WO2007057762A2 - Osmotic bi-layer tablet - Google Patents
Osmotic bi-layer tablet Download PDFInfo
- Publication number
- WO2007057762A2 WO2007057762A2 PCT/IB2006/003255 IB2006003255W WO2007057762A2 WO 2007057762 A2 WO2007057762 A2 WO 2007057762A2 IB 2006003255 W IB2006003255 W IB 2006003255W WO 2007057762 A2 WO2007057762 A2 WO 2007057762A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- layer
- drug
- osmotic
- tablet according
- water
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0002—Galenical forms characterised by the drug release technique; Application systems commanded by energy
- A61K9/0004—Osmotic delivery systems; Sustained release driven by osmosis, thermal energy or gas
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2072—Pills, tablets, discs, rods characterised by shape, structure or size; Tablets with holes, special break lines or identification marks; Partially coated tablets; Disintegrating flat shaped forms
- A61K9/2086—Layered tablets, e.g. bilayer tablets; Tablets of the type inert core-active coat
Definitions
- Osmotic systems for controlled drug-delivery applications are well known in the art.
- Various configurations have been described, including single layer, bi-layer, tri-layer and tablet-in-tablet configurations (for example, see A.G. Thombre et al., J. Controlled Release 94(1), 75-89, 2004).
- the present invention relates to osmotic bi-layer dosage forms. These dosage forms have a core comprising a drug layer and a sweller layer.
- the core is surrounded by a water-permeable coating which is perforated by one or more delivery ports. In use water enters the core through the coating.
- the drug layer is fluidized and the sweller layer expands to push the fluidized drug layer out of the coating through the delivery ports. While such dosage forms have been successfully used for some drugs, there have been difficulties in applying them to low solubility drugs, particularly at high dose levels.
- Suitable solubility/wettability enhancing agents include hydroxyalkyl cellulose derivatives such as hydroxypropyl methyl cellulose (HPMC), hydroxyethyl cellulose (HEC) and hydroxypropyl cellulose (HPC).
- HPMC hydroxypropyl methyl cellulose
- HEC hydroxyethyl cellulose
- HPPC hydroxypropyl cellulose
- HPMC having an average molecular weight of from about 50,000 to about 100,000Da.
- the drug-containing layer includes a drug crystallization inhibiting agent.
- the water-swellable layer has a radial tensile strength (RTS) in the range of from about 0.5 to about 5MPa at 0.3mm/s. In a more preferred embodiment the water-swellable layer has an RTS in the range of from about 0.5 to about 3MPa at 0.3mm/s.
- RTS radial tensile strength
- the intrinsic viscosity of the water-swellable layer when fully hydrated, is preferably from about 10 7 to 10 9 cP.
- Sample volume ⁇ 0.4ml
- Sample gap 500um Flow Procedure: 1 Conditioning: 37°C, wait for correct temperature, 30sec equilibration
- the resulting bi-layer tablet core had a total weight of 422 mg and contained a total of 33.70 wt% sildenafil citrate (142 mg), 18.34 wt% PEO 600,000, 18.34 wt% xylitol, 3.74 wt% HPMCAS, 16.33 wt% PEO 5,000,000, 8.62 wt% microcrystalline cellulose, 0.88 wt% magnesium stearate, and 0.05 wt% blue lake dye.
- Assays of three tablets averaged 103 ⁇ 4.9 mg of active drug (mgA). Coatings were applied by a Vector LDCS-20 pan coater.
- Example 20 released sildenafil at an average rate from two to twelve hours of 6.4 wt%/hour and had released 82 wt% of sildenafil by sixteen hours, and had released 95 wt% by 20 hours.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Medicinal Preparation (AREA)
Abstract
An osmotic bi-layer tablet for the controlled release of a low-solubility drug comprises an inner core containing a drug-containing layer and a water-swellable layer, and a water-permeable outer coating including at least one delivery port, wherein the drug- containing layer comprises a drug substance, polyethylene oxide polymer having an average molecular weight from about 200,000 to 600,000Da, and an osmogen, and the water-swellable layer comprises polyethylene oxide polymer having a weight average molecular weight of from about 4,000,000 to about 7,000,000Da and a sweller layer strengthening agent.
Description
CONTROLLED-RELEASE DOSAGE FORM
The invention relates to an osmotic bi-layer dosage form for the controlled-release of low-solubility drugs.
BACKGROUND
Osmotic systems for controlled drug-delivery applications are well known in the art. Various configurations have been described, including single layer, bi-layer, tri-layer and tablet-in-tablet configurations (for example, see A.G. Thombre et al., J. Controlled Release 94(1), 75-89, 2004). The present invention relates to osmotic bi-layer dosage forms. These dosage forms have a core comprising a drug layer and a sweller layer. The core is surrounded by a water-permeable coating which is perforated by one or more delivery ports. In use water enters the core through the coating. The drug layer is fluidized and the sweller layer expands to push the fluidized drug layer out of the coating through the delivery ports. While such dosage forms have been successfully used for some drugs, there have been difficulties in applying them to low solubility drugs, particularly at high dose levels.
Conventional osmotic bilayer tablet systems which incorporate high levels of low- solubility drugs can demonstrate poor delivery of drug substance from the tablet into the use environment. Because the drug does not dissolve it is necessary to include one or more agents in the drug layer to ensure an appropriate degree of fluidity to entrain the solid drug particles. The extruded material initially forms a plume, and these additional agents must also allow for rapid plume dispersion. The components of the sweller layer must provide sufficient swelling capacity to ensure complete extrusion of the drug layer, while the mechanical properties of the sweller layer must be adapted to the viscosity of the fluidized drug layer.
Appel et al., International Publication No. WO 01/47500, relates to osmotic bilayer formulations and teaches that improved drug layer dissolution properties or internal drug layer concentration effects are possible via inclusion into the drug layer of a significant amount of a polymer such as hydroxypropyl methyl cellulose acetate succinate or hydroxypropyl methyl cellulose. The document also considers a variety of swelling materials, including PEO and microcrystalline cellulose. In particular, it
teaches the use of using highly swelling materials such as sodium cross carmellose or sodium starch glycolate in the sweller layer in order to achieve good release of drug from osmotic tablets containing low-solubility drugs
US Patent No. 4,765,989 discloses an osmotic bilayer tablet in which the sweller layer (second osmotic composition) comprises an osmopolymer 19 and an optional osmogen 18. (CoI. 5, line 42). This patent states, at column 5, lines 57-68, that
"Osmopolymers 17 and 19 preferably are mixed with [osmogen] 16 and 18 for imbibing the maximum volume of external fluid into compartment 14. This imbibed fluid is available to osmopolymers 17 and 19 to optimize the volumetric rate and for total expansion of osmopolymer 17 and 19. That is, osmopolymers
17 and 19 absorb fluid imbibed into compartment 14 by the osmotic imbibition action of osmopolymers 17 and 19 supplemented by the osmotic imbibition action of [osmogens] 16 and 18 for effecting the maximum expansion of osmopolymers 17 and 19 from a rested to an enlarged state."
As such, US 4,765,989 patent teaches that the sweller layer composition should be chosen to effect maximum expansion of the sweller layer.
These publications do not provide a complete solution to the problems of providing osmotic bilayer tablets comprising low solubility drugs. There remains a need for osmotic bilayer tablets that provide effective and complete delivery of low solubility drugs into the use environment, particularly at high drug loading. The tablets should preferably have good mechanical properties such as low friability and resistance to bursting and delamination.
SUMMARY OF THE INVENTION
The present invention provides an osmotic bi-layer tablet comprising (A) an inner core containing a drug-containing layer and a water-swellable layer, and (B) a water- permeable outer coating including at least one delivery port, wherein:
(i) said drug-containing layer comprises:
(a) a drug substance or a pharmaceutically acceptable salt thereof, having a solubility in water of no more than 140 mg/mL at any pH in the range 2 to 7.5, at from about 40% to about 80% by weight of drug-containing layer; (b) polyethylene oxide polymer having an average molecular weight from about 200,000 to 600,000Da at from about 10% to about 30% by weight of drug-containing layer; and (c) osmogen at from about 10% to 40% by weight of drug-containing layer;
(ii) said water-swellable layer comprises:
(a) polyethylene oxide polymer having a weight average molecular weight of from about 4,000,000 to about 7,000,000Da at from about 35% to about 80% by weight of water-swellable layer; (b) a sweller layer strengthening agent at from about 5% to about
60% by weight of water-swellable layer; and
(iii) the weight ratio of said drug-containing layer to said water-swellable layer ranges from about 2:1 to about 4.5:1.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 depicts a typical osmotic bilayer tablet. The tablet 10 comprises a core 12 comprising a drug layer 14 and a sweller layer 16. The core 12 is surrounded by a water-permeable, substantially drug-impermeable coating 18. After introduction to an aqueous use environment, the core 12 imbibes water through the coating 18, causing the sweller layer 16 to expand. The sweller layer 16 pushes the drug layer 14 out of a delivery port 20 into the aqueous use environment.
Figure 2 illustrates the formation of a "drug plume" during the release of a drug from a bilayer tablet in a use environment.
Figure 3 illustrates drug layer plume formation and dispersal according to Study C.
Figure 4 illustrates the results of a crowning study (see Study K).
DETAILED DESCRIPTION OF THE INVENTION
Tablet The present invention provides an osmotic bi-layer tablet that is adapted to providing good delivery of a low-solubility drug, and particularly of a high dose of such a drug, while providing excellent mechanical properties. As is conventional in the art, the tablet of the invention comprises a bi-layer inner core and a water permeable outer coating. One or more delivery ports are provided in the coating to permit delivery of the drug to the use environment.
(A) Inner Core
The inner core is composed of a drug-containing layer and a water-swellable layer. The weight ratio of the drug-containing layer to the water-swellable layer is in the range of from about 2:1 to about 4.5:1. Preferably the weight ratio is in the range of from about 2:1 to about 3:1.
(i) Drug-Containing Layer
The drug-containing layer comprises the low-solubility drug, polyethylene oxide polymer, and an osmogen.
(a) Low-Solubility Drug
The low-solubility drug is present at from about 40% to about 80% by weight of the drug-containing layer. Preferably it is present at from about 40% to about 65% by weight of drug-containing layer. More preferably, it is present at from about 45% to about 60% by weight of drug-containing layer. Most preferably, it is present at from about 45% to about 55% by weight of drug-containing layer.
As used herein, the term "low-solubility drug" refers, unless otherwise indicated, to a drug substance or a pharmaceutically acceptable salt thereof, having a solubility in water of no more than 140 mg/mL at any pH in the range 2 to 7.5. The determination of drug solubility is a routine operation in the art.
The osmotic bi-layer tablet is particularly suitable for drug substances having a solubility in water of no more than 80 mg/mL at any pH in the range 2 to 7.5, more particularly 40 mg/mL, and most particularly 20 mg/mL.
The term "drug substance" includes both pharmacologically active substances and prodrugs thereof.
The use of any low-solubility drug is contemplated. The drug may be employed in its neutral (e.g., free acid, free base or zwitterion) form, or in the form of a pharmaceutically acceptable salt, and may be in anhydrous, hydrated, or solvated form. The drug may be in either crystalline or amorphous. Where the drug is crystalline, it may be present as a single polymorph or as a mixture of two or more polymorphs. The drug may also be in the form of a solid dispersion.
Preferred classes of drugs include, but are not limited to, antihypertensives, antidepressants, antianxiety agents, anticlotting agents, anticonvulsants, blood glucose-lowering agents, decongestants, antihistamines, antitussives, antiinflammatories, antipsychotic agents, cognitive enhancers, cholesterol-reducing agents, cholesterol ester transfer protein inhibitors, high-density lipoprotein enhancers, antiobesity agents, autoimmune disorders agents, anti-impotence agents, antibacterial and antifungal agents, hypnotic agents, anti-Parkinsonism agents, antibiotics, antiviral agents, anti-neoplasties, barbituates, sedatives, nutritional agents, beta blockers, emetics, anti-emetics, diuretics, anticoagulants, cardiotonics, androgens, corticoids, anabolic agents, growth hormone secretagogues, anti-infective agents, coronary vasodilators, carbonic anhydrase inhibitors, antiprotozoals, gastrointestinal agents, serotonin antagonists, anesthetics, hypoglycemic agents, dopaminergic agents, anti- Alzheimer's Disease agents, anti-ulcer agents, platelet inhibitors and glycogen phosphorylase inhibitors.
Specific examples of the above and other classes of drugs and therapeutic agents deliverable by the invention are set forth below, by way of example only. Specific examples of antihypertensives include prazosin, nifedipine, trimazosin, amlodipine, and doxazosin mesylate; a specific example of an antianxiety agent is hydroxyzine; a specific example of a blood glucose lowering agent is glipizide; a specific example of an anti-impotence agent is sildenafil citrate; specific examples of anti-neoplasties
include chlorambucil, lomustine and echinomycin; specific examples of antiinflammatory agents include betamethasone, prednisolone, piroxicam, aspirin, flurbiprofen and (+)-N-{4-[3-(4fluorophenoxy)phenoxy]-2-cyclopenten-1 -yl}-N- hyroxyurea; a specific example of a barbituate is phenobarbital; specific examples of antivirals include acyclovir, nelfinavir, and virazole; specific examples of vitamins/nutritional agents include retinol and vitamin E; specific examples of a β- blocker include timolol and nadolol; a specific example of an emetic is apomorphine; specific examples of a diuretic include chlorthalidone and spironolactone; a specific example of an anticoagulant is dicumarol; specific examples of cardiotonic include digoxin and digitoxin; specific examples of an androgen include 17-methyltestosterone and testosterone; a specific example of a mineral corticoid is desoxycorticosterone; a specific example of a steroidal hypnotic/anesthetic is alfaxalone; specific examples of an anabolic agent include fluoxymesterone and methanstenolone; specific examples of antidepression agents include fluoxetine, pyroxidine, venlafaxine, sertraline, paroxetine, sulpiride, [3,6-dimethyl-2-(2,4,6-trimethyl-phenoxy)-pyridin-4-yl]-(l- ethylpropyl)-amine and 3,5-dimethyl-4-(3'-pentoxy)-2-(2',4',6'-trimethylphenoxy)- pyridine; specific examples of an antibiotic include ampicillin and penicillin G; specific examples of an anti-infective include benzalkonium chloride and chlorhexidine; specific examples of a coronary vasodilator include nitroglycerin and mioflazine; a specific example of a hypnotic is etomidate; specific examples of a carbonic anhydrase inhibitor include acetazolamide and chlorzolamide; specific examples of an antifungal include econazole, terconazole, fluconazole, voriconazole and griseofulvin; a specific example of an antiprotozoal is metronidazole; a specific example of an imidazole-type antineoplastic is tubulazole; specific examples of an anthelmintic agent include thiabendazole, oxfendazole and morantel; specific examples of an antihistaminic include astemizole, levocabastine, cetirizine, and cinnarizine; a specific example of a decongestant is pseudoephedrine; specific examples of antipsychotics include fluspirilene, penfluridol, risperidone and ziprasidone; specific examples of a gastrointestinal agent include loperamide and cisapride; specific examples of a serotonin antagonist include ketanserin and mianserin; a specific example of an anesthetic is lidocaine; a specific example of a hypoglycemic agent is acetohexamide; a specific example of an anti-emetic is dimenhydrinate; a specific example of an antibacterial is cotrimoxazole; a specific example of a dopaminergic agent is L-DOPA; specific examples of anti-Alzheimer agents are THA and donepezil; a specific example of an anti-ulcer agent/H2 antagonist is famotidine; specific examples of a
sedative/hypnotic include chlordiazepoxide and triazolam; a specific example of a vasodilator is alprostadil; a specific example of a platelet inhibitor is prostacyclin; specific examples of an ACE inhibitor/antihypertensive include enalaprilic acid and lisinopril; specific examples of a tetracycline antibiotic include oxytetracycline and minocycline; specific examples of a macrolide antibiotic include azithromycin, clarithromycin, erythromycin and spiramycin; specific examples of glycogen phosphorylase inhibitors include [R-(R*S*)]-5-chloro-N-[2-hydroxy-3-
{methoxymethylamino}-3-oxo-l-(phenylmethyl)-propyl]-IH-indole-2-carboxamide and 5- chloro-1 H-indole-2-carboxylic acid [(1 S)-benzyl-(2R)-hydroxy-3((3R,4S)dihydroxy- pyrrolidin-1 -yl-)-oxypropyl]amide.
Further examples of drugs deliverable by the invention are the glucose-lowering drug chlorpropamide, the anti-fungal fluconazole, the anti-hypercholesterolemic atorvastatin, the antipsychotic thiothixene, the anxiolytics hydroxyzine and doxepin, the anti- hypertensive amlodipine, the antiinflammatories piroxicam, celicoxib, valdicoxib and carprofen, and the antibiotics carbenicillin indanyl, bacampicillin, troleandomycin, and doxycycline.
(b) Polyethylene oxide polymer A second component of the drug-containing layer is a polyethylene oxide polymer (PEO polymer). The PEO polymers suitable for use in the drug layers of the osmotic bilayer tablets according to the present invention become sufficiently viscous in the dosage form to entrain the drug but remain sufficiently fluid to be pushed out of the delivery port.
The polyethylene oxide polymer is present at from about 10% to about 30% by weight of drug-containing layer. Preferably it is present at from about 15% to about 25% by weight of drug-containing layer. More preferably it is present at from about 18% to about 23% by weight of drug-containing layer. Most preferably it is present at about 20% by weight of drug-containing layer.
