WO2007085136A1 - 1,3-benzodioxolecyclopentene derivates, preparation process and medical uses thereof - Google Patents
1,3-benzodioxolecyclopentene derivates, preparation process and medical uses thereof Download PDFInfo
- Publication number
- WO2007085136A1 WO2007085136A1 PCT/CN2006/000373 CN2006000373W WO2007085136A1 WO 2007085136 A1 WO2007085136 A1 WO 2007085136A1 CN 2006000373 W CN2006000373 W CN 2006000373W WO 2007085136 A1 WO2007085136 A1 WO 2007085136A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- ring
- compound
- formula
- acetic acid
- benzodioxole
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
Definitions
- the present invention relates to a novel benzodioxole derivative capable of activating a human peroxisome proliferator-activated receptor (hPPAR), a process for preparing the same, a pharmaceutical composition comprising the above compound, and Use of the compounds for the manufacture of a medicament for the treatment and/or prevention of a hPPAR mediated disease or condition.
- hPPAR human peroxisome proliferator-activated receptor
- the peroxisome proliferator-activated receptor (abbreviated as PP AR) is a ligand-dependent transcription factor that belongs to the nuclear receptor superfamily equivalent to the glucocorticoid receptor, retinoic acid receptor, and thyroxine receptor.
- PPARs have been found to have three subtypes: alpha, gamma, and delta (also known as beta), which are encoded by different genes.
- PPAR y is divided into two isoforms due to different promoters and splicing methods: ⁇ , ⁇ 2 , which differ only in the terminal sequence (Vida Bu Puig, J. Cl in. Inves t. , 97: 2553-2561 , 1996).
- PPARs When activated by a specific small molecule, PPARs interact with the PPARs response element (PPRE) of the target gene promoter region to regulate expression of the gene. PPARs are important transcriptional regulators of glucose, lipids, and cholesterol metabolism in the body.
- PPRE PPARs response element
- PPARa is mainly expressed in tissues with very high catabolic activity in lipids such as brown adipose tissue and liver, followed by kidney, heart and skeletal muscle (Endocnnology, 1995, 137, 354). It can positively or negatively control genes involved in fatty acid metabolism and intracellular delivery (such as acyl-CoA synthetase, fatty acid-binding protein and lipoprotein lipase) and apolipoproteins (AI) involved in the metabolism of cholesterol and neutral lipids. , Al l , CI II) gene expression. ⁇ is mainly found in adipose tissue, and is also present in small amounts in skeletal muscle, liver, colon, retina, and immune system.
- PPAR is highly expressed in macrophages, including atherosclerotic foam cells. Its Among them, PPAR is mainly expressed in adipose tissue, while PPAR yi is found in various tissues and has the highest expression in kidney, intestine and heart. ⁇ major regulation involves adipocyte differentiation and expression of insulin-sensitive genes (L Lipid. Res., 1996, 37, 907). PPARS is widely distributed and expressed in many tissues, with the highest expression in the intestine, kidney and heart. Activation of PPARS has been shown to cause an increase in HDL levels, a decrease in LDL and VLDL levels.
- Thiazolidinediones such as troglitazone and rosiglitazone have been clinically shown to enhance insulin action in patients with type 2 diabetes and reduce serum glucose. Thiazolidinediones have been reported to be potent and selective activators of ⁇ and bind directly to ⁇ receptors (L M. Lehmann et al., J. Biol. Chem. 12953-12956, 270 (1995)).
- Firbrates have been widely used as therapeutic agents for hyperlipidemia, which can lower serum triglycerides (20-50%), LDLc (10-15%), and increase HDLc (10-15%). ).
- PPARa PPARa-activated PPARa
- Activation of PPARa causes transcription of enzymes that increase fatty acid catabolism and reduce the re-synthesis of fatty acids in the liver, causing triglyceride synthesis and reduced VLDL production/secretion.
- PPARo activation reduces the production of apoC-I I I.
- the reduction in apoC-I I I increases the clearance of VLDL (J. Auwerx et al, Atheroscleros i s, J59-S37, 124 (Suppl), (1996)).
- PPAR PPAR involves many biological processes and disease states, including hypercholesterolemia, dyslipidemia and diabetes. However, current drugs are not ideal due to toxic side effects, etc. Therefore, a safe and effective PPAR agonist is needed. Optionally activate a subtype or activate multiple subtypes simultaneously. Summary of the invention
- the object of the present invention is to find and develop a small molecule with PPAR agonistic activity Compound for the treatment of hPPAR-mediated diseases, risk factors or conditions such as dyslipidemia, hyperlipidemia, hypercholesterolemia, atherosclerosis, hyperglycemia, type 1 diabetes, type II diabetes, insulin resistance , Diabetes complications, glucose insufficiency, X syndrome, heart failure, cardiovascular disease, regulation of appetite and food absorption in patients with obesity, anorexia, bulimia and anorexia nervosa.
- diseases such as dyslipidemia, hyperlipidemia, hypercholesterolemia, atherosclerosis, hyperglycemia, type 1 diabetes, type II diabetes, insulin resistance , Diabetes complications, glucose insufficiency, X syndrome, heart failure, cardiovascular disease, regulation of appetite and food absorption in patients with obesity, anorexia, bulimia and anorexia nervosa.
- the invention provides a compound of formula I, a racemate or an optical isomer thereof, or a pharmaceutically acceptable salt or solvate thereof:
- a linear or branched alkyl group selected from H, dC 6 , optionally a benzyl group substituted by one or more on the phenyl ring, said substituent being selected from a linear or branched alkoxy group of ( ⁇ ) d - c a straight-chain or branched-chain alkyl group of 6, c 2 - C 6 alkenyl group, linear or branched, c 3 - C 6 cycloalkyl, halogen, nitrile, trifluoromethyl, trifluoromethoxy a Oxylate
- R 2 is selected from H, halogen, linear or branched alkyl of dC 6 , C 2 - C 6 straight or branched alkenyl, C 3 -cycloalkyl, 5- to 6-membered aromatic carbocyclic or aromatic a ring, the aromatic heterocyclic ring containing 1 to 3 hetero atoms selected from 0, S, N, and the aromatic carbocyclic ring or aromatic heterocyclic ring may be optionally substituted by 1 to 5 substituents selected from the group consisting of: , nitro, hydroxy, hydroxydecyl, trifluoromethyl, trifluoromethoxy, dC 6 straight or branched alkyl, C 2 -C 6 straight or branched alkenyl, Ci-C alkoxy , C 2 - (alkenyloxy, phenoxy, benzyloxy, carboxy or amino;
- m and n are each independently selected from 0, 1, 2, 3, 4, 5 and 6;
- A is an aromatic carbocyclic ring or an aromatic heterocyclic ring, wherein the ring may be a monocyclic ring, a bicyclic ring or a tricyclic ring; each ring is composed of 5 to 6 atoms, and the aromatic heterocyclic ring includes 1 to 4 selected from 0, N a hetero atom of S, and the aromatic carbocyclic or aromatic heterocyclic ring may be optionally substituted by 1 to 5 substituents selected from the group consisting of halogen, nitro, hydroxy, hydroxymethyl, trifluoromethyl, trifluoro Methoxy, d-C 6 straight or branched alkyl, C 2 -C 6 straight or branched alkenyl, d-alkoxy, C 2 -C 4 alkenoxy, phenoxy, benzyloxy Base, carboxyl or amino group;
- Ar 2 is selected from a H atom, an aromatic carbocyclic ring or an aromatic heterocyclic ring, wherein the ring may be a monocyclic ring, a bicyclic ring or a tricyclic ring; each ring is composed of 5 to 6 atoms, and the aromatic heterocyclic ring includes 1 to 4 a hetero atom selected from 0, N, S, and the aromatic carbocyclic or aromatic heterocyclic ring may be optionally substituted with 1 to 5 substituents selected from the group consisting of: 3 ⁇ 4, nitro, hydroxy, hydroxymethyl, trifluoromethyl, trifluoromethoxy, d-C 6 straight or branched alkyl, -( ⁇ straight or branched alkenyl, d-alkoxy Base, C 2 -C 4 alkenyloxy, phenoxy, benzyloxy, carboxy or amino.
- the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula (I) according to the invention, which comprises at least one compound of formula I, its racemate or optical isomer or a pharmaceutically acceptable salt or solvate thereof And one or more pharmaceutically acceptable carriers or excipients.
- the invention is also directed to a process for the preparation of a compound of formula I or a pharmaceutically acceptable salt or solvate thereof.
- the invention relates to the use of a compound of formula I for the manufacture of a medicament for the treatment and/or prevention of a hPPAR mediated disease or condition.
- the invention provides a method of treating and/or preventing a hPPAR mediated disease, risk factor or disorder comprising administering to a subject in need thereof a therapeutically and/or prophylactically effective amount of a compound of the invention.
- hPPAR-mediated diseases, risk factors or conditions described in the present invention include dyslipidemia, hyperlipidemia, hypercholesterolemia, atherosclerosis, hyperglycemia, type I diabetes, type II diabetes, insulin resistance, Diabetes complications, glucose insufficiency, X syndrome, heart failure, cardiovascular disease, regulation of appetite and food absorption in patients with obesity, anorexia, bulimia and anorexia nervosa.
- the invention provides a compound of formula I, a racemate or an optical isomer thereof, a pharmaceutically acceptable salt or solvate thereof:
- a straight or branched alkyl group selected from H, dC 6 , optionally substituted or substituted with a benzyl group on the phenyl ring, said substituent being selected from a linear or branched alkoxy group, d-c a straight-chain or branched-chain alkyl group, alkenyl group c 2 C 6 straight-chain or a branched chain, c 3 - cycloalkyl, halogen, nitrile, trifluoromethyl, trifluoromethoxy Yue group;
- R 2 is selected from H, halogen, d-C 6 linear or branched alkyl, -(: 6 straight or branched alkenyl, C 3 -cycloalkyl, 5- to 6-membered aromatic carbocyclic or aromatic A heterocyclic ring, the aromatic heterocyclic ring comprising 1 to 3 hetero atoms selected from 0, S, N, and the aromatic carbocyclic ring or aromatic heterocyclic ring may be optionally substituted with 1 to 5 substituents selected from the group consisting of: , 3 ⁇ 4, nitro, hydroxy, hydroxymethyl, trifluoromethyl, trifluoromethoxy, d-C 6 straight or branched alkyl, C 2 -C 6 straight or branched alkenyl, d - C 4 methoxy, 0 2 -( ⁇ -alkenyloxy, phenoxy, benzyloxy, carboxy or amino;
- n and n are each independently selected from 0, 1, 2, 3, 4, 5 and 6;
- Is an aromatic carbocyclic ring or an aromatic heterocyclic ring wherein the ring may be monocyclic, bicyclic or tricyclic; each ring is composed of 5-6 atoms, and the aromatic heterocyclic ring includes 1-4 selected from 0, N, a hetero atom of S, and the aromatic carbocyclic or aromatic heterocyclic ring may be optionally substituted by from 1 to 5 substituents selected from the group consisting of: ⁇ , nitro, hydroxy, hydroxymethyl, trifluoromethyl, trifluoromethyl Oxy, dC 6 straight or branched alkyl, C 2 -C 6 straight or branched alkenyl, oxy, 0!
- Ar 2 is selected from the group consisting of a halogen atom, an aromatic carbon ring or an aromatic heterocyclic ring, wherein the ring may be a monocyclic ring, a bicyclic ring or a tricyclic ring; each ring is composed of 5 to 6 atoms, and the aromatic heterocyclic ring includes 1 to 4 a hetero atom selected from 0, N, S, and the aromatic carbocyclic or aromatic heterocyclic ring may be optionally substituted with from 1 to 5 substituents selected from the group consisting of: ?3 ⁇ 4, nitro, hydroxy, hydroxy oxime , trifluoromethyl, trifluoromethoxy, straight or branched alkyl, C 2 - straight or branched alkenyl, d-oxy, C 2 -C 4 alkenyl, phenoxy, Benzyloxy, carboxy or amino.
- aromatic carbocyclic ring examples include, but are not limited to, benzene, naphthalene, anthracene, phenanthrene, 1, 3-benzodioxole, hydrazine, hydrazine, hydrazine.
- aromatic heterocycle examples include, but are not limited to, pyridine, pyrrole, furan, thiophene, pyrazole, imidazole, thiazole, oxazole, isoxazole, hydrazine, benzofuran, benzimidazole, hydrazine Azole, oxime, pyrimidine, pyrocoat, quinoline, isoquinoline, indole, phenoxazine, phenoxazine.
- the invention provides a compound represented by the formula ,, including the racemate or optical isomer thereof, or a pharmaceutically acceptable salt or solvate thereof:
- n are independently selected from 0, 1, 2, 3, 4, 5 and 6;
- A is an aromatic carbocyclic ring or an aromatic heterocyclic ring, wherein the ring may be a monocyclic ring, a silent ring or a tricyclic ring; each ring is composed of 5 to 6 atoms, and the aromatic heterocyclic ring includes 1-4 selected from 0, a hetero atom of N, S, and the aromatic carbocyclic or aromatic heterocyclic ring may be optionally substituted by 1 to 5 substituents selected from the group consisting of: halogen, nitro, hydroxy, hydroxymethyl, trifluoromethyl, three Fluoromethoxy, dC 6 straight or branched alkyl, C 2 -C 6 straight or branched alkenyl, d-alkoxy, C 2 - ( 4 alkenyloxy, phenoxy, benzyloxy , carboxyl or amino;
- Ar 2 is selected from a H atom, an aromatic carbocyclic ring or an aromatic heterocyclic ring, wherein the ring may be a monocyclic ring, a bicyclic ring or a tricyclic ring; each ring is composed of 5 to 6 atoms, and the aromatic heterocyclic ring includes 1 to 4 a hetero atom selected from 0, N, S, and the aromatic carbocyclic or aromatic heterocyclic ring may be optionally substituted with from 1 to 5 substituents selected from the group consisting of halogen, nitro, hydroxy, hydroxydecyl, trifluoro Methyl, trifluoromethoxy, dC 6 straight or branched alkyl, 0 2 -(: 6 straight or branched alkenyl, oxy, C 2 -C 4 alkenoxy, phenoxy, benzyl Oxy, carboxyl or amino.
- Preferred compounds of the invention include:
- the above compound of the formula I and / or formula II or a pharmaceutically acceptable salt or solvate thereof can be prepared by the following method, which comprises the steps of:
- X 2 , X 3 , n are as defined for formula I;
- Ar, Ar 2 , X 2 , X 3 , n have the same meanings as in Formula I.
- a , Ar 2 , X 3 , n are as defined in the formula I, and X is a halogen.
- a , Ar 2 , X 3 , n, R 4 , R 5 and R 6 have the same meanings as defined in the formula I, and X is a halogen.
- ⁇ ⁇ , Ar 2 , X 2 , X 3 , n and R 2 have the same meanings as defined in Formula I.
- ⁇ ⁇ , Ar 2 , X 2 , X 3 , n have the same meanings as defined in Formula II.
- the starting material in the first step is simply changed to the corresponding 4-substituted catechol; the thioether in the intermediate VI is oxidized to the sulfone or sulfoxide, and then the above method can be used.
- ⁇ ⁇ , Ar 2 , ⁇ 2 , ⁇ 3 , ⁇ are defined as the same formula I, X is halogen, preferably Br, Cl.
- the compound of the formula VI is deprotected with p-toluenesulfonic acid in a methanol/water reaction system, reacted at room temperature for 12-24 hours, concentrated, and separated by column (eluent: chloroform/methanol system) to give a white to yellow formula. VI I compound.
- a compound of the formula I wherein ra 1, ⁇ is S, 1 is H atom.
- ⁇ ⁇ , Ar 2 , X 2 , X 3 , n and R 3 have the same meanings as in Formula I.
- Ar ⁇ Ar 2 , X 2 , X 3 , n are as defined in the formula II.
- the compound of Formula I or Formula II is present in stereo Heart.
- a compound of formula I or formula II can be prepared using reactants in the form of a single enantiomer in all possible steps, or in the form of a single enantiomer. It is prepared by carrying out the reaction in the presence of a catalyst or by dissolving a mixture of stereoisomers by a conventional method. Some preferred methods include resolution using a microorganism, resolution of a diastereomeric salt formed with any usable acid such as mandelic acid, camphorsulfonic acid, tartaric acid, lactic acid, or the like, or splitting with a hand.
- Salts of diastereomers formed by alkaloids such as brac ine, cinchona alkaloids and derivatives thereof.
- alkaloids such as brac ine, cinchona alkaloids and derivatives thereof.
- a commonly used method is found in "Enant ioraers, Raceraates and Resolution” edited by Jaques et al. (Wiley Intersc ience, 1981).
- the compounds of the invention may also be used in the form of their pharmaceutically acceptable salts or solvates.
- the pharmaceutically acceptable salts of the compounds of formula I include the conventional salts formed with pharmaceutically acceptable inorganic or organic acids or inorganic or organic bases, and the acid addition salts of quaternary ammonium.
- suitable acid salts include hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, nitric acid, perchloric acid, fumaric acid, acetic acid, propionic acid, succinic acid, glycolic acid, formic acid, lactic acid, maleic acid, tartaric acid, Citric acid, citric acid, malonic acid, hydroxymaleic acid, phenylacetic acid, glutamic acid, benzoic acid, salicylic acid, fumaric acid, toluenesulfonic acid, methanesulfonic acid, naphthalene-2-sulfonic acid, a salt of benzenesulfonic acid, hydroxynaphthoic acid, hydroiodic acid, malic acid, citric acid or the like.
- acids such as oxalic, while not in themselves pharmaceutically acceptable, can be used in the preparation of salts useful as intermediates to obtain the compounds of the invention or pharmaceutically acceptable salts thereof.
- suitable base salts include sodium, lithium, bell, magnesium, aluminum, calcium, zinc, N,N,-dibenzylethylenediamine, chloroprocaine, choline, diethanolamine, ethylene Amine, N-decyl glucosamine and procaine salt.
- the present invention also encompasses prodrugs of the compounds of the invention which, upon administration, are chemically converted by metabolic processes and thereafter become active drugs.
- prodrugs are functional derivatives of the compounds of the invention which are readily converted into the body in vivo.
- the desired compound of formula (I) Conventional methods for the selection and preparation of suitable prodrug derivatives are described, for example, in "Des ign Of Prodrugs", H Bund Saard, El Sevier, ed., 1985.
- the invention also includes active metabolites of the compounds of the invention.
- the compounds of the invention activate the hPPAR receptor, and preferably, the compounds of the invention selectively activate the hPPARa receptor.
- the agonistic effect of the compound of the present invention on PPAR can be measured by the following method.
- the functional effects of transient transfection of compounds in 293-T cells were screened to determine their ability to activate PPAR isoforms.
- the effect of the receptor subtype on the transcriptional activity of the same target gene was compared using a pre-established chimeric receptor system, and R luc was used as an internal standard to reduce endogenous effects.
- the human PPARa, ⁇ ⁇ and PPARS ligand binding domains are each fused to the yeast transcription factor GAL4 DNA binding domain.
- the mammalian expression vector pM was ligated to construct three plasmids: pM-hPPARa/GAL4, pM-PPAIly/GAL4 and pM-PPARS/GAL4.
- the GAL4 DNA binding region is ligated to pB4-tk-luc' to constitute pB4-RES-tk-luc (a reporter gene for firefly luciferase containing a GAL4 DNA binding site). Transfection efficiency and endogenous effects were corrected using pRL-CMV-Rluc as an internal standard.
- the 293-T cells were seeded into 48-well plates at a cell density of 2-4xl 0 4 / well, culture was 10% defatted fetal calf serum (FCS) in phenol red-free 1640 medium without antibiotics. After 48 hours, the culture medium was changed to 5% defatted FCS phenol red-free antibiotic-free 1640 medium, and then three subtypes of pM-hPPAR/GAM, pB4-RES-tk-luc and pRL-CMV-, respectively. Three plasmids of Rluc were co-transfected into 293-T cells and administered 24 hours later. Then, the intensity of luciferase was measured 24 hours after administration. The results showed that the compounds of the present invention have strong agonistic effects on various subtypes of PPAR, and the agonistic effect on PPAR6 is particularly strong.
- FCS fetal calf serum
- compositions of the present invention comprise an effective amount of a compound of formula I according to the invention, or a pharmaceutically acceptable salt or hydrate thereof, and one or more suitable pharmaceutically acceptable carriers. It can be used for body Internal treatment and biocompatibility.
- the pharmaceutical compositions can be prepared in a variety of dosage forms as desired for different routes of administration.
- the pharmaceutically acceptable carrier includes, but is not limited to, an ion exchanger, alumina, aluminum stearate, lecithin, serum proteins such as human albumin, buffer substances such as phosphate, glycerin, sorbic acid, sorbic acid clock, Partial glyceride mixture of saturated vegetable fatty acids, water, salt or electrolyte, such as protamine sulfate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salt, colloidal silica, magnesium trisilicate, polyvinylpyrrolidone , Cellulose, Polyethylene Glycol, Sodium Carboxymethyl Cellulose, Polyacrylate, Beeswax, Lanolin.
- an ion exchanger alumina, aluminum stearate, lecithin
- serum proteins such as human albumin
- buffer substances such as phosphate, glycerin, sorbic acid, sorbic acid clock
- Partial glyceride mixture of saturated vegetable fatty acids such
- composition of the compound of the present invention can be administered in any of the following ways: oral, spray inhalation, rectal administration, nasal administration, buccal administration, topical administration, parenteral administration, such as subcutaneous, intravenous, intramuscular, intraperitoneal, sheath Intra, intraventricular, intrasternal, and intracranial injection or input, or by means of an explant reservoir.
- oral, intraperitoneal or intravenous administration is preferred.
- the compounds of the invention may be formulated in any orally acceptable form including, but not limited to, tablets, capsules, aqueous solutions or aqueous suspensions.
- the carrier used for the tablet generally includes lactose and corn starch, and a lubricant such as magnesium stearate may also be added.
- the diluent used in the capsule preparation generally comprises lactose and dried corn starch.
- Aqueous suspension formulations are usually prepared by admixing the active ingredient with a suitable emulsifier or suspension. If desired, some sweetening, aroma or coloring agents may be added to the above oral formulation.
- the compounds of the present invention can be formulated into different topical preparations according to different affected faces or organs.
- the form is specifically described as follows:
- the compound of the present invention can be formulated into a preparation form of a micronized suspension or solution, and the carrier used is an isotonic pH of sterile saline, which may or may not be added.
- Preservatives such as benzyl chloride alkoxide.
- the compound can also be formulated into a bone form such as vaseline.
- the compounds of the invention may be formulated in a suitable cartilage, lotion or cream formulation wherein the active ingredient is suspended or dissolved in one or more carriers.
- Carriers for cartilage preparations include, but are not limited to: mineral oil, liquid petrolatum, white petrolatum, propylene glycol, polyethylene oxide, polypropylene oxide, emulsifying wax and water; lotions or creams may be used including but not limited to: minerals Oil, sorbitan monostearate, Tween 60, cetyl esters wax, hexadecene aryl alcohol, 2-octyldodecanol, benzyl alcohol and water.
- the compounds of the present invention can also be administered in the form of a sterile injectable preparation, including sterile aqueous or oily suspension or sterile injection solutions.
- a sterile injectable preparation including sterile aqueous or oily suspension or sterile injection solutions.
- carriers and solvents which can be used include water, Ringer's solution and isotonic sodium chloride solution.
- sterilized, fixed oils may be employed as a solvent or suspension medium such as a monoglyceride or a diglyceride.
- the dosage and method of use of the compounds of the invention depends on a number of factors, including the age, weight, sex, natural health, nutritional status of the compound, the strength of the compound, the time of administration, the rate of metabolism, the severity of the condition, and Subjective judgment of the doctor.
- a preferred dosage is from 0.01 to 100 mg/kg body weight per day, wherein the optimal dose is from 5 mg/kg to 10 rag/kg body weight per day.
- the melting point of the compound was determined by a YRT-3 type melting point apparatus, and the temperature was not corrected.
- - NMR spectra were determined by a Bruker ARX 400 nuclear magnetic instrument.
- FAB mass spectra were determined by a Zabspec t high resolution magnetic mass spectrometer.
- the title compound was prepared from benzamide as a crude material.
- the title compound was prepared using p-bromobenzamide as a crude material.
- the title compound is based on piperonamide.
- reaction product of (II) is dissolved in 40 ml of 90% aqueous methanol, added After reacting 500 mg of p-toluenesulfonic acid at room temperature for 12-24 hours, concentrating, and separating by column (elution system: chloroform/methanol) to give pale yellow to yellow 4-[(2-aryl-4-mercaptothiazol-5-yl) )Methylthio] - 1, 2-catechol.
- Example 2 Using the preparation method of Example 1, the intermediate 17 was changed to the intermediate 20 to give 2-(5- ⁇ [4-methyl-2-(4-fluorophenyl)-l as a pale yellow amorphous solid. , 3-thiazole-5-yl]-methylthio ⁇ -l,3-benzodioxole)-acetic acid.
- Example 2 Using the preparation method of Example 1, the intermediate 17 was changed to the intermediate 21 to give 2-(5- ⁇ [4-methyl-2-(4-methoxyphenyl) as a pale yellow amorphous solid. -1,3-thiazol-5-yl]-indolyl ⁇ -1,3-benzodioxole)-acetic acid.
- Example 2 Using the preparation method of Example 1, the intermediate 17 was changed to the intermediate 22 to give a pale yellow amorphous solid of 2-(5- ⁇ [4-mercapto-2-(3,5-didecyloxy). Phenyl)-1,3-thiazole-5-yl]-methylthio ⁇ -1,3-benzodioxole)-acetic acid.
- Example 2 Using the preparation method of Example 1, the intermediate 17 was changed to the intermediate 26 to give 2-(5- ⁇ [4-indolyl-2-(thiophen-3-yl)-1) as a pale yellow amorphous solid. , 3-thiazole-5-yl]-indolyl ⁇ -1,3-benzodioxole)-acetic acid.
- Example 12 2-(5- ⁇ [4-Methyl-2-(1,3-benzodioxol-5-yl)- 1, 3 -thiazole-5-yl]-methylthio ⁇ -1,3-benzodioxole)-acetic acid, using the preparation method of Example 1, the intermediate 17 was changed to an intermediate
- Example 2 Using the preparation method of Example 1, the intermediate 17 was changed to the intermediate 29 to give 2-[5-(2- ⁇ 4-[(4-ethyl)phenyl]phenoxy) as a pale yellow amorphous solid. Base ⁇ ethylthio)- 1, 3-benzodioxole]-acetic acid.
- Example 2 Using the preparation method of Example 1, the intermediate 17 was changed to the intermediate 30 to give 2- ⁇ 5-[2-(carbazol-9-yl)ethylthio]-1 as a pale yellow amorphous solid. 3-benzodioxole ⁇ -acetic acid.
- the 293- T cells were seeded into 48-well plates at a cell density of 2-4xl 0 4 / well, culture was 10% defatted fetal calf serum (FCS) in phenol red-free 1640 medium without antibiotics. After 48 hours, the culture medium was changed to 5% defatted FCS phenol red-free antibiotic-free 1640 medium, and then three subtypes of pM-hPPAR/GAL4, pB4-RES-tk-luc and pRL-CMV-, respectively. Rluc three kinds of shield granules were co-transfected into 293-T cells, and administered 24 hours later.
- FCS fetal calf serum
- the double agonist GW409544 was used as a positive control for ⁇ and ⁇ subtypes, and GW501516 was used as a positive control for ⁇ subtype.
- the intensity of luciferase was measured 24 hours after administration, and the results were expressed as PPAR activation energy (value relative to control drug (100%)) and EC 5Q value for PPAR6, as shown in Tables 1 and 2, respectively.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Child & Adolescent Psychology (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description
苯并间二氧杂环戊烯衍生物及其 Benzodioxole derivatives and their
制备方法和医药用途 技术领域 Preparation method and medical use
本发明涉及新颖的能激活人类过氧化物酶体增殖因子活化受 体(简称 hPPAR)的苯并间二氧杂环戊烯衍生物, 这些化合物的制 备方法, 包含上述化合物的药物组合物, 以及所述化合物用于制 备治疗和 /或预防 hPPAR介导的疾病或病症的药物的用途。 背景技术 The present invention relates to a novel benzodioxole derivative capable of activating a human peroxisome proliferator-activated receptor (hPPAR), a process for preparing the same, a pharmaceutical composition comprising the above compound, and Use of the compounds for the manufacture of a medicament for the treatment and/or prevention of a hPPAR mediated disease or condition. Background technique
过氧化物酶体增殖因子活化受体(简称为 PP AR)是与糖皮质 激素受体、 维甲酸受体和甲状腺素受体等同属于核受体超家族的 配体依赖型转录因子。 迄今为止, 已发现 PPARs存在三种亚型: α、 γ和 δ (也称 β ) , 它们被不同的基因编码。 而且, PPAR y由 于启动子和拼接方式不同分为两种同工型: γι、 γ2, 两者仅 Ν端序 列不同(Vida卜 Puig, J. Cl in. Inves t. , 97: 2553-2561, 1996)。 当被特定小分子激活后, PPARs能与靶基因启动子区域的 PPARs 反应元件 (PPRE )相互作用, 从而调节该基因的表达。 PPARs 是 体内葡萄糖、 脂类、 胆固醇代谢的重要转录调节因子。 The peroxisome proliferator-activated receptor (abbreviated as PP AR) is a ligand-dependent transcription factor that belongs to the nuclear receptor superfamily equivalent to the glucocorticoid receptor, retinoic acid receptor, and thyroxine receptor. To date, PPARs have been found to have three subtypes: alpha, gamma, and delta (also known as beta), which are encoded by different genes. Moreover, PPAR y is divided into two isoforms due to different promoters and splicing methods: γι , γ 2 , which differ only in the terminal sequence (Vida Bu Puig, J. Cl in. Inves t. , 97: 2553-2561 , 1996). When activated by a specific small molecule, PPARs interact with the PPARs response element (PPRE) of the target gene promoter region to regulate expression of the gene. PPARs are important transcriptional regulators of glucose, lipids, and cholesterol metabolism in the body.
PPARa 主要在对脂类有极高分解代谢活性的組织如棕色脂肪 组织和肝脏中表达,其次是在肾、心脏、骨骼肌中(Endocnnology, 1995, 137, 354)。 它能正控制或负控制与脂肪酸代谢和胞内输送 有关的基因(例如酰基 CoA合成酶、脂肪酸结合蛋白质和脂蛋白脂 肪酶)以及与胆固醇和中性脂质的代谢有关的载脂蛋白(AI, Al l , CI I I)基因的表达。 ΡΡΑΙΙγ主要存在于脂肪組织中, 也少量存在于 骨骼肌、 肝脏、 结肠、 视网膜、 免疫系统中。 最近研究结果也提 示,其高度表达于巨噬细胞, 包括动脉粥样硬化的泡沫细胞中。其 中, PPAR 主要是在脂肪组织中专一性表达的, 而 PPARy i则在各 种组织中均有发现, 在肾、 肠和心脏表达最高。 ΡΡΑΙΙγ 主要调节 涉及脂肪细胞分化和胰岛素敏感性基因的表达(L Lipid. Res. , 1996, 37, 907)。 PPARS 分布广泛, 在许多组织中均有表达, 其 中肠、肾、心脏中表达最高。 已经证明, PPARS的激活可以引起 HDL 水平的增加、 LDL和 VLDL水平的降低。 PPARa is mainly expressed in tissues with very high catabolic activity in lipids such as brown adipose tissue and liver, followed by kidney, heart and skeletal muscle (Endocnnology, 1995, 137, 354). It can positively or negatively control genes involved in fatty acid metabolism and intracellular delivery (such as acyl-CoA synthetase, fatty acid-binding protein and lipoprotein lipase) and apolipoproteins (AI) involved in the metabolism of cholesterol and neutral lipids. , Al l , CI II) gene expression. ΡΡΑΙΙγ is mainly found in adipose tissue, and is also present in small amounts in skeletal muscle, liver, colon, retina, and immune system. Recent findings also suggest that it is highly expressed in macrophages, including atherosclerotic foam cells. Its Among them, PPAR is mainly expressed in adipose tissue, while PPAR yi is found in various tissues and has the highest expression in kidney, intestine and heart. ΡΡΑΙΙγ major regulation involves adipocyte differentiation and expression of insulin-sensitive genes (L Lipid. Res., 1996, 37, 907). PPARS is widely distributed and expressed in many tissues, with the highest expression in the intestine, kidney and heart. Activation of PPARS has been shown to cause an increase in HDL levels, a decrease in LDL and VLDL levels.
噻唑烷二酮类药物如曲格列酮、 罗格列酮在临床上显示可增 强 II型糖尿病患者的胰岛素作用, 降低血清葡萄糖。 已报道噻唑 烷二酮为 ΡΡΑΙ γ的有效和选择性的激活剂, 并直接结合到 ΡΡΑΙΙγ 受体(L M. Lehmann 等, J. Biol. Chem. 12953-12956 , 270 (1995) )。 Thiazolidinediones such as troglitazone and rosiglitazone have been clinically shown to enhance insulin action in patients with type 2 diabetes and reduce serum glucose. Thiazolidinediones have been reported to be potent and selective activators of ΡΡΑΙγ and bind directly to ΡΡΑΙΙγ receptors (L M. Lehmann et al., J. Biol. Chem. 12953-12956, 270 (1995)).
贝特类(f ibrates)药物一向被广泛用作高脂血症的治疗药, 可降低血清甘油三酯( 20-50 % )、 LDLc ( 10-15 % ) , 并增加 HDLc ( 10-15 % )。实验表明, f ibrates对血清脂质的作用是通过 PPARa 的激活作用介导的,参见例如 B . Stael s 等, Curr. Pharm. Des., 1-14, 3 (1), (1997)。 PPARa的激活引起增加脂肪酸分解代谢 和降低肝脏中脂肪酸再次合成(引起甘油三酯合成和 VLDL 产生 / 分泌减少)的酶的转录。 此外, PPARo激活降低 apoC-I I I的产生。 apoC-I I I (LPL活性的抑制剂)产生的减少增加了 VLDL的清除( J. Auwerx等, Atheroscleros i s, J59-S37, 124 (Suppl) , (1996) )。 Firbrates have been widely used as therapeutic agents for hyperlipidemia, which can lower serum triglycerides (20-50%), LDLc (10-15%), and increase HDLc (10-15%). ). Experiments have shown that the effect of f ibrates on serum lipids is mediated through the activation of PPARa, see for example B. Stael s et al, Curr. Pharm. Des., 1-14, 3 (1), (1997). Activation of PPARa causes transcription of enzymes that increase fatty acid catabolism and reduce the re-synthesis of fatty acids in the liver, causing triglyceride synthesis and reduced VLDL production/secretion. In addition, PPARo activation reduces the production of apoC-I I I. The reduction in apoC-I I I (inhibitor of LPL activity) increases the clearance of VLDL (J. Auwerx et al, Atheroscleros i s, J59-S37, 124 (Suppl), (1996)).
PPAR涉及到许多生物过程和疾病状态, 包括高胆固醇血症、 血脂异常和糖尿病等, 而目前的药物由于毒副作用等原因, 作用 并不是很理想, 因此需要一种安全有效的 PPAR激动药物, 它可选 择性的激活一个亚型, 或同时激活多个亚型。 发明内容 PPAR involves many biological processes and disease states, including hypercholesterolemia, dyslipidemia and diabetes. However, current drugs are not ideal due to toxic side effects, etc. Therefore, a safe and effective PPAR agonist is needed. Optionally activate a subtype or activate multiple subtypes simultaneously. Summary of the invention
本发明的目的是寻找并开发具有 PPAR 激动活性的小分子化 合物, 用来治疗 hPPAR介导的疾病、 危险因子或病症, 如血脂异 常、 高脂血症、 高胆固醇血症、 动脉粥样硬化、 高血糖、 I 型糖 尿病、 II型糖尿病、胰岛素抗性、糖尿病并发症、 耐糖功能不全、 X 综合征、 心力衰竭、 心血管症、 患有如肥胖症、 厌食症、 贪食 症和神经性厌食症患者的食欲和食物吸收的调节。 The object of the present invention is to find and develop a small molecule with PPAR agonistic activity Compound for the treatment of hPPAR-mediated diseases, risk factors or conditions such as dyslipidemia, hyperlipidemia, hypercholesterolemia, atherosclerosis, hyperglycemia, type 1 diabetes, type II diabetes, insulin resistance , Diabetes complications, glucose insufficiency, X syndrome, heart failure, cardiovascular disease, regulation of appetite and food absorption in patients with obesity, anorexia, bulimia and anorexia nervosa.
本发明已经发现通式 I的化合物可以用于治疗或预防 hPPAR 介导的多种疾病、 危险因子或病症。 The present inventors have discovered that compounds of formula I are useful in the treatment or prevention of a variety of diseases, risk factors or conditions mediated by hPPAR.
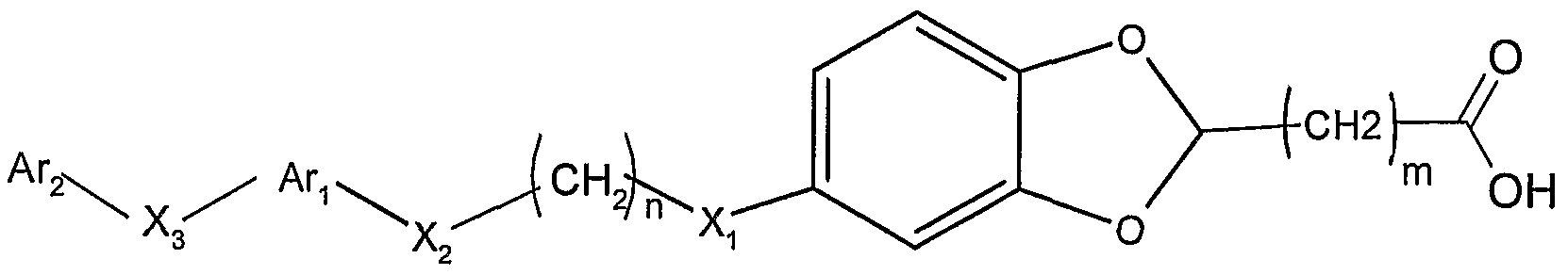
因此, 一方面, 本发明提供通式 I化合物, 其消旋体或旋光 异构体或其药学上可接受的盐和溶剂化物: Thus, in one aspect, the invention provides a compound of formula I, a racemate or an optical isomer thereof, or a pharmaceutically acceptable salt or solvate thereof:

