WO2008064136A2 - Compounds with activity at retinoic acid receptors - Google Patents
Compounds with activity at retinoic acid receptors Download PDFInfo
- Publication number
- WO2008064136A2 WO2008064136A2 PCT/US2007/085027 US2007085027W WO2008064136A2 WO 2008064136 A2 WO2008064136 A2 WO 2008064136A2 US 2007085027 W US2007085027 W US 2007085027W WO 2008064136 A2 WO2008064136 A2 WO 2008064136A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- optionally substituted
- group
- compound
- straight chained
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Definitions
- the present invention is in the field of pharmaceuticals, and in particular in the field of modulators of the retinoic acid receptors and treatment of diseases with these compounds.
- Retinoids are small, lipophilic molecules that derive from the metabolism of vitamin A, a dietary vitamin. Natural and synthetic retinoid derivatives exert pleiotropic effects on cellular growth, differentiation, apoptosis, homeostasis and embryogenesis. A number of non-selective retinoids are currently marketed or undergoing clinical trials for use in dermatology and oncology. For instance, Tretinoin (all-trans-retinoic acid), Isotretinoin (13-cis retinoic acid) and Etretinate (a synthetic retinoic acid analog) are being used successfully in the treatment of acne, psoriasis, photoaging and squamous cell carcinoma. However, acute and chronic toxic side effects (skeletal abnormality, skin toxicity, triglyceride elevation, teratogenesis) are commonly observed which can lead to the discontinuation of the treatment.
- RARs and RXRs are ligand-dependent transcription factors belonging to the steroid nuclear receptor superfamily.
- the retinoid receptors display a modular structure: an N-terminus ligand- independent activation domain (AF-I), a DNA-binding domain (DBD) adjacent to the ligand-dependent domain (LBD) and the ligand-dependent activation domain (AF-2) contiguous to the LBD and located at the C-terminus end.
- AF-I N-terminus ligand- independent activation domain
- DBD DNA-binding domain
- LBD ligand-dependent domain
- AF-2 ligand-dependent activation domain
- RARs and RXRs Three receptors subtypes have been reported for each of the RARs and RXRs, classified as ⁇ , ⁇ , and ⁇ . All six subtypes have reportedly distinct expression patterns in the developing embryo and in the adult, thus, are believed to exhibit specific and non-overlapping functions.
- RAR ⁇ consists of five known isoforms generated from the use of two promoters Pl (RAR ⁇ 1 and RAR ⁇ 3) and P2 (RAR ⁇ 2 and RAR ⁇ 4).
- RAR ⁇ 1 ' a fifth isoform
- the isoforms only differ in the nature of their AF-I transcriptional activation domains located at the very N-terminus.
- RAR ⁇ 1 and RAR ⁇ 3 have very similar AF-I domains. The only difference being the presence of an additional 27 amino acid insert in RAR ⁇ 3.
- RAR ⁇ 2 on the other hand, has a unique AF-I domain, while RAR ⁇ 4 lacks such a domain as well as a portion of its DNA-binding domain (DBD).
- DBD DNA-binding domain
- RAR ⁇ 4 could act as a dominant negative mutant.
- the isoforms have distinct spatial and temporal distribution.
- RAR ⁇ 1 and RAR ⁇ 3 display a relatively restricted pattern - highly present the brain, and in limited amounts in the lung and skin.
- RAR ⁇ 2 is more broadly expressed in the brain and heart, and at much lower levels in the liver, kidney and skeletal muscle. In humans, only the RAR ⁇ 1, 2 and 4 isoforms are expressed.
- RAR ⁇ is expressed in the skin and lungs.
- RAR ⁇ and/or RAR ⁇ modulating compounds may be used to treat cancer.
- a growing body of evidence supports the hypotheses that the RAR ⁇ and/or RAR ⁇ genes are tumor suppressor genes and may have chemopreventive effects on various cancers like head and neck cancer, cervix cancer, lung cancer, skin cancer, pancreatic cancer, liver caner, mammary cancer and cancer in the digestive tract (e.g. oral cavity, stomach, esophagus and colon).
- RAR ⁇ is expressed in normal lung cells and in cells resistant to retinoic acid but is not expressed in lung cancer cells or cells resistant to retinoic acid treatment.
- Use of a RAR ⁇ modulating compound may restore RAR ⁇ expression and thus be efficient for lung cancer treatment.
- RAR ⁇ and/or RAR ⁇ ligands could be used alone or in combination with existing chemo- or radiation therapy. Synergistic cytotoxicity by combination treatment of selective retinoid RAR ⁇ / ⁇ ligands with taxol (Paclitaxel) has already been demonstrated.
- RAR ⁇ and/or RAR ⁇ modulating compounds may be used to treat a variety of neurological disorders. For instance RAR ⁇ null mice exhibit locomotor defects related to dysfunction of the mesolimbic dopamine signaling pathway. Moreover these animals lack hippocampal long-term potentiation (LTP) and long- term depression (LTD), widely studied forms of synaptic plasticity. This results in substantial performance deficits in spatial learning and memory tasks.
- RAR ⁇ 2 is involved in neurite outgrowth from peripheral and central nervous systems.
- RAR ⁇ 2-modulating compounds would be therapeutically relevant to the treatment of neurodegenerative disorders including Parkinson's and Alzheimer's diseases. Because of its involvement in cognitive function, RAR ⁇ 2 would also be relevant to the treatment of neurological disorders where cognition is altered, in particular schizophrenia. Finally, clinical data from the use of Isotretinoin has suggested an association with depression and suicide.
- RAR ⁇ and/or RAR ⁇ modulating compounds may be used to treat a variety of hyperproliferative and inflammatory disorders. Even though RAR ⁇ expression is below detection limits in the skin, RAR ⁇ 2-modulating compounds could act indirectly through transrepression of the activating protein 1 (API) complex, a heterodimeric transcription factor composed of Fos- and Jun-related proteins. API is involved in the expression of metalloproteases, cytokines and other factors which play critical roles in the turnover of extracellular matrix, inflammation and hyperproliferation in diseases such as psoriasis or other associated disorders, rheumatoid arthritis and in tumor metastases.
- API activating protein 1
- the transrepressive effects of retinoids are mediated through a mechanism unrelated to transcriptional activation, involving the RAR-dependent control of transcription factors and cofactor assembly on APl- regulated promoters.
- Relevant therapeutic indications include acne, psoriasis, photoaging and other dermatological disorders.
- RAR ⁇ and/or RAR ⁇ modulating compounds may also be used to treat chronic inflammatory disorders, for example, rheumatoid arthritis.
- retinoids through interaction with the AP-I complex suppress collagenase gene expression.
- the fibroblast interstitial collagenase MMP-I which degrades collagen, is thought to play a critical role in the degradation of the cartilage matrix in arthritis.
- a RAR antagonist improves clinical and histological scores of arthritis.
- RAR ⁇ and/or RAR ⁇ modulating compounds may be used to treat eye disorders/conditions.
- Vitamin A the precursor of natural retinoids
- RAR ⁇ mRNA transcripts are detectable in corneal stroma cells, conjunctival fibroblasts and corneal epithelial cells.
- RAR ⁇ expression is predominantly confined to the periocular mesenchyme and ciliary body.
- retinoic acid further induces the expression of RAR ⁇ in corneal and conjunctival fibroblasts. Knockout of RAR ⁇ indicates that RAR ⁇ is the main RAR subtype involved in modulation of retinal cell populations. In chicken, retinoic acid through its actions on RAR ⁇ is associated with form- deprivation myopia.
- RAR ⁇ and/or RAR ⁇ modulating compounds may be used to treat pulmonary disorders/conditions.
- Retinoic acid suppresses growth of vascular smooth muscle cells (SMCs) from the systemic and pulmonary circulation and inhibits migration of airway smooth muscle cells.
- SMCs vascular smooth muscle cells
- RAR ⁇ and/or RAR ⁇ modulating compounds may be used in treatment of e.g. asthma.
- endogenous retinoids have been implicated in alveologenesis, and exogenous retinoic acid can reverse or partially reverse emphysema and associated pulmonary diseases.
- compounds modulating RAR ⁇ and/or RAR ⁇ may exert the same effects.
- RAR ⁇ and/or RAR ⁇ modulating compounds may also be used for stimulating tropo-elasting gene expression in human lung fibroblasts.
- RAR ⁇ and/or RAR ⁇ modulating compounds may be used to treat dermatological disorders/conditions.
- RAR modulators especially RAR ⁇
- the high level of RAR ⁇ and/or RAR ⁇ in the skin may indicate an ability, for a ligand of either of the two receptors, to exert a therapeutic effect on such dermatological disorders/conditions.
- R 1 , R2, R3, are each independently selected from the group consisting of hydrogen, -OH, -COOH, halogen, optionally substituted C 1 - Cio straight chained or branched alkyl, optionally substituted C 2 -C 1 0 straight chained or branched alkenyl, optionally substituted C 2 -C 1 0 straight chained or branched alkynyl, optionally substituted C 3 -C 9 cycloalkyl, optionally substituted C 3 -C 9 cycloalkylalkyl, optionally substituted C 5 -C 7 cycloalkenyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted aralkyl, and optionally substituted heteroaralkyl;
- X is C or N + ;
- Y is a counter ion and is absent when X is C; and n is an integer from 1 to 5, such as n is 1, 2, 3, 4, or 5, and pharmaceutical composition comprising the same.
- Also disclosed herein is a method of treating, or alleviating the symptoms of, a disease comprising administering to a subject in need thereof a therapeutically effective amount of a compound of Formula I.
- the disease may be selected from the group consisting of cancer, hyperproliferative disorder, inflammatory disorder, neurological disorder, neurodegenerative disorder, eye disorder, eye condition, depression, pulmonary disorders/conditions, and dermatological disorders/conditions.
- DETAILED DESCRIPTION OF THE INVENTION [0014]
- R 1 , R 2 , R3, are each independently selected from the group consisting of hydrogen, - OH, -COOH, halogen, optionally substituted C 1 -C 10 straight chained or branched alkyl, optionally substituted C 2 -C 10 straight chained or branched alkenyl, optionally substituted C 2 -C 10 straight chained or branched alkynyl, optionally substituted C3-C9 cycloalkyl, optionally substituted C5-C7 cycloalkenyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted aralkyl, and optionally substituted heteroaralkyl;
- X is C or N + ;
- Y is a counter ion and is absent when X is C; and n is an integer from 1 to 5, such as n is 1, 2, 3, 4, or 5.
- pharmaceutically acceptable salt refers to a formulation of a compound that does not abrogate the biological activity and properties of the compound.
- Pharmaceutical salts can be obtained by reacting a compound of the invention with inorganic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, methanesulfonic acid, ethanesulfonic acid, p- toluenesulfonic acid, salicylic acid and the like.
- Pharmaceutical salts can also be obtained by reacting a compound of the invention with a base to form a salt such as an ammonium salt, an alkali metal salt, such as a sodium or a potassium salt, an alkaline earth metal salt, such as a calcium or a magnesium salt, a salt of organic bases such as dicyclohexylamine, N-methyl-D-glucamine, tris(hydroxymethyl)methylamine, and salts with amino acids such as arginine, lysine, and the like.
- a salt such as an ammonium salt, an alkali metal salt, such as a sodium or a potassium salt, an alkaline earth metal salt, such as a calcium or a magnesium salt, a salt of organic bases such as dicyclohexylamine, N-methyl-D-glucamine, tris(hydroxymethyl)methylamine, and salts with amino acids such as arginine, lysine, and the like.
- esters refers to a chemical moiety with formula -(R) n -COOR', where R and R' are independently selected from the group consisting of alkyl, cycloalkyl, aryl, heteroaryl (bonded through a ring atom) and heteroalicyclic (bonded through a ring atom), and where n is 0 or 1.
- An "amide” is a chemical moiety with formula -(R) n -C(O)NHR' or -(R) n -NHC(O)R', where R and R' are independently selected from the group consisting of alkyl, cycloalkyl, aryl, heteroaryl (bonded through a ring atom) and heteroalicyclic (bonded through a ring atom), and where n is 0 or 1.
- An amide may be an amino acid or a peptide molecule attached to a molecule of the present invention, thereby forming a prodrug.
- Any amine, hydroxy, or carboxyl side chain on the compounds of the present invention can be esterified or amidified.
- the procedures and specific groups used to achieve this end are known to those of skill in the art and can readily be found in reference sources such as Greene and Wuts, Protective Groups in Organic Synthesis, 3.sup.rd Ed., John Wiley & Sons, New York, N.Y., 1999, which is incorporated herein in its entirety.
- a “prodrug” refers to an agent that is converted into the parent drug in vivo. Prodrugs are often useful because, in some situations, they may be easier to administer than the parent drug. They may, for instance, be bioavailable by oral administration whereas the parent is not. The prodrug may also have improved solubility in pharmaceutical compositions over the parent drug.
- An example, without limitation, of a prodrug would be a compound of the present invention which is administered as an ester (the "prodrug") to facilitate transmittal across a cell membrane where water solubility is detrimental to mobility but which then is metabolically hydrolyzed to the carboxylic acid, the active entity, once inside the cell where water-solubility is beneficial.
- a further example of a prodrug might be a short peptide (polyaminoacid) bonded to an acid group where the peptide is metabolized to reveal the active moiety.
- subsitutent is a group that may be substituted with one or more group(s) individually and independently selected from alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroaryl, heteroalicyclyl, aralkyl, heteroaralkyl, (hetereoalicyclyl)alkyl, hydroxy, protected hydroxyl, alkoxy, aryloxy, acyl, ester, mercapto, alkylthio, arylthio, cyano, halogen, carbonyl, thiocarbonyl, O-carbamyl, N-carbamyl, O-thiocarbamyl, N-thiocarbamyl, C-amido, N-amido, S-sulfonamid
- C m -C n in which "m” and “n” are integers refers to the number of carbon atoms in an alkyl, alkenyl or alkynyl group or the number of carbon atoms in the ring of a cycloalkyl, cycloalkenyl, or aryl group. That is, the alkyl, alkenyl, alkynyl, ring of the cycloalkyl, ring of the cycloalkenyl, or of the aryl can contain from “m” to "n", inclusive, carbon atoms.
- a "Ci-C 4 alkyl” group refers to all alkyl groups having from 1 to 4 carbons, that is, CH 3 -, CH 3 CH 2 -, CH 3 CH 2 CH 2 -, CH 3 CH(CH 3 )-, CH 3 CH 2 CH 2 CH 2 -, CH 3 CH 2 CH(CH 3 )-, and (CH 3 ) 3 CH-. If no "m” and "n” are designated with regard to an alkyl, alkenyl, alkynyl, cycloalkyl or cycloalkenyl group, the broadest range described in these definitions is to be assumed.
- alkyl refers to a straight or branched chain fully saturated (no double or triple bonds) hydrocarbon (all carbon) group.
- alkyl groups include, without limitation, methyl, ethyl, n-propyl, isopropyl, n-butyl, iso-butyl, sec -butyl, tert-butyl, amyl, tert-amyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl and dodecyl.
- an alkyl group of this invention may be substituted or unsubstituted.
- the substituent group(s) is(are) one or more group(s) independently selected from cycloalkyl, aryl, heteroaryl, heteroalicyclyl, hydroxy, alkoxy, aryloxy, mercapto, alkylthio, arylthio, cyano, halo, oxo, carbonyl, thiocarbonyl, O-carbamyl, N-carbamyl, 0-thiocarbamyl, N-thiocarbamyl, C-amido, N-amido, S-sulfonamido, N-sulfonamido, C-carboxy, O-carboxy, isocyanato, thiocyanato, isothiocyanato, nitro, silyl, trihalomethanesulfonyl, -NR a R b , protected hydroxyl, protected
- substituted alkyl groups include, without limitation, 2- oxo-prop-1-yl, 3-oxo-but-l-yl, cyanomethyl, nitromethyl, chloromethyl, hydroxymethyl, tetrahydropyranyloxymethyl, m-trityloxymethyl, propionyloxymethyl, aminomethyl, carboxymethyl, allyloxycarbonylmethyl, allyloxycarbonylaminomethyl, methoxymethyl, ethoxymethyl, t-butoxymethyl, acetoxymethyl, chloromethyl, bromomethyl, iodomethyl, trifluoromethyl, 6- hydroxyhexyl, 2,4-dichlorobutyl, 2-aminopropyl, 1 -chloroethyl, 2-chloroethyl, 1- bromoethyl, 2-chloroethyl, 1 -fluoroethyl, 2-fluoroethyl, 1-iodoethyl,
- alkenyl refers to an alkyl group that contains in the straight or branched hydrocarbon chain one or more double bonds.
- alkenyl group of this invention may be unsubstituted or substituted.
- the substituent(s) may be selected from the same groups disclosed above with regard to alkyl group substitution.
- substituted alkenyl groups include, without limitation, styrenyl, 3-chloro-propen-l-yl, 3-chloro-buten-l-yl, 3-methoxy-propen-2-yl, 3-phenyl-buten-2-yl and 1-cyano-buten- 3-yl.
- alkynyl refers to an alkyl group that contains in the straight or branched hydrocarbon chain one or more triple bonds.
- An alkynyl group of this invention may be unsubstituted or substituted. When substituted, the substituent(s) may be selected from the same groups disclosed above with regard to alkyl group substitution.
- cycloalkyl refers to a completely saturated (no double bonds) hydrocarbon ring. Cycloalkyl groups of this invention may range from C 3 to C9. A cycloalkyl group may be unsubstituted or substituted. If substituted, the substituent(s) may be selected from those indicated above with regard to substitution of an alkyl group.
- the "cycloalkyl” group can be made up of two or more fused rings (rings that share two adjacent carbon atoms). When the cycloalkyl is a fused ring system, then the ring that is connected to the rest of the molecule is a cycloalkyl as defined above. The other ring(s) in the fused ring system may be a cycloalkyl, a cycloalkenyl, an aryl, a heteroaryl, or a heteroalicyclic.
- cycloalkenyl refers to a cycloalkyl group that contains one or more double bonds in the ring although, if there is more than one, they cannot form a fully delocalized pi-electron system in the ring (otherwise the group would be "aryl,” as defined herein).
- a cycloalkenyl group of this invention may unsubstituted or substituted. When substituted, the substituent(s) may be selected from the same groups disclosed above with regard to alkyl group substitution.
- the "cycloalkenyl” group can be made up of two or more fused rings (rings that share two adjacent carbon atoms).
- the ring that is connected to the rest of the molecule is a cycloalkenyl as defined above.
- the other ring(s) in the fused ring system may be a cycloalkyl, a cycloalkenyl, an aryl, a heteroaryl, or a heteroalicyclic.
- acyl groups include, without limitation, formyl, acetyl, propionyl, butyryl, pentanoyl, pivaloyl, hexanoyl, heptanoyl, octanoyl, nonanoyl, decanoyl, undecanoyl, dodecanoyl and benzoyl.
- Presently preferred acyl groups are acetyl and benzoyl.
- An acyl group of this invention may be unsubstituted or substituted. When substituted, the substituent(s) may be selected from the same groups disclosed above with regard to alkyl group substitution.
- Example of substituted acyl groups include, without limitation, 4-phenylbutyroyl, 3- phenylbutyroyl, 3-phenylpropanoyl, 2-cyclohexanylacetyl, cyclohexanecarbonyl, 2- furanoyl and 3-dimethylaminobenzoyl.
- aromatic refers to an aromatic group which has at least one ring having a conjugated pi electron system and includes both carbocyclic aryl (e.g., phenyl) and heterocyclic aryl groups (e.g., pyridine).
- carbocyclic aryl e.g., phenyl
- heterocyclic aryl groups e.g., pyridine
- the term includes monocyclic or fused-ring polycyclic (i.e., rings which share adjacent pairs of carbon atoms) groups.
- carbocyclic refers to a compound which contains one or more covalently closed ring structures, wherein the atoms forming the backbone of the ring are all carbon atoms.
- heteroaryl refers to an aromatic group, which contains at least one heterocyclic ring, which may be optionally substituted.
- aryl refers to a carbocyclic (all carbon) ring that has a fully delocalized pi-electron system.
- the "aryl” group can be made up of two or more fused rings (rings that share two adjacent carbon atoms). When the aryl is a fused ring system, then the ring that is connected to the rest of the molecule has a fully delocalized pi-electron system. The other ring(s) in the fused ring system may or may not have a fully delocalized pi-electron system.
- aryl groups include, without limitation, benzene, naphthalene and azulene.
- heteroaryl refers to a ring that has a fully delocalized pi-electron system and contains one or more heteroatoms selected from the group consisting of nitrogen, oxygen, and sulfur in the ring.
- the "heteroaryl” group can be made up of two or more fused rings (rings that share two adjacent carbon atoms). When the heteroaryl is a fused ring system, then the ring that is connected to the rest of the molecule has a fully delocalized pi-electron system. The other ring(s) in the fused ring system may or may not have a fully delocalized pi- electron system.
- heteroaryl rings include, without limitation, furan, thiophene, phthalazinone, pyrrole, oxazole, thiazole, imidazole, pyrazole, isoxazole, isothiazole, triazole, thiadiazole, pyran, pyridine, pyridazine, pyrimidine, pyrazine and triazine.
- heterocycloalkyl refers to a ring having in the ring system one or more heteroatoms independently selected from nitrogen, oxygen and sulfur.
- the ring may also contain one or more double bonds provided that they do not form a fully delocalized pi- electron system in the rings.
- Heteroalicyclyl groups of this invention may be unsubstituted or substituted.
- the substituent(s) may be one or more groups independently selected from the group consisting of halogen, hydroxy, protected hydroxy, cyano, nitro, alkyl, alkoxy, acyl, acyloxy, carboxy, protected carboxy, amino, protected amino, carboxamide, protected carboxamide, alkylsulfonamido and trifluoromethanesulfonamido.
- the "heterocycloalkyl” group can be made up of two or more fused rings (rings that share two adjacent carbon atoms). When the heterocycloalkyl is a fused ring system, then the ring that is connected to the rest of the molecule is a heterocycloalkyl as defined above.
- the other ring(s) in the fused ring system may be a cycloalkyl, a cycloalkenyl, an aryl, a heteroaryl, or a heteroalicyclic.
- arylalkyl or “aralkyl,” which are used synonymously and interchangeably, refer to an aryl group covalently bonded to an alkyl group, as defined herein.
- a "phenylalkyl” is a species of an aralkyl group, and refers to a phenyl ring covalently bonded to an alkyl group as defined herein. Examples, without limitation, of phenylalkyl groups include, without limitation, benzyl, 2-phenylethyl, 1-phenylpropyl, 4-phenylhexyl, 3-phenylamyl and 3-phenyl-2- methylpropyl.
- phenylalkyl groups are those wherein the phenyl group is covalently bonded to one of the presently preferred alkyl groups.
- a phenyl alkyl group of this invention may be unsubstituted or substituted.
- substituted phenylalkyl groups include, without limitation, 2-phenyl-l-chloroethyl, 2- (4-methoxyphenyl)ethyl, 4-(2,6-dihydroxy phenyl)hexyl, 2-(5-cyano-3- methoxyphenyl)pentyl, 3-(2,6-dimethylphenyl)propyl, 4-chloro-3-aminobenzyl, 6-(4- methoxyphenyl)-3-carboxy(n-hexyl), 5-(4-aminomethylphenyl)-3-
- heteroarylalkyl or “heteroaralkyl,” which are used synonymously and interchangeably, and “heteroalicyclylalkyl” refer to a heteroaryl or a heteroalicyclyl group, respectively, covalently bonded to an alkyl group, as defined herein.
- examples of such groups include, without limitation, 2- pyridylethyl, 3-pyridylpropyl, 4-furylhexyl, 3-piperazylamyl and 3-morpholinylbutyl.
- Presently preferred heteroarylalkyl and heteroalicyclylalkyl groups are those in which a presently preferred heteroaryl or heteroalicyclyl group is covalently bonded to a presently preferred alkyl group as disclosed herein.
- phenyl refers to a 6-member aryl group.
- a phenyl group may be unsubstituted or substituted.
- the substituent(s) is/are one or more, preferably one or two, group(s) independently selected from the group consisting of halogen, hydroxy, protected hydroxy, cyano, nitro, alkyl, alkoxy, acyl, acyloxy, carboxy, protected carboxy, carboxymethyl, protected carboxymethyl, hydroxymethyl, protected hydroxymethyl, -NR a R b wherein R a and R b are as defined above but in addition R a may be an amino protecting group as defined herein, carboxamide, protected carboxamide, N-alkylcarboxamide, protected N- alkylcarboxamide, N,N-dialkylcarboxamide, trifluoromethyl, N-alkylsulfonylamino, N-(phenylsulfonyl)amino and pheny
- substituted phenyl groups include, without limitation, 2, 3 or 4-chlorophenyl, 2,6-dichlorophenyl, 2,5-dichlorophenyl, 3,4-dichlorophenyl, 2, 3 or 4-bromophenyl, 3,4-dibromophenyl, 3-chloro-4-fluorophenyl, 2, 3 and A- fluorophenyl, 2, 3 or 4-hydroxyphenyl, 2,4-dihydroxyphenyl, the protected-hydroxy derivatives thereof, 2, 3 or 4-nitrophenyl; 2, 3 or 4-cyanophenyl; 2, 3 or A- methylphenyl, 2,4-dimethylphenyl, 2, 3 or 4-(iso-propyl)phenyl, 2, 3 or A- ethylphenyl, 2, 3 or 4-(n-propyl)phenyl, 2,6-dimethoxyphenyl, 2, 3 or A- methoxyphenyl, 2, 3 or 4-ethoxyphenyl
- phenylalkoxy refers to a “phenylalkyl-O-" group with “phenyl” and “alkyl” as defined herein.
- a phenylalkoxy group of this invention may be substituted or unsubstituted on the phenyl ring, in the alkyl group or both.
- phenylalkoxy groups include, without limitation, 2-(4- hydroxyphenyl)ethoxy, 4-(4-methoxyphenyl)butoxy, (2R)-3-phenyl-2-amino- propoxy, (2S)-3-phenyl-2-amino-propoxy, 2-indanoxy, 6-phenyl-l-hexanoxy, cinnamyloxy, 2-phenyl-l-propoxy and 2,2-dimethyl-3 -phenyl- 1-propoxy.
- halo and halogen refer to the fluoro, chloro, bromo or iodo atoms. Presently preferred halogens are chloro and fluoro.
- amino protecting group refers to a group commonly employed to keep (i.e., to "block” or “protect”) an amino group from reacting with a reagent while it reacts with an intended target functional group of a molecule.
- a "protected carboxamide” refers to a carboxamide in which the nitrogen is substituted with an amino protecting group.
- amino protecting groups include, without limitation, formyl ("For"), trityl, phthalimido, trichloroacetyl, chloroacetyl, bromoacetyl, iodoacetyl groups, t-butoxycarbonyl ("Boc”), 2-(4-biphenylyl)propyl-2-oxycarbonyl ("Bpoc”), 2-phenylpropyl-2-oxycarbonyl ("Poc”), 2-(4-xenyl)isopropoxycarbonyl, 1 , 1 -diphenylethyl- 1 -oxycarbonyl, 1 , 1 -diphenylpropyl- 1 -oxycarbonyl, 2-(3 ,5 - dimethoxyphenyl)propyl-2-oxycarbonyl (“Ddz”), 2-(p-toluyl)propyl-2-oxycarbonyl, cyclopentanyloxycarbonyl, 1 -methylcycl
- amino-protecting group employed is not critical so long as the derivatized amino group is stable to the conditions of the subsequent reaction(s) and can be removed at the appropriate point without disrupting the remainder of the molecule.
- amino-protecting groups are Boc, Cbz and Fmoc. Descriptions of these and other amino-protecting groups may be found in T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Synthesis," 2nd ed., John Wiley and Sons, New York, N.Y., 1991, Chapter 7, M.
- carboxy protecting group refers to a labile ester commonly used to block or protect a carboxylic acid while reactions are carried out on other functional groups on the compound.
- carboxy protecting groups include, without limitation, t-butyl, 4-nitrobenzyl, 4- methoxybenzyl, 3,4-dimethoxybenzyl, 2,4-dimethoxybenzyl, 2,4,6-trimethoxybenzyl, 2,4,6-trimethylbenzyl, pentamethylbenzyl, 3,4-methylenedioxybenzyl, benzhydryl, 4,4'-dimethoxytrityl, 4,4',4"-trimethoxytrityl, 2-phenylpropyl, trimethylsilyl, t- butyldimethylsilyl, phenacyl, 2,2,2-trichloroethyl, -(trimethylsilyl)ethyl, -(di(n- butyl)methylsily
- the ester employed is not critical so long as it is stable to the conditions of subsequent reaction(s) and can be removed at the appropriate point without disrupting the remainder of the molecule.
- carboxy -protecting groups are found in E. Haslam, "Protective Groups in Organic Chemistry,” J. G. W. McOmie, Ed., Plenum Press, New York, N.Y., 1973, Chapter 5, and T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Synthesis," 2nd ed., John Wiley and Sons, New York, N. Y., 1991, Chapter 5.
- a "hydroxyl protecting group” refers to a readily cleavable group that replaces the hydrogen of the hydroxyl group, such as, without limitation, tetrahydropyranyl, 2-methoxypropyl, 1-ethoxy ethyl, methoxymethyl, 2- methoxyethoxymethyl, methylthiomethyl, t-butyl, t-amyl, trityl, 4-methoxytrityl, 4,4'- dimethoxytrityl, 4,4',4"-trimethoxytrityl, benzyl, allyl, trimethylsilyl, (t- butyl)dimethylsilyl, and 2,2,2-trichloroethoxycarbonyl.
- hydroxyl protecting groups are not critical so long as the derivatized hydroxyl group is stable to the conditions of subsequent reaction(s) and can be removed at the appropriate point without disrupting the remainder of the molecule.
- Further examples of hydroxy- protecting groups are described by C. B. Reese and E. Haslam, "Protective Groups in Organic Chemistry,” J. G. W. McOmie, Ed., Plenum Press, New York, N.Y., 1973, Chapters 3 and 4, respectively, and T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Synthesis," 2nd ed., John Wiley and Sons, New York, N.Y., 1991, Chapters 2 and 3.
- alkylthio refers to an "alkyl-S-" group, with alkyl as defined above.
- alkylthio group include, without limitation, methylthio, ethylthio, n-propylthio, isopropylthio, n-butylthio and t-butylthio.
- alkylsulfinyl refers to an "alkyl-SO-" group, with alkyl as defined above.
- alkylsulfinyl groups include, without limitation, methylsulfinyl, ethylsulfinyl, n-propylsulfinyl, isopropylsulfinyl, n-butylsulfinyl and sec-butylsulfinyl.
- alkylsulfonyl refers to an "alkyl-SO 2 -" group.
- alkylsulfonyl groups include, without limitation, methylsulfonyl, ethylsulfonyl, n-propylsulfonyl, isopropylsulfonyl, n-butylsulfonyl, and t- butylsulfonyl.
- phenylthio refers to a "phenyl-S-,” “phenyl-SO-,” and “phenyl-SO 2 -” group, phenyl as defined herein.
- alkylaminocarbonyl groups include, without limitation, methylaminocarbonyl, ethylaminocarbonyl, propylaminocarbonyl and butylaminocarbonyl.
- substituted alkylaminocarbonyl include, without limitation, methoxymethyl- aminocarbonyl, 2-chloroethylaminocarbonyl, 2-oxopropylaminocarbonyl and A- phenylbutylaminocarbonyl.
- substituted phenylaminocarbonyl groups include, without limitation, 2- chlorophenyl-aminocarbonyl, 3-chlorophenylaminocarbonyl, 2- nitorphenylaminocarbonyl, 4-biphenylaminocarbonyl, and A- methoxyphenylaminocarbonyl.
- alkylaminothiocarbonyl groups include, without limitation, methylaminothiocarbonyl, ethylaminothiocarbonyl, propylaminothiocarbonyl and butylaminothiocarbonyl.
- alkyl-substituted alkylaminothiocarbonyl groups include, without limitation, methoxymethylaminothiocarbonyl, 2- chloroethylaminothiocarbonyl, 2-oxopropylaminothiocarbonyl and A- phenylbutylaminothiocarbonyl.
- phenylaminothiocarbonyl groups include, without limitation, 2- chlorophenylaminothiocarbonyl, 3-chlorophenyl-aminothiocarbonyl, 2- nitrophenylaminothiocarbonyl, 4-biphenylaminothiocarbonyl and A- methoxyphenylaminothiocarbonyl.
- hydroxyl refers to an "-OH” group.
- cyano refers to a "-C ⁇ N” group.
- nitro refers to an "-NO 2 " group.
- a "C-carboxy” group refers to a “-C(O)OR” group with R as defined above.
- acetyl refers to a CH 3 C(O)- group.
- a "trihalomethanesulfonyl” group refers to an "X 3 CSO 2 -" group wherein X is a halogen.
- a "thiocyanato" group refers to a "-CNS” group.
- An "isothiocyanato" group refers to an " -NCS” group.
- a “sulfinyl” group refers to an "-S(O)-R” group with R as defined above.
- S-sulfonamido refers to a "-SO 2 NR" group with R as defined above.
- N-sulfonamido refers to a "RSO 2 NH-" group with R as defined above.
- a "trihalomethanesulfonamido" group refers to an "X 3 CSO 2 NR-" group with X as halogen and R as defined above.
- An "O-carbamyl” group refers to a "-OC(O)-NR" group with R as defined above.
- N-carbamyl refers to an "ROC(O)NH-" group with R as defined above.
- a "C-amido” group refers to a "-C(O)-NR a R b group with R a and R b as defined above.
- N-amido refers to a RC(O)NH- group with R as defined above.
- haloalkyl refers to an alkyl group where one or more of the hydrogen atoms are replaced by halogen.
- groups include but are not limited to , chloromethyl, fluoromethyl, difluoromethyl, trifluoromethyl and l-chloro-2- fluoromethyl, 2-fluoroisobutyl.
- perhaloalkyl refers to an alkyl group in which all the hydrogen atoms are replaced by halogen atoms.
- an “ester” refers to a “-C(O)OR a " group with R a as defined herein.
- an “amide” refers to a “-C(O)NR a R b " group with R a and R as defined herein.
- substituents are not specified (e.g. haloalkyl) there may be one or more substituents presents.
- haloalkyl may include one or more of the same or differents halogens.
- C 1 -C 3 alkoxy phenyl may include one or more of the same of different alkoxygroups containing one, two or three atoms.
- Any unsubstituted or monosubstituted amine group on a compound herein can be converted to an amide, any hydroxyl group can be converted to an ester and any carboxyl group can be converted to either an amide or ester using techniques well-known to those skilled in the art (see, for example, Greene and Wuts, Protective Groups in Organic Synthesis, 3 rd Ed., John Wiley & Sons, New York, NY, 1999). Compounds containing any such converted hydroxyl, amino and/or carboxylic acid groups are within the scope of this invention.
- an “ether” refers to a "-C-O-C-" group wherein either or both carbons may independently be part of an alkyl, alkenyl, alkynyl, aryl, heteroaryl or heteroalicyclyl group.
- halogenated ether refers to an ether in which the groups to either side of the oxygen are both alkyl substituted with halogen.
- amino acid refers to any one of the twenty naturally-occurring L-amino acids, to their non-natural D-enantiomers, to non- naturally occurring amino acids such as, without limitation, norleucine ("NIe”), norvaline (“Nva”), L- or D-naphthalanine, ornithine (“Orn”), homoarginine (homoArg) and to other amino acids well-known in the peptide art such as those described in M.
- NIe norleucine
- Nva norvaline
- Orn ornithine
- homoarginine homoarginine
- contemplated herein is a composition comprising the S enantiomer substantially free of the R enantiomer, or a composition comprising the R enantiomer substantially free of the S enantiomer.
- substantially free it is meant that the composition comprises less than 10%, or less than 8%, or less than 5%, or less than 3%, or less than 1% of the minor enantiomer. If the particular compound comprises more than one chiral center, the scope of the present disclosure also includes compositions comprising a mixture of the various diastereomers, as well as compositions comprising each diastereomer substantially free of the other diastereomers.
- compositions comprising all four diastereomers includes compositions comprising all four diastereomers, compositions comprising the racemic mixture of R,R and S,S isomers, compositions comprising the racemic mixture of R,S and S,R isomers, compositions comprising the R,R enantiomer substantially free of the other diastereomers, compositions comprising the S, S enantiomer substantially free of the other diastereomers, compositions comprising the R, S enantiomer substantially free of the other diastereomers, and compositions comprising the S,R enantiomer substantially free of the other diastereomers.
- Ri is selected from the group consisting of optionally substituted C 1 -C 10 straight chained or branched alkyl, optionally substituted C 3 -C 9 cycloalkyl, optionally substituted aryl, and optionally substituted heteroaryl. In other embodiments, Ri is selected from the group consisting of optionally substituted aryl and optionally substituted heteroaryl.
- the aryl and the heteroaryl of Ri are optionally substituted with one or more substituents selected from the group consisting of alkyl, hydroxy, protected hydroxy, alkoxy, aryloxy, acyl, ester, mercapto, alkylthio, arylthio, cyano, halogen, carbonyl, C-amido, N-amido, C-carboxy, protected C-carboxy, O-carboxy, nitro, silyl, sulfenyl, sulfinyl, sulfonyl, haloalkyl, perhaloalkyl, and amino.
- substituents selected from the group consisting of alkyl, hydroxy, protected hydroxy, alkoxy, aryloxy, acyl, ester, mercapto, alkylthio, arylthio, cyano, halogen, carbonyl, C-amido, N-amido, C-carboxy, protected C-
- the aryl and the heteroaryl of Ri are optionally substituted with one or more substituents selected from the group consisting of hydroxy, alkoxy, aryloxy, cyano, halogen, and amino.
- the aryl and the heteroaryl of Ri are optionally substituted with one or more substituents selected from the group consisting of fluoro, chloro, bromo, iodo, haloalkyl, and perhaloalkyl.
- the aryl is phenyl
- Ri is selected from the group consisting of phenyl, 4-cholorophenyl, 3-cholorophenyl, and 2-cholorophenyl.
- R 2 is selected from the group consisting of hydrogen, optionally substituted C 1 -C 10 straight chained or branched alkyl, optionally substituted C 2 -C 10 straight chained or branched alkenyl, optionally substituted C 2 -C 10 straight chained or branched alkynyl, optionally substituted C 3 -C 9 cycloalkyl, and optionally substituted C5-C7 cycloalkenyl.
- R 2 is selected from the group consisting of hydrogen, optionally substituted Ci-Ci 0 straight chained or branched alkyl.
- R 2 is hydrogen.
- R3 is selected from the group consisting of optionally substituted Ci-Ci 0 straight chained or branched alkyl, optionally substituted C 2 -C 10 straight chained or branched alkenyl, optionally substituted C 2 -C 10 straight chained or branched alkynyl, optionally substituted C 3 -C 9 cycloalkyl, optionally substituted C5-C7 cycloalkenyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted aralkyl, and optionally substituted heteroaralkyl.
- R3 is selected from the group consisting of optionally substituted Ci-Ci 0 straight chained or branched alkyl, optionally substituted C3-C9 cycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted aralkyl, and optionally substituted heteroaralkyl. In further embodiments, R 3 is selected from the group consisting of optionally substituted aralkyl, and optionally substituted heteroaralkyl.
- the aralkyl or heteroaralkyl of R3 is optionally substituted with one or more substituents selected from the group consisting of alkyl, hydroxy, protected hydroxy, alkoxy, aryloxy, acyl, ester, mercapto, alkylthio, arylthio, cyano, halogen, carbonyl, C-amido, N-amido, C-carboxy, protected C-carboxy, O-carboxy, nitro, silyl, sulfenyl, sulfinyl, sulfonyl, haloalkyl, perhaloalkyl, and amino.
- the substituent is selected from the group consisting of alkyl, hydroxy, halogen, carbonyl, C-carboxy, protected C-carboxy, and amino.
- the aralkyl or heteroaralkyl of R3 is optionally substituted with one or more substituents selected from the group consisting of alkyl, halogen, C-carboxy, and protected C-carboxy.
- the alkyl is selected from the group consisting of methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec-butyl, iso-butyl, and tert-butyl.
- the C-carboxy is protected by an alkyl group, which in some embodiments, is selected from the group consisting of methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec-butyl, iso-butyl, and tert-butyl.
- R3 is selected from the group consisting of benzyl, 4- chlorobenzyl, 2-methylbenzyl, 3-methylbenzyl, 4-methylbenzyl, 4-cyanobenzyl, 2,4- dicholorbenzyl, 2,6-dicholorbenzyl, and 4-methylcarboxybenzyl.
- X is N + .
- Y is selected from the group consisting of halide, acetate, tartrate, phosphate, sulfonate, tetrafluoroborate.
- the halide is selected from the group consisting of fluoride, chloride, bromide, and iodide.
- Y is selected from the group consisting of chloride, bromide, and acetate.
