course of treatment in a newly diagnosed cancer patient, and whether or not the cancer has metastasized. The combination may also be used as a second-line treatment strategy e.g., in the treatment of a cancer patient who has been previously treated using at least one other agent but has not responded to the previous agent (s) or has developed a resistance thereto, which may have resulted in termination of the therapy even before it could achieve an appreciable therapeutic efficacy.
[0054] Embodiments of the present invention will now be described by way of the following non-limiting working examples .
[0055] Example 1
[0056] In Vitro Experiments Showing Synergistic Effect of FTS and VPA in Three Different Types of Cancer Cells
[0057] Cell lines
[0058] Human A549, non small cell lung carcinoma cells (CCL- 185, ATCC) , which express oncogenic K-ras, were maintained in Kaighn's modification of Ham's F12 medium (Sigma), supplemented with 1.5g sodium bicarbonate/L, 10% fetal calf serum (FCS) , 100 U/ml penicillin and 100 yg/ml streptomycin.
[0059] DLD1 and SW-480, which are human colon adenocarcinoma cells that also express oncogenic K-ras, were cultured in DMEM
10% FCS, 2mM L-Glutamine, 100 U/ml penicillin and 100 yg/ml streptomycin .
[0060] ARO, thyroid carcinoma cells, wherein wild-type K-Ras is chronically active, and wherein B-Raf is constitutively active, were cultured in RPMI 1640 medium with 10% FCS, 2mM L- Glutamine, 100 U/ml penicillin and 100 yg/ml streptomycin.
[0061] All cells were incubated at 37°C in humidified atmosphere of 95% air and 5% C02.
[0062] Drug Preparation
[0063] FTS was prepared as previously described in Rotbalt, Meth. Enzymol. 439:467-89 (2008) .
[0064] VPA was stored at 4°C as a powder and before each experiment it was weighed and diluted with cell medium to the desired concentration.
[0065] SAHA was reconstituted in DMSO to a concentration of 5 mM, divided into 10 μΐ stocks and stored at 4°C. For each experiment, one stock was diluted to the desired concentration using cell medium.
[0066] Proliferation assay
[0067] To determine cell survival, A549, DLD1, SW-480 and ARO Cells were plated at a density of 8xl03 cells per well in 24-well plates. After 24 hours cells were treated with VPA (Sigma), SAHA (Alexis) or FTS dissolved in 0.1% Me2S04 (Concordia Pharmaceuticals, Ft. Lauderdale, FL) or a combination of FTS with VPA, or a combination of FTS with SAHA, for the indicated periods of time. Control cells were treated with 0.1% Me2S04. In the case of incubation times longer than 3 days the cells were detached, counted and re-plated at a density of 3xl05 cells per 10-cm plate. Below are the concentrations of VPA, SAHA and FTS that were used in the different cell lines:

[ 0068 ] Cell growth was estimated by direct counting of viable cell using Trypan blue. Experiments were preformed twice in quadruplicate .
[ 0069 ] Fluorescence activated call sorter (FACS) analysis
[ 0070 ] A549, DLD1 and ARO cells (4xl05 cells per 10-cm plate) were plated for 24h and then incubated with VPA and FTS for additional 72h in 10% FCS-containing medium. The cells were then collected and resuspended in PBS containing propidium iodide (5C^g/mL; Sigma), 0.1% sodium citrate and 0.1% Triton x- 100 (BDH, Poole, United Kingdom) and incubated overnight in the dark at 4°C.
[ 0071 ] The cells were then subjected to analysis by fluorescence activated cell sorter (FACS Calibur, Becton Dickinson, Los Angeles, CA) . The experiment was preformed twice in duplicate.
[ 0072 ] Western blot analysis
[ 0073 ] To examine the effect of FTS on protein levels in the cultured cells, A549, DLD1 and ARO cells were plated at density of 4xl05 cells per 10-cm plate and grown for 24h. The cells were then treated 0.1% Me2S04 (control), FTS, VPA, SAHA, a combined treatment of VPA plus FTS, or a combined treatment of FTS plus SAHA, for 72h. Next, the cells were lysed in 300μ1 homogenization buffer (50 mmol/L Tris-HCl (pH 7.6), 20mM MgCl2, 200mM NaCl, 0.5% NP40, ImM DTT, and protease inhibitors) centrifuged for 10 min at 14,000 rpm at 4°C and the supernatant was collected. Equal amounts of proteins (50-100yg per lane) were subjected to SDS-PAGE, followed by immunoblotting with rabbit anti-cyclin Dl (1:1,000), mouse anti-pan-Ras antibody Ab (Calbiochem) , rabbit anti-Survivin Ab (Santa Cruz, CA) and rabbit anti- -tubulin Ab (Sigma) . Blots were then exposed to the appropriate secondary peroxidase-coupled IgG (1:5,000) and subjected to enhanced chemiluminescence . Zundelevich, et al . ,
Mol . Cancer Ther. 31:31 (2007) . Protein bands were quantified by densitometry using TINA 2.0 software (Ray Tests) .