选自 H, d-C6的直链或支链烷基, 任选在苯环上被一或多 取代的苄基,所述的取代基选自 (^- 的直链或支链烷氧基、 d - c6的直链或支链烷基、 c2 - c6直链或支链的烯基、 c3 - c6的环烷基、 卤素、 腈基、 三氟甲基、 三氟甲氧基; a linear or branched alkyl group selected from H, dC 6 , optionally a benzyl group substituted by one or more on the phenyl ring, said substituent being selected from a linear or branched alkoxy group of (^) d - c a straight-chain or branched-chain alkyl group of 6, c 2 - C 6 alkenyl group, linear or branched, c 3 - C 6 cycloalkyl, halogen, nitrile, trifluoromethyl, trifluoromethoxy a Oxylate
R2选自 H, 卤素, d-C6的直链或支链烷基, C2- C6直链或支 链烯基, C3- 环烷基, 5 - 6元的芳香碳环或芳香杂环, 所述芳 香杂环包含 1 - 3个选自 0, S, N的杂原子,并且所述芳香碳环或 芳香杂环可任选被 1 - 5个选自下面的取代基取代: 素, 硝基, 羟基, 羟曱基, 三氟甲基, 三氟甲氧基, d-C6直链或支链烷基, C2-C6直链或支链烯基, Ci-C烷氧基, C2- (^烯氧基, 苯氧基, 苄氧基, 羧基或氨基; R 2 is selected from H, halogen, linear or branched alkyl of dC 6 , C 2 - C 6 straight or branched alkenyl, C 3 -cycloalkyl, 5- to 6-membered aromatic carbocyclic or aromatic a ring, the aromatic heterocyclic ring containing 1 to 3 hetero atoms selected from 0, S, N, and the aromatic carbocyclic ring or aromatic heterocyclic ring may be optionally substituted by 1 to 5 substituents selected from the group consisting of: , nitro, hydroxy, hydroxydecyl, trifluoromethyl, trifluoromethoxy, dC 6 straight or branched alkyl, C 2 -C 6 straight or branched alkenyl, Ci-C alkoxy , C 2 - (alkenyloxy, phenoxy, benzyloxy, carboxy or amino;
m和 n各自独立地选自 0, 1, 2, 3, 4, 5和 6; Xi、 X2和 X3各自独立地不存在或为 0、 S、 NR3、 NR4C(=0)、 S0、 S(0) 2, C0、 C(=N-0R5)、 CH(0R6)、 (CH2)Y、 C=C、 C≡C,其中, R3、 R4、 R5和 R6各自独立地选自氢原子或 d- C6的直链或支链烷基, γ 选自 1, 2和 3; m and n are each independently selected from 0, 1, 2, 3, 4, 5 and 6; Xi, X 2 and X 3 are each independently absent or are 0, S, NR 3 , NR 4 C(=0), S0, S(0) 2 , C0, C(=N-0R 5 ), CH ( 0R 6 ), (CH 2 ) Y , C=C, C≡C, wherein R 3 , R 4 , R 5 and R 6 are each independently selected from a hydrogen atom or a linear or branched alkane of d-C 6 Base, γ is selected from 1, 2 and 3;
A 为芳香碳环或芳香杂环, 其中的环可以是单环、 双环或三 环; 每个环由 5 - 6个原子组成, 所述芳香杂环中包括 1 - 4个选 自 0、 N、 S的杂原子,并且所述芳香碳环或芳香杂环可任选被 1 - 5 个选自下面的取代基取代: 卤素, 硝基, 羟基, 羟曱基, 三氟 甲基, 三氟甲氧基, d- C6直链或支链烷基, C2- C6直链或支链烯 基, d- 烷氧基, C2-C4烯氧基, 苯氧基, 苄氧基, 羧基或氨基;A is an aromatic carbocyclic ring or an aromatic heterocyclic ring, wherein the ring may be a monocyclic ring, a bicyclic ring or a tricyclic ring; each ring is composed of 5 to 6 atoms, and the aromatic heterocyclic ring includes 1 to 4 selected from 0, N a hetero atom of S, and the aromatic carbocyclic or aromatic heterocyclic ring may be optionally substituted by 1 to 5 substituents selected from the group consisting of halogen, nitro, hydroxy, hydroxymethyl, trifluoromethyl, trifluoro Methoxy, d-C 6 straight or branched alkyl, C 2 -C 6 straight or branched alkenyl, d-alkoxy, C 2 -C 4 alkenoxy, phenoxy, benzyloxy Base, carboxyl or amino group;
Ar2选自 H原子、芳香碳环或芳香杂环,其中的环可以是单环、 双环或三环; 每个环由 5- 6个原子组成, 所述芳香杂环中包括 1 - 4个选自 0、 N、 S的杂原子,并且所述芳香碳环或芳香杂环可任 选被 1 - 5个选自下面的取代基取代: !¾素,硝基,羟基,羟甲基, 三氟甲基, 三氟甲氧基, d- C6直链或支链烷基, -(^直链或支 链烯基, d- 烷氧基, C2- C4烯氧基, 苯氧基, 苄氧基, 羧基或 氨基。 Ar 2 is selected from a H atom, an aromatic carbocyclic ring or an aromatic heterocyclic ring, wherein the ring may be a monocyclic ring, a bicyclic ring or a tricyclic ring; each ring is composed of 5 to 6 atoms, and the aromatic heterocyclic ring includes 1 to 4 a hetero atom selected from 0, N, S, and the aromatic carbocyclic or aromatic heterocyclic ring may be optionally substituted with 1 to 5 substituents selected from the group consisting of: 3⁄4, nitro, hydroxy, hydroxymethyl, trifluoromethyl, trifluoromethoxy, d-C 6 straight or branched alkyl, -(^ straight or branched alkenyl, d-alkoxy Base, C 2 -C 4 alkenyloxy, phenoxy, benzyloxy, carboxy or amino.
另一方面,本发明提供包含本发明通式( I )化合物的药用组合 物,其含有至少一种通式 I化合物、其消旋体或旋光异构体或其可 药用盐、 溶剂化物, 以及一种或多种药用载体或赋形剂。 In another aspect, the invention provides a pharmaceutical composition comprising a compound of formula (I) according to the invention, which comprises at least one compound of formula I, its racemate or optical isomer or a pharmaceutically acceptable salt or solvate thereof And one or more pharmaceutically acceptable carriers or excipients.
另一方面, 本发明还涉及制备通式 I化合物或者其可药用盐 或溶剂化物的方法。 In another aspect, the invention is also directed to a process for the preparation of a compound of formula I or a pharmaceutically acceptable salt or solvate thereof.
在又一方面,本发明涉及通式 I化合物用于制备治疗和 /或预 防 hPPAR介导的疾病或病症的药物的用途。 In yet another aspect, the invention relates to the use of a compound of formula I for the manufacture of a medicament for the treatment and/or prevention of a hPPAR mediated disease or condition.
在又一方面, 本发明提供了治疗和 /或预防 hPPAR介导的疾 病、 危险因子或病症的方法,包括给予有此需要的对象治疗和 /或 预防有效量的本发明化合物。 本发明中所述的 hPPAR介导的疾病、 危险因子或病症包括血 脂异常、 高脂血症、 高胆固醇血症、 动脉粥样硬化、 高血糖、 I 型糖尿病、 II型糖尿病、 胰岛素抗性、 糖尿病并发症、 耐糖功能 不全、 X综合征、 心力衰竭、 心血管症、 患有如肥胖症、 厌食症、 贪食症和神经性厌食症患者的食欲和食物吸收的调节。 In a further aspect, the invention provides a method of treating and/or preventing a hPPAR mediated disease, risk factor or disorder comprising administering to a subject in need thereof a therapeutically and/or prophylactically effective amount of a compound of the invention. The hPPAR-mediated diseases, risk factors or conditions described in the present invention include dyslipidemia, hyperlipidemia, hypercholesterolemia, atherosclerosis, hyperglycemia, type I diabetes, type II diabetes, insulin resistance, Diabetes complications, glucose insufficiency, X syndrome, heart failure, cardiovascular disease, regulation of appetite and food absorption in patients with obesity, anorexia, bulimia and anorexia nervosa.
在本发明的一个实施方式中, 本发明提供了通式 I化合物、 其消旋体或旋光异构体、 其可药用盐或溶剂化物: In one embodiment of the invention, the invention provides a compound of formula I, a racemate or an optical isomer thereof, a pharmaceutically acceptable salt or solvate thereof:

I I
^选自 H, d-C6的直链或支链烷基, 任选在苯环上被一或多 取代的苄基,所述的取代基选自 直链或支链烷氧基、 d- c6的直链或支链烷基、 c2一 c6直链或支链的烯基、 c3 - 的环烷基、 卤素、 腈基、 三氟甲基、 三氟曱氧基; a straight or branched alkyl group selected from H, dC 6 , optionally substituted or substituted with a benzyl group on the phenyl ring, said substituent being selected from a linear or branched alkoxy group, d-c a straight-chain or branched-chain alkyl group, alkenyl group c 2 C 6 straight-chain or a branched chain, c 3 - cycloalkyl, halogen, nitrile, trifluoromethyl, trifluoromethoxy Yue group;
R2选自 H, 卤素, d- C6的直链或支链烷基, -(:6直链或支 链烯基, C3- 环烷基, 5- 6元的芳香碳环或芳香杂环, 所述芳 香杂环包含 1- 3 个选自 0, S, N的杂原子,并且所述芳香碳环或 芳香杂环可任选被 1-5个选自下面的取代基取代: )¾素, 硝基, 羟基, 羟甲基, 三氟甲基, 三氟甲氧基, d- C6直链或支链烷基, C2-C6直链或支链烯基, d- C4垸氧基, 02-(^烯氧基, 苯氧基, 苄氧基, 羧基或氨基; R 2 is selected from H, halogen, d-C 6 linear or branched alkyl, -(: 6 straight or branched alkenyl, C 3 -cycloalkyl, 5- to 6-membered aromatic carbocyclic or aromatic A heterocyclic ring, the aromatic heterocyclic ring comprising 1 to 3 hetero atoms selected from 0, S, N, and the aromatic carbocyclic ring or aromatic heterocyclic ring may be optionally substituted with 1 to 5 substituents selected from the group consisting of: , 3⁄4, nitro, hydroxy, hydroxymethyl, trifluoromethyl, trifluoromethoxy, d-C 6 straight or branched alkyl, C 2 -C 6 straight or branched alkenyl, d - C 4 methoxy, 0 2 -(^-alkenyloxy, phenoxy, benzyloxy, carboxy or amino;
m和 n各自独立地选自 0, 1, 2, 3, 4, 5和 6; m and n are each independently selected from 0, 1, 2, 3, 4, 5 and 6;
、 X2和 X3各自独立地不存在或为 0、 S、 NR3、 NR4C(=0)、 S0、 S(0)2、 C0、 C(=N-0R5)、 CH(0R6)、 (CH2)Y、 C=C、 C≡C,其中, R3、 R4、 R5和 R6各自独立地选自氢原子或 d- C6的直链或支链烷基, Y 选自 1, 2和 3; , X 2 and X 3 are each independently absent or are 0, S, NR 3 , NR 4 C (=0), S0, S(0) 2 , C0, C(=N-0R 5 ), CH(0R 6 ), (CH 2 ) Y , C=C, C≡C, where R 3 , R 4 , R 5 and R 6 is each independently selected from a hydrogen atom or a linear or branched alkyl group of d-C 6 , and Y is selected from 1, 2 and 3;
为芳香碳环或芳香杂环, 其中的环可以是单环、 双环或三 环; 每个环由 5- 6个原子组成, 所述芳香杂环中包括 1-4个选 自 0、 N、 S的杂原子,并且所述芳香碳环或芳香杂环可任选被 1- 5 个选自下面的取代基取代: 素, 硝基, 羟基, 羟甲基, 三氟 甲基, 三氟甲氧基, d-C6直链或支链烷基, C2- C6直链或支链烯 基, 氧基, 0!2-(4烯氧基, 苯氧基, 苄氧基, 羧基或氨基; Ar2选自 Η原子、芳香碳环或芳香杂环,其中的环可以是单环、 双环或三环; 每个环由 5- 6个原子组成, 所述芳香杂环中包括 1 -4个选自 0、 N、 S的杂原子,并且所述芳香碳环或芳香杂环可任 选被 1-5个选自下面的取代基取代: !¾素,硝基,羟基,.羟曱基, 三氟曱基, 三氟甲氧基, 直链或支链烷基, C2- 直链或支 链烯基, d- 氧基, C2-C4烯氧基, 苯氧基, 苄氧基, 羧基或 氨基。 Is an aromatic carbocyclic ring or an aromatic heterocyclic ring, wherein the ring may be monocyclic, bicyclic or tricyclic; each ring is composed of 5-6 atoms, and the aromatic heterocyclic ring includes 1-4 selected from 0, N, a hetero atom of S, and the aromatic carbocyclic or aromatic heterocyclic ring may be optionally substituted by from 1 to 5 substituents selected from the group consisting of: 素, nitro, hydroxy, hydroxymethyl, trifluoromethyl, trifluoromethyl Oxy, dC 6 straight or branched alkyl, C 2 -C 6 straight or branched alkenyl, oxy, 0! 2 -( 4 alkenyloxy, phenoxy, benzyloxy, carboxy or amino Ar 2 is selected from the group consisting of a halogen atom, an aromatic carbon ring or an aromatic heterocyclic ring, wherein the ring may be a monocyclic ring, a bicyclic ring or a tricyclic ring; each ring is composed of 5 to 6 atoms, and the aromatic heterocyclic ring includes 1 to 4 a hetero atom selected from 0, N, S, and the aromatic carbocyclic or aromatic heterocyclic ring may be optionally substituted with from 1 to 5 substituents selected from the group consisting of: ?3⁄4, nitro, hydroxy, hydroxy oxime , trifluoromethyl, trifluoromethoxy, straight or branched alkyl, C 2 - straight or branched alkenyl, d-oxy, C 2 -C 4 alkenyl, phenoxy, Benzyloxy, carboxy or amino.
根据本发明,术语 "芳香碳环" 的实例包括但不限于苯、 萘、 蒽、 菲、 1, 3-苯并间二氧杂环戊烯、 茚、 芴、 苊。 According to the present invention, examples of the term "aromatic carbocyclic ring" include, but are not limited to, benzene, naphthalene, anthracene, phenanthrene, 1, 3-benzodioxole, hydrazine, hydrazine, hydrazine.
根据本发明,术语 "芳香杂环"的实例包括但不限于吡啶、 吡 咯、 呋喃、 噻吩、 吡唑、 咪唑、 噻唑、 喁唑、 异喁唑、 吲哚、 苯 并呋喃、 苯并咪唑、 咔唑、 哒療、 嘧啶、 吡漆、 喹啉、 异喹啉、 嘌呤、 吩塞嗪、 吩喁嗪。 According to the invention, examples of the term "aromatic heterocycle" include, but are not limited to, pyridine, pyrrole, furan, thiophene, pyrazole, imidazole, thiazole, oxazole, isoxazole, hydrazine, benzofuran, benzimidazole, hydrazine Azole, oxime, pyrimidine, pyrocoat, quinoline, isoquinoline, indole, phenoxazine, phenoxazine.
在本发明的一个优选实施方式中,本发明提供了通式 Π所代 表的化合物, 包括其消旋体或旋光异构体或其可药用盐或溶剂化 物:
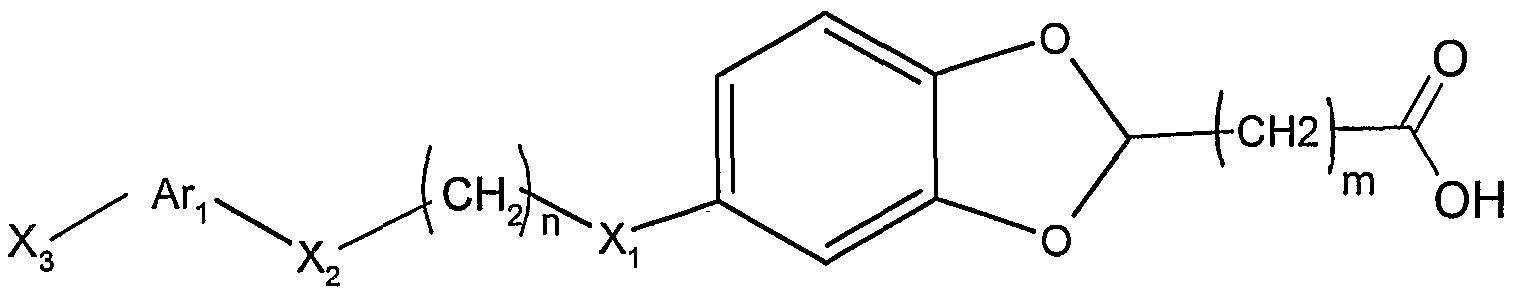
II II
其中: among them:
m、 n独立的选自 0, 1, 2, 3, 4, 5和 6; m, n are independently selected from 0, 1, 2, 3, 4, 5 and 6;
、 X2和 X3各自独立地不存在或为 0、 S、 NR3、 NR4C(-0)、 S0、 S(0)2、 C0、 C(=N-0R5)、 CH(0R6)、 (CH2)Y、 C=C、 C≡C,其中, R3、 R4、 R5和 R6各自独立地选自氢原子或 d- C6的直链或支链烷基, Y 选自 1, 2和 3; , X 2 and X 3 are each independently absent or are 0, S, NR 3 , NR 4 C(-0), S0, S(0) 2 , C0, C(=N-0R 5 ), CH(0R 6 ), (CH 2 ) Y , C=C, C≡C, wherein R 3 , R 4 , R 5 and R 6 are each independently selected from a hydrogen atom or a linear or branched alkyl group of d-C 6 , Y is selected from 1, 2 and 3;
A 为芳香碳环或芳香杂环, 其中的环可以是单环、 默环或三 环; 每个环由 5- 6个原子组成, 所述芳香杂环中包括 1-4个选 自 0、 N、 S的杂原子,并且所述芳香碳环或芳香杂环可任选被 1- 5 个选自下面的取代基取代: 卤素, 硝基, 羟基, 羟甲基, 三氟 甲基, 三氟甲氧基, d-C6直链或支链烷基, C2-C6直链或支链烯 基, d- 烷氧基, C2- (4烯氧基, 苯氧基, 苄氧基, 羧基或氨基;A is an aromatic carbocyclic ring or an aromatic heterocyclic ring, wherein the ring may be a monocyclic ring, a silent ring or a tricyclic ring; each ring is composed of 5 to 6 atoms, and the aromatic heterocyclic ring includes 1-4 selected from 0, a hetero atom of N, S, and the aromatic carbocyclic or aromatic heterocyclic ring may be optionally substituted by 1 to 5 substituents selected from the group consisting of: halogen, nitro, hydroxy, hydroxymethyl, trifluoromethyl, three Fluoromethoxy, dC 6 straight or branched alkyl, C 2 -C 6 straight or branched alkenyl, d-alkoxy, C 2 - ( 4 alkenyloxy, phenoxy, benzyloxy , carboxyl or amino;
Ar2选自 H原子、芳香碳环或芳香杂环,其中的环可以是单环、 双环或三环; 每个环由 5- 6个原子组成, 所述芳香杂环中包括 1 -4个选自 0、 N、 S的杂原子,并且所述芳香碳环或芳香杂环可任 选被 1-5个选自下面的取代基取代: 卤素,硝基,羟基,羟曱基, 三氟甲基, 三氟曱氧基, d-C6直链或支链烷基, 02-(:6直链或支 链烯基, 氧基, C2-C4烯氧基, 苯氧基, 苄氧基, 羧基或 氨基。 Ar 2 is selected from a H atom, an aromatic carbocyclic ring or an aromatic heterocyclic ring, wherein the ring may be a monocyclic ring, a bicyclic ring or a tricyclic ring; each ring is composed of 5 to 6 atoms, and the aromatic heterocyclic ring includes 1 to 4 a hetero atom selected from 0, N, S, and the aromatic carbocyclic or aromatic heterocyclic ring may be optionally substituted with from 1 to 5 substituents selected from the group consisting of halogen, nitro, hydroxy, hydroxydecyl, trifluoro Methyl, trifluoromethoxy, dC 6 straight or branched alkyl, 0 2 -(: 6 straight or branched alkenyl, oxy, C 2 -C 4 alkenoxy, phenoxy, benzyl Oxy, carboxyl or amino.
本发明优选的化合物包括: Preferred compounds of the invention include:
2-{5- [(4-甲基 -2 -苯基 - 1, 3-噻唑 -5-基) -甲硫基] -1, 3-苯并间二 氧杂环戊烯 }-乙酸; 2-{5-[(4-methyl-2-phenyl- 1, 3-thiazol-5-yl)-methylthio]-1,3-benzodioxole}-acetic acid;
2- (5- { [4-曱基 -2- (4-三氟甲基苯基) -1, 3-噻唑 -5-基] -甲硫 基}-1, 3-苯并间二氧杂环戊烯) -乙酸; 2-(5- { [4-mercapto-2-(4-trifluoromethylphenyl)-1,3-thiazol-5-yl]-methyl sulfide }}-1,3-benzodioxole)-acetic acid;
2-(5- {[4-曱基 -2- (4-溴苯基)-1, 3-噻唑 -5-基] -甲硫基}- 1, 3- 苯并间二氧杂环戊烯) -乙酸; 2-(5- {[4-mercapto-2-(4-bromophenyl)-1,3-thiazol-5-yl]-methylthio}- 1, 3-benzodioxole Alkene)-acetic acid;
2-(5-{[4-甲基 -2-(4-氟苯基)-1, 3-噻唑- 5-基] -甲硫基}- 1, 3- 苯并间二氧杂环戊烯) -乙酸; 2-(5-{[4-methyl-2-(4-fluorophenyl)-1,3-thiazole-5-yl]-methylthio}- 1, 3-benzodioxole Alkene)-acetic acid;
2- (5- {[4-甲基- 2- (4-.甲氧基苯基) -1, 3-噻唑- 5-基] -甲硫 基}-1, 3-苯并间二氧杂环戊烯)-乙酸; 2-(5- {[4-Methyl-2-(4-.methoxyphenyl)-1,3-thiazole-5-yl]-methylthio}-1,3-benzodioxane Heterocyclopentene)-acetic acid;
2- (5- { [4-甲基 -2- (3, 5 -二甲氧基苯基) -1, 3-噻唑- 5-基] -甲硫 基}-1, 3-苯并间二氧杂环戊烯)-乙酸; 2-(5- { [4-methyl-2-(3,5-dimethoxyphenyl)-1,3-thiazole-5-yl]-methylthio}-1,3-benzophenan Dioxol)-acetic acid;
2- (5- {[4-甲基- 2-(2,4-二氯苯基)- 1, 3-噻唑- 5-基] -曱硫 基}- 1, 3 -苯并间二氧杂环戊烯)-乙酸; 2-(5- {[4-Methyl-2-(2,4-dichlorophenyl)- 1, 3-thiazole-5-yl]-indolyl}- 1, 3-phenyl-dioxy Heterocyclopentene)-acetic acid;
2 -(5- { [4-甲基- 2- (4-叔丁基苯基) -1, 3-噻唑- 5-基] -甲硫 基}-1, 3-苯并间二氧杂环戊烯)-乙酸; 2-(5- { [4-methyl-2-(4-tert-butylphenyl)-1,3-thiazole-5-yl]-methylthio}-1,3-benzodioxan Cyclopentene)-acetic acid;
2-(5- {[4-曱基 -2- (噻吩 -2-基)- 1, 3-噻唑- 5-基] -甲硫基}-1, 3- 苯并间二氧杂环戊烯) -乙酸; 2-(5- {[4-mercapto-2-(thiophen-2-yl)-1,3-thiazole-5-yl]-methylthio}-1,3-benzodioxole Alkene)-acetic acid;
2-(5- {[4-甲基- 2- (噻吩 -3-基) -1, 3 -噻唑 -5-基]-甲硫基 }-1, 3- 苯并间二氧杂环戊烯) -乙酸; 2-(5- {[4-Methyl-2-(thiophen-3-yl)-1,3-thiazol-5-yl]-methylthio}-1,3-benzodioxole Alkene)-acetic acid;
2-(5-{[4-甲基 -2- (萘- 2-基)- 1, 3 -噻唑 -5-基] -曱硫基}-1, 3 -苯 并间二氧杂环戊烯)-乙酸; 2-(5-{[4-methyl-2-(naphthalen-2-yl)-1,3-thiazol-5-yl]-indolyl}-1,3-benzodioxole Alkene)-acetic acid;
2-(5-{[4-甲基- 2- (1, 3-苯并间二氧杂环戊烯 -5-基)- 1, 3-噻唑 -5-基]-曱硫基 }- 1, 3-苯并间二氧杂环戊烯)-乙酸; 2-(5-{[4-Methyl-2-(1,3-benzodioxole-5-yl)-1,3-thiazol-5-yl]-indenyl}- 1, 3-benzodioxole)-acetic acid;
2- [5- (2- {4- [ (4-乙基)苯基]苯氧基 }乙硫基) -1, 3-苯并间二氧杂 环戊烯]-乙酸; 2-[5-(2- {4- [(4-ethyl)phenyl]phenoxy}ethylthio)-1,3-benzodioxancyclopentene]-acetic acid;
2- {5- [2- (咔唑 -9-基)乙硫基 ]-1, 3-苯并间二氧杂环戊烯 }-乙酸; 2- {5- [2- (吩噻嗪 -10-基)乙硫基 ]-1, 3 -苯并间二氧杂环戊烯} -乙 酸; 2-{5-[2-(carbazol-9-yl)ethylthio]-1,3-benzodioxole}-acetic acid; 2-{5-[2-(phenothiazine) -10-yl)ethylthio]-1,3-benzodioxole}-acetic acid;
或其可药用盐或溶剂化物。 根据本发明,上述通式 I和 /或通式 II化合物或其可药用盐或 溶剂化物可用以下方法制备, 其包括以下步驟: Or a pharmaceutically acceptable salt or solvate thereof. According to the present invention, the above compound of the formula I and / or formula II or a pharmaceutically acceptable salt or solvate thereof can be prepared by the following method, which comprises the steps of:
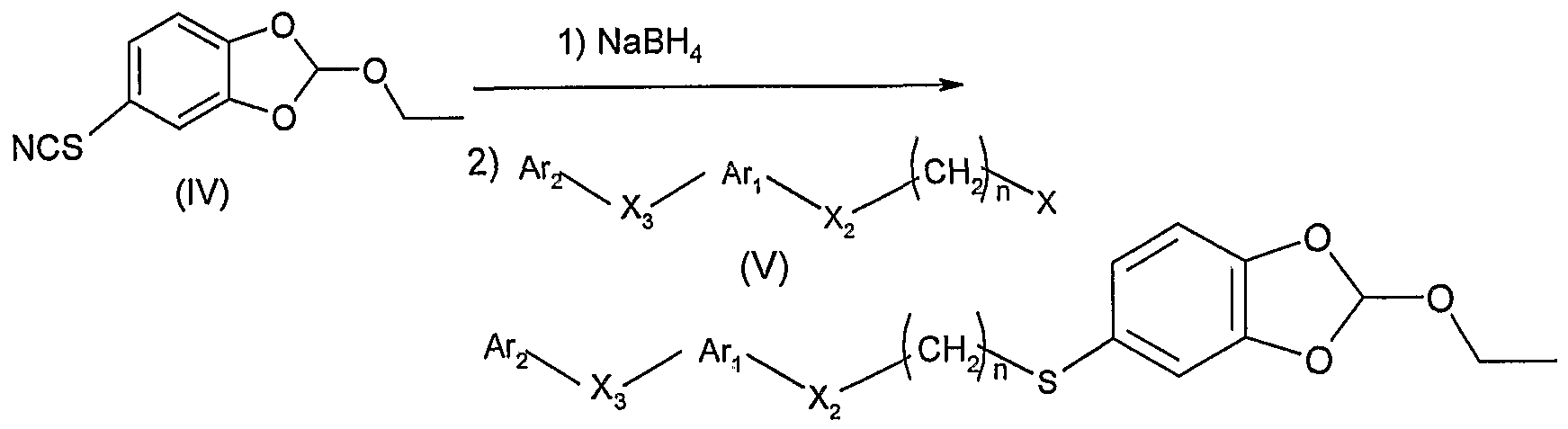
1) 在溴素和溴化钠存在下,将邻苯二酚与硫氰酸钠反应得到 式 III的化合物,将式 III的化合物与原甲酸三乙酯反应,得到式 IV的化合物;