- the compound of Formula I or II is not one of the following compounds:
- the compound of Formula I or II is not one of the following compounds: where Y is a counter ion.
- L is an alkyl spacer optionally substituted with one or more substituents
- Ri and R 3 are each independently optionally substituted aryl or optionally substituted heteroaryl;
- R 2 is selected from the group consisting of hydrogen, -OH, -COOH, halogen, optionally substituted C 1 -C 10 straight chained or branched alkyl, optionally substituted C 2 -C 10 straight chained or branched alkenyl, optionally substituted C 2 -C 10 straight chained or branched alkynyl, optionally substituted C3-C9 cycloalkyl, optionally substituted C5-C7 cycloalkenyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted aralkyl, and optionally substituted heteroaralkyl;
- Y is a counter ion; and n is an integer from 1 to 5, such as n is 1, 2, 3, 4, or 5.
- Ri is optionally substituted phenyl.
- R3 is optionally substituted phenyl.
- L is -CH 2 -.
- a method of modulating the activity of a RAR ⁇ or RAR ⁇ receptor subtypes comprising contacting a RAR ⁇ or RAR ⁇ receptor with at least one compound of Formula I or II.
- the modulating is performed in vivo, whereas in other embodiments, the modulating is performed in vitro.
- the compound of Formula I or II modulates the activity of an RAR ⁇ isoform. In other embodiments, the compound of Formula I or II modulates the activity of an RAR ⁇ isoform.
- a modulator is defined as a compound that is an agonist, a partial agonist, an inverse agonist or an antagonist of one or more RAR ⁇ and/or RAR ⁇ receptors.
- a modulator may increase the activity of the RAR ⁇ or RAR ⁇ receptor subtype, or may decrease the activity of the RAR ⁇ or RAR ⁇ receptor subtype.
- an "agonist” is defined as a compound that increases the basal activity of a receptor (i.e. signal transduction mediated by the receptor).
- An "antagonist” is defined as a compound, which blocks the action of an agonist on a receptor.
- a "partial agonist” is defined as an agonist that displays limited, or less than complete, activity such that it fails to activate a receptor in vitro, functioning as an antagonist in vivo.
- An "inverse agonist” is defined as a compound that decreases the basal activity of a receptor.
- the administered compound modulates the activity of a RAR ⁇ or RAR ⁇ receptor subtypes.
- the compound of Formula I or II is administered in conjunction with at least one chemotherapeutic agent and/or radiation therapy.
- the cancer is associated with malignant tumors.
- the cancer is selected from the group consisting of breast carcinoma and cancers of the skin, head, neck, lung, esophagus, mammary gland, liver, pancreas, cervix and digestive tract (e.g. oral cavity, esophagus, stomach, small intestine, including duodenum, jejunum, and ileum, and colon).
- the term "subject” refers to an animal, preferably a mammal, and most preferably a human, who is the object of treatment, observation or experiment.
- the mammal may be selected from the group consisting of mice, rats, rabbits, guinea pigs, dogs, cats, sheep, goats, cows, primates, such as monkeys, chimpanzees, and apes, and humans.
- terapéuticaally effective amount is used to indicate an amount of an active compound, or pharmaceutical agent, that elicits the biological or medicinal response indicated. This response may occur in a tissue, system, animal or human and includes alleviation of the symptoms of the disease being treated.
- a method of treating, or alleviating the symptoms of, hyperproliferative and inflammatory disorders comprising administering to a subject in need thereof a therapeutically effective amount of at least one compound of Formula I or II, described above.
- the administered compound modulates the activity of a RAR ⁇ or RAR ⁇ receptor subtypes.
- the inflammatory disorder is a chronic inflammatory disorder.
- the inflammatory disorder is rheumatoid arthritis
- a method of treating or alleviating symptoms of a neurological disorder comprising administering to a subject in need thereof a therapeutically effective amount of at least one compound of Formula I or II, described above.
- the administered compound modulates the activity of a RAR ⁇ or RAR ⁇ receptor subtypes.
- the neurological disorder is selected from the group consisting of performance deficits in spatial learning and memory tasks and age-related memory deficit.
- the neurological disorder is a disorder wherein cognition is altered.
- the neurological disorder is schizophrenia.
- a method treating or alleviating symptoms of a neurodegenerative disorder comprising administering to a subject in need thereof a therapeutically effective amount of at least one compound of Formula I or II, described above.
- the administered compound modulates the activity of a RAR ⁇ or RAR ⁇ receptor subtypes.
- the neurodegenerative disorder is Parkinson's disease or Alzheimer's disease.
- the neurodegenerative disorder relates to a method for the treatment of neurodegenerative disorders where nerve regeneration is necessary after, e.g. a spinal cord injury, after a stroke, after damage to the cardiac musles, after damage caused to myelin in multiple sclerosis and damage to islet cells in diabetes.
- a method treating or alleviating symptoms of a hyperproliferative or inflammatory disorder comprising administering to a subject in need thereof a therapeutically effective amount of at least one compound of Formula I or II, described above.
- the administered compound modulates the activity of a RAR ⁇ and/or RAR ⁇ receptor subtypes.
- the inflammatory disorder is a chronic inflammatory disorder.
- the inflammatory disorder is psoriasis or other associated disorders or rheumatoid arthritis.
- a method for treating or alleviating symptoms of an eye disorder or an eye condition comprising administering to a subject in need thereof a therapeutically effective amount of at least one compound of Formula I or II, described above.
- the administered compound modulates the activity of a RAR ⁇ and/or RAR ⁇ receptor subtypes.
- a method for treating or alleviating symptoms of depression comprising administering to a subject in need thereof a therapeutically effective amount of at least one compound of Formula I or II, described above.
- the administered compound modulates the activity of a RAR ⁇ and/or RAR ⁇ receptor subtypes.
- a method for treating or alleviating symptoms of pulmonary disorders/conditions comprising administering to a subject in need thereof a therapeutically effective amount of at least one compound of Formula I or II, described above.
- the administered compound modulates the activity of a RAR ⁇ and/or RAR ⁇ receptor subtypes.
- a method for treating or alleviating symptoms of dermatological disorders/conditions comprising administering to a subject in need thereof a therapeutically effective amount of at least one compound of Formula I or II, described above.
- the administered compound modulates the activity of a RAR ⁇ and/or RAR ⁇ receptor subtypes.
- the dermatological disorder is selected from the group consisting of cystic acne, acne vulgaris and other related acneiform diseases, cutanous disordes of keratinization (e.g.
- a method of identifying a compound that is an agonist, inverse agonist, or antagonist of one or more RAR ⁇ receptors and/or RAR ⁇ receptors comprising contacting RAR ⁇ receptor and/or RAR ⁇ receptor with at least one test compound of Formula I or II and determining any change in activity of the one or more RAR ⁇ receptors and/or RAR ⁇ receptors so as to identify the test compound as an agonist, inverse agonist, or antagonist of one or more RAR ⁇ receptors and/or RAR ⁇ receptors.
- the RAR ⁇ receptor and/or RAR ⁇ receptor is expressed in a cell.
- the cell is a cultured cell.
- the cultured cells overexpress the RAR ⁇ receptor and/or RAR ⁇ receptor.
- the identified agonist, inverse agonist or antagonist is selective for the RAR ⁇ receptor and/or RAR ⁇ receptor.
- the R-SAT test method disclosed in Example 42, may be used. When using this test, a compound is considered to have activity if the pECso is > 5.0 and the %Eff is > 25.
- the present invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising at least one compound of Formula I or II, and a physiologically acceptable component such as a carrier, a diluent, a salt or an excipient, or a combination thereof.
- composition refers to a mixture of a compound disclosed herein with other chemical components, such as diluents or carriers.
- the pharmaceutical composition facilitates administration of the compound to a subject. Multiple techniques of administering a compound exist in the art including, but not limited to, oral, injection, aerosol, parenteral, and topical administration.
- Pharmaceutical compositions can also be obtained by reacting compounds with inorganic or organic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, methanesulfonic acid, ethanesulfonic acid, p-toluenesulfonic acid, salicylic acid and the like.
- carrier defines a chemical compound that facilitates the incorporation of a compound into cells or tissues.
- DMSO dimethyl sulfoxide
- carrier facilitates the uptake of many organic compounds into the cells or tissues of a subject.
- diot defines chemical compounds diluted in water that will dissolve the compound of interest as well as stabilize the biologically active form of the compound. Salts dissolved in buffered solutions are utilized as diluents in the art.
- One commonly used buffered solution is phosphate buffered saline because it mimics the salt conditions of human blood. Since buffer salts can control the pH of a solution at low concentrations, a buffered diluent rarely modifies the biological activity of a compound.
- physiologically acceptable defines a carrier or diluent that does not abrogate the biological activity and properties of the compound.
- Suitable routes of administration may, for example, include oral, rectal, transmucosal, or intestinal administration; parenteral delivery, including intramuscular, subcutaneous, intravenous, intramedullary injections, as well as intrathecal, direct intraventricular, intraperitoneal, intranasal, or intraocular injections.
- compositions disclosed herein may be manufactured in a manner that is itself known, e.g., by means of conventional mixing, dissolving, granulating, dragee-making, levigating, emulsifying, encapsulating, entrapping or tabletting processes.
- compositions for use in accordance with the present disclosure thus may be formulated in a conventional manner using one or more physiologically acceptable carriers comprising excipients and auxiliaries, which facilitate processing of the active compounds into preparations, which can be used pharmaceutically. Proper formulation is dependent upon the route of administration chosen. Any of the well-known techniques, carriers, and excipients may be used as suitable and as understood in the art; e.g., in Remington's Pharmaceutical Sciences, above. [00136]
- the agents disclosed herein may be formulated in aqueous solutions, preferably in physiologically compatible buffers such as Hank's solution, Ringer's solution, or physiological saline buffer.
- physiologically compatible buffers such as Hank's solution, Ringer's solution, or physiological saline buffer.
- penetrants appropriate to the barrier to be permeated are used in the formulation. Such penetrants are generally known in the art.
- the compounds can be formulated readily by combining the active compounds with pharmaceutically acceptable carriers well known in the art.
- Such carriers enable the compounds disclosed herein to be formulated as tablets, pills, dragees, capsules, liquids, gels, syrups, slurries, suspensions and the like, for oral ingestion by a patient to be treated.
- Pharmaceutical preparations for oral use can be obtained by mixing one or more solid excipient with pharmaceutical combination disclosed herein, optionally grinding the resulting mixture, and processing the mixture of granules, after adding suitable auxiliaries, if desired, to obtain tablets or dragee cores.
- Suitable excipients are, in particular, fillers such as sugars, including lactose, sucrose, mannitol, or sorbitol; cellulose preparations such as, for example, maize starch, wheat starch, rice starch, potato starch, gelatin, gum tragacanth, methyl cellulose, hydroxypropylmethyl-cellulose, sodium carboxymethylcellulose, and/or polyvinylpyrrolidone (PVP).
- disintegrating agents may be added, such as the cross-linked polyvinyl pyrrolidone, agar, or alginic acid or a salt thereof such as sodium alginate.
- Dragee cores are provided with suitable coatings.
- suitable coatings may be used, which may optionally contain gum arabic, talc, polyvinyl pyrrolidone, carbopol gel, polyethylene glycol, and/or titanium dioxide, lacquer solutions, and suitable organic solvents or solvent mixtures.
- Dyestuffs or pigments may be added to the tablets or dragee coatings for identification or to characterize different combinations of active compound doses.
- compositions which can be used orally, include push-fit capsules made of gelatin, as well as soft, sealed capsules made of gelatin and a plasticizer, such as glycerol or sorbitol.
- the push-fit capsules can contain the active ingredients in admixture with filler such as lactose, binders such as starches, and/or lubricants such as talc or magnesium stearate and, optionally, stabilizers.
- the active compounds may be dissolved or suspended in suitable liquids, such as fatty oils, liquid paraffin, or liquid polyethylene glycols.
- stabilizers may be added. All formulations for oral administration should be in dosages suitable for such administration.
- compositions may take the form of tablets or lozenges formulated in conventional manner.
- the compounds for use according to the present disclosure are conveniently delivered in the form of an aerosol spray presentation from pressurized packs or a nebulizer, with the use of a suitable propellant, e.g., dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas.
- a suitable propellant e.g., dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas.
- a suitable propellant e.g., dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas.
- a suitable propellant e.g., dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or
- the compounds may be formulated for parenteral administration by injection, e.g., by bolus injection or continuous infusion.
- Formulations for injection may be presented in unit dosage form, e.g., in ampoules or in multi-dose containers, with an added preservative.
- the compositions may take such forms as suspensions, solutions or emulsions in oily or aqueous vehicles, and may contain formulatory agents such as suspending, stabilizing and/or dispersing agents.
- compositions for parenteral administration include aqueous solutions of the active compounds in water-soluble form. Additionally, suspensions of the active compounds may be prepared as appropriate oily injection suspensions. Suitable lipophilic solvents or vehicles include fatty oils such as sesame oil, or synthetic fatty acid esters, such as ethyl oleate or triglycerides, or liposomes. Aqueous injection suspensions may contain substances, which increase the viscosity of the suspension, such as sodium carboxymethyl cellulose, sorbitol, or dextran. Optionally, the suspension may also contain suitable stabilizers or agents, which increase the solubility of the compounds to allow for the preparation of highly, concentrated solutions.
- the active ingredient may be in powder form for constitution with a suitable vehicle, e.g., sterile pyrogen-free water, before use.
- a suitable vehicle e.g., sterile pyrogen-free water
- the compounds may also be formulated in rectal compositions such as suppositories or retention enemas, e.g., containing conventional suppository bases such as cocoa butter or other glycerides.
- the compounds may also be formulated as a depot preparation. Such long acting formulations may be administered by implantation (for example subcutaneously or intramuscularly) or by intramuscular injection.
- the compounds may be formulated with suitable polymeric or hydrophobic materials (for example as an emulsion in an acceptable oil) or ion exchange resins, or as sparingly soluble derivatives, for example, as a sparingly soluble salt.
- a pharmaceutical carrier for the hydrophobic compounds disclosed herein is a cosolvent system comprising benzyl alcohol, a nonpolar surfactant, a water-miscible organic polymer, and an aqueous phase.
- a common cosolvent system used is the VPD co-solvent system, which is a solution of 3% w/v benzyl alcohol, 8% w/v of the nonpolar surfactant Polysorbate 80TM, and 65% w/v polyethylene glycol 300, made up to volume in abs ethanol.
- VPD co-solvent system which is a solution of 3% w/v benzyl alcohol, 8% w/v of the nonpolar surfactant Polysorbate 80TM, and 65% w/v polyethylene glycol 300, made up to volume in abs ethanol.
- the proportions of a co-solvent system may be varied considerably without destroying its solubility and toxicity characteristics.
- co-solvent components may be varied: for example, other low-toxicity nonpolar surfactants may be used instead of POLYSORBATE 80TM; the fraction size of polyethylene glycol may be varied; other biocompatible polymers may replace polyethylene glycol, e.g., polyvinyl pyrrolidone; and other sugars or polysaccharides may be used.
- hydrophobic pharmaceutical compounds may be employed.
- Liposomes and emulsions are well known examples of delivery vehicles or carriers for hydrophobic drugs.
- Certain organic solvents such as dimethylsulfoxide also may be employed, although usually at the cost of greater toxicity.
- the compounds may be delivered using a sustained-release system, such as semipermeable matrices of solid hydrophobic polymers containing the therapeutic agent.
- sustained-release materials have been established and are well known by those skilled in the art. Sustained-release capsules may, depending on their chemical nature, release the compounds for a few weeks up to over 100 days. Depending on the chemical nature and the biological stability of the therapeutic reagent, additional strategies for stabilization may be employed.
- salts may be provided as salts with pharmaceutically compatible counterions.
- Pharmaceutically compatible salts may be formed with many acids, including but not limited to hydrochloric, sulfuric, acetic, lactic, tartaric, malic, succinic, etc. Salts tend to be more soluble in aqueous or other protonic solvents than are the corresponding free acids or base forms.
- compositions suitable for use in the methods disclosed herein include compositions where the active ingredients are contained in an amount effective to achieve its intended purpose. More specifically, a therapeutically effective amount means an amount of compound effective to prevent, alleviate or ameliorate symptoms of disease or prolong the survival of the subject being treated. Determination of a therapeutically effective amount is well within the capability of those skilled in the art, especially in light of the detailed disclosure provided herein.
- the exact formulation, route of administration and dosage for the pharmaceutical compositions disclosed herein can be chosen by the individual physician in view of the patient's condition. (See e.g., Fingl et al. 1975, in "The Pharmacological Basis of Therapeutics", Ch. 1 p. 1).
- the dose about the composition administered to the patient can be from about 0.5 to 1000 mg/kg of the patient's body weight, or 1 to 500 mg/kg, or 10 to 500 mg/kg, or 50 to 100 mg/kg of the patient's body weight.
- the dosage may be a single one or a series of two or more given in the course of one or more days, as is needed by the patient.
- human dosages for treatment of at least some condition have been established.
- the methods disclosed herein will use those same dosages, or dosages that are between about 0.1% and 500%, or between about 25% and 250%, or between 50% and 100% of the established human dosage.
- a suitable human dosage can be inferred from ED50 or ID50 values, or other appropriate values derived from in vitro or in vivo studies, as qualified by toxicity studies and efficacy studies in animals.
- the daily dosage regimen for an adult human patient may be, for example, an oral dose of between 0.1 mg and 500 mg of each ingredient, preferably between 1 mg and 250 mg, e.g. 5 to 200 mg or an intravenous, subcutaneous, or intramuscular dose of each ingredient between 0.01 mg and 100 mg, preferably between 0.1 mg and 60 mg, e.g. 1 to 40 mg of each ingredient of the pharmaceutical compositions disclosed herein or a pharmaceutically acceptable salt thereof calculated as the free base, the composition being administered 1 to 4 times per day.
- compositions disclosed herein may be administered by continuous intravenous infusion, preferably at a dose of each ingredient up to 400 mg per day.
- the total daily dosage by oral administration of each ingredient will typically be in the range 1 to 2000 mg and the total daily dosage by parenteral administration will typically be in the range 0.1 to 400 mg.
- the compounds will be administered for a period of continuous therapy, for example for a week or more, or for months or years.
- Dosage amount and interval may be adjusted individually to provide plasma levels of the active moiety, which are sufficient to maintain the modulating effects, or minimal effective concentration (MEC).
- MEC minimal effective concentration
- the MEC will vary for each compound but can be estimated from in vitro data. Dosages necessary to achieve the MEC will depend on individual characteristics and route of administration. However, HPLC assays or bioassays can be used to determine plasma concentrations.
- Dosage intervals can also be determined using MEC value.
- Compositions should be administered using a regimen, which maintains plasma levels above the MEC for 10-90% of the time, preferably between 30-90% and most preferably between 50-90%.
- the effective local concentration of the drug may not be related to plasma concentration.
- composition administered will, of course, be dependent on the subject being treated, on the subject's weight, the severity of the affliction, the manner of administration and the judgment of the prescribing physician.
- compositions may, if desired, be presented in a pack or dispenser device, which may contain one or more unit dosage forms containing the active ingredient.
- the pack may for example comprise metal or plastic foil, such as a blister pack.
- the pack or dispenser device may be accompanied by instructions for administration.
- the pack or dispenser may also be accompanied with a notice associated with the container in form prescribed by a governmental agency regulating the manufacture, use, or sale of pharmaceuticals, which notice is reflective of approval by the agency of the form of the drug for human or veterinary administration. Such notice, for example, may be the labeling approved by the U.S. Food and Drug Administration for prescription drugs, or the approved product insert.
- Compositions comprising a compound disclosed herein formulated in a compatible pharmaceutical carrier may also be prepared, placed in an appropriate container, and labeled for treatment of an indicated condition.
- the ring closing reaction between an ⁇ -aminoketone and an imino ether followed by an N-alkylation can provide compounds of Formula I or II.
- the ring closing reaction is preferably carried out in a microwave, preferably at a temperature of about 130 0 C and for about 15min.
- Ethanol is preferably used as solvent.
- the N- alkylation is preferably carried out in a microwave, at a temperature about 160 0 C and for about 15min. In one embodiment, the reaction is carried out with acetonitrile as solvent.
- the final product is isolated by conventional means, preferably purified by re-crystallization.
- Rl, R2 and R3 have the definitions as described herein.
- Y is defined as a leaving group e.g. a halide.
- X is defined as a counterion e.g. a halide, acetate or phosphate, n is an integer from 1-5. X can in some cases be identical with
- the cyclic imidazoles are formed in one step using microwave irradiation (Scheme 5).
- R P-CI, 0-CH 3 , m-CH 3 , P-CH 3 , p-CN 0,0-Cl, o, m-CI, o,p-CI
- HPLC/MS analyses were performed on a Waters/Micromass ZQ2000 LC/MS instrument with a Xterra® MS Ci 8 3.5 ⁇ m, 30x4.6 mm ID column with a guard column cartridge system and a 996 Photodiode Array Detector; eluent: A: 10 mM aq. NH 4 OAc; B: 10 mM aq. NH 4 OAc CH 3 CN-H 2 O (95:5); 5 min. gradient starting at 30% B (initial hold for 0.5 min.), over 5 min. to 90% B, hold for 0.5 min., over 0.5 min. to 30% B, hold for 2.5 min. The flow rate was 1 mL/min.
- Example 2 General procedure for the synthesis of 7 member quaternary imidazoles (Procedure B) [00183] The benzylhalide (0.30 mmol), triethylamine (33 ⁇ L, 0.47 mmol) and 3-phenyl-6,7,8,9-tetrahydro-5H-imidazo[l,2-a]azepine (0.24 mmol) in 1 mL dry acetonitrile were microwave irradiated at 160 0 C for 15 min. The reaction mixture was evaporated to dryness. The residue was dissolved in H 2 O and loaded on a hydromatrix. The hydromatrix was washed with Et 2 O and the filtrate was extracted with CH 2 Cl 2 .
- the CH 2 Cl 2 phase was evaporated to dryness and dissolved in H 2 O.
- the water phase was washed 3 times with Et 2 O (limited amounts of brine was added to the water phase), and extracted 3 times with CH 2 Cl 2 .
- the organic phase was dried over Na 2 SO 4 , filtrated and evaporated to dryness.
- This product was added to a solution of hexametylenetetramine (2.12 g, 15 mmol) in 70 mL CHCl 3 and was stirred at 50 0 C for 1 h and 15 min. The mixture was cooled to r.t. and the white precipitate was collected by filtration and washed with CHCI3. The resulting quaternary salt was suspended in a mixture of 95% EtOH (20 mL) and cone. HCl (7.5 mL) and heated at 50 0 C for 3 h. The mixture was cooled to 0 0 C and the resulting white solid was collected by filtration.
- Example 11 3-(4-Chloro-phenyl)-6,7,8,9-tetrahydro-5H-imidazori,2-a1azepine
- 2-Amino-l-(4-chloro-phenyl)-ethanone; hydrochloride (268 mg, 1.30 mmol) and 7-methoxy-3,4,5,6-tetrahydro-2H-azepine (158 mg, 1.24 mmol) were subjected to general procedure A and gave the title compound (293 mg, 96%).
- Example 14 3 -(4-Chloro-phenyl)-5 ,6,7, 8 ,9 , 10-hexahydro-imidazo [ 1 ,2-ai azocine
- the reaction mixture was microwave irradiated at 100 0 C for 5 min, evaporated to dryness, dissolved in 4 mL EtOH and 0.3 mL cone. HCl and then microwave irradiated at 120 0 C for 15 min.
- the product was evaporated to dryness and column chromatographed on silica (heptane/EtOAc 1: 1) which gave the title compound (102 mg, 32%).
- Example 15 3 -(3 -Chloro-phenyl)-5 ,6,7, 8 ,9 , 10-hexahydro-imidazo [ 1 ,2-ai azocine
- the reaction mixture was microwave irradiated at 100 0 C for 5 min, evaporated to dryness, dissolved in 4 mL EtOH and 0.3 mL cone. HCl and then microwave irradiated at 120 0 C for 15 min.
- the product was evaporated to dryness and column chromatographed on silica (heptane/EtOAc 1 : 1) which gave the title compound (59 mg, 18%).
- the reaction mixture was microwave irradiated at 100 0 C for 5 min, evaporated to dryness, dissolved in 4 mL EtOH and 0.3 mL cone. HCl and then microwave irradiated at 120 0 C for 15 min.
- the product was evaporated to dryness and column chromatographed on silica (heptane/EtOAc 1: 1) which gave the title compound (82 mg, 29%).
- Example 17 l-(4-Chloro-benzyl)-3-phenyl-6J,8,9-tetrahydro-5H-imidazori,2- alazepin-l-ium; chloride (compound 1)
- Example 18 1 -(2-Methyl-benzyl)-3-phenyl-6 J,8,9-tetrahydro-5H-imidazor 12- a "
- Example 19 l-(3-Methyl-benzyl)-3-phenyl-6J,8,9-tetrahydro-5H-imidazori,2- alazepin-l-ium; chloride (compound 3)
- Example 20 l-(4-Methyl-benzyl)-3-phenyl-6J,8,9-tetrahydro-5H-imidazori,2- a]azepin-l-ium; chloride (compound 4)
- Example 21 l-(4-Cvano-benzyl)-3-phenyl-6 J,8,9-tetrahydro-5H-imidazori 2- a "
- Example 22 l-(2,6-Dichloro-benzyl)-3-phenyl-6J,8,9-tetrahydro-5H-imidazori,2- a "
- Example 25 3-(4-Chloro-phenyl)-l-(2,4-dichloro-benzyl)-5,6,7,8-tetrahvdro- imidazo
- Example 26 3-(3-Chloro-phenyl)-l-(2,4-dichloro-benzyl)-5,6,7,8-tetrahvdro- imidazori,2-a1pyridin-l-ium; chloride (compound 9)
- Example 30 l-(2 ⁇ -dichloro-benzyl)-3-phenyl-6J,8,9-tetrahvdro-5H-imidazori,2- aiazepin-1-ium; acetate (compound 7)
- Example 32 3 -(3 -chloro-phenyl)- 1 -(4-methoxycarbonyl-benzyl)-5 ,6,7,8-tetrahydro- imidazori.2-a1pyridin-l-ium; acetate (compound 12)
- Example 36 1 -(4-methoxycarbonyl-benzyl)-3-phenyl-6,7,8,9-tetrahvdro-5H- imidazori,2-a1azepin-l-ium; acetate (compound 18)
- R-SAT ® The functional receptor assay, Receptor Selection and Amplification Technology (R-SAT ® ), was used to investigate the pharmacological properties of known and novel RAR ⁇ agonists and antagonists and novel RAR ⁇ agonists and antagonists.
- R-SAT ® is disclosed, for example, in U.S. Patent Nos. 5,707,798, 5,912,132, and 5,955,281, Piu, F., Gauthier, N. K., and Wang, F., (Beta Arrestin 2 modulates the activity of Nuclear Receptor RAR beta 2 through activation of ERK2 kinase) Oncogene 2006, 12:25(2):218-29, Burstein, E.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Disclosed herein is a compound of Formula (I) and pharmaceutical composition comprising the same. Also disclosed herein are methods of treating, or alleviating the symptoms of, a disease comprising administering to a subject in need thereof a therapeutically effective amount of a compound of Formula (I). The disease may be selected from the group consisting of cancer, hyperproliferative disorder, inflammatory disorder, neurological disorder, neurodegenerative disorder, eye disorder, eye condition, depression, pulmonary disorders/conditions, dermatological disorders/conditions, and infertility.
Description
COMPOUNDS WITH ACTIVITY AT RETINOIC ACID RECEPTORS
RELATED APPLICATIONS
[001] The present application claims priority to the U.S. Provisional Patent Application Serial No. 60/866,360, filed on November 17, 2006, by Roger Olsson et al, and entitled "COMPOUNDS WITH ACTIVITY AT RETINOIC ACID RECEPTORS," the entire disclosure of which is incorporated herein by reference, including any drawings.
FIELD OF THE INVENTION
[002] The present invention is in the field of pharmaceuticals, and in particular in the field of modulators of the retinoic acid receptors and treatment of diseases with these compounds.
BACKGROUND
[003] Retinoids are small, lipophilic molecules that derive from the metabolism of vitamin A, a dietary vitamin. Natural and synthetic retinoid derivatives exert pleiotropic effects on cellular growth, differentiation, apoptosis, homeostasis and embryogenesis. A number of non-selective retinoids are currently marketed or undergoing clinical trials for use in dermatology and oncology. For instance, Tretinoin (all-trans-retinoic acid), Isotretinoin (13-cis retinoic acid) and Etretinate (a synthetic retinoic acid analog) are being used successfully in the treatment of acne, psoriasis, photoaging and squamous cell carcinoma. However, acute and chronic toxic side effects (skeletal abnormality, skin toxicity, triglyceride elevation, teratogenesis) are commonly observed which can lead to the discontinuation of the treatment.
[004] The biological effects of retinoids are mediated by two classes of nuclear hormone receptors, the retinoic acid receptors (RARs) and the retinoid X receptors (RXRs). RARs and RXRs are ligand-dependent transcription factors belonging to the steroid nuclear receptor superfamily. Like the majority of nuclear receptors, the retinoid receptors display a modular structure: an N-terminus ligand- independent activation domain (AF-I), a DNA-binding domain (DBD) adjacent to the ligand-dependent domain (LBD) and the ligand-dependent activation domain (AF-2) contiguous to the LBD and located at the C-terminus end. Three receptors subtypes have been reported for each of the RARs and RXRs, classified as α, β, and γ. All six
subtypes have reportedly distinct expression patterns in the developing embryo and in the adult, thus, are believed to exhibit specific and non-overlapping functions.
[005] Moreover, several isoforms have been described for each RAR and RXR subtypes. In particular, RARβ consists of five known isoforms generated from the use of two promoters Pl (RARβ 1 and RARβ 3) and P2 (RARβ 2 and RARβ 4). Recently, a fifth isoform (RARβ 1 ') has been discovered, which is alternatively spliced from RARβ 1. The isoforms only differ in the nature of their AF-I transcriptional activation domains located at the very N-terminus. In particular, RARβ 1 and RARβ 3 have very similar AF-I domains. The only difference being the presence of an additional 27 amino acid insert in RARβ 3. RARβ 2, on the other hand, has a unique AF-I domain, while RARβ 4 lacks such a domain as well as a portion of its DNA-binding domain (DBD). Data from the literature supports the notion that, because of its truncated DBD domain, RARβ 4 could act as a dominant negative mutant. Interestingly, the isoforms have distinct spatial and temporal distribution. For example, in the mouse, RARβ 1 and RARβ 3 display a relatively restricted pattern - highly present the brain, and in limited amounts in the lung and skin. RARβ 2 is more broadly expressed in the brain and heart, and at much lower levels in the liver, kidney and skeletal muscle. In humans, only the RARβ 1, 2 and 4 isoforms are expressed. RARγ is expressed in the skin and lungs.
[006] RARβ and/or RARγ modulating compounds may be used to treat cancer. A growing body of evidence supports the hypotheses that the RARβ and/or RARγ genes are tumor suppressor genes and may have chemopreventive effects on various cancers like head and neck cancer, cervix cancer, lung cancer, skin cancer, pancreatic cancer, liver caner, mammary cancer and cancer in the digestive tract (e.g. oral cavity, stomach, esophagus and colon). . Furthermore, RARβ is expressed in normal lung cells and in cells resistant to retinoic acid but is not expressed in lung cancer cells or cells resistant to retinoic acid treatment. Use of a RARβ modulating compound may restore RARβ expression and thus be efficient for lung cancer treatment. RARβ and/or RARγ ligands could be used alone or in combination with existing chemo- or radiation therapy. Synergistic cytotoxicity by combination treatment of selective retinoid RARα/β ligands with taxol (Paclitaxel) has already been demonstrated.
[007] RARβ and/or RARγ modulating compounds may be used to treat a variety of neurological disorders. For instance RARβ null mice exhibit locomotor defects related to dysfunction of the mesolimbic dopamine signaling pathway. Moreover these animals lack hippocampal long-term potentiation (LTP) and long- term depression (LTD), widely studied forms of synaptic plasticity. This results in substantial performance deficits in spatial learning and memory tasks. Interestingly, the expression of RARβ 2 in the brain strikingly overlaps that of the dopamine Dl and D2 receptors. Other animal studies reveal that deficiency in vitamin A, a precursor of retinoids, results in spatial learning and memory impairment as well as a loss in hippocampal long-term synaptic plasticity. Moreover, age-related relational memory deficit in mouse is associated with decreased expression of RARβ. Administration of retinoic acid, a pan RAR agonist, is accompanied by a complete restoration of the behavioral impairment and associated increase in RARβ expression. These effects can be antagonized by the use of a RAR antagonist. Finally, a growing body of evidence indicates RARβ 2 is involved in neurite outgrowth from peripheral and central nervous systems. Thus, RARβ 2-modulating compounds would be therapeutically relevant to the treatment of neurodegenerative disorders including Parkinson's and Alzheimer's diseases. Because of its involvement in cognitive function, RARβ 2 would also be relevant to the treatment of neurological disorders where cognition is altered, in particular schizophrenia. Finally, clinical data from the use of Isotretinoin has suggested an association with depression and suicide.
[008] RARβ and/or RARγ modulating compounds may be used to treat a variety of hyperproliferative and inflammatory disorders. Even though RARβ expression is below detection limits in the skin, RARβ 2-modulating compounds could act indirectly through transrepression of the activating protein 1 (API) complex, a heterodimeric transcription factor composed of Fos- and Jun-related proteins. API is involved in the expression of metalloproteases, cytokines and other factors which play critical roles in the turnover of extracellular matrix, inflammation and hyperproliferation in diseases such as psoriasis or other associated disorders, rheumatoid arthritis and in tumor metastases. The transrepressive effects of retinoids are mediated through a mechanism unrelated to transcriptional activation, involving the RAR-dependent control of transcription factors and cofactor assembly on APl- regulated promoters. Relevant therapeutic indications include acne, psoriasis,
photoaging and other dermatological disorders. RARβ and/or RARγ modulating compounds may also be used to treat chronic inflammatory disorders, for example, rheumatoid arthritis. For instance, retinoids through interaction with the AP-I complex suppress collagenase gene expression. The fibroblast interstitial collagenase MMP-I, which degrades collagen, is thought to play a critical role in the degradation of the cartilage matrix in arthritis. In animal models of arthritis, a RAR antagonist improves clinical and histological scores of arthritis.
[009] RARβ and/or RARγ modulating compounds may be used to treat eye disorders/conditions. Vitamin A, the precursor of natural retinoids, is essential for the normal development and maintenance of the ocular surface. In the eye, RARβ mRNA transcripts are detectable in corneal stroma cells, conjunctival fibroblasts and corneal epithelial cells. RARβ expression is predominantly confined to the periocular mesenchyme and ciliary body. Moreover, retinoic acid further induces the expression of RARβ in corneal and conjunctival fibroblasts. Knockout of RARβ indicates that RARβ is the main RAR subtype involved in modulation of retinal cell populations. In chicken, retinoic acid through its actions on RARβ is associated with form- deprivation myopia.
[0010] RARβ and/or RARγ modulating compounds may be used to treat pulmonary disorders/conditions. Retinoic acid suppresses growth of vascular smooth muscle cells (SMCs) from the systemic and pulmonary circulation and inhibits migration of airway smooth muscle cells. Thus, RARβ and/or RARγ modulating compounds may be used in treatment of e.g. asthma. Additionally, endogenous retinoids have been implicated in alveologenesis, and exogenous retinoic acid can reverse or partially reverse emphysema and associated pulmonary diseases. Likewise compounds modulating RARβ and/or RARγ may exert the same effects. RARβ and/or RARγ modulating compounds may also be used for stimulating tropo-elasting gene expression in human lung fibroblasts.
[0011] RARβ and/or RARγ modulating compounds may be used to treat dermatological disorders/conditions. RAR modulators (especially RARγ) have been used in the treatment of cystic acne, acne vulgaris and other related acneiform diseases, cutanous disordes of keratinization (e.g. the ichtyoses, Darier's disease, pityriasis rubra pilaris), psoriasis and its pustular and erythrodermic variants, miscellaneous cutaneous diseases (subcorneal pustular dermatosis, discoid lupus
erythematosus, Reiter's syndrome (with and without AIDS), warts, lichen planus, cutaneous sarcoidosis). The high level of RARβ and/or RARγ in the skin may indicate an ability, for a ligand of either of the two receptors, to exert a therapeutic effect on such dermatological disorders/conditions.
SUMMARY OF THE INVENTION [0012] Disclosed herein is a compound of Formula I

