WO2008073461A2 - Ion channel modulators - Google Patents
Ion channel modulators Download PDFInfo
- Publication number
- WO2008073461A2 WO2008073461A2 PCT/US2007/025416 US2007025416W WO2008073461A2 WO 2008073461 A2 WO2008073461 A2 WO 2008073461A2 US 2007025416 W US2007025416 W US 2007025416W WO 2008073461 A2 WO2008073461 A2 WO 2008073461A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- group
- alkyl
- fluorophenyl
- pyridin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *c1c(C(N(*)*[Al]*)=O)nn[n]1* Chemical compound *c1c(C(N(*)*[Al]*)=O)nn[n]1* 0.000 description 8
- ZTSRVXMFPQBBPH-UHFFFAOYSA-N CC(C)(C)OC(N(Cc1c[s]c(N2CCCCC2)n1)c(cc1)ccc1F)=O Chemical compound CC(C)(C)OC(N(Cc1c[s]c(N2CCCCC2)n1)c(cc1)ccc1F)=O ZTSRVXMFPQBBPH-UHFFFAOYSA-N 0.000 description 1
- WFFGQLYKMWGXMU-UHFFFAOYSA-N CC(C)(C)OC(NC1CC1)=O Chemical compound CC(C)(C)OC(NC1CC1)=O WFFGQLYKMWGXMU-UHFFFAOYSA-N 0.000 description 1
- LVCLRVXPUWMWRQ-XEAZBWPWSA-N CC([C@H](C)F)[N]1(C(C)[C@@H]1C)C(OC(C)(C)C)=O Chemical compound CC([C@H](C)F)[N]1(C(C)[C@@H]1C)C(OC(C)(C)C)=O LVCLRVXPUWMWRQ-XEAZBWPWSA-N 0.000 description 1
- HDTFEWPWKHBMDX-UHFFFAOYSA-N CCN(C1CC1)C(OC(C)(C)C)=O Chemical compound CCN(C1CC1)C(OC(C)(C)C)=O HDTFEWPWKHBMDX-UHFFFAOYSA-N 0.000 description 1
- KHGVBURTSBEJGG-UHFFFAOYSA-N CCN(CC)c1cccc(CN(C(c2c[n](Cc3ccccc3)c(C)n2)=O)c(cc2)ccc2F)n1 Chemical compound CCN(CC)c1cccc(CN(C(c2c[n](Cc3ccccc3)c(C)n2)=O)c(cc2)ccc2F)n1 KHGVBURTSBEJGG-UHFFFAOYSA-N 0.000 description 1
- PGNOYARCOJMDDM-UHFFFAOYSA-N CN(c1nc(C(O)=O)c[s]1)c1ccccc1 Chemical compound CN(c1nc(C(O)=O)c[s]1)c1ccccc1 PGNOYARCOJMDDM-UHFFFAOYSA-N 0.000 description 1
- SKLRSBJYSHCISQ-UHFFFAOYSA-N Cc1nc(SC)c[s]1 Chemical compound Cc1nc(SC)c[s]1 SKLRSBJYSHCISQ-UHFFFAOYSA-N 0.000 description 1
- HKVYINROOPCUIP-UHFFFAOYSA-N ClCc1c[s]c(Cl)n1 Chemical compound ClCc1c[s]c(Cl)n1 HKVYINROOPCUIP-UHFFFAOYSA-N 0.000 description 1
- WFNBXZSZIBQEFF-UHFFFAOYSA-N ClCc1c[s]c(N2CCCCC2)n1 Chemical compound ClCc1c[s]c(N2CCCCC2)n1 WFNBXZSZIBQEFF-UHFFFAOYSA-N 0.000 description 1
- KKFZRNANSMPFHM-UHFFFAOYSA-N FC(c1ncc(CNc2ccccc2)cc1)(F)F Chemical compound FC(c1ncc(CNc2ccccc2)cc1)(F)F KKFZRNANSMPFHM-UHFFFAOYSA-N 0.000 description 1
- ZTLZNKQMGKBRIH-UHFFFAOYSA-N Fc(cc1)ccc1NCc1c[s]c(Cl)n1 Chemical compound Fc(cc1)ccc1NCc1c[s]c(Cl)n1 ZTLZNKQMGKBRIH-UHFFFAOYSA-N 0.000 description 1
- YIGDCLWWWZYTAX-UHFFFAOYSA-N Fc(cc1)ccc1NCc1c[s]c(N2CCCCC2)n1 Chemical compound Fc(cc1)ccc1NCc1c[s]c(N2CCCCC2)n1 YIGDCLWWWZYTAX-UHFFFAOYSA-N 0.000 description 1
- KRZCOLNOCZKSDF-UHFFFAOYSA-N Nc(cc1)ccc1F Chemical compound Nc(cc1)ccc1F KRZCOLNOCZKSDF-UHFFFAOYSA-N 0.000 description 1
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Nc1ccccc1 Chemical compound Nc1ccccc1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 1
- WYDKXPVKJAYJHU-UHFFFAOYSA-N O=C(c1c[n](Cc2ccccc2)cn1)N(Cc1cnc(N2CCCCC2)nc1)c(cc1)ccc1F Chemical compound O=C(c1c[n](Cc2ccccc2)cn1)N(Cc1cnc(N2CCCCC2)nc1)c(cc1)ccc1F WYDKXPVKJAYJHU-UHFFFAOYSA-N 0.000 description 1
- GSMYXUWJVOIHGK-UHFFFAOYSA-N O=C(c1c[n](Cc2ccccc2)nn1)N(Cc1cnc(N2CCCCC2)[s]1)c(cc1)ccc1F Chemical compound O=C(c1c[n](Cc2ccccc2)nn1)N(Cc1cnc(N2CCCCC2)[s]1)c(cc1)ccc1F GSMYXUWJVOIHGK-UHFFFAOYSA-N 0.000 description 1
- AFWWKZCPPRPDQK-UHFFFAOYSA-N O=Cc(cc1)cnc1Cl Chemical compound O=Cc(cc1)cnc1Cl AFWWKZCPPRPDQK-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/32—Alcohol-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/34—Tobacco-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/36—Opioid-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/30—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members
- C07D207/34—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/90—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/04—1,2,3-Triazoles; Hydrogenated 1,2,3-triazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/56—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/34—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D307/56—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D307/68—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/34—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D307/56—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D307/70—Nitro radicals
- C07D307/71—Nitro radicals attached in position 5
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/50—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom condensed with carbocyclic rings or ring systems
- C07D333/52—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes
- C07D333/62—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the hetero ring
- C07D333/68—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
- C07D333/70—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- the present teachings relate to certain carboxylic acid amides and related derivatives, processes for their preparation, and their use in therapeutic treatments.
- ion channels that permit these changes are proteinaceous pores consisting of one or multiple subunits, each containing two or more membrane-spanning domains. Most ion channels have selectivity for specific ions, primarily Na + , K + , Ca 2+ , or Cl " , by virtue of physical preferences for size and charge. Electrochemical forces, rather than active transport, drive ions across membranes, thus a single channel may allow the passage of millions of ions per second.
- Channel opening, or "gating" is tightly controlled by changes in voltage or by ligand binding, depending on the subclass of channel. Ion channels are attractive therapeutic targets due to their involvement in so many physiological processes, yet the generation of drugs with specificity for particular channels in particular tissue types remains a major challenge.
- Voltage-gated ion channels open in response to changes in membrane potential. For example, depolarization of excitable cells such as neurons results in a transient influx of Na + ions, which propagates nerve impulses. This change in membrane potential is sensed by voltage-gated K + channels, which then allow an efflux of K + ions. The efflux of K + ions repolarizes the membrane. Other cell types rely on voltage-gated Ca 2+ channels to generate action potentials. Voltage-gated ion channels also perform important functions in non-excitable cells, such as the regulation of secretory, homeostatic, and mitogenic processes.
- Ligand-gated ion channels can be opened by extracellular stimuli such as neurotransmitters (e.g., glutamate, serotonin, and acetylcholine), or intracellular stimuli (e.g., cAMP, Ca 2+ , and phosphorylation).
- extracellular stimuli such as neurotransmitters (e.g., glutamate, serotonin, and acetylcholine), or intracellular stimuli (e.g., cAMP, Ca 2+ , and phosphorylation).
- the Ca v 2 family of voltage-gated calcium channels consists of 3 main subtypes Ca v 2.1 (P or Q-type calcium currents), Ca v 2.2 (N-type calcium currents), and Ca v 2.3 (R-type calcium currents). These currents are found almost exclusively in the central nervous system (CNS), peripheral nervous system (PNS) and neuroendocrine cells, and constitute the predominant forms of presynaptic voltage-gated calcium current. Presynaptic calcium entry is modulated by many types of G-protein coupled receptors (GPCRs) and modulation of Ca v 2 channels is a widespread and highly efficacious means of regulating neurotransmission.
- GPCRs G-protein coupled receptors
- the subunit composition of the Ca v 2 channels is defined by their Ci 1 subunit, which forms the pore and contains the voltage-sensing gates (0 ⁇ 2.1 , c ⁇ i2.2, and ⁇ i2.3, also known as ⁇ i A , ⁇ 1B , and ⁇ 1E , respectively) and the ⁇ and ⁇ 2 subunits.
- ion channel function can have dramatic clinical consequences. Long QT syndrome, epilepsy, cystic fibrosis, and episodic ataxia are a few examples of heritable diseases resulting from mutations in ion channel subunits. Toxic side effects such as arrhythmia and seizure, which can be triggered by certain drugs, can be due to interference with ion channel function (Sirois, J. E. and Atchison, W. D. (1996), Neurotoxicology, 17(1 ): 63-84; Keating, MT. (1996), Science, 272: 681-685).
- Drugs are useful for the therapeutic modulation of ion channel activity, and have applications in treatment of many pathological conditions, including hypertension, angina pectoris, myocardial ischemia, asthma, bladder overactivity, alopecia, pain, heart failure, dysmenorrhea, type Il diabetes, arrhythmia, graft rejection, seizure, convulsions, epilepsy, stroke, gastric hypermotility, psychoses, cancer, muscular dystrophy, and narcolepsy (Coghlan, MJ. et al. (2001 ), J. Med. Chem., 44: 1627-1653; Ackerman, M.J. and Clapham, D. E. (1997), N. Eng. J. Med., 336: 1575-1586).
- pathological conditions including hypertension, angina pectoris, myocardial ischemia, asthma, bladder overactivity, alopecia, pain, heart failure, dysmenorrhea, type Il diabetes, arrhythmia, graft rejection, seizure,
- Therapeutic modulation of Ca v 2 channel activity has applications in treatment of many pathological conditions. All primary sensory afferents provide input to neurons in the dorsal horns of the spinal cord and in dorsal root ganglia neurons in the dorsal horn, and calcium influx through Ca v 2.2 channels triggers the release of neurotransmitters from presynaptic nerve terminals in the spinal cord. Hence, blockade of Ca v 2.2 channels is expected to be broadly efficacious because these channels are in a common pathway downstream from the wide variety of receptors that mediate pain (Julius, D. and Basbaum, A.I. (2001 ), Nature, 413: 203-216).
- Ca v 2.2 channels are found in the periphery and mediate catecholamine release from sympathetic neurons and adrenal chroffin cells. Some forms of hypertension result from elevated sympathetic tone. Ca v 2.2 modulators could be particularly effective in treating this disorder. Although complete block of Ca v 2.2 channels can cause hypotension or impair baroreceptor reflexes, partial inhibition by Ca v 2.2 modulators might reduce hypertension with minimal reflex tachycardia (Uneyama, O. D. (1999), Int. J. MoI. Med., 3: 455-466).
- Overactive bladder is characterized by storage symptoms such as urgency, frequency, and nocturia, with or without urge incontinence, resulting from the overactivity of the detrusor muscle in the bladder. OAB can lead to urge incontinence.
- the etiology of OAB and painful bladder syndrome is unknown, although disturbances in nerves, smooth muscle and urothelium can cause OAB (Steers, W., Rev. Urol., 4: S7-S18). There is evidence to suggest that reduction of bladder hyperactivity may be indirectly effected by inhibition of Ca v 2.2 and/or Ca v 1 channels.
- gabapentin was designed as a metabologically stable GABA mimetic, but most studies find no effect on the GABA receptors.
- the ⁇ 2 ⁇ subunit of voltage-gated calcium channels has been identified as a high affinity binding site for gabapentin in the CNS. There is evidence that suggests that gabapentin could inhibit neurotransmission in the spinal cord by interfering with the function of the ⁇ 2 ⁇ subunits, thereby inhibiting presynaptic calcium currents.
- the present teachings also provide methods of making the compounds of formula (I), and methods of using the compounds of formula (I) for the therapeutic modulation of ion channel function, and treatment of one or more conditions, particularly those mediated by certain calcium channel subtype targets.
- the methods of using the compounds generally include administering a therapeutically effective amount of a compound of formula (I) to a mammal.
- the present teachings also provide methods of making the compounds of formula (II), and methods of using the compounds of formula (II) for the therapeutic modulation of ion channel function, and treatment of one or more conditions, particularly those mediated by certain calcium channel subtype targets.
- the methods of using the compounds generally include administering a therapeutically effective amount of a compound of formula (II) to a mammal.
- the present teachings also provide methods of making the compounds of formula (III), and methods of using the compounds of formula (III) for the therapeutic modulation of ion channel function, and treatment of one or more conditions, particularly those mediated by certain calcium channel subtype targets.
- the methods of using the compounds generally include administering a therapeutically effective amount of a compound of formula (III) to a mammal.
- the present teachings also provide methods of making the compounds of formula (IV), and methods of using the compounds of formula (IV) for the therapeutic modulation of ion channel function, and treatment of one or more conditions, particularly those mediated by certain calcium channel subtype targets.
- the methods of using the compounds generally include administering a therapeutically effective amount of a compound of formula (IV) to a mammal.
- the present teachings also provide methods of making the compounds of formula (V), and methods of using the compounds of formula (V) for the therapeutic modulation of ion channel function, and treatment of one or more conditions, particularly those mediated by certain calcium channel subtype targets.
- the methods of using the compounds generally include administering a therapeutically effective amount of a compound of formula (V) to a mammal.
- the present teachings also provide methods of making the compounds of formula (VI), and methods of using the compounds of formula (Vl) for the therapeutic modulation of ion channel function, and treatment of one or more conditions, particularly those mediated by certain calcium channel subtype targets.
- the methods of using the compounds generally include administering a therapeutically effective amount of a compound of formula (Vl) to a mammal.
- the present teachings relate to compounds of formula (VII): and pharmaceutically acceptable salts, hydrates, and esters thereof, wherein Ar, R 2 , R 3 , p and
- the present teachings also provide methods of making the compounds of formula (VII), and methods of using the compounds of formula (VII) for the therapeutic modulation of ion channel function, and treatment of one or more conditions, particularly those mediated by certain calcium channel subtype targets.
- the methods of using the compounds generally include administering a therapeutically effective amount of a compound of formula (VII) to a mammal.
- Embodiments of the present invention provide compounds that can modulate the activity of ion channels in a mammal, for example, Ca v 2.2 voltage-gated calcium channels, and can treat a variety of pathological conditions, states, disorders or diseases. Unless otherwise indicated, the following terms are held to have the following meanings as used herein.
- “mammal” refers to any warm blooded species, such as a human.
- “ion channel” includes at least voltage-gated calcium channels and voltage- gated sodium channels such as, without limitation, Ca v 1.1 , Ca v 1.2, Ca v 1.3, Ca v 2.1 , Ca v 2.2, Ca v 2.3, Ca v 3.1 , Ca v 3.2, Na «1.1 , Na v 1.2, NaJ .3, Na v 1.7, NaJ .8, and NaJ .9.
- “Ca v 2.2 voltage-gated calcium channel” refers to a voltage-gated calcium channel containing at least one Ca v 2.2 ⁇ i subunit.
- ion channel mediated condition refers to any condition or pathological state of a mammal or any disease present in a mammal that can be treated, or the symptoms of which can be alleviated, by modulation of the activity of one or more ion channels such as Ca v 2.2 voltage-gated calcium channels.
- halo or halogen refers to fluoro, chloro, bromo, and iodo.
- alkyl refers to a straight-chain or branched saturated hydrocarbon group.
- alkyl groups include methyl (Me), ethyl (Et), propyl (e.g., n-propyl and isopropyl), butyl (e.g., n-butyl, isobutyl, s-butyl, t-butyl), pentyl groups (e.g., n-pentyl, isopentyl, neopentyl), and the like.
- a lower alkyl group typically has up to 6 carbon atoms.
- an alkyl group has 1-6 carbon atoms, and is referred to as a "C 1-6 alkyl group.”
- C 1-6 alkyl groups include, but are not limited to, methyl, ethyl, propyl (e.g., n-propyl and isopropyl), butyl (e.g., n-butyl, isobutyl, s-butyl, t-butyl), pentyl (e.g., n-pentyl, neopentyl, isopentyl, t-pentyl), and hexyl groups (e.g., n-hexyl, isohexyl).
- a branched alkyl group has at least 3 carbon atoms (e.g., an isopropyl group), and in various embodiments, has up to 6 carbon atoms, e.g. it is a C 3-6 alkyl group, i.e., a branched lower alkyl group.
- Examples of branched lower alkyl groups include, but are not limited to:
- alkenyl refers to a straight-chain or branched alkyl group having one or more carbon-carbon double bonds.
- alkenyl groups include, but are not limited to, ethenyl, propenyl, butenyl, pentenyl, hexenyl, butadienyl, pentadienyl, hexadienyl groups, and the like.
- the one or more carbon-carbon double bonds can be internal (such as in 2-butene) or terminal (such as in 1-butene).
- a branched alkenyl group has at least 3 carbon atoms, and in various embodiments, has up to 6 carbon atoms, e.g. it is a C 3-6 alkenyl group.
- alkynyl refers to a straight-chain or branched alkyl group having one or more carbon-carbon triple bonds.
- alkynyl groups include, but are not limited to, ethynyl, propynyl, butynyl, pentynyl, and the like.
- the one or more carbon- carbon triple bonds can be internal (such as in 2-butyne) or terminal (such as in 1- butyne).
- the alkynyl group is suitably a C 3-6 alkynyl group.
- alkoxy refers to an -O-alkyl group, wherein the alkyl group may be a straight or branched chain.
- alkoxy groups include, but are not limited to, methoxy, ethoxy, propoxy (e.g., n-propoxy and isopropoxy), t-butoxy groups, and the like.
- haloalkyl refers to an alkyl group having one or more halogen substituents.
- haloalkyl groups include, but are not limited to, -CF 3 , -C 2 F 5 , -CHF 2 , -CH 2 F, -CCI 3 , -CHCI 2 , -CH 2 CI, -C 2 CI 5 , and the like.
- Perhaloalkyl groups i.e., alkyl groups wherein all of the hydrogen atoms are replaced with halogen atoms (e.g., CF 3 and C 2 F 5 ), are included within the definition of "haloalkyl.”
- cycloalkyl refers to a non-aromatic carbocyclic group including cyclized alkyl, alkenyl, and alkynyl groups.
- a cycloalkyl group can be monocyclic (e.g., cyclohexyl) or polycyclic (e.g., containing fused, bridged, and/or spiro ring systems), wherein the carbon atoms are located inside or outside of the ring system.
- a cycloalkyl group has 3-6 carbon atoms, and is referred to as a "C 3-6 cycloalkyl group.”
- C 3-6 cycloalkyl groups include, but are not limited to, cyclopropyl, cyclop ropy I methyl, cyclopropylethyl, cyclopropylpropyl, cyclobutyl, cyclobutylmethyl, cyclobutylethyl, cyclopentyl, cyclopentylmethyl, cyclohexyl, cyclopentenyl, cyclohexenyl, and cyclohexadienyl groups, as well as their homologs, isomers, and the like.
- cycloalkyl groups are moieties that have one or more aromatic rings fused (i.e., having a bond in common with) to the cycloalkyl ring, for example, benzo derivatives of cyclopentane (i.e., an indanyl group), cyclohexane (i.e., a tetrahydronaphthyl group), and the like.
- benzo derivatives of cyclopentane i.e., an indanyl group
- cyclohexane i.e., a tetrahydronaphthyl group
- cycloalkyl groups can be substituted with 1 to 3 substituents independently selected from a Ci -6 alkyl group and a -Y-phenyl group, wherein the phenyl group is optionally substituted with 1 to 3 substituted independently selected from a halogen and a Ci -6 alkoxyl group, and Y is as defined hereinbelow.
- heteroatom refers to an atom of any element other than carbon or hydrogen and includes, for example, nitrogen, oxygen, sulfur, phosphorus, and selenium.
- cycloheteroalkyl refers to a non-aromatic cycloalkyl group having 5-10 ring atoms, among which 1 to 3 ring atoms are heteroatoms independently selected from oxygen (O), nitrogen (N) and sulfur (S), and that optionally contains one or more, e.g., two, double or triple bonds. Also included in the definition of cycloheteroalkyl are moieties that have one or more aromatic rings fused (i.e., have a bond in common with) to the cycloheteroalkyl group.
- cycloheteroalkyl groups include, among others, morpholine, thiomorpholine, pyran, imidazolidine, imidazoline, oxazolidine, pyrazolidine, pyrazoline, pyrrolidine, pyrroline, tetrahydrofuran, tetrahydrothiophene, piperidine, piperazine, tetrahydroisoquinoline, benzimidazoline, chromane, chromene, indolinetetrahydroquinoline, and the like.
- N or S atoms in a cycloheteroalkyl ring can be oxidized (e.g., morpholine N-oxide, thiomorpholine S-oxide, thiomorpholine S,S-dioxide).
- Cycloheteroalkyl groups can also contain one or more oxo groups, such as piperidone, oxazolidinone, pyrimidine-2,4(1/-/,3H)-dione, pyridin-2(1H)-one, and the like.
- a cycloheteroalkyl group can be optionally substituted.
- one or more carbon ring atoms of a cycloheteroalkyl group can bear a substituent independently selected from a halogen, a C 1-6 alkyl group, -C(O)-NR d R e , -Y-OR 0 , -Y-NRdRe, a -Y-phenyl group, a -Y-(5-7 membered cycloheteroalkyl), a - Y-(5-9 membered heteroaryl) group, or a -Y-O-(5-7 membered heteroaryl) group, and/or one or more nitrogen ring atoms of a cycloheteroalkyl group can bear a substituent independently selected from a halogen, a C 1-6 alkyl group, -C(O)R C , -C 2-6 alkyl-OR c , -C 2-6 alkyl-NR d R e , -Y-C(
- each of the phenyl substituents immediately above can be optionally substituted with 1 to 3 substituents independently selected from a halogen, a C 1-6 alkyl group, a C 1-6 haloalkyl group, and a C 1-6 alkoxy group, and each of the 5-7 membered cycloheteroalkyl substituents, the 5-7 membered heteroaryl substituents, and the 5-9 membered heteroaryl substituents immediately above can be optionally substituted with 1 to 3 substituents independently selected from a halogen and a C 1-6 alkyl group.
- aryl refers to an aromatic monocyclic or polycyclic hydrocarbon ring system such as, for example, phenyl, 1-naphthyl, 2-naphthyl, anthracenyl, phenanthrenyl groups, and the like.
- a monocyclic aryl group can have from 6 to 14 carbon atoms and a polycyclic aryl group can have from 8 to 14 carbon atoms. Any suitable ring position of the aryl group can be covalently linked to the defined chemical structure.
- aryl groups optionally contain up to three independently selected substituent groups.
- a phenyl group in some embodiments, can be optionally substituted with 1 to 3 substituents independently selected from a halogen, CN, -C(O)OR 0 , -NR d R e , a C 1-6 alkyl group, a C 1-6 haloalkyl group, and a C 1-6 alkoxy group, wherein R c , R d , and R e are as defined hereinafter.
- the term "heteroaryl” refers to an aromatic monocyclic or polycyclic aromatic ring system containing 5-7 ring atoms, among which 1 to 3 ring atoms are heteroatoms independently selected from oxygen (O), nitrogen (N) and sulfur (S).
- heteroaryl rings do not contain 0-0, S-S, or S-O bonds.
- one or more N or S atoms in a heteroaryl group can be oxidized (e.g., pyridine N-oxide, thiophene S- oxide, thiophene S,S-dioxide).
- heteroaryl groups include, for example, the 5-membered monocyclic and 5-6 bicyclic ring systems shown below:
- K is O, S, NH, or NR"; and R' can be selected from a halogen, a C 1-6 alkyl group, a C(O)R 0 group, a C 2-6 alkyl-OR c group, a C 2-6 alkyl-NR d R e group, a -Y-
- heteroaryl rings include, but are not limited to, pyrrole, furan, thiophene, pyridine, pyrimidine, pyridazine, pyrazine, triazole, tetrazole, pyrazole, imidazole, isothiazole, thiazole, thiadiazole, isoxazole, oxazole, oxadiazole, indole, isoindole, benzofuran, benzothiophene, quinoline, 2- methylquinoline, isoquinoline, quinoxaline, quinazoline, benzotriazole, benzimidazole, benzothiazole, benzisothiazole, benzisoxazole, benzoxadiazole, benzoxazole, cinnoline, 1 H-indazole, 2H-indazole, indolizine, isobenzofuran, naphthyridine, phthalazine,
- heteroaryl groups can be substituted with up to three independently selected substitution groups.
- one or more nitrogen atoms can be substituted with independently selected R 1 groups as defined above, and/or one or more carbon ring atoms of a cycloheteroalkyl group can bear a substituent independently selected from a halogen, a C 1-6 alkyl group, -C(O)-NR d R e , -Y-OR C , - Y-NR d R e , a -Y-phenyl group, a -Y-(5-7 membered cycloheteroalkyl) group, a -Y- (5-9 membered heteroaryl) group, or a -Y-O-(5-7 membered heteroaryl) group, wherein Y, R c , R d , and R e are as defined hereinafter.
- each of the phenyl substituents immediately above can be optionally substituted with 1 to 3 substituents independently selected from a halogen, a C 1-6 alkyl group, a C 1-6 haloalkyl group, and a C 1-6 alkoxy group, and each of the 5-7 membered cycloheteroalkyl substituents, the 5-7 membered heteroaryl substituents, and the 5-9 membered heteroaryl substituents immediately above can be optionally substituted with 1 to 3 substituents independently selected from a halogen and a C 1-6 alkyl group.
- divalent group is defined herein as a linking group capable of forming a covalent bond with two other moieties.
- compounds described herein can include a divalent C 1-6 alkyl group, such as, for example, a methylene group.
- LG refers to a charged or uncharged atom (or group of atoms) that can be displaced as a stable species as a result of, for example, a substitution or elimination reaction.
- leaving groups include, but are not limited to, halide (e.g., Cl, Br, I), tosylate (toluenesulfonyl group, TsO), mesylate (methanesulfonyl group, MsO), brosylate (p-bromobenzenesulfonyl group, BsO), nosylate (4-nitrobenzenesulfonyl group, NsO), water (H 2 O), ammonia (NH 3 ), and triflate (trifluoromethanesulfonyl group, OTf).
- halide e.g., Cl, Br, I
- tosylate toluenesulfonyl group, TsO
- mesylate methanesulfonyl group,
- protecting group refers to modification of a functional group that reduces the reactivity of the functional group in an unwanted reaction.
- protecting groups for amines include, but are not limited to, terf-butyloxycarbonyl (t- BOC), benzyl (Bn), and carbobenzyloxy (Cbz) groups.
- protecting groups for carbonyls include, but are not limited to, acetals and ketals.
- protecting groups for carboxylic acids include, but are not limited to, methyl esters, benzyl esters, te/t-butyl esters, and silyl esters, and are provided in Greene, et al., Protective Groups in Organic Synthesis, 2d. Ed., Wiley & Sons, 1991 , the entire disclosure of which is incorporated by reference herein for all purposes.
- C 1-6 alkyl is specifically intended to individually disclose Ci, C 2 , C 3 , C 4 , C 5 , C 6 , Ci-C 6 , C 1 -C 5 , C 1 -C 4 , C 1 -C 3 , C 1 -C 2 , C 2 -C 6 , C 2 -C 5 , C 2 -C 4 , C 2 -C 3 , C 3 -C 6 , C 3 -C 5 , C 3 -C 4 , C 4 -C 6 , C 4 -C 5 , and C 5 -C 6 alkyl.
- the term "5-9 membered heteroaryl group” is specifically intended to individually disclose a heteroaryl group having 5, 6, 7, 8, 9, 5-9, 5-8, 5-7, 5-6, 6-9, 6- 8, 6-7, 7-9, 7-8, and 8-9 ring atoms.
- X is selected from -NR 0 -, a divalent C 1-6 alkyl group, and a covalent bond;
- R 1 is selected from a C 1 ⁇ alkyl group, a phenyl group, and a 5-7 membered heteroaryl group;
- R 2 is a -C 3-6 cycloalkyl group, a benzyl group, an indole group, or a phenyl group, wherein
- the phenyl group and the benzyl group are each optionally substituted with 1 to 3 substituents independently selected from a halogen, a C 1-6 alkyl group, a C 1-6 alkoxy group, a C 1-6 haloalkyl group, a C 1-6 haloalkoxy group, -OCH 2 -C 6 H 5 , -CN, -C(O)OR C , -OH, and -NR d R e , and the -C 3-6 cycloalkyl group is optionally substituted with 1 to 3 C 1-6 alkyl groups;
- Ar-R 3 is selected from:
- R 3 is selected from a halogen, a C 1-10 alkyl group, a C 1-10 alkoxy group, a C 1-10 haloalkyl group, a C 1-I0 haloalkoxy group, a -C(O)R 0 group, a piperidin-4-yl group, and a -Y-NR f R 9 group; wherein the C 1-10 alkyl group and the C 1-10 alkoxy group are optionally substituted with from 1-3 substitutents selected from a halogen, a phenyl group, and -OH, and the nitrogen ring atom of the piperidin-4-yl group is optionally substituted with -C(O)O-C 1-6 alkyl;
- Y at each occurrence, is independently a divalent C 1-6 alkyl group or a covalent bond
- R 0 , Rd and R e at each occurrence, independently are H, C 1-6 haloalkyl, or a C 1-6 alkyl group
- Rf and R 9 at each occurrence, independently are selected from H 1 -C(O)R 0 , - C 2-6 alkyl-OR c , -C 2-6 alkyl-NR d R e , a C 1-10 alkyl group, a C 3-6 cycloalkyl group, a -Y-phenyl group, a -C(O)-phenyl group, a -Y-(5-7 membered cycloheteroalkyl) group, a -Y-(5-7 membered heteroaryl) group, and a -C 2-6 alkyl-O-Y-(5-7 membered heteroaryl) group, or
- R f and R 9 taken together with the nitrogen atom to which they are bonded form a 5-7 membered cycloheteroalkyl group or a 5-7 membered heteroaryl group, the 5-7 membered cycloheteroalkyl group and the 5-7 membered heteroaryl group containing up to two ring heteroatoms independently selected from nitrogen, oxygen, and sulfur, wherein one or more nitrogen atoms in the ring optionally are independently substituted with -C(O)R 0 , -C 2-6 alkyl-OR c , -C 2-6 alkyl-NR d R e , -Y-C(O)NR d R e , -S(O) 2 -(C 1-6 alkyl), a -C 1-6 alkyl-(phenyl) n group, a -C 2-6 alkyl— (5-7 membered cycloheteroalkyl) group, C 1-6 alkyl, -Y-(
- each of the phenyl groups appearing anywhere in said R f and R g is optionally substituted with 1 to 3 substituents independently selected from halogen, C 1-6 alkyl, C 1-6 haloalkyl, and C 1-6 alkoxy, and each of the 5-7 membered cycloheteroalkyl groups, the 5-7 membered heteroaryl groups, and the 5-9 membered heteroaryl groups appearing anywhere in said R f and R 9 is optionally substituted with 1 to 3 substituents independently selected from halogen and C 1-6 alkyl; n is 1 , 2, or 3;
- q is 1 , 2, or 3;
- p 1 , 2, 3, or 4.
- R 1 can be selected from a methyl group, a phenyl group, and a pyridyl group.
- X can be selected from -NH-, -N(CH 3 )-, -CH 2 -, and a covalent bond.
- p is 1.
- p is 2.
- p is 3.
- R 2 can be a phenyl group optionally substituted with 1-2 substituents independently selected from a halogen, a C 1-6 alkyl group, CN, -
- R 2 can be a 4-fluorophenyl group, a 4-chlorophenyl group, a 4-fluoro-2-methylphenyl group, or a 4-methoxyphenyl group.
- Ar-R 3 can be any organic compound.
- Ar-R 3 can be any organic compound.
- Ar-R 3 can be any organic radical
- R 3 can be NR f R g , wherein R f and Rg are as defined above.
- R 3 can be selected from NH 2 , an NH-C 1-6 alkyl group, an N(Ci -6 alkyl) 2 group wherein the C 1-6 alkyl groups do not need to be the same, wherein the C 1-6 alkyl groups do not need to be the same, an NH-C 3-6 cycloalkyl group, an N(C 1-6 alkyl)— C 3-6 cycloalkyl group, an N(C 1-6 alkyl)— C 2-6 alkyl-OR c group, an N(C 1-6 alkyl)- Y-(5-7 membered cycloheteroalkyl) group, an N(C 1-6 alkyl)-phenyl group, an N(C 1-6 alkyl)-phenyl group, an N(C 1-6 alkyl)-phenyl group, an N(C 1-6 alkyl)-phenyl group
- R 3 can be an optionally substituted 5-7 membered cycloheteroalkyl group or an optionally substituted 5-7 membered heteroaryl group
- R 3 can be selected from a diazepanyl group, an imidazolyl group, a morpholinyl group, a piperidinyl group, a piperazinyl group, a pyridyl group, a pyrrolidyl group, and a thiomorpholinyl group, wherein each of these groups can include a nitrogen ring atom optionally substituted with -C(O)R C , -C 2-6 alkyl-OR c , -C 2-6 alkyl-NR d R e , -Y-C(O)NR d R e , an -S(O) 2 -C 1-6 alkyl group, a - C 2-6 alkyl— (5-7 membered cycloheteroalkyl) group, a C 1-I0 alkyl group, or a 5-7 membered heteroaryl group, a carbon ring atom optionally
- R 3 can be selected from a 1-[1 ,4]diazepanyl group, a 1- imidazolyl group, a 4-morpholinyl group, a 1 -piperidinyl group, a 1 -piperazinyl group, a 4-pyridyl group, a 1 -pyrrolidyl group, and a 4-thiomorpholinyl group, wherein each of these groups can be optionally substituted as described above.
- R 3 can be a 1 -piperazinyl group having a nitrogen atom in the ring optionally substituted with -C(O)R C , -C 2-6 alkyl-OR c , -C 2-6 alkyl-NR d R e , -C 1-6 alkyl-C(O)NR d R e , an S(O) 2 -C 1-6 alkyl group, a -C 2-6 alkyl-(5-7 membered cycloheteroalkyl) group, a C 1-10 alkyl group, or a 5-7 membered heteroaryl group.
- R 3 can be a 4-methyl piperazin-1-yl group.
- R 3 can be a 1 -piperidinyl group having a carbon atom in the ring optionally substituted with -NR d R e , -C(O)-NR d R e , -Y-ORc, a 5-7 cycloheteroalkyl group, a 5-9 membered heteroaryl group, or a -Y-O-(5-7 membered heteroaryl) group.
- Embodiments of the present invention further provide compounds of formula (II):
- R 4 is selected from H, a C 1-6 alkyl group, and a -Y-phenyl group;
- R 5 is selected from H, a d.i 0 alkyl group, a C 3-6 alkenyl group, a C 3-8 cycloalkyl group, a phenyl group, a 5-7 membered heteroaryl group, a -C 1-6 alkyl-phenyl group, a -C 1-6 alkyl— 5-7 membered heteroaryl group, wherein each of the phenyl group and the 5-7 membered heteroaryl group optionally is substituted with 1 to 3 substituents independently selected from a halogen, a C 1-6 alkyl group, a C 1-6 haloalkyl group, and a C 1-6 alkoxy group; provided that at least one of R 4 and R 5 is not H. .
- R 4 can be H, a methyl group, or a benzyl group.
- R 5 can be selected from a C 1-6 alkyl group, C 3-6 alkenyl group, a C 3-6 cycloalkyl group, a -C 1-6 alkyl-phenyl group, and a -C 1-6 alkyl— 5-7 membered heteroaryl group, wherein each of the phenyl group and the 5-7 membered heteroaryl group optionally is substituted with 1 to 3 substituents selected from a halogen or a C 1-6 alkyl group.
- R 5 can be an allyl group, a benzyl group, a 4- chlorobenzyl group, a cyclopentyl group, a cyclopropylmethyl group, a 2,2-dimethyl- propyl group, a methyl group or a 5-methyl-furan-2-yl-methyl group.
- R 2 can be a phenyl group optionally substituted with 1-2 substituents independently selected from a halogen, CN, a C 1-6 alkyl group, - C(O)OR C and -NR d R e , wherein R c , Ra and R e are as defined above.
- R 2 can be a 4-fluorophenyl group, a 2-isopropylphenyl group, or a 4-methylphenyl group.
- p is 1.
- p is 2.
- p is 3.
- Ar-R 3 can be any organic compound.
- Ar-R 3 can be any organic compound.
- Ar-R 3 can be any organic radical
- Ar-R 3 can be any organic radical
- R 3 is as defined above.
- R 3 can be NR f R g , wherein R f and R 9 are as defined above.
- R 3 can be selected from NH 2 , an NH-C 1-6 alkyl group, an N(Ci -6 alkyl) 2 group wherein the C 1-6 alkyl groups do not need to be the same,, an NH-C 3-6 cycloalkyl group, an N(C 1-6 alkyl)— C 3-6 cycloalkyl group, an N(C 1-6 alkyl)— C 2-6 alkyl-OR c group, an N(C 1-6 alkyl)-Y-(5-7 membered cycloheteroalkyl) group, an N(C 1-6 alkyl)-phenyl group, an N(Ci -6 alkyl)- Y-5-7 membered heteroaryl group, and an N(C 1-6 alkyl)-C 2-6 alkyl-O-Y-5-7 membered heteroaryl group, wherein
- R 3 can be a diethylamino group, a dimethylamino group, an ethyl(methyl)amino group, an isopropyl(ethyl)amino group, or a cyclopropyl(ethyl)amino group.
- R 3 can be an optionally substituted 5-7 membered cycloheteroalkyl group or an optionally substituted 5-7 membered heteroaryl group as described herein.
- R 3 can be selected from a diazepanyl group, an imidazolyl group, a morpholinyl group, a piperidinyl group, a piperazinyl group, a pyridyl group, a pyrrolidyl group, and a thiomorpholinyl group, wherein each of these groups can include a nitrogen ring atom optionally substituted with -C(O)R C , -C 2-6 alkyl-OR c , -C 2-6 alkyl-NR d R e , -Y-C(O)NR d R 6 , an -S(O) 2 -C 1-6 alkyl group, a - C 2-6 alkyl— (5-7 membered cycloheteroal
- R 3 can be selected from a 1-[1 ,4]diazepanyl group, a 1- imidazolyl group, a 4-morpholinyl group, a 1 -piperidinyl group, a 1 -piperazinyl group, a 4-pyhdyl group, a 1 -pyrrolidyl group, and a 4-thiomorpholinyl group, wherein each of these groups can be optionally substituted as described above.
- R 3 can be a 1 -piperazinyl group having a nitrogen atom in the ring optionally substituted with -C(O)R C , -C 2-6 alkyl-OR c , -C 2-6 alkyl-NR d R e , -C 1-6 alkyl-C(O)NR d Re, an S(O) 2 -C 1-6 alkyl group, a -C 2-6 alkyl-(5-7 membered cycloheteroalkyl) group, a C 1-10 alkyl group, or a 5-7 membered heteroaryl group.
- R 3 can be a 4-methyl piperazin-1-yl group.
- R 3 can be a 1-piperidinyl group having a carbon atom in the 5 ring optionally substituted with -NR d R e , -C(O)-NR d R 6 , -Y-OR c , a 5-7 cycloheteroalkyl group, a 5-9 membered heteroaryl group, or a -Y-O-(5-7 membered heteroaryl) group.
- R 3 can be a methyl group or a trifluoromethyl group.
- Representative compounds of formula (II) in accordance with embodiments of the present invention include, but are not limited to, the compounds presented in Table 2 below.
- the present teachings additionally provide compounds of formula (III): or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein:
- R 2 , Ar, R 3 , and p are as defined above;
- R 6 is selected from a C 1-10 alkyl group, a phenyl group, a -C 1-6 alkyl-phenyl group, and a 5-7 membered heteroaryl group, wherein each of the phenyl group and the 5-7 membered heteroaryl group optionally is substituted with 1 to 3 substituents independently selected from a halogen, a C 1-6 alkyl group, a C 1-6 haloalkyl group, and a C 1-6 alkoxy group; and
- R 7 is H or a C 1-6 alkyl group.
- p is 1.
- p is 2.
- p is 3.
- R 6 can be a phenyl group, a benzyl group, or a pyridyl group.
- R 6 can be a pyridin-3-yl group.
- R 2 can be a -C 3-6 cycloalkyl group or a phenyl group optionally substituted with 1 to 2 substituents independently selected from a halogen, a C 1-6 alkyl group, a C 1-6 alkoxy group, CN, -C(O)OR 0 , and -NR d R e , wherein R c , R d and R e are as defined above.
- R 2 can be a cyclopropylmethyl group, a phenyl group, a 4-dimethylaminophenyl group, a 4-fluoro-2-methylphenyl group, a 4- methylphenyl group, a 2-isopropylphenyl group, a 4-methoxyphenyl group, a 4- chlorophenyl group or a 4-fluorophenyl group.
- Ar-R 3 can be wherein R 3 is as defined above.
- Ar-R 3 can be any organic radical
- R 3 is as defined above.
- Ar-R 3 can be any organic radical
- R 3 is as defined above.
- R 3 can be NR f R g , wherein R f and R 9 are as defined above.
- R 3 can be selected from NH 2 , an NH-C 1-6 alkyl group, an
- R 3 can be a diethylamino group, a dimethylamino group, or a cyclopropyl(ethyl)amino group.
- R 3 can be an optionally substituted 5-7 membered cycloheteroalkyl group or an optionally substituted 5-7 membered heteroaryl group as described herein.
- R 3 can be selected from a diazepanyl group, an imidazolyl group, a morpholinyl group, a piperidinyl group, a piperazinyl group, a pyridyl group, a pyrrolidyl group, and a thiomorpholinyl group, wherein each of these groups can include a nitrogen ring atom optionally substituted with -C(O)R C , -C 2-6 alkyl-OR c , -C 2-6 alkyl-NR d R e , -Y-C(O)NRdR 6 , an -S(O) 2 -Ci -6 alkyl group, a - C 2-6 alkyl— (5-7 membered cycloheteroalkyl) group, a C 1-10 alkyl group, or a 5-7 membered heteroaryl group, a carbon ring atom optionally substituted with -C(O)- NR
- R 3 can be selected from a 1-[1 ,4]diazepanyl group, a 1- imidazolyl group, a 4-morpholinyl group, a 1 -piperidinyl group, a 1 -piperazinyl group, a 4-pyridyl group, a 1 -pyrrolidyl group, and a 4-thiomorpholinyl group, wherein each of these groups can be optionally substituted as described above.
- R 3 can be a 1 -piperazinyl group having a nitrogen atom in the ring optionally substituted with -C(O)R 0 , -C 2-6 alkyl-OR c , -C 2-6 alkyl-NR d R e , -C 1-6 alkyl-C(O)NR d R e , an S(O) 2 -C 1-6 alkyl group, a -C 2-6 alkyl-(5-7 membered cycloheteroalkyl) group, a Ci -10 alkyl group, or a 5-7 membered heteroaryl group.
- R 3 can be a 4-methyl piperazin-1-yl group.
- R 3 can be a 1 -piperidinyl group having a carbon atom in the ring optionally substituted with -NR d R e , -C(O)-NR d R e , -Y-OR c , a 5-7 cycloheteroalkyl group, a 5-9 membered heteroaryl group, or a -Y-O-(5-7 membered heteroaryl) group.
- Exemplary compounds of formula (III) in accordance with embodiments of the present invention include, but are not limited to, the compounds presented in Table 3 5 below.
- R , Ar, R , R c , and p are as defined above; is selected from the group of
- the 2-furyl and 3-furyl rings are optionally substituted with a -NO 2 group or with 1-2 C 1-6 alkyl groups;
- p is 1 , and R 2 is 4-fluorophenyl, then Ar-R 3 is not 4-dimethylaminophenyl.
- R 2 can be a phenyl group optionally substituted with 1-2 substituents independently selected from a halogen, a C 1-6 alkyl group, and a C 1-6 alkoxy group.
- R 2 can be 4-fluorophenyl group, a 4-chlorophenyl group, a 4-fluoro-2-methylphenyl group, a 4-methylphenyl group, a 2-isopropylphenyl group, or a 4-methoxyphenyl group.
- R 2 can be a -C 3-6 cycloalkyl group optionally substituted with 1 to 3 C 1-6 alkyl groups.
- R 2 can be a 4-tertbutylcyclohexyl group.
- p is 1.
- p is 2.
- p is 3.
- Ar-R 3 can be any organic compound.
- Ar-R 3 can be any organic compound.
- R 3 is as defined above.
- Ar-R 3 can be any organic radical
- Ar-R 3 can be any organic radical
- R 3 is as defined above.
- R 3 can be NR f R g , wherein R f and R 9 are as defined above.
- R 3 can be selected from NH 2 , an NH-Ci -6 alkyl group, an N(Ci -6 alkyl) 2 group wherein the C 1-6 alkyl groups do not need to be the same,, an NH-C 3-6 cycloalkyl group, an N(Ci -6 alkyl)— C 3-6 cycloalkyl group, an N(C 1-6 alkyl)— C 2- 6 alkyl-OR c group, an N(Ci -6 alkyl)-Y-(5-7 membered cycloheteroalkyl) group, an N(C 1-6 alkyl)-phenyl group, an N(Ci -6 alkyl)-Y-5-7 membered heteroaryl group, and an N(Ci -6 alkyl)-C 2-6 alkyl-O-Y-5-7 membered heteroaryl group
- R 3 can be selected from a halogen and a Ci -6 haloalkyl group.
- R 3 can be a chloro group or a thfluoromethyl group.
- R 3 can be an optionally substituted 5-7 membered cycloheteroalkyl group or an optionally substituted 5-7 membered heteroaryl group as described herein.
- R 3 can be selected from a diazepanyl group, an imidazolyl group, a morpholinyl group, a piperidinyl group, a piperazinyl group, a pyridyl group, a pyrrolidyl group, and a thiomorpholinyl group, wherein each of these groups can include a nitrogen ring atom optionally substituted with -C(O)R C , -C 2-6 alkyl-OR c , -C 2-6 alkyl-NR d R e , -Y-C(O)NR d R e , an -S(O) 2 -C 1-6 alkyl group, a - C 2 6 alkyl— (5-7 membered cyclohetero
- R 3 can be selected from a 1-[1 ,4]d ⁇ azepanyl group, a 1- imidazolyl group, a 4-morphol ⁇ nyl group, a 1-p ⁇ pe ⁇ d ⁇ nyl group, a 1-p ⁇ peraz ⁇ nyl group, a 4-pyr ⁇ dyl group, a 1-pyrrol ⁇ dyl group, and a 4-th ⁇ omorphol ⁇ nyl group, wherein each of these groups can be optionally substituted as described above
- R 3 can be a 1-p ⁇ peraz ⁇ nyl group having a nitrogen atom in the ring optionally substituted with -C(O)R C , -C 2 6 alkyl-OR c , -C 2-6 alkyl-NR d R e , -C 1 6 alkyl-C(O)NR d R e , an S(O) 2 -C 1-6 alkyl group, a -C 1-6 alkyl-(phenyl) n group wherein n is 1 , 2, or 3, a -C 2-6 alkyl— (5-7 membered cycloheteroalkyl) group, a C 1 10 alkyl group, a -Y-phenyl group, or a 5-7 membered heteroaryl group
- R 3 can be a 4-methyl p ⁇ peraz ⁇ n-1-yl group, a 4-(4-fluorophenyl)p ⁇ peraz ⁇ n-1-yl group,
- R 3 can be a 1-p ⁇ per ⁇ d ⁇ nyl group having a carbon atom in the ring optionally substituted with -NR d R e , -C(O)-NR d R e , -Y-OR 0 , a 5-7 cycloheteroalkyl group, a 5-9 membered heteroaryl group, or a -Y-O-(5-7 membered heteroaryl) group
- Representative compounds of (IV) in accordance with embodiments of the present invention include, but are not limited to, the compounds presented in 4 below TABLE 4
- Embodiments of the present invention further provide compounds of formula (V):
- R 1 Ar, R J , and p are as defined above; is selected from and ;
- R 8 and R 9 are independently a C 1-6 alkyl group.
- R 8 and R 9 are independently selected from the group of a methyl group and a tert-butyl group.
- p is 1.
- p is 2.
- p is 3.
- Ar-R 3 can be any organic compound.
- Ar-R 3 can be any organic compound.
- Ar-R 3 can be any organic radical
- Ar-R 3 can be any organic radical having the same meaning as defined above.
- Ar-R 3 can be any organic radical having the same meaning as defined above.
- R 3 is as defined above.
- Ar-R 3 can be be 2-morpholin-4-yl-1 ,3-thiazol-4-yl.
- R 2 can be a phenyl group optionally substituted with 1-2 substituents independently selected from a halogen, a C L6 alkyl group, and a C 1-6 alkoxy group.
- R 2 can be 4-fluorophenyl group.
- Representative compounds of Formula (V) in accordance with embodiments of the present invention include, but are not limited to, the compounds presented in Table 5 below.
- R , Ar, R and p are as defined above; and wherein the pyridinyl group is optionally substituted with 1 to 3 substituents independently selected from a halogen and a C 1-6 alkoxy group.
- R 2 can be a phenyl group optionally substituted with 1-2 substituents independently selected from a halogen, a C 1-6 alkyl group, and a C 1 -6 alkoxy group.
- R 2 can be 4-fluorophenyl group, a 4-chlorophenyl group, a 2-methyl-4-fluorophenyl group, a 4-methylphenyl group, a 2-isopropylphenyl group, a 4-methoxyphenyl group, a 3-trifluoromethylphenyl group, a 4-trifluoromethylphenyl group, a 3-tertbutylphenyl group, a 5-chloro-2-methylphenyl group, a 4- hydroxyphenyl group, or a 4-benzyloxyphenyl group.
- R 2 can be a -C 3-6 cycloalkyl group; wherein the -C 3 -6 cycloalkyl group is optionally substituted with 1-3 C 1-6 alkyl groups.
- R 2 can be a cyclohexyl group, a cyclopentyl group, a 4-tertbutylcylcohexyl group, a 2,3- dihydro-1 H-inden-2-yl group, or a cyclopropyl methyl group.
- p is 1.
- p is 2.
- p is 3.
- Ar-R 3 can be wherein R is as defined above.
- Ar-R 3 can be any organic radical
- R 3 is as defined above.
- Ar-R 3 can be any organic radical
- R 3 is as defined above.
- R 3 can be NR f R g , wherein R f and R 9 are as defined above.
- R 3 can be selected from NH 2 , an NH-C 1-6 alkyl group, an
- R 3 can be a diethylamino group or a cyclopropyl(ethyl)amino group.
- R 3 can be an optionally substituted 5-7 membered cycloheteroalkyl group or an optionally substituted 5-7 membered heteroaryl group as described herein.
- R 3 can be selected from a diazepanyl group, an imidazolyl group, a morpholinyl group, a piperidinyl group, a piperazinyl group, a pyridyl group, a pyrrolidyl group, and a thiomorpholinyl group, wherein each of these groups can include a nitrogen ring atom optionally substituted with -C(O)R 0 , -C 2 .6 alkyl-OR c , -C 2-6 alkyl-NR d R e , -Y-C(O)NR d R e , an -S(O) 2 -C 1-6 alkyl group, a - C 2-6 alkyl— (5-7 membered cyclo
- R 3 can be selected from a 1-[1 ,4]diazepanyl group, a 1- imidazolyl group, a 4-morpholinyl group, a 1 -piperidinyl group, a 1 -piperazinyl group, a 4-pyridyl group, a 1 -pyrrolidyl group, and a 4-thiomorpholinyl group, wherein each of these groups can be optionally substituted as described above.
- R 3 can be a 1 -piperazinyl group having a nitrogen atom in the ring optionally substituted with -C(O)R 0 , -C 2-6 alkyl-OR 0 , -C 2 . 6 alkyl-NR d R e , -C 1-6 alkyl-C(O)NR d R e , an S(O) 2 -C 1-6 alkyl group, a -C 2-6 alkyl-(5-7 membered cycloheteroalkyl) group, a C 1-10 alkyl group, or a 5-7 membered heteroaryl group.
- R 3 can be a 4-methyl piperazin-1-yl group.
- R 3 can be a 1 -piperidinyl group having a carbon atom in the ring optionally substituted with -NR 0 Re, -C(O)-NR d R e , -Y-OR 0 , a 5-7 cycloheteroalkyl group, a 5-9 membered heteroaryl group, or a -Y-O-(5-7 membered heteroaryl) group.
- Representative compounds of Formula (Vl) in accordance with embodiments of the present invention include, but are not limited to, the compounds presented in Table 6 5 below.
- R 2 , Ar, R 3 and p are as defined above; and is selected from
- R 2 can be a phenyl group optionally substituted with 1-2 substituents independently selected from a halogen, a C 1 ⁇ alkyl group, and a C 1-6 alkoxy group.
- R 2 can be 4-fluorophenyl group.
- p is 1.
- p is 2.
- p is 3.
- Ar-R 3 can be any organic compound.
- Ar-R 3 can be any organic compound.
- Ar-R 3 can be wherein R 3 is as defined above.
- Ar-R 3 can be any organic radical
- R 3 is as defined above.
- R 3 can be NR f R g , wherein R f and R 9 are as defined above.
- R 3 can be selected from NH 2 , an NH-C 1-6 alkyl group.an N(C 1-6 alkyl) 2 group wherein the C 1-6 alkyl groups do not need to be the same,, an NH-C 3-6 cycloalkyl group, an N(C 1-6 alkyl)— C 3-6 cycloalkyl group, an N(C 1-6 alkyl)— C 2-6 alkyl-OR c group, an N(C 1-6 alkyl)- Y-(5-7 membered cycloheteroalkyl) group, an N(C 1-6 alkyl)-phenyl group, an N(C 1-6 alkyl)-Y-5-7 membered heteroaryl group, and an N(C 1-6 alkyl)-C 2-6 alkyl-O-Y-5-7 membered heteroaryl group, wherein each of
- R 3 can be an optionally substituted 5-7 membered cycloheteroalkyl group or an optionally substituted 5-7 membered heteroaryl group as described herein.
- R 3 can be selected from a diazepanyl group, an imidazolyl group, a morpholinyl group, a piperidinyl group, a piperazinyl group, a pyridyl group, a pyrrolidyl group, and a thiomorpholinyl group, wherein each of these groups can include a nitrogen ring atom optionally substituted with -C(O)R C , -C 2-6 alkyl-OR c , -C 2-6 alkyl-NR d R e , -Y-C(O)NR d R e , an -S(O) 2 -C 1-6 alkyl group, a - C 2-6 alkyl— (5-7 membered cyclohetero
- R 3 can be selected from a 1-[1 ,4]diazepanyl group, a 1- imidazolyl group, a 4-morpholinyl group, a 1-piperidinyl group, a 1-piperazinyl group, a 4-pyridyl group, a 1-pyrrolidyl group, and a 4-thiomorpholinyl group, wherein each of these groups can be optionally substituted as described above.
- R 3 can be a 1-piperazinyl group having a nitrogen atom in the ring optionally substituted with -C(O)R C , -C 2-6 alkyl-OR c , -C 2-6 alkyl-NR d R e , -C 1-6 alkyl-C(O)NR d R e , an S(O) 2 -C 1-6 alkyl group, a -C 2-6 alkyl-(5-7 membered cycloheteroalkyl) group, a C 1-10 alkyl group, or a 5-7 membered heteroaryl group.
- R 3 can be a 4-methyl piperazin-1-yl group.
- R 3 can be a 1-piperidinyl group having a carbon atom in the ring optionally substituted with -NR d R e , -C(O)-NR d R e , -Y-OR c , a 5-7 cycloheteroalkyl group, a 5-9 membered heteroaryl group, or a -Y-O-(5-7 membered heteroaryl) group.
- R 3 can be chloro or a trifluoromethyl group.
- Representative compounds of Formula (Vl) in accordance with embodiments of the present invention include, but are not limited to, the compounds presented in Table 7 below.
- salts of the compounds of formulas (I), (II), (III), (IV), (V), (Vl), and (VII), which can have an acidic moiety can be formed using organic and inorganic bases. Both mono and polyanionic salts are contemplated, depending on the number of acidic hydrogens available for deprotonation.
- Suitable salts formed with bases include metal salts, such as alkali metal or alkaline earth metal salts, for example sodium, potassium, or magnesium salts; ammonia salts and organic amine salts, such as those formed with morpholine, thiomorpholine, piperidine, pyrrolidine, a mono-, di- or th-lower alkylamine (e.g., ethyl-tert-butyl-, diethyl-, diisopropyl-, triethyl-, tributyl- or dimethylpropylamine), or a mono-, di-, or trihydroxy lower alkylamine (e.g., mono-, di- or triethanolamine).
- metal salts such as alkali metal or alkaline earth metal salts, for example sodium, potassium, or magnesium salts
- ammonia salts and organic amine salts such as those formed with morpholine, thiomorpholine, piperidine, pyrrolidine, a mono-
- inorganic bases include NaHCO 3 , Na 2 CO 3 , KHCO 3 , K 2 CO 3 , Cs 2 CO 3 , LiOH, NaOH, KOH, NaH 2 PO 4 , Na 2 HPO 4 , and Na 3 PO 4 .
- Internal salts also can be formed.
- salts can be formed using organic and inorganic acids.
- salts can be formed from the following acids: acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, dichloroacetic, ethenesulfonic, formic, fumaric, gluconic, glutamic, hippuric, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, malonic, mandelic, methanesulfonic, mucic, napthalenesulfonic, nitric, oxalic, pamoic, pantothenic, phosphoric, phthalic, propionic, succinic, sulfuric, tartaric, toluenesulfonic, and as well as other known pharmaceutically acceptable acids.
- esters in the present invention refer to non-toxic esters of the compounds of formulas (I), (II), (III), (IV), (V), (Vl), and (VII) 1 , preferably the alkyl esters such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl or pentyl esters, of which the methyl ester is preferred.
- alkyl esters such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl or pentyl esters, of which the methyl ester is preferred.
- other esters such as phenyl-C 1-5 alkyl may be employed if desired.
- examples of pharmaceutically acceptable esters include, but are not limited to, C 2 -C 6 alkyl esters such as methyl esters and ethyl esters.
- esters include esters made with aliphatic carboxylic acids, preferably those with a linear chain of between two and six carbon atoms, preferably acetic acid, and made with aromatic carboxylic acids, e.g. C 7-I2 acids such as benzoic acid.
- the aliphatic and aromatic acids may optionally be substituted by one or more C 1-4 alkyl groups.
- prodrugs of the compounds disclosed herein As used herein, "prodrug” refers to a moiety that produces, generates or releases a compound of the present teachings when administered to a mammalian subject.
- Prodrugs can be prepared by modifying functional groups present in the compounds in such a way that the modifications are cleaved, either by routine manipulation or in vivo, from the parent compounds.
- Examples of prodrugs include compounds as described herein that contain one or more molecular moieties appended to a hydroxyl, amino, sulfhydryl, or carboxyl group of the compound, and that when administered to a mammalian subject, is cleaved in vivo to form the free hydroxyl, amino, sulfhydryl, or carboxyl group, respectively.
- Examples of prodrugs can include, but are not limited to, acetate, formate and benzoate derivatives of alcohol and amine functional groups in the compounds of the present teachings.
- prodrugs are discussed in T. Higuchi and V. Stella, "Pro-drugs as Novel Delivery Systems,” Vol. 14 of the A.C.S. Symposium Series, and in Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987, the entire disclosures of which are incorporated by reference herein for all purposes.
- Carboxylic acid amide compounds in accordance with the present invention can be prepared as outlined in the schemes below and as illustrated in the examples, from (a) commercially available starting materials, (b) compounds known in the literature, or readily prepared intermediates using literature procedures, or (c) new intermediates described in the schemes and experimental procedures herein.
- Reactions are performed in a solvent appropriate to the reagents and materials employed and suitable for the transformation being effected.
- Suitable solvents typically are substantially nonreactive with the reactants, intermediates, and/or products at the temperatures at which the reactions are carried out, i.e., temperatures that can range from the solvent's freezing temperature to the solvent's boiling temperature.
- a given reaction can be carried out in one solvent or a mixture of more than one solvent.
- suitable solvents for a particular reaction step can be selected.
- suitable solvents for a particular reaction step can be selected.
- suitable solvents One skilled in the art of organic synthesis can readily selected suitable solvents.
- product formation can be monitored by spectroscopic means, such as nuclear magnetic resonance spectroscopy (e.g., 1 H or 13 C), infrared spectroscopy, spectrophotometry (e.g., UV-visible), or mass spectrometry, or by chromatography such as high performance liquid chromatograpy (HPLC) or thin layer chromatography.
- spectroscopic means such as nuclear magnetic resonance spectroscopy (e.g., 1 H or 13 C), infrared spectroscopy, spectrophotometry (e.g., UV-visible), or mass spectrometry
- chromatography such as high performance liquid chromatograpy (HPLC) or thin layer chromatography.
- activated acid derivatives include, for example, acid chlorides, esters, acylimidazoles, anhydrides; these activated acid derivatives can be generated in situ or as isolated compounds.
- Representative activating agents include, but are not limited to, sulfuryl chloride, thionyl chloride, 2-chloro-4,6-dimethoxy-1 ,3,5-thazine, and carbodiimides such as 1-[3-(dimethylamino)propyl]-3-ethyl-carbodiimide and dicyclohexyl carbodiimide; for examples of amide bond formation and acid activation, see Montalbetti C.A.G.N. and Falque, V. (2005), Tetrahedron, 61 (46): 10827-10852.
- thiazole carboxylic acids are commercially available or can be otherwise readily prepared from standard procedures as described in Uchiyama, M. et al. (2005), Chem. Pharm. Bull., 53(4): 437-440.
- Substituted 1 H-imidazole-4-carboxylic acid derivatives can be prepared by methods known in the art, for example, according to the procedures described in Cristalli, G. et al., J. Med. Chem., 34: 1187- 1192 (1991); Bioorg. Med. Chem. Lett., 4(13): 1623-1658 (1994); Ahn, H-S. et. al. , J. Med.
- MIc triazole-4-carboxylic acids
- the amine (b) can be synthesized as described in Scheme 2 below.
- PtG e.g., tert-butyloxycarbonyl LG: e.g., Cl, Br, or I
- alkylation of a protected amine (c) with a compound of formula (d) provides the protected alkylated amine (e).
- Displacement of the leaving group on compound (e) with the appropriate amine (R 3 , wherein R 3 is NR f R g ) provides the corresponding amine-substituted aryl derivative (f).
- alkylation of the protected amine (c) with a compound of formula (g) provides the amine-substituted aryl derivative (f) directly.
- Removal of the protecting group (PtG) under standard conditions provides the desired amine Vl.
- the amine (b) can be synthesized from substituted acid halides, anhydrides or other activated carboxylic acid derivatives (j) or (m), as illustrated in Scheme 3 below.
- Z e.g, halide, acetate LG: e.g, Cl, Br, or I 5 n: 0, 1, 2, or 3
- Certain amines of formula (b) in which Ar is 1 ,3-thiazol-4-yl can be prepared by the reaction of carbothioic acid amides (o), wherein Het is a 5-7 membered cycloheteroalkyl group containing at least one nitrogen atom such as 2-morpholin-4- 5 yl, with malonyl chloride derivatives (n) as provided in Scheme 3b.
- o carbothioic acid amides
- Het is a 5-7 membered cycloheteroalkyl group containing at least one nitrogen atom such as 2-morpholin-4- 5 yl
- malonyl chloride derivatives (n) as provided in Scheme 3b.
- R 3 group can be incorporated in the last step of the synthesis, as illustrated in Scheme 5 below for compounds of formula (III); other compounds in accordance with embodiments of the invention can be prepared in an analogous fashion using acyl chloride or acetate derivative of carboxylic acids (Ia), (Ma), (IVa), (Va), (Via), and (Vila).
- Z e.g., halide or acetate
- LG e.g., Cl, Br, or I
- ion channel mediated condition refers to any condition or pathological state of a mammal or any disease present in a mammal that can be treated, or the symptoms of which can be alleviated, by modulation of the activity of one or more ion channels such as Ca v 2.2 voltage-gated calcium channels.
- An ion channel mediated condition can be attributed to the abnormal functioning of one or more ion channels.
- An ion channel can be functioning abnormally when, for example, the ion channel exhibits abnormally increased or decreased activation.
- ion channel mediated conditions include conditions associated with neuronal hyperexcitability, conditions associated with abnormal glutamate regulation, pain, convulsions, epilepsy, stroke, anxiety disorders, neuronal disorders, traumatic brain injury, angina, hypertension, congestive heart failure, myocardial ischemia, arrhythmia, diabetes, urinary incontinence, hot flush, thermal disregulation, and combinations thereof.
- conditions associated with neuronal hyperexcitability include, but are not limited to, convulsions, including neonatal convulsions, epilepsy, episodic ataxia, myokymia, cerebral ischemia, cerebral palsy, stroke, traumatic brain injury, traumatic spinal cord injury, asphyxia, anoxia, prolonged cardiac surgery, and combinations thereof.
- conditions associated with the abnormal regulation of glutamate include, but are not limited to, hypoglycemia or diseases associated with abnormal glutamate regulation such as, without limitation, Parkinson's disease, Huntingdon's disease, Alzheimer's disease, amyotrophic lateral sclerosis, AIDS-related dementia, and combinations thereof.
- anxiety disorders include, but are not limited to, agoraphobia, panic disorder, specific phobia, social phobia, obsessive compulsive disorder, posttraumatic stress disorder, acute stress disorder, generalized anxiety disorder, separation anxiety disorder, substance-induced anxiety disorder, and anxiety disorder not otherwise specified.
- pain examples include, but are not limited to various types of nociceptic or neuropathic pain, such as, without limitation, inflammatory pain, musculoskeletal pain, bony pain, lumbosacral pain, neck or upper back pain, visceral pain, somatic pain, pain associated with diabetic neuropathy, cancer pain, pain caused by injury or surgery such as burn pain, headaches such as migraines or tension headaches, and combinations of these pains.
- nociceptic or neuropathic pain such as, without limitation, inflammatory pain, musculoskeletal pain, bony pain, lumbosacral pain, neck or upper back pain, visceral pain, somatic pain, pain associated with diabetic neuropathy, cancer pain, pain caused by injury or surgery such as burn pain, headaches such as migraines or tension headaches, and combinations of these pains.
- a pain caused by inflammation can also be visceral or musculoskeletal in nature.
- Other examples of pain include those related to conditions of hyperalgesia, allodynia, or both
- the compounds of the present teachings can be useful for the treatment of a pathological condition, disorder or disease, and the alleviation of a symptom thereof, in a mammal, for example, a human.
- the pathological condition, disorder or disease, or a symptom thereof can be, but is not limited to, one of the various ion channel mediated conditions described above.
- the compounds of the present teachings can be used for pain therapy, including treating, by way of non-limiting examples, the various types of pain described above.
- "treating" refers to partially or completely alleviating, inhibiting, preventing and/or ameliorating the condition.
- the present teachings therefore include use of the compounds disclosed herein as active therapeutic substances for the treatment of a variety of ion channel mediated conditions as well as for pain therapy.
- the compounds of the present teachings can be useful for the preparation of medicaments for the treatment of a pathological condition, disorder or disease, and the alleviation of a symptom thereof, in a mammal, for example, a human.
- the pathological condition, disorder or disease, or a symptom thereof can be one of the various ion channel mediated conditions described herein.
- the compounds disclosed herein can be useful for treating the various conditions associated with neuronal hyperexcitability, the various conditions associated with abnormal glutamate regulation, the various anxiety and neuronal disorders, angina, hypertension, congestive heart failure, myocardial ischemia, arrhythmia, diabetes, urinary incontinence, and combinations thereof, as described above.
- the compounds disclosed herein also can be useful for treating pain, including chronic pain that is neuropathic pain associated with damage to or pathological changes in the peripheral nervous system or the central nervous system; visceral pain associated with, by way of non-limiting examples, the abdominal, pelvic, and/or perineal regions or pancreatitis;, musculoskeletal pain; bony pain associated with, by way of non-limiting examples, bone or joint degenerating disorders such as osteoarthritis, rheumatoid arthritis, or spinal stenosis; cancer pain; musculoskeletal pain associated with, by way of non-limiting examples, the lower or upper back, spine, fibromylagia, temporomandibular joint, or myofascial pain syndrome; headaches such migraine or tension headaches; pain associated with infections such as HIV or shingles, sickle cell anemia, autoimmune disorders, multiple sclerosis, and inflammation in accordance with the methods described herein.
- Inflammatory pain can be associated with a variety of medical conditions such as osteoarthritis, rheumatoid arthritis, surgery, or injury.
- Neuropathic pain may be associated with, for example, diabetic neuropathy, peripheral neuropathy, postherpetic neuralgia, trigeminal neuralgia, lumbar or cervical radiculopathies, fibromyalgia, glossopharyngeal neuralgia, reflex sympathetic dystrophy, casualgia, thalamic syndrome, nerve root avulsion, or nerve damage cause by injury resulting in peripheral and/or central sensitization such as phantom limb pain, reflex sympathetic dystrophy or postthoracotomy pain, cancer, chemical injury, toxins, nutritional deficiencies, or viral or bacterial infections such as shingles or HIV, or combinations thereof.
- the methods of use for compounds of this invention further include treatments in which the neuropathic pain is a condition secondary to metastatic infiltration, adiposis dolorosa, burns, or central pain conditions related to
- Chronic pain may be associated with diabetes, post traumatic pain of amputation, lower back pain, spinal cord damage, cancer, chemical injury, chemotherapy induced peripheral neuropathy, toxins, major surgery, peripheral nerve damage due to traumatic injury, post-herpetic neuralgia, trigeminal neuralgia, lumbar or cervical radiculopathies, fibromyalgia, glossopharyngeal neuralgia, reflex sympathetic dystrophy, causalgia, thalamic syndrome, nerve root avulsion, reflex sympathetic dystrophy or post thoracotomy pain, nutritional deficiencies, viral infection, bacterial infection, metastatic infiltration, adiposis dolorosa, burns, central pain conditions related to thalamic conditions; and any combination thereof.
- chronic pain refers to centralized or peripheral pain that is intense, localized, sharp, or stinging, and/or dull, aching, diffuse, or burning in nature and that occurs for extended periods of time (i.e., persistent and/or regularly reoccurring), including, for the purpose of the present invention, neuropathic pain and cancer pain.
- Chronic pain includes neuropathic pain, hyperalgesia, and/or allodynia.
- the present teachings therefore include methods of administering to a mammal a therapeutically effective amount of a compound disclosed herein.
- administer or “administering” refers to either directly administering a compound of the present teachings or a pharmaceutical composition containing the compound, or administering the compound or pharmaceutical composition indirectly via a prodrug derivative or analog which will form an equivalent amount of the active compound or substance within the body.
- the methods also can include identifying a mammal in need of such treatment, and administering a therapeutically effective amount of a compound disclosed herein to the mammal in need thereof.
- therapeutically effective refers to a substance or an amount that elicits a desirable biological activity or effect.
- the method includes administering to a mammal a pharmaceutical composition that comprises a compound disclosed herein in combination or association with a pharmaceutically acceptable carrier.
- the compound of the present teachings can be administered alone or in combination with other therapeutically effective compounds or therapies for the treatment of such condition(s).
- the other therapeutically effective compounds can include a cardiovascular disease agent and/or a nervous system disease agent.
- a nervous system disease agent can be a peripheral nervous system (PNS) disease agent and/or a central nervous (CNS) disease agent.
- PNS peripheral nervous system
- CNS central nervous
- the present teachings also relate to in vitro or in vivo methods of modulating the activity of ion channels including, but not limited to, Ca v 2.2 voltage-gated calcium channels.
- such methods include contacting a Ca v 2.2 voltage- gated calcium channel with a compound disclosed herein. In certain embodiments, the methods include monitoring the activity of ion channels. In various embodiments, the present teachings relate to methods of modulating the activity of an ion channel such as a Ca v 2.2 voltage-gated calcium channel that include in vitro or in vivo administration of a pharmaceutically effective amount of one or more compounds of formula (I). As used herein, "pharmaceutically effective” refers to an amount that can elicit an intended biological activity or effect.
- an effective dosage can vary depending upon the particular compound utilized, the mode of administration, and severity of the condition being treated, as well as the various physical factors related to the individual being treated.
- a compound of the present teachings can be provided to a patient already suffering from a disease in an amount sufficient to treat the symptoms of the disease and its complications.
- the dosage to be used in the treatment of a specific individual typically must be subjectively determined by the attending physician.
- the variables involved include the specific condition and its state as well as the size, age and response pattern of the patient.
- compositions comprising at least one compound described herein and one or more pharmaceutically acceptable carriers, excipients, or diluents.
- pharmaceutically acceptable carriers are well known to those skilled in the art and can be prepared in accordance with acceptable pharmaceutical procedures, such as, for example, those described in Remington's Pharmaceutical Sciences, 17th edition, ed. Alfonoso R. Gennaro, Mack Publishing Company, Easton, PA (1985), the entire disclosure of which is incorporated by reference herein for all purposes.
- pharmaceutically acceptable refers to a substance that is acceptable for use in pharmaceutical applications from a toxicological perspective and does not adversely interact with the active ingredient.
- pharmaceutically acceptable carriers are those that are compatible with the other ingredients in the formulation and are biologically acceptable.
- Supplementary active ingredients can also be incorporated into the pharmaceutical compositions.
- Compounds of the present teachings can be administered orally or parenterally, neat or in combination with conventional pharmaceutical carriers.
- Applicable solid carriers can include one or more substances which can also act as flavoring agents, lubricants, solubilizers, suspending agents, fillers, glidants, compression aids, binders or tablet-disintegrating agents, or encapsulating materials.
- the compounds can be formulated in conventional manner, for example, in a manner similar to that used for known antiinflammatory agents.
- Oral formulations containing an active compound disclosed herein can comprise any conventionally used oral form, including tablets, capsules, buccal forms, troches, lozenges and oral liquids, suspensions or solutions.
- the carrier can be a finely divided solid, which is an admixture with a finely divided active compound.
- an active compound can be mixed with a carrier having the necessary compression properties in suitable proportions and compacted in the shape and size desired.
- the powders and tablets can contain up to about 99% or greater of the active compound.
- Capsules can contain mixtures of active compound(s) with inert filler(s) and/or diluent(s) such as the pharmaceutically acceptable starches (e.g., corn, potato or tapioca starch), sugars, artificial sweetening agents, powdered celluloses (e.g., crystalline and microcrystalline celluloses), flours, gelatins, gums, and the like.
- inert filler(s) and/or diluent(s) such as the pharmaceutically acceptable starches (e.g., corn, potato or tapioca starch), sugars, artificial sweetening agents, powdered celluloses (e.g., crystalline and microcrystalline celluloses), flours, gelatins, gums, and the like.
- Useful tablet formulations can be made by conventional compression, wet granulation or dry granulation methods and utilize pharmaceutically acceptable diluents, binding agents, lubricants, disintegrants, surface modifying agents (including surfactants), suspending or stabilizing agents, including, but not limited to, magnesium stearate, stearic acid, sodium lauryl sulfate, talc, sugars, lactose, dextrin, starch, gelatin, cellulose, methyl cellulose, microcrystalline cellulose, sodium carboxymethyl cellulose, carboxymethylcellulose calcium, polyvinylpyrrolidine, alginic acid, acacia gum, xanthan gum, sodium citrate, complex silicates, calcium carbonate, glycine, sucrose, sorbitol, dicalcium phosphate, calcium sulfate, lactose, kaolin, mannitol, sodium chloride, low melting waxes, and ion exchange resins.
- pharmaceutically acceptable diluents including
- Surface modifying agents can include nonionic and anionic surface modifying agents.
- Representative examples of surface modifying agents include, but are not limited to, poloxamer 188, benzalkonium chloride, calcium stearate, cetostearyl alcohol, cetomacrogol emulsifying wax, sorbitan esters, colloidol silicon dioxide, phosphates, sodium dodecylsulfate, magnesium aluminum silicate, and triethanolamine.
- Oral formulations herein can utilize standard delay or time-release formulations to alter the absorption of the active compound(s).
- the oral formulation can also consist of administering an active compound in water or fruit juice, containing appropriate solubilizers or emulisifiers as needed.
- Liquid carriers can be used in preparing solutions, suspensions, emulsions, syrups, and elixirs.
- An active compound described herein can be dissolved or suspended in a pharmaceutically acceptable.
- liquid carrier such as water, an organic solvent, or a mixture of both, or pharmaceutically acceptable oils or fats.
- the liquid carrier can contain other suitable pharmaceutical additives such as solubilizers, emulsifiers, buffers, preservatives, sweeteners, flavoring agents, suspending agents, thickening agents, colors, viscosity regulators, stabilizers, and osmo-regulators.
- liquid carriers for oral and parenteral administration include, but are not limited to, water (particularly containing additives as described above, e.g., cellulose derivatives such as a sodium carboxymethyl cellulose solution), alcohols (including monohydric alcohols and polyhydric alcohols, e.g., glycols) and their derivatives, and oils (e.g., fractionated coconut oil and arachis oil).
- the carrier can be an oily ester such as ethyl oleate and isopropyl myristate.
- Sterile liquid carriers are used in sterile liquid form compositions for parenteral administration.
- the liquid carrier for pressurized compositions can be halogenated hydrocarbon or other pharmaceutically acceptable propellants.
- Liquid pharmaceutical compositions which are sterile solutions or suspensions, can be utilized by, for example, intrathecal, intramuscular, intraperitoneal or subcutaneous injection. Sterile solutions can also be administered intravenously.
- Compositions for oral administration can be in either liquid or solid form.
- the pharmaceutical composition is in unit dosage form, for example, as tablets, capsules, powders, solutions, suspensions, emulsions, granules, or suppositories.
- the pharmaceutical composition can be sub-divided in unit dose(s) containing appropriate quantities of the active compound.
- the unit dosage forms can be packaged compositions, for example, packeted powders, vials, ampoules, prefilled syringes or sachets containing liquids.
- the unit dosage form can be a capsule or tablet itself, or it can comprise the appropriate number of any such compositions in package form.
- Such unit dosage form may contain from about 1 mg/kg of active compound to about 500 mg/kg of active compound, and can be given in a single dose or in two or more doses.
- Such doses can be administered in any manner useful in directing the active compound(s) to the recipient's bloodstream, including orally, via implants, parenterally (including intravenous, intraperitoneal and subcutaneous injections), rectally, vaginally, and transdermally.
- Such administrations can be carried out using the compounds of the present teachings including pharmaceutically acceptable salts thereof, in lotions, creams, foams, patches, suspensions, solutions, and suppositories (e.g., rectal and vaginal).
- the compounds of the present teachings can be formulated, for example, into an aqueous or partially aqueous solution.
- Compounds described herein can be administered enterally or parenterally (such as, without limitation, interperitoneal, intramuscular, intravascular, intrathecal, intraarticular or subcuteaneous injection or infusion).
- Solutions or suspensions of these active compounds or pharmaceutically acceptable salts thereof can be prepared in water suitably mixed with a surfactant such as hydroxyl-propylcellulose.
- Dispersions can also be prepared in glycerol, liquid polyethylene glycols, and mixtures thereof in oils. Under ordinary conditions of storage and use, these preparations typically contain a preservative to inhibit the growth of microorganisms.
- the pharmaceutical forms suitable for injection can include sterile aqueous solutions or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersions.
- the form is sterile and its viscosity permits it to flow through a syringe.
- the form preferably is stable under the conditions of manufacture and storage and can be preserved against the contaminating action of microorganisms such as bacteria and fungi.
- the carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (e.g., glycerol, propylene glycol and liquid polyethylene glycol), suitable mixtures thereof, and vegetable oils.
- Compounds described herein can be administered transdermal ⁇ , i.e., administered across the surface of the body and the inner linings of bodily passages including epithelial and mucosal tissues. Such administration can be carried out using the compounds of the present teachings including pharmaceutically acceptable salts thereof, in lotions, creams, foams, patches, suspensions, solutions, and suppositories (e.g., rectal and vaginal). Topical formulations that deliver active compound(s) through the epidermis can be useful for localized treatment of inflammation and arthritis.
- Transdermal administration can be accomplished through the use of a transdermal patch containing an active compound and a carrier that can be inert to the active compound, can be non-toxic to the skin, and can allow delivery of the active compound for systemic absorption into the blood stream via the skin.
- the carrier can take any number of forms such as creams and ointments, pastes, gels, and occlusive devices.
- the creams and ointments can be viscous liquid or semisolid emulsions of either the oil-in-water or water-in-oil type. Pastes comprised of absorptive powders dispersed in petroleum or hydrophilic petroleum containing the active compound can also be suitable.
- occlusive devices can be used to release the active compound into the blood stream, such as a semi-permeable membrane covering a reservoir containing the active compound with or without a carrier, or a matrix containing the active compound.
- Other occlusive devices are known in the literature.
- Compounds described herein can be administered into a body cavity, (e.g., rectally or vaginally) in the form of a conventional suppository.
- Suppository formulations can be made from traditional materials, including cocoa butter, with or without the addition of waxes to alter the suppository's melting point, and glycerin.
- Water-soluble suppository bases such as polyethylene glycols of various molecular weights, can also be used.
- Lipid formulations or nanocapsules can be used to introduce compounds of the present teachings into host cells either in vitro or in vivo.
- Lipid formulations and nanocapsules can be prepared by methods known in the art.
- the compounds described herein can be administered in the form of liposomes.
- liposomes are generally derived from phospholipids or other lipid substances, and are formed by mono or multilamellar hydrated liquid crystals that are dispersed in an aqueous medium. Any nontoxic, pharmacologically acceptable lipid capable of forming liposomes can be used.
- a compound can be desirable to combine a compound with other agents effective in the treatment of the target disease.
- other active compounds i.e., other active ingredients or agents
- active compounds of the present teachings can be administered with active compounds of the present teachings.
- the other agents can be administered at the same time or at different times than the compounds disclosed herein.
- compositions of the present teachings also can consist essentially of, or consist of, the recited components, and that the processes of the present teachings also consist essentially of, or consist of, the recited processing steps.
- asymmetric atom also referred as a chiral center
- some of the compounds can contain one or more asymmetric atoms or centers, which can thus give rise to optical isomers (enantiomers) and diastereomers.
- the present teachings and compounds disclosed herein include such optical isomers (enantiomers) and diastereomers (geometric isomers), as well as the racemic and resolved, enantiomerically pure R and S stereoisomers, as well as other mixtures of the R and S stereoisomers and pharmaceutically acceptable salts thereof.
- Optical isomers can be obtained in pure form by standard procedures known to those skilled in the art, which include, but are not limited to, diastereomeric salt formation, kinetic resolution, and asymmetric synthesis.
- the present teachings also encompass cis and trans isomers of compounds containing alkenyl moieties (e.g., alkenes and imines). It is also understood that the present teachings encompass all possible regioisomers, and mixtures thereof, which can be obtained in pure form by standard separation procedures known to those skilled in the art, and include, but are not limited to, column chromatography, thin-layer chromatography, and high-performance liquid chromatography.
- Amines of formula R 2 NH(CH 2 )pArR 3 can be coupled with various carboxylic acids and acid derivatives to provide compounds of formula (I). (II), (III), (IV), (V), (Vl), and (VII).
- Useful carboxylic acids and activated derivatives include those provided in the following examples as well as those that are commercially available or prepared according to procedures known in the art.
- substitution patterns of the starting materials determines the substitution patterns of the products, and the skilled practioner will be able to exercise routine judgment for the selection of suitable starting materials in order to prepare specific products, the order of synthetic steps, and the need for protecting groups for remote functionalities.
- acyl chlorides are illustrated in the representative schemes as examples of activated acid derivatives useful for acylation of amines, other reagents for amide bond formation as known in the art can be utilized in the preparation of compounds of formulas (I), (II), (III), (IV), (V), (Vl), and (VII) in accordance with the teachings herein.
- the compounds were isolated as hydrochloride salts prepared via standard protocols using anhydrous hydrogen chloride as a gas, or as a solution in dioxane or diethyl ether.
- the protonation state of the test compound is in accordance with the pH of the assay conditions, typically buffered as specified in the assay protocols, and not of the salt form or free base of the compound as synthesized.
- R in the following representative schemes is a generic representation, that R wherever it appears does not have to be the same at each occurrence, and R can be selected from, for example, R fl R g , and substitutents on R 2 and R 3 , among others as appropriate and in accordance with the teachings herein.
- R any of the alkyl, heteroaryl and cylcloalkyl groups may be substituted in accordance with the teachings herein.
- Part IV Preparation of ⁇ 5-[(4-fluorophenylamino)-methyl]-pyridin-2-yl ⁇ -diethyl-amine
- the 6-diethylamino-N-(4-fluorophenyl)-nicotinamide was suspended in a mixture of toluene (5 ml_) and tetrahydrofuran (10 ml_) and stirred at O 0 C.
- To the reaction was slowly added sodium bis(2-methoxyethoxy)aluminum hydride (65 wt.% in toluene, 1.8 ml_). The reaction was allowed to warm to room temperature and stirred for 15 minutes followed by heating at 5O 0 C for 1 hour.
- EXAMPLE 1 B ALTERNATIVE PREPARATION OF ⁇ 5-[(4- FLUOROPHENYLAMINO)-METHYL]-PYRIDIN ⁇ -YLJ-DIETHYL-AMINE
- EXAMPLE 2 PREPARATION OF CYCLOPROPYL- ⁇ 5-[(4- FLUOROPHENYLAMINO)-METHYL]-PYRIDIN ⁇ -YLJ-ETHYL-AMINE
- Flash chromatography (silica gel; 30-75% ethyl acetate in chloroform) provided (6- diethylamino-1-oxy-pyridin-2-ylmethyl)-(4-fluorophenyl)-carbamic acid te/f-butyl ester (230 mg, 0.59 mmol) as a light yellow oil.
- Step III Preparation of [3-(cyclopropyl-ethyl-amino)-benzyl]-(4-fluorophenyl)- carbamic acid terf-butyl ester
- the reaction was warmed to room temperature, diluted with toluene (50 mL) and stirred for 2 hours. The layers were separated and the aqueous phase washed with toluene (50 mL). The organic phases were combined, washed with saturated sodium bicarbonate (30 mL), water (30 mL), and brine (30 mL). The organic phase was filtered through a pad of Celite ® and the Celite ® pad washed with ethyl acetate. The organic filtrates were combined and concentrated under reduced pressure to provide a yellow solid.
- EXAMPLE 8 PREPARATION OF (4-FLUOROPHENYL)- ⁇ -PIPERIDIN-I -YL- PYRIMIDIN-5-YLMETHYL)-AMINE
- reaction mixture was basified with 1 N sodium hydroxide to pH 10 and extracted with dichloromethane (2 x 50 ITiL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure to provide a yellow oil. Flash chromatography (silica gel; 5-30% ethyl acetate in hexanes) provided (4- fluorophenyl)-(2-piperidin-1-yl-pyrimidin-5-ylmethyl)-amine (0.93 g, 3.25 mmol) as a yellow oil.
- 1-Cyclopropylmethyl-2-methyl-1 H-imidazole-4-carboxylic acid trfluoroacetate can be prepared according to procedures described in Example 16, using C-cyclopropyl- methylamine in place of benzyl amine.
- tert-butyl cyclopropyl(ethyl)carbamate (15 g, 0.081 mol) was treated with hydrogen chloride (4N in dioxane, 200 ml_). After 16 h, the reaction was evaporated and the residue triturated with diethylether, then hexane. The solid was then dried under vacuum to afford the product (8.67 g, 72 mmol).
- EXAMPLE 26 PREPARATION OF 2-PYRIDIN-3-YL-THIAZOLE-4-CARBOXYLIC ACID (6-DIETHYLAMINO-PYRIDIN-S-YL-METHYL)- ⁇ -FLUOROPHENYL)-AMIDE
- EXAMPLE 28 PREPARATION OF 1-BENZYL-2-METHYL-1 H-IMIDAZOLE-4- CARBOXYLIC ACID [6-(CYCLOPROPYL-ETHYL-AMINO)-PYRIDIN-S-YL-METHYL]- (4-FLUOROPHENYL)-AMIDE DIHYDROCHLORIDE (COMPOUND NO. 17)
- the free base was treated with ethereal hydrochloric acid to provide 1 -allyl-2-methyl- 1 H-imidazole-4-carboxylic acid [6-(cyclopropyl-ethyl-amino)-pyridin-3-yl-methyl]-(4- fluorophenyl)-amide dihydrochloride (116 mg, 0.23 mmol) as a gummy solid.
- EXAMPLE 35 PREPARATION OF 1 -(2,2-DIMETHYL-PROPYL)-I H-IMIDAZOLE ⁇ - CARBOXYLIC ACID [4-(CYCLOPROPYL-ETHYL-AMINO)-BENZYL]-( ⁇ FLUOROPHENYL)-AMIDE DIHYDROCHLORIDE (COMPOUND NO. 24)
- the free base was treated with ethereal hydrochloric acid to provide 1-(2,2-dimethyl- propyl)-1 H-imidazole-4-carboxylic acid [4-(cyclopropyl-ethyl-amino)-benzyl]-(4- fluorophenyl)-amide dihydrochloride (400 mg, 0.76 mmol) as a colorless oil.
- EXAMPLE 36 PREPARATION OF 1 -(2,2-DIMETHYL-PROPYL)-I H-IMIDAZOLE ⁇ - CARBOXYLIC ACID [3-(CYCLOPROPYL-ETHYL-AMINO)-BENZYL]-( ⁇ FLUOROPHENYL)-AMIDE DIHYDROCHLORIDE (COMPOUND NO. 25)
- the free base was treated with ethereal hydrochloric acid to provide 1-(2,2-dimethyl- propyl)-1 H-imidazole-4-carboxylic acid [3-(cyclopropyl-ethyl-amino)-benzyl]-(4- fluorophenyl)-amide dihydrochloride (235 mg, 0.45 mmol) as a foam.
- Step 3 1 -benzyl-N-((2-(pyrrolidin-1 -yl)thiazol-4-yl)methyl)-N-p-tolyl-1 H-imidazole-4- carboxamide
- EXAMPLE 38 PREPARATION OF 1-BENZYL-1 H-[1 ,2,3]TRIAZOLE-4- CARBOXYLIC ACID (6-DIETHYL ⁇ MINO-PYRIDIN-3-YLMETHYL)-(4- FLUOROPHENYL)-AMIDE HYDROCHLORIDE (COMPOUND NO. 86
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Addiction (AREA)
- Psychiatry (AREA)
- Heart & Thoracic Surgery (AREA)
- Psychology (AREA)
- Pain & Pain Management (AREA)
- Cardiology (AREA)
- Vascular Medicine (AREA)
- Urology & Nephrology (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present teachings provide carboxylic amide compounds that can modulate the activity of ion channels in a mammal. The present teachings also provide processes for producing said compounds and their pharmaceutically acceptable salts, hydrates and esters, and methods of treating a pathological condition or disorder, or alleviating a symptom thereof, using said compounds including their pharmaceutically acceptable salts, hydrates and esters. The compounds can be useful in modulating ion channel activity including treating a variety of conditions associated with the abnormal modulation of one or more voltage-gated calcium channels.
Description
ION CHANNEL MODULATORS
This application claims the benefit of U.S. Provisional Application Ser. No. 60/874,133, filed December 11 , 2006, of U.S. Provisional Application Ser. No. 60/874,152, filed December 11 , 2006, and of U.S. Provisional Application Ser. No. 60/874,179, filed December 11 , 2006, the entire disclosures of all of which are incorporated herein by reference.
FIELD OF THE INVENTION
The present teachings relate to certain carboxylic acid amides and related derivatives, processes for their preparation, and their use in therapeutic treatments.
BACKGROUND OF THE INVENTION
All cells rely on the regulated movement of inorganic ions across cell membranes to perform essential physiological functions. Electrical excitability, synaptic plasticity, and signal transduction are examples of processes in which changes in ion concentration play a critical role. In general, the ion channels that permit these changes are proteinaceous pores consisting of one or multiple subunits, each containing two or more membrane-spanning domains. Most ion channels have selectivity for specific ions, primarily Na+, K+, Ca2+, or Cl", by virtue of physical preferences for size and charge. Electrochemical forces, rather than active transport, drive ions across membranes, thus a single channel may allow the passage of millions of ions per second. Channel opening, or "gating" is tightly controlled by changes in voltage or by ligand binding, depending on the subclass of channel. Ion channels are attractive therapeutic targets due to their involvement in so many physiological processes, yet the generation of drugs with specificity for particular channels in particular tissue types remains a major challenge.
Voltage-gated ion channels open in response to changes in membrane potential. For example, depolarization of excitable cells such as neurons results in a transient influx
of Na+ ions, which propagates nerve impulses. This change in membrane potential is sensed by voltage-gated K+ channels, which then allow an efflux of K+ ions. The efflux of K+ ions repolarizes the membrane. Other cell types rely on voltage-gated Ca2+ channels to generate action potentials. Voltage-gated ion channels also perform important functions in non-excitable cells, such as the regulation of secretory, homeostatic, and mitogenic processes. Ligand-gated ion channels can be opened by extracellular stimuli such as neurotransmitters (e.g., glutamate, serotonin, and acetylcholine), or intracellular stimuli (e.g., cAMP, Ca2+, and phosphorylation).
The Cav2 family of voltage-gated calcium channels consists of 3 main subtypes Cav2.1 (P or Q-type calcium currents), Cav2.2 (N-type calcium currents), and Cav2.3 (R-type calcium currents). These currents are found almost exclusively in the central nervous system (CNS), peripheral nervous system (PNS) and neuroendocrine cells, and constitute the predominant forms of presynaptic voltage-gated calcium current. Presynaptic calcium entry is modulated by many types of G-protein coupled receptors (GPCRs) and modulation of Cav2 channels is a widespread and highly efficacious means of regulating neurotransmission. The subunit composition of the Cav2 channels is defined by their Ci1 subunit, which forms the pore and contains the voltage-sensing gates (0^2.1 , c<i2.2, and αi2.3, also known as αiA, α1B, and α1E, respectively) and the β and α2 subunits.
Genetic or pharmacological perturbations in ion channel function can have dramatic clinical consequences. Long QT syndrome, epilepsy, cystic fibrosis, and episodic ataxia are a few examples of heritable diseases resulting from mutations in ion channel subunits. Toxic side effects such as arrhythmia and seizure, which can be triggered by certain drugs, can be due to interference with ion channel function (Sirois, J. E. and Atchison, W. D. (1996), Neurotoxicology, 17(1 ): 63-84; Keating, MT. (1996), Science, 272: 681-685). Drugs are useful for the therapeutic modulation of ion channel activity, and have applications in treatment of many pathological conditions, including hypertension, angina pectoris, myocardial ischemia, asthma, bladder overactivity, alopecia, pain, heart failure, dysmenorrhea, type Il diabetes, arrhythmia, graft rejection, seizure, convulsions, epilepsy, stroke, gastric hypermotility, psychoses, cancer, muscular dystrophy, and narcolepsy (Coghlan,
MJ. et al. (2001 ), J. Med. Chem., 44: 1627-1653; Ackerman, M.J. and Clapham, D. E. (1997), N. Eng. J. Med., 336: 1575-1586). The growing number of identified ion channels and understanding of their complexity will assist in future efforts at therapies that can modify ion channel function.
Therapeutic modulation of Cav2 channel activity has applications in treatment of many pathological conditions. All primary sensory afferents provide input to neurons in the dorsal horns of the spinal cord and in dorsal root ganglia neurons in the dorsal horn, and calcium influx through Cav2.2 channels triggers the release of neurotransmitters from presynaptic nerve terminals in the spinal cord. Hence, blockade of Cav2.2 channels is expected to be broadly efficacious because these channels are in a common pathway downstream from the wide variety of receptors that mediate pain (Julius, D. and Basbaum, A.I. (2001 ), Nature, 413: 203-216). Indeed, intrathecal injection of the Cav2.2-selective conotoxin ziconitide (SNX-111) has been shown to be effective against both neuropathic pain and inflammatory pain in animals and man (Bowersox, S. S. et al. (1996), J. Pharmacol. Exp. Ther., 279: 1243-1249). Ziconotide has also been shown to be effective as a neuroprotective agent in rat models of global or focal ischemia (Colburne, F. et al. (1999), Stroke, 30: 662-668). Thus, it is reasonable to conclude that modulation of Cav2.2 can have implications for neuroprotection and/or in the treatment of stroke.
Cav2.2 channels are found in the periphery and mediate catecholamine release from sympathetic neurons and adrenal chroffin cells. Some forms of hypertension result from elevated sympathetic tone. Cav2.2 modulators could be particularly effective in treating this disorder. Although complete block of Cav2.2 channels can cause hypotension or impair baroreceptor reflexes, partial inhibition by Cav2.2 modulators might reduce hypertension with minimal reflex tachycardia (Uneyama, O. D. (1999), Int. J. MoI. Med., 3: 455-466).
Overactive bladder (OAB) is characterized by storage symptoms such as urgency, frequency, and nocturia, with or without urge incontinence, resulting from the overactivity of the detrusor muscle in the bladder. OAB can lead to urge incontinence. The etiology of OAB and painful bladder syndrome is unknown,
although disturbances in nerves, smooth muscle and urothelium can cause OAB (Steers, W., Rev. Urol., 4: S7-S18). There is evidence to suggest that reduction of bladder hyperactivity may be indirectly effected by inhibition of Cav2.2 and/or Cav1 channels.
The localization of Cav2.1 channels in the superficial laminae of the dorsal horn of the spinal cord suggests involvement of these channels in the perception and maintenance of certain forms of pain (Vanegas, H. and Schaible, H. (2000), Pain, 85: 9-18). Complete elimination of Cav2.1 calcium currents alters synaptic transmission, resulting in severe ataxia. Gabapentin has been used clinically for many years as an add-on therapy for the treatment of epilepsy. In recent years, it has emerged as a leading treatment of neuropathic pain. Clinical trials have shown gabapentin to be effective for the treatment of post-herpetic neuralgia, diabetic neuropathy, trigeminal neuralgia, migraine and fibromyalgia (Mellegers, P. G. et al. (2001 ), Clin. J. Pain, 17: 284-295). Gabapentin was designed as a metabologically stable GABA mimetic, but most studies find no effect on the GABA receptors. The α2δ subunit of voltage-gated calcium channels has been identified as a high affinity binding site for gabapentin in the CNS. There is evidence that suggests that gabapentin could inhibit neurotransmission in the spinal cord by interfering with the function of the α2δ subunits, thereby inhibiting presynaptic calcium currents.
SUMMARY OF THE INVENTION
The present teachings relate to compounds of formula (I):
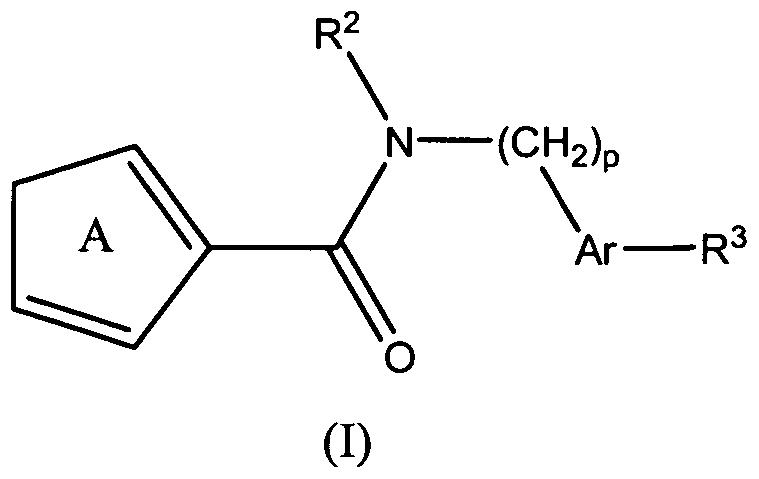