III IV III IV
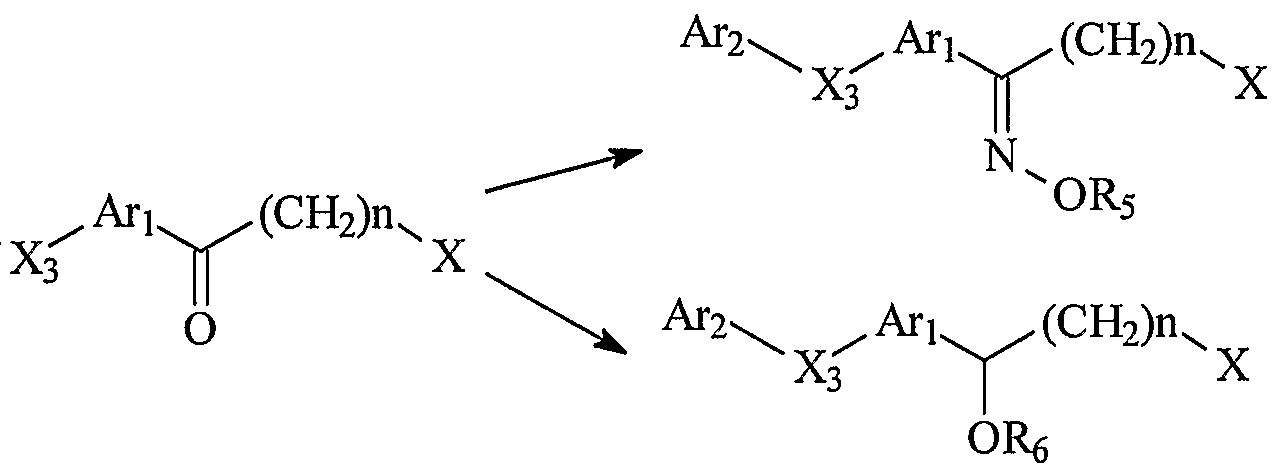
2) 将式 IV的化合物先用硼氢化钠还原, 接着与式 V的中间 体在碱、 例如氢氧化钠、 碳酸铯、 碳酸钾存在下反应,


VI VI
X2, X3, n的定义同通式 I所述; X 2 , X 3 , n are as defined for formula I;
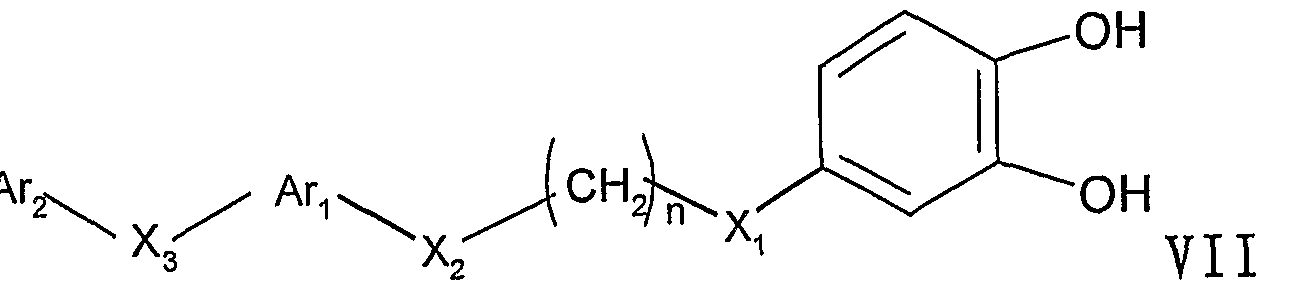
3)使式 VI的化合物在对甲苯磺酸存在下脱保护,得到 1是3 的式 VII化合物。 06000373 3) Deprotecting the compound of formula VI in the presence of p-toluenesulfonic acid to give a compound of formula VII wherein 1 is 3. 06000373
4) 使式 VII的化合物与式 VIII的化合物反应: 4) reacting a compound of formula VII with a compound of formula VIII:
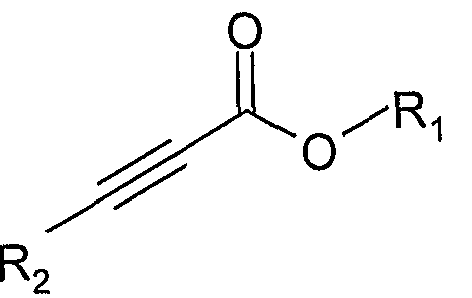
VIII VIII
其中, 1^和 R2如通式 I所定义, 但 Ri不为 H,得到其中 m=l、 不 为 H、 为 S的通式 I的化合物: Wherein 1^ and R 2 are as defined in formula I, but Ri is not H, obtaining a compound of formula I wherein m = 1, not H, and S:
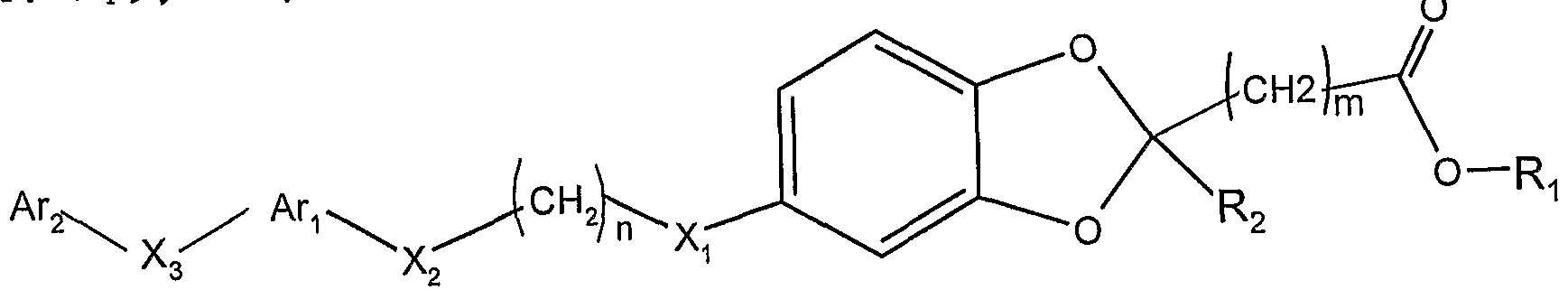
上述反应路线中用到的式 VIII 化合物的合成可参照: Tetrahedron Letters 1989, 30 (34), 4525- 4526中所述。 The synthesis of the compound of the formula VIII used in the above reaction scheme can be referred to: Tetrahedron Letters 1989, 30 (34), 4525-4526.
反应路线中用到的式 V中间体可按下列方法制备: The intermediate of formula V used in the reaction scheme can be prepared as follows:
a) 当 X2为 0, S, NR3, (0=)CNR "时, 可按下述方法制备式 V中间体 a) When X 2 is 0, S, NR 3 , (0=)CNR ", the intermediate of formula V can be prepared as follows
X 、χ X, χ
![]()
![]()

其中, A , Ar2, X3, n的定义同通式 I, X为卤素。 Wherein, A , Ar 2 , X 3 , n are as defined in the formula I, and X is a halogen.
c) 当 X2为 C0、 C(=N-0R5)、 CH(0R6)或- R4NC(=0)时, 可按下述方 法制备 c) When X 2 is C0, C(=N-0R 5 ), CH(0R 6 ) or - R 4 NC(=0), it can be prepared as follows
O O
X^义 X X^义 X
(CH2)r^ (CH 2 )r^
.An —— ^ Ar2 .An —— ^ Ar 2
χ3
、^ N

利用酰 对芳香环 Ar上的胺基进行酰化即可得 X2为 - R4NC (=0)的 式 V中间体。 The acylation of an amine group on the aromatic ring Ar with an acyl group gives an intermediate of the formula V wherein X 2 is - R 4 NC (=0).
其中, A , Ar2,X3,n,R4,R5,R6的定义同通式 I, X为卤素。 Wherein, A , Ar 2 , X 3 , n, R 4 , R 5 and R 6 have the same meanings as defined in the formula I, and X is a halogen.
d) 当 X2不存在或为(CH2)Y、 C=C、 C≡C 时, 则可根据实际情况选 用合适原料先合成 dl化合物, 再对 dl化合物中的羟基! ¾代即可
得式 V中间体。

其中, ^人^ ^!!的定义同通式 ^ X为卤素。 进一步地,可将上述得到的通式 I 的化合物与碱金属氢氧化 物反应或与稀盐酸、三氟醋酸等酸反应,经水解得到 m=l、 ^为 S、 !^为 H的通式 I的化合物: Among them, ^ people ^ ^! ! The definition is the same as the general formula ^ X is halogen. Further, the compound of the above formula I can be reacted with an alkali metal hydroxide or with an acid such as dilute hydrochloric acid or trifluoroacetic acid to obtain m = l and ^ as S, by hydrolysis. ^ Compound of formula I which is H:

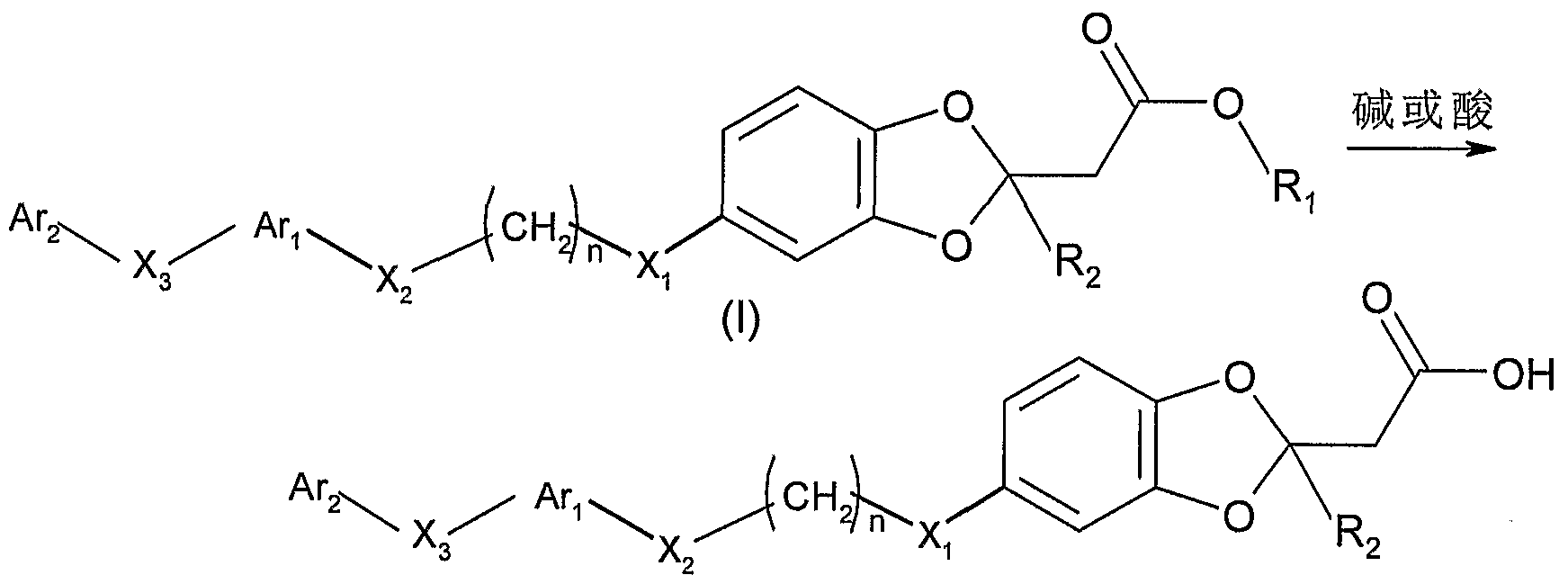
I I
其中,ΑΓι, Ar2, X2, X3, n和 R2的定义同通式 I所述。 Wherein, Α Γι , Ar 2 , X 2 , X 3 , n and R 2 have the same meanings as defined in Formula I.
上述水解产物中, 当 R2为 H时, 即是 m=l、 为 3的通式 II 的化合物

II II
其中, ΑΓι, Ar2, X2, X3, n的定义同通式 II所述。 Wherein, Α Γι , Ar 2 , X 2 , X 3 , n have the same meanings as defined in Formula II.
以上是 1是3的通式 I和 II化合物的制备方法, 当 是^ NR3、 NR,C (=0) (R3和 ^的定义同通式 I) 时仍可按上述方法制备 通式 I和 II化合物, 只需将步骤一中的起始原料 ΙΠ改为相应 4 -取代基的邻苯二酚;将中间体 VI中的硫醚氧化成砜或亚砜后再 按上述方法即可制备 是 S (=0)、 S (=0) 2的通式 I和 11化合物; 对于 是 )、 C(=N-0R5)、 CH(0R6)、 (CH2)Y、 C=C、 C≡C的通式
I和 I I化合物 (R5、 R6和 Y的定义同通式 I ) 的制备则只需先制 备式 VI I化合物, 此后合成路线同上。 具体而言,通式 I化合物的合成反应方案详见下列反应路线: 反应步骤一:

(I I I) (IV) (I I I) (IV)
将 Br2/NaBr的甲醇溶液緩慢滴加到邻苯二酚和硫氰酸钠的 0 。(:的曱醇反应液中, 0°C反应 3-4 小时后, 用水终止反应, 饱和 NaHC03水调至近中性, 乙酸乙酯提取, 干燥。 过滤, 浓缩后柱分 离(洗脱剂: 正己烷 /乙酸乙酯 /乙酸系统), 得白色固体的式 I I I 化合物。 将式 I I I化合物、 原甲酸三乙酯, 酸性离子交换树脂, 4人分子筛在无水苯中回流 72-96小时, 过滤, 浓缩, 柱分离 (硅 胶为常规柱层析用硅胶, 颗粒度 10 - 40μΐη; 洗脱剂: 正己烷 / 乙酸乙酯系统) , 得到浅黄色油状的式 IV化合物。 反应步骤二: A solution of Br 2 /NaBr in methanol was slowly added dropwise to 0 of catechol and sodium thiocyanate. (In the sterol reaction solution, after reacting at 0 ° C for 3-4 hours, the reaction was terminated with water, saturated NaHC0 3 water was adjusted to near neutral, ethyl acetate was extracted, dried. Filtration, concentration and column separation (eluent: Hexane/ethyl acetate/acetic acid system) gave the compound of formula III as a white solid. The compound of formula III, triethyl orthoformate, acidic ion exchange resin, 4 molecular sieves were refluxed in anhydrous benzene for 72-96 hours, filtered , Concentration, column separation (silica gel for conventional column chromatography with silica gel, particle size 10 - 40 μΐ; eluent: n-hexane / ethyl acetate system) to give the compound of formula IV as a pale yellow oil.
(VI) (VI)
其中, ΑΓι, Ar2, Χ2, Χ3, η的定义同通式 I, X为卤素, 优选 Br、 Cl。 Wherein, Α Γι , Ar 2 , Χ 2 , Χ 3 , η are defined as the same formula I, X is halogen, preferably Br, Cl.
将式 IV的化合物和硼氢化钠在乙醇中回流 10-15分钟后,冷 却, 加入碱(碳酸铯、 碳酸钾或氢氧化钠)后滴加式 V的化合物, 室温反应过夜或回溜 3-8小时, 过滤, 浓缩, 柱分离 (洗脱剂: 正己烷 /乙酸乙酯系统) , 得到黄色油状的式 VI化合物。 反应步骤三: The compound of the formula IV and sodium borohydride are refluxed in ethanol for 10-15 minutes, then cooled, and the compound of the formula V is added dropwise after adding a base (barium carbonate, potassium carbonate or sodium hydroxide), and the reaction is carried out overnight at room temperature or back to 3- After 8 hours, filtration, concentration, and column chromatography (eluent: hexane/ethyl acetate system) gave compound of formula VI as a yellow oil. Reaction step three:

(VI I) (VI I)
将式 VI 的化合物在甲醇 /水反应体系中, 用对甲苯磺酸脱保 护, 室温反应 12-24小时后, 浓缩, 柱分离 (洗脱剂: 氯仿 /甲醇 系统) , 得到白色至黄色的式 VI I化合物。 The compound of the formula VI is deprotected with p-toluenesulfonic acid in a methanol/water reaction system, reacted at room temperature for 12-24 hours, concentrated, and separated by column (eluent: chloroform/methanol system) to give a white to yellow formula. VI I compound.
反应步錄四:
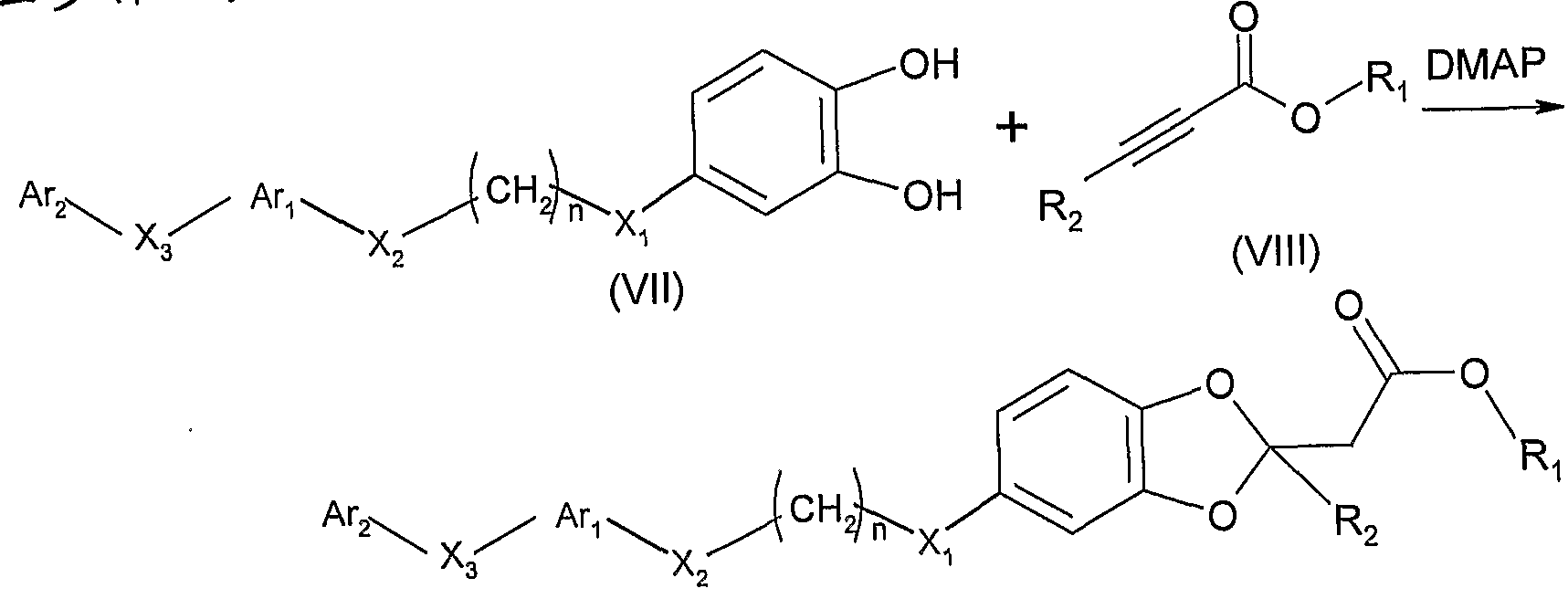
I I
以 4- (N,N-二甲基胺基)吡啶 (DMAP ) 为催化剂, 使式 VI I 化合物与式 VI I I化合物在无水乙腈或丙酮中室温反应 2-5小时, 浓缩后柱分离 (洗脱剂: 正己烷 /乙酸乙酯系统), 得到浅黄色至 黄色的油状通式 I的化合物,其中, 1、^不为 11、^为 S原子, ΑΓι, Ar2, X2, X3, R2和 n的定义同通式 I。 Using 4-(N,N-dimethylamino)pyridine (DMAP) as a catalyst to formulate VI I The compound is reacted with a compound of the formula VI II in anhydrous acetonitrile or acetone at room temperature for 2-5 hours, concentrated and separated by a column (eluent: n-hexane / ethyl acetate system) to give a pale yellow to yellow oily compound of formula I Wherein, 1, ^ is not 11, ^ is an S atom, Α Γι , Ar 2 , X 2 , X 3 , R 2 and n are the same as Formula I.
进一步地,可将上述 m=l、 ^不为 H、 为 S原子的通式 I的 化合物与碱金属氢氧化物反应或与稀盐酸、 三氟醋酸等酸反应 2-6小时, 经水解得到 ra=l、 ^为 S、 1^为 H原子的通式 I的化合 物。 Further, the above compound of the formula I wherein m=l, ^ is not H, is an S atom, can be reacted with an alkali metal hydroxide or with an acid such as dilute hydrochloric acid or trifluoroacetic acid for 2-6 hours, and hydrolyzed. A compound of the formula I wherein ra = 1, ^ is S, 1 is H atom.
I I
其中, ΑΓι, Ar2, X2, X3, n和 R3的定义同通式 I。 Wherein, Α Γι , Ar 2 , X 2 , X 3 , n and R 3 have the same meanings as in Formula I.
上述水解产物中, 当 R2为 H时, 即为 m=l、 为 3的通式 I I化合 物
II II
其中, Ar^ Ar2, X2, X3, n的定义同通式 I I。 Wherein, Ar^Ar 2 , X 2 , X 3 , n are as defined in the formula II.
本领域技术人员应该认识到式 I 或式 I I化合物存在立体中 心。 当需要式 I或式 I I化合物为单一的对映体时, 可以使用在所 有可能的步骤中均处于单一对映异构体形式的反应物来制备, 或 者在单一对映异构体形式的试剂或催化剂的存在下进行反应来制 备, 或者通过常规方法拆分立体异构体混合物来制备。 一些优选 的方法包括使用微生物进行拆分, 拆分与手性酸如扁桃酸、 樟脑 磺酸、 酒石酸、乳酸等任何可使用的酸形成的非对映异构体的盐, 或者拆分与手性碱如番木鳖碱(brac ine)、金鸡纳树生物碱及其衍 生物等形成的非对映异构体的盐。常用的方法见 Jaques等人编辑 的 "Enant ioraers , Raceraates and Resolut ion" (Wi ley Intersc ience, 1981)。 Those skilled in the art will recognize that the compound of Formula I or Formula II is present in stereo Heart. When a compound of formula I or formula II is desired as a single enantiomer, it can be prepared using reactants in the form of a single enantiomer in all possible steps, or in the form of a single enantiomer. It is prepared by carrying out the reaction in the presence of a catalyst or by dissolving a mixture of stereoisomers by a conventional method. Some preferred methods include resolution using a microorganism, resolution of a diastereomeric salt formed with any usable acid such as mandelic acid, camphorsulfonic acid, tartaric acid, lactic acid, or the like, or splitting with a hand. Salts of diastereomers formed by alkaloids such as brac ine, cinchona alkaloids and derivatives thereof. A commonly used method is found in "Enant ioraers, Raceraates and Resolution" edited by Jaques et al. (Wiley Intersc ience, 1981).
本领域技术人员应该意识到, 本发明化合物也可以以其可药 用盐或溶剂化物的形式使用。 式 I化合物的可药用盐包括与药学 上可接受的无机酸或有机酸或者无机碱或有机碱形成的常规的盐 以及季铵的酸加成盐。 合适的酸盐的具体的例子包括盐酸、 氢溴 酸、 硫酸、 磷酸、 硝酸、 高氯酸、 富马酸、 乙酸、 丙酸、 琥珀酸、 羟基乙酸、 甲酸、 乳酸、 马来酸、 酒石酸、 拧檬酸、 朴酸、 丙二 酸、 羟基马来酸、 苯乙酸、 谷氨酸、 苯曱酸、 水杨酸、 富马酸、 甲苯磺酸、 甲磺酸、 萘- 2 -磺酸、 苯磺酸、 羟基萘曱酸、 氢碘酸、 苹果酸、 鞣酸等的盐。 其它的酸, 如草酸, 虽然其本身并非药学 上可接受的, 但可以用于制备用作中间体的盐, 以获得本发明化 合物或其可药用盐。 合适的碱盐的具体的例子包括钠、 锂、 钟、 镁、 铝、 钙、 锌、 N,N,-二苄基乙二胺、 氯代普鲁卡因、 胆碱、 二 乙醇胺、 乙二胺、 N-曱基葡糖胺和普鲁卡因盐。 此后涉及到本发 明的化合物时, 包括式 I化合物及其可药用盐和溶剂化物。 Those skilled in the art will appreciate that the compounds of the invention may also be used in the form of their pharmaceutically acceptable salts or solvates. The pharmaceutically acceptable salts of the compounds of formula I include the conventional salts formed with pharmaceutically acceptable inorganic or organic acids or inorganic or organic bases, and the acid addition salts of quaternary ammonium. Specific examples of suitable acid salts include hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, nitric acid, perchloric acid, fumaric acid, acetic acid, propionic acid, succinic acid, glycolic acid, formic acid, lactic acid, maleic acid, tartaric acid, Citric acid, citric acid, malonic acid, hydroxymaleic acid, phenylacetic acid, glutamic acid, benzoic acid, salicylic acid, fumaric acid, toluenesulfonic acid, methanesulfonic acid, naphthalene-2-sulfonic acid, a salt of benzenesulfonic acid, hydroxynaphthoic acid, hydroiodic acid, malic acid, citric acid or the like. Other acids, such as oxalic, while not in themselves pharmaceutically acceptable, can be used in the preparation of salts useful as intermediates to obtain the compounds of the invention or pharmaceutically acceptable salts thereof. Specific examples of suitable base salts include sodium, lithium, bell, magnesium, aluminum, calcium, zinc, N,N,-dibenzylethylenediamine, chloroprocaine, choline, diethanolamine, ethylene Amine, N-decyl glucosamine and procaine salt. Hereinafter, when referring to the compounds of the present invention, the compounds of formula I, and pharmaceutically acceptable salts and solvates thereof, are included.
本发明还包括本发明化合物的前药, 该前药一经给药, 即通 过代谢过程进行化学转化, 之后变成具有活性的药物。 通常, 这 类前药是本发明化合物的功能性衍生物, 其在体内容易转化成所 需的式(I)的化合物。 例如, 在" Des ign Of Prodrugs", H Bund Saard, El sevier编辑, 1985中描述了选择和制备适宜前药衍生 物的常规方法。 The present invention also encompasses prodrugs of the compounds of the invention which, upon administration, are chemically converted by metabolic processes and thereafter become active drugs. Typically, such prodrugs are functional derivatives of the compounds of the invention which are readily converted into the body in vivo. The desired compound of formula (I). Conventional methods for the selection and preparation of suitable prodrug derivatives are described, for example, in "Des ign Of Prodrugs", H Bund Saard, El Sevier, ed., 1985.
本发明也包括本发明化合物的活性代谢物。 The invention also includes active metabolites of the compounds of the invention.
本发明化合物可激活 hPPAR受体,优选地,本发明化合物选择 性激活 hPPARa受体。 本发明的化合物对 PPAR的激动作用可以采 用下述方法进行测定。 The compounds of the invention activate the hPPAR receptor, and preferably, the compounds of the invention selectively activate the hPPARa receptor. The agonistic effect of the compound of the present invention on PPAR can be measured by the following method.
对化合物在 293- T细胞中瞬时转染的功能效应进行筛选, 以 测定它们激活 PPAR亚型的能力。采用预先建立嵌合受体系统比较 受体亚型对相同的靶基因转录活性的影响,以 R luc作为内标减少 内源性影响。 将人 PPARa、 ΡΡΑΙ γ和 PPARS配体结合域各自与酵 母转录因子 GAL4DNA结合域融合。 再连接到哺乳动物的表达载体 pM上, 构建 pM- hPPARa/GAL4、 pM-PPAIly/GAL4和 pM-PPARS/GAL4 三种质粒。 把 GAL4DNA 结合区与 pB4- tk-luc 连接', 构成 pB4-RES-tk-luc (一个含有 GAL4DNA结合位点的萤火虫荧光素酶 的报告基因)。 以 pRL-CMV- Rluc作为内标校正转染效率以及内源 性影响。 The functional effects of transient transfection of compounds in 293-T cells were screened to determine their ability to activate PPAR isoforms. The effect of the receptor subtype on the transcriptional activity of the same target gene was compared using a pre-established chimeric receptor system, and R luc was used as an internal standard to reduce endogenous effects. The human PPARa, γ γ and PPARS ligand binding domains are each fused to the yeast transcription factor GAL4 DNA binding domain. Then, the mammalian expression vector pM was ligated to construct three plasmids: pM-hPPARa/GAL4, pM-PPAIly/GAL4 and pM-PPARS/GAL4. The GAL4 DNA binding region is ligated to pB4-tk-luc' to constitute pB4-RES-tk-luc (a reporter gene for firefly luciferase containing a GAL4 DNA binding site). Transfection efficiency and endogenous effects were corrected using pRL-CMV-Rluc as an internal standard.
具体而言,将 293-T细胞接种入 48孔板,细胞密度为 2-4xl 04 个 /孔, 培养液为 10%脱脂胎牛血清 (FCS ) 的无酚红无抗生素的 1640培养基。 48小时后, 将培养液更换为 5%脱脂 FCS的无酚红 无抗生素的 1640培养基,然后分别把三种亚型的 pM- hPPAR/GAM、 pB4-RES-tk-luc和 pRL- CMV- Rluc三种质粒共转染到 293-T细胞 中, 24小时后给药。然后,在给药后 24小时检测荧光素酶的强度。 结果表明, 本发明化合物对 PPAR各亚型都有很强的激动作用,对 PPAR6的激动作用尤为强。 Specifically, the 293-T cells were seeded into 48-well plates at a cell density of 2-4xl 0 4 / well, culture was 10% defatted fetal calf serum (FCS) in phenol red-free 1640 medium without antibiotics. After 48 hours, the culture medium was changed to 5% defatted FCS phenol red-free antibiotic-free 1640 medium, and then three subtypes of pM-hPPAR/GAM, pB4-RES-tk-luc and pRL-CMV-, respectively. Three plasmids of Rluc were co-transfected into 293-T cells and administered 24 hours later. Then, the intensity of luciferase was measured 24 hours after administration. The results showed that the compounds of the present invention have strong agonistic effects on various subtypes of PPAR, and the agonistic effect on PPAR6 is particularly strong.
本发明的药物组合物包括有效剂量的本发明式 I 化合物或其 可药用盐或水合物和一种或多种适宜的可药用载体。 其可用于体 内治疗并具有生物相容性。 所述药物组合物可以根据不同给药途 径的需要制备成各种剂型。 The pharmaceutical compositions of the present invention comprise an effective amount of a compound of formula I according to the invention, or a pharmaceutically acceptable salt or hydrate thereof, and one or more suitable pharmaceutically acceptable carriers. It can be used for body Internal treatment and biocompatibility. The pharmaceutical compositions can be prepared in a variety of dosage forms as desired for different routes of administration.
所述的药用载体包括但不限于: 离子交换剂, 氧化铝, 硬脂 酸铝, 卵磷脂, 血清蛋白如人血白蛋白, 緩冲物质如磷酸盐, 甘 油, 山梨酸, 山梨酸钟, 饱和植物脂肪酸的部分甘油酯混合物, 水, 盐或电解质, 如硫酸鱼精蛋白, 磷酸氢二钠, 磷酸氢钾, 氯 化钠, 锌盐, 胶态氧化硅, 三硅酸镁, 聚乙烯吡咯烷酮, 纤维素 物质, 聚乙二醇, 羧甲基纤维素钠, 聚丙烯酸酯, 蜂蜡, 羊毛脂。 The pharmaceutically acceptable carrier includes, but is not limited to, an ion exchanger, alumina, aluminum stearate, lecithin, serum proteins such as human albumin, buffer substances such as phosphate, glycerin, sorbic acid, sorbic acid clock, Partial glyceride mixture of saturated vegetable fatty acids, water, salt or electrolyte, such as protamine sulfate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salt, colloidal silica, magnesium trisilicate, polyvinylpyrrolidone , Cellulose, Polyethylene Glycol, Sodium Carboxymethyl Cellulose, Polyacrylate, Beeswax, Lanolin.
本发明化合物的药物组合物可以以下面的任意方式施用: 口 服, 喷雾吸入, 直肠用药, 鼻腔用药, 颊部用药, 局部用药, 非 肠道用药, 如皮下、 静脉、 肌内、 腹膜内、 鞘内、 心室内、 胸骨 内和颅内注射或输入,或借助一种外植储器用药。其中优选口服、 腹膜内或静脉内给药方式。 The pharmaceutical composition of the compound of the present invention can be administered in any of the following ways: oral, spray inhalation, rectal administration, nasal administration, buccal administration, topical administration, parenteral administration, such as subcutaneous, intravenous, intramuscular, intraperitoneal, sheath Intra, intraventricular, intrasternal, and intracranial injection or input, or by means of an explant reservoir. Among them, oral, intraperitoneal or intravenous administration is preferred.
当口服用药时,本发明化合物可制成任意口服可接受的制剂 形式,包括但不限于片剂、 胶囊、 水溶液或水悬浮液。 其中, 片剂 使用的载体一般包括乳糖和玉米淀粉, 另外也可加入润滑剂如硬 脂酸镁。 胶嚢制剂使用的稀释剂一般包括乳糖和干燥玉米淀粉。 水悬浮液制剂则通常是将活性成分与适宜的乳化剂和悬浮剂混合 使用。 如果需要, 以上口服制剂形式中还可加入一些甜味剂、 芳 香剂或着色剂。 When administered orally, the compounds of the invention may be formulated in any orally acceptable form including, but not limited to, tablets, capsules, aqueous solutions or aqueous suspensions. Among them, the carrier used for the tablet generally includes lactose and corn starch, and a lubricant such as magnesium stearate may also be added. The diluent used in the capsule preparation generally comprises lactose and dried corn starch. Aqueous suspension formulations are usually prepared by admixing the active ingredient with a suitable emulsifier or suspension. If desired, some sweetening, aroma or coloring agents may be added to the above oral formulation.
当局部用药时, 特别是治疗局部外敷容易达到的患面或器官, 如眼睛、 皮肤或下肠道神经性疾病时, 可根据不同的患面或器官 将本发明化合物制成不同的局部用药制剂形式, 具体说明如下: 当眼部局部施用时, 本发明化合物可配制成一种微粉化悬浮 液或溶液的制剂形式, 所使用载体为等渗的一定 pH的无菌盐水, 其中可加入也可不加防腐剂如氯化苄基烷醇盐。 对于眼用, 也可 将化合物制成骨剂形式如凡士林骨。 当皮肤局部施用时, 本发明化合物可制成适当的软骨、 洗剂 或霜剂制剂形式, 其中将活性成分悬浮或溶解于一种或多种载体 中。 软骨制剂可使用的载体包括但不限于: 矿物油, 液体凡士林, 白凡士林, 丙二醇, 聚氧化乙烯, 聚氧化丙烯, 乳化蜡和水; 洗 剂或霜剂可使用的载体包括但不限于: 矿物油, 脱水山梨糖醇单 硬脂酸酯, 吐温 60, 十六烷酯蜡, 十六碳烯芳醇, 2-辛基十二垸 醇, 苄醇和水。 In the case of topical administration, especially in the treatment of facial surfaces or organs easily accessible by topical application, such as eye, skin or lower intestinal neurological diseases, the compounds of the present invention can be formulated into different topical preparations according to different affected faces or organs. The form is specifically described as follows: When the eye is topically applied, the compound of the present invention can be formulated into a preparation form of a micronized suspension or solution, and the carrier used is an isotonic pH of sterile saline, which may or may not be added. Preservatives such as benzyl chloride alkoxide. For ophthalmic use, the compound can also be formulated into a bone form such as vaseline. When the skin is applied topically, the compounds of the invention may be formulated in a suitable cartilage, lotion or cream formulation wherein the active ingredient is suspended or dissolved in one or more carriers. Carriers for cartilage preparations include, but are not limited to: mineral oil, liquid petrolatum, white petrolatum, propylene glycol, polyethylene oxide, polypropylene oxide, emulsifying wax and water; lotions or creams may be used including but not limited to: minerals Oil, sorbitan monostearate, Tween 60, cetyl esters wax, hexadecene aryl alcohol, 2-octyldodecanol, benzyl alcohol and water.
本发明化合物还可以无菌注射制剂形式用药, 包括无菌注射 水或油悬浮液或无菌注射溶液。 其中, 可使用的载体和溶剂包括 水、 林格氏溶液和等渗氯化钠溶液。 另外, 灭菌的非挥发油也可 用作溶剂或悬浮介质, 如单甘油酯或二甘油酯。 The compounds of the present invention can also be administered in the form of a sterile injectable preparation, including sterile aqueous or oily suspension or sterile injection solutions. Among them, carriers and solvents which can be used include water, Ringer's solution and isotonic sodium chloride solution. Alternatively, sterilized, fixed oils may be employed as a solvent or suspension medium such as a monoglyceride or a diglyceride.
另外需要指出, 本发明化合物的使用剂量和使用方法取决于 诸多因素, 包括患者的年龄、 体重、 性别、 自然健康状况、 营养 状况、 化合物的活性强度、 服用时间、 代谢速率、 病症的严重程 度以及诊治医师的主观判断。 优选的使用剂量介于 0. 01 ~ 100mg/kg体重 /天, 其中最优剂量在 5mg/kg- 10rag/kg体重 /天。 具体实施方式 It should also be noted that the dosage and method of use of the compounds of the invention depends on a number of factors, including the age, weight, sex, natural health, nutritional status of the compound, the strength of the compound, the time of administration, the rate of metabolism, the severity of the condition, and Subjective judgment of the doctor. A preferred dosage is from 0.01 to 100 mg/kg body weight per day, wherein the optimal dose is from 5 mg/kg to 10 rag/kg body weight per day. detailed description
下述的实施例将进一步说明本发明, 但这些实施例不构成对 本发明的任何限制。 The invention is further illustrated by the following examples, which are not intended to limit the invention.
化合物熔点由 YRT-3型熔点仪测定, 温度未经校正。 - NMR 光谱由 Bruker ARX 400型核磁仪测定。 FAB质谱由 Zabspec t 高 分辨磁质谱仪测定。 中间体的制备 The melting point of the compound was determined by a YRT-3 type melting point apparatus, and the temperature was not corrected. - NMR spectra were determined by a Bruker ARX 400 nuclear magnetic instrument. FAB mass spectra were determined by a Zabspec t high resolution magnetic mass spectrometer. Preparation of intermediates
由芳香曱酰胺制备 2-芳香基 -4-甲基 -5-羟甲基噻唑的一般 操作 A

(a)在 100ml干甲苯中加入 2mmolP4S10, 20mraolNaHC03, 加热 回流 30分钟, 加入 1 Q讓 ol酰胺, l-2h后反应完全, 过滤, 浓缩 柱分离(洗脱体系:正己烷 /乙酸乙酯)得固体的硫代芳香甲酰胺。 (a) Add 2mmol of P 4 S 10 , 20mraolNaHC0 3 to 100ml of dry toluene, heat under reflux for 30 minutes, add 1 Q to let ol amide, l-2h after the reaction is complete, filter, and concentrate column separation (elution system: n-hexane / acetic acid Ethyl ester) gives a solid thioaromatic formamide.
(b)在 100ml 乙醇中加入 l Ommol 取代的硫代芳香甲酰胺、 l lmmo l 2-氯 -3-氧代-丁酸乙酯 (购自 ACR0S ) , 加热回流 12-24 小时, 浓缩, 柱分离 (洗脱体系: 正己烷 /乙酸乙酯)得(2 -取代 芳香基 -4-甲基噻唑 -5-基) 甲酸乙酯。 (b) Add 10 mmol of substituted thioaromatic formamide, l lmmo l 2-chloro-3-oxo-butyric acid ethyl ester (purchased from ACR0S) in 100 ml of ethanol, heat under reflux for 12-24 hours, concentrate, column Separation (elution system: n-hexane/ethyl acetate) gave (2-substituted aryl-4-methylthiazole-5-yl) ethyl formate.
(c)将 10腿 ol ( 2-取代芳香基- 4-甲基噻唑 -5-基) 甲酸乙酯 的 100ml干燥四氢呋喃溶液滴加到 0°C的 l lmmo l四氢铝锂的 30ml 干燥四氢呋喃反应液中, 滴加完毕, 室温反应 2-4小时后, 滴加 少许水、 15 %氢氧化钠水溶液、 水终止反应, 无水硫酸镁干燥, 过滤, 蒸干即得产品 2-芳香基 -4-甲基 -5-羟曱基噻唑。 (c) A solution of 10 ol (2-substituted aryl-4-methylthiazole-5-yl)carboxylate in 100 ml of dry tetrahydrofuran was added dropwise to 30 ml of dry THF of 1 lmmol of lithium aluminum hydride at 0 °C. In the reaction solution, after the dropwise addition is completed, the reaction is carried out at room temperature for 2-4 hours, the reaction is stopped by adding a little water, 15% aqueous sodium hydroxide solution and water, dried over anhydrous magnesium sulfate, filtered, and evaporated to give the product 2-aryl- 4-methyl-5-hydroxymercaptothiazole.
中间体 1 Intermediate 1

标题化合物以苯甲酰胺为原料如一般操作 A所述制备, 得白 色固体。 The title compound was prepared from benzamide as a crude material.
MS [M] +=205. 2 ra/e; ^-NMR (400MHz, DMSO) δ 7. 89 - 7. 87 (m, 2H) , 7. 50 - 7. 44 (m, 3H) , 5. 55 (t, 1H) , 4. 64 (d, 2H) , 2. 35 (s, 3H) 。 MS [M] += 205. 2 ra/e; ^-NMR (400MHz, DMSO) δ 7. 89 - 7. 87 (m, 2H), 7. 50 - 7. 44 (m, 3H), 5. 55 (t, 1H) , 4. 64 (d, 2H) , 2. 35 (s, 3H).
中间体 2

标题化合物以对三氟甲基苯甲酰胺为原料如一般操作 A所述 制备, 得白色固体。 mp: 120. 5 - 122°C The title compound was prepared using p-trifluoromethylbenzamide as a crude material. Mp: 120. 5 - 122°C
MS [M] +=273. 1 m/e; 'H-NMR (400MHz, DMSO) δ 8. 10 (d, 2H), 7. 83 (d, 2H), 5. 66 (t, 1H), 4. 67 (d, 2H), 2. 37 (s, 3H)。 MS [M] +=273. 1 m/e; 'H-NMR (400MHz, DMSO) δ 8. 10 (d, 2H), 7. 83 (d, 2H), 5. 66 (t, 1H), 4. 67 (d, 2H), 2. 37 (s, 3H).
中间体 3 Intermediate 3

标题化合物以对溴苯甲酰胺为原料如一般操作 A所述制备, 得浅黄色固体。 The title compound was prepared using p-bromobenzamide as a crude material.
MS [M] +=284. 1 m/e; 'H-NMR (400MHz, DMSO) δ 7. 82 (d, 2H) 7. 67 (d, 2H), 5. 58 (t, 1H), 4. 63 (d, 2H), 2. 34 (s, 3H)。 MS [M] +=284. 1 m/e; 'H-NMR (400MHz, DMSO) δ 7. 82 (d, 2H) 7. 67 (d, 2H), 5. 58 (t, 1H), 4 63 (d, 2H), 2. 34 (s, 3H).
中间体 4 Intermediate 4

标题化合物以对氟苯曱酰胺为原料如一般操作 A所述制备, 得白色固体。 MS [M] +=223. 3 m/e The title compound was prepared using p-fluorobenzoic acid amide as a crude material. MS [M] += 223. 3 m/e
中间体 5 Intermediate 5

中间体 6 Intermediate 6

标题化合物以 2,4-二甲氧基苯甲酰胺为原料如一般操作 A所 述制备, 得浅黄色固体 MS [M] +=265. 3 m/e The title compound was obtained from the title compound (m.p.
中间体 7 Intermediate 7
标题化合物以 2, 4-二氯苯甲酰胺为原料如一般操作 A所述制 备, 得黄色固体。 MS [M] +=274. 2 m/e The title compound was obtained as a crude material. MS [M] += 274. 2 m/e
中间体 8 Intermediate 8

标题化合物以对叔丁基苯甲酰胺为原料如一般操作 A所述制 备, 得浅黄色固体。 MS [M] +=261. 4 ra/e The title compound was prepared from p-tert-butyl benzamide as a general procedure A to give a pale yellow solid. MS [M] +=261. 4 ra/e
中间体 9
中间体 10

标题化合物以 3-噻吩曱酰胺为原料如一般操作 A所述制备: 得浅黄色固体。 MS [M] +=211. 3 m/e The title compound was prepared using 3-thiophene amide as a crude material. MS [M] +=211. 3 m/e
中间体 11 Intermediate 11
标题化合物以 2-萘甲酰胺为原料如一般操作 A所述制备, 得 浅黄色固体。 MS [M] +=255. 3 ra/e The title compound was prepared using 2-naphthylcarboxamide as a crude material. MS [M] +=255. 3 ra/e
中间体 12 Intermediate 12