The PEO polymer of the drug-containing layer is a polyethylene oxide polymer having an average molecular weight from about 200,000 to 600,000Da. Preferably the polyethylene oxide polymer has an average molecular weight from about 300,000 to about 600,000Da. More preferably the polyethylene oxide polymer has an average
molecular weight from about 400,000 to about 600,000Da. Most preferably the polyethylene oxide polymer has an average molecular weight of about 600,000Da.
In a preferred embodiment, the PEO polymer is a non-cross linked polyethylene oxide polymer. Such non-cross linked polyethylene oxide polymers are alternatively referred to as linear chain polyethylene oxide polymers.
Suitable PEO polymers include "POLYOX™ Water-Soluble Resins NF" such as WSR N-80, WSR N-750 and WSR N-205, which are available from The Dow Chemical Company. Of these, WSR N-205 is especially preferred.
In another embodiment, the polyethylene oxide polymer in the drug-containing layer is present as a mixed system with another suitable material. Examples of such suitable materials include polyols, and oligomers of polyethers, such as ethylene glycol oligomers or propylene glycol oligomers. In addition, mixtures of polyfunctional organic acids and cationic materials such as amino acids or multivalent salts, such as calcium salts may be used. Of particular utility are polymers such as polyvinyl alcohol, polyvinyl pyrrolidone (PVP), cellulosics such as hydroxyethyl cellulose (HEC), hydroxypropylcellulose (HPC), hydroxypropyl methyl cellulose (HPMC), methyl cellulose (MC), carboxymethyl cellulose (CMC) and carboxyethyl cellulose (CEC), gelatin, xanthan gum or any other water-soluble polymer that forms an aqueous solution with a viscosity similar to that of the polymers listed above. In such a mixed system the further material should have a molecular weight that is in a range sufficient to yield viscosities similar to the viscosities achieved by the polyethylene oxide polymers defined hereinbefore.
(c) Osmogen
A third component of the drug-containing layer is an osmogen.
Osmogens, alternatively known as osmotically effective agents or simply osmagents, are materials that imbibe water from the use environment to form an aqueous solution within the interior of the tablet such that its osmotic pressure exceeds that of the use environment, thereby providing an osmotic pressure driving force for permeation of water from the use environment into the tablet core.
The osmogen is present at from about 10% to about 40% by weight of drug-containing layer.
Suitable osmogens are those with a sufficiently low molecular weight and high solubility that the amount present in the drug-containing layer results in the required osmotic pressure. The osmotic pressure of a material can be calculated using the van't Hoff equation. (See, e.g., Thermodynamics and the Free Energy of Chemical Substances, Lewis, G.N. and Randall, M., revised by Pitzer, K.S. and Brewer, L, McGraw-Hill, New York, 1961).
Suitable osmogens include urea, water-soluble salts such as magnesium sulfate, magnesium chloride, calcium chloride, sodium chloride, lithium chloride, potassium sulfate, sodium carbonate, sodium sulfite, lithium sulfate, potassium chloride and sodium sulfate, and sugars such as xylitol, d-mannitol, sorbitol, inositol, raffinose, sucrose, glucose, fructose and lactose. Mixtures of osmogens may also be used.
Preferably the osmogen is a sugar. A particularly preferred osmogen is xylitol.
(d) Additional components The drug-containing layer may optionally include one or more additional components such as a solubility/wettability enhancing agent, a drug crystallization inhibiting agent, a lubricant, a disintegrant and a dye.
In one embodiment, the drug-containing layer includes a solubility/wettability enhancing agent which is present at from about 5% to about 20% by weight of drug-containing layer. In a preferred embodiment the solubility/wettability enhancing agent is present at from about 5% to about 15% by weight of drug-containing layer. In a more preferred embodiment the solubility/wettability enhancing agent is present at from about 8% to about 12% by weight of drug-containing layer. In a most preferred embodiment the solubility/wettability enhancing agent is present at about 10% by weight of drug-containing layer.
Suitable solubility/wettability enhancing agents include hydroxyalkyl cellulose derivatives such as hydroxypropyl methyl cellulose (HPMC), hydroxyethyl cellulose (HEC) and hydroxypropyl cellulose (HPC). A preferred solubility/wettability enhancing
agent is HPMC having an average molecular weight of from about 50,000 to about 100,000Da.
In certain embodiments, the solubility/wettability enhancing agent and the low-solubility drug are intimately mixed during the manufacture of the drug-containing layer.
In another embodiment, the drug-containing layer includes an organic acid solubilising agent at from about 0.1% to about 30%, preferably at from about 1% to about 25% by weight of drug-containing layer. The organic acid solubilising agent is selected from aspartic acid, tartaric acid, adipic acid, ascorbic acid, benzoic acid, citric acid, fumaric acid, glutamic acid, malic acid, sorbic acid and toluenesulfonic acid. A preferred solubilising agent is aspartic acid at a level of from about 5% to about 25% by weight of drug-containing layer. Highly preferred is aspartic acid at a level of about 12% by weight of drug containing layer.
In a further embodiment the drug-containing layer includes a drug crystallization inhibiting agent.
The drug crystallization inhibiting agent may be present at from about 5% to about 20% by weight of drug containing layer. Preferably the drug crystallization inhibiting agent is present at from about 5% to about 15% by weight of drug containing layer. More preferably the drug crystallization inhibiting agent is present at from about 8% to about 12% weight of drug containing layer. Most preferably the drug crystallization inhibiting agent is present at about 10% by weight of drug containing layer.
The drug crystallization inhibiting agent should be inert, in the sense that it does not chemically react with the drug in an adverse manner, and should have at least some solubility in aqueous solution at physiologically relevant pHs (e.g. 1-8). Suitable drug crystallization inhibiting agents include hydroxyalkyl cellulose derivatives such as hydroxypropyl methyl cellulose (HPMC). A preferred for drug crystallization inhibiting agent is HPMC having a an average molecular weight which is substantially less than 5000Da, and wherein the typical viscosity value for 2% (w/v) aqueous solution is less than 100 mPa.S. Suitable examples are described in "Hydroxypropyl Methylcellulose", Handbook of Pharmaceutical Excipients, 3rd Edition, Ed. Arthur H. Kibbe, Pharmaceutical Press 2000, pages 252 to 255,. and include Methocel E3, E5, E6, E15
and E50, which are available from the Dow Chemical Company. A particularly preferred agent is Methocel E3.
The use of alternative and/or additional drug crystallization inhibiting agents is also contemplated. A suitable alternative and/or additional drug crystallization inhibiting agent should be inert, in the sense that it does not chemically react with the drug in an adverse manner, and should have at least some solubility in aqueous solution at physiologically relevant pHs (e.g. 1-8). Almost any neutral or ionizable polymer that has an aqueous solubility of at least 0.1 mg/mL over at least a portion of the pH range of 1-8 may be suitable as an optional additional agent. Preferred optional and/or additional drug crystallization inhibiting agents include hydroxypropylmethyl cellulose acetate succinate (HPMCAS), hydroxypropyl methyl cellulose phthalate (HPMCP), cellulose acetate phthalate (CAP), cellulose acetate trimellitate (CAT), and polyvinylpyrrolidone (PVP). A preferred alternative crystallization inhibiting agent is hydroxypropyl methyl cellulose acetate succinate (HPMCAS). A particularly preferred alternative crystallization inhibiting agent is the H grade of HPMCAS. HPMCAS is commercially available from Shin-Etsu Chemical (Tokyo, Japan), known by the trade name "AQOAT". By the "H grade" of HPMCAS is meant HPMCAS in which the methoxyl content is from about 22.0 to about 26.0 wt%, the hydroxypropoxyl content is from about 6.0 to about 10.0wt%, the acetyl content is from about 10.0 to about 14.0wt%, and the succinoyl content is from about 4.0 to about 8.0wt%.
The drug-containing layer may additionally comprise a lubricant at a level of up to about 5%, preferably less than about 3% and especially at from about 1 to 2% by weight of the drug containing layer. Suitable lubricants include magnesium stearate, polyethylene glycol and sodium lauryl sulphate.
The drug-containing layer may additionally comprise a disintegrant at a level of up to about 7%, preferably less than about 6% and especially at from about 3 to about 5% by weight of the drug containing layer. Suitable for use as disintegrants herein are sodium starch glycolate (sold under the trade name Explotab), croscarmellose sodium (sold under the trade name Ac-Di-SoI)1 starch, cross-linked polyvinylpyrrolidone (crospovidone), and amberlite resin.
The drug containing layer may additionally comprise a dye at a level of up to about 0.3%, preferably less than about 0.2% and especially at about 0.1% by weight of the drug containing layer. A particular advantage of the inclusion of a dye is to aid in tablet drilling.
(ii) Water-Swellable Layer
The water-swellable layer comprises polyethylene oxide polymer and a sweller layer strengthening agent. It may also include an osmogen and other pharmaceutically acceptable excipients.
The weight ratio of the polyethylene oxide polymer to the sweller layer strengthening agent may range from about 35:60 to about 80:5. Preferably the ratio is in the range from about 50:50 to about 70:30. More preferably the ratio is about 65:35.
(a) Polyethylene Oxide Polymer
The water-swellable layer includes polyethylene oxide polymer at from about 35% to about 80% by weight of water-swellable layer. Preferably, the polyethylene oxide is present at from about 50% to about 70% by weight of water-swellable layer. More preferably, it is present at about 65% by weight of water-swellable layer.
Suitable polyethylene oxide polymers for use in the water-swellable layer are those having a weight average molecular weight of from about 4,000,000 to about 7,000,000Da. Preferably the polyethylene oxide polymer in the water-swellable layer has a weight average molecular weight of about 5,000,000Da.
(b) Sweller Layer Strengthening Agent
The water-swellable layer includes sweller layer strengthening agent at from about 5% to about 60% by weight of water-sweller layer. Preferably the water-swellable layer includes sweller layer strengthening agent at from about from about 20% to about 40% by weight of the water-sweller layer. More preferably the water-swellable layer includes sweller layer strengthening agent at about 35 wt% by weight of the water- sweller layer.
The strengthening agent should be substantially water insoluble and chemically inert in the sense that it does not react with the other agents in the sweller layer in an adverse
manner. It should also have sufficient mechanical strength. A convenient measure of the mechanical strength of a material is its radial tensile strength (RTS).
RTS gives a measure of the strength of a compact. The measurement relies on a compact being formed in a compression event so that the thickness and crushing strength can be measured. RTS values vary for any given material depending on the speed of compression, with an increase in punch velocity generally leading to a reduction in compact hardness.
RTS can be measured according to known methods such as by using an instrumented press (Presster™) or a compaction simulator (Instron™) or similar equipment thereof well known to persons skilled in the art, and is calculated from the formula:
πdT wherein CS = Crushing Strength (N), d = Compact diameter (mm), T = Compact thickness (mm).
Preferred sweller layer strengthening agents have a water solubility of less than or equal to about 0.1mg/ml at ambient temperature and a pH of from about 5 to about 8 and a mechanical strength (radial tensile strength, RTS) of greater than about 4MPa at about 0.3 mm/s. More preferred herein are sweller layer strengthening agents have a water solubility of less than or equal to about 0.1mg/ml at ambient temperature and a pH of from about 5 to about 8 and a mechanical strength of greater than about 7MPa at about 0.3 mm/s.
Suitable sweller layer strengthening agents for use in the water-swellable layers of the osmotic bilayer tablets according to the present invention include microcrystalline cellulose, di basic calcium phosphate (DCP), HPMC and croscarmellose sodium (The Handbook of Pharmaceutical Excipients, 3rd Edn (2000), Pharmaceutical Press, pages 160 - 162, the contents of which are hereby incorporated by reference.
Microcrystalline cellulose materials suitable for use as sweller layer strengthening agents are described in "Cellulose, Microcrystalline", Handbook of Pharmaceutical Excipients, 3rd Edition, Ed. Arthur Kibbe, Pharmaceutical Press, 2000, pages 102 to
105, the contents of which are hereby incorporated by reference. A preferred sweller layer strengthening agent is microcrystalline cellulose having a mean particle size of less than about 200 μm and a moisture content of less than about 5%. The moisture content can be measured via conventional loss-on-drying techniques. More preferably the microcrystalline cellulose has a mean particle size of about 50 to about 100 μm.
Highly preferred sweller layer strengthening agents are Avicel PH-102, Avicel PH-105, Avicel PH-112 and Avicel PH-113, all of which are available from FMC Corporation, USA. (Avicel is a trade mark of FMC Corporation). Mixtures of these agents may also be used. An especially preferred sweller layer strengthening agent is Avicel PH-102, which has a nominal mean particle size of 100 μM and a moisture content of < 5.0% w/w.
(c) Osmogen The use of an osmogen in the water-swellable layer is optional. When an osmogen is used then it should be present at no more than 7% by weight of water-swellable layer.
In one embodiment, the water-swellable layer has no osmogen present.
In another embodiment, up to 7% by weight of an osmogen is present. Preferably, the osmogen should be present at no more than 5% by weight of water-swellable layer, and more preferably it should be present at no more than 3% by weight of water- swellable layer.
Suitable osmogens include urea, inorganic salts such as magnesium sulfate, magnesium chloride, calcium chloride, sodium chloride, lithium chloride, potassium sulfate, sodium carbonate, sodium sulfite, lithium sulfate, potassium chloride and sodium sulfate, and sugars such as d-mannitol, sorbitol, inositol, raffinose, sucrose, glucose, fructose and lactose. Mixtures of osmogens may also be used.
Inorganic salts are a preferred group of osmogens. A particularly preferred osmogen is sodium chloride.
(d) Other excipients
The water-swellable layer may additionally include further excipients such as dye and/or lubricant wherein the total level of such additional excipients is no more than about 2% by weight of the water-swellable layer, and preferably no more than about 1 % by weight of the water-swellable layer.
In one embodiment, the water-swellable layer has a radial tensile strength (RTS) in the range of from about 0.5 to about 5MPa at 0.3mm/s. In a more preferred embodiment the water-swellable layer has an RTS in the range of from about 0.5 to about 3MPa at 0.3mm/s.
In another embodiment, the difference between the radial tensile strength of the drug- containing layer and the radial tensile strength of the water swellable layer is from about 0.5 to about 5Mpa, more preferably from about 0.5 to about 3Mpa and especially from about 0.5 to about 1.5Mpa.
In another embodiment the percent mechanical recovery along the X-axis of the water- swellable layer following compression is less than about 20% relative to its corresponding in-die dimensions over a humidity range of from about 10% to about 75%RH.
The mechanical recovery measurement is typically carried out on a compaction simulator, wherein the dimensions of the powder compact within the die can be recorded as a function of the compression cycle. For example, it is possible to record compact thickness and compact diameter under pressure. By comparing in-die and out-of-die measurements it is possible to calculate the degree of axial and radial recovery (R) a compact undergoes after ejection by using the equation:
D _ Vt min ^Q / XlOO I Imin
wherein Z1mjn = axial or radial length of the compact 1 minute after ejection (mm) and I0 = axial or radial length of the compact under the maximum applied compression force (mm).
The intrinsic viscosity of the water-swellable layer, when fully hydrated, is preferably from about 107 to 109cP.
(B) Outer Coating The outer coating of the osmotic bi-layer tablets according to the present invention has to be sufficiently water-permeable to permit hydration of the inner core, and sufficiently strong to maintain its integrity in view of the pressure arising from the swelling of the water-swellable layer. Provided that these requirements are satisfied then any material or mixture of materials may be used to provide the outer coating. The outer coating is provided with at least one delivery port which allows the drug substance to be released into the use environment.
In a preferred embodiment, the outer coating comprises a water-permeable component and a water-insoluble component. More preferably, the water-permeable component is polyethylene glycol having a molecular weight of about 3350Da and the water insoluble component is cellulose acetate. Most preferably, the weight ratio of the polyethylene glycol having a molecular weight of about 3350Da and the cellulose acetate is from about 10:90 to about 40:60.
In another embodiment the solid content of the outer coating is less than 20% by weight relative to the weight of the inner bilayer core of the tablet.
The tablet may be any conventional shape or size. Typically, the tablet size ranges from about 0.5 cm to about 2.5 cm for its longest dimension. Preferably the osmotic bi- layer tablets according to the present invention have an aspect ratio of from about 1.3 to about 2.1. By aspect ratio is meant the ratio of the diameter of the tablet to the height of the tablet.
Preferably the osmotic bi-layer tablets according to the present invention have a hardness of greater than about 4kP. According to a highly preferred embodiment the tablets of the invention have a hardness of greater than about 4kP and a diameter of from about 5 to 20mm, preferably 10 to 15mm and especially about 12mm.
The osmotic bi-layer tablets according to the present invention are particularly useful for providing controlled-release formulations of sildenafil and its pharmaceutically acceptable salts.
Sildenafil, which is also known as 1-[4-ethoxy~3-(6,7-dihydro-1-methyl-7-oxo-3-propyl- 1 H-pyrazolo[4,3-Gflpyrimidin-5-yl)phenylsulphony]-4-methylpiperazine, has the following formula.