The present teachings also provide methods of making the compounds of formula (I), and methods of using the compounds of formula (I) for the therapeutic modulation of ion channel function, and treatment of one or more conditions, particularly those mediated by certain calcium channel subtype targets. The methods of using the compounds generally include administering a therapeutically effective amount of a compound of formula (I) to a mammal.
The present teachings relate to compounds of formula (II):

The present teachings also provide methods of making the compounds of formula (II), and methods of using the compounds of formula (II) for the therapeutic modulation of ion channel function, and treatment of one or more conditions, particularly those mediated by certain calcium channel subtype targets. The
methods of using the compounds generally include administering a therapeutically effective amount of a compound of formula (II) to a mammal.
The present teachings relate to compounds of formula (III):

The present teachings also provide methods of making the compounds of formula (III), and methods of using the compounds of formula (III) for the therapeutic modulation of ion channel function, and treatment of one or more conditions, particularly those mediated by certain calcium channel subtype targets. The methods of using the compounds generally include administering a therapeutically effective amount of a compound of formula (III) to a mammal.
The present teachings relate to compounds of formula (IV):

(IV) and pharmaceutically acceptable salts, hydrates, and esters thereof, wherein Ar, R2, R3, p and
 are defined as described herein.
are defined as described herein.
The present teachings also provide methods of making the compounds of formula (IV), and methods of using the compounds of formula (IV) for the therapeutic modulation of ion channel function, and treatment of one or more conditions, particularly those mediated by certain calcium channel subtype targets. The methods of using the compounds generally include administering a therapeutically effective amount of a compound of formula (IV) to a mammal.
The present teachings relate to compounds of formula (V):

(V) and pharmaceutically acceptable salts , hydrates, and esters thereof, wherein Ar, R2,
R3, p and

The present teachings also provide methods of making the compounds of formula (V), and methods of using the compounds of formula (V) for the therapeutic modulation of ion channel function, and treatment of one or more conditions,
particularly those mediated by certain calcium channel subtype targets. The methods of using the compounds generally include administering a therapeutically effective amount of a compound of formula (V) to a mammal.
The present teachings relate to compounds of formula (Vl):
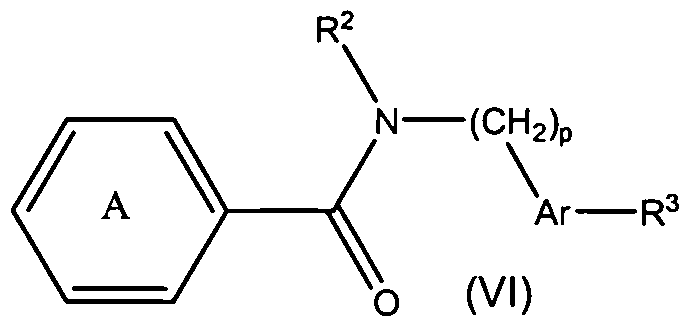

The present teachings also provide methods of making the compounds of formula (VI), and methods of using the compounds of formula (Vl) for the therapeutic modulation of ion channel function, and treatment of one or more conditions, particularly those mediated by certain calcium channel subtype targets. The methods of using the compounds generally include administering a therapeutically effective amount of a compound of formula (Vl) to a mammal.
The present teachings relate to compounds of formula (VII):
 and pharmaceutically acceptable salts, hydrates, and esters thereof, wherein Ar, R2, R3, p and
and pharmaceutically acceptable salts, hydrates, and esters thereof, wherein Ar, R2, R3, p and

The present teachings also provide methods of making the compounds of formula (VII), and methods of using the compounds of formula (VII) for the therapeutic modulation of ion channel function, and treatment of one or more conditions, particularly those mediated by certain calcium channel subtype targets. The methods of using the compounds generally include administering a therapeutically effective amount of a compound of formula (VII) to a mammal.
DETAILED DESCRIPTION OF THE INVENTION
Embodiments of the present invention provide compounds that can modulate the activity of ion channels in a mammal, for example, Cav2.2 voltage-gated calcium channels, and can treat a variety of pathological conditions, states, disorders or diseases.
Unless otherwise indicated, the following terms are held to have the following meanings as used herein.
The term "mammal" refers to any warm blooded species, such as a human. As used herein, "ion channel" includes at least voltage-gated calcium channels and voltage- gated sodium channels such as, without limitation, Cav1.1 , Cav1.2, Cav1.3, Cav2.1 , Cav2.2, Cav2.3, Cav3.1 , Cav3.2, Na«1.1 , Nav1.2, NaJ .3, Nav1.7, NaJ .8, and NaJ .9. As used herein, "Cav2.2 voltage-gated calcium channel" refers to a voltage-gated calcium channel containing at least one Cav2.2 αi subunit. As used herein, "ion channel mediated condition" refers to any condition or pathological state of a mammal or any disease present in a mammal that can be treated, or the symptoms of which can be alleviated, by modulation of the activity of one or more ion channels such as Cav2.2 voltage-gated calcium channels.
The term "halo" or "halogen" refers to fluoro, chloro, bromo, and iodo.
The term "oxo" refers to a double-bonded oxygen (i.e., =O).
The term "alkyl" refers to a straight-chain or branched saturated hydrocarbon group. Examples of alkyl groups include methyl (Me), ethyl (Et), propyl (e.g., n-propyl and isopropyl), butyl (e.g., n-butyl, isobutyl, s-butyl, t-butyl), pentyl groups (e.g., n-pentyl, isopentyl, neopentyl), and the like. A lower alkyl group typically has up to 6 carbon atoms. In various embodiments, an alkyl group has 1-6 carbon atoms, and is referred to as a "C1-6 alkyl group." Examples of C1-6 alkyl groups include, but are not limited to, methyl, ethyl, propyl (e.g., n-propyl and isopropyl), butyl (e.g., n-butyl, isobutyl, s-butyl, t-butyl), pentyl (e.g., n-pentyl, neopentyl, isopentyl, t-pentyl), and hexyl groups (e.g., n-hexyl, isohexyl). A branched alkyl group has at least 3 carbon atoms (e.g., an isopropyl group), and in various embodiments, has up to 6 carbon atoms, e.g. it is a C3-6 alkyl group, i.e., a branched lower alkyl group. Examples of branched lower alkyl groups include, but are not limited to:

isopropyl, isobutyl, sec-butyl, tert-butyl, isopentyl, neopentyl, and terf-pentyl.
The term "alkenyl" refers to a straight-chain or branched alkyl group having one or more carbon-carbon double bonds. Examples of alkenyl groups include, but are not limited to, ethenyl, propenyl, butenyl, pentenyl, hexenyl, butadienyl, pentadienyl, hexadienyl groups, and the like. The one or more carbon-carbon double bonds can be internal (such as in 2-butene) or terminal (such as in 1-butene). A branched alkenyl group has at least 3 carbon atoms, and in various embodiments, has up to 6 carbon atoms, e.g. it is a C3-6 alkenyl group.
The term "alkynyl" refers to a straight-chain or branched alkyl group having one or more carbon-carbon triple bonds. Examples of alkynyl groups include, but are not limited to, ethynyl, propynyl, butynyl, pentynyl, and the like. The one or more carbon- carbon triple bonds can be internal (such as in 2-butyne) or terminal (such as in 1- butyne). The alkynyl group is suitably a C3-6 alkynyl group.
The term "alkoxy" refers to an -O-alkyl group, wherein the alkyl group may be a straight or branched chain. Examples of alkoxy groups include, but are not limited to, methoxy, ethoxy, propoxy (e.g., n-propoxy and isopropoxy), t-butoxy groups, and the like.
The term "haloalkyl" refers to an alkyl group having one or more halogen substituents. Examples of haloalkyl groups include, but are not limited to, -CF3, -C2F5, -CHF2, -CH2F, -CCI3, -CHCI2, -CH2CI, -C2CI5, and the like. Perhaloalkyl groups, i.e., alkyl groups wherein all of the hydrogen atoms are replaced with halogen atoms (e.g., CF3 and C2F5), are included within the definition of "haloalkyl."
The term "cycloalkyl" refers to a non-aromatic carbocyclic group including cyclized alkyl, alkenyl, and alkynyl groups. A cycloalkyl group can be monocyclic (e.g., cyclohexyl) or polycyclic (e.g., containing fused, bridged, and/or spiro ring systems), wherein the carbon atoms are located inside or outside of the ring system. Any suitable ring position of the cycloalkyl group can be covalently linked to the defined chemical structure. In various embodiments, a cycloalkyl group has 3-6 carbon atoms, and is referred to as a "C3-6 cycloalkyl group." Examples of C3-6 cycloalkyl groups include, but are not limited to, cyclopropyl, cyclop ropy I methyl, cyclopropylethyl, cyclopropylpropyl, cyclobutyl, cyclobutylmethyl, cyclobutylethyl, cyclopentyl, cyclopentylmethyl, cyclohexyl, cyclopentenyl, cyclohexenyl, and cyclohexadienyl groups, as well as their homologs, isomers, and the like. Also included in the definition of cycloalkyl groups are moieties that have one or more aromatic rings fused (i.e., having a bond in common with) to the cycloalkyl ring, for example, benzo derivatives of cyclopentane (i.e., an indanyl group), cyclohexane (i.e., a tetrahydronaphthyl group), and the like. In some embodiments, cycloalkyl groups can be substituted with 1 to 3 substituents independently selected from a Ci-6 alkyl group and a -Y-phenyl group, wherein the phenyl group is optionally substituted with 1 to 3 substituted independently selected from a halogen and a Ci-6 alkoxyl group, and Y is as defined hereinbelow.
The term "heteroatom" refers to an atom of any element other than carbon or hydrogen and includes, for example, nitrogen, oxygen, sulfur, phosphorus, and selenium.
The term "cycloheteroalkyl" refers to a non-aromatic cycloalkyl group having 5-10 ring atoms, among which 1 to 3 ring atoms are heteroatoms independently selected from oxygen (O), nitrogen (N) and sulfur (S), and that optionally contains one or more, e.g., two, double or triple bonds. Also included in the definition of cycloheteroalkyl are moieties that have one or more aromatic rings fused (i.e., have a bond in common with) to the cycloheteroalkyl group. Examples of cycloheteroalkyl groups include, among others, morpholine, thiomorpholine, pyran, imidazolidine, imidazoline, oxazolidine, pyrazolidine, pyrazoline, pyrrolidine, pyrroline, tetrahydrofuran, tetrahydrothiophene, piperidine, piperazine, tetrahydroisoquinoline,
benzimidazoline, chromane, chromene, indolinetetrahydroquinoline, and the like. One or more N or S atoms in a cycloheteroalkyl ring can be oxidized (e.g., morpholine N-oxide, thiomorpholine S-oxide, thiomorpholine S,S-dioxide). Cycloheteroalkyl groups can also contain one or more oxo groups, such as piperidone, oxazolidinone, pyrimidine-2,4(1/-/,3H)-dione, pyridin-2(1H)-one, and the like. A cycloheteroalkyl group can be optionally substituted. For example, in some embodiments, one or more carbon ring atoms of a cycloheteroalkyl group can bear a substituent independently selected from a halogen, a C1-6 alkyl group, -C(O)-NRdRe, -Y-OR0, -Y-NRdRe, a -Y-phenyl group, a -Y-(5-7 membered cycloheteroalkyl), a - Y-(5-9 membered heteroaryl) group, or a -Y-O-(5-7 membered heteroaryl) group, and/or one or more nitrogen ring atoms of a cycloheteroalkyl group can bear a substituent independently selected from a halogen, a C1-6 alkyl group, -C(O)RC, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, -Y-C(O)NRdRe, an -S(O)2-C1-6 alkyl group, a -C2-6 alkyl— (5-7 membered cycloheteroalkyl) group, or a 5-7 membered heteroaryl group, wherein Y, Rc, Rd, and Re are as defined hereinbelow. Further, each of the phenyl substituents immediately above can be optionally substituted with 1 to 3 substituents independently selected from a halogen, a C1-6 alkyl group, a C1-6 haloalkyl group, and a C1-6 alkoxy group, and each of the 5-7 membered cycloheteroalkyl substituents, the 5-7 membered heteroaryl substituents, and the 5-9 membered heteroaryl substituents immediately above can be optionally substituted with 1 to 3 substituents independently selected from a halogen and a C1-6 alkyl group.
The term "aryl" refers to an aromatic monocyclic or polycyclic hydrocarbon ring system such as, for example, phenyl, 1-naphthyl, 2-naphthyl, anthracenyl, phenanthrenyl groups, and the like. In some embodiments, a monocyclic aryl group can have from 6 to 14 carbon atoms and a polycyclic aryl group can have from 8 to 14 carbon atoms. Any suitable ring position of the aryl group can be covalently linked to the defined chemical structure. In some embodiments, aryl groups optionally contain up to three independently selected substituent groups. For example, a phenyl group, in some embodiments, can be optionally substituted with 1 to 3 substituents independently selected from a halogen, CN, -C(O)OR0, -NRdRe, a C1-6 alkyl group, a C1-6 haloalkyl group, and a C1-6 alkoxy group, wherein Rc, Rd, and Re are as defined hereinafter.
The term "heteroaryl" refers to an aromatic monocyclic or polycyclic aromatic ring system containing 5-7 ring atoms, among which 1 to 3 ring atoms are heteroatoms independently selected from oxygen (O), nitrogen (N) and sulfur (S). Generally, heteroaryl rings do not contain 0-0, S-S, or S-O bonds. However, one or more N or S atoms in a heteroaryl group can be oxidized (e.g., pyridine N-oxide, thiophene S- oxide, thiophene S,S-dioxide). Examples of heteroaryl groups include, for example, the 5-membered monocyclic and 5-6 bicyclic ring systems shown below:

C(O)NRdRe group, an S(O)2-C1-6 alkyl group, a 5-7 membered heteroaryl group, and a C2-6 alkyl— 5-7 membered cycloheteroalkyl group, where Y, Rc, Rd and Rθ are as defined hereinafter. Examples of such heteroaryl rings include, but are not limited to, pyrrole, furan, thiophene, pyridine, pyrimidine, pyridazine, pyrazine, triazole, tetrazole, pyrazole, imidazole, isothiazole, thiazole, thiadiazole, isoxazole, oxazole, oxadiazole, indole, isoindole, benzofuran, benzothiophene, quinoline, 2- methylquinoline, isoquinoline, quinoxaline, quinazoline, benzotriazole, benzimidazole, benzothiazole, benzisothiazole, benzisoxazole, benzoxadiazole, benzoxazole, cinnoline, 1 H-indazole, 2H-indazole, indolizine, isobenzofuran, naphthyridine, phthalazine, pteridine, purine, oxazolopyridine, thiazolopyridine, imidazopyridine, furopyridine, thienopyridine, pyridopyrimidine, pyridopyrazine, pyridopyridazine, thienothiazole, thienoxazole, and thienoimidazole. In some embodiments, heteroaryl
groups can be substituted with up to three independently selected substitution groups. For example, in some embodiments, one or more nitrogen atoms can be substituted with independently selected R1 groups as defined above, and/or one or more carbon ring atoms of a cycloheteroalkyl group can bear a substituent independently selected from a halogen, a C1-6 alkyl group, -C(O)-NRdRe, -Y-ORC, - Y-NRdRe, a -Y-phenyl group, a -Y-(5-7 membered cycloheteroalkyl) group, a -Y- (5-9 membered heteroaryl) group, or a -Y-O-(5-7 membered heteroaryl) group, wherein Y, Rc, Rd, and Re are as defined hereinafter. Further, each of the phenyl substituents immediately above can be optionally substituted with 1 to 3 substituents independently selected from a halogen, a C1-6 alkyl group, a C1-6 haloalkyl group, and a C1-6 alkoxy group, and each of the 5-7 membered cycloheteroalkyl substituents, the 5-7 membered heteroaryl substituents, and the 5-9 membered heteroaryl substituents immediately above can be optionally substituted with 1 to 3 substituents independently selected from a halogen and a C1-6 alkyl group.
The term "divalent group" is defined herein as a linking group capable of forming a covalent bond with two other moieties. For example, compounds described herein can include a divalent C1-6 alkyl group, such as, for example, a methylene group.
The term a "leaving group" ("LG") refers to a charged or uncharged atom (or group of atoms) that can be displaced as a stable species as a result of, for example, a substitution or elimination reaction. Examples of leaving groups include, but are not limited to, halide (e.g., Cl, Br, I), tosylate (toluenesulfonyl group, TsO), mesylate (methanesulfonyl group, MsO), brosylate (p-bromobenzenesulfonyl group, BsO), nosylate (4-nitrobenzenesulfonyl group, NsO), water (H2O), ammonia (NH3), and triflate (trifluoromethanesulfonyl group, OTf).
The term a "protecting group" ("PtG") refers to modification of a functional group that reduces the reactivity of the functional group in an unwanted reaction. Examples of protecting groups for amines include, but are not limited to, terf-butyloxycarbonyl (t- BOC), benzyl (Bn), and carbobenzyloxy (Cbz) groups. Examples of protecting groups for carbonyls include, but are not limited to, acetals and ketals. Examples of protecting groups for carboxylic acids include, but are not limited to, methyl esters,
benzyl esters, te/t-butyl esters, and silyl esters, and are provided in Greene, et al., Protective Groups in Organic Synthesis, 2d. Ed., Wiley & Sons, 1991 , the entire disclosure of which is incorporated by reference herein for all purposes.
At various places in the present specification, substituents of compounds are disclosed in groups or in ranges. It is specifically intended that the description include each and every individual combination of the members of such groups and ranges. For example, the term "C1-6 alkyl" is specifically intended to individually disclose Ci, C2, C3, C4, C5, C6, Ci-C6, C1-C5, C1-C4, C1-C3, C1-C2, C2-C6, C2-C5, C2-C4, C2-C3, C3-C6, C3-C5, C3-C4, C4-C6, C4-C5, and C5-C6 alkyl. By way of another example, the term "5-9 membered heteroaryl group" is specifically intended to individually disclose a heteroaryl group having 5, 6, 7, 8, 9, 5-9, 5-8, 5-7, 5-6, 6-9, 6- 8, 6-7, 7-9, 7-8, and 8-9 ring atoms.
The present teachings provide compounds of formula (I)

(I) and pharmaceutically acceptable salts, hydrates, and esters thereof, wherein:


X is selected from -NR0-, a divalent C1-6 alkyl group, and a covalent bond;
R1 is selected from a C1^ alkyl group, a phenyl group, and a 5-7 membered heteroaryl group;
R2 is a -C3-6 cycloalkyl group, a benzyl group, an indole group, or a phenyl group, wherein
the phenyl group and the benzyl group are each optionally substituted with 1 to 3 substituents independently selected from a halogen, a C1-6 alkyl group, a C1-6 alkoxy group, a C1-6 haloalkyl group, a C1-6 haloalkoxy group, -OCH2-C6H5, -CN, -C(O)ORC, -OH, and -NRdRe, and the -C3-6 cycloalkyl group is optionally substituted with 1 to 3 C1-6 alkyl groups;
Ar-R3 is selected from:

R3 is selected from a halogen, a C1-10 alkyl group, a C1-10 alkoxy group, a C1-10 haloalkyl group, a C1-I0 haloalkoxy group, a -C(O)R0 group, a piperidin-4-yl group, and a -Y-NRfR9 group; wherein the C1-10 alkyl group and the C1-10 alkoxy group are optionally substituted with from 1-3 substitutents selected from a halogen, a phenyl group, and -OH, and the nitrogen ring atom of the piperidin-4-yl group is optionally substituted with -C(O)O-C1-6alkyl;
Y, at each occurrence, is independently a divalent C1-6 alkyl group or a covalent bond;
R0, Rd and Re, at each occurrence, independently are H, C1-6 haloalkyl, or a C1-6 alkyl group;
Rf and R9, at each occurrence, independently are selected from H1 -C(O)R0, - C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, a C1-10 alkyl group, a C3-6 cycloalkyl group, a -Y-phenyl group, a -C(O)-phenyl group, a -Y-(5-7 membered cycloheteroalkyl) group, a -Y-(5-7 membered heteroaryl) group, and a -C2-6 alkyl-O-Y-(5-7 membered heteroaryl) group, or
alternatively, Rf and R9 taken together with the nitrogen atom to which they are bonded form a 5-7 membered cycloheteroalkyl group or a 5-7 membered heteroaryl group, the 5-7 membered cycloheteroalkyl group and the 5-7 membered heteroaryl group containing up to two ring heteroatoms independently selected from nitrogen, oxygen, and sulfur, wherein one or more nitrogen atoms in the ring optionally are independently substituted with -C(O)R0, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, -Y-C(O)NRdRe, -S(O)2-(C1-6 alkyl), a -C1-6 alkyl-(phenyl)n group, a -C2-6 alkyl— (5-7 membered cycloheteroalkyl) group, C1-6 alkyl, -Y-(phenyl)q, C3-8 cycloalkyl, or a 5-7 membered heteroaryl group; one or more carbon atoms in the ring optionally are independently substituted with -C(O)-NRdRe, -Y-OR0, -Y-NRdRe, -Y- (phenyl)q, -Y-(5-7 membered cycloheteroalkyl), a -Y-(5-9 membered heteroaryl) group, an oxo group, CN, C3-8 cycloalkyl, or a -Y-O-(5-7 membered heteroaryl) group; and any sulfur atom in the ring is optionally substituted with 1 to 2 oxo groups;
wherein each of the phenyl groups appearing anywhere in said Rf and Rg is optionally substituted with 1 to 3 substituents independently selected from halogen, C1-6 alkyl, C1-6 haloalkyl, and C1-6 alkoxy, and each of the 5-7 membered cycloheteroalkyl groups, the 5-7 membered heteroaryl groups, and the 5-9 membered heteroaryl groups appearing anywhere in said Rf and R9 is optionally substituted with 1 to 3 substituents independently selected from halogen and C1-6 alkyl;
n is 1 , 2, or 3;
q is 1 , 2, or 3; and
p is 1 , 2, 3, or 4.
According to some embodiments,
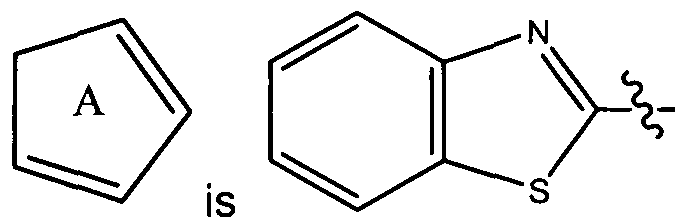
In accordance with some embodiments,


R1 can be selected from a methyl group, a phenyl group, and a pyridyl group.
In some embodiments, X can be selected from -NH-, -N(CH3)-, -CH2-, and a covalent bond.
In accordance with some embodiments, p is 1.
In some embodiments, p is 2.
In certain embodiments, p is 3.
In some embodiments, R2 can be a phenyl group optionally substituted with 1-2 substituents independently selected from a halogen, a C1-6 alkyl group, CN, -
C(O)ORc, and -NRdRe, wherein Rc, Rd and Re are as defined above. For example,
- 19 -
R2 can be a 4-fluorophenyl group, a 4-chlorophenyl group, a 4-fluoro-2-methylphenyl group, or a 4-methoxyphenyl group.
In certain preferred embodiments, Ar-R3 can be
kNAR3 R3 KJ wherein R is as defined above.
In accordance with some preferred embodiments, Ar-R3 can be
T S VR3 i 3nd T ^N /VR3 ; wherein R3 is as defined above. In some embodiments, R3 can be NRfRg, wherein Rf and Rg are as defined above. In particular embodiments, R3 can be selected from NH2, an NH-C1-6 alkyl group, an N(Ci-6 alkyl)2 group wherein the C1-6 alkyl groups do not need to be the same, wherein the C1-6 alkyl groups do not need to be the same, an NH-C3-6 cycloalkyl group, an N(C1-6 alkyl)— C3-6 cycloalkyl group, an N(C1-6 alkyl)— C2-6 alkyl-ORc group, an N(C1-6 alkyl)- Y-(5-7 membered cycloheteroalkyl) group, an N(C1-6 alkyl)-phenyl group, an N(C1-6 alkyl)-Y-(5-7 membered heteroaryl) group, and an N(C1-6 alkyl)-C2-6 alkyl-O-Y-(5-7 membered heteroaryl) group, wherein each of the phenyl group, the 5-7 membered cycloheteroalkyl group, and the 5-7 membered heteroaryl group immediately above is optionally substituted with 1 to 3 substituents independently selected from a halogen and a C1-6 alkyl group; and Y and Rc are as defined above. For example, R3 can be a diethylamino group or a cyclopropyl ethyl amino group.
In other embodiments, R3 can be an optionally substituted 5-7 membered cycloheteroalkyl group or an optionally substituted 5-7 membered heteroaryl group
- 20 -
as described herein. In certain embodiments, R3 can be selected from a diazepanyl group, an imidazolyl group, a morpholinyl group, a piperidinyl group, a piperazinyl group, a pyridyl group, a pyrrolidyl group, and a thiomorpholinyl group, wherein each of these groups can include a nitrogen ring atom optionally substituted with -C(O)RC, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, -Y-C(O)NRdRe, an -S(O)2-C1-6 alkyl group, a - C2-6 alkyl— (5-7 membered cycloheteroalkyl) group, a C1-I0 alkyl group, or a 5-7 membered heteroaryl group, a carbon ring atom optionally substituted with -C(O)- NRdRe, -Y-ORc, -Y-NRdRe, a -Y-phenyl group, a -Y-(5-7 membered cycloheteroalkyl) group, a -Y-(5-9 membered heteroaryl) group, or a -Y-O-(5-7 membered heteroaryl) group, and/or a sulfur ring atom optionally substituted with 1 or 2 oxo groups, wherein each of the phenyl groups immediately above is optionally substituted with 1 to 3 substituents independently selected from a halogen, a Ci-6 alkyl group, a C1-6 haloalkyl group, and a C1-6 alkoxy group, and each of the 5-7 membered cycloheteroalkyl groups, the 5-7 membered heteroaryl groups, and the 5- 9 membered heteroaryl groups immediately above is optionally substituted with 1 to 3 substituents independently selected from a halogen and a C1-6 alkyl group, wherein Y, Rc, Rd and Re are as defined above.
In particular embodiments, R3 can be selected from a 1-[1 ,4]diazepanyl group, a 1- imidazolyl group, a 4-morpholinyl group, a 1 -piperidinyl group, a 1 -piperazinyl group, a 4-pyridyl group, a 1 -pyrrolidyl group, and a 4-thiomorpholinyl group, wherein each of these groups can be optionally substituted as described above.
In some embodiments, R3 can be a 1 -piperazinyl group having a nitrogen atom in the ring optionally substituted with -C(O)RC, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, -C1-6 alkyl-C(O)NRdRe, an S(O)2-C1-6 alkyl group, a -C2-6 alkyl-(5-7 membered cycloheteroalkyl) group, a C1-10 alkyl group, or a 5-7 membered heteroaryl group. For example, R3 can be a 4-methyl piperazin-1-yl group.
In other embodiments, R3 can be a 1 -piperidinyl group having a carbon atom in the ring optionally substituted with -NRdRe, -C(O)-NRdRe, -Y-ORc, a 5-7 cycloheteroalkyl group, a 5-9 membered heteroaryl group, or a -Y-O-(5-7 membered heteroaryl) group.
- 21 -
Representative compounds of formula (I) in accordance with embodiments of the present invention include, but are not limited to, the compounds presented in Table 1 below.
TABLE 1.
Cpd. No. Structure Jame
5 4L 2-(Methyl-phenyl-amino)-thiazole-4-carboxylic acic
OX NXN.. diethylamino-pyridin-3-ylmethyl)-(4-fluorophenyl)-a
)=N k
O
2-Benzyl-thiazole-4-carboxylic acid (6-diethylaminc
N N
\ Ii pyridin-3-ylmethyl)-(4-fluorophenyl)-amide s-1 k
V. /)
 2-Methyl-thiazole-4-carboxylic acid (6-diethylaminc 3-ylmethyl)-(4-fluorophenyl)-amide r k
2-Methyl-thiazole-4-carboxylic acid (6-diethylaminc 3-ylmethyl)-(4-fluorophenyl)-amide r k
o^^r o N N 2-Pyridin-4-yl-thiazole-4-carboxylic acid (6-diethyla
>N k^ pyridin-3-ylmethyl)-(4-fluorophenyl)-amide
 'it '
'it '
s- -7 o Oα ^N".N" 2-Pyridin-3-yl-thiazole-4-carboxylic acid (6-diethyla pyridin-3-ylmethyl)-(4-fluorophenyl)-amide

- 22 -
N-(4-chlorophenyl)-N-{[6-(diethylamino)pyridin-2- yl]methyl}-2-methyl-1 ,3-thiazole-4-carboxamide

 ^N N-(4-fluoro-2-methylphenyl)-2-methyl-N-[(2-piperidin-
^N N-(4-fluoro-2-methylphenyl)-2-methyl-N-[(2-piperidin-
KK ylpyrimidin-5-yl)methyl]-1 ,3-thiazole-4-carboxamide
Y* O
N-{4-[cyclopropyl(ethyl)amino]benzyl}-N-(4- ethoxyphenyl)-2-methyl-1 ,3-thiazole-4-carboxamid€
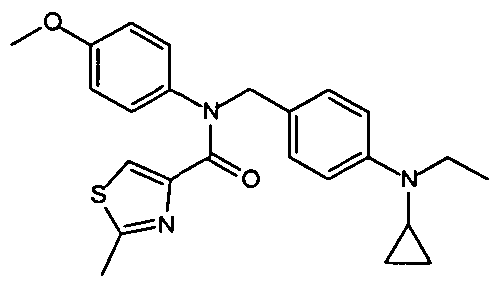 A
A
N-(4-fluorophenyl)-N-{[6-(4-methylpiperazin-1-yl)pyric yl] methyl}- 1 , 3-benzoth iazole-2-ca rboxa mide

^l N-{[6-(diethylamino)pyridin-3-yl]methyl}-N-(4-fluoroph 1 ,3-benzothiazole-2- carboxamide

Embodiments of the present invention further provide compounds of formula (II):
R2

■N O
R4 J \
(H) or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein:
- 23 -
R2, Ar, R3, Y, and p are as defined above;
R4 is selected from H, a C1-6 alkyl group, and a -Y-phenyl group; and
R5 is selected from H, a d.i0 alkyl group, a C3-6 alkenyl group, a C3-8 cycloalkyl group, a phenyl group, a 5-7 membered heteroaryl group, a -C1-6 alkyl-phenyl group, a -C1-6 alkyl— 5-7 membered heteroaryl group, wherein each of the phenyl group and the 5-7 membered heteroaryl group optionally is substituted with 1 to 3 substituents independently selected from a halogen, a C1-6 alkyl group, a C1-6 haloalkyl group, and a C1-6 alkoxy group; provided that at least one of R4 and R5 is not H. .
In accordance with some embodiments, R4 can be H, a methyl group, or a benzyl group.
In some embodiments, R5 can be selected from a C1-6 alkyl group, C3-6 alkenyl group, a C3-6 cycloalkyl group, a -C1-6 alkyl-phenyl group, and a -C1-6 alkyl— 5-7 membered heteroaryl group, wherein each of the phenyl group and the 5-7 membered heteroaryl group optionally is substituted with 1 to 3 substituents selected from a halogen or a C1-6 alkyl group. For example, R5 can be an allyl group, a benzyl group, a 4- chlorobenzyl group, a cyclopentyl group, a cyclopropylmethyl group, a 2,2-dimethyl- propyl group, a methyl group or a 5-methyl-furan-2-yl-methyl group.
In some embodiments, R2 can be a phenyl group optionally substituted with 1-2 substituents independently selected from a halogen, CN, a C1-6 alkyl group, - C(O)ORC and -NRdRe, wherein Rc, Ra and Re are as defined above. For example, R2 can be a 4-fluorophenyl group, a 2-isopropylphenyl group, or a 4-methylphenyl group.
In accordance with some embodiments, p is 1.
In some embodiments, p is 2.
- 24 -
In certain embodiments, p is 3.
In certain preferred embodiments, Ar-R3 can be
/> ,N
/' /vN< -R3

N^R3
 R3
R3
 wherein R is as defined above.
wherein R is as defined above.
In accordance with some preferred embodiments, Ar-R3 can be
wherein R is as defined above.
In accordance with some preferred embodiments, Ar-R3 can be
■Vs\
wherein R3 is as defined above.
In some embodiments, R3 can be NRfRg, wherein Rf and R9 are as defined above. In particular embodiments, R3 can be selected from NH2, an NH-C1-6 alkyl group, an N(Ci-6 alkyl)2 group wherein the C1-6 alkyl groups do not need to be the same,, an NH-C3-6 cycloalkyl group, an N(C1-6 alkyl)— C3-6 cycloalkyl group, an N(C1-6 alkyl)— C2-6 alkyl-ORc group, an N(C1-6 alkyl)-Y-(5-7 membered cycloheteroalkyl) group, an N(C1-6 alkyl)-phenyl group, an N(Ci-6 alkyl)- Y-5-7 membered heteroaryl group, and an N(C1-6 alkyl)-C2-6 alkyl-O-Y-5-7 membered heteroaryl group, wherein each of the phenyl group, the 5-7 membered cycloheteroalkyl group, and the 5-7 membered
- 25 -
heteroaryl group immediately above is optionally substituted with 1 to 3 substituents independently selected from a halogen and a C1-6 alkyl group, and Y and R0 are as defined above. For example, R3 can be a diethylamino group, a dimethylamino group, an ethyl(methyl)amino group, an isopropyl(ethyl)amino group, or a cyclopropyl(ethyl)amino group.
In other embodiments, R3 can be an optionally substituted 5-7 membered cycloheteroalkyl group or an optionally substituted 5-7 membered heteroaryl group as described herein. In certain embodiments, R3 can be selected from a diazepanyl group, an imidazolyl group, a morpholinyl group, a piperidinyl group, a piperazinyl group, a pyridyl group, a pyrrolidyl group, and a thiomorpholinyl group, wherein each of these groups can include a nitrogen ring atom optionally substituted with -C(O)RC, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, -Y-C(O)NRdR6, an -S(O)2-C1-6 alkyl group, a - C2-6 alkyl— (5-7 membered cycloheteroalkyl) group, a C1-10 alkyl group, or a 5-7 membered heteroaryl group, a carbon ring atom optionally substituted with -C(O)- NRdRθ, -Y-ORC, -Y-NRdRe, a -Y-phenyl group, a -Y-(5-7 membered cycloheteroalkyl) group, a -Y-(5-9 membered heteroaryl) group, or a -Y-O-(5-7 membered heteroaryl) group, and/or a sulfur ring atom optionally substituted with 1 or 2 oxo groups, wherein each of the phenyl groups immediately above is optionally substituted with 1 to 3 substituents independently selected from a halogen, a C1-6 alkyl group, a C1-6 haloalkyl group, and a C1-6 alkoxy group, and each of the 5-7 membered cycloheteroalkyl groups, the 5-7 membered heteroaryl groups, and the 5- 9 membered heteroaryl groups immediately above is optionally substituted with 1 to 3 substituents independently selected from a halogen and a C1-6 alkyl group, wherein Y, Rc, Rd and R6 are as defined above.
In particular embodiments, R3 can be selected from a 1-[1 ,4]diazepanyl group, a 1- imidazolyl group, a 4-morpholinyl group, a 1 -piperidinyl group, a 1 -piperazinyl group, a 4-pyhdyl group, a 1 -pyrrolidyl group, and a 4-thiomorpholinyl group, wherein each of these groups can be optionally substituted as described above.
In some embodiments, R3 can be a 1 -piperazinyl group having a nitrogen atom in the ring optionally substituted with -C(O)RC, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, -C1-6
alkyl-C(O)NRdRe, an S(O)2-C1-6 alkyl group, a -C2-6 alkyl-(5-7 membered cycloheteroalkyl) group, a C1-10 alkyl group, or a 5-7 membered heteroaryl group. For example, R3 can be a 4-methyl piperazin-1-yl group.
In other embodiments, R3 can be a 1-piperidinyl group having a carbon atom in the 5 ring optionally substituted with -NRdRe, -C(O)-NRdR6, -Y-ORc, a 5-7 cycloheteroalkyl group, a 5-9 membered heteroaryl group, or a -Y-O-(5-7 membered heteroaryl) group.
In accordance with some embodiments, R3 can be a methyl group or a trifluoromethyl group.
10 Representative compounds of formula (II) in accordance with embodiments of the present invention include, but are not limited to, the compounds presented in Table 2 below.
TABLE 2.
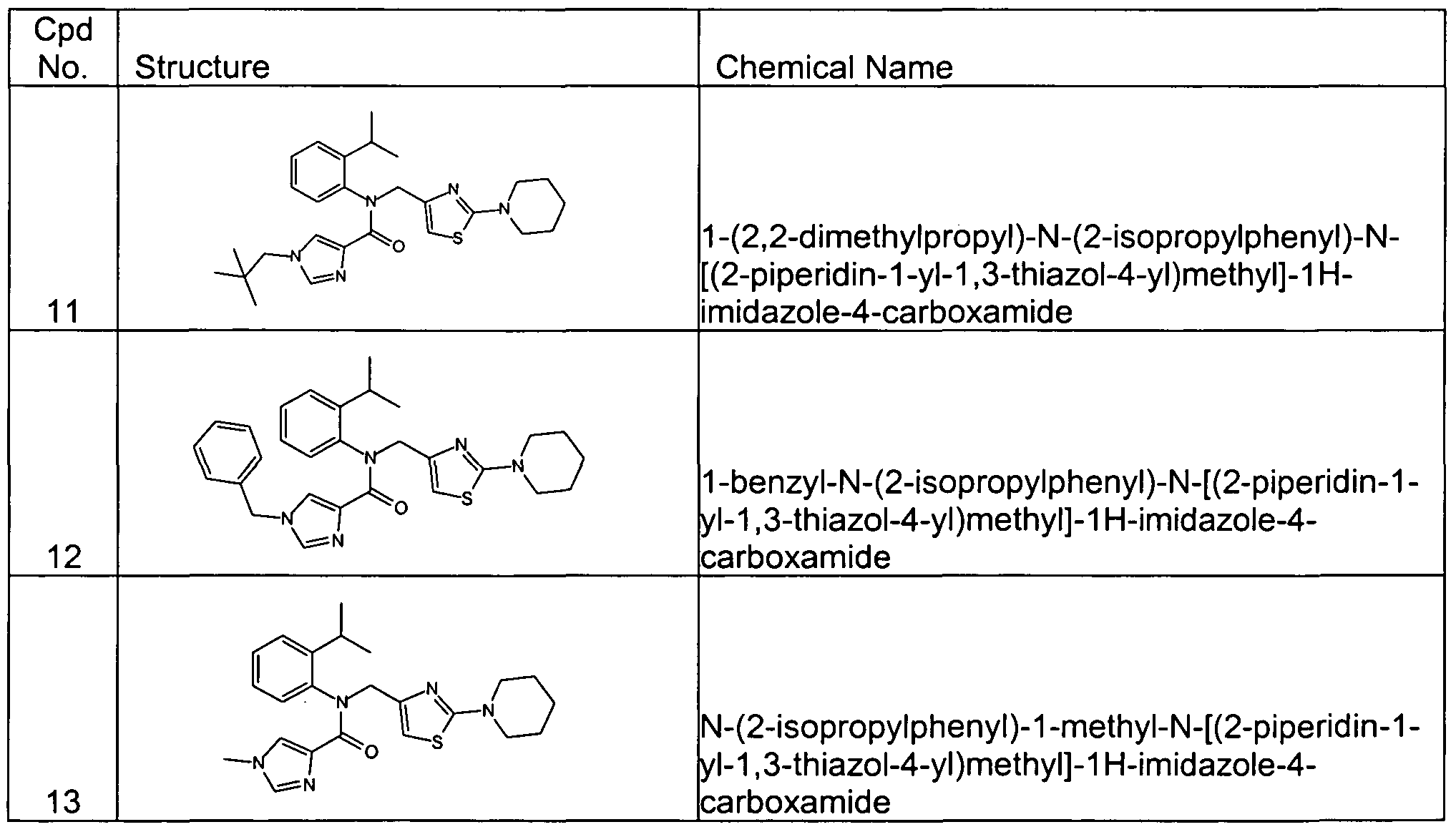
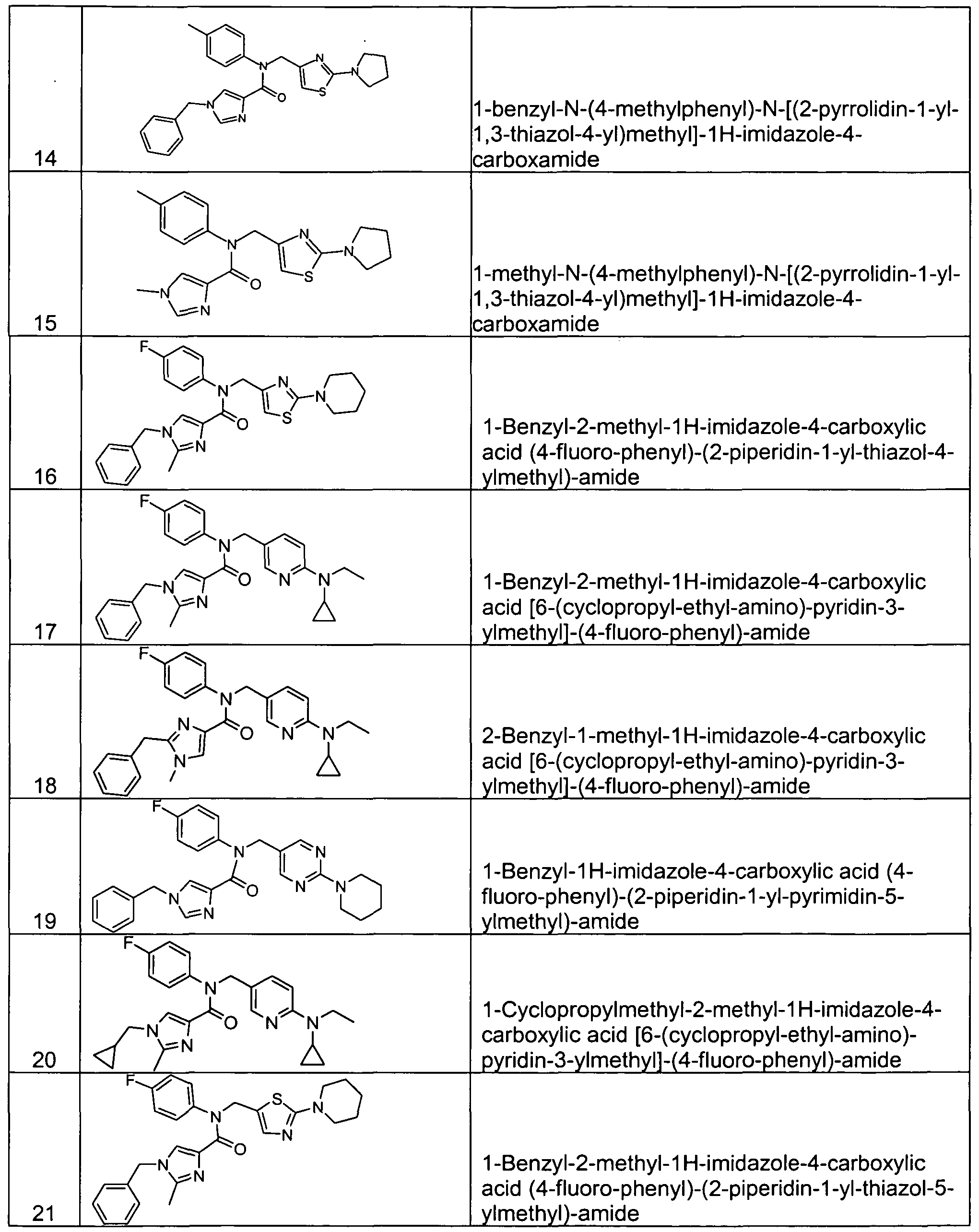