标题化合物以胡椒基酰胺为原料如 The title compound is based on piperonamide.
浅黄色固体。 MS [M] +=249. 2 ra/e Light yellow solid. MS [M] +=249. 2 ra/e
中间体 13 Intermediate 13
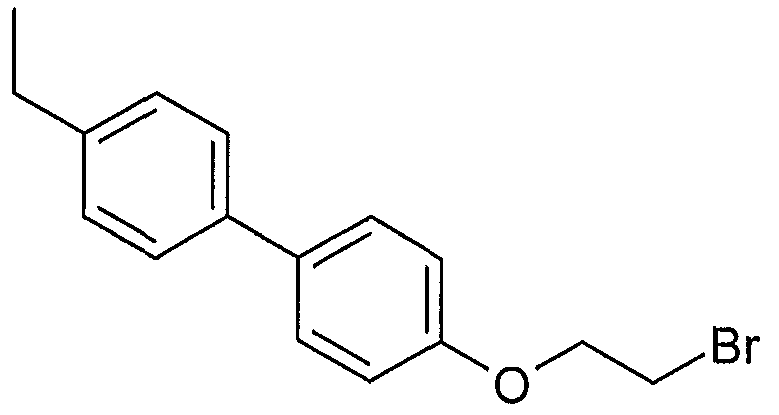
在 2. 42g (l Oramol) 4-对乙基苯基苯酚的 50ml干燥丙酮溶液中 加入 15mmol的 KOH后, 緩慢滴加 1.88g(10腿 ol)l, 2 -二溴乙烷, 50°C反应 8小时后过滤, 浓缩, 柱分离 (洗脱体系: 正己烷 /乙酸 乙酯) , 得油状上述中间体 13, 收率 45%。 MS [M] +=305.2 m/e 中间体 14 In a solution of 2.42 g (l Oramol) 4-p-ethylphenylphenol in 50 ml of dry acetone After adding 15 mmol of KOH, 1.88 g (10 lbs) of 1,2-dibromoethane was slowly added dropwise, and reacted at 50 ° C for 8 hours, followed by filtration, concentration, and column separation (elution system: n-hexane / ethyl acetate) The intermediate 13 was obtained as an oil, and the yield was 45%. MS [M] +=305.2 m/e Intermediate 14
1.99g(10mmol)的吩噻嗪溶于 50ral 无水丁酮, 加入 0.94g (16.7mmol)氢氧化钾粉末, 冷却, 保持 0°C左右 2小时加入溶有 0.79g(18mmol)环氧乙烷的 15ml无水丁酮, (TC反应 2小时后升温 至 50-60°C反应 2小时, 冷后, 过滤, 浓缩, 柱分离 (洗脱体系: 正己烷 /乙酸乙酯)得 1.85 g产品, 收率 76 %。 MS [M] +=243.3 m/ e 中间体 15

8.8g(80mmol)邻苯二酚和 20.8g (256mmol)硫氰酸钠的 100ml 甲醇溶液降至 0°C后, 緩慢滴加 4.4ml 溴素 (80mmol)和 8.24g(480mmol)溴化钠的 100ml 甲醇溶液, 保持 0。C反应 3小时 后, 加入 300ml水终止反应, 饱和 NaHC03水溶液调至近中性, 乙 酸乙酯提取, 干燥, 浓缩, 柱分离 (洗脱体系: 正己烷 /乙酸乙酯 /乙酸),得白色固体产品 12.73g,收率 95.3%, rap: 133.0-134.5°C。 After 8.8 g (80 mmol) of catechol and 20.8 g (256 mmol) of sodium thiocyanate in 100 ml of methanol were dropped to 0 ° C, 4.4 ml of bromine (80 mmol) and 8.24 g (480 mmol) of sodium bromide were slowly added dropwise. 100 ml of methanol solution, kept at 0. C After 3 hours, 300ml of water was added to terminate the reaction, a saturated NaHC0 3 aqueous solution was adjusted near neutral, extracted with ethyl acetate, dried, concentrated, and column separation (elution system: n-hexane / ethyl acetate / acetic acid) to give a white solid The product was 12.73 g, the yield was 95.3%, and the rap: 133.0-134.5 °C.
MS [M]+=167.1 m/e; 'H-NMR (400MHz, DMSO) δ 9.66 (d, 2H), 7.00 (s, 1H), 6.93 (dd, 1H), 6.84 (d, 1H)。 MS [M]+=167.1 m/e; 'H-NMR (400 MHz, DMSO) δ 9.66 (d, 2H), 7.00 (s, 1H), 6.93 (dd, 1H), 6.84 (d, 1H).
中间体 16
在 150ml 圆底瓶中加入 4-硫腈基邻苯二酚(中间体 15) 7.5g(44.9mraol) , 原甲酸三乙酯 .98g(67.4mraol) , 酸性离子交 换树脂 0.5g, 4A分子筛 0.8g, 100ml无水苯, 回流 72-96小时, 过滤, 浓缩, 柱分离 (洗脱体系: 正己垸 /乙酸乙酯 =25:1) , 得 7.24g 浅黄油状产品, 收率 72.3%。 MS [M] +=223.2 m/e; 'H-NMR (400MHz, CDC13) δ 7.12 (d, 1H), 7.11 (s, 1H), 6.93 (s, 1H), 6.90 (dd, 1H), 3.73 (q, 2H), 1.28 (t, 3H)。 In a 150ml round bottom bottle, add 4-sulfonitrile catechol (intermediate 15) 7.5g (44.9mraol), triethyl orthoformate. 98g (67.4mraol), acidic ion exchange resin 0.5g, 4A molecular sieve 0.8 g, 100 ml of anhydrous benzene, refluxing for 72-96 hours, filtration, concentration, and column separation (eluting system: n-hexane/ethyl acetate = 25:1) yielding 7.24 g of lightly buttery product, yield 72.3%. MS [M] +=223.2 m/e; 'H-NMR (400MHz, CDC1 3 ) δ 7.12 (d, 1H), 7.11 (s, 1H), 6.93 (s, 1H), 6.90 (dd, 1H), 3.73 (q, 2H), 1.28 (t, 3H).
由 2-芳香基 -4-甲基- 5-羟曱基噻唑制备 4- [(2-芳香基 -4-甲 基噻唑 -5-基)甲硫基 ]-1, 2-邻苯二酚的一般操作 B Preparation of 4-[(2-aryl-4-methylthiazol-5-yl)methylthio]-1,2-catechol from 2-aryl-4-methyl-5-hydroxymercaptothiazole General operation B

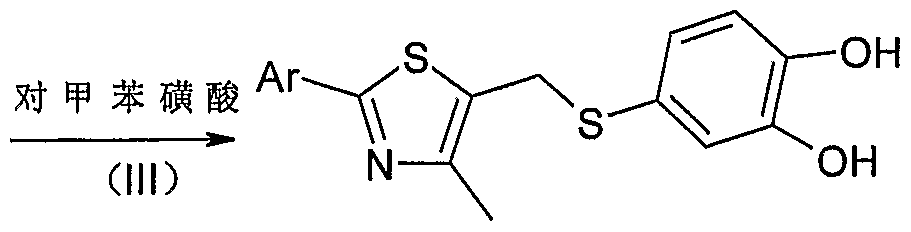
(I) 将 2 -芳香基- 4-甲基- 5-羟曱基噻唑(5匪 ol)溶于 20ml干燥氯 仿中, 滴加 5mlSOCl2,回溜 3-5h后蒸馏, 减压蒸干, 产品待用。 (I) 2-Aromatic 4-methyl-5-hydroxymercaptothiazole (5 匪ol) was dissolved in 20 ml of dry chloroform, 5 ml of SOCl 2 was added dropwise, and after 3-5 hours, it was distilled, and evaporated to dryness under reduced pressure. The product is ready for use.
(II) 在 100ml 圆底瓶中加入原甲酸三乙酯保护的 6腿 ol 4-氰硫 基邻苯二酚(中间体 16), 35ml 绝对乙醇, N2保护, 緩慢加入 NaBH4(6.6mmol),回流 10-15分钟后降至室温, 加入碳酸铯后, 滴 加(I)的反应产物, 室温、 N2反应过夜, 过滤, 乙酸乙酯洗涤, 浓 缩, 柱分离 (洗脱体系: 正己烷 /乙酸乙酯) , 得黄色油状产物。 (II) Add 6-leg ol 4-cyanothio catechol (intermediate 16) protected with triethyl orthoformate in a 100 ml round bottom flask, 35 ml absolute ethanol, N 2 protection, slowly add NaBH 4 (6.6 mmol) After refluxing for 10-15 minutes, the temperature is lowered to room temperature. After adding cesium carbonate, the reaction product of (I) is added dropwise, reacted at room temperature, N 2 overnight, filtered, washed with ethyl acetate, concentrated, and separated by column (elution system: Alkyl/ethyl acetate) gave the product as a yellow oil.
(III) 将(II)的反应产物溶于 40ml 90%甲醇水溶液中, 加入 500mg对甲苯磺酸后室温反应 12-24小时, 浓缩, 柱分离 (洗脱 体系: 氯仿 /甲醇) , 得浅黄至黄色的 4- [ (2-芳香基 -4 -曱基噻唑 -5 -基)甲硫基 ] - 1, 2-邻苯二酚。 (III) The reaction product of (II) is dissolved in 40 ml of 90% aqueous methanol, added After reacting 500 mg of p-toluenesulfonic acid at room temperature for 12-24 hours, concentrating, and separating by column (elution system: chloroform/methanol) to give pale yellow to yellow 4-[(2-aryl-4-mercaptothiazol-5-yl) )Methylthio] - 1, 2-catechol.
中间体 17 Intermediate 17

标题化合物以中间体 1为原料如一般操作 B所述制备, 得浅 黄色固体。 MS [M] +=329. 4 m/e; 'H-NMR (400MHz, DMSO) δ 9. 10 (d, 2Η),δ 7. 84 (in, 2H), 7. 46 (ra, 3H) , 6. 77 (s, 1H), 6. 67 (s, 2H), 4. 20 (s, 2H), 2. 12 (s, 3H)。
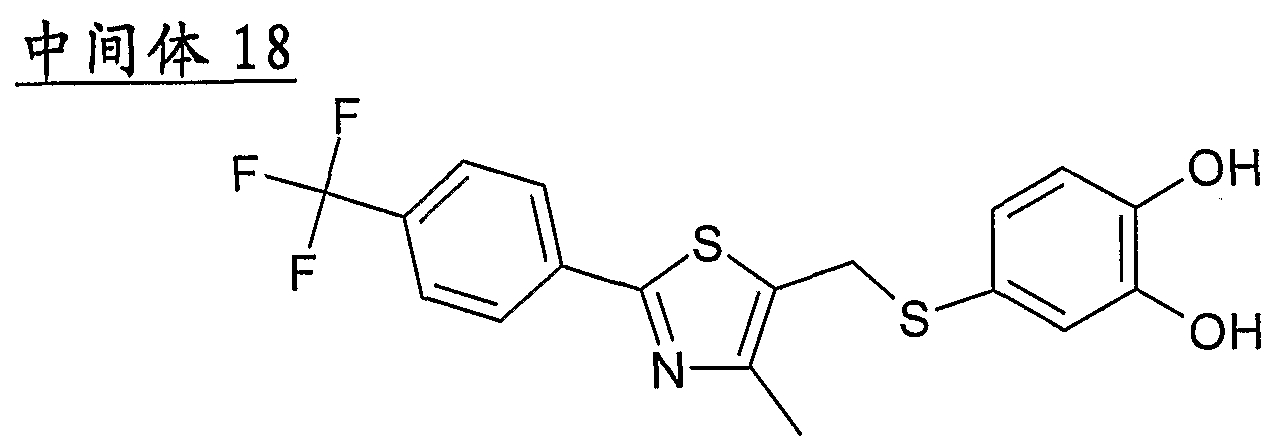
标题化合物以中间体 2为原料如一般操作 B所述制备, 得淡 黄色固体。 MS [M] +=397. 3 m/e; 'H-NMR (400MHz, DMSO) δ 9. 11 (d, 2Η),δ 8. 05 (d, 2H), 7. 82 (d, 2H), 6. 76 (s, 1H), 6. 66 (s, 2H), 4. 21 (s, 2H) , 2. 12 (s, 3H)。 The title compound was prepared from Intermediate 2 as a crude material. MS [M] +=397. 3 m/e; 'H-NMR (400MHz, DMSO) δ 9. 11 (d, 2Η), δ 8. 05 (d, 2H), 7. 82 (d, 2H) , 6. 76 (s, 1H), 6. 66 (s, 2H), 4. 21 (s, 2H), 2. 12 (s, 3H).
中间体 19 Intermediate 19

标题化合物以中间体 3为原料如一般操作 B所述制备, 得淡 黄色固体。 MS [M] +=408. 3 m/e; 'H-NMR (400MHz, DMSO) δ 9. 11 (d, 2Η),δ 7. 77 (d, 2H), 7. 66 (d, 2H) , 6. 77 (s, 1H), 6. 67 (s 2H), 4. 21 (s, 2H) , 2. 14 (s, 3H)。 The title compound was prepared from Intermediate 3 using mp. MS [M] += 408. 3 m/e; 'H-NMR (400 MHz, DMSO) δ 9. 11 (d, 2Η), δ 7. 77 (d, 2H), 7. 66 (d, 2H), 6. 77 (s, 1H), 6. 67 (s 2H), 4. 21 (s, 2H), 2. 14 (s, 3H).
中间体 20 Intermediate 20

标题化合物以中间体 4为原料如一般操作 B所述制备, 得淡 黄色固体。 MS [M] +=347. 4 m/e; ^-NMR (400MHz, DMSO) δ 9. 11 (d, 2Η) , δ 7. 88 (m, 2H), 7. 31 (m, 2H), 6. 77 (s, 1H), 6. 67 (s, 2H), 4. 20 (s, 2H), 2. 12 (s, 3H)。 The title compound was prepared from Intermediate 4 using EtOAc m. MS [M] +=347. 4 m/e; ^-NMR (400MHz, DMSO) δ 9. 11 (d, 2Η), δ 7. 88 (m, 2H), 7. 31 (m, 2H), 6. 77 (s, 1H), 6. 67 (s, 2H), 4. 20 (s, 2H), 2. 12 (s, 3H).
中间体 21 Intermediate 21
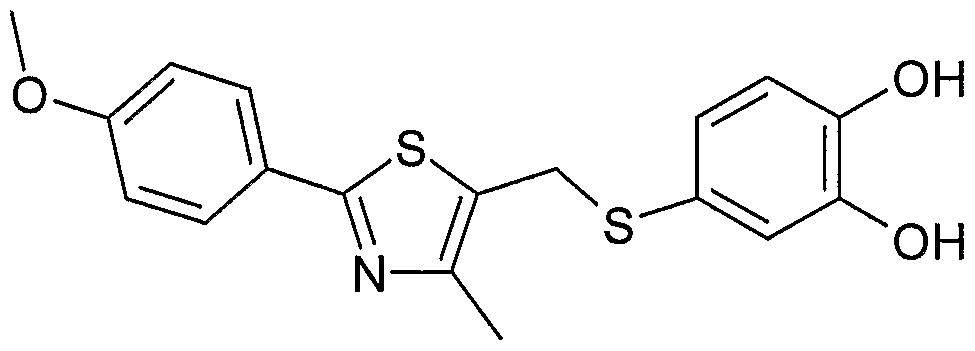
标题化合物以中间体 5为原料如一般操作 B所述制备, 得淡 黄色固体。 MS [M] +=359. 5 m/e The title compound was prepared from Intermediate 5 using EtOAc (EtOAc) MS [M] +=359. 5 m/e
中间体 22 Intermediate 22
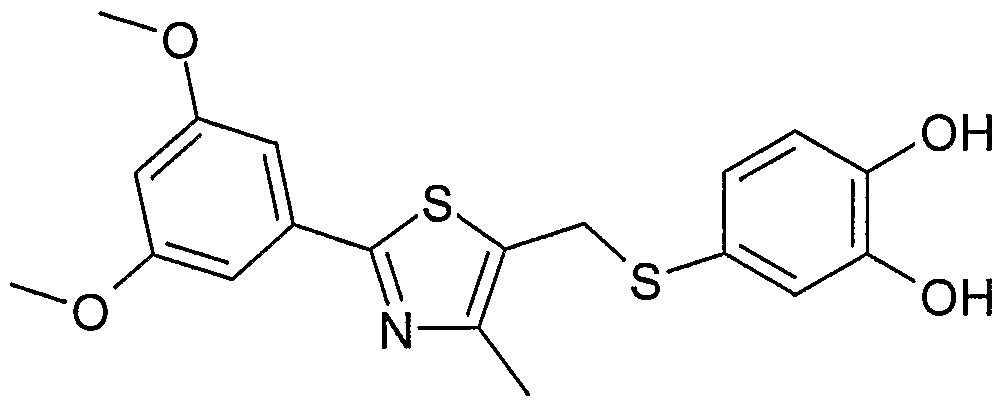
标题化合物以中间体 6为原料如一般操作 B所述制备, 得淡 黄色固体。 MS [M] +=389. 3 m/e The title compound was prepared from Intermediate 6 using EtOAc (EtOAc) MS [M] +=389. 3 m/e
中间体 23

标题化合物以中间体 7为原料如一般操作 B所述制备, 得淡 黄色固体。 MS [M] +=398. 3 m/e The title compound was prepared from Intermediate 7 using EtOAc (EtOAc) MS [M] +=398. 3 m/e
中间体 24 Intermediate 24

标题化合物以中间体 8为原料如一般操作 B所述制备, 得白 色固体。 MS [M] +=385. 5 m/e The title compound was prepared from Intermediate 8 using EtOAc (m.). MS [M] +=385. 5 m/e
中间体 25

标题化合物以中间体 9为原料如一般操作 B所述制备, 得白 色固体。 MS [M] +=335. 4 ra/e The title compound was prepared from Intermediate 9 using EtOAc (m.) MS [M] +=335. 4 ra/e
中间体 26
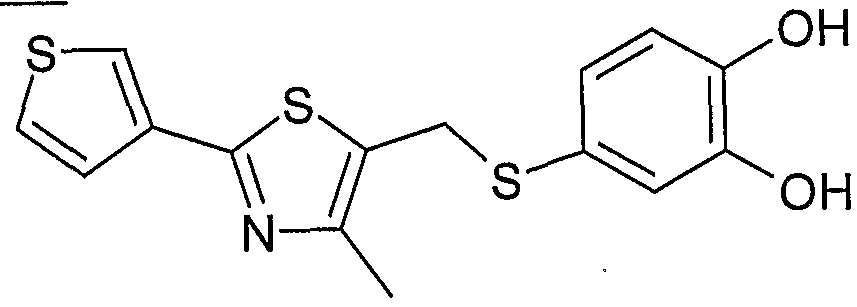
标题化合物以中间体 10为原料如一般操作 B所述制备,得淡 黄色固体。 MS [M] +-335. 4 m/e The title compound was prepared from Intermediate 10 using EtOAc (EtOAc) MS [M] +-335. 4 m/e
中间体 27 Intermediate 27
-28-

标题化合物以中间体 11为原料如一般操作 B所述制备,得淡 黄色固体。 MS [M] +=379.5 m/e The title compound was prepared from Intermediate 11 using EtOAc (EtOAc) MS [M] +=379.5 m/e
中间体 28 Intermediate 28
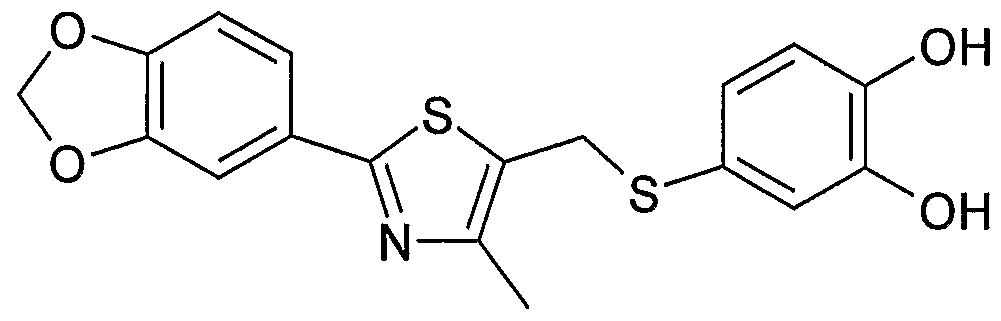
标题化合物以中间体 12为原料如一般操作 B所述制备,得淡 黄色固体。 MS [M] +=373.4 ra/e The title compound was obtained from the title compound (yield: m. MS [M] +=373.4 ra/e
中间体 29 Intermediate 29
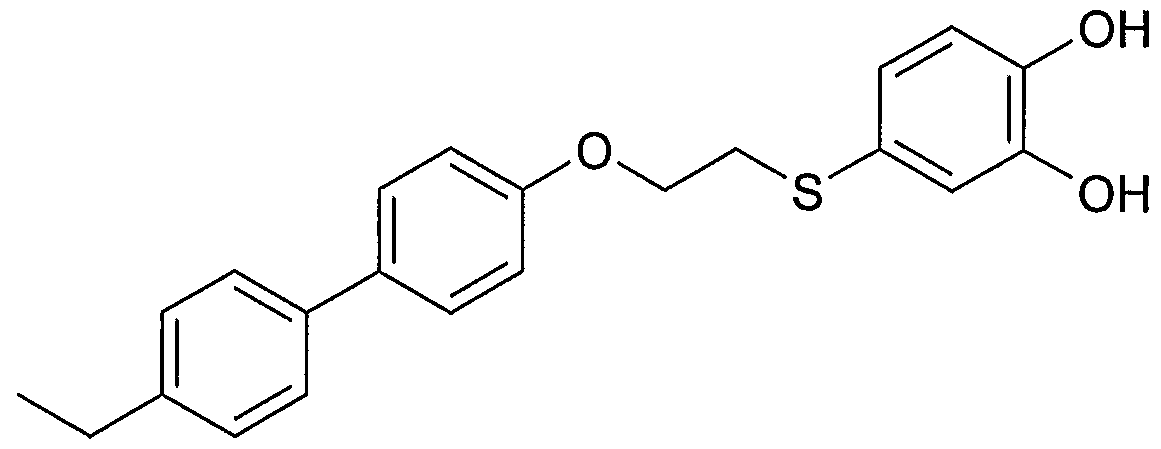
在 100ml圆底瓶中加入原甲酸三乙酯保护的 1.34g(6mmol)4- 氰硫基邻苯二酚(中间体 16), 35ml绝对乙醇, N2保护, 緩慢加入 NaBH4(0.24g,6.6mmol), 回流 10 分钟后降至室温, 加入 0.55g(9.9mmol) K0H后, 滴加 1.52g (5mmol)中间体 13的氯仿溶 液, N2保护回流 8反应小时后, 过滤, 乙酸乙酯洗涤, 浓缩, 柱 分离 (洗脱体系: 正己烷 /乙酸乙酯) , 得黄色油状产物(即原甲 酸三乙酯保护得中间体 29)。将此油状物溶于 40ml的 90%甲醇水 溶液,加入 500rag对曱苯磺酸,室温 24小时后,浓缩,柱分离(洗 脱体系: 氯仿 /甲醇), 得淡黄色固体 0.712g。 MS [M] +=366.3 m/e Add 1.34 g (6 mmol) of 4-cyanothio catechol (intermediate 16) protected with triethyl orthoformate to a 100 ml round bottom flask, protect with 35 ml of absolute ethanol, N 2 and slowly add NaBH 4 (0.24 g, 6.6 mmol), after refluxing for 10 minutes, the temperature was lowered to room temperature. After adding 0.55 g (9.9 mmol) of K0H, 1.52 g (5 mmol) of chloroform solution of intermediate 13 was added dropwise, and the mixture was refluxed with N 2 and then filtered, ethyl acetate. Washing, concentrating, and column chromatography (eluting: hexane/ethyl acetate) afforded product as yellow oil (yield of triethyl orthoformate to afford intermediate 29). This oil was dissolved in 40 ml of a 90% aqueous methanol solution, and 500 rag of p-toluenesulfonic acid was added, and the mixture was concentrated at room temperature for 24 hours, and then concentrated to give a pale yellow solid (yield: 0.712 g). MS [M] +=366.3 m/e