R1, R2, R3, are each independently selected from the group consisting of hydrogen, -OH, -COOH, halogen, optionally substituted C1- Cio straight chained or branched alkyl, optionally substituted C2-C10 straight chained or branched alkenyl, optionally substituted C2-C10 straight chained or branched alkynyl, optionally substituted C3-C9 cycloalkyl, optionally substituted C3-C9 cycloalkylalkyl, optionally substituted C5-C7 cycloalkenyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted aralkyl, and optionally substituted heteroaralkyl; X is C or N+;
Y is a counter ion and is absent when X is C; and n is an integer from 1 to 5, such as n is 1, 2, 3, 4, or 5, and pharmaceutical composition comprising the same.
[0013] Also disclosed herein is a method of treating, or alleviating the symptoms of, a disease comprising administering to a subject in need thereof a therapeutically effective amount of a compound of Formula I. The disease may be selected from the group consisting of cancer, hyperproliferative disorder, inflammatory disorder, neurological disorder, neurodegenerative disorder, eye disorder, eye condition, depression, pulmonary disorders/conditions, and dermatological disorders/conditions.
DETAILED DESCRIPTION OF THE INVENTION [0014] In one aspect, disclosed herein is a compound of Formula I

Y is a counter ion and is absent when X is C; and n is an integer from 1 to 5, such as n is 1, 2, 3, 4, or 5.
[0015] The term "pharmaceutically acceptable salt" refers to a formulation of a compound that does not abrogate the biological activity and properties of the compound. Pharmaceutical salts can be obtained by reacting a compound of the invention with inorganic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, methanesulfonic acid, ethanesulfonic acid, p- toluenesulfonic acid, salicylic acid and the like. Pharmaceutical salts can also be obtained by reacting a compound of the invention with a base to form a salt such as an ammonium salt, an alkali metal salt, such as a sodium or a potassium salt, an alkaline earth metal salt, such as a calcium or a magnesium salt, a salt of organic bases such as dicyclohexylamine, N-methyl-D-glucamine, tris(hydroxymethyl)methylamine, and salts with amino acids such as arginine, lysine, and the like.
[0016] The term "ester" refers to a chemical moiety with formula -(R)n-COOR', where R and R' are independently selected from the group consisting of alkyl, cycloalkyl, aryl, heteroaryl (bonded through a ring atom) and heteroalicyclic (bonded through a ring atom), and where n is 0 or 1.
[0017] An "amide" is a chemical moiety with formula -(R)n-C(O)NHR' or -(R)n-NHC(O)R', where R and R' are independently selected from the group
consisting of alkyl, cycloalkyl, aryl, heteroaryl (bonded through a ring atom) and heteroalicyclic (bonded through a ring atom), and where n is 0 or 1. An amide may be an amino acid or a peptide molecule attached to a molecule of the present invention, thereby forming a prodrug.
[0018] Any amine, hydroxy, or carboxyl side chain on the compounds of the present invention can be esterified or amidified. The procedures and specific groups used to achieve this end are known to those of skill in the art and can readily be found in reference sources such as Greene and Wuts, Protective Groups in Organic Synthesis, 3.sup.rd Ed., John Wiley & Sons, New York, N.Y., 1999, which is incorporated herein in its entirety.
[0019] A "prodrug" refers to an agent that is converted into the parent drug in vivo. Prodrugs are often useful because, in some situations, they may be easier to administer than the parent drug. They may, for instance, be bioavailable by oral administration whereas the parent is not. The prodrug may also have improved solubility in pharmaceutical compositions over the parent drug. An example, without limitation, of a prodrug would be a compound of the present invention which is administered as an ester (the "prodrug") to facilitate transmittal across a cell membrane where water solubility is detrimental to mobility but which then is metabolically hydrolyzed to the carboxylic acid, the active entity, once inside the cell where water-solubility is beneficial. A further example of a prodrug might be a short peptide (polyaminoacid) bonded to an acid group where the peptide is metabolized to reveal the active moiety.
[0020] Whenever a group of this invention is described as being "optionally substituted" that group may be unsubstituted or substituted with one or more of the substituents described for that group. Likewise, when a group is described as being "unsubstituted or substituted," if substituted, the substituent may be selected from the same group of substituents. Unless otherwise indicated, when a substituent is deemed to be "optionally subsituted," or "substituted" it is meant that the subsitutent is a group that may be substituted with one or more group(s) individually and independently selected from alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, cycloalkynyl, aryl, heteroaryl, heteroalicyclyl, aralkyl, heteroaralkyl, (hetereoalicyclyl)alkyl, hydroxy, protected hydroxyl, alkoxy, aryloxy, acyl, ester, mercapto, alkylthio, arylthio, cyano, halogen, carbonyl, thiocarbonyl, O-carbamyl, N-carbamyl, O-thiocarbamyl, N-thiocarbamyl, C-amido, N-amido, S-sulfonamido,
N-sulfonamido, C-carboxy, protected C-carboxy, O-carboxy, isocyanato, thiocyanato, isothiocyanato, nitro, silyl, sulfenyl, sulfinyl, sulfonyl, haloalkyl, haloalkoxy, trihalomethanesulfonyl, trihalomethanesulfonamido, and amino, including mono- and di-substituted amino groups, and the protected derivatives thereof. The protecting groups that may form the protective derivatives of the above substituents are known to those of skill in the art and may be found in references Greene and Wuts, Protective Groups in Organic Synthesis, 3rd Ed., John Wiley & Sons, New York, NY, 1999, which is hereby incorporated by reference in its entirety.
[0021] As used herein, "Cm-Cn" in which "m" and "n" are integers refers to the number of carbon atoms in an alkyl, alkenyl or alkynyl group or the number of carbon atoms in the ring of a cycloalkyl, cycloalkenyl, or aryl group. That is, the alkyl, alkenyl, alkynyl, ring of the cycloalkyl, ring of the cycloalkenyl, or of the aryl can contain from "m" to "n", inclusive, carbon atoms. Thus, for example, a "Ci-C4 alkyl" group refers to all alkyl groups having from 1 to 4 carbons, that is, CH3-, CH3CH2-, CH3CH2CH2-, CH3CH(CH3)-, CH3CH2CH2CH2-, CH3CH2CH(CH3)-, and (CH3)3CH-. If no "m" and "n" are designated with regard to an alkyl, alkenyl, alkynyl, cycloalkyl or cycloalkenyl group, the broadest range described in these definitions is to be assumed.
[0022] As used herein, "alkyl" refers to a straight or branched chain fully saturated (no double or triple bonds) hydrocarbon (all carbon) group. An alkyl group of this invention may comprise from 1 - 20 carbon atoms, that is, "m" = 1 and "n" = 20, designated as a "C1 to C20 alkyl." In some embodiments, "m" = 1 and "n":= 12 (Ci to Ci2 alkyl). In other embodiments, that "m" = 1 and "n" = 6 (Ci to C6 alkyl). Examples of alkyl groups include, without limitation, methyl, ethyl, n-propyl, isopropyl, n-butyl, iso-butyl, sec -butyl, tert-butyl, amyl, tert-amyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl and dodecyl.
[0023] An alkyl group of this invention may be substituted or unsubstituted. When substituted, the substituent group(s) is(are) one or more group(s) independently selected from cycloalkyl, aryl, heteroaryl, heteroalicyclyl, hydroxy, alkoxy, aryloxy, mercapto, alkylthio, arylthio, cyano, halo, oxo, carbonyl, thiocarbonyl, O-carbamyl, N-carbamyl, 0-thiocarbamyl, N-thiocarbamyl, C-amido, N-amido, S-sulfonamido, N-sulfonamido, C-carboxy, O-carboxy, isocyanato, thiocyanato, isothiocyanato, nitro, silyl, trihalomethanesulfonyl, -NRaRb, protected hydroxyl, protected amino, protected carboxy and protected amido groups.
[0024] Examples of substituted alkyl groups include, without limitation, 2- oxo-prop-1-yl, 3-oxo-but-l-yl, cyanomethyl, nitromethyl, chloromethyl, hydroxymethyl, tetrahydropyranyloxymethyl, m-trityloxymethyl, propionyloxymethyl, aminomethyl, carboxymethyl, allyloxycarbonylmethyl, allyloxycarbonylaminomethyl, methoxymethyl, ethoxymethyl, t-butoxymethyl, acetoxymethyl, chloromethyl, bromomethyl, iodomethyl, trifluoromethyl, 6- hydroxyhexyl, 2,4-dichlorobutyl, 2-aminopropyl, 1 -chloroethyl, 2-chloroethyl, 1- bromoethyl, 2-chloroethyl, 1 -fluoroethyl, 2-fluoroethyl, 1-iodoethyl, 2-iodoethyl, 1- chloropropyl, 2-chloropropyl, 3-chloropropyl, 1-bromopropyl, 2-bromopropyl, 3- bromopropyl, 1-fluoropropyl, 2-fluoropropyl, 3-fluoropropyl, 1 -iodopropyl, 2- iodopropyl, 3-iodopropyl, 2-aminoethyl, 1 -aminoethyl, N-benzoyl-2-aminoethyl, N- acetyl-2-aminoethyl, N-benzoyl-1 -aminoethyl, N-acetyl-1 -aminoethyl, and cyclopropylmethyl.
[0025] As used herein, "alkenyl" refers to an alkyl group that contains in the straight or branched hydrocarbon chain one or more double bonds. Examples of alkenyl groups include, without limitation, vinyl (CH2=CH-), allyl (CH3CH=CH2-), 1- propenyl, 2-propenyl, 1-butenyl, 2-butenyl; 1-pentenyl, 2-pentenyl, 3-pentenyl, 4- pentenyl, 3 -methyl- 1-butenyl, and the various isomers of hexenyl, heptenyl, octenyl, nonenyl, decenyl undecenyl and dodecenyl.
[0026] An alkenyl group of this invention may be unsubstituted or substituted. When substituted, the substituent(s) may be selected from the same groups disclosed above with regard to alkyl group substitution. Examples of substituted alkenyl groups include, without limitation, styrenyl, 3-chloro-propen-l-yl, 3-chloro-buten-l-yl, 3-methoxy-propen-2-yl, 3-phenyl-buten-2-yl and 1-cyano-buten- 3-yl.
[0027] As used herein, "alkynyl" refers to an alkyl group that contains in the straight or branched hydrocarbon chain one or more triple bonds.
[0028] An alkynyl group of this invention may be unsubstituted or substituted. When substituted, the substituent(s) may be selected from the same groups disclosed above with regard to alkyl group substitution.
[0029] As used herein, "cycloalkyl" refers to a completely saturated (no double bonds) hydrocarbon ring. Cycloalkyl groups of this invention may range from C3 to C9. A cycloalkyl group may be unsubstituted or substituted. If substituted, the substituent(s) may be selected from those indicated above with regard to substitution
of an alkyl group. The "cycloalkyl" group can be made up of two or more fused rings (rings that share two adjacent carbon atoms). When the cycloalkyl is a fused ring system, then the ring that is connected to the rest of the molecule is a cycloalkyl as defined above. The other ring(s) in the fused ring system may be a cycloalkyl, a cycloalkenyl, an aryl, a heteroaryl, or a heteroalicyclic.
[0030] As used herein, "cycloalkenyl" refers to a cycloalkyl group that contains one or more double bonds in the ring although, if there is more than one, they cannot form a fully delocalized pi-electron system in the ring (otherwise the group would be "aryl," as defined herein). A cycloalkenyl group of this invention may unsubstituted or substituted. When substituted, the substituent(s) may be selected from the same groups disclosed above with regard to alkyl group substitution. The "cycloalkenyl" group can be made up of two or more fused rings (rings that share two adjacent carbon atoms). When the cycloalkenyl is a fused ring system, then the ring that is connected to the rest of the molecule is a cycloalkenyl as defined above. The other ring(s) in the fused ring system may be a cycloalkyl, a cycloalkenyl, an aryl, a heteroaryl, or a heteroalicyclic.
[0031] As used herein, "acyl" refers to an "RC(=O)O-" Examples of acyl groups include, without limitation, formyl, acetyl, propionyl, butyryl, pentanoyl, pivaloyl, hexanoyl, heptanoyl, octanoyl, nonanoyl, decanoyl, undecanoyl, dodecanoyl and benzoyl. Presently preferred acyl groups are acetyl and benzoyl.
[0032] An acyl group of this invention may be unsubstituted or substituted. When substituted, the substituent(s) may be selected from the same groups disclosed above with regard to alkyl group substitution. Example of substituted acyl groups include, without limitation, 4-phenylbutyroyl, 3- phenylbutyroyl, 3-phenylpropanoyl, 2-cyclohexanylacetyl, cyclohexanecarbonyl, 2- furanoyl and 3-dimethylaminobenzoyl.
[0033] The term "aromatic" refers to an aromatic group which has at least one ring having a conjugated pi electron system and includes both carbocyclic aryl (e.g., phenyl) and heterocyclic aryl groups (e.g., pyridine). The term includes monocyclic or fused-ring polycyclic (i.e., rings which share adjacent pairs of carbon atoms) groups. The term "carbocyclic" refers to a compound which contains one or more covalently closed ring structures, wherein the atoms forming the backbone of the ring are all carbon atoms. The term "heteroaromatic" or "heteroaryl" refers to an
aromatic group, which contains at least one heterocyclic ring, which may be optionally substituted.
[0034] As used herein, "aryl" refers to a carbocyclic (all carbon) ring that has a fully delocalized pi-electron system. The "aryl" group can be made up of two or more fused rings (rings that share two adjacent carbon atoms). When the aryl is a fused ring system, then the ring that is connected to the rest of the molecule has a fully delocalized pi-electron system. The other ring(s) in the fused ring system may or may not have a fully delocalized pi-electron system. Examples of aryl groups include, without limitation, benzene, naphthalene and azulene.
[0035] As used herein, "heteroaryl" refers to a ring that has a fully delocalized pi-electron system and contains one or more heteroatoms selected from the group consisting of nitrogen, oxygen, and sulfur in the ring. The "heteroaryl" group can be made up of two or more fused rings (rings that share two adjacent carbon atoms). When the heteroaryl is a fused ring system, then the ring that is connected to the rest of the molecule has a fully delocalized pi-electron system. The other ring(s) in the fused ring system may or may not have a fully delocalized pi- electron system. Examples of heteroaryl rings include, without limitation, furan, thiophene, phthalazinone, pyrrole, oxazole, thiazole, imidazole, pyrazole, isoxazole, isothiazole, triazole, thiadiazole, pyran, pyridine, pyridazine, pyrimidine, pyrazine and triazine.
[0036] As used herein, "heterocycloalkyl," "heteroalicyclic," or "heteroalicyclyl" refers to a ring having in the ring system one or more heteroatoms independently selected from nitrogen, oxygen and sulfur. The ring may also contain one or more double bonds provided that they do not form a fully delocalized pi- electron system in the rings. Heteroalicyclyl groups of this invention may be unsubstituted or substituted. When substituted, the substituent(s) may be one or more groups independently selected from the group consisting of halogen, hydroxy, protected hydroxy, cyano, nitro, alkyl, alkoxy, acyl, acyloxy, carboxy, protected carboxy, amino, protected amino, carboxamide, protected carboxamide, alkylsulfonamido and trifluoromethanesulfonamido. The "heterocycloalkyl" group can be made up of two or more fused rings (rings that share two adjacent carbon atoms). When the heterocycloalkyl is a fused ring system, then the ring that is connected to the rest of the molecule is a heterocycloalkyl as defined above. The
other ring(s) in the fused ring system may be a cycloalkyl, a cycloalkenyl, an aryl, a heteroaryl, or a heteroalicyclic.
[0037] As used herein, "arylalkyl" or "aralkyl," which are used synonymously and interchangeably, refer to an aryl group covalently bonded to an alkyl group, as defined herein. A "phenylalkyl" is a species of an aralkyl group, and refers to a phenyl ring covalently bonded to an alkyl group as defined herein. Examples, without limitation, of phenylalkyl groups include, without limitation, benzyl, 2-phenylethyl, 1-phenylpropyl, 4-phenylhexyl, 3-phenylamyl and 3-phenyl-2- methylpropyl. Presently preferred phenylalkyl groups are those wherein the phenyl group is covalently bonded to one of the presently preferred alkyl groups. A phenyl alkyl group of this invention may be unsubstituted or substituted. Examples of substituted phenylalkyl groups include, without limitation, 2-phenyl-l-chloroethyl, 2- (4-methoxyphenyl)ethyl, 4-(2,6-dihydroxy phenyl)hexyl, 2-(5-cyano-3- methoxyphenyl)pentyl, 3-(2,6-dimethylphenyl)propyl, 4-chloro-3-aminobenzyl, 6-(4- methoxyphenyl)-3-carboxy(n-hexyl), 5-(4-aminomethylphenyl)-3-
(aminomethyl)pentyl and 5-phenyl-3-oxo-pent-l-yl.
[0038] As used herein, "heteroarylalkyl" or "heteroaralkyl," which are used synonymously and interchangeably, and "heteroalicyclylalkyl" refer to a heteroaryl or a heteroalicyclyl group, respectively, covalently bonded to an alkyl group, as defined herein. Examples of such groups include, without limitation, 2- pyridylethyl, 3-pyridylpropyl, 4-furylhexyl, 3-piperazylamyl and 3-morpholinylbutyl. Presently preferred heteroarylalkyl and heteroalicyclylalkyl groups are those in which a presently preferred heteroaryl or heteroalicyclyl group is covalently bonded to a presently preferred alkyl group as disclosed herein.
[0039] As used herein, "phenyl" refers to a 6-member aryl group. A phenyl group may be unsubstituted or substituted. When substituted the substituent(s) is/are one or more, preferably one or two, group(s) independently selected from the group consisting of halogen, hydroxy, protected hydroxy, cyano, nitro, alkyl, alkoxy, acyl, acyloxy, carboxy, protected carboxy, carboxymethyl, protected carboxymethyl, hydroxymethyl, protected hydroxymethyl, -NRaRb wherein Ra and Rb are as defined above but in addition Ra may be an amino protecting group as defined herein, carboxamide, protected carboxamide, N-alkylcarboxamide, protected N- alkylcarboxamide, N,N-dialkylcarboxamide, trifluoromethyl, N-alkylsulfonylamino, N-(phenylsulfonyl)amino and phenyl (resulting in the formation of a biphenyl group).
[0040] Examples of substituted phenyl groups include, without limitation, 2, 3 or 4-chlorophenyl, 2,6-dichlorophenyl, 2,5-dichlorophenyl, 3,4-dichlorophenyl, 2, 3 or 4-bromophenyl, 3,4-dibromophenyl, 3-chloro-4-fluorophenyl, 2, 3 and A- fluorophenyl, 2, 3 or 4-hydroxyphenyl, 2,4-dihydroxyphenyl, the protected-hydroxy derivatives thereof, 2, 3 or 4-nitrophenyl; 2, 3 or 4-cyanophenyl; 2, 3 or A- methylphenyl, 2,4-dimethylphenyl, 2, 3 or 4-(iso-propyl)phenyl, 2, 3 or A- ethylphenyl, 2, 3 or 4-(n-propyl)phenyl, 2,6-dimethoxyphenyl, 2, 3 or A- methoxyphenyl, 2, 3 or 4-ethoxyphenyl, 2, 3 or 4-(isopropoxy)phenyl, 2, 3 or 4-(t- butoxy)phenyl, 3-ethoxy-4-methoxyphenyl; 2, 3 or 4-trifluoromethylphenyl; 2, 3 or A- carboxyphenyl or 2,4-di(protected carboxy)phenyl; 2, 3, or 4-(protected hydroxymethyl)phenyl or 3,4-di(hydroxymethyl)phenyl; 2, 3 or A- (aminomethyl)phenyl or 2,4-(protected aminomethyl)phenyl; and 2, 3 or 4-(N- (methylsulfonylamino))phenyl.
[0041] As used herein, "phenylalkoxy" refers to a "phenylalkyl-O-" group with "phenyl" and "alkyl" as defined herein. A phenylalkoxy group of this invention may be substituted or unsubstituted on the phenyl ring, in the alkyl group or both. Examples of phenylalkoxy groups include, without limitation, 2-(4- hydroxyphenyl)ethoxy, 4-(4-methoxyphenyl)butoxy, (2R)-3-phenyl-2-amino- propoxy, (2S)-3-phenyl-2-amino-propoxy, 2-indanoxy, 6-phenyl-l-hexanoxy, cinnamyloxy, 2-phenyl-l-propoxy and 2,2-dimethyl-3 -phenyl- 1-propoxy.
[0042] As used herein, "halo" and "halogen" refer to the fluoro, chloro, bromo or iodo atoms. Presently preferred halogens are chloro and fluoro.
[0043] As used herein, "amino protecting group" refers to a group commonly employed to keep (i.e., to "block" or "protect") an amino group from reacting with a reagent while it reacts with an intended target functional group of a molecule.
[0044] As used herein, a "protected carboxamide" refers to a carboxamide in which the nitrogen is substituted with an amino protecting group.
[0045] Examples of amino protecting groups include, without limitation, formyl ("For"), trityl, phthalimido, trichloroacetyl, chloroacetyl, bromoacetyl, iodoacetyl groups, t-butoxycarbonyl ("Boc"), 2-(4-biphenylyl)propyl-2-oxycarbonyl ("Bpoc"), 2-phenylpropyl-2-oxycarbonyl ("Poc"), 2-(4-xenyl)isopropoxycarbonyl, 1 , 1 -diphenylethyl- 1 -oxycarbonyl, 1 , 1 -diphenylpropyl- 1 -oxycarbonyl, 2-(3 ,5 - dimethoxyphenyl)propyl-2-oxycarbonyl ("Ddz"), 2-(p-toluyl)propyl-2-oxycarbonyl,
cyclopentanyloxycarbonyl, 1 -methylcyclopentanyloxycarbonyl, cyclohexanyloxy- carbonyl, l-methylcyclohexanyloxycarbonyl, 2-methylcyclohexanyloxycarbonyl, 2- (4-toluylsulfonyl)-ethoxycarbonyl, 2-(methylsulfonyl)ethoxycarbonyl, 2-
(triphenylphosphino)-ethoxycarbonyl, 9-fluorenylmethoxycarbonyl ("Fmoc"), 2- (trimethylsilyl)ethoxycarbonyl, allyloxycarbonyl, 1 -(trimethylsilylmethyl)prop- 1 - enyloxycarbonyl, 5-benzisoxalylmethoxycarbonyl, 4-acetoxybenzyl-oxycarbonyl, 2,2,2-trichloroethoxycarbonyl, 2-ethynyl-2-propoxycarbonyl, cyclopropyl- methoxycarbonyl, isobornyloxycarbonyl, 1 -piperidyloxycarbonyl, benzyloxycarbonyl ("Cbz"), 4-phenylbenzyloxycarbonyl, 2-methylbenzyloxy-carbonyl, -2,4,5,- tetramethylbenzyloxycarbonyl ("Tmz"), 4-methoxybenzyloxy- carbonyl, 4- fluorobenzyloxycarbonyl, 4-chlorobenzyloxycarbonyl, 3-chlorobenzyloxycarbonyl, 2- chlorobenzyloxycarbonyl, 2,4-dichlorobenzyl-oxycarbonyl, 4- bromobenzyloxycarbonyl, 3-bromobenzyloxycarbonyl, 4-nitrobenzyloxy -carbonyl, 4- cyanobenzyloxycarbonyl, 4-(decyloxy) benzyloxycarbonyl, benzoylmethylsulfonyl, dithiasuccinoyl ("Dts"),2-(nitro)phenylsulfenyl ("Nps"), and diphenyl-phosphine oxide. The species of amino-protecting group employed is not critical so long as the derivatized amino group is stable to the conditions of the subsequent reaction(s) and can be removed at the appropriate point without disrupting the remainder of the molecule. Presently preferred amino-protecting groups are Boc, Cbz and Fmoc. Descriptions of these and other amino-protecting groups may be found in T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Synthesis," 2nd ed., John Wiley and Sons, New York, N.Y., 1991, Chapter 7, M. Bodanzsky, "Principles of Peptide Synthesis," 1st and 2nd revised ed., Springer-Verlag, New York, N.Y., 1984 and 1993, and Stewart and Young, "Solid Phase Peptide Synthesis," 2nd ed., Pierce Chemical Co., Rockford, III, 1984.
[0046] As used herein, the term "carboxy protecting group" refers to a labile ester commonly used to block or protect a carboxylic acid while reactions are carried out on other functional groups on the compound. Examples of carboxy protecting groups include, without limitation, t-butyl, 4-nitrobenzyl, 4- methoxybenzyl, 3,4-dimethoxybenzyl, 2,4-dimethoxybenzyl, 2,4,6-trimethoxybenzyl, 2,4,6-trimethylbenzyl, pentamethylbenzyl, 3,4-methylenedioxybenzyl, benzhydryl, 4,4'-dimethoxytrityl, 4,4',4"-trimethoxytrityl, 2-phenylpropyl, trimethylsilyl, t- butyldimethylsilyl, phenacyl, 2,2,2-trichloroethyl, -(trimethylsilyl)ethyl, -(di(n- butyl)methylsilyl)ethyl, p-toluenesulfonylethyl, 4-nitrobenzylsulfonylethyl, allyl,
cinnamyl, and l-(trimethylsilylmethyl)-propenyl. The ester employed is not critical so long as it is stable to the conditions of subsequent reaction(s) and can be removed at the appropriate point without disrupting the remainder of the molecule. Further examples of carboxy -protecting groups are found in E. Haslam, "Protective Groups in Organic Chemistry," J. G. W. McOmie, Ed., Plenum Press, New York, N.Y., 1973, Chapter 5, and T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Synthesis," 2nd ed., John Wiley and Sons, New York, N. Y., 1991, Chapter 5.
[0047] As used herein, a "hydroxyl protecting group" refers to a readily cleavable group that replaces the hydrogen of the hydroxyl group, such as, without limitation, tetrahydropyranyl, 2-methoxypropyl, 1-ethoxy ethyl, methoxymethyl, 2- methoxyethoxymethyl, methylthiomethyl, t-butyl, t-amyl, trityl, 4-methoxytrityl, 4,4'- dimethoxytrityl, 4,4',4"-trimethoxytrityl, benzyl, allyl, trimethylsilyl, (t- butyl)dimethylsilyl, and 2,2,2-trichloroethoxycarbonyl. The species of hydroxyl protecting groups is not critical so long as the derivatized hydroxyl group is stable to the conditions of subsequent reaction(s) and can be removed at the appropriate point without disrupting the remainder of the molecule. Further examples of hydroxy- protecting groups are described by C. B. Reese and E. Haslam, "Protective Groups in Organic Chemistry," J. G. W. McOmie, Ed., Plenum Press, New York, N.Y., 1973, Chapters 3 and 4, respectively, and T. W. Greene and P. G. M. Wuts, "Protective Groups in Organic Synthesis," 2nd ed., John Wiley and Sons, New York, N.Y., 1991, Chapters 2 and 3.
[0048] As used herein, "alkylthio" refers to an "alkyl-S-" group, with alkyl as defined above. Examples of alkylthio group include, without limitation, methylthio, ethylthio, n-propylthio, isopropylthio, n-butylthio and t-butylthio.
[0049] As used herein, "alkylsulfinyl" refers to an "alkyl-SO-" group, with alkyl as defined above. Examples of alkylsulfinyl groups include, without limitation, methylsulfinyl, ethylsulfinyl, n-propylsulfinyl, isopropylsulfinyl, n-butylsulfinyl and sec-butylsulfinyl.
[0050] As used herein, "alkylsulfonyl" refers to an "alkyl-SO2-" group. Examples of alkylsulfonyl groups include, without limitation, methylsulfonyl, ethylsulfonyl, n-propylsulfonyl, isopropylsulfonyl, n-butylsulfonyl, and t- butylsulfonyl.
[0051] As used herein, "phenylthio," "phenylsulfinyl," and "phenylsulfonyl" refer to a "phenyl-S-," "phenyl-SO-," and "phenyl-SO2-" group, phenyl as defined herein.
[0052] As used herein, "alkylaminocarbonyl" refers to an "alkylNHC(=O)-" group, with alkyl as defined herein. Examples of alkylaminocarbonyl groups include, without limitation, methylaminocarbonyl, ethylaminocarbonyl, propylaminocarbonyl and butylaminocarbonyl. Examples of substituted alkylaminocarbonyl include, without limitation, methoxymethyl- aminocarbonyl, 2-chloroethylaminocarbonyl, 2-oxopropylaminocarbonyl and A- phenylbutylaminocarbonyl.
[0053] As used herein, "alkoxycarbonyl" refers to an "alkyl-OC(=O)-" group, with alkyl as defined above.
[0054] As used herein, "phenylaminocarbonyl" refers to a "phenyl-NHC(=O)-" group, with phenyl as defined above. Examples of substituted phenylaminocarbonyl groups include, without limitation, 2- chlorophenyl-aminocarbonyl, 3-chlorophenylaminocarbonyl, 2- nitorphenylaminocarbonyl, 4-biphenylaminocarbonyl, and A- methoxyphenylaminocarbonyl.
[0055] As used herein, "alkylaminothiocarbonyl" refers to an "alkyl- NHC(=O)-" group, with alkyl as defined above. Examples of alkylaminothiocarbonyl groups include, without limitation, methylaminothiocarbonyl, ethylaminothiocarbonyl, propylaminothiocarbonyl and butylaminothiocarbonyl.
[0056] Examples of alkyl-substituted alkylaminothiocarbonyl groups include, without limitation, methoxymethylaminothiocarbonyl, 2- chloroethylaminothiocarbonyl, 2-oxopropylaminothiocarbonyl and A- phenylbutylaminothiocarbonyl.
[0057] As used herein, "phenylaminothiocarbonyl" refers to a "phenyl- NHC(=S)-" group, with phenyl as defined above. Examples of phenylaminothiocarbonyl groups include, without limitation, 2- chlorophenylaminothiocarbonyl, 3-chlorophenyl-aminothiocarbonyl, 2- nitrophenylaminothiocarbonyl, 4-biphenylaminothiocarbonyl and A- methoxyphenylaminothiocarbonyl.
[0058] As used herein, "carbamoyl" refers to an "-NCO-" group.
[0059] As used herein, "hydroxyl" refers to an "-OH" group.
[0060] As used herein, "cyano" refers to a "-C≡N" group.
[0061] As used herein, "nitro" refers to an "-NO2" group.
[0062] An "O-carboxy" group refers to a "RC(=O)O-" group with R as defined above.
[0063] A "C-carboxy" group refers to a "-C(O)OR" group with R as defined above.
[0064] An "acetyl" group refers to a CH3C(O)- group.
[0065] A "trihalomethanesulfonyl" group refers to an "X3CSO2-" group wherein X is a halogen.
[0066] An "isocyanato" group refers to an "-NCO" group.
[0067] A "thiocyanato" group refers to a "-CNS" group.
[0068] An "isothiocyanato" group refers to an " -NCS" group.
[0069] A "sulfinyl" group refers to an "-S(O)-R" group with R as defined above.
[0070] An "S-sulfonamido" group refers to a "-SO2NR" group with R as defined above.
[0071] An "N-sulfonamido" group refers to a "RSO2NH-" group with R as defined above.
[0072] A "trihalomethanesulfonamido" group refers to an "X3CSO2NR-" group with X as halogen and R as defined above.
[0073] An "O-carbamyl" group refers to a "-OC(O)-NR" group with R as defined above.
[0074] An "N-carbamyl" group refers to an "ROC(O)NH-" group with R as defined above.
[0075] An "O-thiocarbamyl" group refers to a "-OC(=S)-NR" group with R as defined above.
[0076] "N-thiocarbamyl" group refers to an "ROC(=S)NH-" group with R as defined above.
[0077] A "C-amido" group refers to a "-C(O)-NRaRb group with Ra and Rb as defined above.
[0078] An "N-amido" group refers to a RC(O)NH- group with R as defined above.
[0079] The term "haloalkyl" refers to an alkyl group where one or more of the hydrogen atoms are replaced by halogen. Such groups include but are not limited
to , chloromethyl, fluoromethyl, difluoromethyl, trifluoromethyl and l-chloro-2- fluoromethyl, 2-fluoroisobutyl.
[0080] The term "perhaloalkyl" refers to an alkyl group in which all the hydrogen atoms are replaced by halogen atoms.
[0081] As used herein, an "ester" refers to a "-C(O)ORa" group with Ra as defined herein.
[0082] As used herein, an "amide" refers to a "-C(O)NRaRb" group with Ra and R as defined herein.
[0083] Where the numbers of substituents are not specified (e.g. haloalkyl) there may be one or more substituents presents. For example "haloalkyl" may include one or more of the same or differents halogens. As another example "C1-C3 alkoxy phenyl" may include one or more of the same of different alkoxygroups containing one, two or three atoms.
[0084] Any unsubstituted or monosubstituted amine group on a compound herein can be converted to an amide, any hydroxyl group can be converted to an ester and any carboxyl group can be converted to either an amide or ester using techniques well-known to those skilled in the art (see, for example, Greene and Wuts, Protective Groups in Organic Synthesis, 3rd Ed., John Wiley & Sons, New York, NY, 1999). Compounds containing any such converted hydroxyl, amino and/or carboxylic acid groups are within the scope of this invention.
[0085] As used herein, an "ether" refers to a "-C-O-C-" group wherein either or both carbons may independently be part of an alkyl, alkenyl, alkynyl, aryl, heteroaryl or heteroalicyclyl group.
[0086] As used herein, a "halogenated ether" refers to an ether in which the groups to either side of the oxygen are both alkyl substituted with halogen.
[0087] As used herein, "amino acid" refers to any one of the twenty naturally-occurring L-amino acids, to their non-natural D-enantiomers, to non- naturally occurring amino acids such as, without limitation, norleucine ("NIe"), norvaline ("Nva"), L- or D-naphthalanine, ornithine ("Orn"), homoarginine (homoArg) and to other amino acids well-known in the peptide art such as those described in M. Bodanzsky, "Principles of Peptide Synthesis," 1st and 2nd revised ed., Springer-Verlag, New York, N.Y., 1984 and 1993, and Stewart and Young, "Solid Phase Peptide Synthesis," 2nd ed., Pierce Chemical Co., Rockford, 111.
[0088] Throughout the present disclosure, when a particular compound comprises a chiral center, the scope of the present disclosure also includes compositions comprising the racemic mixture of the two enantiomers, as well as compositions comprising each enantiomer individually substantially free of the other enantiomer. Thus, for example, contemplated herein is a composition comprising the S enantiomer substantially free of the R enantiomer, or a composition comprising the R enantiomer substantially free of the S enantiomer. By "substantially free" it is meant that the composition comprises less than 10%, or less than 8%, or less than 5%, or less than 3%, or less than 1% of the minor enantiomer. If the particular compound comprises more than one chiral center, the scope of the present disclosure also includes compositions comprising a mixture of the various diastereomers, as well as compositions comprising each diastereomer substantially free of the other diastereomers. The recitation of a compound, without reference to any of its particular diastereomers, includes compositions comprising all four diastereomers, compositions comprising the racemic mixture of R,R and S,S isomers, compositions comprising the racemic mixture of R,S and S,R isomers, compositions comprising the R,R enantiomer substantially free of the other diastereomers, compositions comprising the S, S enantiomer substantially free of the other diastereomers, compositions comprising the R, S enantiomer substantially free of the other diastereomers, and compositions comprising the S,R enantiomer substantially free of the other diastereomers.
[0089] In some embodiments, Ri is selected from the group consisting of optionally substituted C1-C10 straight chained or branched alkyl, optionally substituted C3-C9 cycloalkyl, optionally substituted aryl, and optionally substituted heteroaryl. In other embodiments, Ri is selected from the group consisting of optionally substituted aryl and optionally substituted heteroaryl.
[0090] In some of these embodiments, the aryl and the heteroaryl of Ri are optionally substituted with one or more substituents selected from the group consisting of alkyl, hydroxy, protected hydroxy, alkoxy, aryloxy, acyl, ester, mercapto, alkylthio, arylthio, cyano, halogen, carbonyl, C-amido, N-amido, C-carboxy, protected C-carboxy, O-carboxy, nitro, silyl, sulfenyl, sulfinyl, sulfonyl, haloalkyl, perhaloalkyl, and amino. In other embodiments, the aryl and the heteroaryl of Ri are optionally substituted with one or more substituents selected from the group consisting of hydroxy, alkoxy, aryloxy, cyano, halogen, and amino. In yet other
embodiments, the aryl and the heteroaryl of Ri are optionally substituted with one or more substituents selected from the group consisting of fluoro, chloro, bromo, iodo, haloalkyl, and perhaloalkyl.
[0091] In some embodiments, the aryl is phenyl.
[0092] In some embodiments, Ri is selected from the group consisting of phenyl, 4-cholorophenyl, 3-cholorophenyl, and 2-cholorophenyl.
[0093] In some embodiments, R2 is selected from the group consisting of hydrogen, optionally substituted C1-C10 straight chained or branched alkyl, optionally substituted C2-C10 straight chained or branched alkenyl, optionally substituted C2-C10 straight chained or branched alkynyl, optionally substituted C3-C9 cycloalkyl, and optionally substituted C5-C7 cycloalkenyl. In other embodiments, R2 is selected from the group consisting of hydrogen, optionally substituted Ci-Ci0 straight chained or branched alkyl. In some embodiments, R2 is hydrogen.
[0094] In some embodiments, R3 is selected from the group consisting of optionally substituted Ci-Ci0 straight chained or branched alkyl, optionally substituted C2-C10 straight chained or branched alkenyl, optionally substituted C2-C10 straight chained or branched alkynyl, optionally substituted C3-C9 cycloalkyl, optionally substituted C5-C7 cycloalkenyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted aralkyl, and optionally substituted heteroaralkyl. In other embodiments, R3 is selected from the group consisting of optionally substituted Ci-Ci0 straight chained or branched alkyl, optionally substituted C3-C9 cycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted aralkyl, and optionally substituted heteroaralkyl. In further embodiments, R3 is selected from the group consisting of optionally substituted aralkyl, and optionally substituted heteroaralkyl.
[0095] In some embodiments, the aralkyl or heteroaralkyl of R3 is optionally substituted with one or more substituents selected from the group consisting of alkyl, hydroxy, protected hydroxy, alkoxy, aryloxy, acyl, ester, mercapto, alkylthio, arylthio, cyano, halogen, carbonyl, C-amido, N-amido, C-carboxy, protected C-carboxy, O-carboxy, nitro, silyl, sulfenyl, sulfinyl, sulfonyl, haloalkyl, perhaloalkyl, and amino. In other embodiments, the substituent is selected from the group consisting of alkyl, hydroxy, halogen, carbonyl, C-carboxy, protected C-carboxy, and amino.
[0096] In some embodiments, the aralkyl or heteroaralkyl of R3 is optionally substituted with one or more substituents selected from the group consisting of alkyl, halogen, C-carboxy, and protected C-carboxy.
[0097] In some embodiments, the alkyl is selected from the group consisting of methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec-butyl, iso-butyl, and tert-butyl.
[0098] In some embodiments, the C-carboxy is protected by an alkyl group, which in some embodiments, is selected from the group consisting of methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec-butyl, iso-butyl, and tert-butyl.
[0099] In some embodiments, R3 is selected from the group consisting of benzyl, 2-chlorobenzyl, 3-chlorobenzyl, 4-chlorobenzyl, 2-methylbenzyl, 3-methylbenzyl, 4-methylbenzyl, 2-cyanobenzyl, 3-cyanobenzyl, 4-cyanobenzyl, 2,3-dicholorbenzyl, 2,4-dicholorbenzyl, 2,6-dicholorbenzyl, 2-methylcarboxybenzyl (H3COC(=O)-C6H4-CH2-), 3-methylcarboxybenzyl, and 4-methylcarboxybenzyl. In other embodiments, R3 is selected from the group consisting of benzyl, 4- chlorobenzyl, 2-methylbenzyl, 3-methylbenzyl, 4-methylbenzyl, 4-cyanobenzyl, 2,4- dicholorbenzyl, 2,6-dicholorbenzyl, and 4-methylcarboxybenzyl.
[00100] In some embodiments, X is N+.
[00101] In some embodiments, Y is selected from the group consisting of halide, acetate, tartrate, phosphate, sulfonate, tetrafluoroborate. In some embodiments, the halide is selected from the group consisting of fluoride, chloride, bromide, and iodide. In some embodiments, Y is selected from the group consisting of chloride, bromide, and acetate.
[00102] In some embodiments, the compound of Formula I or II is not one of the following compounds:

[00103] In some embodiments, the compound of Formula I or II is not one of the following compounds:
 where Y is a counter ion.
where Y is a counter ion.
[00104] In another aspect, disclosed herein is a compound of Formula II

L is an alkyl spacer optionally substituted with one or more substituents,
Ri and R3 are each independently optionally substituted aryl or optionally substituted heteroaryl;
R2 is selected from the group consisting of hydrogen, -OH, -COOH, halogen, optionally substituted C1-C10 straight chained or branched alkyl, optionally substituted C2-C10 straight chained or branched alkenyl, optionally substituted C2-C10 straight chained or branched alkynyl, optionally substituted C3-C9 cycloalkyl, optionally substituted C5-C7 cycloalkenyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted aralkyl, and optionally substituted heteroaralkyl;
Y is a counter ion; and n is an integer from 1 to 5, such as n is 1, 2, 3, 4, or 5.
[00105] In some embodiments, Ri is optionally substituted phenyl. [00106] In some embodiments, R3 is optionally substituted phenyl. [00107] In some embodiments, L is -CH2-. [00108] In another aspect, disclosed herein is a compound selected from the group consisting of:



[00109] In another aspect, disclosed herein is a method of modulating the activity of a RARβ or RARγ receptor subtypes, comprising contacting a RARβ or RARγ receptor with at least one compound of Formula I or II. In some embodiments, the modulating is performed in vivo, whereas in other embodiments, the modulating is performed in vitro. In some embodiments, the compound of Formula I or II modulates the activity of an RARβ isoform. In other embodiments, the compound of Formula I or II modulates the activity of an RARγ isoform.
[00110] In the context of the present disclosure, a "modulator" is defined as a compound that is an agonist, a partial agonist, an inverse agonist or an antagonist of one or more RARβ and/or RARγ receptors. A modulator may increase the activity of the RARβ or RARγ receptor subtype, or may decrease the activity of the RARβ or RARγ receptor subtype. In the context of the present disclosure, an "agonist" is defined as a compound that increases the basal activity of a receptor (i.e. signal transduction mediated by the receptor). An "antagonist" is defined as a compound, which blocks the action of an agonist on a receptor. A "partial agonist" is defined as an agonist that displays limited, or less than complete, activity such that it fails to activate a receptor in vitro, functioning as an antagonist in vivo. An "inverse agonist" is defined as a compound that decreases the basal activity of a receptor.
[00111] In another aspect, disclosed herein is a method of treating cancer or for alleviating cancer symptoms, comprising administering to a subject in need thereof a therapeutically effective amount of at least one compound of Formula I or II. Preferably, the administered compound modulates the activity of a RARβ or RARγ receptor subtypes. In some embodiments, the compound of Formula I or II is administered in conjunction with at least one chemotherapeutic agent and/or radiation therapy. The term "in conjunction with" means given prior, concurrently, or subsequently to the other treatment. In some embodiments, the cancer is associated with malignant tumors. In certain embodiments, the cancer is selected from the group consisting of breast carcinoma and cancers of the skin, head, neck, lung, esophagus, mammary gland, liver, pancreas, cervix and digestive tract (e.g. oral cavity, esophagus, stomach, small intestine, including duodenum, jejunum, and ileum, and colon).
[00112] The term "subject" refers to an animal, preferably a mammal, and most preferably a human, who is the object of treatment, observation or experiment. The mammal may be selected from the group consisting of mice, rats, rabbits, guinea pigs, dogs, cats, sheep, goats, cows, primates, such as monkeys, chimpanzees, and apes, and humans.
[00113] The term "therapeutically effective amount" is used to indicate an amount of an active compound, or pharmaceutical agent, that elicits the biological or medicinal response indicated. This response may occur in a tissue, system, animal or human and includes alleviation of the symptoms of the disease being treated.
[00114] In another aspect, disclosed herein is a method of treating, or alleviating the symptoms of, hyperproliferative and inflammatory disorders, comprising administering to a subject in need thereof a therapeutically effective amount of at least one compound of Formula I or II, described above. Preferably, the administered compound modulates the activity of a RARβ or RARγ receptor subtypes. In some embodiments, the inflammatory disorder is a chronic inflammatory disorder. In certain embodiments, the inflammatory disorder is rheumatoid arthritis
[00115] In another aspect, disclosed herein is a method of treating or alleviating symptoms of a neurological disorder, comprising administering to a subject in need thereof a therapeutically effective amount of at least one compound of Formula I or II, described above. Preferably, the administered compound modulates
the activity of a RARβ or RARγ receptor subtypes. In some embodiments, the neurological disorder is selected from the group consisting of performance deficits in spatial learning and memory tasks and age-related memory deficit. In other embodiments, the neurological disorder is a disorder wherein cognition is altered. In further embodiments, the neurological disorder is schizophrenia.
[00116] In another aspect, disclosed herein is a method treating or alleviating symptoms of a neurodegenerative disorder, comprising administering to a subject in need thereof a therapeutically effective amount of at least one compound of Formula I or II, described above. Preferably, the administered compound modulates the activity of a RARβ or RARγ receptor subtypes. In some embodiments, the neurodegenerative disorder is Parkinson's disease or Alzheimer's disease.
[00117] In another aspect, disclosed herein is a method for the treatment of or for alleviating symptoms of a neurodegenerative disorder, comprising administering to a subject a therapeutically effective amount of at least one compound described above, wherein the a compound described above that has activity at RARβ and/or RARγ receptor subtypes. In one aspect, the neurodegenerative disorder relates to a method for the treatment of neurodegenerative disorders where nerve regeneration is necessary after, e.g. a spinal cord injury, after a stroke, after damage to the cardiac musles, after damage caused to myelin in multiple sclerosis and damage to islet cells in diabetes.
[00118] In another aspect, disclosed herein is a method treating or alleviating symptoms of a hyperproliferative or inflammatory disorder, comprising administering to a subject in need thereof a therapeutically effective amount of at least one compound of Formula I or II, described above. Preferably, the administered compound modulates the activity of a RARβ and/or RARγ receptor subtypes. In some embodiments, the inflammatory disorder is a chronic inflammatory disorder. In some embodiments, the inflammatory disorder is psoriasis or other associated disorders or rheumatoid arthritis.
[00119] In another aspect, disclosed herein is a method for treating or alleviating symptoms of an eye disorder or an eye condition, comprising administering to a subject in need thereof a therapeutically effective amount of at least one compound of Formula I or II, described above. Preferably, the administered compound modulates the activity of a RARβ and/or RARγ receptor subtypes.
[00120] In another aspect, disclosed herein is a method for treating or alleviating symptoms of depression, comprising administering to a subject in need thereof a therapeutically effective amount of at least one compound of Formula I or II, described above. Preferably, the administered compound modulates the activity of a RARβ and/or RARγ receptor subtypes.
[00121] In another aspect, disclosed herein is a method for treating or alleviating symptoms of pulmonary disorders/conditions, e.g. alveolar damage, emphysema and asthma, comprising administering to a subject in need thereof a therapeutically effective amount of at least one compound of Formula I or II, described above. Preferably, the administered compound modulates the activity of a RARβ and/or RARγ receptor subtypes.
[00122] In another aspect, disclosed herein is a method for treating or alleviating symptoms of dermatological disorders/conditions, comprising administering to a subject in need thereof a therapeutically effective amount of at least one compound of Formula I or II, described above. Preferably, the administered compound modulates the activity of a RARβ and/or RARγ receptor subtypes. In some embodiments, the dermatological disorder is selected from the group consisting of cystic acne, acne vulgaris and other related acneiform diseases, cutanous disordes of keratinization (e.g. the ichtyoses, Darier's disease, pityriasis rubra pilaris), psoriasis and its pustular and erythrodermic variants, miscellaneous cutaneous diseases (subcorneal pustular dermatosis, discoid lupus erythematosus, Reiter's syndrome (with and without AIDS), warts, lichen planus, cutaneous sarcoidosis).
[00123] In another aspect, disclosed herein is a method of identifying a compound that is an agonist, inverse agonist, or antagonist of one or more RARβ receptors and/or RARγ receptors, comprising contacting RARβ receptor and/or RARγ receptor with at least one test compound of Formula I or II and determining any change in activity of the one or more RARβ receptors and/or RARγ receptors so as to identify the test compound as an agonist, inverse agonist, or antagonist of one or more RARβ receptors and/or RARγ receptors.
[00124] In some embodiments, the RARβ receptor and/or RARγ receptor is expressed in a cell. In some of these embodiments, the cell is a cultured cell. In certain embodiments, the cultured cells overexpress the RARβ receptor and/or RARγ receptor. In other embodiments, the identified agonist, inverse agonist or antagonist
is selective for the RARβ receptor and/or RARγ receptor. In order to decide if a compound has activity at RARβ receptor subtype or has activity at the retinoic acid receptor subtype β isoform 2, the R-SAT test method, disclosed in Example 42, may be used. When using this test, a compound is considered to have activity if the pECso is > 5.0 and the %Eff is > 25.
[00125] The compounds of Formula I or II elevating sperm counts and increase sperm mobility and fertilization rates. Thus, in another aspect, disclosed herein is a method of treating infertility caused by sperm immobility, or a method of treating subfertility, comprising administering to a subject in need thereof a therapeutically effective amount of at least one compound of Formula I or II, described above. Preferably, the administered compound modulates the activity of a RARβ and/or RARγ receptor subtypes.
[00126] In one aspect, the present invention relates to a pharmaceutical composition comprising at least one compound of Formula I or II, and a physiologically acceptable component such as a carrier, a diluent, a salt or an excipient, or a combination thereof.
[00127] The term "pharmaceutical composition" refers to a mixture of a compound disclosed herein with other chemical components, such as diluents or carriers. The pharmaceutical composition facilitates administration of the compound to a subject. Multiple techniques of administering a compound exist in the art including, but not limited to, oral, injection, aerosol, parenteral, and topical administration. Pharmaceutical compositions can also be obtained by reacting compounds with inorganic or organic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, methanesulfonic acid, ethanesulfonic acid, p-toluenesulfonic acid, salicylic acid and the like.
[00128] The term "carrier" defines a chemical compound that facilitates the incorporation of a compound into cells or tissues. For example dimethyl sulfoxide (DMSO) is a commonly utilized carrier as it facilitates the uptake of many organic compounds into the cells or tissues of a subject.
[00129] The term "diluent" defines chemical compounds diluted in water that will dissolve the compound of interest as well as stabilize the biologically active form of the compound. Salts dissolved in buffered solutions are utilized as diluents in the art. One commonly used buffered solution is phosphate buffered saline because it
mimics the salt conditions of human blood. Since buffer salts can control the pH of a solution at low concentrations, a buffered diluent rarely modifies the biological activity of a compound.
[00130] The term "physiologically acceptable" defines a carrier or diluent that does not abrogate the biological activity and properties of the compound.
[00131] The pharmaceutical compositions described herein can be administered to a subject per se, or in pharmaceutical compositions where they are mixed with other active ingredients, as in combination therapy, or suitable carriers or excipient(s). Techniques for formulation and administration of the compounds of the instant application may be found in "Remington's Pharmaceutical Sciences," Mack Publishing Co., Easton, PA, 18th edition, 1990.
[00132] Suitable routes of administration may, for example, include oral, rectal, transmucosal, or intestinal administration; parenteral delivery, including intramuscular, subcutaneous, intravenous, intramedullary injections, as well as intrathecal, direct intraventricular, intraperitoneal, intranasal, or intraocular injections.
[00133] Alternatively, one may administer the compound in a local rather than systemic manner, for example, via injection of the compound directly into the area of pain, often in a depot or sustained release formulation. Furthermore, one may administer the drug in a targeted drug delivery system, for example, in a liposome coated with a tissue-specific antibody. The liposomes will be targeted to and taken up selectively by the organ.
[00134] The pharmaceutical compositions disclosed herein may be manufactured in a manner that is itself known, e.g., by means of conventional mixing, dissolving, granulating, dragee-making, levigating, emulsifying, encapsulating, entrapping or tabletting processes.
[00135] Pharmaceutical compositions for use in accordance with the present disclosure thus may be formulated in a conventional manner using one or more physiologically acceptable carriers comprising excipients and auxiliaries, which facilitate processing of the active compounds into preparations, which can be used pharmaceutically. Proper formulation is dependent upon the route of administration chosen. Any of the well-known techniques, carriers, and excipients may be used as suitable and as understood in the art; e.g., in Remington's Pharmaceutical Sciences, above.
[00136] For injection, the agents disclosed herein may be formulated in aqueous solutions, preferably in physiologically compatible buffers such as Hank's solution, Ringer's solution, or physiological saline buffer. For transmucosal administration, penetrants appropriate to the barrier to be permeated are used in the formulation. Such penetrants are generally known in the art.
[00137] For oral administration, the compounds can be formulated readily by combining the active compounds with pharmaceutically acceptable carriers well known in the art. Such carriers enable the compounds disclosed herein to be formulated as tablets, pills, dragees, capsules, liquids, gels, syrups, slurries, suspensions and the like, for oral ingestion by a patient to be treated. Pharmaceutical preparations for oral use can be obtained by mixing one or more solid excipient with pharmaceutical combination disclosed herein, optionally grinding the resulting mixture, and processing the mixture of granules, after adding suitable auxiliaries, if desired, to obtain tablets or dragee cores. Suitable excipients are, in particular, fillers such as sugars, including lactose, sucrose, mannitol, or sorbitol; cellulose preparations such as, for example, maize starch, wheat starch, rice starch, potato starch, gelatin, gum tragacanth, methyl cellulose, hydroxypropylmethyl-cellulose, sodium carboxymethylcellulose, and/or polyvinylpyrrolidone (PVP). If desired, disintegrating agents may be added, such as the cross-linked polyvinyl pyrrolidone, agar, or alginic acid or a salt thereof such as sodium alginate.
[00138] Dragee cores are provided with suitable coatings. For this purpose, concentrated sugar solutions may be used, which may optionally contain gum arabic, talc, polyvinyl pyrrolidone, carbopol gel, polyethylene glycol, and/or titanium dioxide, lacquer solutions, and suitable organic solvents or solvent mixtures. Dyestuffs or pigments may be added to the tablets or dragee coatings for identification or to characterize different combinations of active compound doses.
[00139] Pharmaceutical preparations, which can be used orally, include push-fit capsules made of gelatin, as well as soft, sealed capsules made of gelatin and a plasticizer, such as glycerol or sorbitol. The push-fit capsules can contain the active ingredients in admixture with filler such as lactose, binders such as starches, and/or lubricants such as talc or magnesium stearate and, optionally, stabilizers. In soft capsules, the active compounds may be dissolved or suspended in suitable liquids, such as fatty oils, liquid paraffin, or liquid polyethylene glycols. In addition,
stabilizers may be added. All formulations for oral administration should be in dosages suitable for such administration.
[00140] For buccal administration, the compositions may take the form of tablets or lozenges formulated in conventional manner.
[00141] For administration by inhalation, the compounds for use according to the present disclosure are conveniently delivered in the form of an aerosol spray presentation from pressurized packs or a nebulizer, with the use of a suitable propellant, e.g., dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas. In the case of a pressurized aerosol the dosage unit may be determined by providing a valve to deliver a metered amount. Capsules and cartridges of, e.g., gelatin for use in an inhaler or insufflator may be formulated containing a powder mix of the compound and a suitable powder base such as lactose or starch.
[00142] The compounds may be formulated for parenteral administration by injection, e.g., by bolus injection or continuous infusion. Formulations for injection may be presented in unit dosage form, e.g., in ampoules or in multi-dose containers, with an added preservative. The compositions may take such forms as suspensions, solutions or emulsions in oily or aqueous vehicles, and may contain formulatory agents such as suspending, stabilizing and/or dispersing agents.
[00143] Pharmaceutical formulations for parenteral administration include aqueous solutions of the active compounds in water-soluble form. Additionally, suspensions of the active compounds may be prepared as appropriate oily injection suspensions. Suitable lipophilic solvents or vehicles include fatty oils such as sesame oil, or synthetic fatty acid esters, such as ethyl oleate or triglycerides, or liposomes. Aqueous injection suspensions may contain substances, which increase the viscosity of the suspension, such as sodium carboxymethyl cellulose, sorbitol, or dextran. Optionally, the suspension may also contain suitable stabilizers or agents, which increase the solubility of the compounds to allow for the preparation of highly, concentrated solutions.
[00144] Alternatively, the active ingredient may be in powder form for constitution with a suitable vehicle, e.g., sterile pyrogen-free water, before use.
[00145] The compounds may also be formulated in rectal compositions such as suppositories or retention enemas, e.g., containing conventional suppository bases such as cocoa butter or other glycerides.
[00146] In addition to the formulations described previously, the compounds may also be formulated as a depot preparation. Such long acting formulations may be administered by implantation (for example subcutaneously or intramuscularly) or by intramuscular injection. Thus, for example, the compounds may be formulated with suitable polymeric or hydrophobic materials (for example as an emulsion in an acceptable oil) or ion exchange resins, or as sparingly soluble derivatives, for example, as a sparingly soluble salt.
[00147] A pharmaceutical carrier for the hydrophobic compounds disclosed herein is a cosolvent system comprising benzyl alcohol, a nonpolar surfactant, a water-miscible organic polymer, and an aqueous phase. A common cosolvent system used is the VPD co-solvent system, which is a solution of 3% w/v benzyl alcohol, 8% w/v of the nonpolar surfactant Polysorbate 80™, and 65% w/v polyethylene glycol 300, made up to volume in abs ethanol. Naturally, the proportions of a co-solvent system may be varied considerably without destroying its solubility and toxicity characteristics. Furthermore, the identity of the co-solvent components may be varied: for example, other low-toxicity nonpolar surfactants may be used instead of POLYSORBATE 80™; the fraction size of polyethylene glycol may be varied; other biocompatible polymers may replace polyethylene glycol, e.g., polyvinyl pyrrolidone; and other sugars or polysaccharides may be used.
[00148] Alternatively, other delivery systems for hydrophobic pharmaceutical compounds may be employed. Liposomes and emulsions are well known examples of delivery vehicles or carriers for hydrophobic drugs. Certain organic solvents such as dimethylsulfoxide also may be employed, although usually at the cost of greater toxicity. Additionally, the compounds may be delivered using a sustained-release system, such as semipermeable matrices of solid hydrophobic polymers containing the therapeutic agent. Various sustained-release materials have been established and are well known by those skilled in the art. Sustained-release capsules may, depending on their chemical nature, release the compounds for a few weeks up to over 100 days. Depending on the chemical nature and the biological stability of the therapeutic reagent, additional strategies for stabilization may be employed.
[00149] Many of the compounds used in the pharmaceutical combinations disclosed herein may be provided as salts with pharmaceutically compatible
counterions. Pharmaceutically compatible salts may be formed with many acids, including but not limited to hydrochloric, sulfuric, acetic, lactic, tartaric, malic, succinic, etc. Salts tend to be more soluble in aqueous or other protonic solvents than are the corresponding free acids or base forms.
[00150] Pharmaceutical compositions suitable for use in the methods disclosed herein include compositions where the active ingredients are contained in an amount effective to achieve its intended purpose. More specifically, a therapeutically effective amount means an amount of compound effective to prevent, alleviate or ameliorate symptoms of disease or prolong the survival of the subject being treated. Determination of a therapeutically effective amount is well within the capability of those skilled in the art, especially in light of the detailed disclosure provided herein.
[00151] The exact formulation, route of administration and dosage for the pharmaceutical compositions disclosed herein can be chosen by the individual physician in view of the patient's condition. (See e.g., Fingl et al. 1975, in "The Pharmacological Basis of Therapeutics", Ch. 1 p. 1). Typically, the dose about the composition administered to the patient can be from about 0.5 to 1000 mg/kg of the patient's body weight, or 1 to 500 mg/kg, or 10 to 500 mg/kg, or 50 to 100 mg/kg of the patient's body weight. The dosage may be a single one or a series of two or more given in the course of one or more days, as is needed by the patient. Note that for almost all of the specific compounds mentioned in the present disclosure, human dosages for treatment of at least some condition have been established. Thus, in most instances, the methods disclosed herein will use those same dosages, or dosages that are between about 0.1% and 500%, or between about 25% and 250%, or between 50% and 100% of the established human dosage. Where no human dosage is established, as will be the case for newly discovered pharmaceutical compounds, a suitable human dosage can be inferred from ED50 or ID50 values, or other appropriate values derived from in vitro or in vivo studies, as qualified by toxicity studies and efficacy studies in animals.
[00152] Although the exact dosage will be determined on a drug-by-drug basis, in most cases, some generalizations regarding the dosage can be made. The daily dosage regimen for an adult human patient may be, for example, an oral dose of between 0.1 mg and 500 mg of each ingredient, preferably between 1 mg and 250 mg, e.g. 5 to 200 mg or an intravenous, subcutaneous, or intramuscular dose of each ingredient between 0.01 mg and 100 mg, preferably between 0.1 mg and 60 mg, e.g. 1
to 40 mg of each ingredient of the pharmaceutical compositions disclosed herein or a pharmaceutically acceptable salt thereof calculated as the free base, the composition being administered 1 to 4 times per day. Alternatively the compositions disclosed herein may be administered by continuous intravenous infusion, preferably at a dose of each ingredient up to 400 mg per day. Thus, the total daily dosage by oral administration of each ingredient will typically be in the range 1 to 2000 mg and the total daily dosage by parenteral administration will typically be in the range 0.1 to 400 mg. Suitably the compounds will be administered for a period of continuous therapy, for example for a week or more, or for months or years.
[00153] Dosage amount and interval may be adjusted individually to provide plasma levels of the active moiety, which are sufficient to maintain the modulating effects, or minimal effective concentration (MEC). The MEC will vary for each compound but can be estimated from in vitro data. Dosages necessary to achieve the MEC will depend on individual characteristics and route of administration. However, HPLC assays or bioassays can be used to determine plasma concentrations.
[00154] Dosage intervals can also be determined using MEC value. Compositions should be administered using a regimen, which maintains plasma levels above the MEC for 10-90% of the time, preferably between 30-90% and most preferably between 50-90%.
[00155] In cases of local administration or selective uptake, the effective local concentration of the drug may not be related to plasma concentration.
[00156] The amount of composition administered will, of course, be dependent on the subject being treated, on the subject's weight, the severity of the affliction, the manner of administration and the judgment of the prescribing physician.
[00157] The compositions may, if desired, be presented in a pack or dispenser device, which may contain one or more unit dosage forms containing the active ingredient. The pack may for example comprise metal or plastic foil, such as a blister pack. The pack or dispenser device may be accompanied by instructions for administration. The pack or dispenser may also be accompanied with a notice associated with the container in form prescribed by a governmental agency regulating the manufacture, use, or sale of pharmaceuticals, which notice is reflective of approval by the agency of the form of the drug for human or veterinary administration. Such notice, for example, may be the labeling approved by the U.S.
Food and Drug Administration for prescription drugs, or the approved product insert. Compositions comprising a compound disclosed herein formulated in a compatible pharmaceutical carrier may also be prepared, placed in an appropriate container, and labeled for treatment of an indicated condition.
SYNTHESIS OF THE COMPOUNDS
[00158] The schemes, set forth below, provide examples of reaction schemes for the synthesis of the compounds of Formula I or II disclosed herein. For example compounds of Formula I or II may be synthesized according to the method depicted in Scheme 1.