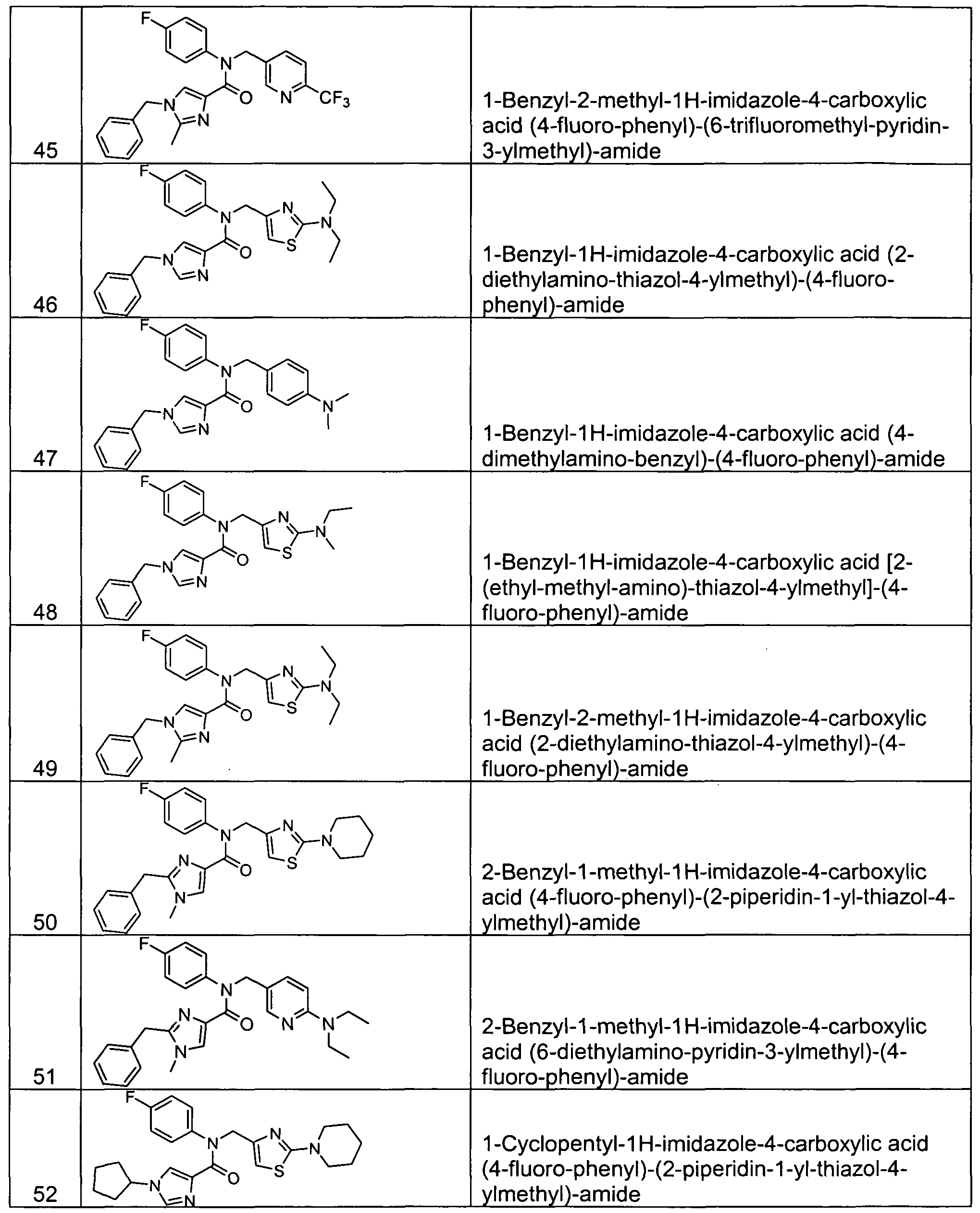

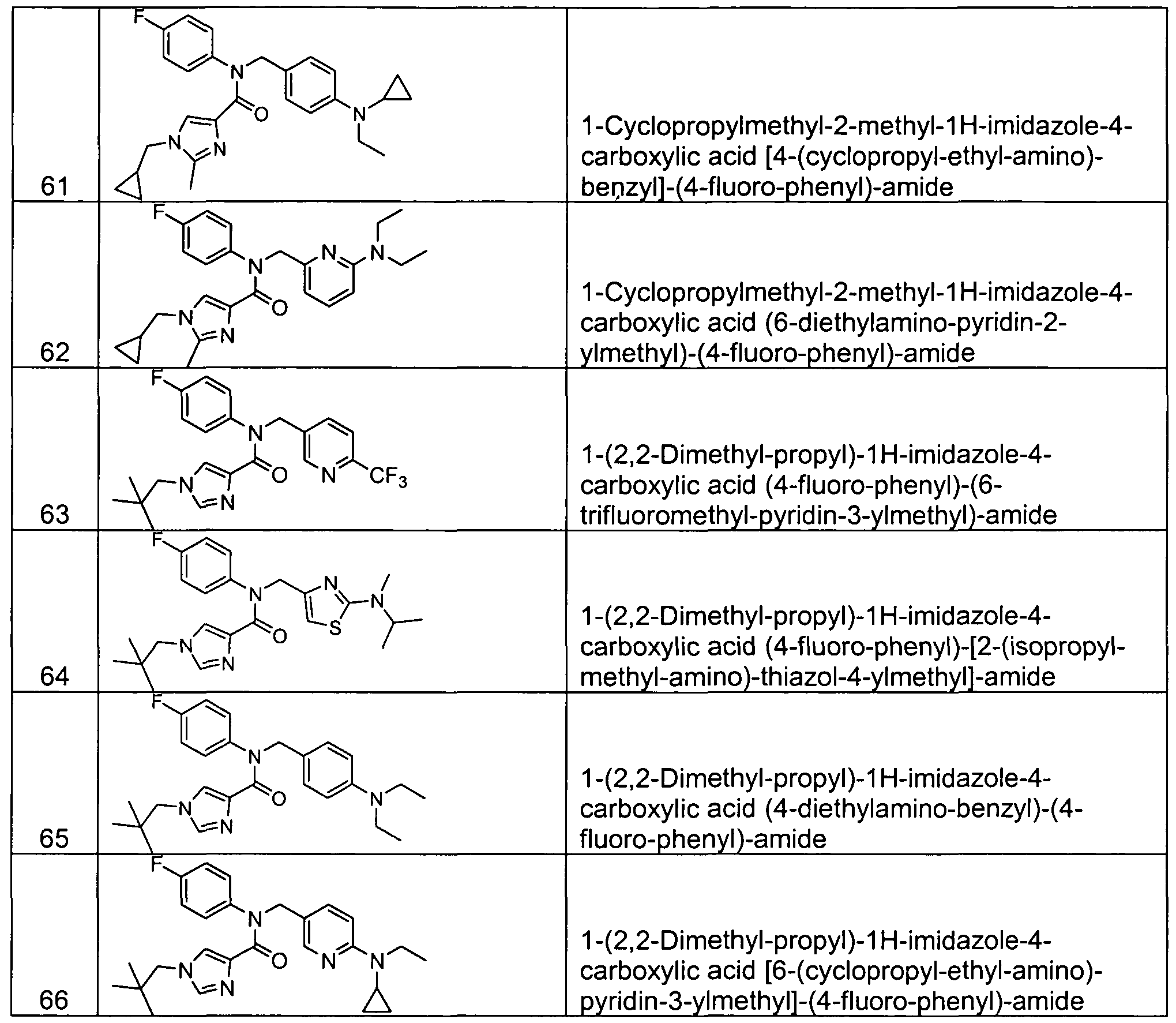
The present teachings additionally provide compounds of formula (III):
 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein:
or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein:
R2, Ar, R3, and p are as defined above;
R6 is selected from a C1-10 alkyl group, a phenyl group, a -C1-6 alkyl-phenyl group, and a 5-7 membered heteroaryl group, wherein each of the phenyl group and the 5-7 membered heteroaryl group optionally is substituted with 1 to 3 substituents independently selected from a halogen, a C1-6 alkyl group, a C1-6 haloalkyl group, and a C1-6alkoxy group; and
R7 is H or a C1-6 alkyl group.
In accordance with some embodiments, p is 1.
According to some embodiments, p is 2.
In some embodiments, p is 3.
In some embodiments, R6 can be a phenyl group, a benzyl group, or a pyridyl group. For example, R6 can be a pyridin-3-yl group.
In some embodiments, R2 can be a -C3-6 cycloalkyl group or a phenyl group optionally substituted with 1 to 2 substituents independently selected from a halogen, a C1-6 alkyl group, a C1-6 alkoxy group, CN, -C(O)OR0, and -NRdRe, wherein Rc, Rd and Re are as defined above. For example, R2 can be a cyclopropylmethyl group, a phenyl group, a 4-dimethylaminophenyl group, a 4-fluoro-2-methylphenyl group, a 4- methylphenyl group, a 2-isopropylphenyl group, a 4-methoxyphenyl group, a 4- chlorophenyl group or a 4-fluorophenyl group.
In certain preferred embodiments, Ar-R3 can be
 wherein R3 is as defined above.
wherein R3 is as defined above.
In accordance with some preferred embodiments, Ar-R3 can be

In accordance with some preferred embodiments, Ar-R3 can be

In some embodiments, R3 can be NRfRg, wherein Rf and R9 are as defined above. In particular embodiments, R3 can be selected from NH2, an NH-C1-6 alkyl group, an
N(Ci-6 alkyl)2 group wherein the C1-6 alkyl groups do not need to be the same,, an
NH-C3-6 cycloalkyl group, an N(C1-6 alkyl)— C3-6 cycloalkyl group, an N(C1-6 alkyl)-C2.6 alkyl-ORc group, an N(C1-6 alkyl)-Y-(5-7 membered cycloheteroalkyl) group, an
N(C1-6 alkyl)-phenyl group, an N(C1-6 alkyl)- Y-5-7 membered heteroaryl group, and an N(C1-6 alkyl)-C2-6 alkyl-O- Y-5-7 membered heteroaryl group, wherein each of the phenyl group, the 5-7 membered cycloheteroalkyl group, and the 5-7 membered heteroaryl group immediately above is optionally substituted with 1 to 3 substituents independently selected from a halogen and a C1-6 alkyl group, and Y and Rc are as defined above. For example, R3 can be a diethylamino group, a dimethylamino group, or a cyclopropyl(ethyl)amino group.
In other embodiments, R3 can be an optionally substituted 5-7 membered cycloheteroalkyl group or an optionally substituted 5-7 membered heteroaryl group as described herein. In certain embodiments, R3 can be selected from a diazepanyl group, an imidazolyl group, a morpholinyl group, a piperidinyl group, a piperazinyl group, a pyridyl group, a pyrrolidyl group, and a thiomorpholinyl group, wherein each of these groups can include a nitrogen ring atom optionally substituted with -C(O)RC, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, -Y-C(O)NRdR6, an -S(O)2-Ci-6 alkyl group, a - C2-6 alkyl— (5-7 membered cycloheteroalkyl) group, a C1-10 alkyl group, or a 5-7 membered heteroaryl group, a carbon ring atom optionally substituted with -C(O)- NRdRe, -Y-ORC, -Y-NRdRe, a -Y-phenyl group, a -Y-(5-7 membered cycloheteroalkyl) group, a -Y-(5-9 membered heteroaryl) group, or a -Y-O-(5-7 membered heteroaryl) group, and/or a sulfur ring atom optionally substituted with 1 or 2 oxo groups, wherein each of the phenyl groups immediately above is optionally substituted with 1 to 3 substituents independently selected from a halogen, a C1-6 alkyl group, a C1-6 haloalkyl group, and a C1-6 alkoxy group, and each of the 5-7 membered cycloheteroalkyl groups, the 5-7 membered heteroaryl groups, and the 5- 9 membered heteroaryl groups immediately above is optionally substituted with 1 to 3 substituents independently selected from a halogen and a C1-6 alkyl group, wherein Y, Rc, Rd and Re are as defined above.
In particular embodiments, R3 can be selected from a 1-[1 ,4]diazepanyl group, a 1- imidazolyl group, a 4-morpholinyl group, a 1 -piperidinyl group, a 1 -piperazinyl group, a 4-pyridyl group, a 1 -pyrrolidyl group, and a 4-thiomorpholinyl group, wherein each of these groups can be optionally substituted as described above.
In some embodiments, R3 can be a 1 -piperazinyl group having a nitrogen atom in the ring optionally substituted with -C(O)R0, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, -C1-6 alkyl-C(O)NRdRe, an S(O)2-C1-6 alkyl group, a -C2-6 alkyl-(5-7 membered cycloheteroalkyl) group, a Ci-10 alkyl group, or a 5-7 membered heteroaryl group. For example, R3 can be a 4-methyl piperazin-1-yl group.
In other embodiments, R3 can be a 1 -piperidinyl group having a carbon atom in the ring optionally substituted with -NRdRe, -C(O)-NRdRe, -Y-ORc, a 5-7
cycloheteroalkyl group, a 5-9 membered heteroaryl group, or a -Y-O-(5-7 membered heteroaryl) group.
Exemplary compounds of formula (III) in accordance with embodiments of the present invention include, but are not limited to, the compounds presented in Table 3 5 below.
TABLE 3
Cpd
Structure Chemical Name No.
1-benzyl-N-{3-[cyclopropyl(ethyl)amino]benzyl}-N-(4-
67 uorophenyl)-1 H-1 ,2,3-triazole-4-carboxamide
1 -benzyl-N-(4-fluoro-2-methylphenyl)-N-[(2-piperidin-1 ■ ylpyrimidin-5-yl)methyl]-1 H-1 ,2,3-triazole-4-
68 carboxamide
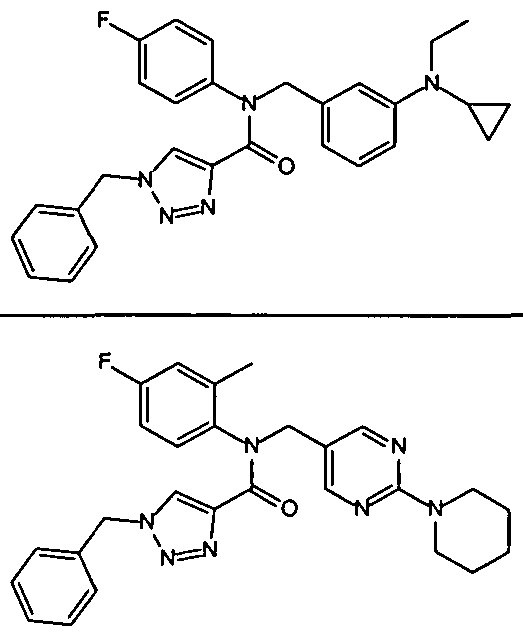
1 -benzyl-N-(4-methylphenyl)-N-[(2-pyrrolidin-1 -yl-1 ,3-
69 thiazol-5-yl)methyl]-1 H-1 ,2,3-triazole-4-carboxamide
1-benzyl-N-{[6-(diethylamino)pyridin-3-yl]methyl}-N-(4- fluorophenyl)-5-methyl-1 H-1 ,2,3-triazole-4-
70 carboxamide
1-benzyl-N-{3-[cyclopropyl(ethyl)amino]benzyl}-N-(4- Jfluorophenyl)-5-methyl-1 H-1 ,2,3-triazole-4-
71 carboxamide
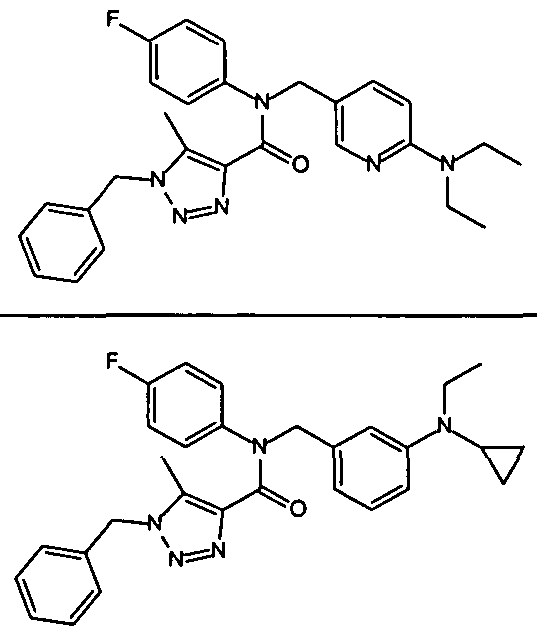
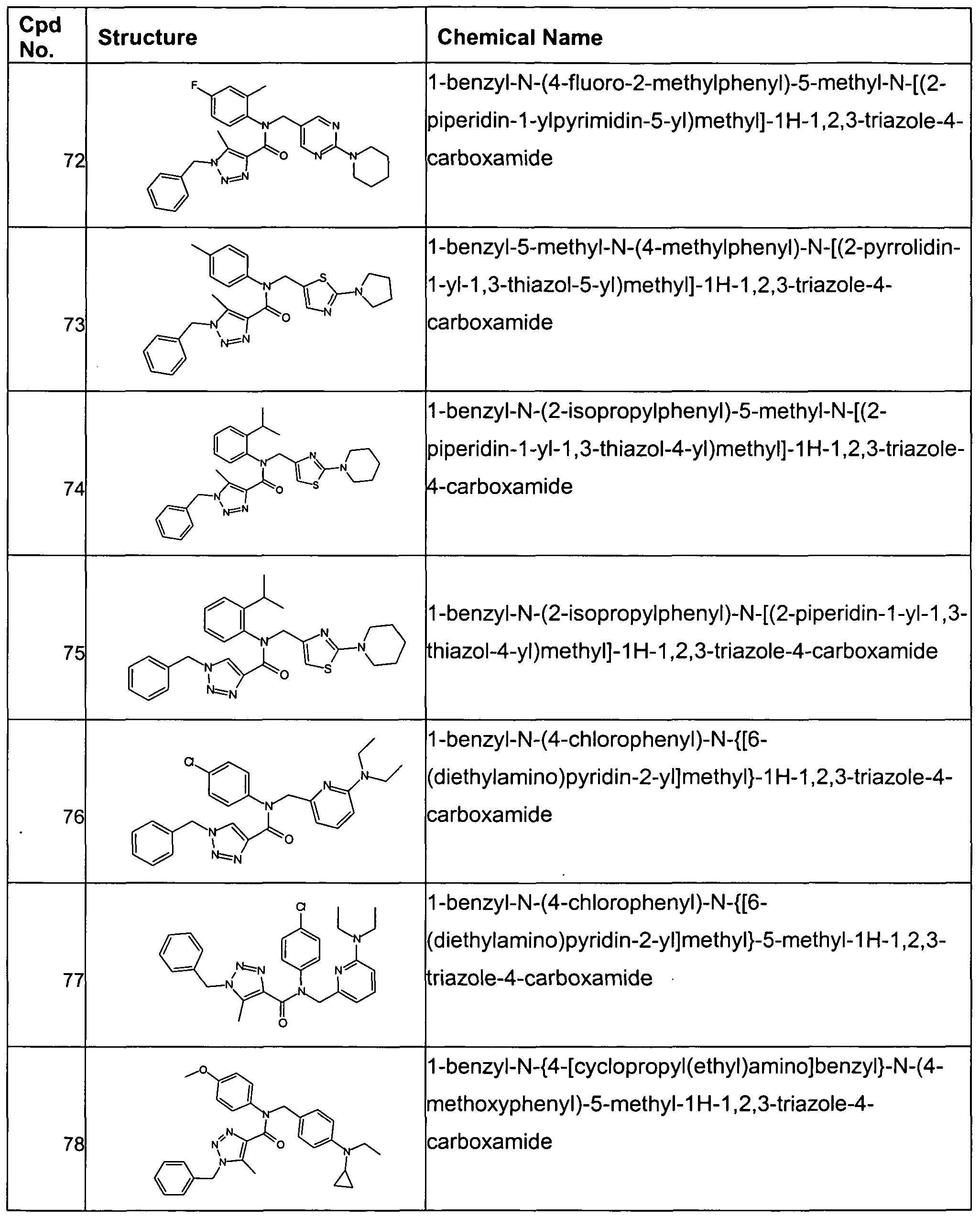 Cpd
Cpd
Structure Chemical Name No.
1-benzyl-N-{4-[cyclopropyl(ethyl)amino]benzyl}-N-(4-
79 methoxyphenyl)-1 H-1 ,2,3-triazole-4-carboxamide
N-{[6-(diethylamino)pyridin-3-yl]methyl}-N-(4- fluorophenyl)-1 -phenyl-1 H-1 ,2,3-triazole-4-
80 carboxamide
1-benzyl-N-(4-fluorophenyl)-N-{[6-(4-methylpiperazin- 1-yl)pyridin-3-yl]methyl}-1 H-1 ,2,3-triazole-4-
81 carboxamide
1-benzyl-N-(4-fluorophenyl)-N-({2- [methyl(phenyl)amino]-1 ,3-thiazol-4-yl}methyl)-1 H-
82 1 ,2,3-triazole-4-carboxamide
1 -benzyl-N-(4-fluorophenyl)-N-[2-(2-morpholin-4-yl-1 ,3-
83 hiazol-4-yl)ethyl]-1 H-1 ,2,3-triazole-4-carboxamide
1 -benzyl-N-(4-fluorophenyl)-N-[3-(2-morpholin-4-yl-1 ,3-
84 hiazol-4-yl)propyl]-1 H-1 ,2,3-triazole-4-carboxamide
1 -benzyl-N-(4-fluorophenyl)-N-[(2-morpholin-4-yl-1 ,3-
85 hiazol-4-yl)methyl]-1 H-1 ,2,3-triazole-4-carboxamide





The present teachings also provide compounds of formula (IV):

(IV) or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein:
R , Ar, R , Rc, and p are as defined above;
 is selected from the group of
is selected from the group of

the 2-furyl and 3-furyl rings are optionally substituted with a -NO2 group or with 1-2 C1-6 alkyl groups;
with the proviso when

p is 1 , and R2 is 4-fluorophenyl, then Ar-R3 is not 4-dimethylaminophenyl.
According to some embodiments,
 is 2-furyl or 3-furyl, optionally substituted with a -NO2 group. In some embodiments,
is 2-furyl or 3-furyl, optionally substituted with a -NO2 group. In some embodiments,
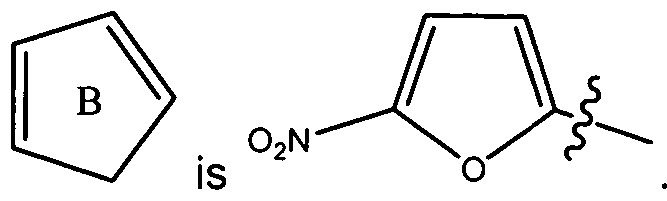

In some embodiments, R2 can be a phenyl group optionally substituted with 1-2 substituents independently selected from a halogen, a C1-6 alkyl group, and a C1-6 alkoxy group. For example, R2 can be 4-fluorophenyl group, a 4-chlorophenyl group, a 4-fluoro-2-methylphenyl group, a 4-methylphenyl group, a 2-isopropylphenyl group, or a 4-methoxyphenyl group.
In some embodiments, R2 can be a -C3-6 cycloalkyl group optionally substituted with 1 to 3 C1-6 alkyl groups. For example, R2 can be a 4-tertbutylcyclohexyl group.
In accordance with some embodiments, p is 1.
In some embodiments, p is 2.
In certain embodiments, p is 3.
In certain preferred embodiments, Ar-R3 can be
wherein R3 is as defined above.
In accordance with some preferred embodiments, Ar-R3 can be

In accordance with some preferred embodiments, Ar-R3 can be
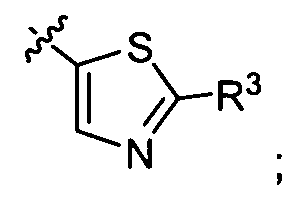
In some embodiments, R3 can be NRfRg, wherein Rf and R9 are as defined above. In particular embodiments, R3 can be selected from NH2, an NH-Ci-6 alkyl group, an N(Ci-6 alkyl)2 group wherein the C1-6 alkyl groups do not need to be the same,, an NH-C3-6 cycloalkyl group, an N(Ci-6 alkyl)— C3-6 cycloalkyl group, an N(C1-6 alkyl)— C2-6 alkyl-ORc group, an N(Ci-6 alkyl)-Y-(5-7 membered cycloheteroalkyl) group, an N(C1-6 alkyl)-phenyl group, an N(Ci-6 alkyl)-Y-5-7 membered heteroaryl group, and an N(Ci-6 alkyl)-C2-6 alkyl-O-Y-5-7 membered heteroaryl group, wherein each of the phenyl group, the 5-7 membered cycloheteroalkyl group, and the 5-7 membered heteroaryl group immediately above is optionally substituted with 1 to 3 substituents independently selected from a halogen and a C1-6 alkyl group, and Y and Rc are as defined above. For example, R3 can be a diethylamino group or a cyclopropyl(ethyl)amino group.
In other embodiments, R3 can be selected from a halogen and a Ci-6 haloalkyl group. For example, R3 can be a chloro group or a thfluoromethyl group.
In other embodiments, R3 can be an optionally substituted 5-7 membered cycloheteroalkyl group or an optionally substituted 5-7 membered heteroaryl group as described herein. In certain embodiments, R3 can be selected from a diazepanyl group, an imidazolyl group, a morpholinyl group, a piperidinyl group, a piperazinyl group, a pyridyl group, a pyrrolidyl group, and a thiomorpholinyl group, wherein each of these groups can include a nitrogen ring atom optionally substituted with -C(O)RC, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, -Y-C(O)NRdRe, an -S(O)2-C1-6 alkyl group, a -
C26 alkyl— (5-7 membered cycloheteroalkyl) group, a C1 10 alkyl group, a -Y-phenyl group, or a 5-7 membered heteroaryl group, a carbon ring atom optionally substituted with -C(O)-NRdRe, -Y-OR01 -Y-NRdRe, a -Y-phenyl group, a -Y-(5-7 membered cycloheteroalkyl) group, a -Y-(5-9 membered heteroaryl) group, or a -Y-O-(5-7 membered heteroaryl) group, and/or a sulfur ring atom optionally substituted with 1 or 2 oxo groups, wherein each of the phenyl groups immediately above is optionally substituted with 1 to 3 substituents independently selected from a halogen, a C1-6 alkyl group, a C1-6 haloalkyl group, and a C1-6 alkoxy group, and each of the 5-7 membered cycloheteroalkyl groups, the 5-7 membered heteroaryl groups, and the 5- 9 membered heteroaryl groups immediately above is optionally substituted with 1 to 3 substituents independently selected from a halogen and a C1-6 alkyl group, wherein Y, R0, Rd and Re are as defined above
In particular embodiments, R3 can be selected from a 1-[1 ,4]dιazepanyl group, a 1- imidazolyl group, a 4-morpholιnyl group, a 1-pιpeπdιnyl group, a 1-pιperazιnyl group, a 4-pyrιdyl group, a 1-pyrrolιdyl group, and a 4-thιomorpholιnyl group, wherein each of these groups can be optionally substituted as described above
In some embodiments, R3 can be a 1-pιperazιnyl group having a nitrogen atom in the ring optionally substituted with -C(O)RC, -C2 6 alkyl-ORc, -C2-6 alkyl-NRdRe, -C1 6 alkyl-C(O)NRdRe, an S(O)2-C1-6 alkyl group, a -C1-6 alkyl-(phenyl)n group wherein n is 1 , 2, or 3, a -C2-6 alkyl— (5-7 membered cycloheteroalkyl) group, a C1 10 alkyl group, a -Y-phenyl group, or a 5-7 membered heteroaryl group For example, R3 can be a 4-methyl pιperazιn-1-yl group, a 4-(4-fluorophenyl)pιperazιn-1-yl group, a bιs(4- fluorophenyl)methyl]pιperazιn-1-yl group, or a 4-(2-phenylethyl)pιperazιn-1-yl
In other embodiments, R3 can be a 1-pιperιdιnyl group having a carbon atom in the ring optionally substituted with -NRdRe, -C(O)-NRdRe, -Y-OR0, a 5-7 cycloheteroalkyl group, a 5-9 membered heteroaryl group, or a -Y-O-(5-7 membered heteroaryl) group
Representative compounds of (IV) in accordance with embodiments of the present invention include, but are not limited to, the compounds presented in 4 below
TABLE 4
Cpd
Structure Chemical Name No.
N-{[6-(diethylamino)pyridin-3-yl]methyl}-N-(4-
107 fluorophenyl)-2-furamide
M-{[6-(diethylamino)pyridin-3-yl]methyl}-N-(4-
108 πuorophenyl)-5-nitro-2-furamide
N-{[6-(diethylamino)pyridin-3-yl]methyl}-N-(4- pluorophenyl)-1-benzothiophene-2-
109 carboxamide
|N-(4-fluoro-2-methylphenyl)-N-[(2-piperidin-1-
1 10 /lpyrimidin-5-yl)methyl]-2-furamide
M-(4-fluoro-2-methylphenyl)-5-nitro-N-[(2-
Diperidin-1-ylpyrimidin-5-yl)methyl]-2-
111 Furamide
N-(4-methylphenyl)-5-nitro-N-[(2-pyrrolidin-1-
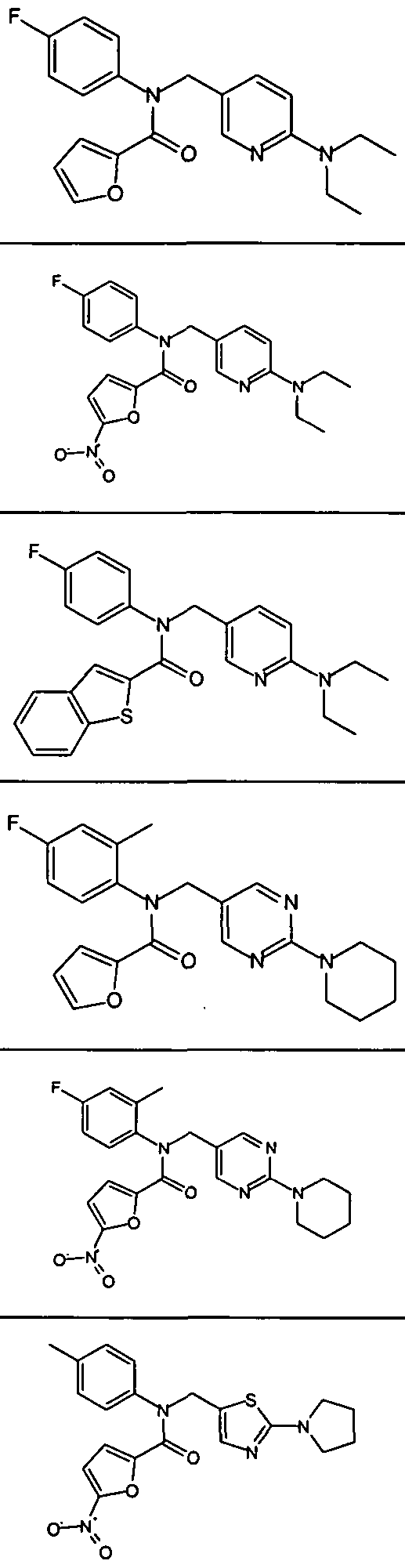
112 yl-1 ,3-thiazol-5-yl)methyl]-2-furamide
Cpd
Structure Chemical Name No.
|N-(4-methylphenyl)-N-[(2-pyrrolidin-1 -yl-1 ,3-13 hiazol-5-yl)methyl]thiophene-2-carboxamide
N-(4-methylphenyl)-N-[(2-pyrrolidin-1 -yl-1 ,3- thiazol-5-yl)methyl]-1-benzothiophene-2-14 _:arboxamide
N-(4-methylphenyl)-N-[(2-pyrrolidin-1 -yl-1 ,3-15 thiazol-5-yl)methyl]-3-furamide
N-(2-isopropylphenyl)-N-[(2-piperidin-1-yl-16 1 ,3-thiazol-4-yl)methyl]-2-furamide
N-(2-isopropylphenyl)-5-nitro-N-[(2-piperidin-17 1-yl-1 ,3-thiazol-4-yl)methyl]-2-furamide
N-(2-isopropylphenyl)-N-[(2-piperidin-1-yl-
1 ,3-thiazol-4-yl)methyl]thiophene-2-18 carboxamide
N-(2-isopropylphenyl)-N-[(2-piperidin-1-yl- 1 ,3-thiazol-4-yl)methyl]-1 -benzothiophene-2-
 19 carboxamide
19 carboxamide
 Cpd
Cpd
Structure Chemical Name No.
N-(4-chlorophenyl)-N-{[6- diethylamino)pyridin-2-yl]methyl}thiophene-
127 3-carboxamide
|N-{4-[cyclopropyl(ethyl)amino]benzyl}-N-(4-
128 iethoxyphenyl)-5-nitro-2-furamide
N-{3-[cyclopropyl(ethyl)amino]benzyl}-N-(4- fluorophenyl)-1 -methyl-1 H-pyrrole-2-
129 carboxamide
N-{3-[cyclopropyl(ethyl)amino]benzyl}-N-(4-
130 fluorophenyl)-2-furamide
N-{3-[cyclopropyl(ethyl)amino]benzyl}-N-(4-
131 Huorophenyl)-5-nitro-2-furamide
N-{3-[cyclopropyl(ethyl)amino]benzyl}-N-(4-
132 fluorophenyl)-3-furamide
N-(4-fluoro-2-methylphenyl)-N-[(2-piperidin-1- ylpyrimidin-5-yl)methyl]thiophene-2-

133 carboxamide
Cpd
Structure Chemical Name No.
N-(4-fluoro-2-methylphenyl)-N-[(2-piperidin-1 ylpyrimidin-5-yl)methyl]thiophene-3-
134 carboxamide
1-methyl-N-(4-methylphenyl)-N-[(2-pyrrolidin-
1-yl-1 ,3-thiazol-5-yl)methyl]-1 H-pyrrole-2-
135 carboxamide
N-(4-methylphenyl)-N-[(2-pyrrolidin-1 -yl-1 ,3-
136 thiazol-5-yl)methyl]thiophene-3-carboxamide
N-(2-isopropylphenyl)-N-[(2-piperidin-1-yl-
1 ,3-thiazol-4-yl)methyl]thiophene-3-
137 carboxamide
N-(4-chlorophenyl)-N-{[6-
138 (diethylamino)pyridin-2-yl]methyl}-2-furamide
N-(4-chlorophenyl)-N-{[6-
(diethylamino)pyridin-2-yl]methyl}-1-
139 benzothiophene-2-carboxamide

140 (diethylamino)pyridin-2-yl]methyl}-3-furamide
 Cpd
Cpd
Structure Chemical Name No.
N-(4-fluorophenyl)-N-{[6-(4-methylpiperazin-
148 1-yl)pyridin-3-yl]methyl}-3-furamide
N-(4-fluorophenyl)-1-methyl-N-{[6- trifluoromethyl)pyridin-3-yl]methyl}-1 H-
149 pyrrole-2-carboxamide
1-methyl-1 H-pyrrole-2-carboxamide N-(4- fluorophenyl)-N-({6-[4-(4- huorophenyl)piperazin-1-yl]pyridin-3-
150 JyJ Ijmethyl)-
N-(4-fluorophenyl)-1-methyl-N-{[6-(4-pyridin- 2-ylpiperazin-1 -yl)pyridin-3-yl]methyl}-1 H-
151 pyrrole-2-carboxamide
N-(4-fluorophenyl)-1-methyl-N-({6-[4-(2- phenylethyl)piperazin-1-yl]pyridin-3-
152 yl}methyl)-1 H-pyrrole-2-carboxamide
N-[(6-{4-[bis(4-fluorophenyl)methyl]piperazin- 1 -yl}pyridin-3-yl)methyl]-N-(4-fluorophenyl)-1 -
153 methyl-1 H-pyrrole-2-carboxamide
N-(4-fluorophenyl)-N-({6-[4-(4- fluorophenyl)piperazin-1-yl]pyridin-3-
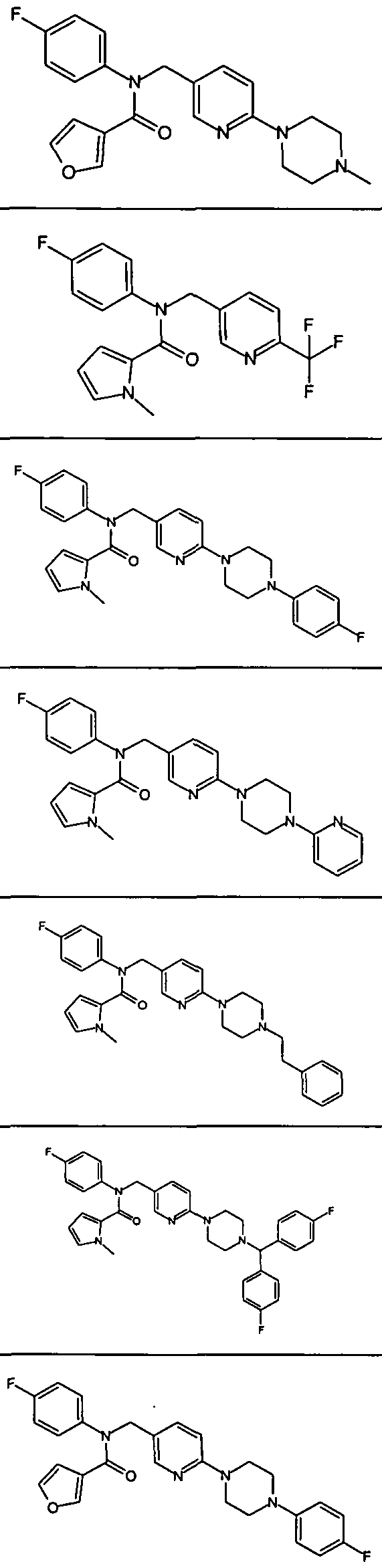
154 yl}methyl)-3-furamide
Cpd
Structure Chemical Name No.
N-(4-fluorophenyl)-N-{[6-(4-pyridin-2- ylpiperazin-1-yl)pyridin-3-yl]methyl}-3-
155 uramide
N-(4-fluorophenyl)-N-({6-[4-(2- phenylethyl)piperazin-1-yl]pyridin-3-
156 yl}methyl)-3-furamide
N-[(6-{4-[bis(4-fluorophenyl)methyl]piperazin- 1-yl}pyridin-3-yl)methyl]-N-(4-fluorophenyl)-3-
157 uramide
N-(4-fluorophenyl)-N-{[6- (trifluoromethyl)pyridin-3-yl]methyl}-3-
158 uramide
N-(4-tert-butylcyclohexyl)-N-[(6-chloropyridin- 3-yl)methyl]-1 -methyl-1 H-pyrrole-2-
159 carboxamide
N-(4-tert-butylcyclohexyl)-1-methyl-N-{[6-(4- methylpiperazin-1 -yl)pyridin-3-yl]methyl}-1 H-
160 py rrole-2-ca rboxa m id e
N-{[6-(diethylamino)pyridin-3-yl]methyl}-N-(4-

161 luorophenyl)thiophene-2-carboxamide
 Cpd
Cpd
Structure Chemical Name No.
N-(4-fluorophenyl)-N-({6-[4- hydroxymethyl)piperidin-1-yl]pyridin-3-
286 yl}methyl)furan-2-carboxamide
N-({6-[4-(2-cyclohexylethyl)piperazin-1- yl]pyridin-3-yl}methyl)-N-(4-
287 fluorophenyl)furan-2-carboxamide
N-(4-fluorophenyl)-N-[(6-pyrrolidin-1- ylpyridin-3-yl)methyl]furan-2-carboxamide
N-(4-fluorophenyl)-N-[(6-piperidin-1-ylpyridin-
289 3-yl)methyl]furan-2-carboxamide
N-{[6-(4-cyclohexylpiperazin-1-yl)pyridin-3- yl]methyl}-N-(4-fluorophenyl)furan-2-
290 carboxamide
N-(4-fluorophenyl)-N-({6-[4-(2- phenylethyl)piperazin-1-yl]pyridin-3-
291 yl}methyl)thiophene-3-carboxamide
N-(4-fluorophenyl)-N-({6-[4-(4-
 luorophenyl)piperazin-1-yl]pyridin-3-
luorophenyl)piperazin-1-yl]pyridin-3-
292 yl}methyl)thiophene-3-carboxamide
Cpd
Structure Chemical Name No.
[N-(4-fluorophenyl)-N-{[6-(4-phenylpiperazin- 1-yl)pyridin-3-yl]methyl}thiophene-3-
293 arboxamide
N-{[6-(4-cyclohexylpiperazin-1-yl)pyridin-3- yl]methyl}-N-(4-fluorophenyl)thiophene-3-
294 carboxamide
N-(4-fluorophenyl)-N-{[6-(4-pyridin-2- ylpiperazin-1 -yl)pyridin-3-
295 yl]methyl}thiophene-3-carboxamide
N-(4-fluorophenyl)-N-{[6-(4-pyrimidin-2- ylpiperazin-1 -yl)pyridin-3-
296 yl]methyl}thiophene-3-carboxamide
N-({6-[4-(diphenylmethyl)piperazin-1- l]pyridin-3-yl}methyl)-N-(4-
297 uorophenyl)thiophene-3-carboxamide
|N-{[6-(4-benzylpiperazin-1-yl)pyridin-3- /l]methyl}-N-(4-fluorophenyl)thiophene-3-
298 lcarboxamide
|N-({6-[4-(2-cyclohexylethyl)piperazin-1-
 /l]pyridin-3-yl}methyl)-N-(4-
/l]pyridin-3-yl}methyl)-N-(4-
299 luorophenyi)thiophene-3-carboxamide

Embodiments of the present invention further provide compounds of formula (V):

R 1 Ar, RJ, and p are as defined above;
 is selected from
is selected from
 and ; wherein
and ; wherein
R8 and R9 are independently a C1-6 alkyl group.
In accordance with some embodiments, R8 and R9 are independently selected from the group of a methyl group and a tert-butyl group.
In accordance with some embodiments, p is 1.
In some embodiments, p is 2.
In certain embodiments, p is 3.
In certain preferred embodiments, Ar-R3 can be
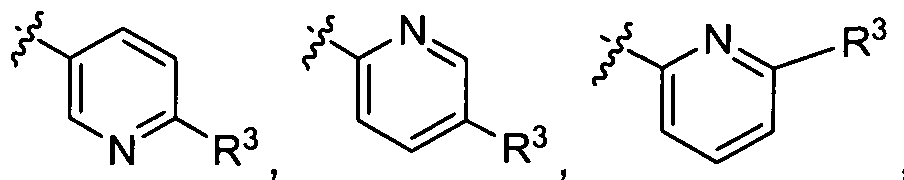
In accordance with some preferred embodiments, Ar-R3 can be
*
T >-R3
wherein R is as defined above.
In accordance with some preferred embodiments, Ar-R3 can be

For example, Ar-R3 can be be 2-morpholin-4-yl-1 ,3-thiazol-4-yl.
In some embodiments, R2 can be a phenyl group optionally substituted with 1-2 substituents independently selected from a halogen, a CL6 alkyl group, and a C1-6 alkoxy group. For example, R2 can be 4-fluorophenyl group.
Representative compounds of Formula (V) in accordance with embodiments of the present invention include, but are not limited to, the compounds presented in Table 5 below.
TABLE 5
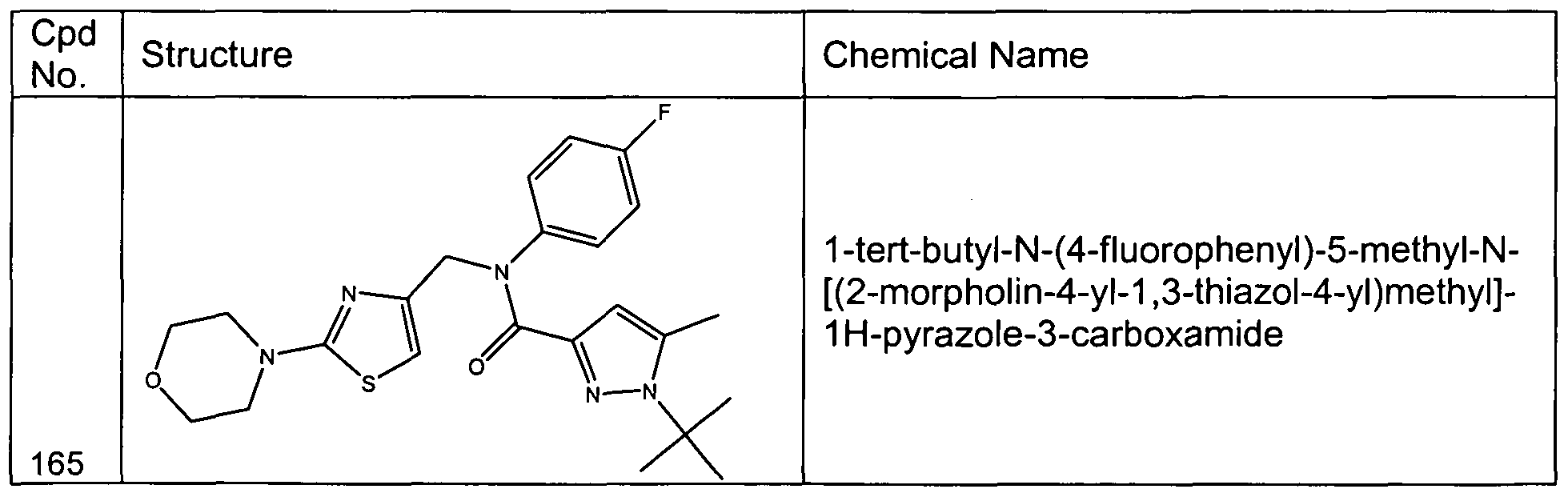

The present teachings provide compounds of formula (Vl)
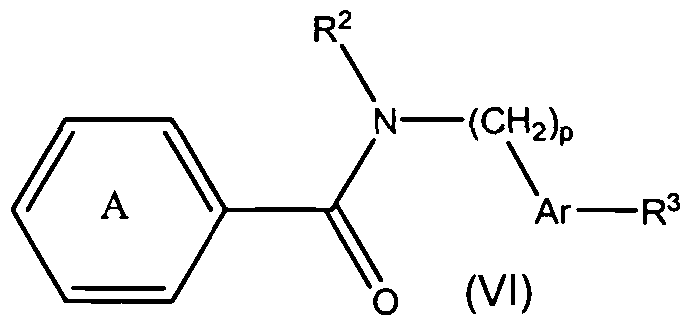
R , Ar, R and p are as defined above;

 and
and
 wherein the pyridinyl group is optionally substituted with 1 to 3 substituents independently selected from a halogen and a C1-6 alkoxy group.
wherein the pyridinyl group is optionally substituted with 1 to 3 substituents independently selected from a halogen and a C1-6 alkoxy group.
In some embodiments,
 can be 2-chloropyridin-3-yl, 6-chloropyridin-
can be 2-chloropyridin-3-yl, 6-chloropyridin-
3-yl, 2,5-dichloropyridin-3-yl, 4-pyridinyl, 3-pyridinyl, and 2-pyridinyl, or 2-chloro-6- methoxypyridin-3-yl.
In some embodiments, R2 can be a phenyl group optionally substituted with 1-2 substituents independently selected from a halogen, a C1-6 alkyl group, and a C1-6 alkoxy group. For example, R2 can be 4-fluorophenyl group, a 4-chlorophenyl group, a 2-methyl-4-fluorophenyl group, a 4-methylphenyl group, a 2-isopropylphenyl group, a 4-methoxyphenyl group, a 3-trifluoromethylphenyl group, a 4-trifluoromethylphenyl group, a 3-tertbutylphenyl group, a 5-chloro-2-methylphenyl group, a 4- hydroxyphenyl group, or a 4-benzyloxyphenyl group.
In certain embodiments, R2 can be a -C3-6 cycloalkyl group; wherein the -C3-6 cycloalkyl group is optionally substituted with 1-3 C1-6 alkyl groups. For example, R2 can be a cyclohexyl group, a cyclopentyl group, a 4-tertbutylcylcohexyl group, a 2,3- dihydro-1 H-inden-2-yl group, or a cyclopropyl methyl group.
In accordance with some embodiments, p is 1.
In some embodiments, p is 2.
In certain embodiments, p is 3.
In certain preferred embodiments, Ar-R3 can be
 wherein R is as defined above.
wherein R is as defined above.
In accordance with some preferred embodiments, Ar-R3 can be

In accordance with some preferred embodiments, Ar-R3 can be

In some embodiments, R3 can be NRfRg, wherein Rf and R9 are as defined above. In particular embodiments, R3 can be selected from NH2, an NH-C1-6 alkyl group, an
N(C1-6 alkyl)2 group wherein the C1-6 alkyl groups do not need to be the same,, an
NH-C3-6 cycloalkyl group, an N(C1^ alkyl)-C3-6 cycloalkyl group, an N(C1-6 alkyl)-C2-6 alkyl-ORc group, an N(C1-6 alkyl)-Y-(5-7 membered cycloheteroalkyl) group, an
N(C1-6 alkyl)-phenyl group, an N(C1-6 alkyl)-Y-5-7 membered heteroaryl group, and an N(C1-6 alkyl)-C2-6 alkyl-O-Y-5-7 membered heteroaryl group, wherein each of the phenyl group, the 5-7 membered cycloheteroalkyl group, and the 5-7 membered heteroaryl group immediately above is optionally substituted with 1 to 3 substituents independently selected from a halogen and a C1-6 alkyl group, and Y and Rc are as defined above. For example, R3 can be a diethylamino group or a cyclopropyl(ethyl)amino group.
In other embodiments, R3 can be an optionally substituted 5-7 membered cycloheteroalkyl group or an optionally substituted 5-7 membered heteroaryl group as described herein. In certain embodiments, R3 can be selected from a diazepanyl group, an imidazolyl group, a morpholinyl group, a piperidinyl group, a piperazinyl group, a pyridyl group, a pyrrolidyl group, and a thiomorpholinyl group, wherein each of these groups can include a nitrogen ring atom optionally substituted with -C(O)R0, -C2.6 alkyl-ORc, -C2-6 alkyl-NRdRe, -Y-C(O)NRdRe, an -S(O)2-C1-6 alkyl group, a - C2-6 alkyl— (5-7 membered cycloheteroalkyl) group, a C1-I0 alkyl group, or a 5-7 membered heteroaryl group, a carbon ring atom optionally substituted with -C(O)- NRdRe, -Y-ORC, -Y-NRdRe, a -Y-phenyl group, a -Y-(5-7 membered cycloheteroalkyl) group, a -Y-(5-9 membered heteroaryl) group, or a -Y-O-(5-7 membered heteroaryl) group, and/or a sulfur ring atom optionally substituted with 1 or 2 oxo groups, wherein each of the phenyl groups immediately above is optionally substituted with 1 to 3 substituents independently selected from a halogen, a C1-6 alkyl group, a C1-6 haloalkyl group, and a C1-6 alkoxy group, and each of the 5-7 membered cycloheteroalkyl groups, the 5-7 membered heteroaryl groups, and the 5- 9 membered heteroaryl groups immediately above is optionally substituted with 1 to 3 substituents independently selected from a halogen and a C1-6 alkyl group, wherein Y, Rc, Rd and Re are as defined above.
In particular embodiments, R3 can be selected from a 1-[1 ,4]diazepanyl group, a 1- imidazolyl group, a 4-morpholinyl group, a 1 -piperidinyl group, a 1 -piperazinyl group, a 4-pyridyl group, a 1 -pyrrolidyl group, and a 4-thiomorpholinyl group, wherein each of these groups can be optionally substituted as described above.
In some embodiments, R3 can be a 1 -piperazinyl group having a nitrogen atom in the ring optionally substituted with -C(O)R0, -C2-6 alkyl-OR0, -C2.6 alkyl-NRdRe, -C1-6 alkyl-C(O)NRdRe, an S(O)2-C1-6 alkyl group, a -C2-6 alkyl-(5-7 membered cycloheteroalkyl) group, a C1-10 alkyl group, or a 5-7 membered heteroaryl group. For example, R3 can be a 4-methyl piperazin-1-yl group.
In other embodiments, R3 can be a 1 -piperidinyl group having a carbon atom in the ring optionally substituted with -NR0Re, -C(O)-NRdRe, -Y-OR0, a 5-7
cycloheteroalkyl group, a 5-9 membered heteroaryl group, or a -Y-O-(5-7 membered heteroaryl) group.
Representative compounds of Formula (Vl) in accordance with embodiments of the present invention include, but are not limited to, the compounds presented in Table 6 5 below.
TABLE 6



Structure Chemical Name No.
1 ,3-thiazol-4-yl}methyl)amino}piperidin-1 ■86 yQcarbonyljpiperidine-i-carboxylate
N-[4-(benzyloxy)phenyl]-N-({2- [cyclopropyl(ethyl)amino]-1 ,3-thiazol-4-87 yl}methyl)nicotinamide
N-({2-[cyclopropyl(ethyl)amino]-1 ,3-thiazol-4-88 yl}methyl)-N-(4- hydroxyphenyQnicotinamide
N-(3-tert-butylphenyl)-N-({2- [cyclopropyl(ethyl)amino]-1 ,3-thiazol-4-89 yl}methyl)nicotinamide
N-(5-chloro-2-methylphenyl)-N-({2- [cyclopropyl(ethyl)amino]-1 ,3-thiazol-4-90 yl}methyl)nicotinamide
N-{[6-(4-ethylpiperazin-1-yl)pyridin-3-91 yl]methyl}-N-(4-fluorophenyl)nicotinamide
N-(4-fluorophenyl)-N-{[6-(4-methyl-1 ,4- diazepan-1 -yl)pyridin-3-
 92 yl]methyl}nicotinamide
Cpd
92 yl]methyl}nicotinamide
Cpd
Structure Chemical Name No.
N-(4-fluorophenyl)-N-{[6-(3-oxopiperazin-1-
193 yl)pyridin-3-yl]methyl}nicotinamide
N-(4-fluorophenyl)-N-({6-[4- hydroxymethyl)piperidin-1-yl]pyridin-3-
194 yl}methyl)nicotinamide
N-(4-fluorophenyl)-N-{[6-(4-hydroxypiperidin-
195 1-yl)pyridin-3- yl]methyl}nicotinamide
N-(4-fluorophenyl)-N-{[6-(3-hydroxypiperidin-
196 1 -yl)pyridin-3- yl]methyl}nicotinamide
N-(4-fluorophenyl)-N-{[6-(3-hydroxypyrrolidin-
197 1 -yl)pyridin-3- yl]methyl}nicotinamide
N-(4-fluorophenyl)-N-({6-[4-(2- hydroxyethyl)piperidin-1-yl]pyridin-3-
198 yl}methyl)nicotinamide
N-(4-fluorophenyl)-N-({6-[4-(2-
 hydroxyethyl)piperazin-1-yl]pyridin-3-
hydroxyethyl)piperazin-1-yl]pyridin-3-
199 yl}methyl)nicotinamide
Cpd
Structure Chemical Name No.
N-(4-fluorophenyl)-N-({6-[(2- hydroxyethyl)(methyl)amino]pyridin-3-
200 yl}methyl)nicotinamide
N-(4-fluorophenyl)-N-{[6-(4-morpholin-4- ylpiperidin-1 -yl)pyridin-3-
201 yl]methyl}nicotinamide
N-{[6-(3-carbamoylpiperidin-1-yl)pyridin-3-
202 [yl]methyl}-N-(4- fluoroprιenyl)nicotinamide
N-{[6-(4-carbamoylpiperidin-1-yl)pyridin-3-
203 yl]methyl}-N-(4- fluorophenyl)nicotinamide
N-(4-fluorophenyl)-N-({6-[3- (hydroxymethyl)piperidin-1-yl]pyridin-3-
204 yl}methyl)nicotinamide
N-[(2-bromo-1 ,3-thiazol-4-yl)methyl]-N-(4-
205 Huorophenyl)nicotinamide
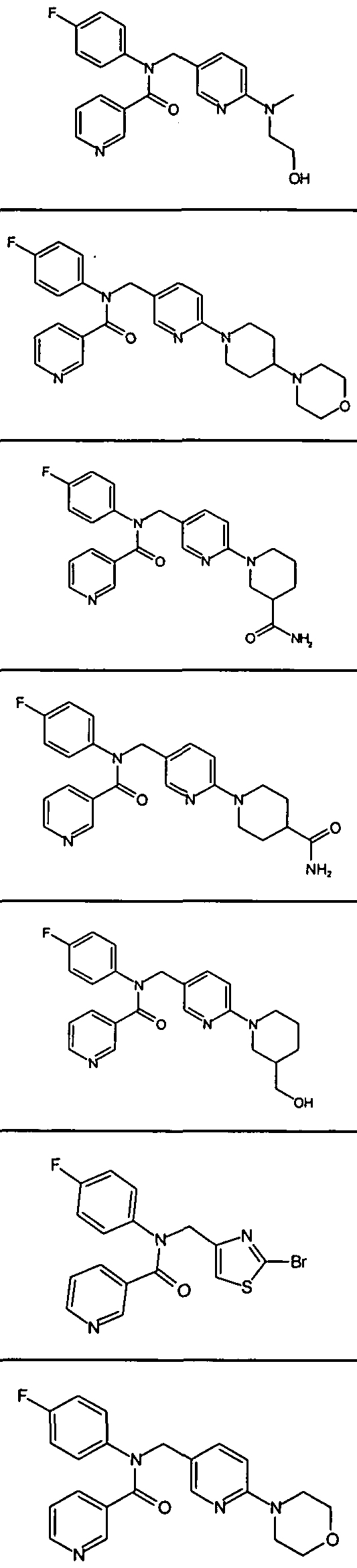
206 /lpyridin-3-yl)methyl]nicotinamide
 Cpd
Cpd
Structure Chemical Name No.