[0074] Immunofluorescence and confocal microscopy
[0075] A549 cells were plated on glass cover slips placed in 10-cm plates at a density of 4xl05 cells/plate for 24h before adding 0.1% Me2S04, 75μΜ FTS or VPA 0.8mM or VPA+FTS for an additional 24 or 72 hours. Afterwards, the cells were fixed with formaldehyde at room temperature for 30 min and then treated with 0.2% Triton X-100. After washing three times with phosphate-buffered saline (PBS) , slides were immersed in 1% bovine serum albumin (BSA) with 200yg/mL naive goat IgG (Jackson ImmunoResearch) for 30 min and then incubated with rabbit anti-Aurora A (1:50, Cell Signaling, Danvers, MA), anti- Aurora B (1:50, Bethyl Labs Montgomery, TX) or mouse anti- phosphor-H3 (1:50, Upstate, Charlottesville, VA) overnight at 4°C. After three additional washings with PBS, the cells were incubated with goat anti-mouse Cy3-conjugated Ab or with donkey anti-rabbit Cy2-conjugated Ab (1:200, Jackson ImmunoResearch) for 1 h in the dark. Then the cells were washed twice in PBS, and further incubated with anti-a-tubulin FITC Ab (1:50, sigma, F2168) for lh at room temperature. Finally, the cells were counterstained with Hoechst 33258 (Fluka AG, CH9470), and examined by fluorescent microscopy at 60x magnification with an LSM 510 META microscope.
[0076] Total RNA purification
[0077] A549 cells were treated for 72 h with either 75μΜ FTS, 0.8mM VPA, 0.1% vehicle (control) or 75μΜ FTS plus 0.8mM VPA. Total RNA was isolated from cultured cells using protocols and reagents contained in the RNeasy Plus Mini Kit (Qiagene) . The concentration of the RNA samples was determined by measuring the absorbance at 260 nm (A260) in a spectrophotometer. Purified RNA was stored at -70°C in RNase- free water. The purified RNA was used for real-time PCR.
[0078] Real-time PCR analysis
[0079] Extracts of total RNA (1 yg) were reverse-transcribed in a total volume of 20yL using the Verso™ RT-PCR Kits (ABgene) .
lpL cDNA samples were then used for real-time PCR (QPCR SYBR Green Mix Plus ROX Vial, ABgene) . The primers used targeted Survivin, AurK-A and NuSap genes, and the housekeeping gene GAPDH. Primer sequences used for these experiments are set forth in the following table.

[ 0080 ] Statistical analysis
[ 0081 ] The data are expressed as the mean ± SD . Statistical significance of the difference in mean values was assessed with one-way ANOVA followed by Tukey post test. A P value ≤ 0.05 was considered to be significant.
[ 0082 ] Results
[ 0083 ] VPA and FTS synergistically in inhibit growth of cells with active Ras pathways.
[ 0084 ] To examine the effect of combined treatment of VPA and FTS on cell growth and death A549, DLD1 and ARO cells were incubated with or without each of the two inhibitors and with their combination. Cells were incubated with the inhibitors for 72 hours and then imaged and counted directly. Each of the inhibitors caused a significant decrease in cell number; the combination caused a much greater reduction in cell number than either VPA or FTS alone (Fig. 1) . Specifically, in A549 cells each of the inhibitors alone caused 50% reductions and the combination caused a 90% decrease in cell number. Similar results were obtained with DLD1 and ARO cells (Fig. 1) . These results suggested that the HDAC inhibitor and the Ras inhibitor exerted a concerted inhibition of cell growth in cells with active Ras pathways.
[ 0085 ] Unexpectedly, when such cells, e.g. A549 (Fig. 2A) and DLD1 (Fig. 2B) were incubated with the inhibitors for
periods of times longer than 72 h (5 days), a marked growth inhibitory effect was observed. While the VPA or FTS-treated cells continued to grow at a relatively slow pace as compared with the controls, cells that were treated with the two inhibitors together surprisingly stopped growing and underwent total growth arrest (Figs. 2A and B) . Moreover, FACS analysis of VPA plus FTS treated cells (72 hours treatment) showed a synergistic reduction of cells in S-phase (Fig. 2C) .
[0086] We thus conclude that VPA and FTS act synergistically to inhibit growth of NSCLC cells with active Ras pathways.
[0087] VPA and FTS synergistically down-regulate survivin in A549 and DLD1 cells
[0088] Next we examined the combined effect of VPA and FTS on Ras and on known downstream targets of Ras. A549 and DLD1 cells were treated for 72 h with VPA or FTS alone, or with both drugs. The impact of the treatment on the levels of Ras, Cyclin Dl, and survivin proteins was then determined by Western immunoblotting with specific Abs . Survivin is known to act as an anti-apoptotic factor through its inhibitory action on a number of caspases as well as an important factor for cell cycle progression through its association with the mitotic spindle. Blum, et al . , Mol . Cancer Ther. 5:2337-47 (2006); Delacour-Larose, et al . , Med. Sci (Paris) 24:828-32 (2008). Survivin is considered to be an important subunit of the chromosomal passenger complex (CPC) , which also contains aurora B, INCENP and borealin. Ruchaud et al . , Mol. Cell Biol. 8: 798-812 (2007). The CPC complex controls critical aspects in mitosis. (Vagnarelli, Chromosoma 113:211-22 (2004). Cycline Dl is critical for cells to enter the S-phase of the cell cycle. It serves as a cell cycle regulatory switch in actively proliferating cells. Stacey, Curr Opin Cell Biol. 15 (2) : 158-63 (2003) .