Scheme 1
[00159] The ring closing reaction between an α-aminoketone and an imino ether followed by an N-alkylation can provide compounds of Formula I or II. The ring closing reaction is preferably carried out in a microwave, preferably at a temperature of about 130 0C and for about 15min. Ethanol is preferably used as solvent. The N- alkylation is preferably carried out in a microwave, at a temperature about 160 0C and for about 15min. In one embodiment, the reaction is carried out with acetonitrile as solvent. The final product is isolated by conventional means, preferably purified by re-crystallization. Rl, R2 and R3 have the definitions as described herein. Y is defined as a leaving group e.g. a halide. X is defined as a counterion e.g. a halide, acetate or phosphate, n is an integer from 1-5. X can in some cases be identical with Y.
[00160] The following strategy was used to synthesize two of the amino ketones used in further synthesis (Scheme 2).
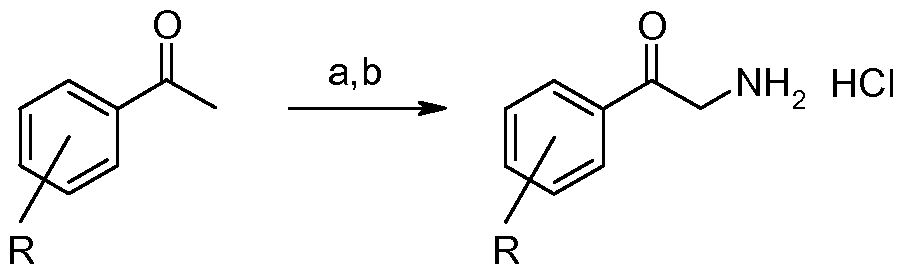
3 R=p-CI
4 R=m-CI
Scheme 2
[00161] Reagents and conditions: a) Br2, CHCl3, r.t. 15-30 min; b) (CH2)6N4, CHCl3, 50 0C, 1,3-1,5 h then cone HCl, 95% EtOH, 50 0C, 3 h.
[00162] Imino ethers were synthesized from the corresponding lactams (Scheme 3).

Scheme 3
[00163] Reagents and conditions: a) (CH3O)2SO2, 60 0C, 23 h. [00164] Two different methods were used to synthesize cyclic imidazoles. The first method is shown in scheme 4.

Scheme 4
[00165] Reagents and conditions: a) abs EtOH, 2 days, r.t.; b) cone. HCl, reflux, 2 h.
[00166] In the second method, the cyclic imidazoles are formed in one step using microwave irradiation (Scheme 5).

R=P-CI, m-CI, H
Scheme 5
[00167] Reagents and conditions: a) abs. EtOH, MW: 130 0C, 15 min.
[00168] The synthesized cyclic imidazoles were reacted with different substituted benzyl halides, yielding three libraries of potential RARγ agonists. In the first library, 7 were reacted with different benzyl chlorides (Scheme 6).

R=P-CI, 0-CH3, m-CH3, P-CH3, p-CN 0,0-Cl, o, m-CI, o,p-CI
Scheme 6
[00169] Reagents and conditions: dry CH3CN, Et3N, MW, 160 0C, 15 min. [00170] In the second library, 6-8 member cyclic imidazoles were reacted with 2,4-dichlorobenzyl chloride (Scheme 7).

[00171] Reagents and conditions: dry CH3CN, Et3N, MW, 160 0C, 15 min. [00172] In the third library, 6-8 member cyclic imidazoles were reacted with methyl-4-(chloromethyl) benzoate (Scheme 8).