214 1 -yl)-1 ,3-thiazol-4-yl]methyl}nicotinamide
V^
Q-CtHIH1 CH N-(4-fluorophenyl)-N-({2-[4- (hydroxymethyl)piperidin-i -yl]-1 ,3-thiazol-4-
215 yl}methyl)nicotinamide
N-(4-fluorophenyl)-N-({2-[3- (hydroxymethyl)piperidin-i -yl]-1 ,3-thiazol-4-
216 yl}methyl)nicotinamide
N-[(6-chloropyridin-3-yl)methyl]-N-
217 cyclopentylnicotinamide
N-[(6-chloropyridin-3-yl)methyl]-N-
218 cyclohexylnicotinamide
-chloro-N-(4-fluorophenyl)-N-[(2-morpholin-
219 4-yl-1 ,3-thiazol-4-yl)methyl]nicotinamide

220 thiazol-4-yl)methyl]quinoline-2-carboxamide
Cpd
Structure Chemical Name
No.
N-cyclopentyl-N-({6-[4- hydroxymethyl)piperidin-1-yl]pyridin-3-
221 yl}methyl)nicotinamide
N-cyclohexyl-N-{[6-(4-methylpiperazin-1-
222 /l)pyridin-3-yl]methyl}nicotinamide
N-cyclohexyl-N-({6-[4- (hydroxymethyl)piperidin-1-yl]pyridin-3-
223 yl}methyl)nicotinamide
N-[(6-chloropyridin-3-yl)methyl]-N-(4-
224 fluorophenyQnicotinamide
N-({2-[cyclopropyl(ethyl)amino]-1 ,3-thiazol-4-
225 yl}methyl)-N-(4- fluorophenyl)isonicotinamide
N-cyclopentyl-N-{[6-(4-methylpiperazin-1-
226 yl)pyridin-3-yl]methyl}nicotinamide
|N-[(2-bromo-1 ,3-thiazol-4-yl)methyl]-N-(4-

227 luorophenyl)isonicotinamide

 Cpd
Cpd
Structure Chemical Name No.
N-[(6-chloropyridin-3-yl)methyl]-N-[3-
242 (trifluoromethyl)phenyl]nicotinarnide
N-[(6-chloropyridin-3-yl)methyl]-N-[4-
243 (trifluoromethyl)phenyl]nicotinamide
lN-(4-fluorophenyl)-N-{[6-(1 -hydroxy-1 -
244 ethylethyl)pyridin-3- yl]methyl}nicotinamide
N-(4-fluorophenyl)-N-{[6-(1 -hydroxy-1 ,2- dimethylpropyl)pyridin-3-
245 yl]methyl}nicotinamide
N-(4-fluorophenyl)-N-({6-[1-(4-fluorophenyl)- 1 -hydroxyethyl]pyridin-3-
246 yl}methyl)nicotinamide
N-(4-fluorophenyl)-N-{[6-(2,2,2- rtrifluoroethoxy)pyridin-3-
247 |yl]methyl}nicotinamide
N-(4-tert-butylcyclohexyl)-N-{[6-(4-
 methylpiperazin-1 -yl)pyridin-3-
methylpiperazin-1 -yl)pyridin-3-
248 ylimethyllnicotinamide



The present teachings further provide compounds of formula (VII)

R2, Ar, R3 and p are as defined above; and
 is selected from
is selected from

In some embodiments, R2 can be a phenyl group optionally substituted with 1-2 substituents independently selected from a halogen, a C1^ alkyl group, and a C1-6 alkoxy group. For example, R2 can be 4-fluorophenyl group.
In accordance with some embodiments, p is 1.
In some embodiments, p is 2.
In certain embodiments, p is 3.
In certain preferred embodiments, Ar-R3 can be

In accordance with some preferred embodiments, Ar-R3 can be
 wherein R3 is as defined above.
wherein R3 is as defined above.
In accordance with some preferred embodiments, Ar-R3 can be

In some embodiments, R3 can be NRfRg, wherein Rf and R9 are as defined above. In particular embodiments, R3 can be selected from NH2, an NH-C1-6 alkyl group.an N(C1-6 alkyl)2 group wherein the C1-6 alkyl groups do not need to be the same,, an NH-C3-6 cycloalkyl group, an N(C1-6 alkyl)— C3-6 cycloalkyl group, an N(C1-6 alkyl)— C2-6 alkyl-ORc group, an N(C1-6 alkyl)- Y-(5-7 membered cycloheteroalkyl) group, an N(C1-6 alkyl)-phenyl group, an N(C1-6 alkyl)-Y-5-7 membered heteroaryl group, and an N(C1-6 alkyl)-C2-6 alkyl-O-Y-5-7 membered heteroaryl group, wherein each of the phenyl group, the 5-7 membered cycloheteroalkyl group, and the 5-7 membered heteroaryl group immediately above is optionally substituted with 1 to 3 substituents independently selected from a halogen and a C1-6 alkyl group, and Y and Rc are as defined above. For example, R3 can be a diethylamino group or a cyclopropyl(ethyl)amino group.
In other embodiments, R3 can be an optionally substituted 5-7 membered cycloheteroalkyl group or an optionally substituted 5-7 membered heteroaryl group as described herein. In certain embodiments, R3 can be selected from a diazepanyl group, an imidazolyl group, a morpholinyl group, a piperidinyl group, a piperazinyl group, a pyridyl group, a pyrrolidyl group, and a thiomorpholinyl group, wherein each of these groups can include a nitrogen ring atom optionally substituted with -C(O)RC, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, -Y-C(O)NRdRe, an -S(O)2-C1-6 alkyl group, a -
C2-6 alkyl— (5-7 membered cycloheteroalkyl) group, a C1-10 alkyl group, or a 5-7 membered heteroaryl group, a carbon ring atom optionally substituted with -C(O)- NRdRe, -Y-OR0, -Y-NRdRe, a -Y-phenyl group, a -Y-(5-7 membered cycloheteroalkyl) group, a -Y-(5-9 membered heteroaryl) group, or a -Y-O-(5-7 membered heteroaryl) group, and/or a sulfur ring atom optionally substituted with 1 or 2 oxo groups, wherein each of the phenyl groups immediately above is optionally substituted with 1 to 3 substituents independently selected from a halogen, a C1-6 alkyl group, a C1-6 haloalkyl group, and a C1-6 alkoxy group, and each of the 5-7 membered cycloheteroalkyl groups, the 5-7 membered heteroaryl groups, and the 5- 9 membered heteroaryl groups immediately above is optionally substituted with 1 to 3 substituents independently selected from a halogen and a C1-6 alkyl group, wherein Y, Rc, Rd and Re are as defined above.
In particular embodiments, R3 can be selected from a 1-[1 ,4]diazepanyl group, a 1- imidazolyl group, a 4-morpholinyl group, a 1-piperidinyl group, a 1-piperazinyl group, a 4-pyridyl group, a 1-pyrrolidyl group, and a 4-thiomorpholinyl group, wherein each of these groups can be optionally substituted as described above.
In some embodiments, R3 can be a 1-piperazinyl group having a nitrogen atom in the ring optionally substituted with -C(O)RC, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, -C1-6 alkyl-C(O)NRdRe, an S(O)2-C1-6 alkyl group, a -C2-6 alkyl-(5-7 membered cycloheteroalkyl) group, a C1-10 alkyl group, or a 5-7 membered heteroaryl group. For example, R3 can be a 4-methyl piperazin-1-yl group.
In other embodiments, R3 can be a 1-piperidinyl group having a carbon atom in the ring optionally substituted with -NRdRe, -C(O)-NRdRe, -Y-ORc, a 5-7 cycloheteroalkyl group, a 5-9 membered heteroaryl group, or a -Y-O-(5-7 membered heteroaryl) group.
In accordance with some embodiments, R3 can be chloro or a trifluoromethyl group.
Representative compounds of Formula (Vl) in accordance with embodiments of the present invention include, but are not limited to, the compounds presented in Table 7 below.
TABLE 7

259 yl)pyridin-3-yl]methyl}pyrimidine-5-carboxamide
N-(4-fluorophenyl)-N-{[6-(4-methylpiperazin-1-
260 l)pyridin-3-yl]methyl}pyridazine-4-carboxamide
N-(4-fluorophenyl)-N-{[6-(trifluoromethyl)pyιϊdin-3-
261 yl]methyl}pyridazine-4-carboxamide
N-(4-fluorophenyl)-N-[(2-morpholin-4-yl-1 ,3-thiazol-
262 4-yl)methyl]pyridazine-4-carboxamide
N-(4-fluorophenyl)-N-[4-(2-morpholin-4-yl-1 ,3-
263 thiazol-4-yl)butyl]pyridazine-4-carboxamide
N-(4-fluorophenyl)-N-({2-[4-(2-phenylethyl)piperazin- 1 -yl]-1 ,3-thiazol-4-yl}methyl)pyridazine-4-
264 carboxamide
N-({2-[4-(diphenylmethyl)piperazin-1 -yl]-1 ,3-thiazol-
 4-yl}methyl)-N-(4-fluorophenyl)pyridazine-4-
4-yl}methyl)-N-(4-fluorophenyl)pyridazine-4-
265 carboxamide
N-{[2-(4-benzylpiperazin-1-yl)-1 ,3-thiazol-4-
M]methyl}-N-(4-fluorophenyl)pyridazine-4-
266 carboxamide
|N-(4-fluorophenyl)-N-{[2-(4-pyridin-2-ylpiperazin-1 -
267 l)-1 ,3-thiazol-4-yl]methyl}pyridazine-4-carboxamide
N-(4-fluorophenyl)-N-{[2-(4-methylpiperazin-1-yl)-
268 1 ,3-thiazol-4-yl]methyl}pyridazine-4-carboxamide
N-(4-fluorophenyl)-N-{[2-(4-pyrimidin-2-ylpiperazin-1-
269 /l)-1 ,3-thiazol-4-yl]methyl}pyridazine-4-carboxamide
N-(4-fluorophenyl)-N-{[2-(4-phenylpiperazin-1-yl)-
270 1 ,3-thiazol-4-yl]methyl}pyridazine-4-carboxamide
N-[(2-{4-[2-(dimethylamino)ethyl]piperazin-1 -yl}-1 ,3- thiazol-4-yl)methyl]-N-(4-fluorophenyl)pyridazine-4-
271 carboxamide
N-{[2-(4-cyclohexylpiperazin-1 -yl)-1 ,3-thiazol-4- yl]methyl}-N-(4-fluorophenyl)pyridazine-4-

272 carboxamide
N-({6-[4-(diphenylmethyl)piperazin-1-yl]pyridin-3-
M}methyl)-N-(4-fluorophenyl)pyridazine-4-
273 carboxamide
N-(4-fluorophenyl)-N-({6-[4-(2-phenylethyl)piperazin-
274 1-yl]pyridin-3-yl}methyl)pyridazine-4-carboxamide
4-carboxamide
N-{[6-(4-benzylpiperazin-1-yl)pyridin-3-yl]methyl}-N-
275 (4-fluorophenyl)pyridazine-
N-(4-fluorophenyl)-N-{[6-(4-pyridin-2-ylpiperazin-1-
276 yl)pyridin-3-yl]methyl}pyridazine-4-carboxamide
N-{[6-(3,5-dimethylpiperidin-1-yl)pyridin-3-yl]methyl}-
277 N-(4-fluorophenyl)pyridazine-4-carboxamide
N-(4-fluorophenyl)-N-{[6-(4-phenylpiperazin-1-
278 yl)pyridin-3-yl]methyl}pyridazine-4-carboxamide
|N-(4-fluorophenyl)-N-[(6-piperazin-1-ylpyridin-3-
279
 /l)methyl]pyridazine-4-carboxamide
/l)methyl]pyridazine-4-carboxamide
 Pharmaceutically acceptable salts of the compounds of formulas (I), (II), (III), (IV), (V), (Vl), and (VII), which can have an acidic moiety, can be formed using organic and inorganic bases. Both mono and polyanionic salts are contemplated, depending on the number of acidic hydrogens available for deprotonation. Suitable salts formed with bases include metal salts, such as alkali metal or alkaline earth metal salts, for example sodium, potassium, or magnesium salts; ammonia salts and organic amine salts, such as those formed with morpholine, thiomorpholine, piperidine, pyrrolidine, a mono-, di- or th-lower alkylamine (e.g., ethyl-tert-butyl-, diethyl-, diisopropyl-, triethyl-, tributyl- or dimethylpropylamine), or a mono-, di-, or trihydroxy lower alkylamine (e.g., mono-, di- or triethanolamine). Specific non-limiting examples of inorganic bases include NaHCO3, Na2CO3, KHCO3, K2CO3, Cs2CO3, LiOH, NaOH, KOH, NaH2PO4, Na2HPO4, and Na3PO4. Internal salts also can be formed. Similarly, when a compound disclosed herein contains a basic moiety, salts can be formed using organic and inorganic acids. For example, salts can be formed from the following acids: acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, dichloroacetic, ethenesulfonic, formic, fumaric, gluconic, glutamic, hippuric, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, malonic, mandelic, methanesulfonic, mucic, napthalenesulfonic, nitric, oxalic, pamoic, pantothenic, phosphoric, phthalic, propionic, succinic, sulfuric, tartaric, toluenesulfonic, and as well as other known pharmaceutically acceptable acids.
Pharmaceutically acceptable salts of the compounds of formulas (I), (II), (III), (IV), (V), (Vl), and (VII), which can have an acidic moiety, can be formed using organic and inorganic bases. Both mono and polyanionic salts are contemplated, depending on the number of acidic hydrogens available for deprotonation. Suitable salts formed with bases include metal salts, such as alkali metal or alkaline earth metal salts, for example sodium, potassium, or magnesium salts; ammonia salts and organic amine salts, such as those formed with morpholine, thiomorpholine, piperidine, pyrrolidine, a mono-, di- or th-lower alkylamine (e.g., ethyl-tert-butyl-, diethyl-, diisopropyl-, triethyl-, tributyl- or dimethylpropylamine), or a mono-, di-, or trihydroxy lower alkylamine (e.g., mono-, di- or triethanolamine). Specific non-limiting examples of inorganic bases include NaHCO3, Na2CO3, KHCO3, K2CO3, Cs2CO3, LiOH, NaOH, KOH, NaH2PO4, Na2HPO4, and Na3PO4. Internal salts also can be formed. Similarly, when a compound disclosed herein contains a basic moiety, salts can be formed using organic and inorganic acids. For example, salts can be formed from the following acids: acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, dichloroacetic, ethenesulfonic, formic, fumaric, gluconic, glutamic, hippuric, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, malonic, mandelic, methanesulfonic, mucic, napthalenesulfonic, nitric, oxalic, pamoic, pantothenic, phosphoric, phthalic, propionic, succinic, sulfuric, tartaric, toluenesulfonic, and as well as other known pharmaceutically acceptable acids.
Pharmaceutically acceptable esters in the present invention refer to non-toxic esters of the compounds of formulas (I), (II), (III), (IV), (V), (Vl), and (VII)1, preferably the alkyl esters such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl or pentyl esters, of which the methyl ester is preferred. However, other esters such as phenyl-C1-5 alkyl may be employed if desired. Examples of pharmaceutically acceptable esters include, but are not limited to, C2-C6 alkyl esters such as methyl esters and ethyl esters. Pharmaceutically acceptable esters include esters made with aliphatic carboxylic acids, preferably those with a linear chain of between two and six carbon atoms, preferably acetic acid, and made with aromatic carboxylic acids, e.g. C7-I2 acids such as benzoic acid. The aliphatic and aromatic acids may optionally be substituted by one or more C1-4 alkyl groups.
Also provided in accordance with the present teachings are prodrugs of the compounds disclosed herein. As used herein, "prodrug" refers to a moiety that produces, generates or releases a compound of the present teachings when administered to a mammalian subject. Prodrugs can be prepared by modifying functional groups present in the compounds in such a way that the modifications are cleaved, either by routine manipulation or in vivo, from the parent compounds. Examples of prodrugs include compounds as described herein that contain one or more molecular moieties appended to a hydroxyl, amino, sulfhydryl, or carboxyl group of the compound, and that when administered to a mammalian subject, is cleaved in vivo to form the free hydroxyl, amino, sulfhydryl, or carboxyl group, respectively. Examples of prodrugs can include, but are not limited to, acetate, formate and benzoate derivatives of alcohol and amine functional groups in the compounds of the present teachings. Preparation and use of prodrugs is discussed in T. Higuchi and V. Stella, "Pro-drugs as Novel Delivery Systems," Vol. 14 of the A.C.S. Symposium Series, and in Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987, the entire disclosures of which are incorporated by reference herein for all purposes.
Carboxylic acid amide compounds in accordance with the present invention can be prepared as outlined in the schemes below and as illustrated in the examples, from (a) commercially available starting materials, (b) compounds known in the literature, or readily prepared intermediates using literature procedures, or (c) new intermediates described in the schemes and experimental procedures herein.
Standard synthetic methods and procedures for the preparation of organic molecules and functional group transformations and manipulations can be readily obtained from the relevant scientific literature or from standard textbooks in the field. It will be appreciated that where typical or preferred process conditions (i.e., reaction temperatures, times, mole ratios of reactants, solvents, pressures, etc.) are given, other process conditions can also be used unless otherwise stated. Optimum reaction conditions may vary with the particular reactants or solvent used, but one skilled in the art can determine such conditions by routine optimization procedures. Those skilled in the art of organic synthesis will recognize that the nature and order
of the synthetic steps presented may be varied for the purpose of optimizing the formation of the compounds described herein.
Reactions are performed in a solvent appropriate to the reagents and materials employed and suitable for the transformation being effected. Suitable solvents typically are substantially nonreactive with the reactants, intermediates, and/or products at the temperatures at which the reactions are carried out, i.e., temperatures that can range from the solvent's freezing temperature to the solvent's boiling temperature. A given reaction can be carried out in one solvent or a mixture of more than one solvent. Depending on the particular reaction step, suitable solvents for a particular reaction step can be selected. One skilled in the art of organic synthesis can readily selected suitable solvents.
It is understood by those skilled in the art of organic synthesis that the various functionalities present on the molecule must be consistent with the chemical transformation proposed. This may necessitate routine judgment as to the order of synthetic steps, and the need for protecting groups for remote functionalities. One skilled in the art can readily determine the need for protection and deprotection and select appropriate protecting groups. The chemistry of protecting groups can be found, for example, in Greene, et al., Protective Groups in Organic Synthesis, 2d. Ed., Wiley & Sons, 1991.
The processes described herein can be monitored according to any suitable method known in the art. For example, product formation can be monitored by spectroscopic means, such as nuclear magnetic resonance spectroscopy (e.g., 1H or 13C), infrared spectroscopy, spectrophotometry (e.g., UV-visible), or mass spectrometry, or by chromatography such as high performance liquid chromatograpy (HPLC) or thin layer chromatography.
One method for preparing compounds of formulas (I) to (VII) involves the coupling of a carboxylic acid represented by formula (a), or alternatively an activated acid derivative, with an appropriate amine of formula (b) as shown in Scheme 1 below:
SCHEME 1

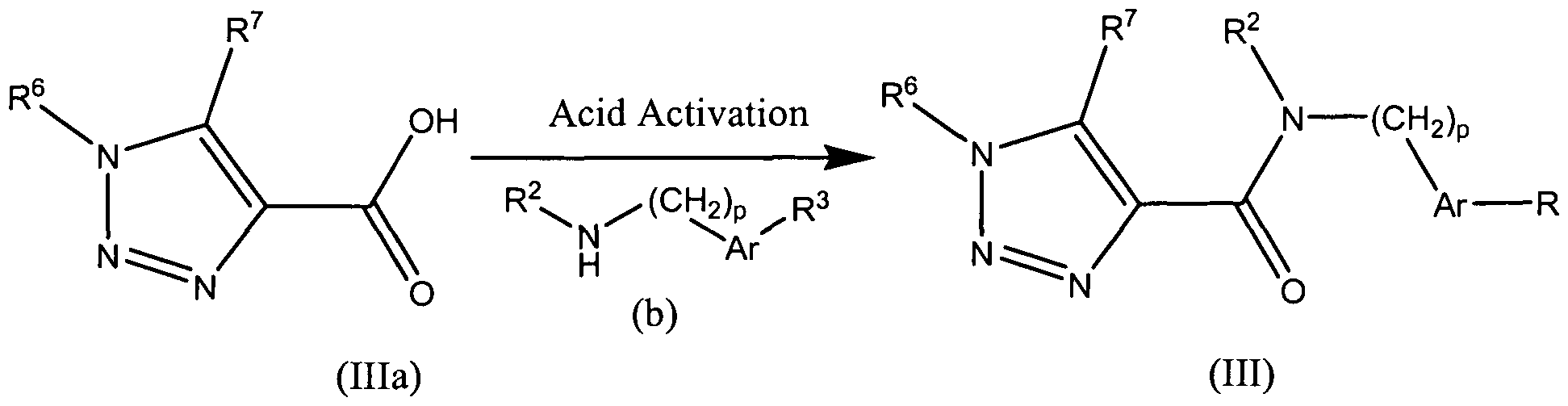

(IVa) (IV)


(Vila) (b) (VII)
Examples of activated acid derivatives include, for example, acid chlorides, esters, acylimidazoles, anhydrides; these activated acid derivatives can be generated in situ or as isolated compounds. Representative activating agents include, but are not limited to, sulfuryl chloride, thionyl chloride, 2-chloro-4,6-dimethoxy-1 ,3,5-thazine, and carbodiimides such as 1-[3-(dimethylamino)propyl]-3-ethyl-carbodiimide and dicyclohexyl carbodiimide; for examples of amide bond formation and acid activation, see Montalbetti C.A.G.N. and Falque, V. (2005), Tetrahedron, 61 (46): 10827-10852.
Many thiazole carboxylic acids (Ia) are commercially available or can be otherwise readily prepared from standard procedures as described in Uchiyama, M. et al. (2005), Chem. Pharm. Bull., 53(4): 437-440. Substituted 1 H-imidazole-4-carboxylic acid derivatives (lia) can be prepared by methods known in the art, for example,
according to the procedures described in Cristalli, G. et al., J. Med. Chem., 34: 1187- 1192 (1991); Bioorg. Med. Chem. Lett., 4(13): 1623-1658 (1994); Ahn, H-S. et. al. , J. Med. Chem., 40: 2196-2210 (1997); and Delest, B. et al., Tetrahedron, 60(29): 6079- 6083 (2004). Substituted [1 ,2,3] triazole-4-carboxylic acids (MIc) can be prepared by methods known in the art, for example, according to the procedures described by Rostovtsev, V.V. et al. (2002), Agnew. Chem. Ed., 41 : 2569-2599; Science of Synthesis (2004), 13: 415-601 ; and Japan Kokai Tokkyo Koho (1981 ), JP-56127363.
The amine (b) can be synthesized as described in Scheme 2 below.
SCHEME 2
(CH2)p PtG
LG ^Ar-LG
H
(d) ■ N- ^Ar-LG
R2' "PtG R2 ' *(CH2)p

(B) (f) (h)
PtG: e.g., tert-butyloxycarbonyl LG: e.g., Cl, Br, or I
In this scheme, alkylation of a protected amine (c) with a compound of formula (d) provides the protected alkylated amine (e). Displacement of the leaving group on compound (e) with the appropriate amine (R3, wherein R3 is NRfRg) provides the corresponding amine-substituted aryl derivative (f). Alternatively, alkylation of the protected amine (c) with a compound of formula (g) provides the amine-substituted aryl derivative (f) directly. Removal of the protecting group (PtG) under standard conditions provides the desired amine Vl.
Alternatively, the amine (b) can be synthesized from substituted acid halides, anhydrides or other activated carboxylic acid derivatives (j) or (m), as illustrated in Scheme 3 below.
SCHEME 3

Z: e.g, halide, acetate LG: e.g, Cl, Br, or I 5 n: 0, 1, 2, or 3
In this scheme, a substituted acid halide, anhydride or activated carboxylic acid derivative (j) or (m) is reacted with an amine of formula R2-NH2 to provide the amide. Displacement of the leaving group (LG) with the appropriate amine (R3, wherein R3 is 0 NRfRg) provides the desired amide. Finally, the amide is reduced under standard conditions to provide the amine (b).
Certain amines of formula (b) in which Ar is 1 ,3-thiazol-4-yl can be prepared by the reaction of carbothioic acid amides (o), wherein Het is a 5-7 membered cycloheteroalkyl group containing at least one nitrogen atom such as 2-morpholin-4- 5 yl, with malonyl chloride derivatives (n) as provided in Scheme 3b.
SCHEME 3B.

A third approach commences with a substituted aryl compound (q), as illustrated in Scheme 4 below.
SCHEME 4
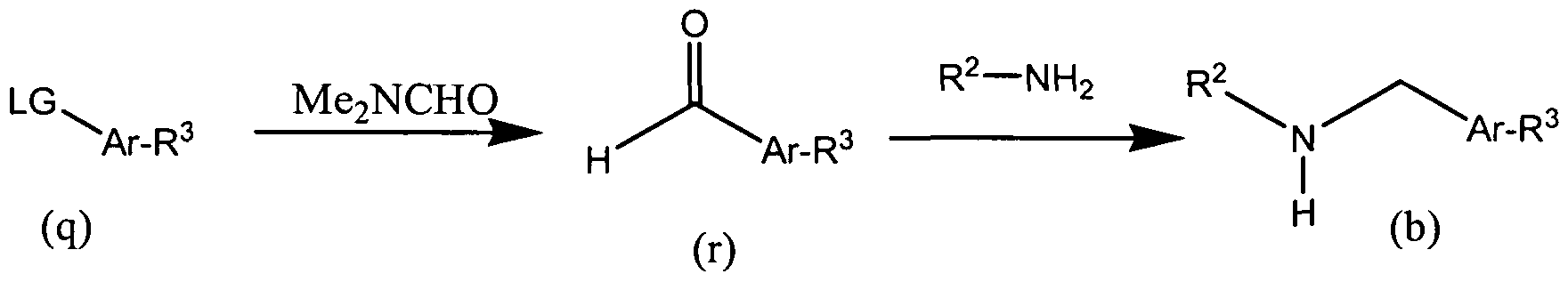
More specifically, conversion of a compound of formula (q) to the corresponding organometallic derivative and treatment with dimethylformamide (Me2NCHO) provides the aryl aldehyde (r). Reductive amination with the appropriate amine (R2- NH2) provides the desired amine (b).
Alternatively the R3 group can be incorporated in the last step of the synthesis, as illustrated in Scheme 5 below for compounds of formula (III); other compounds in accordance with embodiments of the invention can be prepared in an analogous fashion using acyl chloride or acetate derivative of carboxylic acids (Ia), (Ma), (IVa), (Va), (Via), and (Vila).
SCHEME 5

Z: e.g., halide or acetate LG: e.g., Cl, Br, or I
Evaluation of representative compounds according to embodiments of this invention indicated that the compounds of the present teachings can modulate the activity of ion channels in a mammal, for example, Cav2.2 voltage-gated calcium channels.
A variety of pathological conditions, states, disorders or diseases can be treatedby modulating the activity of certain ion channels. As used herein, "ion channel mediated condition" refers to any condition or pathological state of a mammal or any disease present in a mammal that can be treated, or the symptoms of which can be alleviated, by modulation of the activity of one or more ion channels such as Cav2.2 voltage-gated calcium channels. An ion channel mediated condition can be attributed to the abnormal functioning of one or more ion channels. An ion channel can be functioning abnormally when, for example, the ion channel exhibits abnormally increased or decreased activation.
By way of non-limiting examples, ion channel mediated conditions include conditions associated with neuronal hyperexcitability, conditions associated with abnormal glutamate regulation, pain, convulsions, epilepsy, stroke, anxiety disorders, neuronal disorders, traumatic brain injury, angina, hypertension, congestive heart failure, myocardial ischemia, arrhythmia, diabetes, urinary incontinence, hot flush, thermal disregulation, and combinations thereof.
Examples of conditions associated with neuronal hyperexcitability include, but are not limited to, convulsions, including neonatal convulsions, epilepsy, episodic ataxia, myokymia, cerebral ischemia, cerebral palsy, stroke, traumatic brain injury, traumatic spinal cord injury, asphyxia, anoxia, prolonged cardiac surgery, and combinations thereof.
Examples of conditions associated with the abnormal regulation of glutamate include, but are not limited to, hypoglycemia or diseases associated with abnormal glutamate regulation such as, without limitation, Parkinson's disease, Huntingdon's disease, Alzheimer's disease, amyotrophic lateral sclerosis, AIDS-related dementia, and combinations thereof.
Examples of anxiety disorders include, but are not limited to, agoraphobia, panic disorder, specific phobia, social phobia, obsessive compulsive disorder, posttraumatic stress disorder, acute stress disorder, generalized anxiety disorder, separation anxiety disorder, substance-induced anxiety disorder, and anxiety disorder not otherwise specified.
Examples of pain include, but are not limited to various types of nociceptic or neuropathic pain, such as, without limitation, inflammatory pain, musculoskeletal pain, bony pain, lumbosacral pain, neck or upper back pain, visceral pain, somatic pain, pain associated with diabetic neuropathy, cancer pain, pain caused by injury or surgery such as burn pain, headaches such as migraines or tension headaches, and combinations of these pains. One skilled in the art will recognize that these pain types can overlap one another. For example, a pain caused by inflammation can also be visceral or musculoskeletal in nature. Other examples of pain include those
related to conditions of hyperalgesia, allodynia, or both. The types of pain listed above can be acute (short duration) or chronic (regularly reoccuring or persistent), centralized or peripheral, and can be with or without peripheral or central sensitization.
Accordingly, the compounds of the present teachings can be useful for the treatment of a pathological condition, disorder or disease, and the alleviation of a symptom thereof, in a mammal, for example, a human. The pathological condition, disorder or disease, or a symptom thereof, can be, but is not limited to, one of the various ion channel mediated conditions described above. In some embodiments, the compounds of the present teachings can be used for pain therapy, including treating, by way of non-limiting examples, the various types of pain described above. As used herein, "treating" refers to partially or completely alleviating, inhibiting, preventing and/or ameliorating the condition. The present teachings therefore include use of the compounds disclosed herein as active therapeutic substances for the treatment of a variety of ion channel mediated conditions as well as for pain therapy.
Accordingly, the compounds of the present teachings can be useful for the preparation of medicaments for the treatment of a pathological condition, disorder or disease, and the alleviation of a symptom thereof, in a mammal, for example, a human. The pathological condition, disorder or disease, or a symptom thereof, can be one of the various ion channel mediated conditions described herein.
For example, the compounds disclosed herein can be useful for treating the various conditions associated with neuronal hyperexcitability, the various conditions associated with abnormal glutamate regulation, the various anxiety and neuronal disorders, angina, hypertension, congestive heart failure, myocardial ischemia, arrhythmia, diabetes, urinary incontinence, and combinations thereof, as described above.
The compounds disclosed herein also can be useful for treating pain, including chronic pain that is neuropathic pain associated with damage to or pathological changes in the peripheral nervous system or the central nervous system; visceral
pain associated with, by way of non-limiting examples, the abdominal, pelvic, and/or perineal regions or pancreatitis;, musculoskeletal pain; bony pain associated with, by way of non-limiting examples, bone or joint degenerating disorders such as osteoarthritis, rheumatoid arthritis, or spinal stenosis; cancer pain; musculoskeletal pain associated with, by way of non-limiting examples, the lower or upper back, spine, fibromylagia, temporomandibular joint, or myofascial pain syndrome; headaches such migraine or tension headaches; pain associated with infections such as HIV or shingles, sickle cell anemia, autoimmune disorders, multiple sclerosis, and inflammation in accordance with the methods described herein.
Inflammatory pain can be associated with a variety of medical conditions such as osteoarthritis, rheumatoid arthritis, surgery, or injury. Neuropathic pain may be associated with, for example, diabetic neuropathy, peripheral neuropathy, postherpetic neuralgia, trigeminal neuralgia, lumbar or cervical radiculopathies, fibromyalgia, glossopharyngeal neuralgia, reflex sympathetic dystrophy, casualgia, thalamic syndrome, nerve root avulsion, or nerve damage cause by injury resulting in peripheral and/or central sensitization such as phantom limb pain, reflex sympathetic dystrophy or postthoracotomy pain, cancer, chemical injury, toxins, nutritional deficiencies, or viral or bacterial infections such as shingles or HIV, or combinations thereof. The methods of use for compounds of this invention further include treatments in which the neuropathic pain is a condition secondary to metastatic infiltration, adiposis dolorosa, burns, or central pain conditions related to thalamic conditions.
Chronic pain may be associated with diabetes, post traumatic pain of amputation, lower back pain, spinal cord damage, cancer, chemical injury, chemotherapy induced peripheral neuropathy, toxins, major surgery, peripheral nerve damage due to traumatic injury, post-herpetic neuralgia, trigeminal neuralgia, lumbar or cervical radiculopathies, fibromyalgia, glossopharyngeal neuralgia, reflex sympathetic dystrophy, causalgia, thalamic syndrome, nerve root avulsion, reflex sympathetic dystrophy or post thoracotomy pain, nutritional deficiencies, viral infection, bacterial infection, metastatic infiltration, adiposis dolorosa, burns, central pain conditions related to thalamic conditions; and any combination thereof.
As used herein, the term "chronic pain" refers to centralized or peripheral pain that is intense, localized, sharp, or stinging, and/or dull, aching, diffuse, or burning in nature and that occurs for extended periods of time (i.e., persistent and/or regularly reoccurring), including, for the purpose of the present invention, neuropathic pain and cancer pain. Chronic pain includes neuropathic pain, hyperalgesia, and/or allodynia.
One skilled in the art will also recognize that at least some of the types of pain described above can be attributed to a condition associated with the abnormal activity of one or more ion channels such as, but not limited to, the abnormal regulation of glutamate.
The present teachings therefore include methods of administering to a mammal a therapeutically effective amount of a compound disclosed herein. As used herein, "administer" or "administering" refers to either directly administering a compound of the present teachings or a pharmaceutical composition containing the compound, or administering the compound or pharmaceutical composition indirectly via a prodrug derivative or analog which will form an equivalent amount of the active compound or substance within the body. The methods also can include identifying a mammal in need of such treatment, and administering a therapeutically effective amount of a compound disclosed herein to the mammal in need thereof. As used herein, "therapeutically effective" refers to a substance or an amount that elicits a desirable biological activity or effect.
In some embodiments, the method includes administering to a mammal a pharmaceutical composition that comprises a compound disclosed herein in combination or association with a pharmaceutically acceptable carrier. The compound of the present teachings can be administered alone or in combination with other therapeutically effective compounds or therapies for the treatment of such condition(s). For example, the other therapeutically effective compounds can include a cardiovascular disease agent and/or a nervous system disease agent. A nervous system disease agent can be a peripheral nervous system (PNS) disease agent and/or a central nervous (CNS) disease agent.
The present teachings also relate to in vitro or in vivo methods of modulating the activity of ion channels including, but not limited to, Cav2.2 voltage-gated calcium channels. In some embodiments, such methods include contacting a Cav2.2 voltage- gated calcium channel with a compound disclosed herein. In certain embodiments, the methods include monitoring the activity of ion channels. In various embodiments, the present teachings relate to methods of modulating the activity of an ion channel such as a Cav2.2 voltage-gated calcium channel that include in vitro or in vivo administration of a pharmaceutically effective amount of one or more compounds of formula (I). As used herein, "pharmaceutically effective" refers to an amount that can elicit an intended biological activity or effect.
When administered for the treatment or inhibition of a particular disease state or disorder, it is understood that an effective dosage can vary depending upon the particular compound utilized, the mode of administration, and severity of the condition being treated, as well as the various physical factors related to the individual being treated. In therapeutic applications, a compound of the present teachings can be provided to a patient already suffering from a disease in an amount sufficient to treat the symptoms of the disease and its complications. The dosage to be used in the treatment of a specific individual typically must be subjectively determined by the attending physician. The variables involved include the specific condition and its state as well as the size, age and response pattern of the patient.
The present teachings also provide pharmaceutical compositions comprising at least one compound described herein and one or more pharmaceutically acceptable carriers, excipients, or diluents. Examples of such carriers are well known to those skilled in the art and can be prepared in accordance with acceptable pharmaceutical procedures, such as, for example, those described in Remington's Pharmaceutical Sciences, 17th edition, ed. Alfonoso R. Gennaro, Mack Publishing Company, Easton, PA (1985), the entire disclosure of which is incorporated by reference herein for all purposes. As used herein, "pharmaceutically acceptable" refers to a substance that is acceptable for use in pharmaceutical applications from a toxicological perspective and does not adversely interact with the active ingredient. Accordingly, pharmaceutically acceptable carriers are those that are compatible with the other
ingredients in the formulation and are biologically acceptable. Supplementary active ingredients can also be incorporated into the pharmaceutical compositions. Compounds of the present teachings can be administered orally or parenterally, neat or in combination with conventional pharmaceutical carriers. Applicable solid carriers can include one or more substances which can also act as flavoring agents, lubricants, solubilizers, suspending agents, fillers, glidants, compression aids, binders or tablet-disintegrating agents, or encapsulating materials. The compounds can be formulated in conventional manner, for example, in a manner similar to that used for known antiinflammatory agents. Oral formulations containing an active compound disclosed herein can comprise any conventionally used oral form, including tablets, capsules, buccal forms, troches, lozenges and oral liquids, suspensions or solutions. In powders, the carrier can be a finely divided solid, which is an admixture with a finely divided active compound. In tablets, an active compound can be mixed with a carrier having the necessary compression properties in suitable proportions and compacted in the shape and size desired. The powders and tablets can contain up to about 99% or greater of the active compound. Capsules can contain mixtures of active compound(s) with inert filler(s) and/or diluent(s) such as the pharmaceutically acceptable starches (e.g., corn, potato or tapioca starch), sugars, artificial sweetening agents, powdered celluloses (e.g., crystalline and microcrystalline celluloses), flours, gelatins, gums, and the like.
Useful tablet formulations can be made by conventional compression, wet granulation or dry granulation methods and utilize pharmaceutically acceptable diluents, binding agents, lubricants, disintegrants, surface modifying agents (including surfactants), suspending or stabilizing agents, including, but not limited to, magnesium stearate, stearic acid, sodium lauryl sulfate, talc, sugars, lactose, dextrin, starch, gelatin, cellulose, methyl cellulose, microcrystalline cellulose, sodium carboxymethyl cellulose, carboxymethylcellulose calcium, polyvinylpyrrolidine, alginic acid, acacia gum, xanthan gum, sodium citrate, complex silicates, calcium carbonate, glycine, sucrose, sorbitol, dicalcium phosphate, calcium sulfate, lactose, kaolin, mannitol, sodium chloride, low melting waxes, and ion exchange resins. Surface modifying agents can include nonionic and anionic surface modifying agents. Representative examples of surface modifying agents include, but are not limited to,
poloxamer 188, benzalkonium chloride, calcium stearate, cetostearyl alcohol, cetomacrogol emulsifying wax, sorbitan esters, colloidol silicon dioxide, phosphates, sodium dodecylsulfate, magnesium aluminum silicate, and triethanolamine. Oral formulations herein can utilize standard delay or time-release formulations to alter the absorption of the active compound(s). The oral formulation can also consist of administering an active compound in water or fruit juice, containing appropriate solubilizers or emulisifiers as needed.
Liquid carriers can be used in preparing solutions, suspensions, emulsions, syrups, and elixirs. An active compound described herein can be dissolved or suspended in a pharmaceutically acceptable. liquid carrier such as water, an organic solvent, or a mixture of both, or pharmaceutically acceptable oils or fats. The liquid carrier can contain other suitable pharmaceutical additives such as solubilizers, emulsifiers, buffers, preservatives, sweeteners, flavoring agents, suspending agents, thickening agents, colors, viscosity regulators, stabilizers, and osmo-regulators. Examples of liquid carriers for oral and parenteral administration include, but are not limited to, water (particularly containing additives as described above, e.g., cellulose derivatives such as a sodium carboxymethyl cellulose solution), alcohols (including monohydric alcohols and polyhydric alcohols, e.g., glycols) and their derivatives, and oils (e.g., fractionated coconut oil and arachis oil). For parenteral administration, the carrier can be an oily ester such as ethyl oleate and isopropyl myristate. Sterile liquid carriers are used in sterile liquid form compositions for parenteral administration. The liquid carrier for pressurized compositions can be halogenated hydrocarbon or other pharmaceutically acceptable propellants.
Liquid pharmaceutical compositions, which are sterile solutions or suspensions, can be utilized by, for example, intrathecal, intramuscular, intraperitoneal or subcutaneous injection. Sterile solutions can also be administered intravenously. Compositions for oral administration can be in either liquid or solid form.
Preferably the pharmaceutical composition is in unit dosage form, for example, as tablets, capsules, powders, solutions, suspensions, emulsions, granules, or suppositories. In such form, the pharmaceutical composition can be sub-divided in
unit dose(s) containing appropriate quantities of the active compound. The unit dosage forms can be packaged compositions, for example, packeted powders, vials, ampoules, prefilled syringes or sachets containing liquids. Alternatively, the unit dosage form can be a capsule or tablet itself, or it can comprise the appropriate number of any such compositions in package form. Such unit dosage form may contain from about 1 mg/kg of active compound to about 500 mg/kg of active compound, and can be given in a single dose or in two or more doses. Such doses can be administered in any manner useful in directing the active compound(s) to the recipient's bloodstream, including orally, via implants, parenterally (including intravenous, intraperitoneal and subcutaneous injections), rectally, vaginally, and transdermally. Such administrations can be carried out using the compounds of the present teachings including pharmaceutically acceptable salts thereof, in lotions, creams, foams, patches, suspensions, solutions, and suppositories (e.g., rectal and vaginal).
In some cases, it may be desirable to administer a compound directly to the airways of the patient in the form of a dry powder or an aerosol. For administration by intranasal or intrabronchial inhalation, the compounds of the present teachings can be formulated, for example, into an aqueous or partially aqueous solution.
Compounds described herein can be administered enterally or parenterally (such as, without limitation, interperitoneal, intramuscular, intravascular, intrathecal, intraarticular or subcuteaneous injection or infusion). Solutions or suspensions of these active compounds or pharmaceutically acceptable salts thereof can be prepared in water suitably mixed with a surfactant such as hydroxyl-propylcellulose. Dispersions can also be prepared in glycerol, liquid polyethylene glycols, and mixtures thereof in oils. Under ordinary conditions of storage and use, these preparations typically contain a preservative to inhibit the growth of microorganisms.
The pharmaceutical forms suitable for injection can include sterile aqueous solutions or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersions. In preferred embodiments, the form is sterile and its viscosity permits it to flow through a syringe. The form preferably is stable under
the conditions of manufacture and storage and can be preserved against the contaminating action of microorganisms such as bacteria and fungi. The carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (e.g., glycerol, propylene glycol and liquid polyethylene glycol), suitable mixtures thereof, and vegetable oils.
Compounds described herein can be administered transdermal^, i.e., administered across the surface of the body and the inner linings of bodily passages including epithelial and mucosal tissues. Such administration can be carried out using the compounds of the present teachings including pharmaceutically acceptable salts thereof, in lotions, creams, foams, patches, suspensions, solutions, and suppositories (e.g., rectal and vaginal). Topical formulations that deliver active compound(s) through the epidermis can be useful for localized treatment of inflammation and arthritis.
Transdermal administration can be accomplished through the use of a transdermal patch containing an active compound and a carrier that can be inert to the active compound, can be non-toxic to the skin, and can allow delivery of the active compound for systemic absorption into the blood stream via the skin. The carrier can take any number of forms such as creams and ointments, pastes, gels, and occlusive devices. The creams and ointments can be viscous liquid or semisolid emulsions of either the oil-in-water or water-in-oil type. Pastes comprised of absorptive powders dispersed in petroleum or hydrophilic petroleum containing the active compound can also be suitable. A variety of occlusive devices can be used to release the active compound into the blood stream, such as a semi-permeable membrane covering a reservoir containing the active compound with or without a carrier, or a matrix containing the active compound. Other occlusive devices are known in the literature.
Compounds described herein can be administered into a body cavity, (e.g., rectally or vaginally) in the form of a conventional suppository. Suppository formulations can be made from traditional materials, including cocoa butter, with or without the addition of waxes to alter the suppository's melting point, and glycerin. Water-soluble
suppository bases, such as polyethylene glycols of various molecular weights, can also be used.
Lipid formulations or nanocapsules can be used to introduce compounds of the present teachings into host cells either in vitro or in vivo. Lipid formulations and nanocapsules can be prepared by methods known in the art. For example, the compounds described herein can be administered in the form of liposomes. As is known in the art, liposomes are generally derived from phospholipids or other lipid substances, and are formed by mono or multilamellar hydrated liquid crystals that are dispersed in an aqueous medium. Any nontoxic, pharmacologically acceptable lipid capable of forming liposomes can be used.
To increase the effectiveness of compounds of the present teachings, it can be desirable to combine a compound with other agents effective in the treatment of the target disease. For inflammatory diseases, other active compounds (i.e., other active ingredients or agents) effective in their treatment, and particularly in the treatment of asthma and arthritis, can be administered with active compounds of the present teachings. The other agents can be administered at the same time or at different times than the compounds disclosed herein.
Throughout the description, where compositions are described as having, including, or comprising specific components, or where processes are described as having, including, or comprising specific process steps, it is contemplated that compositions of the present teachings also can consist essentially of, or consist of, the recited components, and that the processes of the present teachings also consist essentially of, or consist of, the recited processing steps.
In the application, where an element or component is said to be included in and/or selected from a list of recited elements or components, it should be understood that the element or component can be any one of the recited elements or components and can be selected from a group consisting of two or more of the recited elements or components.
The use of the singular herein includes the plural (and vice versa) unless specifically stated otherwise. In addition, where the use of the term "about" is before a quantitative value, the present teachings also include the specific quantitative value itself, unless specifically stated otherwise.
It should be understood that the order of steps or order for performing certain actions is immaterial so long as the present teachings remain operable. Moreover, two or more steps or actions may be conducted simultaneously.
Compounds described herein can contain an asymmetric atom (also referred as a chiral center), and some of the compounds can contain one or more asymmetric atoms or centers, which can thus give rise to optical isomers (enantiomers) and diastereomers. The present teachings and compounds disclosed herein include such optical isomers (enantiomers) and diastereomers (geometric isomers), as well as the racemic and resolved, enantiomerically pure R and S stereoisomers, as well as other mixtures of the R and S stereoisomers and pharmaceutically acceptable salts thereof. Optical isomers can be obtained in pure form by standard procedures known to those skilled in the art, which include, but are not limited to, diastereomeric salt formation, kinetic resolution, and asymmetric synthesis. The present teachings also encompass cis and trans isomers of compounds containing alkenyl moieties (e.g., alkenes and imines). It is also understood that the present teachings encompass all possible regioisomers, and mixtures thereof, which can be obtained in pure form by standard separation procedures known to those skilled in the art, and include, but are not limited to, column chromatography, thin-layer chromatography, and high-performance liquid chromatography.
Throughout the specification, structures may or may not be presented with chemical names. Where any question arises as to nomenclature, the structure prevails.
Aspects of the present teachings can be further understood in light of the following examples, which should not be construed as limiting the scope of the present teachings in any way.
More specifically, the following examples illustrate various synthetic routes that can be used to prepare reagents and intermediates, including appropriate amines and carboxylic acids, that can be used to prepare compounds of formulas (I) to (VII).
EXAMPLES
Amines of formula R2NH(CH2)pArR3, including those provided in the following examples and others commercially available or prepared according to procedures known in the art, can be coupled with various carboxylic acids and acid derivatives to provide compounds of formula (I). (II), (III), (IV), (V), (Vl), and (VII). Useful carboxylic acids and activated derivatives include those provided in the following examples as well as those that are commercially available or prepared according to procedures known in the art.
Compound Numbers 1-317 were prepared using the appropriate starting materials in accordance with Representative Schemes 1-31 and the procedures described for examples 1-54 of analogous compounds. Selected compounds are shown in Table 8 below.
It is understood by those skilled in the art of organic synthesis that the substitution patterns of the starting materials determines the substitution patterns of the products, and the skilled practioner will be able to exercise routine judgment for the selection of suitable starting materials in order to prepare specific products, the order of synthetic steps, and the need for protecting groups for remote functionalities.
While certain acyl chlorides are illustrated in the representative schemes as examples of activated acid derivatives useful for acylation of amines, other reagents for amide bond formation as known in the art can be utilized in the preparation of compounds of formulas (I), (II), (III), (IV), (V), (Vl), and (VII) in accordance with the teachings herein.
In some cases, the compounds were isolated as hydrochloride salts prepared via standard protocols using anhydrous hydrogen chloride as a gas, or as a solution in dioxane or diethyl ether. Those skilled in the art will also appreciate that the protonation state of the test compound is in accordance with the pH of the assay conditions, typically buffered as specified in the assay protocols, and not of the salt form or free base of the compound as synthesized.
One of skill in the art of organic chemistry would recognize that reference to R in the following representative schemes is a generic representation, that R wherever it appears does not have to be the same at each occurrence, and R can be selected from, for example, Rfl Rg, and substitutents on R2 and R3, among others as appropriate and in accordance with the teachings herein. In the following schemes, any of the alkyl, heteroaryl and cylcloalkyl groups may be substituted in accordance with the teachings herein.
When reference is made to HPLC retention time, the following HPLC conditions were used:
HPLC A: Waters Xterra RP18, 3.5u, 150 x 4.6 mm; Temperature 40 0C; Flow Rate1.2 mL/min; Mobile Phase Comp. 85/15-5/95 (Ammon. Form. Buff. Ph=3.5/ACN+MeOH) for 10min, hold 4min; Injection Volume 5 μL; Detector Wavelength 210-37O nM.
HPLC B: Nucleodur C18 EC, 4.6 x 250 mm, Mobile phase: A = MeCN, B = 0.1 % aqueous formic acid, Time/%B: 0/90, 3/90, 8/20, 15/20, 18/90, 20/90;Flow: 1.0 mL/min; Temperature 50 CC; Diluent: MeOH; and
HPLC C: Mobile phase gradient = 5% acetonitrile / 95% ammonium acetate (10 mM) to 95% acetonitrile / 5% ammonium acetate (10 mM) over 2.5 min, hold for 1.5 min, then re-equilibrate. Column = Keystone Aquasil™ C18 column (2 x 50 mm, 5 mM). Detection = 214 nm and 254 nm.
REPRESENTATIVE SCHEME 1

REPRESENTATIVE SCHEME 2
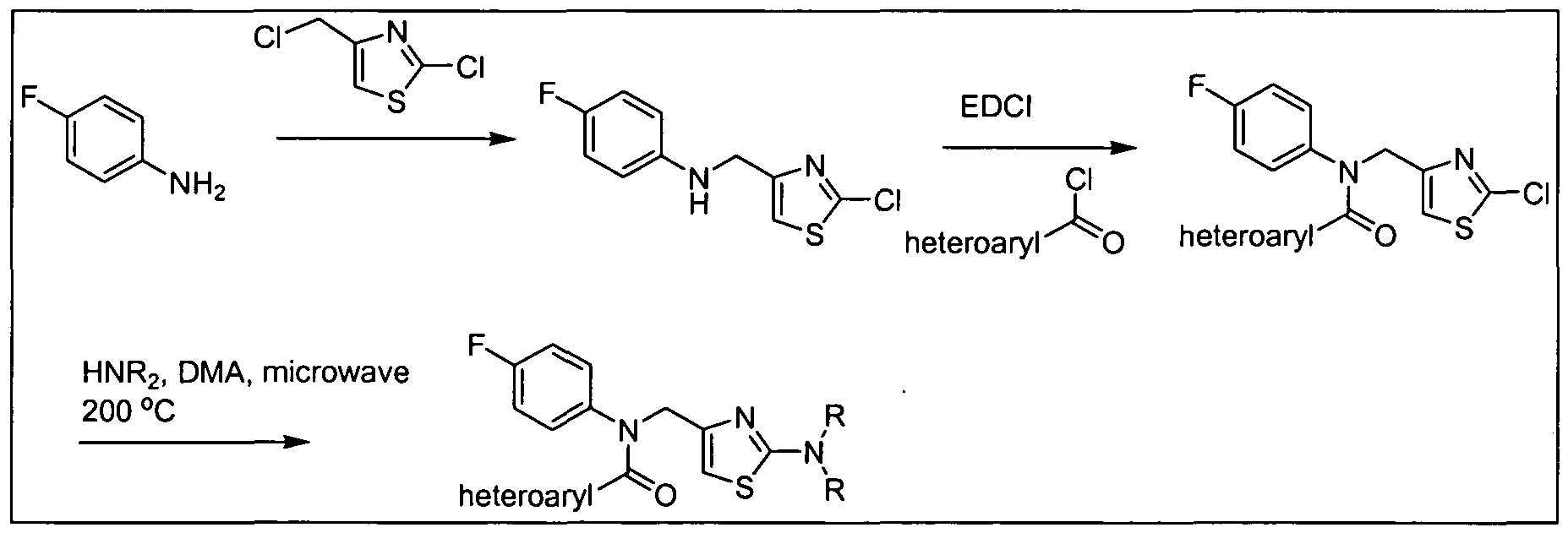 REPRESENTATIVE SCHEME 3
REPRESENTATIVE SCHEME 3
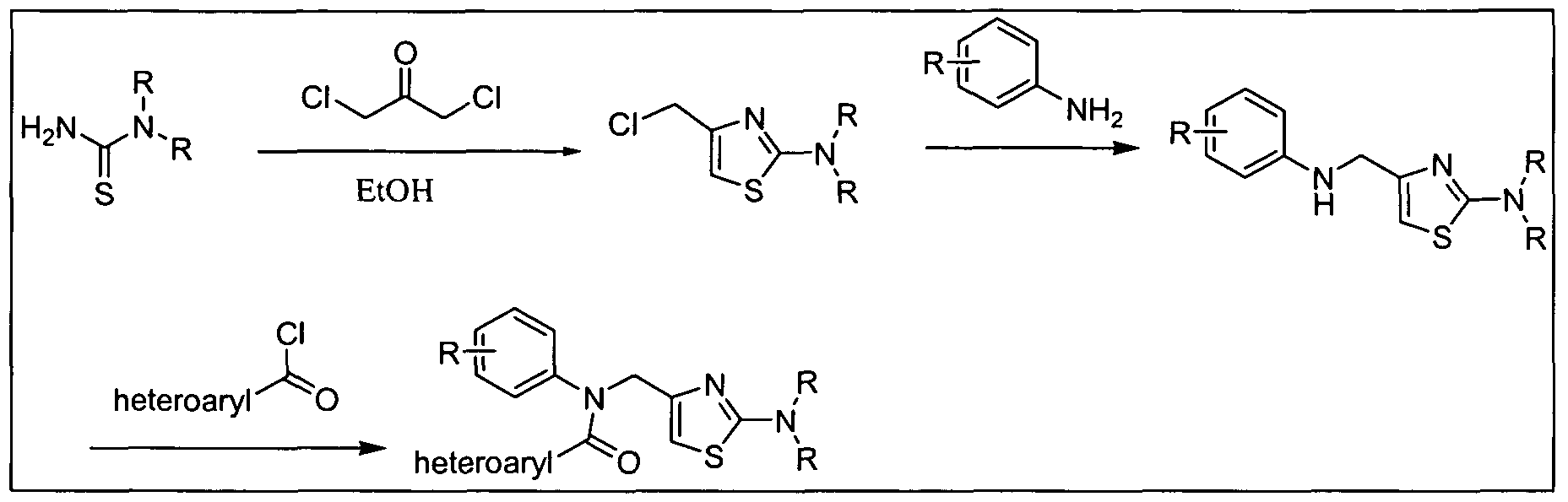
REPRESENTATIVE SCHEME 4

REPRESENTATIVE SCHEME 5

REPRESENTATIVE SCHEME 7

REPRESENTATIVE SCHEME 8

REPRESENTATIVE SCHEME 9
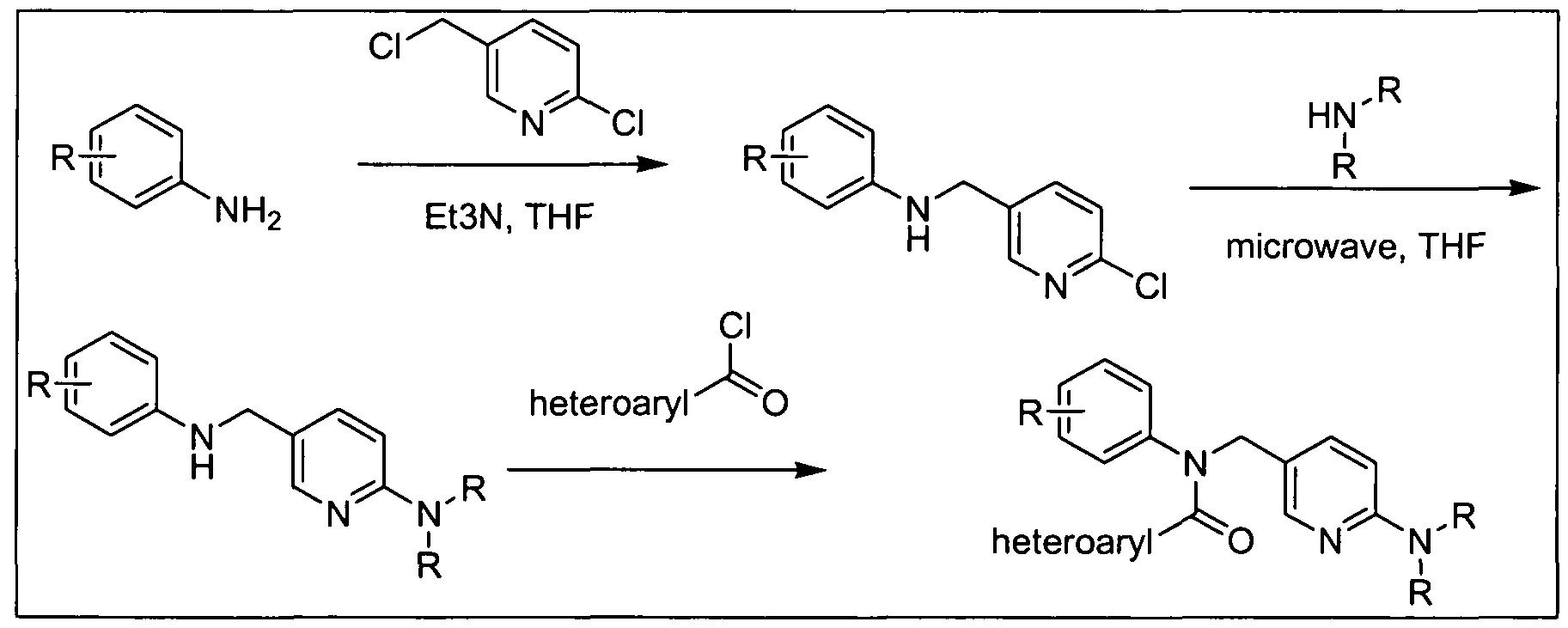 REPRESENTATIVE SCHEME 10
REPRESENTATIVE SCHEME 10

REPRESENTATIVE SCHEME 11

REPRESENTATIVE SCHEME 12
 REPRESENTATIVE SCHEME 13:
REPRESENTATIVE SCHEME 13:

REPRESENTATIVE SCHEME 14

REPRESENTATIVE SCHEME 15
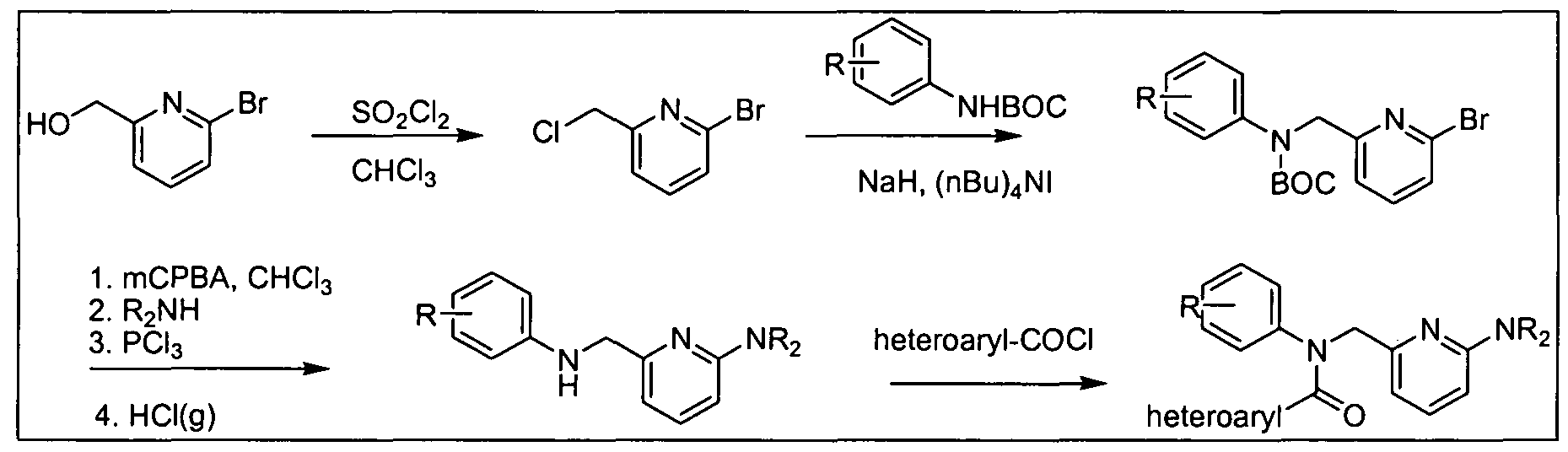
REPRESENTATIVE SCHEME 16
 REPRESENTATIVE SCHEME 17
REPRESENTATIVE SCHEME 17
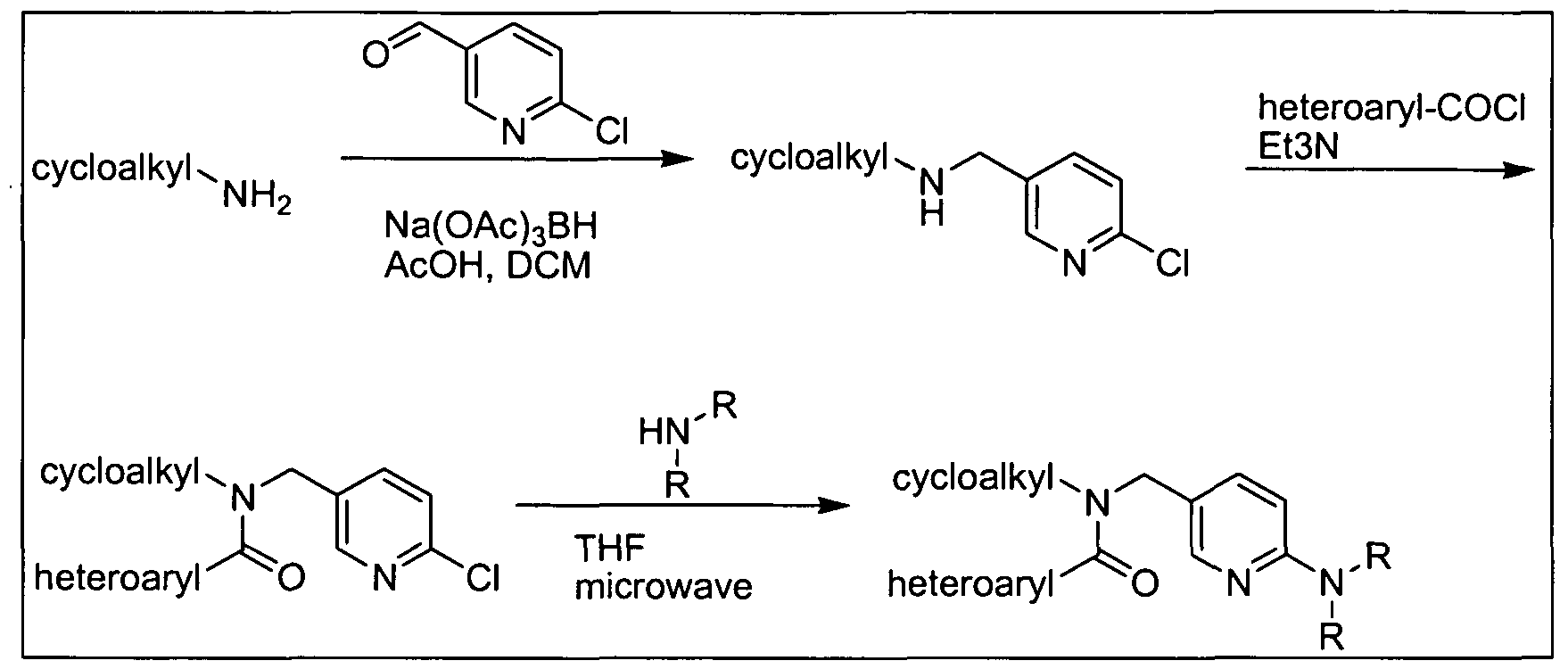
REPRESENTATIVE SCHEME 18
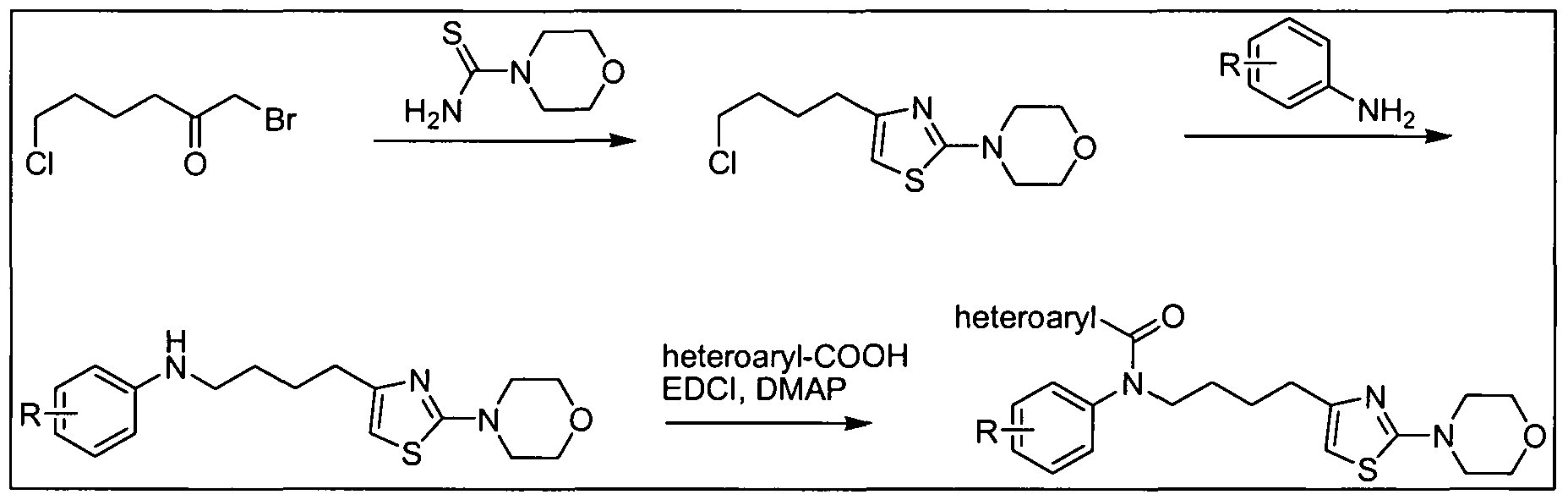
REPRESENTATIVE SCHEME 19

REPRESENTATIVE SCHEME 20
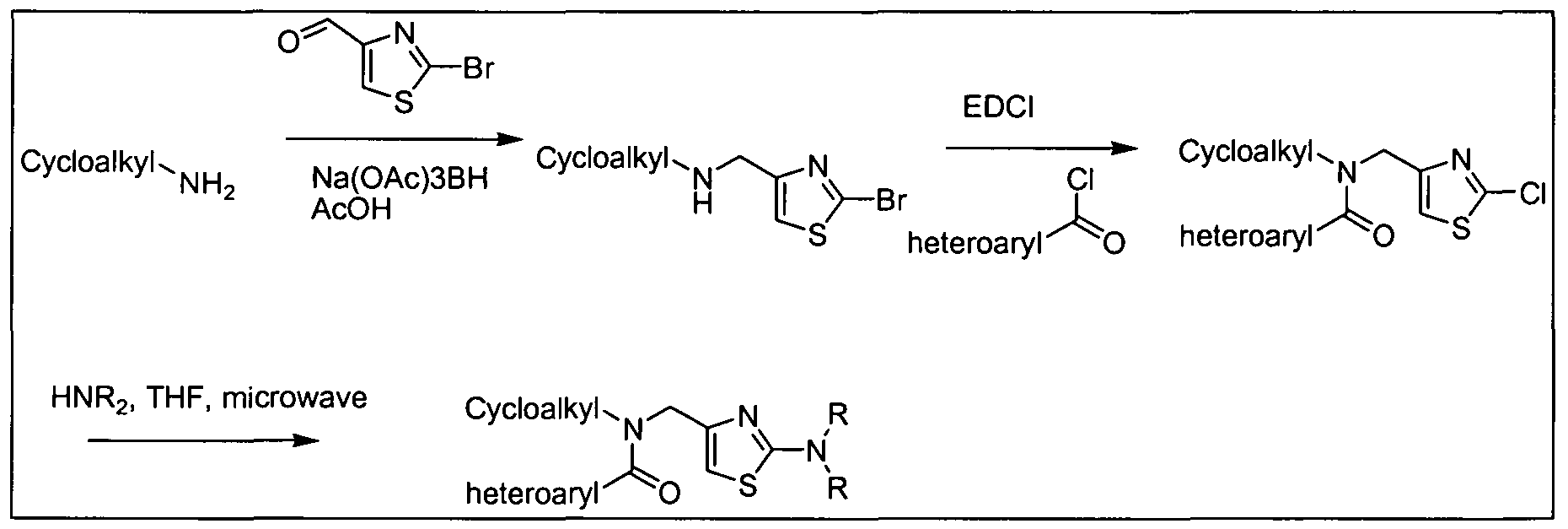
REPRESENTATIVE SCHEME 21

REPRESENTATIVE SCHEME 22

REPRESENTATIVE SCHEME 23

REPRESENTATIVE SCHEME 24

REPRESENTATIVE SCHEME 25
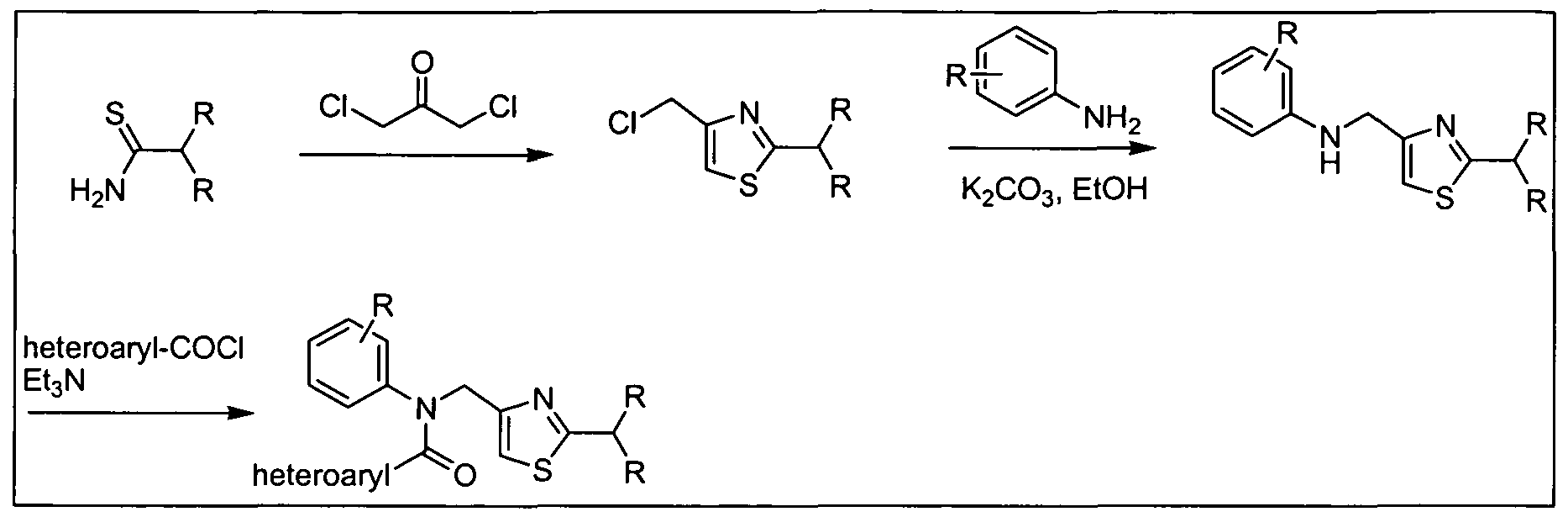
REPRESENTATIVE SCHEME 26

REPRESENTATIVE SCHEME 27
 REPRESENTATIVE SCHEME 28
REPRESENTATIVE SCHEME 28

REPRESENTATIVE SCHEME 29

REPRESENTATIVE SCHEME 30
 REPRESENTATIVE SCHEME 31
REPRESENTATIVE SCHEME 31

EXAMPLE 1A: PREPARATION OF (5-[(4-FLUOROPHENYLAMINO)-METHYL]-
PYRIDIN-2-YLJ-DIETHYLAMINE

NaAIH(OCH2CH2OMe)2
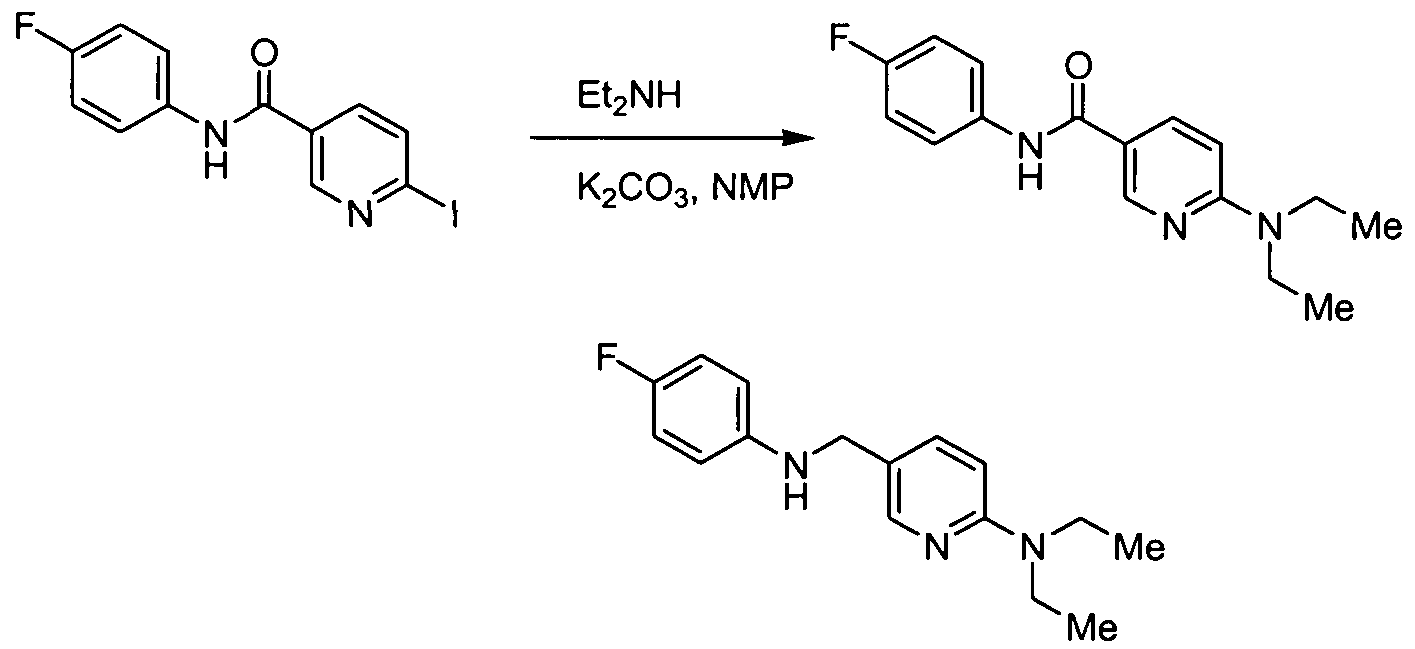
Part I: Preparation of 6-chloro-N-(4-fluorophenyl)-nicotinamide
To a solution of 4-fluoroaniline (15.8 g, 142 mmol) in dichloromethane (450 mL) at 0°C was slowly added a solution of 6-chloro-nicotinoyl chloride (25 g, 142 mmol) in dichloromethane (50 mL), followed by triethylamine (23.7 mL, 170 mmol). After the addition was complete, the reaction was stirred at O0C for 30 minutes, followed by warming to room temperature. After stirring for 30 minutes, the resulting solid was filtered, washed with water and dried under reduced pressure to provide 6-chloro-N- (4-fluorophenyl)-nicotinamide (35 g, 139.6 mmol) as a white solid.
Part II: Preparation of N-(4-fluorophenyl)-6-iodo-nicotinamide
To a solution of 6-chloro-N-(4-fluorophenyl)-nicotinamide (6.4 g, 25.5 mmol) in acetone (130 mL) was added sodium iodide (38.2 g, 255.3 mmol) followed by the dropwise addition of acetyl chloride (7.3 mL, 102 mmol). The yellow mixture was heated at reflux for 1 hour. The reaction mixture was cooled to room temperature and concentrated to dryness under reduced pressure. The residue was partitioned between ethyl acetate (10 mL) and 1 N sodium hydroxide (10 mL). The organic phase was washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to provide N-(4-fluorophenyl)-6-iodo- nicotinamide (6.76 g, 19.8 mmol) as a white solid.
Part III: Preparation of 6-diethylamino-N-(4-fluorophenyl)-nicotinamide
A mixture of N-(4-fluorophenyl)-6-iodo-nicotinamide (684 mg, 2 mmol), diethylamine hydrochloride (0.326 g, 4 mmol), and potassium carbonate (91 1 mg, 6.6 mmol) in 1- methyl-2-pyrrolidinone (2 mL) was heated at 140°C in a sealed tube for 65 hours.
After cooling to room temperature, a saturated aqueous sodium bicarbonate solution
(5 mL) was added, followed by extraction into ethyl acetate (5 mL). The organic phase was dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to provide 6-diethylamino-N-(4-fluorophenyl)-nicotinamide as a solid which was used directly in the next reaction without further purification.
Part IV: Preparation of {5-[(4-fluorophenylamino)-methyl]-pyridin-2-yl}-diethyl-amine
The 6-diethylamino-N-(4-fluorophenyl)-nicotinamide was suspended in a mixture of toluene (5 ml_) and tetrahydrofuran (10 ml_) and stirred at O0C. To the reaction was slowly added sodium bis(2-methoxyethoxy)aluminum hydride (65 wt.% in toluene, 1.8 ml_). The reaction was allowed to warm to room temperature and stirred for 15 minutes followed by heating at 5O0C for 1 hour. The mixture was cooled to room temperature and quenched by the slow addition of an aqueous saturated sodium bicarbonate solution (10 mL) and 6N sodium hydroxide (10 mL) followed by extraction into ethyl acetate (30 mL). The organic phase was dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to provide {5-[(4- fluorophenylamino)-methyl]-pyridin-2-yl}diethyl-amine (300 mg, 1.2 mmol) as an oil.
EXAMPLE 1 B: ALTERNATIVE PREPARATION OF {5-[(4- FLUOROPHENYLAMINO)-METHYL]-PYRIDIN^-YLJ-DIETHYL-AMINE
DIHYDROCHLORIDE