[0089] Typical results of these experiments (Fig. 3A) indicated that FTS but not VPA caused a significant reduction in the levels of Ras and cyclin Dl in A549 and DLD1 cells. These results are in line with the known anti-Ras activity of
FTS . Erlich, et al . , Biochem. Pharmacol. 72:427-36 (2006); Blum, et al . , Int. J. Cancer 119:527-38 (2006). VPA did not itself affect the levels of Ras and Cyclin Dl (Fig. 3A) . However, the effect of the combined treatment on Ras, Cyclin Dl, and survivin was clearly stronger than the effect of either drug alone (Fig. 3A) . Previous studies showed that Ras inhibition by FTS down-regulated Cyclin Dl protein due to the inhibition of the Ras-dependent Raf-MEK-ERK and PI3K-Akt pathways lead to enhanced proteosomal degradation of Cyclin Dl in cells that exhibit active Ras. Erlich, et al . , Biochem. Pharmacol. 72:427-36 (2006); Blum, et al . , Int. J. Cancer 119:527-38 (2006) . This explains the observed reduction in Ras and Cyclin Dl in A549 and DLD1 cells. Similarly, we had shown that Ras positively regulates the transcription of survivin in glioblastoma. Blum, et al., Mol . Cancer Ther. 5:2337-47 (2006) . The positive control of Ras on survivin expression can be relieved by FTS (or by dominant-negative Ras) as manifested by a decrease in the level of survivin mRNA and protein in FTS- treated cells. Blum, et al . , Mol. Cancer Ther. 5:2337-47 (2006) . The present results show a similar strong reduction in survivin protein level by FTS in A549 and DLD1 (Fig. 3A) . This result was unexpected at least from the standpoint that in addition to the cell growth cycle, the two drugs affected unrelated cellular pathways .
[ 0090 ] VPA and FTS synergistically inhibit Aurora kinase A transcription blocking mitosis in A549 cells
[ 0091 ] A very strong synergistic effect of VPA and FTS was detected in the level of survivin. In the presence of the two drugs, no survivin protein could be detected in any of the cell lines (Fig. 3A) . Indeed, RT-PCR for the determination of survivin mRNA suggested a strong synergism of VPA and FTS in reducing the level of survivin transcripts (Fig. 3B) . This could be a result of inhibition of survivin transcription or a decrease in mRNA stability mediated by a small RNA.
[ 0092 ] These data suggested that the observed synergistic effects of FTS and VPA leading to total loss of survivin is associated with the cell cycle arrest observed in the presence of the two drugs (Fig. 2A) . We assumed that the loss of survivin results mainly in defects in mitosis. We therefore examined the impact of VPA, FTS, and of their combination on aurora A expression in A549 cells that were incubated for 72 h with the inhibitors (Fig. 4) . VPA alone had a very small (10% ± 10) inhibitory effect on aurora A expression; FTS had a stronger effect of 57% ±7 inhibition; and the combination of VPA and FTS synergistically completely blocked aurora A expression (96% ± 1.5 inhibition, Fig. 4) .
[ 0093 ] In light of these results we then examined the impact of the drugs on metaphase cells using triple fluorescent confocal microscopy to detect the chromosomes (labeled with Hoechst, blue fluorescence) , the aurora A (anti-aurora A Ab and Cy 3-Ab, red fluorescence), and the a-tubulin that strongly labels in these cells the spindle poles (FITC-labeled anti-a-microtubule Ab, green fluorescence) . In these and in the subsequent experiments, the cells were incubated for 24 h with the drugs so as to enable detection of M-phase cells. The images we obtained (not shown) for a control cell are in agreement with earlier reports (Zhang, et al . , Cancer Biol. Ther. 7:1388-97 (2008)) . Namely, they demonstrated typical metaphase chromosomes (blue) aligned on the spindle equator and show that aurora A is localized to the spindle poles. Treatment of VPA alone had relatively no effect on the level of aurora A in the spindle poles (15% ± 10 reduction, n= 30, p> 0.05), FTS treatment had a stronger effect (60% ± 12 reduction, n= 30, p< 0.001), and the combined treatment synergistically resulted in a almost a complete loss of aurora A in the spindle poles (95% ± 6 reduction, n= 30, p< 0.001) . These results are consistent with the RT-PCR data (Fig. 4) . Moreover, in agreement with previous studies, Vader, Biochim. Biophys . Acta. 1786:60-12 (2008), they demonstrate that the chromosomes of the metaphase control cells are localized in between the spindle
poles which colocalize with aurora A. The drug treatments did not cause destruction of these structures but caused reduced levels or complete disappearance of aurora A. Because aurora A is critical for accurate bipolar spindle assembly, these results are consistent with the total growth arrest observed in the presence of VPA and FTS (Fig. 2A) .
[0094] VPA and FTS synergistically block Aurora kinase B expression and histone-H3 phosphorylation
[0095] Aurora B is localized to the kinetechores during metaphase and aberrant aurora B expression coincides with reduced levels of phospho histone- H3 (serlO) which is critical for mitosis. Vader, Biochim. Biophys . Acta. 1786:60-12 (2008) . Our previous gene profiling analysis in FTS-treated A549 cells (75μΜ FTS for 72 h) (Blum, et al . , Cancer Res. 57:3320-8 (2007)) showed a decrease in aurora B expression. Other studies showed that HDAC inhibitors reduce the level of aurora B. Zhang, et al . , Cancer Biol. Ther. 7:1388-97 (2008) . Thus, we next examined the combined impact of VPA and FTS on aurora B and on the levels of phospho-histone-H3 (serlO) in A549 cells. Triple fluorescent confocal microscopy was used to detect the chromosomes (labeled with Hoechst, blue fluorescence) , the aurora B (anti-aurora B Ab and Cy3-labeled Ab, red fluorescence), and the phospho-histone-H3 ( ser 10) (labeled with anti phospho-histone-H3 ( ser 10) Ab and Cy2 labeled Ab, green fluorescence) (photos not shown) . We found that after 24h treatment VPA caused a reduction in aurora B (30% ± 8 inhibition) and in phospho-histone-H3 and FTS caused 30% ± 7 inhibition of aurora B. The combined treatment caused, unexpectedly, a complete disappearance of aurora B (90% ± 5 inhibition) . The reduction in aurora B levels was accompanied by reduced chromosome condensation in agreement with early studies. Shannon, Curr . Biol. 12:R458-60 (2002); Vas, et al . , Cell Cycle 7:293-6 (2008) .