Scheme 8
[00173] Reagents and conditions: dry CH3CN, Et3N, MW, 160 0C, 15 min.
[00174] It will be understood by those of skill in the art that numerous and various modifications can be made without departing from the spirit of the present disclosure. Therefore, it should be clearly understood that the forms disclosed herein are illustrative only and are not intended to limit the scope of the present disclosure.
EXAMPLES
[00175] The following examples are provided as an illustration of the present invention, but should in no way be considered as limiting the scope of invention
General information
[00176] Chemicals were used as purchased unless otherwise stated. Ethanol was dried over 4 A molecular sieves. Acetonitrile was obtained from Fluka and was dried over 4 A molecular sieves. Column chromatographic separations were performed by using SiC^ 60 A (35-70 μm). TLC analysis was performed on precoated
Merck Silica gel 60 F254 and the spots were visualized by staining with a solution of KMnO4 (3 g), K2CO3 (20 g), 5% aq. NaOH (5 mL) and 300 mL H2O.
[00177] NMR spectra were recorded on a Varian NMR 400 using CDCl3, CD3OD or D2O as solvents, which appear in NMR spectra at following chemicals shifts: 1H, 400 MHz; CDCl3 δ= 7.26 ppm, CD3OD δ= 3.31 ppm, D2O δ= 4.79 ppm and 13C, 100 MHz; CDCl3 δ: 77.16 ppm, CD3OD δ= 49.00, if nothing else is stated.
[00178] Automated extractions were made on a Myriad Allex. The microwave-assisted reactions were carried out using a SmithCreator™ single mode cavity; producing continuous irradiation at 2450 MHz.
[00179] HPLC/MS analyses were performed on a Waters/Micromass ZQ2000 LC/MS instrument with a Xterra® MS Ci8 3.5μm, 30x4.6 mm ID column with a guard column cartridge system and a 996 Photodiode Array Detector; eluent: A: 10 mM aq. NH4OAc; B: 10 mM aq. NH4OAc CH3CN-H2O (95:5); 5 min. gradient starting at 30% B (initial hold for 0.5 min.), over 5 min. to 90% B, hold for 0.5 min., over 0.5 min. to 30% B, hold for 2.5 min. The flow rate was 1 mL/min. Purifications by Preparative HPLC/MS were performed on a Waters/Micromass LC/ZMD instrument with a Xterra® Prep MS C18 5 μm, 19x100 mm ID column and a 996 Photodiode Array Detector; eluent: A: 10 mM aq. NH4OAc; B: 10 mM aq. NH4OAc CH3CN-H2O (95:5); 8.5 min. gradient starting at 30% B (initial hold for 2.5 min.), over 8.5 min. to 100% B, over 0.5 min. to 30% B, hold for 0.5 min. The flow rate was 17 L/min.
[00180] As used herein, the abbreviations for any protective groups, amino acids and other compounds are, unless indicated otherwise in accord with their common usage, recognized abbreviations or the IUPAC-IUB Commision on Biochemical Nomenclature (Biochem., 1972, 11, 942-944).
[00181] As employed herein, the following terms have their accepted meaning in the chemical literature: Abs Absolute
AcOH acetic acid anhydr anhydrous aq aqueous
CDCl3 deuterated chloroform CDI l,l '-carbonyldiimidazole
DCM dichloromethane
DIPA diisopropylamine
DIPEA diisoproylethylamine
DMAP 4-dimethylaminopyridine
DMDO dimethyldioxirane
DMF N,N-dimethylformamide
DMSO dimethyl sulfoxide
EDCLHCl 1 -ethyl-3 -(3 -dimethylaminopropyl)carbodiimide hydrochloride
Et2O diethyl ether
EtOAc ethyl acetate
EtOH ethanol
HOBt 1 -hydroxybenzotπazole
LG leaving group
MeOH methanol
MW microwave reactor
NMP N-methylpyrrolidine
NH4OAc ammonium acetate o.n. over night
Pd/C palladium on actived carbon r.t. room temperature
TBAI tetrabutylammonium iodide
TEA triethylamine
THF tetrahydrofuran
Example 1: General procedure for the synthesis of 6, 7 and 8 member cyclic imidazoles (Procedure A)
[00182] The α-aminoacetophenone hydrochloride (1.30 mmol) and the imino ether (1.24 mmol) in 2 mL dry EtOH were microwave irradiated at 130 0C for 15 min. 3 equivalents of MP-carbonate was added to the vial and the vial was shaken on a shake board for 2h. The product was purified on a SCX column (MeOH as eluent) and evaporated to yield the cyclic imidazole.
Example 2: General procedure for the synthesis of 7 member quaternary imidazoles (Procedure B)
[00183] The benzylhalide (0.30 mmol), triethylamine (33 μL, 0.47 mmol) and 3-phenyl-6,7,8,9-tetrahydro-5H-imidazo[l,2-a]azepine (0.24 mmol) in 1 mL dry acetonitrile were microwave irradiated at 160 0C for 15 min. The reaction mixture was evaporated to dryness. The residue was dissolved in H2O and loaded on a hydromatrix. The hydromatrix was washed with Et2O and the filtrate was extracted with CH2Cl2. The CH2Cl2 phase was evaporated to dryness and dissolved in H2O. The water phase was washed 3 times with Et2O (limited amounts of brine was added to the water phase), and extracted 3 times with CH2Cl2. The organic phase was dried over Na2SO4, filtrated and evaporated to dryness.
Example 3: General procedure for the synthesis of alkylated imidazoles (Procedure
Q
[00184] The benzylchloride (0.30 mmol), triethylamine (66 μL, 0.47 mmol) and the imidazole (0.24 mmol) in 1 mL dry acetonitrile were microwave irradiated at 160 0C for 15 min. The reaction mixture was evaporated to dryness and the residue was dissolved in H2O and an automated workup was performed where the aq. phase was washed 3 times with Et2O. The aq. phase was then extracted 3 times with 2- butanone and 3 times with CH2Cl2. The 2-butanone and CH2Cl2 phases were combined and dried over Na2SO4, filtrated and evaporated to dryness.
Example 4: 2-Amino-l-(4-chloro-phenyl)-ethanone; hydrochloride
[00185] Bromine (1.03 mL, 20 mmol) in 7.5 mL CHCl3 was added portion wise to a solution of 4'-chloroacetophenone (2.60 mL, 20 mmol) in 15 mL CHCl3 with stirring at r.t. After 15 min at r.t, the mixture was diluted with sat. aq. Na2S2O3 and the mixture were extracted with CH2Cl2. The organic phase was washed with H2O, dried over Na2SO4, filtrated and evaporated to give yellow crystals. This product was added to a solution of hexametylenetetramine (2.12 g, 15 mmol) in 70 mL CHCl3 and was stirred at 50 0C for 1 h and 15 min. The mixture was cooled to r.t. and the white precipitate was collected by filtration and washed with CHCI3. The resulting quaternary salt was suspended in a mixture of 95% EtOH (20 mL) and cone. HCl (7.5 mL) and heated at 50 0C for 3 h. The mixture was cooled to 0 0C and the resulting white solid was collected by filtration. The solid was recrystallized from H2O and dried in vacuo to give the title compound as pale yellow crystals (1.27 g, 31%). 1H
NMR (D2O) δ: 8.05-7.99 (m, 2H); 7.68-7.62 (m, 2H); 4,75 (s, 2H). 13C NMR (D2O) δ: 193.1; 141.2; 131.8; 12.0; 129.6; 45.4. Purity by LC/MS (UV/MS): 100/100.
Example 5: 2-Amino-l-(3-chloro-phenyl)-ethanone; hydrochloride
[00186] Bromine (0.63 mL, 12 mmol) in 7.5 mL CHCI3 was added portion wise to a solution of 3'-chloroacetophenone (1.60 mL, 12 mmol) in 15 mL CHCI3 with stirring at r.t. After 30 min at r.t, the mixture was diluted with sat. aq. Na2S2Os and extracted with CH2Cl2. The organic phase was washed with H2O, dried over Na2SO4, filtrated and evaporated to give a yellow oil. To this oil was added hexamethylenetetramine (1.72 g, 12 mmol) and 50 mL of CHCI3. This solution was allowed to stir at 50 0C for 1.5 h. The reaction mixture was allowed to cool to r.t. and the white precipitate was collected by filtration and washed with CHCI3. The resulting quaternary salt was suspended in a mixture of 95% EtOH (25 mL) and cone. HCl (7 mL) and heated at 50 0C for 3 h. The mixture was cooled to 0 0C and the white solid was collected by filtration. The solid was recrystallized from H2O and dried in vacuo to give the title compound as pale yellow crystals (0.93 g, 37%). 1H NMR (D2O) δ: 8.12-8.07 (m, IH); 8.02-7.96 (m, IH); 7.86-7.80 (m, IH), 7.68-7.61 (m, IH); 4.77 (s, 2H). 13C NMR (D2O) δ: 192.5; 136.7; 136.3; 135.5; 131.9; 129.0; 127.8; 46.5. Purity by LC/MS (UV/MS): 99/99.
Example 6: (E)-8-Methoxy-2.3.4.5.6.7-hexahydro-azocine
[00187] Dimethyl sulfate (1.42 mL, 15 mmol) and 2-azacyclooctanone (1.91 g, 15 mmol) were added to a dried, argon flushed 2-necked round bottomed flask and the mixture was heated at 60 0C for 23 h. The mixture was then cooled to r.t. and added to a saturated aq. solution of Na2Cθ3 (100 mL). This solution was extracted with ether, dried over Na2SO4, filtrated and evaporated to give a brownish oil of the title compound (1.71 g, 81%). 1H NMR (CDCl3) δ: 3.62 (s, 3H); 3.44-3.40 (m, 2H); 2.34-2.30 (m, 2H); 1-71-1.65 (m, 2H); 1.64-1.58 (m, 2H); 1.50-1.44 (m, 2H); 1.42-1.35 (m, 2H).
Example 7: 3-Phenyl-2,5,6,7,8,9-hexahydro-3H-imidazori,2-a1azepin-3-ol
[00188] A solution of α-aminoacetophenone hydrochloride (3.43 g, 20 mmol) in abs EtOH (20 mL) was treated with 7-methoxy-3,4,5,6-tetrahydro-2H- azepine (2.90 mL, 20 mmol). The reaction mixture was shaken on a shake board at r.t.
for 2 days. 7.55 g of crude product was obtained. 1.20 g of the crude product was recrystallized two times from acetone/MeOH to give 0.098 g of white crystals and the second crop gave 0.27 g (0.37 g, 30% in total yield). 0.95 g of the crude product was washed with acetone to give 0.59 g of the title compound as white crystals (62%). 1H NMR (CD3OD) δ: 7.60-7.54 (m, 2H); 7.50-7.40 (m, 3H); 4.08-3.99 (m, 2H); 3.35 (s, 2H); 2.92-2.86 (m, 2H); 1.96-1.77 (m, 4H); 1.65-1.58 (m, 2H). 13C NMR (CD3OD) δ: 173.4; 141.4; 131.4; 130.8; 128.0; 97.7; 60.7; 44.6; 30.7; 29.2; 28.6; 24.3. Purity by LC/MS (UV/MS): 95/90.
Example 8: 3-(4-Chloro-phenyl)-5,6,7,8-tetrahydro-imidazori ,2-aipyridine
[00189] 2-Amino-l-(4-chloro-phenyl)-ethanone; hydrochloride (268 mg, 1.30 mmol) and o-methylvalerolactim (140 mg, 1.24 mmol) were subjected to general procedure A and gave the title compound (240 mg, 83%). 1H NMR (CD3OD) δ: 7.63-
7.51 (m, 5H); 4.12-4.06 (m, 2H); 3.16-3.11 (m, 2H); 2.14-2.04 (m, 4H). 13C NMR (CD3OD) δ: 146.7; 137.2; 134.3; 132.1; 130.4; 126.0; 117.2; 46.3; 22.8; 22.8; 18.9. Purity by LC/MS (UV/MS): 97/82.
Example 9: 3-(3-Chloro-phenyl)-5,6 J,8-tetrahydro-imidazori ,2-aipyridine
[00190] 2-Amino-l-(3-chloro-phenyl)-ethanone; hydrochloride (268 mg, 1.30 mmol) and o-methylvalerolactim (140 mg, 1.24 mmol) were subjected to general procedure A and gave the title compound (396 mg, 137%). 1H NMR (CD3OD) δ: 7.64-7.62 (m, IH); 7.59 (s, IH); 7.57-7.52 (m, 3H); 4.13-4.08 (m, 2H); 3.17-3.12 (m, 2H); 2.14-2.04 (m, 4H). Purity by LC/MS (UV/MS): 89/88.
Example 10: 3-Phenyl-5,6J,8-tetrahvdro-imidazori,2-a1pyridine
[00191] α-Aminoacetophenone hydrochloride (223 mg, 1.30 mmol) and o- methylvalerolactim (140 mg, 1.24 mmol) were subjected to general procedure A and gave the title compound (255 mg, 104%). 1H NMR (CD3OD) δ: 7.56-7.54 (m, 5H),
7.52 (s, IH); 4.12-4.38 (m, 2H); 3.16-3.11 (m, 2H); 2.12-2.06 (m, 4H). 13C NMR (CD3OD) δ: 146.4; 135.5; 131.1; 130.5; 130.2; 127.3; 116.8; 46.2; 22.8; 22.8; 19.0. Purity by LC/MS (UV/MS): 95/81.
Example 11 : 3-(4-Chloro-phenyl)-6,7,8,9-tetrahydro-5H-imidazori,2-a1azepine
[00192] 2-Amino-l-(4-chloro-phenyl)-ethanone; hydrochloride (268 mg, 1.30 mmol) and 7-methoxy-3,4,5,6-tetrahydro-2H-azepine (158 mg, 1.24 mmol) were subjected to general procedure A and gave the title compound (293 mg, 96%). 1H NMR (CD3OD) δ: 7.60-7.57 (m, 2H); 7.50-7.45 (m, 2H); 7.46 (s, IH); 4.20-4.16 (m, 2H); 3.25-3.19 (m, 2H); 2.04-1.98 (m, 2H); 1.90-1.83 (m, 4H). 13C NMR (CD3OD) δ: 151.3; 137.5; 135.7; 132.8; 130.5; 126.1; 116.7; 48.3; 30.7; 28.0; 27.3; 25.0. Purity by LC/MS (UV/MS): 94/77.
Example 12: 3-(3-Chloro-phenyl)-6J,8,9-tetrahydro-5H-imidazori,2-a1azepine
[00193] 2-Amino-l-(3-chloro-phenyl)-ethanone; hydrochloride (268 mg, 1.30 mmol) and 7-methoxy-3,4,5,6-tetrahydro-2H-azepine (158 mg, 1.24 mmol) were subjected to general procedure A and gave the title compound (426 mg, 139%). 1H NMR (CD3OD) δ: 7.60-7.55 (m, 3H); 7.50 (s, IH); 7.47-7.44 (m, IH); 4.22-4.18 (m, 2H); 3.30-3.24 (m, 2H); 2.05-1.97 (m, 2H); 1.88-1.81 (m, 4H). Purity by LC/MS (UV/MS): 80/57.
Example 13 : 3-Phenyl-6,7,8,9-tetrahydro-5H-imidazori ,2-aiazepine
[00194] α-Aminoacetophenone hydrochloride (223 mg, 1.30 mmol) and 7- methoxy-3,4,5,6-tetrahydro-2H-azepine (158 mg, 1.24 mmol) were subjected to general procedure A and gave the title compound (287 mg, 1090Zo)-1H NMR (CDCl3) δ: 7.54-7.49 (m, 3H); 7.33-7.29 (m, 2H); 7.13-7.11 (m, IH); 4.12-4.06 (m, 2H); 3.46- 3.39 (m, 2H); 2.02-1.94 (m, 2H); 1.88-1.78 (m, 4H). 13C NMR (CDCl3) δ: 149.5; 134.8; 130.5; 130.0; 129.5; 125.9; 115.3; 47.1; 30.1; 27.6; 26.3; 24.4. Purity by LC/MS (UV/MS): 93/88.
Example 14: 3 -(4-Chloro-phenyl)-5 ,6,7, 8 ,9 , 10-hexahydro-imidazo [ 1 ,2-ai azocine
[00195] 2-Amino-l-(4-chloro-phenyl)-ethanone; hydrochloride (268 mg, 1.30 mmol) and (E)-8-methoxy-2, 3,4,5, 6,7-hexahydro-azocine (175 mg, 1.24 mmol) were subjected to general procedure A. The obtained product was dissolved in 3 mL abs EtOH followed by the addition of 5 drops of cone. HCl. The reaction mixture was microwave irradiated at 100 0C for 5 min, and evaporated to dryness. The dried mixture was dissolved in 3 mL abs EtOH followed by the addition of 5 drops of cone. HCl. The reaction mixture was microwave irradiated at 100 0C for 5 min, evaporated to dryness, dissolved in 4 mL EtOH and 0.3 mL cone. HCl and then microwave
irradiated at 120 0C for 15 min. The product was evaporated to dryness and column chromatographed on silica (heptane/EtOAc 1: 1) which gave the title compound (102 mg, 32%). 1H NMR (CD3OD) δ: 7.45-7.41 (m, 2H); 7.38-7.34 (m, 2H); 6.91 (s, IH); 4.11-4.04 (m, 2H); 2.90-2.84 (m, 2H); 1.82-1.74 (m, 2H); 1.56-1.42 (m, 4H); 1.24- 1.16 (m, 2H). 13C NMR (CD3OD) δ: 152.7; 135.1; 132.9; 131.5; 130.3; 130.0; 42.8; 32.3; 27.2; 26.6; 24.7. Purity by LC/MS (UV/MS): 96/82.
Example 15: 3 -(3 -Chloro-phenyl)-5 ,6,7, 8 ,9 , 10-hexahydro-imidazo [ 1 ,2-ai azocine
[00196] 2-Amino-l-(3-chloro-phenyl)-ethanone; hydrochloride (268 mg, 1.30 mmol) and (E)-8-methoxy-2, 3,4,5, 6,7-hexahydro-azocine (175 mg, 1.24 mmol) were subjected to general procedure A. The obtained product was dissolved in 3 mL abs EtOH followed by the addition of 5 drops of cone. HCl. The reaction mixture was microwave irradiated at 100 0C for 5 min. The reaction mixture was evaporated to dryness, dissolved in 3 mL abs EtOH followed by the addition of 5 drops of cone. HCl. The reaction mixture was microwave irradiated at 100 0C for 5 min, evaporated to dryness, dissolved in 4 mL EtOH and 0.3 mL cone. HCl and then microwave irradiated at 120 0C for 15 min. The product was evaporated to dryness and column chromatographed on silica (heptane/EtOAc 1 : 1) which gave the title compound (59 mg, 18%). 1H NMR (CD3OD) δ: 7.45-7.37 (m, 3H); 7.34-7.30 (m, IH); 6.94 (s, IH); 4.14-4.07 (m, 2H); 2.91-2.85 (m, 2H); 1.82-1.74 (m, 2H); 1.58-1.46 (m, 4H); 1.24- 1.16 (m, 2H). 13C NMR (CD3OD) δ: 152.9; 135.6; 133.6; 132.7; 131.4; 129.8; 129.2; 128.4; 127.2; 32.3 (2C); 27.2; 26.6; 24.7. Purity by LC/MS (UV/MS): 96/78.
Example 16: 3-Phenyl-5,6,7,8,9,10-hexahvdro-imidazori,2-a1azocine
[00197] α-Aminoacetophenone hydrochloride (223 mg, 1.30 mmol) and (E)-8-methoxy-2,3,4,5,6,7-hexahydro-azocine (175 mg, 1.24 mmol) were subjected to general procedure A. The obtained product was dissolved in 3 mL abs EtOH followed by the addition of 5 drops of cone. HCl. The reaction mixture was microwave irradiated at 100 0C for 5 min. The reaction mixture was evaporated to dryness dissolved in 3 mL abs EtOH followed by the addition of 5 drops of cone. HCl. The reaction mixture was microwave irradiated at 100 0C for 5 min, evaporated to dryness, dissolved in 4 mL EtOH and 0.3 mL cone. HCl and then microwave irradiated at 120 0C for 15 min. The product was evaporated to dryness and column chromatographed on silica (heptane/EtOAc 1: 1) which gave the title compound (82
mg, 29%). 1H NMR (CD3OD) δ: 7.45-7.39 (m, 2H); 7.39-7.33 (m, 3H); 6.87 (s, IH); 4.09-4.04 (m, 2H); 2.89-2.83 (m, 2H); 1.82-1.74 (m, 2H); 1.54-1.44 (m, 4H); 1.23- 1.17 (m, 2H). 13C NMR (CD3OD) δ: 152.2; 134.2; 131.7; 130.1; 129.8; 129.2; 126.5; 42.8; 32.3; 27.2; 26.6; 24.7. Purity by LC/MS (UV/MS): 93/87.
Example 17: l-(4-Chloro-benzyl)-3-phenyl-6J,8,9-tetrahydro-5H-imidazori,2- alazepin-l-ium; chloride (compound 1)
[00198] 4-Chlorobenzyl chloride (47 mg, 0.30 mmol), triethylamine (33 μL, 0.24 mmol) and 3-phenyl-6,7,8,9-tetrahydro-5H-imidazo[l,2-a]azepine (50 mg, 0.24 mmol) were subjected to procedure B and gave the title compound (49 mg, 56%). 1H NMR (CDCl3) δ: 7.60 (s, IH); 7.46-7.41 (m, 5H); 7.35 (m, 2H); 7.33-7.27 (m, 2H), 5.76 (s, 2H); 4.21 (m, 2H); 3.46-3.40 (m, 2H); 1.95-1.87 (m, 2H); 1.78-1.72 (m, 2H); 1.70-1.62 (m, 2H). 13C NMR (CDCl3) δ: 149.7; 135.0; 134.3; 132.8; 130.4; 130.1; 130.0; 129.5; 129.3; 125.3; 119.3; 51.6; 47.6; 29.4; 27.1; 25.5; 23.7. Purity by LC/MS (UV/MS): 99/97.
Example 18: 1 -(2-Methyl-benzyl)-3-phenyl-6 J,8,9-tetrahydro-5H-imidazor 12- a"|azepin-l-ium; chloride (compound 2)
[00199] α-Bromo-o-xylene (56 mg, 0.30 mmol), triethylamine (33 μL. 0.24 mmol) and 3-phenyl-6,7,8,9-tetrahydro-5H-imidazo[l,2-a]azepine (50 mg, 0.24 mmol) were subjected to procedure B and gave the title compound (60 mg, 72%). 1H NMR (CDCl3) δ: 7.48-7.42 (m, 3H); 7.39-7.35 (m, 2H); 7.26-7.18 (m, 3H); 7.11-7.06 (m, IH); 7.04 (s, IH); 5.61 (s, 2H); 4.34-4.30 (m, 2H); 3.51-3.46 (m, 2H); 2.35 (s, 3H); 2.03-1.96 (m, 2H); 1.84-1.72 (m, 4H). 13C NMR (CDCl3) δ: 150.2; 136.7; 134.5; 131.5; 131.3; 130.5; 130.2; 129.4; 128.5; 127.0; 125.4; 118.6; 51.0; 47.8; 29.4; 27.2; 23.9; 19.6. Purity by LC/MS (UV/MS): 99/98
Example 19: l-(3-Methyl-benzyl)-3-phenyl-6J,8,9-tetrahydro-5H-imidazori,2- alazepin-l-ium; chloride (compound 3)
[00200] α-Bromo-m-xylene (56 mg, 0.30 mmol), triethylamine (33 μL, 0.24 mmol) and 3-phenyl-6,7,8,9-tetrahydro-5H-imidazo[l,2-a]azepine (50 mg, 0.24 mmol) were subjected to procedure B and gave the title compound (47 mg, 56%). 1H NMR (CDCl3) δ: 7.45-7.41 (m, 3H); 7.38 (s, IH); 7.36-7.33 (m, 2H); 7.26-7.19 (m, IH); 7.16-7.08 (m, 3H); 5.58 (s, 2H); 4.26-4.22 (m, 2H); 3.45-3.40 (m, 2H); 2.30 (s,
3H); 1.98-1.91 (m, 2H); 1.78-1.72 (m, 2H); 1.70-1.64 (m, 2H). 13C NMR (CDCl3) δ: 149.7; 139.3; 134.3; 133.6; 130.4; 130.1; 129.8; 129.3; 128.9; 125.3; 125.2; 119.1; 52.6; 47.6; 29.3; 27.1; 25.5; 23.6; 21.4. Purity by LC/MS (UV/MS): 99/93.
Example 20: l-(4-Methyl-benzyl)-3-phenyl-6J,8,9-tetrahydro-5H-imidazori,2- a]azepin-l-ium; chloride (compound 4)
[00201] α-Bromo-p-xylene (56mg, 0.30 mmol), triethylamine (33 μL, 0.24 mmol) and 3-phenyl-6,7,8,9-tetrahydro-5H-imidazo[l,2-a]azepine (50 mg, 0.24 mmol) were subjected to procedure B and gave the title compound (45 mg, 53%). 1H NMR (CDCl3) δ: 7.45-7.40 (m, 3H); 7.37-7.32 (m, 3H); 7.26-7.22 (m, 2H); 7.16-7.12 (m, 2H); 5.57 (s, 2H); 4.26-4.21 (m, 2H); 3.46-3.41 (m, 2H); 2.28 (s, 3H); 1.97-1.90 (m, 2H); 1.77-1.63 (m, 4H). 13C NMR (CDCl3) δ: 149.6; 139.0; 134.2; 130.7; 130.4; 130.1; 130.1; 129.3; 128.3; 125.4; 119.0; 52.4; 47.5; 29.3; 27.1; 25.5; 23.6; 21.2. Purity by LC/MS (UV/MS): 99/96.
Example 21 : l-(4-Cvano-benzyl)-3-phenyl-6 J,8,9-tetrahydro-5H-imidazori 2- a"|azepin-l-ium; chloride (compound 5)
[00202] 4-Cyanobenzyl chloride (46 mg, 0.30 mmol), triethylamine (33 μL, 0.24 mmol) and 3-phenyl-6,7,8,9-tetrahydro-5H-imidazo[l,2-a]azepine (50 mg, 0.24 mmol) ) in 1 mL dry acetonitrile were microwave irradiated at 160 0C for 15 min. The reaction mixture was evaporated to dryness. The dried residue was triturated with Et2O and evaporated to dryness. The residue was dissolved in H2CVMeC1H 95:5, washed 3 times with Et2O (limited amounts of brine was added to the water phase) and extracted 3 times with CH2Cl2. The organic phase was dried over Na2SO4, filtrated and evaporated to dryness which gave the title compound (49 mg, 57%). 1H NMR (CDCl3) δ: 7.78 (s, IH); 7.63-7.59 (m, 2H); 7.57-7.53 (m, 2H); 7.46-7.36 (m, 3H); 7.34-7.30 (m, 2H); 5.92 (s, 2H); 4.19-4.14 (m, 2H); 3.38-3.31 (m, 2H); 1.90-1.82 (m, 2H); 1.78-1.68 (m, 2H); 1.64-1.56(m, 2H). 13C NMR (CDCl3) δ: 149.8; 139.7; 134.5; 133.0; 130.5; 130.1; 129.3; 127.2; 125.2; 119.7; 118.3; 112.5; 51.5; 47.6; 29.4; 27.0; 25.4; 23.6. Purity by LC/MS (UV/MS): 98/94.