The structure of sildenafil is disclosed, inter alia, in commonly assigned US patent No. 5,250,534 issued October 5th 1993, herein incorporated by reference. Sildenafil is an inhibitor of phosphodiesterase type V (PDE-V) and is currently approved for the treatment of male erectile dysfunction and pulmonary arterial hypertension.
Sildenafil may be present as the free base or a pharmaceutically acceptable salt. The sildenafil or salt thereof may be present in any of its pharmaceutically acceptable forms. By "pharmaceutically acceptable forms" is meant any pharmaceutically acceptable derivative or variation, including tautomers, solvates, hydrates, isomorphs, polymorphs and pseudomorphs.
The sildenafil or a pharmaceutically acceptable salt thereof is present in the dosage form in a therapeutic amount. In a preferred embodiment, the amount is in the range of from 100 to 400 mgA, where "mgA" refers to mg of the active species sildenafil. In a more preferred embodiment the amount of sildenafil or a pharmaceutically acceptable salt thereof is from about IOOmgA to about 200mgA. In a most preferred embodiment the amount of sildenafil or a pharmaceutically acceptable salt thereof is from about 10OmgA to about 150mgA.
Suitable pharmaceutically acceptable salts of sildenafil include the acid addition salts such as the hydrochloride, hydrobromide, sulphate, bisulphate, phosphate, hydrogen phosphate, acetate, citrate, fumarate, gluconate, lactate, maleate, succinate, tartrate and mesylate, and the base addition salts such as the sodium and potassium salts.
Preferred sildenafil salts are sildenafil citrate and sildenafil mesylate. A highly preferred sildenafil salt is sildenafil citrate.
In a preferred embodiment, the weight ratio of the active species sildenafil : polyethylene oxide polymer in the drug layer is from about 3 : 1 to about 1.2 : 1 , more preferably from about 2 : 1 to about 1.2 : 1 , yet more preferably from about 2 : 1 to about 1.4 : 1 and especially from about 1.6 : 1 to about 1.75 : 1.
Where the sildenafil is present as sildenafil citrate these ratios provide the weight ratio of sildenafil citrate : polyethylene oxide polymer in the drug-containing layer is preferably from about 4 : 1 to about 1.7 : 1 , more preferably from about 3 : 1 to about 1.8 : 1 , yet more preferably from about 2.9 : 1 to about 2 : 1 and especially from about 2.4 : 1 to about 2.5 : 1.
The use of a crystallization inhibitor in combination with sildenafil is particularly advantageous. Collectively, sildenafil and the drug crystallisation inhibiting agent may make up a significant fraction of the drug layer. Sildenafil and the drug crystallisation inhibiting agent may collectively be present in an amount of at least 45 wt% of the drug layer. The utility of the invention increases as the fraction of the drug layer that is sildenafil and crystallisation inhibiting agent increases. In a preferred aspect herein sildenafil and the crystallisation inhibiting agent may be at least about 51 wt% of the drug layer, at least about 55 wt% of the drug layer, or even at least about 60 wt% of the drug layer.
Method of Manufacture
To form the tablet, the ingredients comprising the drug-containing layer and the water- swellable layer are first mixed or blended using processes known in the art. See for example, Lachman, et al., 'The Theory and Practice of Industrial Pharmacy" (Lea & Febiger, 1986). These mixtures or blends are then combined to provide the bi-layer core. The cores are then coated and finally the delivery ports are introduced.
Formation of the Drug-Containing Layer
For example, a portion of the ingredients of the drug layer 14 may first be blended, then wet granulated, dried, milled, and then blended with additional excipients prior to tableting.
In a preferred embodiment, the drug-containing layer is granulated. The inventors have found that drug layers that are blended but not granulated tend to exhibit poor flow characteristics, and that they stick to the die surfaces during pre-compression. The inventors believe that the sticking may result from the substantial quantity of drug in the drug layer. Drug exposed to the die surface may stick to the die during compression. Granulating the drug layer may reduce the tendency of the drug layer to stick by covering the drug in other excipients, thus reducing the amount of drug exposed to the die surface. Granulating methods include wet granulation, dry granulation, roller compaction, and melt granulation.
It has also been found that, when the drug-containing layer is formed by granulation, the final tablets show fewer coating defects. The inventors determined that a potential source of coating defects was delamination of the drug-containing layer from the water- swellable layer when tabletting was effected via direct compression. Delamination occurs when the drug layer and sweller layer are not securely bonded to one another. Such poor adhesion between the drug and sweller layer can result in cracks or gaps in the core near the interface of the drug layer and sweller layer which can lead to the layers splitting apart. The inventors solved the delamination problem by granulating the drug layer. Without wishing to be bound by any particular theory, the inventors believe that delamination was caused by over-compression of the drug layer during tamping. Drug layer blends for directly compressed formulations stuck to the upper punch during tamping (before addition of the sweller layer). Overcoming the sticking issue by tamping the drug layer with more force, however, created enough force to form a hard drug layer. When the sweller layer was added to the die and then compressed, a relatively low compression force was used; otherwise, crowning would occur due. However, the use of a relatively low compression force resulted in inadequate bonding between the drug layer and sweller layer, and subsequent delamination of the drug and sweller layers. In contrast, wet granulating the drug layer decreased the incidence of delamination of the drug layer from the sweller layer.
The frequency of the delamination occurrences in tablet lots was significantly reduced when a granulated drug layer was used. The granulated drug layer did not need to be overcompressed to manufacture acceptable bilayer tablets because the granulation exhibited much less sticking to the punch compared with the directly compressed drug
layer. When the drug layer was granulated, tablets were manufactured that had no coating defects and good overall robustness.
Similar processes can be used to form the sweller layer.
Method for Forming the Tablet
Once the materials are properly mixed, the core 12 is formed using procedures known in the art, such as compression or extrusion. For example, to form cores in the form of tablets, the desired amount of drug-containing composition 14 is placed in a tablet press and leveled by lightly tamping with the press. The desired amount of water- swellable composition 16 is then added, and the tablet formed by compression. Alternatively, the water-swellable composition may be added to the tablet press first, followed by the drug-containing composition. The amount of force used to compress the tablet core will depend on the size of the dosage form, as well as the compressibility and flow characteristics of the compositions. Typically, a pressure is used that results in a tablet with a strength of 3 to 20 Kp/cm2 wherein strength is the hardness of the compact in Kp as measured with a Schleuniger Tablet Hardness Tester, model 6D, divided by its maximum cross-sectional area normal to the direction of force in cm2.
Tablet Coating
Following formation of the core 12, coating 18 is applied. Coating 18 should have both a sufficiently high water permeability that the drug can be delivered within the desired time frame, and high strength, while at the same time be easily manufactured. Coatings with these characteristics can be obtained using hydrophilic polymers such as plasticized and unplasticized cellulose esters, ethers, and ester-ethers. Particularly suitable polymers include cellulose acetate ("CA"), cellulose acetate butyrate, and ethyl cellulose. A particularly preferred set of polymers are cellulose acetates having acetyl contents of 25 to 42%. A preferred polymer is CA having an acetyl content of 39.8%, and specifically, CA 398-10 manufactured by Eastman of Kingsport, Tennessee, having an average molecular weight of about 40,000 daltons. Another preferred CA having an acetyl content of 39.8% is high molecular weight CA having an average molecular weight greater than about 45,000, and specifically, CA 398-30 (Eastman) reported to have an average molecular weight of 50,000 daltons. The high molecular
weight CA provides superior coating strength, which allows thinner coatings and thus higher permeability.
Coating is conducted in conventional fashion by first forming a coating solution and then coating by dipping, fluidized bed coating, or preferably by pan coating. To accomplish this, a coating solution is formed comprising the coating polymer and a solvent. Typical solvents useful with the cellulosic polymers noted above include acetone, methyl acetate, ethyl acetate, isopropyl acetate, n-butyl acetate, methyl isobutyl ketone, methyl propyl ketone, ethylene glycol monoethyl ether, ethylene glycol monoethyl acetate, methylene dichloride, ethylene dichloride, propylene dichloride, nitroethane, nitropropane, tetrachloroethane, 1,4-dioxane, tetrahydrofuran, diglyme, and mixtures thereof. A particularly preferred solvent is acetone. The coating solution typically will contain from about 1 to about 15 wt% of the polymer (cellulose acetate), preferably from about 2 to about 10 wt%, most preferably from about 3 to about 7 wt%.
A "half-solids" solution can be used to improve coating uniformity, such as illustrated by the coating in Examples 12 to 15 (3.5/1.5/3/92 CA/PEG/water/acetone and 4/1/3/92 CA/PEG/water/acetone).
The coating solution may also comprise pore-formers, non-solvents, or plasticizers in any amount so long as the polymer remains substantially soluble at the conditions used to form the coating and so long as the coating remains water-permeable and has sufficient strength. Pore-formers and their use in fabricating coatings are described in U.S. Patent Nos. 5,612,059 and 5,698,220, the pertinent disclosures of which are incorporated herein. The term "pore former," as used herein, refers to a material added to the coating solution that has low or no volatility relative to the solvent such that it remains as part of the coating following the coating process but that is sufficiently water swellable or water soluble such that, in the aqueous use environment it provides a water-filled or water-swollen channel or "pore" to allow the passage of water thereby enhancing the water permeability of the coating. Suitable pore-formers include polyethylene glycol (PEG), PVP, PEO, HEC, HPMC and other aqueous-soluble cellulosics, water-soluble acrylate or methacrylate esters, polyacrylic acid and various copolymers and mixtures of these water soluble or water swellable polymers. Enteric polymers such as cellulose acetate phthalate (CAP) and HPMCAS are included in this class of polymers. A particularly preferred pore former is PEG having an average
molecular weight from 1000 to 8000 daltons. A particularly preferred PEG is one having a molecular weight of 3350 daltons. The inventors have found that to obtain a combination of high water permeability and high strength when PEG is used as a pore former, the weight ratio of CA: PEG should range from about 6.5 : 3.5 to about 9 : 1 and are especially in the range of from about 3.5 :1 to about 4 : 1.
The addition of a non-solvent to the coating solution results in exceptional performance. By "non-solvent" is meant any material added to the coating solution that substantially dissolves in the coating solution and reduces the solubility of the coating polymer or polymers in the solvent. In general, the function of the non-solvent is to impart porosity to the resulting coating. As described below, porous coatings have higher water permeability than an equivalent weight of a coating of the same composition that is not porous and this porosity, when the pores are gas filled, as is typical when the non-solvent is volatile, is indicated by a reduction in the density of the coating (mass/volume). Although not wishing to be bound by any particular mechanism of pore formation, it is generally believed that addition of a non-solvent imparts porosity to the coating during evaporation of solvent by causing the coating solution to undergo liquid-liquid phase separation prior to solidification. As described below for the case of using water as the non-solvent in an acetone solution of cellulose acetate, the suitability and amount of a particular candidate material can be evaluated for use as a non-solvent by progressively adding the candidate non-solvent to the coating solution until it becomes cloudy. If this does not occur at any addition level up to about 50 wt% of the coating solution, it generally is not appropriate for use as a non-solvent. When clouding is observed, termed the "cloud point," an appropriate level of non-solvent for maximum porosity is the amount just below the cloud point. When lower porosities are desired, the amount of non-solvent can be reduced as low as desired. It has been found that suitable coatings can be obtained when the concentration of non-solvent in the coating solution is greater than about 20% of the non-solvent concentration that results in the cloud point.
Suitable non-solvents are any materials that have appreciable solubility in the solvent and that lower the coating polymer solubility in the solvent. The preferred non-solvent depends on the solvent and the coating polymer chosen. In the case of using a volatile polar coating solvent such as acetone or methyl ethyl ketone, suitable non-solvents include water, glycerol, ethylene glycol and its low molecular-weight oligomers (e.g.,
less than about 1 ,000 daltons), propylene glycol and its low molecular weight oligomers (e.g., less than about 1 ,000 daltons), C1 to C4 alcohols such as methanol or ethanol, ethylacetate, acetonitrile and the like.
In general, to maximize its effect, (e.g., formation of pores), the non-solvent should have similar or less volatility than the coating solution solvent such that, during initial evaporation of the solvent during the coating process, sufficient non-solvent remains to cause phase separation to occur. In many cases, where a coating solution solvent such as acetone is used, water is a suitable non-solvent. For acetone solutions comprising 7 wt% CA and 3 wt% PEG, the cloud point at room temperature is at about 23 wt% water. Thus the porosity and in turn the water permeability (which increases with increasing porosity) can be controlled by varying the water concentration up to near the cloud point. For acetone solutions comprising CA and PEG with a total concentration of about 10 wt%, it is desired that the coating solution contain at least 4 wt% water to obtain a suitable coating. When a higher porosity, and thus a higher water permeability is desired (to obtain a faster release rate), the coating solution should contain at least about 15 wt% water.
In one embodiment of the invention, the coating solution is homogeneous, in that when the polymer, solvent, and any pore formers or non-solvents are mixed, the solution comprises a single phase. Typically, a homogenous solution will be clear, and not be cloudy as discussed above.
When using CA 398-10, exemplary coating solution weight ratios of CA:PEG 3350:water are 7:3:6, 8:2:6, and 9:1 :6, with the remainder of the solution comprising a solvent such as acetone. Thus, for example, in a solution having a weight ratio of CA:PEG 3350:water of 7:3:6, CA comprises 7 wt% of the solution, PEG 3350 comprises 3 wt% of the solution, water comprises 6 wt% of the solution, and acetone comprises the remaining 84 wt%.
A further example of a suitable coating system, in a solution having a weight ratio of CA : PEG 3350 : water of 7 : 3 : 6, CA comprises 3.5 wt% of the solution, PEG 3350 comprises 1.5 wt% of the solution, water comprises 3 wt% of the solution, and acetone comprises the remaining 92 wt%. Preferred examples of coating systems herein are 3.5/1.5/3/92 CA/PEG/water/acetone and 4/1/3/92 CA/PEG/water/acetone.
Preferred coatings are generally porous even in the dry state (prior to delivery to the aqueous use environment). By "porous" is meant that the coating has a dry-state density less than the density of the nonporous coating material. By "nonporous coating material" is meant a coating material formed by using a coating solution containing no non-solvent, or the minimum amount of non-solvent required to produce a homogeneous coating solution. The coating in the dry state has a density that is less than 0.9 times, and more preferably less than 0.75 times that of the nonporous coating material. The dry-state density of the coating can be calculated by dividing the coating weight (determined from the weight gain of the tablets before and after coating) by the coating volume (calculated by multiplying the coating thickness, as determined by optical or scanning electron microscopy, by the tablet surface area). The porous nature of the coating is one of the factors that leads to the combination of high water permeability and high strength of the coating.
The coatings may also be asymmetric, meaning that there is a gradient of density throughout the coating thickness. Generally, the outside surface of the coating will have a higher density than the coating nearest the core.
The coating can optionally include a plasticizer. A plasticizer generally swells the coating polymer such that the polymer's glass transition temperature is lowered, its flexibility and toughness increased and its permeability altered. When the plasticizer is hydrophilic, such as polyethylene glycol, the water permeability of the coating is generally increased. When the plasticizer is hydrophobic, such as diethyl phthalate or dibutyl sebacate, the water permeability of the coating is generally decreased.
It should be noted that additives can function in more than one way when added to the coating solution. For example, PEG can function as a plasticizer at low levels while at higher levels it can form a separate phase and act as a pore former. In addition, when a non-solvent is added, PEG can also facilitate pore formation by partitioning into the non-solvent-rich phase once liquid-liquid phase separation occurs.
The weight of the coating around the core depends on the composition and porosity of the coating, the surface to volume ratio of the dosage form, and the desired drug release rate, but generally should be present in an amount ranging from about 3 to