[0096] Mitosis defects in FTS-treated A549 cells
[0097] Next we examined the impact of FTS on aurora B in A549 cells that were exposed to the drug for longer period of
time (72 h) . The cells were labeled as detailed above then imaged using triple fluorescent confocal microscopy. As pointed out above, FTS had 30% effect on aurora B in cells treated for 24 h with the drug as detected in M-phase cells (photo not shown) . When we examined M-phase cells after 72 h of FTS we found a strong decrease in aurora B (60% ± 7, not shown) which is in line with the previous gene profiling analysis. Blum, et al . , Cancer Res. 57:3320-8 (2007) . Moreover we detected a strong effect of FTS on localization of aurora B during cytokinesis. In line with previous experiments, Terada, Cell Struct. Funct . 25:653-7 (2001), control cells in telophase exhibited a-tubulin labeled spindle midbody in which aurora B was localized as well. By marked contrast, aurora B was completely mislocalized from the midbody in the FTS-treated cells (photos not shown) . These results showed that the long- term effect of FTS is also manifested in a defect in cytokinesis that can not be completed properly with a mislocalized aurora B. Kollareddy, et al., Bomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub . 152:27-33 (2008) . Unlike the normal spindle structure of the control M-phase cells, the spindle of the FTS-treated M-phase cells was defective (not shown) . Moreover, in the FTS-treated cells in G2 phase, triple polar spindles were detected in contrast to the normal bipolar spindle structure of vehicle treated controls (not shown) . We could not detect a combined effect of VPA and FTS because under these conditions (72 h with FTS), the cells were completely growth-arrested.
[ 0098 ] Recent experiments have shown that the nucleolar and spindle associated protein (NuSAP) is a novel microtubule- associated protein involved in mitotic spindle organization. Fujiwara, et al., Br. J. Haematol. 135:583-90 (2006); Ribbeck, et al., Curr. Biol. 17:230-6 (2007) . Indeed siRNA to NuSap disrupted formation of a normal spindle. Raemaekers, et al., J. Cell. Biol. 152:1017-29 (2003). We therefore examined the impact of VPA and FTS combination on the levels of NuSAP
transcription in A549 cells. As shown in Fig. 5, VPA alone had a minor effect (10 % ± 10 inhibition) , FTS alone had a strong effect (66% ± 8 inhibition) and the combined treatment caused almost a complete inhibition of NuSAP expression (95% ± 1 inhibition) . These results, along with the observations that the combination of VPA and FTS synergized in reducing survivin and aurora kinases, indicate that the combined treatment leads to a mitotic catastrophe which explains the observed cell growth arrest (Fig. 2A) .
[ 0099 ] SAHA and FTS Synergistically Inhibit Growth of A549 and SW-480 Cell Lines.
[ 0100 ] We examined the combination of SAHA plus FTS treatment on A549 and SW-480 cells. The cells were incubated with or without each of these agents the inhibitor and with their combination for 72h, and then imaged and counted directly (photos not shown) . Each of the inhibitors caused a significant decrease in cell numbers. The combination caused a much greater reduction in cell number than either SAHA or FTS alone. These results indicate that the synergistic effect of FTS can be achieved with other different HDAC inhibitors.
[ 0101 ] When the cells were incubated with the inhibitors for periods of times longer than 72h, treatment of SAHA alone caused a marked growth inhibitory effect but the cells continued to grow at a very slow pace. The combined treated cells stopped growing and underwent total growth arrest (Figs. 6A and B) . The mechanism of SAHA plus FTS treatment seems to be a bit different then that of VPA plus FTS since treatment with SAHA elevated apoptosis. In the combined treated cells, two populations were observed, namely, a first population of apoptotic cells and a second population of cells which escaped apoptosis and underwent a complete growth arrest.
[ 0102 ] Next, we examined the effect of the combined treatment of SAHA and FTS on Survivin and Aurora A protein levels in A549 cell line. SAHA treatment caused a decrease in Aurora A and Survivin (data not shown) . The combined treatment of SAHA plus FTS caused a greater decrease in Aurora A protein
levels and a similar decrease in Survivin protein levels as the treatment with SAHA alone.
[ 0103 ] Example 2
[ 0104 ] Tablets containing FTS (200 mg) and Sodium Valproate (230 mg, equivalent to 200 mg VPA)
[ 0105 ] FTS active pharmaceutical ingredient (2.0 kg), sodium valproate active pharmaceutical ingredient (2.3 kg), microcrystalline cellulose (2.0 kg), croscarmellose sodium (0.2 kg), and magnesium stearate (0.1 kg) are blended to homogeneity and compressed into tablets with a tablet press. Assuming a 5% loss on material transfers and tablet press start-up, adjustment, and shut-down, approximately 9,500 tablets containing FTS (200 mg) and sodium valproate (230 mg) are yielded .
[ 0106 ] Example 3
[ 0107 ] Hard Gelatin Capsules containing FTS (100 mg) and Sodium Valproate (230 mg, equivalent to 200 mg VPA)
[ 0108 ] FTS active pharmaceutical ingredient (1.0 kg), sodium valproate (2.3 kg), microcrystalline cellulose (2.0 kg), croscarmellose sodium (0.2 kg), and magnesium stearate (0.1 kg) are blended to homogeneity and filled into hard gelatin capsules with an encapsulation machine. Assuming a 5% loss on material transfer and encapsulating machine start-up, adjustment, and shut-down, approximately 9,500 capsules are yielded .