将 2- (咔唑 -9-基)乙醇(购自 ACR0S) (1.06g, 5mmol)溶于 2 Oral 干燥氯仿中, 滴加 5ml S0C12,回流 4h后蒸馏, 减压蒸干, 产品待 用。 2-(carbazol-9-yl)ethanol (purchased from ACR0S) (1.06g, 5mmol) was dissolved in 2 Oral dry chloroform, 5ml of S0C1 2 was added dropwise, refluxed for 4h, distilled, evaporated under reduced pressure, the product was ready for use .
在 100ml 圆底瓶中加入原甲酸三乙酯保护的 1.34g(6mmol) 4-氰硫基邻苯二酚(中间体 16), 35ml绝对乙醇, N2保护, 緩慢加 入 NaBE O.24g,6.6mmol),回流 10分钟后降至室温, 加入 0.55g (9.9mmol) K0H和 50mgKI后, 滴加上述得到的氯代产物, N2保护 回流 6小时, 过滤, 乙酸乙酯洗涤, 浓缩, 柱分离 (洗脱体系: 正己烷 /乙酸乙酯), 得黄色油状产物(即原曱酸三乙酯保护的中 间体 30) 。 Add 1.34 g (6 mmol) of 4-cyanothiocatechol (intermediate 16) protected with triethyl orthoformate in a 100 ml round bottom flask, 35 ml absolute ethanol, N 2 protection, slowly add NaBE O.24 g, 6.6 Ment), after refluxing for 10 minutes, the temperature was lowered to room temperature. After adding 0.55 g (9.9 mmol) of K0H and 50 mg of KI, the chloro product obtained above was added dropwise, N 2 was refluxed for 6 hours, filtered, washed with ethyl acetate, concentrated, and separated. (elution system: n-hexane / ethyl acetate) to give the product as a yellow oil (yield intermediate 30 protected by triethyl orthanoate).
将上述得到的黄色产物溶于 40ral 90%甲醇水溶液中, 加入 500mg对甲苯磺酸后室温反应 24小时, 浓缩,柱分离(洗脱体系: 氯仿 /甲醇) , 得黄色的 4- [2- (咔唑 -9-基)乙硫基 ] -1,2-邻苯二 酚 0.605g。 MS [M] +=335.4 ra/e The yellow product obtained above was dissolved in a 40 rpm 90% aqueous methanol solution, and after adding 500 mg of p-toluenesulfonic acid, the reaction was carried out at room temperature for 24 hours, concentrated, and separated by column (elution system: chloroform/methanol) to obtain a yellow 4-[2-( Oxazol-9-yl)ethylthio]-1,2-catechol 0.605 g. MS [M] +=335.4 ra/e
中间体 31 Intermediate 31

以中间体 14为原料按中间体 30的制备方法操作, 得黄色固 体 4- [2- (吩噻嗪 -10 -基)乙硫基 ]- 1, 2-邻苯二酚。 MS [M] +=367.5 m/e The intermediate 14 was used as a starting material to give the title compound (yield: 4-[2-(phenothiazine-10-yl)ethylthio]- 1, 2-catechol. MS [M] +=367.5 m/e
中间体 32

2.8g(40mmol)丙炔酸和 6.9g (50mmol)对甲氧基苄醇溶于 4 Oral 干燥二氯甲烷中 , 降温至 -20 °C后 , 緩慢滴加 9.28g(45mraol)DCC 和 0.37g (3腿 ol) DMAP ( 4- N, N-二甲基胺基 吡啶)的 100ml干燥二氯甲烷溶液, 30分钟后, 升至室温反应 12 小时, 过滤, 浓缩, 柱分离 (洗脱体系: 正己烷 /乙酸乙酯) , 得 浅黄色油状物 4.95g, 收率 65.1%。 2.8 g (40 mmol) of propiolic acid and 6.9 g (50 mmol) of p-methoxybenzyl alcohol were dissolved in 4 Oral dry dichloromethane, and after cooling to -20 ° C, 9.28 g (45 mraol) DCC and 0.37 g were slowly added dropwise. (3 leg ol) DMAP (4-N, N-dimethylaminopyridine) in 100 ml of dry dichloromethane, after 30 minutes, warm to room temperature for 12 hours, filter, concentrate, and column separation (elution system: N-hexane/ethyl acetate) gave 4.95 g of pale yellow oil, yield 65.1%.
^-NMR (400MHz, CDC13) δ 7.32 (d, 2Η), 6.90 (d, 2H), 5.16 (s, 2H) , 3.82 (s, 3H), 2.87 (s, 1H)。 实施例 ^-NMR (400MHz, CDC1 3 ) δ 7.32 (d, 2Η), 6.90 (d, 2H), 5.16 (s, 2H), 3.82 (s, 3H), 2.87 (s, 1H). Example
实施例 1: 2-{5-[(4-甲基 -2 -苯基 -1, 3-噻唑 -5-基)-甲硫 基] -1, 3-苯并间二氧杂环戊烯 } -乙酸 Example 1: 2-{5-[(4-Methyl-2-phenyl-1,3-thiazol-5-yl)-methylthio]-1,3-benzodioxole } - acetic acid
将 0· 4g(l.22mmol) 中间体 17 和 0.223g (1.83mmol) DMAP ( 4-N,N-二甲基胺基吡啶) 溶于 20ml 干燥乙腈中, 緩慢滴加 0.254g(l.34腿 ol)丙炔酸对曱氧基苄酯(中间体 32)的 5ral干燥 乙腈溶液, 室温反应 2h后, 浓缩, 柱分离 (洗脱体系: 正己烷 / 乙酸乙酯) , 得 0.573g 油状的 2- {5- [(4-甲基 -2-苯基- 1, 3-噻 唑 -5-基) -甲硫基] -1, 3-苯并间二氧杂环戊烯 } -乙酸对甲氧基苄 酯, 收率 90.2°/。。 0.44 g (1.22 mmol) of intermediate 17 and 0.223 g (1.83 mmol) of DMAP (4-N,N-dimethylaminopyridine) were dissolved in 20 ml of dry acetonitrile, and 0.254 g (l.34) was slowly added dropwise. Leg ol) a solution of propiolic acid to benzyloxybenzyl ester (intermediate 32) in 5 liters of dry acetonitrile, which was reacted at room temperature for 2 h, concentrated and purified by column (eluting system: n-hexane / ethyl acetate) to give 0.573 g of oil. 2-{5-[(4-Methyl-2-phenyl- 1, 3-thiazol-5-yl)-methylthio]-1,3-benzodioxole}-acetic acid Methoxybenzyl ester, yield 90.2 ° /. .
将 0.573g(l. lmmol)2-{5-[(4-甲基- 2-苯基 -1, 3 -噻唑 -5- 基)-甲硫基 ]-1, 3-苯并间二氧杂环戊烯 } -乙酸对甲氧基苄酯溶于 16ml 二氯甲烷中, 加入 0.82ml (llramol)三氟醋酸后, 室温反应 4.5h, 浓缩, 柱分离 (洗脱体系: 二氯曱烷 /曱醇) , 得浅黄色无 定形固体的 2- {5- [(4-甲基- 2-苯基- 1, 3-噻唑 -5-基) -甲硫 基]- 1, 3-苯并间二氧杂环戊烯 } -乙酸 0.325g , 收率 74.0%。 0.573 g (1.1 mmol) of 2-{5-[(4-methyl-2-phenyl-1,3-thiazol-5-yl)-methylthio]-1,3-benzodioxine dioxol} - acetic acid was dissolved in 16ml methoxybenzyl ester in dichloromethane was added 0. 82 ml (llramol) trifluoroacetic acid was stirred at rt 4.5h, concentration, column separation (elution system: methylene chloride / decyl alcohol), 2-{5-[(4-methyl-2-phenyl-1, 3-thiazole) as a pale yellow amorphous solid -5-yl)-methylthio]- 1, 3-benzodioxole}-acetic acid 0.325 g, yield 74.0%.
MS [M]+= 400. Ora/e; ^-NMR (400MHz, DMS0-d6) δ 12.79 (brs, 1H,), 7.85 一 7.82 (m, 2H), 7.48 - 7.45 (m, 3H), 7.04 (d, 1H), 6.85 (d, 2H), 6.52 (t, 1H), 4.35 (s, 2H), 2.96 (d, 2H), 2.20 (s, 3H) 实施例 2: 2-(5-{[4-甲基 -2-(4-三氟甲基苯基)- 1, 3-噻唑 - 5- 基]-甲硫基 } -1 , 3-苯并间二氧杂环戊烯) -乙酸 MS [M]+= 400. Ora/e; ^-NMR (400MHz, DMS0-d 6 ) δ 12.79 (brs, 1H,), 7.85 - 7.82 (m, 2H), 7.48 - 7.45 (m, 3H), 7.04 (d, 1H), 6.85 (d, 2H), 6.52 (t, 1H), 4.35 (s, 2H), 2.96 (d, 2H), 2.20 (s, 3H) Example 2: 2-(5- {[4-Methyl-2-(4-trifluoromethylphenyl)-1, 3-thiazole-5-yl]-methylthio}-1,3-benzodioxole) -acetic acid
釆用实施例 1 的制备方法, 将其中的中间体 17 改为中间体 Using the preparation method of Example 1, the intermediate 17 was changed to an intermediate.
18, 得浅黄色无定形固体的 2-(5- {[4 -甲基 - 2-(4-三氟曱基苯 基)- 1, 3-噻唑- 5-基]-甲硫基 }-1, 3 -苯并间二氧杂环戊烯)-乙酸。 18, 2-(5-{[4-methyl-2-(4-trifluoromethylphenyl)-1, 3-thiazole-5-yl]-methylthio}- 1, 3-benzodioxole)-acetic acid.
MS [M]+= 468. Ora/e; ^- MR (400MHz, DMS0-d6) δ 12, 77 (brs, 1H,), 8.05 (d, 2H), 7.82 (d, 2H) , 7.04 (d, 1H), 6.85 (ra, 2H), 6.52 (t, 1H), 4.38 (s, 2H), 2.96 (d, 2H), 2.23 (s, 3H) 实施例 3: 2- (5- {[4-曱基 -2- (4-溴苯基)-1, 3-噻唑 -5-基] -甲硫 基}-1, 3-苯并间二氧杂环戊烯)-乙酸 MS [M]+= 468. Ora/e; ^- MR (400MHz, DMS0-d 6 ) δ 12, 77 (brs, 1H,), 8.05 (d, 2H), 7.82 (d, 2H) , 7.04 ( d, 1H), 6.85 (ra, 2H), 6.52 (t, 1H), 4.38 (s, 2H), 2.96 (d, 2H), 2.23 (s, 3H) Example 3: 2- (5- {[ 4-mercapto-2-(4-bromophenyl)-1,3-thiazol-5-yl]-methylthio}-1,3-benzodioxole)-acetic acid
采用实施例 1 的制备方法, 将其中的中间体 17 改为中间体 Using the preparation method of Example 1, the intermediate 17 was changed to an intermediate.
19, 得浅黄色无定形固体的:2- (5- {[4-甲基 -2-(4-溴苯基)-l,3- 噻唑-5-基]-甲硫基}-l, 3-苯并间二氧杂环戊烯)-乙酸。 19, obtained as a pale yellow amorphous solid: 2-(5-{[4-methyl-2-(4-bromophenyl)-l,3-thiazol-5-yl]-methylthio}-l, 3-benzodioxole)-acetic acid.
MS [M]+= 478. Om/e; ^-NMR (400MHz, DMS0-d6) 7.78 (d, 2H), 7.65 (d, 2H), 6.96 (d, 1H), 6.82—6.76 (m, 2H), 6.46 (s, 1H), 4.32 (s, 2H), 2.64 (s, 2H), 2.18 (s, 3H) 实施例 4: 2-(5-{[4-甲基-2-(4-氟苯基)-1,3-噻唑-5-基]-曱硫 基}-1, 3-苯并间二氧杂环戊烯)-乙酸 MS [M]+= 478. Om/e; ^-NMR (400MHz, DMS0-d 6 ) 7.78 (d, 2H), 7.65 (d, 2H), 6.96 (d, 1H), 6.82-6.76 (m, 2H), 6.46 (s, 1H), 4.32 (s, 2H), 2.64 (s, 2H), 2.18 (s, 3H) Example 4: 2-(5-{[4-methyl-2-(4) -fluorophenyl)-1,3-thiazol-5-yl]-indole sulfur }}-1,3-benzodioxole)-acetic acid
采用实施例 1 的制备方法, 将其中的中间体 17 改为中间体 20, 得浅黄色无定形固体的 2-(5- {[4-甲基 -2-(4-氟苯基)-l,3- 噻唑-5-基]-甲硫基}-l, 3 -苯并间二氧杂环戊烯)-乙酸。 Using the preparation method of Example 1, the intermediate 17 was changed to the intermediate 20 to give 2-(5-{[4-methyl-2-(4-fluorophenyl)-l as a pale yellow amorphous solid. , 3-thiazole-5-yl]-methylthio}-l,3-benzodioxole)-acetic acid.
MS[M]+= 418. Om/e; ^-NMR (400MHz, DMS0-d6) 12.67 (brs, 1H), 7.88 (m, 2H), 7.29 (ra, 2H) , 7.03 (d, 1H), 6.84 (d, 2H),MS[M]+= 418. Om/e; ^-NMR (400MHz, DMS0-d 6 ) 12.67 (brs, 1H), 7.88 (m, 2H), 7.29 (ra, 2H) , 7.03 (d, 1H) , 6.84 (d, 2H),
6.52 (t, 1H), 4.34 (s, 2H), 2.96 (d, 2H) , 2.19 (s, 3H) 实施例 5: 2- (5- {[4 -甲基 -2- (4-甲氧基苯基) -1, 3-噻唑- 5-基] - 甲硫基 }-1, 3-苯并间二氧杂环戊烯) -乙酸 6.52 (t, 1H), 4.34 (s, 2H), 2.96 (d, 2H), 2.19 (s, 3H) Example 5: 2- (5- {[4-methyl-2-(4-methoxy) Phenyl) -1,3-thiazole-5-yl]-methylthio}-1,3-benzodioxole)-acetic acid
采用实施例 1 的制备方法, 将其中的中间体 17 改为中间体 21, 得浅黄色无定形固体的 2- (5- {[4-甲基 -2-(4-甲氧基苯 基) -1, 3-噻唑 -5-基]-曱硫基 }-1, 3-苯并间二氧杂环戊烯) -乙酸。 Using the preparation method of Example 1, the intermediate 17 was changed to the intermediate 21 to give 2-(5-{[4-methyl-2-(4-methoxyphenyl) as a pale yellow amorphous solid. -1,3-thiazol-5-yl]-indolyl}-1,3-benzodioxole)-acetic acid.
MS [M]+= 430. Om/e; ^-NMR (400MHz, DMS0-d6) 7:77 (d, 2H),MS [M]+= 430. Om/e; ^-NMR (400MHz, DMS0-d 6 ) 7:77 (d, 2H),
7.01 (m, 3H), 6.83 (m, 2H) , 6.50 (s, 1H), 4.31 (s, 2H), 3.80 (s, 3H), 2.87 (s, 2H), 2.16 (s, 3H) 实施例 6: 2- (5- {[4 -甲基 -2- (3, 5-二甲氧基苯基)-1, 3-噻唑- 5- 基]-甲硫基 }- 1, 3-苯并间二氧杂环戊烯) -乙酸 7.1 (m, 3H), 6.83 (m, 2H) 6: 2-(5- {[4 -Methyl-2-(3,5-dimethoxyphenyl)-1,3-thiazole-5-yl]-methylthio}- 1, 3-benzene P-dioxole)-acetic acid
采用实施例 1 的制备方法, 将其中的中间体 17 改为中间体 22, 得浅黄色无定形固体的 2-(5-{[4-曱基-2-(3,5-二曱氧基苯 基) -1, 3-噻唑- 5-基]-甲硫基 }-1, 3-苯并间二氧杂环戊烯)-乙酸。 Using the preparation method of Example 1, the intermediate 17 was changed to the intermediate 22 to give a pale yellow amorphous solid of 2-(5-{[4-mercapto-2-(3,5-didecyloxy). Phenyl)-1,3-thiazole-5-yl]-methylthio}-1,3-benzodioxole)-acetic acid.
MS [M] += 460. lm/e; ^-NMR (400MHz, DMS0-d6) 7.01 (s, 1H), 6.94 (d, 2H), 6.82 (s, 2H) , 6.58 (t, 1H), 6.50 (s, 1H), 4.33 (s, 2H), 3.80 (s, 6H), 2.86 (s, 2H) , 2.19 (s, 3H) 实施例 7: 2- (5- {[4-曱基 -2- (2, 4-二氯苯基)- 1, 3 -噻唑 -5-基〗 - 曱硫基 }- 1, 3-苯并间二氧杂环戊烯) -乙酸 MS [M] += 460. lm/e; ^-NMR (400MHz, DMS0-d 6 ) 7.01 (s, 1H), 6.94 (d, 2H), 6.82 (s, 2H) , 6.58 (t, 1H) , 6.50 (s, 1H), 4.33 (s, 2H), 3.80 (s, 6H), 2.86 (s, 2H), 2.19 (s, 3H) Example 7: 2- (5- {[4-曱-2- (2, 4-dichlorophenyl)- 1, 3 -thiazol-5-yl- 曱thio}- 1, 3-benzodioxole)-acetic acid
采用实施例 1 的制备方法, 将其中的中间体 17改为中间体 Using the preparation method of Example 1, the intermediate 17 was changed to an intermediate.
23, 得浅黄色无定形固体的 2- (5-{[4-甲基- 2- (2,4-二氯苯 基)- 1, 3-噻唑- 5-基]-甲硫基 }-1, 3-苯并间二氧杂环戊烯)-乙酸。 23, 2-(5-{[4-methyl-2-(2,4-dichlorophenyl)- 1, 3-thiazole-5-yl]-methylthio}- 1, 3-benzodioxole)-acetic acid.
MS [M]+= 468. Om/e; 'H-NMR (400MHz, DMSO-d6) 8.17 (d, 1H), 7.79 (d, 1H), 7.54 (dd, 1H), 6.96 (d, 1H), 6.81 (dd, 1H), 6.77 (d, 1H), 6.46 (s, 1H), 4.36 (s, 2H) , 2.67 (s, 2H), 2.23 (s, 3H) 实施例 8: 2- (5- {[4-甲基- 2- (4-叔丁基苯基) -1, 3-噻唑- 5-基] - 曱硫基 3-苯并间二氧杂环戊烯) -乙酸 MS [M]+= 468. Om/e; 'H-NMR (400MHz, DMSO-d 6 ) 8.17 (d, 1H), 7.79 (d, 1H), 7.54 (dd, 1H), 6.96 (d, 1H) ), 6.81 (dd, 1H), 6.77 (d, 1H), 6.46 (s, 1H), 4.36 (s, 2H), 2.67 (s, 2H), 2.23 (s, 3H) Example 8: 2- ( 5- {[4-Methyl-2-(4-tert-butylphenyl)-1,3-thiazole-5-yl]-indolyl 3-benzodioxole)-acetic acid
采用实施例 1 的制备方法, 将其中的中间体 17改为中间体 Using the preparation method of Example 1, the intermediate 17 was changed to an intermediate.
24, 得浅黄色无定形固体的 2- (5-{[4-曱基 -2-(4-叔丁基苯 基)- 1, 3-噻唑 -5-基] -曱硫基}-1, 3-苯并间二氧杂环戊烯)-乙酸。 24, 2-(5-{[4-indolyl-2-(4-tert-butylphenyl)-1,3-thiazol-5-yl]-indenylthio}-1 as a pale yellow amorphous solid , 3-benzodioxole)-acetic acid.
MS [M] += 456. Om/e; ^-NMR (400MHz, DMSO-d6) 7.75 (d, 2H), 7.47 (d, 2H), 6.97 (s, 1H), 6.83 - 6.77 (m, 2H), 6.47 (s, 1H), 4.31 (s, 2H), 2.71 (s, 2H), 2.18 (s, 3H) , 1.29 (s, 9H) 实施例 9: 2- (5- {[4-甲基 -2- (噻吩 -2-基)- 1, 3-噻唑- 5 -基] -甲硫 基}-1, 3-苯并间二氧杂环戊烯) -乙酸 MS [M] += 456. Om/e; ^-NMR (400MHz, DMSO-d 6 ) 7.75 (d, 2H), 7.47 (d, 2H), 6.97 (s, 1H), 6.83 - 6.77 (m, 2H), 6.47 (s, 1H), 4.31 (s, 2H), 2.71 (s, 2H), 2.18 (s, 3H), 1.29 (s, 9H) Example 9: 2- (5- {[4- Methyl-2-(thiophen-2-yl)- 1, 3-thiazol-5-yl]-methylthio}-1,3-benzodioxole)-acetic acid
采用实施例 1 的制备方法, 将其中的中间体 17改为中间体 Using the preparation method of Example 1, the intermediate 17 was changed to an intermediate.
25,得浅黄色无定形固体的 2- (5- {[4-甲基- 2- (噻吩 -2-基)- 1, 3- 噻唑 -5-基]-甲硫基 }- 1, 3-苯并间二氧杂环戊烯) -乙酸。 25, 2-(5-{[4-methyl-2-(thiophen-2-yl)-1,3-thiazol-5-yl]-methylthio}- 1, 3 as a pale yellow amorphous solid - benzodioxole) - acetic acid.
MS [M]+= 406. Om/e; 'H-NMR (400MHz, DMSO-d6) 7.66 (d, 1H) , 7.55 (d, 1H), 7.12 (t, 1H), 6.98 (s, 1H) , 6.83-6.78 (m, 2H), 6.47 (s, 1H), 4.30 (s, 2H), 2.73 (s, 2H), 2.13 (s, 3H) 实施例 10: 2- (5- {[4-甲基- 2- (噻吩- 3-基) - 1, 3-噻唑 -5-基] -曱 硫基 }- 1, 3-苯并间二氧杂环戊烯) -乙酸 MS [M]+= 406. Om/e; 'H-NMR (400MHz, DMSO-d 6 ) 7.66 (d, 1H), 7.55 (d, 1H), 7.12 (t, 1H), 6.98 (s, 1H) ), 6.83-6.78 (m, 2H), 6.47 (s, 1H), 4.30 (s, 2H), 2.73 (s, 2H), 2.13 (s, 3H) Example 10: 2-(5- {[4-Methyl-2-(thiophen-3-yl)-1,3-thiazol-5-yl]-indenyl}} 1,1-benzophenanthrene Oxephthalene)-acetic acid
采用实施例 1 的制备方法, 将其中的中间体 17改为中间体 26,得浅黄色无定形固体的 2- (5-{ [4 -曱基 -2- (噻吩 -3-基)- 1,3- 噻唑 -5-基] -曱硫基 } -1 , 3-苯并间二氧杂环戊烯) -乙酸。 Using the preparation method of Example 1, the intermediate 17 was changed to the intermediate 26 to give 2-(5-{[4-indolyl-2-(thiophen-3-yl)-1) as a pale yellow amorphous solid. , 3-thiazole-5-yl]-indolyl}-1,3-benzodioxole)-acetic acid.
MS [M]+= 406. Oin/e; 'H-NMR (400MHz, D S0-d6) 8.03 (s, 1H),MS [M]+= 406. Oin/e; 'H-NMR (400MHz, D S0-d 6 ) 8.03 (s, 1H),
7.65 (m, 1H), 7.48 (d, 1H) , 6.97 (s, 1H), 6.83-6.77 (m, 2H), 6.47 (s, 1H), 4.30 (s, 2H), 2.74 (s, 2H), 2.15 (s, 3H) 实施例 11: 2- (5- {[4-甲基 -2- (萘 -2-基) -1, 3-噻唑- 5-基]-甲硫 基}-1, 3-苯并间二氧杂环戊烯)-乙酸 7.65 (m, 1H), 7.48 (d, 1H), 6.97 (s, 1H), 6.83-6.77 (m, 2H), 6.47 (s, 1H), 4.30 (s, 2H), 2.74 (s, 2H) , 2.15 (s, 3H) Example 11: 2-(5- {[4-Methyl-2-(naphthalen-2-yl)-1,3-thiazole-5-yl]-methylthio}-1 , 3-benzodioxole)-acetic acid
采用实施例 1 的制备方法, 将其中的中间体 17 改为中间体 Using the preparation method of Example 1, the intermediate 17 was changed to an intermediate.
27,得浅黄色无定形固体的 2-(5-{[4-曱基-2-(蔡-2-基)-1,3-噻 唑- 5-基]-甲硫基 } -1, 3-苯并间二氧杂环戊烯) -乙酸。 27, 2-(5-{[4-indolyl-2-(cai-2-yl)-1,3-thiazole-5-yl]-methylthio}-1, 3 as a pale yellow amorphous solid - benzodioxole) - acetic acid.
MS [M] += 450. Om/e; 'H-NMR ( 00MHz, DMS0-d6) 8.43 (s, 1H),MS [M] += 450. Om/e; 'H-NMR ( 00MHz, DMS0-d 6 ) 8.43 (s, 1H),
8.08-7.94 (m, 4H) , 7.57 (ra, 2H), 7.06 (s, 1H), 6.89-6.84 (m, 2H), 6.52 (t, 1H) , 4.38 (s, 2H) , 2.97 (d, 2H) , 2.24 (s, 3H) 实施例 12: 2-(5- {[4-甲基- 2- (1, 3-苯并间二氧杂环戊烯 -5- 基)- 1, 3-噻唑- 5-基]-甲硫基 }-1, 3 -苯并间二氧杂环戊烯)-乙酸 采用实施例 1 的制备方法, 将其中的中间体 17 改为中间体8.08-7.94 (m, 4H), 7.57 (ra, 2H), 7.06 (s, 1H), 6.89-6.84 (m, 2H), 6.52 (t, 1H), 4.38 (s, 2H), 2.97 (d, 2H), 2.24 (s, 3H) Example 12: 2-(5- {[4-Methyl-2-(1,3-benzodioxol-5-yl)- 1, 3 -thiazole-5-yl]-methylthio}-1,3-benzodioxole)-acetic acid, using the preparation method of Example 1, the intermediate 17 was changed to an intermediate
28, 得浅黄色无定形固体的 2-(5-{[4-甲基- 2-(1, 3-苯并间二氧 杂环戊烯- 5-基)- 1, 3-噻唑- 5-基]-甲硫基 }-1, 3-苯并间二氧杂环 戊烯) -乙酸。 28, a pale yellow amorphous solid of 2-(5-{[4-methyl-2-(1,3-benzodioxole-5-yl)-1, 3-thiazole-5 -yl]-methylthio}-1,3-benzodioxole)-acetic acid.
MS [M] += 444. Om/e; 'H-NMR (400MHz, DMS0-d6) 12.77 (brs, 1H), 7.35-7.33 (m, 2H), 7.02-6.97 (ra, 2H), 6.84 (s, 2H), 6.52 (t, 1H), 6.09 (s, 2H), 4.31 (s, 2H), 2.96 (d, 2H), 2.16 (s, 3H) 实施例 13: 2- [5- (2- {4-〖(4-乙基)苯基]苯氧基 }乙硫基)-1, 3 -苯 并间二氧杂环戊烯] -乙酸 MS [M] += 444. Om/e; 'H-NMR (400MHz, DMS0-d 6 ) 12.77 (brs, 1H), 7.35-7.33 (m, 2H), 7.02-6.97 (ra, 2H), 6.84 (s, 2H), 6.52 (t, 1H), 6.09 (s, 2H), 4.31 (s, 2H), 2.96 (d, 2H), 2.16 (s, 3H) Example 13: 2- [5- (2- {4-〖 (4-ethyl)phenyl]phenoxy}ethylthio)-1,3-benzodioxole]-acetic acid
采用实施例 1 的制备方法, 将其中的中间体 17 改为中间体 29,得浅黄色无定形固体的 2- [5- (2- {4- [(4-乙基)苯基]苯氧基 } 乙硫基)- 1, 3-苯并间二氧杂环戊烯]-乙酸。 Using the preparation method of Example 1, the intermediate 17 was changed to the intermediate 29 to give 2-[5-(2-{4-[(4-ethyl)phenyl]phenoxy) as a pale yellow amorphous solid. Base} ethylthio)- 1, 3-benzodioxole]-acetic acid.
MS [M]+= 437. Om/e; JH-NMR (400MHz, DMS0-d6) 7.44-7.36 (m 4H), 7.15 (d, 2H), 6.98 (s, 1H), 6.85-6.79 (ra, 4H) , 6.47 (s, 1H), 4.26 (t, 2H), 3.15 (t, 2H) , 2.74 (s, 2H) , 2.55 (q, 2H) , 1.18 (t, 3H) 实施例 14: 2- {5- [2- (咔唑 -9-基)乙硫基 ]-1,3-苯并间二氧杂环 戊烯 } -乙酸 MS [M]+= 437. Om/e; J H-NMR (400MHz, DMS0-d 6 ) 7.44-7.36 (m 4H), 7.15 (d, 2H), 6.98 (s, 1H), 6.85-6.79 ( Ra, 4H), 6.47 (s, 1H), 4.26 (t, 2H), 3.15 (t, 2H), 2.74 (s, 2H), 2.55 (q, 2H), 1.18 (t, 3H) Example 14: 2-{5- [2-(carbazol-9-yl)ethylthio]-1,3-benzodioxole}-acetic acid
采用实施例 1 的制备方法, 将其中的中间体 17 改为中间体 30, 得浅黄色无定形固体的 2- {5- [2- (咔唑 -9-基)乙硫基 ]-1, 3- 苯并间二氧杂环戊烯 }-乙酸。 Using the preparation method of Example 1, the intermediate 17 was changed to the intermediate 30 to give 2-{5-[2-(carbazol-9-yl)ethylthio]-1 as a pale yellow amorphous solid. 3-benzodioxole}-acetic acid.
MS[M]+= 406. Ora/e; 'H-NMR ( 00MHz, CDC13) 8.07 (m, 2H), 7.47 (ra, 4H), 7.25 (m, 2H) , 6.96(s, 1H), 6.80 (ra, 2H) , 6.44 (s, 1H), 4.21 (t, 2H), 3.28 (t, 2H), 2.70 (s, 2H) 实施例 15: 2- {5- [2- (吩噻嗪 -10-基)乙硫基 ]- 1, 3-苯并间二氧杂 环戊烯 } -乙酸 MS[M]+= 406. Ora/e; 'H-NMR ( 00MHz, CDC1 3 ) 8.07 (m, 2H), 7.47 (ra, 4H), 7.25 (m, 2H), 6.96(s, 1H), 6.80 (ra, 2H), 6.44 (s, 1H), 4.21 (t, 2H), 3.28 (t, 2H), 2.70 (s, 2H) Example 15: 2- {5- [2- (phenothiazine) -10-yl)ethylthio]- 1, 3-benzodioxole}-acetic acid
采用实施例 1 的制备方法, 将其中的中间体 17 改为中间体 31, 得浅黄色无定形固体的 2- {5-[2- (吩噻嗪- 10-基) 乙硫 基] -1, 3-苯并间二氧杂环戊烯 }-乙酸。 MS [M] += 438. Om/e; ^-NMR (400MHz, CDC13) 7. 38-7. 03 (m, 8H), 6. 97 (s, 1H) , 6. 82 (m, 2H) , 6. 46 (s, 1H), 4. 18 (t, 2H) , 3. 24 (t, 2H) , 2. 72 (s, 2H)。 实施例 16: 部分本发明化合物对 PPAR的激动作用 Using the preparation method of Example 1, the intermediate 17 was changed to the intermediate 31 to give 2-{5-[2-(phenothiazine-10-yl)ethylthio]-1 as a pale yellow amorphous solid. , 3-benzodioxole}-acetic acid. MS [M] += 438. Om/e; ^-NMR (400MHz, CDC1 3 ) 7. 38-7. 03 (m, 8H), 6. 97 (s, 1H) , 6. 82 (m, 2H ), 6. 46 (s, 1H), 4. 18 (t, 2H), 3. 24 (t, 2H), 2. 72 (s, 2H). Example 16: Partial agonistic effects of the compounds of the invention on PPAR
将 293- T细胞接种入 48孔板, 细胞密度为 2-4xl 04个 /孔, 培养液为 10%脱脂胎牛血清 (FCS ) 的无酚红无抗生素的 1640培 养基。 48小时后,将培养液更换为 5%脱脂 FCS的无酚红无抗生素 的 1640 培养基, 然后分别把三种亚型的 pM-hPPAR/GAL4、 pB4-RES-tk-luc和 pRL-CMV- Rluc三种盾粒共转染到 293-T细胞 中, 24小时后给药, 以双重激动剂 GW409544作为 α和 γ亚型的 阳性对照药,以 GW501516作为 δ亚型的阳性对照药。 给药后 24 小时检测荧光素酶的强度,结果以 PPAR 活化能(相对于对照药物 (100%)的值)以及对 PPAR6的 EC5Q值表示,分别见表 1和表 2。 The 293- T cells were seeded into 48-well plates at a cell density of 2-4xl 0 4 / well, culture was 10% defatted fetal calf serum (FCS) in phenol red-free 1640 medium without antibiotics. After 48 hours, the culture medium was changed to 5% defatted FCS phenol red-free antibiotic-free 1640 medium, and then three subtypes of pM-hPPAR/GAL4, pB4-RES-tk-luc and pRL-CMV-, respectively. Rluc three kinds of shield granules were co-transfected into 293-T cells, and administered 24 hours later. The double agonist GW409544 was used as a positive control for α and γ subtypes, and GW501516 was used as a positive control for δ subtype. The intensity of luciferase was measured 24 hours after administration, and the results were expressed as PPAR activation energy (value relative to control drug (100%)) and EC 5Q value for PPAR6, as shown in Tables 1 and 2, respectively.
表 1.部分本发明化合物对 PPAR的激动作用 Table 1. Some of the agonistic effects of the compounds of the invention on PPAR