about 30 wt%, preferably from about 5 to about 25 wt% and more preferably from about 5% to about 20% based on the weight of the uncoated core. However, a coating weight of at least about 5 wt% is generally preferred so as to assure sufficient strength for reliable performance.
While porous coatings based on CA, PEG, and water yield excellent results, other pharmaceutically acceptable materials may be used so long as the coating has the requisite combination of high water permeability, high strength, and ease of manufacture. Further, such coatings may be dense, or asymmetric, having one or more dense layers and one or more porous layers, as described in U.S. Patent Nos. 5,612,059 and 5,698,220.
The coating 18 must also contain at least one delivery port 20 in communication with the interior and exterior of the coating to allow for release of the drug-containing composition to the exterior of the dosage form. The delivery port can range in size from about the size of the drug particles, and thus could be as small as 1 to 100 microns in diameter and may be termed pores, up to about 5000 microns in diameter. The shape of the port may be substantially circular, in the form of a slit, or other convenient shape to ease manufacturing and processing. The port(s) may be formed by post-coating mechanical or thermal means or with a beam of light (e.g., a laser), a beam of particles, or other high-energy source, or may be formed in situ by rupture of a small portion of the coating. Such rupture may be controlled by intentionally incorporating a relatively small weak portion into the coating. Delivery ports may also be formed in situ by erosion of a plug of water-soluble material or by rupture of a thinner portion of the coating over an indentation in the core. Delivery ports may be formed by coating the core such that one or more small regions remains uncoated. In addition, the delivery port can be a large number of holes or pores that may be formed during coating, as in the case of asymmetric membrane coatings of the type disclosed in U.S. Patent Nos. 5,612,059 and 5,698,220, the disclosures of which are incorporated by reference. When the delivery pathways are pores there can be a multitude of such pores that range in size from 1 μm to greater than 100 μm. During operation, one or more of such pores may enlarge under the influence of the hydrostatic pressure generated during operation. The number of delivery ports 20 may vary from 1 to 10 or more. At least one delivery port should be formed on the side of the coating that is adjacent to the drug-containing composition, so that the drug-
containing composition will be extruded out of the delivery port by the swelling action of the water-swellable composition. It is recognized that some processes for forming delivery ports may also form holes or pores in the coating adjacent to the water- swellable composition. In aggregate, the total surface area of core exposed by delivery ports is less than 5%, and more typically less than 1%.
Drug Release Characteristics
The tablets of the present invention provide controlled-release of the low-solubility drug, and release a substantial portion of the drug.
Reference to the "release" of the low-solubility drug as used herein means (1) transport of the low-solubility drug from the interior of the dosage form to its exterior such that it contacts the fluid within a mammal's gastrointestinal (Gl) tract following ingestion or (2) transport of the low-solubility drug from the interior of the dosage form to its exterior such that it contacts an in vitro test medium for evaluation of the dosage form by an in vitro test as described below. Reference to a "use environment" or "environment of use" can thus be either to in vivo Gl fluids or to in vitro test media. "Introduction" to a use environment includes either by ingestion or swallowing, where the use environment is in vivo, or being placed in a test medium where the use environment is in vitro.
Reference to "controlled-release" as used herein means that the dosage form does not provide immediate release of the low-solubility drug. The dosage form releases less than 70 wt% of the low-solubility drug in the dosage form to the use environment within the first hour after administration to the use environment.
Reference to "a substantial portion" as used herein means at least 80% of the drug substance present in the tablet is released within 20 hours. Preferably, the dosage forms release at least 85 wt%, more preferably at least 90 wt%, and even more preferably at least 95 wt% within 20 hours after administration.
Preferably, the osmotic bi-layer tablets according to the present invention release at least 80% of the low-solubility drug into the use environment within 6 to 20 hours after introduction into the use environment. More preferably, the osmotic bi-layer tablets according to the present invention release at least 80% of the low-solubility drug into the use environment within 8 to 20 hours after introduction into the use environment.
In one embodiment, the dosage form provides controlled release of sildenafil over a twelve hour release period. Sildenafil is released to a use environment at an average rate of from 5.5 to 10 wt% per hour from the second to the twelfth hour. Preferably, the dosage form releases less than about 25 wt% of sildenafil during the first 2 hours, and more preferably less than about 15 wt% during the first two hours. In addition, the dosage form releases at least 70 wt%, preferably 80 wt%, and more preferably 90 wt%, by the twelfth hour. For the twelve hour release embodiment, average release rates of drug are wt% of sildenafil averaged over the period beginning at the end of the second hour and ending at the end of the twelfth hour. Thus, for example, if 20 wt% sildenafil is released by the end of hour 2 and 90 wt% is released by the end of hour 12, then the average "release rate" is (90-20)÷10 or 7.0 wt%/hour.
In another embodiment, sildenafil is released to a use environment at an average rate of from 4 to 7 wt% per hour from the fourth to the eighteenth hour. Preferably, the dosage form releases less than about 35 wt% of sildenafil during the first 4 hours, and more preferably less than about 25wt% during the first four hours. In addition, the dosage form releases at least 70 wt%, preferably at least 80 wt%, and more at least preferably 90 wt% by the eighteenth hour. For the eighteen hour release embodiment, unless otherwise specified, average release rates of drug are wt% of sildenafil averaged over the period beginning at the end of the fourth hour and ending at the end of the eighteenth hour. Thus, for example, if 20 wt% sildenafil is released by the end of hour 4 and 90 wt% is released by the end of hour 18, then the average "release rate" is (90-20)÷14 or 5 wt%/hour.
Preferred herein are tablets capable of releasing: greater than about 80 wt% sildenafil by the end of hour 16; greater than about 85% by the end of hour 18; greater than about 90% by the end of hour 20. Especially preferred are tablets capable of releasing: greater than about 85 wt% sildenafil by the end of hour 16; greater than about 90% by the end of hour 18; greater than about 95% by the end of hour 20.
Preferred herein are tablets capable of delivering an average release over 4 to 18 hours of greater than about 4 wt%/hour, preferably more than about 4.2 wt%/hour, more preferably more than about 4.4 wt%/hour, highly preferably more than about 4.6 wt%/hour and especially more than about 4.8 wt%/hour.
An in vitro test may be used to determine whether a dosage form provides a release profile within the scope of the present invention. Two types of in vitro tests may be used, a residual test and a direct test, although other conventional tests known in the art used to measure drug release may also be used.
In the residual test, the dosage form is first placed into a stirred USP type 2 dissoette flask containing 900 ml_ of a buffer solution simulating gastric fluid (0.01 N HCI, 0.12 M NaCI, pH 2.0, 261 mθsm/kg) for 2 hours, then removed, rinsed with deionized water, and transferred to a stirred flask containing 900 mL of a buffer solution simulating the contents of the small intestine (0.03 M NaCI, 0.006 M KH2PO4, 0.06 M KCI, adjusted with 30% NaOH to pH 7.5 or 6.5, 210 mθsm/kg). In both flasks the dosage form is placed in a wire support to keep the tablet off of the bottom of the flask, so that all surfaces are exposed to the solution and the solutions are stirred using paddles that rotate at 50 to 100 revolutions per minute. At each time interval, a single tablet is removed from the solution and placed in 100 mL of a recovery solution (50/50 wt/wt ethanol/water, pH 3), and stirred vigorously at ambient temperature overnight in a flask, stirring to dissolve the drug remaining in the tablet. Samples of the recovery solution containing the dissolved drug are filtered using a Gelman nylon Acrodisc® 13, 0.45 μm pore size, and placed in an HPLC vial and capped. Residual drug is analyzed by HPLC. Drug concentration is calculated byan appropriate method such as, for example, comparing UV absorbance at a wavelength characteristic of the drug to the absorbance of drug controls. The amount remaining in the tablets is subtracted from the total drug originally in the tablet to obtain the amount released at each time interval.
In the direct test, samples of the dosage form are placed into a stirred USP type 2 dissoette flask containing 900 mL of a receptor solution. The receptor solution is 0.01 N HCI, 7 g/L sodium chloride. Samples are taken at periodic intervals using a VanKel VK8000 autosampling dissoette with automatic receptor solution replacement. Tablets are placed in a wire support as above, paddle height is adjusted, and the dissoette flasks stirred at 50 to 100 rpm at 37°C. Periodically, the autosampler removes a sample of the receptor solution, and the concentration of drug in the receptor solution is analyzed directly by HPLC. Since the drug is usually extruded from the dosage form as a suspension in an entraining polymer, there is often a time lag between when the drug is released and when it is dissolved in the test media, and thus, measured in the
direct test. This time lag depends on the solubility of the drug, the test media, and the ingredients of the drug-containing composition, but typically is on the order of 30 to 90 minutes. Accordingly, results of the direct test may tend to underestimate the amount of drug actually released.
Alternatively, an in vivo test may be used to determine whether a dosage form provides a release profile within the scope of the present invention. Dosage forms are dosed to a group of humans, dogs or other suitable mammals and dosage form release and drug absorption is monitored either by (1 ) periodically withdrawing blood and measuring the serum or plasma concentration of drug or (2) measuring the amount of drug remaining in the dosage form (residual drug) following its exit from the anus or (3) both (1) and (2). In the second method, residual drug is measured by recovering the tablet upon exit from the anus of the test subject and measuring the amount of drug remaining in the dosage form using the same procedure described above for the in vitro residual test. The difference between the amount of drug in the original dosage form and the amount of residual drug is a measure of the amount of drug released during the mouth-to-anus transit time. This test has limited utility since it provides only a single drug release time point but is useful in demonstrating the correlation between in vitro and in vivo release.
In one in vivo method, the serum or plasma drug concentration is plotted along the ordinate (y-axis) against the blood sample time along the abscissa (x-axis). The data may then be analyzed to determine drug release rates using any conventional analysis, such as the Wagner-Nelson or Loo-Riegelman analysis. See also, Welling, "Pharmacokinetics: Processes and Mathematics," ACS Monograph 185, Amer. Chem. Soc. (1986). It should be noted that such a procedure may underestimate the amount of drug released, particularly at later time points if the drug is relatively less soluble at higher pH, because much of the drug released from the dosage form may remain undissolved in the lower Gl tract. Therefore, the in vitro tests previously described are preferred for determining whether the release profile of a dosage form is within the scope of the present invention.
As noted above, the inventors have observed that, when applied to low-solubility drugs, conventional osmotic bi-layer tablets can retain significant quantities of the drug even after extended periods. Because the drug is extruded from the tablet, to some extent
at least, in solid form, it is generally necessary to include a polymeric entraining agent in the drug-containing layer. The inclusion of this polymer results in the formation of a viscous fluid when the tablet imbibes water from the environment. However, if the fluid is too viscous then the exudate forms a plume from which the drug does not disperse.
The art teaches that it is desirable to provide a sweller layer that has a high capacity for expansion in order to force the viscous fluidized drug-containing layer through the delivery port. Without being bound by any particular theory, the inventors hypothesize that in situations where the viscosity of the drug-containing layer is reduced then sweller layer "breakthrough" or "push around" can occur. In this situation the drug- containing layer fractures and fragments are trapped by the expanding sweller layer. The drug in these fragments is isolated from the delivery port and remains in the tablet.
In an alternative hypothesis, it may be postulated that in order to extrude the drug layer, the sweller layer should have good mechanical strength at high water content. Without sufficient mechanical strength, the sweller layer will be unable to push the viscous drug layer out of the dosage form, resulting in a high residual amount of drug. The inventors have determined that for swelling agents containing too much osmogen, the sweller layer is not capable of extruding all of the drug out of the tablet. The sweller layer of the present invention comprising PEO and a strengthening agent may function by reducing the osmotic pressure and decreasing the wetting rate of the sweller layer, allowing complete delivery of the drug layer before the viscosity of the sweller layer becomes too low.
The tablets of the present invention provide for: o the use of selected polymers in the drug-containing layer and water-swellable layer to optimize performance; o the inclusion of a strengthening agent but little or no osmogen in the sweller layer, which reduces the capacity for expansion of this layer; and o the inclusion of an osmogen in the drug-containing layer.
Table 3 shows that, for representative low-solubility drugs (sertraline HCI, propranolol
HCI and theophylline) the tablets of the present invention provide for complete extrusion of the drug whereas more conventional osmotic bi-layer tablets retain part of the dose. The results obtained with tablets containing verapamil HCI, which is a more
soluble drug, are also shown, and illustrate that incomplete drug delivery is less of a problem with the drug is soluble. Details of the methodology are provided in Study A of the Examples below.
For each drug, the performance of a tablet according to the present invention, having a sweller layer that is 65wt% polyethylene oxide / 34.4wt% Avicel PH102 ("New Formulation"), is compared to the performance of a tablet with a 65wt% polyethylene oxide / 34.3wt% sodium chloride sweller layer ("Control Formulation"). For the relatively soluble drug verapamil HCI, it can be seen from Table 3 that both formulations provide good release of the drug. Table 3 further shows that the less soluble drugs theophylline, propranolol HCI and sertraline HCI are retained in the control tablets (-25%, -30% and ~60% release respectively), while the new formulation gave essentially complete release. For the less soluble drugs, visual inspection of the tablets after the test was consistent with sweller layer breakthrough occurring in the control tablets but not the new tablets.
Sweller layers with 40wt% and 80wt% polyethylene oxide have also been investigated in conjunction with these drugs (see Study B), and again the performance of the tablets according to the present invention was better than that of the control tablets.
Study C illustrates the improved release of another low-solubility drug, sildenafil citrate, using the tablets of the present invention.
As well as having improved drug-release properties, the tablets of the present invention are amenable to manufacture. The inventors found that the use of highly swelling materials, such as those disclosed in Appel et. al., including sodium starch glycolate
(ExPlotab), sodium starch glycolate and Ac-Di-SoI (croscarmellose sodium), can give rise to compression difficulties leading to delamination due to mechanical relaxation or friability which can significantly effect coating performance. The inventors found that some coating defects resulted from overcompressing cores containing sodium cross carmellose or sodium starch glycolate. Highly swelling materials such as sodium cross carmellose or sodium starch glycolate tend to be friable. Compressing such materials at high compression force can decrease the friability of the cores. However, the inventors found that the large compression force caused crowning of the drug layer. Crowning occurs during compression of the core when material from one layer flows
between the spaces between the die wall and the punch. The result is a core that has circumferential ridges around one of the faces. The ridges tend to create stresses in the coating, which may lead to coatings which burst. As illustrated in Table 21 , replacing the highly swelling material with polyethylene oxide solved this problem, since polyethylene oxide has been found to form non-friable cores at lower compression forces in tablets according to the present invention.
The inventors also surprisingly found that polyethylene oxide sweller layers were less susceptible to relaxation. Relaxation is the tendency of a material to increase in friability and/or size following compression. Highly swelling materials such as sodium croscarmellose or sodium starch glycolate tend to be susceptible to relaxation. In contrast, polyethylene oxide is less subject to relaxation. The data in Table 22 illustrates that a sweller layer according to the invention, Compact C, displays less relaxation when compared to conventional sweller layers.
Other features and embodiments of the invention will become apparent from the following examples which are given for illustration of the invention rather than for limiting its intended scope.
EXAMPLES
Study A: Improved Release of Representative Low-Solubility Drugs (65wt% PEO Sweller Layer)
The low-solubility compounds selected for assessment were: Sertraline HCI (C17H17CI2N-HCI)
l l