Part I: Preparation of (6-chloro-1-oxy-pyridin-3-ylmethyl)-(4-fluorophenyl)-carbamic acid terf-butyl ester
To a solution of (6-chloro-pyridin-3-ylmethyl)-(4-fluorophenyl)-carbamic acid tert-butyl ester (1 g, 2.97 mmol) in chloroform (10 mL) was added m-chloroperbenzoic acid (1 g, 4.5 mmol) and the reaction heated at 50°C for 6 hours. (6-Chloro-pyridin-3- ylmethyl)-(4-fluorophenyl)-carbamic acid tert-butyl ester can be prepared analogously to (6-bromo-pyridin-2-ylmethyl)-(4-fluorophenyl)-carbamic acid tert-butyl ester following the procedures described in Example 3 infra. The reaction was cooled to room temperature, diluted with dichloromethane (6 mL), and washed with 3N sodium hydroxide (6 mL). The organic layer was dried over anhydrous sodium sulfate, filtered and evaporated to dryness under reduced pressure. Purification by chromatography (silica gel; 3:7 ethyl acetate:hexane) provided (6-chloro-1-oxy- pyridin-3-ylmethyl)-(4-fluorophenyl)-carbamic acid tert-butyl ester (1 g, 2.8 mmol) as a colorless oil.
Part II: Preparation of (6-diethylamino-1-oxy-pyridin-3-ylmethyl)-(4-fluorophenyl)- carbamic acid te/t-butyl ester
(6-Chloro-1-oxy-pyridin-3-ylmethyl)-(4-fluorophenyl)-carbamic acid te/f-butyl ester (1 g, 2.8 mmol) and diethyl amine (2.9 mL, 28.4 mmol) were combined in a sealed tube. The reaction was heated at 130°C overnight. The reaction was cooled to room temperature and concentrated under reduced pressure. Purification by chromatography (silica gel; ethyl acetate) provided (6-diethylamino-1-oxy-pyridin-3- ylmethyl)-(4-fluorophenyl)-(4-fluorophenyl)-carbamic acid tert-butyl ester (1 g, 2.5 mmol) as a brown oil.
Part III: Preparation of (6-diethylamino-pyridin-3-ylmethyl)-(4-fluorophenyl)-carbamic acid tert-butyl ester
(6-Diethylamino-1-oxy-pyridin-3-ylmethyl)-(4-fluorophenyl)-(4-fluorophenyl)-carbamic acid te/f-butyl ester (1 g, 2.5 mmol) was dissolved in chloroform (10 mL). Phosphorous trichloride (336 μL, 3.85 mmol) was added, and the reaction was stirred at room temperature for 45 minutes. The reaction mixture was diluted with dichloromethane (10 mL) and washed with 3N sodium hydroxide (20 mL). The organic layer was dried over anhydrous sodium sulfate, filtered and evaporated to
dryness under reduced pressure. Purification by chromatography (silica gel; 1 :9 ethyl acetate:hexane) provided (6-diethylamino-pyridin-3-ylmethyl)-(4-fluorophenyl)- carbamic acid te/f-butyl ester (920 mg, 2.5 mmol) as a colorless oil.
Part IV: Preparation of {5-[(4-fluorophenylamino)-methyl]-pyridin-2-yl}-diethyl-amine dihydrochloride
To a solution of (6-diethylamino-pyridin-3-ylmethyl)-(4-fluorophenyl)-carbamic acid te/f-butyl ester (900 mg, 2.4 mmol) in methanol (10 mL) was added gaseous hydrochloric acid at 00C. The reaction was allowed to warm to room temperature and ■ stirred for 30 minutes. The reaction was concentrated to provide {5-[(4- fluorophenylamino)-methyl]-pyridin-2-yl}-diethyl-amine dihydrochloride (665 mg, 2.4 mmol) as a white solid.
EXAMPLE 2: PREPARATION OF CYCLOPROPYL-{5-[(4- FLUOROPHENYLAMINO)-METHYL]-PYRIDIN^-YLJ-ETHYL-AMINE
DIHYDROCHLORIDE

Part I: Preparation of 6-cyclopropylamino-nicotinic acid ethyl ester
A mixture of ethyl-6-chloro-nicotinate (10 g, 53.9 mmol) in cyclopropyl amine (10 mL) was heated in a sealed tube at 800C for 12 hours. The reaction was cooled and the mixture purified by chromatography (silica gel; ethyl acetate : hexane gradient
elution) to provide 6-cyclopropylamino-nicotinic acid ethyl ester (6.9 g, 32.2 mmol) as an oil.
Part II: Preparation of 6-(cyclopropyl-ethyl-amino)-nicotinic acid ethyl ester
To a solution of 6-cyclopropylamino-nicotinic acid ethyl ester (6.9 g, 32.2 mmol) in anhydrous tetrahydrofuran (80 mL) containing dimethyl formamide (50 μl_) at 00C was added sodium hydride (60% dispersion in mineral oil, 1.85 g, 48.3 mmol). The reaction was allowed to warm to room temperature and stirred for 30 minutes. The reaction was treated with ethyl iodide (3.0 mL, 48.3 mmol) and the reaction allowed to stir overnight. The reaction was quenched by the addition of water (10 mL), followed by extraction with ethyl acetate (2 x 50 mL). The organic phases were combined, washed with saturated sodium bicarbonate, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. Purification by chromatography (silica gel; ethyl acetate:hexane gradient elution) provided 6- (cyclopropyl-ethyl-amino)-nicotinic acid ethyl ester (6.45 g, 27.5 mmol) as an oil.
Part III: Preparation of 6-(cyclopropyl-ethyl-amino)-N-(4-fluorophenyl)-nicotinamide
To a solution of 4-fluoroaniline (4.65 mL, 35.8 mmol) in toluene (50 mL) was slowly added 2M trimethylaluminum in toluene (16.5 mL, 33 mmol) and the reaction allowed to stir for 1 hour. A solution of 6-(cyclopropyl-ethyl-amino)-nicotinic acid ethyl ester (6.45g, 27.5 mmol) in toluene (25 mL) was added and the reaction heated to 600C. After 12 hours, the reaction was cooled to room temperature and quenched by the dropwise addition of methanol. The reaction was concentrated under reduced pressure and the residue taken up into ethyl acetate (100 mL). The organic phase was washed with saturated sodium bicarbonate (25 mL), saturated potassium- sodium tartrate (25 mL), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. Purification by chromatography (silica gel; ethyl acetate:hexane gradient elution) provided 6-(cyclopropyl-ethyl-amino)-N-(4- fluorophenyl)-nicotinamide (7.25 g, 24.1 mmol).
Part IV: Preparation of cyclopropyl-{5-[(4-fluorophenylamino)-methyl]-pyridin-2-yl}- ethyl-amine dihydrochloride
To a mixture of 6-(cyclopropyl-ethyl-amino)-N-(4-fluorophenyl)-nicotinamide (7.25 g, 24.1 mmol) in toluene (20 mL) and anhydrous tetrahydrofuran (40 ml.) at O0C was slowly added sodium bis(2-methoxyethoxy)aluminum hydride (65 wt.% in toluene, 16 mL). The reaction was allowed to warm to room temperature and stirred for 15 minutes followed by heating at 500C for 1 hour. The mixture was cooled to room temperature and quenched by the slow addition of an aqueous saturated sodium bicarbonate solution (20 mL) and 6N sodium hydroxide (20 mL) followed by extraction into ethyl acetate (60 mL). The organic phase was dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. Purification by chromatography (silica gel; ethyl acetate:hexane gradient elution) provided cyclopropyl-{5-[(4-fluorophenylamino)-methyl]-pyridin-2-yl}-ethyl-amine (8.0 g, 23.1 mmol).
The free base was treated with ethereal hydrochloric acid to provide cyclopropyl-{5- [(4-fluorophenylamino)-methyl]-pyridin-2-yl}-ethyl-amine dihydrochloride (6.8 g, 23.1 mmol) as a white solid.
EXAMPLE 3: PREPARATION OF DIETHYL-{6-[(4-FLUOROPHENYLAMINO)- METHYL]-PYRIDIN^-YL)-AMINE DIHYDROCHLORIDE
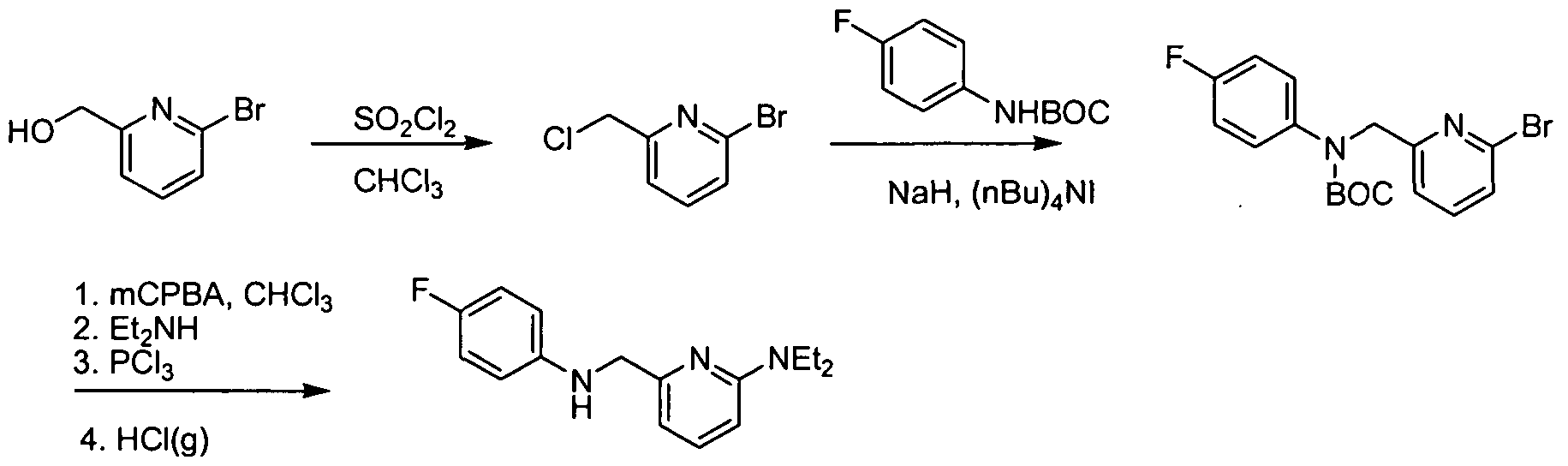
Part I: Preparation of 2-bromo-6-chloromethyl-pyridine
To a solution of (6-bromo-pyridin-2-yl)-methanol (1.5 g, 8.0 mmol) in chloroform (10 ml_) was added dropwise sulfuryl choride (1.29 ml_, 16 mmol) and the reaction stirred overnight. The reaction was concentrated under reduced pressure to provide a yellow semi-solid. The material was triturated with diethyl ether/hexanes and the solid collected to provide 2-bromo-6-chloromethyl-pyridine (900 mg, 4.37 mmol) as a sticky white solid.
Part II: Preparation of (6-bromo-pyridin-2-ylmethyl)-(4-fluorophenyl)-carbamic acid tert-butyl ester
To a solution of (4-fluorophenyl)-carbamic acid tert-butyl ester (60144-53-8, 750 mg, 3.55 mmol) in tetrahydrofuran (10 ml_) was added sodium hydride (60% dispersion in mineral oil, 150 mg, 3.9 mmol). After 30 minutes, tetra-n-butylammonium iodide (51 mg, 0.36 mmol) and 2-bromo-6-chloromethyl-pyridine (804 mg, 3.9 mmol) was added to the reaction and the mixture was heated to 70°C. After 1 hour, the reaction was cooled to room temperature, quenched with saturated sodium bicarbonate (10 mL) and extracted with ethyl acetate (2 x 15 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. Flash chromatography (silica gel; 10% ethyl acetate in hexanes) provided (6-bromo-pyridin-2-ylmethyl)-(4-fluorophenyl)-carbamic acid terf-butyl ester (700 mg, 1.84 mmol) as an oil which solidified upon standing.
Part III: Preparation of (6-bromo-1-oxy-pyridin-2-ylmethyl)-(4-fluorophenyl)-carbamic acid tert-butyl ester
To a solution of (6-bromo-pyridin-2-ylmethyl)-(4-fluorophenyl)-carbamic acid tert-butyl ester (700 mg, 1.84 mmol) in chloroform (8 mL) was added m-chloroperbenzoic acid (477 mg, 2.76 mmol) and the reaction heated to 500C. After stirring overnight, the reaction was cooled to room temperature, diluted with chloroform (10 mL) and washed with 3N sodium hydroxide (5 mL). The layers were separated and the organic phase was dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to provide a yellow solid upon standing. Purification by chromatography (silica gel; 10-20% ethyl acetate in chloroform) provided (6-bromo-1-
oxy-pyridin-2-ylmethyl)-(4-fluorophenyl)-carbamic acid terf-butyl ester (400 mg, 1.0 mmol) as a white solid.
Part IV: Preparation of (6-diethylamino-1-oxy-pyridin-2-ylmethyl)-(4-fluorophenyl)- carbamic acid tert-butyl ester
A suspension of (6-bromo-1-oxy-pyridin-2-ylmethyl)-(4-fluorophenyl)-carbamic acid terf-butyl ester (400 mg, 1.0 mmol) in diethylamine (7 mL) was heated to 13O0C in a sealed tube. After stirring overnight, the reaction was cooled to room temperature and partitioned between brine (10 mL) and ethyl acetate (15 mL). The layers were separated and the organic phase was dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to provide a dark liquid. Flash chromatography (silica gel; 30-75% ethyl acetate in chloroform) provided (6- diethylamino-1-oxy-pyridin-2-ylmethyl)-(4-fluorophenyl)-carbamic acid te/f-butyl ester (230 mg, 0.59 mmol) as a light yellow oil.
Part V: Preparation of diethyl-{6-[(4-fluorophenylamino)-methyl]-pyridin-2-yl}-amine dihydrochloride
To a solution of (6-diethylamino-1-oxy-pyridin-2-ylmethyl)-(4-fluorophenyl)-carbamic acid te/t-butyl ester (230 mg, 0.59 mmol) in chloroform (2 mL) was added phosphorous trichloride (121 mg, 0.89 mmol). After stirring for 1 hour, the reaction was diluted with chloroform (10 mL) and washed with 3N sodium hydroxide (5 mL). The organic phase was dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to provide a yellow oil. The oil was dissolved in chloroform (2 mL) and treated with trifluoroacetic acid (1 mL) and allowed to stir for 1 hour. The reaction was concentrated under reduced pressure and the residue treated with ethereal hydrochloric acid. The resulting solid was collected to provide diethyl-{6-[(4-fluorophenylamino)-methyl]-pyridin-2-yl}-amine dihydrochloride (182 mg, 0.59 mmol) as a white solid.
EXAMPLE 4: PREPARATION OF ETHYL-CYCLOPROPYL-{[4-(4- FLUOROPHENYLAMINO)-METHYL]-PHENYL}-AMINE

Part I: Preparation of (4-fluorophenyl)-carbamic acid terf-butyl ester
A mixture of 4-fluoroaniline (4.2 mL, 44.1 mmol) and carbonic acid di-te/f-butyl ester (11.55 g, 52.9 mmol) in toluene (100 mL) was heated at reflux overnight. The reaction was cooled to room temperature and . the solvent was removed under reduced pressure. The residue was triturated with hexanes to provide (4- fluorophenyl)-carbamic acid terf-butyl ester (8.4 g, 39.8 mmol) as an off-white solid.
Part II: Preparation of (4-fluorophenyl)-(4-iodo-benzyl)-carbamic acid tert-butyl ester
A solution of (4-fluorophenyl)-carbamic acid terf-butyl ester (9.98 g, 47.3 mmol) in anhydrous tetrahydrofuran (150 mL) was cooled to 00C and treated with sodium hydride (60% dispersion in mineral oil, 2.3 g, 56.8 mmol). The mixture was warmed to room temperature and stirred for 30 minutes. To the reaction was added 1- bromomethyl-4-iodo-benzene (14.0 g, 47.3 mmol) and the mixture was allowed to stir at room temperature overnight. The reaction was diluted with water (50 mL) and extracted with ethyl acetate (3 x 50 mL). The organic phases were combined,
washed with brine (50 ml_), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. Purification by chromatography (silica gel; 5% ethyl acetate in hexanes) provided (4-fluorophenyl)-(4-iodo-benzyl)-carbamic acid tert-butyl ester (18 g, 42.1 mmol) as a colorless oil.
Part III: Preparation of (4-cyclopropylamino-benzyl)-(4-fluorophenyl)-carbamic acid tert-butyl ester
A mixture of (4-fluorophenyl)-(4-iodo-benzyl)-carbamic acid tert-butyl ester (10 g, 23.4 mmol), cyclopropylamine (4.86 ml_, 70.2 mmol), copper (I) iodide (445 mg, 2.34 mmol), potassium carbonate (6.5 g, 46.8 mmol), and L-proline (540 mg, 4.68 mmol) were combined in dimethylsulfoxide (100 ml.) and heated at 800C for 5 hours. The reaction mixture was cooled, diluted with water (50 ml_), and extracted with ethyl acetate (2 x 100 ml_). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate, filtered and evaporated to dryness under reduced pressure. The (4-cyclopropylamino-benzyl)-(4-fluorophenyl)-carbamic acid te/t-butyl ester was used in the next step without further purification.
Part IV: Preparation of [4-(cyclopropyl-ethyl-amino)-benzyl]-(4-fluorophenyl)-carbamic acid terf-butyl ester
To the (4-cyclopropylamino-benzyl)-(4-fluorophenyl)-carbamic acid te/t-butyl ester from the previous step in dichloromethane (100 ml_) was added acetaldehyde (1.44 ml_, 25.7 mmol) and acetic acid (1.6 mL, 28.1 mmol). The solution was stirred at room temperature for 30 minutes, followed by the addition of sodium triacetoxyborohydride (2.0 g, 9.4 mmol). After 30 minutes, another portion of sodium triacetoxyborohydride (2.0 g, 9.4 mmol) was added. A third portion of sodium triacetoxyborohydride (2.0 g, 9.4 mmol) was added and the reaction stirred for 30 minutes. The reaction mixture was basified with 1 N sodium hydroxide to pH 10 and extracted with dichloromethane (2 x 50 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure to provide a yellow oil. Flash chromatography (silica gel; 10% ethyl acetate
in hexanes) provided [4-(cyclopropyl-ethyl-amino)-benzyl]-(4-fluorophenyl)-carbamic acid tert-butyl ester a yellow oil, which was used directly in the next reaction.
Part V: Preparation of ethyl-cyclopropyl-{[4-(4-fluorophenylamino)-methyl]-phenyl}- amine
To a solution of [4-(cyclopropyl-ethyl-amino)-benzyl]-(4-fluorophenyl)-carbamic acid tert-butyl ester from the previous step in dichloromethane (20 mL) was added trifluoroacetic acid (20 mL) at 0°C. The solution was warmed up to room temperature and stirred for 30 minutes. The reaction was concentrated to dryness under reduced pressure and the residue was dissolved in dichloromethane (20 mL). The organic layer was washed with 3N sodium hydroxide (10 mL), dried over anhydrous sodium sulfate, filtered and evaporated to dryness under reduced pressure. Flash chromatography (silica gel; 10% ethyl acetate in hexanes) provided ethyl- cyclopropyl-{[4-(4-fluorophenylamino)-methyl]-phenyl}-amine (6 g, 21.1 mmol) as a yellow oil.
EXAMPLE 5: PREPARATION OF DIETHYL-{[4-(4-FLUOROPHENYLAMINO)-
METHYL]-PHENYLJ-AMINE DIHYDROCHLORIDE

Part I: Preparation of 4-diethylamino-N-(4-fluorophenyl)-benzamide
To a solution of 4-diethylamino-benzoic acid (1.0 g, 5.2 mmol) and N-(3- dimethylaminopropyl)-N'-ethylcarbodiimide (1.4 g, 9.4 mmol) in pyridine (10 ml.) was added 4-fluoroaniline (446 μl_, 4.7 mmol) and the reaction stirred overnight. The reaction was concentrated under reduced pressure to provide a red oil, which was partitioned between saturated sodium bicarbonate, and hexanes and flash ethyl acetate. The resulting precipitate was collected and dried under reduced pressure to provide 4-diethylamino-N-(4-fluorophenyl)-benzamide (1.2 g, 4.2 mmol) as a white solid.
Part II: Preparation of diethyl-{[4-(4-fluorophenylamino)-methyl]-phenyl}-amine dihydrochloride
To a solution of 4-diethylamino-N-(4-fluorophenyl)-benzamide (600 mg, 2.1 mmol) in anhydrous tetrahydrofuran (10 ml_) was added dropwise 1 M borane tetrahydrofuran complex (6.3 ml_, 6.3 mmol). The reaction was heated to reflux and stirred for 3 hours. The reaction was cooled to room temperature and treated with saturated hydrochloric acid in methanol (6 ml.) and heated to reflux for 3 hours. The reaction was cooled to room temperature and the resulting precipitate filtered and dried to provide diethyl-{[4-(4-fluorophenylamino)-methyl]-phenyl}-amine dihydrochloride (622 mg, 1.8 mmol) as a white solid.
EXAMPLE 6: PREPARATION OF ETHYL-CYCLOPROPYL-{[3-(4- FLUOROPHENYLAMINO)-METHYL]-PHENYL)}AMINE
ine



Part I: Preparation of (4-fluorophenyl)-(3-iodo-benzyl)-carbamic acid terf-butyl ester
A solution of (4-fluorophenyl)-carbamic acid tert-butyl ester (5 g, 47.3 mmol) in anhydrous tetrahydrofuran (80 mL) was cooled to O0C and treated with sodium hydride (60% dispersion in mineral oil, 1.1 g, 28.4 mmol). The mixture was warmed to room temperature and stirred for 30 minutes. To the reaction was added 1- bromomethyl-3-iodo-benzene (7.0 g, 23.7 mmol) and the mixture was allowed to stir at room temperature overnight. The reaction was diluted with water (50 mL) and extracted with ethyl acetate (3 x 50 mL). The organic phases were combined, washed with brine (50 mL), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. Purification by chromatography (silica gel; 5% ethyl acetate in hexanes) provided (4-fluorophenyl)-(3-iodo-benzyl)-carbamic acid tert-butyl ester (9 g, 21.1 mmol) as a colorless oil, which was contaminated with residual (4-fluorophenyl)-carbamic acid terf-butyl ester.
Part II: Preparation of (3-cyclopropylamino-benzyl)-(4-fluorophenyl)-carbamic acid tert-butyl ester
A mixture of (4-fluorophenyl)-(3-iodo-benzyl)-carbamic acid tert-butyl ester (5.8 g,
13.6 mmol), cyclopropylamine (3.8 mL, 54.4 mmol), copper (I) iodide (260 mg, 1.36 mmol), potassium carbonate (7.5 g, 54.4 mmol), and L-proline (313 mg, 2.72 mmol) were combined in dimethylsulfoxide (60 mL) and heated at 800C for 4 hours. The reaction mixture was cooled, diluted with water (50 mL), and extracted with ethyl acetate (2 x 100 mL). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate, filtered and evaporated to dryness under reduced pressure. The (3-cyclopropylamino-benzyl)-(4-fluorophenyl)-carbamic acid te/t-butyl ester was used in the next step without further purification.
Step III: Preparation of [3-(cyclopropyl-ethyl-amino)-benzyl]-(4-fluorophenyl)- carbamic acid terf-butyl ester
To the (3-cyclopropylamino-benzyl)-(4-fluorophenyl)-carbamic acid terf-butyl ester from the previous step in dichloromethane (30 mL) was added acetaldehyde (840 μL,
15 mmol) and acetic acid (933 μL, 16.3 mmol). The solution was stirred at room temperature for 30 minutes, followed by the addition of sodium triacetoxyborohydride
(3.5 g, 16.32 mmol). After 30 minutes, another portion of sodium triacetoxyborohydride (3.5 g, 16.32 mmol) was added. A third portion of sodium triacetoxyborohydride (3.5 g, 16.32 mmol) was added and the reaction stirred for 30 minutes. The reaction mixture was basified with 1 N sodium hydroxide to pH 10 and extracted with dichloromethane (2 x 50 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure to provide a yellow oil. Flash chromatography (silica gel; 10% ethyl acetate in hexanes) provided (3-(cyclopropyl-ethyl-amino)-benzyl)-(4-fluorophenyl)-carbamic acid tert-butyl ester a yellow oil, which was used directly in the next reaction.
Part IV: Preparation of ethyl-cyclopropyl-{[3-(4-fluorophenylamino)-methyl]-phenyl}- amine
To a solution of [3-(cyclopropyl-ethyl-amino)-benzyl]-(4-fluorophenyl)-carbamic acid tert-butyl ester from the previous step in dichloromethane (20 ml.) was added trifluoroacetic acid (20 ml_) at O0C. The solution was warmed to room temperature and stirred for 30 minutes. The reaction was concentrated to dryness under reduced pressure and the residue was dissolved in dichloromethane (20 ml_). The organic layer was washed with 3N sodium hydroxide (10 ml_), dried over anhydrous sodium sulfate, filtered and evaporated to dryness under reduced pressure. Flash chromatography (silica gel; 10% ethyl acetate in hexanes) provided ethyl- cyclopropyl-{[3-(4-fluorophenylamino)-methyl]-phenyl}-amine (3.4 g, 12 mmol) as a yellow oil.
EXAMPLE 7: PREPARATION OF (4-FLUOROPHENYL)-[4-(4-METHYL-PIPERAZIN-
1-YL)-BENZYL]-AMINE
PhMe


Part I: Preparation of N-(4-fluorophenyl)-4-(4-methyl-piperazin-1-yl)-benzamide
To a solution of 4-fluoroaniline (15.8 g, 142 mmol) in dichloromethane (200 mL) at 0cC was added dropwise a solution of 4-(4-methylpiperazin-1-yl)-benzoyl chloride (25 g, 142 mmol). As a precipitate formed, the reaction was slowly diluted with additional dichloromethane (300 mL). Triethylamine (23.7 mL, 170 mmol) was added and the reaction was stirred for 30 minutes. The reaction was then warmed to room temperature and stirred for 30 minutes. The resulting precipitate was filtered and washed with water. The filtrate was treated with water, upon which additional
precipitates formed. The precipitate was collected and combined with the previously obtained precipitate. The material was dried under reduced pressure overnight to provide N-(4-fluorophenyl)-4-(4-methyl-piperazin-1-yl)-benzamide (35 g, 140 mmol) as a white solid.
Part II: Preparation of (4-fluorophenyl)-[4-(4-methyl-piperazin-1-yl)-benzyl]-amine
To a solution of N-(4-fluorophenyl)-4-(4-methyl-piperazin-1-yl)-benzamide (7.6 g, 24.2 mmol) in anhydrous toluene (50 ml_) and anhydrous terahydrofuran (25 ml.) at 00C was added dropwise sodium bis(2-methoxyethoxy)aluminum hydride (65 wt.% in toluene, 22 ml_). After the addition was complete the reaction was heated to reflux and stirred for 1 hour. The reaction was cooled to 00C and treated by dropwise addition of 6N sodium hydroxide (50 ml_). The reaction was warmed to room temperature, diluted with toluene (50 mL) and stirred for 2 hours. The layers were separated and the aqueous phase washed with toluene (50 mL). The organic phases were combined, washed with saturated sodium bicarbonate (30 mL), water (30 mL), and brine (30 mL). The organic phase was filtered through a pad of Celite® and the Celite® pad washed with ethyl acetate. The organic filtrates were combined and concentrated under reduced pressure to provide a yellow solid. The material was treated with dichloromethane and hexanes to provide (4-fluorophenyl)-[4-(4- methyl-piperazin-1-yl)-benzyl]-amine (5.7 g, 19 mmol) as a white solid.
EXAMPLE 8: PREPARATION OF (4-FLUOROPHENYL)-^-PIPERIDIN-I -YL- PYRIMIDIN-5-YLMETHYL)-AMINE
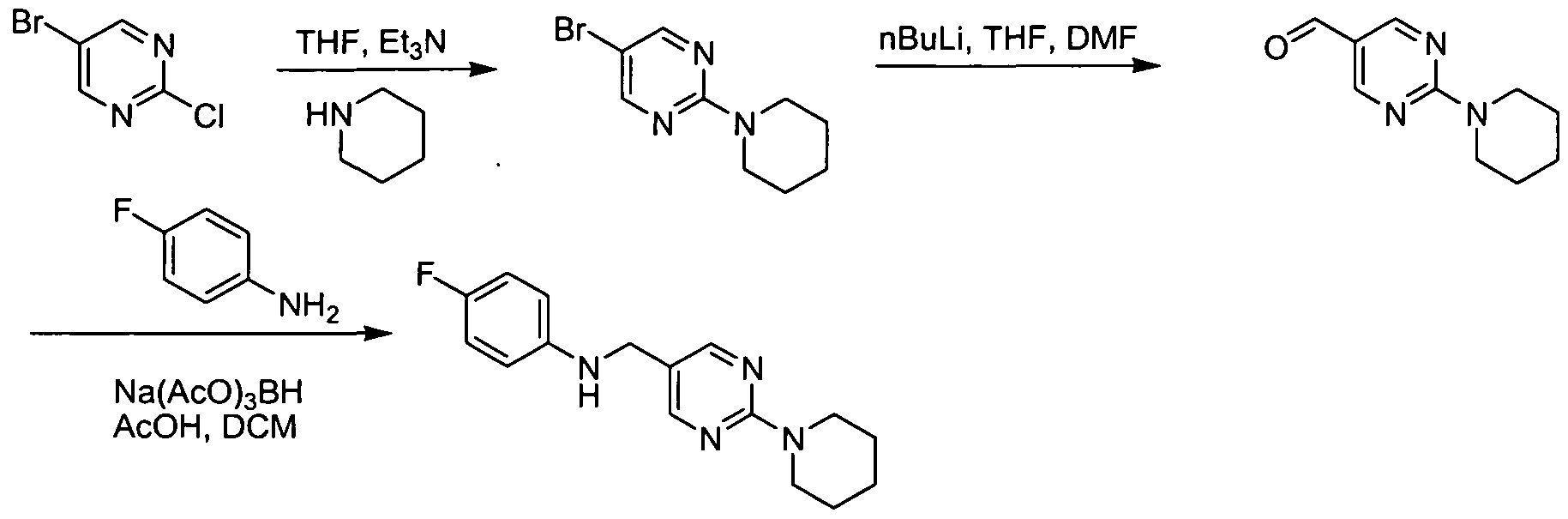
Part I: Preparation of 5-bromo-2-piperidin-1-yl-pyrimidine
To solution of 5-bromo-2-chloro-pyrimidine (3.0 g, 15.5 mmol) in dichloromethane (30 imL) at room temperature was added piperidine (1.53 mL, 15.5 mmol) followed by the dropwise addition of triethylamine (3.23 mL, 23.3 mmol). The reaction was stirred at room temperature overnight. The reaction was diluted with dichloromethane (20 mL), washed with a saturated aqueous sodium bicarbonate solution (50 mL), followed by brine (50 mL). The organic phase was dried with anhydrous sodium sulfate, filtered and concentrated under reduced pressure. Flash chromatography (silica gel; 5% ethyl acetate in hexanes) provided 5-bromo-2-piperidin-1-yl-pyrimidine as a white solid (3.74g, 15.5 mmol).
Part II: Preparation of 2-piperidin-1-yl-pyrimidine-5-carbaldehyde
To a solution of 5-bromo-2-piperidin-1-yl-pyrimidine (1.3 g, 5.4 mmol) in anhydrous tetrahydrofuran (30 mL) at -78°C was added 1.6 M n-butyl lithium in hexanes (3.7 mL, 5.93 mmol). The mixture was stirred at a temperature below -70°C for 1 hour. Dimethylformamide (4.2 mL, 53.9 mmol) was added dropwise and the reaction was stirred at a temperature below -70°C for 1 hour. The reaction was quenched with saturated ammonium chloride (10 mL), diluted with water (20 mL) and extracted with
ethyl acetate (2 x 20 ml_). The organic phases were combined, dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure to provide a viscous brown oil. Flash chromatography (silica gel; 5-30% ethyl acetate in hexanes) provided 2-piperidin-1-yl-pyrimidine-5-carbaldehyde (0.76 g, 4.0 mmol) as a white solid.
Part III: Preparation of (4-fluorophenyl)-(2-piperidin-1-yl-pyrimidin-5-ylmethyl)-amine
2-Piperidin-1-yl-pyrimidine-5-carbaldehyde (0.76 g, 4.0 mmol), 4-fluoroaniline (0.76 ml_, 8.0 mmol), and acetic acid (0.25 ml_, 4.4 mmol) were combined in dichloromethane (8 ml_). The solution was stirred at room temperature for 30 minutes, followed by the addition of sodium triacetoxyborohydride (0.28 mg, 1.32 mmol). After 30 minutes, another portion of sodium triacetoxyborohydride (0.28 mg, 1.32 mmol) was added. A third portion of sodium triacetoxyborohydride (0.28 mg, 1.32 mmol) was added and the reaction stirred for 30 minutes. The reaction mixture was basified with 1 N sodium hydroxide to pH 10 and extracted with dichloromethane (2 x 50 ITiL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure to provide a yellow oil. Flash chromatography (silica gel; 5-30% ethyl acetate in hexanes) provided (4- fluorophenyl)-(2-piperidin-1-yl-pyrimidin-5-ylmethyl)-amine (0.93 g, 3.25 mmol) as a yellow oil.
EXAMPLE 9: PREPARATION OF (4-FLUOROPHENYL)-(2-PYRROLIDIN-1-YL- THIAZOL-5-YLMETHYL)-AMINE DIHYDROCHLORIDE

Part I: Preparation of 2-chloro-5-chloromethyl thiazole
To a solution of 2-chloro-thiazol-5-yl-methanol (1 g, 6.7 mmol) in chloroform (10 mL) was added thionyl chloride (1.6g, 13.4 mmol), and the reaction was allowed to stir at room temperature overnight. The solvent was removed under reduced pressure to afford a cloudy oil (1.1g, 6.5 mmol) which was used directly in the next reaction without further purification.
Part II: Preparation of 2-chloro-thiazol-5-ylmethyl-4-fluorophenylcarbamic acid tert- butyl ester
To a solution of 4-fluorophenylcarbamic acid terf-butyl ester (1.3 g, 6.2 mmol) in anhydrous tetrahydrofuran (15 mL) was added sodium hydride (60% dispersion in mineral oil, 261 mg, 6.8 mmol). After the initial gas evolution had ceased, the reaction was allowed to stir for 15 minutes. Tetra-n-butylammonium iodide (227 mg, 0.6 mmol) was then added followed by addition of the 2-chloro-5-chloromethyl thiazole prepared above. The mixture was heated to reflux for 1 hour. After cooling, the reaction was carefully neutralized with cold saturated sodium bicarbonate (10 mL) and extracted with ethyl acetate (2 x 20 mL). The organic layers were combined, dried over anhydrous sodium sulfate, filtered and the solvent removed
under reduced pressure to provide a dark oil. Flash chromatography (silica gel; 5%- 10% ethyl acetate in hexanes) provided 2-chloro-thiazol-5-ylmethyl-4- fluorophenylcarbamic acid terf-butyl ester (1.5 g, 4.4 mmol) as a yellow oil.
Part III: Preparation of 4-fluorophenyl-2-pyrrolidin-1-yl-thiazol-5-ylmethylcarbamic acid terf-butyl ester
A solution of 2-chloro-thiazol-5-ylmethyl-4-fluorophenylcarbamic acid tert-butyl ester (1.5 g, 4.4 mmol) in pyrrolidine (1.6 ml_, 22 mmol) was heated in a sealed tube to 13O0C and stirred overnight. After cooling, the reaction was partitioned between water and ethyl acetate. The organic layer was separated, dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure to provide 4-fluorophenyl-2-pyrrolidin-1-yl-thiazol-5-ylmethylcarbamic acid tert-butyl ester as a yellow oil.
Part IV: Preparation of (4-fluorophenyl)-(2-pyrrolidin-1-yl-thiazol-5-ylmethyl)-amine dihydrochloride
To the free base of 4-fluorophenyl-2-pyrrolidin-1-yl-thiazol-5-ylmethylcarbamic acid tert-butyl ester in dichloromethane (10 mL) was added trifluoroacetic acid (4 ml.) and the reaction was stirred at room temperature for 3 hours. After removing the solvent under reduced pressure, the resulting oil was dissolved in diethyl ether and treated with excess ethereal hydrochloric acid. The resulting solid was collected by filtration and dried to provide (4-fluorophenyl)-(2-pyrrolidin-1-yl-thiazol-5-ylmethyl)-amine dihydrochloride (274 mg, 1.2 mmol) as a white solid.
EXAMPLE 10: PREPARATION OF (4-FLUOROPHENYL)-^-PIPERIDIN-I -YL- THIAZOL-^YLMETHYL)-AMINE DIHYDROCHLORIDE
H2N

Part I: Preparation of 1-(4-chloromethyl-thiazol-2-yl)-piperidine
A suspension of piperidine-1-carbothioamide (1.0 g, 6.9 mmol) and 1 ,3-dichloro- propan-2-one (876 mg, 6.9 mmol) in ethanol (10 mL) was heated to 800C and the reaction monitored by liquid chromatography (LC)/mass spectrometry (MS). After 1 hour, the reaction was cooled and the solvent was removed under reduced pressure to provide a pinkish-violet liquid. The liquid was dissolved into ice water (10 mL) and slowly treated with solid sodium bicarbonate, upon which a white precipitate formed. The solid was collected by filtration and dried under reduced pressure. The solid was triturated with hexane and filtered. The filtrate was collected, dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure to afford 1- (4-chloromethyl-thiazol-2-yl)-piperidine (1.1 g, 5.1 mmol) as an off-white solid.
Part II: Preparation of (4-fluorophenyl)-(2-piperidin-1-yl-thiazol-4-ylmethyl)-carbamic acid terf-butyl ester
To a solution of 4-fluorophenylcarbamic acid terf-butyl ester (1.02 g, 4.6 mmol) in tetrahydrofuran (15 mL) was added sodium hydride (60% dispersion in mineral oil, 206 mg, 5.1 mmol). After the initial gas evolution had ceased, the reaction was allowed to stir for 15 minutes. Tetra-n-butylammonium iodide (189 mg, 0.5 mmol)
was then added followed by the addition of 1-(4-chloromethyl-thiazol-2-yl)-piperidine (1.1 g, 5.1 mmol) prepared above. The mixture was heated to reflux for 1 hour. After cooling, the reaction was carefully neutralized with cold saturated sodium bicarbonate (10 ml_) and extracted with ethyl acetate (2 x 20 mL). The organic layers were combined, dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure to provide an oil. Flash chromatography (silica gel; 10% ethyl acetate in hexanes) provided (4-fluorophenyl)-(2-piperidin-1-yl-thiazol-4- ylmethyl)-carbamic acid te/t-butyl ester (1.25 g, 3.2 mmol) as a white solid.
Part III: Preparation of (4-fluorophenyl)-(2-piperidin-1-yl-thiazol-4-ylmethyl)-amine dihydrochloride
To a solution of (4-fluorophenyl)-(2-piperidin-1-yl-thiazol-4-ylmethyl)-carbamic acid tert-butyl ester (1.25 g, 3.2 mmol) in dichloromethane (15 mL) was added trifluoroacetic acid (4 mL) and the reaction was stirred at room temperature for 1.5 hours. After removing the solvent under reduced pressure, the resulting oil was dissolved in diethyl ether and treated with excess ethereal hydrochloric acid. The resulting solid was collected by filtration and dried to provide (4-fluorophenyl)-(2- piperidin-1-yl-thiazol-4-ylmethyl)-amine dihydrochloride (1.05 g, 3.2 mmol) as a white solid.
EXAMPLE 11 : PREPARATION OF (4-FLUOROPHENYL)-^-PIPERIDIN-I -YL- THIAZOL-δ-YLMETHYLVAMINE DIHYDROCHLORIDE

Part I: Preparation of (2-chloro-thiazol-5-ylmethyl)-(4-fluorophenyl)-carbamic acid te/f-butyl ester
To a solution of 2-chloro-thiazol-5-yl-methanol (1 g, 6.7 mmol) in chloroform (10 mL) was added thionyl chloride (1.6 g, 13.4 mmol), and the reaction was allowed to stir at room temperature overnight. The solvent was removed under reduced pressure to afford a cloudy oil (1.1 g, 6.5 mmol) which was used directly in the next reaction without further purification.
To a solution of (4-fluorophenyl)-carbamic acid te/f-butyl ester (1.3 g, 6.2 mmol) in anhydrous tetrahydrofuran (15 mL) was added sodium hydride (60% dispersion in mineral oil, 261 mg, 6.8 mmol). After the initial gas evolution had ceased, the reaction was allowed to stir for 15 minutes. Tetra-n-butylammonium iodide (227 mg, 0.6 mmol) was then added followed by addition of 2-chloro-5-chloromethyl thiazole prepared above. The mixture was heated to reflux for 1 hour. After cooling, the reaction was carefully neutralized with cold saturated sodium bicarbonate (10 mL)
and extracted with ethyl acetate (2 x 20 ml_). The organic layers were combined, dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure to provide a dark oil. Flash chromatography (silica gel; 5%-10% ethyl acetate in hexanes) provided (2-chloro-thiazol-5-ylmethyl)-(4-fluorophenyl)-carbamic acid terf-butyl ester as a yellow oil (1.5 g, 4.4 mmol).
Part II: Preparation of (4-fluorophenyl)-(2-piperidin-1-yl-thiazol-5-ylmethyl)-carbamic acid te/t-butyl ester
A solution of (2-chloro-thiazol-5-ylmethyl)-(4-fluorophenyl)-carbamic acid tert-butyl ester (1.5 g, 4.4 mmol) in piperidine (10 ml.) was heated in a sealed tube to 13O0C and stirred overnight. After cooling, the reaction was partitioned between water and ethyl acetate. The organic layer was separated, dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure to obtain a yellow oil. Flash chromatography (silica gel; 10%-20% ethyl acetate in hexanes) provided (4-fluorophenyl)-(2-piperidin-1-yl-thiazol-5-ylmethyl)-carbamic acid te/t-butyl ester as a light yellow oil (981 mg, 2.6 mmol).
Part III: Preparation of (4-fluorophenyl)-(2-piperidin-1-yl-thiazol-5-ylmethyl)-amine dihydrochloride
To a solution of (4-fluorophenyl)-(2-piperidin-1-yl-thiazol-5-ylmethyl)-carbamic acid terf-butyl ester (981 mg, 2.6 mmol) in dichloromethane (10 ml.) was added trifluoroacetic acid (4 ml.) and the reaction was stirred at room temperature for 3 hours. After removing the solvent under reduced pressure, the resulting oil was dissolved in diethyl ether and treated with excess ethereal hydrochloric acid. The resulting solid was collected by filtration and dried to provide (4-fluorophenyl)-(2- piperidin-1-yl-thiazol-5-ylmethyl)-amine dihydrochloride as a white solid (472 mg, 1.7 mmol).
EXAMPLE 12: PREPARATION OF 2-(METHYL-PHENYL-AMINO)-THIAZOLE-4-
CARBOXYLIC ACID

Part I: Preparation of 1-methyl-1 -phenyl-thiourea
To a solution of N-methylaniline (4 mL, 37 mmol) in anhydrous tetrahydrofuran (60 mL) at O0C was added dropwise benzoyl isothiocyanate (7.24 g, 44 mmol). After addition was complete, the reaction was allowed to stir at room temperature for 1 hour, by which time a suspension formed. Diethyl ether (60 mL) was added, and the solid was collected and dried to afford a yellow solid (9.5 g, 35 mmol). The solid was dissolved in ethanol (50 mL) and 1 N sodium hydroxide (25 mL) and the mixture was heated to reflux. After stirring overnight, the reaction was cooled and the organic solvent was removed under reduced pressure. The remaining aqueous phase was extracted with dichloromethane (2 x 50 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure to afford 1-methyl-1 -phenyl-thiourea (3.48 g, 21 mmol). This material was used directly in the next reaction without further purification.
Part II: Preparation of 2-(methyl-phenyl-amino)-thiazole-4-carboxylic acid ethyl ester
A suspension of 1-methyl-1 -phenyl-thiourea (1.0 g, 6 mmol), magnesium sulfate (1 g) and ethyl bromopyruvate (1.2 g, 6 mmol) in acetone (10 mL) was heated to reflux and stirred for 6 hours. After cooling, the reaction was filtered and concentrated under reduced pressure. The residue was dissolved in chloroform (40 mL), washed with saturated sodium bicarbonate, dried over sodium sulfate, filtered and the solvent removed under reduce pressure to afford 2-(methyl-phenyl-amino)-thiazole-4- carboxylic acid ethyl ester (1.5g, 5.7 mmol) as an oil.
Part III: Preparation of 2-(methyl-phenyl-amino)-thiazole-4-carboxylic acid
To a solution of 2-(methyl-phenyl-amino)-thiazole-4-carboxylic acid ethyl ester (1.5 g, 5.7 mmol) in tetrahydrofuran (10 ml_) and water (5 ml.) was added lithium hydroxide monohydrate (361 mg, 8.6 mmol) and the reaction was allowed to stir overnight. The organic phases were removed under reduced pressure and the resulting aqueous phase was acidified with 6N hydrochloric acid, upon which a precipitate formed. The solid was collected and dried under reduced pressure to provide 2-(methyl-phenyl- amino)-thiazole-4-carboxylic acid (1.33 g, 5.7 mmol) as a white solid.
EXAMPLE 13: PREPARATION OF 1-BENZYL-1 H-[1 ,2,3]TRIAZOLE-4-CARBONYL
CHLORIDE

To a suspension of 1-benzyl-1 H-[1 ,2,3]triazole-4-carboxylic acid (175 mg, 0.9 mmol) in chloroform (6 mL) was added thionyl chloride (205 mg, 1.7 mmol) followed by dimethylformamide (5 drops). The reaction was heated to 650C until a clear solution persisted. After cooling, the solvent was removed under reduced pressure to provide 1-benzyl-1 H-[1 ,2,3]triazole-4-carbonyl chloride (199 mg, 0.9 mmol) as a white solid.
EXAMPLE 14: PREPARATION OF I-BENZYL-I H-IMIDAZOLE^-CARBOXYLIC
ACID TRIFLUOROACETATE

A mixture of tert-butyl isonitrileacetate (4 mL, 27.5 mmol) and tert-butoxy- bis(dimethylamino)methane (9.6 g, 55 mmol) was stirred overnight. The reaction was directly chromatographed (silica gel; 5%, 10%, 15%, 30% to 50% ethyl acetate in hexanes) to provide 2-isonitrile-3-dimethylamino-acrylic acid tert-butyl ester (5.1 g, 26 mmol) as a red oil.
Part II: Preparation of i-benzyl-I H-imidazole-4-carboxylic acid tert-butyl ester
A mixture of 2-isonitrile-3-dimethylamino-acrylic acid tert-butyl ester (4.2 g, 21.4 mmol) in benzyl amine (2.3 mL, 21.4 mmol) was stirred at 80°C overnight. The reaction was cooled to room temperature, diluted with ethyl acetate (100 mL), washed with 0.1 N hydrochloric acid (25 mL), and brine (25 mL). The organic phase was dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure to provide an oil. Purification by chromatography (silica gel; 10% to 20% acetone in hexanes) provided 1-benzyl-1 H-imidazole-4-carboxylic acid tert- butyl ester (3.1 g, 12.4 mmol) and recovered 2-cyano-3-dimethylamino-acrylic acid tert-butyl ester (1.2 g, 6.1 mmol).
Part III: Preparation of 1-benzyl-1 H-imidazole-4-carboxylic acid tert-butyl ester trifluoroacetate
A solution of 1-benzyl-1 H-imidazole-4-carboxylic acid tert-butyl ester (3.1 g, 12.4 mmol) in trifluoroacetic acid (4 mL) was heated at 600C overnight. The reaction was cooled to room temperature and concentrated under reduced pressure. The residue was triturated with diethyl ether, filtered, washed with diethyl ether and dried under reduced pressure to provide 1-benzyl-1 H-imidazole-4-carboxylic acid trifluoroacetate (2.6 g, 12.8 mmol) as an off-white solid.
EXAMPLE 15: PREPARATION OF 1 -(2,2-DIMETHYL-PROPYL)-I H-IMIDAZOLE^- CARBOXYLIC ACID TRIFLUOROACETATE
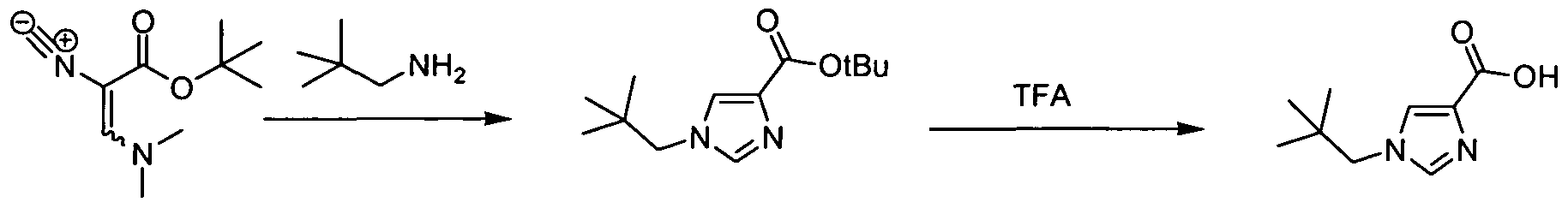
A mixture of 2-cyano-3-dimethylamino-acrylic acid tert-butyl ester (1.4 g, 7.1 mmol) in neopentyl amine (620 mg, 7.1 mmol) was stirred at 800C overnight. The reaction was cooled to room temperature, diluted with ethyl acetate (100 mL), washed with 0.1 N hydrochloric acid (25 mL), and brine (25 mL). The organic phase was dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure to provide an oil. Purification by chromatography (silica gel; 10% to 20% acetone in hexanes) provided 1-(2,2-dimethyl-propyl)-1 H-imidazole-4-carboxylic acid tert-butyl ester (890 mg, 3.0 mmol) as a white solid.
A solution of 1-(2,2-dimethyl-propyl)-1 H-imidazole-4-carboxylic acid tert-butyl ester (890 mg, 3.0 mmol) in trifluoroacetic acid (4 mL) was heated at 600C overnight. The reaction was cooled to room temperature and concentrated under reduced pressure. The residue was triturated with diethyl ether, filtered, washed with diethyl ether and dried under reduced pressure to provide 1-(2,2-dimethyl-propyl)-1 H-imidazole-4- carboxylic acid trifluoroacetate (858 mg, 2.9 mmol) as a white solid.
EXAMPLE 16: PREPARATION OF 1-BENZYL-2-METHYL-1 H-IMIDAZOLE-4- CARBOXYLIC ACID TRIFLUOROACETATE

A solution of amino-cyano-acetic acid tert-butyl ester (13.3 g, 85 mmol), ethyl acetimidate hydrochloride (10.5 g, 85 mmol) and benzyl amine (9.3 ml_, 85 mmol) in chloroform (100 ml_) was heated at 600C overnight. The reaction was cooled to room temperature, washed with saturated sodium bicarbonate (50 ml_), brine (25 ml_), dried over anhydrous magnesium sulfate, filtered and concentrated under reduced pressure. Chromatography of the residue (silica gel; 3% methanol in chloroform) provided S-amino-i-benzyl^-methyl-I H-imidazole^-carboxylic acid tert-butyl ester (12.3 g, 43 mmol).
Part II: Preparation of i-benzyl^-methyl-IH-imidazole^-carboxylic acid
To a solution of S-amino-i-benzyl^-methyl-I H-imidazole^-carboxylic acid tert-butyl ester (3.1 g, 10.8 mmol) in acetone (15 ml.) and water (15 ml.) at -25°C was added 50% hypophosphoric acid (80 ml_). After stirring at -25°C for 6 hours, the reaction was neutralized with saturated sodium bicarbonate and extracted with ethyl acetate (2 x 50 ml_). The organic phases were combined, washed with water (20 mL), brine (20 mL), dried over anhydrous magnesium sulfate, filtered and concentrated under reduced pressure. The residue was dissolved in dichloromethane (15 mL) and trifluoroacetic acid (15 mL) and heated at 600C for 2 hours. The reaction was concentrated under reduced pressure to provide 1-benzyl-2-methyl-1 H-imidazole-4- carboxylic acid (628 mg, 1.9 mmol) as a yellow solid.
EXAMPLE 17: PREPARATION OF I-CYCLOPROPYLMETHYL^-METHYL-I H- IMIDAZOLE-4-CARBOXYLIC ACID TRIFLUOROACETATE

EXAMPLE 18: PREPARATION OF 1-ALLYL-2-METHYL-1 H-IMIDAZOLE-4- CARBOXYLIC ACID TRIFLUOROACETATE

1-Allyl-2-methyl-1 H-imidazole-4-carboxylic acid trfluoroacetate was prepared according to procedures described in Example 16, using allylamine in place of benzyl amine.
EXAMPLE 19: PREPARATION OF 2-BENZYL-1 -METHYL-1 H-IMIDAZOLE-4-
CARBOXYLIC ACID TRFLUOROACETATE

A mixture of amino-cyano-acetic acid tert-butyl ester (789 mg, 5.1 mmol) and 2- phenyl-acetimidic acid methyl ester hydrochloride (917 mg, 4.6 mmol) in chloroform
(20 mL) was heated at reflux for 30 minutes. The reaction was cooled to room temperature and treated with methylamine (2.76 mL, 5.5 mmol, 2M in methanol) and the reaction was heated to reflux and stirred overnight. The reaction was cooled to room temperature and treated with saturated sodium bicarbonate (25 mL). The aqueous phase was extracted with dichloromethane (3 x 50 mL). The organic phases were combined, dried over anhydrous magnesium sulfate, filtered and
concentrated under reduced pressure to provide an oil. Trituration with diethyl ether provided a tan solid which was washed with diethyl ether/hexanes to provide 2- benzyl-1-methyl-1 H-imidazole-4-carboxylic acid tert-butyl ester (400 mg, 1.4 mmol).
A solution of 2-benzyl-1 -methyl-1 H-imidazole-4-carboxylic acid tert-butyl ester (400 mg, 1.4 mmol), dichloromethane (5 mL) and trifluoroacetic acid (5 ml_) was heated at 600C for 2 hours. The reaction was concentrated under reduced pressure to provide 1-benzyl-2-methyl-1 H-imidazole-4-carboxylic acid trifluoroacetate (460 mg, 1.4 mmol).
EXAMPLE 20: PREPARATION OF 1-BENZYL-1 H-[1 ,2,3]TRIAZOLE-4-CARBONYL CHLORIDE

To a suspension of 1-benzyl-1 H-[1 ,2,3]triazole-4-carboxylic acid (175 mg, 0.9 mmol) in chloroform (6 mL) was added thionyl chloride (205 mg, 1.7 mmol) followed by dimethylformamide (5 drops). The reaction was heated to 650C until a clear solution persisted. After cooling, the solvent was removed under reduced pressure to provide 1-benzyl-1 H-[1 ,2,3]triazole-4-carbonyl chloride (199 mg, 0.9 mmol) as a white solid.
EXAMPLE 21. 4-(CHLOROMETHYL)-N-CYCLOPROPYL-N-ETHYL-I ,3-TH IAZOL-
2-AMINE
BOC2O


2. 2N NaOH reflux, 48 hrs.

Step 1. tert-butyl cyclopropylcarbamate
To a solution of cyclopropylamine (6.0 g, 0.10 mol) and triethylamine (14.6 ml, 0.10 mol) in dichloromethane (250 mL) was slowly added di-tertbutyldicarbonate (22.9 g, 0.10 mol) in dichloromethane (100 mL). After stirring overnight at room temperature, the mixture was poured into water, the organic layer was separated, washed with water, dried (MgSO4) and evaporated to afford the product (15.48 g, 0.098 mol).
Step 2. tert-Butyl cyclopropyl(ethyl)carbamate
te/f-Butyl cyclopropylcarbamate (14.24 g, 0.092 mmol) in dry DMF (100 mL) was cooled to 0 0C, and treated with sodium hydride (60% dispersion in oil, 3.79 g, 0.095 mol). After the addition was complete, ethyl iodide (8.0 mL, 100 mmol) was added
dropwise. The mixture was then stirred overnight at room temperature, then pured into water, extracted with diethyl ether, the organic layer was washed with water, dried (MgSO4) and evaporated to afford the product as an oil (17.74 g, 0.092 mol).
Step 3. N-ethylcyclopropanamine hydrochloride
tert-butyl cyclopropyl(ethyl)carbamate (15 g, 0.081 mol) was treated with hydrogen chloride (4N in dioxane, 200 ml_). After 16 h, the reaction was evaporated and the residue triturated with diethylether, then hexane. The solid was then dried under vacuum to afford the product (8.67 g, 72 mmol).
Step 4. 1-cyclopropyl-i-ethylthiourea
To a mixture of cyclohexylethylamine hydrochloride (10.5 g, 87.5 mmol) and triethylamine (12.2 ml_, 87.7 mmol) in tetrahydrofuran was added benzoylisothiocyanate (11.9 ml_, 90.4 mmol) and reluxed 3 hours. The mixture was cooled, and partitioned between water and ethyl acetate. The organic layer was collected, dried over magnesium sulfate, and evaporated. The residue was dissolved in tetrahydrofuran/ethanol (300 ml_); 2N NaOH (60 mL) was added and the mixture refluxed 48 hours. The mixture was partitioned between water and ethyl acetate. The organic layer was collected, dried over magnesium sulfate, and evaporated. The resultant oil was scratched with hexanes/ether to provide 1-cyclopropyl-i- ethylthiourea (9.5 g, 75 %).
Step 5. 4-(chloromethyl)-N-cyclopropyl-N-ethyl-1 ,3-thiazol-2-amine
A mixture of 1-cyclopropyl-i-ethylthiourea (9.5 g, 66.2 mmol) and 1 ,3 dichloroacetone (8.6 g, 68 mmol) in ethanol (100 mL) was refluxed 8 hours. The solvent evaporated under reduced pressure and the residue was partitioned between saturated sodium bicarbonate and ethyl acetate. The organic layer was collected, dried over magnesium sulfate, and concentrated. The residue was purified by
chromatography (silica gel; 1/5 ethylacetate/hexane) to provide 4-(chloromethyl)-N- cyclopropyl-N-ethyl-1 ,3-thiazol-2-amine (8.0 g, 56 %).
EXAMPLE 22: PREPARATION OF 2-(METHYL-PHENYL-AMINO)-THIAZOLE-4-
CARBOXYLIC ACID (6-DIETHYLAMINO-PYRIDIN-S-YL-METHYL)-^- FLUOROPHENYL)-AMIDE (COMPOUND NO. 1)

Part I: Preparation of 2-(methyl-phenyl-amino)-thiazole-4-carbonyl chloride
To 2-(methyl-phenyl-amino)-thiazole-4-carboxylic acid (200 mg, 8.5 mmol, Example 12) in chloroform (8 mL) was added excess sulfuryl chloride (400 μL) and dimethylformamide (100 μL) and the reaction heated to reflux. After 30 minutes, the reaction was cooled to room temperature and the solvent removed under reduced pressure to provide 2-(methyl-phenyl-amino)-thiazole-4-carbonyl chloride as a yellow solid which was used directly in the next reaction.
Part II: Preparation of 2-(methyl-phenyl-amino)-thiazole-4-carboxylic acid (6- diethylamino-pyridin-3-yl-methyl)-(4-fluorophenyl)-amide
To a suspension of (5-((4-fluorophenylamino)-methyl)-pyridin-2-yl)-diethyl-amine (150 mg, 0.43 mmol, Example 1 ) in tetrahydrofuran (4 mL) was added 2-(methyl-phenyl- amino)-thiazole-4-carbonyl chloride (214 mg, 0.85 mmol) and triethylamine (600 μL) and the reaction heated to 600C. After 20 minutes the reaction was cooled to room temperature and quenched with saturated sodium bicarbonate (10 mL). The aqueous phase was extracted with ethyl acetate (2 x 15 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered and the solvent
removed under reduced pressure to afford a yellow oil. Purification by chromatography (silica gel; 20-30% ethyl acetate in dichloromethane) provided 2- (methyl-phenyl-amino)-thiazole-4-carboxylic acid (6-diethylamino-pyridin-3-yl- methyl)-(4-fluorophenyl)-amide as a colorless oil.
Part III: Preparation of 2-(methyl-phenyl-amino)-thiazole-4-carboxylic acid (6- diethylamino-pyridin-3-yl-methyl)-(4-fluorophenyl)-amide dihydrochloride
The free base was treated with ethereal hydrochloric acid to provide 2-(methyl- phenyl-amino)-thiazole-4-carboxylic acid (6-diethylamino-pyridin-3-yl-methyl)-(4- fluorophenyl)-amide dihydrochloride (225 mg, 0.40 mmol) as a white solid. MS (base): m/z 490 [M+H]; HPLC C tr: 3.4 min.
EXAMPLE 23: PREPARATION OF 2-BENZYL-THIAZOLE-4-CARBOXYLIC ACID (6- DIETHYLAMINO-PYRIDIN-S-YL-METHYL)-^-FLUOROPHENYL)-AMIDE
(COMPOUND NO. 2)

To a suspension of 2-benzyl-thiazole-4-carboxylic acid (CAS No. 36916-44-6, 300 mg, 1.37 mmol) in chloroform (4 mL) was added excess sulfuryl chloride (600 μL) and dimethylformamide (100 μL) and the reaction heated to reflux. After 2 hours, the reaction was cooled to room temperature and the solvent removed under reduced pressure to provide 2-benzyl-thiazole-4-carbonyl chloride as a brown oil which solidified upon standing.
Part II: Preparation of 2-benzyl-thiazole-4-carboxylic acid (6-diethylamino-pyridin-3- yl-methyl)-(4-fluorophenyl)-amide
To a suspension of {5-[(4-fluorophenylamino)-methyl]-pyridin-2-yl}-diethyl-amine (100 mg, 0.29 mmol, Example 1 ) in tetrahydrofuran (6 ml_) was added 2-benzyl-thiazole-4- carbonyl chloride (104 mg, 0.44 mmol) and triethylamine (600 μl_). After 1 hour, the reaction was quenched with saturated sodium bicarbonate (2 ml.) and extracted with ethyl acetate (2 x 15 ml_). The organic phases were combined, dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure.
Purification by chromatography (silica gel; 10-40% ethyl acetate in dichloromethane) provided 2-benzyl-thiazole-4-carboxylic acid (6-diethylamino-pyridin-3-yl-methyl)-(4- fluorophenyl)-amide as a colorless oil.
Part III: Preparation of 2-benzyl-thiazole-4-carboxylic acid (6-diethylamino-pyridin-3- yl-methyl)-(4-fluorophenyl)-amide dihydrochloride
The free base was treated with ethereal hydrochloric acid to provide 2-benzyl- thiazole-4-carboxylic acid (6-diethylamino-pyridin-3-yl-methyl)-(4-fluorophenyl)-amide dihydrochloride (50 mg, 0.08 mmol) as a white foam. MS: m/z 475 [M+H]; HPLC C: tr = 3.3 min.
EXAMPLE 24: PREPARATION OF 2-METHYL-THIAZOLE-4-CARBOXYLIC ACID
(6-DIETHYLAMINO-PYRIDIN-S-YL-METHYL)-^-FLUOROPHENYL)-AMIDE (COMPOUND NO. 3)
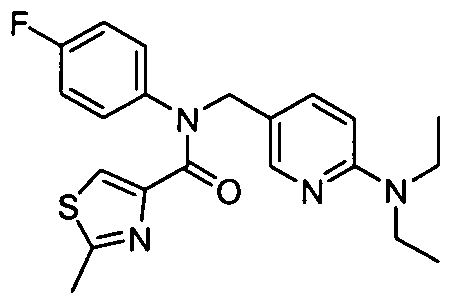
Part I: Preparation of 2-methyl-thiazole-4-carbonyl chloride
To 2-methyl-thiazole-4-carboxylic acid (CAS No. 35272-15-2, 600 mg, 4.2 mmol) in chloroform (4 mL) was added excess sulfuryl chloride (600 μl_) and dimethylformamide (100 μl_) and the reaction heated to reflux. After 2 hours, the reaction was hot filtered and the solvent removed under reduced pressure to provide 2-methyl-thiazole-4-carbonyl chloride as a gummy solid which was used directly in the next reaction.
Part II: Preparation of 2-methyl-thiazole-4-carboxylic acid (6-diethylamino-pyridin-3- yl-methyl)-(4-fluorophenyl)-amide
To a suspension of (5-((4-fluorophenylamino)-methyl)-pyridin-2-yl)-diethyl-amine (150 mg, 0.43 mmol, Example 1 ) in tetrahydrofuran (4 mL) was added 2-methyl-thiazole-4- carbonyl chloride (210 mg, 0.8 mmol) and triethylamine (600 μl_) and the reaction heated to 600C. After 20 minutes the reaction was cooled to room temperature and quenched with saturated sodium bicarbonate (10 mL). The aqueous phase was extracted with ethyl acetate (2 x 15 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure to afford a yellow oil. Purification by chromatography (silica gel; 50-75% ethyl acetate in hexane) provided 2-methyl-thiazole-4-carboxylic acid (6- diethylamino-pyridin-3-yl-methyl)-(4-fluorophenyl)-amide as a colorless oil.
Part III: Preparation of 2-methyl-thiazole-4-carboxylic acid (6-diethylamino-pyridin-3- yl-methyl)-(4-fluorophenyl)-amide dihydrochloride
The free base was treated with ethereal hydrochloric acid to provide 2-methyl- thiazole-4-carboxylic acid (6-diethylamino-pyridin-3-yl-methyl)-(4-fluorophenyl)-amide dihydrochloride (100 mg, 0.18 mmol). MS: m/z 399 [M+H]; HPLC C: tr = 4.1 min.
EXAMPLE 25: PREPARATION OF 2-PYRIDIN-4-YL-THIAZOLE-4-CARBOXYLIC ACID (6-DIETHYLAMINO-PYRIDIN-S-YL-METHYL)-^-FLUOROPHENYL)-AMIDE
(COMPOUND NO. 4)

To 2-pyridin-4-yl-thiazole-4-carboxylic acid (CAS No. 21278-86-4, 500 mg, 2.4 mmol) in chloroform (10 mL) was added excess sulfuryl chloride (400 μL) and dimethylformamide (100 μL) and the reaction heated to reflux. After 30 minutes, the reaction was cooled to room temperature and the solvent removed under reduced pressure to provide 2-pyridin-4-yl-thiazole-4-carbonyl chloride as a solid which was used directly in the next reaction.
Part II: Preparation of 2-pyridin-4-yl-thiazole-4-carboxylic acid (6-diethylarπino- pyridin-3-yl-methyl)-(4-fluorophenyl)-amide
To a suspension of {5-[(4-fluorophenylamino)-methyl]-pyridin-2-yl}-diethyl-amine (150 mg, 0.43 mmol, Example 1 ) in tetrahydrofuran (4 mL) was added 2-pyridin-4-yl- thiazole-4-carbonyl chloride (214 mg, 0.85 mmol) and triethylamine (600 μL) and the reaction heated to 600C. After 20 minutes the reaction was cooled to room temperature and quenched with saturated sodium bicarbonate (10 mL). The aqueous phase was extracted with ethyl acetate (2 x 15 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure. Purification by chromatography (silica gel; ethyl acetate) provided 2-pyridin-4-yl-thiazole-4-carboxylic acid (6-diethylamino-pyridin-3- yl-methyl)-(4-fluorophenyl)-amide as a colorless oil.
Part III: Preparation of 2-pyridin-4-yl-thiazole-4-carboxylic acid (6-diethylamino- pyridin-3-yl-methyl)-(4-fluorophenyl)-amide dihydrochloride
The free base was treated with ethereal hydrochloric acid to provide 2-pyridin-4-yl- thiazole-4-carboxylic acid (6-diethylamino-pyridin-3-yl-methyl)-(4-fluorophenyl)-amide dihydrochloride (160 mg, 0.3 mmol) as a white solid. MS: m/z 462 [M+H]; HPLC C: tr = 3.3 min.
EXAMPLE 26: PREPARATION OF 2-PYRIDIN-3-YL-THIAZOLE-4-CARBOXYLIC ACID (6-DIETHYLAMINO-PYRIDIN-S-YL-METHYL)-^-FLUOROPHENYL)-AMIDE
(COMPOUND NO. 5)