Example 22: l-(2,6-Dichloro-benzyl)-3-phenyl-6J,8,9-tetrahydro-5H-imidazori,2- a"|azepin-l-ium; chloride (compound 6)
[00203] 2,4-Dichlorobenzyle bromide (72 mg, 0.30 mmol), triethylamine (33 μL, 0.24 mmol) and 3-phenyl-6,7,8,9-tetrahydro-5H-imidazo[l,2-a]azepine (50 mg, 0.24 mmol) in 1 mL dry acetonitrile were microwave irradiated at 160 0C for 15 min. The reaction mixtures were evaporated to dryness. The dried residue was triturated with Et2θ and evaporated to dryness. The residue was dissolved in H2O/MeOH 95:5, washed with Et2θ (limited amounts of brine was added to the water phase) and extracted with CH2Cl2. The organic phase was dried over Na2SO4, filtrated and evaporated to dryness which gave the title compound (73 mg, 76%). 1H NMR (CDCl3) δ: 7.45-7.38 (m, 5H); 7.36-7.28 (m, 3H); 6.81 (s, IH); 5.72 (s, 2H); 4.35-4.31 (m, 2H); 3.72-3.66 (m, 2H); 2.06-1.98 (m, 2H); 1.89-1.82 (m, 2H); 1.81-1.74 (m, 2H). 13C NMR (CDCl3) δ: 150.4; 136.7; 135.0; 132.1; 130.6; 130.0; 129.3; 129.3; 128.9; 125.0; 117.0; 47.9; 47.8; 29.1; 27.0; 25.6; 23.9. Purity by LC/MS (UV/MS): 100/95.
Example 23: l-(2.3-Dichloro-benzyl)-3-phenyl-6.7.8.9-tetrahvdro-5H-imidazori.2- alazepin-l-ium; chloride (compound 26)
[00204] 2,3-Dichorobenzyle chloride (59 mg, 0.30 mmol), triethylamine (33 μL, 0.24 mmol) and 3-phenyl-6,7,8,9-tetrahydro-5H-imidazo[l,2-a]azepine (50 mg, 0.24 mmol) in 1 mL dry acetonitrile were microwave irradiated at 160 0C for 15 min. The reaction mixture was evaporated to dryness, dissolved in H2O, washed with 3 times with Et2O (limited amounts of brine was added to the water phase) and extracted 3 times with CH2Cl2. The organic phase was dried over Na2SO4, filtrated and evaporated to dryness. The residue was recrystallized from heptane/CH2Cl2 which gave white crystals of the title compound (53 mg, 56%). 1H NMR (CDCl3) δ: 7.73 (dd, J= 7.6, 1.6 Hz, IH); 7.46-7.37 (m, 4H); 7.34-7.30 (m, 2H); 7.30-7.7.24 (m, 2H); 5.80 (s, 2H); 4.26-4.18 (m, 2H); 3.51-3.36 (m, 2H); 2.00-1.88 (m, 2H); 1.82-1.70 (m, 4H). 13C NMR (CDCl3) δ: 149.9; 134.5; 133.9; 133.4; 132.1; 131.6; 130.5; 130.4; 130.1; 129.3; 128.7; 125.1; 118.9; 50.6; 47.7; 29.4; 27.1; 25.6; 23.8. Purity by LC/MS (UV/MS): 99/94.
Example 24: l-(2,4-Dichloro-benzyl)-3-phenyl-6J,8,9-tetrahvdro-5H-imidazorL2- alazepin-l-ium; chloride (compound 27)
[00205] 2,4-Dichorobenzyle chloride (59 mg, 0.30 mmol), triethylamine (33 μL, 0.24 mmol) and 3-phenyl-6,7,8,9-tetrahydro-5H-imidazo[l,2-a]azepine (50 mg, 0.24 mmol) in 1 mL dry acetonitrile were microwave irradiated at 160 0C for 15
min. The reaction mixture was evaporated to dryness, dissolved in H2O, washed with 3 times with Et2O (limited amounts of brine was added to the water phase) and extracted 3 times with CH2Cl2. The organic phase was dried over Na2SO4, filtrated and evaporated to dryness. The residue was recrystallized from heptane/CH2Cl2 which gave white crystals of the title compound (41 mg, 43%). 1H NMR (CDCl3) δ: 7.87 (d, J=8.2 Hz, IH); 7.46-7.34 (m, 6H); 7.33 (dd, J=8.2, 2.2 Hz, IH); 7.20 (s, IH); 5.77 (s, 2H); 4.27-4.22 (m, 2H); 3.53-3.48 (m, 2H); 2.01-1.93 (m, 2H); 1.82-1.75 (m, 4H). 13C NMR (CDCl3) δ: 149.9; 136.4; 134.7; 134.5; 133.6; 130.5; 130.2; 129.9; 129.9; 129.3; 128.6; 125.3; 118.7; 49.6; 47.7; 29.4; 27.1; 25.6; 23.8. Purity by LC/MS (UV/MS): 100/97.
Example 25: 3-(4-Chloro-phenyl)-l-(2,4-dichloro-benzyl)-5,6,7,8-tetrahvdro- imidazo|"1.2-a"|pyridin-l-ium; chloride (compound 8)
[00206] 2,4-Dichlorobenzyl chloride (42 μL, 0.30 mmol), triethylamine (66 μL, 0.47 mmol) and 3-(4-chloro-phenyl)-5,6,7,8-tetrahydro-imidazo[l,2-a]pyridine (56 mg, 0.24 mmol) were subjected to procedure C. The residue was column chromatographed on silica (CH2Cl2/Me0H 11: 1) which gave the title compound (22 mg, 21%). 1H NMR (CDCl3) δ: 7.93 (d, J=8.4 Hz, IH); 7.46-7.38 (m, 5H); 7.35-7.31 (m, 2H); 5.67 (s, 2H); 4.08-4.03 (m, 2H); 3.37-3.32 (m, 2H); 2.19-2.11 (m, 2H); 2.09- 2.02 (m, 2H). 13C NMR (CDCl3) δ: 145.4; 136.8; 136.5; 134.9; 134.1; 132.7; 131.1; 129.9; 129.6; 129.2; 128.6; 123.7; 119.2; 49.0; 45.6; 22.3; 21.6; 18.0. Purity by LC/MS (UV/MS): 96/100.
Example 26: 3-(3-Chloro-phenyl)-l-(2,4-dichloro-benzyl)-5,6,7,8-tetrahvdro- imidazori,2-a1pyridin-l-ium; chloride (compound 9)
[00207] 2,4-Dichlorobenzyl chloride (42 μL, 0.30 mmol), triethylamine (66 μL, 0.47 mmol) and 3-(3-chloro-phenyl)-5,6,7,8-tetrahydro-imidazo[l,2-a]pyridine (56 mg, 0.24 mmol) were subjected to procedure C. The residue was recrystallized from CH2Cl2/Et0Ac which gave the title compound (16 mg, 16%). 1H NMR (CD3OD) δ: 7.64 (d, J=2.0 Hz, IH); 7.66-7.63 (m, 2H); 7.59-7.42 (m, 5H); 5.49 (s, 2H); 4.15-4.11 (m, 2H); 3.15-3.11 (m, 2H); 2.14-2.06 (m, 4H). 13C NMR (CD3OD) δ: 147.1; 137.2; 136.1; 135.8; 134.3; 132.9; 131.9; 131.4; 131.1; 131.1; 130.5; 129.4; 129.0; 128.7; 120.7; 49.4; 46.9; 22.6; 22.4; 18.8. Purity by LC/MS (UV/MS): 99/98.
Example 27: l-(2,4-Dichloro-benzyl)-3-phenyl-5,6,7,8-tetrahvdro-imidazori,2- alpyridin-l-ium; chloride (compound 10)
[00208] 2,4-Dichlorobenzyl chloride (42 μL, 0.30 mmol), triethylamine (66 μL, 0.47 mmol) and 3-phenyl-5,6,7,8-tetrahydro-imidazo[l,2-a]pyridine (48 mg, 0.24 mmol) were subjected to procedure C. The residue was column chromatographed on silica (CH2CVMeOH 11 : 1) which gave the title compound (35 mg, 36%). 1H NMR (CDCl3) δ: 7.95 (d; J=8.4 Hz, IH); 7.44-7.37 (m, 6H); 7.31 (dd, J=8.2, 2.2 Hz, IH); 7.24 (s, IH); 5.71 (s, 2H); 4.05-4.00 (m, 2H); 3.42-3.36 (m, 2H); 2.17-2.09 (m, 2H); 2.08-2.01 (m, 2H). 13C NMR (CDCl3) δ: 145.1; 136.4; 135.0; 134.1; 133.8; 130.4; 129.8; 129.6; 129.2; 129.2; 128.5; 125.0; 118.8; 48.9; 45.6; 22.3; 21.6; 18.0. Purity by LC/MS (UV/MS): 98/99.
Example 28: 3-(4-Chloro-phenyl)-l-(2.4-dichloro-benzyl)-6.7.8.9-tetrahvdro-5H- imidazori.2-a"|azepin-l-ium; chloride (compound 14)
[00209] 2,4-Dichlorobenzyl chloride (42 μL, 0.30 mmol), triethylamine (66 μL, 0.47 mmol) and 3-(4-chloro-phenyl)-6,7,8,9-tetrahydro-5H-imidazo[l,2- a]azepine (59 mg, 0.24 mmol) were subjected to procedure C. The residue was recrystallized from CH2Cl2ZEtOAc which gave the title compound (25 mg, 24%). 1H NMR (CDCl3) δ: 7.92 (d, J=8.4 Hz, IH); 7.45-7.40 (m, 4H); 7.37-7.32 (m, 3H); 5.80 (s, 2H); 4.27-4.22 (m, 2H); 3.50-3.46 (m, 2H); 2.01-1.94 (m, 2H); 1.84-1.76 (m, 4H). 13C NMR (CDCl3) δ: 150.2; 137.2; 136.5; 134.6; 133.6; 133.4; 131.7; 130.0; 129.9; 129.8; 128.8; 123.8; 119.5; 49.7; 48.0; 29.6; 27.1; 25.8; 23.8. Purity by LC/MS (UV/MS): 100/99.
Example 29: 3-(3-Chloro-phenyl)-l-(2.4-dichloro-benzyl)-6.7.8.9-tetrahvdro-5H- imidazori,2-a1azepin-l-ium; chloride (compound 15)
[00210] 2,4-Dichlorobenzyl chloride (42 μL, 0.30 mmol), triethylamine (66 μL, 0.47 mmol) and 3-(3-chloro-phenyl)-6,7,8,9-tetrahydro-5H-imidazo[l,2- a]azepine (59 mg, 0.24 mmol) were subjected to procedure C. The residue was column chromatographed on silica (CH2Cl2/Me0H 11: 1) which gave the title compound (27 mg, 25%) 1H NMR (CDCl3) δ: 7.90 (d, J=8.4 Hz, IH); 7.46-7.32 (m, 7H); 5.79 (s, 2H); 4.28-4.23 (m, 2H); 3.53-3.48 (m, 2H); 2.02-1.94 (m, 2H); 1.84-1.76 (m, 4H). 13C NMR (CDCl3) δ: 150.3; 136.5; 135.3; 134.7; 133.6; 133.0; 130.7; 130.0;
130.0; 129.7; 128.7; 128.6; 127.0; 119.3; 49.7; 47.9; 29.4; 27.0; 25.7; 23.7. Purity by LC/MS (UV/MS): 97/97.
Example 30: l-(2Λ-dichloro-benzyl)-3-phenyl-6J,8,9-tetrahvdro-5H-imidazori,2- aiazepin-1-ium; acetate (compound 7)
[00211] 2,4-Dichlorobenzyl chloride (42 μL, 0.30 mmol), triethylamine (66 μL, 0.47 mmol) and 3-phenyl-6,7,8,9-tetrahydro-5H-imidazo[l,2-a]azepine (51 mg, 0.24 mmol) were subjected to procedure C. The residue was purified by prep HPLC/MS which gave the title compound (25 mg, 24%). 1H NMR (CDCl3) δ: 7.73 (d, J=8.4 Hz, IH); 7.48-7.42 (m, 4H); 7.37-7.32 (m, 3H); 7.07(s, IH); 5.68 (s, 2H); 4.24-4.18 (m, 2H); 3.47-3.42 (m, 2H); 2.01-1.93 (m, 2H); 1.91 (s, 3H); 1.84-1.75 (m, 4H). 13C NMR (CDCl3) δ: 176.3; 150.1; 136.5; 134.9; 134.4; 133.5; 130.5; 130.1; 130.0; 130.0; 129.4; 128.6; 125.4; 118.5; 47.5; 47.5; 29.6; 27.1; 25.2; 23.7; 23.4. Purity by LC/MS (UV/MS): 100/99.
Example 31 : 3-(4-chloro-phenyl)-l-(4-methoxycarbonyl-benzyl)-5,6J,8-tetrahvdro- imidazori,2-a1pyridin-l-ium; acetate (compound 11)
[00212] Methyl-4-(chloromethyl) benzoate (55 mg, 0.30 mmol), triethylamine (66 μL, 0.47 mmol) and 3-(4-chloro-phenyl)-5,6,7,8-tetrahydro- imidazo[l,2-a]pyridine (56 mg, 0.24 mmol) were subjected to procedure C. The residue was purified by prep HPLC/MS which gave the title compound (17 mg, 16%). 1H NMR (CDCl3) δ: 8.05-8.01 (m, 2H); 7.54 (s, IH); 7.48-4.41 (m, 6H); 5.64 (s, 2H); 4.08-4.03 (m, 2H); 3.90 (s, 3H); 3.21-3.15 (m, 2H); 2.14-2.00 (m, 4H); 1.91 (s, 3H). 13C NMR (CDCl3) δ: 176.5; 166.4; 145.5; 138.2; 136.8; 132.8; 131.0; 131.0; 130.7; 129.6; 128.7; 123.8; 119.6; 52.4; 51.3; 45.5; 23.9; 21.9; 21.6; 17.9. Purity by LC/MS (UV/MS): 99/89.
Example 32 : 3 -(3 -chloro-phenyl)- 1 -(4-methoxycarbonyl-benzyl)-5 ,6,7,8-tetrahydro- imidazori.2-a1pyridin-l-ium; acetate (compound 12)
[00213] Methyl-4-(chloromethyl) benzoate (55 mg, 0.30 mmol), triethylamine (66 μL, 0.47 mmol) and 3-(3-chloro-phenyl)-5,6,7,8-tetrahydro- imidazo[l,2-a]pyridine (56 mg, 0.24 mmol) were subjected to procedure C. The residue was purified by prep HPLC/MS which gave the title compound (9 mg, 9%). 1H NMR (CDCl3) δ: 8,07-8.04 (m, 2H); 7.48-7.42 (m, 7H); 5.66 (s, 2H); 4.10-4.05
(m, 2H); 3.92 (s, 3H); 3.26-3.21 (m, 2H); 2.16-2.02 (m, 4H); 1.93 (s, 3H). 13C NMR (CDCl3) δ: 176.5; 166.4; 145.8; 138.1; 135.3; 132.6; 131.0; 130.7; 130.7; 130.6; 129.5; 128.7; 128.1; 127.1; 119.6; 52.5; 51.4; 45.6; 23.8; 22.0; 21.6; 17.9. Purity by LC/MS (UV/MS): 99/95.
Example 33: l-(4-methoxycarbonyl-benzyl)-3-phenyl-5,6J,8-tetrahydro- imidazori.2-a1pyridin-l-ium; acetate (compound 13)
[00214] Methyl-4-(chloromethyl) benzoate (55 mg, 0.30 mmol), triethylamine (66 μL, 0.47 mmol) and 3-phenyl-5,6,7,8-tetrahydro-imidazo[l,2- a]pyridine (48 mg, 0.24 mmol) were subjected to procedure C. The residue was purified by prep HPLC/MS which gave the title compound (20 mg, 20%). 1H NMR (CDCl3) δ: 8.05-8.01 (m, 2H); 7.48-7.45 (m, 5 H); 7.45-7.41 (m, 2H); 7.35 (s, IH); 5.58 (s, 2H); 4.08-4.03 (m, 2H); 3.89 (s, 3H); 3.09-3.04 (m, 2H); 2.13-1.99 (m, 4H); 1.90 (s, 3H). 13C NMR (CDCl3) δ: 176.4; 166.5; 145.2; 138.3; 134.0; 130.9; 130.6; 130.4; 129.6; 129.3; 128.5; 125.3; 119.1; 52.4; 51.2; 45.6; 23.4; 21.7; 21.6; 17.9. Purity by LC/MS (UV/MS): 100/95.
Example 34: 3-(4-chloro-phenyl)-l-(4-methoxycarbonyl-benzyl)-6J,8,9-tetrahvdro- 5H-imidazori,2-a1azepin-l-ium; acetate (compound 16)
[00215] Methyl-4-(chloromethyl) benzoate (55 mg, 0.30 mmol), triethylamine (66 μL, 0.47 mmol) and 3-(4-chloro-phenyl)-6,7,8,9-tetrahydro-5H- imidazo[l,2-a]azepine (59 mg, 0.24 mmol) were subjected to procedure C. The residue was purified by prep HPLC/MS which gave the title compound (16 mg, 15%). 1H NMR (CDCl3) δ: 8.05-8.01 (m, 2H); 7.52 (s, IH); 7.47-7.35 (m, 6H); 5.73 (s, 2H); 4.22-4.17 (m, 2H); 3.90 (s, 3H); 3.34-3.29 (m, 2H); 1.96-1.89 (m, 5H); 1.81- 1.74 (m, 2H); 1.65-1.59 (m, 2H). 13C NMR (CDCl3) δ: 176.5; 166.4; 150.3; 139.1; 137.0; 133.3; 131.5; 130.9; 130.7; 129.7; 128.0; 123.9; 119.9; 52.4; 52.1; 47.6; 29.5; 27.1; 25.3; 23.8; 23.4. Purity by LC/MS (UV/MS): 99/93.
Example 35: 3-(3-chloro-phenyl)-l-(4-methoxycarbonyl-benzyl)-6J,8,9-tetrahvdro- 5H-imidazori,2-a1azepin-l-ium; acetate (compound 17)
[00216] Methyl-4-(chloromethyl) benzoate (55 mg, 0.30 mmol), triethylamine (66 μL, 0.47 mmol) and 3-(3-chloro-phenyl)-6,7,8,9-tetrahydro-5H- imidazo[l,2-a]azepine (59 mg, 0.24 mmol) were subjected to procedure C. The
residue was purified by prep HPLC/MS which gave the title compound (7 mg, 1H NMR (CDCl3) δ: 8,07-8.03 (m, 2H); 7.52 (s, IH); 7.50-7.34 (m, 6H); 5.79 (s, 2H); 4.25-4.21 (m, 2H); 3.91 (s, 3H); 3.40-3.34 (m, 2H); 2.00-1.92 (m, 5H); 1.84-1.76 (m, 2H); 1.69-1.62 (m, 2H). 13C NMR (CDCl3) δ: 176.7; 166.4; 150.6; 139.1; 135.4; 133.0; 130.9; 130.8; 130.7; 130.7; 130.0; 128.6; 128.1; 127.2; 120.1; 52.4; 52.2; 47.7; 29.5; 27.1; 25.4; 24.1; 23.5. Purity by LC/MS (UV/MS): 100/95.
Example 36: 1 -(4-methoxycarbonyl-benzyl)-3-phenyl-6,7,8,9-tetrahvdro-5H- imidazori,2-a1azepin-l-ium; acetate (compound 18)
[00217] Methyl-4-(chloromethyl) benzoate (55 mg, 0.30 mmol), triethylamine (66 μL, 0.47 mmol) and 3-phenyl-6,7,8,9-tetrahydro-5H-imidazo[l,2- a]azepine (51 mg, 0.24 mmol) were subjected to procedure C. The residue was purified by prep HPLC/MS which gave the title compound (25 mg, 25%). 1H NMR (CDCl3) δ: 8.04-8.01 (m, 2H); 7.47 (m, 3H); 7.42-7.36 (m, 5H); 5.74 (s, 2H); 4.22- 4.18 (m, 2H); 3.89 (s, 3H); 3.35-3.30 (m, 2H); 1.95-1.89 (m, 5H); 1.81-1.73 (m, 2H); 1.66-1.59 (m, 2H). 13C NMR (CDCl3) δ: 176.4; 166.4; 150.1; 139.2; 134.4; 130.8; 130.6; 130.5; 130.1; 129.4; 128.0; 125.3; 119.4; 52.4; 52.0; 47.5; 29.5; 27.1; 25.2; 23.7; 23.5. Purity by LC/MS (UV/MS): 99/93.
Example 37: 3 -(4-chloro-phenyl)- 1 -(2 ,4-dichloro-benzyl)-5 ,6,7,8,9,10-hexahydro- imidazo|"1.2-a"|azocin-l-ium; acetate (compound 19)
[00218] 2,4-Dichlorobenzyl chloride (25 μL, 0.18 mmol), triethylamine (42 μL, 0.30 mmol) and 3-(4-chloro-phenyl)-5, 6,7,8,9, 10-hexahydro-imidazo[l,2- ajazocine (40 mg, 0.15 mmol) in 1 mL dry acetonitrile were microwave irradiated at 160 0C for 15 min. The reaction mixture was evaporated to dryness and the residue was dissolved in H2O, washed 3 times with Et2θ and extracted 3 times with CH2CI2. The organic phase was dried over Na2SO4, filtrated and evaporated to dryness. The residue was purified by prep HPLC/MS which gave the title compound (15 mg, 21%). 1H NMR (CDCl3) δ: 7.80 (d, J=8,2 Hz, IH); 7.46-7.41 (m, 3H); 7.41-7.38 (m, 2H), 7.36 (dd, J=8.3, 2.1 Hz, IH); 7.19 (s, IH); 5.72 (s, 2H); 4.26-4.21 (m, 2H); 3.32-3.27 (m, 2H); 1.93 (s, 3H); 1.73-1.58 (m, 4H); 1.54-1.47 (m, 2H); 1.36-1.29 (m, 2H). 13C NMR (CDCl3) δ: 176.4; 149.1; 137.2; 136.7; 135.1; 133.9; 132.5; 131.6; 130.1; 129.7; 129.4; 128.7; 123.9; 120.0; 49.7; 44.6; 30.6; 28.5; 25.3; 23.8; 23.7; 23.7. Purity by LC/MS (UV/MS): 100/96.
Example 38: S-^-chloro-phenviyi-^-methoxycarbonyl-benzylVS.όJ^gjO- hexahydro-imidazori,2-a1azocin-l-ium; acetate (compound 20)
[00219] Methyl-4-(chloromethyl) benzoate (33 mg, 0.18 mmol), triethylamine (42 μL, 0.30 mmol) and 3-(4-chloro-phenyl)-5,6,7,8,9,10-hexahydro- imidazo[l,2-a]azocine (40 mg, 0.15 mmol) in 1 mL dry acetonitrile were microwave irradiated at 160 0C for 15 min and then at 160 0C for 10 min. The reaction mixture was evaporated to dryness and the residue was dissolved in H2O, washed 3 times with Et2O and extracted 3 times with CH2Cl2. The organic phase was dried over Na2SO4, filtrated and evaporated to dryness. The residue was purified by prep HPLC/MS which gave 35 (15 mg, 22%). 1H NMR (CDCl3) δ: 8.06-8.02 (m, 2H); 7.50 (s, IH); 7.47-7.43 (m, 4H); 7.43-7.39 (m, 2H); 5.66 (s, 2H); 4.25-4.20 (m, 2H); 3.32-3.26 (m, 2H); 1.87 (s, 3H); 1.64-1.56 (m, 2H); 1.48-1.36 (m, 4H); 1.30-1.22 (m, 2H). 13C NMR (CDCl3) δ: 176.7; 166.4; 149.3; 139.2; 137.1; 132.8; 131.6; 130.9; 130.7; 129.7; 128.1; 123.9; 120.9; 52.4; 51.9; 44.7; 30.5; 28.6; 25.2; 23.8; 23.6; 23.4. Purity by LC/MS (UV/MS): 100/98.
Example 39: 3 -(3 -chloro-phenyl)- 1 -(2 ,4-dichloro-benzyl)-5 ,6,7,8,9,10-hexahydro- imidazori,2-a1azocin-l-ium; acetate (compound 21)
[00220] 2,4-Dichlorobenzyl chloride (25 μL, 0.18 mmol), triethylamine (42 μL, 0.30 mmol) and 3-(3-chloro-phenyl)-5, 6,7,8,9, 10-hexahydro-imidazo[l,2- ajazocine (40 mg, 0.15 mmol) in 1.5 mL dry acetonitrile were microwave irradiated at 160 0C for 15 min and twice at 160 0C for 10 min. The reaction mixture was evaporated to dryness and the residue was dissolved in H2O, washed 3 times with Et2O and extracted 3 times with CH2Cl2. The organic phase was dried over Na2SO4, filtrated and evaporated to dryness. The residue was purified by prep HPLC/MS which gave the title compound (24 mg, 34%). 1H NMR (CDCl3) δ: 7.76 (d, J=8.2 Hz, IH); 7.48-7.34 (m, 6H); 7.05 (s, IH); 5.54 (s, 2H); 4.28-4.22 (m, 2H); 3.42-3.26 (m, 2H); 1.82 (s, 3H); 1.74-1.60 (m, 4H); 1.55-1.48 (m, 2H); 1.36-1.29 (m, 2H). 13C NMR (CDCl3) δ: 176.4; 149.3; 136.7; 135.3; 135.2; 134.0; 132.2; 130.8; 130.7; 130.2; 130.0; 129.4; 128.7; 128.5; 127.2; 119.9; 49.8; 44.7; 30.6; 28.4; 25.3; 23.7; 23.6; 23.6. Purity by LC/MS (UV/MS): 100/99.
Example 40: l-(2,4-dichloro-benzyl)-3-phenyl-5,6,7,8,9,10-hexahydro-imidazori,2- aiazocin-1-ium; acetate (compound 22)
[00221] 2,4-Dichlorobenzyl chloride (22 μL, 0.16 mmol), triethylamine (37 μL, 0.26 mmol) and 3-phenyl-5, 6,7,8,9, 10-hexahydro-imidazo[l,2-a]azocine (30 mg, 0.13 mmol) in 1 mL dry acetonitrile were microwave irradiated at 160 0C for 15 min and then at 160 0C for 10 min. The reaction mixture was evaporated to dryness and the residue was dissolved in H2O, washed 3 times with Et2O and extracted 3 times with CH2Cl2. The organic phase was dried over Na2SO4, filtrated and evaporated to dryness. The residue was purified by prep HPLC/MS which gave the title compound (18 mg, 30%). 1H NMR (CDCl3) δ: 7.74 (d, J=8.2 Hz, IH); 7.49-7.34 (m, 7H); 7.05 (s, IH); 5.64 (s, 2H); 4.27-4.22 (m, 2H); 3.42-3.36 (m, 2H); 1.90 (s, 3H); 1.74-1.58 (m, 4H); 1.54-1.46 (m, 2H); 1.36-1.28 (m, 2H). 13C NMR (CDCl3) δ: 176.5; 148.9; 136.6; 135.2; 133.9; 133.8; 130.6; 130.1; 129.5; 129.3; 128.6; 125.4; 119.4; 49.7; 44.6; 30.6; 28.5; 25.3; 23.7; 23.6; 23.5. Purity by LC/MS (UV/MS): 100/93.
Example 41 : l-(4-methoxycarbonyl-benzyl)-3-phenyl-5,6J,8,9J0-hexahvdro- imidazori,2-a1azocin-l-ium; acetate (compound 23)
[00222] Methyl-4-(chloromethyl) benzoate (30 mg, 0.16 mmol), triethylamine (37 μL, 0.26 mmol) and 3-phenyl-5,6,7,8,9,10-hexahydro-imidazo[l,2- a]azocine (30 mg, 0.13 mmol) in 1 mL dry acetonitrile were microwave irradiated at 160 0C for 15 min and then at 160 0C for 10 min. The reaction mixture was evaporated to dryness and the residue was dissolved in H2O, washed 3 times with Et2O and extracted 3 times with CH2Cl2. The organic phase was dried over Na2SO4, filtrated and evaporated to dryness. The residue was purified by prep HPLC/MS which gave 38 (10 mg, 17%). 1H NMR (CDCl3) δ: 8.08-8.04 (m, 2H); 7.52-7.43 (m, 8H); 5.78 (s, 2H); 4.28-4.22 (m, 2H); 3.92 (s, 3H); 3.41-3.35 (m, 2H); 1.94 (3H); 1.68-1.62 (m, 2H); 1.55-1.41 (m, 4H); 1.34-1.28 (m, 2H); 13C NMR (CDCl3) δ: 176.4; 166.5; 149.1; 139.1; 134.0; 131.0; 130.8; 130.7; 130.2; 129.4; 128.2; 125.4; 120.5; 52.5; 52.0; 44.8; 30.7; 28.7; 25.3; 23.8; 23.7; 23.6. Purity by LC/MS (UV/MS): 100/100.
Example 42: Receptor Selection and Amplification Technology Assay
[00223] The functional receptor assay, Receptor Selection and Amplification Technology (R-SAT®), was used to investigate the pharmacological
properties of known and novel RARβ agonists and antagonists and novel RARγ agonists and antagonists. R-SAT® is disclosed, for example, in U.S. Patent Nos. 5,707,798, 5,912,132, and 5,955,281, Piu, F., Gauthier, N. K., and Wang, F., (Beta Arrestin 2 modulates the activity of Nuclear Receptor RAR beta 2 through activation of ERK2 kinase) Oncogene 2006, 12:25(2):218-29, Burstein, E. S., Piu, F., Ma, J-N., Weissman, J. T., Currier, E.A., Nash, N. R., Weiner, D. M., Spalding, T. A., Schiffer, H. H., Del Tredici, A. L., Brann, M. R. (Integrative Functional Assays, Chemical Genomics and High Throughput Screening: Harnessing signal transduction pathways to a common HTS readout) Curr Pharm Des. 2006 12(14): 1717-29, all of which are hereby incorporated herein by reference in their entirety, including any drawings.
[00224] These experiments have provided a molecular profile, or fingerprint, for each of these agents at the human RAR and RXR receptors. As can be seen in Table 1, compounds of Formula I modulate the RARγ and/or the RARβ receptor.
TABLE 1