/ml
 g/ml
g/ml
Theophylline (C7HaN4 O2) 2
g/ml g/ml

Verapamil HCI was used as a more soluble comparator. Verapamil HCI (C27H38N2O4-HCI)
l

Formulations were prepared according to Table 1.
Table 1

(0 Dissolution testing
Dissolution was assessed at 372C using a 40 mesh basket rotating at IOOrpm in 900ml of medium. Measurements were taken every 30 minutes for 24 hours using on-line UV detection. The composition of the medium and the detection wavelength λ are set out in Table 2.
Table 2

The results are summarized in Tables 3 and 4.
Table 3


Table 4

For all three low-solubility drugs, almost complete drug release was obtained with the formulation according to the present invention (examples 1 , 2 and 3) whereas only about 45% of the sertraline HCI (comparator 1), about 18% of the propranolol HCI (comparator 2), and about 24% of the theophylline (comparator 3) was released from the reference formulation.
For the more soluble comparator drug verapamil HCI, both formulations showed good release profiles.
(ii) Rheoloqy investigation
The viscosity of the drug-containing layers of the examples and comparators was investigated.
Instrument. TA Instruments AR 500 Geometry. 2cm stainless sell hatched plate, with hatched base plate.
Sample preparation: 1 g of blend plus 5g of water. Rotate in a vial at 400C for 72 hours. Load onto Rheometer table using a syringe. Allow sample to rest for 1 minute before processing.
Sample volume: ~0.4ml Sample gap: 500um Flow Procedure: 1 Conditioning: 37°C, wait for correct temperature, 30sec equilibration
2 Continuous ramp: 0-1 (1/s), sheer rate, linear mode, duration 1 minute, sample points 3, 37°C.
3 Steady state flow: 1-1 (1/s), linear mode, sample points 6, 37°C, 10sec sample period, steady state tolerances 5% tolerances, 3 consecutive points within tolerance, up to 1 minute.
The results are summarized in Table 5.
Table 5
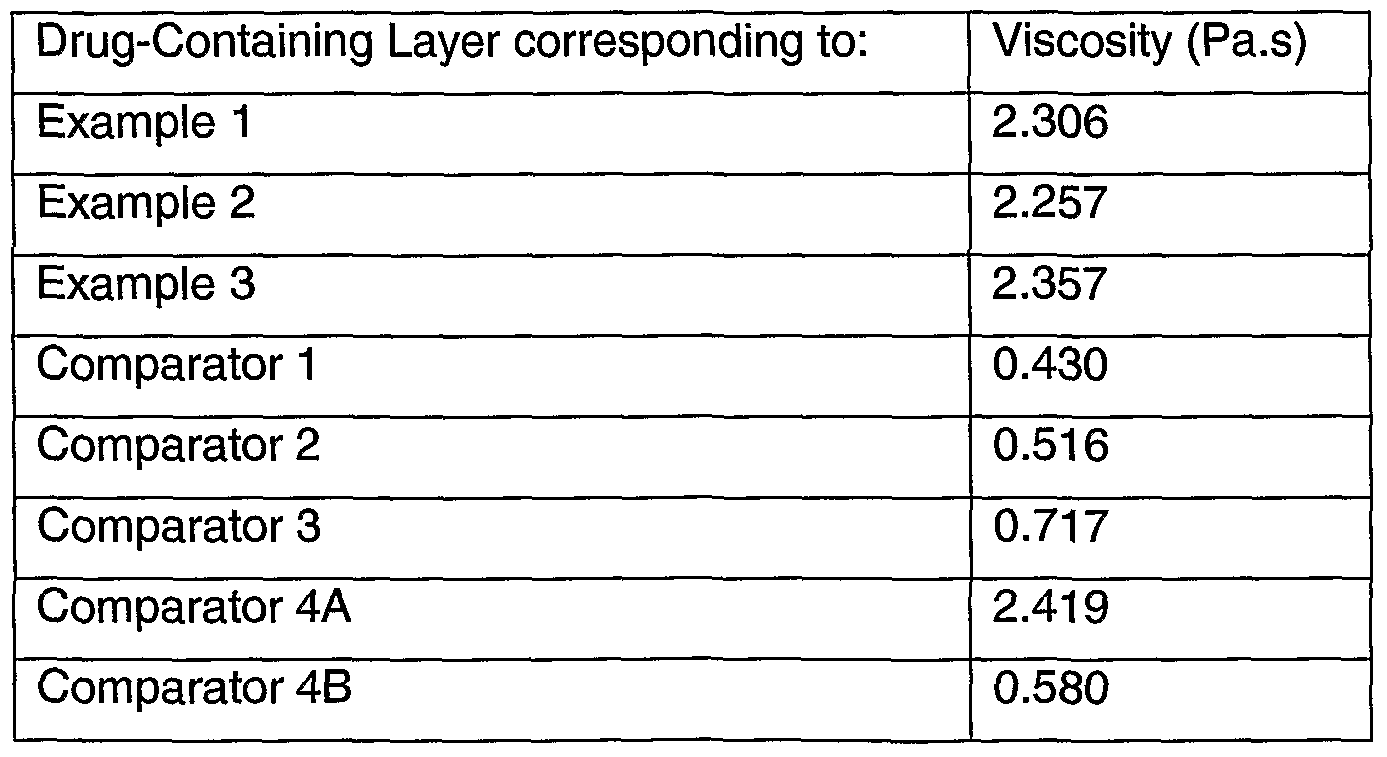
Study B: Improved Release of Representative Low-Solubility Drugs (40 and 80wt% PEO Sweller Layer)
Further tablets were prepared using water-swellable layers with increased or decreased PEO content (Table 6).
Table 6