Part I: Preparation of 2-pyridin-3-yl-thiazole-4-carbonyl chloride
To 2-pyridin-3-yl-thiazole-4-carboxylic acid (CAS No. 39067-29-3, 500 mg, 2.4 mmol) in chloroform (10 mL) was added excess sulfuryl chloride (400 μL) and dimethylformamide (100 μL) and the reaction heated to reflux. After 30 minutes, the reaction was cooled to room temperature and the solvent removed under reduced pressure to provide 2-pyridin-3-yl-thiazole-4-carbonyl chloride as a solid which was used directly in the next reaction.
Part II: Preparation of 2-pyridin-3-yl-thiazole-4-carboxylic acid (6-diethylamino- pyridin-3-yl-methyl)-(4-fluorophenyl)-amide
To a suspension of {5-[(4-fluorophenylamino)-methyl]-pyridin-2-yl}-diethyl-amine (150 mg, 0.43 mmol, Example 1 ) in tetrahydrofuran (4 mL) was added 2-pyridin-3-yl-
thiazole-4-carbonyl chloride (214 mg, 0.85 mmol) and triethylamine (600 μl_) and the reaction heated to 600C. After 20 minutes the reaction was cooled to room temperature and quenched with saturated sodium bicarbonate (10 ml_). The aqueous phase was extracted with ethyl acetate (2 x 15 ml_). The organic phases were combined, dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure. Purification by chromatography (silica gel; ethyl acetate) provided 2-pyridin-3-yl-thiazole-4-carboxylic acid (6-diethylamino-pyridin-3- yl-methyl)-(4-fluorophenyl)-amide as a colorless oil.
Part III: Preparation of 2-pyridin-3-yl-thiazole-4-carboxylic acid (6-diethylamino- pyridin-3-yl-methyl)-(4-fluorophenyl)-amide dihydrochloride
The free base was treated with ethereal hydrochloric acid to provide 2-pyridin-3-yl- thiazole-4-carboxylic acid (6-diethylamino-pyridin-3-yl-methyl)-(4-fluorophenyl)-amide dihydrochloride (110 mg, 0.2 mmol) as a white solid. MS: m/z 462 [M+H]; HPLC C: tr = 3.2 min.
EXAMPLE 27: PREPARATION OF 1 -BENZYL-2-METHYL-1 H-IMIDAZOLE-4-
CARBOXYLIC ACID (4-FLUOROPHENYLH2-PIPERIDIN-1-YL-THIAZOL-4-YL- METHYL)-AMIDE DIHYDROCHLORIDE (COMPOUND NO. 16)

(4-Fluorophenyl)-(2-piperidin-1-yl-thiazol-4-ylmethyl)-amine dihydrochloride (189 mg, 0.58 mmol), 1-benzyl-2-methyl-1 H-imidazole-4-carboxylic acid trifluoroacetate (229
mg, 0.69 mmol), and 1-[3-(dimethylamino)propyl]-3-ethyl-carbodiimide hydrochloride (222 mg, 1.16 mmol) were combined in pyridine (2 mL) and the mixture was stirred overnight. The reaction mixture was cooled and evaporated to dryness. The residue was partitioned between 1 N sodium hydroxide (10 mL) and dichloromethane (20 mL). The aqueous phase was separated and washed with dichloromethane (10 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure. Purification by reverse phase chromatography and isolation of the free base provided 1-benzyl-2-methyl-1 H- imidazole-4-carboxylic acid (4-fluorophenyl)-(2-piperidin-1 -yl-thiazol-4-yl-methyl)- amide.
The free base was treated with ethereal hydrochloric acid to provide 1-benzyl-2- methyl-1 H-imidazole-4-carboxylic acid (4-fluorophenyl)-(2-piperidin-1-yl-thiazol-4-yl- methyl)-amide dihydrochloride (87 mg, 0.15 mmol) as a white solid.
EXAMPLE 28: PREPARATION OF 1-BENZYL-2-METHYL-1 H-IMIDAZOLE-4- CARBOXYLIC ACID [6-(CYCLOPROPYL-ETHYL-AMINO)-PYRIDIN-S-YL-METHYL]- (4-FLUOROPHENYL)-AMIDE DIHYDROCHLORIDE (COMPOUND NO. 17)

Cyclopropyl-{5-[(4-fluorophenylamino)-methyl]-pyridin-2-yl}-ethyl-amine dihydrochloride (120 mg, 0.37 mmol), 1-benzyl-2-methyl-1 H-imidazole-4-carboxylic acid trifluoroacetate (75 mg, 0.37 mmol), and 1-[3-(dimethylamino)propyl]-3-ethyl- carbodiimide hydrochloride (96 mg, 0.5 mmol) were combined in pyridine (5 mL). The mixture was heated to 60°C and stirred overnight. The reaction mixture was cooled and evaporated to dryness. The residue was partitioned between 1 N sodium hydroxide (10 mL) and dichloromethane. The aqueous phase was separated and washed with dichloromethane (10 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced
pressure to provide a yellow oil. Flash chromatography (silica gel; 5% methanol in dichloromethane) provided 1-benzyl-2-methyl-1 H-imidazole-4-carboxylic acid [6- (cyclopropyl-ethyl-amino)-pyridin-3-yl-methyl]-(4-fluorophenyl)-amide.
The free base was treated with ethereal hydrochloric acid to provide 1-benzyl-2- methyl-1 H-imidazole-4-carboxylic acid [6-(cyclopropyl-ethyl-amino)-pyridin-3-yl- methyl]-(4-fluorophenyl)-amide dihydrochloride as a white solid (32 mg, 0.06 mmol).
EXAMPLE 29: PREPARATION OF 2-BENZYL-1-METHYL-1 H-IMIDAZOLE-4-
CARBOXYLIC ACID [6-(CYCLOPROPYL-ETHYL-AMINO)-PYRIDIN-S-YL-METHYL]-
(4-FLUOROPHENYL)-AMIDE DIHYDROCHLORIDE (COMPOUND NO. 18)

Cyclopropyl-{5-[(4-fluorophenylamino)-methyl]-pyridin-2-yl}-ethyl-amine dihydrochloride (111 mg, 0.31 mmol), 2-benzyl-1-methyl-1 H-imidazole-4-carboxylic acid trifluoroacetate (123 mg, 0.37 mmol), and 1-[3-(dimethylamino)propyl]-3-ethyl- carbodiimide hydrochloride (119 mg, 0.62 mmol) were combined in pyridine (2 mL) and stirred overnight. The reaction mixture was cooled and evaporated to dryness. The residue was partitioned between 1 N sodium hydroxide (10 mL) and ethyl acetate (15 mL). The aqueous phase was separated and washed with ethyl acetate (2 x 10 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure. Chromatography (silica gel; 3% methanol in dichloromethane) provided 2-benzyl-1-methyl-1 H-imidazole-4- carboxylic acid [6-(cyclopropyl-ethyl-amino)-pyridin-3-yl-methyl]-(4-fluorophenyl)- amide.
The free base was treated with ethereal hydrochloric acid to provide 2-benzyl-1- methyl-1 H-imidazole-4-carboxylic acid [6-(cyclopropyl-ethyl-amino)-pyridin-3-yl- methyl]-(4-fluorophenyl)-amide dihydrochloride (53 mg, 0.1 mmol) as a white solid.
EXAMPLE 30: PREPARATION OF I-BENZYL-IH-IMIDAZOLE^-CARBOXYLIC
ACID (4-FLUOROPHENYL)-^-PIPERIDIN-I -YL-PYRIMIDIN-S-YL-METHYL)-AMIDE
DIHYDROCHLORIDE (COMPOUND NO. 19)

(4-Fluorophenyl)-(2-piperidin-1-yl-pyrimidin-5-yl-methyl)-amine (135 mg, 0.47 mmol), 1-benzyl-1 H-imidazole-4-carboxylic acid trifluoroacetate (150 mg, 0.47 mmol), and 1- [3-(dimethylamino)propyl]-3-ethyl-carbodiimide hydrochloride (180 mg, 0.94 mmol) were combined in pyridine (4 mL). The mixture was heated to 60°C and stirred overnight. The reaction mixture was cooled and evaporated to dryness. The residue was partitioned between 1 N sodium hydroxide (10 mL) and dichloromethane (20 mL). The aqueous phase was separated and washed with dichloromethane (10 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure to provide a yellow oil. Flash chromatography (silica gel; ethyl acetate to 8% methanol in dichloromethane) provided 1-benzyl-1 H-imidazole-4-carboxylic acid (4-fluorophenyl)-(2-piperidin-1-yl- pyrimidin-5-yl-methyl)-amide (120 mg, 0.26 mmol) as a colorless oil.
The resulting oil was dissolved in diethyl ether (10 mL) and a saturated hydrochloric acid solution in diethyl ether (6 mL) was added. The solvent was removed under reduced pressure to obtain 1-benzyl-1 H-imidazole-4-carboxylic acid (4-fluorophenyl)- (2-piperidin-1-yl-pyrimidin-5-yl-methyl)-amide dihydrochloride (120 mg, 0.24 mmol) as a white solid.
EXAMPLE 31 : PREPARATION OF I-CYCLOPROPYLMETHYL^-METHYL-I H- IMIDAZOLE-4-CARBOXYLIC ACID [6-(CYCLOPROPYL-ETHYL-AMINO)-PYRIDIN- 3-YL-METHYL]-(4-FLUOROPHENYL)-AMIDE DIHYDROCHLORIDE (COMPOUND
NO. 30)

Cyclopropyl-{5-[(4-fluorophenylamino)-methyl]-pyridin-2-yl}-ethyl-amine dihydrochloride (183 mg, 0.51 mmol), 1-cyclopropylmethyl-2-methyl-1 H-imidazole-4- carboxylic acid trifluoroacetate (181 mg, 0.61 mmol), and 1-[3- (dimethylamino)propyl]-3-ethyl-carbodiimide hydrochloride (196 mg, 1.0 mmol) were combined in pyridine (2 mL) and stirred overnight. The reaction mixture was cooled and evaporated to dryness. The residue was partitioned between 1 N sodium hydroxide (10 mL) and ethyl acetate (15 mL). The aqueous phase was separated and washed with ethyl acetate (2 x 10 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure to provide 1-cyclopropylmethyl-2-methyl-1 H-imidazole-4-carboxylic acid (4- fluorophenyl)-(2-piperidin-1-yl-thiazol-5-yl-methyl)-amide.
The free base was treated with ethereal hydrochloric acid to provide 1 -cyclopropyl methyl-2-methyl-1 H-imidazole-4-carboxylic acid [6-(cyclopropyl-ethyl-amino)-pyridin- 3-yl-methyl]-(4-fluorophenyl)-amide dihydrochloride (87 mg, 0.17 mmol) as a white solid.
EXAMPLE 32: PREPARATION OF 1-BENZYL-2-METHYL-1 H-IMIDAZOLE-4-
CARBOXYLIC ACID (4-FLUOROPHENYL)-(2-PIPERIDIN-1-YL-THIAZOL-5-YL-
METHYL)-AMIDE DIHYDROCHLORIDE (COMPOUND NO. 21 )

The free base was treated with ethereal hydrochloric acid to provide 1-benzyl-2- methyl-1 H-imidazole-4-carboxylic acid (4-fluorophenyl)-(2-piperidin-1-yl-thiazol-5-yl- methyl)-amide dihydrochloride (30 mg, 0.5 mmol) as a white solid.
EXAMPLE 33: PREPARATION OF 1-ALLYL-2-METHYL-1 H-IMIDAZOLE-4-
CARBOXYLIC ACID (6-(CYCLOPROPYL-ETHYL-AMINO)-PYRIDIN-S-YL-
METHYL)-(4-FLU0R0PHENYL)-AMIDE DIHYDROCHLORIDE (COMPOUND NO.
22)

Cyclopropyl-iS-^-fluorophenylaminoJ-methyll-pyridin^-ylJ-ethyl-amine dihydrochloride (195 mg, 0.54 mmol), 1-allyl-2-methyl-1 H-imidazole-4-carboxylic acid trifluoroacetate (182 mg, 0.65 mmol), and 1-[3-(dimethylamino)propyl]-3-ethyl- carbodiimide hydrochloride (208 mg, 1.1 mmol) were combined in pyridine (2 mL) and stirred overnight. The reaction mixture was cooled and evaporated to dryness. The residue was partitioned between 1 N sodium hydroxide (10 mL) and dichloromethane (15 mL). The aqueous phase was separated and washed with dichloromethane (2 x 10 mL). The organic phases were combined, dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure to provide a brown oil. Purification by reverse phase chromatography and isolation of the free base provided 1-allyl-2-methyl-1 H-imidazole-4-carboxylic acid [6- (cyclopropyl-ethyl-amino)-pyridin-3-yl-methyl]-(4-fluorophenyl)-amide trifluoroacetate.
The free base was treated with ethereal hydrochloric acid to provide 1 -allyl-2-methyl- 1 H-imidazole-4-carboxylic acid [6-(cyclopropyl-ethyl-amino)-pyridin-3-yl-methyl]-(4- fluorophenyl)-amide dihydrochloride (116 mg, 0.23 mmol) as a gummy solid.
EXAMPLE 34: PREPARATION OF 1-BENZYL-2-METHYL-1 H-IMIDAZOLE-4-
CARBOXYLIC ACID (6-DIETHYLAMINO-PYRIDIN-2-YL-METHYL)-(4- FLUOROPHENYL)-AMIDE DIHYDROCHLORIDE (COMPOUND NO. 23)
l ne



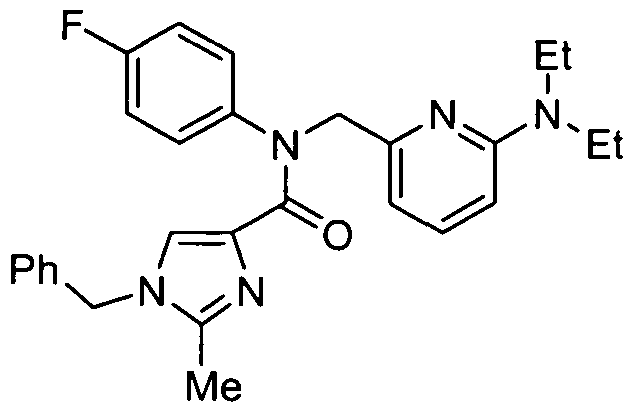
To a suspension of i-benzyl^-methyl-I H-imidazole^-carboxylic acid trifluoroacetate (120 mg, 0.56 mmol) in anhydrous tetrahydrofuran (4 mL) was added 2-chloro-4,6- dimethoxy-1 ,3,5-triazine (118 mg, 0.67 mmol) and N-methylmorpholine (187 μL, 1.7 mmol) and the reaction stirred for 1 hour. To the reaction was added diethyl-{6-[(4- fluorophenylamino)-methyl]-pyridin-2-yl}-amine dihydrochloride (167 mg, 0.56 mmol). The reaction was heated to reflux and allowed to stir overnight. The reaction was cooled to room temperature and partitioned between ethyl acetate (20 mL) and saturated sodium bicarbonate (10 mL). The organic phase was separated, dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure. Purification by chromatography (silica gel; 10-20% methanol in ethyl acetate with 1% ammonium hydroxide) provided 1-benzyl-2-methyl-1H-imidazole-4- carboxylic acid (6-diethylamino-pyridin-2-yl-methyl)-(4-fluorophenyl)-amide.
The free base was treated with ethereal hydrochloric acid to provide 1-benzyl-2- methyl-1 H-imidazole-4-carboxylic acid (6-diethylamino-pyridin-2-yl-methyl)-(4- fluorophenyl)-amide dihydrochloride (70 mg, 0.13 mmol) as a white solid.
EXAMPLE 35: PREPARATION OF 1 -(2,2-DIMETHYL-PROPYL)-I H-IMIDAZOLE^- CARBOXYLIC ACID [4-(CYCLOPROPYL-ETHYL-AMINO)-BENZYL]-(^ FLUOROPHENYL)-AMIDE DIHYDROCHLORIDE (COMPOUND NO. 24)

The free base was treated with ethereal hydrochloric acid to provide 1-(2,2-dimethyl- propyl)-1 H-imidazole-4-carboxylic acid [4-(cyclopropyl-ethyl-amino)-benzyl]-(4- fluorophenyl)-amide dihydrochloride (400 mg, 0.76 mmol) as a colorless oil.
EXAMPLE 36: PREPARATION OF 1 -(2,2-DIMETHYL-PROPYL)-I H-IMIDAZOLE^- CARBOXYLIC ACID [3-(CYCLOPROPYL-ETHYL-AMINO)-BENZYL]-(^ FLUOROPHENYL)-AMIDE DIHYDROCHLORIDE (COMPOUND NO. 25)

The free base was treated with ethereal hydrochloric acid to provide 1-(2,2-dimethyl- propyl)-1 H-imidazole-4-carboxylic acid [3-(cyclopropyl-ethyl-amino)-benzyl]-(4- fluorophenyl)-amide dihydrochloride (235 mg, 0.45 mmol) as a foam.
EXAMPLE 37: 1-BENZYL-N-((2-(PYRROLIDIN-1-YL)THIAZOL-4-YL)METHYL)-N-P- TOLYL-1 H-IMIDAZOLE^-CARBOXAMIDE (COMPOUND NO. 14)
Step 1. i-benzyl-N-p-tolyl-I H-imidazole-4-carboxamide
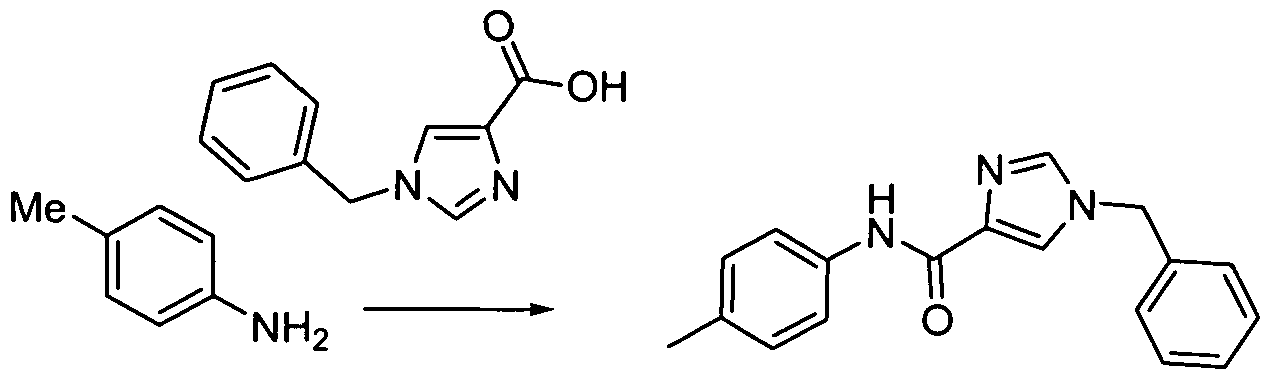
To a solution of 4-methylaniline (0.2 g, 1.86 mmol) in THF (5 mL), triethylamine (0.52 ml, 3.73 mmol) followed by DCC (0.42 g, 2.03 mmol), HOBt (0.26 g, 1.92 mmol) and i-benzyl-I H-imidazole-4-carboxylic acid were added at room temperature, and the reaction mixture was stirred for overnight under nitrogen atmosphere. The reaction mass was filtered and the filtrate was concentrated under reduced pressure. The residue was dissolved in ethyl acetate and washed with plenty of water. The organic layer was dried over sodium sulphate and concentrated under reduced pressure. The mixture was purified by column chromatography using 25% ethyl acetate in pet ether as eluent, to afford the product, 50% yield.
Step 2. 1-benzyl-N-((2-chlorothiazol-4-yl)methyl)-N-p-tolyl-1 H-imidazole-4- carboxamide

To a solution of i-benzyl-N-p-tolyl-I H-imidazole-4-carboxamide (0.2 g, 0.68 mmol) in anhydrous tetrahydrofuran (4 mL), was added sodium hydride (0.03 g, 1.25 mmol). After the initial gas evolution had ceased, the reaction mixture was allowed to stir for 15 minutes. tetra-n-Butyl ammonium iodide (0.05 g, 0.137 g) was then added followed by addition of 2-chloro-5-chloromethyl thiazole (0.05 g, 0.137 mmol). The mixture was heated to reflux for 4 h. After cooling, the reaction was carefully
neutralized with cold saturated sodium bicarbonate and extracted with ethyl acetate. The organic layer was washed with water, dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure to provide yellow solid. The residue was purified by column chromatography over silica gel (60-120 mesh) using 5 % methanol in ethyl acetate as eluent to afford the product (40 % yield).
Step 3. 1 -benzyl-N-((2-(pyrrolidin-1 -yl)thiazol-4-yl)methyl)-N-p-tolyl-1 H-imidazole-4- carboxamide

A solution of 1-benzyl-N-((2-chlorothiazol-4-yl)methyl)-N-p-tolyl-1 H-imidazole-4- carboxamide (0.1 g, 0.236 mmol) in DMF (4 ml_), pyrrolidine (0.077 mL) was added at rt and the mixture heated to 110 0C and stirred overnight. The DMF was distilled under reduced pressure and the residue was partitioned between water and ethyl acetate. The organic layer was separated, dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure. The residue was purified by column chromatography over silica gel using 10% methanol in ethyl acetate as eluent, to afford the product (35 % yield).
EXAMPLE 38: PREPARATION OF 1-BENZYL-1 H-[1 ,2,3]TRIAZOLE-4- CARBOXYLIC ACID (6-DIETHYL^MINO-PYRIDIN-3-YLMETHYL)-(4- FLUOROPHENYL)-AMIDE HYDROCHLORIDE (COMPOUND NO. 86

Treatment of the free base with ethereal hydrochloric acid provided 1-benzyl-1 H- [1 ,2,3]triazole-4-carboxylic acid (6-diethylamino-pyridin-3-ylmethyl)-(4-fluorophenyl)- amide hydrochloride (397 mg, 0.88 mmol) as a white solid.
EXAMPLE 39: PREPARATION OF 1-BENZYL-1 H-[1 ,2,3]TRIAZOLE-4- CARBOXYLIC ACID (4-FLUOROPHENYLH2-PIPERIDIN-1-YL-THIAZOL-4- YLMETHYL)-AMIDE HYDROCHLORIDE (COMPOUND NO. 87

To a solution of 1-benzyl-1 H-[1 ,2,3]triazole-4-carboxylic acid (1.6 g, 8.04 mmol), 1-[3- (dimethylamino)propyl]-3-ethyl-carbodiimide hydrochloride (1.85 g, 9.65 mmol) and a catalytic amount of 4-dimethylaminopyridine in chloroform (32 ml.) was added 4- fluoroaniline (0.9 mL, 9.65 mmol) and the reaction was allowed to stir overnight. The reaction was treated with saturated sodium bicarbonate (10 mL) and extracted with ethyl acetate (2 x 10 mL). The organic phases were combined, washed with brine (10 mL), dried over sodium sulfate, filtered and the solvent removed under reduced pressure. Purification by chromatography (silica gel; 5% ethyl acetate in dichloromethane) provided 1-benzyl-1 H-[1 ,2,3]triazole-4-carboxylic acid (4- fluorophenyl)-amide (1.41 g, 4.8 mmol) as a white solid.
Part II: Preparation of 1-benzyl-1 H-[1 ,2,3]triazole-4-carboxylic acid (4-fluorophenyl)- (2-piperidin-1 -yl-thiazol-4-ylmethyl)-amide hydrochloride
To a suspension of sodium hydride (60% dispersion in mineral oil, 55 mg, 1.38 mmol) in anhydrous tetrahydrofuran (5 mL) was added a solution of 1-benzyl-1 H- [1 ,2,3]triazole-4-carboxylic acid (4-fluorophenyl)-amide (375 mg, 1.27 mmol) in tetrahydrofuran (5 mL). After the addition was complete, dimethylformamide (1 mL) was added and vigorous gas evolution occurred. After 15 minutes, tetra-n- butylammonium iodide (189 mg, 0.5 mmol) and 1-(4-chloromethyl-thiazol-2-yl)- piperidine (250 mg, 1.15 mmol) were added to the reaction. The mixture was heated to 600C and allowed to stir overnight. After cooling, the reaction was quenched with water and the organics removed under reduced pressure. The residue was diluted with saturated sodium bicarbonate (10 mL) and extracted with ethyl acetate (2 x 20 mL). The organic layers were combined, washed with brine (10 mL), dried over anhydrous sodium sulfate, filtered and the solvent removed under reduced pressure to provide an oil. Flash chromatography (silica gel; 10% ethyl acetate in dichloromethane) provided 1-benzyl-1 H-[1 ,2,3]triazole-4-carboxylic acid (4- fluorophenyl)-(2-piperidin-1-yl-thiazol-4-ylmethyl)-amide (284 g, 0.66 mmol) as a white solid.
Treatment of the free base with ethereal hydrochloric acid provided 1-benzyl-1 H- [1 ,2,3]triazole-4-carboxylic acid (4-fluorophenyl)-(2-piperidin-1-yl-thiazol-4-ylmethyl)- amide hydrochloride (225mg, 0.44 mmol) as a white solid.
EXAMPLE 40: PREPARATION OF 1-BENZYL-1 H-[1 ,2,3]TRIAZOLE-4-
CARBOXYLIC ACID (4-FLUOROPHENYL)-^-PIPERIDIN-I -YL-THIAZOL-S-
YLMETHYL)-AMIDE (COMPOUND NO. 88

1-Benzyl-1 H-[1 ,2,3]triazole-4-carbonyl chloride (199 mg, 0.9 mmol) was dissolved in tetrahydrofuran (2 mL) and added dropwise to a suspension of (4-fluorophenyl)-(2- piperidin-1-yl-thiazol-5-ylmethyl)-amine dihydrochloride (251 mg, 0.9 mmol) in tetrahydrofuran (6 mL) followed by the addition of triethyl amine (261 μL, 2.6 mmol). The reaction was heated to 6O0C and stirred for 30 minutes followed by cooling to room temperature. The reaction was neutralized with saturated sodium bicarbonate (4 mL) and extracted with ethyl acetate (4 mL). The organic phases were combined, dried over sodium sulfate, filtered and the solvent removed under reduced pressure to provide a white solid. The solid was triturated with 9:1 hexane:ethyl acetate, followed by suspension in methanol (2 mL). The mixture was treated with excess ethereal hydrochloric acid to provide a clear solution. The reaction was concentrated under reduced pressure to provide 1-benzyl-1-H[1 ,2,3]triazole-4-carboxylic acid (4- fluorophenyl)-(piperidin-1-yl-thiazol-5-ylmethyl)-amide hydrochloride as a white foam (300 mg, 0.6 mmol). MS (base): m/z 477 [M+H]; HPLC C (base): tr = 3.1 min.
EXAMPLE 41 : PREPARATION OF 1-BENZYL-1H-[1 ,2,3]TRIAZOLE-4- CARBOXYLIC ACID (4-DIETHYLAMINO-BENZYL)-(4-FLUOROPHENYL)-AMIDE
(COMPOUND NO. 89

Treatment of the free base with ethereal hydrochloric acid provided 1-benzyl-1 H- [1 ,2,3]triazole-4-carboxylic acid (4-diethylamino-benzyl)-(4-fluorophenyl)-amide hydrochloride (680 mg, 1.38 mmol) as a white solid.
EXAMPLE 42: PREPARATION OF 1-BENZYL-1 H-[1 ,2,3]TRIAZOLE-4- CARBOXYLIC ACID (4-FLUOROPHENYL)-^-PIPERIDIN-I -YL-PYRIMIDIN-S- YLMETHYL)-AMIDE (COMPOUND NO. 90

EXAMPLE 43: 1-BENZYL-N-(4-FLUOROPHENYL)-N-[2-(2-MORPHOLIN-4-YL-1 ,3- THIAZOL-4-YL)ETHYL]-1 H- 1 ,2,3-TRIAZOLE-4-CARBOXAMIDE (COMPOUND NO.
83

LiAIH4 diethylether

Part I: Preparation of ethyl (2-morpholin-4-yl-1 ,3-thiazol-4-yl)acetate
A mixture of ethylmalonylchloride (4.76 mL, 35 mmol) and morpholine-4-carbothioic acid amide (5.0 g, 34.2 mmol) in ethanol (40 mL) was heated to 80 0C for 3 hours. The mixture was cooled, ethanol evaporated, and diluted with saturated sodium bicarbonate, and extracted with ethyl acetate. The organic layers were dried over magnesium sulfate and the reaction mixture purified by chromatography (silica gel; ethylacetate/hexane, 1 :1 ) to provide ethyl (2-morpholin-4-yl-1 ,3-thiazol-4-yl)acetate (9 g, quantitative)
Part II: Preparation of N-(4-fluorophenyl)-2-(2-morpholin-4-yl-1 ,3-thiazol-4- yl)acetamide
A mixture of ethyl (2-morpholin-4-yl-1 ,3-thiazol-4-yl)acetate (1.0 g, 4.65 mmol) and A- fluoroaniline(0.55 mL, 5.8 mmol) was dissolved in toluene (13 ml_) at room temperature. 2M Trimethylaluminum (2.5 mL, 5 mmol) was added dropwise, and the mixture was heated to 60 0C for 3 hours. The mixture was cooled, quenched with methanol, and the solvent evaporated under reduced pressure. The residue was partitioned between saturated sodium bicarbonate and ethyl acetate. The organic layer was collected, washed with saturated potassium-sodium tartrate, dried over magnesium sulfate, and concentrated. The residue was purified by chromatography (silica gel; 10%-50% ethylacetate/hexane, gradient elution) to provide N-(4-fluorophenyl)-2-(2-morpholin-4-yl-1 ,3-thiazol-4-yl)acetamide (0.93Og, 62%).
Part III: Preparation of 4-fluoro-N-[2-(2-morpholin-4-yl-1 ,3-thiazol-4-yl)ethyl]aniline
To a mixture of N-(4-fluorophenyl)-2-(2-morpholin-4-yl-1 ,3-thiazol-4-yl)acetamide (0.90Og, 2.8 mmol) in anhydrous diethylether (30 mL) at 0 0C was slowly added 1 M lithium aluminum hydride (5 mL). The reaction was allowed to warm to room temperature and stirring continued 7 hours. The mixture was quenched sequentially with water (0.2 mL), 2N NaOH (.2 mL), and water (0.6 mL). The mixture was filtered through celite and the filtrate concentrated. The residue was purified by chromatography (silica gel; 20%-50% ethylacetate/hexane, gradient elution) to provide 4-fluoro-N-[2-(2-morpholin-4-yl-1 ,3-thiazol-4-yl)ethyl]aniline (0.52Og, 60%).
Part IV: Preparation of 1-benzyl-N-(4-fluorophenyl)-N-[2-(2-morpholin-4-yl-1 ,3- thiazol-4-yl)ethyl]-1 H- 1 ,2,3-triazole-4-carboxamide
To a mixture of 4-fluoro-N-[2-(2-morpholin-4-yl-1 ,3-thiazol-4-yl)ethyl]aniline (0.100g, 0.325 mmol), N-methylmorpholine (0.071 mL, 0.65 mmol), and 1-benzyl-1 H-1 ,2,3- triazole-4-carboxylic acid (0.132g, .65 mmol) in anhydrous tetrahydrofuran (2.5 mL)
at room temperature was added 2-chloro-4,6-dimethoxytriazine (0.114 g, 0.65 mmol). The reaction was allowed to stir 72 hours. The reaction was partitioned between saturated sodium bicarbonate and ethyl acetate. The organic layer was collected, dried over magnesium sulfate, and concentrated. The residue was purified by chromatography (silica gel; 30%-100% ethylacetate/hexane, gradient elution) to provide 1-benzyl-N-(4-fluorophenyl)-N-[2-(2-morpholin-4-yl-1 ,3-thiazol-4- yl)ethyl]-1 H- 1 ,2,3-triazole-4-carboxamide (0.13Og, 0.265 mmol).
The resulting oil was dissolved in diethylether and 1 N HCI/diethylether was added until precipitation stopped. The solvent was removed under reduced pressure to obtain 1 -benzyl-N-(4-fluorophenyl)-N-[2-(2-morpholin-4-yl-1 ,3-thiazol-4-yl)ethyl]-1 H- 1 ,2,3-triazole-4-carboxamide hydrochloride (0.13Og, 76%).
EXAMPLE 44: Λ/-(4-FLUOROPHENYL)-1-METHYL-Λ/-{[6-
(TRIFLUOROMETHYL)PYRIDIN-S-YL]METHYL)-I H-PYRROLE^-CARBOXAMIDE
(COMPOUND NO. 149

Part 1 : To a stirred solution of 6-(trifluoromethyl)pyridine-3-carboxaldehyde (1.0 g, 5.71 mmol) and 4-fluoroaniline (0.64 g, 5.71 mmol) in dichloromethane (2 mL) was added acetic acid (1.2 mL) and the resulting solution was stirred overnight at room temperature. Sodium thacetoxyborohydride (2.4 g, 1 1.4 mmol) was added and the solution was stirred an additional hour. The reaction was quenched with 1 N NaOH solution and extracted with dichloromethane. The organic layer was concentrated and flash column separation using 0-20% ethyl acetate/ hexane to give 4-fluoro-N- {[6-(trifluoromethyl)pyridin-3-yl]methyl}aniline as an oil. (0.98 g, 64%)
Part 2: To a stirred solution of i-methyl-2-pyrrolecarboxylic acid (0.05 g, 0.40 mmol) in dichloromethane (2 ml.) was added oxalyl chloride (0.35 ml_, 4.0 mmol) and the resulting mixture was stirred overnight at room temperature. The mixture was then concentrated, redissolved in chloroform (3 mL) and reconcentrated. This crude material was dissolved in THF (2 mL) and to it was added triethylamine (0.15 mL, 1.0 mmol) and 4-fluoro-N-{[6-(trifluoromethyl)pyridin-3-yl]methyl}aniline (0.09 g, 0.33 mmol). The resulting solution was stirred overnight, washed with water and concentrated. Flash column separation using 0-30% ethyl acetate/ hexane gave Λ/- (4-fluorophenyl)-1-methyl-Λ/-{[6-(trifluoromethyl)pyridin-3-yl]methyl}-1 /-/-pyrrole-2- carboxamide as an oil (0.096 g, 77%)
EXAMPLE 45: Λ/-({2-[CYCLOPROPYL(ETHYL)AMINO]-1 ,3-THIAZOL-4- YL}METHYL)-Λ/-1 H-INDOL-5-YLNICOTINAMIDE (COMPOUND NO. 253)
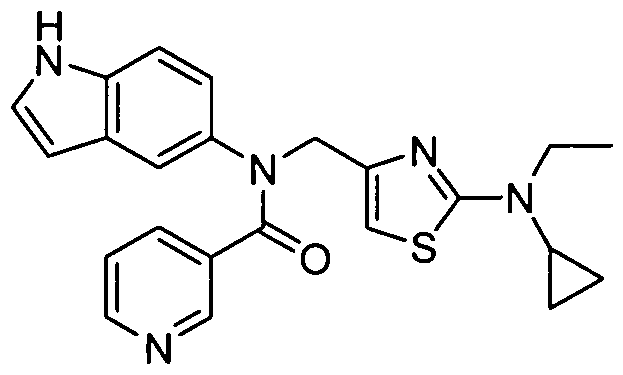
Part 1 : To a stirred solution of 2-[cyclopropyl(ethyl)amino]-1 ,3-thiazole-4- carbaldehyde (0.19 g, 0.97 mmol) and 5-aminoindole (0.13 g, 1.00 mmol) in THF (3 mL) was added acetic acid (0.1 mL) and the resulting solution was stirred overnight at room temperature. Sodium triacetoxyborohydride (0.41 g, 1.95 mmol) was added and the solution was stirred an additional hour. The reaction was quenched with sat. sodium bicarbonate solution and extracted with ethyl acetate. The organic layer was concentrated and flash column separation using 10-50% ethyl acetate / hexane gave
N-({2-[cyclopropyl(ethyl)amino]-1 ,3-thiazol-4-yl}methyl)-1 H-indol-5-amine as a white solid. (0.21 g, 56%)
Part 2: To a stirred solution of N-({2-[cyclopropyl(ethyl)amino]-1 ,3-thiazol-4- yl}methyl)-1 H-indol-5-amine (0.06 g, 0.16 mmol) and triethylamine (0.2 mL, 1.40 mmol) in THF (3 mL) was added nicotinoyl chloride hydrochloride (0.028 g, 0.15
mmol) and the resulting solution was stirred several hours at room temperature. The reaction as washed with water and concentrated. Flash column separation using 30- 100% ethyl acetate/ hexane gave Λ/-({2-[cyclopropyl(ethyl)amino]-1 ,3-thiazol-4- yl}methyl)-Λ/-1H-indol-5-ylnicotinamide as a white solid. (0.049 g, 62%).
EXAMPLE 46: Λ/-{[6-(4-ETHYLPIPERAZIN-1-YL)PYRIDIN-3-YL]METHYL}-Λ/-(4- FLUOROPHENYL)NICOTINAMIDE (COMPOUND NO. 191

To a stirred solution of N-[(6-chloropyridin-3-yl)methyl]-N-(4- fluorophenyl)nicotinamide (0.10 g, 0.29 mmol) in THF (0.5 mL) was added N- ethylpiperizine (0.4 mL, 3.15 mmol) and the resulting solution was microwave irradiated to 1700C for 90 minutes. The crude mixture was concentrated and flash column separation using 0-10% methanol/ methylene chloride gave Λ/-{[6-(4- ethylpiperazin-1-yl)pyridin-3-yl]methyl}-Λ/-(4-fluorophenyl)nicotinamide as an off-white solid. (0.014 g, 12%).
EXAMPLE 47 Λ/-(4-FLUOROPHENYL)-Λ/-{[2-(4-METHYLPIPERAZIN-1-YL)-1 ,3-
THIAZOL-4-YL]METHYL}NICOTINAMIDE (COMPOUND NO. 207

Preparation of Λ/-(4-fluorophenyl)-Λ/-{[2-(4-methylpiperazin-1-yl)-1 ,3-thiazol-4- yl]methyl}nicotinamide
To a stirred solution of N-[(2-bromo-1 ,3-thiazol-4-yl)methyl]-N-(4- fluorophenyl)nicotinamide (0.10 g, 0.26 mmol) in THF (0.5 mL) was added N- methylpiperizine (0.3 mL, 2.7 mmol) and the resulting solution was microwave irradiated to 1500C for 10 minutes. The resulting mixture was concentrated and flash column separation using 0-5% methanol/ methylene chloride gave Λ/-(4- fluorophenyl)-Λ/-{[2-(4-methylpiperazin-1 -yl)-1 ,3-thiazol-4-yl]methyl}nicotinamide as a white solid. (0.103 g, 77%)
EXAMPLE 48 Λ/-[(6-CHLOROPYRIDIN-3-YL)METHYL]-/v- CYCLOPENTYLNICOTINAMIDE (COMPOUND NO. 217

Preparation of Λ/-[(6-chloropyridin-3-yl)methyl]-Λ/-cyclopentylnicotinamide
Part 1 : To a stirred solution of 6-chloronicotinaldehyde (1.0 g, 7.0 mmol) and cyclopentylamine (0.7 mL, 7.0 mmol) in dichloromethane (25 mL) was added acetic acid (1.2 mL) and the resulting solution was stirred overnight at room temperature. Sodium triacetoxyborohydride (3.0 g, 14.12 mmol) was added and the solution was stirred an additional hour. The reaction was quenched with sat. sodium bicarbonate solution and extracted with dichloromethane. The organic layer was concentrated and flash column separation using 0-10% methanol/ methylene chloride gave N-[(6- chloropyridin-3-yl)methyl]cyclopentanamine as a clear oil. (0.89 g, 60%).
Part 2: To a stirred solution of N-Ke-chloropyridin-S-yOmethylJcyclopentanamine (0.25 g, 1.18 mmol) and triethylamine (0.5 mL, 3.58 mmol) in THF (3 mL) was added nicotinoyl chloride hydrochloride (0.27 g, 1.52 mmol) and the resulting solution was stirred several hours at room temperature. The reaction as washed with water and concentrated. Flash column separation using 0-80% ethyl acetate/ hexane gave N-
[(6-chloropyridin-3-yl)methyl]-Λ/-cyclopentylnicotinamide as a white solid. (0.30 g, 81%)
EXAMPLE 49: Λ/-[(6-ACETYLPYRIDIN-3-YL)METHYL]-Λ/-(4- FLUOROPHENYL)NICOTINAMIDE (COMPOUND NO. 234

To a stirred solution of N-[(6-chloropyridin-3-yl)methyl]-N-(4- fluorophenyl)nicotinamide (1.15 g, 3.36 mmol) in toluene (22 mL) was added tributyl(i-ethoxyvinyl) tin (1.15 mL, 3.45 mmol) and tetrakis(triphenylphosphine) palladium (0) (0.19 g, 0.16 mmol) and the resulting solution was heated to 1000C overnight. The mixture was partitioned between 0.5 N NaOH solution and ethyl acetate. The organic phase was concentrated. The crude organic was dissolved in THF (15 mL) and 2N HCI solution (7 mL) was added. This mixture was stirred overnight, neutralized with 1 N NaOH solution and extracted with ethyl acetate. Flash column separation using 10-100% ethyl acetate/ hexane gave Λ/-[(6-acetylpyridin-3- yl)methyl]-Λ/-(4-fluorophenyl)nicotinamide as a white solid. (0.81 g, 69 %)
EXAMPLE 50: Λ/-(4-FLUOROPHENYL)-Λ/-{[6-(TRIFLUOROMETHYL)PYRIDIN-3- YL]METHYL}PYRIDINE-2-CARBOXAMIDE (COMPOUND NO. 235

EXAMPLE 51 : Λ/-(4-FLUOROPHENYL)-Λ/-{[6-(1-HYDROXY-1- METHYLETHYL)PYRIDIN-S-YL]METHYLJNICOTINAMIDE (COMPOUND NO. 244

To a stirred solution of Λ/-[(6-acetylpyridin-3-yl)methyl]-Λ/-(4- fluorophenyl)nicotinamide (0.08 g, 0.23 mmol) in THF (2 mL) at 00C was added methyl magnesium bromide (3M in ethyl ether, 0.1 mL, 0.3 mmol). The resulting solution was allowed to warm to room temperature and stirred one hour. The reaction was quenched with water and extracted with ethyl acetate. The organic layer was concentrated and flash column separation using 20-100% ethyl acetate/ hexane gave Λ/-(4-fluorophenyl)-Λ/-{[6-(1 -hydroxy- 1 -methylethyl)pyridin-3- yl]methyl}nicotinamide as an off-white solid. (0.021 g, 25%).
EXAMPLE 52: Λ/-(4-FLUOROPHENYL)-Λ/-{[6-(2,2,2-
TRIFLUOROETHOXY)PYRIDIN-S-YL]METHYL)NICOTINAMIDE (COMPOUND NO.
247

Step 2: To a stirred solution of 2,2,2-trifluoroethanol (0.42 g, 4.2 mmol) in DMF (3 mL) at 00C was added sodium hydride (0.17 g, 4.20 mmol) and the resulting solution was stirred 15 minutes. To this was added N-[(6-chloropyridin-3-yl)methyl]-4- fluoroaniline (0.20 g, 0.85 mmol) and the resulting solution was heated over 3 days at 60°C. The reaction was allowed to cool, diluted with water and extracted several times with ethyl acetate. The combined organic phase was concentrated and flash column separation using 0-20% ethyl acetate/ hexane gave 4-fluoro-N-((6-(2,2,2- trifluoroethoxy)pyridin-3-yl)methyl)aniline as an oil. (0.16 g, 65%)
Step 3: To a stirred solution of 4-fluoro-N-((6-(2,2,2-trifluoroethoxy)pyridin-3- yl)methyl)aniline (0.08 g, 0.27 mmol) and triethylamine (0.2 mL, 1.4 mmol) in dichloromethane (2 mL) was added nicotinoyl chloride hydrochloride (0.06 g, 0.34 mmol) and the resulting solution was stirred several hours at room temperature. The reaction as washed with 1 N NaOH and concentrated. Flash column separation using
0-60% ethyl acetate/ hexane gave Λ/-(4-fluorophenyl)-Λ/-{[6-(2,2,2- trifluoroethoxy)pyridin-3-yl]methyl}nicotinamide as a white solid. (0.062 g, 53%)
EXAMPLE 53: Λ/-(4-FLUOROPHENYL)-Λ/-{[6-(TRIFLUOROMETHYL)PYRIDIN-3- YL]METHYL}PYRIDAZINE-4-CARBOXAMIDE (COMPOUND NO. 261

To a stirred solution of 4-pyridazinecarboxylic acid (0.05 g, 0.40 mmol) and DMAP (0.049 g, 0.40 mmol) in dichloromethane (2 mL) was added EDC (0.082 g, 0.43 mmol) and the resulting solution was stirred room temperature. After 20 minutes, a solution of 4-fluoro-N-{[6-(trifluoromethyl)pyridin-3-yl]methyl}aniline (0.09g, 0.33 mmol) in dichloromethane (2 mL) was added and the combined solution was stirred overnight at room temperature. The mixture was then washed with 1.0N NaOH solution and concentrated. Flash column separation using 10-100% ethyl acetate/ hexane gave Λ/-(4-fluorophenyl)-Λ/-{[6-(trifluoromethyl)pyridin-3-yl]methyl}pyridazine- 4-carboxamide as an oil. (0.075 g, 60%).
EXAMPLE NO. 54 N-((6-BROMOPYRIDIN-3-YL)METHYL)-N-(4-
FLUOROPHENYL)PYRIDAZINE^-CARBOXAMIDE (COMPOUND NO. 304

10 TABLE 8A










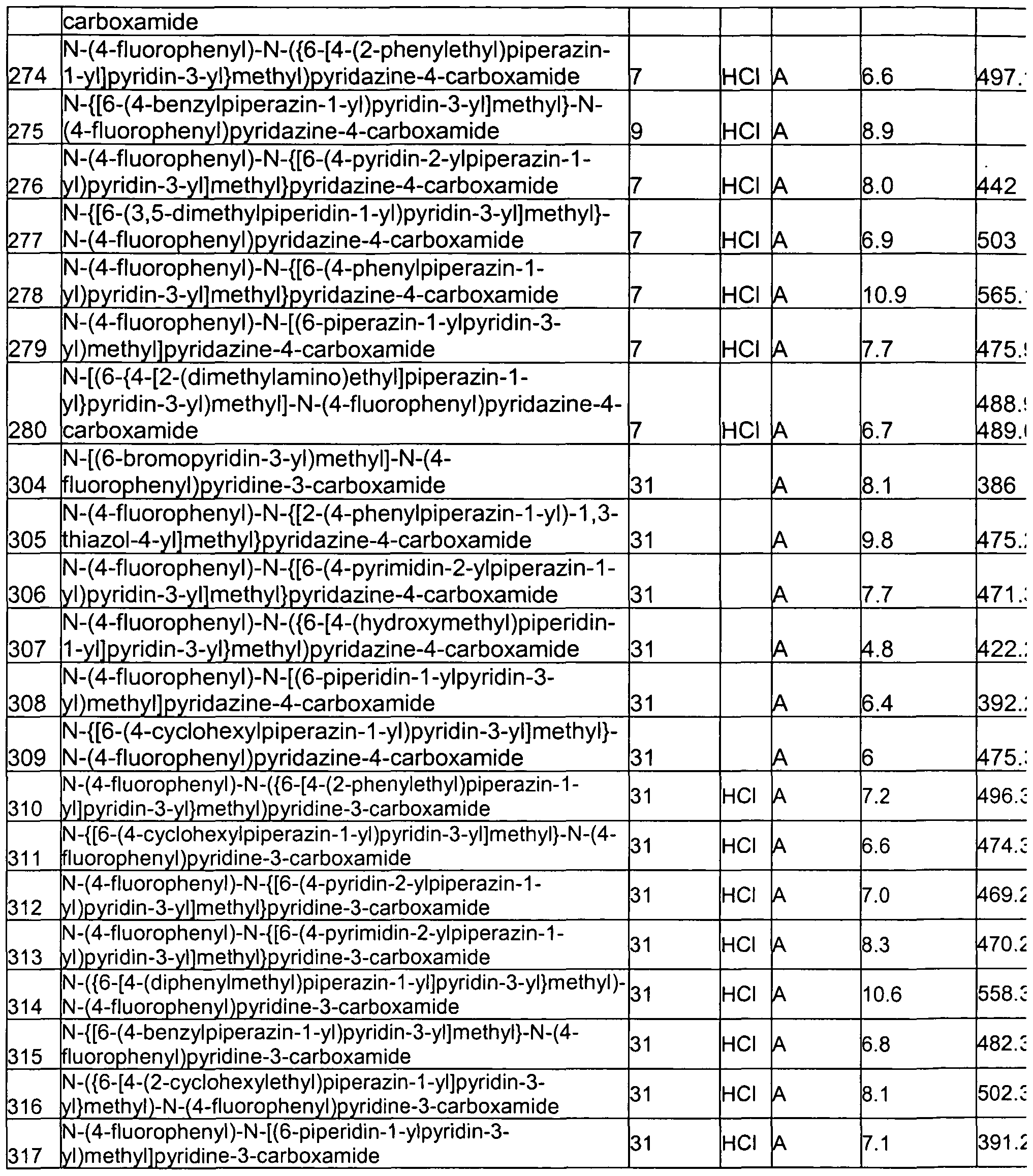

EXAMPLE 55: PHARMACOLOGICAL TESTING
Representative compounds of formulas (I) to (VII) were screened for activity against calcium channel targets in several standard pharmacological test procedures. Based on the activity shown in the standard pharmacological test procedures, the compounds of the present teachings can be useful as ion channel modulators.
Oocyte Assay
This assay was essentially performed as described in Lin et al. (1997), Neuron 18(11 ): 153-166; Pan J. and Lipsombe D. (2000), J. Λ/eurosc/.,20(13): 4768-75; and Xu W. and Lipscombe D. (2001), J. Neurosci., 21(16): 5944-5951 , the entire disclosures of which are herein incorporated by reference, using Xenopus oocyte heterologeous expression system. The assay was performed on various calcium channels (e.g., Cav2.2 subfamily) whereby the modulation of the calcium channel was measured for each tested compound. For measuring compound potency on Cav2.2, a train of five depolarizing pulses of 20-30 ms to about +10 mV was applied at a frequency of 5 Hz from a holding potential of -100 mV every 30 seconds. The 50% inhibitory concentration (IC50) of the test compounds was calculated by measuring the current obtained at the fifth pulse (P5).
HEK Assay
HEK-293T/17 cells were transiently transfected in a similar manner as described in FuGENE 6 Package Insert Version 7, April 2002, Roche Applied Science, Indianapolis, IN. The cells were plated at 2.5 x 105 cells in 2 mL in a 6-well plate, incubated for one night, and achieved a -30-40% confluence. In a small sterile tube, sufficient serum-free medium was added as diluent for FuGENE Transfection Reagent (Roche Applied Science, Indianapolis, IN) to a total volume of 100 μl_. To this medium was added 3 μl_ of FuGENE 6 Reagent. The mixture was tapped gently to mix. To the prediluted FuGENE 6 Reagent was added 2 μg of DNA solution (0.8- 2.0 μg/μL). The DNA/Fugene 6 mixture was gently pipeted to mix the contents and incubated for about 15 minutes at room temperature. The complex mixture was then added to the HEK-293T/17 cells, distributed around the well, and swirled to ensure even dispersal. The cells were returned to the incubator for 24 hours. The transfected cells were then replated at density 2.5 x 105 in a 35 mm dish with 5 glass coverslips and grew in low serum (1%) media for 24 hours. Coverslips with isolated cells were then transferred into a chamber, and calcium channel (e.g., L-type, N-type, etc.) current or other currents for counter screening were recorded from the transiently transfected HEK-293T/17 cells.
The whole-cell voltage clamp configuration of the patch clamp technique was employed to evaluate voltage-dependent calcium currents essentially as described by Thompson and Wong (1991 ), J. Physiol., 439: 671-689, the entire disclosure of which is herein incorporated by reference. To record calcium channel (e.g., L-type, N-type, etc.) currents for evaluation of inhibitory potency of compounds (steady-state concentration-response analysis), five pulses of 20-30 ms voltage steps to about +10 mV (the peak of the current voltage relationship) were delivered at five Hz every 30 second from a holding potential at -10OmV. In order to obtain an estimate of the degree of use-dependent block of the test compounds, IC50 values were determined at the first and fifth pulse of the train (P1 and P5, respectively, in Table 2). Compound evaluations were carried out essentially as described by Sah D. W. and Bean B. P. (1994), MoI. Pharmacol., 45:84-92, the entire disclosure of which is herein incorporated by reference.
FLIPR Assay
TSA201 cells stably transfected with human Cav2.2 (composed of the subunits α1 , β3 and α2δ) and human Kir2.3 to enhance the FLIPR Ca2+ signal were used. These cells were plated on 384-well collagen-coated plates (BD Bioscience, Franklin Lakes, NJ) at a density of 2x104 cells/well one day prior to the FLIPR assay. For each assay plate, FLUO-4 dye (Invitrogen, Carlsbad, CA) was diluted in 12 mL of Dulbecco's Modified Eagle's Medium (Invitrogen, Carlsbad, CA) to a final concentration of 4μM in the presence of Pluronic F-127 (Invitrogen, Carlsbad, CA). Culture media is removed and replaced with 25μl of the FLUO-4 dye solution and incubated for one hour at room temperature. The cell plate is then placed on the FLIPR where the dye is aspirated off and replaced with 25μl of HBSS (Invitrogen, Carlsbad, CA). The HBSS is then aspirated and replaced with 25Dl of compound which is diluted in HBSS with 1% DMSO. Compounds are incubated on the cells for 15 minutes. The cells are then depolarized with 25μl of 14OmM KCI solution (also containing 2mM CaCI2 and 1OmM HEPES). The final concentration of KCI on the cells is 7OmM.
The results for selected compounds are summarized in Tables 9 and 10 below. Data presented represent the average value when one or more samples were tested.
TABLE 9

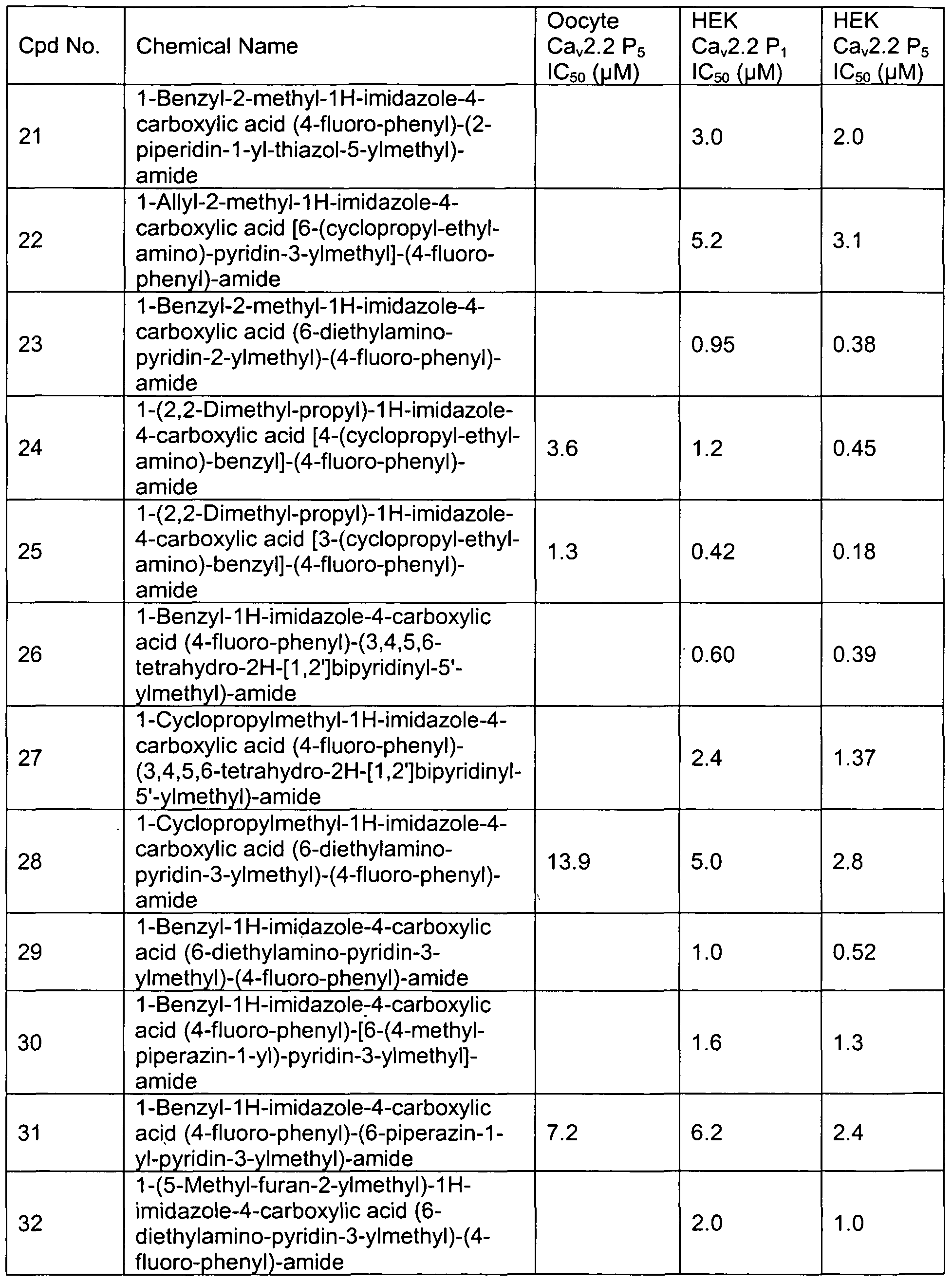



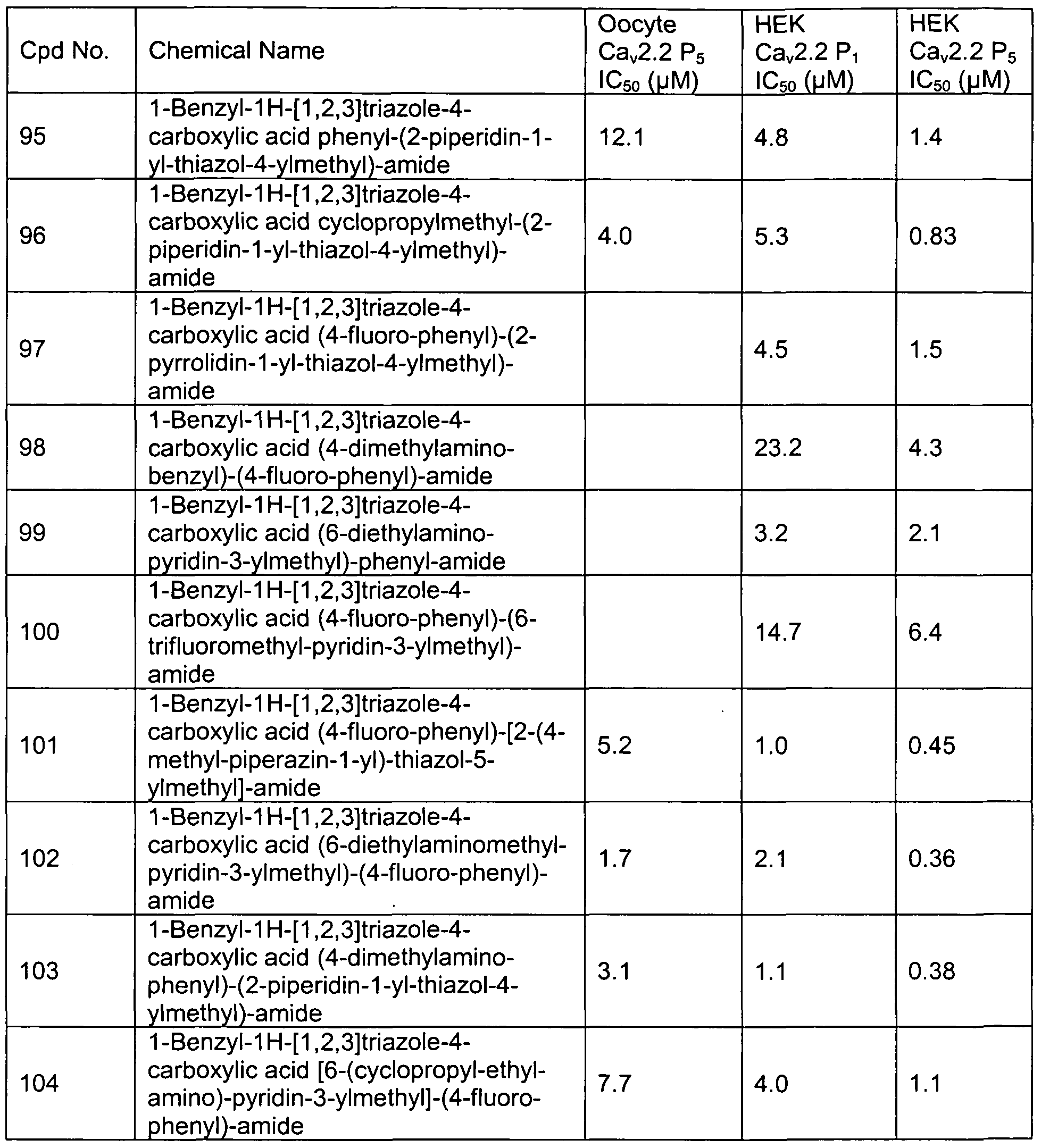
TABLE 10



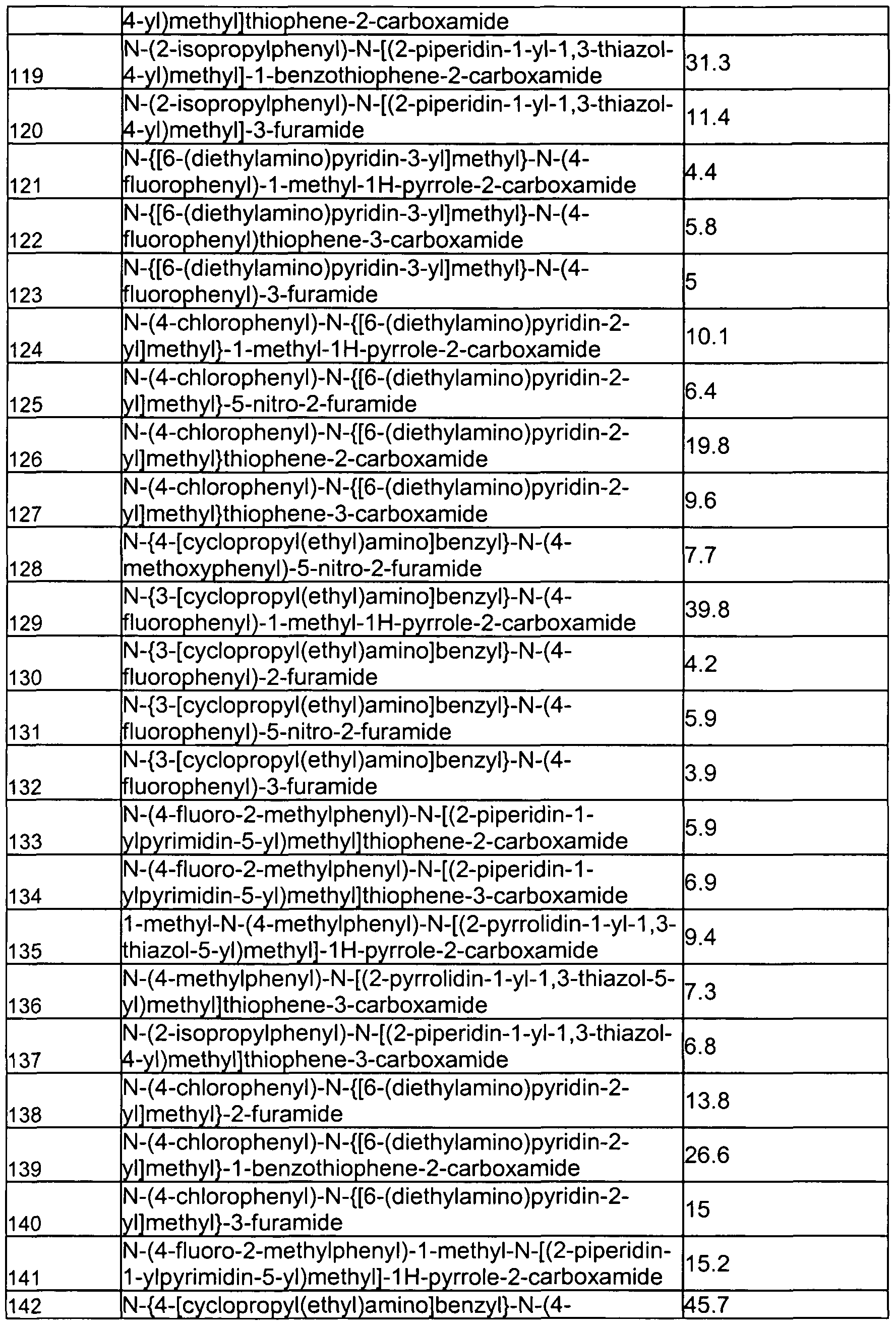






Variations, modifications, and other implementations of what is described herein will occur to those of ordinary skill in the art without departing from the spirit and the essential characteristics of the present teachings. It is not intended that the present invention be limited to the illustrated embodiments but rather by the following claims, and all changes that come within the meaning and range of equivalency of the claims are intended to be embraced therein.
Claims
1. A compound of formula (I),

(I) or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein

X is selected from -NRC-, a divalent C1-6 alkyl group, and a covalent bond;
R1 is selected from C1-6 alkyl, phenyl, and a 5-7 membered heteroaryl group;
R2 is selected from C3-6 cycloalkyl, indole, benzyl, and phenyl, wherein
phenyl and benzyl are each optionally substituted with 1 to 3 substituents independently selected from halogen, C1-6 alkyl, C1-6 alkoxy, C1-6 haloalkyl, C1-6 haloalkoxy, -CN, -C(O)ORC, -OH, and -NRdRe; and
the -C3-6 cycloalkyl is optionally substituted with 1 to 3 C1-6 alkyl groups;
Ar-R3 is selected from the group consisting of

R3 is selected from halogen, CM0 alkyl, C1.10 alkoxy, C1-10 haloalkyl, C1-10 haloalkoxy, C(O)R0, piperidin-4-yl, and a -Y-N RfRg group, wherein
the C1-10 alkyl and the C1-10 alkoxy are optionally substituted with 1-3 substitutents selected from halogen, phenyl, and -OH; and
the nitrogen ring atom of the piperidin-4-yl is optionally substituted with -C(O)O-C1-6alkyl;
Y, at each occurrence, is independently a divalent C1-6 alkyl group or a covalent bond;
Rc, Rd and Re, at each occurrence, are independently H, C1-6 haloalkyl, or C1-6 alkyl;
Rf and R9, at each occurrence, are independently selected from the group consisting of -H, -C(O)RC, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, C1-10 alkyl, C3-6 cycloalkyl, -Y-phenyl, -C(O)-phenyl, -Y-(5-7 membered cycloheteroalkyl), -
Y-(5-7 membered heteroaryl), and -C2-6 alkyl-O-Y-(5-7 membered heteroaryl); or
alternatively, Rf and R9 taken together with the nitrogen atom to which they are bonded form a 5-7 membered cycloheteroalkyl group or a 5-7 membered heteroaryl group, the 5-7 membered cycloheteroalkyl group and the 5-7 membered heteroaryl group each containing up to two ring heteroatoms independently selected from nitrogen, oxygen, and sulfur, wherein
any sulfur atom in the ring is optionally substituted with 1 to 2 oxo groups,
one or more nitrogen atoms in the ring optionally are independently substituted with -C(O)RC, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, -Y-
C(O)NRdRe,
 alkyl), a -C1-6 alkyl-(phenyl)n group, a -C2-6 alkyl— (5-7 membered cycloheteroalkyl) group, C1-6 alkyl, -Y-(phenyl)q, C3.
alkyl), a -C1-6 alkyl-(phenyl)n group, a -C2-6 alkyl— (5-7 membered cycloheteroalkyl) group, C1-6 alkyl, -Y-(phenyl)q, C3.
8 cycloalkyl, or a 5-7 membered heteroaryl group;
one or more carbon atoms in the ring optionally are independently substituted with -C(O)-NRdRe, -Y-ORc, -Y-NRdRe, -Y-(phenyl)q, -Y-(5-7 membered cycloheteroalkyl), a -Y-(5-9 membered heteroaryl) group, an oxo group, CN, C3-8 cycloalkyl, or a -Y-O-(5-7 membered heteroaryl) group;
each of the phenyl groups appearing anywhere in said Rf and R9 is optionally substituted with 1 to 3 substituents independently selected from halogen, C1-6 alkyl, C1-6 haloalkyl, and C1-6 alkoxy, and
each of the 5-7 membered cycloheteroalkyl groups, the 5-7 membered heteroaryl groups, and the 5-9 membered heteroaryl groups appearing anywhere in said Rf and R9 is optionally substituted with 1 to 3 substituents independently selected from halogen and C1-6 alkyl;
n is 1 , 2, or 3;
q is 1 , 2, or 3; and
p is 1 , 2, 3, or 4.
2. The compound of claim 1 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein

3. A compound according to claim 2 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R1 is selected from methyl, phenyl, and pyridyl.
4. A compound according to claim 2 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein X is selected from -NH-, -N(CH3)-, -CH2-, and a covalent bond.
5. The compound of claim 1 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein

7. The compound of claim 1 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R2 is phenyl optionally substituted with 1 to 2 substituents independently selected from halogen, C1-6 alkyl, CN, -C(O)ORC, and -NRdRe.
8. The compound of claim 7 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R2 is selected from the group of a 4-fluorophenyl group, a 4- chlorophenyl group, a 4-fluoro-2-methylphenyl group, and a 4-methoxyphenyl group.
9. The compound of claim 1 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein Ar-R3 is selected from the group consisting of

wherein R3 is as defined in claim 1.
10. The compound of claim 1 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R3 is NRfRg.
1 1. The compound of claim 1 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R3 is selected from NH2, NH-(C1-6 alkyl), N(C1-6 alkyl)2, NH-(C3-6 cycloalkyl), N(C1-6 alkyl)-(C3-6 cycloalkyl), N(C1-6 alkyl)-(C2-6 alkyl)-ORc group, N(C1-6 alkyl)-Y-(5-7 membered cycloheteroalkyl), N(C1-6 alkyl)-phenyl, N(C1-6 alkyl)-Y-(5-7 membered heteroaryl), and N(C1-6 alkyl)-C2-6 alkyl-O-Y-(5-7 membered heteroaryl); wherein each of the phenyl group, the 5-7 membered cycloheteroalkyl group, and the 5-7 heteroaryl group are optionally substituted with 1 to 3 substituents independently selected from halogen and C1-6 alkyl.
12. The compound of claim 11 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R3 is a diethylamino group or a cyclopropyl(ethyl)amino group.
13. The compound of claim 1 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R3 is an optionally substituted 5-7 membered cycloheteroalkyl group or an optionally substituted 5-7 membered heteroaryl group.
14. The compound of claim 13 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R3 is selected from a diazepanyl group, an imidazolyl group, a morpholinyl group, a piperidinyl group, a piperazinyl group, a pyridyl group, a pyrrolidyl group, and a thiomorpholinyl group, wherein
each of these groups optionally includes a nitrogen ring atom substituted with -
C(O)R0, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, -Y-C(O)NRdRe, -S(O)2-(C1-6 alkyl),— C2-6 alkyl— (5-7 membered cycloheteroalkyl, C1-6 alkyl, or 5-7 membered heteroaryl; a carbon ring atom substituted with -C(O)-NRdRe, -Y-
ORc, -Y-NRdRe, -Y-phenyl, -Y-(5-7 membered cycloheteroalkyl),-Y-(5-9 membered heteroaryl), or-Y-O-(5-7 membered heteroaryl); and/or a sulfur ring atom substituted with 1 or 2 oxo groups,
wherein each of the phenyl groups is optionally substituted with 1 to 3 substituents independently selected from halogen, C1-6 alkyl, C1-6 haloalkyl, and C1-6 alkoxy, and each of the 5-7 membered cycloheteroalkyl groups, the 5-7 membered heteroaryl groups, and the 5-9 membered heteroaryl groups is optionally substituted with 1 to 3 substituents independently selected from halogen and C1-6 alkyl.
15. The compound of claim 13 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R3 is selected from the group consisting of a 1-[1 ,4]diazepanyl group, a 1-imidazolyl group, a 4-morpholinyl group, a 1-piperidinyl group, a 1- piperazinyl group, a 4-pyridyl group, a 1-pyrrolidyl group, a 4-thiomorpholinyl group, and a 4-methyl piperazin-1-yl group.
16. The compound of claim 13 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R3 is a 1-piperazinyl group having a nitrogen atom in the ring optionally substituted with -C(O)R0, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, -C1-6 alkyl— C(O)NRdRe, S(O)2-C1-6 alkyl,-C2-6 alkyl-(5-7 membered cycloheteroalkyl), C1-10 alkyl, or 5-7 membered heteroaryl.
17. The compound of claim 16 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R3 is an N-methyl 1-piperazinyl group.
18. The compound of claim 1 selected from
2-(Methyl-phenyl-amino)-thiazole-4-carboxylic acid (6-diethylamino-pyridin-3- ylmethyl)-(4-fluorophenyl)-amide;
2-Benzyl-thiazole-4-carboxylic acid (6-diethylamino-pyridin-3-ylmethyl)-(4- fluorophenyl)-amide;
2-Methyl-thiazole-4-carboxylic acid (6-diethylamino-pyridin-3-ylmethyl)-(4- fluorophenyl)-amide;
2-Pyridin-4-yl-thiazole-4-carboxylic acid (6-diethylamino-pyridin-3-ylmethyl)-(4- fluorophenyl)-amide;
2-Pyridin-3-yl-thiazole-4-carboxylic acid (6-diethylamino-pyridin-3-ylmethyl)-(4- fluorophenyl)-amide;
N-(4-chlorophenyl)-N-{[6-(diethylamino)pyridin-2-yl]methyl}-2-methyl-1 ,3-thiazole- 4-carboxamide;
N-(4-fluoro-2-methylphenyl)-2-methyl-N-[(2-piperidin-1-ylpyrimidin-5-yl)methyl]- 1 ,3-thiazole-4-carboxamide;
N-{4-[cyclopropyl(ethyl)amino]benzyl}-N-(4-methoxyphenyl)-2-methyl-1 ,3- thiazole-4-carboxamide;
N-(4-fluorophenyl)-N-{[6-(4-methylpiperazin-1-yl)pyridin-3-yl]methyl}-1 ,3- benzothiazole-2-carboxamide;
N-{[6-(diethylamino)pyridin-3-yl]methyl}-N-(4-fluorophenyl)-1 ,3-benzothiazole-2- carboxamide; and
pharmaceutically acceptable salts, hydrates, and esters thereof.
19. A pharmaceutical composition comprising the compound of any of claims 1-18 or a pharmaceutically acceptable salt, hydrate or ester thereof and a pharmaceutically acceptable carrier or excipient.
20. The pharmaceutical composition of claim 19, further comprising an additional therapeutic agent.
21. A method of treating a disease or disease symptom selected from angina, hypertension, congestive heart failure, myocardial ischemia, and arrhythmia, the method comprising administering to a subject a therapeutically effective amount of the compound of any of claims 1-18 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
22. A method of treating a disease or disease symptom selected from stroke, convulsion, epilepsy, traumatic brain injury, and neuronal disorder, the method comprising administering to a subject a therapeutically effective amount of the compound of any of claims 1-18 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
23. A method of treating a disease or disease symptom selected from diabetes, urinary incontinence, hot flush, and thermal disregulation, the method comprising administering to a subject a therapeutically effective amount of the compound of any of claims 1-18 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
24. A method of treating pain in a subject, the method comprising administering to a subject a therapeutically effective amount of the compound of any of claims 1-18 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
25. A method of claim 24 wherein said pain is chronic pain.
26. A method of claim 25 wherein said chronic pain is associated with diabetes, post traumatic pain of amputation, lower back pain, spinal cord damage, cancer, chemical injury, chemotherapy induced peripheral neuropathy, toxins, major surgery, peripheral nerve damage due to traumatic injury, post-herpetic neuralgia, trigeminal neuralgia, lumbar or cervical radiculopathies, fibromyalgia, glossopharyngeal neuralgia, reflex sympathetic dystrophy, causalgia, thalamic syndrome, nerve root avulsion, reflex sympathetic dystrophy or post thoracotomy pain, nutritional deficiencies, viral infection, bacterial infection, metastatic infiltration, adiposis dolorosa, burns, central pain conditions related to thalamic conditions, or a combination thereof.
27. A method of claim 24 wherein said pain is chronic back pain.
28. A method of claim 24 wherein said pain is neuropathic pain.
29. A method of claim 24 wherein said pain is associated with diabetic neuropathy.
30. A method of claim 24 wherein said pain is associated with post-herpetic neuropathy.
31. A method of claim 24 wherein said pain is associated with post-herpetic fibromyalgia.
32. A method of treating a disease or disease symptom modulated by calcium channel Cav2, the method comprising administering to a subject a therapeutically effective amount of the compound of any of claims 1-18 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
33. The method of claim 32, wherein the disease or disease symptom is modulated by calcium channel Cav2.2.
34. A method of modulating calcium channel activity in a subject, the method comprising administering the compound of any one of claims 1-18, or a pharmaceutically acceptable salt, hydrate or ester thereof, to a subject.
35. The method of any one of claims 21-34, wherein the subject is a mammal.
36. A method for making a compound of formula (I) according to claim 1 and pharmaceutically acceptable salts thereof, comprising
(a) reacting a carboxylic acid compound of formula (Ia)

(b) coupling the resultant activated acid with an amine of formula (b)
N ^Ar
(b)
37. The method of claim 36, wherein the activating agent is selected from thionyl chloride, 2-chloro-4,6-dimethoxy-1 ,3,5-triazine, 1-[3-(dimethylamino)propyl]-3-ethyl- carbodiimide and dicyclohexyl carbodiimide.
38. A compound of formula (II)
R\
(H) or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein
R4 is selected from H, C1-6 alkyl, and -Y-phenyl;
R5 is selected from H, C1-10 alkyl, C3-6 alkenyl, C3-8 cycloalkyl, phenyl, 5-7 membered heteroaryl, a -C1-6 alkyl-phenyl group, a -C1-6 alkyl— 5-7 membered heteroaryl group,
wherein each of the phenyl group and the 5-7 membered heteroaryl group optionally is substituted with 1 to 3 substituents independently selected from halogen, C1-6 alkyl, C1-6 haloalkyl, and C1-6 alkoxy; provided that at least one of R4 and R5 is not H
R2 is selected from C3-6 cycloalkyl, indole, benzyl, and phenyl, wherein
phenyl and benzyl are each optionally substituted with 1 to 3 substituents independently selected from halogen, Ci-6 alkyl, C1-6 alkoxy, C1-6 haloalkyl, C1-6 haloalkoxy, -CN, -C(O)ORC, -OH, and -NRdRe; and
the -C3-6 cycloalkyl is optionally substituted with 1 to 3 C1-6 alkyl groups;
Ar-R3 is selected from the group consisting of

R3 is selected from halogen, CL10 alkyl, C1-10 alkoxy, C1-I0 haloalkyl, C1-10 haloalkoxy, C(O)R0, piperidin-4-yl, and a -Y-NRfRggroup, wherein
the C1-I0 alkyl and the C1-10 alkoxy are optionally substituted with 1-3 substitutents selected from halogen, phenyl, and -OH; and
the nitrogen ring atom of the piperidin-4-yl is optionally substituted with -C(O)O-C1-6alkyl;
Y, at each occurrence, is independently a divalent C1-6 alkyl group or a covalent bond;
Rc, Rd and Re, at each occurrence, are independently H, C1-6 haloalkyl, or C1-6 alkyl; Rf and R9, at each occurrence, are independently selected from the group consisting of -H, -C(O)RC, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, C1-10 alkyl, C3-6 cycloalkyl, -Y-phenyl, -C(O)-phenyl, -Y-(5-7 membered cycloheteroalkyl), -Y-(5-7 membered heteroaryl), and -C2-6 alkyl-O-Y-(5-7 membered heteroaryl); or
alternatively, Rf and R9 taken together with the nitrogen atom to which they are bonded form a 5-7 membered cycloheteroalkyl group or a 5-7 membered heteroaryl group, the 5-7 membered cycloheteroalkyl group and the 5-7 membered heteroaryl group each containing up to two ring heteroatoms independently selected from nitrogen, oxygen, and sulfur, wherein
any sulfur atom in the ring is optionally substituted with 1 to 2 oxo groups,
one or more nitrogen atoms in the ring optionally are independently substituted with -C(O)R0, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, -Y-
C(O)NRdRe, -S(O)2-(Ci-6 alkyl), a -C1-6 alkyl-(phenyl)n group, a -C2-6 alkyl— (5-7 membered cycloheteroalkyl) group, C1-6 alkyl, -Y-(phenyl)q, C3. 8 cycloalkyl, or a 5-7 membered heteroaryl group;
one or more carbon atoms in the ring optionally are independently substituted with -C(O)-NRdRe, -Y-ORC, -Y-NRdRe, -Y-(phenyl)q, -Y-(5-7 membered cycloheteroalkyl), a -Y-(5-9 membered heteroaryl) group, an oxo group, CN, C3-8 cycloalkyl, or a -Y-O-(5-7 membered heteroaryl) group; and
each of the phenyl groups appearing anywhere in said Rf and R9 is optionally substituted with 1 to 3 substituents independently selected from halogen, C1-6 alkyl, C1-6 haloalkyl, and C1-6 alkoxy, and
each of the 5-7 membered cycloheteroalkyl groups, the 5-7 membered heteroaryl groups, and the 5-9 membered heteroaryl groups appearing anywhere in said Rf and R9 is optionally substituted with 1 to 3 substituents independently selected from halogen and C1-6 alkyl;
n is 1 , 2, or 3;
q is 1 , 2, or 3; and
p is 1 , 2, 3, or 4.
39. The compound of claim 38 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein p is 1.
40. The compound of claim 38 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R4 is selected from H, methyl, and benzyl.
41. The compound of claim 38 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R5 is selected from the group consisting of an allyl group, benzyl, a 4-chlorobenzyl group, a cyclopentyl group, a cyclopropylmethyl group, a 2,2- dimethyl-propyl group, methyl and a 5-methyl-furan-2-yl-methyl group.
42. The compound of claim 38 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R2 is phenyl optionally substituted with 1 to 2 substituents independently selected from halogen, C1-6 alkyl, CN, -C(O)ORC, and -NRdRe.
43. The compound of claim 42, wherein R2 is selected from the group of a 4- fluorophenyl group, a 2-isopropylphenyl group, and a 4-methylphenyl.
44. The compound of claim 38 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein Ar-R3 is selected from the group consisting of

45. The compound of claim 38 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R3 is NRfRg.
46. The compound of claim 45 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R3 is selected from NH2, NH-(Ci-6 alkyl), N(C1-6 alkyl)2, NH-(C3-6 cycloalkyl), N(C1-6 alkyl)-(C3-6 cycloalkyl), N(C1-6 alkyl)-(C2-6 alkyl)-ORc group, N(C1-6 alkyl)-Y-(5-7 membered cycloheteroalkyl), N(C1-6 alkyl)-phenyl, N(C1-6 alkyl)-Y-(5-7 membered heteroaryl), and N(C1-6 alkyl)-C2-6 alkyl-O-Y-(5-7 membered heteroaryl); wherein each of the phenyl , the 5-7 membered cycloheteroalkyl, and the 5-7 heteroaryl are optionally substituted with 1 to 3 substituents independently selected from halogen and C1-6 alkyl.
47. The compound of claim 46 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R3 is selected from the group consisting of a diethylamino group, a dimethylamino group, an ethyl(methyl)amino group, an isopropyl(ethyl)amino group, and a cyclopropyl(ethyl)amino group.
48. The compound of claim 45 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R3 is an optionally substituted 5-7 membered cycloheteroalkyl group or an optionally substituted 5-7 membered heteroaryl group.
49. The compound of claim 48 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R3 is selected from a diazepanyl group, an imidazolyl group, a morpholinyl group, a piperidinyl group, a piperazinyl group, a pyridyl group, a pyrrolidyl group, and a thiomorpholinyl group, wherein each of these groups optionally includes a nitrogen ring atom substituted with -C(O)R0, -C2.6 alkyl-ORc, - C2-6 alkyl-NRdRe, -Y-C(O)NRdRe, -S(O)2-(Ci-6 alkyl), -C2-6 alkyl-(5-7 membered cycloheteroalkyl), C1-6 alkyl, or 5-7 membered heteroaryl, a carbon ring atom substituted with -C(O)-NRdRe, -Y-OR0, -Y-NRdR6, -Y-phenyl, -Y-(5-7 membered cycloheteroalkyl), -Y-(5-9 membered heteroaryl), or -Y-O-(5-7 membered heteroaryl), and/or a sulfur ring atom substituted with 1 or 2 oxo groups, wherein each of the phenyl groups is optionally substituted with 1 to 3 substituents independently selected from halogen, Ci-6 alkyl, C1-6 haloalkyl, and C1-6 alkoxy, and each of the 5-7 membered cycloheteroalkyl groups, the 5-7 membered heteroaryl groups, and the 5-9 membered heteroaryl groups immediately above is optionally substituted with 1 to 3 substituents independently selected from halogen and C1-6 alkyl.
50. The compound of claim 49 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R3 is selected from the group consisting of a 1-[1 ,4]diazepanyl group, a 1 -imidazolyl group, a 4-morpholinyl group, a 1 -piperidinyl group, a 1- piperazinyl group, a 4-pyridyl group, a 1 -pyrrolidyl group, and a 4-thiomorpholinyl group.
51. The compound of claim 49 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R3 is a 1-piperazinyl group having a nitrogen atom in the ring optionally substituted with -C(O)RC, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, -d.6 alkyl- C(O)NRdRe, S(O)2-C1-6 alkyl, -C2-6 alkyl-(5-7 membered cycloheteroalkyl), C1-6 alkyl, or 5-7 membered heteroaryl, wherein Rc, Rd and Re are as defined in claim 1.
52. The compound of claim 51 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R3 is an N-methyl 1-piperazinyl group.
53. The compound of claim 38 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R3 is methyl or a trifluoromethyl group.
54. The compound of claim 38 selected from
1 -(2,2-dimethylpropyl)-N-(2-isopropylphenyl)-N-[(2-piperidin-1 -yl-1 ,3-thiazol-4- yl)methyl]-1 H-imidazole-4-carboxamide;
1-benzyl-N-(2-isopropylphenyl)-N-[(2-piperidin-1-yl-1 ,3-thiazol-4-yl)methyl]-1 H- imidazole-4-carboxamide;
N-(2-isopropylphenyl)-1-methyl-N-[(2-piperidin-1-yl-1 ,3-thiazol-4-yl)methyl]-1 H- imidazole-4-carboxamide;
1-benzyl-N-(4-methylphenyl)-N-[(2-pyrrolidin-1-yl-1 ,3-thiazol-4-yl)methyl]-1 H- imidazole-4-carboxamide;
1 -methyl-N-(4-methylphenyl)-N-[(2-pyrrolidin-1 -yl-1 ,3-thiazol-4-yl)methyl]-1 H- imidazole-4-carboxamide;
1-Benzyl-2-methyl-1 H-imidazole-4-carboxylic acid (4-fluoro-phenyl)-(2-piperidin- 1 -yl-thiazol-4-ylmethyl)-amide;
1-Benzyl-2-methyl-1 H-imidazole-4-carboxylic acid [6-(cyclopropyl-ethyl-amino)- pyridin-3-ylmethyl]-(4-fluoro-phenyl)-amide;
2-Benzyl-1 -methyl-1 H-imidazole-4-carboxylic acid [6-(cyclopropyl-ethyl-amino)- pyridin-3-ylmethyl]-(4-fluoro-phenyl)-amide;
1-Benzyl-1 H-imidazole-4-carboxylic acid (4-fluoro-phenyl)-(2-piperidin-1-yl- pyrimidin-5-ylmethyl)-amide;
1-Cyclopropylmethyl-2-methyl-1 H-imidazole-4-carboxylic acid [6-(cyclopropyl- ethyl-amino)-pyridin-3-ylmethyl]-(4-fluoro-phenyl)-amide;
1-Allyl-2-methyl-1 H-imidazole-4-carboxylic acid [6-(cyclopropyl-ethyl-amino)- pyridin-3-ylmethyl]-(4-fluoro-phenyl)-amide;
1-Benzyl-2-methyl-1 H-imidazole-4-carboxylic acid (6-diethylamino-pyridin-2- ylmethyl)-(4-fluoro-phenyl)-amide;
1 -(2, 2-Dimethyl-propyl)-1 H-imidazole-4-carboxylic acid [4-(cyclopropyl-ethyl- amino)-benzyl]-(4-fluoro-phenyl)-amide;
1-(2,2-Dimethyl-propyl)-1 H-imidazole-4-carboxylic acid [3-(cyclopropyl-ethyl- amino)-benzyl]-(4-fluoro-phenyl)-amide;
1-Benzyl-1 H-imidazole-4-carboxylic acid (4-fluoro-phenyl)-(3,4,5,6-tetrahydro-2H- [i ^'Jbipyridinyl-S'-ylmethyO-amide;
1 -Cyclopropylmethyl-1 H-imidazole-4-carboxylic acid (4-fluoro-phenyl)-(3,4,5,6- tetrahydro-2H-[1 ^'ibipyridinyl-S'-ylmethyO-amide;
1 -Cyclopropylmethyl-1 H-imidazole-4-carboxylic acid (6-diethylamino-pyridin-3- ylmethyl)-(4-fluoro-phenyl)-amide;
1 -Benzyl-1 H-imidazole-4-carboxylic acid (6-diethylamino-pyridin-3-ylmethyl)-(4- fluoro-phenyl)-amide;
' 1 -Benzyl-1 H-imidazole-4-carboxylic acid (4-fluoro-phenyl)-[6-(4-methyl-piperazin- 1-yl)-pyridin-3-ylmethyl]-amide;
1 -Benzyl-1 H-imidazole-4-carboxylic acid (4-fluoro-phenyl)-(6-piperazin-1-yl- pyridin-3-ylmethyl)-amide;
1 -(5-Methyl-furan-2-ylmethyl)-1 H-imidazole-4-carboxylic acid (6-diethylamino- pyridin-3-ylmethyl)-(4-fluoro-phenyl)-amide;
1-(4-Chloro-benzyl)-1 H-imidazole-4-carboxylic acid (4-fluoro-phenyl)-(6-piperazin- 1 -yl-pyridin-3-ylmethyl)-amide;
1-(4-Chloro-benzyl)-1 H-imidazole-4-carboxylic acid (4-fluoro-phenyl)-[6-(4- methyl-piperazin-1-yl)-pyridin-3-ylmethyl]-amide;
1 -Benzyl-1 H-imidazole-4-carboxylic acid (4-fluoro-phenyl)-(6-trifluoromethyl- pyridin-3-ylmethyl)-amide;
1-Cyclopentyl-1 H-imidazole-4-carboxylic acid (6-diethylamino-pyridin-3-ylmethyl)-
(4-fluoro-phenyl)-amide;
1 -Benzyl-1 H-imidazole-4-carboxylic acid (4-fluoro-phenyl)-(6-morpholin-4-yl- pyridin-3-ylmethyl)-amide;
1-Benzyl-2-methyl-1 H-imidazole-4-carboxylic acid (4-fluoro-phenyl)-(6-morpholin- 4-yl-pyridin-3-ylmethyl)-amide;
1 -Benzyl-1 H-imidazole-4-carboxylic acid (4-fluoro-phenyl)-(2-piperidin-1-yl- thiazol-4-ylmethyl)-amide;
1-Benzyl-1 H-imidazole-4-carboxylic acid (6-dimethylamino-pyridin-3-ylmethyl)-(4- fluoro-phenyl)-amide;
1-Benzyl-1 H-imidazole-4-carboxylic acid (4-fluoro-phenyl)-(6-methyl-pyridin-3- ylmethyl)-amide;
1-Benzyl-1 H-imidazole-4-carboxylic acid (4-fluoro-phenyl)-(2-pyrrolidin-1-yl- thiazol-4-ylmethyl)-amide;
1-Benzyl-1 H-imidazole-4-carboxylic acid (4-fluoro-phenyl)-(2-morpholin-4-yl- thiazol-4-ylmethyl)-amide;
1 -Benzyl-2-methyl-1 H-imidazole-4-carboxylic acid (6-diethylamino-pyridin-3- ylmethyl)-(4-fluoro-phenyl)-amide;
1 -Benzyl-2-methyl-1 H-imidazole-4-carboxylic acid (4-fluoro-phenyl)-(6- trifluoromethyl-pyridin-3-ylmethyl)-amide;
1-Benzyl-1 H-imidazole-4-carboxylic acid (2-diethylamino-thiazol-4-ylmethyl)-(4- fluoro-phenyl)-amide;
1-Benzyl-1 H-imidazole-4-carboxylic acid (4-dimethylamino-benzyl)-(4-fluoro- phenyl)-amide;
1 -Benzyl-1 H-imidazole-4-carboxylic acid [2-(ethyl-methyl-amino)-thiazol-4- ylmethyl]-(4-fluoro-phenyl)-amide;
1-Benzyl-2-methyl-1 H-imidazole-4-carboxylic acid (2-diethylamino-thiazol-4- ylmethyl)-(4-fluoro-phenyl)-amide;
2-Benzyl-1-methyl-1 H-imidazole-4-carboxylic acid (4-fluoro-phenyl)-(2-piperidin-
1 -yl-thiazol-4-ylmethyl)-amide;
2-Benzyl-1-methyl-1 H-imidazole-4-carboxylic acid (6-diethylamino-pyridin-3- ylmethyl)-(4-fluoro-phenyl)-amide;
1-Cyclopentyl-1 H-imidazole-4-carboxylic acid (4-fluoro-phenyl)-(2-piperidin-1-yl- thiazol-4-ylmethyl)-amide;
1-Allyl-2-methyl-1 H-imidazole-4-carboxylic acid (6-diethylamino-pyridin-3- ylmethyl)-(4-fluoro-phenyl)-amide;
1-Benzyl-1 H-imidazole-4-carboxylic acid (2-diethylamino-pyrimidin-5-ylmethyl)-(4- fluoro-phenyl)-amide;
1-Allyl-2-methyl-1 H-imidazole-4-carboxylic acid (4-fluoro-phenyl)-(2-piperidin-1-yl- thiazol-4-ylmethyl)-amide;
1-Benzyl-2-methyl-1 H-imidazole-4-carboxylic acid (4-fluoro-phenyl)-(2-piperidin- 1-yl-pyrimidin-5-ylmethyl)-amide;
1-Benzyl-2-methyl-1 H-imidazole-4-carboxylic acid (2-diethylamino-pyrimidin-5- ylmethyl)-(4-fluoro-phenyl)-amide;
1-Benzyl-2-methyl-1 H-imidazole-4-carboxylic acid [2-(cyclopropyl-ethyl-amino)- thiazol-4-ylmethyl]-(4-fluoro-phenyl)-amide;
1-Benzyl-2-methyl-1 H-imidazole-4-carboxylic acid (4-diethylamino-benzyl)-(4- fluoro-phenyl)-amide;
1-Cyclopropylmethyl-2-methyl-1 H-imidazole-4-carboxylic acid (4-diethylamino- benzyl)-(4-fluoro-phenyl)-amide;
1-Cyclopropylmethyl-2-methyl-1 H-imidazole-4-carboxylic acid [4-(cyclopropyl- ethyl-amino)-benzyl]-(4-fluoro-phenyl)-amide;
i-Cyclopropylmethyl-2-methyl-i H-imidazole-4-carboxylic acid (6-diethylamino- pyridin-2-ylmethyl)-(4-fluoro-phenyl)-amide;
1 -(2,2-Dimethyl-propyl)-1 H-imidazole-4-carboxylic acid (4-fluoro-phenyl)-(6- trifluoromethyl-pyridin-3-ylmethyl)-amide;
1 -(2, 2-Dimethyl-propyl)-1 H-imidazole-4-carboxylic acid (4-fluoro-phenyl)-[2-
(isopropyl-methyl-amino)-thiazol-4-ylmethyl]-amide;
1 -(2,2-Dimethyl-propyl)-1 H-imidazole-4-carboxylic acid (4-diethylamino-benzyl)- (4-fluoro-phenyl)-amide;
1-(2,2-Dimethyl-propyl)-1 H-imidazole-4-carboxylic acid [6-(cyclopropyl-ethyl- amino)-pyridin-3-ylmethyl]-(4-fluoro-phenyl)-amide; and
pharmaceutically acceptable salts, hydrates, and esters thereof.
55. A pharmaceutical composition comprising the compound of any one of claims 38- 54 or a pharmaceutically acceptable salt, hydrate or ester thereof and a pharmaceutically acceptable carrier or excipient.
56. The pharmaceutical composition of claim 55, further comprising an additional therapeutic agent.
57. A method of treating a disease or disease symptom selected from angina, hypertension, congestive heart failure, myocardial ischemia, and arrhythmia, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 38-54 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
58. A method of treating a disease or disease symptom selected from stroke, convulsion, epilepsy, traumatic brain injury, and neuronal disorder, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 38-54 or a pharmaceutically acceptable salt, hydrate,
5 or ester thereof.
59. A method of treating a disease or disease symptom selected from diabetes, urinary incontinence, hot flush, and thermal disregulation, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 38-54 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
10.
60. A method of treating pain in a subject, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 38- 54 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
61. A method of claim 60 wherein said pain is chronic pain.
62. A method of claim 61 wherein said chronic pain is associated with diabetes, post 15 traumatic pain of amputation, lower back pain, spinal cord damage, cancer, chemical injury, chemotherapy induced peripheral neuropathy, toxins, major surgery, peripheral nerve damage due to traumatic injury, post-herpetic neuralgia, trigeminal neuralgia, lumbar or cervical radiculopathies, fibromyalgia, glossopharyngeal neuralgia, reflex sympathetic dystrophy, causalgia, thalamic syndrome, nerve root 0 avulsion, reflex sympathetic dystrophy or post thoracotomy pain, nutritional deficiencies, viral infection, bacterial infection, metastatic infiltration, adiposis dolorosa, burns, central pain conditions related to thalamic conditions, or a combination thereof.
63. A method of claim 60 wherein said pain is chronic back pain. 5
64. A method of claim 60 wherein said pain is neuropathic pain.
65. A method of claim 60 wherein said pain is associated with diabetic neuropathy.
66. A method of claim 60 wherein said pain is associated with post-herpetic neuropathy.
67. A method of claim 60 wherein said pain is associated with post-herpetic fibromyalgia.
68. A method of treating a disease or disease symptom modulated by calcium channel Cav2, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 38-54 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
69. The method of claim 68, wherein the disease or disease symptom is modulated by calcium channel Cav2.2.
70. The method of any one of claims 57-70, wherein the subject is a mammal.
71. A method of modulating calcium channel activity in a subject, the method comprising administering the compound of any one of claims 38-54, or a pharmaceutically acceptable salt, hydrate or ester thereof, to a subject.
72. A method for making a compound of formula (II) of claim 38 and pharmaceutically acceptable salts thereof, comprising
(a) reacting a carboxylic acid compound of formula (Ma)

with an activating agent; and (b) coupling the resultant activated acid with an amine of formula (b)
N ^Ar (b)
73. The method according to claim 72, wherein the activating agent is selected from thionyl chloride, 2-chloro-4,6-dimethoxy-1 ,3,5-triazine, 1-[3-(dimethylamino)propyl]-3- ethyl-carbodiimide and dicyclohexyl carbodiimide.
74. A compound of formula (III)

(III) and pharmaceutically acceptable salts thereof, wherein
R6 is selected from C1.10 alkyl, phenyl, a -C1-6 alkyl-phenyl group, and 5-7 membered heteroaryl, wherein
each of the phenyl group and the 5-7 membered heteroaryl group optionally is substituted with 1 to 3 substituents independently selected from halogen, C1-6 alkyl, Ci-6 haloalkyl, and Ci-6 alkoxy;
R7 is H or C1-6 alkyl;
R2 is selected from C3-6 cycloalkyl, indole, benzyl, and phenyl, wherein
phenyl and benzyl are each optionally substituted with 1 to 3 substituents independently selected from halogen, C1-6 alkyl, C1-6 alkoxy, C1-6 haloalkyl, C1. β haloalkoxy, -CN, -C(O)ORC, -OH, and -NRdRe; and
the -C3-6 cycloalkyl is optionally substituted with 1 to 3 C1-6 alkyl groups;
Ar-R3 is selected from the group consisting of

R3 is selected from halogen, C1-10 alkyl, Ci-10 alkoxy, C1-10 haloalkyl, C1-10 haloalkoxy, C(O)RC, piperidin-4-yl, and a -Y-NRfRg group, wherein the C1- 10 alkyl and the C1-10 alkoxy are optionally substituted with 1-3 substitutents selected from halogen, phenyl, and -OH; and
the nitrogen ring atom of the piperidin-4-yl group is optionally substituted with -C(O)O-Ci-6alkyl;
Y, at each occurrence, is independently a divalent C1-6 alkyl group or a covalent bond;
Rc, Rd and Re, at each occurrence, are independently H or C1-6 alkyl;
Rf and R9, at each occurrence, are independently selected from the group consisting of -H, -C(O)Rc, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, C1-6 alkyl, a C3.
6 cycloalkyl, -Y-phenyl, -C(O)-phenyl, -Y-(5-7 membered cycloheteroalkyl), -Y-(5-7 membered heteroaryl), and -C2-6 alkyl-O-Y-(5-7 membered heteroaryl), or
alternatively, Rf and R9 taken together with the nitrogen atom to which they are bonded form a 5-7 membered cycloheteroalkyl group or a 5-7 membered heteroaryl group, the 5-7 membered cycloheteroalkyl group and the 5-7 membered heteroaryl group containing up to two ring heteroatoms independently selected from nitrogen, oxygen, and sulfur; wherein
any sulfur atom in the ring is optionally substituted with 1 to 2 oxo groups,
one or more nitrogen atoms in the ring optionally are independently substituted with -C(O)RC, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, -Y- C(O)NRdRe, -S(O)2-(C1-6 alkyl), a -C1-6 alkyl-(phenyl)n group, a -C2-6 alkyl— (5-7 membered cycloheteroalkyl) group, Ci-6 alkyl, -Y-(phenyl)q, C3.
8 cycloalkyl, or a 5-7 membered heteroaryl group;
one or more carbon atoms in the ring optionally are independently substituted with -C(O)-NRdRe, -Y-OR0, -Y-NRdRe, -Y-(phenyl)q, -Y-(5-7 membered cycloheteroalkyl), a -Y-(5-9 membered heteroaryl) group, an oxo group, CN, C3-8 cycloalkyl, or a -Y-O-(5-7 membered heteroaryl) group; and
each of the phenyl groups appearing anywhere in said Rf and R9 is optionally substituted with 1 to 3 substituents independently selected from halogen, C1-6 alkyl, C1-6 haloalkyl, and C1-6 alkoxy, and
each of the 5-7 membered cycloheteroalkyl groups, the 5-7 membered heteroaryl groups, and the 5-9 membered heteroaryl groups appearing anywhere in said Rf and R9 is optionally substituted with 1 to 3 substituents independently selected from halogen and C1-6 alkyl;
n is 1 , 2, or 3;
q is 1 , 2, or 3; and
p is 1 , 2, 3, or 4.
75. The compound of claim 74, wherein p is 1.
76. The compound of claim 74, wherein p is 2.
77. The compound of claim 74, wherein p is 3.
78. The compound of claim 74, wherein R6 is selected from phenyl, benzyl, and pyridin-3-yl.
79. The compound of claim 74, wherein R2 is phenyl optionally substituted with 1 to 2 substituents independently selected from halogen, C1-6 alkyl, CN, -C(O)ORC, and - NRdRe.
80. The compound of claim 79, wherein R2 is selected from the group of phenyl, a 4- dimethylaminophenyl group, a 4-fluoro-2-methylphenyl group, a 4-methylphenyl group, a 2-isopropylphenyl group, a 4-methoxyphenyl group, a 4-chlorophenyl group a 4-fluorophenyl group, and a 2-isopropylphenyl group.
81. The compound of claim 74, wherein Ar-R3 is selected from the group consisting of

82. The compound of claim 74, wherein Ar-R3 is

83. The compound of claim 74, wherein R3 is NRfRg.
84. The compound of claim 83, wherein R3 is selected from the group of a diethylamino group, a dimethylamino group, an ethyl(methyl)amino group, an isopropyl(ethyl)amino group, a cyclopropyl(ethyl)amino group, a 1-[1 ,4]diazepanyl group, a 1-imidazolyl group, a 4-morpholinyl group, a 1-piperidinyl group, a 1- piperazinyl group, a 4-pyridyl group, a 1-pyrrolidyl group, a 4-thiomorpholinyl group, and a 4-methyl piperazin-1-yl group.
85. The compound of claim 74, wherein R3 is methyl or trifluoromethyl.
86. The compound of claim 74 selected from
1 -benzyl-N-{3-[cyclopropyl(ethyl)amino]benzyl}-N-(4-fluorophenyl)-1 H-1 ,2,3- triazole-4-carboxamide;
1-benzyl-N-(4-fluoro-2-methylphenyl)-N-[(2-piperidin-1-ylpyrimidin-5-yl)methyl]-
1 H-1 ,2,3-triazole-4-carboxamide;
1-benzyl-N-{[6-(diethylamino)pyridin-3-yl]methyl}-N-(4-fluorophenyl)-5-methyl-1 H- 1 ,2,3-triazole-4-carboxamide;
1-benzyl-N-{3-[cyclopropyl(ethyl)amino]benzyl}-N-(4-fluorophenyl)-5-methyl-1 H- 1 ,2,3-triazole-4-carboxamide;
1-benzyl-N-(4-fluoro-2-methylphenyl)-5-methyl-N-[(2-piperidin-1-ylpyrimidin-5- yl)methyl]-1 H-1 ,2,3-triazole-4-carboxamide;
1 -benzyl-N-(2-isopropylphenyl)-5-methyl-N-[(2-piperidin-1 -yl-1 ,3-thiazol-4- yl)methyl]-1 H-1 ,2,3-triazole-4-carboxamide;
1 -benzyl-N-(2-isopropylphenyl)-N-[(2-piperidin-1 -yl-1 ,3-thiazol-4-yl)methyl]-1 H-
1 ,2,3-triazole-4-carboxamide;
1 -benzyl-N-(4-chlorophenyl)-N-{[6-(diethylamino)pyridin-2-yl]methyl}-1 H-1 ,2,3- triazole-4-carboxamide;
1-benzyl-N-(4-chlorophenyl)-N-{[6-(diethylamino)pyridin-2-yl]methyl}-5-methyl-1 H- 1 ,2,3-triazole-4-carboxamide;
1-benzyl-N-{4-[cyclopropyl(ethyl)amino]benzyl}-N-(4-methoxyphenyl)-5-methyl- 1 H-1 ,2,3-triazole-4-carboxamide;
1 -benzyl-N-{4-[cyclopropyl(ethyl)amino]benzyl}-N-(4-methoxyphenyl)-1 H-1 ,2,3- triazole-4-carboxamide;
N-{[6-(diethylamino)pyridin-3-yl]methyl}-N-(4-fluorophenyl)-1 -phenyl-1 H-1 ,2,3- triazole-4-carboxamide;
1-benzyl-N-(4-fluorophenyl)-N-{[6-(4-methylpiperazin-1-yl)pyridin-3-yl]methyl}-1 H- 1 ,2,3-triazole-4-carboxamide;
1-benzyl-N-(4-fluorophenyl)-N-({2-[methyl(phenyl)amino]-1 ,3-thiazol-4-yl}methyl)- 1 H-1 ,2,3-triazole-4-carboxamide;
1 -benzyl-N-(4-fluorophenyl)-N-[2-(2-morpholin-4-yl-1 ,3-thiazol-4-yl)ethyl]-1 H-
1 ,2,3-triazole-4-carboxamide;
1 -benzyl-N-(4-fluorophenyl)-N-[3-(2-morpholin-4-yl-1 ,3-thiazol-4-yl)propyl]-1 H- 1 ,2,3-triazole-4-carboxamide;
1 -benzyl-N-(4-fluorophenyl)-N-[(2-morpholin-4-yl-1 ,3-thiazol-4-yl)methyl]-1 H- 1 ,2,3-triazole-4-carboxamide ;
1-Benzyl-1 H-[1 ,2,3]triazole-4-carboxylic acid (6-diethylamino-pyridin-3-ylmethyl)-
(4-fluoro-phenyl)-amide;
1 -Benzyl-1 H-[1 ,2,3]triazole-4-carboxylic acid (4-fluoro-phenyl)-(2-piperidin-1 -yl- thiazol-4-ylmethyl)-amide;
1 -Benzyl-1 H-[1 ,2,3]triazole-4-carboxylic acid (4-diethylamino-benzyl)-(4-fluoro- phenyl)-amide;
1 -Benzyl-1 H-[1 ,2,3]triazole-4-carboxylic acid (4-fluoro-phenyl)-(2-piperidin-1 -yl- pyrimidin-5-ylmethyl)-amide;
1-Pyridin-3-yl-1 H-[1 ,2,3]triazole-4-carboxylic acid (6-diethylamino-pyridin-3- ylmethyl)-(4-fluoro-phenyl)-amide;
1-Phenyl-1 H-[1 ,2,3]triazole-4-carboxylic acid (6-diethylamino-pyridin-3-ylmethyl)-
(4-fluoro-phenyl)-amide;
1 -Benzyl-1 H-[1 ,2,3]triazole-4-carboxylic acid (4-fluoro-phenyl)-[6-(4-methyl- piperazin-1-yl)-pyridin-3-ylmethyl]-amide;
1 -Benzyl-1 H-[1 ,2,3]triazole-4-carboxylic acid (4-fluoro-phenyl)-(2-morpholin-4-yl- thiazol-4-ylmethyl)-amide;
1 -Benzyl-1 H-[1 ,2,3]triazole-4-carboxylic acid phenyl-(2-piperidin-1-yl-thiazol-4- ylmethyl)-amide;
1 -Benzyl-1 H-[1 ,2,3]triazole-4-carboxylic acid cyclopropylmethyl-(2-piperidin-1 -yl- thiazol-4-ylmethyl)-amide;
1 -Benzyl-1 H-[1 ,2,3]triazole-4-carboxylic acid (4-fluoro-phenyl)-(2-pyrrolidin-1-yl- thiazol-4-ylmethyl)-amide;
1 -Benzyl-1 H-[1 , 2, 3]triazole-4-carboxylic acid (4-dimethylamino-benzyl)-(4-fluoro- phenyl)-amide;
1 -Benzyl-1 H-[1 ,2,3]triazole-4-carboxylic acid (6-diethylamino-pyridin-3-ylmethyl)- phenyl-amide;
1 -Benzyl-1 H-[1 ,2,3]triazole-4-carboxylic acid (4-fluoro-phenyl)-(6-trifluoromethyl- pyridin-3-ylmethyl)-amide;
1 -Benzyl-1 H-[1 ,2,3]triazole-4-carboxylic acid (6-diethylaminomethyl-pyridin-3- ylmethyl)-(4-fluoro-phenyl)-amide;
1 -Benzyl-1 H-[1 ,2,3]triazole-4-carboxylic acid (4-dimethylamino-phenyl)-(2- piperidin-1-yl-thiazol-4-ylmethyl)-amide;
1 -Benzyl-1 H-[1 , 2, 3]triazole-4-carboxylic acid [6-(cyclopropyl-ethyl-amino)-pyridin-
3-ylmethyl]-(4-fluoro-phenyl)-amide;
1 -Benzyl-1 H-[1 ,2,3]triazole-4-carboxylic acid (4-fluoro-phenyl)-[2-(methyl-phenyl- amino)-thiazol-4-ylmethyl]-amide;
1 -Benzyl-1 H-[1 ,2,3]triazole-4-carboxylic acid (2-diethylamino-pyrimidin-5- ylmethyl)-(4-fluoro-phenyl)-amide; and
pharmaceutically acceptable salts, hydrates, and esters thereof.
87. A pharmaceutical composition comprising the compound of any one of claims 74- 86 or a pharmaceutically acceptable salt, hydrate or ester thereof and a pharmaceutically acceptable carrier or excipient.
88. The pharmaceutical composition of claim 87, further comprising an additional therapeutic agent.
89. A method of treating a disease or disease symptom selected from angina, hypertension, congestive heart failure, myocardial ischemia, and arrhythmia, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 74-86or a pharmaceutically acceptable salt, hydrate, or ester thereof.
90. A method of treating a disease or disease symptom selected from stroke, convulsion, epilepsy, traumatic brain injury, and neuronal disorder, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 74-86or a pharmaceutically acceptable salt, hydrate, or ester thereof.
91. A method of treating a disease or disease symptom selected from diabetes, urinary incontinence, hot flush, and thermal disregulation, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 74-86 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
92. A method of treating pain in a subject, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 74- 86 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
93. A method of claim 92 wherein said pain is chronic pain.
94. A method of claim 93 wherein said chronic pain is associated with diabetes, post traumatic pain of amputation, lower back pain, spinal cord damage, cancer, chemical injury, chemotherapy induced peripheral neuropathy, toxins, major surgery, peripheral nerve damage due to traumatic injury, post-herpetic neuralgia, trigeminal neuralgia, lumbar or cervical radiculopathies, fibromyalgia, glossopharyngeal neuralgia, reflex sympathetic dystrophy, causalgia, thalamic syndrome, nerve root avulsion, reflex sympathetic dystrophy or post thoracotomy pain, nutritional deficiencies, viral infection, bacterial infection, metastatic infiltration, adiposis dolorosa, burns, central pain conditions related to thalamic conditions, or a combination thereof.
95. A method of claim 92 wherein said pain is chronic back pain.
96. A method of claim 92 wherein said pain is neuropathic pain.
97. A method of claim 92 wherein said pain is associated with diabetic neuropathy.
98. A method of claim 92 wherein said pain is associated with post-herpetic neuropathy.
99. A method of claim 92 wherein said pain is associated with post-herpetic fibromyalgia.
100. A method of treating a disease or disease symptom modulated by calcium channel Cav2, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 74-86 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
101. The method of claim 100, wherein the disease or disease symptom is modulated by calcium channel Cav2.2.
102. The method of any one of claims 89-101 , wherein the subject is a mammal.
103. A method of modulating calcium channel activity in a subject, the method comprising administering the compound of any one of claims 74-86, or a pharmaceutically acceptable salt, hydrate or ester thereof, to a subject.
104. A method for making a compound of formula (III) according to claim 74 and pharmaceutically acceptable salts thereof, comprising
(a) reacting a carboxylic acid compound of formula (Ilia)

with an activating agent; and (b) coupling the resultant activated acid with an amine of formula (b)
105. The method according to claim 104, wherein the activating agent is selected from thionyl chloride, 2-chloro-4,6-dimethoxy-1 ,3,5-triazine, 1-[3- (dimethylamino)propyl]-3-ethyl-carbodiimide and dicyclohexyl carbodiimide.
106. A compound of formula (IV):

(IV) and pharmaceutically acceptable salts thereof, wherein


R2 is selected from C3-6 cycloalkyl, indole, benzyl, and phenyl, wherein
phenyl and benzyl are each optionally substituted with 1 to 3 substituents independently selected from halogen, C1-6 alkyl, C1-6 alkoxy, C1-6 haloalkyl, C1-6 haloalkoxy, -CN, -C(O)ORC, -OH, and -NRdRe; and
the -C3-6 cycloalkyl is optionally substituted with 1 to 3 C1-6 alkyl groups;
Ar-R3 is selected from the group consisting of

R3 is selected from halogen, Ci-10 alkyl, C1-10 alkoxy, C1-10 haloalkyl, C1--I0 haloalkoxy, C(O)RC, piperidin-4-yl, and a -Y-NRfR9 group, wherein
the C1-10 alkyl and the C1-10 alkoxy are each optionally substituted with 1-3 substitutents selected from halogen, phenyl, and -OH; and
the nitrogen ring atom of the piperidin-4-yl is optionally substituted with -C(O)O-C1-6alkyl;
Y, at each occurrence, is independently a divalent C1-6 alkyl group or a covalent bond;
Rc, Rd and Re, at each occurrence, are independently H, C1-6 haloalkyl, or C1-6 alkyl;
Rf and R9, at each occurrence, are independently selected from the group consisting of -H, -C(O)RC, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, C1-10 alkyl, C3-6 cycloalkyl, -Y-phenyl, -C(O)-phenyl, -Y-(5-7 membered cycloheteroalkyl), -
Y-(5-7 membered heteroaryl), and -C2-6 alkyl-O-Y-(5-7 membered heteroaryl); or
alternatively, Rf and R9 taken together with the nitrogen atom to which they are bonded form a 5-7 membered cycloheteroalkyl group or a 5-7 membered heteroaryl group, the 5-7 membered cycloheteroalkyl group and the 5-7 membered heteroaryl group each containing up to two ring heteroatoms independently selected from nitrogen, oxygen, and sulfur, wherein
any sulfur atom in the ring is optionally substituted with 1 to 2 oxo groups,
one or more nitrogen atoms in the ring optionally are independently substituted with -C(O)R0, -C2^ alkyl-ORc, -C2-6 alkyl-NRdRe, -Y-
C(O)NRdRe, -S(O)2-(C1-6 alkyl), a -C1-6 alkyl-(phenyl)n group, a -C2-6 alkyl— (5-7 membered cycloheteroalkyl) group, C1-6 alkyl, -Y-(phenyl)q, C3. a cycloalkyl, or a 5-7 membered heteroaryl group;
one or more carbon atoms in the ring optionally are independently substituted with -C(O)-NRdR6, -Y-ORc, -Y-NRdRe, -Y-(phenyl)q, -Y-(5-7 membered cycloheteroalkyl), a -Y-(5-9 membered heteroaryl) group, an oxo group, CN, C3-8 cycloalkyl, or a -Y-O-(5-7 membered heteroaryl) group; and
each of the phenyl groups appearing anywhere in said Rf and R9 is optionally substituted with 1 to 3 substituents independently selected from halogen, Ci-6 alkyl, C1-6 haloalkyl, and C1-6 alkoxy, and
each of the 5-7 membered cycloheteroalkyl groups, the 5-7 membered heteroaryl groups, and the 5-9 membered heteroaryl groups appearing anywhere in said Rf and R9 is optionally substituted with 1 to 3 substituents independently selected from halogen and C1-6 alkyl;
n is 1 , 2, or 3;
q is 1 , 2, or 3; and
p is 1 , 2, 3, or 4,
with the proviso that when

Fc is 4-fluorophenyl, and p is 1 , then Ar-R is not 4-dimethylaminophenyl
107. The compound of claim 106, wherein

108. The compound of claim 106, wherein

109. The compound of claim 106, wherein p is 1.
110. The compound of claim 106, wherein R2 is phenyl optionally substituted with 1 to 3 substituents independently selected from halogen, C1-6 alkyl, CN, -C(O)OR0, and -NRdRe.
111. The compound of claim 110, wherein R2 is selected from the group of phenyl, a 4-dimethylaminophenyl group, a 4-fluoro-2-methylphenyl group, a 4-methylphenyl group, a 2-isopropylphenyl group, a 4-methoxyphenyl group, a 4-chlorophenyl group a 4-fluorophenyl group, and a 2-isopropylphenyl group.
1 12. The compound of claim 106, wherein R2 is C3.6 cycloalkyl optionally substituted with 1 to 3 C1-6 alkyl groups.
113. The compound of claim 106, wherein R2 is a 4-tertbutylcyclohexyl group.
114. The compound of claim 106, wherein Ar-R3 is selected from the group consisting of

115. The compound of claim 106, wherein Ar-R3 is
T S y&
116. The compound of claim 106, wherein R3 is NRfRg.
117. The compound of claim 116, wherein R3 is selected from the group of a diethylamino group, a dimethylamino group, an ethyl(methyl)amino group, an isopropyl(ethyl)amino group, a cyclopropyl(ethyl)amino group, a 1-[1 ,4]diazepanyl group, a 1-imidazolyl group, a 4-morpholinyl group, a 1-piperidinyl group, a 1- piperazinyl group, a 4-pyridyl group, a 1-pyrrolidyl group, a 4-thiomorpholinyl group, and a 4-methyl piperazin-1-yl group.
118. The compound of claim 106, wherein R3 is methyl or trifluoromethyl.
1 19. The compound of claim 106 selected from
N-{[6-(diethylamino)pyridin-3-yl]methyl}-N-(4-fluorophenyl)-2-furamide;
N-{[6-(diethylamino)pyridin-3-yl]methyl}-N-(4-fluorophenyl)-5-nitro-2-furamide;
N-{[6-(diethylamino)pyridin-3-yl]methyl}-N-(4-fluorophenyl)-1-benzothiophene-2- carboxamide;
N-(4-fluoro-2-methylphenyl)-N-[(2-piperidin-1-ylpyrimidin-5-yl)methyl]-2-furarnide;
N-(4-fluoro-2-methylphenyl)-5-nitro-N-[(2-piperidin-1-ylpyrimidin-5-yl)methyl]-2- furamide;
N-(2-isopropylphenyl)-N-[(2-piperidin-1-yl-1 ,3-thiazol-4-yl)methyl]-2-furamide;
N-(2-isopropylphenyl)-5-nitro-N-[(2-piperidin-1 -yl-1 ,3-thiazol-4-yl)methyl]-2- furamide;
N-(2-isopropylphenyl)-N-[(2-piperidin-1-yl-1 ,3-thiazol-4-yl)methyl]thiophene-2- carboxamide;
N-(2-isopropylphenyl)-N-[(2-piperidin-1-yl-1 ,3-thiazol-4-yl)methyl]-1- benzothiophene-2-carboxamide;
N-(2-isopropylphenyl)-N-[(2-piperidiπ-1-yl-1 ,3-thiazol-4-yl)methyl]-3-furamide;
N-{[6-(diethylamino)pyridin-3-yl]methyl}-N-(4-fluorophenyl)-1-methyl-1 H-pyrrole-2- carboxamide;
N-{[6-(diethylamino)pyridin-3-yl]methyl}-N-(4-fluorophenyl)thiophene-3- carboxamide;
N-{[6-(diethylamino)pyridin-3-yl]methyl}-N-(4-fluorophenyl)-3-furamide;
N-(4-chlorophenyl)-N-{[6-(diethylamino)pyridin-2-yl]methyl}-1-methyl-1 H-pyrrole- 2-carboxamide;
N-(4-chlorophenyl)-N-{[6-(diethylamino)pyridin-2-yl]methyl}-5-nitro-2-furamide;
N-C^chlorophenyO-N-^θ-CdiethylaminoJpyridin^-yljmethylJthiophene^- carboxamide;
N-(4-chlorophenyl)-N-{[6-(diethylamino)pyridin-2-yl]methyl}thiophene-3- carboxamide;
N-{4-[cyclopropyl(ethyl)amino]benzyl}-N-(4-methoxyphenyl)-5-nitro-2-furamide;
N-{3-[cyclopropyl(ethyl)amino]benzyl}-N-(4-fluorophenyl)-1-methyl-1 H-pyrrole-2- carboxamide;
N-{3-[cyclopropyl(ethyl)amino]benzyl}-N-(4-fluorophenyl)-2-furamide;
N-{3-[cyclopropyl(ethyl)amino]benzyl}-N-(4-fluorophenyl)-5-nitro-2-furamide;
N-{3-[cyclopropyl(ethyl)amino]benzyl}-N-(4-fluorophenyl)-3-furamide;
N-(4-fluoro-2-methylphenyl)-N-[(2-piperidin-1-ylpyrimidin-5-yl)methyl]thiophene-2- carboxamide;
N-(4-fluoro-2-methylphenyl)-N-[(2-piperidin-1-ylpyrimidin-5-yl)methyl]thiophene-3- carboxamide;
N-(2-isopropylphenyl)-N-[(2-piperidin-1-yl-1 ,3-thiazol-4-yl)methyl]thiophene-3- carboxamide;
N-(4-chlorophenyl)-N-{[6-(diethylamino)pyridin-2-yl]methyl}-2-furamide;
N-(4-chlorophenyl)-N-{[6-(diethylamino)pyridin-2-yl]methyl}-1-benzothiophene-2- carboxamide;
N-(4-chlorophenyl)-N-{[6-(diethylamino)pyridin-2-yl]methyl}-3-furamide;
N-(4-fluoro-2-methylphenyl)-1-methyl-N-[(2-piperidin-1-ylpyrimidin-5-yl)methyl]- 1 H-pyrrole-2-carboxamide;
N-{4-[cyclopropyl(ethyl)amino]benzyl}-N-(4-methoxyphenyl)-1-benzothiophene-2- carboxamide;
N-(4-fluorophenyl)-N-{[6-(4-methylpiperazin-1-yl)pyridin-3-yl]methyl}thiophene-2- carboxamide;
N-(4-fluorophenyl)-1 -methyl-N-{[6-(4-methylpiperazin-1 -yl)pyridin-3-yl]methyl}-1 H- pyrrole-2-carboxamide;
N-(4-fluorophenyl)-N-{[6-(4-methylpiperazin-1 -yl)pyridin-3-yl]methyl}-2-furamide;
N-(4-fluorophenyl)-N-{[6-(4-pyridin-2-ylpiperazin-1-yl)pyridin-3-yl]methyl}-2- furamide;
5-tert-butyl-N-(4-fluorophenyl)-2-methyl-N-[(2-morpholin-4-yl-1 ,3-thiazol-4- . yl)methyl]-3-furamide;
N-(4-fluorophenyl)-N-{[6-(4-methylpiperazin-1 -yl)pyridin-3-yl]methyl}-3-furamide;
N-(4-fluorophenyl)-1-methyl-N-{[6-(trifluoromethyl)pyridin-3-yl]methyl}-1 H-pyrrole- 2-carboxamide;
1 -methyl-1 H-pyrrole-2-carboxamide N-(4-fluorophenyl)-N-({6-[4-(4- fluorophenyl)piperazin-1-yl]pyridin-3-yl}methyl)-;
N-(4-fluorophenyl)-1-methyl-N-{[6-(4-pyridin-2-ylpiperazin-1-yl)pyridin-3- yl]methyl}-1 H-pyrrole-2-carboxamide;
N-(4-fluorophenyl)-1-methyl-N-({6-[4-(2-phenylethyl)piperazin-1-yl]pyridin-3- yl}methyl)-1 H-pyrrole-2-carboxamide;
N-[(6-{4-[bis(4-fluorophenyl)methyl]piperazin-1-yl}pyridin-3-yl)methyl]-N-(4- fluorophenyl )-1 -methyl-1 H-pyrrole-2-carboxamide;
N-(4-fluorophenyl)-N-({6-[4-(4-fluorophenyl)piperazin-1-yl]pyridin-3-yl}methyl)-3- furamide;
N-(4-fluorophenyl)-N-{[6-(4-pyridin-2-ylpiperazin-1-yl)pyridin-3-yl]methyl}-3- furamide;
N-(4-fluorophenyl)-N-({6-[4-(2-phenylethyl)piperazin-1-yl]pyridin-3-yl}methyl)-3- furamide;
N-[(6-{4-[bis(4-fluorophenyl)methyl]piperazin-1-yl}pyridin-3-yl)methyl]-N-(4- fluorophenyl)-3-furamide;
N-(4-fluorophenyl)-N-{[6-(trifluoromethyl)pyridin-3-yl]methyl}-3-furamide;
N-(4-tert-butylcyclohexyl)-N-[(6-chloropyridin-3-yl)methyl]-1-methyl-1 H-pyrrole-2- carboxamide;
N-(4-tert-butylcyclohexyl)-1-methyl-N-{[6-(4-methylpiperazin-1-yl)pyridin-3- yl]methyl}-1 H-pyrrole-2-carboxamide;
N-{[6-(diethylamino)pyridin-3-yl]methyl}-N-(4-fluorophenyl)thiophene-2- carboxamide;
N-(4-fluorophenyl)-1 -methyl-N-{[6-(4-methylpiperazin-1 -yl)pyridin-3-yl]methyl}-1 H- pyrrole-2-carboxamide;
N-(4-fluorophenyl)-N-{[6-(4-pyridin-2-ylpiperazin-1-yl)pyridin-3-yl]methyl}-2- furamide;
N-(4-fluorophenyl)-N-{[6-(4-methylpiperazin-1-yl)pyridin-3-yl]methyl}-2-furamide;
N-(4-fluorophenyl)-N-{[6-(4-phenylpiperazin-1-yl)pyridin-3-yl]methyl}furan-2- carboxamide;
N-(4-fluorophenyl)-N-({6-[4-(2-phenylethyl)piperazin-1-yl]pyridin-3- yl}methyl)furan-2-carboxamide;
N-({6-[4-(diphenylmethyl)piperazin-1 -yl]pyridin-3-yl}methyl)-N-(4- fluorophenyl)furan-2-carboxamide;
N-(4-fluorophenyl)-N-{[6-(4-pyrimidin-2-ylpiperazin-1-yl)pyridin-3-yl]methyl}furan- 2-carboxamide;
N-{[6-(4-benzylpiperazin-1-yl)pyridin-3-yl]methyl}-N-(4-fluorophenyl)furan-2- carboxamide;
N-(4-fluorophenyl)-N-({6-[4-(hydroxymethyl)piperidin-1-yl]pyridin-3- yl}methyl)furan-2-carboxamide;
N-({6-[4-(2-cyclohexylethyl)piperazin-1-yl]pyridin-3-yl}methyl)-N-(4- fluorophenyl)furan-2-carboxamide;
N-(4-fluorophenyl)-N-[(6-pyrrolidin-1-ylpyridin-3-yl)methyl]furan-2-carboxamide;
N-(4-fluorophenyl)-N-[(6-piperidin-1-ylpyridin-3-yl)methyl]furan-2-carboxamide;
N-{[6-(4-cyclohexylpiperazin-1-yl)pyridin-3-yl]methyl}-N-(4-fluorophenyl)furan-2- carboxamide;
N-(4-fluorophenyl)-N-({6-[4-(2-phenylethyl)piperazin-1-yl]pyridin-3- yl}methyl)thiophene-3-carboxamide;
N-(4-fluorophenyl)-N-({6-[4-(4-fluorophenyl)piperazin-1-yl]pyridin-3- yl}methyl)thiophene-3-carboxamide;
N-(4-fluorophenyl)-N-{[6-(4-phenylpiperazin-1-yl)pyridin-3-yl]methyl}thiophene-3- carboxamide;
N-{[6-(4-cyclohexylpiperazin-1 -yl)pyridin-3-yl]methyl}-N-(4- fluorophenyl)thiophene-3-carboxamide;
N-(4-fluorophenyl)-N-{[6-(4-pyridin-2-ylpiperazin-1-yl)pyridin-3- yl]methyl}thiophene-3-carboxamide;
N-(4-fluorophenyl)-N-{[6-(4-pyrimidin-2-ylpiperazin-1-yl)pyridin-3- yl]methyl}thiophene-3-carboxamide;
N-({6-[4-(diphenylmethyl)piperazin-1-yl]pyridin-3-yl}methyl)-N-(4- fluorophenyl)thiophene-3-carboxamide;
N-{[6-(4-benzylpiperazin-1-yl)pyridin-3-yl]methyl}-N-(4-fluorophenyl)thiophene-3- carboxamide;
N-({6-[4-(2-cyclohexylethyl)piperazin-1-yl]pyridin-3-yl}methyl)-N-(4- fluorophenyl)thiophene-3-carboxamide;
N-{[6-(3,5-dimethylpiperidin-1-yl)pyridin-3-yl]methyl}-N-(4-fluorophenyl)thiophene- 3-carboxamide;
N-(4-fluorophenyl)-N-[(6-piperidin-1-ylpyridin-3-yl)methyl]thiophene-3- carboxamide;
N-[(6-bromopyridin-3-yl)methyl]-N-(4-fluorophenyl)thiophene-3-carboxamide;
and pharmaceutically acceptable salts, hydrates, and esters thereof..
120. A pharmaceutical composition comprising the compound of any one of claims 106-1 19, or a pharmaceutically acceptable salt, hydrate or ester thereof and a pharmaceutically acceptable carrier or excipient.
121. The pharmaceutical composition of claim 106, further comprising an additional therapeutic agent.
122. A method of treating a disease or disease symptom selected from angina, hypertension, congestive heart failure, myocardial ischemia, and arrhythmia, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 106-1 19 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
123. A method of treating a disease or disease symptom selected from stroke, convulsion, epilepsy, traumatic brain injury, and neuronal disorder, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 106-119 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
124. A method of treating a disease or disease symptom selected from diabetes, urinary incontinence, hot flush, and thermal disregulation, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 106-1 19 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
125. A method of treating pain in a subject, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 106-1 19 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
126. A method of claim 125 wherein said pain is chronic pain.
127. A method of claim 126 wherein said chronic pain is associated with diabetes, post traumatic pain of amputation, lower back pain, spinal cord damage, cancer, chemical injury, chemotherapy induced peripheral neuropathy, toxins, major surgery, peripheral nerve damage due to traumatic injury, post-herpetic neuralgia, trigeminal neuralgia, lumbar or cervical radiculopathies, fibromyalgia, glossopharyngeal neuralgia, reflex sympathetic dystrophy, causalgia, thalamic syndrome, nerve root avulsion, reflex sympathetic dystrophy or post thoracotomy pain, nutritional deficiencies, viral infection, bacterial infection, metastatic infiltration, adiposis dolorosa, burns, central pain conditions related to thalamic conditions, or a combination thereof.
128. A method of claim 125 wherein said pain is chronic back pain.
129. A method of claim 125 wherein said pain is neuropathic pain.
130. A method of claim 125 wherein said pain is associated with diabetic neuropathy.
131. A method of claim 125 wherein said pain is associated with post-herpetic neuropathy.
132. A method of claim 125 wherein said pain is associated with post-herpetic fibromyalgia.
133. A method of treating a disease or disease symptom modulated by calcium channel Cav2, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 106-119 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
134. The method of claim 133, wherein the disease or disease symptom is modulated by calcium channel Cav2.2.
135. The method of any one of claims 122-134, wherein the subject is a mammal.
136. A method of modulating calcium channel activity in a subject, the method comprising administering the compound of any one of claims 106-119, or a pharmaceutically acceptable salt, hydrate or ester thereof, to a subject.
137. A method for making a compound of formula (IV) according to claim 106 and pharmaceutically acceptable salts thereof, comprising
(a) reacting a carboxylic acid compound of formula (IVa)

with an activating agent; and (b) coupling the resultant activated acid with an amine of formula (b)

(b)
138. The method according to claim 137, wherein the activating agent is selected from thionyl chloride, 2-chloro-4,6-dimethoxy-1 ,3,5-triazine, 1-[3- (dimethylamino)propyl]-3-ethyl-carbodiimide and dicyclohexyl carbodiimide.
139. A compound of formula (V):
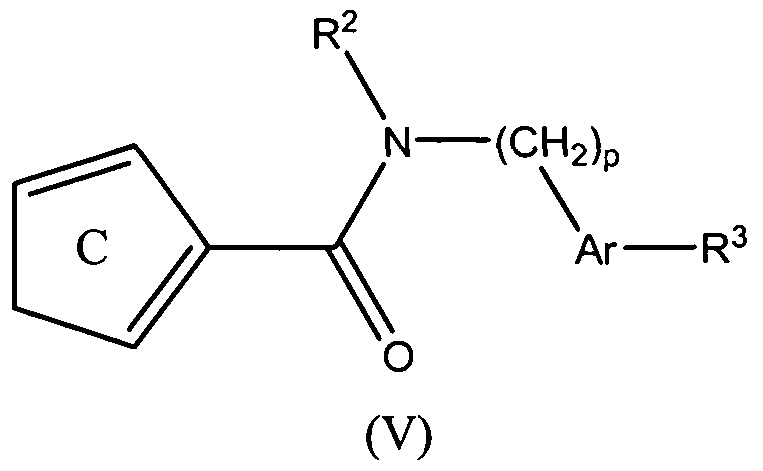


R2 is selected from C3-6 cycloalkyl, indole, benzyl, and phenyl, wherein
phenyl and benzyl are each optionally substituted with 1 to 3 substituents independently selected from halogen, C1-6 alkyl, C1-6 alkoxy, C1-6 haloalkyl, C1-6 haloalkoxy, -CN, -C(O)OR0, -OH, and -NRdRe; and
the -C3-6 cycloalkyl is optionally substituted with 1 to 3 C1-6 alkyl groups;
Ar-R3 is selected from the group consisting of

R3 is selected from halogen, C1-10 alkyl, C1-10 alkoxy, C1-10 haloalkyl, C1-10 haloalkoxy, C(O)RC, piperidin-4-yl, and a -Y-NRfRg group, wherein
the C1-10 alkyl and the C1-10 alkoxy are optionally substituted with 1-3 substitutents selected from halogen, phenyl, and -OH; and
the nitrogen ring atom of the piperidin-4-yl is optionally substituted with -C(O)O-C1-6alkyl; -
Y, at each occurrence, is independently a divalent C1-6 alkyl group or a covalent bond;
Rc, Rd and Re, at each occurrence, are independently H, C1-6 haloalkyl, or C1-6 alkyl; Rf and R9, at each occurrence, are independently selected from the group consisting of -H, -C(O)R0, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, C1-10 alkyl, C3-6 cycloalkyl, -Y-phenyl, -C(O)-phenyl, -Y-(5-7 membered cycloheteroalkyl), -Y-(5-7 membered heteroaryl), and -C2-6 alkyl-O-Y-(5-7 membered heteroaryl); or
alternatively, Rf and R9 taken together with the nitrogen atom to which they are bonded form a 5-7 membered cycloheteroalkyl group or a 5-7 membered heteroaryl group, the 5-7 membered cycloheteroalkyl group and the 5-7 membered heteroaryl group each containing up to two ring heteroatoms independently selected from nitrogen, oxygen, and sulfur, wherein
any sulfur atom in the ring is optionally substituted with 1 to 2 oxo groups,
one or more nitrogen atoms in the ring optionally are independently substituted with -C(O)RC, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, -Y-
C(O)NRdRe, -S(O)2-(Ci-6 alkyl), a -C1-6 alkyl-(phenyl)n group, a -C2-6 alkyl— (5-7 membered cycloheteroalkyl) group, C1-6 alkyl, -Y-(phenyl)q, C3- 8 cycloalkyl, or a 5-7 membered heteroaryl group;
one or more carbon atoms in the ring optionally are independently substituted with -C(O)-NRdRe, -Y-OR01 -Y-NRdRe, -Y-(phenyl)q, -Y-(5-7 membered cycloheteroalkyl), a -Y-(5-9 membered heteroaryl) group, an oxo group, CN, C3-8 cycloalkyl, or a -Y-O-(5-7 membered heteroaryl) group; and
each of the phenyl groups appearing anywhere in said Rf and R9 is optionally substituted with 1 to 3 substituents independently selected from halogen, Ci-6 alkyl, Ci-6 haloalkyl, and Ci-6 alkoxy, and
each of the 5-7 membered cycloheteroalkyl groups, the 5-7 membered heteroaryl groups, and the 5-9 membered heteroaryl groups appearing anywhere in said Rf and R9 is optionally substituted with 1 to 3 substituents independently selected from halogen and Ci-6 alkyl;
n is 1 , 2, or 3;
q is 1 , 2, or 3; and
p is 1 , 2, 3, or 4.
140. A compound of claim 139, wherein p is 1.
141. A compound of claim 139, wherein one of R8 and R9 is methyl, and the other of R8 and R9 is tert-butyl.
142. A compound of claim 139, wherein R2 is phenyl optionally substituted with 1 to 2 substituents independently selected from halogen, C1-6 alkyl, CN, -C(O)OR0, and -NRdRe.
143. The compound of claim 142, wherein R2 is a 4-fluorophenyl group.
144. A compound of claim 139, wherein Ar-R3 is 2-morpholin-4-yl-1 ,3-thiazol-4-yl.
145. A compound of claim 139 selected from
1-tert-butyl-N-(4-fluorophenyl)-5-methyl-N-[(2-morpholin-4-yl-1 ,3-thiazol-4- yl)methyl]-1 H-pyrazole-3-carboxamide;
1-tert-butyl-N-(4-fluorophenyl)-3-methyl-N-[(2-morpholin-4-yl-1 ,3-thiazol-4- yl)methyl]-1 H-pyrazole-5-carboxamide;
and pharmaceutically acceptable salts, hydrates, and esters thereof..
146. A pharmaceutical composition comprising the compound of any one of claims 139-145 or a pharmaceutically acceptable salt, hydrate or ester thereof and a pharmaceutically acceptable carrier or excipient.
147. The pharmaceutical composition of claim 146, further comprising an additional therapeutic agent.
148. A method of treating a disease or disease symptom selected from angina, hypertension, congestive heart failure, myocardial ischemia, and arrhythmia, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 139-145 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
149. A method of treating a disease or disease symptom selected from stroke, convulsion, epilepsy, traumatic brain injury, and neuronal disorder, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 139-145 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
150. A method of treating a disease or disease symptom selected from diabetes, urinary incontinence, hot flush, and thermal disregulation, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 139-145 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
151. A method of treating pain in a subject, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 139-145 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
152. A method of claim 151 wherein said pain is chronic pain.
153. A method of claim 152 wherein said chronic pain is associated with diabetes, post traumatic pain of amputation, lower back pain, spinal cord damage, cancer, chemical injury, chemotherapy induced peripheral neuropathy, toxins, major surgery, peripheral nerve damage due to traumatic injury, post-herpetic neuralgia, trigeminal neuralgia, lumbar or cervical radiculopathies, fibromyalgia, glossopharyngeal neuralgia, reflex sympathetic dystrophy, causalgia, thalamic syndrome, nerve root avulsion, reflex sympathetic dystrophy or post thoracotomy pain, nutritional deficiencies, viral infection, bacterial infection, metastatic infiltration, adiposis dolorosa, burns, central pain conditions related to thalamic conditions, or a combination thereof.
154. A method of claim 151 wherein said pain is chronic back pain.
155. A method of claim 151 wherein said pain is neuropathic pain.
156. A method of claim 151 wherein said pain is associated with diabetic neuropathy.
157. A method of claim 151 wherein said pain is associated with post-herpetic neuropathy.
158. A method of claim 151 wherein said pain is associated with post-herpetic fibromyalgia.
159. A method of treating a disease or disease symptom modulated by calcium channel Cav2, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 139-145 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
160. The method of claim 159, wherein the disease or disease symptom is modulated by calcium channel Cav2.2.
161. A method of modulating calcium channel activity in a subject, the method comprising administering the compound of any one of claims 139-145, or a pharmaceutically acceptable salt, hydrate or ester thereof, to a subject.
162. The method of any one of claims 148-161 , wherein the subject is a mammal.
163. A method for making a compound of formula (V) according to claim 139 and pharmaceutically acceptable salts thereof, comprising
(a) reacting a carboxylic acid compound of formula (Va)

with an activating agent; and (b) coupling the resultant activated acid with an amine of formula (b)
(b)
164. The method according to claim 162, wherein the activating agent is selected from thionyl chloride, 2-chloro-4,6-dimethoxy-1 ,3,5-triazine, 1-[3- (dimethylamino)propyl]-3-ethyl-carbodiimide and dicyclohexyl carbodiimide.
165. A compound of formula (Vl)
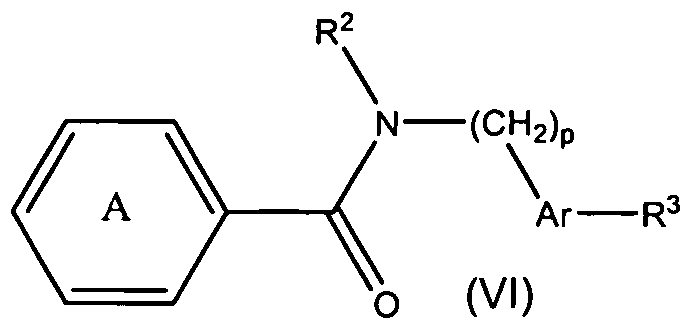 and pharmaceutically acceptable salts thereof, wherein
and pharmaceutically acceptable salts thereof, wherein