[ 0109 ] Example 4
[ 0110 ] Hard Gelatin Capsules containing FTS (100 mg) and vorinostat (100 mg)
[ 0111 ] FTS active pharmaceutical ingredient (2.0 kg), vorinostat active pharmaceutical ingredient (2.0 kg), microcrystalline cellulose (2.0 kg), croscarmellose sodium (0.2 kg), and magnesium stearate (0.1 kg) are blended to homogeneity and filled into hard gelatin capsules with an encapsulation machine. Assuming a 5% loss on material transfer and
encapsulating machine start-up, adjustment, and shut-down, approximately 19,000 capsules are yielded.
[ 0112 ] Example 5
[ 0113 ] Soft-Gelatin Capsules containing FTS (100 mg) and VPA (100 mg)
[ 0114 ] FTS active pharmaceutical ingredient (2.0 kg) and VPA active pharmaceutical ingredient (2.0 kg) and are dissolved in corn oil (4.0 kg) and stirred to form a homogeneous solution. The solution is filled into soft gelatin capsules with a form- fill-seal encapsulation machine. Assuming a 5% loss on material transfers and soft-gelatin encapsulating machine start-up, adjustment, and shut-down, approximately 19,000 capsules containing FTS (100 mg) and VPA (100 mg) are yielded.
[ 0115 ] All patent publications and non-patent publications are indicative of the level of skill of those skilled in the art to which this invention pertains. All these publications are herein incorporated by reference to the same extent as if each individual publication were specifically and individually indicated as being incorporated by reference.
[ 0116 ] Although the invention herein has been described with reference to particular embodiments, it is to be understood that these embodiments are merely illustrative of the principles and applications of the present invention. It is therefore to be understood that numerous modifications may be made to the illustrative embodiments and that other arrangements may be devised without departing from the spirit and scope of the present invention as defined by the appended claims .
Claims
composition for treating a cancer patient, a Ras antagonist represented by the formula

2. The composition of claim 1, wherein the Ras antagonist is FTS .
3. The composition of claim 1, wherein the Ras antagonist is an FTS analog selected from the group consisting of 5-chloro- FTS, 5-fluoro-FTS, FTS-methyl ester, FTS-amide, FTS- methylamide, FTS-dimethylamide, methoxymethyl S- farnesylthiosalicylate , methoxymethyl S- geranylgeranylthiosalicylate, methoxymethyl 5-fluoro-S- farnesylthiosalicylate , and ethoxymethyl S- farnesylthiosalicyate .
4. The composition of any of claims 1-3, wherein the HDAC inhibitor is valproic acid.
5. The composition of any of claims 1-3, wherein the HDAC inhibitor is vorinostat .
6. The composition of any of claims 1-3, wherein the HDAC inhibitor is SNDX-275.
7. The composition of any preceding claim, wherein the pharmaceutical carrier is a liquid.
8. The composition of any preceding claim, wherein the pharmaceutically acceptable carrier is solid.
9. The composition of claim 8, which is in the form of a tablet or a capsule.
10. A method for treating a cancer patient, comprising coadministering to the patient therapeutically effective amounts of a Ras antagonist represented by the formula

11. The method of claim 10, wherein the Ras antagonist and the HDAC inhibitor are administered in the same dosage form.
12. The method of claim 10, wherein the co-administering is via oral delivery.
13. The method of claim 10, wherein the cancer is selected from the group consisting of renal carcinoma, skin cancer, pancreatic cancer, colorectal cancer, NSCLC, ovarian cancer, hepatic cancer, thyroid cancer, seminoma, skin cancer, endometrial cancer, melanoma, leukemia, myelodysplastic syndrome, lymphoma, prostate cancer, bladder and urinary cancers, breast cancer, and brain metastases of said cancers and carcinomas, primary brain cancers and head and neck cancers .
14. The method of claim 10, wherein the cancer is thyroid cancer .
15. The method of claim 10, wherein the cancer is colon cancer .
16. The method of claim 10, wherein the cancer is lung cancer.
17. The method of any of claims 10-16, wherein the Ras antagonist is FTS .
18. The method of any of claims 10-16, wherein the Ras antagonist is an FTS analog selected from the group consisting of 5-chloro-FTS, 5-fluoro-FTS, FTS-methyl ester, FTS-amide, FTS-methylamide, FTS-dimethylamide , methoxymethyl S- farnesylthiosalicylate , methoxymethyl S- geranylgeranylthiosalicylate, methoxymethyl 5-fluoro-S- farnesylthiosalicylate , and ethoxymethyl S- farnesylthiosalicyate .
19. The method of any of claims 10-18, wherein the HDAC inhibitor is valproic acid.
20. The method of any of claims 10-18, wherein the HDAC inhibitor is vorinostat .
21. The method of any of claims 10-18, wherein the HDAC inhibitor is SNDX-275.
22. A kit for use in treating cancer, comprising a first dosage form containing therapeutically effective amounts of the Ras antagonist defined by the formula herein, and an HDAC inhibitor, or separate dosage forms containing the Ras antagonist and the HDAC inhibitor respectively, and optionally, written instructions for using the dosage form(s) to treat a cancer patient.
23. A combination of active agents of effective amounts of a Ras antagonist represented by the formula

24. The combination of active agents for use according to claim 23, wherein the Ras antagonist and the HDAC inhibitor are for co-administration in the same dosage form.
25. The combination of active agents for use according to claim 23, wherein the Ras antagonist and the HDAC inhibitor are for oral co-administration.