The tablets were manufactured following the method described for Study A. Automated dissolution testing and visual sweller layer breakthrough assessment of the resulting SCT was conducted over a 24 hour period. The results are summarized in Table 7.
Table 7
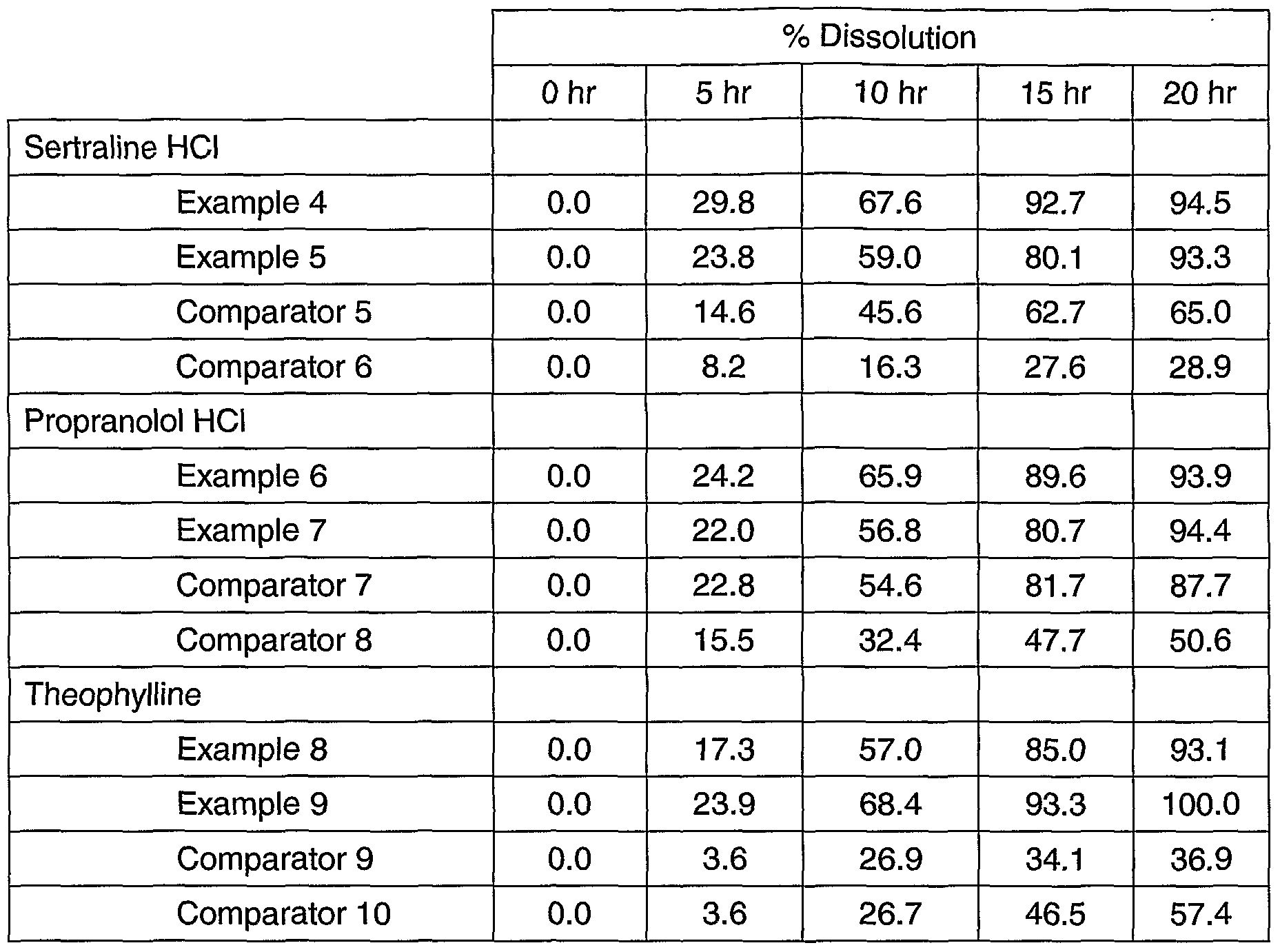
For all the tablets according to the present invention (i.e. Examples 4 to 9) drug release was essentially complete by the end of the study period. For the comparator formulations, the formulations with 80% PEO in the sweller layer performed reasonably well in the case of sertraline HCI (Comparator 5) and very well in the case of propranolol HCI (Comparator 7), but less well for theophylline (Comparator 9). The comparator formulations with 40% PEO generally did not completely release the drug (Comparators 6, 8 and 10). No release problems were observed with any of the formulations of the more soluble drug verapamil HCI (comparators 11A - 11 D).
Study C: Improved Release of Sildenafil Citrate
Sildenafil citrate tablets according to the invention were made with improved sweller layers containing polyethylene oxide (PEO) and a strengthening agent, and compared to a control formulation with the same amount of PEO and with an osmogen, sodium chloride, substituted for the strengthening agent (Comparator 12). Example 10 had a
sweller layer containing microcrystalline cellulose as the strengthening agent, while Example 11 had a sweller layer containing dibasic calcium phosphate as the strengthening agent. Comparator 12 was the same as Examples 10 and 11 except that it contained the osmogen sodium chloride instead of a strengthening agent. The composition of each formulation is shown in Table 8. Table 8


To form the drug layers of Examples 10 and 11 , and Comparator 12, the drug layer ingredients were first combined without the magnesium stearate and blended for 20 minutes in a TURBULA mixer. The drug layer included the crystallization inhibiting agent hydroxypropyl methyl cellulose (HPMC) grade Methocel E3 (Dow Chemicals). This blend was pushed through a 20 mesh sized screen, then blended again for 20 minutes in the same mixer. Next, magnesium stearate was added and the drug layer was blended again for 4 minutes in the same mixer.
To form the sweller layer, the ingredients were first combined without the magnesium stearate and blended for 20 minutes in a TURBULA mixer. Next, the magnesium stearate was added. All ingredients were pushed through a 20 mesh sized screen, then blended again for 4 minutes in the same mixer.
Tablet cores were formed by placing 570 mg of the drug layer composition in a 7/16 inch diameter standard round concave (SRC) die and gently leveling with the press. Then, 200 mg of the sweller layer composition was placed in the die on top of the drug layer. The tablet core was then compressed to a hardness of about 8 kP. The so- formed tablet contained 200mgA sildenafil.
Coatings were applied by a Vector LDCS-20 pan coater. The coating solution contained cellulose acetate (CA) (CA 398-10 from Eastman Fine Chemical, Kingsport, Tennessee), polyethylene glycol (PEG 3350, Union Carbide), water, and acetone in a weight ratio of 7/3/6/84 (wt%). The flow rate of the inlet heated drying air of the pan coater was set at 40 ft3/min with the outlet temperature set at 250C. Nitrogen at 20 psi was used to atomize the coating solution from the spray nozzle, with a nozzle-to-bed distance of 2 inches. The pan rotation was set to 20 rpm. The so-coated tablets were dried at 500C in a convection oven. The final dry coating weight amounted to about 11 wt% of the uncoated tablet core. One 900 μm diameter hole was then laser-drilled in the coating on the drug layer side of the tablet to provide a delivery port.
The tablets of Examples 10 and 11 and Comparator 12 were tested in vitro using direct drug analysis, as detailed hereinbefore, in gastric medium (0.01 M HCI, 7 g/L NaCI) to
determine their relative drug release rates. To perform a "direct test" , each dosage form was first placed into a stirred USP type 2 dissoette flask containing 900 ml_ of a buffer solution simulating the contents of the stomach (0.01 M HCI, 7 g/L NaCI). In the flasks, the dosage form was placed in a wire support to keep the dosage form off of the bottom of the flask, so that all surfaces were exposed to the moving release solution and the solutions were stirred using paddles that rotate at a rate of 100 rpm. At each time interval, samples were taken from the flask automatically, with media replacement. Samples were analyzed by HPLC using a Waters Symmetry C18 column. The mobile phase consisted of 0.05 M triethanolamine (pH 3)/ methanol/ acetonitrile in a volume ratio of 58/25/17. Drug concentration was calculated by comparing UV absorbance at 290 nm to the absorbance of sildenafil standards. The relative rates of drug release from the three tablets is illustrated by the data in Table 9. The results illustrated in Table 3 demonstrate that the use of a strengthening agent in the sweller layer improves performance of the tablets relative to Comparator 12 which contained PEO/NaCl and no strengthening agent.
Table 9

The tablets of Examples 10 and 11 provided improved release of sildenafil versus the release from the tablet of Comparator 12. Example 10 released sildenafil at an average rate from four to eighteen hours of 4.9 wt%/hour, and Example 11 released sildenafil at an average rate from four to eighteen hours of 4.4 wt%/hour; Comparator
12 released sildenafil at an average rate from four to eighteen hours of only 3.5 wt%/hour. In addition, both Examples 10 and 11 released greater than 90wt% sildenafil by 20 hours, whilst Comparator 12 released 74 wt%.
A further control tablet, Comparator 13 was prepared according to the methodology described herein before for Examples 10 and 11.

Figures 3a and 3b illustrate the plume dispersion of formulations containing 10% HPMC in the drug layer (3) - as illustrated by the formulation of Example 10 and no HPMC in the drug layer (4) as illustrated by the formulation of Comparator 13. The
images illustrated in Figures 3a and 3b were taken after 4 hours following the start of the dissolution testing and were recorded via a standard 35mm camera (AXIOCAM, Germany).
Study D
Examples 12-13
Additional examples of sildenafil citrate tablets according to the invention were made with improved sweller layers containing PEO and the strengthening agent microcrystalline cellulose in varying ratios. The composition of each formulation is shown in Table 10. Ingredients were blended, tablets were pressed, and coatings were applied using procedures described above. The so-formed tablets each contained 200 mgA sildenafil.
Table 10

The tablets of Examples 12 and13 were tested in vitro using direct drug analysis as described hereinbefore, in gastric medium (0.01 M HCI, 7 g/L NaCI). The relative rates of drug release from the tablets of Examples 12 and 13 are illustrated in Table 11.
Table 11
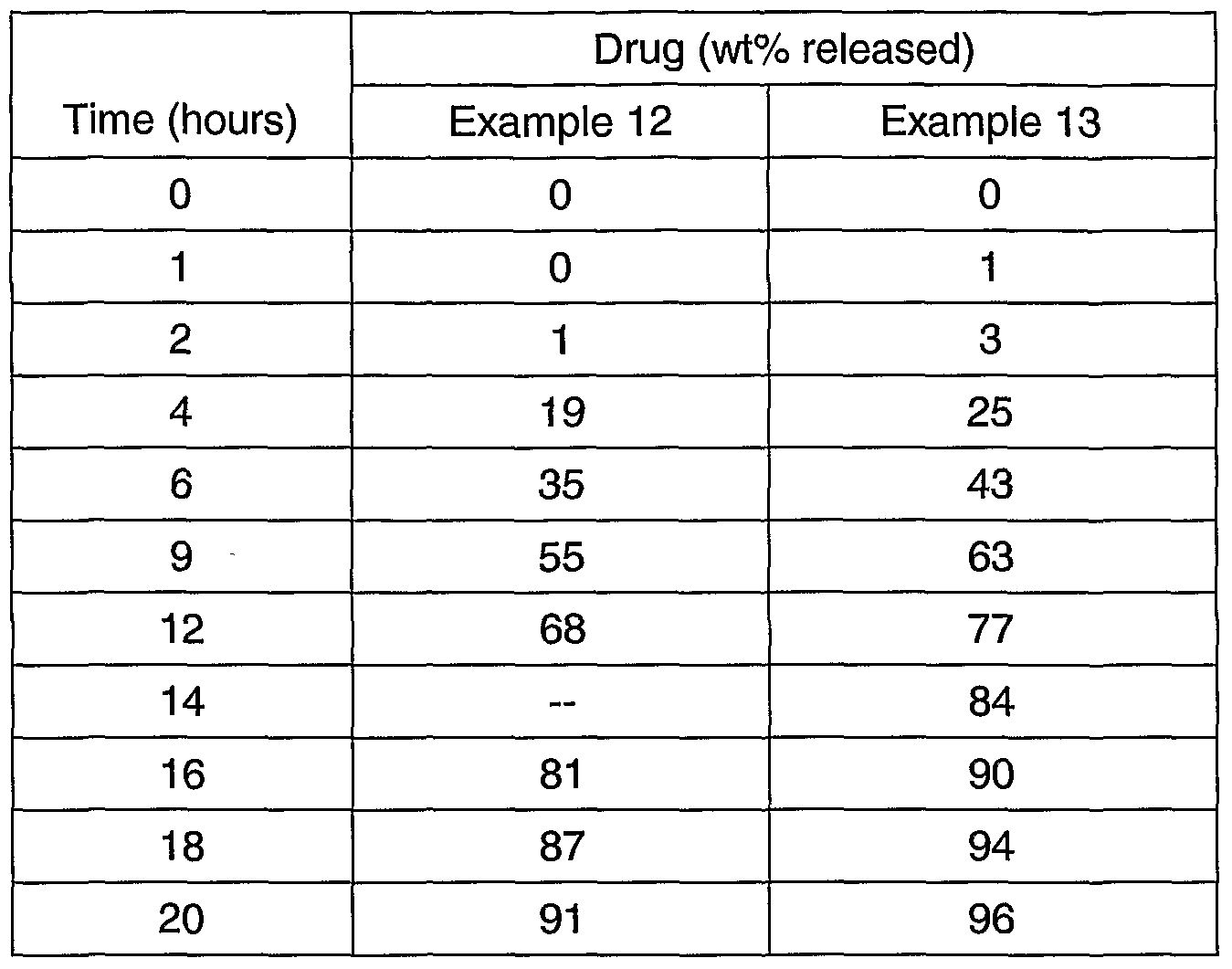
The tablet of Example 12 released sildenafil at an average rate from four to eighteen hours of 4.9 wt%/hour, and the tablet of Example 13 released sildenafil at an average rate from four to eighteen hours of 4.9 wt%/hour. In addition, the tablets of Examples 12 and 13 released greater than 90 wt% sildenafil by 20 hours.
Study E
Example 14
Exemplary tablets according to the invention were made in which the drug layer was processed using an extrusion-granulation procedure to reduce delamination of the drug layer and sweller layer.
To form the drug layer the following materials were blended: 50.3 wt% of sildenafil citrate, 17.2 wt% PEO with an average molecular weight of 600,000, 17.5 wt% xylitol (Xylitab 200), 10.0 wt% of the crystallization inhibiting agent HPMC, 4 wt% of the disintegrant sodium starch glycolate (Explotab), and 1 wt% of the lubricant magnesium stearate. The drug-containing composition ingredients were first combined without the magnesium stearate, and this preblend feed was delivered to a B&P 19-mm twin-screw extruder at a rate of 25 g/min. A mixture of isopropyl alcohol: water (85:15) was added
to the extruder at a rate of 6 g/min. The screw speed was set at 80 rpm. The particles formed by the extruder were placed in a shallow tray and dried in a 400C convection oven for 16 hours. Next, the particles were passed through a hammer mill (Fitz model L1A), with knives forward at 1500 rpm, using a 0.03" screen. The collected granulation was weighed, magnesium stearate was added, and the drug-containing composition was blended for 4 minutes in a TURBULA mixer.
To form the water-swellable composition, the following materials were blended: 65 wt% PEO with an average molecular weight of 5,000,000, 34.3 wt% of the strengthening agent microcrystalline cellulose (Avicel PH 200), 0.5 wt% magnesium stearate, and 0.2 wt% Blue Lake #2. The tablet was formed using 570 mg of the drug layer composition and 200 mg of the sweller layer composition, with 12 mm standard round concave tooling. The tablets were coated as described above, and one 900 μm diameter hole was mechanically-drilled in the coating on the drug-containing composition side of the tablet. The coating was 8 wt% of the uncoated tablet core weight. The so-formed tablet contained 200 mgA sildenafil.
In vitro tests were performed using direct drug analysis, as described hereinbefore, in gastric medium (0.01 M HCI, 7 g/L NaCI). Drug release is illustrated in Table 12.
Table 12
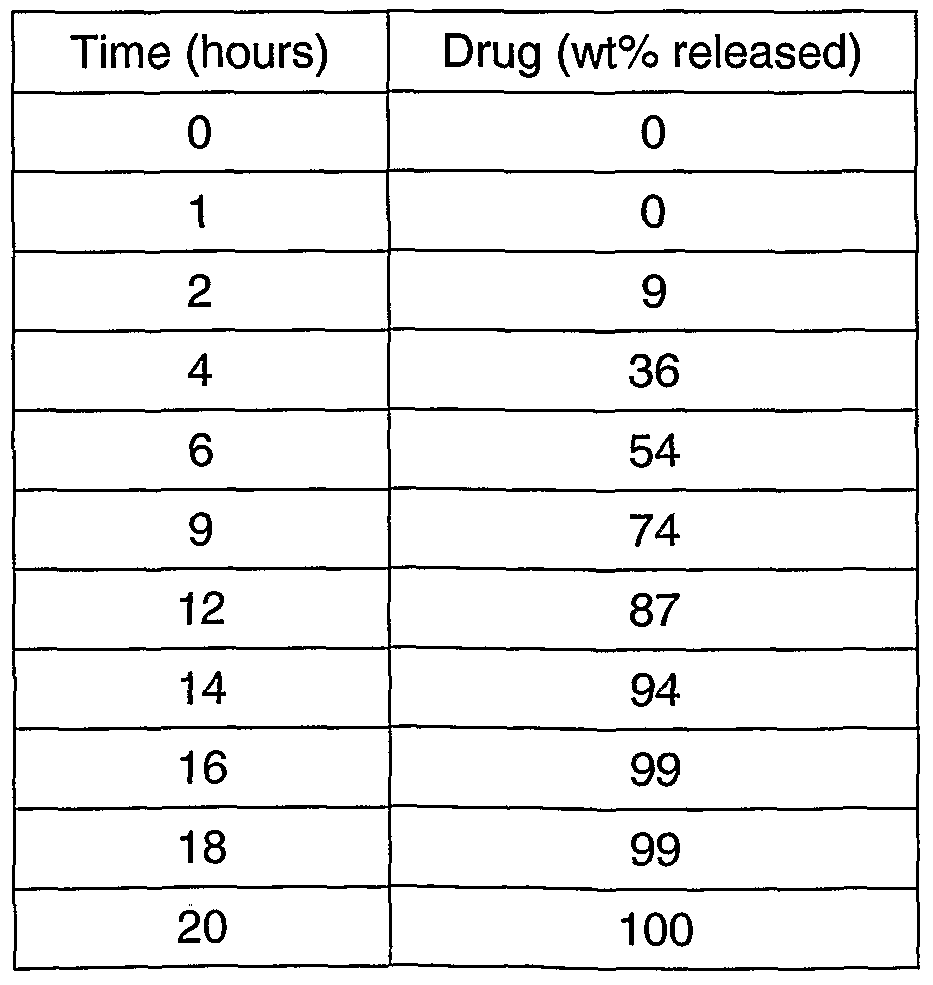
Study F Example 15
The tablets of Example 15 were made with the crystallization inhibiting agent hydroxy propylmethyl cellulose acetate succinate (HPMCAS)(AQOAT-HF manufactured by Shin Etsu; the high designation (H) refers to the relative pH of dissolution, and the fine designation (F) refers to the powder form).
To form the drug layer the following materials were blended: 45 wt% of sildenafil citrate, 24.5 wt% PEO with an average molecular weight of 600,000, 24.5 wt% xylitol (Xylitab 200), 5 wt% HPMCAS-HF, and 1 wt% magnesium stearate. The drug layer ingredients were first combined without the magnesium stearate and blended for 20 minutes in a TURBULA mixer. This blend was pushed through a #20 screen, then blended again for 20 minutes in the same mixer. Next, magnesium stearate was added and the drug layer was blended again for 4 minutes in the same mixer. To form the sweller layer, the following materials were blended: 65 wt% PEO with an average molecular weight of 5,000,000, 34.3 wt% of the strengthening agent microcrystalline cellulose (Avicel PH 200), 0.5 wt% magnesium stearate, and 0.2 wt% Blue Lake #2. The PEO, microcrystalline cellulose, and Blue Lake dye were combined and blended for 20 minutes in a TURBULA mixer. Next, the magnesium stearate was added. All ingredients were pushed through a screen, then blended again for 4 minutes in the same mixer.
The tablet cores were formed by placing 316 mg of the drug layer composition in a 3/8 inch standard round concave die and gently leveling with the press. Then, 106 mg of the sweller layer composition was placed in the die on top of the drug layer. The tablet core was then compressed to a hardness of about 8 Kp. The resulting bi-layer tablet core had a total weight of 422 mg and contained a total of 33.70 wt% sildenafil citrate (142 mg), 18.34 wt% PEO 600,000, 18.34 wt% xylitol, 3.74 wt% HPMCAS, 16.33 wt% PEO 5,000,000, 8.62 wt% microcrystalline cellulose, 0.88 wt% magnesium stearate, and 0.05 wt% blue lake dye. Assays of three tablets averaged 103 ± 4.9 mg of active drug (mgA).
Coatings were applied by a Vector LDCS-20 pan coater. The coating solution contained CA (CA 398-10 from Eastman Fine Chemical, Kingsport, Tennessee), polyethylene glycol (PEG 3350, Union Carbide), water, and acetone in a weight ratio of 3.5/1.5/3/92 (wt%). The flow rate of the inlet heated drying air of the pan coater was set at 40 ft3/min with the outlet temperature set at 25°C. Nitrogen at 20 psi was used to atomize the coating solution from the spray nozzle, with a nozzle-to-bed distance of 2 inches. The pan rotation was set to 20 rpm. The so-coated tablets were dried at 500C in a convection oven. The final dry coating weight amounted to 10 wt% of the uncoated tablet core. One 900 μm diameter hole was then laser-drilled in the coating on the drug-containing composition side of the tablet to provide a delivery port.
In vitro tests were performed using direct drug analysis to determine drug release rates and the results are illustrated in Table 13.
Table 13