R2 is selected from C3-6 cycloalkyl, indole, benzyl, and phenyl, wherein
phenyl and benzyl are each optionally substituted with 1 to 3 substituents independently selected from halogen, C1-6 alkyl, Ci-6 alkoxy, C1-6 haloalkyl, Ci-6 haloalkoxy, -CN, -C(O)ORC, -OH, and -NRdRe; and
the -C3-6 cycloalkyl is optionally substituted with 1 to 3 C1-6 alkyl groups;
Ar-R3 is selected from the group consisting of

R3 is selected from halogen, C,.1O alkyl, C1-10 alkoxy, C1-10 haloalkyl, C1-10 haloalkoxy, C(O)RC, piperidin-4-yl, and a -Y-N Rf R9 group, wherein
the C1-10 alkyl and the C1-10 alkoxy are optionally substituted with 1-3 substitutents selected from halogen, phenyl, and -OH; and
the nitrogen ring atom of the piperidin-4-yl is optionally substituted with -C(O)O-C1-6alkyl;
Y, at each occurrence, is independently a divalent C1-6 alkyl group or a covalent bond;
Rc, Rd and R6, at each occurrence, are independently H, C1-6 haloalkyl, or C1-6 alkyl; Rf and R9, at each occurrence, are independently selected from the group consisting of -H, -C(O)RC, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, C1-10 alkyl, C3-6 cycloalkyl, -Y-phenyl, -C(O)-phenyl, -Y-(5-7 membered cycloheteroalkyl), -Y-(5-7 membered heteroaryl), and -C2-6 alkyl-O-Y-(5-7 membered heteroaryl); or
alternatively, Rf and R9 taken together with the nitrogen atom to which they are bonded form a 5-7 membered cycloheteroalkyl group or a 5-7 membered heteroaryl group, the 5-7 membered cycloheteroalkyl group and the 5-7 membered heteroaryl group each containing up to two ring heteroatoms independently selected from nitrogen, oxygen, and sulfur, wherein
any sulfur atom in the ring is optionally substituted with 1 to 2 oxo groups,
one or more nitrogen atoms in the ring optionally are independently substituted with -C(O)Rc, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, -Y-
C(O)NRdRe, -S(O)2-(C1-6 alkyl), a -C1-6 alkyl-(phenyl)n group, a -C2-6 alkyl— (5-7 membered cycloheteroalkyl) group, Ci-6 alkyl, -Y-(phenyl)q, C3. 8 cycloalkyl, or a 5-7 membered heteroaryl group;
one or more carbon atoms in the ring optionally are independently substituted with -C(O)-NRdRe, -Y-ORc -Y-NRdRe, -Y-(phenyl)q, -Y-(5-7 membered cycloheteroalkyl), a -Y-(5-9 membered heteroaryl) group, an oxo group, CN, C3-8 cycloalkyl, or a -Y-O-(5-7 membered heteroaryl) group; and
each of the phenyl groups appearing anywhere in said Rf and R9 is optionally substituted with 1 to 3 substituents independently selected from halogen, Ci-6 alkyl, Ci-6 haloalkyl, and C1-6 alkoxy, and
each of the 5-7 membered cycloheteroalkyl groups, the 5-7 membered heteroaryl groups, and the 5-9 membered heteroaryl groups appearing anywhere in said Rf and R9 is optionally substituted with 1 to 3 substituents independently selected from halogen and C1-6 alkyl;
n is 1 , 2, or 3;
q is 1 , 2, or 3; and
p is 1 , 2, 3, or 4..
166. The compound of claim 165, wherein

167. The compound of claim 165, wherein p is 1.
168. The compound of claim 165, wherein R2 is phenyl optionally substituted with 1 to 2 substituents independently selected from halogen, C1-6 alkyl, CN, -C(O)OR0, and
169. The compound of claim 168, wherein R2 is selected from a 4-fluorophenyl group, a 4-chlorophenyl group, a 2-methyl-4-fluorophenyl group, a 4-methylphenyl group, a 2-isopropylphenyl group, a 4-methoxyphenyl group, a 3- trifluoromethylphenyl group, a 4-trifluoromethylphenyl group, a 3-tertbutylphenyl group, a 5-chloro-2-methylphenyl group, a 4-hydroxyphenyl group, and a 4- benzyloxyphenyl group.
170. The compound of claim 165, wherein R2 is -C3-6 cycloalkyl is optionally substituted with 1 -3 C1-6 alkyl groups.
171. The compound of claim 170, wherein R2 is selected from cyclohexyl, cyclopentyl, 4-tert-butylcylcohexyl, 2,3-dihydro-1 H-inden-2-yl, and cyclopropylmethyl.
172. The compound of claim 165, wherein Ar-R3 is selected from the group consisting of

173. The compound of claim 165, wherein Ar-R3 is

174. The compound of claim 165, wherein R3 is NRfRg.
175. The compound of claim 174 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R3 is selected from NH2, NH-(C1-6 alkyl), N(Ci-6 alkyl)2, NH- (C3-6 cycloalkyl), N(C1-6 alkyl)-(C3^ cycloalkyl), an N(C1-6 alkyl)-(C2-6 alkyl)-ORc group, N(C1-6 alkyl)-Y-(5-7 membered cycloheteroalkyl), N(C1-6 alkyl)-phenyl, N(C1-6 alkyl)-Y-(5-7 membered heteroaryl), and N(C1-6 alkyl)-C2-6 alkyl-O-Y-(5-7 membered heteroaryl) , wherein each of the phenyl , the 5-7 membered cycloheteroalkyl, and the 5-7 heteroaryl are optionally substituted with 1 to 3 substituents independently selected from halogen and C1-6 alkyl.
176. The compound of claim 175, wherein R3 is selected from the group of a diethylamino group, a dimethylamino group, an ethyl(methyl)amino group, an isopropyl(ethyl)amino group, and a cyclopropyl(ethyl)amino group,.
177. The compound of claim 174 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R3 is an optionally substituted 5-7 membered cycloheteroalkyl group or an optionally substituted 5-7 membered heteroaryl group.
178. The compound of claim 177 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R3 is selected from a diazepanyl group, an imidazolyl group, a morpholinyl group, a piperidinyl group, a piperazinyl group, a pyridyl group, a pyrrolidyl group, and a thiomorpholinyl group, wherein each of these groups optionally includes a nitrogen ring atom substituted with -C(O)RC, -C2-6 alkyl-ORc, - C2-6 alkyl-NRdRe, -Y-C(O)NRdRe, -S(O)2-(Ci-6 alkyl), -C2-6 alkyl-(5-7 membered cycloheteroalkyl), C1-6 alkyl, or 5-7 membered heteroaryl, a carbon ring atom substituted with -C(O)-NRdRe, -Y-ORC, -Y-NRdR6, -Y-phenyl, -Y-(5-7 membered cycloheteroalkyl), -Y-(5-9 membered heteroaryl), or -Y-O-(5-7 membered heteroaryl), and/or a sulfur ring atom substituted with 1 or 2 oxo groups, wherein each of the phenyl groups is optionally substituted with 1 to 3 substituents independently selected from halogen, C1-6 alkyl, C1-6 haloalkyl, and C1-6 alkoxy, and each of the 5-7 membered cycloheteroalkyl, the 5-7 membered heteroaryl, and the 5- 9 membered heteroaryl immediately above is optionally substituted with 1 to 3 substituents independently selected from halogen and C1-6 alkyl.
179. The compound of claim 178 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R3 is selected from the group consisting of a 1-[1 ,4]diazepanyl group, a 1 -imidazolyl group, a 4-morpholinyl group, a 1 -piperidinyl group, a 1- piperazinyl group, a 4-pyridyl group, a 1 -pyrrolidyl group, and a 4-thiomorpholinyl group.
180. The compound of claim 165 selected from
N-(4-fluorophenyl)-N-{[6-(4-methylpiperazin-1-yl)pyridin-3-yl]methyl}nicotinamide;
N-(4-fluorophenyl)-N-{[6-(4-methylpiperazin-1-yl)pyridin-3- yl]methyl}isonicotinamide;
2-chloro-N-(4-fluorophenyl)-N-{[6-(4-methylpiperazin-1-yl)pyridin-3- yl]methyl}nicotinamide;
N-benzyl-6-chloro-N-({2-[cyclopropyl(ethyl)amino]-1 ,3-thiazol-4- yl}methyl)nicotinamide;
6-chloro-N-cyclohexyl-N-({2-[cyclopropyl(ethyl)amino]-1 ,3-thiazol-4- yl}methyl)nicotinamide;
6-chloro-N-cyclohexyl-N-[(2-morpholin-4-yl-1 ,3-thiazol-4-yl)methyl]nicotinamide;
N-benzyl-6-chloro-N-[(2-morpholin-4-yl-1 ,3-thiazol-4-yl)methyl]nicotinamide;
N-[(2-cyclohexyl-1 ,3-thiazol-4-yl)methyl]-N-(4-fluorophenyl)isonicotinamide;
2-chloro-N-(4-fluorophenyl)-N-[(2-morpholin-4-yl-1 ,3-thiazol-4- yl)methyl]nicotinamide;
tert-butyl 4-(4-{[(4-fluorophenyl)(isonicotinoyl)amino]methyl}-1 ,3-thiazol-2- yl)piperidine-1 -carboxylate;
N-cyclohexyl-N-({2-[cyclopropyl(ethyl)amino]-1 ,3-thiazol-4-yl}methyl)nicotinamide;
N-(4-fluorophenyl)-N-[(2-piperidin-4-yl-1 ,3-thiazol-4-yl)methyl]isonicotinamide;
6-chloro-N-({2-[cyclopropyl(ethyl)amino]-1 ,3-thiazol-4-yl}methyl)-N-(4- fluorophenyl)nicotinamide;
N-cyclohexyl-N-({2-[cyclopropyl(ethyl)amino]-1 ,3-thiazol-4-yl}methyl)quinoline-2- carboxamide;
2-chloro-N-cyclohexyl-N-({2-[cyclopropyl(ethyl)amino]-1 ,3-thiazol-4- yl}methyl)nicotinamide;
2,5-dichloro-N-cyclohexyl-N-({2-[cyclopropyl(ethyl)amino]-1 ,3-thiazol-4- yl}methyl)nicotinamide;
2-chloro-N-cyclohexyl-N-({2-[cyclopropyl(ethyl)amino]-1 ,3-thiazol-4- yl}methyl)isonicotinamide;
2-chloro-N-cyclohexyl-N-({2-[cyclopropyl(ethyl)amino]-1 ,3-thiazol-4-yl}methyl)-6- methoxyisonicotinamide;
N-cyclohexyl-N-({2-[cyclopropyl(ethyl)amino]-1 ,3-thiazol-4-yl}methyl)pyridine-2- carboxamide;
1 ,3-thiazol-4-yl}methyl)amino}piperidin-1-yl)carbonyl]piperidine-1-carboxylate;
N-^-CbenzyloxyJphenyll-N-^-fcyclopropyKethyOaminol-i ,3-thiazol-4- yl}methyl)nicotinamide;
N-({2-[cyclopropyl(ethyl)amino]-1 ,3-thiazol-4-yl}methyl)-N-(4- hydroxyphenyl)nicotinamide;
N-(3-tert-butylphenyl)-N-({2-[cyclopropyl(ethyl)amino]-1 ,3-thiazol-4- yl}methyl)nicotinamide;
N-(5-chloro-2-methylphenyl)-N-({2-[cyclopropyl(ethyl)amino]-1 ,3-thiazol-4- yl}methyl)nicotinamide;
N-{[6-(4-ethylpiperazin-1-yl)pyridin-3-yl]methyl}-N-(4-fluorophenyl)nicotinamide;
N-(4-fluorophenyl)-N-{[6-(4-methyl-1 ,4-diazepan-1-yl)pyridin-3- yl]methyl}nicotinamide;
N-(4-fluorophenyl)-N-{[6-(3-oxopiperazin-1-yl)pyridin-3-yl]methyl}nicotinamide;
N-(4-fluorophenyl)-N-({6-[4-(hydroxymethyl)piperidin-1-yl]pyridin-3- yl}methyl)nicotinamide;
N-(4-fluorophenyl)-N-{[6-(4-hydroxypiperidin-1-yl)pyridin-3- yl]methyl}nicotinamide;
N-(4-fluorophenyl)-N-{[6-(3-hydroxypiperidin-1-yl)pyridin-3- yl]methyl}nicotinamide;
N-(4-fluorophenyl)-N-{[6-(3-hydroxypyrrolidin-1-yl)pyridin-3- yl]methyl}nicotinamide;
N-(4-fluorophenyl)-N-({6-[4-(2-hydroxyethyl)piperidin-1 -yl]pyridin-3- yl}methyl)nicotinamide;
N-(4-fluorophenyl)-N-({6-[4-(2-hydroxyethyl)piperazin-1-yl]pyridin-3- yl}methyl)nicotinamide;
N-(4-fluorophenyl)-N-({6-[(2-hydroxyethyl)(methyl)amino]pyridin-3- yl}methyl)nicotinamide;
N-(4-fluorophenyl)-N-{[6-(4-morpholin-4-ylpiperidin-1-yl)pyridin-3- yl]methyl}nicotinamide;
N-{[6-(3-carbamoylpiperidin-1-yl)pyridin-3-yl]methyl}-N-(4- fluorophenyl)nicotinamide;
N-{[6-(4-carbamoylpiperidin-1 -yl)pyridin-3-yl]methyl}-N-(4- fluorophenyl)nicotinamide;
N-(4-fluorophenyl)-N-({6-[3-(hydroxymethyl)piperidin-1-yl]pyridin-3- yl}methyl)nicotinamide;
N-[(2-bromo-1 ,3-thiazol-4-yl)methyl]-N-(4-fluorophenyl)nicotinamide;
N-(4-fluorophenyl)-N-[(6-morpholin-4-ylpyridin-3-yl)methylInicotinamide;
N-(4-fluorophenyl)-N-{[2-(4-methylpiperazin-1-yl)-1 ,3-thiazol-4- yl]methyl}nicotinamide;
N-(4-fluorophenyl)-N-{[2-(4-methyl-1 ,4-diazepan-1 -yl)-1 ,3-thiazol-4- yl]methyl}nicotinamide;
N-(4-fluorophenyl)-N-[(2-morpholin-4-yl-1 ,3-thiazol-4-yl)methyl]nicotinamide;
N-{[2-(diethylamino)-1 ,3-thiazol-4-yl]methyl}-N-(4-fluorophenyl)nicotinamide;
N-{[2-(4-cyanopiperidin-1 -yl)-1 ,3-thiazol-4-yl]methyl}-N-(4- fluorophenyl)nicotinamide;
N-(4-fluorophenyl)-N-({2-[(2-hydroxyethyl)(methyl)amino]-1 ,3-thiazol-4- yl}methyl)nicotinamide;
N-(4-fluorophenyl)-N-{[2-(4-hydroxypiperidin-1 -yl)-1 ,3-thiazol-4- yl]methyl}nicotinamide;
N-(4-fluorophenyl)-N-{[2-(3-hydroxypiperidin-1-yl)-1 ,3-thiazol-4- yl]methyl}nicotinamide;
N-(4-fluorophenyl)-N-({2-[4-(hydroxymethyl)piperidin-1-yl]-1 ,3-thiazol-4- yl}methyl)nicotinamide;
N-(4-fluorophenyl)-N-({2-[3-(hydroxymethyl)piperidin-1-yl]-1 ,3-thiazol-4- yl}methyl)nicotinamide;
N-[(6-chloropyridin-3-yl)methyl]-N-cyclopentylnicotinamide;
N-[(6-chloropyridin-3-yl)methyl]-N-cyclohexylnicotinamide;
6-chloro-N-(4-fluorophenyl)-N-[(2-morpholin-4-yl-1 ,3-thiazol-4- yl)methyl]nicotinamide;
N-(4-fluorophenyl)-N-[(2-morpholin-4-yl-1 ,3-thiazol-4-yl)methyl]quinoline-2- carboxamide;
N-cyclopentyl-N-({6-[4-(hydroxymethyl)piperidin-1-yl]pyridin-3- yl}methyl)nicotinamide;
N-cyclohexyl-N-{[6-(4-methylpiperazin-1-yl)pyridin-3-yl]methyl}nicotinamide;
N-cyclohexyl-N-({6-[4-(hydroxymethyl)piperidin-1-yl]pyridin-3- yl}methyl)nicotinamide;
N-^θ-chloropyridin-S-yOmethyll-N^-fluorophenyOnicotinamide;
N-({2-[cyclopropyl(ethyl)amino]-1 ,3-thiazol-4-yl}methyl)-N-(4- fluorophenyl)isonicotinamide;
N-cyclopentyl-N-{[6-(4-methylpiperazin-1-yl)pyridin-3-yl]methyl}nicotinamide;
N-[(2-bromo-1 ,3-thiazol-4-yl)methyl]-N-(4-fluorophenyl)isonicotinamide;
N-(4-tert-butylcyclohexyl)-N-({2-[cyclopropyl(ethyl)amino]-1 ,3-thiazol-4- yl}methyl)nicotinamide;
N-(4-fluorophenyl)-N-{[2-(4-methylpiperazin-1-yl)-1 ,3-thiazol-4- yl]methyl}isonicotinamide;
N-(4-fluorophenyl)-N-({2-[4-(hydroxymethyl)piperidin-1 -yl]-1 ,3-thiazol-4- yl}methyl)isonicotinamide;
N-(4-tert-butylcyclohexyl)-N-{[2-(4-methylpiperazin-1-yl)-1 ,3-thiazol-4- yl]methyl}nicotinamide;
N-{[6-(cyclopentylamino)pyridin-3-yl]methyl}-N-(4-fluorophenyl)nicotinamide;
N-({6-[(4-tert-butylcyclohexyl)amino]pyridin-3-yl}methyl)-N-(4- fluorophenyl)nicotinamide;
N-[(6-acetylpyridin-3-yl)methyl]-N-(4-fluorophenyl)nicotinamide;
Λ/-(4-fluorophenyl)-A/-{[6-(1-hydroxy-1-methylethyl)pyridin-3- yl]methyl}nicotinamide;
N-(4-fluorophenyl)-N-{[6-(trifluoromethyl)pyridin-3-yl]methyl}nicotinamide;
N-(2,3-dihydro-1 H-inden-2-yl)-N-{[6-(4-methylpiperazin-1-yl)pyridin-3- yl]methyl}nicotinamide;
N-(2,3-dihydro-1 H-inden-2-yl)-N-({6-[4-(4-fluorophenyl)piperazin-1-yl]pyridin-3- yl}methyl)nicotinamide;
N-(2,3-dihydro-1 H-inden-2-yl)-N-{[6-(4-pyridin-2-ylpiperazin-1-yl)pyridin-3- yl]methyl}nicotinamide;
N-(4-tert-butylcyclohexyl)-N-[(6-chloropyridin-3-yl)methyl]nicotinamide;
N-[(6-chloropyridin-3-yl)methyl]-N-(4-fluorophenyl)pyridine-2-carboxamide;
N-^θ-chloropyridin-S-yOmethyll-N-^trifluoromethyOphenylJnicotinamide;
N-[(6-chloropyridin-3-yl)methyl]-N-[4-(trifluoromethyl)phenyl]nicotinamide;
N-(4-fluorophenyl)-N-{[6-(1-hydroxy-1-methylethyl)pyridin-3- yl]methyl}nicotinamide;
N-(4-fluorophenyl)-N-{[6-(1-hydroxy-1 ,2-dimethylpropyl)pyridin-3- yl]methyl}nicotinamide;
N-(4-fluorophenyl)-N-({6-[1-(4-fluorophenyl)-1-hydroxyethyl]pyridin-3- yl}methyl)nicotinamide;
N-(4-fluorophenyl)-N-{[6-(2,2,2-trifluoroethoxy)pyridin-3-yl]methyl}nicotinamide;
N-(4-tert-butylcyclohexyl)-N-{[6-(4-methylpiperazin-1-yl)pyridin-3- yl]methyl}nicotinamide;
N-(4-fluorophenyl)-N-{[6-(4-methylpiperazin-1-yl)pyridin-3-yl]methyl}pyridine-2- carboxamide;
(trifluoromethyl)phenyl]nicotinamide N-{[6-(4-methylpiperazin-1-yl)pyridin-3- yl]methyl}-N-[3-;
[4- (trifluoromethyl)phenyl]nicotinamide N-{[6-(4-methylpiperazin-1 -yl)pyridin-3- yl]methyl}-N-;
2,6-dichloro-N-(4-fluorophenyl)-N-[(2-morpholin-4-yl-1 ,3-thiazol-4- yl)methyl]isonicotinamide;
N-({2-[cyclopropyl(ethyl)amino]-1 , 3-th iazol-4-yl}methy I)-N-I H-indol-5- ylnicotinamide;
N-(4-fluorophenyl)-N-({6-[4-(2-phenylethyl)piperazin-1-yl]pyridin-3- yl}methyl)pyridine-3-carboxamide;
N-{[6-(4-cyclohexylpiperazin-1-yl)pyridin-3-yl]methyl}-N-(4-fluorophenyl)pyridine- 3-carboxamide;
N-(4-fluorophenyl)-N-{[6-(4-pyridin-2-ylpiperazin-1-yl)pyridin-3-yl]methyl}pyridine- 3-carboxamide;
N-(4-fluorophenyl)-N-{[6-(4-pyrimidin-2-ylpiperazin-1-yl)pyridin-3- yl]methyl}pyridine-3-carboxamide;
N-({6-[4-(diphenylmethyl)piperazin-1 -yl]pyridin-3-yl}methyl)-N-(4- fluorophenyl)pyridine-3-carboxamide;
N-{[6-(4-benzylpiperazin-1-yl)pyridin-3-yl]methyl}-N-(4-fluorophenyl)pyridine-3- carboxamide;
N-({6-[4-(2-cyclohexylethyl)piperazin-1-yl]pyridin-3-yl}methyl)-N-(4- fluorophenyl)pyridine-3-carboxamide;
N-(4-fluorophenyl)-N-[(6-piperidin-1-ylpyridin-3-yl)methyl]pyridine-3-carboxamide;
and pharmaceutically acceptable salts, hydrates, and esters thereof.
181. A pharmaceutical composition comprising the compound of any one of claims 165-180, or a pharmaceutically acceptable salt, hydrate or ester thereof and a pharmaceutically acceptable carrier or excipient.
182. The pharmaceutical composition of claim 181 , further comprising an additional therapeutic agent.
183. A method of treating a disease or disease symptom selected from angina, hypertension, congestive heart failure, myocardial ischemia, and arrhythmia, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 165-180 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
184. A method of treating a disease or disease symptom selected from stroke, convulsion, epilepsy, traumatic brain injury, and neuronal disorder, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 165-180 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
185. A method of treating a disease or disease symptom selected from diabetes, urinary incontinence, hot flush, and thermal disregulation, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 165-180 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
186. A method of treating pain in a subject, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 165-180 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
187. A method of claim 186 wherein said pain is chronic pain.
188. A method of claim 187 wherein said chronic pain is associated with diabetes, post traumatic pain of amputation, lower back pain, spinal cord damage, cancer, chemical injury, chemotherapy induced peripheral neuropathy, toxins, major surgery, peripheral nerve damage due to traumatic injury, post-herpetic neuralgia, trigeminal neuralgia, lumbar or cervical radiculopathies, fibromyalgia, glossopharyngeal neuralgia, reflex sympathetic dystrophy, causalgia, thalamic syndrome, nerve root avulsion, reflex sympathetic dystrophy or post thoracotomy pain, nutritional deficiencies, viral infection, bacterial infection, metastatic infiltration, adiposis dolorosa, burns, central pain conditions related to thalamic conditions, or a combination thereof.
189. A method of claim 186 wherein said pain is chronic back pain.
190. A method of claim 186 wherein said pain is neuropathic pain.
191. A method of claim 186 wherein said pain is associated with diabetic neuropathy.
192. A method of claim 186 wherein said pain is associated with post-herpetic neuropathy.
193. A method of claim 186 wherein said pain is associated with post-herpetic fibromyalgia.
194. A method of treating a disease or disease symptom modulated by calcium channel Cav2, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 165-180 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
195. The method of claim 152, wherein the disease or disease symptom is modulated by calcium channel Cav2.2.
196. A method of modulating calcium channel activity in a subject, the method comprising administering the compound of any one of claims 165-180, or a pharmaceutically acceptable salt, hydrate or ester thereof, to a subject.
197. The method of any one of claims 183-197 wherein the subject is a mammal.
198. A method for making a compound of formula (Vl) according to claim 165 and pharmaceutically acceptable salts thereof, comprising
(a) reacting a carboxylic acid compound of formula (Via)

with an activating agent; and (b) coupling the resultant activated acid with an amine of formula (b)
(b)
199. The method according to claim 198, wherein the activating agent is selected from thionyl chloride, 2-chloro-4,6-dimethoxy-1 ,3,5-triazine, 1-[3- (dimethylamino)propyl]-3-ethyl-carbodiimide and dicyclohexyl carbodiimide.
200. A compound of formula (VII):

(VII) and pharmaceutically acceptable salts thereof, wherein
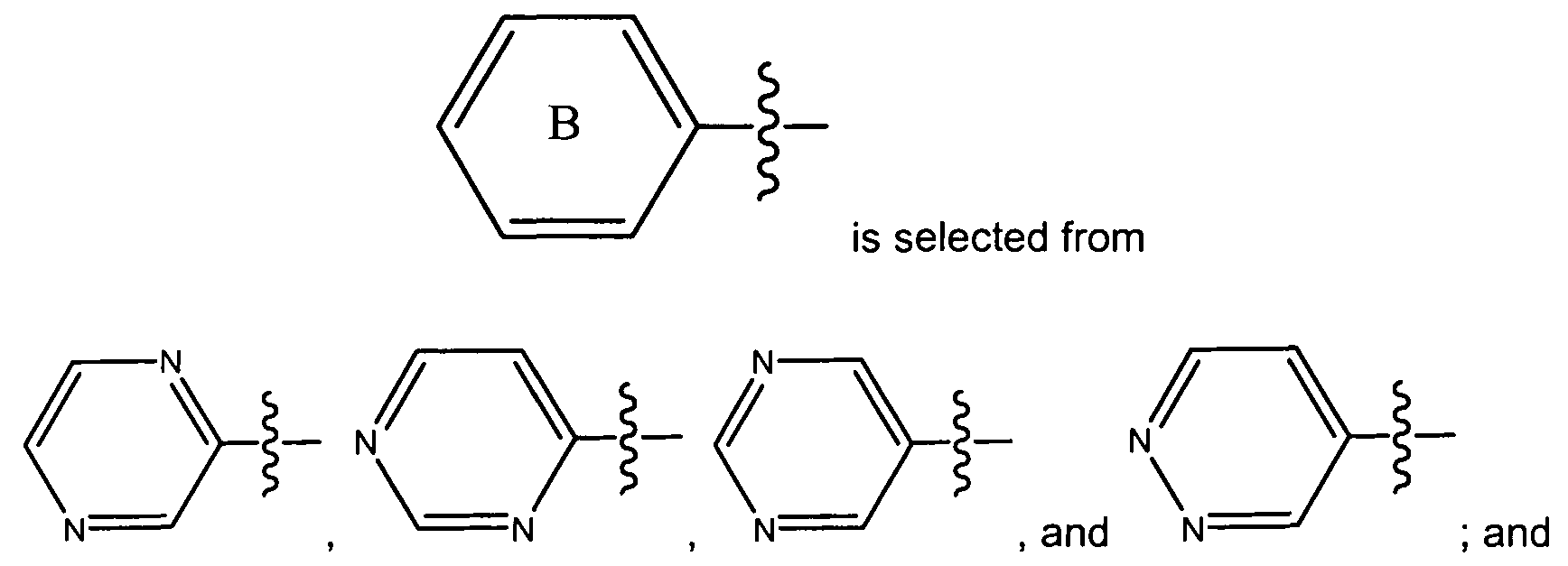
phenyl and benzyl are each optionally substituted with 1 to 3 substituents independently selected from halogen, Ci-6 alkyl, C1-6 alkoxy, C1-6 haloalkyl, C1. 6 haloalkoxy, -CN, -C(O)ORC, -OH, and -NRdRe; and
the -C3-6 cycloalkyl is optionally substituted with 1 to 3 C1-6 alkyl groups;
Ar-R3 is selected from the group consisting of

R3 is selected from halogen, C1-10 alkyl, C1-10 alkoxy, C1-10 haloalkyl, C1-10 haloalkoxy, C(O)R0 piperidin-4-yl, and a -Y-NRfRg group, wherein the C1- 1o alkyl and the C1--I0 alkoxy are optionally substituted with 1-3 substitutents selected from halogen, phenyl, and -OH; and
the nitrogen ring atom of the piperidin-4-yl group is optionally substituted with -C(O)O-C1-6alkyl;
Y, at each occurrence, is independently a divalent C1-6 alkyl group or a covalent bond;
Rc, Rd and Re, at each occurrence, are independently H or Ci-6 alkyl;
Rf and R9, at each occurrence, are independently selected from the group consisting of -H, -C(O)Rc, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, C1-6 alkyl, a C3- 6 cycloalkyl, -Y-phenyl, -C(O)-phenyl, -Y-(5-7 membered cycloheteroalkyl), -Y-(5-7 membered heteroaryl), and -C2-6 alkyl-O-Y-(5-7 membered heteroaryl), or
alternatively, Rf and R9 taken together with the nitrogen atom to which they are bonded form a 5-7 membered cycloheteroalkyl group or a 5-7 membered heteroaryl group, the 5-7 membered cycloheteroalkyl group and the 5-7 membered heteroaryl group containing up to two ring heteroatoms independently selected from nitrogen, oxygen, and sulfur; wherein
any sulfur atom in the ring is optionally substituted with 1 to 2 oxo groups,
one or more nitrogen atoms in the ring optionally are independently substituted with -C(O)RC, -C2-6 alkyl-ORc, -C2-6 alkyl-NRdRe, -Y-
C(O)NRdRe, -S(O)2-(C1-6 alkyl), a -C1-6 alkyl-(phenyl)n group, a -C2-6 alkyl— (5-7 membered cycloheteroalkyl) group, C1-6 alkyl, -Y-(phenyl)q, C3- β cycloalkyl, or a 5-7 membered heteroaryl group;
one or more carbon atoms in the ring optionally are independently substituted with -C(O)-NRdRe, -Y-ORC, -Y-NRdRe, -Y-(phenyl)q, -Y-(5-7 membered cycloheteroalkyl), a -Y-(5-9 membered heteroaryl) group, an oxo group, CN, C3-8 cycloalkyl, or a -Y-O-(5-7 membered heteroaryl) group; and
each of the phenyl groups appearing anywhere in said Rf and R9 is optionally substituted with 1 to 3 substituents independently selected from halogen, C1-6 alkyl, C1-6 haloalkyl, and C1-6 alkoxy, and
each of the 5-7 membered cycloheteroalkyl groups, the 5-7 membered heteroaryl groups, and the 5-9 membered heteroaryl groups appearing anywhere in said Rf and R9 is optionally substituted with 1 to 3 substituents independently selected from halogen and C1-6 alkyl;
n is 1 , 2, or 3;
q is 1 , 2, or 3; and
p is 1 , 2, 3, or 4.
201. The compound of claim 200, wherein p is 1.
202. The compound of claim 200, wherein R2 is phenyl optionally substituted with 1 to 2 substituents independently selected from halogen, C1-6 alkyl, CN, -C(O)ORC, and -NRdRe.
203. The compound of claim 202, wherein R2 is a 4-fluorophenyl group.
204. The compound of claim 200, wherein Ar-R3 is selected from the group consisting of

206. The compound of claim 200 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R3 is selected from NH2, NH-(C1-6 alkyl), N(C1-6 alkyl)2> NH- (C3-6 cycloalkyl), N(C1-6 alkyl)-(C3-6 cycloalkyl), an N(C1-6 alkyl)-(C2-6 alkyl)-ORc group, N(C1-6 alkyl)-Y-(5-7 membered cycloheteroalkyl), N(C1-6 alkyl)-phenyl, N(C1-6 alkyl)-Y-(5-7 membered heteroaryl), and N(C1-6 alkyl)-C2-6 alkyl-O-Y-(5-7 membered heteroaryl) , wherein each of the phenyl , the 5-7 membered cycloheteroalkyl, and the 5-7 heteroaryl are optionally substituted with 1 to 3 substituents independently selected from halogen and C1-6 alkyl.
207. The compound of claim 206, wherein R3 is selected from the group of a diethylamino group, a dimethylamino group, an ethyl(methyl)amino group, an isopropyl(ethyl)amino group, a cyclopropyl(ethyl)amino group.
208. The compound of claim 200 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R3 is an optionally substituted 5-7 membered cycloheteroalkyl group or an optionally substituted 5-7 membered heteroaryl group.
209. The compound of claim 208 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R3 is selected from a diazepanyl group, an imidazolyl group, a morpholinyl group, a piperidinyl group, a piperazinyl group, a pyridyl group, a pyrrolidyl group, and a thiomorpholinyl group, wherein each of these groups optionally includes a nitrogen ring atom substituted with -C(O)R0, -C2-6 alkyl-ORC) - C2-6 alkyl-NRdRe, -Y-C(O)NRdRe, -S(O)2-(Ci-6 alkyl), -C2-6 alkyl-(5-7 membered cycloheteroalkyl), C1-6 alkyl, or 5-7 membered heteroaryl, a carbon ring atom substituted with -C(O)-NRdRe, -Y-ORC, -Y-NRdR6, -Y-phenyl, -Y-(5-7 membered cycloheteroalkyl), -Y-(5-9 membered heteroaryl), or -Y-O-(5-7 membered heteroaryl), and/or a sulfur ring atom substituted with 1 or 2 oxo groups, wherein each of the phenyl groups is optionally substituted with 1 to 3 substituents independently selected from halogen, Ci-6 alkyl, C1-6 haloalkyl, and
 alkoxy, and each of the 5-7 membered cycloheteroalkyl, the 5-7 membered heteroaryl, and the.5- 9 membered heteroaryl immediately above is optionally substituted with 1 to 3 substituents independently selected from halogen and Ci-6 alkyl..
alkoxy, and each of the 5-7 membered cycloheteroalkyl, the 5-7 membered heteroaryl, and the.5- 9 membered heteroaryl immediately above is optionally substituted with 1 to 3 substituents independently selected from halogen and Ci-6 alkyl..
210. The compound of claim 209 or a pharmaceutically acceptable salt, hydrate or ester thereof, wherein R3 is selected from the group consisting of a 1-[1 ,4]diazepanyl group, a 1 -imidazolyl group, a 4-morpholinyl group, a 1 -piperidinyl group, a 1- piperazinyl group, a 4-pyridyl group, a 1 -pyrrolidyl group, a 4-thiomorpholinyl group, and a 4-methyl piperazin-1-yl group.
211. The compound of claim 200, wherein R3 is chloro or trifluoromethyl.
212. The compound of claim 200 selected from
N-[(6-chloropyridin-3-yl)methyl]-N-(4-fluorophenyl)pyrazine-2-carboxamide;
N-(4-fluorophenyl)-N-{[6-(4-methylpiperazin-1-yl)pyhdin-3-yl]methyl}pyrazine-2- carboxamide;
N-[(6-chloropyridin-3-yl)methyl]-N-(4-fluorophenyl)pyrimidine-4-carboxamide;
N-[(6-chloropyridin-3-yl)methyl]-N-(4-fluorophenyl)pyrimidine-5-carboxamide;
N-(4-fluorophenyl)-N-{[6-(4-methylpiperazin-1-yl)pyridin-3-yl]methyl}pyrimidine-4- carboxamide;
N-(4-fluorophenyl)-N-{[6-(4-methylpiperazin-1-yl)pyridin-3-yl]methyl}pyrimidine-5- carboxamide;
N-(4-fluorophenyl)-N-{[6-(4-methylpiperazin-1-yl)pyridin-3-yl]methyl}pyridazine-4- carboxamide;
N-(4-fluorophenyl)-N-{[6-(trifluoromethyl)pyridin-3-yl]methyl}pyridazine-4- carboxamide;
N-(4-fluorophenyl)-N-[(2-morpholin-4-yl-1 ,3-thiazol-4-yl)methyl]pyridazine-4- carboxamide;
N-(4-fluorophenyl)-N-[4-(2-morpholin-4-yl-1 ,3-thiazol-4-yl)butyl]pyridazine-4- carboxamide;
N-(4-fluorophenyl)-N-({2-[4-(2-phenylethyl)piperazin-1-yl]-1 ,3-thiazol-4- yl}methyl)pyridazine-4-carboxamide;
N-({2-[4-(diphenylmethyl)piperazin-1-yl]-1 ,3-thiazol-4-yl}methyl)-N-(4- fluorophenyl)pyridazine-4-carboxamide;
N-{[2-(4-benzylpiperazin-1-yl)-1 ,3-thiazol-4-yl]methyl}-N-(4- fluorophenyl)pyridazine-4-carboxamide;
N-(4-fluorophenyl)-N-{[2-(4-pyridin-2-ylpiperazin-1 -yl)-1 ,3-thiazol-4- yl]methyl}pyridazine-4-carboxamide;
N-(4-fluorophenyl)-N-{[2-(4-methylpiperazin-1-yl)-1 ,3-thiazol-4- yl]methyl}pyridazine-4-carboxamide;
N-(4-fluorophenyl)-N-{[2-(4-pyrimidin-2-ylpiperazin-1-yl)-1 ,3-thiazol-4- yl]methyl}pyridazine-4-carboxamide;
N-(4-fluorophenyl)-N-{[2-(4-phenylpiperazin-1 -yl)-1 ,3-thiazol-4- yl]methyl}pyridazine-4-carboxamide;
N-[(2-{4-[2-(dimethylamino)ethyl]piperazin-1-yl}-1 ,3-thiazol-4-yl)methyl]-N-(4- fluorophenyl)pyridazine-4-carboxamide;
N-{[2-(4-cyclohexylpiperazin-1-yl)-1 ,3-thiazol-4-yl]methyl}-N-(4- fluorophenyl)pyridazine-4-carboxamide;
N-({6-[4-(diphenylmethyl)piperazin-1-yl]pyridin-3-yl}methyl)-N-(4- fluorophenyl)pyridazine-4-carboxamide;
N-(4-fluorophenyl)-N-({6-[4-(2-phenylethyl)piperazin-1-yl]pyridin-3- yl}methyl)pyridazine-4-carboxamide;
N-{[6-(4-benzylpiperazin-1-yl)pyridin-3-yl]methyl}-N-(4-fluorophenyl)pyridazine-4- carboxamide;
N-(4-fluorophenyl)-N-{[6-(4-pyridin-2-ylpiperazin-1-yl)pyridin-3- yl]methyl}pyridazine-4-carboxamide;
N-{[6-(3,5-dimethylpiperidin-1-yl)pyridin-3-yl]methyl}-N-(4-fluorophenyl)pyridazine- 4-carboxamide;
N-(4-fluorophenyl)-N-{[6-(4-phenylpiperazin-1-yl)pyridin-3-yl]methyl}pyridazine-4- carboxamide;
N-(4-fluorophenyl)-N-[(6-piperazin-1-ylpyridin-3-yl)methyl]pyridazine-4- carboxamide;
N-((6-bromopyridin-3-yl)methyl)-N-(4-fluorophenyl)pyridazine-4-carboxamide;
N-(4-fluorophenyl)-N-{[2-(4-phenylpiperazin-1-yl)-1 ,3-thiazol-4- yl]methyl}pyridazine-4-carboxamide;
N-(4-fluorophenyl)-N-{[6-(4-pyrimidin-2-ylpiperazin-1-yl)pyridin-3- yl]methyl}pyridazine-4-carboxamide;
N-(4-fluorophenyl)-N-({6-[4-(hydroxymethyl)piperidin-1-yl]pyridin-3- yl}methyl)pyridazine-4-carboxamide;
N-(4-fluorophenyl)-N-[(6-piperidin-1 -ylpyridin-3-yl)methyl]pyridazine-4- carboxamide;
N-{[6-(4-cyclohexylpiperazin-1-yl)pyridin-3-yl]methyl}-N-(4- fluorophenyl)pyridazine-4-carboxamide;
and pharmaceutically acceptable salts, hydrates, and esters thereof..
213. A pharmaceutical composition comprising the compound of any one of claims 200-212, or a pharmaceutically acceptable salt, hydrate or ester thereof and a pharmaceutically acceptable carrier or excipient.
214. The pharmaceutical composition of claim 213, further comprising an additional therapeutic agent.
215. A method of treating a disease or disease symptom selected from angina, hypertension, congestive heart failure, myocardial ischemia, and arrhythmia, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 200-212 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
216. A method of treating a disease or disease symptom selected from stroke, convulsion, epilepsy, traumatic brain injury, and neuronal disorder, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 200-212 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
217. A method of treating a disease or disease symptom selected from diabetes, urinary incontinence, hot flush, and thermal disregulation, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 200-212 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
218. A method of treating pain in a subject, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 200-212 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
219. A method of claim 218 wherein said pain is chronic pain.
220. A method of claim 219 wherein said chronic pain is associated with diabetes, post traumatic pain of amputation, lower back pain, spinal cord damage, cancer, chemical injury, chemotherapy induced peripheral neuropathy, toxins, major surgery, peripheral nerve damage due to traumatic injury, post-herpetic neuralgia, trigeminal neuralgia, lumbar or cervical radiculopathies, fibromyalgia, glossopharyngeal neuralgia, reflex sympathetic dystrophy, causalgia, thalamic syndrome, nerve root avulsion, reflex sympathetic dystrophy or post thoracotomy pain, nutritional deficiencies, viral infection, bacterial infection, metastatic infiltration, adiposis dolorosa, burns, central pain conditions related to thalamic conditions, or a combination thereof.
221. A method of claim 218 wherein said pain is chronic back pain.
222. A method of claim 218 wherein said pain is neuropathic pain.
223. A method of claim 218 wherein said pain is associated with diabetic neuropathy.
224. A method of claim 218 wherein said pain is associated with post-herpetic neuropathy.
225. A method of claim 218 wherein said pain is associated with post-herpetic fibromyalgia.
226. A method of treating a disease or disease symptom modulated by calcium channel Cav2, the method comprising administering to a subject a therapeutically effective amount of the compound of any one of claims 200-212 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
227. The method of claim 226, wherein the disease or disease symptom is modulated by calcium channel Cav2.2.
228. A method of modulating calcium channel activity in a subject, the method comprising administering the compound of any one of claims 200-212, or a pharmaceutically acceptable salt, hydrate or ester thereof, to a subject.
229. The method of any one of claims 215-228, wherein the subject is a mammal.
230. A method for making a compound of formula (VII) according to claim 200 and pharmaceutically acceptable salts thereof, comprising
(a) reacting a carboxylic acid compound of formula (Vila)

(Vila)
with an activating agent; and (b) coupling the resultant activated acid with an amine of formula (b)
(b)
231. The method according to claim 230, wherein the activating agent is selected from thionyl chloride, 2-chloro-4,6-dimethoxy-1 ,3,5-triazine, 1-[3- (dimethylamino)propyl]-3-ethyl-carbodiimide and dicyclohexyl carbodiimide.
232. A pharmaceutical composition comprising a compound of formula (VIII),

(VIII) or a pharmaceutically acceptable salt, hydrate or ester thereof and a pharmaceutically acceptable carrier or excipient, wherein
Het is selected from the group consisting of

A is O, S, or NRC;
D is O or S;
E, G, and M are each CH or N, wherein one of E, G, and X is N, and the other two of E, G, and X are CH;
E1, G1, and M' are each CH or N, wherein two of E, G1 and X is N, and the other of E, G, and X is CH;
T is CH or N;
R2, R3, R4, R5, R6, R7, R8, R9, Rc, X, and p are as defined in claim 1.
233. The pharmaceutical composition of claim 232, further comprising an additional therapeutic agent.
234. A method of treating a disease or disease symptom selected from angina, hypertension, congestive heart failure, myocardial ischemia, and arrhythmia, the method comprising administering to a subject a therapeutically effective amount of the compound of formula (VIII) as defined in claim 232 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
235. A method of treating a disease or disease symptom selected from stroke, convulsion, epilepsy, traumatic brain injury, and neuronal disorder, the method comprising administering to a subject a therapeutically effective amount of the compound of formula (VIII) as defined in claim 232 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
236. A method of treating a disease or disease symptom selected from diabetes, urinary incontinence, hot flush, and thermal disregulation, the method comprising administering to a subject a therapeutically effective amount of the compound of formula (VIII) as defined in claim 232 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
237. A method of treating pain in a subject, the method comprising administering to a subject a therapeutically effective amount of compound of formula (VIII) as defined in claim 232 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
238. A method of claim 237 wherein said pain is chronic pain.
239. A method of claim 238 wherein said chronic pain is associated with diabetes, post traumatic pain of amputation, lower back pain, spinal cord damage, cancer, chemical injury, chemotherapy induced peripheral neuropathy, toxins, major surgery, peripheral nerve damage due to traumatic injury, post-herpetic neuralgia, trigeminal neuralgia, lumbar or cervical radiculopathies, fibromyalgia, glossopharyngeal neuralgia, reflex sympathetic dystrophy, causalgia, thalamic syndrome, nerve root avulsion, reflex sympathetic dystrophy or post thoracotomy pain, nutritional deficiencies, viral infection, bacterial infection, metastatic infiltration, adiposis dolorosa, burns, central pain conditions related to thalamic conditions, or a combination thereof.
240. A method of claim 237 wherein said pain is chronic back pain.
241. A method of claim 237 wherein said pain is neuropathic pain.
242. A method of claim 237 wherein said pain is associated with diabetic neuropathy.
243. A method of claim 237 wherein said pain is associated with post-herpetic neuropathy.
244. A method of claim 237 wherein said pain is associated with post-herpetic fibromyalgia.
245. A method of treating a disease or disease symptom modulated by calcium channel Cav2, the method comprising administering to a subject a therapeutically effective amount of the compound of formula (VIII) as defined in claim 232 or a pharmaceutically acceptable salt, hydrate, or ester thereof.
246. The method of claim 245, wherein the disease or disease symptom is modulated by calcium channel Cav2.2.
247. A method of modulating calcium channel activity in a subject, the method comprising administering compound of formula (VIII) as defined in claim 232, or a pharmaceutically acceptable salt, hydrate or ester thereof, to a subject.
248. The method of any one of claims 234-247, wherein the subject is a mammal.
249. The method of any one of claims 234-247, wherein the compound of formula (VIII) is selected from
1-Benzyl-2-methyl-1 H-imidazole-4-carboxylic acid (4-fluoro-phenyl)-(2-piperidin- 1 -yl-thiazol-5-ylmethyl)-amide;
1 -benzyl-N-(4-methylphenyl)-N-[(2-pyrrolidin-1 -yl-1 ,3-thiazol-5-yl)methyl]-1 H- 1 ,2,3-triazole-4-carboxamide;
1-benzyl-5-methyl-N-(4-methylphenyl)-N-[(2-pyrrolidin-1-yl-1 ,3-thiazol-5- yl)methyl]-1 H-1 ,2,3-triazole-4-carboxamide;
1-Benzyl-1 H-[1 ,2,3]triazole-4-carboxylic acid (4-fluoro-phenyl)-(2-piperidin-1-yl- thiazol-5-ylmethyl)-amide;
1-Benzyl-1 H-[1 ,2,3]triazole-4-carboxylic acid (4-fluoro-phenyl)-[2-(4-methyl- piperazin-1-yl)-thiazol-5-ylmethyl]-amide;
N-(4-methylphenyl)-5-nitro-N-[(2-pyrrolidin-1-yl-1 ,3-thiazol-5-yl)methyl]-2- furamide;
N-(4-methylphenyl)-N-[(2-pyrrolidin-1-yl-1 ,3-thiazol-5-yl)methyl]thiophene-2- carboxamide;
N-(4-methylphenyl)-N-[(2-pyrrolidin-1 -yl-1 ,3-thiazol-5-yl)methyl]-1 - benzothiophene-2-carboxamide;
N-(4-methylphenyl)-N-[(2-pyrrolidin-1-yl-1 ,3-thiazol-5-yl)methyl]-3-furamide;
1 -methyl-N-(4-methylphenyl)-N-[(2-pyrrolidin-1 -yl-1 ,3-thiazol-5-yl)methyl]-1 H- pyrrole-2-carboxamide;
N-(4-methylphenyl)-N-[(2-pyrrolidin-1-yl-1 ,3-thiazol-5-yl)methyl]thiophene-3- carboxamide;
and pharmaceutically acceptable salts, hydrates, and esters thereof.
250. A compound as defined in any one of claims 1 to 18 for use in the treatment of a disease or disease symptom selected from angina, hypertension, congestive heart failure, myocardial ischemia, arrhythmia, stroke, convulsion, epilepsy, traumatic brain injury, neuronal disorder, urinary incontinence, hot flush, thermal disregulation, pain or a disease or disease symptom modulated by calcium channel Cav2, or the modulation of calcium channel activity in a subject.
251. Use of a compound as defined in any one of claims 1 to 18 in the preparation of a medication for the treatment of a disease or disease symptom selected from angina, hypertension, congestive heart failure, myocardial ischemia, arrhythmia, stroke, convulsion, epilepsy, traumatic brain injury, neuronal disorder, urinary incontinence, hot flush, thermal disregulation, pain or a disease or disease symptom modulated by calcium channel Cav2, or the modulation of calcium channel activity in a subject.
252. A compound as defined in any one of claims 38 to 54 for use in the treatment of a disease or disease symptom selected from angina, hypertension, congestive heart failure, myocardial ischemia, arrhythmia, stroke, convulsion, epilepsy, traumatic brain injury, neuronal disorder, urinary incontinence, hot flush, thermal disregulation, pain or a disease or disease symptom modulated by calcium channel Cav2, or the modulation of calcium channel activity in a subject.
253. Use of a compound as defined in any one of claims 38 to 54 in the preparation of a medication for the treatment of a disease or disease symptom selected from angina, hypertension, congestive heart failure, myocardial ischemia, arrhythmia, stroke, convulsion, epilepsy, traumatic brain injury, neuronal disorder, urinary incontinence, hot flush, thermal disregulation, pain or a disease or disease symptom modulated by calcium channel Cav2, or the modulation of calcium channel activity in a subject.
254. A compound as defined in any one of claims 74 to 86 for use in the treatment of a disease or disease symptom selected from angina, hypertension, congestive heart failure, myocardial ischemia, arrhythmia, stroke, convulsion, epilepsy, traumatic brain injury, neuronal disorder, urinary incontinence, hot flush, thermal disregulation, pain or a disease or disease symptom modulated by calcium channel Cav2, or the modulation of calcium channel activity in a subject.
255. Use of a compound as defined in any one of claims 74 to "86 in the preparation of a medication for the treatment of a disease or disease symptom selected from angina, hypertension, congestive heart failure, myocardial ischemia, arrhythmia, stroke, convulsion, epilepsy, traumatic brain injury, neuronal disorder, urinary incontinence, hot flush, thermal disregulation, pain or a disease or disease symptom modulated by calcium channel Cav2, or the modulation of calcium channel activity in a subject.
256. A compound as defined in any one of claims 106 to 1 19 for use in the treatment of a disease or disease symptom selected from angina, hypertension, congestive heart failure, myocardial ischemia, arrhythmia, stroke, convulsion, epilepsy, traumatic brain injury, neuronal disorder, urinary incontinence, hot flush, thermal disregulation, pain or a disease or disease symptom modulated by calcium channel Cav2, or the modulation of calcium channel activity in a subject.
257. Use of a compound as defined in any one of claims 106 to 119 in the preparation of a medication for the treatment of a disease or disease symptom selected from angina, hypertension, congestive heart failure, myocardial ischemia, arrhythmia, stroke, convulsion, epilepsy, traumatic brain injury, neuronal disorder, urinary incontinence, hot flush, thermal disregulation, pain or a disease or disease symptom modulated by calcium channel Cav2, or the modulation of calcium channel activity in a subject.
258. A compound as defined in any one of claims 139 to 145 for use in the treatment of a disease or disease symptom selected from angina, hypertension, congestive heart failure, myocardial ischemia, arrhythmia, stroke, convulsion, epilepsy, traumatic brain injury, neuronal disorder, urinary incontinence, hot flush, thermal disregulation, pain or a disease or disease symptom modulated by calcium channel Cav2, or the modulation of calcium channel activity in a subject.
259. Use of a compound as defined in any one of claims 139 to 145 in the preparation of a medication for the treatment of a disease or disease symptom selected from angina, hypertension, congestive heart failure, myocardial ischemia, arrhythmia, stroke, convulsion, epilepsy, traumatic brain injury, neuronal disorder, urinary incontinence, hot flush, thermal disregulation, pain or a disease or disease symptom modulated by calcium channel Cav2, or the modulation of calcium channel activity in a subject.
260. A compound as defined in any one of claims 165 to 180 for use in the treatment of a disease or disease symptom selected from angina, hypertension, congestive heart failure, myocardial ischemia, arrhythmia, stroke, convulsion, epilepsy, traumatic brain injury, neuronal disorder, urinary incontinence, hot flush, thermal disregulation, pain or a disease or disease symptom modulated by calcium channel Cav2, or the modulation of calcium channel activity in a subject.
261. Use of a compound as defined in any one of claims 165 to 180 in the preparation of a medication for the treatment of a disease or disease symptom selected from angina, hypertension, congestive heart failure, myocardial ischemia, arrhythmia, stroke, convulsion, epilepsy, traumatic brain injury, neuronal disorder, urinary incontinence, hot flush, thermal disregulation, pain or a disease or disease symptom modulated by calcium channel Cav2, or the modulation of calcium channel activity in a subject.
262. A compound as defined in any one of claims 200 to 212 for use in the treatment of a disease or disease symptom selected from angina, hypertension, congestive heart failure, myocardial ischemia, arrhythmia, stroke, convulsion, epilepsy, traumatic brain injury, neuronal disorder, urinary incontinence, hot flush, thermal disregulation, pain or a disease or disease symptom modulated by calcium channel Cav2, or the modulation of calcium channel activity in a subject.
263. Use of a compound as defined in any one of claims 200 to 212 in the preparation of a medication for the treatment of a disease or disease symptom selected from angina, hypertension, congestive heart failure, myocardial ischemia, arrhythmia, stroke, convulsion, epilepsy, traumatic brain injury, neuronal disorder, urinary incontinence, hot flush, thermal disregulation, pain or a disease or disease symptom modulated by calcium channel Cav2, or the modulation of calcium channel activity in a subject.
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US87417906P | 2006-12-11 | 2006-12-11 | |
| US87413306P | 2006-12-11 | 2006-12-11 | |
| US87415206P | 2006-12-11 | 2006-12-11 | |
| US60/874,133 | 2006-12-11 | ||
| US60/874,179 | 2006-12-11 | ||
| US60/874,152 | 2006-12-11 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2008073461A2 true WO2008073461A2 (en) | 2008-06-19 |
| WO2008073461A3 WO2008073461A3 (en) | 2008-09-12 |
Family
ID=39284027
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/025416 Ceased WO2008073461A2 (en) | 2006-12-11 | 2007-12-11 | Ion channel modulators |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2008073461A2 (en) |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009060053A1 (en) * | 2007-11-09 | 2009-05-14 | Smithkline Beecham Corporation | 1, 2, 3-triazole derivatives for use as stearoyl-coa desaturase inhibitors |
| WO2010012793A1 (en) * | 2008-08-01 | 2010-02-04 | Bayer Cropscience Sa | Fungicide aminothiazole derivatives |
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2014120994A1 (en) * | 2013-01-31 | 2014-08-07 | The Johns Hopkins University | Rufinamide and derivatives and their use in modulating the gating process of human voltage-gated sodium channels |
| EP2774927A1 (en) * | 2008-12-03 | 2014-09-10 | Presidio Pharmaceuticals, Inc. | Inhibitors of HCV NS5A |
| EP2942346A1 (en) * | 2009-02-17 | 2015-11-11 | Syntrix Biosystems, Inc. | Pyridine- and pyrimidinecarboxamides as cxcr2 modulators |
| JP2016128401A (en) * | 2009-10-22 | 2016-07-14 | フィブロテック セラピューティクス プロプライエタリー リミテッド | Anti-fibrotic agents of fused ring analogues |
| WO2017146128A1 (en) * | 2016-02-26 | 2017-08-31 | 大日本住友製薬株式会社 | Imidazolylamide derivative |
| US9828362B2 (en) | 2015-06-22 | 2017-11-28 | Sumitomo Dainippon Pharma Co., Ltd. | 1,4-disubstituted imidazole derivative |
| US9951086B2 (en) | 2013-12-19 | 2018-04-24 | Bayer Pharma Aktiengesellschaft | Indazolecarboxamides, processes for their preparation, pharmaceutical preparations comprising them and their use for producing medicaments |
| JP2018100270A (en) * | 2016-12-20 | 2018-06-28 | 大日本住友製薬株式会社 | Medicine composed of 1,4-disubstituted imidazole derivative |
| WO2018202524A1 (en) * | 2017-05-04 | 2018-11-08 | Bayer Cropscience Aktiengesellschaft | 2-{[2-(phenyloxymethyl)pyridin-5-yl]oxy}-ethanamin-derivatives and related compounds as pest-control agents e.g. for the protection of plants |
| US10294237B2 (en) | 2015-06-22 | 2019-05-21 | Sumitomo Dainippon Pharma Co., Ltd. | Bicyclic heterocyclic amide derivative |
| JPWO2018117127A1 (en) * | 2016-12-20 | 2019-10-24 | 大日本住友製薬株式会社 | Remover of undifferentiated iPS cells |
| USRE47740E1 (en) | 2010-08-23 | 2019-11-26 | Syntrix Biosystems Inc. | Aminopyridinecarboxamides as CXCR2 modulators |
| US11014873B2 (en) | 2017-02-03 | 2021-05-25 | Certa Therapeutics Pty Ltd. | Anti-fibrotic compounds |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2006120549A (en) * | 2003-11-14 | 2007-12-27 | Вертекс Фармасьютикалз Инкорпорейтед (Us) | THIAZOLES AND OXAZOLES USED AS MODULATORS OF ART-BINDING CASSETTE TRANSPORTERS |
| DE102004051277A1 (en) * | 2004-10-21 | 2006-04-27 | Merck Patent Gmbh | Heterocyclic carbonyl compounds |
-
2007
- 2007-12-11 WO PCT/US2007/025416 patent/WO2008073461A2/en not_active Ceased
Cited By (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9051281B2 (en) | 2007-11-09 | 2015-06-09 | Glaxosmithkline Llc | Compounds |
| JP2011503031A (en) * | 2007-11-09 | 2011-01-27 | グラクソスミスクライン・リミテッド・ライアビリティ・カンパニー | 1,2,3-triazole derivatives for use as stearoyl-CoA desaturase inhibitors |
| EA016621B1 (en) * | 2007-11-09 | 2012-06-29 | ГЛЭКСОСМИТКЛАЙН ЭлЭлСи | 1,2,3-triazole derivatives for use as stearoyl-coa desaturase inhibitors |
| WO2009060053A1 (en) * | 2007-11-09 | 2009-05-14 | Smithkline Beecham Corporation | 1, 2, 3-triazole derivatives for use as stearoyl-coa desaturase inhibitors |
| US8486977B2 (en) | 2007-11-09 | 2013-07-16 | Glaxosmithkline Llc | 1,2,3-triazole derivatives for use as stearoyl-CoA desaturase inhibitors |
| CN101918376B (en) * | 2007-11-09 | 2013-11-06 | 葛兰素史密斯克莱有限责任公司 | 1,2,3-Triazole derivatives useful as stearoyl-CoA desaturase inhibitors |
| WO2010012793A1 (en) * | 2008-08-01 | 2010-02-04 | Bayer Cropscience Sa | Fungicide aminothiazole derivatives |
| US9120779B2 (en) | 2008-12-03 | 2015-09-01 | Presidio Pharmaceuticals, Inc. | Inhibitors of HCV NS5A |
| EP2774927A1 (en) * | 2008-12-03 | 2014-09-10 | Presidio Pharmaceuticals, Inc. | Inhibitors of HCV NS5A |
| USRE47267E1 (en) | 2009-02-17 | 2019-03-05 | Syntrix Biosystems, Inc. | Pyridinecarboxamides as CXCR2 modulators |
| EP2942346A1 (en) * | 2009-02-17 | 2015-11-11 | Syntrix Biosystems, Inc. | Pyridine- and pyrimidinecarboxamides as cxcr2 modulators |
| USRE47415E1 (en) | 2009-02-17 | 2019-06-04 | Syntrix Biosystems, Inc. | Pyrimidinecarboxamides as CXCR2 modulators |
| JP2016128401A (en) * | 2009-10-22 | 2016-07-14 | フィブロテック セラピューティクス プロプライエタリー リミテッド | Anti-fibrotic agents of fused ring analogues |
| US9951087B2 (en) | 2009-10-22 | 2018-04-24 | Fibrotech Therapeutics Pty Ltd | Fused ring analogues of anti-fibrotic agents |
| USRE47740E1 (en) | 2010-08-23 | 2019-11-26 | Syntrix Biosystems Inc. | Aminopyridinecarboxamides as CXCR2 modulators |
| USRE48547E1 (en) | 2010-08-23 | 2021-05-11 | Syntrix Biosystems Inc. | Aminopyrimidinecarboxamides as CXCR2 modulators |
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2014120994A1 (en) * | 2013-01-31 | 2014-08-07 | The Johns Hopkins University | Rufinamide and derivatives and their use in modulating the gating process of human voltage-gated sodium channels |
| US9951086B2 (en) | 2013-12-19 | 2018-04-24 | Bayer Pharma Aktiengesellschaft | Indazolecarboxamides, processes for their preparation, pharmaceutical preparations comprising them and their use for producing medicaments |
| US9828362B2 (en) | 2015-06-22 | 2017-11-28 | Sumitomo Dainippon Pharma Co., Ltd. | 1,4-disubstituted imidazole derivative |
| US10294237B2 (en) | 2015-06-22 | 2019-05-21 | Sumitomo Dainippon Pharma Co., Ltd. | Bicyclic heterocyclic amide derivative |
| US11661397B2 (en) | 2015-06-22 | 2023-05-30 | Sumitomo Pharma Co., Ltd. | 1,4-disubstituted imidazole derivative |
| US10807945B2 (en) | 2015-06-22 | 2020-10-20 | Sumitomo Dainippon Pharma Co., Ltd. | 1,4-disubstituted imidazole derivative |
| JPWO2017146128A1 (en) * | 2016-02-26 | 2018-12-20 | 大日本住友製薬株式会社 | Imidazolylamide derivatives |
| WO2017146128A1 (en) * | 2016-02-26 | 2017-08-31 | 大日本住友製薬株式会社 | Imidazolylamide derivative |
| US10898469B2 (en) | 2016-02-26 | 2021-01-26 | Sumitomo Dainippon Pharma Co., Ltd. | Imidazolylamide derivative |
| JP2018100270A (en) * | 2016-12-20 | 2018-06-28 | 大日本住友製薬株式会社 | Medicine composed of 1,4-disubstituted imidazole derivative |
| JP7123809B2 (en) | 2016-12-20 | 2022-08-23 | 住友ファーマ株式会社 | Removal agent for undifferentiated iPS cells |
| JPWO2018117127A1 (en) * | 2016-12-20 | 2019-10-24 | 大日本住友製薬株式会社 | Remover of undifferentiated iPS cells |
| US11014873B2 (en) | 2017-02-03 | 2021-05-25 | Certa Therapeutics Pty Ltd. | Anti-fibrotic compounds |
| US11603349B2 (en) | 2017-02-03 | 2023-03-14 | Certa Therapeutics Pty Ltd | Anti-fibrotic compounds |
| TWI779027B (en) * | 2017-05-04 | 2022-10-01 | 德商拜耳作物科學股份有限公司 | Novel heterocyclic compounds as pesticides |
| IL270314A (en) * | 2017-05-04 | 2019-12-31 | ||
| JP2020518635A (en) * | 2017-05-04 | 2020-06-25 | バイエル・クロップサイエンス・アクチェンゲゼルシャフト | New heterocyclic compounds as insecticides |
| CN110869349A (en) * | 2017-05-04 | 2020-03-06 | 拜耳作物科学股份公司 | 2-{[2-(phenoxymethyl)pyridin-5-yl]oxy}ethanamine derivatives and related compounds as, for example, pest control agents for protecting plants |
| JP7241028B2 (en) | 2017-05-04 | 2023-03-16 | バイエル・クロップサイエンス・アクチェンゲゼルシャフト | Novel heterocyclic compounds as insecticides |
| IL270314B2 (en) * | 2017-05-04 | 2023-05-01 | Bayer Cropscience Ag | 2-{[2-(phenyloxymethyl)pyridin-5-yl]oxy}-ethanamin-derivatives and related compounds as pest-control agents e.g. for the protection of plants |
| WO2018202524A1 (en) * | 2017-05-04 | 2018-11-08 | Bayer Cropscience Aktiengesellschaft | 2-{[2-(phenyloxymethyl)pyridin-5-yl]oxy}-ethanamin-derivatives and related compounds as pest-control agents e.g. for the protection of plants |
| US11827616B2 (en) | 2017-05-04 | 2023-11-28 | Discovery Purchaser Corporation | Heterocyclic compounds as pesticides |
| CN110869349B (en) * | 2017-05-04 | 2025-07-22 | 发现号收购集团 | 2- { [2- (Phenoxymethyl) pyridin-5-yl ] oxy } ethanamine derivatives and related compounds as pest control agents, for example, for protecting plants |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2008073461A3 (en) | 2008-09-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2008073461A2 (en) | Ion channel modulators | |
| US11773110B2 (en) | Heterocycle amines and uses thereof | |
| EP2099754A1 (en) | Ion channel modulators | |
| EP2097379A1 (en) | Carboxamide derivatives as ion channel modulators | |
| AU2006205920B2 (en) | Thiazole-4-carboxamide derivatives as mGluR5 antagonists | |
| AP388A (en) | Inhibition of the replicateion of HIV and related viruses using thiourea derivative compounds or salts thereof. | |
| JP6013375B2 (en) | Thiazolylphenyl-benzenesulfonamide derivatives as kinase inhibitors | |
| CN112384500B (en) | Immunomodulator, composition and preparation method thereof | |
| CZ299836B6 (en) | Aryl- and heteroaryl-substituted heterocyclic ureas, their use and pharmaceutical compositions in which the ureas are comprised | |
| EP2342190A1 (en) | Bicyclic kinase inhibitors | |
| JPWO2003062233A1 (en) | 2-acylaminothiazole derivatives or salts thereof | |
| JP4767975B2 (en) | Pyridine-2-carboxamide derivatives as mGluR5 antagonists | |
| CA2717388A1 (en) | Pim kinase inhibitors and methods of their use | |
| JP6896701B2 (en) | Imidazolylamide derivative | |
| CN117561238A (en) | Phenylurea derivatives | |
| MX2014015691A (en) | 1-[m-carboxamido(hetero)aryl-methyl]-heterocyclyl-carboxamide derivatives. | |
| AU2011299904A1 (en) | Prostaglandin D synthase inhibitory piperidine compounds | |
| AU2005300734A1 (en) | Nicotinamide pyridinureas as vascular endothelial growth factor (VEGF) receptor kinase inhibitors | |
| WO2008073934A1 (en) | Ion channel modulators |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07862821 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 07862821 Country of ref document: EP Kind code of ref document: A2 |