26. The combination of active agents for use according to claim 23, wherein the cancer is selected from the group consisting of renal carcinoma, skin cancer, pancreatic cancer, colorectal cancer, NSCLC, ovarian cancer, hepatic cancer, thyroid cancer, seminoma, skin cancer, endometrial cancer, melanoma, leukemia, myelodysplastic syndrome, lymphoma, prostate cancer, bladder and urinary cancers, breast cancer, and brain metastases of said cancers and carcinomas, primary brain cancers and head and neck cancers.
27. The combination of active agents for use according to claim 23, wherein the cancer is thyroid cancer.
28. The combination of active agents for use according to claim 23, wherein the cancer is colon cancer.
29. The combination of active agents for use according to claim 23, wherein the cancer is lung cancer.
30. The combination of active agents for use according to any of claims 23-29, wherein the Ras antagonist is FTS .
31. The combination of active agents for use according to any of claims 23-29, wherein the Ras antagonist is an FTS analog selected from the group consisting of 5-chloro-FTS, 5-fluoro- FTS, FTS-methyl ester, FTS-amide, FTS-methylamide, FTS- dimethylamide, methoxymethyl S-farnesylthiosalicylate, methoxymethyl S-geranylgeranylthiosalicylate, methoxymethyl 5- fluoro-S-farnesylthiosalicylate, and ethoxymethyl S- farnesylthiosalicyate .
32. The combination of active agents for use according to any of claims 23-31, wherein the HDAC inhibitor is valproic acid.
33. The combination of active agents for use according to any of claims 23-31, wherein the HDAC inhibitor is vorinostat .
34. The combination of active agents for use according to any of claims 23-31, wherein the HDAC inhibitor is SNDX-275.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012536940A JP2013508458A (en) | 2009-10-26 | 2010-10-26 | Cancer treatment using a combination of FTS and HDAC inhibitor |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US25487209P | 2009-10-26 | 2009-10-26 | |
| US61/254,872 | 2009-10-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011056542A1 true WO2011056542A1 (en) | 2011-05-12 |
Family
ID=43480463
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2010/054042 Ceased WO2011056542A1 (en) | 2009-10-26 | 2010-10-26 | Cancer therapy with combinations of fts with hdac inhibitors |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP2013508458A (en) |
| WO (1) | WO2011056542A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103113274A (en) * | 2013-01-25 | 2013-05-22 | 南通大学 | Ras and HDAC dual inhibitor and its preparation method and use |
| WO2024246114A1 (en) * | 2023-05-31 | 2024-12-05 | Boehringer Ingelheim International Gmbh | Survivin as a biomarker for predicting the responsiveness of cancer treatment |
Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5369108A (en) | 1991-10-04 | 1994-11-29 | Sloan-Kettering Institute For Cancer Research | Potent inducers of terminal differentiation and methods of use thereof |
| US5608108A (en) | 1988-11-14 | 1997-03-04 | Sloan-Kettering Institute For Cancer Research | Potent inducers of terminal differentiation and method of use thereof |
| US6174905B1 (en) | 1996-09-30 | 2001-01-16 | Mitsui Chemicals, Inc. | Cell differentiation inducer |
| USRE38506E1 (en) | 1991-10-04 | 2004-04-20 | Sloan-Kettering Institute For Cancer Research | Potent inducers of terminal differentiation and methods of use thereof |
| US6897220B2 (en) | 2001-09-14 | 2005-05-24 | Methylgene, Inc. | Inhibitors of histone deacetylase |
| WO2006023639A1 (en) * | 2004-08-18 | 2006-03-02 | Concordia Pharmaceuticals, Inc. | Methods and compositions for oral delivery of fts |
| WO2007064448A2 (en) * | 2005-11-28 | 2007-06-07 | Ramot At Tel Aviv University, Ltd. | Cancer treatment using fts and 2-deoxyglucose |
| US7399787B2 (en) | 2002-03-04 | 2008-07-15 | Merck Hdac Research, Llc | Methods of treating cancer with HDAC inhibitors |
| US7456219B2 (en) | 2002-03-04 | 2008-11-25 | Merck Hdac Research, Llc | Polymorphs of suberoylanilide hydroxamic acid |
| WO2008143947A1 (en) * | 2007-05-14 | 2008-11-27 | University Of South Florida | Farnesylamine derivatives and methods of use |
| WO2009067500A1 (en) * | 2007-11-19 | 2009-05-28 | Syndax Pharmaceuticals, Inc. | Administration of an inhibitor of hdac |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090298843A1 (en) * | 2006-04-11 | 2009-12-03 | Ramot At Tel Aviv University Ltd. | Treatment of hematological malignancies with fts and a bcr-abl tyrosine kinase inhibitor |
| WO2008075342A1 (en) * | 2006-12-19 | 2008-06-26 | Ramot At Tel Aviv University Ltd. | Treatment of lung cancer |