The tablet of Example 15 released sildenafil at an average rate from two to twelve hours of 7.8 wt%/hour. The tablet of Example 15 released 97 wt% of sildenafil by twelve hours, and had released 97 wt% by 20 hours.
Studv G
Example 16
The tablet of Example 16 was made with a drug-containing composition that included hydroxypropyl methyl cellulose (HPMC)(E3 Prem Methocel®, manufactured by Dow Chemical Company, Midland, Michigan) as the crystallization inhibiting agent.
To form the drug layer the following materials were blended as described hereinbefore: 49.6 wt% of sildenafil citrate, 17.9 wt% PEO with an average molecular weight of 600,000, 17.5 wt% xylitol (Xylitab 200), 10.0 wt% HPMC, 4 wt% sodium starch glycolate (Explotab), and 1 wt% magnesium stearate. The sweller layer was also made as described above, and consisted of the following materials: 65 wt% PEO with an average molecular weight of 5,000,000, 34.3 wt% microcrystalline cellulose (Avicel PH 200), 0.5 wt% magnesium stearate, and 0.2 wt% Blue Lake #2. Tablets were formed using 570 mg of the drug layer composition and 200 mg of the sweller layer composition, with 15/32" standard round concave tooling. Tablets were coated as described above, and the coating was 11 wt% of the uncoated tablet core weight.
In vitro tests were performed using direct drug analysis to determine drug release rates. Samples were analyzed by HPLC using the method described hereinbefore, and the results are illustrated in Table 14.
Table 14


The tablet of Example 16 released sildenafil at an average rate from two to twelve hours of 7.6 wt%/hour. The tablet of Example 16 released 82 wt% of sildenafil by twelve hours, and had released 98 wt% by 20 hours.
Study H
Examples 17-19
Further Tablets according to the present invention were made using drug-entraining agents of various molecular weights. The composition of each formulation is shown in Table 15. Ingredients were blended, tablets were pressed, and coatings were applied using procedures described hereinbefore. In Example 17, the drug-entraining agent was PEO with a molecular weight of 200,000; in Example 18, the drug-entraining agent was PEO with a molecular weight of 400,000; and in Example 19, the drug-entraining agent was PEO with a molecular weight of 600,000.
Table 15

Table 16

As illustrated by the data in Table 16, PEO 400,000 and PEO 600,000 in the drug layer gave similar performance. However, PEO 200,000 resulted in less release of drug from the formulation.
The tablet of Example 17 released sildenafil at an average rate from two to twelve hours of 7.1 wt%/hour; the tablet of Example 18 released sildenafil at an average rate from two to twelve hours of 7.9 wt%/hour; and the tablet of Example 19 released sildenafil at an average rate from two to twelve hours of 7.6 wt%/hour. The tablet of Example 17 released 89 wt% of sildenafil by twelve hours, and had released 90 wt% by 20 hours. The tablet of Example 18 released 86 wt% of sildenafil by twelve hours, and had released 100 wt% by 20 hours. The tablet of Example 19 released 82 wt% of sildenafil by twelve hours, and had released 98 wt% by 20 hours.
Studyl
Examples 20 and 21
Tablets containing 100 mgA of sildenafil citrate are prepared with the composition shown in Table 17. The ingredients were blended, the tablets were pressed and the coatings were applied using the procedures described hereinbefore. The 100-mgA tablet cores are made with 9.5-mm-diameter standard round concave tooling and weighed 385 mg.
Table 17

* LEO designates a low ethylene oxide grade of PEO.
The tablet of Example 20 was coated to achieve a twelve hour release. A coating solution was prepared containing 53.9 mg cellulose acetate (CA 398-10), 23.1 mg polyethylene glycol (PEG 3350), 46.2 mg water and 1416.8 mg acetone (for a weight ratio of 3.5/1.5/3/92 of CA/PEG/water/acetone). The coating was applied in an amount of 77 mg, or 20 wt% of the uncoated core weight.
The tablet of Example 21 was coated to achieve an eighteen hour release. A coating solution was prepared containing 30.8 mg cellulose acetate (CA 398-10), 7.7 mg polyethylene glycol (PEG 3350), 23.1 mg water and 708.4 mg acetone (for a weight
ratio of 4/1/3/92 of CA/PEG/water/acetone). The coating was applied in an amount of 38.5 mg, or 10 wt% of the uncoated core weight.
The tablet of Example 20 was tested in vitro using direct drug analysis as described hereinbefore, in gastric medium (0.01 M HCI, 7 g/L NaCI) and the rate of drug released over time is illustrated in Table 18.
Table 18

The tablet of Example 20 released sildenafil at an average rate from two to twelve hours of 6.4 wt%/hour and had released 82 wt% of sildenafil by sixteen hours, and had released 95 wt% by 20 hours.
Study J
Examples 22 and 23
Tablets containing 200 mgA of sildenafil citrate were prepared with the composition shown in Table 19. The ingredients were blended, the tablets were pressed, and the coating was applied using the procedures described hereinbefore. The 200-mgA tablet cores were made with 12-mm-diameter standard round concave tooling and weighed 770 mg.
Table 19

The tablet of Example 22 was coated to achieve a twelve hour release. A coating solution was prepared containing 43.1 mg cellulose acetate (CA 398-10), 18.5 mg polyethylene glycol (PEG 3350), 37.0 mg water and 1133.4 mg acetone (for a weight ratio of 3.5/1.5/3/92 of CA/PEG/water/acetone). The coating was applied in an amount of 61.6 mg, or 8.0 wt% of the uncoated core weight.
The tablet of Example 23 was coated to achieve an eighteen hour release. A coating solution was prepared containing 80.9 mg cellulose acetate (CA 398-10), 34.6 mg polyethylene glycol (PEG 3350), 69.3 mg water and 2125.2 mg acetone (for a weight ratio of 3.5/1.5/3/92 of CA/PEG/water/acetone). The coating was applied in an amount of 115.5 mg, or 14.3 wt% of the uncoated core weight.
The tablet of Example 22 was tested in vitro using direct drug analysis as described hereinbefore, in gastric medium (0.01 M HCI, 7 g/L NaCI) and the rate of drug released over time is illustrated in Table 20.
Table 20

The tablet of Example 22 released sildenafil at an average rate from four to eighteen hours of 6.4 wt%/hour and had released 88 wt% of sildenafil by sixteen hours, 94 wt% by eighteen hours and had released 97 wt% by 20 hours.
Study K
Tablet compacts were formed to evaluate the hardness and friability of two different sweller layer compositions. Compact A, a sweller layer according to the present invention, contained PEO and the strengthening agent microcrystalline cellulose (Avicel). Reference Compact B, for comparison, contained the highly swelling material sodium starch glycolate (Explotab) and silicified microcrystalline cellulose (Prosolv).
To form the compacts, ingredients were weighed and blended for 20 minutes using a Turbula mixer. Compact A contained 65.0 wt% PEO (Polyox WSR Coagulant, available from Dow Chemical Co., Midland, Ml), 34.3 wt% Avicel PH102 (available from FMC Corp., Philadelphia, PA), 0.2 wt% Blue #2 Lake, and 0.5 wt% magnesium stearate. Reference Compact B contained 50 wt% Explotab (available from JRS Pharma, Patterson, NY) and 50 wt% Prosolv 90 (also available from JRS Pharma). Using a Killian press, single station, manual fill, with a 25,000 tab/hr setting, and a 13/32" SRC die, 400 mg compacts A and B were individually weighed and pressed.
Hardness was measured using a Schleuniger hardness tester, and friability was measured using an Electrolab Friabilator EF2, with a setting of 100 revolutions. The results are shown in Table 21.
Table 21

The results illustrated in Table 21 show improved hardness and friability with Compact A, the sweller layer according to the present invention.
The data in Table 22 compares the % recovery Compact C, a sweller layer according to the present invention, and Reference Compacts D and E.
Table 22

The data in Table 22 refers to the percentage recovery of the tabletted sweller layer formulations in the axial and radial planes. The reference sweller layers comprising NaCI/PEO and Explotab/Prosolv show much greater relaxation (particularly in the axial plane) compared to the Avicel/PEO sweller layer formulation according to the present invention.
In a further study, crowning during compaction of the sweller layer was investigated. Crowning produces a pronounced ridge that is likely to chip off during tumbling. The formulations listed in Table 23 were compressed at a known compression force and crowning was assessed visually by observing any sharp edges around the tablet rim.
Table 23