Efficacy is relative to the maximal response of the reference ligand, Am-580. * Na means no activity
Claims
WHAT IS CLAIMED IS: 1. A compound of Formula I

R1, R2, R3, are each independently selected from the group consisting of hydrogen, -OH, -COOH, halogen, optionally substituted C1- Cio straight chained or branched alkyl, optionally substituted C2-C10 straight chained or branched alkenyl, optionally substituted C2-C10 straight chained or branched alkynyl, optionally substituted C3-C9 cycloalkyl, optionally substituted C3-C9 cycloalkylalkyl, optionally substituted C5-C7 cycloalkenyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted aralkyl, and optionally substituted heteroaralkyl; X is C or N+;
Y is a counter ion and is absent when X is C; and n is an integer from 1 to 5, such as n is 1, 2, 3, 4, or 5.
2. The compound of claim 1, wherein Ri is selected from the group consisting of optionally substituted C1-C10 straight chained or branched alkyl, optionally substituted C3-C9 cycloalkyl, optionally substituted aryl, and optionally substituted heteroaryl.
3. The compound of claim 1, wherein Ri is selected from the group consisting of optionally substituted aryl and optionally substituted heteroaryl.
4. The compound of claim 3, wherein the aryl and the heteroaryl are optionally substituted with one or more substituents selected from the group consisting of alkyl, hydroxy, protected hydroxy, alkoxy, aryloxy, acyl, ester, mercapto, alkylthio, arylthio, cyano, halogen, carbonyl, C-amido, N-amido, C-carboxy, protected C-carboxy, O-carboxy, nitro, silyl, sulfenyl, sulfinyl, sulfonyl, haloalkyl, perhaloalkyl, and amino.
5. The compound of claim 3, wherein the aryl and the heteroaryl are optionally substituted with one or more substituents selected from the group consisting of hydroxy, alkoxy, aryloxy, cyano, halogen, and amino.
6. The compound of claim 3, wherein the aryl and the heteroaryl are optionally substituted with one or more substituents selected from the group consisting of fluoro, chloro, bromo, iodo, haloalkyl, and perhaloalkyl.
7. The compound of claim 1, wherein the aryl is phenyl.
8. The compound of claim 1, wherein Ri is selected from the group consisting of phenyl, 4-cholorophenyl, 3-cholorophenyl, and 2-cholorophenyl.
9. The compound of claim 1, wherein R2 is selected from the group consisting of hydrogen, optionally substituted C1-C10 straight chained or branched alkyl, optionally substituted C2-C10 straight chained or branched alkenyl, optionally substituted C2-C10 straight chained or branched alkynyl, optionally substituted C3-C9 cycloalkyl, and optionally substituted C5-C7 cycloalkenyl.
10. The compound of claim 1, wherein R2 is selected from the group consisting of hydrogen, optionally substituted Ci-Ci0 straight chained or branched alkyl.
11. The compound of claim 1, wherein R2 is hydrogen.
12. The compound of claim 1, wherein R3 is selected from the group consisting of optionally substituted Ci-Ci0 straight chained or branched alkyl, optionally substituted C2-Ci0 straight chained or branched alkenyl, optionally substituted C2-Ci0 straight chained or branched alkynyl, optionally substituted C3-C9 cycloalkyl, optionally substituted C5-C7 cycloalkenyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted aralkyl, and optionally substituted heteroaralkyl.
13. The compound of claim 1, wherein R3 is selected from the group consisting of optionally substituted Ci-Ci0 straight chained or branched alkyl, optionally substituted C3-C9 cycloalkyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted aralkyl, and optionally substituted heteroaralkyl.
14. The compound of claim 1, wherein R3 is selected from the group consisting of optionally substituted aralkyl, and optionally substituted heteroaralkyl.
15. The compound of claim 14, wherein the aralkyl or heteroaralkyl is optionally substituted with one or more substituents selected from the group consisting of alkyl, hydroxy, protected hydroxy, alkoxy, aryloxy, acyl, ester, mercapto, alkylthio, arylthio, cyano, halogen, carbonyl, C-amido, N-amido, C-carboxy, protected C-carboxy, O-carboxy, nitro, silyl, sulfenyl, sulfinyl, sulfonyl, haloalkyl, perhaloalkyl, and amino.
16. The compound of claim 14, wherein the aralkyl or heteroaralkyl is optionally substituted with one or more substituents selected from the group consisting of alkyl, hydroxy, halogen, carbonyl, C-carboxy, protected C-carboxy, and amino.
17. The compound of claim 14, wherein the aralkyl or heteroaralkyl is optionally substituted with one or more substituents selected from the group consisting of alkyl, halogen, C-carboxy, and protected C-carboxy.
18. The compound of claim 17, wherein the alkyl is selected from the group consisting of methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec -butyl, iso-butyl, and tert-butyl.
19. The compound of claim 14, wherein the C-carboxy is protected by an alkyl group.
20. The compound of claim 19, wherein the alkyl group is selected from the group consisting of methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec -butyl, iso- butyl, and tert-butyl.
21. The compound of claim 1, wherein R3 is selected from the group consisting of benzyl, 2-chlorobenzyl, 3-chlorobenzyl, 4-chlorobenzyl, 2- methylbenzyl, 3-methylbenzyl, 4-methylbenzyl, 2-cyanobenzyl, 3-cyanobenzyl, A- cyanobenzyl, 2,3-dicholorbenzyl, 2,4-dicholorbenzyl, 2,6-dicholorbenzyl, 2- methylcarboxybenzyl (H3COC(=O)-C6H4-CH2-), 3-methylcarboxybenzyl, and A- methylcarboxybenzyl.
22. The compound of claim 1, wherein R3 is selected from the group consisting of benzyl, 4-chlorobenzyl, 2-methylbenzyl, 3-methylbenzyl, A- methylbenzyl, 4-cyanobenzyl, 2,4-dicholorbenzyl, 2,6-dicholorbenzyl, and A- methylcarboxybenzyl.
23. The compound of claim 1, wherein X is N+.
24. The compound of claim 1, wherein Y is selected from the group consisting of halide, acetate, tartrate, phosphate, sulfonate, tetrafluoroborate.
25. The compound of claim 24, wherein the halide is selected from the group consisting of fluoride, chloride, bromide, and iodide.
26. The compound of claim 1, wherein Y is selected from the group consisting of chloride, bromide, and acetate.
27. A compound of Formula II
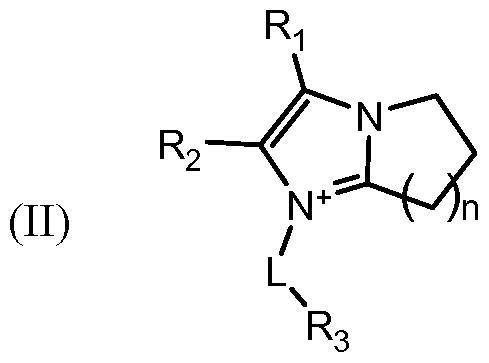
L is an alkyl spacer optionally substituted with one or more substituents,
Ri and R3 are each independently optionally substituted aryl or optionally substituted heteroaryl;
R2 is selected from the group consisting of hydrogen, -OH, -COOH, halogen, optionally substituted C1-C10 straight chained or branched alkyl, optionally substituted C2-C10 straight chained or branched alkenyl, optionally substituted C2-C10 straight chained or branched alkynyl, optionally substituted C3-C9 cycloalkyl, optionally substituted C5-C7 cycloalkenyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted aralkyl, and optionally substituted heteroaralkyl; Y is a counter ion; and n is an integer from 1 to 5, such as n is 1, 2, 3, 4, or 5.
28. The compound of claim 27, wherein Ri is optionally substituted phenyl.
29. The compound of claim 27, wherein R3 is optionally substituted phenyl.
30. The compound of claim 27, wherein L is -CH2-.
31. A compound selected from the group consisting of:
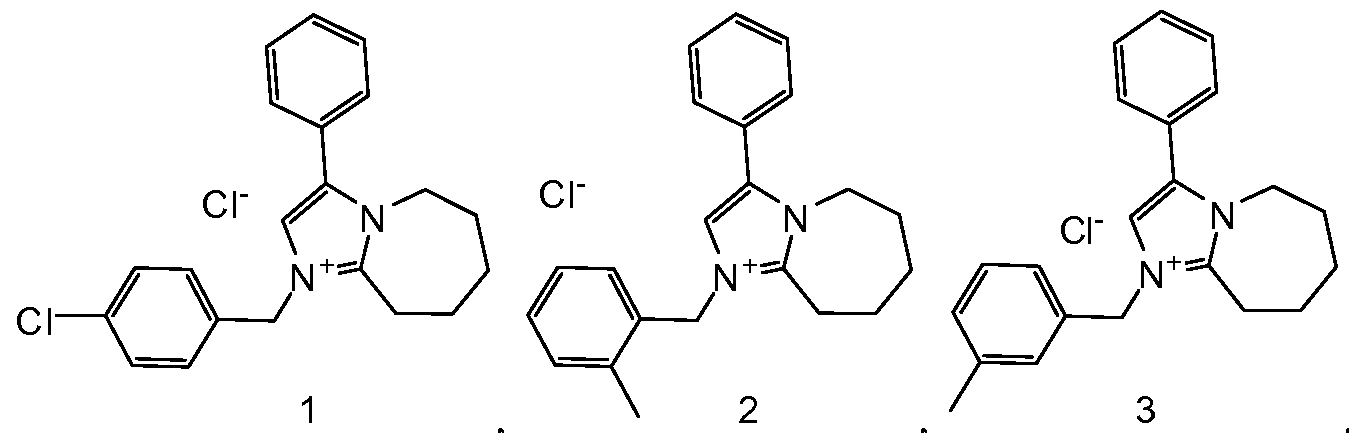



32. A method of modulating the activity of a RARβ or RARγ receptor subtypes, comprising contacting a RARβ or RARγ receptor with a compound of Formula I.
33. The method of claim 32, wherein the modulating is performed in vivo.
34. The method of claim 32, wherein the modulating is performed in vitro.
35. A method of treating, or alleviating the symptoms of, a disease comprising administering to a subject in need thereof a therapeutically effective amount of a compound of Formula I

Ri, R2, R3, are each independently selected from the group consisting of hydrogen, -OH, -COOH, halogen, optionally substituted Ci- Cio straight chained or branched alkyl, optionally substituted C2-C10 straight chained or branched alkenyl, optionally substituted C2-C10 straight chained or branched alkynyl, optionally substituted C3-C9 cycloalkyl, optionally substituted C5-C7 cycloalkenyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted aralkyl, and optionally substituted heteroaralkyl; X is C or N+;
Y is a counter ion and is absent when X is C; and n is an integer from 1 to 5, such as n is 1, 2, 3, 4, or 5.
36. The method of claim 35, wherein the disease is selected from the group consisting of cancer, hyperproliferative disorder, inflammatory disorder, neurological disorder, neurodegenerative disorder, eye disorder, eye condition, depression, pulmonary disorders/conditions, and dermatological disorders/conditions.
37. The method of claim 36, wherein disease is cancer and the compound of Formula I is administered in conjunction with at least one chemotherapeutic agent and/or radiation therapy.
38. The method of claim 36, wherein the cancer is selected from the group consisting of cancer of breast, head, skin, neck, lung, esophagus, mammary gland, liver, pancreas, cervix and digestive tract.
39. The method of claim 34, wherein the cancer of the digestive tract is a cancer is oral cavity cancer, esophagus cancer, stomach cancer, duodenum cancer, jejunum cancer, ileum cancer, and colon cancer.
40. The method of claim 35, wherein the inflammatory disorder is a chronic inflammatory disorder.
41. The method of claim 35, wherein the inflammatory disorder is rheumatoid arthritis
42. The method of claim 35, wherein the inflammatory disorder is psoriasis.
43. The method of claim 35, wherein, the neurological disorder is a performance deficit in spatial learning and memory tasks, age-related memory deficit.
44. The method of claim 35, wherein the neurological disorder is a disorder in which cognition is altered.
45. The method of claim 35, wherein the neurological disorder is schizophrenia.
46. The method of claim 35, wherein, the neurodegenerative disorder is Parkinson's disease or Alzheimer's disease.
47. The method of claim 35, wherein nerve regeneration is necessary to treat the neurodegenerative disorder.
48. The method of claim 46, wherein the neurodegenerative disorder is caused by a condition selected from the group consisting of spinal cord injury, stroke, damage to the cardiac muscles, damage caused to myelin in multiple sclerosis, and damage to islet cells in diabetes.
49. The method of claim 35, wherein the pulmonary disorders/conditions is selected from the group consisting of alveolar damage, emphysema and asthma.
50. The method of claim 35, wherein the dermatological disorders/conditions is selected from the group consisting of cystic acne, acne vulgaris, acneform disease, cutanous disorder of keratinization psoriasis and its pustular and erythrodermic variants, and cutaneous disease.
51. The method of claim 50, wherein the cutaneous disorder of keratinization is selected from the group consisting of the ichtyoses, Darier's disease, pityriasis rubra pilaris.
52. The method of claim 50, wherein the cutaneous disease is selected from the group consisting of subcorneal pustular dermatosis, discoid lupus erythematosus, Reiter's syndrome (with or without AIDS), warts, lichen planus, and cutaneous sarcoidosis.
53. A method of treating a condition selected from infertility caused by sperm immobility and subfertility, comprising administering to a subject in need thereof a therapeutically effective amount of a compound of Formula I

Ri, R2, R3, are each independently selected from the group consisting of hydrogen, -OH, -COOH, halogen, optionally substituted Ci- Cio straight chained or branched alkyl, optionally substituted C2-C10 straight chained or branched alkenyl, optionally substituted C2-C10 straight chained or branched alkynyl, optionally substituted C3-C9 cycloalkyl, optionally substituted C5-C7 cycloalkenyl, optionally substituted aryl, optionally substituted heteroaryl, optionally substituted aralkyl, and optionally substituted heteroaralkyl; X is C or N+;
Y is a counter ion and is absent when X is C; and n is an integer from 1 to 5, such as n is 1, 2, 3, 4, or 5.
54. A method of identifying a compound that is an agonist, inverse agonist, or antagonist of a RARβ receptor or RARγ receptor, comprising contacting the RARβ receptor or RARγ receptor with at least one test compound of Formula I, and determining any change in activity of the RARβ receptor or RARγ receptor, thereby identifying the test compound as an agonist, inverse agonist, or antagonist of the RARβ receptor or RARγ receptor.
55. The method of claim 54, wherein the RARβ receptor or RARγ receptor is expressed in a cell.
56. The method of claim 55, wherein the cell is a cultured cell.
57. The method of claim 55, wherein the cultured cell overexpresses the RARβ receptor or RARγ receptor.
58. The method of claim 54, wherein the identified agonist, inverse agonist or antagonist is selective for the RARβ receptor
59. The method of claim 54, wherein the identified agonist, inverse agonist or antagonist is selective for the RARγ receptor.
60. A pharmaceutical composition comprising a compound of Formula I, and a physiologically acceptable carrier, diluent, salt, or an excipient, or a combination thereof.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US86636006P | 2006-11-17 | 2006-11-17 | |
| US60/866,360 | 2006-11-17 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2008064136A2 true WO2008064136A2 (en) | 2008-05-29 |
| WO2008064136A3 WO2008064136A3 (en) | 2008-07-10 |
Family
ID=39430527
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/085027 Ceased WO2008064136A2 (en) | 2006-11-17 | 2007-11-16 | Compounds with activity at retinoic acid receptors |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2008064136A2 (en) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011012897A1 (en) | 2009-07-31 | 2011-02-03 | Astrazeneca Ab | New combinations for the treatment of asthma |
| WO2011061527A1 (en) | 2009-11-17 | 2011-05-26 | Astrazeneca Ab | Combinations comprising a glucocorticoid receptor modulator for the treatment of respiratory diseases |
| EP2329817A1 (en) * | 2009-09-04 | 2011-06-08 | Ernst-Moritz-Arndt-Universität Greifswald | Retinoic acid receptor antagonists, miR-10a inhibitors and inhibitors of HOXB1 or HOXB3 repressors for treating pancreatic cancer |
| WO2012085583A1 (en) | 2010-12-23 | 2012-06-28 | Astrazeneca Ab | New compound |
| WO2012085582A1 (en) | 2010-12-23 | 2012-06-28 | Astrazeneca Ab | Compound |
| WO2013192301A1 (en) * | 2012-06-22 | 2013-12-27 | The Board Of Trustees Of The Leland Stanford Junior University | Imidazo bicyclic imminium compounds as antitumor agents |
| WO2015109231A1 (en) | 2014-01-17 | 2015-07-23 | Cornell University | Methods of treating metabolic syndrome related conditions using retinoic acid receptor agonists |
| US20160289228A1 (en) * | 2013-12-09 | 2016-10-06 | Ucb Biopharma Sprl | Tetrahydroimidazopyridine Derivatives as Modulators of TNF Activity |
| US9512407B2 (en) | 2009-09-07 | 2016-12-06 | Genome Research Limited | Cells and methods for obtaining them |
| EP3563683A1 (en) | 2014-03-10 | 2019-11-06 | Cornell University | Combination therapy for head and neck cancer |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0889032A4 (en) * | 1996-03-18 | 2000-01-05 | Eisai Co Ltd | Fused-ring carboxylic acid derivatives |
| US6521814B1 (en) * | 1997-12-22 | 2003-02-18 | Institut National De La Santa Et De La Recherche Medicale | Use of ligands for treatment of diseases responsive to retinoids |
| KR20010053282A (en) * | 1998-07-01 | 2001-06-25 | 다케다 야쿠힌 고교 가부시키가이샤 | Retinoid-associated receptor regulators |
| EP1280757B1 (en) * | 2000-05-02 | 2005-08-17 | F. Hoffmann-La Roche Ag | New gamma selective retinoids |
| AU2002364082A1 (en) * | 2001-12-19 | 2003-07-09 | Bristol-Myers Squibb Company | Fused heterocyclic compounds and analogs thereof: modulators of nuclear hormone receptor function |
| CN100418965C (en) * | 2003-08-15 | 2008-09-17 | 万有制药株式会社 | imidazopyridine derivatives |
| EP1812798A2 (en) * | 2004-11-19 | 2007-08-01 | Acadia Pharmaceuticals Inc. | Enabling tools to identify ligands for hormone nuclear receptors |
-
2007
- 2007-11-16 WO PCT/US2007/085027 patent/WO2008064136A2/en not_active Ceased
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011012897A1 (en) | 2009-07-31 | 2011-02-03 | Astrazeneca Ab | New combinations for the treatment of asthma |
| WO2011026912A3 (en) * | 2009-09-04 | 2011-08-25 | Ernst-Moritz-Arndt-Universität Greifswald | Retinoic acid receptor antagonists, mir-10a inhibitors and inhibitors of hoxb1 or hoxb3 repressors for treating pancreatic cancer |
| EP2329817A1 (en) * | 2009-09-04 | 2011-06-08 | Ernst-Moritz-Arndt-Universität Greifswald | Retinoic acid receptor antagonists, miR-10a inhibitors and inhibitors of HOXB1 or HOXB3 repressors for treating pancreatic cancer |
| US9512407B2 (en) | 2009-09-07 | 2016-12-06 | Genome Research Limited | Cells and methods for obtaining them |
| WO2011061527A1 (en) | 2009-11-17 | 2011-05-26 | Astrazeneca Ab | Combinations comprising a glucocorticoid receptor modulator for the treatment of respiratory diseases |
| WO2012085583A1 (en) | 2010-12-23 | 2012-06-28 | Astrazeneca Ab | New compound |
| WO2012085582A1 (en) | 2010-12-23 | 2012-06-28 | Astrazeneca Ab | Compound |
| WO2013192301A1 (en) * | 2012-06-22 | 2013-12-27 | The Board Of Trustees Of The Leland Stanford Junior University | Imidazo bicyclic imminium compounds as antitumor agents |
| US9611276B2 (en) | 2012-06-22 | 2017-04-04 | The Board Of Trustees Of The Leland Stanford Junior University | Imidazo bicyclic iminium compounds as antitumor agents |
| US20160289228A1 (en) * | 2013-12-09 | 2016-10-06 | Ucb Biopharma Sprl | Tetrahydroimidazopyridine Derivatives as Modulators of TNF Activity |
| US9969728B2 (en) * | 2013-12-09 | 2018-05-15 | Ucb Biopharma Sprl | Tetrahydroimidazopyridine derivatives as modulators of TNF activity |
| WO2015109231A1 (en) | 2014-01-17 | 2015-07-23 | Cornell University | Methods of treating metabolic syndrome related conditions using retinoic acid receptor agonists |
| US11160769B2 (en) | 2014-01-17 | 2021-11-02 | Cornell University | Methods of treating metabolic syndrome related conditions using retinoic acid receptor agonists |
| EP3563683A1 (en) | 2014-03-10 | 2019-11-06 | Cornell University | Combination therapy for head and neck cancer |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2008064136A3 (en) | 2008-07-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2008064136A2 (en) | Compounds with activity at retinoic acid receptors | |
| EP2380881B1 (en) | Novel bicyclic heterocyclic compound | |
| JP6573704B2 (en) | Arylamide kinase inhibitor | |
| EP2720546B1 (en) | Spiro-cyclic trpv4 antagonists | |
| BR112020000863A2 (en) | MAGL INHIBITORS | |
| CA2696725A1 (en) | Pyridine derivatives and methods of use thereof | |
| EP4332102A1 (en) | Isoquinolone compound and use thereof | |
| EP3388428B1 (en) | Five-membered heterocyclic amides wnt pathway inhibitor | |
| JP2024517498A (en) | Compound | |
| WO2022240966A1 (en) | Compounds and methods for yap/tead modulation and indications therefor | |
| WO2013169631A2 (en) | Wnt protein signalling inhibitors | |
| KR102725123B1 (en) | compositions and methods for treating neurodegenerative diseases | |
| WO2021202900A1 (en) | 1,6-naphthyridine compounds and methods for csk modulation and indications therefor | |
| CN104530038A (en) | Amide imidazole derivative and application thereof | |
| TW202432509A (en) | Methods and compounds useful for the preparation of orexin-2 receptor antagonists and levofloxacin with low impurities | |
| EP3041835B1 (en) | Liver x receptor (lxr) modulators | |
| EP4337207A1 (en) | Compounds and methods for yap/tead modulation and indications therefor | |
| AU2019251363B2 (en) | Morpholine derivates as inhibitors of Vps34 | |
| US8048877B2 (en) | Guanidine derivatives and their medical use | |
| WO2016064682A1 (en) | Selective inhibitors of constitutive androstane receptor | |
| CN117384170A (en) | Wnt pathway inhibitor compound | |
| EP4428121A1 (en) | Modulators of sortilin activity | |
| JP7713954B2 (en) | Compounds active against nuclear receptors | |
| EP4654969A1 (en) | Compounds and methods for yap/tead modulation and indications therefor | |
| WO2024159103A1 (en) | Compounds and methods for yap/tead modulation and indications therefor |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07864565 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 07864565 Country of ref document: EP Kind code of ref document: A2 |