EC50 (uM) 0. 396 0. 084 0. 092 0. 416 0. 149 化合物 实施例 7 实施例 9 实施例 10 实施例 11 实施例 12EC50 (uM) 0. 396 0. 084 0. 092 0. 416 0. 149 Compound Example 7 Example 9 Example 10 Example 11 Example 12
BC50 (uM) 0. 147 1. 042 0. 684 0. 117 0. 454 BC50 (uM) 0. 147 1. 042 0. 684 0. 117 0. 454
Claims
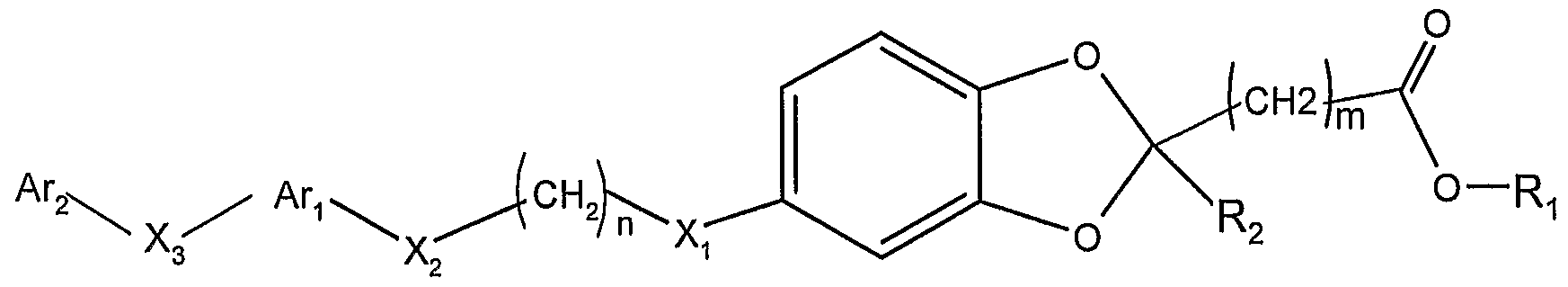


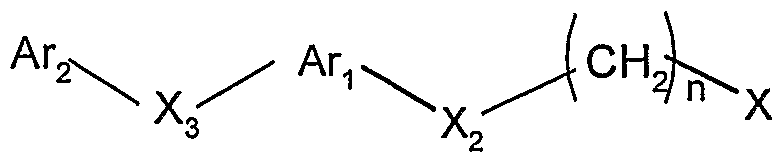





Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN200610002081.4 | 2006-01-24 | ||
| CN2006100020814A CN101007798B (en) | 2006-01-24 | 2006-01-24 | Benzodioxole derivatives and their preparation method and medicinal uses |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007085136A1 true WO2007085136A1 (en) | 2007-08-02 |
Family
ID=38308838
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2006/000373 Ceased WO2007085136A1 (en) | 2006-01-24 | 2006-03-13 | 1,3-benzodioxolecyclopentene derivates, preparation process and medical uses thereof |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN101007798B (en) |
| WO (1) | WO2007085136A1 (en) |
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US10328053B2 (en) | 2016-08-26 | 2019-06-25 | Gilead Sciences, Inc. | Substituted pyrrolizine compounds and uses thereof |
| US10689371B2 (en) | 2018-04-18 | 2020-06-23 | Constellation Pharmaceuticals, Inc. | Modulators of methyl modifying enzymes, compositions and uses thereof |
| US10836769B2 (en) | 2018-02-26 | 2020-11-17 | Gilead Sciences, Inc. | Substituted pyrrolizine compounds and uses thereof |
| US11919912B2 (en) | 2018-05-21 | 2024-03-05 | Constellation Pharmaceuticals, Inc. | Modulators of methyl modifying enzymes, compositions and uses thereof |
| US12516040B2 (en) | 2019-07-24 | 2026-01-06 | Constellation Pharmaceuticals, Inc. | Crystalline forms of 7-chloro-2-(4-3-methoxyazetidin-1-yl)cyclohexyl)-2,4-dimethyl-N-((6-methyl-4-(methylthio)-2-oxo-1,2-dihydropyridin-3-yl)methyl)benzo[d][1,3]dioxole-5-carboxamide |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0107622A1 (en) * | 1982-10-22 | 1984-05-02 | Ciba-Geigy Ag | Benzodioxole derivatives, process for their preparation and pharmaceutical compositions containing them |
| JPS6239583A (en) * | 1985-08-14 | 1987-02-20 | Yamanouchi Pharmaceut Co Ltd | Benzodioxazole derivative and production thereof |
| US6924276B2 (en) * | 2001-09-10 | 2005-08-02 | Warner-Lambert Company | Diacid-substituted heteroaryl derivatives as matrix metalloproteinase inhibitors |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6875780B2 (en) * | 2002-04-05 | 2005-04-05 | Warner-Lambert Company | Compounds that modulate PPAR activity and methods for their preparation |
-
2006
- 2006-01-24 CN CN2006100020814A patent/CN101007798B/en not_active Expired - Fee Related
- 2006-03-13 WO PCT/CN2006/000373 patent/WO2007085136A1/en not_active Ceased
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0107622A1 (en) * | 1982-10-22 | 1984-05-02 | Ciba-Geigy Ag | Benzodioxole derivatives, process for their preparation and pharmaceutical compositions containing them |
| JPS6239583A (en) * | 1985-08-14 | 1987-02-20 | Yamanouchi Pharmaceut Co Ltd | Benzodioxazole derivative and production thereof |
| US6924276B2 (en) * | 2001-09-10 | 2005-08-02 | Warner-Lambert Company | Diacid-substituted heteroaryl derivatives as matrix metalloproteinase inhibitors |
Cited By (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US10328053B2 (en) | 2016-08-26 | 2019-06-25 | Gilead Sciences, Inc. | Substituted pyrrolizine compounds and uses thereof |
| US10874640B2 (en) | 2016-08-26 | 2020-12-29 | Gilead Sciences, Inc. | Substituted pyrrolizine compounds and uses thereof |
| US12161625B2 (en) | 2016-08-26 | 2024-12-10 | Gilead Sciences, Inc. | Substituted pyrrolizine compounds and uses thereof |
| US10836769B2 (en) | 2018-02-26 | 2020-11-17 | Gilead Sciences, Inc. | Substituted pyrrolizine compounds and uses thereof |
| US11420974B2 (en) | 2018-02-26 | 2022-08-23 | Gilead Sciences, Inc. | Substituted pyrrolizine compounds and uses thereof |
| US10689371B2 (en) | 2018-04-18 | 2020-06-23 | Constellation Pharmaceuticals, Inc. | Modulators of methyl modifying enzymes, compositions and uses thereof |
| US11274095B2 (en) | 2018-04-18 | 2022-03-15 | Constellation Pharmaceuticals, Inc. | Modulators of methyl modifying enzymes, compositions and uses thereof |
| US11919912B2 (en) | 2018-05-21 | 2024-03-05 | Constellation Pharmaceuticals, Inc. | Modulators of methyl modifying enzymes, compositions and uses thereof |
| US12516040B2 (en) | 2019-07-24 | 2026-01-06 | Constellation Pharmaceuticals, Inc. | Crystalline forms of 7-chloro-2-(4-3-methoxyazetidin-1-yl)cyclohexyl)-2,4-dimethyl-N-((6-methyl-4-(methylthio)-2-oxo-1,2-dihydropyridin-3-yl)methyl)benzo[d][1,3]dioxole-5-carboxamide |
Also Published As
| Publication number | Publication date |
|---|---|
| CN101007798B (en) | 2011-01-26 |
| CN101007798A (en) | 2007-08-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007085136A1 (en) | 1,3-benzodioxolecyclopentene derivates, preparation process and medical uses thereof | |
| JP4345230B2 (en) | Carboxylic acid derivatives and drugs containing the derivatives as active ingredients | |
| JP3490704B2 (en) | Thiazole and oxazole derivatives and their pharmaceutical use | |
| US7553867B2 (en) | Furan or thiophene derivative and medicinal use thereof | |
| TWI404712B (en) | Benzoazepin-oxy-acetic acid derivatives as ppar-delta agonists used for the increase of hdl-c, lower ldl-c and lower cholesterol | |
| JP2000507216A (en) | Substituted 4-hydroxyphenylalkanoic acid derivatives having agonist activity on PPAR-γ | |
| JPWO1999046232A1 (en) | Carboxylic acid derivatives and drugs containing the derivatives as active ingredients | |
| TW200400027A (en) | Compounds that modulate PPAR activity | |
| JP2008528628A (en) | Antidiabetic bicyclic compound | |
| EP2001844A2 (en) | Bicyclic carboxylic acid derivatives useful for treating metabolic disorders | |
| JP2013067652A (en) | Activating agent for peroxisome proliferator activated receptor delta | |
| JPWO2002046176A1 (en) | Activator of peroxisome proliferator-activated receptor | |
| WO2009012650A1 (en) | Aryl pyrimidine derivatives, preparation methods and pharmaceutical uses thereof | |
| CN101007804B (en) | 1,3-benzodioxole-2,2-dicarboxylic acid derivatives and their preparation method and medicinal uses | |
| CN100357280C (en) | Dihydronaphthalene devivatives and drugs contanining these compounds as active ingrdeient | |
| JP4986927B2 (en) | Medicine | |
| JP4832897B2 (en) | Ester derivatives and their pharmaceutical uses | |
| WO2004011446A1 (en) | Indane, dihydrobenzofuran, and tetrahydronaphthalene carboxylic acid derivatives and their use as antidiabetics | |
| US7241784B2 (en) | Carboxylic acid derivative and a pharmaceutical composition containing the derivative as active ingredient | |
| JP2009514973A (en) | Compounds and compositions as PPAR modulators | |
| JPWO2012081570A1 (en) | Lactam compound or salt thereof and PPAR activator | |
| JP2005035966A (en) | Furan or thiophene derivative and its pharmaceutical use | |
| WO2005115998A1 (en) | TYROSINE DERIVATIVES SUBSTITUTED BY ALKANOYLAS hPPARα & hPPARϜ ANGONISTS | |
| WO2007085202A1 (en) | Oxamic acid derivatives,preparation methods and medical uses thereof | |
| KR960008245B1 (en) | Rhodanine derivatives and pharmaceutical compositions |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 06722031 Country of ref document: EP Kind code of ref document: A1 |