| RU2446796C2 (en) * | 2006-12-26 | 2012-04-10 | Фармасайкликс, Инк. | Method for using histone deacetylase and biomarker monitoring in combined therapy |
-
2010
- 2010-10-26 JP JP2012536940A patent/JP2013508458A/en active Pending
- 2010-10-26 WO PCT/US2010/054042 patent/WO2011056542A1/en not_active Ceased
Patent Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5608108A (en) | 1988-11-14 | 1997-03-04 | Sloan-Kettering Institute For Cancer Research | Potent inducers of terminal differentiation and method of use thereof |
| US5369108A (en) | 1991-10-04 | 1994-11-29 | Sloan-Kettering Institute For Cancer Research | Potent inducers of terminal differentiation and methods of use thereof |
| US6087367A (en) | 1991-10-04 | 2000-07-11 | Sloan-Kettering Institute For Cancer Research | Potent inducers of terminal differentiation and methods of use thereof |
| USRE38506E1 (en) | 1991-10-04 | 2004-04-20 | Sloan-Kettering Institute For Cancer Research | Potent inducers of terminal differentiation and methods of use thereof |
| US6174905B1 (en) | 1996-09-30 | 2001-01-16 | Mitsui Chemicals, Inc. | Cell differentiation inducer |
| US6897220B2 (en) | 2001-09-14 | 2005-05-24 | Methylgene, Inc. | Inhibitors of histone deacetylase |
| US7399787B2 (en) | 2002-03-04 | 2008-07-15 | Merck Hdac Research, Llc | Methods of treating cancer with HDAC inhibitors |
| US7456219B2 (en) | 2002-03-04 | 2008-11-25 | Merck Hdac Research, Llc | Polymorphs of suberoylanilide hydroxamic acid |
| WO2006023639A1 (en) * | 2004-08-18 | 2006-03-02 | Concordia Pharmaceuticals, Inc. | Methods and compositions for oral delivery of fts |
| WO2007064448A2 (en) * | 2005-11-28 | 2007-06-07 | Ramot At Tel Aviv University, Ltd. | Cancer treatment using fts and 2-deoxyglucose |
| WO2008143947A1 (en) * | 2007-05-14 | 2008-11-27 | University Of South Florida | Farnesylamine derivatives and methods of use |
| WO2009067500A1 (en) * | 2007-11-19 | 2009-05-28 | Syndax Pharmaceuticals, Inc. | Administration of an inhibitor of hdac |
Non-Patent Citations (56)
| Title |
|---|
| "Remington's Pharmaceutical Sciences, The Science and Practice of Pharmacy", 2000 |
| ANDREWS ET AL., INT. J. PARASITOL, vol. 30, 2000, pages 761 - 8 |
| BARBACID, BIOCHEM., vol. 56, 1987, pages 779 - 827 |
| BLUM ET AL., CANCER RES., vol. 67, 2007, pages 3320 - 8 |
| BLUM ET AL., INT. J. CANCER, vol. 119, 2006, pages 527 - 38 |
| BLUM ET AL., MOL. CANCER THER., vol. 5, 2006, pages 2337 - 47 |
| BOGUSKI ET AL., NATURE, vol. 366, 1993, pages 643 - 654 |
| BOS, CANCER RES., vol. 49, 1989, pages 4682 - 4689 |
| BOS, CANCER RES., vol. 49, 1989, pages 4682 - 89 |
| BOX, EUR. J. CANCER, vol. 31, 1995, pages 1051 - 1054 |
| CELL STRUCT. FUNCT., vol. 26, 2001, pages 653 - 7 |
| COX ET AL., CURR. OPIN. CELL BIOL., vol. 4, 1992, pages 1008 - 1016 |
| COX ET AL., MOL. CELL. BIOL., vol. 12, 1992, pages 2606 - 2615 |
| DELACOUR-LAROSE ET AL., MED. SCI, vol. 24, 2008, pages 828 - 32 |
| DOWNWARD, J., NAT. REV. CANCER, vol. 3, 2003, pages 11 - 22 |
| ELAD-SFADIA ET AL., J. BIOL. CHEM., vol. 277, 2002, pages 37169 - 37175 |
| ELAD-SFADIA ET AL., J. BIOL. CHEM., vol. 279, 2004, pages 34922 - 34930 |
| ERLICH ET AL., BIOCHEM. PHARMACOL., vol. 72, 2006, pages 427 - 36 |
| FUJIWARA ET AL., BR. J. HAEMATOL., vol. 135, 2006, pages 583 - 90 |
| FURUMAI ET AL., PNAS, vol. 98, no. 1, 2001, pages 87 - 92 |
| GANA-WEISZ, M. ET AL., CLIN. CANCER RES., vol. 8, 2002, pages 555 - 65 |
| GOODMANGILMAN: "Pergamon; and Remington's, Pharmaceutical Sciences", MACK PUBLISHING CO., article "The Pharmacological Basis of Therapeutics" |
| GREENBLATT DAVID YU ET AL: "Valproic acid activates notch-1 signaling and regulates the neuroendocrine phenotype in carcinoid cancer cells.", THE ONCOLOGIST AUG 2007 LNKD- PUBMED:17766653, vol. 12, no. 8, August 2007 (2007-08-01), pages 942 - 951, XP002620461, ISSN: 1083-7159 * |
| HAKLAI ET AL., BIOCHEMISTRY, vol. 37, no. 5, 1998, pages 1306 - 14 |
| HANCOCK ET AL., CELL, vol. 57, 1989, pages 1167 - 1177 |
| HANCOCK ET AL., EMBO J., vol. 10, 1991, pages 4033 - 4039 |
| JOHNSTONE, NATURE REVIEWS DRUG DISOVERY, vol. 1, 2002, pages 287 - 99 |
| KOGHE ET AL., BIOCHEM. PHARMACOL., vol. 56, 1998, pages 1359 - 64 |
| KOLLAREDDY ET AL., BOMED. PAP. MED., vol. 152, 2008, pages 27 - 33 |
| LEE ET AL., INT. J. ONCOL., vol. 33, no. 4, 2008, pages 767 - 76 |
| LOPREVITE ET AL., ONCOL RES., vol. 15, no. 1, 2005, pages 39 - 48 |
| MARKS PAUL A ET AL: "Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug.", NATURE BIOTECHNOLOGY JAN 2007 LNKD- PUBMED:17211407, vol. 25, no. 1, January 2007 (2007-01-01), pages 84 - 90, XP002620460, ISSN: 1087-0156 * |