PEO Coagulant = Polyox WSR Coagulant (5,000,000 MW), Dow Chemical, Midland,
2 DiCaI = calcium phosphate dibasic, anhydrous ("ATab") Rhodia Specialty Phosphates (Cranbury, NJ)
The results of the tests are illustrated in Figure 4. Tablets prepared with the compacts according to the present invention could be compressed to significantly greater hardness before crowning was observed in comparison to the tablets prepared with the reference compact.
Claims
1. An osmotic bi-layer tablet comprising (A) an inner core containing a drug- containing layer and a water-swellable layer, and (B) a water-permeable outer coating including at least one delivery port, wherein:
(i) said drug-containing layer comprises:
(a) a drug substance or a pharmaceutically acceptable salt thereof, having a solubility in water of no more than 140 mg/mL at any pH in the range 2 to 7.5, at from about 40% to about 80% by weight of drug-containing layer;
(b) polyethylene oxide polymer having an average molecular weight from about 200,000 to 600,000Da at from about 10% to about 30% by weight of drug-containing layer; and
(c) osmogen at from about 10% to 40% by weight of drug-containing layer;
(ii) said water-swellable layer comprises:
(a) polyethylene oxide polymer having a weight average molecular weight of from about 4,000,000 to about 7,000,000Da at from about 35% to about
80% by weight of water-swellable layer;
(b) a sweller layer strengthening agent at from about 5% to about 60% by weight of water-swellable layer; and
(iii) the weight ratio of said drug-containing layer to said water-swellable layer is in the range of from about 2:1 to about 4.5:1.
2. An osmotic bi-layer tablet according to claim 1 wherein the drug substance or pharmaceutically acceptable salt thereof has a solubility in water of no more than 80 mg/mL at any pH in the range 2 to 7.5.
3. An osmotic bi-layer tablet according to claim 2 wherein the drug substance or pharmaceutically acceptable salt thereof has a solubility in water of no more than 40 mg/mL at any pH in the range 2 to 7.5.
4. An osmotic bi-layer tablet according to claim 3 wherein the drug substance or pharmaceutically acceptable salt thereof has a solubility in water of no more than 20 mg/mL at any pH in the range 2 to 7.5.
5. An osmotic bi-layer tablet according to any of the preceding claims wherein the drug substance or pharmaceutically acceptable salt thereof is present at from about 45% to about 60% by weight of drug containing layer.
6. An osmotic bi-layer tablet according to claim 5 wherein the drug substance or pharmaceutically acceptable salt thereof is present at from about 45% to about
55% by weight of drug containing layer.
7. An osmotic bi-layer tablet according to any of the preceding claims wherein the polyethylene oxide polymer in the drug layer is present at from about 15% to about 25% by weight of the drug containing layer.
8. An osmotic bi-layer tablet according to claim 7 wherein the polyethylene oxide polymer in the drug layer is present at from about 18% to about 23% by weight of the drug containing layer.
9. An osmotic bi-layer tablet according to claim 8 wherein the polyethylene oxide polymer in the drug layer is present at about 20% by weight of the drug containing layer.
10. An osmotic bi-layer tablet according to any of the preceding claims wherein the polyethylene oxide polymer in the drug layer has an average molecular weight from about 400,000 to 600,000Da.
11. An osmotic bi-layer tablet according to any of the preceding claims wherein the osmogen in the drug-containing layer is a sugar.
12. An osmotic bi-layer tablet according to claim 11 wherein the osmogen in the drug-containing layer is xylitol.
13. An osmotic bi-layer tablet according to any of the preceding claims wherein the drug containing layer additionally comprises a drug solubility/wettability enhancing agent at from about 5% to about 20% by weight of drug containing layer.
14. An osmotic bi-layer tablet according to claim 13 wherein the drug containing layer additionally comprises a drug solubility/wettability enhancing agent at from about 5% to about 15% by weight of drug containing layer.
15. An osmotic bi-layer tablet according to claim 14 wherein the drug containing layer additionally comprises a drug solubility/wettability enhancing agent at from about 8% to about 12% of drug containing layer.
16. An osmotic bi-layer tablet according to claim 15 wherein the drug containing layer additionally comprises a drug solubility/wettability enhancing agent at about 10% by weight of drug containing layer.
17. An osmotic bi-layer tablet according to any one of claims 13 to 16 wherein the drug solubility/wettability enhancing agent in the drug containing layer is a hydroxyalkyl cellulose derivative.
18. An osmotic bi-layer tablet according to claim 17 wherein the drug solubility/wettability enhancing agent in the drug containing layer is hydroxypropyl methyl cellulose (HPMC) having a an average molecular weight of from about 50,000 to about 100,000Da.
19. An osmotic bi-layer tablet according to any one of claims 13 to 18 wherein the drug substance or pharmaceutically acceptable salt thereof and the drug solubility/wettability enhancing agent are intimately mixed during manufacture of the drug containing layer.
20. An osmotic bi-layer tablet according to any of the preceding claims wherein the weight ratio of polyethylene oxide polymer to sweller layer strengthening agent in the water-swellable layer ranges from about 50:50 to 70:30.
21. An osmotic bi-layer tablet according to claim 20 wherein the weight ratio of polyethylene oxide polymer to sweller layer strengthening agent in the water- swellable layer is about 65:35.
22. An osmotic bi-layer tablet according to any of the preceding claims wherein the polyethylene oxide polymer in the water-swellable layer has a weight average molecular weight of about 5,000,000Da.
23. An osmotic bi-layer tablet according to any of the preceding claims wherein the sweller layer strengthening agent in the water-swellable layer has a water solubility of less than or equal to about 0.1mg/ml at ambient temperature and a pH of from about 5 to about 8 and has a mechanical strength of greater than about 4Mpa at about 0.3 mm/s.
24. An osmotic bi-layer tablet according to claim 23 wherein the sweller layer strengthening agent in the water-swellable layer has a mechanical strength of greater than about 7Mpa at about 0.3 mm/s.
25. An osmotic bi-layer tablet according to any of the preceding claims wherein the sweller layer strengthening agent in the water-swellable layer is selected from: microcrystalline cellulose, di calcium phosphate (DCP), HPMC, croscarmellose sodium.
26. An osmotic bi-layer tablet according to claim 25 wherein the sweller layer strengthening agent in the water-swellable layer is microcrystalline cellulose having a mean particle size of less than about 200 μm and a moisture content of less than about 5%.
27. An osmotic bi-layer tablet according to claim 26 wherein microcrystalline cellulose has a mean particle size of about 50 to about 100 μm.
28. An osmotic bi-layer tablet according to claim 27 wherein the weight ratio of polyethylene oxide polymer to microcrystalline cellulose in the water-swellable layer ranges from about 50:50 to about 70:30.
29. An osmotic bi-layer tablet according to claim 28 wherein the weight ratio of polyethylene oxide polymer to microcrystalline cellulose in the water-swellable layer is about 65:35.
30. An osmotic bi-layer tablet according to claim 25 wherein the sweller layer strengthening agent in the water-swellable layer is microcrystalline cellulose with a nominal mean particle size of 100 μM and a moisture content of less than about 5.0 %w/w.
31. An osmotic bi-layer tablet according to any of the preceding claims wherein the water-swellabe layer further comprises an osmogen.
32. An osmotic bi-layer tablet according to claim 31 wherein the osmogen in the water-swellable layer is an inorganic salt.
33. An osmotic bi-layer tablet according to claim 32 wherein the osmogen in the water-swellable layer is sodium chloride.
34. An osmotic bi-layer tablet according to any of the preceding claims wherein the radial tensile strength of the water-swellable layer is from about 0.5 to about
5MPa.
35. An osmotic bi-layer tablet according to claim 34 wherein the radial tensile strength of the water-swellable layer is from about 0.5 to about 3MPa.
36. An osmotic bi-layer tablet according to any of the preceding claims wherein the percent mechanical recovery along the X-axis of the water-swellable layer following compression is less than about 20% relative to its corresponding in-die dimensions over a humidity range of from about 10% to about 75%RH.
37. An osmotic bi-layer tablet according to any of the preceding claims wherein the intrinsic viscosity of the water-swellable layer, when fully hydrated, is from about 107 to 109cP.
38. An osmotic bi-layer tablet according to any of the preceding claims wherein the weight ratio of said drug-containing layer to said water-swellable layer is in the range of from about 2:1 to about 3:1.
39. An osmotic bi-layer tablet according to any of the preceding claims wherein the difference between the radial tensile strength of the drug-containing layer and the radial tensile strength of the water swellable layer is less than about 1.5MPa.
40. An osmotic bi-layer tablet according to any of the preceding claims wherein the outer coating comprises a water-permeable component and a water-insoluble component.
41. An osmotic bi-layer tablet according to claim 40 wherein the outer coating comprises polyethylene glycol having a molecular weight of about 3350Da as the water-permeable component and cellulose acetate as the water-insoluble component.
42. An osmotic bi-layer tablet according to claim 41 wherein the outer coating the weight ratio of the polyethylene glycol having a molecular weight of about
3350Da and cellulose acetate is from about 10:90 to about 40:60.
43. An osmotic bi-layer tablet according to any of the preceding claims wherein the solid content of the outer coating is less than 20% by weight relative to the weight of the inner bilayer core of the tablet.
44. An osmotic bi-layer tablet according to any of the preceding claims having an aspect ratio of from about 1.3 to about 2.1.
45. An osmotic bi-layer tablet according to any of the preceding claims wherein the hardness of the tablet is greater than about 4kP.
46. An osmotic bi-layer tablet according to any of the preceding claims wherein at least 80% of said low solubility drug is released to the use environment within 6 to 20 hours after introduction to said use environment.
47. An osmotic bi-layer tablet according to any of the preceding claims wherein the drug substance or pharmaceutically acceptable salt thereof is sildenafil free- base or a pharmaceutically acceptable salt thereof.
48. An osmotic bi-layer tablet according to claim 47 wherein the total amount of sildenafil free-base or a pharmaceutically acceptable salt thereof is from about 10Omg to about 400mg.
49. An osmotic bi-layer tablet according to claim 48 wherein total amount of sildenafil free-base or a pharmaceutically acceptable salt thereof is from about 100mg to about 200mg.
50. An osmotic bi-layer tablet according to claim 49 wherein the total amount of sildenafil free-base or a pharmaceutically acceptable salt thereof is from about
100mg to about 150mg.
51. An osmotic bi-layer tablet according to any of claims 47 to 50 containing sildenafil citrate.
52. An osmotic bi-layer tablet according to any of claims 47 to 50 containing sildenafil mesylate.
53. An osmotic bi-layer tablet according to claim 51 wherein the weight ratio of sildenafil citrate : polyethylene oxide polymer in the drug layer is from about 4 :
1 to about 1.7 : 1.
54. An osmotic bi-layer tablet according to claim 53 wherein the weight ratio of sildenafil citrate : polyethylene oxide polymer in the drug layer is about 3.1 : 1 to about 1.8 : 1.
55. An osmotic bi-layer tablet according to any of claims 47 to 53 wherein the weight ratio of sildenafil : polyethylene oxide polymer in the drug layer is about 3.1 : 1 to about 1.2 : 1.
56. An osmotic bi-layer tablet according to claim 55 wherein the weight ratio of sildenafil : polyethylene oxide polymer in the drug layer is about 2.1 : 1 to about 1.2 : 1.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US73770305P | 2005-11-16 | 2005-11-16 | |
| US60/737,703 | 2005-11-16 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007057762A2 true WO2007057762A2 (en) | 2007-05-24 |
| WO2007057762A3 WO2007057762A3 (en) | 2008-04-10 |
Family
ID=38049018
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2006/003255 Ceased WO2007057762A2 (en) | 2005-11-16 | 2006-11-10 | Osmotic bi-layer tablet |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2007057762A2 (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009000216A3 (en) * | 2007-06-28 | 2009-07-16 | Osmotica Costa Rica Sa | A rupturing controlled release device comprising a subcoat |
| WO2009122431A3 (en) * | 2008-02-15 | 2010-01-07 | Sun Pharma Advanced Research Company Ltd., | Oral controlled release tablet with reduced burst effect |
| WO2010115576A1 (en) * | 2009-04-06 | 2010-10-14 | Ratiopharm Gmbh | Melting tablet containing a sildenafil salt |
| WO2011039686A1 (en) * | 2009-09-30 | 2011-04-07 | Pfizer Inc. | Latrepirdine oral sustained release dosage forms |
| WO2014147526A1 (en) * | 2013-03-16 | 2014-09-25 | Pfizer Inc. | Tofacitinib oral sustained release dosage forms |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6764697B1 (en) * | 1991-06-27 | 2004-07-20 | Alza Corporation | System for delaying drug delivery up to seven hours |
| WO2001047498A2 (en) * | 1999-12-23 | 2001-07-05 | Pfizer Products Inc. | Hydrogel-driven layered drug dosage form comprising sertraline |
| GEP20043334B (en) * | 1999-12-23 | 2004-03-10 | Pfizer Prod Inc | Hydrogel-Driven Drug Dosage Form |
| US20050058707A1 (en) * | 2003-08-06 | 2005-03-17 | Iran Reyes | Uniform delivery of topiramate over prolonged period of time with enhanced dispersion formulation |
| DE102005031577A1 (en) * | 2005-07-06 | 2007-01-11 | Bayer Healthcare Ag | Pharmaceutical dosage forms containing a combination of nifedipine and / or nisoldipine and an angiotensin II antagonist |
-
2006
- 2006-11-10 WO PCT/IB2006/003255 patent/WO2007057762A2/en not_active Ceased
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009000216A3 (en) * | 2007-06-28 | 2009-07-16 | Osmotica Costa Rica Sa | A rupturing controlled release device comprising a subcoat |
| US8637080B2 (en) | 2007-06-28 | 2014-01-28 | Osmotica Kereskedelmi és Szolgáltató, KFT | Rupturing controlled release device comprising a subcoat |
| WO2009122431A3 (en) * | 2008-02-15 | 2010-01-07 | Sun Pharma Advanced Research Company Ltd., | Oral controlled release tablet with reduced burst effect |
| WO2010115576A1 (en) * | 2009-04-06 | 2010-10-14 | Ratiopharm Gmbh | Melting tablet containing a sildenafil salt |
| WO2011039686A1 (en) * | 2009-09-30 | 2011-04-07 | Pfizer Inc. | Latrepirdine oral sustained release dosage forms |
| WO2014147526A1 (en) * | 2013-03-16 | 2014-09-25 | Pfizer Inc. | Tofacitinib oral sustained release dosage forms |
| US9937181B2 (en) | 2013-03-16 | 2018-04-10 | Pfizer Inc. | Tofacitinib oral sustained release dosage forms |
| US10639309B2 (en) | 2013-03-16 | 2020-05-05 | Pfizer Inc. | Tofacitinib oral sustained release dosage forms |
| EP2968155B1 (en) | 2013-03-16 | 2021-03-03 | Pfizer Inc | Tofacitinib oral sustained release dosage forms |
| EP3854400A1 (en) * | 2013-03-16 | 2021-07-28 | Pfizer Inc. | Tofacitinib oral sustained release dosage forms |
| US11253523B2 (en) | 2013-03-16 | 2022-02-22 | Pfizer Inc. | Tofacitinib oral sustained release dosage forms |
| EP4249055A3 (en) * | 2013-03-16 | 2023-12-27 | Pfizer Inc. | Tofacitinib oral sustained release dosage forms |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2007057762A3 (en) | 2008-04-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1242055B1 (en) | Hydrogel-driven drug dosage form | |
| US20040052845A1 (en) | Hydrogel-driven drug dosage form | |
| EP1027888B1 (en) | Osmotic system for delivery of solid amorphous dispersions of drugs | |
| US7550158B2 (en) | Controlled release by extrusion of solid amorphous dispersions of drugs | |
| EP1027887B1 (en) | Matrix controlled release device | |
| US20040121015A1 (en) | Controlled-Release of an active substance into a high fat environment | |
| TW200302748A (en) | Osmotic delivery system | |
| WO2007057762A2 (en) | Osmotic bi-layer tablet | |
| WO2021014360A1 (en) | Oral modified release dosage forms |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 06809236 Country of ref document: EP Kind code of ref document: A2 |