| MARSHALL, CURR. OPIN. CELL BIOL., vol. 8, 1996, pages 197 - 204 |
| MITIN, N. ET AL., CURR. BIOL., vol. 15, 2005, pages R563 - 74 |
| PARK ET AL., CLIN. CANCER RES., vol. 10, 2004, pages 5271 |
| RAEMAEKERS ET AL., J. CELL. BIOL., vol. 162, 2003, pages 1017 - 29 |
| REMISZEWSKI ET AL., J. MED. CHEM., vol. 46, no. 21, 2003, pages 4609 - 23 |
| REUTHER ET AL., CURR. OPIN. CELL BIOL., vol. 12, 2000, pages 157 - 65 |
| RIBBECK ET AL., CURR. BIOL., vol. 17, 2007, pages 230 - 6 |
| RICHON ET AL., PNAS, vol. 95, 1998, pages 3003 - 7 |
| ROTBALT, METH. ENZYMOL., vol. 439, 2008, pages 467 - 89 |
| ROTBLAT ET AL., J. BIOL. CHEM., vol. 64, 2004, pages 3112 - 3118 |
| RUCHAUD ET AL., MOL. CELL BIOL., vol. 8, 2007, pages 798 - 812 |
| SAVICKIENE ET AL., EUR J PHARMACOL., vol. 549, no. 1-3, 2006, pages 9 - 18 |
| SCHEFFZEK ET AL., SCIENCE, vol. 277, 1997, pages 333 - 338 |
| SCHEFFZEK ET AL., SCIENCE, vol. 277, 1997, pages 333 - 7 |
| SHANNON, CURR. BIOL., vol. 12, 2002, pages R458 - 60 |
| SHIELDS, J. M. ET AL., TRENDS CELL BIOL., vol. 10, 2000, pages 147 - 541 |
| STACEY, CURR OPIN CELL BIOL., vol. 15, no. 2, 2003, pages 158 - 63 |
| TAKAI ET AL., GYNECOL ONCOL, vol. 101, 2006, pages 108 - 113 |
| TAN ET AL., J HEMATOL ONCOL., vol. 3, 2010, pages 5 |
| VADER, BIOCHIM. BIOPHYS. ACTA, vol. 1786, 2008, pages 60 - 72 |
| VAGNARELLI, CHROMOSOMA, vol. 113, 2004, pages 211 - 22 |
| VAS ET AL., CELL CYCLE, vol. 7, 2008, pages 293 - 6 |
| WEISZ, B. ET AL., ONCOGENE, vol. 18, 1999, pages 2579 - 2588 |
| ZHANG ET AL., CANCER BIOL. THER., vol. 7, 2008, pages 1388 - 97 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103113274A (en) * | 2013-01-25 | 2013-05-22 | 南通大学 | Ras and HDAC dual inhibitor and its preparation method and use |
| WO2024246114A1 (en) * | 2023-05-31 | 2024-12-05 | Boehringer Ingelheim International Gmbh | Survivin as a biomarker for predicting the responsiveness of cancer treatment |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2013508458A (en) | 2013-03-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Martínez-Iglesias et al. | Histone deacetylase inhibitors: mechanism of action and therapeutic use in cancer | |
| KR100537707B1 (en) | Analgesic Compositions Comprising Anti-epileptic Compounds and Methods of Using Same | |
| US20060009526A1 (en) | Method of treating TRX mediated diseases | |
| US20150283136A1 (en) | Pharmaceutical combination comprising a b-raf inhibitor and a histone deacetylase inhibitor and their use in the treatment of proliferative diseases | |
| KR20150013476A (en) | Pharmaceutical compositions for combination therapy | |
| US8093220B2 (en) | Combination of an HDAC inhibitor and an antimetabolite | |
| WO2015184279A1 (en) | Compositions and methods for treating kabuki syndrome and related disorders | |
| WO2008154382A1 (en) | Hdac inhibitors and hormone targeted drugs for the treatment of cancer | |
| JP2006290863A (en) | Treatment of gastrointestinal disorder | |
| US20170014376A1 (en) | Systems, methods, and formulations for treating cancer | |
| Vane et al. | Mechanism of action of anti-inflammatory drugs | |
| KR101354237B1 (en) | Use of hdac inhibitors for the treatment of myeloma | |
| AU2019261718A1 (en) | Cancer treatment | |
| WO2019040319A1 (en) | Methods of improving cancer chemotherapy | |
| WO2011056542A1 (en) | Cancer therapy with combinations of fts with hdac inhibitors | |
| KR20120090060A (en) | Cancer cell apoptosis | |
| EP2344253B1 (en) | Use of histone deacetylase inhibitors for the care of philadelphia-negative myeloproliferative syndromes | |
| Wolfson et al. | Enhancing FTS (Salirasib) efficiency via combinatorial treatment | |
| EP1296665B1 (en) | Analgesic and anti-inflammatory compositions containing celecoxib and ibuprofen | |
| US20140370080A1 (en) | Histone deacetylase (hdac) inhibitors for treatment of post-surgical adhesions | |
| WO2013085902A1 (en) | Combination therapy methods for treating an inflammatory breast cancer | |
| JP7332589B2 (en) | Combining an MDM2 inhibitor and an inhibitor of ERK to treat cancer | |
| US8217079B2 (en) | Method for treating Philadelphia-negative myeloproliferative syndromes | |
| US20090306098A1 (en) | Combination of roscovitine and a hdca inhibitor to treat proliferative diseases | |
| US20060100286A1 (en) | Treatment of canine hemangiosarcoma with a histone deacetylase inhibitor |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10774375 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012536940 Country of ref document: JP |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 10774375 Country of ref document: EP Kind code of ref document: A1 |